
Die Bedeutung des Asymmetrischen Dimethylarginins für die ... fileAus der Medizinischen Klinik IV...
73
Aus der Medizinischen Klinik IV der Friedrich-Alexander-Universität Erlangen-Nürnberg Direktor: Prof. Dr. K.-U. Eckardt Die Bedeutung des Asymmetrischen Dimethylarginins für die Pathogenese der Endorganschäden bei Angiotensin II-induzierter Hypertonie der Maus Inaugural-Dissertation zur Erlangung der Doktorwürde der medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg vorgelegt von Kilian Sebastian Koch aus Wörth a.d. Donau
Transcript of Die Bedeutung des Asymmetrischen Dimethylarginins für die ... fileAus der Medizinischen Klinik IV...

Aus der Medizinischen Klinik IV
der
Friedrich-Alexander-Universität Erlangen-Nürnberg
Direktor: Prof. Dr. K.-U. Eckardt
Die Bedeutung des Asymmetrischen Dimethylarginins für die Pathogenese der Endorganschäden bei Angiotensin II-induzierter
Hypertonie der Maus
Inaugural-Dissertation
zur Erlangung der Doktorwürde
der medizinischen Fakultät
der
Friedrich-Alexander-Universität
Erlangen-Nürnberg
vorgelegt von
Kilian Sebastian Koch
aus Wörth a.d. Donau

Gedruckt mit Erlaubnis derMedizinischen Fakultät der Friedrich-Alexander-Universität
Erlangen-Nürnberg
Dekan: Prof. Dr. med. Dr. h.c. Jürgen Schüttler
Referent: Priv. Doz. Dr. med. Johannes Jacobi
Korreferent: Prof. Dr. med. Renke Maas
Tag der mündlichen Prüfung: 27. 10. 2010

Gewidmet meinen Großeltern

Inhaltsverzeichnis
1 Zusammenfassung.........................................................................................11.1 Hintergrund und Ziele................................................................................1
1.2 Methoden...................................................................................................1
1.3 Ergebnisse und Beobachtungen................................................................1
1.4 Praktische Schlussfolgerungen.................................................................1
2 Summary..........................................................................................................22.1 Background and aim of the study..............................................................2
2.2 Methods.....................................................................................................2
2.3 Results.......................................................................................................2
2.4 Conclusions...............................................................................................2
3 Einleitung.........................................................................................................34 Fragestellung – Hypothesen........................................................................165 Material und Methoden.................................................................................17
5.1 Versuchstiere...........................................................................................17
5.2 Genotypisierung der Mäuse.....................................................................18
5.3 Induktion der Hypertonie mittels Ang II....................................................19
5.4 Metabolische Käfige...............................................................................20
5.5 Blutentahme durch retroorbitale Phlebotomie.........................................21
5.6 Blutdruckmessung...................................................................................21
5.7 Blut- und Organentnahme.......................................................................22
5.8 Färbung und Auswertung histologischer Schnitte in PAS-Technik.........24
5.9 Immunhistochemische Färbungen .........................................................25
5.10 Oxidativer Stress (Dihydroethidium-Fluoreszenz).................................27
5.11 Messung der ADMA-Spiegel.................................................................27
5.12 Bestimmung der Aldosteronspiegel.......................................................28
5.13 Bestimmung der mRNA-Expression mittels Real-Time RT-PCR..........29
5.14 Statistik...................................................................................................31
6 Ergebnisse.....................................................................................................336.1 Tiere.........................................................................................................33
6.2 Effekte von Ang II auf Blutdruck und Herzfrequenz................................33
Metabolische Käfige......................................................................................36
6.3 Herzhypertrophie und vaskuläre Hypertrophie........................................36

6.4 Plasma-Aldosteron-Spiegel unter Ang II-Infusion...................................36
6.5 ADMA-Plasmaspiegel unter Ang II-Infusion............................................39
6.6 Glomerulosklerose, interstitielle Fibrose und Inflammation.....................44
6.7 Oxidativer Stress......................................................................................46
6.8 PRMT/DDAH mRNA Expression in der Niere.........................................48
Diskussion ......................................................................................................49 Literaturverzeichnis........................................................................................58 Abkürzungsverzeichnis..................................................................................65 Danksagungen.................................................................................................67

1 Zusammenfassung
1.1 Hintergrund und Ziele
Asymmetrisches Dimethylarginin (ADMA) ist ein endogen gebildeter,
kompetitiver Hemmstoff der NO-Synthase. Erhöhte ADMA-Spiegel gelten als
kardiovaskulärer Risikofaktor und sind mit Herz-Kreislauferkrankungen
assoziiert. Ziel der vorliegenden Arbeit war es, im Tierversuch einen möglichen
Zusammenhang von ADMA und Angiotensin II (Ang II) bei der Pathogenese der
Hypertonie und ihrer Endorganschäden zu untersuchen.
1.2 Methoden
Die Versuche wurden an transgenen (TG) Mäusen durchgeführt, die die
humane Form des ADMA abbauenden Enzyms DDAH1 überexprimieren. Die
TG Mäuse sowie Wildtyp (WT) Geschwistertiere erhielten Ang II (1.0µg/kg/min
oder 3.0µg/kg/min) bzw. gepufferte Kochsalzlösung (PBS) als subkutane
Infusion über eine Dauer von vier Wochen.
1.3 Ergebnisse und Beobachtungen
Die Infusion von Ang II erzeugte bei allen Mäusen eine Hypertonie sowie
hypertoniebedingte Endorganschäden. Allerdings bestanden keine signifikanten
Unterschiede zwischen den Genotypen, lediglich das Ausmaß der renalen
interstitiellen Fibrose und des oxidativen Stress in der Aorta war bei DDAH1
transgenen Mäusen vermindert.. Die transgenen Mäuse wiesen insgesamt
deutlich niedrigere ADMA-Spiegel auf. Die Infusion von Ang II hatte keinerlei
Einfluss auf die ADMA-Spiegel. In der RT-PCR zeigte sich eine gesteigerte
Expression der Enzyme DDAH2 und PRMT1 in der Niere nach Ang II Gabe.
1.4 Praktische Schlussfolgerungen
Die in dieser Arbeit erhobenen Daten sprechen insgesamt gegen einen engen
Zusammenhang zwischen ADMA und Ang II bei der Genese hypertensiver
Organschäden. Interessant scheint der Einfluss von ADMA auf die renale
interstitielle Fibrose sowie den vaskulären oxidativen Stress. Hierin liegen
Ansatzpunkte für weiterführende Experimente zur Aufklärung der
pathophysiologischen Relevanz von ADMA.

2 Summary
2.1 Background and aim of the study
Asymmetric dimethylarginine (ADMA) is an endogenous competitive inhibitor of
NO-synthase. Elevated ADMA levels are predictive of cardiovascular risk and
are associated with cardiovascular disease. The aim of the present study was to
elucidate a possible link between ADMA and angiotensin II (Ang II) in the
pathogenesis of hypertension and resulting target organ damage.
2.2 Methods
Transgenic (TG) mice overexpressing the human isoform 1 of the ADMA
degrading enzyme DDAH as well as wildtype (WT) littermates were treated with
Ang II (1.0µg/kg/min or 3.0µg/kg/min) or phosphate buffered saline solution
(PBS) via subcutaneous infusion using osmotic minipumps over a period of four
weeks.
2.3 Results
Infusion of Ang II caused hypertension and target organ damage in all mice.
Except for renal interstitial fibrosis and vascular oxidative stress that were less
pronounced in TG mice no differences between the two genotypes were
observed. As expected, TG mice had markedly lower ADMA levels.
Interestingly, infusion of Ang II had no effect on plasma ADMA levels. RT-PCR
revealed an increased renal expression of ADMA degrading (DDAH2) and
ADMA generating (PRMT1) enzymes following Ang II infusion.
2.4 Conclusions
The data presented herein do not support a clear link between ADMA and Ang
II in the pathogenesis of hypertension or hypertensive target organ damage.
Interestingly, TG mice were resistant to Ang II induced renal interstitial fibrosis
and vascular oxidative stress. This observation needs further exploration in
future studies to investigate the pathophysiological relevance of ADMA.

3 Einleitung
Stickstoffmonoxid (NO) bzw. EDRF (endothelium derived relaxing factor) ist der
potenteste Vasodilatator, der bis heute bekannt ist (61). Dieses in
Endothelzellen gebildete vasoaktive Autakoid entsteht aus dem physiologischen
Substrat, der Aminosäure L-Arginin, über die Oxidation von L-Arginin in zwei
Schritten zu L-Citrullin. Die Oxidation der Aminosäure und Bildung des
Effektormoleküls NO wird über das Enzym NO-Synthase katalysiert (siehe
Abbildung 1).
Von dem Enzym NO-Synthase gibt es 3 verschiedene Isoformen, die neuronale
(nNOS oder NOS I), induzierbare (iNOS oder NOS II) und endotheliale NOS
(eNOS oder NOS III) (21, 27, 50, 51). Die Isoformen werden in Geweben
unterschiedlich stark exprimiert und haben unterschiedliche Funktionen. Die
nNOS wird in neuronalen Geweben exprimiert und wurde zuerst im Kleinhirn
von Ratte und Schwein nachgewiesen (7). Die eNOS kommt im Gefäßendothel
vor, nicht dagegen in der glatten Gefäßmuskulatur (61). Während nNOS und
eNOS konstitutiv exprimiert werden, ist die iNOS eine induzierbare Isoform, die
z.B. durch stimulierte Makrophagen aktiviert wird (89).
Die Effekte von NO im Organismus sind vielfältig. Im Endothel gebildetes NO
diffundiert in die umliegenden glatten Muskelzellen und aktiviert dort die lösliche
Guanylatzyklase. Dies führt zu einem Anstieg von zyklischem
Guanosinmonophosphat (cGMP), einem Botenstoff („second messenger“), der
letztendlich eine Vasodilatation der glatten Muskulatur der Gefäße induziert
(50). Auf diese Weise reguliert NO den Blutdruck und Gefäßwiderstand im
Organismus (66, 80). Neben der Beeinflussung des Vasotonus ist NO das
wichtigste anti-atherogene Molekül im menschlichen Körper. So hemmt NO die
Proliferation glatter Muskelzellen, die Plättchenaggregation, die Expression von
Adhäsionsmolekülen und pro-inflammatorischen Zytokinen, um nur einige
weitere wichtige Funktionen dieses Moleküls zu nennen (23, 35, 64, 83). Eine
verminderte NO-Synthese spielt somit eine Schlüsselrolle in der Pathogenese
der Atherosklerose.
Die Bioverfügbarkeit von NO im menschlichen Organismus unterliegt einer
komplexen Regulation, an der die Methylarginine als endogene Hemmstoffe
der NO-Synthase entscheidend beteiligt sind. Asymmetrisches Dimethylarginin

(ADMA), der wichtigste Vertreter der Methylarginine, ist ein kompetitiver
Inhibitor aller drei NO-Synthasen (siehe Abbildung 2). Dieses Molekül
konkurriert mit der Aminosäure L-Arginin um das Enzym und vermindert somit
die Bioverfügbarkeit von NO (81).
Abbildung 1: ADMA, ein endogener kompetitiver Inhibitor der NO-Synthase
Neben ADMA gibt es weitere beim Menschen vorkommende Methylarginine,
die sich durch die Anlagerung von Methylgruppen am Guanidinostickstoffatom
von der Aminosäure L-Arginin unterscheiden. Durch Substitution eines
Wasserstoffatoms gegen eine Methylgruppe am Guanidinostickstoffatom
entsteht NG-Monomethyl-L-Arginin (L-NMMA). Eine weitere Methylierung an
diesem Stickstoffatom führt zur Bildung von NG,NG-Dimethyl-L-Arginin
(asymmetrisches Dimethylarginin oder auch ADMA). Eine Substitution zweier
Wasserstoffatome durch Methylgruppen an den beiden unterschiedlichen

Stickstoffatomen der Guanidinogruppe führt zur Bildung von NG,NG`-Dimethyl-L-
Arginin (symmetrisches Dimethylarginin oder auch SDMA). Lediglich L-NMMA
und ADMA sind dabei kompetitive Inhibitoren der NO-Synthase, wohingegen
SDMA aufgrund der sterischen Konformation inert gegenüber dem Enzym ist
(siehe Abbildung 2).
Abbildung 2: Endogen beim Menschen vorkommende Methylarginine
Wenngleich SDMA die Aktivität von NOS nicht hemmt, so konkurriert dieses
Molekül ebenso wie L-NMMA und ADMA mit der Aminosäure L-Arginin um die
zelluläre Aufnahme über den kationischen Aminosäuretransporter (y+Carrier
hCAT-2B) (13). Dieser Transporter reguliert ca. 70% der intrazellulären
Aufnahme von L-Arginin. Die Präsenz aller drei Methylarginine (L-NMMA,
ADMA und SDMA) vermindert somit die intrazelluläre Bereitstellung von L-
Arginin. Die Nomenklatur von ADMA bzw. SDMA ist aus chemischer Sicht
unglücklich, da ADMA kein chirales bzw. asymmetrisches Stickstoffatom

aufweist bzw. die chemische Struktur von SDMA kein Symmetriezentrum
beinhaltet. Begrifflich sind ADMA und SDMA somit keine Stereoisomere,
sondern Strukturisomere, in der Literatur hat sich jedoch fälschlicherweise die
Bezeichnung asymmetrisches bzw. symmetrisches Dimethylarginin
durchgesetzt.
Erstmalig wurden Methylarginine 1970 von Kakimoto und Akazawa im Urin
mittels Ionenaustauschchromatographie beim Menschen detektiert (32).
Interessanterweise konnten die Autoren zeigen, dass die Urinkonzentrationen
von einer exogenen L-Arginingabe unbeeinflusst blieben, was als erster
Hinweis gedeutet wurde, dass Methylarginine nicht durch Methylierung von
freiem L-Arginin entstehen (32). Ihre Biosynthese wurde indes erst 1988 durch
Ghosh et al. aufgeklärt (24). Methylarginine entstehen durch die
posttranslationelle Methylierung von Argininresten in Proteinen. Erst durch
anschließende Proteolyse im Rahmen des natürlichen Proteinumsatzes der
Zelle erfolgt eine Freisetzung der nun modifizierten Aminosäuren in die
Zirkulation. Die Methylierung von L-Arginin erfolgt dabei durch Enzyme aus der
Familie der Proteinmethylasen (siehe Abbildung 3), welche sich in drei
verschiede Gruppen unterteilt, die wie folgt benannt wurden:
• Protein Methylase I (Protein-Arginin-Methyltransferase) (58)
• Protein Methylase II (Protein-Carboxyl-Methyltransferase) (33)
• Protein Methylase III (Protein-Lysin-Methyltransferase) (59)
Für die Synthese der Methylarginine ist lediglich die Proteinmethylase I
(Protein-Arginin-N-Methyltransferase oder kurz PRMT) von Bedeutung. Bis dato
wurden 9 PRMT-Gene (PRMT 1-9) identifiziert. Diese werden in Abhängigkeit
von ihrer Substratspezifität in 2 Typen (PRMT vom Typ 1 bzw. 2) eingeteilt (24,
34, 75). PRMTs vom Typ 1 produzieren ADMA und L-NMMA durch
Methylierung von Seitenketten in Histonen und Ribonukleinsäure (RNA)-
bindenden Proteinen, davon insbesondere in hnRNPs (Heterogeneous nuclear
ribonucleoproteins) (5, 53). PRMTs vom Typ 2 generieren SDMA und L-NMMA
und verwenden u.a. myelin-basisches Protein als Substrat (79).

Abbildung 3: Protein-Methylasen; PRMT=Protein-Arginin-N-Methyltransferase
Die Tatsache, dass beide Enzymgruppen L-NMMA produzieren, dessen
Konzentration im Plasma jedoch ca. 10-fach unter der von ADMA liegt, lässt
vermuten, dass L-NMMA in-vivo nur als Zwischenprodukt bei der Bildung von
ADMA und SDMA anfällt (79, 82). Hinzu kommt, dass bis heute keine PRMT
bekannt ist, die ausschließlich L-NMMA produziert (34).
Die Menge an ADMA, die mittels dieses Stoffwechselweges beim Menschen
generiert wird, wurde auf ca. 300µmol pro Tag geschätzt (2). Initial ging man
davon aus, dass Methylarginine unverändert renal eliminiert werden, passend
hierzu wurden die höchsten ADMA-Plasmaspiegel bei Patienten mit terminaler
Niereninsuffizienz gemessen, bei denen dieses Molekül akkumuliert (82).
Untersuchungen mit radioaktiv markierten Methylargininen am Kaninchen durch
Protein-Methylasen
Protein-Methylase I
(PRMT)
Protein-
Methylase II
Protein-
Methylase III
PRMT Typ I PRMT Typ II Nicht klassifiziert
PRMT 1,3,4,6
und 8
PRMT 5, 7
und 9
PRMT 2
Produkte:
NMMA,
ADMA
Produkte:
NMMA,
SDMA

McDermott et al. aus dem Jahre 1976 sprachen jedoch früh dafür, dass
Methylarginine im Organismus weiter verstoffwechselt werden (45). So lag die
Wiederfindungsrate im Urin für L-NMMA und ADMA lediglich bei 0.14% bzw.
5.1%, die für SDMA hingegen bei 66%. Dies legte nahe, dass insbesondere L-
NMMA und ADMA über weitere Abbauwege aus dem Körper eliminiert werden.
Erst 1987 gelang einer japanischen Arbeitsgruppe der Nachweis eines Enzyms,
welches L-NMMA und ADMA metabolisiert (55, 56). Das Enzym
Dimethylarginin-Dimethylaminohydrolase (DDAH) katalysiert dabei die
Hydrolyse von ADMA zu L-Citrullin und Dimethylamin, bzw. im Falle von L-
NMMA zu L-Citrullin und Monomethylamin (siehe Abbildung 4).
Abbildung 4: Synthese und Metabolismus der Methylarginine
SAM = S-Adenosyl-Methionin; SAH = S-Adenosyl-Homozystein; PRMT = Protein-Arginin-N-Methyltransferase; DDAH = Dimethylarginin-Dimethylaminohydrolase (modifiziert nach (30))

Bei dem Enzym DDAH handelt es sich um ein Protein mit einem
Molekulargewicht von ca. 33 kDA. Im Jahre 1999 gelang Leiper der Nachweis
einer zweiten Isoform dieses Enzyms (DDAH1 und DDAH2) (38). Die
Sequenzhomologie beider Isoenzyme liegt bei 62%. Entscheidende
Unterschiede zwischen den Isoformen ergeben sich in ihrem
Expressionsmuster in den verschiedenen Geweben. DDAH1 überwiegt dabei in
Geweben, in denen die neuronale Form der NO-Synthase (nNOS) exprimiert
wird, wohingegen DDAH2 in Geweben dominiert, in denen vornehmlich die
endotheliale Isoform der NO-Synthase (eNOS) anzutreffen ist (38, 78).
Im Gegensatz zu L-NMMA und ADMA ist SDMA kein Substrat von DDAH, was
wiederum an der sterischen Konformation (Methylierung beider Stickstoffatome
der Guanidinogruppe) des Moleküls liegt. Für SDMA wurde neben der
unveränderten Ausscheidung über die Nieren bis dato kein weiterer
Eliminationsweg beschrieben, L-NMMA und ADMA werden zu 10-15%
unverändert über den Urin ausgeschieden, der weitaus größte Teil (85-90%)
wird über DDAH abgebaut (2, 45, 56, 81) (siehe Abbildung 4).
Für die Aktivität sowohl von DDAH1 als auch von DDAH2 sind die drei
Aminosäuren (Cystein, Aspartat und Histidin) im katalytisch aktiven Zentrum
des Enzyms von Bedeutung. Unter diesen drei ist offenbar das Cystein mit
seiner Sulfhydrylgruppe entscheidend (52). Dies wurde durch die endgültige
Strukturaufklärung der humanen Isoform DDAH1 mittels Kristallographie
bestätigt (52). Substanzen, die eine hohe Affinität zu Sulfhydrylgruppen
aufweisen, wie z.B. Quecksilberchlorid, bzw. divalente Metallionen (z.B.
Cadmium, Zink oder Kupfer) führen zu einer potenten Hemmung der DDAH-
Enzymaktivität in-vitro (56).
Der synthetische DDAH-Inhibitor S-2-amino-4(3-methylguanidino)butansäure
(4124W) führt in-vitro zu einem Anstieg von ADMA, nicht jedoch von SDMA,
zudem bewirkt diese Substanz eine Vasokonstriktion isolierter Aortenringe von
Ratten, welche durch Gabe von L-arginin antagonisierbar ist (43).
Neben den beschriebenen Hemmern des Enzyms DDAH führt eine
Nitrosylierung der Sulfhydrylgruppe im Cystein des katalytischen Zentrums von
DDAH zu einer Hemmung der Enzymaktivität (36). Hierdurch besteht ein
direkter Rückkopplungsmechanismus („feedback“), indem NO durch
Nitrosylierung von DDAH zu einer Hemmung der DDAH-Aktivität und einem

