
Darstellung und Charakterisierung metallbeladener Nafion ... · Säuren sind wässrige Säuren, wie...
173
Darstellung und Charakterisierung metallbeladener NafionP ® P/Silikat-Verbundwerkstoffe DISSERTATION zur Erlangung des Grades eines Doktors der Naturwissenschaften der Abteilung Chemie der Ruhr-Universität Bochum vorgelegt von Diplom-Chemikerin KATRIN FREITAG aus Zwickau Mülheim an der Ruhr 2003
Transcript of Darstellung und Charakterisierung metallbeladener Nafion ... · Säuren sind wässrige Säuren, wie...

Darstellung und Charakterisierung metallbeladener Nafion P
®P/Silikat-Verbundwerkstoffe
DISSERTATION
zur Erlangung des Grades eines Doktors der Naturwissenschaften
der Abteilung Chemie der Ruhr-Universität Bochum
vorgelegt von Diplom-Chemikerin
KATRIN FREITAG
aus Zwickau
Mülheim an der Ruhr
2003

Meinen Eltern

Referent: Professor Dr. Manfred T. Reetz
Korreferent: Professor Dr. Martin Feigel
Tag der mündlichen Prüfung: 29. Januar 2004

Die vorliegende Arbeit wurde in der Zeit von Dezember 2000 bis Dezember
2003 am Max-Planck-Institut für Kohlenforschung in Mülheim/Ruhr unter der
Leitung von Herrn Prof. Dr. M. T. Reetz angefertigt.
Herrn Prof. Dr. M. T. Reetz danke ich herzlich für die interessante und
herausfordernde Themenstellung, die gewährte Freiheit bei der Durchführung
der Arbeit, die hervorragenden Arbeitsbedingungen und die stetige
Gesprächsbereitschaft.
Ich danke Herrn Prof. Dr. M. Feigel für die Übernahme des Korreferats sowie
Herrn Prof. Dr. W. S. Sheldrick für sein Auftreten als Drittprüfer.
Der Max-Planck-Gesellschaft zur Förderung der Wissenschaften danke ich für
das mir gewährte Promotionsstipendium.
Allen Mitgliedern des Arbeitskreises danke ich für die gute Zusammenarbeit,
die ständige Hilfsbereitschaft und das gute Arbeitsklima. Ein besonderer Dank
gilt meinen Bürokollegen C. Torre, A. Pletsch und T. Schneider für viele
anregende Diskussionen bei literweise Kaffee und die generell angenehme
Atmosphäre im Büro 3.31. Den Mitarbeitern aus Box 2, G. Mehler,
K. Sommer, P. Tielmann und W. Wiesenhöfer, danke ich für die gute
Zusammenarbeit.
Frau A. Rathofer und Frau E. Enk danke ich herzlich für ihre Hilfe und Unter-
stützung in jeglicher Situation.
Herrn Professor Dr. G. Fink, Frau D. Ferrari, Herrn U. Blumenthal und Herrn
P. Jödicke danke ich für die Zusammenarbeit auf dem Gebiet der Polymeri-
sationskatalysatoren.
Mein besonderer Dank gilt Herrn Dr. B. Tesche, dem Leiter der Abteilung für
Elektronenmikroskopie am Max-Planck-Institut für Kohlenforschung. Frau
M. Germann und Herrn H. J. Bongard danke ich herzlich für die Einführung in
die Welt der Mikrotom-Schnitte. H. J. Bongard danke ich außerdem für die

SEM-Aufnahmen. Weiterer herzlicher Dank gilt Herrn B. Splithof und Herrn
A. Dreier für die gewissenhafte Anfertigung zahlreicher TEM-Aufnahmen.
Frau Dr. C. Weidenthaler und Frau S. Palm aus der Abteilung für Kristallo-
graphie des Max-Planck-Instituts für Kohlenforschung danke ich herzlich für
die Durchführung zahlreicher XPS- und XRD-Messungen.
Für die Anfertigung der BET-Messungen und die Hilfe bei den thermo-
gravimetrischen Analysen bedanke ich mich herzlich bei Herrn K. Schlichte.
Den analytischen Abteilungen des Max-Planck-Instituts für Kohlenforschung
gilt mein Dank für das Engagement und die zahlreichen Untersuchungen, die
zum Gelingen dieser Arbeit beigetragen haben.
Herzlich bedanke ich mich bei den Mitarbeitern der Werkstätten und des
Drucktechnikums für die technische Unterstützung.
Für die Hilfe bei EDV-Problemen danke ich unseren Administratoren, den
Herren M. Hermes und H. Lenk.
Den Hauptkorrektoren, T. Schneider und C. Torre, gilt mein besonderer Dank
für das zügige und gewissenhafte Lesen sowie die kritischen und hilfreichen
Anregungen. Frau Dr. C. Weidenthaler danke ich für die Korrektur der Ab-
schnitte zu den XRD- und XPS-Untersuchungen.
Herzlich bedanke ich mich bei meinen Eltern und meiner Schwester für die
Unterstützung während meines Studiums, ohne die diese Arbeit nicht möglich
gewesen wäre.

I
TInhaltsverzeichnisT
1 Einleitung.......................................................................................1
1.1 Die Supersäure NafionP®P ..................................................................1
1.2 Vergleich der homogenen und heterogenen Katalyse ....................4
1.3 Immobilisierung mittels des Sol-Gel-Prozesses ..............................5
1.4 Synthese mesoporöser Materialien.................................................7
1.5 Trägerung von Metallen auf Silikat................................................10
2 Aufgabenstellung........................................................................12
3 Synthese und Charakterisierung Nafion P®P-haltiger Silikate .....14
3.1 Synthese ungeordneter Nafion P®P-haltiger Silikate ..........................14
3.1.1 Immobilisierung mittels des Sol-Gel-Verfahrens ...........................14
3.1.2 Trägerung mittels Imprägnierung ..................................................15
3.1.3 Trägerung mittels kovalenter Fixierung.........................................15
3.2 Synthese geordneter Nafion P®P-haltiger Silikate ..............................16
3.3 Eigene Arbeiten ............................................................................16
3.3.1 Synthese ungeordneter Nafion P®P-haltiger Silikate ..........................16
3.3.2 Synthese geordneter Nafion P®P-haltiger Silikate ..............................18
3.4 Charakterisierung Nafion P®P-haltiger Silikate ...................................23
3.4.1 Elementaranalyse .........................................................................23
3.4.2 IR-Spektroskopie ..........................................................................24
3.4.3 Thermogravimetrische Analyse.....................................................26
3.4.4 Ionenaustauschkapazität ..............................................................27
3.4.5 Stickstoff-Sorptions-Messungen ...................................................30
3.4.6 XRD-Messungen...........................................................................36
3.4.7 Elektronenmikroskopische Untersuchungen .................................38
4 Platinsysteme..............................................................................46
4.1 Synthese .......................................................................................47
4.2 Charakterisierung..........................................................................48
4.2.1 Elementaranalyse .........................................................................48
4.2.2 XRD-Untersuchungen...................................................................50
4.2.3 XPS-Untersuchungen ...................................................................52

II
4.2.4 TEM-Untersuchungen...................................................................61
4.3 Katalysen mit Platin-beladenen Nafion P®P-haltigen Silikaten ...........69
4.3.1 Hydrierung von Cycloocten ...........................................................69
5 Zirconiumsysteme ......................................................................75
5.1 Synthese .......................................................................................76
5.2 Charakterisierung..........................................................................78
5.2.1 Elementaranalyse .........................................................................78
5.2.2 SEM-Untersuchungen...................................................................79
5.3 Katalysen ......................................................................................81
5.3.1 Polymerisationen von Ethen .........................................................81
6 Kupfersysteme ............................................................................95
6.1 Synthese .......................................................................................96
6.2 Charakterisierung..........................................................................98
6.2.1 IR-Spektroskopie ..........................................................................98
6.2.2 Elementaranalysen .......................................................................99
6.2.3 XPS-Untersuchungen ...................................................................99
6.3 Katalysen ....................................................................................101
6.3.1 Cyclopropanierungen..................................................................101
7 Kombination von Säure- und Enzym-Katalyse.......................109
7.1 Grundlagen der Enzymkatalyse ..................................................109
7.2 Eigene Untersuchungen..............................................................113
7.2.1 Racemisierung von (S)-Phenylethanol durch Nafion P®P-Systeme..114
7.2.2 Reaktion der NafionP®P-Systeme mit (R)-Phenylethylacetat ..........121
7.2.3 Kombination von immobilisierter CaL B und NR 50 ....................124
7.2.4 Doppel-Immobilisierung des Nafions P®P und CaL B.......................126
8 Zusammenfassung und Ausblick............................................129
9 Experimenteller Teil ..................................................................132
9.1 Allgemeine Hinweise...................................................................132
9.2 Chemikalien ................................................................................133
9.3 Analytik .......................................................................................134
9.3.1 Elementaranalyse .......................................................................134

III
9.3.2 Gaschromatographie (GC)..........................................................134
9.3.3 GC/MS-Kopplung........................................................................134
9.3.4 Infrarotspektroskopie (IR)............................................................134
9.3.5 Kernresonanzspektroskopie (NMR) ............................................135
9.3.6 Transmissionselektronenmikroskopie (TEM) ..............................135
9.3.7 Rasterelektronenmikroskopie (REM) ..........................................136
9.3.8 BET-Messungen .........................................................................136
9.3.9 Röntgen-Photoelektronen-Spektroskopie (XPS).........................136
9.3.10 Röntgen-Pulver-Diffraktometrie (XRD)........................................136
9.3.11 Gelpermeationschromatographie (GPC).....................................136
9.3.12 Bestimmung der Säurestärke der NafionP®P-haltigen Silikate ........137
9.4 Darstellung der NafionP®P-haltigen Silikate ....................................137
9.4.1 AAV 1: Darstellung ungeordneter NafionP®P-haltiger Silikate .........137
9.4.2 AAV 2: Darstellung geordneter, mesoporöser Nafion P®P-haltiger
Silikate ........................................................................................138
9.5 Umsetzung der NafionP®P-haltigen Silikate mit Metallverbindungen
....................................................................................................139
9.5.1 AAV 3: Platinbeladung der NafionP®P-haltigen Silikate...................139
9.5.2 AAV 4: Zirconiumbeladung der Nafion P®P-haltigen Silikate ............140
9.5.3 AAV 5: Kupferbeladung der NafionP®P-haltigen Silikate .................141
9.5.4 AAV 6: Wasserstoff-Reduktion der Platin-beladenen Systeme...142
9.6 Katalysen ....................................................................................142
9.6.1 AAV 7: Hydrierung von Olefinen .................................................142
9.6.2 AAV 8: Cyclopropanierungen......................................................143
9.6.3 Durchführung der Polymerisation................................................144
9.7 Untersuchungen zur Kombination von Säure- und Enzymkatalyse
....................................................................................................148
9.7.1 AAV 9: Racemisierung des (S)-Phenylethanol............................148
9.7.2 AAV 10: Untersuchung der Stabilität des (R)-Esters...................149
9.7.3 Doppel-Immobilisierung von Nafion P®P-Polymer und Candida
antarctica Lipase B .....................................................................150
9.7.4 AAV 11: Kombinierte Säure- und Enzymkatalyse .......................151

IV
10 Anhang.......................................................................................153
10.1 Abkürzungsverzeichnis ...............................................................153
10.2 TBenennung der Sol-Gel-Immobilisate .........................................155
11 Literaturverzeichnis..................................................................157

Einleitung 1
1 Einleitung
Säure-katalysierte Umsetzungen gehören zu den meist genutzten und am
besten untersuchten Reaktionen. Dabei können organische Substrate mit
nukleophilen Reagenzien und einer TLEWIST-Säure als Katalysator in einer
Vielzahl von Umsetzungen reagieren. P
[1]P Klassische Beispiele für TLEWIST-
Säuren sind wässrige Säuren, wie HF und HB2 BSO B4 B, und Metallhalogenide wie
AlClB3 B.
Während der Katalyse kommt es zur Bildung eines Säure-Base-Addukts aus
dem Katalysator und Produkt bzw. Edukt, das nach beendeter Reaktion
zerstört werden muss, um das Produkt freizusetzen. Dadurch wird normaler-
weise die TLEWIST-Säure zerstört und große Mengen an anorganischen Salzen
als Abfall produziert. Da die Reaktionsprodukte meist stärkere Basen als die
Edukte sind, reichen katalytische Mengen der TLEWIST-Säure oft nicht aus,
sondern es müssen sogar stöchiometrische Mengen der Säure eingesetzt
werden. Viele der eingesetzten TLEWIST-Säuren sind außerdem sehr stark
korrosiv.
Daher geht in den letzten Jahren der Trend verstärkt zum Ersatz der
homogenen durch heterogene TLEWIST-Säuren. Durch Einsatz heterogener
Katalysatoren können die Abfallmengen verringert, die Katalysatoren oft
recycelt und Sicherheitsrisiken minimiert werden. Erste Beispiele für den
Einsatz heterogener TLEWIST-Säure-Katalysatoren wurden bereits zu Beginn
des 20. Jahrhunderts in der Petrolchemie beschrieben.P
[2]P
Im Folgenden soll ein heterogener Säure-Katalysator, das Ionenaustauscher-
harz NafionP
®P, und verschiedene Aspekte der heterogenen Katalyse, wie
verschiedene Heterogenisierungsmethoden, beschrieben werden.
1.1 Die Supersäure NafionP
®P
Nafion P
®P, ein perfluoriertes Ionenaustauscherpolymer mit Sulfonsäuregruppen,
wurde in den 1960igern bei TDuPont T durch eine Variation des Teflon P
®P-
Polymers entwickelt. P
[3]P Es handelt sich um ein Copolymer aus Tetrafluorethen
und Perfluor-2-(fluorsulfonylethoxy)propyl-vinylether (vgl. Abbildung 1).

Einleitung 2
CFF2C CF2CF
CF3
O CF2CF2O SO2FF2C CF2
Abbildung 1: Monomere des NafionP
®P-Polymers.
Nafion P
®P war das erste synthetische Polymer, das auch ionische Eigen-
schaften aufwies und begründete damit die Klasse der so genannten
Ionomere. Es weist ein hydrophobes Perfluorpolyethylen-Grundgerüst auf, an
das über Etherbrücken hydrophile Perfluorethylensulfonsäure-Gruppen
gebunden sind. Die chemische Struktur ist in Abbildung 2 dargestellt. Das
Polymer weist eine Äquivalentmasse von etwa 1070 auf.
CF2F2C CF CF2
O CF2 CF CF3
O CF2 CF2 SO3H
n
xmm = 6, 7x ~ 1000n = 1, 2, 3
Abbildung 2: Das perfluorierte Ionenaustauscherpolymer NafionP
®P.
Die exakte Struktur des NafionP
®P-Polymers ist nicht bekannt, es wurden jedoch
verschiedene Modelle entwickelt, um die Anordnung der ionischen Gruppen
innerhalb des Polymers zu beschreiben. So wird angenommen, dass die
ionischen Gruppen durch elektrostatische Wechselwirkungen zu dicht
gepackten Regionen, so genannten Clustern von etwa 30 bis 50 Å,
aggregieren. Diese Cluster sind durch Kanäle miteinander verbunden.P
[4]P
Eine vereinfachte Darstellung nach YEAGERT zeigt diskrete hydrophobe
Regionen, die durch das Fluorkohlenstoff-Rückgrat des Polymers aufgebaut
sind, und hydrophile Regionen, in denen die Sulfonsäure-Gruppen und
Gegenionen zu finden sind (vgl. Abbildung 3). Zwischen diesen Regionen
wird eine Übergangsregion postuliert, in der sich wenige Seitenketten,
Wasser und einige Sulfonat-Gruppen, die nicht in Clustern angeordnet sind,
befinden. P
[5]P

Einleitung 3
Abbildung 3: Schematischer Aufbau des NafionP
®P-Polymers anhand des Drei-Phasen-
Modells nach TYEAGERT (hydrophobe Region: hellgrau, Übergangsregion: grau, hydrophile Region: weiß).P
[5]P
Aufgrund des Fluorkohlenstoff-Rückgrats ist NafionP
®P, ähnlich wie TeflonP
®P,
sehr widerstandsfähig gegen chemische Angriffe. Laut TDuPont T kann es nur
durch Alkalimetalle, besonders Natrium, bei normalem Druck und Temperatur
zersetzt werden. Es kann in Anwendungen bis etwa 190 °C ohne Zersetzung
verwendet werden. Es weist eine hohe Ionenleitfähigkeit auf, wirkt als
Kationenaustauscherharz und stark saurer Katalysator. Die Sulfonsäure-
Gruppen im Fluorkohlenstoff-Rückgrat wirken als extrem starke Protonen-
Donoren aufgrund von Stabilisierungseffekten durch die große Polymermatrix.
Für die Säurestärke der Säurefunktionen des Nafion P
®P-Polymers wurde eine
Hammett-Acidität von 12 angegeben. P
[6]P Dies entspricht der Säurestärke
100 %iger Schwefelsäure.
NafionP
®P-Polymer findet aufgrund seiner Eigenschaften vier Hauptan-
wendungen. So wird es zur Herstellung von Ionenaustauschermembranen für
die Synthese von Chlorgas und Natriumhydroxid in der Elektrolyse von
Salzwasser genutzt, in der Trocknung oder Befeuchtung von Gasen zum
Beispiel für Anästhesiegase verwendet, als Protonenaustauschmembran in

Einleitung 4
Polymer-Elektroden-Brennstoffzellen und als supersaurer Katalysator in der
Herstellung von Feinchemikalien eingesetzt. Bei TExxonT wird zum Beispiel
Formaldehyd mit CO/HB2 BO bei 150 °C mit einer NafionP
®P-Membran zu 70 %
Glykolsäure umgesetzt. P
[7]P
NafionP
®P ist in verschiedenen Formen kommerziell erhältlich. Die Produktion
erfolgt als Salzform mit neutralisierten Sulfonsäure-Gruppen. In dieser Form
ist das Polymer thermoplastisch, d. h. dehnbar und formbar unter Hitze und
Druck, aber nicht chemisch aktiv. Anschließend wird es normalerweise zu
Folien oder dünnen Rohren geformt und durch Überführung in die Säureform
chemisch aktiviert.
Inzwischen sind auch Lösungen des Nafion P
®P-Polymers in wässriger
alkoholischer Lösung mit verschiedenen Wasser- bzw. Alkoholanteilen erhält-
lich. P
[8]P Die Äquivalentmasse des Polymers beträgt 1025 bis 1500. Zur
Gewinnung der Lösung wird das Nafion P
®P-Polymer in beliebiger Form in dem
entsprechenden Lösungsmittel-Gemisch auf Temperaturen zwischen 180 und
300 °C im Autoklaven erhitzt. Für eine bei 240 °C in 18 Stunden dargestellte
Lösung in einem Wasser/TnT-Propanol-Gemisch wurden durch Röntgen-
diffraktion kolloidale Partikel mit einer Größe von 50 Å beobachtet. P
[8]P
1.2 Vergleich der homogenen und heterogenen Katalyse
Klassische heterogene Katalysatoren sind eher beschränkt in der Natur ihrer
aktiven Zentren und der Bandbreite der Reaktionen, die sie katalysieren
können. Lösliche organische Katalysatoren können dagegen eine deutlich
höhere Anzahl von Reaktionstypen katalysieren, können jedoch meist nicht
wiedergewonnen werden. Das Hauptproblem der homogenen Katalyse ist
damit das Abtrennen des meist teuren Katalysators aus der Reaktions-
mischung nach der Umsetzung. Im Gegensatz dazu können heterogene
Katalysatoren durch Filtrieren oder Zentrifugieren vom Reaktionsmedium
getrennt und potentiell für weitere Katalysen eingesetzt werden.P
[9]P Immerhin
basieren 90 % der Katalysen in der chemischen Industrie auf heterogenen
Verfahren. P
[10]P
Um die Vorteile der homogenen Katalysatoren, wie Vielseitigkeit, Flexibilität
der Synthese und hohe Aktivität, mit den Vorteilen der heterogenen

Einleitung 5
Katalysatoren, wie die gute Morphologie, geringeres Reaktor- TFoulingT und
hohe Schüttdichte zu kombinieren, bietet sich die Trägerung homogener
Katalysatoren an. P
[11]P
Heterogene Katalysatoren müssen für industrielle Anwendungen bestimmte
Anforderungen erfüllen. Dazu gehört die thermische, chemische und
mechanische Stabilität des Trägermaterials während der Reaktion. Die
Struktur des Katalysators muss eine hohe Verteilung der katalytisch-aktiven
Zentren auf der Oberfläche und die gute Zugänglichkeit dieser Zentren
gewährleisten. Daher sollte der Katalysator eine spezifische Oberfläche von
über 100 m P
2 PgP
-1P und Porengrößen größer 20 Å aufweisen, so dass die
Diffusion des Substrats zu den aktiven Zentren erleichtert wird. P
[12]P
Als Träger kommen anorganische Materialien, wie Siliciumdioxid, Zeolithe,
Aluminiumoxid, Zirconiumoxid, Zinkoxid oder Ton, aber auch organische
Polymere in Frage.
1.3 Immobilisierung mittels des Sol-Gel-Prozesses
Eines der häufigsten verwendeten Trägermaterialien in der heterogenen
Katalyse sind Silikate. Sie können durch den so genannten Sol-Gel-Prozess
durch Hydrolyse und Polykondensation von Alkoxysilanen dargestellt werden
(vgl. Abbildung 4). P
[13]P Dabei werden zunächst aus dem monomeren Silan-
Precursor durch die Hydrolyse und Polykondensation kolloidale Partikel
gebildet, die als Sol bezeichnet werden. Im Weiteren erhält man durch die
fortschreitende Kondensation und die damit verbundene Vernetzung der
Partikel das so genannte Gel, ein hochviskoses, dreidimensionales Netzwerk.
Erlaubt man die Polykondensation über einen längeren Zeitraum (Alterung),
kommt es zur Bildung des hochvernetzten, amorphen Gelkörpers. Durch das
anschließende Trocknen werden der Alkohol und das Wasser entfernt.

Einleitung 6
Si(OR)4 + H2O (RO)3Si OH + ROH..
.
.
(HO)3Si OR + H2O Si(OH)4 + ROH
Si OR + HO Si
R = H, Alkyl
Si O Si + ROH
n Si(OR)4 + 2n H2OH+ oder OH-
(SiO2)n + 4n ROH
Abbildung 4: Sol-Gel-Prozess mit Hydrolyse und Polykondensation.
Als Katalysator für den Sol-Gel-Prozess werden sowohl Säuren, wie HCl,
HB2 BSO B4 B und HNO B3 B, als auch Basen wie Amine oder NaOH, eingesetzt. Das
Verhältnis von Hydrolyse- und Kondensationsgeschwindigkeit und damit die
Morphologie des Silikates sind abhängig vom Katalysator. So weisen im
basischen Milieu gebildete Silikate hochkondensierte, kolloidale Sol-Gel-
Partikel auf, die im sauren Milieu gebildeten Silikate kleine, hochverzweigte
Sol-Gel-Partikel. P
[13]P
Silikate können je nach Synthesemethode verschiedene Porositäten
aufweisen. Generell unterteilt man in mikro- (Porendurchmesser bis 2 nm),
meso- (2 bis 50 nm) und makroporöse (über 50 nm) Materialien. Es ist eine
möglichst einheitliche Porengröße erwünscht.
Das bekanntes Beispiel für mikroporöse Materialien sind die kristallinen
Zeolithe. Sie finden aufgrund ihrer einheitlichen Porengröße Anwendung in
der selektiven Abtrennung kleiner Moleküle aus Mischungen mit größeren
Molekülen und werden daher auch Molekularsieb genannt. Zeolithe haben
eine breite Anwendung in der industriellen Katalyse gefunden, vor allem in der
Hydrolyse:
Polykondensation:
Gesamtreaktion:

Einleitung 7
Petrolchemie und Raffination. P
[14]P Jedoch erwiesen sie sich für Reaktionen
größerer Moleküle aufgrund der geringen Porengröße als ungeeignet. So
kann es zur Blockierung der Poren und damit der Deaktivierung des
Katalysators kommen.P
[15]P Daher werden seit einigen Jahren Anstrengungen
zur Synthese eines den Zeolithen ähnlichen Materials mit Poren im meso-
porösen Bereich unternommen.
1.4 Synthese mesoporöser Materialien
Die Darstellung geordneter, mesoporöser Silikate und Alumosilikate mit
gleichmäßiger Porenverteilung (M41S-Materialien) wurde zum ersten Mal
1992 durch die TMobil Oil Corporation T veröffentlicht.P
[16-18]P Wahrscheinlich
wurden solche Materialien jedoch bereits 1971 dargestellt, allerdings nicht als
solche erkannt. P
[19]P Weitere frühere Arbeiten durch andere Arbeitsgruppen sind
beschrieben, erreichten jedoch nicht die entsprechende Aufmerksamkeit.P
[20, 21]P
Inzwischen wurde eine Vielzahl mesoporöser Materialien mit gleichmäßiger
Porengrößenverteilung beschrieben. Je nach Mechanismus, pH-Wert und des
benutzten Templats in der Synthese, ihrer Topologie und Porenarchitektur
unterscheidet man sie in Materialen, wie MCM-41, MCM-48, MSU, HMS,
FMS-16, und verschiedene SAB-Materialien.
Das berühmteste Beispiel ist das durch die TMobilT-Forscher beschriebene
MCM-41.P
[16-18]P Es weist eine hexagonale Anordnung von einheitlichen, meso-
porösen (15 bis 100 Å) Kanälen auf, die in transmissionselektronenmikro-
skopischen Aufnahmen deutlich zu sehen sind (vgl. Abbildung 5).
Abbildung 5: TEM-Aufnahme des durch TMobilT-Forscher beschriebenen MCM-41P
[17]P.

Einleitung 8
Zur Synthese dieser Materialien werden Silikat- und eventuell auch
Aluminium-Precursor und ein Tensid, wie quaternäre Ammoniumsalze, einge-
setzt. Es handelt sich daher um eine basische Synthese. Das zunächst
gebildete (Alumo-)Silikat-Gel kann anschließend durch Calcinieren vom
Templat befreit werden. Die Größe der Poren wird durch die Wahl des
Lösungsmittels, der Reaktionsbedingungen, der Kettenlänge des Tensids
oder zugesetzter organischer Hilfsstoffe beeinflusst.
Untersuchungen zur Bildung dieser Materialien zeigten Analogien zu der von
Flüssigkristall-Phasen, weshalb sie durch einen Flüssigkristall-Templat-
Mechanismus beschrieben wird (vgl. Abbildung 6). P
[18]P Zunächst kommt es zur
hexagonalen Anordnung zylindrischer Micellen. Dabei zeigen die polaren
Gruppen der Tensidmoleküle nach außen und die Silikat-Spezies füllen den
Platz zwischen den Zylindern. Beim Calcinieren wird dann das Tensid, das als
Templat in der Synthese diente, herausgebrannt und es bleibt ein
anorganisches Material mit der typischen MCM-41-Struktur.P
[17]P
Abbildung 6: Flüssigkristall-Templat-Mechanismus der Bildung von MCM-41-Materialien.P
[18]P
Dieser Mechanismus wurde jedoch später widerlegt, da sich eine hexagonale
Flüssigkristall-Phase im verwendeten Tensid-Wasser-System mit CB16 BTMACl
bei 25 °C erst bei 40 % Konzentration an C B16 BTMACl bildet, aber MCM-41
auch schon bei 1 Gew.% an Tensid gebildet wird. Bei dieser geringen
Konzentration existieren zwar Micellen in der Lösung, sie bilden aber keine
hexagonale Anordnung aus.
Eine weitere Ursache für die Bildung der hexagonalen Phase könnte nach
TMobilT in den Silikat-Spezies liegen. P
[17, 18]P Untersuchungen zeigten, dass

Einleitung 9
zunächst zufällig verteilte stäbchenförmige Tensid-Micellen gebildet werden,
die mit den Silikat-Oligomeren wechselwirken. Auf diese Weise werden
zufällig geordnete Micellen umgeben von einer Silikatschicht geformt. Durch
die basenkatalysierte Kondensation zwischen den Silikat-Spezies benach-
barter Stäbchen wird eine weitreichende hexagonale Anordnung ausgelöst,
da dies eine energetisch günstigen Konfiguration darstellt. P
[22]P
Abbildung 7: Aggregation von Silikat-umhüllten Stäbchen.P
[22]P
Inzwischen sind eine große Anzahl mesoporöser, geordneter Materialien
beschrieben. Sie weisen zum Beispiel verschiedene Zusammensetzungen,
Porengrößen und Wanddicken auf, je nach verwendeter Synthesemethode.
Eine häufig verwendete Einordnung richtet sich nach dem eingesetzten
Templat. Für die M41S-FamilieP
[17, 18]P, zu der MCM-41 und MCM-48 gehört,
werden als Templat langkettige Alkylammonium-Tenside, für HMSP
[23]P-
Materialien langkettige aliphatische Amine, für MSU- P
[24]P und SBAP
[25-27]P-
Materialien Poly(ethylenoxid)-Tenside oder deren Copolymer benutzt.
Es sind nicht nur Silikate mit uniformen Mesoporen, sondern auch andere
Metalloxide wie AlB2 BO B3 B, ZrOB2 B, TiOB2 B, aber auch SnSB2 B und SnO B2 B bekannt. P
[28]P Aber
nicht nur mesoporöse Metalloxide, sondern auch Metalllegierungen und
organisch-anorganische Hybridmaterialien wurden inzwischen beschrieben.
Auch die Darstellung kontrollierter Morphologien, zum Beispiel in Form von
Stäbchen oder Kugeln ist möglich.P
[29]P
Die geordneten, mesoporösen Materialien fanden bisher Anwendung in der
Katalyse, dem Ionenaustausch und der Separation, zum Beispiel als
stationäre Phase in der HPLC. P
[30]P Für den Einsatz in der heterogenen
Katalyse kommen zum einen die reinen (Alumo-)Silikaten in Frage. Zum
anderen können weitere Metalle aufgebracht werden, um ein breites
Spektrum an katalytischen Reaktionen zu ermöglichen. P
[31, 32]P

Einleitung 10
1.5 Trägerung von Metallen auf Silikat
Die Heterogenisierung klassischer homogener Metall-Katalysatoren ermög-
licht die leichtere Rückgewinnung für weitere Katalysen und verhindert die
Produktverschmutzung durch den Katalysator. In der Literatur ist eine Vielzahl
an Methoden zur Heterogenisierung beschrieben, die in vier Typen eingeteilt
werden können. So kann die katalytisch-aktive Spezies mittels Adsorption
oder Ionenpaar-Bildung in den Poren des Trägermaterials immobilisiert
werden, eine kovalente Bindung der katalytischen Funktionalität an den
Träger erfolgen, die Spezies direkt während der Synthese in das
Trägermaterial eingeschlossen oder schrittweise in den Poren des Trägers
aufgebaut werden (Tship in the bottleT).P
[9, 12, 31, 32]P
Die Adsorption von Metall-Spezies auf die Silikatoberfläche gehört zu den
klassischen Methoden der Trägerung. Dabei wird das Trägermaterial mit einer
Lösung eines Metall-Precursern imprägniert und anschließend calciniert. Das
Hauptproblem dieser Methode ist das unerwünschte Cluster-Wachstum
während der Ablagerung der Metall-Spezies. Um höhere Dispersionen zu
erreichen wurden Ablagerungen von leicht-flüchtigen Metall-Precursern aus
der Gasphase ( TChemical Vapor DepositionT, CVD) beschrieben.P
[33]P
Der Einschluss von Nanopartikeln in ein Silikat kann durch die direkte
Immobilisierung während des Sol-Gel-Prozesses erreicht werden. Jedoch ist
auf diese Weise nicht unbedingt eine gute Zugänglichkeit der katalytischen
Spezies gewährleistet, da sie durch Silikat verdeckt sein kann. Außerdem
wird häufig die Bildung von Partikeln gleichmäßiger Größe verhindert.P
[34]P
Durch die Silanolgruppen auf der Silikatoberfläche können katalytische
Spezies über eine kovalente Bindung auf das Trägermaterial aufgebracht
werden. Dabei können die katalytisch-aktiven Spezies direkt mit den
Silanolgruppen reagieren oder zunächst organische Gruppen fixiert werden,
mit denen der Metall-Precursor in einem weiteren Schritt reagiert.
Das Aufbringen wohl definierter, metallorganischer Spezies auf Silikat-
oberflächen wurde vor allem durch TBASSET T im Rahmen der so genannten
organometallischen Oberflächenchemie untersucht.P
[10]P In dieser Arbeitsgruppe
wurden verschiedene Metalle über die Oberflächen-Silanolgruppen auf Silikat
aufgebracht (vgl. Abbildung 8) und durch eine Vielzahl von Analysemethoden

Einleitung 11
charakterisiert. Zum Verständnis der Prozesse während der Trägerung
werden molekulare Modellsysteme mit Silanolgruppen eingesetzt. Durch die
so ermöglichte, genaue Charakterisierung der katalytisch-aktiven Zentren
können der Verlauf der Katalyse untersucht, Struktur-Aktivitäts-Beziehungen
und Elementarschritte der Reaktion aufgeklärt werden.
Träger
OH
+ RxMLn
Träger
OM
Lm
Rx-1
RH
Abbildung 8: Fixierung metallorganischer Spezies auf der Silikatoberfläche.
Das Aufbringen organischer Gruppen auf Silikate hat sich inzwischen zu einer
wichtigen Methode zur Fixierung von Metall-Spezies entwickelt. Es wurde
eine Vielzahl von Funktionen beschrieben, die nach dem in Abbildung 9
gezeigten Prinzip auf das Silikat fixiert werden. Als organische Funktionen
kommen auch chirale Spezies in Frage, so dass eine enantioselektive
Katalyse ermöglicht wird.
TrägerOH +
OH
OH(RO)3Si X
TrägerOO
OSi X
3 ROH
Abbildung 9: Funktionalisierung der Silikatoberfläche.

Aufgabenstellung 12
2 Aufgabenstellung
In der klassischen heterogenen Katalyse werden als Trägermaterialien oder
Katalysatoren auf Silikatbasis häufig Zeolithe eingesetzt. Diese zeigen jedoch
aufgrund ihrer geringen Porengröße ein auf verhältnismäßig kleine Edukt- und
Produktmoleküle beschränktes Substratspektrum. Außerdem können durch
Ablagerungen in den Poren eine Blockierung und damit eine Deaktivierung
der katalytisch-aktiven Zentren auftreten. Um diese Probleme zu umgehen,
werden seit einigen Jahren Silikatsysteme mit Poren im meso- oder makro-
porösen Bereich eingesetzt.
Eine weitere Unzulänglichkeit der klassischen Silikatsysteme ist die geringe
Austauschbarkeit durch Metallspezies und damit geringere erzielte Metall-
beladungen. Methoden zur besseren Fixierung von Metallspezies auf
Silikaten sind zum Beispiel der Austausch von Silicium-Atomen des Silikat-
gerüsts durch andere Atome, wie zum Beispiel Aluminium, oder das
nachträgliche Anbringen funktioneller Gruppen auf der Silikatoberfläche, die
mit den Metallspezies reagieren können. P
[9, 12, 31, 32]P
Im Rahmen dieser Arbeit soll ein alternativer Weg zur Fixierung von
Metallspezies auf Silikat beschritten werden. Dazu sollen verschiedene
NafionP
®P-haltige Silikate mit mikroporösen und mesoporösen Eigenschaften
synthetisiert werden. Die hochverteilten Sulfonsäurereste des Nafion P
®P-
Polymers sollen dann zur Fixierung verschiedener Metallspezies dienen.
Dazu sollen Alkylmetall-Komplexe unter Freisetzung des Alkans mit den
Protonen der Sulfonsäurereste reagieren. Bei Verwendung von Methyl-
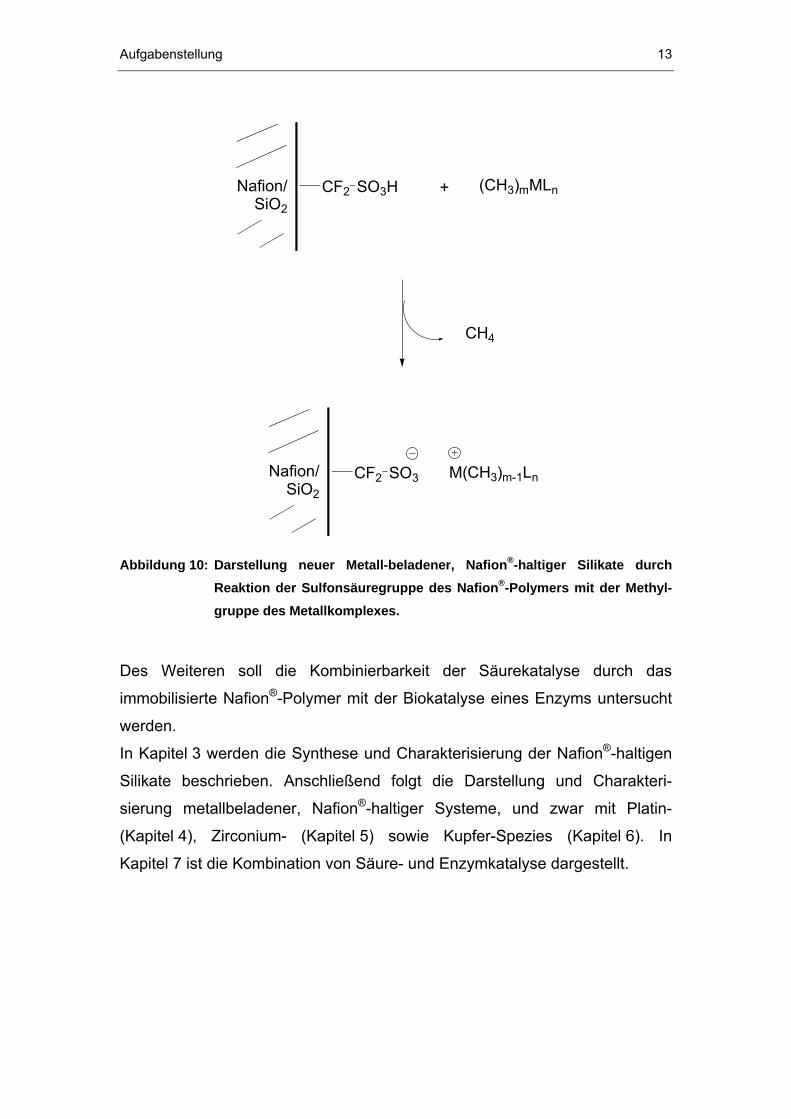
Komplexen wird als Nebenprodukt Methan freigesetzt (vgl. Abbildung 10).
Durch Trägerung über die funktionellen Gruppen des Polymers und nicht der
Oberflächen-Silanolgruppen des Silikats sollen, im Vergleich zu Literatur-
beschriebenen Methoden zur Fixierung von Metall-Spezies, genau definierte
Zentren mit einer hohen Verteilung erzeugt werden.
Die so gewonnenen metallbeladenen, Nafion P
®P-haltigen Silikate sollen mittels
verschiedener analytischer Methoden charakterisiert werden. Erste Katalysen
sollen einen Aufschluss über die Einsatzmöglichkeiten dieser Systeme in der
heterogenen Katalyse geben.
Aufgabenstellung 13
CF2 SO3HNafion/SiO2
+ (CH3)mMLn
CH4
CF2 SO3Nafion/SiO2
M(CH3)m-1Ln
Abbildung 10: Darstellung neuer Metall-beladener, NafionP
®P-haltiger Silikate durch
Reaktion der Sulfonsäuregruppe des NafionP
®P-Polymers mit der Methyl-
gruppe des Metallkomplexes.
Des Weiteren soll die Kombinierbarkeit der Säurekatalyse durch das
immobilisierte Nafion P
®P-Polymer mit der Biokatalyse eines Enzyms untersucht
werden.
In Kapitel 3 werden die Synthese und Charakterisierung der NafionP
®P-haltigen
Silikate beschrieben. Anschließend folgt die Darstellung und Charakteri-
sierung metallbeladener, Nafion P
®P-haltiger Systeme, und zwar mit Platin-
(Kapitel 4), Zirconium- (Kapitel 5) sowie Kupfer-Spezies (Kapitel 6). In
Kapitel 7 ist die Kombination von Säure- und Enzymkatalyse dargestellt.

Synthese und Charakterisierung Nafion-haltiger Silikate 14
3 Synthese und Charakterisierung Nafion-haltiger Silikate
Das perfluorierte Polymer NafionP
®P ist zwar ein starker heterogener Säure-
katalysator, zeigt allerdings vor allem in apolaren Lösungsmitteln und in
Gasphasenreaktionen eine sehr niedrige Reaktivität aufgrund der geringen
spezifischen Oberfläche der Polymer-Körner von weniger als 0.02 mP
2 PgP
-1P. Um
die katalytischen Eigenschaften des Nafions P
®P zu verbessern, wird die
Immobilisierung einer dispergierten Form dieses Polymers in Silikat genutzt.
In der Literatur wurden dazu bereits verschiedene Verfahren beschrieben, die
hier zunächst kurz vorgestellt werden sollen.
3.1 Synthese ungeordneter NafionP
®P-haltiger Silikate
3.1.1 Immobilisierung mittels des Sol-Gel-Verfahrens
Bereits 1996 wurde eine Silikat-immobilisierte Form des Nafions P
®P durch
TDuPont T-Forscher beschrieben und patentiert. P
[35, 36]P Dabei wird eine NafionP
®P-
Lösung mit Hilfe des Sol-Gel-Verfahrens umgesetzt. Das verwendete Ver-
fahren ähnelt dem sogenannten Zwei-Schritt-Verfahren (sauer/basisch) für die
Darstellung reinen Silikats. Zunächst wird das als lösliche Siliciumquelle
eingesetzte Alkoxysilan einer sauren Vorhydrolyse unterzogen, dann folgt ein
basischer Schritt –hier unter Zugabe des NafionP
®P-Polymers–, der die
Kondensation zum Gel beschleunigt. Nach Trocknen des Gels wird ein
glasartiger mikroporöser Verbundwerkstoff gewonnen, in dem das NafionP
®P-
Polymer hoch dispergiert vorliegt. Je nach Verhältnis von Polymer und
Siliciumquelle können diese Systeme 0.1 bis 90 Gew.% des Polymers
enthalten. Ein 10 bis 15 Gew.%iger Verbundwerkstoff ist als sogenanntes
SAC-13 kommerziell erhältlich. Typische Eigenschaften dieses Systems,
bestimmt durch BET-Messungen, sind eine spezifische Oberfläche von ca.
350 m P
2 PgP
-1P, ein Porenvolumen von 0.85 cm P
3 PgP
-1P und ein Porendurchmesser
von 9.8 nm. Das hochverteilte und durch die poröse Struktur des Silikats sehr
gut zugängliche NafionP
®P-Polymer wurde zum Beispiel als Katalysator in der
Dimerisierung von TαT-Methylstyrol, Isomerisierungsreaktionen, Friedel-Crafts-
Alkylierungen und Acylierungen eingesetzt. P
[35, 37-40]P

Synthese und Charakterisierung Nafion-haltiger Silikate 15
3.1.2 Trägerung mittels Imprägnierung
TTÖRÖKT et al. synthetisierten ein NafionP
®P-imprägniertes Silikat zum Vergleich
mit den unter Kapitel 3.1.1 nach TDuPont T dargestellten Systemen. Dazu wurde
ein kommerziell erhältliches Silikat mit einer Nafion P
®P-haltigen Propanol/
Wasser-Lösung behandelt. P
[41]P Untersuchungen zeigten für dieses System
jedoch eine deutlich niedrigere katalytische Aktivität. Mit Hilfe der Pyridin-
Adsorption wurde eine geringere Anzahl an katalytisch aktiven Säurezentren
auf der Oberfläche dieses Systems nachgewiesen.
3.1.3 Trägerung mittels kovalenter Fixierung
Die kovalente Fixierung einer Perfluorsulfonsäure auf einer Silikatoberfläche
ist eine weitere Möglichkeit, ein stark saures heterogenes Katalysatorsystem
zu generieren. In der Literatur wurde die Platin-katalysierte Fixierung der
[Si(CHB2 B) B3 B(CFB2 B) B2 B(O)(CF B2 B) B2 BSO B3 BH]-Einheit an Silanol-Gruppen der Silikatober-
fläche beschrieben (vgl. Abbildung 11). P
[42]P
SiO2
OOO
Si (CF2)2O(CF2)2SO3H
Abbildung 11: Oberflächenfixierte Perfluorsulfonsäure nach THARMER T et al. P
[42]P
Dieses System zeigt eine ähnliche Säurestärke und katalytische Eigen-
schaften wie der in Kapitel 3.1.1 genannte Katalysator SAC-13. Allerdings ist
die Synthese der perfluorierten Vorstufe recht komplex und nicht sehr wirt-
schaftlich, weshalb dieses Katalysatorsystem bisher keine weitere An-
wendung fand.

