
Inaugural-Dissertation - docserv.uni-duesseldorf.de · Abbildung 3: Dreidimensionale Struktur der...
152
Entwicklung eines rekombinanten Ganzzellsystems-- Klonierung, Coexpression und Mutagenese der Phenylalanin-Dehydrogenase aus Rhodococcus sp. M4 und des malic enzymes aus E.coli K12 Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität vorgelegt von Shukrallah Na’amnieh aus Düsseldorf Jülich 2002
Transcript of Inaugural-Dissertation - docserv.uni-duesseldorf.de · Abbildung 3: Dreidimensionale Struktur der...

Entwicklung eines rekombinanten Ganzzellsystems--
Klonierung, Coexpression und Mutagenese der Phenylalanin-Dehydrogenase
aus Rhodococcus sp. M4 und des malic enzymes aus E.coli K12
Inaugural-Dissertation
zur Erlangung des Doktorgrades der
Mathematisch-Naturwissenschaftlichen Fakultät
der Heinrich-Heine-Universität
vorgelegt von
Shukrallah Na’amnieh
aus Düsseldorf
Jülich 2002

Die vorliegende Arbeit wurde in der Zeit von September 1998 bis März 2002 am Institut für
Enzymtechnologie der Heinrich-Heine-Universität Düsseldorf im Forschungszentrum Jülich
unter der Leitung von Herrn Priv. Doz.- Dr. W. Hummel durchgeführt.
Die Arbeit wurde von Degussa AG und Minerva Stiftung der Max-Planck-Gesellschaft zur Förderung der Wissenschaften gefördert.

Gedruckt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf. Referent: Priv.- Doz. Dr. W. Hummel Korreferent: Prof. Dr. Cornelis P. Hollenberg

Inhaltsverzeichnis
1 Einleitung ................................................................................................................................................. 1 1.1 BEDEUTUNG VON ENZYMEN ................................................................................................................... 1 1.2 ENZYME IN DER INDUSTRIE .................................................................................................................... 2 1.3 DIE PHENYLALANIN DEHYDROGENASE AUS RHODOCOCCUS SP. M4 ....................................................... 4 1.3.1 Malic enzyme aus E. coli K12............................................................................................................... 8 1.3.2 L-Phenylalanin-Synthese unter Coenzymregeneration ........................................................................... 8 1.3.3 Expression rekombinanter Proteine ....................................................................................................... 9 1.4 PROTEIN-DESIGN.................................................................................................................................. 11 1.4.1 Gezielte Mutagenese........................................................................................................................... 11 1.4.2 Zufallsmutagenese.............................................................................................................................. 13 1.5 HOCHZELLDICHTE-FERMENTATION (HZD) ......................................................................................... 14 2 Problemstellung und Zielsetzung........................................................................................................... 15 3 Material .................................................................................................................................................. 17 4 Methoden................................................................................................................................................ 20 4.1 MOLEKULARBIOLOGIE ......................................................................................................................... 20 4.1.1 Isolierung der genomischen DNA ....................................................................................................... 20 4.1.2 Auftrennung von DNA-Fragmenten über Elektrophorese .................................................................... 21 4.1.3 Ethanolfällung.................................................................................................................................... 21 4.1.4 Polymerase Ketten Reaktion [PCR] .................................................................................................... 22 4.1.5 Berechnen der Schmelztemperatur der Primer ..................................................................................... 23 4.1.6 Isolierung von DNA- Fragmenten aus Agarosegelen ........................................................................... 23 4.1.7 Dephosphorylierung linearisierter Plasmid-DNA................................................................................. 24 4.1.8 Ligation.............................................................................................................................................. 24 4.1.9 Herstellung kompetenter Bakterienzellen ............................................................................................ 25 4.1.10 Transformation................................................................................................................................. 26 4.1.11 Schnellisolierung von Plasmid-DNA aus E.coli (modifiziert nach Birnboim und Doly) ...................... 26 4.1.12 Plasmidisolierung [Biorad] ............................................................................................................... 27 4.1.13 Plasmidisolierung [Qiagen]............................................................................................................... 27 4.1.14 Restriktion........................................................................................................................................ 28 4.1.15 Sequenzierung.................................................................................................................................. 28 4.1.16 Gezielte Mutagenese......................................................................................................................... 28 4.1.17 Zufallsmutagenese ............................................................................................................................ 29 4.1.18 Sättigungsmutagenese....................................................................................................................... 31 4.2 BIOCHEMISCHE METHODEN ................................................................................................................. 32 4.2.1 Zellaufschluß und Rohextraktgewinnung ............................................................................................ 32 4.2.2 Aktivitätstest für die Phenylalanin Dehydrogenase .............................................................................. 33 4.2.3 Screening und Aktivitätstest in Titerplatten......................................................................................... 34 4.2.4 Aktivitätstest für das NAD-abhängige malic enzyme........................................................................... 35 4.2.5 Gekoppelte enzymatische Synthese..................................................................................................... 35 4.2.6 Bestimmung des L-Phenylalanin mittels HPLC................................................................................... 35 4.2.7 Bestimmung der Acetatkonzentration.................................................................................................. 36 4.2.8 Ganzzell Biotransformation ................................................................................................................ 36 4.2.9 Proteinaufreinigung ............................................................................................................................ 36 4.2.10 Proteinbestimmung nach Bradford .................................................................................................... 37 4.2.11 Entsalzen von Proteinlösungen.......................................................................................................... 37 4.2.12 Konzentrierung von Proteinlösungen................................................................................................. 37 4.2.13 SDS-Polyacrylamidgelelektrophorese (SDS-PAGE).......................................................................... 38 4.2.14 Färbung............................................................................................................................................ 39 4.3 MIKROBIOLOGISCHE METHODEN ........................................................................................................ 40 4.3.1 Verwendete Organismen..................................................................................................................... 40 4.3.2 Anzuchtbedingungen und Medien....................................................................................................... 40 4.3.3 Plattenkulturen ................................................................................................................................... 41 4.3.4 Schüttelkolbenkulturen ....................................................................................................................... 41 4.3.5 Konservieren von mikrobiologischen Stämmen................................................................................... 41 4.3.6 Fermentation der rekombinanten PheDH in Hochdichte Medium (HZD) ............................................. 41 4.3.7 Bestimmung der optischen Dichte....................................................................................................... 44 5 Ergebnisse .............................................................................................................................................. 45 5.1 KLONIERUNG DER PHENYLALANIN DEHYDROGENASE (PHEDH).......................................................... 45 5.1.1 Präparation genomischer DNA............................................................................................................ 45 5.1.2 Klonierung des PheDH-Gens mittels PCR........................................................................................... 46 5.1.3 Klonierung des PheDH Gens in den pUC-18....................................................................................... 47 5.1.4 Sequenzierung des phedh-Gens........................................................................................................... 49

5.1.5 Expression des recPheDH-Gens in E.coli ........................................................................................... 50 5.1.5.1 Klonierung der PheDH im Expressionsvektor PET16b ..................................................................... 50 5.1.5.2 Expression der PheDH im PET-System............................................................................................ 52 5.1.5.3 Klonierung des PheDH-Gens in pkk223-3........................................................................................ 53 5.1.5.4 Expression der recPheDH im pKK223-3 .......................................................................................... 56 5.1.6 Optimierung der Induktionsparameter................................................................................................. 57 5.1.7 Reinigung der recPheDH aus E. coli JM105........................................................................................ 60 5.1.8 Substratspektrum und KM-Werte......................................................................................................... 62 5.1.9 Temperaturstabilität............................................................................................................................ 63 5.1.10 Temperaturoptimum ......................................................................................................................... 64 5.1.11 pH-Optimum .................................................................................................................................... 64 5.2 HOCHZELLDICHTE FERMENTATION (HZD).......................................................................................... 66 5.2.1 Zuführung von Nährstoffen, Nebenproduktbildung und Wachstum...................................................... 66 5.2.2 Bestimmung des Induktionszeitpunkts ................................................................................................ 68 5.3 KLONIERUNG DES MALIC ENZYMES ...................................................................................................... 70 5.3.1 Präparation genomischer DNA............................................................................................................ 70 5.3.2 Genisolierung und Klonierung des malic enzymes im recPhe-pKK-223-3............................................ 70 5.4 KONSTRUKTION EINES EXPRESSIONSVEKTORS MIT HETEROLOGER EXPRESSION................................. 75 5.4.1 Coexpression der PheDH und des malic enzymes................................................................................ 77 5.4.2 Optimierung der Aktivität................................................................................................................... 78 5.4.3 Km-Wertbestimmung ......................................................................................................................... 81 5.4.4 Gekoppelte L-Phenylalanin Synthese unter Regeneration des Coenzyms NADH ................................. 81 5.5 GANZZELLUMSETZUNG ........................................................................................................................ 85 5.6 GEZIELTE MUTAGENESE ...................................................................................................................... 87 5.6.1 Mutation des Lysin 66 ........................................................................................................................ 88 5.6.2 Sequenzierung der K66I ..................................................................................................................... 94 5.6.3 Expression der PheDH-Mutante K66I ................................................................................................. 94 5.6.4 Herstellung weiterer Muteine.............................................................................................................. 96 5.6.5 Mutation von Lysin 66 zu Arginin und Lysin 78 zu Isoleucin .............................................................. 97 5.6.6 PCR der K66R- Mutanten und deren Klonierung................................................................................. 98 5.6.7 Aktivitätsnachweis des PheDH-Muteins K66R und K78I .................................................................... 99 5.7 ZUFALLSMUTAGENESE ....................................................................................................................... 101 5.8 ENTWICKLUNG EINES FARBTESTES ALS SCREENINGSMETHODE ......................................................... 102 5.8.1 K66R –Muteine................................................................................................................................ 104 5.8.2 Biochemische Charakterisierung des K66R-8-Muteins ...................................................................... 107 5.8.3 Synthese von Phenyllactat mit dem Mutein K66R-8.......................................................................... 109 5.8.4 Austausch K66I................................................................................................................................ 112 6 Diskussion............................................................................................................................................. 114 7 Zusammenfassung................................................................................................................................ 127 8 Literaturverzeichnis............................................................................................................................. 131

Tabellenverzeichnis
Tabelle 1: Industriell genutzte Enzyme, ihr Ursprungsorganismus, aus dem sie isoliert werden und mögliche Verwendungszwecke................................................................................................................................. 2
Tabelle 2: PCR-Zyklen ................................................................................................................................... 46 Tabelle 3: PCR Protokoll zur Amplifizierung des PheDH-Gens. Variiert wurde die Konzentration der Template-
DNA....................................................................................................................................................... 46 Tabelle 4: Ligationsansatz zur Klonierung der PheDH im pUC18-Vektor ........................................................ 48 Tabelle 5: Expressionsresultate der rekombinanten E.coli Stämme. Die Aktivitäten wurden im Rohextrakt „30
%iger Aufschluss“ gemessen................................................................................................................... 57 Tabelle 6: Zusammenfassung der Reinigung der recPheDH aus E.coli-Rohextrakt........................................... 61 Tabelle 7: Untersuchte Substrate der Rec-PheDH. ........................................................................................... 62 Tabelle 8: Kinetische Parameter für die Rec-PheDH........................................................................................ 62 Tabelle 9: PCR Protokoll zur Amplifizierung des malic enzymes aus dem rekombinanten pUC18-Plasmid.
Variiert wurden die Konzentrationen der Template-DNA......................................................................... 76 Tabelle 10: Aktivitätsbestimmung der exprimierten Enzyme in einem 10 L Fermenter mit LB-Medium als batch-
Fermentation........................................................................................................................................... 78 Tabelle 11: Aktivitätsvergleich in Abhängigkeit des Aufschlusspuffers ........................................................... 79 Tabelle 12: Proteinbestimmung beider exprimierten Enzymen, PheDH und malic enzyme, nach Variation der
Aufschlusszeit. Die Zellen wurden nach einer 60-, 30-sekundigen Behandlung zwischenzeitlich 30Sekunden abgekühlt. ........................................................................................................................... 81
Tabelle 13: Aufreinigung des rekombinanten malic enzymes........................................................................... 81 Tabelle 14: Vergleich der PheDH- bzw. der malic enzyme-Aktivität in verschiedenen Reaktionspuffern. Die
Aktivitäten sind in Prozent vom Optimalen zu sehen................................................................................ 84 Tabelle 15: Möglichkeiten zur Substratumsetzung nach einer Mutagenese. Durch den Austausch des Lysins in
Isoleucin (neutrale Aminosäure) könnte die Methylgruppe des Ketons akzeptiert werden und somit das Keton zum Amin umsetzen. Es wird eine direkte Hydrierung der Ketosäure zur Hydroxysäure bei einer Mutagense des Lysin 66 in Arginin erwartet. ........................................................................................... 88
Tabelle 16: Zusammenfassung der hergestellten Mutanten und deren neue Eigenschaften . (O) getestet aber keine Aktivität, ( -) nicht getestet. Die Restaktivität bezieht sich auf der rec-PheDH. ....................................... 100
Tabelle 17: Km-Werte des Muteins K66R-8. Untersucht wurde Phenylpyruvat sowohl bei einer reduktiven Aminierung als auch bei einer Reduktion ohne Ammoniumionen........................................................... 108
Tabelle 18: Vergleich der Reaktionsmechanismen verschiedener Muteine. Die 3 Mutationen wurden einzeln rückgängig mutiert. Die drei Mutationen sind als Kombination für die neue Eigenschaft der PheDH verantwortlich. Die Substitution der einzelnen Mutationen in die ursprünglichen Codons der PheDH führte zum Verlust der LDH-Aktivität. ............................................................................................................ 111

Abbildungsverzeichnis
Abbildung 1: Reaktionsschema der Phenylalanin Dehydrogenase. ..................................................................... 4 Abbildung 2: Reaktionsmechanismus zur Aminierung von Ketosäuren und die dabei beteiligten Aminosäuren in
der NAD-abhängigen Phenylalanin Dehydrogenase. .................................................................................. 5 Abbildung 3: Dreidimensionale Struktur der NAD-abhängigen Phenylalanin Dehydrogenase aus Rhodococcus
sp. M4. Dargestellt ist das Enzym mit dem Substrat Phenylpyruvat ohne Coenzym und Inhibitor. Die Pfeile zeigen auf die Aminosäure Lysin 66, die im aktiven Zentrum beteiligt ist, sowie auf das Substrat im Substratkanal. Die tiefe Spalte ist ein typischer Substratkanal für Aminosäure Dehydrogenasen. ................ 6
Abbildung 4: Die beteiligten Aminosäuren im aktiven Zentrum der PheDH. Die Aminosäure Lysin 66 ist die Bindungsstelle zur Carboxylgruppe im Phenylpyruvat ............................................................................... 7
Abbildung 5: Schema zur Synthese von L-Phenylalanin unter Regeneration des Coenzyms NADH mittels Formiat Dehydrogenase............................................................................................................................. 9
Abbildung 6: Protein-Design Zyklus zur gezielten Mutagenese eines Enzyms.................................................. 12 Abbildung 7: Prinzip der gezielten Mutagenese mittels überlappender Fragmente............................................ 29 Abbildung 8: Vorgehensweise der Zufallsmutagenese ..................................................................................... 31 Abbildung 9: Schema zum Screeningsvorgang ................................................................................................ 34 Abbildung 10: genomische DNA aus Rhodococcus sp. M4 nach Elektrophorese im 0.5 %igem Agarosegel ..... 45 Abbildung 11: Ergebnis der PCR Reaktionen, die Abbildung zeigt ein Fragment mit einer Größe von etwa 1100
bp, als Marker wurde die Gibco KB-Leiter verwendet. Von den PCR-Ansätze wurden 7 µl auf ein 0.8 % Agarosegel aufgetragen, 1-3 entsprechen den Ansätze in Tabelle 2 .......................................................... 47
Abbildung 12: Vektorkarte des pUC18 -PheDH .............................................................................................. 48 Abbildung 13: DNA-Sequenz der Phenylalanin Dehydrogenase aus Rhodococcus sp. M4 mit zwei Stopcodons.
............................................................................................................................................................... 49 Abbildung 14: Fließschema zur Klonierung des recPheDH-Gens in die Expressionsvektoren PET11a oder PET
16b ......................................................................................................................................................... 51 Abbildung 15: Agarose-Gel zur Überprüfung der Restriktion der recpET-16b-und 11a-Vektoren. Bei einer Größe
von etwa 1100bp tritt eine Bande des geschnittenen Inserts auf Bahn 2 pET-16b und Bahn 5 pET-11a. Als Marker wurde eine KB-Leiter (Gibco) verwendet Bahn 1. Der Ansätze wurden mit NdeI/BamHI behandelt............................................................................................................................................................... 51
Abbildung 16: SDS-Gel zur Überprüfung der Expression. Bei einer Größe von etwa 39 kDa tritt eine Bande auf. Bahn 1: Rohextrakt aus dem rec-pET16b in E. coli BL21 das Rohextrakt wurde nicht abzentrifugiert. Bahn 2: Überstand des abzentrifugierten Rohextrakts aus rec-pET16b in E. coli BL21. Bahn 3: Rohextrakt aus dem rec-pET11a in E. coli BL21. Bahn 4: Überstand des abzentrifugierten Rohextrakts aus rec-pET11a in E. coli BL21. 5: Marker........................................................................................................................... 52
Abbildung 17: Vektorkarte des pKK-223-3recPheDH. Das PheDH-Gen wurde an der SmaI-Schnittstelle im Expressionsvektor pKK-223-3 mit einem Abstand zur Ribosomenbindungsstelle von 14bp ligiert............ 54
Abbildung 18: rec-pKK223-3-PheDH verdaut mit EcoRI. 1: KB-Marker, 2-6 recPhe-pKK223-3 verdaut mit EcoRI (verschiedene Konzentrationen). Nach einer Restriktionsanalyse taucht eine Bande mit der erwarteten Größe von ca. 800bp auf......................................................................................................... 56
Abbildung 19: Variation der IPTG-Konzentration bei der Induktion von pkk-223-3-PheDH/ JM105. Die Kulturen wurden mit verschiedenen IPTG-Konzentrationen zwischen 0 und 3 mM induziert. Die spezifische Aktivität wurde im Rohextrakt gemessen............................................................................... 58
Abbildung 20: Wachstumsverhalten für E. coli nach der Induktion .................................................................. 59 Abbildung 21: Variation des Induktionszeitpunktes in Expressionskulturen von pkk-223-3-PheDH/ JM105. Die
Kulturen wurden zu unterschiedlichen Zeitpunkten mit 1 mM IPTG induziert. Die spezifische Aktivität wurde im Rohextrakt gemessen. .............................................................................................................. 60
Abbildung 22: SDS-PAGE (12.5 %, Silberfärbung) zur Überprüfung der Reinheit der recPheDH nach DEAE-Sepharose FF –Chromatographiegel. 1 Marker, 2+3 recPheDH nach DEAE-Sepharose FF, 4 Rohextrakt aus E. coli ..................................................................................................................................................... 61
Abbildung 23: Temperaturstabilität der rec-PheDH im Vergleich zur WT-PheDH im Rohextrakt..................... 63 Abbildung 24: Temperaturoptimum der recPheDH im Vergleich zur WT-PheDH, Enzym-präparate aus dem
Rohextrakt JM105. 100 % bei der rec-PheDH entsprechen 270 U/mg, und im WT-PheDH 24 U/mg. ....... 64 Abbildung 25: pH-Optimum der rec-PheDH für die reduktive Aminierung. 100 % bei der rec-PheDH
entsprechen 190 U/mg, und im WT-PheDH 16 U/mg. Die Messung wurde bei 30°C durchgeführt. .......... 65 Abbildung 26: Wachstumsverhalten und Acetatbildung durch E. coli JM105 [rec-pKK-223-3-PheDH] bei
linearer Erhöhung der Glucosezufuhr in HZD-Medium mit Hefeextraktzusatz bei 30°C. Die Acetatbildung wurde mittels HPLC bestimmt................................................................................................................. 66
Abbildung 27: Wachstumsverhalten und Acetatbildung durch E. coli JM105 [rec-pKK223-3-PheDH] in HZD-Medium mit Hefeextraktzusatz bei 30°C und exponentieller Erhöhung der Glucosezufuhr ....................... 67
Abbildung 28: Entwicklung der PheDH-Volumenaktivität in Abhängigkeit vom Induktionszeitpunkt während der Zufütterungsphase ............................................................................................................................. 68
Abbildung 29: 0.8 %iges Agarosegel zum Nachweis der Konzentrationszunahme an rec-pKK-223-3 bei Induktion 1h nach Beginn der Glucose-Zuführung während der Fermentation.......................................... 69

Abbildungsverzeichnis
Abbildung 30: Zu- und Abnahme der Plasmidkopienzahl bei Induktion 5 h nach Beginn der Glucosezuführung69 Abbildung 31: Neue PCR-Strategie zur Amplifikation des malic enzymes. Es wurde ein Einzelstrang
amplifiziert, der als Template für eine zweite PCR dient .......................................................................... 71 Abbildung 32: DNA-Sequenz des malic enzymes aus E. coli K12 ................................................................... 72 Abbildung 33: Proteinsequenz des malic enzymes aus E. coli K12 abgeleitet von der DNA-Sequenz ............... 73 Abbildung 34: Alignment der Aminosäuresequenz verschiedener malic enzymes. NADP- Salmonella enterica
subsp. enterica serovar Typhi (Parkhill et al., 2001), Mdh Pasteurella multocida (May et al., 2001), NAD-Brucella melitensis (DelVecchio et al., 2002), NADP-E.coli (Blattner et al., 1997). ................................. 75
Abbildung 35: Konstruktion des Plasmids für eine heterologe Expression zur L-Phe Synthese mittels Ganzzellumsetzung ................................................................................................................................. 75
Abbildung 36: PCR-Ausbeute des malic enzyme-Gens bei verschiedenen Konzentrationen an Template-DNA (rec-pUC18)............................................................................................................................................ 76
Abbildung 37: Vektorkarte des rec-pKK-223-3 PheDH-Malic. Malic enzyme wurde 3’ zur PheDH an der Pst1- und HindIII-Schnittstelle mit eigener Ribosomenbindungsstelle kloniert. ................................................. 77
Abbildung 38: Stabilitätsbestimmung der PheDH bzw. des malic enzymes nach verschiedenen Aufschlusszeiten mittels Ultraschalls. Die Zellen wurden nach einer 60-, 30-sekundigen Behandlung zwischenzeitlich 30 Sekunden abgekühlt. ............................................................................................................................... 80
Abbildung 39: pH-Optimum von malic enzyme und der PheDH. Die Aktivität beider Enzymen nimmt bei Zunahme des pH-Wertes zu. Die Messungen wurden mit partiell gereinigtem Enzym durchgeführt. Für die Phenylalanin Dehydrogenase wurde die reduktive Aminierung gemessen................................................. 82
Abbildung 40: Temperaturoptimum. Die Messungen wurden bei pH 8,5 und in 0,1M Hepes-Puffer durchgeführt................................................................................................................................................................ 83
Abbildung 41: HPLC-Analytik zur Nachweis von in situ Regenerationsystems gildeten L-Phenylalanin .......... 84 Abbildung 42: Bildungskinetik für L-Phe. Die Bildung von L-Phe wurde in situ durchgeführt. ........................ 85 Abbildung 43: Produktnachweis nach einer 5 stündigen Umsetzung mit ganzen rekombinanten E. coli Zellen . 86 Abbildung 44: Ganzzellumsetzung zur Produktion von L-Phe. Die Bestimmung von L-Phe erfolgte mittels
HPLC ..................................................................................................................................................... 86 Abbildung 45: Reaktionsmechanismus der reduktiven Aminierung von Phenylalanin mittels PheDH............... 87 Abbildung 46: Schema zur Mutagenese mittels PCR nach der Methode der „overlapping extension“,
ausgetauscht wurde das Lysin66 gegen Isoleucin ..................................................................................... 89 Abbildung 47: Klonierungsstrategie zur Einführung der Mutation PheDHK66I. Im ersten Schritt wurden zwei
Fragmente amplifiziert, die die gleiche Mutation tragen; beim zweiten Schritt hybridisierten sie zum ganzen Gen. Somit konnte das amplifizierte Gen in den Expressionsvektor ligiert werden.................................... 90
Abbildung 48: Agarosegelanalyse der PCR zur K66I- Mutagenese. Spur 1: kb-Leiter; Spur 2 unspezifische Amplifikation der Teilfragmente; Spur 3 und 4 sind gewünschte Amplifikate nach einer Optimierung der PCR- Bedingungen mit einer Größe von ca. 200 bp bzw. 900 bp.............................................................. 93
Abbildung 49: Sauberes PCR-Produkt nach der Fusion der beiden Fragmente (Abbildung 48; Bahn 3+4) mit einer Größe von 1.1 kb ............................................................................................................................ 93
Abbildung 50: Gensequenz des K66I- Muteins................................................................................................ 94 Abbildung 51: Das aktive Zentrum der PheDH und der Effekt des Lysin66 durch den Austausch in Isoleucin. (A)
Wechselwirkung zwischen Lysin66 und der Carboxylgruppe des Substrates in der PheDH, (B) Mutein K66I, Lysin wurde gegen Isoleucin ausgetauscht, das keine Wasserstoffbindung zur Carboxylgruppe des Substrates bildet. Wasserstoffbindungen sind grün dargestellt. ................................................................. 95
Abbildung 52: Reaktionsmechanismus der reduktive Aminierung von Phenylaceton....................................... 95 Abbildung 53: Klonierungsstrategie der PheDH-Mutanten am Beispiel der PheDH-K66R. Das ganze
rekombinante Plasmid wird mittels Mutagenese-Primer amplifiziert und in E.coli-Zellen transformiert ... 98 Abbildung 54: Reaktionsmechanismus zur Umsetzung der Ketosäure Phenylpyruvat zur Hydroxysäure
Phenyllactat. ........................................................................................................................................... 99 Abbildung 55: Reaktionsschema zum Verlauf des Farbtests........................................................................... 103 Abbildung 56: Farbtest auf Agarplatten mit ganzen Zellen. Die gelben Zellen enthalten PheDH-Aktivität...... 103 Abbildung 57: Bestimmung der Aktivität der K66R-Muteine. Gezeigt werden einige Muteine mit den restlichen
Aktivitäten im Vergleich zur Wt-PheDH. Bei zwei Muteinen (Mutein 2 und 8) konnte der natürliche Reaktionsmechanismus verändert und somit neue Eigenschaften erzielt werden..................................... 104
Abbildung 58: Aminosäurensequenz des Mutein K66R-8 im Vergleich zur PheDH-AS-Sequenz. 3 Mutationen K66R, T208A, E225D sind in diesem Mutein für die neue Aktivität verantwortlich................................ 105
Abbildung 59: Dreidimensionale Struktur des K66R-8-Mutein. Gezeigt sind die durch die Mutagenese ausgetauschten Aminosäuren. Die Mutationen K66R, T208A und E225D sind hervorgehoben. Wasserstoffbindungen sind grün dargestellt. .......................................................................................... 106
Abbildung 60: Das aktive Zentrum der PheDH und der Effekt des Lysin66 durch den Austausch in Arginin. (A) Wechselwirkung zwischen Lysin66 und der Carboxylgruppe des Substrates, (B) Arginin wird anders orientiert und geht keine Wechselwirkung mit der Carboxylgruppe des Substrates ein. Wasserstoffbindungen sind grün dargestellt. .......................................................................................... 107

Abbildungsverzeichnis
Abbildung 61: Reaktionsschema des K66R-8-Mutein.................................................................................... 107 Abbildung 62: pH-Optimum des Muteins K66R-8 im Vergleich zur Wildtyp-PheDH. Der Aktivitätstest
(Reduktion des Phenlpyruvat ohne Ammoniumionen) wurde bei verschiedenen pH-Werten zwischen pH 5 und pH 10 gemessen ............................................................................................................................. 108
Abbildung 63: Temperaturoptimum des K66R-8-Muteins im Vergleich zur Wildtyp-PheDH. Die Ansätze enthielten Tris/HCl Puffer (100 mM pH 8.0 + Phenylpyruvat und NADH) wurden auf den jeweiligen Meßtemperaturen (25-50°C) in der Küvette temperiert und gemessen .................................................... 109
Abbildung 64a: GC-Chromatogramm des Syntheseansatzes der K66R-8 nach zweistündiger Inkubation. Eingesetzt wurden 30 mM Phenylpyruvat, 100 mM HEPES-Puffer, 100 mM Formiat, 2 mM NAD+, 2 mM MgCl2, 20 U K66R-8 und 30 U Formiat Dehydrogenase. ....................................................................... 110
Abbildung 65: Einfluss der Mutationen K66R, T208A und E225D, auf die Enantioselektivität der Reduktion von Phenylpyruvat (30 mM) in Abhängigkeit von der Zeit..................................................................... 111
Abbildung 66 Produktnachweis nach einer 9 stündigen Umsetzung mit dem Mutein K66I. Zur Regenerierung des Coenzyms NADH wurden 40 U Formiat Dehydrogenase eingesetzt. Die reduktive Aminierung erfolgte mit (NH4)2SO4 und 30 mM Phenylpyruvat als Substrat. (L-Phe = 23 min; der Peak bei 29 min ist nicht identifiziert). ......................................................................................................................................... 113
Abbildung 67: Alignment der Aminosäurensequenzen der Wt-PheDH aus Rhodococcus sp. M4 (Brunhuber et al., 1994) mit der LDH aus Bifidobacterium longum (Iwata et al., 1994) und der Mutante K66R-8. Rot: Identische Aminosäuren; grün: ähnliche Aminosäuren; Durch Fettdruck hervorgehoben: Mutationen; blaue Pfeile: wichtige Reste zur Bildung der Rossmann fold. .......................................................................... 125

Abkürzungsverzeichnis
Abb. Abbildung
ALF Automated Laser Fluorescent Sequencer
Amp Ampicillin
APS Ammoniumpersulfat
ATP Adenosintriphosphat
BSA Bovine serum albumine
C-Quelle Kohlenstoffquelle
CTAB N-cetyl-N,N,N-trimethylammonium Bromid
C-Terminus Carboxy-Terminus
Da Dalton
DEAE Diethylaminoethyl
dATP Desoxyadenosintriphosphat
dCTP Desoxycytidintriphosphat
dGTP Desoxyguanosintriphosphat
dNTP Desoxyribonukleotidtriphosphat
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
DTT Dithiothreitol
EDTA Ethylendiamintetraessigsäure
FDH Formiatdehydrogenase
FF Fast Flow
h Stunde
IB-L-C N-Iobutyryl-L-Cystein
IEF Isoelektrische Fokussierung
IEP Isoelektrischer Punkt
IPTG Isopropylthiogalactosid
kb Kilobasen
kDa Kilodalton
Kpi Kaliumphosphatpuffer
Km Michaelis-Menten Konstante
L Liter
LB Luria Bertani
min Minute
M molar

Abbildungsverzeichnis
mM Millimolar
MOPS 3-N-Morpholino-propansulfonsäure
NAD Nicotinamidadenindinukleotid (oxidierte Form)
NADH Nicotinamidadenindinucleotid (reduzierte Form)
N-Terminus Amino-Terminus
OD optische Dichte
OPA o-Phtaldialdehyd
PAA Polyacrylamid
PAGE Polyacrylamidgelelektrophorese
PCR Polymerase chain reaction
PEG Polyethylenglycol
PheDH Phenylalanin Dehydrogenase
Rec-PheDH rekombinante wt-PheDH
SDS Natriumlaurylsulfat
TBE Tris-Borsäure-EDTA Puffer
TE Tris-EDTA-Puffer
TEA Triethanolamin
TEMED N,N,N,N-Tetramethylethylendiamin
Tris N-Tris-(hydroxymethyl)-aminomethan
U Unit (Enzymeinheit)
Upm Umdrehung pro Minute
UV Ultraviolett
Vmax maximale Reaktionsgeschwindigkeit
v/v Volumenanteil pro Volumenanteil
w/v Gewichtsanteil pro Volumenanteil
w/w Gewichtsanteil pro Gewichtsanteil
WT Wildtyp

Einleitung 1
1 Einleitung 1.1 Bedeutung von Enzymen
In den meisten Publikationen über die Biokatalyse findet man die Aussage, dass die
chemische Industrie durch die Biokatalyse inhärent bessere Verfahren zu erwarten hat. Dabei
sollte allerdings nicht übersehen werden, dass seit 200 Jahren verschiedenste Verbindungen
von Arzneimitteln bis zu Haushaltsreinigern ohne Biokatalysatoren effizient hergestellt
werden. Daher besteht kein Grund zu der Annahme, dass die Chemie in den nächsten
Jahrhunderten nicht ebenso erfolgreich sein wird. Die großen Vorteile chemischer Verfahren
sind Einfachheit, Geschwindigkeit (gesteigert durch den Einsatz von Hitze und Druck) und
niedrige Kosten, warum also sollten Biokatalysatoren interessant sein? Die genannten
Vorteile der chemischen Verfahren kommen manchmal nicht zum Tragen, wenn für das
gewünschte Produkt regio- oder stereospezifische Reaktionen erforderlich sind, wenn Edukte
oder das Produkt instabil sind oder wenn Verunreinigungen durch Nebenreaktionen ein ernst-
haftes Problem sind. Biokatalysatoren sind attraktiv, weil zu ihren Eigenschaften Chemo-,
Regio- und Stereoselektivität, eine beeindruckende Katalyseeffizienz und die Fähigkeit zur
Reaktion in wässrigen Medien gehören.
So besitzen Enzyme eine hohe Chemo- und Regioselektivität, die Voraussetzungen für die
Steuerung der komplexen Vorgänge in einer Zelle sind. Enzyme beschleunigen Reaktionen
um Faktoren von wenigstens einer Million. Ohne sie würden die meisten Reaktionen in
biologischen Systemen nicht in wahrnehmbarem Umfang ablaufen.
Eine erhebliche Bedeutung hat die Biotechnologie in der Qualitätsverbesserung von
Nahrungsmitteln in Bezug auf Struktur und Geschmack (Uhlig, 1991) sowie in der
Herstellung von Aminosäuren erlangt. Aufgrund der hohen Spezifität und Selektivität werden
Enzyme in zunehmendem Maße in die klassische Organische Chemie integriert. Für
Pharmaka und Agrochemikalien sind enantiomerenreine Wirkstoffe zunehmend gefordert, um
unerwünschte Nebenwirkungen auszuschließen, wie im bekanntesten Fall beim Contergan,
bei dem das (R)-Enantiomer beruhigend, das (S)-Enantiomer aber teratogen wirkt (Faber,
1997). Die Synthese von enantiomerenreinen Verbindungen ist daher von besonderem
Interesse.
Dass Enzyme nicht immer bei industriellen Prozessen zum Einsatz kommen, liegt
hauptsächlich daran, dass Enzyme unter harten industriellen Bedingungen eingesetzt werden
sollten, die zu deren Denaturierung führen. Dazu gehören vor allem hohe Temperaturen und
die Anwesenheit organischer Lösungsmittel. Dennoch haben biotechnologische Verfahren es
ermöglicht, einige klassische chemische Prozesse zu ersetzen.
Einleitung 2
1.2 Enzyme in der Industrie
Wegen der strengen Umweltauflagen für die chemische Industrie suchen die Groß-Industrien,
aber auch mittelständige Betriebe nach umweltfreundlichen Methoden. Dadurch entwickeln
sich biotechnologische Verfahren rasch und es werden immer mehr Enzyme in der Industrie
eingesetzt.
Die Pharmaindustrie hat enorme Aufwendungen in die Herstellung enantiomerenreiner
Wirkstoffe gesteckt, um pharmakologisch wirksame Substanzen in der geforderten Reinheit
zu erhalten. Mittlerweile wird die Forderung nach enantiomerenreinen Wirkstoffen auch für
die Entwicklung neuer Agrochemikalien intensiv geprüft (Ernst&Young, 1998). Zu erwarten
ist, dass dieses Qualitätskriterium eingehalten werden muss, sobald der Stand der Technik
dies zulässt.
Industriell genutzte Enzyme sind z.B. die bei der Fruchtsaftklärung verwendeten Pektinasen
oder die als enzymatische Wirkstoffkomponenten in Waschmitteln und in der
Textilverarbeitung zum Einsatz kommenden Lipasen, Proteasen, und Cellulasen (Tabelle 1)
(Jaag, 1968).
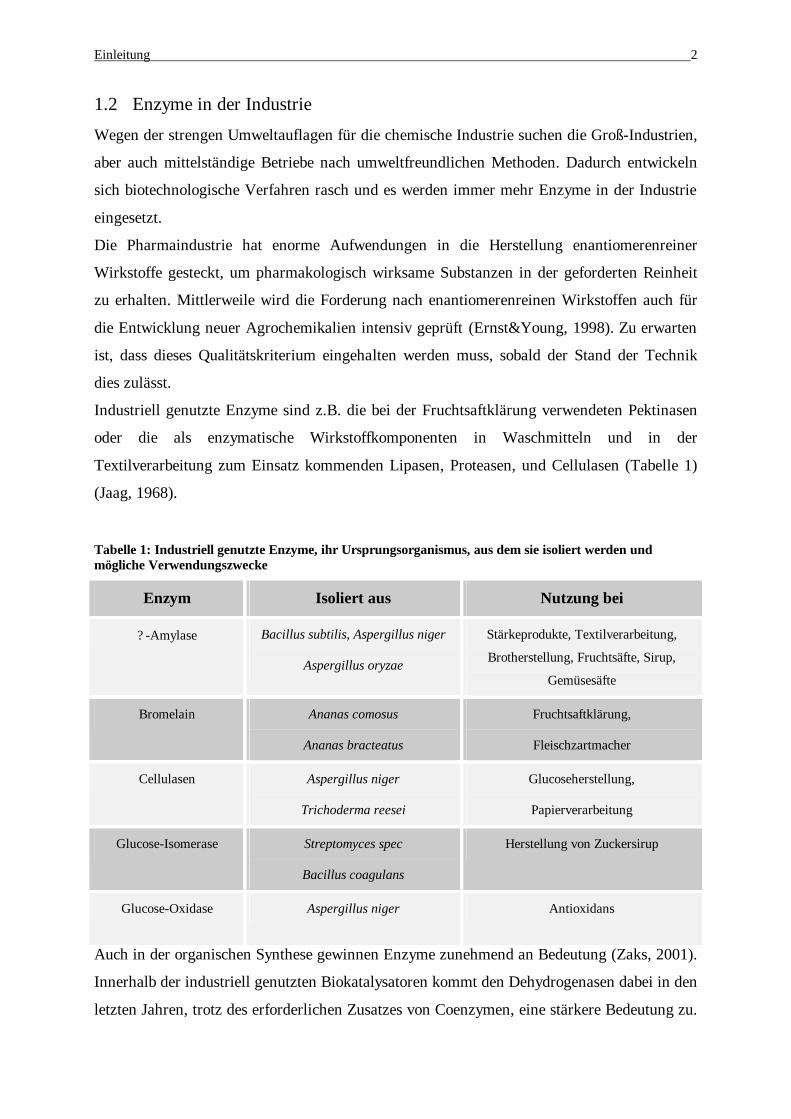
Tabelle 1: Industriell genutzte Enzyme, ihr Ursprungsorganismus, aus dem sie isoliert werden und mögliche Verwendungszwecke
Enzym Isoliert aus Nutzung bei
? -Amylase Bacillus subtilis, Aspergillus niger
Aspergillus oryzae
Stärkeprodukte, Textilverarbeitung,
Brotherstellung, Fruchtsäfte, Sirup,
Gemüsesäfte
Bromelain Ananas comosus
Ananas bracteatus
Fruchtsaftklärung,
Fleischzartmacher
Cellulasen Aspergillus niger
Trichoderma reesei
Glucoseherstellung,
Papierverarbeitung
Glucose-Isomerase Streptomyces spec
Bacillus coagulans
Herstellung von Zuckersirup
Glucose-Oxidase Aspergillus niger Antioxidans
Auch in der organischen Synthese gewinnen Enzyme zunehmend an Bedeutung (Zaks, 2001).
Innerhalb der industriell genutzten Biokatalysatoren kommt den Dehydrogenasen dabei in den
letzten Jahren, trotz des erforderlichen Zusatzes von Coenzymen, eine stärkere Bedeutung zu.

Einleitung 3
(Carrea et al., 1996). Da Coenzyme kostenintensiv sind, wenn sie in äquimolaren Mengen in
der enzymatischen Synthese im industriellen Maßstab eingesetzt werden, wurden eine Reihe
von Verfahren entwickelt, die Coenzyme in situ zu regenerieren (Kometani et al., 1994;
Leonida et al., 1998). Dabei stellen enzymatische Verfahren die effizienteste Methode zur
Coenzymregenerierung dar.
So stellen Dehydrogenasen ideale Biokatalysatoren für asymmetrische Synthesen chiraler,
enantiomerenreiner Substanzen aus prochiralen Vorstufen dar (Hummel & Kula, 1989).
Einsatzgebiete sind derzeit vor allem die Produktion von Pharmazeutika, Feinchemikalien und
Lebensmittelzusatzstoffen (Drauz et al., 1994). Kommerziell gehandelte Enzyme wie zum
Beispiel Alkohol Dehydrogenasen, Phenylalanin Dehydrogenase (Hummel, 1997), D- oder L-
Lactat Dehydrogenasen oder D-Hydroxyisocapronsäure Dehydrogenasen (Lerch et al., 1989),
wurden für die Präparation von optisch aktiven Substanzen mit Erfolg eingesetzt (Hanson et
al., 2000).
Am Beispiel der Synthese von enantiomerenreinen Aminosäuren durch die enzymatische
reduktive Aminierung prochiraler ? -Ketosäuren werden Aminosäure Dehydrogenasen wie die
Leucin Dehydrogenase (LeuDH) aus Bacillus sp. (Bommarius & Drauz, 1994; Bommarius et
al., 1995; Wichmann et al., 1981) und die Phenylalanin Dehydrogenase (PheDH) aus
Rhodococcus sp. M4 mit Erfolg eingesetzt (Hummel et al., 1987).
Eine Vielzahl neuer aliphatischen und aromatischen ? -Aminosäurederivate, kann aufgrund
der breiten Substratspektren der LeuDH und der PheDH mit hoher Enantioselektivität
hergestellt werden (Krix et al., 1997). Erfolgreich wurde die NAD-abhängige Leucin
Dehydrogenase aus Bacillus sp. für die Synthese optisch aktiver Aminosäuren mit
ungewöhnlichen Resten wie (S)-tert.-Leucin eingesetzt (Bommarius et al., 1995). Das meist
breite Substratspektrum dieser für die chemische Synthese einsetzbaren Enzyme wirkt sich
vorteilhaft aus, da meistens die Enzyme für mehr als ein Substrat verwendbar sein sollten.
Das bei Dehydrogenasen notwendige Coenzym beeinträchtigt die Synthese nicht, da NAD+
über einen längeren Zeitraum stabil (Janssen et al., 1987; Oppenheimer & Kaplan, 1974;
Wong & Whitesides, 1981) und durch die Regenerierung mit Formiat kostengünstig
einsetzbar ist.

Einleitung 4
1.3 Die Phenylalanin Dehydrogenase aus Rhodococcus sp. M4
Die Phenylalanin Dehydrogenase (PheDH) aus Rhodococcus sp. M4 katalysiert die oxidative
Desaminierung von L-Phenylalanin zum Phenylpyruvat und Ammonium, dabei wird das
Coenzym NAD+ zum NADH reduziert (Abbildung 1) (Hummel et al., 1987).
O C COOH
CH 2
H2NHC COOH
CH 2PheDH+ NH4
+ + NADH + NAD+ + H20
Phenylpyruvat L-Phenylalanin
Abbildung 1: Reaktionsschema der Phenylalanin Dehydrogenase.
Das Enzym benötigt NAD+ als natürliches Coenzym und wurde bei einigen gram-positiven,
aeroben Bakterien gefunden. Anfänglich wurde die PheDH in Brevibacterium sp (Hummel et
al., 1984) und später in anderen Bakterienstämmen wie Bacillus, Sporosarcina (Asano et al.,
1987), Nocardia (de Boer et al., 1989), Thermoactinomyces (Ohshima et al., 1991), und
Rhodococcus (Hummel et al., 1987) gefunden. Die Phenylalanin Dehydrogenase gehört zu
einer großen Aminosäure-Dehydrogenasen-Familie, zu der die Glutamat Dehydrogenase,
Alanin Dehydrogenase, Leucin Dehydrogenase, und die selten vorkommende Lysin-
Dehydrogenase gehören. Die PheDH kann für die Synthese von optisch aktivem L-
Homophenylalanin, einem Baustein vom Angiotensin, für die Behandlung von Bluthochdruck
(Hypertension) und Herzfehlern (Abrams et al., 1984; Ondetti & Cushman, 1981) und
ebenfalls für die Herstellung von Allysin als Vasopeptidase Inhibitor (Hanson et al., 2000)
sowie zur industriellen Synthese von L-Phenylalanin als Komponente von Aspartam
(Wichmann et al., 1981) verwendet werden. Ebenso kann das Enzym für den Nachweis der
Phenylketonurie (Centerwall & Centerwall, 2000) mittels eines Biosensors eingesetzt werden
(Wendel et al., 1991; Wendel et al., 1990).
Die Phenylalanin Dehydrogenase aus Rhodococcus sp. M4 ist im Gegensatz zu den octameren
Phenylalanin Dehydrogenasen aus Bacillus sphaericus (Okazaki et al., 1988) und
Sporosarcina urea (Asano et al., 1987) ein Tetramer von 39.5 kDa pro Monomer (Brunhuber
et al., 1994). Die Phenylalanin Dehydrogenase aus Thermoactinomyces liegt als Hexamer vor
(Takada et al., 1991). In Abbildung 2 ist der Reaktionsmechanismus der Phenylalanin
Dehydrogenase dargestellt (Brunhuber et al., 2000). Die reduktive Aminierung des Phenyl-
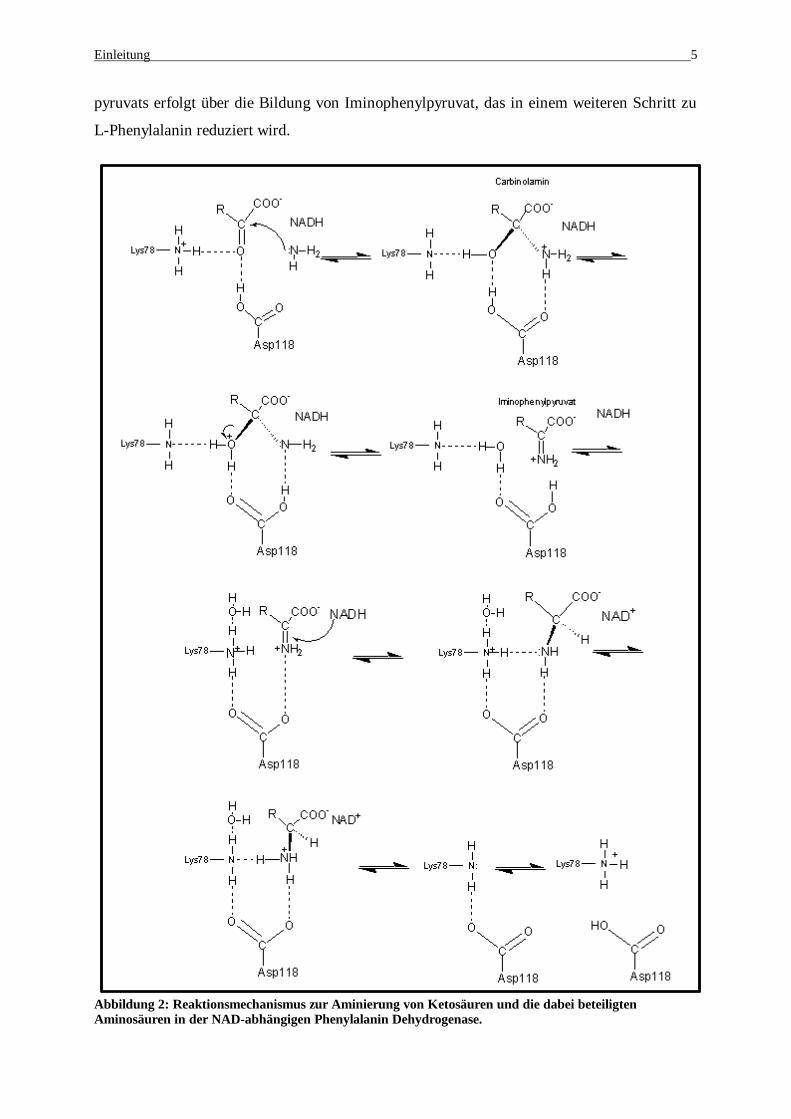
Einleitung 5
pyruvats erfolgt über die Bildung von Iminophenylpyruvat, das in einem weiteren Schritt zu
L-Phenylalanin reduziert wird.
Abbildung 2: Reaktionsmechanismus zur Aminierung von Ketosäuren und die dabei beteiligten Aminosäuren in der NAD-abhängigen Phenylalanin Dehydrogenase.

Einleitung 6
Die Kristallisation und Aufklärung der PheDH aus Rhodococcus sp. M4 wurde durch
Vanhooke et al (Vanhooke et al., 1999) ermöglicht. Basierend auf den Daten der
dreidimensionalen Struktur konnte die PheDH mittels 3D-Programm (Swiss PDB Viewer)
modelliert werden (Abbildung 3). Hervorgehoben sind die Aminosäure Lysin an Position 66
im aktiven Zentrum, die Lage des Substrates Phenylpyruvat und das Coenzym NAD+.
Abbildung 3: Dreidimensionale Struktur der NAD-abhängigen Phenylalanin Dehydrogenase aus Rhodococcus sp. M4. Dargestellt ist das Enzym mit dem Substrat Phenylpyruvat ohne Coenzym und Inhibitor. Die Pfeile zeigen auf die Aminosäure Lysin 66, die im aktiven Zentrum beteiligt ist, sowie auf das Substrat im Substratkanal. Die tiefe Spalte ist ein typischer Substratkanal für Aminosäure Dehydrogenasen.
Lysin 66
Phenylpyruvat
NAD+
Einleitung 7
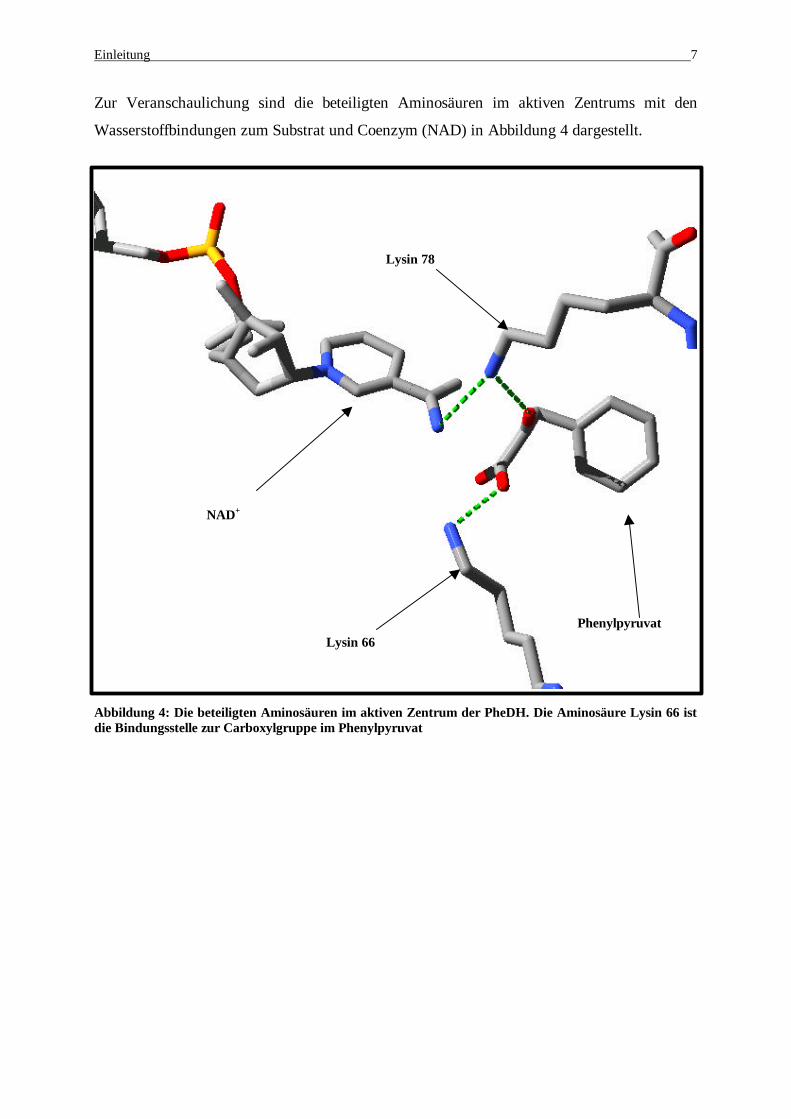
Zur Veranschaulichung sind die beteiligten Aminosäuren im aktiven Zentrums mit den
Wasserstoffbindungen zum Substrat und Coenzym (NAD) in Abbildung 4 dargestellt.
Abbildung 4: Die beteiligten Aminosäuren im aktiven Zentrum der PheDH. Die Aminosäure Lysin 66 ist die Bindungsstelle zur Carboxylgruppe im Phenylpyruvat
NAD+
Phenylpyruvat Lysin 66
Lysin 78

Einleitung 8
1.3.1 Malic enzyme aus E. coli K12
Das NAD abhängige malic enzyme ist in der Natur weit verbreitet und katalysiert die
oxidative Decarboxylierung von L-Malat. Dieses Enzym gehört zur Klasse der
Oxidoreduktasen und kann für die kolorimetrische Bestimmung von Malat und ebenfalls für
die NADH-Regenerierung im Enzymreaktor eingesetzt werden (Nakamura et al., 1986).
Das malic enzyme wurde sehr umfangreich in verschiedenen Organismen, in Bakterien
(Sulfolobus solfataricus (Iwakura et al., 1978), Clostridium thermocellum (Lamed & Zeikus,
1981), Bacillus subtilis (Diesterhaft & Freese, 1973), Pseudomonas fluorescens (Knichel &
Radler, 1982), in Pflanzen Zea mays, Solanum tuberosum (Hausler et al., 1987; Willeford &
Wedding, 1987), und ebenfalls in höheren Organismen, Rattenleber (Bagchi et al., 1987;
Magnuson et al., 1986; Winberry et al., 1983) untersucht.
Das in dieser Arbeit klonierte NAD abhängige E. coli K12 malic enzyme zeigt Ähnlichkeit im
Molekulargewicht (50 kDa) zum malic enzyme aus Bacillus stearothermophilus (Kobayashi
et al., 1989) aber einen Unterschied zum NADP-abhängigen malic enzyme aus E. coli (62
kDa) (Nakamura et al., 1986).
1.3.2 L-Phenylalanin-Synthese unter Coenzymregeneration
Verschiedene Wege führen zur Produktion der Aminosäure L-Phenylalanin. Man kann dies
durch chemische Synthesen, Extraktion von Protein-Hydrolysaten, fermentative oder
enzymatische Methoden erreichen. Eine direkte Fermentation zur Produktion von L-
Phenylalanin ist mit Corynebacterium glutamicum (Nakayama et al., 1978) oder Bacillus
subtilis (McEvoy & Joyce, 1974) beschrieben. Dennoch konnten mittels der Phenylalanin
Dehydrogenase aus Rhodococcus sp. M4 in einem Enzym-Membran-Reaktor 456 g L-1·d-1
L- Phenylalanin erzielt werden (Hummel et al., 1987). Die Synthese des L- Phenylalanins
wurde durch die PheDH unter Regeneration des Coenzyms NADH mit der Formiat
Dehydrogenase (FDH) katalysiert. Phenylpyruvat wird in diesem System durch reduktive
Aminierung mit Hilfe von PheDH in L-Phenylalanin umgewandelt. Gleichzeitig sorgt die
FDH für die Regenerierung des NADH durch Formiat (Abbildung 5) (Hummel et al., 1987).
Für die Regenerierung des NADH im industriellen Maßstab wird bis dato die NAD-abhängige
Formiat Dehydrogenase eingesetzt (Kula & Wandrey, 1987; Shaked & Whitesides, 1980).
Das Verfahren konnte großtechnisch angewandt werden (Kragl et al., 1992).

