
Hybrid DNA virus in Chinese patients with seronegative hepatitis
9
Corrections MICROBIOLOGY Correction for “Hybrid DNA virus in Chinese patients with se- ronegative hepatitis discovered by deep sequencing,” by Baoyan Xu, Ning Zhi, Gangqing Hu, Zhihong Wan, Xiaobin Zheng, Xiaohong Liu, Susan Wong, Sachiko Kajigaya, Keji Zhao, Qing Mao, and Neal S. Young, which appeared in issue 25, June 18, 2013, of Proc Natl Acad Sci USA (110:10264–10269; first pub- lished May 28, 2013; 10.1073/pnas.1303744110). The editors note that on September 10, 2013, PNAS received a letter to the editor from Naccache et al. (1), which raised concerns about a possible contaminant affecting the results of this paper. The authors were asked to provide a response to these concerns, but while we were awaiting their comments, the Office of Research Integrity (ORI) published a case summary (2) for Baoyan Xu on December 30, 2013. Based on a limited ad- mission by Xu, ORI found that Baoyan Xu had engaged in re- search misconduct. ORI recommended a correction to Fig. 6 and Fig. S4. After evaluation of the authors’ response (3) to Naccache et al. and the detailed information supplied below from the au- thors, the Editorial Board approved the following correction: The authors wish to note, “Correspondence from Stephen Korsman, Medical Virologist at Groote Schuur National Health Laboratory Service, South Africa, suggested that some immuno- blots in the above article contained multiple copied images, in which images were reproduced for several different patients. Using graphics-editing software (varying contrast and lighting of individual lanes of the blots), we confirmed that the images had been ex- tensively manipulated. This finding led to a formal complaint to and subsequent investigation by the Department of Health and Human Services ORI. ORI has formally concluded that one au- thor engaged in research misconduct (2). Misconduct appeared to be restricted to assembly of the final, complex figures. “We were able to retrieve and reconstruct digital images from the original experiments; only immunoblots from patients with seronegative hepatitis, in Fig. 6B and Fig. S4A, had been ma- nipulated. We further repeated immunoblotting experiments, using existing samples, and we used these data to replace both Fig. 6 and Fig. S4. This correction does not affect the original findings of the serology studies. We have evaluated additional healthy controls from different ethnicities, which we add to the Supporting Information as Fig. S4C. “We reproduced the experiments using sera samples from 77 Chinese hepatitis patients by immunoblotting as described in our paper. Fifteen patient sera samples that were used in the original study were unavailable. In addition, we tested sera samples from 28 healthy controls with origins from the United States, France, Korea, Brazil, China, Vietnam, Japan, and Puerto Rico. By immunoblot, 86% (66/77) of seronegative hepatitis patients were positive for NIH-CQV IgG, and 16% (12/77) were positive for IgM (Fig. 6B and Fig. S4A). Of healthy controls from different ethnicities, 78% (13/28) were positive for IgG (Fig. S4C). These data confirm the presence of an antibody response against recombinant capsid protein of NIH-CQV in the cohort of Chinese seronegative hep- atitis patients, and also in healthy individuals. Reactivity between the polyhistidine-tag and samples also was evaluated and excluded. However, whether the antibody response against recombinant NIH-CQV viral capsid protein detected by immunoblot was induced by infection with NIH-CQV or derives from cross re- activity with an epitope present on another microorganism remains to be clarified. Although we excluded cross reactivity between NIH-CQV and other common human parvoviruses, additional confirmatory experiments based on serologic reactivity to mul- tiple, nonoverlapping fragments derived from NIH-CQV capsid protein are in progress and the results will be reported.” The corrected figure and its corrected legend appear below. 1. Naccache SN, Hackett J, Delwart EL, Chiu CY (2014) Concerns over the origin of NIH- CQV, a novel virus discovered in Chinese patients with seronegative hepatitis. Proc Natl Acad Sci USA 111:E976. 2. http://ori.hhs.gov/content/case-summary-xu-baoyan. Accessed January 30, 2014. 3. Zhi N, et al. (2014) Reply to Naccache et al: Novel NIH-CQV virus a contaminant of DNA extraction method. Proc Natl Acad Sci USA 111:E977. 4344–4346 | PNAS | March 18, 2014 | vol. 111 | no. 11 www.pnas.org
Transcript of Hybrid DNA virus in Chinese patients with seronegative hepatitis

Corrections
MICROBIOLOGYCorrection for “Hybrid DNA virus in Chinese patients with se-ronegative hepatitis discovered by deep sequencing,” by BaoyanXu, Ning Zhi, Gangqing Hu, Zhihong Wan, Xiaobin Zheng,Xiaohong Liu, Susan Wong, Sachiko Kajigaya, Keji Zhao, QingMao, and Neal S. Young, which appeared in issue 25, June 18,2013, of Proc Natl Acad Sci USA (110:10264–10269; first pub-lished May 28, 2013; 10.1073/pnas.1303744110).The editors note that on September 10, 2013, PNAS received
a letter to the editor from Naccache et al. (1), which raisedconcerns about a possible contaminant affecting the results ofthis paper. The authors were asked to provide a response tothese concerns, but while we were awaiting their comments, theOffice of Research Integrity (ORI) published a case summary (2)for Baoyan Xu on December 30, 2013. Based on a limited ad-mission by Xu, ORI found that Baoyan Xu had engaged in re-search misconduct. ORI recommended a correction to Fig. 6 andFig. S4. After evaluation of the authors’ response (3) to Naccacheet al. and the detailed information supplied below from the au-thors, the Editorial Board approved the following correction:The authors wish to note, “Correspondence from Stephen
Korsman, Medical Virologist at Groote Schuur National HealthLaboratory Service, South Africa, suggested that some immuno-blots in the above article contained multiple copied images, inwhich images were reproduced for several different patients. Usinggraphics-editing software (varying contrast and lighting of individuallanes of the blots), we confirmed that the images had been ex-tensively manipulated. This finding led to a formal complaint toand subsequent investigation by the Department of Health andHuman Services ORI. ORI has formally concluded that one au-thor engaged in research misconduct (2). Misconduct appeared tobe restricted to assembly of the final, complex figures.“We were able to retrieve and reconstruct digital images from
the original experiments; only immunoblots from patients withseronegative hepatitis, in Fig. 6B and Fig. S4A, had been ma-nipulated. We further repeated immunoblotting experiments,
using existing samples, and we used these data to replace bothFig. 6 and Fig. S4. This correction does not affect the originalfindings of the serology studies. We have evaluated additionalhealthy controls from different ethnicities, which we add to theSupporting Information as Fig. S4C.“We reproduced the experiments using sera samples from 77
Chinese hepatitis patients by immunoblotting as described in ourpaper. Fifteen patient sera samples that were used in the originalstudy were unavailable. In addition, we tested sera samples from 28healthy controls with origins from the United States, France, Korea,Brazil, China, Vietnam, Japan, and Puerto Rico. By immunoblot,86% (66/77) of seronegative hepatitis patients were positive forNIH-CQV IgG, and 16% (12/77) were positive for IgM (Fig. 6Band Fig. S4A). Of healthy controls from different ethnicities, 78%(13/28) were positive for IgG (Fig. S4C). These data confirm thepresence of an antibody response against recombinant capsidprotein of NIH-CQV in the cohort of Chinese seronegative hep-atitis patients, and also in healthy individuals. Reactivity betweenthe polyhistidine-tag and samples also was evaluated and excluded.However, whether the antibody response against recombinantNIH-CQV viral capsid protein detected by immunoblot wasinduced by infection with NIH-CQV or derives from cross re-activity with an epitope present on another microorganism remainsto be clarified. Although we excluded cross reactivity betweenNIH-CQV and other common human parvoviruses, additionalconfirmatory experiments based on serologic reactivity to mul-tiple, nonoverlapping fragments derived from NIH-CQV capsidprotein are in progress and the results will be reported.”The corrected figure and its corrected legend appear below.