Synthese und Charakterisierung Nafion-haltiger Silikate 16
3.2 Synthese geordneter NafionP
®P-haltiger Silikate
TFUJIWARAT et al. beschrieben im Jahr 2000 die Synthese eines Nafion P
®P/Silikat-
Verbundwerkstoffs mit geordneter mesoporöser MCM-41-Struktur.P
[43]P Dazu
wird eine ethanolische NafionP
®P-Lösung zu einer Suspension von Tetramethyl-
ammoniumhydroxid und Cetyltrimethylammoniumbromid (CTAB) in Wasser
gegeben und die Siliciumquelle Tetraethoxysilan zugesetzt. Das CTAB wirkt
dabei als Templat für die während des Alterns bei 130 °C ausgebildete
wabenförmige Struktur, die anschließend durch saure Extraktion vom Templat
befreit wird (vgl. Kapitel 1.3.3). Katalytische Untersuchungen zeigten eine
selektivere Reaktion dieses Systems in der Dimerisierung von TαT-Methylstyrol
als andere Nafion P
®P-Katalysatoren, wie zum Beispiel NR 50 oder SAC-13.
3.3 Eigene Arbeiten
Im Rahmen dieser Arbeit wurden verschiedene NafionP
®P/Silikat-Verbundwerk-
stoffe entsprechend der in Abschnitt 3.1 und 3.2 beschriebenen, literatur-
bekannten Methoden dargestellt, um diese in weiteren Schritten in einer
Kombination von Säurekatalyse und Metall- bzw. Enzymkatalyse einzusetzen.
Da sich im Laufe der Untersuchungen einige Methoden zur Herstellung der
NafionP
®P/Silikat-Verbundwerkstoffe als nicht direkt reproduzierbar erwiesen,
wurden Variationen und neue Syntheserouten bearbeitet. In den folgenden
Abschnitten werden zunächst die Synthese und danach die Charakterisierung
dieser Systeme im Vergleich zu den Systemen aus der Literatur beschrieben.
3.3.1 Synthese ungeordneter Nafion P
®P-haltiger Silikate
Die Darstellung der ungeordneten Nafion P
®P/Silikat-Verbundstoffe erfolgte
analog der Methode nach TDuPont TP
[35]P (vgl. Kapitel 3.1.1). Dazu wird zunächst
eine 45-minütige Vorhydrolyse der Siliciumquelle Tetramethoxysilan mit einer
stark verdünnten Salzsäure-Lösung durchgeführt. Gleichzeitig wird eine
kommerzielle 5 Gew.%ige Nafion P
®P-Lösung mit Natriumhydroxid-Lösung ver-
setzt und diese stark basische Lösung mit der Silicium-haltigen Lösung unter
starkem Rühren vermischt. Nach wenigen Sekunden geliert diese Mischung

Synthese und Charakterisierung Nafion-haltiger Silikate 17
unter Bildung eines trüben, weißen Gels, das für mehrere Tage bei 95 °C
getrocknet wird. Der auf diese Weise gewonnene weiße, glasartige Feststoff
wird anschließend gemörsert, die neutralisierten Sulfonsäurereste mit Säure
regeneriert und das Produkt bei 120 °C getrocknet. In Abbildung 12 ist ein
Schema der Darstellung der ungeordneten Nafion P
®P/Silikat-Verbundstoffe
gezeigt.
Neutralisation des Polymers
Nafion®-Lösung + NaOH (0.4 N)
Saure Vorhydrolyse
TMOS + HCl (0.04 N)
Gelbildung
Trocknen bei 95 °C
Trocknen bei 120 °C
Mörsern
Regenerieren der Säurereste
HCl (3.5 M)
HNO3 (25 Gew.%)
Neutralisation des Polymers
Nafion®-Lösung + NaOH (0.4 N)
Saure Vorhydrolyse
TMOS + HCl (0.04 N)
Gelbildung
Trocknen bei 95 °C
Trocknen bei 120 °C
Mörsern
Regenerieren der Säurereste
HCl (3.5 M)
HNO3 (25 Gew.%)
Abbildung 12: Schema der Darstellung der ungeordneten NafionP
®P/Silikat-Verbund-
stoffe.
Im Rahmen dieser Arbeit wurden Verbundstoffe mit verschiedenen Gewichts-
anteilen an Nafion P
®P-Polymer dargestellt. Es wurden Systeme mit 5, 10, 13,
15, 20, 25 und 40 Gew.% Nafion P
®P gewonnen. Dabei wurde die Nafion P
®P-
Menge im Verhältnis zur Siliciumquelle variiert, die Säuremenge für die Vor-
Synthese und Charakterisierung Nafion-haltiger Silikate 18
hydrolyse jedoch konstant gehalten. Die Natriumhydroxid-Menge wurde
proportional mit der Nafion P
®P-Menge variiert. Es ergaben sich die in Tabelle 1
gezeigten Massenverhältnisse.
Tabelle 1: Massenverhältnisse der Edukte in der Synthese der ungeordneten NafionP
®P/Silikat-Verbundstoffe.
Produkt TMOS NafionP
®P HCl NaOH
SAC/Naf5.3 46600 1026 1 362
SAC/Naf10 46600 2052 1 476
SAC/Naf13 46600 2740 1 548
SAC/Naf15 46600 3246 1 605
SAC/Naf20 46600 4609 1 750
SAC/Naf25 46600 6141 1 915
SAC/Naf40 46250 12182 1 1316
3.3.2 Synthese geordneter Nafion P
®P-haltiger Silikate
Zur Darstellung eines geordneten Nafion P
®P-haltigen Silikats wurde zunächst
die von TFUJIWARAT et al. beschriebene Methode P
[43]P verwendet (vgl. Kapitel 3.2).
Allerdings gelang es bei direkter Umsetzung des literaturbeschriebenen Ver-
fahrens nicht, eine geordnete Phase herzustellen.
Deshalb wurden im Weiteren verschiedene Parameter, wie Temperatur,
Templat, Base bzw. Säure, die verwendete Extraktionsmethode und die
Zusammensetzung der NafionP
®P-Lösung, variiert.
TVariation der Temperatur
In der Literatur von TFUJIWARAT et al. P
[43]P wird die Synthese der geordneten
Phase bei einer Temperatur von 130 °C durchgeführt. Diese Bedingung
wurde im Rahmen dieser Arbeit zunächst beibehalten. Nachdem es auf diese
Weise nicht gelang, ein geordnetes Silikat zu gewinnen, wurden Versuche der
Darstellung einer geordneten Phase bei Raumtemperatur unternommen.
Dazu wurden die eingesetzten Substanzen zunächst vermischt und das dabei

Synthese und Charakterisierung Nafion-haltiger Silikate 19
gebildete Gel mindestens 24 Stunden bei Raumtemperatur gealtert. An-
schließend wurde das Gel in einer Lyophille gefriergetrocknet.
Variation des Templats
Anstelle des in der LiteraturP
[43]P verwendeten Templats CTAB wurde in
weiteren Versuchen D-Glucose eingesetzt, die bereits durch TWEI T et al. in der
Darstellung geordneter Enzym/Silikat-Verbundstoffe benutzt wurde. P
[44, 45]P In
diesen Versuchen wurde bei Raumtemperatur gearbeitet und die Trocknung
des Gels mittels einer Lyophille durchgeführt.
Variation der Extraktionsmethode
Die ursprünglich in der LiteraturP
[43]P verwendete Extraktion zur Entfernung des
Templats wurde mittels mehrstündigem Erhitzens in einer ca. 1 Vol.%igen
Lösung von konz. HB2BSO B4B in Ethanol unter Rückfluss und anschließendem
zweifachen Erhitzens in reinem Ethanol durchgeführt.
In der Literatur sind Fälle beschrieben, in denen zwar zunächst eine
geordnete Silikatphase gebildet werden konnte, diese aber während der
Extraktion des Templats kollabierte. P
[46]P Um ausschließen zu können, dass die
Art der Extraktion die Gewinnung der geordneten Phase verhindert, wurde
statt der sauren Extraktion, durch Erhitzen in einer sauren ethanolischen
Lösung unter Rückfluss, die schonendere Soxhlet-Extraktion eingesetzt.
Dabei wurde die Probe vor bzw. nach der Extraktion mit HCl-Lösung be-
handelt, um die Säurereste zu regenerieren.
Proben, die mit D-Glucose als Templat dargestellt wurden, wurden alternativ
bei Raumtemperatur mit einer 0.01 N wässrigen HCl-Lösung extrahiert.
Weitere Variationen
Des Weiteren wurde die Siliciumquelle von Tetraethoxy- zu Tetramethoxy-
silan bzw. Natriumsilikat-Lösung, die Base von Tetramethylammonium-
hydroxid zu Natriumhydroxid und die Säure von Salz- zu Schwefelsäure
variiert.
In Tabelle 2 sind die verschiedenen Kombinationen der Parameter zu-
sammengefasst, die im Rahmen dieser Arbeit untersucht wurden. Bei keiner
Synthese und Charakterisierung Nafion-haltiger Silikate 20
der Varianten konnte jedoch die Bildung einer geordneten Silikatmatrix
beobachtet werden.
Tabelle 2: Parameter des Versuchs der Darstellung geordneter NafionP
®P-haltiger
Silikate bei Raumtemperatur (Nafion P
®P gewonnen aus kommerzieller
Lösung durch Gefriertrocknung).
[Si]-Quelle Templat Säure Base T Extraktion
TEOS CTAB – MeB4BNOH 130 °CHB2BSO B4B+EtOH/
Rückfluss
TEOS CTAB – MeB4BNOH 130 °CMeOH/Soxhlet
HCl*
TEOS CTAB – MeB4BNOH 130 °CHCl+MeOH/Soxhlet
HCl*
TEOS CTAB – NaOH 130 °CHCl+EtOH/
Rückfluss
TEOS CTAB HCl – RT HCl+EtOH/
Rückfluss
TMOS CTAB HCl NaOH RT HB2BSO B4B+EtOH/
Rückfluss
TMOS CTAB HCl NaOH RT MeOH/Rückfluss
HCl*
Natrium-
silikat-Lsg. CTAB HB2BSO B4B – RT **
TMOS D-Glucose HCl – RT HCl BaqB/RT
TMOS D-Glucose HCl NaOH RT MeOH/Rückfluss
HCl*
TMOS D-Glucose HCl NaOH RT MeOH/Soxhlet
HCl*
* Im 2. Schritt sauer regeneriert.
** Keine regelmäßige Gelbildung, daher nicht weiter bearbeitet.
Synthese und Charakterisierung Nafion-haltiger Silikate 21
Variation der Nafion P
®P-Lösung
In der Vorschrift nach TFUJIWARAT et al. wird das NafionP
®P-Polymer isoliert und
für die Darstellung des Verbundstoffs in Ethanol gelöst. P
[43]P In den im Rahmen
dieser Arbeit durchgeführten Untersuchungen wurde das NafionP
®P zunächst
aus einer kommerziellen 5 Gew.%igen Lösung durch Gefriertrocknung in
einer Lyophille gewonnen und dann in Ethanol gelöst.
Nachdem Variationen verschiedener Parameter keinen Erfolg in der Dar-
stellung eines geordneten Silikats zeigten, wurde in späteren Untersuchungen
direkt die kommerzielle Lösung, in niederen Alkoholen und Wasser, in der
Synthese eingesetzt und die Menge des zugesetzten Wassers entsprechend
verringert. Anhand dieser Methode war es möglich, eine geordnete Phase
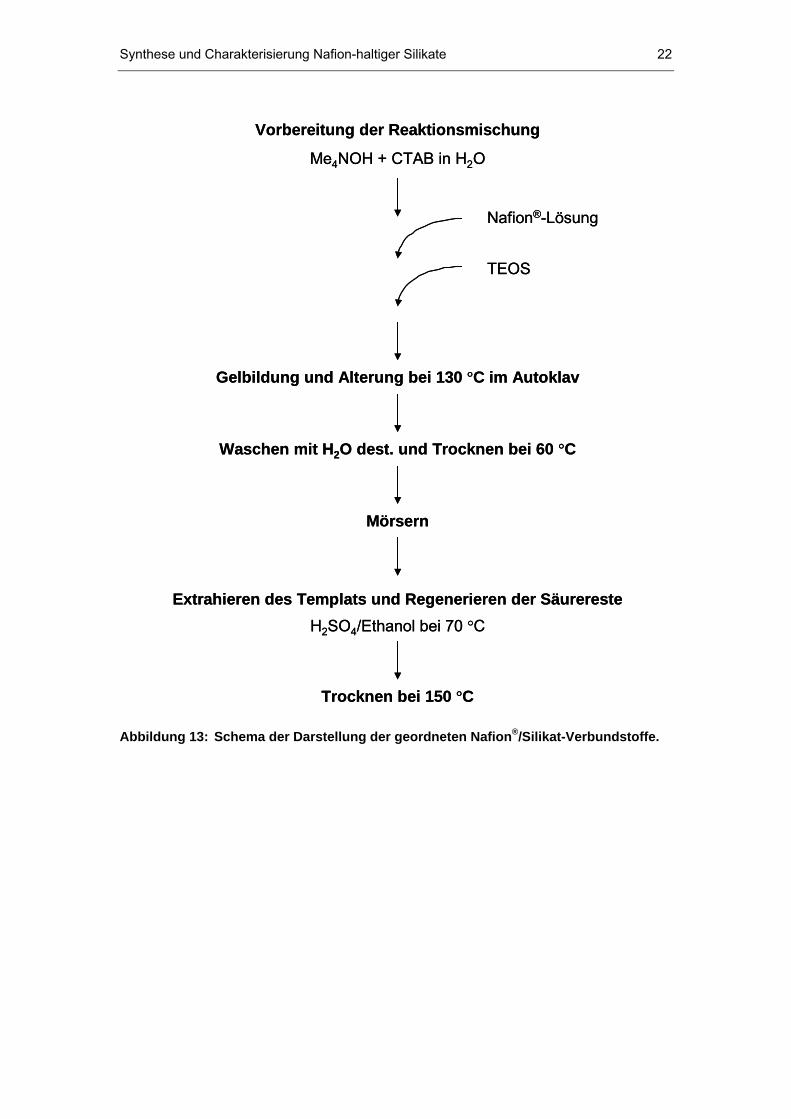
darzustellen. Abbildung 13 zeigt schematisch, wie die geordneten Nafion P
®P/
Silikat-Systeme gewonnen wurden.
Auch bei Erhöhung des NafionP
®P-Gehalts von 5.3 auf 10, 13, 15 bzw.
20 Gew.% wurde eine geordnete Phase nachgewiesen. Erst bei Erhöhung
des Gehalts auf 25 Gew.% Nafion P
®P blieb die Bildung einer geordneten
Struktur aus.
Die Massenverhältnisse der Edukte für die dargestellten, geordneten Ver-
bundstoffe sind in Tabelle 3 gezeigt. Dies entspricht einem Molverhältnis von
3.8:1:1 von TEOS zu CTAB und Tetramethylammoniumhydroxid.
Tabelle 3: Massenverhältnisse der Edukte in der Synthese der geordneten NafionP
®P/Silikat-Verbundstoffe.
Produkt TEOS NafionP
®P CTAB MeB4 BNOH
MCM/Naf5.3 4282 68 1123 1000
MCM/Naf10 4282 137 1123 1000
MCM/Naf13 4282 184 1123 1000
MCM/Naf15 4282 218 1123 1000
MCM/Naf20 4282 308 1123 1000
MCM/Naf25 4282 412 1123 1000
Synthese und Charakterisierung Nafion-haltiger Silikate 22
Gelbildung und Alterung bei 130 °C im Autoklav
Waschen mit H2O dest. und Trocknen bei 60 °C
Trocknen bei 150 °C
Mörsern
Extrahieren des Templats und Regenerieren der SäureresteH2SO4/Ethanol bei 70 °C
Vorbereitung der Reaktionsmischung
Me4NOH + CTAB in H2O
Nafion®-Lösung
TEOS
Gelbildung und Alterung bei 130 °C im Autoklav
Waschen mit H2O dest. und Trocknen bei 60 °C
Trocknen bei 150 °C
Mörsern
Extrahieren des Templats und Regenerieren der SäureresteH2SO4/Ethanol bei 70 °C
Vorbereitung der Reaktionsmischung
Me4NOH + CTAB in H2O
Nafion®-Lösung
TEOS
Abbildung 13: Schema der Darstellung der geordneten NafionP
®P/Silikat-Verbundstoffe.

Synthese und Charakterisierung Nafion-haltiger Silikate 23
3.4 Charakterisierung NafionP
®P-haltiger Silikate
Im Folgenden werden die verschiedenen Charakterisierungsmethoden für die
dargestellten Nafion P
®P-haltigen Silikate und die Ergebnisse für die ver-
schiedenen Verbundwerkstoffe vorgestellt.
Das ungeordnete System, das analog der Vorschrift von DuPont dargestellt
wurde und als ca. 15 Gew.%iges System unter dem Namen SAC-13 kommer-
ziell erhältlich ist, wurde bereits ausführlich durch diese Forschergruppe
untersucht und beschrieben. Diese Ergebnisse sind in den folgenden
Abschnitten zum Vergleich aufgeführt. Außerdem sind die ungeordneten
Systeme mit den verschiedenen Nafion P
®P-Anteilen beschrieben.
TFUJIWARAT et al. P
[43]P geben in ihrer Veröffentlichung für das geordnete ca.
5 Gew.%ige System nur wenige Charakterisierungen, wie XRD-, BET- und
IR-Daten an. Daher wird dieses System und die geordneten Systeme mit
höherem NafionP
®P-Gehalt im Folgenden ausführlich behandelt.
3.4.1 Elementaranalyse
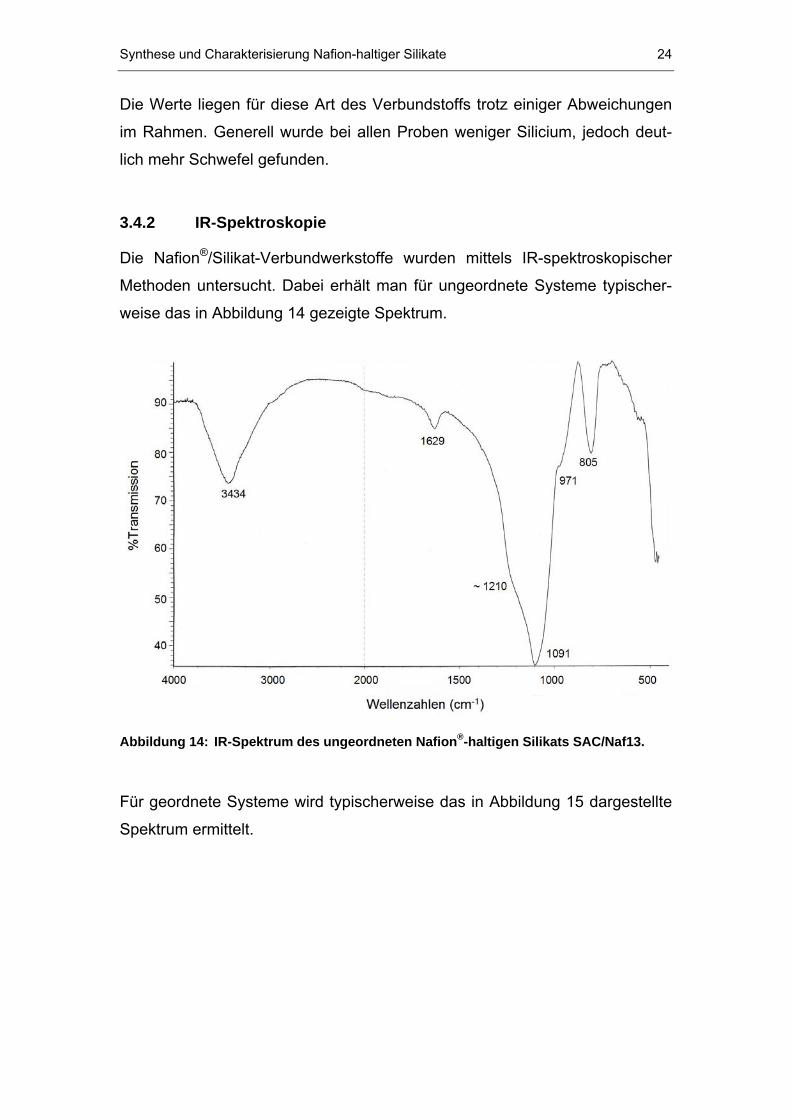
Die Elementaranalyse der Verbundstoffe für die Elemente Silicium, Fluor und
Schwefel führte zu den in Tabelle 4 gezeigten Ergebnissen. Für die Werte ist
ein relativer Fehler von 5 % anzunehmen. Zusätzlich sind die Ergebnisse aus
der Literatur für das vergleichbare ungeordnete Nafion P
®P/Silikat-System von
DuPont P
[35]P angegeben.
Tabelle 4: Ergebnisse der Elementaranalyse für ausgewählte NafionP
®P-haltige
Silikate (in Klammern sind die berechneten Anteile angegeben).
Probe Gew.% Si Gew.% F Gew.% S
SAC/Naf10 39.0 (42.1) 6.3 (6.8) 0.47 (0.26)
MCM/Naf10 36.9 (42.1) 8.6 (6.8) 0.65 (0.26)
SAC/Naf13 39.2 (40.7) 8.3 (8.8) 0.60 (0.34)
MCM/Naf13 34.9 (40.7) 9.4 (8.8) 0.64 (0.34)
SAC-13P
[35]P 37.5 (38.7) 7.4 (8.9) 0.39 (0.35)
Synthese und Charakterisierung Nafion-haltiger Silikate 24
Die Werte liegen für diese Art des Verbundstoffs trotz einiger Abweichungen
im Rahmen. Generell wurde bei allen Proben weniger Silicium, jedoch deut-
lich mehr Schwefel gefunden.
3.4.2 IR-Spektroskopie
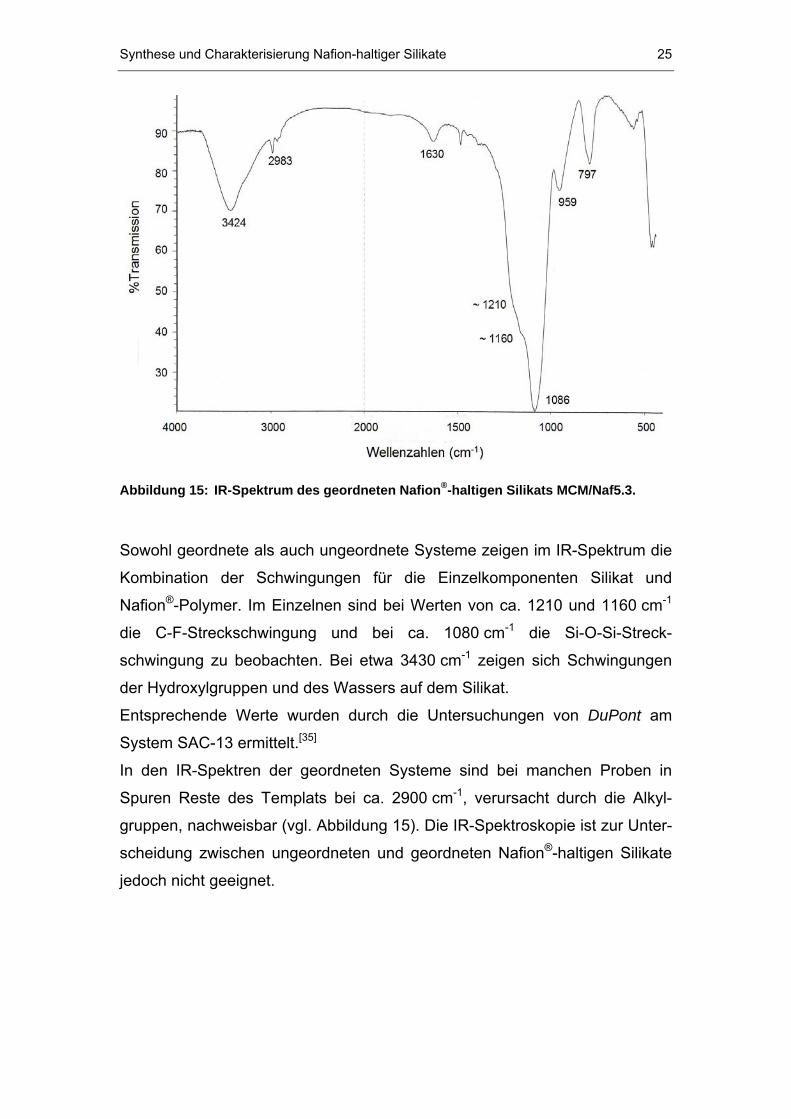
Die Nafion P
®P/Silikat-Verbundwerkstoffe wurden mittels IR-spektroskopischer
Methoden untersucht. Dabei erhält man für ungeordnete Systeme typischer-
weise das in Abbildung 14 gezeigte Spektrum.
Abbildung 14: IR-Spektrum des ungeordneten Nafion P
®P-haltigen Silikats SAC/Naf13.
Für geordnete Systeme wird typischerweise das in Abbildung 15 dargestellte
Spektrum ermittelt.
Synthese und Charakterisierung Nafion-haltiger Silikate 25
Abbildung 15: IR-Spektrum des geordneten NafionP
®P-haltigen Silikats MCM/Naf5.3.
Sowohl geordnete als auch ungeordnete Systeme zeigen im IR-Spektrum die
Kombination der Schwingungen für die Einzelkomponenten Silikat und
NafionP
®P-Polymer. Im Einzelnen sind bei Werten von ca. 1210 und 1160 cm P
-1P
die C-F-Streckschwingung und bei ca. 1080 cm P
-1P die Si-O-Si-Streck-
schwingung zu beobachten. Bei etwa 3430 cm P
-1P zeigen sich Schwingungen
der Hydroxylgruppen und des Wassers auf dem Silikat.
Entsprechende Werte wurden durch die Untersuchungen von DuPont am
System SAC-13 ermittelt.P
[35]P
In den IR-Spektren der geordneten Systeme sind bei manchen Proben in
Spuren Reste des Templats bei ca. 2900 cm P
-1P, verursacht durch die Alkyl-
gruppen, nachweisbar (vgl. Abbildung 15). Die IR-Spektroskopie ist zur Unter-
scheidung zwischen ungeordneten und geordneten Nafion P
®P-haltigen Silikate
jedoch nicht geeignet.
Synthese und Charakterisierung Nafion-haltiger Silikate 26
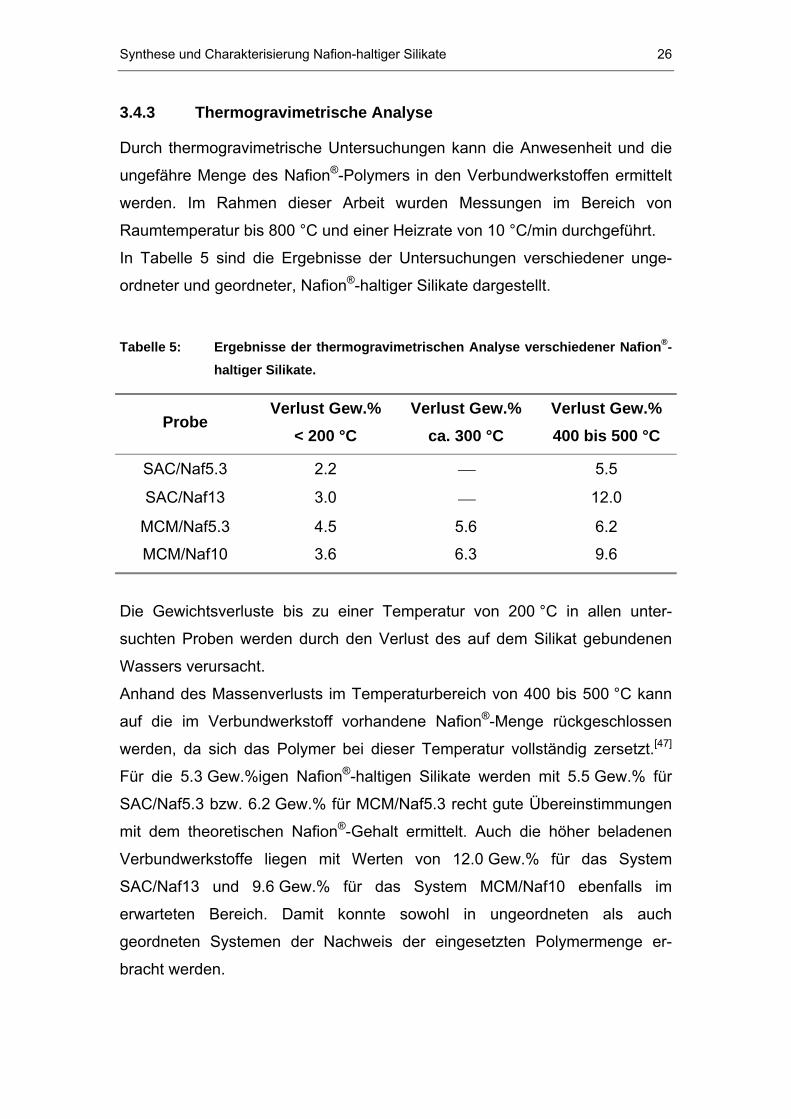
3.4.3 Thermogravimetrische Analyse
Durch thermogravimetrische Untersuchungen kann die Anwesenheit und die
ungefähre Menge des Nafion P
®P-Polymers in den Verbundwerkstoffen ermittelt
werden. Im Rahmen dieser Arbeit wurden Messungen im Bereich von
Raumtemperatur bis 800 °C und einer Heizrate von 10 °C/min durchgeführt.
In Tabelle 5 sind die Ergebnisse der Untersuchungen verschiedener unge-
ordneter und geordneter, NafionP
®P-haltiger Silikate dargestellt.
Tabelle 5: Ergebnisse der thermogravimetrischen Analyse verschiedener NafionP
®P-
haltiger Silikate.
Probe Verlust Gew.%
< 200 °C Verlust Gew.%
ca. 300 °C Verlust Gew.% 400 bis 500 °C
SAC/Naf5.3 2.2 ⎯ 5.5
SAC/Naf13 3.0 ⎯ 12.0
MCM/Naf5.3 4.5 5.6 6.2
MCM/Naf10 3.6 6.3 9.6
Die Gewichtsverluste bis zu einer Temperatur von 200 °C in allen unter-
suchten Proben werden durch den Verlust des auf dem Silikat gebundenen
Wassers verursacht.
Anhand des Massenverlusts im Temperaturbereich von 400 bis 500 °C kann
auf die im Verbundwerkstoff vorhandene NafionP
®P-Menge rückgeschlossen
werden, da sich das Polymer bei dieser Temperatur vollständig zersetzt.P
[47]P
Für die 5.3 Gew.%igen NafionP
®P-haltigen Silikate werden mit 5.5 Gew.% für
SAC/Naf5.3 bzw. 6.2 Gew.% für MCM/Naf5.3 recht gute Übereinstimmungen
mit dem theoretischen Nafion P
®P-Gehalt ermittelt. Auch die höher beladenen
Verbundwerkstoffe liegen mit Werten von 12.0 Gew.% für das System
SAC/Naf13 und 9.6 Gew.% für das System MCM/Naf10 ebenfalls im
erwarteten Bereich. Damit konnte sowohl in ungeordneten als auch
geordneten Systemen der Nachweis der eingesetzten Polymermenge er-
bracht werden.

Synthese und Charakterisierung Nafion-haltiger Silikate 27
Durch DuPont wurde für das kommerziell erhältliche System SAC-13 ein
Gewichtsverlust von 12.8 Gew.% im Temperaturbereich von 400 bis 500 °C
veröffentlicht.P
[35]P Dies bestätigt die hier gefundene recht gute Überein-
stimmung der theoretischen Werte mit den experimentell ermittelten Daten.
Bei den geordneten, Nafion P
®P-haltigen Silikaten wird ein zusätzlicher Gewichts-
verlust von 5 bis 6 % bei einer Temperatur von ca. 300 °C beobachtet. Dies
kann durch Reste des organischen Templats in der Silikatmatrix begründet
werden.
3.4.4 Ionenaustauschkapazität
3.4.4.1 Methodenbeschreibung
Zur Charakterisierung von Ionenaustauscherharzen und sauren bzw.
basischen heterogenen Katalysatoren wird häufig die so genannte Ionenaus-
tauschkapazität herangezogen. Die einfachste Methode zur Bestimmung
dieser Größe ist die Behandlung der Probe mit einer Salzlösung, wie zum
Beispiel einer Natriumchlorid-Lösung, und anschließender Titration der freige-
setzten Protonen bzw. Hydroxid-Ionen. Bei den hier untersuchten Systemen
werden entsprechend die Kationenaustauschkapazität CEC (Cation exchange
capacity), hier der Natrium-Ionen des Natriumchlorids, und die freigesetzten
Protonen durch Titration gegen Natriumhydroxid-Lösung bestimmt. Dabei wird
die CEC in meq/g angegeben, wobei ein meq dem Milliäquivalent an Kation,
berechnet mittels der Molmasse des Kations und seiner Ladung (vgl.
Gleichung 1), entspricht und pro Gramm Feststoff angegeben wird.
Gl. 1 ( )KationLadungKationMKationeÄquivalent )()( =
Zum Vergleich sind in Tabelle 6 die Werte für die CEC einiger fester Per-
fluorsulfonsäuren, wie NafionP
®P-Polymer und das durch DuPont vertriebene
NafionP
®P/Silikat-System SAC-13, angegeben. Der Wert für das kommerziell
erhältliche SAC-13 konnte mit 0.15 meq/g sowohl in Form der käuflichen
„Sticks“ als auch gemörsert in Form eines Pulvers reproduziert werden.

Synthese und Charakterisierung Nafion-haltiger Silikate 28
Tabelle 6: Ionenaustauschkapazitäten einiger fester Säuren.
Beispiel CEC meq/g
NafionP
®P NR50P
[48]P 0.89
SAC-13P
[35]P 0.14
AmberliteP
® P IRP-64P
[49]P 10
3.4.4.2 CEC-Bestimmungen für die NafionP
®P-haltigen Silikate
Entsprechend der oben beschriebenen Methode wurde die Ionenaustausch-
kapazität der im Rahmen dieser Arbeit dargestellten Nafion P
®P-haltigen Silikate
bestimmt. In Tabelle 7 sind zunächst die Ergebnisse der ungeordneten
Systeme aufgeführt.
Tabelle 7: Ermittelte Ionenaustauschkapazitäten für ungeordnete NafionP
®P-haltige
Silikate.
Probe CEC meq/g
SAC/Naf5.3 0.06
SAC/Naf10 0.11
SAC/Naf13 0.13
SAC/Naf15 0.17
SAC/Naf20 0.22
SAC/Naf25 0.29
SAC/Naf40 0.46
Aus den ermittelten Werten ist eine kontinuierliche Zunahme der Kationen-
austauschkapazität proportional zur Zunahme an NafionP
®P-Polymer im Ver-
bundwerkstoff festzustellen. Die 13 bzw. 15 Gew.%igen Systeme weisen mit
0.13 meq/g bzw. 0.17 meq/g einen vergleichbaren Wert zu den 0.15 meq/g
des kommerziellen Systems mit durchschnittlich bis zu 15 Gew.% NafionP
®P
auf.

Synthese und Charakterisierung Nafion-haltiger Silikate 29
In Tabelle 8 sind die Ergebnisse für die geordneten NafionP
®P/Silikat-Systeme
angegeben. Durch TFUJIWARAT et al. wurden diese Untersuchungen für den
analogen, durch sie synthetisierten 5 Gew.%igen Katalysator nicht veröffent-
licht. P
[43]P
Tabelle 8: Ermittelte Ionenaustauschkapazitäten für geordnete NafionP
®P-haltige
Silikate.
Probe CEC meq/g
MCM/Naf5.3 0.02
MCM/Naf10 0.05
MCM/Naf13 0.06
MCM/Naf15 0.06
MCM/Naf20 0.09
MCM/Naf25 0.09
Auch bei den Proben mit geordneter Struktur wurden mit ansteigendem
NafionP
®P-Gehalt zunehmende Kationenaustauschkapazitäten gefunden. Im
Vergleich zu den ungeordneten Systemen wurden jedoch für die geordneten
NafionP
®P/Silikat-Verbundstoffe deutlich niedrigere Ionenaustauschkapazitäten
beobachtet. Dies deutet darauf hin, dass nicht alle vorhandenen Säure-
funktionen in diesen Untersuchungen für den Austausch durch die Natrium-
Ionen zugänglich sind. Die deutlich stärkere Hydrophobie der geordneten
Proben, die eine gleichmäßige Benetzung durch die wässrige Lösung trotz
Zugabe von Methanol erschwerte, könnte eine Erklärung für diese Be-
obachtung sein. Bei der späteren Funktionalisierung der Sulfonsäuregruppen
mit Metall-Spezies in organischen Lösungsmitteln konnte jedoch eine
vollständige Beladung und damit die Zugänglichkeit der Säurefunktionen
gezeigt werden.

Synthese und Charakterisierung Nafion-haltiger Silikate 30
3.4.5 Stickstoff-Sorptions-Messungen
3.4.5.1 Methodenbeschreibung
Die Porengröße und -form heterogener Katalysatoren zeigt oft einen Einfluss
auf den Reaktionsverlauf und den Transport von Edukten und Produkten
während der Katalyse. Die Art der Porosität und die damit verbundenen
Eigenschaften des Materials, wie spezifische Oberfläche, Porengröße und
-volumen, werden daher für solche Systeme untersucht. Standardmäßig wird
zur Untersuchung poröser Materialien die TBRUNAUER-EMMETT-TELLERT-
Methode (BET) herangezogen, die diese Größen anhand der Physisorption
eines Gases, meist Stickstoff bei 77 K, bestimmt.P
[50]P
Die Physisorption eines Gases tritt immer dann auf, wenn ein Gas auf eine
feste Oberfläche trifft. Nach Gleichung 2 besteht eine Relation zwischen der
Menge an adsorbiertem Gas nP
aP und dem relativem Druck p/pB0 B bei einer
bestimmten Temperatur
Gl. 2 ( )Ta ppfn 0/= .
Die Ergebnisse einer solchen Messung werden als Isotherme in Form einer
Auftragung von adsorbierter Gasmenge nP
aP geteilt durch die Masse des
adsorbierenden Feststoffs mP
sP gegen den relativen Druck gezeigt. Dabei treten
je nach untersuchtem Feststoff typische, nach IUPAC klassifizierte Isotherme
auf (vgl. Abbildung 16).P
[51]P

Synthese und Charakterisierung Nafion-haltiger Silikate 31
Abbildung 16: Gasphysisorptionsisotherme nach IUPAC.P
[50]P
Anhand der ermittelten Isotherme können unter Voraussetzung bestimmter
Bedingungen mittels vereinfachter Berechnungen die spezifische Oberfläche
und die Porosität des Feststoffs ermittelt werden. Für die dafür häufig ver-
wendete BET-Methode muss zunächst der so genannte Sättigungswert bei
monomolekularer Bedeckung amn (monolayer capacity) bestimmt werden, der
definiert ist als die nötige Menge an adsorbiertem Gas, um eine vollständige
monomolekulare Schicht an Adsorbat auf der Oberfläche einer Masseneinheit
des Feststoffs zu bilden. Die spezifische Oberfläche aBs B (BET) kann unter
Annahme der durchschnittlichen Oberfläche aBmB, die durch das Adsorbat ein-
genommen wird, nach Gleichung 3 berechnet werden. Dabei ist L die
Avogadro-Konstante.
Gl. 3 ( ) mams aLnBETa ⋅⋅=
Unter der Annahme, dass nach der Adsorption der ersten monomolekularen
Schicht an Gasmolekülen, die Bedingungen für die Adsorption und Desorption
für alle weiteren Molekülschichten gleich sind, dabei die Adsorptionsenergie
gleich der Kondensationsenergie ist und bei p = pB0 B die multimolekulare
Schicht eine unendliche Dicke aufweist, konntenT BRUNAUER T, TEMMETT T und
TTELLERT die Menge des adsorbierten Gases nach Gleichung 4 annähern.

Synthese und Charakterisierung Nafion-haltiger Silikate 32
Gl. 4 ( ) 00
0 11/1
/pp
cnc
cnppnpp
am
am
a ⋅⋅−
+⋅
=−
Dabei ist c eine Konstante. Sie steht exponentiell mit der Adsorptionsenergie
der ersten Schicht im Zusammenhang und gibt einen Hinweis auf die Form
der Isotherme und die Größenordnung der Gas-Feststoff-Wechselwirkungen.
Nach Gleichung 4 können die Werte für amn und c aus der Steigung und dem
Achsenabschnitt der linearen BET-Auftragung ermittelt werden. Allerdings
sind nur wenige Abschnitte der Isotherme im linearen Bereich und nur der
Bereich, in dem eine monomolekulare Schicht vorliegt, kann zum richtigen
Wert für amn führen.
In der Praxis hat sich Stickstoff bei 77 K als das beste Adsorptiv für die
Bestimmung der spezifischen Oberfläche von nichtporösen, meso- und
makroporösen Feststoffen herausgestellt. Für die durch den Stickstoff bei
dichtester Packung eingenommene Oberfläche aBmB wird ein Wert von
0.162 nm P
2P angenommen.
Obwohl die molekularen Dimensionen in mikroporösen Feststoffen das
Auftreten der Monoschicht-Mehrschichten-Adsorption verhindern und die
BET-Methode damit keine exakten Ergebnisse liefert, werden die Werte aus
den BET-Messungen trotzdem in Patenten und der wissenschaftlichen
Literatur zum Vergleich angegeben. Sie können allerdings je nach ver-
wendeter Methode stark von den realen Werten abweichen.
Für mesoporöse Stoffe wird eine so genannte Typ IV-Isotherme (nach
IUPAC) beobachtet. Zunächst verläuft diese Isotherme (vgl. Abbildung 17)
wie bei einem nichtporösen Feststoff unter Adsorption der monomolekularen
Schicht und anschließend der ersten Stufen der Bildung einer multi-
molekularen Schicht. Bei einem bestimmten p/pB0 B-Verhältnis weicht der Verlauf
der Isotherme jedoch ab und es tritt ein starker Anstieg zu höheren
adsorbierten Volumen auf. Dieses Verhalten wird mit dem Füllen der
Mesoporen durch Kapillarkondensation erklärt. Da die Kapillarkondensation
mit einer irreversiblen Entropieänderung verbunden ist, tritt notwendigerweise
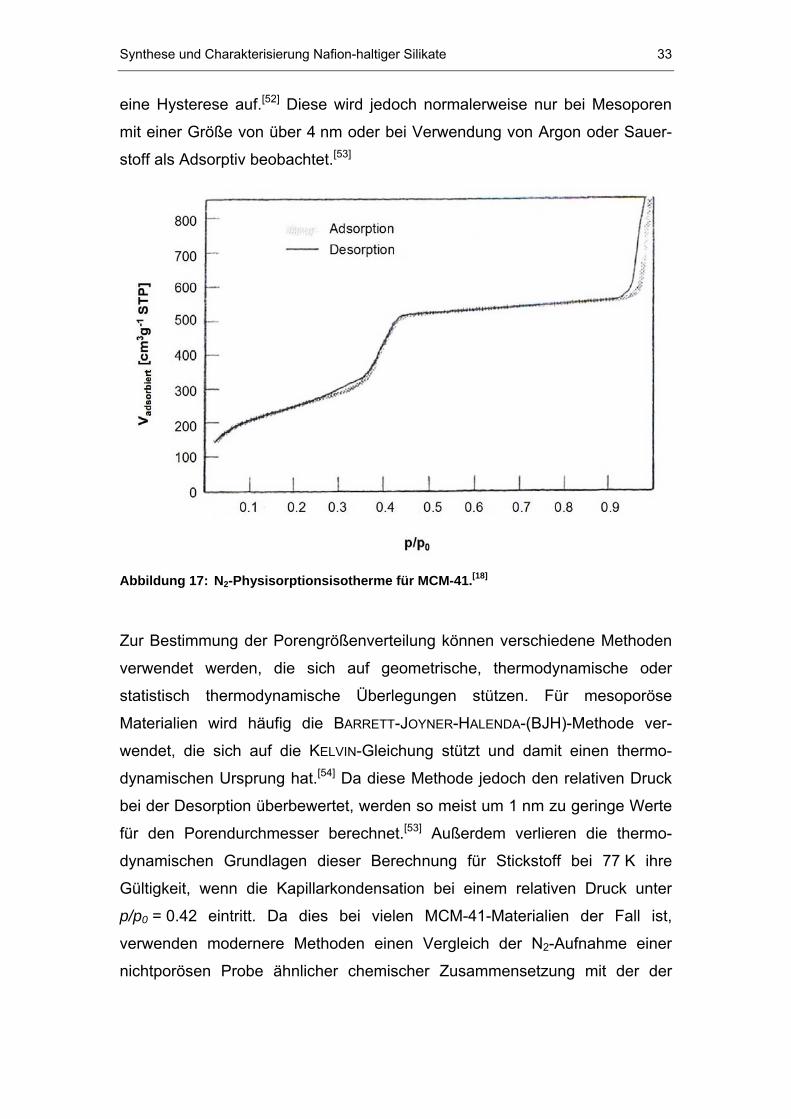
Synthese und Charakterisierung Nafion-haltiger Silikate 33
eine Hysterese auf.P
[52]P Diese wird jedoch normalerweise nur bei Mesoporen
mit einer Größe von über 4 nm oder bei Verwendung von Argon oder Sauer-
stoff als Adsorptiv beobachtet. P
[53]P
Abbildung 17: NB2B-Physisorptionsisotherme für MCM-41.P
[18]P
Zur Bestimmung der Porengrößenverteilung können verschiedene Methoden
verwendet werden, die sich auf geometrische, thermodynamische oder
statistisch thermodynamische Überlegungen stützen. Für mesoporöse
Materialien wird häufig die TBARRETT-JOYNER-HALENDAT-(BJH)-Methode ver-
wendet, die sich auf die TKELVINT-Gleichung stützt und damit einen thermo-
dynamischen Ursprung hat. P
[54]P Da diese Methode jedoch den relativen Druck
bei der Desorption überbewertet, werden so meist um 1 nm zu geringe Werte
für den Porendurchmesser berechnet. P
[53]P Außerdem verlieren die thermo-
dynamischen Grundlagen dieser Berechnung für Stickstoff bei 77 K ihre
Gültigkeit, wenn die Kapillarkondensation bei einem relativen Druck unter
p/pB0 B = 0.42 eintritt. Da dies bei vielen MCM-41-Materialien der Fall ist,
verwenden modernere Methoden einen Vergleich der NB2B-Aufnahme einer
nichtporösen Probe ähnlicher chemischer Zusammensetzung mit der der
Synthese und Charakterisierung Nafion-haltiger Silikate 34
mesoporösen Probe und anschließenden Modellrechnungen, wie der Monte
Carlo-Simulation oder der Density Functional Theory.
3.4.5.2 BET-Messungen für die Nafion P
®P-haltigen Silikate
Im Rahmen der vorliegenden Arbeit wurden die dargestellten NafionP
®P-haltigen
Silikate mit Hilfe eines Oberflächen- und Porengrößenanalysengeräts der
Firma Quantachrome Instruments auf ihre Porosität untersucht. Dazu wurde
die Stickstoffsorption bei 77 K eingesetzt.
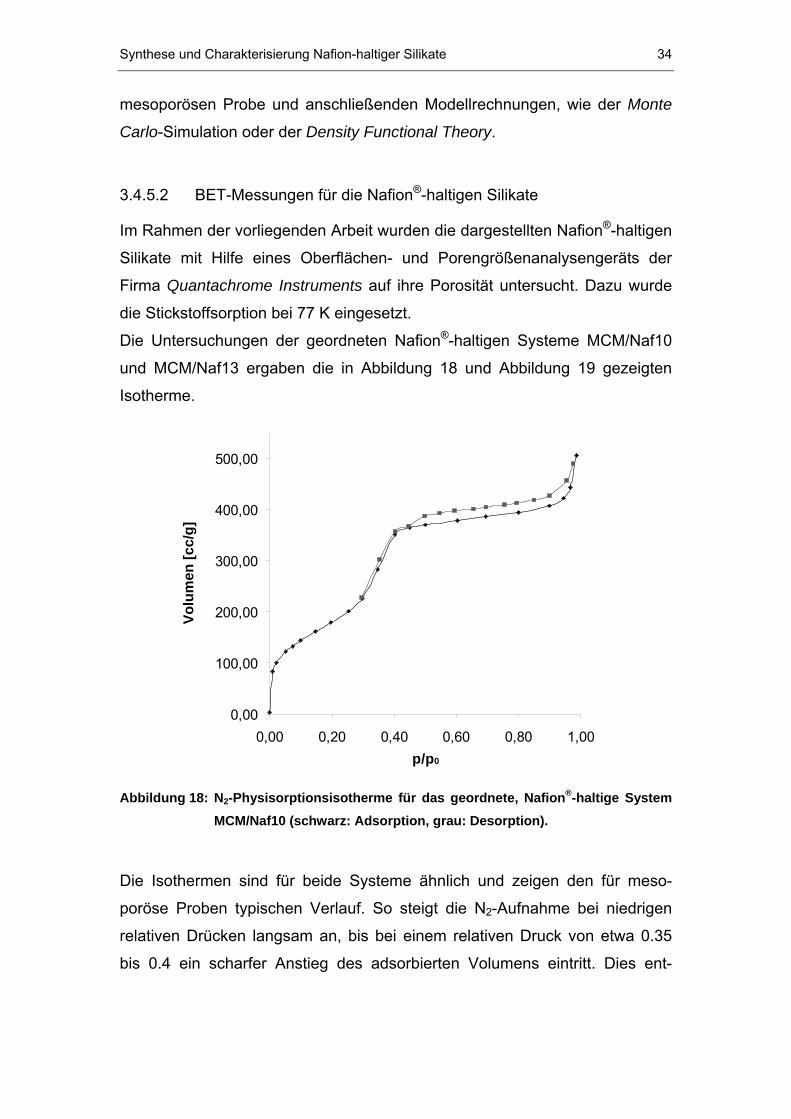
Die Untersuchungen der geordneten NafionP
®P-haltigen Systeme MCM/Naf10
und MCM/Naf13 ergaben die in Abbildung 18 und Abbildung 19 gezeigten
Isotherme.
0,00
100,00
200,00
300,00
400,00
500,00
0,00 0,20 0,40 0,60 0,80 1,00p/p0
Volu
men
[cc/
g]
Abbildung 18: NB2B-Physisorptionsisotherme für das geordnete, NafionP
®P-haltige System
MCM/Naf10 (schwarz: Adsorption, grau: Desorption).
Die Isothermen sind für beide Systeme ähnlich und zeigen den für meso-
poröse Proben typischen Verlauf. So steigt die NB2B-Aufnahme bei niedrigen
relativen Drücken langsam an, bis bei einem relativen Druck von etwa 0.35
bis 0.4 ein scharfer Anstieg des adsorbierten Volumens eintritt. Dies ent-
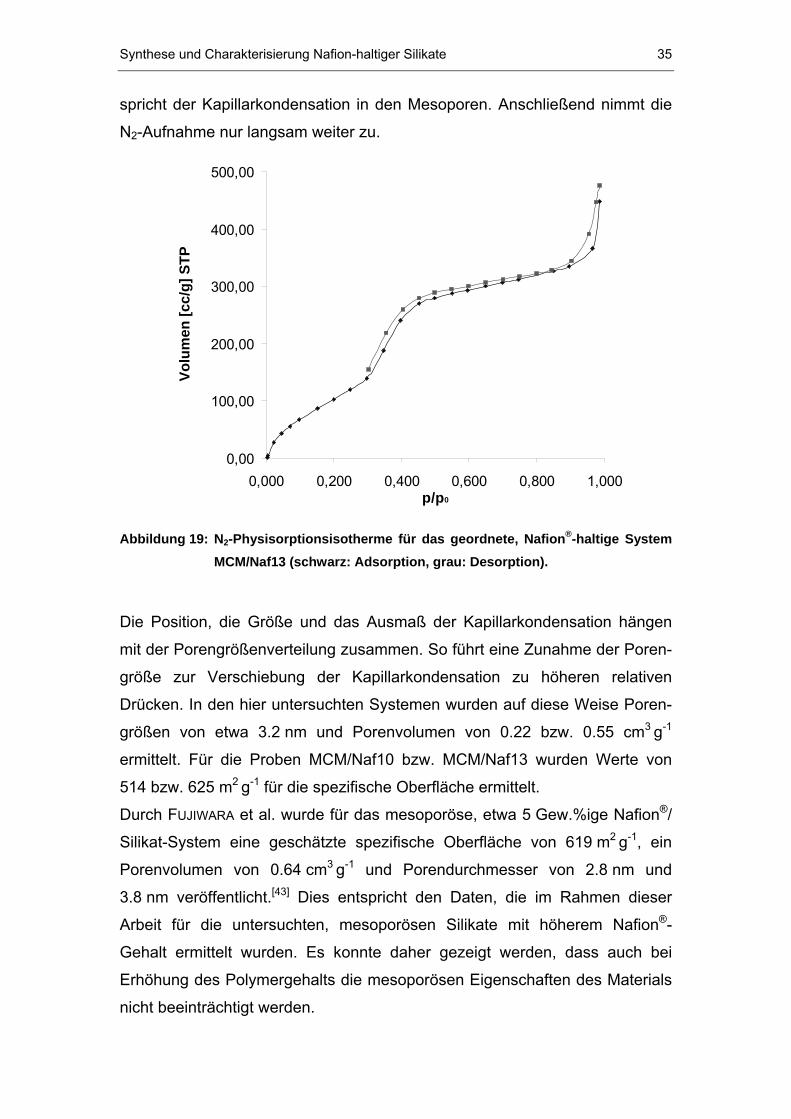
Synthese und Charakterisierung Nafion-haltiger Silikate 35
spricht der Kapillarkondensation in den Mesoporen. Anschließend nimmt die
NB2B-Aufnahme nur langsam weiter zu.
0,00
100,00
200,00
300,00
400,00
500,00
0,000 0,200 0,400 0,600 0,800 1,000p/p0
Volu
men
[cc/
g] S
TP
Abbildung 19: NB2B-Physisorptionsisotherme für das geordnete, NafionP
®P-haltige System
MCM/Naf13 (schwarz: Adsorption, grau: Desorption).
Die Position, die Größe und das Ausmaß der Kapillarkondensation hängen
mit der Porengrößenverteilung zusammen. So führt eine Zunahme der Poren-
größe zur Verschiebung der Kapillarkondensation zu höheren relativen
Drücken. In den hier untersuchten Systemen wurden auf diese Weise Poren-
größen von etwa 3.2 nm und Porenvolumen von 0.22 bzw. 0.55 cm P
3 PgP
-1P
ermittelt. Für die Proben MCM/Naf10 bzw. MCM/Naf13 wurden Werte von
514 bzw. 625 m P
2 PgP
-1P für die spezifische Oberfläche ermittelt.
Durch TFUJIWARAT et al. wurde für das mesoporöse, etwa 5 Gew.%ige NafionP
®P/
Silikat-System eine geschätzte spezifische Oberfläche von 619 mP
2 PgP
-1P, ein
Porenvolumen von 0.64 cm P
3 PgP
-1P und Porendurchmesser von 2.8 nm und
3.8 nm veröffentlicht. P
[43]P Dies entspricht den Daten, die im Rahmen dieser
Arbeit für die untersuchten, mesoporösen Silikate mit höherem NafionP
®P-
Gehalt ermittelt wurden. Es konnte daher gezeigt werden, dass auch bei
Erhöhung des Polymergehalts die mesoporösen Eigenschaften des Materials
nicht beeinträchtigt werden.