Einleitung 9
Abbildung 5: Schema zur Synthese von L-Phenylalanin unter Regeneration des Coenzyms NADH mittels Formiat Dehydrogenase.
1.3.3 Expression rekombinanter Proteine
Die Möglichkeit, Proteine, die normalerweise entweder gar nicht oder nur in extrem geringen
Mengen verfügbar waren, in großen Mengen herzustellen, beeindruckte Wissenschaftler,
Ärzte und Geschäftsleute gleichermaßen. Im Jahre 1976 hielt die Gentechnik Einzug in die
Biotechnologie. Erstmals wandte man die Verfahren der DNA-Klonierung, Oligonucleotid-
synthese und Genexpression in einem einzelnen Experiment an, bei dem man ein
menschliches Protein durch die Expression einer rekombinierten DNA gewann. Es handelte
sich um den aus 14 Aminosäuren bestehenden Peptidneurotransmitter Somatostatin (Itakura et
al., 1977). Das Gen des entsprechenden Proteins wurde in einem Plasmid kloniert und in E.
coli exprimiert. Wenig später gelang die Expression des menschlichen Insulins (Nossal,
1980), das zur Behandlung des Diabetes dient und zum ersten kommerziell genutzten Produkt
der Gentechnik wurde.
Solche Meisterleistungen beruhten auf Erfolgen in allen Bereichen der Molekularbiologie,
unter anderem der Oligonucleotidsynthese, der Isolierung der DNA-spaltenden und
verknüpfenden Enzyme, der Charakterisierung bakterieller Plasmide und der Erforschung der
Genexpression.
Eine Vielfalt an Expressionsplasmiden ist kommerziell erhältlich. Die Minimal-Ausstattung
solcher Plasmide besteht aus einem Promotor, einer Ribosomenbindungsstelle, dem Origin of
Replication, und einem Selektionsmarker. Dennoch führt der Abbau oder die chemische
Modifizierung des Antibiotikums durch das Produkt des Resistenzgens oder den Wirtsstamm
oft zur Reduktion der Plasmidkopienzahl in der Zelle und begünstigen die Entstehung
heterogener Organismenpopulationen (Bentley et al., 1993).
Formiat
CO2
NAD +
NADH
L-Phenylalanin + H 2O
Phenylpyruvat + NH4+
PheDHFDH
Regeneration des Coenzymes NADH

Einleitung 10
Für die Verbesserung solcher Systeme wurden spezielle DNA-Sequenzen bezüglich
Plasmidstabilität, mRNA-Stabilität, effizienter Translationsinitiation, und rechtszeitiger
Transkriptionstermination entwickelt (Balbas & Bolivar, 1990). Beispiele solcher Sequenzen
bilden Repressorgene, Terminatoren, Antiterminatoren, Sekretionsgene und Reportgene
(Stader, 1995). Die Stabilität des Fremdgens oder Plasmids (Segregationsstabilität), wird
durch hohe Kopienzahl, par-Sequenzen und Selektionsmarker gefördert (Balbas & Bolivar,
1990). Durch die Einführung einer par-Sequenz kann die Segregationsstabilität eines
Expressionsvektors erhöht werden (Zurita et al., 1984). Einfluß üben außerdem die
Transkriptionsaktivität des Fremdgens und die genetischen Eigenschaften des
Wirtsorganismus (Stader, 1995) sowie die Zusammensetzung des Kulturmediums aus (Mason
& Bailey, 1989).
Die Klonierung des Gens der cDNA eines Proteins ist nur der erste von vielen Schritten, um
gentechnisch ein Protein für medizinische oder industrielle Anwendungen herzustellen. Der
nächste Schritt besteht darin, das Gen in eine Wirtszelle einzuschleusen und es darin zu
exprimieren. Die bekanntesten Expressionssysteme sind die Bakterien E.coli und Bacillus
subtilis, die Hefe, sowie kultivierte Insekten- und Säugetierzellen. Welches System geeignet
ist, hängt von den Projektzielen und den Eigenschaften des zu produzierenden Proteins ab.
Bakterienzellen sind einfache Systeme mit kurzer Generationszeit, hoher Ausbeute und
niedrigen Kosten. Die Zellen, insbesondere von B. subtilis, lassen sich induzieren und
schleusen das Produkt in das Kulturmedium aus, was die Reinigung des Proteins
außerordentlich vereinfacht. Allerdings haben prokaryotische Zellen auch einige Nachteile.
Manche Proteine werden zwar extrem stark exprimiert (sie können mehr als 20 % der Masse
aller Bakterienproteine ausmachen), aber oft falten sie sich nicht korrekt, und bilden dann
unlösliche Einschlusskörper (inclusion bodies) (Hibino et al., 1994). Diese Proteine kann man
zwar durch Extraktion gewinnen, sie sind allerdings in aller Regel biologisch inaktiv. Kleine
Proteine lassen sich unter Umständen in ihre aktive Form überführen (Babbitt et al., 1990;
Huang et al., 1993), bei großen Produkten ist das jedoch normalerweise nicht möglich. Ein
zweites Problem ist, dass Fremdproteine für Bakterien manchmal toxisch sind wie z.B.
Aminosäure-Oxidasen, sodass Kulturen, die das Protein herstellen, nicht in hoher Dichte
wachsen können. Diese Schwierigkeit lässt sich oft durch einen induzierbaren Promotor
umgehen. Wenn die Kultur dicht gewachsen ist, schaltet man den Promotor an, der daraufhin
mit der Transkription des Gens für das toxische Fremdprotein beginnt.

Einleitung 11
Drittens fehlen den Bakterien jene Enzyme, die Proteine in Eukaryotenzellen nach der
Translation modifizieren, indem sie ihnen beispielsweise Phosphatgruppen oder Zuckerreste
anhängen. Derartige chemische Modifikationen sind häufig notwendig, damit die Proteine
Aktivität zeigen. Eine Möglichkeit zur posttranslationalen Modifikation besteht darin, die
modifizierenden eukaryotischen Enzyme zu reinigen und mit ihnen bakteriell exprimierte
Proteine nachträglich abzuwandeln.
1.4 Protein-Design
1.4.1 Gezielte Mutagenese
Die Nutzung von Mikroorganismen und der daraus gewonnenen Enzyme erhalten neue
Aspekte durch die Gentechnik, mit denen sowohl neue Produkte herstellbar werden, als auch
erhebliche Verbesserungen in Ausbeute und Qualität bei konventionellen Produkten zu
erwarten sind.
Mittels NMR-Techniken und Röntgenstrukturanalyse wurde die Ermittlung der
dreidimensionalen Struktur eines Proteins ermöglicht, und durch den Einsatz von
Computertechnik gezielt Veränderungen vorgenommen.
Über die gezielte Verknüpfung dieser Strukturaufklärung mittels Computertechnik mit den
neuen Kenntnissen der Biochemie und der Molekularbiologie können wirtschaftlich
interessante Enzyme untersucht und modifiziert werden. So wurden Enzymvarianten
hergestellt, die sich durch überlegene Eigenschaften, wie z.B. höhere Aktivität, bessere
Stabilität, erweitertes Substratspektrum oder Coenzymspezifität auszeichnen (Galkin et al.,
1997; Slusarczyk et al., 2000). Einige veränderte Enzyme können so technisch interessante
Chemikalien bis zu 10000x besser umsetzen, als die natürliche Form des Enzyms (Wilkinson
et al., 1984). Statt tausender von Proteinen auf mögliche Wirksamkeit zu testen, versucht man
mit Hilfe des Protein-Designs eine neue maßgeschneiderte Struktur am Computer zu
entwerfen. Diese neumodifizierten Enzyme werden dann mit molekularbiologischen
Methoden hergestellt und hinsichtlich Struktur und neuer Eigenschaften untersucht. Für den
Einsatz eines erfolgreichen rationalen Designs müssen vorab tiefgehende Informationen über
Struktur und Reaktionsmechanismus vorliegen. Abbildung 6 zeigt den Zyklus, wie er
normalerweise durchgeführt wird.

Einleitung 12
W ild ty p
D N A -Is o la t io n
G e n b a n k
K lo n ie ru n g
E x p re s s io n
P ro te in
P r im ä rs tru k tu r
D N A -S y n th e s e
P ro te in is o lie ru n g
N M R
m o le k u la re s M o d e ll
K r is ta llis a t io n
R a u m s tru k tu r
R K S A
E ig e n s c h a f te n
M o d if ik a t io n
Abbildung 6: Protein-Design Zyklus zur gezielten Mutagenese eines Enzyms
(RKSA = Röntgenstrukturanalyse)
Die Überlegung, dass durch die Senkung der Entfaltungsentropie mittels ausgewählter
Aminosäurensubstitutionen die Konformation des T4-Lysozyms stabilisiert werden kann,
haben Mathews et al. (Matthews et al., 1987) durch die Erzeugung einer Doppelmutante
nachgewiesen und somit die Thermostabilität dieses Proteins erhöht. In einer folgenden
Arbeit wurde durch die Einführung von zwei Disulfidbrücken an definierten Stellen im
Lysozym, das heißt Austausch von vier Aminosäuren, die strukturell günstig für eine
Ausbildung lagen, die Thermostabilität von 41°C auf 58°C erhöht, wobei das neu entstandene
Enzym die volle Aktivität besaß (Matsumura et al., 1989).
Vacca et al. (Vacca et al., 1995) konnten durch den Austausch von Tryptophan 140 gegen
Histidin mittels gezielter Mutagenese die Reaktions- bzw. Substratspezifität der
Aspartataminotransferase verändern. Dadurch kann Alanin siebenmal schneller als durch das
Wildtypenzym racemisieren, während die Aktivität des Muteins gegenüber Alanin sechsfach
abnahm.
Die benötigte exakte Aufklärung der Struktur eines Enzyms ist ein Nachteil der gezielten
Mutagenese, da sich die Strukturen bekannter Proteine nicht auf andere mit ähnlichen
Sequenzmotiven übertragen lassen.

Einleitung 13
1.4.2 Zufallsmutagenese
Die Methode der „Zufallsmutagense“ (random mutagenesis), bei der ein großer Pool an
Veränderungen in ein Enzym eingeführt wird, ist eine gute Alternative zum rationalen
Protein-Design. Um das Screening der durch die Zufallsmutagenese hergestellten
Enzymvarianten- Bibliothek ermöglichen zu können, sind geeignete und effektive
Selektionsmechanismen notwendig. Mit geeigneten Methoden können die gewünschten
Eigenschaften detektiert werden. Es wird eine Art künstlicher Evolution herbeigeführt
(Arnold & Moore, 1997), bei der man den Selektionsdruck zur Verbesserung der
Eigenschaften zu Hilfe nimmt (Bornscheuer et al., 1998; Bornscheuer et al., 1999).
Diese Methode wurde zur Erhöhung der Temperaturstabilität (Bryan et al., 1986) bzw. der
pH-Stabilität (Cunningham & Wells, 1987) von Enzymen oder zur Erhöhung der Resistenz
(Oliphant & Struhl, 1989) erfolgreich angewandt. Durch den Austausch von Asparagin 218
gegen Serin konnte die Thermostabilität der Serin-Protease Subtilisin aus Bacillus
amyloliquefaciens im Vergleich zum Wildtyp um das vierfache gesteigert werden (Bryan et
al., 1986). Hier erfolgte die Mutagenese durch Zugabe von Natriumbisulfit, das zur
Desaminierung von Cytosin zu Uracil führte. Das Screening nach diesem Mutein erfolgte
über ein geeignetes Subtilisinsubstrat und einen pH-Indikator auf Agarplatten, so dass beim
Abbau des Substrates eine pH-Senkung stattfand und sich somit die Farbe des Indikators
änderte. Zhou et al. verwendeten für die Erhöhung der Thermostabilität des Subtilisins die
Polymerase- Ketten- Reaktion (PCR) (Zhou et al., 1996), konnten damit allerdings die
gleichen Punktmutationen wie Bryan et al. nachweisen. In einer anschließenden gezielten
Mutagenese wurde eine weitere Aminosäure substituiert und somit eine Doppelmutante
erzeugt, die sowohl thermostabil als auch oxidationsresistent ist. Arnold et al. bewirkte eine
Verbesserung der Thermostabilität der Bacillus subtilis-Protease – Subtilisin E und erhielten
somit eine ähnliche Thermostabilität wie bei der Thermoactinomyces vulgaris- Thermitase
(Zhao & Arnold, 1999). Mit der „error-prone“ Methode konnte ebenfalls die Aktivität vom
Subtilisin E auf das 16 fache erhöht werden (You & Arnold, 1996).
Die gerichtete Evolution als erfolgreiches Konzept zeigen die folgenden Beispiele:
p-NB-Esterase: 3 Mutationen / höhere Aktivität in org. Lösungsmittel (Moore et al., 1997)
Subtilisin E: 8 Mutationen / 1000 x höhere Halbwertszeit bei 65°C (Zhao et al., 1998)

Einleitung 14
Die Zufallsmutagenese hat folgende Vorteile:
1. keine Strukturinformation nötig
2. schnellerer Optimierungsprozeß
3. mehrere unspezifische Mutationen/Generationen sind möglich
1.5 Hochzelldichte-Fermentation (HZD)
Um die durch kontinuierliche Zuführung ein oder mehrerer Nährstoffe während der
Fermentation auftretende Katabolit-Repression und Anreicherung toxischer Substanzen
vermeiden zu können, erfolgt die Kultivierung rekombinanter Mikroorganismen in der Regel
mittels der fed-batch-Technologie.
Die HZD-Fermentation dient zur Verbesserung der Produktivität mikrobieller Systeme, der
Reduktion von Produktions- Investitionskosten und Kulturvolumina (Knorre et al., 1991).
Ebenso können Sauerstoffverbrauch und die Wärmeentwicklung durch die Limitierung der
Wachstumsrate kontrolliert werden (Riesenberg, 1991). Dennoch führen die
Substratlimitation und -inhibition, Limitierung der Sauerstoffversorgung und Wärmetransfer,
CO2 –Konzentration sowie die Bildung metabolischer Nebenprodukte zu Problemen bei der
HZD-Fermentation (Riesenberg et al., 1991).
Metabolische Nebenprodukte: Die Bildung von Nebenprodukten hängt von der
Zusammensetzung des Mediums, dem Wirtsstamm und der spezifischen Wachstumsrate ab
(Pan et al., 1987). Unvollständig oxidierte Nebenprodukte wie Acetat, Lactat, Pyruvat,
Ethanol und Isobutyrat werden von E. coli Kulturen, deren Dichte über 40 g Biomasse pro
Liter Medium beträgt, akkumuliert (Landwall & Holme, 1977; Paalme et al., 1990). Dieser
Effekt ist durch die Bildung von Salzen im Medium zu beobachten (Jensen & Carlsen, 1990).
Nach Lee et al. führt insbesondere das Nebenprodukt Acetat zur Inhibition des Wachstums,
infolgedessen wird die Produktion des heterologen Proteins vermindert (Lee, 1996).
CO2-Konzentration: Verminderte Wachstumsrate sowie die Bildung metabolischer
Nebenprodukte können bei CO2-Partialdrücke oberhalb 0,3 atm beobachtet werden (Pan et
al., 1987).

Einleitung 15
Sauerstofftransfer: Da das Wachstum rekombinanter E.coli-Zellen unter aeroben
Bedingungen erfolgt, sollte der Sauerstoff-Partialdruck nicht unter 10 % sinken. Dennoch
wächst das Problem der Sauerstoff-Transferrate bei großen Fermentervolumina aufgrund der
Löslichkeit von Sauerstoff in Wasser, die auf maximal 200 mmol O2 pro Liter limitiert ist
(Riesenberg, 1991).
Substratinhibition: Die Inhibierung des Wachstums wird durch Substratkonzentrationen
oberhalb bestimmter Grenzwerte verursacht. Hefeextrakt oder Pepton, die komplexe
Verbindungen darstellen, sind nur in geringer Konzentration löslich (Riesenberg & Guthke,
1999).
Fermenter-Durchmischung: Bei mangelhafter Durchmischung großer Fermenter sind Zellen,
die sich nahe der Nährstoff-Injektionsstelle befinden, erhöhten Nährstoffkonzentration
ausgesetzt, während an anderen Stellen Nährstoffmangel herrschen kann. Sowohl der Mangel
als auch der Überschuss bewirken eine Verringerung der Zellausbeute (Neubauer et al., 1995)
2 Problemstellung und Zielsetzung Das erste Ziel der vorliegenden Arbeit war, sowohl die NAD-abhängige Phenylalanin
Dehydrogenase (PheDH) aus Rhodococcus sp. M4 als auch das malic enzyme aus E.coli K12
zu klonieren. Die Klonierung dieser zwei Enzyme im gleichen Plasmid sollte die Grundlage
für die reduktive Aminierung von ? -Ketosäuren und gleichzeitig die Regeneration des
Coenzyms mittels Ganzzell-Biotransformation ermöglichen. Ein weiteres Ziel war die
Erweiterung des Substratspektrums der PheDH durch Mutagenese.
Der Faktor, der die Effektivität der NADH-Regeneration im Produktionsprozeß herabsetzt, ist
die schwache Aktivität und die mangelnde Stabilität des üblicherweise verwendeten
Regenerationsenzym Formiat-Dehydrogenase (FDH). Die erforderliche Nachdosierung des
Coenzyms während des Prozesses erhöht die Produktionskosten signifikant. Um die
Wirtschaftlichkeit des Verfahren zu erhöhen, wird in dieser Arbeit das malic enzyme aus E.
coli K12 als eine Alternative zur FDH für die Regeneration kloniert und überprüft.
Die Klonierung des zweiten Enzyms – die rec-PheDH - soll eine Grundlage für die
Mutagenese schaffen. Die Erweiterung des Substratspektrum der PheDH, wird mittels
gezielter Mutagenese bzw. Zufallsmutagenese durchgeführt. Die Sequenz der PheDH wird
mit bekannten Aminosäurensequenzen verglichen und die Bindungsstellen sowie die

Einleitung 16
Interaktionen zwischen den Aminosäurenresten im aktiven Zentrum der PheDH zum Substrat
bestimmt. Mit Hilfe der Kristalllisationsdaten der PheDH wird die PheDH modelliert und ein
Proteindesign durchgeführt.
Eine weitere Zielsetzung dieser Arbeit war, Einblicke in den Reaktionsmechanismus der
PheDH und Aufschluss über die Struktur–Funktionsbeziehungen dieses Enzyms zu gewinnen.
Hierbei sollte das aktive Zentrum der PheDH mit dem der Lactatdehydrogenase verglichen
und eventuelle Mutagenese durchgeführt werden.
Für die Erweiterung des Substratspektrums werden Mutationen in der PheDH mit Hilfe
verschiedener Methoden eingeführt, um somit die Konstruktion einer neuen Amin-
Dehydrogenase für die Herstellung von aromatischen Aminen aus aromatischen Ketonen zu
erreichen.

Material
17
3 Material Geräte
Analytik-Apparaturen
Gaschromatograph Shimadzu
HBLC-System Gynkotek (Germering)
GC-9A Shimadzu (Düsseldorf)
NMR-ARX500 Bruker
Robocycler Stratagene
Bildverarbeitung
Eagle Eye II, Videosystem Stratagene (Heidelberg)
Fraktionssammler Frac 100 (Pharmacia)
Disintegration
Schwingkugelmühle (Retsch)
Disintegrator S IMA
Ultraschallgerät Branson
Dialyse und Ultrafiltration
Ultrafiltrationskammer 8050, 8010 Amicon
Fermentation
20 L-Bioreaktor Biostat Braun
Contifuge 300 MD Heraeus Christ
Elektrophorese
Gel elektrophorese GNA 100 Pharmacia (Freiburg)
Gelelektrophorese Horizon 11.14 Life Technologies (Eggenstein)
Prep Cell Biorad
Küvetten
Mikroküvetten (50 µl), Quarzglas Hellma (Müllheim/Baden)
Halbmikroküvetten (1 ml), optisches Glas Hellma (Müllheim/Baden)
Fluoreszenzhalbmikroküvetten(1 ml), Quarzglas Hellma (Müllheim/Baden)
Photometer
UV/Vis-Spektralphotometer 16 A Shimadzu (Duisburg)
UV/Vis-Spektralphotometer DU 650 Beckmann (Düsseldorf)
Fluoreszenzphotometer LS 50 B Perkin Elmer (Düsseldorf

Material
18
Zentrifugen
Eppendorf-Zentrifuge 5415 C Eppendorf (Hamburg)
Kühlzentrifuge Sorvall RC-5B Du Pont Instruments
(Bad Homburg)
Vacuumzentrifuge (Univapo 150 H)
+Kühlfalle (Unicryo MC1L)
Chemikalien
Alle nicht aufgeführten Chemikalien für Lösungen und Puffer waren mindestens von
analytischer Qualität (p.a.) und wurden in der Regel von Fluka, Sigma, Roth oder Merck
bezogen.
Nährmedienbestandteile waren von Merck oder Difco, die Coenzyme von Bts.
Enzyme für die Molekularbiologie wurden von NEB, Pharmacia, Biozym, Stratagene oder
Roche bezogen. Die Chemikalien für molekularbiologische Untersuchungen waren von
höchster Qualität. Die genannten Chemikalien wurden in „ultra pure“ Qualität eingesetzt.
Acetonitril Applied biosystems
(Weiterstadt)
EDTA Pharmacia (Freiburg)
Acrylamid Biorad (München)
Bis-Acrylamid Biorad (München)
APS Biorad (München)
Nucleotide/Nucleinsäuren
dATP, dGTP, dTTP, dCTP Pharmacia (Freiburg)
DNA-Molekulargewichtsmarker:
A. DNA-Marker IV [Boehringer]
B. DNA-Marker VI [Boehringer]
C. DNA-Marker VII [Boehringer]
D. DNA-Leiter [Gibco]

Material
19
Enzyme
BamHI, NcoI StuI, EcoRI, HindIII,pStI, NdeI, SmaI Biolabs
RNaseA Roth (Karlsruhe)
Taq-DNA-Polymerase (10 U/µl) Finnzyme
Ligase Roche
Alkalische Phosphatase Promega (Heidelberg)
Mikroorganismen
E. coli BL21 [hsdS gal (? cIts857 ind1Sam7 nin5 lacUV5)]
E. coli DH5? [supE44 ? lacU169 ? 80lacZ? M15) hsdR17 recA1 gyrA96 thi-
1relA1]
E. coli HB101 [supE44 hsdS20 (rB- mB-) recA13 ara-14 proA2 lacY1 galK2
rpsL20xyl-5mtl-1]
E. coli JM105 [subE endA sbcB15 hsdR4 rpsL(strr) thi ? (lac-proAB)
F(traD36proAB+ lacIq lac? ZM15)]
E. coli JM109 [recA1 sup E44 endA1 hsdR17 gyrA96 relA1 thi ? (lac-proAB
F’(traD36proAB+ lacIq lacZ? M15)]
E. coli XL1-Blue [supE44 hsdR17 recA1 endA1 gyrA46 thi relA1 lac- F’[proAB+ lacIq
lacZ? M15 Tn10(tetr)]
Vektoren
pKK223-3 [4,584kb, ColE1-Replicon, Ptac, Ampr]
pTRC99a [4,176 kb, ColE1-Replicon, Ptrc, lacIq, AmpR]
pUC18 [2,686 kb, ColE1-Replicon, lacZ’, lacI, AmpR]
pET-16b [5,711 kb, His.Tag, T7, Amp]
Kits für die Molekularbiolgie
Sure clone Kit Pharmacia
Plasmid Präparation Qiagen
DNA Extraktion Qiagen
Auto Read Sequencing Kit Pharmacia
Rapid DNA Ligation Kit Roche

Methoden
20
4 Methoden 4.1 Molekularbiologie
4.1.1 Isolierung der genomischen DNA
Sowohl die genomische DNA von Rhodococcus sp. M4 für die Isolierung der Phenylalanin
Dehydrogenase als auch die des E. coli K12 für malic enzyme, wurden nach van Soolingen et
al. isoliert (van Soolingen et al., 1991). Dafür wurden Kulturen mit einer einzelnen Kolonie
angeimpft und bei 30°C und 120 rpm bis zu einer optischen Dichte OD600 ca. 0.7 wachsen
gelassen. Die Kultur wurde für 30 min bei 80°C erhitzt und dann abzentrifugiert. Das Pellet wurde in
10 ml TE-Puffer resuspendiert. Nach dem Resuspendieren wurden die Zellen mit 0.5 ml
Lysozym (10 mg/ml) behandelt, und für 30 min bei 37°C inkubiert.
1.5 ml 10 %iges SDS und 120 µl Protease K (10 mg/ml) wurden zugefügt und für 10 min bei
65°C inkubiert. Durch diese Behandlung konnten die Proteine denaturiert und ausgefällt
werden. Um die restlichen Proteine und Lipide zu fällen, wurde die Lösung mit 1 ml 5 M
NaCl und 1.5 ml N-Cetyl-N,N,N-trimethylammonium Bromid/NaCl, (4.1 g NaCl, 10 g N-
cetyl-N,N,N-trimethylammonium Bromid in 100 ml H2O), versetzt. Der ganze Ansatz wurde
gut vermischt und für 20 min bei 65°C inkubiert.
Die DNA wurde durch ein äquivalentes Volumen von CHCl3 / Isoamyl Alkohol (24:1) nach
Zentrifugation extrahiert. Der Vorgang wurde zweimal durchgeführt, um die DNA vollständig
zu isolieren.
Die DNA aus der wässrigen Phase konnte durch 0.6 Vol. Isopropanol bei -20°C für 30 min
gefällt werden. Danach wurde sie abzentrifugiert und das erhaltene DNA-Pellet mit 2 ml
70%igem kalten Ethanol gewaschen.
Da Ethanol spätere Reaktionen inhibieren könnte, wurde die DNA durch Zentrifugation im
Vakuum (vacuum speed univapo) getrocknet.
Zum Nachweis der Reinheit der DNA können zwei Methoden eingesetzt werden:
1. Absorption bei 260 und 280 nm
2. durch Gelelektrophorese ( 0.5 % Agarose)

Methoden
21
4.1.2 Auftrennung von DNA-Fragmenten über Elektrophorese
Die Agarose wurde durch Aufkochen in der gewünschten Konzentration (0,8-1.2 %) in
1xTBE-Puffer gelöst und nach dem Abkühlen auf ca. 60°C mit 1/20000 Volumen an
Ethidiumbromid (10 mg/ml) versetzt und in horizontale Kammern gegossen. Die Proben
wurden vor dem Auftragen mit 1/6 Volumen Probenpuffer gemischt. Die Elektrophorese
erfolgte in einer mit 1xTBE-Puffer gefüllten Kammer. Die Ethidiumbromidfluoreszenz wurde
im UV-Durchlicht (312 nm) mit dem Videosystem „Eagle Eye II“ (Stratagene) aufgezeichnet.
6X Probenpuffer: 30 % Glycerin, 0,25 % Bromphenolblau
TBE-Puffer: 90 mM Tris-HCL, 90 mM Borsäure, 2 mM EDTA, pH8,2
Als Größenstandard für die DNA- Gelelektrophorese wurde die kB-Leiter der Fa. Gibco
benutzt.
10X TBE-Puffer:
Tris 121.12 g
Borsäure 51.32 g
EDTA 3.72 g
Probenvorbereitung:
Die zu trennende DNA-Lösung wurde mit einfach DNA-Probenpuffer [Endkonzentration] pH
8.0 versetzt.
DNA-Probenpuffer [6fach]:
6xTBE; 50 % (v/v) Glycerin; 0.25 % (w/v) Bromphenolblau
4.1.3 Ethanolfällung
Zur Konzentrierung und Reinigung von DNA-Lösungen wurden diese mit 2 Vol. Ethanol und
1/10 Vol. 3 M Natriumacetat, pH 4.8 versetzt, 30 min. bei –20°C inkubiert und 20 min. bei
4°C und 13000 rpm abzentrifugiert. Die sedimentierte DNA wurde zweimal mit 70 % Ethanol
(v/v) gewaschen, in einer Vakuumzentrifuge getrocknet und in Aq. bidest oder TE-Puffer,
pH8.0 (50 mM Tris-HCl, 50 mM EDTA) resuspendiert.

Methoden
22
4.1.4 Polymerase Ketten Reaktion [PCR]
Die Polymerase Ketten Reaktion (PCR) bietet die Möglichkeit, gezielt spezifische DNA-
Fragmente zu amplifizieren. Durch entsprechende Geräte [Robocycler, Stratagene] ist die
Reaktion komplett automatisiert. Durch Zugabe von genomischer DNA oder Plasmiden
zusammen mit für ein DNA-Stück spezifischen Primern wird dieses DNA-Stück durch die
Taq-Polymerase [Thermus aquaticus] amplifiziert. Da die Taq-Polymerase die hohen
Denaturierungstemperaturen von 94°C übersteht und diese Temperatur für die Aufspaltung
der doppelsträngigen DNA benötigt wird, ist es möglich, über viele Zyklen eine
Amplifikation zu erreichen. Innerhalb jedes Zyklus erhöht sich exponentiell die Zahl des
Template, da jedes neu gebildete Template Ausgangsmaterial für das nächste wird. Ein
Zyklus besteht aus einem Denaturierungsschritt [94°C], einem Annealingschritt, in dem die
Primer an das Template binden [Temperaturvariabel] und einem Amplifizierungsschritt
[72°C], bei dem die Polymerase aktiv ist.
Der Reaktionsansatz aus:
10 mM Tris pH 8.3
50 mM KCl
1.5 mM MgCl2
jeweils 200 µM dATP, dCTP, dTTP, dGTP
20-40 pmol jedes Oligonucleotid-Primers
2.5 U Taq-Polymerase pro 100 µl Ansatz
10 ng Plasmid-DNA als PCR-Template
Die PCR Amplifikation wurde auf einem automatischen DNA-Thermal-Cycler (Robocycler,
Fa. Stratagene) nach folgendem Programm durchgeführt.
??5 min Denaturierung bei 94°C (zu Anfang des Programms 1x)
??1.2 min Annealingsschritt, in dem die Primer an das Template binden
??1.5 min Extension (Verlängerung der Primer durch die Taq-Polymerase) bei 72°C
[Temperaturvariabel].
??1 min Denaturierung bei 95°C
??Zyklisches Wiederholen der letzten drei Schritte (25-30x)
??5 min Extension (nach Abschluß der PCR, um eine vollständige Verlängerung
eventuell vorher abgebrochener DNA-Produkte zu gewährleisten)

Methoden
23
4.1.5 Berechnen der Schmelztemperatur der Primer
Die Schmelztemperatur Tm der Primer wurde nach Thein und Wallace (Thein & Wallace,
1988) abgeschätzt:
Schmelztemperatur Tm[°C] = 2x(Basenzahl A+T)+3x(Basenzahl G+C)
Diese Bezeichnung gilt für Primerlängen ? 24 bp.
Für Primerlängen ? 14 bp gilt [MWG-Biotech]:
Schmelztemperatur Tm [°C] = 69.3°C + 0.41 x (GC-Gehalt [%]) – 650 / Primerlänge
Annealingtemperatur Ta [°C] = Tm + 3°C
4.1.6 Isolierung von DNA- Fragmenten aus Agarosegelen
Die aus dem Gel ausgeschnittenen DNA-Banden können auch über spezielle Membransäulen
aus dem Gel eluiert werden. Das dreifache Volumen des Gelstückgewichts an Puffer QX1
[keine Herstellangaben] wird hinzugegeben, und die Probe unter einmaligem Schütteln 10min
bei 50°C inkubiert. Die Lösung wird auf eine Qiaquick-Zentrifugationssäule gegeben und
60sec bei 13000 rpm zentrifugiert. Das Filtrat wird verworfen und die Säule durch erneute
Zentrifugation mit 0.75 ml Puffer PE gewaschen und durch nochmalige Zentrifugation Reste
des Puffers von der Membran der Säule entfernt. Abweichend von den Herstellerangaben
wurde jedoch zur Elution 50 µl H2O (pH 8.0) verwendet. Die eluierte DNA wurde über
Einengen in der Vakuumzentrifuge auf die gewünschte Konzentration gebracht und
weiterverarbeitet. Eine zweite Methode zur Isolierung der DNA- Fragmente erfolgte ebenso
nach einem Qiagen Kit, mit dem man die komplette PCR-Lösung mit fünffachem Puffer PB
[keine Herstellungsangaben] verdünnt und in eine Qiaquick Säule pipettiert. Die Säule wird
zusammen mit einem Auffanggefäß 60 sec bei 13000 rpm zentrifugiert. Die DNA kann nun
mit 0.75 ml PE-Puffer [Ethanol] gewaschen. Dabei werden durch nochmalige Zentrifugation
(60 sec., 13000 rpm) sämtliche Reste des PE entfernt. Anschließend wird die DNA mit 50µl
1mM Tris bzw. H2O pH 8.0 [1min, 13000 rpm] von der Säule eluiert und über
Vakuumzentrifugation auf das gewünschte Volumen zur Weiterverarbeitung eingeengt.

Methoden
24
4.1.7 Dephosphorylierung linearisierter Plasmid-DNA
Linearisierte Plasmide, deren Religationsrate aufgrund des Vorliegens komplementärer Enden
besonders groß war, wurden unter Verwendung Alkalischer Phosphatase an ihren 5’-Termini
dephosphoryliert. Dazu wurden 40µl des Restriktionsansatzes mit 1U alkalischer Phosphatase
aus Shrimps (Shrimp Alkaline Phosphotase, SAP, Boehringer Mannheim) versetzt, mit dem
zugehörigen, Zn2+-haltigen Reaktionspuffer und Aq. bidest. auf ein Endvolumen von 50µl
gebracht und 30 min. bei 37°C inkubiert. Eine weitere Unit des Enzyms wurde nach Ablauf
dieser Periode zugefügt und die Inkubation um zusätzliche 30 min. verlängert. Das Enzym
wurde mit einem Vol. Phenol-Isoamylalkohol-Chloroform (25:24:1; v/v/v) aus dem Ansatz
extrahiert und die dephosphorylierte Vektor-DNA durch Ethanolfällung gereinigt.
4.1.8 Ligation
Die Ligation der geschnittenen und isolierten Gene erfolgte sowohl mit Hilfe der T4-DNA-
Ligase der Firma Roche als auch mit dem „sure clone kit“ der Fa. Pharmacia. Je nach
Schnittenden wurde für Ligationen mit überhängenden Enden (sticky-ends) ein molares
Verhältnis von Vektor zu Insert von 1:3 bzw. glatten Enden (blunt-end) von 1:5 gewählt.
Nach vorschriftmäßiger Reaktion mit Klenowenzym und Phosphonucleotidkinase (zur
Auffüllung und Phosphorylierung der PCR-Enden) wurden die Fragmente von den Proteinen
mittels Agarosegel aufgereinigt.
Genfragment 1-16 µl [50-200 ng]
Klenow-Fragment 1 µl
10 x Puffer 2 µl
Polynucleotidkinase 1 µl
Der Ansatz wurde 30 min bei 37°C inkubiert, dann ein äquivalentes Volumen an Phenol-
Chloroform zugegeben, intensiv vermischt [Vortex] und zur Phasentrennung bei 13000 rpm
2min abzentrifugiert. Die obere Phase wurde abgenommen und auf ein Agarosegel auf-
getragen. Das Genfragment wurde aus dem Gel isoliert. Anschließend wurde eine Ligation
der glatten Enden durchgeführt.
Ligation nach „sure clone kit Pharmacia“
Genfragment 50-200 ng
pUC18 Vektor 50 ng
2 x Ligationspuffer 10 ng
DTT 1 µl

Methoden
25
T4-Ligase 1 µl
H2O ad 20 µl
Das Reaktionsgemisch wurde für 4 h bei 16°C inkubiert und dann kompetente E.coli Zellen
[verschiedene Stämme] mit dem ligierten Plasmid transformiert.
Ligation nach “rapid ligation kit Roche”
Genfragment 6 µl (200 ng)
Plasmid (geschnitten) 2 µl (50 ng)
Dilution Puffer (keine Herstellerangaben) 2 µl
Ligation Puffer (keine Herstellerangaben) 10 µl
T4-Ligase 1 µl
Das Reaktionsgemisch wurde für 5 min bei Raumtemperatur inkubiert und dann kompetente
E. coli Zellen [verschiedene Stämme] mit dem ligierten Plasmid transformiert.
4.1.9 Herstellung kompetenter Bakterienzellen
Die Herstellung kompetenter E. coli XL1 Blue und JM 105 sowie HB101 erfolgte nach der
Methode von Hanahan (Hanahan, 1983). Die Lagerung erfolgte bei –80°C.
Aus dem konservierten E. coli Stamm, wurden 5 ml LB-Medium 5 % angeimpt und über
Nacht wachsen gelassen. 200 µl davon werden in 300 ml LB-Medium überimpft, das dann bis
zu einer OD550 0.5 bei 37°C inkubiert wird. Die Kultur wird dann in eine SS34-
Zentrifugenbecher überführt, 15 min auf Eis gekühlt und dann bei 4°C abzentrifugiert. ( 5000
rpm, 10 min. ).
Die Zellen werden mit einer Lösung von 100 mM RbCl, 50 mM MnCl2, 30 mM Kalium-
Acetat pH 5.8, 10 mM CaCl2, 15 % (v/v) Glycerin resuspendiert und erneut für 1h auf Eis
gestellt. Anschließend werden die Zellen nochmals abzentrifugiert und in einer Lösung von
10 mM MOPS pH 7.0, 10 mM RbCl, 15 mM CaCl2, 15 % (v/v) Glycerin aufgenommen und
15 min auf Eis inkubiert. Diese Zellsuspension wird dann in Aliquots von 100 µl schock-
gefroren (flüssiger Stickstoff) und bei -80°C aufbewahrt.

Methoden
26
4.1.10 Transformation
Die Standardtransformation wurde nach dem Protokoll von Hanahan (Hanahan, 1983)
durchgeführt. Hierzu wurden 100-200 µl kompetenter E. coli –Zellen auf Eis aufgetaut und 40
ng DNA aus dem Ligationsansatz zugegeben. Die Plasmidzellsuspension wurde 30 min auf
Eis gekühlt und anschließend für 90 sec auf 42°C erwärmt und sofort wieder auf Eis gekühlt.
Nach Zugabe von 300 µl LB - Medium wurden die Zellen zur Regeneration etwa 45 min bei
37°C inkubiert. Anschließend wurden 200 µl dieser Kultur auf einer Antibiotikahaltigen LB-
Platte ausgestrichen und über Nacht bei 37°C inkubiert.
4.1.11 Schnellisolierung von Plasmid-DNA aus E.coli (modifiziert nach
Birnboim und Doly)
Für die Schnellisolierung von Plasmiden werden die Zellen einem osomotischen Schock
ausgesetzt und die Proteine danach ausgefällt, wobei die Zellen mit osmotischen Lösungen
behandelt werden, die zum Platzen der Zellen führen (Birnboim & Doly, 1979).
1.5ml einer Übernachtkultur werden abzentrifugiert und mit 100µl Lsg. A + 100 µg/ml RNase
sowie 200 µl Lsg. B versetzt (für 5 ml ÜN-Kultur werden alle Mengen bis zur Isopropanol-
Fällung vervierfacht).
Der Ansatz wird dann 5 min auf Eis gelegt, gefolgt von 150 µl Lsg. C. Nach der Zugabe von
Lsg. C wird der Ansatz wieder auf Eis für 15 min inkubiert und anschließend für 10 min bei
13000 rpm abzentrifugiert.
Der Überstand (400µl) wird mit 260µl Isopropanol versetzt und bei -20°C für 30min absetzen
gelassen. Danach wird der Ansatz für 5 min bei 13000 rpm abzentrifugiert und 2x mit
70%igem kaltem Ethanol gewaschen. Über der Speedvacuumzentrifuge werden die Plasmide
getrocknet und in 50 µl TE-Puffer resuspendiert und bei -20°C aufbewahrt.
Lsg. A: 50 mM Glucose
10 mM EDTA
2.5 mM Tris pH 8.0
Lsg. B: 0.2 M NaOH
1 % SDS
Lsg. C: 3 M NaAC pH 8.0 (mit HCl titriert)

Methoden
27
4.1.12 Plasmidisolierung [Biorad]
Für die saubere Plasmidreinigung wurde das Quantum Prep? (Plasmid Miniprep kit [Biorad])
verwendet. Die bindende Matrix ist ein Silicon dioxid exoskeleton aus Diatome.
Für die Reinigung der Plasmide wurden 2 ml transformierte Zellen bei 13000 rpm für 30 sec.
abzentrifugiert und den Überstand entfernt. Dazu wurden 200µl Cell-Resuspension zu-
pipettiert und gut resuspendiert (Vortex).
Auf die Zellsuspension wurden 250 µl Cell-Lysis-Solution zugegeben und gut vermischt.
Nach der Behandlung mit der Lyse-Lösung wurden 250 µl „Neutralization Solution“
zugegeben und vorsichtig vermischt (nicht vortexen).
Die Zellen wurden dann abzentrifugiert (5 min bei 13000 rpm).
Die wäßrige Phase wurde entfernt und mit 200 µl Matrix-Suspension vermischt. Die Lösung
wurde dann in einen Spin-Filter angebracht und bei 13000 rpm für 5’ abzentrifugiert.
Der Bodensatz wurde entfernt und der Ansatz (im Spin-Filter) wurde mit 500µl „Wash-
Solution“ durch Zentrifugation gewaschen. Der Waschvorgang wurde 2x wiederholt.
Der Bodensatz wurde wieder entfernt und die Plasmid-DNA mit 56°C heißem deionisiertem
H2O durch Zentrifugation für 30 sek. eluiert.
Die Plasmid-DNA wurde bei -20°C aufbewahrt.
4.1.13 Plasmidisolierung [Qiagen]
Um die Plasmide aus den Bakterienzellen zu isolieren, wurde der Plasmid-Präparations-Kit
der Fa. Qiagen verwendet. Dieser beruht auf dem Prinzip der alkalischen Lyse in Verbindung
mit einer Anionenaustauschchromatographie. Zur Vermeidung der Kopräzipitation störender
Salze wurde die anschließende Isopropanol- Fällung bei Raumtemperatur durchgeführt und
auch das 2 malige Ethanolwaschen (70 %) erfolgte bei Raumtemperatur. Nach der Trocknung
in der Speed-Vac wurde das erhaltene Pellet in 10 µl 1 mM Tris, pH 8.0 aufgenommen. Die
genaue Konzentration wurde mittels Restriktion eines 1µl Aliquots gelelektrophoretisch
bestimmt.

Methoden
28
4.1.14 Restriktion
Analytische Restriktionen erfolgten in einem Volumen von 30 µl, präparative in einem
Volumen von 50µl. Die für die einzelnen Restriktionsenzyme nötigen Puffer-Konzentrationen
folgten den Herstellerempfehlungen. Die DNA-Menge für den präparativen Verdau lag in
einem Bereich von 3-4 µg/Ansatz. Die Vektoren wurden noch zusätzlich 1h mit 5 Units
alkalischer Phosphatase bei 37°C inkubiert, um die 5’-Phosphatgruppe zu entfernen und so
die Religation des Vektors zu verhindern.
4.1.15 Sequenzierung
Die DNA- Sequenzierung erfolgte per Auftrag durch die Firma SequiServe, Vaterstetten, oder
mittels eines Automated Laser Fluoreszenz- (A.L.F.) Sequenzierautomaten (Fa. Phasrmacia )
in einem 6 %igen Polyacrylamidgel. Die Sequenzreaktion nach Sanger et al. (Sanger et al.,
1977) mittels T7- DNA-Polymerase wurde gemäß Herstellerangaben durchgeführt [AutoRead TM Sequencing Kit, Fa. Pharmacia]. Pro Sequenzierung wurden 7 µg hochreine Plasmid-DNA
benötigt. Sie erfolgten mit den im Kit enthaltenen vektorspezifischen Primern (pUC universal
und reversal). Die Daten der Sequenz wurden mit dem Programm DNAsis ausgewertet.
4.1.16 Gezielte Mutagenese
Die gezielte Mutagenese erfolgte nach dem Prinzip der überlappenden PCR (Ho et al., 1989).
Hierbei werden Primer verwendet, die zwei DNA- Fragmente mit überlappenden Enden
erzeugen. In einer zweiten PCR hybridisieren diese Fragmente. Das überlappende 3’-Ende
jeden Stranges dient als Primer für die Synthese des Gegenstranges. Werden nun in die
Oligonucleotiden Mutationen eingeführt, finden sich diese auch 100 % im Produkt wieder.
In einer zweiten PCR hybridisieren diese Fragmente, das überlappende 3’-Ende jeden
Stranges dient als Primer für die Synthese des Gegenstranges.
Das resultierende Fusionsprodukt wurde durch weitere PCR mit den „äußeren“ Primern, die
den gesamten Bereich einschließen, amplifiziert.
Vorteil dieser Methode ist, dass weder auf das Vorhandensein einer Restriktionsschnittstelle
geachtet werden muss, noch das gesamte Plasmid amplifiziert wird. Das Prinzip der PCR-
Mutagenese ist in Abbildung 7 dargestellt.

Methoden
29
5'
5'
5'Templat-DNA
*
* 5'P1'
K66
P1P2
P2'
2.Produkt
1.Produkt ** 5'
5'PCR mit den PrimernP2+P2'
PCR mit denPrimern P1+P1'
PCR mit denPrimern P1+P2'und Produkt 1+2als Templat
* 5'
5'
Abbildung 7: Prinzip der gezielten Mutagenese mittels überlappender Fragmente.
4.1.17 Zufallsmutagenese
Für die Herstellung verbesserter Enzymvarianten existieren zwei Strategien:
1. Asexuelle Evolution (Kuchner & Arnold, 1997), dabei wird durch zufällige
Mutagenese, die möglichst auf das für das gewünschte Protein codierende
Ausgangsgen beschränkt wird, eine Enzymbibliothek hergestellt und anschließend auf
verbesserte Eigenschaften durchmustert. Die in der ersten Generation gefundenen
besten Enzyme können in folgenden Mutationsrunden optimiert werden. Dies setzt
jedoch eine weitere Verbesserung der Varianten der ersten Generation durch
Einführung neuer und Deletion sich als negativ erweisender Mutationen voraus.
2. Die sexuelle Evolution hingegen geht von einem Pool homologer Ausgangsgene aus.
Diese können aus einer asexuellen Evolution hervorgegangen oder nah verwandten
natürlichen Sequenzen sowie Enzymvarianten abgeleitet sein, die durch rationales
Design hergestellt wurden. Das Prinzip dieses DNA shuffling oder gene shuffling
(Stemmer, 1994) basiert auf einem partiellen DNAseI-Verdau und einer
nachfolgenden Rekombination der Bruchstücke durch eine Polymerasekettenreaktion

Methoden
30
(PCR). Nachteilig sind die nicht immer gegebenen Verfügbarkeiten homologer Gene,
die Kontrolle des DNAse-Verdaus zur Herstellung von Bruchstücken optimaler Größe
und Probleme mit der Ligationseffizienz bei der PCR (Bornscheuer et al., 1998).
Die wohl wichtigste Methode zur asexuellen Evolution ist die Fehlerhafte Polymerase-
kettenreaktion (error-prone PCR) (Rice et al., 1992).
Durch Einstellung nicht-optimaler Reaktionsbedingungen in der PCR kann die Fehler-
häufigkeit der Polymerase gesteigert werden
??Die Einflußgrößen in der „error prone PCR“ sind:
??Fehlerrate der Taq- Polymerase
??Konzentration der Taq-Polymerase
??molare Verhältnisse der Desoxynukleotide
??Konzentration der Ziel- DNA (Template)
??PCR- Zyklenzahl
??c [MgCl2]
??c [MnCl2]
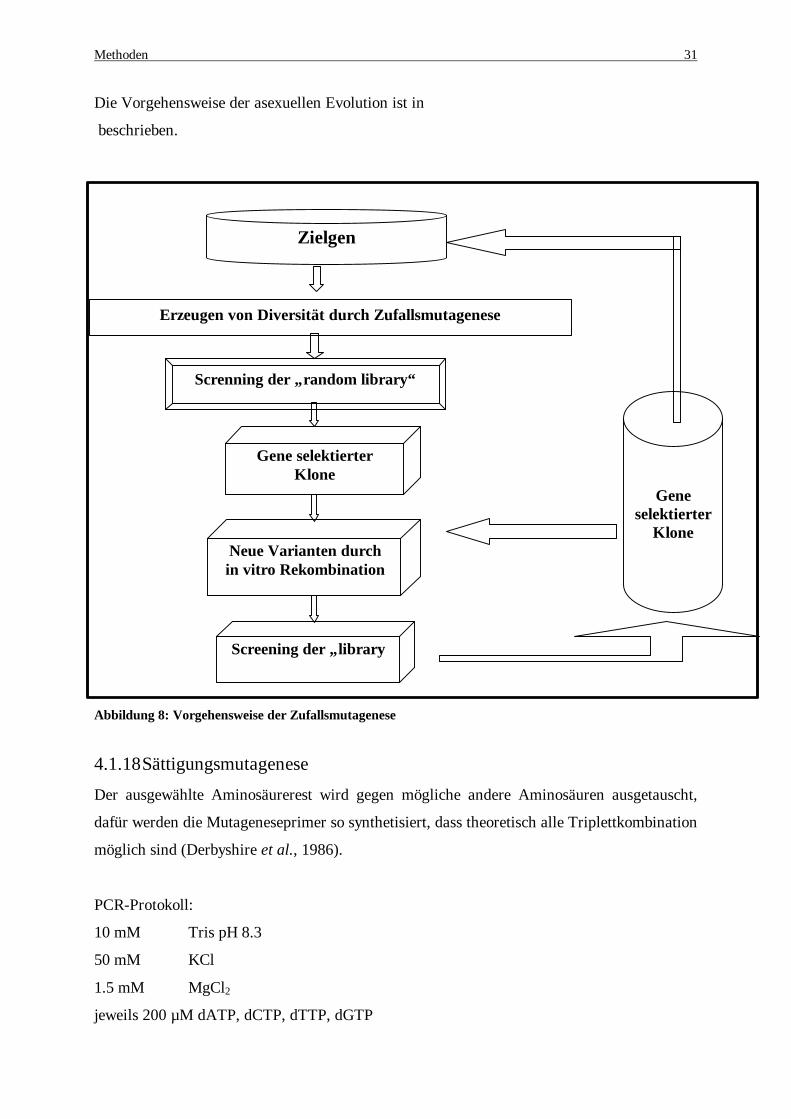
Methoden
31
Die Vorgehensweise der asexuellen Evolution ist in
beschrieben.
Abbildung 8: Vorgehensweise der Zufallsmutagenese
4.1.18 Sättigungsmutagenese
Der ausgewählte Aminosäurerest wird gegen mögliche andere Aminosäuren ausgetauscht,
dafür werden die Mutageneseprimer so synthetisiert, dass theoretisch alle Triplettkombination
möglich sind (Derbyshire et al., 1986).
PCR-Protokoll:
10 mM Tris pH 8.3
50 mM KCl
1.5 mM MgCl2
jeweils 200 µM dATP, dCTP, dTTP, dGTP
Erzeugen von Diversität durch Zufallsmutagenese
Zielgen
Screnning der „random library“
Gene selektierter Klone
Neue Varianten durch in vitro Rekombination
Screening der „library
Gene selektierter
Klone

Methoden
32
20-40 pmol jedes Mutagenese-Primers
2.5 U Polymerase-pfu – Stratagene pro 100 µl Ansatz
20 ng Plasmid-DNA als PCR-Template
Die PCR-Amplifikation wurde auf einen automatischen DNA-Thermal-Cycler (Robocycler,
Fa Stratagene) nach folgendem Programm durchgeführt.
??3 min Denaturierung bei 94°C (zu Anfang des Programms 1x)
??1 min Annealingsschritt, in dem die Primer an das Template binden.
??Extension bei 72°C (Zeit hängt von der Größe des Plasmids ab. 1 min/ kb)
??1 min Denaturierung bei 95°C
??Zyklisches Wiederholen der letzten drei Schritte (25x)
??6 min Extension (nach Abschluß der PCR)
4.2 Biochemische Methoden
4.2.1 Zellaufschluß und Rohextraktgewinnung
Da das Protein in kleinen Mengen für den qualitativen Nachweis gebraucht wurde, wurden
die Zellen durch eine Schwingkugelmühle aufgeschlossen [Retsch]. Dafür wurde ein
Verhältnis von Zellen zum Aufschlußpuffer [ 0.1 M Kpi pH 7.0, 1 mM MgCl2 , 10 Tropfen
Ucolup/L) von 1:3 eingesetzt, und 1,2g Glasperlen (0.3 ? ) pro 600 µl dieser Lösung zu-
gegeben.
Die Eppendorfgefäße wurden in die Schwingkugelmühlenzylinder gestellt und dann die
Zellen in der Schwingkugelmühle für 10 min. disintegriert. Anschließend wurden die Proben
5 min. auf Eis abgekühlt und danach 3 min. bei 13000 rpm abzentrifugiert.
Der Überstand wurde abpipettiert. Der Aufschluß wurde ein zweites Mal wiederholt, indem
die Aufschlußpuffer auf die Eppendorfgefäße verteilt und die restlichen, nicht
aufgeschlossenen Zellen durch die Schwingkugelmühle disintegriert worden sind.
Um die große Zahl an Mutanten schnell und effektiv zu überprüfen, wurden die Kolonien in
kleinem Maßstab (1,2 ml) angezüchtet und mit der unten geschilderten Methode
aufgeschlossen.
Die Transformanten der Zufallsmutagenese wurden einzeln mit Zahnstochern abgepickt und
in 150 µl LB-amp angeimpft. Das Wachstum der Vorkultur erfolgte über Nacht. 50 µl aus
dieser Vorkultur wurden in 1,2 ml LB-amp als Hauptkultur angeimpft und nach einem

Methoden
33
Wachstum von etwa 5 h (OD600 0,7) wurden die Kulturen mit 1 mM IPTG induziert, um die
Expression zu starten.
Die Zellen werden nach dem folgenden Protokoll geerntet:
??1,2 ml Kultur abzentrifugieren
??600 µl Überstand wegwerfen und den Rest resuspendieren
??50 µl Lysis-Puffer dazu und noch mal resuspendieren
??30-45’ bei 37°C schütteln
??Aus dem Ansatz der lysierten Zellen werden 50µl zur photometrischen
Messung benötigt
Lysis-Puffer:
0,2 % Triton X-100 10 ml
20 mM EDTA 40 ml
200 mM Kpi-Puffer 50 ml pH 7.0
4.2.2 Aktivitätstest für die Phenylalanin Dehydrogenase
Die Bestimmung der enzymatischen rekombinanten PheDH erfolgte mittels photometrischer
Tests anhand der Extinktionsabnahme bei 340 nm. Dies basiert auf dem Verbrauch von
NADH im Zuge der Umsetzung der Ketosäure Phenylpyruvat (Hummel et al., 1987)
Phenylpyruvat + NADH + NH4+ L-Phe + NAD+ + H2O
Die Extinktsionsabnahme bei 340nm wurde über 1min verfolgt. Die Berechnung der Aktivität
als U/ml (U entsprecht 1 µmol Substratumsatz/min) erfolgte über das Lambert-Beersche
Gesetz. Der Aktivitätstest wurde bei 30°C und bei pH 8.5 durchgeführt
Der Testansatz setzte sich wie folgt zusammen:
Tris/HCl 0.1 M
(NH4)2SO4 0,15 M
Phenylpyruvat 3 mM
NADH 0.3 mM
Die Reaktion wurde durch Zugabe von 10 µl Enzym gestartet.

Methoden
34
4.2.3 Screening und Aktivitätstest in Titerplatten
Die Aktivitätbestimmung der hergestellten Muteine in Bezug auf Phenylaceton bzw
Phenylpyruvat wurde photometrisch in Titerplatten durchgeführt.
Dafür wurden Mutanten durch eine schnelle Methode isoliert (4.2.1) und auf Aktivität
überprüft.
Vorkultur im 150µl-Maßstab
Hauptkultur und Expression im 1 ml- Maßstab
Zellernte und Permeabilisierung
Bestimmung der Aktivität
Selektion aktiver Mutanten
Abbildung 9: Schema zum Screeningsvorgang
Reaktionsansatz:
120 µl Substratansatz
50 µl Rohextrakt
Messung bei 340 nm
Substratansatz:
100 mM (NH4)2SO4
100 mM (Tris-Puffer) pH-Wert 8.5
3 mM Phenylpyruvat bzw. Phenylaceton
0,3 mM NADH
Für die Reaktion wurden 120 µl davon verwendet und mit 50 µl Rohextrakt gestartet.

