
Expression und Funktion von SNARE-Proteinen in humanen ... fileAus der Abteilung für...
90
Aus der Abteilung für Gastroenterologie, Hepatologie und Endokrinologie des Zentrums Innere Medizin der Medizinischen Hochschule Hannover Expression und Funktion von SNARE-Proteinen in humanen intestinalen Mastzellen Dissertation zur Erlangung des Doktorgrades der Medizin an der Medizinischen Hochschule Hannover vorgelegt von Seza Bolat aus Celle Hannover, 2006
Transcript of Expression und Funktion von SNARE-Proteinen in humanen ... fileAus der Abteilung für...

Aus der Abteilung für Gastroenterologie, Hepatologie und Endokrinologie
des Zentrums Innere Medizin der
Medizinischen Hochschule Hannover
Expression und Funktion von SNARE-Proteinen in
humanen intestinalen Mastzellen
Dissertation
zur Erlangung des Doktorgrades der Medizin
an der Medizinischen Hochschule Hannover
vorgelegt von
Seza Bolat
aus Celle
Hannover, 2006

2
Angenommen vom Senat der Medizinischen Hochschule Hannover am 25.02.2008 Gedruckt mit Genehmigung der Medizinischen Hochschule Hannover
Präsident: Prof. Dr. Dieter Bitter-Suermann
Betreuer: Prof. Dr. Stephan C. Bischoff
Referent/Referentin: Prof. Dr. med. Reinhard Pabst
Koreferentin: Prof.`in Dr. med. Bettina Wedi
Tag der mündlichen Prüfung: 25.02.2008
Promotionsausschussmitglieder: Prof. Dr. Alexander Kapp
Prof. Dr. Burkhard Wippermann
Prof. Dr. Stefan Kubicka

3
Inhaltverzeichnis
1. Einleitung................................................................................................................ 8
1.1 Allergie .............................................................................................................. 8
1.2 Mastzellen......................................................................................................... 8
1.2.1 Morphologie, Entwicklung und Heterogenität............................................... 8
1.2.3 Funktion..................................................................................................... 10
1.2.4 Mediatoren................................................................................................. 11
1.2.5 Aktivierung................................................................................................. 13
1.2.6 Hemmung .................................................................................................. 14
1.3 Exozytose und SNARE-Proteine..................................................................... 15
1.4 Botulinum Neurotoxin...................................................................................... 18
1.5 Adenoviren...................................................................................................... 20
1.6 Fragestellung .................................................................................................. 21
2. Materialien und Methoden .................................................................................... 22
2.1 Materialien ...................................................................................................... 22
2.1.1 Chemikalien und Reagenzien .................................................................... 22
2.1.2 Antikörper .................................................................................................. 23
2.1.3 Kommerzielle Kits ...................................................................................... 24
2.1.4 Enzyme...................................................................................................... 24
2.1.5 Puffer, Lösungen und Medien.................................................................... 24
2.1.6 Verwendete Zelllinien ................................................................................ 26
2.1.7 Klonierungsvektoren .................................................................................. 27
2.1.8 Rekombinante Adenoviren......................................................................... 28
2.2. Molekularbiologische Methoden..................................................................... 28
2.2.1 Konzentrationsbestimmung von DNA ........................................................ 28
2.2.2 Transformation........................................................................................... 29
2.2.3 Mini-Plasmidpräparation ............................................................................ 30
2.2.4 Maxi-Plasmidpräparation ........................................................................... 30
2.2.5 DNA-Spaltung mit Restriktionsendonukleasen .......................................... 31
2.2.6 Phenol-Chloroform-Extraktion.................................................................... 31
2.2.7 RNA-Isolation und Reverse Transkriptase-Polymerase-Kettenreaktion .... 32
2.2.8 Agarosegelelektrophorese......................................................................... 34

4
2.3 Proteinbiochemische Methoden...................................................................... 35
2.3.1 Zellextraktion ............................................................................................. 35
2.3.2 Western Blot .............................................................................................. 35
2.4 Virale Methoden.............................................................................................. 36
2.4.1 Zellkultur .................................................................................................... 36
2.4.2 Transfektion............................................................................................... 36
2.4.3 Virusisolierung ........................................................................................... 37
2.4.4 Vermehrung und Präparation von Adenoviren........................................... 37
2.4.5 Titerbestimmung ........................................................................................ 38
2.4.6 Transduktion von Primärzellen .................................................................. 39
2.5 Zellbiologische Methoden ............................................................................... 39
2.5.1 Isolation von Mastzellen aus menschlichem Darmgewebe........................ 39
2.5.2 Mastzellanreicherung durch MACS (Magnetic beads-activated cell sorting)
.................................................................................................................. 40
2.5.3 Kultur menschlicher Darm-Mastzellen ....................................................... 42
2.5.4 Zellzählung und Differenzierung ................................................................ 42
2.5.5 Mastzellaktivierung/ Mediatormessung...................................................... 42
2.5.6 Permeabilisierung mit Streptolysin O (SLO) .............................................. 43
2.5.7 Konfokale Immunfluoreszenz-Mikroskopie ................................................ 44
2.6 Statistik ........................................................................................................... 45
3. Ergebnisse............................................................................................................ 46
3.1 Nachweis von SNARE-Proteinen in menschlichen Darm- Mastzellen ............ 46
3.1.1 SNARE-mRNA Nachweis mittels RT-PCR ................................................ 46
3.1.2 SNARE-Protein Nachweis mittels Western Blot......................................... 48
3.2 Untersuchung des Einflusses von Botulinum Neurotoxin auf die
Mastzelldegranulation..................................................................................... 49
3.2.1 Herstellung adenoviraler Konstrukte.......................................................... 49
3.2.2 Virusgenerierung ....................................................................................... 52
3.2.3 Transduktion der Mastzellen mit rekombinierten Adenoviren .................... 55
3.2.4 Untersuchung der Exozytose adenoviral transduzierter Mastzellen .......... 57
3.3 Funktionelle Bedeutung der gefundenen SNARE-Proteine............................. 59
3.3.1 Intrazelluläre Lokalisation der SNARE-Proteine ........................................ 59
3.3.2 VAMP-7 transloziert während der Exozytose............................................. 60

5
3.3.3 Die Inhibition des SNARE-Proteins VAMP-7 bewirkt eine
Degranulationshemmung .......................................................................... 62
4. Diskussion ............................................................................................................ 64
4.1 Expression von SNARE-Proteinen in humanen intestinalen Mastzellen ......... 64
4.2 Hemmung der Mastzelldegranulation durch Botulinum Neurotoxin................. 65
4.3 Bedeutung von VAMP-7 für die Mastzelldegranulation ................................... 69
4.4 Ausblick........................................................................................................... 72
5. Zusammenfassung ............................................................................................... 73
6. Literaturverzeichnis............................................................................................... 75
7. Danksagung ......................................................................................................... 87
8. Lebenslauf ............................................................................................................ 89
9. Erklärung nach §2 Abs. 2 Nr. 5 und 6 PromO ...................................................... 90

6
Abkürzungsverzeichnis
ACC Acetylcystein
BoNT Botulinumneurotoxin CAR Coxsackie Adenovirus Receptor
CD Cluster of Differentiation
cDNA komplementäre Desoxyribonukleinsäure
cys-LT cysteinyl-Leukotriene
DNA Desoxyribonukleinsäure
EDTA Ethylendiamintetraacetat
Fc konstante Region der Immunglobuline (Fragment crystallizable)
FCS fetales Kälberserum
FcεR Immunglobulin E Rezeptor
GAPDH Glycerinaldehyd-3-Phosphatdehydrogenase GFP Green Fluorescent Protein
HMC-1 Humane Mastzellinie-1
Ig Immunglobuline
IP3 Inositol Trisphosphat LB Luria Broth
MACS magnetische Zellseparation (magnetic-activated cell sorting)
mAk monoklonaler Antikörper
MC Mastzelle/n MCS Multiple Cloning Site
MCT Tryptase positive und Chymase negative Mastzelle
MCTC Tryptase und Chymase positive Mastzelle MHC Major Histocompatibility Complex
MOI Multiplicity of Infection (Anzahl infektiöser Partikel pro Zelle)
mRNA Boten-Ribonukleinsäure (messenger RNA)
NSF N-ethylmaleimide-sensitive factor
PBS Phosphat-gepufferte Kochsalzlösung (phosphate buffered saline)
PCR Polymerase-Kettenreaktion (polymerase chain reaction) PDT Protein Transduction Domains
PFU Plaque Forming Unit
PGD2 Prostaglandin D2 PMA Phorbol-Myristat-Acetat
RBL-2H3 rat basophilic leukemia cells
rh rekombinant, human
RIA Radioimmunoassay rpm Rotationen pro Minute

7
RT-PCR Reverse Transkriptase Polymerase-Kettenreaktion
SCF Stem Cell Factor (c-kit Ligand, Stammzellfaktor)
SDS Sodiumdodecylsulfat
SLO Streptolysin O
SNAP-25 Synaptosome Associated Protein with molecular weight of 25 kd
SNARE Soluble N-ethylmaleimide-Sensitive Factor Attachment Protein Receptor
TNF-α Tumornekrosefaktor-α (tumor nekrosis factor-α)
VAMP Vesicle-associated membrane protein
α-, β-, γ-SNAP α-, β-, γ-Soluble NSF Attachment Protein

Einleitung
8
1. Einleitung
1.1 Allergie
Der Begriff Allergie bezeichnet eine erworbene Hypersensibilität des Körpers.
Coombs und Gell unterteilten die Überempfindlichkeitsreaktionen des Immunsystems
in vier Typen, wobei die Immunglobulin E–vermittelte Sofortreaktion als Typ I
bezeichnet wurde. Durch Kontakt mit im Grunde harmlosen Antigenen, z.B.
Blütenpollen oder Hausstaub, kommt es zu einer überschießenden Immunantwort.
Effektorzellen dieser Reaktion sind zuvor durch Immunglobulin E sensibilisierte
Mastzellen, die verschiedene Mediatoren, unter anderem Histamin, freisetzen,
wodurch es zu Gewebeentzündungen und Organdysfunktionen kommt [1-3]. Die
Prävalenz der durch IgE und Mastzellmediatoren verursachten Erkrankungen, wie
Heuschnupfen und allergisches Asthma, liegt bei 20% [4;5].
1.2 Mastzellen
1.2.1 Morphologie, Entwicklung und Heterogenität
Ihren Namen verdankt die Mastzelle Paul Ehrlich. Er beschrieb 1877 erstmals die
Mastzelle, die wegen ihrer großen, reichhaltigen Granula wie „gemästet“ aussah [6].
Lichtmikroskopisch betrachtet besitzt die Mastzelle eine rundliche, spindelige,
zuweilen dendritisch verzweigte Form [7]. Aufgrund ihres runden Zellkerns kann man
die Mastzelle von den morphologisch und funktionell sehr ähnlichen basophilen
Granulozyten unterscheiden, die einen segmentierten Kern aufweisen.
Charakteristisch für beide Zellarten ist das Phänomen der Metachromasie.
Verwendet man einen basischen Farbstoff, so nehmen die das Zytoplasma dicht
ausfüllenden Granula eine Farbe an, die sich von der Kernfärbung unterscheidet.
Aus dem Knochenmark stammende myeloische Mastzell-Vorläuferzellen gelangen
über die Blutbahn durch Interaktion mit verschiedenen Adhäsionmolekülen
(z.B. VLA-4, VCAM-1) in die Zielorgane [8;9]. Dort differenzieren sie mit Hilfe des auf

Einleitung
9
der Oberfläche exprimierten Stammzellfaktor-(SCF-)Rezeptors c-kit . Im Gegensatz
zu anderen Zellen bleibt c-kit auf Mastzellen auch nach der Differenzierung
lebenslang erhalten [10]. Neben der Reifung ist der Stammzellfaktor (SCF) auch bei
anderen, die Mastzellfunktion modulierenden Effekten beteiligt. Aktivierung zur
Mediator-Freisetzung, Induktion von Chemotaxis und Regulation der Adhäsion an
extrazelluläre Matrixproteine werden durch SCF reguliert [8;11]. In hohen
Konzentrationen induziert SCF die Mastzell-Proliferation und wirkt in niedrigen
Konzentrationen überlebensfördernd. So lassen sich reife, aus Lunge und Darm
isolierte, humane Mastzellen nur in Anwesenheit von SCF kultivieren [12-16].
Nach Ausreifung finden sich Mastzellen bevorzugt in Geweben mit
Barrierefunktion gegen die Außenwelt, wie Lunge, Haut oder Gastrointestinaltrakt.
Aber auch in vielen anderen Organen, z.B. Herz, Niere, Bauchspeicheldrüse und
Gebärmutter [7;10;17], sind Mastzellen eng assoziiert mit Blutgefässen, Nerven und
Epithelien [7;18-20].
Viele Erkenntnisse über Mastzellen beruhen auf Untersuchungen im murinen
System. Bei vergleichenden Untersuchungen zeigten sich jedoch erhebliche
Unterschiede zwischen Mastzellen aus Menschen und Mäusen oder Ratten. Daher
lassen sich an Nagern gewonnene Erkenntnisse nicht ohne Weiteres auf das
menschliche System übertragen. So konnte der Stammzellfaktor (SCF) als
essentieller Wachstumsfaktor für Mastzellen aller Spezies nachgewiesen werden,
während das Zytokin Interleukin-3 zwar im Nagermodell, nicht aber im humanen
System, eine wichtige Rolle beim Wachstum zu spielen scheint [21;22]. Jedoch
konnten nicht nur Unterschiede zwischen dem murinen und humanen System
aufgezeigt werden. Auch Mastzellen aus Menschen bilden keine homologen
Gruppen, sondern differenzieren abhängig vom Mikromilieu der jeweiligen Gewebe.
Vergleichbar mit dem Nagersystem, haben humane Mastzellen unterschiedliche
Ansprüche an die Anwesenheit von Wachstumsfaktoren, wie Zytokine. Humane
Mastzellen weisen Unterschiede in Fixierungseigenschaft, granulärer Ultrastruktur
und Stimulierbarkeit durch Agonisten (Substanz P, Compound 48/80) auf
[7;17;18;23]. Nach ihrem Gehalt an neutralen Proteasen lassen sich zwei Subtypen
abgrenzen. MCTC kommen in Haut, Tonsillen, Tela Submukosa des Darms vor und
enthalten die Proteasen Tryptase und Chymase. MCT enthalten nur Tryptase und
finden sich eher in der Schleimhaut von Lunge und Gastrointestinaltrakt [17;23]. Das

Einleitung
10
organspezifische Gewebemilieu scheint, unabhängig von den Subtypen, die
Mastzell-Funktion zu beeinflussen. Mastzellen der Haut - es kommen fast nur MCTC
vor - sind durch Substanz P und Compound 48/80 aktivierbar. Obwohl die Mastzell-
Population des Darms sowohl MCTC als auch MCT enthält, ist die Stimulation durch
Agonisten nicht möglich. Bei Subtypenspezifität müsste zumindest ein Teil der
Mastzellen reagieren. Aus verschiedenen Geweben isolierte Mastzellen
unterscheiden sich auch in ihrer Fähigkeit, nach IgE-abhängiger Aktivierung
Lipidmediatoren wie cysteinyl-Leukotrien (cys-LT) oder Prostaglandin D2 (PGD2) zu
synthetisieren [24;25]. Diese Heterogenität unterschiedlicher Mastzell-Populationen
lässt auf eine gewisse Plastizität an Reaktionsmustern schließen.
1.2.3 Funktion
Aufgrund ihrer weiten Verbreitung in Geweben und ihrer Fähigkeit, eine große Anzahl
verschiedener Mediatoren freizusetzen, sind Mastzellen an unterschiedlichen
physiologischen und pathophysiologischen Ereignissen beteiligt. So spielen sie eine
Rolle bei der Regulation neurophysiologischer Prozesse, wie Sekretion und Motilität,
und fördern Wundheilung und Angiogenese. Außerdem unterstützen sie die Abwehr
gegen infektiöse Mikroorganismen (Bakterien, Parasiten) [7;26-27]. Weiterhin sind
Mastzellen als gewebeständige Immunzellen in der Lage, mittels Histocompatibility II
(MHC II) Molekülen spezifischen Lymphozyten (T-Zellen) Antigene zu präsentieren
[28-30] und die Produktion von IgE-Antikörpern in B-Zellen bzw. Plasmazellen zu
fördern [31]. Wenn es zu einer pathophysiologischen Fehlregulation kommt, sind
Mastzellen auch an fibrotischen Prozessen in Geweben, chronischen Entzündungen
und Tumorentwicklung beteiligt [27;32-34]. Die IgE-abhängige Aktivierung von
Mastzellen ist ein zentrales Ereignis der Überempfindlichkeitsreaktion vom Soforttyp
(Allergie Typ I nach Coombs und Gell). Dazu gehören atopische Erkrankungen, wie
allergische Rhinitis, allergisches Asthma, atopisches Ekzeme, allergische
Gastroenteritis und anaphylaktische Erkrankungen, z.B. Urtikaria, Angioödem,
generalisierte Anaphylaxie.

Einleitung
11
1.2.4 Mediatoren
Generell unterscheidet man zwei Gruppen von Mediatoren. Histamin, Proteasen und
Tumornekrosefaktor-α (TNFα) können nach Stimulation schnell freigesetzt werden,
da sie in den Granula gespeichert vorliegen. Im Gegensatz dazu können Lipid-
Glykoprotein-Mediatoren (cys-LT, PGD2) und diverse Zytokine erst nach
Neusynthese abgegeben werden [18;26;35;36].
Einen wesentlichen Anteil an den Allergie-Symptomen wie Juckreiz, Ödeme und
Rötung hat der Mastzell-Mediator Histamin, der Schleimsekretion, Kontraktion glatter
Muskulatur, Gefäßerweiterung und –permeabilisierung vermittelt. Gleichzeitig
scheinen Histamin und Lipidmediatoren eine Rolle bei der Regulation
neurophysiologischer Prozesse, wie Sekretion und Motilität, zu spielen [18;37].
Histamin entsteht im Golgi Apparat nach Decarboxylierung der Aminosäure Histidin
und wird, gebunden an Proteoglykane und Proteine, gespeichert. Eine humane
Mastzelle enthält ungefähr 1 bis 8 pg Histamin.
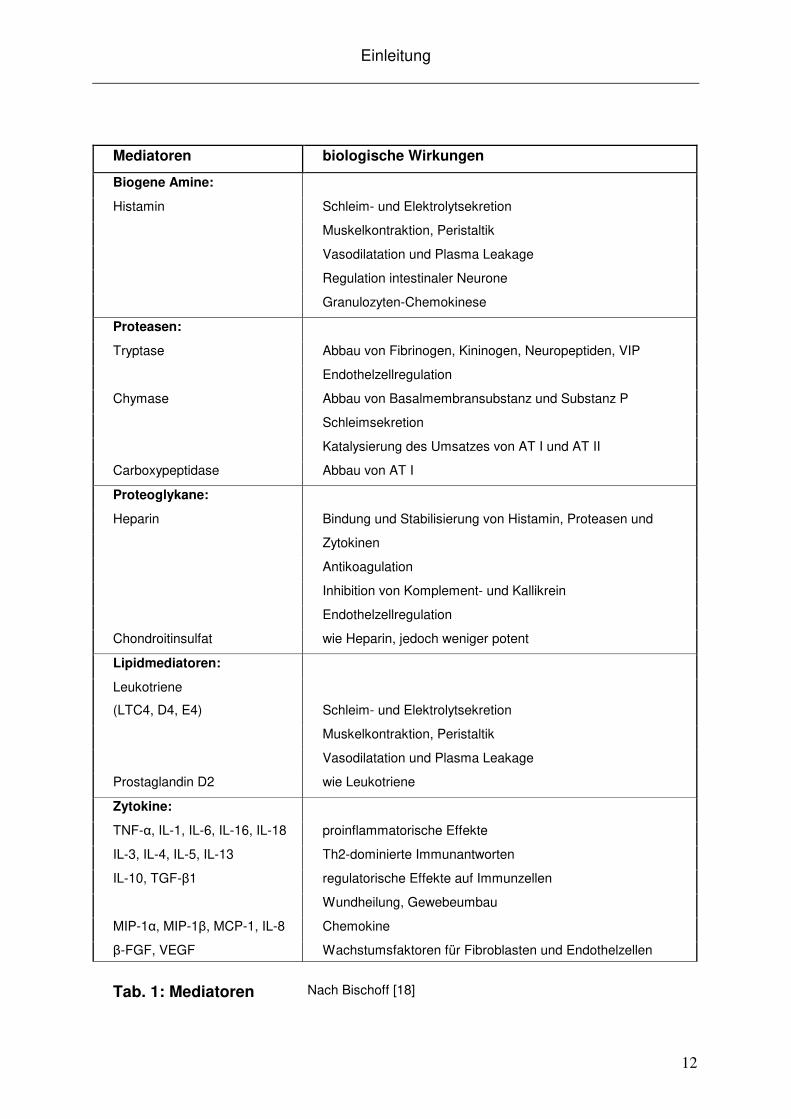
Die biologischen Effekte diverser Mastzell-Mediatoren sind zusammengefasst in
Tabelle 1 dargestellt.
Einleitung
12
Mediatoren biologische Wirkungen
Biogene Amine:
Histamin Schleim- und Elektrolytsekretion
Muskelkontraktion, Peristaltik
Vasodilatation und Plasma Leakage
Regulation intestinaler Neurone
Granulozyten-Chemokinese
Proteasen:
Tryptase Abbau von Fibrinogen, Kininogen, Neuropeptiden, VIP
Endothelzellregulation
Chymase Abbau von Basalmembransubstanz und Substanz P
Schleimsekretion
Katalysierung des Umsatzes von AT I und AT II
Carboxypeptidase Abbau von AT I
Proteoglykane:
Heparin Bindung und Stabilisierung von Histamin, Proteasen und
Zytokinen
Antikoagulation
Inhibition von Komplement- und Kallikrein
Endothelzellregulation
Chondroitinsulfat wie Heparin, jedoch weniger potent
Lipidmediatoren:
Leukotriene
(LTC4, D4, E4) Schleim- und Elektrolytsekretion
Muskelkontraktion, Peristaltik
Vasodilatation und Plasma Leakage
Prostaglandin D2 wie Leukotriene
Zytokine:
TNF-α, IL-1, IL-6, IL-16, IL-18 proinflammatorische Effekte
IL-3, IL-4, IL-5, IL-13 Th2-dominierte Immunantworten
IL-10, TGF-β1 regulatorische Effekte auf Immunzellen
Wundheilung, Gewebeumbau
MIP-1α, MIP-1β, MCP-1, IL-8 Chemokine
β-FGF, VEGF Wachstumsfaktoren für Fibroblasten und Endothelzellen
Tab. 1: Mediatoren Nach Bischoff [18]

Einleitung
13
1.2.5 Aktivierung
Zwei Fcε-Rezeptoren für IgE sind bekannt, die von verschiedenen Zelltypen
präsentiert werden. Während der niedrigaffine Rezeptor Fcε-RII von zahlreichen
Entzündungszellen, wie Makrophagen, eosinophilen Granulozyten, Thrombozyten, B-
und T-Lymphozyten exprimiert wird, findet sich der hochaffine FcεRI vor allem auf
Mastzellen und basophilen Granulozyten [38;39]. FcεRI besteht aus vier
transmembranen Polypeptidketten – jeweils eine α- und β-Kette, sowie zwei γ-
Ketten. Während die α-Kette, mit ihrem aus der Zellmembran herausragenden Teil,
für die IgE-Bindung zuständig ist, vermitteln β- und γ-Kette die Signaltransduktion
über ITAMs (immunreceptor tyrosine based activation motif) [40-43].
Da der größte Anteil des monomeren IgE an die α-Kette des hochaffinen FcεRI
bindet, befindet sich freies IgE nur zu geringen Anteilen im Serum. Wenn IgE an den
Rezeptor bindet, steigt die Expression von Zytokinen und des Rezeptors FcεRI [44].
Ohne die gesamte Entzündungsreaktion mit ihren möglicherweise schädlichen
Folgen zu starten, soll das Immunsystem bei schwächeren Angriffen in Bereitschaft
versetzt werden, gegebenenfalls schnell weitere Prozesse einzuleiten.
Zur Degranulation der Mastzelle kommt es erst, wenn benachbarte, mit ihrem Fc-
Teil an den Rezeptor gebundene IgE-Moleküle durch polyvalente Antigene
kreuzvernetzt werden. Diese Brückenbildung verändert durch eine Annäherung der
Rezeptoren die Oberfläche und setzt somit die Signaltransduktion in Gang [40;45].
Daran sind an β- und γ-Kette sitzende Tyrosinkinasen, wie Lyn und Syk, beteiligt, die
durch Phosphorylierungen eine Kaskade komplexer biochemischer Reaktionen
einleiten [46]. Über die Bildung von IP3 kommt es zur Erhöhung der intrazellulären
Calciumkonzentration mit anschließender Freisetzung gespeicherter Substanzen aus
den Vesikeln oder Neusynthese verschiedener Mediatoren.
Neben der klassischen Aktivierung durch IgE-Rezeptor Kreuzvernetzung sind
unterschiedliche Agonisten bekannt, die IgE-unabhängig die Mastzelle stimulieren
können. Zu den Aktivatoren humaner Mastzellen gehören das Zytokin SCF,
Neuropeptide (Substanz P, Vasoaktives Intestinales Peptid – VIP, Neuropeptid),
Morphin, Anaphylatoxin (C5a), basische Substanzen (Compound 48/80), bakterielle
Produkte, wie Lipopolysaccharid oder FimH, sowie Calciumionophor (bewirkt
Calciumeinstrom) und PMA (aktiviert Proteinkinase C). Das Ausmaß der

Einleitung
14
Aktivierbarkeit durch verschiedene Substanzen hängt, wie bereits in Kapitel 1.2.1
„Morphologie, Entwicklung und Heterogenität“ beschrieben, von der anatomischen
Lokalisation, dem Differenzierungsgrad und nicht zuletzt von der Spezies ab [40;45-
46].
1.2.6 Hemmung
Die Mastzelle als Effektorzelle IgE-abhängiger Überempfindlichkeitsreaktionen des
„Soforttyps“ scheint ein geeignetes therapeutisches Ziel für die Behandlung von
Allergien zu sein. Zahlreiche Substanzen wirken auf Mastzellfunktionen, z.B. um die
Freisetzung oder Synthese von Mediatoren direkt zu hemmen. Verschiedene
Medikamente, wie Lodoxamid, die Cromone Dinatriumcromoglycat und Nedocromil-
Natrium, der Bromhexin-Metabolit Ambroxol [47], das Methylxanthin Theophyllin und
diverse Immunsuppressiva (Glukokortikoide, Cyclosporin A und Tacrolismus) werden
zur Hemmung der IgE-abhängigen Mediatorfreisetzung angewendet [48-50]. H1-
Antihistaminika der zweiten Generation besitzen nicht nur wie ihre Vorgänger eine
antagonistische Wirkung auf H1-Rezeptoren, sondern hemmen in unterschiedlichem
Ausmaß die Histaminfreisetzung aus Mastzellen [51;52]. Die Wirkmechanismen der
verschiedenen Wirkstoffe sind zum großen Teil noch ungeklärt. Man nimmt an, dass
die Cromone durch Blockade von Chloridkanälen den für die Degranulation
notwendigen initialen Calciumeinstrom verhindern [53]. Ähnliche Mechanismen
nimmt man auch für Lodoxamid an, während die mastzellstabilisierende Wirkung des
Theophyllins auf einer Hemmung der Phosphodieesterase mit anschließender cAMP-
Erhöhung beruht [54].
Neuere Therapieansätze gegen allergische Erkrankungen bieten Antikörper, die
zirkulierende IgE-Moleküle abfangen und der Eliminierung zuführen oder direkt den
Fcε-RI blockieren. So bildet der gegen IgE gerichtete Antikörper Omalizumab mit IgE
einen Komplex, der nicht mehr an den Fcε-RI-Rezeptor binden und daher auch keine
Degranulation mehr einleiten kann [55].

