
EudraVigilance NEU - AGES€¦ · EudraVigilance neu • Legal Requirements • Process Change...
41
Mag. pharm. Katharina Weber BASG/AGES Medizinmarktaufsicht Institut Überwachung - Abteilung Blut, Gewebe und Vigilanz EudraVigilance NEU
Transcript of EudraVigilance NEU - AGES€¦ · EudraVigilance neu • Legal Requirements • Process Change...

Mag. pharm. Katharina Weber
BASG/AGES Medizinmarktaufsicht Institut Überwachung - Abteilung Blut, Gewebe und Vigilanz
EudraVigilance NEU

EudraVigilance neu • Legal Requirements • Process Change Management • EV System Implementation plan • Stakeholder Implementation planning
Addressed questions: • Reporting of medication errors • Reporting of off-label-use • Reporting of issues in combination with medical devices • PV Inspections
Supporting Information
AGENDA
2

Management Board endorsement starts countdown for stakeholders to get ready for launch of improved system in November 2017
The new version of EudraVigilance will go live on
22 November 2017
with enhanced functionalities for reporting and analysing suspected adverse reactions.
http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2017/05/news_detail_002752.jsp&mid=WC0b01ac058004d5c1 http://www.ema.europa.eu/docs/en_GB/document_library/Other/2017/05/WC500228158.pdf
EudraVigilance neu
3

Regulation (EC) No 726/2004 (amended 1235/2010) Community procedures for the authorisation and supervision of
medicinal products for human and veterinary use and establishing a European Medicines Agency
Directive 2001/83/EC (amended 2010/84/EU)
Community code relating to medicinal products for human use
Implementing Regulation (EU) No 520/2012
on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council
Legal Requirements
4

The main changes to electronic reporting requirements are:
• Usage of ISO standards in the reporting of ICSRs, ISO ICSR standard 27953-2:2011
• Usage of ISO IDMP terminologies, once available, in the submission of ISO ICSR messages
• EMA medical literature monitoring service • The audit of the EudraVigilance system • Simplification of reporting of ICSRs in the EU and forwarding of
national cases to the relevant NCA.
Legal Requirements
5
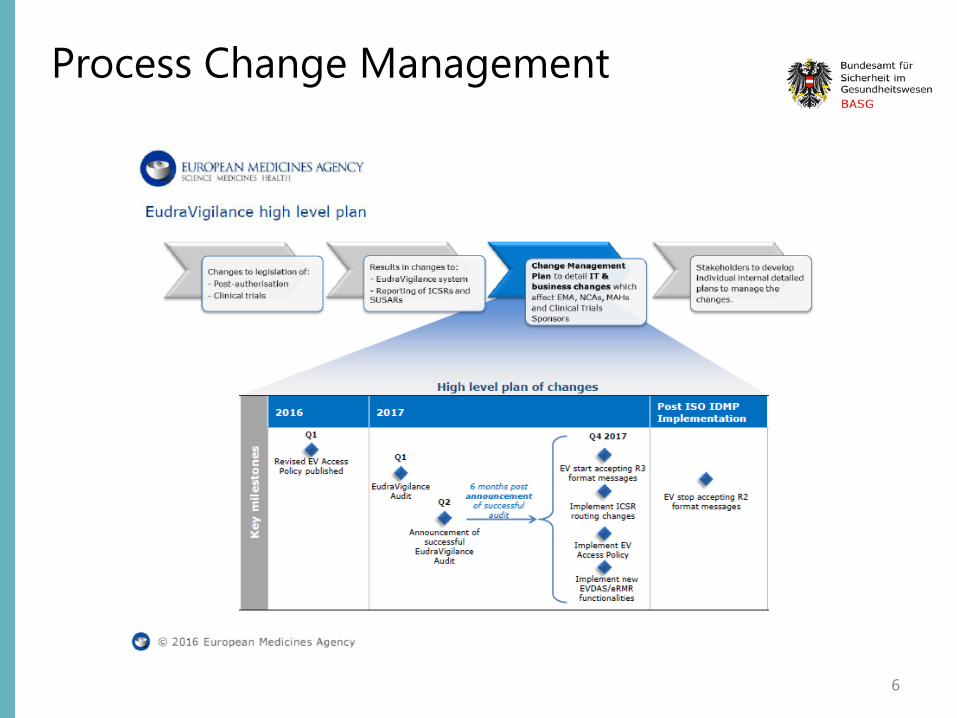
Process Change Management
6

