
Design, Synthese und Charakterisierung peptidischer ... · Abbildung 2: Prinzip der somatischen...
145
Design, Synthese und Charakterisierung peptidischer Paratop-Mimetika von HIV-1 neutralisierenden Antikörpern Der Naturwissenschaftlichen Fakultät / dem Fachbereich Chemie und Pharmazie der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Christina Haußner aus Nürnberg
Transcript of Design, Synthese und Charakterisierung peptidischer ... · Abbildung 2: Prinzip der somatischen...

Design, Synthese und Charakterisierung peptidischer Paratop-Mimetika
von HIV-1 neutralisierenden Antikörpern
Der Naturwissenschaftlichen Fakultät /
dem Fachbereich Chemie und Pharmazie
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur Erlangung des Doktorgrades Dr. rer. nat.
vorgelegt von
Christina Haußner
aus Nürnberg


Als Dissertation genehmigt
von der Naturwissenschaftlichen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung: 15.04.2016
Vorsitzender des Promotionsorgans: Prof. Dr. Jörn Wilms
Gutachterinnen: Prof. Dr. Jutta Eichler (FAU)
Prof. Dr. Barbara Schmidt (IMHR)


Meinen Eltern.


i
Inhaltsverzeichnis
Inhaltsverzeichnis ................................................................................................................. i Abkürzungsverzeichnis ...................................................................................................... iii 1. Einleitung ......................................................................................................................... 1
1.1 Antikörper und Antikörper-Antigen Interaktion .............................................................. 1
1.2 Peptidische Paratop-Mimetika ...................................................................................... 6
1.3 Synthetische Peptide in der Wirkstoffforschung ............................................................ 7
1.4 Humanes Immundefizienz-Virus (HIV) ........................................................................ 10
1.5 Neutralisierende Antikörper gegen HIV-1 ................................................................... 15
1.6 Kristallstrukturen von Fab b12 und Fab X5 in Komplex mit gp120 .............................. 17
2. Zielsetzung ..................................................................................................................... 23
3. Material und Methoden .................................................................................................. 24
3.1 Material ...................................................................................................................... 24
3.1.2 Chemikalien ......................................................................................................... 25
3.1.3 Verbrauchsmaterialien ......................................................................................... 30
3.2 Peptidsynthese ........................................................................................................... 31
3.2.1 Reaktionen an der festen Phase .......................................................................... 32
3.2.2 Präparative Gesamtabspaltung ............................................................................ 35
3.2.3 Oxidation zum Disulfid ......................................................................................... 36
3.2.4 Ellmans Test ........................................................................................................ 36
3.3 Reinigung der Peptide mittels semipräparativer HPLC ............................................... 37
3.4 Analytik mittels HPLC-gekoppelter Massenspektrometrie (HPLC/MS) ....................... 39
3.3 Enzyme-linked Immunosorbent Assay (ELISA) .......................................................... 40
3.3.1 Detektion verschiedener gp120-Proteine ............................................................. 40
3.3.2 Bindung von Peptiden an gp120 (EC50-Wert-Bestimmung) – Format A ................ 41
3.3.3 Streptavidin-ELISA – Format B ............................................................................ 41
3.3.4 Kompetitiver ELISA – Format C ........................................................................... 42
3.4 Zellinfektionsassays ................................................................................................... 42
3.4.1 Herstellung der Virusstocklösungen ..................................................................... 42
3.4.2 SEAP-Zellinfektionsassay mit HIV-1 .................................................................... 42
3.4.3 GHOST-Zellinfektionsassay mit HIV-1 ................................................................. 44
3.4.4 Vero-Zellinfektionsassay mit HSV-1 ..................................................................... 45
3.4.5 Langzeitinkubations-Versuche mit Peptiden ......................................................... 46
3.5 Auswertungen ............................................................................................................ 47
4. Ergebnisse ..................................................................................................................... 49
4.1 Design des b12-Antikörper-Mimetikums HAM ............................................................ 49

ii
4.2 Synthese von HAM ..................................................................................................... 52
4.3 Bindung von HAM an HIV-1 gp120 ............................................................................. 54
4.4 Kompetitionsexperimente mit HAM ............................................................................. 58
4.5 Virusneutralisation durch HAM ................................................................................... 60
4.6 HAM: b12-Ähnlichkeit und Optimierungsansätze ........................................................ 62
4.6.1 Einzel-, Doppel- und Triple-Alanin-Varianten........................................................ 63
4.6.2. Scrambled HAM .................................................................................................. 65
4.6.3 Lineare Varianten von HAM ................................................................................. 66
4.6.4 Verkürzte HAM-Varianten .................................................................................... 67
4.7 Langzeitinkubation und Kreuzresistenzexperimente mit HIV-1(NL4-3) ....................... 71
4.7.1 Langzeitinkubation von HIV-1(NL4-3) mit b12 bzw. Peptiden ............................... 72
4.7.2 Kreuzresistenz-Testung im SEAP-Zellinfektionsassay ......................................... 76
4.8 Anwendung des Design-Ansatzes von HAM auf einen anderen Antikörper ................ 78
4.8.1 Design von HAMX ................................................................................................ 78
4.8.2 Synthese von HAMX ............................................................................................ 80
4.8.3 Bindung von HAMX an gp120 .............................................................................. 82
4.8.4 Virusneutralisation durch HAMX........................................................................... 84
4.8.5 Scrambled und lineare Varianten von HAMX ....................................................... 85
4.9 Inhibition der HSV-Infektion durch HAM/HAMX-Peptide ............................................. 86
5. Diskussion und Ausblick............................................................................................... 88
5.1 HAM – ein Mimetikum von b12? ................................................................................. 88
5.2 Optimierung von HAM ................................................................................................ 95
5.3 Peptidische Paratop-Mimetika anderer Antikörper .................................................... 102
5.4 Welche Möglichkeiten eröffnen peptidische Mimetika von Anti-HIV-1-Antikörpern? .. 104
6. Zusammenfassung / Summary ................................................................................... 106
6.1 Zusammenfassung ................................................................................................... 106
6.2 Summary .................................................................................................................. 109
7. Anhang ......................................................................................................................... 112
7.1 Analytik der Peptide .................................................................................................. 112
7.2 Sequenz von gp120(ZM96) .......................................................................................... 120
7.3 Abbildungsverzeichnis .............................................................................................. 121
7.4 Tabellenverzeichnis .................................................................................................. 123
Referenzen ............................................................................................................................ I Danksagung ........................................................................................................................IX

iii
Abkürzungsverzeichnis
A492nm Absorption bei 492 nm
Abb. Abbildung
Ac2O Essigsäureanhydrid
AcOH Essigsäure
AE Absorptionseinheiten
Ahx 6-(Fmoc-amino)hexansäure
AIDS erworb. Immundefizienz-Syndrom (aquired immune deficiency syndrome)
Alloc Allyloxycarbonyl
aq in wässriger Lösung
ART Antiretrovirale Therapie
AS Aminosäure
B β-Alanin
Boc/tBoc tert-Butyloxycarbonyl
BSA bovines Albumin-Serum (bovine serum albumin)
Bzl Benzyl
calc berechnet (calculated)
CCR5 CC-Motiv-Chemokin-Rezeptor 5
CD4 cluster of differentiation 4
CD4i CD4-induziert
cDNA complementary DNA
CDR complementarity determining region
CPP cell penetrating peptide
CXCR4 CXC-Motiv-Chemokin-Rezeptor 4
D diversity
DAD Diodenarray-Detektor
Dap/Dpr 2,3-Diaminopropionsäure
DCM Dichlormethan
DIC N,N'-Diisopropylcarbodiimid
DMEM Dulbecco's Modified Eagle's Medium
DMF Dimethylformamid
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure (desoxyribonucleic acid)

iv
DTNB 5,5′-Dithiobis-2-nitrobenzoesäure
EDTA Ethylendiamintetraacetat
ELISA enzyme-linked immunosorbent assay
env envelope glycoprotein
ESI Elektrospray-Ionisation
Fab fragment antigen binding
FACS fluorescence-activated cell sorting
Fc fragment crystallizable
FI Fusions-Inhibitors
FKS fötales Kalbsserum
Fmoc Fluorenylmethoxycarbonyl
FR framework region
gag group-specific antigen
GFP grün fluoreszierendes Protein (green fluorescent protein)
gp120 Glykoprotein 120 kDa
gp41 Glykoprotein 41 kDa
H schwer (heavy)
h Stunde
HAM human antibody (b12) mimic
HAMX human antibody (X5) mimic
HCV Hepatitis C Virus
HER2 human epidermal growth factor receptor 2
HIV Humanes Immundefizienz-Virus
HOBt 1-Hydroxybenzotriazol
HOS human osteosarcoma
HPLC high performance liquid chromatography
HPW hochreines Wasser (engl. high purity water)
HRP horseradish peroxidase
HSV Herpes Simplex Virus
IC50 mittlere inhibitorische Konzentration
Ig Immunglobulin
INSTI Integrase-Inhibitor
ivDde 6,6-Dimethyl-3-(2-methylpropyl)-4-oxo-4,5,6,7-tetrahydro-1H-indazol
J junction
v
L light
LTR long terminal repeat
mAb monoklonaler Antikörper (monoclonal antibody)
MeCN Acetonitril
min Minute
MPER membrane-proximal external region
MS Massen-Spektrometrie
nef negative regulating factor
NGF Nervenwachstums-Faktor (nerve growth factor)
NNRTI nicht-nukleosidischer Reverse-Transkriptase-Inhibitor
NRTI nukleosidischer Reverse-Trankskriptase-Inhibitor
NS3 nonstructural protein 3 (HCV)
OAll Allyl
OPD o-Phenylendiamin
p Protein
Pbf 2,2,4,6,7-Pentamethyldihydrobenzofuran-5-sulfonyl
PBS Phosphatgepufferte Salzlösung (phosphate buffered saline)
PCR Polymerase-Kettenreaktion (polymerase chain reaction)
PDB protein data bank
PI Protease-Inhibitor
pol polymerase
rev RNA-splicing-regulator
RPMI Roswell Park Memorial Institute
s Sekunde
SEAP sekretierte alkaline Phosphatase (secreted alkaline phosphatase)
SIV simianes Immundefizienz-Virus
SPPS Festphasenpeptidsynthese (solid phase peptide synthesis)
Tab. Tabelle
tat Transaktivator-Protein
tBME tert-Butylmethylether tBu tert-Butyl
TFA Trifluoressigsäure
TIPS Triisopropylsilan
TRIS Tris(hydroxymethyl)-aminomethan
Trt Trityl

vi
V variable Region
v/v Volumen bezogen auf Volumen
vif viral infectivity protein
vpr viral protein r
vpu viral protein u
vpx viral protein x
w/v Gewicht bezogen auf Volumen
WT Wildtyp
z.B. zum Beispiel

- Einleitung -
1
1. Einleitung
1.1 Antikörper und Antikörper-Antigen Interaktion
Antikörper, oder auch Immunglobuline (Igs), sind hoch entwickelte Waffen unseres
Immunsystems gegen Krankheitserreger jeder Art und stellen einen Teil der humoralen, also
der nicht-zellgebundenen, Immunantwort dar. Sie sind, ebenso wie die T-Lymphozyten, Teil
der spezifischen Immunabwehr des Körpers. Sie werden von den B-Zellen produziert und
erkennen nur ein ganz bestimmtes Antigen. Vereinfacht ausgedrückt, werden körperfremde
Antigene über den Antikörper-ähnlichen B-Zell-Rezeptor an eine ruhende B-Zelle gebunden.
Dadurch, und durch zusätzliche Wechselwirkung mit einer antigenspezfischen T-Helferzelle
und dendritischen Zelle, wird die B-Zelle aktiviert und zur klonalen Proliferation angeregt.
Durch die anschließende Differenzierung entwickelt sich die aktivierte B-Zelle entweder zu
einer B-Gedächtniszelle oder zu einer Antikörper sezernierenden Plasmazelle, wobei die
sezernierten Antikörper dieselbe Bindungsspezifität besitzen, wie der antigenbindende
B-Zell-Rezeptor. Die Antigen-spezifischen Antikörper können dann die entsprechenden
Antigene erkennen und markieren, damit sie vom körpereigenen Immunsystem unschädlich
gemacht werden. 1–3
Antikörper bestehen in der Regel aus zwei identischen leichten und zwei identischen
schweren Polypeptidketten, die über Disulfidbrücken miteinander verbunden sind. Die
schweren Ketten bestimmen den Isotyp eines Immunglobulins und werden beim Menschen
in μ, δ, γ, α und ε -Ketten unterteilt. Man unterscheidet folglich fünf Isotypen: IgM, IgD, IgG,
IgA und IgE. IgG-Antikörper sind diejenigen, die am häufigsten im Blut vorkommen und die
gegen Viren und Bakterien gerichtet sind. 4
Ein IgG-Antikörper ist ein Molekül mit einer charakteristischen Y-förmigen Struktur. Er kann
durch proteolytische Spaltung durch das Enzym Papain in zwei identische, antigenbindende
Fragmente (Fab-Fragmente; engl. fragment antigen binding) und in ein drittes Fragment, das
Fc-Fragment (engl. fragment crystallizable), gespalten werden (siehe Abbildung (Abb.) 1) 5.
Ein Fab-Fragment enthält eine vollständige leichte und eine halbe schwere Kette. Die
variable Region wird aus den aminoterminalen Teilen der leichten und der schweren Kette
gebildet und setzt sich aus hypervariablen Bereichen und Gerüstregionen (FR, engl.
framework regions) zusammen 1. Die hypervariablen Bereiche bestimmen die Spezifität für
das Antigen, das gebunden werden kann, und werden deshalb auch als complementarity
determining regions (CDRs) bezeichnet 1 (Abb. 1). Die Gerüstregion dagegen wird durch
konservierte Nukleinsäuresequenzen codiert und legt die räumliche Struktur der
Antigenbindungsstelle fest 6. Das Fc-Fragment ist die konstante Region eines Antikörpers

- Einleitung -
2
und wird ausschließlich durch Anteile der schweren Ketten gebildet 1. Das Fc-Fragment dient
beispielsweise dazu, dass Phagozyten die Antikörperbeladenen Antigene erkennen und
anschließend vernichten können 1,7.
Abbildung 1: Aufbau eines IgG-Antikörpers.
Der erwachsene Mensch hat durch den Kontakt mit zahlreichen körperfremden Antigenen
ca. 10 12 verschiedene Antikörper entwickelt. Die Summe der verschiedenen Antikörper eines
Individuums bezeichnet man als Antikörperrepertoire. Die große Vielfalt der Antikörper
erfordert bei der Biosynthese spezielle Mechanismen, da die Kapazität des Genoms nicht
ausreichen würde, um jeden Antikörper durch ein bestimmtes Gen zu codieren. Auch die
Tatsache, dass die Antigenbindungsstellen durch unterschiedliche Paarungen von leichter
und schwerer Kette gebildet werden, reicht allein nicht als Erklärung für die große Anzahl
verschiedener Antikörperspezifitäten. Ein wichtiger Mechanismus zur Generierung der
Antikörpervielfalt beruht auf der Tatsache, dass die Gene, die die leichte und schwere Kette
codieren, aus verschiedenen Gensegmenten bestehen (siehe Abb. 2). Die Gensegmente,

- Einleitung -
3
die die variable Region der leichten Kette codieren, werden mit variable (V) bzw. joining (J)
bezeichnet. Das V-Segment bestimmt dabei die CDR L1- und CDR L2-Region der
Antigenbindungsstelle. Die CDR L3-Region wird durch das V- und das J-Segment festgelegt.
Die variable Region der schweren Kette wird durch drei Gensegmente codiert. Zusätzlich zu
V und J gibt es hier noch das diversity-Segment (D), das zwischen dem V- und dem
J-Segment liegt (siehe Abb. 2). 4,8
Abbildung 2: Prinzip der somatischen Rekombination und Expression von B-Zell-Rezeptoren. 1
Um die Vielfalt des genetischen Codes zu erhöhen, liegen diese Gensegmente in mehreren
Kopien vor und können aufgrund ähnlicher Desoxyribonukleinsäure(DNA)-Sequenzen in
Familien eingeteilt werden. Bei der Verknüpfung in der B-Zelle kommt es dann zu einer
zufälligen Rekombination von V-, D- und J-Segment (somatische Rekombination, siehe
Abb. 2), wobei an den Verknüpfungstellen eine zufällige Anzahl von Nukleotiden eingefügt
wird, ein weiterer Mechanismus zur Erhöhung der Antikörpervielfalt. Bindet eine B-Zelle an
ihrem B-Zell-Rezeptor ein Antigen, wird sie aktiviert und beginnt zu proliferieren. In dieser
Phase kann man den Prozess der somatischen Hypermutation beobachten. Es kommt dabei
zu einer großen Anzahl von Punktmutationen im gesamten Bereich der variablen Region.
Dies hat zur Folge, dass es auch zu Veränderungen an den antigenbindenden

- Einleitung -
4
B-Zellrezeptoren kommt, so dass einige Mutanten das Antigen mit höherer Affinität binden
als der ursprüngliche B-Zellrezeptor. Diesen Vorgang nennt man auch Affinitätsreifung.
Durch diese Selektion auf stärkere Affinität findet man in den CDRs der schweren Kette eine
hohe Anzahl an Austauschmutationen, während in den Gerüstregionen vorwiegend stille
Mutationen zu finden sind. Auch dieser Mechanismus trägt dazu bei, die Diversität der
Antikörper zu erhöhen. So entsteht die große Vielfalt an Antikörpern, die notwendig ist, um
eine hohe Spezifität und Affinität für alle denkbaren Antigene bereit zu halten. 8,9
Das Gegenstück zu einem Epitop auf einem Antigen bezeichnet man als Paratop eines
Antikörpers, d.h. es umfasst die Teilsequenzen, die die Hauptinteraktion eines Antikörpers
mit seinem Antigen ausmachen. Oft besteht ein Paratop aus den sechs Antikörper-CDRs,
drei der schweren Kette und drei der leichten Ketten, aber nicht immer machen alle CDRs
direkten Kontakt 10. Paratope sind diskontinuierliche Bindungsstellen und werden jeweils
einmal auf den beiden Fab-Fragmenten eines Antikörpers präsentiert.
An der Bindung zwischen Epitop und Paratop sind diverse nicht-kovalente Interaktionen
beteiligt, z.B. hydrophobe und elektrostatische Wechselwirkungen 10–12. Es gibt theoretisch
gegen jedes potentielle Antigen einen Antikörper, der dieses äußerst spezifisch und mit
hoher Affinität erkennt. Immer mehr Protein-, Antikörper- und Komplex-Strukturdaten sind in
den letzten Jahren durch technische Entwicklung verfügbar geworden und erlauben uns ein
tieferes Verständnis der Antikörper-Antigen-Erkennung. Wissenschaftler sind sich
heutzutage allerdings uneinig, ob Antikörper-Antigen-Interaktionen generell als Modelle für
Protein-Protein-Interaktionen genutzt werden können, oder ob sie doch zu speziell
sind 10,13–15. Als Werkzeuge zur Vorhersage von Antikörper-Epitopen werden verschiedene
Methoden verwendet: Röntgenkristallstrukturanalyse, Pepscan, Phage Display,
Mutationsanalyse, Massenspektrometrie, partielle Proteolyse oder die Untersuchung von
exprimierten Antikörper-Fragmenten 10. Auch gibt es bereits computergestützte Ansätze zur
Vorhersage von Epitopen, wobei sich bisher keine Methode als absolut zuverlässig und
eindeutig herausgestellt hat 16. Das Hauptproblem ist, dass es sehr schwer ist, die zum
Epitop beitragenden Reste eindeutig als solche zu identifizieren, da die Gestaltung des
Epitops sehr stark vom entsprechenden Antikörper abhängt. Teile eines Antigens können
auch erst in Anwesenheit eines bestimmten Antikörpers und unter bestimmten Bedingungen
zu Teilen eines Epitops werden 17–19. Dadurch kann es natürlich bei der Epitop-Vorhersage
neben falsch-negativen auch leicht zu falsch-positiven Interpretationen kommen. Daher
macht es Sinn, Epitop-Vorhersagen auf Antigenen nur im Bezug auf einen bestimmten
Antikörper zu machen und keine generellen Aussagen treffen zu wollen. Am Beispiel der
Bindungsstelle für den cluster of differentiation 4 (CD4) auf dem HIV-1 Oberflächenprotein
gp120, an welcher eine der ersten Interaktionen zwischen Virus und Wirtszelle stattfindet,
kann man sehr schön sehen, wie sich auf ein und derselben Bindungsregion auf einem

- Einleitung -
5
Antigen die Epitope verschiedener neutralisierender Antikörper (F105, b12, b13) überlagern.
Die Beteiligung der Reste variiert allerdings von Antikörper zu Antikörper und auch der
CD4-Rezeptor wird von einem individuellen Muster erkannt (Abb. 3) 20.
Abbildung 3: HIV-1 gp120 (grau) mit der entsprechenden Bindungsstelle für CD4, bzw. den Epitopen für die Antikörper F105, b12 und b13 (orange Linie). Hydrophobe Reste von gp120 sind
hervorgehoben (grün). 20
Es ist üblich sich der Identifikation eines Antikörper-Paratops durch die Identifizierung seiner
CDRs zu nähern 10. Kabat et al. führten dazu als erstes einen systematischen Ansatz ein, der
darauf aufbaute, dass die CDRs eines Antikörpers die variabelsten Bereiche im Vergleich mit
anderen Antikörpern sind 21,22. Dadurch wurde eine allgemein gültige Nummerierung der
Reste von Antikörpern geschaffen, die zwar inzwischen erweitert wurde, aber noch heute zur
Orientierung angewendet wird 23. Alternativ wird oft auch die über Jahre hinweg entwickelte
Nummerierung von IgGs nach Chothia et al. benutzt 24,25, welche auf der von Kabat et al.
basiert. Erschwert wird eine korrekte Nummerierung dadurch, dass gerade die CDR-
Bereiche nicht nur in ihrer Sequenz, sondern auch in ihrer Länge stark variieren können.
Daher wurden mit der Zeit bestimmte Zusatz-Chiffren für Insertionen eingeführt, um diese
kenntlich zu machen und trotzdem die allgemein gültige Nummerierung der übrigen Reste
beibehalten zu können. Es hat sich herausgestellt, dass bei vielen Antikörpern vor allem die
CDR H3 eine wichtige Rolle bei der Interaktion mit dem Antigen spielt und auch oft
überdurchschnittlich lang ist 26. Zum Beispiel wurden Rinder-Antikörper gefunden und deren
Kristallstrukturen analysiert, bei denen die CDR H3 sogar länger war als 60 Aminosäuren
(AS) (normal wären nach Kabat 8 AS) 27, wodurch eine korrekte Identifizierung der
CDR-Regionen durch simples Abzählen der Reste nicht möglich war.
In den meisten Fällen interagieren nicht alle Reste der direkt Kontakt machenden CDRs mit
dem Antigen. Neben den CDRs können aber auch FRs entscheidend für die Interaktion mit
dem Antigen sein, entweder durch direkten Kontakt oder durch die richtige konformationelle
Präsentation des Paratops 28,6. MacCallum et al. postulierten 1996 11, dass Aminosäure-
Reste, die direkten Kontakt mit dem Antigen machen, vermehrt im Zentrum des
Interaktionsbereichs (engl. antigen combining site) zu finden sind, während Reste der CDRs,

- Einleitung -
6
die nicht direkt das Antigen kontaktieren, mit anderen Bereichen des Antikörpers
zusammenspielen, die für den Erhalt der richtigen strukturellen Präsentation der
hypervariablen Schleifen verantwortlich sind.
Monoklonale Antikörper werden heutzutage bereits auf verschiedenen Gebieten als
Therapeutika eingesetzt, z. B. zur Behandlung von Autoimmun-, Herz-Kreislauf- oder
Tumorerkrankungen 29. Doch es treten beim Einsatz von Antikörpern verschiedene Probleme
auf, wie eine ungünstigen Pharmakokinetik, geringe Gewebegängigkeit oder eine mindere
Interaktion mit dem Immunsystem 29. Neben den Immunglobulinen und ihren Fab- oder Fc-
Fragmenten wurden deshalb inzwischen diverse Formen von Antikörper-Mimetika entwickelt,
die ebenso wie Antikörper in der Lage sind, Antigene zu binden. Beispiele sind von Protein A
abgeleitete Peptide (Affibodys) 30, von Protease-Inhibtioren des Kunitz-Typs abgeleitete
Domänen-Peptide (Kunitz-Domänenpeptide) 31,32, die Enzym-abgeleiteten Fynomere 32, auf
einem Gerüst einer Fibronectin type III Domäne aufgebaute Monobodys 33, von Lipocalinen
abgeleitet Anticaline 34, aber auch peptidische Paratop-Mimetika von Antikörpern, welche in
großer Zahl als neue Möglichkeiten zur Therapie oder als Werkzeuge für Diagnostik und
Forschung dienen sollen.
1.2 Peptidische Paratop-Mimetika
Während der letzten 25 Jahre wurde bereits von verschiedenen Forschergruppen Literatur
veröffentlicht, die die Nachahmung von Antikörper-Paratopen bzw. deren CDRs durch
synthetische Peptide beschreiben. Dazu zählen Antikörper gegen diverse Antigene, wie zum
Beispiel CD4, HER2-Rezeptor, die NS3-Protease-Domäne von HCV oder auch HIV-1 gp120
(einige Beispiele in Tabelle 1). Das Fazit der meisten Autoren war, dass die erhaltenen
Peptide das entsprechende Antigen tatsächlich erkennen. Auch sind in vielen Fällen
Kompetitions- oder Inhibitionsexperimente durchgeführt worden, die auf eine Interaktion der
Peptide im Bereich des entsprechenden Antikörper-Epitops hinweisen und eine Mimikry der
Antikörper durch die synthetischen Moleküle wahrscheinlich machen. Die Peptide, deren
Inhibitionsvermögen untersucht wurde, werden in der Literatur mit IC50-Werten von ca.
5-20 µM beschrieben, wobei Zyklisierungen zur Einschränkungen der konformationellen
Freiheit der flexiblen Peptide die IC50-Werte in der Regel verbesserten.

- Einleitung -
7
Tabelle 1: Beispiele CDR-abgeleiteter peptidischer Antikörper-Mimetika. 35–42
Die Gruppe um Timmerman et al., publizierte 2009 eine Methode zur Präsentation von CDRs
am Beispiel mehrerer Anti-Gastrin17-Antikörper auf einem synthetischen Gerüst und
untersuchte diese Peptide und Peptid-Varianten hinsichtlich ihrer Bindungsaffinitäten zum
Antigen und ihrer biologischen Aktivität 43,35. Nach der Auswertung der Ergebnisse äußerten
sich die Autoren jedoch kritisch gegenüber der Echtheit der Antikörper-Mimikry durch die
CDR-abgeleiteten Peptide. Vor allem die großen Unterschiede zwischen den Affinitäten von
Antikörpern und Peptiden zu ihrem Antigen und die Importanz der Anwesenheit aromatischer
Reste und einer bestimmten Gesamt-Ladung der Peptide führten die Autoren als Hinweise
für eine eher unspezifische Interaktion zwischen Peptiden und Antigen auf.
Timmermann et al. konnten in ihrem Fall eindeutig zeigen, dass der Antigenbindung der
synthetischen Peptide andere Bindungsmechanismen zugrunde lagen als der der Eltern-
Antikörper. Daher stellten sie in Frage, dass monoklonale Antikörper eine gute Basis sind,
um davon synthetische Peptide als Antigen-Binder bzw. Inhibitoren abzuleiten.
Es ist also noch nicht geklärt, ob es tatsächlich möglich ist, mit CDR-abgeleiteten,
synthetischen Peptiden ein Antikörper-Paratop in seiner Funktion nachzuahmen, d.h.
Affinität, Spezifität und Bindungsmechanismus. Weitere Studien an verschiedenen
Antikörpern sind notwendig, um das Potential von Peptiden auf diesem Gebiet abschätzen
zu können.
1.3 Synthetische Peptide in der Wirkstoffforschung
Arzneistoffe, die aufgrund ihrer Größe nicht mehr zu den small molecules (bis ca. 800 g/mol)
zählen, werden als Biologika bezeichnet und werden in der Regel rekombinant hergestellt.
Dazu zählen vor allem diverse Proteine, wie z.B. Antikörper. Peptidische Arzneistoffe

- Einleitung -
8
werden, wie Proteine, teilweise rekombinant hergestellt, aber ein großer Teil mit Hilfe der
heute zur Verfügung stehenden Methoden auch synthetisch. Der Anteil an peptidischen
Arzneistoffen auf dem weltweiten Markt steigt seit Jahren stetig an 44. Aktuell stehen nahezu
140 neue peptidische Therapeutika in klinischen Studien auf dem Prüfstand 45. Ein sehr
bekannter Vertreter der erfolgreich als Medikament von Hoffmann-La Roche eingesetzten
peptidischen Arzneistoffe ist Enfuvirtid (auch bekannt als T-20 oder Fuzeon), ein 36-mer, das
die Fusion von HIV-1-Partikeln mit den Wirtszellen und somit eine weitere Infektion von
Immunzellen im Patienten verhindert 46,47. Auch zur Behandlung von Diabetes-Patienten
kommen synthetische Peptide zum Einsatz, z. B. Inkretin-Mimetika wie Exenatid (auch
Byetta, von Eli Lilly) oder Liraglutid (auch Victoza, von Novo Nordisk), welche in Kombination
mit anderen Arzneimitteln zur Behandlung von Diabetes-Typ-2 verabreicht werden 44,48.
Ursprünglich war das Interesse an Peptiden in der Wirkstoffforschung eher mäßig, da sie
aufgrund ihrer geringen oralen Bioverfügbarkeit und ihrer Tendenz, schnell metabolisiert zu
werden, nicht besonders gut geeignet schienen. Vor allem die Tatsache, dass Peptide dem
Patienten nicht oral verabreicht werden können, sondern ins Gewebe injiziert werden
müssen, schreckte die Arzneimittelforscher ab. Aufgrund ihrer Hydrophilie und ihrer
Molekülgröße sind Peptide zudem nur unter Umständen in der Lage, physiologische
Barrieren zu überwinden. Auch ihre hohe konformationelle Flexibilität wird oft als Manko
gesehen, da sie leicht zu einer niedrigen Selektivität der peptidischen Binder führen kann.
Auf der Suche nach neuen Produkten und alternativen Therapieformen investierten aber
einige Firmen in der letzten Zeit mehr und mehr in die Peptidforschung 49. Abhängig von ihrer
Größe werden Peptide heutzutage auf verschiedene Weise gewonnen: durch chemische
Synthese, rekombinante DNA-Technologie, in zellfreien Expressionssystemen, transgenen
Pflanzen und Tiere oder auch durch enzymatische Synthese 44. Besonders die chemische
Synthese ermöglicht durch den Einsatz unnatürlicher Aminosäuren und Bausteine, bzw.
durch den Einbau von pseudo-Peptidbindungen, eine große chemische Vielfalt an
Verbindungen, die anders nicht zugänglich wären. Einen besonders großen Fortschritt auf
dem Gebiet brachte die Entwicklung der Festphasenpeptidsynthese (SPPS), die erstmals
1963 von Bruce Merrifield beschrieben wurde 50. Heute sind die automatisierte tBoc/Bzl- und
Fmoc/tBu-SPPS am prominentesten, wobei die Unterschiede vor allem in der Stabilität der
verwendeten Schutzgruppen liegen.
Im Laufe der Jahre wurden unterschiedliche Strategien entwickelt, um die biologische
Aktivität, Spezifität und Stabilität synthetischer Peptide zu verbessern, z.B.
Zyklisierungsstrategien, Vereinfachungen/Verkürzungen nach Alanin-Scans zur Identifikation
der relevanten Reste, Modifikationen des Peptid-Rückgrats, Modifikationen von N- und C-
Terminus, und viele mehr. Mit diesen Werkzeugen kann auch das pharmakokinetische Profil
eines Peptids gezielt beeinflusst und optimiert werden.

- Einleitung -
9
Peptide bieten als Wirkstoffe aber auch viele Vorteile gegenüber anderen Molekülen. Ihre
geringe Größe im Vergleich zu anderen Biologika erlaubt es ihnen deutlich weiter ins
Gewebe vorzudringen und sie können auch leichter sterisch abgeschirmte Zielorte
erreichen. Außerdem sind therapeutische Peptide deutlich weniger immunogen als
rekombinant hergestellte Proteine oder Antikörper 51. Die Kosten zur synthetischen
Generierung von Peptiden im Vergleich zur rekombinanten Produktion werden inzwischen
als geringer diskutiert, Peptide besitzen eine höhere Aktivität pro Masse, sind länger stabil
bei der Lagerung und können leichter in Organe oder Tumoren eindringen 44,52, z.B. in Form
von cell penetrating peptides (CPPs) 53. Im Vergleich mit kleinen organischen Molekülen als
Wirkstoffen weisen Peptide häufig eine höhere Spezifität, Selektivität und Wirksamkeit auf,
da sie meist den funktionellen, kleinen Teil eines Proteins präsentieren, also ihr Vorbild in der
Natur haben 54. Darüber hinaus sind die metabolischen Abbauprodukte von Peptiden in der
Regel Aminosäuren, die für den menschlichen Körper nicht toxisch sind 45. Peptide
akkumulieren aufgrund ihrer kurzen Halbwertszeit nicht im Gewebe oder in Organen,
wodurch die Gefahr von Komplikationen bei der Behandlung verringert wird 55. Desweiteren
sind viele peptidischen Arzneistoffe Agonisten 56 und es bedarf nur einer geringen Menge
davon, um die Zielrezeptoren zu aktivieren 52.
Peptide werden aber auch oft als Inhibitoren von Protein-Protein-Interaktionen entworfen.
Dabei wird die Bindungsstelle eines Proteins, welche sowohl als kontinuierliche als auch
diskontinuierliche Sequenz vorliegen kann, im Peptid nachgeahmt. Ein peptidisches
Mimetikum entsteht 57. Dieser „Ausschnitt“ des Proteins ist dann in der Lage, die Interaktion
zwischen den beiden „Volllängen-Proteinen“ zu stören (Abb. 4). Diese Methode wird auch
gerne zur Untersuchung von Protein-Protein-Interaktionen verwendet, z.B. um
Bindungsmechanismen aufzuklären 58.
Abbildung 4: Wirkungsprinzip peptidischer Mimetika: Die Interaktion zweier Proteine (blau und grün) wird durch ein peptidisches Mimetikum (rot) verhindert (bearbeitet nach 57).

- Einleitung -
10
Für den Entwurf solcher Peptide eignen sich unterschiedliche Ansätze: das rationale,
strukturbasierte Design, für das als Grundlage eine Struktur des Proteins mit seinem
entsprechenden Liganden zur Verfügung stehen muss, oder ein kombinatorischer Ansatz,
bei dem eine Bibliothek aus diversen Molekülen verwendet wird, um einen idealen Inhibitor
zu finden 57. Neben der Vorstellung einer starren Schlüssel-Schloss-artigen Interaktion
zwischen Peptiden und Proteinen, bei welcher die relativ hohe Flexibilität von Peptiden ein
Nachteil sein kann, gibt es auch die Theorie, dass Peptide nach dem Induced-Fit-Prinzip
interagieren 59. Die Induced-Fit-Theorie wurde zunächst für Enzym-Substrat-Interaktionen
beschrieben und besagt, dass die konformationelle Passform (der „fit“) des Enzyms erst
durch die Bindung des Substrats induziert wird 60. Für die Peptid-Protein-Interaktion würde
das direkt übersetzt bedeuten, dass das Protein seine ideale Konformation erst bei Bindung
des Peptids einnimmt. Man geht aber eher davon aus, dass das deutlich flexiblere Peptid bei
Bindung an das Zielmolekül eine optimale Konformation einnimmt, oder dass bei beiden
Bindungspartnern, Peptid und Protein, konformationelle Umlagerungen induziert
werden 59,61,62.
Zu den inhibitorischen mimetischen Peptiden zählt auch der bereits oben erwähnte HIV-1-
Fusions-Inhibitor Enfuvirtid. Im Fall von Enfuvirtid findet die zu unterdrückende Interaktion
allerdings nicht zwischen zwei verschiedenen Proteinen sondern zwischen zwei
Proteindomänen desselben Proteins (gp41) statt 63. Das HI-Virus hat sich seit den 1980er
Jahren zu einer Pandemie entwickelt und ist nach wie vor ein weltweites Problem, vor allem
in ärmeren Ländern, in denen teure, moderne Therapie-Formen für die Patienten nicht
zugänglich sind.
1.4 Humanes Immundefizienz-Virus (HIV)
Das Humane Immundefizienz-Virus ist der Erreger der tödlich verlaufenden Krankheit
Acquired Immunedeficiency Syndrome (AIDS), die 1981 zum ersten Mal geschildert wurde.
Zwei Jahre später wurde der Erreger der Erkrankung im Labor von Luc Montagnier
identifiziert und beschrieben 64. Das HI-Virus verbreitete sich rund um die Welt, derzeit gibt
es ca. 35 Millionen Infizierte (WHO). Jeden Tag infizieren sich etwa 7000 Menschen neu und
nach Schätzungen der Vereinten Nationen starben bislang mehr als 30 Millionen Menschen
an AIDS, alleine 2014 starben 1,2 Millionen Menschen an der Krankheit 65. Bis heute, mehr
als 30 Jahre nach der Entdeckung des Virus, gibt es keinen Impfstoff, der der hohen Zahl an

- Einleitung -
11
Neuinfektionen entgegen wirken könnte. Es gibt verschiedene Typen des HI-Virus, HIV-1
und HIV-2, wobei in Europa vor allem der Typus HIV-1 verbreitet ist 66.
Das Virus zählt zu den Retroviren, einer Gruppe der Familie der Lentiviren. Die Viruspartikel
haben einen Durchmesser von ca. 100 bis 150 nm und sind von einer Lipoproteinhülle
umgeben, die von der Wirtszelle stammt. In diese Virusmembran sind die Hüllproteine gp41
eingelagert, das äußere Hüll-Glykoprotein 120 (gp120) ist nicht kovalent an das Hüll-
Glykoprotein 41 (gp41) gebunden. Die Membran eines reifen HI-Viruspartikels ist mit
p17-Matrixproteinen ausgekleidet. Das kegelförmige Kapsid besteht aus dem
p24-Core-Antigen und enthält das diploide virale RNA-Genom welches mit dem
Nukleoprotein, den Enzymen Reverse Transkriptase und Protease und einer speziellen
tRNA, die als Primer bei der reversen Transkriptase dient, assoziiert ist. Das HIV-Genom
besteht aus zwei Kopien einer einzelsträngigen +Strang-RNA (+ssRNA) von je ca.
9,4 (HIV-1) bzw. 9,7 (HIV-2) Kilobasen. 67
Abbildung 5: Aufbau eines HI-Viruspartikels (bearbeitet nach 68).
Das Genom von HIV-1 ist im Vergleich zu anderen Retroviren ungewöhnlich komplex
aufgebaut. Vom 5‘- zum 3‘-Ende seines Genoms findet man die Erbinformation für die
Proteine Gag (group-specific antigen), Pol (polymerase) und Env (envelope glycoprotein),
aus denen die wichtigsten Proteine zum Aufbau und zur Replikation der Viruspartikel
entstehen 68. Zusätzlich kodiert HIV-1 eine Anzahl an regulatorischen und akzessorischen

- Einleitung -
12
Proteinen: Tat, Rev, Vpu, Vif, Vpr und Nef 68. Tabelle 2 zeigt eine Übersicht der HIV-Proteine
und ihre entsprechenden Funktionen.
Die Replikation des Virus läuft in einem schrittweisen Prozess ab. Zunächst bindet das Virus
mit seinem Hüllprotein gp120 an den zellulären Rezeptor CD4 69, welcher als erster
retroviraler Rezeptor identifiziert wurde 70. Dadurch wird eine konformationelle Umlagerung in
gp120 ausgelöst und die Corezeptor-Bindungsstelle freigelegt 71. Somit kann das Virus, je
nach Corezeptor-Tropismus, im zweiten Schritt unter Zuhilfenahme der V3-Schleife an einen
der transmembranären Corezeptoren der Wirtszelle, CXC-Motiv-Chemokin-Rezeptor 4
(CXCR4) oder CC-Motiv-Chemokin-Rezeptor 5 (CCR5), binden 72–74. Fusionspeptide aus
gp41 dringen dann in die Zellmembran ein und es kommt zur Bildung sogenannter „6-Helix-
Bündel“, welche die Virusmembran dicht an die Zellmembran heranziehen und die Fusion
von Zelle und Viruspartikel ermöglichen (siehe Abb. 6) 75.
Tabelle 2: HIV-Proteine und ihr Funktion, im roten Kasten hervorgehoben die Proteine des viralen Spikes. 76
Nach der Fusion wird das Kapsid in das Zytoplasma der Wirtszelle entlassen und es findet
das uncoating statt, d.h. das Kapsid wird entfernt, das virale Genom wird von der reversen

- Einleitung -
13
Transkriptase in komplementäre DNA (cDNA) umgeschrieben, und diese anschließend in
den Zellkern transportiert und dort in das Genom der Wirtszelle integriert 77.
Abbildung 6: Das Andocken eines HIV-Partikels an eine Wirtszelle bis zur Fusion ist ein mehrstufiger Prozess (bearbeitet nach 75).
Danach kommt es zur Expression der viralen Proteine, welche nach komplexen
Transkriptions- und Splicing-Abläufen fertig sind für den Zusammenbau vieler neuer Viren an
der Membran der Wirtzelle 78. Der letzte Schritt im Prozess der HIV-Replikation ist die
Knospung, d.h. die neu gebildeten Viruspartikel werden nach dem Ausstülpen der
Wirtszellmembran an dieser Stelle aus der Zelle entlassen 78. Wenn die abschließenden
Reifungsprozesse im inneren der Partikel abgeschlossen sind, können sie andere
CD4-tragende Zelle befallen und somit die Schwächung des Immunsystems des Patienten
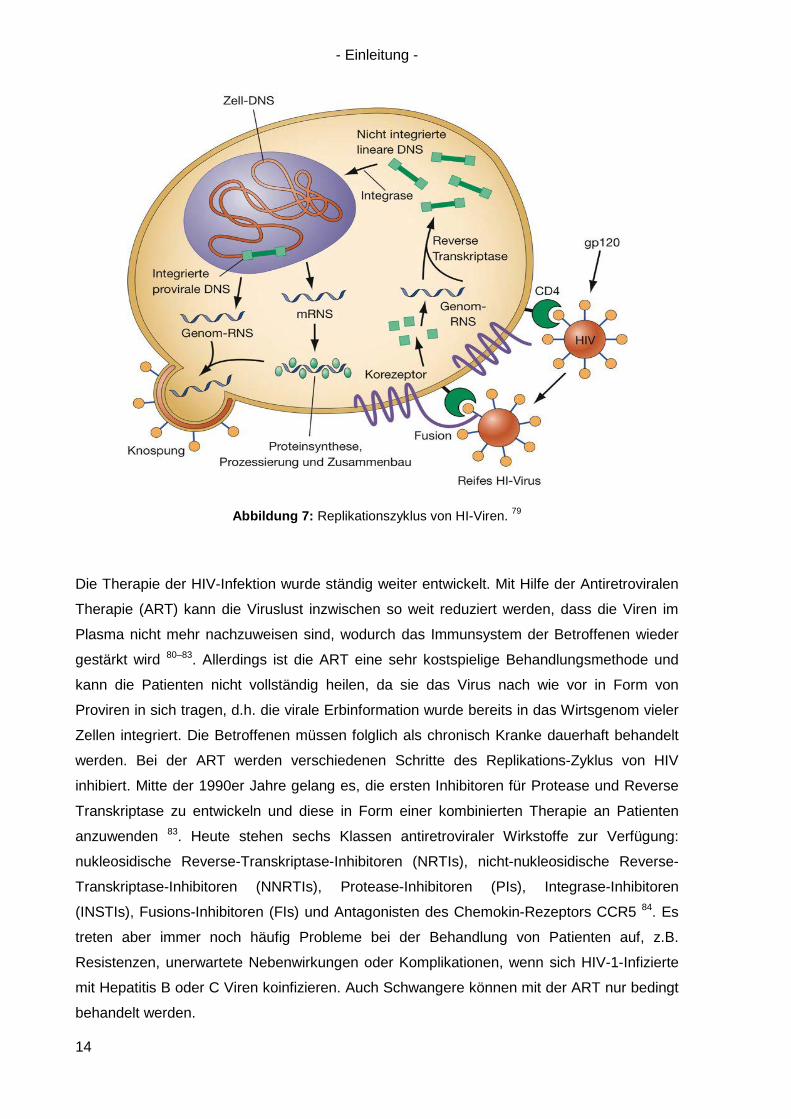
voran treiben. Ein vollständiger HIV-1 Replikationsszyklus ist in Abb. 7 dargestellt.
- Einleitung -
14
Abbildung 7: Replikationszyklus von HI-Viren. 79
Die Therapie der HIV-Infektion wurde ständig weiter entwickelt. Mit Hilfe der Antiretroviralen
Therapie (ART) kann die Viruslust inzwischen so weit reduziert werden, dass die Viren im
Plasma nicht mehr nachzuweisen sind, wodurch das Immunsystem der Betroffenen wieder
gestärkt wird 80–83. Allerdings ist die ART eine sehr kostspielige Behandlungsmethode und
kann die Patienten nicht vollständig heilen, da sie das Virus nach wie vor in Form von
Proviren in sich tragen, d.h. die virale Erbinformation wurde bereits in das Wirtsgenom vieler
Zellen integriert. Die Betroffenen müssen folglich als chronisch Kranke dauerhaft behandelt
werden. Bei der ART werden verschiedenen Schritte des Replikations-Zyklus von HIV
inhibiert. Mitte der 1990er Jahre gelang es, die ersten Inhibitoren für Protease und Reverse
Transkriptase zu entwickeln und diese in Form einer kombinierten Therapie an Patienten
anzuwenden 83. Heute stehen sechs Klassen antiretroviraler Wirkstoffe zur Verfügung:
nukleosidische Reverse-Transkriptase-Inhibitoren (NRTIs), nicht-nukleosidische Reverse-
Transkriptase-Inhibitoren (NNRTIs), Protease-Inhibitoren (PIs), Integrase-Inhibitoren
(INSTIs), Fusions-Inhibitoren (FIs) und Antagonisten des Chemokin-Rezeptors CCR5 84. Es
treten aber immer noch häufig Probleme bei der Behandlung von Patienten auf, z.B.
Resistenzen, unerwartete Nebenwirkungen oder Komplikationen, wenn sich HIV-1-Infizierte
mit Hepatitis B oder C Viren koinfizieren. Auch Schwangere können mit der ART nur bedingt
behandelt werden.

- Einleitung -
15
Zur Verhinderung des initialen Schritts des Replikationszyklus von HI-Viren, der Anheftung
an die Wirtszelle (Abb. 6) mittels CD4-Bindung, wurde bisher kein Wirkstoff gefunden, der
die klinischen Studien überstand 85. Die Anlagerung an die Wirtszelle kann jedoch
theoretisch von Anti-HIV-1-Antikörpern unterbunden werden. Das Antigen solcher Antikörper
ist das bereits oben erwähnte virale Hüllprotein Env 86,87, welches aus einem Trimer aus
Heterodimeren, zusammengesetzt aus den Glykoproteinen gp120 und gp41, besteht 88. Es
wird in seiner Gesamtheit auch als viraler Spike bezeichnet.
1.5 Neutralisierende Antikörper gegen HIV-1
HIV-1 wendet verschiedene, äußerst effiziente Mechanismen an, um den viralen Spike vor
einem Angriff durch das humane Immunsystem zu schützen. So werden die Hüllproteine
durch ein Schild aus Zuckerresten geschützt (Abb. 8), hoch variable immundominante
Schleifen und konformationelle Maskierung erschweren den Zugang zu den viralen
Schlüsselepitopen 87,89,90. Alle durch potentielle Vakzine induzierten Antikörper waren bisher
nicht in der Lage, die meisten der sich im Umlauf befindenden primären HIV-Isolate zu
neutralisieren 91–93.
Um die Interaktion zwischen Virus und menschlichem Immunsystem besser zu verstehen,
wird intensive Forschung betrieben. Ein Weg, um an Informationen zu gelangen, ist die
Isolierung und Untersuchung des Bindungsverhalten von breit neutralisierenden Anti-HIV-1
Antikörpern 94. Diese Antikörper werden aus den Seren von HIV-positiven Individuen
gewonnen, frühestens 2 Jahre nach deren Infektion. Man geht bislang zwar davon aus, dass
diese Antikörper den entsprechenden Patienten nur einen kleinen Vorteil im Kampf gegen
das Virus verschaffen 95,96, aber in Tiermodellen konnte mit der Verabreichung dieser
Antikörper eine Infektion mit HIV komplett verhindert werden 97,98. Daher sind diese
Antikörper überaus interessant für die HIV-Forschung.
Es sind bisher fünf Regionen des viralen Spikes bekannt, die von neutralisierenden
Antikörpern erkannt werden: die CD4-Bindungsstelle auf gp120 (erkannt durch z.B. b12,
VRC01, HJ16), die variablen Regionen V1, V2 und V3 von gp120 (erkannt durch z.B. PG9,
PG16), bestimmte Glykanreste auf Env (erkannt durch z.B. 2G12) und die membrane-
proximal external region (MPER) von gp41 (erkannt durch z.B. 2F5, 4E10, Z13e1) (siehe
Abb. 8) 99. Außerdem gibt es auch Antikörper, die die Corezeptor-Bindungsstelle auf gp120
erkennen (z.B. 17b, X5). Da diese Epitope aber erst freigelegt und somit vollständig
zugänglich werden, nachdem CD4 gebunden hat, nennt man diese Antikörper
CD4-induzierte (CD4i)-Antikörper 100–102.