Synthese und Charakterisierung Nafion-haltiger Silikate 36
Wie erwartet, zeigt das System SAC/Naf10 eine für eine mikroporöse Probe
typische Isotherme. Die anhand der experimentellen Daten ermittelte
spezifische Oberfläche beträgt 366 m P
2 PgP
-1P. DuPont veröffentlichte für das
kommerziell erhältliche System SAC-13 eine geschätzte spezifische Ober-
fläche von 344 m P
2 PgP
-1P und damit einen fast identischen Wert.
3.4.6 XRD-Messungen
3.4.6.1 Methodenbeschreibung
Die Röntgendiffraktometrie (X-Ray Diffraction) wird genutzt, um Informationen
über Kristallstruktur, Partikelgröße, Phasenzusammensetzung und Gitter-
konstanten eines Feststoffs zu erhalten.
Röntgendiffraktogramme erhält man durch Messung der Winkel, bei denen
ein Röntgenstrahl einer Wellenlänge Tλ T an der Probe gebeugt wird. Die Lage,
Intensität, Form und Breite der Beugungslinien ermöglichen Aussagen über
die Natur der Probe. P
[28, 50]P Dabei ist der Abstand d zwischen zwei Netzebenen
(hkl) durch die Bragg-Gleichung (vgl. Gleichung 5) mit dem Beugungswinkel
2TθT verbunden
Gl. 5 θλ sin2dn = .
Für Einkristalle kann so durch die Ermittlung der diskreten Bragg-Reflexe die
Kristallstruktur ermittelt werden.
Die im Rahmen dieser Arbeit beschriebenen XRD-Untersuchungen wurden
von Frau Dr. C. Weidenthaler in der Abteilung für Kristallographie am Max-
Planck-Institut für Kohlenforschung in Mülheim durchgeführt. Dazu wurde ein
Transmissions-Pulverdiffraktometer der Firma Stoe STAPI P mit einem Cu-
KTBα T1B-Primärmonochromator und einem ortsempfindlichen linearen Detektor
eingesetzt.
Synthese und Charakterisierung Nafion-haltiger Silikate 37
3.4.6.2 XRD-Untersuchung der geordneten Nafion P
®P-haltigen Silikate
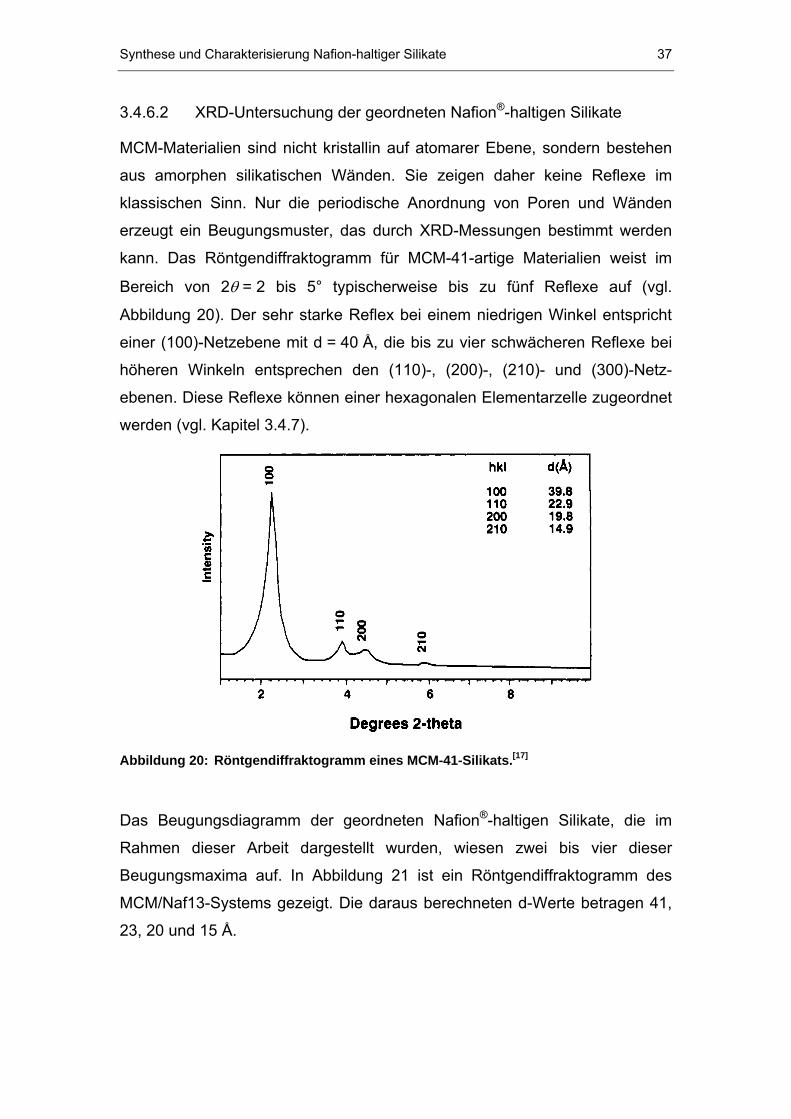
MCM-Materialien sind nicht kristallin auf atomarer Ebene, sondern bestehen
aus amorphen silikatischen Wänden. Sie zeigen daher keine Reflexe im
klassischen Sinn. Nur die periodische Anordnung von Poren und Wänden
erzeugt ein Beugungsmuster, das durch XRD-Messungen bestimmt werden
kann. Das Röntgendiffraktogramm für MCM-41-artige Materialien weist im
Bereich von 2 TθT = 2 bis 5° typischerweise bis zu fünf Reflexe auf (vgl.
Abbildung 20). Der sehr starke Reflex bei einem niedrigen Winkel entspricht
einer (100)-Netzebene mit d = 40 Å, die bis zu vier schwächeren Reflexe bei
höheren Winkeln entsprechen den (110)-, (200)-, (210)- und (300)-Netz-
ebenen. Diese Reflexe können einer hexagonalen Elementarzelle zugeordnet
werden (vgl. Kapitel 3.4.7).
Abbildung 20: Röntgendiffraktogramm eines MCM-41-Silikats.P
[17]P
Das Beugungsdiagramm der geordneten Nafion P
®P-haltigen Silikate, die im
Rahmen dieser Arbeit dargestellt wurden, wiesen zwei bis vier dieser
Beugungsmaxima auf. In Abbildung 21 ist ein Röntgendiffraktogramm des
MCM/Naf13-Systems gezeigt. Die daraus berechneten d-Werte betragen 41,
23, 20 und 15 Å.
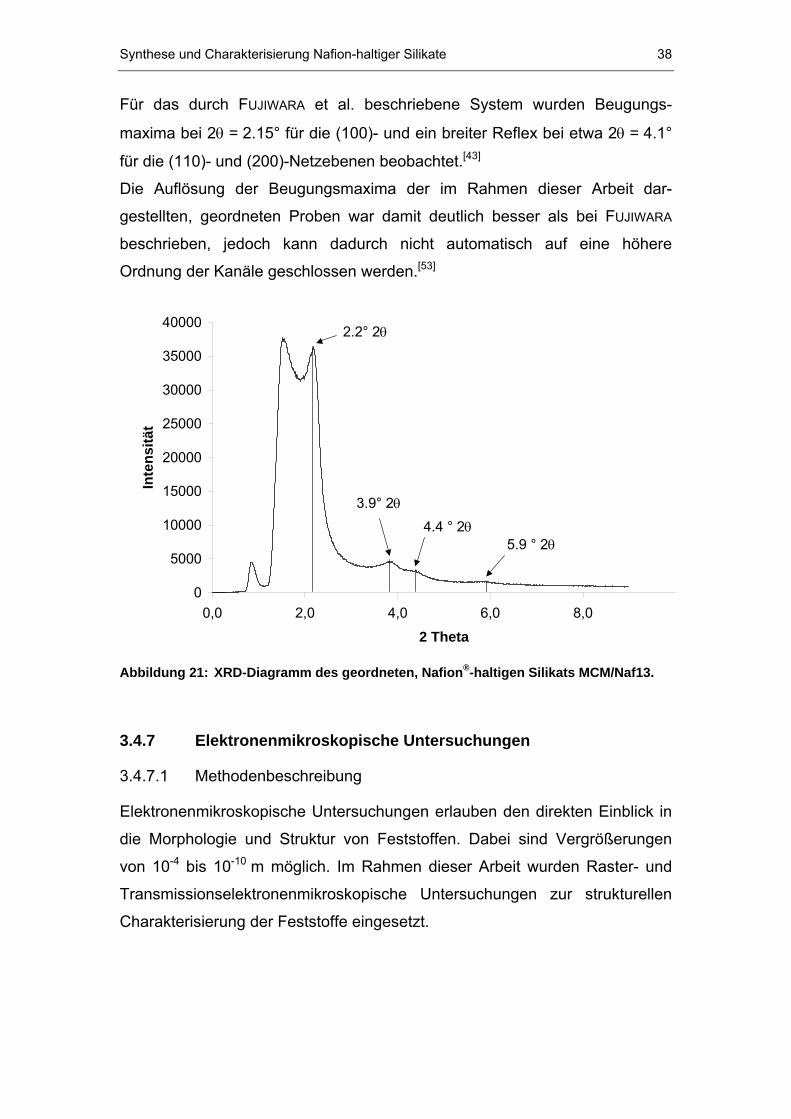
Synthese und Charakterisierung Nafion-haltiger Silikate 38
Für das durch TFUJIWARAT et al. beschriebene System wurden Beugungs-
maxima bei 2 TθT = 2.15° für die (100)- und ein breiter Reflex bei etwa 2Tθ T = 4.1°
für die (110)- und (200)-Netzebenen beobachtet.P
[43]P
Die Auflösung der Beugungsmaxima der im Rahmen dieser Arbeit dar-
gestellten, geordneten Proben war damit deutlich besser als bei TFUJIWARA T
beschrieben, jedoch kann dadurch nicht automatisch auf eine höhere
Ordnung der Kanäle geschlossen werden. P
[53]P
0
5000
10000
15000
20000
25000
30000
35000
40000
0,0 2,0 4,0 6,0 8,0
2 Theta
Inte
nsitä
t
2.2° 2θ
3.9° 2θ4.4 ° 2θ
5.9 ° 2θ
Abbildung 21: XRD-Diagramm des geordneten, Nafion P
®P-haltigen Silikats MCM/Naf13.
3.4.7 Elektronenmikroskopische Untersuchungen
3.4.7.1 Methodenbeschreibung
Elektronenmikroskopische Untersuchungen erlauben den direkten Einblick in
die Morphologie und Struktur von Feststoffen. Dabei sind Vergrößerungen
von 10P
-4P bis 10 P
-10P m möglich. Im Rahmen dieser Arbeit wurden Raster- und
Transmissionselektronenmikroskopische Untersuchungen zur strukturellen
Charakterisierung der Feststoffe eingesetzt.

Synthese und Charakterisierung Nafion-haltiger Silikate 39
SEM-Untersuchungen
Bei der Rasterelektronenmikroskopie (SEM, scanning electron microscopy)
wird ein Elektronenstrahl durch Linsen vor der Probe so fokussiert, dass nur
eine sehr kleine Elektronenstrahlmikrosonde auf die Probe gerichtet ist. Die
Wechselwirkung zwischen Elektronenstrahlmikrosonde und Atomen in der
kleinen bestrahlten Region der Probe erzeugt verschiedene Arten von
Signalen, die durch spezifische Detektoren aufgenommen und für analytische
Zwecke und Abbildungen genutzt werden können. In der SEM wird das Bild
der Probe durch Registrierung der Sekundärelektronen oder rückgestreuter
Elektronen der bestrahlten Fläche erzeugt. Dazu ist ein Elektronendetektor
seitlich vor der Probe angebracht. Die Oberfläche der Probe muss elektronen-
leitend sein und wird daher meist durch Sputtern mit einer leitenden Metall-
schicht bedampft. Die Anzahl an emittierten Elektronen hängt stark vom
Winkel, mit dem der Elektronenstrahl auf die Probenoberfläche und dann auf
den Detektor trifft, ab. Zum Beispiel wird durch Bestrahlung einer vertikalen
Fläche eine große Anzahl an Sekundärelektronen freigesetzt und so erscheint
diese Fläche im SEM-Bild heller. Umgekehrt werden durch eine horizontale
Oberfläche weniger Sekundärelektronen freigesetzt und dieser Bereich wird
weniger hell abgebildet. Daher erscheinen die erzeugten Bilder der Proben-
oberfläche dreidimensional. Allerdings wird in der Rasterelektronenmikro-
skopie nur eine verhältnismäßig niedrige Auflösung von 5 bis 10 nm
erreicht.P
[50]P
TEM-Untersuchungen
Das nützlichste Werkzeug zur Untersuchung von wenigen Nanometer großen
Partikeln und Strukturen ist das Transmissionselektronenmikroskop (TEM),
besonders das hochauflösende TEM (HRTEM, high resolution transmission
electron microscope).
Das Prinzip der Bilderzeugung mittels Transmissionselektronenmikroskopie
ist in Abbildung 22 gezeigt. Die Elektronen und Streuelektronen werden mit
der Linse in der hinteren Brennebene fokussiert, wo das Beugungsbild
erzeugt wird. Nachdem die gestreuten und ungestreuten Wellen durch diese
Ebene hindurchgetreten sind, interferieren sie unter Ausbildung eines Bildes
in der Bildebene. Die Projektionslinsen können entweder auf das Beugungs-

Synthese und Charakterisierung Nafion-haltiger Silikate 40
bild oder auf das Zwischenbild fokussiert, und so das fertige Beugungsbild
oder Bild auf einem Fluoreszenzschirm abgebildet werden. Das erzeugte Bild
ist eine Projektion der untersuchten Struktur auf die Ebene senkrecht zum
Elektronenstrahl, d. h. die Partikel werden in nur zwei Dimensionen abge-
bildet.
Abbildung 22: Prinzip der Bilderzeugung in der Transmissionselektronenmikroskopie: (1) Partikel im parallelen Elektronenstrahl, (2) Objektivlinse, (3) hintere Brennebene, (4) Bildebene, (5) Öffnung.P
[50]P
EDX
Zur Aufklärung der chemischen Zusammensetzung der Probe kann die
Energy dispersive X-Ray-Analyse (EDX) genutzt werden. Dazu wird in einem
punktförmigen Spot von ca. 2 nm Größe der Elektronenstrahl auf die Probe
fokussiert. Die im Elektronenmikroskop beschleunigten Elektronen regen die
kernnahen Elektronen der Objektatome an und bewirken eine Ionisierung der
Probe und damit die Emission von Röntgenstrahlen. Die emittierten Röntgen-
quanten werden durch einen EDX-Detektor aufgefangen und gezählt. Da
jedes Element ein einzigartiges Muster an Röntgenlinien der K-, L- und M-
Schale aufweist, ist so der Nachweis vorhandener Elemente möglich. Durch
den punktförmigen Messbereich können die Werte für die Anteile an den
untersuchten Elementen jedoch nur als Richtwert dienen, eine Angabe der
exakten Menge ist nur begrenzt möglich.

Synthese und Charakterisierung Nafion-haltiger Silikate 41
3.4.7.2 Elektronenmikroskopische Untersuchung Nafion P
®P-haltiger Silikate
Für die im Rahmen dieser Arbeit dargestellten NafionP
®P-haltigen Silikate
wurden Rasterelektronenaufnahmen erstellt und verglichen.
So wurde für das ungeordnete System SAC/Naf10 die in Abbildung 23
gezeigten SEM-Aufnahmen erhalten. Es sind bei kleineren Vergrößerungen
von 1 k splitterförmige Partikel verschiedener Größen zu erkennen. Bei
höherer Vergrößerung von 10 k zeigt sich eine geschuppte Oberflächen-
struktur, die typisch für basisch gewonnene Silikate ist.P
[13]P
Abbildung 23: SEM-Aufnahmen des ungeordneten Systems SAC/Naf10 bei einer Ver-größerung von 1 k und 10 k.
Die Verteilung der im Verbundwerkstoff enthaltenen Elemente wurde mittels
Elemental Mapping (EDX) untersucht. Dazu wurde das Pulver in Epoxydharz
eingebettet und angeschnitten. So ist ein Einblick ins Innere des Verbund-
werkstoffs möglich. Es zeigte sich eine gleichmäßige Verteilung der Elemente
Kohlenstoff, Sauerstoff, Silicium und Fluor in den Partikeln. In Abbildung 24
sind Übersichtsaufnahmen für die Elemente Sauerstoff, Silicium und Fluor
gezeigt. Da Silicium und Fluor in der gesamten Probe gleichmäßig verteilt vor-
liegen, kann von einer hohen Dispersion des Nafion P
®P-Polymers im Silikat
ausgegangen werden.

Synthese und Charakterisierung Nafion-haltiger Silikate 42
Abbildung 24: Übersichtsaufnahme für die Elemente Sauerstoff, Silicium und Fluor im ungeordneten System SAC/Naf10.
Durch DuPont wurden SEM- bzw. EDX-Untersuchungen an einem
40 Gew.%igem NafionP
®P-haltigen Silikat bzw. am kommerziell erhältlichen
System SAC-13 beschrieben. P
[35]P Auch diese Proben zeigten eine hohe Ver-
teilung des Polymers im Silikat.
Das geordnete NafionP
®P-haltige System MCM/Naf10 zeigt im Gegensatz zum
ungeordneten System bei einer Vergrößerung von 2.5 k keine splitterförmige,
sondern eine kugelförmige Struktur. Diese kugelförmigen Partikel sind zu
Haufen agglomeriert. Bei weiterer Vergrößerung bis auf 10 k erkennt man
eine beerenförmige Unterstruktur. Die SEM-Aufnahmen sind in Abbildung 25
dargestellt.
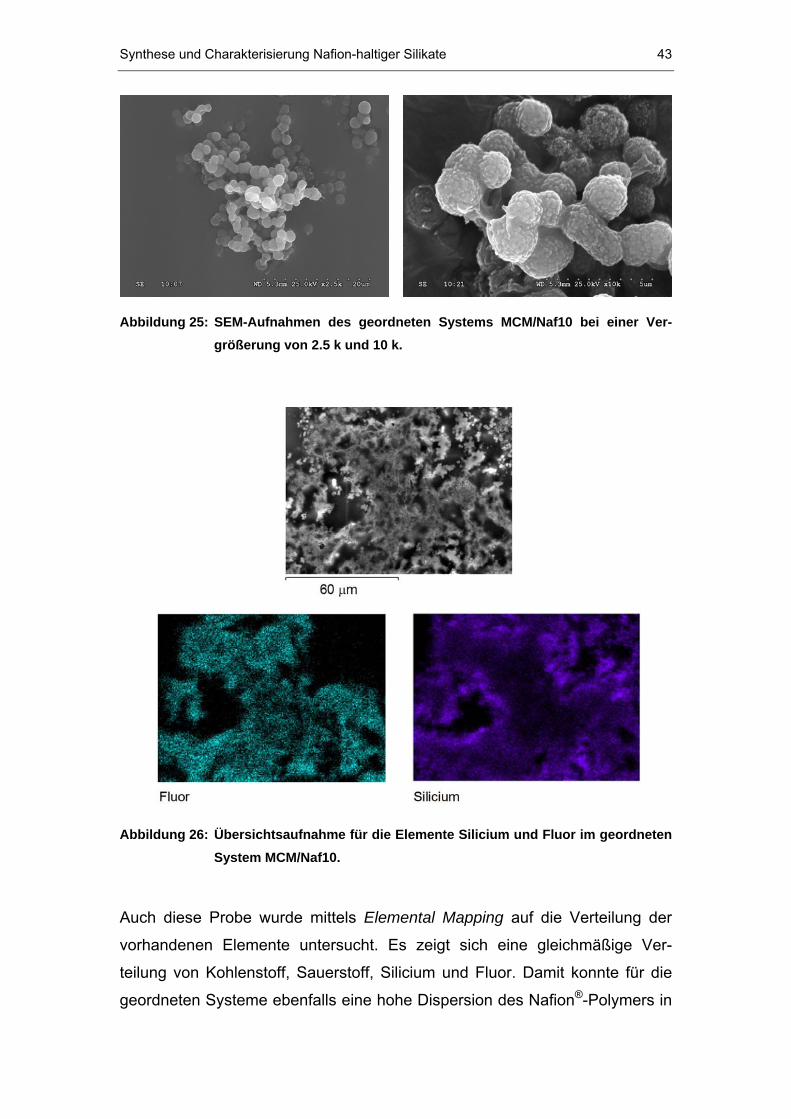
Synthese und Charakterisierung Nafion-haltiger Silikate 43
Abbildung 25: SEM-Aufnahmen des geordneten Systems MCM/Naf10 bei einer Ver-größerung von 2.5 k und 10 k.
Abbildung 26: Übersichtsaufnahme für die Elemente Silicium und Fluor im geordneten System MCM/Naf10.
Auch diese Probe wurde mittels Elemental Mapping auf die Verteilung der
vorhandenen Elemente untersucht. Es zeigt sich eine gleichmäßige Ver-
teilung von Kohlenstoff, Sauerstoff, Silicium und Fluor. Damit konnte für die
geordneten Systeme ebenfalls eine hohe Dispersion des Nafion P
®P-Polymers in

Synthese und Charakterisierung Nafion-haltiger Silikate 44
der Silikatmatrix nachgewiesen werden. In Abbildung 26 sind die Verteilungen
von Silicium und Fluor dargestellt.
Durch TFUJIWARAT et al. wurden für das geordnete, etwa 5 Gew.%ige System
keine rasterelektronenmikroskopischen Untersuchungen veröffentlicht. P
[43]P
Transmissionselektronenmikroskopische Aufnahmen (vgl. Abbildung 27)
zeigen für MCM-41-Materialien eine gleichmäßige, hexagonale Anordnung
von gleichgroßen Kanälen, ähnlich dem Aufbau von Bienenwaben.
Abbildung 27: TEM-Aufnahme des durch Mobil-Forscher dargestellten MCM-41. P
[17]P
Für das im Rahmen dieser Arbeit dargestellte, geordnete Nafion P
®P-haltige
System MCM/Naf10 wurden die in Abbildung 28 gezeigten TEM-Aufnahmen
erhalten. In diesen Aufnahmen ist die regelmäßige, wabenförmige Struktur
erkennbar. Je nach Betrachtungswinkel sind die Waben von oben oder die
Kanäle entlang der Schnittflächen zu sehen. Der geschätzte Durchmesser der
Kanäle beträgt etwa 3 bis 4 nm.

Synthese und Charakterisierung Nafion-haltiger Silikate 45
Abbildung 28: TEM-Aufnahmen des geordneten NafionP
®P-haltigen Silikats MCM/Naf10
(Vergrößerungen 50000 und 100000).
Erst die Kombination von elektronenmikroskopischen Aufnahmen, den Ergeb-
nissen der Stickstoffsorptionsmessungen und den XRD-Untersuchungen
lassen eine Aussage über den geordneten, mesoporösen Charakter einer
MCM-41-artigen Probe zu. P
[53]P Damit konnte nachgewiesen werden, dass für
die MCM/Naf-Proben trotz der Immobilisierung des Nafion P
®P-Polymers im
Silikat eine geordnete Struktur erhalten wird.

Platinsysteme 46
4 Platinsysteme
Für die Darstellung von Platin-beladenen Silikaten wurden bereits viele
Untersuchungen in der Literatur beschrieben. Dazu können verschiedene
Methoden eingesetzt werden, wie zum Beispiel der IonenaustauschP
[55-61]P, die
so genannte Incipient Wetness Impregnation (IWI) P
[60, 62-64]P, die Gleich-
gewichtsadsorption (Equilibrium Adsorption, EA) P
[65]P oder das Aufbringen
flüchtiger Platinverbindungen in der Gasphase (Vapor Phase Deposition) P
[65]P.
Dabei häufig eingesetzte Platinverbindungen sind ionische Precursor, wie die
Tetrammin-Platinsalze Pt(NHB3B) B4B(NO B3B) B2B, Pt(NHB3B) B4B(OH) B2B, Pt(NHB3B) B4BClB2B oder die
Hexachloroplatinsäure H B2BPtClB6B, und neutrale Vorläufer, wie Platinacetyl-
acetonat Pt(acac) B2B.
Eine weitere Möglichkeit zur Fixierung von metallorganischen Spezies ist die
Bildung einer kovalenten Bindung zwischen Träger und Metall-Precursor.
Dabei können sowohl die Oberflächensilanolgruppen des Silikats reagieren
als auch Spacergruppen mit reaktiven Endgruppen, die auf das Silikat aufge-
bracht wurden (vgl. Kapitel 1.5). Wird bei der Umsetzung des Metall-
Precursors und der funktionellen Gruppen des Trägers ein Gas gebildet, so
kann bei milden Bedingungen ein quantitativer Umsatz erreicht und eine
Rückreaktion verhindert werden. In der Literatur ist beispielsweise die
Umsetzung von N,N-Dialkylcarbamato-Komplexen mit den sauren Silanol-
gruppen eines Silikats unter Freisetzung von Kohlenstoffdioxid und einem
sekundären Amin beschrieben. P
[66]P Die auf diese Weise dargestellten Platin-
beladenen Silikate wurden nach Reduktion des Edelmetalls in der Hydrierung
von Cyclohexen bei Raumtemperatur eingesetzt.
Im Rahmen der vorliegenden Arbeit sollte die Reaktion der sauren Sulfon-
säurereste des NafionP
®P-Polymers mit einem Dimethyl-Platin-Komplex unter
Freisetzung des Methans untersucht werden. Durch die gezielte Reaktion der
Säurefunktionalitäten sollte eine hohe Verteilung des Platins auf dem Träger-
material erreicht werden. Die so dargestellten Platin-beladenen Systeme
sollten nach der Reduktion in der Katalyse eingesetzt werden, wie zum
Beispiel in Hydrierungsreaktionen.

Platinsysteme 47
Im Folgenden ist zunächst die Synthese der Platin-beladenen Nafion P
®P-
haltigen Silikate beschrieben und anschließend die analytische und kataly-
tische Charakterisierung dieser Systeme.
4.1 Synthese
Zur Darstellung der Platin-beladenen Nafion P
®P-haltigen Silikate wurde ein
NafionP
®P-haltiges Silikat in Lösungsmittel suspendiert und dann die ge-
wünschte Menge an ( TηTP
4P-1,5-Cyclooctadien)dimethyl-Platin(II)-Komplex, gelöst
im selben Lösungsmittel, bei Raumtemperatur zur Suspension gegeben (vgl.
Abbildung 29). Man lässt die Reaktionsmischung für mehrere Stunden rühren
und gewinnt nach Filtration und Waschen das weiße pulverförmige Produkt.
CF2 SO3H
Nafion/SiO2
+
H3C
Pt
H3CCF2 SO3H
CF2 SO3
Nafion/SiO2 CF2 SO3
2 CH4
Pt(COD)2+
(COD)
Abbildung 29: Darstellung der Platin-beladenen, NafionP
®P-haltigen Silikate.
Als Träger wurden sowohl geordnete als auch ungeordnete Nafion P
®P-haltige
Silikate verwendet. In der Reaktion wurden verschiedene Lösungsmittel, wie
Aceton, Dichlormethan, Toluol, Pentan und MTBE, eingesetzt. Außerdem
wurde die Menge des Platins variiert und Systeme mit Beladungen von 0.3 bis

Platinsysteme 48
2.0 Gew.% Platin hergestellt. Das Platin in diesen Systemen wurde in einer
Wasserstoffatmosphäre (1.4 bar) bei Raumtemperatur reduziert.
Eine Zusammenstellung der dargestellten Systeme ist in Tabelle 9 aufgeführt.
Tabelle 9: Dargestellte Platin-beladene NafionP
®P-haltige Systeme.
Ungeordnete Systeme Geordnete Systeme
SAC/Naf10/Pt1.3 MCM/Naf10/Pt0.3 bis 2.0
SAC/Naf13/Pt1.7 MCM/Naf13/Pt0.8 bis 2.3
SAC/Naf40/Pt4.3
Des Weiteren wurden die Nafion P
®P-haltigen Silikate mit Dichloro( TηTP
4P-cyclo-
octadien)platin(II) umgesetzt, um zu überprüfen, ob auch ohne die Alkyl-
gruppen im Platin-Precursor eine Reaktion mit den sauren Sulfonsäureresten
des NafionP
®P-Polymers eintritt. Die Produkte enthielten jedoch keine nachweis-
baren Mengen an Platin und zeigten auch keine Reaktivität in der Katalyse
(vgl. Kapitel 4.3.1).
4.2 Charakterisierung
Im Weiteren wird die Charakterisierung der Platin-beladenen Systeme be-
schrieben. Da sich die Beladung an Platin für einige Methoden als zu niedrig
erwies und eine Charakterisierung erschwerte, wurde als weiteres Träger-
material ein 40 Gew.%iges Nafion P
®P-haltiges Silikat (SAC/Naf40) mit dem
Platin-Precursor behandelt und das Produkt als Modellsystem untersucht.
4.2.1 Elementaranalyse
Die Platin-beladenen Systeme wurden mittels Elementaranalyse auf ihren
Platingehalt untersucht. Für die angegebenen Werte ist ein relativer Fehler
von 5 % anzunehmen.
Nach Umsetzung des ungeordneten Trägermaterials SAC/Naf10 in ver-
schiedenen Lösungsmitteln mit einer Platin-Precursor-Menge, die zu
1.3 Gew.% Platin im Produkt führen sollte, wurden die in Tabelle 10 auf-
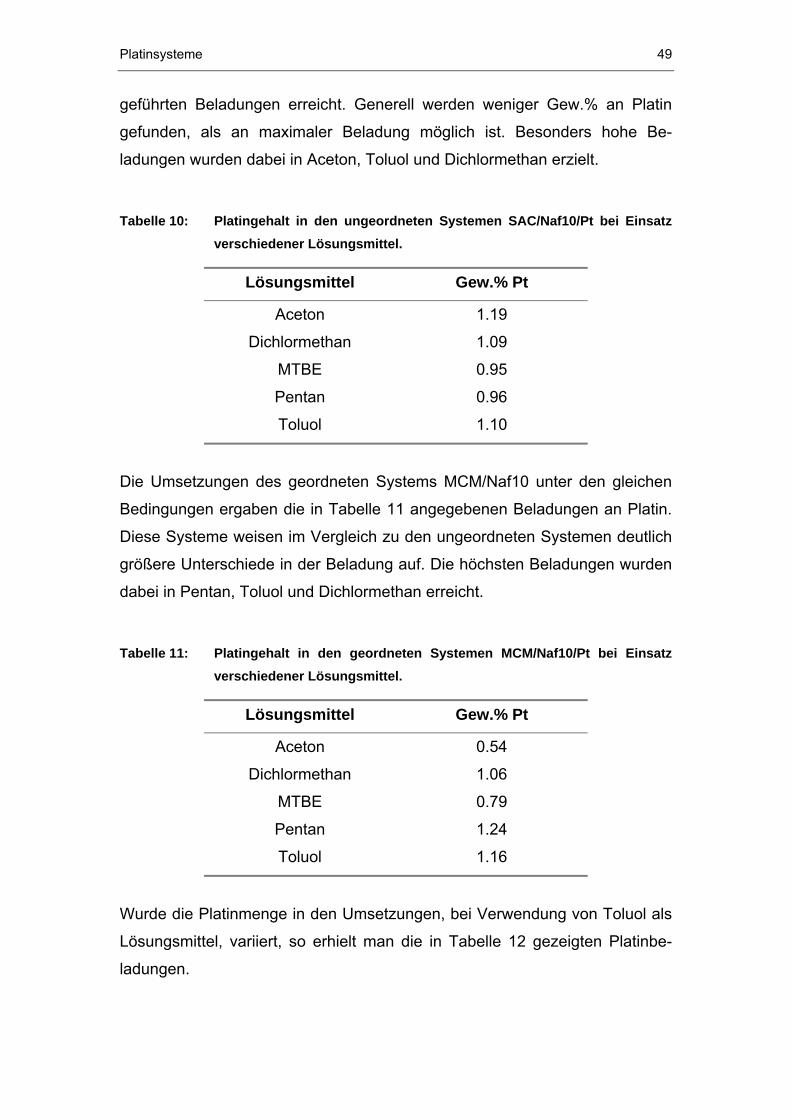
Platinsysteme 49
geführten Beladungen erreicht. Generell werden weniger Gew.% an Platin
gefunden, als an maximaler Beladung möglich ist. Besonders hohe Be-
ladungen wurden dabei in Aceton, Toluol und Dichlormethan erzielt.
Tabelle 10: Platingehalt in den ungeordneten Systemen SAC/Naf10/Pt bei Einsatz verschiedener Lösungsmittel.
Lösungsmittel Gew.% Pt
Aceton 1.19
Dichlormethan 1.09
MTBE 0.95
Pentan 0.96
Toluol 1.10
Die Umsetzungen des geordneten Systems MCM/Naf10 unter den gleichen
Bedingungen ergaben die in Tabelle 11 angegebenen Beladungen an Platin.
Diese Systeme weisen im Vergleich zu den ungeordneten Systemen deutlich
größere Unterschiede in der Beladung auf. Die höchsten Beladungen wurden
dabei in Pentan, Toluol und Dichlormethan erreicht.
Tabelle 11: Platingehalt in den geordneten Systemen MCM/Naf10/Pt bei Einsatz verschiedener Lösungsmittel.
Lösungsmittel Gew.% Pt
Aceton 0.54
Dichlormethan 1.06
MTBE 0.79
Pentan 1.24
Toluol 1.16
Wurde die Platinmenge in den Umsetzungen, bei Verwendung von Toluol als
Lösungsmittel, variiert, so erhielt man die in Tabelle 12 gezeigten Platinbe-
ladungen.

Platinsysteme 50
Tabelle 12: Platingehalt in den geordneten Systemen bei Umsatz in Toluol unter Variation der Menge an Platin-Precursor.
Theoretischer Pt-GehaltGew.%
Pt-Gehalt nach EA Gew.%
2.0 1.96
1.3 1.16
1.0 1.18
0.7 1.02
0.3 0.50
Dabei zeigt sich eine starke Abweichung der experimentell ermittelten von
den theoretischen Platinbeladungen. Diese sind deutlich größer als der ange-
gebene relative Fehler von 5 %. Bei den niedrigen Beladungen, ab einem
theoretischen Wert von 1.0 Gew.% Platin, wurden in allen Fällen größere
Werte als maximal möglich ermittelt. Daher wurde in den durchgeführten
Katalysen (vgl. Kapitel 4.3) jeweils mit der theoretisch berechneten Menge an
Platin gearbeitet und nicht die experimentell ermittelten Werte verwendet.
4.2.2 XRD-Untersuchungen
Um zu überprüfen, ob die Platin-Beladung der geordneten Systeme die
geordnete Struktur beeinträchtigt, wurden an verschiedenen geordneten,
Platin-beladenen, Nafion P
®P-haltigen Silikaten XRD-Messungen (vgl. Ab-
schnitt 3.4.6) durchgeführt.
Für alle untersuchten Proben konnte gezeigt werden, dass auch nach der
Platin-Beladung eine geordnete Silikatphase vorliegt. Als Beispiel ist in Ab-
bildung 30 das XRD-Beugungsdiagramm des Systems MCM/Naf10/Pt1.2
dargestellt.
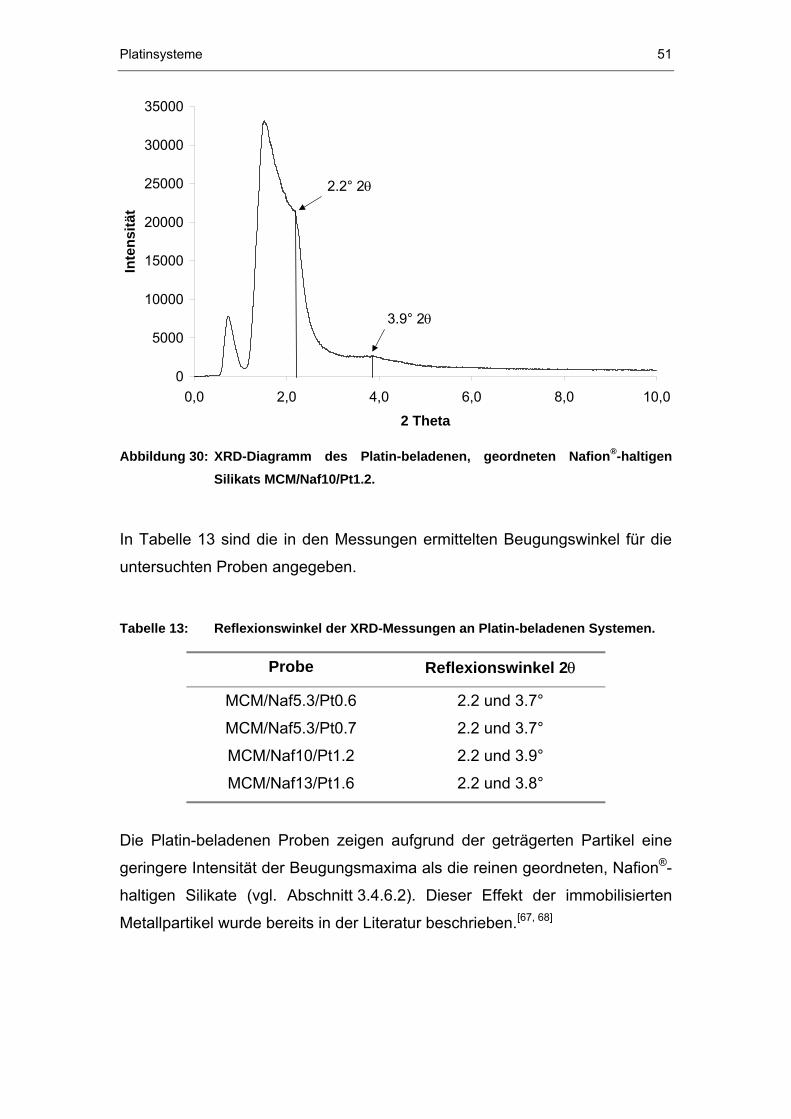
Platinsysteme 51
0
5000
10000
15000
20000
25000
30000
35000
0,0 2,0 4,0 6,0 8,0 10,0
2 Theta
Inte
nsitä
t
2.2° 2θ
3.9° 2θ
Abbildung 30: XRD-Diagramm des Platin-beladenen, geordneten Nafion P
®P-haltigen
Silikats MCM/Naf10/Pt1.2.
In Tabelle 13 sind die in den Messungen ermittelten Beugungswinkel für die
untersuchten Proben angegeben.
Tabelle 13: Reflexionswinkel der XRD-Messungen an Platin-beladenen Systemen.
Probe Reflexionswinkel 2Tθ T
MCM/Naf5.3/Pt0.6 2.2 und 3.7°
MCM/Naf5.3/Pt0.7 2.2 und 3.7°
MCM/Naf10/Pt1.2 2.2 und 3.9°
MCM/Naf13/Pt1.6 2.2 und 3.8°
Die Platin-beladenen Proben zeigen aufgrund der geträgerten Partikel eine
geringere Intensität der Beugungsmaxima als die reinen geordneten, Nafion P
®P-
haltigen Silikate (vgl. Abschnitt 3.4.6.2). Dieser Effekt der immobilisierten
Metallpartikel wurde bereits in der Literatur beschrieben. P
[67, 68]P

Platinsysteme 52
Es wurde außerdem versucht, die Platin-Spezies mittels XRD-Messungen zu
untersuchen. Allerdings konnte aufgrund der niedrigen Platin-Beladung und
der Position der Platin-Spezies im Inneren der Kanäle (vgl. Abschnitt 4.2.4)
keine Beugung beobachtet werden. In der Literatur wurde dieselbe
Beobachtung für Platin-Partikel in kubischen, mesoporösen Silikaten erzielt,
für die ebenfalls keine Platin-Linien nachgewiesen werden konnten. P
[69]P
4.2.3 XPS-Untersuchungen
4.2.3.1 Methodenbeschreibung
Die Röntgen-Photoelektronen-Spektroskopie (X-ray photoelectron spectro-
scopy, XPS) gibt Informationen über die chemische Zusammensetzung von
Oberflächen von Metall-Komplexen, atomaren Verbindungen oder anorga-
nischen Festkörpern.P
[50]P So kann zum Beispiel anhand der gemessenen
Spektren die Oxidationsstufe der vorhandenen Elemente ermittelt werden.
Allerdings handelt es sich bei dieser Spektroskopiemethode um eine reine
Oberflächenmethode, so dass keine Aussagen über das Material insgesamt,
sondern nur über die Feststoffoberfläche getroffen werden kann.
Messgeräte für die Aufzeichnung von XP-Spektren bestehen prinzipiell aus
einer Quelle für Röntgenstrahlung und einem Energieanalysator. Als
Röntgenquelle werden Aluminium-, Magnesium- oder Chrom-Anoden
verwendet, die eine Röntgenstrahlung von 1486.6 eV, 1253.6 eV beziehungs-
weise 5420 eV emittieren. P
[70]P Beim Auftreffen der Röntgenstrahlung auf die
Probe werden Photoelektronen freigesetzt. Die kinetische Energie EBkin B dieser
austretenden Photoelektronen wird experimentell gemessen.
Durch die experimentell ermittelte kinetische Energie EBkin B der austretenden
Photoelektronen kann nach Gleichung 6 die Ionisierungsenergie EBi B der
Rumpfelektronen berechnet werden.
Gl. 6 ikin EhmvE −== ν2
21
Dabei gibt hTνT die Energie der verwendeten Röntgenstrahlung an. Die
Ionisierungsenergie EBi B kann nach dem Koopmann´schen Theorem in guter

Platinsysteme 53
Näherung mit der Bindungsenergie EBb B gleichgesetzt und so die Bindungs-
energie EBb B der emittierten Photoelektronen ermittelt werden.
Gl. 7 kinb EhE −= ν
Die im Rahmen dieser Arbeit aufgeführten XPS-Messungen wurden von Frau
Dr. C. Weidenthaler in der Abteilung für Kristallographie des Max-Planck-
Instituts für Kohlenforschung in Mülheim/Ruhr mit einem Spektrometer vom
Typ Axis HSi der Firma Kratos Analytical unter Verwendung mono-
chromatisierter Al- TΚBα TB-Strahlung durchgeführt. Das Gerät arbeitet im Hybrid-
Linsenmodus, d. h. mit einer Kombination aus magnetischen und elektro-
statischen Linsen, und einer Anodeleistung von 15 kV und 15 mA. Die Pass
Energy (PE), das Energiefenster während der Untersuchung, variiert von
160 eV für Übersichtsspektren bis 40 eV für hochaufgelöste Spektren. Zur
Korrektur von Aufladungseffekten wurden die Werte auf das Signal der C 1s-
Elektronen bei 285.0 eV oder auf das Signal der Si 2p-Elektronen des Silikats
bei 103.4 eV bezogen. Sämtliche Substanzen wurden in Pulverform
gemessen. Die gemessenen Daten wurden einem theoretischen Diagramm
angepasst. Erfolgte die beste Anpassung mit mehreren chemischen Spezies,
so wurde der Massenanteil angegeben, mit dem die Spezies in die An-
näherung des experimentellen Spektrums eingingen.
4.2.3.2 XPS-Messungen der Platin-beladenen Systeme
MCM/Naf10/Pt
Zunächst wurden geordnete Systeme des Typs MCM/Naf10/Pt mit ver-
schiedenen Platinbeladungen untersucht und die Ergebnisse mit denen des
Trägermaterials MCM/Naf10 verglichen. Die ermittelten Werte sind in Tabelle
14 aufgeführt.
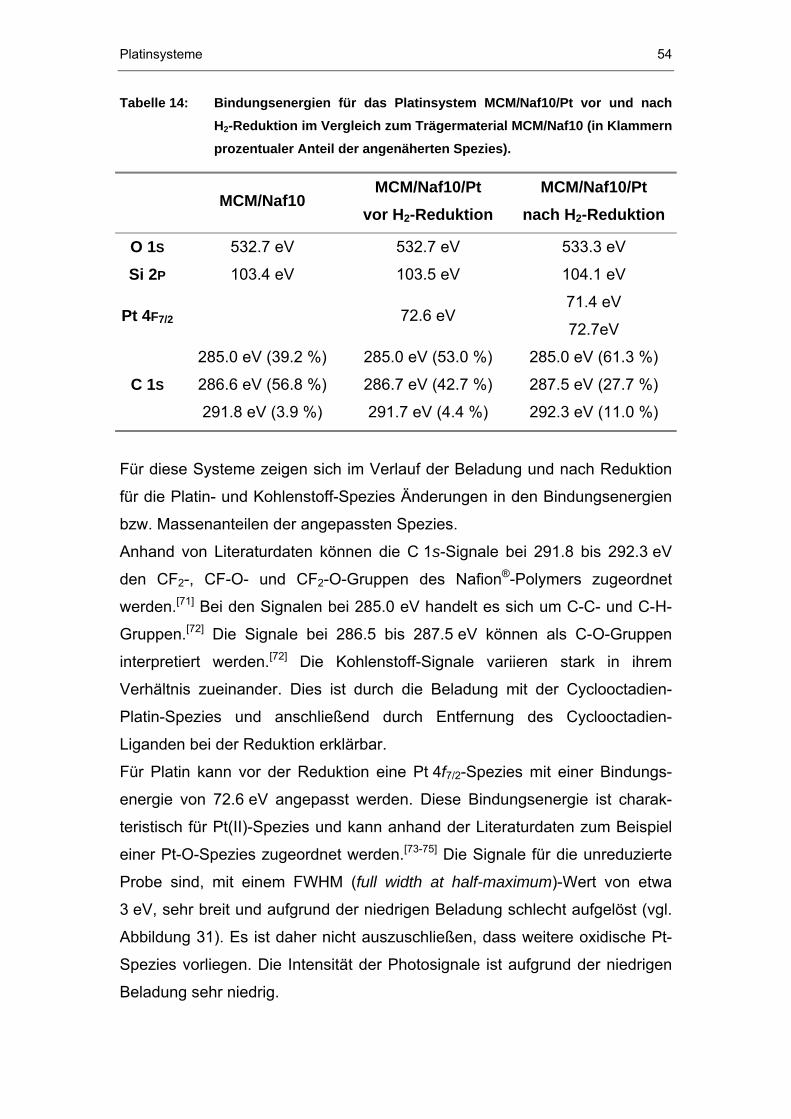
Platinsysteme 54
Tabelle 14: Bindungsenergien für das Platinsystem MCM/Naf10/Pt vor und nach HB2B-Reduktion im Vergleich zum Trägermaterial MCM/Naf10 (in Klammern prozentualer Anteil der angenäherten Spezies).
MCM/Naf10 MCM/Naf10/Pt
vor HB2B-Reduktion MCM/Naf10/Pt
nach HB2B-Reduktion
O 1TST 532.7 eV 532.7 eV 533.3 eV
Si 2TPT 103.4 eV 103.5 eV 104.1 eV
Pt 4TFTB7/2B 72.6 eV 71.4 eV
72.7eV
285.0 eV (39.2 %) 285.0 eV (53.0 %) 285.0 eV (61.3 %)
286.6 eV (56.8 %) 286.7 eV (42.7 %) 287.5 eV (27.7 %) C 1TST
291.8 eV (3.9 %) 291.7 eV (4.4 %) 292.3 eV (11.0 %)
Für diese Systeme zeigen sich im Verlauf der Beladung und nach Reduktion
für die Platin- und Kohlenstoff-Spezies Änderungen in den Bindungsenergien
bzw. Massenanteilen der angepassten Spezies.
Anhand von Literaturdaten können die C 1s-Signale bei 291.8 bis 292.3 eV
den CFB2B-, CF-O- und CFB2B-O-Gruppen des Nafion P
®P-Polymers zugeordnet
werden. P
[71]P Bei den Signalen bei 285.0 eV handelt es sich um C-C- und C-H-
Gruppen. P
[72]P Die Signale bei 286.5 bis 287.5 eV können als C-O-Gruppen
interpretiert werden. P
[72]P Die Kohlenstoff-Signale variieren stark in ihrem
Verhältnis zueinander. Dies ist durch die Beladung mit der Cyclooctadien-
Platin-Spezies und anschließend durch Entfernung des Cyclooctadien-
Liganden bei der Reduktion erklärbar.
Für Platin kann vor der Reduktion eine Pt 4f B7/2B-Spezies mit einer Bindungs-
energie von 72.6 eV angepasst werden. Diese Bindungsenergie ist charak-
teristisch für Pt(II)-Spezies und kann anhand der Literaturdaten zum Beispiel
einer Pt-O-Spezies zugeordnet werden. P
[73-75]P Die Signale für die unreduzierte
Probe sind, mit einem FWHM (full width at half-maximum)-Wert von etwa
3 eV, sehr breit und aufgrund der niedrigen Beladung schlecht aufgelöst (vgl.
Abbildung 31). Es ist daher nicht auszuschließen, dass weitere oxidische Pt-
Spezies vorliegen. Die Intensität der Photosignale ist aufgrund der niedrigen
Beladung sehr niedrig.
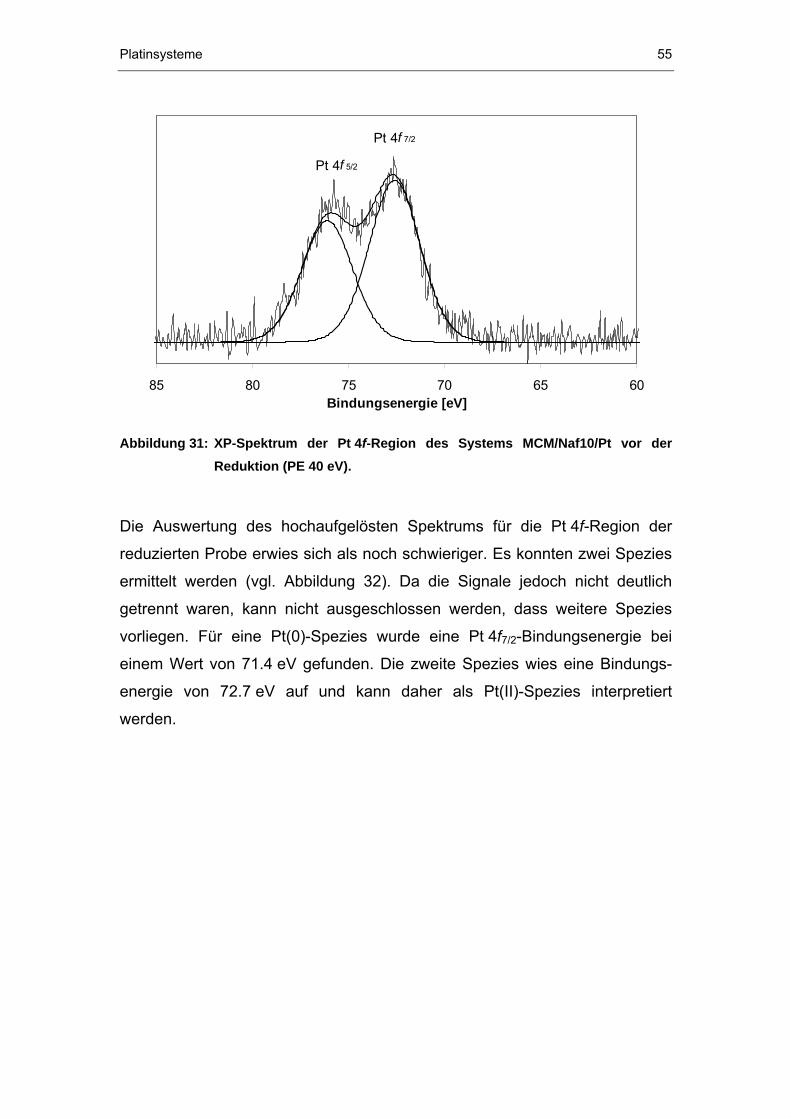
Platinsysteme 55
606570758085Bindungsenergie [eV]
Pt 4f 7/2
Pt 4f 5/2
Abbildung 31: XP-Spektrum der Pt 4f-Region des Systems MCM/Naf10/Pt vor der Reduktion (PE 40 eV).
Die Auswertung des hochaufgelösten Spektrums für die Pt 4f-Region der
reduzierten Probe erwies sich als noch schwieriger. Es konnten zwei Spezies
ermittelt werden (vgl. Abbildung 32). Da die Signale jedoch nicht deutlich
getrennt waren, kann nicht ausgeschlossen werden, dass weitere Spezies
vorliegen. Für eine Pt(0)-Spezies wurde eine Pt 4f B7/2B-Bindungsenergie bei
einem Wert von 71.4 eV gefunden. Die zweite Spezies wies eine Bindungs-
energie von 72.7 eV auf und kann daher als Pt(II)-Spezies interpretiert
werden.
Platinsysteme 56
606570758085Bindungsenergie [eV]
Pt 4f 7/2
Pt 4f 5/2
Abbildung 32: XP-Spektrum der Pt 4f-Region des Systems MCM/Naf10/Pt nach der Reduktion (PE 40 eV).
Generell weisen die hier untersuchten Systeme verhältnismäßig schwache
Signale auf, was sehr gut im Übersichtsspektrum (vgl. Abbildung 33) ersicht-
lich wird. Daher wurde im Weiteren ein deutlich höher beladenes Modell-
system für die XPS-Messungen eingesetzt.
Abbildung 33: Übersichtsspektrum der XPS-Messung der Probe MCM/Naf10/Pt.
Platinsysteme 57
SAC/Naf40/Pt
Als Modellsystem wurde das System SAC/Naf40/Pt in XPS-Messungen
untersucht. Im Übersichtsspektrum (vgl. Abbildung 34) sind für das un-
reduzierte System deutlich mehr Spezies zu erkennen als für das niedriger
beladene System MCM/Naf10/Pt (vgl. Abbildung 33).
Abbildung 34: Übersichtsspektrum der XPS-Messung der Probe SAC/Naf40/Pt.
Auch bei diesen Untersuchungen wurde zunächst das Trägermaterial
SAC/Naf40 gemessen und mit dem Platin-beladenen System vor und nach
der Wasserstoff-Reduktion verglichen. Bei diesen Untersuchungen ergaben
sich die in Tabelle 15 aufgeführten Verhältnisse von Silicium zu Sauerstoff
bzw. Kohlenstoff.
Tabelle 15: Si/O- und Si/C-Verhältnisse des Systems SAC/Naf40/Pt vor und nach HB2B-Reduktion im Vergleich zum Trägermaterial SAC/Naf40.
Probe Si/O Si/C
SAC/Naf40 29 : 71 56 : 44
SAC/Naf40/Pt vor Reduktion 30 : 70 37 : 63
SAC/Naf40/Pt nach Reduktion 28 :72 42 : 58

Platinsysteme 58
Durch die Beladung des Nafion P
®P-haltigen Silikats SAC/Naf40 mit der Cyclo-
octadien-Platin-Spezies kommt es zunächst zu einer starken Zunahme an
Kohlenstoff. Durch die anschließende Reduktion der Platin-beladenen Probe
SAC/Naf40/Pt zeigt sich dann eine Verringerung des Kohlenstoffgehalts im
Vergleich zur unreduzierten Probe. Dieser Verlust an Kohlenstoff ist erklärbar
durch die Abgabe des Cyclooctadien-Liganden bei Reduktion des Platins.
Das Verhältnis von Silicium zu Sauerstoff ändert sich nur innerhalb der
Fehlergrenzen. Es sollte sich durch die Platin-Beladung selbst nicht ver-
ändern, da weder durch den Platin-Komplex noch durch Reduktion mit
Wasserstoff Silicium- und Sauerstoff-Anteile hinzukommen oder verloren
gehen können.
Für die Ermittlung der Bindungsenergien der verschiedenen Elemente wurde
das Si 2p-Signal auf 103.4 eV festgelegt und die anderen Werte ent-
sprechend korrigiert.
Aus den C 1s-Spektren konnten mehrere Spezies abgeleitet werden. Ein
Vergleich der drei Spektren für das reine Trägermaterial SAC/Naf40, die
unreduzierte und die reduzierte Platin-beladenen Proben SAC/Naf40/Pt ist in
Abbildung 35 dargestellt.
Im Spektrum des Trägermaterials SAC/Naf40 wird das Hauptsignal bei etwa
291.4 eV anhand von Literaturdaten als eine Überlagerung von CF B2B-, CF-O-
und CFB2B-O-Gruppen der Polymerketten des Nafions P
®P interpretiert.P
[71]P Die
Komponente bei 293.1 eV wird als CF B3B-Gruppe diskutiert. Das Signal bei
289.1 eV könnte einer Carboxyl-Gruppe entsprechen. Die drei schwachen
Signale bei 284.2, 285.8 und 287.1 eV könnten C-C-, C-OH-, C=OH-Gruppen
sein. P
[72]P
In der Probe SAC/Naf40/Pt, also nach der Beladung mit Platin beobachtet
man das Hauptsignal bei 291.3 eV, was erneut als Überlappung ver-
schiedener CF-Spezies interpretiert wird. Das Signal bei 284.8 eV wird dem
neuen Kohlenstoff-Fragment, dem Cyclooctadien-Liganden des Platins, zuge-
ordnet. Das Signal bei 285.5 eV könnte wieder eine C-O-Gruppe darstellen.
Die C 1s-Spezies bei niedrigen Bindungsenergien von 283.8 eV könnte zu
isolierten Kohlenstoff-Spezies gehören. P
[72]P
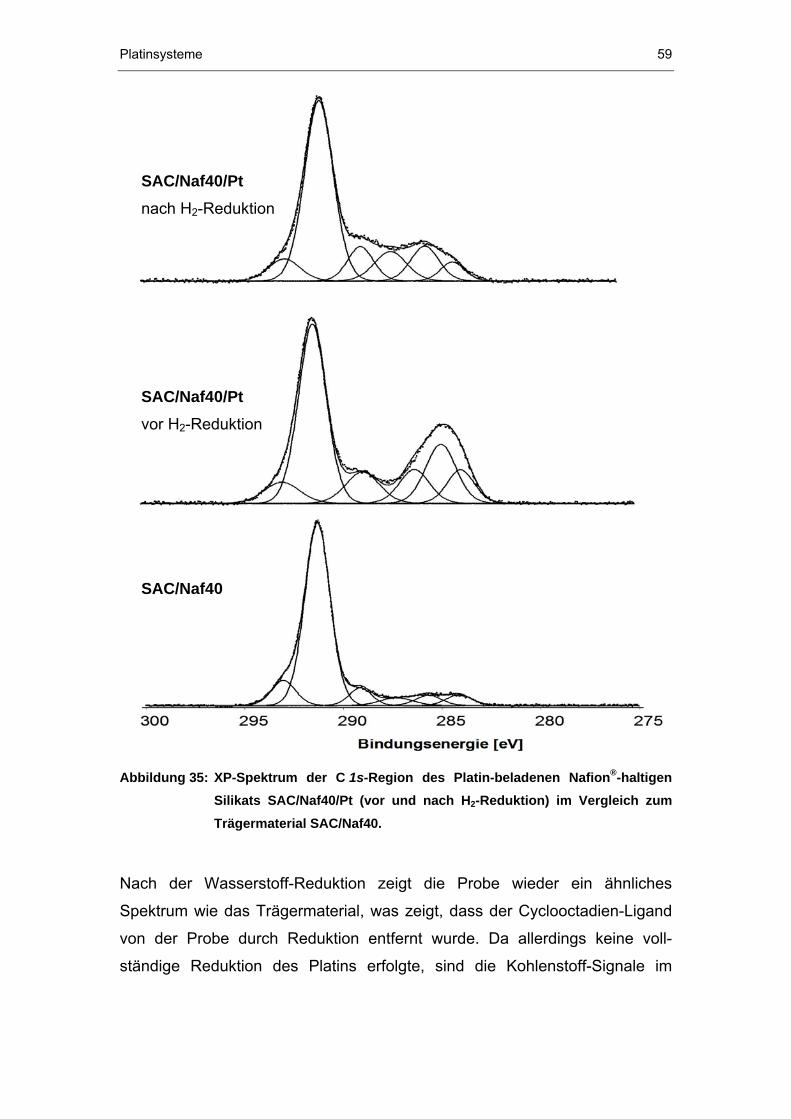
Platinsysteme 59
Abbildung 35: XP-Spektrum der C 1s-Region des Platin-beladenen NafionP
®P-haltigen
Silikats SAC/Naf40/Pt (vor und nach HB2B-Reduktion) im Vergleich zum Trägermaterial SAC/Naf40.
Nach der Wasserstoff-Reduktion zeigt die Probe wieder ein ähnliches
Spektrum wie das Trägermaterial, was zeigt, dass der Cyclooctadien-Ligand
von der Probe durch Reduktion entfernt wurde. Da allerdings keine voll-
ständige Reduktion des Platins erfolgte, sind die Kohlenstoff-Signale im
SAC/Naf40/Pt nach HB2B-Reduktion
SAC/Naf40/Pt vor HB2B-Reduktion
SAC/Naf40

Platinsysteme 60
Bereich von 284 bis 289 eV jedoch etwas stärker ausgeprägt als für das
Ausgangsmaterial.
In Tabelle 16 sind die angepassten Signale der C 1s-Spezies mit ihrem
prozentualen Anteil für die drei verglichenen Proben aufgelistet.
Tabelle 16: Korrigierte Werte für die C 1s-Region des Trägermaterials SAC/Naf40 und der Platin-beladenen Probe SAC/Naf40/Pt vor und nach HB2B-Reduktion.
SAC/Naf40 SAC/Naf40/Pt
vor HB2B-Reduktion SAC/Naf40/Pt
nach HB2B-Reduktion
284.2 eV (4 %) 283.8 eV (9 %) 284.2 eV (5 %)
285.8 eV (5 %) 284.8 eV (17 %) 285.6 eV (11 %)
287.2 eV (2 %) 285.5 eV (10 %) 287.3 eV (10 %)
289.1 eV (8 %) 288.7 eV (10 %) 288.8 eV (10 %)
291.4 eV (72 %) 291.3 eV (47 %) 291.0 eV (57 %)
293.1 eV (9 %) 292.8 eV (7 %) 292.7 eV (7 %)
Für Platin können in der unreduzierten Probe zwei Spezies angepasst
werden, mit einer Bindungsenergie von 72.6 und 73.7 eV. Das erste Signal
könnte sehr wahrscheinlich zum Platin-Cyclooctadien-Fragment gehören, das
zweite Signal könnte ein Pt-O- oder ein weiterer Pt-metallorganischer
Komplex sein. So zeigte zum Beispiel der eingangs erwähnte Carbamato-
Platin(II)-Komplex auf Silikat Bindungsenergien von 72.4 bis 72.8 eV. P
[66]P
In der reduzierten Probe sind sogar drei Spezies zu beobachten, eine
reduzierte Platin-Spezies und die zwei Ausgangsspezies. Dies lässt auf eine
unvollständige Reduktion schließen. Die reduzierte Spezies weist dabei eine
Bindungsenergie von 71.5 eV auf. Die korrigierten Werte der Pt 4fB7/2B-Region
und ihre Anteile für beide Proben sind in Tabelle 17 angegeben.