Anstieg von ADMA führt, welches dann konsekutiv durch Hemmung der NOS-
Aktivität eine verminderte Bioverfügbarkeit von NO zur Folge hat (siehe
Abbildung 5).
Durch diesen Mechanismus wird bei exzessiver NO-Produktion (wie z.B. im
Rahmen der Sepsis durch Stimulation der induzierbaren NO-Synthase) über
eine Hemmung des ADMA abbauenden Enzyms die NO-Synthese gedrosselt.
Abbildung 5: Nitrosylierung von DDAH durch NO
Die Michaelis-Menten Konstante (Km), welche die Substratkonzentration bei
halbmaximaler Reaktionsgeschwindigkeit eines Enzyms beschreibt, liegt für das
Enzym DDAH bei ca. 180µM und somit sehr viel höher als die im Plasma von
Gesunden ermittelten ADMA-Spiegel, die im Bereich von ca. 0.3-0.5µM liegen
(46). Der Km-Wert des Enzyms DDAH suggeriert, dass die intrazellulären
ADMA-Konzentrationen deutlich über den im Plasma gemessenen Werten
liegen. Hierzu gibt es bis heute leider nur sehr wenige Daten. In-vitro
Untersuchungen an bovinen aortalen Endothelzellen (BAECs) ergaben jedoch
intrazelluläre ADMA-Konzentrationen, die ca. 10-fach über den im Plasma
gemessenen Spiegeln lagen (8). Dennoch wird angezweifelt, dass die im
Plasma bzw. intrazellulär gemessenen ADMA-Konzentrationen ausreichen, um
eine relevante Hemmung der NO-Synthase zu bewirken, da die im Plasma
gemessenen physiologischen Konzentrationen von L-Arginin mit ca. 50-60µM
ca. 100fach über denen von ADMA liegen.
Eine ähnliche, auch als L-Arginin-Paradoxon bezeichnete Problematik ergibt
sich bei näherer Betrachtung der L-Arginin Konzentrationen, die sowohl
intrazellulär (ca. 100-800µmol/l) als auch extrazellulär deutlich über dem am

aufgereinigten Enzym eNOS in-vitro ermitteltem Km-Wert von 2.9µM liegen (21,
63). Selbst unter pathophysiologischen Bedingungen mit vermindertem
Substratangebot liegen die L-Argininkonzentrationen immer noch deutlich über
dem Km-Wert, so dass das Enzym gesättigt sein sollte und eine exogene
Substratzufuhr keinen Effekt auf die NO-Produktion haben sollte.
Dies widerspricht jedoch in-vivo Befunden am Kaninchen (14) und beim
Menschen (19), in denen die exogene Gabe von L-Arginin zu einer
Verbesserung der endothelabhängigen Vasodilatation führte. Neuere
Untersuchungen lassen jedoch berechtigte Zweifel am Nutzen einer L-
Argininsubstitution aufkommen. So musste eine plazebo-kontrollierte Studie an
Patienten mit stattgehabten Myokardinfarkt, bei denen der Effekt einer additiven
Therapie mit L-Arginin (3x3g/Tag) getestet wurde, vorzeitig wegen einer
gehäuften Sterblichkeit in der Verumgruppe abgebrochen werden (68). Eine
weitere Studie an 133 Patienten mit peripherer arterieller Verschlusskrankheit,
die zusätzlich zur Standardmedikation entweder über 6 Monate L-Arginin
(3g/Tag) oder Plazebo erhielten, zeigte eine geringere Verbesserung der
schmerzfreien Gehstrecke unter L-Argininsubstiution (88).
Die bis dato vorliegenden Untersuchungen erlauben somit keine klare Aussage,
ob eine Substratzufuhr zu einer gesteigerten Bioverfügbarkeit von NO führt.
Eine plausible Erklärung für das L-Arginin-Paradoxon könnte die NOS-
Hemmung durch Methylarginine darstellen. Allerdings stellt sich hier, wie bereits
oben erwähnt, die Frage, ob die im Plasma gemessenen ADMA-Spiegel beim
Gesunden (0.3-0.5µM) bzw. 10fach höhere Werte im Intrazellulärraum
ausreichen, um signifikant in Konkurrenz zu L-Arginin (extra- und intrazelluläre
Konzentrationen s.o.) die Enzymaktivität der NO-Synthase zu beeinflussen. In-
vitro Untersuchungen an bovinen aortalen Endothelzellen (BAECs) belegen,
dass physiologische Konzentrationen von ADMA zu keiner Hemmung von NOS
führen, pathophysiologisch relevante Konzentrationen (5µM) führen indes bei
Abwesenheit von L-Arginin zu einer 38%igen Hemmung der NO-Produktion, in
Anwesenheit physiologischer L-Argininkonzentrationen zu einer 12%igen
Hemmung des Enzyms (8).
Unabhängig von diesen stöchiometrischen Überlegungen wird die
pathophysiologische Relevanz erhöhter ADMA-Plasmaspiegel seit der
klinischen Erstbeschreibung beim Menschen durch Vallance et al. (82) nicht

angezweifelt. Zahlreiche klinische Studien, die seitdem publiziert wurden,
zeigen eine enge Assoziation erhöhter ADMA-Spiegel mit allen etablierten
kardiovaskulären Risikofaktoren, wie Hypercholesterinämie,
Hypertriglyzeridämie, Hyperhomozysteinämie, Diabetes mellitus,
Bluthochdruck, Nikotinabusus, aber auch für Inflammationsmarker wie z.B. CRP
(1, 3, 6, 31, 41, 71, 73, 86, 93). Erhöhte ADMA-Plasmaspiegel wurden zudem
bei Patienten mit manifester koronarer Herzkrankheit (KHK), zerebraler
Ischämie (Apoplex) und peripherer arterieller Verschlusskrankheit (pAVK)
gemessen (6, 47-49, 90). Hierbei handelt es sich jedoch meistens um
Querschnittstudien mit kleiner Patientenfallzahl, große, randomisierte,
prospektive Studien mit harten Endpunkten zu dieser Thematik sind rar. Neuere
klinische Studien belegen jedoch, dass ADMA ein unabhängiger Risikofaktor für
kardiovaskuläre Ereignisse ist. Dies haben Zoccali et al. in mehreren Studien an
Patienten mit Niereninsuffizienz untersucht (91-95). Auch in nierengesunden
Patientenkollektiven sind erhöhte ADMA-Spiegel ein Prädiktor für
kardiovaskuläre Ereignisse, wie z.B. Schlaganfälle (84) oder Myokardinfarkte
(47, 67, 86). ADMA wird daher zunehmend als prognostisch relevanter
Biomarker diskutiert.
Aufgrund der mangelnden Vergleichbarkeit vorliegender Studien, die auf der
großen Variabilität gemessener ADMA-Spiegel beruht (30), ist eine
abschließende Beurteilung der pathophysiologischen Relevanz von ADMA zum
jetzigen Zeitpunkt nicht möglich. Hauptproblem ist dabei eine Standardisierung
der ADMA-Analytik und die Etablierung von Referenzwerten an definierten
Patientenkollektiven. Die vorliegenden klinischen Studien erlauben
insbesondere nicht die Beantwortung der Frage, ob erhöhte ADMA-Spiegel
kausal an der Pathogenese der Atherosklerose beteiligt sind, oder erst infolge
der Gefäßschädigung ansteigen. Zur Klärung dieser Frage lag es nahe,
genetische Mausmodelle zu etablieren, die mit einer Modulation der ADMA-
Spiegel einhergehen, um die funktionelle Relevanz dieses Moleküls zu
beweisen. Betrachtet man Synthese und Metabolismus der Methylarginine,
bietet sich das Enzym DDAH in idealer Weise als Target an.
Bis dato gibt es verschiedene Tiermodelle, in denen das Enzym DDAH
entweder ausgeschaltet (DDAH knock-out) oder überexprimiert wird. Die
Arbeitsgruppe von Leiper generierte DDAH1 defiziente Mäuse (37). Der

homozygote knock-out dieser Isoform führte zu einem embryonal letalen
Phänotyp, was die Bedeutung dieses Enyzms in der Embryonalentwicklung
unterstreicht. Heterozygote Tiere sind lebensfähig und weisen gegenüber
Wildtyptieren erhöhte ADMA-Plasma- und Gewebespiegel auf. Die DDAH-
Aktivität ist um ca. 50% reduziert. Zudem weisen die Tiere einen signifikant
höheren Blutdruck und eine gestörte Endothelfunktion auf (37). Den genau
umgekehrten Phänotyp haben DDAH1 transgene Mäuse, in denen die humane
Isoform 1 dieses Enzyms unter Kontrolle eines β-actin Pomotors ubiquitär
überexprimiert wird (16). Diese Tiere haben signifikant niedrigere ADMA-
Plasmaspiegel (40%), zudem weisen diese Mäuse einen tendenziell niedrigeren
arteriellen Mitteldruck (ca. 10%) sowie eine verbesserte endothelabhängige
Vasodilatation auf (16, 17) (siehe Abbildung 6).
Abbildung 6: Effekte einer Defizienz bzw. Überexpression von DDAH 1 (nach
(37), (16, 17)
Zum jetzigen Zeitpunkt ist eine weitere Charakterisierung dieser Mausstämme
zur Klärung der Bedeutung erhöhter ADMA-Spiegel notwendig. Die bis dato
vorliegenden Ergebnisse sprechen jedoch für eine funktionelle Relevanz
erhöhter ADMA-Spiegel im Rahmen von Gefäßschäden.

Gestützt durch die enge Assoziation erhöhter ADMA-Plasmaspiegel mit
etablierten kardiovaskulären Risikofaktoren stellt sich die Frage, ob ADMA-
Spiegel therapeutisch gesenkt werden können und ob dies mit einer Besserung
funktioneller bzw. struktureller Gefäßschäden einhergeht. Bis dato gibt es keine
Substanz, die ADMA-Spiegel spezifisch zu senken vermag. Betrachtet man die
Synthese und den Metabolismus von ADMA, so wäre eine Senkung der ADMA-
Spiegel durch verminderte Bildung über PRMTs, gesteigerte Ausscheidung
über die Nieren bzw. gesteigerten enzymatischen Abbau durch DDAH denkbar.
Da DDAH als Schlüsselenzym den Metabolismus von ADMA reguliert, ergibt
sich hier ein Hauptangriffspunkt für die Entwicklung pharmakologischer
Substanzen, die dieses Enzym in seiner Aktivität stabilisieren bzw. induzieren.
Die Entwicklung derartiger Substanzen befindet sich derzeit noch in klinischer
Erprobung.
Zum jetzigen Zeitpunkt wurden zahlreiche Substanzen, die durch Modulation
etablierter Risikofaktoren Ihren Stellenwert in der Reduktion kardiovaskulärer
Ereignisse klinisch eindeutig unter Beweis gestellt haben, dahingehend
untersucht, ob diese auch zu einer Beeinflussung der ADMA-Plasmaspiegel
führen. Hierzu zählen insbesondere die Klasse der
Cholesterinsyntheseenzymhemmer (CSE-Hemmer oder Statine) bzw.
Bluthochdruckmedikamente (Antihypertensiva), hierbei insbesondere
Medikamente, welche durch Hemmung des Renin-Angiotensin-Systems (RAS)
die Bildung (Angiotensin Converting Enzym Hemmer oder kurz ACE-Hemmer)
oder die pharmakologische Wirkung (Angiotensin II-Rezeptorblocker oder kurz
AT1-Blocker) von Angiotensin II (Ang II) hemmen. Betrachtet man die Gruppe
der Statine, so zeigen die vorliegenden klinischen Studien trotz signifikanter
Senkung der Cholesterinspiegel überraschenderweise keinen Effekt auf ADMA-
Plasmaspiegel (42). Einzige Ausnahme hiervon scheint der in Deutschland für
die Behandlung gerade zugelassene CSE-Hemmer Rosuvastatin zu sein,
vermutlich handelt es sich dabei um einen substanzspezifischen Effekt (40).
Anders sieht die Datenlage für die Gruppe der ACE-Hemmer bzw. AT1-Blocker
aus. In einer randomisierten, plazebokontrollierten cross-over Studie konnten
Delles et al. 2002 in einem Kollektiv von 20 jungen, männlichen Patienten mit
milder essentieller Hypertonie zeigen, dass sowohl durch den ACE-Hemmer
Enalapril als auch durch den AT1-Rezeptorblocker (ARB) Eprosartan bzw. eine

Kombination beider Substanzen ADMA-Plasmaspiegel signifikant gesenkt
werden konnten (18). Dieser Effekt wurde nach nur einwöchiger Therapie mit
den entsprechenden Substanzen beobachtet und war unabhängig von der
Blutdrucksenkung, da die Monotherapie beider Substanzen zu keiner
signifikanten Blutdrucksenkung führte (18). In der Folge konnte auch für andere
Erkrankungen gezeigt werden, dass eine Intervention des RAS die ADMA-
Spiegel senkt, so bei dialysepflichtiger Niereninsuffizienz (4), bei Syndrom X
(10) und Diabetes mellitus Typ II (28).
Tierexperimentelle Daten und in-vitro Befunde an Endothelzellen sprechen für
eine Assoziation erhöhter ADMA-Spiegel mit dem RAS. In diesem
Zusammenhang untersuchten Chen und Kollegen Zellkulturen aus humanen
Nabelschnurendothelzellen (HUVECs), die über 24 Stunden mit Angiotensin II
(1µM) inkubiert wurden (11). Im Kulturmedium zeigte sich ein signifikanter
Anstieg von ADMA um ca. 100%, welcher mit einer Hemmung der DDAH-
Aktivität von 75% einherging (11). Neben diesen in-vitro-Befunden gibt es auch
in-vivo Daten in einem DDAH2 transgenen Mausmodell, die zeitgleich mit den
im Rahmen dieser Doktorarbeit durchgeführten Versuchen von einer
japanischen Arbeitsgruppe veröffentlicht wurden (26). Hier führte die
kontinuierliche Infusion von Ang II in einer Konzentration von 1.0µg/kg/min über
2 Wochen mittels osmotischer Minipumpen zu einer Verdoppelung der ADMA-
Plasmaspiegel bei Wildtypmäusen, bei DDAH2 transgenen Tieren war dieser
Anstieg signifikant vermindert (26). Weitgehend unklar ist, über welchen
Mechanismus Ang II zu einem Anstieg von ADMA führt.
Ziel dieser Doktorarbeit war es, die Rolle von ADMA bzw. seines abbauenden
Enzyms DDAH bei Ang II induzierter Hypertonie in einem Mausmodell unter
Verwendung DDAH1 transgener Mäuse näher zu untersuchen.

4 Fragestellung – Hypothesen
Im Rahmen dieser Doktorarbeit wurde der Frage nachgegangen, welche Rolle
ADMA bzw. sein abbauendes Enzym DDAH im Rahmen eines Ang II
induzierten Hochdruckmodells der Maus spielt. Zur Klärung dieser
Fragestellung wurde der Effekt einer chronischen Infusion von Ang II auf
ADMA-Plasmaspiegel, Bluthochdruck und Endorganschädigung an
Wildtypmäusen sowie DDAH1 transgenen Mäusen untersucht.
Hypothesen:
Die chronische Infusion von Ang II führt zu einem Anstieg der ADMA-
Plasmaspiegel.
ADMA ist von pathophysiologischer Relevanz hinsichtlich Ang II-induzierter
Endorganschäden.
Die Überexpression von DDAH1 geht mit erniedrigten ADMA-Plasmaspiegeln
einher und schützt vor Ang II-induzierten Endorganschäden.

5 Material und Methoden
5.1 Versuchstiere
Bei den für die Versuche verwendeten DDAH1 transgenen (TG) Mäusen
handelte es sich um Tiere, an deren Generierung und Charakterisierung Herr
PD Dr. J. Jacobi im Rahmen eines Postdoktorandenstipendiums der Deutschen
Forschungsgemeinschaft in den USA bei Prof. John P. Cooke (Dept. of
Cardiovascular Medicine, Stanford University) beteiligt war (16). Einige
transgene Tiere wurden von Herrn Prof. Cooke freundlicherweise zur Verfügung
gestellt und nach Deutschland importiert.
Im Biotechnologischen Entwicklungslabor (BTE) der Universität Erlangen-
Nürnberg erfolgte zunächst die Sanierung der Kolonie mittels Embryotransfer,
um die Tiere anschließend in die Barrierehaltung überführen zu können.
DDAH1 TG Mäuse exprimieren zusätzlich zur murinen Form die humane
Isoform DDAH1 heterozygot auf einem C57BL/6J Hintergrund (hDDAH1+/-). Die
Expression des Transgens unterliegt dabei der Kontrolle eines β-Actin-
Promotors, wodurch eine konstitutive Expression in allen Zellen erfolgt. Die
Phänotypisierung der Tiere in den USA zeigte, dass die Überexpression von
DDAH mit erniedrigten systemischen ADMA-Plasmaspiegeln und konsekutiv
mit einer gesteigerten NOS-Aktivität einherging (16). Zudem waren der arterielle
Mitteldruck und der systemische Gefäßwiderstand bei transgenen Mäusen
tendenziell vermindert (16). In unserer Arbeit wurden 4 Monate alte, männliche,
heterozygote DDAH1 TG Mäuse und deren Wildtyp-Geschwistertiere (WT) aus
dem gleichen Wurf verwendet. Für den Versuch wurden je Genotyp drei
Behandlungsgruppen untersucht.
Gruppe 1: WT und DDAH1 TG Mäuse (je n=7) mit Infusion von Phosphat-
gepufferter Kochsalzlösung (PBS) über 4 Wochen (Kontrollgruppe)
Gruppe 2: WT und DDAH1 TG Mäuse (je n=13) mit Infusion von Ang II in einer
Dosierung von 1.0µg/kg/min über 4 Wochen
Gruppe 3: WT (n=9) und DDAH1 TG Mäuse (n=10) mit Infusion von Ang II in
einer Dosierung von 3.0µg/kg/min über 4 Wochen (Hochdosis Ang II)