Methoden
35
4.2.4 Aktivitätstest für das NAD-abhängige malic enzyme
HEPES pH 7.5 0.1 M
L-Malat 2 mM
NAD+ 0.5 mM
Enzym (Rohextrakt) 10 µl
4.2.5 Gekoppelte enzymatische Synthese
Das Reaktionsmedium für die kontinuierliche Produktion des L-Phenylalanin wurde
folgendermaßen zusamenpipettiert:
0,1 M (NH4)2SO4
0,1 M Hepes (pH 8.0)
30 mM Phenylpyruvat
60 mM Malat
2 mM MgCl2
1,5 g/l BSA
2 mM NAD+
Das gesamte Volumen des Reaktionsmediums beträgt 10 ml und wurde mit 50 U PheDH und
70 U malic enzyme gestartet. Die L-Phenylalaninkonzentration wurde zu unterschiedlichen
Reaktionszeiten mittels HPLC bestimmt.
4.2.6 Bestimmung des L-Phenylalanin mittels HPLC
Die Derivatisierung der Proben für die HPLC-Analytik wurde wie folgt durchgeführt:
OPA-Derivatisierungsansatz: 140 µl Na-Borat-Puffer + 40 µl Probe bzw. Standard + 20 µl
OPA/IBLC-Reagenz. Die Mischung wurde sofort kräftig gerührt.
OPA/IBLC oder IBDC: 260 mM IBLC oder IBDC + 170 mM OPA werden in Na-Borat-
Puffer ( 0,1 M, pH 10,5) gelöst.
Na-Borat-Puffer: 0,1 M Na2B4O7 ·10 H20 in Wasser gelöst und mit NaOH auf pH 10,5
eingestellt.

Methoden
36
4.2.7 Bestimmung der Acetatkonzentration
Die Bestimmung der Acetatkonzentration in Fermenterlösungen erfolgte über HPLC auf einer
Aminex HPX 87 H Säule (Biorad) mit einem Fluß von 0,5 ml/min unter Verwendung von
0,2N Schwefelsäure als Laufmittel. Das Injektionsvolumen betrug 100 µl und die Temperatur
40°C, die Detektion erfolgte bei einer Wellenlänge von 215nm. Die absoluten Acetat-
konzentrationen der Proben, aus denen die Biomasse durch Zentrifugation abgetrennt worden
war, wurden anhand einer Eichung mittels Acetatlösung ermittelt.
4.2.8 Ganzzell Biotransformation
In 200 ml Reaktionsansatz wurden 1 g rekombinante Zellen für die Produktion von L-Phenyl-
alanin verwendet.
Reaktionsansatz für L-Phenylalanin-Synthese:
0,1 M (NH4)2SO4
0,1 M HEPES-Puffer pH8.5
0,1 M Malat
2 mM MgCl2
2 mM NAD+
40 mM Phenylpyruvat
Die Synthese erfolgte bei 30°C unter langsamen Rühren. Für kleinere Ansätze wurden die
Zellmengen entsprechend abgewogen.
4.2.9 Proteinaufreinigung
Das durch den Aufschluß erhaltene Rohextrakt wurde auf eine DEAE-Sepharose-FF Säule
(Volumen 7.1 ml, Fluß 1 ml/min.) aufgetragen. Die Säule wurde vorher mit 5 fachem Säulen-
volumen eines 0.1 M Kpi-Puffers (8.94 g K2HPO4 + 0.21 g KH2PO4) gespült. Anschließend
wurde die Konzentration von NaCl über einen linearen Gradienten bis auf 100 % zum
Eluieren erhöht.
Dadurch wurden die Proteine aufgrund ihrer unterschiedlichen anionischen Bindungsstärke
bei verschiedenen Natriumchloridkonzentrationen eluiert und in Fraktionen aufgefangen. Die
Fraktionen wurden anhand des Aktivitätstests auf PheDH-Aktivität überprüft. Diejenigen mit
der höchsten Aktivität wurden vereinigt, und für weitere Untersuchungen aufbewahrt.

Methoden
37
4.2.10 Proteinbestimmung nach Bradford
Der Proteingehalt wurde photometrisch nach Bradford (Bradford, 1976) bestimmt.
Diese Methode beruht auf der Farbverschiebung des Farbstoffes im Gegenwart von Proteinen.
Es bildet sich ein blau gefärbter Komplex, der bei 595 nm detektiert werden kann.
Die Proteinbestimmung nach Bradford ist sehr empfindlich und für geringe Proteinmengen
geeignet. Das in der Reaktion eingesetzte Bradfordreagenz wurde selbst hergestellt:
100 mg Coomassie Brillant Blue G-250 wurden mit 50 ml Ethanol versetzt und über Nacht
gerührt. Danach erfolgte die Zugabe von 100 ml 85 %iger Phosphorsäure, und anschließend
wurde die Lösung auf 1 L mit Wasser aufgefüllt. Die fertige Lösung wurde einige Zeit
gerührt, anschließend filtriert und unter Lichtabschluß aufbewahrt.
Testansatz:
900 µl Bradfordreagenz
100 µl Proteinlösung
Die Proben wurden 5 min. bei Raumtemperatur inkubiert und anschließend bei 595 nm
gemessen.
4.2.11 Entsalzen von Proteinlösungen
Die Entsalzung von Proteinen erfolgte entweder über Gelfiltration oder Ultrafiltration. Hierbei
wurden 2.5 ml der Proteinlösung auf eine vorher mit dem gewünschten Puffer äquilibrierte
PD10 Sephadex G 25 Säule [Pharmacia] aufgetragen. Die Proteine wurden anschließend
durch Elution der PD10 mit 3.5 ml des gewünschten Puffers wiedergewonnen und damit
umgepuffert.
4.2.12 Konzentrierung von Proteinlösungen
Die Enzympräparate wurden, je nach Bedarf, mittels Ultrafiltration aufkonzentriert. Es
wurden entweder YM20-Membranen in Rührzellen [Fa. Amicon] oder Zentrifugations-
röhrchen [Fa. Centricon] verwendet.

Methoden
38
4.2.13 SDS-Polyacrylamidgelelektrophorese (SDS-PAGE)
Die Polyacrylamidgelelektrophorese (PAGE) gehört zu den in der biochemischen Analytik
meistverwendeten Elektrophoresemethoden. Sie verwendet als Träger polymerisiertes und
quervernetztes Acrylamid, dessen Porengröße in einem weiten Bereich variierbar ist.
Diese Methode liefert scharf getrennte Banden und dient bei entsprechender Eichung zur
Bestimmung der molaren Masse bzw. des stoke’schen Radius von nichtglobulären
Makromolekülen.
Die Geldichte, auch Gelkonzentration genannt, wird durch die Größen T und C bestimmt. T
ist gleich dem Prozentanteil an Acrylamid und Methylen-bis-Acrylamid am Gesamtvolumen,
C ist der prozentuale Anteil von N,N,-Methylen-bis-acrylamid (dem quervernetzenden
Agens). Die Gele wurden nach Laemmli (Laemmli, 1970) gegossen.
Verwendete Stammlösungen:
Trennpuffer 1.5 M Tris/HCl pH 8.8
Sammelgelpuffer 0.5 M Tris/HCl pH 6.8
SDS 10 % (w/v)
Glycerin 85 % (w/v)
APS 10 % (w/v)
Sammelgelzusammensetzung:
Sammelgelpuffer 0.125 M
Acrylamid 5 %
SDS 0.10 % (w/v)
APS 0.02 % (w/v)
TEMED 0.25 % (w/v)
Elektrophoresepuffer pH 8.0
Tris 0.025 M
Glycerin 0.192 M
SDS 0.1 % (w/v)

Methoden
39
Probenpuffer:
Glycerin 10 % (v/v)
SDS 2 % (w/v)
Tris/HCl pH 6.8 0.063 M
Bromphenolblau 0.1 % (w/v)
? -Mercaptoethanol 10 % (v/v)
Um eine Denaturierung zu erreichen, wurden die Proben mit 20 µl Probenpuffer versetzt und
4 min bei 95°C aufgekocht. Anschließend wurden die Proben auf Eis abgekühlt.
4.2.14 Färbung
Silberfärbung von Proteinen (Blum et al., 1987):
Das Gel wurde in eine Fixierlösung überführt und unter ständigem Schütteln (3x30 min.)
fixiert. Nach der Fixierung wurde das Gel 10 min mit 20 % (v/v) Ethanol inkubiert und direkt
10 min mit Millipore Wasser gewaschen. Nach dem Waschen wurde das Gel silbergefärbt.
Um das Gel zu färben und sichtbar zu machen, wurde es für 1min in der Sensitivierungs-
lösung (0.02 % (w/v) Natriumthiosulfat x H2O) geschüttelt, dann kurz mit Wasser gespült und
anschließend 20-30 min in der Färbelösung [2 % (v/v) AgNO3 , 750 µl/L (v/v) 37 % HCHO]
inkubiert. Nach Ende der Inkubation wurde das Gel wieder kurz mit Wasser gespült und dann
entwickelt [6 %(w/v) Na2CO3, 500 µl/l (v/v) 37 % HCHO, 0.0005 % (w/v) Natriumsulfat x
H2O].
Die Fixierreaktion wurde abgestoppt, sobald die Banden sichtbar waren.
Fixierlösung
40 %(v/v) Ethanol, vergällt (Methylethylketon)
10 %(v/v) Essigsäure konz.
Coomassie Färbung:
Färbelösung 0.1 % Coomassie blue R-250 (w/v)
40 % Ethanol, vergällt [Methylethylketon] (v/v)
10 % Essigsäure (v/v)
Entfärber 40 % Ethanol, vergällt [Methylethylketon] (v/v)
10 % Essigsäure (v/v)

Methoden
40
Nach erfolgter Elektrophorese wurde das Gel für 20 min in der Färbelösung inkubiert und
anschließend die Banden mit Hilfe des Entfärbers [mehrmals wechseln] unter Schütteln
sichtbar gemacht. Um einen klaren Hintergrund zu erzielen, konnte das Gel nach Einsatz des
Entfärbers über Nacht in 0.5 M NaCl oder Wasser inkubiert werden.
4.3 Mikrobiologische Methoden
4.3.1 Verwendete Organismen
In der vorliegenden Arbeit wurde mit dem Stamm Rhodococcus sp. M4, sowie mit E.coli
XL1-blue, E.coli k12, HB101, JM105, SG13009, UT5600 und BL21 gearbeitet.
4.3.2 Anzuchtbedingungen und Medien
Rhodococcus sp. M4
Für den Rhodococcus sp. M4 wurde das M4-Medium (Hummel et al., 1987) nach folgender
Zusammensetzung verwendet:
KH2PO4 4 g
L-Phenylalanin 10 g
Hefeextrakt 1 g
HVK 2 ml
Thiamin/HCl (nach dem Autoklavieren) 200 µg add 1L H2O
Der pH-Wert des Mediums wurde auf 7.5 mit NaOH eingestellt.
Als Vorkultur wurden 200ml dieses Mediums mit einer einzelnen Kolonie des Rhodococcus
sp. M4 angeimpft. Die Zellen wurden für 48h bis zur stationären Phase unter aeroben Beding-
ungen (Kolben mit Schikanen) bei 120 rpm und 30°C kultiviert.
Davon wurde die Hauptkultur 5 %ig angeimpft und unter gleichen Bedingungen kultiviert.
E. coli
Für den E. coli wurde das LB-Medium (Luria-Bertani) nach folgender Zusammensetzung
verwendet:
Bacto-Trypton 1 %
Bacto-Yeast Extrakt 0.5 %
NaCl 0.5 % pH 7.5

Methoden
41
Für feste Nährböden wurde 1.5 % Bactoagar (Difco) zugesetzt. Der Selektionsdruck wurde
durch Zugabe von Ampicillin (100 µg/ml) nach dem Autoklavieren eingestellt.
Die Anzucht von E. coli erfolgte bei 37°C als Schüttelkultur in den entsprechenden Medien.
Vorkulturen wurden in Reagenzgläsern, größere Kulturen in Schüttelkolben mit Schikanen
inkubiert.
4.3.3 Plattenkulturen
Die Inkubation fester Medien erfolgte aerob bei 37°C, bis Einzelkolonien erkennbar waren.
Bewachsene Platten wurden bis zu vier Wochen bei 4°C gelagert.
4.3.4 Schüttelkolbenkulturen
Flüssigmedien wurden mit Einzelkolonien, Glycerin-Stocks oder 0,1-10 Vol.-% anderer
Flüssigkulturen beimpft und auf einem Rundschüttelgerät mit einer Frequenz von 110-160
rpm bei geeigneter Temperatur inkubiert.
4.3.5 Konservieren von mikrobiologischen Stämmen
Von allen Stämmen wurden 1 ml einer stationären Kultur mit 5 % DMSO [steril] versetzt und
in entsprechenden, sterilen Kryoröhrchen (Nunc) bei -80°C konserviert.
4.3.6 Fermentation der rekombinanten PheDH in Hochdichte Medium (HZD)
5 L bzw. 20 L HZD-Medium wurden mit 1 % exponentiell wachsender HZDamp Flüssigkultur
beimpft und bei 37°C inkubiert. Rührerdrehzahl und Sauerstoffeintrag wurden so angepasst,
dass ein Sauerstoffpartialdruck von 20 % nicht unterschritten wurde. Die maximalen Werte
betrugen 800 rpm Rührerdrehzahl und 5 L·min-1 Lufteintrag. Bei Bedarf wurde ein Überdruck
von bis zu 1 bar angelegt. Die Einstellung des pH-Werts auf 7.0 erfolgte unter Verwendung
von 25 %igem Ammoniakwasser und 1 N Phosphorsäure. Sobald die vorgelegte C-Quelle
erschöpft war, wurde Feedlösung computergesteuert zugesetzt. Die Induktion der Expression
erfolgte innerhalb der Feedphase durch IPTG. War der Sauerstoffpartialdruck irreversibel
unter 20 % gesunken, wurde die Fermentation abgebrochen.
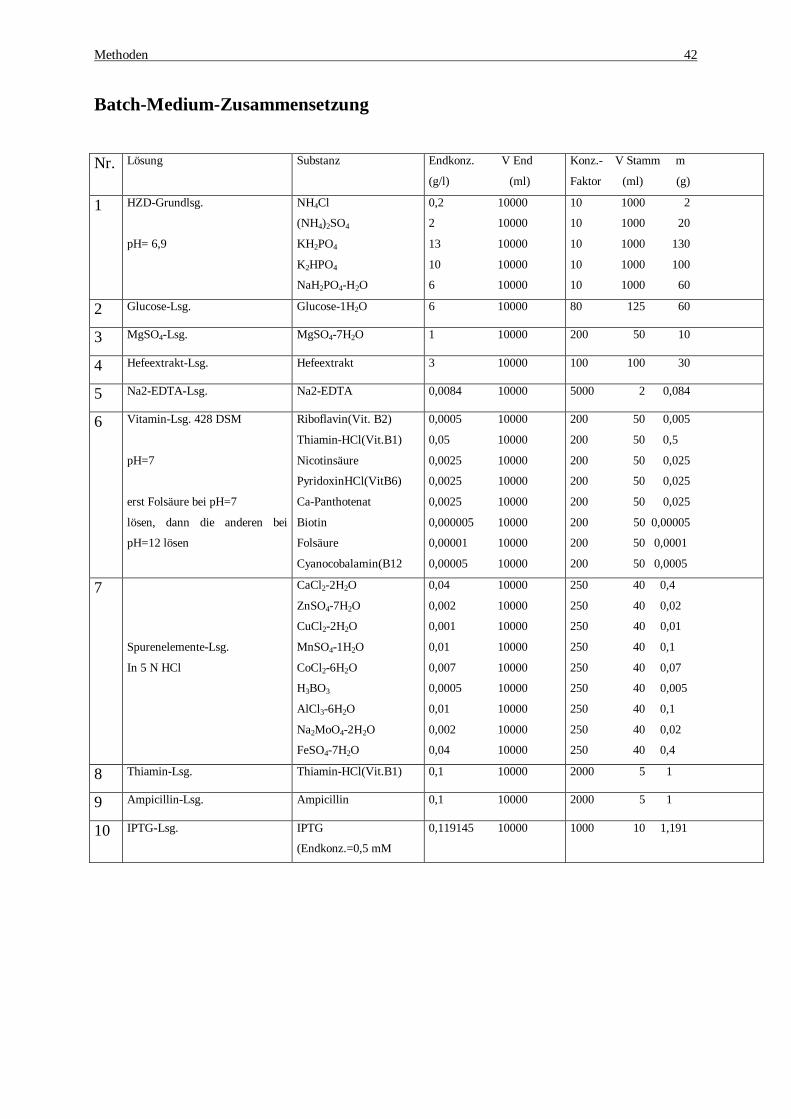
Methoden
42
Batch-Medium-Zusammensetzung
Nr. Lösung Substanz Endkonz. V End
(g/l) (ml)
Konz.- V Stamm m
Faktor (ml) (g)
1 HZD-Grundlsg.
pH= 6,9
NH4Cl
(NH4)2SO4
KH2PO4
K2HPO4
NaH2PO4-H2O
0,2 10000
2 10000
13 10000
10 10000
6 10000
10 1000 2
10 1000 20
10 1000 130
10 1000 100
10 1000 60
2 Glucose-Lsg. Glucose-1H2O 6 10000 80 125 60
3 MgSO4-Lsg. MgSO4-7H2O 1 10000 200 50 10
4 Hefeextrakt-Lsg. Hefeextrakt 3 10000 100 100 30
5 Na2-EDTA-Lsg. Na2-EDTA 0,0084 10000 5000 2 0,084
6 Vitamin-Lsg. 428 DSM
pH=7
erst Folsäure bei pH=7
lösen, dann die anderen bei
pH=12 lösen
Riboflavin(Vit. B2)
Thiamin-HCl(Vit.B1)
Nicotinsäure
PyridoxinHCl(VitB6)
Ca-Panthotenat
Biotin
Folsäure
Cyanocobalamin(B12
0,0005 10000
0,05 10000
0,0025 10000
0,0025 10000
0,0025 10000
0,000005 10000
0,00001 10000
0,00005 10000
200 50 0,005
200 50 0,5
200 50 0,025
200 50 0,025
200 50 0,025
200 50 0,00005
200 50 0,0001
200 50 0,0005
7
Spurenelemente-Lsg.
In 5 N HCl
CaCl2-2H2O
ZnSO4-7H2O
CuCl2-2H2O
MnSO4-1H2O
CoCl2-6H2O
H3BO3
AlCl3-6H2O
Na2MoO4-2H2O
FeSO4-7H2O
0,04 10000
0,002 10000
0,001 10000
0,01 10000
0,007 10000
0,0005 10000
0,01 10000
0,002 10000
0,04 10000
250 40 0,4
250 40 0,02
250 40 0,01
250 40 0,1
250 40 0,07
250 40 0,005
250 40 0,1
250 40 0,02
250 40 0,4
8 Thiamin-Lsg. Thiamin-HCl(Vit.B1) 0,1 10000 2000 5 1
9 Ampicillin-Lsg. Ampicillin 0,1 10000 2000 5 1
10 IPTG-Lsg. IPTG
(Endkonz.=0,5 mM
0,119145 10000 1000 10 1,191
Methoden
43
Fed-Batch-Medium- Zusammensetzung
Nr. Lösung Substanz Endkonz. V End
(g/l) (ml)
Konz. V Stamm m
faktor (ml) (g)
1 HZD-Grundlsg. Stammlsg von Batch 4000 10 400
2 Glucoselsg. Glucose 600 4000 1,28 3124 2400
3 MgSO4-Lsg. Stammlsg von Batch 12 4000 16,6666 240 48
4 Hefeextrakt-Lsg. Stammlsg von Batch 18 4000 16,6666 240 72
5 Vitaminlsg428DSM Stammlsg von Batch 4000 200 20
6 Spurenelemente-Lsg. Stammlsg von Batch 4000 250 16
7 Thiamin-Lsg. Thiamin-HCl (Vit.B1 1 4000 40 4
Batch-Medium: Im Fermenter
HZD-Grundlsg. 1000 ml
Glucose-Lsg. 125 ml
MgSO4-Lsg. 50 ml
Hefeextrakt-Lsg. 100 ml
Na2-EDTA-Lsg. 1 ml
Vitamin-Lsg. 428 (DSM) 50 ml
Spurenelemente-Lsg. 40 ml
Thiamin-Lsg. 5..ml
Ampicillin-Lsg. 5 ml
IPTG-Lsg. 10 ml
Antischaum 4 ml
Inokulum 100 ml
ad Aqua dest 8510 ml
Gesamt 10000
Fed-Batch-Medium: Im Fermenter
HZD-Grundlsg. 400 ml
Glucose-Lsg. 3044 ml
MgSO4-Lsg. 240 ml
Hefeextrakt-Lsg. 240 ml
Vitamin-Lsg. 428 (DSM) 20 ml

Methoden
44
Spuremelemente-Lsg. 16 ml
Thiamin-Lsg. 40 ml
Gesamt 4000 ml
Die rekombinaten Zellen wurden bei einer OD600 von 60 mit 1 mM IPTG induziert. Unter
Sauerstoff- sowie pH-Regulation sind die Zellen bis zu einer OD600 von 170 gewachsen.
4.3.7 Bestimmung der optischen Dichte
Mikrobielle Zellen, die in einen Lichtstrahl gebracht werden, bewirken einen
Intensitätsverlust, der unter Berücksichtigung des Lambert-Beer’schen Gesetzes beschrieben
werden kann (Bergter, 1983): E=? .c.d. Bei konstanten Extinktionskoeffizienten (? ) und
konstanter Küvettenschichtdicke (d) besteht für definierte Bereiche eine direkte proportionale
Beziehung zwischen Extinktion und Zellkonzentration (c) (Bursch et al., 1970).
Innerhalb einer Population wachsende Zellen kann über spektrophotometrische Messung der
Extinktion im Wellenlängenbereich von 550 nm bis 610 nm die zeitabhängige Zunahme der
Zellkonzentration bestimmt werden. Dies erleichtert die Reproduzierbarkeit wachstums-
abhängiger Versuchsergebnisse, erfordert jedoch konstante Versuchsbedingungen, da der
jeweilige Extinktionskoeffizient von Größe, Form, Brechungsindex und Wachstums-
bedingungen der untersuchten Spezies abhängig ist. Aussagen über absolute Zell-
konzentrationen sind unter mikroskopischer Bestimmung der Zellzahl erstellt worden.
Bei jeder OD 600 wurden die Zellsuspensionen durch Zugabe vom Medium soweit verdünnt,
dass die Extinktion unterhalb von 0,5 lag. Darüber besteht keine lineare Beziehung mehr
zwischen Extinktion und Konzentration.

Ergebnisse
45
5 Ergebnisse 5.1 Klonierung der Phenylalanin Dehydrogenase (PheDH)
5.1.1 Präparation genomischer DNA
Das phedh-Gen aus Rhodococcus sp. M4 (Hummel et al., 1987) wurde mittels PCR mit
genomischer DNA als Templat amplifiziert. Die genomische DNA wurde nach der Methode
von van Soolingen (van Soolingen et al., 1991) aus dem Stamm Rhodococcus sp. M4 isoliert.
Diese Methode beinhaltet die enzymatische Vorbehandlung der Zellwand mit Lysozym und
die Fällung der Proteine mit CTAB und Proteinase K als Denaturierungsmittel. Die Zugabe
von CHCl3/Isoamylalkohol (23:1) führt zur Abtrennung der genomischen DNA.
Das DNA-Pellet wurde mit kaltem 70 %igem Ethanol gewaschen und in TE-Puffer bei 4°C
aufbewahrt. (bei –20°C Lagerung würde die DNA beim Auftauen durch die Scherkräfte
gespalten).
Die Reinheit sowie die Ausbeute der DNA wurde durch ein 0.5 %iges Agarose Gel mit
Ethidiumbromid überprüft. Aus Abbildung 10 geht hervor, dass die nach dieser Methode
präparierte genomische DNA hochmolekular und intakt war.
Abbildung 10: genomische DNA aus Rhodococcus sp. M4 nach Elektrophorese im 0.5 %igem Agarosegel.
genom. DNA 1µl = 70 ng
9 kb 5 kb

Ergebnisse
46
5.1.2 Klonierung des PheDH-Gens mittels PCR
Die benötigten Primer für die 5’- und 3’-Enden des zu klonierenden phedh-Gens wurden aus
der veröffentlichten DNA-Sequenz (Brunhuber et al., 1994) abgeleitet.
5’Primer (5’Phe-for)
5’ ATG AGT ATC GAC AGC GCA CTG AAC
3’Primer (5’Phe-rev)
5’ CTA CTA GGC AGT CGC TGT CGT TGT
An den 3’Primer wurden hinter die letzte Aminosäure des C-Terminus 2 Stopcodons
angefügt, um eine effiziente Termination der Transkription zu gewährleisten.
Für die PCR wurde eine Verdünnungsreihe des DNA-Templates durchgeführt, um die
optimale Ausgangskonzentration zu bestimmen. Dafür wurden zwei Versuchsreihen
angesetzt. Die PCR-Ansätze wurden 7 min bei 94°C denaturiert, anschließend wurden 30
Zyklen wie aus Tabelle 2 ersichtlich durchgeführt. Die verwendeten Ansatzgrößen sind in
Tabelle 3 dargestellt.
Tabelle 2: PCR-Zyklen
Zyklusschritt Zeit [min] Temperatur [°C]
Annealing 2 65
Polymerisation 1.5 72
Denaturierung der Stränge 2 94
Tabelle 3: PCR Protokoll zur Amplifizierung des PheDH-Gens. Variiert wurde die Konzentration der Template-DNA.
Template DNA 5’-ende 3’-ende dNTP Puffer Taq-Polymerase DMSO H2O
100 ng/µl 1µl 1µl 2µl 10µl 1µl 3 81µl
70 ng/µl 1µl 1µl 2µl 10µl 1µl 3 81µl
30 ng/µl 1µl 1µl 2µl 10µl 1µl 3 81µl
Durch die Verwendung der obengenanten Primer (5’Phe-for und 5’Phe-rev), die aus der N-
terminalen bzw. C-terminalen Sequenz abgeleitet wurden, konnte ein spezifisches 1.1 kb-
Fragment bei einer Annealing-Temperatur von 65°C amplifiziert werden (Abbildung 11). Um
eventuelle Fehler, die bei Verwendung der Taq-Polymerase auftreten können zu minimieren,

Ergebnisse
47
wurde zur Klonierung des PheDH-Gens in der PCR eine DNA-Polymerase mit ‚proof
reading’-Funktion (high fidelity®- Polymerase) (Roche), Fehlerrate 1.3*10-6 verwendet.
Abbildung 11: Ergebnis der PCR Reaktionen, die Abbildung zeigt ein Fragment mit einer Größe von etwa 1100 bp, als Marker wurde die Gibco KB-Leiter verwendet. Von den PCR-Ansätze wurden 7 µl auf ein 0.8 % Agarosegel aufgetragen, 1-3 entsprechen den Ansätze in Tabelle 2.
5.1.3 Klonierung des PheDH Gens in den pUC-18
Das phedh- Gen wurde in die „multiple cloning site“ des pUC18-Vektors über glatte Enden
an der SmaI- Restriktionsschnittstelle (CCCGGG) kloniert. Das Verhältnis zwischen Vektor
und Insert lag bei 1:3. Für eine erfolgreiche Ligation wurden 200 ng DNA eingesetzt.
Der Vektor wurde an der SmaI -Schnittstelle geschnitten und um eine Religation des linearen
Plasmids zu verhindern, wurden die 5'-Phosphatgruppen abgespalten. Die Dephosphory-
lierung wurde durch alkalische Phosphatase (SAP, Shrimp Alkaline Phosphatase)
durchgeführt, und in die Ligation eingesetzt (Abbildung 12).
1.0 kb
1.6 kb
1 2 3
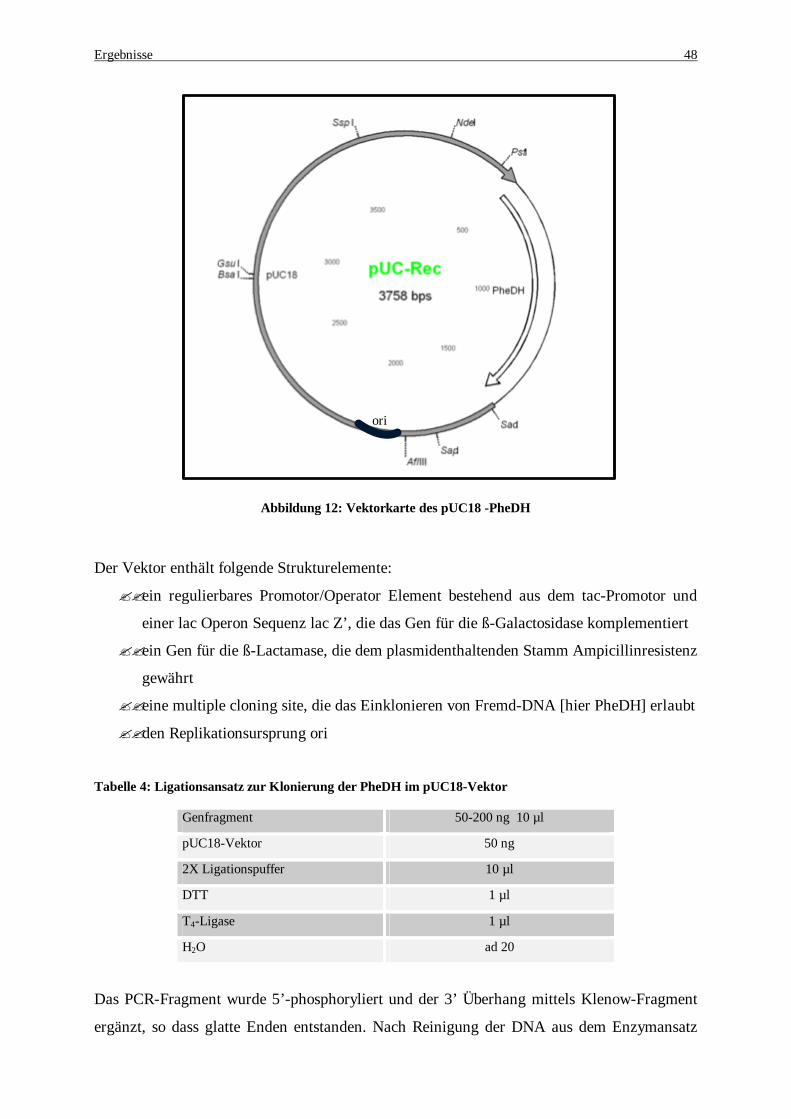
Ergebnisse
48
Abbildung 12: Vektorkarte des pUC18 -PheDH
Der Vektor enthält folgende Strukturelemente:
??ein regulierbares Promotor/Operator Element bestehend aus dem tac-Promotor und
einer lac Operon Sequenz lac Z’, die das Gen für die ß-Galactosidase komplementiert
??ein Gen für die ß-Lactamase, die dem plasmidenthaltenden Stamm Ampicillinresistenz
gewährt
??eine multiple cloning site, die das Einklonieren von Fremd-DNA [hier PheDH] erlaubt
??den Replikationsursprung ori
Tabelle 4: Ligationsansatz zur Klonierung der PheDH im pUC18-Vektor
Genfragment 50-200 ng 10 µl
pUC18-Vektor 50 ng
2X Ligationspuffer 10 µl
DTT 1 µl
T4-Ligase 1 µl
H2O ad 20
Das PCR-Fragment wurde 5’-phosphoryliert und der 3’ Überhang mittels Klenow-Fragment
ergänzt, so dass glatte Enden entstanden. Nach Reinigung der DNA aus dem Enzymansatz
ori

Ergebnisse
49
wurde eine Ligation durchgeführt und in kompetente E. coli XL1 Blue Zellen transformiert.
Nach Ausplattierung auf einer LBamp Agarplatte wurden ca. 150 Kolonien erhalten.
5.1.4 Sequenzierung des phedh-Gens
Von den durch die Transformation erhaltenen Kolonien wurden exemplarisch 20 Kolonien
ausgewählt und über Nacht in 5ml LBamp Medium inkubiert. Aus der hochgewachsenen
Kultur wurde eine Schnellisolierung der Plasmid-DNA durchgeführt. Mit der isolierten
Plasmid-DNA wurde eine Restriktionsanalyse mit SacI und BamHI durchgeführt, die auf
einem 0.8 % Agarosegel elektrophoretisch aufgetrennt wurde.
Die Sequenz der PheDH aus Rhodococcus sp. M4 ist in Abbildung 13 dargestellt.
Abbildung 13: DNA-Sequenz der Phenylalanin Dehydrogenase aus Rhodococcus sp. M4 mit zwei Stopcodons.
ATGAGTATCGACAGCGCACTGAACTGGGACGGGGAAATGACGGTCACCCGATTCGACCGGGAGACTGGTGCCCATTTCGTCATTCGACTCGATTCGACCCAACTCGGACCGGCGGCCGGAGGCACCAGAGCCGCACAGTACTCACAGCTGGCGGACGCCCTCACCGACGCCGGCAAATTGGCGGGGGCGATGACGTTGAAGATGGCAGTGAGCAACCTTCCGATGGGCGGGGGCAAATCCGTCATTGCGCTTCCTGCGCCGCGTCATTCGATCGATCCGAGCACGTGGGCACGCATCCTCCGAATCCACGCCGAGAACATCGACAAGTTGTCCGGCAACTACTGGACCGGACCGGACGTCAACACCAATTCGGCAGACATGGATACTCTGAACGACACCACCGAGTTCGTGTTCGGACGGTCGCTCGAACGCGGCGGCGCGGGTTCGAGCGCGTTCACCACCGCCGTTGGCGTGTTCGAGGCGATGAAGGCGACCGTCGCGCACCGTGGGCTGGGCTCACTCGACGGTTTGACGGTCCTGGTCCAAGGACTGGGGGCAGTCGGAGGATCATTGGCATCCCTGGCCGCCGAAGCGGGTGCGCAACTCCTGGTGGCAGACACCGACACCGAGCGAGTAGCGCACGCTGTTGCGTTGGGCCACACAGCGGTTGCCCTCGAGGACGTTCTGTCCACCCCGTGTGATGTCTTCGCACCCTGCGCAATGGGCGGCGTCATCACCACCGAGGTGGCGCGAACACTCGACTGTTCCGTCGTGGCCGGTGCCGCCAACAACGTCATCGCCGACGAGGCCGCCTCGGACATCCTGCACGCACGCGGAATTCTGTACGCTCCCGACTTCGTGGCCAACGCCGGCGGTGCCATCCACCTCGTAGGCCGGGAGGTTCTCGGTTGGTCCGAGTCGGTTGTCCACGAACGAGCAGTTGCCATAGGCGACACCCTGAATCAGGTCTTCGAGATCTCCGACAACGACGGCGTCACCCCGGACGAGGCCGCCCGCACTCTCGCTGGACGGCGCGCCCGCGAGGCCTCGACAACGACAGCGACTGCCTAGTAA

Ergebnisse
50
5.1.5 Expression des recPheDH-Gens in E.coli
5.1.5.1 Klonierung der PheDH im Expressionsvektor PET16b
Zur Klonierung des recPheDH-Gens in einen Expressionsvektor wurden durch
entsprechendes Primerdesign Restriktionsschnittstellen an beiden Enden des Gens eingeführt.
Diese Vorgehensweise ermöglicht eine Klonierung des Gens in der gewünschten Orientierung
downstream des Promotors. Außerdem kann durch die Wahl der Restriktionsschnittstelle am
5’-Ende des Gens der Abstand des Startcodons ATG zum Promotor und damit auch zur
Ribosomenbindungsstelle auf dem Plasmid beeinflusst werden. Die verwendeten
Expressionsvektoren pET11a und pET16b tragen eine Nde1 Restriktionstelle vor dem
Startcodon des lacZ-Gens. Daher wurde an das 5’-Ende des recPheDH-Gens eine Nde1-
Restriktionsschnittstelle eingefügt. An das 3’-Ende des Gens wurde eine BamHI-Restriktions-
schnittstelle hinter dem Stopcodon angehängt.
Der pET16b-Vektor enthält neben einem starken Promotor einen His-Tag upstream der MCS.
Durch die Klonierung der PheDH über die Schnittstellen NdeI/BamHI wird der His-Tag am
3’-Ende des Gens angefügt, sodass die exprimierte PheDH einen His-Tag am N-Terminus
besitzt. Dafür wurden folgende Primer für eine PCR konstruiert:
5’-For NdeI-Schnittstelle
CGGCATATGAGTATCGACAGCGCACTG
5’-Rev BamHI-Schnittstelle
GCGGATCCCTACTAGGCAGTCGCTGTC
Mit den konstruierten Primern wurde das Insert aus dem rec-pUC18 amplifiziert und in die
Expressionsvektoren pET-16b und pET-11a an der NdeI/BamHI ligiert. Die Strategie zur
Expression der PheDH ist in Abbildung 14 dargestellt.
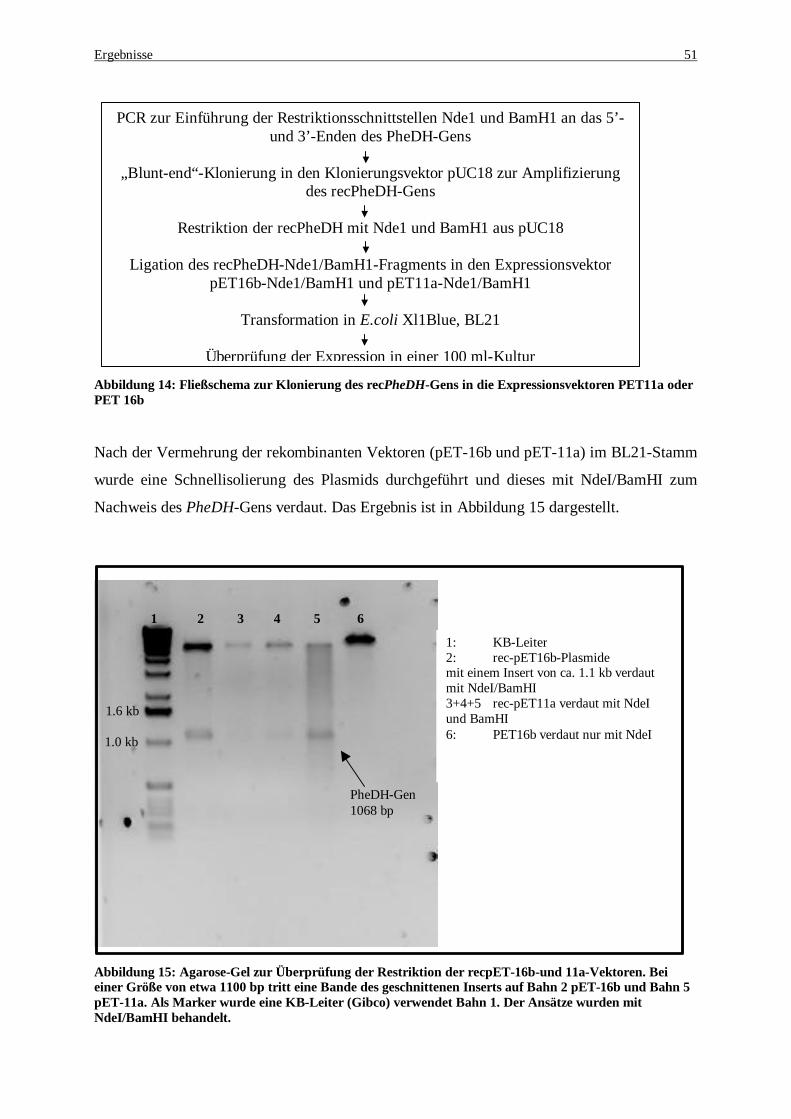
Ergebnisse
51
Abbildung 14: Fließschema zur Klonierung des recPheDH-Gens in die Expressionsvektoren PET11a oder PET 16b
Nach der Vermehrung der rekombinanten Vektoren (pET-16b und pET-11a) im BL21-Stamm
wurde eine Schnellisolierung des Plasmids durchgeführt und dieses mit NdeI/BamHI zum
Nachweis des PheDH-Gens verdaut. Das Ergebnis ist in Abbildung 15 dargestellt.
Abbildung 15: Agarose-Gel zur Überprüfung der Restriktion der recpET-16b-und 11a-Vektoren. Bei einer Größe von etwa 1100 bp tritt eine Bande des geschnittenen Inserts auf Bahn 2 pET-16b und Bahn 5 pET-11a. Als Marker wurde eine KB-Leiter (Gibco) verwendet Bahn 1. Der Ansätze wurden mit NdeI/BamHI behandelt.
1.6 kb
1.0 kb
PCR zur Einführung der Restriktionsschnittstellen Nde1 und BamH1 an das 5’- und 3’-Enden des PheDH-Gens
„Blunt-end“-Klonierung in den Klonierungsvektor pUC18 zur Amplifizierung
des recPheDH-Gens
Restriktion der recPheDH mit Nde1 und BamH1 aus pUC18
Ligation des recPheDH-Nde1/BamH1-Fragments in den Expressionsvektor pET16b-Nde1/BamH1 und pET11a-Nde1/BamH1
Transformation in E.coli Xl1Blue, BL21
Überprüfung der Expression in einer 100 ml-Kultur
1 2 3 4 5 6
1: KB-Leiter 2: rec-pET16b-Plasmide mit einem Insert von ca. 1.1 kb verdaut mit NdeI/BamHI 3+4+5 rec-pET11a verdaut mit NdeI und BamHI 6: PET16b verdaut nur mit NdeI
PheDH-Gen 1068 bp

Ergebnisse
52
5.1.5.2 Expression der PheDH im PET-System
Die Induktion der rekombinanten Stämme erfolgte bei einer OD600 sowohl mit 0.5 als auch
1.5 mM IPTG bei 37°C. Das SDS-Gel (Abbildung 16) zeigte eine breite Bande bei 39 kDa,
die der PheDH entspricht. Es wurde aber keine Enzymaktivität nachgewiesen. Eine Analyse
des exprimierten Proteins auf einem SDS-Gel (12.5 %ig) zeigte die Bildung von
Einschlusskörpern (inclusion bodies).
Abbildung 16: SDS-Gel zur Überprüfung der Expression. Bei einer Größe von etwa 39 kDa tritt eine Bande auf. Bahn 1: Rohextrakt aus dem rec-pET16b in E. coli BL21 das Rohextrakt wurde nicht abzentrifugiert. Bahn 2: Überstand des abzentrifugierten Rohextrakts aus rec-pET16b in E. coli BL21. Bahn 3: Rohextrakt aus dem rec-pET11a in E. coli BL21. Bahn 4: Überstand des abzentrifugierten Rohextrakts aus rec-pET11a in E. coli BL21. 5: Marker
Um die Bildung von inclusion bodies zu minimieren, wurde das PheDH-Gen in weitere
Plasmide ohne His-Tag (pTRC99a, pBtac, pKK223-3) kloniert. Auch die Klonierung in diese
Vektoren führte zu inclusion bodies. Die Induktion der rekombinanten Zellen durch
niedrigere IPTG-Konzentrationen bzw. die Anzucht bei erniedrigten Temperaturen (bis 25°C)
konnte dieses Problem nicht lösen. Ein weiterer Ansatzpunkt war die Manipulation auf
genetischer Ebene. Inclusion bodies könnten durch eine schwächere Transkription bzw.
Translation verhindert oder zumindest minimiert werden. Diesem Gedanken folgend wurde
die PheDH einerseits in einen anderen Expressionsvektor mit schwächerem Promotor
97.400 Da
39.200 Da
66.200 Da
1 2 3 4 5
26.600 Da
14.400 Da

Ergebnisse
53
umkloniert. Andererseits wurde der Abstand zwischen dem Startcodon und der
Ribosomenbindungsstelle variiert.
5.1.5.3 Klonierung des PheDH-Gens in pkk223-3
Die optimale Expression eines Enzyms erreicht man normalerweise, wenn das Startcodon des
dazugehörigen Gens zwischen 7 und 9 bp von der Ribosomenbindungsstelle entfernt ist
(Esipov et al., 1999; Tedin et al., 1997). Diese Klonierungsstrategie wurde für die PheDH in
dieser Arbeit auch mit mehreren Vektoren, pTRC99A, pET16b, pET11a und pBtac,
durchgeführt. Die Klonierungen führten in allen Fällen zu einer starken Expression des
Proteins, das aber, wie oben dargestellt, in unlöslicher Form (inclusion bodies) vorlag.
Unlösliche Proteine können durch eine zu schnelle Expression gebildet werden, die den
Proteinen nicht genügend Zeit und Raum zur Faltung lässt. Für die Vermeidung der Bildung
von unlöslichen Proteinen wurden physikalische bzw. biochemische Methoden (Temperatur,
Induktion, Schüttelgeschwindigkeit) (Shin et al., 1997; Xu et al., 1994; Yang et al., 1997)
während des Wachstums variiert. Dafür wurden die rekombinanten Zellen bei 25°C bzw.
30°C inkubiert sowie eine Inkubation bei einer Schüttelgeschwindigkeit von 80 rpm, die
eindeutig niedriger liegt als üblicherweise mit 140 rpm, durchgeführt. Durch diese Parameter
konnte das Wachstum negativ beeinflusst werden, das aber nicht zur Vermeidung der
„inclusion bodies“ führte. Eine Induktion mit verschiedenen IPTG- Konzentrationen (0,1mM-
1,5 mM) erzielte ebenso keine positiven Ergebnisse.
Wie bereits oben angedeutet kann durch entsprechende Manipulation auf genetischer Ebene
eine schwächere Transkription bzw. Translation erreicht werden, was zu einer langsameren
und schwächeren Expression führt. Diesen bleibt dann genug Zeit und Raum zur Faltung und
man erhält zwar weniger, dafür aber aktives Enzym. Um dies zu erreichen, erfolgte die
Klonierung an der SmaI-Schnittstelle im Expressionsvektor pKK223-3 mit einer „nicht
optimalen“ Entfernung von 14 bp des Startcodons zur Ribosomenbindungsstelle. Dadurch ist
die Translation schwächer mit dem bereits oben erklärten Effekt. Zusätzlich dazu besitzt das
verwendete Plasmid pKK223-3 einen schwächeren Promotor als beispielsweise die oben
verwendeten pET Systeme, was zu einer weiteren Abschwächung der Expression führt. In
Abbildung 17 ist die Vektorkarte dargestellt.

Ergebnisse
54
Abbildung 17: Vektorkarte des pKK-223-3recPheDH. Das PheDH-Gen wurde an der SmaI-Schnittstelle im Expressionsvektor pKK-223-3 mit einem Abstand zur Ribosomenbindungsstelle von 14bp ligiert.
Der Vektor enthält folgende Strukturelemente:
??ein regulierbares Promotor/Operator Element bestehend aus dem tac-Promotor.
Dadurch kann die Expression über IPTG, das den Repressor des lac-Operons
inaktiviert, induziert werden
??abwärts der Klonierungsstelle den rrnB Transkriptionsterminator, der das Ende der
Transkription signalisiert
??eine „multiple cloning site“ für die Insertion von Fremd-DNA [hier rec-PheDH],
dadurch wird das lacZ-Gen inaktiviert und eine blau-weiß-Selektion rekombinanter
Klone ermöglicht
??den Replikationsursprung [Ori] und das Gen für die ? -Lactamase, die die
Ampicillinresistenz bereitstellt
??die lacZ Ribosomenbindungsstelle

Ergebnisse
55
Um die Klonierung des PheDH-Gens an einer „nicht optimalen“ Entfernung des Startcodons
zur ribosomalen Bindungsstelle durchführen zu können, wurde das recPheDH-Gen in die
„multiple cloning site“ des pKK223-3 Expressionsvektors über glatte Schnittstellen an der
SmaI Restriktionsschnittstelle [CCCGGG] ligiert. Dabei wurde zunächst der Vektor mit dem
Restriktionsenzym SmaI geschnitten und anschließend mit einer alkalischen Phosphatase
inkubiert, um eine 5’-Dephosphorylierung zu erreichen. Das amplifizierte recPheDH-Gen
wurde mit dem Klenow-Fragment aufgefüllt und mittels Agarosegelsauftrennung gereinigt.
Die benötigten Primer für die 5’- und 3’-Enden sind:
5’Primer (5’Phe-for)
5’ ATG AGT ATC GAC AGC GCA CTG AAC
3’Primer (5’Phe-rev)
5’ CTA CTA GGC AGT CGC TGT CGT TGT
An den 3’-Primer wurden hinter die letzte Aminosäure des C-Terminus 2 Stopcodons
angefügt, um eine effiziente Termination der Translation zu gewährleisten.
Die Vektor-DNA konnte über Phenol/Chloroform Fällungen wiedergewonnen werden und
wurde für die Ligation mit dem aufgereinigten recPheDH-Gens eingesetzt. Für die
anschließende Transformation wurden kompetente E. coli HB101 bzw. JM-105 Zellen
hergestellt (Hanahan, 1983) und verwendet.
Einige der Transformanden wurden über Nacht in 5 ml LBamp inkubiert, und aus der
hochgewachsenen Kultur Plasmid-DNA isoliert. Mit den durch Schnellisolierung
gewonnenen Plasmiden wurde eine Restriktionsanalyse mit EcoRI durchgeführt. 5 µL des
Ansatzes wurden auf einem Agarosegel (0,8 %) elektrophoretisch aufgetrennt (Abbildung
18).
In der „multiple cloning site“ des pKK223-3-Vektor befindet sich vor der SmaI-Schnittstelle,
über die die PheDH kloniert wurde, eine EcoRI-Schnittstelle. Da sich im Gen an Position 821
ebenfalls eine EcoRI-Schnittstelle befindet, führt ein Verdau mit EcoRI zu einem Fragment
von etwa 830 bp.