Process Change Management EudraVigilance 8
7

Submission von ICSRs within EEA
Note: in E2B(R3), seriousness is reported at event level rather than at case level in E2B(R2) => if any event is considered serious then the whole case should be considered serious.
Process Change Management
MAH Procedure Origin Adverse reaction type Destination Time frame
• Centralised
• Mutual recogintion, decentralised or subject to referral
• purely national
EU
All serious Eudravigilance Database 15 days
All non serious Eudravigilance Database 90 days
Non EU All serious Eudravigilance Database 15 days
8

Clinical trial SUSAR reporting • Until the clinical trials regulation is applicable there is no change to
the current process for the submission of SUSARs for clinical trials.
EEA case submissions to the WHO UMC
• Following the implementation of the new EudraVigilance system, ICSR submissions for cases occurring in the EEA should only be made by the EudraVigilance system to WHO-UMC.
Process Change Management
9

Signal Management • Authorised users of the EudraVigilance Data Analysis System
(EVDAS) will be expected to retrieve data and use the available predefined reports included in the newly implemented functionalities in order to meet their data analysis and signal detection obligations.
User Manual and training materials Revised EudraVigilance Access Policy Revised GVP Module IX (planned approval by PRAC in 07/2017 – coming into
force in 11/2017)
Module IX Addendum I – Methodological Aspects of Signal Detection from
Spontaneous Reports of Suspected Adverse Reactions (planned approval by PRAC in
07/2017
Process Change Management
10

Signal Management Definition according to the “Council for International Organisations of Medical Sciences WG VIII “:
Information that arises from one or multiple sources (including observations and experiments), which suggests a new potentially causal association, or a new aspect of a known association, between an intervention and an event or set of related events, either adverse (or beneficial), that is judged to be of sufficient likelihood to justify verificatory action. Performance of signal detection activities required by the EU pharmacovigilance legislation*
Process Change Management
11 *COMMISSION IMPLEMENTING REGULATION (EU) No 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council
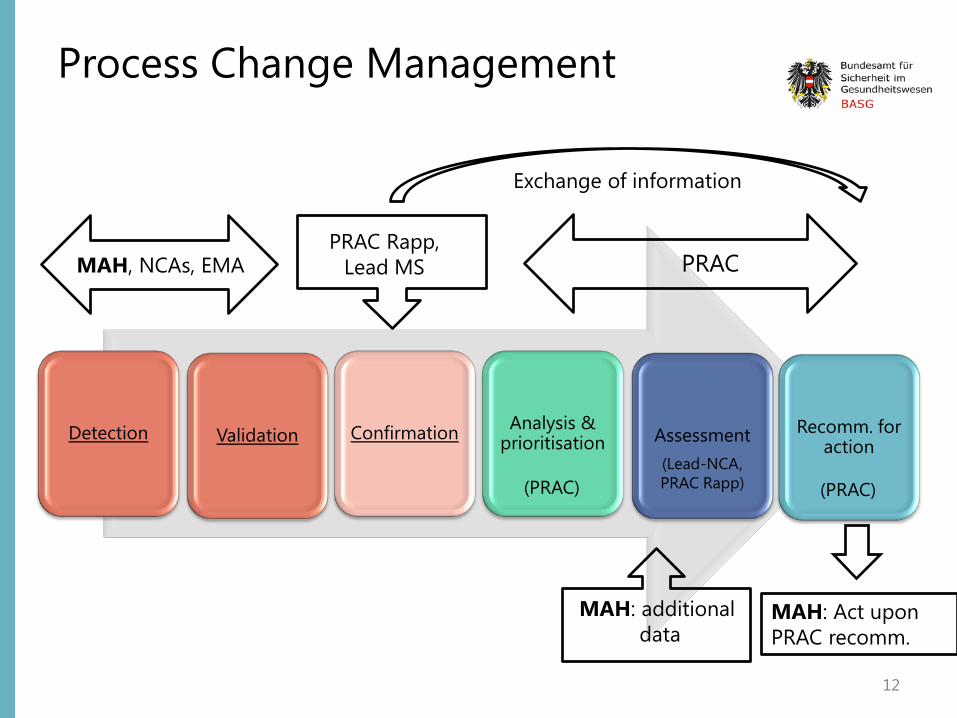
Process Change Management
12
Detection Validation Confirmation Analysis & prioritisation Assessment Recomm. for
action
MAH, NCAs, EMA PRAC Rapp,
Lead MS PRAC
Exchange of information
MAH: additional data
MAH: Act upon PRAC recomm.
(PRAC) (Lead-NCA, PRAC Rapp) (PRAC)