Das Versuchsprotokoll wurde von der Tierschutzkommission der Regierung von
Mittelfranken (Bezirksregierung Mittelfranken, AZ 54-2531.31-1/06) genehmigt.
Die Haltung der Versuchstiere erfolgte in einem klimatisierten Tierstall, die
Temperatur betrug konstant 22 +/- 2°C bei einer Luftfeuchtigkeit von 50-60%.
Über eine automatische Zeitschaltuhr wurde ein zwölfstündiger Tag-Nacht
Rhythmus erzeugt. Die Versuchstiere hatten freien Zugang zu Leitungswasser
und Futter (Standard-Diät Nr. 1324, Altromin, Lage).
5.2 Genotypisierung der Mäuse
Die Genotypisierung der Mäuse erfolgte mittels Polymerase-Kettenreaktion
(PCR) aus isolierter Schwanz-DNA unter Verwendung transgen-spezifischer
Primer (s.u.). Die Isolierung der DNA erfolgte mit Hilfe eines DNA-Isolationskits
(NucleoSpin Tissue-Kit, Macherey-Nagel GmbH & Co KG, Düren). Hierbei
erfolgte zunächst ein Gewebeverdau im Wasserbad (56°C) unter Inkubation mit
Proteinase K. Nach anschließender Zentrifugation wurde die an der Membran
des Filters befindliche DNA in einem zweiten Zentrifugationsschritt eluiert und
die DNA-Konzentration mittels Photometer (Ultrospec 3000 pro, Amersham
Pharmacia Biotech, Piscataway, NJ, USA) gemessen. Hieraus wurde die
einzusetzende DNA-Menge für die PCR errechnet.
Mittels PCR wurde ein 282 Basenpaar-Fragment des humanen DDAH1-Gens
amplifiziert. Die Primersequenzen wurden dabei so gewählt, dass die Maus-
Isoform des Enzyms nicht amplifiziert wurde. Hierzu wurden die
Primersequenzen mittels Basic Local Alignment Search Tool (BLAST) des
National Center for Biotechnical Information (NCBI) auf ihre Spezifität hin
überprüft. Die PCR erfolgte über einen Thermocycler (Biometra GmbH,
Göttingen). Folgende Primer (MWG Biotech AG, Ebersberg) wurden verwendet:
Tabelle 1. Primersequenzen für die Genotypisierung
Primer SequenzDDAH forward 5’-ATG-CAA-CTT-TAG-ATG-GCG-GAG-3’DDAH reverse 5’-TCA-TCA-GGC-ACA-GTG-AGT-TTG-3’
Die Zyklen der PCR waren wie folgt aufgebaut:

Denaturieren: 15 min bei 95°C
Insgesamt 35 Amplifikationszyklen, bestehend aus
Denaturieren: 1 min bei 94° C
Annealing: 1 min bei 55° C
Extension: 1 min bei 72° C
Schlussphase: 10 min bei 72° C
Ende: Kühlung bei 4° C bis zur Entnahme der Proben
Im Anschluss an die PCR erfolgte eine Gelelektrophorese (1% Agarose-Gel,
Spannung 70 Volt, NEEO Ultra-Qualität, Carl Roth GmbH & Co KG, Karlsruhe).
Mittels Ethidiumbromid (Carl Roth GmbH & Co KG, Karlsruhe) wurden die
Banden im ultravioletten Licht sichtbar gemacht. Zur Skalierung diente eine 100
Basenpaar- (bp) DNA-Leiter (New England Biolabs, Ipswich, MA, USA).
5.3 Induktion der Hypertonie mittels Ang II
Im Alter von 16 Wochen erfolgte die Implantation osmotischer Minipumpen
(Alzet, Modell 2004, DURECT Corporation, Cupertino, CA), welche mit PBS
bzw. Ang II (1.0 bzw. 3.0µg/kg/min) befüllt wurden.
Das Funktionsprinzip der osmotischen Minipumpen beruht darauf, dass sie in
verschiedene Kammern aufgeteilt sind. In der Hauptkammer befindet sich das
Reservoir für die zu infundierende Substanz, welches komprimierbar ist. Das
Reservoir ist durch eine impermeable Membran von einer Kammer getrennt,
welche eine hochosmolare Salzmischung enthält. Diese ist wiederum nach
außen hin durch eine semipermeable Membran begrenzt. Nach Implantation
der Pumpe wird durch Osmose Wasser aus der Umgebung in die Salzkammer
gezogen. Dadurch nimmt das Volumen letzterer zu und komprimiert das
Reservoir der Hauptkammer, wodurch der Inhalt mit einer konstanten Rate
herausgedrückt wird (76).
Für die Infusion von Ang II (Bachem AG, Weil a. Rhein) wurden die Minipumpen
wie im Folgenden beschrieben befüllt. Zunächst wurde unter Berücksichtigung
der Förderrate der Pumpe (6µl/Tag) für jedes Tier einzeln die Menge an Ang II

(in mg) berechnet, die in die jeweilige Pumpe appliziert werden muss, damit
jedes Tier die angestrebte Infusionsrate von 1.0 oder 3.0µg/kg/min erhält.
Zur Befüllung der Pumpen wurde alsdann eine Stammlösung hergestellt, indem
100mg Ang II in 4ml PBS aufgelöst wurden, die Konzentration der
Stammlösung lag somit bei 25mg/ml bzw. 25µg/µl. Dividiert man nun die für
jedes Tier spezifische Menge an einzusetzendem Ang II (in mg) durch obige
Konzentration, erhält man das Volumen an Stammlösung, welches in den
Ansatz zum Befüllen der Pumpe gegeben werden muss. Dieses Volumen füllt
man mit PBS auf, bis ein Gesamtvolumen von 250µl vorliegt. Das Füllvolumen
der Pumpe liegt bei 200µl, aufgrund des Restvolumens im Spritzenansatz
wurde bewusst ein Volumen von 250µl zur Befüllung der Pumpen angesetzt.
Bei den Kontrolltieren wurden die Pumpen mit reiner PBS-Lösung befüllt.
Die Implantation der Minipumpen erfolgte unter Inhalationsnarkose (2%
Isofluran, Abbot GmbH & Co KG, Wiesbaden) mittels eines entsprechenden
Verdampfersystems (Völker GmbH, Kaltenkirchen). Zur Narkoseeinleitung
wurden die Tiere in eine Plexiglaskammer mit Narkosegasanschluss gelegt, zur
Aufrechterhaltung der Narkose diente eine Kunststoffmaske. Am narkotisierten
Tier wurde zwischen den Schulterblättern die Haut rasiert, dann erfolgte ein
Hautquerschnitt über ca. 5mm mittels Skalpell, durch anschließende stumpfe
Präparation mit Nadelhalterklemmen wurde atraumatisch eine Tasche im
Subkutangewebe präpariert, um die Pumpen widerstandslos und steril
implantieren zu können. Der Wundverschluss erfolgte durch Einzelknopfnähte
unter Verwendung eines Fadens der Stärke 5-0 (Ethicon Prolene, Johnson &
Johnson GmbH, Düsseldorf).
5.4 Metabolische Käfige
Eine Woche vor Pumpenimplantation sowie drei Tage vor der Organentnahme
wurden die Mäuse für 24 Stunden in metabolischen Stoffwechselkäfigen
gehalten (STT 100, Ebeco GmbH, Castrop-Rauxel).
Leitungswasser und gemahlene Standard-Diät waren ad libitum vorhanden. Der
24h Urin der Tiere wurde in einem Sammelbehälter aufgefangen. Mittels einer
Präzisionswaage (BP 2100, Sartorius AG, Göttingen) wurde die absolute
Urinmenge der Mäuse bestimmt, aus der später die Diurese errechnet wurde.
Anschließend wurde der Urin zentrifugiert, der Überstand abpipettiert, aliquotiert

und umgehend bei -20°C zur späteren Bestimmung von Albumin und Kreatinin
tiefgefroren. Auch Wasser- und Futterbehälter der Mäuse wurden vor und nach
Einsetzen in den Käfig zur Bestimmung der Trink- und Futtermenge gewogen.
Die Albuminkonzentration im Urin wurde mit einem ELISA (Enzyme linked
immunosorbent assay) gemäß den Herstellerangaben gemessen (Mouse
Albumin Elisa Quantification Kit, Bethyl Laboratories Inc., Montgomery, Texas).
Die Absorption des Substrats Tetramethylbenzidin (TMB) wurde bei 450nm
photometrisch gemessen. Bei allen Urinproben erfolgte eine
Doppelbestimmung, aus der anschließend der Mittelwert berechnet wurde.
Die Kreatininkonzentration im Urin wurde im Zentrallabor der Kinder- und
Jugendklinik der Friedrich-Alexander-Universität Erlangen-Nürnberg bestimmt.
5.5 Blutentahme durch retroorbitale Phlebotomie
Bei den Tieren, die Ang II in der höheren Dosierung (3.0µg/kg/min) erhielten,
wurden die ADMA-Plasmaspiegel sowohl vor als auch nach Infusion von Ang II
gemessen. Die Bestimmung der basalen ADMA-Plasmaspiegel (vor Ang II
Infusion) erfolgte aus Blutproben, welche mittels retroorbitaler Phlebotomie
gewonnen wurden. Die Blutentnahme fand dabei unter Inhalationsnarkose (2%
Isofluran) statt. Es wurden kleine Glaskapillaren (Hettich Zentrifugen,
Tuttlingen) verwendet, mit denen durch eine langsam kreisende Bewegung
nach Luxation des Auges aus dem Bulbus der retroorbitale Venenplexus
vorsichtig eröffnet wurde. Das in die Kapillare aufgesogene Blut (ca. 150µl)
wurde in ein Eppendorf-Gefäß überführt, umgehend zentrifugiert und das
Plasma anschließend bei -20° C gelagert.
5.6 Blutdruckmessung
Die Blutdruckmessung erfolgte nicht-invasiv mittels Schwanzplethysmographie
über ein vollautomatisches Messgerät (Blood Pressure 209000, TSE Systems,
Bad Homburg). Um die Tiere an die Meßmethode zu gewöhnen, wurde mit den
Messungen bereits vier Wochen vor dem eigentlichen Versuchsbeginn
begonnen, dabei erfolgten alternierende Blutdruckmessungen in Abständen von
2-3 Tagen.

Zur Blutdruckmessung wurden die Tiere auf eine auf Körpertemperatur
vorgeheizte Wärmeplatte gesetzt und mittels magnetisch haftender Restrainer
an der Flucht gehindert. Dann wurde der Schwanz der Mäuse durch die
automatisch aufblasbare Manschette geführt, in einen optischen Pulssensor
eingelegt und mit einem Stück Pflaster fixiert, um Bewegungsartefakte zu
vermeiden. Das Blutdruckmessgerät verfügte über sechs Kanäle, die eine
simultane Datenerfassung von sechs Tieren zuließen. Das Messprotokoll wurde
für diesen Versuch wie folgt gewählt.
Nach fünfminütiger Akklimatisationszeit wurden zunächst 10 Testmessungen
vorgenommen, erst im Anschluss wurden 20 weitere Messungen durchgeführt,
die in die Versuchsauswertung gelangten. Während der Blutdruckmessung
wurde der Cuff computergesteuert aufgeblasen, bis der Blutfluss in der
Schwanzarterie sistierte. Dann wurde der Cuff-Druck automatisch schrittweise
abgesenkt, bis mittels des optischen Lichtsensors eine Pulskurve detektiert
werden konnte. Der Cuff-Druck, bei dem diese Kurve gemessen wurde,
entsprach dem systolischen Blutdruck. Zusätzlich wurde die Herzfrequenz zu
den jeweiligen Messungen aufgezeichnet.
Die Werte aus den Einzelmessungen wurden in eine Excel-Datentabelle (Excel,
Version 2003, Microsoft Corporation, Redmond, WA, USA)
überführt und der Durchschnitt für Mitteldruck und Herzfrequenz aus 20
Messungen errechnet.
Die für die Auswertung verwendeten Blutdruckmessungen erfolgten am Tag vor
Implantation der osmotischen Minipumpen, sowie an den Tagen 2, 3, 7, 14, 21
und 28.
5.7 Blut- und Organentnahme
Vier Wochen nach Pumpenimplantation (Tag 28) erfolgte die Tötung der Tiere
zum Zwecke der Gewinnung von Blut- und Organproben. Nach einer
abschließenden Blutdruckmessung und Erfassung des Körpergewichtes
wurden die Tiere in Narkose (2% Isofluran) versetzt, um einen arteriellen
Katheter für die Blutentnahme zu legen. Die Anlage der arteriellen Katheter
erfolgte, um eine verlässliche und standardisierte Bestimmung der Plasma-
Aldosteronspiegel durchführen zu können.

Zur Katheteranlage wurde am narkotisierten Tier ein medianer Hautschnitt am
Hals gesetzt, anschließend erfolgte eine Feinpräparation der Arteria carotis
sowie des begleitenden Nervus vagus und der Vena jugularis. Als nächstes
wurde die Arterie proximal mit einem dünnen Faden ligiert, während das
Gefäßlumen kaudal mit einer Gefäßklemme vorübergehend okkludiert wurde.
Mit einer Irisschere wurde ein Loch in die Arterienwand geschnitten, welches
gerade groß genug war, um einen Polyethylenkatheter (Innendurchmesser
0.28mm, Außendurchmesser 0.61mm, NeoLab, Migge Laborbedarf-Vertriebs
GmbH, Heidelberg) mit einer Führungspinzette einzuführen. Der Katheter
wurde im Gefäß bis zur Klemme vorgeschoben und mit zwei Fäden fixiert. Dann
wurde der arterielle Blutsstrom durch Öffnen der Klemme freigegeben.
Nachdem sich eine pulsatile Blutsäule im Katheter zeigte, wurde dieser mit
einem Metallstift verschlossen. Über einen weiteren Hautschnitt hinter dem Ohr
wurde ein subkutaner Tunnel präpariert, durch den der Katheter zur weiteren
Fixierung geführt wurde.
Nach erfolgreicher Katheteranlage wurden die Tiere in tiefer Narkose (5%
Isofluran) über den arteriellen Katheter entblutet. Hierzu wurde ein 2ml
Eppendorf-Gefäß (Eppendorf AG, Hamburg) verwendet, in welches zunächst
30µl Ethylendiamintetraacetat (pEDTA, Merck, Darmstadt) pipettiert wurden.
Das gewonnene Blut wurde umgehend zentrifugiert, das Plasma in Aliquots
aufgeteilt und bei -80°C tiefgefroren.
Unmittelbar im Anschluss erfolgte die Organentnahme. Hierfür wurden mit einer
Schere Abdomen und Thorax eröffnet, der rechte Vorhof mittels Pinzette
rupturiert und der linke Ventrikel mit einer Butterflykanüle (21 Gauge) kanüliert,
über die der Kreislauf mit 4°C kalter physiologischer Kochsalzlösung (0.9%
NaCl, Berlin Chemie AG, Berlin) perfundiert wurde. Alsdann erfolgte die
Entnahme und Gewichtsbestimmung der Nieren. Von der linken Niere wurde
die obere und untere Polkappe in flüssigem Stickstoff zur späteren Isolierung
von messenger RNA (mRNA) eingefroren. Der Rest der Niere wurde in zwei
gleich große Stücke unterteilt, die in Methylcarnoylösung (MC) bzw.
Gefriermedium (TissueTek, Sakura, Zoeterwoude, Niederlande) eingebettet
wurden. Anschließend wurde das Herz im Ganzen entnommen und gewogen.
Daraufhin erfolgte die Präparation der Aorta bis zum Diaphragma zur Entnahme
verschiedener Teilstücke. Die Aorta aszendens (distal der Aortenwurzel) und

deszendens (in Höhe des Diaphragmas) wurden für spätere Gefrierschnitte in
TissueTek-Medium eingebettet und sofort bei -80°C tiefgefroren. Ein weiteres
Stück der Aorta abdominalis (unterhalb des Diaphragma) wurde in MC
eingebettet.
Die für die Fixation der Gewebe benötigte MC-Lösung bestand aus einem
Gemisch aus 60% Methanol, 30% Chloroform und 10% Eisessig, welches vor
der Organentnahme frisch angesetzt wurde. Nach Fixation der Gewebeproben
in MC-Lösung und Prozessierung in einer aufsteigenden Methanolreihe (70-
100%) und Isopropanol (beide Merck, Darmstadt) erfolgte eine
Gewebeeinbettung in Paraffin. Aus den Gewebeblöcken wurden mit Hilfe eines
Mikrotoms (HM 335 E, Microm International GmbH, Walldorf) 2µm dicke
Schnitte angefertigt und auf Objektträger aufgebracht.
5.8 Färbung und Auswertung histologischer Schnitte in PAS-Technik
Paraffinschnitte der linken Niere sowie der Aorta deszendens wurden nach
Entparaffinisierung und anschließender Rehydratation einer PAS-Färbung
(Perjodsäure-Leukofuchsin-Färbung) unterzogen.
Als Maß für die Gefäßhypertrophie wurde das Wanddicken-Lumen-Verhältnis
(wall to lumen ratio) der Aorta lichtmikroskopisch bestimmt (Mikroskop: Nikon
Eclipse 80i, Nikon, Tokio, Japan). Mittels einer an das Mikroskop
angeschlossenen Kamera (Spot RT, Diagnostic Instruments Inc., Sterling
Heights, MI, USA) wurden unter 40-facher Vergrößerung Bilder der Aorten
gespeichert. Die Berechnung des Wanddicken-Lumen-Verhältnis erfolgte
flächenbasiert nach Kalibrierung für das entsprechende Objektiv. Für die
Eichung wurde ein entsprechender Objektträger mit Linealmarkierung
verwendet. Die Berechnung der Wand- und Lumenfläche erfolgte
computergestützt mit dem Bildbearbeitungsprogramm MetaVue (Version 4.6r9;
Molecular Devices, Sunnyvale, CA, USA), der Quotient aus beiden Flächen
(Wanddicken-Lumen-Verhältnis) wurde in einer Excel-Tabelle gespeichert.
An PAS-gefärbten Nierenschnitten wurde das Ausmaß der glomerulären
Schädigung mittels eines standardisierten Glomerulosklerose-Scores
semiquantitativ erfasst. In jeder Niere wurden 100 Glomeruli zufällig
ausgewählt. Je nach Schweregrad der Glomerulosklerose wurde jedem

