
Evolution der Proteinstruktur - uni-frankfurt.de
62
Evolution der Proteinstruktur Strukturelle Bioinformatik WS15/16 Dr. Stefan Simm, 09.12.2015 [email protected]
Transcript of Evolution der Proteinstruktur - uni-frankfurt.de

Evolution der Proteinstruktur
Strukturelle Bioinformatik WS15/16
Dr. Stefan Simm, 09.12.2015 [email protected]

DOMÄNEN UND MOTIVE (EINE KURZE WIEDERHOLUNG)
Evolution der Proteinstruktur
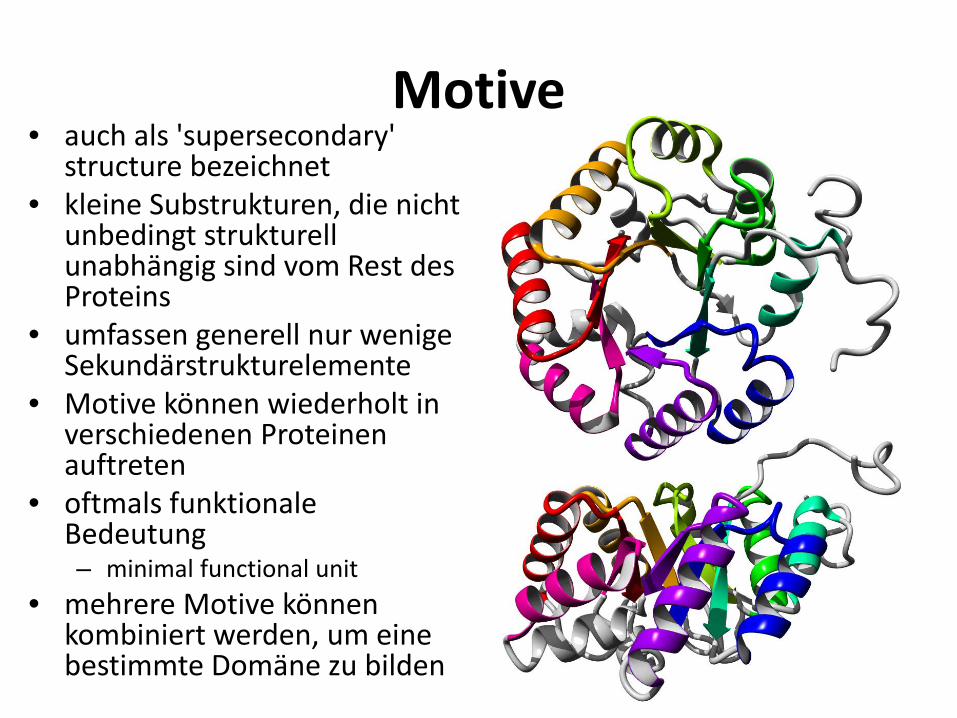
Motive • auch als 'supersecondary'
structure bezeichnet • kleine Substrukturen, die nicht
unbedingt strukturell unabhängig sind vom Rest des Proteins
• umfassen generell nur wenige Sekundärstrukturelemente
• Motive können wiederholt in verschiedenen Proteinen auftreten
• oftmals funktionale Bedeutung – minimal functional unit
• mehrere Motive können kombiniert werden, um eine bestimmte Domäne zu bilden
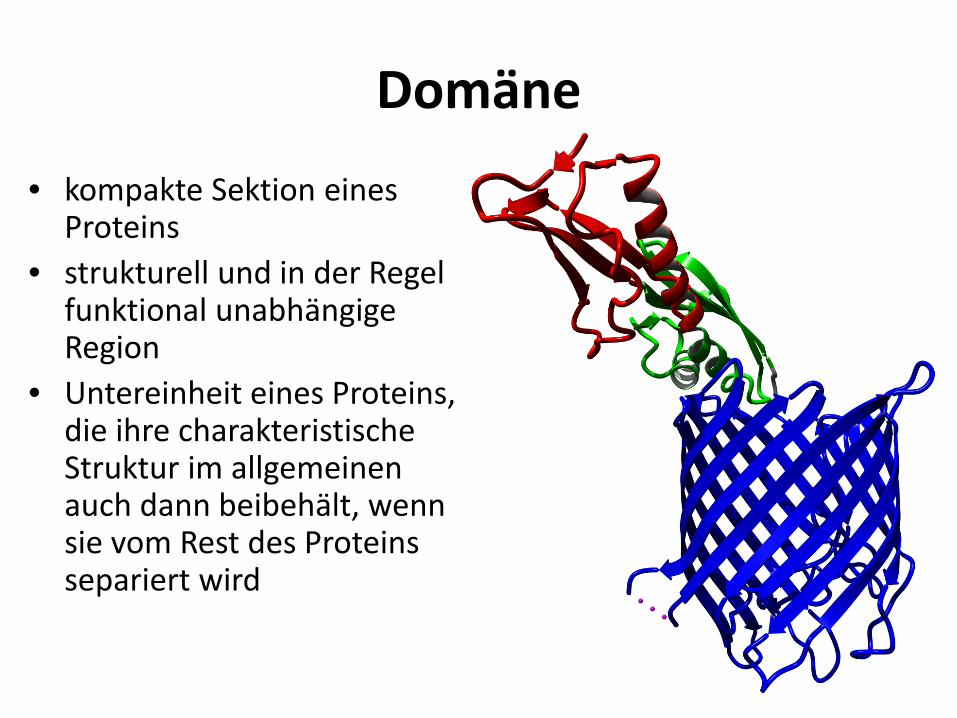
Domäne • kompakte Sektion eines
Proteins • strukturell und in der Regel
funktional unabhängige Region
• Untereinheit eines Proteins, die ihre charakteristische Struktur im allgemeinen auch dann beibehält, wenn sie vom Rest des Proteins separiert wird

Fold
• die finale 3D Tertiärstruktur eines Proteins • ein Fold kann verschiedene Domänen und
Motive enthalten

Fold
• umreißt 3 Hauptaspekte der 3D Struktur von Proteinen – Sekundärstrukturen – relative Anordnung der Sekundärstrukturen – Pfad der Polypeptidkette durch die Struktur
• definiert durch – Komposition – Architektur – Topologie

Fold
• homologe Proteine mit ähnlicher Sequenz besitzen denselben Fold
• derselbe Fold kann auch von Proteinen mit unähnlicher Sequenz ausgebildet werden
• Frage: Bedeutet strukturelle Ähnlichkeit zweier unterschiedlicher Proteine entfernte Homologie oder ist sie das Resultat der grundlegenden Eigenschaften von Physik und Chemie?

SCOP
• Structural Classification of Proteins database (1994)
• SCOP umgeht diese Frage mit der Einführung einer neuen Kategorie – Superfamilie
• diese Kategorie gruppiert Proteine, die vielleicht von einem gemeinsamen Vorfahren abstammen, deren Sequenzen aber dergestalt evolviert sind, daß sie nicht mehr als ähnlich zu erkennen sind (Murzin et al. 1995)

Fold
• Idee möglicher entfernter Homologie lockerte die Definition eines Folds – ‘consensus’ fold, der einer Menge von evolutionär
verwandten Proteinen gemein ist • ein Fold kann sich im Laufe der Evolution
verändern • Veränderungen können nicht nur periphere
sondern auch zentrale Elemente der Struktur betreffen

HOMOLOGIE Evolution der Proteinstruktur
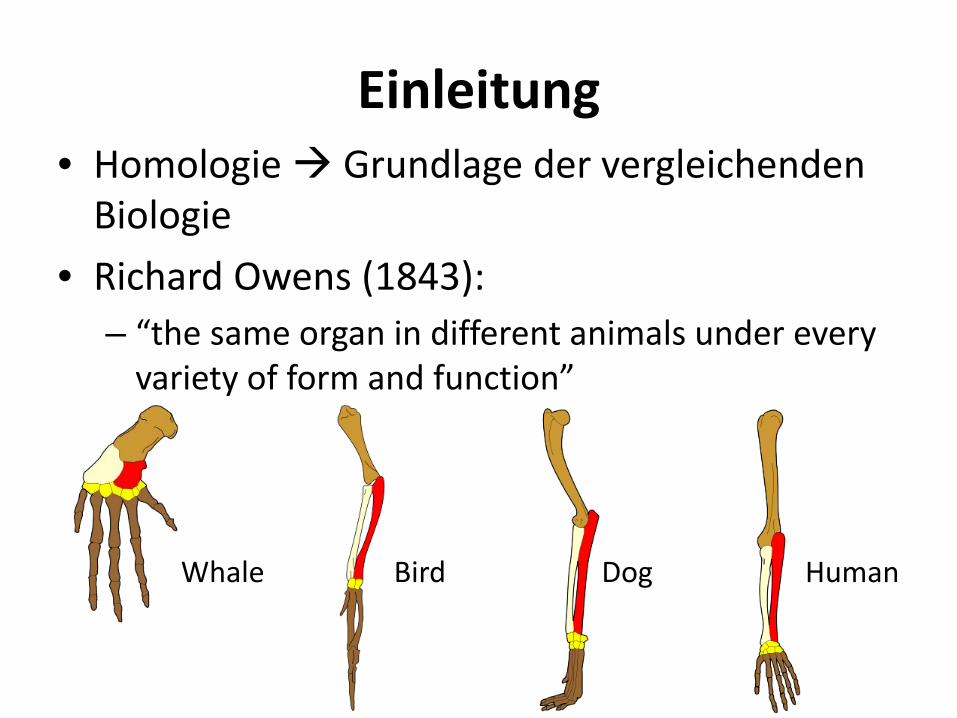
Einleitung • Homologie Grundlage der vergleichenden
Biologie • Richard Owens (1843):
– “the same organ in different animals under every variety of form and function”
Human Dog Whale Bird
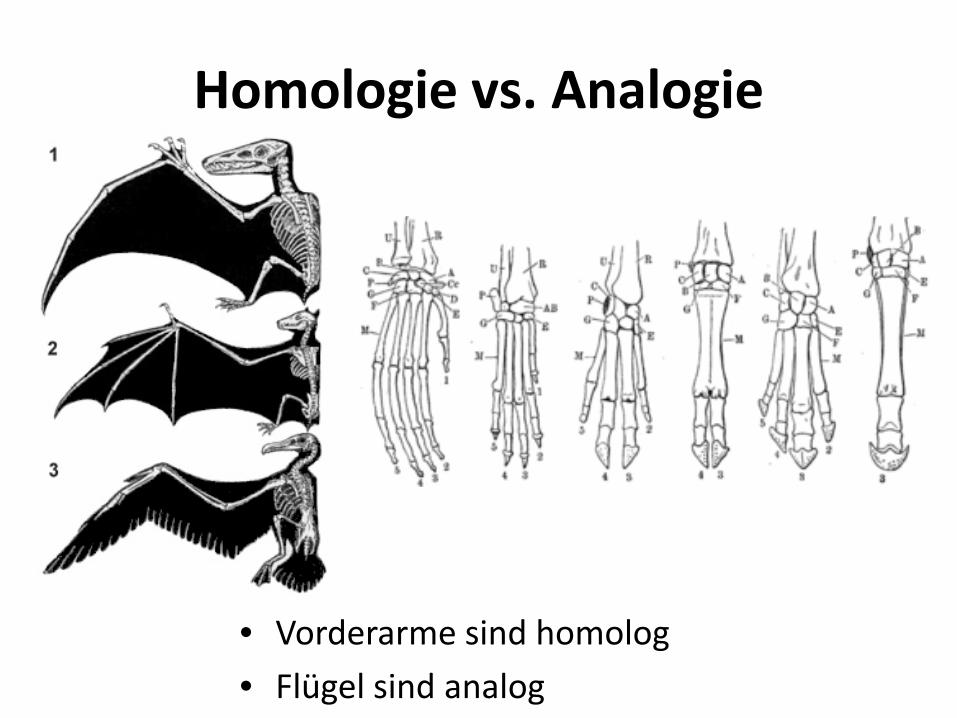
Homologie vs. Analogie
• Vorderarme sind homolog • Flügel sind analog
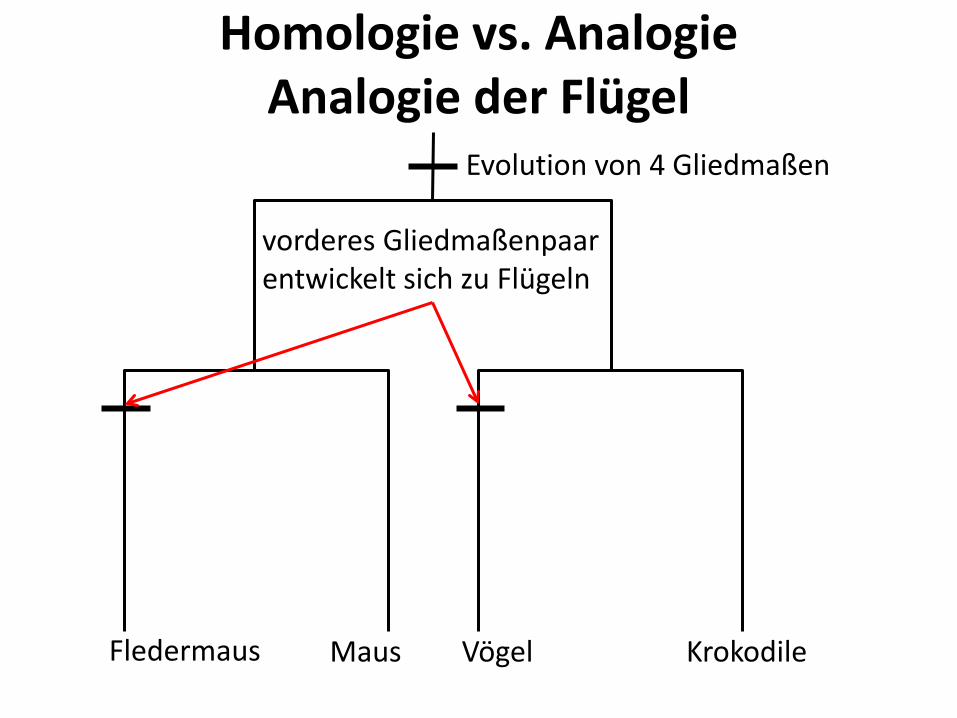
Evolution von 4 Gliedmaßen
Krokodile Vögel Maus Fledermaus
vorderes Gliedmaßenpaar entwickelt sich zu Flügeln
Homologie vs. Analogie Analogie der Flügel