- Einleitung -
16
Abbildung 8: HIV-1 neutralisierende Antikörper und ihre Angriffspunkte auf dem viralen Spike, Glykosylierungen in blau/grün. 103
Außerdem publizierten Kwong et al. 2014 die Entdeckung eines Antikörpers, der besonders
potent und breit HIV-1-Isolate neutralisiert und der ein bisher nicht beschriebenes,
konserviertes Epitop von Env erkennt, welches sich sowohl über gp120 als auch gp41
erstreckt 104. Der Antikörper trägt die Bezeichnung „35O22“ und zeigt, dass es
möglicherweise noch andere, unentdeckte Angriffspunkte auf gp120 bzw. für Immunogen-
Design geeignete Bereiche auf dem viralen Spike gibt. Eine andere aktuelle Veröffentlichung
von Caskey et al. 2015 macht Hoffnung darauf, dass die Immuntherapie mit Anti-HIV-1-
Antikörpern im Menschen bald als zusätzliche Therapieform genutzt werden kann. Bisher
war diese Behandlungsmethode immer in den vorklinischen oder klinischen Studien
gescheitert 105,106. Neue Entwicklungen auf dem Gebiet der Antikörper-Klonierung durch
Kultivierung einzelner B-Zellen ermöglichen seit kurzem die Generierung deutlich
wirksamerer Antikörper, einer von ihnen ist 3BNC117, ein CD4-Bindungsstellen-Antikörper 107. Die Ergebnisse der ersten klinischen Studien zeigen, dass die Viruslast von infizierten
Patienten durch eine einmalige Gabe des Antikörpers über 4 Wochen hinweg deutlich

- Einleitung -
17
gesenkt werden konnte. Außerdem wurde die Behandlung von allen teilnehmenden
Patienten relativ gut vertragen und das Präparat zeigte eine gute Pharmakokinetik 107.
Einen anderen möglichen Weg, um Individuen durch Antikörper vor einer Infektion mit HIV-1
zu schützen, beschrieben Johnson et al. 2009 108: sie brachten mit Hilfe der Gentransfer-
Technologie generierte Adeno-assoziierte Vektoren in Muskelzellen von Affen ein, wodurch
diese Antikörper oder Antikörper-ähnliche Immunoadhesine mit einer vorbestimmten
Spezifität für SIV produzierten. Somit wurde das adaptive Immunsystem in den Tieren
überbückt und man fand in den Seren der ansonsten nativen Affen eine langfristig generierte
biologische Aktivität gegen SIV. Sechs der neun Tiere waren nach der Behandlung immun
gegen SIV, wohingegen alle nicht immunisierten Tiere vom Virus infiziert wurden. Diese
Form der Therapie bringt allerdings einen entscheidenden Nachteil: die Vektoren, welche die
entsprechende Information zur Expression der schützenden Antikörper tragen, wurden hier
irreversibel in die Zellen der Tiere eingebracht. Die Auswirkungen einer permanenten, nicht
regulierbaren Expression von körperfremden Molekülen und die dauerhafte Anwesenheit der
Vektoren auf das natürliche Immunsystem oder auch andere Körperfunktionen können nur
schwer abgeschätzt werden und bergen unter Umständen große Risiken im weiteren Verlauf
des Lebens dieser Tiere 109.
1.6 Kristallstrukturen von Fab b12 und Fab X5 in Komplex mit gp120
Einer der bereits am längsten bekannten Vertreter der breit neutralisierenden Anti-HIV-1-
Antikörper ist b12. Er wurde in Form eines Fab-Fragmentes 1991 aus dem Knochenmark
eines Infizierten isoliert, bei dem auch nach Jahren keine Symptome auftraten 110. Der größte
Unterschied zwischen b12 und anderen bekannten CD4-Bindungsstellen-Antikörpern war,
dass er als einziger nicht nur das monomere gp120 erkannte, sondern auch seine native
trimere Form und dadurch in der Lage war, Viruspartikel zu neutralisieren 88. IgG b12 ist
passiv verabreicht sogar in der Lage, Makaken vor einer HIV-Infektion zu schützen 111. Der
Antikörper zeichnet sich durch eine hohe Affinität zu seinem Antigen gp120 aus 110 und er
war einer der ersten breit neutralisierende Antikörper (75% der damals getesteten
HIV-Isolate) 112, dessen Bindungseigenschaften sehr detailliert charakterisiert wurden 113. Er
adressiert die CD4-Bindungsstelle auf gp120 112 und verhindert durch Bindung daran den
ersten Schritt der HIV-1-Infektion CD4-tragender Zellen.
2001 wurde die Kristallstruktur von b12 publiziert und man postulierte, dass vor allem das
Tryptophan an der Spitze des aus dem Arm des Antikörpers herausragenden CDR H3
(18 AS) eine wichtige Rolle spielt. Man ging anhand eines Docking Models von b12 mit

- Einleitung -
18
Kern-gp120 (core gp120) 114 davon aus, dass der Indol-Rest in die hydrophobe
Bindungstasche der CD4-Bindungsstelle auf gp120 hineingreift, ähnlich dem prominenten
Phenylalanin 43 von CD4 115. Es wurden daraufhin ausgiebige Mutationsstudien, sowohl auf
Antikörper- als auch auf Antigen(gp120)-Seite, durchgeführt, um das Epitop von b12 und die
an der Interaktion beteiligten Reste möglichst detailliert zu beschrieben 116,113. Die
Ergebnisse zeigten, dass vor allem die CDR H3-Sequenz, das Tyrosin (Y53) an der Spitze
der CDR H2 und die CDR L1-Sequenz entscheidend zur b12-gp120 Bindung beitragen 113.
Abbildung 9: Kristallstruktur (PDB: 2NY7) von b12 (Fab) (grau) in Komplex mit HIV-1 gp120 (gelb), farbig hervorgehoben sind die CDR Hs von b12 CDR H3 (rot), CDR H2 (blau) und CDR H1 (grün). 117
2007 war es Zhou et al. endlich gelungen, b12 (Fab) in Komplex mit gp120 zu kristallisieren.
Zum Erhalt der bis heute einzigen verfügbaren Komplex-Kristallstruktur dieses Antikörpers
mit gp120 musste die hohe konformationelle Flexibilität von HIV-1 gp120 durch
Verkürzungen und Stabilisierungen deutlich eingeschränkt werden, um überhaupt Komplex-
Kristalle zu erhalten 117. Die Struktur zeigt, dass b12, anders als vorher angenommen, nur
mit den CDRs seiner schweren Kette direkt mit dem Kern-gp120 interagiert. Die am
nächsten gelegene leichte Kette liegt 10 Å von gp120 entfernt. Außerdem ist zu sehen, dass
der CDR H3 über die CD4-Bindungsstelle greift um mit dem Tryptophan an seiner Spitze den
Stamm der V4-Schleife von gp120 zu kontaktieren, also nicht die gleiche Tasche adressiert
wird wie von CD4 118. Drei Reste, einer in jeder der drei CDR Hs, machen zusammen 40 %
der Kontaktfläche von b12 mit gp120 aus. Diese drei Reste sind Asparagin 31 (CDR H1),
und die bereits vorher als wichtig erachteten Positionen Tyrosin 53 (CDR H2) und

- Einleitung -
19
Tryptophan 100 (CDR H3), welche an den Spitzen der beiden CDR-Schleifen aus der
Struktur heraus zeigen in Richtung gp120 117, siehe Abb. 10.
Abbildung 10: CDR Hs (rot, blau, grün) von b12 mit den drei Hauptinteraktionsaminosäuren (magenta) 117; Ausschnitt aus der Kristallstruktur in Komplex mit gp120 (PDB: 2NY7).
Aus vergleichenden Studien mit CD4 und VRC01, einem 2010 erstmals beschriebenen, hoch
potenten CD4-Bindungsstellen-Antikörper 119, lässt sich inzwischen aus thermodynamischen
Daten ableiten, dass b12, im Gegensatz zu CD4 und VRC01, bei Bindung an gp120 kaum
konformationelle Änderungen auslöst und dass die Corezeptor-Bindungsstelle dann nicht
vollständig freigelegt wird 120,89,117. Unterschiedliche Antikörper binden folglich an der
gleichen Bindungsstelle, aber an unterschiedlichen Epitopen und lösen unterschiedliche
Effekte beim Antigen (HIV-1 gp120) aus.
Die Eigenschaften b12-resistenter HIV-1-Stämme und die Bedeutung von Resistenzen
gegen neutralisierende Antikörper für die Vakzin-Forschung wurden bereits ausführlich
untersucht 121. 2010 beschrieben Bunnik et al. den Verlauf von HIV-1-Infektionen in vivo mit
Blick auf die Entstehung von b12-resistenten Spezies, welche vor allem erst in einem späten
Stadium der Infektion auftraten, aber bei fast der Hälfte der beobachteten Individuen 122. Sie
postulierten, dass Resistenzbildungen gegen CD4-Bindungsstellen-Antikörper erst dann
auftreten, wenn es nur noch wenige Wirtzellen (CD4-tragende Zellen) gibt und die virale
Replikationskinetik sich beschleunigt 122. Darüber hinaus konnte kein direkter
Zusammenhang zwischen der Anwesenheit von CD4-Bindungsstellen-erkennenden
Antikörpern im Serum der Patienten und einer b12-Resistenzbildung gefunden werden 122.
Daraus lässt sich ableiten, dass eine Medikation mit Antikörpern oder Antikörper-

- Einleitung -
20
abgeleiteten Peptiden, zumindest in einem früheren Stadium der Infektion, vermutlich nicht
den Nebeneffekt einer besonders schnellen Resistenzbildung hätte.
Ein weiterer breit neutralisierender Anti-HIV-1-Antikörper, von dem eine Kristallstruktur in
Komplex mit gp120 zur Verfügung steht, ist X5 102. Wie bereits beschrieben ist er ein
CD4i-Antikörper und adressiert den Bereich um die Corezeptor-Bindungsstelle auf gp120 123.
Es konnte gezeigt werden, dass im Fall von X5, anders als bei anderen Antikörpern, das
Fab-Fragment deutlich potenter ist als das komplette IgG. Dies ist vermutlich sterischen
Einschränkungen beim Zugang zu seinem Epitop auf gp120 während der Infektion
zuzuschreiben 124. Da X5 erst nach CD4-Bindung Zugang zu seinem Epitop hat, ist der
Raum zwischen Virus und Wirtszelle bereits begrenzt und kleinere Moleküle haben einen
deutlichen Vorteil. Die Kristallstruktur von X5 (Fab) in Komplex mit gp120 und CD4 ist in
Abb. 11 gezeigt.
Abbildung 11: Kristallstruktur (PDB:2B4C) von X5 (Fab) (grau) in Komplex mit HIV-1 gp120 (gelb) und CD4 (violett), farbig hervorgehoben sind die CDR Hs von X5 CDR H3 (rot), CDR H2 (blau) und
CDR H1 (grün). 102
Auch X5 besitzt, ebenso wie b12, eine vergleichsweise lange CDR H3 (22 AS), welche
hervorragt und an der Spitze eine Art Haken bildet, durch den vermutlich die hohe Affinität zu
gp120 zu Stande kommt 102. Leider ist der Bereich an der Spitze der CDR H3 in der
Kristallstruktur nicht vollständig aufgelöst, jedoch sind zwei aromatische Reste zu erkennen,
- Einleitung -
21
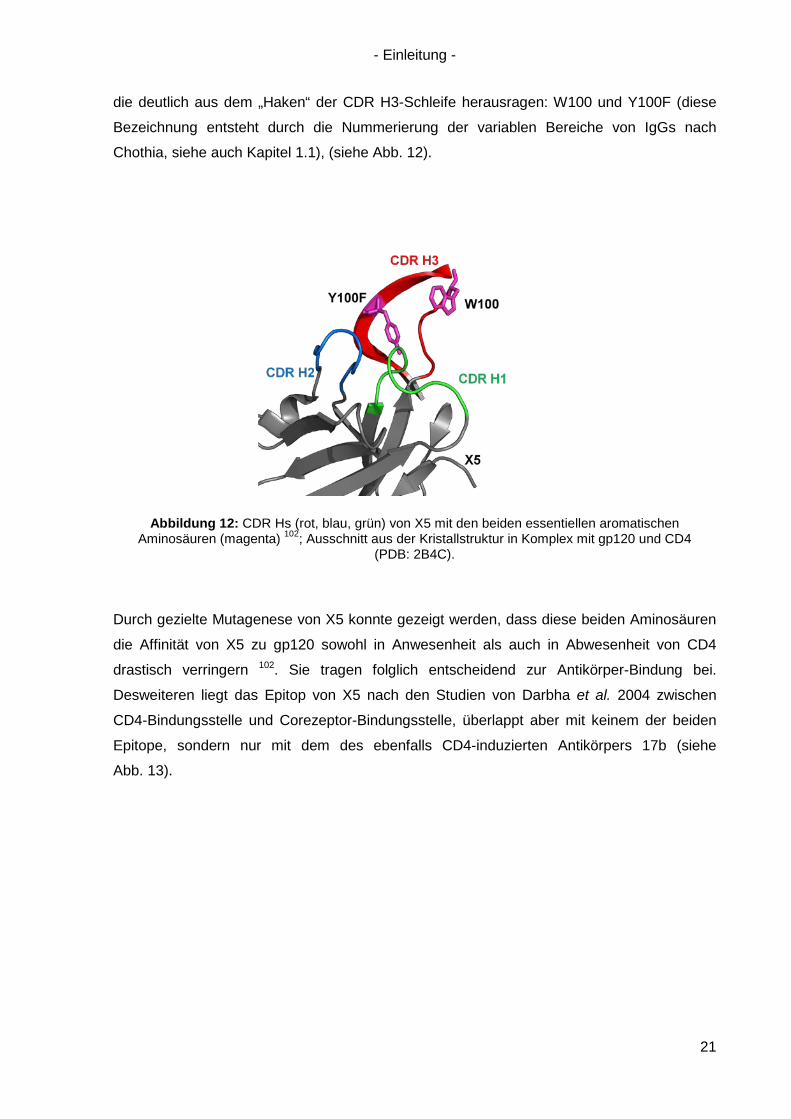
die deutlich aus dem „Haken“ der CDR H3-Schleife herausragen: W100 und Y100F (diese
Bezeichnung entsteht durch die Nummerierung der variablen Bereiche von IgGs nach
Chothia, siehe auch Kapitel 1.1), (siehe Abb. 12).
Abbildung 12: CDR Hs (rot, blau, grün) von X5 mit den beiden essentiellen aromatischen Aminosäuren (magenta) 102; Ausschnitt aus der Kristallstruktur in Komplex mit gp120 und CD4
(PDB: 2B4C).
Durch gezielte Mutagenese von X5 konnte gezeigt werden, dass diese beiden Aminosäuren
die Affinität von X5 zu gp120 sowohl in Anwesenheit als auch in Abwesenheit von CD4
drastisch verringern 102. Sie tragen folglich entscheidend zur Antikörper-Bindung bei.
Desweiteren liegt das Epitop von X5 nach den Studien von Darbha et al. 2004 zwischen
CD4-Bindungsstelle und Corezeptor-Bindungsstelle, überlappt aber mit keinem der beiden
Epitope, sondern nur mit dem des ebenfalls CD4-induzierten Antikörpers 17b (siehe
Abb. 13).

- Einleitung -
22
Abbildung 13: „Fußabdrücke“ von X5, 17b, CCR5 und CD4 (rot) auf HIV-1 gp120 (grau). 102
Die Autoren der Publikation postulierten bereits damals, dass sich der X5-Antikörper, im
Besonderen sein langer CDR H3, möglicherweise sehr gut zum Ableiten kleiner HIV-
Inhibitor-Moleküle eignen könnte. Bisher sind aber keine von X5 abgeleiteten Inhibitoren
beschrieben worden.

- Zielsetzung -
23
2. Zielsetzung
Das Ziel meiner Arbeit war es, peptidische Antikörper-Mimetika von Anti-HIV-1-Antikörpern
zu synthetisieren, zu charakterisieren, und mit den erworbenen Kenntnissen zu optimieren.
Als Eltern-Antikörper wurde zunächst b12 ausgewählt, ein breit HIV-1 neutralisierender
Antikörper, der auf molekularer Ebene ausführlich charakterisiert wurde und von dem eine
Kristallstruktur im Komplex mit seinem Antigen zur Verfügung steht. b12 erkennt die
Bindungsstelle für den humanen Zellrezeptor CD4 auf dem Hüllprotein gp120 von HIV-1 und
verhindert durch Bindung an gp120 die erste Interaktion von Viruspartikel und Wirtszelle,
bevor es zu einer Infektion der Zelle kommt.
Das konstruierte Leitpeptid sollte strukturbasiert vom Paratop des b12-Antikörpers, den drei
CDRs seiner schweren Kette, abgeleitet werden und zyklisiert werden, um die
schleifenähnliche Struktur der CDRs, wie sie in der Kristallstruktur des Antikörpers zu
erkennen ist, nachzuahmen. Mit den in dieser Arbeit durchgeführten Experimenten sollte
zum einen untersucht werden, inwieweit das Peptid sich vergleichbar zu seinem Eltern-
Antikörper b12 verhält. Zum anderen sollte untersucht werden, welche Teile des Konstrukts
für seine biologische Aktivität notwendig sind, bzw. inwieweit sie die Antikörper-ähnliche
Interaktion des Peptids mit dem Antigen gp120 beeinflussen.
Zusätzlich sollte der gewählte Ansatz auf einen weiteren Anti-HIV-1-Antikörper angewendet
werden, der nicht die CD4-Bindungsstelle, sondern die Corezeptor-Bindungsstelle auf gp120
erkennt. Damit sollte eine Grundlage zur Herstellung peptidischer Hybridmoleküle
geschaffen werden, die in der Lage sind, gleichzeitig die beiden wichtigsten Angriffsstellen
des HI-Virus bei einer Infektion der Wirtszelle zu blockieren.
Die Mimikry von Antikörper-Paratopen durch Peptide kann einerseits neue Möglichkeiten auf
dem Gebiet der HIV-Forschung und -Therapie eröffnen. Andererseits sollen die in dieser
Arbeit gewonnenen Erkenntnisse dazu beitragen, mehr über das Potential von Peptiden als
Antikörper-Paratop-Mimetika zu erfahren.

- Material und Methoden -
24
3. Material und Methoden 3.1 Material
3.1.1 Geräte
Die folgenden Geräte und die entsprechende Software kamen bei der Durchführung der
beschriebenen Methoden zum Einsatz.
Gerät Hersteller
Analytische HPLC/MS
Agilent Series 1100 HP
Degasser, Binäre Pumpe; Säulenofen, DAD
Autosampler Series 200 Perkin Elmer
Kinetex C18 Säule, 2,6 µ, 100 Å, 50 mm x 2,1 mm Phenomenex
Massenspektrometer API2000 mit ESI-Quelle AppliedBiosystems
CO2-Inkubator CB150 Binder
FACS: LSR II Durchflusszytometer BD
Gefrierschrank Innova ULT laboratory freezer (-80 °C) New Brunswick Scientific
Gerfriertrocknungsanlage ALPHA 1-4 Christ
mit Hybridvakuumpumpe RC6 Vacuubrand
Konfokalmikroskop LSM 710 Zeiss
Luminometer Orion Microplate Luminometer Titertek-Berthold
Mikroskop Leica Leitz DM IL Leica Microsystems
Millipore Synergy 185 Reinstwasseranlage (Simpak 2-Kartusche) Millipore
Neubauer improved Zählkammer Brand
pH-Meter Seven Easy mit InLab Micro Pro-Elektrode Mettler Toledo
Photometer (Fluorophotometer)
Plattenlesegerät Infinite Reader F200 Tecan
NanoPhotometer Implen
Pipetten

- Material und Methoden -
25
Einkanalpipetten Gilson
12-Kanal Pipetten Eppendorf
Präparative HPLCs
4 Channel Stand Alone Vacuum Degasser Rheodyne
Pumpe L-6200 Intelligent Pump Merck Hitachi
Detektor L-4250 UV/Vis-Detektor (Anlage 1) Merck Hitachi
Variable Wavelength Detektor (Anlage 2) Knauer
Fraktionssammler Super Frac GE Healthcare
Kinetex C18 Säule, 5 µm, 100 Å, 100 x 21,2 mm (Anl. 1) Phenomenex
Reprosil C18 Säule, 5 µm, 100 Å, 250 x 25 mm (Anl. 2) Dr. Maisch
Schüttler
Titramax 101 Heidolph
VTX-3000L LMS Harmony
PS M3D Grant Instruments
Sterilbank LaminAir HB2448 Heraeus
Syntheseroboter Syro I Multisyntech
Transmissions-Elektronenmikroskop EM10 Zeiss
Waage CPA225D Sartorius
Zetasizer Nano ZS ZEN3600 Malvern Instruments
3.1.2 Chemikalien
Standard-Syntesebausteine der verwendeten proteinogenen Aminosäuren mit für die
Festphasenpeptidsynthese geeigneten Fmoc- und Seitenketten-Schutzgruppen:
Synthesebaustein Hersteller Aminosäure 3- / 1-Buchstabencode
Fmoc-L-Ala-OH Iris Biotech L-Alanin Ala A
Fmoc-L-Cys(Trt)-OH Iris Biotech L-Cystein Cys C
Fmoc-L-Asp(OtBu)-OH Iris Biotech L-Asparaginsäure Asp D

- Material und Methoden -
26
Fmoc-L-Glu(OtBu)-OH * H2O Iris Biotech L-Glutaminsäure Glu E
Fmoc-L-Phe-OH Iris Biotech L-Phenylalanin Phe F
Fmoc-Gly-OH Iris Biotech Glycin Gly G
Fmoc-L-His(Trt)-OH Iris Biotech L-Histidin His H
Fmoc-L-Ile-OH Iris Biotech L-Isoleucin Ile I
Fmoc-L-Lys(Boc)-OH Iris Biotech L-Lysin Lys K
Fmoc-L-Leu-OH Iris Biotech L-Leucin Leu L
Fmoc-L-Met-OH Iris Biotech L-Methionin Met M
Fmoc-L-Asn(Trt)-OH Iris Biotech L-Asparagin Asn N
Fmoc-L-Pro-OH Iris Biotech L-Prolin Pro P
Fmoc-L-Gln(Trt)-OH Iris Biotech L-Glutamin Gln Q
Fmoc-L-Arg(Pbf)-OH Iris Biotech L-Arginin Arg R
Fmoc-L-Ser(tBu)-OH Iris Biotech L-Serin Ser S
Fmoc-L-Thr(tBu)-OH Iris Biotech L-Threonin Thr T
Fmoc-L-Val-OH Iris Biotech L-Valin Val V
Fmoc-L-Trp(Boc)-OH Iris Biotech L-Tryptophan Trp W
Fmoc-L-Tyr(tBu)-OH Iris Biotech L-Tyrosin Tyr Y
Besondere Synthesebausteine und Platzhalteraminosäuren:
Synthesebaustein Hersteller Beschreibung / Besonderheit Code
Fmoc-Ahx-OH Iris Biotech ε-Aminohexansäure X
Fmoc-β-Ala-OH Iris Biotech Konstitutionsisomer von L-Ala B
Fmoc-L-Asp(OAll)-OH Iris Biotech orthogonal geschütztes L-Asp
Fmoc-L-Dpr(Alloc)-OH Iris Biotech orthogonal geschütztes Dpr
Fmoc-L-Glu(OAll)-OH Iris Biotech orthogonal geschütztes L-Glu

- Material und Methoden -
27
Fmoc-L-Lys(ivDde)-OH Iris Biotech orthogonal geschütztes Lys
Fmoc-L-Lys(Biotin)-OH Iris Biotech biotinyliertes Lys
Weitere Reagenzien und Chemikalien
Der verwendeten Chemikalien hatten einen hohen Reinheitsgrad von ≥ 99 %, welcher
mindestens mit pro analysis ausgezeichnet war.
Reagenzien und Chemikalien Hersteller Produktnummer
1,2-Ethandithiol Sigma Aldrich 02390
2-Mercaptoethanol Sigma Aldrich 63689
Ac2O Carl Roth CP28.2
AcOH Merck Chemicals 1.00063
Ar Linde
Cyclohexan Carl Roth 6570.3
DCM Fisher Chemical D/1856/17
DIC Iris Biotech RL-1015
DMF Sigma Aldrich D8654
DMSO Sigma Aldrich D8418
Geneticinsulfat-Lösung 50 µg/mL Carl Roth CP11.3
L-Glutamin Sigma Aldrich G3126
Glycin Sigma Aldrich 50046
H2SO4 konz. Sigma Aldrich 258105
HOBt * H2O Sigma Aldrich 54802
KCl Sigma Aldrich P9333
K2HPO4 * 3 H2O Merck Chemicals 1.05099
KH2PO4 Carl Roth 3904.3
MeCN Fisher Chemical A/0627/17
N2 gasförmig 5.0 Linde

- Material und Methoden -
28
NaCl Sigma Aldrich S5886
NaHCO3 Acros A0301775
Na2CO3 Carl Roth A135.2
Na2HPO4 * 2 H2O Sigma Aldrich 71643
Natrium-N, N-Diethyldithiocarbamat*3 H2O Sigma Aldrich 228680
OPD * 2HCl Sigma Aldrich P8287
Pd(PPh3)4 Sigma Aldrich 216666
Penicillin-Streptomycin Sigma Aldrich P4333
Phenol Carl Roth 0040.1
Piperidin Sigma Aldrich 80640
Pyridin Acros 339420010
TBME Merck Chemicals 1.01843
TentaGel SRAM Harz Rapp Polymere S30 023
TentaGel XV RAM Harz Rapp Polymere XV18130.023
TFA Carl Roth P088.2
Thioanisol Sigma Aldrich 88470
TIPS Sigma Aldrich 233781
Tris Sigma Aldrich T6066
Tween 20 Carl Roth 9127.1
NaOAc Sigma Aldrich S2889
HCl(aq), rauchend, 37 % Carl Roth 4625.2
HAT Medienzusatz 50x Sigma Aldrich H0262
HT Medienzusatz 50x Sigma Aldrich H0137
NH3(aq) 30 – 33 % Sigma Aldrich 05002
BSA Sigma Aldrich A7906

- Material und Methoden -
29
Medien und Puffer
Name / Abkürzung Herstellung / Zusammensetzung
DMEM Medium Gibco
RPMI 1640 Medium Gibco
PBSo siehe PBS 0,1M Carbonat-Puffer pH 9,5 mit HPW auf 100 mL aufgefüllt
6,67 mL 1 M NaHCO3(aq)
3,33 mL 1 M Na2CO3(aq)
PBS pH 7,4 mit HPW auf 100 mL aufgefüllt, mit HCl(aq) eingestellt
8,18 g NaCl
0,20 g KCl
1,77 g Na2HPO4 * 2 H2O
0,24 g KH2PO4
Phosphatpuffer pH 7,4 mit HPW auf 1 L aufgefüllt
71,7 mL 1 M KH2PO4(aq)
28,3 mL 1 M K2HPO4(aq)
Waschpuffer Phosphatpuffer pH 7,4 mit
0,01 % (w/v) Tween 20
Assaypuffer Phosphatpuffer pH 7,4 mit
0,1 % (w/v) BSA
0,01 % (w/v) Tween 20
Verwendete Proteine und Kits
Name Hersteller Produktnummer
mAb b12 Polymun Scientific AB011
mAb HJ16 NIH AIDS Research Program 12138
mAb VRC01 NIH AIDS Research Program 12033
mAb X5 Dennis Burton Laboratory
mAb 17b Dr. James E. Robinson Laboratory

- Material und Methoden -
30
mAb 2G12 Polymun AB002
Anti-V3 (3869) NIH AIDS Research Program 12039
Anti-NGF Biozol GTX10513
human CD4 Sino Biological 10400-H08H
gp120 HIV-1 IIIB (Euk) Immuno Diagnostics 1001
gp120 HIV-1 HXBc2 Immune Technology IT-001-0022p
gp120 HIV-1 BAL NIH AIDS Reagent Program 4961
gp120 HIV-1 ADA Immune Technology IT-001-0023p
gp120 HIV-1 PVO Immune Technology IT-001-RB3p
gp120 HIV-1 ZM96 AG Dr. Ralf Wagner, Universität Regensburg
NovaBright™ Secreted Placental
Alkaline Phosphatase (SEAP)
Enzyme Reporter Gene Chemi-
luminescent Detection System Thermo Fisher T1015
Streptavidin Thermo Fisher 21122
BSA Roth 3737.1
3.1.3 Verbrauchsmaterialien Verbrauchsmaterial Hersteller Produktnummer
UV/Vis-Küvetten Brand 759210
0,2 µM Membran-Spritzenfilter Merck Millipore SLLG013SL
0,5 mL Reaktionsgefäße Sarstedt 72.704
1,5 mL Reaktionsgefäße Sarstedt 72.706
2 mL Reaktionsgefäße Sarstedt 72.695.500
2 mL Polypropylen-Spritzen B.Braun 4606027V
Amicon Ultra 0,5 mL Zentrifugenfilter Merck Millipore UFC501024
konisches 15-mL Zentrifugenrohr Sarstedt 62.554.502
konisches 50-mL Zentrifugenrohr Sarstedt 62.547.254

- Material und Methoden -
31
Mikrotiterplatten
Immulon 2HB Thermo Scientific 3455
für Zellkultur Greiner bio-one
Nunc™ F96 MicroWell™ White Thermo Scientific 236105
Pipettenspitzen Eppendorf
Serologische Pipetten Greiner bio-one
FACS-Röhrchen BD Falcon
3.2 Peptidsynthese
Alle in dieser Arbeit vorgestellten Peptide wurden mittels Festphasenpeptidsynthese (SPPS)
nach der Fmoc/tBu-Strategie hergestellt. Als feste Phase diente dabei ein Polystyrolharz,
welches mit einem Rinkamid-Linker funktionalisiert ist (TentaGel S RAM, Rapp Polymere)
und dessen Beladung 0,24 mmol/g beträgt. Die Ansatzgröße betrug bei allen Synthesen
24 µmol und wurde in 2 mL-Polypropylenspritzen als Reaktoren durchgeführt, welche mit
einer Fritte versehen waren. Somit konnten die Reagenzien und Lösungen schnell und ohne
Verlust von Teilen der festen Phase einfach abgesaugt werden. Die Synthese erfolgte am
Harz vom C- zum N-Terminus. Zum orthogonalen Schutz der N-terminalen Aminosäure
wurde die Fmoc-Schutzgruppe verwendet, welche im Basischen selektiv abgespalten
werden kann. Die Funktionen der Aminosäure-Seitenketten und die Bindung zum Harz über
den Rinkamid-Linker sind dagegen säurelabil. Vor Beginn der Synthese konnte das Harz
stets mindestens für 2 h in 1 ml DMF quellen.
Größenteils erfolgte die Peptidsynthese automatisiert mit Hilfe eines Syntheseroboters.
Kostspielige Synthesebausteine, wie Aminosäuren mit besonderen orthogonalen
Schutzgruppen oder Biotin-Markierung und Abstandshalter-Bausteine, wurden in einer
manuellen Kupplung eingefügt, um den Reagenzienüberschuss zu verringern und die
Kupplungszeit entsprechend verlängern zu können. Ebenso wurden manuell
Lactamzyklisierungen, Abspaltungen orthogonaler Schutzgruppen und die N-terminalen
Acetylierungen durchgeführt.
- Material und Methoden -
32
3.2.1 Reaktionen an der festen Phase
3.2.1.1 Automatisierte Peptidsynthese
Der für die automatisierte Peptidsynthese verwendete Roboter war ein Syro 1 (Multisyntech),
der die Aminosäurekupplungen in sich wiederholenden Zyklen nach einem vorher
festgelegten Syntheseprotokoll durchführte. Die vorbereiteten Lösungen wurden mit einer
Nadel von oben in die mit Harz befüllten Reaktoren pipettiert, für eine vorgegebene
Zeitspanne dort inkubiert und anschließend mittels Unterdruck wieder abgesaugt. Ein solcher
Zyklus wird in Tabelle 3 beschrieben, wobei die Fmoc-Entschützung und
Aminosäurekupplung stets zweifach durchgeführt wurde und ein ca. fünffacher Überschuss
an Fmoc-Aminosäure eingesetzt wurde.
Tabelle 3: Zyklus bei der automatisierten SPPS
Schritt Beschreibung Zeit [min] Wieder-holungen
Volumen [µL]
Reagenzien (gelöst in)
1 Erste Fmoc-Entschützung
5 1 500 20 % (v/v) Piperidin (DMF)
Zweite Fmoc-Entschützung
15 1 500 20 % Piperidin (DMF)
Waschen 1 5 800 DMF 2 Erste Kupplung 60 1 350 0,33 M Fmoc-Aminosäure
(0,5 M HOBt*H2O in DMF) 150 20 % (v/v) DIC (DMF)
Waschen 1 2 800 DMF 3 Zweite Kupplung 60 1 350 0,33 M Fmoc-Aminosäure
(0,5 M HOBt*H2O in DMF) 150 20 % (v/v) DIC (DMF)
Waschen 1 5 800 DMF 4 Capping 30 1 500 33,3 % (v/v) Pyridin und
16,7 % (v/v) Ac2O (DMF) Waschen 1 5 800 DMF
Nach den Kupplungsschritten (2 und 3 in Tabelle 3) wurden die noch freien Aminofunktionen
mit einem Gemisch aus DMF, Pyridin und Essigsäureanhydrid (Ac2O) 3:2:1 acetyliert, um die
Entstehung unerwünschter Aminosäuresequenzen zu unterbinden und somit die Diversität
der Nebenprodukte möglichst gering zu halten (Capping). Neben den Standardaminosäuren
wurde auch Fmoc-ß-Ala-OH am Syntheseroboter gekuppelt.
3.2.1.2 Manuelle Kupplung von Synthesebausteinen
Zur manuellen Kupplung der Synthesebausteine Fmoc-Ahx-OH, Fmoc-L-Asp(OAll)-OH,
Fmoc-L-Dpr(Alloc)-OH, Fmoc-L-Glu(OAll)-OH, Fmoc-L-Lys(ivDde)-OH und Fmoc-L-
Lys(Biotin)-OH wurden die Reaktoren mit einem Stempel und einer verschließbaren Kanüle
- Material und Methoden -
33
versehen, um die Reagenzien und Lösungen in den Reaktor aufziehen und wieder
ausdrücken zu können. Während der Reaktionszeiten wurden die Reaktoren verschlossen
und auf einem Schüttler platziert. Der Hauptunterschied zwischen einem manuellen und
einem automatisierten Synthesezyklus bestand darin, dass die Kupplung des
Synthesebausteines nur mit einem Reagenzienüberschuss von zwei Äquivalenten
durchgeführt wurde, dafür aber über Nacht. Pro eingesetztem Äquivalent Synthesebaustein
wurden 1,2 Äquivalente DIC zur Aktivierung hinzugefügt um eine mögliche Hydrolyse der
aktivierten Spezies während der langen Reaktionszeit zu vermeiden. Alle Lösungen wurden
in Eppis frisch vorbereitet und dann direkt auf den Reaktor aufgezogen. Der Ablauf einer
solchen manuellen Kupplung ist in Tabelle 4 dargestellt.
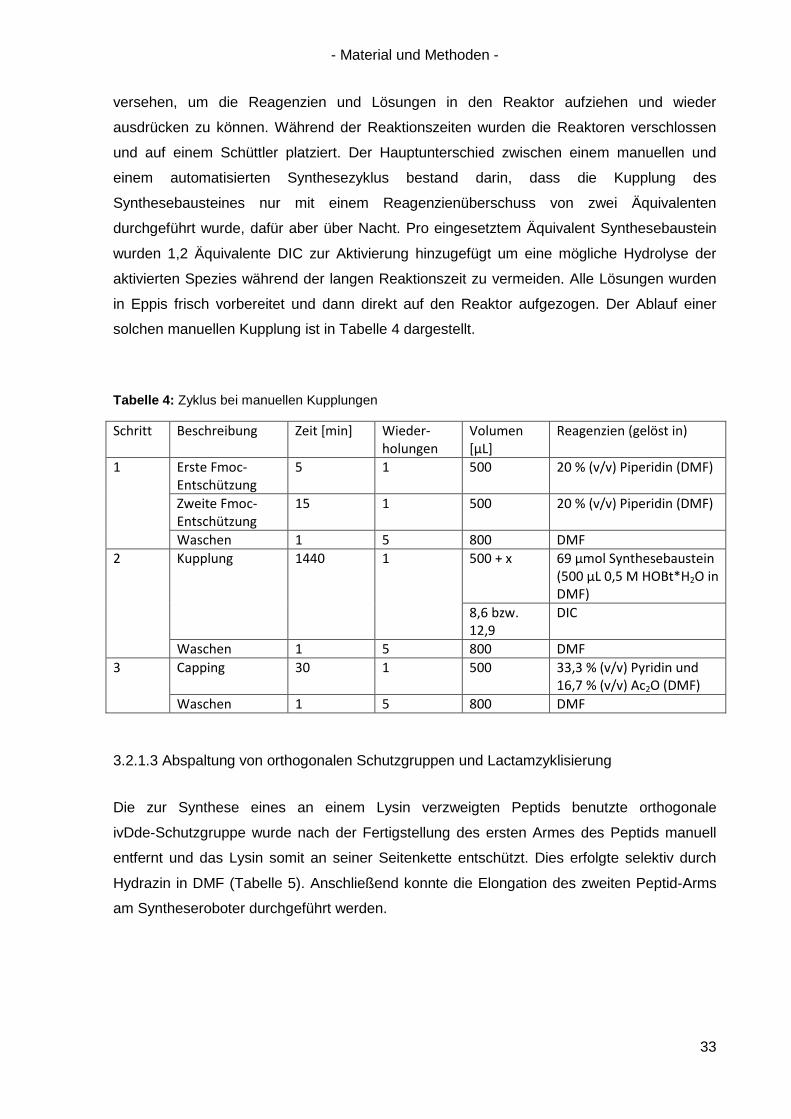
Tabelle 4: Zyklus bei manuellen Kupplungen
Schritt Beschreibung Zeit [min] Wieder-holungen
Volumen [µL]
Reagenzien (gelöst in)
1 Erste Fmoc-Entschützung
5 1 500 20 % (v/v) Piperidin (DMF)
Zweite Fmoc-Entschützung
15 1 500 20 % (v/v) Piperidin (DMF)
Waschen 1 5 800 DMF 2 Kupplung 1440 1 500 + x 69 µmol Synthesebaustein
(500 µL 0,5 M HOBt*H2O in DMF)
8,6 bzw. 12,9
DIC
Waschen 1 5 800 DMF 3 Capping 30 1 500 33,3 % (v/v) Pyridin und
16,7 % (v/v) Ac2O (DMF) Waschen 1 5 800 DMF
3.2.1.3 Abspaltung von orthogonalen Schutzgruppen und Lactamzyklisierung
Die zur Synthese eines an einem Lysin verzweigten Peptids benutzte orthogonale
ivDde-Schutzgruppe wurde nach der Fertigstellung des ersten Armes des Peptids manuell
entfernt und das Lysin somit an seiner Seitenkette entschützt. Dies erfolgte selektiv durch
Hydrazin in DMF (Tabelle 5). Anschließend konnte die Elongation des zweiten Peptid-Arms
am Syntheseroboter durchgeführt werden.
- Material und Methoden -
34
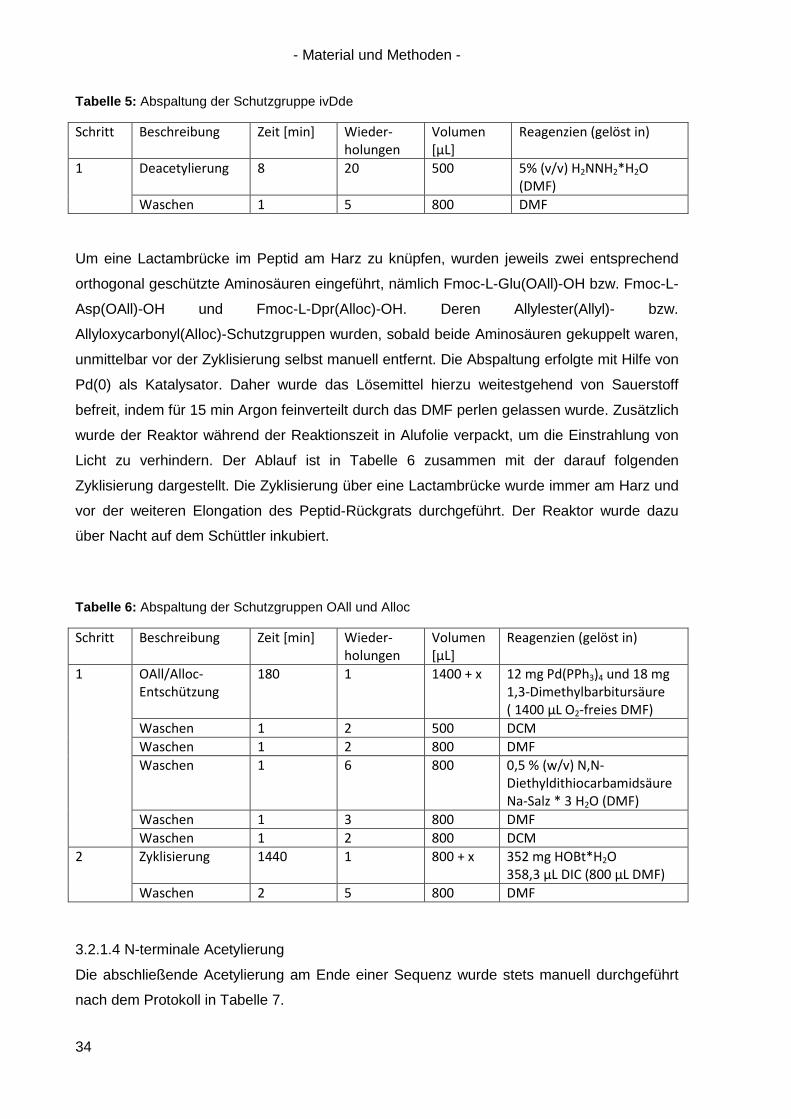
Tabelle 5: Abspaltung der Schutzgruppe ivDde
Schritt Beschreibung Zeit [min] Wieder-holungen
Volumen [µL]
Reagenzien (gelöst in)
1 Deacetylierung 8 20 500 5% (v/v) H2NNH2*H2O (DMF)
Waschen 1 5 800 DMF
Um eine Lactambrücke im Peptid am Harz zu knüpfen, wurden jeweils zwei entsprechend
orthogonal geschützte Aminosäuren eingeführt, nämlich Fmoc-L-Glu(OAll)-OH bzw. Fmoc-L-
Asp(OAll)-OH und Fmoc-L-Dpr(Alloc)-OH. Deren Allylester(Allyl)- bzw.
Allyloxycarbonyl(Alloc)-Schutzgruppen wurden, sobald beide Aminosäuren gekuppelt waren,
unmittelbar vor der Zyklisierung selbst manuell entfernt. Die Abspaltung erfolgte mit Hilfe von
Pd(0) als Katalysator. Daher wurde das Lösemittel hierzu weitestgehend von Sauerstoff
befreit, indem für 15 min Argon feinverteilt durch das DMF perlen gelassen wurde. Zusätzlich
wurde der Reaktor während der Reaktionszeit in Alufolie verpackt, um die Einstrahlung von
Licht zu verhindern. Der Ablauf ist in Tabelle 6 zusammen mit der darauf folgenden
Zyklisierung dargestellt. Die Zyklisierung über eine Lactambrücke wurde immer am Harz und
vor der weiteren Elongation des Peptid-Rückgrats durchgeführt. Der Reaktor wurde dazu
über Nacht auf dem Schüttler inkubiert.
Tabelle 6: Abspaltung der Schutzgruppen OAll und Alloc
Schritt Beschreibung Zeit [min] Wieder-holungen
Volumen [µL]
Reagenzien (gelöst in)
1 OAll/Alloc-Entschützung
180 1 1400 + x 12 mg Pd(PPh3)4 und 18 mg 1,3-Dimethylbarbitursäure ( 1400 µL O2-freies DMF)
Waschen 1 2 500 DCM Waschen 1 2 800 DMF Waschen 1 6 800 0,5 % (w/v) N,N-
Diethyldithiocarbamidsäure Na-Salz * 3 H2O (DMF)
Waschen 1 3 800 DMF Waschen 1 2 800 DCM
2 Zyklisierung 1440 1 800 + x 352 mg HOBt*H2O 358,3 µL DIC (800 µL DMF)
Waschen 2 5 800 DMF
3.2.1.4 N-terminale Acetylierung
Die abschließende Acetylierung am Ende einer Sequenz wurde stets manuell durchgeführt
nach dem Protokoll in Tabelle 7.
- Material und Methoden -
35
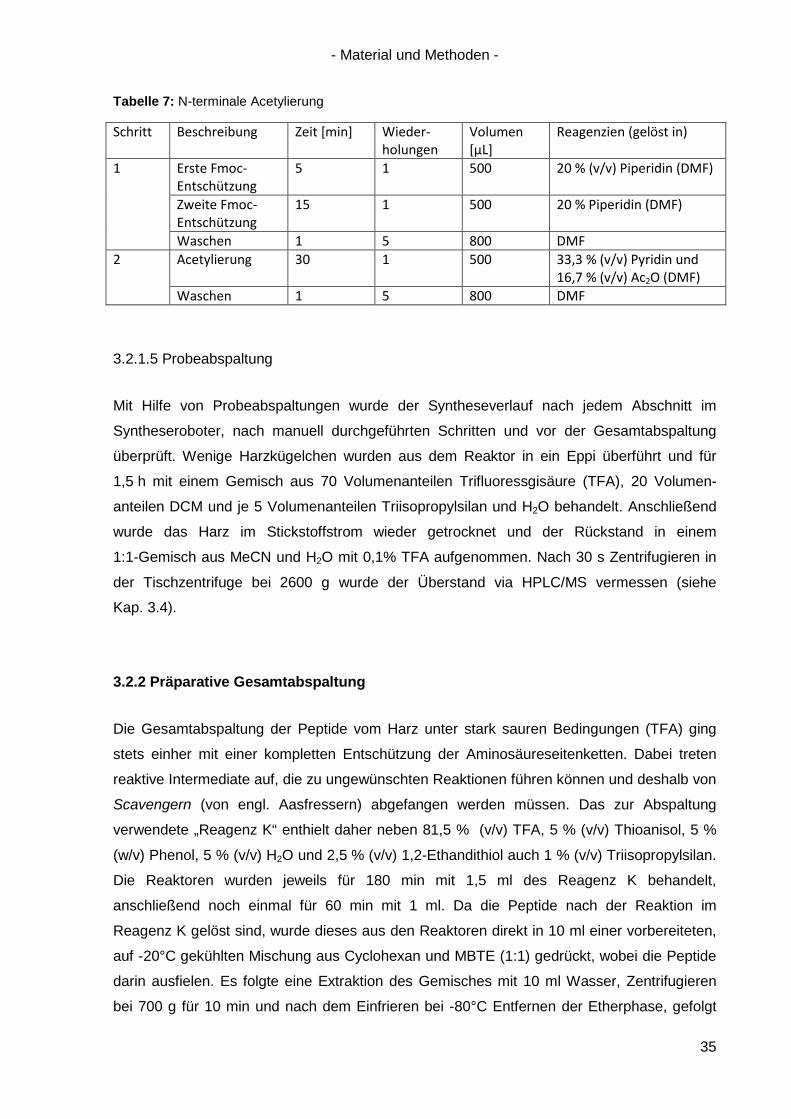
Tabelle 7: N-terminale Acetylierung
Schritt Beschreibung Zeit [min] Wieder-holungen
Volumen [µL]
Reagenzien (gelöst in)
1 Erste Fmoc-Entschützung
5 1 500 20 % (v/v) Piperidin (DMF)
Zweite Fmoc-Entschützung
15 1 500 20 % Piperidin (DMF)
Waschen 1 5 800 DMF 2 Acetylierung 30 1 500 33,3 % (v/v) Pyridin und
16,7 % (v/v) Ac2O (DMF) Waschen 1 5 800 DMF
3.2.1.5 Probeabspaltung
Mit Hilfe von Probeabspaltungen wurde der Syntheseverlauf nach jedem Abschnitt im
Syntheseroboter, nach manuell durchgeführten Schritten und vor der Gesamtabspaltung
überprüft. Wenige Harzkügelchen wurden aus dem Reaktor in ein Eppi überführt und für
1,5 h mit einem Gemisch aus 70 Volumenanteilen Trifluoressgisäure (TFA), 20 Volumen-
anteilen DCM und je 5 Volumenanteilen Triisopropylsilan und H2O behandelt. Anschließend
wurde das Harz im Stickstoffstrom wieder getrocknet und der Rückstand in einem
1:1-Gemisch aus MeCN und H2O mit 0,1% TFA aufgenommen. Nach 30 s Zentrifugieren in
der Tischzentrifuge bei 2600 g wurde der Überstand via HPLC/MS vermessen (siehe
Kap. 3.4).
3.2.2 Präparative Gesamtabspaltung
Die Gesamtabspaltung der Peptide vom Harz unter stark sauren Bedingungen (TFA) ging
stets einher mit einer kompletten Entschützung der Aminosäureseitenketten. Dabei treten
reaktive Intermediate auf, die zu ungewünschten Reaktionen führen können und deshalb von
Scavengern (von engl. Aasfressern) abgefangen werden müssen. Das zur Abspaltung
verwendete „Reagenz K“ enthielt daher neben 81,5 % (v/v) TFA, 5 % (v/v) Thioanisol, 5 %
(w/v) Phenol, 5 % (v/v) H2O und 2,5 % (v/v) 1,2-Ethandithiol auch 1 % (v/v) Triisopropylsilan.
Die Reaktoren wurden jeweils für 180 min mit 1,5 ml des Reagenz K behandelt,
anschließend noch einmal für 60 min mit 1 ml. Da die Peptide nach der Reaktion im
Reagenz K gelöst sind, wurde dieses aus den Reaktoren direkt in 10 ml einer vorbereiteten,
auf -20°C gekühlten Mischung aus Cyclohexan und MBTE (1:1) gedrückt, wobei die Peptide
darin ausfielen. Es folgte eine Extraktion des Gemisches mit 10 ml Wasser, Zentrifugieren
bei 700 g für 10 min und nach dem Einfrieren bei -80°C Entfernen der Etherphase, gefolgt

- Material und Methoden -
36
von Lyophilisieren bei 0,1 mbar. Man erhielt am Ende das Rohpeptid, welches noch zweimal
aus einer 1:1-Mischung aus MeCN und H2O versetzt mit 0,1 % TFA lyophilisiert wurde.
3.2.3 Oxidation zum Disulfid
Alle Peptide, die zwei Cysteine zur Zyklisierung über ein Disulfid enthielten, wurden in
Lösung nach einem ersten Reinigungsschritt an der HPLC (siehe Kapitel 3.3) im Basischen
mit Hilfe von Luftsauerstoff oxidiert. Hierfür wurde das Peptid, unabhängig von seinem
Reinheitsgrad, zu 0,3 mg/ml in 0,1M NH4OAc-Puffer gelöst, der zu gleichen Volumenanteilen
aus MeCN und H2O bestand und mit NH3 auf pH 8,0 eingestellt wurde. Diese niedrige
Peptidkonzentration sollte die Wahrscheinlichkeit der Bildung von intermolekularen
Disulfiden möglichst gering halten und die intramolekulare Reaktion begünstigen. Die
Ansätze wurden so für 3 Tage bei Raumtemperatur auf dem Schüttler inkubiert und
regelmäßig belüftet, um genügend Luftsauerstoff bereit zu stellen. Der Reaktionsverlauf
wurde mit Hilfe des Ellmans Tests (Kap. 3.2.4) kontrolliert, indem Proben zu Beginn der
Oxidation sowie nach drei Tagen genommen und nach Umsetzung mit DTNB photometrisch
verglichen wurden. Konnte mit bloßem Auge vor der Oxidation noch eine deutliche
Gelbfärbung erkannt werden, war diese nach vollendeter Oxidation nicht mehr zu
beobachten. Durch Ansäuern mit 25% (v/v) AcOH (aq) wurde die Reaktion gestoppt, das
Volumen der Probe wurde dabei auf das 1,5-fache erhöht. Die Probe wurde schließlich bei
-80 °C eingefroren und bei 0,1 mbar lyophilisiert. Der Rückstand wurde noch zwei weitere
Male aus 10 mL eines Gemisches aus gleichen Volumenteilen MeCN und H2O lyophilisiert,
welches mit 0,1% (v/v) TFA angesäuert wurde.
3.2.4 Ellmans Test
Zur Überprüfung der Vollständigkeit der Oxidation von Cysteinen zu Disulfidbrücken wurde
zu Beginn und am Ende der Inkubationszeit ein Ellmans Test mit Ellmans Reagenz
durchgeführt 125. Dieser Test detektiert freie Thiole. Es wurden hierzu in einer Küvette ein
Gemisch aus 325 µl H2O, 50 µl Tris (aq) pH 8,0 und 25 µl 2 mM DTNB in 50 mM NaOAc
vorgelegt. Dort hinein wurden 100 µl Peptidlösung (0,3 mg/ml) pipettiert, alles gut vermischt
und die Absorption nach 5 min bei 412 nm im Nanophotometer vermessen. Die
Hintergrundmessung erfolgte vor der eigentlichen Messung ohne Peptid, stattdessen wurden
100 µl reiner Oxidationspuffers (siehe oben) zugegeben.