Glomerulum ein Score von 0 (keine Glomerulosklerose) bis 4 (schwerste
Glomerulosklerose) zugeordnet (25). Nachdem erste Auswertungen an Ang II
infundierten Mäusen lediglich eine leichtgradige glomeruläre Schädigung
erkennen ließen (Score von 0-2), beschlossen wir, anstelle des Scores den
Anteil geschädigter Glomeruli in Prozent anzugeben.
5.9 Immunhistochemische Färbungen
Zum Nachweis der Ang II-induzierten Inflammation in der Niere wurde die
Makrophageninfiltration mittels spezifischer Antikörper analysiert. Für
Paraffinschnitte erfolgte eine Färbung unter Verwendung eines Antikörpers
gegen das Antigen F4/80 (Ratte anti Maus, Klon CI:A3-1, Verdünnung 1:100,
AbD Serotec, MorphoSys AG, Planegg). Dieser Klon erkennt das Maus F4/80
Antigen, ein 160kD Glykoprotein, welches von Makrophagen der Maus
exprimiert wird.
Vor der Inkubation erfolgte ein hitzeinduziertes Antigen-Retrieval durch
Aufkochen der Objektträger in einer Zitratpufferlösung. Nach Inkubation mit
dem Primärantikörper über Nacht erfolgte die Inkubation mit einem Biotin-
markierten Sekundärantikörper (Pferd-anti-Ratte, Verdünnung 1:1000, Vector
Laboratories, Inc., Burlingame, CA, USA) über eine Stunde. Anschließend
wurden die Objektträger mit Avidin-D (Verdünnung 1:2000, Vector Laboratories,
Inc., Burlingame, CA, USA) inkubiert. Avidin D ist an das Enzym Peroxidase
aus der Meerrettich-Pflanze (Horseradish peroxidase) gekoppelt und bindet mit
hoher Affinität an die Biotinylgruppe der Sekundärantikörper. Mittels
Diaminobenzidin (DAB-Kit, Vector Laboratories, Inc., Burlingame, CA, USA)
wurde die Färbung entwickelt. Die im Kit enthaltenen H2O2 –Moleküle werden
dabei von der Peroxidase des Avidin umgesetzt. Es entstehen Protonen, die
das Diaminobenzidin oxidieren und so braun färben. Die Spezifität der Färbung
wurde bei jedem Tier durch Weglassen des Primärantikörpers
(Negativkontrolle) überprüft.
Neben der lichtmikroskopischen Immunhistochemie wurde für F4/80 auch eine
Immunfluoreszenz durchgeführt. Die Schnitte wurden hierfür mit dem
identischen Primärantikörper analog zur Lichtmikroskopie über einen Zeitraum
von zwei Stunden inkubiert. Als Sekundärantikörper wurde ein

fluoreszenzmarkierter Antikörper (Alexa-555, Ziege anti Ratte, Verdünnung
1:250, Molecular Probes, Inc., Eugene, OR, USA) verwendet. Bei dem
verwendeten Sekundärantikörper erscheinen die Makrophagen unter dem
Immunfluoreszenzmikroskop in Rotfluoreszenz. Zellkerne wurden mittels
Hoechst 33342 (Verdünnung 1:1000, Molecular Probes, Inc., Eugene, OR,
USA) angefärbt. Dieser Farbstoff bindet an doppelsträngige DNA und emittiert
eine Blaufluoreszenz. Zur Färbung glatter Muskelzellen erfolgte zudem eine Ko-
Inkubation mit einem Fluoreszeinisothiocyanat (FITC)-markierten Antikörper
gegen α-Actin glatter Muskelzellen der Gefäße (Verdünnung 1:500, Sigma
Chemical Co., St. Louis, MO, USA). Glatte Muskelzellen der Gefäße erscheinen
so unter dem Immunfluoreszenzmikroskop in Grünfluoreszenz. Die Analyse und
Auswertung erfolgte unter dem Fluoreszenzmikroskop (Nikon Eclipse 80i,
Nikon, Tokio, Japan) unter Verwendung der für die Emission der verwendeten
Sekundärantikörper benötigten Filter. Mittels der am Mikroskop integrierten
Digitalkamera (Spot RT, Diagnostic Instruments Inc., Sterling Heights, MI, USA)
wurde die Zahl der F4/80 positiven Zellen pro Gesichtsfeld bestimmt. Sowohl
für die Licht- als auch die Fluoreszenzmikroskopie wurde die Anzahl F4/80
positiver Zellen an 20 zufällig gewählten, nicht überlappenden Kortexschnitten
ausgezählt.
Zusätzlich zur F4/80 Färbung erfolgte an Gefrierschnitten eine Färbung mit dem
Maus-Makrophagenmarker MOMA-2 (Verdünnung 1:100, BMA Biochemicals
AG, Augst, Switzerland). Die Färbung wurde floureszenzmikroskopisch
dargestellt, eine weitere Quantifizierung wurde nicht vorgenommen.
Zur Erfassung der Ang II-induzierten renalen interstitiellen Fibrose sowie
Glomerulosklerose erfolgte eine immunhistochemische Färbung für Kollagen I
(Verdünnung 1:100, Biogenesis Ltd., Poole, England, UK) und Kollagen IV
(Verdünnung 1:500, Southern Biotechnology Associates Inc., Birmingham, AL,
USA) als Proteine des Bindegewebes.
Die Expression von Kollagen I in der Niere dient als Marker für das Ausmaß
einer renalen interstitiellen Fibrose. Die semiquantitative Auswertung erfolgte
mittels Lichtmikroskopie unter Verwendung eines Okulars, in welches ein
Rastergitter bestehend aus 100 Kästchen eingearbeitet ist. Mittels dieses
Rasters wurde die Kollagenfärbung im Interstitium des Nierenkortex (Anzahl
positiver Felder von 100) quantifiziert. Die Auswertung erfolgte an insgesamt 20

nicht überlappenden Gesichtsfeldern unter 400-facher Vergrößerung. Auf diese
Weise wurde für jedes Tier das Ausmaß der Fibrose im Interstitium der Niere
semiquantitativ abgeschätzt.
Die Auswertung der Kollagen IV-Färbung als Maß der glomerulären Sklerose
erfolgte computergestützt mit dem Bildbearbeitungsprogramm MetaVue
(Hersteller und Version s.o.). Pro Nierenschnitt wurden bei dieser Technik 30
Glomeruli zufällig ausgewählt und zunächst deren Fläche bestimmt. Für die
Braunfärbung, die Kollagen IV anzeigt, wurde ein Schwellenwert, ab dem diese
von der Software detektiert wurde, definiert. Im Anschluss wurde der
prozentuale Anteil von Kollagen IV an der Gesamtfläche eines jeden
Glomerulus bestimmt. Zur besseren Vergleichbarkeit der Messergebnisse
wurden für alle Schnitte die gleichen Geräteeinstellungen verwendet.
5.10 Oxidativer Stress (Dihydroethidium-Fluoreszenz)
Unfixierte Gefrierschnitte (10µm) der Aorta aszendens (distal der Aortenwurzel)
und Aorta deszendens (in Höhe des Diaphragma) wurden mit dem Fluorophor
Dihydroethidium (DHE, 8µM, Molecular Probes, Inc., Eugene, OR, USA) über
eine Stunde bei 37°C unter Lichtabschirmung inkubiert.
In Anwesenheit reaktiver Sauerstoffradikale (z.B. Superoxidanionen) wird
Dihydroethidium zu Ethidiumbromid oxidiert, welches mit der DNA der
entsprechenden Zellen interkaliert und nach Anregung bei einer Wellenlänge
von 488nm im Fluoreszenzmikroskop Licht der Wellenlänge 610nm emittiert
(Rotfluoreszenz).
Um die Spezifität der Fluoreszenz zu verifizieren, wurden zur Kontrolle einige
Schnitte vor Inkubation mit Dihydroethidium mit dem Enzym Polyethylen-Glycol-
Superoxid-Dismutase (Sigma Chemical Co., St. Louis, MO, USA) vorbehandelt,
welches freie Sauerstoffradikale degradiert.
Die Auswertung des Fluoreszenzsignals erfolgte unter dem
Fluoreszenzmikroskop (Nikon Eclipse 80i, Nikon, Tokio, Japan) mit integrierter
Digitalkamera (Spot RT, Diagnostic Instruments Inc., Sterling Heights, MI,
USA). Mit Hilfe des Programms MetaVue (Hersteller und Version s.o.) wurde für
jeden Schnitt das Fluoreszenzsignal detektiert und das Flächenverhältnis von
fluoreszierenden Pixeln zur Gesamtpixelzahl der Wandfläche des Gefäßes

bestimmt. Um Vergleichbarkeit zu gewährleisten, wurden für alle Schnitte die
gleichen Einstellungen verwendet.
5.11 Messung der ADMA-Spiegel
Die Bestimmung der ADMA-Plasmaspiegel erfolgte mittels eines kommerziellen
ELISA Assays (DLD Diagnostika GmbH, Hamburg). Dabei handelt es sich um
einen kompetitiven ELISA, bei dem das Antigen in der Probe mit in der
Festphase gebundenem Antigen um eine feste Anzahl freier
Antikörperbindungsstellen konkurriert. Die ADMA-Konzentration der
Plasmaproben verhält sich somit umgekehrt proportional zu der Anzahl der
gemessenen Antigen-Antikörper-Komplexe.
In einem ersten Reaktionsschritt wurde ADMA durch Inkubation mit einem
Acylierungsreagenz in N-Acyl-ADMA überführt. Die entsprechend
vorbehandelten Plasmaproben, Kontrollen und Standards wurden anschließend
mit Antiserum (Kaninchen-anti-N-Acyl-ADMA Antikörper) inkubiert. Die
Antikörper binden kompetitiv entweder an das freie Antigen der Proben, oder an
das gebundene Antigen, mit dem die Platten beschichtet sind. Die Inkubation
erfolgte über Nacht im Kühlschrank bei 4°C.
Am Folgetag wurde das Enzym-Konjugat (Anti-Kaninchen-IgG-Peroxidase)
hinzugefügt und der ELISA mittels Substratlösung (Tetramethylbenzidin)
entwickelt. Nach Inkubation über einen Zeitraum von 30 Minuten wurde die
Reaktion durch eine Stopplösung beendet und die Absorption bei einer
Wellenlänge von 450nm photometrisch gemessen.
Alle Proben wurden doppelt bestimmt und der Mittelwert berechnet. Der
Variationskoeffizient innerhalb des Assays lag bei 8.7%.
Bei den Tieren, die Ang II in der Dosis 3.0µg/kg/min erhielten, wurden die
ADMA-Spiegel zusätzlich zum ELISA auch mittels
Tandemmassenspektrometrie (LC-MS) bestimmt, und zwar vor und nach Ang
II-Gabe. Die Analyse erfolgte in Kollaboration mit Herrn Prof. Rainer Böger
(Institut für experimentelle und klinische Pharmakologie, Universitätsklinik
Hamburg-Eppendorf) nach der von Schwedhelm et al. beschriebenen Methode
(69).

5.12 Bestimmung der Aldosteronspiegel
Im Rahmen der Tötung der Mäuse wurde den Tieren u.a.
Ethylendiamintetraessigsäure (EDTA) - Plasma zur Bestimmung der Plasma-
Aldosteronspiegel entnommen. Dies diente der Kontrolle, ob Ang II erfolgreich
infundiert wurde.
Die Bestimmung der Aldosteronspiegel erfolgte durch einen
Radioimmunoassay (RIA; Adaltis Italia S.p.A., Bologna, Italien, Handelsname
Aldosteron MAIA). Hierbei handelt es sich um einen kompetitiven Assay, bei
dem unmarkiertes, „kaltes“ Antigen der zu messenden Probe und radioaktiv
markiertes, „heißes“ Antigen (Tracer) um eine definierte Anzahl an
Antikörperbindungsstellen konkurrieren. Mittels eines magnetisch markierten
Sekundärantikörpers können gebundenes und ungebundenes Antigen getrennt
werden.
Nach Inkubation mit dem Tracer in einem Reagenzröhrchen und Zugabe des
magnetischen Antikörpers werden die Proben auf eine Magnetplatte gebracht.
Dann wird der Überstand dekantiert, während die Röhrchen noch auf der Platte
sind. Die Antigen-Antikörper-Komplexe verbleiben so im Röhrchen, das
ungebundene Antigen wird entfernt. Die Messung der Radioaktivität erfolgt über
eine Minute im Gammacounter (1261 Multigamma, LKB Wallac, Turku,
Finnland). Die counts per minute (cpm)-Werte der doppelt gemessenen
Ansätze wurden gemittelt.
Da der Assay kompetitiv ist, ist die gemessene Radioaktivität der Proben
umgekehrt proportional zum Aldosterongehalt der Probe. Alle gemessenen
Werte wurden auf den Nullstandard (B0) bezogen, der die höchste Radioaktiviät
aufwies, die mit 100% gleichgesetzt wurde. Die so normierten Werte der
mitgeführten Standards wurden anschließend gegen B0 logarithmisch
aufgetragen. Mittels dieser Standardkurve konnte den Messwerten der Proben
eine absolute Aldosteronkonzentration zugeordnet werden.
5.13 Bestimmung der mRNA-Expression mittels Real-Time RT-PCR
Aus den Polkappen der linken Niere von Mäusen, die PBS oder Ang II in der
Dosis 1.0µg/kg/min erhielten, wurde die mRNA-Expression verschiedener
Proteine mittels Real-Time RT-PCR bestimmt. Von den ADMA-generierenden

Enzymen wurde die Expression von PRMT1 und PRMT3 näher untersucht, von
den ADMA-abbauenden Enzymen die murine Expression von DDAH1 und
DDAH2. Des Weiteren wurde die Kollagen I mRNA Expression in der Niere
gemessen.
Die RNA-Extraktion erfolgte nach der Methode von Chomczynski (12). Hierbei
handelt es sich um ein 1-Schritt-Verfahren, welches nach dem Prinzip einer
Flüssigphasenseparation abläuft. Zunächst wird das Gewebe mechanisch
homogenisiert und mit TriFast-Reagenz (peqGOLD, Peqlab, Erlangen)
versetzt. Es bildet sich eine wässrige Phase, die die RNA enthält, welche
abpipettiert und weiter verarbeitet werden kann. Nach adäquater Verdünnung
der gewonnenen RNA mit RNAse freiem Wasser wird diese in copy DNA
(cDNA) transkribiert. Hierfür wird eine Reverse-Transkriptase-PCR (RT-PCR)
durchgeführt, die ihrem Prinzip nach einer normalen PCR ähnelt, außer dass
der Master-Mix eine reverse Transkriptase enthält. Es wurden Kontrollen
sowohl ohne reverse Transkriptase als auch ohne RNA mitgeführt. Als Primer
dienten Random Hexamere, es wurde die Multiscribe Reverse Transcriptase
verwendet. Der Master-Mix (TaqMan Reverse Transcription Reagents, Applied
Biosystems by Roche Molecular Systems Inc., Branchburg, NJ, USA) wurde
gemäß Herstellerangaben angesetzt, die Volumina wurden so gewählt, dass
sich ein Probenvolumen von 20µl ergab.
Zur Quantifizierung der so gewonnenen und adäquat verdünnten cDNA wurde
als nächstes die eigentliche Real-Time-PCR durchgeführt (je 40 Zyklen). Zur
Normierung der Ergebnisse wurde 18s ribosomale RNA (rRNA) als endogene
Kontrolle mitgeführt. Folgende Primer (MWG Biotech AG, Ebersberg) wurden
eingesetzt:

Tabelle 2. Primer für die Real-Time-PCR
Zum Primerdesign wurde die Software Primer Express (Version 2.0, Applied
Biosystems, Foster City, CA, USA) verwendet. Die Primersequenzen wurden
mit Hilfe des Basic Local Alignment Search Tool (BLAST) des National Center
for Biotechnical Information (NCBI) auf ihre Spezifität überprüft. Die DDAH1
Primer wurden spezifisch für die Maus-Isoform des Enzyms konzipiert, um eine
Kreuzreaktion mit dem humanen Transgen auszuschließen.
Zur Erzeugung des Fluoreszenzsignals wurde der SYBR-Green Master-Mix von
Applied Biosystems (Roche Molecular Systems Inc., Branchburg, NJ, USA)
verwendet. Als Gerät diente das Sequence Detection System 7000 (Applied
Biosystems, Foster City, CA, USA) mit ABI Prism 7000 SDS-Software (gleicher
Hersteller). Es wurden 96-Well Platten verwendet, alle Proben wurden doppelt
bestimmt. Durch anschließende Schmelzkurvenanalyse wurde die Spezifität
des SYBR-Green-Signals verifiziert.
Zur Berechnung der Ergebnisse wurde anhand mitgeführter Kalibratoren eine
Eichgerade erstellt, mittels der die cycle of threshold (ct)-Werte der Proben in
Kopienzahlen umgerechnet wurden. Die geschah sowohl für die 18s rRNA als
auch für die spezifischen mRNAs, um im Anschluss die Ergebnisse als relative
Kopienzahl bezogen auf 18s rRNA anzugeben.
5.14 Statistik
Die statistische Auswertung erfolgte mit dem Statistikprogramm SPSS (Version
16.0, SPSS Inc., Chicago, IL, USA). Initial erfolgte eine Überprüfung aller
Variablen auf Normalverteilung mittels Kolmogorov-Smirnov Test.
Für die anschließende statistische Auswertung kam ein univariates, allgemein-
lineares Modell zum Einsatz, um Unterschiede zwischen den Genotypen und
Behandlungsgruppen zu ermitteln. Dafür wurden Genotyp (WT versus TG) und
Behandlungsgruppen (PBS versus Ang II 1.0µg/kg/min. versus Ang II
Gen Forward-Primer Reverse-PrimerDDAH1 CATGTCTTGCTGCACCGAAC GACCTTTGCGCTTTCTGGDDAH2 GGTTGATGGAGTGCGTAAAGC TCCACAATTCGGAGTCCCAAPRMT1 AATGGGATGAGCCTCCAGC TGCTTGGCCACAGGAAACTTPRMT3 TTACCCTGAGAACCACAAAGACG AGTACCCAGCAACTGCCGTGKollagen I TCACCTACAGCACCCTTGTGG CCCAAGTTCCGGTGTGACTC

3.0µg/kg/min.) als feste Faktoren gewählt, was für jeden der Faktoren einen
eigenen p-Wert zur Überprüfung der Nullhypothese lieferte. Bei multiplem
Testen (Behandlungseffekte) wurde ein Bonferroni-Test als Post-Hoc-Test
durchgeführt, um das Signifikanzniveau zu adjustieren.
Bei einem p-Wert von <0.05 wurde ein Ergebnis als statistisch signifikant
akzeptiert. Die Messwerte sind als Mittelwert ± SEM (Standardfehler des
Mittelwertes) angegeben.
Bei Box-plot Abbildungen repräsentiert der obere und untere Rahmen die 75.
bzw. 25. Perzentile, die horizontale Linie in der Box die 50. Perzentile, welche
dem Median entspricht.
Korrelationskoeffizienten zwischen zwei Variablen wurden mittels Pearson Test
berechnet. Die Konkordanz der beiden Meßmethoden für die ADMA-Analytik
(ELISA versus LC-MS) wurde mittels Bland-Altman Plot (Auftragen der
Differenz beider Meßmethoden gegen den gemeinsamen Mittelwert) sowie
durch Ermittelung des Konkordanz-Korrelationskoeffizienten nach Lin (39) wie
folgt berechnet:
Hierbei sind µx und µy die Mittelwerte beider Variablen, σ2x und σ2
y die
korrespondierenden Varianzen, und ρ der bivariate Korrelationskoeffizient
zwischen beiden Variablen.
ρc=
2ρσxσy
σ2x + σ2
y + (µ
x - µ
y)2
6 Ergebnisse
6.1 Tiere
Insgesamt verstarben 3 Tiere unter Ang II Behandlung vor Abschluss des
Versuchs. Hierbei handelte es sich um ein WT-Tier, welches mit Ang II in der
Dosierung 1.0µg/kg/min infundiert wurde, sowie zwei TG Mäuse aus der
Hochdosisgruppe (3.0µg/kg/min). Das WT Tier starb zwei Wochen nach
Pumpenimplantation, eine postmortale Obduktion ergab keinen eindeutigen
Hinweis auf die zugrunde liegende Todesursache. Die beiden TG Mäuse
verstarben vier bzw. sechs Tage nach Pumpenimplantation, die Autopsie zeigte
bei beiden Tieren einen Hämatothorax, als wahrscheinlichste Todesursache
kommt somit eine Aortendissektion in Betracht.
Bei den Kontrollmäusen (PBS-Infusion) kam es zu einem signifikanten Anstieg
des Körpergewichtes (korrigiert auf das Pumpengewicht) während der
vierwöchigen Behandlung, wohingegen bei den Ang II infundierten Tieren ein
signifikanter Gewichtsverlust auftrat (Tabelle 3).
Tabelle 3. Gewichtsverlauf unter Ang II Infusion
WT TG
PBSAng II
1.0µg/kg/min
Ang II 3.0µg/kg/
minPBS
Ang II 1.0µg/kg/mi
n
Ang II 3.0µg/kg/mi
n
Gewicht Tag 0 (g) 25.9 ±0.4
27.9 ±0.5
30.6 ±0.4 25.9±1.0 26.4±0.4 30.3±0.6
Gewicht Tag 28 (g) 28.3 ±0.3
26.0 ±0.6
28.2 ±0.6 27.0±0.7 25.9±0.6 28.0±0.7
∆Gewicht Tag 28-Tag 0 (g)
2.4 ±0.4*
-1.9 ±0.6*
-2.4 ±0.4‡ 1.2±0.4* -0.6±0.6 -2.3±0.6†
* p<0.05, † p<0.01, ‡ p<0.001
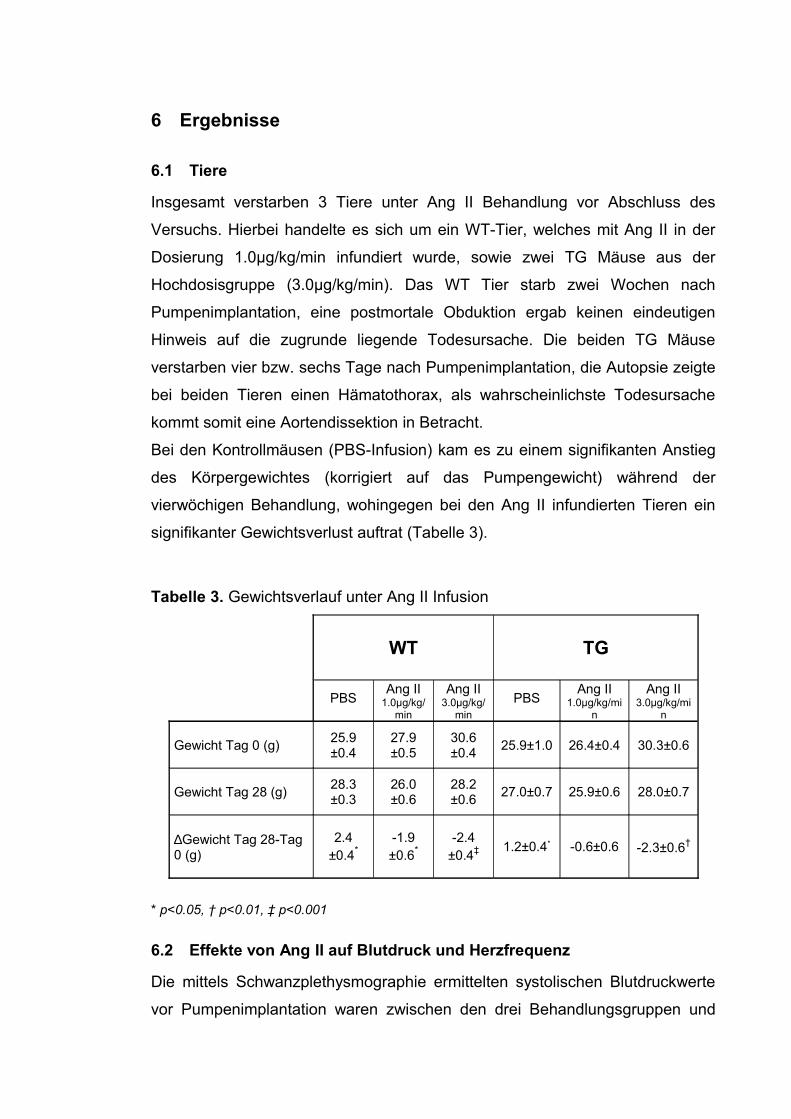
6.2 Effekte von Ang II auf Blutdruck und Herzfrequenz
Die mittels Schwanzplethysmographie ermittelten systolischen Blutdruckwerte
vor Pumpenimplantation waren zwischen den drei Behandlungsgruppen und