1. Signal Detection Identifying signals using data from any source (MAH, EudraVigilance, national ADR-databases, non-EU regulatory agencies, combination of statistical methods and review of ICSRs and scientific literature)
2. Signal Validation Process of evaluating in order to verify that the available documentation contains sufficient evidence demonstrating the existence of a new potentially causal association or a new aspect of a known association (Justification for further analysis, incl. clinical significance of the signal, its previous awareness, the biological and temporal plausibility and any relevant sources of information)
EMA and NCA EPITT* MAH Emerging Safety Issue
Process Change Management
13 * European Pharmacovigilance Information Tracking Tool

VI.C.2.2.6. Emerging safety issues Events may occur, which do not fall within the definition of reportable valid ICSRs, and thus are not subject to the reporting requirements, even though they may lead to changes in the known risk-benefit balance of a medicinal product and/or impact on public health. • major safety findings from a newly completed non-clinical study; • major safety concerns identified in the course of a non-interventional post-authorisation study or of a clinical
trial; • signal of a possible teratogen effect or of significant hazard to public health; • safety issues published in the scientific and medical literature; • safety issues arising from the signal detection activity (see Module IX) or emerging from a new ICSR and which
impact on the risk-benefit balance of the medicinal product and/or have implications for public health; • safety issues related to the use outside the terms of the marketing authorisation; • safety issues due to misinformation in the product information; • marketing authorisation withdrawal, non-renewal, revocation or suspension outside the EU for safety-related
reasons; • urgent safety restrictions outside the EU; • safety issues in relation to the supply of raw material; • lack of supply of medicines.
Process Change Management
14

VI.C.2.2.6. Emerging safety issues / Signal Validation These events/observations, which may affect the risk-benefit balance of a medicinal product should be notified as emerging safety issues in writing to the competent authorities in Member States where the medicinal product is authorised and to the Agency via email ([email protected]);
This should be done immediately when becoming aware of them. MAHs should provide a precise description of the safety issue, including the available evidence and the proposed regulatory action(s). All other validated signals should be handled according to available guidelines (see Module IX – Signal management of the GVP) Validated signals should also be presented in the relevant sections of the periodic safety update report (PSUR) (see also Module VII - Periodic safety update report (Rev 1) of the GVP).
Process Change Management
15