- Material und Methoden -
37
3.3 Reinigung der Peptide mittels semipräparativer HPLC Die Peptide wurden in der Regel zweimal via Hochleistungsflüssigkeitschromatographie
(HPLC) gereinigt, einmal als Rohpeptide direkt nach der Abspaltung vom Harz und danach
entweder noch im Zwischenschritt oxidiert zu Disulfiden oder direkt ein zweites Mal gereinigt.
Die Peptide wurden dazu in ca. 1mL eines Gemisches aus gleichen Volumenanteilen
Eisessig und Wasser, oder falls dies nicht möglich war in TFA, gelöst. Es wurden immer
RP18-Säulen mit Vorsäule verwendet. Die Reinigung der Rohpeptide erfolgte auf einer
Dr. Maisch Reprosil Säule bei einem Fluss von 9 mL/min, die zweite Reinigung auf einer
Phenomenex Kinetex Säule bei einem Fluss von 3 mL/min.
Die HPLC-Läufe zur ersten Reinigung wurden für alle Peptide hinsichtlich
Anfangskonzentration, Steigung und Dauer des Gradienten gleich durchgeführt, wie in
Tabelle 8 dargestellt. Bei der zweiten Reinigung wurden Startbedingung und Gradient dem
jeweiligen Peptid angepasst, um ein möglichst sauberes Produkt zu erhalten, Wasch- und
Konditionierungsschritte wurden wie bei der ersten Reinigung durchgeführt. Die Details zu
den zweiten HPLC-Läufen für die einzelnen Peptide sind in Tabelle 9 aufgeführt. Am Ende
jedes Laufes wurde die Säule mit 95 % MeCN gespült und die Anlage anschließend auf die
Anfangsbedingungen rekonditioniert. Die Extinktion wurde während der Läufe bei 214 nm
und 280 nm mit einem UV-Detektor aufgezeichnet.
Tabelle 8: HPLC-Lauf zur Reinigung der Peptide
Dauer [min] MeCN-Anteil 5 Startbedingung 20 % MeCN 80 Linear 20 % - 60 % MeCN 5 60 %linear auf 95 % MeCN 10 95 % MeCN (waschen) 5 95 % MeCN linear auf 20% MeCN 10 20 % MeCN
Die Sequenzen der einzelnen Peptide und deren Ausbeuten sind im Ergebnisteil dieser
Arbeit in Tabelle 12 bzw. Tabelle 16 zu finden.
Mit Hilfe eines Fraktionensammlers wurde das Eluat nach der Säule pro Minute in einem
Reagenzglas gesammelt und die Fraktionen, welche Peptid enthielten, anschließend via
HPLC/MS analysiert (siehe Kapitel 3.4). Die Fraktionen, die das gesuchte Produkt enthielten
wurden bei vergleichbarem Reinheitsgrad vereinigt oder entsprechend getrennt bei -80°C
eingefroren und danach bei 0,1 mbar lyophilisiert. In diesem Zustand wurden die Peptide in
der Regel entweder bei -20°C aufbewahrt oder zur biologischen Testung in gleichen
Volumenanteilen von MeCN und H2O mit 0,1 % TFA zu 2,5 mM Stammlösungen gelöst,
welche ebenfalls bei -20°C gelagert wurden.
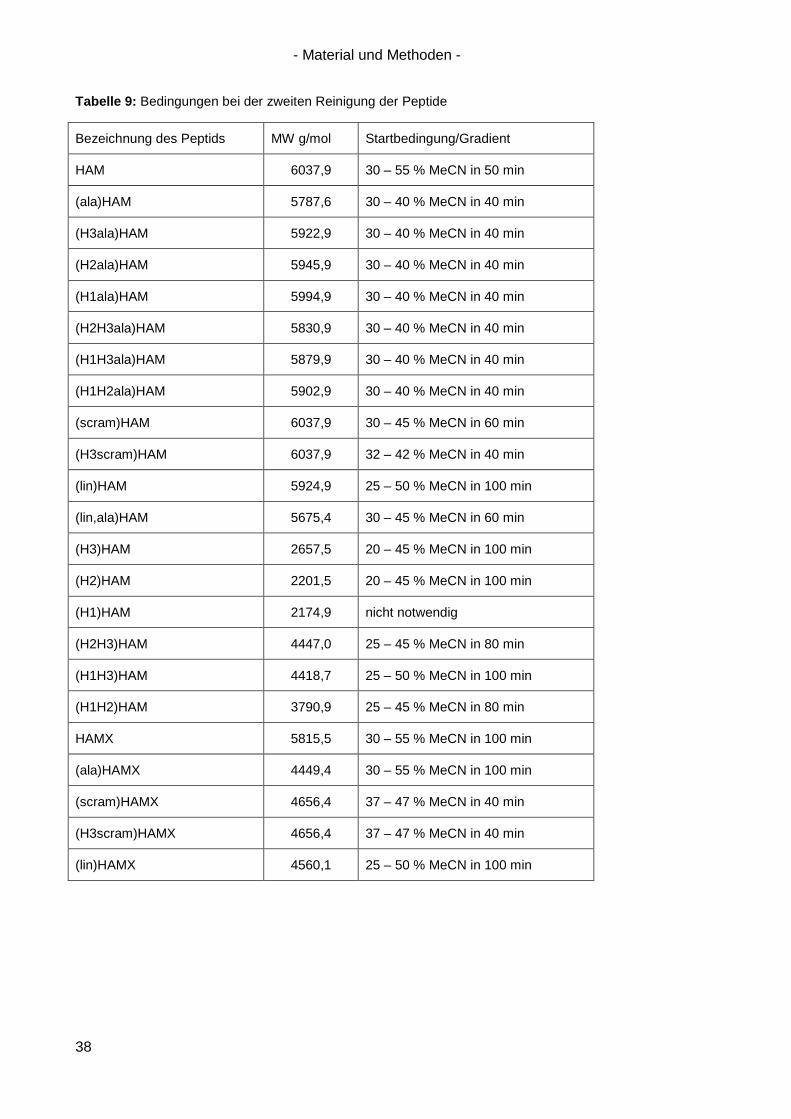
- Material und Methoden -
38
Tabelle 9: Bedingungen bei der zweiten Reinigung der Peptide
Bezeichnung des Peptids MW g/mol Startbedingung/Gradient
HAM 6037,9 30 – 55 % MeCN in 50 min
(ala)HAM 5787,6 30 – 40 % MeCN in 40 min
(H3ala)HAM 5922,9 30 – 40 % MeCN in 40 min
(H2ala)HAM 5945,9 30 – 40 % MeCN in 40 min
(H1ala)HAM 5994,9 30 – 40 % MeCN in 40 min
(H2H3ala)HAM 5830,9 30 – 40 % MeCN in 40 min
(H1H3ala)HAM 5879,9 30 – 40 % MeCN in 40 min
(H1H2ala)HAM 5902,9 30 – 40 % MeCN in 40 min
(scram)HAM 6037,9 30 – 45 % MeCN in 60 min
(H3scram)HAM 6037,9 32 – 42 % MeCN in 40 min
(lin)HAM 5924,9 25 – 50 % MeCN in 100 min
(lin,ala)HAM 5675,4 30 – 45 % MeCN in 60 min
(H3)HAM 2657,5 20 – 45 % MeCN in 100 min
(H2)HAM 2201,5 20 – 45 % MeCN in 100 min
(H1)HAM 2174,9 nicht notwendig
(H2H3)HAM 4447,0 25 – 45 % MeCN in 80 min
(H1H3)HAM 4418,7 25 – 50 % MeCN in 100 min
(H1H2)HAM 3790,9 25 – 45 % MeCN in 80 min
HAMX 5815,5 30 – 55 % MeCN in 100 min
(ala)HAMX 4449,4 30 – 55 % MeCN in 100 min
(scram)HAMX 4656,4 37 – 47 % MeCN in 40 min
(H3scram)HAMX 4656,4 37 – 47 % MeCN in 40 min
(lin)HAMX 4560,1 25 – 50 % MeCN in 100 min

- Material und Methoden -
39
3.4 Analytik mittels HPLC-gekoppelter Massenspektrometrie (HPLC/MS)
Die HPLC/Massenspektrometrie(MS)-Analytik wurde sowohl zur Kontrolle des
Syntheseverlaufes, als auch für die Endanalytik der synthetisierten Peptide verwendet. Dabei
konnte zu jedem Zeitpunkt das Produkt im Chromatogramm mit Hilfe der Massenspur
identifiziert werden. Die Peptide wurden zunächst chromatographisch getrennt und durch die
Kupplung der HPLC an das ESI-Massenspektrometer konnten die Peaks des
Chromatogramms über die entsprechenden festgestellten Molekülionen einer Verbindung
zugeordnet werden. Zur Auftrennung der Proben wurden die binären Gradienten aus MeCN
und H2O, welche beide 0,1 % TFA enthielten, jeweils so angepasst, dass die benötigte
Information zu diesem Zeitpunkt erhalten werden konnte. Zur Prozesskontrolle während der
Synthese wurde ein Gradient verwendet, welcher in 10 min linear von 5 % MeCN + 0,1 %
TFA auf 95 % MeCN + 0,1 % TFA gesteigert wurde. Für die Endanalytik der Peptide wurde
ein Gradient verwendet, der für die Erhöhung auf 95 % MeCN + 0,1 % TFA 15 min benötigte.
Zur raschen Identifikation und Analytik der produkthaltigen Fraktionen, die bei der
semipräparativen HPLC aufgefangen wurden, wurde ein Gradient verwendet, welcher für die
gleiche Steigerung des MeCN-Anteils im Laufmittel nur 5 min benötigte (Tabelle 10).
Tabelle 10: Gradienten bei der HPLC/MS-Analytik
Verwendungszweck Zeitpunkte [min] Laufmittelzusammensetzung Fraktionsanalytik 0 5 % MeCN 0 - 5 Linear auf 95 % MeCN 5 -7 95 % MeCN 7 - 8 Linear auf 5 % MeCN 8 -10 5 % MeCN Prozesskontrolle 0 5 % MeCN 0 - 10 Linear auf 95 % MeCN 10 -12 95 % MeCN 12 - 13 Linear auf 5 % MeCN 13 -15 5 % MeCN Endanalytik 0 5 % MeCN 0 - 15 Linear auf 95 % MeCN 15 -17 95 % MeCN 17 - 18 Linear auf 5 % MeCN 18 - 20 5 % MeCN
Die Endanalytiken der verschiedenen Peptide und deren Retentionszeiten sind im Anhang
aufgeführt (siehe Kapitel 7.1).

- Material und Methoden -
40
3.3 Enzyme-linked Immunosorbent Assay (ELISA)
Die Bindungsexperimente wurden alle in Form von ELISAs durchgeführt, wobei die
jeweiligen Liganden mit entsprechenden Detektionsantikörper detektiert wurden.
Grundsätzlich wurde einer der beiden Bindungspartner oder Streptavidin über Nacht
auf 96-Loch-Mikrotiterplatte (Immulon 2 HB) immobilisiert. Dazu wurden die Proteine in
Carbonat-Puffer mit pH 9,5 gelöst und die Platte, nachdem die entsprechenden Vertiefungen
mit je 100 µl der Proteinlösung befüllt worden waren, über Nacht bei 4 °C im Kühlschrank
inkubiert. Am nächsten Morgen wurde die Lösung aus den Platten geklopft und die noch
freien Bindungsstellen mit 200 µl pro Loch einer einprozentigen Lösung von bovinem
Serumalbumin (BSA) in Phosphatpuffer pH = 7,4 für eine Stunde bei Raumtemperatur
(300 rpm) blockiert. Anschließend wurden die Vertiefungen zweimal mit je 300 µl
Waschpuffer gewaschen. Als Liganden wurden Proteine in Assaypuffer gelöst, Peptide
wurden aus 2,5 mM Stammlösungen (MeCN:H2O 1:1 mit 0,1% TFA) in Assaypuffer
verdünnt, und die Platte mit 100 µl der Lösung pro Loch für 2-3 h bei Raumtemperatur
(300 rpm) inkubiert. Danach wurden alle Vertiefungen wieder mit jeweils 300 µl Waschpuffer
viermal gewaschen. Als letztes wurden die entsprechenden Detektionsantikörper
(ausschließlich monoklonale Antikörper) für jeweils eine Stunde aufgebracht, ebenfalls
100 µl/Loch und nach der Inkubationszeit viermal mit Waschpuffer gewaschen. Der letzte
Antikörper war stets mit Meerrettichperoxidase (Horse Raddish Peroxidase, HRP) markiert.
Am Ende wurden alle Vertiefungen mit 100 µl einer einprozentigen Lösung von
O-Phenylendiamin (OPD) versehen, die kurz vor dem Einpipettieren zusätzlich mit 0,3 %
Wasserstoffperoxid versetzt wurde. Im Dunklen wurde die Mikrotiterplatte für 15 Minuten
inkubiert und dann die Reaktion in allen Vertiefungen mit je 50 µl einer zweimolaren
Schwefelsäure abgestoppt. Die Lösung in den einzelnen Ansätzen färbte sich daraufhin gelb
bis dunkelorange und die unterschiedlichen Absorptionen der einzelnen Ansätze wurden mit
Hilfe eines Plattenlesegeräts bei einer Wellenlänge von 492 nm ausgelesen.
In der Regel wurden alle Messungen dreimal im Duplikat durchgeführt. Die Einzelheiten zu
den unterschiedlichen verwendeten ELISA-Formaten sind in den Kapiteln 3.3.1 – 3.3.4
beschrieben.
3.3.1 Detektion verschiedener gp120-Proteine
Die gp120-Proteine wurden in einer Konzentration von 0,5 µg/ml über Nacht auf der ELISA-
Platte immobilisiert. Anschließend erfolgte direkt die Detektion über ein duales Antikörper-
System, zunächst Schaf-Anti-gp120 (Aalto, erkennt den C-Terminus von gp120) gefolgt von

- Material und Methoden -
41
Kaninchen-Anti-Schaf-HRP (Dianova). Als Leerwerte dienten Ansätze, bei denen entweder
der erste oder der zweite Detektionsantikörper durch Pufferlösung ersetzt wurde. Der
Mittelwert der Leerwerte wurde vor der Auswertung der Daten von allen gemessenen Werten
subtrahiert.
3.3.2 Bindung von Peptiden an gp120 (EC50-Wert-Bestimmung) – Format A
Die EC50-Werte der Peptide wurden stets für die Bindung an HIV-1 gp120(IIIB) bestimmt. Dafür
wurde das Protein über Nacht in einer Konzentration von 0,5 µg/ml auf der Mikrotiterplatte
immobilisiert. Die Peptide wurden als Liganden ab einer Konzentration von 30, 60 oder
100 µM in 1:1-Verdünnungsreihen aufgetragen, für 3 h inkubiert und anschließend mit Hilfe
ihrer Biotin-Markierung und eines Ziege-Anti-Biotin-Antikörpers (Calbiochem) detektiert, der
selbst mit HRP markiert war. Als Leerwerte dienten Ansätze, die anstelle von Peptid-Lösung
nur mit Assaypuffer inkubiert wurden. Der Mittelwert der Leerwerte wurde vor der
Auswertung der Daten von allen gemessenen Werten subtrahiert.
3.3.3 Streptavidin-ELISA – Format B
Um die Messung der Bindung verschiedener Liganden an verschiedene Peptide im Vergleich
zu untersuchen, wurde ein Streptavidin-basiertes ELISA-Format verwendet. Dazu wurde
Streptavidin über Nacht bei einer Konzentration von 4 µg/ml in Coating-Puffer auf der
Mikrotiter-Platte immobilisiert. Nach dem Behandeln mit BSA wurde das zu analysierende
Peptid in einer Konzentration von 5 µM in Assaypuffer für 2 h auf die Platte gegeben, damit
es nach der Erkennung der Biotin-Markierung durch Streptavidin im ELISA auf der Platte
präsentiert wurde. Die Liganden wurden im nächsten Schritt in einer Konzentration von
1 µg/ml oder zur EC50-Wert-Bestimmung in 1:1-Verdünnungsreihen ab 4 µg/ml aufgebracht.
Für vergleichende EC50-Wert-Bestimmungen an b12 wurde der Antikörper über Nacht in
einer Konzentration von 0,5 µg/ml in Coating-Puffer auf der Mikrotiterplatte immobilisert und
nach dem Blockieren direkt mit den Liganden fortgefahren. CD4 als Induktor wurde immer
äquimolar zu gp120 eingesetzt. Alle gp120-Proteine wurden mittels eines dualen
Antikörpersystems detektiert, nämlich Schaf-Anti-gp120 (Aalto) und Kaninchen-Anti-Schaf-
HRP (Dianova). Alle Anti-HIV-1-Antikörper, die als Liganden eingesetzt wurden, wurden über
einen Ziege-Anti-Mensch-HRP-Antikörper (Sigma Aldrich) erkannt, CD4 über einen Maus-
Anti-His6-HRP (Roche) und Anti-NGF mit Hilfe eines dualen Systems aus Maus-Anti-NGF
(Biozol) und Ziege-Anti-Maus-HRP (Abcam). Die Leerwerte waren in diesem Fall Ansätze, in
denen keine Proteine oder Antikörper als Liganden aufgetragen wurden, sondern nur

- Material und Methoden -
42
Assaypuffer. Der Mittelwert der Leerwerte wurde vor der Auswertung der Daten von allen
gemessenen Werten subtrahiert.
3.3.4 Kompetitiver ELISA – Format C
Die Kompetitionsexperimente wurden ebenso an gp120(IIIB) durchgeführt wie die EC50-Wert-
Bestimmungen für Peptide (siehe Kap. 3.3.2). Als Ligand wurde das Peptid in gleich
bleibender Konzentration (0,25 µM) eingesetzt und der Kompetitor ab einer Konzentration
von 0,2 µM in einer 1:1-Verdünnungsreihe zugesetzt. Das Endvolumen in den Vertiefungen
betrug auch hier 100 µl, die Ansätze wurden so für 3 h inkubiert. Am Ende wurde die
gebundene Peptidmenge mit einem Ziege-Anti-Biotin-Antikörper (Calbiochem) ermittelt. Als
Leerwerte dienten Ansätze, die anstelle der Peptid-Inhibitor-Lösung nur mit Assaypuffer
inkubiert wurden. Der Mittelwert der Leerwerte wurde vor der Auswertung der Daten von
allen gemessenen Werten subtrahiert. Als maximal erreichbarer Wert (0% Inhibition), auf den
sich die berechnete Inhibition in Prozent bezieht, dienten Ansätze ohne Inhibitor.
3.4 Zellinfektionsassays
3.4.1 Herstellung der Virusstocklösungen
Das für die HIV-1-Zellinfektionsassays verwendete Virus war HIV-1 Clade B vom Stamm
NL4-3 (HIV-1(NL4-3)). Das Plasmid HIV-1pBRNL4.3 (F. Kirchhoff, Ulm) wurde in XL-1 Blue
kompetenten Zellen (Stratagene, Waldbronn) transformiert, anschließend aufgereinigt und
mittels FuGENE HD(Roche, Mannheim) in 293T-Zellen transfiziert. Nach der Vermehrung
des Virus auf CEMx174 SEAP-Zellen wurden die Überstände am Höhepunkt der viralen
Replikation geerntet, mit Sterilfiltern (Porengröße 0,2 µm) gereinigt und in Aliquots bei -80°C
gelagert. Die Viruskonzentration der Stocklösungen wurde mit Hilfe eines p24-Assays
bestimmt.
3.4.2 SEAP-Zellinfektionsassay mit HIV-1
Die Infektionsassays wurden mit CEMx174-SEAP-Zellen, freundlicherweise bereitgestellt
von R. E. Means und R. C. Desrosiers, und HIV-1 Cade B NL4-3 (HIV-1(NL4-3))
durchgeführt 126. Die Zellen wurden in 1640-Medium mit 10% durch Hitze (60 min bei 65°C)

- Material und Methoden -
43
inaktiviertem fötalem Kalbsserum (FKS), 1% Penicillin, 1% Streptomycin und 2% L-Glutamin
kultiviert und 2-mal wöchentlich in frisches Medium umgesetzt. Außerdem wurden zur
ausschließlichen Selektion der SEAP-exprimierenden Zellen bei jeder Passage à 50 ml
500 µl Geneticin zugegeben. Die SEAP-Zellen wurden ab Passage 5 bis Passage 20 für den
Assay verwendet.
Die zu testenden Analyten, Antikörper oder Peptide, wurden in einer zweiten 96-Vertiefungs-
Platte in 1640-Medium mit Zusätzen vorverdünnt. Für die Verdünnungsreihe wurden in der
ersten Vertiefung 240 µl der entsprechenden Konzentration an Analyt vorbereitet und in den
Vertiefungen für die weiteren Verdünnungsstufen jeweils 160 µl Medium vorgelegt. Es folgte
in der Regel eine 1-zu-3-Verdünnung über 5 Verdünnungsstufen, indem jeweils 80 µl
übertragen wurden. Die Peptide wurden dabei maximal ab Konzentrationen von 60 µM
(Endkonzentration im Assay) eingesetzt. Anschließend wurden aus jeder Vertiefung der
Mikrotiterplatte mit den vorverdünnten Analyten jeweils 100 µl Lösung in einer frischen
Mikrotiterplatte vorgelegt. Freie Felder auf der Platte wurden mit 200 µl PBSo belegt. Unter
S3-Bedingungen wurde anschließend das Virus zugegeben. Hierzu wurde zunächst ein
Aliquot des Virus-Stocks (bei -80°C gelagert), mit einer HIV-1(NL4-3) Konzentration von
6,7 µg/ml, aufgetaut, mit 1640-Medium mit Zusätzen zu 0,4 µg/ml verdünnt und davon
jeweils 30 µl in jede Vertiefung der Platte mit Analyt pipettiert und die Platte bei 37°C und 7%
CO2 für ca. 30 min vorinkubiert.
Währenddessen wurden die Zellen zuerst für 10 min bei 1200 rpm abzentrifugiert und das
alte Medium verworfen. Ein Zellpellet aus 20 ml Zellmedium wurde dann in 10 ml
1640-Medium mit Zusätzen resuspendiert und die darin enthaltene Zellzahl mittels einer
Neubauerschen Zählkammer bestimmt. Mit 1640-Medium mit Zusätzen wurde die
Konzentration der Zellen auf 250.000 Zellen/ml eingestellt. Unter S3-Bedingungen wurden
dann in die vorbereitete Mikrotiterplatte mit Analyt in Medium und Virus jeweils 100 µl
Zellsuspension pipettiert, so dass am Ende in allen Vertiefungen 200 µl Flüssigkeit vorlagen.
Positivkontrolle: CEMx174-SEAP-Zellen mit HIV-1(NL4-3), Analyt-Lösung ersetzt durch
1640-Medium mit Zusätzen
Negativkontrolle: CEMx174-SEAP-Zellen allein: alles andere durch 1640-Medium mit
Zusätzen ersetzt
Toxizitätstest: CEMx174-SEAP-Zellen mit Peptid (in der höchsten Konzentration); hier nur
optische Auswertung unter dem Mikrospkop
Die Platte wurde dann für 72 Stunden bei 37°C und 7% CO2 inkubiert. Unter dem Mikroskop
wurde anschließend optisch festgestellt, ob die Zellen in allen Vertiefungen, insbesondere in
Positiv-, Negativ- und Toxizitätskontrollen, am Leben waren und keinerlei ungewöhnliche

- Material und Methoden -
44
Verformungen angenommen hatten. Wenn dies der Fall war, wurde die SEAP-Aktivität in
den Überständen jeder einzelnen Vertiefung bestimmt.
Die Messung der Platte erfolgte mit Hilfe von NovaBrightTM Secreted Placental Alkaline
Phosphatase (SEAP) Enzyme Reporter Gene Chemiluminescenct Detection System
(Thermo Fisher). Das Kit wurde nach den Angaben des Herstellers verwendet. Als erstes
wurden jeweils 12 µl 1x Dilution Buffer in den Vertiefungen einer Luminometerplatte für jede
verwendete Vertiefung auf der 96-Vertiefungs-Assay-Platte vorgelegt. Unter
S3-Bedingungen wurden dann vom jedem Kulturüberstand der Vertiefungen auf der
Mikrotiterplatte 12 µl zu je 12 µl vorgelegtem 1x Dilution Buffer hinzugefügt und die
Luminometerplatte für 30 min bei 65°C inkubiert, um das infektiöse Virus zu inaktivieren.
Nach dem Abkühlen der Luminometerplatte auf Raumtemperatur wurden pro Ansatz 22 µl
Assaybuffer zugegeben und für 5 Minuten bei Raumtemperatur inkubiert. Als letztes wurden
jeweils 22 µl Working Reaction Buffer hinzupipettiert und nach exakt 20 Minuten die Platte im
Luminometer gemessen.
Die SEAP-Zellinfektionsassays wurden in der Regel dreimal im Triplikat durchgeführt. Die in
meiner Doktorarbeit gezeigten SEAP-Zellinfektionsassay-Daten habe ich zu großen Teilen
selbst in der Virologie Erlangen im Arbeitskreis meiner Co-Mentorin Prof. Dr. Barbara
Schmidt generiert. Die restlichen Messungen wurden nach deren Umzug in das Institut für
Mikrobiologie und Hygiene in Regensburg vor Ort dankenswerterweise von ihr und ihren
Mitarbeitern nach dem Standardprotokoll durchgeführt, jedoch selbständig ausgewertet.
3.4.3 GHOST-Zellinfektionsassay mit HIV-1
Zu Beginn wurden adhärente 5x104 GHOST(3) CXCR4+-Zellen (NIH AIDS reagent program,
3685) auf 12-Loch-Mikrotiterplatten in 500 µl Medium (DMEM), mit 10% durch Hitze (60 min
bei 65°C) inaktiviertem fötalem Kalbsserum (FKS), 1% Penicillin, 1% Streptomycin und 2%
L-Glutamin kultiviert und 2-mal wöchentlich in frisches Medium umgesetzt. Diese GHOST-
Zellen exprimieren bei einer Infektion mit HIV-1 grün fluoreszierendes Protein (GFP). Es
wurden nur Zellen der Passagen 3-10 verwendet. Die Zellen wurden so über Nacht im
Brutschrank bei 37°C und 7% CO2 inkubiert.
Am nächsten Tag wurden die Inhibitoren (Antikörper oder Peptid) entsprechend vorverdünnt
(1:3-Verdünnungsreihen, ab 15 µM (Peptid) bzw. 1 µg/ml (Antikörper), in möglichst wenig
Medium (Endvolumen 3µl). Dann wurden unter S3-Bedingungen in jeden Ansatz 10 µl des
HIV-1(NL4-3)-Virusstocks pipettiert und die Lösung für 1h vorinkubiert, um den Inhibitoren
etwas Vorlauf für die Kontaktzeit mit den Viruspartikeln zu geben, vergleiche SEAP-

- Material und Methoden -
45
Zellinfektionsassays. Als Positivkontrolle und zur Bestimmung des 100%-Wertes (max.
mögliche Infektion) ohne Einfluss von Peptiden/Inhibitoren wurden Ansätze nur mit 10 µl
Virusstock-Lösung versehen, als Negativkontrolle dienten Ansätze ohne Inhibitor und ohne
Virus.
Die Zellen wurden zunächst mit Medium gewaschen, um nicht adhärente Zellen zu
entfernen. Dann wurde frisches Medium (500 µl) auf die Zellen gegeben und unter
S3-Bedingungen die entsprechenden Ansätze aus Inhibitor und Virus zu den Zellen
pipettiert. Die Platten wurden kurz zum Mischen geschwenkt und dann im Brutschrank bei
37 °C und 7% CO2 für 48 h inkubiert.
Dann wurde das Medium abgenommen und die Zellen einmal mit 200 µl PBSo gewaschen.
Anschließend folgte die Zugabe von 200 µl kaltem PBSo mit 1% FKS und 1 mM EDTA und
die Platten wurden für 15 min auf Eis gestellt, um die Zellen abzulösen. Nach gründlichem
Resuspendieren wurden die Zell-Suspensionen jeweils in ein FACS-Röhrchen überführt und
im Anschluss bei 2000 rpm für 10 min zentrifugiert. Die Überstände wurden abgenommen
und 200 µl 4%iges Paraformaldehyd zugegeben, nach 1 h wurden die Proben via
Durchflusszytometrie vermessen und mit Hilfe des Programms FCS Express V3
ausgewertet.
Die GHOST-Zellinfektionsassays wurden in der Regel 3-mal wiederholt, die
Durchflusszytometrie wurde von meinem Kollege Norbert Donhauser im Institut für klinische
und molekulare Virologie des Universitäts-Klinikums Erlangen durchgeführt und ausgewertet.
3.4.4 Vero-Zellinfektionsassay mit HSV-1
Das Herpes-simplex-Virus 1 (HSV-1) Isolat 166v (G.Elliott, Lehrstuhl für Virologie,
Medizinische Fakultät, Imperial College London und P.O’Hare, Maire Curie Research
Institute, The Chart, Oxted, Surrey) expremiert ein GFP-fusioniertes VP22-Protein. Die Vero-
Zellen wurden mit drei verschiedenen infektiösen Dosen von HSV-1 166v (MOI: 0,05, 0,005
und 0,0005) für eine Stunde infiziert. Die Peptide wurden als potentielle Inhibitoren in einer
Konzentration von 10 µM zugesetzt. Nach wiederholtem Einfrieren und Auftauen der
infizierten Vero-Zellen wurden die Überstände geerntet und durch eine Saccharose-
Dichtegradientenzentrifugation gereinigt. Nach zwei Waschschritten wurden die Zellen
resuspendiert, für drei Tage kultiviert und mittels Durchflusszytometrie vermessen
(Auswertungsprogramm FCS Express V3). Die Versuche wurden in vierfach-Bestimmungen
durchgeführt.

- Material und Methoden -
46
3.4.5 Langzeitinkubations-Versuche mit Peptiden
Selektion resistenter Viren
HIV-1(NL4-3) wurde auf CEMx174 SEAP-Zellen mit Verdünnungsreihen von HAM-Peptiden
oder b12-Antikörper (siehe Kapitel 3.4.2, SEAP-Zellinfektionsassay) kultiviert. Abhängig von
der viralen Replikationsrate wurden nach drei bis sechs Tagen Inkubationszeit 25 µl
Zellkulturüberstand in die nächste Platte überführt. In den frischen Vertiefungen wurden
immer steigende Konzentrationen des Inhibitors vorgelegt, um den Selektionsdruck in
Richtung resistenter Viren zu erhöhen. Als Kontrolle wurden Ansätze mit Virus ohne Inhibitor
parallel inkubiert. Zur erfolgreichen Weiterkultivierung von HIV-1(NL4-3) in Gegenward
ansteigender Konzentrationen des b12-Antikörpers war es gelegentlich notwendig auch
infizierte Zellen zu transferieren, was bei der Auswertung nicht als zusätzliche Passage
gezählt wurde. Vor dem Einfrieren der Viren als Stocklösungen wurden sie noch einmal in
Abwesenheit des Inhibitors auf MT-2-Zellen (NIH Reagent Program, Nr. 237) passagiert.
Mutationsanalyse Mit Hilfe des QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) wurde die virale RNA
aus den Zellkulturüberständen isoliert. Das Kit wurde den Herstellerangaben entsprechend
verwendet. Die eluierte RNA wurde bei 65°C für 3 min denaturiert, danach auf Eis abgekühlt
und anschließend mit Zufallshexameren und MuLV reverser Transkriptase (Applied
Biosystems/Life Technologies) innerhalb von 30 min bei 42°C und weiteren 5 min bei 95°C in
die entsprechende cDNA überschrieben. Die HIV-1 gp120-Sequenzen wurden mit Hilfe der
Expand High Fidelity taq Polymerase (Roche, Mannheim, Germany) amplifiziert um vier
überlappende Polymerase-Kettenreaktions(PCR)-Fragmente mit den Primer-Paaren env01
(5’-AGAAAGAGCAGAAGACAGTG-3’) und env02 (5’-ATCATTCTCCCGCTACTACT-3’),
env1 (5’-GACATGGTAGAACAGATGCA-3’) und env2 (5’-TTTAGAATCGCAAAACCAGC-3’),
env3 (5’-TATCCTTTGAGCCAATTCCC-3’) und env4 (5’-CTCCTGAGGATTGCTTAAAGA-3’),
und env5 (5’-CAGTTTTAATTGTGGAGGGG-3’) oder env5a (5’-
GCCAATTTCACAGACAATGCT-3’) und env6 (5’-TTCACTTCTCCAATTGTCCC-3’) zu
generieren. Hierzu wurden die Proben zunächst bei 95°C für 5 min denaturiert. Die cDNA
wurde in 40 Zyklen folgender Inkubationszeiten vermehrt: 30 s bei 95°C, 30 s bei 50 °C, 75 s
bei 72 °C und abschließend 5 min bei 72°C. Die PCR-Produkte wurden auf einem
zweiprozentigen Agarosegel sichtbar gemacht und mit dem MinElute PCR Purification Kit
(Qiagen) aufgereinigt, welches nach den Angaben des Herstellers verwendet wurde. Die
DNA-Konzentration wurden mit Hilfe eines NanoDrop-ND-1000 Spektrophotometer (Peqlab
Biotechnologie GmbH, Erlangen, Germany) bestimmt. Die PCR-Fragmente wurden sowohl
vorwärts als auch rückwärts sequenziert und mit der Software SeqMan (DNASTAR,
Madison, WI) analysiert.

- Material und Methoden -
47
Alle für die Langzeitinkubationsversuche notwendigen Schritte wurden im Institut für
Mikrobiologie und Hygiene Regensburg unter der Leitung von Prof. Dr. Barbara Schmidt von
ihren Mitarbeitern Anette Rohrhofer, Dominik Damm und Sandra Nirschl durchgeführt.
3.5 Auswertungen
Zum direkten Vergleich verschiedener Peptid-Bindungsassays im Streptavidin-ELISA
(Format B) wurde bei allen Experimenten das Leitpeptid als Referenz mitgeführt und zur
Auswertung die Absorption des Leitpeptids A492nm = 2,0 festgesetzt. Die Ergebnisse wurden
mit dem entsprechenden Faktor für das Leitpeptid umgerechnet.
Zur Auswertung von IC50-Wert-Bestimmungen (Zellinfektionsassays) wurden die folgenden
Gleichungen verwendet:
Inhibition [%] 1001%100
×
−
−−=
− LeerwertWert
Leerwert
ExtinktionExtinktionExtinktionExtinktion
Dargestellt werden die Mittelwerte und deren Standardabweichung, welche mit Hilfe der
folgenden Gleichungen bestimmt wurden:
Mittelwert ∑=
=n
nix
nx
1
1
Standardabweichung des Mittelwerts( )
( )11
2
−
−=∆∑=
nn
xxx
n
ni
Wenn es möglich war, wurde zur EC50-Wert-Bestimmung im ELISA und zur IC50-Wert-
Bestimmung aus den Zellinfektionsassays eine Regression der gemessenen Daten mit der
Software SigmaPlot der Firma Systat nach der folgenden logistischen Funktion mit 4
Parametern durchgeführt:
Regressionsfunktion b
xx
ayy
+
+=
0
0
1

- Material und Methoden -
48
Darin beschreibt y0 die untere waagrechte Asymptote, a die Differenz aus der oberen und
der unteren waagrechten Asymptote, x0 die Konzentration am Wendepunkt und b die Hill-
Steigung der Kurve. Das Bestimmtheitsmaß dieser Regression wird mittels R2 ausgedrückt
und beschreibt die Abweichung der gemittelten Messwerte von der zugrunde gelegten
Regressionsfunktion. Der EC50-Wert bzw. IC50-Wert berechnet sich aus deren
Umkehrfunktion für y = 50:
by
axIC 150 0
050 −−
=

- Ergebnisse -
49
4. Ergebnisse
4.1 Design des b12-Antikörper-Mimetikums HAM
Der gegen HIV-1 gerichtete Antikörper b12 wurde für dieses Projekt ausgewählt um von
dessen Paratop (siehe Kapitel 1.1) ein peptidisches Mimetikum abzuleiten. Wie bereits in der
Einleitung beschrieben (siehe Kapitel 1.6), überlappt das Epitop für b12 mit der
Bindungsstelle für den zellulären Rezeptor CD4 auf dem HIV-Oberflächenprotein gp120. Der
breit neutralisierende Antikörper verhindert somit die initiale Interaktion des Virus mit der
Wirtszelle, wobei die Bindung von b12, im Gegensatz zur Bindung von CD4, an gp120 keine
konformationelle Änderung im gp120 auslöst, die Corezeptor-Bindungsstelle wird nicht
freigelegt.
Der Antikörper b12 wurde ausgewählt, da er zu Beginn dieses Projektes einer der am
ausführlichsten in der Literatur beschriebenen Antikörper und außerdem einer der am
breitesten neutralisierenden bekannten Anti-HIV-1-Antikörper war. Zusätzlich war von b12 im
Komplex mit gp120(HXBc2) bereits eine Kristallstruktur verfügbar (PDB: 2NY7). Die in der
Kristallstruktur sichtbare Hauptinteraktion von gp120 mit b12 wird durch die CDRs der
schweren Kette ausgebildet. Die mit 16 Aminosäuren verhältnismäßig lange CDR H3-
Sequenz liegt zu einer Schleife geformt vor und ragt sichtbar aus dem Arm des Antikörpers
heraus (Abb. 14B).
Das peptidisches Paratop-Mimetikum HAM wurde basierend auf der in der Literatur
beschriebenen Annahme, dass die CDR H3 von b12 die wichtigste der CDRs für die
Interaktion des Antikörpers mit gp120 ist 113, und dass in der Regel bei Antikörpern CDR H2
und H1 in Kombination mit CDR H3 wesentlich zur Affinität und Selektivität beitragen 10,
entworfen. Das Peptid enthält daher die Sequenzen aller drei CDRs der schweren Kette von
b12 (Abb. 14D). Die Sequenz-Abschnitte von H2 und H3 wurden allerdings bewusst etwas
länger, bzw. kürzer gewählt als die nach Chothia als CDRs definierten Abschnitte im
Antikörper 113,25, nämlich bis zu den Aminosäuren, deren Cα-Atome in den beiden gebildeten
Schleifen am nächsten beieinander liegen (H2: Cα1-Cα2 = 4,2 Å, H3: Cα1-Cα2 = 5,2 Å) (Abb.
14C). Die H1-Sequenz wurde ebenfalls verlängert, bis zu der Aminosäure, die am nächsten
an den Stämmen der H2- und H3-Schleife liegt. Im Folgenden werden die entsprechenden
Abschnitte im Peptid (siehe Tabelle 11) dennoch als CDRs (H1, H2 und H3) bezeichnet.
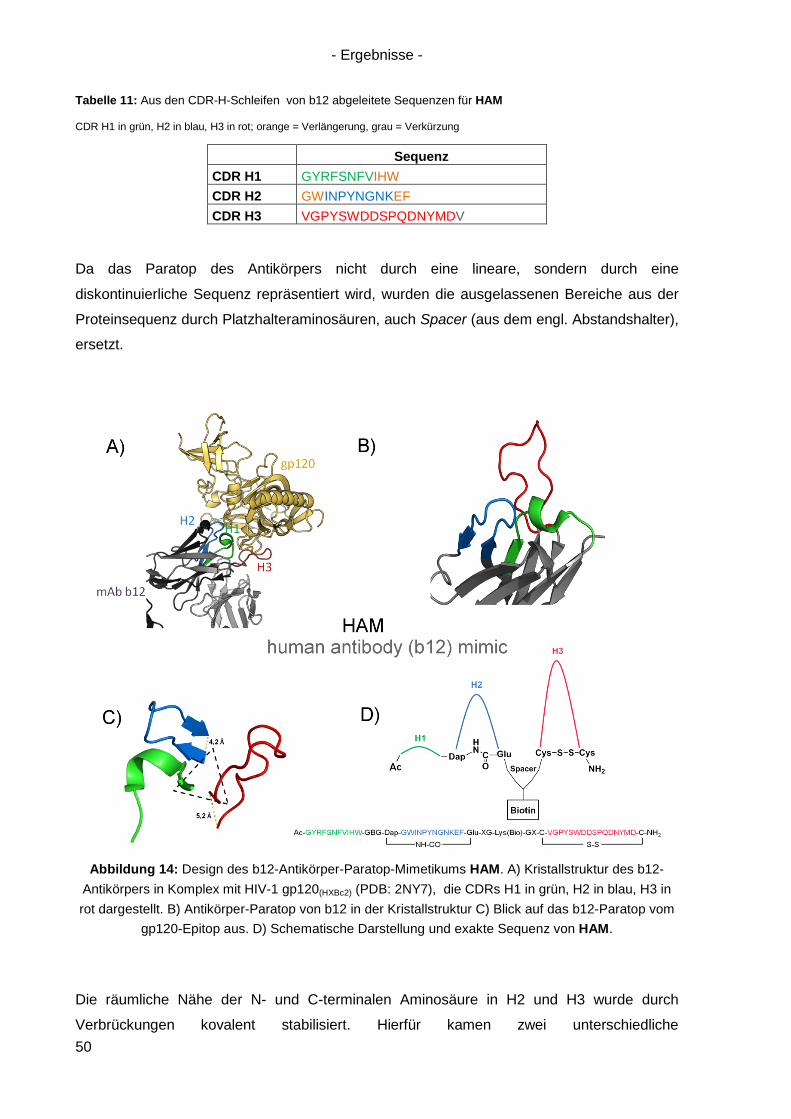
- Ergebnisse -
50
Tabelle 11: Aus den CDR-H-Schleifen von b12 abgeleitete Sequenzen für HAM
CDR H1 in grün, H2 in blau, H3 in rot; orange = Verlängerung, grau = Verkürzung
Sequenz CDR H1 GYRFSNFVIHW CDR H2 GWINPYNGNKEF CDR H3 VGPYSWDDSPQDNYMDV
Da das Paratop des Antikörpers nicht durch eine lineare, sondern durch eine
diskontinuierliche Sequenz repräsentiert wird, wurden die ausgelassenen Bereiche aus der
Proteinsequenz durch Platzhalteraminosäuren, auch Spacer (aus dem engl. Abstandshalter),
ersetzt.
Abbildung 14: Design des b12-Antikörper-Paratop-Mimetikums HAM. A) Kristallstruktur des b12-
Antikörpers in Komplex mit HIV-1 gp120(HXBc2) (PDB: 2NY7), die CDRs H1 in grün, H2 in blau, H3 in rot dargestellt. B) Antikörper-Paratop von b12 in der Kristallstruktur C) Blick auf das b12-Paratop vom
gp120-Epitop aus. D) Schematische Darstellung und exakte Sequenz von HAM.
Die räumliche Nähe der N- und C-terminalen Aminosäure in H2 und H3 wurde durch
Verbrückungen kovalent stabilisiert. Hierfür kamen zwei unterschiedliche

- Ergebnisse -
51
Zyklisierungsmethoden zum Einsatz: CDR H2 wurde über ein Lactam verbrückt, die deutlich
längere CDR H3 mittels Disulfid über zwei Cysteine. Die Sequenz der CDR H1 wurde linear
dargestellt, da sie auch im b12-Anitkörper keine Schleifen-Struktur aufweist (siehe Abb.
14B). Da die CDRs der schweren Kette des b12-Antikörpers nahezu in einem Dreieck
angeordnet sind (Abb. 14C), lässt sich ableiten, dass die Abfolge der drei CDRs H1, H2 und
H3 im Peptid keine entscheidende Rolle spielen sollte. Da die Sequenzen des gp120 in den
Komplexkristallstrukturen auch keinen freien N-Terminus besitzen, wurden die
synthetisierten Peptide N-terminal acetyliert.
Das Antikörper-Paratop-Mimetikum wurde zur Detektion bzw. zur Präsentation auf
Streptavidin-ELISA-Platten mit Biotin markiert. Als human antibody (b12) mimic (HAM), und
somit als Leitpeptid meiner Arbeit, wird im Folgenden das Konstrukt bezeichnet, welches alle
drei CDRs, H1, H2 und H3 enthält, wobei H3 und H2 zyklisiert vorlagen und das Peptid
zwischen H2 und H3 eine Biotin-Markierung enthielt (siehe Abb. 14D). Die Spacer zwischen
den einzelnen CDRs bestanden aus Glycin (G), ß-Alanin (B) und/oder 6-Aminohexansäure
(X). Die Biotin-Markierung zwischen H2 und H3 wurde ebenfalls räumlich abgetrennt, damit
das Biotin möglichst ohne Einfluss auf das Bindungsverhalten des Peptids genutzt werden
konnte.
In jeder der drei CDRs von b12 gibt es eine Aminosäure-Seitenkette, die als besonders
essentiell für die Interaktion mit gp120 beschrieben wurde 117. Diese sind W100 in CDR H3,
Y53 in CDR H2 und N31 in CDR H1 (siehe Abb. 10, Kapitel 1.6). Durch den Austausch
dieser drei Aminosäuren durch Alanin im Peptid entstand die Variante (ala)HAM, welche als
Negativkontrolle bei den Aktivitätstests bzw. zur ersten Bestätigung des Peptid-Designs
dienen sollte.
HAM Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2
(ala)HAM
Ac-GYRFSAFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2