Ergebnisse
56
Abbildung 18: rec-pKK223-3-PheDH verdaut mit EcoRI. 1: KB-Marker, 2-6 recPhe-pKK223-3 verdaut mit EcoRI (verschiedene Konzentrationen). Nach einer Restriktionsanalyse taucht eine Bande mit der erwarteten Größe von ca. 800bp auf.
5.1.5.4 Expression der recPheDH im pKK223-3
Das beschriebene Expressionssystem wurde zunächst in weitere E. coli Stämme (Tabelle 5)
transformiert und hinsichtlich ihrer Expressionsrate sowie der nachweisbaren spezifischen
Aktivität im zellfreien Rohextrakt untersucht. Das Ergebnis der Expressionsuntersuchung ist
in Tabelle 5 dargestellt. Es wurden zwei weitere Stämme ausgewählt [JM105 und UT5600]
und in 100 ml Kulturen [+amp] kultiviert. Beim Erreichen einer OD600 von 0.5 wurden sie mit
1 mM IPTG induziert. Nach 7 h wurde die Expression abgebrochen und die Zellen mittels
Ultraschall aufgeschlossen. Aus den Daten wird ersichtlich, dass in allen untersuchten
Stämmen eine Expression der NAD-abhängigen Phenylalanin Dehydrogenase aus
Rhodococcus sp. M4 nachzuweisen war. Die höchsten spezifischen Aktivitäten im zellfreien
Rohextrakt konnten mit dem E. coli JM105 erzielt werden. Infolgedessen wurde dieser
Stamm für alle weiteren Untersuchungen und für die Klonierung und Expression der
Mutanten verwendet.
1 2 3 4 5 6
PKK-223-3
Fragment aus dem PheDH-Gen ca.800 bp
3 kb
1 kb
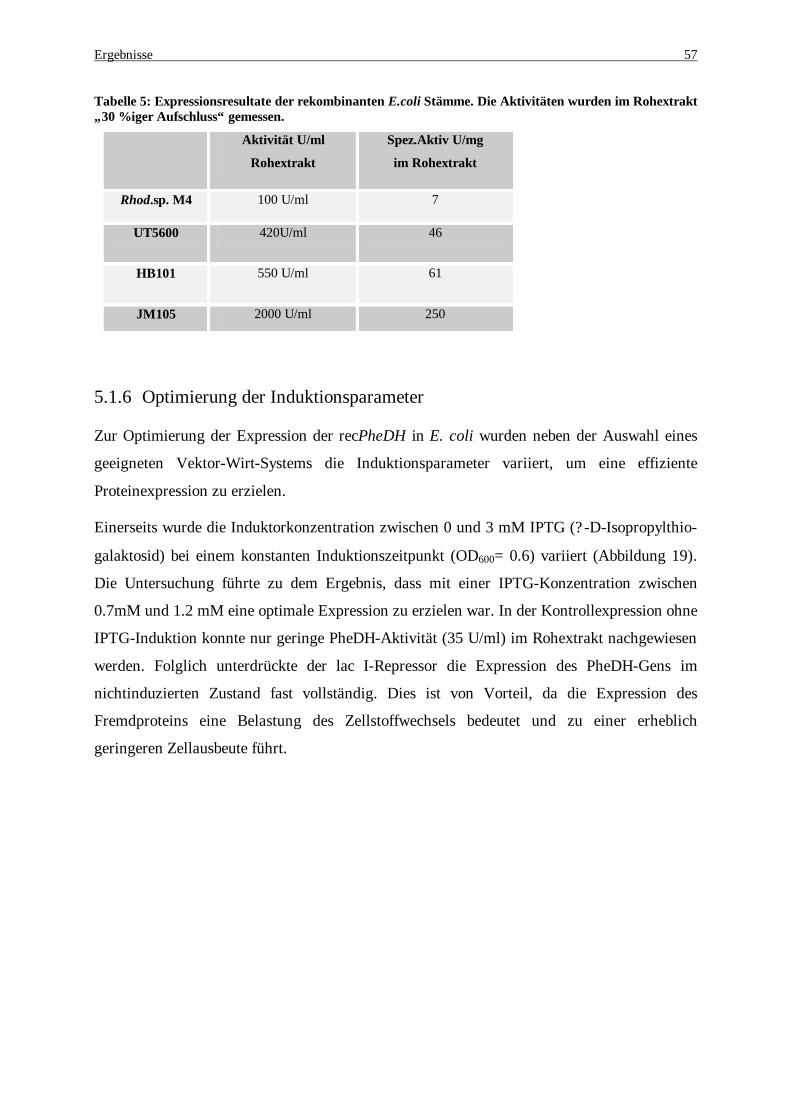
Ergebnisse
57
Tabelle 5: Expressionsresultate der rekombinanten E.coli Stämme. Die Aktivitäten wurden im Rohextrakt „30 %iger Aufschluss“ gemessen.
Aktivität U/ml
Rohextrakt
Spez.Aktiv U/mg
im Rohextrakt
Rhod.sp. M4 100 U/ml 7
UT5600 420U/ml 46
HB101 550 U/ml 61
JM105 2000 U/ml 250
5.1.6 Optimierung der Induktionsparameter
Zur Optimierung der Expression der recPheDH in E. coli wurden neben der Auswahl eines
geeigneten Vektor-Wirt-Systems die Induktionsparameter variiert, um eine effiziente
Proteinexpression zu erzielen.
Einerseits wurde die Induktorkonzentration zwischen 0 und 3 mM IPTG (? -D-Isopropylthio-
galaktosid) bei einem konstanten Induktionszeitpunkt (OD600= 0.6) variiert (Abbildung 19).
Die Untersuchung führte zu dem Ergebnis, dass mit einer IPTG-Konzentration zwischen
0.7mM und 1.2 mM eine optimale Expression zu erzielen war. In der Kontrollexpression ohne
IPTG-Induktion konnte nur geringe PheDH-Aktivität (35 U/ml) im Rohextrakt nachgewiesen
werden. Folglich unterdrückte der lac I-Repressor die Expression des PheDH-Gens im
nichtinduzierten Zustand fast vollständig. Dies ist von Vorteil, da die Expression des
Fremdproteins eine Belastung des Zellstoffwechsels bedeutet und zu einer erheblich
geringeren Zellausbeute führt.
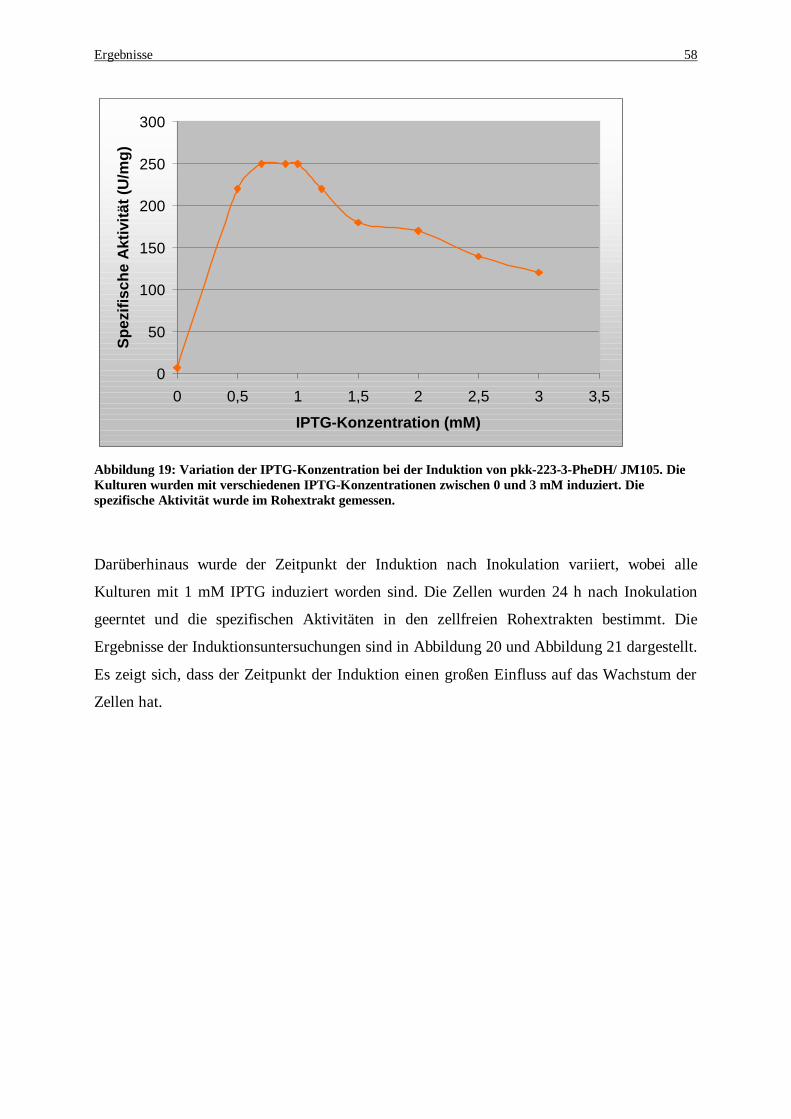
Ergebnisse
58
0
50
100
150
200
250
300
0 0,5 1 1,5 2 2,5 3 3,5
IPTG-Konzentration (mM)
Spe
zifis
che
Akt
ivitä
t (U
/mg)
Abbildung 19: Variation der IPTG-Konzentration bei der Induktion von pkk-223-3-PheDH/ JM105. Die Kulturen wurden mit verschiedenen IPTG-Konzentrationen zwischen 0 und 3 mM induziert. Die spezifische Aktivität wurde im Rohextrakt gemessen.
Darüberhinaus wurde der Zeitpunkt der Induktion nach Inokulation variiert, wobei alle
Kulturen mit 1 mM IPTG induziert worden sind. Die Zellen wurden 24 h nach Inokulation
geerntet und die spezifischen Aktivitäten in den zellfreien Rohextrakten bestimmt. Die
Ergebnisse der Induktionsuntersuchungen sind in Abbildung 20 und Abbildung 21 dargestellt.
Es zeigt sich, dass der Zeitpunkt der Induktion einen großen Einfluss auf das Wachstum der
Zellen hat.
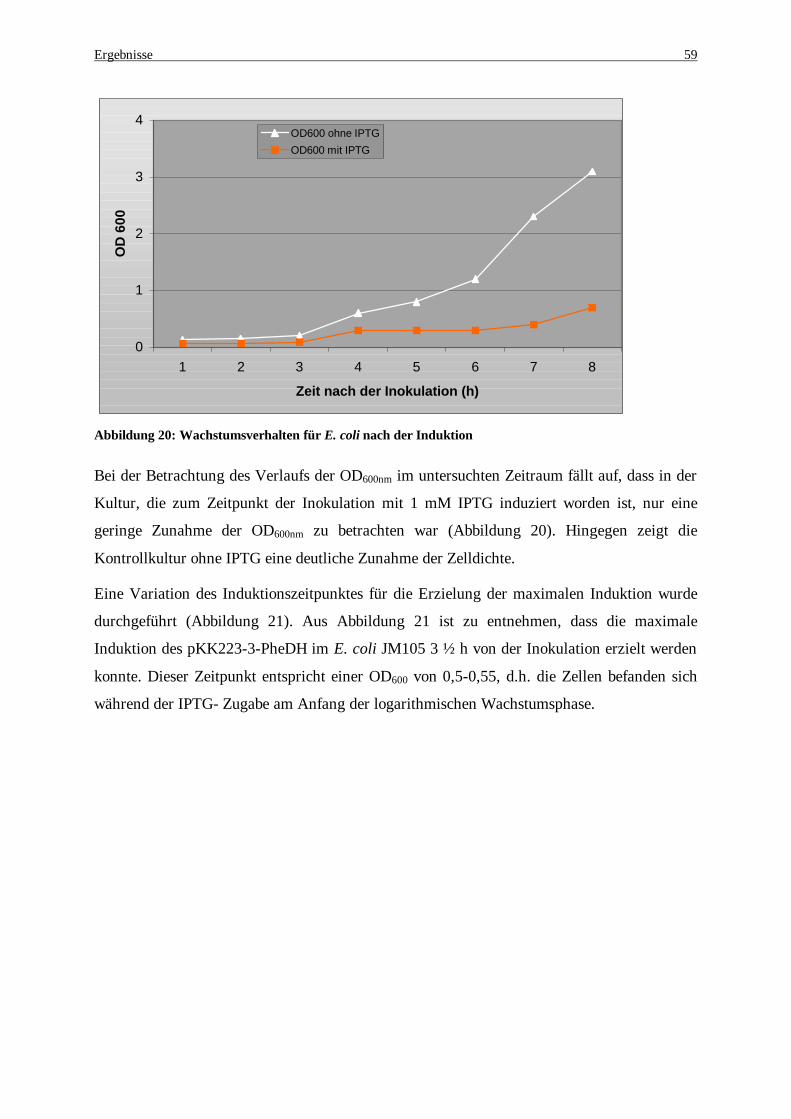
Ergebnisse
59
0
1
2
3
4
1 2 3 4 5 6 7 8
Zeit nach der Inokulation (h)
OD
600
OD600 ohne IPTGOD600 mit IPTG
Abbildung 20: Wachstumsverhalten für E. coli nach der Induktion
Bei der Betrachtung des Verlaufs der OD600nm im untersuchten Zeitraum fällt auf, dass in der
Kultur, die zum Zeitpunkt der Inokulation mit 1 mM IPTG induziert worden ist, nur eine
geringe Zunahme der OD600nm zu betrachten war (Abbildung 20). Hingegen zeigt die
Kontrollkultur ohne IPTG eine deutliche Zunahme der Zelldichte.
Eine Variation des Induktionszeitpunktes für die Erzielung der maximalen Induktion wurde
durchgeführt (Abbildung 21). Aus Abbildung 21 ist zu entnehmen, dass die maximale
Induktion des pKK223-3-PheDH im E. coli JM105 3 ½ h von der Inokulation erzielt werden
konnte. Dieser Zeitpunkt entspricht einer OD600 von 0,5-0,55, d.h. die Zellen befanden sich
während der IPTG- Zugabe am Anfang der logarithmischen Wachstumsphase.
Ergebnisse
60
0
50
100
150
200
250
0 1 2 3 4 5 6 7
Induktionszeitpunkt (h)
Spe
zifis
che
Akt
ivitä
t (U
/mg)
Spezifische Aktivität
Abbildung 21: Variation des Induktionszeitpunktes in Expressionskulturen von pkk-223-3-PheDH/ JM105. Die Kulturen wurden zu unterschiedlichen Zeitpunkten mit 1 mM IPTG induziert. Die spezifische Aktivität wurde im Rohextrakt gemessen.
5.1.7 Reinigung der recPheDH aus E. coli JM105
Die Reinigung der recPheDH aus E. coli-Rohextrakten erfolgte in Anlehnung an das
Reinigungsprotokoll der PheDH aus Rhodococcus sp. M4 (Hummel et al., 1987). Zunächst
wurden die Bakterienzellen durch Disintegration mit Glasperlen aufgeschlossen. Im Anschluß
daran erfolgte ein zusätzlicher Reinigungsschritt zu den Literaturangaben durch eine
Hitzedenaturierung der Begleitproteine in Gegenwart von 5 % (w/v) L-Phenylalanin und
Entfernung des L-Phenylalanin mittels Gelfiltrationschromatographie. Die nach dieser
Methode präparierte recPheDH-Fraktion konnte über Anionenaustauschchromatographie an
DEAE-Sepharose FF bis zur Homogenität gereinigt werden. Abbildung 22 (Bahn 1+2) zeigt
die Auftrennung der gereinigten recPheDH in einem SDS-Polyacrylamidgel nach
Silberfärbung.
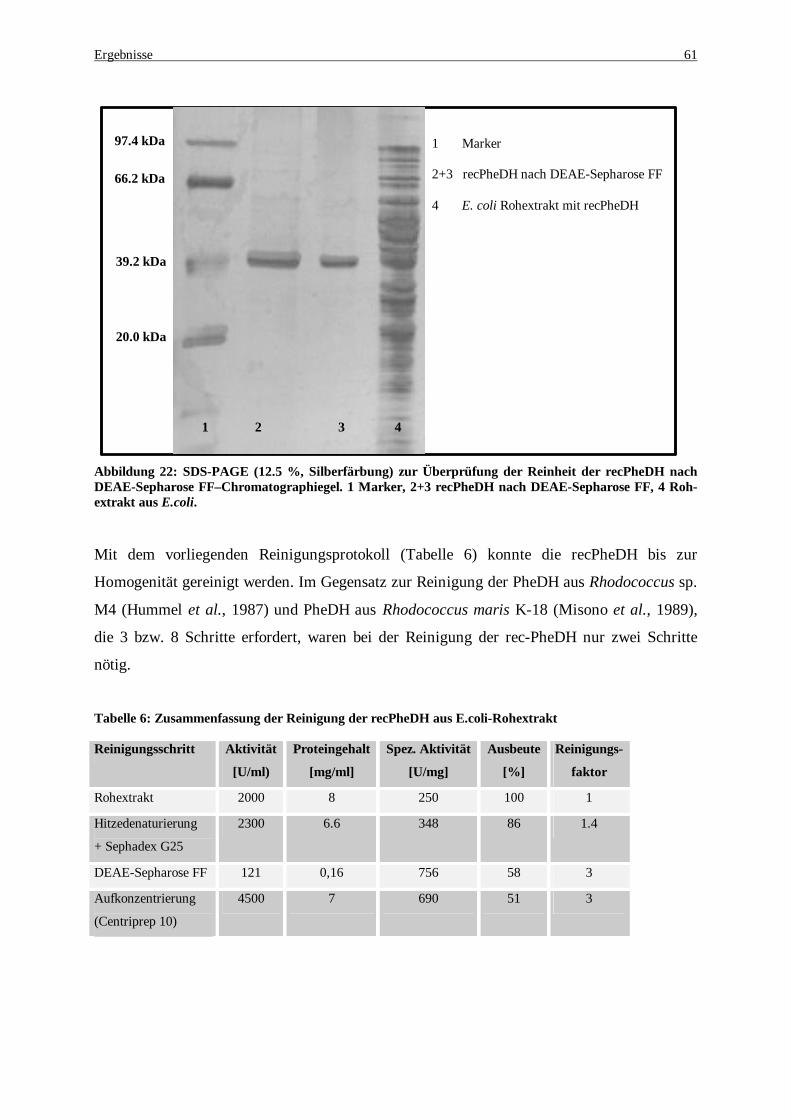
Ergebnisse
61
Abbildung 22: SDS-PAGE (12.5 %, Silberfärbung) zur Überprüfung der Reinheit der recPheDH nach DEAE-Sepharose FF–Chromatographiegel. 1 Marker, 2+3 recPheDH nach DEAE-Sepharose FF, 4 Roh-extrakt aus E.coli.
Mit dem vorliegenden Reinigungsprotokoll (Tabelle 6) konnte die recPheDH bis zur
Homogenität gereinigt werden. Im Gegensatz zur Reinigung der PheDH aus Rhodococcus sp.
M4 (Hummel et al., 1987) und PheDH aus Rhodococcus maris K-18 (Misono et al., 1989),
die 3 bzw. 8 Schritte erfordert, waren bei der Reinigung der rec-PheDH nur zwei Schritte
nötig.
Tabelle 6: Zusammenfassung der Reinigung der recPheDH aus E.coli-Rohextrakt
Reinigungsschritt Aktivität
[U/ml)
Proteingehalt
[mg/ml]
Spez. Aktivität
[U/mg]
Ausbeute
[%]
Reinigungs-
faktor
Rohextrakt 2000 8 250 100 1
Hitzedenaturierung
+ Sephadex G25
2300 6.6 348 86 1.4
DEAE-Sepharose FF 121 0,16 756 58 3
Aufkonzentrierung
(Centriprep 10)
4500 7 690 51 3
97.4 kDa
39.2 kDa
1 Marker 2+3 recPheDH nach DEAE-Sepharose FF 4 E. coli Rohextrakt mit recPheDH
1 2 3 4
66.2 kDa
20.0 kDa
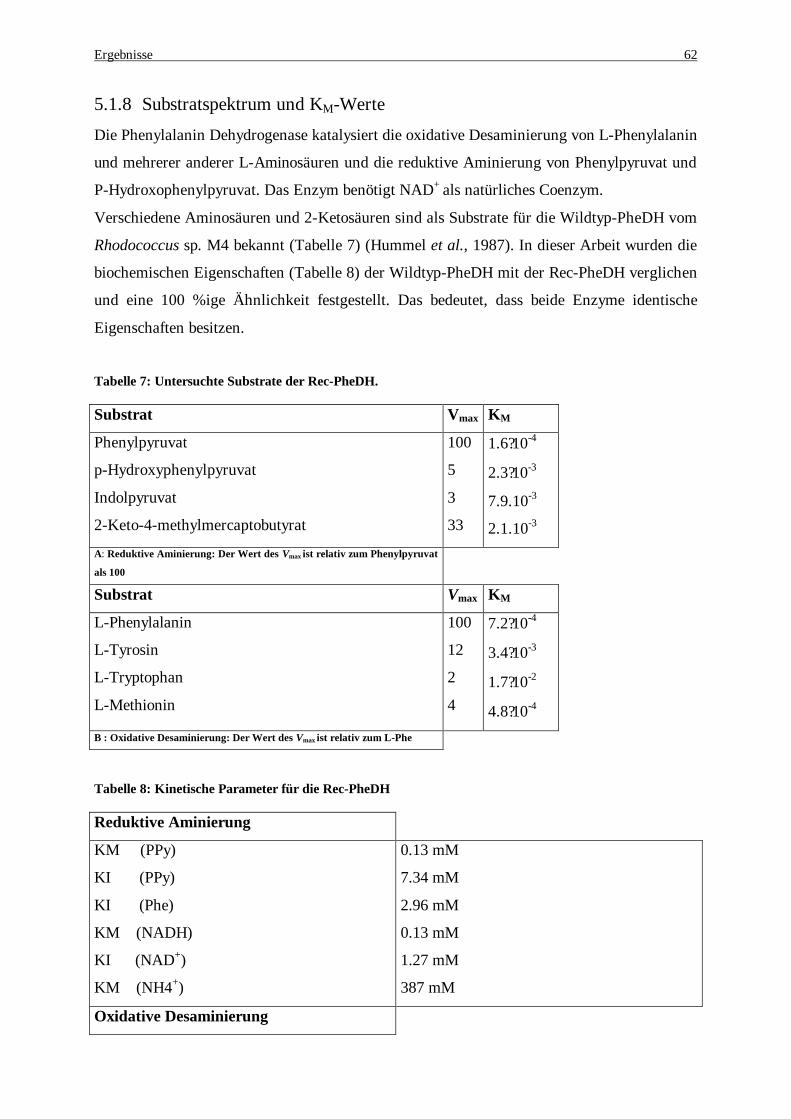
Ergebnisse
62
5.1.8 Substratspektrum und KM-Werte
Die Phenylalanin Dehydrogenase katalysiert die oxidative Desaminierung von L-Phenylalanin
und mehrerer anderer L-Aminosäuren und die reduktive Aminierung von Phenylpyruvat und
P-Hydroxophenylpyruvat. Das Enzym benötigt NAD+ als natürliches Coenzym.
Verschiedene Aminosäuren und 2-Ketosäuren sind als Substrate für die Wildtyp-PheDH vom
Rhodococcus sp. M4 bekannt (Tabelle 7) (Hummel et al., 1987). In dieser Arbeit wurden die
biochemischen Eigenschaften (Tabelle 8) der Wildtyp-PheDH mit der Rec-PheDH verglichen
und eine 100 %ige Ähnlichkeit festgestellt. Das bedeutet, dass beide Enzyme identische
Eigenschaften besitzen.
Tabelle 7: Untersuchte Substrate der Rec-PheDH.
Substrat Vmax KM
Phenylpyruvat
p-Hydroxyphenylpyruvat
Indolpyruvat
2-Keto-4-methylmercaptobutyrat
100
5
3
33
1.6?10-4
2.3?10-3
7.9.10-3
2.1.10-3 A: Reduktive Aminierung: Der Wert des Vmax ist relativ zum Phenylpyruvat
als 100
Substrat Vmax KM
L-Phenylalanin
L-Tyrosin
L-Tryptophan
L-Methionin
100
12
2
4
7.2?10-4
3.4?10-3
1.7?10-2
4.8?10-4 B : Oxidative Desaminierung: Der Wert des Vmax ist relativ zum L-Phe
Tabelle 8: Kinetische Parameter für die Rec-PheDH
Reduktive Aminierung
KM (PPy)
KI (PPy)
KI (Phe)
KM (NADH)
KI (NAD+)
KM (NH4+)
0.13 mM
7.34 mM
2.96 mM
0.13 mM
1.27 mM
387 mM
Oxidative Desaminierung
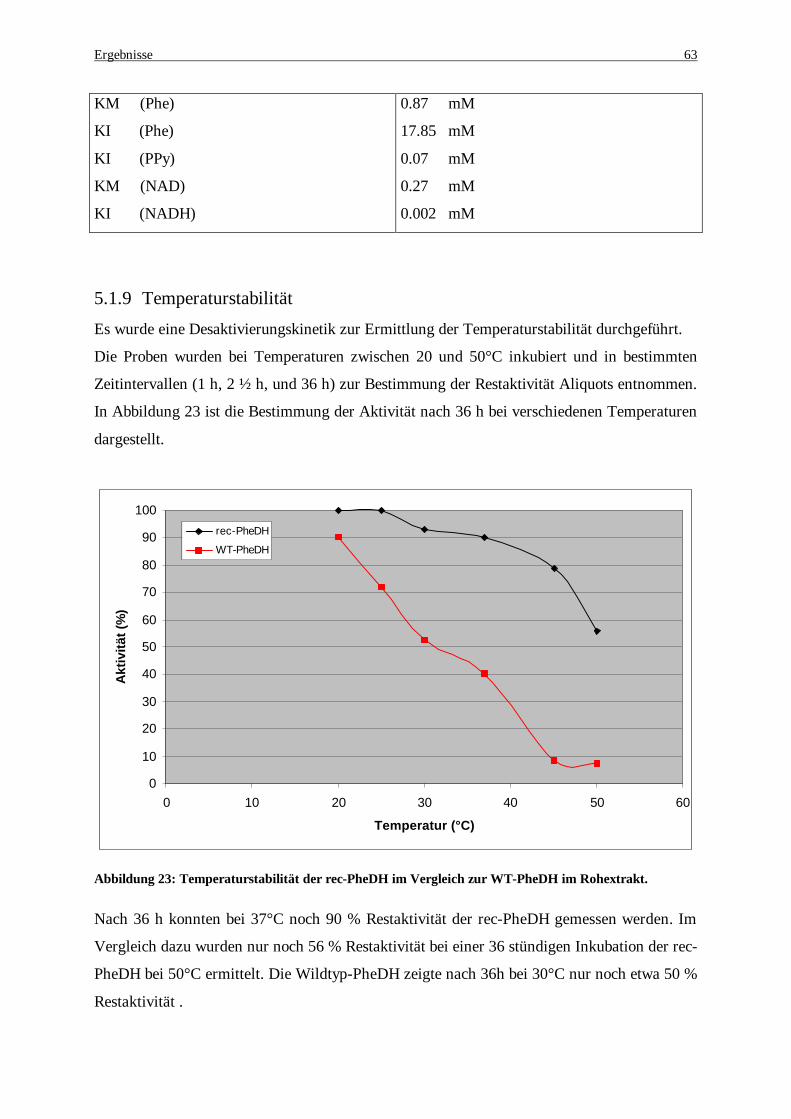
Ergebnisse
63
KM (Phe)
KI (Phe)
KI (PPy)
KM (NAD)
KI (NADH)
0.87 mM
17.85 mM
0.07 mM
0.27 mM
0.002 mM
5.1.9 Temperaturstabilität
Es wurde eine Desaktivierungskinetik zur Ermittlung der Temperaturstabilität durchgeführt.
Die Proben wurden bei Temperaturen zwischen 20 und 50°C inkubiert und in bestimmten
Zeitintervallen (1 h, 2 ½ h, und 36 h) zur Bestimmung der Restaktivität Aliquots entnommen.
In Abbildung 23 ist die Bestimmung der Aktivität nach 36 h bei verschiedenen Temperaturen
dargestellt.
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60
Temperatur (°C)
Akt
ivitä
t (%
)
rec-PheDH
WT-PheDH
Abbildung 23: Temperaturstabilität der rec-PheDH im Vergleich zur WT-PheDH im Rohextrakt.
Nach 36 h konnten bei 37°C noch 90 % Restaktivität der rec-PheDH gemessen werden. Im
Vergleich dazu wurden nur noch 56 % Restaktivität bei einer 36 stündigen Inkubation der rec-
PheDH bei 50°C ermittelt. Die Wildtyp-PheDH zeigte nach 36h bei 30°C nur noch etwa 50 %
Restaktivität .
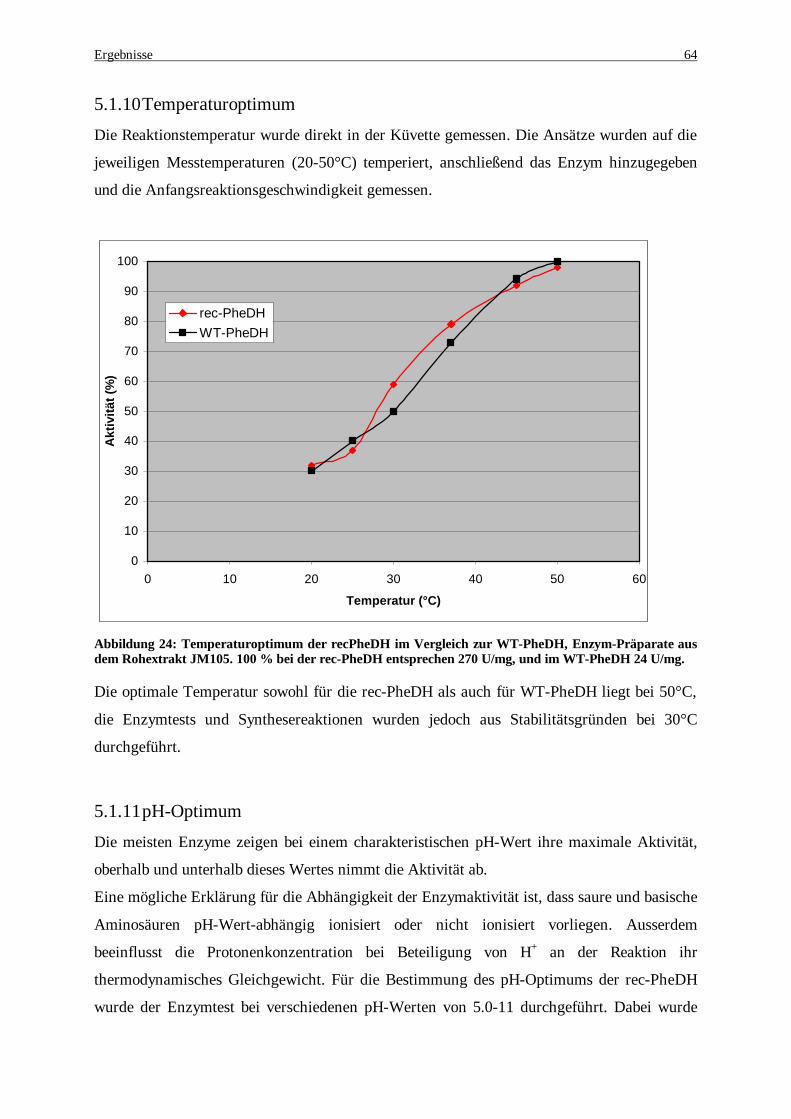
Ergebnisse
64
5.1.10 Temperaturoptimum
Die Reaktionstemperatur wurde direkt in der Küvette gemessen. Die Ansätze wurden auf die
jeweiligen Messtemperaturen (20-50°C) temperiert, anschließend das Enzym hinzugegeben
und die Anfangsreaktionsgeschwindigkeit gemessen.
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60
Temperatur (°C)
Akt
ivitä
t (%
)
rec-PheDHWT-PheDH
Abbildung 24: Temperaturoptimum der recPheDH im Vergleich zur WT-PheDH, Enzym-Präparate aus dem Rohextrakt JM105. 100 % bei der rec-PheDH entsprechen 270 U/mg, und im WT-PheDH 24 U/mg.
Die optimale Temperatur sowohl für die rec-PheDH als auch für WT-PheDH liegt bei 50°C,
die Enzymtests und Synthesereaktionen wurden jedoch aus Stabilitätsgründen bei 30°C
durchgeführt.
5.1.11 pH-Optimum
Die meisten Enzyme zeigen bei einem charakteristischen pH-Wert ihre maximale Aktivität,
oberhalb und unterhalb dieses Wertes nimmt die Aktivität ab.
Eine mögliche Erklärung für die Abhängigkeit der Enzymaktivität ist, dass saure und basische
Aminosäuren pH-Wert-abhängig ionisiert oder nicht ionisiert vorliegen. Ausserdem
beeinflusst die Protonenkonzentration bei Beteiligung von H+ an der Reaktion ihr
thermodynamisches Gleichgewicht. Für die Bestimmung des pH-Optimums der rec-PheDH
wurde der Enzymtest bei verschiedenen pH-Werten von 5.0-11 durchgeführt. Dabei wurde
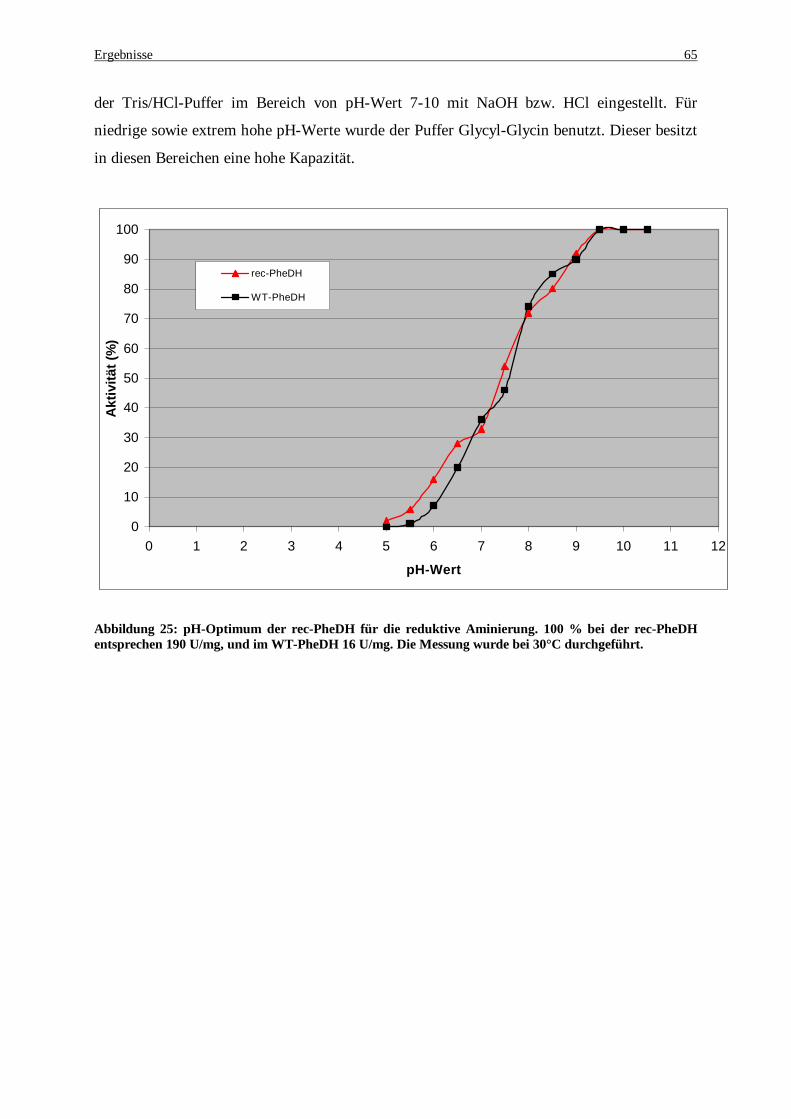
Ergebnisse
65
der Tris/HCl-Puffer im Bereich von pH-Wert 7-10 mit NaOH bzw. HCl eingestellt. Für
niedrige sowie extrem hohe pH-Werte wurde der Puffer Glycyl-Glycin benutzt. Dieser besitzt
in diesen Bereichen eine hohe Kapazität.
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7 8 9 10 11 12
pH-Wert
Akt
ivitä
t (%
)
rec-PheDH
WT-PheDH
Abbildung 25: pH-Optimum der rec-PheDH für die reduktive Aminierung. 100 % bei der rec-PheDH entsprechen 190 U/mg, und im WT-PheDH 16 U/mg. Die Messung wurde bei 30°C durchgeführt.
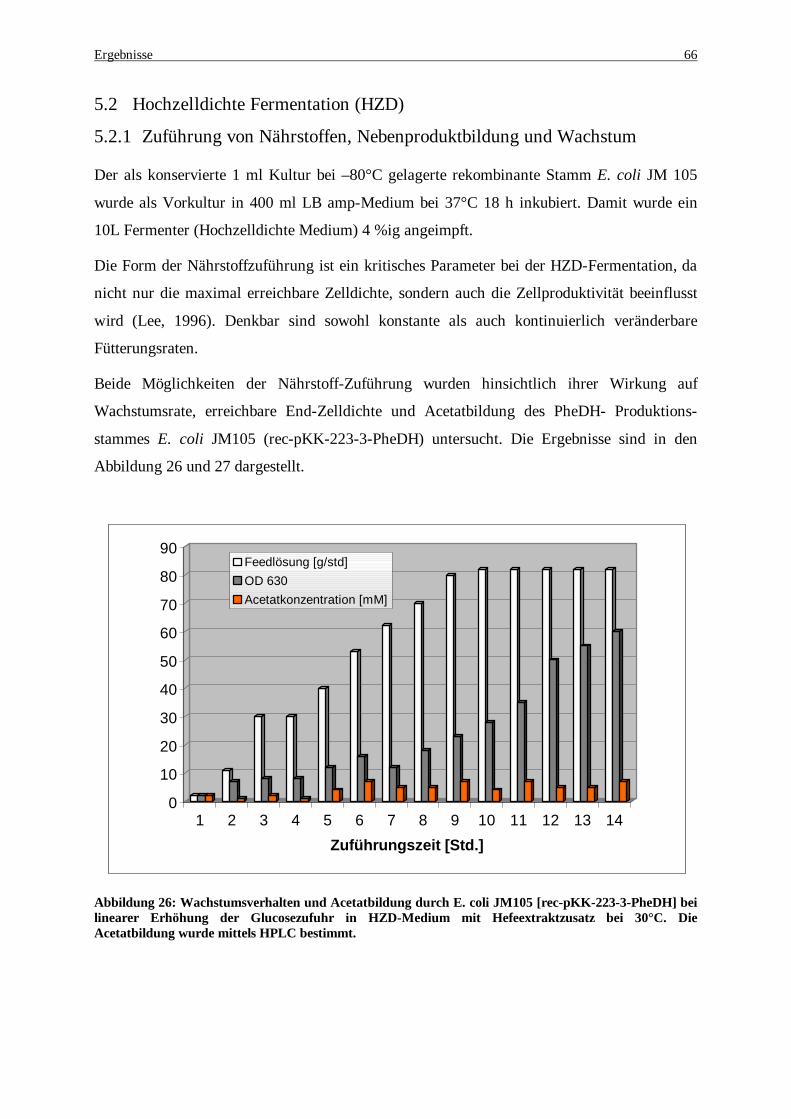
Ergebnisse
66
5.2 Hochzelldichte Fermentation (HZD)
5.2.1 Zuführung von Nährstoffen, Nebenproduktbildung und Wachstum
Der als konservierte 1 ml Kultur bei –80°C gelagerte rekombinante Stamm E. coli JM 105
wurde als Vorkultur in 400 ml LB amp-Medium bei 37°C 18 h inkubiert. Damit wurde ein
10L Fermenter (Hochzelldichte Medium) 4 %ig angeimpft.
Die Form der Nährstoffzuführung ist ein kritisches Parameter bei der HZD-Fermentation, da
nicht nur die maximal erreichbare Zelldichte, sondern auch die Zellproduktivität beeinflusst
wird (Lee, 1996). Denkbar sind sowohl konstante als auch kontinuierlich veränderbare
Fütterungsraten.
Beide Möglichkeiten der Nährstoff-Zuführung wurden hinsichtlich ihrer Wirkung auf
Wachstumsrate, erreichbare End-Zelldichte und Acetatbildung des PheDH- Produktions-
stammes E. coli JM105 (rec-pKK-223-3-PheDH) untersucht. Die Ergebnisse sind in den
Abbildung 26 und 27 dargestellt.
0
10
20
30
40
50
60
70
80
90
1 2 3 4 5 6 7 8 9 10 11 12 13 14
Zuführungszeit [Std.]
Feedlösung [g/std]OD 630Acetatkonzentration [mM]
Abbildung 26: Wachstumsverhalten und Acetatbildung durch E. coli JM105 [rec-pKK-223-3-PheDH] bei linearer Erhöhung der Glucosezufuhr in HZD-Medium mit Hefeextraktzusatz bei 30°C. Die Acetatbildung wurde mittels HPLC bestimmt.
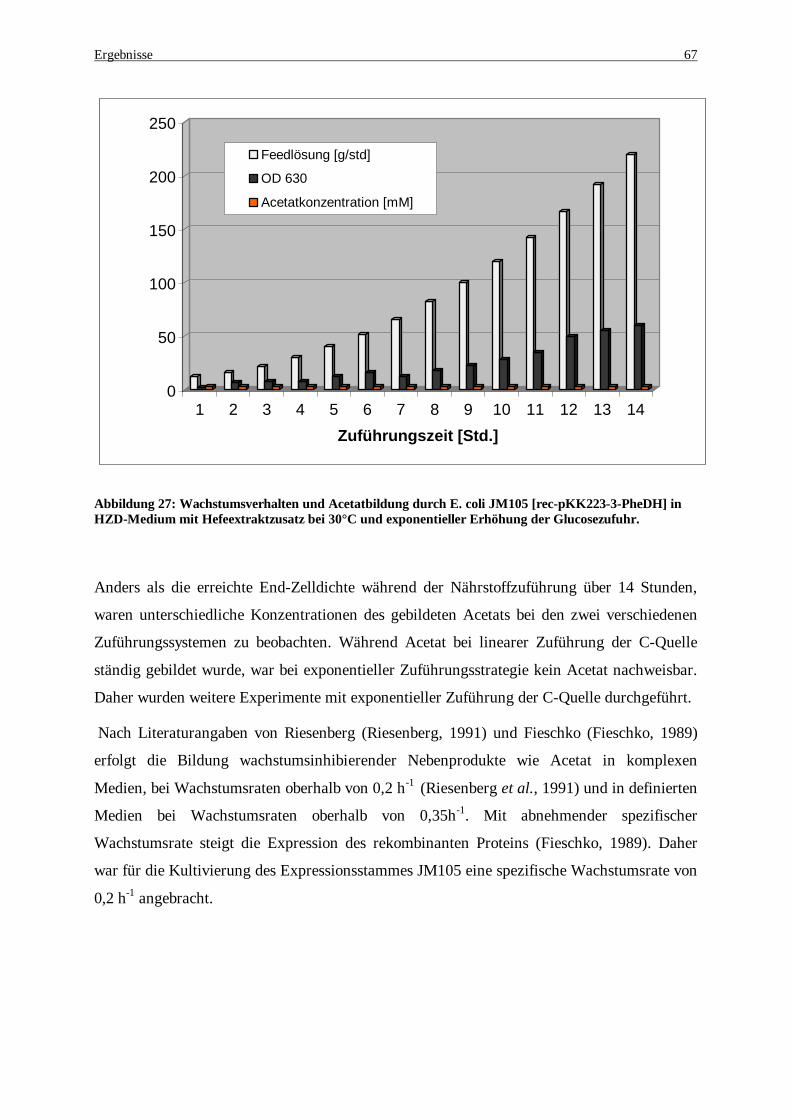
Ergebnisse
67
0
50
100
150
200
250
1 2 3 4 5 6 7 8 9 10 11 12 13 14
Zuführungszeit [Std.]
Feedlösung [g/std]
OD 630
Acetatkonzentration [mM]
Abbildung 27: Wachstumsverhalten und Acetatbildung durch E. coli JM105 [rec-pKK223-3-PheDH] in HZD-Medium mit Hefeextraktzusatz bei 30°C und exponentieller Erhöhung der Glucosezufuhr.
Anders als die erreichte End-Zelldichte während der Nährstoffzuführung über 14 Stunden,
waren unterschiedliche Konzentrationen des gebildeten Acetats bei den zwei verschiedenen
Zuführungssystemen zu beobachten. Während Acetat bei linearer Zuführung der C-Quelle
ständig gebildet wurde, war bei exponentieller Zuführungsstrategie kein Acetat nachweisbar.
Daher wurden weitere Experimente mit exponentieller Zuführung der C-Quelle durchgeführt.
Nach Literaturangaben von Riesenberg (Riesenberg, 1991) und Fieschko (Fieschko, 1989)
erfolgt die Bildung wachstumsinhibierender Nebenprodukte wie Acetat in komplexen
Medien, bei Wachstumsraten oberhalb von 0,2 h-1 (Riesenberg et al., 1991) und in definierten
Medien bei Wachstumsraten oberhalb von 0,35h-1. Mit abnehmender spezifischer
Wachstumsrate steigt die Expression des rekombinanten Proteins (Fieschko, 1989). Daher
war für die Kultivierung des Expressionsstammes JM105 eine spezifische Wachstumsrate von
0,2 h-1 angebracht.
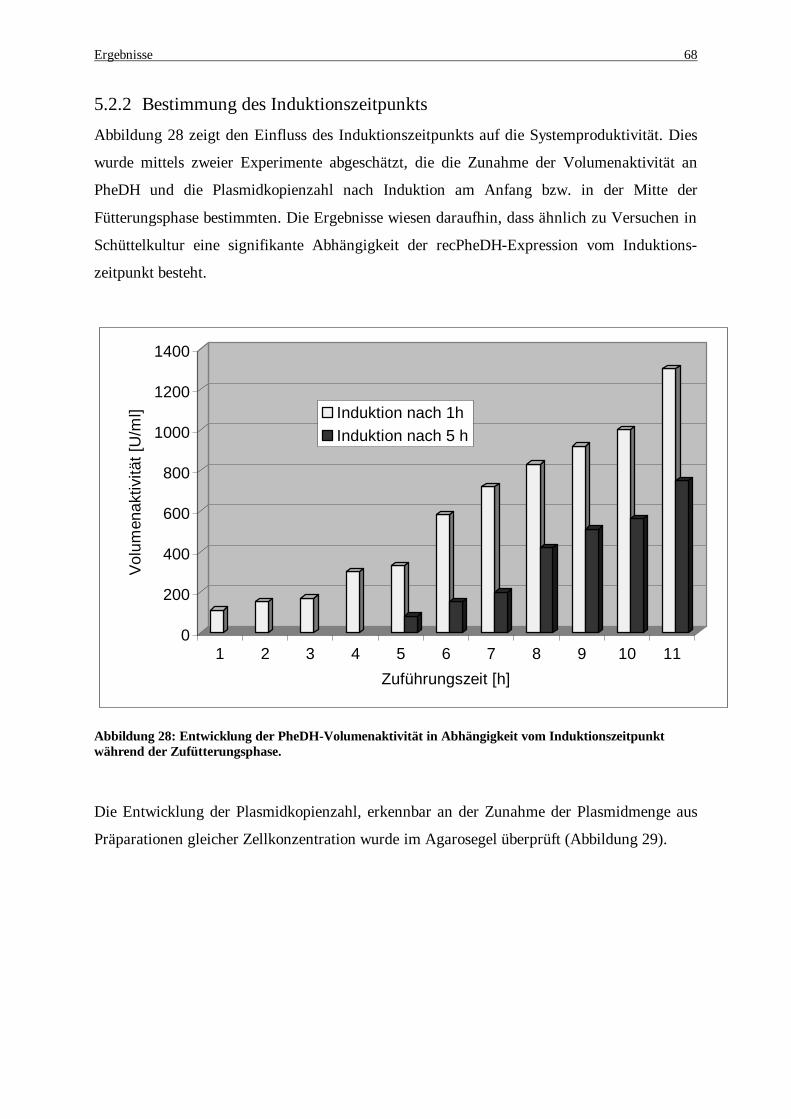
Ergebnisse
68
5.2.2 Bestimmung des Induktionszeitpunkts
Abbildung 28 zeigt den Einfluss des Induktionszeitpunkts auf die Systemproduktivität. Dies
wurde mittels zweier Experimente abgeschätzt, die die Zunahme der Volumenaktivität an
PheDH und die Plasmidkopienzahl nach Induktion am Anfang bzw. in der Mitte der
Fütterungsphase bestimmten. Die Ergebnisse wiesen daraufhin, dass ähnlich zu Versuchen in
Schüttelkultur eine signifikante Abhängigkeit der recPheDH-Expression vom Induktions-
zeitpunkt besteht.
0
200
400
600
800
1000
1200
1400
Vol
umen
aktiv
ität [
U/m
l]
1 2 3 4 5 6 7 8 9 10 11Zuführungszeit [h]
Induktion nach 1hInduktion nach 5 h
Abbildung 28: Entwicklung der PheDH-Volumenaktivität in Abhängigkeit vom Induktionszeitpunkt während der Zufütterungsphase.
Die Entwicklung der Plasmidkopienzahl, erkennbar an der Zunahme der Plasmidmenge aus
Präparationen gleicher Zellkonzentration wurde im Agarosegel überprüft (Abbildung 29).
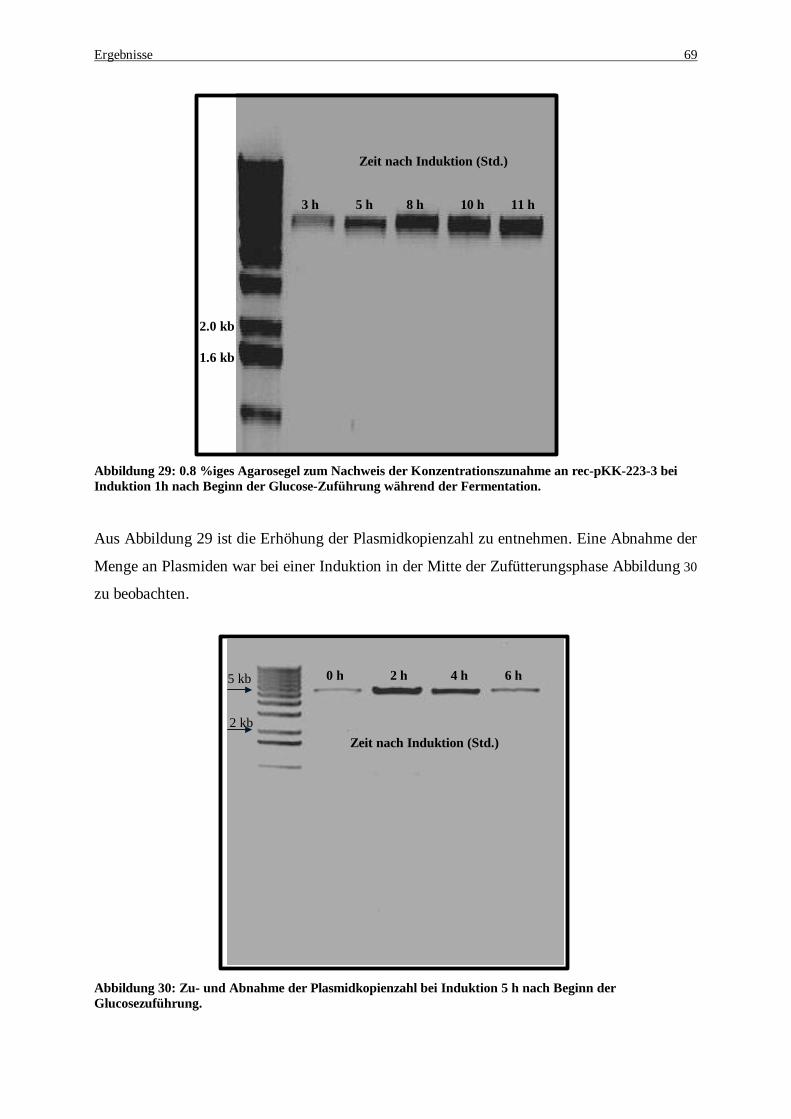
Ergebnisse
69
Abbildung 29: 0.8 %iges Agarosegel zum Nachweis der Konzentrationszunahme an rec-pKK-223-3 bei Induktion 1h nach Beginn der Glucose-Zuführung während der Fermentation.
Aus Abbildung 29 ist die Erhöhung der Plasmidkopienzahl zu entnehmen. Eine Abnahme der
Menge an Plasmiden war bei einer Induktion in der Mitte der Zufütterungsphase Abbildung 30
zu beobachten.
Abbildung 30: Zu- und Abnahme der Plasmidkopienzahl bei Induktion 5 h nach Beginn der Glucosezuführung.
2.0 kb 1.6 kb
3 h 5 h 8 h 10 h 11 h
Zeit nach Induktion (Std.)
0 h 2 h 4 h 6 h
2 kb
5 kb
Zeit nach Induktion (Std.)

Ergebnisse
70
5.3 Klonierung des malic enzymes
5.3.1 Präparation genomischer DNA
Das malic enzyme- Gen wurde mittels PCR mit genomischer DNA als Template amplifiziert.
Die genomische DNA aus E. coli K12 wurde durch enzymatische Vorbehandlung der
Zellwand mit Lysozym und Fällung der Proteine mit CTAB gewonnen. Die Reinheit der
DNA wurde durch ein 0.5 % Agarose Gel überprüft. Die präparierte genomische DNA war
hochmolekular und intakt.
Das DNA-Pellet wurde in TE-Puffer bei 4°C aufbewahrt.
5.3.2 Genisolierung und Klonierung des malic enzymes im recPhe-pKK-223-3
Für die Amplifikation des malic enzymes aus der genomischen DNA des E. coli K12, wurde
der 5’Forward-Primer aus der bekannten N-Terminus-Sequenz abgeleitet (Mahajan et al.,
1990; Stols & Donnelly, 1997). Da der C-Terminus des NAD-abhängigen malic enzymes aus
E. coli K12 (zum Zeitpunkt der Durchführung dieser Arbeiten) nicht veröffentlicht war,
wurde mit Hilfe des 5’Forward Primers ein Einzelstrang ampilifiziert, und die genomische
DNA, die als Template im PCR-Ansatz vorlag, mit dem Restriktionsenzym DpnI verdaut.
Somit konnte der Einzelstrang erhalten und gewonnen werden, das dann für eine zweite PCR
verwendet werden konnte. Da die Taq-Polymerase ca. ein Kb pro Minute amplifiziert, wurde
eine Elongationszeit im PCR-Zyklus von 1,7 min gewählt. Somit konnte ein ca. 1,6-1,8 Kb
großer Einzelstrang, der der Größe des malic enzymes entspricht, amplifiziert werden. In
einer zweiten PCR wurden drei Ansätze vorbereitet, bei denen als Primer sowohl der 5’-
Forward-Primer als auch je ein 5’-Reverse-Primer (siehe unten) Verwendung fanden.
1: 5’-Forward: N’-Malic-pst
5’ CTGCAGAGCCCAGGGATGGATATTC 3’
2: 5’-Reverse A
TTATTATTATTATTA
3: 5’-Reverse B
CTACTACTACTACTA
4: 5’-Reverse C
TCATCATCATCATCA

Ergebnisse
71
Konzentration jeweils 100 pmol/µl
Die Primer für den C-Terminus wurden aus den Stopcodons abgeleitet. Da das Stopcodon des
malic enzymes zum Zeitpunkt der Klonierung nicht bekannt war, wurden 3 verschiedene
Primer, denen die 3 Stopcodons entsprechen, konstruiert (2-4). Mit einer Kombinationen aus
dem bekannten 5’-Forward-Primer (1) und je einem Reverse-Primer (5’-ReverseA-C) wurden
3 PCR-Ansätze durchgeführt (Abbildung 31).
Denaturierung 94°C
5'-ForwardAnnealing 59°C
5'-Rev A
5'-Rev B
5'-Rev C
Annealing 50°C
genom. DNA5' 3'
5'3'
5' 3'
5'3'
5' 3'
5' 3'
3'
3'
5'
5'
Amplifizierung 72°C 1,7 min
Verdau der genomischen DNA und Gewinnung des Einzelstranges
5' 3'
5' 3'Kein Produkt
Kein Produkt
malic enzyme
Abbildung 31: Neue PCR-Strategie zur Amplifikation des malic enzymes. Es wurde ein Einzelstrang amplifiziert, der als Template für eine zweite PCR dient.