Platinsysteme 61
Tabelle 17: Korrigierte Werte für die Pt 4fB7/2B-Region der Platin-beladenen Probe SAC/Naf40/Pt vor und nach HB2B-Reduktion.
SAC/Naf40/Pt vor HB2B-Reduktion
SAC/Naf40/Pt nach HB2B-Reduktion
71.5 eV (33 %)
72.6 eV (47 %) 72.6 eV (35 %)
73.7 eV (53 %) 73.7 eV (32 %)
In der Literatur ist für bulk-Platin für die Pt 4fB7/2B-Bindungsenergien ein Wert
von 71.0 eV angegeben. P
[69]P Für Platin(0) auf Silikat wurden jedoch höhere
Bindungsenergien von 71.3 eV beobachtet. P
[76, 77]P Diese Verschiebung zu
höheren Bindungsenergien und die häufig simultan auftretende deutliche
Linienverbreiterung wurde bereits 1974 von TROSST et al. mit einer hohen
Dispersion des Platins in Zusammenhang gebracht. P
[78]P
Das O 1s-Signal kann mit Hilfe von zwei Spezies mit einer Bindungsenergie
von 532.6 und 534.3 eV angenähert werden.
Für Fluor wurde für die Proben SAC/Naf40 und das Platin-beladene System
vor der Reduktion ein Wert von 688.4 eV, für das reduzierte Platin-System ein
Wert von 688.0 eV ermittelt.
Anhand der XPS-Untersuchungen konnte somit die erfolgreiche Trägerung
der (Cyclooctadien)Platin-Spezies auf den Nafion P
®P-haltigen Silikaten und die
anschließende Reduktion dieser Spezies unter Freisetzung des Cyclo-
octadien-Liganden gezeigt werden.
4.2.4 TEM-Untersuchungen
Anhand von transmissionselektronenmikroskopischen Aufnahmen (vgl.
Kapitel 3.4.7) wurde die Struktur der geordneten Systeme und die Platinver-
teilung in den Platin-beladenen Proben ermittelt. Es sollte untersucht werden,
ob durch die Variation des Lösungsmittels die Immobilisierung der Platin-
Spezies zu beeinflussen ist. Außerdem sollte überprüft werden, ob durch eine
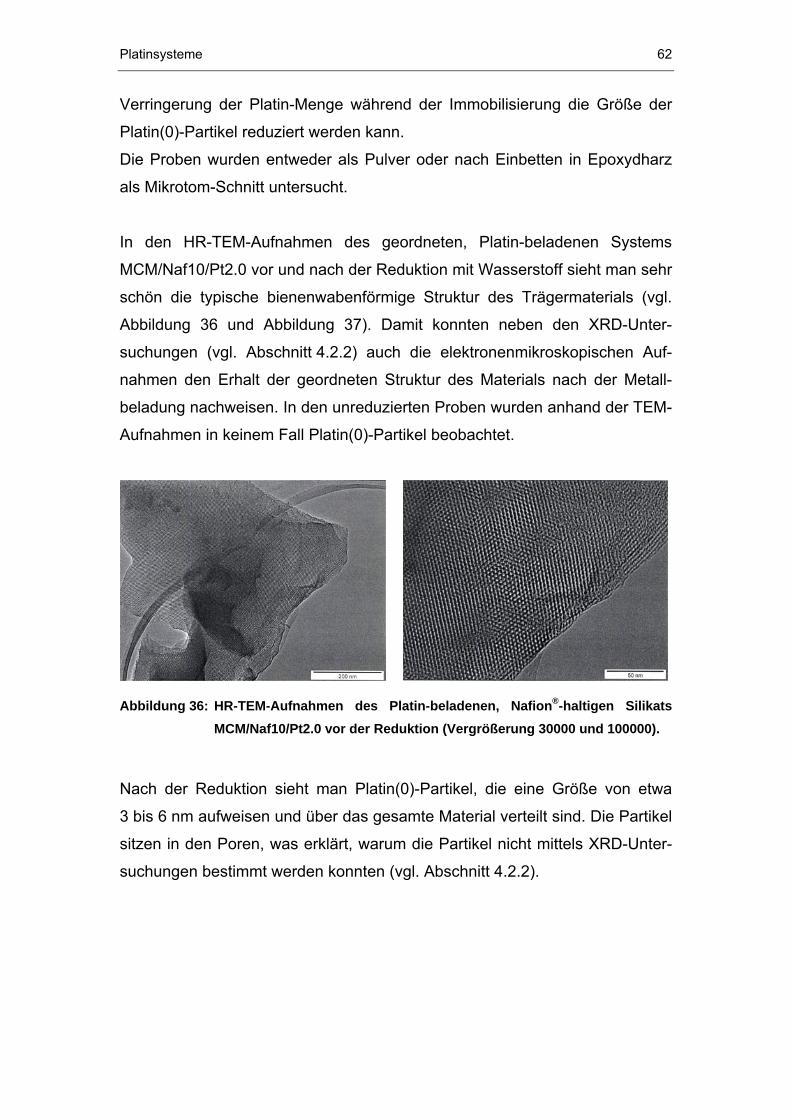
Platinsysteme 62
Verringerung der Platin-Menge während der Immobilisierung die Größe der
Platin(0)-Partikel reduziert werden kann.
Die Proben wurden entweder als Pulver oder nach Einbetten in Epoxydharz
als Mikrotom-Schnitt untersucht.
In den HR-TEM-Aufnahmen des geordneten, Platin-beladenen Systems
MCM/Naf10/Pt2.0 vor und nach der Reduktion mit Wasserstoff sieht man sehr
schön die typische bienenwabenförmige Struktur des Trägermaterials (vgl.
Abbildung 36 und Abbildung 37). Damit konnten neben den XRD-Unter-
suchungen (vgl. Abschnitt 4.2.2) auch die elektronenmikroskopischen Auf-
nahmen den Erhalt der geordneten Struktur des Materials nach der Metall-
beladung nachweisen. In den unreduzierten Proben wurden anhand der TEM-
Aufnahmen in keinem Fall Platin(0)-Partikel beobachtet.
Abbildung 36: HR-TEM-Aufnahmen des Platin-beladenen, NafionP
®P-haltigen Silikats
MCM/Naf10/Pt2.0 vor der Reduktion (Vergrößerung 30000 und 100000).
Nach der Reduktion sieht man Platin(0)-Partikel, die eine Größe von etwa
3 bis 6 nm aufweisen und über das gesamte Material verteilt sind. Die Partikel
sitzen in den Poren, was erklärt, warum die Partikel nicht mittels XRD-Unter-
suchungen bestimmt werden konnten (vgl. Abschnitt 4.2.2).
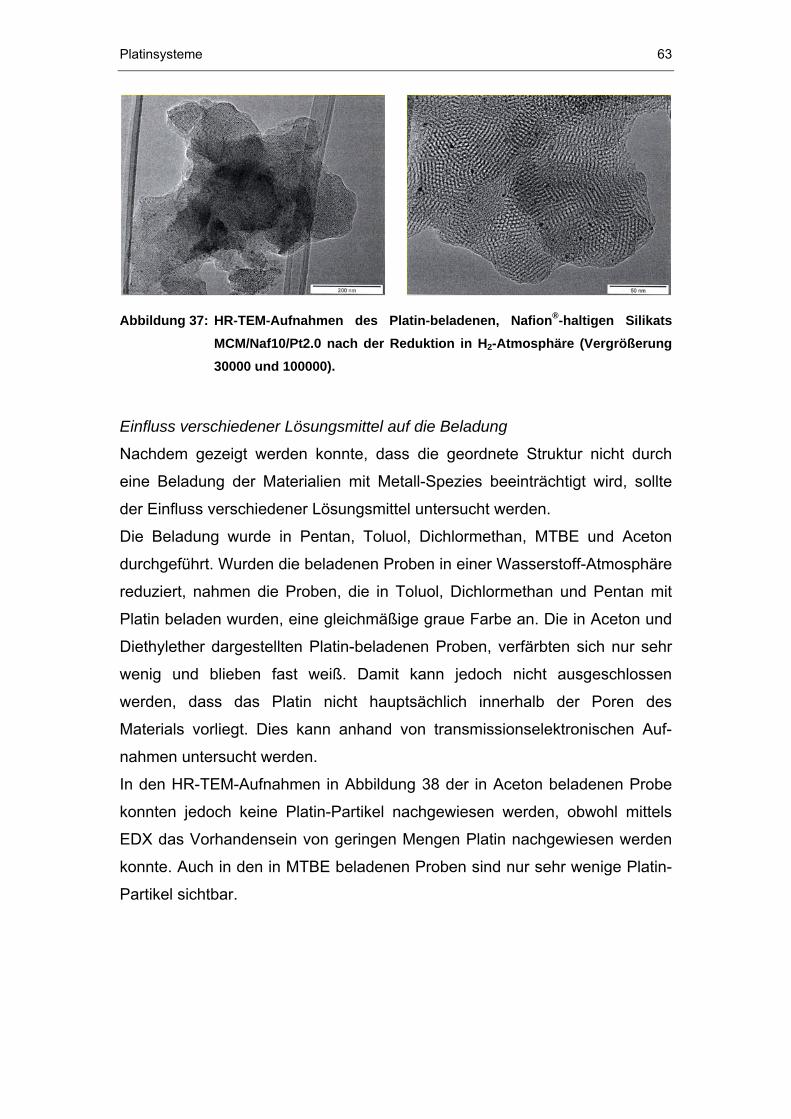
Platinsysteme 63
Abbildung 37: HR-TEM-Aufnahmen des Platin-beladenen, NafionP
®P-haltigen Silikats
MCM/Naf10/Pt2.0 nach der Reduktion in HB2B-Atmosphäre (Vergrößerung 30000 und 100000).
Einfluss verschiedener Lösungsmittel auf die Beladung
Nachdem gezeigt werden konnte, dass die geordnete Struktur nicht durch
eine Beladung der Materialien mit Metall-Spezies beeinträchtigt wird, sollte
der Einfluss verschiedener Lösungsmittel untersucht werden.
Die Beladung wurde in Pentan, Toluol, Dichlormethan, MTBE und Aceton
durchgeführt. Wurden die beladenen Proben in einer Wasserstoff-Atmosphäre
reduziert, nahmen die Proben, die in Toluol, Dichlormethan und Pentan mit
Platin beladen wurden, eine gleichmäßige graue Farbe an. Die in Aceton und
Diethylether dargestellten Platin-beladenen Proben, verfärbten sich nur sehr
wenig und blieben fast weiß. Damit kann jedoch nicht ausgeschlossen
werden, dass das Platin nicht hauptsächlich innerhalb der Poren des
Materials vorliegt. Dies kann anhand von transmissionselektronischen Auf-
nahmen untersucht werden.
In den HR-TEM-Aufnahmen in Abbildung 38 der in Aceton beladenen Probe
konnten jedoch keine Platin-Partikel nachgewiesen werden, obwohl mittels
EDX das Vorhandensein von geringen Mengen Platin nachgewiesen werden
konnte. Auch in den in MTBE beladenen Proben sind nur sehr wenige Platin-
Partikel sichtbar.
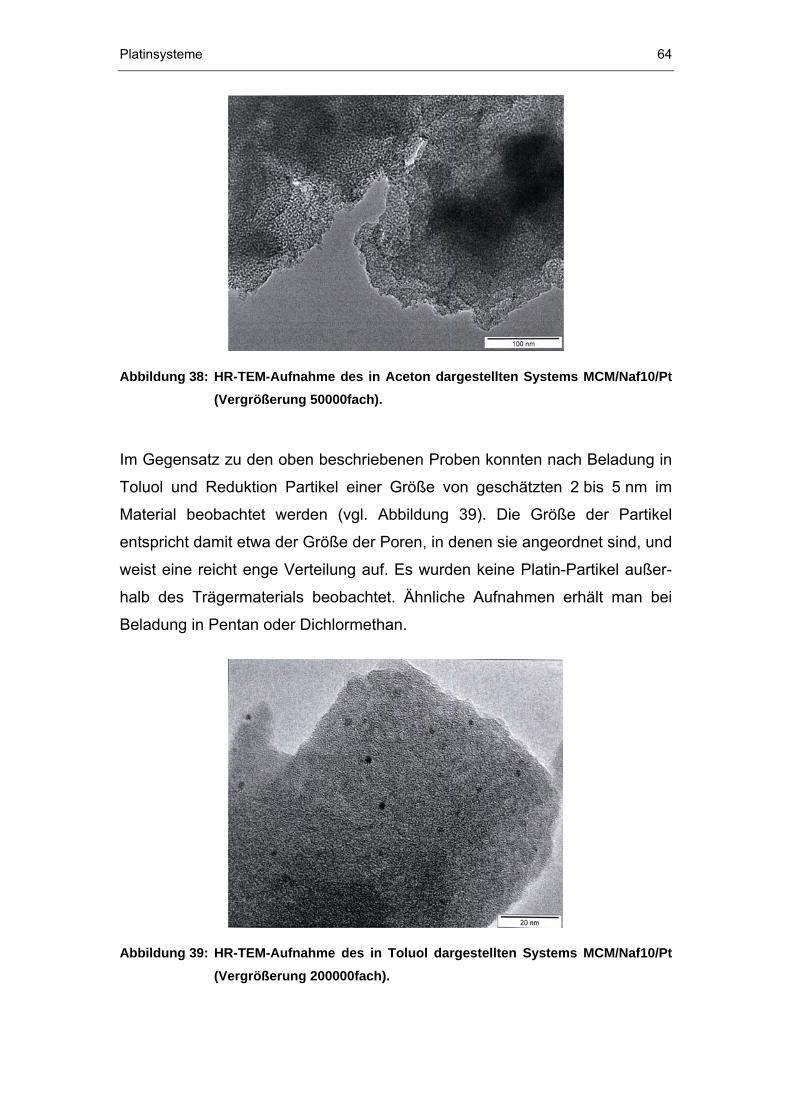
Platinsysteme 64
Abbildung 38: HR-TEM-Aufnahme des in Aceton dargestellten Systems MCM/Naf10/Pt (Vergrößerung 50000fach).
Im Gegensatz zu den oben beschriebenen Proben konnten nach Beladung in
Toluol und Reduktion Partikel einer Größe von geschätzten 2 bis 5 nm im
Material beobachtet werden (vgl. Abbildung 39). Die Größe der Partikel
entspricht damit etwa der Größe der Poren, in denen sie angeordnet sind, und
weist eine reicht enge Verteilung auf. Es wurden keine Platin-Partikel außer-
halb des Trägermaterials beobachtet. Ähnliche Aufnahmen erhält man bei
Beladung in Pentan oder Dichlormethan.
Abbildung 39: HR-TEM-Aufnahme des in Toluol dargestellten Systems MCM/Naf10/Pt (Vergrößerung 200000fach).
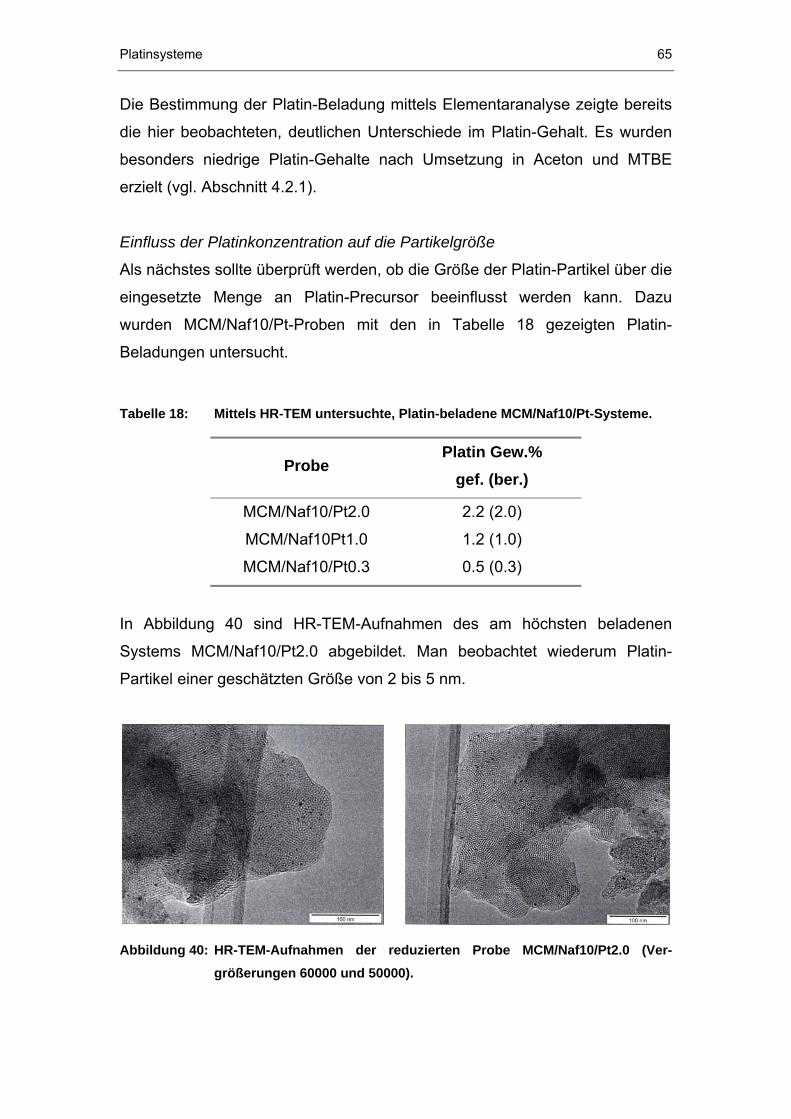
Platinsysteme 65
Die Bestimmung der Platin-Beladung mittels Elementaranalyse zeigte bereits
die hier beobachteten, deutlichen Unterschiede im Platin-Gehalt. Es wurden
besonders niedrige Platin-Gehalte nach Umsetzung in Aceton und MTBE
erzielt (vgl. Abschnitt 4.2.1).
Einfluss der Platinkonzentration auf die Partikelgröße
Als nächstes sollte überprüft werden, ob die Größe der Platin-Partikel über die
eingesetzte Menge an Platin-Precursor beeinflusst werden kann. Dazu
wurden MCM/Naf10/Pt-Proben mit den in Tabelle 18 gezeigten Platin-
Beladungen untersucht.
Tabelle 18: Mittels HR-TEM untersuchte, Platin-beladene MCM/Naf10/Pt-Systeme.
Probe Platin Gew.%
gef. (ber.)
MCM/Naf10/Pt2.0 2.2 (2.0)
MCM/Naf10Pt1.0 1.2 (1.0)
MCM/Naf10/Pt0.3 0.5 (0.3)
In Abbildung 40 sind HR-TEM-Aufnahmen des am höchsten beladenen
Systems MCM/Naf10/Pt2.0 abgebildet. Man beobachtet wiederum Platin-
Partikel einer geschätzten Größe von 2 bis 5 nm.
Abbildung 40: HR-TEM-Aufnahmen der reduzierten Probe MCM/Naf10/Pt2.0 (Ver-größerungen 60000 und 50000).

Platinsysteme 66
Betrachtet man im Vergleich eine Aufnahme der nur halb so hoch beladenen
Probe MCM/Naf10/Pt1.0 (vgl. Abbildung 41), sieht man etwa gleich große
Partikel wie in der Probe MCM/Naf10/Pt2.0, jedoch in deutlich geringeren
Mengen. In beiden Bildern ist exakt der gleiche Bereich der Probe, jedoch in
verschiedener Fokussierung aufgenommen, so dass links das Trägermaterial
und rechts die Platin-Partikel deutlicher zu sehen sind.
Abbildung 41: HR-TEM-Aufnahmen der reduzierten Probe MCM/Naf10/Pt1.0 mit unter-schiedlicher Fokussierung (Vergrößerung 100000).
Eine noch stärkere Abnahme der Anzahl der Platin-Partikel wird in den
Aufnahmen der Probe MCM/Naf10/Pt0.3 ersichtlich (vgl. Abbildung 42). Die
Größe der Partikel ändert sich jedoch nicht. Auch hier wurde die
Fokussierung variiert, um die Position und Anzahl der Platin-Partikel zu ver-
deutlichen.
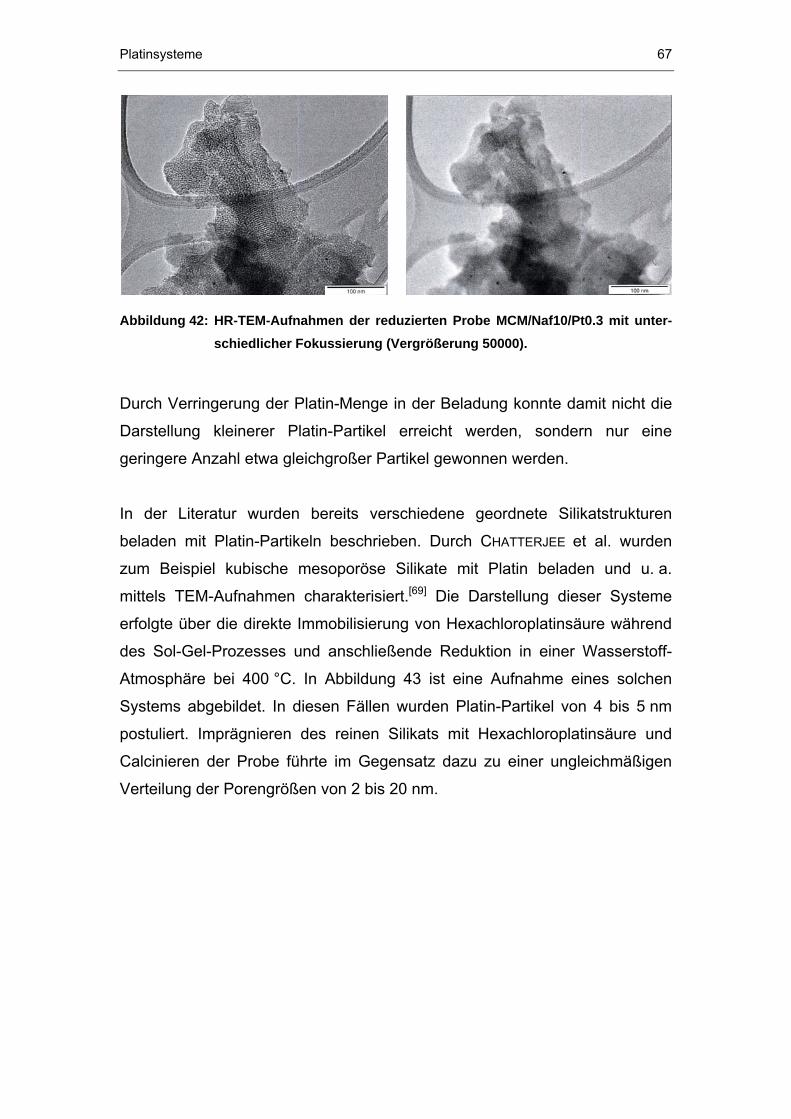
Platinsysteme 67
Abbildung 42: HR-TEM-Aufnahmen der reduzierten Probe MCM/Naf10/Pt0.3 mit unter-schiedlicher Fokussierung (Vergrößerung 50000).
Durch Verringerung der Platin-Menge in der Beladung konnte damit nicht die
Darstellung kleinerer Platin-Partikel erreicht werden, sondern nur eine
geringere Anzahl etwa gleichgroßer Partikel gewonnen werden.
In der Literatur wurden bereits verschiedene geordnete Silikatstrukturen
beladen mit Platin-Partikeln beschrieben. Durch TCHATTERJEE Tet al. wurden
zum Beispiel kubische mesoporöse Silikate mit Platin beladen und u. a.
mittels TEM-Aufnahmen charakterisiert.P
[69]P Die Darstellung dieser Systeme
erfolgte über die direkte Immobilisierung von Hexachloroplatinsäure während
des Sol-Gel-Prozesses und anschließende Reduktion in einer Wasserstoff-
Atmosphäre bei 400 °C. In Abbildung 43 ist eine Aufnahme eines solchen
Systems abgebildet. In diesen Fällen wurden Platin-Partikel von 4 bis 5 nm
postuliert. Imprägnieren des reinen Silikats mit Hexachloroplatinsäure und
Calcinieren der Probe führte im Gegensatz dazu zu einer ungleichmäßigen
Verteilung der Porengrößen von 2 bis 20 nm.

Platinsysteme 68
T Abbildung 43: TEM-Aufnahme eines kubischen, mesoporösen Silikats (MCM-48)
beladen mit Platin-Partikeln.P
[69]P
Geordnete, mesoporöse MCM-41-Silikate beladen mit Platin-Partikeln wurden
durch SCHÜTH et al. veröffentlicht.P
[62]P In den TEM-Aufnahmen dieser Systeme
konnten je nach Synthesemethode Partikel-Größen von 2 bis 20 nm be-
obachtet werden. Die direkte Immobilisierung während des Sol-Gel-
Prozesses führte zu Partikeln einer Größe von 4 bis 10 nm. Durch Ionen-
austausch dargestellte Proben zeigten eine bimodale Verteilung der Partikel-
größen mit einer Größe von 2 und 20 nm.
Im Vergleich zeigten die im Rahmen dieser Arbeit dargestellten, geträgerten
Platin-Partikel eine recht enge Größenverteilung von 2 bis maximal 5 nm. Sie
werden nur in den Poren des Materials und in einer recht gleichmäßigen
Verteilung auf dem mesoporösen System gewonnen.

Platinsysteme 69
4.3 Katalysen mit Platin-beladenen NafionP
®P-haltigen Silikaten
4.3.1 Hydrierung von Cycloocten
Die Hydrierung von Olefinen oder Aromaten ist eine Standardreaktion zur
Bewertung der katalytischen Aktivität von geträgerten Platin-Systemen. So
findet man in der Literatur eine Vielzahl an Untersuchungen zur Hydrierung,
zum Beispiel von CyclohexenP
[15, 79, 80]P, CyclooctenP
[15]P, NitrobenzolP
[15]P und
Naphthalen P
[81]P.
Im Rahmen der vorliegenden Arbeit wurde die Hydrierung von Cycloocten für
die Untersuchung der hergestellten Platin-beladenen, Nafion P
®P-haltigen
Silikate eingesetzt. Es sollte der Einfluss des Lösungsmittels während der
Platin-Beladung, des Trägermaterials und die Variation der Beladung be-
trachtet werden. Zum Vergleich der Systeme wurden die relativen Aktivitäten
berechnet und untereinander verglichen.
Die Reaktionen wurden in Methanol bei einem konstanten Wasserstoff-Druck
von 1.4 bar und Raumtemperatur durchgeführt.
Einfluss verschiedener Lösungsmittel
Die im Rahmen dieser Arbeit dargestellten ungeordneten und geordneten
NafionP
®P-haltigen Silikate SAC/Naf10 und MCM/Naf10 wurden nach der
Beladung mit Platin in verschiedenen Lösungsmitteln in der Hydrierung von
Cycloocten eingesetzt. Es wurde jeweils die gleiche Menge an Katalysator in
der Reaktion verwendet.
Der Verlauf der Reaktionen mit den geordneten Systemen MCM/Naf10/Pt1.3
ist anhand der Auftragung von Umsatz gegen die Reaktionszeit in Abbildung
44 dargestellt.
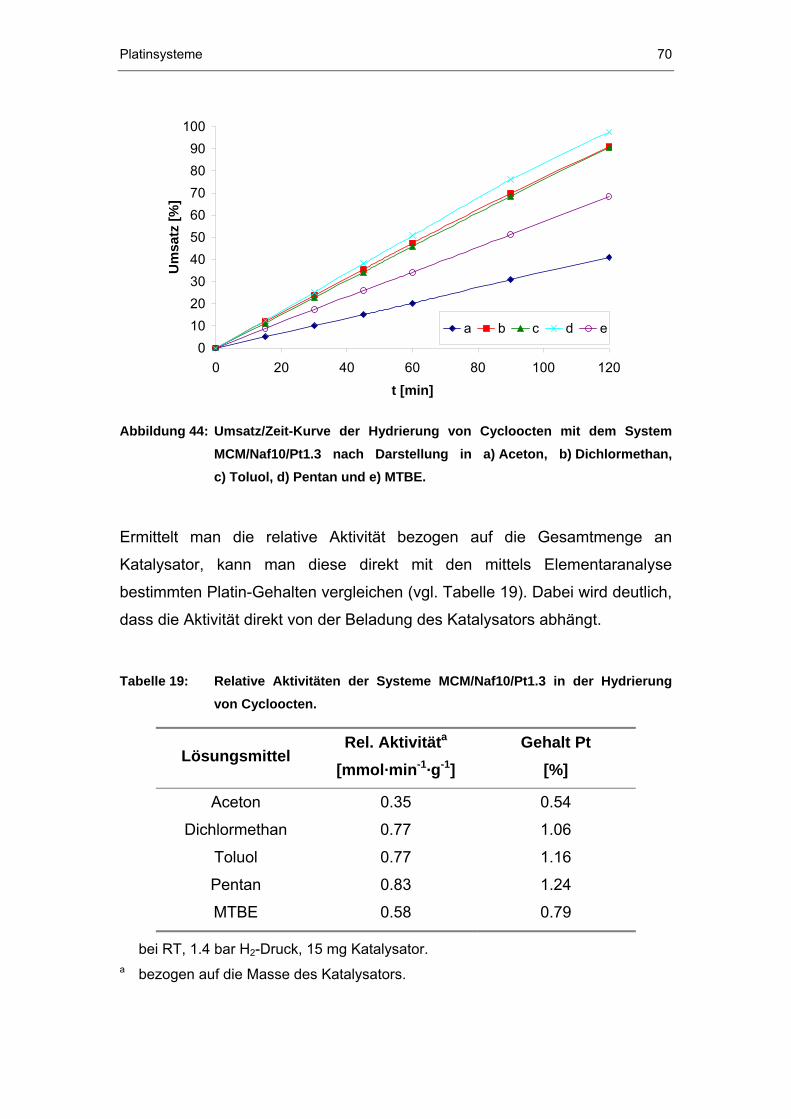
Platinsysteme 70
0102030405060708090
100
0 20 40 60 80 100 120
t [min]
Um
satz
[%]
a b c d e
Abbildung 44: Umsatz/Zeit-Kurve der Hydrierung von Cycloocten mit dem System MCM/Naf10/Pt1.3 nach Darstellung in a) Aceton, b) Dichlormethan, c) Toluol, d) Pentan und e) MTBE.
Ermittelt man die relative Aktivität bezogen auf die Gesamtmenge an
Katalysator, kann man diese direkt mit den mittels Elementaranalyse
bestimmten Platin-Gehalten vergleichen (vgl. Tabelle 19). Dabei wird deutlich,
dass die Aktivität direkt von der Beladung des Katalysators abhängt.
Tabelle 19: Relative Aktivitäten der Systeme MCM/Naf10/Pt1.3 in der Hydrierung von Cycloocten.
Lösungsmittel Rel. AktivitätP
aP
[ TmTmol·minP
-1P·gP
-1P]
Gehalt Pt [%]
Aceton 0.35 0.54
Dichlormethan 0.77 1.06
Toluol 0.77 1.16
Pentan 0.83 1.24
MTBE 0.58 0.79
bei RT, 1.4 bar HB2B-Druck, 15 mg Katalysator.
P
aP bezogen auf die Masse des Katalysators.
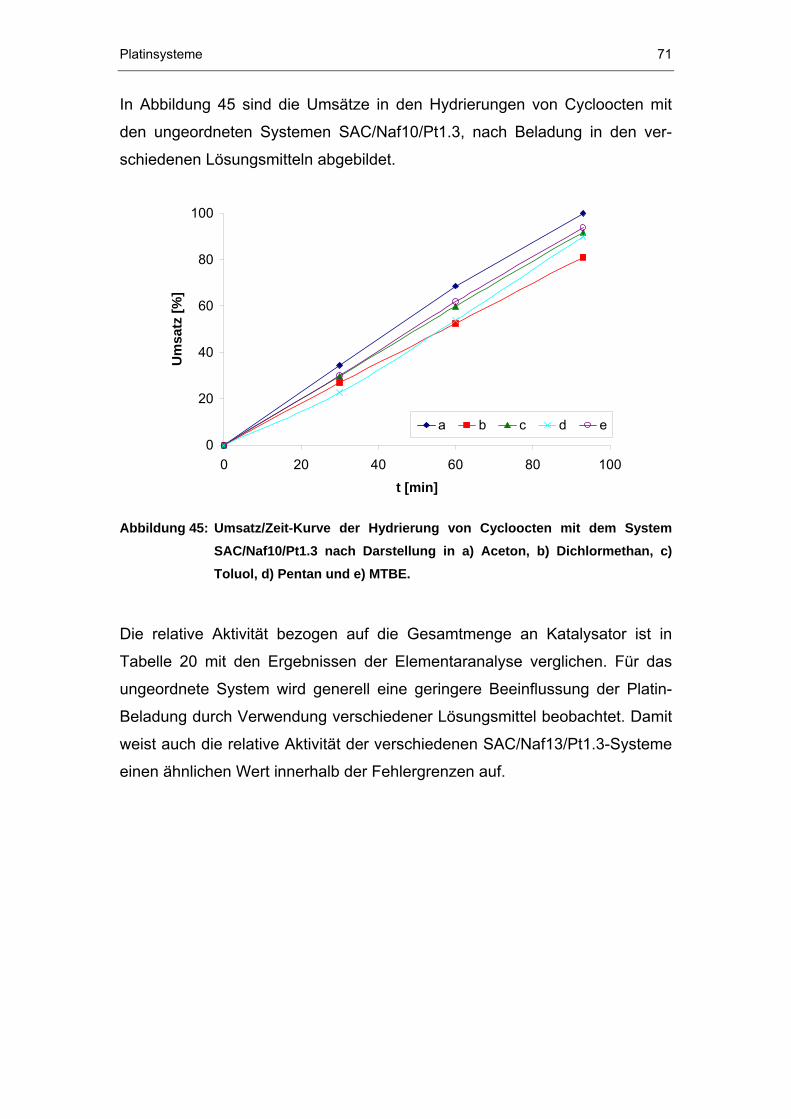
Platinsysteme 71
In Abbildung 45 sind die Umsätze in den Hydrierungen von Cycloocten mit
den ungeordneten Systemen SAC/Naf10/Pt1.3, nach Beladung in den ver-
schiedenen Lösungsmitteln abgebildet.
0
20
40
60
80
100
0 20 40 60 80 100
t [min]
Um
satz
[%]
a b c d e
Abbildung 45: Umsatz/Zeit-Kurve der Hydrierung von Cycloocten mit dem System SAC/Naf10/Pt1.3 nach Darstellung in a) Aceton, b) Dichlormethan, c) Toluol, d) Pentan und e) MTBE.
Die relative Aktivität bezogen auf die Gesamtmenge an Katalysator ist in
Tabelle 20 mit den Ergebnissen der Elementaranalyse verglichen. Für das
ungeordnete System wird generell eine geringere Beeinflussung der Platin-
Beladung durch Verwendung verschiedener Lösungsmittel beobachtet. Damit
weist auch die relative Aktivität der verschiedenen SAC/Naf13/Pt1.3-Systeme
einen ähnlichen Wert innerhalb der Fehlergrenzen auf.

Platinsysteme 72
Tabelle 20: Relative Aktivitäten der Systeme SAC/Naf10/Pt1.3 in der Hydrierung von Cycloocten.
Lösungsmittel Rel. AktivitätP
aP
[ TmTmol·minP
-1P·gP
-1P]
Gehalt Pt [%]
Aceton 0.87 1.19
Dichlormethan 0.67 1.09
Toluol 0.76 1.10
Pentan 0.74 0.96
MTBE 0.77 0.95
bei RT, 1.4 bar HB2B-Druck, 20 mg Katalysator.
P
aP bezogen auf die Masse des Katalysators.
Ein Vergleich beider Trägermaterialien in der Hydrierung zeigt für Systeme
mit etwa gleich hoher Platin-Beladung eine etwa vergleichbare relative
Aktivität. Zum Beispiel wird bei einer Beladung von etwa 1.1 Gew.% eine
relative Aktivität von ca. 0.7 bis 0.8 Tm Tmol·minP
-1P·gP
-1P gefunden. Damit wird die
Hydrierung wahrscheinlich nicht durch das Trägermaterial beeinflusst.
Einfluss verschiedener Beladungen
Als nächstes sollte untersucht werden, ob bei einer geringeren Platin-
Beladung kleinere Partikel nach der Reduktion gebildet werden. Mittels TEM-
Untersuchungen (vgl. Abschnitt 4.2.4) wurde gezeigt, dass die Partikelgröße
nicht durch die Menge des Platin-Precursors beeinflusst wird. Allerdings kann
nicht ausgeschlossen werden, dass zusätzlich zu den sichtbaren Partikeln
von 2 bis 5 nm sehr viel kleinere Partikel vorliegen.
In den katalytischen Untersuchungen wurden die in Tabelle 21 aufgeführten
Katalysatorsysteme und Mengen eingesetzt. Es wurde bei gleichen effektiven
Platin-Mengen gearbeitet, um Konzentrationseffekte ausschließen zu können.
Der Verlauf der Hydrierungen ist anhand der Auftragungen von Umsatz gegen
Zeit für die Katalysatoren in Abbildung 46 dargestellt.
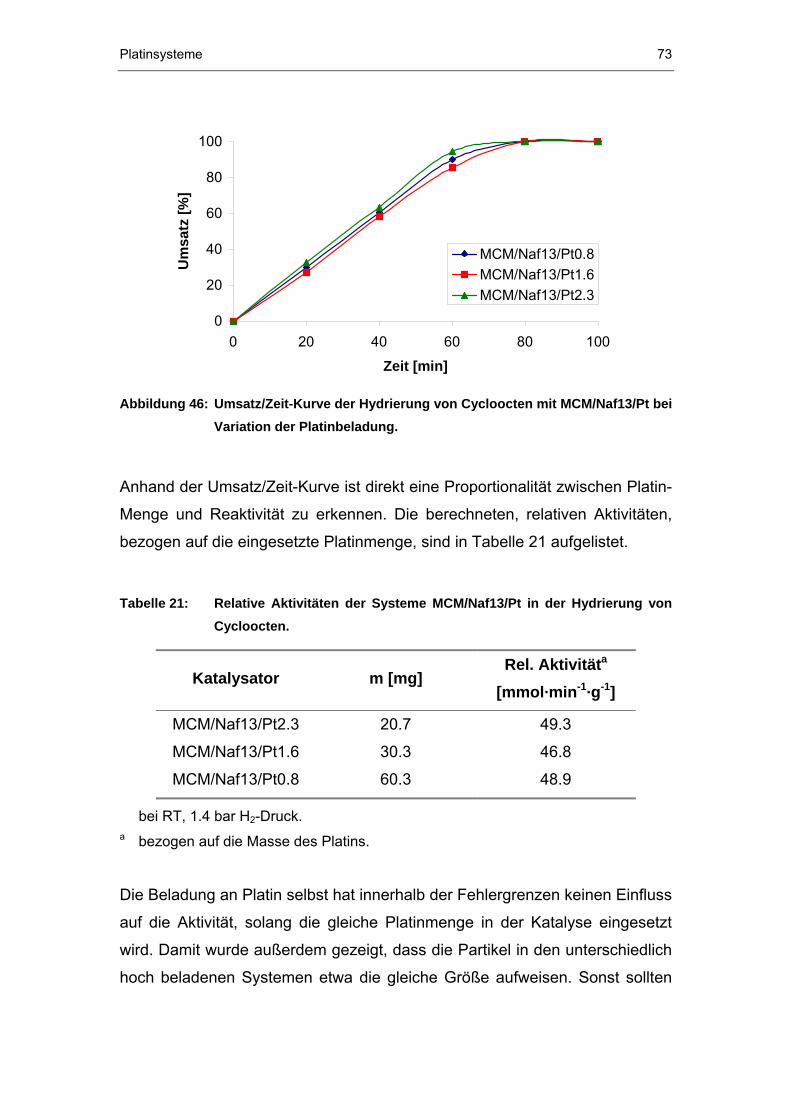
Platinsysteme 73
0
20
40
60
80
100
0 20 40 60 80 100
Zeit [min]
Um
satz
[%]
MCM/Naf13/Pt0.8MCM/Naf13/Pt1.6MCM/Naf13/Pt2.3
Abbildung 46: Umsatz/Zeit-Kurve der Hydrierung von Cycloocten mit MCM/Naf13/Pt bei Variation der Platinbeladung.
Anhand der Umsatz/Zeit-Kurve ist direkt eine Proportionalität zwischen Platin-
Menge und Reaktivität zu erkennen. Die berechneten, relativen Aktivitäten,
bezogen auf die eingesetzte Platinmenge, sind in Tabelle 21 aufgelistet.
Tabelle 21: Relative Aktivitäten der Systeme MCM/Naf13/Pt in der Hydrierung von Cycloocten.
Katalysator m [mg] Rel. AktivitätP
aP
[ TmTmol·minP
-1P·gP
-1P]
MCM/Naf13/Pt2.3 20.7 49.3
MCM/Naf13/Pt1.6 30.3 46.8
MCM/Naf13/Pt0.8 60.3 48.9
bei RT, 1.4 bar HB2B-Druck.
P
aP bezogen auf die Masse des Platins.
Die Beladung an Platin selbst hat innerhalb der Fehlergrenzen keinen Einfluss
auf die Aktivität, solang die gleiche Platinmenge in der Katalyse eingesetzt
wird. Damit wurde außerdem gezeigt, dass die Partikel in den unterschiedlich
hoch beladenen Systemen etwa die gleiche Größe aufweisen. Sonst sollten

Platinsysteme 74
die Aktivitäten aufgrund einer unterschiedlichen effektiven Platin-Oberfläche
deutlichere Unterschiede zeigen.
Die hier ermittelten relativen Aktivitäten von etwa 47 bis 49 Tm Tmol·minP
-1P·gP
-1P,
bezogen auf die Menge an Platin, sind im Vergleich zu Literaturdaten recht
gut. So weisen die oben erwähnten Platin-Silikat-Katalysatoren, gewonnen
durch Trägerung mittels eines N,N-Dialkylcarbamato-Platin-Komplexes, nur
relative Aktivitäten von etwa 27 Tm Tmol·minP
-1P·gP
-1P in der Hydrierung von Cyclo-
hexen auf. P
[66]P Cycloocten ist in der Hydrierung um eine Größenordnung
weniger reaktiv als Cyclohexen P
[15]P, womit das literaturbeschriebene System
eine noch deutlich geringere Aktivität in der Hydrierung von Cycloocten
zeigen müsste. Aufgrund der Vielzahl von beschriebenen Reaktions-
bedingungen, wie höherer Druck oder Temperatur, die in der Literatur
gefunden werden, wird der Vergleich mit weiteren literaturbekannten
Systemen erschwert.
Einfluss verschiedener Precursor
Abschließend sollte überprüft werden, ob die mit dem Dichloro(TηTP
4P-cyclo-
octadien)platin(II)-Komplex behandelten Nafion P
®P-haltigen Silikate eine
Aktivität in der Hydrierung und damit eine Beladung an Platin aufweisen.
Dazu wurden die gleichen Bedingungen wie in den oben beschriebenen
Versuchen verwendet.
Es zeigte sich keinerlei Aktivität in der Hydrierung von Cycloocten und damit
die bereits durch Elementaranalyse gezeigte erfolglose Beladung der Nafion P
®P-
haltigen Silikate mit dem Dichloro-Platin-Komplex.

Zirconiumsysteme 75
5 Zirconiumsysteme
In der homogenen Katalyse wurden bereits vor einigen Jahren kationische
Zirconocen-Komplexe mit Trifluormethansulfonat-Gegenionen als LEWIS-
Säure-Katalysatoren beschrieben. Sie wurden zum Beispiel in der DIELS-
ALDER-ReaktionP
[82-85]P und der gekreuzten MUKAIYAMA-Aldol-ReaktionP
[84, 86]P ein-
gesetzt.
So beschrieben BOSNICH et al. 1995 die DIELS-ALDER-Reaktion von Acryloyl-2-
oxazolidinon mit Cyclopentadien (vgl. Abbildung 47), in der bei Einsatz von
CpB2BZr(OTf)B2B unter 92 % Ausbeute ein endo:exo-Verhältnis von 4.6 erzielt
werden konnte. P
[87]P
N
O O
O +
Cp2Zr(OTf)2< 10 mol%
CH2Cl20 °C
O N
O
O
92 %, endo:exo 4.6:1
Abbildung 47: DIELS-ALDER-Reaktion von Acryloyl-2-oxazolidinon mit Cyclopentadien nach BOSNICH et al.P
[87]P
Mit chiralen, verbrückten Indenyl-Liganden ist es möglich, eine asymmet-
rische Induktion mit maximal 91 % ee bezogen auf das (2R)-endo-Stereo-
isomer und ein endo:exo-Verhältnis von 11.5 zu erreichen. Dazu muss in
Nitroalkanen als Lösungsmittel, bei tiefen Temperaturen und bei niedrigen
Katalysatormengen (1 mol%) gearbeitet werden.P
[85]P
Eine der wichtigsten Katalysen, die durch Metallocen-Systeme ermöglicht
wird, ist die Olefin-Polymerisation nach ZIEGLERP
[88]P und NATTAP
[89]P. In der
Patentliteratur ist unter anderem der Einsatz der Verbindung Cp* B2BZr(Me)(OTf)
nach Aktivierung mit MAO für die Ethenpolymerisation veröffentlicht.P
[90]P

Zirconiumsysteme 76
Zur Einführung des Triflat-Liganden sind in der Literatur verschiedene
Umsetzungen beschrieben. Zum einen kann das Silbersalz AgCF B3BSO B3B mit
dem gewünschten Zirconocen-Dichlorid P
[82]P oder aber die Trifluormethansulfon-
säure mit dem entsprechenden Zirconocen-Dimethyl-Komplex in Toluol P
[91]P
oder MTBEP
[85]P zur Reaktion gebracht werden.
Im Rahmen der vorliegenden Arbeit sollten analoge heterogene Systeme
dargestellt werden, die statt des Triflat-Gegenions die Sulfonsäurereste des
NafionP
®P-Polymers aufweisen. Dazu sollte die Umsetzung des Dimethyl-
Komplexes, Cp B2BZrMeB2B, mit einem ungeordneten, NafionP
®P-haltigen Silikat im
Verhältnis 1 : 1 bzw. 1 : 2 erfolgen. Ziel war dabei die Darstellung und Unter-
suchung der mono- und disubstituierten Varianten.
5.1 Synthese
Zur Darstellung der Zirconium-beladenen, Nafion P
®P-haltigen Silikate wurde der
Dimethyl-Zirconocen-Komplex Cp B2BZrMeB2B mit dem NafionP
®P-haltigen Silikat
SAC/Naf10 in Toluol umgesetzt. Die Edukte wurden dazu unter Argon bei
-78 °C suspendiert und die Reaktion durch langsames Auftauen auf Raum-
temperatur ermöglicht. Dabei kommt es zur Reaktion der Sulfonsäurereste
des immobilisierten Nafion P
®P-Polymers mit den Methylgruppen des Zirconium-
Komplexes. Je nach molekularem Verhältnis von Sulfonsäureresten zum
Zirconocen-Komplex wird ein mono- und disubstituiertes Triflat-Zirconium-
System erhalten.
In Abbildung 48 ist die Umsetzung bei einem Verhältnis von 1 : 1 abgebildet.
Das dabei gebildete System wird im Weiteren als SAC/Naf10/Zr/Cp B2BMe
bezeichnet.