- Ergebnisse -
52
4.2 Synthese von HAM
Mit Hilfe der Fmoc-basierten Festphasenpeptidsynthese konnten das Peptid HAM und seine
Varianten synthetisch dargestellt werden. Die Sequenzen, molaren Massen und Synthese-Ausbeuten aller HAM-Peptide sind in Tabelle 12 aufgelistet.
Dabei wurde immer die entsprechende Aminosäuresequenz am Harz (TentaGel SRAM)
synthetisiert und Lactamzyklisierungen erfolgten ebenfalls am Harz direkt nach Kupplung
von Fmoc-Dap(Alloc)-OH und anschließender Abspaltung der orthogonalen Schutzgruppen
(Allyl/Alloc). Oxidation der Cysteine zu Disulfidbrücken wurde erst nach Abspaltung der
Peptide vom Harz und einer ersten Reinigung mittels HPLC durchgeführt, wobei in einem
Peptid nie mehr als zwei Cysteine enthalten waren, sodass nur eine Disulfidbrücke
ausgebildet werden konnte. Nach der Oxidation folgte in der Regel eine zweite Reinigung.
Die Ausbeuten bei den HAM-Peptiden waren sehr unterschiedlich (siehe Tabelle 12). Ein
besonders limitierender Schritt bei der Synthese war die Lactamzyklisierung: bei Peptiden,
die eine Lactambrücke enthalten, war bereits die Menge an Rohpeptid im Durchschnitt nur
halb so groß wie bei Varianten ohne Lactambrücke. Die Ausbeute an reinem Peptid belief
sich in diesen Fällen dann nur auf 0,3 – 3,0 %, was bei einer Ansatzgröße von 100 mg Harz,
mit einer Beladung von 0,24 mmol/g (24 µmol) je nach molarer Masse 0,5 – 1,5 mg Peptid
entsprach.
Der Einsatz eines anderen, ebenso funktionalisierten Harzes (TentaGel XV RAM, Beladung
0,21 mmol/g), dessen Partikel geringer beladen sind und bessere Quelleigenschaften
besitzen, um dadurch mehr Abstand zwischen den einzelnen Peptidsträngen während der
Synthese zu schaffen, brachte keine Verbesserung der Ausbeuten. Der Versuch eine
Variante von HAM zu synthetisieren, in der die drei essentiellen L-Aminosäuren durch die
entsprechende D-Aminosäure ersetzt werden, scheiterte ebenfalls an der Lactamzyklisierung
und auch ein Fragment-basierter Ansatz zur Synthese einer D-Aminosäure-Variante von HAM blieb erfolglos.
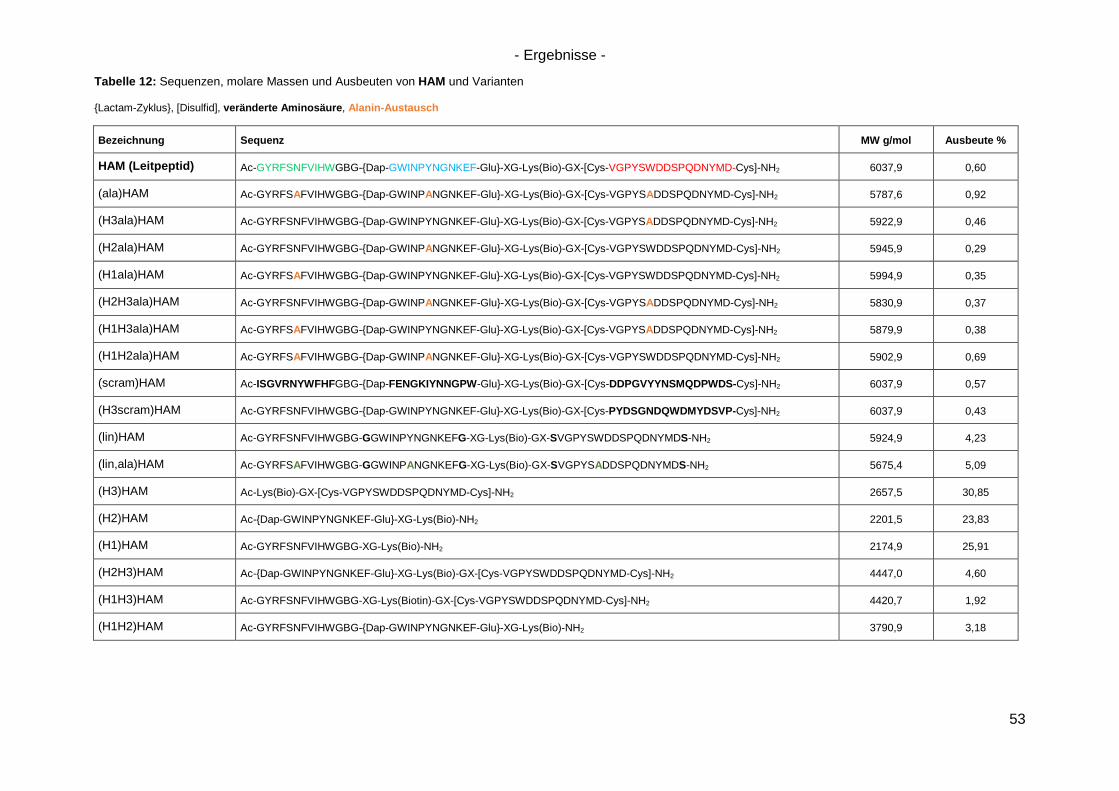
- Ergebnisse -
53
Tabelle 12: Sequenzen, molare Massen und Ausbeuten von HAM und Varianten
{Lactam-Zyklus}, [Disulfid], veränderte Aminosäure, Alanin-Austausch
Bezeichnung Sequenz MW g/mol Ausbeute %
HAM (Leitpeptid) Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 6037,9 0,60
(ala)HAM Ac-GYRFSAFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2 5787,6 0,92
(H3ala)HAM Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2 5922,9 0,46
(H2ala)HAM Ac-GYRFSNFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 5945,9 0,29
(H1ala)HAM Ac-GYRFSAFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 5994,9 0,35
(H2H3ala)HAM Ac-GYRFSNFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2 5830,9 0,37
(H1H3ala)HAM Ac-GYRFSAFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2 5879,9 0,38
(H1H2ala)HAM Ac-GYRFSAFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 5902,9 0,69
(scram)HAM Ac-ISGVRNYWFHFGBG-{Dap-FENGKIYNNGPW-Glu}-XG-Lys(Bio)-GX-[Cys-DDPGVYYNSMQDPWDS-Cys]-NH2 6037,9 0,57
(H3scram)HAM Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-PYDSGNDQWDMYDSVP-Cys]-NH2 6037,9 0,43
(lin)HAM Ac-GYRFSNFVIHWGBG-GGWINPYNGNKEFG-XG-Lys(Bio)-GX-SVGPYSWDDSPQDNYMDS-NH2 5924,9 4,23
(lin,ala)HAM Ac-GYRFSAFVIHWGBG-GGWINPANGNKEFG-XG-Lys(Bio)-GX-SVGPYSADDSPQDNYMDS-NH2 5675,4 5,09
(H3)HAM Ac-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 2657,5 30,85
(H2)HAM Ac-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-NH2 2201,5 23,83
(H1)HAM Ac-GYRFSNFVIHWGBG-XG-Lys(Bio)-NH2 2174,9 25,91
(H2H3)HAM Ac-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 4447,0 4,60
(H1H3)HAM Ac-GYRFSNFVIHWGBG-XG-Lys(Biotin)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2 4420,7 1,92
(H1H2)HAM Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-NH2 3790,9 3,18
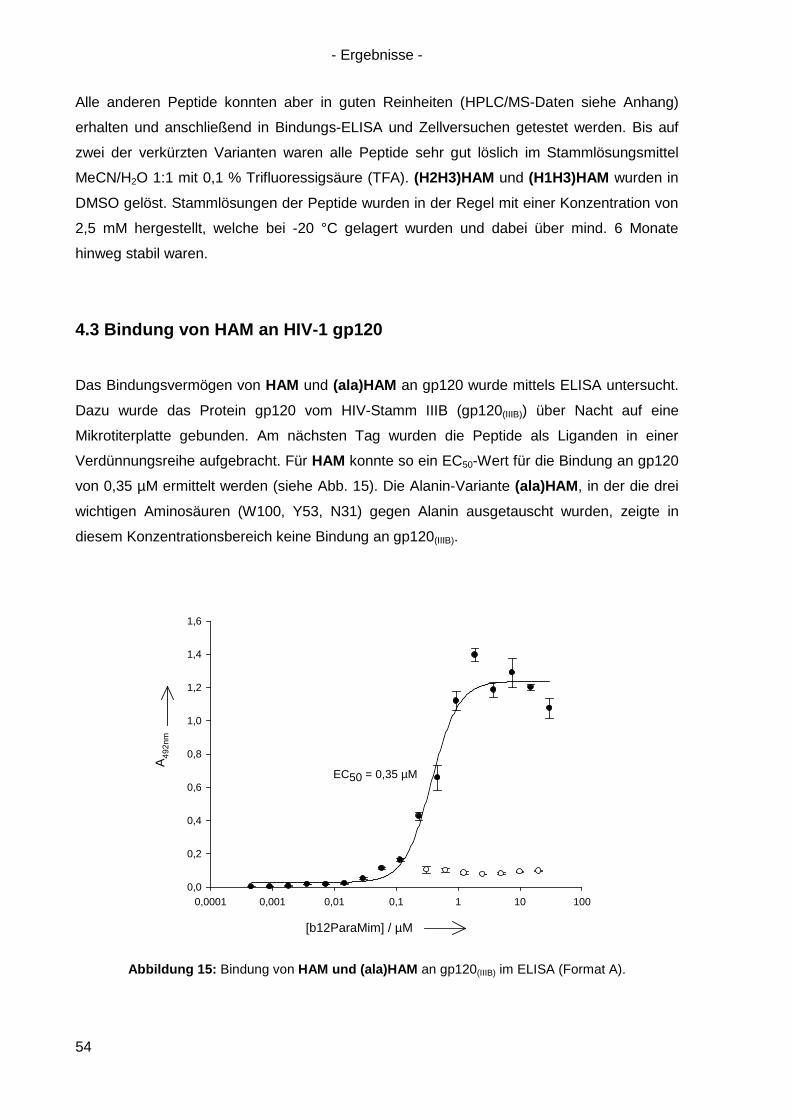
- Ergebnisse -
54
Alle anderen Peptide konnten aber in guten Reinheiten (HPLC/MS-Daten siehe Anhang)
erhalten und anschließend in Bindungs-ELISA und Zellversuchen getestet werden. Bis auf
zwei der verkürzten Varianten waren alle Peptide sehr gut löslich im Stammlösungsmittel
MeCN/H2O 1:1 mit 0,1 % Trifluoressigsäure (TFA). (H2H3)HAM und (H1H3)HAM wurden in
DMSO gelöst. Stammlösungen der Peptide wurden in der Regel mit einer Konzentration von
2,5 mM hergestellt, welche bei -20 °C gelagert wurden und dabei über mind. 6 Monate
hinweg stabil waren.
4.3 Bindung von HAM an HIV-1 gp120
Das Bindungsvermögen von HAM und (ala)HAM an gp120 wurde mittels ELISA untersucht.
Dazu wurde das Protein gp120 vom HIV-Stamm IIIB (gp120(IIIB)) über Nacht auf eine
Mikrotiterplatte gebunden. Am nächsten Tag wurden die Peptide als Liganden in einer
Verdünnungsreihe aufgebracht. Für HAM konnte so ein EC50-Wert für die Bindung an gp120
von 0,35 µM ermittelt werden (siehe Abb. 15). Die Alanin-Variante (ala)HAM, in der die drei
wichtigen Aminosäuren (W100, Y53, N31) gegen Alanin ausgetauscht wurden, zeigte in
diesem Konzentrationsbereich keine Bindung an gp120(IIIB).
[b12ParaMim] / µM
0,0001 0,001 0,01 0,1 1 10 100
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
EC50 = 0,35 µM
Abbildung 15: Bindung von HAM und (ala)HAM an gp120(IIIB) im ELISA (Format A).
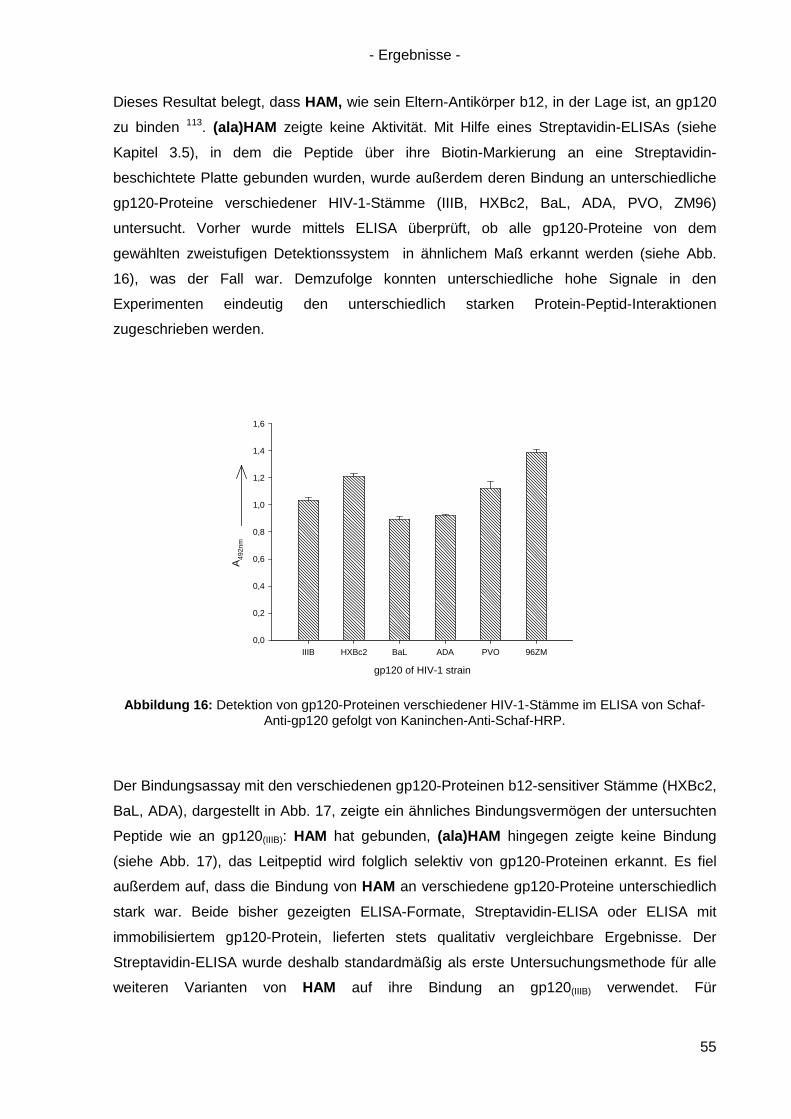
- Ergebnisse -
55
Dieses Resultat belegt, dass HAM, wie sein Eltern-Antikörper b12, in der Lage ist, an gp120
zu binden 113. (ala)HAM zeigte keine Aktivität. Mit Hilfe eines Streptavidin-ELISAs (siehe
Kapitel 3.5), in dem die Peptide über ihre Biotin-Markierung an eine Streptavidin-
beschichtete Platte gebunden wurden, wurde außerdem deren Bindung an unterschiedliche
gp120-Proteine verschiedener HIV-1-Stämme (IIIB, HXBc2, BaL, ADA, PVO, ZM96)
untersucht. Vorher wurde mittels ELISA überprüft, ob alle gp120-Proteine von dem
gewählten zweistufigen Detektionssystem in ähnlichem Maß erkannt werden (siehe Abb.
16), was der Fall war. Demzufolge konnten unterschiedliche hohe Signale in den
Experimenten eindeutig den unterschiedlich starken Protein-Peptid-Interaktionen
zugeschrieben werden.
gp120 of HIV-1 strain
IIIB HXBc2 BaL ADA PVO 96ZM
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Abbildung 16: Detektion von gp120-Proteinen verschiedener HIV-1-Stämme im ELISA von Schaf-Anti-gp120 gefolgt von Kaninchen-Anti-Schaf-HRP.
Der Bindungsassay mit den verschiedenen gp120-Proteinen b12-sensitiver Stämme (HXBc2,
BaL, ADA), dargestellt in Abb. 17, zeigte ein ähnliches Bindungsvermögen der untersuchten
Peptide wie an gp120(IIIB): HAM hat gebunden, (ala)HAM hingegen zeigte keine Bindung
(siehe Abb. 17), das Leitpeptid wird folglich selektiv von gp120-Proteinen erkannt. Es fiel
außerdem auf, dass die Bindung von HAM an verschiedene gp120-Proteine unterschiedlich
stark war. Beide bisher gezeigten ELISA-Formate, Streptavidin-ELISA oder ELISA mit
immobilisiertem gp120-Protein, lieferten stets qualitativ vergleichbare Ergebnisse. Der
Streptavidin-ELISA wurde deshalb standardmäßig als erste Untersuchungsmethode für alle
weiteren Varianten von HAM auf ihre Bindung an gp120(IIIB) verwendet. Für
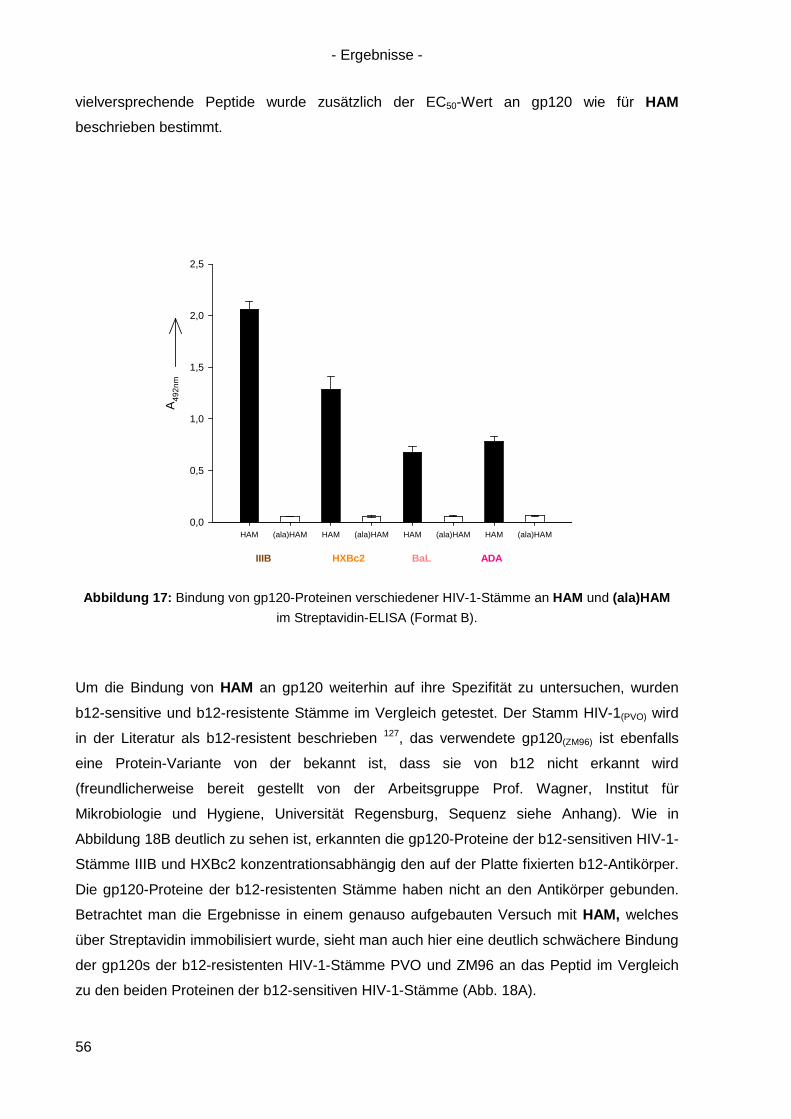
- Ergebnisse -
56
vielversprechende Peptide wurde zusätzlich der EC50-Wert an gp120 wie für HAM
beschrieben bestimmt.
HAM (ala)HAM HAM (ala)HAM HAM (ala)HAM HAM (ala)HAM
A 492n
m
0,0
0,5
1,0
1,5
2,0
2,5
IIIB HXBc2 BaL ADA
Abbildung 17: Bindung von gp120-Proteinen verschiedener HIV-1-Stämme an HAM und (ala)HAM im Streptavidin-ELISA (Format B).
Um die Bindung von HAM an gp120 weiterhin auf ihre Spezifität zu untersuchen, wurden
b12-sensitive und b12-resistente Stämme im Vergleich getestet. Der Stamm HIV-1(PVO) wird
in der Literatur als b12-resistent beschrieben 127, das verwendete gp120(ZM96) ist ebenfalls
eine Protein-Variante von der bekannt ist, dass sie von b12 nicht erkannt wird
(freundlicherweise bereit gestellt von der Arbeitsgruppe Prof. Wagner, Institut für
Mikrobiologie und Hygiene, Universität Regensburg, Sequenz siehe Anhang). Wie in
Abbildung 18B deutlich zu sehen ist, erkannten die gp120-Proteine der b12-sensitiven HIV-1-
Stämme IIIB und HXBc2 konzentrationsabhängig den auf der Platte fixierten b12-Antikörper.
Die gp120-Proteine der b12-resistenten Stämme haben nicht an den Antikörper gebunden.
Betrachtet man die Ergebnisse in einem genauso aufgebauten Versuch mit HAM, welches
über Streptavidin immobilisiert wurde, sieht man auch hier eine deutlich schwächere Bindung
der gp120s der b12-resistenten HIV-1-Stämme PVO und ZM96 an das Peptid im Vergleich
zu den beiden Proteinen der b12-sensitiven HIV-1-Stämme (Abb. 18A).
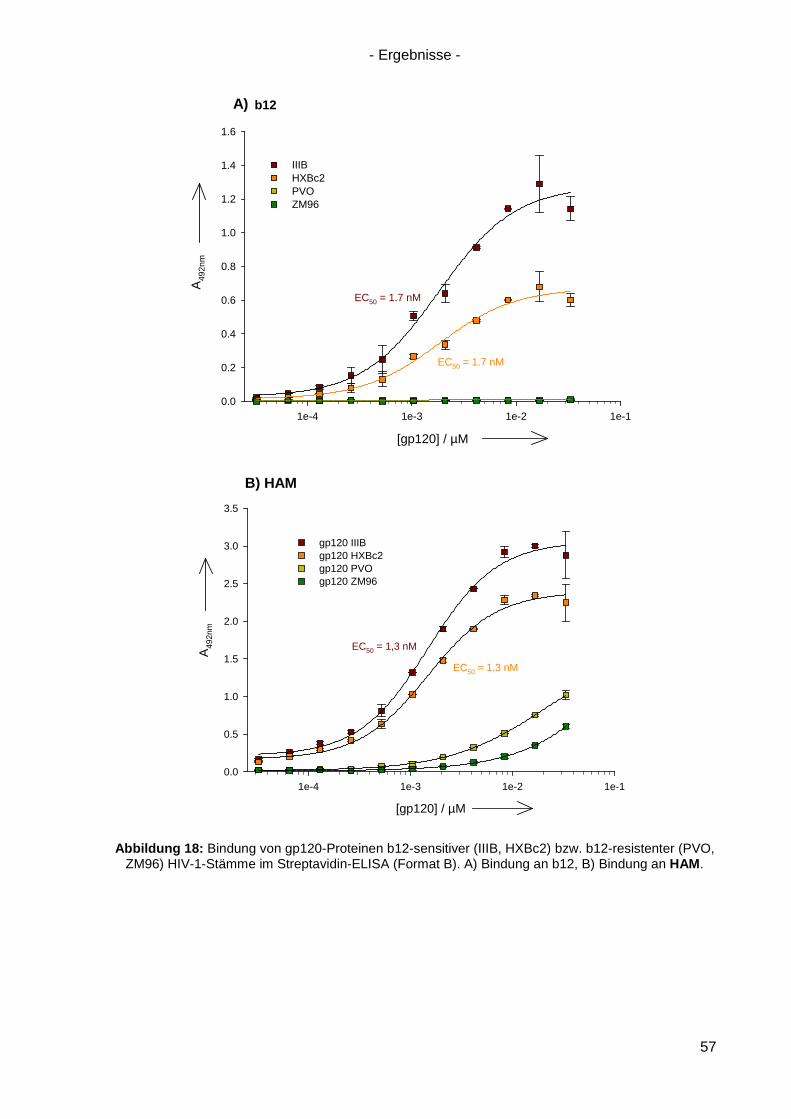
- Ergebnisse -
57
b12
[gp120] / µM
1e-4 1e-3 1e-2 1e-1
A 492n
m
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
IIIBHXBc2PVOZM96
EC50 = 1.7 nM
EC50 = 1.7 nM
A)
B) HAM
[gp120] / µM
1e-4 1e-3 1e-2 1e-1
A 492n
m
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
gp120 IIIBgp120 HXBc2gp120 PVOgp120 ZM96
EC50 = 1,3 nM
EC50 = 1,3 nM
Abbildung 18: Bindung von gp120-Proteinen b12-sensitiver (IIIB, HXBc2) bzw. b12-resistenter (PVO, ZM96) HIV-1-Stämme im Streptavidin-ELISA (Format B). A) Bindung an b12, B) Bindung an HAM.
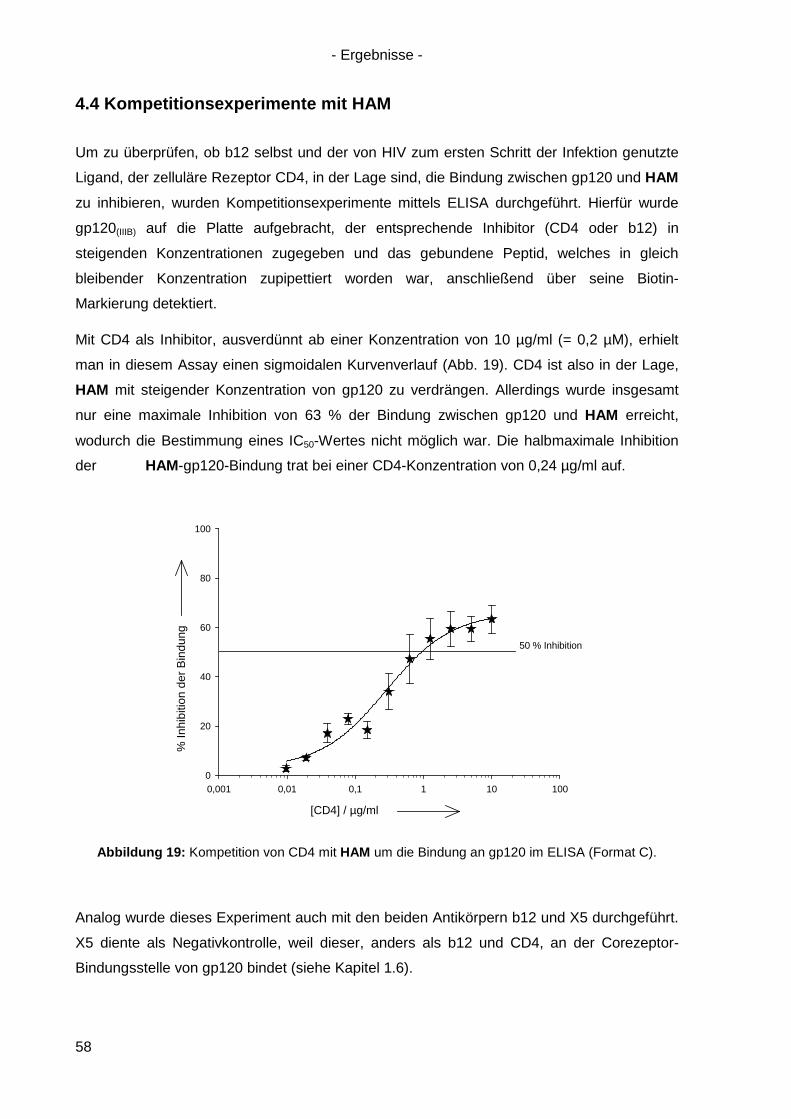
- Ergebnisse -
58
4.4 Kompetitionsexperimente mit HAM
Um zu überprüfen, ob b12 selbst und der von HIV zum ersten Schritt der Infektion genutzte
Ligand, der zelluläre Rezeptor CD4, in der Lage sind, die Bindung zwischen gp120 und HAM
zu inhibieren, wurden Kompetitionsexperimente mittels ELISA durchgeführt. Hierfür wurde
gp120(IIIB) auf die Platte aufgebracht, der entsprechende Inhibitor (CD4 oder b12) in
steigenden Konzentrationen zugegeben und das gebundene Peptid, welches in gleich
bleibender Konzentration zupipettiert worden war, anschließend über seine Biotin-
Markierung detektiert.
Mit CD4 als Inhibitor, ausverdünnt ab einer Konzentration von 10 µg/ml (= 0,2 µM), erhielt
man in diesem Assay einen sigmoidalen Kurvenverlauf (Abb. 19). CD4 ist also in der Lage,
HAM mit steigender Konzentration von gp120 zu verdrängen. Allerdings wurde insgesamt
nur eine maximale Inhibition von 63 % der Bindung zwischen gp120 und HAM erreicht,
wodurch die Bestimmung eines IC50-Wertes nicht möglich war. Die halbmaximale Inhibition der HAM-gp120-Bindung trat bei einer CD4-Konzentration von 0,24 µg/ml auf.
[CD4] / µg/ml
0,001 0,01 0,1 1 10 100
% In
hibi
tion
der B
indu
ng
0
20
40
60
80
100
50 % Inhibition
Abbildung 19: Kompetition von CD4 mit HAM um die Bindung an gp120 im ELISA (Format C).
Analog wurde dieses Experiment auch mit den beiden Antikörpern b12 und X5 durchgeführt.
X5 diente als Negativkontrolle, weil dieser, anders als b12 und CD4, an der Corezeptor-
Bindungsstelle von gp120 bindet (siehe Kapitel 1.6).
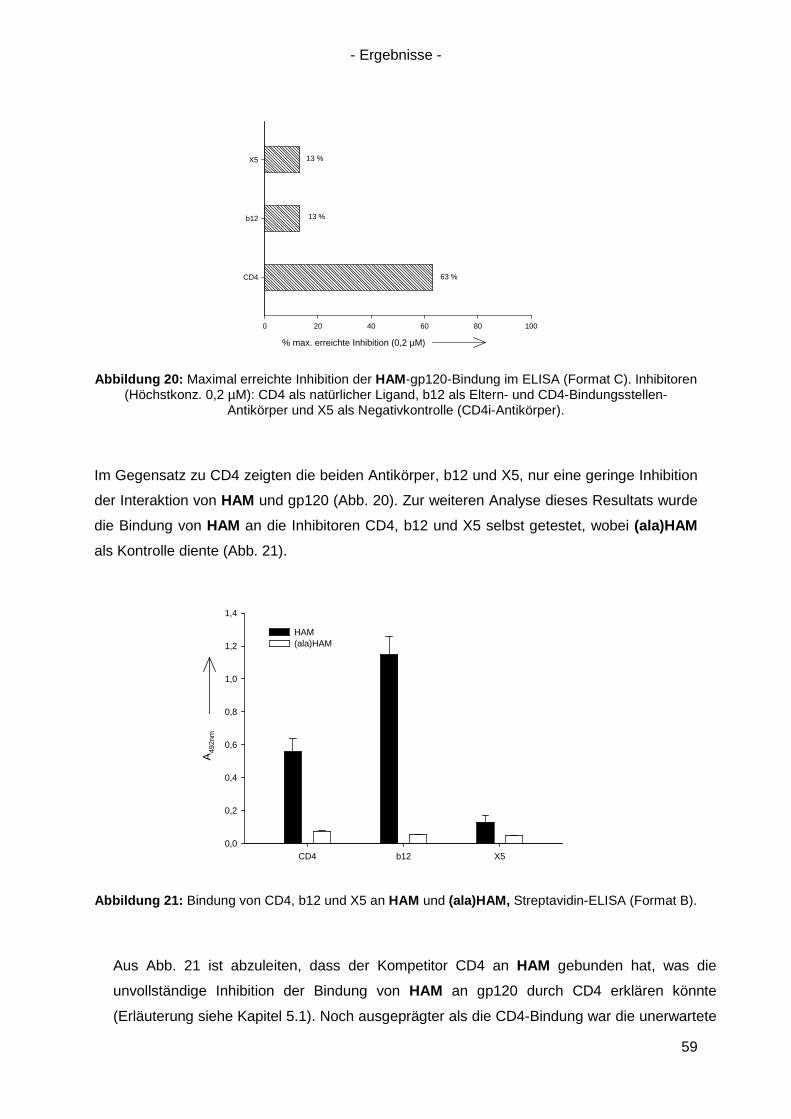
- Ergebnisse -
59
% max. erreichte Inhibition (0,2 µM)
0 20 40 60 80 100
CD4
b12
X5 13 %
13 %
63 %
Abbildung 20: Maximal erreichte Inhibition der HAM-gp120-Bindung im ELISA (Format C). Inhibitoren (Höchstkonz. 0,2 µM): CD4 als natürlicher Ligand, b12 als Eltern- und CD4-Bindungsstellen-
Antikörper und X5 als Negativkontrolle (CD4i-Antikörper).
Im Gegensatz zu CD4 zeigten die beiden Antikörper, b12 und X5, nur eine geringe Inhibition
der Interaktion von HAM und gp120 (Abb. 20). Zur weiteren Analyse dieses Resultats wurde
die Bindung von HAM an die Inhibitoren CD4, b12 und X5 selbst getestet, wobei (ala)HAM
als Kontrolle diente (Abb. 21).
CD4 b12 X5
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
HAM(ala)HAM
Abbildung 21: Bindung von CD4, b12 und X5 an HAM und (ala)HAM, Streptavidin-ELISA (Format B).
Aus Abb. 21 ist abzuleiten, dass der Kompetitor CD4 an HAM gebunden hat, was die
unvollständige Inhibition der Bindung von HAM an gp120 durch CD4 erklären könnte
(Erläuterung siehe Kapitel 5.1). Noch ausgeprägter als die CD4-Bindung war die unerwartete
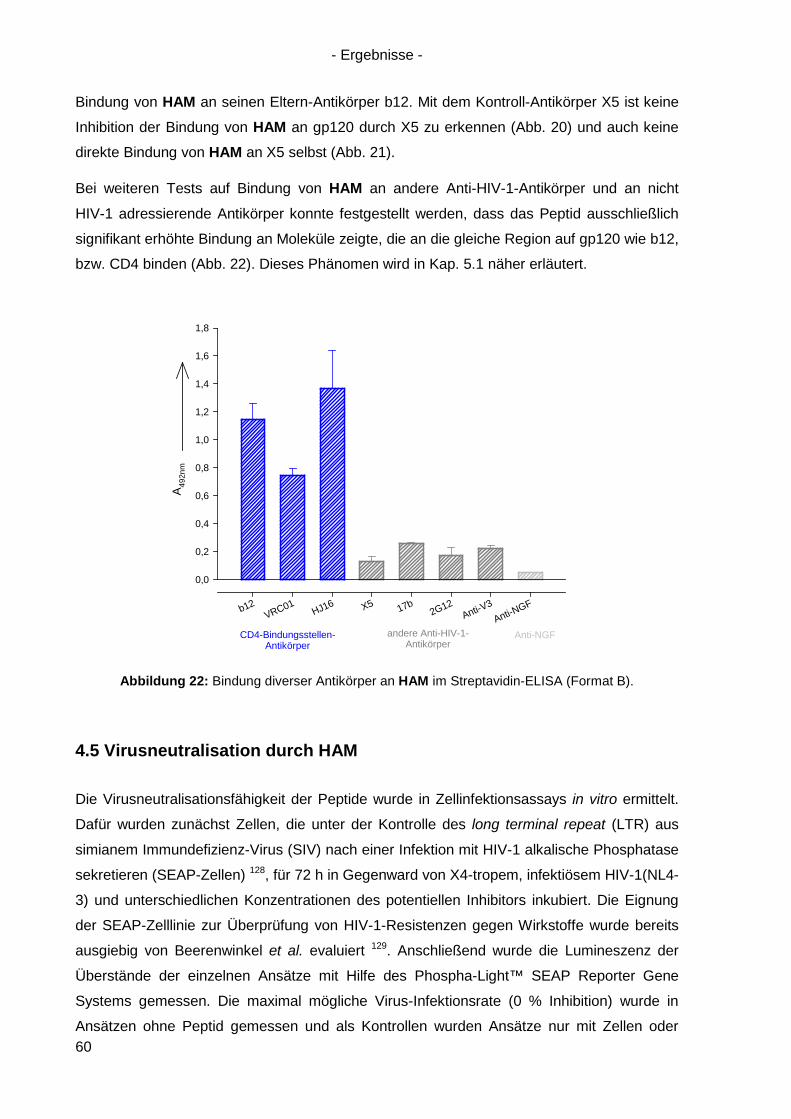
- Ergebnisse -
60
Bindung von HAM an seinen Eltern-Antikörper b12. Mit dem Kontroll-Antikörper X5 ist keine
Inhibition der Bindung von HAM an gp120 durch X5 zu erkennen (Abb. 20) und auch keine
direkte Bindung von HAM an X5 selbst (Abb. 21).
Bei weiteren Tests auf Bindung von HAM an andere Anti-HIV-1-Antikörper und an nicht
HIV-1 adressierende Antikörper konnte festgestellt werden, dass das Peptid ausschließlich
signifikant erhöhte Bindung an Moleküle zeigte, die an die gleiche Region auf gp120 wie b12,
bzw. CD4 binden (Abb. 22). Dieses Phänomen wird in Kap. 5.1 näher erläutert.
b12VRC01
HJ16 X5 17b2G12
Anti-V3Anti-NGF
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
CD4-Bindungsstellen-Antikörper
andere Anti-HIV-1-Antikörper
Anti-NGF
Abbildung 22: Bindung diverser Antikörper an HAM im Streptavidin-ELISA (Format B).
4.5 Virusneutralisation durch HAM
Die Virusneutralisationsfähigkeit der Peptide wurde in Zellinfektionsassays in vitro ermittelt.
Dafür wurden zunächst Zellen, die unter der Kontrolle des long terminal repeat (LTR) aus
simianem Immundefizienz-Virus (SIV) nach einer Infektion mit HIV-1 alkalische Phosphatase
sekretieren (SEAP-Zellen) 128, für 72 h in Gegenward von X4-tropem, infektiösem HIV-1(NL4-
3) und unterschiedlichen Konzentrationen des potentiellen Inhibitors inkubiert. Die Eignung
der SEAP-Zelllinie zur Überprüfung von HIV-1-Resistenzen gegen Wirkstoffe wurde bereits
ausgiebig von Beerenwinkel et al. evaluiert 129. Anschließend wurde die Lumineszenz der
Überstände der einzelnen Ansätze mit Hilfe des Phospha-Light™ SEAP Reporter Gene
Systems gemessen. Die maximal mögliche Virus-Infektionsrate (0 % Inhibition) wurde in
Ansätzen ohne Peptid gemessen und als Kontrollen wurden Ansätze nur mit Zellen oder
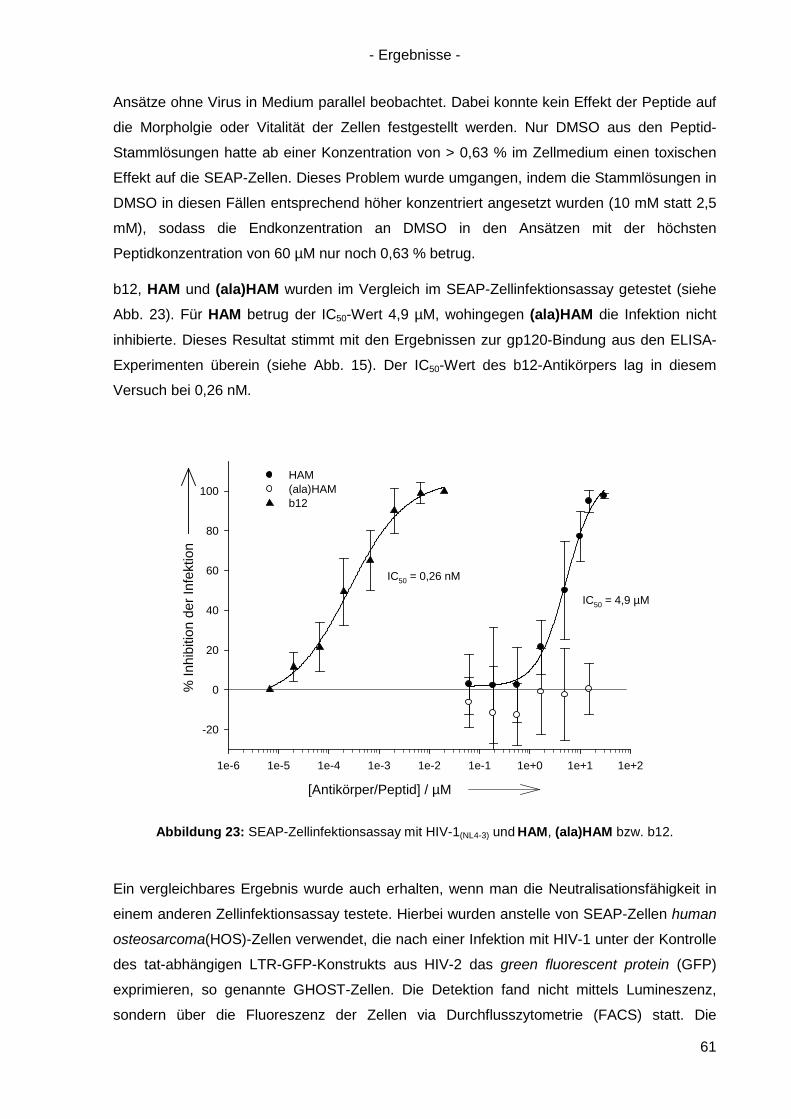
- Ergebnisse -
61
Ansätze ohne Virus in Medium parallel beobachtet. Dabei konnte kein Effekt der Peptide auf
die Morpholgie oder Vitalität der Zellen festgestellt werden. Nur DMSO aus den Peptid-
Stammlösungen hatte ab einer Konzentration von > 0,63 % im Zellmedium einen toxischen
Effekt auf die SEAP-Zellen. Dieses Problem wurde umgangen, indem die Stammlösungen in
DMSO in diesen Fällen entsprechend höher konzentriert angesetzt wurden (10 mM statt 2,5
mM), sodass die Endkonzentration an DMSO in den Ansätzen mit der höchsten
Peptidkonzentration von 60 µM nur noch 0,63 % betrug.
b12, HAM und (ala)HAM wurden im Vergleich im SEAP-Zellinfektionsassay getestet (siehe
Abb. 23). Für HAM betrug der IC50-Wert 4,9 µM, wohingegen (ala)HAM die Infektion nicht
inhibierte. Dieses Resultat stimmt mit den Ergebnissen zur gp120-Bindung aus den ELISA-
Experimenten überein (siehe Abb. 15). Der IC50-Wert des b12-Antikörpers lag in diesem
Versuch bei 0,26 nM.
[Antikörper/Peptid] / µM
1e-6 1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100HAM(ala)HAMb12
IC50 = 0,26 nM
IC50 = 4,9 µM
Abbildung 23: SEAP-Zellinfektionsassay mit HIV-1(NL4-3) und HAM, (ala)HAM bzw. b12.
Ein vergleichbares Ergebnis wurde auch erhalten, wenn man die Neutralisationsfähigkeit in
einem anderen Zellinfektionsassay testete. Hierbei wurden anstelle von SEAP-Zellen human
osteosarcoma(HOS)-Zellen verwendet, die nach einer Infektion mit HIV-1 unter der Kontrolle
des tat-abhängigen LTR-GFP-Konstrukts aus HIV-2 das green fluorescent protein (GFP)
exprimieren, so genannte GHOST-Zellen. Die Detektion fand nicht mittels Lumineszenz,
sondern über die Fluoreszenz der Zellen via Durchflusszytometrie (FACS) statt. Die
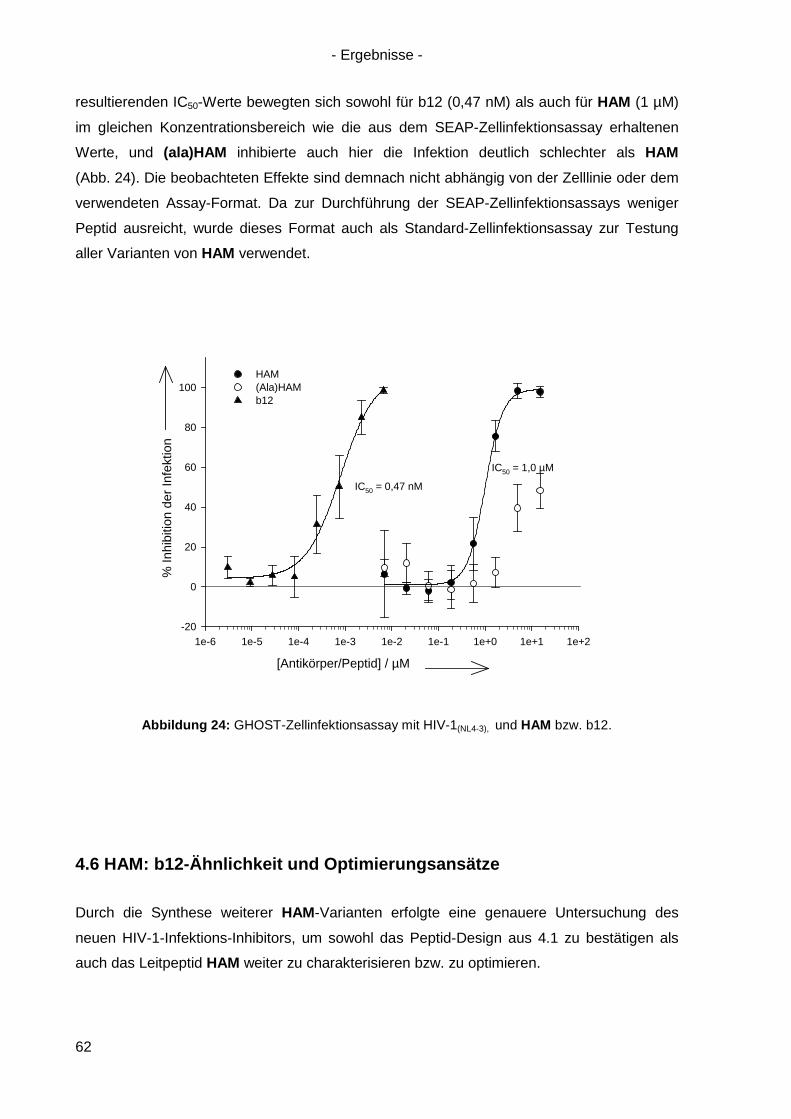
- Ergebnisse -
62
resultierenden IC50-Werte bewegten sich sowohl für b12 (0,47 nM) als auch für HAM (1 µM)
im gleichen Konzentrationsbereich wie die aus dem SEAP-Zellinfektionsassay erhaltenen
Werte, und (ala)HAM inhibierte auch hier die Infektion deutlich schlechter als HAM (Abb. 24). Die beobachteten Effekte sind demnach nicht abhängig von der Zelllinie oder dem
verwendeten Assay-Format. Da zur Durchführung der SEAP-Zellinfektionsassays weniger
Peptid ausreicht, wurde dieses Format auch als Standard-Zellinfektionsassay zur Testung
aller Varianten von HAM verwendet.
Abbildung 24: GHOST-Zellinfektionsassay mit HIV-1(NL4-3), und HAM bzw. b12.
4.6 HAM: b12-Ähnlichkeit und Optimierungsansätze
Durch die Synthese weiterer HAM-Varianten erfolgte eine genauere Untersuchung des
neuen HIV-1-Infektions-Inhibitors, um sowohl das Peptid-Design aus 4.1 zu bestätigen als auch das Leitpeptid HAM weiter zu charakterisieren bzw. zu optimieren.
[Antikörper/Peptid] / µM
1e-6 1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100HAM(Ala)HAMb12
IC50 = 0,47 nM
IC50 = 1,0 µM
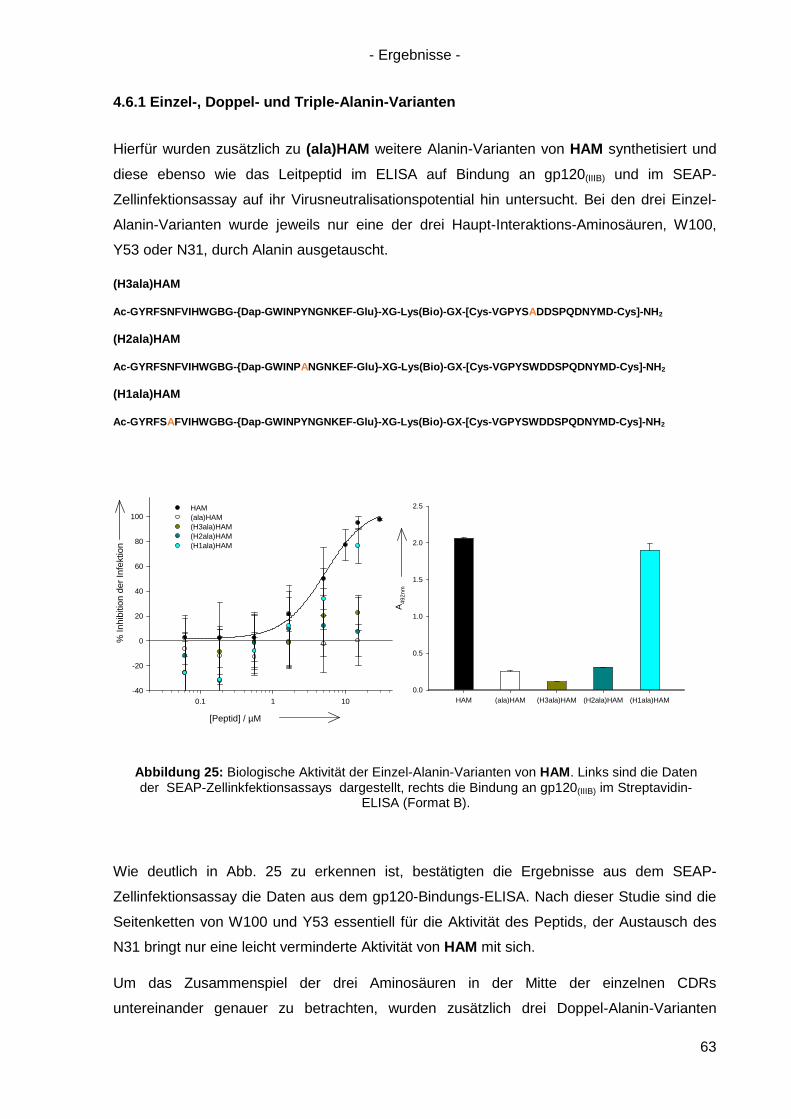
- Ergebnisse -
63
4.6.1 Einzel-, Doppel- und Triple-Alanin-Varianten
Hierfür wurden zusätzlich zu (ala)HAM weitere Alanin-Varianten von HAM synthetisiert und
diese ebenso wie das Leitpeptid im ELISA auf Bindung an gp120(IIIB) und im SEAP-
Zellinfektionsassay auf ihr Virusneutralisationspotential hin untersucht. Bei den drei Einzel-
Alanin-Varianten wurde jeweils nur eine der drei Haupt-Interaktions-Aminosäuren, W100,
Y53 oder N31, durch Alanin ausgetauscht.
(H3ala)HAM
Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2
(H2ala)HAM
Ac-GYRFSNFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2
(H1ala)HAM
Ac-GYRFSAFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2
Wie deutlich in Abb. 25 zu erkennen ist, bestätigten die Ergebnisse aus dem SEAP-
Zellinfektionsassay die Daten aus dem gp120-Bindungs-ELISA. Nach dieser Studie sind die
Seitenketten von W100 und Y53 essentiell für die Aktivität des Peptids, der Austausch des
N31 bringt nur eine leicht verminderte Aktivität von HAM mit sich.
Um das Zusammenspiel der drei Aminosäuren in der Mitte der einzelnen CDRs
untereinander genauer zu betrachten, wurden zusätzlich drei Doppel-Alanin-Varianten
[Peptid] / µM
0.1 1 10 100
% In
hibi
tion
der I
nfek
tion
-40
-20
0
20
40
60
80
100HAM(ala)HAM(H3ala)HAM(H2ala)HAM(H1ala)HAM
Abbildung 25: Biologische Aktivität der Einzel-Alanin-Varianten von HAM. Links sind die Daten der SEAP-Zellinkfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-
ELISA (Format B).
HAM (ala)HAM (H3ala)HAM (H2ala)HAM (H1ala)HAM
A 492n
m
0.0
0.5
1.0
1.5
2.0
2.5
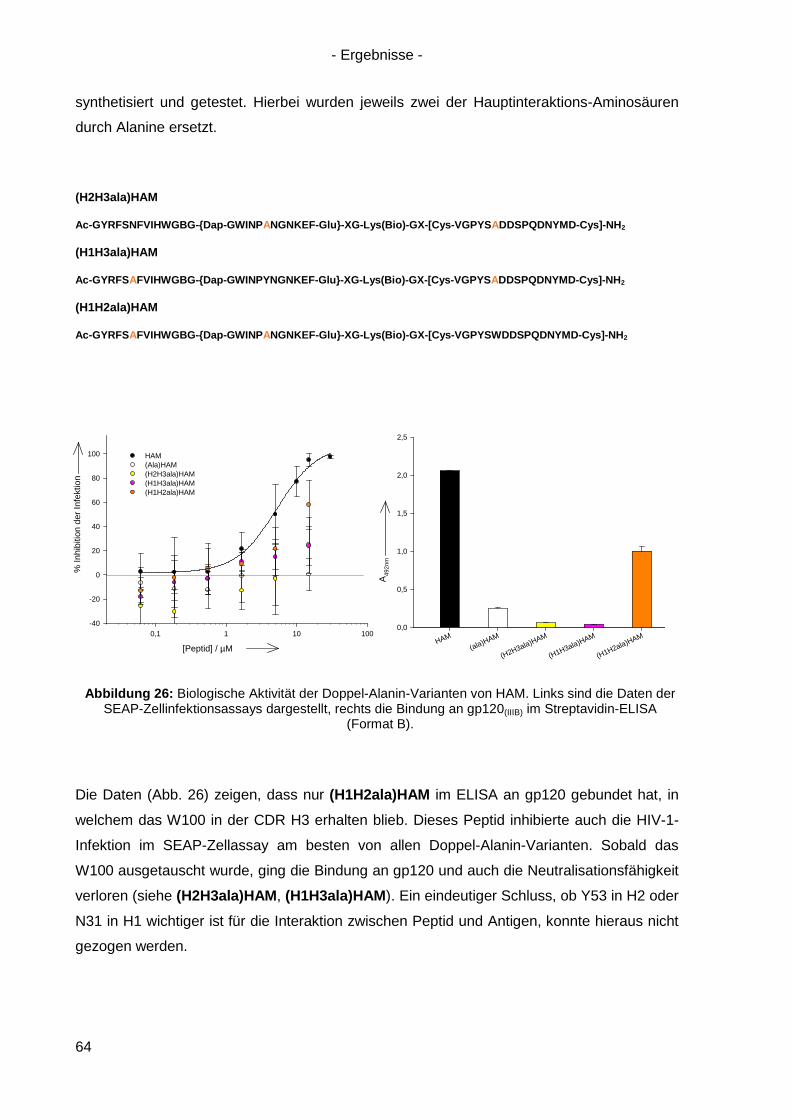
- Ergebnisse -
64
synthetisiert und getestet. Hierbei wurden jeweils zwei der Hauptinteraktions-Aminosäuren
durch Alanine ersetzt.
(H2H3ala)HAM
Ac-GYRFSNFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2
(H1H3ala)HAM
Ac-GYRFSAFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSADDSPQDNYMD-Cys]-NH2
(H1H2ala)HAM
Ac-GYRFSAFVIHWGBG-{Dap-GWINPANGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-VGPYSWDDSPQDNYMD-Cys]-NH2
Die Daten (Abb. 26) zeigen, dass nur (H1H2ala)HAM im ELISA an gp120 gebundet hat, in
welchem das W100 in der CDR H3 erhalten blieb. Dieses Peptid inhibierte auch die HIV-1-
Infektion im SEAP-Zellassay am besten von allen Doppel-Alanin-Varianten. Sobald das
W100 ausgetauscht wurde, ging die Bindung an gp120 und auch die Neutralisationsfähigkeit
verloren (siehe (H2H3ala)HAM, (H1H3ala)HAM). Ein eindeutiger Schluss, ob Y53 in H2 oder
N31 in H1 wichtiger ist für die Interaktion zwischen Peptid und Antigen, konnte hieraus nicht
gezogen werden.
[Peptid] / µM
0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
-40
-20
0
20
40
60
80
100 HAM(Ala)HAM(H2H3ala)HAM(H1H3ala)HAM(H1H2ala)HAM
HAM(ala)HAM
(H2H3ala)HAM
(H1H3ala)HAM
(H1H2ala)HAM
A 492n
m
0,0
0,5
1,0
1,5
2,0
2,5
Abbildung 26: Biologische Aktivität der Doppel-Alanin-Varianten von HAM. Links sind die Daten der SEAP-Zellinfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA
(Format B).
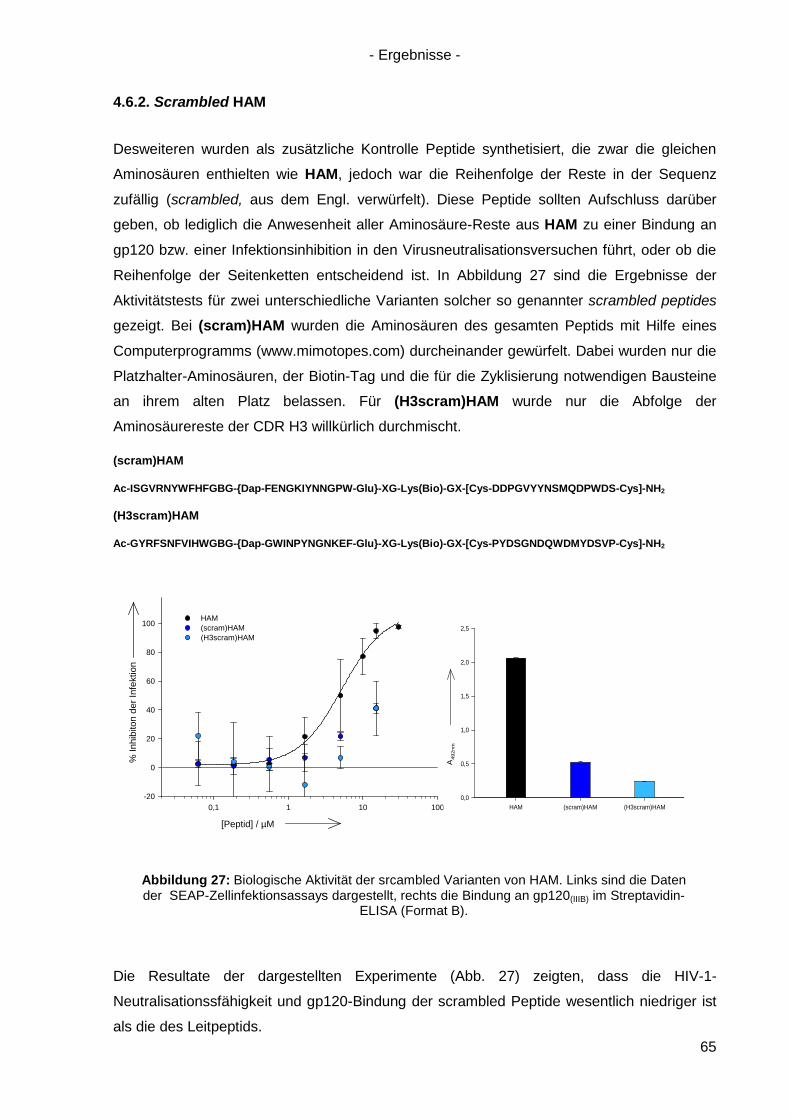
- Ergebnisse -
65
4.6.2. Scrambled HAM
Desweiteren wurden als zusätzliche Kontrolle Peptide synthetisiert, die zwar die gleichen
Aminosäuren enthielten wie HAM, jedoch war die Reihenfolge der Reste in der Sequenz
zufällig (scrambled, aus dem Engl. verwürfelt). Diese Peptide sollten Aufschluss darüber
geben, ob lediglich die Anwesenheit aller Aminosäure-Reste aus HAM zu einer Bindung an
gp120 bzw. einer Infektionsinhibition in den Virusneutralisationsversuchen führt, oder ob die
Reihenfolge der Seitenketten entscheidend ist. In Abbildung 27 sind die Ergebnisse der
Aktivitätstests für zwei unterschiedliche Varianten solcher so genannter scrambled peptides
gezeigt. Bei (scram)HAM wurden die Aminosäuren des gesamten Peptids mit Hilfe eines
Computerprogramms (www.mimotopes.com) durcheinander gewürfelt. Dabei wurden nur die
Platzhalter-Aminosäuren, der Biotin-Tag und die für die Zyklisierung notwendigen Bausteine
an ihrem alten Platz belassen. Für (H3scram)HAM wurde nur die Abfolge der
Aminosäurereste der CDR H3 willkürlich durchmischt.
(scram)HAM
Ac-ISGVRNYWFHFGBG-{Dap-FENGKIYNNGPW-Glu}-XG-Lys(Bio)-GX-[Cys-DDPGVYYNSMQDPWDS-Cys]-NH2
(H3scram)HAM
Ac-GYRFSNFVIHWGBG-{Dap-GWINPYNGNKEF-Glu}-XG-Lys(Bio)-GX-[Cys-PYDSGNDQWDMYDSVP-Cys]-NH2
Die Resultate der dargestellten Experimente (Abb. 27) zeigten, dass die HIV-1-
Neutralisationssfähigkeit und gp120-Bindung der scrambled Peptide wesentlich niedriger ist
als die des Leitpeptids.
[Peptid] / µM
0,1 1 10 100
% In
hibi
ton
der I
nfek
tion
-20
0
20
40
60
80
100 HAM(scram)HAM(H3scram)HAM
HAM (scram)HAM (H3scram)HAM
A49
2nm
0,0
0,5
1,0
1,5
2,0
2,5
Abbildung 27: Biologische Aktivität der srcambled Varianten von HAM. Links sind die Daten der SEAP-Zellinfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-
ELISA (Format B).
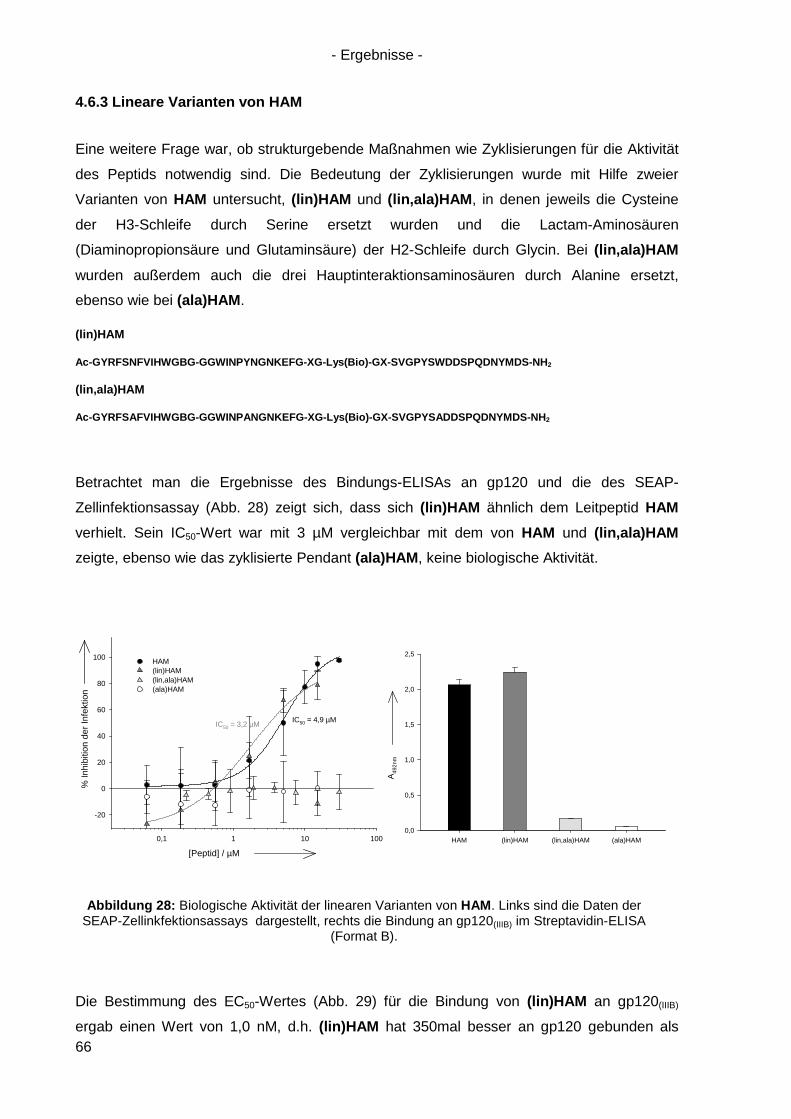
- Ergebnisse -
66
4.6.3 Lineare Varianten von HAM
Eine weitere Frage war, ob strukturgebende Maßnahmen wie Zyklisierungen für die Aktivität
des Peptids notwendig sind. Die Bedeutung der Zyklisierungen wurde mit Hilfe zweier
Varianten von HAM untersucht, (lin)HAM und (lin,ala)HAM, in denen jeweils die Cysteine
der H3-Schleife durch Serine ersetzt wurden und die Lactam-Aminosäuren
(Diaminopropionsäure und Glutaminsäure) der H2-Schleife durch Glycin. Bei (lin,ala)HAM
wurden außerdem auch die drei Hauptinteraktionsaminosäuren durch Alanine ersetzt, ebenso wie bei (ala)HAM.
(lin)HAM
Ac-GYRFSNFVIHWGBG-GGWINPYNGNKEFG-XG-Lys(Bio)-GX-SVGPYSWDDSPQDNYMDS-NH2
(lin,ala)HAM
Ac-GYRFSAFVIHWGBG-GGWINPANGNKEFG-XG-Lys(Bio)-GX-SVGPYSADDSPQDNYMDS-NH2
Betrachtet man die Ergebnisse des Bindungs-ELISAs an gp120 und die des SEAP-
Zellinfektionsassay (Abb. 28) zeigt sich, dass sich (lin)HAM ähnlich dem Leitpeptid HAM
verhielt. Sein IC50-Wert war mit 3 µM vergleichbar mit dem von HAM und (lin,ala)HAM
zeigte, ebenso wie das zyklisierte Pendant (ala)HAM, keine biologische Aktivität.
Die Bestimmung des EC50-Wertes (Abb. 29) für die Bindung von (lin)HAM an gp120(IIIB)
ergab einen Wert von 1,0 nM, d.h. (lin)HAM hat 350mal besser an gp120 gebunden als
[Peptid] / µM0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100 HAM (lin)HAM(lin,ala)HAM(ala)HAM
IC50 = 3,2 µM IC50 = 4,9 µM
HAM (lin)HAM (lin,ala)HAM (ala)HAM
A 492n
m
0,0
0,5
1,0
1,5
2,0
2,5
Abbildung 28: Biologische Aktivität der linearen Varianten von HAM. Links sind die Daten der SEAP-Zellinkfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA
(Format B).
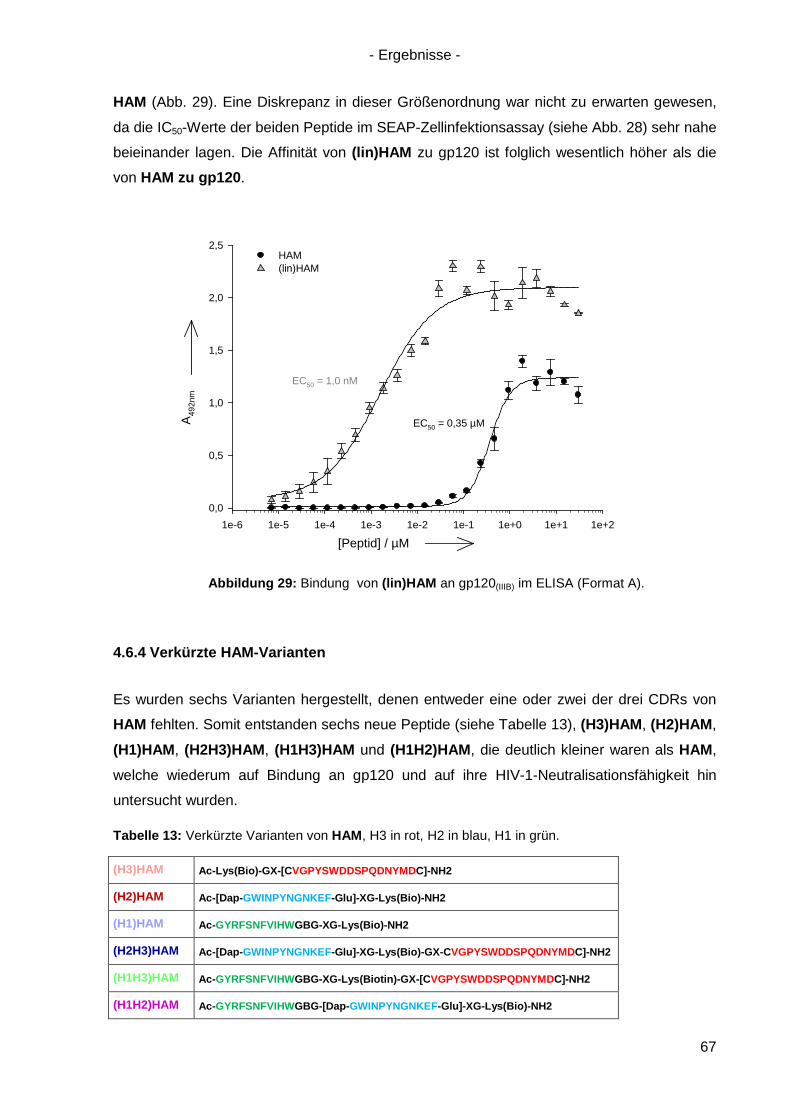
- Ergebnisse -
67
HAM (Abb. 29). Eine Diskrepanz in dieser Größenordnung war nicht zu erwarten gewesen,
da die IC50-Werte der beiden Peptide im SEAP-Zellinfektionsassay (siehe Abb. 28) sehr nahe
beieinander lagen. Die Affinität von (lin)HAM zu gp120 ist folglich wesentlich höher als die
von HAM zu gp120.
[Peptid] / µM1e-6 1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2
A 492n
m
0,0
0,5
1,0
1,5
2,0
2,5HAM(lin)HAM
EC50 = 1,0 nM
EC50 = 0,35 µM
4.6.4 Verkürzte HAM-Varianten
Es wurden sechs Varianten hergestellt, denen entweder eine oder zwei der drei CDRs von
HAM fehlten. Somit entstanden sechs neue Peptide (siehe Tabelle 13), (H3)HAM, (H2)HAM,
(H1)HAM, (H2H3)HAM, (H1H3)HAM und (H1H2)HAM, die deutlich kleiner waren als HAM,
welche wiederum auf Bindung an gp120 und auf ihre HIV-1-Neutralisationsfähigkeit hin
untersucht wurden.
Tabelle 13: Verkürzte Varianten von HAM, H3 in rot, H2 in blau, H1 in grün.
(H3)HAM Ac-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H2)HAM Ac-[Dap-GWINPYNGNKEF-Glu]-XG-Lys(Bio)-NH2
(H1)HAM Ac-GYRFSNFVIHWGBG-XG-Lys(Bio)-NH2
(H2H3)HAM Ac-[Dap-GWINPYNGNKEF-Glu]-XG-Lys(Bio)-GX-CVGPYSWDDSPQDNYMDC]-NH2
(H1H3)HAM Ac-GYRFSNFVIHWGBG-XG-Lys(Biotin)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H1H2)HAM Ac-GYRFSNFVIHWGBG-[Dap-GWINPYNGNKEF-Glu]-XG-Lys(Bio)-NH2
Abbildung 29: Bindung von (lin)HAM an gp120(IIIB) im ELISA (Format A).
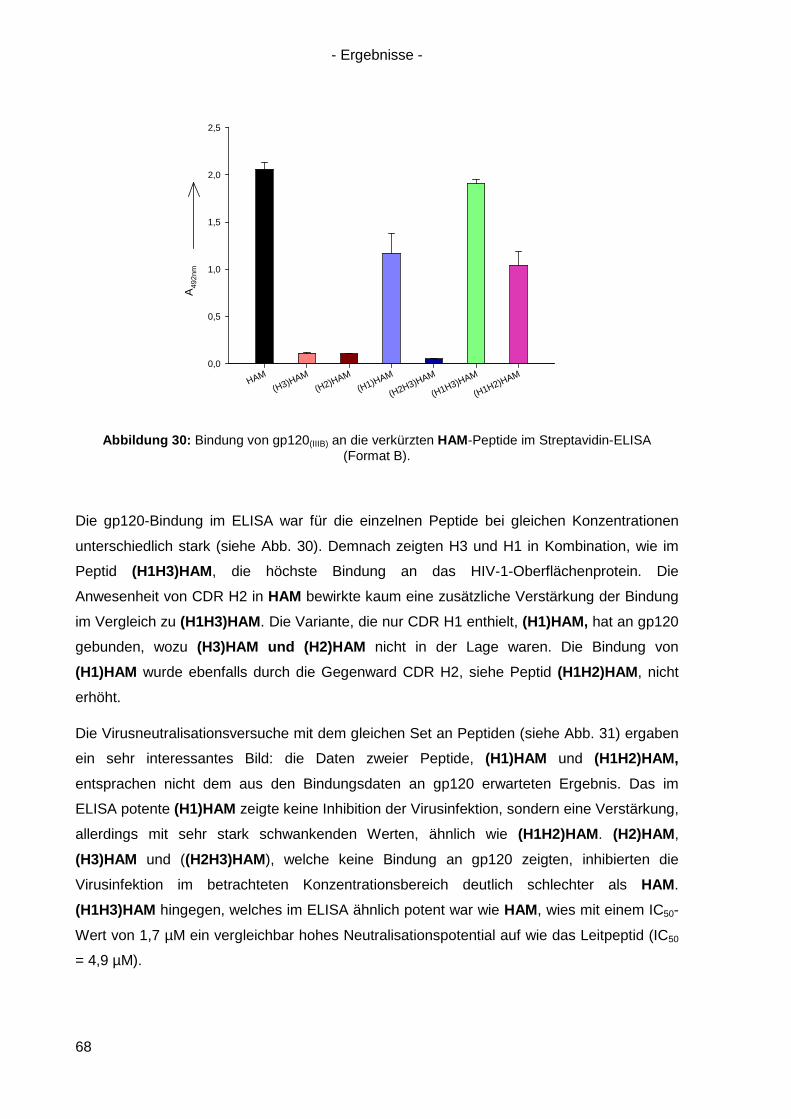
- Ergebnisse -
68
HAM(H3)HAM
(H2)HAM(H1)HAM
(H2H3)HAM
(H1H3)HAM
(H1H2)HAM
A 492n
m
0,0
0,5
1,0
1,5
2,0
2,5
Abbildung 30: Bindung von gp120(IIIB) an die verkürzten HAM-Peptide im Streptavidin-ELISA (Format B).
Die gp120-Bindung im ELISA war für die einzelnen Peptide bei gleichen Konzentrationen
unterschiedlich stark (siehe Abb. 30). Demnach zeigten H3 und H1 in Kombination, wie im
Peptid (H1H3)HAM, die höchste Bindung an das HIV-1-Oberflächenprotein. Die
Anwesenheit von CDR H2 in HAM bewirkte kaum eine zusätzliche Verstärkung der Bindung
im Vergleich zu (H1H3)HAM. Die Variante, die nur CDR H1 enthielt, (H1)HAM, hat an gp120
gebunden, wozu (H3)HAM und (H2)HAM nicht in der Lage waren. Die Bindung von
(H1)HAM wurde ebenfalls durch die Gegenward CDR H2, siehe Peptid (H1H2)HAM, nicht
erhöht.
Die Virusneutralisationsversuche mit dem gleichen Set an Peptiden (siehe Abb. 31) ergaben
ein sehr interessantes Bild: die Daten zweier Peptide, (H1)HAM und (H1H2)HAM, entsprachen nicht dem aus den Bindungsdaten an gp120 erwarteten Ergebnis. Das im
ELISA potente (H1)HAM zeigte keine Inhibition der Virusinfektion, sondern eine Verstärkung,
allerdings mit sehr stark schwankenden Werten, ähnlich wie (H1H2)HAM. (H2)HAM,
(H3)HAM und ((H2H3)HAM), welche keine Bindung an gp120 zeigten, inhibierten die
Virusinfektion im betrachteten Konzentrationsbereich deutlich schlechter als HAM.
(H1H3)HAM hingegen, welches im ELISA ähnlich potent war wie HAM, wies mit einem IC50-
Wert von 1,7 µM ein vergleichbar hohes Neutralisationspotential auf wie das Leitpeptid (IC50
= 4,9 µM).
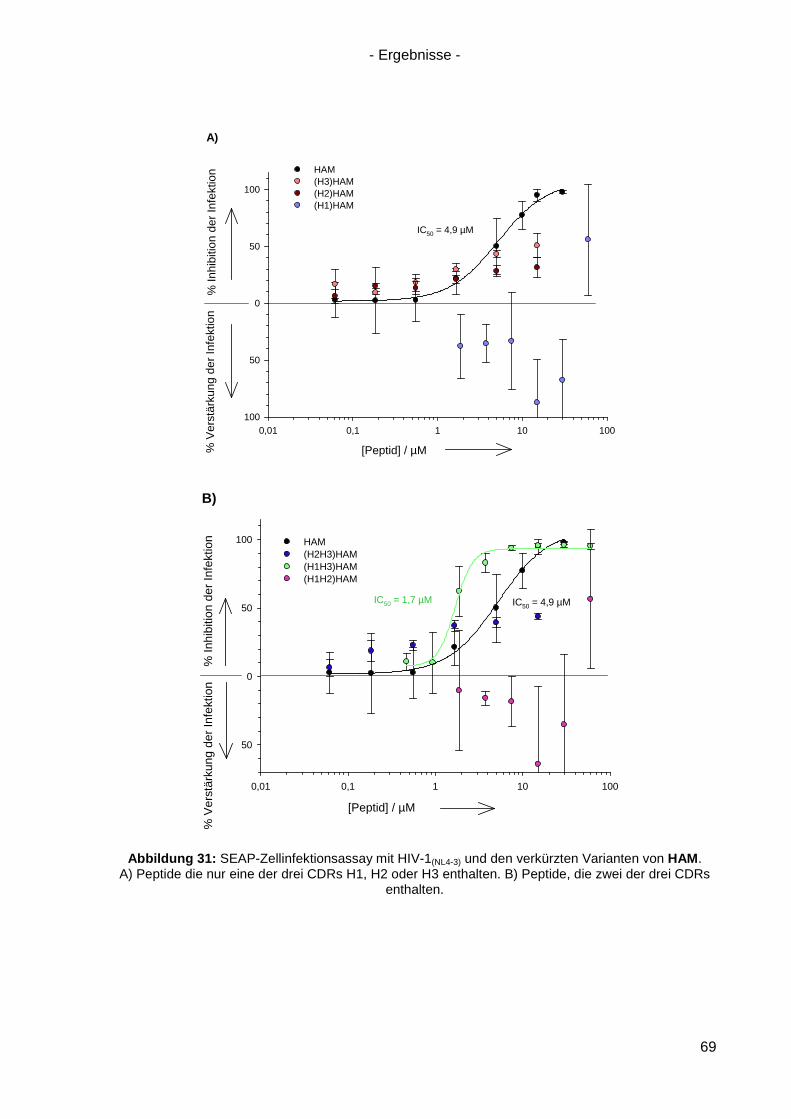
- Ergebnisse -
69
[Peptid] / µM
0,01 0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
100
50
0
50
100
HAM(H3)HAM (H2)HAM(H1)HAM
% V
erst
ärku
ng d
er In
fekt
ion
A)
IC50 = 4,9 µM
[Peptid] / µM
0,01 0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
50
0
50
100 HAM(H2H3)HAM(H1H3)HAM(H1H2)HAM
% V
erst
ärku
ng d
er In
fekt
ion
B)
IC50 = 4,9 µMIC50 = 1,7 µM
Abbildung 31: SEAP-Zellinfektionsassay mit HIV-1(NL4-3) und den verkürzten Varianten von HAM. A) Peptide die nur eine der drei CDRs H1, H2 oder H3 enthalten. B) Peptide, die zwei der drei CDRs
enthalten.
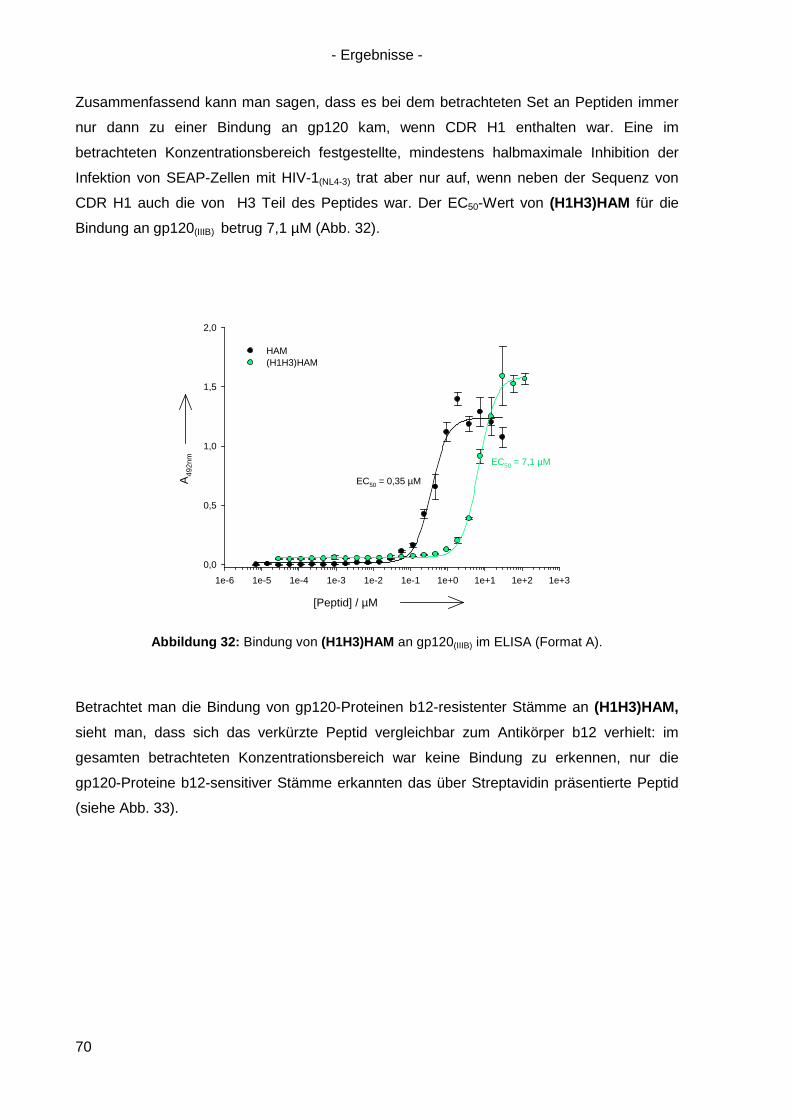
- Ergebnisse -
70
Zusammenfassend kann man sagen, dass es bei dem betrachteten Set an Peptiden immer
nur dann zu einer Bindung an gp120 kam, wenn CDR H1 enthalten war. Eine im
betrachteten Konzentrationsbereich festgestellte, mindestens halbmaximale Inhibition der
Infektion von SEAP-Zellen mit HIV-1(NL4-3) trat aber nur auf, wenn neben der Sequenz von
CDR H1 auch die von H3 Teil des Peptides war. Der EC50-Wert von (H1H3)HAM für die
Bindung an gp120(IIIB) betrug 7,1 µM (Abb. 32).
[Peptid] / µM
1e-6 1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2 1e+3
A 492n
m
0,0
0,5
1,0
1,5
2,0
HAM(H1H3)HAM
EC50 = 0,35 µM
EC50 = 7,1 µM
Abbildung 32: Bindung von (H1H3)HAM an gp120(IIIB) im ELISA (Format A).
Betrachtet man die Bindung von gp120-Proteinen b12-resistenter Stämme an (H1H3)HAM, sieht man, dass sich das verkürzte Peptid vergleichbar zum Antikörper b12 verhielt: im
gesamten betrachteten Konzentrationsbereich war keine Bindung zu erkennen, nur die
gp120-Proteine b12-sensitiver Stämme erkannten das über Streptavidin präsentierte Peptid
(siehe Abb. 33).
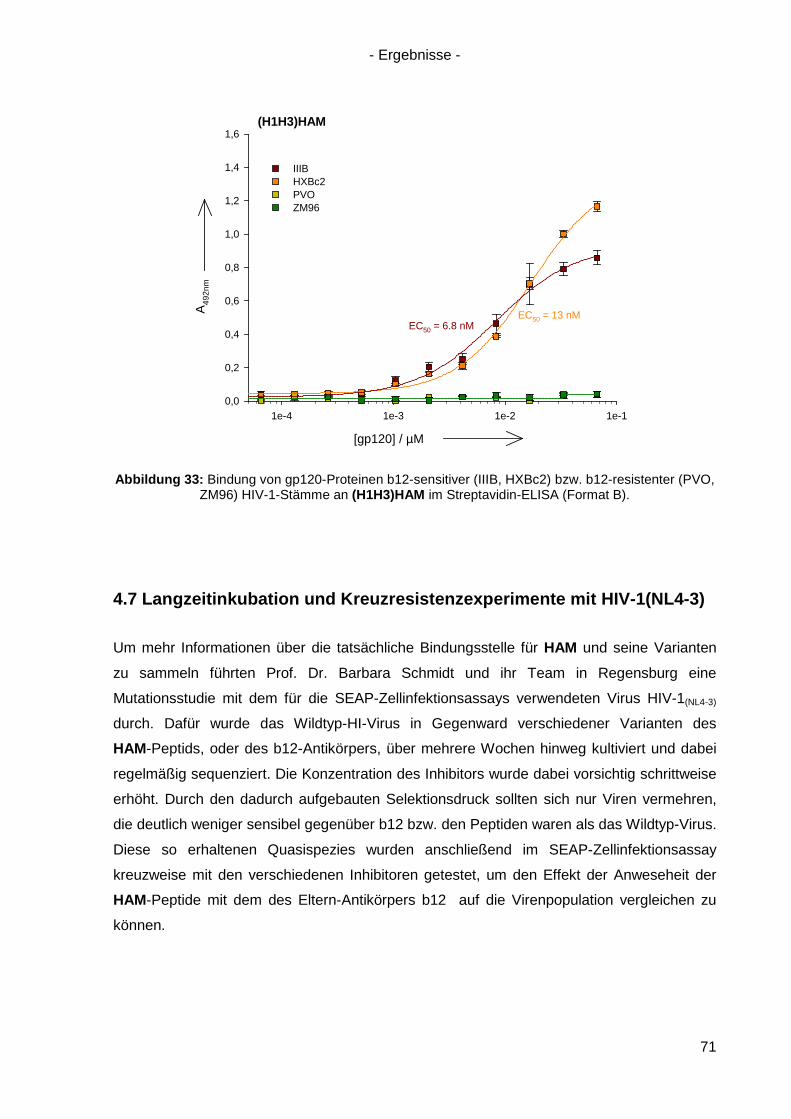
- Ergebnisse -
71
[gp120] / µM
1e-4 1e-3 1e-2 1e-1
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
IIIB HXBc2 PVO ZM96
(H1H3)HAM
EC50 = 13 nMEC50 = 6.8 nM
Abbildung 33: Bindung von gp120-Proteinen b12-sensitiver (IIIB, HXBc2) bzw. b12-resistenter (PVO, ZM96) HIV-1-Stämme an (H1H3)HAM im Streptavidin-ELISA (Format B).
4.7 Langzeitinkubation und Kreuzresistenzexperimente mit HIV-1(NL4-3)
Um mehr Informationen über die tatsächliche Bindungsstelle für HAM und seine Varianten
zu sammeln führten Prof. Dr. Barbara Schmidt und ihr Team in Regensburg eine
Mutationsstudie mit dem für die SEAP-Zellinfektionsassays verwendeten Virus HIV-1(NL4-3)
durch. Dafür wurde das Wildtyp-HI-Virus in Gegenward verschiedener Varianten des
HAM-Peptids, oder des b12-Antikörpers, über mehrere Wochen hinweg kultiviert und dabei
regelmäßig sequenziert. Die Konzentration des Inhibitors wurde dabei vorsichtig schrittweise
erhöht. Durch den dadurch aufgebauten Selektionsdruck sollten sich nur Viren vermehren,
die deutlich weniger sensibel gegenüber b12 bzw. den Peptiden waren als das Wildtyp-Virus.
Diese so erhaltenen Quasispezies wurden anschließend im SEAP-Zellinfektionsassay
kreuzweise mit den verschiedenen Inhibitoren getestet, um den Effekt der Anweseheit der
HAM-Peptide mit dem des Eltern-Antikörpers b12 auf die Virenpopulation vergleichen zu
können.
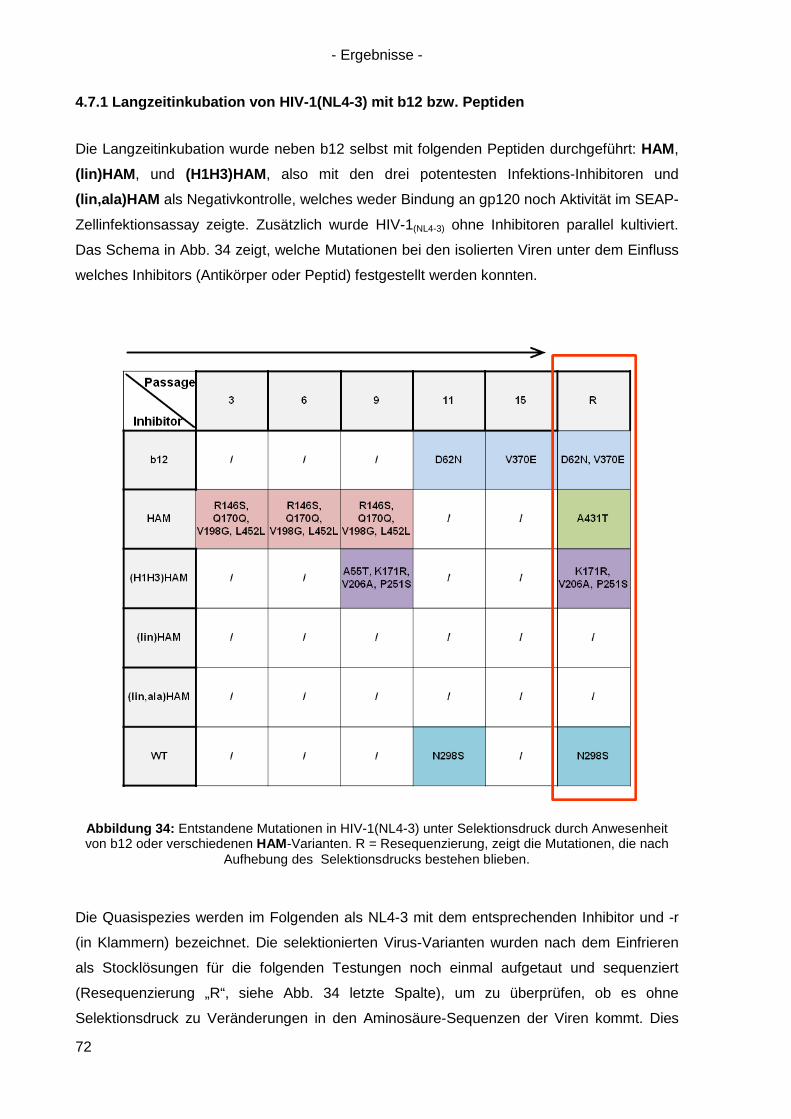
- Ergebnisse -
72
4.7.1 Langzeitinkubation von HIV-1(NL4-3) mit b12 bzw. Peptiden
Die Langzeitinkubation wurde neben b12 selbst mit folgenden Peptiden durchgeführt: HAM,
(lin)HAM, und (H1H3)HAM, also mit den drei potentesten Infektions-Inhibitoren und
(lin,ala)HAM als Negativkontrolle, welches weder Bindung an gp120 noch Aktivität im SEAP-
Zellinfektionsassay zeigte. Zusätzlich wurde HIV-1(NL4-3) ohne Inhibitoren parallel kultiviert.
Das Schema in Abb. 34 zeigt, welche Mutationen bei den isolierten Viren unter dem Einfluss
welches Inhibitors (Antikörper oder Peptid) festgestellt werden konnten.
Abbildung 34: Entstandene Mutationen in HIV-1(NL4-3) unter Selektionsdruck durch Anwesenheit von b12 oder verschiedenen HAM-Varianten. R = Resequenzierung, zeigt die Mutationen, die nach
Aufhebung des Selektionsdrucks bestehen blieben.
Die Quasispezies werden im Folgenden als NL4-3 mit dem entsprechenden Inhibitor und -r
(in Klammern) bezeichnet. Die selektionierten Virus-Varianten wurden nach dem Einfrieren
als Stocklösungen für die folgenden Testungen noch einmal aufgetaut und sequenziert
(Resequenzierung „R“, siehe Abb. 34 letzte Spalte), um zu überprüfen, ob es ohne
Selektionsdruck zu Veränderungen in den Aminosäure-Sequenzen der Viren kommt. Dies
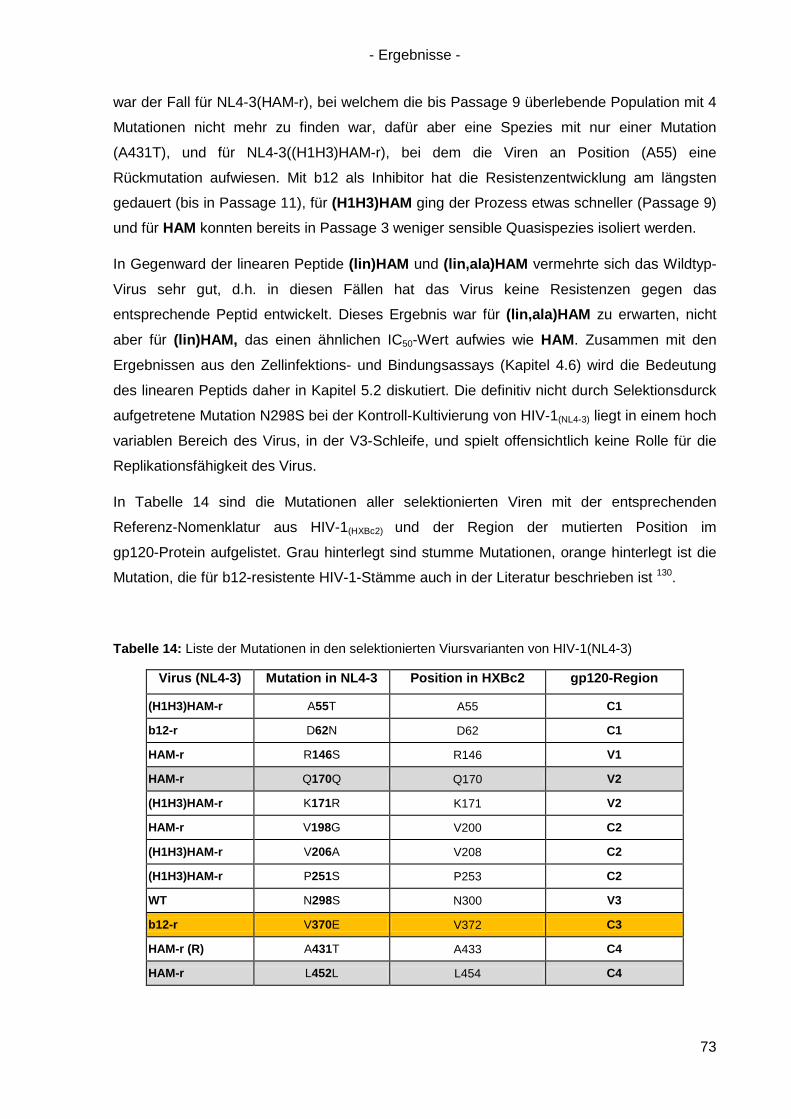
- Ergebnisse -
73
war der Fall für NL4-3(HAM-r), bei welchem die bis Passage 9 überlebende Population mit 4
Mutationen nicht mehr zu finden war, dafür aber eine Spezies mit nur einer Mutation
(A431T), und für NL4-3((H1H3)HAM-r), bei dem die Viren an Position (A55) eine
Rückmutation aufwiesen. Mit b12 als Inhibitor hat die Resistenzentwicklung am längsten
gedauert (bis in Passage 11), für (H1H3)HAM ging der Prozess etwas schneller (Passage 9)
und für HAM konnten bereits in Passage 3 weniger sensible Quasispezies isoliert werden.
In Gegenward der linearen Peptide (lin)HAM und (lin,ala)HAM vermehrte sich das Wildtyp-
Virus sehr gut, d.h. in diesen Fällen hat das Virus keine Resistenzen gegen das
entsprechende Peptid entwickelt. Dieses Ergebnis war für (lin,ala)HAM zu erwarten, nicht
aber für (lin)HAM, das einen ähnlichen IC50-Wert aufwies wie HAM. Zusammen mit den
Ergebnissen aus den Zellinfektions- und Bindungsassays (Kapitel 4.6) wird die Bedeutung
des linearen Peptids daher in Kapitel 5.2 diskutiert. Die definitiv nicht durch Selektionsdurck
aufgetretene Mutation N298S bei der Kontroll-Kultivierung von HIV-1(NL4-3) liegt in einem hoch
variablen Bereich des Virus, in der V3-Schleife, und spielt offensichtlich keine Rolle für die
Replikationsfähigkeit des Virus.
In Tabelle 14 sind die Mutationen aller selektionierten Viren mit der entsprechenden
Referenz-Nomenklatur aus HIV-1(HXBc2) und der Region der mutierten Position im
gp120-Protein aufgelistet. Grau hinterlegt sind stumme Mutationen, orange hinterlegt ist die
Mutation, die für b12-resistente HIV-1-Stämme auch in der Literatur beschrieben ist 130.
Tabelle 14: Liste der Mutationen in den selektionierten Viursvarianten von HIV-1(NL4-3)
Virus (NL4-3) Mutation in NL4-3 Position in HXBc2 gp120-Region
(H1H3)HAM-r A55T A55 C1
b12-r D62N D62 C1
HAM-r R146S R146 V1
HAM-r Q170Q Q170 V2
(H1H3)HAM-r K171R K171 V2
HAM-r V198G V200 C2
(H1H3)HAM-r V206A V208 C2
(H1H3)HAM-r P251S P253 C2
WT N298S N300 V3
b12-r V370E V372 C3
HAM-r (R) A431T A433 C4
HAM-r L452L L454 C4
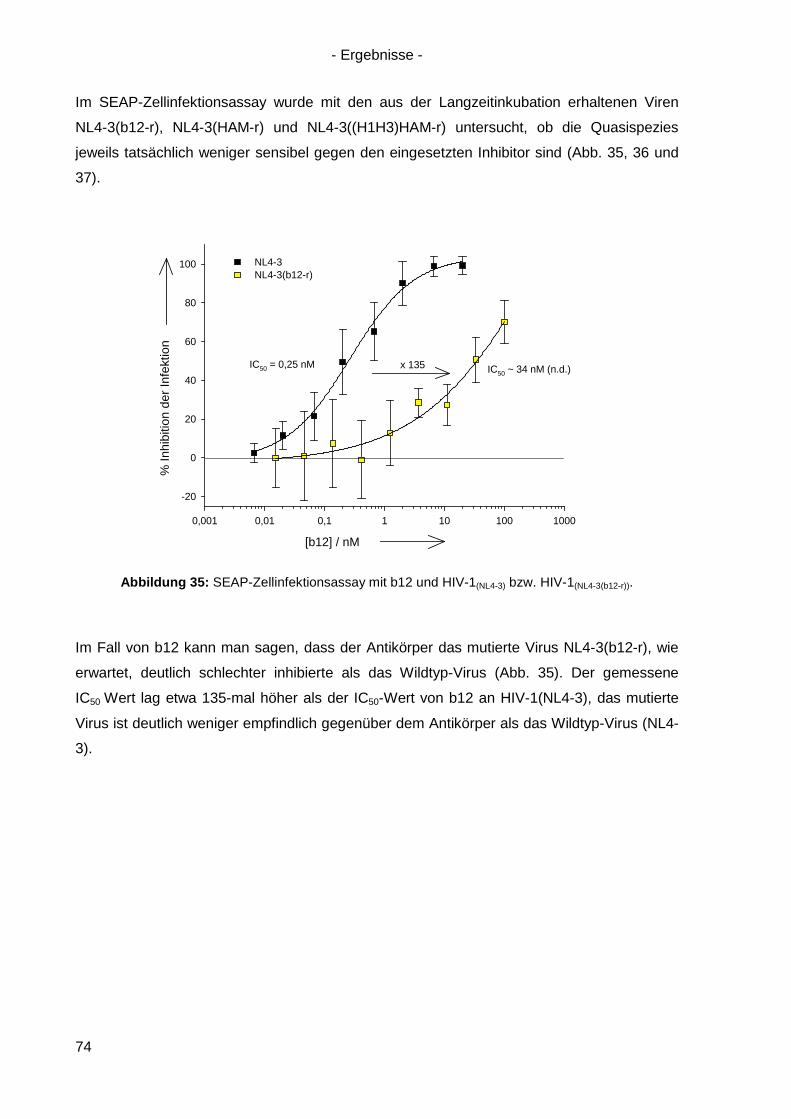
- Ergebnisse -
74
Im SEAP-Zellinfektionsassay wurde mit den aus der Langzeitinkubation erhaltenen Viren
NL4-3(b12-r), NL4-3(HAM-r) und NL4-3((H1H3)HAM-r) untersucht, ob die Quasispezies
jeweils tatsächlich weniger sensibel gegen den eingesetzten Inhibitor sind (Abb. 35, 36 und
37).
[b12] / nM
0,001 0,01 0,1 1 10 100 1000
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100 NL4-3NL4-3(b12-r)
IC50 = 0,25 nM IC50 ~ 34 nM (n.d.)x 135
Abbildung 35: SEAP-Zellinfektionsassay mit b12 und HIV-1(NL4-3) bzw. HIV-1(NL4-3(b12-r)).
Im Fall von b12 kann man sagen, dass der Antikörper das mutierte Virus NL4-3(b12-r), wie
erwartet, deutlich schlechter inhibierte als das Wildtyp-Virus (Abb. 35). Der gemessene
IC50 Wert lag etwa 135-mal höher als der IC50-Wert von b12 an HIV-1(NL4-3), das mutierte
Virus ist deutlich weniger empfindlich gegenüber dem Antikörper als das Wildtyp-Virus (NL4-
3).
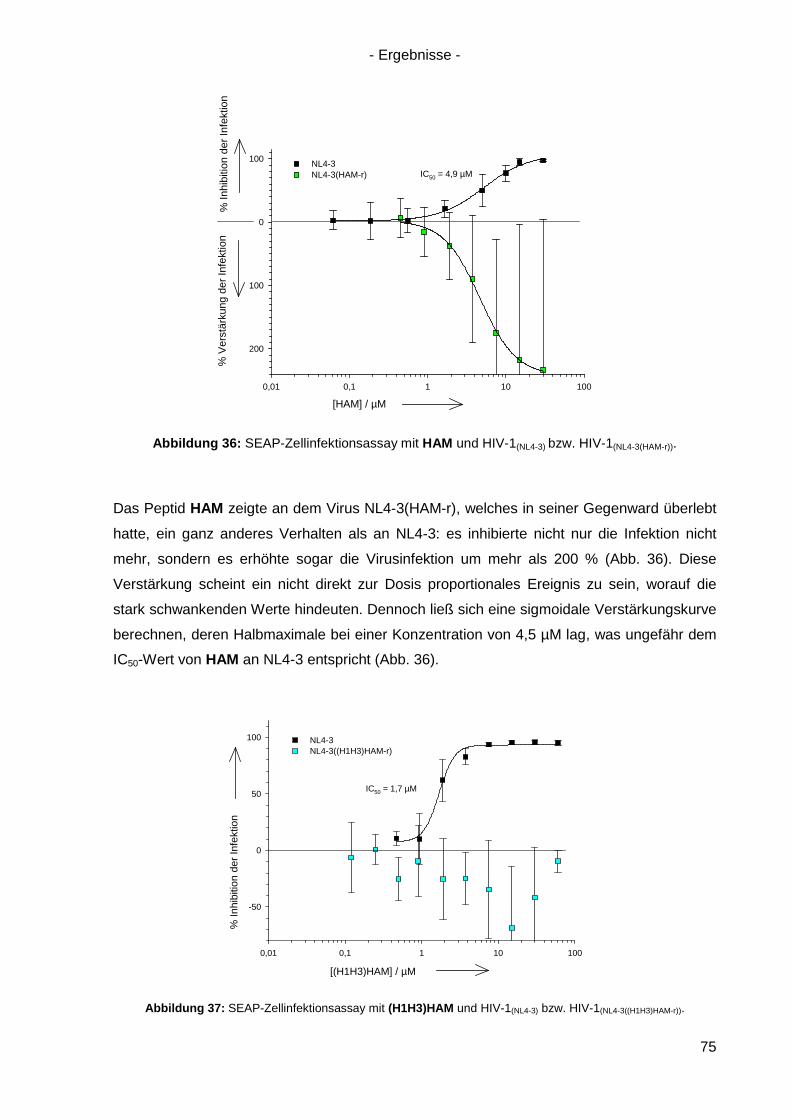
- Ergebnisse -
75
[HAM] / µM0,01 0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
200
100
0
100 NL4-3NL4-3(HAM-r) IC50 = 4,9 µM
% V
erst
ärku
ng d
er In
fekt
ion
Abbildung 36: SEAP-Zellinfektionsassay mit HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3(HAM-r)).
Das Peptid HAM zeigte an dem Virus NL4-3(HAM-r), welches in seiner Gegenward überlebt
hatte, ein ganz anderes Verhalten als an NL4-3: es inhibierte nicht nur die Infektion nicht
mehr, sondern es erhöhte sogar die Virusinfektion um mehr als 200 % (Abb. 36). Diese
Verstärkung scheint ein nicht direkt zur Dosis proportionales Ereignis zu sein, worauf die
stark schwankenden Werte hindeuten. Dennoch ließ sich eine sigmoidale Verstärkungskurve
berechnen, deren Halbmaximale bei einer Konzentration von 4,5 µM lag, was ungefähr dem IC50-Wert von HAM an NL4-3 entspricht (Abb. 36).
[(H1H3)HAM] / µM
0,01 0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
-50
0
50
100 NL4-3NL4-3((H1H3)HAM-r)
IC50 = 1,7 µM
Abbildung 37: SEAP-Zellinfektionsassay mit (H1H3)HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3((H1H3)HAM-r)).
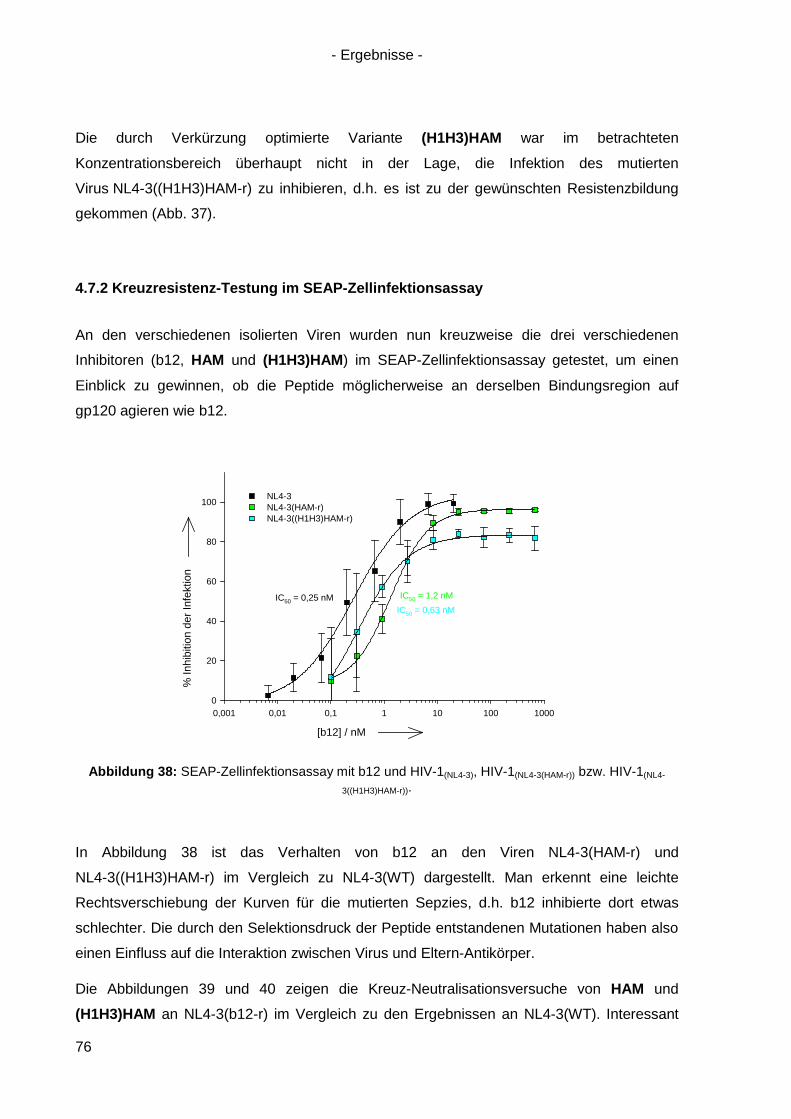
- Ergebnisse -
76
Die durch Verkürzung optimierte Variante (H1H3)HAM war im betrachteten
Konzentrationsbereich überhaupt nicht in der Lage, die Infektion des mutierten
Virus NL4-3((H1H3)HAM-r) zu inhibieren, d.h. es ist zu der gewünschten Resistenzbildung
gekommen (Abb. 37).
4.7.2 Kreuzresistenz-Testung im SEAP-Zellinfektionsassay
An den verschiedenen isolierten Viren wurden nun kreuzweise die drei verschiedenen
Inhibitoren (b12, HAM und (H1H3)HAM) im SEAP-Zellinfektionsassay getestet, um einen
Einblick zu gewinnen, ob die Peptide möglicherweise an derselben Bindungsregion auf
gp120 agieren wie b12.
[b12] / nM
0,001 0,01 0,1 1 10 100 1000
% In
hibi
tion
der I
nfek
tion
0
20
40
60
80
100 NL4-3NL4-3(HAM-r) NL4-3((H1H3)HAM-r)
IC50 = 0,25 nM IC50 = 1,2 nMIC50 = 0,63 nM
Abbildung 38: SEAP-Zellinfektionsassay mit b12 und HIV-1(NL4-3), HIV-1(NL4-3(HAM-r)) bzw. HIV-1(NL4-
3((H1H3)HAM-r)).
In Abbildung 38 ist das Verhalten von b12 an den Viren NL4-3(HAM-r) und
NL4-3((H1H3)HAM-r) im Vergleich zu NL4-3(WT) dargestellt. Man erkennt eine leichte
Rechtsverschiebung der Kurven für die mutierten Sepzies, d.h. b12 inhibierte dort etwas
schlechter. Die durch den Selektionsdruck der Peptide entstandenen Mutationen haben also
einen Einfluss auf die Interaktion zwischen Virus und Eltern-Antikörper.
Die Abbildungen 39 und 40 zeigen die Kreuz-Neutralisationsversuche von HAM und
(H1H3)HAM an NL4-3(b12-r) im Vergleich zu den Ergebnissen an NL4-3(WT). Interessant
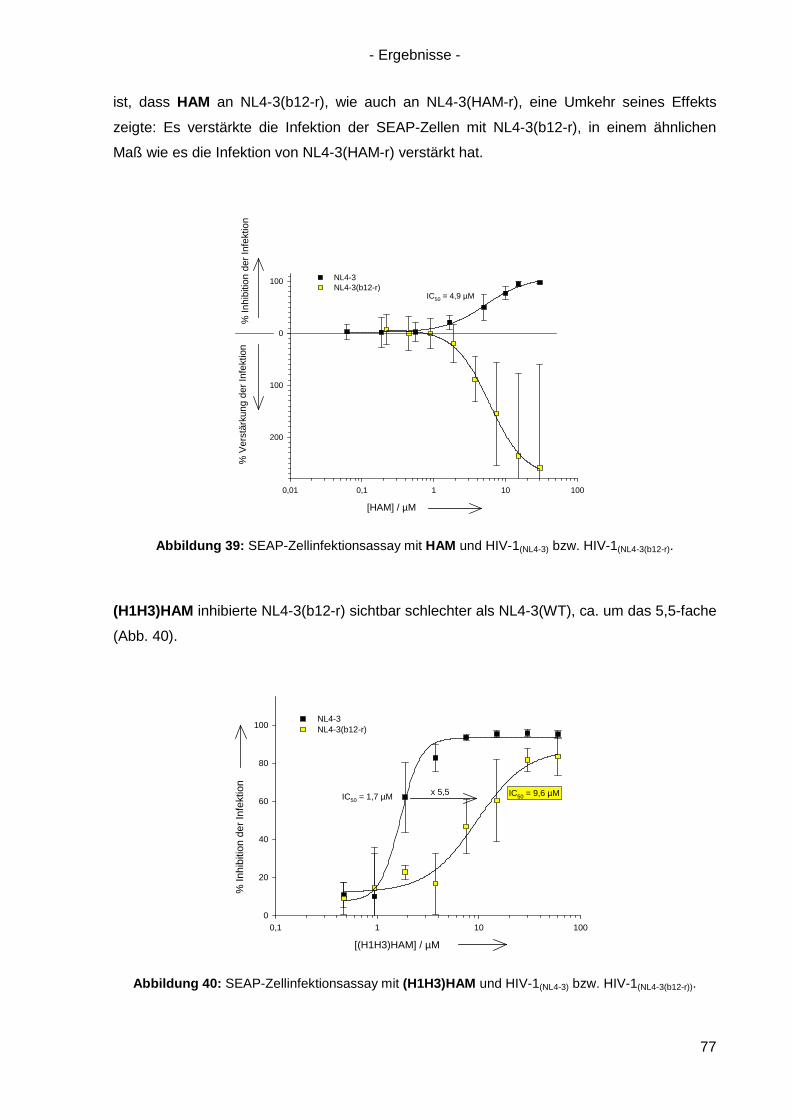
- Ergebnisse -
77
ist, dass HAM an NL4-3(b12-r), wie auch an NL4-3(HAM-r), eine Umkehr seines Effekts
zeigte: Es verstärkte die Infektion der SEAP-Zellen mit NL4-3(b12-r), in einem ähnlichen
Maß wie es die Infektion von NL4-3(HAM-r) verstärkt hat.
[HAM] / µM
0,01 0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
200
100
0
100 NL4-3 NL4-3(b12-r)
IC50 = 4,9 µM
% V
erst
ärku
ng d
er In
fekt
ion
Abbildung 39: SEAP-Zellinfektionsassay mit HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3(b12-r).
(H1H3)HAM inhibierte NL4-3(b12-r) sichtbar schlechter als NL4-3(WT), ca. um das 5,5-fache
(Abb. 40).
[(H1H3)HAM] / µM
0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
0
20
40
60
80
100NL4-3NL4-3(b12-r)
IC50 = 1,7 µM IC50 = 9,6 µMx 5,5
Abbildung 40: SEAP-Zellinfektionsassay mit (H1H3)HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3(b12-r)).