3. Signal Confirmation Communication via EPITT, within 30 days of its receipt by the Rapporteur, the lead Member State or a national competent authority that the validated signal is confirmed or not confirmed. Any confirmed signal should be analysed and prioritised by the Pharmacovigilance Risk Assessment Committee (PRAC).
4. Signal Analysis and Priorisation Prepared by the confirmer, for endorsement by the PRAC; taking into account the potential impact of the signal on the benefit-risk profile of the involved medicine(s);
5. Signal Assessment scientific evaluation of all the evidence available, including additional data from MAHs; responsibility of the PRAC. At the start of an evaluation, the PRAC appoints a Rapporteur who takes the lead for the assessment of all collected data.
Process Change Management
16

6. Recommendation for action Responsibility of the PRAC possible actions are: • no need for further evaluation or action at this point of time; • need for additional information:
the MAH, Member States and/or the EMA, where relevant, should monitor any relevant emerging information on the signal as it becomes available;
the MAH should address the signal in the following PSUR or submit an ad-hoc PSUR; the MAH should submit additional data; the EMA or Member States, as relevant, should collect further information (e.g. via a nonurgent information
request) or perform additional analyses in EudraVigilance or other data sources; the MAH(s) should conduct a post-authorisation safety study;
• need for regulatory action: the product information and/or risk management plan (RMP) should be updated through a variation; the Member States or the Commission, as appropriate, should initiate a referral procedure; urgent safety restrictions should be imposed.
These actions may be accompanied by additional communication measures, e.g. a Direct Healthcare Professional Communication (DHPC).
• pharmacovigilance inspection should take place.
Process Change Management
17

6. Recommendation for action Responsibility of the PRAC When applicable the PRAC recommendation includes the timeline for the
action(s) and a brief rationale. PRAC recommendations to provide additional data are directly actionable by the
concerned MAHs. PRAC recommendations for regulatory action are submitted to the Committee for Medicinal Products for Human Use (CHMP), in case of CAPs inclusion, and to the Co-ordination group for Mutual recognition and Decentralised procedures – human (CMDh) for information in the case of NAPs, before implementation by the concerned MAHs. All PRAC recommendations on signals are published on the EMA website within a month of the relevant PRAC meeting and are further reflected in the meeting minutes.
Process Change Management
18

Signal Management – place for improvement • Criteria for quality management • Description of the whole process (detection / validation / communication to NCAs /
implementation of recommendation) • Method of signal detection (sources, statistics incl. algorithm and rationale) • Frequency of signal detection (risk based approach) • Method of causality assessment and outcome • Communication to NCAs and EMA • Qualified staff (trainings, documentation of courses) • standardised documentation of all steps (incl. tracking) • Regular audits
Process Change Management
19