Ergebnisse
72
Da das Triplett für die Stopcodons mehrmals im Gen vorkommt, wurden durch diese Methode
bei der Amplifikation des Einzelstranges entsprechend mehrere Fragmente amplifiziert.
Aufgrund der bekannten Größe des malic enzyme-Gens konnte ein Fragment aus dem Gel
isoliert und für die Ligation eingesetzt werden. Die restlichen Banden sind Teilfragmente vom
malic enzyme.
Mit dieser vollkommen neuen Methode konnte ein doppelsträngiges Fragment amplifiziert
werden, das dann an der SmaI-Schnittstelle im pUC18 blunt ends ligiert und sequenziert
wurde.
Abbildung 32: DNA-Sequenz des malic enzymes aus E. coli K12.
1 ATGGATATTC AAAAAAGAGT GAGTGACATG GAACCAAAAA CAAAAAAACA GCGTTCGCTT 61 TATATCCCTT ACGCTGGCCC TGTACTGCTG GAATTTCCGT TGTTGAATAA AGGCAGTGCC 121 TTCAGCATGG AAGAACGCCG TAACTTCAAC CTGCTGGGGT TACTGCCGGA AGTGGTCGAA 181 ACCATCGAAG AACAAGCGGA ACGAGCATGG ATCCAGTATC AGGGATTCAA AACCGAAATC 241 GACAAACACA TCTACCTGCG TAACATCCAG GACACTAACG AAACCCTCTT CTACCGTCTG 301 GTAAACAATC ATCTTGATGA GATGATGCCT GTTATTTATA CCCCAACCGT CGGCGCAGCC 361 TGTGAGCGTT TTTCTGAGAT CTACCGCCGT TCACGCGGCG TGTTTATCTC TTACCAGAAC 421 CGGCACAATA TGGACGATAT TCTGCAAAAC GTGCCGAACC ATAATATTAA AGTGATTGTG 481 GTGACTGACG GTGAACGCAT TCTGGGGCTT GGTGACCAGG GCATCGGCGG GATGGGCATT 541 CCGATCGGTA AACTGTCGCT CTATACCGCC TGTGGCGGCA TCAGCCCGGC GTATACCCTT 601 CCGGTGGTGC TGGATGTCGG AACGAACAAC CAACAGCTGC TTAACGATCC GCTGTATATG 661 GGCTGGCGTA ATCCGCGTAT CACTGACGAC GAATACTATG AATTCGTTGA TGAATTTATC 721 CAGGCTGTGA AACAACGCTG GCCAGACGTG CTGTTGCAGT TTGAAGACTT TGCTCAAAAA 781 AATGCGATGC CGTTACTTAA CCGCTATCGC AATGAAATTT GTTCTTTTAA CGATGACATT 841 CAGGGCACTG CGGCGGTAAC AGTCGGCACA CTGATCGCAG CAAGCCGCGC GGCAGGTGGT 901 CAGTTAAGCG AGAAAAAAAT CGTCTTCCTT GGCGCAGGTT CAGCGGGATG CGGCATTGCC 961 GAAATGATCA TCTCCCAGAC CCAGCGCGAA GGATTAAGCG AGGAAGCGGC GCGGCAGAAA 1021 GTCTTTATGG TCGATCGCTT TGGCTTGCTG ACTGACAAGA TGCCGAACCT GCTGCCTTTC 1081 CAGACCAAAC TGGTGCAGAA GCGCGAAAAC CTCAGTGACT GGGATACCGA CAGCGATGTG 1141 CTGTCACTGC TGGATGTGGT GCGCAATGTA AAACCAGATA TTCTGATTGG CGTCTCAGGA 1201 CAGACCGGGC TGTTTACGGA AGAGATCATC CGTGAGATGC ATAAACACTG TCCGCGTCCG 1261 ATCGTGATGC CGCTGTCTAA CCCGACGTCA CGCGTGGAAG CCACACCGCA GGACATTATC 1321 GCCTGGACCG AAGGTAACGC GCTGGTCGCC ACGGGCAGCC CGTTTAATCC AGTGGTATGG 1381 AAAGATAAAA TCTACCCTAT CGCCCAGTGT AACAACGCCT TTATTTTCCC GGGCATCGGC 1441 CTGGGTGTTA TTGCTTCCGG CGCGTCACGT ATCACCGATG AGATGCTGAT GTCGGCAAGT 1501 GAAACGCTGG CGCAGTATTC ACCATTGGTG CTGAACGGCG AAGGTATGGT ACTGCCGGAA 1561 CTGAAAGATA TTCAGAAAGT CTCCCGCGCA ATTGCGTTTG CGGTTGGCAA AATGGCGCAG 1621 CAGCAAGGCG TGGCGGTGAA AACCTCTGCC GAAGCCCTGC AACAGGCCAT TGACGATAAT 1681 TTCTGGCAAG CCGAATACCG CGACTACCGC CGTACCTCCA TCTAA
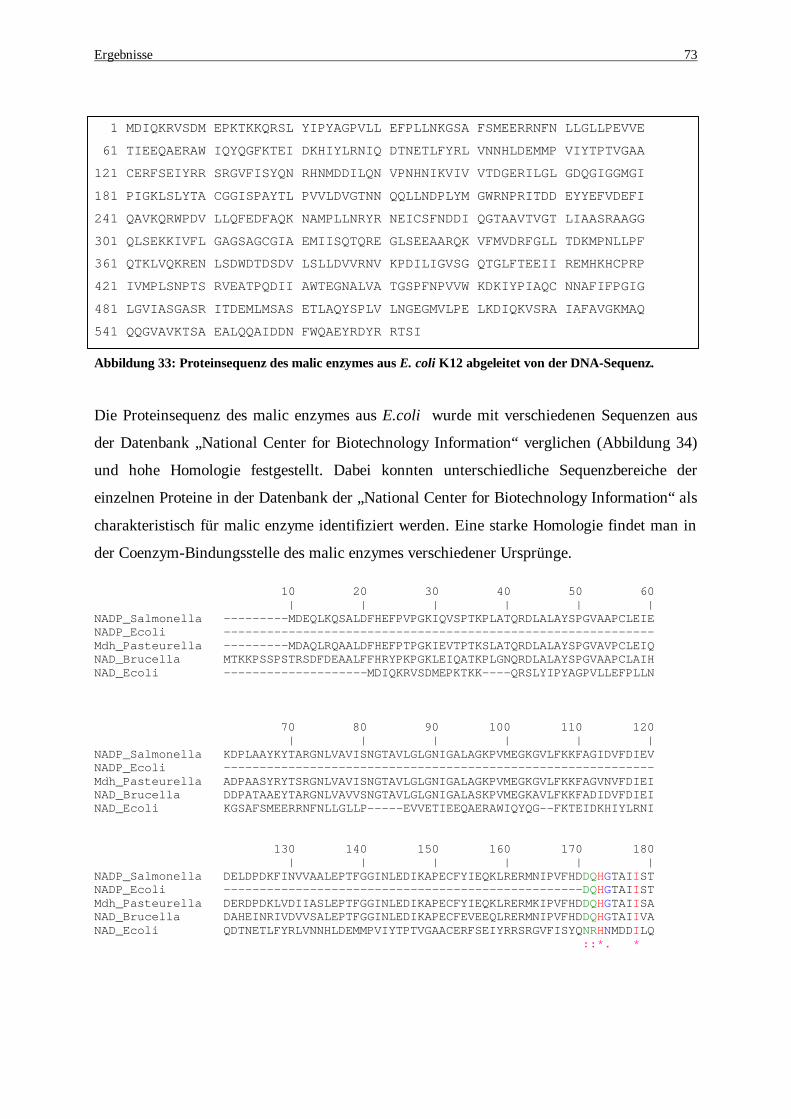
Ergebnisse
73
1 MDIQKRVSDM EPKTKKQRSL YIPYAGPVLL EFPLLNKGSA FSMEERRNFN LLGLLPEVVE
61 TIEEQAERAW IQYQGFKTEI DKHIYLRNIQ DTNETLFYRL VNNHLDEMMP VIYTPTVGAA
121 CERFSEIYRR SRGVFISYQN RHNMDDILQN VPNHNIKVIV VTDGERILGL GDQGIGGMGI
181 PIGKLSLYTA CGGISPAYTL PVVLDVGTNN QQLLNDPLYM GWRNPRITDD EYYEFVDEFI
241 QAVKQRWPDV LLQFEDFAQK NAMPLLNRYR NEICSFNDDI QGTAAVTVGT LIAASRAAGG
301 QLSEKKIVFL GAGSAGCGIA EMIISQTQRE GLSEEAARQK VFMVDRFGLL TDKMPNLLPF
361 QTKLVQKREN LSDWDTDSDV LSLLDVVRNV KPDILIGVSG QTGLFTEEII REMHKHCPRP
421 IVMPLSNPTS RVEATPQDII AWTEGNALVA TGSPFNPVVW KDKIYPIAQC NNAFIFPGIG
481 LGVIASGASR ITDEMLMSAS ETLAQYSPLV LNGEGMVLPE LKDIQKVSRA IAFAVGKMAQ
541 QQGVAVKTSA EALQQAIDDN FWQAEYRDYR RTSI
Abbildung 33: Proteinsequenz des malic enzymes aus E. coli K12 abgeleitet von der DNA-Sequenz.
Die Proteinsequenz des malic enzymes aus E.coli wurde mit verschiedenen Sequenzen aus
der Datenbank „National Center for Biotechnology Information“ verglichen (Abbildung 34)
und hohe Homologie festgestellt. Dabei konnten unterschiedliche Sequenzbereiche der
einzelnen Proteine in der Datenbank der „National Center for Biotechnology Information“ als
charakteristisch für malic enzyme identifiziert werden. Eine starke Homologie findet man in
der Coenzym-Bindungsstelle des malic enzymes verschiedener Ursprünge. 10 20 30 40 50 60 | | | | | | NADP_Salmonella ---------MDEQLKQSALDFHEFPVPGKIQVSPTKPLATQRDLALAYSPGVAAPCLEIE NADP_Ecoli ------------------------------------------------------------ Mdh_Pasteurella ---------MDAQLRQAALDFHEFPTPGKIEVTPTKSLATQRDLALAYSPGVAVPCLEIQ NAD_Brucella MTKKPSSPSTRSDFDEAALFFHRYPKPGKLEIQATKPLGNQRDLALAYSPGVAAPCLAIH NAD_Ecoli --------------------MDIQKRVSDMEPKTKK----QRSLYIPYAGPVLLEFPLLN 70 80 90 100 110 120 | | | | | | NADP_Salmonella KDPLAAYKYTARGNLVAVISNGTAVLGLGNIGALAGKPVMEGKGVLFKKFAGIDVFDIEV NADP_Ecoli ------------------------------------------------------------ Mdh_Pasteurella ADPAASYRYTSRGNLVAVISNGTAVLGLGNIGALAGKPVMEGKGVLFKKFAGVNVFDIEI NAD_Brucella DDPATAAEYTARGNLVAVVSNGTAVLGLGNIGALASKPVMEGKAVLFKKFADIDVFDIEI NAD_Ecoli KGSAFSMEERRNFNLLGLLP-----EVVETIEEQAERAWIQYQG--FKTEIDKHIYLRNI 130 140 150 160 170 180 | | | | | | NADP_Salmonella DELDPDKFINVVAALEPTFGGINLEDIKAPECFYIEQKLRERMNIPVFHDDQHGTAIIST NADP_Ecoli --------------------------------------------------DQHGTAIIST Mdh_Pasteurella DERDPDKLVDIIASLEPTFGGINLEDIKAPECFYIEQKLRERMKIPVFHDDQHGTAIISA NAD_Brucella DAHEINRIVDVVSALEPTFGGINLEDIKAPECFEVEEQLRERMNIPVFHDDQHGTAIIVA NAD_Ecoli QDTNETLFYRLVNNHLDEMMPVIYTPTVGAACERFSEIYRRSRGVFISYQNRHNMDDILQ ::*. *
Ergebnisse
74
190 200 210 220 230 240 | | | | | | NADP_Salmonella AAILNGLRVVEKNISDVRMVVSGAGAAAIACMNLLVALGMQKHNIVVCDSKGVIYKGREP NADP_Ecoli AAILNGLRVVEKNISDVRMVVSGAGAAAIACMNLLVALGLQKHNIVVCDSKGVIYQGREP Mdh_Pasteurella AAILNGLRIVKKDIAKVKLIASGAGAASIACLNLLVSLGLPRENIIVCDSKGVIFHGRDE NAD_Brucella SAVLNGLELAGKKIEEAKIVTSGAGAAALACLNLLVNLGAKRENIWVCDIEGVVYDGRNT NAD_Ecoli NVPNHNIKVIVVTDGERILGLGDQGIGGMGIPIGKLSLYTACGGISPAYTLPVVLDVG-- . :.:.: . : .. * ..:. : * .* . *: . 250 260 270 280 290 300 | | | | | | NADP_Salmonella NMAETKAAYAVDDSGKRTLDEVIDGADIFLGCSGPKVLTQEMVKKMARAPMILALANPEP NADP_Ecoli NMAETKAAYAVVDDGKRTLDDVIEGADIFLGCSGPKVLTQEMVKKMARAPMILALANPEP Mdh_Pasteurella RMDETKKLYAIEDNGKRTLAEVINDADIFLGCSAAGTLTQDMVKTMAANPLILALANPDP NAD_Brucella LMDRWKEVYAQKTD-ARTLADVIGGADVFLGLSAAGVLKPELLKQMAEKPLIMALANPKP NAD_Ecoli -----------------TNNQQLLNDPLYMGWRNPRITDDEYYEFVD--EFIQAVKQRWP * : : . :::* . : : : :* *: : * 310 320 330 340 350 360 | | | | | | NADP_Salmonella EILPPLAKEVRPDAIICTGRSDYPNQVNNVLCFPFIFRGALDVGATAINEEMKLAAVRAI NADP_Ecoli EILPPLAKEVRPDAIICTGRSDYPNQVNNVLCFPFIFRGALDVGATAINEEMKLAAVRAI Mdh_Pasteurella EILPPLAKAVRPDAIVCTGRSDYPNQVNNVLCFPFIFRGALDVSATAINEEMKLAAVHAI NAD_Brucella EIMPEEARAARADAMICTGRSDFPNQVNNVLCFPYIFRGALDVGATVINEEMKLAAVRAI NAD_Ecoli DVLLQFEDFAQKNAMPLLNR------YRNEICS---FNDDIQGTAAVTVGTLIAASRAAG ::: .: :*: .* .* :* *.. :: *:. : *: * 370 380 390 400 410 420 | | | | | | NADP_Salmonella AELAHAEQSEVVASAYGDQDLSFGPEYIIPKPFDPRLIVKIAPAVAKAAMDSGVATRPIA NADP_Ecoli AELAHAEQSEVVASAYGDQDLSFGPEYIIPKPFDPRLIVKIAPAVAKAAMESGVATRPIA Mdh_Pasteurella ADLALAEQSEVVTSAYGETELSFGPEYLIPKPFDPRLIVKIAPAVAKAAMDSGVATRPIK NAD_Brucella ASLAREEPSDVAARAYSGDTPTFGPDYIIPSPFDQRLILRIAPAVAKAAMETGVATRPIE NAD_Ecoli GQLSEKKIVFLGAGSAG---------------------CGIAEMIISQTQREGLSEEAAR ..*: : : : : . ** : . : *:: .. 430 440 450 460 470 480 | | | | | | NADP_Salmonella DFDAYIDKLTEFVYKTNLFMKPIFSQARKD--PKRVVLPEGEEARVLHATQELITLGLAK NADP_Ecoli DFDVYIDKLTEFVYKTNLFMKPIFSQARKA--PKRVVLPEGEEARVLHATQELVTLGLAK Mdh_Pasteurella DFDAYIEKLTQFVYKTNLFMKPVFAQAKQN--KKRVLLTDGEESRVLHAVQEIATLGIAT NAD_Brucella DMEAYLDRLNRFVFRSGLIMKPVFAAARTQGAPMRVIYADGEDERVLRAAQVVIEERIAA NAD_Ecoli QKVFMVDRFGLLTDKMPNLLP--FQTKLVQ---KRENLSDWDTDSDVLSLLDVVRN-VKP : :::: :. : :: * * .: : : : : : 490 500 510 520 530 540 | | | | | | NADP_Salmonella PILIGRPSVIEMRIQKLGLQIKAGVDFEIVNNESDPRFKEYWSEYYQIMKRRGVTQEQAQ NADP_Ecoli PILIGRPNVIEMRIQKLGLQIKAGVDFEIVNNESDPRFKEYWTEYFQIMKRRGVTQEQAQ Mdh_Pasteurella PILLGRPSVIAQKIKQLGLHIQEGRDFELVDIENNPYFEECYKTYHNLLKRKGITPAGAQ NAD_Brucella PILVGRPQVVETRLRRYGLKIRPGTDFELINPEDDPRYRDYVDLYLSYTGRQGVTPEAAR NAD_Ecoli DILIGVSGQTGLFTEEIIREMHKHCPRPIVMPLSNP------------------T----- **:* . .. .:: :: .:* * 550 560 570 580 590 600 | | | | | | NADP_Salmonella RAMIGNHTAIGAIMVQRGEADAMICGTIGDYHEHFSVVKAVFGYRDGVHTAGAMNALLLP NADP_Ecoli RALISNPTVIGAIMVQRGEADAMICGTVGDYHEHFSVVKNVFGYRDGVHTAGAMNALLLP Mdh_Pasteurella RKMLHNPTVIGATLLQLGKADAMLCGLVGPYASHLSNIKEVIGVQPCVPTPATVNGLVLP NAD_Brucella TIVRTNSTAIAALAVMRDEADAMICGLEGRFERHLRVVRQIIGKVPGIRDYSALSLLISQ NAD_Ecoli ----SRVEATPQDIIAWTEGNALVATGSPFNPVVWKDKIYPIAQCNNAFIFPGIGLGVIA . . : :.:*::. :. :. :

Ergebnisse
75
610 620 630 640 650 660 | | | | | | NADP_Salmonella SGNTFIADTYVNEDPTPEQLAEIAVMAAETVRRFGIEPKVALLSHSNFGSSNSLSASKMR NADP_Ecoli SGNTFIADTYVNDEPDAEELAEITLMAAETVRRFGIEPRVALLSHSNFGSSDCPSSSKMR Mdh_Pasteurella TGNLFITDTFVNHNPTAQELAEITIMAANEVSRFGIEPKVALVSHSNFGTFDDPSAVKMR NAD_Brucella RGALFLTDTYVNAEPDANEIAEMAVLAAREISRFGIEPKAALVSHSNFGSKESASAEKMR NAD_Ecoli SGASRITD-------------EMLMSASETLAQY---S-PLVLNGEGMVLPELKDIQKVS * ::* *: : *:. : :: . ::. ..: : . *: 670 680 690 700 710 720 | | | | | | NADP_Salmonella ETLERVRERAPDLMIDGEMHGDAALVESIRNDRMPDSPLKGAANILVMPNMEAARISYNL NADP_Ecoli QALELVRERAPELMIDGEMHGDAALVEAIRNDRMPDSSLKGSANILVMPNMEAARISYNL Mdh_Pasteurella EVLHLVKEKAPDLIIDGEMHCDVALNEKLRQDIMPDSPLKGAANLLVMPNMEAARISLNL NAD_Brucella QAAAILAEKAPDLESDGEMHGDAALSQTLRDRVFPNSRLKGEANLLVFPNLDAANITLNV NAD_Ecoli RAIAFAVGKMAQQQGVAVKTSAEALQQAIDDNFWQAEYRDYRRTSI-------------- .. : .: . ** : : : . . . : 730 740 750 760 770 | | | | | NADP_Salmonella LRVSSSEGVTVGPVLMGVSKPVHVLTPIASVRRIVNMVALAVVEAQTTPL------ NADP_Ecoli LRVSSSEGVTVGPVLMGVAKPVHVLTPIASVRRIVNMVALAVVEAQTQPL------ Mdh_Pasteurella LQGTATP-ITVGPILMGMNKPVHILTSASSVRRIINMVAIAAANVEPTCK------ NAD_Brucella VKAVTDA-LHVGPILLGATRPAHILTPSVTSRGVVNMTALAVVEALQKNASNRRQN NAD_Ecoli --------------------------------------------------------
Abbildung 34: Alignment der Aminosäuresequenz verschiedener malic enzymes. NADP- Salmonella enterica subsp. enterica serovar Typhi (Parkhill et al., 2001), Mdh Pasteurella multocida (May et al., 2001), NAD-Brucella melitensis (DelVecchio et al., 2002), NADP-E.coli (Blattner et al., 1997).
5.4 Konstruktion eines Expressionsvektors mit heterologer Expression
Aus der Sequenz des amplifizierten Fragmentes (Abbildung 32) wurden Primer mit
integrierten Restriktionsschnittstellen und ein Codon für die ribosomale Bindungsstelle
konstruiert. Nach der Amplifikation des malic enzymes aus dem rekombinanten pUC18
wurde das PCR-Fragment in den rekombinanten recPhe-pKK-223-3-Expressionsvektor hinter
der PheDH-Sequenz an der PstI und HindIII-Restriktionsschnittstellen kloniert (Abbildung 35
und Abbildung 37).
Abbildung 35: Konstruktion des Plasmids für eine heterologe Expression zur L-Phe Synthese mittels Ganzzellumsetzung.
RBS ATG PheDH TAG RBS ATG ME TAA
PstI HindIII
Ptac
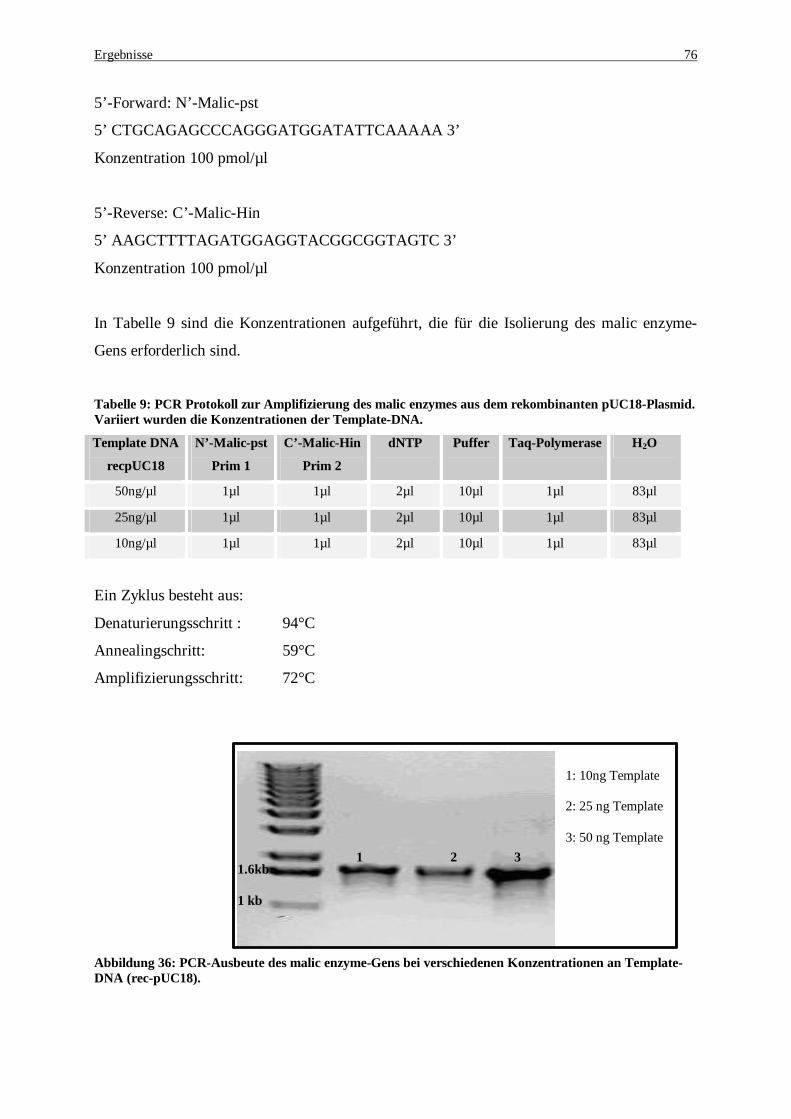
Ergebnisse
76
5’-Forward: N’-Malic-pst
5’ CTGCAGAGCCCAGGGATGGATATTCAAAAA 3’
Konzentration 100 pmol/µl
5’-Reverse: C’-Malic-Hin
5’ AAGCTTTTAGATGGAGGTACGGCGGTAGTC 3’
Konzentration 100 pmol/µl
In Tabelle 9 sind die Konzentrationen aufgeführt, die für die Isolierung des malic enzyme-
Gens erforderlich sind.
Tabelle 9: PCR Protokoll zur Amplifizierung des malic enzymes aus dem rekombinanten pUC18-Plasmid. Variiert wurden die Konzentrationen der Template-DNA.
Template DNA
recpUC18
N’-Malic-pst
Prim 1
C’-Malic-Hin
Prim 2
dNTP Puffer Taq-Polymerase H2O
50ng/µl 1µl 1µl 2µl 10µl 1µl 83µl
25ng/µl 1µl 1µl 2µl 10µl 1µl 83µl
10ng/µl 1µl 1µl 2µl 10µl 1µl 83µl
Ein Zyklus besteht aus:
Denaturierungsschritt : 94°C
Annealingschritt: 59°C
Amplifizierungsschritt: 72°C
Abbildung 36: PCR-Ausbeute des malic enzyme-Gens bei verschiedenen Konzentrationen an Template-DNA (rec-pUC18).
1 2 3 1.6kb 1 kb
1: 10ng Template 2: 25 ng Template 3: 50 ng Template

Ergebnisse
77
Abbildung 37: Vektorkarte des rec-pKK-223-3 PheDH-malic. malic enzyme wurde 3’ zur PheDH an der Pst1- und HindIII-Schnittstelle mit eigener Ribosomenbindungsstelle kloniert.
Das neue Konstrukt des rekombinanten Plasmids wurde in kompetente E. coli-Zellen JM 105
bzw. HB 101 transformiert.
Mit der Klonierung des malic enzyme 3’ zur PheDH an der Pst1- und HindIII-Schnittstelle
mit eigener ribosomalen Bindungsstelle konnte eine Expression beider Enzyme gleichzeitig
erfolgen.
5.4.1 Coexpression der PheDH und des malic enzymes
Von den positiven Klonen wurden einige zur Expression ausgewählt. Eine Einzelkolonie der
jeweiligen Klone wurde in 5 ml LBamp –Medium überimpft, und nach Erreichen von OD580 =
0,6 mit 1 mM IPTG induziert. Die Induktion erfolgte über Nacht und die geernteten Zellen
wurden mit Ultraschall aufgeschlossen.
pstI
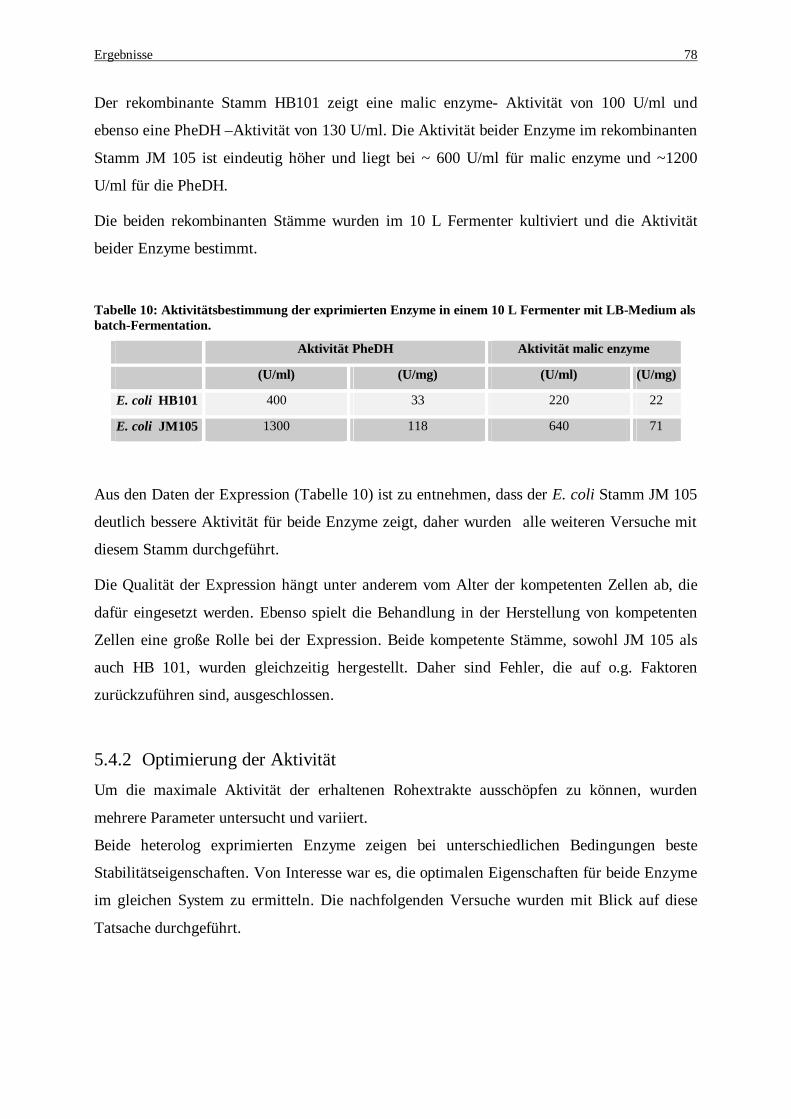
Ergebnisse
78
Der rekombinante Stamm HB101 zeigt eine malic enzyme- Aktivität von 100 U/ml und
ebenso eine PheDH –Aktivität von 130 U/ml. Die Aktivität beider Enzyme im rekombinanten
Stamm JM 105 ist eindeutig höher und liegt bei ~ 600 U/ml für malic enzyme und ~1200
U/ml für die PheDH.
Die beiden rekombinanten Stämme wurden im 10 L Fermenter kultiviert und die Aktivität
beider Enzyme bestimmt.
Tabelle 10: Aktivitätsbestimmung der exprimierten Enzyme in einem 10 L Fermenter mit LB-Medium als batch-Fermentation.
Aktivität PheDH Aktivität malic enzyme
(U/ml) (U/mg) (U/ml) (U/mg)
E. coli HB101 400 33 220 22
E. coli JM105 1300 118 640 71
Aus den Daten der Expression (Tabelle 10) ist zu entnehmen, dass der E. coli Stamm JM 105
deutlich bessere Aktivität für beide Enzyme zeigt, daher wurden alle weiteren Versuche mit
diesem Stamm durchgeführt.
Die Qualität der Expression hängt unter anderem vom Alter der kompetenten Zellen ab, die
dafür eingesetzt werden. Ebenso spielt die Behandlung in der Herstellung von kompetenten
Zellen eine große Rolle bei der Expression. Beide kompetente Stämme, sowohl JM 105 als
auch HB 101, wurden gleichzeitig hergestellt. Daher sind Fehler, die auf o.g. Faktoren
zurückzuführen sind, ausgeschlossen.
5.4.2 Optimierung der Aktivität
Um die maximale Aktivität der erhaltenen Rohextrakte ausschöpfen zu können, wurden
mehrere Parameter untersucht und variiert.
Beide heterolog exprimierten Enzyme zeigen bei unterschiedlichen Bedingungen beste
Stabilitätseigenschaften. Von Interesse war es, die optimalen Eigenschaften für beide Enzyme
im gleichen System zu ermitteln. Die nachfolgenden Versuche wurden mit Blick auf diese
Tatsache durchgeführt.
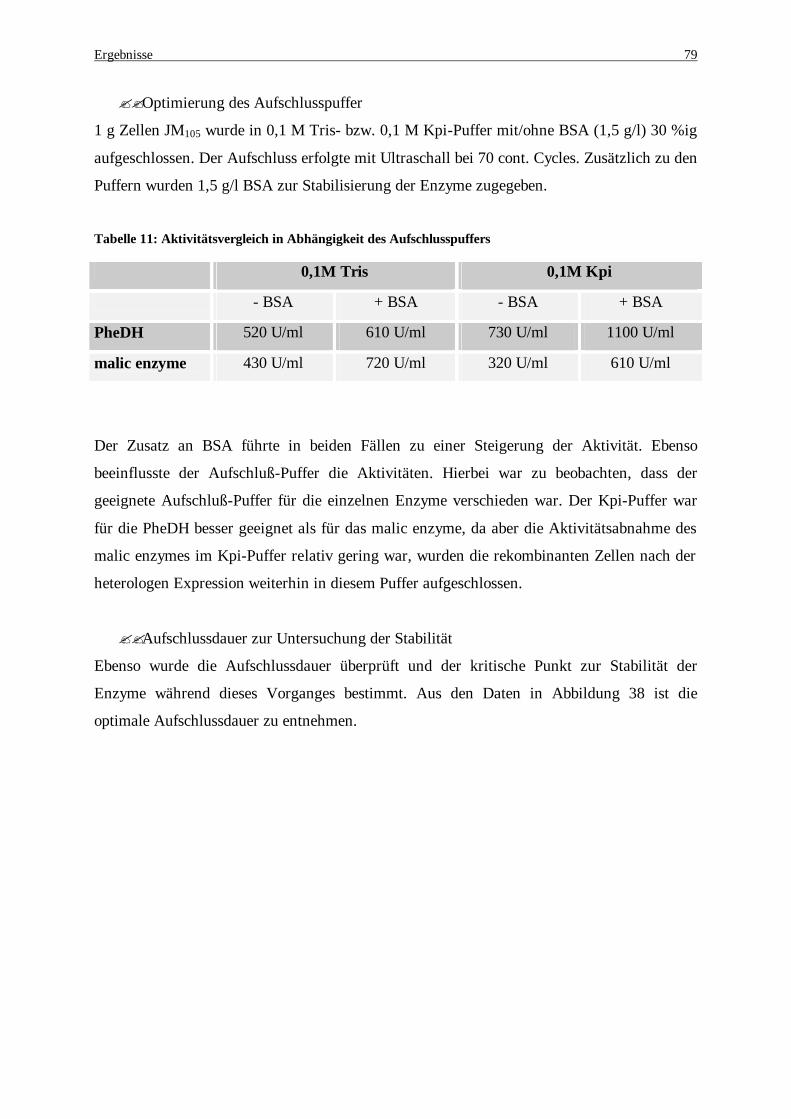
Ergebnisse
79
??Optimierung des Aufschlusspuffer
1 g Zellen JM105 wurde in 0,1 M Tris- bzw. 0,1 M Kpi-Puffer mit/ohne BSA (1,5 g/l) 30 %ig
aufgeschlossen. Der Aufschluss erfolgte mit Ultraschall bei 70 cont. Cycles. Zusätzlich zu den
Puffern wurden 1,5 g/l BSA zur Stabilisierung der Enzyme zugegeben.
Tabelle 11: Aktivitätsvergleich in Abhängigkeit des Aufschlusspuffers
0,1M Tris 0,1M Kpi
- BSA + BSA - BSA + BSA
PheDH 520 U/ml 610 U/ml 730 U/ml 1100 U/ml
malic enzyme 430 U/ml 720 U/ml 320 U/ml 610 U/ml
Der Zusatz an BSA führte in beiden Fällen zu einer Steigerung der Aktivität. Ebenso
beeinflusste der Aufschluß-Puffer die Aktivitäten. Hierbei war zu beobachten, dass der
geeignete Aufschluß-Puffer für die einzelnen Enzyme verschieden war. Der Kpi-Puffer war
für die PheDH besser geeignet als für das malic enzyme, da aber die Aktivitätsabnahme des
malic enzymes im Kpi-Puffer relativ gering war, wurden die rekombinanten Zellen nach der
heterologen Expression weiterhin in diesem Puffer aufgeschlossen.
??Aufschlussdauer zur Untersuchung der Stabilität
Ebenso wurde die Aufschlussdauer überprüft und der kritische Punkt zur Stabilität der
Enzyme während dieses Vorganges bestimmt. Aus den Daten in Abbildung 38 ist die
optimale Aufschlussdauer zu entnehmen.
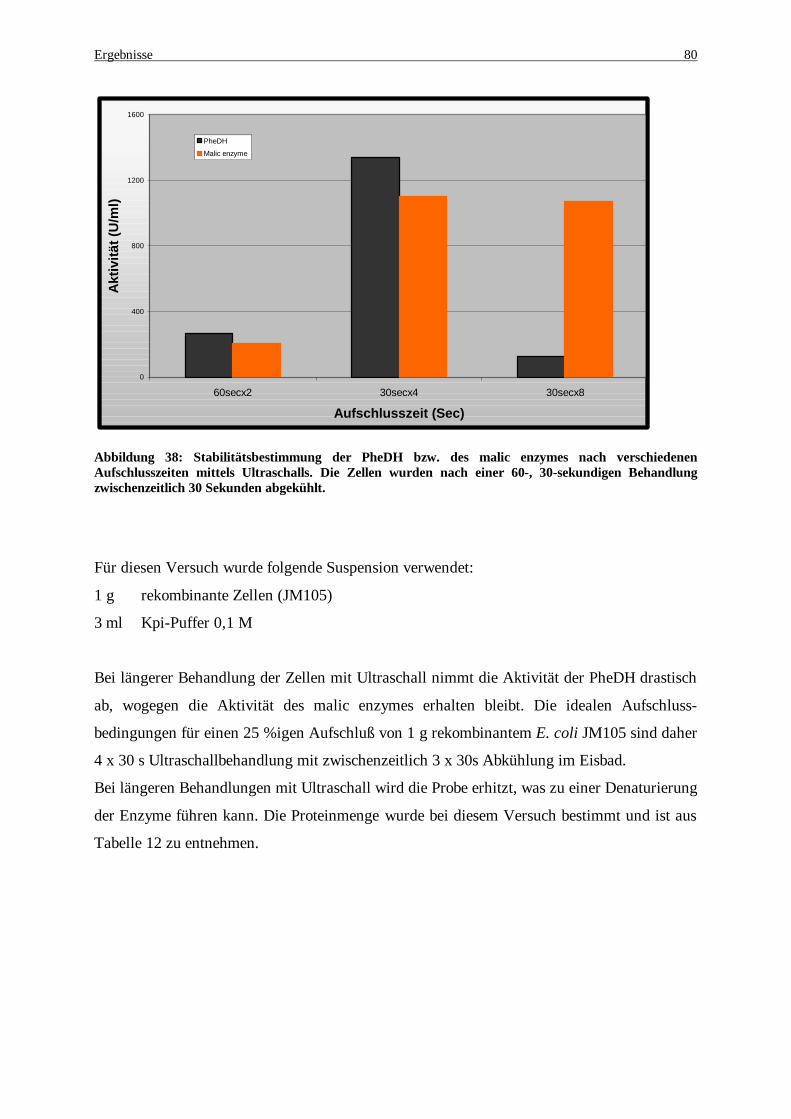
Ergebnisse
80
0
400
800
1200
1600
60secx2 30secx4 30secx8
Aufschlusszeit (Sec)
Akt
ivitä
t (U
/ml)
PheDH
Malic enzyme
Abbildung 38: Stabilitätsbestimmung der PheDH bzw. des malic enzymes nach verschiedenen Aufschlusszeiten mittels Ultraschalls. Die Zellen wurden nach einer 60-, 30-sekundigen Behandlung zwischenzeitlich 30 Sekunden abgekühlt.
Für diesen Versuch wurde folgende Suspension verwendet:
1 g rekombinante Zellen (JM105)
3 ml Kpi-Puffer 0,1 M
Bei längerer Behandlung der Zellen mit Ultraschall nimmt die Aktivität der PheDH drastisch
ab, wogegen die Aktivität des malic enzymes erhalten bleibt. Die idealen Aufschluss-
bedingungen für einen 25 %igen Aufschluß von 1 g rekombinantem E. coli JM105 sind daher
4 x 30 s Ultraschallbehandlung mit zwischenzeitlich 3 x 30s Abkühlung im Eisbad.
Bei längeren Behandlungen mit Ultraschall wird die Probe erhitzt, was zu einer Denaturierung
der Enzyme führen kann. Die Proteinmenge wurde bei diesem Versuch bestimmt und ist aus
Tabelle 12 zu entnehmen.
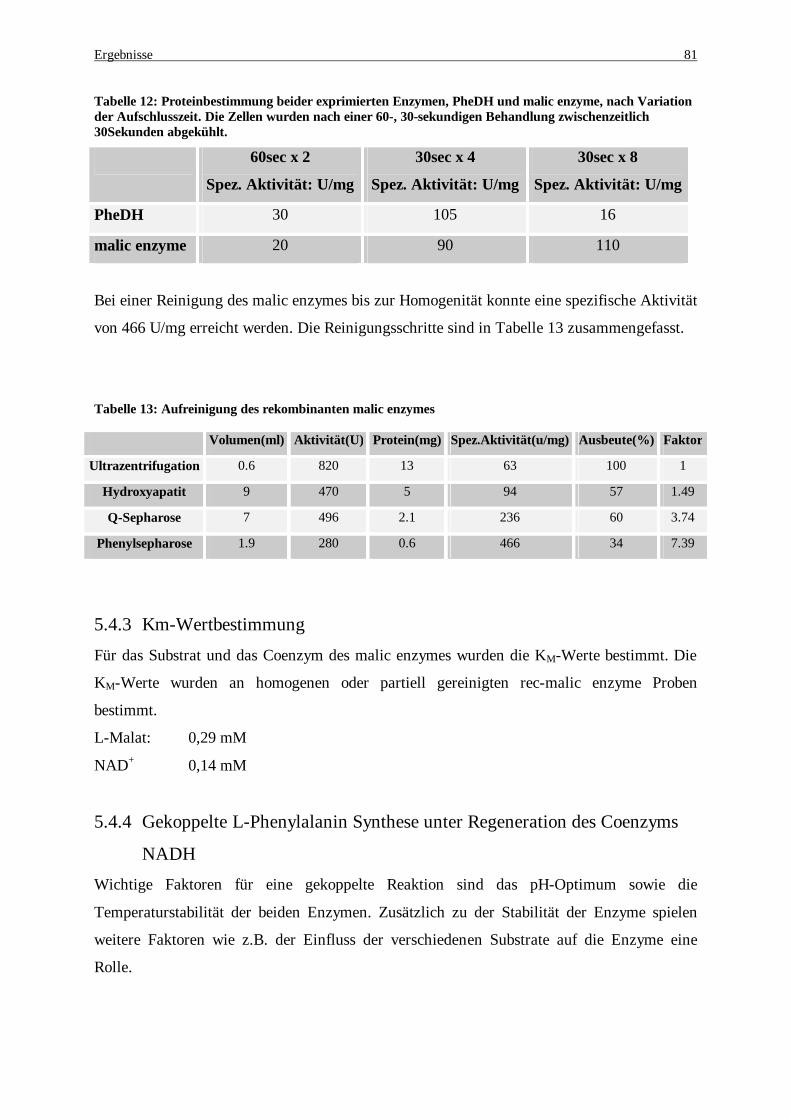
Ergebnisse
81
Tabelle 12: Proteinbestimmung beider exprimierten Enzymen, PheDH und malic enzyme, nach Variation der Aufschlusszeit. Die Zellen wurden nach einer 60-, 30-sekundigen Behandlung zwischenzeitlich 30Sekunden abgekühlt.
60sec x 2
Spez. Aktivität: U/mg
30sec x 4
Spez. Aktivität: U/mg
30sec x 8
Spez. Aktivität: U/mg
PheDH 30 105 16
malic enzyme 20 90 110
Bei einer Reinigung des malic enzymes bis zur Homogenität konnte eine spezifische Aktivität
von 466 U/mg erreicht werden. Die Reinigungsschritte sind in Tabelle 13 zusammengefasst.
Tabelle 13: Aufreinigung des rekombinanten malic enzymes
Volumen(ml) Aktivität(U) Protein(mg) Spez.Aktivität(u/mg) Ausbeute(%) Faktor
Ultrazentrifugation 0.6 820 13 63 100 1
Hydroxyapatit 9 470 5 94 57 1.49
Q-Sepharose 7 496 2.1 236 60 3.74
Phenylsepharose 1.9 280 0.6 466 34 7.39
5.4.3 Km-Wertbestimmung
Für das Substrat und das Coenzym des malic enzymes wurden die KM-Werte bestimmt. Die
KM-Werte wurden an homogenen oder partiell gereinigten rec-malic enzyme Proben
bestimmt.
L-Malat: 0,29 mM
NAD+ 0,14 mM
5.4.4 Gekoppelte L-Phenylalanin Synthese unter Regeneration des Coenzyms
NADH
Wichtige Faktoren für eine gekoppelte Reaktion sind das pH-Optimum sowie die
Temperaturstabilität der beiden Enzymen. Zusätzlich zu der Stabilität der Enzyme spielen
weitere Faktoren wie z.B. der Einfluss der verschiedenen Substrate auf die Enzyme eine
Rolle.
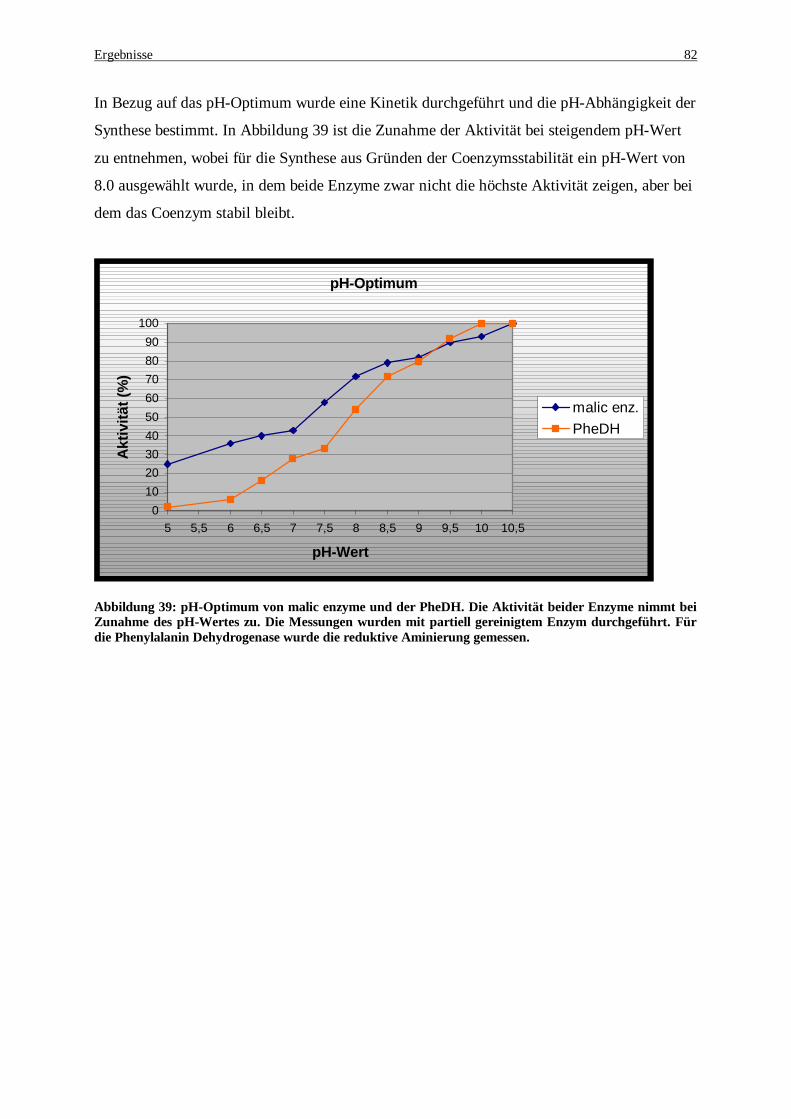
Ergebnisse
82
In Bezug auf das pH-Optimum wurde eine Kinetik durchgeführt und die pH-Abhängigkeit der
Synthese bestimmt. In Abbildung 39 ist die Zunahme der Aktivität bei steigendem pH-Wert
zu entnehmen, wobei für die Synthese aus Gründen der Coenzymsstabilität ein pH-Wert von
8.0 ausgewählt wurde, in dem beide Enzyme zwar nicht die höchste Aktivität zeigen, aber bei
dem das Coenzym stabil bleibt.
pH-Optimum
0102030405060708090
100
5 5,5 6 6,5 7 7,5 8 8,5 9 9,5 10 10,5
pH-Wert
Akt
ivitä
t (%
)
malic enz.PheDH
Abbildung 39: pH-Optimum von malic enzyme und der PheDH. Die Aktivität beider Enzyme nimmt bei Zunahme des pH-Wertes zu. Die Messungen wurden mit partiell gereinigtem Enzym durchgeführt. Für die Phenylalanin Dehydrogenase wurde die reduktive Aminierung gemessen.
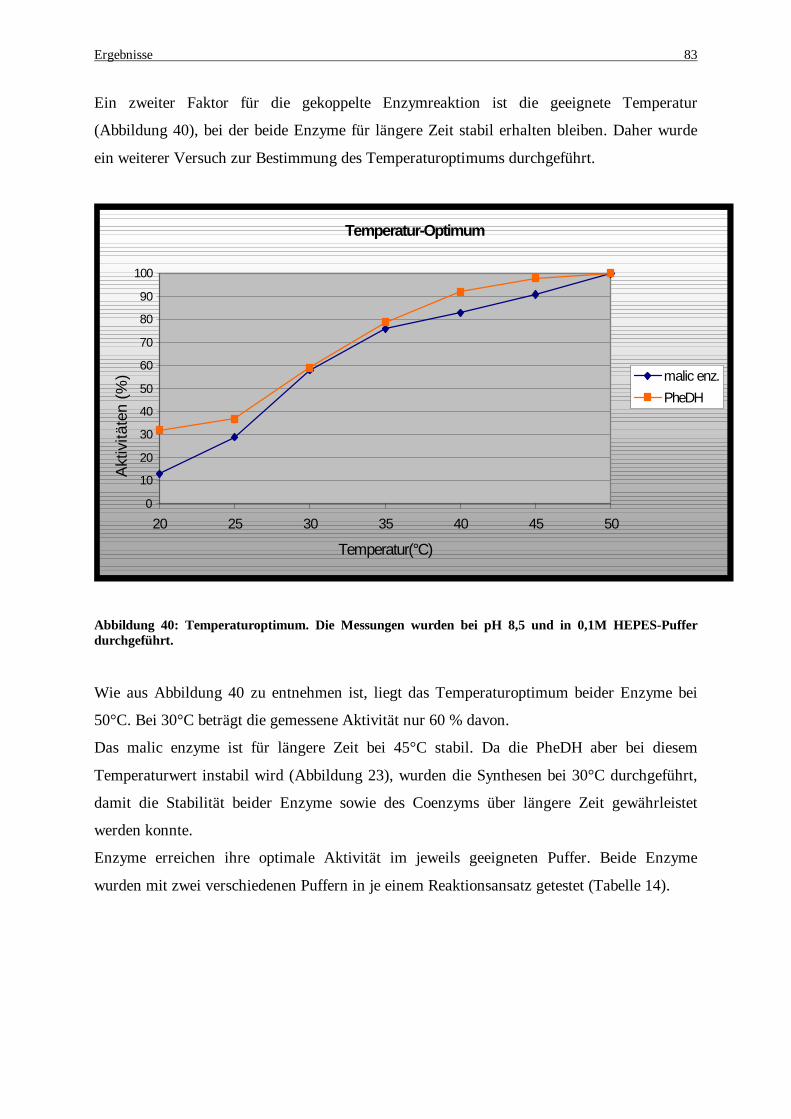
Ergebnisse
83
Ein zweiter Faktor für die gekoppelte Enzymreaktion ist die geeignete Temperatur
(Abbildung 40), bei der beide Enzyme für längere Zeit stabil erhalten bleiben. Daher wurde
ein weiterer Versuch zur Bestimmung des Temperaturoptimums durchgeführt.
Temperatur-Optimum
0
10
20
30
40
50
60
70
80
90
100
20 25 30 35 40 45 50
Temperatur(°C)
Akt
ivitä
ten
(%) malic enz.
PheDH
Abbildung 40: Temperaturoptimum. Die Messungen wurden bei pH 8,5 und in 0,1M HEPES-Puffer durchgeführt.
Wie aus Abbildung 40 zu entnehmen ist, liegt das Temperaturoptimum beider Enzyme bei
50°C. Bei 30°C beträgt die gemessene Aktivität nur 60 % davon.
Das malic enzyme ist für längere Zeit bei 45°C stabil. Da die PheDH aber bei diesem
Temperaturwert instabil wird (Abbildung 23), wurden die Synthesen bei 30°C durchgeführt,
damit die Stabilität beider Enzyme sowie des Coenzyms über längere Zeit gewährleistet
werden konnte.
Enzyme erreichen ihre optimale Aktivität im jeweils geeigneten Puffer. Beide Enzyme
wurden mit zwei verschiedenen Puffern in je einem Reaktionsansatz getestet (Tabelle 14).
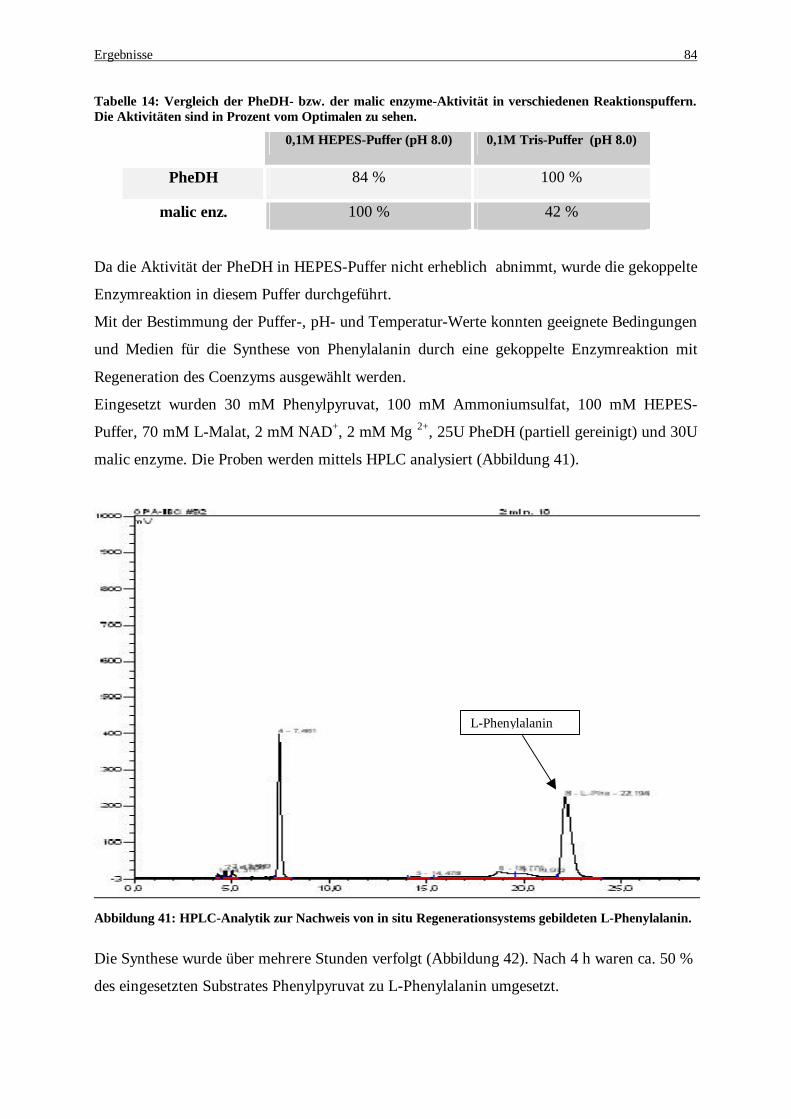
Ergebnisse
84
Tabelle 14: Vergleich der PheDH- bzw. der malic enzyme-Aktivität in verschiedenen Reaktionspuffern. Die Aktivitäten sind in Prozent vom Optimalen zu sehen.
0,1M HEPES-Puffer (pH 8.0) 0,1M Tris-Puffer (pH 8.0)
PheDH 84 % 100 %
malic enz. 100 % 42 %
Da die Aktivität der PheDH in HEPES-Puffer nicht erheblich abnimmt, wurde die gekoppelte
Enzymreaktion in diesem Puffer durchgeführt.
Mit der Bestimmung der Puffer-, pH- und Temperatur-Werte konnten geeignete Bedingungen
und Medien für die Synthese von Phenylalanin durch eine gekoppelte Enzymreaktion mit
Regeneration des Coenzyms ausgewählt werden.
Eingesetzt wurden 30 mM Phenylpyruvat, 100 mM Ammoniumsulfat, 100 mM HEPES-
Puffer, 70 mM L-Malat, 2 mM NAD+, 2 mM Mg 2+, 25U PheDH (partiell gereinigt) und 30U
malic enzyme. Die Proben werden mittels HPLC analysiert (Abbildung 41).
Abbildung 41: HPLC-Analytik zur Nachweis von in situ Regenerationsystems gebildeten L-Phenylalanin.
Die Synthese wurde über mehrere Stunden verfolgt (Abbildung 42). Nach 4 h waren ca. 50 %
des eingesetzten Substrates Phenylpyruvat zu L-Phenylalanin umgesetzt.
L-Phenylalanin
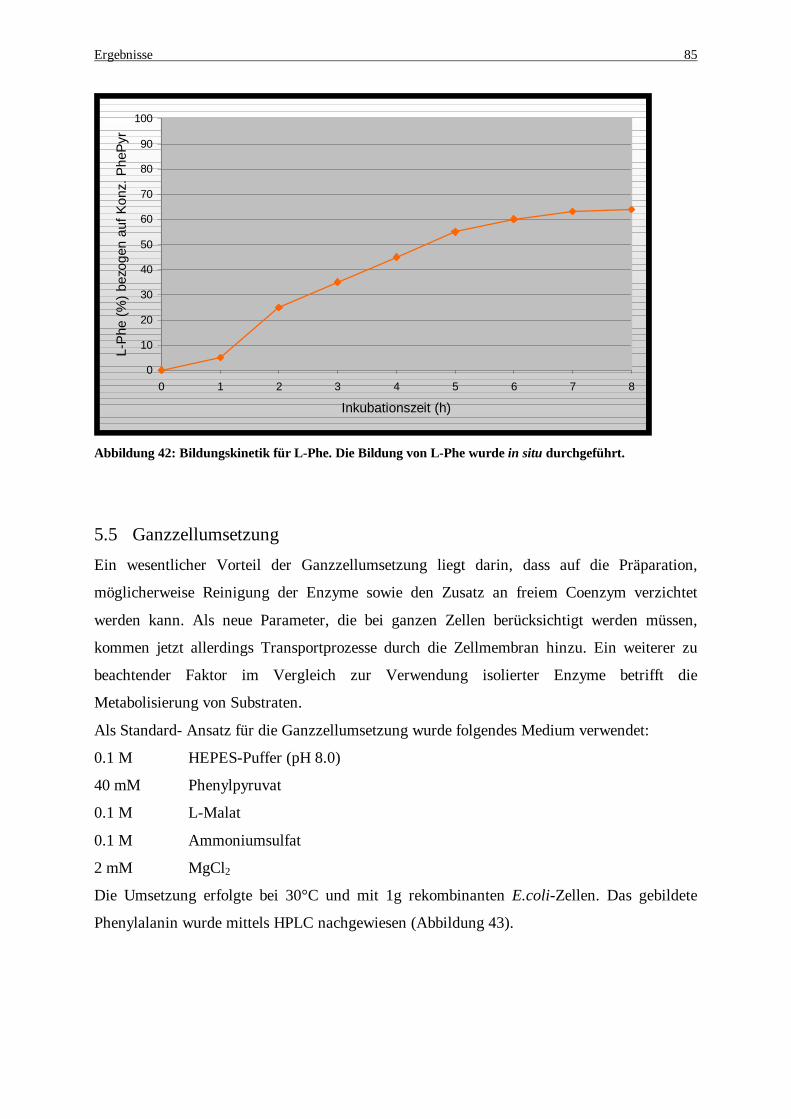
Ergebnisse
85
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7 8
Inkubationszeit (h)
L-P
he (
%)
bezo
gen
auf K
onz.
Phe
Pyr
Abbildung 42: Bildungskinetik für L-Phe. Die Bildung von L-Phe wurde in situ durchgeführt.
5.5 Ganzzellumsetzung
Ein wesentlicher Vorteil der Ganzzellumsetzung liegt darin, dass auf die Präparation,
möglicherweise Reinigung der Enzyme sowie den Zusatz an freiem Coenzym verzichtet
werden kann. Als neue Parameter, die bei ganzen Zellen berücksichtigt werden müssen,
kommen jetzt allerdings Transportprozesse durch die Zellmembran hinzu. Ein weiterer zu
beachtender Faktor im Vergleich zur Verwendung isolierter Enzyme betrifft die
Metabolisierung von Substraten.
Als Standard- Ansatz für die Ganzzellumsetzung wurde folgendes Medium verwendet:
0.1 M HEPES-Puffer (pH 8.0)
40 mM Phenylpyruvat
0.1 M L-Malat
0.1 M Ammoniumsulfat
2 mM MgCl2
Die Umsetzung erfolgte bei 30°C und mit 1g rekombinanten E.coli-Zellen. Das gebildete
Phenylalanin wurde mittels HPLC nachgewiesen (Abbildung 43).
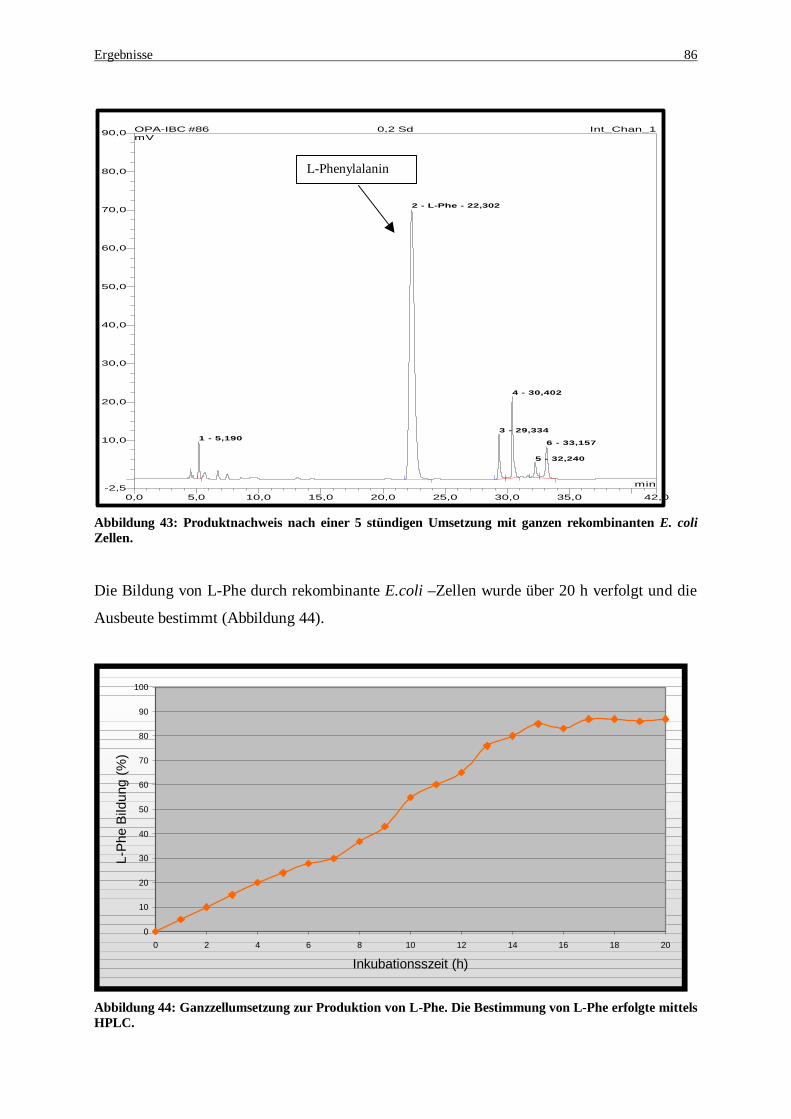
Ergebnisse
86
-2,5
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
0,0 5,0 10,0 15,0 20,0 25,0 30,0 35,0 42,0
mV
min
OPA-IBC #86 0,2 Sd Int_Chan_1
1 - 5,190
2 - L-Phe - 22,302
3 - 29,334
4 - 30,402
5 - 32,240
6 - 33,157
Abbildung 43: Produktnachweis nach einer 5 stündigen Umsetzung mit ganzen rekombinanten E. coli Zellen.
Die Bildung von L-Phe durch rekombinante E.coli –Zellen wurde über 20 h verfolgt und die
Ausbeute bestimmt (Abbildung 44).
0
10
20
30
40
50
60
70
80
90
100
0 2 4 6 8 10 12 14 16 18 20
Inkubationsszeit (h)
L-P
he B
ildun
g (%
)
Abbildung 44: Ganzzellumsetzung zur Produktion von L-Phe. Die Bestimmung von L-Phe erfolgte mittels HPLC.
L-Phenylalanin

Ergebnisse
87
Eine Metabolisierung des entstandenen Produktes L-Phe konnte nach 20h Inkubation nicht
nachgewiesen werden.
5.6 Gezielte Mutagenese
Im Rahmen der Dissertation sollten weiterhin PheDH- Mutanten erzeugt werden, die ein
deutlich verändertes Substratspektrum als das der PheDH aufweisen.
Als Ausgangsgen für eine Mutagenese zur Amin- bzw. Lactat- Dehydrogenase wurde das
Phenylalanin Dehydrogenase - Gen aus den folgenden Gründen ausgewählt:
??Gen ist verfügbar
??gutes Expressionsystem verfügbar
??Enzym zeigt eine hohe Aktivität (>2000 U/ml, 250 U/mg)
??Raumstruktur bekannt
??breites Substratspektrum
??Ergebnisse übertragbar auf LeuDH, AlaDH, GluDH
In Abbildung 45 ist der Reaktionsmechanismus der PheDH katalysierten reduktiven
Aminierung von Phenylpyruvat dargestellt.
COOH
O
NH4+
PheDH
NADH +
COOH
NH2
H2O
NAD+Phenylpyruvat L-Phenylalanin
Abbildung 45: Reaktionsmechanismus der reduktiven Aminierung von Phenylalanin mittels PheDH.
Für die Durchführung einer gezielten Mutagenese wurde die räumliche Struktur der PheDH
mittels Computereinsatz modelliert und die Position des Substrates bestimmt (Abbildung 3).
Ziel ist die Erweiterung des Substratspektrums durch den Austausch von Lysin 66 durch
weitere Aminosäuren, die sowohl in Größe, Hydrophobizität als auch Ladung variieren.
Durch eine gezielte Mutagenese von Lysin 66 gegen Isoleucin bzw. Arginin sowie Lysin 78
gegen Isoleucin wurde versucht, der Phenylalanin Dehydrogenase neue Aktivitäten auf
weitere Substrate (Tabelle 15) zu verleihen. Die Lysinreste an Position 66 und 78 sind im

Ergebnisse
88
aktiven Kanal der PheDH lokalisiert. Interessant ist dieses, da Lysin 66 die Bindungsstelle für
die Carboxylgruppe und Lysin 78 für die Ketogruppe im Substrat sind (Vanhooke et al.,
1999). Für die Umsetzung ähnlicher Substrate, welche aber eine Methylgruppe anstatt
Carboxylgruppe besitzen, könnte der Austausch des basischen Lysins gegen eine neutrale
Aminosäure wie z.B. Isoleucin zum Erfolg führen.
Tabelle 15: Möglichkeiten zur Substratumsetzung nach einer Mutagenese. Durch den Austausch des Lysins in Isoleucin (neutrale Aminosäure) könnte die Methylgruppe des Ketons akzeptiert werden und somit das Keton zum Amin umsetzen. Es wird eine direkte Hydrierung der Ketosäure zur Hydroxysäure bei einer Mutagense des Lysin 66 in Arginin erwartet.
Substrat Produkt
Keton = Substrat
CH3
O
Reduktive Aminierung des Phenylacetons
Amin
CH3
NH2
Ketosäure = Substrat
COOH
O
Reduktion der Ketosäure Phenylpyruvat
Hydroxysäure
COOH
OH
5.6.1 Mutation des Lysin 66
Um eine veränderte Phenylalanin Dehydrogenase herstellen zu können, musste die Sequenz
auf DNA-Ebene verändert werden. Die gezielte Mutagenese erfolgte nach dem Prinzip der
überlappenden PCR (Ho et al., 1989). Hierbei wurden Primer verwendet, die zwei DNA-
Fragmente mit überlappenden Enden erzeugten. In einer zweiten PCR hybridisierten diese
Fragmente, wobei das überlappende 3’-Ende jedes Stranges als Primer für die Synthese des
Gegenstranges diente (Abbildung 46). Die Mutationen, die in die Oligonucleotid-Primer
eingeführt wurden, fanden sich zu 100 % im Produkt wieder. Das resultierende Fusions-
produkt wurde durch weitere PCR mit den „äußeren“ Primern, die den gesamten Bereich
einschließen, amplifiziert.

