Die Auswirkungen des „21 CFR Part 4“ auf einen ...CFR+Part+4.pdf · II Danksagung: Ich möchte...
87
Die Auswirkungen des „21 CFR Part 4“ auf einen europäischen Hersteller von Kombinationsprodukten mit besonderem Fokus auf die Entwicklung Autor: Carsten Jähnisch (Matrikelnummer: 40789049) Dotzheimer Straße 122 65197 Wiesbaden Eingereichte Abschlussarbeit zur Erlangung des Grades Master of Business Administration im Studiengang MBA für Umweltschutz- und Qualitätsmanagementsysteme an der Karl-Scharfenberg-Fakultät der Ostfalia Hochschule für angewandte Wissenschaften Erster Prüfer: Professor Mathias Portugall Eingereicht am: 04.Okt.2016 Zweiter Prüfer: Dr. Oliver Hien
Transcript of Die Auswirkungen des „21 CFR Part 4“ auf einen ...CFR+Part+4.pdf · II Danksagung: Ich möchte...

Die Auswirkungen des „21 CFR Part 4“ auf einen europäischen Hersteller von Kombinationsprodukten mit
besonderem Fokus auf die Entwicklung
Autor: Carsten Jähnisch (Matrikelnummer: 40789049)
Dotzheimer Straße 122 65197 Wiesbaden
Eingereichte Abschlussarbeit
zur Erlangung des Grades
Master of Business Administration
im Studiengang
MBA für Umweltschutz- und Qualitätsmanagementsysteme
an der
Karl-Scharfenberg-Fakultät
der Ostfalia Hochschule für angewandte Wissenschaften
Erster Prüfer: Professor Mathias Portugall Eingereicht am: 04.Okt.2016 Zweiter Prüfer: Dr. Oliver Hien

II
Danksagung:
Ich möchte Claudia und Cara dafür danken, dass sie mich unterstützt haben und
damit die Masterarbeit möglich gemacht haben.
Herrn Professor Portugal sowie Herrn Dr. Hien möchte ich für die fachliche
Unterstützung bei der Formulierung des Themas, sowie für die wertvollen Hinweise
während der Erstellung der Masterarbeit danken.
Ferner möchte ich Frau Kuball und Frau Hölzel vom Prüfungssekretariat sowie Frau
Mödecker für die Tipps und Hilfestellung rund um Studium und Masterarbeit danken.

III
Inhaltsverzeichnis
1 Einleitung ............................................................................................................................ 1
2 Akronyme und Abkürzungen .............................................................................................. 3
3 Begriffsdefinition ................................................................................................................ 6
4 Situationsbeschreibung ...................................................................................................... 8
4.1 Beschreibung der Beispielorganisation ....................................................................... 8
4.2 Produkte .................................................................................................................... 11
4.3 Märkte und deren Anforderungen ............................................................................ 12
4.3.1 Der US - amerikanische Markt ........................................................................... 13
4.3.2 Der europäische Markt ...................................................................................... 13
5 Stand der Gesetzgebung / Regulatorische Definitionen ................................................. 16
5.1 Pharmagesetzgebung der USA .................................................................................. 16
5.1.1 Struktur der Gesetzgebung ................................................................................ 17
5.1.2 Zulassungsverfahren für Arzneimittel und Medizinprodukte ............................ 19
5.1.3 Der 21 CFR Part 4................................................................................................ 22
5.2 Pharmagesetzgebung der EU .................................................................................... 29
5.2.1 Struktur der Gesetzgebung ................................................................................ 30
5.2.2 Gesetzgebung zu Kombinationsprodukten in Europa ....................................... 31
6 Auswirkungen und Umsetzung in der Produktrealisierung mit besonderem Fokus auf die
Entwicklung .............................................................................................................................. 33
6.1 Verantwortung der obersten Leitung ........................................................................ 33
6.1.1 Beauftragter der obersten Leitung .................................................................... 34
6.2 Beschaffung ............................................................................................................... 35
6.2.1 Generelle Anforderungen an die Beschaffung ................................................... 35
6.2.2 Anforderungen an ausgelagerte Prozesse ......................................................... 36
6.2.3 Auswahl von Lieferanten .................................................................................... 38
6.2.4 Evaluierung und Freigabe von Lieferanten ........................................................ 40
6.2.5 Überwachung und Re-Evaluierung von Lieferanten .......................................... 40
6.2.6 Fazit zu den Anforderungen im Bereich Lieferantenmanagement .................... 42
6.3 Design und Entwicklungslenkung .............................................................................. 43
6.3.1 Generelle Anforderungen an die Design und Entwicklungslenkung ................. 43

IV
6.3.2 Entwicklungsplanung .......................................................................................... 46
6.3.3 Entwicklungseingaben ........................................................................................ 49
6.3.4 Entwicklungsergebnisse ..................................................................................... 51
6.3.5 Entwicklungsverifizierung .................................................................................. 53
6.3.6 Entwicklungsvalidierung ..................................................................................... 55
6.3.7 Entwicklungsbewertung ..................................................................................... 57
6.3.8 Lenkung von Entwicklungsänderungen ............................................................. 60
6.3.9 Transfer der Entwicklungsaktivität..................................................................... 62
6.3.10 Design History File (Entwicklungsakte) .............................................................. 63
6.3.11 Fazit zu den Anforderungen im Bereich Entwicklungslenkung .......................... 65
6.4 Korrektur und Vorbeugungsmaßnahmen ................................................................. 65
6.5 Tätigkeiten bei der Installation .................................................................................. 70
6.6 Tätigkeiten bei der Instandhaltung ........................................................................... 71
7 Zusammenfassung und Fazit ............................................................................................ 73
8 Verzeichnis der Tabellen .................................................................................................. 78
9 Verzeichnis der Abbildungen ........................................................................................... 79
10 Bibliographie .................................................................................................................... 80
11 Eidesstattliche Erklärung .................................................................................................. 83

Einleitung
1
1 Einleitung
Die folgende Arbeit soll die Auswirkung des US-amerikanischen Gesetzes „21 CFR Part 4“ auf
einen europäischen Hersteller von Kombinationsprodukten mit besonderem Fokus auf die
Entwicklung darlegen.
Die Auswirkungen des 21 CFR Part 4 werden für einen hypothetischen europäischen
Arzneimittelhersteller mit einem Pharma-GMP Qualitätsmanagementsystem aufgezeigt. Die
Situationsbeschreibung umfasst die Darstellung der Organisation, der Produkte sowie der
Zielmärkte und deren Anforderungen. In einem eigenen Kapitel werden der Stand der
Gesetzgebung sowie die regulatorischen Anforderungen dargelegt. Dabei wird insbesondere
auf die Unterschiede der europäischen Gesetzgebung zur US-amerikanischen Gesetzgebung
eingegangen. Im Rahmen der US-amerikanischen Gesetzgebung wird speziell der 21 CFR Part
4 analysiert und dargelegt. Nach diesen vorbereitenden Kapiteln folgt die Analyse der
Auswirkungen auf das GMP Qualitätsmanagementsystem, um dann auf Lösungsvorschläge
mit besonderem Fokus auf die Produktentwicklung einzugehen. Im letzten Kapitel des
Hauptteils dieser Masterarbeit werden die neuesten Entwicklungen zu diesem Thema
erörtert und mit einem Ausblick abgeschlossen.
Kombinationsprodukte sind, wie der Name schon sagt, eine Verbindung von Arzneimittel
und Medizinprodukt. Medizinprodukte wirken rein physikalisch, während Arzneimittel
physiologisch wirken. Die US-amerikanische Regulierungsbehörde Food and Drug
Administration (FDA) hat den Auftrag sicherzustellen, dass nur sichere und wirksame
Produkte in den USA in den Verkehr gebracht werden. Durch das erhöhte Aufkommen von
Kombinationsprodukten sah die FDA die Notwendigkeit, eine eigenständige Gesetzgebung
zu etablieren. Die FDA hat ein spezielles Interesse, innovative Produkte zügig für den US-
amerikanischen Markt zuzulassen. Jedoch sollen die Anforderungen an die Sicherheit und die
Wirksamkeit der zugelassen Produkte kontinuierlich sichergestellt werden. Der 21 CFR Part
4 definiert als Gesetz die Anforderungen an die „Gute Herstellungs Praxis“ inklusive der
Produktentwicklung für Kombinationsprodukte. Der 21 CFR Part 4 ist eine wertvolle

Einleitung
2
Hilfestellung für Arzneimittelhersteller zur Umsetzung und Erfüllung der regulatorischen
Anforderungen an Kombinationsprodukte.
Der 21 CFR Part 4 eröffnet die Möglichkeit, die Konformität des Kombinations-produktes mit
den Anforderungen an das Qualitätsmanagementsystem mit dem „Streamlined Approach“
nachzuweisen. Der „Streamlined Approach“ bietet dem Hersteller die Möglichkeit, den
Aufwand zur Implementierung der Anforderungen an das Qualitätssystem für
Medizinprodukte zu reduzieren.
Akronyme und Abkürzungen
3
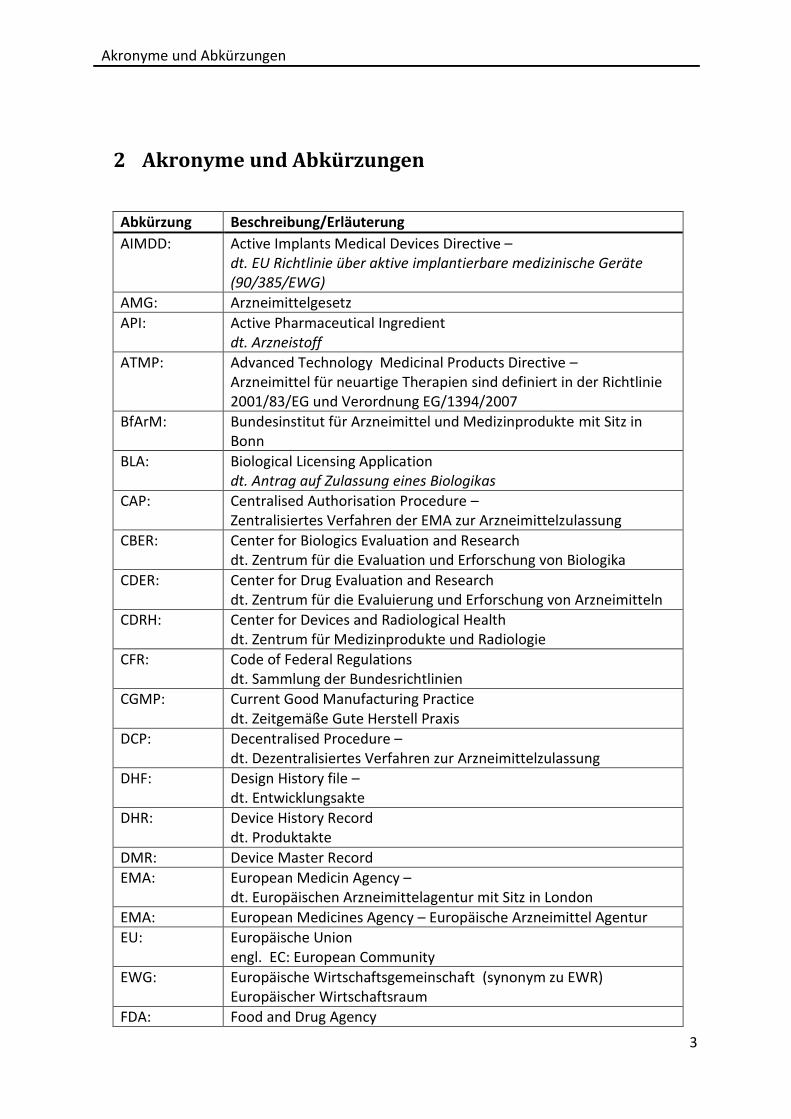
2 Akronyme und Abkürzungen
Abkürzung Beschreibung/Erläuterung
AIMDD: Active Implants Medical Devices Directive – dt. EU Richtlinie über aktive implantierbare medizinische Geräte (90/385/EWG)
AMG: Arzneimittelgesetz
API: Active Pharmaceutical Ingredient dt. Arzneistoff
ATMP: Advanced Technology Medicinal Products Directive – Arzneimittel für neuartige Therapien sind definiert in der Richtlinie 2001/83/EG und Verordnung EG/1394/2007
BfArM: Bundesinstitut für Arzneimittel und Medizinprodukte mit Sitz in Bonn
BLA: Biological Licensing Application dt. Antrag auf Zulassung eines Biologikas
CAP: Centralised Authorisation Procedure – Zentralisiertes Verfahren der EMA zur Arzneimittelzulassung
CBER: Center for Biologics Evaluation and Research dt. Zentrum für die Evaluation und Erforschung von Biologika
CDER: Center for Drug Evaluation and Research dt. Zentrum für die Evaluierung und Erforschung von Arzneimitteln
CDRH: Center for Devices and Radiological Health dt. Zentrum für Medizinprodukte und Radiologie
CFR: Code of Federal Regulations dt. Sammlung der Bundesrichtlinien
CGMP: Current Good Manufacturing Practice dt. Zeitgemäße Gute Herstell Praxis
DCP: Decentralised Procedure – dt. Dezentralisiertes Verfahren zur Arzneimittelzulassung
DHF: Design History file – dt. Entwicklungsakte
DHR: Device History Record dt. Produktakte
DMR: Device Master Record
EMA: European Medicin Agency – dt. Europäischen Arzneimittelagentur mit Sitz in London
EMA: European Medicines Agency – Europäische Arzneimittel Agentur
EU: Europäische Union engl. EC: European Community
EWG: Europäische Wirtschaftsgemeinschaft (synonym zu EWR) Europäischer Wirtschaftsraum
FDA: Food and Drug Agency
Akronyme und Abkürzungen
4
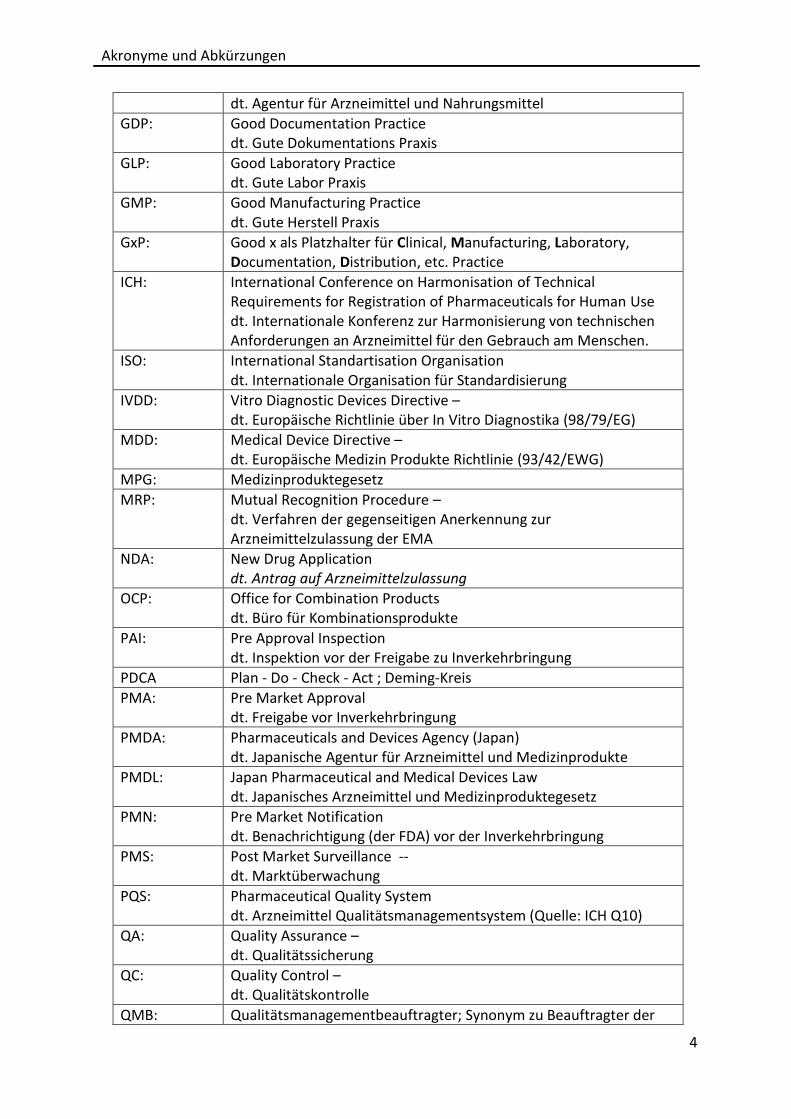
dt. Agentur für Arzneimittel und Nahrungsmittel
GDP: Good Documentation Practice dt. Gute Dokumentations Praxis
GLP: Good Laboratory Practice dt. Gute Labor Praxis
GMP: Good Manufacturing Practice dt. Gute Herstell Praxis
GxP: Good x als Platzhalter für Clinical, Manufacturing, Laboratory, Documentation, Distribution, etc. Practice
ICH: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use dt. Internationale Konferenz zur Harmonisierung von technischen Anforderungen an Arzneimittel für den Gebrauch am Menschen.
ISO: International Standartisation Organisation dt. Internationale Organisation für Standardisierung
IVDD: Vitro Diagnostic Devices Directive – dt. Europäische Richtlinie über In Vitro Diagnostika (98/79/EG)
MDD: Medical Device Directive – dt. Europäische Medizin Produkte Richtlinie (93/42/EWG)
MPG: Medizinproduktegesetz
MRP: Mutual Recognition Procedure – dt. Verfahren der gegenseitigen Anerkennung zur Arzneimittelzulassung der EMA
NDA: New Drug Application dt. Antrag auf Arzneimittelzulassung
OCP: Office for Combination Products dt. Büro für Kombinationsprodukte
PAI: Pre Approval Inspection dt. Inspektion vor der Freigabe zu Inverkehrbringung
PDCA Plan - Do - Check - Act ; Deming-Kreis
PMA: Pre Market Approval dt. Freigabe vor Inverkehrbringung
PMDA: Pharmaceuticals and Devices Agency (Japan) dt. Japanische Agentur für Arzneimittel und Medizinprodukte
PMDL: Japan Pharmaceutical and Medical Devices Law dt. Japanisches Arzneimittel und Medizinproduktegesetz
PMN: Pre Market Notification dt. Benachrichtigung (der FDA) vor der Inverkehrbringung
PMS: Post Market Surveillance -- dt. Marktüberwachung
PQS: Pharmaceutical Quality System dt. Arzneimittel Qualitätsmanagementsystem (Quelle: ICH Q10)
QA: Quality Assurance – dt. Qualitätssicherung
QC: Quality Control – dt. Qualitätskontrolle
QMB: Qualitätsmanagementbeauftragter; Synonym zu Beauftragter der

Akronyme und Abkürzungen
5
obersten Leitung eng. Management Representative
QMS: Qualitätsmanagementsystem
QSIT: Quality System Inspection Technique dt. Qualitätssystem Audit Verfahren der FDA
Begriffsdefinition
6
3 Begriffsdefinition
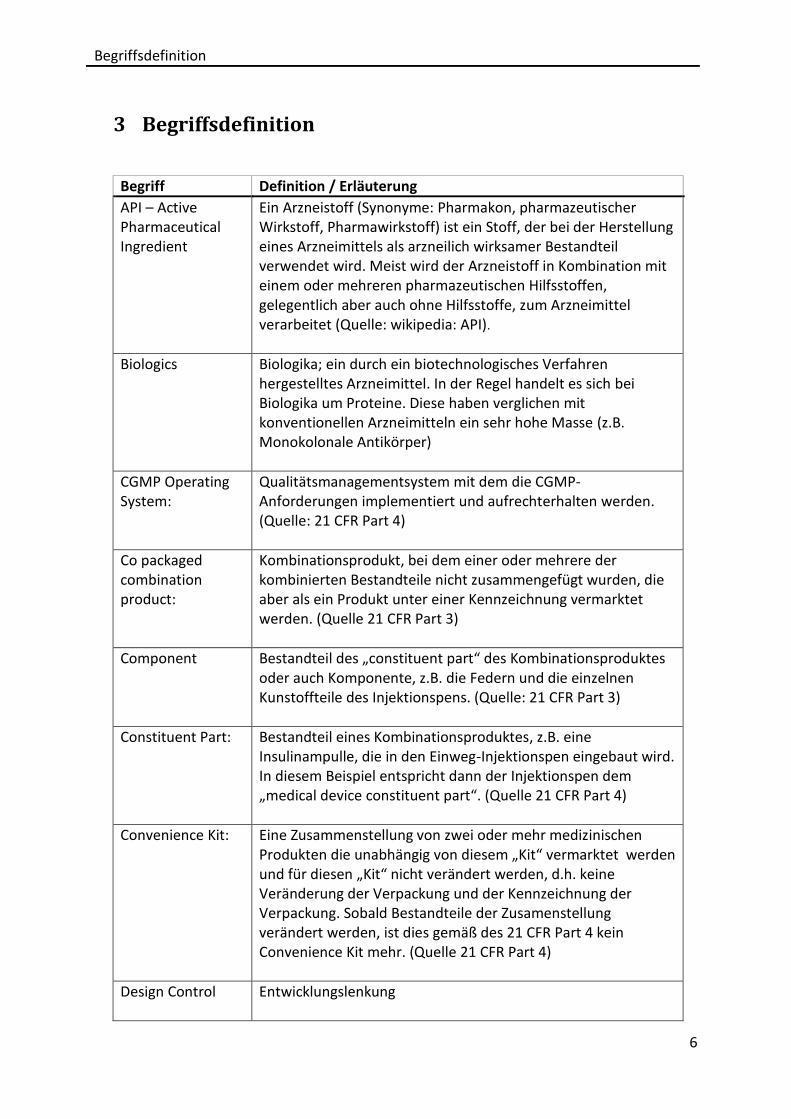
Begriff Definition / Erläuterung
API – Active Pharmaceutical Ingredient
Ein Arzneistoff (Synonyme: Pharmakon, pharmazeutischer Wirkstoff, Pharmawirkstoff) ist ein Stoff, der bei der Herstellung eines Arzneimittels als arzneilich wirksamer Bestandteil verwendet wird. Meist wird der Arzneistoff in Kombination mit einem oder mehreren pharmazeutischen Hilfsstoffen, gelegentlich aber auch ohne Hilfsstoffe, zum Arzneimittel verarbeitet (Quelle: wikipedia: API).
Biologics Biologika; ein durch ein biotechnologisches Verfahren hergestelltes Arzneimittel. In der Regel handelt es sich bei Biologika um Proteine. Diese haben verglichen mit konventionellen Arzneimitteln ein sehr hohe Masse (z.B. Monokolonale Antikörper)
CGMP Operating System:
Qualitätsmanagementsystem mit dem die CGMP-Anforderungen implementiert und aufrechterhalten werden. (Quelle: 21 CFR Part 4)
Co packaged combination product:
Kombinationsprodukt, bei dem einer oder mehrere der kombinierten Bestandteile nicht zusammengefügt wurden, die aber als ein Produkt unter einer Kennzeichnung vermarktet werden. (Quelle 21 CFR Part 3)
Component Bestandteil des „constituent part“ des Kombinationsproduktes oder auch Komponente, z.B. die Federn und die einzelnen Kunstoffteile des Injektionspens. (Quelle: 21 CFR Part 3)
Constituent Part: Bestandteil eines Kombinationsproduktes, z.B. eine Insulinampulle, die in den Einweg-Injektionspen eingebaut wird. In diesem Beispiel entspricht dann der Injektionspen dem „medical device constituent part“. (Quelle 21 CFR Part 4)
Convenience Kit: Eine Zusammenstellung von zwei oder mehr medizinischen Produkten die unabhängig von diesem „Kit“ vermarktet werden und für diesen „Kit“ nicht verändert werden, d.h. keine Veränderung der Verpackung und der Kennzeichnung der Verpackung. Sobald Bestandteile der Zusamenstellung verändert werden, ist dies gemäß des 21 CFR Part 4 kein Convenience Kit mehr. (Quelle 21 CFR Part 4)
Design Control Entwicklungslenkung