Sequenzhomologie • definitiert im Sinne gemeinsamer Abstammung
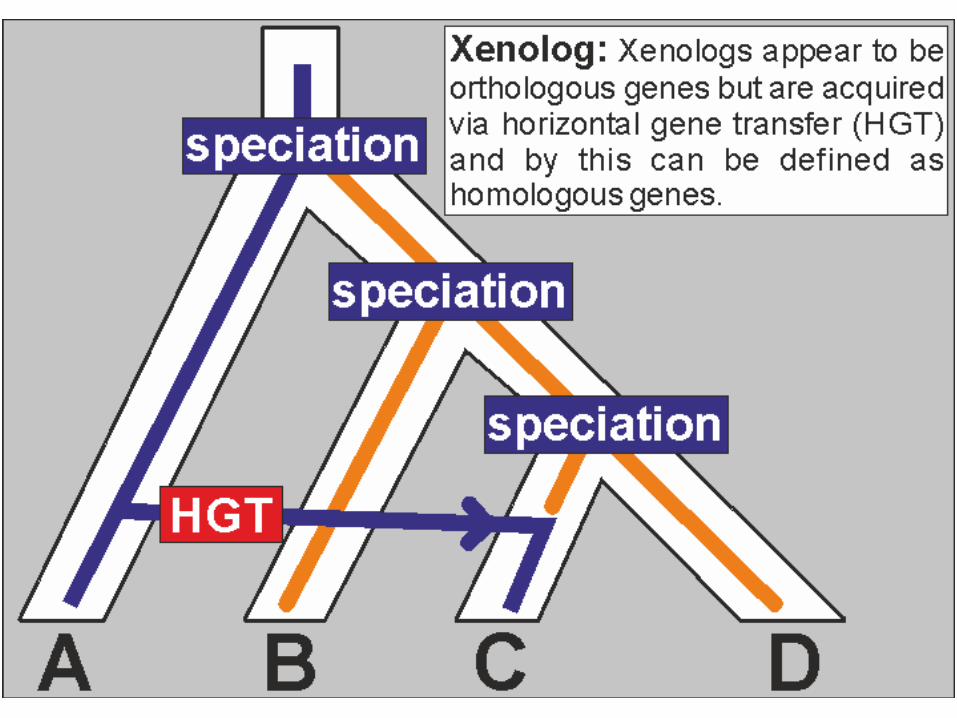
– Artenbildung (Orthologe) – Duplikationsereignis (Paraloge) – Verlust von Genen – horizontaler Gentransfer (HGT) – Fusion, Spaltung und andere Neuanordnungen
• Homologie von Proteinen oder DNA oftmals inkorrekt anhand einer
Sequenzähnlichkeit hergeleitet: – konvergente Evolution (lange Sequenzen) – Zufall (kurze Sequenzen)
• Datenbanken zur Homologie:
– Vertebraten (HOVERGEN, HOMOLENS, HOGENOM) – Bakterien (HOBACGEN)
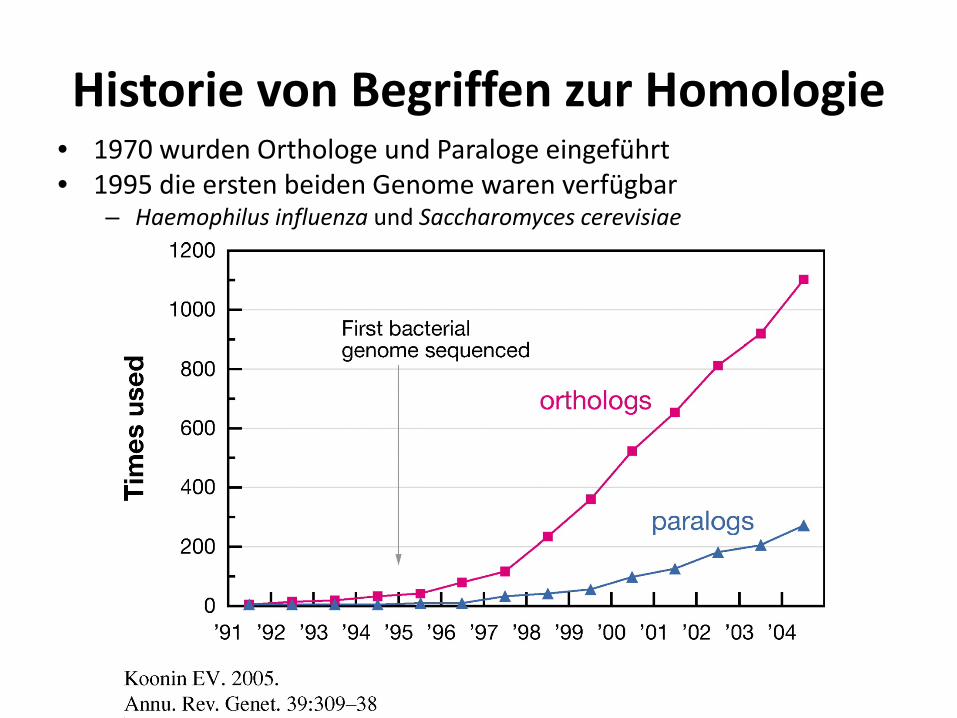
Historie von Begriffen zur Homologie • 1970 wurden Orthologe und Paraloge eingeführt • 1995 die ersten beiden Genome waren verfügbar
– Haemophilus influenza und Saccharomyces cerevisiae

Paralogy
• genes related via duplication • Requirements:
– first duplication in same organism – major functional connotation
• Not necessary: – resided in same genome – fixed timepoint – equal function
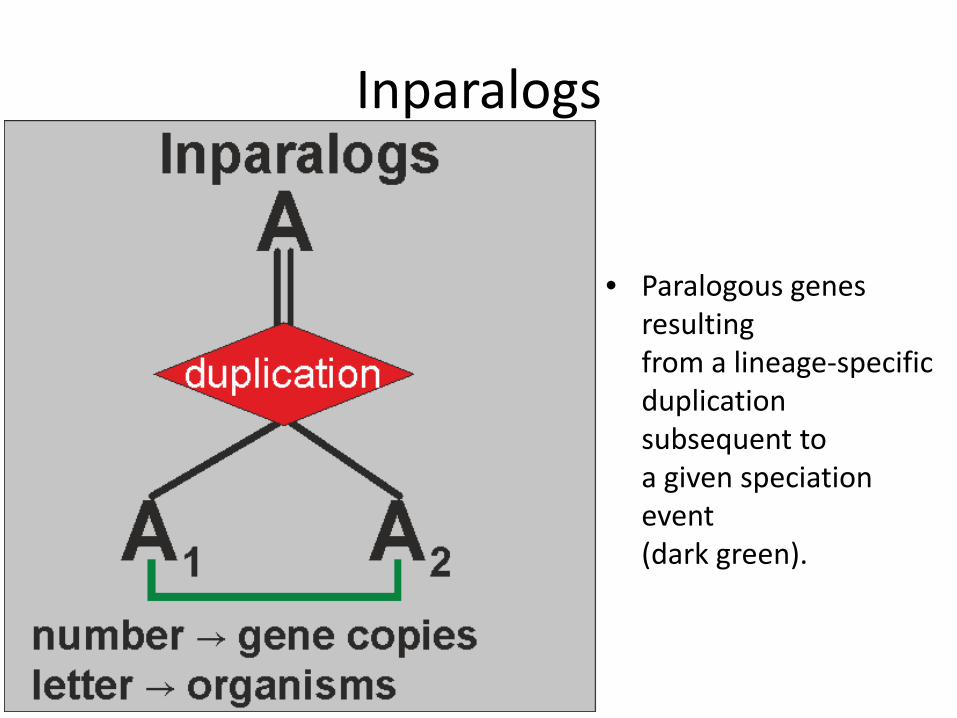
Inparalogs
• Paralogous genes
resulting from a lineage-specific duplication subsequent to a given speciation event (dark green).
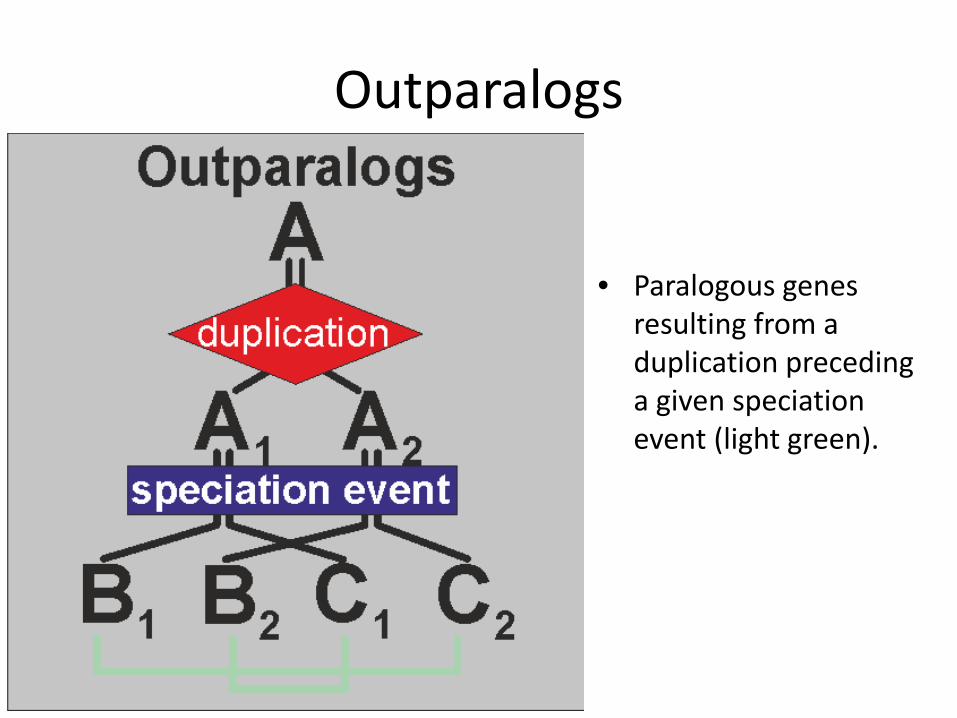
Outparalogs
• Paralogous genes resulting from a duplication preceding a given speciation event (light green).