Ergebnisse
89
5'
5'
5'Templat-DNA
*
* 5'P1'
K66
P1P2
P2'
2.Produkt
1.Produkt ** 5'
5'PCR mit den PrimernP2+P2'
PCR mit denPrimern P1+P1'
PCR mit denPrimern P1+P2'und Produkt 1+2als Template
* 5'
5'Fusionsprodukt
Abbildung 46: Schema zur Mutagenese mittels PCR nach der Methode der „overlapping extension“, ausgetauscht wurde das Lysin 66 gegen Isoleucin.
Mittels dieser Technologie wurde die Aminosäure Lysin 66 gegen Isoleucin ausgetauscht.
Das erhaltene mutierte DNA-Fragment wurde „blunt ends“ in den pKK-223-3 an der SmaI-
Schnittstelle kloniert (Abbildung 47). Die Auswahl dieses Plasmids und die Klonierung an der
SmaI- Schnittstelle, die etwa 13 bp von der Ribosomenbindungsstelle entfernt ist, wurde zur
Vermeidung von inclusion bodies gewählt, da bei Klonierungen der PheDH in anderen
Plasmiden solche aufgetreten sind (5.1.5.3).
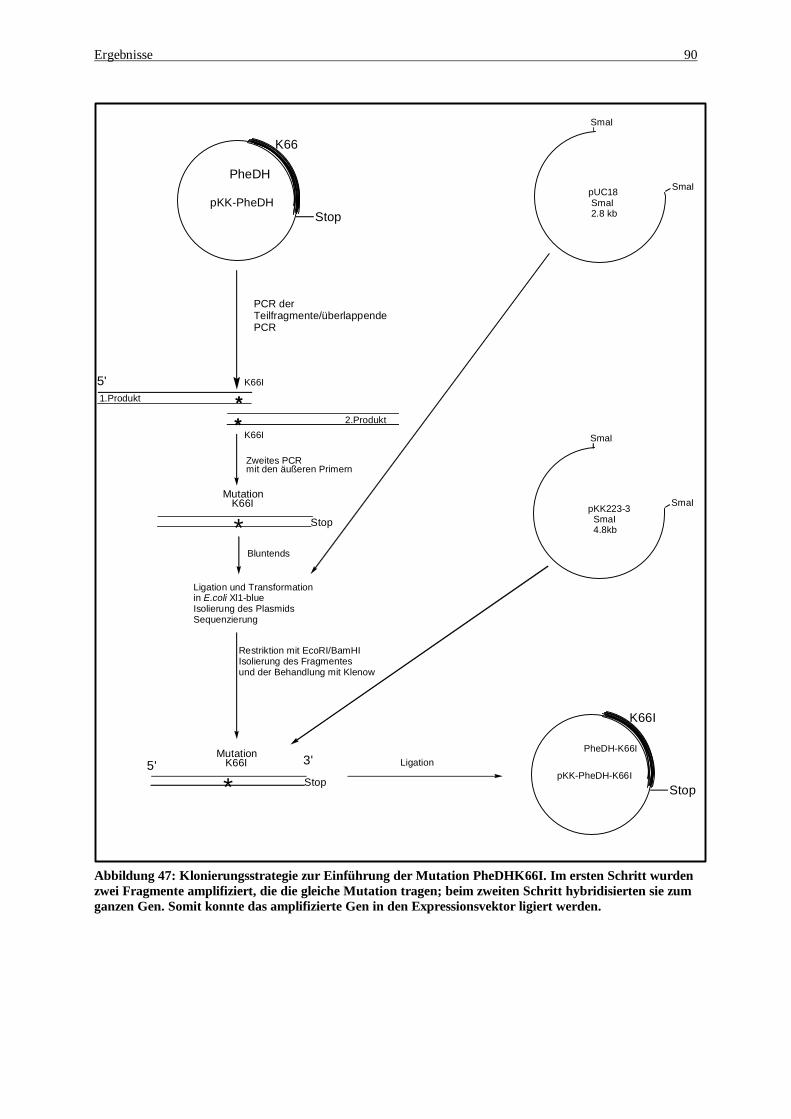
Ergebnisse
90
pKK-PheDHStop
K66
PheDH
PCR der Teilfragmente/überlappendePCR
* Mutation K66I
Stop
SmaI
SmaIpUC18 SmaI 2.8 kb
Ligation und Transformationin E.coli Xl1-blueIsolierung des PlasmidsSequenzierung
SmaI
SmaIpKK223-3 SmaI 4.8kb
Mutation K66I
Stop*Ligation
pKK-PheDH-K66I
Stop
K66I
PheDH-K66I
2.Produkt
1.Produkt **
5' K66I
K66I
Zweites PCR mit den äußeren Primern
Bluntends
Restriktion mit EcoRI/BamHIIsolierung des Fragmentesund der Behandlung mit Klenow
5' 3'
Abbildung 47: Klonierungsstrategie zur Einführung der Mutation PheDHK66I. Im ersten Schritt wurden zwei Fragmente amplifiziert, die die gleiche Mutation tragen; beim zweiten Schritt hybridisierten sie zum ganzen Gen. Somit konnte das amplifizierte Gen in den Expressionsvektor ligiert werden.

Ergebnisse
91
Auswahl der Primer
Dass Codons, die für die gleiche Aminosäure codieren, in den einzelnen Spezies
unterschiedlich häufig verwendet werden, liegt an der unterschiedlichen Basenzusammen-
setzung in den DNAs verschiedener Organismen (Sharp et al., 1988). Selbst innerhalb eines
Organismus existieren zum Teil beträchtliche Unterschiede zwischen der Codonnutzung in
Genen, die stark exprimiert werden, und solchen, die weniger häufig exprimiert werden.
Diese Unterschiede können zur Genregulierung genutzt werden. Es gibt z.B. für selten
genutzte Codons in der E. coli-Zelle auch nur geringere Mengen der passenden tRNA. Das
hat zur Folge, dass mRNA von Genen mit vielen „seltenen“ Codons auch verzögert
translatiert wird (Grantham et al., 1981). Dies ist bei mutierten Proteinen, die überexprimiert
werden sollen, nicht erwünscht.
Die ausgesuchten Codons wurden hinsichtlich dieses Kriteriums, wie oft dieses Codon in der
Wt-PheDH genutzt wird, überprüft.
Für den Austausch von Lysin in Isoleucin wurde das Codon ATT unter Berücksichtigung der
Codon-Usage von E. coli verwendet.
Mutagenese-Primer
P1': GCT CAC TGC CAT AAT CAA CGT CAT CGC CCC
P2 : GGG GCG ATG ACG TTG ATT ATG GCA GTG AGC
Äußere Primer
P1 : ATC AAG GGG TAC ATC ATG AGT ATC GAC AGC
P2': TTA CTA GGC AGT CGC TGT CGT TGT CGA GGC
Bei der Herstellung der beiden Teilfragmente der PheDH, die die gewünschten Mutationen
tragen, bestand der Reaktionsansatz aus:
10 mM Tris pH 8.3
50 mM KCl
1.5 mM MgCl2
jeweils 200 µM dATP, dCTP, dTTP, dGTP
20-40 pmol jedes Oligonucleotid-Primers
2.5 U Taq-Polymerase pro 100 µl Ansatz

Ergebnisse
92
10 ng Plasmid-DNA als PCR-Template (pKK-223-PheDH)
Die PCR Amplifikation wurde auf einem automatischen DNA-Thermal-Cycler (Robocycler,
Fa. Stratagene) nach folgendem Programm durchgeführt:
??3 min Denaturierung bei 94°C (zu Anfang des Programms)
??1 min Denaturierung bei 94°C
??1 min Annealing der Primer bei 65°C
??1 min Extension (Verlängerung der Primer durch die Taq-Polymerase) bei 72°C
??zyklisches Wiederholen der letzten drei Schritte (25 x)
??5 min Extension (nach Abschluss der Zyklen, um eine vollständige Verlängerung
eventuell vorher abgebrochener DNA-Produkte zu gewährleisten)
Nach der PCR wurden die erhaltenen Fragmente gelelektrophoretisch aufgetrennt und isoliert
(Abbildung 48). Die zweite PCR zur Überlappung der Fragmente enthielt equimolare Mengen
beider Fragmente, wobei von dem kürzeren ca. 60ng eingesetzt wurden (die Menge des
größeren Fragmentes wurde dementsprechend berechnet). Die Annealing- Temperatur richtete
sich nach der Schmelztemperatur der resultierenden Überlappungsregion. Somit konnte ein
großes Fragment von 1,1 kb amplifiziert werden (Abbildung 49).
Alle hergestellten Muteine wurden sequenziert und die Mutationen bestimmt. Die gezielte
Mutagenese nach Ho et al. (Ho et al., 1989) führte in den meisten Fällen sowohl zu niedriger
Ausbeute des gewünschten Produktes als auch zu unerwünschten Mutationen. Nach einer
Optimierung des PCR-Ansatzes in Bezug auf das Verhältnis der zu fusionierenden
Fragmenten sowie der dNTP- Konzentrationen konnten sowohl gute Ausbeuten als auch
gewünschte Mutationen erreicht und unerwünschte Mutationen vermieden werden.
Zur Einführung der Mutationen wurde nicht das ganze PheDH-Gen (1068 bp) amplifiziert,
sondern nur ca. 891 bp bzw. 213 bp große Fragmente und dann hybridisiert und amplifiziert.
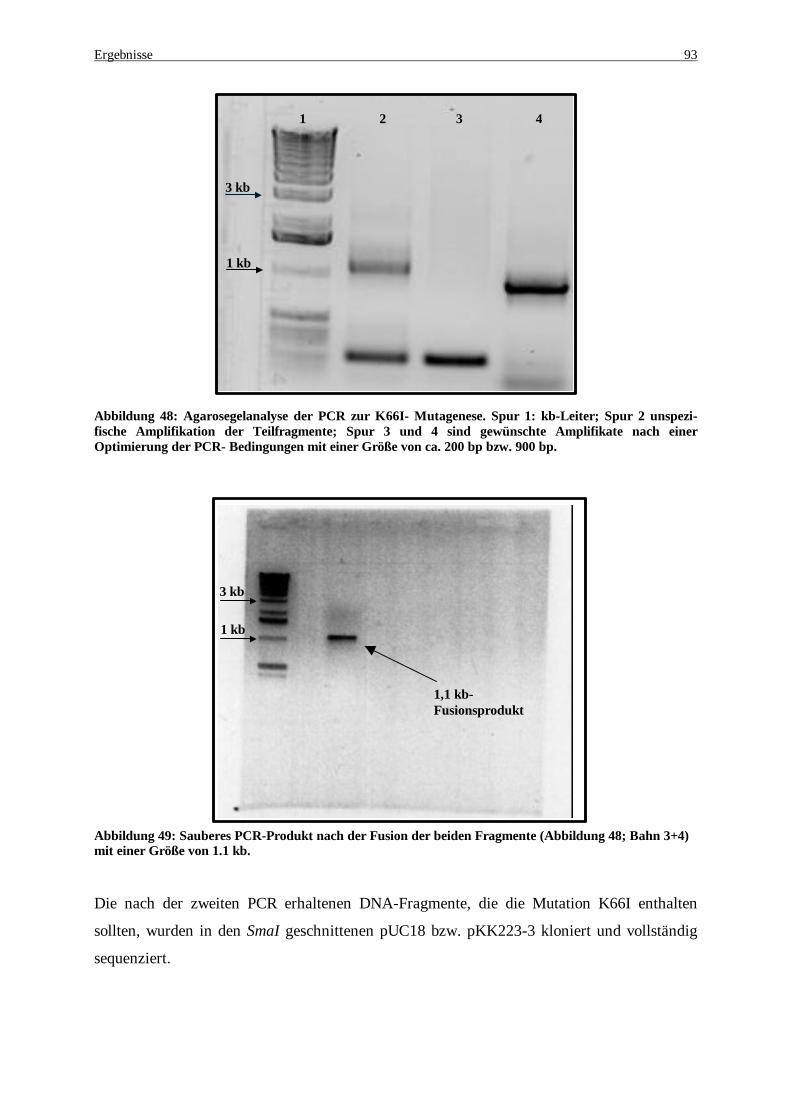
Ergebnisse
93
Abbildung 48: Agarosegelanalyse der PCR zur K66I- Mutagenese. Spur 1: kb-Leiter; Spur 2 unspezi-fische Amplifikation der Teilfragmente; Spur 3 und 4 sind gewünschte Amplifikate nach einer Optimierung der PCR- Bedingungen mit einer Größe von ca. 200 bp bzw. 900 bp.
Abbildung 49: Sauberes PCR-Produkt nach der Fusion der beiden Fragmente (Abbildung 48; Bahn 3+4) mit einer Größe von 1.1 kb.
Die nach der zweiten PCR erhaltenen DNA-Fragmente, die die Mutation K66I enthalten
sollten, wurden in den SmaI geschnittenen pUC18 bzw. pKK223-3 kloniert und vollständig
sequenziert.
3 kb
1 kb
1,1 kb-Fusionsprodukt
1 2 3 4
3 kb
1 kb

Ergebnisse
94
Die rekombinanten Klone wurden mittels Restriktionsanalyse auf das Vorhandensein und die
richtige Orientierung des Inserts geprüft. Mit einem positiven Klon wurde anschließend der
Expressionsstamm JM105 transformiert und das PheDH-Mutein exprimiert.
5.6.2 Sequenzierung der K66I
Das neukonstruierte Mutein wurde sequenziert und den Austausch des Lysin 66 zu Isoleucin
bestimmt.
ATGAGTATCGACAGCGCACTGAACTGGGACGGGGAAATGACGGTCACCCGATTCGACCGGGAGAC
TGGTGCCCATTTCGTCATTCGACTCGATTCGACCCAACTCGGACCGGCGGCCGGAGGCACCAGAGC
CGCACAGTACTCACAGCTGGCGGACGCCCTCACCGACGCCGGCAAATTGGCGGGGGCGATGACGT
TGATTATGGCAGTGAGCAACCTTCCGATGGGCGGGGGCAAATCCGTCATTGCGCTTCCTGCGCCGC
GTCATTCGATCGATCCGAGCACGTGGGCACGCATCCTCCGAATCCACGCCGAGAACATCGACAAGT
TGTCCGGCAACTACTGGACCGGACCGGACGTCAACACCAATTCGGCAGACATGGATACTCTGAACG
ACACCACCGAGTTCGTGTTCGGACGGTCGCTCGAACGCGGCGGCGCGGGTTCGAGCGCGTTCACCA
CCGCCGTTGGCGTGTTCGAGGCGATGAAGGCGACCGTCGCGCACCGTGGGCTGGGCTCACTCGACG
GTTTGACGGTCCTGGTCCAAGGACTGGGGGCAGTCGGAGGATCATTGGCATCCCTGGCCGCCGAAG
CGGGTGCGCAACTCCTGGTGGCAGACACCGACACCGAGCGAGTAGCGCACGCTGTTGCGTTGGGC
CACACAGCGGTTGCCCTCGAGGACGTTCTGTCCACCCCGTGTGATGTCTTCGCACCCTGCGCAATG
GGCGGCGTCATCACCACCGAGGTGGCGCGAACACTCGACTGTTCCGTCGTGGCCGGTGCCGCCAAC
AACGTCATCGCCGACGAGGCCGCCTCGGACATCCTGCACGCACGCGGAATTCTGTACGCTCCCGAC
TTCGTGGCCAACGCCGGCGGTGCCATCCACCTCGTAGGCCGGGAGGTTCTCGGTTGGTCCGAGTCG
GTTGTCCACGAACGAGCAGTTGCCATAGGCGACACCCTGAATCAGGTCTTCGAGATCTCCGACAAC
GACGGCGTCACCCCGGACGAGGCCGCCCGCACTCTCGCTGGACGGCGCGCCCGCGAGGCCTCGAC
AACGACAGCGACTGCCTAGTAA
Abbildung 50: Gensequenz des K66I- Muteins.
5.6.3 Expression der PheDH-Mutante K66I
Da E. coli JM-105 sich als geeigneter Stamm für die Expression der PheDH erwiesen hat,
wurde das neuhergestellte PheDH-Mutein „pKK-223-K66I“ in diesen Stamm transformiert
und zur Expression eingesetzt. Die Anzucht erfolgte in 250 ml LB-amp, induziert wurde mit
1mM IPTG bei einer OD590 von 0,6. Der Aktivitätstest dieses Muteins erfolgte sowohl mit
Phenylpyruvat (um die Restaktivität der PheDH zu überprüfen) als auch für die gewünschte
Aktivität mit Phenylaceton als Substrat.
Erwartungsgemäß besitzt das Mutein keine Aktivität mehr gegenüber Phenylpyruvat als
Substrat. Da die Bindungsstelle Lysin66 im aktiven Zentrum der PheDH zur Carboxylgruppe
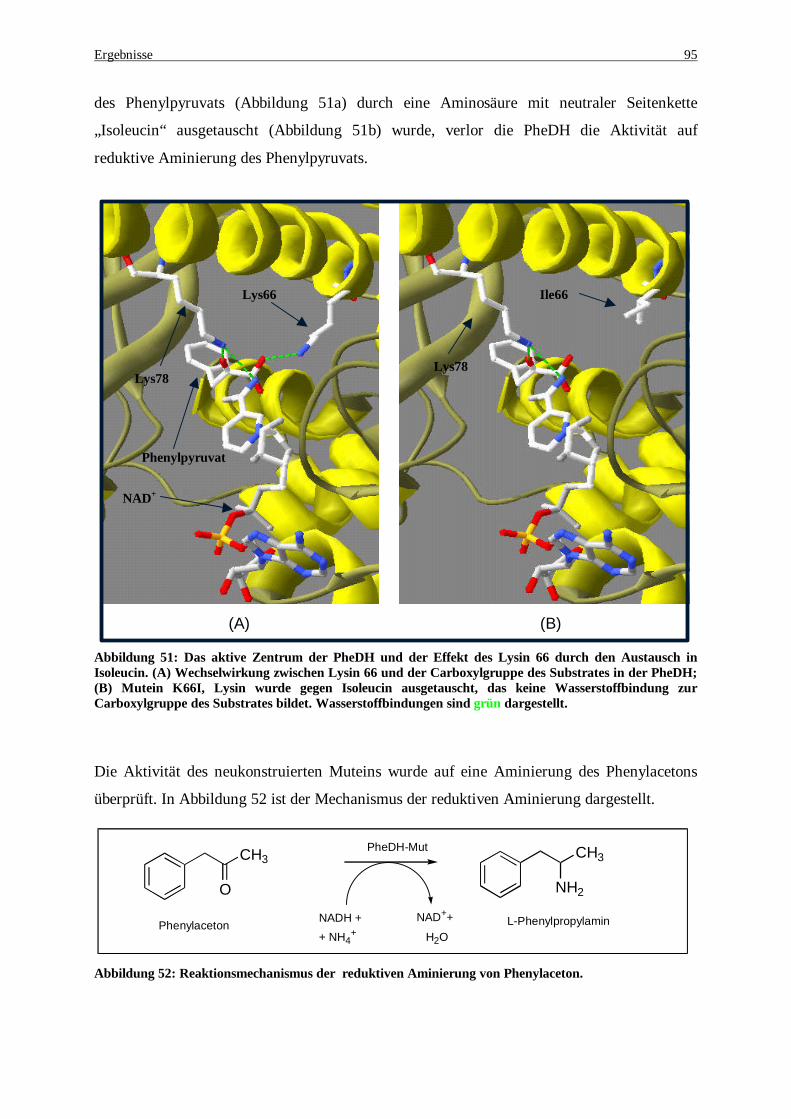
Ergebnisse
95
des Phenylpyruvats (Abbildung 51a) durch eine Aminosäure mit neutraler Seitenkette
„Isoleucin“ ausgetauscht (Abbildung 51b) wurde, verlor die PheDH die Aktivität auf
reduktive Aminierung des Phenylpyruvats.
(A) (B)
Abbildung 51: Das aktive Zentrum der PheDH und der Effekt des Lysin 66 durch den Austausch in Isoleucin. (A) Wechselwirkung zwischen Lysin 66 und der Carboxylgruppe des Substrates in der PheDH; (B) Mutein K66I, Lysin wurde gegen Isoleucin ausgetauscht, das keine Wasserstoffbindung zur Carboxylgruppe des Substrates bildet. Wasserstoffbindungen sind grün dargestellt.
Die Aktivität des neukonstruierten Muteins wurde auf eine Aminierung des Phenylacetons
überprüft. In Abbildung 52 ist der Mechanismus der reduktiven Aminierung dargestellt.
CH3
O
+ NH4+
PheDH-Mut
NADH +
CH3
NH2
H2O
NAD++Phenylaceton L-Phenylpropylamin
Abbildung 52: Reaktionsmechanismus der reduktiven Aminierung von Phenylaceton.
Lys78
Ile66
Lys78
Lys66
Phenylpyruvat
NAD+

Ergebnisse
96
Eine Aktivität bezüglich des Phenylacetons konnte nicht nachgewiesen werden. Durch diesen
Austausch konnte zwar die Ladung der Bindungsstelle zum Substrat verändert werden, aber
für die Akzeptanz eines neuen Substrates, könnten neben der Mutagenese von Lysin 66 noch
weitere Aminosäurenaustausche notwendig sein. Da die räumliche Struktur ähnlicher Amin
Dehydrogenasen wie die der gewünschten nicht bekannt sind, war es nicht möglich die
weitere Optimierung durch gezielten Aminosäurenaustausch durchzuführen.
Das Mutein K66I trägt die gewünschte Mutation (5.6.2), trotzdem konnte keine Aktivität
bezüglich Phenylaceton festgestellt werden. Dies könnte konformationsbedingt sein, was zu
der Überlegung führte, die Sequenz dieses Muteins ungezielt mittels Zufallsmutagenese
(directed evolution) zu verändern. Daher wurde dieses Mutein als Template benutzt und für
die weitere Optimierung eingesetzt.
5.6.4 Herstellung weiterer Muteine
Aufgrund der Überlegung, basische Aminosäuren durch neutrale auszutauschen, um eine
Aminosäure Dehydrogenase in eine Amin Dehydrogenase umzuwandeln (zu diesem Zeit-
punkt war die Kristallstruktur noch unbekannt), wurden die Muteine R210I bzw. R140I
hergestellt.
Bei der Expression der Mutante R210I konnte kein aktives Protein nachgewiesen werden. Da
Arginin bei dem benutzten pH-Wert eine positive Ladung aufweist und dadurch Interaktionen
mit anderen Aminosäuren hervorruft, kann ein Abstoßungseffekt bei Austausch gegen eine
neutrale Aminosäure (Isoleucin) auftreten.
Es wurde eine verminderte PheDH-Aktivität (83 % der urprünglichen PheDH-Aktivität gegen
Phenylpyruvat) bei dem exprimierten Protein der Mutante R140I beobachtet. Durch diesen
Austausch konnte die Proteinfaltung verlangsamt und dadurch ein proteolytischer Abbau
begünstigt werden. Vereinfacht: Das Mutein ist für Proteasen angreifbarer. Die gewünschte
Amin Dehydrogenase Aktivität gegenüber Phenylaceton war nicht nachzuweisen.
Das Mutein PheDH-R210I zeigt keine Aktivität, weder die ursprüngliche PheDH Aktivität
noch eine neue Amin Dehydrogenase Aktivität. Obwohl Arginin 210 nicht im aktiven
Zentrum beteiligt ist, führt eine Deletion bzw. Substitution zum Verlust der PheDH-Akitivität.

Ergebnisse
97
5.6.5 Mutation von Lysin 66 zu Arginin und Lysin 78 zu Isoleucin
Ein Austausch von Lysin 66 gegen Arginin könnte theoretisch eine direkte Reduktion der
Ketogruppe von Phenylpyruvat verursachen und somit eine Ähnlichkeit zum Lactat
Dehydrogenase-Mechanismus führen (Hart et al., 1987).
Dieser Fall einer direkten Reduktion der Ketogruppe ist bei Lactat-Dehydrogenasen zu
beobachten. Um einen ähnlichen Mechanismus wie bei der Lactat-Dehydrogenase und damit
eine Reduktion der Ketogruppe des Phenylpyruvats durch die PheDH zu erreichen, wurden
die Bindungsstellen zum Substrat beider Enzymen verglichen. In der Lactat Dehydrogenase
ist Arginin für die Bindung der Carboxylgruppe verantwortlich (Luyten et al., 1989),
wogegen in der PheDH wie bei allen Aminosäure-Dehydrogenasen Lysin diesen Part
übernimmt.
Ein weiterer Unterschied ist, dass eine direkte Reduktion der Ketogruppe im Phenylpyruvat
nicht stattfinden kann, weil die Entfernung zwischen dem C4 des Nicotinamidrings und der
Ketogruppe am C? des Phenylpyruvat größer als 6 Å ist. Diese Entfernung wird durch die
Bindung von Lysin 78 sowie Lysin 66 an die Ketogruppe bzw. an die Carboxylgruppe des
Substrates, das dann umorientiert und von NADH entfernt wird, verursacht. Der dadurch
vergrößerte Abstand verhindert einen direkten Hydridtransfer. Infolgedessen entsteht im
ersten Schritt ein Imin, das erst im nächsten Schritt reduziert werden kann (Vanhooke et al.,
1999). Anders als bei der PheDH ist bei der Lactat-Dehydrogenase für die Bindung zur
Ketogruppe des Substrates Histidin verantwortlich. Luyten et al. haben postuliert, dass durch
den Autausch der Aminosäure Arginin 171 (Bindungsstelle zur Carboxylgruppe im Substrat)
in der Lactat-Dehydrogenase gegen eine neutrale Aminosäure, die Reduktion des Substrates
vermieden werden kann. Dieser Austausch Arginin 171 gegen Tyrosin bzw. Tryptophan
führte nicht zur Verlust der LDH –Aktivität. Überraschenderweise war die Aktivität in diesen
zwei Muteinen höher als in einem Mutein mit dem Austausch der basischen Aminosäure
Arginin gegen das basische Lysin (Luyten et al., 1989).
Es besteht eine Ähnlichkeit in der PheDH zur LDH in Bezug auf die Bindung zur
Carboxylgruppe bzw. Ketogruppe des Substrates. Sowohl in der LDH als auch in der PheDH
sind die beteiligten Aminosäuren basisch. Da nach Luyten et al. der Austausch der basischen
Aminosäure (Bindungsstelle für die Carboxylgruppe im Substrat) keinen Einfluss auf die
Orientierung brachte, wurde in dieser Arbeit ebenso die Bindung zur Ketogruppe untersucht.
Durch die Mutation des Lysin 78 gegen Isoleucin könnte obiger Abstand verringert und die
Umorientierung des Substrates verhindert werden, so das eine Hydrierung der Ketogruppe
stattfinden könnte.
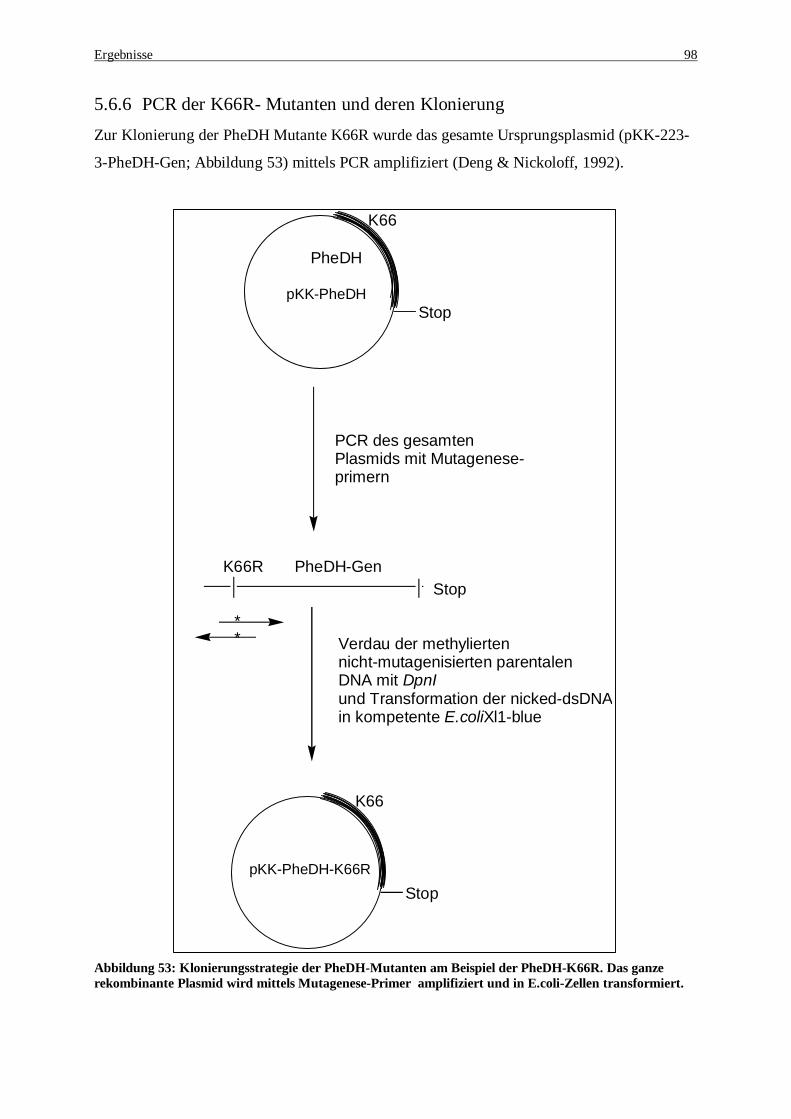
Ergebnisse
98
5.6.6 PCR der K66R- Mutanten und deren Klonierung
Zur Klonierung der PheDH Mutante K66R wurde das gesamte Ursprungsplasmid (pKK-223-
3-PheDH-Gen; Abbildung 53) mittels PCR amplifiziert (Deng & Nickoloff, 1992).
pKK-PheDHStop
K66
PCR des gesamtenPlasmids mit Mutagenese-primern
K66R PheDH-GenStop
PheDH
Verdau der methyliertennicht-mutagenisierten parentalenDNA mit DpnIund Transformation der nicked-dsDNAin kompetente E.coliXl1-blue
**
pKK-PheDH-K66R
Stop
K66
Abbildung 53: Klonierungsstrategie der PheDH-Mutanten am Beispiel der PheDH-K66R. Das ganze rekombinante Plasmid wird mittels Mutagenese-Primer amplifiziert und in E.coli-Zellen transformiert.

Ergebnisse
99
Beide Primer beinhalten die gewünschte Mutation.
P1: GCT CAC TGC CAT TCT CAA CGT CAT CGC CCC
P2: GGG GCG ATG ACG TTG AGA ATG GCA GTG AGC
Im Anschluß an die PCR wird die parentale Template-DNA mit dem Restriktionsenzym DpnI
abgebaut. DpnI erkennt ausschließlich methylierte DNA. Die verbliebene mutierte DNA wird
in kompetente E. coli Xl1 Blue bzw. JM105 transformiert, wobei eventuelle Einzel-
strangbrüche vom bakteriellen Reparatursystem behoben werden. Der Anteil mutierter
Plasmide ist ? 80 % (Stratagene).
Positive Klone wurden sequenziert und ein fehlerfreies mutiertes Plasmid zur Expression
eingesetzt.
5.6.7 Aktivitätsnachweis des PheDH-Muteins K66R und K78I
Die Muteine PheDH-K66R und K78I wurden in 200 ml Maßstab kultiviert und die Ex-
pression erfolgte ausschließlich in E. coli JM105. Während die Expression der rec-PheDH
(pKK223-3-PheDH) in E. coli JM105 zu einem Anteil von ca. 13 % PheDH des löslichen
Gesamtproteins führte, beträgt der Anteil der PheDH-K66R nur 10 % und der K78I etwa 4 %
des löslichen Zellproteins.
Das natürliche Substrat der wt-PheDH ist Phenylpyruvat, das mit Zusatz von
Ammoniumionen durch eine reduktive Aminierung zum Phenylalanin umgesetzt wird. Bei
der Aktivitätsbestimmung des Muteins PheDH-K66R bzw. K78I wurde neben der reduktiven
Aminierung auch ein Parallelversuch ohne Ammoniumzusatz durchgeführt, um zu prüfen, ob
eine direkte Reduktion stattfinden kann, was auf einen Reaktionsmechanismus analog der
Lactat Dehydrogenase hinweisen würde.
COOH
O
NADH
PheDH-K66R
COOH
OH
Abbildung 54: Reaktionsmechanismus zur Umsetzung der Ketosäure Phenylpyruvat zur Hydroxysäure Phenyllactat.
Eine Veränderung der PheDH durch den Austausch des Lysin 66 in Arginin führte nicht zur
drastischen Abnahme der PheDH-Aktivität. Bei diesem Mutein K66R waren etwa 90 % der
PheDH Aktivität noch nachweisbar. Die gewünschte Erweiterung des Substratspektrum durch
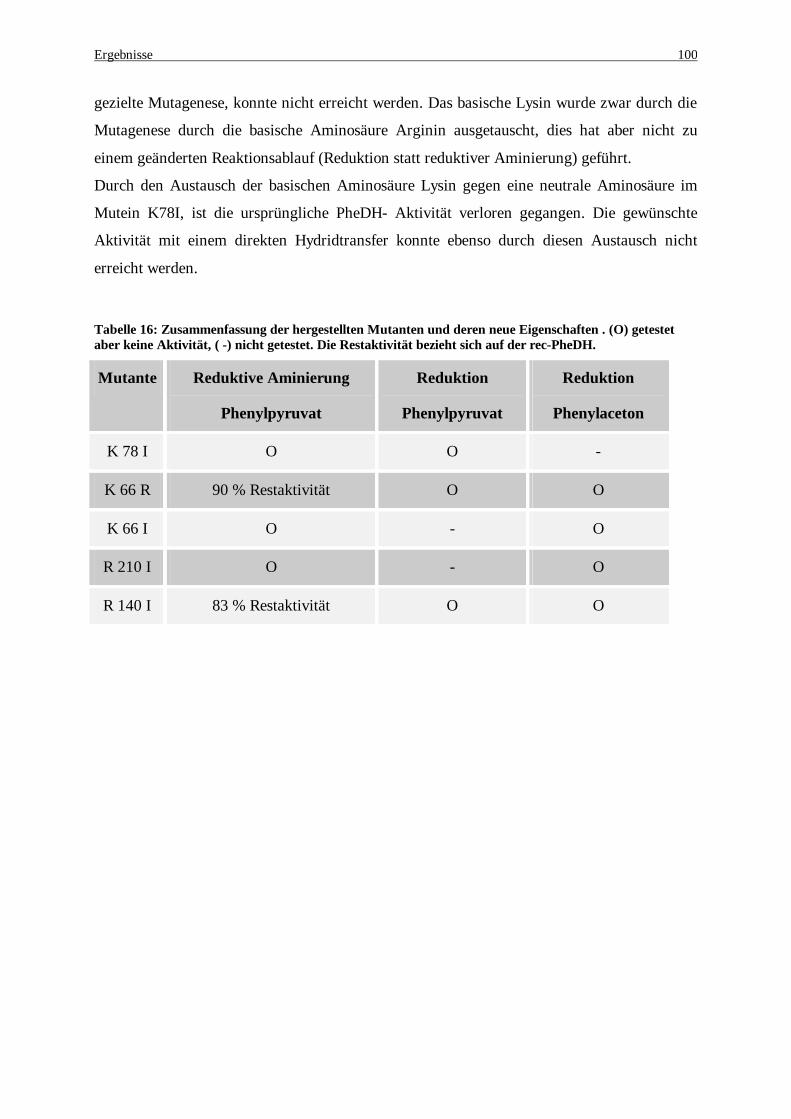
Ergebnisse
100
gezielte Mutagenese, konnte nicht erreicht werden. Das basische Lysin wurde zwar durch die
Mutagenese durch die basische Aminosäure Arginin ausgetauscht, dies hat aber nicht zu
einem geänderten Reaktionsablauf (Reduktion statt reduktiver Aminierung) geführt.
Durch den Austausch der basischen Aminosäure Lysin gegen eine neutrale Aminosäure im
Mutein K78I, ist die ursprüngliche PheDH- Aktivität verloren gegangen. Die gewünschte
Aktivität mit einem direkten Hydridtransfer konnte ebenso durch diesen Austausch nicht
erreicht werden.
Tabelle 16: Zusammenfassung der hergestellten Mutanten und deren neue Eigenschaften . (O) getestet aber keine Aktivität, ( -) nicht getestet. Die Restaktivität bezieht sich auf der rec-PheDH.
Mutante Reduktive Aminierung
Phenylpyruvat
Reduktion
Phenylpyruvat
Reduktion
Phenylaceton
K 78 I O O -
K 66 R 90 % Restaktivität O O
K 66 I O - O
R 210 I O - O
R 140 I 83 % Restaktivität O O

Ergebnisse
101
5.7 Zufallsmutagenese
In dieser Arbeit wurde bereit die gezielte Mutation der K66I, K66R und K78I beschrieben.
Diese Austausche führten zu keiner Erweiterung des Substratspektrums bzw. detektierbaren
Expression eines aktiven Enzyms. Da die gezielte Mutagenese nicht zu den gewünschten
Aktivitäten geführt hat, wurde eine Zufallsmutagenese durchgeführt. Als Template wurden
sowohl die rekombinante PheDH als auch in einer zweiten Versuchsreihe die Muteine K66I
und K66R als DNA-Template eingesetzt.
Die error-prone PCR-Methode für die Zufallsmutagenese wurde für die Erzeugung der
Mutanten verwendet. Dafür wurde das folgende PCR-Protokoll verwendet:
10 µl 10x Mutagenese Puffer inkl. MgCl2
10 µl 10x Mutagenese-dNTP-mix
0-10 µl 5 mM MnCl2 (0-0.5 mM Endkonz.)
2 fmol Template-DNA (ca. 7.5 ng eines 5.7 kb Plasmid)
je 40 pmol upstream and downstream primer
1µl Taq- Polymerase
ad 100µl Millipor- A-dest
Mit Mineralöl überschichten
Mutagenese Puffer:
70 mM MgCl2
500 mM KCl
100 mM Tris-HCl, pH 8.3 bei 25°C
0,1 %(W/V) Gelatine
Mutagenese dNTP-mix (10x):
2 mM dGTP
2 mM dATP
10 mM dTTP
10 mM dCTP
Mit der Optimierung des PCR-Protokolls und dem Einsatz bestimmter Konzentrationen an
dNTPs bzw. MnCl2 konnten 2-4 Mutationen eingeführt werden. Falsche Konzentrationen an
MgCl2 führten bei der PheDH-Amplifikation aufgrund des GC-Gehaltes zu unkontrollierter
Zahl an Mutationen.

Ergebnisse
102
Es wurden etwa 12.000 Kolonien aus der Zufallsmutagenese sowohl gegen Phenylaceton als
auch Phenylpyruvat getestet. Die Aktivität gegen Phenylpyruvat wurde sowohl bezüglich
reduktiver Aminierung (mit (NH4)2SO4) als auch bezüglich einfacher Reduktion ohne
(NH4)2SO4 überprüft. Aufgrund der hohen Zahl an erzeugten Muteinen wurde ein schneller
Farbtest zum Screening der Klone entwickelt.
5.8 Entwicklung eines Farbtestes als Screeningsmethode
Bei der Zufallsmutagenese produziert man eine große Zahl an Enzymvarianten, die einzeln
auf Aktivität getestet werden müssen. Eine einfache Methode ist eine Überprüfung der
Enzymaktivität direkt in den Zellen. Dafür wurde ein Farbtest entwickelt.
Er beruht auf der Aufnahme und Umsetzung von Farbstoffen durch ganze lebende
Mikroorganismen. Das Ausstreichen bzw. Sprühen von Farbstofflösungen auf Agarplatten mit
Bakterienkolonien führt zur Aufnahme dieses Farbstoffes, der dann umgesetzt wird. Durch
den Farbumschlag, der durch die folgende Umsetzung verursacht wird, können positive
Zellen erkannt werden.
NAD+ bzw. NADP+ -abhängige Enzyme sind durch diese Methode nachweisbar.
Eine Mischung aus INT und PES (siehe unten) wird auf eine Agarplatte, die die zu
untersuchenden Bakterien enthält, gegeben. Das Substrat befindet sich im Agar. Für die
Induktion ist IPTG erforderlich. Nach Zugabe der Farbstofflösung wurde die Agarplatte mit
Öl beschichtet (um die Re-Oxidation des gebildeten reduzierten Tetrazoliums zu vermeiden)
und für 4 Stunden bei 30°C inkubiert. Der Farbstoff wird dadurch aufgenommen und um-
gesetzt. Der Farbumschlag ist ein Nachweis für die Aktivität des Enzyms.
Substrat (L-Phenylalanin) 20 mM
INT (oder TNBT) 25 mg
PES 2 mg/ 100ml Puffer
TEA pH 7.0 50 mM
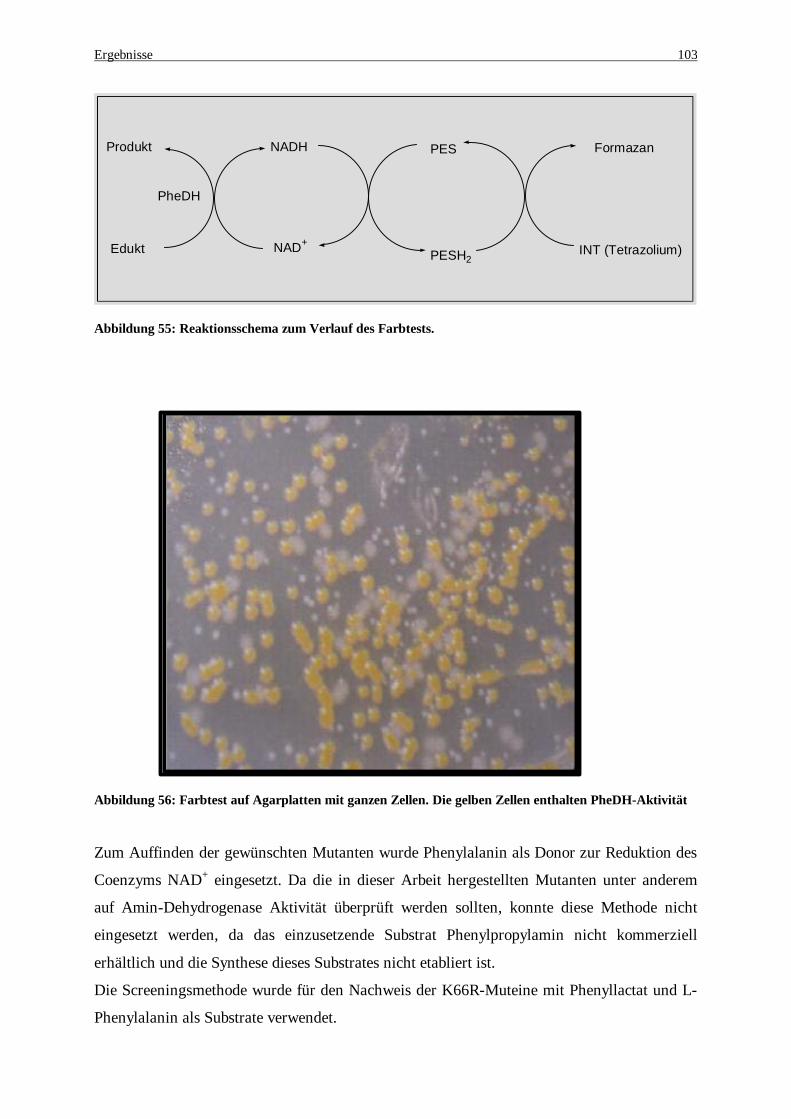
Ergebnisse
103
Produkt
Edukt
PheDH
NAD+
NADH PES
PESH2
Formazan
INT (Tetrazolium)
Abbildung 55: Reaktionsschema zum Verlauf des Farbtests.
Abbildung 56: Farbtest auf Agarplatten mit ganzen Zellen. Die gelben Zellen enthalten PheDH-Aktivität
Zum Auffinden der gewünschten Mutanten wurde Phenylalanin als Donor zur Reduktion des
Coenzyms NAD+ eingesetzt. Da die in dieser Arbeit hergestellten Mutanten unter anderem
auf Amin-Dehydrogenase Aktivität überprüft werden sollten, konnte diese Methode nicht
eingesetzt werden, da das einzusetzende Substrat Phenylpropylamin nicht kommerziell
erhältlich und die Synthese dieses Substrates nicht etabliert ist.
Die Screeningsmethode wurde für den Nachweis der K66R-Muteine mit Phenyllactat und L-
Phenylalanin als Substrate verwendet.
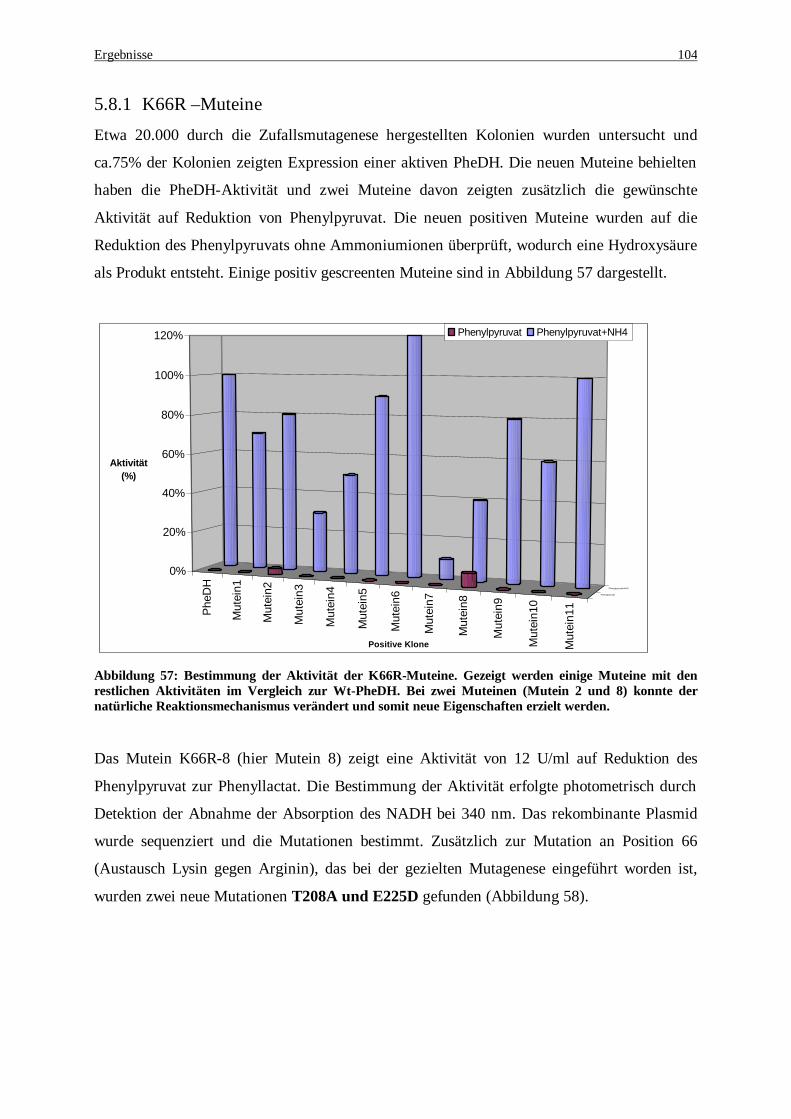
Ergebnisse
104
5.8.1 K66R –Muteine
Etwa 20.000 durch die Zufallsmutagenese hergestellten Kolonien wurden untersucht und
ca.75% der Kolonien zeigten Expression einer aktiven PheDH. Die neuen Muteine behielten
haben die PheDH-Aktivität und zwei Muteine davon zeigten zusätzlich die gewünschte
Aktivität auf Reduktion von Phenylpyruvat. Die neuen positiven Muteine wurden auf die
Reduktion des Phenylpyruvats ohne Ammoniumionen überprüft, wodurch eine Hydroxysäure
als Produkt entsteht. Einige positiv gescreenten Muteine sind in Abbildung 57 dargestellt.
Phe
DH
Mut
ein1
Mut
ein2
Mut
ein3
Mut
ein4
Mut
ein5
Mut
ein6
Mut
ein7
Mut
ein8
Mut
ein9
Mut
ein1
0
Mut
ein1
1
Phenylpyruvat
Phenylpyruvat+NH4
0%
20%
40%
60%
80%
100%
120%
Positive Klone
Phenylpyruvat Phenylpyruvat+NH4
Aktivität (%)
Abbildung 57: Bestimmung der Aktivität der K66R-Muteine. Gezeigt werden einige Muteine mit den restlichen Aktivitäten im Vergleich zur Wt-PheDH. Bei zwei Muteinen (Mutein 2 und 8) konnte der natürliche Reaktionsmechanismus verändert und somit neue Eigenschaften erzielt werden.
Das Mutein K66R-8 (hier Mutein 8) zeigt eine Aktivität von 12 U/ml auf Reduktion des
Phenylpyruvat zur Phenyllactat. Die Bestimmung der Aktivität erfolgte photometrisch durch
Detektion der Abnahme der Absorption des NADH bei 340 nm. Das rekombinante Plasmid
wurde sequenziert und die Mutationen bestimmt. Zusätzlich zur Mutation an Position 66
(Austausch Lysin gegen Arginin), das bei der gezielten Mutagenese eingeführt worden ist,
wurden zwei neue Mutationen T208A und E225D gefunden (Abbildung 58).
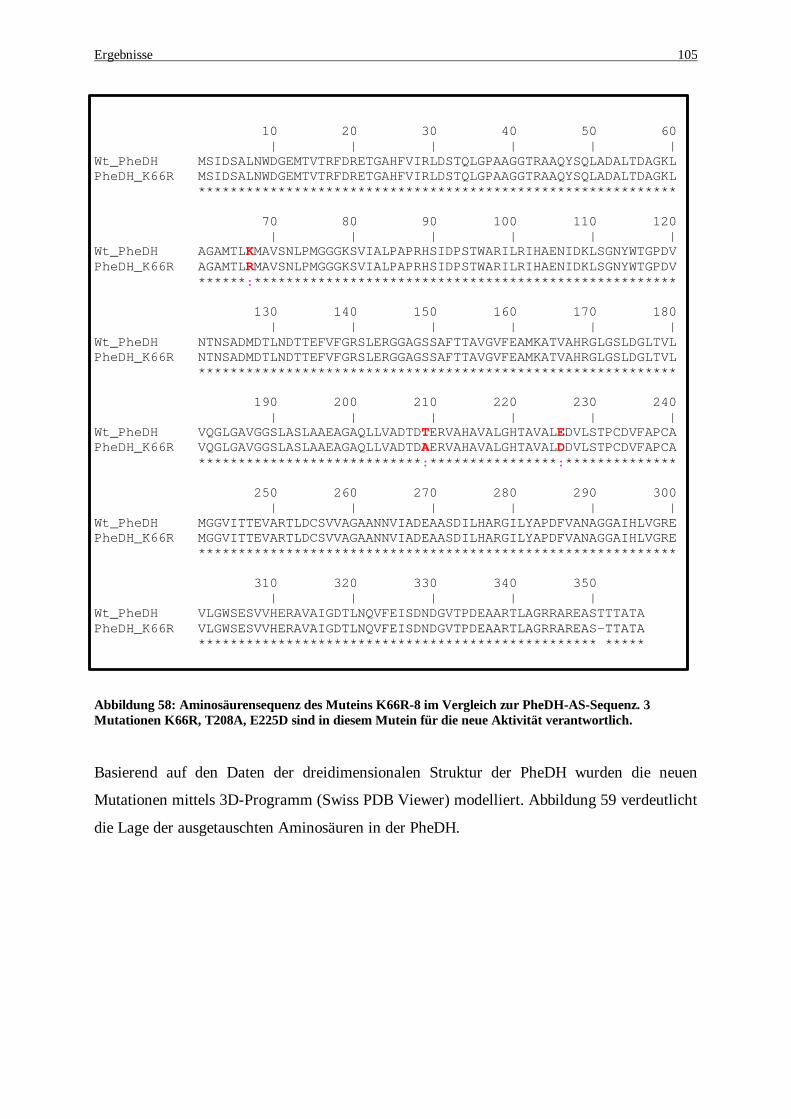
Ergebnisse
105
10 20 30 40 50 60 | | | | | | Wt_PheDH MSIDSALNWDGEMTVTRFDRETGAHFVIRLDSTQLGPAAGGTRAAQYSQLADALTDAGKL PheDH_K66R MSIDSALNWDGEMTVTRFDRETGAHFVIRLDSTQLGPAAGGTRAAQYSQLADALTDAGKL ************************************************************ 70 80 90 100 110 120 | | | | | | Wt_PheDH AGAMTLKMAVSNLPMGGGKSVIALPAPRHSIDPSTWARILRIHAENIDKLSGNYWTGPDV PheDH_K66R AGAMTLRMAVSNLPMGGGKSVIALPAPRHSIDPSTWARILRIHAENIDKLSGNYWTGPDV ******:***************************************************** 130 140 150 160 170 180 | | | | | | Wt_PheDH NTNSADMDTLNDTTEFVFGRSLERGGAGSSAFTTAVGVFEAMKATVAHRGLGSLDGLTVL PheDH_K66R NTNSADMDTLNDTTEFVFGRSLERGGAGSSAFTTAVGVFEAMKATVAHRGLGSLDGLTVL ************************************************************ 190 200 210 220 230 240 | | | | | | Wt_PheDH VQGLGAVGGSLASLAAEAGAQLLVADTDTERVAHAVALGHTAVALEDVLSTPCDVFAPCA PheDH_K66R VQGLGAVGGSLASLAAEAGAQLLVADTDAERVAHAVALGHTAVALDDVLSTPCDVFAPCA ****************************:****************:************** 250 260 270 280 290 300 | | | | | | Wt_PheDH MGGVITTEVARTLDCSVVAGAANNVIADEAASDILHARGILYAPDFVANAGGAIHLVGRE PheDH_K66R MGGVITTEVARTLDCSVVAGAANNVIADEAASDILHARGILYAPDFVANAGGAIHLVGRE ************************************************************ 310 320 330 340 350 | | | | | Wt_PheDH VLGWSESVVHERAVAIGDTLNQVFEISDNDGVTPDEAARTLAGRRAREASTTTATA PheDH_K66R VLGWSESVVHERAVAIGDTLNQVFEISDNDGVTPDEAARTLAGRRAREAS-TTATA ************************************************** *****
Abbildung 58: Aminosäurensequenz des Muteins K66R-8 im Vergleich zur PheDH-AS-Sequenz. 3 Mutationen K66R, T208A, E225D sind in diesem Mutein für die neue Aktivität verantwortlich.
Basierend auf den Daten der dreidimensionalen Struktur der PheDH wurden die neuen
Mutationen mittels 3D-Programm (Swiss PDB Viewer) modelliert. Abbildung 59 verdeutlicht
die Lage der ausgetauschten Aminosäuren in der PheDH.