Begriffsdefinition
7
Device Class I: Klassifizierung für Medizinprodukt: Geringstes Risiko für den Anwender /Patienten, General controls with exemption. (Quelle: https://en.wikipedia.org/wiki/Medical_device)
Device Class II: Klassifizierung für Medizinprodukt: Mittleres Risiko für den Anwender / Patienten. (Quelle: https://en.wikipedia.org/wiki/Medical_device)
Device Class III: Klassifizierung für Medizinprodukt: Höchstes Risiko für den Anwender /Patienten. (Quelle: https://en.wikipedia.org/wiki/Medical_device)
Disposable Pen Injektionspen, der nach Bestimmungsgemäßen Gebrauch entsorgt werden soll
Inverkehrbringung: Prozess der Vermarktung eines Kombinationsproduktes inklusive der Zulassung durch die beauftragte Behörde oder Körperschaft.
Manufacture: Herstellung
Manufacturer: Hersteller
Mode of action Wirkung: Die Art und Weise, über die der therapeutische Nutzen eines Bestandteils des Kombinationsproduktes erreicht wird. (Quelle: 21 CFR Part 4)
PDCA-Zyklus (Deming Kreis)
Qualitätsprinzip des zyklischen Planen – Umsetzen – Überprüfen(der Maßnahme) – Agieren/Handeln (Definieren und Umsetzen einer Gegenmaßnahme), um Situationen und Prozesse kontinuierlich zu verbessern (Quelle: Iso 9001:2008)
Primary mode of action
Hauptwirkung: Wirkung über die der Therapeutische Nutzen des Kombinationsproduktes erreicht wird. (Quelle: 21 CFR Part 3)
Primary Packaging: Primäres Packmittel: im Falle eines injezierbaren Arzneimittels ist dies die Ampulle in welche das Arzneimittel gefüllt wird. (Quelle: 21 CFR Part 4)
Reusable Injection Pen
Wiederverwendbarer Injektionspen
Single entity combination product:
Kombinationsprodukt, bei dem die beiden kombinierten Bestandteile (z.B. Arznei und Medizinprodukt) zusammengebaut wurden und das als Einheit vermarktet wird. (Quelle: 21 CFR Part 3)

Situationsbeschreibung
8
4 Situationsbeschreibung
Bevor die Anforderungen an Kombinationsprodukte analysiert und daraus die notwendigen
Änderungen für die zugrundeliegende Organisation beleuchtet werden, muss zunächst die
Organisation selbst definiert werden. Daher werden im folgenden Kapitel das Unternehmen,
seine Organisation, die Produkte, sowie kurz die Zielmärkte und deren Anforderungen
beschrieben.
4.1 Beschreibung der Beispielorganisation Das für die Zwecke dieser Masterarbeit angeführte Unternehmen ist ein global operierendes
europäisches Pharmaunternehmen. Es vertreibt mehrere Medikamente zur Behandlung von
Diabetes, mit Schwerpunkt auf injezierbare Insuline. Zur Darreichung der Insuline bietet das
Unternehmen unter anderem Einweg- Injektionspens an.
In Abbildung 1 ist die Herstellungsabfolge für einen Insulin-Injektionspen dargestellt und
wird im Folgenenden ausführlicher erläutert.
Herstellung des Arzneimittels
QMS: GMP
Abfüllung QMS: GMP
EndmontageQMS :GMP
Legal Manufacturer
Herstellung APIQMS: GMP
Die Insuline werden an einem Standort in Deutschland hergestellt und in Ampullen abgefüllt.
Die Herstellung der Arznei kann bis zur Abfüllung grob in drei Schritte unterschieden
werden. In einem vierten Schritt wird das Arzneimittel mit dem Medizinprodukt kombiniert.
Abbildung 1: Fertigungssequenz

Situationsbeschreibung
9
1. Herstellung API/Wirkstoff:
Für moderne Arzneimittel wird häufig ein biotechnologisches Verfahren angewandt,
bei dem mit Hilfe von Bakterien oder Mikroorganismen der Wirkstoff erzeugt wird.
Dieser wird gereinigt und getrocknet. Das Endprodukt der Wirkstoffproduktion ist der
Wirkstoff in Reinform (getrocknetes Pulver). Die Erzeugung der Reinform ist
notwendig, um später den Wirkstoff besser dosieren zu können. Diese Arbeitsschritte
sind in der Arzneistoff-Fertigung (API-Fertigung) angesiedelt.
2. Herstellung Arzneimittel:
Im nächsten Schritt wird der getrocknete Wirkstoff in Lösung gebracht, um eine
injezierbare Lösung zu erzeugen. Zu diesem Zweck wird der Wirkstoff in Wasser
gelöst, zusätzlich werden Konservierungsstoffe, Stabilisatoren, Farbstoffe und Stoffe
zum Einstellen der Viskosität zugefügt. Das Endprodukt dieses Prozesses ist eine
Lösung mit einer bestimmten Dosis (Wirkstoff pro Volumeneinheit), einer definierten
Haltbarkeit und einer definierten Viskosität, um die Injezierbarkeit zu gewährleisten.
3. Abfüllung Arzneimittel:
Die Lösung wird in einem sterilen Prozess auf Ampullen gefüllt. Die Ampullen sind das
sogenannte primäre Packmittel.
4. Endmontage Kombinationsprodukt:
Im letzten Arbeitsschritt werden die sterilen Ampullen unter anderem in Einwegpens
verbaut, entsprechend gekennzeichnet, verpackt, eingelagert und vertrieben. Durch
das Verbauen der Ampulle in den Einweg-Injektionspen wird ein (Arznei-
Medizinprodukt) Kombinationsprodukt hergestellt.
Die Herstellung und das Abfüllen der Insuline in die Ampullen unterliegen in Deutschland
dem Arzneimittelgesetz. Hier kommen die sogenannten "Good Manufacturing Practices"
(GMP) als Qualitätsanforderungen zum Tragen, welche in Deutschland durch die jeweilige
Überwachungsbehörde(z.B. Regierungspräsidium etc.) des Bundeslandes überwacht werden.
Allgemeine Beschreibung des Qualitätssystemes der Beispielorganisation:

Situationsbeschreibung
10
Das bestehende Qualitätsmanagementsystem der Organisation ist ein „Pharmaceutical
Quality System“ (PQS) gemäß ICH Q10.
Die ICH ist die “International Conference for Harmonisation of technical requirements for
registration of pharmaceuticals for Human Use”. Die Zielsetzung des ICH Q10 besteht darin,
die Regionalen GMP Anforderungen auf eine gemeinsame Basis zu stellen und dadurch zu
harmonisieren. Die FDA hat den ICH Q10 in einer „Guideline for Industry“ im April 2009
veröffentlicht, somit stellt diese Richtlinie die Erwartungshaltung der FDA an ein
Qualitätsmanagementsystem eines Arzneimittelherstellers dar1.
Gemäß des ICH Q10 beinhaltet das PQS Anforderungen für:
1. Pharmaceutical Quality System
2. Management Responsibility
3. Continual improvement of Process Performance and Product Quality
4. Continual Improvement of the Pharmaceutical Quality system
Die Beispielorganisation erfüllt die Anforderungen des ICH Q10 für ein PQS und führt den
Nachweis bei den regelmäßigen Inspektionen entweder der lokalen Behörden oder der FDA.
Abbildung 2: Pharmaceutical Quality Management System gemäß ICH Q102
1 Vgl. 5.1.1 Struktur der Gesetzgebung
2 Vgl. FDA, o.V. (2009): Guidance for Industry Q10 Pharmaceutical Quality System, S. 19

Situationsbeschreibung
11
Jedoch ist in der Beispielorganisation der Bereich „Pharmaceutical Development“ also die
Arzneistoffentwicklung ausgenommen. Dieser Bereich wird von einer getrennten Einheit
abgedeckt, welche über ein eigenes QMS verfügt. Ebenso wird der Bereich der
Medizinprodukteentwicklung von einer separierten Einheit abgedeckt, die ebenfalls über ein
eigenes QMS verfügt. Die Organisationseinheit, welche die Injektionspens herstellt und die
abgefüllten Insulinampullen in die Einweg-Injektionspens einbaut, ist qualitätsmäßig von den
davorliegenden Organisationseinheiten getrennt und verfügt über ein eigenständiges QMS
gemäß den besonderen Anforderungen der GMP. Diese Organisationseinheit ist zugleich der
Inverkerkehrbringer des mit der Insulinampulle bestückten Einweg-Injektionspens.
4.2 Produkte Das Unternehmen entwickelt, produziert und vertreibt diverse Arzneimittel weltweit unter
Berücksichtigung der länderspezifischen Gesetze und Richtlinien.
Die Organisation vertreibt sowohl Einweg-Injektionspens bzw. Auto-Injektoren mit fest
verbauter Kartusche, die dem Arzneimittelgesetz unterliegen, als auch mehrfach
verwendbare Injektionspens mit CE Zeichen, die dem Medizinproduktegesetz unterliegen.
Wie später noch dargelegt wird, handelt es sich bei den Einweg-Injektionspens
(Injektionspens mit fest verbauter Kartusche) im europäischen Rechtsraum nicht um ein
Medizinprodukt (Medical Device) im Sinne der europäischen Medizin Produkte Richtlinie
(93/42/EWG). In Europa wird der Einweg-Injektionspen als sogenanntes Medicinal Product
(Arzneimittel) gewertet, da die Hauptwirkung des Produktes die Wirkung des Insulins ist.
Somit findet das Arzneimittelgesetz (AMG) Anwendung. Der europäische Rechtraum
definiert keine Kombinationsprodukte und stellt hierfür auch keine separate Richtlinie oder
Verordnung bereit.
In den USA sind im Sinne des 21 CFR Part 4 Einweg-Injektionspens (Disposable Injection
Pens) mit fest verbauter Kartusche, je nach enthaltenem Arzneimittel entweder „Drug-
Device Combination Products“ oder „Biologic-Device Combination Products“. Im Sinne des
Gesetzes handelt es sich bei Insulinen und Insulin-Analoga um Arzneimittel, somit ist der
Insulin Einweg Injektionspen ein Drug Device Combination Product (DDC)

Situationsbeschreibung
12
„Für Proteinmedikamente wie Insulin) gilt, dass sie bei einer oralen Aufnahme nicht
bzw. nicht in ausreichender Menge in den Blutkreislauf aufgenommen (resorbiert)
werden. Diese Wirkstoff werden durch Enzyme des Verdauungstrakts abgebaut oder
durch die im Magen vorhandene Salzsäure denaturiert und damit funktionsunfähig
gemacht“.3
Für eine wirksame Therapie mit proteinbasierenden Arzneimitteln mussten daher andere
Darreichungsformen gefunden werden als die übliche orale Aufnahme in Form von Tabletten
oder Kapseln. Neben der respiratorischen Aufnahme durch Inhalatoren, ist dies sehr häufig
die Darreichung per Injektion. Die Injektion ist eine im klinischen Bereich durch Fachpersonal
ausgeführte etablierte Methode. Insbesondere die subkutane Injektion ist hier
hervorzuheben. Diese ist im Gegensatz zur intramuskulären oder intravenösen Injektion
komplikationsarm und einfach durchzuführen
Handelsübliche Spritzen, bestehend aus Zylinder, Kolben und Kanüle, konnten durch die
Entwicklung und Zulassung von vorgefüllten Einweg-Injektionspens ersetzt werden.
Injektionspens bieten die Vorteile, dass sie einfacher zu dosieren sind und die subkutane
Injektion auch für Patienten ohne spezielle medizinische Ausbildung sicher durchführbar ist.
Dadurch, dass die Injektion nun wesentlich einfacher und sicherer durchzuführen ist, kann
diese durch den Patienten selbst erfolgen. Dies bringt diverse Vorteile für den Patienten mit
sich, wie zum Beispiel:
- Erhöhte Flexibilität
- Verbesserte Integration in den Tagesablauf des Patienten
- Unabhängigkeit von Fachpersonal
4.3 Märkte und deren Anforderungen Vor einer Inverkehrbringung müssen Arzneien, Medizinprodukte oder deren Kombinationen
für den jeweiligen Markt zugelassen werden. Es gibt in diesem Bereich keine einheitliche
marktübergreifende Gesetzgebung, daher wird den Entwicklungsprojekten im
Anforderungsprofil für das zu entwickelnde Produkt der Zielmarkt mitgegeben. In aller Regel
3 Vgl. Wikipedia, o.V.(o.J.): Injektion Medizin

Situationsbeschreibung
13
werden die Entwicklungen auf eine Inverkehrbringung auf dem US-amerikanischen-, den
europäischen- sowie den japanischen- Markt ausgerichtet. Im Folgenden werden die
Anforderungen für den US-amerikanischen Markt sowie den europäischen Markt
zusammenfassend erläutert.
4.3.1 Der US - amerikanische Markt
Die Anforderungen zur Inverkehrbringung von Kombinationsprodukten auf den US-
amerikanischen Markt gelten als besonders streng. Die Marktüberwachungsbehörde ist die
„Food and Drug Administration“ oder kurz die FDA. Die FDA ist eine Bundesbehörde der USA
und verfügt über ähnliche Befugnisse wie eine Strafverfolgungsbehörde und ist mit ca. 10
000 Mitarbeitern die größte regulierende Behörde für Arzneimittel und Medizinprodukte
weltweit. Die Arbeit, das Vorgehen und die Befugnisse der FDA sind im Food, Drug and
Cosmetic Act geregelt. Für Arzneimittel überprüft die FDA die Einhaltung des 21 CFR Part 210
& 211, welche die Good Manufacturing Practises (GMP) oder anders gesagt die
Qualitätsanforderungen für die Arzneimittelindustrie beschreiben. Für Medizinprodukte sind
dies die Quality Systems Regulations (QSR), welche im 21 CFR 820 verankert sind. Verstöße
gegen den 21 CFR 820 sind dem zufolge Gesetzesverstöße und müssen als solche behandelt
werden. Die Überprüfung der Einhaltung wird von Behördenvertreten im Rahmen von
Inspektionen des Unternehmens durchgeführt. Da Kombinationsprodukte, wie der Name
schon andeutet, eine Kombination aus einem Medizinprodukte und einer Arznei sein
können, hat die FDA auch die Aufsicht über diese Produktklasse.
4.3.2 Der europäische Markt
Für die Inverkehrbringung von Kombinationsprodukten auf dem europäischen Markt ist es
zunächst wichtig zu wissen, dass es keine Gesetzgebung zu Kombinationsprodukten gibt. Im
europäischen Rechtsraum sind Kombinations-produkte nicht definiert. Da es solche
Produkte aber offensichtlich gibt und diese reguliert werden müssen, wurde in der EU der
Begriff des „Borderline“- Produktes geprägt. Dieser Begriff ist so zu verstehen, dass bei
solchen Produkten die Grenzen zwischen zwei oder mehreren Produktkategorien
überschritten werden. Um den daraus resultierenden Rechtsunsicherheiten zu begegnen,
werden Kombinations-produkte für die Marktzulassung in Europa zunächst entweder als

Situationsbeschreibung
14
Arzneimittel oder als Medizinprodukt klassifiziert. Wird das Kombinationsprodukt als
Arzneimittel klassifiziert, sind die Arzneimittelrichtlinien anwendbar, bzw. ist in der
Bundesrepublik Deutschland das AMG mit seinen Paragraphen 21 Abs.1 sowie den
Paragraphen 22 - 24 anwendbar.
Wird das Kombinationsprodukt als Medizinprodukt klassifiziert ist, muss es das in der
jeweiligen Richtlinie beschriebene Konformitätsbewertungsverfahren durchlaufen. Die
anwendbaren Richtlinien sind
- Richtlinie über 98/79/EG über In-Vitro-Diagnostika
- Richtlinie90/385/EWG über aktive implantierbare medizinische Geräte, ergänzt mit
Richtlinie 2007/47/EG
- Richtlinie 93/42/EWG über Medizinprodukte, ergänzt mit Richtlinie 2007/47/EG
- In der BRD sind die obengenannten Richtlinien in das Medizinproduktegesetz
überführt worden.
4.3.2.1 Zulassungsverfahren für Arzneimittel und Medizinprodukte
Das Zulassungsverfahren für Arzneimittel ist in der Richtlinie 2001/83/EG
„Gemeinschaftskodex für Humanarzneimittel“ beschrieben. Aufgrund der komplexen
Struktur der europäischen Union gibt es mehrere Verfahren, die auf unterschiedliche Weise
die einzelnen Nationalstaaten einbinden. Folgende Verfahren werden unterschieden:
MRP: Mutual Recognition Procedure – Verfahren der gegenseitigen Anerkennung zur
Arzneimittelzulassung der EMA.
DCP: Decentralised Procedure – Dezentralisiertes Verfahren der EMA zur
Arzneimittelzulassung.
CAP: Centralised Authorisation Procedure – Zentralisiertes Verfahren der EMA zur
Arzneitmittelzulassung.
Alle genannten Verfahren werden mehr oder weniger von der EMA European Medizin
Agency mit Sitz in London koordiniert oder direkt durchgeführt.
Es gibt noch die Möglichkeit einer nationalen Zulassung, wie sie in Deutschland vom
Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) gemäß des AMG mit seinen

Situationsbeschreibung
15
Paragraphen 21 Abs. 1 und den Paragraphen 22-24 durchgeführt werden kann. Ein solche
Zulassung hat aber den Nachteil, dass sie nur für Deutschland gilt, d. h. das Medikament ist
im Rest der EU nicht zugelassen.
Für Medizinprodukte ist in der Medical Device Directive 93/42/EWG festgelegt, dass für die
Zulassung eine Konformitätsbewertung durchzuführen ist. Im Rahmen des
Konformitätsbewertungsverfahrens wird die Einhaltung der sogenannten „grundlegenden
Anforderungen“ der jeweils anwendbaren Richtlinie, sowie die Wirksamkeit seines
Marküberwachungssystems (Post Market Surveillance – PMS) geprüft. Je nach Klasse des
Medizingerätes kann die Konformitätsbewertung selbst durchgeführt werden oder muss von
einer sogenannte „benannten Stelle“ durchgeführt werden. Nach erfolgreicher Bewertung
erklärt der Hersteller seine Konformität mit den Anforderungen der Richtlinie und bringt das
CE Zeichen auf seinem Produkt an. Zum besseren Überblick werden die Zulassungsverfahren
für Arzneimittel und für Medizinprodukte in der folgenden Tabelle 4.1: Zulassungsverfahren
EU, zusammengefasst.
Tabelle 4.1: Zulassungsverfahren EU
Pharma Medizinprodukte
Stoffklasse Verfahren Kategorie Verfahren
Arzneimittel zentralisiertes Verfahren (CAP) Verfahren der gegenseitigen Anerkennung (MRP), Dezentralisiertes Verfahren (DCP)
Class I CE Zeichen Konformitätserklärung als Selbstdeklaration
Biologic Werden durch die EMA im zentralisierten Verfahren (CAP) zugelassen
Class II CE-Zeichen Konformitätserklärung durch die benannte Stelle
N/A N/A Class III CE-Zeichen Konformitätserklärung durch die benannte Stelle

Stand der Gesetzgebung / Regulatorische Definitionen
16
5 Stand der Gesetzgebung / Regulatorische Definitionen
Arzneimittel und Medizingeräte sind regulierte Produkte. Die Herstellung, der
Vertrieb/Vermarktung sowie die Entwicklung unterliegen gesetzlichen Auflagen. Diese sind
von Markt zu Markt unterschiedlich, wodurch ein erheblicher Aufwand entsteht, diese
unterschiedlichen Anforderungen zu erfüllen. Im Folgenden sollen die Anforderungen an
Kombinationsprodukte in den USA im Vergleich mit den Anforderungen im europäischen
Rechtsraum dargestellt werden.
5.1 Pharmagesetzgebung der USA Arzneimittel und Medizinprodukte sowie deren Kombinationen sind auch in den USA
regulierte Produkte und unterliegen demnach den Gesetzen der USA. Der Food Drug and
Cosmetic Act von 1938 ist das grundlegende Gesetz für die Regulierung von Arzneimitteln
und Medizinprodukten. Der Food, Drug and Cosmetics Act beauftragt die FDA mit der
Überwachung. Die FDA ist eine Behörde des US-amerikanischen Gesundheitsministeriums
(Department of Health and Human Services) und ist damit ein Bundesbehörde. Es folgt ein
kurzer Abriss über die Entstehung der des Food, Drug and Cosmetic Act und der FDA.
1862: Das Bureau of Chemistey wird gegründet und ist die Vorgängerorganisation
der FDA.
1906: Der Food and Drug Act wird durch den amerikanischen Kongress
verabschiedet.
1938: Der Food, Drug and Cosmetics (FDC) Act wird verabschiedet.
1962: Kefauver-Harris Drug Amendment. Einführung des Wirksamkeitskriteriums
sowie die Einführung der heutigen Zulassungsprozedur, im Lichte des Contergan-
Skandals in Europa.
1976: Die Medical Device Amendments werden verabschiedet. Es besteht nun die
grundlegende Anforderung, dass Hersteller von Medizinprodukten sich bei der FDA

Stand der Gesetzgebung / Regulatorische Definitionen
17
registrieren und Qualitätskontrollverfahren einführen. Bestimmte Produkte müssen
freigegeben werden, andere müssen Leistungsstandards erfüllen.
1988: Food and Drug Administration Act von 1988 etabliert die FDA als Teil des
Department of Health and Human Services.
1990: Safe Medical Devices Act wird verabschiedet. Hersteller sind nun verpflichtet
eine Marktbeobachtung – Post Market Surveillance einzuführen.
2002: Unter anderem wird der Medical Device User Fee and Modernization Act
verabschiedet.
Das Office of Combination Products (OCP) wird innerhalb des Büros des
Commissioners gegründet, um die Überprüfung von Produkten, die in mehrere
Produktklassenfallen, zu betreuen.
Die current Good Manufacturing Practice (cGMP) Initiative wird gestartet. Die Ziele
der Initiative besteht darin die größten Risiken in Herstellungsprozessen zu ermitteln
und sicherzustellen, dass Prozess und Produktqualitätsstandards nicht die
Weiterentwicklung behindern, sowie eine einheitliche Herangehensweise der FDA an
diese Aufgaben zu garantieren.
5.1.1 Struktur der Gesetzgebung
In den USA ist das höchste Gesetz die Verfassung (Constitution), darunter ist das sogenannte
Gesetzes Recht oder auch Kodifizierte Recht (Statutory Law) aufgehängt. Für den
medizinischen Bereich (Arzneimittel, Medizinprodukte und Kombinationsprodukte) ist dies
der bereits erwähnte, 1938 vom Kongress der Vereinigten Staaten von Amerika
verabschiedete Federal Food, Drug and Cosmetic Act.4
Die dritte Ebene bildet das Verwaltungsrecht (Administrative Law), welches für den
medizinischen Bereich im Titel 21 des Code of Federal Regulations (CFR) niedergelegt ist. Das
Verwaltungsrecht wird nicht direkt vom Kongress verabschiedet, sondern von der
beauftragten Bundesbehörde, in diesem Fall die FDA. Die CGMP Regulations werden von
4 Vgl. Johner, C.; Hölzer-Klüpfel,M.; Wittorf, S. (2011): Basiswissen Medizinische Software; S. 41

Stand der Gesetzgebung / Regulatorische Definitionen
18
der FDA herausgegeben und regelmäßig überprüft. In den USA kommt zu den GMP s noch
der Buchstabe C hinzu. Das C steht für „Current“ und stellt die Anforderung der FDA dar,
dass bei der Herstellung von Arzneimitteln der „Stand der Technik“ berücksichtigt werden
muss. Insgesamt gesehen ist der Unterschied zwischen den USA und der EU in diesem Falle
aber nicht wesentlich, da auch in Europa der Stand der Technik berücksichtigt werden muss.
Was zu Verwirrung führen kann, ist das die Schreibweise des C vor dem GMP nicht eindeutig
geklärt wurde. Es gibt also die Schreibweisen „cGMP“ und „CGMP“, beide bedeuten
„Current“ und beide verlangen die Berücksichtigung des „Standes der Technik“.
Die Struktur der Gesetze wird zum besseren Verständnis in der folgenden Grafik dargestellt.
Abbildung 3: Gesetzesstruktur USA5
Die FDA gibt auch sogenannte Guidance-Dokumente heraus die, zwar als nicht rechtlich
bindend bezeichnet werden jedoch ein hohe Relevanz besitzen. Guidance- Dokumente
stellen nach Aussage der FDA das sogenannte „Current Thinking“ der FDA zu einem
bestimmten Thema dar. Dadurch definieren sie die Erwartungshaltung der FDA zu diesem
Thema. Ähnlich den Harmonisierten Normen in Europa, berücksichtigt die FDA „Recognized
5 Johner, C.; Hölzer-Klüpfel, M.; Wittorf, S. (2011): Basiswissen Medizinische Software; S. 42
FDA Guidance
Dokumente
Recognized
consensus
Standards
Administratives Recht
Code of Federal Regulations, Titel 21 Food and Drug
Gesetzes Recht
Federal Food, Drug and Cosmetics Act
Verfassung
Nicht gesetzlich
bindend
Erstellt durch
Fachgremien
US
Ko
ngr
ess
Beh
örd
e FD
A

