
Fluoreszenz-Spektroskopie · Spin-Verbot: Der Gesamtspin bzw. die Multiplizität darf sich während...
19
Fluoreszenz-Spektroskopie Physikalisches Fortgeschrittenenpraktikum FP28 Dr. Katharina Förg Lehrstuhl für Festkörperchemie Raum 3004 (R) Stand: 10/2016
Transcript of Fluoreszenz-Spektroskopie · Spin-Verbot: Der Gesamtspin bzw. die Multiplizität darf sich während...

Fluoreszenz-Spektroskopie Physikalisches Fortgeschrittenenpraktikum FP28
Dr. Katharina Förg
Lehrstuhl für Festkörperchemie
Raum 3004 (R)
Stand: 10/2016

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
1
Inhaltsverzeichnis
1 Spektroskopische Methoden und Lumineszenzprozesse .................................... 2
2 Leuchtstoffe ......................................................................................................... 4
3 Seltenerd-Elemente ............................................................................................. 6
4 Emission von Lanthanoid-Ionen ........................................................................... 8
4.1 Eu3+-Lumineszenz ......................................................................................... 9
4.2 Eu2+-Lumineszenz ....................................................................................... 10
4.3 Ce3+-Lumineszenz ....................................................................................... 11
4.4 Tb3+-Lumineszenz ....................................................................................... 11
5 Ligand-Metall Charge-Transfer-Übergänge ....................................................... 12
5.1 CaWO4 ........................................................................................................ 12
5.2 CdWO4 ........................................................................................................ 13
6 Versuchsdurchführung ....................................................................................... 14
7 Auswertung ........................................................................................................ 15
8 Literatur .............................................................................................................. 16

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
2
1 Spektroskopische Methoden und Lumineszenzprozesse
Spektroskopische Methoden sind ein wichtiger Teil der modernen Analytik. Sie beruhen auf
der energieaufgelösten Wechselwirkung elektromagnetischer Strahlung mit Materie. Diese
Wechselwirkung lässt sich mit der Bohr’schen Gleichung beschreiben. Die Energiedifferenz
(∆E) zwischen Anfangs- und Endzustand ist dann von der Strahlungsfrequenz (ν) und dem
Planckschen Wirkungsquantum (h = 6.626·10-34 Js) abhängig:
Gleichung 1: Bohr’sche Frequenzbedingung.
Die elektromagnetische Strahlung im UV-Vis Bereich hat eine Wellenlänge von 200–800 nm
(500000–12000 cm-1). Dies entspricht der Energiedifferenz zweier elektronischer Zustände.
Trifft elektromagnetische Strahlung auf eine Substanz, deren Energie der Energiedifferenz
zwischen Grund- und angeregtem Zustand (Gleichung 1) exakt entspricht, so können
Elektronen unter Absorption der entsprechenden Frequenzen in das höhere Energieniveau
angeregt werden.
Selbst bei passender Energie tritt der Übergang nicht automatisch ein, sondern hängt von
der elektrischen Feldkomponente des Lichtes ab. Die Wahrscheinlichkeit, dass ein Elektron
einen Übergang vollzieht, wird quantenmechanisch durch das beschrieben, welches die
Änderung des Dipolmomentes während des Überganges (proportional zur Signalintensität)
beschreibt. Aus dem Drehimpulserhaltungssatz lassen sich Gesetze ableiten, die eine
Abschätzung des Übergangsmomentes erlauben.
Übergangsverbote [1,2,3,4]:
Spin-Verbot: Der Gesamtspin bzw. die Multiplizität darf sich während eines
Überganges nicht ändern. Der Spin s eines Elektrons ist entweder +½ oder -½.
Symmetrie-Verbot: Das Ausgangs- und Endorbital eines Überganges dürfen nicht
orthogonal (Integral des Produktes = 0) sein. Übergänge zwischen Orbitalen gleicher
Parität (g→g, u→u) sind verboten.
Überlappungs-Verbot: Übergänge sind nur zwischen überlappenden Molekülorbitalen
möglich.
Die Vorgänge während der Anregung und den möglichen Emissionen von Photonen sind im
Jablonski Diagramm (Abbildung 1) zusammengefasst [2,5,6].
hEEE mn

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
3
Abbildung 1: Jablonski Diagramm. A: Anregung, F: Fluoreszenz, P: Phosphoreszenz, S0: Singulett
Grundzustand, S1, S2: angeregte Singulett Zustände, T: Triplett Zustand, IC: Internal Conversion, ISC:
Intersystem Crossing, R: Strahlungslose Relaxation, ν: Schwingungszustand, gerade Pfeile: optische
Übergänge, geschwungene Pfeile: strahlungslose Übergänge [7].
Durch Absorption eines energetisch geeigneten Photons wird ein Elektron in einen
energetisch höheren, unbesetzten Zustand angeregt. Dieser Vorgang findet innerhalb von
10-15 s statt. Abhängig von der Spin-Orientierung des angehobenen Elektrons ist der
angeregte Zustand ein Triplett- (T1, parallel, Gesamtspin = 1) oder ein Singulett-Zustand
(S1, antiparallel, Gesamtspin = 0), wobei letzterer wahrscheinlicher ist. Der Triplett-Zustand
liegt energetisch immer etwas niedriger als der Singulett-Zustand. Nach weiteren 10-12 s ist
das Elektron über Schwingungsrelaxation (R) und internal conversion (IC) aus energetisch
höheren elektronischen Schwingungszuständen (S2) (ν > 0) (Franck-Condon
Anregungszustand) in den niedrigsten, den nullten Schwingungszustand (ν = 0) des ersten
angeregten elektronischen Zustands (S1) zurückgegangen. Die Energieabstände
benachbarter Schwingungszustände liegen bei 0.1 eV, die der rotatorischen Unterniveaus
eine Größenordnung niedriger, weshalb sie im Jablonski Diagramm nicht mehr dargestellt
sind. Das angeregte Elektron befindet sich nun im thermischen Gleichgewicht, aus dem die
Emission eines Photons (Photolumineszenz) stattfinden kann.
Photolumineszenzprozesse werden in Fluoreszenz (F) und Phosphoreszenz (P) unterteilt.
Die Kriterien der Aufteilung sind allerdings nicht immer einheitlich. Die Phosphoreszenz
beschreibt Prozesse bei denen die Emission nach der Anregung noch lange anhält. Oft
verwendet man 1 ms als zeitliche Untergrenze. Fluoreszenz beschreibt demzufolge schnelle
Prozesse mit weniger als 1 μs Lebensdauer. Da diese Definition einerseits eine Lücke von
drei Größenordnungen lässt und andererseits keinen physikalischen Bezug zum