1. Naccache SN, Hackett J, Delwart EL, Chiu CY (2014) Concerns over the origin of NIH-CQV, a novel virus discovered in Chinese patients with seronegative hepatitis. Proc NatlAcad Sci USA 111:E976.
2. http://ori.hhs.gov/content/case-summary-xu-baoyan. Accessed January 30, 2014.3. Zhi N, et al. (2014) Reply to Naccache et al: Novel NIH-CQV virus a contaminant of DNA
extraction method. Proc Natl Acad Sci USA 111:E977.
4344–4346 | PNAS | March 18, 2014 | vol. 111 | no. 11 www.pnas.org

www.pnas.org/cgi/doi/10.1073/pnas.1402288111
Fig. 6. Immunoblot of seronegative hepatitis patient using recombinant capsid protein (rCP) of NIH-CQV. (A) Specificity test for the rCP of NIH-CQV. The celllysates derived from the cells transfected with the plasmids that expressed capsid proteins of AAV2, Parv4, HBoV, and B19V and purified rCP were subjected toSDS/PAGE, and then transferred to a nitrocellulose membrane. After blocking with 5% (wt/vol) nonfat milk for 2 h, the membrane was incubated withrespective antisera at 1:1,000 dilution. (B and C) Detection of specific antibodies against the rCP of NIH-CQV. Samples subjected to SDS/PAGE consisted of25 ng of affinity-purified rCP. These proteins were transferred to a nitrocellulose membrane and incubated with a 1:1,000 dilution of individual serum. Abovethe lanes are the identification numbers of individual patients and healthy controls. Only representative results are shown; see Fig. S4 A and B for completedata. Mouse monoclonal antibody against the polyhistidine tag was used as a positive control. The numbers on the left indicate molecular masses inkilodaltons based on the PageRuler Prestained Protein Ladder (Fermentas).
PNAS | March 18, 2014 | vol. 111 | no. 11 | 4345
CORR
ECTIONS

SYSTEMS BIOLOGYCorrection for “mChIP-KAT-MS, amethod tomap protein interactions and acetylation sites for lysine acetyltransferases,” by LeslieMitchell,SylvainHuard,Michael Cotrut, Roghayeh Pourhanifeh-Lemeri, Anne-Lise Steunou,Akil Hamza, Jean-Philippe Lambert,Hu Zhou, ZhibinNing, Amrita Basu, Jacques Côté, Daniel A. Figeys, and Kristin Baetz, which appeared in issue 17, April 23, 2013, ofProc Natl AcadSci USA (110:E1641–E1650; first published April 9, 2013; 10.1073/pnas.1218515110).The authors note “that a mistake was made generating the Epl1 mutant EPcA-Q shown in Fig. 3 B and C and Fig. S4. Instead of
mutating K39, 345, 376 and 379 to Q (glutamine, Gln) these sites were mutated to E (glutamic acid, Glu). Through gene synthesis, wehave generated the correct mutant and assessed its impact on NuA4. As anticipated, detectable acetylation signal on Epl1 mutantsEPcA-R and EpcA-Q was decreased (corrected Fig. 3B). We next sought to determine the function of Epl1 acetylation within the EPcAdomain. Although the EPcA-Q and EpCA-R mutants do not impact coimmunopurification with Eaf5-TAP, global histone H4acetylation was reproducibly decreased in esa1-L254P and EPcA-R (corrected Fig. 3B, lanes 2 and 3), in contrast EPcA-Q displayedglobal histone H4 acetylation similar to wild type Epl1 strains (corrected Fig. 3B, lanes 1 and 4). In agreement with this finding, thestrain expressing EPcA-Q does not display temperature sensitivity (corrected Fig. S4). In conclusion, while mutating K39, 345,376, and 379 to acidic glutamic acid has negative consequences for NuA4 catalytic activity (original Fig. 3), mutating these sites toglutamine does not. Therefore, as reported for other MYST KATs (1), autoacetylation does contribute to NuA4 catalytic activity.The authors apologize for this error.Please see the table below for new strains.”
As a result of these errors, the authors note that the following section should be added to the Materials and Methods.
“Cloning and Mutagenesis of Enhancer of Polycomb Like 1. Acetylation point mutants mimicking acetylated (K-to-Q) state for each acet-ylated lysine were generated by synthesizing a custom-designed DNA fragment (Genscript) of 857 base pairs carrying the mutationK345Q, K376Q, and K379Q and corresponding to nucleotides 600–1456 of EPL1. Following double digestion with BspE1/Nru1 and gelpurification (Qiagen; 28704), this mutated DNA fragment was ligated into pRS415-EPL1-HA3 (pKB41) that was also digested withBspE1/NruI, subjected to calf intestinal phosphatase treatment (New England Biolabs; M0290) and gel purified. Ligation was carried outovernight at 16 °C with T4 DNA ligase (New England Biolabs; M0202S), and then transformed into competent Escherichia coli DH5αcells. K39Q was then generated by using a site-directed mutagenesis kit (Stratagene; 200528). The successful introduction of allpoint mutations pRS415-epl1-EPcA-Q-HA3 (pKB264) was confirmed by sequencing and expression confirmed by Westernblot analysis.”
The authors also note that Fig. 3 and its corresponding legend appeared incorrectly. The corrected figure and its corrected legendappear below.
www.pnas.org/cgi/doi/10.1073/pnas.1401790111
Strain list
Strain Genotype Source
YKB3402 MATα his3 leu2 lys2 ura3 trp1 epl1Δ::kanMX EAF5-TAP::HIS3 [epl1-K39Q-K345Q-K376Q-K379Q-3HA::LEU2] Present studyYKB 3579 MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 epl1ΔKAN [epl1-K39Q-K345Q-K376Q-K379Q-3HA::LEU2] Present study
Fig. 3. NuA4 subunits are acetylated in vivo and in vitro.(A) Radioactive KAT assay to assess NuA4 auto-acetylationin vitro. [3H]-acetyl CoA was added directly to bead matrixof a stringently immunopurified NuA4 preparation [Esa1-TAP(YKB440)] versus an untagged control (YPH499) immuno-purification. Histone proteins were added to the reaction toserve as a positive control for acetylation. The reactions wereseparated on a gradient gel, Coomassie-stained to visualizeproteins (Right), treated for fluorography, and finally ex-posed to film for 2 wk (Left). Proteins are identified on theright side and protein size is indicated on the left (kDa).(B) Epl1 acetylation is dependent on Esa1 in vivo. NuA4was purified from cells grown at 25 °C through Eaf5-TAP.Epl1 was expressed as a C-terminal HA fusion proteinin epl1Δ background strain in the presence (lane 2,YKB2876) or absence (lane 1, YKB2862) of the acetyl-transferase-deficient esa1-L254P allele, or as a lysine-to-arginine or -glutamine multipoint mutant [EPcA-R;EPcA-Q (K39,345,376,379R or Q) YKB2781 and YKB3402,lanes 3 and 4, respectively]. Immunopurified (IP) productsand whole cell extract (WCE) were separated by SDS/PAGE(7.5%) and subjected to Western blot using the indicatedantibodies: antiacetyl lysine (α-AcK), antihistone H4 acetyllysine (α-AcK H4), antiglyceraldehyde 6-phosphate de-hydrogenase (α-G6PDH). Image is a representative ofthree independent replicates. (C ) Epl1-EPcA-R reducesNuA4 in vitro KAT activity. NuA4 was purified through Eaf5-TAP (lanes 2–4) relative to an untagged control sample (lane 1, YPH499). Epl1 was expressed fromits endogenous locus (lane 2, YKB1042) or as a C-terminal HA fusion protein of wild type EPL1 (lane 3, YKB2862) or epl1-EPcA-R (lane 4, YKB2781). KAT assayswere performed using NuA4 preparations equalized for Esa1 and nucleosome purified from HeLa cells. Error bars represent SD from duplicate reactions.NuA4 complexes used in assay were separated by SDS/PAGE and subjected to Western blot using the indicated antibody anti-Esa1 (α-Esa1) (Lower).
1. Yuan H, et al. (2012) MYST protein acetyltransferase activity requires active site lysine autoacetylation. EMBO J 31(1):58–70.