Zirconiumsysteme 77
CF2 SO3
CF2 SO3H ZrH3C
H3C
ZrH3C
Nafion/SiO2
+
- CH4
Nafion/SiO2
Abbildung 48: Darstellung des Zirconocen-beladenen, NafionP
®P-haltigen Silikats
SAC/Naf10/Zr/CpB2BMe.
Wird das Verhältnis von Zirconium-Komplex zu Sulfonsäureresten auf 1 : 2
verringert, wird die vollständige Substitution der Methylgruppen durch die
Sulfonsäurereste des Nafion P
®P-Polymers ermöglicht (vgl. Abbildung 49).
Dieses System wird im Folgenden als SAC/Naf10/Zr/CpB2B bezeichnet.

Zirconiumsysteme 78
CF2 SO3
CF2 SO3H
ZrH3C
H3C
Zr
Nafion/SiO2
+
- 2 CH4
Nafion/SiO2
CF2 SO3H
CF2 SO3
Abbildung 49: Darstellung des Zirconocen-beladenen, NafionP
®P-haltigen Silikats
SAC/Naf10/Zr/CpB2B.
5.2 Charakterisierung
Für die Charakterisierung der dargestellten Systeme SAC/Naf10/Zr/Cp B2BMe
und SAC/Naf10/Zr/Cp B2B wurden Methoden zur Bestimmung der Zirconium-
Beladung, wie EA und das Elemental Mapping durch die Rasterelektronen-
mikroskopie, eingesetzt.
5.2.1 Elementaranalyse
Anhand der Elementaranalyse wurde die Beladung der Nafion P
®P-haltigen
Silikate an Zirconium ermittelt. Für das monosubstituierte System
SAC/Naf10/Zr/Cp B2BMe und das disubstituierte System SAC/Naf10/Zr/Cp B2 B
wurden die in Tabelle 22 angegebenen Gewichtsprozente an Zirconium er-
mittelt.

Zirconiumsysteme 79
Tabelle 22: Ermittelte Zirconium-Beladungen der Systeme SAC/Naf10/Zr/CpB2BMe und SAC/Naf10/Zr/CpB2B (in Klammern berechneter Wert).
Probe Gew. % Zr
SAC/Naf10/Zr/Cp B2BMe 0.87 (0.94)
SAC/Naf10/Zr/Cp B2B 0.61 (0.47)
5.2.2 SEM-Untersuchungen
Die Verteilung des Zirconiums auf dem Trägermaterial wurde für das System
SAC/Naf10/Zr/Cp B2BMe mittels rasterelektronenmikroskopischer Untersuchun-
gen (SEM) und EDX betrachtet (vgl. Kapitel 3.4.7).
Die Morphologie des Trägermaterials entspricht auch nach dem Trägern der
Zirconium-Spezies der des Ausgangsmaterials SAC/Naf10. In Abbildung 50
erkennt man wiederum splitterförmige Partikel, die bei einer Vergrößerung
von 10 k die typische Unterstruktur basisch-dargestellter Silikate zeigen.
Abbildung 50: SEM-Aufnahmen des Zirconium-beladenen, Nafion P
®P-haltigen Systems
SAC/Naf10/Zr/CpB2BMe bei einer Vergrößerung von 1 k und 10 k.
Elemental Mapping zeigt eine gleichmäßige Verteilung des Zirconiums auf
dem Trägermaterial. In Abbildung 51 sind die Übersichtsspektren für die im
System vorhandenen Elemente Kohlenstoff, Sauerstoff, Silicium, Fluor und
Zirconium abgebildet. Durch die Trägerung wurde die homogene Verteilung
der anderen Elemente nicht verändert.

Zirconiumsysteme 80
Abbildung 51: Übersichtsspektren für die Elemente Sauerstoff, Kohlenstoff, Silicium, Fluor und Zirconium im System SAC/Naf10/Zr/CpB2BMe.
Bereits bei diesem System erwies sich die Zirconium-Beladung als recht
niedrig für diese Analysemethode, weshalb das deutlich niedriger beladene
System SAC/Naf10/Zr/CpB2B nicht untersucht werden konnte.
Nachdem gezeigt wurde, dass die Zirconium-Spezies auf dem Nafion P
®P-
haltigen Silikat hochverteilt geträgert werden konnten, sollten als nächstes die
katalytischen Eigenschaften dieser Systeme untersucht werden.

Zirconiumsysteme 81
5.3 Katalysen
Die hier dargestellten Zirconium-beladenen, Nafion P
®P-haltigen Silikate wurden
in der Polymerisation von Ethen eingesetzt und ihre katalytischen Eigen-
schaften untersucht.
5.3.1 Polymerisationen von Ethen
5.3.1.1 Literatur und Hintergrund
Die weltweite Produktion von Polyolefinen wie Polyethylen (PE) und Poly-
propylen (PP) und ihrer Copolymere nimmt wegen der hervorragenden
Produkteigenschaften und ihrer Umweltfreundlichkeit ständig zu. Diese
Polymere werden als Packungsmaterialien, Kunststofffolien, Fasern und in
Komponenten für die Automobil- und Elektroindustrie eingesetzt. Die welt-
weite Produktion von PE und PP betrug im Jahr 1996 40 bzw. 20 Millionen
Tonnen. P
[11]P Im Jahr 1999 belief sich die Produktion von PE bereits auf
49 Millionen Tonnen, die von PP im Jahr 2000 auf 29 Millionen Tonnen.P
[7]P
Industriell werden Polyolefine durch klassische heterogene ZIEGLER-
Katalysatoren auf MgCl B2B-Trägern oder Chrom-Katalysatoren auf SiO B2B- oder
AlB2BO B3B-Trägern, so genannte PHILLIPS-Katalysatoren, eingesetzt. Der Einsatz
modernerer Metallocen-Systeme blieb jedoch lange auf wissenschaftliche
Untersuchungen beschränkt. Erst im Jahr 1995 führte Exxon Chemical
Achieve, einen Metallocen-Katalysator, für die Darstellung von Polypropylen
ein. P
[92]P
Generell erfolgt die Olefinpolymerisation durch eine Kombination eines
Übergangsmetall-Komplexes, dem Katalysator-Precursor, mit einer organo-
metallischen Hauptgruppen-Verbindung, dem Cokatalysator.
Als Cokatalysator für die Olefinpolymerisation mit Titanocen- und Zirconocen-
Verbindungen wird häufig Methylalumoxan (MAO) verwendet.P
[93]P Natürlich
sind in der Literatur eine Vielzahl von Cokatalysatoren beschrieben P
[94]P, im
Weiteren soll jedoch nur auf das im Rahmen dieser Arbeit eingesetzte MAO
eingegangen werden.
MAO wird durch die kontrollierte Hydrolyse von Methylaluminium in
toluolischer Lösung hergestellt. Die Zusammensetzung beträgt [–Al(Me)–O–] BnB
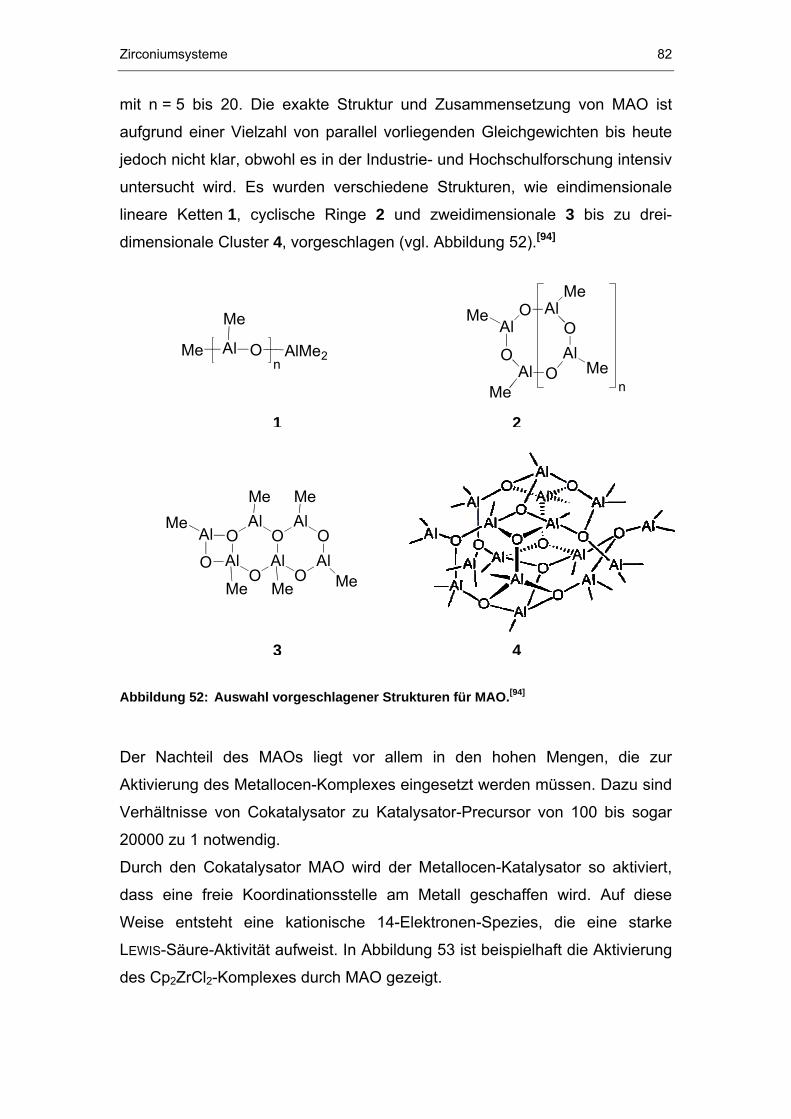
Zirconiumsysteme 82
mit n = 5 bis 20. Die exakte Struktur und Zusammensetzung von MAO ist
aufgrund einer Vielzahl von parallel vorliegenden Gleichgewichten bis heute
jedoch nicht klar, obwohl es in der Industrie- und Hochschulforschung intensiv
untersucht wird. Es wurden verschiedene Strukturen, wie eindimensionale
lineare Ketten T1T, cyclische Ringe T2T und zweidimensionale T3T bis zu drei-
dimensionale Cluster T4, Tvorgeschlagen T(vgl. TAbbildung 52 T). P
[94]PT
Me Al
Me
O AlMe2n
Al
OAl O
AlO
AlOMe
MeMe
Me
n
Al
O AlO
OAlO
Al
OAlO
AlMe
Me
Me Me
Me Me
Abbildung 52: Auswahl vorgeschlagener Strukturen für MAO. P
[94]P
Der Nachteil des MAOs liegt vor allem in den hohen Mengen, die zur
Aktivierung des Metallocen-Komplexes eingesetzt werden müssen. Dazu sind
Verhältnisse von Cokatalysator zu Katalysator-Precursor von 100 bis sogar
20000 zu 1 notwendig.
Durch den Cokatalysator MAO wird der Metallocen-Katalysator so aktiviert,
dass eine freie Koordinationsstelle am Metall geschaffen wird. Auf diese
Weise entsteht eine kationische 14-Elektronen-Spezies, die eine starke
LEWIS-Säure-Aktivität aufweist. In Abbildung 53 ist beispielhaft die Aktivierung
des CpB2BZrClB2B-Komplexes durch MAO gezeigt.
1 2
3 4

Zirconiumsysteme 83
ZrCl
Cl
MAOZr
Me MAO
Abbildung 53: Aktivierung des Metallocenkomplexes CpB2BZrClB2B durch MAO.P
[95]P
Der Mechanismus der Aktivierung gliedert sich in die Methylierung des
Zentralmetalls und Abstraktion eines Carbanions unter Bildung des
Metallocen-Monomethyl-Kations T5T als katalytisch aktive Spezies.P
[96, 97]P
Die stabilisierende MAO-Spezies besteht wahrscheinlich aus einem Käfig aus
sechsgliedrigen Ringen, die aus MeAlO-Einheiten aufgebaut sind. Im Käfig
soll sich ein monomeres AlMe B3B-Molekül befinden (vgl. Abbildung 54).P
[98]P Das
formal gebildete Kontaktionenpaar ist der eigentliche Katalysator in der
Olefinpolymerisation. Die Käfigform erklärt auch den nötigen MAO-Über-
schuss.
Abbildung 54: Modell des MAO-Käfig-stabilisierten Metallocen-Komplexes.P
[98]P
Der Reaktionsmechanismus der Olefinpolymerisation gliedert sich in die
Koordination des Olefins an das LEWIS-saure Metallzentrum als ersten Schritt
und in die formale Ethen-Insertion in die Metall-Kohlenstoff-Bindung als
Kettenwachstumsschritt (vgl. Abbildung 55). Der Insertionsschritt besteht aus
der Alkylwanderung zum Olefin-Ligand unter gleichzeitiger Bildung einer
neuen freien Koordinationsstelle. Anschließend findet die Koordination des
nächsten Olefinmoleküls an dieser Stelle statt.
T5

Zirconiumsysteme 84
ZrMe
MAO
ZrMe
MAO
Zr
MAO
Abbildung 55: Kettenwachstum in der Ethenpolymerisation.P
[95]P
Als Kettenabbruchreaktion kommt hauptsächlich die Tβ T-Hydrideliminierung hin
zum Monomer in Frage, so dass am Ende der Polymer-Kette eine Doppel-
bindung vorliegt. Diese kann wiederum in der Olefinpolymerisation reagieren,
wodurch Verzweigungen in der Polymerkette auftreten können. Weitere
mögliche Abbruchsreaktionen sind die Kettenübertragung auf ein Aluminium
bzw. ein Monomer oder die Tβ T-Alkylabspaltung. P
[95]P
Für industrielle Anwendungen werden aus mehreren Gründen hetero-
genisierte Katalysatorsysteme eingesetzt. So basieren bereits existierende
Anlagen normalerweise auf festen Katalysatoren, die die Polymerisation in
Gasphasen- oder Suspensionsprozessen katalysieren. Außerdem können
durch heterogene Systeme die Polymermorphologie besser kontrolliert, und
der Reaktorverschleiß und die MAO-Menge verringert werden. P
[99]P
Silikat ist das meist genutzte Trägermaterial für Polymerisationskatalysatoren.
In der Literatur sind der Einsatz von Silikaten mit verschiedenen Partikel-
größen, Oberflächen und Porenvolumen, und eine Vielzahl an Methoden zur
Immobilisierung der katalytisch aktiven Spezies beschrieben. So kann
zunächst das MAO auf den Träger aufgebracht und dieses dann mit der
Metallocen-Spezies zur Reaktion gebracht oder genau umgekehrt erst das
Metallocen und dann der Cokatalysator immobilisiert werden. Alternativ
können zunächst Metallocen und MAO in Lösung vorreagieren und die
gebildete aktive Spezies geträgert werden. P
[100]P

Zirconiumsysteme 85
5.3.1.2 Eigene Untersuchungen
Im Rahmen der vorliegenden Arbeit sollten die gewonnenen Zirconium-
beladenen, Nafion P
®P-haltigen Silikate auf ihre katalytische Aktivität in der
Polymerisation von Ethen untersucht werden.
Die beschriebenen Polymerisationsexperimente wurden im Arbeitskreis von
Prof. Dr. G. Fink am Max-Planck-Institut für Kohlenforschung in Mülheim/Ruhr
durchgeführt. Die Experimente wurden als Suspensionspolymerisationen
geführt. Für die Untersuchungen wurde ein Laborrührautoklav der Firma
Büchi AG verwendet. Das Fließschema der Polymerisationsanlage ist in
Abbildung 56 dargestellt. Weitere Details zur Durchführung und technischen
Daten der Anlage sind in Abschnitt 9.6.3 beschrieben.
Zirconiumsysteme 86
1 Flaschendruckminderer M1 Rührmotor 2 Leitungsdruckminderer M2 Getriebestellmotor 3-10 Ventile a Manometer (mech.) R1 Molekularsiebsäule b Füllöffnung R2 NaAlEt B4B-Säule c Pt100-Thermofühler R3 BTS-Katalysatorsäule d Flügelrührer R4 Molekularsiebsäule e Temperiermantel P Druckminderventil E Einspritzsystem F1 Durchflussmesser 0-100 ml/min PR Flachbettschreiber F2 Durchflussmesser 0-1000 ml/min TM Temperaturmessgerät F3 Durchflussmesser 0-5000 ml/min PG Druckanzeigegerät D Drehzahlregler und -anzeige VG Vakuummessgerät und -anzeige M Messumformer VP Vakuumpumpe S Umschalter für F1, F2, F3 T Thermostat
Abbildung 56: Fließschema der Polymerisationsanlage.

Zirconiumsysteme 87
SAC/Naf10/Zr/Cp B2 BMe
Zunächst sollte untersucht werden, ob das System SAC/Naf10/Zr/CpB2BMe (vgl.
Abbildung 57) unter Einsatz von Standardbedingungen polymerisationsaktiv
ist. Dazu wurde der Katalysator SAC/Naf10/Zr/CpB2BMe zur Aktivierung mit
1.7 M MAO-Lösung in einem Al/Zr-Verhältnis von 2000 zu 1 versetzt und
20 Minuten gerührt. Die Polymerisation wurde bei 50 °C, mit einem Ethen-
druck von 2 bar und Rühren mit 1200 U/min durchgeführt.
CF2 SO3
ZrH3C
Nafion/SiO2
Abbildung 57: Zirconium-System SAC/Naf10/Zr/CpB2BMe für die Polymerisation von Ethen.
Im ersten Versuch wurden 55.4 mg des Katalysators SAC/Naf10/Zr/Cp B2BMe
eingesetzt. Dabei kam es zu einer stark exothermen Reaktion. Die
Suspension heizte sich dabei soweit auf, dass der Katalysator schnell zerstört
und mit nur 2.8 g recht wenig Polymer isoliert wurde. Bei einer stark
exothermen Polymerisationsreaktion kommt es durch die geringe Wärme-
abfuhr zu lokalen Überhitzungen und zur Bildung so genannter Hotspots.
Dadurch kann das Polymer schmelzen und große Klumpen oder Filme
gebildet werden.
Daher wurde in einer weiteren Reaktion die Katalysatormenge auf 16.8 mg
reduziert. Diese Reaktion war zwar leicht exotherm, allerdings verlief diese
Reaktion kontrolliert und es konnte mehr Polymer gewonnen werden. In
Abbildung 58 ist der Verlauf der Reaktion durch Auftragung des Ethenflusses
gegen die Zeit beschrieben. Der Ethenfluss entspricht der Polymerisations-
geschwindigkeit. Man beobachtet, dass die Polymerisation relativ schnell
anspringt. Es folgt dann ein Anstieg der Polymerisationsgeschwindigkeit in
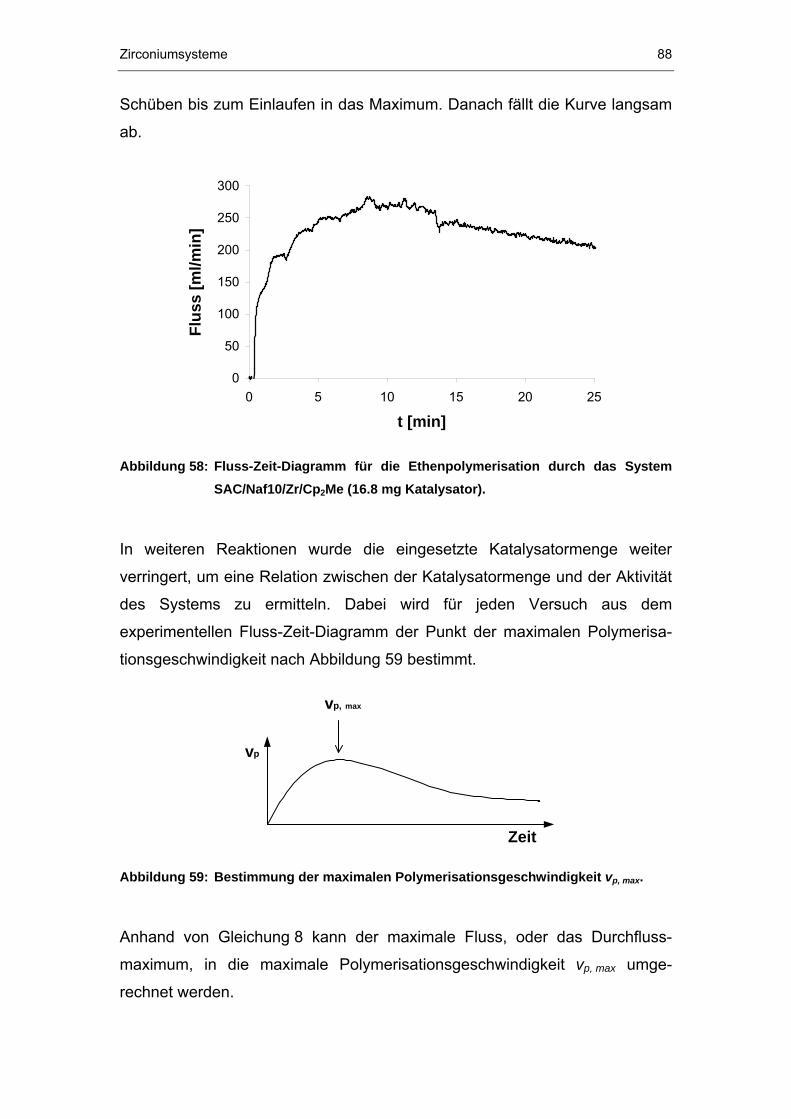
Zirconiumsysteme 88
Schüben bis zum Einlaufen in das Maximum. Danach fällt die Kurve langsam
ab.
0
50
100
150
200
250
300
0 5 10 15 20 25
t [min]
Flus
s [m
l/min
]
Abbildung 58: Fluss-Zeit-Diagramm für die Ethenpolymerisation durch das System SAC/Naf10/Zr/CpB2BMe (16.8 mg Katalysator).
In weiteren Reaktionen wurde die eingesetzte Katalysatormenge weiter
verringert, um eine Relation zwischen der Katalysatormenge und der Aktivität
des Systems zu ermitteln. Dabei wird für jeden Versuch aus dem
experimentellen Fluss-Zeit-Diagramm der Punkt der maximalen Polymerisa-
tionsgeschwindigkeit nach Abbildung 59 bestimmt.
vp
Zeit
vp, max
Abbildung 59: Bestimmung der maximalen Polymerisationsgeschwindigkeit v Bp, maxB.
Anhand von Gleichung 8 kann der maximale Fluss, oder das Durchfluss-
maximum, in die maximale Polymerisationsgeschwindigkeit vBp, max B umge-
rechnet werden.

Zirconiumsysteme 89
Gl. 8 60max, ⋅⋅
=molL
p vvDMv
Dabei ist DM das aus dem Diagramm ermittelte Durchflussmaximum in
ml·minP
-1P, vBL B das Lösungsvolumen im Reaktor in ml, vBmol B das Molvolumen des
Monomergases Ethen bei 1 bar und 0 °C, d. h. 22.246 mP
3P·kmolP
-1P.
In Tabelle 23 sind die eingesetzten Katalysatormengen und die auf diese
Weise ermittelten maximalen Polymerisationsgeschwindigkeiten vBp, max B der
Versuche aufgelistet.
Tabelle 23: Katalysatormengen, Zirconium-Konzentrationen und ermittelte maximale Polymerisationsgeschwindigkeiten des Systems SAC/Naf10/Zr/CpB2BMe mit MAO als Cokatalysator.
m Katalysator [mg]
c Zr [mmol·lP
-1P]
DM [ml·minP
-1P]
vBp, maxB · 10P
3P
[mol·lP
-1P·sP
-1P]
16.8 12.3 284 29
9.3 6.8 123 13
4.9 3.6 29 3.0
4.9 3.6 21 2.1
Trägt man die eingesetzte Zirconium-Konzentration gegen die maximale
Polymerisationsgeschwindigkeit der verschiedenen Versuche auf, ergibt sich
der in Abbildung 60 gezeigte Graph.

Zirconiumsysteme 90
v p, m
ax⋅1
03[m
ol⋅l-
1 s-1
]
c Zr [mmol⋅l-1]
0
5
10
15
20
25
30
0 2 4 6 8 10 12 14
v p, m
ax⋅1
03[m
ol⋅l-
1 s-1
]
c Zr [mmol⋅l-1]
0
5
10
15
20
25
30
0 2 4 6 8 10 12 14
Abbildung 60: Auftragung der maximalen Polymerisationsgeschwindigkeit vBp, maxB gegen die eingesetzte Zirconium-Konzentration für das System SAC/Naf10/Zr/CpB2BMe.
In Abbildung 60 wird ein linearer Zusammenhang zwischen vBp, max B und der
Zirconium-Konzentration deutlich. Dies weist auf eine homogene Verteilung
der katalytisch aktiven Spezies, aber auch auf eine reproduzierbare Arbeits-
weise hin.
Es fällt auf, dass der Schnittpunkt der Geraden mit der x-Achse nicht beim
Koordinatenursprung liegt. Dies ist erklärbar, da bei den eingesetzten, hoch
empfindlichen Zirconocen-Spezies immer zuerst Verunreinigungen in der
Reaktionsmischung mit dem Katalysator „titriert“ werden. Es kommt also zur
teilweisen Zerstörung oder Deaktivierung des Katalysators, was eine minimal
notwendige Katalysatorkonzentration erfordert, bei der überhaupt erst eine
Polymerisationsaktivität eintritt. Für das System SAC/Naf10/Zr/CpB2BMe liegt die
minimal erforderliche Katalysatormenge bei ca. 3.7 mg Katalysator,
0.36 mmol Zirconium, bzw. 2.7 mmol·lP
-1P Zirconium in der Reaktion.
Für die durchgeführten Polymerisationen können relative Aktivitäten im Be-
reich von 2000 bis 12000 g·mmol P
-1P Zr·hP
-1P ermittelt werden. Jedoch können
diese Werte nicht direkt miteinander verglichen werden, da durch die unter-
schiedliche Polymerisationsdauer bei manchen System nur die hoch aktive
Anfangsphase in die Berechnungen einging.

Zirconiumsysteme 91
Als nächstes sollte die Heterogenität der Reaktion überprüft werden, indem
etwa 100 mg des Katalysators für 24 Stunden in Toluol gerührt und abfiltriert
wurden. Das Filtrat wurde anschließend mit MAO als Cokatalysator versetzt
und in der Ethenpolymerisation getestet. Es zeigte sich keine Polymeri-
sationsaktivität, was darauf schließen lässt, dass die Zirconium-Spezies nicht
durch Toluol abgelöst wird.
Im Weiteren sollte die Wirkung der Aktivierung des MAO auf das System
SAC/Naf10/Zr/Cp B2BMe untersucht werden, da bei Zugabe des MAOs zum
geträgerten Zirconium-System Gasentwicklung und eine leichte Entfärbung
des hellgelben Pulvers eintrat. Dazu wurden ca. 20 mg des Katalysators mit
MAO behandelt und nach der Aktivierungszeit von 20 Minuten abfiltriert. Das
Filtrat wurde in der Polymerisation eingesetzt und zeigte dabei eine ähnliche
Polymerisationsaktivität wie das in toluolischem MAO suspendierte System
SAC/Naf10/Zr/Cp B2BMe. Der Rückstand selbst war ebenfalls polymerisations-
aktiv, jedoch nur nach Zugabe von weiterem MAO zur Polymerisationslösung.
Daher muss zusammenfassend festgestellt werden, dass das Katalysator-
system SAC/Naf10/Zr/CpB2BMe mit MAO keine rein heterogene Katalyse zeigt.
Nachdem für das System die Polymerisationsaktivität bei Einsatz klassischer
Reaktionsbedingungen nachgewiesen werden konnte, sollte untersucht
werden, ob das System SAC/Naf10/Zr/CpB2BMe auch ohne die Aktivierung
durch den Cokatalysator MAO Aktivität zeigt. Dabei wurde eine Analogie des
Systems SAC/Naf10/Zr/CpB2BMe mit kationischer Zirconocen-Spezies und
einem schwach koordinierenden Anion, dem Sulfonsäurerest des NafionP
®P-
Polymers, zu den in der Literatur beschriebenen, polymerisationsaktiven
Metallocen-Borat-Systemen T6T nach MARKSP
[101]P und EWENP
[102]P vermutet (vgl.
Abbildung 61). Allerdings zeigten Versuche ohne MAO-Aktivierung trotz
Verwendung von bis zu 100 mg Katalysator keinerlei Aktivität in der Ethen-
polymerisation. Auch das literaturbeschriebene, ähnliche homogene System
Cp* B2BZr(Me)(OTf) zeigte erst nach MAO-Aktivierung Polymerisations-
aktivität.P
[90]P

Zirconiumsysteme 92
CF2 SO3
ZrH3C
Nafion/SiO2 Zr
CH3
CH3 B(C6F5)3
Abbildung 61: Vergleich des Systems SAC/Naf10/Zr/CpB2BMe und des Metallocen-Borat-Systems (6) nach MarksP
[101]P und EwenP
[102]P.
Als alternativer Cokatalysator wurde in weiteren Versuchen Triisobutyl-
aluminium (TIBA) eingesetzt. Es zeigte sich jedoch bei Katalysatormengen
von bis zu 11 mg Katalysator keinerlei Polymerisationsaktivität. Auch die
Variation des Lösungsmittels zu Heptan, unter Verwendung von TIBA als
Cokatalysator, führte nicht zum Erfolg.
In Untersuchungen nach Lagerung des Katalysators SAC/Naf10/Zr/Cp B2BMe bei
Raumtemperatur unter Argon für etwa einen Monat zeigte sich, dass der
Katalysator eine stark herabgesetzte Polymerisationsaktivität auf etwa ein
Viertel der ursprünglichen Aktivität aufwies. Eine genaue Erklärung für diese
Deaktivierung des Systems konnte leider nicht gefunden werden. Denkbar ist
ein Verlust der Methylgruppe des Zirconium-Komplexes unter Reaktion des
Zirconiums mit Silanolgruppen des Trägermaterials (vgl. Abbildung 62). Auf
diese Weise auf Silikat geträgerte Dimethyl-Zirconocen-Spezies weisen eine
sehr geringe Polymerisationsaktivität auf.P
[103]P
CF2 SO3
ZrH3C
Nafion/SiO2
OH
CF2 SO3
Zr
Nafion/SiO2
OCH4
Abbildung 62: Mögliche Deaktivierungsreaktion des Systems SAC/Naf10/Zr/CpB2BMe.
6

Zirconiumsysteme 93
SAC/Naf10/Zr/Cp B2 B
Des Weiteren sollte das System SAC/Naf10/Zr/Cp B2B in der Ethenpolymerisa-
tion untersucht werden. Dazu wurden die gleichen Bedingungen wie für das
oben beschriebene System SAC/Naf10/Zr/Cp B2BMe angewendet.
Zunächst wurde die Reaktion mit 49.6 mg des Katalysators nach Aktivierung
mit MAO durchgeführt. Das System zeigte Polymerisationsaktivität, jedoch
aufgrund der niedrigeren Zirconium-Beladung eine niedrigere Aktivität.
Danach wurde direkt die Heterogenität der Reaktion überprüft, wozu das
System SAC/Naf10/Zr/CpB2B mit der entsprechenden Menge MAO behandelt
und nach 20 Minuten filtriert wurde. Das Filtrat zeigte in der Polymerisation
von Ethen Aktivität. Da auch dieses System unter den hier gewählten
Bedingungen nicht eine rein heterogene Katalyse zeigte, wurden keine
weiteren Versuche durchgeführt.
Charakterisierung der gewonnenen Polymere mittels GPC
Das im Rahmen dieser Arbeit gewonnene Polyethylen war in allen Fällen
farblos. Die Polymerpartikel wiesen eine flockige Struktur und eine niedrige
Schüttdichte auf. Allerdings bildete sich bei höheren Zirconium-Konzentra-
tionen in der Reaktion oft ein Film an der Reaktorwand bzw. am Rührer, was
auf lokale Überhitzung und ein Schmelzen des Polymers hinweist.
Zur Charakterisierung der Polymere wurden sie mittels Gelpermeations-
chromatographie (GPC) auf die Molmassenverteilung untersucht. Die Ergeb-
nisse ausgewählter Polymere sind in Tabelle 24 wiedergegeben. Jede
Polymerprobe wurde zweifach vermessen und der Mittelwert beider
Messungen angegeben.

Zirconiumsysteme 94
Tabelle 24: Ergebnisse der GPC-Messungen für die gewonnenen Polymere (Durch-schnittswerte aus zwei Messungen).
Katalysator MBwB [g·molP
-1P] MBwB/MBnB
SAC/Naf10/Zr/Cp B2BMe 3.39·10P
5P 3.1
Filtrat (SAC/Naf10/Zr/CpB2BMe + MAO) 3.78·10P
5P 2.5
Rückstand (SAC/Naf10/Zr/Cp B2BMe + MAO) 3.42·10P
5P 2.6
SAC/Naf10/Zr/Cp B2BMe 3.96·10P
5P 2.8
Filtrat (SAC/Naf10/Zr/CpB2B + MAO) 3.59·10P
5P 2.6
Alle untersuchten Polymerproben zeigten Molekulargewichte M BwB im Bereich
von etwa 3.4 bis 4.0·10 P
5P. Dies liegt etwa im Bereich, der für das homogene
System CpB2BZrClB2B mit MAO und einem Ethen-Druck von 10 bar und einer
Temperatur von 60 °C erreicht wird. P
[95]P Die Molekulargewichtsverteilung
M BwB/MBn B der Polyethylene ist etwas breiter als für analoge homogene
Katalysatoren typisch (M BwB/MBn B = 2.0 bis 2.5).
Die Unterschiede in den Molekulargewichten zwischen den Polymeren, die
mit den heterogenen Katalysator-Systemen SAC/Naf10/Zr/CpB2BMe und
SAC/Naf10/Zr/Cp B2BMe, bzw. den Filtraten als Katalysator gewonnen wurden,
sind zu gering, um eine Aussage über die katalytisch-aktiven Spezies zu
treffen. Wahrscheinlich sind diese jedoch einander recht ähnlich, da in der
Literatur bei Untersuchungen von Katalysator und Filtrat bei Ablösung
katalytisch-aktiver Spezies Unterschiede von mehreren Größenordnungen in
den Molekulargewichten und bis zu einer Größenordnung bei den Molekular-
gewichtsverteilungen zeigten. P
[104]P

Kupfersysteme 95
6 Kupfersysteme
In den letzten zehn Jahren wurden die chiralen Bis(oxazolin)-Liganden
erfolgreich in der asymmetrischen Katalyse eingesetzt. Da normalerweise ein
hohes Katalysator/Substrat-Verhältnis von 1 bis 10 mol% für diese Um-
setzungen notwendig ist, wurden in den letzten Jahren verschiedene Möglich-
keiten der Heterogenisierung der katalytisch aktiven Spezies untersucht.
Dabei wurde sowohl die Trägerung durch kovalente als auch nicht-kovalente
Bindungen an das Trägermaterial und die Verwendung von organischen und
anorganischen Trägern beschrieben. Bisher wurden die heterogenen Bis-
(oxazolin)-Systeme in Cyclopropanierungen, allylischen Substitutionen, DIELS-
ALDER-, Aziridinierungs-, En- und MUKAYAMA-Aldol-Reaktionen eingesetzt. P
[105]P
Hier soll speziell auf die in der Cyclopropanierung eingesetzten Kupfer-
Bis(oxazolin)-Systeme eingegangen werden. Eine Möglichkeit der Immo-
bilisierung ist die kovalente Verankerung des chiralen Kupfer-Komplexes auf
dem Träger über einen bifunktionellen Linker (vgl. Kapitel 1.5).P
[106-108]P Es
zeigte sich jedoch, dass in manchen Fällen durch die kovalente Bindung die
Reaktant-Katalysator-Übergangsspezies beeinflusst und auf diese Weise oft
die asymmetrische Induktion geringer wird. Daher wurde durch MAYORAL et al.
vorgeschlagen, die Bis(oxazolin)-Kupfer-Spezies nicht durch eine kovalente
Bindung sondern durch Kationenaustausch auf dem Trägermaterial zu
fixieren. P
[109]P Dafür nutzten sie die Austauschkapazität von Tonen oder
anionischen Austauscherharzen. P
[109-113]P Unter anderem wurde NafionP
®P-
Polymer in Form des reinen Polymers NR 50 und des Nafion P
®P-haltigen
Silikats SAC-13 eingesetzt, da sich in der homogenen Katalyse das analoge
Triflat-Anion als geeignetes Gegenion erwies. Zunächst wurde ein homogener
Komplex aus Kupfer(II)-triflat und dem entsprechenden Bis(oxazolin) (vgl.
Abbildung 63) hergestellt und damit das Trägermaterial in Methanol aus-
getauscht. Es wurden auch weitere Lösungsmittel, wie Nitroethan, und
Kupfer-Precursor, wie Kupfer(II)-chlorid, untersucht. Jedoch führte dies zu
deutlich geringeren Kupfermengen auf dem Trägermaterial.

Kupfersysteme 96
N
OO
N
R
MeMe
R
R = Ph, CH2Ph, tBu
Cu
2+
Abbildung 63: Eingesetzte Bis(oxazolin)-Liganden für die elektrostatische Immobili-sierung von Kupfer-Spezies auf Tonen und anionischen Austauscher-harzen durch MAYORAL et al.P
[109]P
Im Rahmen dieser Arbeit wurden die Untersuchungen von MAYORAL et al. auf
geordnete NafionP
®P-haltige Silikate als Trägermaterial ausgedehnt und im
Folgenden untersucht, ob durch Veränderung der Struktur des Träger-
materials die katalytischen Eigenschaften beeinflusst werden.
6.1 Synthese
Die Darstellung der heterogenen Kupferkatalysatoren erfolgte analog der
Methode von MAYORAL. Dazu wurde zunächst, durch Behandlung mit
Natriumchlorid-Lösung, das Natrium-ausgetauschte Nafion P
®P-haltige Silikat
hergestellt und mit einer methanolischen Lösung des gewünschten Liganden
und des Kupfer-Precursors, Kupfer(II)triflat, behandelt (vgl. Abbildung 64). Die
Produkte wurden in Form eines türkisblauen Pulvers gewonnen.
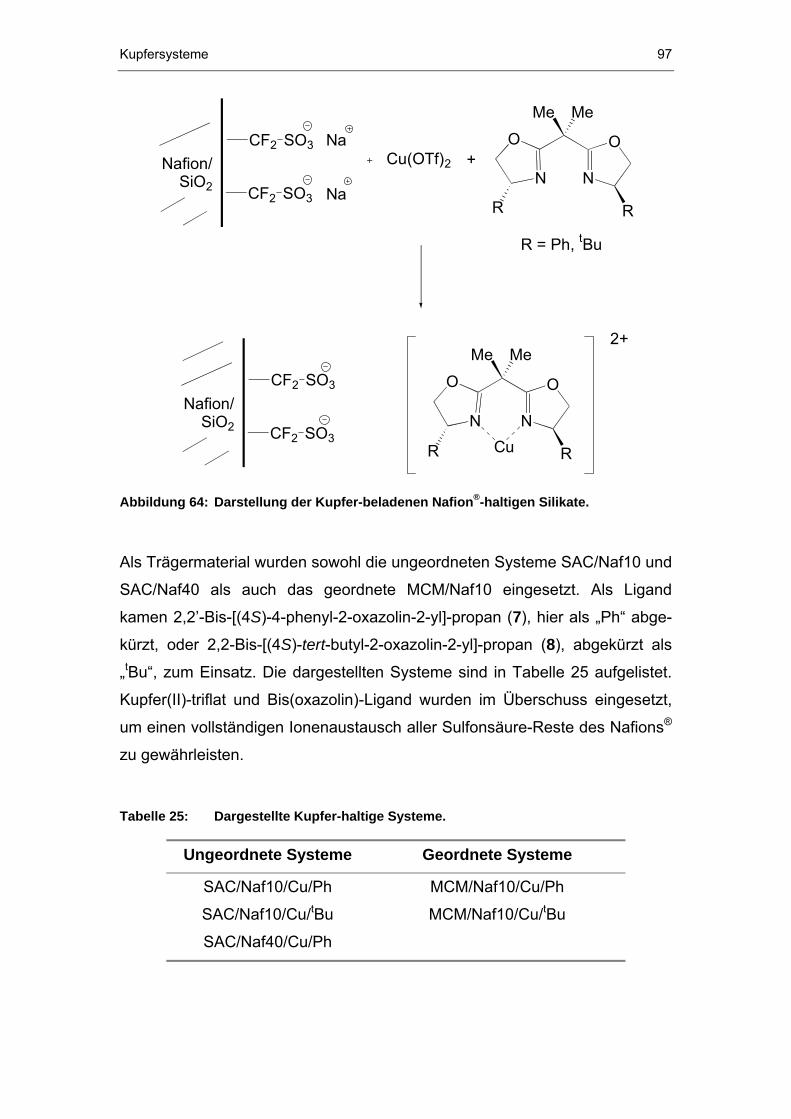
Kupfersysteme 97
CF2 SO3
+
Nafion/SiO2 CF2 SO3
CF2 SO3
Nafion/SiO2 CF2 SO3
Na
Na
Cu(OTf)2 +
N
OO
N
R
MeMe
R
R = Ph, tBu
Cu
2+
N
OO
N
R
MeMe
R
Abbildung 64: Darstellung der Kupfer-beladenen NafionP
®P-haltigen Silikate.
Als Trägermaterial wurden sowohl die ungeordneten Systeme SAC/Naf10 und
SAC/Naf40 als auch das geordnete MCM/Naf10 eingesetzt. Als Ligand
kamen 2,2’-Bis-[(4S)-4-phenyl-2-oxazolin-2-yl]-propan (7), hier als „Ph“ abge-
kürzt, oder 2,2-Bis-[(4S)-tert-butyl-2-oxazolin-2-yl]-propan (8), abgekürzt als
„ P
tPBu“, zum Einsatz. Die dargestellten Systeme sind in Tabelle 25 aufgelistet.
Kupfer(II)-triflat und Bis(oxazolin)-Ligand wurden im Überschuss eingesetzt,
um einen vollständigen Ionenaustausch aller Sulfonsäure-Reste des Nafions P
®P
zu gewährleisten.
Tabelle 25: Dargestellte Kupfer-haltige Systeme.
Ungeordnete Systeme Geordnete Systeme
SAC/Naf10/Cu/Ph MCM/Naf10/Cu/Ph
SAC/Naf10/Cu/ P
tPBu MCM/Naf10/Cu/ P
tPBu
SAC/Naf40/Cu/Ph
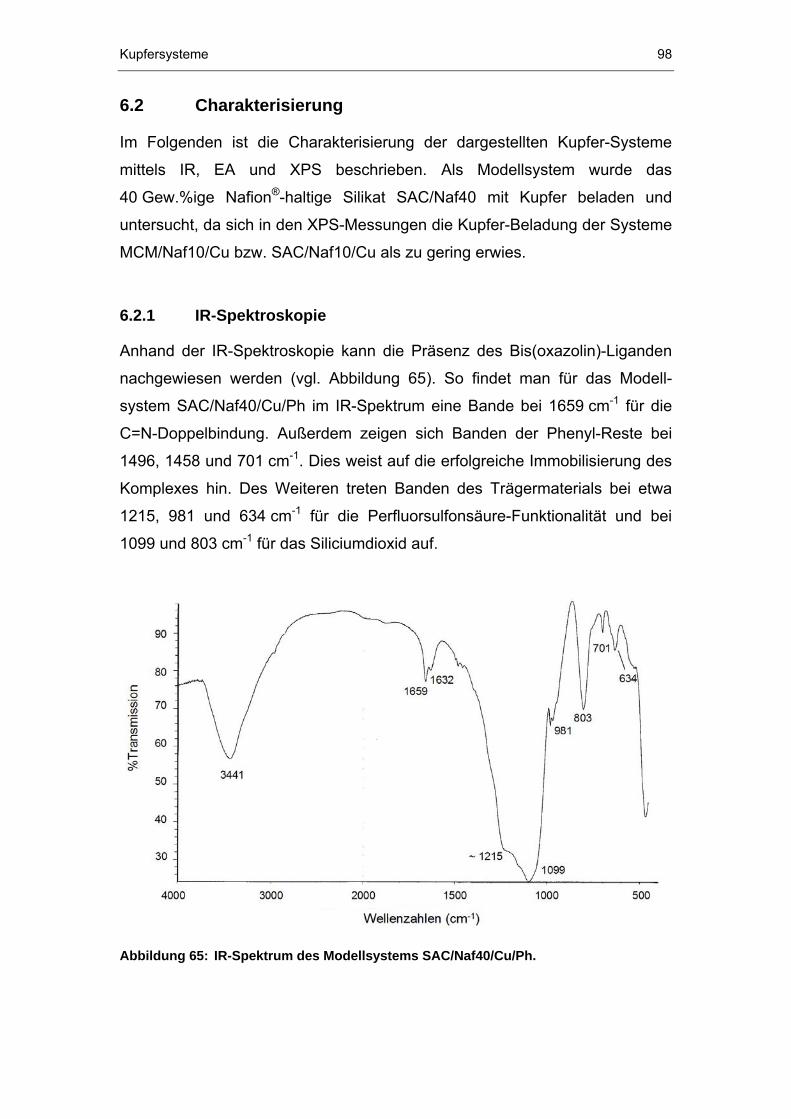
Kupfersysteme 98
6.2 Charakterisierung
Im Folgenden ist die Charakterisierung der dargestellten Kupfer-Systeme
mittels IR, EA und XPS beschrieben. Als Modellsystem wurde das
40 Gew.%ige Nafion P
®P-haltige Silikat SAC/Naf40 mit Kupfer beladen und
untersucht, da sich in den XPS-Messungen die Kupfer-Beladung der Systeme
MCM/Naf10/Cu bzw. SAC/Naf10/Cu als zu gering erwies.
6.2.1 IR-Spektroskopie
Anhand der IR-Spektroskopie kann die Präsenz des Bis(oxazolin)-Liganden
nachgewiesen werden (vgl. Abbildung 65). So findet man für das Modell-
system SAC/Naf40/Cu/Ph im IR-Spektrum eine Bande bei 1659 cmP
-1P für die
C=N-Doppelbindung. Außerdem zeigen sich Banden der Phenyl-Reste bei
1496, 1458 und 701 cm P
-1P. Dies weist auf die erfolgreiche Immobilisierung des
Komplexes hin. Des Weiteren treten Banden des Trägermaterials bei etwa
1215, 981 und 634 cm P
-1P für die Perfluorsulfonsäure-Funktionalität und bei
1099 und 803 cm P
-1P für das Siliciumdioxid auf.
Abbildung 65: IR-Spektrum des Modellsystems SAC/Naf40/Cu/Ph.

Kupfersysteme 99
MAYORAL et al. veröffentlichten für das NafionP
®P-System NR 50 eine Bande für
die C=N-Doppelbindung bei 1662 cm P
-1P und für das SAC-13-System bei
1660 cm P
-1P. Dies entspricht auch den homogenen Bis(oxazolin)-triflat-Kupfer-
Komplexen, die diese Bande bei 1660 cm P
-1P aufweisen. P
[109]P
6.2.2 Elementaranalysen
Für die dargestellten Systeme SAC/Naf10/Cu bzw. MCM/Naf10/Cu wurden
mittels Elementaranalyse Kupfer-Beladungen zwischen 0.3 bis 0.5 Gew.%
gefunden (vgl. Tabelle 26). Die Anteile an Schwefel und Stickstoff waren in
allen Proben zu gering (< 0.1 Gew.%), um genau bestimmt werden zu
können.
Tabelle 26: Ergebnisse der Elementaranalyse für die Kupfer-beladenen Systeme.
Probe Gew.% Cu
SAC/Naf10/Cu/Ph 0.30
SAC/Naf10/Cu/ P
tPBu 0.42
MCM/Naf10/Cu/Ph 0.38
MCM/Naf10/Cu/ P
tPBu 0.51
Durch MAYORAL et al. wurden je nach eingesetztem Ligand für Kupfer-
beladene SAC-13-Systeme 0.05 bis 0.07 mmol·g P
-1P Kupfer gefunden; dies ent-
spricht 0.3 bis 0.4 Gew.% Kupfer.P
[109]P Die Stickstoffwerte waren auch in
diesen Fällen zu niedrig, um reproduzierbar bestimmt werden zu können.
6.2.3 XPS-Untersuchungen
Zunächst wurden die Proben mit 10 Gew.% NafionP
®P und damit 0.3 bis
0.5 Gew.% Kupfer in XPS-Untersuchungen eingesetzt. Jedoch erwies sich
der Gehalt an Kupfer als zu gering, um die vorhandenen Kupfer-Spezies zu
charakterisieren.
Daher wurde in weiteren Messungen das Modellsystem SAC/Naf40/Cu/Ph
betrachtet. Für dieses System konnten zwar die Cu 2p-Bindungsenergien
ermittelt werden, jedoch nicht die LLM-(Auger)-Signale, die zur Charak-
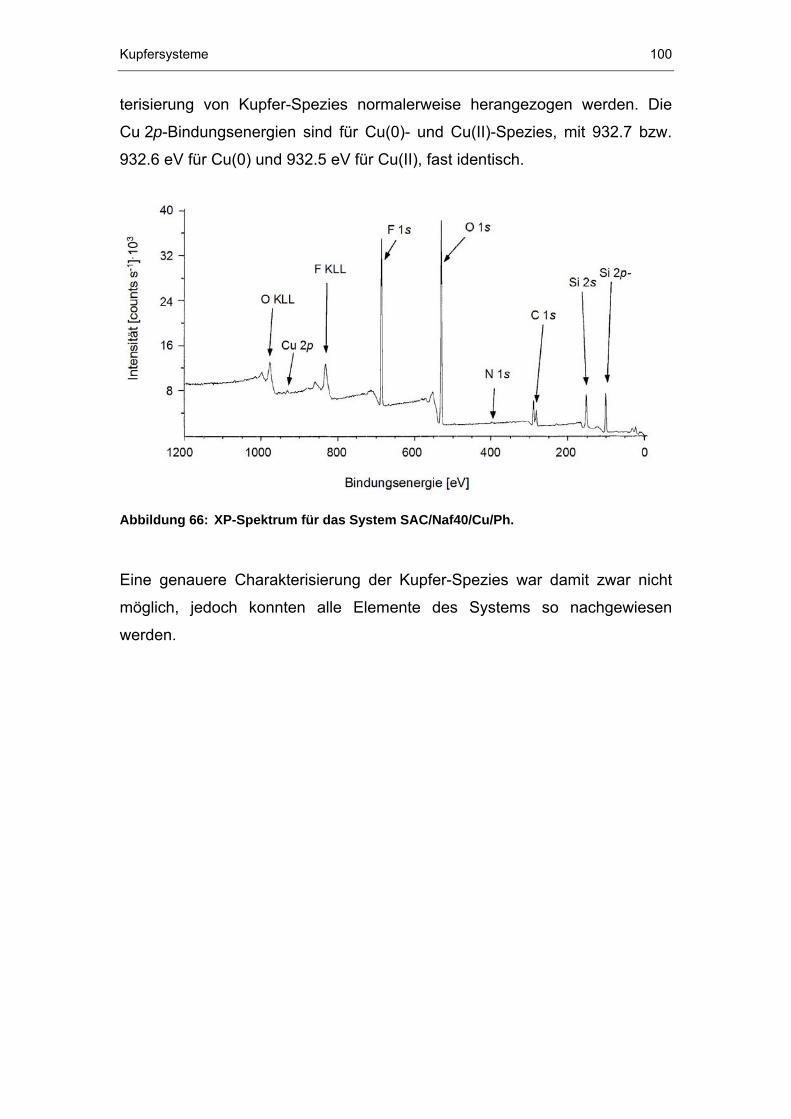
Kupfersysteme 100
terisierung von Kupfer-Spezies normalerweise herangezogen werden. Die
Cu 2p-Bindungsenergien sind für Cu(0)- und Cu(II)-Spezies, mit 932.7 bzw.
932.6 eV für Cu(0) und 932.5 eV für Cu(II), fast identisch.
Abbildung 66: XP-Spektrum für das System SAC/Naf40/Cu/Ph.
Eine genauere Charakterisierung der Kupfer-Spezies war damit zwar nicht
möglich, jedoch konnten alle Elemente des Systems so nachgewiesen
werden.