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
4
zugrundeliegenden Mechanismus aufweist, verwendet man heute häufiger die beteiligten
elektronischen Zustände als Definition. Hier beschreibt die Fluoreszenz alle erlaubten
Übergänge zwischen Niveaus ohne Spinänderung (∆S = 0), während die Phosphoreszenz
alle Prozesse mit Spinänderung (∆S ≠ 0), z. B. durch intersystem crossing (ISC), beschreibt
[2].
2 Leuchtstoffe
Leuchtstoffe (engl.: phosphors) sind heute aufgrund der Vielzahl ihrer Anwendungen aus
dem alltäglichen Leben nicht mehr wegzudenken. Hauptsächlich finden sie in Bildschirmen
und Leuchtstofflampen, aber auch in Röntgendetektoren Einsatz, wobei intensive Forschung
in den letzten Jahrzehnten zu einer stetigen Verbesserung der Lumineszenzeigenschaften
wie der spektralen Energieverteilung und der Quantenausbeute geführt hat [8-10].
Die Geschichte der Leuchtstoffe ist noch relativ jung, da für die Lichterzeugung in frühester
Zeit noch keine Leuchtstoffe eingesetzt wurden. Zwar waren Stoffe, wie der Phosphor mit
seinen phosphoreszierenden Eigenschaften, bereits im 17. Jahrhundert bekannt [11]. Zur
Beleuchtung wurden allerdings bis zum 20. Jahrhundert direkte Lichtquellen wie Fackeln, Öl,
oder Petroleumlampen, in der Straßenbeleuchtung auch Gaslampen, verwendet. In den 20er
und 30er Jahren des letzten Jahrhunderts kamen Leuchtstoffe erstmals in der Beleuchtung
zum Einsatz. Die verwendeten Quecksilberentladungslampen waren eine vielversprechende
Lichtquelle, doch emittieren diese einen Großteil ihres Lichtes im UV-Bereich. Diese
Leuchtstofflampen basieren auf der Quecksilber-Niederdruckentladung (quecksilber-
dampfgesättigte Edelgasatmosphäre mit wenigen mbar Druck), die im ersten Schritt die
elektrische Leistung in UV-C-Strahlung bei 254 nm umwandelt (dabei entsteht noch ca. 15 %
Strahlung bei 185 nm). Im zweiten Schritt wird diese Strahlung durch Leuchtstoffe in
sichtbares Licht transformiert [10].
Eine Quecksilber-Hochdruckentladung verschiebt die Emission auf die sichtbaren Linien
zwischen 250 und 550 nm. Doch fehlt bei diesen immer noch der Rotanteil, um es als weiße
Lichtquelle mit ausreichender Farbwiedergabe und Effizienz zu verwenden. Als Lösung fand
sich der Einsatz von Leuchtstoffen [8].
1937 wurde die erste Fluoreszenzlampe vorgestellt, die aus einer Mischung von MgWO4 und
M2SiO4:Mn2+ (M = Zn, Be) bestand [12]. Ein Nachteil des Einsatzes dieser Mischung in einer
Niederdruck-Entladungslampe ist jedoch, dass sie leicht Quecksilber aus dem Gas aufnimmt
und unter UV-Licht leicht zerfällt. Die in den 1940er Jahren entwickelte Halophosphatlampe,
die auf mit Sb3+ oder Mn2+ dotiertem Halophosphat Ca5(PO4)3X (X = F, Cl) basiert [13-15],
war ein bedeutender Durchbruch auf dem Gebiet von Leuchtstofflampen. Diese Leuchtstoffe

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
5
können sowohl im blauen (Sb3+) als auch im orangefarbenen Spektralbereich (Mn2+)
emittieren, wodurch bei additiver Farbmischung weißes Licht entsteht. Halophosphatlampen
haben noch heute einen hohen Marktanteil. Aktuell versucht man ihre Lebensdauer zu
erhöhen sowie eine Verschiebung des Farbpunktes während der Lebensphase zu
vermeiden.
In den 1970er Jahren entwickelten Opstelten und Koedam das Drei-Banden-Konzept [16],
welches die Grundlage der heute gebräuchlichen Fluoreszenzlampen bildet [17]. Die
Kombination der blau, rot und grün emittierenden Leuchtstoffe BaMgAl10O17:Eu2+ (BAM),
Y2O3:Eu3+ sowie LaPO4:Ce3+,Tb3+, CeMgAl11O17:Tb3+ und GdMgB5O10:Tb3+ ermöglicht die
Erzeugung von weißem Licht mit sehr guten Quantenausbeuten [18].
Mit der Entwicklung der blau emittierenden Leuchtdiode (LED, light emitting diode) auf der
Basis von InGaN Anfang der 1990er Jahre [19] und der stetigen Verbesserung ihrer Effizienz
in der Folgenzeit [20] ergaben sich neue Möglichkeiten auf dem Beleuchtungsmarkt. Wegen
ihrer hohen Energieeffizienz von mehr als 100 lm/W, ihrer Helligkeit, der kompakten
Bauweise und der langen Lebensdauer revolutionieren Hochleistungs-LEDs den
Beleuchtungsmarkt [8]. Mit dem EU-weiten Verbot herkömmlicher Glühlampen mit
Wolframdrahtwendel bis 2012 hat die Entwicklung und Optimierung alternativer weißer
Lichtquellen wie Fluoreszenzlampen und warm-weißen LEDs zusätzlich vorangetrieben.
Ein wichtiger LED-Leuchtstoff ist YAG:Ce3+ [19], der sich wegen seiner breiten grün-gelben
Bande und seiner hohen Quantenausbeute als sehr effizient erwies. Als LED-Leuchtstoffe
werden seit kurzem auch Leuchtstoffe auf Nitrid-Basis eingesetzt. Das Prinzip der drei
Farben-LEDs basiert auf der Beschichtung eines blau emittierenden GaInN-Chip mit dem
orange leuchtenden Nitridosilicat Sr2Si5N8:Eu2+ [21] und den grün fluoreszierenden
Oxonitridosilicaten CaSi2O2N2 Eu2+ [22] oder SrSi2O2N2:Eu2+ [21].
Inzwischen sind so genannte UV-LEDs ((Al,Ga)N) möglich, die mit drei unterschiedlichen
Leuchtstoffen (blau, rot, grün) Emissionswellenlängen bis 630 nm ermöglichen [23]. Die
große Herausforderung besteht darin, drei verschiedene Leuchtstoffe auf einem LED-Chip
zu positionieren. Dieses Problem versucht man nun mit Zwei-Farben-Leuchtstoffen zu lösen.
Dabei wird ein geeignetes Wirtgitter mit zwei Aktivatoren dotiert, um mindestens zwei
Wellenlängen zu emittieren. Ein Beispiel hierfür ist α-Sr(PO3)2:Eu2+,Mn2+, das unter Anregung
mit UV-Licht weißes Licht emittiert [24].
Es gibt weiterhin große Forschungsanstrengungen zur Entwicklung neuer lanthanoidhaltiger
Leuchtstoffe. Insbesondere die Verbesserung der Lebensdauer sowie der spektralen
Stabilität neben einer Anpassung an die voranschreitende Miniaturisierung
optoelektronischer Bauteile und nicht zuletzt die Suche nach kostengünstigen Alternativen
stehen im Vordergrund.