4346 | www.pnas.org

Hybrid DNA virus in Chinese patients with seronegativehepatitis discovered by deep sequencingBaoyan Xua,b,1, Ning Zhia,1,2, Gangqing Huc,1, Zhihong Wana, Xiaobin Zhengd, Xiaohong Liua, Susan Wonga,Sachiko Kajigayaa, Keji Zhaoc,3, Qing Maob,2, and Neal S. Younga,3
aHematology Branch and cSystems Biology Center, National Heart, Lung, and Blood Institute, Bethesda, MD 20892; bInstitute of Infectious Disease, SouthwestHospital, Third Military Medical University, Chongqing 400038, China; dDepartment of Embryology, Carnegie Institution for Science, Baltimore, MD 21218
Edited* by Harvey Alter, National Institutes of Health, Bethesda, MD, and approved March 19, 2013 (received for review March 4, 2013)
Seronegative hepatitis—non-A, non-B, non-C, non-D, non-E hepati-tis—is poorly characterized but strongly associated with seriouscomplications. We collected 92 sera specimens from patients withnon-A–E hepatitis in Chongqing, China between 1999 and 2007. Tensera pools were screened by Solexa deep sequencing. We discov-ered a 3,780-bp contig present in all 10 pools that yielded BLASTx Escores of 7e-05–0.008 against parvoviruses. The complete sequenceof the in silico-assembled 3,780-bp contig was confirmed by geneamplification of overlapping regions over almost the entire ge-nome, and the virus was provisionally designated NIH-CQV. Furtheranalysis revealed that the contig was composed of two major ORFs.By protein BLAST, ORF1 and ORF2 were most homologous to thereplication-associated protein of bat circovirus and the capsid pro-tein of porcine parvovirus, respectively. Phylogenetic analysis indi-cated that NIH-CQV is located at the interface of Parvoviridae andCircoviridae. Prevalence of NIH-CQV in patients was determined byquantitative PCR. Sixty-three of 90 patient samples (70%) were pos-itive, but all those from 45 healthy controls were negative. Averagevirus titer in the patient specimens was 1.05 e4 copies/μL. Specificantibodies against NIH-CQV were sought by immunoblotting.Eighty-four percent of patients were positive for IgG, and 31%were positive for IgM; in contrast, 78% of healthy controls werepositive for IgG, but all were negative for IgM. Although morework is needed to determine the etiologic role of NIH-CQV in hu-man disease, our data indicate that a parvovirus-like virus is highlyprevalent in a cohort of patients with non-A–E hepatitis.
hepatitis-associated aplastic anemia | liver failure | Solexa sequencing |virological diagnosis
Most viral hepatitis is secondary to infection by knownhepatotropic viruses: hepatitis A virus (HAV) (1), hepatitis
B virus (HBV) (2), hepatitis C virus (HCV) (3), hepatitis deltavirus (HDV) (4), and hepatitis E virus (HEV) (5). Other hepa-titis-associated viruses, including CMV (6), Epstein–Barr virus(EBV) (7), herpes simplex virus 1 and 2 (8), varicella-zoster vi-rus, human herpesvirus 6 (9), human parvovirus B19V (B19V)(10), and adenoviruses (11), may cause liver injury, ranging frommild and transient elevation of aminotransferases to acute hep-atitis and occasionally acute liver failure. Despite the number ofhepatitis viruses and hepatitis-associated viruses, etiology cannotbe determined in 10–20% of the cases of acute hepatitis (12), in30% of cases of cryptogenic chronic liver diseases (13), in hep-atitis-associated aplastic anemia (14), and in a large proportionof cases of acute liver failure (15). Patients with acute seroneg-ative hepatitis have fewer parenteral risk factors, more severebilirubin and transaminase elevations, and worse hepatocel-lular synthetic function than do patients with hepatitis C (16).Moreover, seronegative hepatitis is more strongly associatedwith serious complications, especially bone marrow failure (14)and childhood fulminant hepatitis (17).Next-generation sequencing (NGS) technology has had an in-
creasing impact on biological research and clinical diagnosis, be-cause it provides high-resolution genome-scale data rapidly andreliably. The first application of NGS for pathogen discovery in
a patient who died of a febrile illness after solid-organ trans-plantation led to the identification of an arenavirus (18). NGS-based platforms, such as 454 and Solexa deep sequencing, havebeen applied successfully to the investigation of emerging humanpathogens, resulting in major discoveries of new human viruses(19–21).In the present study, we established an experimental and an-
alytic procedure for identifying virus identification in humanspecimens based on Solexa deep sequencing. We applied themethod to screen for potential viral infection in human seraspecimens and discovered a human virus in Chinese patients whohad seronegative hepatitis. From its genome organization andphylogeny, the virus is unusual, because it is evolutionarily at theinterface of the parvovirus and circovirus families, perhapshaving emerged from interfamilial recombination. The virus washighly prevalent in a patient population, and active infection inseronegative hepatitis cases was suggested by the presence ofviral DNA in the blood and serology. However, more work isneeded to establish a firm disease association.
ResultsDetection of a Virus in Patients with Seronegative Hepatitis. Tenpools were made of sera from 92 patients with non-A–E hepatitisand were screened for potential pathogens by Solexa deep se-quencing. After filter sterilization, the samples were digestedwith DNase and RNase to eliminate host nucleotide contami-nation, and the remaining nucleic acids were extracted usingcarrier RNA [synthetic poly(A)]. cDNA was synthesized fromextracted viral nucleic acids with non-poly(A) random hexamers,which were designed specifically to block reverse transcription ofthe carrier RNA. With non-poly(A) random hexamers, redundantsequence tags derived from the carrier RNA were reduced toa level of <0.001%. High capacity enabled multiplex sequencingof more than 70 individual samples in a single Solexa run. ShortDNA sequences were analyzed and filtered at the nucleotide levelto exclude potential DNA contamination from human cells, mousecells, and known bacteria. For detection of known viruses, non-redundant reads were mapped against virus genomes from theNational Center for Biotechnology Information (NCBI) Reference
Author contributions: B.X., N.Z., K.Z., Q.M., and N.S.Y. designed research; B.X., N.Z., G.H.,Z.W., X.Z., X.L., S.W., and S.K. performed research; B.X., N.Z., G.H., and X.Z. analyzed data;Z.W. and Q.M. collected the clinical specimens; and B.X., N.Z., and N.S.Y. wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Freely available online through the PNAS open access option.
Data deposition: The sequence reported in this paper has been deposited in the GenBankdatabase (accession no. KC617868).1B.X., N.Z., and G.H. contributed equally to this work.2To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
3K.Z. and N.S.Y. contributed equally to this work.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303744110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1303744110 PNAS Early Edition | 1 of 6
MICRO
BIOLO
GY

Sequence (RefSeq) database using the short sequence alignmenttool Bowtie. HAV and HCV sequences were detected in sam-ples derived from sera pools 2 and 4, respectively. Individualsamples from these two pools then were sequenced to clarifythe source of the hepatitis viruses, and high copy numbers ofHAV or HCV genomes were detected in one patient in eachpool (these two samples were excluded from further analysis ofseronegative hepatitis cases). Furthermore, sequences fromAdenoviridae, Herpesviridae, Parvoviridae, and Anelloviridae werealso commonly detected. For virus discovery, filtered short se-quences were subjected to de novo assembly. Contigs fromdifferent samples were clustered based on nucleotide-level sim-ilarities. The resulting contigs were further compared with theNCBI nonredundant (NR) protein database using tBLASTx. Acontig was predicted as viral origin if the best tBLASTx hit wasfrom the kingdom of viruses, and a cluster was defined as acandidate if it contained a contig with viral origin. Contigs foreach cluster were subjected to multiple alignments to define aconsensus contig for later data analysis and experimental validation.By applying this pipeline to the 10 pooled samples, we identifieda 3,780-bp contig that yielded BLASTx E scores of 7e-05–0.008against parvoviruses (GenBank accession no.: KC617868).