Stand der Gesetzgebung / Regulatorische Definitionen
19
Consensus Standards“. Diese sind von unterschiedlichen Fachgremien erarbeitet
Dokumente welche den Stand der Technik darstellen und zur Erfüllung von gesetzlichen
Forderungen herangezogen werden.6
Innerhalb der FDA werden Arzneimittel und Medizinprodukte und deren Kombinationen
von vier Zentren betreut.
Diese Zentren sind :
- Das CBER Center of Biologics Evaluation and Research.
- Das CDER Center for Drug Evaluation and Research.
- Das CDRH Center for Devices and Radiological Health.
- Und das OCP Office for Combination Products als koordinierende Stelle für die
Zulassung und Überwachung von Kombinationsprodukten.
5.1.2 Zulassungsverfahren für Arzneimittel und Medizinprodukte
Bevor eine Arznei, ein Medizinprodukt oder ein Kombinationsprodukt in den USA auf den
Markt gebracht werden darf, muss das Produkt gemäß den geltenden Gesetzen durch die
FDA freigegeben bzw. genehmigt werden. Für Arzneimittel geschieht dies auf Grundlage des
New Drug Application (NDA) Verfahrens und für Biologika auf Grundlage des Biologic
Licensing Application (BLA) Verfahrens.
Beide Verfahren zielen darauf ab, zu ergründen und zu dokumentieren ob:
- das Arzneimittel bei bestimmungsgemäßem Gebrauch sicher und wirksam ist und der
pharmazeutische Nutzen die Risiken überwiegt?
- die Gebrauchsinformation (Packungsbeilage) sowie die Packungsbeschriftung von
ausreichender Qualität sind
- die verwendeten Herstellungsmethoden (Good Manufacturing Practices, GMP) sowie
die Kontrollen, die zur Kontrolle des Arzneimittels eingesetzt werden, geeignet sind,
um Identität, Stärke, Qualität und Reinheit zu gewährleisten
6 Vgl. Johner, C.; Hölzer-Klüpfel,M.; Wittorf, S. (2011): Basiswissen Medizinische Software; S. 41

Stand der Gesetzgebung / Regulatorische Definitionen
20
Für Medizinprodukte bestimmt das Risiko, welches aus dem Medizinprodukt für den
Anwender bzw. Patienten resultiert, das Zulassungsverfahren sowie den Kontrollaufwand.
Im Food, Drug and Cosmetic Act (FD&CA) werden drei Klassen von Medizinprodukten
definiert. Diese definieren den Kontrollaufwand, welcher notwendig ist, um die Sicherheit
und Effektivität des Medizinproduktes sicherzustellen. Im Folgenden wird eine Übersicht
über die drei Klassen, ihre Kontrollen und Zulassungsverfahren gegeben:
Class I, General Controls:
Klasse I Medizinprodukte erfordern den geringsten regulatorischen Aufwand und sind den
„General Controls“ unterworfen. Als Klasse I Medizinprodukte werden solche Produkte
eingestuft, die nicht dafür notwendig sind, Leben zu erhalten oder wesentlich dafür sind, der
Verletzung von menschlicher Gesundheit vorzubeugen. Zusätzlich dürfen Klasse 1
Medizinprodukte kein unnötiges Risiko für den Patienten bzw. Anwender darstellen. Die
„General Controls“ umfassen unter anderem Anforderungen in Bezug auf Verfälschung,
Kennzeichnung, Produktregistrierung, GMP, Reparatur, Pre Market Notification, Ersatz ,
Aufzeichnungen und Berichte. Viele „Class I“ Medizinprodukte sind vom „Pre Market
Notification“-Prozess ausgenommen. Beispiele für Klasse I Medizinprodukte sind elastische
Bandagen, Untersuchungshandschuhe, chirurgische Handinstrumente.7
Class II, General Controls with Special Controls:
Klasse II Medizinprodukte sind neben den „General Controls“ auch „Special Controls“
unterworfen. Klasse II Medizinprodukte müssen so entwickelt sein, dass sie wie beschrieben
funktionieren, ohne dem Patienten Schaden oder Verletzungen zuzufügen. Klasse II
Medizinprodukte werden stärker geprüft als Klasse I Medizinprodukte. Die „Special Controls“
beinhalten spezielle Kennzeichnungsanforderungen, verpflichtende Leistungsstandards und
Marktüberwachung (PMS). Klasse II Produkte werden im Allgemeinen durch eine Pre Market
Notification (510(k)) zugelassen, jedoch sind wenige Klasse II Medizinprodukte von der „Pre
7 Vgl. FDA, o.V. (2015): Classify your Medical Device; Vgl. FDA, o.V. (2016): Premarket Notification
510(k); FDA, o.V. (2014): General and Special Controls; Vgl. Wikipedia, o.V.(o.J.): Medical Devices

Stand der Gesetzgebung / Regulatorische Definitionen
21
Market Notification“ ausgenommen und durchlaufen das „Pre Market Approval“-Verfahren
(PMA). Beispiele für Klasse II Produkte sind Infusionspumpen, Akkupunkturnadeln oder
elektrische Rollstühle.8
Class III, General Controls, Special Controls and Premarket Approval:
Klasse III Medizinprodukte sind solche Medizinprodukte, für die nicht ausreichend
Informationen vorliegen, um die Sicherheit oder Wirksamkeit alleine durch die „General
Controls“ und „Special Controls“ nachzuweisen. Dies ist insbesondere der Fall wenn es sich
um ein „De Novo“ Medizinprodukt handelt, also kein Vergleichsprodukt vorhanden ist oder
um ein Hochrisikoprodukt. Klasse III Medizinprodukte müssen das „Pre Market Approval“ –
Verfahren durchlaufen. Beispiele für Klasse III sind implantierbare Herzschrittmacher,
Hüftimplantate und HIV-Tests.9
Nachfolgend wird ein kurzer Abriss der beiden möglichen Zulassungsverfahren gegeben.
Für wenige Klasse I und viele Klasse II Produkte wird die Zulassung über die „Pre Market
Notification“ (PMN) durchgeführt. Ziel des Prozesses ist die sogenannte „Clearance“ des
Produktes, welche im 21 CFR Part 510(k) beschrieben ist. Für die „Clearance“ muss der
Nachweis geführt werden, dass das zu vermarktende Medizinprodukt einem bereits
zugelassenen Medizinprodukt substantiell gleicht, und Sicherheit und Wirksamkeit des
Medizinproduktes sichergestellt sind.10
Für wenige Klasse II Produkte ist das Pre Market Approval (PMA) anzuwenden, wenn für das
zu vermarktende Medizinprodukt nicht ausreichend Informationen vorhanden sind, um die
Sicherheit und Wirksamkeit zu gewährleisten. Für alle Klasse III Produkte ist das PMA
Verfahren durchzuführen. Ein PMA beinhaltet neben administrativen Informationen,
Informationen zum Produkt, den Herstellungsprozessen, die Qualitätssicherungsverfahren
8 Vgl. FDA, o.V. (2015): Classify your Medical Device; Vgl. FDA, o.V. (2016): Premarket Notification
510(k); FDA, o.V. (2016): Premarket Approval PMA FDA, o.V. (2014): General and Special Controls; Vgl. Wikipedia, o.V.(o.J.): Medical Devices. 9 Vgl. FDA, o.V. (2015): Classify your Medical Device; FDA, o.V. (2016): Premarket Approval PMA; Vgl.
FDA, o.V. (2014): General and Special Controls; Vgl. Wikipedia, o.V.(o.J.): Medical Devices 10
Vgl. FDA, o.V. (2016): Premarket Notification 510(k); Vgl. Wikipedia, o.V.(o.J.): Medical Devices.
Stand der Gesetzgebung / Regulatorische Definitionen
22
und Ergebnisse aus klinischen Tests. All dies wird durch die FDA gefordert und geprüft, um
die Sicherheit und Wirksamkeit des Klasse III Medizinproduktes zu gewährleisten.11
In der folgenden Tabelle 5.1 ist eine Übersicht der anzuwendenden Zulassungsverfahren für
Arzneimittel und Medizinprodukte dargestellt.
Tabelle 5.1: Zulassungsverfahren USA
Arzneimittel Medizinprodukte
Stoffklasse Verfahren Kategorie Verfahren
Arzneimittel NDA Class I Pre Market Notification: 510(k) wenn nicht davon ausgenommen
Biologika BLA Class II Pre Market Notification: 510(k) wenn nicht davon ausgenommen
N/A N/A Class III Pre Market Approval
5.1.3 Der 21 CFR Part 4
Das Gesetze über Kombinationsprodukte soll klarzustellen, welche CGMP Anforderungen
anzuwenden sind, wenn ein Arzneimittel und/oder Biologikum mit einem Medizinprodukt zu
einem Kombinationsprodukt zusammengeführt wird. Dies wurde nach Aussage der FDA im
Vorwort zum 21CFR Part 4 getan, um die regulatorischen Unsicherheiten für Hersteller zu
reduzieren und durch klare Anforderungen, Hemmnisse für Innovationen zu reduzieren und
dadurch die Versorgung der US amerikanischen Bevölkerung mit modernen und innovativen
Therapiemöglichkeiten sicherzustellen.12
Zunächst ist es essentiell zu wissen, dass Kombinationsprodukte im 21 CFR Part 3 definiert
werden. Der 21 CFR Part 3 legt fest, dass ein Kombinationsprodukt aus zwei oder mehr
Komponenten besteht, welche durch Gesetze reguliert werden und als Einheit in den
Verkehr gebracht werden. Der 21 CFR Part 4 nimmt direkten Bezug auf die Definitionen aus
dem 21 CFR Part 3.
11
Vgl. FDA, o.V. (2016): Premarket Approval PMA; Vgl. Wikipedia, o.V.(o.J.): Medical Devices 12
Vgl. FDA, o.V. (2013): 21 CFR Part 4 Final Rule, Vorwort

Stand der Gesetzgebung / Regulatorische Definitionen
23
In 21 CFR Part 3 Sektion 3.2 wird unter Absatz (e) Satz (1) definiert, dass ein
Kombinationsprodukt aus mehreren regulierten Komponenten (Constituent Parts)
zusammengesetzt ist, die eine Einheit bilden (single entity combination product). In
Abbildung 4 und Abbildung 5 sind Beispiele für Single Entity Combination Products
dargestellt:
Abbildung 4: Einweg-Injektionspen (Lantus SoloStar® Hersteller Sanofi) mit aufgesetzter Kanüle
Abbildung 5: Autoinjektor (DAI Autoinjektor Hersteller SHL)mit abgezogener Kappe
Im darauffolgenden Satz der Section 3.2, Absatz (e) wird ein Kombinationsprodukte
definiert welches ebenfalls aus 2 oder mehreren regulierten Constituent Parts besteht diese
aber nicht zusammengebaut werden (Beispiel: Mehrfach verwendbarer Injections Pen und
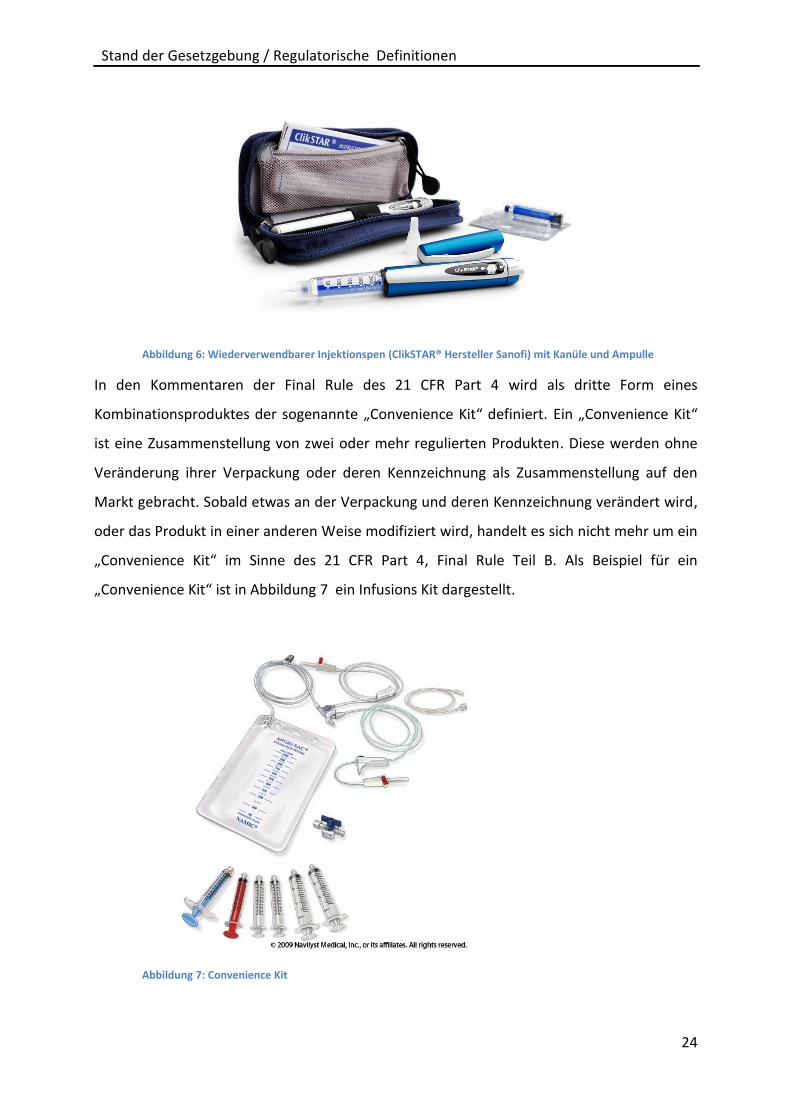
eine beigelegte Ampulle (Abbildung 6: Wiederverwendbarer Injektionspen (ClikSTAR®
Hersteller Sanofi) mit Kanüle und Ampulle). Dies wird als ein „co-packaged combination
product“ bezeichnet:
Stand der Gesetzgebung / Regulatorische Definitionen
24
Abbildung 6: Wiederverwendbarer Injektionspen (ClikSTAR® Hersteller Sanofi) mit Kanüle und Ampulle
In den Kommentaren der Final Rule des 21 CFR Part 4 wird als dritte Form eines
Kombinationsproduktes der sogenannte „Convenience Kit“ definiert. Ein „Convenience Kit“
ist eine Zusammenstellung von zwei oder mehr regulierten Produkten. Diese werden ohne
Veränderung ihrer Verpackung oder deren Kennzeichnung als Zusammenstellung auf den
Markt gebracht. Sobald etwas an der Verpackung und deren Kennzeichnung verändert wird,
oder das Produkt in einer anderen Weise modifiziert wird, handelt es sich nicht mehr um ein
„Convenience Kit“ im Sinne des 21 CFR Part 4, Final Rule Teil B. Als Beispiel für ein
„Convenience Kit“ ist in Abbildung 7 ein Infusions Kit dargestellt.
Abbildung 7: Convenience Kit

Stand der Gesetzgebung / Regulatorische Definitionen
25
5.1.3.1 Wirkung und Hauptwirkung des Kombinationsproduktes
Die Art der Bestandteile eines Kombinationsproduktes hat einen entscheidenden Einfluss auf
die Produktkategorie. Der 21 CFR Part 3 definiert im Abschnitt (e,1) vier Kategorien, welche
für die weitere Definition von Kombinationsprodukten von Bedeutung sind. Dies sind:
- Drug / Device z.B. Einweg-Insulininjektionspen
- Biologic / Device z.B. Injektionspen mit monoklonalem Antikörper
- Drug / Biologic z.B. Interferon – Ribavirin. Interferon ist ein körpereigenes Protein.
Ribavirin ist wirksam gegen Viren. In Kombination werden die beiden Wirkstoffe zur
Behandlung von Hepatitis C eingesetzt.
- Drug / Device / Biologic
Weiterhin ist für die Definition eines Kombinationsproduktes die Wirkung (engl. Mode of
Action) bzw. Hauptwirkung (engl. Primary Mode of Action) von Bedeutung. Dieser
Themenkomplex wird im 21 CFR Part 3 Abschnitt (m) benannt. Durch die Wirkung
/Hauptwirkung wird der therapeutische Nutzeffekt beschrieben. Die Hauptwirkung ist jene,
die den größten therapeutischen Nutzeffekt des Kombinationsproduktes liefert. Im Abschnitt
(k,1-3) des 21 CFR Part 3 werden die folgenden Wirkungen für die einzelnen Bestandteile
(engl. Constituent Parts) eines Kombinationproduktes definiert.
Biological Mode of Action:
Bei einem Auto-Injektor für einen monoklonalen Antikörper zur Behandlung von
rheumatoider Arthritis ist dies die Wirkung des Monoklonalen Antikörpers und nicht die
„Drug Delivery“ durch den Autoinjektor.
Device Mode of Action:
Beschichteter Stent, der ein Medikament abgibt, um Blutverklumpungen am Stent zu
verhindern. Dennoch ist die Hauptwirkung das Aufweiten des Blutgefäßes.
Drug Mode of Action:
Bei einem Insulin-Injektionspen ist dies die Wirkung des Insulins.
Die Hauptwirkung des Kombinationsproduktes legt fest von welchem „Center“ der FDA die
Zulassung federführend bearbeitet wird. Im Falle des Einweg Insulin Injektionspens ist dies

Stand der Gesetzgebung / Regulatorische Definitionen
26
das „CDER“ welches aber die Bearbeitung der Zulassung der Medizinproduktbestandteils an
das „CDRH“ weiterleitet.
5.1.3.2 Welche Gesetze sind anzuwenden
Eine der Hauptaussagen des 21 CFR Part 4 ist es, dass die bereits existierenden Gesetze
anwendbar sind. Der 21 CFR Part 4 führt keine zusätzlichen Anforderungen für die
Bestandteile des Kombinationsprodukts ein. Ein Medizinprodukt fällt also weiterhin unter
den 21 CFR Part 820 und dessen Anforderungen. Für ein Arzneimittel sind weiterhin die Teile
21 CFR Part 210 & 211 und dessen Anforderungen anwendbar, dies wird explizit in der
Präambel der Final Rule unter (I). Background (A). „Rationale for the Rulemaking“ in Absatz 4
dargestellt:
“The constituent Parts of a combination product retain their regulatory status (as a
drug or device, for example) after they are combined.
Accordingly, the CGMP requirements that apply to each of the constituent part
continue to apply when they are combined to make combination products”.13
„Die Bestandteile eines Kombinationsproduktes behalten ihren regulatorischen Status
(z.B. als Arznei oder Medizinprodukt) nachdem sie zu [(Anm. des Verf.)einem
Kombinationsprodukt] verbunden wurden.
Demgemäß sind die CGMP Anforderungen, für die Bestandteile des
Kombinationsproduktes weiterhin anwendbar wenn sie zu einem
Kombinationsprodukt verbunden wurden“.14
Diese Auslegung führt dann durch den §4.3 Absatz a und b zu einem parallelen GMP
Operating System, das heißt für den Arzneibestandteil eines Kombinationsproduktes sind die
CGMP Anforderungen des 21 CFR Part 210 & 211 zu erfüllen. Für den Medizinprodukte-
Bestandteil des Kombinationsproduktes sind die Anforderungen des 21 CFR Part 820 zu
erfüllen. Die Erfüllung von zwei vollständigen QM-Systemen führt zwangsläufig zu
erheblichem Mehraufwand. Um solchen Mehraufwand und die damit verbunden Kosten zu
vermeiden, erlaubt der 21 CFR Part 4 ein sogenanntes „Streamlined“ GMP Operating
13
FDA, o.V. (2013): 21 CFR Part 4, Final Rule I Background A, Absatz4 14
Übersetzung des Verfassers

Stand der Gesetzgebung / Regulatorische Definitionen
27
System. In der „Executive Summary“ und im Abschnitt e des 21 CFR Part 4 wird der
„Streamlined Approach“ zur Umsetzung des Systems beschrieben.
“The rule permits the use of the streamlined approach to demonstrate compliance
with the drug CGMP and device QS regulation requirements when both are applicable
to a facility’s manufacturing activities for a single-entity or co-packaged combination
product (§ 4.4(a) and (b))”.15
„Die „rule” erlaubt den Gebrauch eines „Streamlined Approach“ zum Nachweis der
Übereinstimmung mit dem CGMP und Medizinprodukte Qualitäts System
Anforderungen (QSR; 21 CFR Part 820) wenn beide anwendbar sind für die
Fertigungsaktivitäten eines Herstellers und dessen „single entity“ oder „co –
packaged“ Kombinationsprodukte(§ 4.4(a) and (b))“ 16
Der Hersteller darf, um Übereinstimmung zu demonstrieren, einen „streamlined approach“
verwenden. Dabei ist das Wort „demonstrieren“ wichtig. In diesem Zusammenhang
bedeutet „demonstrieren“, den Nachweis zu erbringen. Es soll der Nachweis erbracht
werden, dass das QMS oder genauer das „GMP Operating System“ den Anforderungen der
anwendbare Teile des Kapitel 21 des „Code of Federal Regulations“ entspricht. Der 21 CFR
Part 4 legt fest, dass die Anforderungen an die einzelnen Bestandteile erhalten bleiben, es
aber ausreicht ein führendes GMP Operating System zu definieren und Ausschnitte aus
dem jeweils anderen GMP Operating System, zu erfüllen.
Der „Streamlined Approach“ ist die Zusammenführung der Anforderungen aus dem 21 CFR
Part 210 & 211 und den Anforderungen aus dem 21 CFR Part 820. Für Arznei-
Medizinprodukt Kombinationsprodukte liefertder 21 CFR Part 4 zwei Möglichkeiten.
1. Verfügt der Hersteller überein Qualitäts Management System aufbauend auf den
Vorgaben des 21 CFR Parts 210 und 211 (Pharma GMP) als führendes GMP Operating
System etabliert, sind noch die Vorgaben aus 6 Abschnitten des 21 CFR 820 Quality
Systems Regulations zu erfüllen.
15
FDA, o.V. (2013): 21 CFR Part 4 Final Rule, Abschnitt e; 16
Übersetzung des Verfassers
Stand der Gesetzgebung / Regulatorische Definitionen
28
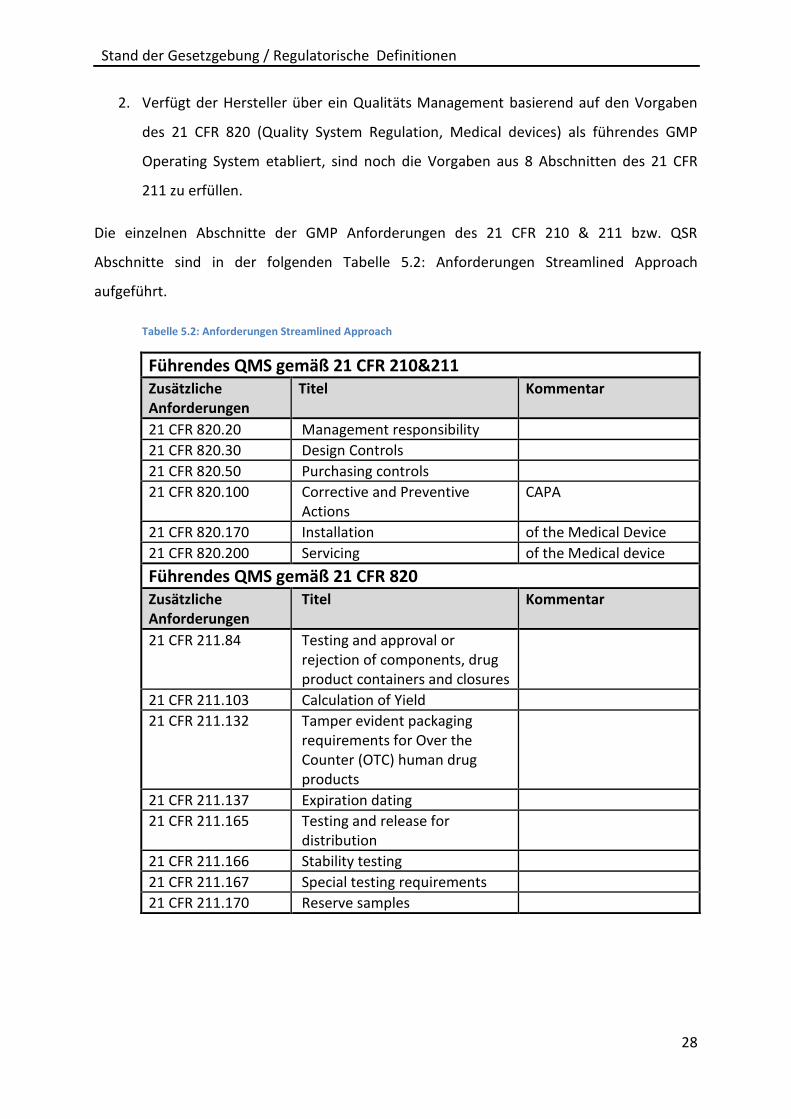
2. Verfügt der Hersteller über ein Qualitäts Management basierend auf den Vorgaben
des 21 CFR 820 (Quality System Regulation, Medical devices) als führendes GMP
Operating System etabliert, sind noch die Vorgaben aus 8 Abschnitten des 21 CFR
211 zu erfüllen.
Die einzelnen Abschnitte der GMP Anforderungen des 21 CFR 210 & 211 bzw. QSR
Abschnitte sind in der folgenden Tabelle 5.2: Anforderungen Streamlined Approach
aufgeführt.
Tabelle 5.2: Anforderungen Streamlined Approach
Führendes QMS gemäß 21 CFR 210&211 Zusätzliche Anforderungen
Titel Kommentar
21 CFR 820.20 Management responsibility
21 CFR 820.30 Design Controls
21 CFR 820.50 Purchasing controls
21 CFR 820.100 Corrective and Preventive Actions
CAPA
21 CFR 820.170 Installation of the Medical Device
21 CFR 820.200 Servicing of the Medical device
Führendes QMS gemäß 21 CFR 820 Zusätzliche Anforderungen
Titel Kommentar
21 CFR 211.84 Testing and approval or rejection of components, drug product containers and closures
21 CFR 211.103 Calculation of Yield
21 CFR 211.132 Tamper evident packaging requirements for Over the Counter (OTC) human drug products
21 CFR 211.137 Expiration dating
21 CFR 211.165 Testing and release for distribution
21 CFR 211.166 Stability testing
21 CFR 211.167 Special testing requirements
21 CFR 211.170 Reserve samples