den beiden untersuchten Genotypen identisch. DDAH transgene Mäuse hatten
tendenziell niedrigere Blutdruckwerte, dieser Effekt war statistisch jedoch nicht
signifikant.
Die Infusion von Ang II führte bei allen behandelten Tieren zu einem raschen
und signifikanten Blutdruckanstieg (Abbildung 7). Das Blutdruckmaximum
wurde nach zwei Wochen erreicht, danach blieb der Blutdruck konstant. Der
Blutdruckanstieg war zwischen den Genotypen identisch, interessanterweise
zeigte sich kein additiver Effekt der höheren Ang II Dosierung auf den
Blutdruck, der mittlere systolische Blutdruckanstieg war in beiden Dosierungen
identisch und lag bei ca. 35-45mmHg (Abbildung 7). Bei Mäusen der
Kontrollgruppe (Infusion von PBS) blieb der Blutdruck über den
Beobachtungszeitraum von vier Wochen konstant.
In der bivariaten Korrelation nach Pearson ergab sich weder für den
Ausgangsblutdruck bzw. Endblutdruck noch für die Blutdruckdifferenz eine
Korrelation mit den ADMA-Plasmaspiegeln (Endblutdruck: r=0.02, p= nicht
signifikant (n.s.); Blutdruckdifferenz: r=-0.17, p=n.s.).
Die Infusion von Ang II führte zu einer Abnahme der Herzfrequenz. Dieser
Effekt war nach zwei Wochen statistisch signifikant (WT Mäuse, PBS vs. Ang II
1.0µg/kg/min vs. Ang II 3.0µg/kg/min: 646 ± 13 vs. 537 ± 15 vs. 530 ± 19
Schläge/min, p<0.001; TG Mäuse, PBS vs. Ang II 1.0µg/kg/min vs. Ang II
3.0µg/kg/min: 629 ± 15 vs. 574 ± 14 vs. 541 ± 31 Schläge/min, p<0.001).
Auch der Genotyp beeinflusste die Herzfrequenz. In einem allgemeinen
linearen Modell für Messwiederholungen mit den verschiedenen
Messzeitpunkten als Innersubjektfaktoren zeigte sich, dass der Einfluss des
Genotyp auf die Herzfrequenz signifikant war (p= 0.013).
Abbi
ldun
g 7.
Effe
kt v
on A
ng II
auf d
en s
ysto
lisch
en B
lutd
ruck
.

Metabolische Käfige
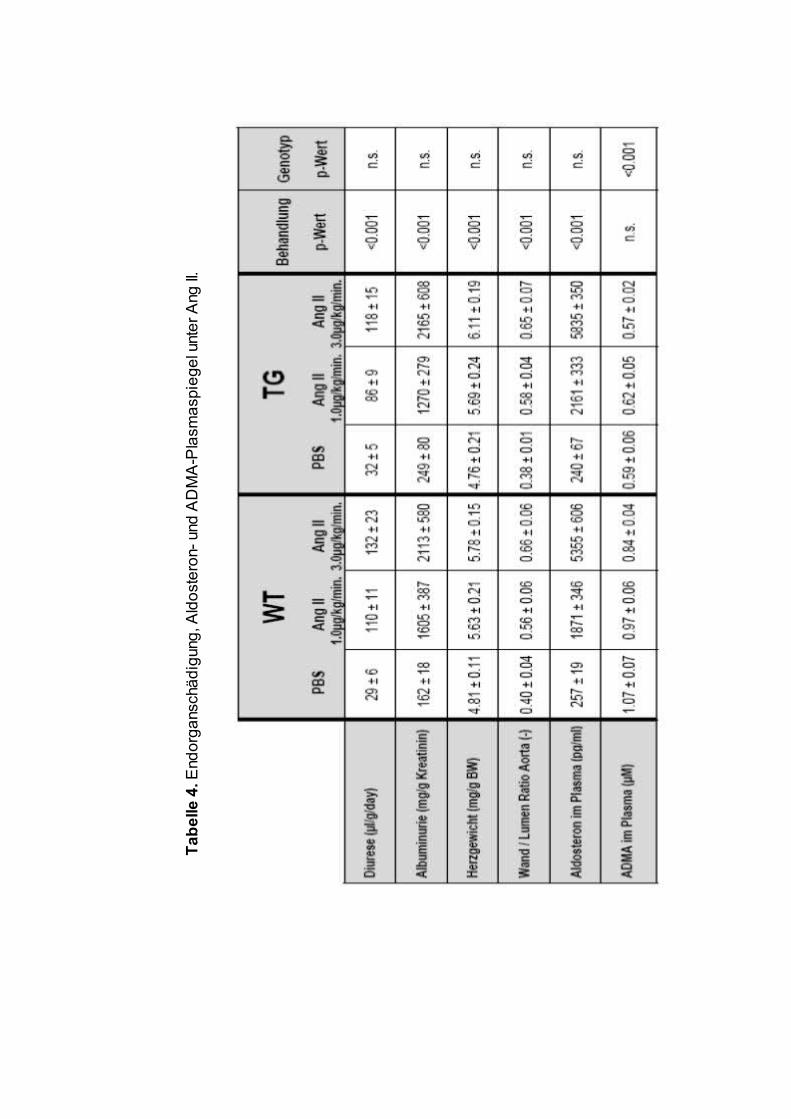
Die Urinausscheidung über 24 Stunden war vor der Pumpenimplantation
zwischen allen Behandlungsgruppen gleich, der Mittelwert lag bei den WT
Mäusen bei 46.6 ± 3.3µl/g/Tag, bei den TG Tieren bei 49.2 ± 4.2µl/g/Tag
(p=n.s.). Die Infusion von Ang II, nicht jedoch von PBS führte zu einem
signifikanten Anstieg der Diurese (Tabelle 4). Die Urinausscheidung lag bei den
mit Ang II behandelten Mäusen in etwa dreifach über der Menge der
Kontrolltiere und war zwischen beiden Ang II Dosierungen vergleichbar.
Unterschiede zwischen den Genotypen fanden sich nicht (Tabelle 4).
Neben einer Steigerung der Diurese führte die Ang II Gabe auch zu einer
signifikanten und vergleichbaren Zunahme der Proteinurie bei beiden
Genotypen.
6.3 Herzhypertrophie und vaskuläre Hypertrophie
Die Infusion von Ang II führte zu einer signifikanten Zunahme des auf die
Körpermasse normierten Herzgewichtes (Tabelle 4). Es zeigte sich dabei kein
dosisabhängiger Effekt von Ang II auf die Herzhypertrophie, des Weiteren
ergaben sich keinerlei Unterschiede zwischen den beiden Genotypen.
Eine analoge Konstellation ergab sich für das Wanddicken-Lumen-Verhältnis
der Aorta. Auch hier zeigte sich eine deutliche Hypertrophie der Gefäßwand
unter Ang II (Tabelle 4, Abbildung 8), wiederum ohne Dosis- bzw. Genotyp-
Effekt.
6.4 Plasma-Aldosteron-Spiegel unter Ang II-Infusion
Die mittels Radioimmunoassay ermittelten Plasma-Aldosteronspiegel waren bei
den mit Ang-II behandelten Tieren gegenüber den PBS-Tieren signifikant erhöht
(Tabelle 4). Hierbei fand sich ein klar dosisabhängiger Effekt von Ang II, der
Anstieg der Plasma-Aldosteronspiegel war zwischen beiden Genotypen
identisch.
Tabe
lle 4
. End
orga
nsch
ädig
ung,
Ald
oste
ron-
und
AD
MA
-Pla
smas
pieg
el u
nter
Ang
II.
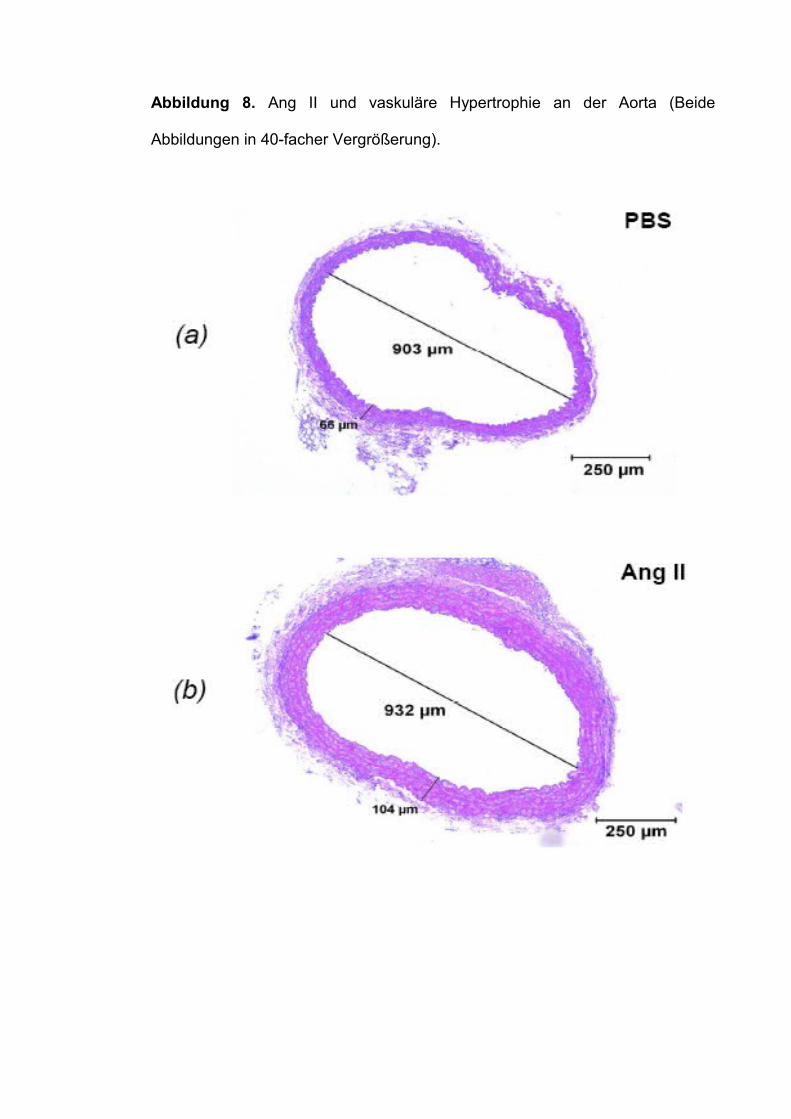
Abbildung 8. Ang II und vaskuläre Hypertrophie an der Aorta (Beide
Abbildungen in 40-facher Vergrößerung).

6.5 ADMA-Plasmaspiegel unter Ang II-Infusion
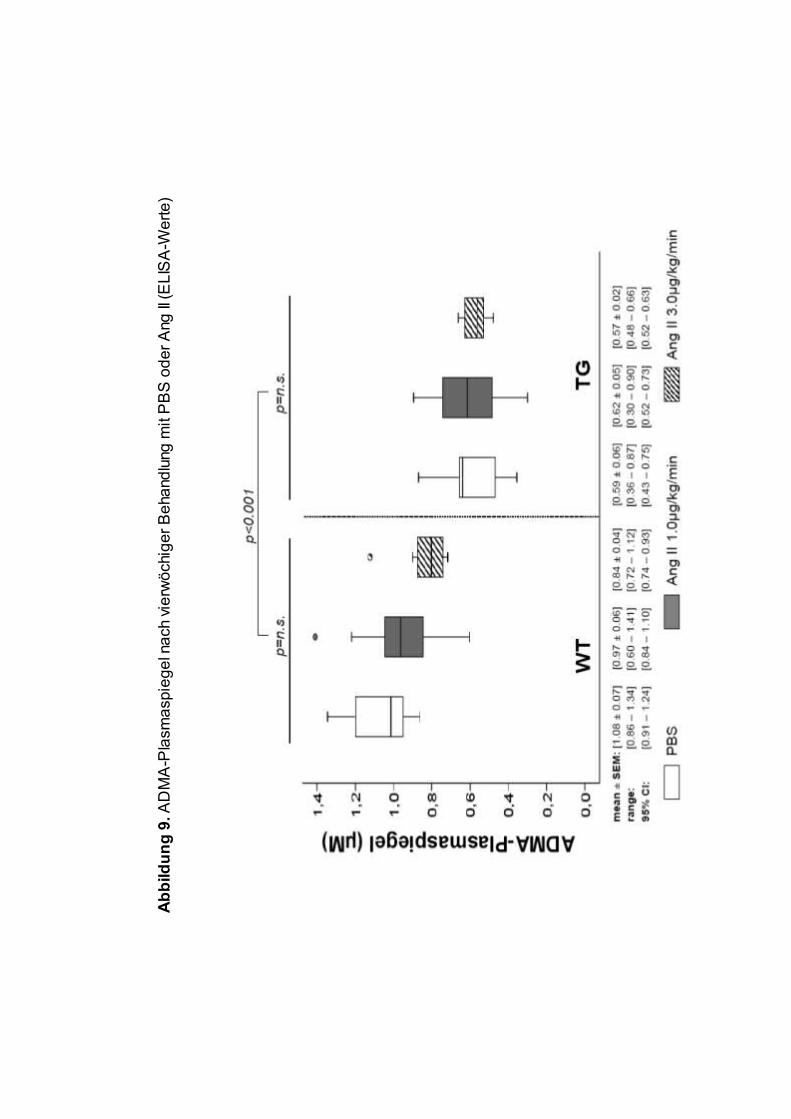
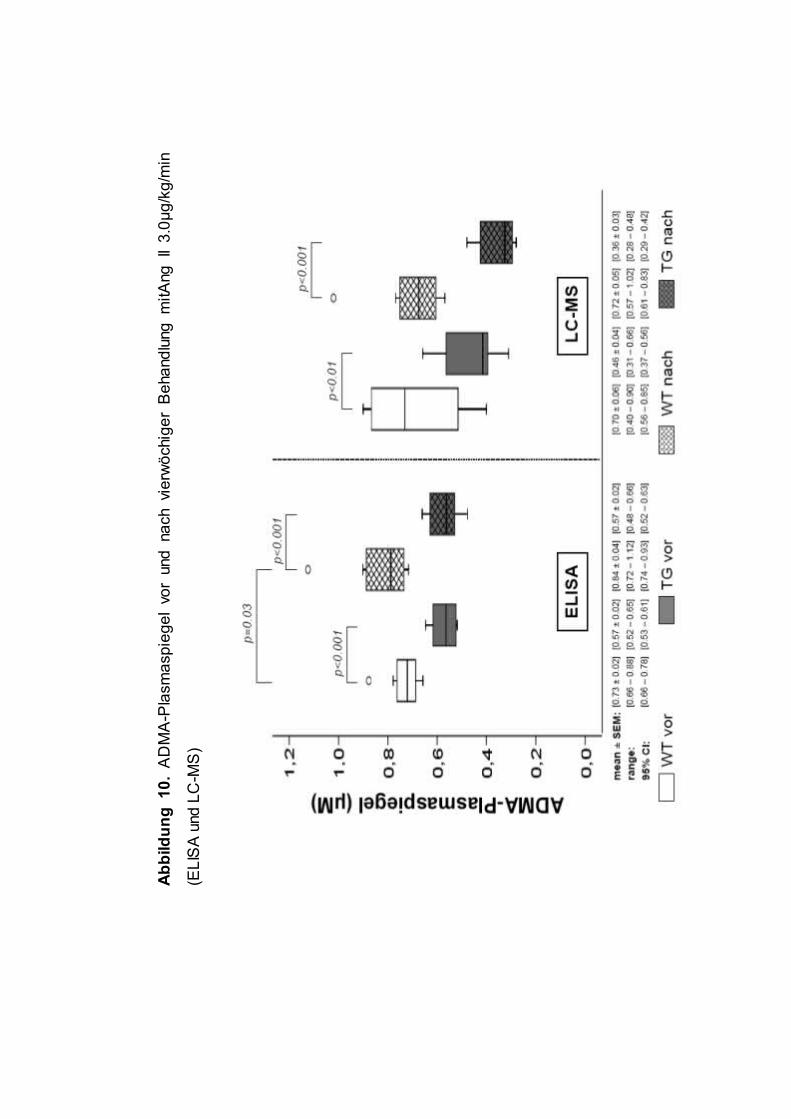
Die gemessenen ADMA-Werte sind Abbildung 9 und Abbildung 10 zu
entnehmen. Die ADMA-Spiegel wurden mit ELISA und LC-MS gemessen,
wobei die Tandemmassenspektrometrie nur bei den Hochdosis-Tieren zur
Anwendung kam. DDAH transgene Mäuse hatten erwartungsgemäß
hochsignifikant niedrigere ADMA-Plasmaspiegel, dies galt für alle untersuchten
Gruppen sowohl vor als auch nach Ang II-Infusion. Der Mittelwert der ADMA
Plasmaspiegel (ELISA) aus allen drei Gruppen lag bei den WT Mäusen bei 0.95
± 0.04µM, bei den DDAH TG Mäusen bei 0.60 ± 0.03µM (WT vs. TG,
p<0.00001), der Effekt des Transgens auf die ADMA-Plasmaspiegel lag somit
bei ca. 37%.
Ein vergleichbarer Unterschied der ADMA-Spiegel von ca. 35% zeigte sich
auch mittels LC-MS in der Hochdosis Ang II Gruppe (WT vs. TG: 0.70 ± 0.06
vs. 0.46 ± 0.04µM, p=0.007). Demgegenüber waren die Werte für SDMA bzw.
L-Arginin zwischen beiden Genotypen identisch (SDMA: 0.17 ± 0.02 vs. 0.17 ±
0.01µM; L-Arginin: 66 ± 4 vs. 76 ± 7µM, p=n.s.).
Entgegen unserer Arbeitshypothese führte die Infusion von Ang II zu keiner
signifikanten Erhöhung der mittels ELISA am Ende der vierwöchigen
Behandlungsphase gemessenen ADMA-Spiegel gegenüber der Kontrollgruppe
(Abbildung 9). In der Hochdosisgruppe Ang II, in der ADMA sowohl mittels
ELISA als auch LC-MS sowohl vor als auch nach Behandlung gemessen
wurde, zeigte sich ebenfalls kein Effekt des Vasopressors auf die ADMA-
Plasmaspiegel (Abbildung 10). Lediglich mittels ELISA fand sich ein marginaler,
wenngleich signifikanter, Anstieg der ADMA-Spiegel im Vorher-Nachher
Vergleich (Abbildung 10). Dieser konnte mittels LC-MS jedoch nicht bestätigt
werden (Abbildung 10), zudem waren die Werte unter Ang II vergleichbar zu
denen der Kontrolltiere.
Die ADMA-Plasmaspiegel wiesen keine Korrelation mit den Aldosteronwerten
(r= -0.22, p=n.s.) auf.
Im direkten Methodenvergleich (ELISA versus LC-MS) zeigte sich eine
exzellente Korrelation der ADMA-Werte (r= 0.95, p<0.001, Abbildung 11),
wenngleich die Werte mittels LC-MS insgesamt niedriger ausfielen. Die mittlere