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
6
Da Lanthanoide neben hervorragenden optischen Eigenschaften auch über interessante
magnetische Eigenschaften verfügen, bietet sich die Möglichkeit multifunktioneller
Werkstoffe [25].
Mit der Entdeckung hochenergetischer Röntgenstrahlen im Jahre 1895 find das Mineral
Scheelit (CaWO4) seit 1896 als Leuchtstoff in der Röntgendiagnostik Verwendung. Der
Einsatz von CaWO4 war erforderlich, da der zuvor verwendete Fotofilm zur Detektion der
Röntgenstrahlung nicht sehr empfindlich war, woraus lange Aufnahmezeiten resultierten. Die
lange Bestrahlungszeit lieferte unscharfe Bilder und führte zu hohen Strahlendosen für den
Menschen, von denen die negativen Einflüsse heutzutage gut bekannt sind. Der Gebrauch
von CaWO4 als Röntgenleuchtstoff im Röntgenschirm diente der Verstärkung der
Signalintensität und führte zu einer Verringerung der Bestrahlungszeit um drei
Größenordnungen. CaWO4 hält mit über 75 Jahren den absoluten Rekord in der Anwendung
als Leuchtstoff, wurde jedoch mit der Entdeckung und Entwicklung von lanthanoidaktivierten
Leuchtstoffsystemen als Szintillatormaterial abgelöst [26]. Neben der historischen
Verwendung von CaWO4 als Szintillatormaterial bilden die Wolframatverbindungen eine
große anorganische Substanzklasse, welche höchst interessante physikalische
Eigenschaften besitzt. So finden sie Verwendung in der Photokatalyse, der
Quantenelektronik [27] und werden als Materialien mit negativen
Temperaturausdehnungskoeffizienten eingesetzt [28].
3 Seltenerd-Elemente
Unter den Seltenerd-Elementen (SE) versteht man die Elemente Scandium (Sc), Yttrium (Y)
sowie die 15 auf das Barium folgenden Elemente, die eine Untergruppe der Nebengruppe
des Periodensystems bilden und als Lanthanide bezeichnet werden. Aufgrund der
besonderen Periodizität in den physikalischen und chemischen Eigenschaften der
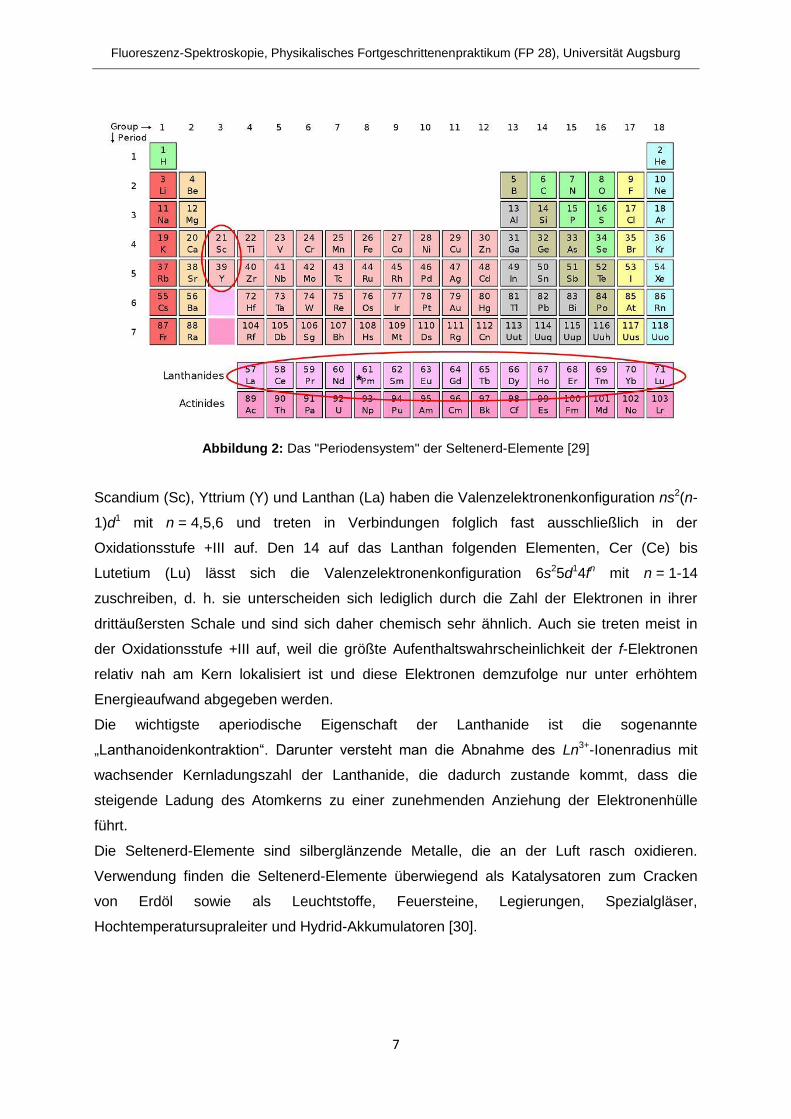
Lanthanide ist es zweckmäßig, die Seltenerd-Metalle gemäß Abbildung 2 in ein eigenes
„Periodensystem“ einzuteilen [1]. Das Promethium (Pm) ist darin mit einem „*“ markiert, weil
es als einziges Seltenerd-Element ausschließlich radioaktive Isotope besitzt.
Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
7
Abbildung 2: Das "Periodensystem" der Seltenerd-Elemente [29]
Scandium (Sc), Yttrium (Y) und Lanthan (La) haben die Valenzelektronenkonfiguration ns2(n-
1)d1 mit n = 4,5,6 und treten in Verbindungen folglich fast ausschließlich in der
Oxidationsstufe +III auf. Den 14 auf das Lanthan folgenden Elementen, Cer (Ce) bis
Lutetium (Lu) lässt sich die Valenzelektronenkonfiguration 6s25d14fn mit n = 1-14
zuschreiben, d. h. sie unterscheiden sich lediglich durch die Zahl der Elektronen in ihrer
drittäußersten Schale und sind sich daher chemisch sehr ähnlich. Auch sie treten meist in
der Oxidationsstufe +III auf, weil die größte Aufenthaltswahrscheinlichkeit der f-Elektronen
relativ nah am Kern lokalisiert ist und diese Elektronen demzufolge nur unter erhöhtem
Energieaufwand abgegeben werden.
Die wichtigste aperiodische Eigenschaft der Lanthanide ist die sogenannte
„Lanthanoidenkontraktion“. Darunter versteht man die Abnahme des Ln3+-Ionenradius mit
wachsender Kernladungszahl der Lanthanide, die dadurch zustande kommt, dass die
steigende Ladung des Atomkerns zu einer zunehmenden Anziehung der Elektronenhülle
führt.
Die Seltenerd-Elemente sind silberglänzende Metalle, die an der Luft rasch oxidieren.
Verwendung finden die Seltenerd-Elemente überwiegend als Katalysatoren zum Cracken
von Erdöl sowie als Leuchtstoffe, Feuersteine, Legierungen, Spezialgläser,
Hochtemperatursupraleiter und Hydrid-Akkumulatoren [30].
*
Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
8
4 Emission von Lanthanoid-Ionen
Emissionslinien mit geringer Halbwertsbreite sind für Lanthanoidionen charakteristisch und
resultieren aus den durch die 5s und 5p Orbitale gut abgeschirmten 4f-4f-
Intrakonfigurationsübergängen [15]. Durch diese Abschirmung ist der Einfluss des Wirtgitters
(= Liganden) auf die spektrale Lage der Linien gering. Die energetische Lage der
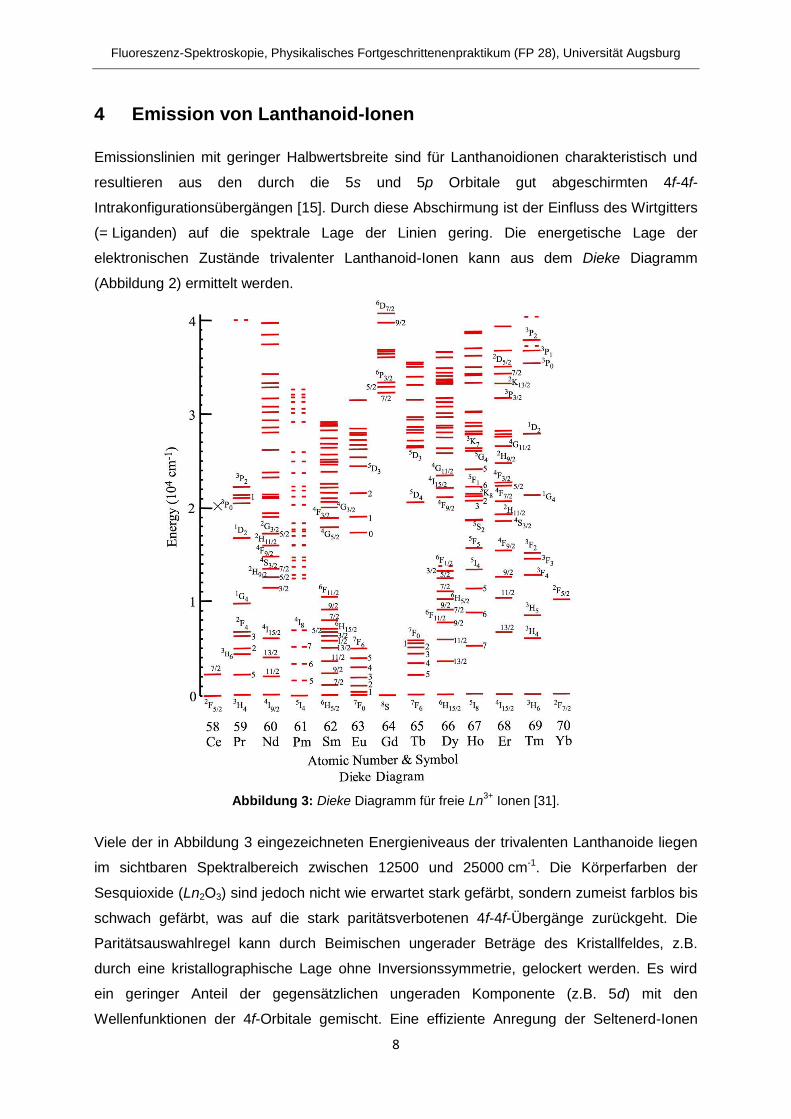
elektronischen Zustände trivalenter Lanthanoid-Ionen kann aus dem Dieke Diagramm
(Abbildung 2) ermittelt werden.
Abbildung 3: Dieke Diagramm für freie Ln3+
Ionen [31].
Viele der in Abbildung 3 eingezeichneten Energieniveaus der trivalenten Lanthanoide liegen
im sichtbaren Spektralbereich zwischen 12500 und 25000 cm-1. Die Körperfarben der
Sesquioxide (Ln2O3) sind jedoch nicht wie erwartet stark gefärbt, sondern zumeist farblos bis
schwach gefärbt, was auf die stark paritätsverbotenen 4f-4f-Übergänge zurückgeht. Die
Paritätsauswahlregel kann durch Beimischen ungerader Beträge des Kristallfeldes, z.B.
durch eine kristallographische Lage ohne Inversionssymmetrie, gelockert werden. Es wird
ein geringer Anteil der gegensätzlichen ungeraden Komponente (z.B. 5d) mit den
Wellenfunktionen der 4f-Orbitale gemischt. Eine effiziente Anregung der Seltenerd-Ionen