Stand der Gesetzgebung / Regulatorische Definitionen
29
5.2 Pharmagesetzgebung der EU Arzneimittel und Medizinprodukte sind in Europa über Richtlinien regulierte Produkte und
unterliegen demnach den Gesetzen der EU bzw. den Gesetzen der Nationalstaaten. Wie
bereits erwähnt, gibt es keine Richtlinie über oder eine direkte Definition von
Kombinationsprodukte/n im europäischen Rechtsraum. Im Folgenden sei ein kurzer Abriss
über die Entstehung der Gesetzgebung zur Regulierung von Arzneimitteln und
Medizinprodukten in der Bundesrepublik Deutschland/Europa dargestellt.
1976: Die Neufassung des Arzneimittelgesetzes erscheint. Beschluss 24.08.1976
1978: Das AMG tritt am 1. Januar 1978 in Kraft.
1985: Die erste Ausgabe der Pharma Betriebs Verordnung erscheint. Sie enthält erste
GMP-Vorgaben für Deutschland.
1987: Einführung der ISO 9000 Normenreihe.
1989: Europa: Der EU-GMP-Leitfaden wird veröffentlicht.
1990: Die EU-Kommission initiiert das ICH-Programm zur internationalen
Harmonisierung von technischen Zulassungsanforderungen.
1994: Die erste Version des Medizinproduktegesetzes wird verabschiedet und tritt
am 1. Januar 1995 in Kraft.
2001: ICH Q 7A (GMP für Wirkstoffhersteller) wird als Draft (Step 2) erstmals
publiziert
2005: ICH Q8 Pharmaceutical Development (Quality by Design) und Q9 Quality Risk
Management werden verabschiedet. Die EU publiziert den neu strukturierten EG
GMP Leitfaden mit Teil 1 Arzneimittel und Teil Wirkstoffe.

Stand der Gesetzgebung / Regulatorische Definitionen
30
2006: Die Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) tritt in Kraft
und ersetzt die Pharma Betriebs Verordnung.
2007: Die ICH publiziert die Guide ICH Q 10 „Pharmaceutical Quality System“ als Step
2 Dokument.
2009: Die 15. AMG-Novelle wird verabschiedet.
2012: Wesentliche Revision im EU GMP Leitfaden durch die Erweiterung des Kapitel 1
in Bezug auf „Pharmaceutical Quality Systems“.
2013: EU GDP Guideline Revision: GMP und GDP wachsen zusammen.
2015: Neuer Annex 15 zum EU GMP Guide zu Qualifizierung und Validierung.
5.2.1 Struktur der Gesetzgebung
Eine europäische Richtlinie ist ein europäisches Gesetz, welches von den Mitgliedsstaaten
binnen einer festgelegten Zeitspanne in nationales Recht umgesetzt werden muss. Im
Gegensatz dazu ist eine europäische Verordnung ein direkt wirksames europäisches Gesetz,
das für die Mitgliedsstaaten bindend ist aber von den Mitgliedsstaaten nicht in nationales
Recht umgesetzt werden muss. Die Verordnung muss binnen einer festgelegten Frist im
europäischen Rechtsraum angewendet werden.
Für den Arzneimittelbereich, beziehungsweise den Medizinproduktebereich sind die
regulatorischen Anforderungen in europäischen Richtlinien niedergelegt. In Europa sind die
sogenannten Good Manufacturing Practices (GMP dt. Gute Herstellungs Praxis) in den EU
GMPs gesetzlich fixiert. Die folgende Aufzählung stellt die zugrunde liegenden Europäischen
Richtlinien dar:
- Richtlinie 91/356/EWG der Kommission vom 13. Juni 1991 zur Festlegung der
Grundsätze und Leitlinien der Guten Herstellungspraxis für zur Anwendung beim
Menschen bestimmte Arzneimittel
- Richtlinie 91/412/EWG der Kommission vom 23. Juli 1991 zur Festlegung der
Grundsätze und Leitlinien der Guten Herstellungspraxis für Tierarzneimittel.

Stand der Gesetzgebung / Regulatorische Definitionen
31
- Richtlinie 2003/94/EG der Kommission vom 8. Oktober 2003 zur Festlegung der
Grundsätze und Leitlinien der Guten Herstellungspraxis für Humanarzneimittel und
für zur Anwendung beim Menschen bestimmte Prüfpräparate sowie detaillierte
Richtlinien
In der BRD wurden die obengenannten Europäischen Richtlinien im Deutschen Arzneimittel
Gesetz (AMG) von 1978 umgesetzt.
Die Regelungen für Medizinprodukte sind in Europa in mehreren Richtlinien niedergelegt.
- Richtlinie 90/385/ EWG des Rates vom 20.Juni 1990 zur Angleichung der
Rechtsvorschriften der Mitgliedstaaten über aktive implantierbare medizinische
Geräte (AIMDD)
- Richtlinie 93/42/EWG des Rates vom 14.Juni 1993 über Medizinprodukte
- Richtlinie 98/79/EG des Europäischen Parlaments und des Rates vom 27.Oktober
1998 über In-vitro-Diagnostika
In der BRD wurden diese Richtlinien im Deutschen Medizinproduktegesetz MPG von 1994
umgesetzt.
5.2.2 Gesetzgebung zu Kombinationsprodukten in Europa
Es gibt keine eigenständige Gesetzgebung für Kombinationsprodukte in der EU. Daher teilt
die Europäische Gesetzgebung diese Produkte entweder in Arzneimittel oder
Medizinprodukte ein. Diese Einteilung der Kombinationsprodukte wird durch Einschlüsse
bzw. Ausschlüsse in den Richtlinien über Arzneimittel 2001/83/EG und Medizinprodukte
93/42/EWG erreicht. Die Richtlinie über Arzneimittel 2001/83/EG (MPD) definiert
Arzneimittel und die Richtlinie über Medizinprodukte 93/42/EWG (MDD) definiert
Medizinprodukte. In der MDD wird in den Artikeln 1(3-4) festgelegt wann ein
Kombinationsprodukt entweder als Medizinprodukt oder als Arzneimittel zu behandeln ist.
Dabei wird besonders auf die Hauptwirkung des Kombinationsproduktes eingegangen und
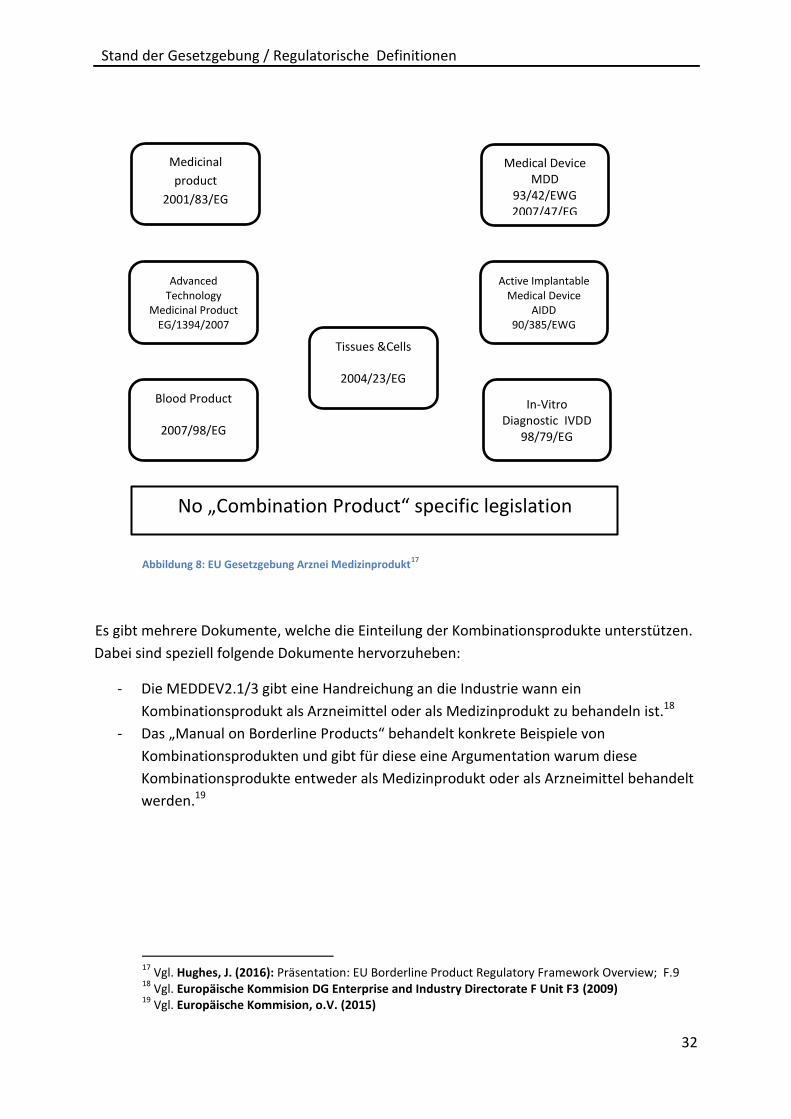
diese als Unterscheidungshilfe hervorgehoben. Zum besseren Verständnis folgt eine
grafische Darstellung (Abbildung 7) der Richtlinien, die sich mit Kombinationsprodukten
beschäftigen.
Stand der Gesetzgebung / Regulatorische Definitionen
32
Abbildung 8: EU Gesetzgebung Arznei Medizinprodukt17
Es gibt mehrere Dokumente, welche die Einteilung der Kombinationsprodukte unterstützen.
Dabei sind speziell folgende Dokumente hervorzuheben:
- Die MEDDEV2.1/3 gibt eine Handreichung an die Industrie wann ein
Kombinationsprodukt als Arzneimittel oder als Medizinprodukt zu behandeln ist.18
- Das „Manual on Borderline Products“ behandelt konkrete Beispiele von
Kombinationsprodukten und gibt für diese eine Argumentation warum diese
Kombinationsprodukte entweder als Medizinprodukt oder als Arzneimittel behandelt
werden.19
17
Vgl. Hughes, J. (2016): Präsentation: EU Borderline Product Regulatory Framework Overview; F.9 18
Vgl. Europäische Kommision DG Enterprise and Industry Directorate F Unit F3 (2009) 19
Vgl. Europäische Kommision, o.V. (2015)
Medicinal
product
2001/83/EG
Advanced Technology
Medicinal Product EG/1394/2007
Blood Product
2007/98/EG
Tissues &Cells
2004/23/EG
Medical Device MDD
93/42/EWG 2007/47/EG
In-Vitro Diagnostic IVDD
98/79/EG
Active Implantable Medical Device
AIDD 90/385/EWG
No „Combination Product“ specific legislation

Auswirkungen und Umsetzung in der Produktrealisierung
33
6 Auswirkungen und Umsetzung in der Produktrealisierung
mit besonderem Fokus auf die Entwicklung Wie bereits dargelegt, muss ein Hersteller von Arzneimitteln für die Inverkehrbringung in
den USA die Einhaltung der Teile 21 CFR Part 210 & 211 nachweisen. Soll jedoch ein
Medizinprodukt in Verkehr gebracht werden, ist der Nachweis der Einhaltung der
Anforderungen des 21 CFR Part 820 notwendig. Soll ein Kombinationsprodukt in den USA auf
den Markt gebracht werden, kann gemäß des 21 CFR Part 4 der „Streamlined Approach“
genutzt werden. Das Qualitätssystem der Beispielorganisation genügt den Anforderungen
des 21 CFR Part 210 & 211, beziehungsweise den Anforderungen des ICH Q10
Pharmaceutical Quality System. Entsprechend des 21 CFR Part 4 müssen für ein
Kombinationsprodukt noch die zusätzlichen Anforderungen aus dem 21 CFR Part 820 erfüllt
werden (Siehe Tabelle 6.1: Forderungen “Streamlined Approach“ bei führendem
Qualitätssystem für Arzneimittel).
Tabelle 6.1: Forderungen “Streamlined Approach“ bei führendem Qualitätssystem für Arzneimittel
Führendes QMS gemäß 21 CFR 210&211 Zusätzliche Anforderungen
Titel Kommentar
21 CFR 820.20 Management Responsibility
21 CFR 820.30 Design Controls
21 CFR 820.50 Purchasing Controls
21 CFR 820.100 Corrective and Preventive Actions
Entspricht CAPA
21 CFR 820.170 Installation Installation des Medizinproduktes
21 CFR 820.200 Servicing Wartung und Instandhaltung am Medizinprodukt
6.1 Verantwortung der obersten Leitung Im 21 CFR Part 210 & 211 gibt es keine Anforderungen zur Verantwortung der obersten
Leitung. Im ICH Q10 wird der Verantwortung der obersten Leitung im Vergleich dazu viel
Platz eingeräumt. Die Forderungen im ICH Q10 an die oberste Leitung entsprechen
weitestgehend den Forderungen des 21 CFR Part 820.20. Mit einer einzigen Ausnahme, das
die Benennung eines „Management Representative“ oder Beauftragten der obersten Leitung

Auswirkungen und Umsetzung in der Produktrealisierung
34
(QMB) nicht gefordert wird. Die Normenreihe der ISO 9000 (z. B. ISO 9001, ISO13485)
fordert ebenfalls die Benennung eines Beauftragten der obersten Leitung.20
6.1.1 Beauftragter der obersten Leitung
Der originale Text des 21 CFR 820.20(b. 3) ist In Tabelle 7.2 Verantwortung der obersten
Leitung abgebildet. Der Beauftragte der obersten Leitung (QMB) muss ein Mitglied der
obersten Leitung der Organisation sein und wird von dieser beauftragt (Tabelle 6.2).
Tabelle 6.2: Verantwortung der obersten Leitung
820.20(b,3,i,ii)
(3)Management representative. Management with executive responsibility shall appoint, and document such appointment of, a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility for: (i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this part; and (ii) Reporting on the performance of the quality system to management with executive responsibility for review.
(3)Managementvertreter: Das Management mit Führungsverantwortung muss ein Mitglied des Managements ernennen (und diese Ernennung dokumentieren), das unabhängig von anderen Verantwortungen, die etablierte Befugnis und Verantwortung für Folgendes hat: (i) Absicherung, dass die Qualitätssystemanforderungen gemäß den in diesem Teil aufgeführten Vorschriften effektiv festgelegt wurden und aufrechterhalten werden und (ii) Bericht über die Leistung des Qualitätssystems an das Management mit Führungsverantwortung zur Überprüfung.21
Der QMB muss unabhängig von anderen Aktivitäten sein und über die Autorität und die
Verantwortung verfügen, das Qualitätssystem einzuführen, zu erhalten und über die
Leistung des Systems an die oberste Leitung zu berichten [820.20 (b.3, i, ii)]. Die
Erwartungshaltung der FDA besteht darin, dass ein QMB schriftlich benannt und dies in der
Organisation kommuniziert wird.
20
Vgl. ISO, o.V. (2012): ISO 9001:2008 Kap.: 5.5.2; Vgl. ISO, o.V. (2016): ISO 13485:2016; Kap. 5.5.2 21
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.20(b,3,i,ii)

Auswirkungen und Umsetzung in der Produktrealisierung
35
6.2 Beschaffung
Aus dem Wirtschaftlichkeitsprinzip ergibt sich, dass nur mit einwandfreien Rohstoffen und
Materialien einwandfreie Produkte hergestellt werden können. Dieser Umstand hat auch in
den verschiedenen Qualitätsrichtlinien, Normen, Verordnungen und Gesetzen Niederschlag
gefunden.
Im 21 CFR Part 211 werden Forderungen an Lieferanten bzw. an geliefertes Material in den
Abschnitten 211.84 (b; d, 2, 3) beschrieben. Dabei geht es jedoch nur indirekt um das
Lieferantenmanagement, sondern primär um die Prüfung der gelieferten Materialien, die zur
Freigabe oder Zurückweisung der Materialien führen können. Im ICH Q10 2.7 “Management
of Outsourced Processes and Purchased Materials“ sind die Anforderungen an das
Lieferantenmanagement beschrieben. Im 21 CFR Part 820 beschreibt der Abschnitt 50(a, b)
„Purchasing Controls“ die Anforderungen an das Lieferantenmanagement.
6.2.1 Generelle Anforderungen an die Beschaffung
In den Abschnitten 2.7(a-d); 4.1(b.2) des ICH Q10 wird festgelegt, dass die Verantwortung für
geliefertes Material und für ausgelagerte Prozesse beim Arzneimittelhersteller verbleibt. Es
muss eine Eignungsprüfung des Lieferanten bzw. des gelieferten Materials durchgeführt
werden. Des Weiteren wird die Forderung gestellt Qualitätssicherungsvereinbarungen mit
Lieferanten abzuschließen und eine Überwachung und Re-Evaluierung für Lieferanten
durchzuführen. Im Bereich der Beschaffung gehen die Anforderungen des 21 CFR 820.50
über die Forderungen des ICH Q10 hinaus und sind detaillierter. Generell fordert der 21 CFR
820.50, dass dokumentierte Verfahren eingeführt und erhalten werden, um sicherzustellen,
dass eingekauftes oder anderweitig beschafftes Material bzw. Dienstleistungen den
spezifizierten Anforderungen und den anwendbaren regulatorischen Anforderungen
entsprechen. Der 21 CFR 820.50 definiert
- verschiedene Typen von Lieferanten,
- verschiedene Phasen der Lieferantenauswahl,
- die Art und Weise der Lieferantenüberwachung auf dem Ergebnis der
Lieferantenauswahl zu basieren,

Auswirkungen und Umsetzung in der Produktrealisierung
36
- Aufzeichnungen zu freigegeben Lieferanten zu erstellen,
- Spezifikationen des gelieferten Gutes klar zu beschreiben oder zu referenzieren,
- schriftliche Übereinkünfte zwischen Lieferant und Hersteller zu erstellen, die den
Lieferanten verpflichten, den Hersteller über Änderungen am Produkt oder an der
Dienstleistung zu informieren.
6.2.2 Anforderungen an ausgelagerte Prozesse
Im Folgenden wird zunächst auf die Forderungen des ICHQ10 2.7(a, b) (original text und
Übersetzung Tabelle 7.3: ICH Q10 2.7(a, b) und ICH Q10 4.1(b, 2)) eingegangen (Tabelle 6.3).
Tabelle 6.3: Anforderungen an ausgelagerte Prozesse
ICH Q10 2.7(a,b);
The pharmaceutical quality system, including the management responsibilities described in this section, extends to the control and review of any outsourced activities and quality of purchased materials. The pharmaceutical company is ultimately responsible to ensure processes are in place to assure the control of outsourced activities and quality of purchased materials. These processes should incorporate quality risk management and include: (a) Assessing prior to outsourcing operations or selecting material suppliers, the suitability and competence of the other party to carry out the activity or provide the material using a defined supply chain (e.g., audits, material evaluations, qualification). (b) Defining the responsibilities and communication processes for quality-related activities of the involved parties. For outsourced activities, this should be included in a written agreement between the contract giver and contract acceptor.
Das Arzneimittel Qualitätssystem, inclusive der Verantwortung der Obersten Leitung welches in diesem Abschnitt beschrieben wird, erstreckt sich auf die Überprüfung und Lenkung jeglicher ausgelagerter Prozesses und die Qualität beschaffter Materialien. Das herstellende Unternehmen ist final dafür verantwortlich sicherzustellen das Verfahren erstellt und aufrechterhalten werden die die Lenkung von ausgelagerten Aktivitäten und Qualität von Beschafften Materialien sicherstellen. Diese Verfahren sollten Risiko Management anwenden und beinhalten: (a) Die Angemessenheit und Kompetenz der anderen Partei, daraufhin zu
bewerten bevor die Auslagerungsaktivität oder die Auswahl von Material Lieferanten gestartet wird, diese Aktivität auszuführen oder Material mittels einer definierten Lieferkette zur Verfügung zu stellen (z.B. Audit,

Auswirkungen und Umsetzung in der Produktrealisierung
37
Materialevaluierung, Qualifizierung). (b) Festlegen der Verantwortlichkeiten und des Kommunikationsprozesses für
Qualitätsrelevante Aktivitäten der involvierten Parteien. Dies sollte schriftliche Vereinbarung zwischen den Vertragsparteien beinhalten.22
ICH Q10 4.1(b.2)
Management should have a formal process for reviewing the pharmaceutical quality system on a periodic basis. The review should include: (a) Measurement of achievement of pharmaceutical quality system objectives (b) Assessment of performance indicators that can be used to monitor the effectiveness of processes within the pharmaceutical quality system, such as: (1) Complaint, deviation, CAPA and change management processes (2) Feedback on outsourced activities
Die Oberste Leitung sollte einen formalen Prozess haben, um das Arzneimittel Qualitätssystem in regelmäßigen Abständen zu bewerten. Diese Bewertungen sollten beinhalten : (a)Die Messung der Qualitätsziele des Arzneimittel Qualitätssystems. (b)Bewertung von Leistungsindikatoren welche für die Überwachung der Effektivität von Prozessen genutzt werden können, wie: (1)Beanstandungen, Abweichungen, Korrektur und Vorbeugemaßnahmen und die Lenkung von Änderungen. (2)Rückmeldung zu ausgelagerten Prozessen23
Es ist zu klären, was ein ausgelagerter Prozess ist. Hier stellt die ISO 9001:2008 in Anhang B1,
Abschnitt 4.1, Anmerkung 2 eine Definition zur Verfügung:
„Ein ausgegliederter Prozess ist ein Prozess, den die Organisation[(Anm. des. Verf.)das
Unternehmen] für ihr Qualitätsmanagementsystem benötigt und bei dem sie
entschieden hat, dass sie ihn durch eine externe Partei ausführen lässt.“24
Es ist also ein Prozess der in die Wertschöpfungskette des Herstellers eingegliedert ist
und/oder durch das QMS gefordert wird. Im Pharmaumfeld handelt es sich dabei häufig um
Verpackungsprozesse, im Medizinprodukteumfeld um Sterilisierungs-prozesse. Ein weiterer
typischer ausgelagerter Prozess stellt die Kalibrierung von Messgeräten dar. Wird ein
Prozess oder eine Aktivität an einen Lieferanten ausgelagert, verbleibt die Verantwortung
für diesen Prozess dennoch beim Unternehmen. Das Unternehmen hat die Aufgabe,
geeignete dokumentierte Verfahren einzuführen und zu erhalten.
22
Übersetzung durch Verfasser 23
Übersetzung durch Verfasser 24
ISO, o.V. (2012): ISO 9001:2008; Kap. 4.1 Anm. 2