Einleitung
15
1.3 Exozytose und SNARE-Proteine
Grundsätzlich lassen sich zwei Formen der Exozytose unterscheiden: Konstitutive
und regulierte Exozytose [56;57]. In allen eukaryoten Zellen lässt sich konstitutive
Exozytose beobachten, die kontinuierlich und ohne äußeren Stimulus abläuft. Dabei
werden verschiedene Substanzen, beispielsweise Plasmamembranbestandteile zur
Aufrechterhaltung der Zellintegrität, in den Extrazellulärraum abgegeben. Abhängig
von der Zellart werden auch extrazelluläre Matrixkomponenten (Kollagene,
Proteoglykane), aber auch IgE, über die Plasmamembran ausgeschleust.
Besonders spezialisierte Zellen, wie Neuronen, endokrine Zellen oder Mastzellen
sind in der Lage, nach einem Stimulus, größere Mengen ihres Vesikelinhalts
auszuschütten. Dabei kann es entweder zu einer absoluten Verschmelzung von
Vesikel- und Plasmamembran kommen (total fusion) oder während der Fusion bildet
sich, unter Beibehaltung der Vesikelintegrität, nur eine Öffnung zwischen beiden
Strukturen (transient fusion; „kiss and run“). Dadurch sind die Zellen in der Lage, die
Mediatorauschüttung der jeweiligen Situation bzw. dem Bedarf entsprechend zu
regulieren [58;59].
Zur Fusion von Granula- bzw. Vesikel- mit der Plasmamembran kommt es durch
eine Reihe komplexer Prozesse. Zunächst gelangt das Vesikel durch Diffusion oder
Transport über das Zytoskelett in die Zellperipherie und bindet an die aktive Zone
der Plasmamembran. ATP-abhängige Reifeprozesse bereiten das Vesikel vor für die
Ca2+-getriggerte Membranfusion mit anschließender Freisetzung des Vesikelinhalts.
Anschließend werden die Vesikel internalisiert und recycled [59;60]. Obwohl es,
abhängig von der individuellen physiologischen Funktion der Zelle, einige
Unterschiede im Ablauf der Exozytose gibt (Dauer, Größe der Pore oder des
Vesikels), hat der Prozess der Membranverschmelzung eine grundlegende
Gemeinsamkeit: spezifische Interaktionen zwischen evolutionär konservierten
SNARE-Proteinen (soluble N-ethylmaleimide-sensitive factor attachment receptor)
[61-64].
SNARE-Proteine können, nach Ausbildung eines multimolekularen Komplexes,
die Fusion zweier gegenüberliegender Membranen katalysieren. Durch die
Komplexbildung ändern die SNARE-Isoformen ihre Struktur, d.h. die Proteinfaltung,
und ihre Anordnung zueinander. Dabei wird genug Energie freigesetzt, um die

Einleitung
16
normalerweise abstoßenden Kräfte zweier Membranen zu überwinden [65;66]. Der
sogenannte „core“-Komplex ist extrem stabil und resistent gegenüber Denaturierung
durch Temperaturen bis 950C und Einwirkung von Detergenzen [67].
Entlang des exozytotischen aber auch endozytotischen Pfades kommen SNARE-
Proteine in bestimmten Kompartimenten vor. Sie regeln nicht nur die Fusion, sondern
auch in welche Richtung die Membranverschmelzung erfolgt [68]. Eine mögliche
Einteilung erfolgt danach, ob ein Arginin- (R-SNARE) oder Glutamin-Rest (Q-
SNARE) zu der zentralen Region des Komplexes beigesteuert wird [69]. Die
alternative Klassifikation richtet sich nach der Lage der SNARE-Proteine. Zu den
Vesikel-assoziierten v-SNAREs (vesicle) gehören die homologen Proteine des
Synaptobrevin, das auch als VAMP (vesicle-associated membrane protein)
bezeichnet wird [70;71]. Die t-SNAREs (target) sind an die Plasmamembran
gebunden. Diese Gruppe umfasst die Syntaxine und SNAP-Proteine (synaptosome
associated protein with molecular weight of 25, 23 or 29 kd) [72-77].
Neben den SNARE-Isoformen haben auch andere zytosolische Proteine eine
wichtige Bedeutung für die Exozytose. Während des Reifeschrittes der Exozytose
wird der SNARE-Komplex, bestehend aus je einem Molekül Synaptobrevin, Syntaxin
und SNAP, gebildet [78-80]. Bei Neuronen handelt es sich dabei um VAMP-2,
Syntaxin-1A und SNAP-25 [79]. An diesen Kern- oder 7S-Komplex bindet SNAP
(soluble NSF attachment protein), wodurch sich die ATPase NSF (N-ethylmaleimide-
sensitive factor) anlagern kann und der 20S-Komplex entsteht [81-83]. Durch
Hydrolyse von ATP durch NSF werden Proteininteraktionen gestört, so dass der
SNARE-Komplex dissoziiert und beide Membranen verschmelzen [84-86]. Auch am
Recycling der SNARE-Proteine in ihrer jeweiligen Kompartimente sind NSF und
SNAP, man unterscheidet α-, β- und γ-SNAP, beteiligt [87]. Eine Voraussetzung für
die Bindung von NSF und SNAP an den 7S-Komplex ist die calciumabhängige
Synaptotagmin-Ablösung. Nach extrazellulärer Stimulation mit anschließendem
intrazellulären Calciumanstieg bindet Synaptotagmin Calcium und löst sich vom
SNARE-Komplex [88-90]. Auch andere akzessorische Proteine, wie Sec1/Munc18-
Proteine, Rab-Proteine (ras-like GTPasen) und Synaptophysin scheinen die SNARE-
Komplex-Bildung zu regulieren. Ein Schema der an der Komplex-Bildung beteiligten
SNARE-Proteine und ihrer Kofaktoren ist in Abbildung 1 dargestellt.

Einleitung
17
Zuerst beschrieben wurden SNARE-Proteine an Hefezellen und Neuronen und
sind hier besonders gut untersucht. Aber auch in anderen, sekretorisch aktiven
Zellen konnten SNARE-Isoformen nachgewiesen werden [91-93]. Obwohl SNARE-
Proteine grundlegende Prozesse der Membranfusion vermitteln, ist nicht hinreichend
geklärt, wie die einzelnen Isoformen in den verschiedenen Zellarten verteilt sind.
Deutliche Unterschiede konnten zwischen neuronalen und nicht-neuronalen
Geweben festgestellt werden. Bestimmte Isoformen, wie SNAP-25 oder Syntaxin-1
sind Neuronen-spezifisch, während SNAP-23 ubiquitär vorkommt.
Die Expression von SNARE-Isoformen in Mastzellen wurde überwiegend an der
Rattenmastzelllinie RBL-2H3 (rat basophilic leukemia cells) untersucht. Es zeigte
sich, dass VAMP-8, Syntaxin-4 und SNAP-23, ein SNAP-25 Homologon, in die
Exozytose involviert sind [94-96]. Nach Stimulation wird SNAP-23 von der
Plasmamembran in das Zellinnere verlagert. Wird der Lagewechsel verhindert,
kommt es zu einer Hemmung der Exozytose [94]. Die v-SNARE Isoformen VAMP-2,
VAMP-3, VAMP-8 (endobrevin) und VAMP-7 (tetanus insensitive VAMP) konnten in
der Vesikelmembran nachgewiesen werden [97;98], wobei die Neurotoxin-
insensitiven Synaptobrevine VAMP-7 und VAMP-8 in endozytotischen
Kompartimenten nicht-neuronaler Zellen beschrieben sind [97;99-102]. Außerdem
wird eine Beteiligung von VAMP-7 bei der Exozytose sekretorischer Lysosome aus
hämatopoetischen Zellen diskutiert [97;103-106]. Des Weiteren konnte die FcεRI-
abhängige Exozytose durch Überexpression von Syntaxin-4, nicht aber von Syntaxin-
2 oder-3, gehemmt werden. Als weitere Regulatoren der FcεRI abhängigen
Exozytose wurden in RBL-2H3-Zellen und RPMCs (rat peritoneal mast cells) die
Synaptotagmin-Homologe Synaptotagmin I und II gefunden. Während die
Transfektion von RBL-2H3-Zellen mit Synaptotagmin I die Ca2+-getriggerte Exozytose
begünstigt, scheint Synaptotagmin II die lysosomale Sekretion der Zellen zu
vermindern [107;108].
Von humanen Mastzellen sind bisher keine Informationen über Expression und
Funktion der SNARE-Proteine oder ihrer regulatorischen Ko-Faktoren vorhanden.
Einen Ansatz zur Analyse und Beeinflussung SNARE-vermittelter Exozytose-
Prozesse bieten SNARE-spezifische Proteasen, wie das Botulinum Neurotoxin.

Einleitung
18
Abb. 1: Schematische Darstellung des Exozytose vermittelnden SNARE-
Komplexes. Die SNARE-Proteine VAMP, Syntaxin und SNAP bilden den Komplex.
An der Membranfusion sind weiterhin akzessorische Proteine beteiligt, wie
Synaptophysin, Rab3a, Munc-18 oder das Calcium-abhängige Synaptotagmin. Die
Exozytose kann durch verschiedene Isotypen von Botulinum-Neurotoxin gestört
werden, die in der Lage sind, SNARE-Proteine zu spalten. Abbildung modifiziert nach
Shukla et al [139].
1.4 Botulinum Neurotoxin
Botulinum Neurotoxin, ein Toxin des anaeroben Bakteriums Clostridium Botulinum,
ist in der Lage, die Exozytose in Neuronen zu hemmen. Durch Spaltung von SNARE-
Proteinen wird sowohl die Zusammenlagerung zu „core“-Komplexen, als auch die
spätere Fusion von Vesikeln mit der Plasmamembran verhindert. Insgesamt werden
sieben Isoformen (BoNT-A – BoNT-G) des Botulinum Neurotoxins unterschieden,
Vesikel
Synaptotagmin
Plamamembran
Munc-18
Synaptophysin
Rab3a
Botulinum
NeurotoxinCa2+
Ca2+
VA
MP
SN
AP
-25
Syn
taxi
n

Einleitung
19
das unter den bekannten biologischen Giften eines der potentesten ist. Eine
intravenöse Dosis von 1-5 ng/kg Körpergewicht kann bereits tödlich sein [109;110]. In
der Regel stammen Vergiftungen mit Botulinum Neurotoxin, auch als Botulismus
bezeichnet, vom Verzehr verdorbener Lebensmittel. Über den Gastrointestinaltrakt
gelangt das Toxin in die Blutbahn und von dort an die motorischen Endplatten
quergestreifter Muskulatur, wo sie durch Hemmung der Neurotransmitterfreisetzung
(Acetylcholin) eine schlaffe Lähmung bewirken. Da Botulinum Neurotoxin die Blut-
Hirn-Schranke nicht passiert, sind die Auswirkungen nur peripher sichtbar.
Das Neurotoxin wird als einkettiges Protein synthetisiert und durch bakterielle
Proteasen in eine schwere (HC: heavy chain; 100 kDa) und eine leichte
Polypeptidkette (LC: light chain; 50 kDa) gespalten, die durch eine Disulfidbücke
miteinander verbunden sind [111]. Nach einer spezifischen Bindung der HC an
Ganglioside, die in der Plasmamembran von neuronalen Zellen angereichert sind,
kommt es zu einer Rezeptor-vermittelten Endozytose [112-116]. Durch die
Translokationsdomäne der HC gelangt das Toxin unter Porenbildung aus dem
sauren Milieu des Endosomens ins Zytosol. Anschließend kommt es zu einer
enzymatischen Abtrennung der leichten von der schweren Kette. Erst nach der
Trennung von der HC, die Bindung und Translokation vermittelt, kann die LC ihre
proteolytische Aktivität entfalten. Extrazellulär sind beide Ketten für sich allein nicht
toxisch [116].
Zwar hemmen alle sieben Subtypen des Toxins die Acetylcholinfreisetzung in
Neuronen, haben aber unterschiedliche Zielproteine und Wirkstärken [117]. So
konnte für BoNT-A, -G und -E eine Spaltung von SNAP-25 nachgewiesen werden.
BoNT-B, -D und -F sind spezifische Proteasen für Synaptobrevin/VAMP, während
Bont-C1 die SNARE-Proteine Syntaxin und SNAP-25 spaltet [118-122].
Durch Hemmung der Exozytose von Acetylcholin aus Nervenendigungen kommt
es zu einer Lähmung der quergestreiften Muskulatur, aber auch von
parasympathischen Funktionen. Daher ist Botulinum Neurotoxin, vor allem BoNT-A
und -B, nicht nur ein tödliches Toxin, sondern wird seit den achtziger Jahren
vermehrt für therapeutische Zwecke eingesetzt. Botulinum Neurotoxin wird bei der
Behandlung von vorwiegend neurologischen Erkrankungen, wie dystone
Bewegungsstörungen und Spastizität, aber auch bei autonomen Störungen
(Blasenfunktionsstörung, Achalasie) verwendet [123;124]. In zahlreichen

Einleitung
20
Fachgebieten wird das Neurotoxin bereits genutzt. Dazu gehören Augenheilkunde,
Gastroenterologie, Orthopädie, Urologie und Dermatologie. Bekannt geworden ist
Botulinum Neurotoxin vor allem als BOTOX (BoNT-A) im kosmetischen Bereich, wo
es als Substanz zur Faltenreduktion durch Lähmung der Muskelaktivität im Gesicht
verwendet wird.
Dennoch wird das Potenzial der Neurotoxine noch nicht vollständig genutzt.
Zahlreiche weitere Anwendungsgebiete werden untersucht. So könnte Botulinum
Neurotoxin nicht nur die Acetylcholinfreisetzung aus Synapsen hemmen, sondern
auch die SNARE-vermittelte, IgE-abhängige Mediator-Freisetzung aus Mastzellen
einschränken.
1.5 Adenoviren
Eines der effektivsten Behelfsmittel, Fremd-DNA in Säugetierzellen einzuschleusen,
besteht in der Verwendung adenoviraler Vektoren. Adenoviren sind hüllenlose DNA-
Viren, deren Capsid aus verschiedenen Proteinen besteht. Zu Beginn einer Infektion
binden bestimmte adenovirale Oberflächenstrukturen, die sogenannten fiber knobs,
an den Zelloberflächenrezeptor CAR (Coxsackievirus und Adenovirus Rezeptor)
[125]. Die physiologische Funktion des CAR besteht in der Vermittlung von Zell-Zell
Kontakten [126], woraufhin es zu Wechselwirkungen zwischen Integrinen der
Zielzelle und Oberflächenproteinen des Adenovirus kommt. Das Ausmaß der
adenoviralen Transduzierbarkeit einer Zelle hängt von ihrem Rezeptorstatus ab.
Anschließend wird der Adenovirus durch rezeptorvermittelte Endozytose
aufgenommen und gelangt an den Kern, wo die Transkription und Replikation der
viralen DNA beginnt. Innerhalb von 18 h nach Infektion ist die Transgenexpression
detektierbar und erreicht ihr Maximum nach 48-72 Stunden [127]. Der Abschluß des
viralen Zyklus löst eine Zelllyse und damit die Freisetzung der vermehrten Viren aus.
Die Zellyse wird durch das "adenovirus death protein" E3 ausgelöst [128].
Adenoviren (Fam. Adenoviridae) des humanen Serotyps 5 sind einer der am
häufigsten verwendeten Vektortypen für den Gentransfer in Säugerzellen. Sie
können ein breites Spektrum an Zelltypen und Geweben, sowohl ruhende als auch
proliferierende Zellen, infizieren [129]. Außerdem lässt sich das adenovirale Genom

Einleitung
21
(ca 36 kb) einfach modifizieren. In adenovirale Vektoren können bis zu 8 kb große
DNA-Abschnitte kloniert und anschließend in hohen Mengen produziert werden
[130;131]. Im Infektionszyklus erfolgt die Expression der Gene zeitlich versetzt, so
dass zwischen „frühen“ und „späten“ Genen unterschieden wird. Während Gene der
späten Phase hauptsächlich für virale Strukturproteine kodieren, sind Gene der
frühen Phase (E1, E2, E3 und E4) für die Genexpression und Virusreplikation
essentiell. Fremde DNA kann durch Deletion verschiedener Regionen früher Gene in
das Virusgenom eingebracht werden [132]. Adenovirale Vektoren mit E1- und E3-
Deletion (pAdEasy-1) werden häufig verwendet. Die Deletion von E1 bewirkt, dass
sich die Zellen nicht mehr replizieren und auch keine infektiösen Viruspartikel in den
Zielzellen produzieren können. Das E3-Gen ist zur Umgehung einer Immunantwort
nötig. Somit können für den Laborgebrauch sichere, replifikationsdefiziente
adenovirale Vektoren hergestellt werden. Die Präparation der rekombinanten
Adenoviren erfolgt in entsprechend stabil transfizierten Zellen, die fehlende Faktoren
für die virale Replikation transkomplementieren [133].
1.6 Fragestellung
Mastzellen sind in der Lage, nach extrazellulärer Stimulation größere Mengen
unterschiedlicher Mediatoren mittels Exozytose freizusetzen, darunter Histamin,
welches mit verantwortlich ist für die pathophysiologische Rolle der Mastzelle als
Effektorzelle IgE-abhängiger Überempfindlichkeitsreaktionen vom Soforttyp (Typ I
Allergie nach Coombs und Gell).
SNARE-Proteine vermitteln die regulierte Exozytose von Neuronen und anderen
sekretorisch aktiven Zellen. Bisher sind Daten über die Expression von SNARE-
Proteinen in Mastzellen überwiegend anhand der Rattenmastzelllinie RBL-2H3
gewonnen worden. Ziel der vorliegenden Arbeit ist es aufzuzeigen, welche SNARE-
Isoformen in humanen Mastzellen vorkommen und welche Bedeutung sie für die IgE-
vermittelte Mastzell-Exozytose haben. Außerdem soll mit Hilfe adenoviraler
Transduktion geklärt werden, ob Botulinum-Neurotoxin in humanen Mastzellen die
Exozytose hemmen kann, was möglicherweise ein neuer Ansatz für die Therapie
allergischer Symptome sein könnte.

Materialien und Methoden
22
2. Materialien und Methoden
2.1 Materialien
2.1.1 Chemikalien und Reagenzien
Acrylamid/Bisacrylmid (30%/80%) (Rotiphorese® Gel 30) Roth
Agarose Gibco
Albumin Roche
Ammoniumperoxidsulfat (10%) Roth
Amphotericin B Gibco
Ampicillin Sigma
Ampuwa (steriles, pyrogenfeies Wasser) Boehringer
Bactotrypton Gibco
Bio Rad Protein Assays Bio Rad Laboratories
Bovines Serumalbumin (BSA, Fraktion 4, fettsäurefrei) Boehringer
Cäsiumchlorid Sigma
DiffQuick Dade Behring Marburg GmbH
DMEM (Dulbecco´s Modified Eagle Medium) mit Glutamax Gibco
dNTPs Invitrogen
EDTA Merck
Essigsäure J.T.Baker
Ethanol J.T.Baker
Ethidiumbromid Sigma
Fetal Calf Serum (FCS) Gibco
Gelatine Merck
Gentamycin Gibco
Glucose Sigma
Hanks Basic Salt Solution (HBSS) Gibco
Hefeextrakt Gibco
HEPES Sigma
Isopropanol J.T.Baker
Kanamycin Merck
Lipofectamin Invitrogen
Low Range Maker BioRad

Materialien und Methoden
23
Magermilchpulver Merck
Magnesiumchlorid Merck
Methanol J.T.Baker
Metronidazol Fresenius
Natriumacetat Fluka
Natriumdodecysulfat (SDS) Sigma
OptiMEM Gibco
PBS 10 x Gibco
Penicillin/Streptomycin Biochrom
Phosphate Buffered Saline (PBS) Gibco
Protease Inhibitor Cocktail CompleteTM Mini Roche
2.1.2 Antikörper
Isotype mouse IgG Serotec, Oxford, UK
Isotype rabbit IgG Serotec, Oxford, UK
anti-SNAP-25 (SMI 81), monoclonal mouse antibody Sternberger Monoclonals Incorporated
anti-SNAP-23, polyclonal rabbit antibody Synaptic Systems, Germany
anti-VAMP-2 , polyclonal rabbit antibody Synaptic Systems, Germany
anti-VAMP-2 (Synaptobrevin), monoclonal mouse antibody Synaptic Systems, Germany
anti-VAMP-3 (Cellubrevin), polyclonal rabbit antibody Affinity BioReagents
ani-VAMP-7 (TI-VAMP), monoclonal mouse antibody T. Galli, Institut Jacques Monod, Paris
anti-VAMP-8 (Endobrevin), polyclonal rabbit antibody abcam, Cambridge, Uk
anti-Syntaxin-1A, polyclonal rabbit antibody Synaptic Systems, Germany
anti-Syntaxin-1B, polyclonal rabbit antibody Synaptic Systems, Germany
anti-Syntaxin-2, polyclonal rabbit antibody StressgGen Biotechnologies Corp
anti-Syntaxin-3, polyclonal rabbit antibody abcam, Cambridge, Uk
anti-Syntaxin-4, monoclonal mouse antibody BD Biosciences Pharmingen
Alexa Fluor® goat anti-mouse 488 Caltag Laboratories
Alexa Fluor® donkey anti-rabbit 594 Caltag Laboratories
ECL anti-mouse IgG Amersham Biosciences
ECL anti-rabbit IgG Amersham Biosciences
IgE-Fc-BoNT/B-LcHN Biotecon, Potsdam
anti-SCF-Rezeptor c-kit (anti-CD 117, mAk YB5.B8) Pharmingen, Hamburg
anti-IgE-Rezeptor α-chain (mA 22E7) Hoffmann-La Roche, Nutely, USA

Materialien und Methoden
24
2.1.3 Kommerzielle Kits
Rneasy Mini Kit Qiagen
Qiagen Plasmid Maxi Kit Qiagen
Qiagen Endofree Maxi Kit Qiagen
Qiaquick PCR Purification Kit Qiagen
Histamin-Radioimmunoassay (RIA) Coulter-Immunotech, Hamburg
2.1.4 Enzyme
Acetylcystein (ACC) Sigma
Chymopapain Sigma
Collagenase D Roche
DNAse I Roche
Lysozym Sigma
Platinum Taq Polymerase Invitrogen
Pronase Roche
Restriktionsendonukleasen Pme I, Pac I, NEB
RNAse freie DNAse Promega
Superscript TM Reverse Transkriptase Invitrogen
Trypsin Gibco
2.1.5 Puffer, Lösungen und Medien
Nährmedien
Für die Mastzellkultur wurde RPMI 1640 (mit 25 mM HEPES, 2 mM L-Glutamin) ohne
Phenolrot verwendet, das versetzt war mit 10 % (v/v) FCS, 0,5 µg/ml Amphotericin,
100 µg/ml Gentamycin, 100 U/ml Penicillin und 100 µg/ml Streptomycin.
Die adhärente Zelllinie HEK-293 wurde in DMEM-Medium, versetzt mit 10% FCS,
100U/ml Penicillin und 100 µg/ml Streptomycin, kultiviert. Für die Infektionsversuche
mit Adenoviren wurde der FCS-Anteil auf 2% reduziert. OptiMEM wurde für die
Transfektionen benutzt.