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
9
kann durch 4f→5d-Übergänge und über Ladungstransfer (engl. charge transfer (CT)) vom
Wirtgitter oder Sensibilisatoren auf den Aktivator stattfinden. Allgemein gilt, dass CT-
Übergänge bei relativ leicht reduzierbaren Seltenerd-Ionen, wie Sm3+, Eu3+ und Yb3+, und
4fn→4fn-15d-Übergänge bei leicht oxidierbaren Ionen wie Ce3+, Pr3+und Tb3+ stattfinden.
Beide Arten dieser Übergänge sind erlaubt und sowohl im Anregungs- als auch im
Emissionsspektrum treten breite Banden auf.
Die Kristallfeldaufspaltung der f-Orbitale (Strichbreite der Energiezustände) der Seltenen
Erden ist mit ca. 100 cm-1 verhältnismäßig klein im Vergleich zu den Übergangsmetallionen
(5dn), bei denen sie etwa 10000 cm-1 beträgt. Dies ist ebenfalls auf die gute Abschirmung
durch die 5s und 5p Orbitale zurückzuführen.
Zur Vermeidung einer Löschung der Emission durch Energieüberträge zwischen den
einzelnen Aktivatoren sollten diese in der Regel bei verbotenen elektronischen Dipol-
Übergängen mindestens 5-8 Å voneinander im Wirtgitter entfernt sein. Man spricht in diesem
Fall auch von konzentrationsbedingter Löschung. Dabei handelt es sich um einen
Energietransfer von Aktivator zu Aktivator, der schließlich bei Defektstellen im Kristallgitter
enden kann und so zur Auslöschung führt. Diese Art des Energieverlustes ist bevorzugt, da
dieser Prozess eine um einige Größenordnungen (~104) schnellere Übertragungsrate besitzt
als die Relaxation vom angeregten in den Grundzustand. Ein Energietransfer über größere
Distanzen als 5–8 Å (bis einige 10 Å) ist möglich, jedoch ineffektiv. Bei Aktivator-Abständen
von kleiner als 5 Å werden Energietransportvorgänge sehr effizient und führen so schon oft
zur Löschung der Emission bei Dotierungsgraden von wenigen Prozent.
4.1 Eu3+-Lumineszenz
Eu3+ ([Xe]4f6) ist ein typischer f-f Linienemitter im roten Spektralbereich und besitzt daher
Anwendungen in der Beleuchtungs- und Displaytechnologie [20,32,33,34]. Neben der
stabileren Oxidationsstufe +III besitzt Europium auch die zweiwertige Oxidationsstufe Eu2+
[1] (Kapitel 4.2).
Im Grundzustand besitzt Eu3+ das Termsymbol 7F0. Durch die Absorption verschiedener
Energien elektromagnetischer Strahlung ist eine Anregung in die 5DJ, 5LJ
5HJ und
Energieniveaus möglich [35,31,36]. Wie bereits erwähnt finden strahlungslose
Relaxationsprozesse zum niedrigsten Schwingungszustand 5D0 statt, nach denen die
Emission im sichtbaren Bereich zu den elektronischen Grundzuständen 7FJ folgt. Die
ursprünglich spin- und paritätsverbotenen Übergänge führen durch Beimischen
entgegengesetzter Parität zu den bekannten Emissionen von 5D0 zu den 7FJ Energieniveaus
(J = 0,1,2,3,4) bzw. den entsprechenden Wellenlängen um 580, 590, 620, 650 und 695 nm.