Auswirkungen und Umsetzung in der Produktrealisierung
38
Der 21 CFR 820 macht keinen großen Unterschied, ob es sich um einen ausgelagerten
Prozess oder ein zugekauftes Produkt handelt. Zwar werden ausgelagerte Prozesse durch die
Erwähnung des Lieferantentyps „Contractor“ erfasst (siehe Tabelle 6.4: Anforderungen an
die Auswahl von Lieferanten Spalte 820.50(a, 1)). Es wird jedoch für alle Lieferantentypen
(von Material, Dienstleistung, ausgelagerten Prozessen usw.) gefordert, dass die
geforderten Spezifikationen erfüllt werden und dass sich der Kontrollaufwand für Auswahl,
Evaluierung, Überwachung und Re-Evaluierung an der Kritikalität des Produktes oder der zu
erbringenden Dienstleistung orientiert.
6.2.3 Auswahl von Lieferanten
Betreffend der Auswahl von geeigneten Lieferanten sind die Anforderungen des ICH Q10
(2.7(a, b)) den Anforderungen des 21 CFR 820.50 (a, 1) ähnlich, (siehe Tabelle 6.4:
Anforderungen an die Auswahl von Lieferanten). Beide Regelwerke fordern, dass Lieferanten
initial beurteilt werden. Basis der Beurteilung ist ihrer Fähigkeit, die (spezifizierten)
Anforderungen, welche aus dem Material, der Dienstleistung oder dem Prozess resultieren,
zu erfüllen. Außerdem fordern beide Regularien, dass die initiale Beurteilung vor dem
Gebrauch solcher Materialien, Dienstleistung oder Prozesse durchzuführen ist (Tabelle 6.4).
Tabelle 6.4: Anforderungen an die Auswahl von Lieferanten
ICH Q10 2.7(a,b) 820.50(a,1);
Siehe Tabelle 7.3 Anforderungen an ausgelagerte Prozesse
Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements. (a)Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and maintain the requirements, including quality requirements, that must be met by suppliers, contractors, and consultants. Each manufacturer shall: (1) Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability to meet specified requirements, including quality

Auswirkungen und Umsetzung in der Produktrealisierung
39
requirements. The evaluation shall be documented.
Jeder Hersteller muss Verfahren festlegen und erhalten, um sicherzustellen, dass alle eingekauften oder auf andere Weise erhaltenen Produkte und Dienstleistungen den spezifizierten Anforderungen entsprechen. (a) Bewertung von Lieferanten, Auftragnehmern und Beratern. Jeder Hersteller muss die Anforderungen einschließlich der von Lieferanten, Auftragnehmern und Beratern einzuhaltenden Anforderungen festlegen und aufrechterhalten. Jeder Hersteller muss: (1) potenzielle Lieferanten, Auftragnehmer und Berater auf der Grundlage ihrer Fähigkeit zur Erfüllung der spezifischen Anforderungen einschließlich der Qualitätsanforderungen bewerten und auswählen. Die Bewertung muss dokumentiert werden.25
Für die Auswahl von Lieferanten besteht kein Handlungsbedarf von Anpassungen an den 21
CFR Part 820, wenn das PQS den Abschnitt 2.7 (a, b) des ICH Q10 erfüllt.
25
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.50(a.1)

Auswirkungen und Umsetzung in der Produktrealisierung
40
6.2.4 Evaluierung und Freigabe von Lieferanten
Hinsichtlich Evaluierung und Freigabe von Lieferanten sind die Forderungen der Regelwerke
ICH Q10 und 21 CFR Part 820 recht ähnlich (Tabelle 6.5).
Tabelle 6.5: Anforderungen an die Auswahl und Freigabe von Lieferanten
ICH Q10 2.7(a,b) 820.50(a,1)
Siehe Tabelle 6.3: Anforderungen an ausgelagerte Prozesse
Siehe Tabelle 6.4: Anforderungen an die Auswahl von Lieferanten
NA 820.50.(a.3)
Establish and maintain records of acceptable suppliers, contractors, and consultants.
(3) Aufzeichnungen von akzeptablen Lieferanten, Auftragnehmern und Beratern erstellen und aufrechterhalten.26
Der 21 CFR Part 820.50 ist jedoch präziser in der Formulierung seiner Forderungen. So wird
unter 820.50 (a) gefordert, dass Evaluationskriterien, darunter auch Qualitätskriterien
festgelegt und dokumentiert werden. Des Weiteren wird unter 820.50 (a.3) gefordert, dass
Aufzeichnungen über freigegebene Lieferanten geführt werden müssen. Obwohl die
Anforderungen nicht in Regularien definiert sind, ist es in Pharmaunternehmen üblich, den
Auswahl- und Evaluierungsprozess von Lieferanten zu dokumentieren. Im
Medizinproduktesektor sind konkrete Forderungen zum Lieferantenmanagement im QSR
definiert. Zur Umsetzung des „streamlined Approach“ sind in der Praxis kaum Anpassungen
notwendig. Um jedoch den Forderung des 21 CFR Part 820.50 vollständig zu entsprechen,
sollte zumindest auf Verfahrensebene geprüft werden, ob alle Forderungen umgesetzt sind.
6.2.5 Überwachung und Re-Evaluierung von Lieferanten
Die konkreten Forderungen zur Überwachung und Re-Evaluierung sind in Tabelle 6.6:
Anforderungen an die Überwachung und Re-Evaluierung von Lieferanten“ dargestellt. Im
Arzneimittelbereich ist der Qualitätskontrollgedanke noch sehr stark verankert und wird
auch explizit in den Regularien gefordert. Dies spiegelt sich auch im ICH Q10 2.7(c, d) wieder.
26
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.50(a.3)
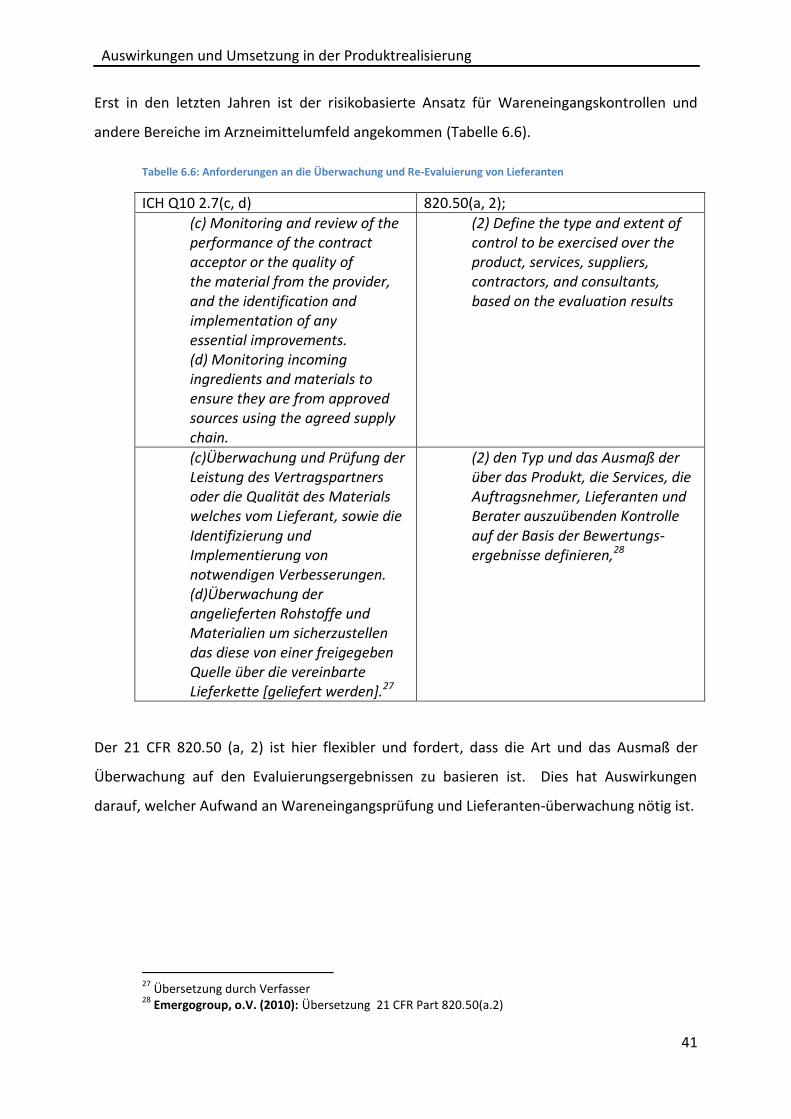
Auswirkungen und Umsetzung in der Produktrealisierung
41
Erst in den letzten Jahren ist der risikobasierte Ansatz für Wareneingangskontrollen und
andere Bereiche im Arzneimittelumfeld angekommen (Tabelle 6.6).
Tabelle 6.6: Anforderungen an die Überwachung und Re-Evaluierung von Lieferanten
ICH Q10 2.7(c, d) 820.50(a, 2);
(c) Monitoring and review of the performance of the contract acceptor or the quality of the material from the provider, and the identification and implementation of any essential improvements. (d) Monitoring incoming ingredients and materials to ensure they are from approved sources using the agreed supply chain.
(2) Define the type and extent of control to be exercised over the product, services, suppliers, contractors, and consultants, based on the evaluation results
(c)Überwachung und Prüfung der Leistung des Vertragspartners oder die Qualität des Materials welches vom Lieferant, sowie die Identifizierung und Implementierung von notwendigen Verbesserungen. (d)Überwachung der angelieferten Rohstoffe und Materialien um sicherzustellen das diese von einer freigegeben Quelle über die vereinbarte Lieferkette [geliefert werden].27
(2) den Typ und das Ausmaß der über das Produkt, die Services, die Auftragsnehmer, Lieferanten und Berater auszuübenden Kontrolle auf der Basis der Bewertungs-ergebnisse definieren,28
Der 21 CFR 820.50 (a, 2) ist hier flexibler und fordert, dass die Art und das Ausmaß der
Überwachung auf den Evaluierungsergebnissen zu basieren ist. Dies hat Auswirkungen
darauf, welcher Aufwand an Wareneingangsprüfung und Lieferanten-überwachung nötig ist.
27
Übersetzung durch Verfasser 28
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.50(a.2)

Auswirkungen und Umsetzung in der Produktrealisierung
42
6.2.6 Fazit zu den Anforderungen im Bereich Lieferantenmanagement
Wie mehrfach erwähnt sind sich die Qualitätsanforderungen des ICH Q10 und des QSR im
Bereich Lieferanten Management / Beschaffung ähnlich. Der QSR ist an manchen Stellen
sehr viel detaillierter und ist stärker am Risikobasierten Ansatz orientiert als die
entsprechenden Kapitel des ICH Q10. Im Arzneimittelbereich ist im Gegensatz dazu der
Ansatz zur Kontrolle der Qualität durch Prüfung noch wesentlich stärker verankert. Bei
Umsetzung der Anforderungen des ICH Q10 werden die Forderungen des 21 CFR Part 4 / 21
CFR Part 820 weitestgehend erfüllt. Jedoch sollten die etablierten Verfahrensanweisungen
dahingehend überprüft werden, ob sie dem höheren Detailierungsgrad des 21CFR Part
820.50 Rechnung tragen.

Auswirkungen und Umsetzung in der Produktrealisierung
43
6.3 Design und Entwicklungslenkung
Die wohl größte Anpassung, um in einem ICH Q10 konformen QMS den „Streamlined
Approach“ für Kombinationsprodukte gemäß 21 CFR Part 4 umzusetzen, besteht in der
Forderung, einen Design Control Prozess gemäß 21 CFR Part 820.30 zu implementieren und
zu erhalten. Es gibt zwar mit dem ICH Q8 „Pharmaceutical Development“ ein GMP-
Dokument, welches sich mit der Arzneimittelentwicklung beschäftigt, jedoch ist dieser sehr
speziell auf die Arzneimittelzulassung gerichtet. Es behandelt vor allem die
Entwicklungsdaten, welche bei einer Einreichung benötigt werden, als den eigentlichen
Entwicklungsprozess29. Im Gegensatz dazu, ist der 21 CFR Part 820.30 auf den konkreten
Entwicklungsprozess ausgerichtet. Die Anforderungen sind sehr viel ausführlicher formuliert.
Im Folgenden werden die Forderungen 21 CFR Part 820.30 dargelegt und Beispiele zur
Umsetzung gegeben.
6.3.1 Generelle Anforderungen an die Design und Entwicklungslenkung
Die allgemeinen Forderungen des 21CFR Part 820 zum Entwicklungslenkungsprozess sind in
den Absätzen 820.30(a, 1-2) formuliert (Tabelle 6.7)
Tabelle 6.7: Anforderungen an die Design und Entwicklungslenkung
820.30(a,1-2)
(a)General (1) Each manufacturer of any class III or class II device, and the class I devices listed in paragraph (a)(2) of this section, shall establish and maintain procedures to control the design of the device in order to ensure that specified design requirements are met.
(a)Allgemein(1) Jeder Hersteller von Medizinprodukten der Klasse III oder II sowie der Klasse l, die in §(a)(2) dieses Abschnitts aufgeführt sind, muss Verfahren zur Kontrolle der Entwicklung des Medizinprodukts festlegen und aufrechterhalten, um die Erfüllung der spezifizierten Anforderungen sicherzustellen.30
Die Forderungen des 21 CFR Part 820.30 sollen den Design- und
Entwicklungslenkungsprozess strukturieren und planbar gestalten. Der Design- und
Entwicklungslenkungsprozess ist gegen Forschungsaktivitäten (z.B. Machbarkeits-studien,
29
Vgl. ICH, o.V. (2009): ICH Pharmaceutical Development Q8; Kap. 1.1 & 1.2 30
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(a, 1-2)

Auswirkungen und Umsetzung in der Produktrealisierung
44
Entwicklung von Konzepten usw.) abgegrenzt und diesen nachgelagert.31 Ziel ist es, den
Entwicklungsprozess geplant und strukturiert, von der Entscheidung für ein bestimmtes
Design bis zur abgeschlossenen Übergabe an die Fertigung, zu durchlaufen. Nach der
Übergabe an die Fertigung muss bei Verbesserungen oder Veränderungen am Design wieder
in die Entwicklungsphase zurückgekehrt werden. Ein solches Vorgehen fällt unter das
sogenannte Life Cycle Management (LCM).
Am Anfang steht immer die Entscheidung, welches Produkt entwickelt werden soll. Sind die
Anforderungen an das zu entwickelnde Produkt klar, kann am Ende des
Entwicklungsprozesses bzw. des Entwicklungsprojektes festgestellt werden, ob das Projekt
erfolgreich war. Der 21 CFR Part 820.30 definiert, die Anforderungen an das zu
dokumentierende Verfahren zwischen Startpunkt (User Needs = Nutzer-anforderungen) und
dem Endpunkt (Medical Device) des Entwicklungsprozesses. Die Elemente, welche gemäß
der Anforderungen des 820.30 enthalten sein müssen sind in Abbildung 9: Wasserfall
Modell, Schlüsselelemente des Design Control Prozess (Quelle: Health Canada)dargestellt.
Der Entwicklungsprozess setzt sich aus mehreren Teilprozessen zusammen, die
nacheinander zu durchlaufen sind. Diese Teileprozesse sind:
- Festlegung der Anforderungen (User needs)
- Entwicklungseingaben (Design Input)
- Entwicklung (Design Process)
- Entwicklungsergebnisse (Design output)
- Entwicklungsverifzierung (Verification)
- Realisierung des Medizinproduktes (Medical Device)
- Entwicklungsvalidierung (Validation)
- Entwicklungsbewertung (Review)
- Transfer der Entwicklungsaktivitäten (Design Transfer); nicht abgebildet
- Entwicklungsänderungen (Design Changes); nicht abgebildet
31
AAMI (2007): Section IV. Design Controls, S.69

Auswirkungen und Umsetzung in der Produktrealisierung
45
Abbildung 9: Wasserfall Modell, Schlüsselelemente des Design Control Prozess (Quelle: Health Canada)
Ein weiteres Modell, das den Entwicklungsprozess anschaulich darzustellt, ist das
sogenannte „V-Modell“ der Entwicklung. Es ist in seinen Elementen identisch mit dem
Wasserfallmodell (siehe Abbildung 10: V Modell der Entwicklung). Es folgt ebenfalls dem
sequentiellen Entwicklungsprozess.

Auswirkungen und Umsetzung in der Produktrealisierung
46
Bestimmungsgemäßer Gebrauch
STAGE 1
Entwicklungseingaben
STAGE 2
Technische Spezifikation
Verifikations Dokumente
STAGE 4
Validierungs Dokumente
STAGE 4
Legende: TRM - Trace Matrix RM – Risikomanagement HFE - Human Factors engineering
Verifizierung
Validierung
TRACE
MATR
IX
HFE &
RM
TRACE MATRIX
HFE &
RM
Bewertung
TRACE MATRIX TR
ACE M
ATRIX
HFE &
RM
HFE &
RM
Bewertung
Bewertung
Bewertung
Bewertung
Abbildung 10: V Modell der Entwicklung
Was beide Modelle nicht darstellen, sind die weiteren Anforderungen des 21 CFR Part
820.30. Dies sind im Einzelnen:
- dass die Entwicklung geplant werden muss 820. 30(b),
- der eigentliche Entwicklungsprozess und
- die Dokumentation des Entwicklungsprozesses in einer Entwicklungsakte, dem
Design History File (DHF) 820.30(j)und
- wie mit Entwicklungsänderungen umzugehen ist 820.30(i).
6.3.2 Entwicklungsplanung
Die Anforderungen zur Planung einer Entwicklung sind im Abschnitt 820.30(b) des QSR
beschrieben (Tabelle 6.8).

Auswirkungen und Umsetzung in der Produktrealisierung
47
Tabelle 6.8: Anforderungen an die Entwicklungsplanung
820.30(b)
(b)Design and development planning. Each manufacturer shall establish and maintain plans that describe or reference the design and development activities and define responsibility for implementation. The plans shall identify and describe the interfaces with different groups or activities that provide, or result in, input to the design and development process. The plans shall be reviewed, updated, and approved as design and development evolves.
(b)Entwicklungsplanung. Jeder Hersteller muss Pläne festlegen und aufrecht-erhalten, in denen die Entwicklungsaktivitäten beschrieben oder referenziert sind und die Verantwortung für deren Implementierung definiert ist. Die Pläne müssen die Schnittstellen mit verschiedenen Gruppen oder Aktivitäten identifizieren und beschreiben, die als Grundlagen für den Entwicklungs-prozess verwendet werden oder aus denen diese Grundlagen hervorgehen. Die Pläne müssen im Laufe der Entwicklung geprüft, aktualisiert und genehmigt werden.32
Im 820.30(b) „Design and Development Planning“ fordern die QSR, dass Design- und
Entwicklungspläne welche die Entwicklungsaktivitäten beschreiben, erstellt, erhalten und
dokumentiert werden müssen. Der Plan muss im Laufe des Entwicklungs-projektes auf den
neuesten Stand gebracht werden und ist zu überprüfen und frei-zugeben. Der Plan sollte vor
dem Beginn der eigentlichen Entwicklungstätigkeit erstellt werden und alle Funktionen und
Gruppen, die zur Umsetzung des Planes beitragen, sowie deren Schnittstellen definieren.
Der Plan soll der Komplexität des zu entwickelnden Produktes Rechnung tragen und kann
aus einem oder mehreren Dokumenten bestehen (z.B. Softwareentwicklung und
Hardwareentwicklung). Der Plan soll alle Verfahrensanweisungen referenzieren, die für den
Entwicklungsprozess relevant sind. Die FDA erwartet, dass die folgenden Elemente im
Entwicklungsplan gezeigt werden:
- Aufgaben und Verantwortlichkeiten
- Mindestanzahl der Designbewertungen (pro Entwicklungsphase)
- Standartverfahren
- Qualitätsverfahren welche zur Anwendung kommen und die Verifikationsmethoden
so diese bekannt sind.
- Methoden zur Erstellung von Dokumenten und Aufzeichnungen
- Ressourcenbedarf
32
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(b)

Auswirkungen und Umsetzung in der Produktrealisierung
48
- Risikobewertungsmethoden
- Zeitplan33
Dem Entwicklungsplan kann enorme regulatorische Bedeutung zukommen, da mit Freigabe
dieses Dokuments der Start für ein Entwicklungsprojekt und somit der Start für die
Anwendung der Entwicklungslenkung dokumentiert werden kann. Weiterhin dokumentiert
er die angewendeten Verfahren und Methoden. Bei dem Entwicklungs-plan handelt sich um
ein dynamisches Dokument, da er dem Fortschreiten des Entwicklungsprojektes angepasst
werden muss. Der Entwicklungsplan ist hilfreich bei internen Bewertungen, Audits und
Inspektionen, da er einen guten Überblick über den Status des Projektes gibt und die
Besonderheiten des Projektes definiert.
Es soll an dieser Stelle auf zwei essentielle Einflussfaktoren hingewiesen werden, die
entscheidenden Einfluss auf den erfolgreichen Abschluss eines Entwicklungsprojektes haben,
und die im Design- und Entwicklungsplan adressiert werden können. Zum einen sollte im
Entwicklungsplan eine klare Beschreibung des Projektes zu finden sein. Erfolgreich Projekte
sind dadurch gekennzeichnet, dass der Auftrag präzise formuliert ist, der zeitliche Rahmen
definiert, das Ziel beschrieben ist und die benötigten Ressourcen adressiert sind. Je besser
ein Projekt definiert ist umso mehr Sicherheit gibt es bei der Planung der
Entwicklungsaktivitäten und dem Einsatz der Ressourcen. Zum anderen kann über den
Entwicklungsplan das Zusammenwirken der involvierten Abteilungen und die
Kommunikationsstruktur im Projekt klar definiert werden. Der Design und Entwicklungsplan
kann daher als Werkzeug benutzt werden, um das Projekt auf eine solide Grundlage zu
stellen.34
Es gibt keine regulatorische Anforderung, welche Abteilung oder Funktion im Unternehmen
den Design und Entwicklungsplan erstellen soll. Es ist lediglich gefordert, dass das Dokument
geprüft und freigegeben werden muss. Es ist hilfreich, wenn die Projektleitung an der
Erstellung und den Änderungen des Entwicklungsplans beteiligt ist.
33
Vgl. AAMI (2007): Section IV. Design Controls, S.71 34
Vgl. AAMI (2007): Section IV. Design Controls, S.72

Auswirkungen und Umsetzung in der Produktrealisierung
49
6.3.3 Entwicklungseingaben
Im 21 CFR Part 820.30 (c) (Tabelle 6.9) werden die Anforderungen an den „Design Input“
bzw. die Entwicklungseingaben beschrieben. Die Entwicklungseingaben sind die Produkt-
spezifischen Übersetzungen der „User Needs“ bzw. des „Intended Use“.
Tabelle 6.9: Anforderungen an die Entwicklungseingaben
820.30(c)
(c)Design input. Each manufacturer shall establish and maintain procedures to ensure that the design requirements relating to a device are appropriate and address the intended use of the device, including the needs of the user and patient. The procedures shall include a mechanism for addressing incomplete, ambiguous, or conflicting requirements. The design input requirements shall be documented and shall be reviewed and approved by a designated individual(s). The approval, including the date and signature of the individual(s) approving the requirements, shall be documented.
(c) Entwicklungseingabe. Jeder Hersteller muss Verfahren entwickeln und aufrechterhalten, um sicherzustellen, dass die Auslegungsanforderungen für ein Medizinprodukt angemessen sind und der beabsichtigten Zweckbestimmung des Produkts entsprechen, einschließlich der Bedürfnisse des Anwenders und des Patienten. Zu diesen Prozeduren gehört ein Mechanismus für den Umgang mit unvollständigen, zweideutigen oder widersprüchlichen Anforderungen. Die Anforderungen der Entwicklungseingabe müssen dokumentiert und von einer oder mehreren ernannten Personen geprüft und bestätigt werden. Die Genehmigung einschließlich des Datums und der Unterschrift der die Anforderungen genehmigende(n) Person(en) müssen dokumentiert werden35
Die Entwicklungseingaben müssen den „Intended Use“ (dt. bestimmungsgemäßen
Gebrauch) und die „User Needs“ (dt. Nutzeranforderungen) vollständig erfüllen und auf den
bestimmungsgemäßen Gebrauch bzw. die Nutzeranforderungen rückführbar sein. Nur wenn
die Entwicklungseingaben vollständig und korrekt sind, ist man in die Lage versetzt, ein
sicheres und wirksames Kombinationsprodukt zu entwickeln, d.h. seinen
bestimmungsgemäßen Gebrauch und alle weiteren, von Behörden und Gesetzen
geforderten Eigenschaften zu erfüllen. Es ist daher nur folgerichtig, dass die FDA die
Vollständigkeit und Korrektheit der Entwicklungseingaben überprüft.
In der Design-Verifikation werden die Resultate des Design und Entwicklungsprozess und
damit das eigentliche Design geprüft. Die Design-Verifikation muss die Entwicklungseingaben
35
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(c)

Auswirkungen und Umsetzung in der Produktrealisierung
50
abdecken und die einzelnen Tests müssen auf die Entwicklungseingaben rückführbar sein. Es
ist daher notwendig, dass die Entwicklungsvorgaben eindeutig (nicht missverständlich) und
quantifizierbar (prüfbar) sind. Selbstverständlich müssen die Entwicklungseingaben
dokumentiert werden. Häufig sind die Entwicklungseingaben in einem oder mehreren
Dokumenten erfasst. Daher sollte jede einzelne Eingabe ein eindeutiges
Identifikationsmerkmal erhalten, um die Rückführbarkeit zu gewährleisten.
Änderungen an den Entwicklungseingaben müssen gemäß 21 CFR Part 820.30(i) (s. Kapitel
7.3.8) über eine formale Änderungskontrolle vorgenommen werden. Hierbei muss überprüft
werden, welche anderen Dokumente (Inhalte) von der Änderung betroffen sind und ggf.
auch geändert werden müssen. Alle betroffenen Parteien und Funktionen müssen Informiert
werden und ggf. ihre Bewertung abgeben. Dies ist notwendig, um sicherzustellen, dass kein
nachteiliger Effekt durch die Änderung entsteht.
Besondere Aufmerksamkeit muss dem Transferprozess der Nutzer-Anforderungen in die
Entwicklungseingaben gewidmet werden. Zunächst gibt es ein Anforderungsprofil mit
einzelnen Anforderungen aus dem Unternehmen, es gibt aber noch weitere Quellen für
Entwicklungseingaben:
- Ergebnisse der Marktbeobachtung
o Trendanalysen
o Beschwerden aus dem Feld
o Beobachtung von ähnlichen Produkten auch von Mitbewerbern
- Erfahrungen des Unternehmens mit ähnlichen Produken
- Studien zur Gebrauchstauglichkeit (Human Factors Engineering)
- Ergebnisse aus dem Risikomanagement
Besonderes Gewicht kommt während des Transferprozess dem „Human Factos
Engineering“- und dem Risikomanagementprozess zu. „Human Factors Engineering“ gemäß
DIN EN ISO 62366 behandelt die Gebrauchstauglichkeit. Sie ist bei allen Produkten mit einem
User Interface (dt. Nutzer-Schnittstelle) eine wichtige Eingabe für die Entwicklung und
ergänzt das Risiko Management gemäß DIN EN ISO 14971 als Übersetzungswerkzeug für die
Entwicklungsvorgaben. Daher sind definierte Prozesse für „Human Factors Engineering“ und
Risikomanagement wichtige Werkzeuge, um den Entwicklungsprozess zu strukturieren.