Ergebnisse
106
Abbildung 59: Dreidimensionale Struktur des K66R-8-Muteins. Gezeigt sind die durch die Mutagenese ausgetauschten Aminosäuren. Die Mutationen K66R, T208A und E225D sind hervorgehoben. Wasser-stoffbindungen sind grün dargestellt.
Die Aminosäure Asparaginsäure, wurde durch Glutaminsäure mittels Zufallsmutagenese an
Position 225 substituiert und zeigt keine Wechselwirkung mit dem Coenzym bzw. mit den am
Coenzym bindenden Aminosäuren (Asp 205). Aus Abbildung 59 ist eine Wechselwirkung
zwischen Ala 208 zum Asp 205 bzw. zum Asp 207 ersichtlich. An einer direkten Wechsel-
wirkung zum Coenzym ist diese zweite Mutation T208A nicht beteiligt, dennoch geht sie eine
Wasserstoffbindung mit Asparaginsäure 205 ein. Asp 205 zeigt eine Wasserstoffbindung zum
Coenzym (Abbildung 59).
Die Modellierung der 3D-Struktur der Wt-PheDH zeigt eine Wasserstoffbindung zwischen
Lysin 66 zur Carboxylgruppe des Substrates (Phenylpyruvat) (Abbildung 60a). Nach dem
Austausch von Lysin 66 gegen Arginin konnte keine Wechselwirkung mehr festgestellt
werden (Abbildung 60b).
Ala 208
Asp 225
Arg 66
Asp 205 Thr 206
Asp 207
NAD+
Phenylpyruvat
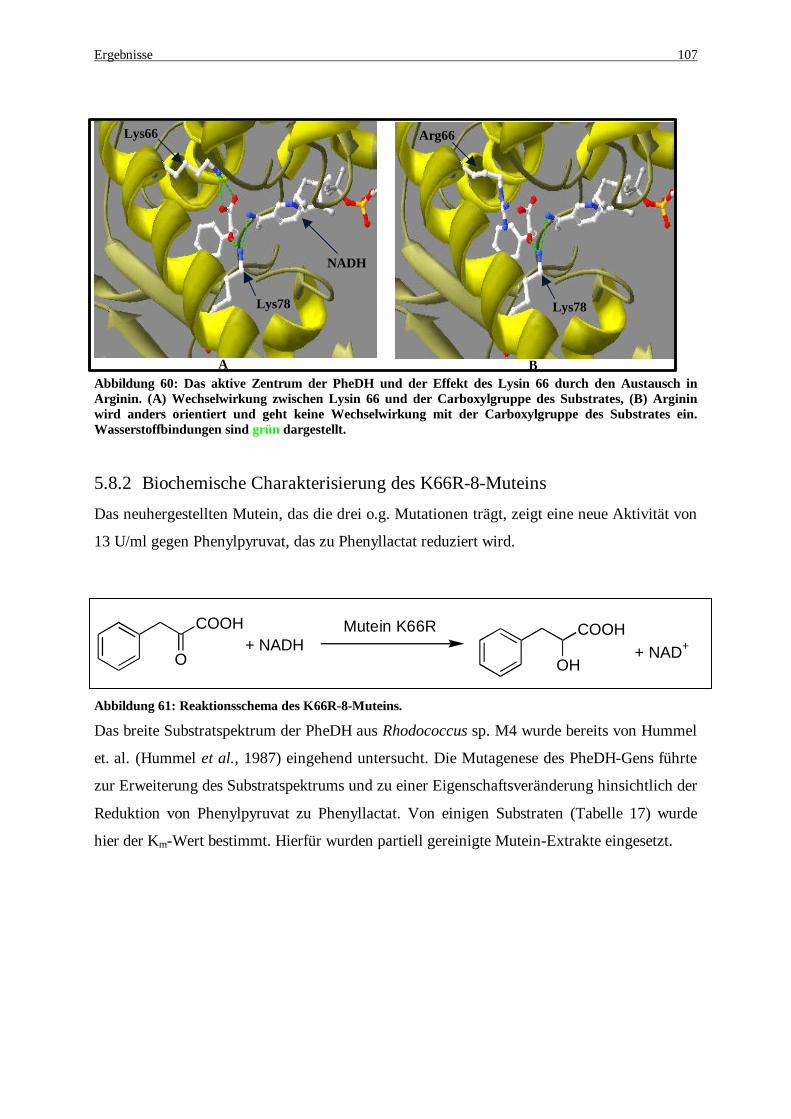
Ergebnisse
107
Abbildung 60: Das aktive Zentrum der PheDH und der Effekt des Lysin 66 durch den Austausch in Arginin. (A) Wechselwirkung zwischen Lysin 66 und der Carboxylgruppe des Substrates, (B) Arginin wird anders orientiert und geht keine Wechselwirkung mit der Carboxylgruppe des Substrates ein. Wasserstoffbindungen sind grün dargestellt.
5.8.2 Biochemische Charakterisierung des K66R-8-Muteins
Das neuhergestellten Mutein, das die drei o.g. Mutationen trägt, zeigt eine neue Aktivität von
13 U/ml gegen Phenylpyruvat, das zu Phenyllactat reduziert wird.
COOH
O+ NADH
Mutein K66R COOH
OH+ NAD+
Abbildung 61: Reaktionsschema des K66R-8-Muteins.
Das breite Substratspektrum der PheDH aus Rhodococcus sp. M4 wurde bereits von Hummel
et. al. (Hummel et al., 1987) eingehend untersucht. Die Mutagenese des PheDH-Gens führte
zur Erweiterung des Substratspektrums und zu einer Eigenschaftsveränderung hinsichtlich der
Reduktion von Phenylpyruvat zu Phenyllactat. Von einigen Substraten (Tabelle 17) wurde
hier der Km-Wert bestimmt. Hierfür wurden partiell gereinigte Mutein-Extrakte eingesetzt.
A B
Lys78
NADH
Lys66
Lys78
Arg66
Lys78
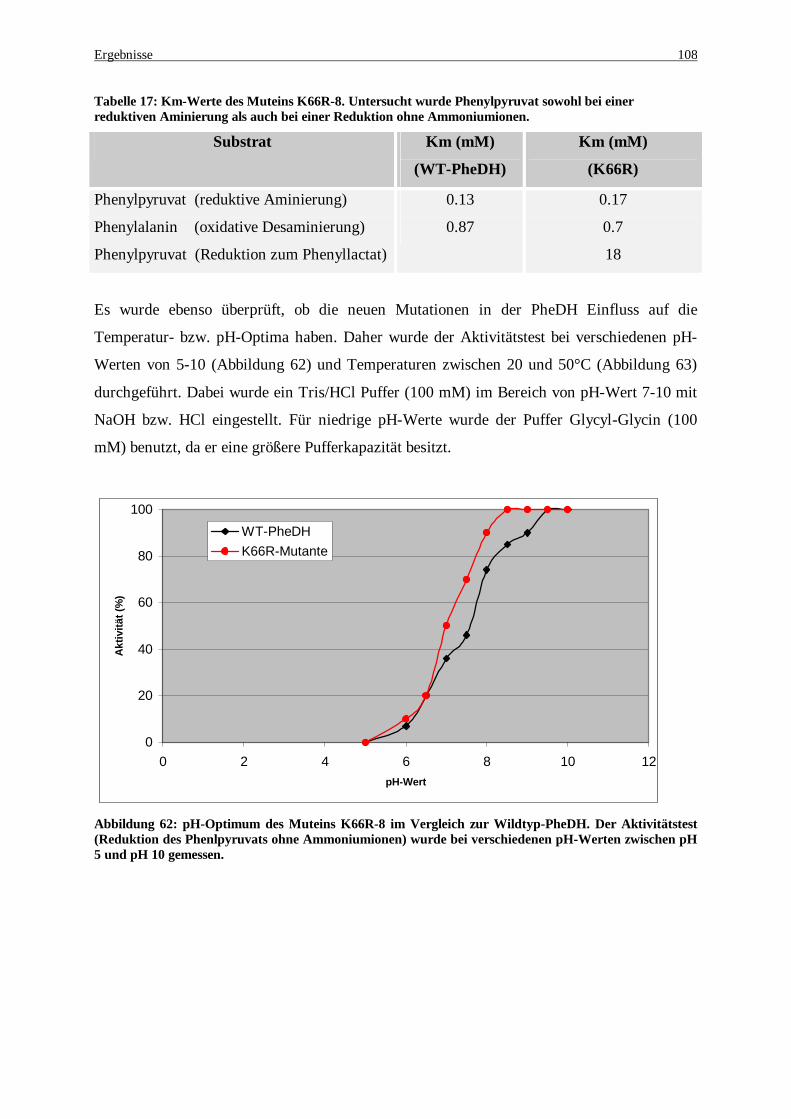
Ergebnisse
108
Tabelle 17: Km-Werte des Muteins K66R-8. Untersucht wurde Phenylpyruvat sowohl bei einer reduktiven Aminierung als auch bei einer Reduktion ohne Ammoniumionen.
Substrat Km (mM)
(WT-PheDH)
Km (mM)
(K66R)
Phenylpyruvat (reduktive Aminierung)
Phenylalanin (oxidative Desaminierung)
Phenylpyruvat (Reduktion zum Phenyllactat)
0.13
0.87
0.17
0.7
18
Es wurde ebenso überprüft, ob die neuen Mutationen in der PheDH Einfluss auf die
Temperatur- bzw. pH-Optima haben. Daher wurde der Aktivitätstest bei verschiedenen pH-
Werten von 5-10 (Abbildung 62) und Temperaturen zwischen 20 und 50°C (Abbildung 63)
durchgeführt. Dabei wurde ein Tris/HCl Puffer (100 mM) im Bereich von pH-Wert 7-10 mit
NaOH bzw. HCl eingestellt. Für niedrige pH-Werte wurde der Puffer Glycyl-Glycin (100
mM) benutzt, da er eine größere Pufferkapazität besitzt.
0
20
40
60
80
100
0 2 4 6 8 10 12pH-Wert
Akt
ivitä
t (%
)
WT-PheDHK66R-Mutante
Abbildung 62: pH-Optimum des Muteins K66R-8 im Vergleich zur Wildtyp-PheDH. Der Aktivitätstest (Reduktion des Phenlpyruvats ohne Ammoniumionen) wurde bei verschiedenen pH-Werten zwischen pH 5 und pH 10 gemessen.
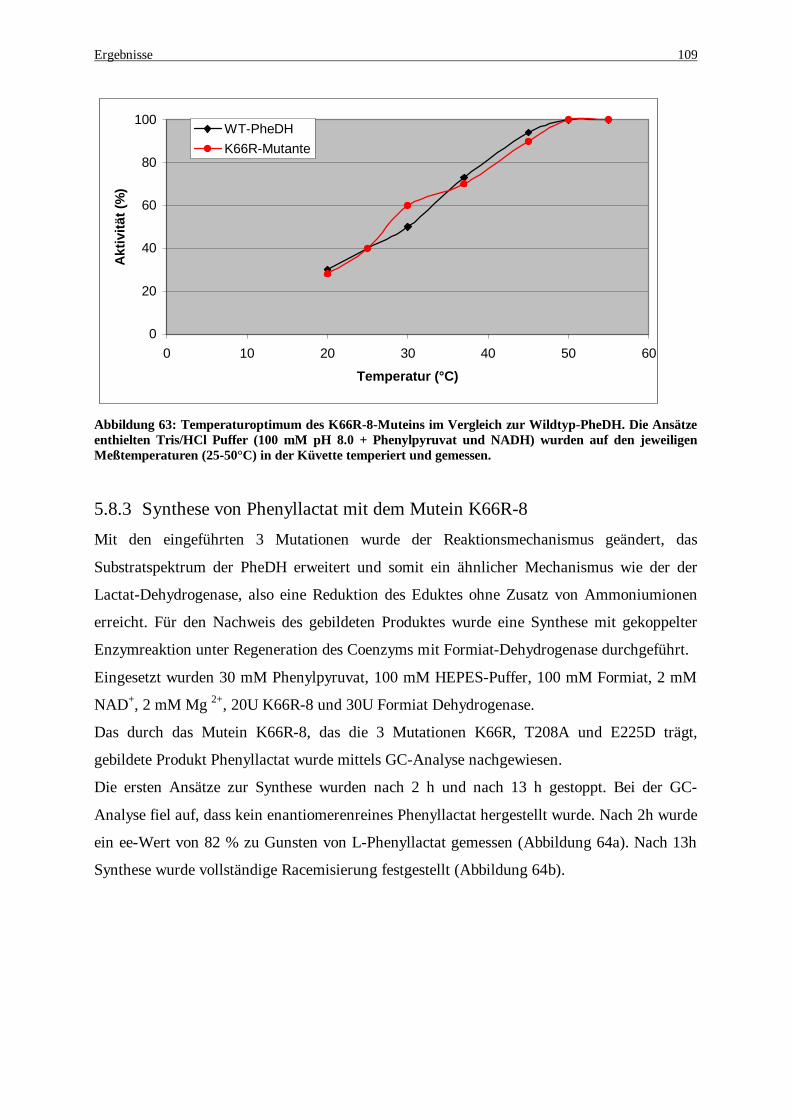
Ergebnisse
109
0
20
40
60
80
100
0 10 20 30 40 50 60
Temperatur (°C)
Akt
ivitä
t (%
)WT-PheDHK66R-Mutante
Abbildung 63: Temperaturoptimum des K66R-8-Muteins im Vergleich zur Wildtyp-PheDH. Die Ansätze enthielten Tris/HCl Puffer (100 mM pH 8.0 + Phenylpyruvat und NADH) wurden auf den jeweiligen Meßtemperaturen (25-50°C) in der Küvette temperiert und gemessen.
5.8.3 Synthese von Phenyllactat mit dem Mutein K66R-8
Mit den eingeführten 3 Mutationen wurde der Reaktionsmechanismus geändert, das
Substratspektrum der PheDH erweitert und somit ein ähnlicher Mechanismus wie der der
Lactat-Dehydrogenase, also eine Reduktion des Eduktes ohne Zusatz von Ammoniumionen
erreicht. Für den Nachweis des gebildeten Produktes wurde eine Synthese mit gekoppelter
Enzymreaktion unter Regeneration des Coenzyms mit Formiat-Dehydrogenase durchgeführt.
Eingesetzt wurden 30 mM Phenylpyruvat, 100 mM HEPES-Puffer, 100 mM Formiat, 2 mM
NAD+, 2 mM Mg 2+, 20U K66R-8 und 30U Formiat Dehydrogenase.
Das durch das Mutein K66R-8, das die 3 Mutationen K66R, T208A und E225D trägt,
gebildete Produkt Phenyllactat wurde mittels GC-Analyse nachgewiesen.
Die ersten Ansätze zur Synthese wurden nach 2 h und nach 13 h gestoppt. Bei der GC-
Analyse fiel auf, dass kein enantiomerenreines Phenyllactat hergestellt wurde. Nach 2h wurde
ein ee-Wert von 82 % zu Gunsten von L-Phenyllactat gemessen (Abbildung 64a). Nach 13h
Synthese wurde vollständige Racemisierung festgestellt (Abbildung 64b).
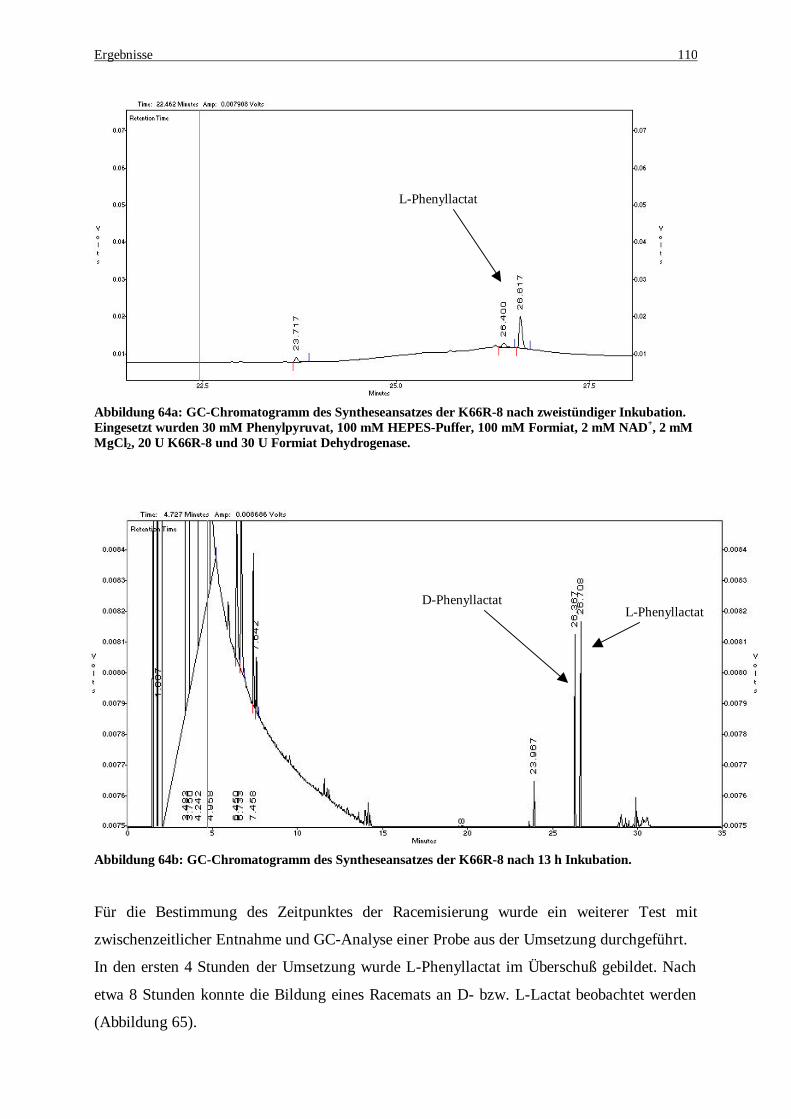
Ergebnisse
110
Abbildung 64a: GC-Chromatogramm des Syntheseansatzes der K66R-8 nach zweistündiger Inkubation. Eingesetzt wurden 30 mM Phenylpyruvat, 100 mM HEPES-Puffer, 100 mM Formiat, 2 mM NAD+, 2 mM MgCl2, 20 U K66R-8 und 30 U Formiat Dehydrogenase.
Abbildung 64b: GC-Chromatogramm des Syntheseansatzes der K66R-8 nach 13 h Inkubation.
Für die Bestimmung des Zeitpunktes der Racemisierung wurde ein weiterer Test mit
zwischenzeitlicher Entnahme und GC-Analyse einer Probe aus der Umsetzung durchgeführt.
In den ersten 4 Stunden der Umsetzung wurde L-Phenyllactat im Überschuß gebildet. Nach
etwa 8 Stunden konnte die Bildung eines Racemats an D- bzw. L-Lactat beobachtet werden
(Abbildung 65).
D-Phenyllactat L-Phenyllactat
L-Phenyllactat
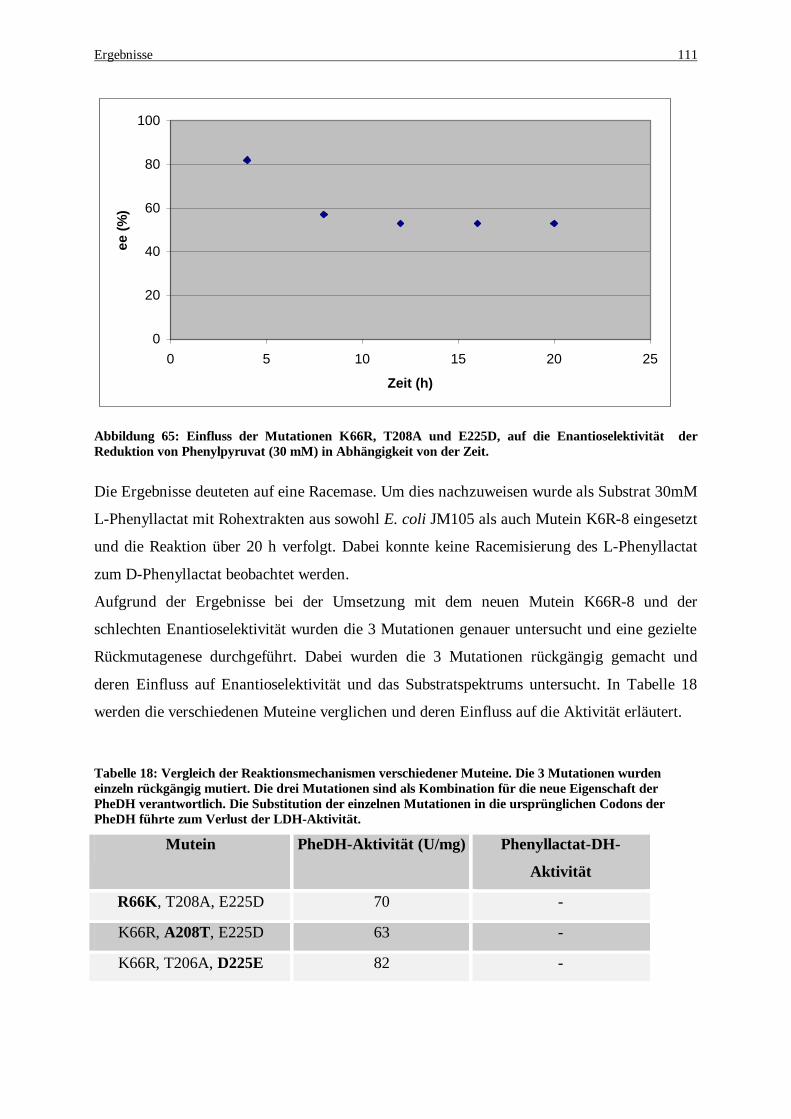
Ergebnisse
111
0
20
40
60
80
100
0 5 10 15 20 25
Zeit (h)
ee (%
)
Abbildung 65: Einfluss der Mutationen K66R, T208A und E225D, auf die Enantioselektivität der Reduktion von Phenylpyruvat (30 mM) in Abhängigkeit von der Zeit.
Die Ergebnisse deuteten auf eine Racemase. Um dies nachzuweisen wurde als Substrat 30mM
L-Phenyllactat mit Rohextrakten aus sowohl E. coli JM105 als auch Mutein K6R-8 eingesetzt
und die Reaktion über 20 h verfolgt. Dabei konnte keine Racemisierung des L-Phenyllactat
zum D-Phenyllactat beobachtet werden.
Aufgrund der Ergebnisse bei der Umsetzung mit dem neuen Mutein K66R-8 und der
schlechten Enantioselektivität wurden die 3 Mutationen genauer untersucht und eine gezielte
Rückmutagenese durchgeführt. Dabei wurden die 3 Mutationen rückgängig gemacht und
deren Einfluss auf Enantioselektivität und das Substratspektrums untersucht. In Tabelle 18
werden die verschiedenen Muteine verglichen und deren Einfluss auf die Aktivität erläutert.
Tabelle 18: Vergleich der Reaktionsmechanismen verschiedener Muteine. Die 3 Mutationen wurden einzeln rückgängig mutiert. Die drei Mutationen sind als Kombination für die neue Eigenschaft der PheDH verantwortlich. Die Substitution der einzelnen Mutationen in die ursprünglichen Codons der PheDH führte zum Verlust der LDH-Aktivität.
Mutein PheDH-Aktivität (U/mg) Phenyllactat-DH-
Aktivität
R66K, T208A, E225D 70 -
K66R, A208T, E225D 63 -
K66R, T206A, D225E 82 -

Ergebnisse
112
Aus Tabelle 18 ist zu entnehmen, dass die drei Mutationen im Mutein K66R-8 als
Kombination für die neue Aktivität der PheDH verantwortlich sind. Die Substitution der
einzelnen Mutationen zu den ursprünglichen Codons der PheDH führte zum Verlust der
LDH-Aktivität.
5.8.4 Austausch K66I
Neben der Mutante K66R ist eine weitere durch gezielte Mutagenese erzeugt worden, bei der
das Lysin 66 gegen Isoleucin (K66I) ausgetauscht worden ist. Auch dieses Mutein ist als
Template für eine Reihe ungerichteter Mutationen verwendet worden.
Mit diesem Template wurde eine Muteinbank von ca. 32000 Varianten konstruiert. Eine
weitere Muteinbank wurde mit der wt-PheDH-DNA konstruiert und überprüft. Alle
konstruierten Muteine wurden auf neue Aktivität gegen Phenylaceton mittels Schnelltest in
Titerplatten überprüft.
Die neu konstruierten Muteine aus der K66I-Enzymvariantenbank zeigten keine neue
Aktivität gegenüber Phenylaceton. Zusätzliche Mutationen zur K66I konnten die Akzeptanz
des Phenylacetons als Substrat nicht positiv beeinflussen. Da das Hauptinteresse an der
Umsetzung von Ketonen zu Aminen bestand, wurde die neukonstruierte Enzymbank nicht auf
neue Eigenschaften gegen weitere Substrate überprüft. Es ist aber von Interesse, diese
Enzymbank zum weiteren Screening zu verwenden und nach Muteinen für neue Umsetzungen
zu suchen.
Etwa 60 % der Muteine aus der zweiten Enzymvariantenbank, die mit der wt-PheDH als
Template hergestellt wurden, zeigten PheDH-Aktivität. Die Einführung der Mutationen in das
PheDH-Gen hat keinen Einfluss auf die Enantioselektivität. Bei der Umsetzung von
Phenylpyruvat (Abbildung 66) war der ee-Wert von dem der PheDH nicht zu unterscheiden
und lag bei >99 %.
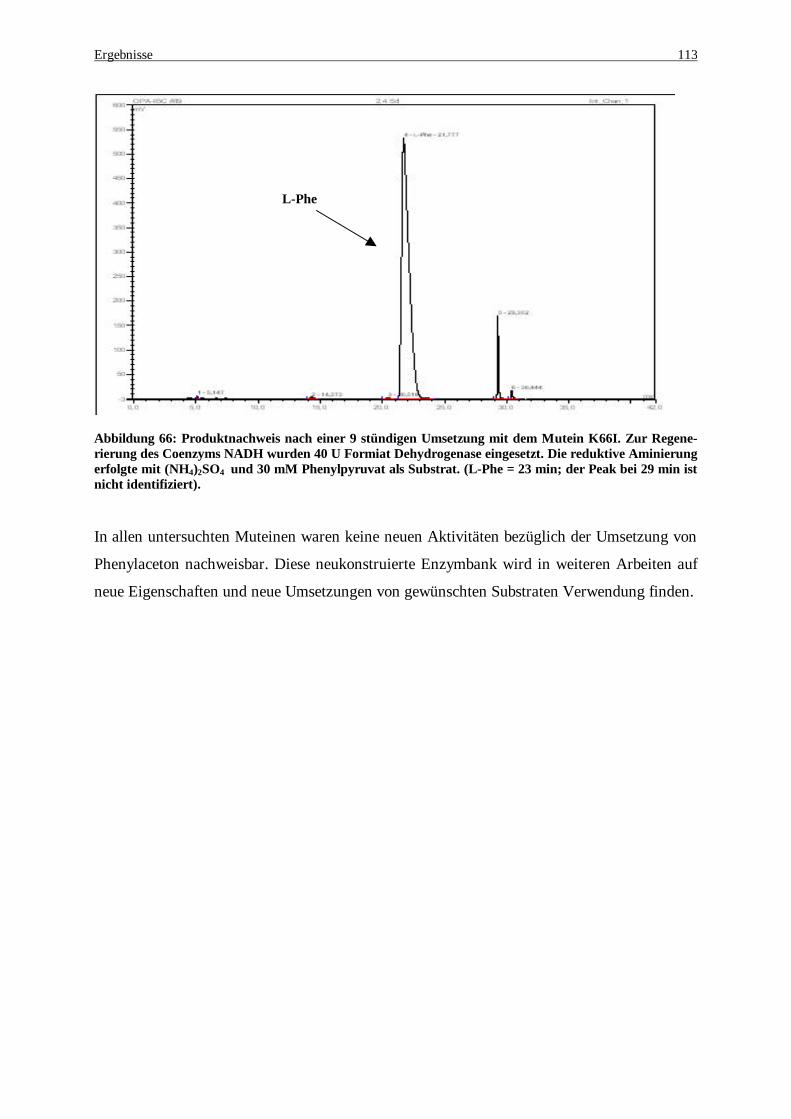
Ergebnisse
113
Abbildung 66: Produktnachweis nach einer 9 stündigen Umsetzung mit dem Mutein K66I. Zur Regene-rierung des Coenzyms NADH wurden 40 U Formiat Dehydrogenase eingesetzt. Die reduktive Aminierung erfolgte mit (NH4)2SO4 und 30 mM Phenylpyruvat als Substrat. (L-Phe = 23 min; der Peak bei 29 min ist nicht identifiziert).
In allen untersuchten Muteinen waren keine neuen Aktivitäten bezüglich der Umsetzung von
Phenylaceton nachweisbar. Diese neukonstruierte Enzymbank wird in weiteren Arbeiten auf
neue Eigenschaften und neue Umsetzungen von gewünschten Substraten Verwendung finden.
L-Phe

Diskussion
114
6 Diskussion Klonierung und Expression der PheDH aus Rhodococcus sp. M4
Das Gen der Phenylalanin Dehydrogenase aus Rhodococcus sp. M4 (Hummel et al., 1987) ist
wie die meisten Gene in Organismen der Actinomyces- Familie (Corynebacteria,
Mycobacteria, und Nocardia) (Watts et al., 2001) hoch GC-reich (Anteil: 65 %) . Das führte
zur Bildung von Sekundärstrukturen (Choi et al., 1999), was die Amplifizierung des PheDH-
Gens mittels PCR erschwert. Durch Zugabe von DMSO (3%) und die Variation der
Annealingstemperatur konnten Amplifikate gewonnen und kloniert werden. Die Klonierung
führte anfänglich zur Bildung von „inclusion bodies“ (Hibino et al., 1994). Der Versuch zur
Vermeidung dieses Problems mittels Klonierungen in verschiedenen Vektoren, pTRC99a,
pET16b, pET11a, führte nicht zur Expression eines aktiven Proteins. Das exprimierte Protein
wurde mittels SDS-Gel-Elektrophorese als „inclusion bodies“ analysiert. Nach
Literaturangaben könnten unlöslichen Proteine durch physikalische bzw. biochemische
Methoden (Temperatur, Induktion, Schüttelgeschwindigkeit) (Shin et al., 1997; Xu et al.,
1994; Yang et al., 1997; Zhang et al., 1995) entfaltet werden, was aber bei der exprimierten
PheDH nicht zu positiven Ergebnissen führte.
Unlösliche Proteine werden durch eine zu schnelle Expression gebildet (Camacho et al.,
2002), die den Proteinen nicht genügend Zeit und Raum zur Faltung lässt. Optimale
Expression eines Enzyms erreicht man normalerweise, wenn das Startcodon des
dazugehörigen Gens mit einem Abstand zwischen 7 und 9 bp von der Ribosomenbindungs-
stelle kloniert wird (Esipov et al., 1999; Vellanoweth & Rabinowitz, 1992).
Da die Klonierung nach dieser Strategie zu inclusion bodies geführt hat, wurde die PheDH an
der SmaI- Schnittstelle im Expressionsvektor pKK223-3 mit einer „nicht optimalen“
Entfernung von 14 bp des Startcodons zur Ribosomenbindungsstelle kloniert. Aufgrund
dieses Abstandes konnte eine schwächere Translation erreicht werden, was zu einer
langsameren und schwächeren Expression führte. Dem gebildeten Enzym bleibt dann genug
Zeit und Raum zur Faltung und man erhält zwar weniger, dafür aber aktives Enzym.
Zusätzlich dazu besitzt das verwendete Plasmid pKK223-3 einen schwächeren Promotor als
beispielsweise die oben verwendeten pET-Systeme, was zu einer weiteren Abschwächung der
Expression führt. Durch die Auswahl eines geeigneten Expressionsstammes E. coli JM105
konnte die Aktivität mit dem gleichen Plasmidkonstrukt auf das 3-fache im Vergleich zum
E.coli -HB101 erhöht werden.

Diskussion
115
Die Charakterisierung zeigt eine stabile rekombinante PheDH, die nach 36 h bei 30°C noch
etwa 95 % Restaktivität besitzt. Eine Denaturierung erfolgt erst bei höhere Temperaturen von
50°C. Nach 36 h konnten bei 37°C noch 90 % Restaktivität der rec-PheDH gemessen werden.
Im Vergleich dazu zeigte die Wildtyp-PheDH nach 36 h bei 30°C nur noch etwa 50 % Rest-
aktivität. Da die Sequenz der Wt-PheDH und die der rec-PheDH gleich sind, kann eine
falsche Faltung des Proteins ausgeschlossen werden. Die schlechte Stabilität der PheDH im
Wildtyp beruht wahrscheinlich auf vorhandenen Proteasen, die das Enzym nach Aufschluss
der Zellen abbauen(De Mot et al., 1998). Durch die Expression der PheDH aus Rhodococcus
sp. M4 in E. coli -JM105 und somit der Vermeidung von PheDH-spezifischen Proteasen,
konnte die Stabilität des Enzyms erhöht werden.
Die optimale Temperatur für eine Enzymreaktion liegt sowohl für die rec-PheDH als auch für
WT-PheDH bei 50 °C, die Enzymtests und Synthesereaktionen wurden jedoch aus Stabilitäts-
gründen bei 30 °C durchgeführt.
In einer ersten Arbeit, in der das Vorkommen der PheDH Rhodococcus sp. M4 beschrieben
wurde, ist eine partielle Reinigung für dieses Enzym bereits veröffentlicht worden (Hummel
et al., 1987). Die Reinigung der recPheDH aus E. coli- Rohextrakten erfolgte in Anlehnung
an dieses Protokoll. Für die Reinigung bis zur Homogenität konnte allerdings ein zusätzlicher
Reinigungsschritt zu den Literaturangaben durch eine Hitzedenaturierung der Begleitproteine
in Gegenwart von 5 % (w/v) L-Phenylalanin eingefügt werden. Die Entfernung des L-Phenyl-
alanins wurde mittels Gelfiltrationschromatographie durchgeführt.
Optimierung und Fermentation der PheDH
Neben der Auswahl eines geeigneten Vektor-Wirt-Systems wurden weitere Optimierungen,
wie Induktionsparameter, variiert, um eine effiziente Proteinexpression zu erzielen. Einerseits
wurde die Induktorkonzentration zwischen 0 und 3 mM IPTG (? -D-Isopropylthiogalaktosid)
bei einem konstanten Induktionszeitpunkt (OD600= 0.6) variiert. Die Untersuchung führte zu
dem Ergebnis, dass mit einer IPTG-Konzentration von 0.7 mM eine optimale Expression zu
erzielen war. In der Kontrollexpression ohne IPTG-Induktion konnte nur geringe PheDH-
Aktivität (35 U/ml) im Rohextrakt nachgewiesen werden. Folglich unterdrückte der lac I-
Repressor die Expression des PheDH-Gens im nicht induzierten Zustand fast vollständig (van
den Bogaard et al., 2000). Dies ist von Vorteil, da die Expression des Fremdproteins eine
Belastung des Zellstoffwechsels bedeutet und zu einer erheblich geringeren Zellausbeute

Diskussion
116
führt. Eine hohe Konzentration an IPTG kann das Wachstum der Zellen hemmen (Rhee et al.,
1997).
Andererseits wurde der Zeitpunkt der Induktion nach Inokulation variiert, wobei alle Kulturen
mit 1 mM IPTG induziert worden sind. Die Zellen wurden 24 h nach Inokulation geerntet und
die spezifischen Aktivitäten in den zellfreien Rohextrakten bestimmt. Es zeigt sich, dass der
Zeitpunkt der Induktion einen großen Einfluss auf das Wachstum der Zellen hat. Der Einfluss
des Induktionszeitpunkts auf die Systemproduktivität wurde mittels zweier Experimente
abgeschätzt, die die Zunahme der Volumenaktivität an PheDH nach Induktion am Anfang
bzw. in der Mitte der Fütterungsphase bestimmten. Die Ergebnisse, die in Abbildung 21
dargestellt sind, wiesen daraufhin, dass, ähnlich zu Versuchen in Schüttelkultur, eine
signifikante Abhängigkeit der recPheDH- Expression vom Induktionszeitpunkt besteht. Eine
zu frühe Induktion kann die Zellen unter Stressfaktoren setzen, was zu einer Störung des
Expressionsapparats führen kann (Donovan et al., 2000). Eine Induktion in der stationären
Phase, in der die Wachstums- und Sterberate gleich sind, kann nicht zu einer produktiven
Expression führen.
Nach Riesenberg (Riesenberg, 1991) und Fieschko (Fieschko, 1989) erfolgt die Bildung von
wachstumsinhibierender Nebenprodukte wie Acetat in komplexen Medien bei Wachstums-
raten oberhalb von 0,2 h-1 (Riesenberg et al., 1991) und in definierten Medien bei
Wachstumsrate oberhalb von 0,35 h-1. Mit abnehmender spezifischer Wachstumsrate steigt
die Expression des rekombinanten Proteins (Fieschko, 1989). Da in dieser Arbeit die
Kultivierung in komplexen Medien erfolgte, war für die Kultivierung des Expressions-
stammes JM105 eine spezifische Wachstumsrate von 0,2 h-1 angebracht.
Die Form der Nährstoffzuführung war ein kritischer Parameter bei der HZD-Fermentation;
dadurch kann sowohl die Zelldichte als auch Zellproduktivität beeinflusst werden (Lee, 1996).
Möglich waren konstante als auch kontinuierliche Fütterungsraten. Konstante Fütterungsraten
wurden für das Expressionssystem E. coli JM105 nicht in Erwägung gezogen, da dies nach
Lee et al. zur Verminderung der Wachstumsrate führt (Lee, 1996) und damit zur Vermin-
derung von Produktbildung des Expressionssystems führt (Riesenberg, 1991).
Die Wachstumsrate, Acetatbildung und End-Zelldichte wurden sowohl bei konstanter als auch
linearer Zufütterungsrate untersucht. Nach Paalme et al. kann eine konstante Wachstumsrate
durch eine exponentiell gesteigerte Zuführung der C-Quelle erreicht werden (Paalme et al.,
1997). Eine lineare Erhöhung der Nährstoffzuführung ist mit geringeren Schwankungen der
Wachstumsrate verbunden (Yee & Blanch, 1992).

Diskussion
117
Die Untersuchungen in dieser Arbeit zeigten, dass Acetat bei linearer Zuführung der C-Quelle
ständig gebildet wurde und im Gegensatz hierzu kein Acetat bei exponentieller Zuführung
nachweisbar war. Daher wurden weitere Experimente mit exponentieller Zuführung
durchgeführt.
In der HZD-Fermentation wird zwar eine hohe Biomasse erzielt, dies wurde aber durch eine
geringe spezifische Aktivität erkauft. Durch die Zugabe von Vitaminen, Spurenelementen und
optimiertem Medium mit Nährstoffen können die Zellen schnell wachsen und eine hohe
optische Dichte erreichen. Somit werden die Zellen aber unter Stress gesetzt, so dass die
Expression nicht ideal verläuft, was sich auf die spezifische Aktivität niederschlägt.
Bei einer ersten Fermentation der PheDH im Batch war die spezifische Aktivität geringer als
bei der Anzucht im Kolben. Der Grund für die schwächere Aktivität lag möglicherweise im
späten Erntezeitpunkt, da die Lyse der Zellen zur Desaktivierung der Enzyme führen kann.
Erwartet war eine bessere Aktivität als im Kolben aufgrund der besseren Sauerstoff-
versorgung sowie der pH-Regulierung.
Coexpression der PheDH und des malic enzymes
Das in dieser Arbeit klonierte NAD abhängige E. coli malic enzyme zeigt Ähnlichkeit im
Molekulargewicht (50 kDa) zum malic enzyme aus Bacillus stearothermophilus (Kobayashi
et al., 1989), aber einen Unterschied zum NADP-abhängigen malic enzyme aus E.coli
(62kDa) (Nakamura et al., 1986). Da der C-Terminus des NAD-abhängigen malic enzymes
aus E. coli zum Zeitpunkt der Klonierung nicht zugänglich war, wurde mit Hilfe des 5’-
Forward Primers ein Einzelstrang ampilifiziert. Mit der Gewinnung des Einzelstranges konnte
eine zweite PCR durchgeführt und ein doppelsträngiges Fragment amplifiziert werden. Mit
dieser vollkommen neuen Methode war das gesamte Gen amplifizierbar. Die Klonierung von
Genen ist somit wesentlich schneller und vereinfacht. Schwierigkeiten bei dieser Methode
haben sich in GC-reichen Sequenzen ergeben. Ein Vergleichsversuch mit der PheDH ist
anfänglich gescheitert, dennoch konnte durch Zusatz von DMSO im PCR-Ansatz eine
vollständige Amplifikation GC-reicher Sequenzen erreicht werden. Hier sollte beachtet
werden, dass Einzelstränge nicht stabil sind und daher der Umgang mit Einzelsträngen
schonend durchgeführt werden muß.
Für die Klonierung des malic enzymes 3’ zur PheDH an der PstI- und HindIII-Schnittstelle
war eine eigene Ribosomenbindungsstelle notwendig, da am C-Terminus der PheDH ein
Stopcodon angehängt war. Somit konnte eine Expression beider Enzyme gleichzeitig

Diskussion
118
erfolgen. Interessant wäre, aus den zwei Enzymen (PheDH / malic enzyme) ein Fusions-
protein herzustellen. Dies kann durch die Deletion des Stopcodons am C-Terminus der
PheDH sowie der Ribosomenbindungsstelle vor dem malic enzyme erreicht werden. Eine
katalysierte Reaktion der PheDH kann theoretisch erfolgen, da das aktive Zentrum nicht am
C-Terminus der PheDH liegt (Vanhooke et al., 1999). Somit kann sowohl die Umsetzung des
Substrates als auch die Regeneration des Coenzyms im gleichen fusionierten Protein erfolgen.
Da keine Kristallstruktur vom malic enzyme aus E. coli K12 vorliegt und noch nicht bekannt
ist, wo das aktive Zentrum sich befindet, wäre eine erfolgreiche Umsetzung durch ein
Fusionsprotein nur hypothetisch. Fraglich ist dabei auch, mit welcher Struktur die beiden
Enzyme als Fusionsprotein exprimiert werden.
Die Aktivität beider rekombinanten Enzyme im E. coli- Stamm JM 105 ist höher als im
HB101 und liegt bei 300 U/ml für malic enzyme und 400 U/ml für die PheDH. Die Aktivität
des malic enzymes in diesem Konstrukt mit einer exprimierten PheDH ist eindeutig niedriger
als ein homolog exprimiertes malic enzyme allein. Es liegt daran, dass der Haushalt an
Aminosäuren während der Translation auf mehrere Enzymen verteilt ist, sodass in dem Fall
weniger Enzymmenge hergestellt wird als wenn die Aminosäuren einem exprimierten Enzym
allein zur Verfügung ständen.
Ganzzell-Umsetzung
Als neue Parameter, die bei ganzen Zellen berücksichtigt werden müssen, kommen jetzt
allerdings Transportprozesse durch die Zellmembran hinzu (Andersen et al., 2001; Sanden et
al., 2002). Ein Versuch zur Immobilisierung der Zellen mit CTAB, um sie durchlässig zu
machen, führte nicht zu Steigerung des Umsatzes. Das liegt daran, dass eine Aufnahme des
Substrates Phenylpyruvat ohne Immobilisierung der Zellwand möglich war. Die Überlegung,
dass die Substrate Phenylpyruvat und Phenylalanin als C-Quelle metabolisiert werden
könnten, hat sich nicht bestätigt. Es konnten zwar nach der Umsetzung nur 90% des
erwarteten Produktes L-Phenylalanin festgestellt werden, das ist aber nicht unbedingt auf
Metabolisierung zurückzuführen ist. Ursache könnten die Reaktionsbedingungen sein, da die
Reaktion im Batch Ansatz durchgeführt wurde und das Produkt nicht der Reaktion entzogen
worden ist.
Eine weitere Überlegung war, dass L-Malat über eine E. coli eigene Malat-Dehydrogenase
metabolisiert wird. Aus diesem Grund wurde L-Malat im Überschuss zugegeben und somit
dieses Problem vermieden. Im Vergleich zur Umsetzung mit isolierten Enzymen war es
allerdings wahrscheinlich, dass Pyruvat, das durch die Umsetzung von L-Malat für die NADH

Diskussion
119
Regenerierung entsteht, als leicht verwertbare C-Verbindung in den Primärstoffwechsel
einfließt und nicht mehr als störendes Nebenprodukt, wie bei der Verwendung isolierter
Enzyme vorliegt. Bei E. coli und anderen Bakterien wird Pyruvat durch Phosphoenolpyruvat-
Synthetase direkt phosphoryliert und fließt somit in den Stoffwechsel der Zellen ein (Geerse
et al., 1989).
Pyruvat + ATP + H2O Phosphoenolpyruvat + AMP + Pi
PEP-Synthetase
Der Vorteil der Formiat Dehydrogenase, die sich bis dato für die Regenerierung des
Coenzyms NADH in situ durchgesetzt hat, liegt darin, dass bei der Regeneration durch
Formiat nur CO2 und keine weiteren Nebenprodukte entstehen (Schütte et al., 1976). Da aber
in dieser Arbeit nachgewiesen wurde, dass das malic enzyme ein durchaus erfolgreiches
System darstellt und im Ganzellsystem keine Nebenprodukte das gewünschte Produkt
verunreinigen, könnten solche in dieser Arbeit entwickelten Konstrukte technisch und
wirtschaftlich interessanter sein. Dazu kommt der wirtschaftliche Faktor, da L-Malat im
Vergleich zum Na-Formiat vergleichbar kostengünstig ist. Ein weiterer wesentlicher Vorteil
der Ganzzellumsetzung liegt darin, dass auf die Präparation, möglicherweise Reinigung der
Enzyme sowie den Zusatz an freiem Coenzym verzichtet werden kann.
Für eine gekoppelte Reaktion zweier Enzyme wurden bestimmte Parameter für die Stabilität
der beiden Enzymen untersucht, um optimale Verhältnisse zu schaffen. Wichtige Faktoren
sind das pH-Optimum sowie die Temperaturstabilität der beiden Enzyme. Zusätzlich zu der
Stabilität der Enzyme spielen weitere Faktoren, wie z.B. der Einfluss der verschiedenen
Substrate auf die Enzyme eine Rolle (Brunhuber et al., 2000; Solovjeva & Kochetov, 1999).
Die PheDH könnte durch Malat inhibiert werden bzw. malic enzyme durch das
Phenylpyruvat. In Bezug auf diese Coexpression der Phenylalanin Dehydrogenase aus
Rhodococcus sp. M4 und des malic enzymes aus E. coli konnte keine Inhibierung festgestellt
werden. Vor der Übertragung dieser Technik auf weitere Enzyme sollte aber eine
Untersuchung bezüglich der Inhibierung durchgeführt werden.
Es wurde für einen wirtschaftlichen Einsatz der Ganzzell-Umsetzung untersucht, ob das malic
enzyme durch D- Malat inhibiert wird. Eine Inhibierung war nicht festzustellen, daher ist es
von wirtschaftlicher Bedeutung, das kostengünstigere racemische D,L- Malat für die
Regenerierung des Coenzyms einzusetzen.

Diskussion
120
Bei einer in situ Regenerierung führte der Zusatz an BSA zu einer Steigerung der Aktivität.
Ebenso beeinflusste der Aufschluß-Puffer die Aktivitäten. Hierbei war zu beobachten, dass
der geeignete Aufschluß- Puffer für die einzelnen Enzyme unterschiedlich war. Der Kpi-
Puffer war für die PheDH besser geeignet als für das malic enzyme, da aber die
Aktivitätsabnahme des malic enzyme im Kpi-Puffer nicht drastisch war, wurden die
rekombinanten Zellen nach der heterologen Expression weiterhin im Kpi-Puffer
aufgeschlossen.
Dieses Konstrukt mit dem malic enzyme als Regenerationssystem eröffnet neue Möglich-
keiten für Coexpressionen mit weiteren Dehydrogenasen und damit Ganzzell-Umsetzungen
von verschiedenen Substraten.
Entwicklung einer Screeningsmethode
Um das Screening der durch die Zufallsmutagenese hergestellten Enzymvarianten- Bibliothek
ermöglichen zu können, sind geeignete und effektive Selektionsmechanismen notwendig. Mit
geeigneten Methoden können die gewünschten Eigenschaften detektiert werden. Zahlreiche
erfolgreiche Screeningsmethoden wurden bereits entwickelt. Beispielsweise wurden pH-
Indikatoren auf Agarplatten eingesetzt, so dass beim Abbau des Substrates eine Abnahme des
pH-Wertes stattfand und sich die Farbe des Indikators dadurch ändert (Zhou et al., 1996). Da
aber bei vielen Umsetzungen (unter anderem bei reduktiver Aminierung) die Änderung des
pH-Wertes nicht detektierbar wäre und man auf die Isolierung des Enzyms für photometrische
Tests angewiesen ist, wurde in dieser Arbeit eine effektivere Screeningsmethode für den
direkten Nachweis der positiven Klone entwickelt. Die neue Methode beruht auf dem
Farbumschlag von Tetrazolium. Die Reduktion von Tetrazolium führt zur Veränderung seines
Absorptionsspektrums, was eine schnelle Detektierung positiver Kolonien ermöglicht. Für die
Evaluierung dieser Methode, wurde Phenylalanin als Donor zur Reduktion des Coenzyms
NAD+ eingesetzt. Anfänglich haben sich alle Klone durch den Luftsauerstoff gefärbt. Die
Vermeidung dieses Problems erfolgte durch die Überschichtung der Zellen mit Öl bzw. mit
Agar. Dass die neue Methode nicht direkt für das Screening nach Muteinen, die Amin
Dehydrogenase Aktivität zeigen, eingesetzt wurde, liegt am Mangel des einzusetzenden
Substrates Phenylpropylamin. Phenylpropylamin ist nicht kommerziell erhältlich und seine
Synthese noch nicht etabliert.
Diese neue Screeningsmethode ist zum Screening von Dehydrogenasen soweit optimiert und
somit eine schnelle und effektive Methode.