Materialien und Methoden
25
Für die Bakterienkultur wurden 10 g Bactotrypton, 5 g Hefeextrakt und 10 g NaCl
in 1 l destilliertem Wasser gelöst und autoklaviert (LB-Medium). Zur
plasmidspezifischen Selektion wurde unmittelbar vor Inkulturnahme das
entsprechende Antibiotikum hinzugefügt. Zum Gießen von Agarplatten wurden
15 g Agar in 1l LB-Medium gegeben und autoklaviert. Erst nach Abkühlung auf unter
500 C wurde das Antibiotikum hinzugefügt.
Nach der Elektroporation wurden die kompetenten Bakterien in SOC-Medium
aufgenommmen. Für das SOC-Medium wurden 20 g Tryptone, 5 g Hefeextrakt,
10 mM NaCl und 2,5 mM KCl mit destilliertem Wasser auf 1 l aufgefüllt. Nach dem
Autoklavieren wurden 10 mM MgCl2, 10 mM MgSO4 und 20 mM Glucose steril
zugegeben.
Puffer für die Mastzellaufarbeitung
Sämtliche Lösungen und Puffer wurden mit sterilem, pyrogenfreiem Wasser („Aqua
Spüllösung“, Delta Select GmbH, Dreieich) angesetzt, sterilfiltriert und bei 4°C
aufbewahrt.
ACC-Lösung (20 µg ACC in 50 ml Tyrode (1x) -EDTA-Puffer); CE-Lösung:
1,5 mg/ml Collagenase D in TGMD-Puffer); HA-Puffer (2,5 mg/ml BSA in HEPES-
Puffer); HEPES-Puffer (20 mM HEPES; 5 mM NaCl; 0,5 mM D-Glucose; 5 mM KCl);
10 x PBS-Puffer (Gibco Life Technologies, GB); PCh-Lösung (3 mg/ml Pronase,
0,75 mg/ml Chymopapain in TE-Puffer); TE-Puffer (500 mM EDTA in Tyrode-Puffer);
TEA-Puffer (0,2 % (v/v) Ampicillin; 0,4 % (v/v) Gentamycin; 4 % (v/v) Metronidazol,
in TE-Puffer); TGMD-Puffer (1,23 mM MgCl2; 0,015 mg/ml DNAse; 5 % (v/v) 2 %-ige
Gelatine, in 10 x Tyrode-Puffer); Tyrode-Puffer (37 mM NaCl, 2,7 mM KCl,
0,36 mM Na2HPO4, 5,55 mM D-Glucose)
Puffer für Westernblot
Acrylamidlösung (30 %ige Acrylamidlösung mit 0,8 % Bisacrylmid); Extraktionspuffer
(25 mM Tris-HCl pH 7,4; 0,5 mM EDTA; 0,5 mM EGTA; 0,05 % Triton x-100; 100 mM
β-Mercaptoethanol; 1 µg/ml Leupeptin; 1 µg/ml Aprotinin; für den Gebrauch: 0,5 ml
100 mM PMSF in 100 % Ethanol auf 100 ml Extraktions-Puffer geben);
10 x Laufpuffer (30 g Tris; 144 g Glycin; 100 ml SDS 10 %ig mit H2O auf 1 l

Materialien und Methoden
26
auffüllen); PBS-Tween (PBS; 0,25 % (v/v) Tween); 2 x Probenpuffer für Westernblot/
Laemmli-Puffer (0,2 mg/ml Glycerin; 20 ml 10 %iges SDS; 25 ml SGP; 20 ml DTT
[1 mol]; 0,5 mg Bromphenolblau, mit H2O auf 100 ml auffüllen pH 8,8); Sammelgel
(0,75 ml Sammelgelpuffer; 0,45 ml Acrylamidlösung; 1,8 ml H2O; 30 µl 10%-iges
Ammoniumperoxidsulfat; 3 µl TEMED); 4 x Sammelgelpuffer (18,18 g Tris; 12 ml
SDS 10%ig; mit H2O auf 100 ml auffüllen, pH 8,8); 10 x SDS-Page-Puffer (151,4 g
Tris; 720 g Glycin; 50 g SDS; pH 8,8); Trenngel (2,5 ml Trenngelpuffer; 4 ml
Acrylamidlösung; 3,4 ml H2O; 100 µl 10 %iges Ammoniumperoxidsulfat (0,1 g in 1 ml
H2O); 10 µl TEMED); 4 x Trenngelpuffer (18,18 g Tris, 4 ml SDS 10 %ig; mit H2O auf
100 ml auffüllen, pH 8,8); Westernblot-Puffer (2,9 g Glycin; 5,8 g Tris; 0,37 g SDS;
200 ml Methanol, mit H2O auf 100 ml auffüllen; pH 8,3)
Puffer für die Herstellung rekombinanter Adenoviren
Lyse-Puffer (10 mM Tris pH 8,0; 10 mM MgCl2; 50 mM NaCl2); CsCl-Gradient,
Dichte 1,2 g/ml (26,5 g CsCl2; 1 M Tris-HCl ph 7,5, mit H2O auf 100 ml auffüllen);
CsCl-Gradient, Dichte 1,4 g/ml (53,5 g CsCl2; 1 M Tris-HCl ph 7,5, mit H2O auf 100
ml auffüllen); 2 x Lagerungspuffer für Adenoviren (10 mM Tris-HCl ph 8,0;
100 mM NaCl; 1 mM MgCl2; 50% Glycerol; 0,1% BSA)
2.1.6 Verwendete Zelllinien
Prokaryote Zelllinien
BJ 5183 ist ein modifizierter E. coli Stamm, der für Rekombinationen geeignet ist. In
den verwendeten elektrokompetenten Zellen lag der adenovirale Backbone-Vektor
AdEasy-1 bereits vor (BJ5183-AD1 Electrocompetent Cells, Stratagene), so dass nur
noch pShuttle-CMV eingeschleust werden musste. Die Ausbeute an Plasmid-DNA ist
in diesen Zellen jedoch gering.
Der modifizierte E. coli Stamm XL10-Gold eignet sich zur stabilen Replikation von
Plasmiden in hoher Kopienzahl. Auch größere Plasmide können in die chemisch
kompetenten Zellen eingeschleust werden (XL10-Gold® Kan´ Ultracompetent Cells,
Stratagene).

Materialien und Methoden
27
Eukaryote Zelllinien
Die humane, embryonale Nierenzelllinie HEK 293 (human endocrine kidney cells;
ATCC CRL-1573) verfügt durch Transformation über die adenovirale E1 Region.
Daher eignen sich diese Zellen zur Vermehrung E1 deletierter rekombinanter
Adenoviren.
2.1.7 Klonierungsvektoren
pShuttle-CMV
In die Multiple Cloning Site dieses Shuttle Vektors wurden die cDNA der leichten
Ketten von BoNT-A, -B und –C1 kloniert. Da der Vektor keine zusätzliche
Expressionskassette für das Reportergen GFP (green fluorescent protein) enhält,
wurde die cDNA als N-terminaler Fusionspartner von eGFP Kloniert. Der Vektor
enthält ein Kanamycinresistenzgen zur Selektion in E. coli (pShuttle-CMV freundlich
überlassen von B. Vogelstein, John Hopkins Oncology Center, Baltimore, USA). Die
im Rahmen dieser Arbeit konstruierten Plasmide pBoNT-A, -B und -C sind in Kapitel
3.2.1 ausführlich beschrieben.
pAdTrack-CMV
Der Vektor pAdTrack-CMV wurde als Kontrollplasmid eingesetzt. Er enthält eine
Expressionskassette für das Reportergen GFP (green fluorescent protein), wodurch
das Ausmaß der Genexpression direkt in den Zellen mittels Fluoreszenzmikroskopie
(Wellenlänge 456 nm) erkennbar ist. Der Vektor enthält ein Kanamycinresistenzgen
(freundlich überlassen von B. Vogelstein, Johns Hopkins Oncology Center,
Baltimore, USA).
pAD-Easy-1
Der Backbone-Vektor pAd-Easy-1 (33414 bp) enthält einen Großteil des
adenoviralen Genoms, mit Ausnahme der E1 und E3 Gene. Diese Deletionen
ermöglichen den Einbau von bis zu 8 kb fremder DNA und verhindern die Entstehung
replikationsfähiger Viren außerhalb bestimmter Zellen (z.B. HEK 293), die das

Materialien und Methoden
28
fehlende E1 Gen supplementieren. Der Backbone-Vektor befand sich bereits in den
BJ5183-AD1 Zellen (Stratagene), die für die Transformation durch Elektroporation
verwendet wurden.
2.1.8 Rekombinante Adenoviren
Für sämtliche Versuche wurden nur durch E1 Deletion replikationsdefiziente
Adenoviren des humanen Serotyps 5 verwendet. Die Vermehrung der Viren erfolgte
in der Zelllinie HEK 293, die das fehlende E1 Gen transkomplementierte.
Der Virus AdGFP wurde wegen seiner starken GFP-Signale als Kontrollvirus
eingesetzt (freundlicherweise überlassen von Dr. B. Fleischmann-Mundt).
Im Rahmen dieser Arbeit wurden die Adenoviren AdBoNT-A, AdBoNT-B und
AdBoNT-C generiert. Für diese Adenoviren wurden die von Biotecon konstruierte
nPlasmide pBoNT-A, pBoNT-B und pBoNT-C mit dem adenoviralen Genom in
pAdEasy-1 rekombiniert (vgl. Kapitel 3.2.1).
2.2. Molekularbiologische Methoden
2.2.1 Konzentrationsbestimmung von DNA
Die Konzentration der DNA wurde photometrisch bestimmt. Dazu wurde die
Absorption der DNA-Lösung bei 260 nm gemessen. Eine Extinktion von 1,0
entspricht hierbei etwa 50 µg/ml doppelsträngiger DNA. Für jede Probe wurde die
Absorption bei einer Wellenlänge von 280 nm gemessen , um eine Aussage über die
Reinheit der Nukleinsäuren treffen zu können. Der Absorptionsquotient E280/E260
beträgt für eine hinreichend reine DNA-Lösung 1,7 bis 1,9. Entsprechend tiefer liegt
der Absorptionsquotient bei größeren Proteinverunreinigungen.

Materialien und Methoden
29
2.2.2 Transformation
Transformation chemisch kompetenter Bakterien
Ein Aliquot der chemisch kompetenten Zellen wurde vorsichtig auf Eis aufgetaut. Von
der Bakteriensuspension wurden 50µl genommen, mit etwa 15 ng zirkulärer Plasmid-
DNA vermischt und 30 min auf Eis inkubiert. Nach einem Hitzeschock (90 sec bei
420C) regenerierten die Bakterien für 2 min auf Eis, wurden in 100µl SOC-Medium
aufgenommen und für eine Stunde bei 37°C und 225 rp m in den Heizblock gestellt.
Anschließend wurde der Transformationsansatz auf einer Kanamycin (10 mg/ml)
enthaltenden Agarplatte ausgestrichen und über Nacht bei 370C inkubiert. Das
Antibiotikum diente zur Selektion der transformierten von den nicht-transformierten
Bakterienzellen.
Transformation durch Elektroporation
Besonders für größere Plasmiden, z.B. Adenovirusplasmide, ist das Verfahren der
Elektroporation geeignet, da es eine höhere Effizienz aufweist als die Transformation
in chemisch kompetente Zellen. Die elektrokompetenten Bakterien wurden auf Eis
aufgetaut, 40µl der Bakteriensuspension mit 1-2µl der DNA (5-50 ng in Wasser
gelöst) versetzt und in eine vorgekühlte Elektroporationsküvette überführt. Die
Elektroporation erfolgte bei 2,5 kV, 200 und 25 µF. Direkt im Anschluss an den
Elektroimpuls wurden 900 µl SOC-Medium hinzugefügt und der
Transformationsansatz für eine Stunde unter Schütteln inkubiert. Verschiedene
Volumina der Bakteriensuspension wurden auf Kanamycin enthaltenden Agarplatten
ausgestrichen und über Nacht bei 370C inkubiert.

Materialien und Methoden
30
2.2.3 Mini-Plasmidpräparation
Einzelne, transformierte Klone konnten durch Mini-Präparation und anschließendem
Restriktonsverdau relativ schnell überprüft werden.
Dazu wurden einzelne Kolonien von der Agarplatte gepickt und zur Vermehrung
der plasmidtragenden Klone in 3 ml LB-Medium mit dem ensprechenden
Antibiotikum bei 370C über Nacht unter Schütteln (225 rpm) inkubiert. Von der über
Nacht inkubierten Bakterien-Vorkultur wurden 1,5 ml in Eppendorfgefäße überführt
und 2 min bei 10.000 g zentrifugiert. Der Überstand wurde verworfen bis auf einen
kleinen Rest zum Resuspendieren. Nach Zugabe von 300 µl Lyse-Puffer und 10 µl
Lysozym (10 mg/ml) wurde die Suspension für 90 sec bei 950C erhitzt, 20 min
(10.000 g, 40C) zentrifugiert und das Pellet entfernt. Es folgte eine Fällung der DNA
im Überstand mit 400 µl Isopropanol für 10 min und Zentrifugation (15 min, 10.000 g,
40C). Das Pellet wurde mit 70%-igem Ethanol gewaschen, nochmals zentrifugiert und
anschließend 15 min luftgetrocknet. Nach Lösung in 50µl TE-Puffer wurde die
erhaltene DNA bei –200C gelagert.
2.2.4 Maxi-Plasmidpräparation
Größere Mengen endotoxinfreier DNA wurden mit dem Plasmid Maxi Kit bzw. dem
Endofree Maxi Kit nach einem modifizierten Protokoll des Herstellers gewonnen. Die
Effizienz der Transfektion wird durch die Verwendung endotoxinfreier DNA deutlich
erhöht.
Nach der Überprüfung einzelner, transformierter Klone durch Mini-Präparation und
anschließenden Restriktionsverdau, wurde der Rest der Bakterienkultur mit
antibiotikahaltigem LB-Medium auf 500 ml aufgefüllt und für etwa 16 h unter
Schütteln inkubiert (370C, 225 rpm). Am folgenden Morgen wurde die Kultur 30 min
mit 3.800 g bei 4°C zentrifugiert, das Bakteriensed iment in 10 ml Puffer P1
resuspendiert und nach Zugabe von 10 ml Puffer P2 unter Kippen des
Zentrifugenröhrchens 5 min lysiert. Auf eine 20 min Inkubation mit dem Puffer P3 (10
ml) folgte eine zweimalige Zentrifugation (30 min, 4°C, 20000 g), um genomische
DNA, Protein und den Zelldetritus von der gelösten Plasmid-DNA zu trennen. Zu dem

Materialien und Methoden
31
klaren Überstand wurden 2,5 ml ER-Puffer hinzugefügt, 30 min auf Eis inkubiert und
dann auf eine mit 10 ml Puffer QBT äquilibrierte Qiagen-tip Säule gegeben. Die
dabei gebundene DNA wurde mit 15 ml QN-Puffer gelöst, nachdem RNA, Proteine
und Endotoxine durch zweimaliges Waschen mit jeweils 30 l QC-Puffer entfernt
wurden. Die gewonnene DNA wurde mit 10,5 ml Isopropanol gefällt, durch 30 min
Zentrifugation bei 10000 g und 4°C sedimentiert und das Pellet mit 5 ml 70%-igem
Ethanol gewaschen. Nach erneuter Zentrifugaton wurde das Pellet getrocknet und in
100-200 µl TE-Puffer aufgenommen.
2.2.5 DNA-Spaltung mit Restriktionsendonukleasen
Die enzymatische Spaltung von Plasmid-DNA wurde nach den vom Hersteller der
Restriktionsenzyme (NEB) angegebenen Bedingungen durchgeführt. Zur Spaltung
von DNA aus Mini-Plasmidpräparationen wurden 10-15 µl DNA-Lösung mit 3 bis 6
Units des entsprechenden Enzyms inkubiert. Für präparative Restriktionen wurde
das Enzym im 2- bis 3-fachen Überschuss eingesetzt und der Ansatz inkubierte über
Nacht bei 370C.
2.2.6 Phenol-Chloroform-Extraktion
Zur Aufreinigung von DNA wurde eine Phenol-Chloroform-Extraktion mit
anschließender Ethanolfällung zum Konzentrieren der DNA durchgeführt. Die gelöste
Plasmid-DNA wurde mit dem gleichen Volumen Phenol (Roth) versetzt, durch
Schütteln gemischt und anschließend zur Phasentrennung 1 min bei 10000 g
zentrifugiert. Die wässrige, DNA-enthaltende obere Schicht wurde in ein neues
Eppendorfgefäß überführt, ohne dabei die denaturierten Proteine (z.B.
Restriktionsenzyme) enthaltende Interphase zwischen der wässrigen Phase und der
Phenolphase zu stören. Zur Beseitigung des restlichen Phenols wurde die wässrige
Phase mit dem gleichen Volumen Chloroform versetzt, gemischt und zur
Phasentrennung wiederum 1 min bei 10000 g zentrifugiert. Zu dem Überstand
wurden das dreifache Volumen Ethanol (97 %) und 1/10 des Volumens 3 mol/l

Materialien und Methoden
32
Natriumacetat gegeben. Nach gutem Durchmischen wurde die DNA über Nacht bei -
20°C gefällt und anschließend durch 10 min Zentrifu gation bei 10000 g sedimentiert.
Das Sediment wurde mit 200 µl Ethanol (70 %) gewaschen, um mitgefällte Salze zu
lösen. Es folgte eine erneute Zentrifugation von 10 min bei 10000 g. Nachdem das
entstandene Sediment getrocknet war, wurde es in 20 µl TE-Puffer aufgenommen.
2.2.7 RNA-Isolation und Reverse Transkriptase-Polymerase-Kettenreaktion
Zur Isolation von RNA aus kultivierten Mastzellen wurden 1-2 x 105 Zellen in 335µl
RLT-Puffer aufgenommen und bei –800 C gelagert bis zur Präparation der Gesamt-
RNA mit Hilfe des RNeasy Mini Kits. Durch reverse Transkription erfolgte
anschließend eine Umschreibung in cDNA. Die genomische DNA wurde durch
Inkubation mit 1 U RNAse-freier DNAse für 15 min bei 37°C verdaut und nach
Zugabe von 25 mM EDTA die RNA bei 70°C für 10 min d enaturiert. Durch
Superscript III Reverse Transcriptase und Zugabe von 20 pmol Oligo-dT-Primern
wurde die cDNA synthetisiert (60 min, 49 °C). Nebe n humanen intestinalen
Mastzellen wurden auch Proben mit in vitro entwickelten Mastzellen aus
Nabelschnurblut (CBMC) und der Mastzellinie HMC-1, sowie aus menschlichem Blut
isolierten eosinophilen Granulozyten, verglichen. HMC-1 ist eine Mastzelllinie mit
einem defekten Fcε-Rezeptor-I und einer Mutation am SCF-Rezeptor c-kit. Als
Kontrolle diente die humane Neuroblastom Zelllinie MHH-NB-11.
Für eine PCR-Reaktion wurden 1/20 bzw. 1/33 der erhaltenen cDNA-Menge
verwendet. Das 25 bzw. 50 µl große Reaktionsvolumen enthielt 1,25 bzw. 2,5 U
Platinum Taq Polymerase und 20 pmol der entsprechenden Primerpaare. Die
Amplifikation der cDNA-Fragmente fand in einem Peltier-Thermal-Cycler (PTC200,
MJ Research, USA) nach folgendem Schema statt:
1. Schritt: Aktivierung der Polymerase bei 94°C für 5 min.
2. Schritt: Denaturierung der DNA bei 94°C für 30 s ec.
3. Schritt: Annealing der Primer an die DNA bei 54-58°C für 45 sec.
4. Schritt: Synthese der neuen DNA-Stränge bei 72°C für 35 sec.
Materialien und Methoden
33
5. Schritt: 31-37 mal Wiederholung der Schritte 2 bis 4
6. Schritt: Elongation bei 72°C für 2 min.
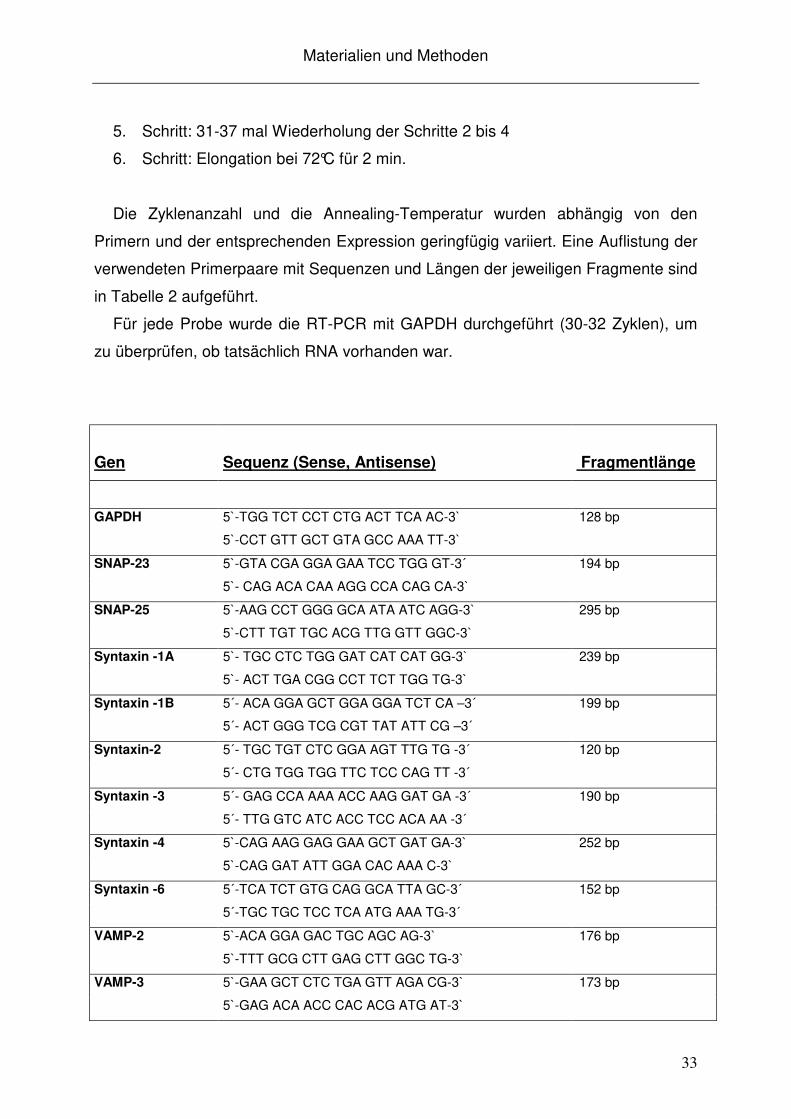
Die Zyklenanzahl und die Annealing-Temperatur wurden abhängig von den
Primern und der entsprechenden Expression geringfügig variiert. Eine Auflistung der
verwendeten Primerpaare mit Sequenzen und Längen der jeweiligen Fragmente sind
in Tabelle 2 aufgeführt.
Für jede Probe wurde die RT-PCR mit GAPDH durchgeführt (30-32 Zyklen), um
zu überprüfen, ob tatsächlich RNA vorhanden war.
Gen
Sequenz (Sense, Antisense)
Fragmentlänge
GAPDH 5`-TGG TCT CCT CTG ACT TCA AC-3` 128 bp
5`-CCT GTT GCT GTA GCC AAA TT-3`
SNAP-23 5`-GTA CGA GGA GAA TCC TGG GT-3´ 194 bp
5`- CAG ACA CAA AGG CCA CAG CA-3`
SNAP-25 5`-AAG CCT GGG GCA ATA ATC AGG-3` 295 bp
5`-CTT TGT TGC ACG TTG GTT GGC-3`
Syntaxin -1A 5`- TGC CTC TGG GAT CAT CAT GG-3` 239 bp
5`- ACT TGA CGG CCT TCT TGG TG-3`
Syntaxin -1B 5´- ACA GGA GCT GGA GGA TCT CA –3´ 199 bp
5´- ACT GGG TCG CGT TAT ATT CG –3´
Syntaxin-2 5´- TGC TGT CTC GGA AGT TTG TG -3´ 120 bp
5´- CTG TGG TGG TTC TCC CAG TT -3´
Syntaxin -3 5´- GAG CCA AAA ACC AAG GAT GA -3´ 190 bp
5´- TTG GTC ATC ACC TCC ACA AA -3´
Syntaxin -4 5`-CAG AAG GAG GAA GCT GAT GA-3` 252 bp
5`-CAG GAT ATT GGA CAC AAA C-3`
Syntaxin -6 5´-TCA TCT GTG CAG GCA TTA GC-3´ 152 bp
5´-TGC TGC TCC TCA ATG AAA TG-3´
VAMP-2 5`-ACA GGA GAC TGC AGC AG-3` 176 bp
5`-TTT GCG CTT GAG CTT GGC TG-3`
VAMP-3 5`-GAA GCT CTC TGA GTT AGA CG-3` 173 bp
5`-GAG ACA ACC CAC ACG ATG AT-3`

Materialien und Methoden
34
VAMP-5 5`-CGT AAC AAC TTC GGC AAG GT-´3 235 bp
5´-TGT CAC TGC TCT GAG GGA GA-´3
VAMP-7 5´ -AAC GTT CCC GAG CCT TTA AT -´3 242 bp
5´- TCC TCG CTG AGC TAC CAG AT -´3
VAMP-8 5`-GTG CGG AAC CTG CAA AGT GA-3` 260 bp
5`-GAA GGC ACC AGT GGC AAA GA-3`
Synaptotagmin-1 5`-GAA GAT GGA TGT GGG TGG CT-3` 279 bp
5`- CAT GTC TGA CCA GTG TCG CA-3`
Synaptotagmin-2 5`-TCA AGG TGC CAT ACC AGG AG-3` 330 bp
5`-CCT CTT GCC ATT CTG CAT CA-3`
Synaptotagmin-4 5`-AAG TGA ACC TGT ACC ATG CC-3` 202 bp
5`-CTG CCC GAT TAC CTC ATT TC-3`
BoNT-A 5´-CAC CAC CAG AAG CAA AAC AA-3´ 360 bp
5´- CCA TAA CCA TTT CGC GTA AGA-3
BoNT-B 5´-CGT GAA CAA GCT CAT CTC CA-3´ 398 bp
5´- TAC AGC TCC TCA GCC TGG AT-3´
BoNT-C 5´-GGC ACA AGA AGG ATT TGG TG-3´ 343 bp
5´-ATC CAA TGC CTT TTC CTC AA-3´
GFP
5´-ACC ACC TCG AGT GAG GCC TCA CCG GTC GCC
ACG ATG GTG AGC AAG GGC GAG G-3´
770 bp
5´-GAG AAT AAG CTT AAC GCG TCT TGT ACA GCT
CGT CCA TG-3´
Tabelle 2: Verwendete Primersequenzen, Fragmentlängen
2.2.8 Agarosegelelektrophorese
Die Proben wurden ihrer Größe entsprechend in einem 1 %igen Agarosegel mit 500
ng/ml Ethidiumbromid in TAE-Puffer aufgetrennt und dann unter UV-Licht fotografiert.
Zur Bildung der Markerbanden wurde ein 1 kb plus DNA-Ladder verwendet.

Materialien und Methoden
35
2.3 Proteinbiochemische Methoden
2.3.1 Zellextraktion
Für die Western Blot Analysen mussten Zelllysate gewonnen werden. Pro Zelllysat
wurden 5 x 105 Zellen einer hochreinen, 2-3 Wochen kultivierten Mastzell-Kultur
eingesetzt. Nach Zentrifugation (5 min, 300 g) wurde der Überstand abgenommen
und das Zellpellet in 60-100 µl Extraktionspuffer mit Zusatz von Proteaseinhibitoren
(Protease inhibitor cocktail CompleteTM Mini) lysiert. Anschließend wurde das Lysat
nochmals zentrifugiert (14000 g, 40C), der proteinhaltige Überstand abgenommen,
aliquotiert und bei –800C aufbewahrt.
2.3.2 Western Blot
Um gleiche Mengen an Protein einsetzen zu können, wurde die Proteinkonzentration
mit Hilfe des Bio Rad Protein Assays (Bio Rad Laboratories) bestimmt. Es wurden je
Spur 10-25 µg Protein im Verhältnis 1:1 mit 2 x Probenpuffer (Laemmli-Puffer)
angesetzt, für 5 min bei 950C denaturiert und auf ein 4%-iges SDS-Acrylamid-
Sammelgel aufgetragen. Zur Bildung der Markerbanden, um die Proteingröße der
später auf dem Film ersichtlichen Banden erkennen zu können, wurde der Bio Rad
Low Range Marker eingesetzt. Nach dem Auftrag auf das Sammelgel erfolgte die
Auftrennung von Proben und Marker in einem 12%-igen SDS-Acrylamid-Trenngel (90
min, 120 V). Die im Trenngel enthaltenen Proteine wurden auf eine
Nitrozellulosemembran durch den Bau eines „Sandwiches“ nach dem Prinzip „Fast
Blot/ Semi-Dry“ transferiert (Kathode, 3 x Filterpapier, Acrylamid-Trenngel,
Nitrozellulosemembran, 3 x Filterpapier, Anode). Der Transfer erfolgte in einer mit
Westernblot-Puffer gefüllten Blotkammer (Mini Blot Kammer, BioRad) für 4h bei 40 V
und einer Temperatur von 40C. Anschließend inkubierte die Nitrozellulosemembran
über Nacht bei 40C mit 5%-iger Magermilchlösung in PBS-Tween, um unspezifische
Bindungen des im Folgenden Verwendeten Antikörpers zu blockieren.