Characterization of a Human Virus Genome. The 3,780-bp contig wascomposed of three major ORFs (Fig. 1 and Fig. S1). Protein Blast(BLASTp) searches showed that ORF1 encoded a 45-kDa pro-tein, which contained a domain (GxxxxGK[T/S]) (22) for a phos-phate-binding loop (P-loop) that is conserved among differentDNA viruses (Table S1); ORF1 was homologous to the replica-tion-associated protein (Rep) of bat circovirus with E values of 7e-04. ORF2 encoded a 55-kDa protein which was homologous toParvovirus coat protein 1 (VP1) of porcine parvovirus (PPV) andgoose parvovirus, with E scores of 2e-05. Because of low homol-ogy, only the first 87 amino acid residues encoded by ORF2 couldbe aligned reliably to the first 100 amino acids of VP1 of PPV. Thefirst 100 amino acids of the N-terminal region of the PPV VP1region constituted an active phospholipase A2 (PLA2), which ishighly conserved among the members of the Parvoviridae and isessential for parvovirus infection (23). The putative capsid protein(CP) encoded by ORF2 included a PLA2-like motif that is highlyconserved among the members of the Parvoviridae (Table S2).ORF3 was located on the left side of the genome on the viralnegative strand and encoded a 15-kDa protein. There was nohomology at the nucleic acid level between the 3,780-bp contigand known viruses in GenBank.Sequence analysis also revealed inverted terminal repeats (ITR)
at both ends of the genome (Fig. S1). The ITR was 156 nucleotideslong. The first 75 nucleotides complemented nucleotides 82–156,allowing the formation of a stem of a hairpin, whereas nucleotides76–81 were mismatched and could form a loop. By sequence anal-ysis of the intergenic region between ORF1 and ORF3, we foundmultiple direct repeats and TATA boxes (Fig. S1). In silico pro-moter prediction suggested that consensus sequences for a bi-directional eukaryote promoter were present in this region (www.fruitfly.org/seq_tools/promoter.html) and that the NIH-CQV mighthave an ambisense genome.
To confirm the sequence of the de novo-assembled 3,780-bpcontig, eight pairs of PCR primers (Table S3) were designed,based on the sequence predicted and were used to amplifyoverlapping DNA fragments from the pooled patient samples.Eight amplified DNA fragments were of the expected lengths(Fig. S2), and their sequences could be assembled into a 3,629-bpcontig. Sequencing analysis revealed that the 3,629-bp contig,composed of the entire coding region plus about half of the ITRsfrom both 5′ and 3′ termini, aligned exactly with that of the denovo-assembled 3,780-bp contig, indicating that the contig con-tained the nearly complete virus genome, was indeed present inthe patient samples, and was not an artifact generated by mis-assembly. The virus was provisionally designated NIH-CQ virus(NIH-CQV), because the samples were collected in a hospital inChongqing and the laboratory experiments were conducted atthe National Institutes of Health (NIH).Because ORF1 of NIH-CQV encodes a putative Rep protein
homologous to circovirus Rep, we speculated that the virus mighthave a circular form of its genome. To test this hypothesis, weperformed inverse PCR with a primer pair (Table S3) orientedoutwardly with respect to each other. Amplification with theinverted-primer pair generated an amplicon of 116 bp from two ofthree patient samples tested. Sequencing and alignment of theinverse PCR product showed a junction region between the 5′ and3′ termini in a head-to-tail orientation. In comparison with theliner genome of NIH-CQV, a 419-bp region composed of both 5′and 3′ ITRs present in the linear virus genome was absent in theinverse PCR product (Fig. S3), indicating a closed circular form ofthe virus genome of 3,361 bp present in the patient samples.Circularization of the viral genome resulted in an extension ofthe ORF3 at the left end of the genome, from 369 bp to 435 bp,with predicted encoding of a 17-kDa protein. The circular form ofNIH-CQV was designated NIH-CQV-Co.
Phylogenetic and Evolutionary Analyses of NIH-CQV. Because ORF1and ORF2 of NIH-CQV were homologous to different viralfamilies, phylogenetic analysis of the two ORFs was performedseparately by comparison with members of Circoviridae andParvoviridae. Although BLASTp searches showed that the aminoacid sequences encoded by ORF1 were homologous to Rep of batcircovirus, by gene tree analysis NIH-CQVwas not closely relatedto any known circoviruses (Fig. 2A). For ORF2, the amino acidsimilarity between NIH-CQV and known parvoviruses was below20%. Because of low homology, only the first 87 amino acid
P-loop
cp
PLA 2
5’ ITR 3’ ITR
rep15 kDa
1 kb
Fig. 1. Schematic diagram of the NIH-CQV genome. The putative ORFs arediagramed in boxes, and the arrows indicate the orientation of the ORFs.The conserved P-loop is shown as a shaded box in rep, and the conservedPLA2-like motifs are shown as shaded boxes with the Ca2+ binding loop inblack and the catalytic residues in gray.
FiCVStCVRaCVCaCVCoCVGuCVBFDVBaCVGoCVMusDCVDuCVMulDCVPoCV-1PoCV-2NIH-CQV
AAV2
AAV1AAV4BAAVAAV5AAAVGPVMDPVBPV
ParV4
NIH-CQV
BmDNVAalDNV
AMDVMVM
AAV3
CnMV
B19V
GDNVJcDNVPfDNV
AeDNV
Dependovirus
Bocavirus
Erythrovirus
DensovirusPefudensovirusIteravirusBrevidensovirusAmdovirusParvovirus
100
92100
100
90
58
7291
7497
55
76
100
100
98
94
82
67
43
100
57
97
84100
100
98100
90
53
67
100
BA
Fig. 2. Phylogenetic analysis of Rep and CP of NIH-CQV. (A) Phylogeneticrelationship of the Rep of NIH-CQV and other related circoviruses based onan alignment of amino acid sequences. (B) Phylogenetic relationship of theCP of NIH-CQV and other related parvoviruses based on an alignment ofamino acid sequences. The phylogenetic trees were constructed by neighborjoining (NEIGHBOR program from PHYLIP) based on alignments generatedwith CLUSTAL V, and 1,000 bootstrap replications were performed. Allsequences were downloaded from GenBank and SwissProt with a BioPerlscript. Detailed information is available in Dataset S1A.
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1303744110 Xu et al.