Orthology
• genes derived from single ancestral gene in last common ancestor
• Requirements: – single ancestral gene – presence in last common ancestor
• Not necessary: – one-to-one relationship – equal function
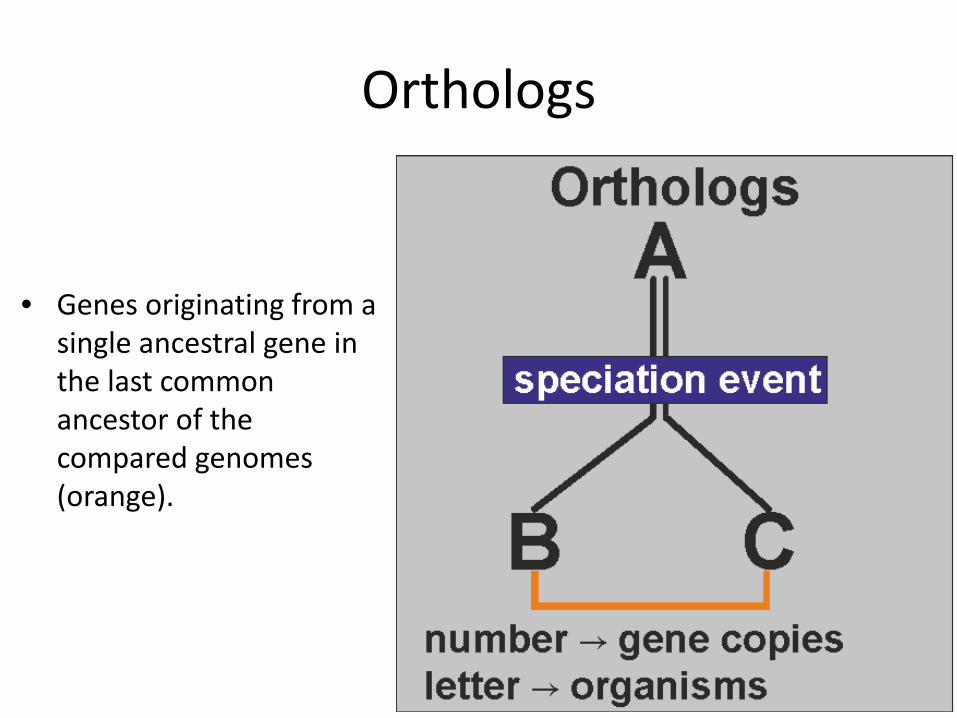
Orthologs
• Genes originating from a single ancestral gene in the last common ancestor of the compared genomes (orange).

Co-Orthologs • Two or more genes in
one lineage that are, collectively, orthologous to one or more genes in another lineage due to a lineage-specific duplication. Members of a co-orthologous gene set are inparalogs relative to the respective speciation event (yellow).

NEUTRALE THEORIE DER MOLEKULAREN EVOLUTION
Evolution der Proteinstruktur

Neutrale Theorie der Molekularen Evolution
• große Mehrheit der evolutionären Veränderungen auf molekularer Ebene erfolgt durch zufällige Drifts selektiv neutraler Mutanten (Fitneß nicht beeinflußt; Kimura 1983)
• kompatibel mit Darwins Theorie von Evolution durch natürliche Auslese: – adaptive Veränderungen werden anerkannt und sind
wichtig, aber es wird hypothetisiert, daß sie nur eine Minderheit aller Änderungen wären, die in der DNA Sequenz fixiert werden (Kimura 1986)

Neutrale Theorie der Molekularen Evolution
• eine Aminosäure wird von mehreren Basentripletts kodiert
• stille (silent, neutral) Mutationen verändern das Triplet aber nicht die kodierte Aminosäure
http://www.nature.com/scitable/topicpage/the-information-in-dna-determines-cellular-function-6523228
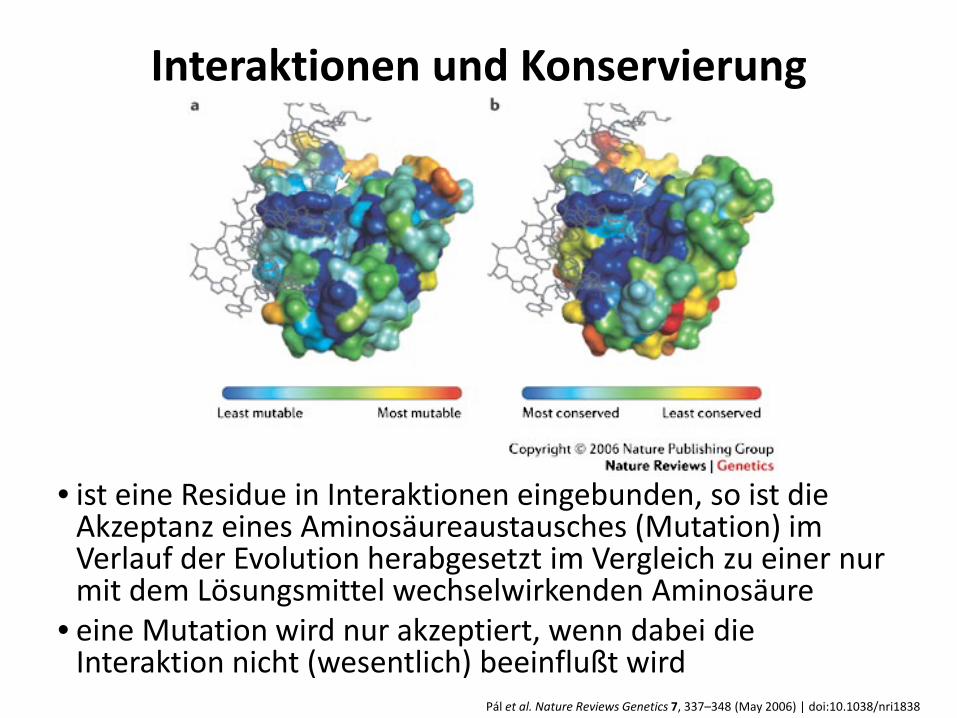
Pál et al. Nature Reviews Genetics 7, 337–348 (May 2006) | doi:10.1038/nri1838
Interaktionen und Konservierung
• ist eine Residue in Interaktionen eingebunden, so ist die Akzeptanz eines Aminosäureaustausches (Mutation) im Verlauf der Evolution herabgesetzt im Vergleich zu einer nur mit dem Lösungsmittel wechselwirkenden Aminosäure
• eine Mutation wird nur akzeptiert, wenn dabei die Interaktion nicht (wesentlich) beeinflußt wird

COEVOLUTION VON INTERAGIERENDEN RESIDUEN
Evolution der Proteinstruktur
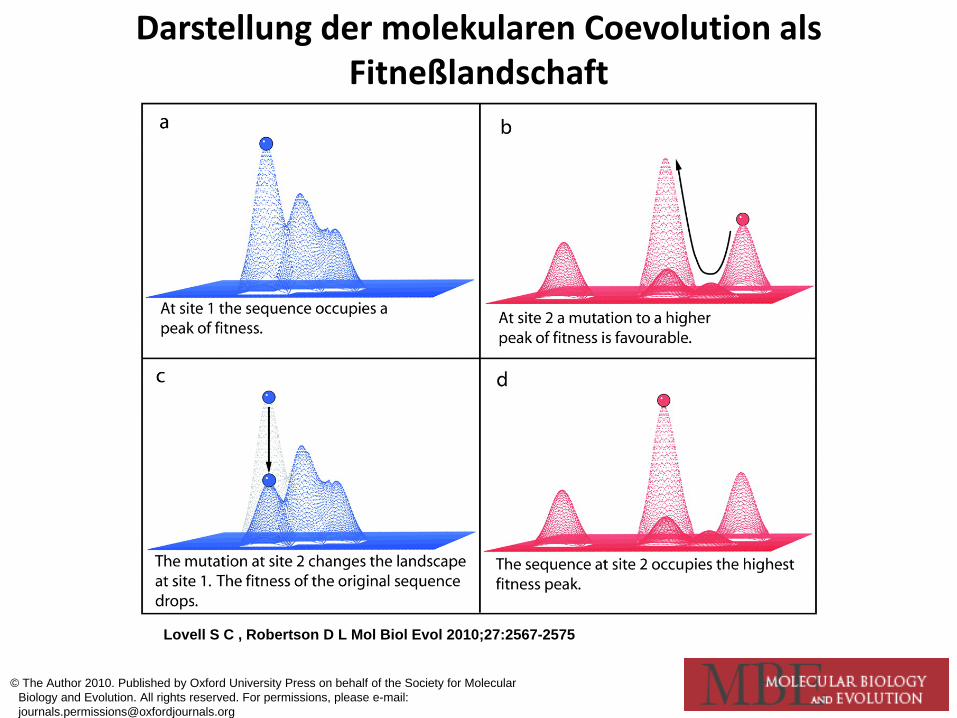
Darstellung der molekularen Coevolution als Fitneßlandschaft
Lovell S C , Robertson D L Mol Biol Evol 2010;27:2567-2575
© The Author 2010. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: [email protected]
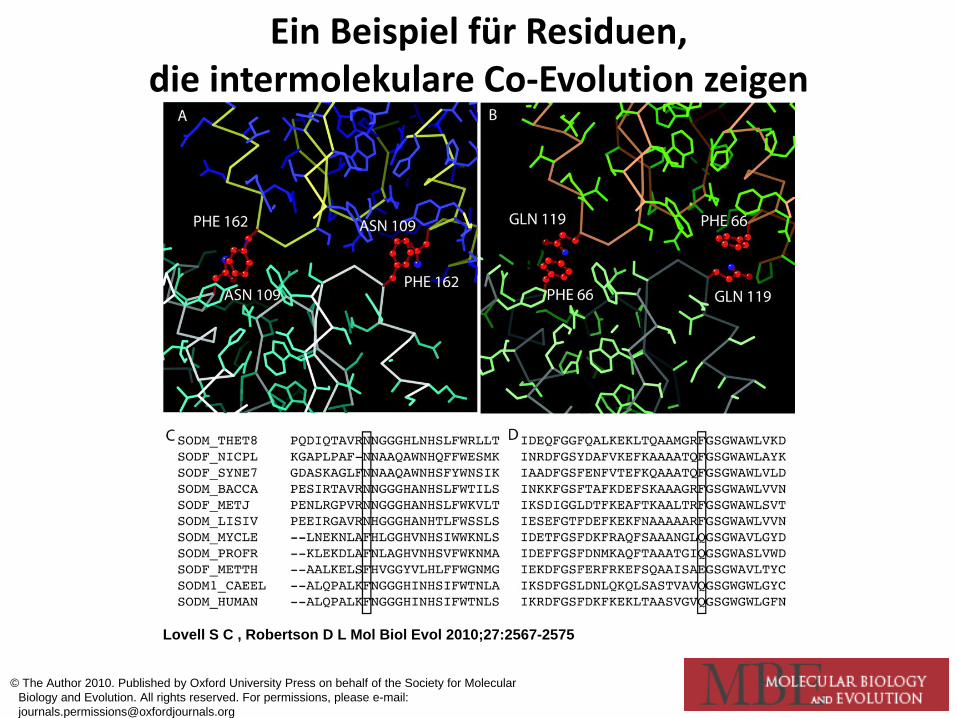
Ein Beispiel für Residuen, die intermolekulare Co-Evolution zeigen
Lovell S C , Robertson D L Mol Biol Evol 2010;27:2567-2575
© The Author 2010. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: [email protected]