Materialien und Methoden
36
Nach zweimaligem Waschen mit PBS-Tween inkubierte die Membran mit dem
Primärantikörper, seiner Spezifität entsprechend verdünnt, für 90 min bei
Raumtemperatur. Es folgten zweimaliges Waschen mit PBS-Tween und Inkubation
mit einem entsprechend verdünnten, Peroxidase-gekoppelten Sekundärantikörper
für 90 min bei Raumtemperatur. Danach wurde die Nitrozellulosemembran zur
Entfernung unspezifischer Bindungen dreimal mit PBS-Tween gewaschen.
Anschließend konnte der Sekundärantikörper mittels Chemilumineszenz Reagenz
(Western Lightning Chemiluminescence Reagent Plus) auf einem hochempfindlichen
Röntgenfilm dargestellt werden.
2.4 Virale Methoden
2.4.1 Zellkultur
Die adhärent wachsende Zelllinie HEK-293 wurde in 75 cm² Gewebekulturflaschen
mit DMEM (versetzt mit 10% FCS und 1% Penicillin/Streptomycin) bei 370C und 5%
CO2 kultiviert. Bei Erreichen der Konfluenz des Zellrasens wurden die Zellen
passagiert. Dazu wurde das Medium abgesaugt, die Zellen mit PBS gewaschen und
trypsiniert. Zum Lösen des Zellrasens wurden die Zellen mit 1 x Trypsin/EDTA
bedeckt und inkubierten 2-5 min bei 370C. Durch Abklopfen ließen sich die Zellen
vom Untergrund ablösen und wurden nach Resuspension in frischem Medium in
einem Verhältnis 1:4 bis 1:6 ausgesät.
2.4.2 Transfektion
Um ausreichende Mengen des Adenovirusplasmids in HEK 293 einzuschleusen,
wurde das Liposomenreagenz LipofectamineTM für die Transfektion verwendet. Es
bildet sich ein Liposomen/DNA-Komplex, der mit der Plasmamembran fusioniert und
die DNA ins Cytoplasma transferiert.

Materialien und Methoden
37
Für die Transfektion wurden in 25 cm ² Flaschen kultivierte HEK 293-Zellen mit
einer Konfluenz von 80% verwendet. Zu einem vorbereiteten Ansatz aus 5 µg zu
transfizierender DNA und 500µl OptiMEM wurden 20 µl LipofectamineTM gegeben
und 30 min bei Raumtemperatur inkubiert. Währenddessen wurden die Zellen mit
PBS gewaschen und mit 2,5 ml OptiMem versorgt. Nach 30 min wurde der
LipofectamineTM/OptiMem-Mix für 5 h auf die Zellen gegeben.
Unter dem Fluoreszenzmikroskop ließ sich der Transfektionserfolg beobachten,
der an der GFP-Expression und den abgerundeten lytischen Zellen (zytopathischer
Effekt) erkennbar war. Nach ca. 5-7 Tagen konnte die erste Virusisolation erfolgen.
2.4.3 Virusisolierung
Nach etwa sieben Tagen konnte der primäre Virusstock aus den transfizierten Zellen
isoliert werden. Der Zellrasen wurde durch Auf- und Abpipettieren des Mediums
resuspendiert, 10 min bei 500 g zentrifugiert und das Zellpellet in PBS
aufgenommen. Zur vollständigen Zelllyse wurde die Suspension in flüssigem
Stickstoff tiefgefroren, sofort wieder aufgetaut und kräftig auf dem Tischrüttler
durchmischt (freeze/thaw). Um eine möglichst hohe Ausbeute an infektiösen
Partikeln zu erhalten, wurde dieser Vorgang dreimal wiederholt. Die Suspension
wurde zentrifugert (700 g, 20 min, 40C), der Überstand in ein neues Gefäß überführt
und für die Transduktion weiterer Zellen verwendet.
2.4.4 Vermehrung und Präparation von Adenoviren
Für die Gewinnung größerer Virusmengen wurden HEK 293-Zellen bis zu einer
Konfluenz von 90% kultiviert. Vor der Transduktion wurde das zur Kultivierung
verwendete Medium (DMEM, 10% FCS, 1% Penicillin/Streptomycin) gegen Medium
mit einem reduzierten Serumgehalt (DMEM, 2% FCS, 1% Penicillin/Streptomycin)
ersetzt, da hierdurch die Transduktionseffizienz gesteigert wird. Der Zellüberstand
einer Virusisolierung wurde auf den Zellrasen der Kulturflasche gegeben. Der
Infektionserfolg konnte mikroskopisch bei GFP-exprimierenden Viren unter UV-Licht,

Materialien und Methoden
38
vor allem aber über die Entwicklung eines zytopathogenen Effekts (CPE) verfolgt
werden. Dieser zeigte sich nach etwa 48 h – erkennbar an den abgerundeten Zellen,
die sich großflächig von der Zellkulturflaschenoberfläche lösten. Nach der Ernte der
Zellen fand die Virusisolation (vgl. Kap. 2.4.3) statt. Anschließend konnten die Viren
in größerem Maßstab präpariert werden.
Die Anreicherung und Reinigung der Viruspräparation erfolgt über
Dichtegradienten-Zentrifugation. Hierzu wurde zunächst ein Cäsiumchlorid-
Stufengradient aus jeweils 3,5 ml CsCl2 mit den Dichten 1,4 g/ml und 1,2 g/ml
erstellt. Das Virusisoltat von 20-30 transduzierten Zellkulturflaschen (75 cm²) wurde
in einem Volumen von 6 ml auf den Gradienten geschichtet. Die
Dichtegradientenzentrifugation erfolgte mit 27.000 rpm (Beckman-Ultrazentrifuge,
SW-28 Rotor) über 4 h und bei 4°C. Durch seitliches Einstechen in das
Zentrifugenröhrchen mit einer Kanüle (Durchmesser 0,8 mm) wurde die als trüber
Ring sichtbare Virusbande aus dem Gradienten abgezogen. Um eine größere
Reinheit des Virusmaterials zu erhalten, wurde die Suspension mit PBS auf 6 ml
aufgefüllt, erneut auf einen CsCl2–Gradienten geladen und für weitere 2 h
zentrifugiert. Die entstandene Virusbande wurde in einem möglichst kleinen Volumen
abgezogen, mit 1 Vol. 2 x Lagerungspuffer versetzt und bei -20°C gelagert.
2.4.5 Titerbestimmung
Eine Titerbestimmung mit dem Immunoassay Adeno-X™ Rapid Titer Kit (BD
Biosciences) wurde nach Herstellerangaben durchgeführt, um Aussagen über die
Infektiösität der Viruspräparation zu erhalten. Mit Hilfe eines spezifischen
Primärantikörpers gegen das Protein Hexon und einem Peroxidase-konjugierten
Sekundärantikörper wurden adenoviral infizierte Zellen markiert. Somit konnte
berechnet werden, wie viele infektiöse Einheiten pro Milliliter Viruspräparation
enthalten waren (infectious units per milliliter; ifu/ml). Daraus wurde die MOI
(multiplicity of infection) berechnet, die im statistischen Mittel die vorhandene Anzahl
der infektiösen Partikel pro Zielzelle angibt.

Materialien und Methoden
39
2.4.6 Transduktion von Primärzellen
Humane intestinale Darmastzellen wurden in Mastzell-Kulturmedium mit reduzierter
FCS-Konzentration aufgenommen. Der auf 2% gesenkte FCS-Gehalt steigerte die
Infektionseffizienz. Da sich die Mastzellen schlecht transduzieren ließen, wurde das
CAR-PDT-Fusionprotein CARex-VP22 (coxsackievirus adenovirus receptor; protein
transduction domains) in einer Konzentration von etwa 400 ng/ml hinzugefügt, das
als eine Art Adaptor zwischen Mastzellen und Viren fungierte (feundlich überlassen
von Dr. F. Kühnel, MHH).
Nach einer Inkubation von 4 h wurde SCF (50ng/ml) hinzugefügt und 24 h später
wurden die Zellen in Kulturmedium mit 10 % FCS aufgenommen. Die Ernte mit
anschließenden Stimulationsexperimenten erfolgte ca. 48 h nach Infektion als etwa
50-60 % der Matzellen infiziert waren (s. Kapitel 2.5.5. Mastzellaktivierung).
2.5 Zellbiologische Methoden
2.5.1 Isolation von Mastzellen aus menschlichem Darmgewebe
Alle Untersuchungen an menschlichem Darmgewebe wurden durch die
Ethikkommission der Medizinischen Hochschule genehmigt (Nr. 2880). Die Isolation
von Mastzellen aus menschlichem Darmgewebe war durch eine Zusammenarbeit mit
der Abteilung für Viszeral- und Transplantationschirurgie der Medizinischen
Hochschule Hannover möglich. Es erfolgte eine zeitnahe Entnahme von
makroskopisch unauffälligen, tumorfreien Proben (10-50 cm² Schleimhaut) aus
chirurgischen Resektaten, die hauptsächlich aufgrund von Tumoren im Dünn- oder
Dickdarm, aber auch anderer Erkrankungen, wie M.Crohn, Divertikulose oder
Pankreas-Karzinom, operativ entfernt werden mussten. Für die Weiterverarbeitung
wurde die Probe sofort in TEA-Puffer bei 40 C über Nacht gelagert. Die weiteren
Schritte der Zellisolation erfolgten am Folgetag in einer Laminair-Werkbank und
einem 370 C Wasserschüttelbad:

Materialien und Methoden
40
1. Zuerst wurden Mukosa und Submukosa mit Hilfe einer Schere von der
Muskularis abpräpariert.
2. Der aus den Becherzellen freigesetzte Schleim wurde durch eine Inkubation
für 20 min in 50 ml ACC-Lösung und die Epithelzellen durch eine 30 min
Inkubation in Tyrode-Puffer mit 5mM EDTA bei 370C in einem
Wasserschüttelbad entfernt.
3. In einer Petrischale wurde das Gewebe unter Zusatz einer Enzymlösung
(PCh-Lösung: Pronase/Chymopapain) mit einer Schere zu einer Suspension
von ca. 1 mm² großen Gewebestücken zerkleinert und durch einen 250 µm
Nybold-Filter unter Spülung mit TE-Puffer filtriert. Anschließend inkubierte die
Gewebesuspension für 30 min in 25 ml PCh-Lösung.
4. Nach erneuter Filtration wurde das Filtrat wiederum verworfen und das
Gewebe inkubierte für 30 min in Collagenase-Lösung. Das nach dem Spülen
mit TGMD-Puffer gewonnene Filtrat wurde abzentrifugiert (300g 10 min,
20°C) und das Zellpellet in Kulturmedium aufgenomme n. Es erfolgte eine
Wiederholung dieses Schrittes mit dem auf dem Filter verbleibenden Gewebe.
5. Beide Filtrate wurden vereinigt und durch einen 100 µm Nybold-Filter gegeben
(Spülen mit 50ml Medium)
6. Die Zellen wurden wieder zentrifugiert (300 g, 10 min, 20°C), das Pellet in 20
ml Kulturmedium aufgenommen, gezählt (Trypanblau-Methode und Neubauer-
Zählkammer) und dann differenziert (DiffQuick-gefärbte Zytospins).
Die gewonnene Einzelzellsuspension ( 17±8*106 Zellen pro Gramm Mucosa) wurde
in einer Dichte von 2,5-4*106 Zellen/ml über Nacht in Kulturmedium ohne Zytokin-
Zusatz bei 370C kultiviert und am Folgetag der Mastzell-Aufreinigung mittels
magnetischer Zellseparation (Magnetic Cell Sorting, MACS) zugeführt.
2.5.2 Mastzellanreicherung durch MACS (Magnetic beads-activated cell sorting)
Aus der Darmzellsuspension wurden die Mastzellen nach dem Prinzip der
Positivselektion unter Verwendung des SCF-Rezeptors c-kit aufgereinigt. Die
Magnetseparation ermöglicht es, aus der heterogenen Zellsuspension einen

Materialien und Methoden
41
speziellen Zelltyp zu isolieren. Durch monoklonale Antikörper gegen zelltypspezifisch
exprimierte Oberflächenantigene kann die Zelle markiert werden. An den
Primärantikörper erfolgt die Bindung eines mit superparamagnetischen
Eisenkomplexen gekoppelten Sekundärantikörpers. Wird die mit den Antikörpern
inkubierte Zellsuspension durch eine sich in einem Magnetfeld befindliche Säule
gegeben, so werden die antikörpermarkierten Zellen im Magnetfeld zurückgehalten
(Positivfraktion), während mangels der zelltypspezifischen Oberflächenantigene nicht
markierte Zellen das Magnetfeld durch die Säule passieren können (Negativfraktion).
Nach Entfernung der Säule aus dem Magnetfeld kann die mastzellenthaltende
Positivfraktion durch Spülen gewonnen werden.
1. Die über Nacht kultivierte Darmzellsuspension wurde abzentrifugiert (10 min
bei 300 g), in HA-Puffer aufgenommen (1 x 108 Zellen/250µl) und mit dem
Primärantikörper gegen c-kit (YB5.B8; Maus IgG1; 5 ng/ml) für 15 min unter
leichtem Schütteln bei 40 C inkubiert.
2. Die Zellsuspension in 50 ml HA-EDTA-Puffer aufgenommen, abzentrifugiert
und das Pellet in HA-EDTA-Puffer (1 x 108 Zellen/250µl) resuspendiert.
3. Nach Zugabe eines mit magnetischen Beads gekoppelten
Sekundärantikörpers (Ziege-anti-Maus IgG, 50µl Antikörperlösung/ 250 µl
Zellsuspension) inkubierte die Zellsuspension 15 min unter leichtem
Schütteln bei 40 C, wurde danach in 50 ml HA-EDTA-Puffer aufgenommen
und abzentrifugiert.
4. Das Pellet wurde in 50 ml HA-EDTA-Puffer resuspendiert, über einen 30 µl
Nybold-Filter gegeben, erneut zentrifugiert und das Pellet in 1 x 108 Zellen/15
ml aufgenommen.
5. Nach Spülen einer Macs-Säule mit HA-EDTA-Puffer in einem Magnetfeld
(MidiMacs Seperator) wurde die Zellsuspension über die Säule gegeben und
anschließend die Negativfraktion herausgespült. Außerhalb des
Magnetfeldes wurde die Säule noch mal gespült und die MC-enthaltende
Positivfraktion (60-95% Mastzellen) unter den im Folgenden beschriebenen
Bedingungen kultiviert.

Materialien und Methoden
42
2.5.3 Kultur menschlicher Darmmastzellen
Die Mastzellen wurden in RPMI-Medium+Glutamax ( mit Zusatz von 10% FCS,
100µg/ml Gentamycin, 100µg/ml Streptamycin, 100U/l Penicillin und 0,5µg/ml
Amphotericin) mit Zusatz von SCF (50 ng/ml) bei 370 C, 5% CO2 und max.
Luftfeuchtigkeit kultiviert. Bei ungereinigten Zellkuturen (ohne MACS) betrug die
Zelldichte 1-4 x 106 Zellen/ml, bei aufgereinigten Kulturen 0,1- 0,4 x 106 Zellen/ml.
Wöchentlicher Wechsel des Mediums erfolgte unter Zugabe von SCF. Nach 21
Tagen Kultur betrug die Reinheit der Mastzellen 98 ± 2 %.
2.5.4 Zellzählung und Differenzierung
Es wurden 20 µl Zellsuspension abgenommen, mit 20 µl Trypan-Blau gefärbt und in
eine Neubauer- oder Fuchs-Rosenthal-Zählkammer überführt. Tote Zellen färbten
sich blau und erschienen dunkel. Mindestens 100 lebendige Zellen wurden gezählt,
die keine Farbe aufnahmen und hell leuchten. Ein Zählquadrat entspricht einem
Volumen von 0,1 µl (Neubauer) bzw. 0,2 µl (Fuchs-Rosenthal). Dadurch kann die
Gesamtzellzahl bestimmt werden.
Zur Differenzierung der Zellen wurden Zytospins aus 80 – 200 µl der
Zellsuspension mit einer Zytozentrifuge (Shandon, Pitsburg, PA, 500 rpm für 3 Min)
hergestellt. Danach folgte eine Färbung der Zytospins mit DiffQuick oder mit May-
Grünwald /Giemsa nach Pappenheim sowie eine differenzierte Auszählung von 200 -
500 Zellen. Aufgrund ihrer metachromischen Granula konnten die Mastzellen leicht
erkannt werden.
2.5.5 Mastzellaktivierung/ Mediatormessung
Nach mindestens 14-tägiger Kultur in Gegenwart von SCF wurden die Mastzellen
gewaschen, gezählt und in einer Dichte von 105MC/ml in Kulturmedium
aufgenommen. Es folgte eine Inkubation mit verschiedenen Substanzen
(Leichtketten der Botulinum Neurotoxine, rekombinierte Adenoviren, Antikörper

Materialien und Methoden
43
gegen SNAREs: Vamp2, Vamp7), welche die Mediatorausschüttung der Mastzellen
verringern sollten. Anschließend wurden die Mastzellen mit Hilfe des gegen die α-
Kette des IgE-Rezeptors gerichteten Antikörpers 22E7 (100 ng/ml) aktiviert, der die
IgE-Rezeptoren auf der Mastzelloberfläche kreuzvernetzt. Der PKC-Aktivator PMA
(Phorbol-Myristat-Acetat) und das Calciumionophor Ionomycin wurden als weitere
Mastzellaktivatoren angewendet. Als Messgröße für die Mediatorausschüttung diente
Histamin. Nach 60 min Inkubation mit den aktivierenden Substanzen im
Wasserschüttelbad bei 370 C wurden die MC abzentrifugiert (5 min, 350 g) und die
Überstände bis zur Histaminmessung bei –200 C gelagert. Zusätzlich wurde pro
Bedingung die basale, ohne Stimulation stattfindende Histaminfreisetzung
(Überstände mit PBS behandelter Mastzellen) und das intrazelluär gespeicherte,
präformierte Histamin bestimmt. Der Gehalt des Mastzell-Mediators Histamin in den
Überständen wurde in Relation zu dem präformierten Histamin (Mastzell-Lysate,
Aufnahme in Wasser 1:1, 2 Einfrier-Auftau-Zyklen) gesetzt. Die Histaminmessung
erfolgte mit einem kommerziellen Radioimmunoassay (RIA, Coulter-Immunotech,
Krefeld). Zur Darstellung der Histaminfreisetzung wurde jeweils die spezifische
Freisetzung (Subtraktion der basalen von der induzierten Freisetzung) angegeben.
2.5.6 Permeabilisierung mit Streptolysin O (SLO)
Zur Permeabilisierung der MC wurde Streptolysin O, ein Toxin ß-hämolysierender
Streptokokken, verwendet. Mit Hilfe von Cholesterol bindet Streptolysin O an die
Plasmamembran, wodurch es über die Bildung von Oligomeren zu über 12nm
großen Poren kommt. Durch das Streptolysin O werden die Zellen durchlässig für
Ionen und auch größere Moleküle wie Immunglobuline bzw. Antikörper [134].
Zur Permeabilisierung inkubierten die Mastzellen (1 x 105 MC/ml) 15 min bei 370C in
calciumfreier HBSS-Lösung, der 30 mM Hepes ph 7,2, SLO (10 µg/ml) und die
entsprechenden Antikörper zugefügt wurden (anti-VAMP-7, anti-VAMP-2; beide
Antikörper wurden 1:250 verdünnt). Zum Verschluss der Poren wurden die
Mastzellen nach Zugabe von calciumhaltigen RPMI-Medium für 60 min auf Eis
gestellt. Anschließend wurden die Zellen zentrifugiert ( 300 g, 5 min), resuspendiert
und für 60 min mit 22E7 stimuliert (s. Kapitel 2.5.5. Mastzellaktivierung).

Materialien und Methoden
44
Durch Anfärbung mit Trypanblau wurde die der Permeabilisierungserfolg (etwa
60 - 80 %) durch Auszählen der dunkler gefärbten Zellen lichtmikroskopisch
überprüft.
2.5.7 Konfokale Immunfluoreszenz-Mikrosopie
Zur Intrazellulärfärbung wurden je 1 x 105 Mastzellen in Kulturmedium aufgenommen
und für 60 min mit PMA/Ionomycin (10-6 M) stimuliert. Nach der Fixierung für 30 min
bei 40 C in 2%-iger Paraformaldehydlösung wurden die Zellen in 10 ml PBS
gewaschen, abzentrifugiert und für 10 min bei Raumtemperatur in Blockingreagenz
mit 0,3% Saponin aufgenommen. Saponin permeabilisierte die Zellen und das
Blockingreagenz (PBS + goat-serum + swine-serum + human IgG, je 1:100)
verhinderte unspezifische Bindungen des Primärantikörpers. Es folgte eine 30 min
Inkubation mit je zwei Primärantikörpern verschiedener Spezies (mouse antibody
anti-VAMP-7 (1:250)/ rabbit antibody anti-SNAP-23 (1:200); mouse antibody anti-
Syntaxin-4 (1:50)/ rabbit antibody anti-VAMP-3 (1:100)) und eine erneute
Zentrifugation. Danach inkubierten die MC in Blockingreagenz mit 0,3% Saponin und
den entsprechenden Sekundärantikörpern (Alexa Fluor® goat anti-mouse 488/ Alexa
Fluor® donkey anti-rabbit 594; je 1:200 verdünnt) abgedunkelt für 30 min. Nach
erneutem Waschen wurden die Zellen in 10 µl PBS aufgenommen, auf einen
Objektträger gegeben und nach Absinken der Mastzellen mit Hilfe eines
Konfokalmikroskops betrachtet. Für die Doppelfärbung VAMP-7/ Syntaxin-3
(1:250/1:250) erfolgte eine zusätzliche Kernfärbung mit To-Pro 3 (Molecular Probes)
zur besseren Lokalisierbarkeit der SNARE-Proteine. Für die Kernfärbung inkubierten
die Zellen für 3 min in To-Pro-3 (1:1000 in PBS) und wurden anschließend 10 min in
PBS gewaschen. Die Konfokalmikroskopie wurde mit Hilfe von Herrn Tibor Veres im
Frauenhofer-Institut für Toxikologie und experimentelle Medizin, Abteilung
Allergologie und Immunologie durchgeführt.

Materialien und Methoden
45
2.5.8 Patch-Clamp-Messungen
Die Patch-Clamp-Technik ermöglicht es, die Membranströme von Zellen in vivo zu
messen.
Zunächst wurde eine Suspension aus humanen Mastzellen auf eine mit
Fibronectin (BD Biosciences) gecoatete Petrischale gebracht. Nach einer
einstündigen Inkubation mit SCF (100 ng/ml) adhärierten die Mastzellen auf der
Oberfläche [8]. Anschließend wurde eine dünn ausgezogene Glaskapillare auf eine
intakte Zelle gedrückt. Damit bei der hier angewendeten cell-attached-Konfiguration
die Zellmembran fest an der Glaswand der Pipette anlag und somit ein kleiner
Abschnitt der Zellmembran (Patch) elektrisch von seiner Umgebung isoliert war,
wurde durch leichtes Ansaugen ein Unterdruck ausgeübt. Die Isotypen der Botulinum
Neurotoxin-Leichtketten sollten nun ihre proteolytische Wirkung in der Zelle entfalten
(mind. 5 – 10 min), um anschließend die Degranulation bzw. die
Degranulationshemmung zu messen (mind. 10-15 min). Jedoch erwiesen sich die
menschlichen Mastzellen als zu instabil. Die Zellintegrität konnte nicht bis zum Ende
der Messungen aufrechterhalten werden. Die Patch-Clamp-Messungen fanden in
Zusammenarbeit mit F. Wegner von der Neurologischen Klinik der Universität
Leipzig statt.
2.6 Statistik
Die Daten des Ergebnisteils wurden als Mittelwert ± Standardabweichung (SD)
dargestellt. Signifikante Differenzen wurden mittels des one tailed t-Tests bestimmt.
Eine Irrtumswahrscheinlichkeit von P < 0,05 wurde als signifikant bewertet.

Ergebnisse
46
3. Ergebnisse
3.1 Nachweis von SNARE-Proteinen in menschlichen Darm-
Mastzellen
3.1.1 SNARE-mRNA Nachweis mittels RT-PCR
Als Vermittler der Exozytose in Neuronen, aber auch in verschiedenen sekretorisch
aktiven Zellen, ist eine Gruppe hochkonservierter Proteine, die sogenannten SNARE-
Proteine, beschrieben worden. Bisherige Untersuchungen in Zellen des
Immunsystems zeigten, abhängig von der Zellart, unterschiedliche
Expressionsmuster der SNARE-Isoformen. Die Kenntnisse über das SNARE-
Vorkommen in Mastzellen beruhen hauptsächlich auf der Rattenmastzelllinie RBL-
2H3. Da sich im Nagersystem gewonnene Daten nicht ohne Weiteres auf das
menschliche System übertragen lassen, erfolgte eine Untersuchung verschiedener
Vertreter der VAMP-, Syntaxin- und SNAP-Familien in humanen Mastzellen. Dazu
wurden menschliche Darm-Mastzellen (Abb.2A) mit der humanen Neuroblastom-
Zelllinie MHH-NB-11 (Abb.2B), als Kontrolle für neuronale SNARE-Proteine
verglichen. Weitere Analysen erfolgten an in vitro entwickelten Mastzellen aus
Nabelschnurblut (CBMC) und der Mastzellinie HMC-1. Anhand von eosinophilen
Granulozyten, wurden die Mastzellen mit einem weiteren Vertreter sekretorisch
aktiver Immunzellen verglichen.
Die mRNA der SNARE-Isoformen SNAP-23, Syntaxin-2, -3, -4 und –5, sowie
VAMP-3, -5, -7 und –8 ließ sich in allen untersuchten Zellen zeigen. Dagegen konnte
SNAP-25, das als neuronale Isoform des ubiqutär nachgewiesenen SNAP-23
angesehen wird, nicht in Mastzellen dargestellt werden. Außerdem wurden neben
SNAP-25 auch Syntaxin-1A und Synatxin-1B nur in Neuronen detektiert. Für SNAP-
25 und Synatxin-1B zeigte sich eine zusätzliche, wenn auch schwächere Expression
in HMC-1. In primären humanen Mastzellen waren keine Signale für diese Gene
nachweisbar (Abb.2 C-F).