Differenz zwischen ELISA und LC-MS gemessenen ADMA-Spiegeln lag bei
0.12µM bzw. 17% (0.685 vs. 0.569µM, p=0.008). Im Bland-Altman Plot wurde
die Differenz aus beiden Meßmethoden (ELISA minus LC-MS) gegen den
gemeinsamen Mittelwert (ELISA + LC-MS / 2) aufgetragen. Hierbei lagen
nahezu alle Messwerte innerhalb der zweifachen Standardabweichung der
mittleren Differenz, was für eine hinreichende Übereinstimmung beider
Meßmethoden spricht (Abbildung 12).
Der Konkordanz-Korrelations-Koeffizient (CCC) lag bei 0.68, was einer starken
Konkordanz beider Meßmethoden entspricht (Tabelle 5).
Tabelle 5: Interpretation des Konkordanz-Korrelations-Koeffizienten (CCC),
nach (39).
CCC-Wert Konkordanzniveau<0,10 Keine Konkordanz
0.10-0.40 Schwach
0.41-0.60 Erheblich
0.61-0.80 Stark
0.81-1.00 Nahezu vollständig
Abbi
ldun
g 9.
AD
MA
-Pla
smas
pieg
el n
ach
vier
wöc
hige
r Beh
andl
ung
mit
PB
S o
der A
ng II
(ELI
SA
-Wer
te)
Abbi
ldun
g 10
. A
DM
A-P
lasm
aspi
egel
vor
und
nac
h vi
erw
öchi
ger
Beh
andl
ung
mitA
ng I
I 3.
0µg/
kg/m
in
(ELI
SA
und
LC
-MS
)
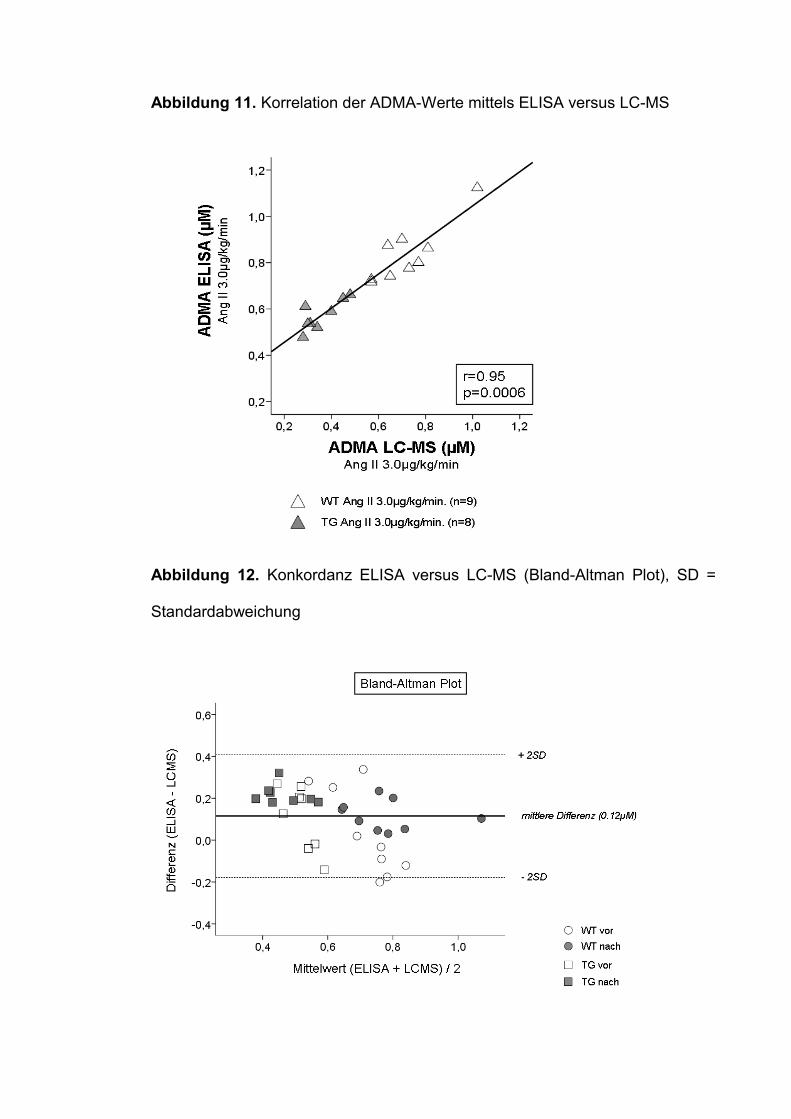
Abbildung 11. Korrelation der ADMA-Werte mittels ELISA versus LC-MS
Abbildung 12. Konkordanz ELISA versus LC-MS (Bland-Altman Plot), SD =
Standardabweichung

6.6 Glomerulosklerose, interstitielle Fibrose und Inflammation
Das Ausmaß der Glomerulosklerose wurde an PAS gefärbten Schnitten der
Niere bestimmt. Unter Infusion von Ang II kam es zu einem signifikanten
Anstieg der Glomerulosklerose, wobei die Anzahl bzw. das Ausmaß der
geschädigten Glomeruli milde war (Abbildung 13a+c). Das Schädigungsmuster
der Niere war zwischen beiden untersuchten Genotypen identisch. Dieser
Befund konnte immunhistochemisch mittels Kollagen IV Färbung bestätigt
werden, hierbei zeigte sich nur in der Hochdosisgruppe ein signifikanter Anstieg
der Kollagen IV Expression in der Niere (WT: PBS vs. Ang II 1.0µg/kg/min vs.
Ang II 3.0µg/kg/min: 10.9 ± 0.8% vs. 11.2 ± 0.9% vs. 18.3 ± 0.8%; TG: PBS vs.
Ang II 1.0µg/kg/min vs. Ang II 3.0 µg/kg/min: 9.6 ± 0.5% vs. 9.6 ± 0.7% vs. 18.6
± 0.6%; p-Wert Behandlung: p<0.001, p-Wert Genotyp: p=n.s.).
Bei Kollagen I als Marker für die renale interstitielle Fibrose war eine
signifikante Erhöhung bei Behandlung mit Ang II festzustellen (Abbildung 13b).
Interessanterweise war das Ausmaß der interstitiellen Fibrose bei den DDAH
transgenen Mäusen signifikant geringer ausgeprägt (Abbildung 13b). Der
immunhistochemische Befund konnte mittels RT-PCR auch auf
transkriptioneller Ebene bestätigt werden. Hier zeigte sich eine verstärkte
mRNA-Expression für Kollagen I (siehe Tabelle 6).
Die F4/80-Färbung zur Makrophagendetektion zeigte ein perivaskuläres und
periglomeruläres Verteilungsmuster für diesen Marker, die Glomeruli blieben
weitestgehend ausgespart. Die Ang II Infusion war mit einer deutlichen
Makrophageninfiltration assoziiert, ein Unterschied zwischen den Genotypen
fand sich dabei nicht (Abbildung 14a-c).

Abbildung 13. Glomerulosklerose und interstitielle Fibrose, (Vergrößerung in
Abbildung 13c: 400-fach)

Abbildung 14. Makrophageninfiltration in der Niere (F4/80 Immunhistochemie);
(Vergrößerung in Abb. 14b 200fach, in Abb. 14c 400fach), HPF= high power
field
6.7 Oxidativer Stress
Der oxidative Stress in der Aorta wurde mittles DHE-Fluoreszenzmikroskopie
ermittelt. Die Gabe von Ang II führte zu einer Zunahme der DHE-Fluoreszenz
und damit des oxidativen Stress in der Aorta. Dieser Effekt war dosisabhängig
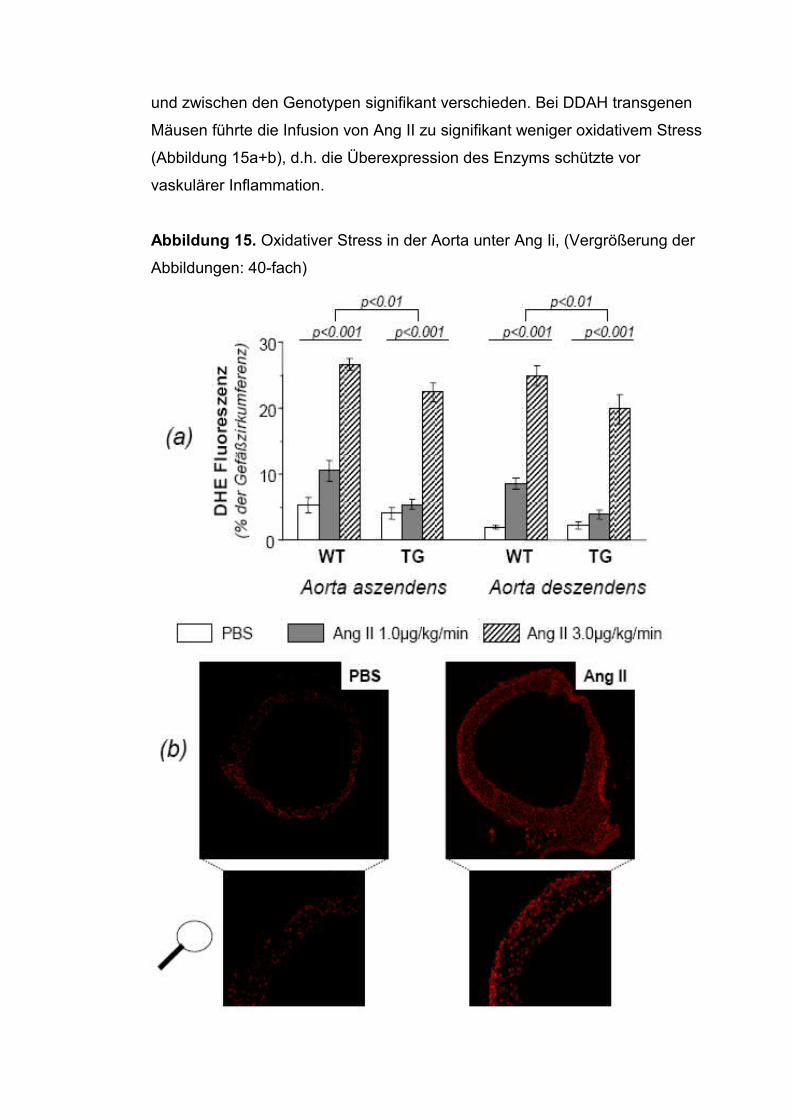
und zwischen den Genotypen signifikant verschieden. Bei DDAH transgenen
Mäusen führte die Infusion von Ang II zu signifikant weniger oxidativem Stress
(Abbildung 15a+b), d.h. die Überexpression des Enzyms schützte vor
vaskulärer Inflammation.
Abbildung 15. Oxidativer Stress in der Aorta unter Ang Ii, (Vergrößerung der
Abbildungen: 40-fach)

6.8 PRMT/DDAH mRNA Expression in der Niere
Mittels RT-PCR wurde die mRNA Expression ADMA generierender (PRMT1
und PRMT3) sowie ADMA abbauender Enzyme (DDAH1 und DDAH2) in der
Niere analysiert (Tabelle 6). Hierbei zeigte sich unter Ang II Infusion eine
Hochregulation der Expression von DDAH2 und PRMT1, während die
Expression der murinen DDAH1 und PRMT3 Expression unbeeinflusst blieb.
Die Expression von Kollagen I bestätigte den immunhistochemisch erhobenen
Befund einer verminderten renalen interstitiellen Fibrose bei Mäusen mit
Überexpression von DDAH.
Tabelle 6. Expression ADMA generierender und abbauender Enzyme

Diskussion
Die wichtigsten Ergebnisse dieser Dissertation lauten wie folgt. Die vierwöchige
Infusion von Ang II geht mit deutlicher Hypertonie und Endorganschädigung im
Mausmodell einher, führt jedoch zu keiner signifikanten Erhöhung der
systemischen ADMA-Plasmaspiegel. Die Überexpression des ADMA
abbauenden Enzyms DDAH1 schützt vor Ang II induzierter interstitieller Fibrose
in der Niere und vor vaskulärem oxidativem Stress.
Der fehlende Effekt der Ang II Infusionen auf die ADMA-Plasmaspiegel war für
uns überraschend und entsprach nicht unserer Arbeitshypothese. Die
Ergebnisse stehen somit in klarem Widerspruch zu Untersuchungen einer
japanischen Arbeitsgruppe, deren Daten zeitgleich mit den hier vorliegenden
veröffentlicht wurden (26).
In jener Studie wurde Ang II ebenfalls mittels osmotischer Minipumpen bei WT
und DDAH2 TG Mäusen infundiert. Die Autoren verwendeten dabei die gleiche
Dosierung (1.0µg/kg/min) wie sie bei einem Teil unserer Versuchstiere appliziert
wurde, die Infusionsdauer war mit zwei Wochen jedoch nur halb so lang. Im
Gegensatz zu unseren Ergebnissen führte die Ang II Infusion bei WT Mäusen
zu einer Verdoppelung der ADMA-Spiegel gegenüber Kontrolltieren, die eine
Kochsalzlösung infundiert bekamen (26). Der Anstieg der ADMA-Plasmaspiegel
war bei DDAH2 TG Mäusen nur halb so hoch wie bei den WT Tieren.
Wir fanden bei gleicher Dosierung von Ang II weder bei WT noch DDAH1 TG
Mäusen eine Änderung der ADMA-Plasmaspiegel. Lediglich unter
Hochdosisinfusion (3.0µg/kg/min) zeigte sich mittels ELISA ein marginaler
Anstieg der ADMA-Spiegel im Plasma. Hierbei handelte es sich aber nur um
einen Vergleich der ADMA-Spiegel vor und nach Applikation von Ang II, also
innerhalb der Behandlungsgruppe. Gegenüber den mit PBS behandelten
Kontrolltieren war kein Anstieg der ADMA-Plasmaspiegel nachweisbar. Zudem
ließ sich der im ELISA gemessene Anstieg nicht mittels
Tandemmassenspektrometrie (LC-MS), dem Goldstandard der ADMA-Analytik,
verifizieren. In jedem Falle steht der im ELISA gemessene Anstieg der ADMA-
Plasmaspiegel von 14% in klarem Kontrast zu dem von der japanischen
Arbeitsgruppe beobachteten Anstieg von 100%.

Eine schlüssige Erklärung für die diskrepanten Befunde gibt es nicht,
insbesondere bei den genetisch identischen WT Tieren (C57Bl/6J Hintergrund)
in beiden Studien sollten reproduzierbare Effekte von Ang II auf die ADMA-
Spiegel nachweisbar sein.
Einen möglichen Erklärungsansatz könnten die unterschiedlichen
Messmethoden darstellen. Während unsere Gruppe auf einen ELISA und die
LC-MS zurückgriff, verwendeten Hasegawa et al. die Hochdruck-
Flüssigkeitschromatographie (HPLC) ohne anschließende
Massenspektrometrie zur Messung der ADMA-Spiegel (26).
Eine weitere Erklärung könnte die unterschiedliche Dauer der Ang II-Infusion in
beiden Arbeiten darstellen, da bei uns Ang II über vier Wochen infundiert
wurde, bei Hasegawa et al. dagegen nur zwei Wochen. Es wäre denkbar, dass
Ang II einen passageren Anstieg der ADMA-Spiegel provoziert, der nach zwei
Wochen nachweisbar ist, nach vier Wochen allerdings bereits wieder
abgeklungen ist. Als Ursache hierfür kämen beispielsweise gegenregulatorische
Genexpressionsmuster ADMA modulierender Enzyme in Frage, wie wir sie in
der RT-PCR messen konnten.
Wir halten die von uns erhobenen Befunde für valide und glaubhaft, da wir
durch diverse Maßnahmen mögliche Störgrößen kontrolliert haben. Die von uns
gewählten Gruppengrößen der mit Ang II behandelten Tiere lagen deutlich
höher als in der japanischen Arbeit, in der lediglich fünf Tiere je Gruppe
untersucht wurden, so dass eine höhere statistische Irrtumswahrscheinlichkeit
gegeben ist. Zudem haben wir den fehlenden Effekt von Ang II durch Gabe
einer höheren Dosis (3.0µg/kg/min) verifiziert und dabei sowohl ADMA-
Plasmaspiegel vor als auch nach Behandlung gemessen. Schlussendlich haben
wir dabei zwei unterschiedliche Meßmethoden zur ADMA-Analytik gewählt, die
letztlich keinen signifikanten Effekt von Ang II auf systemische ADMA-Spiegel
nachweisen konnten. Trotz leichter Differenzen beider Methoden, die am
ehesten auf der Präanalytik der Proben beruhen, zeigte sich eine gute
Konkordanz zwischen ELISA und LC-MS. Zudem ergab sich nahezu keine
Überlappung der ADMA-Werte zwischen WT und DDAH TG Mäusen, was die
Messgenauigkeit der Verfahren zusätzlich untermauert.
Ein technisches Versagen der Infusionspumpen erscheint sehr
unwahrscheinlich, da alle Ang II behandelten Tiere eine Hypertonie und

entsprechende Endorganschäden aufwiesen und zudem ein dosisabhängiger
Anstieg der Aldosteronspiegel im Blut zu verzeichnen war.
Trotz der eingangs erwähnten Diskrepanzen unserer Ergebnisse mit denen der
Gruppe von Hasegawa et al. (26), gab es auch zahlreiche Gemeinsamkeiten. In
beiden Arbeiten wurde mittels Schwanzplethysmographie unabhängig vom
Genotyp ein vergleichbarer Anstieg der Blutdruckwerte unter Ang II Infusion
gemessen, der den Erwartungen an dieses Hypertoniemodell entsprach.
Während die DDAH-Überexpression im Ang II Hypertoniemodell keinen Schutz
vor Glomerulosklerose, renaler Makrophageninfiltration und vaskulärer
Hypertrophie bot, konnte ein Effekt auf den oxidativen Stress in der Aortenwand
sowie auf die renale interstitielle Fibrose nachgewiesen werden. Die
letztgenannten Befunde decken sich mit den Ergebnissen von Hasegawa et al.,
die bei DDAH2 transgenen Mäusen unter Ang II ebenfalls einen verminderten
oxidativen Stress und eine geringere perivaskuläre Fibrose beschrieben (26).
Eine Abnahme der interstitiellen Fibrose in der Niere durch DDAH wurde zudem
auch in einem subtotalen Nephrektomiemodell der Ratte beobachtet (44). Hier
führte die Überexpression von DDAH1 mittels eines adenoviralen Vektors
neben einer verminderten Fibrose auch zu einer Abnahme der
Kapillarrarefizierung und somit zu einer verbesserten Organprotektion (44). Die
Überexpression von DDAH ging dabei mit einer verminderten Expression des
Fibrosemarkers TGF-β (transforming growth factor β) einher. Gleiche Effekte
wurden von den Autoren auch in einem Diabetesmodell der Ratte beschrieben
(70).
Im Gegensatz dazu waren im vorliegenden Hypertoniemodell die signifikant
niedrigeren ADMA-Spiegel der DDAH1 TG Mäuse nicht protektiv im Hinblick auf
die hypertensiven Endorganschäden an Niere und Gefäßen
(Glomerulosklerose, renale Inflammation, vaskuläre Hypertrophie). In diesem
Zusammenhang ist es erstaunlich, dass die von Hasegawa et al. untersuchten
DDAH2 TG Tiere unter Ang II Infusionen signifikant weniger Schäden an den
Koronargefäßen - gemessen an Wandverdickung und perivaskulärer Fibrose -
im Vergleich zu WT Mäusen aufwiesen (26).
Aufschluss über diesen scheinbaren Widerspruch könnte eine wegweisende
Arbeit von Wang et al. geben (85). Diese Arbeitsgruppe untersuchte Ratten, bei
denen mittels RNA-Interferenz ein selektiver Knockdown von DDAH1 bzw.