Kupfersysteme 101
6.3 Katalysen
Im Folgenden werden die Ergebnisse der mit den Kupfer-beladenen
Systemen durchgeführten Katalysen aufgeführt und mit der Literatur ver-
glichen.
6.3.1 Cyclopropanierungen
6.3.1.1 Mechanismus und Hintergrund
Cyclopropanierungen gehören zu den ersten Reaktionen, die enantioselektiv
durchgeführt wurden, obwohl zunächst nur bis zu 10 % Enantiomeren-
überschuss erreicht werden konnte. P
[114]P Ein Durchbruch in der asymmet-
rischen Induktion dieser Reaktion wurde 1988 durch PFALTZ et al. erreicht, die
Semicorrin-Kupfer(II)-Komplexe als Katalysatoren einsetzten. P
[115]P Da Bis-
(oxazoline) in ihrer Struktur und ihrem katalytischen Verhalten den Semi-
corrinen entsprechen und synthetisch einfacher zugänglich sind, wurden sie
in der Folgezeit ebenfalls erfolgreich als Liganden verwendet. P
[116-118]P
Üblicherweise wird zur Untersuchung und Beurteilung der Katalysatorsysteme
die Cyclopropanierungsreaktion zwischen Styrol und Ethyldiazoacetat be-
trachtet (vgl. Abbildung 67). In dieser Reaktion wurden unter homogener
Katalyse mit Bis(oxazolin)-Kupfer(II)-Komplexen, je nach Substituent am
Bis(oxazolin)-Ring, bis zu 99 % ee für die trans- und 97 % ee für die cis-
Produkte erreicht. Das trans:cis-Verhältnis betrug dabei etwa 70 : 30.P
[116, 117]P
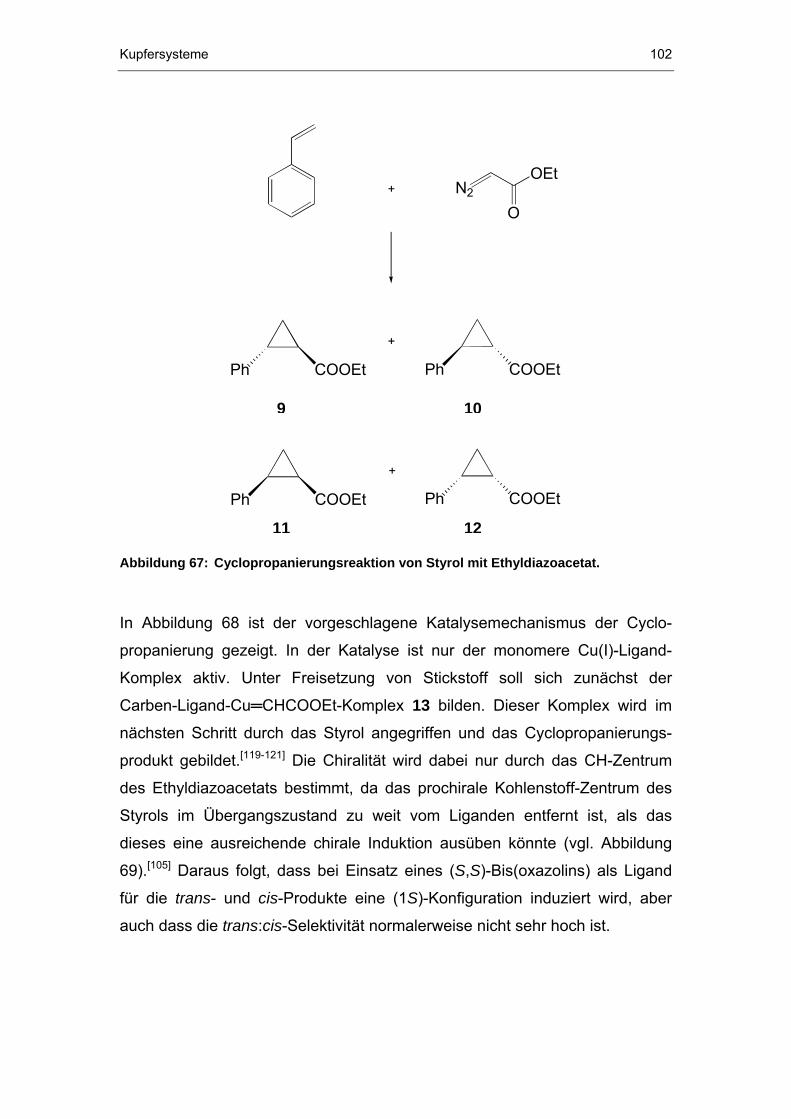
Kupfersysteme 102
Ph COOEt
Ph COOEt Ph COOEt
Ph COOEt
+
+
+
N2OEt
O
Abbildung 67: Cyclopropanierungsreaktion von Styrol mit Ethyldiazoacetat.
In Abbildung 68 ist der vorgeschlagene Katalysemechanismus der Cyclo-
propanierung gezeigt. In der Katalyse ist nur der monomere Cu(I)-Ligand-
Komplex aktiv. Unter Freisetzung von Stickstoff soll sich zunächst der
Carben-Ligand-Cu═CHCOOEt-Komplex T13T bilden. Dieser Komplex wird im
nächsten Schritt durch das Styrol angegriffen und das Cyclopropanierungs-
produkt gebildet. P
[119-121]P Die Chiralität wird dabei nur durch das CH-Zentrum
des Ethyldiazoacetats bestimmt, da das prochirale Kohlenstoff-Zentrum des
Styrols im Übergangszustand zu weit vom Liganden entfernt ist, als das
dieses eine ausreichende chirale Induktion ausüben könnte (vgl. Abbildung
69). P
[105]P Daraus folgt, dass bei Einsatz eines (S,S)-Bis(oxazolins) als Ligand
für die trans- und cis-Produkte eine (1S)-Konfiguration induziert wird, aber
auch dass die trans:cis-Selektivität normalerweise nicht sehr hoch ist.
9 10
11 12
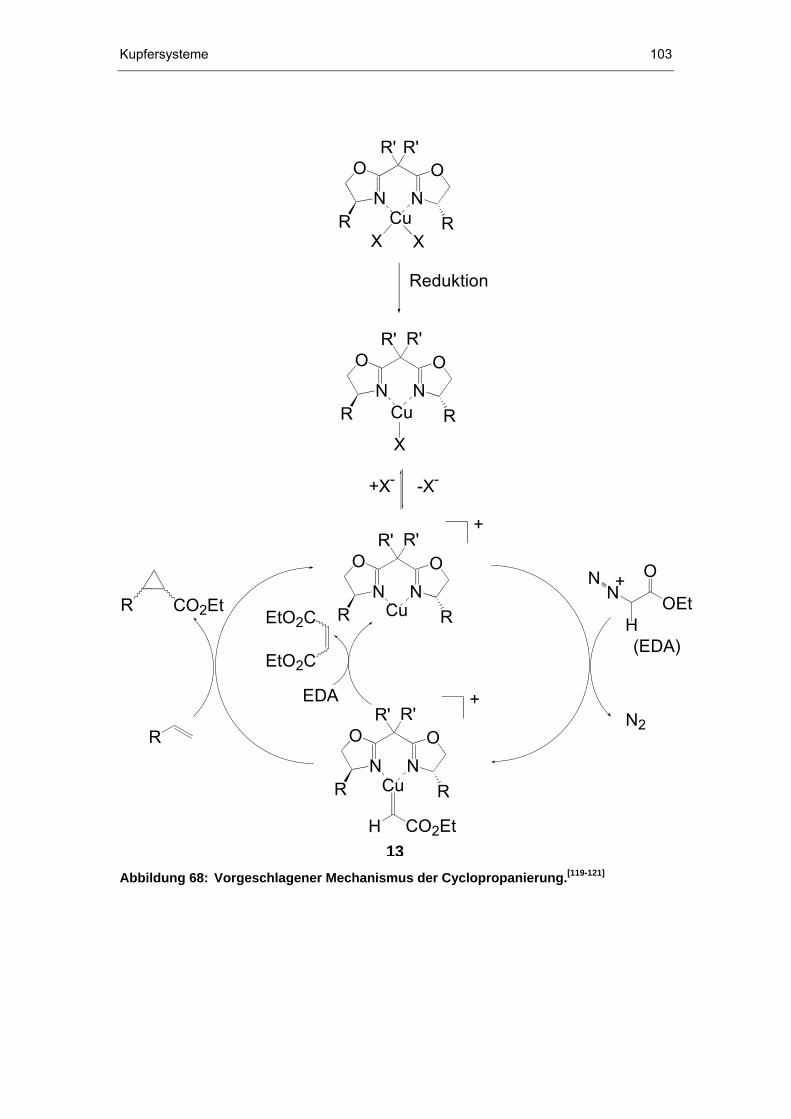
Kupfersysteme 103
NOO
NR RCu
+
NOO
NR RCu
+
NOO
NR
R'R'
RCuX X
NOO
NR
R'
RCu
X
Reduktion
+X- -X-
H CO2Et
EDA
EtO2C
EtO2C
R
R CO2Et OEt
O
H
NN +
(EDA)
N2
R'
R' R'
R' R'
Abbildung 68: Vorgeschlagener Mechanismus der Cyclopropanierung.P
[119-121]P
13

Kupfersysteme 104
N
OO
N
R RCu
CO2EtH
R1 R2
R1, R2 = Ph, H oder H, PhEnantioselektive Kontrolle
R' R'
Abbildung 69: Vorgeschlagener Übergangszustand in der Cyclopropanierung.P
[115]P
Seit 1997 wurde die Untersuchung der Cyclopropanierung auf heterogene
Systeme ausgeweitet. Dabei wurden sowohl kovalent als auch nicht-kovalent
gebundene (Bis)oxazolin-Kupfer-Systeme beschrieben. Für alle untersuchten
Systeme wurden jedoch deutlich geringere Enantioselektivitäten als für die
analogen homogenen Systeme nachgewiesen.
Im Rahmen der vorliegenden Arbeit sollten die Untersuchungen Kupfer-
beladener NafionP
®P-haltiger Systeme von MAYORAL et al. P
[109-113]P auf die hier
dargestellten, geordneten NafionP
®P-haltigen Silikate ausgedehnt und der Ein-
fluss des Trägermaterials betrachtet werden.
6.3.1.2 Eigene Arbeiten
Die hier dargestellten Systeme SAC/Naf10/Cu/Ph, SAC/Naf10/Cu/ P
tPBu,
MCM/Naf10/Cu/Ph und MCM/Naf10/Cu/ P
tPBu wurden in der Cyclopropanie-
rungsreaktion von Styrol und Ethyldiazoacetat zu 2-Phenyl-cyclopropan-
carboxylsäureethylester eingesetzt. Dazu wurden die Katalysatoren in
Dichlormethan bei 25 °C unter einer Argon-Atmosphäre mit den Reaktanten
umgesetzt. Außerdem wurden die entsprechenden homogenen Katalysatoren
untersucht. Die verwendeten Mengen und Bedingungen sind in Ab-
schnitt 9.6.2 angegeben. Die Ergebnisse der Umsetzungen sind in Tabelle 27
aufgeführt.
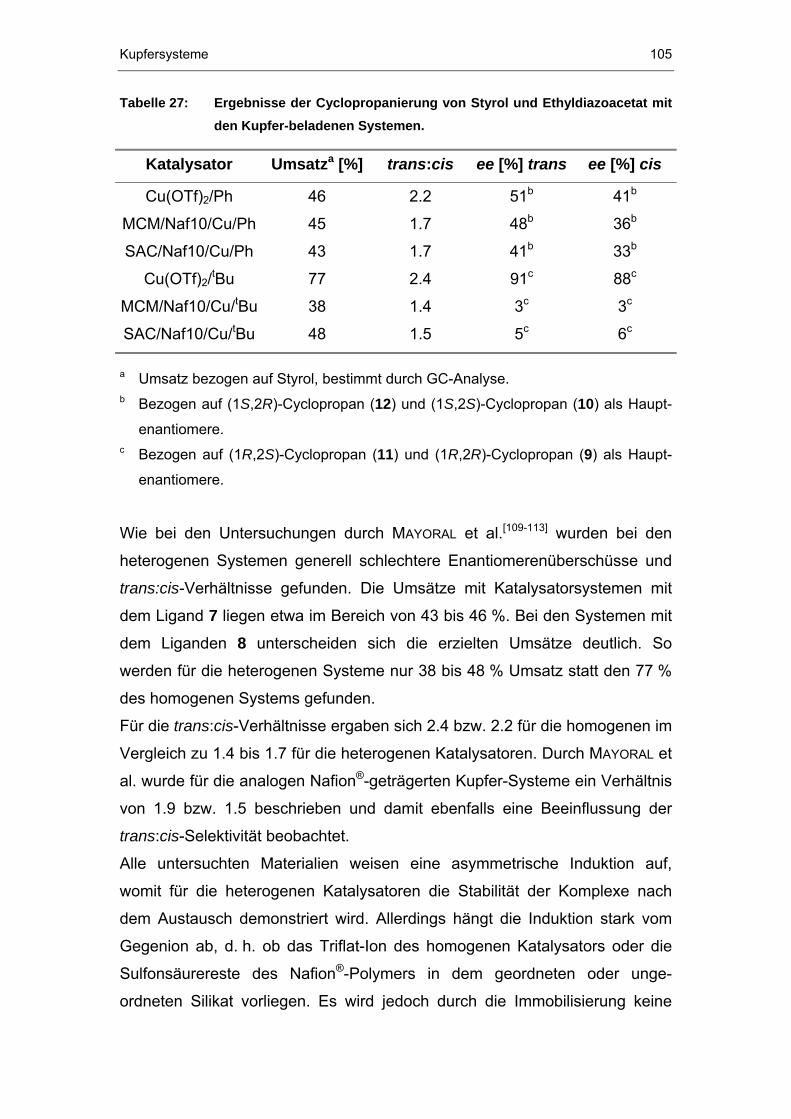
Kupfersysteme 105
Tabelle 27: Ergebnisse der Cyclopropanierung von Styrol und Ethyldiazoacetat mit den Kupfer-beladenen Systemen.
Katalysator UmsatzP
aP [%] trans:cis ee [%] trans ee [%] cis
Cu(OTf)B2B/Ph 46 2.2 51 P
bP 41P
bP
MCM/Naf10/Cu/Ph 45 1.7 48 P
bP 36P
bP
SAC/Naf10/Cu/Ph 43 1.7 41 P
bP 33P
bP
Cu(OTf)B2B/ P
tPBu 77 2.4 91P
cP 88P
cP
MCM/Naf10/Cu/ P
tPBu 38 1.4 3P
cP 3P
cP
SAC/Naf10/Cu/ P
tPBu 48 1.5 5P
cP 6P
cP
P
aP Umsatz bezogen auf Styrol, bestimmt durch GC-Analyse.
P
bP Bezogen auf (1S,2R)-Cyclopropan ( T12) Tund (1S,2S)-Cyclopropan ( T10)T als Haupt-
enantiomere.
P
cP Bezogen auf (1R,2S)-Cyclopropan ( T11) Tund (1R,2R)-Cyclopropan ( T9) Tals Haupt-
enantiomere.
Wie bei den Untersuchungen durch MAYORAL et al. P
[109-113]P wurden bei den
heterogenen Systemen generell schlechtere Enantiomerenüberschüsse und
trans:cis-Verhältnisse gefunden. Die Umsätze mit Katalysatorsystemen mit
dem Ligand T7T liegen etwa im Bereich von 43 bis 46 %. Bei den Systemen mit
dem Liganden T8T unterscheiden sich die erzielten Umsätze deutlich. So
werden für die heterogenen Systeme nur 38 bis 48 % Umsatz statt den 77 %
des homogenen Systems gefunden.
Für die trans:cis-Verhältnisse ergaben sich 2.4 bzw. 2.2 für die homogenen im
Vergleich zu 1.4 bis 1.7 für die heterogenen Katalysatoren. Durch MAYORAL et
al. wurde für die analogen NafionP
®P-geträgerten Kupfer-Systeme ein Verhältnis
von 1.9 bzw. 1.5 beschrieben und damit ebenfalls eine Beeinflussung der
trans:cis-Selektivität beobachtet.
Alle untersuchten Materialien weisen eine asymmetrische Induktion auf,
womit für die heterogenen Katalysatoren die Stabilität der Komplexe nach
dem Austausch demonstriert wird. Allerdings hängt die Induktion stark vom
Gegenion ab, d. h. ob das Triflat-Ion des homogenen Katalysators oder die
Sulfonsäurereste des Nafion P
®P-Polymers in dem geordneten oder unge-
ordneten Silikat vorliegen. Es wird jedoch durch die Immobilisierung keine

Kupfersysteme 106
Änderung der Richtung der asymmetrischen Induktion beobachtet, also die
gleichen Hauptenantiomere in homogener und heterogener Katalyse ge-
funden.
Mit dem Bis(oxazolin)-Ligand T7T verringern sich die Enantiomerenüberschüsse
der Systeme für die trans-Produkte, bezogen auf die (1S)-Konfiguration, von
51 % des homogenen Katalysators auf 48 % für das geordnete Silikat bis zu
41 % des ungeordneten Silikats. Für die cis-Produkte ergaben sich, bezogen
auf die (1S)-Konfiguration, für diese Systeme 41, 36 bzw. 33 % ee. Im
Vergleich dazu wurden in den Untersuchungen von MAYORAL et al. als bestes
Ergebnis 57 und 46 % ee für die trans- bzw. cis-Produkte veröffentlicht.
Obwohl das dort verwendete System, basierend auf dem kommerziellen,
NafionP
®P-haltigen Silikat SAC-13, analog dem hier beschriebenen System
SAC/Naf10 ist, konnten diese Enantiomerenüberschüsse, trotz gleicher
Reaktionsbedingungen, nicht erreicht werden.
Die Systeme mit dem Bis(oxazolin)-Liganden T8T zeigten mit 3 % für trans- und
cis-Produkte mit dem geordneten MCM-System und 5 bzw. 6 % Enantio-
merenüberschuss für das ungeordnete SAC-System eine sehr geringe
asymmetrische Induktion. Im Gegensatz dazu zeigt das homogene System
sehr gute Werte mit 91 bzw. 88 % ee. MAYORAL et al. beobachteten einen
ähnlich geringen Enantiomerenüberschuss von 5 bzw. 7 % für das reine
NafionP
®P-Polymer NR 50 als Trägermaterial, jedoch 23 bzw. 19 % ee für das
NafionP
®P-haltige Silikat SAC-13. Diese Ergebnisse wurden jedoch nur bei
starker Verkürzung der Reaktionszeit und damit bei geringem Umsatz er-
reicht.
Die unterschiedlichen Enantiomerenüberschüsse können durch Annahme
verschiedener Spezies erklärt werden. So kann ein freier Bis(oxazolin)-Ligand
vorliegen, der jedoch keine katalytische Aktivität zeigen sollte. Außerdem
wäre ein [Bis(oxazolin)]B2B-Kupfer-Komplex möglich, der sofort zu einem
weiteren Komplex, nämlich einem Bis(oxazolin)-Kupfer-Komplex, zerfallen
würde. Bis(oxazolin)-Kupfer-Spezies sind die einzig katalytisch aktiven
Spezies, die enantioselektiv wirken. Des Weiteren könnte eine Kupfer-
Spezies ohne Bis(oxazolin)-Ligand vorliegen, die nur racemisch katalysieren
kann und damit zur Verringerung der Enantioselektivität führen muss. Von
MAYORAL et al. wurde zum Beispiel mittels IR-Spektroskopie ein nicht-chiraler

Kupfersysteme 107
Komplex des Kupfers mit dem als Nebenprodukt gebildeten Diethylmaleat
nachgewiesen (vgl. Abbildung 70).P
[109]P
N
OO
N
R RCu
Me Me 2+
Träger
+
O O
OEtEtO
Träger
N
OO
N
R R
Me Me
+
OEtEtO
OO
Cu
2+
Abbildung 70: Bildung des nicht-chiralen, katalytisch aktiven Kupfer-Komplexes mit Diethylmaleat.P
[109]P
Die Unterschiede der Enantiomerenüberschüsse mit den zwei untersuchten
Bis(oxazolin)-Liganden können durch die unterschiedlich stark auftretende
Dissoziation des Liganden vom Trägermaterial erklärt werden. Das Auftreten
einer abstoßenden Kraft zwischen Ligand und Trägermaterial ist dabei wohl
eher sterischer Natur, da die Liganden etwa gleiche elektrostatische Eigen-
schaften aufweisen. P
[113]P Für den Liganden T8T mit dem sperrigen tert-Butyl-Rest
muss dabei die größte sterische Wechselwirkung auftreten, was die niedrige
Enantioselektivität dieser heterogenen Systeme erklärt.

Kupfersysteme 108
N
OO
N
R RCu
Me Me
2+
Träger
sterischeWechsel-wirkungen
elektrostatischeWechselwirkungen
Abbildung 71: Wechselwirkungen zwischen chiralem Komplex und Trägermaterial.P
[113]P
Nachdem die heterogenen Katalysatoren mit dem Liganden T8T bereits
während des ersten Katalyseversuchs geringe Enantioselektivitäten aufgrund
des Verlusts des Liganden zeigten, wurden nur die Systeme mit dem
Liganden T7T in weiteren Katalyseversuchen eingesetzt. Dazu wurde die
Reaktionsmischung nach der Katalyse filtriert, das Filtrat isoliert und der
Katalysator mit Dichlormethan gewaschen und getrocknet.
Es wurde ein Verlust an katalytischer Aktivität der heterogenen Katalysatoren
beim zweiten Durchgang beobachtet, der durch Leaching bzw. Vergiftung der
katalytisch aktiven Spezies erklärt werden kann. Die Verringerung der
Enantioselektivität ist durch Bildung weiterer nicht-chiraler Zentren nach
Abbildung 70 erklärbar.
Die Reaktionsmischungen, die durch Filtration nach der Katalyse vom
heterogenen Katalysator befreit waren, wurden in weiteren Versuchen mit
einer weiteren Portion von Ethyldiazoacetat versetzt. Da diese Lösungen
weiterhin eine katalytische Aktivität zeigten und die Menge an Produkt
zunahm, muss davon ausgegangen werden, dass die Katalyse neben
geträgerten auch durch freie Kupfer-Spezies erfolgte. Es werden damit nicht
nur inaktive Kupfer-Komplexe freigesetzt, wie durch MAYORAL et al. ver-
öffentlicht P
[109]P. Die Untersuchungen der Kupfer-beladenen Nafion P
®P-haltigen
Systeme wurden aufgrund dieser Ergebnisse an dieser Stelle abgebrochen.

Kombination von Säure- und Enzym-Katalyse 109
7 Kombination von Säure- und Enzym-Katalyse
Im Folgenden sollen die Ergebnisse einer Kombination von klassischer
Säurekatalyse durch NafionP
®P-Polymere mit der Enzymkatalyse durch eine
Lipase beschrieben werden. Diese Arbeiten wurden in Zusammenarbeit mit
W. Wiesenhöfer im eigenen Arbeitskreis durchgeführt.
7.1 Grundlagen der Enzymkatalyse
Die Biokatalyse hat sich inzwischen neben der homogenen und heterogenen
(Metall)katalyse zu einer Standardmethode in der organischen Synthese
entwickelt. Enzyme sind besonders attraktiv aufgrund ihrer hohen Effizienz
und meist exzellenten Chemo-, Regio-, Diastereo- und Enantioselektivität. So
finden sie industrielle Anwendung in der Nahrungsmittel-, Waschmittel- und
Papierindustrie, sowie in der Duftstoff- und Aromaherstellung. Besonders
interessant sind Enzyme für den so genannten chiralen Markt, zum Beispiel
für die Darstellung pharmazeutischer Produkte.
Enzyme werden in verschiedene Klassen unterteilt, wovon die Hydrolasen die
für die organische Chemie wichtigste Gruppe darstellt. P
[122]P Zu den Hydrolasen
gehören die Lipasen (Triacylglycerinhydrolasen, E.C.3.1.1.3). Sie werden
durch Bakterien, Pilze und alle höheren Organismen hergestellt. Im Stoff-
wechsel des Organismus bewirken sie die hydrolytische Spaltung von Fett-
säureglyceriden (vgl. Abbildung 72).P
[123]P
OO
O
R2
R1
R3
O
O
O
LipaseH2O OH
HOOH
+
R1COOH
R3COOH
R2COOH
Abbildung 72: Lipasen-katalysierte hydrolytische Spaltung von Triacylglycerinen.
Der große Nutzen der Lipasen erklärt sich aus ihrem breiten Substrat-
spektrum. Sie können nicht nur Hydrolysen, sondern auch Veresterungen,
Umesterungen mit Alkoholen, sowie Acylierungen von Aminen und Oximen

Kombination von Säure- und Enzym-Katalyse 110
katalysieren. In Abbildung 73 ist der katalytische Mechanismus Lipasen-
katalysierter Reaktionen dargestellt.
Das katalytisch aktive Zentrum der Lipasen besteht aus einer katalytischen
Triade, die aus drei speziell angeordneten Aminosäuren aufgebaut ist. Ent-
scheidend ist dabei nicht nur die Position dieser Aminosäuren in der Protein-
sequenz, sondern die durch die Konformation des Enzyms erzeugte räum-
liche Anordnung.
In der Katalyse wird zunächst die Nucleophilie des Serin-Restes durch
Bildung einer Wasserstoffbrücke mit der Imidazolgruppe eines Histidins
erhöht. Diese Wechselwirkung wird zusätzlich durch die Carboxylgruppe
eines Aspartats stabilisiert. Anschließend kann die Hydroxylgruppe des Serins
den Carbonylkohlenstoff eines Esters oder einer Carbonsäure angreifen.
Unter Freisetzung eines Alkohol- bzw. Wassermoleküls wird der Acyl-Enzym-
Komplex T14T ausgebildet. Die dazu durchlaufene tetraedrische Zwischenstufe
wird über zwei Wasserstoffbrückenbindungen im so genannten Oxyanion
Hole stabilisiert. Als nächstes erfolgt der Angriff eines Nucleophils am
Carbonylkohlenstoff des Acyl-Enzym-Komplexes. Über eine zweite tetraedri-
sche Zwischenstufe wird abschließend das Produkt freigesetzt.
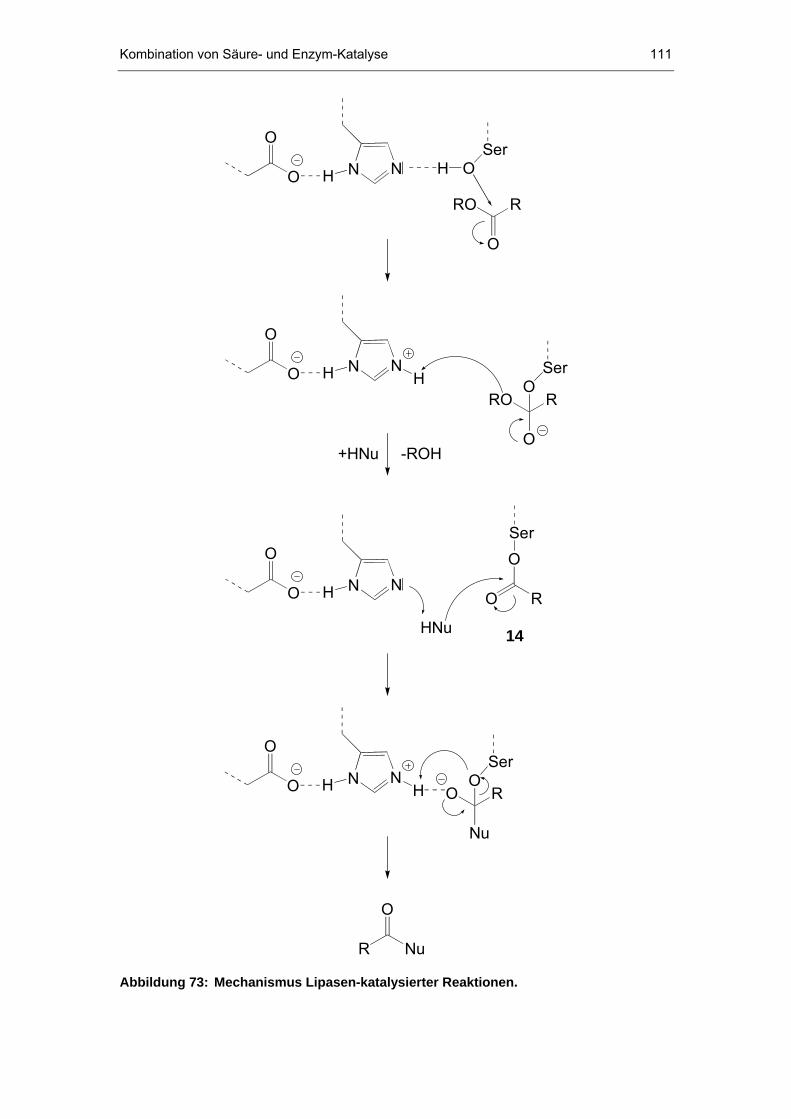
Kombination von Säure- und Enzym-Katalyse 111
O
O NNH H OSer
RO R
O
O
O NNH H O
O
RRO
+HNu -ROH
Ser
O
O NNH
SerO
RO
HNu
O
O NNH H O
Nu
RO
Ser
R Nu
O
Abbildung 73: Mechanismus Lipasen-katalysierter Reaktionen.
T14

Kombination von Säure- und Enzym-Katalyse 112
Für industrielle Anwendungen wird meist die Immobilisierung der Enzyme
gewünscht, um die mechanische, chemische und thermische Stabilität der
Enzyme zu erhöhen und sie nach der Reaktion leichter abtrennen und auch
wieder verwenden zu können. P
[122, 124]P Generell stehen zwei verschiedene
Methoden zur Immobilisierung von Enzymen zur Verfügung. Zum einen
können die Enzyme durch An- und Verknüpfung oder durch Einschluss des
Enzyms geträgert werden. Durch Adsorption an die Oberfläche des Träger-
materials werden oft nicht ausreichend stabile Materialien gewonnen, von
denen das Enzym nach wenigen Reaktionscyclen vollständig herunter-
gewaschen wird. Der Einschluss in organischen Polymeren oder Membranen
führt meist zu recht stabilen und recycelfähigen Systemen, allerdings wird in
manchen Fällen eine Verringerung der Aktivität im Vergleich zum freien
Enzym beobachtet. In den letzten Jahren erwies sich der Einschluss in Sol-
Gel-Materialien als die beste Immobilisierungsmethode für Lipasen. Die erste
Veröffentlichung zur erfolgreichen Immobilisierung mit Erhalt der teilweisen
Aktivität des Enzyms erfolgte bereits im Jahr 1971. P
[125]P Seitdem wurden
verschiedene Enzyme mit vollständigem Erhalt der Aktivität in Sol-Gel-
Materialien immobilisiert und erfolgreich in der Biokatalyse eingesetzt.
Eine Weiterentwicklung wurde 1995 durch REETZ et al. durch die Immobili-
sierung von Lipasen in Sol-Gel-Materialien mit hydrophoben Gruppen
erzielt.P
[126]P Bei der Synthese werden statt des reinen Tetramethoxysilans
Alkyltrimethoxysilane eingesetzt, deren organischer Rest während der
Polymerisation nicht hydrolisiert wird. Diese Hybridmaterialien zeigten erst-
mals deutlich verbesserte Aktivitäten in organischen Medien, die durch die
Variation des Alkylrestes weiter erhöht werden konnten.
Die auf diese Weise immobilisierten Lipasen wurden zum Beispiel in der
kinetischen Racematspaltung von 1-Phenylethanol mit Acetanhydrid als
Acylierungsreagenz untersucht und eine Verbesserung des ee-Werts von
92 % auf über 99 % für Edukt und Produkt beobachtet. Eine systematische
Untersuchung von Sol-Gel-immobilisierten Lipasen in der kinetischen
Racematspaltung unterschiedlicher Alkohole mit Vinylacetat in verschiedenen
Lösungsmitteln zeigte ebenfalls eine häufig gesteigerte Aktivität und gleich
bleibende bis verbesserte Enantioselektivität (vgl. Abbildung 74).P
[127]P

Kombination von Säure- und Enzym-Katalyse 113
OHLipase
O
O
+ OHVinylacetat
Abbildung 74: Lipasen-katalysierte Racematspaltung von (1)-Phenylethanol mit Vinyl-acetat zum (R)-Ester und zum (S)-Alkohol.
7.2 Eigene Untersuchungen
Da bei der kinetischen Racematspaltung einer racemischen Mischung
generell nie mehr als 50 % des Produkts gewonnen werden können, liegt es
nahe, das unerwünschte Enantiomer durch eine Racemisierung ebenfalls der
Reaktion zur Verfügung zu stellen und so einen Umsatz von über 50 % zu
erzielen. Diese Kombination von kinetischer Racematspaltung mit einer
in situ-Racemisierung wird als dynamisch-kinetische Racematspaltung be-
zeichnet. Für die Racemisierung des 1-Phenylethanols kommen zum Beispiel
saure Katalysatoren, wie das im Rahmen dieser Arbeit untersuchte NafionP
®P-
Polymer, in Frage. Ein ähnliches System wurde durch JACOBS et al.
untersucht, in dem saure Zeolithe in Kombination mit einer Lipase in einem
Zwei-Phasen-System die dynamisch-kinetische Racematspaltung von
(1)-Phenylethanol bewirken.P
[128]P
Es sollte daher untersucht werden, ob die im Rahmen dieser Arbeit ver-
wendeten NafionP
®P-Katalysatoren in Kombination mit der Lipase CaL B die
dynamisch-kinetische Racematspaltung von 1-Phenylethanol ermöglichen
können (vgl. Abbildung 75).

Kombination von Säure- und Enzym-Katalyse 114
OHLipase
O
O
+ OHVinylacetat
Nafion
Abbildung 75: Versuch der dynamisch-kinetischen Racematspaltung des (1)-Phenyl-ethanols durch Kombination von Lipase- und Säurekatalyse.
Die Untersuchungen wurden in verschiedenen Lösungsmitteln, wie Hexan,
Dichlormethan und Wasser, bei Raumtemperatur, 40 °C oder 60 °C durch-
geführt. Als saurer Katalysator kamen das kommerzielle NafionP
®P-System
NR 50 und die im Rahmen dieser Arbeit dargestellten NafionP
®P-haltigen
Silikate MCM/Naf10 und SAC/Naf13 zum Einsatz. Als Biokatalysator wurde
Candida antarctica Lipase B (CaL B) immobilisiert in einer hydrophoben
Silikat-Matrix verwendet.
7.2.1 Racemisierung von (S)-Phenylethanol durch NafionP
®P-Systeme
Zunächst sollte untersucht werden, unter welchen Bedingungen die einge-
setzten Nafion P
®P-Systeme die Racemisierung von (S)-Phenylethanol mit den
besten Ergebnissen katalysieren. Dazu wurden die Katalysatoren NR 50 und
SAC/Naf13 in Hexan, Dichlormethan oder Wasser bei Raumtemperatur und
60 °C eingesetzt. Nach zwei und 24 Stunden wurde die Reaktionsmischung
auf ihre Zusammensetzung und die ee-Werte des 1-Phenylethanols unter-
sucht.
In Tabelle 28 und Tabelle 29 sind die Ergebnisse für das reine NafionP
®P-
Polymer NR 50 bei Raumtemperatur bzw. 60 °C aufgelistet.
?
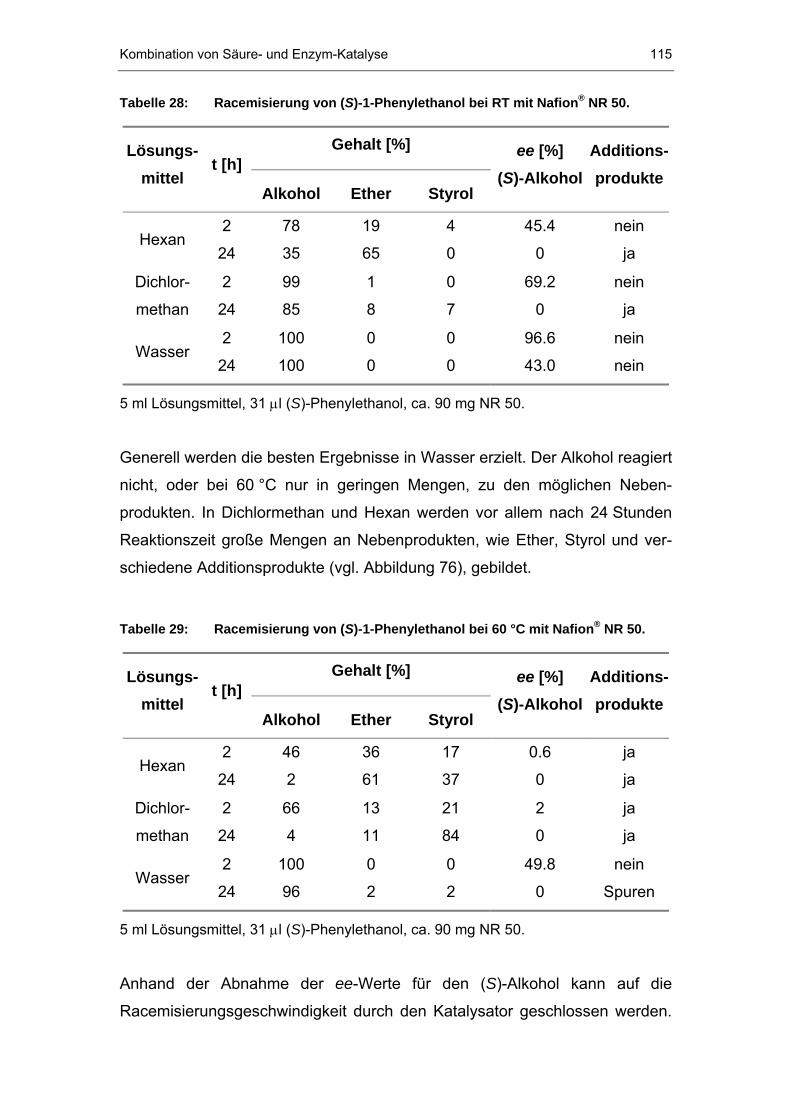
Kombination von Säure- und Enzym-Katalyse 115
Tabelle 28: Racemisierung von (S)-1-Phenylethanol bei RT mit NafionP
®P NR 50.
Gehalt [%] Lösungs-mittel
t [h]
Alkohol Ether Styrol
ee [%] (S)-Alkohol
Additions-produkte
Hexan 2
24
78
35
19
65
4
0
45.4
0
nein
ja
Dichlor-
methan
2
24
99
85
1
8
0
7
69.2
0
nein
ja
Wasser 2
24
100
100
0
0
0
0
96.6
43.0
nein
nein
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg NR 50.
Generell werden die besten Ergebnisse in Wasser erzielt. Der Alkohol reagiert
nicht, oder bei 60 °C nur in geringen Mengen, zu den möglichen Neben-
produkten. In Dichlormethan und Hexan werden vor allem nach 24 Stunden
Reaktionszeit große Mengen an Nebenprodukten, wie Ether, Styrol und ver-
schiedene Additionsprodukte (vgl. Abbildung 76), gebildet.
Tabelle 29: Racemisierung von (S)-1-Phenylethanol bei 60 °C mit NafionP
®P NR 50.
Gehalt [%] Lösungs-mittel
t [h]
Alkohol Ether Styrol
ee [%] (S)-Alkohol
Additions-produkte
Hexan 2
24
46
2
36
61
17
37
0.6
0
ja
ja
Dichlor-
methan
2
24
66
4
13
11
21
84
2
0
ja
ja
Wasser 2
24
100
96
0
2
0
2
49.8
0
nein
Spuren
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg NR 50.
Anhand der Abnahme der ee-Werte für den (S)-Alkohol kann auf die
Racemisierungsgeschwindigkeit durch den Katalysator geschlossen werden.

Kombination von Säure- und Enzym-Katalyse 116
In Wasser zeigt sich im Vergleich zu Hexan und Dichlormethan die mit
Abstand geringste Reaktionsgeschwindigkeit. Bei 60 °C verläuft die
Racemisierung in allen Lösungsmitteln schneller und ist in Hexan und
Dichlormethan bereits nach zwei Stunden abgeschlossen.
O
O
Abbildung 76: Mögliche Nebenprodukte bei der Racemisierung von (S)-Phenylethanol mit NafionP
®P-Katalysatoren (ermittelt mittels GC-MS).
In Tabelle 30 und Tabelle 31 sind die Ergebnisse der Racemisierung für das
Katalysatorsystem SAC/Naf13 bei Raumtemperatur und 40 °C dargestellt.
Das NafionP
®P-System SAC/Naf13 erwies sich als deutlich reaktiver als das
reine NafionP
®P-Polymer NR 50, trotz der geringeren effektiven Nafion P
®P-
Beladung im Silikatsystem SAC/Naf13. Dies wird auf die hohe Dispersion des
NafionP
®P-Polymers in der Silikatmatrix und die hohe spezifische Oberfläche
zurückgeführt. Auch hier zeigte Wasser die besten Eigenschaften als
Lösungsmittel, da keine Nebenprodukte gebildet wurden. In Dichlormethan
und Hexan entstehen als Nebenprodukte bei Raumtemperatur meist Ether,
bei 60 °C Styrol und die Additionsprodukte. Die Racemisierungsgeschwindig-
keiten sind auch hier besonders hoch in Dichlormethan und Hexan.
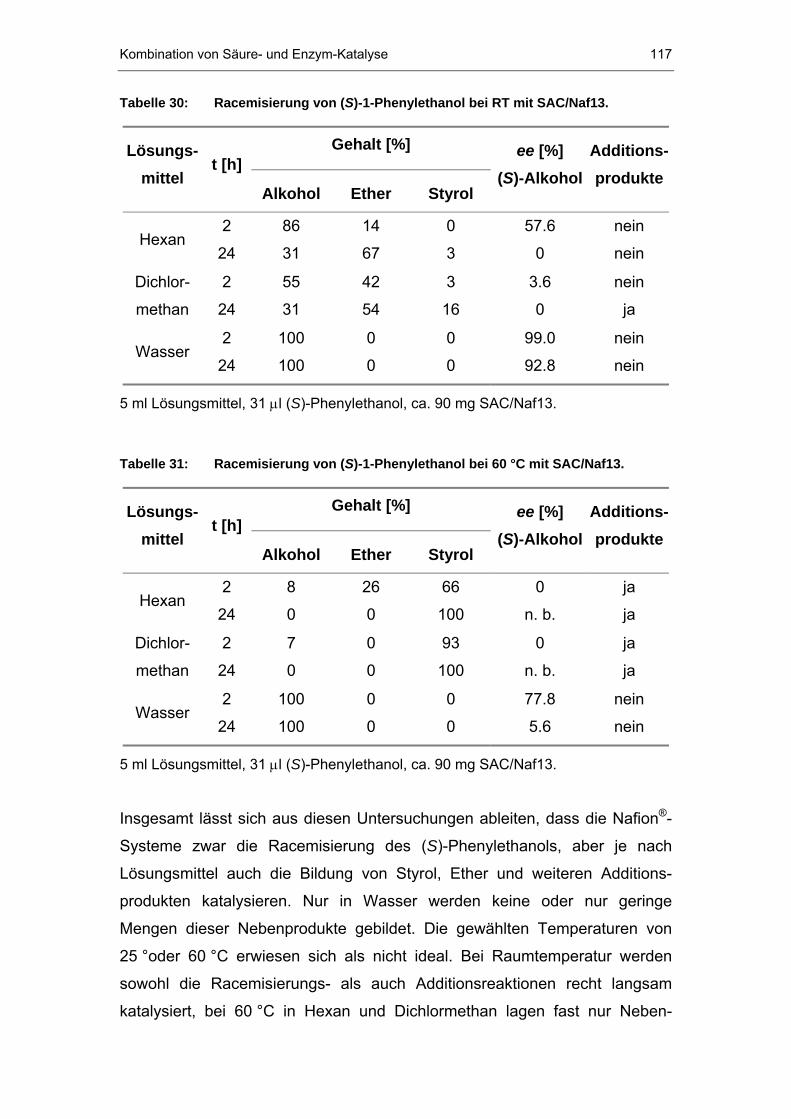
Kombination von Säure- und Enzym-Katalyse 117
Tabelle 30: Racemisierung von (S)-1-Phenylethanol bei RT mit SAC/Naf13.
Gehalt [%] Lösungs-mittel
t [h]
Alkohol Ether Styrol
ee [%] (S)-Alkohol
Additions-produkte
Hexan 2
24
86
31
14
67
0
3
57.6
0
nein
nein
Dichlor-
methan
2
24
55
31
42
54
3
16
3.6
0
nein
ja
Wasser 2
24
100
100
0
0
0
0
99.0
92.8
nein
nein
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg SAC/Naf13.
Tabelle 31: Racemisierung von (S)-1-Phenylethanol bei 60 °C mit SAC/Naf13.
Gehalt [%] Lösungs-mittel
t [h]
Alkohol Ether Styrol
ee [%] (S)-Alkohol
Additions-produkte
Hexan 2
24
8
0
26
0
66
100
0
n. b.
ja
ja
Dichlor-
methan
2
24
7
0
0
0
93
100
0
n. b.
ja
ja
Wasser 2
24
100
100
0
0
0
0
77.8
5.6
nein
nein
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg SAC/Naf13.
Insgesamt lässt sich aus diesen Untersuchungen ableiten, dass die Nafion P
®P-
Systeme zwar die Racemisierung des (S)-Phenylethanols, aber je nach
Lösungsmittel auch die Bildung von Styrol, Ether und weiteren Additions-
produkten katalysieren. Nur in Wasser werden keine oder nur geringe
Mengen dieser Nebenprodukte gebildet. Die gewählten Temperaturen von
25 °oder 60 °C erwiesen sich als nicht ideal. Bei Raumtemperatur werden
sowohl die Racemisierungs- als auch Additionsreaktionen recht langsam
katalysiert, bei 60 °C in Hexan und Dichlormethan lagen fast nur Neben-
Kombination von Säure- und Enzym-Katalyse 118
produkte vor. Um einen Kompromiss zwischen beiden Reaktionswegen zu
finden, sollten weitere Versuche bei 40 °C durchgeführt werden.
Für die nächsten Versuche wurde bei 40 °C mit den Katalysator-Systemen
NR 50 und MCM/Naf10 in Wasser, Dichlormethan oder Hexan gearbeitet. Die
Ergebnisse sind in Tabelle 32 und Tabelle 33 dargestellt. Für das System
MCM/Naf10 wurden in Dichlormethan und Hexan nur Proben bis zu vier
Stunden Reaktionszeit genommen.
Tabelle 32: Racemisierung von (S)-1-Phenylethanol bei 40 °C mit NR 50.
Lösungs-mittel
t [h] ee [%]
(S)-AlkoholEther Styrol
Additions-produkte
Hexan 2
24
1.4
0
ja
ja
nein
ja
ja
ja
Dichlor-
methan
2
24
11.2
0
ja
ja
ja
ja
ja
ja
Wasser 2
24
86.6
2
nein
nein
nein
nein
nein
nein
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg NR 50.
Tabelle 33: Racemisierung von (S)-1-Phenylethanol bei 40 °C mit MCM/Naf10.
Gehalt [%] Lösungs-mittel
t [h]
Alkohol Ether Styrol
ee [%] (S)-Alkohol
Additions-produkte
Hexan 2
4
91.0
n. b.
6.4
n. b.
0
n. b.
67.0
36.4
nein
n. b.
Dichlor-
methan
2
4
94.5
n. b.
2.6
n. b.
3.0
n. b.
41.6
23.4
ja
n. b.
Wasser 2
24
100
100
0
0
0
0
99.3
98.6
nein
nein
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg MCM/Naf10.
Kombination von Säure- und Enzym-Katalyse 119
Wie bereits aus den ersten Untersuchungen hervorgegangen, zeigen die
Katalysatoren in Wasser die geringste Aktivität. Vor allem das stark hydro-
phobe System MCM/Naf10 wies kaum Aktivität in Wasser auf. Für die
Lösungsmittel Dichlormethan und Hexan gibt es keinen klaren Trend. So
katalysiert NR 50 besser in Hexan als in Dichlormethan und MCM/Naf10
besser in Dichlormethan als in Hexan. Leider zeigte sich für das System
NR 50 durch Verringerung der Reaktionstemperatur von 60 ° auf 40 °C keine
Verringerung der isolierten Nebenprodukte bei Durchführung in Hexan und
Dichlormethan.
Da im Verlauf dieser Reaktion alle 15 Minuten Proben der Reaktions-
mischungen genommen wurden, konnte so der Reaktionsverlauf mittels der
ee-Werte beobachtet werden. In Abbildung 77 ist zum Beispiel der Verlauf für
das Katalysatorsystem MCM/Naf10 in den verschiedenen Lösungsmitteln
abgebildet.
0
10
20
30
40
50
60
70
80
90
100
0 0,5 1 1,5 2 2,5 3 3,5 4t [h]
ee [%
]
Abbildung 77: Zeitlicher Verlauf des ee-Wertes in der Racemisierung von (S)-Phenyl-ethanol mit dem Katalysator MCM/Naf10 in Dichlormethan (rot), Hexan (blau) und Wasser (grün) bei 40 °C.
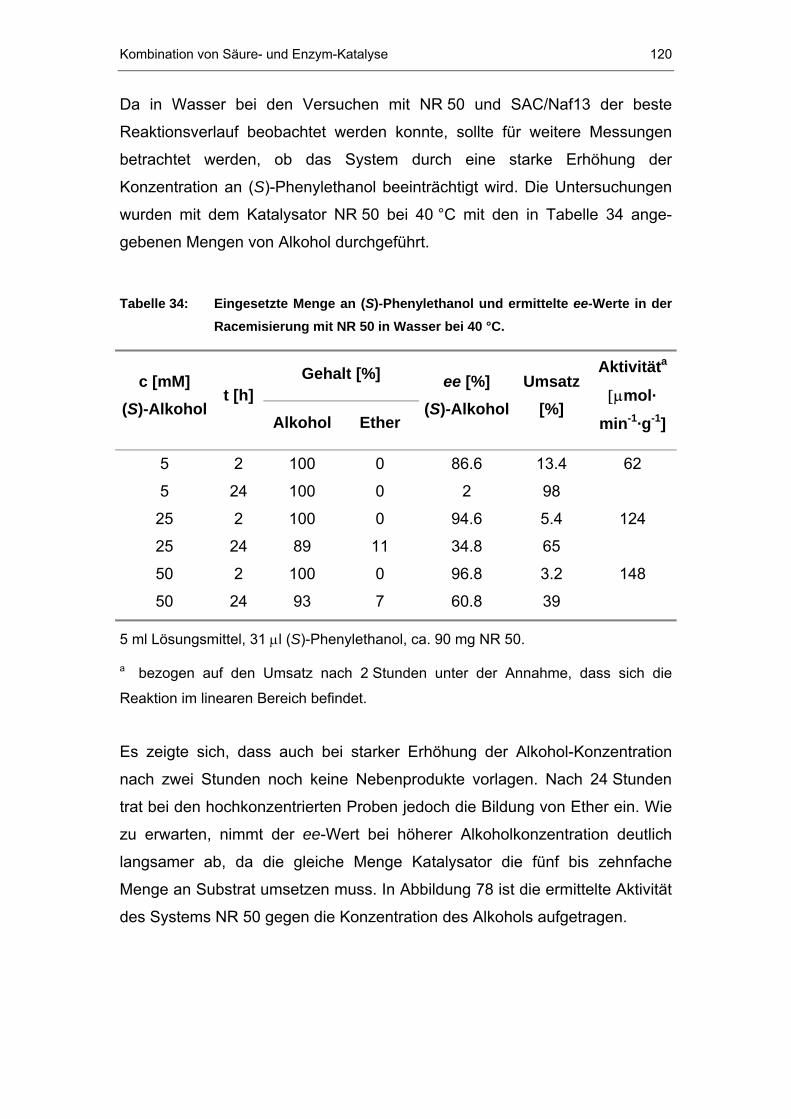
Kombination von Säure- und Enzym-Katalyse 120
Da in Wasser bei den Versuchen mit NR 50 und SAC/Naf13 der beste
Reaktionsverlauf beobachtet werden konnte, sollte für weitere Messungen
betrachtet werden, ob das System durch eine starke Erhöhung der
Konzentration an (S)-Phenylethanol beeinträchtigt wird. Die Untersuchungen
wurden mit dem Katalysator NR 50 bei 40 °C mit den in Tabelle 34 ange-
gebenen Mengen von Alkohol durchgeführt.
Tabelle 34: Eingesetzte Menge an (S)-Phenylethanol und ermittelte ee-Werte in der Racemisierung mit NR 50 in Wasser bei 40 °C.
Gehalt [%] c [mTMT] (S)-Alkohol
t [h] Alkohol Ether
ee [%] (S)-Alkohol
Umsatz [%]
AktivitätP
a
T[µTmol·
minP
-1P·gP
-1P]
5 2 100 0 86.6 13.4 62
5 24 100 0 2 98
25 2 100 0 94.6 5.4 124
25 24 89 11 34.8 65
50 2 100 0 96.8 3.2 148
50 24 93 7 60.8 39
5 ml Lösungsmittel, 31 TµTl (S)-Phenylethanol, ca. 90 mg NR 50.
P
aP bezogen auf den Umsatz nach 2 Stunden unter der Annahme, dass sich die
Reaktion im linearen Bereich befindet.
Es zeigte sich, dass auch bei starker Erhöhung der Alkohol-Konzentration
nach zwei Stunden noch keine Nebenprodukte vorlagen. Nach 24 Stunden
trat bei den hochkonzentrierten Proben jedoch die Bildung von Ether ein. Wie
zu erwarten, nimmt der ee-Wert bei höherer Alkoholkonzentration deutlich
langsamer ab, da die gleiche Menge Katalysator die fünf bis zehnfache
Menge an Substrat umsetzen muss. In Abbildung 78 ist die ermittelte Aktivität
des Systems NR 50 gegen die Konzentration des Alkohols aufgetragen.