Ergebnisse
47
Abb.2: Darstellung der Expression von SNARE-mRNA in verschiedenen Mastzellkulturen und Neuronen mittles RT-PCR (C-F). Lichtmikroskopische Bilder humaner intestinaler Mastzellen (A) und der Neuronenzelllinie MHH-NB-11 (B). C GAPDH; D SNAP-23, -25; E Syntaxin-1A, -1B, -2, -3, -4, -6; F VAMP-2, -3, -5, -7, -8; L 1 kb plus ladder zur Fragmentgrößen-Bestimmung, Banden verlaufen im Abstand von 100 bp; 1 - 3 MC1 - MC3; 4 CBMC; 5 HMC-1; 6 eosinophile Granulozyten; 7 humane Neuroblastom-Zelllinie MHH-NB-11. Als Kontrolle wurde die Expression von GAPDH als „house-keeping gene“ dargestellt. Zusätzlich wurde MHH-NB-11 als Positivkontrolle verwendet.
L 1 2 3 4 5 6 7
GA
PD
H
C
SN
AP
-25
SN
AP
-23
D L 1 2 3 4 5 6 7
Stx
-1B
Stx
-2
Stx
-3S
tx-4
Stx
-6
Stx
-1A
E L 1 2 3 4 5 6 7 L 1 2 3 4 5 6 7
VA
MP
-2
L 1 2 3 4 5 6 7
VA
MP
-5
VA
MP
-7
VA
MP
-3
VA
MP
-8
L 1 2 3 4 5 6 7 F
A
B

Ergebnisse
48
3.1.2 SNARE-Protein Nachweis mittels Western Blot
Die Analyse der SNARE-Expression auf Protein-Ebene erfolgte mittels Western Blot.
Für die Western Blots wurden Lysate aus humanen intestinalen Mastzellen (5 x105
Zellen) verwendet. Als Kontrolle diente dabei eine Präparation aus dem
Hirnhomogenat einer Maus. In humanen Mastzellen konnten die t-SNAREs SNAP-
23, Syntaxin-2, Syntaxin-3, Syntaxin-4 und die v-SNAREs VAMP-3, VAMP-7, VAMP-
8 nachgewiesen werden. SNAP-25 und Syntaxin-1A waren in Mastzellen nicht
vorhanden, jedoch gelang in der Postivkontrolle der Nachweis für diese neuronalen
Isoformen (Abb.3). Die mittels Western Blot gewonnenen Daten bestätigten auf
Protein-Ebene die Ergebnisse aus der RT-PCR.
Unterschiede fanden sich dagegen für die SNARE-Proteine VAMP-2 und
Syntaxin-1B. Während VAMP-2 auf mRNA-Ebene nocht starke Signale aufwies, war
es im Western Blot mittels eines monoklonalen Antikörpers nicht darstellbar. Erst bei
Verwendung eines polyklonalen Antikörpers ließ sich eine, wenn auch schwache,
Expression für VAMP-2 nachweisen. Die Positivkontrolle zeigte in beiden Fällen eine
starke Bande. Während Syntaxin-1B in der RT-PCR nicht detektiert wurde, konnte
man auf Protein-Ebene schwache Signale in Mastzellen sehen.
Abb. 3: Verschiedene an der Vesikel- (v-SNAREs) oder Plasmamembran (t-SNAREs) lokalisierte SNARE-Proteine konnten in humanen Darm-Mastzellen nachgewiesen werden. (VAMP-2*: monoklonaler Antikörper; VAMP-2**: polyklonaler Antikörper)
SNAP-23 SNAP-25 Syntaxin-1A Syntaxin-2 Syntaxin-3 Syntaxin-4
Kontrolle
(Maushirn-Extrakt)
Humane
Darm-Mastzellen
VAMP-2* VAMP-2** VAMP-3 VAMP-7 VAMP-8
Kontrolle
(Maushirn-Extrakt)
Humane
Darm-Mastzellen
t-SN
AR
Es
v-S
NA
RE
s
Syntaxin-1B

Ergebnisse
49
3.2 Untersuchung des Einflusses von Botulinum Neurotoxin auf die
Mastzelldegranulation
3.2.1 Herstellung adenoviraler Konstrukte
Durch Einschleusung von Tetanus-Toxin in eosinophile Granulozyten konnte die
Bedeutung des SNARE-Proteins VAMP-2 für die Exozytose gezeigt werden [135].
VAMP-2 dient sowohl Tetanus als auch Botulinum Neurotoxin Typ B als Substrat und
nach Proteolyse zeigte sich eine Exozytosehemmung in eosinophilen Granulozyten.
Mehrere Ansätze zur Einschleusung der proteolytisch wirksamen Leichketten von
Botulinum Neurotoxin erwiesen sich als nicht praktikabel, da nicht genug Mastzellen
überlebten, um anschließende Stimulationsexperimente durchzuführen. Dazu
gehörten Gangliosidbehandlung, Elektroporation und Patch Clamp-Technik. Ein
weiterer Ansatz bestand in der Verwendung adenoviraler Vektoren. Nachdem in
Vorarbeiten durch Lorentz et al. (unpublizierte Daten) die Infektion von Mastzellen mit
Adenoviren erfolgreich durchgeführt werden konnte, wurden zur Analyse der
Degranulationshemmung durch SNARE-spaltende Neurotoxine adenovirale
Konstrukte verwendet. Im Rahmen dieser Arbeit wurde die Aktivität der proteolytisch
aktiven, leichten Ketten von Botulinum-Neurotoxin Typ A, B und C1 untersucht. Dazu
kamen Adenoviren, welche die gewünschten Gene enthielten, sowie ein Kontrollvirus
ohne zusätzliche genetische Information zum Einsatz.
Zur Herstellung der verwendeten Plasmide pBoNT-A, pBoNT-B, pBoNT-C1
wurden die leichten Ketten der Botulinum-Neurotoxine Typ A, B und C1 als N-
terminale Fusionspartner von eGFP in die Multiple Cloning Site des pShuttle-CMV-
Vektors kloniert (Schema in Abb.4). Zunächst wurde das Gen für eGFP über XhoI /
HindIII eingefügt, im zweiten Schritt dann die Gene für die jeweiligen leichten Ketten
über Sal I / Stu I, wobei letztere Restriktionsschnittstelle durch die Klonierung von
eGFP eingebracht wurde. Anschließend erfolgte eine Umklonierung der drei
Konstrukte in den bakteriellen Expressionsvektor über NcoI / MluI. Die Kontrolle des
Klonierungserfolges durch Überprüfen der Größe der produzierten Proteine und der
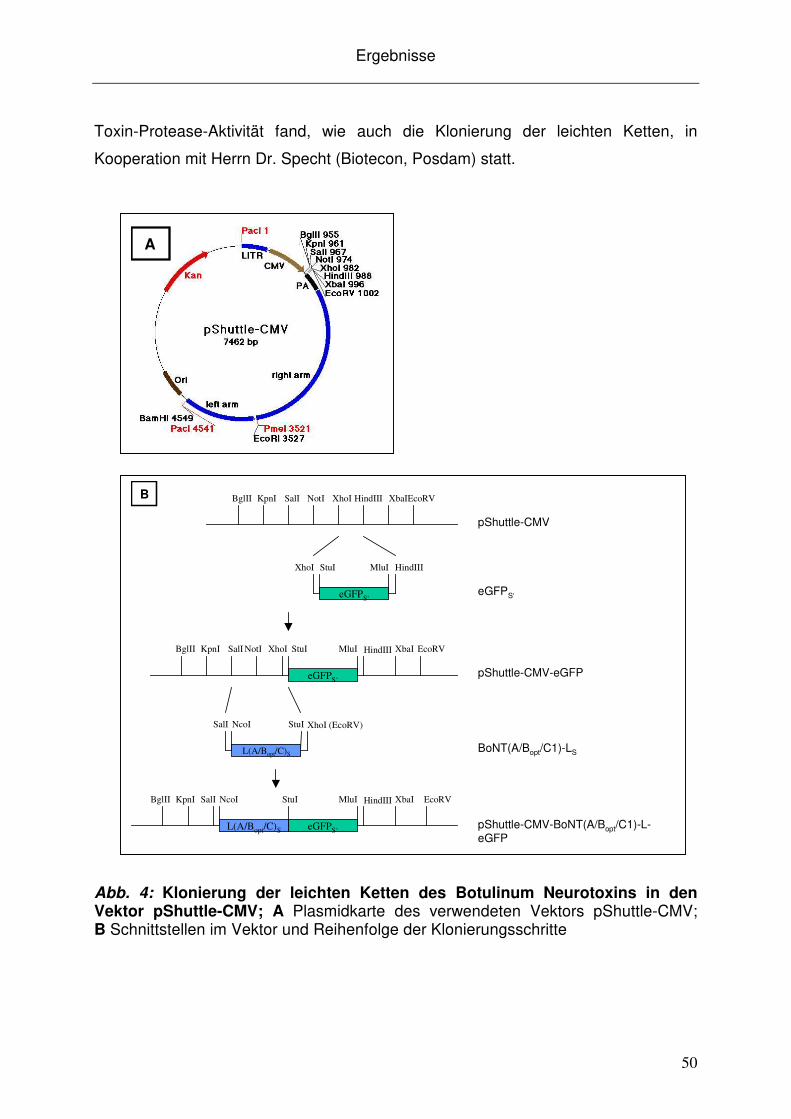
Ergebnisse
50
Toxin-Protease-Aktivität fand, wie auch die Klonierung der leichten Ketten, in
Kooperation mit Herrn Dr. Specht (Biotecon, Posdam) statt.
Abb. 4: Klonierung der leichten Ketten des Botulinum Neurotoxins in den Vektor pShuttle-CMV; A Plasmidkarte des verwendeten Vektors pShuttle-CMV; B Schnittstellen im Vektor und Reihenfolge der Klonierungsschritte
HindIII XbaISalI NotI XhoIKpnI
pShuttle-CMV
eGFPS‘
StuI HindIII
BglII
eGFPS‘
pShuttle-CMV-eGFP
XhoI (EcoRV)StuINcoISalI
BoNT(A/Bopt/C1)-LS
EcoRV
XhoI MluI
XbaISalI NotIKpnIBglII EcoRV
eGFPS‘
StuI HindIIIXhoI MluI
L(A/Bopt/C)S
XbaIKpnIBglII EcoRV
eGFPS‘
HindIIIMluIStuINcoISalI
L(A/Bopt/C)SpShuttle-CMV-BoNT(A/Bopt/C1)-L-eGFP
B
A
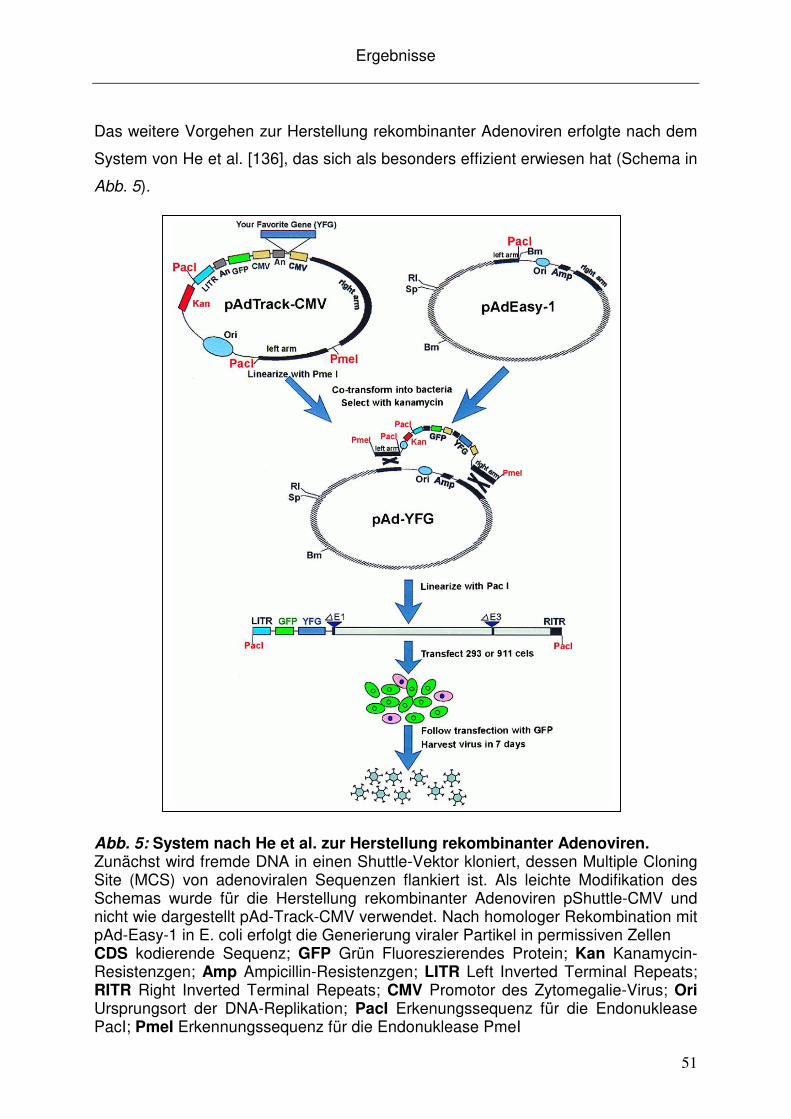
Ergebnisse
51
Das weitere Vorgehen zur Herstellung rekombinanter Adenoviren erfolgte nach dem
System von He et al. [136], das sich als besonders effizient erwiesen hat (Schema in
Abb. 5).
Abb. 5: System nach He et al. zur Herstellung rekombinanter Adenoviren. Zunächst wird fremde DNA in einen Shuttle-Vektor kloniert, dessen Multiple Cloning Site (MCS) von adenoviralen Sequenzen flankiert ist. Als leichte Modifikation des Schemas wurde für die Herstellung rekombinanter Adenoviren pShuttle-CMV und nicht wie dargestellt pAd-Track-CMV verwendet. Nach homologer Rekombination mit pAd-Easy-1 in E. coli erfolgt die Generierung viraler Partikel in permissiven Zellen
CDS kodierende Sequenz; GFP Grün Fluoreszierendes Protein; Kan Kanamycin-Resistenzgen; Amp Ampicillin-Resistenzgen; LITR Left Inverted Terminal Repeats; RITR Right Inverted Terminal Repeats; CMV Promotor des Zytomegalie-Virus; Ori Ursprungsort der DNA-Replikation; PacI Erkenungssequenz für die Endonuklease PacI; PmeI Erkennungssequenz für die Endonuklease PmeI

Ergebnisse
52
Unter Nutzung einer homologen Rekombination konnten adenovirale Konstrukte in
Bakterien kloniert werden, da sowohl das verwendete Shuttle Plasmid als auch das
Backbone Plasmid zwei homologe Bereiche enthielten. Zunächst mussten die
Plasmide pBoNT-A, pBoNT-B und pBoNT-C1 durch den Verdau mit der
Restriktionsendonuklease PmeI linearisiert werden. Die linearisierten adenoviralen
Plasmide wurden durch Phenol-Chloroform-Extraktion mit anschließender
Ethanolfällung aufgereinigt, konzentriert und durch Elektroporation in E. coli der
Zelllinie BJ 5183-Ad1, in denen der adenovirale Backbone Vektor bereits vorlag,
transformiert. Nach einer DNA-Minipräparation konnten die gewünschten Klone
durch Restriktionsverdau mit PacI identifiziert und anschließend in den chemisch
kompetenten E. coli –Stamm XL10-Gold transformiert werden, um größere Mengen
an Plasmid zu erhalten. Nach einer endotoxinfreien Maxi-Plasmidpräparation wurde
durch PCR das Vorliegen des klonierten Gens überprüft (Abb.6).
Abb. 6: PCR zur Überprüfung der klonierten Gene; A BoNT-A; B BoNT-B; C BoNT-C (A-C: die PCR wurde mit den entsprechenden Primern durchgeführt); D-F GFP (in der Reihenfolge BoNT-A, BoNT-B, BoNT-C aufgetragen)
3.2.2 Virusgenerierung
Zur Herstellung infektiöser Viren aus Adenovirusplasmiden wurden Viren des
humanen Serotyps 5 verwendet, die aufgrund einer Deletion der E1- und E3-Region
nur in bestimmten Zelllinien, wie HEK-293, vermehrt werden können. Diese Zellen
transkomplementieren den viralen Defekt, da sie in ihrem Genom die stabil integrierte
E1-Region enthalten, womit eine Virusvermehrung ermöglicht wird. Zunächst erfolgte
eine Linearisierung der Adenovirusplasmide mit der Restriktionsendonuklease PacI,
A B C D E F

Ergebnisse
53
um den für die Herstellung infektiöser Viren notwendigen rechten und linken ITR
(Inverted Terminal Repeat) freizulegen. Dabei wurden gleichzeitig für die stabile
Replikation in E. coli notwendige DNA-Abschnitte entfernt. Nach Aufreinigung und
Konzentration durch Phenol-Chloroform-Extraktion mit Ethanolfällung wurden HEK
293-Zellen mit rekombiniertem Ad-Easy-1 transfiziert, um den primären Virusstock zu
erstellen. Nach siebentägiger Inkubation fanden die ersten Virusisolationen mit
anschließender Vermehrung und Präparation der Viren statt (vgl. Kap. 2.4.4).
Nicht nur mit Hilfe des zytopathischen Effekts, sondern auch durch die GFP-
Expression sollte die Transfektionseffizienz und später der Transduktionserfolg
abgeschätzt werden können. Es zeigten sich aber bereits während der Transduktion
von HEK 293-Zellen Unterschiede in der GFP-Expression. So wies das Kontrollvirus
AdGFP ein intensiveres GFP-Signal als die anderen rekombinierten Adenoviren auf.
Aber auch untereinander zeigten AdBoNT-A, AdBoNT-B und AdBoNT-C stark
unterschiedliche GFP-Signale (Abb. 7). Als Nachweis für die Replikationfähigkeit und
Infektiösität der Viren diente der bei allen Konstrukten etwa gleich starke
zytopathische Effekt. Er trat etwa zwei bis drei Tage nach Infektion auf - erkennbar
an der Abrundung und Ablösung der Zellen. Abschließend wurde eine
Titerbestimmung der präparierten Viren zur genauen Bestimmung der Infektiösität
durchgeführt. Die Titerbestimmung erfolgte über das Capsid-Protein Hexon, das nur
in infizierten, die E1-Deletion des Adenovirus transkomplementierenden Zellen, wie
HEK 293, produziert wird. In allen drei Fällen konnte ein Titer bestimmt werden.

Ergebnisse
54
Abb. 7: Darstellung der GFP-Expression der verwendeten Adenoviren in HEK
293-Zellen. Alle Bilder wurden mit den gleichen Lichtverhältnissen photographiert,
um die verschiedenen GFP-Signale vergleichen zu können; A AdGFP; B AdBoNT-A;
C AdBoNT-B; D AdBoNT-C;
* Auflicht
A
B
A*
B*
C C*
D D*

Ergebnisse
55
3.2.3 Transduktion der Mastzellen mit rekombinierten Adenoviren
Zur Einschätzung der Transduzierbarkeit von Mastzellen wurden verschiedene
Virustiter verwendet. Erst als etwa 500 infektiöse Viruspartikel pro Zelle eingesetzt
wurden (MOI 500), konnten 1-5 % der Mastzellen mit dem Kontrolvirus AdGFP
transduziert werden. Höhere Viruskonzentrationen erzielten kein wesentlich besseres
Ergebnis, da mit steigender MOI auch der zytopathische Effekt auf die Mastzellen
zunahm. So waren bei Verwendung höherer Titer etwa 70% der Mastzellen nach
72 h abgestorben, während weiterhin keine nennenswerte Infektion beobachtet
werden konnte. Der Transduktionserfolg wurde dabei mittels eines
Fluoreszenzmikroskops anhand der GFP-Expression von AdGFP abgeschätzt. Jede
GFP-produzierende Zelle konnte als erfolgreich infiziert gewertet werden. Aufgrund
der niedrigen Infektionsraten lag die Vermutung nahe, dass bestimmte Strukturen,
die den für die Virusaufnahme notwendigen initialen Kontakt zwischen Zielzelle und
Adenovirus vermitteln, auf Mastzellen nicht exprimiert werden. Das Fehlen dieser für
die Transduktion notwendigen Strukuren konnte durch ein CAR-PDT-Fusionprotein
(coxsackievirus adenovirus receptor; protein transduction domains) behoben werden
[137]. Bestimmte Proteine, wie VP22 aus dem Herpes simplex Virus, können über
rezeptorunabhängige Wege von Säugetierzellen aufgenommen werden.
Verantwortlich für diese Prozesse sind Protein Transduktions Domänen. So bindet
der CAR-Anteil des Fusionsprotein (CARex-VP22) an bestimme
Oberflächenstrukturen der Adenoviren, die sogenannten fiber knobs.
Währenddessen dockt der VP22-Anteil mit seiner PDT-Domäne an die Zielzellen an,
so dass es zu dem notwendigen initialen Kontakt zwischen Zelle und Virus kommen
kann. Die weiteren Schritte, die die Virusaufnahme zur Folge haben, werden durch
verschiedene Integrine vermittelt. Mit Hilfe des Fusionproteins CARex-VP22, das als
Adaptor zwischen den Mastzellen und dem Virus dient, konnte die Effizienz der
adenoviralen Infektion deutlich verbessert werden. Außerdem wurde der
zytopathische Effekt durch den Einsatz niedrigerer Viruskonzentrationen gesenkt.
Nach 48 h waren bei Verwendung einer MOI von 50 etwa 50-60 % der Mastzellen
infiziert. Zu diesem Zeitpunkt waren noch etwa 95 –100 % der eingesetzten Zellen
vital. Mit längeren Inkubationszeiten nahm der zytopathische Effekt der Adenoviren
auf die Mastzellen zu, so dass nach 72 h nur 70-80 % der Zellen überlebten,

Ergebnisse
56
während die Infektionsrate nicht wesentlich weiter stieg. Die GFP-Intensität der
konstruierten Adenoviren zeigte, wie bereits in den HEK 293-Zellen, starke
Unterschiede und war in den Mastzellen sogar heruntergesetzt (Abb.8). Während für
AdBoNT-A und AdBoNT-C, die auch in den HEK 293-Zellen schwache Signale
aufwiesen, in den Mastzellen kein GFP darstellbar war, wiesen AdBoNT-B-infizierte
Zellen nur sehr schwaches GFP auf. Jedoch wurden für alle Konstrukte gleiche
Viruskonzentrationen eingesetzt. Da die Transduktion über Oberflächenproteine von
Viren und Zellen vermittelt wird, die durch die eingefügten Gene nicht beeinflusst
werden, konnte für pBoNT-A, pBoNT-B und pBoNT-C1 von einer dem Kontrollvirus
entsprechenden Infektionsrate ausgegangen werden [137].
Abb. 8: Darstellung der unterschiedlichen GFP-Expression in infizierten
Mastzellen. Alle Bilder wurden mit der gleichen Beleuchtungsstärke photographiert,
um die verschiedenen GFP-Signale vergleichen zu können; A AdGFP; B AdBoNT-B;
In mit AdBoNT-A und AdBoNT-C transduzierten Zellen waren kaum GFP-Signale
detektierbar
* Auflicht
A A*
B B*

Ergebnisse
57
3.2.4 Untersuchung der Exozytose adenoviral transduzierter Mastzellen
Da der Transduktionserfolg unter dem Fluoreszenzmikroskop anhand der GFP-
Signale des Kontrollvirus abgeschätzt werden konnte, erfolgte die Ernte nach etwa
zwei Tagen, als 50-60 % der AdGFP-behandelten Mastzellen mit Hilfe des
Fusionsproteins CARex-VP22 infiziert waren. Zu diesem Zeitpunkt sollten auch die
leichten Ketten der Botulinum-Neurotoxine, deren cDNA in die Adenoviren kloniert
wurde, in den Zellen exprimiert werden. Nach zweimaligem Waschen erfolgte eine
Aktivierung der Zellen. Dazu wurden die Zellen sowohl mit dem gegen die α-Kette
des IgE-Rezeptors gerichteten Antikörper 22E7, als auch mit PMA und Ionomycin für
1 h inkubiert. Nach PMA/Ionomycin-Stimulierung konnte eine
Degranulationshemmung für BoNT-B und BoNT-C festgestellt werden, wobei die
spezifische Histaminfreisetzung mit etwa 50 % insgesamt auf einem hohen Niveau
lag. Zur besseren Anschaulichkeit wurde die Kontrollbedingung, die nur mit dem
Fusionsprotein CARex-VP22 inkubierte Zellen enthielt, als 100 % gesetzt (Abb.9A).
Mit dem Adenovirus AdBoNT-B infizierte Mastzellen zeigten im Vergleich zu AdGFP-
transduzierten Zellen einen um 24,6 ± 23,0 % gesenkten Histaminrelease. Der
hemmende Effekt des Neurotoxins auf die Degranulation war bei BoNT-C
exprimierenden Zellen noch stärker ausgeprägt. Hier wurden nach Stimulation 31,4 ±
25,7 % weniger Histamin freigesetzt. Dagegen konnte für BoNT-A keine signifikante
Reduktion festgestellt werden. Während nach einer PMA/Ionomycin-Aktivierung der
sekretionshemmende Effekt von BoNT-B und –C gezeigt wurde, konnten nach
Stimulation mit 22E7 keine signifikanten Unterschiede durch Botulinum-Neurotoxin-
Einwirkung dargestellt werden. Die spezifische Histaminfreisetzung war hierbei
generell niedriger als nach PMA/Iono-Aktivierung. Sie lag bei etwa 25 %. Auch hier
wurde die Kontrollbedingung als 100% gesetzt (Abb.9B). Auffällig war eine
allgemeine Hemmung der Exozytosefähigkeit, die 22E7-aktivierte Zellen nach
adenoviraler Transduktion zeigten. Für den Kontrollvirus ergab sich eine Reduktion
der Histamin-Freisetzung von 26,8 ± 24,8 %.