EV System Implementation plan EV system is being updated to be ISO ICSR 27953-2:2011 (E2B(R3)) compliant Before the move to simplified reporting, the new EudraVigilance system has to undergo an independent audit that will check that the required functionalities.
several changes in EudraVigilance: • adapting the EudraVigilance Gateway, the secure electronic communication tool to exchange ICSRs, to accept
ICH E2B(R2) as well as (R3) files; • adapting the EudraVigilance database management system (EDBMS) to support the storage and processing of
reports submitted with the new ICH ICSR format • migrating all existing ICH E2B(R2) ICSR data to the new ICH ICSR format; • redesign of EVWEB, the web application for the electronic reporting and management of ICSRs in the ISO ICSR
format, using a new technology; • introducing new functionalities in the EudraVigilance data analysis system (EVDAS), including the adrreports.eu
portal, to support analysis of data and support signal management activities
In addition, EMA will add the following two new functionalities to the system: • EudraVigilance rerouting functionality, which defines the rules for the forwarding of ICSRs to the national
competent authority (NCA) where the adverse reaction occurred (rules can be set by NCAs according to their needs and preferences);
• ICSR download functionality to enable marketing authorisation holders to download ICSRs concerning their products or ICSRs reported with a substance for which they hold a marketing authorisation in the EEA.
EV System Implementation plan
20
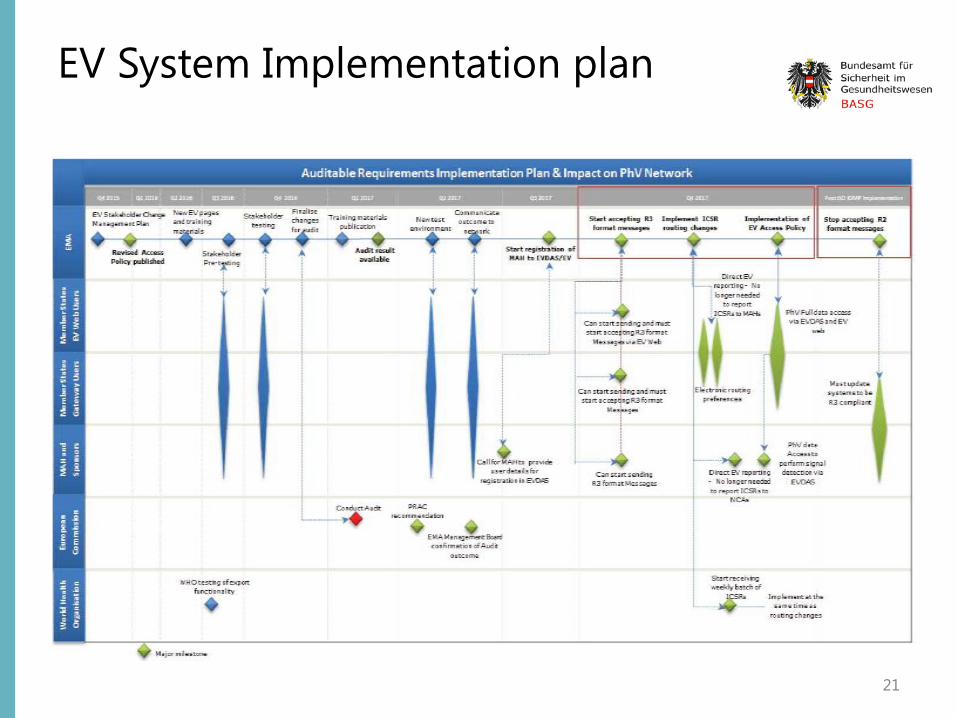
EV System Implementation plan
EV System Implementation plan
21

22
EV database management system • The EudraVigilance database management system will have the most
significant changes as the database will be updated to support the E2B(R3) structure and fields. All existing E2B(R2) data will be migrated into this new database.
• The XML parser2 will be replaced and will be able to process both E2B(R2) and E2B(R3) messages. If an E2B(R2) message is sent to the EudraVigilance system, an E2B(R2) acknowledgement message will be returned. Likewise, an E2B(R3) acknowledgment will be returned for E2B(R3) ICSR messages.
• EudraVigilance applications for Medicinal Product Recoding and Duplicate Detection and management will also be amended to support E2B(R3). Master cases that are produced by the EMA in response to identifying duplicated ICSR in the EudraVigilance system will be produced in E2B(R3) format only.
EV System Implementation plan

EV data analysis system (EVDAS): • The EudraVigilance data analysis system (EVDAS) will be updated
to the new E2B(R3) data structure. a) Healthcare professionals and general public
The current European database of suspected adverse drug reaction reports (http://www.adrreports.eu ) will be updated and improved to include: • Weekly refresh of published data; • Publication of data for non-serious cases received in EudraVigilance; • Publication of ‘country’ data (in aggregated dashboard); • Publication of additional data elements via Line listing reports and the new ICSR form
(replacing the existing CIOMS I form).
b) Marketing Authorisation Holders (for Signal detection)
• The electronic Reaction Monitoring Report (eRMR) based on the eRMR used within the EU network and the EMA for signal detection activities;
• The individual case line listing and ICSR form.
EV System Implementation plan
23