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
10
Im Vergleich dazu besitzen die 5D0→7F5 und 5D0→
7F6 Übergänge schwache Intensitäten und
liegen im nahen IR Bereich bei ca. 750 und 810 nm [36].
Untersuchungen von Blasse et al. zeigten variierende Verhältnisse der Hauptübergänge
5D0→7F1 und 5D0→
7F2. In Verbindungen mit einem Inversionszentrum dominiert der 5D0→7F1
Übergang, da er unabhängig von der Lagesymmetrie ist [37]. Da elektrische Dipol-
Übergänge strikt verboten sind, muss dieser Übergang ein magnetischer Dipol-Übergang
sein. Liegt Eu3+ auf einer Lage ohne Inversionszentrum, dominiert der 5D0→7F2 Übergang
durch Beimischen von Zuständen entgegengesetzter Parität. Diese Übergänge sind jetzt
erlaubt und werden als „hypersensitiv“ bezeichnet. Oft führen kleine Abweichungen von der
Inversionssymmetrie zu 5D0→7F1 und 5D0→
7F2 Übergängen mit ähnlicher Intensität [38,39].
4.2 Eu2+-Lumineszenz
Aufgrund seiner paritätserlaubten f-d-Übergänge im sichtbaren Bereich hat sich Eu2+ als
effizienter Absorber und Emitter in Verbindungen etabliert, die als Leuchtstoffe für
Fluoreszenzlampen verwendet werden (z.B. BaMgAl10O17:Eu2+ (BAM) [40], Sr4Al14O25: Eu2+
und Sr5(PO4)3Cl:Eu2+ [15]). Verbindungen, die Sr2+, Ca2+ und Ba2+ Ionen enthalten, eigenen
sich zur Dotierung mit Eu2+ aufgrund ihrer ähnlichen Ionenradien (rEu2+ = 1.17 pm,
rCa2+ = 1.00 pm, rSr
2+ = 1.18 pm, rBa2+ = 1.35 pm) [41,42]. Im Grundzustand besitzt Eu2+ das
Termsymbol 8S7/2 und die elektronische Konfiguration [Xe]4f75d06s0. Die Koordinationssphäre
hat einen starken Einfluss auf die Ligandenfeldaufspaltung der 5d Orbitale. Je stärker die
Wechselwirkung der Liganden mit Eu2+, desto stärker ist die Ligandenfeldaufspaltung der 5d
Orbitale (Abbildung 4).
Abbildung 4: Schematische Darstellung der f-d Ligandenfeldaufspaltung in Abhängigkeit von der
Ligandenstärke [7].