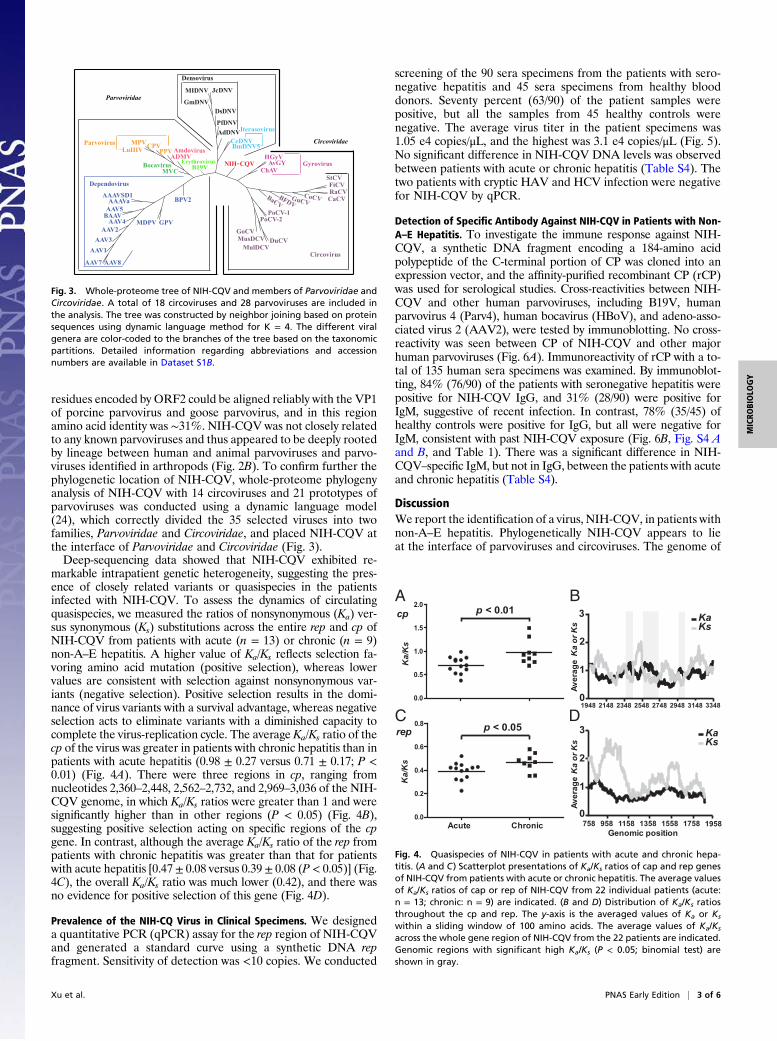
residues encoded by ORF2 could be aligned reliably with the VP1of porcine parvovirus and goose parvovirus, and in this regionamino acid identity was ∼31%. NIH-CQV was not closely relatedto any known parvoviruses and thus appeared to be deeply rootedby lineage between human and animal parvoviruses and parvo-viruses identified in arthropods (Fig. 2B). To confirm further thephylogenetic location of NIH-CQV, whole-proteome phylogenyanalysis of NIH-CQV with 14 circoviruses and 21 prototypes ofparvoviruses was conducted using a dynamic language model(24), which correctly divided the 35 selected viruses into twofamilies, Parvoviridae and Circoviridae, and placed NIH-CQV atthe interface of Parvoviridae and Circoviridae (Fig. 3).Deep-sequencing data showed that NIH-CQV exhibited re-
markable intrapatient genetic heterogeneity, suggesting the pres-ence of closely related variants or quasispecies in the patientsinfected with NIH-CQV. To assess the dynamics of circulatingquasispecies, we measured the ratios of nonsynonymous (Ka) ver-sus synonymous (Ks) substitutions across the entire rep and cp ofNIH-CQV from patients with acute (n = 13) or chronic (n = 9)non-A–E hepatitis. A higher value of Ka/Ks reflects selection fa-voring amino acid mutation (positive selection), whereas lowervalues are consistent with selection against nonsynonymous var-iants (negative selection). Positive selection results in the domi-nance of virus variants with a survival advantage, whereas negativeselection acts to eliminate variants with a diminished capacity tocomplete the virus-replication cycle. The average Ka/Ks ratio of thecp of the virus was greater in patients with chronic hepatitis than inpatients with acute hepatitis (0.98 ± 0.27 versus 0.71 ± 0.17; P <0.01) (Fig. 4A). There were three regions in cp, ranging fromnucleotides 2,360–2,448, 2,562–2,732, and 2,969–3,036 of the NIH-CQV genome, in which Ka/Ks ratios were greater than 1 and weresignificantly higher than in other regions (P < 0.05) (Fig. 4B),suggesting positive selection acting on specific regions of the cpgene. In contrast, although the average Ka/Ks ratio of the rep frompatients with chronic hepatitis was greater than that for patientswith acute hepatitis [0.47± 0.08 versus 0.39 ± 0.08 (P < 0.05)] (Fig.4C), the overall Ka/Ks ratio was much lower (0.42), and there wasno evidence for positive selection of this gene (Fig. 4D).
Prevalence of the NIH-CQ Virus in Clinical Specimens. We designeda quantitative PCR (qPCR) assay for the rep region of NIH-CQVand generated a standard curve using a synthetic DNA repfragment. Sensitivity of detection was <10 copies. We conducted
screening of the 90 sera specimens from the patients with sero-negative hepatitis and 45 sera specimens from healthy blooddonors. Seventy percent (63/90) of the patient samples werepositive, but all the samples from 45 healthy controls werenegative. The average virus titer in the patient specimens was1.05 e4 copies/μL, and the highest was 3.1 e4 copies/μL (Fig. 5).No significant difference in NIH-CQV DNA levels was observedbetween patients with acute or chronic hepatitis (Table S4). Thetwo patients with cryptic HAV and HCV infection were negativefor NIH-CQV by qPCR.
Detection of Specific Antibody Against NIH-CQV in Patients with Non-A–E Hepatitis. To investigate the immune response against NIH-CQV, a synthetic DNA fragment encoding a 184-amino acidpolypeptide of the C-terminal portion of CP was cloned into anexpression vector, and the affinity-purified recombinant CP (rCP)was used for serological studies. Cross-reactivities between NIH-CQV and other human parvoviruses, including B19V, humanparvovirus 4 (Parv4), human bocavirus (HBoV), and adeno-asso-ciated virus 2 (AAV2), were tested by immunoblotting. No cross-reactivity was seen between CP of NIH-CQV and other majorhuman parvoviruses (Fig. 6A). Immunoreactivity of rCP with a to-tal of 135 human sera specimens was examined. By immunoblot-ting, 84% (76/90) of the patients with seronegative hepatitis werepositive for NIH-CQV IgG, and 31% (28/90) were positive forIgM, suggestive of recent infection. In contrast, 78% (35/45) ofhealthy controls were positive for IgG, but all were negative forIgM, consistent with past NIH-CQV exposure (Fig. 6B, Fig. S4 Aand B, and Table 1). There was a significant difference in NIH-CQV–specific IgM, but not in IgG, between the patients with acuteand chronic hepatitis (Table S4).
DiscussionWe report the identification of a virus, NIH-CQV, in patients withnon-A–E hepatitis. Phylogenetically NIH-CQV appears to lieat the interface of parvoviruses and circoviruses. The genome of
BPV2
GPVMDPV
AAV8AAV7
AAV1AAV3
AAV2AAV4
BAAVAAV5AAAVa
AAAVSD1
MVC
LuIIIVMPVCPV
PPVADMV
GmDNV
MIDNV JcDNV
DsDNV
PfDNVAdDNV
CeDNVBmDNV5
HGyVAvGY
ChAVStCVFiCVRaCVCaCVCoCVGuCV
BFDVBaCV
PoCV-2PoCV-1
DuCVMulDCV
MusDCVGoCV
AmdovirusParvovirus
B19VErythrovirus
Densovirus
Iterasovirus
Circoviridae
Parvoviridae
Circovirus
Dependovirus
Bocavirus Gyrovirus NIH-CQV
Fig. 3. Whole-proteome tree of NIH-CQV and members of Parvoviridae andCircoviridae. A total of 18 circoviruses and 28 parvoviruses are included inthe analysis. The tree was constructed by neighbor joining based on proteinsequences using dynamic language method for K = 4. The different viralgenera are color-coded to the branches of the tree based on the taxonomicpartitions. Detailed information regarding abbreviations and accessionnumbers are available in Dataset S1B.
Acute Chronic
Ka/
Ks
p < 0.05
Ka/
Ks
cp
rep
0
2
3
Genomic position758 958 1158 1358 1558 1758
KaKs
Aver
age
Ka
or K
sAv
erag
e K
a or
Ks
1958
1948 2148 2348 2548 2748 2948 3148
KaKs
0
1
2
3
1
BA
DC3348
p < 0.01
0.0
0.2
0.4
0.6
0.8
0.0
0.5
1.0
1.5
2.0
Fig. 4. Quasispecies of NIH-CQV in patients with acute and chronic hepa-titis. (A and C) Scatterplot presentations of Ka/Ks ratios of cap and rep genesof NIH-CQV from patients with acute or chronic hepatitis. The average valuesof Ka/Ks ratios of cap or rep of NIH-CQV from 22 individual patients (acute:n = 13; chronic: n = 9) are indicated. (B and D) Distribution of Ka/Ks ratiosthroughout the cp and rep. The y-axis is the averaged values of Ka or Ks
within a sliding window of 100 amino acids. The average values of Ka/Ks
across the whole gene region of NIH-CQV from the 22 patients are indicated.Genomic regions with significant high Ka/Ks (P < 0.05; binomial test) areshown in gray.