DDAH2 induziert wurde (85). Die Autoren beobachteten, dass DDAH2
vorrangig in der Gefäßwand exprimiert wird und dort vermutlich über die lokalen
ADMA-Spiegel die NO-vermittelte Vasodilatation reguliert, während die
systemischen ADMA-Spiegel maßgeblich von der Aktivität der DDAH1
beeinflusst werden, welche vorwiegend in Leber und Nierenkortex exprimiert
wird. Hieraus lässt sich für die vorliegende Arbeit ableiten, dass für die
hypertensiven Gefäßschäden ebenfalls die lokalen, durch DDAH2 modulierten
ADMA-Spiegel von Bedeutung sind, die von DDAH1 regulierten systemischen
ADMA-Spiegel in der Pathophysiologie dagegen eine untergeordnete Rolle zu
spielen scheinen. Der Befund von Wang et al. erklärt ferner, warum die ADMA-
Plasmaspiegel, die vornehmlich durch die Aktivität bzw. Expression von DDAH1
reguliert werden, durch Überexpression der DDAH2 in Mäusen nur um etwa
25% gesenkt werden, während der Effekt bei DDAH1 TG Mäusen bei ca. 40-
50% liegt (16, 26). Eine Limitation unserer Arbeit wie auch der Studien von
Hasegawa et al. und Wang et al. (26, 85) stellt die Tatsache dar, dass ADMA
nur im Plasma nicht jedoch im Gewebe bzw. Organen gemessen wurde.
Allgemein muss festgehalten werden, dass aufgrund der unterschiedlichen
ADMA-Analytik ein direkter Vergleich mit Ergebnissen anderer Arbeitsgruppen
generell schwierig ist. In der Literatur finden sich bei Verwendung der am
breitesten zum Einsatz kommenden HPLC als Meßmethode stark schwankende
ADMA-Werte in vergleichbaren, gesunden Kontrollkollektiven. Dies hat zur
Folge, dass es bis dato keine validierten bzw. verbindlichen Normwerte für
ADMA gibt. Exemplarisch sei hier auf eine Publikation hingewiesen, in der
zunächst die Streuung der ADMA-Werte in der Literatur näher beleuchtet wird
und die im Folgenden den Versuch unternimmt, Normwerte für Gesunde zu
ermitteln (46). Eine Zusammenstellung publizierter ADMA-Werte an gesunden
Kontrollkollektiven aus zwölf klinischen Studien zeigt dabei eine Streuung der
ADMA-Spiegel zwischen 0.12-2.77µM. Da in zahlreichen klinischen Studien
bereits leichte Anstiege von ADMA im Bereich von 0.2-0.5µM mit erhöhter
Morbidität und Mortalität assoziiert sind (47, 67), ist diese Variabilität nicht
hinnehmbar und unterstreicht die Wichtigkeit der Analytik dieses im
mikromolaren Bereich anzutreffenden Moleküls.
Zur Etablierung von Normwerten für die HPLC-Methode untersuchten Meinitzer
et al. in der Folge ein Kollektiv von 292 Männern im Alter von 20-75 Jahren. Der

Median der ADMA-Werte variierte je nach Altersquartile zwischen 0.58-0.64µM
und stieg somit nur marginal im Alter an (46). Normwerte für Nierengesunde
dürften somit im Bereich von 0.5µM liegen.
Die von Meinitzer et al. monierte Streuung der ADMA-Werte in der Literatur
findet sich auch bei Patienten mit eingeschränkter Nierenfunktion, bei denen
ADMA akkumuliert. Eine systematische Analyse aller publizierten Daten zu
diesem Thema, in welche 17 Studien eingeschlossenen wurden, ergab
Mittelwerte zwischen 0.36-1.40µM bei nierengesunden Kontrollpersonen bzw.
0.46-4.20µM bei Patienten mit chronischer, nicht dialysepflichtiger
Niereninsuffizienz (30). Hieraus ergibt sich eine deutliche Überlappung der
Werte nierengesunder und nierenkranker Populationen. Aufgrund dieser
Streubreite hat die routinemäßige Bestimmung von ADMA trotz zahlreicher, viel
versprechender epidemiologischer Studien bis dato keinen Eingang in die Klinik
gefunden.
Ob tatsächlich ein Zusammenhang zwischen Ang II und ADMA besteht, ist
anhand bisher vorliegender Daten zu diesem Thema nicht klar ersichtlich. Da
Ang II zu einer endothelialen Dysfunktion mit verminderter Bioverfügbarkeit von
NO führt, liegt die Vermutung nahe, dass ein Anstieg des endogenen NOS
Inhibitors ADMA eine entscheidende pathophysiologische Rolle spielen könnte.
Bis heute gibt es hierzu nur wenig veröffentlichte Daten.
In-vitro führt die Inkubation von Nabelschnurendothelzellen mit Ang II zu einem
Anstieg der ADMA-Spiegel im Kulturmedium (11). Parallel beobachteten die
Autoren dabei eine Abnahme der DDAH-Aktivität in den Zellen. Abgesehen von
der Tatsache, dass es sich hierbei um einen in-vitro Befund handelt, wurden die
Zellen mit supraphysiologischen Konzentrationen von Ang II inkubiert (1µM).
Auch die DDAH-Aktivitätsmessung erfolgte unter supraphysiologischer
Inkubation mit exogen zugeführtem ADMA (500µM), so dass diese Befunde
nicht ohne weiteres auf in-vivo Bedingungen übertragbar sind (11). Interessant
an dieser Veröffentlichung ist jedoch, dass die Inkubation mit Ang II und ADMA
mit einer Erhöhung freier Sauerstoffradikale im Medium einherging (11), was an
den von uns erhobenen Befund eines vermehrten oxidativen Stress unter Ang II
Infusion erinnert.
An dieser Stelle soll auch die Möglichkeit erwähnt werden, dass eine
umgekehrte Abhängigkeit nicht ausgeschlossen werden kann, d.h. dass ADMA

seinerseits die Ang II Spiegel bzw. das RAS beeinflusst. Das vorliegende
Tierexperiment eignete sich nicht zur Überprüfung dieser Hypothese, allerdings
existiert eine Publikation, in der die Infusion von ADMA bei eNOS-defizienten
Mäusen zu einer gesteigerten Expression des Angiotensin-Converting-Enzyms
(ACE) führte (72).
Abgesehen von der Arbeit von Hasegawa et al. gibt es bisher keine weiteren in-
vivo Experimente, die den Zusammenhang zwischen Ang II induzierter
Hypertonie und ADMA näher untersucht haben. In der vorliegenden in-vivo
Studie fanden wir keinen Zusammenhang zwischen Hypertonie und ADMA-
Plasmaspiegeln.
Dies steht in Widerspruch zu klinischen Studien, die eine Assoziation zwischen
Bluthochdruck und ADMA zeigen konnten (15, 29, 62, 73). Hierbei handelt es
sich jedoch um Studien mit sehr kleinen Fallzahlen. In prospektiven
Kohortenstudien an weit mehr als tausend Patienten konnte diese Assoziation
hingegen nicht bestätigt werden (47, 67). Unterstützt werden die Befunde einer
fehlenden Assoziation von Blutdruck und ADMA-Plasmaspiegeln zudem durch
Ergebnisse einer kleineren Studie (n=67 Patienten), in der 24h-
Langzeitblutdruckmessungen durchgeführt wurden (60). Auch hier fand sich
keine Assoziation zwischen ADMA und Blutdruck. Zum Teil können die
diskrepanten Befunde auf die bereits diskutierte unterschiedliche ADMA-
Analytik zurückgeführt werden. Damit muss eine abschließende Bewertung zu
dieser Thematik abgewartet werden, die Evidenz für eine enge Assoziation
zwischen Bluthochdruck und ADMA-Plasmaspiegeln ist zum jetzigen Zeitpunkt
nicht überzeugend.
Ebenfalls ungeklärt ist, inwiefern das Renin-Angiotensin-System bzw. eine
Blockade desselbigen ADMA-Spiegel beim Menschen moduliert.
Hier wurde insbesondere der Effekt einer RAS-Blockade mittels Inhibitoren des
Angiotensin-Converting-Enzyms (ACE-Hemmer) bzw. Angiotensin-
Rezeptorblockern (AT1-Blocker) näher untersucht. In einer kleinen, doppel-blind
randomisierten cross-over Studie an n=20 Hypertonikern untersuchten Delles et
al. den Effekt einer einwöchigen Gabe des ACE-Hemmers Enalapril (20mg/Tag)
oder des AT1-Blockers Eprosartan (600mg/Tag) bzw. einer Kombination beider
Medikamente (Enalapril 10mg/Tag + Eprosartan 300mg/Tag) auf die ADMA-
Plasmaspiegel (18). In einer vierten Phase wurden die Patienten mit einem

Plazebo behandelt (Kontrolle). Zwischen den jeweiligen Behandlungsphasen
erfolgte eine zweiwöchige Auswaschphase der Medikamente. In der
Plazebophase lagen die ADMA-Spiegel bei 1.69µM, unter Enalapril
Monotherapie bei 1.41µM, unter Eprosartan Monotherapie bei 1.42µM und in
der Kombination bei 1.38µM. Die Reduktion der Plasmaspiegel war dabei
blutdruckunabhängig (18). Ähnliche Befunde wurden bei Hypertonikern von
anderen Autoren beschrieben (29, 54).
Eine Reduktion von ADMA durch RAS-Blockade wurde auch bei Patienten mit
dialysepflichtiger Niereninsuffizienz (4), bei Syndrom X (10) und Diabetes
mellitus Typ II (28) gefunden. Bei den genannten Studien, die eine Reduktion
von ADMA unter RAS-Blockade fanden, ist kritisch anzumerken, dass es sich
um kleine Fallzahlen handelt. Zudem zeigt sich auch hier eine hohe Variabilität
der publizierten ADMA-Werte.
Prospektive Studien mit größeren Fallzahlen, in denen ADMA mittels moderner
Analytik gemessen wurde, konnten eine Beeinflussung der ADMA-
Plasmaspiegel durch RAS-Blockade indes nicht bestätigen (20, 22, 87).
Trotz fehlenden Effekts von Ang II auf die systemischen ADMA-Plasmaspiegel
sahen wir in unserem Experiment einen vom Genotyp unabhängigen Anstieg
der mRNA Expression von PRMT1 und DDAH2 unter Ang II-Infusion in der
Niere. Dieser Sachverhalt könnte einen Grund für das Ausbleiben eines ADMA-
Anstiegs darstellen, da eine gesteigerte ADMA-Produktion durch PRMT1 durch
einen vermehrten Abbau über DDAH2 kompensiert werden könnte, so dass
sich zwar der ADMA-Umsatz erhöht, nicht aber die resultierenden
Serumspiegel. Einschränkend muss jedoch hinzugefügt werden, dass das
genannte Expressionsmuster nicht auf Proteinebene verifiziert wurde.
Es existieren bereits andere Studien, die die Expression von DDAH und PRMT
unter pathophysiologischen Bedingungen untersucht haben, und deren Befunde
den hier erhobenen ähneln. In einem Diabetesmodell der Ratte, welches mit
erhöhten Ang II und ADMA-Spiegeln einhergeht, führte die Gabe des AT1-
Blockers Telmisartan zu einer Herabregulation der Proteinexpression von
DDAH2 und PRMT1 in der Niere, d.h. die RAS-Blockade lieferte ein gegenüber
unseren Versuchen spiegelbildliches Expressionsmuster (57). In einem
Salzmodell der Ratte führte die Kochsalzrestriktion zu einer Aktivierung des
RAS und einer vermehrten DDAH-Expression in der Niere. Letztere erfolgte

Ang II abhängig und konnte durch einen AT1-Blocker gehemmt werden (77).
Ähnliche Beobachtungen wurden bei Ratten unter kochsalzreicher Diät mit Ang
II Infusion gemacht (77). Die von uns und anderen erhobenen Befunde einer
Änderung der Expression einzelner DDAH- und PRMT-Isoenzyme durch
Modulation des RAS bedürfen jedoch weiterer experimenteller Studien.
Interessanterweise beobachteten wir in unseren Versuchen, dass die
Überexpression von DDAH mit einem verminderten vaskulären oxidativen
Stress in der Aorta unter Ang II einherging. Ang II vermag eine gefäßständige
NADH/NADPH-Oxidase zu aktivieren, was zu einer verstärkten Produktion
freier Sauerstoffradikale (•O2-) führt. Dies erklärt den erhöhten vaskulären
oxidativen Stress bei Ang II induzierten Schädigungsmodellen (65). Ferner führt
Ang II über Sauerstoffradikale zu einer Herabregulation der
Dihydrofolatreduktase, was zu einer Abnahme der Konzentration von
Tetrahydrobiopterin führt, welches als Co-Substrat für die eNOS dient (9). Fehlt
der eNOS das Cosubstrat, überträgt sie Elektronen nicht auf ihr eigentliches
Substrat Arginin, sondern auf molekularen Sauerstoff, d.h. eNOS produziert nun
selbst Sauerstoffradikale. Dieses Phänomen wird als Entkopplung der NOS
(NOS-uncoupling) bezeichnet (9). Der Vorgang wird offenbar durch ADMA
begünstigt, welches ebenfalls eine Entkopplung der NOS zu induzieren vermag,
indem es das Substrat Arginin vom aktiven Zentrum der NOS verdrängt und zu
einem funktionellen Substratmangel führt (74). Vor diesem Hintergrund
erscheint unser Befund plausibel, dass DDAH1 TG Tiere aufgrund ihrer
niedrigeren ADMA-Spiegel weniger anfällig für oxidativen Stress durch Ang II
sind.
Als weiterer Erklärungsansatz könnte die Tatsache dienen, dass die DDAH ein
redox-sensitives Enzym ist, d.h. in ihrer Aktivität durch oxidativen Stress
gedämpft wird. Für die katalytische Aktivität der DDAH sind drei Aminosäuren
(Cystein, Aspartat und Histidin) im aktiven Zentrum des Enzyms verantwortlich
(36). Von diesen ist offenbar Cystein entscheidend, sowohl für die Katalyse als
auch für die Redoxsensitiviät (36, 37, 52). Präziser ausgedrückt wird davon
ausgegangen, dass eine Oxidation der Sulfhydrylgruppe durch
Sauerstoffradikale die DDAH-Aktivität vermindert.
Abschließend stellt sich die Frage, warum in unserer Studie offenbar die
niedrigeren ADMA-Spiegel der TG Tiere einen positiven Effekt auf den

oxidativen Stress haben konnten, wenn doch die Spiegel, die wir bei den WT
Tieren vor und nach Ang II messen konnten, weitestgehend normal waren
(<1.0µM). Es kann nicht ausgeschlossen werden, dass auch diese „normalen“
ADMA-Spiegel zur Pathogenese von Gefäßschäden beitragen, wenn andere
Noxen, wie in diesem Fall ein Ang II induzierter Hypertonus, hinzukommen.
Unter diesen Umständen mögen physiologische ADMA-Spiegel relativ zu hoch
sein, man könnte hier von einer permissiven Rolle von ADMA für
Gefäßschäden sprechen.
Zusammenfassend zeigen unsere Befunde, dass die Infusion von Ang II in
pathophysiologisch relevanten Konzentrationen zu keiner Beeinflussung der
systemischen ADMA-Plasmaspiegel führt. Die Überexpression des ADMA-
abbauenden Enzyms DDAH schützte jedoch vor Ang II induzierter interstitieller
Fibrose im Nierenstromgebiet und vor vaskulärem oxidativen Stress in der
Aorta.

Literaturverzeichnis
1. Abbasi F, Asagmi T, Cooke JP, Lamendola C, McLaughlin T, Reaven GM, Stuehlinger M, and Tsao PS. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am J Cardiol 88: 1201-1203, 2001.
2. Achan V, Broadhead M, Malaki M, Whitley G, Leiper J, MacAllister R, and Vallance P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol 23: 1455-1459, 2003.
3. Altinova AE, Arslan M, Sepici-Dincel A, Akturk M, Altan N, and Toruner FB. Uncomplicated type 1 diabetes is associated with increased asymmetric dimethylarginine concentrations. J Clin Endocrinol Metab 92: 1881-1885, 2007.
4. Aslam S, Santha T, Leone A, and Wilcox C. Effects of amlodipine and valsartan on oxidative stress and plasma methylarginines in end-stage renal disease patients on hemodialysis. Kidney Int 70: 2109-2115, 2006.
5. Boffa LC, Karn J, Vidali G, and Allfrey VG. Distribution of NG, NG,-dimethylarginine in nuclear protein fractions. Biochem Biophys Res Commun 74: 969-976, 1977.
6. Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, and Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98: 1842-1847, 1998.
7. Bredt DS and Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A 87: 682-685, 1990.
8. Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, and Zweier JL. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem 282: 879-887, 2007.
9. Chalupsky K and Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 102: 9056-9061, 2005.
10. Chen JW, Hsu NW, Wu TC, Lin SJ, and Chang MS. Long-term angiotensin-converting enzyme inhibition reduces plasma asymmetric dimethylarginine and improves endothelial nitric oxide bioavailability and coronary microvascular function in patients with syndrome X. Am J Cardiol 90: 974-982, 2002.
11. Chen MF, Xie XM, Yang TL, Wang YJ, Zhang XH, Luo BL, and Li YJ. Role of asymmetric dimethylarginine in inflammatory reactions by angiotensin II. J Vasc Res 44: 391-402, 2007.
12. Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 15: 532-534, 536-537, 1993.