Kombination von Säure- und Enzym-Katalyse 121
Die Aktivität entwickelt sich bei Erhöhung der Alkohol-Konzentration nicht
linear und es kommt bei längerer Reaktionszeit zur Bildung des Ethers als
Nebenprodukt. Die Konzentration an Alkohol wurde daher bei 5 mM beibe-
halten.
0
20
40
60
80
100
120
140
160
0 10 20 30 40 50
c [mM]
Akt
ivitä
t [µ
mol
/(min
*g)]
Abbildung 78: Aktivität des Katalysators NR 50 bei Variation der Alkoholkonzentration.
7.2.2 Reaktion der NafionP
®P-Systeme mit (R)-Phenylethylacetat
Des Weiteren sollte untersucht werden, wie sich das Produkt der kinetischen
Racematspaltung von (1)-Phenylethanol mit Vinylacetat in der Gegenwart von
NafionP
®P-Polymer verhält. Dazu wurde (R)-Phenylethylacetat mit den
Katalysatoren in den verschiedenen Lösungsmitteln bei 40 °C umgesetzt und
die Stabilität des (R)-Esters untersucht.
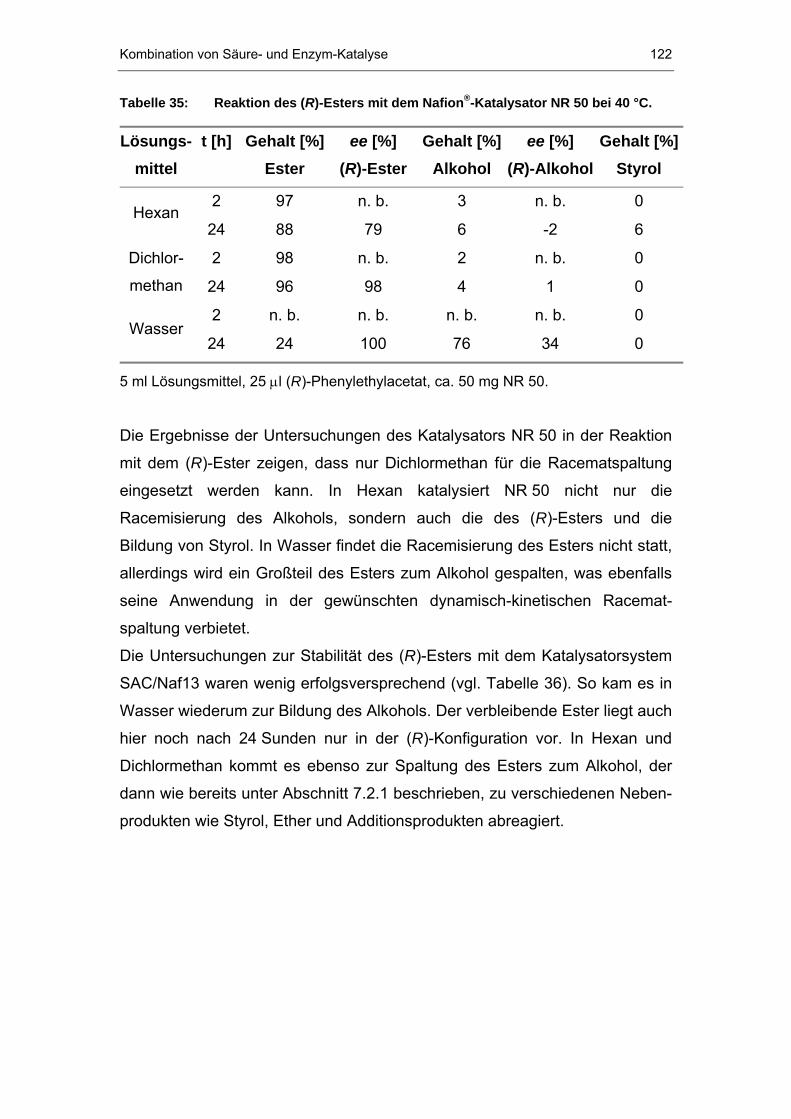
Kombination von Säure- und Enzym-Katalyse 122
Tabelle 35: Reaktion des (R)-Esters mit dem Nafion P
®P-Katalysator NR 50 bei 40 °C.
Lösungs-mittel
t [h] Gehalt [%] Ester
ee [%] (R)-Ester
Gehalt [%]Alkohol
ee [%] (R)-Alkohol
Gehalt [%]Styrol
2 97 n. b. 3 n. b. 0 Hexan
24 88 79 6 -2 6
2 98 n. b. 2 n. b. 0 Dichlor-
methan 24 96 98 4 1 0
2 n. b. n. b. n. b. n. b. 0 Wasser
24 24 100 76 34 0
5 ml Lösungsmittel, 25 TµTl (R)-Phenylethylacetat, ca. 50 mg NR 50.
Die Ergebnisse der Untersuchungen des Katalysators NR 50 in der Reaktion
mit dem (R)-Ester zeigen, dass nur Dichlormethan für die Racematspaltung
eingesetzt werden kann. In Hexan katalysiert NR 50 nicht nur die
Racemisierung des Alkohols, sondern auch die des (R)-Esters und die
Bildung von Styrol. In Wasser findet die Racemisierung des Esters nicht statt,
allerdings wird ein Großteil des Esters zum Alkohol gespalten, was ebenfalls
seine Anwendung in der gewünschten dynamisch-kinetischen Racemat-
spaltung verbietet.
Die Untersuchungen zur Stabilität des (R)-Esters mit dem Katalysatorsystem
SAC/Naf13 waren wenig erfolgsversprechend (vgl. Tabelle 36). So kam es in
Wasser wiederum zur Bildung des Alkohols. Der verbleibende Ester liegt auch
hier noch nach 24 Sunden nur in der (R)-Konfiguration vor. In Hexan und
Dichlormethan kommt es ebenso zur Spaltung des Esters zum Alkohol, der
dann wie bereits unter Abschnitt 7.2.1 beschrieben, zu verschiedenen Neben-
produkten wie Styrol, Ether und Additionsprodukten abreagiert.
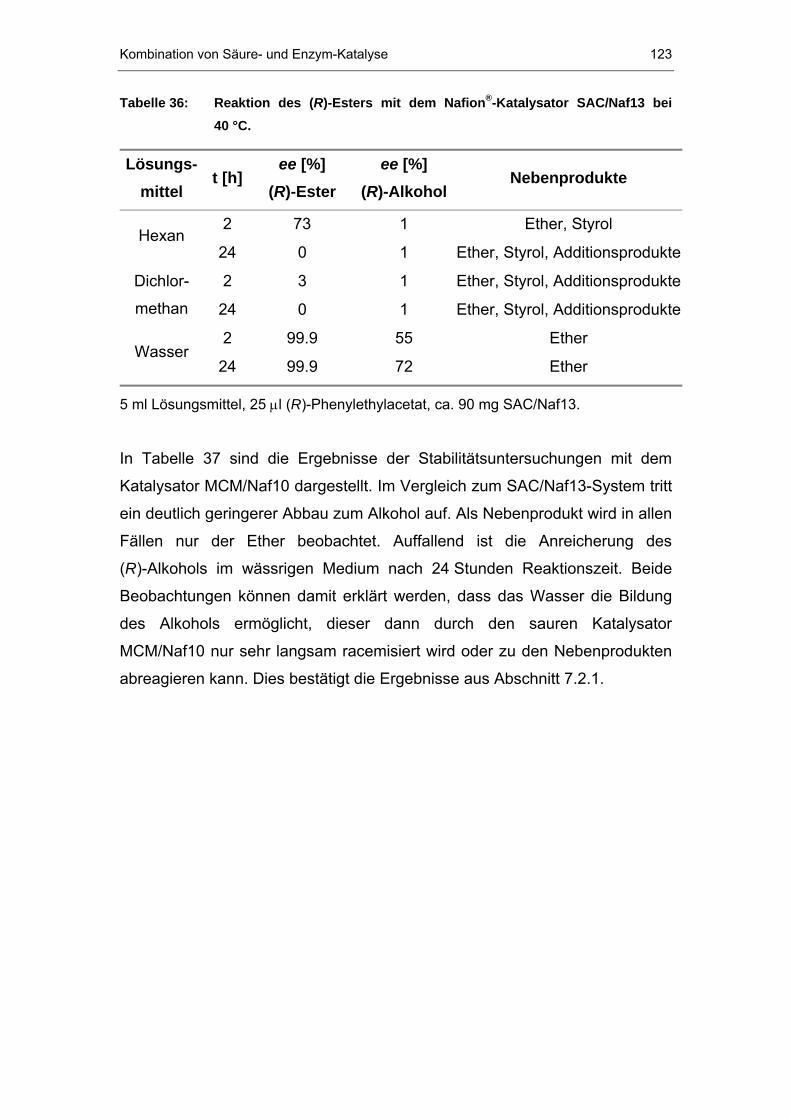
Kombination von Säure- und Enzym-Katalyse 123
Tabelle 36: Reaktion des (R)-Esters mit dem NafionP
®P-Katalysator SAC/Naf13 bei
40 °C.
Lösungs-mittel
t [h] ee [%]
(R)-Ester ee [%]
(R)-AlkoholNebenprodukte
2 73 1 Ether, Styrol Hexan
24 0 1 Ether, Styrol, Additionsprodukte
2 3 1 Ether, Styrol, AdditionsprodukteDichlor-
methan 24 0 1 Ether, Styrol, Additionsprodukte
2 99.9 55 Ether Wasser
24 99.9 72 Ether
5 ml Lösungsmittel, 25 TµTl (R)-Phenylethylacetat, ca. 90 mg SAC/Naf13.
In Tabelle 37 sind die Ergebnisse der Stabilitätsuntersuchungen mit dem
Katalysator MCM/Naf10 dargestellt. Im Vergleich zum SAC/Naf13-System tritt
ein deutlich geringerer Abbau zum Alkohol auf. Als Nebenprodukt wird in allen
Fällen nur der Ether beobachtet. Auffallend ist die Anreicherung des
(R)-Alkohols im wässrigen Medium nach 24 Stunden Reaktionszeit. Beide
Beobachtungen können damit erklärt werden, dass das Wasser die Bildung
des Alkohols ermöglicht, dieser dann durch den sauren Katalysator
MCM/Naf10 nur sehr langsam racemisiert wird oder zu den Nebenprodukten
abreagieren kann. Dies bestätigt die Ergebnisse aus Abschnitt 7.2.1.
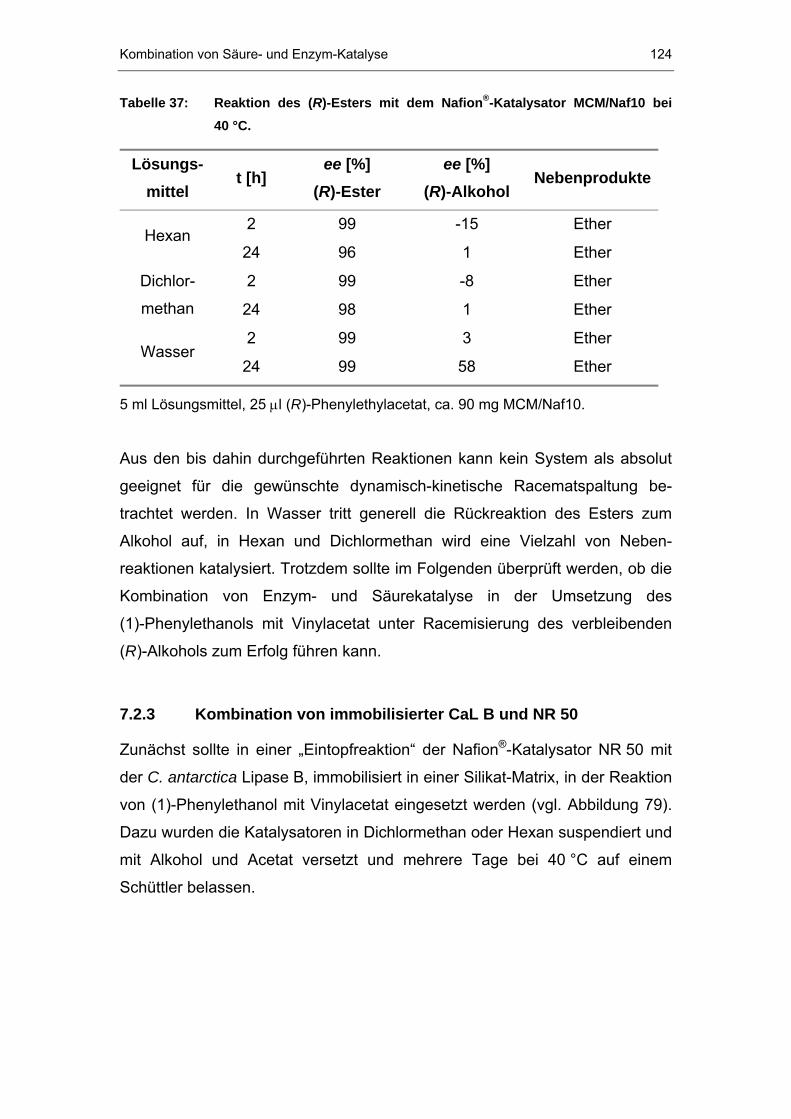
Kombination von Säure- und Enzym-Katalyse 124
Tabelle 37: Reaktion des (R)-Esters mit dem NafionP
®P-Katalysator MCM/Naf10 bei
40 °C.
Lösungs-mittel
t [h] ee [%]
(R)-Ester ee [%]
(R)-Alkohol Nebenprodukte
2 99 -15 Ether Hexan
24 96 1 Ether
2 99 -8 Ether Dichlor-
methan 24 98 1 Ether
2 99 3 Ether Wasser
24 99 58 Ether
5 ml Lösungsmittel, 25 TµTl (R)-Phenylethylacetat, ca. 90 mg MCM/Naf10.
Aus den bis dahin durchgeführten Reaktionen kann kein System als absolut
geeignet für die gewünschte dynamisch-kinetische Racematspaltung be-
trachtet werden. In Wasser tritt generell die Rückreaktion des Esters zum
Alkohol auf, in Hexan und Dichlormethan wird eine Vielzahl von Neben-
reaktionen katalysiert. Trotzdem sollte im Folgenden überprüft werden, ob die
Kombination von Enzym- und Säurekatalyse in der Umsetzung des
(1)-Phenylethanols mit Vinylacetat unter Racemisierung des verbleibenden
(R)-Alkohols zum Erfolg führen kann.
7.2.3 Kombination von immobilisierter CaL B und NR 50
Zunächst sollte in einer „Eintopfreaktion“ der NafionP
®P-Katalysator NR 50 mit
der C. antarctica Lipase B, immobilisiert in einer Silikat-Matrix, in der Reaktion
von (1)-Phenylethanol mit Vinylacetat eingesetzt werden (vgl. Abbildung 79).
Dazu wurden die Katalysatoren in Dichlormethan oder Hexan suspendiert und
mit Alkohol und Acetat versetzt und mehrere Tage bei 40 °C auf einem
Schüttler belassen.

Kombination von Säure- und Enzym-Katalyse 125
OH O
O
+O
O CaL B, NR 50
40 °C
1 äq 3 äq
Abbildung 79: Reaktion von (1)-Phenylethanol mit Vinylacetat bei 40 °C mit NR 50 und immobilisierter CaL B.
In Tabelle 38 und Tabelle 39 sind die Ergebnisse für die Reaktion in Hexan
bzw. Dichlormethan dargestellt. In beiden Lösungsmitteln wurden in großen
Mengen Nebenprodukte wie Styrol, Ether und Additionsprodukte beobachtet.
Anscheinend wird die Umsetzung zu den Nebenprodukten schneller
katalysiert, als entstehender (R)-Alkohol durch die Lipase zum Ester umge-
setzt werden kann. Die Enantiomerenüberschüsse des Esters sind in beiden
Systemen deutlich schlechter als bei der Umsetzung mit der Lipase allein, da
es zu einer Racemisierung des Esters durch den NafionP
®P-Katalysator kommt.
Tabelle 38: Ergebnisse der Reaktion von (1)-Phenylethanol mit Vinylacetat bei 40 °C in Hexan.
Gehalt [%] t [h]
Styrol Alkohol Ester Ether Addition
ee [%] (S)-Alkohol
ee [%] (R)-Ester
13 0.7 10.1 64.5 9.0 12.3 98.7 84.9
20 0.6 9.9 63.3 9.3 12.6 97.9 84.9
85 0.8 9.0 65.2 8.8 12.1 100.0 84.1
5 ml Hexan, 31 TµTl (1)-Phenylethanol, 69 TµTl Vinylacetat, 10.8 mg Lipase, 25.6 mg
NR 50.
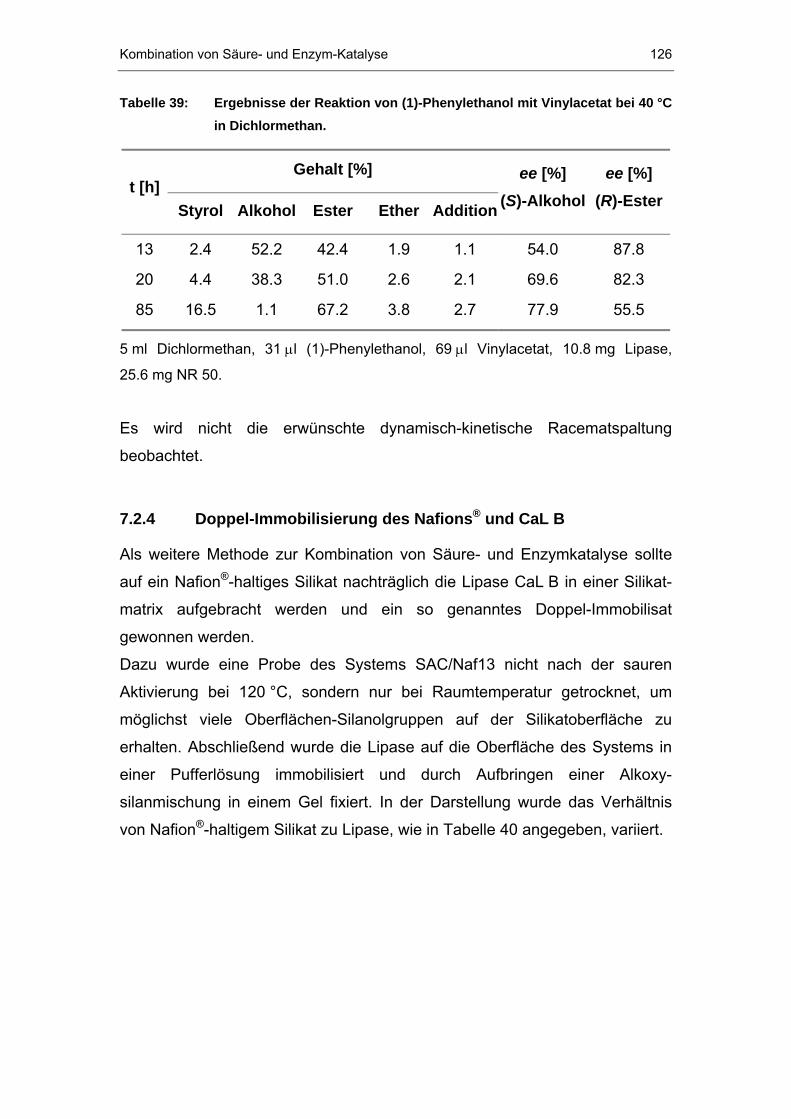
Kombination von Säure- und Enzym-Katalyse 126
Tabelle 39: Ergebnisse der Reaktion von (1)-Phenylethanol mit Vinylacetat bei 40 °C in Dichlormethan.
Gehalt [%] t [h]
Styrol Alkohol Ester Ether Addition
ee [%] (S)-Alkohol
ee [%] (R)-Ester
13 2.4 52.2 42.4 1.9 1.1 54.0 87.8
20 4.4 38.3 51.0 2.6 2.1 69.6 82.3
85 16.5 1.1 67.2 3.8 2.7 77.9 55.5
5 ml Dichlormethan, 31 TµTl (1)-Phenylethanol, 69 TµTl Vinylacetat, 10.8 mg Lipase,
25.6 mg NR 50.
Es wird nicht die erwünschte dynamisch-kinetische Racematspaltung
beobachtet.
7.2.4 Doppel-Immobilisierung des NafionsP
®P und CaL B
Als weitere Methode zur Kombination von Säure- und Enzymkatalyse sollte
auf ein Nafion P
®P-haltiges Silikat nachträglich die Lipase CaL B in einer Silikat-
matrix aufgebracht werden und ein so genanntes Doppel-Immobilisat
gewonnen werden.
Dazu wurde eine Probe des Systems SAC/Naf13 nicht nach der sauren
Aktivierung bei 120 °C, sondern nur bei Raumtemperatur getrocknet, um
möglichst viele Oberflächen-Silanolgruppen auf der Silikatoberfläche zu
erhalten. Abschließend wurde die Lipase auf die Oberfläche des Systems in
einer Pufferlösung immobilisiert und durch Aufbringen einer Alkoxy-
silanmischung in einem Gel fixiert. In der Darstellung wurde das Verhältnis
von NafionP
®P-haltigem Silikat zu Lipase, wie in Tabelle 40 angegeben, variiert.

Kombination von Säure- und Enzym-Katalyse 127
Tabelle 40: Eingesetzte SAC/Naf13- und CaL B-Mengen in der Darstellung der Doppel-Immobilisate.
Probe m [mg]
SAC/Naf13 m [mg] CaL B
SAC/Naf13B10B/CaL B 10 25
SAC/Naf13B20B/CaL B 20 25
SAC/Naf13B50B/CaL B 50 25
SAC/Naf13B100B/CaL B 100 25
SAC/Naf13B200B/CaL B 200 25
Diese Doppel-Immobilisate wurden anschließend in der Reaktion von
(1)-Phenylethanol mit Vinylacetat bei 40 °C in verschiedenen Lösungsmitteln
eingesetzt (vgl. Abbildung 80). Verwendet wurden Hexan, MTBE, Toluol und
Dichlormethan.
OH O
O
+O
O SAC/Naf13/CaL B
40 °C
1 äq 2 äq
Abbildung 80: Reaktion von (1)-Phenylethanol mit Vinylacetat bei 40 °C mit den Doppel-Immobilisaten SAC/Naf13/CaL B.
Bei allen Umsetzungen mit den Doppel-Immobilisaten wurden für (R)-Phenyl-
ethylacetat ee-Werte größer 99 % ermittelt. Die ee-Werte des Alkohols
variierten sehr stark, jedoch ohne einen klaren Trend. Bei einem Umsatz von
50 % kamen die Reaktionen zum Stillstand, so dass anzunehmen ist, dass
durch diese Katalysatorsysteme keine dynamisch-kinetische Racemat-
spaltung erreicht werden kann. Aufgrund dieser Beobachtungen wird
angenommen, dass das NafionP
®P-Polymer im Immobilisat deaktiviert vorliegt.
Dies wird durch die geringe Menge an gebildeten Nebenprodukten unter-
stützt. So liegen Styrol und Ether nur in Spuren vor.
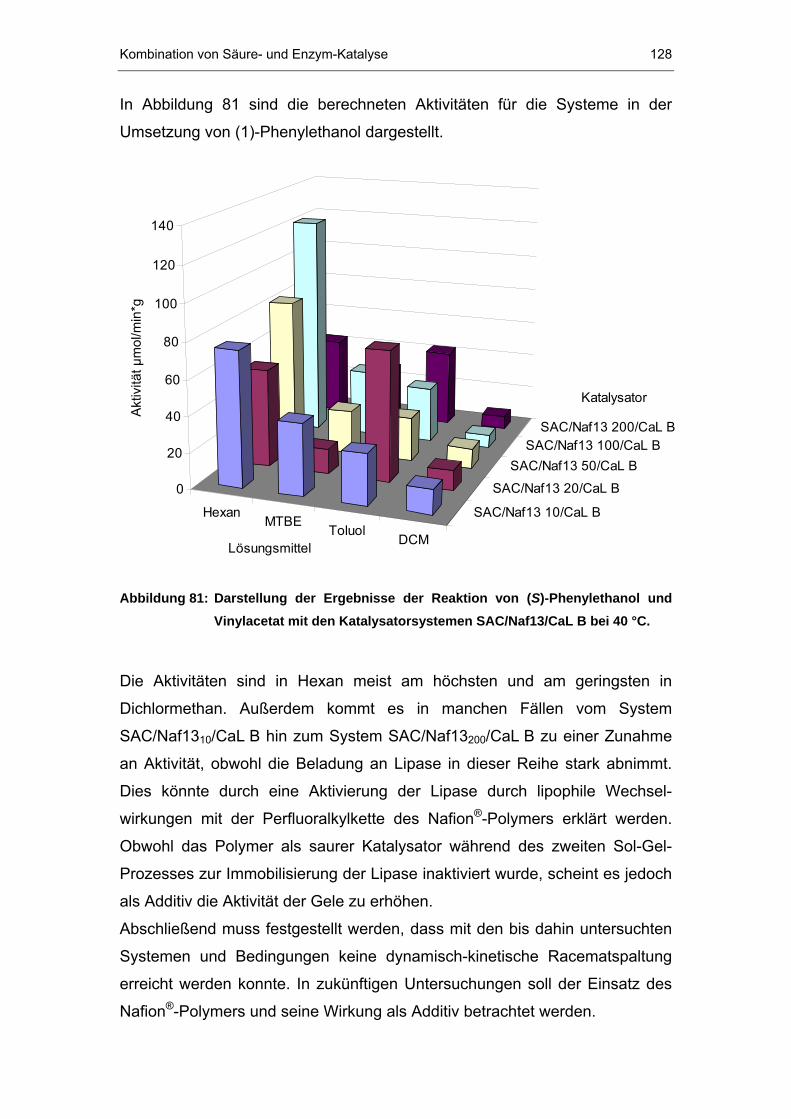
Kombination von Säure- und Enzym-Katalyse 128
In Abbildung 81 sind die berechneten Aktivitäten für die Systeme in der
Umsetzung von (1)-Phenylethanol dargestellt.
HexanMTBE
ToluolDCM
SAC/Naf13 10/CaL B
SAC/Naf13 20/CaL B
SAC/Naf13 50/CaL BSAC/Naf13 100/CaL B
SAC/Naf13 200/CaL B
0
20
40
60
80
100
120
140
Akt
ivitä
t µm
ol/m
in*g
Lösungsmittel
Katalysator
Abbildung 81: Darstellung der Ergebnisse der Reaktion von (S)-Phenylethanol und Vinylacetat mit den Katalysatorsystemen SAC/Naf13/CaL B bei 40 °C.
Die Aktivitäten sind in Hexan meist am höchsten und am geringsten in
Dichlormethan. Außerdem kommt es in manchen Fällen vom System
SAC/Naf13B10B/CaL B hin zum System SAC/Naf13B200B/CaL B zu einer Zunahme
an Aktivität, obwohl die Beladung an Lipase in dieser Reihe stark abnimmt.
Dies könnte durch eine Aktivierung der Lipase durch lipophile Wechsel-
wirkungen mit der Perfluoralkylkette des Nafion P
®P-Polymers erklärt werden.
Obwohl das Polymer als saurer Katalysator während des zweiten Sol-Gel-
Prozesses zur Immobilisierung der Lipase inaktiviert wurde, scheint es jedoch
als Additiv die Aktivität der Gele zu erhöhen.
Abschließend muss festgestellt werden, dass mit den bis dahin untersuchten
Systemen und Bedingungen keine dynamisch-kinetische Racematspaltung
erreicht werden konnte. In zukünftigen Untersuchungen soll der Einsatz des
NafionP
®P-Polymers und seine Wirkung als Additiv betrachtet werden.

Zusammenfassung und Ausblick 129
8 Zusammenfassung und Ausblick
Im Rahmen dieser Arbeit konnten erfolgreich Nafion P
®P-haltige Silikate mit
ungeordneten, mikroporösen und geordneten, mesoporösen Strukturen dar-
gestellt werden. Dabei konnten unterschiedliche Polymer-Beladungen in den
Systemen erzielt werden. Die Systeme wurden mittels EA, IR, XPS, XRD,
BET, SEM und TEM-Untersuchungen charakterisiert.
Im Weiteren wurden diese Systeme über die Reaktion der Sulfonsäurereste
des immobilisierten Nafion P
®P-Polymers mit Platin- und Zirconium-Methyl-
Spezies beladen (vgl. Abbildung 82). Auf diese Weise wurde eine hohe
Verteilung der Metall-Spezies auf dem Trägermaterial erreicht.
CF2 SO3H
Nafion/SiO2
+
H3C
MLn
H3CCF2 SO3H
CF2 SO3
Nafion/SiO2 CF2 SO3
2 CH4
MLn
2+
CH4
CF2 SO3
Nafion/SiO2 CF2 SO3H
M(CH3)Ln
Abbildung 82: Metall-Beladung der NafionP
®P-haltigen Silikate unter Reaktion einer Alkyl-
metall-Spezies mit den sauren Sulfonsäuregruppen.

Zusammenfassung und Ausblick 130
Des Weiteren wurden Kupfer-Systeme über den Ionenaustausch mit den
Natriumsalzen der NafionP
®P-Systeme gewonnen (vgl. Abbildung 83).
CF2 SO3
+
Nafion/SiO2 CF2 SO3
CF2 SO3
Nafion/SiO2 CF2 SO3
Na
Na
MLn X2
2 NaX
MLn2+
Abbildung 83: Metall-Beladung der NafionP
®P-haltigen Silikate durch Ionenaustausch.
Diese Metall-beladenen Systeme wurden mittels EA, IR, XPS, XRD, SEM und
TEM-Untersuchungen analysiert. Des Weiteren wurden sie in ersten Test-
reaktionen eingesetzt, um das katalytische Potential dieser Systeme zu
erschließen.
Die reduzierten Platin-Systeme wiesen eine hohe Verteilung der Platin-
Partikel und recht hohe Aktivitäten in der Hydrierung von Cycloocten als
Testreaktion auf. Dabei wurde die Aktivität nicht durch die Beladung des
Systems beeinflusst.
Die Zirconium-Systeme wurden in der Polymerisation von Ethen untersucht.
Unter Aktivierung von MAO zeigten diese Systeme eine recht gute
Polymerisationsaktivität. Jedoch wurde in weiteren Untersuchungen nach-
gewiesen, dass die Katalyse keinen rein heterogenen Charakter aufweist.
Die Kupfer-Systeme wurden in der Cyclopropanierung eingesetzt und zeigten
eine moderate bis schlechte Enantioselektivität. Außerdem wurde ein Aus-
bluten der Kupfer-Spezies beobachtet.

Zusammenfassung und Ausblick 131
Nachdem generell das Prinzip der Metall-Beladung eines Nafion P
®P-haltigen
Silikats über die Sulfonsäurereste gezeigt werden konnte, können in der
Zukunft weitere Metall-beladene Spezies durch Umsatz mit einer Vielzahl von
Metall-Vorläufern dargestellt werden.
In den Untersuchungen zur Kombination von Säure- und Enzymkatalyse
wurde zunächst die Racemisierung eines Alkohols durch die NafionP
®P-Systeme
betrachtet. Weitere Untersuchungen zeigten, dass auch eine Racemisierung
des durch die Enzymkatalyse gebildeten Esters erfolgt. Daher wurde die
Kombination von kinetischer Racematspaltung durch die Lipase mit einer
Racemisierung des verbleibenden Edukts durch die Nafion P
®P-Systeme leider
nicht erreicht. Sowohl Versuche mit einer Kombination beider Katalysatoren,
als auch mit so genannten Doppel-Immobilisaten verliefen negativ (vgl.
Abbildung 84). Allerdings zeigte sich in einigen Versuchen eine Aktivierung
der Lipase durch das NafionP
®P-Polymer, die wahrscheinlich auf Wechsel-
wirkungen mit der Perfluoralkylkette des Polymers zurückzuführen ist. Weitere
Untersuchungen müssen zeigen, ob das Nafion P
®P-Polymer als Additiv generell
zu einer Aktivierung der Lipase führt.
OH O
O
+O
O Nafion/CaL B
Abbildung 84: Versuch der dynamisch-kinetischen Racematspaltung durch Kombina-tion von Säure- und Enzymkatalyse.

Experimenteller Teil 132
9 Experimenteller Teil
9.1 Allgemeine Hinweise
Der überwiegende Teil der im Rahmen dieser Arbeit dargestellten
Substanzen ist luft- und feuchtigkeitsstabil. In den Fällen, in denen die
Anwendung von Schutzgastechniken erforderlich war, wird dies in den
Versuchsbeschreibungen explizit angeführt. Die verwendeten Chemikalien
wurden, soweit nicht anders angegeben, in ihrer kommerziell erhältlichen
Reinheit eingesetzt. Die Aufbewahrung der luft- und feuchtigkeitsempfind-
lichen Substanzen erfolgte in getrockneten und mit Argon befüllten Glas-
gefäßen. Die Entnahme erfolgte unmittelbar vor ihrem Einsatz unter Berück-
sichtigung der notwendigen Vorsichtsmaßnahmen.
Schutzgastechnik
Soweit die Anwendung von Schutzgastechniken erforderlich war, wurden die
verwendeten Glasgefäße vor ihrer Benutzung mehrfach unter Vakuum
(10P
-3P mbar) ausgeheizt und nach dem Abkühlen mit Argon belüftet. Die
Zugabe der Edukte erfolgte bei Feststoffen im Argongegenstrom und bei
Flüssigkeiten mittels Hebertechnik oder durch zuvor mehrfach mit Argon
gespülte Spritzen. Geringe Flüssigkeitsmengen wurden mit Einwegspritzen
über ein Septum zugegeben.
Temperaturen
Die angegebenen Temperaturen beziehen sich auf das jeweils verwendete
Heizmedium (Ölbad oder Aluminiumblock).

Experimenteller Teil 133
9.2 Chemikalien
Lösungsmittel
Die verwendeten organischen Lösungsmittel wurden nach literaturbekannten
Verfahren getrocknet und anschließend in Vorratsflaschen mit Steilbrust-
aufsatz unter Argon gelagert.
Aceton Refluxieren und Destillieren über Phosphorpentoxid
Dichlormethan Refluxieren und Destillieren über Calciumhydrid
Ethanol Refluxieren und Destillieren über Magnesium
Methanol Refluxieren und Destillieren über Magnesium
Tetrahydrofuran Refluxieren und Destillieren über LiAlHB4B
Toluol Refluxieren und Destillieren über LiAlHB4B
Wasser vollentsalzt mit Ionenaustauscher
Reagenzien
5 Gew.%ige Nafion P
®P 117-Lösung (Fluka), Nafion P
®P NR 50 Beads (Aldrich),
Nafion SAC-13 (Aldrich), Tetramethoxysilan (Fluka), Tetraethoxysilan
(Aldrich), Cetyltrimethylammoniumbromid (Acros), Tetramethylammonium-
hydroxid (Acros), Kupfertrifluormethansulfonat (Fluka), Dicyclopentadienyl-
dimethylzirconium (Strem), 2,2’-Bis-[(4S)-4-phenyl-2-oxazolin-2-yl]-propan
(Aldrich), 2,2-Bis-[(4S)-tert.-butyl-2-oxazolin-2-yl]-propan (Aldrich), Methyl-
alumoxan 1.7 M (Witco)
Im Rahmen der vorliegenden Arbeit wurden die bereits in der Literatur
beschriebenen Verbindungen Pt(COD)Cl B2BP
[129]P und Pt(COD)MeB2BP
[130]P syntheti-
siert.

Experimenteller Teil 134
Gase
Argon: 0.5 bis 0.8 ppm Sauerstoff, Messer-Griesheim
Wasserstoff: 99.9 %, Messer-Griesheim
Ethen: 3.0, Messer-Griesheim
9.3 Analytik
9.3.1 Elementaranalyse
Sämtliche Elementaranalysen wurden vom Mikroanalytischen Laboratorium
H. Kolbe in Mülheim an der Ruhr durchgeführt. Der relative Fehler der Edel-
metallbestimmung beträgt dabei ± 5 Gew.%.
9.3.2 Gaschromatographie (GC)
Die Bestimmungen der Umsätze der katalytischen Testreaktionen erfolgten
mit einem Gaschromatographen vom Typ HP 5890 der Firma Hewlett-
Packard, der mit einer 30 m HP-5-Säule ausgestattet war. Als Trägergas
wurde Wasserstoff verwendet.
9.3.3 GC/MS-Kopplung
Zur Identifizierung der Reaktionsprodukte der katalytischen Testreaktionen
wurde ein gekoppeltes GC/MS-System vom Typ SATURN 2100T der Firma
Varian verwendet.
9.3.4 Infrarotspektroskopie (IR)
IR-Messungen wurden an einem Magna-IR 750-Spektrometer der Firma
Nicolet durchgeführt. Feststoffe wurden als KBr-Pressling präpariert.

Experimenteller Teil 135
9.3.5 Kernresonanzspektroskopie (NMR)
Die Aufnahme von P
1PH- und P
13PC-NMR-Spektren wurden mit einem
AvanceP
TMP 300-Spektrometer der Firma Bruker bei 300.1 bzw. 75.5 MHz
aufgenommen. Chemische Verschiebungen sind in ppm relativ zu Tetra-
methylsilan angegeben. Als interner Standard wurden die Restwasserstoff-
und Kohlenstoffsignale des verwendeten Lösungsmittels herangezogen.
9.3.6 Transmissionselektronenmikroskopie (TEM)
Die elektronenmikroskopischen Untersuchungen wurden in der Mikroskopie-
Abteilung des Max-Planck-Instituts für Kohlenforschung in Mülheim an der
Ruhr durchgeführt. Dazu wurde ein Transmissionselektronenmikroskop der
Baureihe ELMISKOP 102 der Firma Siemens bei 80 kV sowie ein
HF-7500 TEM der Firma Hitachi bei 100 kV eingesetzt. EDX-Analysen
erfolgten mit einem Noran EDX-Analysator mit einem Lithium-dotierten
Siliciumkristall.
Trockene Präparation
Als Probenträger kamen Kupfer-Netzchen (Maschenweite 400 Mesh/inch)
zum Einsatz, die mit einer ca. 30 Å dicken Kohlenstoffschicht bedampft
wurden. Für die Probenpräparation gibt man 2 bis 3 mg der zu vermessenden
Substanz direkt auf die Kohlenstoffschicht des Trägers. Ein Überschuss an
Substanz wurde durch vorsichtiges Abschlagen des an einer Pinzette fixierten
Objektträgers entfernt.
Mikrotom-Schnitte
Mesoporöse Substanzen wurden zunächst in Epoxydharz eingebettet und für
24 Stunden bei 60 °C gehärtet. Anschließend wurden die eingebetteten
Proben mit Hilfe eines Diamantmessers der Firma Drukker geschnitten. Die
Mikrotom-Schnitte wurden auf Kupfer-Netzchen der Maschenweite 400 Mesh/
inch aufgebracht und mit einer ca. 30 Å dicken Kohlenstoffschicht bedampft.

Experimenteller Teil 136
9.3.7 Rasterelektronenmikroskopie (REM)
Die rasterelektronischenmikroskopischen Untersuchungen erfolgten in der
elektronenmikroskopischen Abteilung des Max-Planck-Instituts für Kohlen-
forschung in Mülheim an der Ruhr. Dazu wurde ein S-3500N-Raster-
elektronenmikroskop der Firma Hitachi eingesetzt.
9.3.8 BET-Messungen
Die Bestimmung der Porendurchmesser, -oberflächen und -volumina erfolgte
durch BET-Messungen mit einem NOVA 3000e Oberflächen- und Poren-
größenmessgerät der Firma Quantachrome Instruments. Die Auswertung der
Isothermen wurde mit dem Programm Autosorb for Windows P
®P Version 1.24
durchgeführt.
9.3.9 Röntgen-Photoelektronen-Spektroskopie (XPS)
Die XP-Spektren wurden mit einem Spektrometer des Typs AXIS HSI der
Firma Kratos Analytical unter Verwendung monochromatischer Al-KTBα TB-
Strahlung (1486.6 eV, 15 kV, 15 mA) aufgenommen.
9.3.10 Röntgen-Pulver-Diffraktometrie (XRD)
Die Röntgenpulverdiffraktogramme wurden mit einem Transmissions-Pulver-
diffraktometer der Firma Stoe STAPI P aufgenommen. Das Gerät verfügt über
einen Cu-KTBα T1B-Primärmonochromator und einen ortsempfindlichen linearen
Detektor.
9.3.11 Gelpermeationschromatographie (GPC)
Die GPC-Messungen wurden an einem Millipore 150-C ALC-GPC-Gerät der
Firma Waters im Arbeitskreis von Prof. Dr. Fink am Max-Planck-Institut für
Kohlenforschung durchgeführt.

Experimenteller Teil 137
9.3.12 Bestimmung der Säurestärke der NafionP
®P-haltigen Silikate
Ca. 100 mg des NafionP
®P-haltigen Silikats werden in 5 ml 2 M Natriumchlorid-
Lösung und 0.5 ml Methanol suspendiert und bei RT für 24 Stunden gerührt.
Anschließend wird die Suspension filtriert, der Rückstand mit ca. 70 ml dest.
Wasser gewaschen und die Lösung mit 0.0025 N Natriumhydroxid-Lösung mit
Hilfe einer pH-Elektrode titriert.
9.4 Darstellung der NafionP
®P-haltigen Silikate
9.4.1 AAV 1: Darstellung ungeordneter NafionP
®P-haltiger Silikate
Die Darstellung erfolgt ähnlich der Vorschrift von HARMERP
[35]P.
Eine Mischung aus 14 ml (14.4 g, 94.7 mmol) Tetramethoxysilan, 2.3 ml dest.
Wasser und 212 µl einer 0.04 N HCl-Lösung wird für 45 Minuten gerührt. Zu
einer kommerziellen NafionP
®P-Lösung (5 Gew.% in einem Wasser-Alkohol-
Gemisch) gibt man tropfenweise unter starkem Rühren eine 0.4 N NaOH-
Lösung (eingesetzte Mengen vgl. Tabelle 41). Anschließend gibt man schnell
die Silicium-haltige Lösung unter starkem Rühren zu. Nach wenigen
Sekunden geliert die Mischung unter Bildung eines trüben weißen Feststoffs.
Das Gel wird für etwa drei Tage bei 95 °C getrocknet. Der gewonnene weiße,
glasartige Feststoff wird gemörsert, in 3.5 M HCl-Lösung gerührt, filtriert und
mit dest. Wasser gewaschen. Dieser Schritt wird viermal wiederholt. Das
Silikat wird abschließend über Nacht bei ca. 75 °C mit 25 Gew.%iger HNO B3B-
Lösung behandelt. Nach Filtrieren, Waschen mit dest. Wasser bis zur
Neutralität und Trocknen für 24 Stunden bei 120 °C gewinnt man einen
weißen Feststoff.

Experimenteller Teil 138
Tabelle 41: NafionP
®P- und Natriumhydroxid-Mengen für die Darstellung ver-
schiedener NafionP
®P-haltiger Silikate
Gew.% NafionP
®P
NafionP
®P-Lösung
(5 Gew.%) NaOH (0.4 N)
5.3 7.3 ml (6.3 g) 7.0 ml
10 14.6 ml (12.7 g) 9.2 ml
13 19.5 ml (16.9 g) 10.6 ml
15 23.1 ml (20.1 g) 11.7 ml
20 32.8 ml (28.5 g) 14.5 ml
25 43.7 ml (38.0 g) 17.7 ml
Ein Silikat mit 40 Gew.% NafionP
®P wurde analog dieser Vorschrift hergestellt.
Dazu wurden 5.9 ml (40 mmol) TMOS, 970 TµTl dest. Wasser, 90 TµTl 0.04 N
Salzsäure, 36.8 ml der kommerziellen Nafion P
®P-Lösung und 10.8 ml 0.4 N
Natriumhydroxid-Lösung eingesetzt.
9.4.2 AAV 2: Darstellung geordneter, mesoporöser NafionP
®P-haltiger
Silikate
Die Darstellung erfolgt ähnlich der Vorschrift von FUJIWARAP
[43]P.
Zu einer Suspension von 0.97 g (5.3 mmol) Tetramethylammoniumhydroxid
Pentahydrat und 1.09 g (3.0 mmol) Cetyltrimethylammoniumbromid in dest.
Wasser (Menge vgl. Tabelle 42) tropft man unter starkem Rühren eine
kommerzielle Nafion P
®P-Lösung (5 Gew.% in einem Wasser-Alkohol-Gemisch).
Dabei lösen sich alle Bestandteile der Suspension. Anschließend gibt man zu
dieser Mischung unter starkem Rühren tropfenweise 4.4 ml (20 mmol) Tetra-
ethoxysilan. Die Mischung wird in einen Teflon-Behälter überführt und in
einem Autoklav für 24 Stunden auf 130 °C erhitzt. Der resultierende weiße
Feststoff wird zweimal mit 200 ml dest. Wasser gewaschen und bei 60 °C im
Trockenschrank getrocknet. Zur Entfernung des Templats und zur
Regeneration des Nafions P
®P wird das Rohprodukt in einer Mischung aus
200 ml Ethanol und 2 ml konz. Schwefelsäure für 18 Stunden unter Rückfluss

Experimenteller Teil 139
erhitzt. Anschließend wird zweimal für je 18 Stunden in 200 ml Ethanol unter
Rückfluss erhitzt. Nach Trocknen bei 120 °C für 24 Stunden gewinnt man
einen weißen Feststoff.
Tabelle 42: NafionP
®P- und Wassermengen für die Darstellung verschiedener
geordneter, mesoporöser NafionP
®P-haltiger Silikate
Gew.% NafionP
®P
NafionP
®P-Lösung
(5 Gew.%) dest. Wasser
5.3 1.5 ml (1.3 g) 9.5 ml
10 3.1 ml (2.7 g) 8.0 ml
13 4.1 ml (3.6 g) 6.9 ml
15 4.9 ml (4.2 g) 6.2 ml
20 6.9 ml (6.0 g) 4.1 ml
25 9.2 ml (8.0 g) 1.6 ml
9.5 Umsetzung der NafionP
®P-haltigen Silikate mit Metallver-
bindungen
9.5.1 AAV 3: Platinbeladung der NafionP
®P-haltigen Silikate
Das NafionP
®P-haltige Silikat wird bei Raumtemperatur zu einer Lösung von
( TηTP
4P-Cyclooctadien)dimethyl-Platin gegeben (insgesamt 2 ml Lösungsmittel je
100 mg Verbundstoff). Nach 24 Stunden Rühren lässt man die Mischung
absetzen, filtriert und wäscht das Produkt zweimal mit wenig Lösungsmittel.
Das weiße, pulverförmige Produkt wird im Vakuum bei Raumtemperatur
getrocknet.
Folgende Lösungsmittel kamen zum Einsatz: Aceton, Dichlormethan, MTBE,
Pentan oder Toluol. Die Menge des Platin-Precursors wurde von
ca. 5 bis 35 mg (16 bis 105 TµTmol) ( TηTP
4P-1,5-Cyclooctadien)dimethyl-Platin je
100 mg Nafion P
®P variiert. In weiteren Versuchen wurde als Platin-Precursor
Dichloro( Tη TP
4P-cyclooctadien)platin(II) eingesetzt.

Experimenteller Teil 140
Tabelle 43: Mengen der Edukte für die Darstellung der Platinbeladenen NafionP
®P-
haltigen Silikate.
Produkt Träger m [mg]
Pt(COD)MeB2B
m [mg]
SAC/Naf10/Pt1.3 102 2.2
SAC/Naf13/Pt1.7 118 3.4
SAC/Naf40/Pt4.3 305 23.1
MCM/Naf10/Pt0.3 151 0.78
MCM/Naf10/Pt0.7 151 1.7
MCM/Naf10/Pt1.0 150 2.5
MCM/Naf10/Pt1.3 151 3.4
MCM/Naf10/Pt2.0 150 5.1
MCM/Naf13/Pt0.8 150 2.0
MCM/Naf13/Pt1.6 150 4.1
MCM/Naf13/Pt2.3 151 6.1
9.5.2 AAV 4: Zirconiumbeladung der NafionP
®P-haltigen Silikate
Das NafionP
®P-haltige Silikat wird für sechs Stunden bei 100 °C in einem
Vakuumofen getrocknet. Anschließend wird das Trägermaterial unter Argon in
Toluol abs. suspendiert und im Aceton/Trockeneis-Bad gekühlt. Dicyclopenta-
dienyldimethylzirconium wird in ca. 5 ml Toluol abs. gelöst und die farblose
Lösung zur gekühlten Suspension gegeben. Die Mischung wird unter Rühren
auf Raumtemperatur aufgetaut und insgesamt etwa 16 Stunden gerührt. Man
lässt absitzen und filtriert das Lösungsmittel ab, wäscht zweimal mit Toluol
abs. und trocknet im Vakuum. Man erhält ein hellgelbes Pulver.
Es wurden Zirconium-beladene Materialien mit einem Zirconium/Sulfonsäure-
rest-Verhältnis von 1 : 1 (0.9 Gew.%) und 1 : 2 (0.5 Gew.%) hergestellt. Dazu
wurden die in Tabelle 44 angegebenen Mengen an Trägermaterial und
Zirconium-Precursor umgesetzt.

Experimenteller Teil 141
Tabelle 44: Mengen der Edukte für die Darstellung der Zirconium-beladenen, NafionP
®P-haltigen Silikate.
Produkt Trägermaterial
m [g]
CpB2BZrMeB2B
m [mg]
(n [TµTmol])
SAC/Naf10/Zr/Cp B2BMe (1:1) 6.94 183 (728)
SAC/Naf10/Zr/Cp B2B (1:2) 2.13 28 (112)
9.5.3 AAV 5: Kupferbeladung der NafionP
®P-haltigen Silikate
Das NafionP
®P-haltige Silikat wird zunächst in einer Säule bis zur pH-Neutralität
mit 2 M Natriumchlorid-Lösung behandelt, anschließend mit dest. Wasser
gewaschen und für vier Stunden im Vakuum bei 150 °C getrocknet.
Das NaP
+P-ausgetauschte Material wird zu einer methanolischen, türkisfarbenen
Lösung des Bis(oxazolins) und Kupfer(II)-trifluormethansulfonats gegeben
und für 24 Stunden bei Raumtemperatur gerührt. Man lässt absitzen, filtriert
und wäscht gründlich mit Methanol und Dichlormethan. Nach Trocknen im
Vakuum bei Raumtemperatur erhält man ein hellblaues Pulver.
Als Ligand wurde 2,2’-Bis-[(4S)-4-phenyl-2-oxazolin-2-yl]-propan oder 2,2-Bis-
[(4S)-tert.-butyl-2-oxazolin-2-yl]-propan verwendet. Die eingesetzten Mengen
an Trägermaterial, Ligand bzw. Kupfer-Precursor sind in Tabelle 45 aufge-
listet.
Experimenteller Teil 142
Tabelle 45: Mengen der Edukte für die Darstellung der Kupferbeladenen NafionP
®P-
haltigen Silikate.
Produkt Trägermaterial
m [mg]
Cu(OTf)B2B
m [mg]
(n [TµTmol])
Ligand m [mg]
(n [TµTmol])
SAC/Naf10/Cu/Ph 500 19.9 (55) 18.4 (55) T7T
MCM/Naf10/Cu/Ph 500 19.9 (55) 18.4 (55) T7T
SAC/Naf40/Cu/Ph 300 43.4 (120) 40.1 (120) T7T
SAC/Naf10/Cu/ P
tPBu 500 19.9 (55) 16.2 (55) T8T
MCM/Naf10/Cu/ P
tPBu 500 19.9 (55) 16.2 (55) T8T
9.5.4 AAV 6: Wasserstoff-Reduktion der Platin-beladenen Systeme
Das gewünschte Platin-beladene Nafion P
®P-haltige Silikat wird als Feststoff bei
Raumtemperatur in einer Wasserstoffatmosphäre (1.4 bar) reduziert. Dazu
wird das Reaktionsgefäß dreimal evakuiert und anschließend mit Wasserstoff
belüftet. Man belässt den Feststoff für mindestens 60 Minuten in der Wasser-
stoffatmosphäre. Dabei wechselt die Farbe des Materials von weiß zu grau.
9.6 Katalysen
9.6.1 AAV 7: Hydrierung von Olefinen
In einem 6-ml-Reaktionsgefäß legt man 15 bis 60 mg des festen Katalysators
vor, verschließt mit einer Septumkappe und reduziert zunächst das Edelmetall
nach AAV 6. Dann gibt man unter Druckausgleich 2 ml Methanol abs. und
200 TµTl (169 mg, 1.54 mmol) Cycloocten zu und rührt bei Raumtemperatur und
1.4 bar Wasserstoffdruck. Zur Analyse des Cycloocten-Umsatzes werden in
regelmäßigen Abständen 50 TµTl-Proben in 2 ml Dichlormethan zur Entfernung
des Metalls über Kieselgel filtriert, mit Magnesiumsulfat getrocknet und mittels
GC analysiert.