PROTEIN-EVOLUTION Evolution der Proteinstruktur
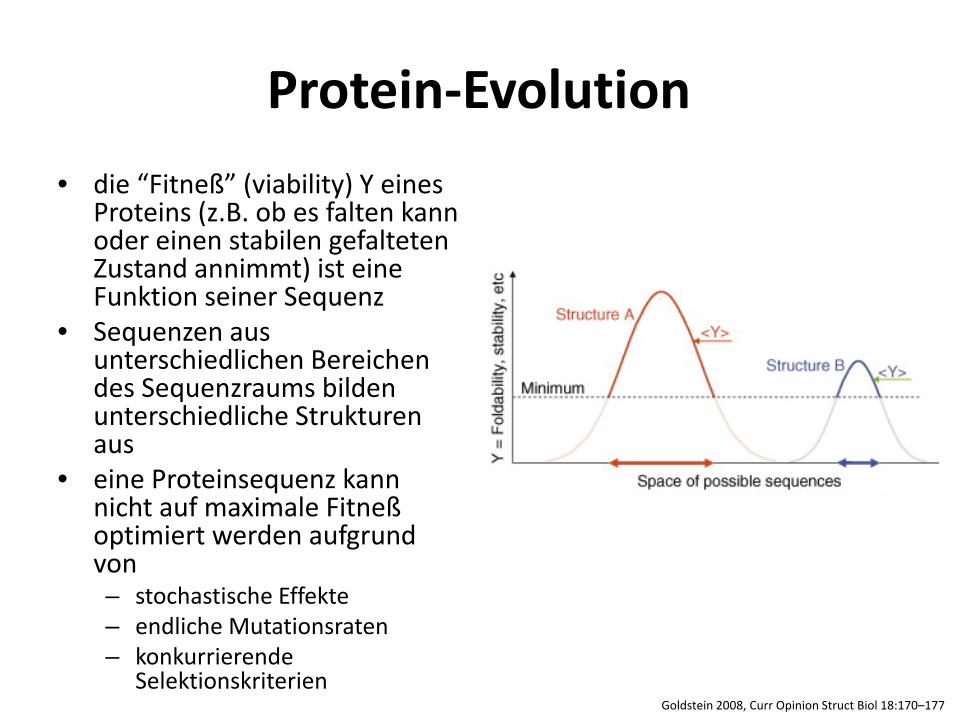
Protein-Evolution • die “Fitneß” (viability) Y eines
Proteins (z.B. ob es falten kann oder einen stabilen gefalteten Zustand annimmt) ist eine Funktion seiner Sequenz
• Sequenzen aus unterschiedlichen Bereichen des Sequenzraums bilden unterschiedliche Strukturen aus
• eine Proteinsequenz kann nicht auf maximale Fitneß optimiert werden aufgrund von – stochastische Effekte – endliche Mutationsraten – konkurrierende
Selektionskriterien Goldstein 2008, Curr Opinion Struct Biol 18:170–177

Neutrales Netzwerk
• Proteinevolution wird oft als Bewegung in einem Sequenzraum modelliert
• die möglichen Sequenzen können als Knoten eines Graphen dargestellt werden
• Nachbarn unterscheiden sich durch eine Mutation und sind über eine Kante verbunden
• “neutral” bedeutet, daß alle Mutationen in diesem Netzwerk die Struktur nicht verändern
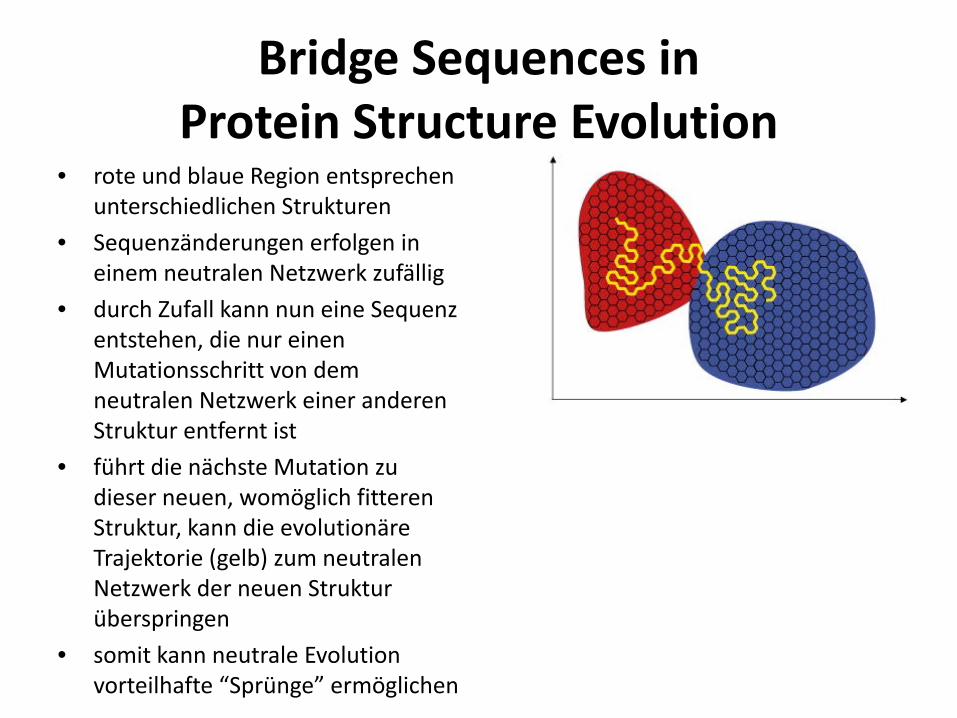
Bridge Sequences in Protein Structure Evolution
• rote und blaue Region entsprechen unterschiedlichen Strukturen
• Sequenzänderungen erfolgen in einem neutralen Netzwerk zufällig
• durch Zufall kann nun eine Sequenz entstehen, die nur einen Mutationsschritt von dem neutralen Netzwerk einer anderen Struktur entfernt ist
• führt die nächste Mutation zu dieser neuen, womöglich fitteren Struktur, kann die evolutionäre Trajektorie (gelb) zum neutralen Netzwerk der neuen Struktur überspringen
• somit kann neutrale Evolution vorteilhafte “Sprünge” ermöglichen

Bridge Sequences in Protein Structure Evolution
• aber in Wirklichkeit bildet jede Sequenz eine Verteilung von Strukturen bei physiologischer Temperatur aus
• es gibt zunehmend Hinweise, daß ein einzelnes Protein unterschiedliche Funktionen in unterschiedlicher Struktur ausüben könnte (James und Tawfik 2003)
• dadurch verwischen die Grenzen zwischen neutralen Netzwerken und der Prozeß der strukturellen Evolution wird beschleunigt (Wroe et al. 2007)
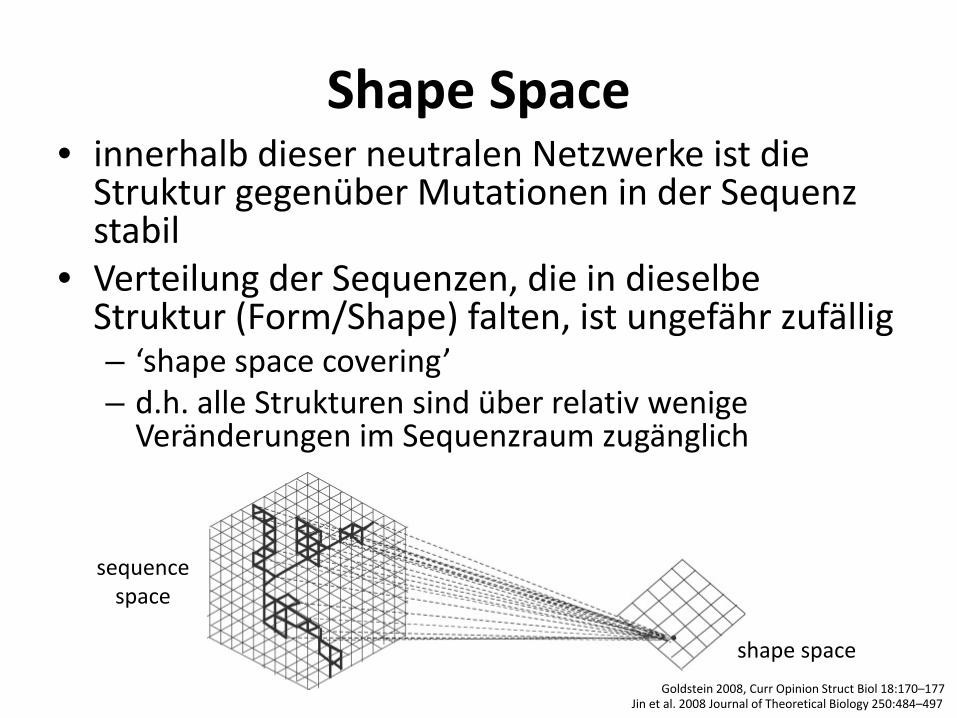
Shape Space • innerhalb dieser neutralen Netzwerke ist die
Struktur gegenüber Mutationen in der Sequenz stabil
• Verteilung der Sequenzen, die in dieselbe Struktur (Form/Shape) falten, ist ungefähr zufällig – ‘shape space covering’ – d.h. alle Strukturen sind über relativ wenige
Veränderungen im Sequenzraum zugänglich
shape space
sequence space
Jin et al. 2008 Journal of Theoretical Biology 250:484–497 Goldstein 2008, Curr Opinion Struct Biol 18:170–177

RNA vs. Protein • so wie auch Proteine bilden viele RNA Sequenzen dieselbe
Struktur aus • interessanter Weise ist die Beziehung zwischen Sequenz
und Struktur sehr unterschiedlich • der RNA Sequenzraum gleicht einer “Schüssel mit
Spaghettie” – sehr unterschiedliche Sequenzen können die gleiche Struktur
ausbilden – die Veränderung einer einzelnen Base wandelt die Struktur in
nahezu jede andere Struktur – unterschiedliche Sequenzen mit der gleichen Struktur besitzen
extrem unterschiedliche “benachbarte Strukturen” • also Strukturen, die mittels der Mutation einer einzelnen Base
zugänglich sind

RNA vs. Protein
• umgekehrt verhalten sich Proteine eher wie ein “Pflaumenkuchen” – die Größe einer jeden Pflaume ist begrenzt und bildet
womöglich nur wenige Kontakte mit anderen Pflaumen aus
• RNA: die große Mehrheit der Sequenzen bildet gefaltete Strukturen aus
• Proteine: die große Mehrheit der Sequenzen entspricht ungefalteten Proteinen
• in dieser Hinsicht stellen Proteine und RNA womöglich zwei unterschiedliche Extreme dar