13. Closs EI, Basha FZ, Habermeier A, and Forstermann U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide 1: 65-73, 1997.
14. Cooke JP, Singer AH, Tsao P, Zera P, Rowan RA, and Billingham ME. Antiatherogenic effects of L-arginine in the hypercholesterolemic rabbit. J Clin Invest 90: 1168-1172, 1992.
15. Curgunlu A, Uzun H, Bavunoglu I, Karter Y, Genc H, and Vehid S. Increased circulating concentrations of asymmetric dimethylarginine (ADMA) in white coat hypertension. J Hum Hypertens 19: 629-633, 2005.
16. Dayoub H, Achan V, Adimoolam S, Jacobi J, Stuehlinger MC, Wang BY, Tsao PS, Kimoto M, Vallance P, Patterson AJ, and Cooke JP. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation 108: 3042-3047, 2003.
17. Dayoub H, Rodionov R, Lynch C, Cooke JP, Arning E, Bottiglieri T, Lentz SR, and Faraci FM. Overexpression of dimethylarginine dimethylaminohydrolase inhibits asymmetric dimethylarginine-induced endothelial dysfunction in the cerebral circulation. Stroke 39: 180-184, 2008.
18. Delles C, Schneider MP, John S, Gekle M, and Schmieder RE. Angiotensin converting enzyme inhibition and angiotensin II AT1-receptor blockade reduce the levels of asymmetrical N(G), N(G)-dimethylarginine in human essential hypertension. Am J Hypertens 15: 590-593, 2002.
19. Drexler H, Zeiher AM, Meinzer K, and Just H. Correction of endothelial dysfunction in coronary microcirculation of hypercholesterolaemic patients by L-arginine. Lancet 338: 1546-1550, 1991.
20. Fliser D, Wagner KK, Loos A, Tsikas D, and Haller H. Chronic angiotensin II receptor blockade reduces (intra)renal vascular resistance in patients with type 2 diabetes. J Am Soc Nephrol 16: 1135-1140, 2005.
21. Forstermann U, Schmidt HH, Pollock JS, Sheng H, Mitchell JA, Warner TD, Nakane M, and Murad F. Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochem Pharmacol 42: 1849-1857, 1991.
22. Galle J, Schwedhelm E, Pinnetti S, Boger RH, and Wanner C. Antiproteinuric effects of angiotensin receptor blockers: telmisartan versus valsartan in hypertensive patients with type 2 diabetes mellitus and overt nephropathy. Nephrol Dial Transplant 23: 3174-3183, 2008.
23. Garg UC and Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83: 1774-1777, 1989.
24. Ghosh SK, Paik WK, and Kim S. Purification and molecular identification of two protein methylases I from calf brain. Myelin basic protein- and histone-specific enzyme. J Biol Chem 263: 19024-19033, 1988.
25. Hartner A, Cordasic N, Klanke B, Veelken R, and Hilgers KF. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant 18: 1999-2004, 2003.
26. Hasegawa K, Wakino S, Tatematsu S, Yoshioka K, Homma K, Sugano N, Kimoto M, Hayashi K, and Itoh H. Role of asymmetric

dimethylarginine in vascular injury in transgenic mice overexpressing dimethylarginie dimethylaminohydrolase 2. Circ Res 101: e2-10, 2007.
27. Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu Rev Pharmacol Toxicol 30: 535-560, 1990.
28. Ito A, Egashira K, Narishige T, Muramatsu K, and Takeshita A. Angiotensin-converting enzyme activity is involved in the mechanism of increased endogenous nitric oxide synthase inhibitor in patients with type 2 diabetes mellitus. Circ J 66: 811-815, 2002.
29. Ito A, Egashira K, Narishige T, Muramatsu K, and Takeshita A. Renin-angiotensin system is involved in the mechanism of increased serum asymmetric dimethylarginine in essential hypertension. Jpn Circ J 65: 775-778, 2001.
30. Jacobi J and Tsao PS. Asymmetrical dimethylarginine in renal disease: limits of variation or variation limits? A systematic review. Am J Nephrol 28: 224-237, 2008.
31. Jiang DJ, Jia SJ, Yan J, Zhou Z, Yuan Q, and Li YJ. Involvement of DDAH/ADMA/NOS pathway in nicotine-induced endothelial dysfunction. Biochem Biophys Res Commun 349: 683-693, 2006.
32. Kakimoto Y and Akazawa S. Isolation and identification of N-G,N-G- and N-G,N'-G-dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. J Biol Chem 245: 5751-5758, 1970.
33. Kim S and Paik WK. Purification and properties of protein methylaase II. J Biol Chem 245: 1806-1813, 1970.
34. Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, and Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther 113: 50-87, 2007.
35. Kubes P, Suzuki M, and Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A 88: 4651-4655, 1991.
36. Leiper J, Murray-Rust J, McDonald N, and Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A 99: 13527-13532, 2002.
37. Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, and Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med 13: 198-203, 2007.
38. Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, and Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J 343 Pt 1: 209-214, 1999.
39. Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics 45: 255-268, 1989.
40. Lu TM, Ding YA, Leu HB, Yin WH, Sheu WH, and Chu KM. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am J Cardiol 94: 157-161, 2004.

41. Lundman P, Eriksson MJ, Stuhlinger M, Cooke JP, Hamsten A, and Tornvall P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J Am Coll Cardiol 38: 111-116, 2001.
42. Maas R. Pharmacotherapies and their influence on asymmetric dimethylargine (ADMA). Vasc Med 10 Suppl 1: S49-57, 2005.
43. MacAllister RJ, Parry H, Kimoto M, Ogawa T, Russell RJ, Hodson H, Whitley GS, and Vallance P. Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br J Pharmacol 119: 1533-1540, 1996.
44. Matsumoto Y, Ueda S, Yamagishi S, Matsuguma K, Shibata R, Fukami K, Matsuoka H, Imaizumi T, and Okuda S. Dimethylarginine dimethylaminohydrolase prevents progression of renal dysfunction by inhibiting loss of peritubular capillaries and tubulointerstitial fibrosis in a rat model of chronic kidney disease. J Am Soc Nephrol 18: 1525-1533, 2007.
45. McDermott JR. Studies on the catabolism of Ng-methylarginine, Ng, Ng-dimethylarginine and Ng, Ng-dimethylarginine in the rabbit. Biochem J 154: 179-184, 1976.
46. Meinitzer A, Puchinger M, Winklhofer-Roob BM, Rock E, Ribalta J, Roob JM, Sundl I, Halwachs-Baumann G, and Marz W. Reference values for plasma concentrations of asymmetrical dimethylarginine (ADMA) and other arginine metabolites in men after validation of a chromatographic method. Clin Chim Acta 384: 141-148, 2007.
47. Meinitzer A, Seelhorst U, Wellnitz B, Halwachs-Baumann G, Boehm BO, Winkelmann BR, and Marz W. Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (the Ludwigshafen Risk and Cardiovascular Health study). Clin Chem 53: 273-283, 2007.
48. Mittermayer F, Krzyzanowska K, Exner M, Mlekusch W, Amighi J, Sabeti S, Minar E, Muller M, Wolzt M, and Schillinger M. Asymmetric dimethylarginine predicts major adverse cardiovascular events in patients with advanced peripheral artery disease. Arterioscler Thromb Vasc Biol 26: 2536-2540, 2006.
49. Miyazaki H, Matsuoka H, Cooke JP, Usui M, Ueda S, Okuda S, and Imaizumi T. Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis. Circulation 99: 1141-1146, 1999.
50. Moncada S and Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 329: 2002-2012, 1993.
51. Moncada S, Palmer RM, and Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43: 109-142, 1991.
52. Murray-Rust J, Leiper J, McAlister M, Phelan J, Tilley S, Santa Maria J, Vallance P, and McDonald N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat Struct Biol 8: 679-683, 2001.
53. Najbauer J, Johnson BA, Young AL, and Aswad DW. Peptides with sequences similar to glycine, arginine-rich motifs in proteins interacting with RNA are efficiently recognized by methyltransferase(s) modifying arginine in numerous proteins. J Biol Chem 268: 10501-10509, 1993.

54. Napoli C, Sica V, de Nigris F, Pignalosa O, Condorelli M, Ignarro LJ, and Liguori A. Sulfhydryl angiotensin-converting enzyme inhibition induces sustained reduction of systemic oxidative stress and improves the nitric oxide pathway in patients with essential hypertension. Am Heart J 148: e5, 2004.
55. Ogawa T, Kimoto M, and Sasaoka K. Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-L-arginine to L-citrulline in rats. Biochem Biophys Res Commun 148: 671-677, 1987.
56. Ogawa T, Kimoto M, and Sasaoka K. Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J Biol Chem 264: 10205-10209, 1989.
57. Onozato ML, Tojo A, Leiper J, Fujita T, Palm F, and Wilcox CS. Expression of NG,NG-dimethylarginine dimethylaminohydrolase and protein arginine N-methyltransferase isoforms in diabetic rat kidney: effects of angiotensin II receptor blockers. Diabetes 57: 172-180, 2008.
58. Paik WK and Kim S. Protein methylase I. Purification and properties of the enzyme. J Biol Chem 243: 2108-2114, 1968.
59. Paik WK and Kim S. Solubilization and partial purification of protein methylase 3 from calf thymus nuclei. J Biol Chem 245: 6010-6015, 1970.
60. Paiva H, Laakso J, Kahonen M, Turjanmaa V, Koobi T, Majahalme S, Lehtimaki T, Ruokonen I, and Laaksonen R. Asymmetric dimethylarginine and hemodynamic regulation in middle-aged men. Metabolism 55: 771-777, 2006.
61. Palmer RM, Ferrige AG, and Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524-526, 1987.
62. Perticone F, Sciacqua A, Maio R, Perticone M, Maas R, Boger RH, Tripepi G, Sesti G, and Zoccali C. Asymmetric dimethylarginine, L-arginine, and endothelial dysfunction in essential hypertension. J Am Coll Cardiol 46: 518-523, 2005.
63. Pollock JS, Forstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, and Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci U S A 88: 10480-10484, 1991.
64. Radomski MW, Palmer RM, and Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 2: 1057-1058, 1987.
65. Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, and Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916-1923, 1996.
66. Rees DD, Palmer RM, and Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci U S A 86: 3375-3378, 1989.
67. Schnabel R, Blankenberg S, Lubos E, Lackner KJ, Rupprecht HJ, Espinola-Klein C, Jachmann N, Post F, Peetz D, Bickel C, Cambien F, Tiret L, and Munzel T. Asymmetric dimethylarginine and the risk of

cardiovascular events and death in patients with coronary artery disease: results from the AtheroGene Study. Circ Res 97: e53-59, 2005.
68. Schulman SP, Becker LC, Kass DA, Champion HC, Terrin ML, Forman S, Ernst KV, Kelemen MD, Townsend SN, Capriotti A, Hare JM, and Gerstenblith G. L-arginine therapy in acute myocardial infarction: the Vascular Interaction With Age in Myocardial Infarction (VINTAGE MI) randomized clinical trial. JAMA 295: 58-64, 2006.
69. Schwedhelm E, Maas R, Tan-Andresen J, Schulze F, Riederer U, and Boger RH. High-throughput liquid chromatographic-tandem mass spectrometric determination of arginine and dimethylated arginine derivatives in human and mouse plasma. J Chromatogr B Analyt Technol Biomed Life Sci 851: 211-219, 2007.
70. Shibata R, Ueda S, Yamagishi SI, Kaida Y, Matsumoto Y, Fukami K, Hayashida A, Matsuoka H, Kato S, Kimoto M, and Okuda S. Involvement of asymmetric dimethylarginine (ADMA) in tubulointerstitial ischaemia in the early phase of diabetic nephropathy. Nephrol Dial Transplant, 2008.
71. Stuhlinger MC, Oka RK, Graf EE, Schmolzer I, Upson BM, Kapoor O, Szuba A, Malinow MR, Wascher TC, Pachinger O, and Cooke JP. Endothelial dysfunction induced by hyperhomocyst(e)inemia: role of asymmetric dimethylarginine. Circulation 108: 933-938, 2003.
72. Suda O, Tsutsui M, Morishita T, Tasaki H, Ueno S, Nakata S, Tsujimoto T, Toyohira Y, Hayashida Y, Sasaguri Y, Ueta Y, Nakashima Y, and Yanagihara N. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase-deficient mice: involvement of renin-angiotensin system and oxidative stress. Arterioscler Thromb Vasc Biol 24: 1682-1688, 2004.
73. Surdacki A, Nowicki M, Sandmann J, Tsikas D, Boeger RH, Bode-Boeger SM, Kruszelnicka-Kwiatkowska O, Kokot F, Dubiel JS, and Froelich JC. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol 33: 652-658, 1999.
74. Sydow K and Munzel T. ADMA and oxidative stress. Atheroscler Suppl 4: 41-51, 2003.
75. Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, and Herschman HR. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem 275: 7723-7730, 2000.
76. Theeuwes F and Yum SI. Principles of the design and operation of generic osmotic pumps for the delivery of semisolid or liquid drug formulations. Ann Biomed Eng 4: 343-353, 1976.
77. Tojo A, Kimoto M, and Wilcox CS. Renal expression of constitutive NOS and DDAH: separate effects of salt intake and angiotensin. Kidney Int 58: 2075-2083, 2000.
78. Tran CT, Fox MF, Vallance P, and Leiper JM. Chromosomal localization, gene structure, and expression pattern of DDAH1: comparison with DDAH2 and implications for evolutionary origins. Genomics 68: 101-105, 2000.
79. Tran CT, Leiper JM, and Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl 4: 33-40, 2003.

80. Vallance P, Collier J, and Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 2: 997-1000, 1989.
81. Vallance P and Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol 24: 1023-1030, 2004.
82. Vallance P, Leone A, Calver A, Collier J, and Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339: 572-575, 1992.
83. von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, Kaneda Y, Cooke JP, and Dzau VJ. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci U S A 92: 1137-1141, 1995.
84. Wanby P, Teerlink T, Brudin L, Brattstrom L, Nilsson I, Palmqvist P, and Carlsson M. Asymmetric dimethylarginine (ADMA) as a risk marker for stroke and TIA in a Swedish population. Atherosclerosis 185: 271-277, 2006.
85. Wang D, Gill PS, Chabrashvili T, Onozato ML, Raggio J, Mendonca M, Dennehy K, Li M, Modlinger P, Leiper J, Vallance P, Adler O, Leone A, Tojo A, Welch WJ, and Wilcox CS. Isoform-specific regulation by N(G),N(G)-dimethylarginine dimethylaminohydrolase of rat serum asymmetric dimethylarginine and vascular endothelium-derived relaxing factor/NO. Circ Res 101: 627-635, 2007.
86. Wang J, Sim AS, Wang XL, Salonikas C, Naidoo D, and Wilcken DE. Relations between plasma asymmetric dimethylarginine (ADMA) and risk factors for coronary disease. Atherosclerosis 184: 383-388, 2006.
87. Warnholtz A, Ostad MA, Heitzer T, Thuneke F, Frohlich M, Tschentscher P, Schwedhelm E, Boger R, Meinertz T, and Munzel T. AT1-receptor blockade with irbesartan improves peripheral but not coronary endothelial dysfunction in patients with stable coronary artery disease. Atherosclerosis 194: 439-445, 2007.
88. Wilson AM, Harada R, Nair N, Balasubramanian N, and Cooke JP. L-arginine supplementation in peripheral arterial disease: no benefit and possible harm. Circulation 116: 188-195, 2007.
89. Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, and Nathan C. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 256: 225-228, 1992.
90. Yoo JH and Lee SC. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 158: 425-430, 2001.
91. Zoccali C. ADMA: a critical cardio-renal link in heart failure? Eur J Clin Invest 33: 361-362, 2003.
92. Zoccali C. Traditional and emerging cardiovascular and renal risk factors: an epidemiologic perspective. Kidney Int 70: 26-33, 2006.
93. Zoccali C, Benedetto FA, Maas R, Mallamaci F, Tripepi G, Malatino LS, and Boger R. Asymmetric dimethylarginine, C-reactive protein, and carotid intima-media thickness in end-stage renal disease. J Am Soc Nephrol 13: 490-496, 2002.
94. Zoccali C, Bode-Boger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, and Boger R.

Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 358: 2113-2117, 2001.
95. Zoccali C, Mallamaci F, Maas R, Benedetto FA, Tripepi G, Malatino LS, Cataliotti A, Bellanuova I, and Boger R. Left ventricular hypertrophy, cardiac remodeling and asymmetric dimethylarginine (ADMA) in hemodialysis patients. Kidney Int 62: 339-345, 2002.

Abkürzungsverzeichnis
ACE-Hemmer Angiotensin Converting Enzyme-HemmerADMA Asymmetrisches Dimethylarginin; Synonym:
NG,NG-Dimethyl-L-ArgininAng II Angiotensin IIARB Angiotensin II-RezeptorblockerAT1-Blocker Angiotensin II-RezeptorblockerBAECs bovine aortale EndothelzellenBLAST Basic Local Alignment Search Toolbp BasenpaareBTE Biotechnologisches EntwicklungslaborCCC Konkordanz-Korrelations-KoeffizientcDNA copy DNAcGMP zyklisches Guanosinmonophosphatcpm counts per minuteCSE-Hemmer Cholesterinsyntheseenzymhemmerct cycle of thresholdDDAH Dimethylarginin-DimethylaminohydrolaseDHE DihydroethidiumDNA DesoxyribonukleinsäureEDRF endothelium derived relaxing factorEDTA EthylendiamintetraessigsäureELISA enzyme linked immunosorbent assayeNOS endotheliale NO-SynthaseFITC FloureszeinisothiocyanathnRNPs heterogeneous nuclear ribonucleoproteinsHPF high power fieldHPLC Hochdruck-FlüssigkeitschromatographieHUVECs humane NabelschnurendothelzelleniNOS induzierbare NO-SynthaseKHK koronare HerzkrankheitKm Michaelis-Menten KonstanteLC-MS TandemmassenspektrometrieL-NMMA NG-Monomethyl-L-ArgininMC MethylcarnoymRNA messenger RNAn.s. nicht signifikantNCBI National Center for Biotechnical InformationnNOS neuronale NO-SynthaseNO StickstoffmonoxidNOS NO-SynthasePAS-Färbung Perjodsäure-Leukofuchsin-FärbungpAVK periphere arterielle VerschlusskrankheitPBS Phosphat-gepufferte KochsalzlösungPCR Polymerase-KettenreaktionPRMT Protein-Arginin-N-Methyltransferase

r Korrelationskoeffizient RAS Renin-Angiotensin-SystemRIA RadioimmunoassayRNA RibonukleinsäurerRNA ribosomale RNART-PCR Reverse Transkriptase-Polymerase-
KettenreaktionSAH S-Adenosyl-HomozysteinSAM S-Adenosyl-MethioninSD StandardabweichungSDMA NG,NG`-Dimethyl-L-Arginin; Synonym:
Symmetrisches DimethylargininSEM Standardfehler des MittelwertesSH- Sulfhydryl-TG TransgenTGF-β transforming growth factor βTMB TetramethylbenzidinWT Wildtyp

Danksagungen
Herrn PD Dr. med. Johannes Jacobi danke ich für die vielen Hilfestellungen und
die hervorragende und unkomplizierte Betreuung bei allen Aspekten der
Doktorarbeit.
Ich möchte mich bei Herrn Prof. Dr. med. Karl F. Hilgers für die Überlassung
des Themas und die Unterstützung bei der Planung und Durchführung der
Arbeit bedanken.
Frau Michaela Arend danke ich für die äußerst kompetente Unterstützung bei
der Durchführung der Arbeiten im Labor.
Danken möchte ich ferner Frau Dr. Nada Cordasic für die Hilfe bei den
operativen Eingriffen und den weiteren Teammitgliedern der AG Hilgers für die
Unterstützung bei diversen Aufgaben im Rahmen der Labortätigkeiten.
Für die Tätigkeit als Korreferent möchte ich mich bei Prof. Dr. med. Renke
Maas bedanken.
Herrn Prof. Dr. med K.-U. Eckardt danke ich für die Arbeitsmöglichkeit in den
Laboreinrichtungen der Medizinischen Klinik IV sowie für die Möglichkeit der
Teilnahme an Doktorandenseminaren.


