Experimenteller Teil 143
9.6.2 AAV 8: Cyclopropanierungen
In einem 6-ml-Reaktionsgefäß gibt man den festen Katalysator vor, trocknet
für 2 Stunden bei RT im Vakuum und belüftet mit Argon. Dann gibt man 2 ml
Dichlormethan abs., 50 TµTl n-Decan und 229 TµTl (2.0 mmol, 208 mg) Styrol zu.
Über zwei Stunden werden unter Rühren bei 25 °C zu dieser Mischung 105 TµTl
(1.0 mmol, 114 mg) Ethyldiazoacetat in 0.2 ml Dichlorethan mittels einer
Spritzenpumpe zugetropft. Der Reaktionsverlauf wird mittels GC kontrolliert.
Sobald das Ethyldiazoacetat aufgebraucht ist, gibt man in gleicher Weise
weitere 105 TµTl Ethyldiazoacetat in 0.2 ml Dichlorethan zu. Nach abge-
schlossener Reaktion werden ca. 50 TµTl der Reaktionsmischung zur Ent-
fernung der Metallanteile über Kieselgel filtriert und mittels chiraler GC
analysiert.
Um die Katalysatoren in einer weiteren Katalyse einzusetzen, werden sie
abfiltriert, mit Dichlorethan gewaschen und im Vakuum getrocknet.
In Tabelle 46 sind die eingesetzten Katalysatormengen und Reaktionszeiten
der Katalyse aufgelistet.
Tabelle 46: Eingesetzte Katalysatormengen und Reaktionszeiten für die Cyclo-propanierung.
Katalysator Einwaage [mg]
Reaktionszeit [h]
SAC/Naf10/Cu/Ph 99 8
MCM/Naf10/Cu/Ph 101 8
SAC/Naf10/Cu/ P
tPBu 100 7
MCM/Naf10/Cu/ P
tPBu 100 7
Außerdem wurde der entsprechende homogene Katalysator in der Reaktion
eingesetzt. Dazu werden 2.2 mg (6 TµTmol) Kupfer(II)-trifluormethansulfonat zu
einer Lösung von 2.0 mg (6 TµTmol) des Liganden T7T bzw. von 1.8 mg (6 TµTmol)
des Liganden T8T in 2 ml Dichlorethan gegeben und zunächst für zwei Stunden
bei RT unter Argon gerührt. Anschließend wird diese Lösung wie oben

Experimenteller Teil 144
beschrieben in der Cyclopropanierung eingesetzt. Die Reaktionszeit betrug
acht bzw. neun Stunden.
9.6.3 Durchführung der Polymerisation
Die Ethen-Polymerisationen wurden im Arbeitskreis von Prof. Dr. G. Fink am
Max-Planck-Institut für Kohlenforschung durchgeführt. Es kam ein Laborrühr-
autoklav der Firma Büchi AG (Uster) zum Einsatz.
Der Glasreaktor besteht aus einem doppelwandigen, temperierbaren Glas-
gefäß aus Borosilikatglas, das bis 12 bar geprüft ist. Die Temperatur im
Reaktor wird mit einem glasummantelten Widerstandsthermometer (PT 100)
und der Druck durch einen piezoresistiven Druckaufnehmer der Firma Jumo
Mess- und Regeltechnik gemessen. Das Rühren der Reaktionsmischung
erfolgt durch ein stopfbuchsenloses Rührwerk, an das ein Glasflügelrührer
angeschraubt ist. Der Verbrauch des Monomers während der Polymerisation
wird über eine Durchflussmessung bestimmt, die mit Hilfe eines thermischen
Massendurchflussmessers (Firma Brooks Instruments B. V., Holland) verfolgt
und durch einen Schreiber aufgezeichnet wird. Zusätzlich werden Fluss und
Temperatur gegen die Zeit an einen angeschlossenen Computer übermittelt
und können mit dem Windows-Programm Excel ausgewertet werden. Es
stehen drei Durchflussbereiche von 0 bis100 ml/min, 0 bis 1000 ml/min oder 0
bis 5000 ml/min zur Auswahl. Der Massendurchflussmesser ist geeicht gegen
2 bar Ethen bei 20 °C.
Vor dem Polymerisationsversuch wird das Reaktorgefäß im Trockenschrank
bei ca. 95 °C ausgeheizt, noch heiß an das Gestell angeflanscht und mehr-
mals evakuiert und mit Argon belüftet. Anschließend werden im Argongegen-
strom in den Reaktor das Lösungsmittel und, falls verwendet, der Co-
katalysator vorgelegt. Der Reaktor wird verschlossen und auf die gewünschte
Reaktionstemperatur gebracht. Man lässt den Argonüberdruck ab und presst
Ethen mit einem Druck von 2 bar auf. Dieser Druck bleibt während der
Polymerisation konstant. Nach vollständiger Sättigung des Lösungsmittels mit
Ethen wird der Katalysator, suspendiert in Lösungsmittel und Cokatalysator,

Experimenteller Teil 145
mit Hilfe eines Argon-Überdrucks über ein Einspritzsystem in den Reaktor
gegeben und mit Lösungsmittel nachgespült. Die genauen Bedingungen der
Polymerisation sind in Tabelle 47 angegeben.
Tabelle 47: Bedingungen für die Polymerisation von Ethen.
Ethendruck Temperatur Rührer Voraktivierung
2 bar 50 °C 1200 U/min 20 min
Nach beendeter Reaktion wird der Ethendruck abgelassen und der
Katalysator durch vorsichtige Zugabe von Methanol zerstört. Die Polymer-
suspension wird in ein Becherglas überführt, mit 300 ml Methanol und ca.
10 ml konz. HCl versetzt und gerührt. Das Polymer wird dann über einen
Büchner-Trichter abfiltriert, mit frischem Methanol versetzt und erneut gerührt.
Das Polymer wird abfiltriert und im Vakuumofen bei 70 °C für ca. 48 Stunden
getrocknet.
9.6.3.1 Katalysatorsystem SAC/Naf10/Zr/CpB2BMe
Im Folgenden sind die eingesetzten Mengen an Katalysator
SAC/Naf10/Zr/Cp B2BMe, die Reaktionszeiten und die isolierten Polymermengen
angegeben.
Experimenteller Teil 146
Tabelle 48: Eingesetzte Katalysatormengen und Reaktionsbedingungen in der Ethen-Polymerisation mit dem Katalysatorsystem SAC/Naf10/Zr/CpB2BMe.
SAC/Naf10/Zr/CpB2BMe m [mg]
Lösungs-mittelP
aP
Co-katalysator
Polymer m [g]
Reaktions-zeit [min]
55.4 Toluol MAO P
bP 2.80 4.5
16.8 Toluol MAO P
bP 8.14 25
9.3 Toluol MAO P
bP 9.05 72
4.9 Toluol MAO P
bP 1.71 140
4.9 Toluol MAO P
bP 1.18 55
50.3 Toluol ⎯ 0 50
11.1 Toluol TIBA P
cP 0 35
11.0 Heptan TIBAP
cP 0 90
13.5P
dP Toluol MAO 1.67 40
19.6P
dP Toluol MAO 3.77 35.5
P
aP Insgesamt ca. 130 ml.
P
b PAl/Zr-Verhältnis 2000:1, 1.7 M MAO-Lösung in Toluol.
P
c PAl/Zr-Verhältnis 2000:1, 100 %iges TIBA.
P
dP Katalysator nach drei Wochen Lagerung bei RT unter Argon.
Heterogenitätsuntersuchungen
Zur Überprüfung des heterogenen Charakters der Reaktion wurden etwa
100 mg des Katalysators für 24 Stunden bei RT in 5 ml Toluol gerührt und der
Feststoff abfiltriert. Anschließend wurde das Filtrat getrennt in einem
Polymerisationsversuch mit MAO als Cokatalysator eingesetzt.
Des Weiteren wurden zu 348 mg des Katalysators 42 ml MAO gegeben, es
trat Gasentwicklung auf und eine Entfärbung des gelben Feststoffs. Nach
20 Minuten Rühren wurde die Suspension abfiltriert und Teile des farblosen
Filtrats und des mit Toluol gewaschenen Feststoffs in getrennten Poly-
merisationsversuchen eingesetzt.
Experimenteller Teil 147
Tabelle 49: Eingesetzte Katalysatormengen und Reaktionsbedingungen in der Ethen-Polymerisation bei Einsatz von Rückstand bzw. Filtrat nach Behandlung des Katalysatorsystems SAC/Naf10/Zr/CpB2BMe mit MAO.
Katalysator Lösungs-
mittelP
aP
Co-katalysator
Polymer m [g]
Reaktions-zeit [min]
Rückstand 55.0 mg Toluol MAO P
bP 2.41 50
Rückstand 54.9 mg Toluol ⎯ 0 28
Filtrat (von 20.1 mg) Toluol MAOP
bP 2.14 30
P
aP Insgesamt ca. 130 ml.
P
b PAl/Zr-Verhältnis 2000:1, 1.7 M MAO-Lösung in Toluol.
9.6.3.2 Katalysatorsystem SAC/Naf10/Zr/CpB2B
Das Katalysatorsystem SAC/Naf10/Zr/CpB2B wurde ebenfalls in der Polymeri-
sation von Ethen untersucht. Dabei wurden die in Tabelle 50 aufgeführten
Bedingungen verwendet.
Tabelle 50: Eingesetzte Katalysatormengen und Reaktionsbedingungen in der Ethen-Polymerisation mit dem Katalysatorsystem SAC/Naf10/Zr/CpB2BMe.
SAC/Naf10/Zr/CpB2BMe m [mg]
Lösungs-mittelP
aP
Co-katalysator
Polymer m [g]
Reaktions-zeit [min]
49.6 Toluol MAO P
bP 3.30 28
Filtrat (von 50.6 mg) Toluol MAOP
bP 2.59 30
P
aP Insgesamt ca. 130 ml.
P
b PAl/Zr-Verhältnis 2000:1, 1.7 M MAO-Lösung in Toluol.
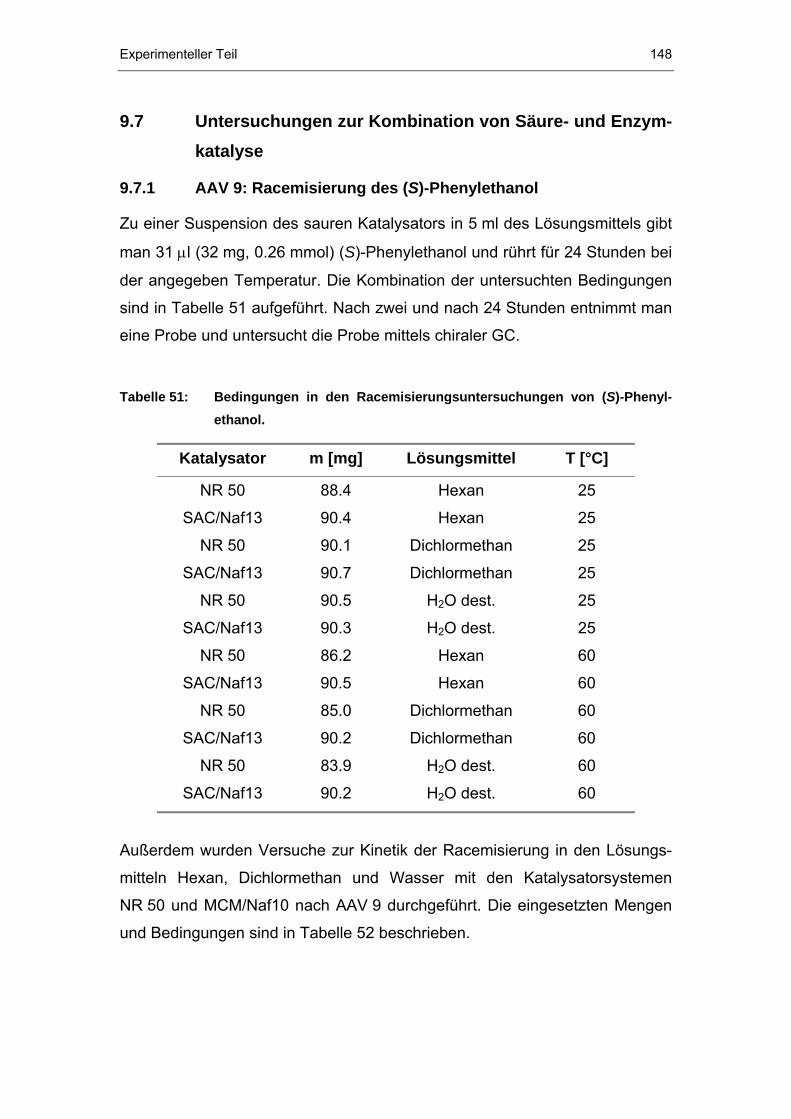
Experimenteller Teil 148
9.7 Untersuchungen zur Kombination von Säure- und Enzym-katalyse
9.7.1 AAV 9: Racemisierung des (S)-Phenylethanol
Zu einer Suspension des sauren Katalysators in 5 ml des Lösungsmittels gibt
man 31 TµTl (32 mg, 0.26 mmol) (S)-Phenylethanol und rührt für 24 Stunden bei
der angegeben Temperatur. Die Kombination der untersuchten Bedingungen
sind in Tabelle 51 aufgeführt. Nach zwei und nach 24 Stunden entnimmt man
eine Probe und untersucht die Probe mittels chiraler GC.
Tabelle 51: Bedingungen in den Racemisierungsuntersuchungen von (S)-Phenyl-ethanol.
Katalysator m [mg] Lösungsmittel T [°C]
NR 50 88.4 Hexan 25
SAC/Naf13 90.4 Hexan 25
NR 50 90.1 Dichlormethan 25
SAC/Naf13 90.7 Dichlormethan 25
NR 50 90.5 HB2BO dest. 25
SAC/Naf13 90.3 HB2BO dest. 25
NR 50 86.2 Hexan 60
SAC/Naf13 90.5 Hexan 60
NR 50 85.0 Dichlormethan 60
SAC/Naf13 90.2 Dichlormethan 60
NR 50 83.9 HB2BO dest. 60
SAC/Naf13 90.2 HB2BO dest. 60
Außerdem wurden Versuche zur Kinetik der Racemisierung in den Lösungs-
mitteln Hexan, Dichlormethan und Wasser mit den Katalysatorsystemen
NR 50 und MCM/Naf10 nach AAV 9 durchgeführt. Die eingesetzten Mengen
und Bedingungen sind in Tabelle 52 beschrieben.
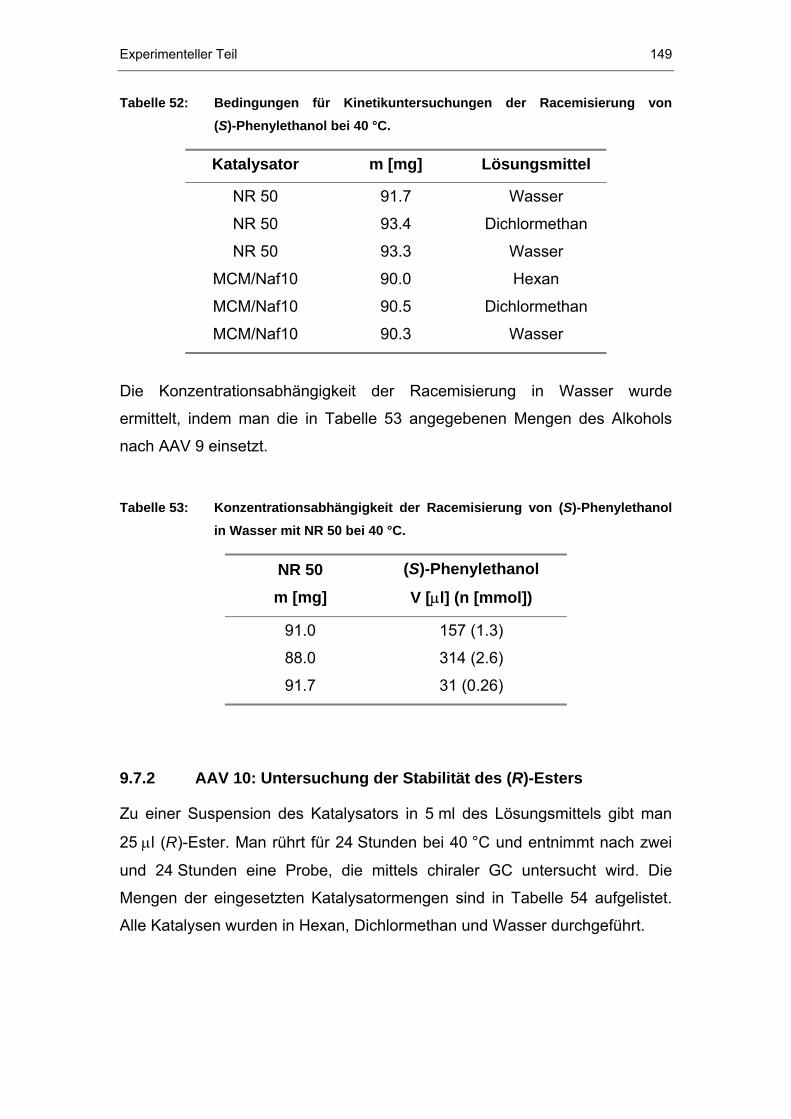
Experimenteller Teil 149
Tabelle 52: Bedingungen für Kinetikuntersuchungen der Racemisierung von (S)-Phenylethanol bei 40 °C.
Katalysator m [mg] Lösungsmittel
NR 50 91.7 Wasser
NR 50 93.4 Dichlormethan
NR 50 93.3 Wasser
MCM/Naf10 90.0 Hexan
MCM/Naf10 90.5 Dichlormethan
MCM/Naf10 90.3 Wasser
Die Konzentrationsabhängigkeit der Racemisierung in Wasser wurde
ermittelt, indem man die in Tabelle 53 angegebenen Mengen des Alkohols
nach AAV 9 einsetzt.
Tabelle 53: Konzentrationsabhängigkeit der Racemisierung von (S)-Phenylethanol in Wasser mit NR 50 bei 40 °C.
NR 50 m [mg]
(S)-Phenylethanol
V [ TµTl] (n [mmol])
91.0 157 (1.3)
88.0 314 (2.6)
91.7 31 (0.26)
9.7.2 AAV 10: Untersuchung der Stabilität des (R)-Esters
Zu einer Suspension des Katalysators in 5 ml des Lösungsmittels gibt man
25 TµTl (R)-Ester. Man rührt für 24 Stunden bei 40 °C und entnimmt nach zwei
und 24 Stunden eine Probe, die mittels chiraler GC untersucht wird. Die
Mengen der eingesetzten Katalysatormengen sind in Tabelle 54 aufgelistet.
Alle Katalysen wurden in Hexan, Dichlormethan und Wasser durchgeführt.

Experimenteller Teil 150
Tabelle 54: Menge der NafionP
®P-Systeme bei der Umsetzung mit (R)-Ester.
Katalysator m [mg]
NR 50 50
SAC/Naf13 90
MCM/Naf10 90
9.7.3 Doppel-Immobilisierung von NafionP
®P-Polymer und Candida
antarctica Lipase B
Für die Doppel-Immobilisierung von NafionP
®P und Candida antarctica Lipase B
in Silikat wurde der 13 Gew.%ige Nafion P
®P-Silikat-Verbundstoff SAC/Naf13
nach TAAV 1T (Kapitel 9.4.1) hergestellt, jedoch nach dem Ansäuern und
Waschen nur bei Raumtemperatur getrocknet.
Zu diesem Verbundwerkstoff werden 128 TµTl der Stammlösung I (vgl. Tabelle
55) zugegeben, die Mischung auf dem Vortex-Mixer vermischt und 110 TµTl der
Stammlösung II (vgl. Tabelle 55) zupipettiert und weitere 15 Sekunden auf
dem Vortex-Mixer vermischt.
Tabelle 55: Zusammensetzung der Stammlösung I und II.
Stammlösung I Stammlösung II
125 mg Candida antartica Lipase B
390 TµTl Tris-Puffer
100 TµTl PVA-Lösung
50 TµTl NaF-Lösung
100 TµTl Isopropanol
477 TµTl Isobutyltrimethoxy-Silan
74 TµTl TMOS
Es wurden verschiedene Verbundwerkstoff-Mengen eingesetzt. Die Mengen
und Beobachtungen bei der Immobilisierung sind in Tabelle 56 aufgeführt.
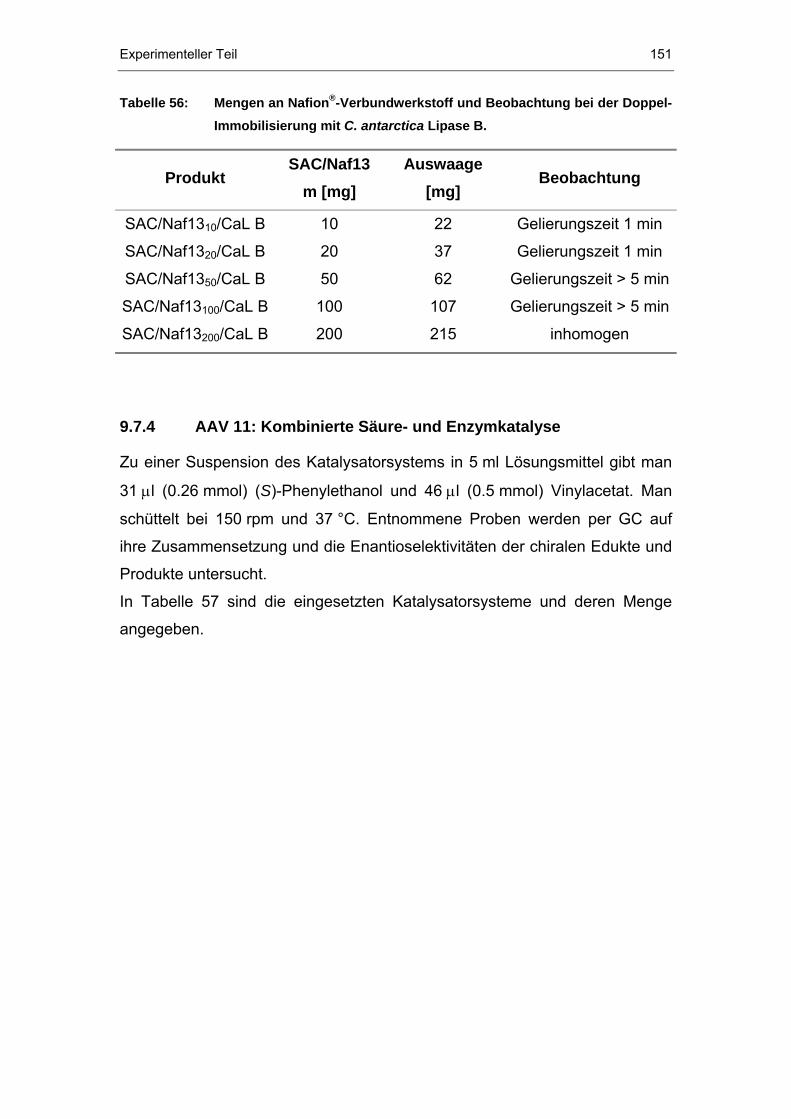
Experimenteller Teil 151
Tabelle 56: Mengen an NafionP
®P-Verbundwerkstoff und Beobachtung bei der Doppel-
Immobilisierung mit C. antarctica Lipase B.
Produkt SAC/Naf13
m [mg] Auswaage
[mg] Beobachtung
SAC/Naf13B10B/CaL B 10 22 Gelierungszeit 1 min
SAC/Naf13B20B/CaL B 20 37 Gelierungszeit 1 min
SAC/Naf13B50B/CaL B 50 62 Gelierungszeit > 5 min
SAC/Naf13B100B/CaL B 100 107 Gelierungszeit > 5 min
SAC/Naf13B200B/CaL B 200 215 inhomogen
9.7.4 AAV 11: Kombinierte Säure- und Enzymkatalyse
Zu einer Suspension des Katalysatorsystems in 5 ml Lösungsmittel gibt man
31 TµTl (0.26 mmol) (S)-Phenylethanol und 46 TµTl (0.5 mmol) Vinylacetat. Man
schüttelt bei 150 rpm und 37 °C. Entnommene Proben werden per GC auf
ihre Zusammensetzung und die Enantioselektivitäten der chiralen Edukte und
Produkte untersucht.
In Tabelle 57 sind die eingesetzten Katalysatorsysteme und deren Menge
angegeben.
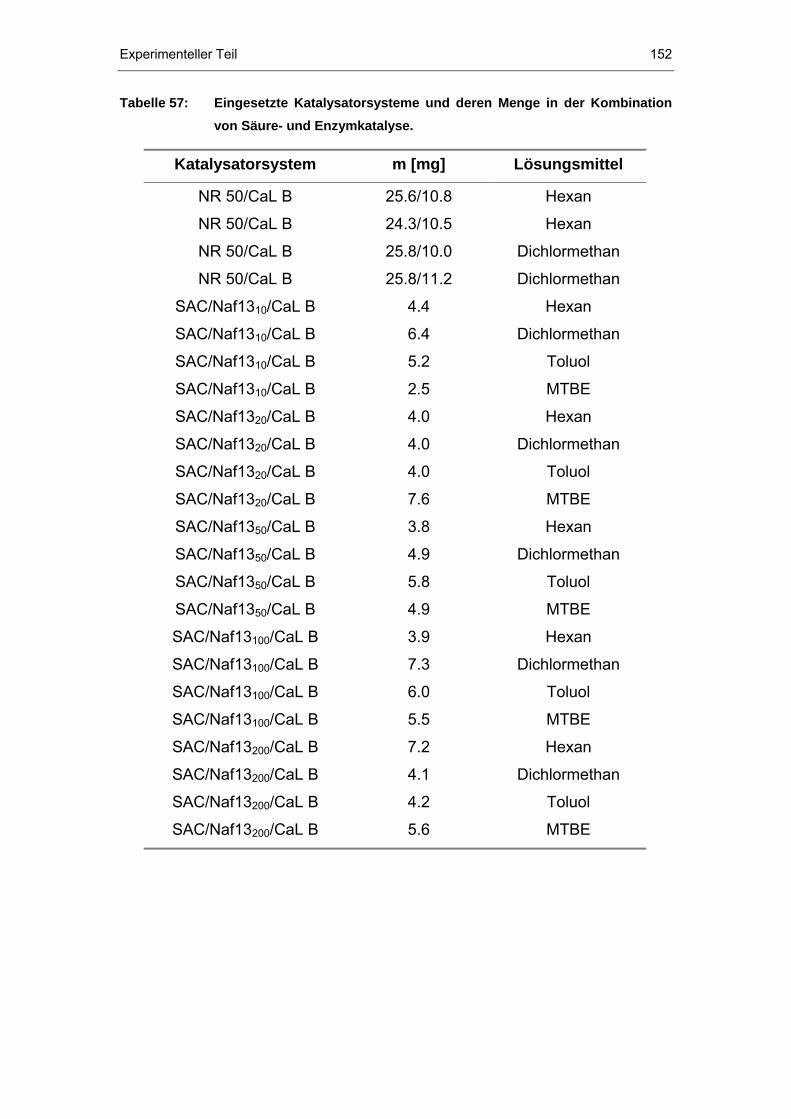
Experimenteller Teil 152
Tabelle 57: Eingesetzte Katalysatorsysteme und deren Menge in der Kombination von Säure- und Enzymkatalyse.
Katalysatorsystem m [mg] Lösungsmittel
NR 50/CaL B 25.6/10.8 Hexan
NR 50/CaL B 24.3/10.5 Hexan
NR 50/CaL B 25.8/10.0 Dichlormethan
NR 50/CaL B 25.8/11.2 Dichlormethan
SAC/Naf13B10B/CaL B 4.4 Hexan
SAC/Naf13 B10B/CaL B 6.4 Dichlormethan
SAC/Naf13B10B/CaL B 5.2 Toluol
SAC/Naf13B10B/CaL B 2.5 MTBE
SAC/Naf13B20B/CaL B 4.0 Hexan
SAC/Naf13 B20B/CaL B 4.0 Dichlormethan
SAC/Naf13B20B/CaL B 4.0 Toluol
SAC/Naf13B20B/CaL B 7.6 MTBE
SAC/Naf13B50B/CaL B 3.8 Hexan
SAC/Naf13 B50B/CaL B 4.9 Dichlormethan
SAC/Naf13B50B/CaL B 5.8 Toluol
SAC/Naf13B50B/CaL B 4.9 MTBE
SAC/Naf13B100B/CaL B 3.9 Hexan
SAC/Naf13 B100B/CaL B 7.3 Dichlormethan
SAC/Naf13B100B/CaL B 6.0 Toluol
SAC/Naf13B100B/CaL B 5.5 MTBE
SAC/Naf13B200B/CaL B 7.2 Hexan
SAC/Naf13 B200B/CaL B 4.1 Dichlormethan
SAC/Naf13B200B/CaL B 4.2 Toluol
SAC/Naf13B200B/CaL B 5.6 MTBE

Anhang 153
10 Anhang
10.1 Abkürzungsverzeichnis
AAV Allgemeine Arbeitsvorschrift
abs. absolut
Äquiv. Äquivalent
bzw. beziehungsweise
c Konzentration
ca. circa
CEC cation exchange capacity
cmc critical micelle concentration
COD Cyclooctadien
CTAB Cetyltrimethylammoniumbromid
d Durchmesser
EBaB Aktivierungsenergie
EA Elementaranalyse
EDX Energy dispersive X-ray analysis
et al. und andere
FWHM Full width at half-maximum
GC-Analyse gaschromatographische Analyse
ges. gesättigt
Gew.% Gewichtsprozent
Gl. Gleichung
GPC Gelpermeationschromatographie
h Stunde
HR-TEM hochauflösende Transmissionselektronenmikroskopie
M Mol pro Liter
MAO Methylalumoxan
min Minute
n Stoffmenge
n. b. nicht bestimmt
p Druck
PE Polyethylen

Anhang 154
PP Polypropylen
R organischer Rest
RT Raumtemperatur
s Sekunde
S/C Substrat/Katalysator
T Temperatur
t Zeit
TEM Transmissionselektronenmikroskopie
TEOS Tetraethoxysilan
THF Tetrahydrofuran
TIBA Triisobutylaluminium
TMEDA N,N,N’,N’-Tetramethylethylendiamin
TMOS Tetramethoxysilan
Triflat Trifluormethansulfonat
UV Ultraviolett
V Volumen
vgl. vergleiche
w Massenanteil
XPS Röntgen-Photoelektronen-Spektroskopie
XRD Röntgen-Pulver-Diffraktometrie

Anhang 155
10.2 TBenennung der Sol-Gel-Immobilisate
Erläuterung des Abkürzungssystems
Für eine kurze und aussagekräftige Benennung der in der Arbeit eingesetzten
Sol-Gel-Immobilisate wurde ein Abkürzungssystem entwickelt, anhand
dessen direkt der verwendete Silikattyp, die eingesetzte Menge des NafionsP
®P
und Typ und Menge eventuell vorhandener Metallspezies ersichtlich wird.
Der Aufbau des Systems generell ist im Folgenden dargestellt:
[Silikattyp]/[NafionP
®P mit Mengenangabe]/[Metall mit Mengenangabe].
An erster Stelle wird der verwendete Silikattyp angegeben und an zweiter
Stelle die eingesetzte NafionP
®P-Menge in Gewichtsprozent. Wurden die Sol-
Gel-Immobilisate mit Metall beladen, folgt die Angabe des Metalls mit
Mengenangabe in Gewichtsprozent an dritter Stelle. Ein geordnetes, meso-
poröses Silikat mit 10 Gew.% NafionP
®P und 0.5 Gew.% Zirconium wird zum
Beispiel wie folgt bezeichnet:
MCM/Naf10/Zr0.5.
Falls Metallspezies mit unterschiedlichen Liganden beziehungsweise Gegen-
ionen eingesetzt werden und eine Unterscheidung notwendig ist, kann dies an
vierter Stelle angegeben werden. Sind die Zirconium-Zentren im oben
genannten Beispiel noch von zwei Cyclopentadienyl-Resten umgeben, so
wird folgende Benennung verwendet:
MCM/Naf10/Zr0.5/Cp B2B.

Anhang 156
Verwendete Abkürzungen
Silikate:
SAC Ungeordnetes Silikat (analog Benennung DuPont)
MCM Geordnetes Silikat (analog Benennung Mobil)
Perfluorsäurespezies:
Naf NafionP
®P-Polymer
Metalle:
Cu Kupfer
Pt Platin
Zr Zirconium
Liganden und Gegenionen:
Cp Cyclopentadienyl
COD Cyclooctadien
Ph 2,2’-Bis-[(4S)-4-phenyl-2-oxazolin-2-yl]-propan
P
tPBu 2,2-Bis-[(4S)-tert.-butyl-2-oxazolin-2-yl]-propan

Literaturverzeichnis 157
11 Literaturverzeichnis
[1] Lewis Acids in Organic Synthesis, Vol. 1 und 2, (Hrsg.: H. Yamamoto),
Wiley VCH, Weinheim, 2000.
[2] A. Corma, H. Garcia, Chem. Rev. 2003, 103, 4307.
[3] J. C. Donald, F. G. William, US Patent 3,282,875, 1964.
[4] G. B. Butler, K. F. O'Driscoll, G. L. Wilkes, JMS-Rev. Macromol. Chem.
Phys. 1994, C34(3), 325.
[5] P. J. Brookman, J. W. Nicholson, in Developments in Ionic Polymers,
Vol. 2 (Hrsg.: A. D. Wilson, H. J. Prosser), Elsevier Applied Science
Publishers, London, 1986, S. 269.
[6] F. J. Waller, R. W. Van Scoyoc, Chemtech 1987, 17, 438.
[7] K. Weissermel, H.-J. Arpe, Industrial Organic Chemistry, 4. Auflage,
Wiley-VCH, Weinheim, 2003.
[8] W. G. Grot, US Patent 4 433 082, 1981.
[9] A. P. Wight, M. E. Davis, Chem. Rev. 2002, 102, 3589.
[10] C. Copéret, M. Chabanas, R. Petroff Saint-Arroman, J.-M. Basset,
Angew. Chem. Int. Ed. 2003, 42, 156.
[11] G. Fink, B. Steinmetz, J. Zechlin, C. Przybyla, B. Tesche, Chem. Rev.
2000, 100, 1377.
[12] C. E. Song, S. Lee, Chem. Rev. 2002, 102, 3495.
[13] C. J. Brinker, G. W. Scherer, Sol-Gel Science, Academic Press, San
Diego, 1990.
[14] A. Corma, Chem. Rev. 1997, 97, 2373.
[15] L. B. Okhlopkova, A. S. Lisitsyn, V. A. Likholobov, M. Gurrath,
H. P. Boehm, Appl. Catal. A-Gen. 2000, 204, 229.
[16] J. S. Beck, U.S. Patent 5 057 296, 1991.
[17] J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge,
K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard,
S. B. McCullen, J. B. Higgins, J. L. Schlenker, J. Am. Chem. Soc.
1992, 114, 10834.
[18] C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli, J. S. Beck,
Nature 1992, 359, 710.
[19] V. Chiola, J. E. Ritsko, C. D. Vanderpool, US Patent 3 556 725, 1971.

Literaturverzeichnis 158
[20] M. R. S. Manton, J. C. Davidtz, J. Catal. 1979, 60, 156.
[21] T. Yanagisawa, T. Shimizu, K. Kuroda, C. Kato, Bull. Chem. Soc. Jpn.
1990, 63, 988.
[22] C.-Y. Chen, S. L. Burkett, H.-X. Li, M. E. Davis, Microporous Mater.
1993, 2, 27.
[23] P. T. Tanev, T. J. Pinnavaia, Science 1995, 267, 865.
[24] S. A. Bagshaw, E. Prouzet, T. J. Pinnavaia, Science 1995, 269, 1242.
[25] A. Monnier, F. Schüth, Q. Huo, D. Kumar, D. Margolese,
R. S. Maxwell, G. D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi,
M. Janicke, B. F. Chmelka, Science 1993, 261, 1299.
[26] D. Zhao, Q. Huo, J. Feng, B. F. Chmelka, G. D. Stucky, J. Am. Chem.
Soc. 1998, 120, 6024.
[27] Q. S. Huo, D. I. Margolese, U. Ciesla, P. Y. Feng, T. E. Gier, P. Sieger,
R. Leon, P. M. Petroff, F. Schüth, G. D. Stucky, Nature 1994, 368, 317.
[28] S. Biz, M. L. Occelli, Catal. Rev.-Sci. Eng. 1998, 40, 329.
[29] A. Stein, Adv. Mater. 2003, 15, 763.
[30] M. E. Davis, Nature 2002, 417, 813.
[31] D. E. De Vos, M. Dams, B. F. Sels, P. A. Jacobs, Chem. Rev. 2002,
102, 3615.
[32] D. Brunel, A. C. Blanc, A. Galarneau, F. Fajula, Catal. Today 2002, 73,
139.
[33] C. P. Mehnert, D. W. Weaver, J. Y. Ying, J. Am. Chem. Soc. 1998,
120, 12289.
[34] L. M. Bronstein, Top. Curr. Chem. 2003, 226, 55.
[35] M. A. Harmer, W. E. Farneth, Q. Sun, J. Am. Chem. Soc. 1996, 118,
7708.
[36] M. A. Harmer, EP Patent 0 739 239 B 1, 1995.
[37] A. Heidekum, M. A. Harmer, W. F. Hoelderich, Stud. Surf. Sci. Catal.
2000, 130, 3441.
[38] Q. Sun, W. E. Farneth, M. A. Harmer, J. Catal. 1996, 164, 62.
[39] Q. Sun, M. A. Harmer, W. E. Farneth, Ind. Eng. Chem. Res. 1997, 36,
5541.
[40] Q. Sun, M. A. Harmer, W. E. Farneth, Chem. Commun. 1996, 1201.
[41] B. Török, I. Kiricsi, A. Molnár, G. A. Olah, J. Catal. 2000, 193, 132.

Literaturverzeichnis 159
[42] M. A. Harmer, Q. Sun, M. J. Michalczyk, Z. Yang, Chem. Commun.
1997, 1803.
[43] M. Fujiwara, K. Kuraoka, T. Yazawa, Q. Xu, M. Tanaka, Y. Souma,
Chem. Commun. 2000, 1523.
[44] Y. Wei, J. Xu, H. Dong, J. H. Dong, K. Qiu, S. A. Jansen-Varnum,
Chem. Mater. 1999, 11, 2023.
[45] Y. Wei, J. Xu, Q. Feng, H. Dong, M. Lin, Mater. Lett. 2000, 44, 6.
[46] I. S. Paulino, U. Schuchardt, Stud. Surf. Sci. Catal. 2002, 141, 93.
[47] S. R. Samms, S. Wasmus, Savinell, J. Electrochem. Soc. 1996, 143,
1498.
[48] Aldrich Katalog, 2003-2004, S. 1441.
[49] Aldrich Katalog, 2003-2004, S. 70.
[50] Handbook of Heterogeneous Catalysis, Vol. 2, (Hrsg.: G. Ertl,
H. Knözinger, J. Weitkamp), Wiley-VCH, Weinheim, 1997.
[51] K. S. W. Sing, D. H. Everett, R. A. Haul, L. Moscou, R. A. Pierotti,
J. Rouquerol, T. Siemieniewska, Pure Appl. Chem. 1985, 57, 603.
[52] D. H. Everett, J. M. Haynes, J. Colloid Interface Sci. 1972, 38, 125.
[53] U. Ciesla, F. Schüth, Microporous Mesoporous Mat. 1999, 27, 131.
[54] E. P. Barrett, L. G. Joyner, P. P. Halenda, J. Am. Chem. Soc. 1951, 73,
373.
[55] M. S. Tzou, B. K. Teo, W. M. H. Sachtler, J. Catal. 1988, 113, 220.
[56] T. R. Felthouse, J. A. Murphy, J. Catal. 1986, 98, 411.
[57] H. A. Benisi, R. M. Curtis, H. P. Studer, J. Catal. 1968, 10, 328.
[58] R. D. Cortright, J. A. Dumesic, J. Catal. 1994, 148, 771.
[59] L. Persuad, A. J. Bard, A. Campion, M. A. Fox, T. E. Mallouk,
S. E. Webber, J. M. White, Inorg. Chem. 1987, 26, 3825.
[60] K. M. Reddy, C. Song, Catal. Today 1996, 31, 137.
[61] R. Ryoo, C. H. Ko, J. M. Kim, R. Howe, Catal. Lett. 1996, 37, 29.
[62] U. Junges, W. Jacobs, I. Voigtmartin, B. Krutzsch, F. Schüth, J. Chem.
Soc.-Chem. Commun. 1995, 2283.
[63] G. Jacobs, C. L. Padro, D. E. Resasco, J. Catal. 1998, 179, 43.
[64] R. Mokaya, W. Jones, S. Moreno, G. Poncelet, Catal. Lett. 1997, 49,
87.

Literaturverzeichnis 160
[65] N. S. Yao, C. Pinckney, S. Y. Lim, C. Pak, G. L. Haller, Microporous
Mesoporous Mat. 2001, 44-45, 377.
[66] L. Abis, D. B. Dell' Amico, C. Busetto, F. Calderazzo, R. Caminiti,
C. Ciofi, F. Garbassi, M. Masciarelli, J. Mater. Chem. 1998, 8, 751.
[67] B. Marler, U. Oberhagemann, S. Vortmann, H. Gies, Microporous
Mater. 1996, 6, 19.
[68] M. Fröba, R. Köhn, G. Bouffaud, Chem. Mater. 1999, 11, 2858.
[69] M. Chatterjee, T. Iwasaki, Y. Onodera, T. Nagase, Catal. Lett. 1999,
61, 199.
[70] G. Wedler, Lehrbuch der Physikalischen Chemie, Vol. 3., VCH,
Weinheim, 1987.
[71] R. Zeng, Z. Pang, H. Zhu, J. Electroanal. Chem. 2000, 490, 102.
[72] N. M. Rodriguez, P. E. Anderson, A. Wootsch, U. Wild, R. Schlögl,
Z. Paál, J. Catal. 2001, 197, 365.
[73] J. Burkhardt, L. D. Schmidt, J. Catal. 1989, 116, 240.
[74] C. Roth, M. Goetz, H. Fuess, J. Appl. Electrochem. 2001, 31, 793.
[75] M. T. Reetz, M. Lopez, W. Grünert, W. Vogel, F. Mahlendorf, J. Phys.
Chem. B 2003, 107, 7414.
[76] M. Che, C. O. Bennett, Adv. Catal. 1989, 36, 55.
[77] S. D. Jackson, J. Willis, G. D. McLellan, G. Webb, M. B. T. Keegan,
R. B. Moyes, S. Simpson, P. B. Wells, R. Whyman, J. Catal. 1993, 139,
191.
[78] N. R. Ross, K. Kinoshita, P. Stonehart, J. Catal. 1974, 32, 163.
[79] E. E. Gonzo, M. Boudart, J. Catal. 1978, 52, 462.
[80] R. J. Madon, J. P. O'Connell, M. Boudart, AIChE Journal 1978, 24,
904.
[81] A. Corma, A. Martinéz, V. Martinéz-Soria, J. Catal. 1997, 169, 480.
[82] T. K. Hollis, N. P. Robinson, B. Bosnich, Organometallics 1992, 11,
2745.
[83] G. V. Bondar, R. Aldea, C. J. Levy, J. B. Jaquith, S. Collins,
Organometallics 2000, 19, 947.
[84] T. K. Hollis, W. Odenkirk, N. P. Robinson, J. Whelan, B. Bosnich,
Tetrahedron 1993, 49, 5415.

Literaturverzeichnis 161
[85] J. B. Jaquith, C. J. Levy, G. V. Bondar, S. T. Wang, S. Collins,
Organometallics 1998, 17, 914.
[86] T. K. Hollis, N. P. Robinson, B. Bosnich, Tetrahedron Letters 1992, 33,
6423.
[87] W. Odenkirk, B. Bosnich, J. Chem. Soc., Chem. Commun. 1995, 1181.
[88] K. Ziegler, E. Holzkamp, H. Breil, H. Martin, Angew. Chem. 1955, 67,
426.
[89] G. Natta, Angew. Chem. 1956, 68, 869.
[90] H. W. Turner, US Patent 4 752 597, 1988.
[91] A. R. Siedle, R. A. Newmark, W. B. Gleason, W. M. Lamanna,
Organometallics 1990, 9, 1290.
[92] Nachr. Chem. Tech. Lab. 1995, 43, 1208.
[93] W. Kaminsky, K. Kulper, H.-H. Brintzinger, F. R. W. P. Wild, Angew.
Chem. Int. Ed. Engl. 1985, 24, 507.
[94] E. Y.-X. Chen, T. J. Marks, Chem. Rev. 2000, 100, 1391.
[95] H. G. Alt, A. Köppl, Chem. Rev. 2000, 100, 1205.
[96] J. J. Eisch, A. M. Piottowski, S. K. Brownstein, E. J. Gabe, F. L. Lee,
J. Am. Chem. Soc. 1985, 107, 7219.
[97] M. Bochmann, L. M. Wilson, J. Chem. Soc., Chem. Commun. 1986,
1610.
[98] H. Sinn, Makromol. Chem. Macromol. Symp. 1995, 97, 27.
[99] H. Schneider, G. T. Puchta, F. A. R. Kaul, G. Raudaschl-Sieber,
F. Lefebvre, G. Saggio, D. Mihalios, W. A. Herrmann, J. M. Basset,
J. Mol. Catal. A-Chem. 2001, 170, 127.
[100] G. G. Hlatky, Chem. Rev. 2000, 100, 1347.
[101] X. Yang, C. L. Stern, T. J. Marks, J. Am. Chem. Soc. 1991, 113, 3623.
[102] J. A. Ewen, M. J. Elder, Patent EU 0 427 697, 1991.
[103] M. Jezequel, V. Dufaud, M. J. Ruiz-Garcia, F. Carrillo-Hermosilla,
U. Neugebauer, G. P. Niccolai, F. Lefebvre, F. Bayard, J. Corker,
S. Fiddy, J. Evans, J.-P. Broyer, J. Malinge, J.-M. Basset, J. Am.
Chem. Soc. 2001, 123, 3520.
[104] M. Bochmann, G. J. Pindado, S. J. Lancaster, J. Mol. Catal. A-Chem.
1999, 146, 179.
[105] D. Rechavi, M. Lemaire, Chem. Rev. 2002, 102, 3467.

Literaturverzeichnis 162
[106] M. I. Burguete, J. M. Fraile, J. I. Garcia, E. Garcia-Verdugo,
C. I. Herrerias, S. V. Luis, J. A. Mayoral, J. Org. Chem. 2001, 66, 8893.
[107] A. Corma, H. García, A. Moussaif, M. J. Sabater, R. Zniber,
A. Redouane, Chem. Commun. 2002, 1058.
[108] J. K. Park, S.-W. Kim, T. Hyeon, B. M. Kim, Tetrahedron: Asymmetry
2001, 12, 2931.
[109] J. M. Fraile, J. I. Garcia, J. A. Mayoral, T. Tarnai, M. A. Harmer,
J. Catal. 1999, 186, 214.
[110] J. M. Fraile, J. I. Garcia, M. A. Harmer, C. I. Herrerias, J. A. Mayoral,
J. Mol. Catal. A-Chem. 2001, 165, 211.
[111] J. M. Fraile, J. I. Garcia, M. A. Harmer, C. I. Herrerias, J. A. Mayoral,
O. Reiser, H. Werner, J. Mater. Chem. 2002, 12, 3290.
[112] J. M. Fraile, J. I. Garcia, J. A. Mayoral, T. Tarnai, Tetrahedron:
Asymmetry 1998, 9, 3997.
[113] M. J. Fernandez, J. M. Fraile, J. I. Garcia, J. A. Mayoral,
M. I. Burguete, E. Garcia-Verdugo, S. V. Luis, M. A. Harmer, Top.
Catal. 2000, 13, 303.
[114] H. Nozaki, S. Moriuti, H. Takaya, R. Noyori, Tetrahedron Lett. 1966,
5239.
[115] H. Fritschi, U. Leutenegger, A. Pfaltz, Helv. Chim. Acta 1988, 71, 1553.
[116] R. E. Lowenthal, A. Abiko, S. Masamune, Tetrahedron Lett. 1990, 31,
6005.
[117] D. A. Evans, K. A. Woerpel, M. M. Hinman, M. M. Faul, J. Am. Chem.
Soc. 1991, 113, 726.
[118] D. Müller, G. Umbricht, B. Weber, A. Pfaltz, Helv. Chim. Acta 1991, 74,
232.
[119] J. M. Fraile, J. I. García, V. Martinez Merino, J. A. Mayoral,
L. Salvatella, J. Am. Chem. Soc. 2001, 123, 7616.
[120] T. Rasmussen, J. F. Jensen, N. Ostergaard, D. Tanner, T. Ziegler,
P. O. Norrby, Chem. Eur. J. 2002, 8, 177.
[121] M. M. Díaz-Requejo, T. R. Belderrain, M. C. Nicasio, F. Prieto,
P. J. Perez, Organometallics 1999, 18, 2601.
[122] K. Faber, Biotransformations in Organic Chemistry, 4. Auflage,
Springer Verlag, Berlin, 1997.

Literaturverzeichnis 163
[123] B. Rubin, E. A. Dennis, Lipases, Part B: Enzyme Characterization and
Utilization, Methods in Enzymology, Vol. 286, Academic Press, San
Diego, 1997.
[124] A. Tanaka, T. Tosa, T. Kobayashi, Industrial Application of Immobilized
Enzymes, Marcel Dekker, New York, 1993.
[125] P. Johnson, T. L. Whateley, J. Colloid Interface Sci. 1971, 557.
[126] M. T. Reetz, A. Zonta, J. Simpelkamp, Angew. Chem. 1995, 107, 373.
[127] W. Wiesenhöfer, Diplomarbeit, Sol-Gel-Immobilisierung von Lipasen,
2001.
[128] S. Wuyts, K. De Temmermann, D. De Vos, P. Jacobs, Chem.
Commun. 2003, 1928.
[129] D. Drew, J. R. Doyle, in Inorganic Synthesis, Vol. 28, 1990, S. 346.
[130] E. Costa, P. G. Pringle, M. Ravetz, in Inorganic Synthesis, Vol. 31,
1997, S. 284.

Lebenslauf
Name Freitag
Vorname Katrin
Geburtsdatum 03.07.1975
Geburtsort Zwickau
Schulausbildung 1982 - 1989 Polytechnische Oberschule, Zwickau
1989 - 1991 Krupp-Gymnasium, Duisburg
1991 - 1992 Gymnasium Petrinum, Dorsten
1992 - 1993 Fairview High School, Fairview, TN, USA
1993 - 1995 Geschwister-Scholl-Gymnasium, Marl
Abschluss mit der Allgemeinen Hochschulreife
Studium 10/1995 - 09/2000 Studium der Chemie an der Westfälischen Wilhelms-
Universität Münster und der Universidad de Alcalá de
Henares, Spanien
10/1997 Diplom-Vorprüfung
04/2000 Diplom-Prüfung
04/2000 - 09/2000 Diplom-Arbeit: Dr. M. Mena (Universidad de Alcalá
de Henares, Spanien)
Fernbetreuung durch Prof. Dr. G. Erker (Universität
Münster)
12/2000 - 01/2004 Promotion bei Prof. Dr. M. T. Reetz am Max-Planck-
Institut für Kohlenforschung, Mülheim an der Ruhr














![Metallorganische Lewis-Säuren, XXXV [1] Bildung und Struktur …zfn.mpdl.mpg.de/data/Reihe_B/43/ZNB-1988-43b-0665.pdf · 2018. 2. 9. · Metallorganische Lewis-Säuren, XXXV [1]](https://static.fdokument.com/doc/165x107/60a5d1e5f1c7977e6b0133b8/metallorganische-lewis-suren-xxxv-1-bildung-und-struktur-zfnmpdlmpgdedatareiheb43znb-1988-43b-0665pdf.jpg)