Ergebnisse
58
Abb. 9: Nach PMA/Ionomycin-Stimulation zeigt sich die
degranulationshemmende Wirkung von BoNT-B und –C; 2 Tage nach Infektion
der CARex-VP22 behandelten Zellen wurden die Mastzellen geerntet und aktiviert;
durch die GFP-Produktion in AdGFP-infizierten Zellen konnte der
Transduktionserfolg exemplarisch abgeschätzt werden; Für die in Abbildung 9A
dargestellte Bedingung betrug die spezifische Histaminfreisetzung der
Kontrollbedingung, die nur mit dem Fusionsprotein CARex-VP22 inkubierte Zellen
enthielt, etwa 50% und für die Bedingung in Abbildung 9B etwa 25%. Zur Besseren
Vergleichbarkeit wurden die Kontrollbedingungen jeweils als 100 % gesetzt. A
spezifischer Histaminrelease nach PMA/Ionomycin-Stimulation (10-6 M/10-6 M); B
spezifischer Histaminrelease nach 22E7-Stimulation (100 ng/ml); Die Mittelwerte (±
Standardabweichung) für A resultieren aus n = 3 Experimenten, für B aus n = 5
Experimenten.
Ap = 0,02
p = 0,01
B p < 0,01
Kontrolle
AdGFP
BoNT-A
BoNT-B
BoNT-C
0
25
50
75
100
125
His
tam
infr
eise
tzu
ng
nac
h I
nfe
ktio
n (
%)
Kontrolle
AdGFP
BoNT-A
BoNT-B
BoNT-C
0
25
50
75
100
125
His
tam
infr
eise
tzu
ng
nac
h I
nfe
ktio
n (
%)

Ergebnisse
59
3.3 Funktionelle Bedeutung der gefundenen SNARE-Proteine
3.3.1 Intrazelluläre Lokalisation der SNARE-Proteine
Zur genaueren Analyse der funktionellen Bedeutung sollte zunächst geklärt werden,
in welchen Kompartimenten die gefundenen SNARE-Isotypen liegen und ob es zu
einer Translokation nach Stimulation kommt.
Dazu wurden die humanen Darm-Mastzellen mit Saponin permeabilisiert, wodurch es
möglich war, Antikörper durch die Zellmembran zu schleusen, um die intrazellulär
gelegenen SNARE-Proteine der Mastzellen anzufärben. Zur genaueren Analyse der
Lokalisation der angefärbten Strukturen wurden die Zellen konfokalmikroskopisch auf
verschiedenen optischen Ebenen betrachtet. Da es sich bei den untersuchten
Mastzellen um nicht-adhärente Zellen handelte, wurde ein möglichst kleines Volumen
von der angefärbten Zellsuspension auf einen Objektträger getropft. Nachdem die
Zellen abgesunken waren, konnte etwas PBS hinzugefügt werden, um ein
Austrocknen der Zellen während des Mikroskopierens zu verhindern. Trockneten die
Zellen ein, so war eine konfokalmikroskopische Betrachtung über verschiedene
Ebenen nicht mehr möglich, da sie sich nun nicht mehr als runde, sondern als ovale,
stark abgeplattete Strukturen zeigten. Ähnliche Beobachtungen ergaben sich auch
für Cytospins oder mit unterschiedlichen Substanzen eingedeckelte Zellen.
In verschiedenen Doppelfärbungen wurden SNARE-Proteine als vesikel- oder
plasmamembranständige Strukturen nachgewiesen. SNAP-23 zeigte eine deutliche
membranständige Färbung, während sich VAMP-7 vorwiegend im Zellinneren befand
und scheinbar vermehrt in der Nähe des Zellkerns vorlag. Des Weiteren war eine
Darstellung des Syntaxin-4 an der Plasmamembran, sowie eine cytoplasmatische
Färbung von VAMP-3 möglich. Auch weitere Vertreter der VAMP-Familie wurden
untersucht, wobei eine Kernfärbung mit To-Pro 3 zur besseren Lokalisierbarkeit der
SNARE-Isoformen erfolgte. In einer Doppelfärbung mit dem
plasmamembranständigen Syntaxin-3 wurde VAMP-7 nochmals dargestellt. Es
zeigte sich eine gleichmäßige Verteilung des SNARE-Proteine im Zellinnneren. Die
BoNT-B Zielstruktur VAMP-2, die bereits im Western Blot nur durch Verwendung
eines polyklonalen Antikörpers (pAk) darzustellen war, konnte in der

Ergebnisse
60
Immunfluoreszenz weder durch den monoklonalen noch den polyklonalen Antikörper
aufgezeigt werden. Darstellung der Immunfluoreszenz-Konfokalmikroskopischen
Bilder in Abbildung 10 A-I.
3.3.2 VAMP-7 transloziert während der Exozytose
Die Lage der verschiedenen SNARE-Proteine wurde nicht nur im Ruhezustand,
sondern auch nach einstündiger PMA/Ionomycin-Stimulation untersucht. Dadurch
sollte gezeigt werden, ob bestimmte SNARE-Isoformen während der Exozytose ihre
Lage wechseln, womit ein Hinweis auf ihre Beteiligung an den stattfindenden
Membranfusionen gegeben wäre. Bis auf VAMP-7 behielten alle untersuchten
SNARE-Proteine (s. Kap. 3.3.1) ihre jeweilige Position bei. Nach Aktvierung der
Mastzellen kam es zu einer Translokation des cytoplasmatisch gelegenen VAMP-7
zur Zellperipherie hin. Somit lagen in stimulierten Mastzellen die SNARE-Isoformen
SNAP-23, VAMP-7, Syntaxin-3 und –4 nach Stimulation an der Plasmamembran vor.
Darstellung der Immunfluoreszenz-Konfokalmikroskopischen Bilder nach Stimulation
in Abbildung 10 D * - I*.

Ergebnisse
61
Abb. 10: Darstellung der intrazellulär gelegenen t- und v-SNAREs durch
Immunfluoreszenz an Mastzellen. Gezeigt werden konfokalmikroskopische
Aufnahmen aus der Zellmitte.
D E F
D * E * F *
G H I
G * H * I *
A B C

Ergebnisse
62
zu Abb. 10: A Syntaxin-4; B VAMP-3; C Syntaxin-4/ VAMP-3 (grün/rot); D VAMP-7,
E SNAP-23, F VAMP-7/ SNAP-23 (grün/rot); G-I Zellkerne mittels nukleärem Marker
To-Pro 3 dargestellt (blau) ; G VAMP-7; H Syntaxin-3; I VAMP-7/Synatxin-3 (grün/rot)
Mit * markierte Abbildungen wurden ihrer Bezeichnung entsprechend angefärbt ,
sind aber vorher noch mit PMA/Ionomycin stimuliert worden
3.3.3 Die Inhibition des SNARE-Proteins VAMP-7 bewirkt eine
Degranulationshemmung
Mit der durch Immunfluoreszenz gezeigten Translokation von VAMP-7 nach
Stimulation war ein Hinweis für die Beteiligung diese SNARE-Proteins an
Exozytoseprozessen gegeben. Eine nähere Analyse mittels spezifischer, Antikörper-
vermittelter Inhibition schloss sich zur Klärung der Bedeutung von VAMP-7 an.
Obwohl VAMP-2 in den funktionell den Mastzellen ähnelnden eosinophilen
Granulozyten eine wichtige Rolle spielt, konnte es in der Immunfluoreszenz mittels
eines monoklonalen Antikörpers nicht angefärbt werden. Somit kann angenommen
werden, dass in den humanen Mastzellen kein VAMP-2 vorhanden ist, an das der
Antikörper hätte binden können. Daher diente der anti-VAMP-2 Antikörper als
Kontrolle. Zur Darstellung des Releases nach VAMP-2 bzw. VAMP-7 Inhibition
wurden die für SLO erhaltenen Werte als 100 % gesetzt. Im weiteren Ablauf wurden
zunächst die Zellwände der Mastzellen durch eine SLO-Behandlung so weit
permeabilisiert, dass spezifische Antikörper eingeschleust werden konnten, um
intrazellulär gelegene SNARE-Proteine zu blockieren (Abb.11). Etwa 60-80 % der
Zellen waren zum Zeitpunkt der Antikörper-Einschleusung permeabilisiert. Nach dem
Verschluss der Poren durch Zugabe von calciumhaltigem Medium und Inkubation auf
Eis, wurden die Mastzellen mit 22E7 stimuliert. Mit einem spezifischen Antikörper
gegen VAMP-7 behandelte Mastzellen konnten zwar noch Histamin freisetzen, doch
war dabei die Degranulation, verglichen mit der Sekretionsfähigkeit der
Negativkontrolle (anti-VAMP-2), um 54,7 ± 15,5 % vermindert. Eine
Sekretionshemmung durch die VAMP-7-Inhibition konnte auch im Vergleich zur
alleinigen SLO-Behandlung festgestellt werden. Die Reduktion des Releases betrug
hierbei 38,6 ± 28,0 %.
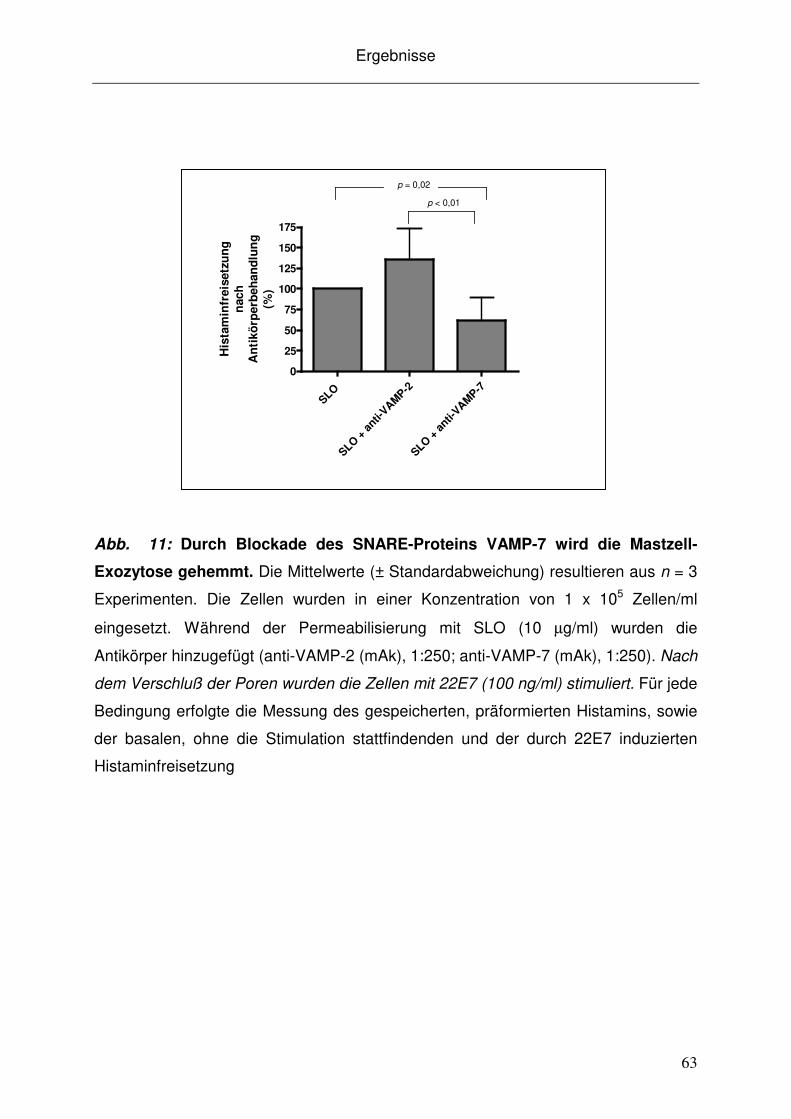
Ergebnisse
63
Abb. 11: Durch Blockade des SNARE-Proteins VAMP-7 wird die Mastzell-
Exozytose gehemmt. Die Mittelwerte (± Standardabweichung) resultieren aus n = 3
Experimenten. Die Zellen wurden in einer Konzentration von 1 x 105 Zellen/ml
eingesetzt. Während der Permeabilisierung mit SLO (10 µg/ml) wurden die
Antikörper hinzugefügt (anti-VAMP-2 (mAk), 1:250; anti-VAMP-7 (mAk), 1:250). Nach
dem Verschluß der Poren wurden die Zellen mit 22E7 (100 ng/ml) stimuliert. Für jede
Bedingung erfolgte die Messung des gespeicherten, präformierten Histamins, sowie
der basalen, ohne die Stimulation stattfindenden und der durch 22E7 induzierten
Histaminfreisetzung
p < 0,01
SLO
SLO + a
nti-VAM
P-2
SLO + a
nti-VAM
P-7
0
25
50
75
100
125
150
175H
ista
min
frei
setz
un
gn
ach
An
tikö
rper
beh
and
lun
g(%
)
p = 0,02

Diskussion
64
4. Diskussion
4.1 Expression von SNARE-Proteinen in humanen intestinalen Mastzellen
SNARE-Proteine wurden als Vermittler grundlegender Prozesse der
Membranfusionen in Neuronen und anderen sekretorisch aktiven Zellen, wie
Mastzellen und weiteren Zellen des Immunsystems, nachgewiesen. Vor allem
zwischen neuronalen und nicht-neuronalen Zellen zeigten sich deutliche
Unterschiede im Expressionsmuster der einzelnen SNARE-Isoformen. Bisher gibt es
wenige Daten über die SNARE-Expression in Mastzellen, wobei die meisten
Kenntnisse anhand der leukämischen Rattenmastzelllinie RBL-2H3 gewonnen
wurden. In der vorliegenden Arbeit wurden primäre humane Mastzellen verwendet,
die bislang nicht unter dem Aspekt der SNARE-Protein vermittelten Exozytose
untersucht worden sind.
Zu den zentralen Vorgängen, die während der Exozytose ablaufen, gehört die
Anlagerung von drei SNARE-Proteinen zum „core“-Komplex, der die Membranfusion
energetisch begünstigt. Je ein Vertreter der SNAP-, Syntaxin-, oder VAMP-Familie ist
beteiligt. Von Neuronen ist bekannt, dass es sich dabei um SNAP-25, Syntaxin-1 und
VAMP-2 handelt [79]. Diese SNARE-Proteine konnten nicht eindeutig in humanen
Mastzellen nachgewiesen werden. Dabei waren die neuronalen Isoformen SNAP-25
und der Isotyp 1A von Syntaxin weder auf mRNA- noch auf Protein-Ebene
vorhanden. Syntaxin-1B konnte auf mRNA-Ebene, trotz starker Signale für die
Positivkontrolle MHH-NB-11, nicht detektiert werden, obwohl wiederholt positive
Ergebnisse auf Protein-Ebene erzielt wurden. Mittels RT-PCR konnte VAMP-2
detektiert werden. Dagegen wurden im Western Blot auch bei Verwendung eines
polyklonalen Antikörpers nur schwache Signale für VAMP-2 in humanen Mastzellen
gezeigt, während in der Rattenmastzelllinie RBL-2H3 die Expression dieses
SNARE-Proteins bereits bestätigt wurde [94]. Obwohl die den neuronalen „core“-
Komplex bildenden Isotypen nicht nachzuweisen waren, konnten verschiedene
andere, möglicherweise Exozytose-vermittelnde SNARE-Proteine, in Mastzellen
dargestellt werden. Dazu wurden humane intestinale Mastzellen mit in vitro
entwickelten Mastzellen aus Nabelschnurblut (CBMC) und der Mastzellinie HMC-1,

Diskussion
65
sowie aus menschlichem Blut isolierten eosinophilen Ganulozyten, als weiteren
Vertreter sekretorisch aktiver Immunzellen, auf mRNA-Ebene verglichen. Die
SNARE-Proteine SNAP-23, Syntaxin-2, -3, -4 und –6 sowie VAMP-3, -5, -7 und –8
konnten in allen untersuchten Zellen detektiert werden. Diese Ergebnisse bestätigten
sich für humane Mastzellen auch im Western Blot auf Protein-Ebene. Die Beteiligung
dieser SNARE-Isoformen an Exozytose-Vorgängen, außer für VAMP-5, ließ sich
bereits an verschiedenen Zellen, darunter auch RBL-2H3-Zellen, zeigen [96]. Bis auf
das zusätzlich vorhandene Syntaxin-1 und VAMP-2 entspricht das
Expressionsmuster der Rattenmastzelllinie RBL-2H3 humanen Mastzellen. In
eosinophilen Granulozyten, die den Mastzellen funktionell sehr ähneln – beides sind
sekretorisch aktive Zellen des Immunsystems - konnte die Expression von SNAP-23,
Syntaxin-3 und –4 sowie VAMP-1 und –2 nachgewiesen werden [138;139]. So
unterscheiden sich humane Mastzellen in ihrem SNARE-Expressionsmuster nicht nur
von neuronalen Zellen, sondern auch von RBL-2H3-Zellen, die bislang als Mastzell-
Modell dienten und von den funktionell nahestehenden eosinophilen Granulozyten.
4.2 Hemmung der Mastzelldegranulation durch Botulinum Neurotoxin
Um nun durch Spaltung und damit Inaktivierung zu zeigen, ob und welche SNARE-
Isotypen für die Exozytose-Vorgänge in Mastzellen notwendig sind, wurden die
Mastzellen mit verschiedenen SNARE-spezifischen Proteasen behandelt. Somit wäre
auch ein therapeutischer Ansatz für die Behandlung mastzellbedingter allergischer
Symptome gegeben, die zum großen Teil durch den ausgeschütteten Mediator
Histamin verursacht werden. Als ein nützliches Hilfsmittel zur Analyse der Bedeutung
der SNARE-Proteine für Degranulationvorgänge erwies sich Botulinum Neurotoxin,
das in Neuronen durch SNARE-Spaltung die Exozytose hemmt. Dabei sind die
bakteriellen Proteasen (Toxine) spezifisch für die jeweiligen SNARE-Subtypen.
Neurotoxine, die von verschiedenen Serotypen der Clostridien gebildet werden,
spalten verschiedene SNARE-Isoformen. Bisherige Erkenntnisse über Expression
und Funktion der SNARE-Proteine, sowie ihre Spaltung durch Neurotoxine, beruhen
größtenteils auf Untersuchungen an Neuronen. Dabei stellte sich heraus, dass
SNAP-25 durch BoNT-A, VAMP-1, -2 (Synaptobrevin) und –3 (Cellubrevin) durch

Diskussion
66
BoNT-B und SNAP-25, als auch Syntaxin-1A, -1B und –2 durch BoNT-C gespalten
werden [121;140;141]. Im Gegensatz dazu erwiesen sich VAMP-7 und VAMP-8 als
Neurotoxin-resistent [96;140]. Eine Hemmung der Exozytose durch SNARE-
spezifische Spaltung mit den verschiedenen BoNT-Isotypen konnte nicht nur in
Neuronen nachgewiesen werden, sondern auch in verschiedenen anderen
sekretorisch aktiven Zellen - unter anderem in azinösen Zellen der Ohrspeicheldrüse,
chromaffinen Zellen des Nebennierenmarks oder beta-Zellen des Pankreas [142-
144]. Dabei sind vor allem SNAP-25, VAMP-2 und Syntaxin-1 als Zielstrukturen von
Botulinum Neurotoxin von Bedeutung. Zur Untersuchung der SNARE-Abhängigkeit
der Mastzelldegranulation kamen Botulinum Neurotoxin Typ A, B und C zum Einsatz.
Botulinum Neurotoxine binden aufgrund ihrer schweren Kette spezifisch an
Neuronen, in denen sie sich nach rezeptorspezifischer Endozytose anreichern. Um
den Einfluss des proteolytisch wirksamen Anteils des Toxins auf Mastzellen zu
untersuchen, wurden in mehreren Vorversuchen verschiedene Möglichkeiten zur
Einschleusung der Botulinum Neurotoxin-Leichtketten getestet.
Nach vorhergehender Gangliosidbeladung sind chromaffinen Zellen in der Lage,
das eigentlich streng neurotrope BoNT-A aufzunehmen [145]. Obwohl dieses
Verfahren bei verschiedenen Zellkulturen erfolgreich war, sorgte die
Gangliosidmischung bei den primären humanen Mastzellen zu einem vorzeitigen
Absterben der Zellkulturen, wie Vorversuche von L. Sander et A. Lorentz
(unpublizierte Daten) zeigten. Dabei konnte eine konzentrations- und zeitabhängige
Toxizität in entsprechenden Versuchsreihen nachgewiesen werden. Auch die
leukämische Mastzelllinie HMC-1, die üblicherweise eine starke Proliferationstendenz
aufweist, starb nach Inkubation mit den Gangliosiden ab.
Eine alternative Methode zur Einschleusung der Toxine bestand in der
Elektroporation. Die Anwendung dieses Verfahrens für primäre humane Mastzellen
wurde von L. Sander et A. Lorentz getestet. Durch Anlegen einer elektrischen
Spannung für wenige ms entstehen kurzfristig große Poren in der Zellmembran,
durch die Toxine aus dem Medium in die Zellen eindringen können. Es zeigte sich,
dass ein großer Teil der eingesetzten Mastzellen in den ersten Tagen nach der
Elektroporation starb. Erste Experimente mit BoNT-A , -B und –C unter Verwendung
einer Spannung, bei der ca. 30-50 % der Mastzellen überlebten, konnten keine
eindeutigen Ergebnisse bezüglich eines Effekts auf die Histaminfreisetzung aus den

Diskussion
67
Mastzellen liefern. Als zusätzliche Kontrolle wurde die Wirkung von Tetanus
Neurotoxin untersucht, das dieselben Zielstrukturen wie BoNT-B besitzt. Weder
durch Elektroporation der Zellen mit Fluoreszenz-markierten Proteinen und
anschließender Betrachtung unter dem Fluoreszenzmikroskop noch mittels
Durchflußzytometrie konnte die Proteinaufnahme in die Zellen eindeutig
nachgewiesen werden. Auch nach Elektroporation mit 125I-markiertem Tetanus Toxin
(aus Clostridium Tetani, ein Protein mit etwa dem gleichen Molekulargewicht wie
Botulinum Neurotoxin) gelang kein eindeutiger Nachweis, da ein Anhaften der
Radioaktivität an die Zelltrümmer nicht auszuschließen war. Obwohl die Toxine in
einigen Experimenten scheinbar eine sekretionsinhibierende Wirkung ausübten,
konnten diese Daten aufgrund der auftretenden Zelllyse und der zum Teil geringen
Histaminfreisetzung nicht zufriedenstellend reproduziert werden.
Elektrophysiologische Experimente mittels Patch Clamp-Technik sollten dieses
Problem umgehen, indem über Membranströme die Degranulationsfähigkeit
einzelner toxinbehandelter Zellen untersucht wurde. Jedoch blieb die Zellmembran
bei dieser Methode nicht lange genug intakt, um die notwendigen Messungen
durchzuführen. Nach etwa 5 min gab die Membran nach und die Messungen konnten
nicht weitergeführt werden.
Nachdem das Einschleusen der proteolytisch aktiven Anteile der Botulinum
Neurotoxine mittels Gangliosiden, Elektroporation und Patch Clamp-Technik nicht
möglich war, wurden in einem weiteren Ansatz Adenoviren verwendet. Adenovirale
Vektoren sind als eines der effektivsten Behelfsmittel, Fremd-DNA in Säugetierzellen
zu bringen, in der Lage, ein breites Spektrum von verschiedenen Zellen zu infizieren.
Dazu gehören sowohl sich teilende als auch ruhende Zellen - Primärzellen konnten
ebenfalls erfolgreich transduziert werden [146]. Mittels adenoviraler Vektoren sollten
die betreffenden Gene nun in primäre humane Mastzellen eingeschleust und
anschließend exprimiert werden. Nachdem die Gene für die Leichtketten von BoNT-
A, -B und –C in einen kleinen Shuttle Vektor kloniert wurden, erfolgte unter
Verwendung des Systems nach He et al. eine Rekombination des konstruierten
Plasmids mit dem adenoviralen Backbone AdEasy-1 [136]. Die Generierung von
viralen Partikeln fand in Packungszellen (HEK 293) statt, die fehlende Faktoren des
adenoviralen Vektors transkomplementierten. Die Expression der Botulinum
Neurotoxin-Leichtketten in den Mastzellen sollte mittels der GFP-Signale der

Diskussion
68
jeweiligen Klone angezeigt werden. Obwohl die Klone alle unterschiedlich hohe GFP-
Signale aufwiesen, konnte anhand der starken GFP-Expression des Kontrollvirus
AdGFP stellvertetend für alle Viren die Infektionsrate abgeschätzt werden [137].
Nachdem die Gene für die proteolytisch wirksamen Leichtketten von BoNT-A, -B
und –C über adenovirale Vektoren in die Mastzellen eingeschleust und anschließend
exprimiert wurden, zeigte sich auch hier eine Senkung der Sekretionsfähigkeit. Dabei
wurde die Mastzelldegranulation durch BoNT-B um 24,6 ± 23,0 % und BoNT-C um
31,4 ± 25,7 % nach Stimulation mit PMA/Ionomycin gesenkt. Ein Effekt für BoNT-A
war nicht nachweisbar. Die Resistenz der Mastzellen gegenüber einer Behandlung
mit BoNT-A lässt sich durch das Fehlen des Substrates SNAP-25 erklären.
Verschiedene SNARE-Proteine werden, wie bereits aufgeführt, in Mastzellen
exprimiert und könnten als Substrate für BoNT-B und –C dienen. Welche SNARE-
Isoformen die Zielstrukturen in Mastzellen darstellen, lässt sich nicht genau sagen.
Da die beiden Neurotoxin Subtypen BoNT-B und -C die Exozytose der Mastzellen
hemmen, scheinen sie bestimmte SNARE-Proteine zu spalten. Die Spaltung von
VAMP, vor allem für die in Neuronen vorkommenden Isoformen VAMP-1 und –2,
aber auch für das ubiquitär exprimierte VAMP-3 (Cellubrevin) konnte für BoNT-B
aufgezeigt werden [140]. Außerdem gelang mittels Tetanus-Toxin eine
Degranulationshemmung in eosinophilen Granulozyten durch VAMP-2-Spaltung
[135]. Eine solche Beteiligung von VAMP-2 scheint in Mastzellen eher
unwahrscheinlich zu sein, da es in der Immunfluoreszenz aufgrund fehlender
Bindungstellen für den spezifischen Antikörper nicht dargestellt werden konnte. So
wäre die Proteolyse des BoNT-B-sensitiven VAMP-3 möglich, während die
Neurotoxin-resistenten Isotypen VAMP-7 und –8 eher keine Bedeutung als BoNT-B-
Substrate haben sollten. Auch VAMP–5 könnte als Zielstruktur eine Rolle spielen.
SNAP-25 und Sxyntaxin-1A, -1B und –2 dienen BoNT-C als Substrate, jedoch
konnten die SNARE-Proteine SNAP-25 und Syntaxin-1A in Mastzellen nicht
nachgewiesen werden, während sich für Syntaxin-1B unterschiedliche Ergebnisse
zeigten. Da im Western-Blot nur eine schwache Expression für Syntaxin-1B detektiert
werden konnten, wäre es möglich, dass die mRNA-Signale zu schwach waren, um
mit der angewandten Methode nachgewiesen zu werden. Auch Syntaxin-2, -3, -4
oder –6 könnten beteiligt sein. Obwohl nicht genau gezeigt werden kann, welche
SNARE-Isoformen für die Mastzellexozytose von unmittelbarer Bedeutung sind und