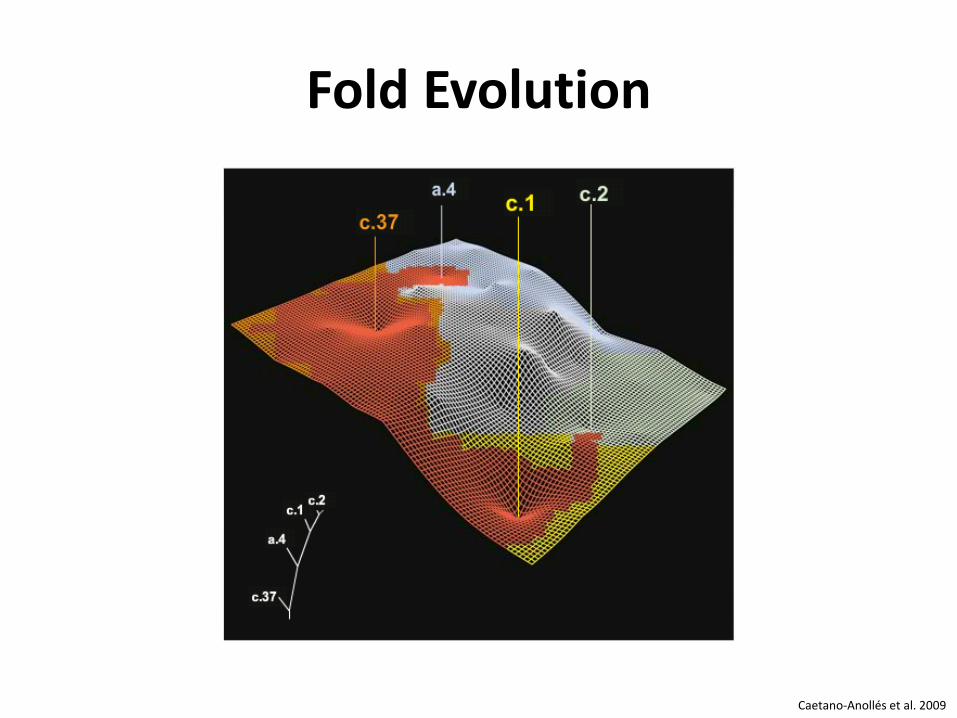
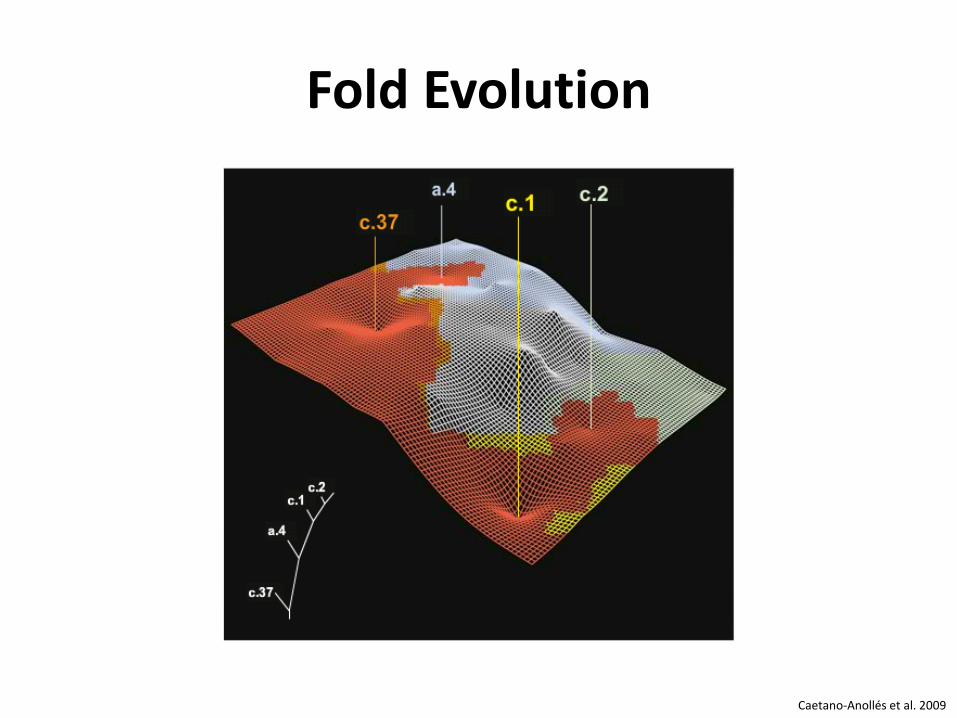
PROTEIN FOLD EVOLUTION Evolution der Proteinstruktur
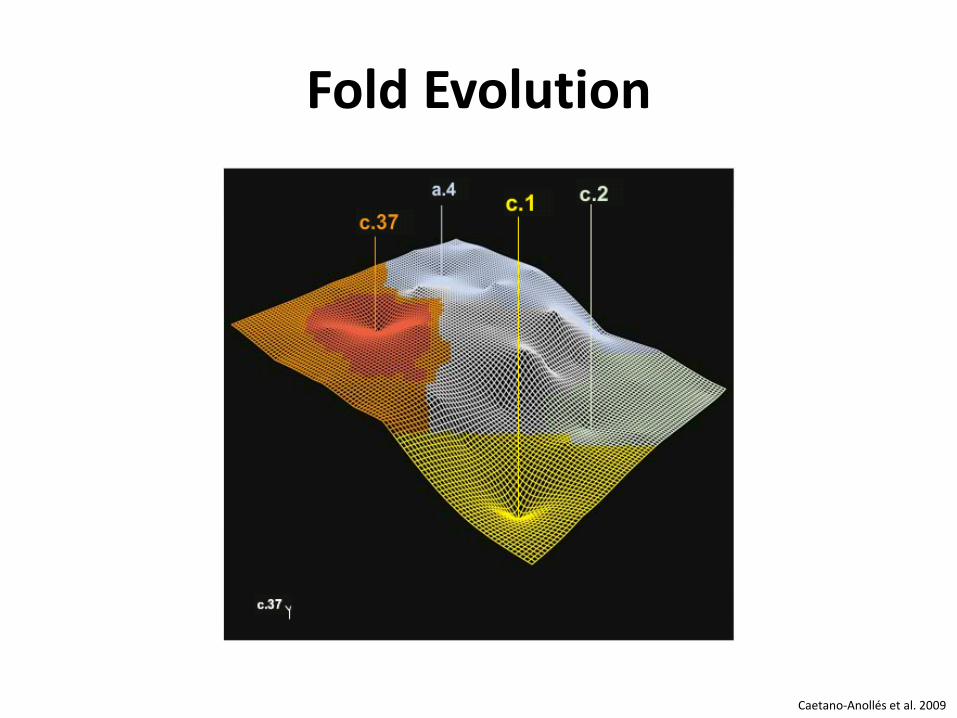
Fold Evolution
Caetano-Anollés et al. 2009
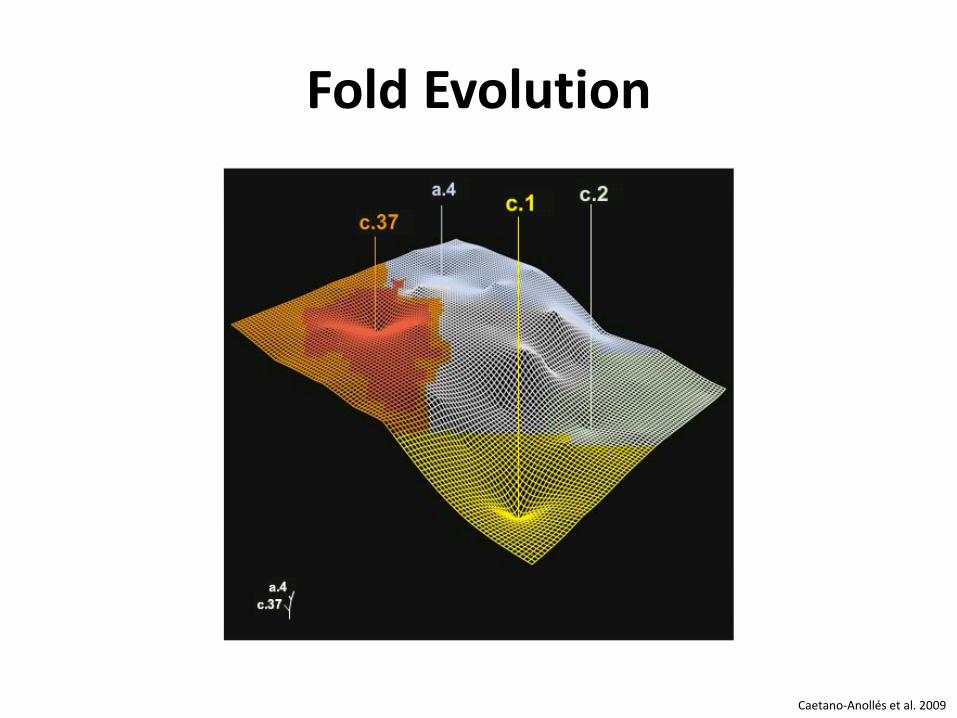
Fold Evolution
Caetano-Anollés et al. 2009
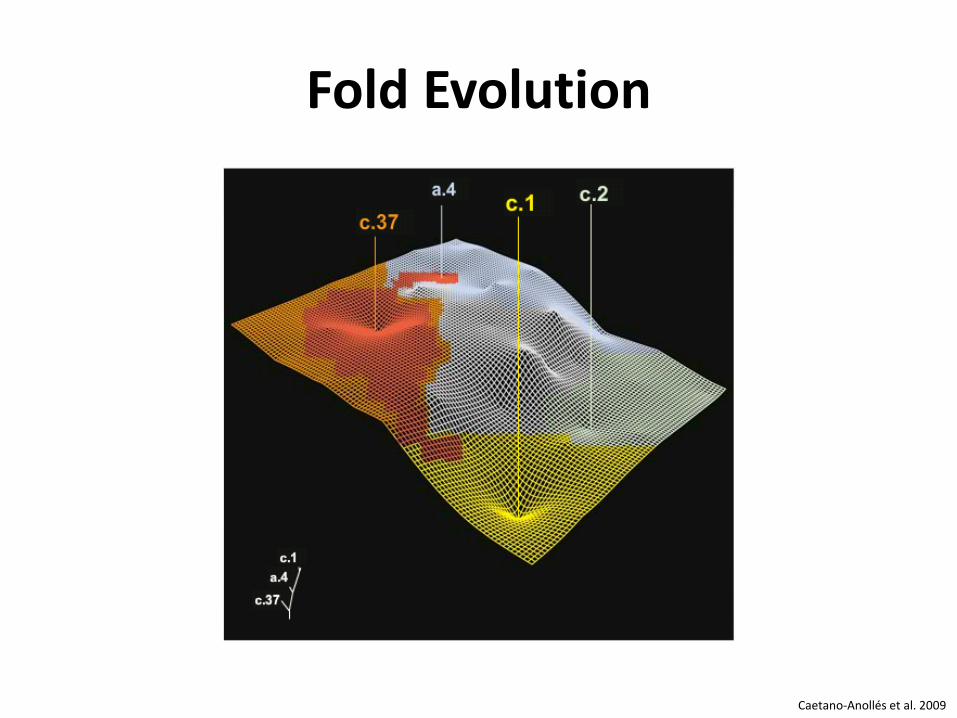
Fold Evolution
Caetano-Anollés et al. 2009
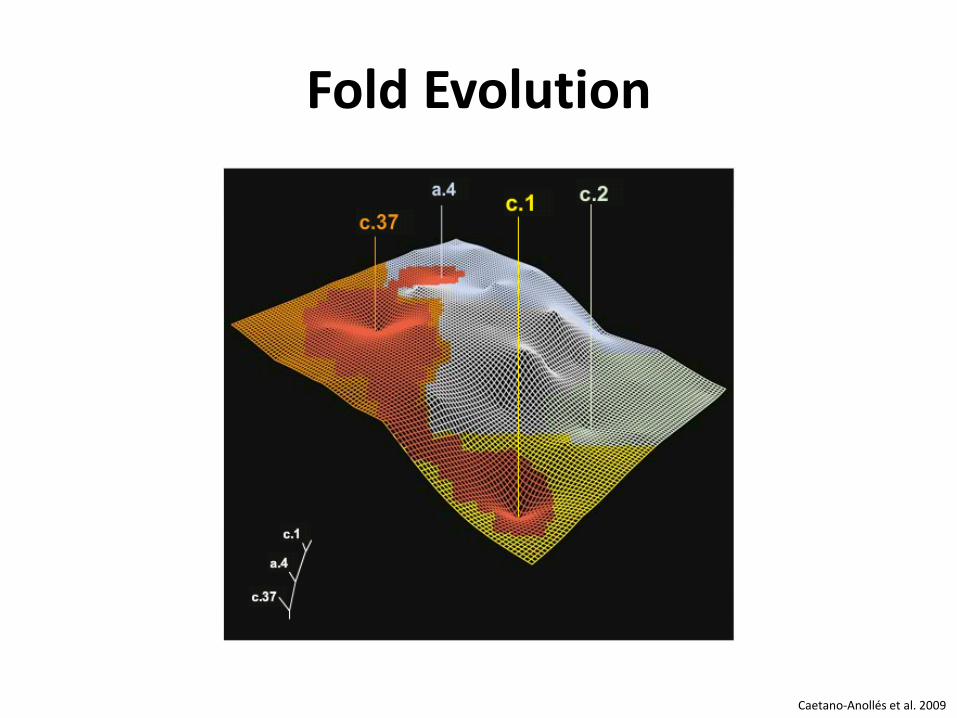
Fold Evolution
Caetano-Anollés et al. 2009
Fold Evolution
Caetano-Anollés et al. 2009
Fold Evolution
Caetano-Anollés et al. 2009
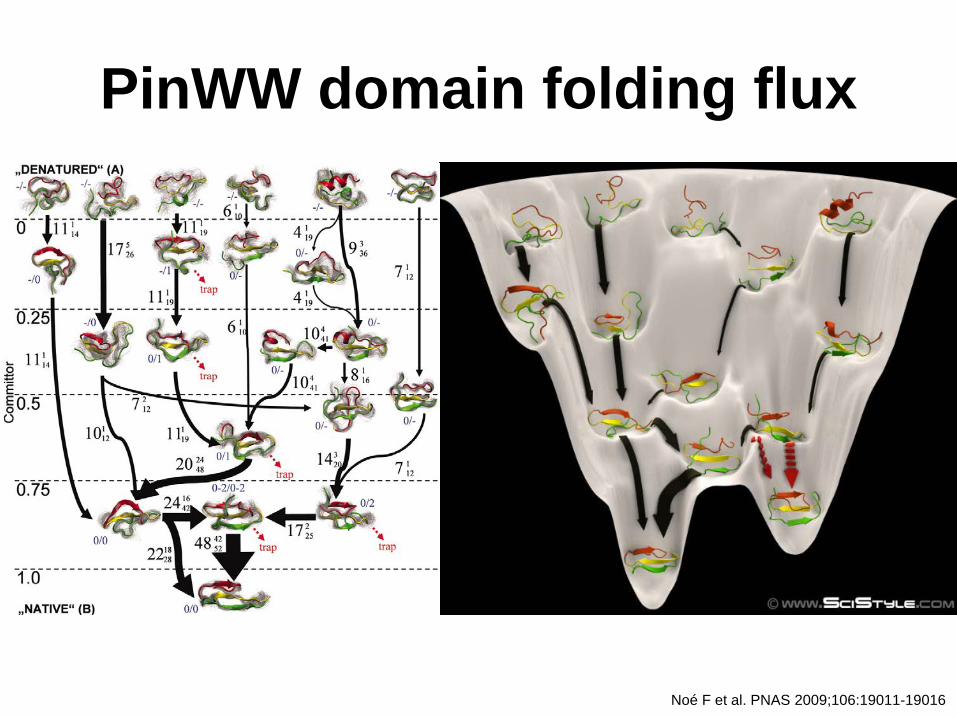
PinWW domain folding flux
Noé F et al. PNAS 2009;106:19011-19016