ICSR download function for MAHs
After the switch to simplified reporting, MAHs will no longer receive ICSRs directly from NCAs. In order for MAHs to receive ICSRs concerning their products download functionality will be made available. It should be noted that the ICSRs that will be provided via this download functionality will only be available in E2B(R3) format. The download function will be made available through the new EVWEB application.
EV System Implementation plan
24

Testing with the new EudraVigilance system and user training • Once the Audit has been completed, all organisations with a registered
Test EudraVigilance account can start sending E2B(R3) Test files and will also be able to download E2B(R3) files based on test data. In addition, EVWEB users will be able to view and use the new EVWEB interface in the Test EudraVigilance system.
• The EMA will publish a user guide, online eLearning, training materials and provide support webinars. The training materials will explain the new E2B(R3) format including the new data fields and the new EVWEB tool.
EV System Implementation plan
25

Stakeholder Implementation planning MAH Change Management Summary
26

Stakeholder Implementation planning MAH Change Management Summary
27

The EudraVigilance helpdesk:
• EudraVigilance Registration • Email - [email protected]
• EMA IT Service Desk • Link - https://servicedesk.ema.europa.eu (For support with EudraVigilance and EMA gateway/webclient)
Stakeholder Implementation planning
28

EudraVigilance neu • Legal Requirements • Process Change Management • EV System Implementation plan • Stakeholder Implementation planning
addressed questions: • Reporting of medication errors • Reporting of off-label-use • Reporting of issues in combination with medical devices • PV Inspections
Supporting Information
AGENDA
29

“A medication error is an unintended failure in the drug treatment process that leads to, or has the potential to lead to, harm to the patient. “
To be reported in the E2B field „reaction“ E2B(R2): B.2.i.1.b = E2B(R3): E.i.2.1 only if really reported as „medication error“ by primary source; otherwise as „Sender‘s diagnosis“ in E2B(R2)-field B.5.3.b = E2B(R3): H.3.r.1.b
Reporting of medication errors
30