- Ergebnisse -
78
4.8 Anwendung des Design-Ansatzes von HAM auf einen anderen Antikörper
Um zu zeigen, dass die gewählte Vorgehensweise zur Herstellung eines peptidischen
Paratop-Mimetikums auch auf andere Antikörper übertragbar ist, wurde ein zweiter Anti-
HIV-1-Antikörper ausgewählt, nämlich X5. Dieser bindet nicht an die CD4-Bindungsstelle
sondern an die Corezeptor-Bindungsstelle von gp120. X5 ist ein sogenannter
CD4-induzierter Antikörper (CD4i), weil durch Bindung an CD4 die Corezeptor-
Bindungsstelle durch konformationelle Umlagerungen besser zugänglich wird 102.
4.8.1 Design von HAMX
Auch von diesem Antikörper stand eine Kristallstruktur im Komplex mit gp120 und zusätzlich
CD4 zur Verfügung, von der der Aufbau des neuen Peptids HAMX (human antibody (X5)
mimic) abgeleitet wurde. HAMX setzt sich, ebenso wie HAM, aus drei CDRs, H1, H2 und H3,
welche die nach der Chothia-Nummerierung als CDRs definierten Sequenzen der schweren
Kette des X5-Antikörpers enthalten (siehe Tabelle 15), zusammen. Die Zyklisierungen zur
Nachahmung der Schleifenform von CDR H2 und H3 wurden analog zu HAM eingeführt und
das Konstrukt wurde ebenfalls mit einem Biotin-Tag versehen.
Tabelle 15: Aus den CDR-H-Schleifen von b12 abgeleitete Sequenzen für HAMX
CDR H1 in grün, H2 in blau, H3 in rot; orange = Verlängerung, grau = Verkürzung
Sequenz CDR H1 GTFSMYGF CDR H2 Asp-IIPIFGTS-Dap CDR H3 C-DFGPDWEDGDSYDGSGRGFFDF-C
Anders als HAM ist HAMX ein verzweigtes Peptid, dessen Gerüst mit Hilfe eines zusätzlich
orthogonal geschützten Fmoc-L-Lys(ivDde)-OH aufgebaut wurde. Der C-Terminus lag in der
CDR H2, wodurch der schwierigste Schritt der Synthese, die Lactamzyklisierung, schon in
einem frühen Stadium des Syntheseprozesses durchgeführt werden konnte. Auch bei X5
liegen die drei CDRs der schweren Kette, wenn man vom b12-Epitop auf gp120 aus darauf
blickt, in einem Dreieck angeordnet vor und die CDR H3 ist mit 22 Aminosäuren die längste
Sequenz, deren oberer Teil zur Schleife geformt deutlich aus dem Arm des Antikörper-Fab-
Fragmentes heraus ragt (Abb. 41B).