Conformational Diversity • Antikörper SPE7 • zwei unterschiedliche
Konformationen erlauben SPE7 die Bindung von völlig unterschiedlichen Substraten – Hapten – Protein
• unterschiedlich gestaltete Bindungsseite
• pre-steady-state kinetics zeigt zwei unterschiedliche Isomere
• jeder Ligand wählt einen anderen komplementären Isomer und verschiebt somit das Gleichgewicht in dessen Richtung
James and Tawfik 2003
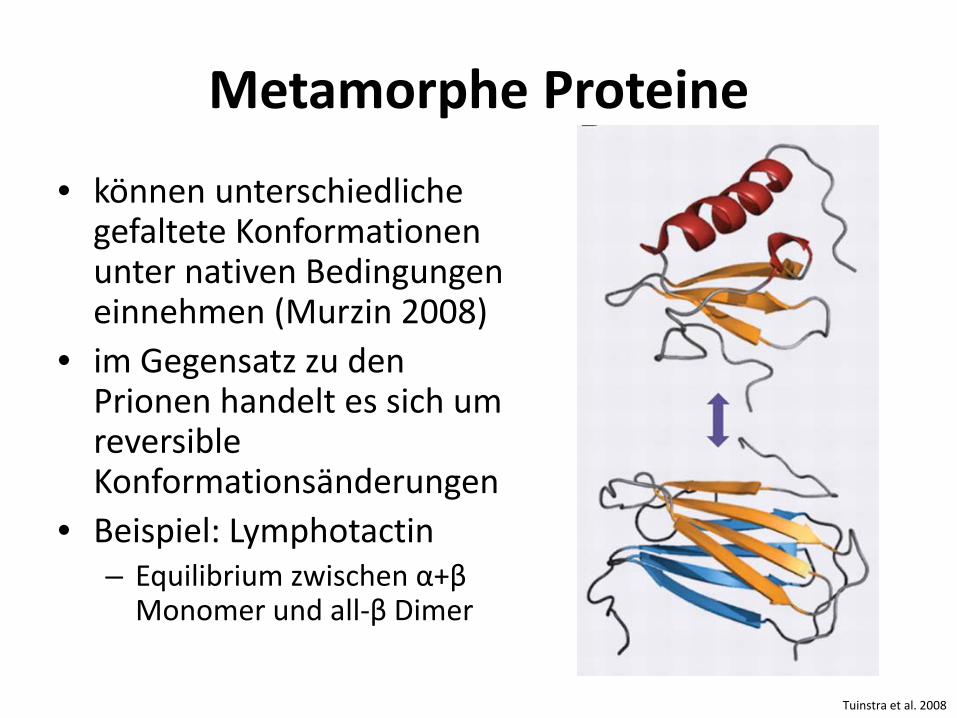
Metamorphe Proteine
• können unterschiedliche gefaltete Konformationen unter nativen Bedingungen einnehmen (Murzin 2008)
• im Gegensatz zu den Prionen handelt es sich um reversible Konformationsänderungen
• Beispiel: Lymphotactin – Equilibrium zwischen α+β
Monomer und all-β Dimer
Tuinstra et al. 2008