Suspected (serious and non-serious) adverse reactions associated with medication errors should be recorded, reported and assessed.
Should be reported as Emerging Safety Issue if important safety concern which impacts on the overall benefit-risk balance of the medicinal product involved or on public health
Reporting of medication errors
31
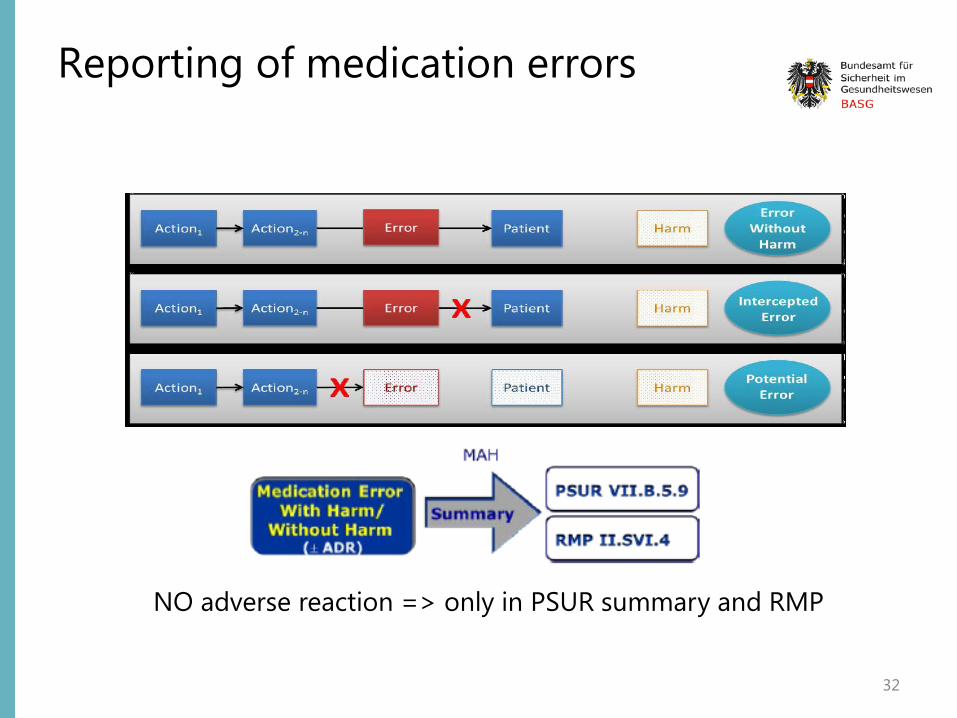
NO adverse reaction => only in PSUR summary and RMP
Reporting of medication errors
32
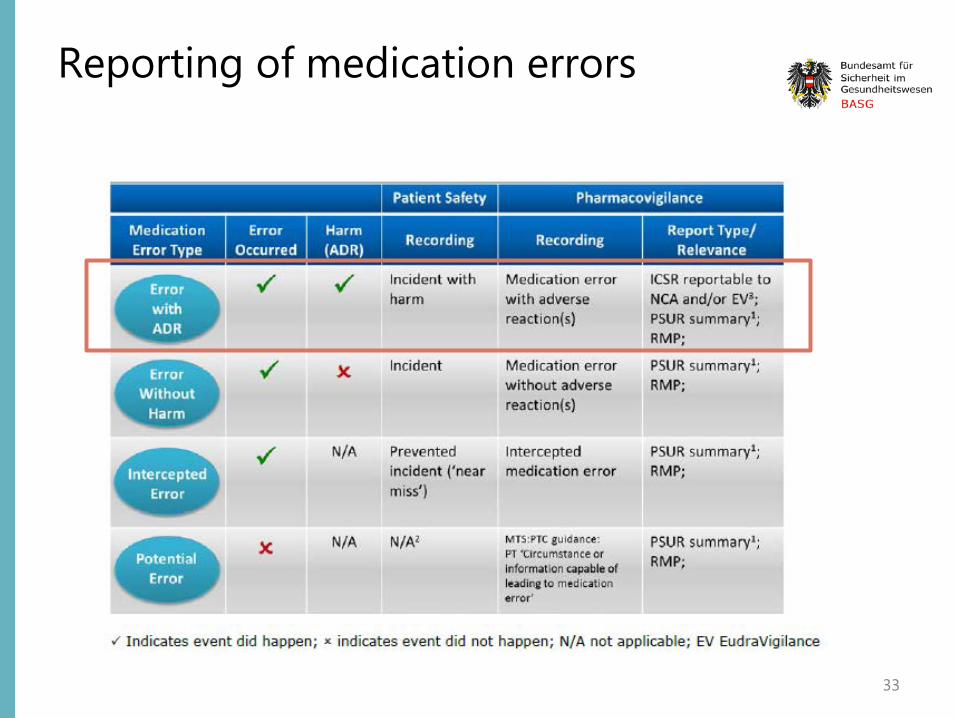
Reporting of medication errors
33

GVP Module VI.A.2.1.2. b. Off-label use This relates to situations where the medicinal product is intentionally used for a medical purpose not in accordance with the authorised product information. VI.B.6.3. : Reports of overdose, abuse, off-label use, misuse, medication error or occupational exposure with no associated adverse reaction should not be reported as ICSRs. They should be considered in periodic safety update reports as applicable. Reports associated with suspected adverse reactions => reported as ICSR
Reporting of off-label-use
34