Xu et al. PNAS Early Edition | 3 of 6
MICRO
BIOLO
GY

NIH-CQV has no homology to any known viruses in GenBank.Although comparative analysis revealed that the Rep and CPproteins of NIH-CQV have limited homologies with circovirusesand parvoviruses, respectively, the overall genome organization ofNIH-CQV has the basic characteristics of viruses in Parvoviridae.NIH-CQV has a small, compact linear DNA genome with two
tandemly arranged major ORFs encoding Rep and CP proteins,respectively, and a pair of ITRs at the left and right ends of thegenome. In silico promoter prediction suggested that transcriptionof the rep and cp was regulated by a single promoter upstreamof the rep, because there was no apparent promoter sequenceimmediately upstream of cp. A pre-mRNA encoded by a single
promoter that is processed by alternative splicing and alternativepolyadenylation to generate multiple mRNAs had been reportedfor other parvoviruses, such as B19V (25) and Aleutian minkdisease virus (26). By amino acid sequence analysis, CP protein ofNIH-CQV is minimally homologous to porcine and goose par-vovirus VP1 (27). The homologous region is mainly in the Nterminus of CP and contains the conserved PLA2-like motif,which is present in the N-terminal extension of the VP1 uniqueregion of members of Parvoviridae (23). The phylogenetic treeconstructed using amino acid sequences of CP of NIH-CQV andVP1 of other representative viruses in Parvoviridae showed thatNIH-CQV represents a deeply rooted lineage between twogroups: (i) human and animal parvoviruses and (ii) insect par-voviruses. Thus, NIH-CQV appears to be a parvovirus-like virus.However, NIH-CQV also has features shared by members of theCircoviridae family (28). First, a BLASTp search revealed that theRep of NIH-CQV is more related to circoviral Rep than to thoseof other viruses. Second, NIH-CQV appears to have an ambi-sense genome because there are bidirectional putative promotersin the intergenic region between the rep and 15-kDa protein andbecause rep and 15-kDa protein arranged head-to-head flank theintergenic region, which is a feature of circoviruses. Finally, se-quencing and alignment of the inverse PCR products indicatedthe existence of the circular genome of NIH-CQV in the clinicalsamples. The circular form of NIH-CQV genomemight representa subviral DNA or an intermediate structure of viral replication,as reported for other parvoviruses (29). Therefore, NIH-CQVappears to be a “hybrid” virus, perhaps formed by interfamilialrecombination between a parvovirus and a circovirus. This ideawas supported further by whole-proteome phylogenetic analysis,which showed NIH-CQV located between Parvoviridae andCircoviridae.Both recombination and a high mutation rate are often cited as
key in evolutionary innovation. For many viruses, recombination
P < 0.0001
Seronegative hepatitis
Blood donors
Cop
ies/
μl
Avg: 1.05 e4 copies/μl
0
1.0 e4
2.0 e4
3.0 e4
4.0 e4
Fig. 5. Detection of NIH-CQV DNA by qPCR. Scatterplot showing virus loadper microliter of sera specimens obtained from patients (n = 90) and healthycontrols (n = 45). Each dot represents one specimen. Bars show the averagecopy numbers of virus genome.
α -NIH-CQV α -AAV2α -Parv4α -HBoVα -B19V
130(kDa)
70
55
40
35
25
15
10
100
130(kDa)
70
55
40
35
25
15
10
100
130(kDa)
70
55
40
35
25
15
10
100
130(kDa)
70
55
40
35
25
15
10
100
130(kDa)
70
55
40
35
25
15
10
100
21 3 4P 6 7 8 9 10 11 12 315 14 15 16 17 18 19 20 21 22 23 24
B6A11 C1 D7 11F01F9F8F6F11E8EP 01A5E C5 C7 C11 D8 E9 F4 G10 H3 H8 C4 A2
IgM
IgG
IgM
IgG
Healthy controls
Chronic infectionAcute infection
A
B
C
Fig. 6. Immunoblotting of sera from patients withnon-A–E hepatitis and from healthy controls usingrCP of NIH-CQV. (A) Specificity test for the rCP of NIH-CQV. The cell lysates derived from the cells trans-fected with the plasmids that expressed capsid pro-teins of AAV2, Parv4, HBoV, and B19V and purifiedrCP were subjected to SDS/PAGE and then weretransferred to a nitrocellulose membrane. Afterblocking with 5% (wt/vol) nonfat milk for 2 h, themembrane was incubated with respective antiseraat 1:1,000 dilution. (B and C) Detection of specificantibodies against the rCP of NIH-CQV. Samples sub-jected to SDS/PAGE consisted of 25 ng of affinity-purified rCP. These proteins were transferred to anitrocellulose membrane and incubated with a 1:1,000dilution of patient sera. The numbers at the top arepatient identification numbers. Only representativesresult are shown; complete data are shown in Fig. S4A and B. Mouse monoclonal antibody against thepolyhistidine tag was used as a positive control. Thenumbers on the left indicate molecular masses inkilodaltons based on the PageRuler PrestainedProtein Ladder (Fermentas).
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1303744110 Xu et al.

allows single-step acquisition of multiple genetic changes, anda high mutation rate enhances adaptation to new hosts (30). Theseprocesses are critical driving forces in viral evolution and lead toviral emergence, host-switching, and adaptation, which often re-sult in disease outbreaks (31, 32). Recent studies had demon-strated that small eukaryotic ssDNA viruses have high rates ofnucleotide substitutions and genetic recombination, which maysurpass rates seen in RNA viruses (33, 34). These observationsmay explain the extensive diversity seen in ssDNA viruses. Par-voviruses (family Parvoviridae) and circoviruses (family Circovir-idae) are both ssDNA viruses that infect a wide variety of animalspecies, including mammals, birds, and insects. Although hori-zontal gene transfer and high mutation rates have been docu-mented in these viruses, cross-species viral transfers resulting frominterfamilial recombination and leading to a infection in the newhost species are not common.A high rate of viral evolution has been reported for emerging
parvoviruses (35). Canine parvovirus (CPV) is one well-charac-terized example (35). CPV emerged from feline panleukopeniaparvovirus or a closely related virus, differing at several amino acidresidues. Sequences of the viruses collected at time points beforeand after host-switching suggest that the emergence of CPV wasdependent on a high mutation rate and positive selection of themajor capsid gene. Our deep-sequencing data showed that NIH-CQV has a high substitution mutation rate throughout its genome.However, the impact of selective pressures, as measured by the Ka/Ks ratio, appeared to vary depending on location. The cp genecontained three regions in which the Ka/Ks ratio exceeded 1, sug-gesting positive selection. These substitutions would be presumedto enhance fitness. In contrast, selective pressure on the rep ofNIH-CQV appeared to be dominated by purifying selection, because theaverage Ka/Ks ratio for the rep from 14 patients was only 0.42. TheKa/Ks ratios for the regions encoding the PLA2 motif in the CPprotein or P-loop nucleoside triphosphatase (NTPase) in the Repproteins were lower than in other regions, as is consistent withstrong purifying selection and functional importance of this regionin viral replication.NIH-CQV exhibited remarkable genetic heterogeneity within
patients, indicating the presence of closely related variants orquasispecies in NIH-CQV–infected individuals. The Ka/Ks ratios ofboth viral cp and rep were greater in patients classified as havingchronic hepatitis than in those with acute hepatitis, suggestinggreater variation or more diversity of quasispecies of NIH-CQVover time. For hepatitis viruses C and E, diversification of qua-sispecies during infection is driven by the interaction between theviruses and host immune response and may be associated withdifferent clinical outcomes (36, 37). With a limited number ofserial samples from a single individual, we were unable to concludewhether quasispecies dynamics of NIH-CQV in patients correlatedwith disease.Despite technological advances in molecular biology, an etiology
cannot be determined in as many as 20% of hepatitis cases. As ofyet, no causative agent may be defined in a significant proportionof patients with chronic liver disease and cirrhosis, whose diseasetherefore is characterized as “cryptogenic.” Additional hepatitisagents have been suspected. Over several decades, several human
viruses, such as GB virus C (38), Torque teno virus (39), and SENvirus (40), have been putatively claimed to be hepatitis viruses,but subsequent investigation revealed no conclusive associationbetween these viruses and liver disease. Therefore, the etiologyof non-A–E hepatitis remains unresolved. In this study, we appliedNGS methodology to virus discovery and indentified a parvovirus-like virus in the sera samples collected from a cohort of patientswith non-A–E hepatitis. Our results showed that 28% of patientswere positive for both NIH-CQV–specific IgM and viral DNA,suggesting recent infection in patients. Most patients and healthycontrols were positive for virus-specific IgG, suggesting that ex-posure was common in the population studied. The patients whoyielded positive specimens suffered from symptoms of hepatitiswith a wide range of liver dysfunction, including acute and chronichepatitis. There was no difference between patients diagnosed withacute versus or chronic hepatitis with respect to detection ofNIH-CQV DNA, but viral DNA was not detected in healthycontrols. Although these findings are suggestive of an etiologicrelationship, our study has limitations. By electron microscopy,we observed virus-like particles in NIH-CQV qPCR-positive serawhich cosedimented with viral DNA, but we were unable to con-firm their identity by immunogold labeling with our antisera. Inaddition, because of the limited availability of samples, we hadinsufficient material for viral propagation and isolation in cellculture, and efforts are in progress to detect viral proteins in afew liver biopsy tissue blocks. Additionally, with the limitednumber of samples available, we cannot exclude the possibility ofreactivation of a latent virus in the course of liver inflammation.In general, parvoviruses cause systemic infection, but clinical
manifestations of the infection are varied and may depend onspecific tissue tropisms as well as on host immunologic compe-tency. For two human pathogenic parvoviruses, B19V (10) andHBoV (41), clinical symptoms range from none to acute systemicmanifestations involving skin and joints to subtle persistence ofinfection; in immunocompromised populations, B19V chronicinfection causes pure red cell aplasia. In addition, coinfection ofparvoviruses with other viruses, especially related DNA viruses,can lead to synergy and more severe clinical symptoms in theaffected host. Because of the complexity of disease associations,more comprehensive epidemiology and in vitro studies are neededto establish a pathogenic role of NIH-CQV in hepatitis.