- Ergebnisse -
79
Abbildung 41: Design des X5-Antikörper-Paratop-Mimetikums HAMX. A) Kristallstruktur von X5 im Komplex mit HIV-1 gp120(JRFL) und CD4 (PDB: 2B4C). H1 in grün, H2 in blau, H3 in rot dargestellt. B) Antikörper-Paratop von X5 in der Kristallstruktur C) Blick auf das X5-Paratop vom gp120-Epitop aus.
D) Schematische Darstellung von HAMX und exakte Sequenz.
Die in der Literatur beschriebenen Hauptinteraktions-Aminosäuren von X5 mit gp120 liegen
beide in der CDR H3 des Antikörpers, nämlich ein Tryptophan (Position 100) und ein Tyrosin
(Position 100F), siehe Abb. 12, Kapitel 1.6. Durch deren Austausch durch Alanine in der
Sequenz von HAMX entstand das Pendant zu (ala)HAM, die Alanin-Variante (ala)HAMX.
HAMX Ac-[CFGPDWDGDSYDGSGRGC]-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-{Asp-IIPIFGTS-Dap}-NH2 (ala)HAMX Ac-[CFGPDWDGDSYDGSGRGC]-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-{Asp-IIPIFGTS-Dap}-NH2

- Ergebnisse -
80
4.8.2 Synthese von HAMX
Das peptidische Mimetikum des Antikörpers X5 konnte, ebenso wie HAM, mittels
Fmoc-gestützter Festphasenpeptidsynthese hergestellt werden. Die Vorgehensweise war
durchweg die gleiche wie bei HAM. Zur Verzweigung wurde mit einem ivDde-geschützten
Lysin gearbeitet, dessen orthogonale Schutzgruppe mit 5 % Hydrazin entfernt wurde, um
anschließend die CDR H1 daran zu synthetisieren. Diese Schritte geschahen vor dem
Abspalten des Peptids vom Harz. Die Peptide der HAMX-Serie wurden in ähnlichen
Ausbeuten und Reinheiten wie die der HAM-Serie erhalten. Alle synthetisierten Peptide sind
mit entsprechender molarer Masse und Ausbeute in Tabelle 16 aufgelistet.

- Ergebnisse -
81
Tabelle 16: Sequenzen, molare Massen und Ausbeuten von HAMX und Varianten
{Lactam-Zyklus}, [Disulfid], veränderte Aminosäuren, Alanin-Austausch
Bezeichnung Sequenz MW g/mol Ausbeute %
HAMX Ac-[Cys-FGPDWDGDSYDGSGRG-Cys]-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-{Asp-IIPIFGTS-Dap}-NH2 4656,4 1,01
(ala)HAMX Ac-[Cys-FGPDADGDSADGSGRG-Cys]-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-{Asp-IIPIFGTS-Dap}-NH2 4449,4 12,17
(scram)HAMX Ac-[Cys-WGDPGDSSYDFGGGDR-Cys]-XG-Lys(Bio)-Lys(Ac-YTFGFGMS)-GB-{Asp-PSFIGTII-Dap}-NH2 4656,4 0,53
(H3scram)HAMX Ac-[Cys-SDGWSGYDGFPGDRDG-Cys]-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-{Asp-IIPIFGTS-Dap}-NH2 4656,4 0,61
(lin)HAMX Ac-SFGPDWDGDSYDGSGRGS-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-GIIPIFGTSG-NH2 4560,1 6,34
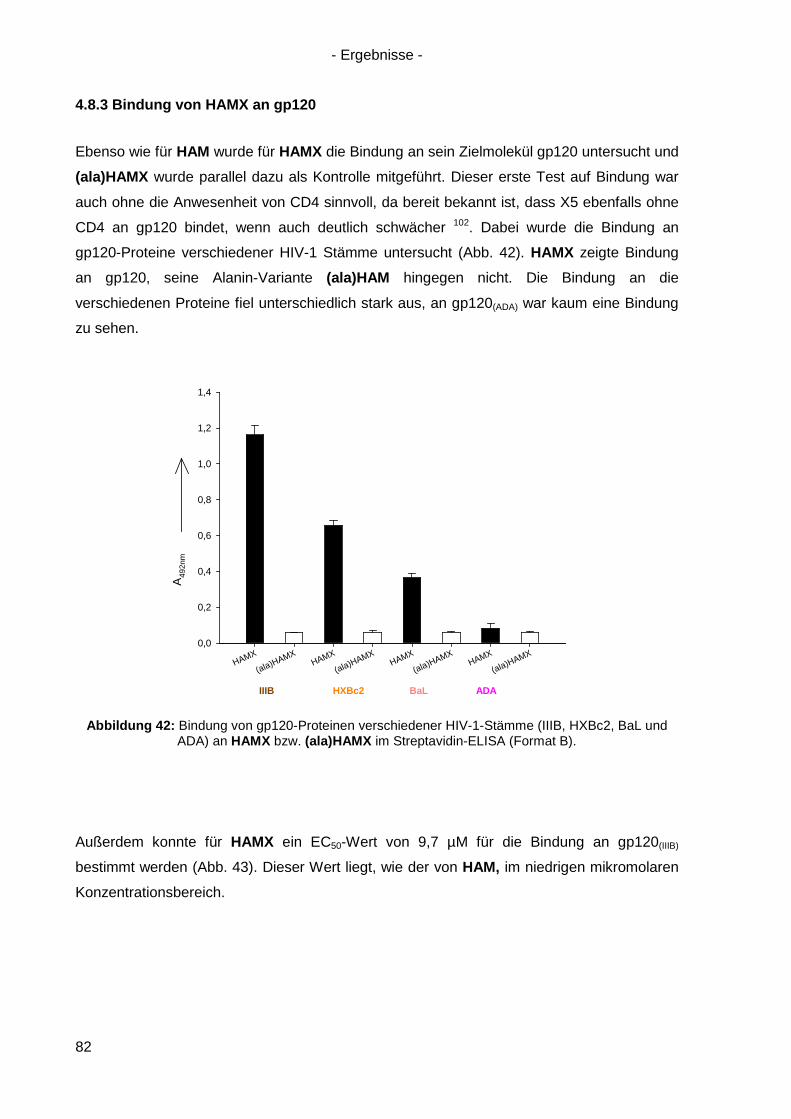
- Ergebnisse -
82
4.8.3 Bindung von HAMX an gp120
Ebenso wie für HAM wurde für HAMX die Bindung an sein Zielmolekül gp120 untersucht und
(ala)HAMX wurde parallel dazu als Kontrolle mitgeführt. Dieser erste Test auf Bindung war
auch ohne die Anwesenheit von CD4 sinnvoll, da bereit bekannt ist, dass X5 ebenfalls ohne
CD4 an gp120 bindet, wenn auch deutlich schwächer 102. Dabei wurde die Bindung an
gp120-Proteine verschiedener HIV-1 Stämme untersucht (Abb. 42). HAMX zeigte Bindung
an gp120, seine Alanin-Variante (ala)HAM hingegen nicht. Die Bindung an die
verschiedenen Proteine fiel unterschiedlich stark aus, an gp120(ADA) war kaum eine Bindung
zu sehen.
HAMX
(ala)HAMXHAMX
(ala)HAMXHAMX
(ala)HAMXHAMX
(ala)HAMX
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
IIIB HXBc2 BaL ADA
Abbildung 42: Bindung von gp120-Proteinen verschiedener HIV-1-Stämme (IIIB, HXBc2, BaL und ADA) an HAMX bzw. (ala)HAMX im Streptavidin-ELISA (Format B).
Außerdem konnte für HAMX ein EC50-Wert von 9,7 µM für die Bindung an gp120(IIIB)
bestimmt werden (Abb. 43). Dieser Wert liegt, wie der von HAM, im niedrigen mikromolaren
Konzentrationsbereich.
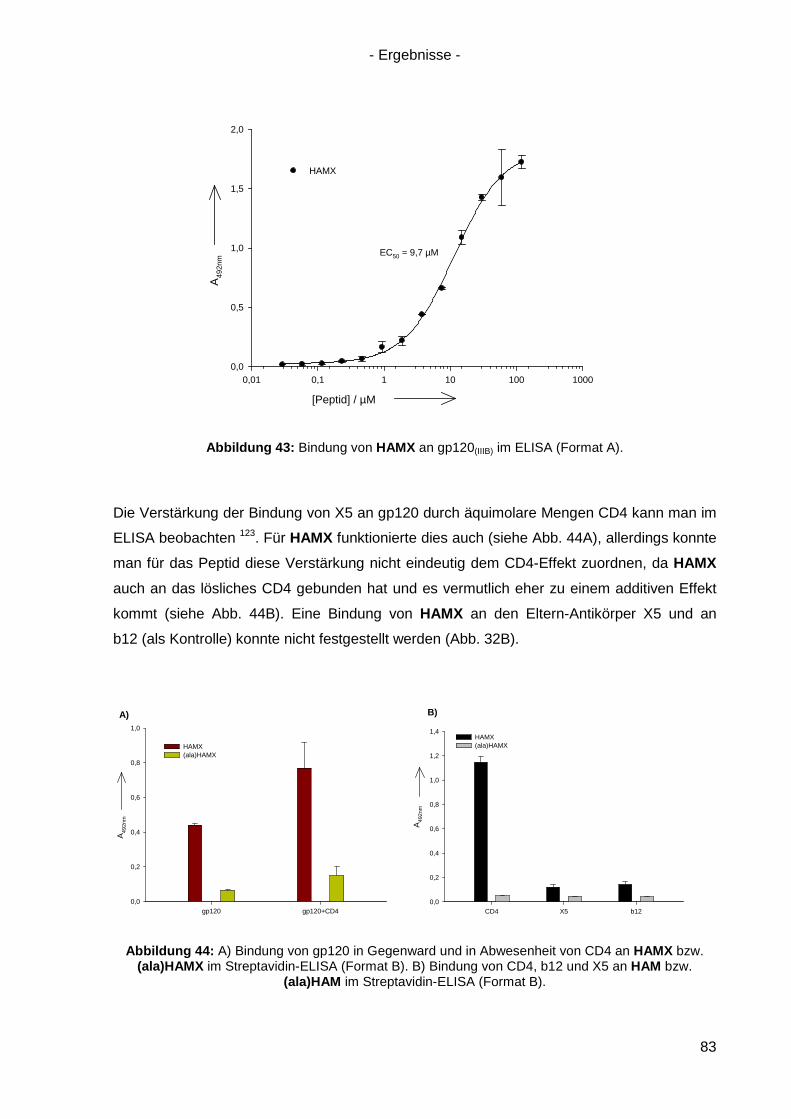
- Ergebnisse -
83
[Peptid] / µM
0,01 0,1 1 10 100 1000
A 492n
m
0,0
0,5
1,0
1,5
2,0
HAMX
EC50 = 9,7 µM
Abbildung 43: Bindung von HAMX an gp120(IIIB) im ELISA (Format A).
Die Verstärkung der Bindung von X5 an gp120 durch äquimolare Mengen CD4 kann man im
ELISA beobachten 123. Für HAMX funktionierte dies auch (siehe Abb. 44A), allerdings konnte
man für das Peptid diese Verstärkung nicht eindeutig dem CD4-Effekt zuordnen, da HAMX
auch an das lösliches CD4 gebunden hat und es vermutlich eher zu einem additiven Effekt
kommt (siehe Abb. 44B). Eine Bindung von HAMX an den Eltern-Antikörper X5 und an
b12 (als Kontrolle) konnte nicht festgestellt werden (Abb. 32B).
gp120 gp120+CD4
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
HAMX (ala)HAMX
A)
CD4 X5 b12
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4HAMX(ala)HAMX
B)
Abbildung 44: A) Bindung von gp120 in Gegenward und in Abwesenheit von CD4 an HAMX bzw. (ala)HAMX im Streptavidin-ELISA (Format B). B) Bindung von CD4, b12 und X5 an HAM bzw.
(ala)HAM im Streptavidin-ELISA (Format B).
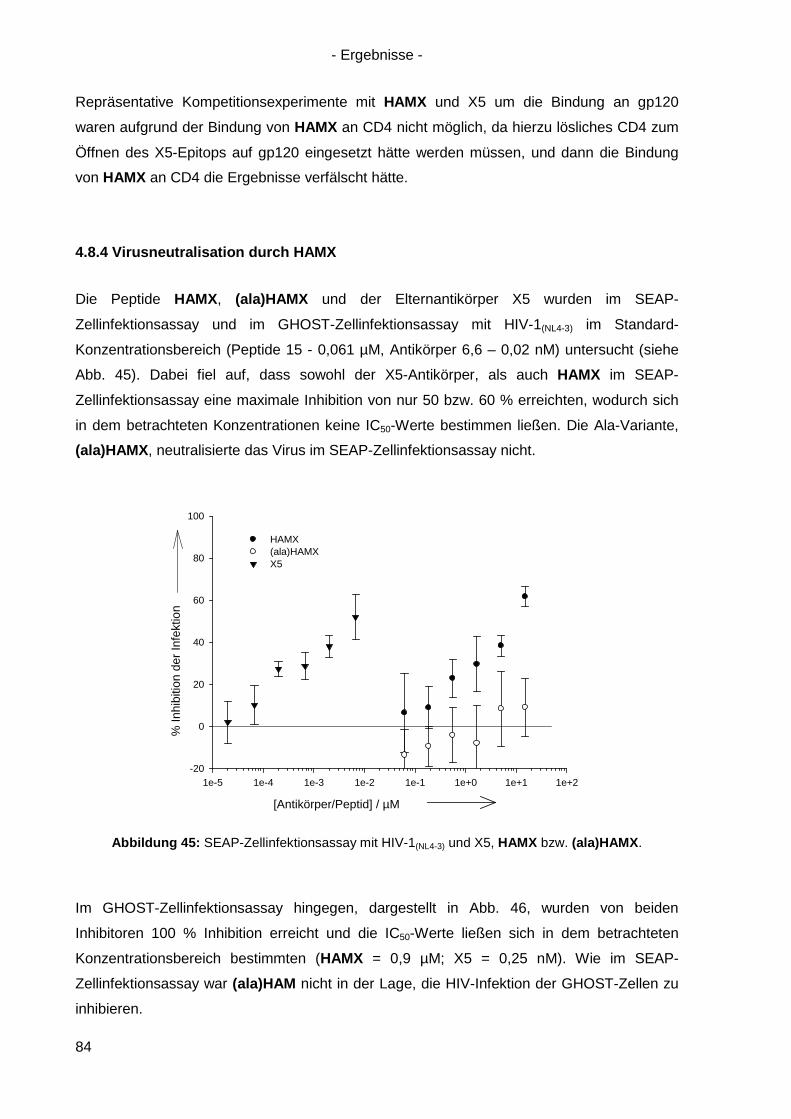
- Ergebnisse -
84
Repräsentative Kompetitionsexperimente mit HAMX und X5 um die Bindung an gp120
waren aufgrund der Bindung von HAMX an CD4 nicht möglich, da hierzu lösliches CD4 zum
Öffnen des X5-Epitops auf gp120 eingesetzt hätte werden müssen, und dann die Bindung von HAMX an CD4 die Ergebnisse verfälscht hätte.
4.8.4 Virusneutralisation durch HAMX
Die Peptide HAMX, (ala)HAMX und der Elternantikörper X5 wurden im SEAP-
Zellinfektionsassay und im GHOST-Zellinfektionsassay mit HIV-1(NL4-3) im Standard-
Konzentrationsbereich (Peptide 15 - 0,061 µM, Antikörper 6,6 – 0,02 nM) untersucht (siehe
Abb. 45). Dabei fiel auf, dass sowohl der X5-Antikörper, als auch HAMX im SEAP-
Zellinfektionsassay eine maximale Inhibition von nur 50 bzw. 60 % erreichten, wodurch sich
in dem betrachteten Konzentrationen keine IC50-Werte bestimmen ließen. Die Ala-Variante, (ala)HAMX, neutralisierte das Virus im SEAP-Zellinfektionsassay nicht.
[Antikörper/Peptid] / µM
1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100
HAMX (ala)HAMX X5
Abbildung 45: SEAP-Zellinfektionsassay mit HIV-1(NL4-3) und X5, HAMX bzw. (ala)HAMX.
Im GHOST-Zellinfektionsassay hingegen, dargestellt in Abb. 46, wurden von beiden
Inhibitoren 100 % Inhibition erreicht und die IC50-Werte ließen sich in dem betrachteten
Konzentrationsbereich bestimmten (HAMX = 0,9 µM; X5 = 0,25 nM). Wie im SEAP-
Zellinfektionsassay war (ala)HAM nicht in der Lage, die HIV-Infektion der GHOST-Zellen zu
inhibieren.
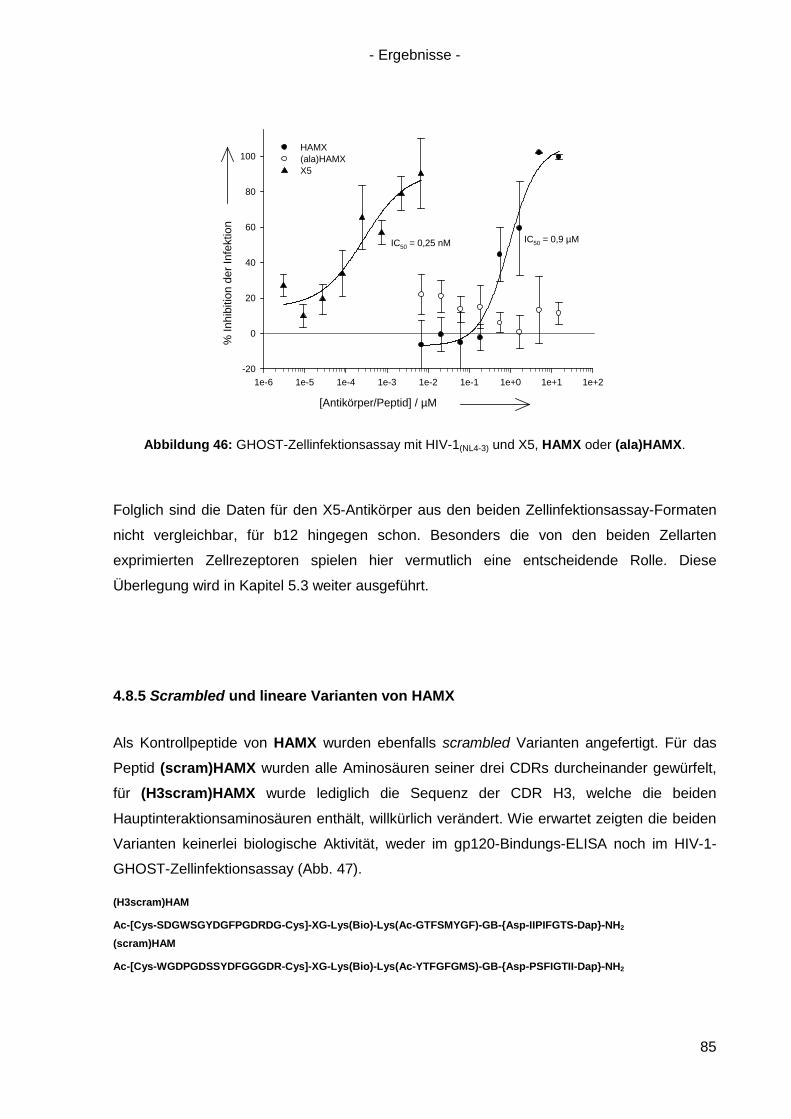
- Ergebnisse -
85
[Antikörper/Peptid] / µM
1e-6 1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100HAMX (ala)HAMX X5
IC50 = 0,9 µMIC50 = 0,25 nM
Abbildung 46: GHOST-Zellinfektionsassay mit HIV-1(NL4-3) und X5, HAMX oder (ala)HAMX.
Folglich sind die Daten für den X5-Antikörper aus den beiden Zellinfektionsassay-Formaten
nicht vergleichbar, für b12 hingegen schon. Besonders die von den beiden Zellarten
exprimierten Zellrezeptoren spielen hier vermutlich eine entscheidende Rolle. Diese
Überlegung wird in Kapitel 5.3 weiter ausgeführt.
4.8.5 Scrambled und lineare Varianten von HAMX
Als Kontrollpeptide von HAMX wurden ebenfalls scrambled Varianten angefertigt. Für das
Peptid (scram)HAMX wurden alle Aminosäuren seiner drei CDRs durcheinander gewürfelt,
für (H3scram)HAMX wurde lediglich die Sequenz der CDR H3, welche die beiden
Hauptinteraktionsaminosäuren enthält, willkürlich verändert. Wie erwartet zeigten die beiden
Varianten keinerlei biologische Aktivität, weder im gp120-Bindungs-ELISA noch im HIV-1-
GHOST-Zellinfektionsassay (Abb. 47).
(H3scram)HAM
Ac-[Cys-SDGWSGYDGFPGDRDG-Cys]-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-{Asp-IIPIFGTS-Dap}-NH2 (scram)HAM
Ac-[Cys-WGDPGDSSYDFGGGDR-Cys]-XG-Lys(Bio)-Lys(Ac-YTFGFGMS)-GB-{Asp-PSFIGTII-Dap}-NH2
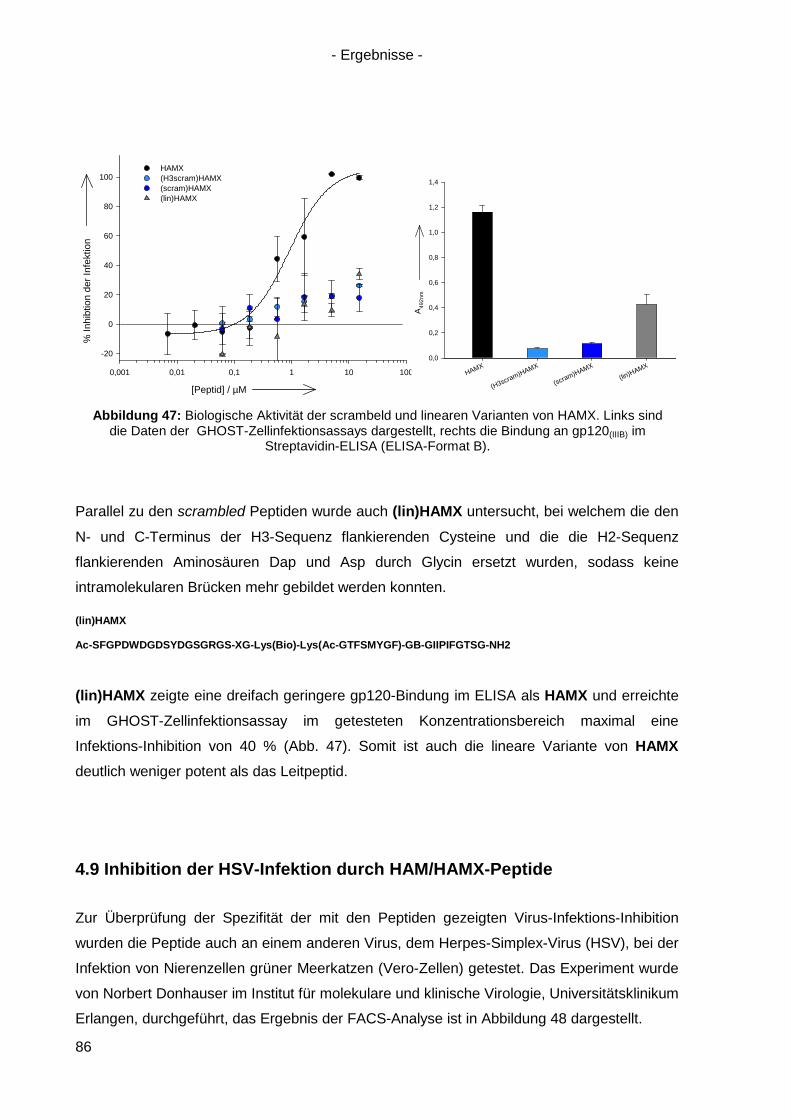
- Ergebnisse -
86
Parallel zu den scrambled Peptiden wurde auch (lin)HAMX untersucht, bei welchem die den
N- und C-Terminus der H3-Sequenz flankierenden Cysteine und die die H2-Sequenz
flankierenden Aminosäuren Dap und Asp durch Glycin ersetzt wurden, sodass keine
intramolekularen Brücken mehr gebildet werden konnten.
(lin)HAMX Ac-SFGPDWDGDSYDGSGRGS-XG-Lys(Bio)-Lys(Ac-GTFSMYGF)-GB-GIIPIFGTSG-NH2
(lin)HAMX zeigte eine dreifach geringere gp120-Bindung im ELISA als HAMX und erreichte
im GHOST-Zellinfektionsassay im getesteten Konzentrationsbereich maximal eine
Infektions-Inhibition von 40 % (Abb. 47). Somit ist auch die lineare Variante von HAMX
deutlich weniger potent als das Leitpeptid.
4.9 Inhibition der HSV-Infektion durch HAM/HAMX-Peptide
Zur Überprüfung der Spezifität der mit den Peptiden gezeigten Virus-Infektions-Inhibition
wurden die Peptide auch an einem anderen Virus, dem Herpes-Simplex-Virus (HSV), bei der
Infektion von Nierenzellen grüner Meerkatzen (Vero-Zellen) getestet. Das Experiment wurde
von Norbert Donhauser im Institut für molekulare und klinische Virologie, Universitätsklinikum
Erlangen, durchgeführt, das Ergebnis der FACS-Analyse ist in Abbildung 48 dargestellt.
[Peptid] / µM
0,001 0,01 0,1 1 10 100
% In
hibt
ion
der I
nfek
tion
-20
0
20
40
60
80
100HAMX (H3scram)HAMX (scram)HAMX (lin)HAMX
HAMX
(H3scram)HAMX
(scram)HAMX(lin)HAMX
A 492n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
Abbildung 47: Biologische Aktivität der scrambeld und linearen Varianten von HAMX. Links sind die Daten der GHOST-Zellinfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im
Streptavidin-ELISA (ELISA-Format B).
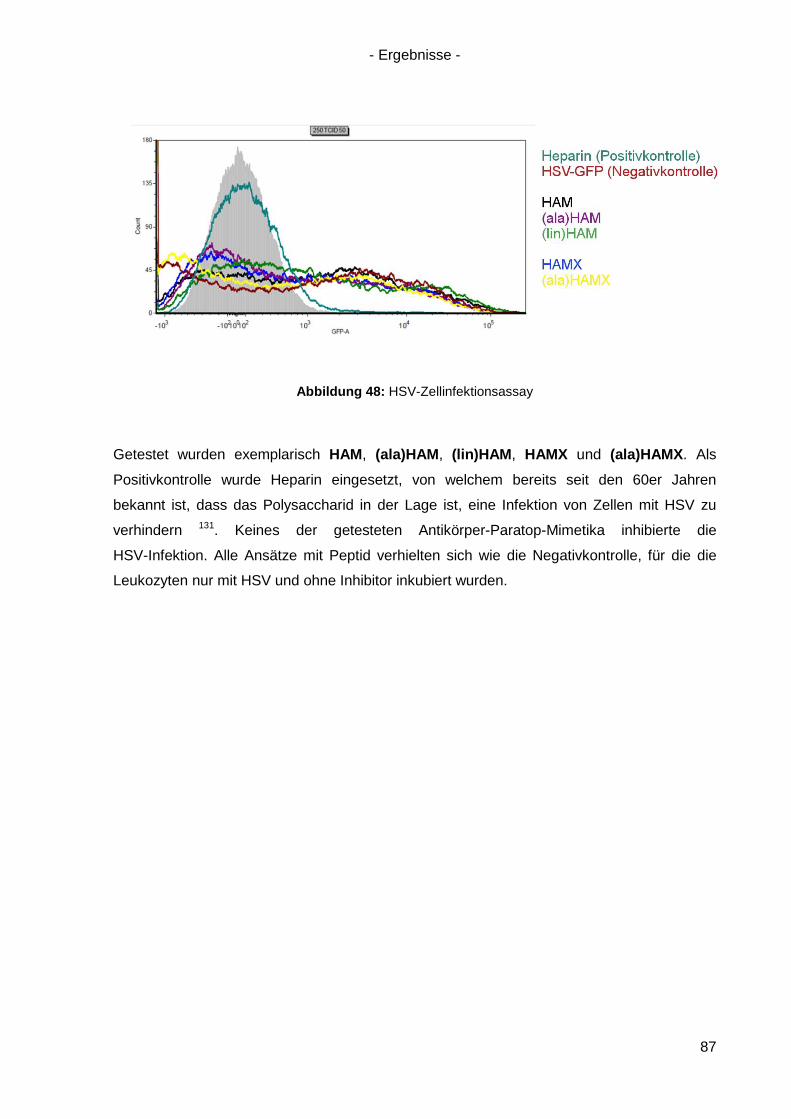
- Ergebnisse -
87
Abbildung 48: HSV-Zellinfektionsassay
Getestet wurden exemplarisch HAM, (ala)HAM, (lin)HAM, HAMX und (ala)HAMX. Als
Positivkontrolle wurde Heparin eingesetzt, von welchem bereits seit den 60er Jahren
bekannt ist, dass das Polysaccharid in der Lage ist, eine Infektion von Zellen mit HSV zu
verhindern 131. Keines der getesteten Antikörper-Paratop-Mimetika inhibierte die
HSV-Infektion. Alle Ansätze mit Peptid verhielten sich wie die Negativkontrolle, für die die
Leukozyten nur mit HSV und ohne Inhibitor inkubiert wurden.

- Diskussion und Ausblick -
88
5. Diskussion und Ausblick
Das in dieser Arbeit vorgestellte und charakterisierte HAM ist ein Paratop-mimetisches
Peptid und präsentiert die CDRs der schweren Kette des Antikörpers b12. Der
b12-Antikörper ist gegen das Oberflächenprotein gp120 von HIV-1 gerichtet und ist in der
Lage, ein breites Spektrum an HIV-1-Stämmen zu neutralisieren, d.h. die Infektion von Zellen
mit dem Virus zu verhindern 115. Es ist mit HAM gelungen, ein Peptid herzustellen, welches
wie sein Eltern-Antikörper in der Lage ist, die Infektion humaner Zellen (CEMx174 SEAP-
Zellen oder GHOST-Zellen) mit HIV-1 zu inhibieren. In der folgenden Diskussion werden b12
und HAM hinsichtlich der untersuchten Eigenschaften im Vergleich betrachtet. Desweiteren
wird das gewählte Design des Peptids diskutiert und das Potential der Varianten
(H1H3)HAM und (lin)HAM, welche eine zu HAM vergleichbare neutralisierende Aktivität im
Zellinfektionsassay aufweisen, im Kontext der erhaltenen Ergebnisse analysiert.
Abschließend werden die Anwendbarkeit des Designs auf andere Antikörper und die sich
durch die Verfügbarkeit effizienter peptidischer Paratop-Mimetika bietenden Möglichkeiten
für die HIV-Forschung erörtert.
5.1 HAM – ein Mimetikum von b12?
Die erste Fragestellung, die beantwortet werden sollte, war, inwieweit das Peptid HAM
tatsächlich ein Mimetikum des b12-Antikörpers ist. Das Peptid HAM bindet gp120-Proteine
diverser HIV-1-Stämme, welche auch vom Antikörper selbst erkannt werden. Die Kontroll-
Variante (ala)HAM hingegen wird nicht von gp120 erkannt. Dies ist eine Bestätigung des
Peptid-Designs und eine eindeutiger Hinweis auf eine spezifische Interaktion von HAM mit
gp120. Darüber hinaus ist das Peptid in der Lage, zwischen b12-sensitiven und
b12-resistenten HIV-1-Stämmen zu unterscheiden, d.h. die Bindung an gp120 b12-
resistenter Stämme (PVO, ZM96) ist deutlich schwächer ausgeprägt. Dieses Resultat belegt
die Fähigkeit von HAM zur Liganden-Selektivität. Allerdings ist im Fall von HAM in den
höchsten gemessenen Konzentrationen ein leichter Anstieg der Absorption im Bindungs-
Assay bei den Proteinen der b12-resistenten HIV-1-Stämme gp120(PVO) und gp120(ZM96) zu
erkennen. Eine mögliche Erklärung hierfür könnte die Größe des Peptids sein: die molare
Masse von HAM ist ca. 25-mal kleiner als die von b12. Das Peptid könnte somit leichter
Zugang zu den sterisch, z.B. durch Glykane, abgeschirmten Epitopen auf gp120 erlangen
und Paratop-Mimetika könnten potentiell also sogar breiter neutralisierend sein als ihre
Eltern-Antikörper. Im Bindungs-ELISA unterscheiden sich Peptid und Antikörper in ihrem
Verhalten an den untersuchten gp120-Proteinen nicht wesentlich, die gemessenen

- Diskussion und Ausblick -
89
EC50-Werte für die Bindung von gp120 an b12 bzw. HAM liegen im niedrigen nanomolaren
Konzentrationsbereich (1,3 bzw. 1,7 nM).
Es war leider nicht möglich, eine Kompetition von b12 und HAM um die Bindung an gp120
zu zeigen, und somit die Interaktion von HAM mit dem b12-Epitop auf gp120 nachzuweisen,
da HAM nicht nur an gp120 bindet, sondern auch an b12 selbst. Im kompetitiven ELISA
kommt es dadurch vermutlich zu einem Sandwich-artigen Auftürmen der Moleküle HAM,
gp120 und b12, wodurch keine Verringerung des Signals mehr detektiert werden kann,
sondern nur ein gleich bleibend hohes Signal. Eine Kompetition mit dem zellulären Rezeptor
CD4, dessen Bindungsstelle auf gp120 zu großen Teilen mit dem b12-Epitop überlappt 116,113,117, konnte bis zu einem gewissen Grad (63% Inhibition) gezeigt werden und ist ein
klarer Hinweis darauf, dass HAM tatsächlich an der gleichen Region auf gp120 bindet wie
CD4. Dadurch, dass HAM auch in einem gewissen Maß an lösliches CD4 bindet, kann
vermutlich keine 100%-ige Inhibition erreicht werden: wenn alle CD4-Bindungsstellen auf
gp120 durch CD4 belegt sind kann man immer noch eine gewisse Menge HAM detektieren.
Dies ist der Anteil an Peptid, der an das gp120-gebundende CD4 bindet. Bei der
Untersuchung, ob HAM auch an andere Antikörper neben b12 im Streptavidin-ELISA bindet,
fiel auf, dass das Peptid selektiv CD4-Bindungsstellen-Antikörper bindet. Bereits 1988
beschrieben Kang et al. 132 ein Peptid, welches die Selbst-Bindung des Anti-
Phosphorylcholin-Antikörpers T15 inhibiert. Die Forscher hatten ein Erkennungs-Modell
mittels Translation codierender und nicht-codierender DNA-Stränge aufgestellt, in dem die
für die Selbsterkennung verantwortlichen Bereiche (CDR H2 und FR 3) bestimmt wurden.
Das Antikörper-abgeleitete Peptid inhibierte die Entstehung von Antikörper-Komplexen durch
Selbst-Bindung fast genauso gut wie dessen Antigen Phosphorylcholin. Für breit
neutralisierende HIV-1-Antikörper wurden Poly- und Autoreaktivität zum ersten Mal von
Haynes et al. 2005 beschrieben 133. In der aktuellen Literatur (Liu et al., 2015) zu breit HIV-1
neutralisierenden Antikörpern findet man auch nähere Beschreibungen bzw.
Untersuchungen zu auftretender Poly- oder Autoreaktivitäten 134, unter anderem bei
CD4-Bindungsstellen-Antikörpern 134. Eine mögliche Erklärung für die auffallend starke
Bindung von HAM an CD4-Bindungsstellen-Antikörper könnte folglich das Phänomen der
Selbst-Bindung von Antikörpern in Kombination mit der Polyreaktivität und Autoreaktivität
von HIV-1-Antikörpern sein.
In den Zellinfektionsassays konnte für das b12-Paratop-Mimetikum HAM ein IC50-Wert von
4,9 µM (SEAP-Zellinfektionsassay) bzw. 1,0 µM (GHOST-Zellinfektionsassay) bestimmt
werden, d.h. das Peptid kann nicht nur an das von b12 adressierte Oberflächenprotein
gp120 binden, sondern tatsächlich Zellen vor einer HIV-1-Infektion schützen. Damit ist es im
Vergleich mit anderen peptidischen Paratop-Mimetika ein durchaus potenter Inhibitor (siehe

- Diskussion und Ausblick -
90
Kapitel 1.2). Die Inhibition der Infektion humaner Zellen mit dem HI-Virus ist der
entscheidende Effekt, der HAM als ein neues antivirales Agens gegen HIV-1 qualifiziert. Der
IC50-Wert für den Eltern-Antikörper b12 liegt mit 0,26 nM (SEAP) bzw. 0,47 nM (GHOST) in
einem viel niedrigeren Konzentrationsbereich als der des Peptids. Das liegt zum einen
vermutlich daran, dass an der außergewöhnlich hohen Bindungs-Affinität und –Spezifität von
Antikörpern zu ihrem Antigen nicht nur die CDRs („hypervariable regions“), sondern
nachweislich auch die umgebenden Bereiche („framework regions“) maßgeblich beteiligt
sein. Diese framework regions kontaktieren das Antigen zwar nicht direkt, aber sie dienen als
Gerüst in Form von ß-Faltblatt-Strukturen, und können so die korrekte Konformation der
CDRs stabilisieren 10,28,6. Zum anderen werden auf einem Antikörper die CDRs stets zweimal
präsentiert. Eine multivalente Präsentation von HAM ist daher auch eine Möglichkeit, die
Avidität zu steigern.
Weitere Experimente, um HAM als ein Mimetikum von b12 zu verifizieren und seine
spezifische Interaktion mit HIV-1 gp120 nachzuweisen, waren die Untersuchung von
scrambled und Alanin-Varianten von HAM auf Bindung an gp120 und
Neutralisationsfähigkeit. Die Notwendigkeit der richtigen Reihenfolge der Aminosäuren in der
Sequenz impliziert eine spezifische, Antikörper-ähnliche Interaktion mit dem HIV-1
Hüllprotein gp120, die nicht durch ein zufällig zusammengesetztes Peptid erreicht werden
kann. Die Peptide (scram)HAM und (H3scram)HAM zeigen beide eine signifikant geringere
Aktivität als HAM, sowohl bei der Bindung an gp120 als auch bei der Virusneutralisation.
Daraus wird klar, dass HAM in seinem ursprünglichen Design durchaus spezifisch agiert,
und dass die CDR H3, welche die längste, hervorstehende CDR-Schleife des b12
repräsentiert, dafür unentbehrlich ist. Dieses Ergebnis steht in Einklang mit der Literatur
(siehe Kapitel 1.6) und wird zusätzlich durch die mit den Alanin-Varianten von HAM erhaltenen Ergebnisse untermauert. Durch die einzelnen oder kombinierten Austausche der
drei Hauptinteraktionsaminosäuren 117 von b12 in der Peptidsequenz von HAM durch Alanin
konnte gezeigt werden, dass diese Reste auch auf Peptidebene eine wichtige Rolle spielen.
Dies gilt vor allem für das Tryptophan an der Spitze der H3-Schleife (W100 in b12) und das
Tyrosin der H2-Schleife (Y53 in b12). Diese beiden Positionen waren bereits vor der
Veröffentlichung der Kristallstruktur durch Mutationsstudien als essentiell für die
gp120-Bindung beschrieben worden 113. Ersetzt man alle drei Aminosäuren, W100, Y53 und
Asn 31, entsprechend im Peptid durch Alanine, zeigt es keine gp120-Bindung mehr und
inhibiert kaum noch die Virusinfektion. Folglich sind die wichtigen Aminosäuren im Eltern-
Antikörper auch für die biologische Aktivität von HAM von Bedeutung, was für eine
Nachahmung der Interaktion von b12 mit gp120 durch HAM spricht.
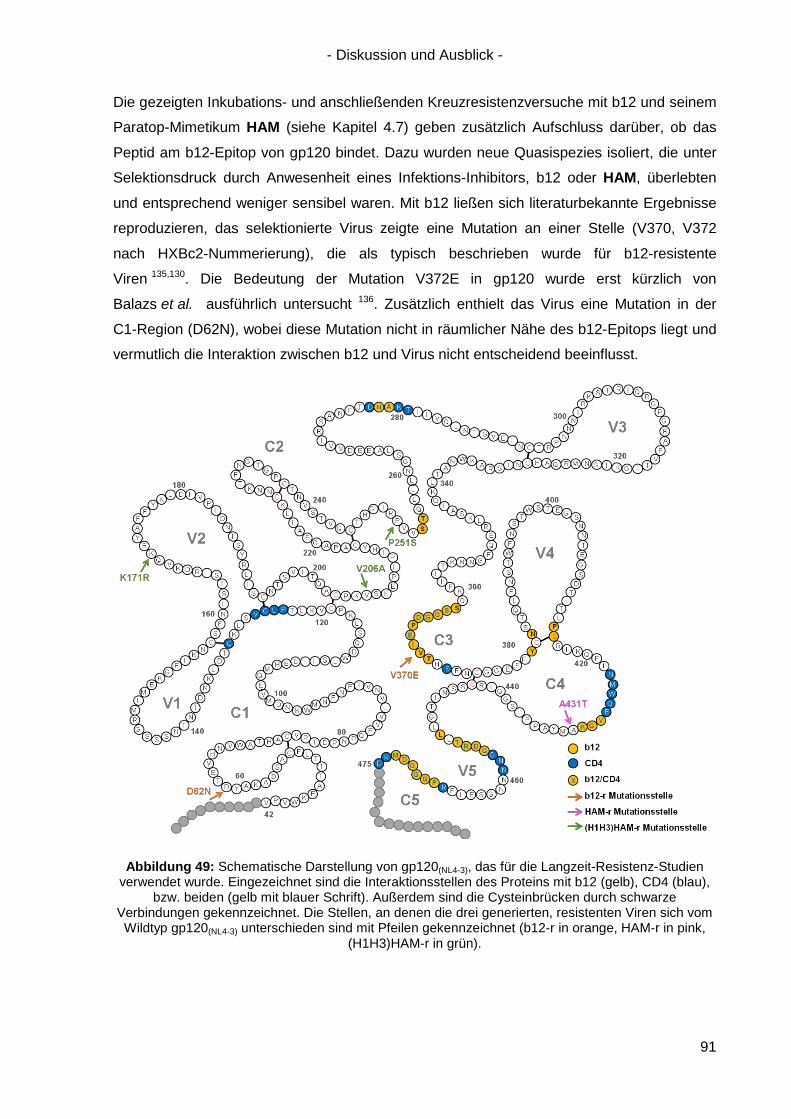
- Diskussion und Ausblick -
91
Die gezeigten Inkubations- und anschließenden Kreuzresistenzversuche mit b12 und seinem
Paratop-Mimetikum HAM (siehe Kapitel 4.7) geben zusätzlich Aufschluss darüber, ob das
Peptid am b12-Epitop von gp120 bindet. Dazu wurden neue Quasispezies isoliert, die unter
Selektionsdruck durch Anwesenheit eines Infektions-Inhibitors, b12 oder HAM, überlebten
und entsprechend weniger sensibel waren. Mit b12 ließen sich literaturbekannte Ergebnisse
reproduzieren, das selektionierte Virus zeigte eine Mutation an einer Stelle (V370, V372
nach HXBc2-Nummerierung), die als typisch beschrieben wurde für b12-resistente
Viren 135,130. Die Bedeutung der Mutation V372E in gp120 wurde erst kürzlich von
Balazs et al. ausführlich untersucht 136. Zusätzlich enthielt das Virus eine Mutation in der
C1-Region (D62N), wobei diese Mutation nicht in räumlicher Nähe des b12-Epitops liegt und
vermutlich die Interaktion zwischen b12 und Virus nicht entscheidend beeinflusst.
Abbildung 49: Schematische Darstellung von gp120(NL4-3), das für die Langzeit-Resistenz-Studien verwendet wurde. Eingezeichnet sind die Interaktionsstellen des Proteins mit b12 (gelb), CD4 (blau),
bzw. beiden (gelb mit blauer Schrift). Außerdem sind die Cysteinbrücken durch schwarze Verbindungen gekennzeichnet. Die Stellen, an denen die drei generierten, resistenten Viren sich vom Wildtyp gp120(NL4-3) unterschieden sind mit Pfeilen gekennzeichnet (b12-r in orange, HAM-r in pink,
(H1H3)HAM-r in grün).

- Diskussion und Ausblick -
92
Nach der Langzeitinkubation von HIV-1(NL4-3) in Anwesenheit steigender Konzentrationen von
HAM stand zunächst ein Virus mit vier Mutationen im Oberflächenprotein gp120 im
Vordergrund. Allerdings war bereits bei der Sequenzierung zu sehen, dass keine reine,
sondern eine Mischpopulation an Viren überlebt hatte. Die Sequenz-Chromatogramme
zeigten nämlich Doppelpeaks an den entsprechenden Mutationsstellen, was darauf
hindeutet, dass die passagierten Überstände eine Mischung von Virusspezies enthielten.
Nach der Aufhebung des Selektionsdrucks blieb vorwiegend eine Spezies mit nur einer
Mutation bestehen, welche in einem konservierten Bereich von gp120 liegt (C4, A431T).
Bemerkenswert ist, dass sich diese Mutation direkt benachbart zu einer Reihe an
Hauptinteraktionsaminosäuren von gp120 mit b12 (und CD4) befindet (Abb. 49), was
ebenfalls dafür spricht, dass HAM im Bereich des b12-Epitops agiert. Darüber hinaus liegt
die mutierte Stelle in der Kristallstruktur ganz dicht beim V4-Loop-Stamm von gp120 (siehe
Abb. 50B), der nach der Publikation von Dueñas-Decamp et al. vom essentiellen W100 des
CDR H3 kontaktiert wird 118,117. Diese Tatsache legt nahe, dass sich b12 und HAM in ihrem
Bindungsverhalten ähneln.
A) B)
Abbildung 50: A) Kristallstruktur von gp120 (PDB: 4ZMJ, kristallisiertes Env-Trimer ohne Ligand). Gezeigt sind gp120 (grau), b12-Bindungseptiop (gelb) und die Mutationsstellen von b12-r (schwarz)
und HAM-r (pink). B) Kristallstruktur von gp120 (PDB: 2NY7, stabilisiertes, b12-gebundenes Kernprotein). Gezeigt sind gp120(gelb), CDR H3 von b12 (rot), Stamm der V4-Schleife (cyan),
Mutationsstelle von b12-r (magenta).

- Diskussion und Ausblick -
93
Ein Virus mit einer einzelnen Mutation ist auch während der Kontroll-Langzeitinkubation von
HIV-1(NL4-3) ohne Inhibitor entstanden. Diese liegt in der besonders langen und hoch
variablen V3-Schleife von gp120 (N298S) und man kann davon ausgehen, dass sie, ebenso
wie K171R, durch die für das Virus typische sehr hohe Mutationsrate entstanden ist, aber
nicht maßgeblich die Replikation des Virus beeinflusst.
Das Virus mit der Mutation A431T, genannt NL4-3(HAM-r), wurde für die SEAP-
Zellinfektionsassays verwendet, bei denen die Resistenzen und Kreuzresistenzen untersucht
wurden. In Abbildung 51 sieht man den Effekt von HAM an NL4-3(Wildtyp), NL4-3(HAM-r)
und NL4-3(b12-r) im Vergleich.
[HAM] / µM0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
300
200
100
0
100 NL4-3 NL4-3(HAM-r) NL4-3(b12-r)
% V
erst
ärku
ng d
er In
fekt
ion
IC50 = 4,9 µM
Abbildung 51: SEAP-Zellinfektionsassay mit HAM und drei verschiedenen Viren; direkter Vergleich von Resistenz- und Kreuzresistenztestung.
Erstaunlicherweise verstärkt HAM bei den beiden neu generierten Viren die Infektion in
gleichem Maß. Bei IC50-Wert-ähnlichen Konzentrationen (4,5 bzw. 5,7 µM) erreicht HAM
seinen halbmaximalen verstärkenden Effekt. Man kann also sagen, dass die beiden Viren
NL4-3(b12-r) und NL4-3(HAM-r) nicht nur resistent sind gegen die Inhibition durch HAM,
sondern dass sie das Peptid sogar nutzen, um die Zellinfektion zu verstärken. Die großen
Fehlerbalken bei den Messungen im negativen Bereich deuten aber darauf hin, dass dieser
Effekt nicht typisch Dosis-Wirkung-abhängig auftritt. Beispiel für Peptide, die gezielt die
Virusinfektion verstärken, sind die gp120-abgeleiteten Peptide ef-c und CD4bsM 137,138. Für
diese Peptide wurde die Infektionsverstärkung ebenfalls im SEAP-Zellinfektionassay

- Diskussion und Ausblick -
94
gemessen und die Streuung der Messwerte war hier in der Regel deutlich geringer. HAM
beeinflusst durch Bindung die Virus-Wirtszell-Interaktion der unter Selektionsdruck von b12
oder HAM isolierten Spezies folglich vergleichbar günstig. Dies ist ein unbeabsichtigter
Effekt. Das ähnliche Verhalten aber von HAM an den beiden Viren NL4-3(HAM-r) und
NL4-3(b12-r) ist ein weiterer Hinweis darauf, dass das Peptid im gleichen Bereich auf gp120
agiert wie sein Elternantikörper b12. Der b12-Antikörper auf der anderen Seite inhibiert das
Peptid-resistente Virus NL4-3(HAM-r) ca. 2,5-fach schlechter als NL4-3(Wildtyp), siehe
Abb. 38, Kapitel 4.7. Die Inkubation der Viren unter HAM-Selektionsdruck führt also zu
Viren-Spezies, die durch b12 schlechter inhibiert werden können.
Zusammenfassend kann man festhalten, dass die gesammelten Indizien auf eine echte
Mimikry des b12-Antikörpers durch das peptidische Paratop-Mimetikum HAM hindeuten.
Eine weitere Steigerung der Affinität zu gp120 und der Selektivität von HAM könnte der
Schlüssel sein, um ungewollte Effekte zu unterbinden, wie z.B. eine Verstärkung der
Virusinfektion. Wie in Kapitel 4.9 gezeigt, bestätigen Tests an anderen Viren wie HSV eine
für HIV-1 spezifische Inhibition durch HAM. Um die Antigen-Spezifität von HAM definitiv
nachzuweisen, und mit der von b12 zu vergleichen, müsste aber noch die Bindung an viele
weitere Antigene und die Neutralisationsfähigkeit bei anderen Viren untersucht werden.
Um die Bindungsstelle für HAM auf gp120 genau zu definieren, wäre eine Kristallstruktur von
HAM in Komplex mit gp120 erstrebenswert. Dies könnte sich aber als ein sehr schwieriges
Unterfangen erweisen, da sich bereits die Kristallisation von b12 in Komplex mit gp120 als
sehr schwierig herausstellte. Es musste hierfür ein stark verkürztes gp120-Kernprotein
eingesetzt werden, da es nicht gelang mit anderen gp120-Proteinen entsprechende Kristalle
zu züchten 117. Die Kristallisation eines gp120-HAM-Komplexes wird aufgrund der Flexibiltät
des Peptids und seiner geringeren Affinität im Vergleich zum b12-Antikörper voraussichtlich
noch schwieriger sein. Alternativ hätte man die Möglichkeit, durch gezielte Mutationsstudien
auf Seiten des gp120-Proteins die Bindungsstelle für HAM auf gp120 zu beschreiben. Es
wäre möglich diverse Reste im b12-Epitop von gp120 einzeln oder auch in Kombination zu
mutieren, um zu sehen, welchen Einfluss die Austausche auf die Bindung von gp120 an
HAM haben. In weiterführenden Experimenten könnte man außerdem die in den
Langzeitinkubationen entstandenen gp120-Proteine exprimieren, um diese im
Bindungsassay zu untersuchen und die Ergebnisse und Annahmen, die aus den
Kreuzresistenzversuchen erhalten wurden, zu verifizieren. So wäre es möglich, weitere
Informationen zur Kartierung der Bindungsstelle für das Peptid auf gp120 zu sammeln.