Auswirkungen und Umsetzung in der Produktrealisierung
51
Dabei definiert HFE z.B. mit einer Analyse der Gebrauchsszenarien mögliche Einflüsse auf das
Produkt, welche wiederum Eingaben in den Risikomanagement Prozess darstellen. Wird
dieser Prozess für jedes Designelement durchlaufen, ist die Rückführbarkeit der
Entwicklungseingaben gewährleistet, und es entstehen keine Lücken bei der Umsetzung
dieser Eingaben.36
Die Dokumentation der Rückführbarkeit wird häufig über eine sogenannte „Trace Matrix“
umgesetzt. Eine solche „Trace Matrix“ kann derart aufgebaut sein, dass sie sowohl vorwärts
(von Entwicklungseingaben über Entwicklungsergebnisse und Verifikation zu Validierung) als
auch rückwärts (von Validierung über Verifikation und Entwicklungsergebnisse zu den
Entwicklungseingaben) funktioniert und dadurch eine gute Übersicht gibt
6.3.4 Entwicklungsergebnisse
Im 21 CFR Part 820.30 (d) werden die Anforderungen an den „Design Output“ bzw. die
Entwicklungsergebnisse beschrieben (Tabelle 6.10).
Tabelle 6.10: Anforderungen an die Entwicklungseingaben
820.30(d)
(d)Design output. Each manufacturer shall establish and maintain procedures for defining and documenting design output in terms that allow an adequate evaluation of conformance to design input requirements. Design output procedures shall contain or make reference to acceptance criteria and shall ensure that those design outputs that are essential for the proper functioning of the device are identified. Design output shall be documented, reviewed, and approved before release. The approval, including the date and signature of the individual(s) approving the output, shall be documented
(d)Entwicklungsergebnis.Jeder Hersteller muss Verfahren zur Definition und Dokumentation des Entwicklungsergebnisses festlegen und aufrechterhalten, so dass auf angemessene Weise beurteilt werden kann, ob die Anforderungen der Entwicklungsgrundlage erfüllt werden. Verfahren für Entwicklungs-ergebnisse müssen Akzeptanzkriterien enthalten oder angeben und sicherstellen, dass die für die einwandfreie Funktion des Medizinprodukts wesentlichen Entwicklungsergebnisse identifiziert werden. Entwicklungsergebnisse müssen vor der Freigabe dokumentiert, geprüft und genehmigt werden. Die Genehmigung einschließlich Datum und Unterschrift der Person oder Personen, die das Ergebnis genehmigen, müssen dokumentiert werden.37
36
Vgl. AAMI (2007): Section IV. Design Controls, S.75 37
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(d)

Auswirkungen und Umsetzung in der Produktrealisierung
52
Die Entwicklungsergebnisse sind definiert als das Resultat der Entwicklungstätigkeit in den
Entwicklungsphasen und als Gesamtergebnis am Ende der Entwicklungstätigkeit. Die
Organisation muss dokumentierte Verfahren einführen und erhalten, die festlegen, wie und
in welcher Form Entwicklungsergebnisse definiert und dokumentiert werden. Wie alle
anderen Dokumente müssen Entwicklungsergebnisse bewertet und freigegeben werden. Die
für den sicheren und ordnungsgemäßen Gebrauch essentiellen Entwicklungsergebnisse
müssen gekennzeichnet und Akzeptanzkriterien definiert sein. Ziel ist es, zu beurteilen, ob
die Entwicklungsergebnisse die Entwicklungseingaben erfüllen. Entwicklungsergebnisse sind
nicht nur Zeichnungen, sondern auch alle Dokumente, die während der
Entwicklungstätigkeit erstellt werden, wie z.B.:
- Spezifikationen
- Schaltpläne
- Layouts
- Stücklisten
- Verifizierungs- und Validierungs-Pläne, sowie deren zugehörigen Berichte
- Risikoanalysen
- Softwarespezifikationen 38
Die Dokumente der Entwicklungsergebnisse sind ein wichtiger Bestandteil der Produktakte
(engl. Device Master Record – DMR). Dies wird im Kapitel 7.3.9 „Design Transfer gemäß
820.30 (h)“ separat behandelt.
Ein wichtiges Kriterium ist, ob alle wesentlichen Entwicklungsergebnisse in ausreichender
Tiefe behandelt wurden. Dafür ist es notwendig, diese zu definieren. Hierfür leistet der
Risikomanagement-Prozess wertvolle Dienste. Das Risikomanagement führt zu
nachvollziehbaren quantitativen Aussagen, und erzeugt außerdem durch seine Struktur
rückführbare Ergebnisse. Dies hat mehrere Vorteile:
- Durch die Rückführbarkeit („Tracebility and Identifcation“) wird sichergestellt, dass
kein Entwicklungsmerkmal vergessen wird.
38
Vgl. AAMI (2007): Section IV. Design Controls, S.76

Auswirkungen und Umsetzung in der Produktrealisierung
53
- Die Entscheidungen, welche zu einem bestimmten Design geführt haben sind
nachvollziehbar.
- Änderungen am Design können besser bewertet und begründet werden.
- Der gesamte Entwicklungsprozess ist dokumentiert.
Insgesamt trägt der Risikomanagement-Prozess stark zur Nachvollziehbarkeit und
Rückführbarkeit der Entwicklung bei. Diese Vorteile sind aber nicht umsonst zu haben.
Risikomanagement ist je nach Komplexität des Produktes ein aufwendiger Prozess, der
ebenfalls eingeführt und erhalten werden muss. Dennoch lohnt sich der Aufwand, da er zum
nachhaltigen Wissensmanagement im Unternehmen beiträgt.
6.3.5 Entwicklungsverifizierung
Die Entwicklungsverifizierung soll sicherstellen, dass die Entwicklungsergebnisse die
Anforderungen aus den Entwicklungseingaben erfüllen. Dies ist grafisch in den beiden
Modellen Abbildung 9 bzw. Abbildung 10 dargestellt. Die Anforderungen an die
Entwicklungsverifizierung sind im 21 CFR Part 820.30(f) formuliert (Tabelle 6.11).
Tabelle 6.11: Anforderungen an die Entwicklungsverifizierung
820.30(f)
(f) Design verification. Each manufacturer shall establish and maintain procedures for verifying the device design. Design verification shall confirm that the design output meets the design input requirements. The results of the design verification, including identification of the design, method(s), the date, and the individual(s) performing the verification, shall be documented in the DHF
(f)Entwicklungsverifizierung. Jeder Hersteller muss Verfahren zur Verifizierung der Produktauslegung festlegen und aufrechterhalten. Die Entwicklungsverifizierung muss bestätigen, dass das Entwicklungsergebnis die Anforderungen der Entwicklungseingabe erfüllt. Die Ergebnisse der Entwicklungsverifizierung einschließlich der Identifikation der Entwicklung, der Methoden, des Datums und der Person oder für Personen, die die Verifizierung durchführen, müssen in der Produktentwicklungsakte dokumentiert werden.39
Der Nachweis der Erfüllung soll durch Untersuchungen den objektiven Beweis erbringen,
dass die spezifizierten Anforderungen umgesetzt wurden.40 Der 21 CFR Part 820.30(f)
fordert, dass die Organisation geeignete dokumentierte Verfahren einführt und erhält, um
39
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(f) 40
Vgl. AAMI (2007): Section IV. Design Controls, S.79

Auswirkungen und Umsetzung in der Produktrealisierung
54
den Verifizierungsprozess sicherzustellen. Die Verifizierungs-dokumentation ist Bestandteil
der Entwicklungsakte (DHF). Hierbei müssen die überprüften Designmerkmale, die
Methoden, die Personen, Zeitpunkte und natürlich die Resultate dokumentiert werden. Die
Verifizierung ist häufig ein komplexer Prozess, der eine umfangreiche Dokumentation nach
sich zieht. Üblicherweise wird zuerst ein Verifikationsplan erstellt. Diese Planung umfasst
unter anderem:
- Musterzug
- Testmethoden und deren Validierung
- benutzte Testgeräte und deren Qualifizierung
- Anforderungen an das Testpersonal
- Zeitplanung
- Umgang mit Abweichungen und Fehlern
- Dokumentation
Nach der Planung sind die entsprechenden Voraussetzungen zu schaffen und all dies
umzusetzen, was im Verifikationsplan gefordert wird. Sind die Vorrausetzungen wie
gefordert geschaffen und die entsprechenden Verifikationsmuster vorhanden, werden die
einzelnen Verifizierungstests durchgeführt und dokumentiert.
Selbst bei sorgfältigster Planung und Entwicklung kann nicht verhindert werden, dass Tests
nicht so funktionieren wie geplant, oder dass alle Verifizierungstests bestanden werden. Hat
ein Entwicklungsergebnis die entsprechende Entwicklungseingabe nicht erfüllt, muss
folgerichtig der Entwicklungsprozess für dieses Entwicklungsmerkmal und alle damit
verbundenen Entwicklungsmerkmale erneut durchlaufen werden. Nach erfolgter Änderung
des Designs (Änderung der Spezifikation) muss die Wirksamkeit der Änderung durch
Verifizierungstests erneut nachgewiesen werden. Wenn alle Verifizierungstests erfolgreich
bestanden wurden, d.h. alle Entwicklungseingaben werden durch die entsprechenden
Entwicklungsergebnisse erfüllt, wird das Gesamtergebnis der Verifizierung in einem
zusammenfassenden Verifikationsbericht dokumentiert. Auch hier ist die Rückführbarkeit
der Verifizierungsergebnisse auf die Entwicklungseingaben zu demonstrieren. Dies kann in
einer übergreifenden „Trace Matrix“ oder in einer kleineren „Trace Matrix“ im
zusammenfassenden Verifizierungsbericht geschehen, welche die Rückführbarkeit von
Entwicklungseingabe bis Verifikationsergebnis darstellt (Abbildung 11: Verifikationssequenz)

Auswirkungen und Umsetzung in der Produktrealisierung
55
Entwicklungs-ergebnis
VerifikationstestVerifikations-
ergebnis
Entwicklungs-eingabe
Abbildung 11: Verifikationssequenz
6.3.6 Entwicklungsvalidierung
Die Entwicklungsvalidierung soll sicherstellen, dass das Produkt die Produktspezifikationen
für die sogenannten Nutzeranforderungen und die Zweckbestimmung des Produktes
erfüllen, d.h. das Produkt kann vom potenziellen Anwender bedient werden. Die
Anforderungen an die Entwicklungsvalidierung werden im 21 CFR Part 820.30(g) formuliert
(Tabelle 6.12)
Tabelle 6.12: Anforderungen an die Entwicklungsvalidierung
820.30(g)
(g) Design validation. Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation, including identification of the design, method(s), the date, and the individual(s) performing the validation, shall be documented in the DHF.
(g) Entwicklungsvalidierung. Jeder Hersteller muss Verfahren zur Validierung der Produktentwicklung festlegen und aufrechterhalten. Die Entwicklungsvalidierung muss unter festgelegten Betriebsbedingungen an den ursprünglichen Produktionseinheiten, Losen oder Chargen oder entsprechenden Elementen durchgeführt werden. Die Entwicklungsvalidierung muss sicherstellen, dass Medizinprodukte die definierten Bedürfnisse des Anwenders und die beabsichtigte Zweckbestimmung erfüllen, und muss einen Test von Produktionseinheiten unter tatsächlichen oder simulierten

Auswirkungen und Umsetzung in der Produktrealisierung
56
Anwendungsbedingungen umfassen. Die Entwicklungsvalidierung muss gegebenenfalls eine Softwarevalidierung und eine Risikoanalyse umfassen. Die Ergebnisse der Entwicklungsvalidierung einschließlich der Identifikation der Entwicklung, der Methode(n), des Datums und Person oder Personen, die die Validierung durchführen, müssen in der Produktentwicklungsakte dokumentiert werden.41
Das Unternehmen muss für die Entwicklungsvalidierung dokumentierte Verfahren einführen
und aufrechterhalten. Die Validierungsdokumente sind Bestandteil der
Entwicklungsakte(DHF).
Die Entwicklungsvalidierung ist kein trivialer Prozess. Zunächst sei darauf hingewiesen, dass
die Entwicklungsverifizierung kein Ersatz für die Entwicklungsvalidierung ist. Für die
Entwicklungsvalidierung sind die folgenden Bedingungen einzuhalten:
- Für die Validierung müssen Produkte verwendet werden, die aus validierten
Herstellprozessen stammen, und zumindest äquivalent zum späteren Serienprodukt
sind.
- Die Validierungstests müssen unter den gleichen Bedingungen wie sie der Patient
oder der Anwender beim bestimmungsgemäßen Gebrauch vorfindet, durchgeführt
werden. Ist eine „Real Life“ Situation nicht verfügbar, können diese Bedingungen
auch simuliert werden.
- Die Entwicklungsvalidierung muss, wo notwendig eine Softwarevalidierung und,
wenn angemessen, eine Risikoanalyse beinhalten.42
Wie die Verifizierung muss die Validierung geplant durchgeführt und dokumentiert werden.
Es empfiehlt sich auch hier, die folgende Dokumentensequenz (Abbildung 12:
Dokumentensequenz Validierung) einzuhalten:
41
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(g) 42
Vgl. AAMI (2007): Section IV. Design Controls, S.79 - 83

Auswirkungen und Umsetzung in der Produktrealisierung
57
Validierungstest
ValidierungsberichtZusammen-fassender
Validierungsbericht
Validierungsplan
Abbildung 12: Dokumentensequenz Validierung
Die Entwicklungsvalidierung ist einer der letzten Schritte in der Entwicklung eines Produktes.
Wenn die Entwicklungsvalidierung nicht bestanden wird, hat das massive Auswirkungen auf
das Entwicklungsprojekt. Das Projekt muss erneut zurück in die Entwicklung mit der
Konsequenz, dass Zeitlinien neu geplant werden müssen und Markteinführungen nicht wie
geplant erfolgen können. Die Entwicklungsvalidierung und alle zugehörigen Prozesse müssen
sorgfältig geplant werden, um den Erfolg weitestgehend sicherzustellen.
Bei Einreichungen an Behörden sind die Validierungsergebnisse und die Prozesse, durch
welche sie erzeugt wurden, von besonderem Interesse. Die Validierungs-ergebnisse
demonstrieren, ob die wesentliche Produktmerkmale und Patienten-bedürfnisse erfüllt
werden. Bei Inspektionen durch Behörden sind speziell die oben genannten Bedingungen für
eine Validierung von Bedeutung und bieten viele Ansatzpunkte zur Überprüfung. Die
Entwicklungsvalidierung kann zwar mit äquivalenten Teilen durchgeführt werden, jedoch
muss die Äquivalenz nachgewiesen werden.
Wie schon erwähnt ist die Entwicklungsvalidierung einer der letzten Schritte im
Entwicklungsprojekt. Fehler die am Anfang des Projektes gemacht wurden machen sich zu
diesem Zeitpunkt besonders schmerzhaft bemerkbar. Zeit, die in die Entwicklungsplanung
investiert wurde, zahlt sich dann in dieser Phase aus.
6.3.7 Entwicklungsbewertung
Formale Entwicklungsbewertungen sind zu geeigneten Zeitpunkten durchzuführen, wie dies
im Wasserfallmodell (Abbildung 9) anschaulich dargestellt wird. Die Forderungen des QSR
zu Entwicklungsbewertungen sind im Abschnitt 820.30 (e) (Tabelle 6.13) formuliert.

Auswirkungen und Umsetzung in der Produktrealisierung
58
Tabelle 6.13: Anforderungen an die Entwicklungsbewertung
820.30(e)
(e) Design review. Each manufacturer shall establish and maintain procedures to ensure that formal documented reviews of the design results are planned and conducted at appropriate stages of the device's design development. The procedures shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed. The results of a design review, including identification of the design, the date, and the individual(s) performing the review, shall be documented in the design history file (the DHF)
(e) Entwicklungsbewertung. Jeder Hersteller muss Verfahren festlegen und erhalten, die sicherstellen, dass formale dokumentierte Überprüfungen der Entwicklungsergebnisse geplant und in geeigneten Phasen der Entwicklung des Medizinprodukts durchgeführt werden. Die Verfahren müssen sicherstellen, dass an jeder Entwicklungsbewertung Vertreter aller von der überprüften Entwicklungsphase betroffenen Funktionen und eine Person oder Personen, die keine direkte Verantwortung für die geprüfte Entwicklungsphase haben sowie andere erforderliche Spezialisten teilnehmen. Die Ergebnisse einer Entwicklungsbewertung einschließlich der Identifikation der Bewertung, des Datums und der Person oder Personen, die die Überprüfung durchführen, müssen in der Produktentwicklungsakte dokumentiert werden.43
Die Organisation hat die Aufgabe, dokumentierte Verfahren zur Durchführung und
Dokumentation von Entwicklungsbewertungen einzuführen und aufrechtzuerhalten. Es wird
weiter gefordert, die Bewertungen zu geeigneten Zeitpunkten durchzuführen und alle
notwendigen Funktionen zu beteiligen. Außerdem soll eine unabhängige Funktion
(„Independent Reviewer“, dt. unabhängiger Prüfer) an diesen Bewertungen beteiligt sein.
Entwicklungsbewertungen sollten entsprechend geplant und gemäß der Planung
durchgeführt werden. Kann eine Entwicklungsbewertung nicht gemäß der Planung
durchgeführt werden, ist es ratsam, eine Begründung zu geben, warum der Plan nicht
eingehalten wurde.
Der 21 CFR Part 820.30(e) spricht eindeutig in der Mehrzahl von Entwicklungsbewertungen
und fordert, diese zu angemessenen Zeitpunkten (engl. Appropriate Stages) im Verlauf einer
43
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(e)

Auswirkungen und Umsetzung in der Produktrealisierung
59
Entwicklung durchzuführen. Dies wird auch eindeutig in (Abbildung 9: Wasserfallmodel der
Entwicklung), welches aus dem FDA Guidance-Dokument „Design Control Guidance for
Medical Device Manufacturers“44 entnommen wurde, so dargestellt. Jedoch ist der QSR
hinsichtlich der Definition der Anzahl der Entwicklungsbewertungen unbestimmt. Es ist
angeraten, am Ende einer Entwicklungsphase eine solche Bewertung durchzuführen, um
diese Phase abzuschließen und in die nächste überzugehen.
Entwicklungsbewertungen sind nicht nur eine Forderung des 21 CFR 820. 30(e), sondern
werden auch im sogenannten NPI (New Product Introduction) Prozess, wie er in der
Automobilbranche praktiziert wird, gefordert. Dort werden Entwicklungsbewertungen als
„Phase Gate Reviews“ bezeichnet.
In einem Entwicklungsprojekt findet ein reger Austausch zwischen den einzelnen Funktionen
statt, da ein Produkt von einzelnen voneinander unabhängigen Funktionen nicht erfolgreich
entwickelt werden kann. Daher sind gemeinsame Bewertungen von hohem Wert für das
Entwicklungsprojekt, da bei solchen Meetings der Stand der Entwicklung abgeglichen und
bewertet werden kann. Dies geschieht, um sicherzustellen, dass die Entwicklungseingaben
vollständig und korrekt sind, oder die Entwicklungsergebnisse die Entwicklungseingaben
erfüllen. Die Forderung nach formellen Entwicklungsbewertungen ist realistisch betrachtet,
daher nicht für das Qualitätssystem essentiell, sondern für das Entwicklungsprojekt von
höchster Wichtigkeit. Die gängige Interpretation von Entwicklungsbewertungen ist:
„A documented, comprehensive, systematic examination of the design to evaluate the
adequacy of the design requirements, to evaluate the capability of the design to meet
these requirements and to identify problems”45
„Eine dokumentierte, umfassende, systematische Untersuchung der Entwicklung zur
Bewertung der Angemessenheit der Entwicklungseingaben, der Fähigkeit der
Entwicklung diese Eingaben zu erfüllen und Probleme zu identifizieren“46
Die Forderung, dass die Bewertung umfassend und systematisch zu erfolgen hat, bedingt,
dass alle an der Entwicklung beteiligten Funktionen, wie z.B. Konstruktion,
44
FDA, o.V. (1997): Design Control Guidance for Medical Device Manufacturers 45
AAMI (2007): Section IV. Design Controls, S.77 46
Übersetzung des Verfassers

Auswirkungen und Umsetzung in der Produktrealisierung
60
Risikomanagement, „Human Factors Engineering“ an der Bewertung teilhaben. Aus dieser
Forderung kann ebenfalls geschlossen werden, dass ein reines Prüfen des Vorliegens von
Dokumenten nicht ausreichend ist.
Entwicklungsbewertungen passen perfekt in den PDCA-Zyklus / Deming-Zyklus47, da es hier
zu einem Abgleich mit dem Plan kommt. Gegebenenfalls werden zusätzliche erforderliche
Maßnahmen definiert und durchgeführt. Spätestens bei der nächsten Bewertung werden
diese Maßnahmen auf Erfolg geprüft. Daher sind Entwicklungsbewertungen auch der
geeignete Zeitpunkt, um die kontinuierliche Verbesserung voranzutreiben, da hier Prozesse
und Schnittstellen bewertet werden, die nicht nur von qualitätstechnischem Interesse,
sondern auch für wirtschaftliche Geschäftsprozesse von Belang sind.
6.3.8 Lenkung von Entwicklungsänderungen
In jedem Entwicklungsprojekt muss mit Entwicklungsänderungen umgegangen werden. Dies
kann je nach Status des Projektes (vor oder nach der Design Verifizierung) und Art der
Änderung (Korrektur von Rechtschreibfehlern vs. Änderung der grundlegenden
Anforderungen an das Kombinationsprodukt) einen unterschiedlich hohen formellen
Aufwand bedeuten. Die Anforderungen des QSR an Entwicklungsänderungen sind im
Abschnitt 820.30(i) dargelegt (Tabelle 6.14).
Tabelle 6.14: Anforderungen an die Lenkung von Entwicklungsänderungen
820.30(i) (i)Design changes. Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.
(i) Entwicklungslenkung. Jeder Hersteller muss Verfahren zur Identifikation, Dokumentation, Validierung und gegebenenfalls zur Verifizierung, Überprüfung und Genehmigung von Entwicklungsänderungen vor ihrer Implementierung festlegen und aufrechterhalten.48
47
Vgl. ISO, o.V. (2012): ISO 9001:2008, Kap.: 0.2 Anm.; Vgl. Wikipedia, o.V.(o.J.): Deming Kreis 48
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(i)