Materials and MethodsStudy Subjects. We studied a total of 92 sera samples from patients with non-A–E hepatitis who were admitted to the Institute of Infectious Disease ofSouthwest Hospital, Third Military Medical University, China, between 1999and 2007. According to the criteria of the Chinese Society of Infectious Diseaseand Parasitology, and the Chinese Society of Hepatology (42), 33 patients werediagnosed with acute hepatitis by clinical and biochemical parameters. Fifty-ninepatients had chronic aggressive hepatitis confirmed by biopsy, and 10 of themhad cirrhosis; eight outpatients with chronic persistent hepatitis did not haveliver biopsies performed. The patients had amedian age of 42 y (range: 12–73 y);among of them, 55 males had a median age of 41 y (range: 12–73 y), and 37females had median age of 43 y (range: 15–70 y). On admission, mean liverfunction tests were as follows: total bilirubin 174 ± 130 μmol/L (range: 14–735μmol/L), alanine aminotransferase 421 ± 623 IU/L (range: 5–3,150 IU/L). Sero-logical tests for anti-HAV IgM (HAV IgM, ELISA Kit; Wantai Biological Pharmacy),
Table 1. Results of real-time PCR and immunoblot analysis
qPCR
Seronegative hepatitis patients Healthy controls
IgM IgG IgM IgG
+ - + - + - + -
+ 27.8% (25/90) 42.2% (38/90) 70.0% (63/90) 0 (0/90) 0 (0/45) 0 (0/45) 0 (0/45) 0 (0/45)- 3.3% (3/90) 26.7% (24/90) 14.4% (13/90) 15.6% (14/90) 0 (0/45) 100% (45/45) 77.8% (35/45) 22.2% (10/45)Total 31.1% (28/90) 68.9% (62/90) 84.4% (76/90) 15.6% (14/90) 0 (0/45) 100% (45/45) 77.8% (35/45) 22.2% (10/45)
Xu et al. PNAS Early Edition | 5 of 6
MICRO
BIOLO
GY

HBV markers (HBsAg and HBeAg; Roche Diagnostic Systems) anti-HCV anti-bodies (HCV ELISA Kit; Wantai Biological Pharmacy), anti-HDV IgM (HDV IgMELISA Kit; Wantai Biological Pharmacy), anti-HEV IgM (HAV IgM ELISA Kit;Wantai Biological Pharmacy), HCV RNA tests (HCV Quantitative RT-PCR Kit;Kehua Bio-engineering), anti-HIV-1 and 2 antibodies (HIV ELISA Kit; WantaiBiological Pharmacy), HIV viral load test (HIV Quantitative RT-PCR Kit; RocheDiagnostic Systems), and serological tests for anti-CMV IgM and (CMV IgMELISA Kit; Wantai Biological Pharmacy) and anti-EBV IgM (EBV IgM Kit; YHLOBiotech) were all negative. Additional tests for antinuclear antibody, rheu-matoid factor, anti-mitochondrial antibody (Microplate ELISA Test Systemsfor Autoimmunity; EUROIMMUN), as well as blood cultures, urine cultures,and throat swabs for bacteria were negative. The research protocol wasapproved by the Human Bioethics Committee of the Third Military MedicalUniversity, and all participants provided written informed consent.
Solexa Deep Sequencing and Phylogenetic and Evolutionary Analysis. Details ofdeep sequencing and phylogenetic and evolutionary analysis are provided inSI Material and Methods.
Overlapping PCR and Reverse PCR. To verify the sequence of the viral genomeassembled from the Solexa data, eight sets of overlapping primer pairs weredesigned (Table S3). Viral DNA was extracted from sera as described in theSolexa deep sequencing. Extracted DNA (5 μL) was used as template for thePCR. The 50-μL reaction mix consisted of 1× Platinum Taq PCR Buffer (Invi-trogen), 1.5 mM MgCl2, each dNTP at 0.2 mM, and 25 pmol each of theprimers. After 2 min at 94 °C, 35 cycles of amplification (94 °C for 1 min, 54 °Cfor 1 min, and 72 °C for 1 min) were performed. To detect circularized viralDNA, inverse PCR with a primer pair (Table S3) that oriented outwardly withrespect to each other was used for amplification. PCR was performed as
described above, and amplified products were visualized on an agarose gel.All PCR products were sequenced.
Diagnostic qPCR. For quantitative PCR, DNA was extracted by the QIAampMinElute Virus Kit (Qiagen), and 5 μL of the resulting DNA was used foranalysis with the NIH-CQV rep primers set (NIH-CQV_Rep-1295F and NIH-CQV_Rep-1470R), and NIH-CQV_Rep probe was used for qPCR (Table S3). Allreactions were performed using the Chromo4 real-time detector (Bio-Rad).The reaction started with activation of the polymerase at 95 °C for 15 min,followed by 45 cycles of 15 s at 94 °C and 1 min at 60 °C. The quantitation ofamplicon was performed by interpolation with the standard curve to thesynthesized rep gene (ORF1) with serial dilutions.
Immunoblotting. To investigate the immune response against NIH-CQV,a synthetic DNA fragment encoding a 184-amino acid polypeptide of theC-terminal portion of CP was cloned into an expression vector, and the af-finity-purified rCP was used for immunoblotting. Cross-reactivities betweenNIH-CQV and other human parvoviruses, including B19V, Parv4, HBoV, andAAV2, were tested by immunoblotting. Details are provided in SI Materialand Methods.
ACKNOWLEDGMENTS. We thank Dr. Jun Zhu at the DNA Sequencing andGenomics Core of the National Heart, Lung, and Blood Institute (NHLBI)for assistance in deep-sequencing and data analysis; Dr. Amit Kapoor ofColumbia University for technical advice; and Genoveffa Franchini of theNational Cancer Institute, Leonid Margolis of the National Institute ofChild Health and Development, Cynthia Dunbar of the NHLBI, and MikeFang in our laboratory for their careful reading of the manuscript. Thiswork was supported by the National Institutes of Health IntramuralResearch Program.