- Diskussion und Ausblick -
95
5.2 Optimierung von HAM
Die chemische Synthese eines 51mer-Peptids ist anspruchsvoll. Ein besonders limitierender
Schritt bei der Synthese von HAM und seinen Varianten war die Lactamzyklisierung der
CDR H2. Wie bereits beschrieben war die Menge an erhaltenem Rohpeptid bei
Lactam-verbrückten Peptiden deutlich geringer als bei linearen Varianten. Der Grund hierfür
sind vermutlich intermolekulare Lactambrücken, die sich anstelle der angestrebten
intramolekularen Brücken so ausbilden, dass es zu einer Vernetzung der einzelnen Peptide
im Harz kommt. Dadurch kann das Peptidnetzwerk nicht mehr aus der festen Phase
ausgewaschen werden und verbleibt im Harz. Man erhält somit nur ein Bruchteil an
Rohpeptid, und der Anteil an gesuchtem Peptid sinkt drastisch durch die Nebenreaktion der
Vernetzung im Harz. Daher wäre auch eine Verkürzung bzw. Vereinfachung von HAM von
Vorteil für weitere Forschungsarbeiten.
In meiner Arbeit habe ich HAM nach der Synthese charakterisiert, um das Peptid mit den
gewonnenen Erkenntnissen optimieren zu können. Vor allem galt es herauszufinden, ob die
Zyklisierungen und tatsächlich alle drei CDRs des Peptids notwendig bzw. günstig sind für
seine biologische Aktivität, insbesondere hinsichtlich b12-ähnlichem Bindungsverhalten und
Affinität.
Zunächst werden sechs verkürzten Varianten von HAM betrachtet (siehe Kapitel 4.6.4):
Eines der Peptide zeigt eine mit HAM vergleichbare biologische Aktivität: (H1H3)HAM. Zwei
der verkürzten Peptide, (H1)HAM und (H1H2)HAM, werden zwar ebenfalls von gp120
erkannt, aber sie zeigen im Zellinfektionsassay keine Inhibition der HIV-1-Infektionsrate,
sondern sogar einen leicht verstärkenden Effekt auf die Infektion der Zellen. Dieser scheint
noch dazu willkürlich aufzutreten, worauf auch hier stark schwankende Messwerte
hindeuten. Alle anderen getesteten Peptide sind deutlich weniger biologisch aktiv als HAM
selbst (IC50-Werte siehe Tabelle 17).
Berücksichtigt man die Ergebnisse mit allen verkürzten Varianten von HAM, lässt sich zu
jeder seiner drei CDRs eine charakterisierende Aussage treffen:
• H1 ist essentiell für die Bindung an gp120.
• Zur Virusneutralisation trägt H3 entscheidend bei, weil im gemessenen
Konzentrationsbereich eine halbmaximale Inhibition der Infektion von SEAP-Zellen
mit HIV-1(NL4-3) nur dann auftritt, wenn H1 und H3 Teil des Peptids sind.
• H2 leistet keinen Beitrag zur biologischen Aktivität.
Somit ist (H1H3)HAM nach diesen Ergebnissen die einzige verkürzte Variante von HAM, die
mindestens ähnlich gute Eigenschaften aufweist wie das Leitpeptid.
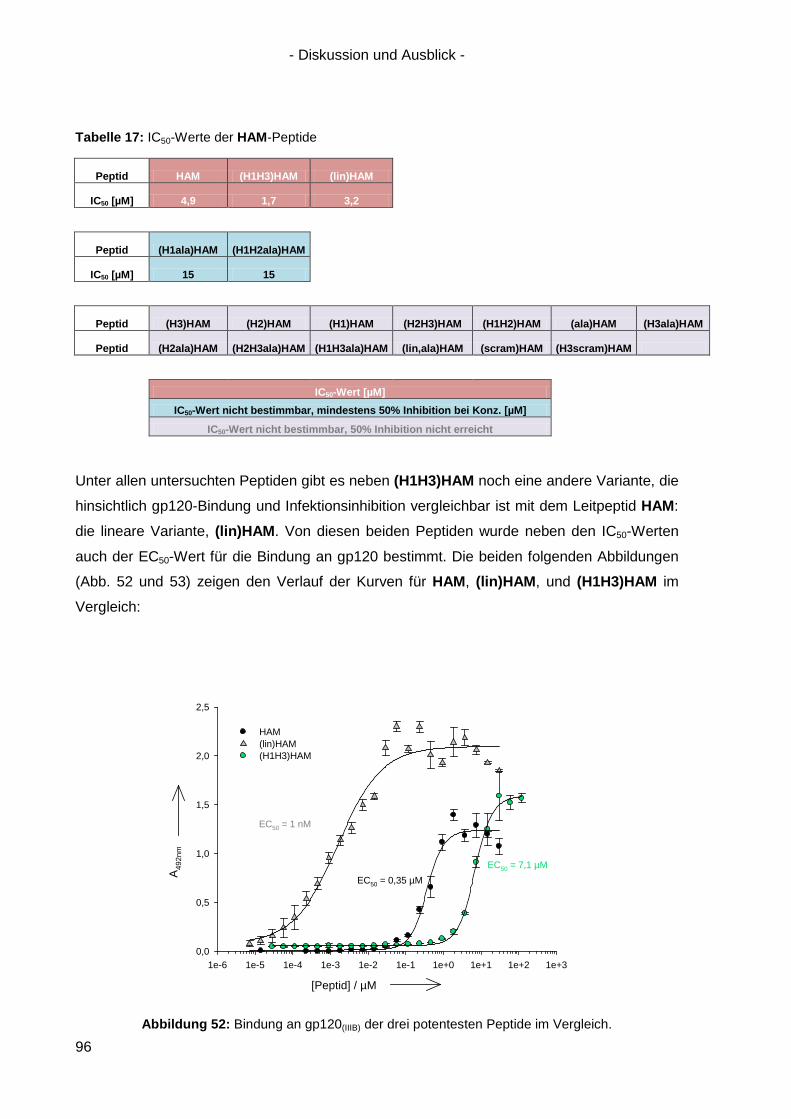
- Diskussion und Ausblick -
96
Tabelle 17: IC50-Werte der HAM-Peptide
Peptid HAM (H1H3)HAM (lin)HAM
IC50 [µM] 4,9 1,7 3,2
Peptid (H1ala)HAM (H1H2ala)HAM
IC50 [µM] 15 15
Peptid (H3)HAM (H2)HAM (H1)HAM (H2H3)HAM (H1H2)HAM (ala)HAM (H3ala)HAM
Peptid (H2ala)HAM (H2H3ala)HAM (H1H3ala)HAM (lin,ala)HAM (scram)HAM (H3scram)HAM
IC50-Wert [µM]
IC50-Wert nicht bestimmbar, mindestens 50% Inhibition bei Konz. [µM]
IC50-Wert nicht bestimmbar, 50% Inhibition nicht erreicht
Unter allen untersuchten Peptiden gibt es neben (H1H3)HAM noch eine andere Variante, die
hinsichtlich gp120-Bindung und Infektionsinhibition vergleichbar ist mit dem Leitpeptid HAM:
die lineare Variante, (lin)HAM. Von diesen beiden Peptiden wurde neben den IC50-Werten
auch der EC50-Wert für die Bindung an gp120 bestimmt. Die beiden folgenden Abbildungen
(Abb. 52 und 53) zeigen den Verlauf der Kurven für HAM, (lin)HAM, und (H1H3)HAM im
Vergleich:
[Peptid] / µM
1e-6 1e-5 1e-4 1e-3 1e-2 1e-1 1e+0 1e+1 1e+2 1e+3
A 492n
m
0,0
0,5
1,0
1,5
2,0
2,5
HAM(lin)HAM(H1H3)HAM
EC50 = 1 nM
EC50 = 0,35 µMEC50 = 7,1 µM
Abbildung 52: Bindung an gp120(IIIB) der drei potentesten Peptide im Vergleich.

- Diskussion und Ausblick -
97
[Peptid] / µM
0,01 0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
-20
0
20
40
60
80
100 HAM(lin)HAM(H1H3)HAM
IC50 = 1,7 µM IC50 = 4,9 µM
IC50 = 3,2 µM
Abbildung 53: SEAP-Zellinfektionsassay der drei potentesten Peptide im Vergleich.
(lin)HAM besitzt mit einem EC50-Wert von 1,0 nM für die Bindung an gp120 offensichtlich
eine wesentlich höhere Affinität zum Protein als HAM oder auch (H1H3)HAM. Das hat
allerdings keinen Einfluss auf die Neutralisationsfähigkeit des Peptids, die IC50-Werte aller
drei Varianten liegen in einem ähnlichen Bereich, wobei (H1H3)HAM mit 1,7 µM sogar den
niedrigsten IC50-Wert aufweist. Eine mögliche Erklärung hierfür könnte sein, dass das lineare
Peptid auch mit anderen Bindungsstellen des Ziel-Proteins interagiert, und nicht nur im
Bereich des b12-Epitops bzw. der CD4-Bindungsstelle. Das entspräche einer unspezifischen
Bindung, bei der man nicht von einer Optimierung des b12-Antikörper-Mimetikums sprechen
kann. Desweiteren erkennt man, dass die sigmoidale Kurve für (lin)HAM im
Virusneutralisationsassay (Abb. 53) nicht bei 0 % Inhibition beginnt, sondern im negativen
Bereich, dass also das Peptid eine leichte Verstärkung der Infektion zeigt. Im Hinblick auf die
Diskrepanz zwischen EC50- und IC50-Wert-Bestimmungen kann man das als weitere
Bestätigung für einen gewissen Anteil an ungewollten Interaktionen von (lin)HAM mit
HIV-1(NL4-3) werten, die in niedrigen Konzentrationen eine Verstärkung der Infektion zur Folge
haben und keine Inhibition. Die Zyklisierungen spielen folglich eine entscheidende Rolle für
eine spezifische Interaktion von HAM mit den Oberflächenproteinen des Virus. Diese
Annahme wird zusätzlich dadurch bestätigt, dass (lin)HAM neben der Negativkontrolle
(lin,ala)HAM der einzige Inhibitor ist, durch dessen Anwesenheit bei den
Langzeitinkubationsversuchen keine mutierte Variante von HIV-1(NL4-3) isoliert wurde, sondern
lediglich Wildtyp-Virus.

- Diskussion und Ausblick -
98
Die verkürzte Variante (H1H3)HAM zeigt ein größeres Potential als (lin)HAM. Da ihr die
CDR H2 mit der Lactambrücke fehlt, ist die synthetische Ausbeute ca. dreimal höher als die
von HAM. Der IC50-Wert liegt mit 1,7 µM noch etwas niedriger als der des Leitpeptids.
Zudem verhält sich (H1H3)HAM bei der Unterscheidung der b12-sensitiven und der
b12-resistenten Stämme im ELISA ebenso selektiv wie der Antikörper selbst, und ist somit in
dieser Eigenschaft b12 noch ähnlicher als sein Vorläufermolekül HAM. Man kann also
(H1H3)HAM als eine optimierte Variante von HAM sehen, die die Infektion von Zellen mit
HIV effizienter hemmt als alle anderen in dieser Arbeit getesteten Peptide.
Durch die Langzeitinkubation von HIV-1(NL4-3) mit (H1H3)HAM konnte ein Virus mit drei
stabilen Mutationen im gp120-Protein isoliert werden. Eine der Mutationen, K171R, liegt weit
ab des b12-Bindungepitops in der variablen V2-Region des Proteins. Die anderen beiden
allerdings, V206A und P251S, liegen in der konstanten C2-Region im Inneren des gp120s,
nahe des b12-Epitops (siehe Abb. 54). Das Virus NL4-3((H1H3)-r) ist im betrachteten
Konzentrationsbereich resistent gegen (H1H3)HAM. Es ist denkbar, dass die Tertiärstruktur
des Glykoproteins durch die Mutationen in seinem Kern so stark verändert wird, dass das
Peptid keinen Zugang mehr zu seiner ursprünglichen Bindungsstelle hat. Es ist auch eine
leichte Kreuzresistenz mit b12 und dem generierten NL4-3(b12-r) entstanden: (H1H3)HAM
inhibiert das b12-resistente Virus 5,5-fach schlechter als das Wildtyp-Virus und b12 inhibiert
das (H1H3)HAM-resistente Virus 5-fach schlechter als das Wildtyp-Virus. Diese Daten
bestärken die Annahme, dass (H1H3)HAM im Bereich des b12-Epitops auf gp120 interagiert
und dass es dadurch in der Lage ist, die Infektion von Zellen mit HIV-1(NL4-3) zu inhibieren.

- Diskussion und Ausblick -
99
Abbildung 54: Kristallstruktur von gp120 (PDB: 4ZMJ, kristallisiertes Env-Trimer ohne Ligand). Gezeigt sind gp120 (grau), b12-Epitop (gelb), b12-r-Mutationsstellen (schwarz) und (H1H3)HAM-r-Mutationsstellen (grün).
Vergleicht man die Langzeitinkubationsversuche von (H1H3)HAM mit denen von HAM, fällt
auf, dass durch die Anwesenheit beider Peptide ganz unterschiedliche Reste im
gp120-Protein mutierten. Aus den Resistenz- und Kreuzresistenz-SEAP-Zellinfektionsassays
wird klar, dass die Verkürzung von HAM zu (H1H3)HAM das Peptid und seine biologische
Aktivität weit mehr verändert, als bis dahin angenommen. Die CDR H2, die bei den anderen
Experimenten als weniger wichtig erschien, scheint hier hinsichtlich der Infektions-
verstärkenden Eigenschaften einen entscheidenden Unterschied zu machen. Ein so
ausgeprägter, die Infektion fördernder Effekt wie von HAM an NL4-3(b12-r) und
NL4-3(HAM-r) wurde für (H1H3)HAM an keinem der Viren beobachtet, man kann lediglich
eine Tendenz zur Infektions-Verstärkung an NL4-3((H1H3)HAM-r) erkennen (Abb. 55).

- Diskussion und Ausblick -
100
[(H1H3)HAM] / µM
0,1 1 10 100
% In
hibi
tion
der I
nfek
tion
-50
0
50
100 NL4-3 NL4-3((H1H3)HAM-r) NL4-3(b12-r)
IC50 = 1,7 µM
IC50 = 9,6 µM
Abbildung 55: SEAP-Zellinfektionsassay vom (H1H3)HAM an drei verschiedenen Viren zum direkten Vergleich von Resistenz- und Kreuzresistenztestung.
Diese Beobachtungen zeigen, dass die vom b12-Paratop abgeleiteten Peptide, sobald die
von ihnen adressierte Bindungsstelle nicht mehr zugänglich ist, die Virusinfektionsrate
erhöhen können. An dieser Stelle lohnt sich noch einmal ein Blick auf die Daten aus den
SEAP-Zellinfektionsassays der verkürzten Varianten von HAM: wie bereits erwähnt, zeigen
zwei der Peptide schon am NL4-3(Wildtyp) eine Verstärkung der Infektion, nämlich (H1)HAM
und (H1H2)HAM. Es ist also wahrscheinlich, dass die CDRs H1 und H2 nicht b12-ähnlich an
gp120 binden, aber auf eine unbekannte Weise mit dem Antigen interagieren. H1 bringt aber
anscheinend die CDR H3 in HAM und (H1H3)HAM in die Nähe des b12-Epitops, erhöht also
die Affinität. Ist die spezifische Bindungsstelle durch Mutationen im Virus nicht mehr
verfügbar, bindet das Peptid trotzdem mit H1 (in (H1H3)HAM) bzw. H1 und H2 (in (HAM)) an
gp120. Dadurch kommt es wiederum zu einer Infektionsverstärkung, wobei diese durch die
Anwesenheit der CDR H2 im Fall von HAM offensichtlich noch gefördert wird, auch wenn
(H2)HAM alleine keine Infektionsverstärkende Wirkung gezeigt hat. (H1H3)HAM zeigt
folglich hinsichtlich der ungewollten Infektionsverstärkung bessere Eigenschaften als HAM:
seine willkürliche Verstärkung der Virusinfektion betrug in den Messungen maximal 80% und
nahm in höheren Konzentrationen wieder ab. HAM verstärkte die Infektion um bis zu 300%
(Abb. 51).
Zusammenfassend kann man sagen, dass (H1H3)HAM als Ausgangsverbindung für weitere
Optimierung herangezogen werden kann. Es wäre für zukünftige Projekte denkbar, anstelle
der CDR H1 eine Einheit an (H3)HAM anzuhängen, die mit einem anderen, nahegelegen
Epitop auf gp120 interagiert. Diese Einheit sollte die Affinitäts-steigernde Rolle von H1 in
HAM übernehmen und darüber hinaus selbst spezifisch mit gp120 interagieren. Hierfür käme

- Diskussion und Ausblick -
101
zum Beispiel ein Paratop-Mimetikum eines anderen Anti-HIV-1-Antikörpers in Frage. Der
mögliche Einsatz solcher Hybrid-Moleküle wird in Kapitel 5.4 noch weiter erläutert. Der
einzige Nachteil, den (H1H3)HAM aufweist, ist eine deutlich geringere Löslichkeit in
wässrigen Lösungsmitteln, was bestimmte Experimente in wässrigen Systemen erschweren
kann. Eigenschaften wie die Löslichkeit einer Verbindung spielen natürlich aus
pharmakologischer Sicht eine entscheidende Rolle. Bei weiteren Optimierungsprozessen
sollten daher die für einen potentiellen neuen Wirkstoff wichtigen Kriterien festgelegt werden
und die Eigenschaften des Paratop-Mimetikums entsprechend verändert werden. Dies ist vor
allem in Bereichen der Peptide möglich, die nicht direkt mit dem Antigen interagieren, z.B.
die Spacer oder die Region mit der Markierung für die ELISA-Detektion. Durch den Einsatz
lipophiler/hydrophiler Spacer könnte zum Beispiel die Löslichkeit der Peptide verändert
werden oder durch die Modifikation von Seitenketten könnten Angriffspunkte für einen
enzymatischen Abbau eliminiert werden.
Weitere Gesichtspunkte, die noch untersucht werden sollten, sind die Längen der Spacer
zwischen den einzelnen Teilen des Peptids oder auch die Länge der H3-Schleife. Es könnte
sein, dass das Peptid durch größere Abstände zwischen den CDRs bzw.
Hauptinteraktionsstellen wichtige Bereiche des b12-Epitops effektiver erreichen kann. Da
man nicht weiß, inwieweit die Ahx-Spacer des Peptids in seiner jeweiligen Umgebung in
gestreckter Form vorliegen, kann man die genauen Abstände schwer abschätzen. Daher
würde ein Variieren der Spacerlänge und des Spacertyps sicherlich Aufschluss geben über
die optimale Abmessung der Spacer für (H1H3)HAM. Möglichweise könnte dadurch der
IC50-Wert des Peptids noch deutlich verbessert werden, wie in der Literatur bereits für andere
Hybridmoleküle beschrieben 139,140. Es könnte sich theoretisch auch herausstellen, dass die
Beweglichkeit des Peptids eher weiter eingeschränkt werden muss, um die Affinität und
Spezifität seiner Interaktion an des b12-Epitops noch zu erhöhen. Eine multivalente
Präsentation der CDR H3 auf einem Gerüst, wie zum Beispiel nach dem Vorbild von
Timmermann et al. 35, wäre möglicherweise ein Weg, um dadurch die Effizienz der CDR H3
zu steigern.
Aus den in dieser Arbeit gezeigten Ergebnissen lässt sich schlussfolgern, dass die CDR H1
vor allem für die Affinität zum gp120 verantwortlich ist, dass CDR H3 aber für eine
b12-ähnliche Interaktion und für eine Blockade der CD4-Bindungsstelle verantwortlich ist. In
jedem Fall ist die Affinität von H3 alleine zu gp120 offensichtlich zu gering. Die gewonnenen
Erkenntnisse bestätigen auch frühere Experimente von Saphire et al., die in ihrer Publikation
zur Kristallstruktur von IgG b12 2001 bereits die Neutralisationsfähigkeit eines an
BSA-konjugierten CDR H3-Peptids, abgeleitet von b12, beschrieben 115. Das Konjugat ist in
der Lage, die Infektion von Zellen mit HIV-1 im nanomolaren Konzentrationsbereich zu

- Diskussion und Ausblick -
102
inhibieren, allerdings konnte, im Unterschied zu HAM und (H1H3)HAM, keinerlei Bindung an
gp120-Proteine im ELISA gemessen werden und somit die Infektionsinhibition nicht einer
spezifischen Interaktion mit dem viralen Spike zugeordnet werden113.
5.3 Peptidische Paratop-Mimetika anderer Antikörper
Zusätzlich zu b12 sollte die beschriebene Vorgehensweise auch an einem anderen Anti-HIV-
1-Antikörper getestet werden. Da aus HAM später ein Hybridmolekül zur simultanen
Inhibition der ersten beiden Schritte im Infektionszyklus von HIV-1 entstehen soll, war der
CD4i-Antikörper X5 ein geeigneter Kandidat, da er die nahe der CD4-Bindungsstelle
gelegene Corezeptor-Bindungsstelle auf gp120 adressiert (siehe Kapitel 1.6). Analog zur
HAM-Studie wurden auch von HAMX Alanin-, scrambled und lineare Varianten synthetisiert
und wie die Varianten des Vorbilds HAM untersucht. Es stellte sich heraus, dass HAMX,
genau wie X5, auch schon ohne CD4 von gp120 gebunden wird, und dass das Peptid die
Infektion von humanen Zellen mit einem ähnlich guten IC50-Wert wie HAM inhibiert,
allerdings nur im GHOST-Zellinfektionsassay. Im SEAP-Zellinfektionsassay sank die
Infektionsrate erst bei deutlich höheren Konzentrationen von HAMX. Die unerwartet
deutliche Diskrepanz zwischen den Ergebnissen aus SEAP-Zellinfektionsassay und GHOST-
Zellinfektionsassay liegt möglicherweise an dem unterschiedlichen Verhältnis zwischen
exprimiertem CD4-Rezeptor und Corezeptoren auf den jeweiligen Zellen. Dieses spielt aber
nur für X5 eine Rolle, und nicht für b12, da X5 nach der CD4-Bindung des Virus mit den
Corezeptoren um die Bindung an gp120 kompetiert. Folglich exprimieren SEAP-Zellen im
Verhältnis zu CD4 vermutlich mehr Corezeptoren als GHOST-Zellen.
Erfreulicherweise zeigen die Daten, dass sich X5-Antikörper und HAMX vergleichbar
verhalten. Die Virusneutralisation findet bei beiden im SEAP-Zellinfektionsassay erst bei
entsprechend höheren Konzentrationen statt. Dies deutet auf eine erfolgreiche Mimikry des
Antikörper-Paratops hin. Auch der Befund, dass (ala)HAMX, bei dem die in der Literatur
beschriebenen Hauptinteraktionsaminosäuren für die Bindung von X5 durch Alanine ersetzt
wurden, keine biologische Aktivität zeigt, ist ein Indiz für eine spezifische Interaktion von
HAMX mit gp120 und bestätigt das Design des Paratop-Mimetikums. Ebenso wie für HAM
konnte auch für HAMX gezeigt werden, dass die Abfolge der Aminosäuren und die
Zyklisierungen der CDR H2 und H3 eine entscheidende Rolle für seine biologische Aktivität
spielen.
Wie in Abbildung 44 gezeigt, bindet HAMX, genauso wie HAM, an das im ELISA verwendete
lösliche CD4. Deshalb war es bisher leider nicht möglich, die Funktionalität von HAMX

- Diskussion und Ausblick -
103
bezüglich der CD4-Induktion der gp120-Bindung eindeutig dem Peptid zuzuschreiben, was
ein eindeutiger Beweis dafür wäre, dass das Peptid, ebenso wie X5, den Bereich der
Corezeptor-Bindungsstelle auf gp120 adressiert. Auch die Synthese und Testung verkürzter
Varianten von HAMX steht noch aus. Es wäre sehr interessant zu sehen, ob eine
Verkürzung zu (H1H3)HAMX, ebenso wie bei (H1H3)HAM, von Vorteil wäre.
Im Vergleich mit den Resultaten zu CDR-abgeleiteten Peptiden von Timmerman et al. 43 und
anderen Arbeitsgruppen (siehe Kapitel 1.2) lassen sich sowohl Parallelen, als auch
Unterschiede feststellen. Die Neutralisationsfähigkeit von HAM und HAMX kann auch erst
bei deutlich höheren Konzentrationen als bei den Eltern-Antikörpern beobachtet werden.
Außerdem spielen auch in unseren Beispielen aromatische Reste eine entscheidende Rolle
für die Interaktion zwischen Peptid und Antigen, was allerdings für den b12-Antikörper selbst
ebenso beschrieben wurde. Die Zyklisierungen hatten für HAM bzw. HAMX einen positiven
Effekt auf die biologische Aktivität, ebenso wie bei den meisten anderen beschriebenen
CDR-abgeleiteten Peptiden. Die Gesamtladung der Peptide hatte allerdings in unseren
Experimenten eindeutig keinen entscheidenden Einfluss auf das Bindungs- oder
Neutralisationsverhalten, da die scrambled-Varianten der Peptide mit der gleichen Ladung
kaum noch Aktivität zeigten. Darüber hinaus konnten verschiedenste Indizien, sowohl im
Bindungsverhalten, der Neutralisationsfähigkeit oder auch in den Virus-Mutationsstudien
gesammelt werden, die auf ein ähnliches Bindungsverhalten von Peptiden und Eltern-
Antikörpern hindeuten.
Um die Selektivität der Peptide auch auf Virusebene zu untersuchen, wurden
Neutralisationsversuche mit einem anderen Virus, dem humanen Herpes-Simplex-Virus
(HSV), durchgeführt (siehe Kap. 4.9). Die Ergebnisse belegen, dass die HAM/HAMX-Peptide
nicht wahllos Viren neutralisieren, sondern selektiv an dem Virus agieren, welches auch von
deren Eltern-Antikörper adressiert wird. Es ist also gelungen, spezifisch agierende,
peptidische Paratop-Mimetika von Antikörpern herzustellen. Antikörper wie b12 oder X5,
deren Hauptinteraktion mit dem Antigen vor allem über die besonders lange CDR H3
stattfindet, sind als Ausgangspunkt für das Design virusneutralisierender Peptide
offensichtlich sehr gut geeignet.

- Diskussion und Ausblick -
104
5.4 Welche Möglichkeiten eröffnen peptidische Mimetika von Anti-HIV-1-Antikörpern?
Einer der Hauptunterschiede zwischen Eltern-Antikörpern und Paratop-Mimetika als
potentielle Arzneistoffe ist die molare Masse der Moleküle. Mit einem Molekulargewicht im
Bereich von 3700 - 6100 Da zählen die HAM/HAMX-Peptide unter den Wirkstoffen nicht zu
den small molecules (bis 800 Da) sondern sind kleinere Biologika (Biologicals). Dennoch
werden sie synthetisch hergestellt und das ermöglicht den Einbau unnatürlicher
Aminosäuren und anderer Bausteine, mit denen die chemischen/pharmakologischen
Eigenschaften und eine spezifische Interaktion mit dem Antigen vielfältig beeinflusst werden
können.
Die überaus hohe Affinität eines Antikörpers für genau ein bestimmtes Epitop auf einem
Antigen ist für ein Peptid allein sicherlich schwer zu erlangen, aber Paratop-Mimetika wie
HAM und HAMX können als Werkzeuge oder Teilbausteine zur Herstellung multivalenter
Wirkstoffe verwendet werden, die eben nicht nur ein, sondern gleich mehrere Epitope auf
dem Target adressieren. Vorbilder für derartige Hybridmoleküle gibt es in der jüngeren
Literatur verschiedene, zum Beispiel CD4 in Kombination mit dem Domänen-Antikörper
m36 139, der ebenso wie X5 die Corezeptor-Bindungsstelle auf gp120 adressiert.
Dimitrov et al. konnten an diesem Beispiel bereits zeigen, dass das Neutralisationspotential
durch die Kombination zweier spezifisch interagierender Moleküle und durch die Wahl der
passenden Spacer-Länge deutlich gesteigert werden kann. Neben Antikörpern, die gp120
adressieren, kommen auch solche in Frage, die gp41 erkennen, z.B. 10E8. Die
membranständige Domäne von gp41 des viralen Spikes ist, ebenso wie gp120, seit einigen
Jahren im Fokus der HIV-Forschung, da auch viele gegen sie gerichtete Antikörper eine
hohe und breite Neutralisationsfähigkeit ausweisen 141,142. Es ist denkbar, dass Hybride aus
Paratop-Mimetika beispielsweise ergänzend in der Anti-retroviralen Therapie (ART)
eingesetzte werden könnten. Wie in Kapitel 1.4 beschrieben, kommen hier bisher
ausschließlich Wirkstoffe zum Einsatz, welche die nachfolgenden Schritte des
Replikationszyklus von HIV-1 stören, nicht aber den initialen Schritt, der zur Fusion von Virus
und Wirtszelle führt. Es stellt sich auch die Frage, ob spezifisch gp120 erkennende
peptidische Paratop-Mimetika nicht als Trägermoleküle für gezielt Viren-vernichtende
Substanzen (siehe z.B. Denton et al., 2014 143) dienen könnten, damit möglichst wenige
Zellen des humanen Immunsystems infiziert werden und die Viruslast im Körper schneller
sinkt.
Abschließend lässt sich festhalten, dass Peptide zur Nachahmung von Antikörpern, einer der
stärksten und sehr hoch entwickelten Waffen unseres Immunsystems, durchaus geeignet
sind. Besonders in Fällen wie HIV, wo sowohl die Impfstoffentwicklung als auch die

- Diskussion und Ausblick -
105
herkömmliche Antikörper-Therapie bisher nicht erfolgreich waren, bieten sich neue
Möglichkeiten durch die Arbeit an Paratop-Mimetika. Auch die Anwendung dieses
Forschungs-Ansatzes auf andere virale Infektionskrankheiten, bzw. die entsprechenden
Antikörper, ist denkbar, wobei die Peptide nicht nur als Wirkstoffe sondern auch zur
Aufklärung von Infektionsmechanismen und Krankheitsverläufen dienen können. Großes
Potential besitzen peptidische Paratop-Mimetika sicherlich als Bausteine zur Entwicklung
von genau auf einen Erreger abgestimmten Hybridmolekülen, die in ihren Eigenschaften
einzigartig sind und in der Lage sind, das Immunsystem des Körpers im Kampf gegen
Krankheitserreger zu unterstützen.

- Zusammenfassung / Summary -
106
6. Zusammenfassung / Summary
6.1 Zusammenfassung Humane Antikörper, die ein breites Spektrum an HIV-1-Stämmen neutralisieren, sind
wertvolle Werkzeuge zur Ermittlung potentieller Schwachstellen von HIV-1-Viruspartikeln und
als Basis zur Entwicklung neuer antiviraler Verbindungen. Viele solcher Antikörper wurden
bereits aus mit HIV-1 infizierten Menschen isoliert. Sie sind in der Regel gegen die
Oberflächenproteine gp120 und/oder gp41 des viralen Spikes gerichtet. Leider ist es bis
heute nicht gelungen, diese neutralisierenden Antikörper als Therapeutika zur Behandlung
von HIV-Patienten einzusetzen. Gerade deshalb sind Anti-HIV-1-Antikörper interessante
Kandidaten, um von Ihnen peptidische Mimetika abzuleiten.
Eine Möglichkeit zur Nachahmung ist, strukturbasiert ein Peptid vom Paratop des
Antikörpers, dem Gegenstück zu seinem Epitop, abzuleiten. Für diese Arbeit wurde zunächst
der Anti-HIV-1-Antikörper b12 als Templat für das Design eines peptidischen
Paratop-Mimetikums verwendet. Der b12-Antikörper adressiert die Bindungsstelle für den
humanen Zellrezeptor CD4 auf dem viralen Hüllprotein gp120, d.h. der Antikörper inhibiert
durch Bindung an sein Epitop die erste Interaktion zwischen Virus und Wirtszelle.
Die Kristallstruktur von b12 im Komplex mit gp120 zeigt, dass der Antikörper nur über die
CDRs seiner schweren Kette direkte Kontakte mit seinem Antigen ausbildet. Drei besonders
wichtige Aminosäuren machen dabei 40 % der Antikörper-Antigen-Kontaktfläche aus: W100,
Y53 und N31, welche jeweils im Zentrum einer CDR H (H3, H2 und H1) liegen. Besonders
das Indolringsystem des W100, welches an der Spitze des lang herausragenden CDR H3
von b12 sitzt, wurde als essentiell für die Bindung von b12 an gp120 beschrieben.
Das in dieser Arbeit beschriebene b12-Paratop-Mimetikum setzt sich folglich auch aus drei
entsprechenden Sequenz-Abschnitten, CDR H1, H2 und H3, zusammen und es wird somit
eine diskontinuierliche Bindungsstelle nachgeahmt. Die Proteinabschnitte zwischen den
CDR-Sequenzen wurden im Peptid durch Spacer ersetzt. Die schleifenartige Struktur der
CDR H3- und H2-Sequenz wurde durch Verbrückungen in Form eines Disulfids bzw. eines
Lactams nachgestellt. Außerdem trug das Peptid eine Biotin-Markierung zur leichteren
Präsentation bzw. Detektion im Bindungsassay (ELISA). So entstand das Leitpeptid HAM (human antibody (b12) mimic). Diese neue, potentiell antivirale Verbindung wurde mittels
Fmoc/tBu-Strategie an der festen Phase synthetisiert und nach der Aufarbeitung auf ihre
biologische Aktivität hin untersucht.
Wie sein Eltern-Antikörper b12 war auch das b12-Paratop-Mimetikum HAM im
Bindungsassay in der Lage, an gp120-Proteine b12-sensitiver HIV-1-Stämme zu binden,
wohingegen gp120-Proteine b12-resistenter HIV-1-Stämme selektiv deutlich schlechter

- Zusammenfassung / Summary -
107
erkannt wurden. Kompetitionsversuche mit b12 scheiterten, da das Peptid an seinen
Eltern-Antikörper selbst bindet, möglicherweise aufgrund einer Art Autoreaktivität, wie sie
häufig für breit neutralisierende Antikörper beschrieben wird. In Kompetitionsexperimenten
mit CD4 konnte jedoch gezeigt werden, dass HAM mit CD4 um die Bindung an gp120
konkurriert, was ein eindeutiges Indiz dafür ist, dass das Peptid den Bereich der CD4-
Bindungsstelle auf gp120, welche mit dem Epitop von b12 überlappt, adressiert.
In verschiedenen HIV-1-Zellinfektionsassays war HAM ein potenter Inhibitor und
neutralisierte HIV-1 mit IC50-Werten zwischen 1,0 µM und 5,0 µM. Alanin-Varianten, bei
denen W100, Y53 und/oder N31 im Peptid durch Alanin ersetzt wurden, belegten, dass für
HAM vor allem das W100 der CDR H3-Sequenz unentbehrlich ist für Bindung an gp120 und
ebenso für seine Neutralisationsfähigkeit, entsprechend dem Eltern-Antikörper b12. Mit der
Testung von scrambled Varianten von HAM konnte die Notwendigkeit der richtigen
Reihenfolge der Aminosäuren im Peptid gezeigt und somit das Peptid-Design zusätzlich
bestätigt werden. Versuche mit einer linearen Variante von HAM konnten beweisen, dass die
Einschränkung der Flexibilität des Peptids die spezifische Interaktion zwischen Peptid und
gp120 deutlich fördert.
Die Daten aus Bindungsassays und Zellinfektionsassays mit sechs verkürzten Varianten von
HAM gaben Aufschluss über die Rolle der einzelnen CDRs im Peptid. Es konnte gezeigt
werden, dass CDR H1 entscheidend ist für die Bindung an gp120, dass aber die CDR H3
essentiell ist für eine Inhibition der Infektion von Zellen mit HIV-1. CDR H2 leistet hingegen
keinen Beitrag zur biologischen Aktivität von HAM. Die verkürzte Variante (H1H3)HAM,
welcher die CDR H2-Sequenz fehlt, stellte sich folglich als optimierte Variante von HAM
heraus, mit vergleichbarem Bindungsverhalten an gp120-Proteine, einem leicht verbesserten
Neutralisationspotential und einem schnelleren und effizienteren Syntheseweg.
Um mehr Informationen zur Bindungsstelle für HAM und (H1H3)HAM auf gp120 zu erhalten
wurde HIV-1 in Gegenward des b12-Antikörpers bzw. der Peptide über mehrere Wochen
hinweg kultiviert, die durch den Selektionsdruck erhaltenen Viren isoliert und anschließend
sequenziert. Unter dem Einfluss des b12-Antikörpers vermehrten sich ausschließlich Viren,
die eine Mutation an einer für b12-Resistenzen typischen Position im b12-Epitop von gp120
trugen (V372). Nach der durch HAM, bzw. (H1H3)HAM erzwungene Selektion wurden Viren
isoliert, welche Mutationen im gp120-Protein aufwiesen, die sich direkt angrenzend an das
b12-Epitop befinden, bzw. im Kern des Proteins. Letzteres verändert die Tertiärstruktur von
gp120 vermutlich entscheidend, sodass eine Neutralisation durch HAM bzw. (H1H3)HAM
deutlich erschwert wird. Die so erhaltenen Viren zeigten sich in den anschließenden
Zellinfektionsassays nämlich deutlich weniger sensibel gegenüber den entsprechend
eingesetzten Inhibitoren als das Wildtyp-HIV-1. Eine kreuzweise Testung im

- Zusammenfassung / Summary -
108
Zellinfektionsassay von b12, HAM bzw, (H1H3)HAM mit den drei mutierten Viren zeigte auf,
dass auch die Neutralisationsfähigkeit der beiden nicht zur Selektion anwesenden Inhibitoren
durch die Mutationen beeinflusst wird. Dieses Resultat lässt darauf schließen, dass die
Peptide an der gleichen Bindungsstelle mit gp120 interagieren wie der b12-Antikörper selbst.
Desweiteren wurde nach dem Vorbild von HAM ein anderes peptidisches Mimetikum
entworfen und untersucht: HAMX (human antibody (X5) mimic). Es ahmt das Paratop des
X5-Antikörpers nach, welcher die Corezeptor-Bindungsstelle auf gp120, ebenfalls mit Hilfe
einer auffällig langen CDR H3-Schleife, adressiert. Es wurden in den durchgeführten
Experimenten mit HAMX ähnliche Ergebnisse erhalten wie mit HAM: auch HAMX bindet an
gp120 und neutralisiert HIV-1 vergleichbar effektiv in den Zellinfektionsassays, diverse
Varianten von HAMX bestätigten auch hier das gewählte Peptid-Design. HAMX könnte in
Zukunft zum Beispiel in Verbindung mit HAM, oder (H1H3)HAM, zur Herstellung von
Hybridmolekülen dienen, die simultan zwei der wichtigsten Bindungsstellen von HIV-1
gp120, die CD4-Bindungsstelle und die Corezeptor-Bindungsstelle, blockieren können.
Neutralisationsversuche mit HAM- und HAMX-Peptiden an einem anderen Virus, HSV,
zeigten, dass die Paratop-Mimetika darüber hinaus selektiv HIV-1 neutralisieren und nicht
willkürlich Virus-Wirtszell-Interaktionen blockieren.
Zusammenfassend lässt sich festhalten, dass die gesammelten Daten alle für eine
erfolgreiche Mimikry von Anti-HIV-1-Antikörpern durch die peptischen Paratop-Mimetika
sprechen. Peptide besitzen demnach großes Potential zur Nachahmung von Antikörpern in
deutlich kleineren Molekülen. In dieser Forschungsarbeit wurden neue, selektiv aktive HIV-1-
neutralisierende Verbindungen entwickelt, die das körpereigene Immunsystem zum Vorbild
haben. HAM, (H1H3)HAM, HAMX und sicherlich auch andere nach ihrem Vorbild
entworfene Peptide können in Zukunft zur weiteren Erforschung von Infektionsmechanismen
und zur Entwicklung neuartiger Virusinfektions-Inhibitoren mit einzigartigen Eigenschaften
dienen.

- Zusammenfassung / Summary -
109
6.2 Summary Human antibodies which neutralize a broad range of HIV-1 isolates are valuable tools for the
discovery of potential points of vulnerability on HIV-1 viral particles and as basis for the
development of new antiviral agents. Many of these antibodies have already been isolated
from HIV-1 infected people and they are mainly directed against the viral spike, consisting of
the surface proteins gp120 and gp41. Unfortunately, scientists have not been successful in
using those neutralizing antibodies for the therapy of HIV-1 infected patients so far.
Therefore, they are interesting candidates for the deduction of mimetic compounds.
One possibility to mimic an antibody is the structure based design of peptides derived from
the antibody’s paratope, which is the counterpart of its epitope. Initially in this work, the anti-
HIV-1 antibody b12 was used as template for the design of a peptide paratope mimic. b12
addresses the binding site for the human cell receptor CD4 on gp120 and thus inhibits
through binding to its epitope the initial interaction between virus and host cell.
The crystal structure of b12 in complex with gp120 shows that the antibody makes only direct
contact with gp120 via the three CDRs of its heavy chain. Three amino acids make up 40 %
of the contact surface between b12 and gp120: W100, Y53 and N31, each in the center of
one CDR (H3, H2 and H1). Especially the indole ring system of W100 at the tip of CDR H3
has been shown to be essential for binding of b12 to gp120.
The herein described b12 paratope mimic thus also consists of three sequential parts, CDR
H1, H2 and H3, and displays a discontinuous binding site. The protein sequences between
these sections were replaced by spacers in the peptide. The loop like structure of CDR H3
und H2 was provided by a disulfide bridge and a lactam bridge, respectively. Furthermore,
the peptide was tagged with biotin to enable an easy presentation or detection in ELISA
experiments. This design approach resulted in the new lead peptide HAM (human antibody
(b12) mimic). The potentially antiviral agent was synthesized via solid phase peptide
synthesis using Fmoc/tBu strategy and its biologic activity was investigated after
reprocessing and purification.
Like its parent antibody b12, HAM bound to gp120 proteins of HIV-1 sensitive strains,
whereas b12 resistant strains were selectively worse recognized. Competition experiments
with b12 for binding to gp120 were not successful as HAM binds to its parent antibody b12
itself, probably due to a kind of autoreactivity which has already been described in literature
for broadly neutralizing antibodies. However, competition experiments with CD4
demonstrated that HAM competes with CD4 for binding to gp120. This indicates that the
peptide addresses the same binding region on gp120 as CD4 which overlaps the b12
epitope on gp120.

- Zusammenfassung / Summary -
110
In different HIV-1 cell infection assays HAM was able to neutralize HIV-1 with IC50 values
ranging from 1.0 µM to 5.0 µM. Replacing the three most important amino acids of b12,
W100, Y53 and/or N31, through alanin on peptide level revealed that also for HAM the W100
residue is most important for gp120 binding and virus neutralization. By testing scrambled
variants of HAM the importance of the correct amino acid sequence and thus the peptide
design could be confirmed. A linear derivative verified that conformational restrictions in
order to lower the peptide’s flexibility at certain positions support a specific interaction between HAM and gp120.
The results of binding assays and cell infection assays applying truncated variants of HAM
gave information about the role of each CDR H for the peptide’s activity. According to the
results, CDR H1 is crucial for binding to gp120 whereas CDR H3 is essential for an effective
neutralization of HIV-1. CDR H2 does not contribute to the biological activity of HAM. The
truncated variant (H1H3)HAM, which lacks the CDR H2, turned out to be an optimized
version of HAM with comparable binding properties, a slightly better neutralization potential
and a faster way of synthesis with higher yields.
In order to obtain further information about the binding site for HAM and (H1H3)HAM on
gp120 virus has been cultivated for several weeks in presence of b12 antibody or a peptide
inhibitor. The viral species surviving under this selective pressure were isolated and
sequenced afterwards. The b12 resistant viruses showed a mutation at a position (V372) in
the antibody’s epitope which has been described for b12-resistant HIV-1 strains earlier.
Under the impact of HAM or HAM(H1H3) the isolated viruses carried mutations at positions
neighboring the b12 epitope or also in the core of gp120, respectively. The latter probably
causes critical changes in the tertiary structure of the protein and thus impedes neutralization
by HAM and (H1H3)HAM. In cell infection assays it could be shown that neutralization of the
mutated viruses was harder for the inhibitors used for selection compared to neutralization of
the wild type virus. Moreover, crosswise testing of the inhibitors b12, HAM and (H1H3)HAM
in cell infection assays with the three isolated viral species depicted that also the
neutralization potential of not for the selection pressure responsible inhibitors has been
reduced. These results indicate that the peptides address the same region on HIV-1 gp120
for binding as their parent antibody b12 does.
Additionally, another peptide paratope mimic following the model HAM has been designed
and analyzed, called HAMX (human antibody (X5) mimic). It displays the paratope of X5, an
antibody addressing the coreceptor binding site on gp120 also involving a long protruding
CDR H3. The results obtained for HAMX were comparable to those for HAM: also HAMX did
bind to gp120, neutralized HIV-1 in cell infection assays in similar concentrations and diverse
variants of HAMX confirmed the peptide’s design. In the future, HAMX can probably be used

- Zusammenfassung / Summary -
111
to construct hybrid molecules in combination with HAM or (H1H3)HAM addressing the two
main binding sites, CD4 binding site and coreceptor binding site, on gp120 simultaneously
and thus effectively blocking viral entry.
Further neutralization experiments with another virus, HSV, showed that the antibody
paratope mimics do not randomly inhibit interactions between viruses and host cells but
selectively inhibit HIV-1 infection.
In summary, all collected data point towards a successful mimicry of anti-HIV-1 antibodies
through peptide paratope mimics. Therefore, peptides exhibit a high potential to be used for
the design of antibody mimics. In this work new selectively active and HIV-1 neutralizing
compounds have been developed, derived from the human immune system. In the future,
HAM, (H1H3)HAM, HAMX and certainly also other peptide compounds designed in this
manner can contribute to further investigation of infection mechanisms and to the
development of new viral entry inhibitors with unique properties.
- Anhang -
112
7. Anhang
7.1 Analytik der Peptide
Gezeigt sind jeweils Sequenz und Endanalytik aller Peptide, welche via HPLC/MS mit dem in
Tabelle 10 beschriebenen Gradienten gemessen wurde.
HAM Ac-GYRFSNFVIHWGBG-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(ala)HAM Ac-GYRFSAFVIHWGBG-[Dap(Alloc)-GWINPANGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX- [CVGPYSADDSPQDNYMDC]-NH2
(H3ala)HAM Ac-GYRFSNFVIHWGBG-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSADDSPQDNYMDC]-NH2
- Anhang -
113
(H2ala)HAM Ac-GYRFSNFVIHWGBG-[Dap(Alloc)-GWINPANGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H1ala)HAM Ac-GYRFSAFVIHWGBG-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H2H3ala)HAM Ac-GYRFSNFVIHWGBG-[Dap(Alloc)-GWINPANGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSADDSPQDNYMDC]-NH2
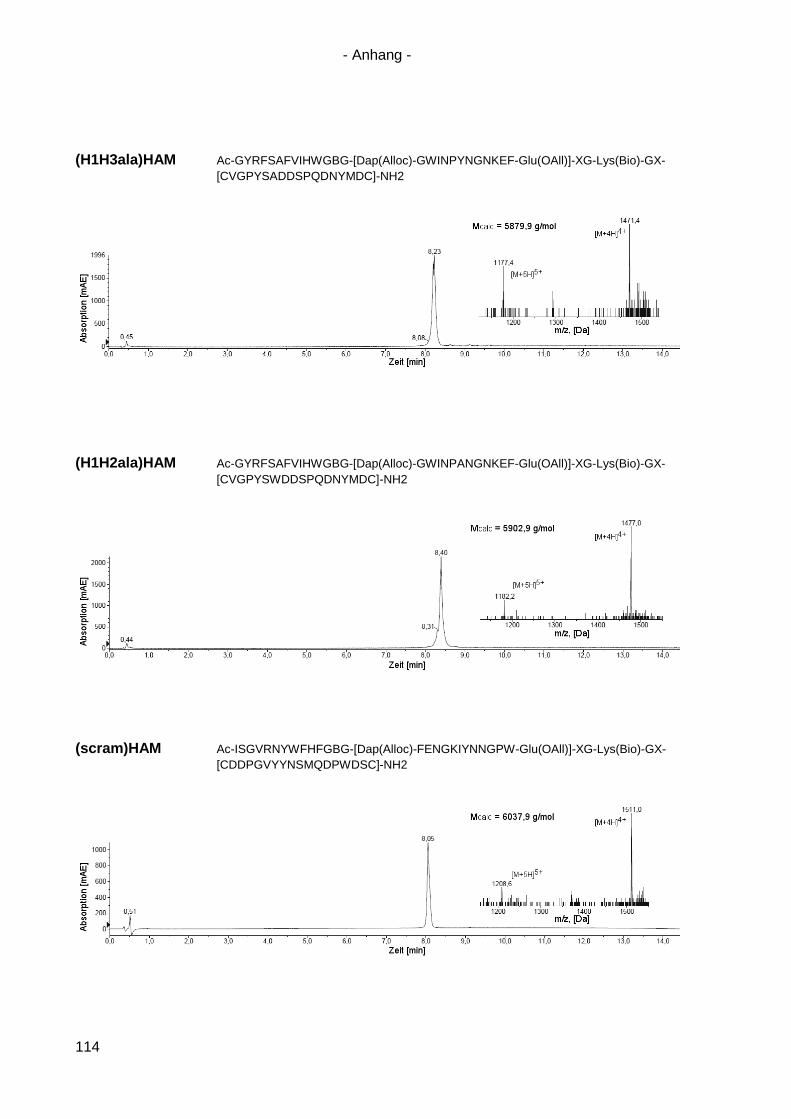
- Anhang -
114
(H1H3ala)HAM Ac-GYRFSAFVIHWGBG-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSADDSPQDNYMDC]-NH2
(H1H2ala)HAM Ac-GYRFSAFVIHWGBG-[Dap(Alloc)-GWINPANGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(scram)HAM Ac-ISGVRNYWFHFGBG-[Dap(Alloc)-FENGKIYNNGPW-Glu(OAll)]-XG-Lys(Bio)-GX-[CDDPGVYYNSMQDPWDSC]-NH2
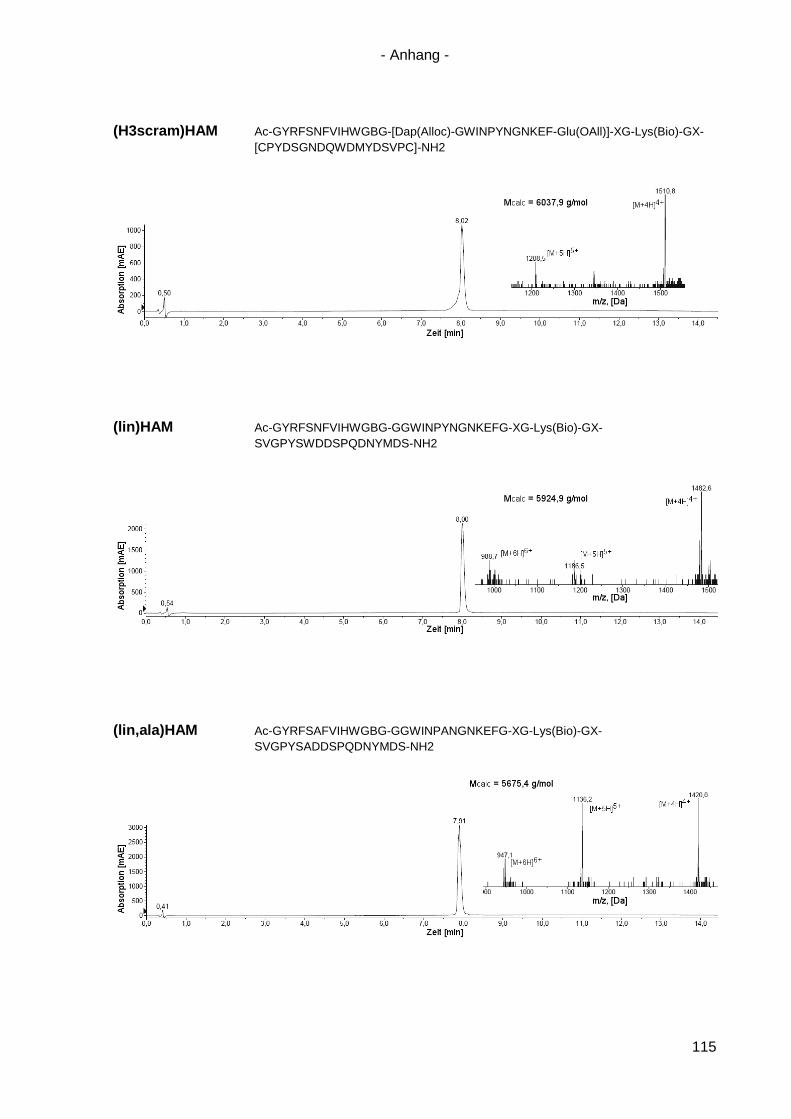
- Anhang -
115
(H3scram)HAM Ac-GYRFSNFVIHWGBG-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CPYDSGNDQWDMYDSVPC]-NH2
(lin)HAM Ac-GYRFSNFVIHWGBG-GGWINPYNGNKEFG-XG-Lys(Bio)-GX-SVGPYSWDDSPQDNYMDS-NH2
(lin,ala)HAM Ac-GYRFSAFVIHWGBG-GGWINPANGNKEFG-XG-Lys(Bio)-GX-SVGPYSADDSPQDNYMDS-NH2
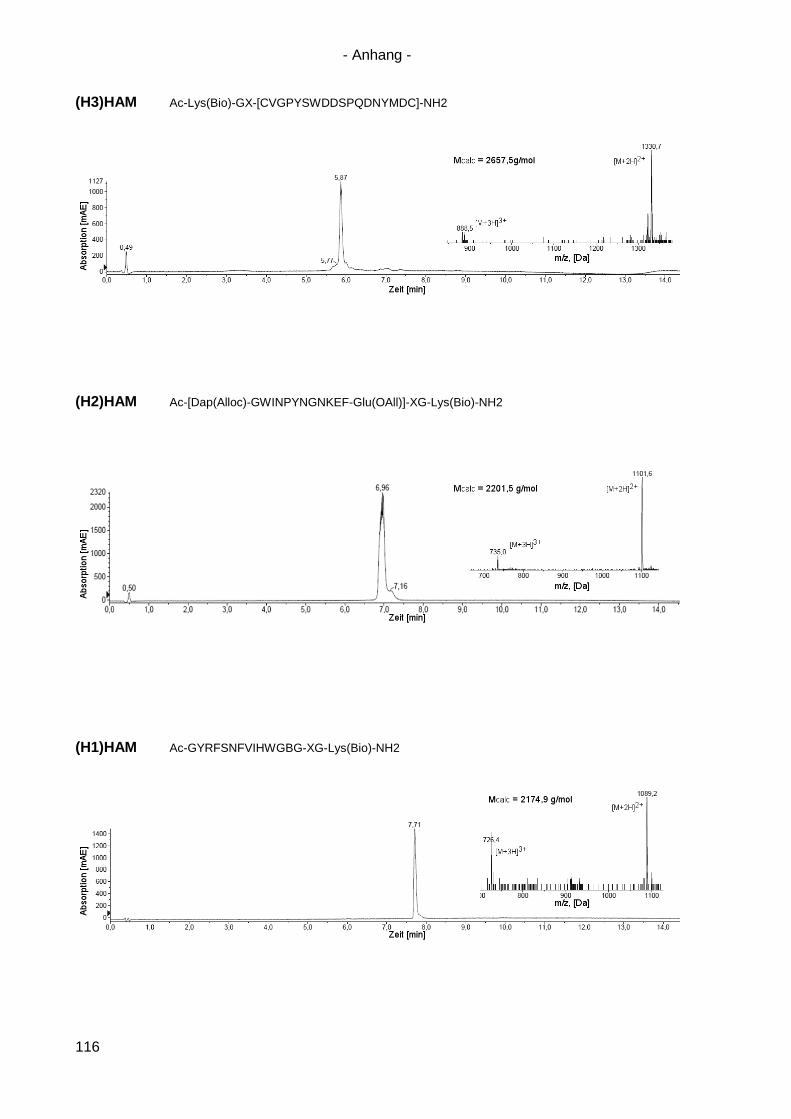
- Anhang -
116
(H3)HAM Ac-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H2)HAM Ac-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-NH2
(H1)HAM Ac-GYRFSNFVIHWGBG-XG-Lys(Bio)-NH2