Auswirkungen und Umsetzung in der Produktrealisierung
61
Für die Lenkung von Entwicklungsänderungen hat die Organisation dokumentierte Verfahren
einzuführen und aufrecht zu erhalten. Die Anwendbarkeit dieses Verfahrens erstreckt sich
vom Beginn des Entwicklungsprozesses über die Entwicklungsergebnisse bis hin zum
Transfer der Entwicklungstätigkeit in die Produktion. Alle Änderungen müssen dokumentiert
und bewertet werden. Jedoch ist nicht immer der gleiche Aufwand zu betreiben. Der
Aufwand sollte im direkten Zusammenhang zum Einfluss der Änderung auf die Entwicklung
bzw. des Produktes stehen. Besondere Aufmerksamkeit muss Änderungen an bereits
eingereichte Produkte gewidmet werden. Hier ist zu prüfen, ob eine Änderung des 510(k)
oder des PMA bei der FDA einzureichen ist.
Wenn eine Änderung eine Spezifikation betrifft, muss die Änderung der Spezifikation durch
den Kreis der Personen / Funktionen freigegeben werden, welche die Spezifikation
ursprünglich freigegeben haben. Wird ein Entwicklungseingaben-dokument geändert muss
geprüft werden, ob eine erneute Verifizierung bzw. Validierung durchgeführt werden muss.
Entwicklungsänderungen könne prinzipiell mit einem einzigen Prozess über den gesamten
Entwicklungszeitraum gelenkt werden. Hierbei sollte beachtet werden, zu welchem
Zeitpunkt im Entwicklungsprozess die Änderung durchgeführt wird oder welchen Umfang die
Änderung besitzt.49
EDV-Systeme für die Bearbeitung und Lenkung von Änderungen können, wenn sie auf den
Prozess abgestimmt sind, eine große Hilfe sein. Mit einem EDV-System kann die
Nachverfolgung der Änderungsbewertung und die Sicherstellung, dass keine Arbeitsschritte
fehlen vereinfacht und automatisiert werden.
Dem Änderungsprozess, sowie den durchzuführenden Änderungen sollte einiges an
Aufmerksamkeit gewidmet werden, da Änderungen bei Behördeninspektionen immer ein
Thema sind. An der Lenkung von Änderungen im realen Projekt lässt sich die Einhaltung von
verschiedenen Verfahren, wie z.B. Dokumentenkontrolle, Verifizierung, Freigaben, gut
prüfen.
49
Vgl. AAMI (2007): Section IV. Design Controls, S.84 - 86

Auswirkungen und Umsetzung in der Produktrealisierung
62
6.3.9 Transfer der Entwicklungsaktivität
Zu den letzten Schritten in einem Entwicklungsprojekt gehört der Transfer des entwickelten
Produktes in die Fertigung. Die Anforderungen an den Entwicklungstransfer aus Sicht des
QSR werden in dem Abschnitt 820.30(h) dargelegt (Tabelle 6.15).
Tabelle 6.15: Anforderungen an den Transfer der Entwicklungsaktivität
820.30(h)
(h) Design transfer. Each manufacturer shall establish and maintain procedures to ensure that the device design is correctly translated into production specifications.
(h) Entwicklungsumsetzung. Jeder Hersteller muss Verfahren festlegen und aufrechterhalten, um sicherzustellen, dass die Produktauslegung korrekt in die Produktspezifikationen umgesetzt wird50
Wie bei allen Schlüsselelementen der Entwicklungslenkung fordert der 21 CFR Part 820.30(h)
auch für den Transfer der Entwicklungstätigkeit dokumentierte Verfahren.
Der Transfer der Entwicklungstätigkeit beschreibt den Übergang des Entwicklungswissens in
Form von Dokumenten an die Produktion, um schlussendlich ein fertigbares Produkt zu
erhalten. Der DMR bildet die Basis für jede weitere Dokumentation von anderen Abteilungen
und Funktionen.
Die Einbindung der Produktion ist bereits in einer frühen Phase des Projektes sinnvoll, um
sicherzustellen, dass alle notwendigen Prozess vorhanden sind und alle notwendigen
Anpassungen durchgeführt wurden, um Kosten zu sparen und Verzögerungen zu vermeiden.
Durch die frühe Einbindung soll sichergestellt werden, dass die notwendigen Prozesse zum
Serienstart bekannt und validiert sind. Die Einbindung der Produktion kann auf
verschiedenste Weise erfolgen. Eine Möglichkeit besteht darin ein „Design Transfer Team“
zu bilden, in dem die relevanten Funktionen präsent sind und welches bereits bei der
Erarbeitung der Entwicklungseingaben auf Aspekte der Produktion eingeht. Die
Qualitätsplanung für die Produktion nimmt im Transfer der Entwicklungstätigkeit ihren
Anfang. Sie hat indirekt Einfluss auf die Fertigbarkeit des Produktes, sowie auf die
entstehenden Qualitätskosten in der Produktion, da ein Großteil der Qualitätsprobleme
bereits im Rahmen der Entwicklung verhindert werden kann.
50
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(h)

Auswirkungen und Umsetzung in der Produktrealisierung
63
Am Ende der Transferphase wird letztmalig eine Entwicklungsbewertung durchgeführt, um
sicherzustellen, dass alle Aspekte berücksichtigt wurden und die Fertigung formal
freigegeben werden kann.
Die Gesamtheit aller Dokument die zur Produktion des Kombinationsproduktes notwendig
ist, im sogenannten „Device Master Record“ zusammengefasst:
- Spezifikationen zur Kennzeichnung des Produktes
- Spezifikationen des Produktes (Baugruppen, Komponenten, Materialien usw.),
- Produktionsspezifikationen (Arbeitsanweisungen, Fertigungsanweisungen usw.)
- Installations- und Instandhaltungsspezifikationen
- Prüfspezifikationen (Prüfpläne, Prüfanweisungen usw.)
Der DMR ist das Ergebnis der Entwicklungstätigkeit. Mit der Freigabe des DMR sind die
Entwicklungsergebnisse abgeschlossen, und der DMR kann transferiert werden. Die
Freigabe des DMR markiert das Ende der Entwicklungstätigkeit und somit kann auch der DHF
fertiggestellt werden.51
6.3.10 Design History File (Entwicklungsakte)
Bei der Entwicklung eines Kombinationsproduktes, müssen die notwendigen Dokumente in
einer Entwicklungsakte abgelegt werden, um später darauf zurückgreifen zu können. Der
QSR beschreibt im Abschnitt 820.30(j), siehe Tabelle 6.16, die Anforderungen an die
Entwicklungsakte (Design History File = DHF).
Tabelle 6.16: Anforderungen an das Design History File (Entwicklungsakte)
820.30(j)
(j) Design history file. Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part.
(j) Produktentwicklungsakte. Jeder Hersteller muss eine Produktentwicklungsakte für jeden Medizinprodukttyp festlegen und aufrechterhalten. Die Produktentwicklungsakte muss die erforderlichen Aufzeichnungen enthalten oder angeben, um nachzuweisen, dass die Entwicklung entsprechend dem genehmigten Entwicklungsplan und den in diesem Teil aufgeführten Anforderungen entwickelt wurde.52
51
Vgl. AAMI (2007): Section IV. Design Controls, S.83 - 84 52
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.30(j)

Auswirkungen und Umsetzung in der Produktrealisierung
64
Der 21 CFR Part 820.30(j) fordert, dass für jedes Produkt eine Entwicklungsakte erstellt und
aufrechterhalten wird. Es gibt zwar keine Forderung nach Verfahren zur Erstellung und
Pflege eines DHF, dennoch ist es gängige Praxis ein solches Verfahren einzuführen und
aufrechtzuerhalten. Im DHF werden alle Dokumente, die während der Entwicklung
entstehen und die notwendig sind, um Einhaltung der gesetzlichen Forderungen
nachzuweisen, abgelegt. Die Struktur ist nicht vorgeschrieben. Es gibt verschiedenste
Umsetzungen, hier seien zwei genannt:
1. Im DHF wird die Struktur der Design Controls gemäß QSR nachgebildet
a. Entwicklungsplanung
b. Entwicklungseingaben
c. Entwicklungsergebnisse
d. Entwicklungsbewertungen
e. Entwicklungsverifizierung
f. Entwicklungsvalidierung
g. Entwicklungsänderungen
h. Entwicklungstransfer53
2. Im DHF wird die Umsetzung der Entwicklungslenkung im organisationsinternen
Entwicklungsprozess abgebildet.
Beide Methoden haben Vor- und Nachteile. Häufig sind jedoch die Unterschiede nicht groß.
Eine Rolle spielt der Inhalt der einzelnen Dokumente und ob ein „roter Faden“ in den
Dokumenten und den Dokumenten untereinander vorhanden ist. Hier sei noch mal auf die
„Trace Matrix“ verwiesen, die sich gerade in diesem Zusammenhang als äußerst nützlich
erweisen kann, da sie die Beziehung von Entwicklungseingabe-Entwicklungsergebnis-
Entwicklungsverifikation und Entwicklungsvalidierung darstellt. Ebenso wichtig wie die
„Trace Matrix“ ist der Entwicklungsplan, da er u.a. Zielstellung, Ablauf und
Qualitätsanforderungen des Projektes beschreibt.
53
Vgl. AAMI (2007): Section IV. Design controls, S.87

Auswirkungen und Umsetzung in der Produktrealisierung
65
6.3.11 Fazit zu den Anforderungen im Bereich Entwicklungslenkung
Die Änderungen für den Bereich Entwicklungslenkung sind die umfangreichsten, die für die
Umsetzung des „Streamlined Aproach“ notwendig sind. Es gilt einen der zentralen Prozesse
des 21 CFR Part 820 von Beginn an umzusetzen. Alle anderen Prozesse sind wenigstens im
Arzneimittel QMS in den Grundzügen vorhanden. Die „Design Controls“ jedoch sind für den
Arzneimittelbereich Neuland und folglich auch entsprechend komplex. Bei der Umsetzung
der Anforderungen sollte den Bereichen „Human Factors Engineering“ im Zusammenspiel
mit dem Risikomanagement und dem eigentlichen Entwicklungsprozess genügend Raum
gegeben werden. Ebenso ist die Rückführbarkeit von Ergebnissen und deren Dokumentation
über den ganzen Prozess geeignet, umzusetzen. Hier bietet sich eine „Trace Matrix“ an. Die
Erfahrung zeigt, dass dies ein Werkzeug ist, um sicherzustellen, dass alle
Produktanforderungen korrekt und vollständig umgesetzt wurden. Darüber hinaus dient es
auch dem Wissens-management für zukünftige Projekte. Der Entwicklungsprozess ist ein
kreativer Prozess, der durch Überregulierung leidet. Es ist daher wichtig, dass richtige Maß in
der Umsetzung zu finden, um Kreativität zu ermöglichen aber auch den regulatorischen
Anforderungen zu genügen.
6.4 Korrektur und Vorbeugungsmaßnahmen Die Anforderungen zu Korrektur und Vorbeugungsmaßnahmen werden im ICH Q10 3.2.2 und
im 21 CFR Part 820. 100 (a, b) beschrieben (Tabelle 6.17)
Tabelle 6.17: Anforderungen an Korrektur und Vorbeugemaßnahmen
ICH Q10 3.2.2 820.100
The pharmaceutical company should have a system for implementing corrective actions and preventive actions resulting from the investigation of complaints, product rejections, non-conformances, recalls, deviations, audits, regulatory inspections and findings, and trends from process performance and product quality monitoring. A structured approach to the investigation process should be used with the objective of
(a)Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for: (1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other

Auswirkungen und Umsetzung in der Produktrealisierung
66
determining the root cause. The level of effort, formality, and documentation of the investigation should be commensurate with the level of risk, in line with ICH Q9. CAPA methodology should result in product and process improvements and enhanced product and process understanding.
quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems; (2) Investigating the cause of nonconformities relating to product, processes, and the quality system; (3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems; (4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device; (5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems; (6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and (7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review. (b) All activities required under this section, and their results, shall be documented.
Das Arzneimittel Unternehmen sollte ein System haben zur Implementierung von Korrekturmaßnahmen und Vorbeugemaßnahmen als Ergebnis von Beschwerden Produktrückweisungen, Produktrückrufen, Abweichungen, Audits, Behördeninspektionen, und
a) Jeder Hersteller muss Verfahren zur Implementierung von Korrektur- und Vor-beugungsmaßnahmen festlegen und aufrechterhalten. Diese Verfahren müssen Anforderungen für Folgendes umfassen: (1) Analyseprozesse, Arbeitsgänge, Konzessionen,

Auswirkungen und Umsetzung in der Produktrealisierung
67
Beobachtungen und Trends von Prozessüberwachung und Produktqualitätsüberwachung . Ein strukturierter Ansatz für den Untersuchungsprozess sollte angewendet werden mit der Zielsetzung die Ursache zu bestimmen. Der Grad des Aufwands, der Formalität, der Dokumentation der Untersuchung sollte im Verhältnis stehen mit dem Grad des Risikos in Übereinstimmung mit dem ICH Q9. Die CAPA Methodik sollte in Produkt und Prozessverbesserungen sowie einem verbesserten Produkt und Prozessverständnis resultieren.54
Qualitätsauditberichte, Qualitätsaufzeichnungen, Serviceaufzeichnungen, Reklamationen, Retouren und andere Quellen von Qualitätsdaten zur Identifikation existierender und potenzieller Ursachen von fehlerhaften Produkten oder anderen Qualitätsproblemen. Gegebenenfalls müssen geeignete statistische Methoden angewandt werden, um wiederholt auftretende Qualitätsprobleme zu ermitteln. (2) Untersuchung der Ursache für die Fehlerhaftigkeit in Verbindung mit dem Produkt, den Prozessen und dem Qualitätssystem (3) Identifikation der erforderlichen Korrektur- und Vorbeugungsmaßnahme(n), um eine Wiederholung des Fehlers und andere Qualitätsprobleme zu vermeiden (4) Verifizierung oder Validierung der Korrektur- oder Vorbeugungsmaßnahme, um sicherzustellen, dass diese Maßnahme wirksam ist und sich nicht nachteilig auf das Endprodukt auswirkt (5) Implementierung und Aufzeichnung von erforderlichen Methoden- und Verfahrensänderungen, um identifizierte Qualitätsprobleme zu korrigieren und zu vermeiden (6) Weiterleitung der Informationen über Qualitätsprobleme oder fehlerhafter Produkte an die für die Qualität dieser Produkte oder die Prävention dieser Probleme direkt verantwortlichen Personen
54
Übersetzung durch Verfasser

Auswirkungen und Umsetzung in der Produktrealisierung
68
und (7) Weiterleitung relevanter Informationen über identifizierte Qualitätsprobleme sowie Korrektur-und Vorbeugungsmaßnahmen für die Managementbewertung (b) Alle in diesem Abschnitt geforderten Aktivitäten und ihre Ergebnisse müssen dokumentiert werden.55
Die Forderungen der beiden Regularien sind im Großen und Ganzen als ähnlich zu
bezeichnen. Jedoch ist der 21 CFR Part 820.100 um einiges ausführlicher und beinhaltet
spezifische Anforderungen, welche in den Verfahren abgebildet werden müssen. Die
Verwendung unterschiedlicher Terminologien in der Arzneimittel-industrie im Vergleich zur
Medizinprodukteindustrie stellt ein nicht zu unterschätzendes Problem dar. So spricht die
Medizinprodukteindustrie von fehlerhaften Produkten oder Non- Konformitäten, während
die Arzneimittelindustrie dies als Abweichungen oder „Deviations“ bezeichnet, obwohl
diese Begriffe das Gleiche meinen. Dieser unterschiedliche Sprachgebrauch ist in den zu
erstellenden Verfahren zu berücksichtigen und möglichst aufzulösen.
Beide Regularien beschreiben die Quellen von Ursachen, welche zu CAPA führen können und
verlangen formale Vorgaben für die Untersuchung zur Ursachenermittlung.
Der ICH Q10 verlangt einen Bezug zum Risikomanagement, während dies ist im 21 CFR Part
820.100 nicht direkt gefordert wird. Jedoch ist die Erwartung der FDA an das dokumentierte
CAPA-Verfahren hier ganz klar; Es muss eine Risikobewertung durchgeführt und
dokumentiert werden. Die Resultate der Risikobewertung haben Einfluss auf die weitere
Behandlung eines CAPA.
Der 21 CFR Part 820.10 fordert, dass Trends, Qualitätsprobleme und deren Korrektur sowie
Vorbeugungsmaßnahmen in der „Bewertung durch die oberste Leitung“ betrachtet werden.
55
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.100

Auswirkungen und Umsetzung in der Produktrealisierung
69
Ein wesentlicher Unterschied zwischen Pharma-GMP und dem QSR ist, dass der 21 CFR Part
820.100 explizit eine Effektivitätsbewertung der Korrektur und Vorbeugungsmaßnahmen
fordert. Die Effektivitätsbewertung ist zu dokumentieren und nach der Umsetzung der
Korrektur bzw. Vorbeugungsmaßnahme durchzuführen. Dies hat zur Folge, dass die
Abweichung oder Non-Konformität solange nicht als abgeschlossen gilt, bis die Effektivität
der Maßnahme nachgewiesen ist. Dies kann ohne weiteres 6 Monate, 12 Monate oder
länger in Anspruch nehmen.
Der Q10 verbindet die Methodik der Korrektur und Vorbeugungsmaßnahmen direkt mit der
Verbesserung von Produkten und Prozessen, wohingegen der 21 CFR Part 820.100 dies nicht
explizit beschreibt. Die Tatsache, dass der QSR unter Abschnitt 820.100 (a.7) fordert, dass
die Ergebnisse aus Korrektur- und Vorbeugungsmaßnahmen Eingang in die Bewertung durch
die oberste Leitung finden, ist ein deutlicher Hinweis darauf, dass das CAPA-System auch in
den Augen der FDA ein Werkzeug zur Verbesserung des Systems darstellt.
Der 21 CFR Part 820.100 fordert zusätzlich, dass das CAPA Verfahren festlegen soll, wer im
Falle von Korrektur- und Vorbeugungsmaßnahmen zu informieren ist. Hierbei ist
sicherzustellen, dass relevante Personen oder Funktionen und Abteilungen über die
Qualitätsprobleme informiert werden.
Korrektur- und Vorbeugungsmaßnahmen sind bei Audits oder Inspektionen eine gute
Möglichkeit für den Auditor bzw. Inspektor, die Wirksamkeit des gesamten QMS zu
untersuchen. Im Leitfaden für Auditoren der FDA (Quality System Inspection Technique
(QSIT)56) wird auf über 30 Seiten dargelegt, wie das CAPA-Subsystem zu prüfen ist. Kein
anderes Subsystem des Qualitätsmanagementsystems erhält in dieser Auditanleitung so viel
Aufmerksamkeit. In den letzten 10 Jahren war das CAPA Subsystem der häufigste Grund für
„Warning Letters“ durch die FDA.57 Das oder die Verfahren für die Beschreibung und
Umsetzung von Korrektur-und Vorbeugungsmaßnahmen gehört ohne Frage zu den
anspruchsvollsten im gesamten QMS. Zudem lässt sich durch die Überprüfung von CAPAs
sehr effektiv die Funktion fast des gesamten QMS überprüfen, weswegen es ein beliebtes
56
Vgl. FDA, o.V. (1999): QSIT Quality system Inspection Technology; S.47 ff 57
Vgl. GMP Navigator, o.V. (2015): Medical Devices Warning Letter Statistik 2015

Auswirkungen und Umsetzung in der Produktrealisierung
70
Thema während Inspektionen ist. Zudem sind CAPAs ein guter Indikator dafür, wie ernst
Qualität im Unternehmen genommen wird.
Zusammengefasst kann man sagen, dass sowohl in der Arzneimitteindustrie als auch in der
Medizinprodukteindustrie CAPA einen gleichwertig hohen Stellenwert besitzt. Jedoch sind
gewisse Anpassungen in den Verfahren vorzunehmen, um die Forderungen des 21 CFR
820.100 umzusetzen. Hier sollte besonders die Forderungen nach der Effektivitätsbewertung
und die unterschiedliche Terminologie berücksichtigt werden.
6.5 Tätigkeiten bei der Installation Wenn Kombinationsprodukte zuerst installiert werden müssen, bevor sie das erste Mal
angewendet werden können, sind die Anforderungen des 21 CFR Part 820.170 (Tabelle 6.18)
einzuhalten.
Tabelle 6.18: Anforderungen an die Tätigkeiten bei der Installation
820.170
(a) Each manufacturer of a device requiring installation shall establish and maintain adequate installation and inspection instructions, and where appropriate test procedures. Instructions and procedures shall include directions for ensuring proper installation so that the device will perform as intended after installation. The manufacturer shall distribute the instructions and procedures with the device or otherwise make them available to the person(s) installing the device. (b) The person installing the device shall ensure that the installation, inspection, and any required testing are performed in accordance with the manufacturer's instructions and procedures and shall document the inspection and any test results to demonstrate proper installation.
(a) Jeder Hersteller eines Medizinprodukts, das eine Installation erfordert, muss geeignete Installations- und Inspektionsanweisungen und gegebenenfalls Testverfahren festlegen und aufrechterhalten. Diese Anweisungen und Verfahren müssen Anleitungen zur Gewährleistung der einwandfreien Installation enthalten, damit das Produkt nach Installation wie beabsichtigt funktioniert. Der Hersteller muss die Anweisungen und Verfahren zusammen mit dem Produkt weitergeben oder der Person bzw. den Personen, die das Produkt installieren, auf andere Weise zur Verfügung stellen. (b) Die Person, die das Produkt installiert, muss sicherstellen, dass die Installation, Inspektion und alle erforderlichen Prüfungen gemäß den Anweisungen und Verfahren des Herstellers ausgeführt werden. Sie muss die Inspektion und alle Prüfungsergebnisse dokumentieren, um die einwandfreie Installation nachzuweisen.58
58
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.170

Auswirkungen und Umsetzung in der Produktrealisierung
71
Die Forderungen zur Installation sind für Arzneimittelhersteller in der Regel unbekannt, da
eine Schachtel Tabletten in aller Regel nicht installiert oder aufgestellt werden muss. In der
Medizinprodukteindustrie gehört die Installation des Medizinproduktes dagegen nicht selten
zum Geschäftskonzept. Es sind Kombinationsprodukte, denkbar die installiert werden
müssen und ein Arzneimittel benötigen, um wie vorgesehen zu funktionieren. In solchen
Fällen hat der Inverkehrbringer die Aufgabe, dokumentierte Verfahren einzuführen und
aufrechtzuerhalten, welche die Installation beschreiben und die Durchführung
dokumentieren. Archivierungsort für die Verfahrensbeschreibung ist der DMR. und
Archivierungsort für den Nachweis der Durchführung der Installation inklusive
Funktionstests, ist die entsprechende zu führende Herstelltakte (DHR = Device History
Record) des Produkts.59
Für Kombinationsprodukte, die keine Installation vor dem Gebrauch benötigen, kann darauf
verzichtet werden. Jedoch sollte dies explizit beschrieben sein, um Missverständnissen
vorzubeugen.
6.6 Tätigkeiten bei der Instandhaltung Wenn Instandhaltungsaktivitäten am Kombinationsprodukt durchgeführt werden müssen,
um die Sicherheit und Wirksamkeit aufrecht zu erhalten, sind die Anforderungen des 21 CFR
Part 820.200 einzuhalten (Tabelle 6.19).
Tabelle 6.19: Anforderungen an die Tätigkeiten bei Instandhaltung
820.200
(a) Where servicing is a specified requirement, each manufacturer shall establish and maintain instructions and procedures for performing and verifying that the servicing meets the specified requirements. (b) Each manufacturer shall analyze service reports with appropriate statistical methodology in accordance with 820.100. (c) Each manufacturer who receives a service report that represents an event which must be reported to FDA under part 803 of this chapter shall automatically consider the report a complaint and shall process it in accordance with the requirements of 820.198. (d) Service reports shall be documented and shall include: (1) The name of the device serviced; (2) Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used; (3) The date of service;
59
Vgl. AAMI (2007): Section VII. Product Controls , S. 154-155