Diskussion
69
gleichzeitig als Botulinum Neurotoxin-Substrate dienen, lassen sich doch einige
potentielle Kandidaten darstellen, die auch in der Exozytose von Rattenmastzellen
beteiligt sind (vgl. Kapitel 4.1). Außerdem senken BoNT-B und BoNT-C die
Sekretionsfähigkeit in Mastzellen zwar um über 20 Prozent, aber eine vollkommene
Hemmung konnte nicht erzielt werden. Dies könnte darin begründet sein, dass die
Stimulationsexperimente schon etwa zwei Tage nach Infektion stattfanden, als der
zytotoxische Effekt der Viren, erkennbar am Zelltod, noch nicht auftrat. Der gewählte
Zeitraum von zwei Tagen könnte zu kurz gewesen sein, um die Toxine in
ausreichenden Mengen zu produzieren und anschließend die jeweiligen SNARE-
Proteine zu spalten. Des Weiteren waren zum Zeitpunkt der Experimente nur etwa
50-60% der Zellen transfiziert. Die nicht-infizierten Zellen sind in ihrer
Sekretionsfähigkeit nicht beeinträchtigt und tragen somit zum recht hohen
Histaminrelease bei. Denkbar wären auch zusätzliche, teilweise kompensierende
Exozytose-Vorgänge, die durch BoNT-resistente SNARE-Proteine vermittelt werden
oder gänzlich SNARE-unabhängig ablaufen. Außerdem entfalten Botulinum
Neurotoxine ihre proteolytische Aktivität nur an unkomplexiert vorliegenden SNARE-
Proteinen [147]. In verschiedenen Zellen liegen Vesikel als Reserve für eine schnelle
Sekretion bereits der Plasmamembran an [59]. Falls dies auch in Mastzellen der
Fall ist und die SNARE-Proteine der aneinander liegenden Membranen bereits
Komplexe gebildet haben, könnten die Neurotoxin-Behandlung die Exozytose dieser
Vesikel nicht verhindern.
Interessanterweise ließ sich eine Degranulationshemmung nur nach Stimulation
mit PMA/Ionomycin darstellen. Eine solche Senkung der Histaminfreisetzung war
nicht aufzuzeigen, wenn eine Aktivierung durch eine 22E7-bedingte Kreuzvernetzung
der IgE-Rezeptoren erfolgte. Da die Histaminfreisetzung nach 22E7-Stimulation
insgesamt auf einem weit tieferen Niveau stattfand, könnte der Release zu niedrig
gewesen sein, um die hemmenden Effekte der Neurotoxine aufzuzeigen.
4.3 Bedeutung von VAMP-7 für die Mastzelldegranulation
Eine Reihe verschiedener Zellen weist bestimmte SNARE-Proteine auf, die an
Membranen spezieller Kompartimente gebunden sind. Dies deutet darauf hin, dass

Diskussion
70
spezifische SNARE-Komplexbildungen die Richtung der Membranfusionen
regulieren [148], wobei die Lage der jeweiligen SNARE-Proteine von Bedeutung ist.
Während die VAMP-Isotypen an den Membranen verschiedener Kompartimente im
Zellinneren sitzen, liegen die Syntaxine und SNAP-25/-23 an der Plasmamembran.
Mittels Immunfluoreszenz ließ sich diese Einteilung auch für die in Mastzellen
untersuchten SNARE-Proteine bestätigen. So lagen SNAP-23 sowie Syntaxin-3 und
–4 an der Plasmamembran vor, während VAMP-7 und –3 in den Membranen
zytoplasmatischer Vesikel lokalisiert waren. Neben den SNARE-Proteinen SNAP-25,
VAMP-2 und Syntaxin-1, die den neuronalen SNARE-Komplex bilden, sind auch
andere Vertreter der drei SNARE-Familien in der Lage, miteinander in Kontakt zu
treten. Dabei müssen nicht immer die gleichen Partner interagieren. So konnte in
RBL-2H3-Zellen gezeigt werden, dass Syntaxin-4 mit SNAP-23 und verschiedenen
VAMP-Isoformen, außer mit VAMP-7, Komplexe bilden kann [96]. In Normal Rat
Kidney-Zellen (NRK cells) wiederum bildet Syntaxin-4 zusammen mit VAMP-7 und
SNAP-23 SNARE-Komplexe [149]. Abhängig von der Zellart und der Richtung der
Membranfusion scheinen unterschiedliche SNARE-Proteine in grundlegende
Prozesse der Exozytose involviert zu sein. Die große Vielfalt der möglichen SNARE-
Komplexbildungen in RBL-2H3-Zellen lässt eine ähnliche Varianz auch für
Mastzellen vermuten. Potentielle SNARE-Komplexbildner für humane Mastzellen
wurden bereits dargestellt, sollten aber näher untersucht werden.
Während sich VAMP-2 in humanen Mastzellen nicht durch Immunfluoreszenz
zeigen ließ, ergab sich anhand des Isotypen VAMP-7 ein Hinweis auf die Beteiligung
von SNARE-Proteinen an der Mastzellexozytose. Nachdem die Mastzellen stimuliert
wurden, um eine Membranfusion im Rahmen der Exozytose zu erreichen, wanderte
das in der Vesikelmembran liegende VAMP-7 vom Zytoplasma in Richtung
Zelloberfläche. Der für VAMP-7 gezeigte Postionswechsel nach Stimulation weist auf
eine Beteiligung an Degranulationsprozessen in Mastzellen hin. Auch in RBL-2H3
Zellen erfolgt eine solche Translokation von VAMP-7 [100]. Das translozierte
VAMP-7 lag zusammen mit SNAP-23 an der Plasmamembran vor. Während
Syntaxin-4 in RBL-2H3-Zellen keine Komplexe mit VAMP-7 bildet, ist nicht
auszuschließen, dass andere Syntaxine als Vertreter der dritten SNARE-Familie
dazu durchaus in der Lage sind. So wäre etwa ein Komplex, bestehend aus VAMP-7,
SNAP-23 und Syntaxin-3, dessen plasmamembranständige Position nachgewiesen

Diskussion
71
wurde, denkbar. Die für VAMP-7 gezeigte Lageveränderung konnte für andere
SNARE-Isoformen, wie das ebenfalls im Zellinneren gelegene und bestimmte
Membranfusionen vermittelnde VAMP-3/ Cellubrevin [150], nicht dargestellt werden.
Ein weiterer Hinweis für die Bedeutung von Vamp-7 ist dessen Anteil an der späten
endosomalen Sekretion von Makrophagen, die nach Hemmung des SNARE-Proteins
sinkt [151].
Zur weiteren Klärung der Funktion von VAMP-7 in sekretorischen Prozessen der
Mastzellen wurde das Neurotoxin-resistente SNARE-Protein mittels spezifischer
Antikörper gehemmt. Durch eine SLO-Permeabilisierung, die aufgrund der
entstehenden Poren den Eintritt größerer Moleküle in die Mastzellen erlaubte,
konnten spezifische Antikörper gegen VAMP-7 und VAMP-2 eingeschleust werden.
Passend zu den bisherigen Befunden – VAMP-2 konnte in humanen Mastzellen
weder durch Western Blot noch durch Immunfluoreszenz zufriedenstellend
dargestellt werden – zeigte anti-VAMP-2 keine Effekte auf die Mastzellexozytose,
während das in cytoplasmatischen Vesikeln gelegene VAMP-2 in eosinophilen
Granulozyten von entscheidender Bedeutung für die Degranulation ist [137]. Daher
wurde der monoklonale anti-VAMP-2 Antikörper, der keine Bindungsstellen in den
humanen Mastzellen haben sollte, als Isotyp-Kontrolle verwendet. Dagegen wurde
durch anti-VAMP-7 eine Degranulationshemmung von 54,7 ± 15,5 % erreicht.
Obwohl nur etwa 60-80 % der untersuchten Zellen überhaupt permeabilisiert und
somit in der Lage waren, die spezifischen Antikörper aufzunehmen, ließ sich eine
hohe Hemmung der Exozytose nachweisen. Wahrscheinlich käme es zu einer
nahezu vollständigen Inhibition der Histaminfreisetzung, wenn alle Zellen
permeabilisiert wären. Aufgrund des Absterbens der Zellen durch zu hohe SLO-
Konzentrationen konnten nicht alle Zellen permeabilisiert und sämtliche VAMP-7-
Proteine blockiert werden. Gelänge dies, so wäre eine noch höhere Hemmung der
Histamin-Freisetzung denkbar. Außerdem ist es möglich, dass neben der VAMP-7
vermittelten Sekretion noch andere Vorgänge stattfinden, die ausgleichend eingreifen
können. Möglich wäre auch eine Kompensation des inhibierten VAMP-7 durch
andere VAMP-Isoypen, die ebenfalls einen zur Exozytose führenden SNARE-
Komplex bilden könnten, wie etwa das ebenfalls Neurotoxin-resistene VAMP-8 oder
das Neurotoxin-sensitive VAMP-3. Eine Beteiligung dieser beiden Isotypen an
Degranulationsvorgängen wird für RBL-2H3-Zellen angenommen. Die Bedeutung der

Diskussion
72
Neurotoxin-sensitiven SNARE-Proteine an der Mastzell-Exozytose konnte bereits
durch Botulinum Neurotoxin-vermittelte Spaltung gezeigt werden. Daher ist es
möglich, dass beide SNARE-Familien, sowohl die Neurotoxin-sensitiven als auch die
insensitiven, Membranfusionen vermitteln, die zur Sekretion führen
4.4 Ausblick
Ein großer Teil der Kenntnisse über Exozytose-Vorgänge und der dabei beteiligten
SNARE-Proteine beruht auf Untersuchungen an Neuronen, wobei deutliche
Unterschiede zu nicht-neuronalen, sekretorischen Zellen bestehen. Die Bedeutung
der SNARE-Isoformen für die Mastzellexozytose ist noch nicht hinreichend geklärt,
weshalb weitere ergänzende Analysen mit humanen Mastzellen durchgeführt werden
sollten. Dazu gehört die Untersuchung der Komplex-bildenden SNARE-Proteine. In
RBL-2H3-Zellen formt Syntaxin-4 als multifunktionales SNARE-Protein mit SNAP-23
und diversen VAMP-Isotypen unterschiedliche Komplexe. VAMP-7 ist dabei jedoch
nicht involviert. So bleibt zu klären, mit welchen SNARE-Proteinen VAMP-7 in
humanen Mastzellen Komplexe formt, da es hier allem Anschein nach während der
Exozytose Membranfusionen vermittelt. Außerdem sollte analysiert werden, welche
SNARE-Proteine in den Mastzellen durch die Botulinum Neurotoxine gespalten
werden und ob somit eine relevante Senkung der Histaminausschüttung erreicht
werden kann. Dazu sollte die Expression der Botulinum Neurotoxine durch den
viralen Ansatz optimiert werden. Die große Vielfalt der gefundenen SNARE-
Isoformen in Mastzellen, sowie die Tatsache, dass sowohl Neurotoxin-sensitive als
auch insensitiven SNARE-Proteine an entscheidenden Exozytosevorgängen in
Mastzellen beteiligt sind, lässt eine Komplexität der Prozesse vermuten, die zum
besseren Verständnis weiter untersucht werden muss. Dabei könnte die
Ausschaltung von SNARE-Isoformen durch spezifische siRNA behilflich sein, die
mittels adenoviraler Vektoren in die Zellen eingeschleust und anschließend
exprimiert wird. Bessere Kenntnisse über die Mastzelldegranulation und der dafür
essentiellen SNARE-Isoformen könnten die Basis für die Generierung neuer
Therapiestrategien bilden.

Zusammenfassung
73
5. Zusammenfassung
Mastzellen sind als Effektorzellen allergischer Reaktionen (Hypersensitivitätsreaktion
vom Typ I) in der Lage, nach Aktivierung innerhalb kurzer Zeit eine Vielzahl von
Mediatoren mittels Exozytose freizusetzen und dadurch verschiedene physiologische
und pathophysiologische Reaktionen des Organismus zu bewirken.
Die exozytotischen Prozesse eukaryoter Zellen werden durch die Interaktion
sogenannter SNARE-Proteine reguliert, die zuerst als Vermittler der neuronalen
Membranfusion beschrieben worden, aber auch an der Degranulation von
Immunzellen beteiligt sind. Bisher gibt es wenige Daten über die Expression und
Funktion der verschiedenen SNARE-Isoformen in Mastzellen, wobei die meisten
Kenntnisse anhand der leukämischen Rattenmastzelllinie RBL-2H3 gewonnen
wurden. In der vorliegenden Arbeit erfolgten die Untersuchungen an primären
humanen Mastzellen, die bislang nicht unter dem Aspekt der SNARE-Protein-
vermittelten Exozytose analysiert worden sind.
Mittels quantitativer RT-PCR und Western Blot zeigte sich, dass humane
Mastzellen die SNARE-Isoformen SNAP-23, Syntaxin-2, -3, -4 und –6, sowie VAMP-
3, -5, -7 und –8 exprimieren. Unterschiede in der SNARE-Expression zeigten sich im
Vergleich humaner Mastzellen zu der Rattenmastzelllinie RBL-2H3 und zu den
funktionell ähnlichen eosinophilen Granulozyten. In Eosinophilen konnte VAMP-2
durch Immunfluoreszenz dargestellt werden, während ein solcher Nachweis in
Mastzellen nicht gelang.
SNARE-Proteine stellen Substrate für Botulinum Neurotoxine dar, wobei
verschiedene Isotypen spezifisch für bestimmte SNARE-Subtypen sind. Nachdem
verschiedene Ansätze, wie Gangliosidbehandlung, Elektroporation und Patch-
Clamp-Technik, zur Einschleusung der Neurotoxine nicht zum Erfolg führten, wurde
in dieser Arbeit ein adenovirales System zur Expression der Botulinum Neurotoxine
Typ A, B und C in humanen Mastzellen verwendet. Zunächst wurden die
gewünschten Gene in einen kleinen Shuttle Vektor kloniert und mit einem
adenoviralen Backbone Vektor rekombiniert. In permissiven Zellen erfolgte
anschließend die Generierung von viralen Partikeln, die zur Transduktion primärer
humaner Mastzellen verwendet wurden. Nach adenoviraler Infektion und

Zusammenfassung
74
anschließender Expression der Neurotoxine zeigte sich eine
Degranulationshemmung für BoNT-B und –C, während eine Behandlung mit BoNT-A
die Mastzellexozytose unbeeinflusst ließ.
Im weiteren Verlauf gelang eine Immunfluoreszenz-Konfokalmikroskopische
Darstellung der an der Zellmembran gelegenen SNARE-Proteine SNAP-23,
Syntaxin-3 und –4 sowie der zytoplasmatischen Lokalistion von VAMP-3 und –7.
Dabei zeigte sich nach Stimulation eine Translokation des Neurotoxin-resistenten
VAMP-7 vom Zellinneren zur –peripherie. Dort lag es in Colokalisation zu
plasmamembranständigen SNARE-Isoformen, wie SNAP-23, Syntaxin-3 und –4, vor.
Zur funktionellen Analyse der Bedeutung von VAMP-7 für die Mastzellexozytose
wurden anti-VAMP-7 Antikörper in SLO-permeabilisierte Zellen eingeschleust. Dabei
zeigte sich eine deutliche Exozytosehemmnung von über 50 % für VAMP-7 inhibierte
Zellen, wobei nur etwa 70 % der Zellen permeabilisiert waren.
Zusammenfassend stellt die vorliegende Studie dar, dass komplexe Prozesse, an
denen sowohl BoNT-sensitive als auch -insensitive SNARE-Proteine beteiligt sind,
die Membranfusionen in Mastzellen vermitteln.

Literaturverzeichnis
75
6. Literaturverzeichnis
(1) Church MK, F.Levi-Schaffer. 1997. The human mast cell. J Allergy Clin
Immunol 99, 155-160.
(2) Bruijnzeel PLB, M.Gebhardt, F.J.av Overveld. 1991. Mastzellen und basophile Granulozyten: Ihre Bedeutung bei allergischen Erkrankungen. Schweiz.Med.Wschr. 121, 1675-1685.
(3) Bischoff SC. 1994. Humane basophile Granulozyten und Mastzellen: Vermittler zwischen allergischer Entzündung und spezifischem Immunsystem. Immun.Infekt 22, 93-103.
(4) Diepgen TL, M.Fartasch, O.P.Hornstein. 1989. Evaluation and relevance of atopic basic and minor features in patients with atopic dermatitis and in general population. Acta Derm Venerol 144, 50-54.
(5) Burr ML. 1993. Epidemiology of clinical allergy. Monographs in Allergy 31, Introduction.
(6) Ehrlich P. 1877. Beiträge zur Kenntnis der Quilinfärbung und ihrer Verwendung in der mikroskopischen Technik . .Alch.Mikros.Anat. 13: 263-67.
(7) Metcalfe D.D., D.Baram, Y.A.Mekori. 1997. Mast cells. Physiol Rev. 77, 1033-1079.
(8) Lorentz A, D.Schuppan, A.Gebert, M.P.Manns, S.C.Bischoff. 2002. Regulatory effects of stem cell factor and interleukin-4 on adhesion od human mast cells to extracellular matrix proteins. Blood 99, 966-972.
(9) Speer W, R.H.Agis, K.Czerwenka, W.Klepetko, G.Kubista, G.Bolitz-Nitulescu, K.Lechner, P.Valent. 1992. Differential expression of cell surface integrins on human mast cells and basophils. Ann.Hematol. 65, 10-16.
(10) Li L, S.W.Reddel, S.A.Krilis. 2000. The phenotypic similarities and differences between human basophils and mast cells. G.Marone; L.M.Lichtenstein; S.J.Galli, eds. Academic press, London. Mast cells and basophils , 97-116.
(11) Galli SJ, M.Tsai, B.K.Wershil. 1993. The c-kit receptor, stem cell factor, and mast cells. What each is teaching us about the others. Am.J.Pathol. 965-974.
(12) Mierke CT, M.Ballmaier, U.Werner, M.P.Manns, K.Welte, S.C.Bischoff. 2000. Human endothelial cells regulate survival and proliferation of human mast cells. J.Exp.Med 192, 801-811.
(13) Bischoff SC, G.Sellge, S.Schwengberg, A.Lorentz, M.P.Manns. 1999. Stem cell factor dependent survival, proliferation and enhanced releasability of

Literaturverzeichnis
76
purified mature mast cells isolated from human intestinal tissue. Int.Arch.Allergy Immunol. 118, 104-107.
(14) Levi-Schaffer F, K.F.Austen, J.P.Caulfield, A.Hein, P.M.Gravallese, R.L.Stevens. 1987. Co-culture of human lung-derived mast cells with mouse 3T3 fibroblasts: morphology and IgE-mediated release of histamine, prostaglandin D2, and leukotrienes. J.Immunol. 139, 494-500.
(15) Schulma ES, J.G.Mohanty. 2001. Long term cultivation of human lung mast cells. J.Allergy Clin.Immunol.
(16) Bischoff SC, S.Schwengberg, R.Raab, M.P. Manns. 1997. Functional properties of human intestinal mast cells cultured in a new culture system: enhancement of IgE receptor-dependent mediator release and response to stem cell factor. The Journal of Immunology; 159(11):5560-5567.
(17) Weidner N. N. and K.F.Austen. 1991. Ultrastructural and immunohistochemical characterization of normal mast cells at multiple body sites. J.Invest Dermatol. 96, 26-30.
(18) Bischoff SC, 2000. Regulation and function of human intestinal mast cells. Mast cells and basophils.G.Marone, L.M.Lichtenstein and S.J.Galli, eds Academic Press , 541-566.
(19) Dvorak AM, R.A.Monahan, J.E.Osage, G.R.Dickersin. 1980. Crohn´s disease: transmission electron microscopic studies. II. Immunologic inflammatory response. Alterations of mast cells, basophils, eosinophils and the microvasculature. Hum Pathol 11, 606-619.
(20) Stead RH, M.F.Dixon, N.H.Bramwell, R.H.Riddel, J.Bienenstock. 1989. Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology , 575-585.
(21) Saito H, K.Hatake, A.M.Dvorak, K.M.Leiferman, A.D.Arai, K.Ishizaka, T.Ishizaka. 1988. Selective differentiation and proliferation of hematopoietic cells induced by recombinant human interleukins. Proc Natl Acad Sci U S A 85, 2288-2292.
(22) Tsuji K, M.Nakahata, T.Takagi, T.Kobayashi, T.Ishiguro, T.Kikuchi, K.Naganuma, K.Koike, A.Miyajima, K.Arai. 1990. Effects of inteleukin-3 and interleukin-4 on the development of "connective tissue-type" mast cells: interleukin-3 supports their survival and interleukin-4 triggers and supports their proliferation synergistically with interleukin-3. Blood 75, 421-427.
(23) Irani AA, N.M Schlechter, S.S.Craig, G.DeBlois, L.B.Schwartz, editors. 1986. Two types of human mast cells that have distinct neutral protease compositions. Proc.Natl.Acad.Sci.U.S.A 83, 4464-4468.
(24) Lowman MA, P.H.Rees, R.C.Benyon, M.K.Church. 1988. Human mast cell heterogenity: histamine release from mast cell dispersed from skin, lung,

Literaturverzeichnis
77
adenoids, tonsils and colon in response to IgE-dependent and nonimmunologic stimuli. J.Allergy Clin.Immunol. 590-597.
(25) Marone G, G.de Crescenzo, F.Patella, F.Granata, L.Verga, E.Arbustini, A.Genovese. 2000. Human heart mast cells: immunological charcterization in situ and in vivo. -Mast cells and basophils; L.M.Lichtenstein and S.J.Galli, eds Academic Press.
(26) Wedemeyer JM, M.Tsai, S.J.Galli. 2000. Roles of mast cells and basophils in innate and acquired immunity. Curr.Opin.Immunol. 12, 624-631.
(27) Wershil BK. 1995. Role of mast cells and basophils in gastrointestinal inflammation. Chem.Immunol 62, 187-203.
(28) Fox CC., S.D.Jewell, C.C.Withacre. 1994. Rat peritoneal mast cells present antigen to a PDD specific T cell line. Cell.Immunol 158; 253-264.
(29) Mekori YA., D.D.Metcalfe. 1999. Mast cell-T cell interactions. J.Allergy Clin.Immunol. 104, 517-523.
(30) Frandji P, C.Oskeritzian, F.Cacaraci, J.Lapeyre, R.Peronet, B.David, JG.Guillet, S.Mecheri. 1993. Antigen-dependent stimulation by bone marrow-derived mast cells of MHC class II-restricted T cell hybridoma. The Journal of Immunology; 151(11):6318-6328.
(31) Pankawar R, M.Okuda, H.Yssel, K.Okumura, C.Ra. 1997. Nasal mast cells in perennial allergic rhinitis exhibit increased expression of the Fc epsilon RI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J Clin Invest 99, 1492-1499.
(32) Gelbmann CM, S.Mestermann, V.Gross, M.Kollinger, J.Scholmerich, W.Falk. 1999. Strictures in Crohn`s disease are characterised by an accumulation of mast cells colocalised with laminin but not with fibronectin or vitronectin. Gut 45, 210-217.
(33) Bischoff SC, J.Wedemeyer, A.Herrmann, P.N.Meier, C.Trautwein, Y.Cetin, H.Maschek, M.Stolte, M.Gebel, M.P.Manns. 1996. Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology 28, 1-13.
(34) Wershil BK. 2000. Mast cell-deficient mice and intestinal biology. Am.J.Physiol.Gastrointest.Liver Physiol. 278, 343-348.
(35) Plaut M, J.H.Pierce, C.J.Watson, J.Hanley-Hyde, R.P.Nordan, W.E.Paul. 1989. Mast cell lines produce lymphokines in response to cross-linkage of Fc epsilon RI or to calcium ionophores. Nature 339, 624-631.
(36) Harvima R, L.Schwartz. 1993. Mast cell derived mediators. Immunopharmacology of mast cells and basophils. J.C.Foreman, ed Academic Press Ltd.London , 115.

Literaturverzeichnis
78
(37) Feger F, S.Varadaradjalou, Z.Gao, S.N.Abraham, M.Arock. 2003. The role of mast cells in host defence and their subversion by bacterial pathogens. Trends Immunol. 23, 151-158.
(38) Maurer D, G.Stingl. 1995. Immunglobulin E-binding structures on antigen-presenting cells present on skin and blood. J.Invest Dermatol. 104, 707-710.
(39) Sihra B, O.Kon, J.Grant, A.Kay. 1997. Expression of high-affinity IgE-receptors on peripheral blod basophils, monocytes, and eosinophils in atopic and nonatopic subjects: Relationship to total Serum IgE-concentrations. J.Allergy Clin.Immunol. 99, 699-706.
(40) Kinet JP. 1999. The high affinity IgE-receptor: from physiology to pathology.Annu.Rev.immunol. 17, 931-972.
(41) Beavil A, R.Beavil, C.Chan, J.Cook, H.Gould, A.Henry, R.Owens, J.Shi, B.Sutton, R.Young. 1993. Structural basis of the IgE-FcåRI interaction. Biochem.Soc.Trans. 21, 968-972.
(42) Blank U, C.Ra, J.P.Kinet. 1991. Characterization of truncated α-chain products from human, rat and mouse high affinity receptor for immunoglobulin E. J.Biol.Chem. 266, 2639-2646.
(43) Hakimi J, C.Seals, J.Kondas, L.Pettine, W.Danho, J.Kochan. 1990. The α-subunit of the human IgE receptor is sufficient for high affinity IgE binding. J.Biol.Chem. 265, 22079-22081.
(44) Blank U, J.Rivera. 2004. The ins and outs of IgE-dependent mast cell exocytosis. Trends Immunol. 25, 266-273.
(45) Grant J, M.Humbert, L.Taborda-Barata, B.Sihra, O.Kon, K.Rajakulasingam, S.Durham, A.Kay. 1997. High-affinity IgE-receptor Fc Epsilon RI Expression in allergic reactions. Int.Arch.Allergy Immunol. 113, 376-378.
(46) Jouvin MH, J.P. Kinet. 2005. Signal transduction through the conserved motifs of the high affinity IgE-receptor FcåRI. Semin.Immunol. 7, 29-35.
(47) Gibbs BF, S.W.Schmutzler, I.B.Vollrath, P.Broshardt, U.Braam, H.H.Wolff, G.Zwadlo-Klarwasser. 1999. Ambroxol inhibits the release of histamine, leukotrienes and cytokines from human leukocytes and mast cells. Inflamm.res. 48, 86-93.
(48) de Paulis AR, A.Cirillo, G.Ciccarelli, A.de Crescenco, A.Oriente, G.Marone.
1991. Characterization of the anti-inflammatory effect of FK-506 on human mast cells. J.Immunol. 147, 4278-4285.
(49) Daeron M, A.R.Sterk, F.Hirata, T.Ishizaka. 1982. Biochemical analysis of glucocorticoid inhibition of IgE-mediated histamine release from mouse mast cells. J.Immunol. 129, 1212-118.