- Anhang -
117
(H2H3)HAM Ac-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H1H3)HAM Ac-GYRFSNFVIHWGBG-XG-Lys(Biotin)-GX-[CVGPYSWDDSPQDNYMDC]-NH2
(H1H2)HAM Ac-GYRFSNFVIHWGBG-[Dap(Alloc)-GWINPYNGNKEF-Glu(OAll)]-XG-Lys(Bio)-NH2
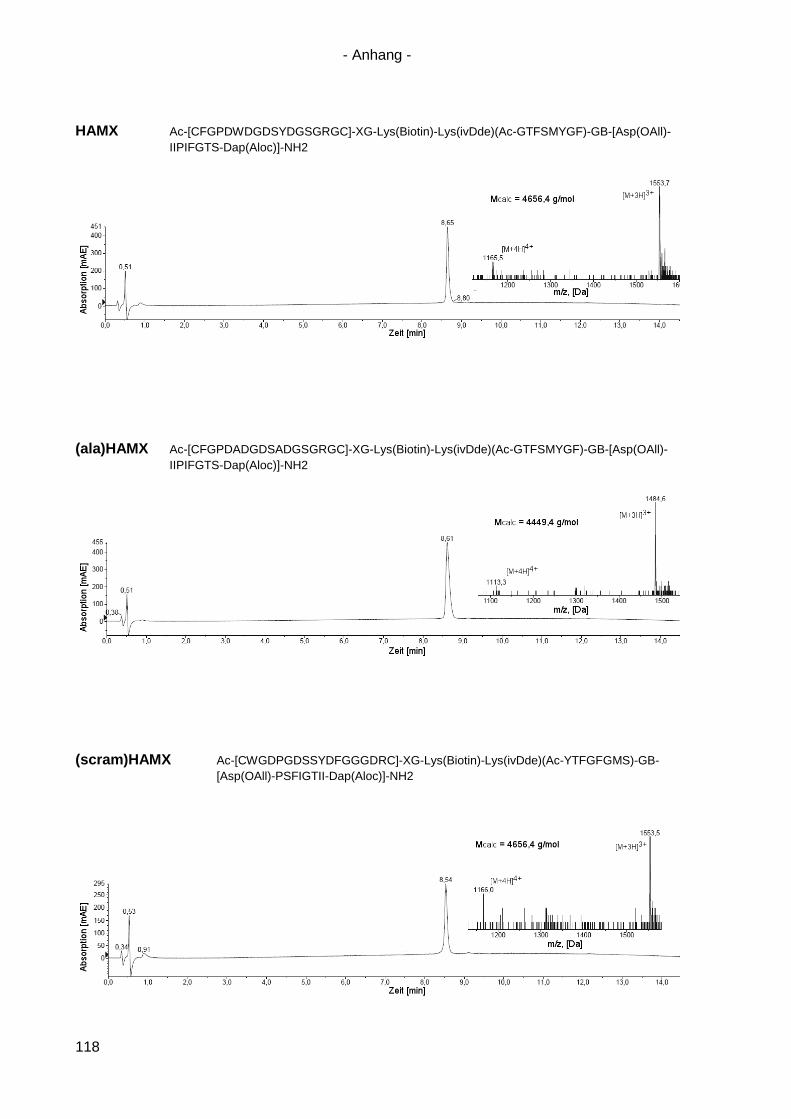
- Anhang -
118
HAMX Ac-[CFGPDWDGDSYDGSGRGC]-XG-Lys(Biotin)-Lys(ivDde)(Ac-GTFSMYGF)-GB-[Asp(OAll)-IIPIFGTS-Dap(Aloc)]-NH2
(ala)HAMX Ac-[CFGPDADGDSADGSGRGC]-XG-Lys(Biotin)-Lys(ivDde)(Ac-GTFSMYGF)-GB-[Asp(OAll)-IIPIFGTS-Dap(Aloc)]-NH2
(scram)HAMX Ac-[CWGDPGDSSYDFGGGDRC]-XG-Lys(Biotin)-Lys(ivDde)(Ac-YTFGFGMS)-GB-[Asp(OAll)-PSFIGTII-Dap(Aloc)]-NH2
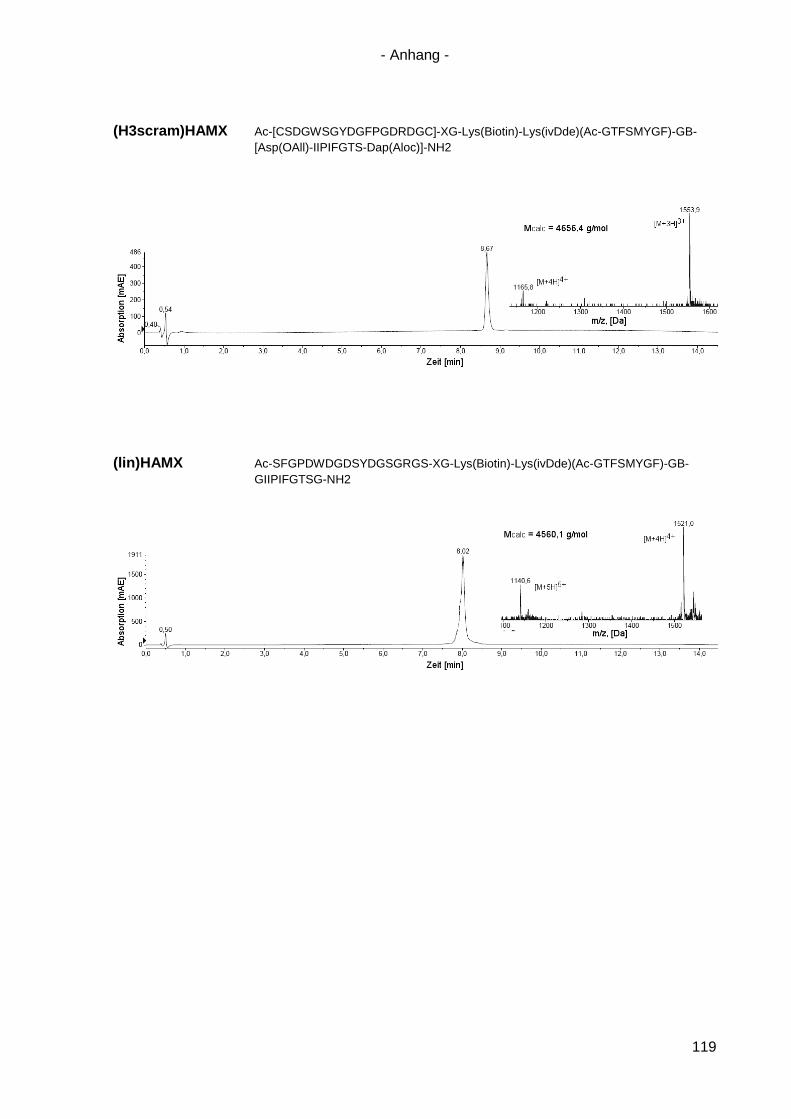
- Anhang -
119
(H3scram)HAMX Ac-[CSDGWSGYDGFPGDRDGC]-XG-Lys(Biotin)-Lys(ivDde)(Ac-GTFSMYGF)-GB-[Asp(OAll)-IIPIFGTS-Dap(Aloc)]-NH2
(lin)HAMX Ac-SFGPDWDGDSYDGSGRGS-XG-Lys(Biotin)-Lys(ivDde)(Ac-GTFSMYGF)-GB-GIIPIFGTSG-NH2

- Anhang -
120
7.2 Sequenz von gp120(ZM96)
Das verwendete Protein gp120(ZM96) wurde dankenswerterweise von Dr. Ralf Wagner und
seinen Kollegen (Institut für Mikrobiologie und Hygiene, Universität Regensburg) zur
Verfügung gestellt.
Aminosäure-Sequenz von gp120(ZM96):
MGVREILRNWQRWWTWGILGFWMLMICNVWGNLWVTVYYGVPVWKEAKTTLFCASDAKSY EKEVHNVWATHACVPTDPNPQEIVLGNVTENFNMWKNDMVDQMHEDIISLWDQSLKPCVK LTPLCVTLNCTEVNVTRNVNNSVVNNTTNVNNSMNGDMKNCSFNITTELKDKKKNVYALF YKLDIVSLNETDDSETGNSSKYYRLINCNTSALTQACPKVSFDPIPIHYCAPAGYAILKC NNKTFNGTGPCHNVSTVQCTHGIKPVVSTQLLLNGSLAEEGIIIRSENLTNNVKTIIVHL NRSIEIVCVRPNNNTRQSIRIGPGQTFYATGDIIGDIRQAHCNISRTNWTKTLREVRNKL REHFPNKNITFKPSSGGDLEITTHSFNCRGEFFYCNTSGLFSINYTENNTDGTPITLPCR IRQIINMWQEVGRAMYAPPIEGNIACKSDITGLLLVRDGGSTNDSTNNNTEIFRPAGGDM RDNWRSELYKYKVVEIKPLGIAPTEAKRRVVEREKR

- Anhang -
121
7.3 Abbildungsverzeichnis
Abbildung 1: Aufbau eines IgG-Antikörpers. ............................................................................................2 Abbildung 2: Prinzip der somatischen Rekombination und Expression von B-Zell-Rezeptoren. 1 ..........3 Abbildung 3: HIV-1 gp120 (grau) mit der entsprechenden Bindungsstelle für CD4, bzw. den Epitopen für die Antikörper F105, b12 und b13 (orange Linie). Hydrophobe Reste von gp120 sind hervorgehoben (grün). 20 ..........................................................................................................................5 Abbildung 4: Wirkungsprinzip peptidischer Mimetika: Die Interaktion zweier Proteine (blau und grün) wird durch ein peptidisches Mimetikum (rot) verhindert (bearbeitet nach 57). ..........................................9 Abbildung 5: Aufbau eines HI-Viruspartikels (bearbeitet nach 68). ........................................................ 11 Abbildung 6: Das Andocken eines HIV-Partikels an eine Wirtszelle bis zur Fusion ist ein mehrstufiger Prozess (bearbeitet nach 75). ................................................................................................................. 13 Abbildung 7: Replikationszyklus von HI-Viren. 79 .................................................................................. 14 Abbildung 8: HIV-1 neutralisierende Antikörper und ihre Angriffspunkte auf dem viralen Spike, Glykosylierungen in blau/grün. 103 ......................................................................................................... 16 Abbildung 9: Kristallstruktur (PDB: 2NY7) von b12 (Fab) (grau) in Komplex mit HIV-1 gp120 (gelb), farbig hervorgehoben sind die CDR Hs von b12 CDR H3 (rot), CDR H2 (blau) und CDR H1 (grün). 117
............................................................................................................................................................... 18 Abbildung 10: CDR Hs (rot, blau, grün) von b12 mit den drei Hauptinteraktionsaminosäuren (magenta) 117; Ausschnitt aus der Kristallstruktur in Komplex mit gp120 (PDB: 2NY7). ........................................ 19 Abbildung 11: Kristallstruktur (PDB:2B4C) von X5 (Fab) (grau) in Komplex mit HIV-1 gp120 (gelb) und CD4 (violett), farbig hervorgehoben sind die CDR Hs von X5 CDR H3 (rot), CDR H2 (blau) und CDR H1 (grün). 102 ......................................................................................................................................... 20 Abbildung 12: CDR Hs (rot, blau, grün) von X5 mit den beiden essentiellen aromatischen Aminosäuren (magenta) 102; Ausschnitt aus der Kristallstruktur in Komplex mit gp120 und CD4 (PDB: 2B4C). ......................................................................................................................................... 21 Abbildung 13: „Fußabdrücke“ von X5, 17b, CCR5 und CD4 (rot) auf HIV-1 gp120 (grau). 102 ............. 22 Abbildung 14: Design des b12-Antikörper-Paratop-Mimetikums HAM. A) Kristallstruktur des b12-Antikörpers in Komplex mit HIV-1 gp120(HXBc2) (PDB: 2NY7), die CDRs H1 in grün, H2 in blau, H3 in rot dargestellt. B) Antikörper-Paratop von b12 in der Kristallstruktur C) Blick auf das b12-Paratop vom gp120-Epitop aus. D) Schematische Darstellung und exakte Sequenz von HAM. .............................. 50 Abbildung 15: Bindung von HAM und (ala)HAM an gp120(IIIB) im ELISA (Format A). .......................... 54 Abbildung 16: Detektion von gp120-Proteinen verschiedener HIV-1-Stämme im ELISA von Schaf-Anti-gp120 gefolgt von Kaninchen-Anti-Schaf-HRP. .................................................................................... 55 Abbildung 17: Bindung von gp120-Proteinen verschiedener HIV-1-Stämme an HAM und (ala)HAM im Streptavidin-ELISA (Format B). ............................................................................................................. 56 Abbildung 18: Bindung von gp120-Proteinen b12-sensitiver (IIIB, HXBc2) bzw. b12-resistenter (PVO, ZM96) HIV-1-Stämme im Streptavidin-ELISA (Format B). A) Bindung an b12, B) Bindung an HAM. . 57 Abbildung 19: Kompetition von CD4 mit HAM um die Bindung an gp120 im ELISA (Format C). ........ 58 Abbildung 20: Maximal erreichte Inhibition der HAM-gp120-Bindung im ELISA (Format C). Inhibitoren (Höchstkonz. 0,2 µM): CD4 als natürlicher Ligand, b12 als Eltern- und CD4-Bindungsstellen-Antikörper und X5 als Negativkontrolle (CD4i-Antikörper). ................................................................... 59 Abbildung 21: Bindung von CD4, b12 und X5 an HAM und (ala)HAM, Streptavidin-ELISA (Format B). ............................................................................................................................................................... 59 Abbildung 22: Bindung diverser Antikörper an HAM im Streptavidin-ELISA (Format B). ..................... 60 Abbildung 23: SEAP-Zellinfektionsassay mit HIV-1(NL4-3) und HAM, (ala)HAM bzw. b12. .................... 61 Abbildung 24: GHOST-Zellinfektionsassay mit HIV-1(NL4-3), und HAM bzw. b12. ................................. 62 Abbildung 25: Biologische Aktivität der Einzel-Alanin-Varianten von HAM. Links sind die Daten der SEAP-Zellinkfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA (Format B). ............................................................................................................................................ 63 Abbildung 26: Biologische Aktivität der Doppel-Alanin-Varianten von HAM. Links sind die Daten der SEAP-Zellinfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA (Format B). ............................................................................................................................................ 64 Abbildung 27: Biologische Aktivität der srcambled Varianten von HAM. Links sind die Daten der SEAP-Zellinfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA (Format B). ............................................................................................................................................ 65

- Anhang -
122
Abbildung 28: Biologische Aktivität der linearen Varianten von HAM. Links sind die Daten der SEAP-Zellinkfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA (Format B). ............................................................................................................................................................... 66 Abbildung 29: Bindung von (lin)HAM an gp120(IIIB) im ELISA (Format A). ........................................... 67 Abbildung 30: Bindung von gp120(IIIB) an die verkürzten HAM-Peptide im Streptavidin-ELISA (Format B). ............................................................................................................................................. 68 Abbildung 31: SEAP-Zellinfektionsassay mit HIV-1(NL4-3) und den verkürzten Varianten von HAM. A) Peptide die nur eine der drei CDRs H1, H2 oder H3 enthalten. B) Peptide, die zwei der drei CDRs enthalten. ............................................................................................................................................... 69 Abbildung 32: Bindung von (H1H3)HAM an gp120(IIIB) im ELISA (Format A). ...................................... 70 Abbildung 33: Bindung von gp120-Proteinen b12-sensitiver (IIIB, HXBc2) bzw. b12-resistenter (PVO, ZM96) HIV-1-Stämme an (H1H3)HAM im Streptavidin-ELISA (Format B). .......................................... 71 Abbildung 34: Entstandene Mutationen in HIV-1(NL4-3) unter Selektionsdruck durch Anwesenheit von b12 oder verschiedenen HAM-Varianten. R = Resequenzierung, zeigt die Mutationen, die nach Aufhebung des Selektionsdrucks bestehen blieben. ............................................................................ 72 Abbildung 35: SEAP-Zellinfektionsassay mit b12 und HIV-1(NL4-3) bzw. HIV-1(NL4-3(b12-r)). ..................... 74 Abbildung 36: SEAP-Zellinfektionsassay mit HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3(HAM-r)). .................. 75 Abbildung 37: SEAP-Zellinfektionsassay mit (H1H3)HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3((H1H3)HAM-r)).75 Abbildung 38: SEAP-Zellinfektionsassay mit b12 und HIV-1(NL4-3), HIV-1(NL4-3(HAM-r)) bzw. HIV-1(NL4-
3((H1H3)HAM-r)). ............................................................................................................................................ 76 Abbildung 39: SEAP-Zellinfektionsassay mit HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3(b12-r). .................... 77 Abbildung 40: SEAP-Zellinfektionsassay mit (H1H3)HAM und HIV-1(NL4-3) bzw. HIV-1(NL4-3(b12-r)). ........ 77 Abbildung 41: Design des X5-Antikörper-Paratop-Mimetikums HAMX. A) Kristallstruktur von X5 im Komplex mit HIV-1 gp120(JRFL) und CD4 (PDB: 2B4C). H1 in grün, H2 in blau, H3 in rot dargestellt. B) Antikörper-Paratop von X5 in der Kristallstruktur C) Blick auf das X5-Paratop vom gp120-Epitop aus. D) Schematische Darstellung von HAMX und exakte Sequenz. ........................................................... 79 Abbildung 42: Bindung von gp120-Proteinen verschiedener HIV-1-Stämme (IIIB, HXBc2, BaL und ADA) an HAMX bzw. (ala)HAMX im Streptavidin-ELISA (Format B). ................................................... 82 Abbildung 43: Bindung von HAMX an gp120(IIIB) im ELISA (Format A). ................................................ 83 Abbildung 44: A) Bindung von gp120 in Gegenward und in Abwesenheit von CD4 an HAMX bzw. (ala)HAMX im Streptavidin-ELISA (Format B). B) Bindung von CD4, b12 und X5 an HAM bzw. (ala)HAM im Streptavidin-ELISA (Format B). ........................................................................................ 83 Abbildung 45: SEAP-Zellinfektionsassay mit HIV-1(NL4-3) und X5, HAMX bzw. (ala)HAMX. ................. 84 Abbildung 46: GHOST-Zellinfektionsassay mit HIV-1(NL4-3) und X5, HAMX oder (ala)HAMX. .............. 85 Abbildung 47: Biologische Aktivität der scrambeld und linearen Varianten von HAMX. Links sind die Daten der GHOST-Zellinfektionsassays dargestellt, rechts die Bindung an gp120(IIIB) im Streptavidin-ELISA (ELISA-Format B). ...................................................................................................................... 86 Abbildung 48: HSV-Zellinfektionsassay ................................................................................................. 87 Abbildung 49: Schematische Darstellung von gp120(NL4-3), das für die Langzeit-Resistenz-Studien verwendet wurde. Eingezeichnet sind die Interaktionsstellen des Proteins mit b12 (gelb), CD4 (blau), bzw. beiden (gelb mit blauer Schrift). Außerdem sind die Cysteinbrücken durch schwarze Verbindungen gekennzeichnet. Die Stellen, an denen die drei generierten, resistenten Viren sich vom Wildtyp gp120(NL4-3) unterschieden sind mit Pfeilen gekennzeichnet (b12-r in orange, HAM-r in pink, (H1H3)HAM-r in grün). ........................................................................................................................... 91 Abbildung 50: A) Kristallstruktur von gp120 (PDB: 4ZMJ, kristallisiertes Env-Trimer ohne Ligand). Gezeigt sind gp120 (grau), b12-Bindungseptiop (gelb) und die Mutationsstellen von b12-r (schwarz) und HAM-r (pink). B) Kristallstruktur von gp120 (PDB: 2NY7, stabilisiertes, b12-gebundenes Kernprotein). Gezeigt sind gp120(gelb), CDR H3 von b12 (rot), Stamm der V4-Schleife (cyan), Mutationsstelle von b12-r (magenta). .................................................................................................... 92 Abbildung 51: SEAP-Zellinfektionsassay mit HAM und drei verschiedenen Viren; direkter Vergleich von Resistenz- und Kreuzresistenztestung. .......................................................................................... 93 Abbildung 52: Bindung an gp120(IIIB) der drei potentesten Peptide im Vergleich. ................................. 96 Abbildung 53: SEAP-Zellinfektionsassay der drei potentesten Peptide im Vergleich. .......................... 97 Abbildung 54: Kristallstruktur von gp120 (PDB: 4ZMJ, kristallisiertes Env-Trimer ohne Ligand). Gezeigt sind gp120 (grau), b12-Epitop (gelb), b12-r-Mutationsstellen (schwarz) und (H1H3)HAM-r-Mutationsstellen (grün). ......................................................................................................................... 99

- Anhang -
123
Abbildung 55: SEAP-Zellinfektionsassay vom (H1H3)HAM an drei verschiedenen Viren zum direkten Vergleich von Resistenz- und Kreuzresistenztestung. ........................................................................ 100
7.4 Tabellenverzeichnis
Tabelle 1: Beispiele CDR-abgeleiteter peptidischer Antikörper-Mimetika. 35–42 .......................................7 Tabelle 2: HIV-Proteine und ihr Funktion, im roten Kasten hervorgehoben die Proteine des viralen Spikes. 76 ............................................................................................................................................... 12 Tabelle 3: Zyklus bei der automatisierten SPPS ................................................................................... 32 Tabelle 4: Zyklus bei manuellen Kupplungen ....................................................................................... 33 Tabelle 5: Abspaltung der Schutzgruppe ivDde .................................................................................... 34 Tabelle 6: Abspaltung der Schutzgruppen OAll und Alloc .................................................................... 34 Tabelle 7: N-terminale Acetylierung ...................................................................................................... 35 Tabelle 8: HPLC-Lauf zur Reinigung der Peptide ................................................................................. 37 Tabelle 9: Bedingungen bei der zweiten Reinigung der Peptide .......................................................... 38 Tabelle 10: Gradienten bei der HPLC/MS-Analytik ............................................................................... 39 Tabelle 11: Aus den CDR-H-Schleifen von b12 abgeleitete Sequenzen für HAM .............................. 50 Tabelle 12: Sequenzen, molare Massen und Ausbeuten von HAM und Varianten .............................. 53 Tabelle 13: Verkürzte Varianten von HAM, H3 in rot, H2 in blau, H1 in grün. ...................................... 67 Tabelle 14: Liste der Mutationen in den selektionierten Viursvarianten von HIV-1(NL4-3) .................. 73 Tabelle 15: Aus den CDR-H-Schleifen von b12 abgeleitete Sequenzen für HAMX ............................ 78 Tabelle 16: Sequenzen, molare Massen und Ausbeuten von HAMX und Varianten ........................... 81 Tabelle 17: IC50-Werte der HAM-Peptide .............................................................................................. 96

- Anhang -
124

- Referenzen -
I
Referenzen 1. Murphy, K. Janeway's immunobiology (Garland Science, 2014).
2. Modrow, S.; Falke, D.; Truyen, U. & Schätzl, H. Molekulare Virologie. 3rd ed. (Spektrum Akademischer Verlag, Heidelberg, 2010).
3. Kaufmann, S. H. E. Basiswissen Immunologie (Imprint: Springer, Berlin, Heidelberg, 2014).
4. Charles A Janeway, JR; Travers, P.; Walport, M. & Shlomchik, M. J. The distribution and functions of immunoglobulin isotypes (Garland Science, 2001).
5. Porter, R. R.: The hydrolysis of rabbit γ-globulin and antibodies with crystalline papain, Biochem. J. 73, 119–127 (1959).
6. Foote, J. & Winter, G.: Antibody framework residues affecting the conformation of the hypervariable loops, Journal of Molecular Biology 224, 487–499 (1992).
7. Quie, P. G.: Phagocytosis in Subacute Bacterial Endocartitis. Localization of the Primary OpsonicC Site to Fc Fragment, Journal of Experimental Medicine 128, 553–570 (1968).
8. Li, Z.; Woo, C. J.; Iglesias-Ussel, M. D.; Ronai, D. & Scharff, M. D.: The generation of antibody diversity through somatic hypermutation and class switch recombination, Genes & development 18, 1–11 (2004).
9. Hawkins, R. E.; Russell, S. J. & Winter, G.: Selection of phage antibodies by binding affinity, Journal of Molecular Biology 226, 889–896 (1992).
10. Sela-Culang, I.; Kunik, V. & Ofran, Y.: The structural basis of antibody-antigen recognition, Frontiers in immunology 4, 302 (2013).
11. MacCallum, R. M.; Martin, A. C. & Thornton, J. M.: Antibody-antigen interactions: contact analysis and binding site topography, Journal of Molecular Biology 262, 732–745 (1996).
12. van Oss, C. J.: Hydrophobic, hydrophilic and other interactions in epitope-paratope binding, Molecular Immunology 32, 199–211 (1995).
13. Keskin, O.: Binding induced conformational changes of proteins correlate with their intrinsic fluctuations: a case study of antibodies, BMC structural biology 7, 31 (2007).
14. Voet, A.; Berenger, F. & Zhang, K. Y. J.: Electrostatic similarities between protein and small molecule ligands facilitate the design of protein-protein interaction inhibitors, PLoS ONE 8, e75762 (2013).
15. Neuvirth, H.; Raz, R. & Schreiber, G.: ProMate: a structure based prediction program to identify the location of protein-protein binding sites, Journal of Molecular Biology 338, 181–199 (2004).
16. Xu, X. et al.: Evaluation of spatial epitope computational tools based on experimentally-confirmed dataset for protein antigens, Chin. Sci. Bull. 55, 2169–2174 (2010).
17. Benjamin, D. C. et al.: The antigenic structure of proteins: a reappraisal, Annual review of immunology 2, 67–101 (1984).
18. Greenbaum, J. A. et al.: Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools, Journal of molecular recognition : JMR 20, 75–82 (2007).
19. Novotny, J. et al.: Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains), Proceedings of the National Academy of Sciences 83, 226–230 (1986).

- Referenzen -
II
20. Chen, L. et al.: Structural Basis of Immune Evasion at the Site of CD4 Attachment on HIV-1 gp120, Science 326, 1123–1127 (2009).
21. WuTT, K.: Ananalysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J ExpMed, 211–250 (1970).
22. Kabat, E.A. Wu, T.T. Bilofsky H.: Sequence of Proteins of Immunological Interest. Bethesda, NIH (1983).
23. Abhinandan, K. R. & Martin, A. C. R.: Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains, Molecular Immunology 45, 3832–3839 (2008).
24. Lesk, A. M. & Chothia, C.: Evolution of proteins formed by β-sheets, Journal of Molecular Biology 160, 325–342 (1982).
25. Al-Lazikani, B.; Lesk, A. M. & Chothia, C.: Standard conformations for the canonical structures of immunoglobulins, Journal of Molecular Biology 273, 927–948 (1997).
26. Kuroda, D.; Shirai, H.; Kobori, M. & Nakamura, H.: Structural classification of CDR-H3 revisited: a lesson in antibody modeling, Proteins 73, 608–620 (2008).
27. Wang, F. et al.: Reshaping antibody diversity, Cell 153, 1379–1393 (2013).
28. Ewert, S.; Honegger, A. & Plückthun, A.: Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering, Methods (San Diego, Calif.) 34, 184–199 (2004).
29. Chames, P.; van Regenmortel, M.; Weiss, E. & Baty, D.: Therapeutic antibodies: successes, limitations and hopes for the future, British journal of pharmacology 157, 220–233 (2009).
30. Löfblom, J. et al.: Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications, FEBS letters 584, 2670–2680 (2010).
31. Dennis, M. S. et al.: Peptide exosite inhibitors of factor VIIa as anticoagulants, Nature 404, 465–470 (2000).
32. Gebauer, M. & Skerra, A.: Engineered protein scaffolds as next-generation antibody therapeutics, Current Opinion in Chemical Biology 13, 245–255 (2009).
33. Wojcik, J. et al.: A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain, Nature structural & molecular biology 17, 519–527 (2010).
34. Gebauer, M. & Skerra, A.: Anticalins small engineered binding proteins based on the lipocalin scaffold, Methods in enzymology 503, 157–188 (2012).
35. Timmerman, P. et al.: A combinatorial approach for the design of complementarity-determining region-derived peptidomimetics with in vitro anti-tumoral activity, The Journal of biological chemistry 284, 34126–34134 (2009).
36. Levi, M. et al.: A complementarity-determining region synthetic peptide acts as a miniantibody and neutralizes human immunodeficiency virus type 1 in vitro, Proceedings of the National Academy of Sciences of the United States of America 90, 4374–4378 (1993).
37. Heap, C. J. et al.: Analysis of a 17-amino acid residue, virus-neutralizing microantibody, The Journal of general virology 86, 1791–1800 (2005).

- Referenzen -
III
38. Park, B. W. et al.: Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo, Nature biotechnology 18, 194–198 (2000).
39. Berezov, A.; Zhang, H. T.; Greene, M. I. & Murali, R.: Disabling erbB receptors with rationally designed exocyclic mimetics of antibodies: structure-function analysis, Journal of medicinal chemistry 44, 2565–2574 (2001).
40. Tsumoto, K. et al.: Inhibition of hepatitis C virus NS3 protease by peptides derived from complementarity-determining regions (CDRs) of the monoclonal antibody 8D4: tolerance of a CDR peptide to conformational changes of a target, FEBS letters 525, 77–82 (2002).
41. Casset, F. et al.: A peptide mimetic of an anti-CD4 monoclonal antibody by rational design, Biochemical and Biophysical Research Communications 307, 198–205 (2003).
42. Feng, J.; Li, Y.; Zhang, W. & Shen, B.: Rational design of potent mimic peptide derived from monoclonal antibody: antibody mimic design, Immunology Letters 98, 311–316 (2005).
43. Timmerman, P. et al.: Binding of CDR-derived peptides is mechanistically different from that of high-affinity parental antibodies, J. Mol. Recognit. 23, 559–568 (2010).
44. Vlieghe, P.; Lisowski, V.; Martinez, J. & Khrestchatisky, M.: Synthetic therapeutic peptides: science and market, Drug Discovery Today 15, 40–56 (2010).
45. Loffet, A.: Peptides as drugs: is there a market?, Journal of peptide science : an official publication of the European Peptide Society 8, 1–7 (2002).
46. Kilby, J. M. et al.: Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry, Nature medicine 4, 1302–1307 (1998).
47. Lalezari, J. P. et al.: A phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy, AIDS (London, England) 17, 691–698 (2003).
48. Nielsen, L. L.: Incretin mimetics and DPP-IV inhibitors for the treatment of type 2 diabetes, Drug Discovery Today 10, 703–710 (2005).
49. Watching Peptide Drugs grow up, Chem. Eng. News 83, 17–24 (2005).
50. Merrifield, R. B.: Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide, J. Am. Chem. Soc. 85, 2149–2154 (1963).
51. McGregor, D. P.: Discovering and improving novel peptide therapeutics, Current opinion in pharmacology 8, 616–619 (2008).
52. Ladner, R. C.; Sato, A. K.; Gorzelany, J. & Souza, M. de: Phage display-derived peptides as therapeutic alternatives to antibodies, Drug Discovery Today 9, 525–529 (2004).
53. Bechara, C. & Sagan, S.: Cell-penetrating peptides: 20 years later, where do we stand?, FEBS letters 587, 1693–1702 (2013).
54. Hummel, G.; Reineke, U. & Reimer, U.: Translating peptides into small molecules, Molecular bioSystems 2, 499–508 (2006).
55. Lebl, M. & Houghten, R. A. Peptides. The wave of the future : proceedings of the Second International and the Seventeenth American Peptide Symposium, June 9-14, 2001, San Diego, California, U.S.A (Springer, Dordrecht, 2001).
56. Hruby, V. J.: Designing peptide receptor agonists and antagonists, Nature reviews. Drug discovery 1, 847–858 (2002).

- Referenzen -
IV
57. Eichler, J.: Peptides as protein binding site mimetics, Current Opinion in Chemical Biology 12, 707–713 (2008).
58. Benyamini, H. & Friedler, A.: Using peptides to study protein-protein interactions, Future medicinal chemistry 2, 989–1003 (2010).
59. Trellet, M.; Melquiond, A. S. J. & Bonvin, Alexandre M J J: A unified conformational selection and induced fit approach to protein-peptide docking, PLoS ONE 8, e58769 (2013).
60. Koshland, D. E.: Enzyme flexibility and enzyme action, J. Cell. Comp. Physiol. 54, 245–258 (1959).
61. Baggio, R. et al.: Induced fit of an epitope peptide to a monoclonal antibody probed with a novel parallel surface plasmon resonance assay, The Journal of biological chemistry 280, 4188–4194 (2005).
62. Dagliyan, O.; Proctor, E. A.; D'Auria, K. M.; Ding, F. & Dokholyan, N. V.: Structural and dynamic determinants of protein-peptide recognition, Structure (London, England : 1993) 19, 1837–1845 (2011).
63. LaBonte, J.; Lebbos, J. & Kirkpatrick, P.: Enfuvirtide, Nature reviews. Drug discovery 2, 345–346 (2003).
64. Barre-Sinoussi, F. et al.: Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS), Science 220, 868–871 (1983).
65. UNAIDS/World Health Organization: United Nations report on the global AIDS epidemic 2013. Accessed 2015 Apr 9 (2015).
66. Perrin, L.; Kaiser, L. & Yerly, S.: Travel and the spread of HIV-1 genetic variants, The Lancet Infectious Diseases 3, 22–27 (2003).
67. Hans W. Doerr, Wolfram H. Gerlich: Medizinische Virologie: Grundlagen, Diagnostik, Prävention und Therapie, Georg Thieme Verlag KG (2002).
68. Freed, E. O.: HIV-1 Replication, Somatic Cell and Molecular Genetics, 167–172 (2002).
69. Dalgleish, A. G. et al.: The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus, Nature 312, 763–767 (1984).
70. S. Bour, R. Geleziunas, M. A. Wainberg: The Human Immunodeficiency Virus Type 1 (HIV-1) CD4 Receptor and Its Central Role in Promotion of HIV-1 Infection, MICROBIOLOGICAL REVIEWS, 63–93 (1995).
71. Sullivan, N. et al.: CD4-Induced Conformational Changes in the Human Immunodeficiency Virus Type 1 gp120 Glycoprotein: Consequences for Virus Entry and Neutralization, J Virol 72, 4694–4703 (1998).
72. Samson, M. et al.: Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene, Nature 382, 722–725 (1996).
73. Deng, H. et al.: Identification of a major co-receptor for primary isolates of HIV-1, Nature 381, 661–666 (1996).
74. Tamamis, P. & Floudas, C. A.: Molecular recognition of CCR5 by an HIV-1 gp120 V3 loop, PLoS ONE 9, e95767 (2014).
75. Wilen, C. B.; Tilton, J. C. & Doms, R. W.: HIV: cell binding and entry, Cold Spring Harbor Perspectives in Medicine 2 (2012).

- Referenzen -
V
76. Bundesgesundheitsbl - Gesundheitsforsch - Gesundheitsschutz: Humanes Immunschwächevirus (HIV), Springer-Verlag, 83–95 (2004).
77. Auewarakul, P.; Wacharapornin, P.; Srichatrapimuk, S.; Chutipongtanate, S. & Puthavathana, P.: Uncoating of HIV-1 requires cellular activation, Virology 337, 93–101 (2005).
78. Sundquist, W. I. & Kräusslich, H.-G.: HIV-1 assembly, budding, and maturation, Cold Spring Harbor Perspectives in Medicine 2, a006924 (2012).
79. Harrisons innere Medizin. 18th ed. (ABW-Wissenschaftsverl. Berlin, 2012).
80. Komanduri, K. V. et al.: Restoration of cytomegalovirus-specific CD4+ T-lymphocyte responses after ganciclovir and highly active antiretroviral therapy in individuals infected with HIV-1, Nat Med 4, 953–956 (1998).
81. Autran, B.: Positive Effects of Combined Antiretroviral Therapy on CD4+ T Cell Homeostasis and Function in Advanced HIV Disease, Science 277, 112–116 (1997).
82. Lederman, M. M. et al.: Immunologic Responses Associated with 12 Weeks of Combination Antiretroviral Therapy Consisting of Zidovudine, Lamivudine, and Ritonavir. Results of AIDS Clinical Trials Group Protocol 315, Journal of Infectious Diseases 178, 70–79 (1998).
83. Arts, E. J. & Hazuda, D. J.: HIV-1 antiretroviral drug therapy, Cold Spring Harbor Perspectives in Medicine 2, a007161 (2012).
84. Williams, I. et al.: British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2012 (Updated November 2013. All changed text is cast in yellow highlight.), HIV medicine 15 Suppl 1, 1–85 (2014).
85. Henrich, T. J. & Kuritzkes, D. R.: HIV-1 entry inhibitors: recent development and clinical use, Current Opinion in Virology 3, 51–57 (2013).
86. Wyatt, R.: The HIV-1 Envelope Glycoproteins. Fusogens, Antigens, and Immunogens, Science 280, 1884–1888 (1998).
87. Pantophlet, R. & Burton, D. R.: GP120: Target for Neutralizing HIV-1 Antibodies, Annu. Rev. Immunol. 24, 739–769 (2006).
88. Pascal Poignard, Erica Ollmann Saphire, Paul WHI Parren, and Dennis R. Burton: GP120: Biologic Aspects of Structural Features.
89. Kwong, P. D. et al.: HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites, Nature 420, 678–682 (2002).
90. Antibody neutralization and escape by HIV-1.
91. Mascola, J. R. & Montefiori, D. C.: The role of antibodies in HIV vaccines, Annual review of immunology 28, 413–444 (2010).
92. McElrath, M. J. & Haynes, B. F.: Induction of immunity to human immunodeficiency virus type-1 by vaccination, Immunity 33, 542–554 (2010).
93. Overbaugh, J. & Morris, L.: The Antibody Response against HIV-1, Cold Spring Harbor Perspectives in Medicine 2, a007039 (2012).
94. Kwong, P. D.; Mascola, J. R. & Nabel, G. J.: Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning, Nature reviews. Immunology 13, 693–701 (2013).

- Referenzen -
VI
95. Sather, D. N. et al.: Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection, Journal of Virology 83, 757–769 (2009).
96. Gray, E. S. et al.: The neutralization breadth of HIV-1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection, Journal of Virology 85, 4828–4840 (2011).
97. Shibata, R. et al.: Neutralizing antibody directed against the HIV-1 envelope glycoprotein can completely block HIV-1/SIV chimeric virus infections of macaque monkeys, Nature medicine 5, 204–210 (1999).
98. Hessell, A. J. et al.: Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers, PLoS pathogens 5, e1000433 (2009).
99. Kwong, P. D. & Mascola, J. R.: Human Antibodies that Neutralize HIV-1: Identification, Structures, and B Cell Ontogenies, Immunity 37, 412–425 (2012).
100. DeVico, A. et al.: Antibodies to CD4-induced sites in HIV gp120 correlate with the control of SHIV challenge in macaques vaccinated with subunit immunogens, Proceedings of the National Academy of Sciences of the United States of America 104, 17477–17482 (2007).
101. BIAuser: HIV-1 Neutralizing Antibody 17b Weakens the Interaction of Envelope Protein gp120 with CD4.
102. Darbha, R. et al.: Crystal Structure of the Broadly Cross-Reactive HIV-1-Neutralizing Fab X5 and Fine Mapping of Its Epitope †,‡, Biochemistry 43, 1410–1417 (2004).
103. Burton, D. R. & Weiss, R. A.: AIDS/HIV. A boost for HIV vaccine design, Science (New York, N.Y.) 329, 770–773 (2010).
104. Huang, J. et al.: Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface, Nature 515, 138–142 (2014).
105. Armbruster, C. et al.: Passive immunization with the anti-HIV-1 human monoclonal antibody (hMAb) 4E10 and the hMAb combination 4E10/2F5/2G12, The Journal of antimicrobial chemotherapy 54, 915–920 (2004).
106. Mehandru, S. et al.: Adjunctive passive immunotherapy in human immunodeficiency virus type 1-infected individuals treated with antiviral therapy during acute and early infection, Journal of Virology 81, 11016–11031 (2007).
107. Caskey, M. et al.: Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117, Nature 522, 487–491 (2015).
108. Johnson, P. R. et al.: Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys, Nature medicine 15, 901–906 (2009).
109. Mingozzi, F. & High, K. A.: Immune responses to AAV vectors: overcoming barriers to successful gene therapy, Blood 122, 23–36 (2013).
110. D R Burton, C F Barbas, 3rd, M A Persson, S Koenig, R M Chanock, and R A Lerner: A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc Natl Acad Sci U S A, 10134–10137 (1991).

- Referenzen -
VII
111. Parren, P. W. H. I. et al.: Antibody Protects Macaques against Vaginal Challenge with a Pathogenic R5 Simian/Human Immunodeficiency Virus at Serum Levels Giving Complete Neutralization In Vitro, Journal of Virology 75, 8340–8347 (2001).
112. Burton, D. et al.: Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody, Science 266, 1024–1027 (1994).
113. Zwick, M. B. et al.: Molecular Features of the Broadly Neutralizing Immunoglobulin G1 b12 Required for Recognition of Human Immunodeficiency Virus Type 1 gp120, Journal of Virology 77, 5863–5876 (2003).
114. Kwong, P. D. et al.: Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody, Nature 393, 648–659 (1998).
115. Saphire, E. O.: Crystal Structure of a Neutralizing Human IgG Against HIV-1: A Template for Vaccine Design, Science 293, 1155–1159 (2001).
116. Pantophlet, R. et al.: Fine Mapping of the Interaction of Neutralizing and Nonneutralizing Monoclonal Antibodies with the CD4 Binding Site of Human Immunodeficiency Virus Type 1 gp120, Journal of Virology 77, 642–658 (2003).
117. Zhou, T. et al.: Structural definition of a conserved neutralization epitope on HIV-1 gp120, Nature 445, 732–737 (2007).
118. Dueñas-Decamp, M. J.; O'Connell, O. J.; Corti, D.; Zolla-Pazner, S. & Clapham, P. R.: The W100 pocket on HIV-1 gp120 penetrated by b12 is not a target for other CD4bs monoclonal antibodies, Retrovirology 9, 9 (2012).
119. Wu, X. et al.: Rational Design of Envelope Identifies Broadly Neutralizing Human Monoclonal Antibodies to HIV-1, Science 329, 856–861 (2010).
120. Li, Y. et al.: Mechanism of Neutralization by the Broadly Neutralizing HIV-1 Monoclonal Antibody VRC01, Journal of Virology 85, 8954–8967 (2011).
121. Wu, X. et al.: Mechanism of Human Immunodeficiency Virus Type 1 Resistance to Monoclonal Antibody b12 That Effectively Targets the Site of CD4 Attachment, Journal of Virology 83, 10892–10907 (2009).
122. Bunnik, E. M. et al.: Emergence of monoclonal antibody b12-resistant human immunodeficiency virus type 1 variants during natural infection in the absence of humoral or cellular immune pressure, The Journal of general virology 91, 1354–1364 (2010).
123. Moulard, M. et al.: Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120-CD4-CCR5 complexes, Proceedings of the National Academy of Sciences 99, 6913–6918 (2002).
124. Labrijn, A. F. et al.: Access of Antibody Molecules to the Conserved Coreceptor Binding Site on Glycoprotein gp120 Is Sterically Restricted on Primary Human Immunodeficiency Virus Type 1, Journal of Virology 77, 10557–10565 (2003).
125. Ellman, G. L.: Tissue sulfhydryl groups, Archives of Biochemistry and Biophysics 82, 70–77 (1959).
126. Means, R. E.; Greenough, T. & Desrosiers, R. C.: Neutralization sensitivity of cell culture-passaged simian immunodeficiency virus, Journal of Virology 71, 7895–7902 (1997).
127. Li, Y. et al.: Broad HIV-1 neutralization mediated by CD4-binding site antibodies, Nat Med 13, 1032–1034 (2007).
128. Neutralization Sensitivity of Cell Culture-Passaged Simian Immunodeficiency Virus.

- Referenzen -
VIII
129. Beerenwinkel, N. et al.: Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from genotype, Proceedings of the National Academy of Sciences of the United States of America 99, 8271–8276 (2002).
130. Wu, X. et al.: Mechanism of Human Immunodeficiency Virus Type 1 Resistance to Monoclonal Antibody b12 That Effectively Targets the Site of CD4 Attachment▿, J Virol 83, 10892–10907 (2009).
131. A. J. Nahmias, S. Kibrick: Inhibtiory effect of heparin on herpes simplex virus, Journal of Bacteriology, 1060–1066 (1964).
132. C.-Y. Kang, T. K. Brunck, T. Kieber-Emmons, J. E. Blalock, H. Kohler: Inhibition of Self-Binding Antibodies (Autobodies) by a Inhibtion of Self-Binding Antibodies (Autobodies) by a VH-derived peptide, Science, 1034–1036 (1988).
133. Haynes, B. F. et al.: Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies, Science (New York, N.Y.) 308, 1906–1908 (2005).
134. Liu, M. et al.: Polyreactivity and autoreactivity among HIV-1 antibodies, Journal of Virology 89, 784–798 (2015).
135. O'Rourke, S. M. et al.: Mutation at a single position in the V2 domain of the HIV-1 envelope protein confers neutralization sensitivity to a highly neutralization-resistant virus, Journal of Virology 84, 11200–11209 (2010).
136. Balazs, A. B. et al.: Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission, Nature medicine 20, 296–300 (2014).
137. Yolamanova, M. et al.: Peptide nanofibrils boost retroviral gene transfer and provide a rapid means for concentrating viruses, Nature nanotechnology 8, 130–136 (2013).
138. Groß, A. et al.: Enhancement and induction of HIV-1 infection through an assembled peptide derived from the CD4 binding site of gp120, Chembiochem : a European journal of chemical biology 16, 446–454 (2015).
139. Chen, W.; Xiao, X.; Wang, Y.; Zhu, Z. & Dimitrov, D. S.: Bifunctional fusion proteins of the human engineered antibody domain m36 with human soluble CD4 are potent inhibitors of diverse HIV-1 isolates, Antiviral Research 88, 107–115 (2010).
140. Feng, Y. et al.: Conjugates of Small Molecule Drugs with Antibodies and Other Proteins, Biomedicines 2, 1–13 (2014).
141. Serrano, S. et al.: Structure and immunogenicity of a peptide vaccine, including the complete HIV-1 gp41 2F5 epitope: implications for antibody recognition mechanism and immunogen design, The Journal of biological chemistry 289, 6565–6580 (2014).
142. Verkoczy, L.; Kelsoe, G. & Haynes, B. F.: HIV-1 envelope gp41 broadly neutralizing antibodies: hurdles for vaccine development, PLoS pathogens 10, e1004073 (2014).
143. Denton, P. W. et al.: Targeted cytotoxic therapy kills persisting HIV infected cells during ART, PLoS pathogens 10, e1003872 (2014).

IX
Danksagung
Mein besonderer Dank geht an meine Betreuerin und Mentorin Frau Prof. Dr. Jutta Eichler, die mir eine Promotion in ihrer Arbeitsgruppe ermöglichte und mich stets in meinen Vorhaben unterstützte und förderte. Die Zusammenarbeit mit ihr war immer sehr konstruktiv und motivierend, ich habe in der Zeit sehr viel gelernt und meinen wissenschaftlichen Horizont deutlich erweitert. Ihre persönliche Betreuung hat dazu beigetragen, dass ich mich in meinem Arbeitsumfeld sehr wohl gefühlt habe und daher auch meine Projekte mit Begeisterung bearbeiten konnte.
Ein herzliches Dankeschön geht außerdem an meine Co-Mentorin Frau Prof. Dr. Barbara Schmidt, die mich bei meinem Projekt vom virologischen Blickwinkel aus immer zuverlässig unterstützte und mir vieles beibrachte. Auch die wissenschaftlichen Gespräche und Diskussionen mit ihr und ihre Ideen zu meiner Forschungsarbeit brachten oft wichtige Impulse und neue Aspekte ein. Vielen Dank also für diese schöne Zusammenarbeit.
Außerdem möchte ich mich bei allen aktuellen und ehemaligen Mitgliedern des AK-Eichlers bedanken, die mich auf meinem Weg begleitet haben: die Zeit mit euch war sowohl auf fachlicher, als auch auf menschlicher Ebene eine Bereicherung. Von vielen von euch habe ich sehr viel gelernt, es war eine wertvolle Zusammenarbeit und ohne euch wäre es nicht halb so schön gewesen.
Desweiteren geht mein Dank für eine stets sehr angenehme Zusammenarbeit an die Arbeitsgruppe von Prof. Dr. Barbara Schmidt. Danke, sowohl für die Unterstützung bei den virologischen Versuchen in Erlangen, hier ganz besonders an Norbert Donhauser, als auch für die zuverlässige Fortsetzung des Projektes nach dem Umzug nach Regensburg.
Sehr viel Unterstützung und Zuspruch über alle Höhen und Tiefen hinweg habe ich natürlich auch von meiner Familie erhalten. Besonders bedanken möchte ich mich heute bei meinem Vater, der mich während meiner Promotion nicht nur liebevoll unterstützt, sondern oft auch zum Nachdenken angeregt hat und bei Entscheidungsfindungen hilfreich war.
Abschließend möchte ich noch ein paar sehr wichtigen Menschen danken für ihre nie endende Unterstützung und wunderbare Freundschaft und Begleitung während der letzten Jahre, egal ob aus der Nähe oder Ferne: Jenny und Lisa (aus der Ferne), Anna, Judy und Rebecca (aus der Nähe)…und ganz besonders: Danke Marek.







![Zum Problem des Genetischen Lehrens - Martin Wagenscheinmartin-wagenschein.de/en/2/W-172.pdf · Martin Wagenschein Zum Problem des Genetischen Lehrens* [W172] Einige Erfahrungen und](https://static.fdokument.com/doc/165x107/5b9f242f09d3f2ab0b8cf3f4/zum-problem-des-genetischen-lehrens-martin-wagenscheinmartin-martin-wagenschein.jpg)