Auswirkungen und Umsetzung in der Produktrealisierung
72
(4) The individual(s) servicing the device; (5) The service performed; and (6) The test and inspection data
(a) Soweit die Wartung spezifizierte Anforderungen darstellt, muss jeder Hersteller Anweisungen und Verfahren festlegen und aufrechterhalten, um zu gewährleisten und zu prüfen, dass die Wartung die spezifizierten Anforderungen erfüllt. (b) Jeder Hersteller muss die Wartungsberichte mit geeigneten statistischen Methoden gemäß §820.100 Korrektur- und Vorbeugungsmaßnahmen analysieren. (c) Jeder Hersteller, der einen Wartungsbericht mit Darstellung eines Vorkommnis erhält, der gemäß Teil 803 dieses Kapitels an die FDA zu melden ist, muss den Bericht automatisch als Reklamation betrachten und gemäß den Anforderungen von §820.198 Reklamationsprotokolle verarbeiten. (d) Wartungsberichte müssen dokumentiert werden und Folgendes enthalten: (1) den Namen des gewarteten Medizinprodukts (2) alle verwendeten Identifikationen und Kontrollnummern des Medizinprodukts (3) das Datum der Wartung (4) die individuelle(n) Wartung(en) des Medizinprodukts (5) die durchgeführte Wartung und (6) die Test-und Inspektionsdaten60
Der Hersteller muss dokumentierte Verfahren zur Wartung und Instandhaltung einführen
und aufrechterhalten. Da Wartungsanfragen häufig mit Problemen des Geräts
zusammenhängen, beinhaltet diese Kapitel auch einige Anforderungen, wie mit
Beschwerden im Zusammenhang mit Wartungsanfragen umzugehen ist. Zusätzlich wird im
Absatz (b) gefordert, dass auf Wartungsanfragen, die ermittelten Fehlerbilder und auf die
Ursachen statistische Techniken angewendet werden, wie dies im Kapitel über Vorbeugungs-
und Korrekturmaßnahmen im 21 CFR Part 820.100 beschrieben ist. Unter Absatz (d) sind
explizit die Mindestanforderungen an einen Wartungsbericht definiert. Wartungsberichte
sind Aufzeichnungen, die in der Herstellakte archiviert werden müssen.
Für Kombinationsprodukte, die keine Wartung und Instandhaltung benötigen, kann darauf
verzichtet werden. Jedoch sollte dies explizit beschrieben sein, um Missverständnissen
vorzubeugen.
60
Emergogroup, o.V. (2010): Übersetzung 21 CFR Part 820.200

Zusammenfassung und Fazit
73
7 Zusammenfassung und Fazit
Im US-amerikanischen Rechtsraum wurde der Begriff der Kombinationsprodukte geprägt
und beschrieben. Die Definition für Kombinationsprodukte ist im 21 CFR Part 3 (Product
Jurisdiction) niedergelegt. Demnach sind dies ein oder mehrere, unabhängig voneinander
gesetzlich regulierte Produkte, welche zu einer Einheit zusammengefügt werden oder als
Einheit benutzt werden sollen. Dies können z.B., ein Injektionssystem mit einer fest
verbauten Arzneimittelampulle oder ein mehrfach verwendbares Injektionssystem sein,
welches nur mit einem bestimmten Arzneimittel benutzt werden kann. Die US-
amerikanische Regulierungsbehörde FDA hat im Jahr 2013 den 21 CFR Part 4 (Current Good
Manufacturing Practice for Combination Products) veröffentlicht. Dieses Gesetz gründet auf
dem 21 CFR Part 3 und führt aus, welche gesetzlichen Anforderungen an die regulierten
Bestandteile der Kombinationsprodukte zu erfüllen sind. Der 21 CFR Part 4 stellt klar, dass
die für die einzelnen regulierten Bestandteile (Arzneimittel, Biologika, Medizinprodukte)
ursprünglich anzuwendenden Gesetze und deren Forderungen erhalten bleiben. Dazu
gehören unter anderem, auch Anforderungen an das jeweils zu implementierende und
aufrechtzuerhaltende Qualitätssystem. Die Anforderungen für den jeweiligen Bereich wie
z.B. Arzneimittel, Biologika oder Medizinprodukte sind unterschiedlich. Die Anforderungen
(Gesetze bzw. Normen) und die daraus resultierenden Qualitätssysteme haben sich zu
verschiedenen Zeiten entwickelt, verwenden unterschiedliche Terminologien und haben
abweichende Ansätze zum Qualitätsmanagement. Man kann sagen, dass im
Arzneimittelbereich der Qualitätskontrollgedanke fest verwurzelt ist. Im Gegensatz dazu
wurde in der Gesetzgebung zu Medizinprodukten von Anfang an das Qualitätsmanagement
implementiert. Die Lücken, die zwischen einem Arzneimittel-QMS und einem
Medizinprodukte-QMS zu überbrücken sind, können je nach Organisation und Umsetzung
erheblich sein. Die Herausforderung besteht darin, wie die Anforderungen aus den
Regularien zu erfüllen und nachzuweisen sind, wenn ein Kombinationsprodukt gefertigt und
auf den Markt gebracht werden soll. Der 21 CFR Part 4 eröffnet hier prinzipiell zwei
Möglichkeiten:

Zusammenfassung und Fazit
74
1. Zwei voll implementierte Qualitätsmanagementsysteme. Ein Arzneimittel QMS
gemäß 21 CFR Part 210 & 211 bzw. ICH Q10 und ein Medizinprodukte QMS gemäß 21
CFR Part 820(QSR).
2. Der „Streamlined Approach“, bei dem bei einem existierenden Arzneimittel QMS
gemäß 21 CFRPart 210 & 211 noch Teile aus dem 21 CFR Part 820 zu erfüllen sind und
umgekehrt, bei einem existierenden QMS gemäß 21 CFR Part 820 noch Teile aus dem
21 CFR Part 210 & 211 zu erfüllen sind.
Beide Möglichkeiten sind in Tabelle 5.2: Anforderungen Streamlined Approach explizit
dargelegt. Es sei in diesem Zusammenhang noch einmal darauf hingewiesen, dass der 21 CFR
Part 4 mit dem „Streamlined Approach“ eine Möglichkeit eröffnet, die gesetzlichen
Forderungen an das implementierte QMS, für den Arzneimittel- Bestandteil bzw. die
gesetzlichen Forderungen für den Medizinprodukte Bestandteil eines
Kombinationsproduktes mit einem auf das Kombinationsprodukt zugeschnittenen
(„Streamlined“) QMS nachzuweisen. Das QMS kann dadurch kompakter werden, nicht aber
die gesetzlichen Forderungen. Die Forderungen aus den unterschiedlichen anwendbaren
Regularien bleiben vollumfänglich erhalten.
Beide oben genannten Möglichkeiten haben ihre Vor- und Nachteile, welche bezüglich der
Unternehmenssituation abgewogen werden müssen. Dies sind zum Beispiel:
1. Zwei voll implementierte QMS (Arzneimittel & Medizinprodukte)
a. Vorteile:
i. Es sind keine Anpassungen notwendig
ii. Es werden keine Sonderaspekte des QMS (z.B. von Arzneimitteln)
vergessen.
iii. Es gibt bei behördlichen Inspektionen keine Probleme
b. Nachteile
i. Hoher Anteil von Redundanzen
ii. Eine Vielzahl von Schnittstellen, die bewusst gelenkt und geprüft werden
müssen, um die Fehlerwahrscheinlichkeit zu reduzieren.

Zusammenfassung und Fazit
75
2. Ein kombiniertes Qualitätsmanagementsystem (Streamlined Approach) wie im 21CFR
Part 4 (Abschnitt e) vorgeschlagen
a. Vorteile:
i. Ein kompaktes QMS mit geringen Redundanzen
ii. Reduzierte Anzahl von Schnittstellen
iii. Das QMS kann auf die jeweilige Unternehmenssituation angepasst
werden.
b. Nachteile:
i. Es sind zum Teil signifikante Anpassungen notwendig.
ii. Bei Inspektionen durch Behörden kann die unterschiedliche Terminologie
zu Verwirrung führen.
In dem in dieser Masterarbeit behandelten Fall eines Inverkehrbringers von Einweg-
Injektionspens mit einem eingeführten Arzneimittel-QMS gemäß 21 CFR Part 210 & 211 bzw.
ICH Q10 sind die notwendigen Anpassungen für ein kombiniertes QMS in der folgenden
Aufzählung zusammengefasst:
1. Verantwortung der Leitung (engl. Management Responsibility)
2. Beschaffung (engl. Purchasing Controls)
3. Entwicklungslenkung (engl. Design Controls)
4. Korrektur- und Vorbeugungsmaßnahmen (engl. Corrective Actions/ Preventive
Actions; CAPA)
Aufgrund nicht notwendiger Installations- bzw. Instandhaltungs- und Wartungstätigkeiten
bei Einweg-Injektionspens können diese beiden Kapitel des QSR bei der Umsetzung in der
Beispielorganisation entfallen.
Auch wenn die Anpassungen auf den ersten Blick minimal aussehen, sind die
vorzunehmenden Anpassungen aufwendig. Dies liegt unter anderem auch daran, dass die
beiden Qualitätssysteme signifikant unterschiedliche Grundlagen haben. Im
Arzneimittelbereich ist der Qualitätskontrollgedanke in den zugrundeliegenden Regularien
verankert, während im Medizinproduktebereich der prozessorientierte Ansatz verankert ist.
Dieses Schisma zu überwinden, bedarf eines wohlüberlegten Vorgehens und läuft auf ein
Projekt hinaus in dem der Ansatz des Change Management integriert ist.

Zusammenfassung und Fazit
76
In der Praxis sind die Unterschiede nicht unüberbrückbar, da sich die positiven Eigenschaften
des prozessorientierten Ansatzes aufgrund von Kostendruck und Sinnfälligkeit nach und nach
durchsetzen. Nichts desto weniger muss auf die unterschiedliche Terminologie, auf
Grundlagen und das Qualitätsverständnis eingegangen werden. Dabei ist zu beachten, dass
die Mitarbeiter in der Implementierungsphase des Projektes abgeholt und angeleitet
werden.
Zu Veränderungen der Regularien im Arzneimittelbereich ist es nicht selten durch Skandale
gekommen. Immer wenn Menschen durch Arzneimittel geschädigt wurden, ob fahrlässig
oder durch Unvermögen wurden in Europa oder Nordamerika Gesetze eingeführt, angepasst
oder verschärft. Als prominentes Beispiel ist hier der Thalidomid-Skandal zu nennen, auch als
„Contergan-Skandal“ bekannt, in dessen Folge sowohl in den USA als auch in Europa die
ersten GMP Regularien eingeführt bzw. verschärft wurden. Mit dem „PIP-Skandal“
(Industriesilikon in Brustimplantaten), liegt nun auch in Europa ein Fall in der
Medizinproduktebranche vor, der zu signifikanten Änderungen in der Gesetzgebung für
Medizinprodukte führen wird. Die derzeit gültige europäische Medizinprodukte-Richtlinie
soll noch in 2016 durch eine europäische Medizinprodukte-Verordnung (MDR – Medical
Device Regulation) ersetzt werden. Die MDR wird neben dem geänderten Status als
europäisches Gesetz einige gravierende Veränderungen mit sich bringen. Bei
Kombinationsprodukten ist zunächst keine regulatorische Veränderung geplant, da die neue
MDR keine Begriffsdefinition für Kombinationsprodukte einführt. Es bleibt bei der Aufteilung
in Medical Devices und Medicinal Products. Nichts desto weniger ändert die EMA, die für die
europaweite Zulassung von Arzneimitteln zuständige europäische Behörde, nach und nach
ihre Praxis. Die EMA verlangt nun auch bei als Arzneimittel deklarierten
Kombinationsprodukten die technische Akte bzw. die Beurteilung durch die benannte Stelle
des Herstellers.
In der Medizinproduktebranche ist es über die Jahre zu einer offensichtlichen Angleichung
der Regularien und der zugrundeliegenden Normen gekommen. Dies wird bei einem
Vergleich des 21 CFR 820 und der ISO 13485:2016 sichtbar. Die ISO 13485 basiert auf der ISO
9001. Die Ausgabe ISO13485:2016 ist klar auf der Struktur der ISO 9001:2008 aufgebaut.
Hier ist es mit der Ausgabe der ISO 9001:2015 zu einem Bruch gekommen. Die ISO 9001 hat
in der neuesten Ausgabe, die selbstauferlegte Struktur, die seit dem Jahr 2000 befolgt

Zusammenfassung und Fazit
77
wurde, signifikant verändert und die ISO 9001:2015 und die ISO13485:2016 unterscheiden
sich zumindest hinsichtlich der Struktur der Kapitel signifikant. Ob die beiden Normen
einander angeglichen werden und wann dies geschieht, ist derzeit ungeklärt. Dies kann auch
in der Praxis Auswirkungen haben. Für Unternehmen, die nach beiden Standards zertifiziert
werden, hat dies größere Auswirkungen zur Folge, da zum Beispiel die Struktur des
Qualitätshandbuchs auf beide Normen angepasst werden muss. Es ist vorstellbar, dass
getrennte Handbücher für beide Normen erstellt werden müssen.
Insgesamt können die Veränderungen, welche durch die Einführung des 21 CFR Part 4
angestoßen wurden, als positiv für die Industrie und die Innovation und Zulassung von
modernen Kombinationsprodukten bezeichnet werden. Es wurden regulatorische Hürden
und Unklarheiten reduziert. Im Lichte dieser Umstände, ist es wahrscheinlich nur eine Frage
der Zeit, bis auf europäischer Ebene die Diskussion entsteht, Kombinationsprodukte
eigenständig zu regulieren.

Verzeichnis der Tabellen
78
8 Verzeichnis der Tabellen
Tabelle 4.1: Zulassungsverfahren EU ....................................................................................... 15
Tabelle 5.1: Zulassungsverfahren USA ..................................................................................... 22
Tabelle 5.2: Anforderungen Streamlined Approach ................................................................ 28
Tabelle 6.1: Forderungen “Streamlined Approach“ bei führendem Qualitätssystem für
Arzneimittel .............................................................................................................................. 33
Tabelle 6.2: Verantwortung der obersten Leitung ................................................................... 34
Tabelle 6.3: Anforderungen an ausgelagerte Prozesse............................................................ 36
Tabelle 6.4: Anforderungen an die Auswahl von Lieferanten ................................................. 38
Tabelle 6.5: Anforderungen an die Auswahl und Freigabe von Lieferanten ........................... 40
Tabelle 6.6: Anforderungen an die Überwachung und Re-Evaluierung von Lieferanten ........ 41
Tabelle 6.7: Anforderungen an die Design und Entwicklungslenkung .................................... 43
Tabelle 6.8: Anforderungen an die Entwicklungsplanung ....................................................... 47
Tabelle 6.9: Anforderungen an die Entwicklungseingaben ..................................................... 49
Tabelle 6.10: Anforderungen an die Entwicklungseingaben ................................................... 51
Tabelle 6.11: Anforderungen an die Entwicklungsverifizierung .............................................. 53
Tabelle 6.12: Anforderungen an die Entwicklungsvalidierung ................................................ 55
Tabelle 6.13: Anforderungen an die Entwicklungsbewertung ................................................. 58
Tabelle 6.14: Anforderungen an die Lenkung von Entwicklungsänderungen ......................... 60
Tabelle 6.15: Anforderungen an den Transfer der Entwicklungsaktivität ............................... 62
Tabelle 6.16: Anforderungen an das Design History File (Entwicklungsakte) ......................... 63
Tabelle 6.17: Anforderungen an Korrektur und Vorbeugemaßnahmen ................................. 65
Tabelle 6.18: Anforderungen an die Tätigkeiten bei der Installation ...................................... 70
Tabelle 6.19: Anforderungen an die Tätigkeiten bei Instandhaltung ...................................... 71

Verzeichnis der Abbildungen
79
9 Verzeichnis der Abbildungen
Abbildung 1: Fertigungssequenz ................................................................................................ 8
Abbildung 2: Pharmaceutical Quality Management System gemäß ICH Q10 ......................... 10
Abbildung 3: Gesetzesstruktur USA ......................................................................................... 18
Abbildung 4: Einweg-Injektionspen (Lantus SoloStar® Hersteller Sanofi) mit aufgesetzter
Kanüle ....................................................................................................................................... 23
Abbildung 5: Autoinjektor (DAI Autoinjektor Hersteller SHL)mit abgezogener Kappe ........... 23
Abbildung 6: Wiederverwendbarer Injektionspen (ClikSTAR® Hersteller Sanofi) mit Kanüle
und Ampulle ............................................................................................................................. 24
Abbildung 7: Convenience Kit .................................................................................................. 24
Abbildung 8: EU Gesetzgebung Arznei Medizinprodukt .......................................................... 32
Abbildung 9: Wasserfall Modell, Schlüsselelemente des Design Control Prozess (Quelle:
Health Canada) ......................................................................................................................... 45
Abbildung 10: V Modell der Entwicklung ................................................................................. 46
Abbildung 11: Verifikationssequenz ......................................................................................... 55
Abbildung 12: Dokumentensequenz Validierung .................................................................... 57

Bibliographie
80
10 Bibliographie
AAMI (2007): The Quality System Compendium, Pelnik. M. T. (Hrsg), 2 Aufl.; Aarlington,
AAMI; isbn: 1-57020-292-3
Emergogroup, o.V. (2010): US FDA Quality System Regulation (QSR) für Medizinprodukte –
21 CFR Part 820, Übersetzung; Hamburg; Emergo Deutschland GmbH;
http://www.emergogroup.com/de/resources/vereinigten-staaten/21-cfr-820; Stand 2016-
09-28
Europäische Kommision DG Enterprise and Industry Directorate F Unit F3, o.V.
(2009):Cosmetics and Medical devices at MEDDEV 2.1/3 Revision 3, “MEDICAL DEVICES:
Guidance document Borderline products, drug-delivery products and medical devices
incorporating, as an integral part, an ancillary medicinal substance or an ancillary human
blood derivative”; o.O.; http://ec.europa.eu/consumers/sectors/medical-
devices/files/meddev/2_7_1rev_3_en.pdf; Stand 2016-09-28
Europäische Kommision, o.V. (2015): Manual On Borderline And Classification In The
Community Regulatory Framework For Medical Devices; o.O.;
file:///C:/Users/de021457/Downloads/borderline_manual_ol_en%20(3).pdf; Stand 2016-09-
28; Stand 2016-09-28
FDA, o.V. (1996): 21 CFR Part 820 Quality System Regulation; o.O.;
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820;
Stand 2016-09-28
FDA, o.V. (1997): Design Control Guidance for Medical Device Manufacturers, o.O.;
http://www.fda.gov/downloads/MedicalDevices/.../ucm070642.pdf; Stand 2016-09-28
FDA, o.V. (1999): QSIT Quality system Inspection Technology; o.O.;
http://www.fda.gov/ICECI/Inspections/InspectionGuides/ucm074883.htm; Stand 2016-09-
28
FDA, o.V. (2006): Guidance for Industry – Quality Systems Approach to Pharmaceutical
CGMP regulations; o.O.;
http://www.fda.gov/downloads/Drugs/.../Guidances/UCM070337.pdf; Stand 2016-09-28
FDA, o.V. (2009): Guidance for Industry Q10 Pharmaceutical Quality System, o.O.;
http://www.fda.gov/downloads/Drugs/.../Guidances/ucm073517.pdf; Stand 2016-09-28
FDA, o.V. (2013): 21 CFR Part 4 Current Good Manufacturing Practice for Combination
Products; o.O.; http://www.fda.gov/downloads/CombinationProducts/UCM336194.pdf;
Stand 2016-09-28

Bibliographie
81
FDA, o.V. (2014): General and Special Controls, o.O.;
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/GeneralandS
pecialControls/default.htm; Stand 2016-09-28
FDA, o.V. (2015): 21 CFR Part 211 Current Good Manufacturing Practice for Finished
Pharmaceuticals; o.O.; http://www.ecfr.gov/cgi-bin/text-
idx?c=ecfr&tpl=/ecfrbrowse/Title21/21cfr211_main_02.tpl; Stand 2016-09-28
FDA, o.V. (2015): Classify your Medical Device o.O.;
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYour
Device/ucm2005371.htm; Stand 2016-09-28
FDA, o.V. (2015): Guidance for Industry and FDA Staff: Current Good Manufacturing Practice
Requirements for Combination Products. (Draft Guidance); o.O.;
http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM429304.pdf; Stand
2016-09-28
FDA, o.V. (2016): 21 CFR Part 3 Product Jurisdiction; o.O.;
https://www.gpo.gov/fdsys/pkg/CFR-2012-title21-vol1/pdf/CFR-2012-title21-vol1-part3.pdf;
Stand 2016-09-28
FDA, o.V. (2016): 21 CFR Part 210 Current Good Manufacturing Practice in Manufacturing,
Processing, Packing, or Holding of Drugs; General; o.O.;
https://www.gpo.gov/fdsys/pkg/CFR-2013-title21-vol4/pdf/CFR-2013-title21-vol4-
part210.pdf; Stand 2016-09-28
FDA, o.V. (2016): Premarket Approval PMA, o.O.;
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevi
ce/PremarketSubmissions/PremarketApprovalPMA/default.htm; Stand 2016-09-28
FDA, o.V. (2016): Premarket Notification 510(k), o.O.;
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevi
ce/PremarketSubmissions/PremarketNotification510k/default.htm; Stand 2016-09-28
GMP Navigator, o.V. (2015): Medical Devices Warning Letter Statistik 2015 - Process
Validation wieder unter den Top 5; Heidelberg, CONCEPT HEIDELBERG GmbH, 2015;
https://www.gmp-navigator.com/dnews_05104_Medical-Devices-Warning-Letter-Statistik-
2015---Process-Validation-wieder-unter-den-Top-5.html; Stand 2016-09-28
Hughes, J, (2016): Unterlagen zum Seminar : “Combination Products: Human Factors-
Usability, Quality Systems and Design Controls”, Präsentation: EU Borderline Product
Regulatory Framework Overview; veranstaltet Amsterdam 14.-16.Juni.2016
ICH, o.V. (2008): ICH Harmonised Tripartite Guideline Pharmaceutical Quality System Q10;
o.O.;

Bibliographie
82
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step
4/Q10_Guideline.pdf; Stand 2016-09-28
ICH, o.V. (2009): ICH Harmonised Tripartite Guideline Pharmaceutical Development Q8; o.O.
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/St
ep4/Q8_R2_Guideline.pdf; Stand 2016-09-28
ISO, o.V. (2012): ISO 9001:2008 Qualitätsmanagementsysteme – Anforderungen; Aufl. 2012,
Berlin, Beuth Verlag
ISO, o.V. (2016): ISO 13485:2016 Medizinprodukte – Qualitätsmanagementsysteme -
Anforderungen für regulatorische Zwecke; Aufl. 2016, Berlin, Beuth Verlag
Johner, C.; Hölzer-Klüpfel,M.; Wittorf, S. (2011): Basiswissen Medizinische Software, 1.Aufl.;
Heidelberg, dpunkt.verlag; isbn: 978-3-89764-688-8
Wikipedia, o.V.(o.J.): Medical Devices, o.O.; https://en.wikipedia.org/wiki/Medical_device;
Stand 2016-09-28
Wikipedia, o.V.(o.J.): Deming Kreis, o.O.; https://de.wikipedia.org/wiki/Demingkreis; Stand
2016-09-28
Wikipedia, o.V.(o.J.): Injektion Medizin, o.O.;
https://de.wikipedia.org/wiki/Injektion_(Medizin); Stand 2016-09-28

83
11 Eidesstattliche Erklärung
Hiermit erkläre ich an Eides Statt, dass ich die vorliegende Arbeit selbstständig
und ohne unerlaubte Hilfe angefertigt, andere als die angegebenen Quellen
nicht benutzt und die den benutzten Quellen wörtlich oder inhaltlich entnommenen
Stellen als solche kenntlich gemacht habe.
Ort, Datum : Wiesbaden, 04.Oktober 2016
_______________
(Unterschrift)