Literaturverzeichnis
79
(50) Stellato C, A.de Paulis, A.Ciccarelli, R.Cirillo, V.Patella, V.Casolaro, G.Marone. 1992. Anti-inflammatory effect of cyclosporin A on human skin mast cells. J.Invest Dermatol. 98, 800-804.
(51) Simons FER. 1992. Clinical importance of inhibition of mediator release by anthistaminics. J.Allergy Clin.Immunol. 90, 699-728.
(52) Kroegel C, V.Herzog, B.Knöchel, P.Julius, D.Wagnetz, J.C.Virchow, W.Luttmann. 1996. Anti-inflammatory actions os histamine H1 receptor antagonists unrelated to H1 receptor blockade. Clin Immunother 5, 449-460.
(53) Norris A, E.Alton. 1996. Chloride transport and the action of sodium cromoglycate and nedocromil sodium in asthma. Clin Exp Allergy 26, 250-253.
(54) Schmutzle W. W.Heppt, H.Renz, M.Röcken, editors. 1998. Antiallergische und antientzündliche Pharmakotherapie. Allergologie, Springer-Verlag, 160-163.
(55) Heusser N, P.Jardieu. 1997. Therapeutic potential of anti-IgE-antibodies. Curr.Opin.Immunol. 9(6), 805-813.
(56) Burgess T, R.B Kelly. 1987. Constitutive and regulated secretion of proteins. Annu Rev Cell Biol 6, 243-293.
(57) Miller S, H.P.Moore. 1990. Regulated secretion. Curr Opin Cell Biol 2, 642-647.
(58) Schneider SW. 2001. kiss and run mechanism in exocytosis. J.Membrane Biol. 181, 67-76.
(59) Burgoyne RD, A.Morgan. 2003. Secretory Granule Exocytosis. Physiol Rev. 83, 581-632.
(60) Südhof T. 1995. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375, 645-653.
(61) Jahn R, T.C. Südhof. 1999. Membrane fusion and exocytosis. Annu Rev Biochem 68, 863-911.
(62) Rothman JE. 1994. Mechanisms of intracellular protein transport. Nature 372, 55-62.
(63) Söllner T, S.Whiteheart, M.Brunner, H.Erdjument-Bromage, S.Geromanos, P.Tempst, J.Rothman. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318-324.
(64) Söllner T, M.Bennett, S.Whiteheart, R.Scheller, J.Rothman. 1993. A protein assembly-disassembly pathway in vitro that may correspondent to sequential steps of synaptic vesicle docking, activation and fusion. Cell 75, 409-418.
(65) Weber T, B.Zemelman, J.McNew, B.Westermann, M.Gmachl, F.Parlati, T.Söllner, J.Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell 92, 759-772.

Literaturverzeichnis
80
(66) Hanson PI, J.E.Heuser, R.Jahn. 1997. Neurotransmitter release - four years of SNARE complexes. Curr.Opin.Neurobiol. 7, 310-315.
(67) Hayashi T, H.McMahon, S.Yamasaki, T.Binz, Y.Hata, T.Südhof, H.Niemann. 1994. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO Journal 13, 5051-5061.
(68) Pelham HR. 1999. SNAREs and the secretory pathway-lessons from yeast. Experimental Cell Research 247, 1-8.
(69) Fasshauer D, R.B.Sutton, A.T.Brunger, R.Jahn. 1998. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc.Natl.Acad.Sci.U.S.A 95, 15781-15786.
(70) Baumert M, P.R.Maycox, F.Navone, C.P.De, R.Jahn. 1989. Synaptobrevin: An integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO Journal 8, 379-384.
(71) Chlicote TJ, T.Galli, O.Mundigl, et al. 1995. Cellubrevin and synaptobrevin: similar subcellular localization and biochemical properties in PC-12 cells. J.Cell.Biol. 129, 219-231.
(72) Bennet MK, N.Calakos, R.H.Scheller. 1992. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255-259.
(73) Kretzschmar S, W.Volknandt, H.Zimmermann. 1996. Colocalization on the same synaptic vesicles of syntaxin and SNAP-25 with synaptic vesicle proteins: a re-evaluation of functional models required? Neurosci Res 26, 141-148.
(74) Bennet MK, J.E.Garcia-Arras, L.A.Elferink. 1993. The syntaxin family of vesicular transport receptors. Cell 74, 863-873.
(75) Ravichandran V, A.Chawla, P.A.Roche. 1996. Identification of a novel syntaxin and synaptobrevin/VAMP-binding protein, SNAP-23, expressed in non-neuronal tissues. J Biol Chem 271, 13300-13303.
(76) Oyler GA, G.A.Higgins, R.A.Hart. 1989. The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J.Cell.Biol. 109, 3039-3052.
(77) Wong PP, N.Daneman, A.Volchuk. 1997. Tissue distribution of SNAP-23 and its sucellular localization in 3T3-LI cells. Biochem Biophys Res Commun 230, 64-68.
(78) Sutton RB, D.Fasshauer, R.Jahn, R.and A.T.Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis. Nature 395, 347-353.
(79) Yuanyuan H, R.H.Scheller. 2001. Three SNARE complexes cooperate to mediate membrane fusion. Proc Natl Acad Sci U S A 98, 8065-8070.

Literaturverzeichnis
81
(80) Rizo J, T.Südhof. 2002. SNAREs and Munc18 in synaptic vesicle fusion. Nat Rev Neurosci 395, 641-653.
(81) Whiteheart S, I.C.Griff, M.Brunner, et al. 1993. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature 362, 353-355.
(82) Hay JC, R.H.Scheller. 1997. SNAREs and NSF in targeted membrane fusion. Curr Opin Cell Biol 9, 505-512.
(83) Chamberlain L, D.Roth, A.Morgan, D.Burgoyne. 1995. Distinct effects of α-SNAP, 14-3-3 proteins and calmodulin on priming and triggering of regulated exocytosis. J.Cell.Biol. 130, 1063-1070.
(84) McMahon H, T.Südhof. 1995. Synaptic core complex of synaptobrevin, syntaxin, and SNAP-25 forms high affinity α-SNAP binding site. J Biol Chem 270, 2213-2217.
(85) Whiteheart S, K.Rossnagel, S.A.Buhrow, M.Brunner, R.Jaenicke, J.Rothman. 1994. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J.Cell.Biol. 126, 945-954.
(86) Söllner T, J.Rothman. 1994. Neurotransmission: harnessing fusion machinery at the synapse. TINS 17, 344-348.
(87) Cremona O, P.DeCamilli. 1997. Synaptic vesicle endocytosis. Curr.Opin.Neurobiol. 7, 323-330.
(88) Brose N, A.G.Petrenko, T.C. Südhof, R.Jahn. 1992. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256, 1021-1025.
(89) Südhof TC, J.Rizo. 1996. C2-Domain proteins that regulate membrane traffic. Neuron 17, 379-388.
(90) Chapman ER. 2002. A Ca2+-sensor that triggers exocytosis? Nat Rev Mol Cell Biol 3, 498-508.
(91) Fujita H, P.Tuma, C.Finnegan, L.Locco, A.Hubbard. 1998. Endegenous syntaxins 2,3, and 4 exhibit distinct but overlapping patterns of expression at the hepatocyte plasma membrane. Biochem.J. 329, 527-538.
(92) Jo I, H.Harris, A.Amendt-Raduege, R.Majewski, T.Hammond. 1995. Rat kidney papilla contains abundant synaptobrevin protein that participates in the fusion of antidiuretic hormone-regulated water channel-containing endosomes in vitro. Proc Natl Acad Sci U S A 92, 1876-1880.
(93) Jöns T, S.Lenhardt, H.Bigalke, H.Heim, G.Ahnert-Hilger. 1999. SNARE-proteins and rab3A contribute to canalicular formation in parietal cells. J.Cell.Biol. 78, 779-786.
(94) Guo Z, C.Turner, D.Castle. 1998. Relocation of the t-SNARE SNAP-23 from lamellipodia-like cell surface projections regulates compound exocytosis in mast cells. 1998;. Cell 94, 537-548.

Literaturverzeichnis
82
(95) Vaidyanathan VV, N.Puri, P.A. Roche. 2001. Related Articles, The last exon of SNAP-23 regulates granule exocytosis from mast cells. J Biol Chem 276(27), 25101-25106.
(96) Paumet F, J.LeMao, S.Martin, T.Galli, B.David, U.Blank, M.Roa. 2000. Soluble NSF attachment protein receptors (SNAREs) in RBL-2H3 mast cells: functional role of syntaxin 4 in exocytosis and identification of a vesicle-associated membrane protein 8-containing secretory compartment. J Immunol 164, 5850-5857.
(97) Baram D, Y.A.Mekori, R.Sagi-Eisenberg. 2005. Synaptotagmin regulates mast cell functions. Immunol Rev 179, 25-34.
(98) Hibi T, N.Hirashima, M.Nakanishi. 2000. Rat basophilic leukemia cells express syntaxin-3 and VAMP-7 in granule membranes. Biochem Biophys Res Commun 271, 36-41.
(99) Galli T, A.Zahraoi, V.V.Vaidyanathan. 1998. A novel tetanus neurotoxin-insensitive vesicle-associated membrane protein in SNARE complexes of the apical plasma membrane of epithelial cells. Mol Biol Cell 146, 1437-1448.
(100) Advani RJ, B.Yang, R.Prekeris, K.C.Lee, J.Klumperman, R.H.Scheller. 1999. VAMP-7 mediates vesicular transport from endosomes to lysosomes. J.Cell.Biol. 146, 765-776.
(101) Wong SH, T.Zhang, Y.XU, V.N.Subramaniam, G.Griffiths, W.Hong. 1998. Endobrevin, a novel synaptobrevin/VAMP-like protein preferentially associated with the early endosome. Mol Biol Cell 9, 1549-1563.
(102) Mullock BM, C.W.Smith, G.Ihrke, N.A.Bright, M.Lindsay, E.J.Parkinson. 2000. Syntaxin 7 is localized to late endosome compartments, associates with VAMP 8, and is required for late endosome-lysosome fusion. Mol Biol Cell 11, 3137-3153.
(103) Blott EJ, G.M.Griffiths. 2002. Secretory lysosomes. Nat Rev Mol Cell Biol 3, 122-131.
(104) Coco S, G.Raposo, S.Martinez, J.J.Fontaine, S.Takamori, A.Zahraoui. 1999. Subcellular localization of tetanus neurotoxin-insensitive vesicle-associated membrane protein (VAMP)/VAMP7 in neuronal cells: evidence for a novel membrane compartment. J Neurosci 19, 9803-9812.
(105) Andrews NW. 2000. Regulated secretion of conventional lysosomes. Trens Cell Biol 10, 316-321.
(106) Stinchcombe JC, G.M.Griffiths. 1999. Regulated secretion from hemopoietic cells. J.Cell.Biol. 147, 1-6.
(107) Baram D, M.Linial, Y.A.Mekori, R.Sagi-Eisenberg. 1998. Ca-dependent exocytosis in mast cells is stimulated by the Ca-sensor, synaptotagmin I. J Immunol 161, 5120-5123.

Literaturverzeichnis
83
(108) Baram D, R.Adachi, O.Medalia, M.Tuvim B.F.Dickey, Y.A.Mekori. 1999. Synaptotagmin II negatively regulates Ca2+-triggered exocytosis of lysosomes in mast cells. J.Exp.Med 189, 1649-1658.
(109) Rigoni M.et al. 2001. Site-directed mutagenesis identifies active-site residues of the light chain of botulinum neurotoxin type A: a report of two cases. Biochem Biophys Res Commun 288, 1231-1237.
(110) Burnett JC, J.J.Schmidt, C.F.McGrath, T.L.Nguyen, A.R.Hermone, R.G.Panchal, J.L.Vennerstrom, K.Kodukula, D.W.Zaharevitz, R.Gussion, S.Bavari. 2005. Conformational sampling of the botulinum neurotoxin serotype A light chain: implications for inhibitor binding. Bioorg Med Chem 13, 333-341.
(111) Dekleva M, B.DasGupta. 1990. Purification and charcterization of a protease from botulinum neurotoxin type A that nicks single chain type A Neurotoxin into the Di-chain form. J.Bacteriol 172, 2498-2503.
(112) Black J, J.Dolly. 1987. Selective location of acceptors for Botulinum Neurotoxin A in the central and peripheral nervous system. Neuroscience 23, 767-779.
(113) Montecucco C, G.Schiavo, Z.Gao, E.Bauerlein, P.Boquet, B.DasGupta. 1988. Interaction of botulinum and tetanus toxins with the lipid bilayer surface. Biochem.J. 251, 379-383.
(114) Black J, J.Dolly. 1986. Interaction of 125 J-labeled botulinum neurotoxins with nerve terminals. Autoradiographic evidence for its uptake into motor nerves by acceptor-mediated endocytosis. J.Cell.Biol. 103, 535-544.
(115) Hanson M, R.Stevens. 2002. Structural view of botulinum neurotoxin in numerous functional states. Scientific and therapeutic aspects of botulinum toxin, In: Brin MF, Hallet M, Jankovic J, eds , 11-28.
(116) Stecher B, U.Weller, E.Habermann, M.Gratzl, G.Ahnert-Hilger. 1989. The light chain but not the heavy chain of botulinum A toxin inhibits exocytosis from permeabilized adrenal chromaffin cells. FEBS Letters 255, 391-394.
(117) Atassi MZ. 2004. Basic immunological aspects of botulinum toxin therapy. Mov.Disord. 19, 68-84.
(118) Blasi J, E.Chapman, S.Yamasaki, T.Binz, H.Niemann, R.Jahn. 1993. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO Journal 12, 4821-4828.
(119) Blasi J, E.Chapman, E.Link, T.Binz, S.Yamasaki, P.De Camilli, T.Südhof, H.Niemann, R.Jahn. 1993. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 365, 160-163.
(120) Schiavo G, O.Rossetto, S.Catsicas, P.Polverino de Laureto, B.DasGupta, F.Benfenati, C.Montecucco. 1993. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A,D and E. J.Biol.Chem. 268, 23784-23787.

Literaturverzeichnis
84
(121) Schiavo G, M.Matteoli, C.Montecucco. 2000. Neurotoxins affecting neuroexocytosis. Physiol Rev. 80, 716-766.
(122) Schiavo G, F.G. van der Goot. 2001. The bacterial toxin toolkit. Nat Rev Mol Cell Biol 2, 530-537.
(123) Jankovic J. 2004. Botulinum toxin in clinical practice. J.Neurol.Neurosurg.Psychiatry 75, 951-957.
(124) Clarke C. 1992. Therapeutic potential of Botulinum Toxin in neurological disorders. Quarterly J.Med. 299, 197-205.
(125) Bergelson JM, J.A.Cunningham, G.Droguett, E.A.Kurt-Jones, A.Krithivas, J.S.Hong, M.S.Horwitz, R.L.Crowell, R.W.Finberg. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5 . Science 275, 1320-1323.
(126) Walters RW, P.Freimuth, T.O.Moninger, I.Ganske, J.Zabner, M.J.Welsh. 2002.
Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape . Cell 110 (6), 789-799.
(127) Greber UF, M.Willetts, P.Webster, A.Helenius. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75, 477-486.
(128) Doronin K, K.Toth, M.Kuppuswamy, P.Krajcsi, A.E.Tollefson, WS Wold. 2003. Overexpression of the ADP (E3-11.6K) Protein Increases Cell Lysis and Spread of Adenovirus . Virology 305, 378-387.
(129) Mitani K., S.Kochanek, C.T.Caskey, F.L.Graham. 1995. Rescue, propagation, and partial purification of a helper virus-dependent adenovirus vector. Proc Natl Acad Sci U S A 92, 3854-3858.
(130) Hitt M.M., Ch.L.Addison, F.L.Graham. 2005. Human Adenovirus Vectors for Gene Transfer into Mammalian Cells. Adv Pharmacol 40, 137-206.
(131) Graham FL, L.Prevec. 1995. Methods for Construction of Adenovirus Vectors. Mol.Biotechn. 3, 207-220.
(132) Chartier C. 1996. Efficient Generation of Recombinant Adenovirus Vectors by Homologous Recombination in Escherichia coli. J.Virol 70(7), 4805-4810.
(133) Graham FL, J.Smiley, W.C.Russell, R.Nairn. 1977. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J.Gen.Virol. 36, 59-72.
(134) Walev I, S.C.Bhakdi, F.Hofmann, F.Djonder, A.Valeva, K.Aktories, S.Bhakdi. 2001. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U S A. 98(6), 3185-3190.

Literaturverzeichnis
85
(135) Hoffmann HJ, T.Bjerke, M.Karawajczyk RD, M.A.Knepper, S.Nielsen. 2001. SNARE proteins are critical for regulated exocytosis of ECP from human eosinophils. Biochem Biopys Res Commun 282, 194-199.
(136) He T-C, S.Zhou, L.T.daCosta, J.Yu, K.W.Kinzler, B.Vogelstein. 1998. A Simplified System for Generating Recombinant Adenoviruses. Proc.Natl.Acad.Sci. 95, 2509-2514.
(137) Kuhnel F, B.Schulte, T.Wirth, N.Woller, S.Schafers, L.Zender, M.Manns, S.Kubicka. 2004. Protein Transduction Domains Fused to Virus Receptors Improve Cellular Virus Uptake and Enhance Oncolysis by Tumor-Specific Replicating Vectors. J Virol; 78(24):13743-13754.
(138) Logan MR, P.Lacy, B.Bablitz, R.Moqbel. 2002. Expression of eosinophil target SNAREs as potential cognate receptors for vesicle-associated membrane protein-2 in exocytosis. J Allergy Clin Immunol 109, 299-306.
(139) Shukla A, L.Berglund, L.P.Nielsen, H.J.Hoffmann, R.Dahl. 2001. Regulated exocytosis in immune function: are SNARE-proteins involved? Respiratory Medicine; 95(10):773-780.
(140) Galli T, A.Zahraoui, V.V.Vaidyanathan, G.Raposo, J.M.Tian, M.Karin, H.Niemann, D.Louvard. 1998. A Novel Tetanus Neurotoxin-insensitive Vesicleassociated Membrane Protein in SNARE Complexes of the Apical Plasma Membrane of Epithelial Cells. Molecular Biology of the Cell 9, 1437-1448.
(141) Schiavo G, C.C.Shone, M.K.Bennett, R.H.Scheller, C.Montecucco. 1995.Botulinum Neurotoxin Type C Cleaves a Single Lys-Ala Bond within the Carboxyl-terminal Region of Syntaxins. J Biol Chem; 270(18):10566-10570.
(142) Fujita-Yoshigaki J, Y.Dohke, M.Hara-Yokoyama, Y.Kamata, S.Kozaki, S.Furuyama, H.Sugiya. 1996. Vesicle-associated Membrane Protein 2 Is Essential for cAMP-regulated Exocytosis in Rat Parotid Acinar Cells. The Inhibition of cAMP-Dependent Amylase Release by Botulinum Neurotoxin B. J Biol Chem; 271(22):13130-13134.
(143) Knight DE. Botulinum toxin types A, B and D inhibit catecholamine secretion from bovine adrenal medullary cells. FEBS Letters 1986; 207(2):222-226.
(144) Lang J, H.Zhang, V.V.Vaidyanathan, K.Sadoul, H.Niemann, C.B.Wollheim. 1997. Transient expression of botulinum neurotoxin C1 light chain differentially inhibits calcium and glucose induced insulin secretion in clonal [beta]-cells. FEBS Letters; 419(1):13-17.
(145) Bartels F, H.Bergel, H.Bigalke, J.Frevert, J.Halpern, J.Middlebrook. 1994. Specific Antibodies against the Zn2+-binding Domain of Clostridial Neurotoxins Restore Exocytosis in Chromaffin Cells Treated with Tetanus or Botulinum A Neurotoxin. Journal of Biological Chemistry 269(11), 8122-8127.

Literaturverzeichnis
86
(146) Massie B, F.Couture, L.Lamoureux, D.D.Mosser, C.Guilbault, P.Jolicoeur, F.Belanger, Y.Langelier. 1998. Inducible Overexpression of a Toxic Protein by an Adenovirus Vector with a Tetracycline-Regulatable Expression Cassette. J Virol; 72(3):2289-2296.
(147) Pellegrini LL, V.O'Connor, F.Lottspeichl, H.Betz. 1995. Clostridial neurotoxins compromise the stability of a low energy SNARE complex mediating NSF activation of synaptic vesicle fusion. The EMBO Journal 14 (19), 4705-4713.
(148) Scales SJ, J.B.Bock, R.H.Scheller. 2000. Cell biology: The specifics of
membrane fusion. Nature; 407(6801):144-146.
(149) Rao SK, C.Huynh, V.Proux-Gillardeaux, T.Galli, N.W.Andrews. 2004. Identification of SNAREs Involved in Synaptotagmin VII-regulated Lysosomal Exocytosis. J Biol Chem; 279(19):20471-20479.
(150) Proux-Gillardeaux V, R.Rudge, T.Galli. 2005. The Tetanus Neurotoxin-Sensitive and Insensitive Routes to and from the Plasma Membrane: Fast and Slow Pathways? Traffic 6, 366-373.
(151) Braun V, V.Fraisier, G.Raposo, I.Hurbain, J.B.Sibarita, P.Chavrier, T.Galli, F.Niedergang. 2004. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. The EMBO Journal 23, 4166-4176.

Danksagung
87
7. Danksagung
Mein vornehmlicher Dank gilt Herrn Professor Michael P. Manns für die
Bereitstellung von Laborflächen und Arbeitsgeräten und die Unterstützung des
Projekts.
Besonders möchte ich mich bei Herrn Professor Stephan C. Bischoff bedanken,
der die vorliegende Disseration betreut und ermöglicht hat.
Des Weiteren danke ich Herrn Dr. Axel Lorentz für die wissenschaftliche
Betreuung und Einführung in wichtige Arbeitsmethoden, sowie für die konstruktiv-
kritische Durchsicht der Arbeit.
Auch Herrn Leif Sander möchte ich danken für die Vorarbeiten in diesem Projekt,
sowie die vielen Anregungen und seine selbstverständliche Diskussionsbereitschaft.
Außerdem danke ich den Mitgliedern der Arbeitsgruppe von Herrn Professor
Kubicka, und Herrn Dr. Florian Kühnel im Besonderen, für ihre freundliche
Unterstützung beim Erlernen viraler Arbeitsmethoden und ihre selbstverständliche
Diskussionsbereitschaft.
Des Weiteren danke ich Herrn Professor Hans Bigalke der Abteilung für
Toxikologie, des Instituts Pharmakologie und Toxikologie, sowie Herrn Dr. Volker
Specht von der Firma Biotecon, Potsdam und Herrn Dr. Florian Wegner von der
Neurologischen Klinik der Universität Leipzig für ihre freundliche Unterstützung und
Anregungen beim experimentellen Teil meiner Arbeit.
Außerdem danke ich Herrn Tibor Veres vom Frauenhofer-Institut für Toxikologie
und experimentelle Medizin, Abteilung Allergologie und Immunologie für seine
freundliche Hilfe beim Erstellen der konfokalmikroskopischen Bilder. Herrn Dr. Tierry
Galli am INSERM, Paris danke ich für die freundliche Überlassung des anti-VAMP-7
Antikörpers.
Ich möchte auch den Mitarbeitern der Abteilung für Viszeral- und Transplantations-
chirurgie der Medizinischen Hochschule unter Leitung von Herrn Professor Jürgen
Klempnauer ausdrücklich für die gute Zusammenarbeit danken.
Einen großen Dank möchte ich den gegenwärtigen und ehemaligen Mitgliedern
unserer Arbeitsgruppe aussprechen. Hervorheben möchte ich die sehr gute

Danksagung
88
Arbeitsatmosphäre, Unterstützung und Zusammenarbeit - auch außerhalb der
Laborarbeit.
Nicht zuletzt möchte ich mich bei meinen Freunden für ihre motivierende und
moralische Unterstützung bedanken.
Meiner Familie und besonders meinem Vater danke ich für die langjährige finanzielle
und moralische Unterstützung meines Studiums und meiner Dissertation. Deswegen
möchte ich die Doktorarbeit meiner Familie widmen.

Lebenslauf
89
8. Lebenslauf
Persönliche Daten
Name: Seza Bolat
Geburtsdatum/-ort: 29.04.1981 in Constanta (RO)
Wohnort: Rautenstrasse 4, 30171 Hannover
Familienstand: ledig
Schulbildung
1987- Einschulung, Celle
2000 Erwerb der Allgemeinen Hochschulreife, Celle
Hochschulausbildung
2000 Beginn des Studiums der Humanmedizin an der
Medizinischen Hochschule Hannover (MHH)
2002 Ärztliche Vorprüfung
2003 1. Abschnitt der Ärztlichen Prüfung
2006 2. Abschnitt der Ärztlichen Prüfung
2006-2007 Praktisches Jahr am Klinikum Braunschweig,
Universitätsklinikum Kapstadt, Südafrika,
Medizinische Hochschule Hanover
Mai 2007 3. Abschnitt der Ärztlichen Prüfung
Berufliche Tätigkeiten
seit Juni 2007 Assistentärztin an der Medizinischen Hochschule
Hannover, Abteilung Neurologie
Hannover, den 28. Februar 2008
Seza Bolat

9. Erklärung nach §2 Abs. 2 Nr. 5 und 6 PromO
90
9. Erklärung nach §2 Abs. 2 Nr. 5 und 6 PromO
Ich erkläre, dass ich die der Medizinischen Hochschule Hannover zur Promotion
eingereichte Dissertation mit dem Titel
„Expression und Funktion von SNARE-Proteinen in humanen intestinalen Mastzellen“
in der Abteilung Gastroenterologie, Hepatologie und Endokrinologie der
Medizinischen Hochschule Hannover
unter der Betreuung von Professor Dr. med. S. C. Bischoff
ohne sonstige Hilfe durchgeführt und bei der Abfassung der Dissertation keine
anderen als die dort aufgeführten Hilfsmittel benutzt habe. Ich habe diese
Dissertation bisher an keiner in- oder ausländischen Hochschule zur Promotion
eingereicht. Weiterhin versichere ich, dass ich den beantragten Titel noch nicht
erworben habe.
Hannover, den 28. Februar 2008
Seza Bolat


