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
11
Dadurch nähern sich die unteren 5d Orbitale den 4f Orbitalen an, wodurch die Anregung
eines Elektrons in die unteren 5d Orbitale in den sichtbaren Bereich geschoben wird. Die
Emissionen Eu2+ enthaltender Verbindungen reichen vom UV (BaFBr:Eu2+ [43]), in den
blauen (BaMgAl10O17:Eu2+ (BAM), λmax = 453 nm [40,44]), grünen (Eu2Si2O2N2:Eu2+,
λmax = 575 nm [21,45]) bis hin in den roten Teil (Ba2Si5N8:Eu2+, λmax = 610 nm [21,45]) des
sichtbaren Spektrums. Dies ermöglicht eine Vielzahl an Möglichkeiten die Emissionsmaxima
entsprechend dem Liganden zu verschieben.
4.3 Ce3+-Lumineszenz
Ce3+ ist ein weiteres Beispiel für einen typischen f-d Emitter. Die Übergänge sind erlaubt und
relativ schnell. Ein sehr bekanntes Beispiel eines gelben Breit-Band-Leuchtstoffes ist
Y3Al5O12:Ce3+ (YAG:Ce). Es absorbiert stark im blauen Spektralbereich um 450 nm und
emittiert gelbes Licht im Bereich um 560 nm. Dieses System zeichnet sich neben seiner
hohen chemischen und thermischen Stabilität durch eine hohe Quantenausbeute aus
[8,15,20,32,40,46]. In Kombination mit einer blauen LED (z.B. GaN) [19] kann weißes Licht
mit einer Effizienz vergleichbar mit Halogenlampen erzeugt werden [8,15,20,32,40,46].
Im Grundzustand besitzt Ce3+ das Termsymbol 2F5/2 mit der Elektronenkonfiguration
[Xe]4f15d06s0. Sowohl Anregungs- als auch Emissionswellenlängen hängen stark von der
Ligandenfeldaufspaltung der 5d Orbitale ab. Stärkere Wechselwirkungen führen zu einer
stärkeren Aufspaltung und damit zu einer Verschiebung der Emissionswellenlänge zu
niedrigerer Energie (größere Wellenlänge). Abhängig von der chemischen Umgebung
können Emissionen zwischen 510 und 280 nm verschoben werden [8].
4.4 Tb3+-Lumineszenz
Wie die meisten der dreiwertigen Lanthanoidionen ist Tb3+ ([Xe]4f8) ein typischer f-f
Linienemitter im gelb-grünen Spektralbereich [15,40]. Im Grundzustand besitzt Tb3+ das
Termsymbol 7F6 [31,35,47]. Anregung erfolgt im UV-Bereich und über strahlungslose
Relaxationsprozesse erreicht das System den untersten Vibrationszustand 5D4. Darauf folgt
die Emission in die elektronischen Grundzustände 7FJ. Die bekannten Emissionen von 5D4 zu
7F6, 7F5,
7F4 und 7F3 im gelb-grünen Bereich des sichtbaren Spektrums liegen bei ca. 490,
543, 583 und 617 nm. Übergänge von 5D4 zu 7F2, 7F1 und 7F0 sind sehr schwach im orange-
roten Bereich und liegen bei ca. 645, 664 und 674 nm [47].

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
12
5 Ligand-Metall Charge-Transfer-Übergänge
Ligand-Metall Charge-Transfer-Übergänge (LMCT-Übergänge) sind elektronische
Übergänge aus den Orbitalen der Liganden in die Orbitale eines Kations. Innerhalb
komplexer Anionen (z.B. WO42-) treten sie auch zwischen Kationen und Anionen auf. Sie
sind nach der Laporte-Auswahlregel erlaubt und darum gelten für sie im Wesentlichen die
gleichen Eigenschaften, die bereits zu den 4fn→4fn-15d1-Übergängen (f-d) herausgestellt
wurden: LMCT-Übergänge sind sehr intensiv, besitzen eine kurze Lebensdauer und
erscheinen in Spektren aufgrund ihrer starken Kopplung mit Liganden-Schwingungen als
breite Banden. Je stärker der nephelauxetische Effekt (Elektronenwolkenerweiternder
Effekt), desto niedriger ist die Übergangsenergie. Formal handelt es sich um eine
vorübergehende Reduktion des Kations bzw. des Zentralatoms in Anionen (z.B. WO42-) als
angeregten Zustand [1,8].
Tendenziell zeigen die Ln3+-Ionen LMCT-Übergänge bei niedrigeren Energien, die dazu
neigen in den zweiwertigen Oxidationszustand überzugehen [48]. Es handelt sich dabei um
Sm3+, Eu3+, Tm3+ und Yb3+, denn durch Aufnahme eines Elektrons nähern sie sich den
besonders stabilen Elektronenkonfigurationen von Gd3+ oder Lu3+ bzw. erreichen sie sogar
[26].
Wie weiter oben bereits angedeutet sind LMCT Banden für d0-Übergangsmetallionen
koordiniert mit Sauerstoff, wie z.B. in VO43-, NbO4
3- und WO42-, charakteristisch. Die spektrale
Position der Absorptionsbande wird durch das Ionisationspotential der d1→d0 Ionisation, der
Anzahl und Polarisierbarkeit der Liganden und von der gemeinsamen Wechselwirkung von
Ionen im Gitter bestimmt [1].
5.1 CaWO4
CaWO4 zeigt eine starke Anregungsbande bei 250 nm, das Absorptionsmaximum von
CaMoO4 hingegen liegt bei 290 nm, trotz identischer Kristallstruktur, Symmetrie,
Bindungslängen und -winkel. Diese Rotverschiebung der Absorptionsbande von CdMoO4 ist
auf das höhere sechste Ionisationspotential von Mo (70 eV) relativ zu W (61 eV)
zurückzuführen. Der Vergleich der Absorption von {WO4}-Tetraedern im CdWO4 mit {WO6}-
Oktaedern im Cd3WO6, zeigt eine Rotverschiebung der Absorptionsbande um ca. 35 nm mit
steigender Ligandenzahl. Die Verbindungen CdWO4 und Cd3WO6 besitzen beide eine weiße
Körperfarbe mit Bandlücken von 250 bzw. 285 nm im ultravioletten Spektralbereich. Die
Bandlücke von WO3 hingegen befindet sich im sichtbaren Spektralbereich, da es eine gelbe
Körperfarbe besitzt. Diese Abweichung erklärt sich durch die unterschiedliche Verknüpfung
der Wolframat-Einheiten. So liegen in CaWO4 und Ca3WO6 die {WO4}-Tetraeder und {WO6}-

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
13
Oktaeder isoliert in der Kristallstruktur vor. Im WO3 hingegen werden die Sauerstoffatome
der {WO6}-Oktaeder geteilt und Wechselwirkungen der optischen Zentren durch
Ladungsübertragung findet statt [48].
5.2 CdWO4
Als Szintillatoren bezeichnet man Materialien, die Teilchenstrahlung z.B. in Form von
Röntgenstrahlung, absorbieren und die dadurch aufgenommene Energie in Form von
elektromagnetischer Strahlung wieder abgeben. Diese liegt meist im optisch sichtbaren
Bereich des elektromagnetischen Spektrums. Diese Werkstoffe zählen zu den ältesten
Nachweismitteln für Röntgen- oder auch radioaktive Strahlung. Einer der am längsten
verwendeten Szintillatoren ist silberdotiertes Zinksulfid (ZnS:Ag), Gd2O2S:Tb3+ [49], CsI:Tl+
[50] sowie CaWO4 [26].
In Verbindungen wie CdWO4 wird ein Elektron aus dem Valenzband in das Leitungsband
angeregt. Das erzeugt ein Loch im normalerweise gefüllten Leitungsband. Durch Relaxation
kehrt das Elektron in das Valenzband zurück. Allerdings entspricht die Energie des
abgestrahlten Lichtquants genau der Energie der Bandlücke, sodass häufig Reabsorption
auftritt, was die Szintillationseffizienz verringert. Der Abstand zwischen Valenz- und
Leitungsband ist meist so groß, dass die Wellenlänge der abgestrahlten Photonen nicht
mehr im sichtbaren sondern im UV-Bereich liegt. Das Emissionsspektrum von Szintillatoren
ohne Aktivator hat sein Maximum meist im UV-Bereich, kann aber auch im sichtbaren
Bereich liegen (z.B. Cadmiumwolframat, CdWO4, 470 nm) [51].