Proteinevolution auf Domänenebene
• Veränderungen in der Domänenarchitektur vorzugsweise an den Proteintermini (Bjorklund et al. 2005, Weiner et al. 2006)
• Termini sind normalerweise – geladen – flexibel – an der Proteinoberfläche
• Addition oder Deletion an Termini mit geringstem Einfluß auf Proteinstruktur
• Frequenz der Deletion oder Addition von Domänen ist nach einer Genduplikation doppelt so hoch – d.h. liegen zwei oder mehr Kopien eines Gens vor, so kann
mehr damit “experimentiert” werden
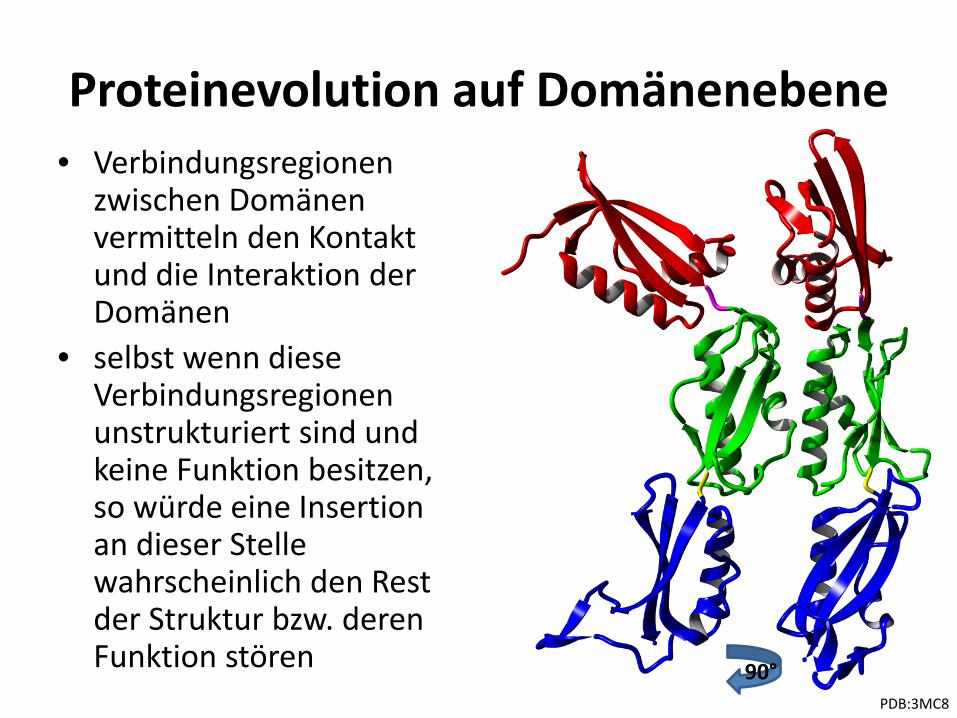
Proteinevolution auf Domänenebene
PDB:3MC8
90°
• Verbindungsregionen zwischen Domänen vermitteln den Kontakt und die Interaktion der Domänen
• selbst wenn diese Verbindungsregionen unstrukturiert sind und keine Funktion besitzen, so würde eine Insertion an dieser Stelle wahrscheinlich den Rest der Struktur bzw. deren Funktion stören
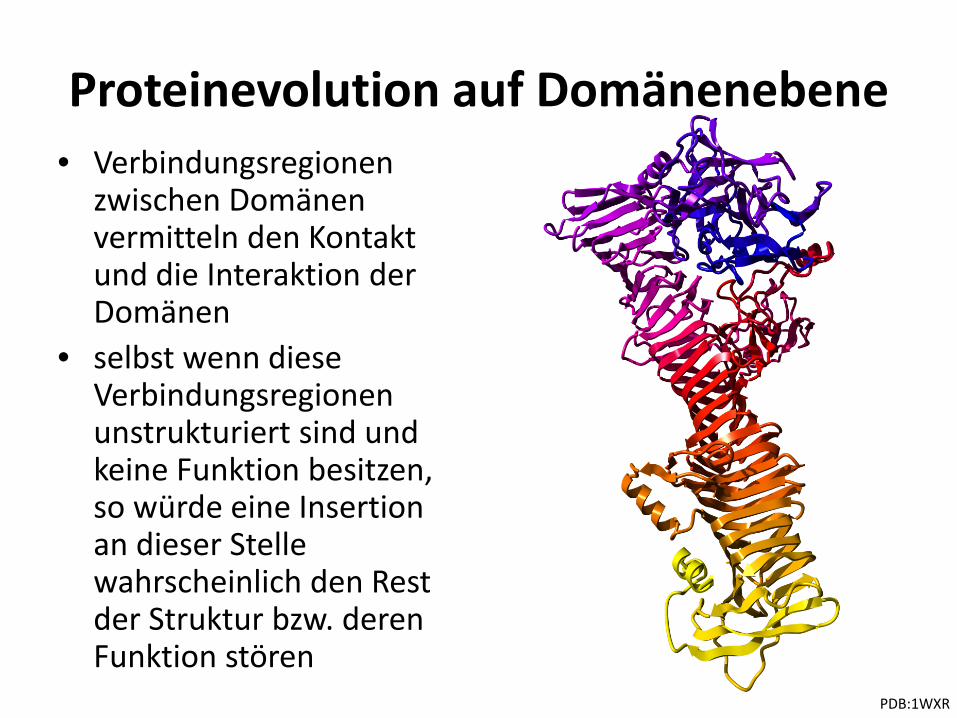
Proteinevolution auf Domänenebene • Verbindungsregionen
zwischen Domänen vermitteln den Kontakt und die Interaktion der Domänen
• selbst wenn diese Verbindungsregionen unstrukturiert sind und keine Funktion besitzen, so würde eine Insertion an dieser Stelle wahrscheinlich den Rest der Struktur bzw. deren Funktion stören
PDB:1WXR
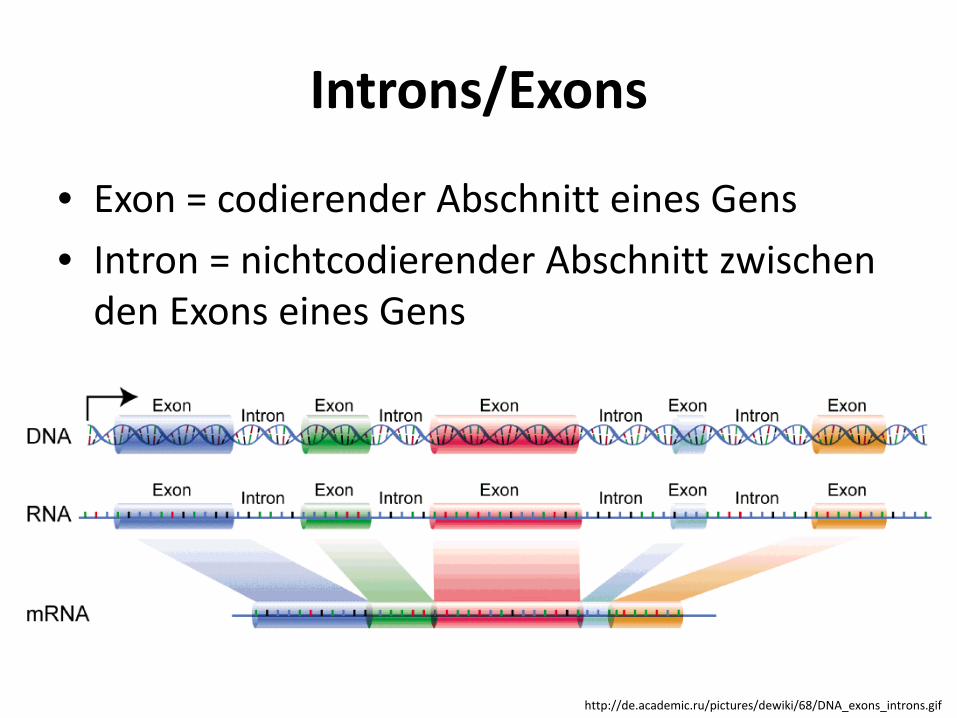
Introns/Exons
• Exon = codierender Abschnitt eines Gens • Intron = nichtcodierender Abschnitt zwischen
den Exons eines Gens
http://de.academic.ru/pictures/dewiki/68/DNA_exons_introns.gif
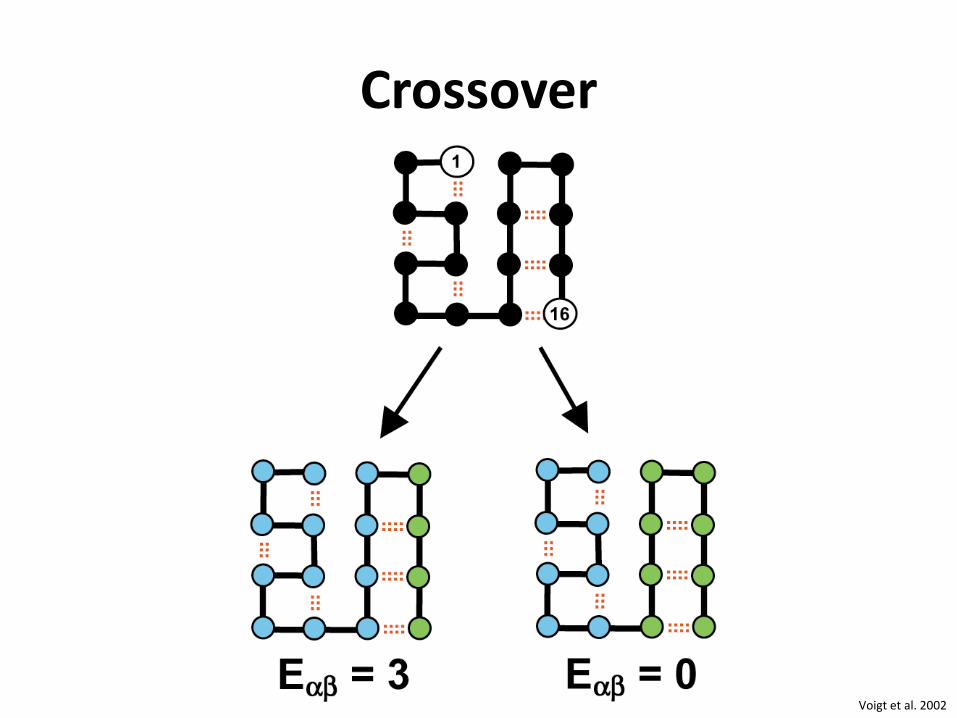
Crossover
Voigt et al. 2002
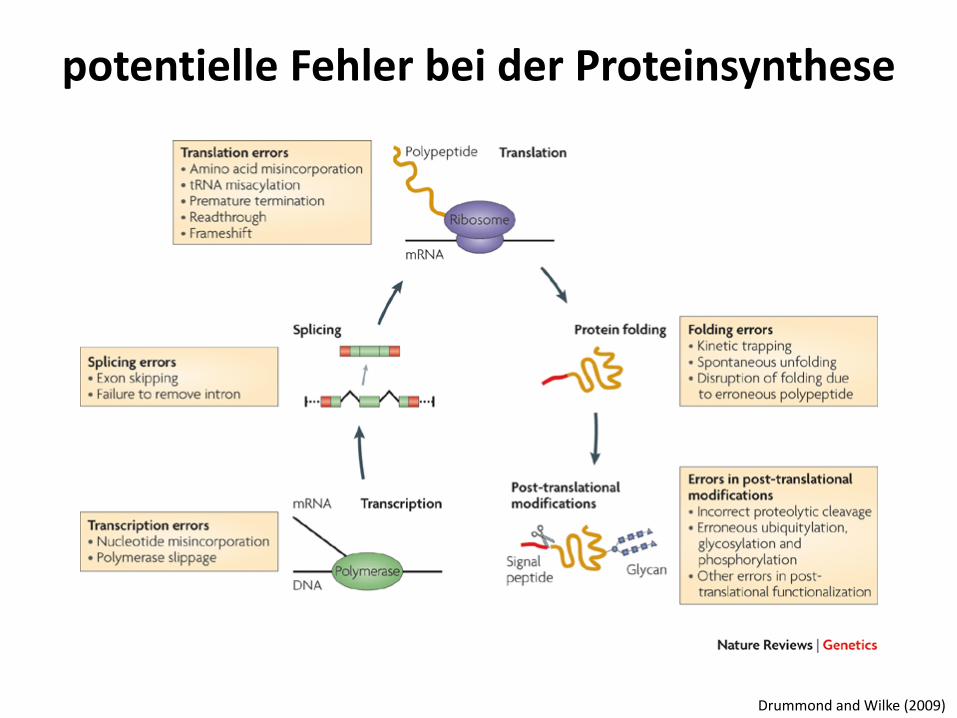
potentielle Fehler bei der Proteinsynthese
Drummond and Wilke (2009)

eine kleine Statistik • 103 bis 105 Proteinsequenzen pro Genom • 107 bis 108 Arten existieren auf der Erde • nur 1010 bis 1013 Varianten im Permutationsraum der
Aminosäuresequenz mit 10321-10469 möglichen Anordnungen im Sequenzraum abgedeckt
• Annahme: keine intraspezifische Sequenzvariation • (4-6)*1030 bakterielle Zellen auf unserem Planeten mit einem
Umsatz von 8x1029 Zellen pro Jahr und Mutationsraten von ~4*10-7 pro Zelle und Generation obere Grenze von ~2*1032 mutationsbedingten
Aminosäureaustauschen in bakteriellen Proteinen in ~4 Mrd Jahren der Evolution
• somit werden global gesehen neue Genotypen in Bruchteilen von Mikrosekunden erzeugt
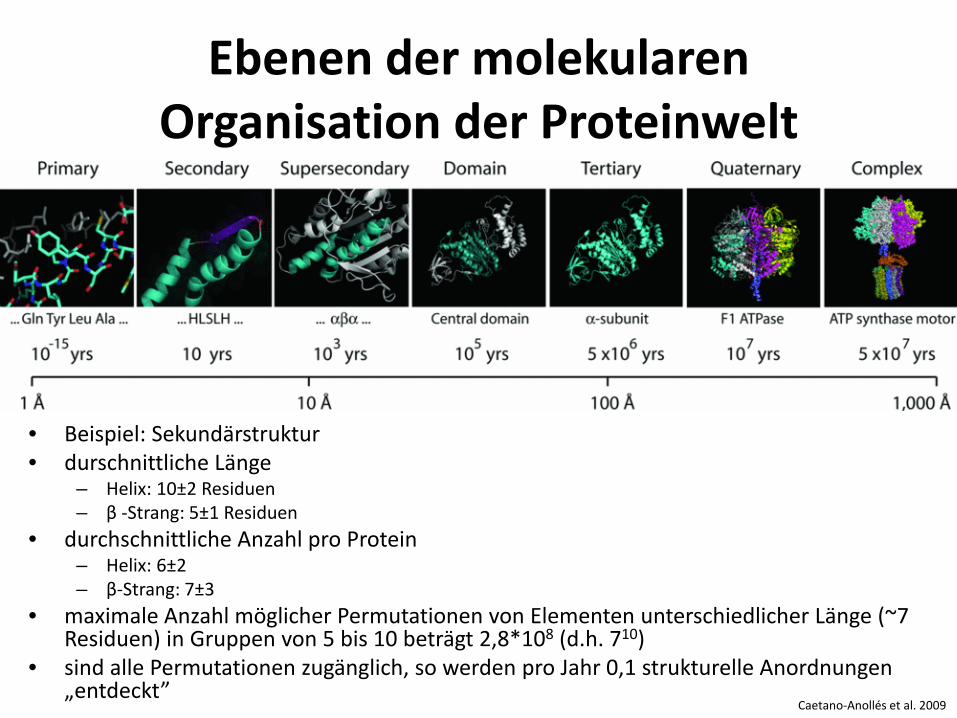
Caetano-Anollés et al. 2009
Ebenen der molekularen Organisation der Proteinwelt
• Beispiel: Sekundärstruktur • durschnittliche Länge
– Helix: 10±2 Residuen – β -Strang: 5±1 Residuen
• durchschnittliche Anzahl pro Protein – Helix: 6±2 – β-Strang: 7±3
• maximale Anzahl möglicher Permutationen von Elementen unterschiedlicher Länge (~7 Residuen) in Gruppen von 5 bis 10 beträgt 2,8*108 (d.h. 710)
• sind alle Permutationen zugänglich, so werden pro Jahr 0,1 strukturelle Anordnungen „entdeckt”
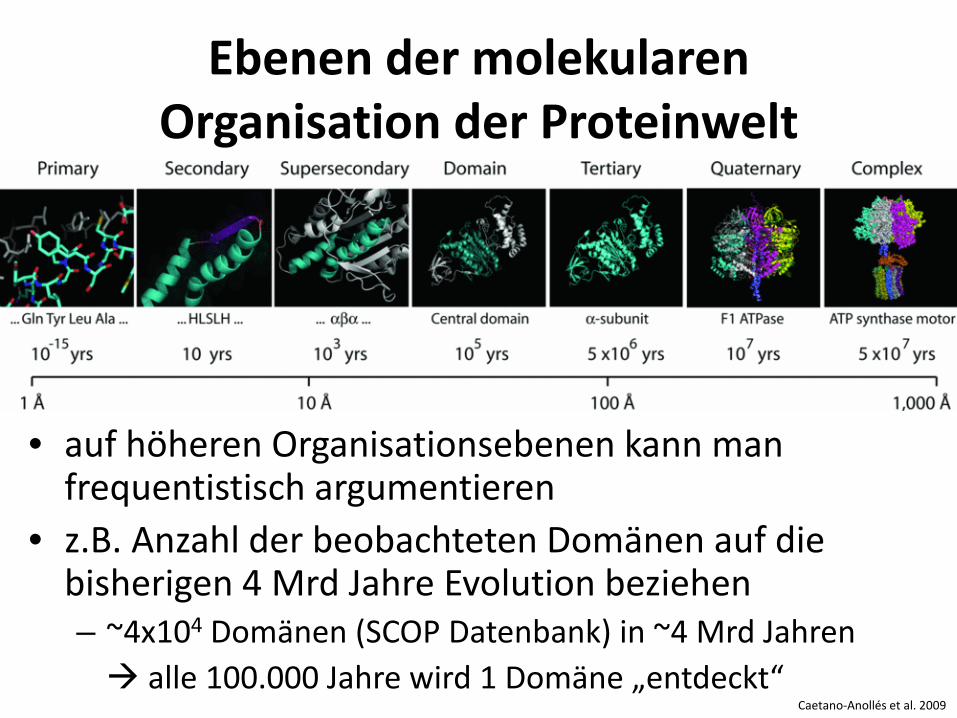
Caetano-Anollés et al. 2009
Ebenen der molekularen Organisation der Proteinwelt
• auf höheren Organisationsebenen kann man frequentistisch argumentieren
• z.B. Anzahl der beobachteten Domänen auf die bisherigen 4 Mrd Jahre Evolution beziehen – ~4x104 Domänen (SCOP Datenbank) in ~4 Mrd Jahren alle 100.000 Jahre wird 1 Domäne „entdeckt“