Medizinproduktegesetz: Abgrenzung zu Arzneimittelregelungen § 5. (1) Medizinprodukte, die im Zeitpunkt der Abgabe kein Arzneimittel enthalten, jedoch dazu bestimmt sind, ein Arzneimittel im Sinne des Arzneimittelgesetzes anzuwenden, unterliegen diesem Bundesgesetz unbeschadet der das Arzneimittel betreffenden Bestimmungen des Arzneimittelgesetzes.
(2) Wird eine Kombination von Medizinprodukt und Arzneimittel so in Verkehr gebracht, dass Medizinprodukt und Arzneimittel ein einheitliches, miteinander verbundenes Produkt bilden, dessen bestimmungsgemäße Hauptwirkung auf der stofflichen Zusammensetzung des Arzneimittels beruht und das ausschließlich zur Verwendung in dieser Verbindung bestimmt und nicht wiederverwendbar ist, so unterliegt dieses Produkt dem Arzneimittelgesetz.
Reporting of Issues in Comb. with MD
35

PV-Systeminspections: Inspection of the PV - Documentation (the complete PV-System) Risked based Inspection-planning (also in accordance with other Agencies) Approx. Duration: 1,5 – 2 d
PV/GMP-Inspectionens Processed in accordance with GMP-INS Procedure (PV-Inspection will be
mentioned in GMP- Inspection - Announcement) „Screening“ of the national PV- scenery preferred for SME Approx. Duration: 0,5 – 1 d
PV - Inspections
36

PV - Inspectionsprogramm in AT Main Focus: MAH with PSMF (QPPV) in AT Inspection of concerned MAHs (risk-based approach: Inspection-Status,
PSMF-Status, and Compliance-Information) After the first Inspection: Definition of Inspection-intervall
enhanced Programm: MAH as locale Affiliate Inspection of all concerned MAHs (risk-based approach : product types,
internal/external triggers, as well as other EU PV– INS–activities) After the first Inspection: Definition of Inspection-intervall (risk-based)
PV - Inspections
37

EV Change Management: • http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q
_and_a/q_and_a_detail_000165.jsp (EudraVigilance Change Management)
• http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2015/10/WC500196029.pdf (revised EudraVigilance Stakeholder Change Management Plan)
• http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2016/04/WC500204567.pdf (EudraVigilance auditable requirement project - EudraVigilance training plan )
• http://www.ema.europa.eu/docs/en_GB/document_library/Other/2015/12/WC500199048.pdf (European Medicines Agency policy on access to EudraVigilance data for medicinal products for human use (EudraVigilance Access Policy))
• http://www.ema.europa.eu/docs/en_GB/document_library/Other/2013/09/WC500150743.pdf (Questions & answers on signal management)
Supporting Information - 01
38

EV Change Management: • http://www.ema.europa.eu/docs/en_GB/document_library/Other/2015/07/
WC500189042.pdf (Safety Signal - Assessment of responses to Request for Supplementary Information
(RSI))
• http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000375.jsp (PRAC recommendations on safety signals)
• http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q_and_a/q_and_a_detail_000166.jsp&mid=WC0b01ac0580a68f78 (EudraVigilance system
overview)
• http://estri.ich.org/ (ISO ICSR standard / ICH E2B(R3) )
• http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/01/news_detail_002252.jsp&mid=WC0b01ac058004d5c1 (New
international standard to improve safety of medicines)
Supporting Information - 02
39
• http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2014/04/WC500165979.pdf (EU Individual Case Safety Report (ICSR) Implementation Guide )
Medication error: • http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/gene
ral/general_content_000570.jsp (Medication errors)
• http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2015/11/WC500196979.pdf (Good practice guide on recording, coding, reporting and assessment of medication errors)
• http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2015/11/WC500196981.pdf (Good practice guide on risk minimisation and prevention of medication errors)
Medizinproduktegesetz: • https://www.ris.bka.gv.at/GeltendeFassung.wxe?Abfrage=Bundesnormen&
Gesetzesnummer=10011003
Supporting Information - 03
40

BASG - Bundesamt für Sicherheit im Gesundheitswesen
www.basg.gv.at
Traisengasse 5 1200 Wien
Gutachterin / Inspektorin Pharmakovigilanz
Mag.pharm. Katharina Weber


