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
14
6 Versuchsdurchführung
Es werden insgesamt sechs Proben am Fluoreszenzspektrometer vermessen
(Anregungs- und Emissionsspektren):
Na2Y(PO4)(WO4):Eu3+
Na2Y(PO4)(WO4):Tb3+
Y3Al5O12:Ce3+
Sr3(PO4)2:Eu2+
Sr(PO3)2:Eu2+,Mn2+
CdWO4
Der Assistent erklärt Ihnen die Probenpräparation, die Funktionsweise (Aufbau
Abbildung 5) und Bedienung des Geräts.
Abbildung 5: Schematischer Aufbau eines Fluoreszenzspektrometers.
Das Gerät ist empfindlich, daher bitte mit Vorsicht benutzen!
Ziel des Versuchs ist neben dem Erlernen der methodischen Vorgehensweise eine
Aussage über die erwartete Anregung und Emission (schwach/intensiv; schmal/breit;
Lage der Banden) machen zu können und zu verstehen weshalb genau diese
Banden auftreten (Kapitel 4).
Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
15
7 Auswertung
Vor dem Versuchstag recherchieren Sie nach Veröffentlichungen zu den oben
genannten Verbindungen. Diese sind alle in Fachjournalen veröffentlicht und online
einsehbar. Bitte bringen Sie die Ausdrucke der Veröffentlichungen zum Versuch mit.
Während der Versuchsdurchführung vergleichen Sie jedes Messergebnis mit den
Literaturdaten. Bei Abweichungen passen Sie den Messvorgang an bzw. diskutieren
dies mit Ihrem Assistenten.
Hinweis zum Protokoll:
Einleitung mit theoretischem Hintergrund, Geräteaufbau schematisch etc.
Versuchsdurchführung (kurz)
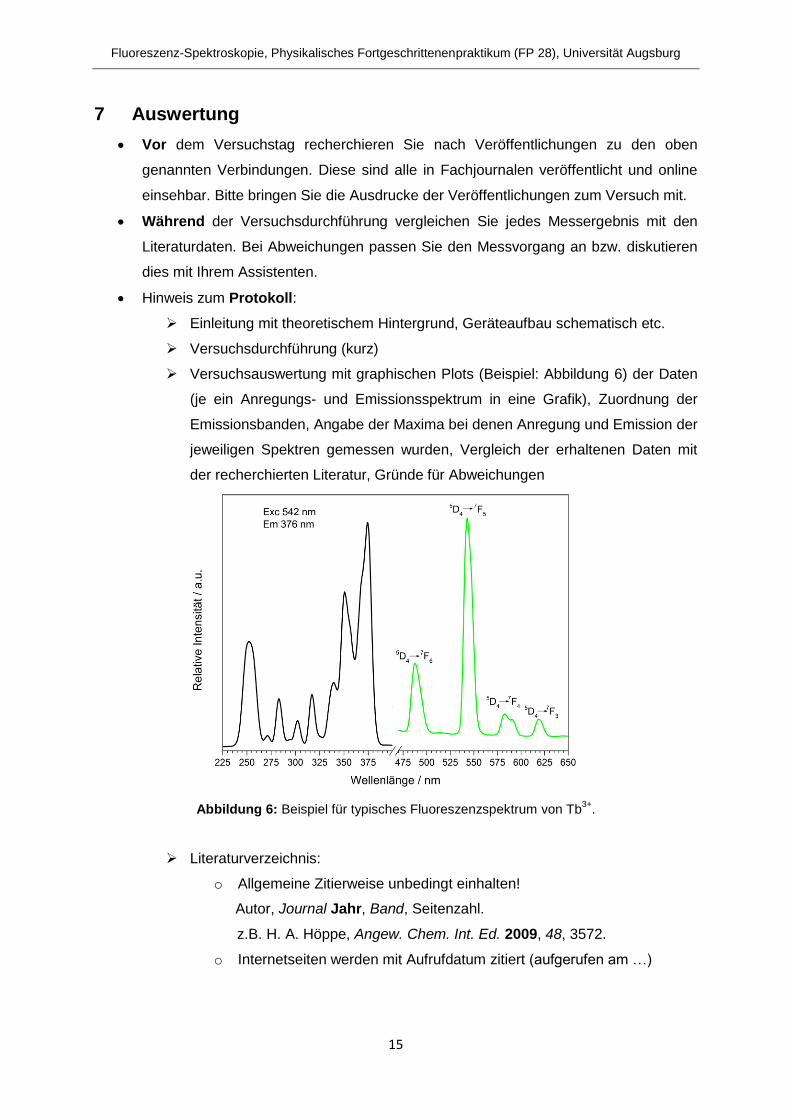
Versuchsauswertung mit graphischen Plots (Beispiel: Abbildung 6) der Daten
(je ein Anregungs- und Emissionsspektrum in eine Grafik), Zuordnung der
Emissionsbanden, Angabe der Maxima bei denen Anregung und Emission der
jeweiligen Spektren gemessen wurden, Vergleich der erhaltenen Daten mit
der recherchierten Literatur, Gründe für Abweichungen
Abbildung 6: Beispiel für typisches Fluoreszenzspektrum von Tb3+
.
Literaturverzeichnis:
o Allgemeine Zitierweise unbedingt einhalten!
Autor, Journal Jahr, Band, Seitenzahl.
z.B. H. A. Höppe, Angew. Chem. Int. Ed. 2009, 48, 3572.
o Internetseiten werden mit Aufrufdatum zitiert (aufgerufen am …)