1. Cohen JI (1989) Hepatitis A virus: Insights from molecular biology. Hepatology 9(6):889–895.
2. Theilmann L, et al. (1988) HBV DNA and other hepatitis B virus markers in sera fromlong-term hemodialysis and kidney transplant patients. Hepatogastroenterology35(4):147–150.
3. Choo QL, et al. (1989) Isolation of a cDNA clone derived from a blood-borne non-A,non-B viral hepatitis genome. Science 244(4902):359–362.
4. Rizzetto M, et al. (1977) Immunofluorescence detection of new antigen-antibodysystem (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAgcarriers. Gut 18(12):997–1003.
5. Reyes GR, et al. (1990) Isolation of a cDNA from the virus responsible for entericallytransmitted non-A, non-B hepatitis. Science 247(4948):1335–1339.
6. Lemonovich TL, Watkins RR (2012) Update on cytomegalovirus infections of thegastrointestinal system in solid organ transplant recipients. Curr Infect Dis Rep 14(1):33–40.
7. Crum NF (2006) Epstein Barr virus hepatitis: Case series and review. South Med J 99(5):544–547.
8. Riediger C, et al. (2009) Herpes simplex virus sepsis and acute liver failure. ClinTransplant 23(Suppl 21):37–41.
9. Razonable RR, Zerr DM; AST Infectious Diseases Community of Practice (2009) HHV-6,HHV-7 and HHV-8 in solid organ transplant recipients. Am J Transplant 9(Suppl 4):S100–S103.
10. Young NS, Brown KE (2004) Parvovirus B19. N Engl J Med 350(6):586–597.11. Lynch JP, 3rd, Fishbein M, Echavarria M (2011) Adenovirus. Semin Respir Crit Care Med
32(4):494–511.12. Alter MJ, et al. (1992) The natural history of community-acquired hepatitis C in the
United States. The Sentinel Counties Chronic non-A, non-B Hepatitis Study Team. NEngl J Med 327(27):1899–1905.
13. Kodali VP, Gordon SC, Silverman AL, McCray DG (1994) Cryptogenic liver disease inthe United States: Further evidence for non-A, non-B, and non-C hepatitis. Am JGastroenterol 89(10):1836–1839.
14. Brown KE, Young NS (1997) Human parvovirus B19: Pathogenesis of disease. Humanparvovirus B19, eds Anderson LJ, Young NS (Karger, Basel), pp 105–119.
15. Ferraz ML, et al. (1996) Fulminant hepatitis in patients undergoing liver trans-plantation: Evidence for a non-A, non-B, non-C, non-D, and non-E syndrome. LiverTranspl Surg 2(1):60–66.
16. Rochling FA, et al. (1997) Acute sporadic non-A, non-B, non-C, non-D, non-D, non-Ehepatitis. Hepatology 25(2):478–483.
17. Lee WS, McKiernan P, Kelly DA (2005) Etiology, outcome and prognostic indicators ofchildhood fulminant hepatic failure in the United kingdom. J Pediatr GastroenterolNutr 40(5):575–581.
18. Palacios G, et al. (2008) A new arenavirus in a cluster of fatal transplant-associateddiseases. N Engl J Med 358(10):991–998.
19. Briese T, et al. (2009) Genetic detection and characterization of Lujo virus, a newhemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog 5(5):e1000455.
20. Yozwiak NL, et al. (2010) Human enterovirus 109: A novel interspecies recombinantenterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua. JVirol 84(18):9047–9058.
21. Feng H, Shuda M, Chang Y, Moore PS (2008) Clonal integration of a polyomavirus inhuman Merkel cell carcinoma. Science 319(5866):1096–1100.
22. Momoeda M, Wong S, Kawase M, Young NS, Kajigaya S (1994) A putative nucleosidetriphosphate-binding domain in the nonstructural protein of B19 parvovirus is re-quired for cytotoxicity. J Virol 68(12):8443–8446.
23. Zádori Z, et al. (2001) A viral phospholipase A2 is required for parvovirus infectivity.Dev Cell 1(2):291–302.
24. Yu ZG, et al. (2010) Whole-proteome phylogeny of large dsDNA viruses and parvo-viruses through a composition vector method related to dynamical language model.BMC Evol Biol 10:192.
25. Liu JM, Green SW, Shimada T, Young NS (1992) A block in full-length transcriptmaturation in cells nonpermissive for B19 parvovirus. J Virol 66(8):4686–4692.
26. Qiu J, Cheng F, Pintel D (2007) The abundant R2 mRNA generated by aleutian minkdisease parvovirus is tricistronic, encoding NS2, VP1, and VP2. J Virol 81(13):6993–7000.
27. Tijssen P, et al. (2011) Family Parvoviridae. Family Parvoviridae Virus Taxonomy, NinthReport of the International Committee on Taxonomy of Viruses, eds King AMQ,Adams MJ, Carstens EB, Lefkowitz EJ (Elsevier Academic, San Diego, CA), pp 405–425.
28. Biagini P, et al. (2011) Family Circoviridae. Family Circoviridae Virus Taxonomy, NinthReport of the International Committee on Taxonomy of Viruses, eds King AMQ,Adams MJ, Carstens EB, Lefkowitz EJ (Elsevler Academic, San Diego, CA), pp 343–349.
29. Kapoor A, et al. (2011) Bocavirus episome in infected human tissue contains non-identical termini. PLoS ONE 6(6):e21362.
30. Woolhouse ME, Haydon DT, Antia R (2005) Emerging pathogens: The epidemiologyand evolution of species jumps. Trends Ecol Evol 20(5):238–244.
31. Keele BF, et al. (2006) Chimpanzee reservoirs of pandemic and nonpandemic HIV-1.Science 313(5786):523–526.
32. Li W, et al. (2006) Animal origins of the severe acute respiratory syndrome corona-virus: Insight from ACE2-S-protein interactions. J Virol 80(9):4211–4219.
33. López-Bueno A, Villarreal LP, Almendral JM (2006) Parvovirus variation for disease: Adifference with RNA viruses? Curr Top Microbiol Immunol 299:349–370.
34. Martin DP, et al. (2011) Recombination in eukaryotic single stranded DNA viruses.Viruses 3(9):1699–1738.
35. Shackelton LA, Parrish CR, Truyen U, Holmes EC (2005) High rate of viral evolution asso-ciated with the emergence of carnivore parvovirus. Proc Natl Acad Sci USA 102(2):379–384.
36. Farci P, et al. (2000) The outcome of acute hepatitis C predicted by the evolution ofthe viral quasispecies. Science 288(5464):339–344.
37. Lhomme S, et al. (2012) Hepatitis E virus quasispecies and the outcome of acutehepatitis E in solid-organ transplant patients. J Virol 86(18):10006–10014.
38. Linnen J, et al. (1996) Molecular cloning and disease association of hepatitis G virus: Atransfusion-transmissible agent. Science 271(5248):505–508.
39. Nishizawa T, et al. (1997) A novel DNA virus (TTV) associated with elevated trans-aminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys ResCommun 241(1):92–97.
40. Tanaka Y, et al. (2001) Genomic and molecular evolutionary analysis of a newlyidentified infectious agent (SEN virus) and its relationship to the TT virus family. JInfect Dis 183(3):359–367.
41. Jartti T, et al. (2012) Human bocavirus-the first 5 years. Rev Med Virol 22(1):46–64.42. Chinese Society of Infectious D & Parasitology (2000) Chinese Society of Hepatology.
Management scheme of diagnostic and therapeutic criteria of viral hepatitis.Zhonghua Gan Zang Bing Za Zhi 8:324–329.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1303744110 Xu et al.















![Transfusionsindikationen bei malignen Erkrankungen · Scenario 1: number of patients Scenario 2: number of patients % of patients (mean) [46] Scenario 1: number of patients Scenario](https://static.fdokument.com/doc/165x107/605ea3d56540f665aa09ac74/transfusionsindikationen-bei-malignen-scenario-1-number-of-patients-scenario-2.jpg)