EVOLUTIONARY ANALYSIS Evolution der Proteinstruktur
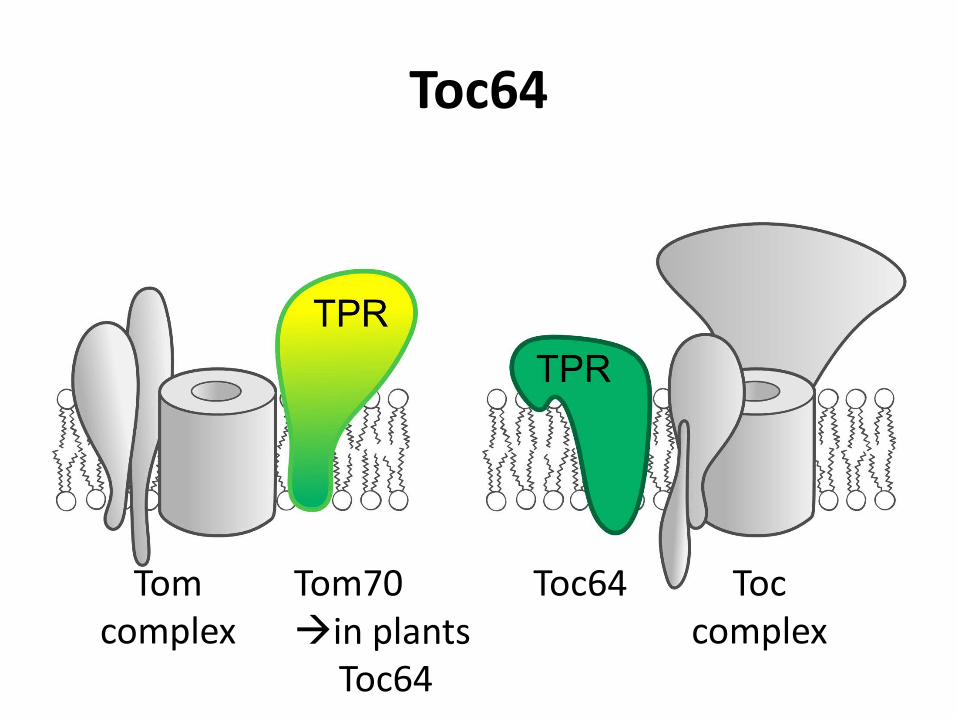
Toc64
Toc complex
Toc64 Tom complex
Tom70 in plants Toc64
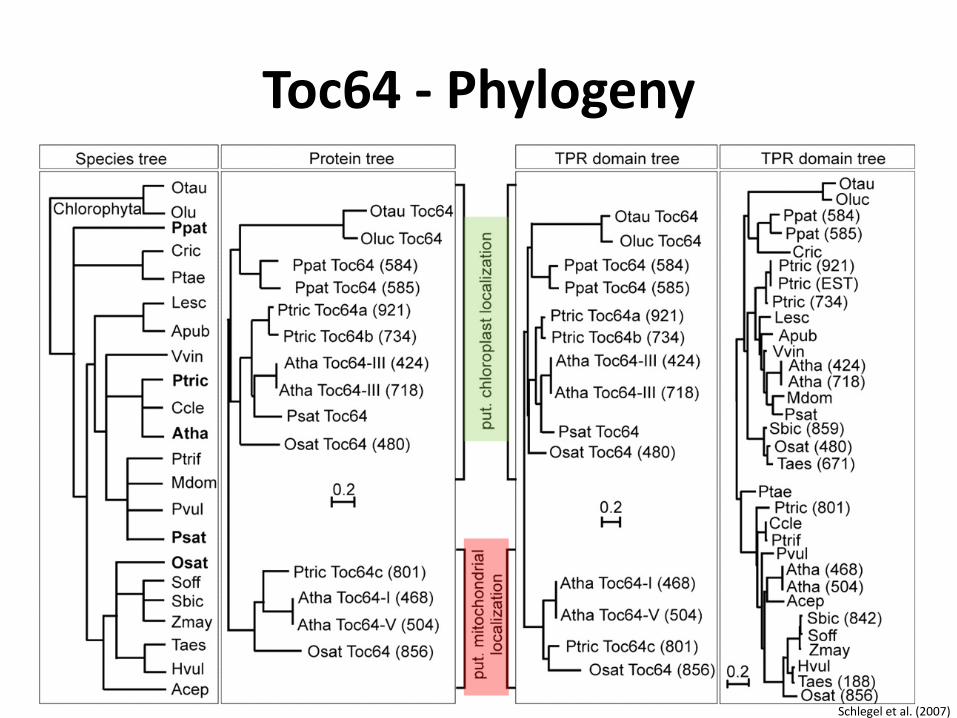
Toc64 - Phylogeny
Schlegel et al. (2007)
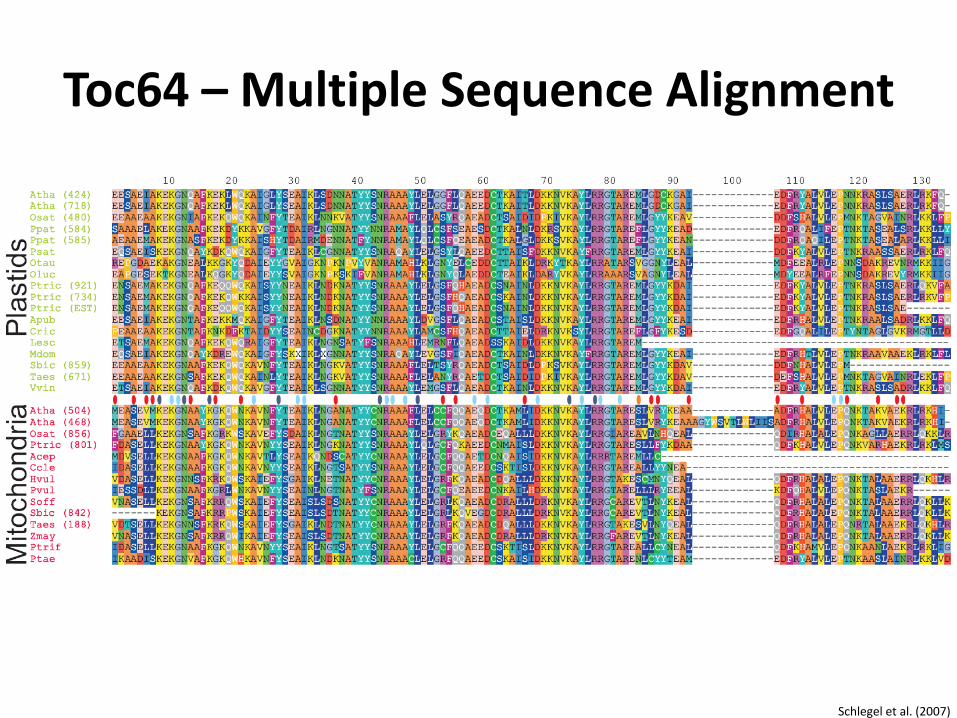
Toc64 – Multiple Sequence Alignment
Schlegel et al. (2007)
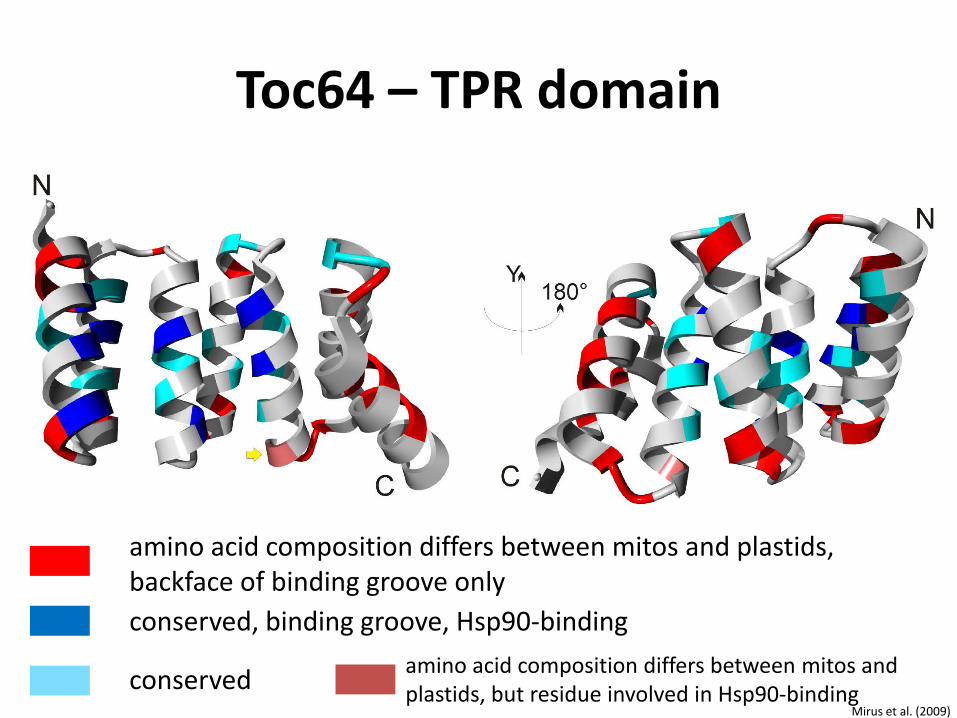
Toc64 – TPR domain
conserved, binding groove, Hsp90-binding
conserved
amino acid composition differs between mitos and plastids, backface of binding groove only
amino acid composition differs between mitos and plastids, but residue involved in Hsp90-binding
Mirus et al. (2009)

Vielen Dank für Eure Aufmerksamkeit!


