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
16
8 Literatur
[1] A. Holleman, E. Wiberg, Lehrbuch der Anorganischen Chemie, deGruyter Berlin/New
York, 102. Ed., 2007.
[2] P. Skrabal, Spektroskopie, vdf Hochschulverlag AG an der ETH Zürich, Zürich, 2009.
[3] C. Janiak, H.-J. Meyer, D. Gudat, R. Alsfasser, Riedel Moderne Anorganische
Chemie, de Gruyter Berlin/Boston, 2012.
[4] J. E. Huheey, E. A. Keiter, R. L. Keiter, Anorganische Chemie, de Gruyter
Berlin/Boston, 2014.
[5] A. Sharma, S. G. Schulman, Introduction to Fluorescence Spectroscopy, Wiley New
York, 1999.
[6] J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science+
Business Media, LLC, 3. Ed., 2006.
[7] K. Förg, Dissertation, Universität Augsburg, 2016.
[8] T. Jüstel, H. Nikol, C. Ronda, Angew. Chem. 1998, 110, 3250; Angew. Chem. Int. Ed.
1998, 37, 3084.
[9] T. Welker, J. Lumin. 1991, 48/49, 49.
[10] P. Maestro, D. Huguenin, J. Alloys Compd. 1995, 225, 520.
[11] A. Oppenheim, H. Brand, Allgemeine Deutsche Biographie (ADB), Bd. 3, Duncker &
Humblot Leipzig, 1876.
[12] G. E. Inman, Trans, Illum. Eng. Soc. 1939, 34, 65; R. N. Thayer, B. T. Barnes, J. Opt.
Soc. Am. 1939, 29, 131.
[13] A. H. McKeag, P. W. Ranby (General Electric Company), GB-B 578.192, 1942.
[14] H. G. Jenkins, A. H. McKeag, P. W. Ranby, J. Electrochem. Soc. 1949, 96, 1.
[15] C. Feldmann, T. Jüstel, C. R. Ronda, P. J. Schmidt, Adv. Funct. Mater. 2003, 13, 511.
[16] M. Koedam, J. J. Opstelten, J. Light. Res. Technol. 1971, 3, 205.
[17] J. M. P. J. Verstegen, D. Radielovic, L E. Vrenken, J. Electrochem. Soc. 1974, 121,
1627.
[18] S. Ekambaram, K. C. Patil, M. Maaza, J. Alloys Compd. 2005, 393, 81.
[19] S. Nakamura, T. Mukai, M. Senoh, Appl. Phys. Lett. 1994, 64, 1687.
[20] H. A. Höppe, Angew. Chem. Int. Ed. 2009, 48, 3572.
[21] H. A. Höppe, H. Lutz, P. Morys, W. Schnick, A. Seilmeier, J. Phys. Chem. Solids 2000,
61, 2001.

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
17
[22] H. A. Höppe, F. Stadler, O. Oeckler, W. Schnick, Angew. Chem. 2004, 116, 5656;
Angew. Chem. Int. Ed. 2004, 43, 5540.
[23] A. Sandhu, Nat. Photonics 2007, 1, 37.
[24] H. A. Höppe, M. Daub, M. C. Bröhmer, Chem. Mater. 2007, 19, 6358.
[25] J. R. Mc Carthy, K. A. Kelly, E. Y. Sun, R. Weissleder, Nanomedicine 2007, 2, 153.
[26] G. Blasse, B. Grabmaier, Luminescent Materials, Springer Berlin, 1994.
[27] F. Lei, B. Yan, J.Phys. Chem C 2009, 113, 1074.
[28] A. Leleckaite, A. Kareiva, H. Bettentrup, T. Jüstel, Z. Anorg. Allg. Chem, 2005, 631,
2987.
[29] http://www.greenmotorsblog.de/wp-content/uploads/2012/02/Seltene-Erden-im-
Periodensystem2.jpg, abgerufen am 03.08.2016.
[30] H. R. Christen, G. Meyer, Allgemeine und Anorganische Chemie, Bd. 2, Otto Salle
Verlag Stuttgart, 1995.
[31] K. Ogasawara, S. Watanabe, H. Toyoshima, M. G. Brik, Handbook on the Physics and
Chemistry of Rare Earths, Bd. 37, Elsevier, 2007.
[32] M. Born, T. Jüstel, Physik Journal 2003, 2, 294.
[33] T. Jüstel, J.-C. Krupa, D. U. Wiechert, J. Lumin. 2001, 93, 179.
[34] M. Born, T. Jüstel, Chemie in unserer Zeit 2006, 40, 294.
[35] G. H. Dieke, H. M. Crosswhite, Appl. Opt. 1963, 2, 675.
[36] W. T. Carnall, P. R. Fields, K. Rajnak, J. Chem. Phys. 1968, 49, 4450.
[37] Y.-C. Li, Y.-H. Chang, Y.-F.Lin, Y.-S. Chang, Y.-J. Lin, J. Alloys Compd. 2007, 439,
367.
[38] G. Blasse, A. Bril, W. Nieuwpoort, J. Phys. Chem. Solids 1966, 27, 1587.
[39] G. Blasse, A. Bril, W. Nieuwpoort, J. Phys. Chem. Solids 1967, 47, 5442.
[40] C. Ronda, T. Jüstel, H. Nikol, J. Alloys Compd. 1998, 275-277, 669.
[41] R. D. Shannon, Acta Crsytallogr. A 1976, 32, 751.
[42] F. D. S. Butement, Trans. Faraday Soc. 1948, 44, 617.
[43] M. Thoms, H. von Seggern, A. Winnacker, Phys. Rev. B 1991, 44, 9240.
[44] P. Dorenbos, J. Lumin. 2003, 104, 239.
[45] F. Stadler, O. Oeckler, H. A. Höppe, M. H. Möller, R. Pöttgen, B. D. Mosel, P. Schmidt,
V. Duppel, A. Simon, W. Schnick, Chem. Eur. J. 2006, 12, 6984.
[46] D. J. Robbins, J. Electrochem. Soc. 1979, 126, 1550.
[47] W. T. Carnall, P. R. Fields, K. Rajnak, J. Chem. Phys. 1968, 49, 4447.
[48] G.Blasse, M. Wiegel, J. Alloys Compd. 1995, 224, 342.
[49] F. J. Garlick, R. A. Fatehally, Phys. Rev. 1949, 75, 1446.

Fluoreszenz-Spektroskopie, Physikalisches Fortgeschrittenenpraktikum (FP 28), Universität Augsburg
18
[50] T. Schmidt, IRE Trans. Nucl. Sci. 1960, 7, 25.
[51] Karim, Herstellung polykristalliner Szintillatoren für die Positronen-Emissions-
Tomographie, Jülich, Forschungsbericht.


