Diskussion
121
Mutagenese
Die Klonierung der PheDH aus Rhodococcus sp. M4 war die Voraussetzung für die gezielte-
bzw. Zufallsmutagenese, um neue Enzymvarianten für die Herstellung von Aminen und
Hydroxysäuren zu konstruieren.
Zum Zeitpunkt der Planung der Mutagenese war die 3D-Struktur der Phenylalanin
Dehydrogenase aus Rhodococcus sp. M4 noch nicht aufgeklärt. Die PheDH sollte in dieser
Arbeit zu diesem Zweck kristallisiert werden, aber gleichzeitig während der Vorbereitung zur
Kristallisation der PheDH wurden auch einige Mutationen durchgeführt. Die Postulierung,
dass die Bindungsstelle zur Carboxylgruppe des Substrates eine basische sein sollte, beruhte
auf Literaturangaben zum Mechanismus (Clarke et al., 1986; Pasquo et al., 1998). Nach
Literaturangaben kommen Arginin oder Lysin in Lactat Dehydrogenasen bzw. Aminosäuren
Dehydrogenasen (Baker et al., 1995; Baker et al., 1997) im aktiven Zentrum vor. Um die
Akzeptanz einer Methylgruppe anstelle der Carboxylgruppe im Substrat zu ermöglichen,
sollte daher eine basische Aminosäure in eine neutrale Aminosäure umgewandelt werden
(Nicholls et al., 1994). Da aber in der PheDH etwa zwanzig Arginine und fünf Lysine
vorkommen, ist eine gezielte Mutagenese zur Erweiterung des Substratspektrums in diesem
Falle nicht praktikabel und eine Zufallsmutagenese deutlich effektiver. Doch aus wissen-
schaftlichen Gründen wurden einige Versuche zur Substitution von Arginin in weitere
neutrale Aminosäuren wie beispielsweise Isoleucin durchgeführt, um den Einfluss der
Substitution von Arginin gegen neutrale Aminosäure zu untersuchen.
Zwei Muteine R210I und R140I wurden konstruiert und auf Aktivität untersucht.
Es wurde eine verminderte Expression und etwa 83 % Restaktivität der PheDH bei der
Mutante R140I beobachtet. Durch diesen Austausch konnte die Proteinfaltung verlangsamt
und dadurch ein proteolytischer Abbau begünstigt werden (Jiang et al., 2001). Vereinfacht:
Das Mutein ist für Proteasen angreifbarer. Die gewünschte Amin Dehydrogenase Aktivität
gegenüber Phenylaceton war nicht nachzuweisen.
Keine Expression konnte bei der Mutante R210I festgestellt werden. Ein Grund dafür könnte
die Ladung des Arginins sein. Da Arginin bei dem benutzten pH-Wert eine positive Ladung
aufweist und dadurch Interaktionen mit anderen Aminosäuren hervorruft, kann ein
Abstoßungseffekt bei Austausch gegen eine neutrale Aminosäure (Isoleucin) auftreten. Das
Mutein PheDH-R210I hat durch diesen Austausch die ursprüngliche Aktivität der PheDH
verloren, zeigt aber keine neue Amin Dehydrogenase- Aktivität.
Zu diesem Zeitpunkt wurde die Strukturaufklärung der PheDH durch Vanhooke (Vanhooke et
al., 1999) durchgeführt und die am aktiven Zentrum beteiligten Aminosäuren bestimmt.

Diskussion
122
Anhand der 3D-Struktur-Daten konnte das Enzym mittels Computereinsatz modelliert und die
Mutationen simuliert werden.
Dass es bei der PheDH nicht zu einem direkten Hydridtransfer auf die Ketogruppe kommen
kann, liegt an der Entfernung zwischen dem C4 des Nicotinamidrings und C? des
Phenylpyruvat, die mehr als 6 Å beträgt. Dieser Abstand kommt dadurch zustande, dass die
Ketogruppe des Substrates eine Bindung zum Lysin 78 eingeht und somit das Substrat vom
NADH weggezogen wird (Vanhooke et al., 1999). Eine weitere Bindung zum Substrat wird
durch Lysin 66 verursacht. Durch die Simulierung der Substitution dieser Aminosäuren
mittels Computereinsatzes konnte festgestellt werden, dass durch den Austausch von Lysin 66
gegen Isoleucin bzw. gegen Arginin keine Wechselwirkung mehr zur Carboxylgruppe des
Substrates stattfinden kann. Um die Akzeptanz von Ketonen, die dem Phenylpyruvat struktur-
ähnlich sind, und deren Umsetzung zu Aminen zu ermöglichen, wurde Lysin 66 gegen die
neutrale Aminosäure Isoleucin substituiert. Weitere Muteine, die sich im Mechanismus von
der PheDH unterscheiden und einen direkten Hydridtransfer verursachen, wurden durch den
Austausch von Lysin 66 zu Arginin und Lysin 78 gegen Isoleucin konstruiert.
Die Durchführung der einzelnen Substitutionen erfolgte nach dem Prinzip der überlappenden
PCR „overlap extension“ (Ho et al., 1989). Dafür wurde das in dieser Arbeit klonierte
PheDH- Gen als Template verwendet. Die Ausbeute der Hybride mittels dieser Technologie
war anfänglich sehr gering. Es ist aus dem Literatur (Ho et al., 1989) zu entnehmen, dass man
equimolare Mengen beider Fragmente einzusetzen hat, was aber in allen Fällen zu niedriger
Ausbeute geführt hat. Das liegt daran, dass dadurch vier Fusionen entstehen können, wobei
nur eine gewünscht ist. Daher erfolgte die Optimierung dieser Methode durch die Ampli-
fikation der zur Hybridisierung gewünschten Einzelstränge aus den jeweiligen beiden Frag-
menten, die dann in einer weiteren PCR eingesetzt wurden. Somit konnte die Ausbeute auf
das 3 fache gesteigert werden.
Im Gegensatz zur Expression der rec-PheDH betrug der Anteil des Muteins K66R nur 10 %
und der K78I etwa 4 % des löslichen Zellproteins, wobei bei der rec-PheDH etwa 13 % des
löslichen Gesamtproteins nachzuweisen waren. Ein Grund dafür könnte eine schlechte, durch
die Mutagenese verursachte, Proteinfaltung sein.
Eine Veränderung der PheDH durch den Austausch des Lysin 66 gegen Arginin führte nicht
zur drastischen Abnahme der PheDH-Aktivität. Bei diesem Mutein K66R waren etwa 90 %
der PheDH Aktivität noch nachweisbar. Die gewünschte Aktivität, ein direkter Hydridtransfer
konnte durch diesen Austausch nicht erreicht werden. Das Lysin wurde zwar durch die
Mutagenese gegen Arginin ausgetauscht, hat aber nicht zum gewünschten Mechanismus

Diskussion
123
geführt. Das deutet darauf hin, dass noch weitere Aminosäuren an dem gewünschten
Mechanismus beteiligt sind und ebenso substituiert werden müssen.
Durch den Austausch der basischen Aminosäure Lysin gegen eine neutrale Aminosäure im
Mutein K78I, ist die ursprüngliche PheDH- Aktivität verloren gegangen, da Isoleucin keine
Wechselwirkung mit der Ketogruppe des Phenylpyruvats mehr eingeht und somit die
reduktive Aminierung nicht mehr stattfinden kann. Ähnliche Mutation wurde durch Sekimoto
et al. (Sekimoto et al., 1994) bei der Leucin Dehydrogenase aus Bacillus stearothermophilus
durchgeführt, wo Lysin 68 im aktiven Zentrum der LeuDH gegen Alanin bzw. Arginin
ausgetauscht wurde. Dadurch verlor die LeuDH ihre Aktivität aufgrund fehlender Bindung
zum Coenzym.
Trotz der erzielten Vermeidung der Wechselwirkung zwischen der Ketogruppe des Substrates
und der Aminosäure und des dadurch bei der PheDH vorhandenen Abstands zwischen dem
C4 des Nicotinamidrings und C? des Phenylpyruvats, konnte der gewünschte Reaktions-
mechanismus und der direkte Hydridtransfer nicht erzielt werden. In diesem Fall spielen nicht
nur die Ladung und die Größe der Aminosäuren für neue Umsetzungen eine Rolle, sondern
auch die räumliche Struktur der PheDH, die wahrscheinlich durch Substitution einzelner
Aminosäuren für eine Erweiterung des Substratspektrums nicht stark genug beeinflusst
werden konnte. Da die räumliche Struktur ähnlicher Amin Dehydrogenasen, wie die
gewünschte nicht bekannt ist, war es nicht möglich, eine Optimierung durch einen gezielten
Aminosäurenaustausch durchzuführen.
Die Muteine K66R und K66I wurden als Templates in der Zufallsmutagenese verwendet,
wobei das K66R für die Herstellung von neuen Varianten mit Phenyllactat Dehydrogenase-
Eigenschaften (Hydridtransfer), und das K66I zur Hersllung von Amin Dehydrogenase-
Varianten vorgesehen waren. Da die vorhandenen Austausche theoretisch wirksam sein
könnten, und an unbekannten Positionen weitere Aminosäurenaustausche für eine
Konformationsänderung notwendig sein könnten, war es durch Zufallsmutagenese möglich,
weitere Mutationen an neuen Positionen einzuführen und gleichzeitig die Veränderung an
Position 66 zu erhalten.
Die durch die „error prone“ Methode neukonstruierten Muteine - ca. 32000 Varianten aus der
K66I-Template - zeigten keine neue Aktivität gegen Phenylaceton. Zusätzliche Mutationen
zur K66I konnten die Akzeptanz des Phenylacetons als Substrat nicht positiv beeinflussen. Da
das Hauptinteresse an der Umsetzung von Ketonen zu Aminen bestand, wurde die neu-
konstruierte Enzymbank nicht besonders umfassend auf andere neue Eigenschaften gegen
weitere Substrate überprüft. Es ist aber von Interesse, in zukünftigen Arbeiten diese

Diskussion
124
Enzymbank für weitere Screenings zu verwenden und nach Muteinen für neue Umsetzungen
zu suchen.
Die Kombination aus zwei verschiedenen Methoden, sowohl der gezielten als auch der
ungezielten Mutagenese, zeigte sich aber als Erfolgskonzept beim K66R- Template, und
führte zu der gewünschten Aktivität. Das neu hergestellte Mutein trägt zwei Mutationen
T208A, E225D zusätzlich zum K66R und setzt Phenylpyruvat zum Phenyllactat um.
Aufgrund der Ergebnisse bei der Umsetzung mit diesem neuen Mutein (K66R-8) und der
schlechten Enantioselektivität wurden die drei Mutationen genauer untersucht und mittels
gezielter Mutagenese einzeln zurück mutiert zu den ursprünglichen Codons der PheDH, um
festzustellen, welche der o.g. drei Mutationen für die Enantioselektivität bzw. für die
Erweiterung des Substratspektrums verantwortlich ist. Die Substitution der einzelnen
Mutationen gegen die ursprünglichen Codons der PheDH führte zum Verlust der neuen LDH-
Aktivität. Aufgrund dessen, sind die drei Mutationen in diesem Mutein (K66R-8) zusammen
als Kombination für die neue Aktivität der PheDH verantwortlich.
Obwohl die zwei neuen Mutationen T208A und E225D nicht am aktiven Zentrum beteiligt
sind, kam es zu Veränderung des PheDH- Mechanismus. Die Aminosäure Alanin wurde
gegen Threonin an Position 208 ausgetauscht, so daß eine Wechselwirkung mit der
benachbarten Asparaginsäure 205 möglich ist, die wiederum eine starke Wechselwirkung zum
Coenzym besitzt. Die Substitution dieser Aminosäure zum ursprünglichen Threonin führte
zum Verlust der neuen Eigenschaft, wobei Threonin keine Wechselwirkung zum Asp 205 ein-
geht.
Eine Suche in den Datenbanken Genbank, Swissprot und Blast-NCBI ergab eine hohe Homo-
logie des K66R-8-Muteins mit der Lactat Dehydrogenase (LDH) aus Bifidobacterium longum
(Iwata et al., 1994). Wie aus dem Sequenzvergleich hervorgeht, zeigen das Mutein K66R-8
und die LDH aus Bifidobacterium longum eine hohe Homologie in den Sequenzen für die
Bildung von charakteristischen Sekundärstrukturen (Abbildung 67). Zwei der drei ein-
geführten Mutationen, nämlich R 66 und A 208, findet man in der LDH aus Bifidobacterium
longum wieder. An der Stelle der Asparaginsäure 225 in der K66R-8-Mutante kommt in der
LDH allerdings ein Alanin vor.
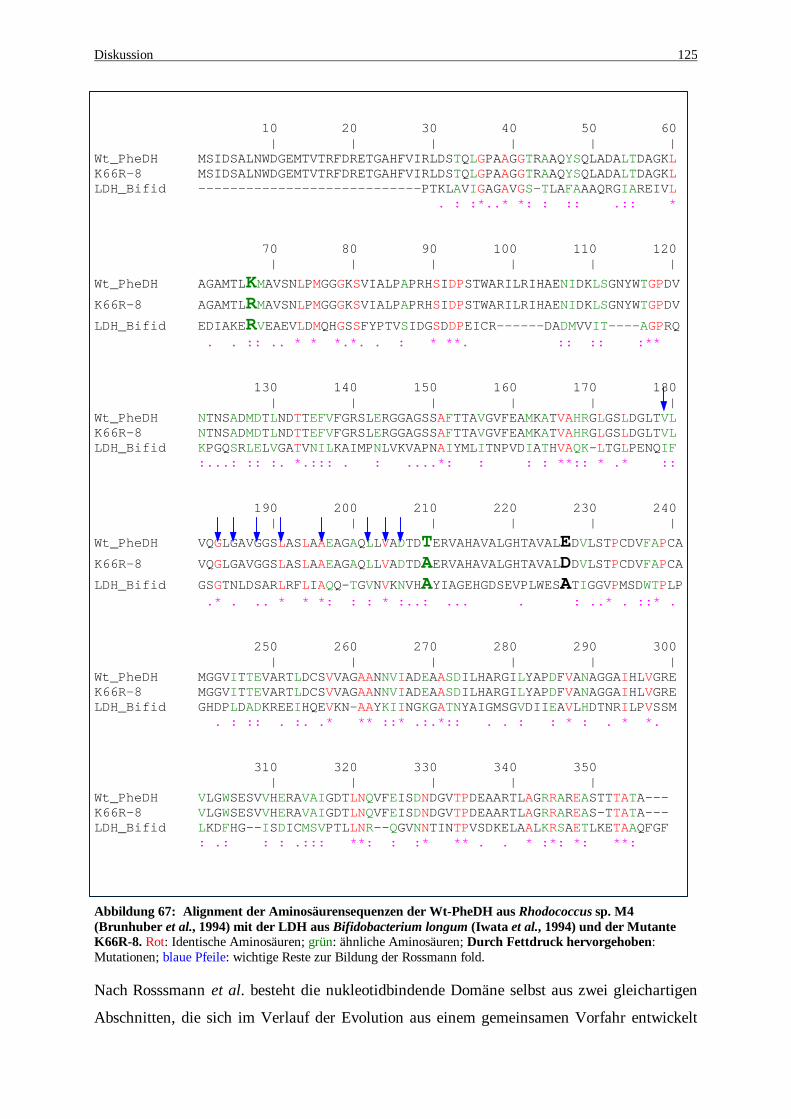
Diskussion
125
10 20 30 40 50 60 | | | | | | Wt_PheDH MSIDSALNWDGEMTVTRFDRETGAHFVIRLDSTQLGPAAGGTRAAQYSQLADALTDAGKL K66R-8 MSIDSALNWDGEMTVTRFDRETGAHFVIRLDSTQLGPAAGGTRAAQYSQLADALTDAGKL LDH_Bifid ----------------------------PTKLAVIGAGAVGS-TLAFAAAQRGIAREIVL . : :*..* *: : :: .:: * 70 80 90 100 110 120 | | | | | | Wt_PheDH AGAMTLKMAVSNLPMGGGKSVIALPAPRHSIDPSTWARILRIHAENIDKLSGNYWTGPDV K66R-8 AGAMTLRMAVSNLPMGGGKSVIALPAPRHSIDPSTWARILRIHAENIDKLSGNYWTGPDV LDH_Bifid EDIAKERVEAEVLDMQHGSSFYPTVSIDGSDDPEICR------DADMVVIT----AGPRQ . . :: .. * * *.*. . : * **. :: :: :** 130 140 150 160 170 180 | | | | | | Wt_PheDH NTNSADMDTLNDTTEFVFGRSLERGGAGSSAFTTAVGVFEAMKATVAHRGLGSLDGLTVL K66R-8 NTNSADMDTLNDTTEFVFGRSLERGGAGSSAFTTAVGVFEAMKATVAHRGLGSLDGLTVL LDH_Bifid KPGQSRLELVGATVNILKAIMPNLVKVAPNAIYMLITNPVDIATHVAQK-LTGLPENQIF :...: :: :. *.::: . : ....*: : : : **:: * .* :: 190 200 210 220 230 240 | | | | | | Wt_PheDH VQGLGAVGGSLASLAAEAGAQLLVADTDTERVAHAVALGHTAVALEDVLSTPCDVFAPCA K66R-8 VQGLGAVGGSLASLAAEAGAQLLVADTDAERVAHAVALGHTAVALDDVLSTPCDVFAPCA LDH_Bifid GSGTNLDSARLRFLIAQQ-TGVNVKNVHAYIAGEHGDSEVPLWESATIGGVPMSDWTPLP .* . .. * * *: : : * :..: ... . : ..* . ::* . 250 260 270 280 290 300 | | | | | | Wt_PheDH MGGVITTEVARTLDCSVVAGAANNVIADEAASDILHARGILYAPDFVANAGGAIHLVGRE K66R-8 MGGVITTEVARTLDCSVVAGAANNVIADEAASDILHARGILYAPDFVANAGGAIHLVGRE LDH_Bifid GHDPLDADKREEIHQEVKN-AAYKIINGKGATNYAIGMSGVDIIEAVLHDTNRILPVSSM . : :: . :. .* ** ::* .:.*:: . . : : * : . * *. 310 320 330 340 350 | | | | | Wt_PheDH VLGWSESVVHERAVAIGDTLNQVFEISDNDGVTPDEAARTLAGRRAREASTTTATA--- K66R-8 VLGWSESVVHERAVAIGDTLNQVFEISDNDGVTPDEAARTLAGRRAREAS-TTATA--- LDH_Bifid LKDFHG--ISDICMSVPTLLNR--QGVNNTINTPVSDKELAALKRSAETLKETAAQFGF : .: : : .::: **: : :* ** . . * :*: *: **:
Abbildung 67: Alignment der Aminosäurensequenzen der Wt-PheDH aus Rhodococcus sp. M4 (Brunhuber et al., 1994) mit der LDH aus Bifidobacterium longum (Iwata et al., 1994) und der Mutante K66R-8. Rot: Identische Aminosäuren; grün: ähnliche Aminosäuren; Durch Fettdruck hervorgehoben: Mutationen; blaue Pfeile: wichtige Reste zur Bildung der Rossmann fold.
Nach Rosssmann et al. besteht die nukleotidbindende Domäne selbst aus zwei gleichartigen
Abschnitten, die sich im Verlauf der Evolution aus einem gemeinsamen Vorfahr entwickelt

Diskussion
126
haben. Die einzelnen Dehydrogenasen (LDH: Lactatdehydrogenase, GAPDH: Glycerin-
aldehydphosphatdehydrogenase, ADH: Alkoholdehydrogenase) unterscheiden sich durch ihre
Substratspezifität. Die wiederum beruht auf der Verknüpfung der nukleotidbindenden
Domänen mit zusätzlichen Abschnitten am C- oder N-terminalen Ende (Eventoff et al., 1976;
Ruzheinikov et al., 2001). Alanin 208 ist nicht direkt an der „Rossmann fold“ beteiligt, liegt
aber angrenzend dazu und geht eine Wechselwirkung mit Asparaginsäure 205 ein, die
wiederum an der Rossmann fold beteiligt ist.
Dass die rückgängige Substitution der Mutation Asparaginsäure 225 zum Verlust der neuen
Aktivität geführt hat, war nicht zu erklären, da die Position 225 nicht an einer Wechsel-
wirkung zum Coenzym bzw. zu benachbarten Aminosäuren oder auch zum Substrat beteiligt
ist.
Unerwarteterweise zeigen die HPLC-Chromatogramme des durch die Dreifach-Mutante
gebildeten Phenyllactats, das die Reduktion nicht stereoselektiv erfolgt. L-Phenyllactat wird
zwar deutlich bevorzugt, aber nicht ausschließlich gebildet. Zusätzlich verändert sich der ee-
Wert noch im Verlauf der weiteren Umsetzung bis hin zum Racemat. Die HPLC-
Chromatogramme zeigen, dass diese nachträglich Veränderung des Enantiomeren-
verhältnisses wahrscheinlich durch einen sekundären Abbau des L-Phenyllactats durch eine E.
coli-eigene Lactat-Dehydrogenase bedingt ist. Auffallend ist aber die schwache Stereo-
selektivität des neuen Muteins. Diese konnte nicht durch Variation der Mutationen verändert
werden, da durch die Rücksubstitutionen gezeigt wurde, dass alle drei Mutationen eine Rolle
für die neue Eigenschaft spielen und die Deletion bzw. Substitution zum Verlust der Aktivität
führte. Eine Erklärung für die schwächere Stereoselektivität wäre, dass Arginin 66, das gegen
Lysin ausgetauscht wurde und im Gegensatz zum Lysin keine Wechselwirkung zur
Carboxylgruppe im Substrat hat, dafür verantwortlich sein könnte. Durch die Bindung von
Lysin 66 zur Carboxylgruppe im Substrat konnte ursprünglich das Substrat fixiert werden und
in der richtigen Orientierung in der Reaktion umgesetzt werden. Da aber im neuen Mutein die
Carboxylgruppe nicht mehr an Arginin bindet, könnte eine Reduktion nicht enantioselektiv
erfolgen.

Zusammenfassung
127
7 Zusammenfassung In der vorliegenden Arbeit wurden zwei NAD-abhängige Enzyme, Phenylalanin Dehydro-
genase (PheDH) aus Rhodococcus sp M4 und das malic enzyme aus E. coli K12 im gleichen
Expressionsvektor kloniert und deren Coexpression durchgeführt. Eine weitere Zielsetzung
dieser Arbeit war die Mutagenese der Phenylalanin Dehydrogenase, die sowohl mittels
Protein Design als auch durch Zufallsmutagenese (“directed evolution”) durchgeführt wurde.
Die Mutagenese diente zur Erweiterung des Substratspektrums der PheDH und um Einblicke
in deren Reaktionsmechanismus sowie Aufschluss über die Struktur – Funktionsbeziehungen
dieses Enzyms zu gewinnen.
Die Coexpression der PheDH und des malic enzymes diente der Ganzzellbiotransformation
zur reduktiven Aminierung von aromatischen Ketosäuren. Somit können Ketosäuren
umgesetzt und eine gleichzeitige Regenerierung des Coenzyms NADH mittels malic enzyme
durchgeführt werden.
Durch die Entwicklung einer neuen Klonierungsstrategie konnte das malic enzyme
erfolgreich amplifiziert, 3’ zur PheDH an der PstI- und HindIII- Schnittstelle kloniert und eine
Coexpression beider Enzyme ermöglicht werden. Mithilfe des bekannten N-Terminus des
malic enzymes konnten Einzelstränge amplifiziert und in weiteren PCR-Ansätze
komplementär verlängert werden.
Die PheDH und das malic enzyme wurden biochemisch umfassend charakterisiert und das
neue Plasmid-Konstrukt in E. coli JM105 für eine Ganzzellbiotransformation zur reduktiven
Aminierung von aromatischen Ketosäuren erfolgreich eingesetzt. Neben der Auswahl eines
geeigneten Vektor-Wirt-Systems wurden weitere Optimierungen, beispielsweise Induktions-
parameter, variiert, um eine effiziente Proteinexpression zu erzielen. Eine Hochzelldichte-
Fermentation führte zwar zu hoher Biomasse, dies wurde aber durch geringere spezifische
Aktivitäten erkauft.
Der Vorteil der Formiat Dehydrogenase, die sich bis dato für die Regenerierung des
Coenzyms NADH in situ durchgesetzt hat, liegt darin, dass bei der Regeneration durch
Formiat nur CO2 und keine weiteren Nebenprodukte entstehen. Da aber in dieser Arbeit nach-
gewiesen wurde, dass das malic enzyme ein durchaus erfolgreiches System darstellt und im
Ganzellsystem keine Nebenprodukte das gewünschte Produkt verunreinigen, könnten solche
in dieser Arbeit entwickelten Konstrukte technisch und wirtschaftlich interessanter sein. Das
L- Malat ist im Vergleich zum Na-Formiat vergleichbar kostengünstig. Ein weiterer
wesentlicher Vorteil der Ganzzellumsetzung liegt darin, dass auf die Präparation, möglicher-
weise Reinigung der Enzyme sowie den Zusatz an freiem Coenzym verzichtet werden kann.

Zusammenfassung
128
Zur Erweiterung des Substratspektrums der Phenylalanin Dehydrogenase aus Rhodococcus sp
M4, wurde die aufgeklärte Kristallstruktur mittels Computereinsatz modelliert und die am
aktiven Zentrum beteiligten Aminosäuren genauer für die Planung der Mutagenese
untersucht. Lysin 66 ist die Bindungsstelle zur Carboxylgruppe im Substrat (Phenylpyruvat
bzw. Phenylalanin). Für die Akzeptanz von Methylgruppen in Ketonen wurde der Austausch
des basischen Lysins 66 gegen eine neutrale Aminosäure mittels gezielter Mutagenese
postuliert. eine weitere Überlegung betraf die Änderung des Reaktionsmechanismus der
PheDH von einer reduktiven Aminierung zu einer Reduktion vom Typ einer Lactat
Dehydrogenase. Hierbei wurden Strukturen von Lactat-Dehydrogenasen verglichen und
Arginin als eine charakteristische Bindungsstelle zu Carboxylgruppe identifiziert, das dann an
der Stelle von Lysin 66 in der PheDH eingeführt worden ist.
Beide Versuche führten mittels gezielter Mutagenese zu negativen Ergebnissen. Durch den
Austausch der basischen Aminosäure Lysin 66 gegen das neutrale Isoleucin, verlor die
PheDH ihre ursprüngliche Aktivität. Beim zweiten Versuch, die Mutation des Lysin 66 gegen
Arginin und somit die Konstruktion einer Phenyllactat Dehydrogenase zu ermöglichen, ist die
PheDH-Aktivität aufgrund des Austausches einer basischen Aminosäure gegen eine basische
Aminosäure erhalten geblieben.
Für die Erweiterung des Substratspektrums war eine Änderung der Ladung im Falle K66I
nicht ausreichend. Daher wurden beide Mutanten, K66I und K66R, als Template in der
Zufallsmutagenese („directed evolution“) eingesetzt. Durch eine Zufallsmutagenese mittels
Variation der Konzentration an MgCl2 bzw. MnCl2 und dNTPs, konnte eine große Bank an
Enzymvarianten mit unterschiedlichen Mutationen hergestellt werden. Ein Screening nach der
gewünschten Phenyllactat Dehydrogenase erfolgte sowohl mittels einem in dieser Arbeit
entwickelten Verfahren als auch photometrisch durch die Absorption des Coenzyms NADH.
Sieben Mutanten aus ca. 20.000 Varianten zeigten Phenyllactat Dehydrogenase Aktivität
wobei nur die aktivste Mutante näher untersucht und zwei Mutationen T208A und E225D
zusätzlich zum K66R festgestellt wurden.
Die Kombination aus zwei verschiedenen Methoden, sowohl der gezielten als auch der
ungezielten Mutagenese, zeigte sich an diesem Beispiel als Erfolgskonzept und kann bei
zukünftigen Arbeiten in Betracht gezogen werden.

Anhang
129
DNA-Sequenz der Phenylalanin Dehydrogenase aus Rhodococcus sp. M4
1 ATGAGTATCG ACAGCGCACT GAACTGGGAC GGGGAAATGA CGGTCACCCG ATTCGACCGG 61 GAGACTGGTG CCCATTTCGT CATTCGACTC GATTCGACCC AACTCGGACC GGCGGCCGGA 121 GGCACCAGAG CCGCACAGTA CTCACAGCTG GCGGACGCCC TCACCGACGC CGGCAAATTG 181 GCGGGGGCGA TGACGTTGAA GATGGCAGTG AGCAACCTTC CGATGGGCGG GGGCAAATCC 241 GTCATTGCGC TTCCTGCGCC GCGTCATTCG ATCGATCCGA GCACGTGGGC ACGCATCCTC 301 CGAATCCACG CCGAGAACAT CGACAAGTTG TCCGGCAACT ACTGGACCGG ACCGGACGTC 361 AACACCAATT CGGCAGACAT GGATACTCTG AACGACACCA CCGAGTTCGT GTTCGGACGG 421 TCGCTCGAAC GCGGCGGCGC GGGTTCGAGC GCGTTCACCA CCGCCGTTGG CGTGTTCGAG 481 GCGATGAAGG CGACCGTCGC GCACCGTGGG CTGGGCTCAC TCGACGGTTT GACGGTCCTG 541 GTCCAAGGAC TGGGGGCAGT CGGAGGATCA TTGGCATCCC TGGCCGCCGA AGCGGGTGCG 601 CAACTCCTGG TGGCAGACAC CGACACCGAG CGAGTAGCGC ACGCTGTTGC GTTGGGCCAC 661 ACAGCGGTTG CCCTCGAGGA CGTTCTGTCC ACCCCGTGTG ATGTCTTCGC ACCCTGCGCA 721 ATGGGCGGCG TCATCACCAC CGAGGTGGCG CGAACACTCG ACTGTTCCGT CGTGGCCGGT 781 GCCGCCAACA ACGTCATCGC CGACGAGGCC GCCTCGGACA TCCTGCACGC ACGCGGAATT 841 CTGTACGCTC CCGACTTCGT GGCCAACGCC GGCGGTGCCA TCCACCTCGT AGGCCGGGAG 901 GTTCTCGGTT GGTCCGAGTC GGTTGTCCAC GAACGAGCAG TTGCCATAGG CGACACCCTG 961 AATCAGGTCT TCGAGATCTC CGACAACGAC GGCGTCACCC CGGACGAGGC CGCCCGCACT 1021 CTCGCTGGAC GGCGCGCCCG CGAGGCCTCG ACAACGACAG CGACTGCCTA GTAG

Anhang
130
DNA-Sequenz des malic enzymes aus E.coli K12
1 ATGGATATTC AAAAAAGAGT GAGTGACATG GAACCAAAAA CAAAAAAACA GCGTTCGCTT 61 TATATCCCTT ACGCTGGCCC TGTACTGCTG GAATTTCCGT TGTTGAATAA AGGCAGTGCC 121 TTCAGCATGG AAGAACGCCG TAACTTCAAC CTGCTGGGGT TACTGCCGGA AGTGGTCGAA 181 ACCATCGAAG AACAAGCGGA ACGAGCATGG ATCCAGTATC AGGGATTCAA AACCGAAATC 241 GACAAACACA TCTACCTGCG TAACATCCAG GACACTAACG AAACCCTCTT CTACCGTCTG 301 GTAAACAATC ATCTTGATGA GATGATGCCT GTTATTTATA CCCCAACCGT CGGCGCAGCC 361 TGTGAGCGTT TTTCTGAGAT CTACCGCCGT TCACGCGGCG TGTTTATCTC TTACCAGAAC 421 CGGCACAATA TGGACGATAT TCTGCAAAAC GTGCCGAACC ATAATATTAA AGTGATTGTG 481 GTGACTGACG GTGAACGCAT TCTGGGGCTT GGTGACCAGG GCATCGGCGG GATGGGCATT 541 CCGATCGGTA AACTGTCGCT CTATACCGCC TGTGGCGGCA TCAGCCCGGC GTATACCCTT 601 CCGGTGGTGC TGGATGTCGG AACGAACAAC CAACAGCTGC TTAACGATCC GCTGTATATG 661 GGCTGGCGTA ATCCGCGTAT CACTGACGAC GAATACTATG AATTCGTTGA TGAATTTATC 721 CAGGCTGTGA AACAACGCTG GCCAGACGTG CTGTTGCAGT TTGAAGACTT TGCTCAAAAA 781 AATGCGATGC CGTTACTTAA CCGCTATCGC AATGAAATTT GTTCTTTTAA CGATGACATT 841 CAGGGCACTG CGGCGGTAAC AGTCGGCACA CTGATCGCAG CAAGCCGCGC GGCAGGTGGT 901 CAGTTAAGCG AGAAAAAAAT CGTCTTCCTT GGCGCAGGTT CAGCGGGATG CGGCATTGCC 961 GAAATGATCA TCTCCCAGAC CCAGCGCGAA GGATTAAGCG AGGAAGCGGC GCGGCAGAAA 1021 GTCTTTATGG TCGATCGCTT TGGCTTGCTG ACTGACAAGA TGCCGAACCT GCTGCCTTTC 1081 CAGACCAAAC TGGTGCAGAA GCGCGAAAAC CTCAGTGACT GGGATACCGA CAGCGATGTG 1141 CTGTCACTGC TGGATGTGGT GCGCAATGTA AAACCAGATA TTCTGATTGG CGTCTCAGGA 1201 CAGACCGGGC TGTTTACGGA AGAGATCATC CGTGAGATGC ATAAACACTG TCCGCGTCCG 1261 ATCGTGATGC CGCTGTCTAA CCCGACGTCA CGCGTGGAAG CCACACCGCA GGACATTATC 1321 GCCTGGACCG AAGGTAACGC GCTGGTCGCC ACGGGCAGCC CGTTTAATCC AGTGGTATGG 1381 AAAGATAAAA TCTACCCTAT CGCCCAGTGT AACAACGCCT TTATTTTCCC GGGCATCGGC 1441 CTGGGTGTTA TTGCTTCCGG CGCGTCACGT ATCACCGATG AGATGCTGAT GTCGGCAAGT 1501 GAAACGCTGG CGCAGTATTC ACCATTGGTG CTGAACGGCG AAGGTATGGT ACTGCCGGAA 1561 CTGAAAGATA TTCAGAAAGT CTCCCGCGCA ATTGCGTTTG CGGTTGGCAA AATGGCGCAG 1621 CAGCAAGGCG TGGCGGTGAA AACCTCTGCC GAAGCCCTGC AACAGGCCAT TGACGATAAT 1681 TTCTGGCAAG CCGAATACCG CGACTACCGC CGTACCTCCA TCTAA

Literaturverzeichnis
131
8 Literaturverzeichnis
Abrams W. B., Davies R. O., and Ferguson R. K. (1984). Overview: the role of angiotensin-converting enzyme inhibitors in cardiovascular therapy. Fed Proc 43: 1314-21.
Andersen D. C., Swartz J., Ryll T., Lin N., and Snedecor B. (2001). Metabolic oscillations in an E. coli fermentation. Biotechnol Bioeng 75: 212-8.
Arnold F. H., and Moore J. C. (1997). Optimizing industrial enzymes by directed evolution. Adv Biochem Eng Biotechnol 58: 1-14.
Asano Y., Nakazawa A., and Endo K. (1987). Novel phenylalanine dehydrogenases from Sporosarcina ureae and Bacillus sphaericus. Purification and characterization. J Biol Chem 262: 10346-54.
Babbitt P. C., West B. L., Buechter D. D., Kuntz I. D., and Kenyon G. L. (1990). Removal of a proteolytic activity associated with aggregates formed from expression of creatine kinase in Escherichia coli leads to improved recovery of active enzyme. Biotechnology (N Y) 8: 945-9.
Bagchi S., Wise L. S., Brown M. L., Bregman D., Sul H. S., and Rubin C. S. (1987). Structure and expression of murine malic enzyme mRNA. Differentiation- dependent accumulation of two forms of malic enzyme mRNA in 3T3-L1 cells. J Biol Chem 262: 1558-65.
Baker P. J., Turnbull A. P., Sedelnikova S. E., Stillman T. J., and Rice D. W. (1995). A role for quaternary structure in the substrate specificity of leucine dehydrogenase. Structure 3: 693-705.
Baker P. J., Waugh M. L., Wang X. G., Stillman T. J., Turnbull A. P., Engel P. C., and Rice D. W. (1997). Determinants of substrate specificity in the superfamily of amino acid dehydrogenases. Biochemistry 36: 16109-15.
Balbas P., and Bolivar F. (1990). Design and construction of expression plasmid vectors in Escherichia coli. Methods Enzymol 185: 14-37.
Bentley J., Hyatt L. S., Ainley K., Parish J. H., Herbert R. B., and White G. R. (1993). Cloning and sequence analysis of an Escherichia coli gene conferring bicyclomycin resistance. Gene 127: 117-20.
Bergter F. (1983). Wachstum von Microorganismen. Verlag Chemie, Weinheim.
Birnboim H. C., and Doly J. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7: 1513-23.
Blattner F. R., Plunkett G., 3rd, Bloch C. A., Perna N. T., Burland V., Riley M., Collado-Vides J., Glasner J. D., Rode C. K., Mayhew G. F., Gregor J., Davis N. W., Kirkpatrick H. A., Goeden M. A., Rose D. J., Mau B., and Shao Y. (1997). The complete genome sequence of Escherichia coli K-12. Science 277: 1453-74.
Blum H., Beier H., and Gross H. J. (1987). Improved Silver Staining of Plant Proteins, RNA and DNA in Polyacrylamide Gels. Electrophoresis 8: 93-99.
Bommarius A. S., and Drauz K. (1994). An enzymatic route to L-ornithine from arginine-activation, selectivity and stabilization of L-arginase. Bioorg Med Chem 2: 617-26.
Bommarius A. S., Schwarm M., Stingl K., Kottenhahn M., Huthmacher K., and Drauz K. (1995). Synthesis and use of enantiomerically pure tert-Leucine. Tetr. Asymm 6: 2841-2888.

Literaturverzeichnis
132
Bornscheuer U. T., Altenbuchner J., and Meyer H. H. (1998). Directed evolution of an esterase for the stereoselective resolution of a key intermediate in the synthesis of epothilones. Biotechnol Bioeng 58: 554-9.
Bornscheuer U. T., Altenbuchner J., and Meyer H. H. (1999). Directed evolution of an esterase: screening of enzyme libraries based on pH-indicators and a growth assay. Bioorg Med Chem 7: 2169-73.
Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-54.
Brunhuber N. M., Banerjee A., Jacobs W. R., Jr., and Blanchard J. S. (1994). Cloning, sequencing, and expression of Rhodococcus L-phenylalanine dehydrogenase. Sequence comparisons to amino-acid dehydrogenases. J Biol Chem 269: 16203-11.
Brunhuber N. M., Thoden J. B., Blanchard J. S., and Vanhooke J. L. (2000). Rhodococcus L-phenylalanine dehydrogenase: kinetics, mechanism, and structural basis for catalytic specificity. Biochemistry 39: 9174-87.
Bryan P. N., Rollence M. L., Pantoliano M. W., Wood J., Finzel B. C., Gilliland G. L., Howard A. J., and Poulos T. L. (1986). Proteases of enhanced stability: characterization of a thermostable variant of subtilisin. Proteins 1: 326-34.
Bursch J., Johs R., and Heintzen P. (1970). [Studies on the validity of the Lambert-Beer law in the x-ray measurement of contrast media density]. Fortschr Geb Rontgenstr Nuklearmed 112: 259-66.
Camacho M., Rodriguez-Arnedo A., and Bonete M. J. (2002). NADP-dependent isocitrate dehydrogenase from the halophilic archaeon Haloferax volcanii: cloning, sequence determination and overexpression in Escherichia coli. FEMS Microbiol Lett 209: 155-60.
Carrea G., Ottolina G., Pasta P., and Riva S. (1996). Synthetic applications of NAD(P)(H)-dependent enzymes. Ann N Y Acad Sci 799: 642-9.
Centerwall S. A., and Centerwall W. R. (2000). The discovery of phenylketonuria: the story of a young couple, two retarded children, and a scientist. Pediatrics 105: 89-103.
Choi J. S., Kim J. S., Joe C. O., Kim S., Ha K. S., and Park Y. M. (1999). Improved cycle sequencing of GC-rich DNA template. Exp Mol Med 31: 20-4.
Clarke A. R., Wigley D. B., Chia W. N., Barstow D., Atkinson T., and Holbrook J. J. (1986). Site-directed mutagenesis reveals role of mobile arginine residue in lactate dehydrogenase catalysis. Nature 324: 699-702.
Cunningham B. C., and Wells J. A. (1987). Improvement in the alkaline stability of subtilisin using an efficient random mutagenesis and screening procedure. Protein Eng 1: 319-25.
de Boer L., van Rijssel M., Euvernik G. J., and Dijkhuizen a. L. (1989). Purification, characterization and regulation of a monomeric L-phenylalanine dehydrogenase from the facultative methylotroph Nocardia sp. 239. Arch Microbiol 153: 12-18.
De Mot R., Nagy I., and Baumeister W. (1998). A self-compartmentalizing protease in Rhodococcus: the 20S proteasome. Antonie Van Leeuwenhoek 74: 83-7.
DelVecchio V. G., Kapatral V., Redkar R. J., Patra G., Mujer C., Los T., Ivanova N., Anderson I., Bhattacharyya A., Lykidis A., Reznik G., Jablonski L., Larsen N., D'Souza M., Bernal A., Mazur M., Goltsman E., Selkov E., Elzer P. H., Hagius S.,

Literaturverzeichnis
133
O'Callaghan D., Letesson J. J., Haselkorn R., Kyrpides N., and Overbeek R. (2002). The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc Natl Acad Sci U S A 99: 443-8.
Deng W. P., and Nickoloff J. A. (1992). Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal Biochem 200: 81-8.
Derbyshire K. M., Salvo J. J., and Grindley N. D. (1986). A simple and efficient procedure for saturation mutagenesis using mixed oligodeoxynucleotides. Gene 46: 145-52.
Diesterhaft M. D., and Freese E. (1973). Role of pyruvate carboxylase, phosphoenolpyruvate carboxykinase, and malic enzyme during growth and sporulation of Bacillus subtilis. J Biol Chem 248: 6062-70.
Donovan R. S., Robinson C. W., and Glick B. R. (2000). Optimizing the expression of a monoclonal antibody fragment under the transcriptional control of the Escherichia coli lac promoter. Can J Microbiol 46: 532-41.
Drauz K., Bommarius A., Knaup G., Kottenhahn M., and Schwarm M. (1994). Synthesis of chiral Building Blocks by Amino Acid Transformation. Chiral Europe 94, Symposium 19.-20. Sept. Nizza, Frankreich: 13-16.
Ernst&Young (1998). European Life Sciences 98. Aufbruchstimmung 1998, Erster Deutscher Biotechnologie Report.
Esipov R. S., Gurevich A. I., Kaiushin A. L., Korosteleva M. D., and Belova M. A. (1999). The dependence of the E.coli gene expression level on the structure of the translation initiation region (TIR). IV. Distal complementary TIR interactions with the mRNA coding region. Bioorg Khim 25: 548-53.
Eventoff W., Tanaka N., and Rossmann M. G. (1976). Crystalline bovine liver catalase. J Mol Biol 103: 799-801.
Faber K. (1997). Biotransformation in organic chemistry, Springer-Verlag Berlin Heidelberg New York, 3rd ed.
Fieschko J. C. (1989). Fermentation Technology Using Recombinant Microorganisms. Biotechnology. Weinheim VCH 7b: 117-140.
Galkin A., Kulakova L., Ohshima T., Esaki N., and Soda K. (1997). Construction of a new leucine dehydrogenase with preferred specificity for NADP+ by site-directed mutagenesis of the strictly NAD+-specific enzyme. Protein Eng 10: 687-90.
Geerse R. H., van der Pluijm J., and Postma P. W. (1989). The repressor of the PEP:fructose phosphotransferase system is required for the transcription of the pps gene of Escherichia coli. Mol Gen Genet 218: 348-52.
Grantham R., Gautier C., Gouy M., Jacobzone M., and Mercier R. (1981). Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res 9: r43-74.
Hanahan D. (1983). Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166: 557-80.
Hanson R. L., Howell J. M., LaPorte T. L., Donovan M. J., Cazzulino D. L., Zannella V. V., Montana M. A., Nanduri V. B., Schwarz S. R., Eiring R. F., Durand S. C., Wasylyk J. M., Parker W. L., Liu M. S., Okuniewicz F. J., Chen B., Harris J. C., Natalie K. J., Ramig K., Swaminathan S., Rosso V. W., Pack S. K., Lotz B. T., Bernot P. J., Rusowicz A., Lust D. A., Tse K. S., Venit J. J., Szarka L. J., and Patel R. N. (2000).

Literaturverzeichnis
134
Synthesis of allysine ethylene acetal using phenylalanine dehydrogenase from Thermoactinomyces intermedius. Enzyme Microb Technol 26: 348-358.
Hart K. W., Clarke A. R., Wigley D. B., Chia W. N., Barstow D. A., Atkinson T., and Holbrook J. J. (1987). The importance of arginine 171 in substrate binding by Bacillus stearothermophilus lactate dehydrogenase. Biochem Biophys Res Commun 146: 346-53.
Hausler R. E., Holtum J. A., and Latzko E. (1987). CO2 is the inorganic carbon substrate of NADP malic enzymes from Zea mays and from wheat germ. Eur J Biochem 163: 619-26.
Hibino T., Misawa S., Wakiyama M., Maeda S., Yazaki K., Kumagai I., Ooi T., and Miura K. (1994). High-level expression of porcine muscle adenylate kinase in Escherichia coli: effects of the copy number of the gene and the translational initiation signals. J Biotechnol 32: 139-48.
Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989). Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51-9.
Huang C. Y., Yuan C. J., Livanova N. B., and Graves D. J. (1993). Expression, purification, characterization, and deletion mutations of phosphorylase kinase gamma subunit: identification of an inhibitory domain in the gamma subunit. Mol Cell Biochem 127-128: 7-18.
Hummel W. (1997). New alcohol dehydrogenases for the synthesis of chiral compounds. Adv Biochem Eng Biotechnol 58: 145-84.
Hummel W., and Kula M. R. (1989). Dehydrogenases for the synthesis of chiral compounds. Eur J Biochem 184: 1-13.
Hummel W., Schütte H., Schmidt E., Wandrey C., and Kula M. R. (1987). Isolation of L-phenylalanine dehydrogenase from Rhodococcus sp. M4 and its application for the production of L-phenylalanine. Appl Microbiol Biotechnol 26: 409-416.
Hummel W., Weiss N., and Kula M. R. (1984). Isolation and characterization of bacterium possessing L-phenylalanine dehydrogenase activity. Arch Microbiol 137: 47-52.
Itakura K., Hirose T., Crea R., Riggs A. D., Heyneker H. L., Bolivar F., and Boyer H. W. (1977). Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 198: 1056-63.
Iwakura M., Tokushige M., Katsuki H., and Muramatsu S. (1978). Studies on regulatory functions of malic enzymes. V. Comparative studies of malic enzymes in bacteria. J Biochem (Tokyo) 83: 1387-94.
Iwata S., Kamata K., Yoshida S., Minowa T., and Ohta T. (1994). T and R states in the crystals of bacterial L-lactate dehydrogenase reveal the mechanism for allosteric control. Nat Struct Biol 1: 176-85.
Jaag H. (1968). Über proteolytische Enzyme, deren Prüfmöglichkeit in der Waschmittelindustrie. Europ. Hochschul Reihe. Verlag Herbert Lang, Bern.
Janssen D., Keuning S., and Witholt B. (1987). Involvment of a Quinoproteine Alcohol Dehydrogenase and NAD-dependent Aldehyde Dehydrogenase in 2-Chlorethanol Metabolism im Xanthobacter autrophicus CJ10. J. Gen. Microbiol 133: 85-92.
Jensen E. B., and Carlsen S. (1990). Production of Recombinant Human Growth Hormone in Escherichia coli: Expression of Different Precursors and Physiological Effects of Glucose, Acetate, and Salts. Biotechnol. Bioeng 36: 1-11.

Literaturverzeichnis
135
Jiang X., Kowalski J., and Kelly J. W. (2001). Increasing protein stability using a rational approach combining sequence homology and structural alignment: Stabilizing the WW domain. Protein Sci 10: 1454-65.
Knichel W., and Radler F. (1982). D-Malic enzyme of Pseudomonas fluorescens. Eur J Biochem 123: 547-52.
Knorre W. A., Deckwer W. D., Korz D., Pohl H. D., Riesenberg D., Ross A., Sanders E., and Schulz V. (1991). High cell density fermentation of recombinant Escherichia coli with computer-controlled optimal growth rate. Ann N Y Acad Sci 646: 300-6.
Kobayashi K., Doi S., Negoro S., Urabe I., and Okada H. (1989). Structure and properties of malic enzyme from Bacillus stearothermophilus. J Biol Chem 264: 3200-5.
Kometani T., Morita Y., Furui H., Yoshii H., and Matsuno R. (1994). NAD(P)H regeneration using ethanol as an energy source in baker's yeast-mediated bioreaction. J. Ferm. Bioeg. 17: 13-16.
Kragl U., Vasic-Racki D., and Wandrey C. (1992). Kontinuierliche Reaktionsführung mit löslichen Enzymen. Chem. -Ing. -Tech 64: 499-509.
Krix G., Bommarius A., Drauz K., Kottenhahn M., Schwarm M., and Kula M. R. (1997). Enzymatic Reductionof Alpha-Keto Acids Leading to L-Amino Acids or Chiral Hydroxy Acid. J. Biotechnol.: 29-39.
Kuchner O., and Arnold F. H. (1997). Directed evolution of enzyme catalysts. Trends Biotechnol 15: 523-30.
Kula M. R., and Wandrey C. (1987). Continuous enzymatic transformation in an enzyme-membrane reactor with simultaneous NADH regeneration. Methods Enzymol 136: 9-21.
Laemmli U. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685.
Lamed R., and Zeikus J. G. (1981). Thermostable, ammonium-activated malic enzyme of Clostridium thermocellum. Biochim Biophys Acta 660: 251-5.
Landwall P., and Holme T. (1977). Removal of inhibitors of bacterial growth by dialysis culture. J Gen Microbiol 103: 345-52.
Lee S. Y. (1996). High cell-density culture of Escherichia coli. Trends Biotechnol 14: 98-105.
Leonida M. D., Sobolov S. B., and Fry A. J. (1998). FAD-mediated enzymatic conversion of NAD+ to NADH: application to chiral synthesis of L-lactate. Bioorg Med Chem Lett 8: 2819-24.
Lerch H. P., Frank R., and Collins J. (1989). Cloning, sequencing and expression of the L-2-hydroxyisocaproate dehydrogenase-encoding gene of Lactobacillus confusus in Escherichia coli. Gene 83: 263-70.
Luyten M. A., Gold M., Friesen J. D., and Jones J. B. (1989). On the effects of replacing the carboxylate-binding arginine-171 by hydrophobic tyrosine or tryptophan residues in the L-lactate dehydrogenase from Bacillus stearothermophilus. Biochemistry 28: 6605-10.
Magnuson M. A., Morioka H., Tecce M. F., and Nikodem V. M. (1986). Coding nucleotide sequence of rat liver malic enzyme mRNA. J Biol Chem 261: 1183-6.

Literaturverzeichnis
136
Mahajan S. K., Chu C. C., Willis D. K., Templin A., and Clark A. J. (1990). Physical analysis of spontaneous and mutagen-induced mutants of Escherichia coli K-12 expressing DNA exonuclease VIII activity. Genetics 125: 261-73.
Mason C. A., and Bailey J. E. (1989). Effects of Plasmid Presence on Growth and Enzyme Activity of Escherichia coli DH5-Alpha. Appl. Microbiol. Biotechnol. 32: 54-60.
Matsumura M., Signor G., and Matthews B. W. (1989). Substantial increase of protein stability by multiple disulphide bonds. Nature 342: 291-3.
Matthews B. W., Nicholson H., and Becktel W. J. (1987). Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc. Natl. Acad. Sci. USA 84: 6663-6667.
May B. J., Zhang Q., Li L. L., Paustian M. L., Whittam T. S., and Kapur V. (2001). Complete genomic sequence of Pasteurella multocida, Pm70. Proc Natl Acad Sci U S A 98: 3460-5.
McEvoy J. J., and Joyce A. (1974). Production of L-phenylalanine by DL-phenylalanine hydroxamate-resistant Tyr- mutants of Bacillus subtilis. Mol Cell Biochem 4: 191-5.
Misono H., Yonezawa J., Nagata S., and Nagasaki S. (1989). Purification and characterization of a dimeric phenylalanine dehydrogenase from Rhodococcus maris K-18. J Bacteriol 171: 30-6.
Moore J. C., Jin H. M., Kuchner O., and Arnold F. H. (1997). Strategies for the in vitro evolution of protein function: enzyme evolution by random recombination of improved sequences. J Mol Biol 272: 336-47.
Nakamura A., Urabe I., and Okada H. (1986). Anchimeric assistance in the intramolecular reaction of glucose- dehydrogenase-polyethylene glycol NAD conjugate. J Biol Chem 261: 16792-4.
Nakayama K., Araki K., and Kase H. (1978). Microbial production of essential amino acid with Corynebacterium glutamicum mutants. Adv Exp Med Biol 105: 649-61.
Neubauer P., Ahman M., Tornkvist M., Larsson G., and Enfors S. O. (1995). Response of guanosine tetraphosphate to glucose fluctuations in fed- batch cultivations of Escherichia coli. J Biotechnol 43: 195-204.
Nicholls D. J., Davey M., Jones S. E., Miller J., Holbrook J. J., Clarke A. R., Scawen M. D., Atkinson T., and Goward C. R. (1994). Substitution of the amino acid at position 102 with polar and aromatic residues influences substrate specificity of lactate dehydrogenase. J Protein Chem 13: 129-33.
Nossal G. J. (1980). Human insulin through recombinant DNA technology. Med J Aust 2: 295-6.
Ohshima T., Takada H., Yoshimura T., Esaki N., and Soda K. (1991). Distribution, purification, and characterization of thermostable phenylalanine dehydrogenase from thermophilic actinomycetes. J Bacteriol 173: 3943-8.
Okazaki N., Hibino Y., Asano Y., Ohmori M., Numao N., and Kondo K. (1988). Cloning and nucleotide sequencing of phenylalanine dehydrogenase gene of Bacillus sphaericus. Gene 63: 337-41.
Oliphant A. R., and Struhl K. (1989). An efficient method for generating proteins with altered enzymatic properties: application to beta-lactamase. Proc Natl Acad Sci U S A 86: 9094-8.

Literaturverzeichnis
137
Ondetti M. A., and Cushman D. W. (1981). Inhibition of the renin-angiotensin system. A new approach to the therapy of hypertension. J Med Chem 24: 355-61.
Oppenheimer N. J., and Kaplan N. O. (1974). Structure of the primary acid rearrangement product of reduced nicotinamide adenine dinucleotide (NADH). Biochemistry 13: 4675-85.
Paalme T., Elken R., Kahru A., Vanatalu K., and Vilu R. (1997). The growth rate control in Escherichia coli at near to maximum growth rates: the A-stat approach. Antonie Van Leeuwenhoek 71: 217-30.
Paalme T., Tiisma K., Kahru A., Vanatalu K., and Vilu R. (1990). Glucose-limited Fed-batch Cultivation of Escherichia coli with Computer-Controlled Fixed Growth Rate. Biotechnol. Bioeng. 35: 312-319.
Pan J. G., Rhee J. S., and Lebeaut J. M. (1987). Physiological Constrains in Increasing Biomass Concentration of Escherichia coli B in Fed-batch Culture. Biotechnol.Lett. 9: 89-94.
Parkhill J., Dougan G., James K. D., Thomson N. R., Pickard D., Wain J., Churcher C., Mungall K. L., Bentley S. D., Holden M. T., Sebaihia M., Baker S., Basham D., Brooks K., Chillingworth T., Connerton P., Cronin A., Davis P., Davies R. M., Dowd L., White N., Farrar J., Feltwell T., Hamlin N., Haque A., Hien T. T., Holroyd S., Jagels K., Krogh A., Larsen T. S., Leather S., Moule S., O'Gaora P., Parry C., Quail M., Rutherford K., Simmonds M., Skelton J., Stevens K., Whitehead S., and Barrell B. G. (2001). Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413: 848-52.
Pasquo A., Britton K. L., Baker P. J., Brearley G., Hinton R. J., Moir A. J., Stillman T. J., and Rice D. W. (1998). Crystallization of NAD+-dependent phenylalanine dehydrogenase from Nocardia sp239. Acta Crystallogr D Biol Crystallogr 54: 269-72.
Rhee J. I., Bode J., Diaz-Ricci J. C., Poock D., Weigel B., Kretzmer G., and Schugler K. (1997). Influence of the medium composition and plasmid combination on the growth of recombinant Escherichia coli JM109 and on the production of the fusion protein EcoRI::SPA. J Biotechnol 55: 69-83.
Rice G. C., Goeddel D. V., Cachianes G., Woronicz J., Chen E. Y., Williams S. R., and Leung D. W. (1992). Random PCR mutagenesis screening of secreted proteins by direct expression in mammalian cells. Proc Natl Acad Sci U S A 89: 5467-71.
Riesenberg D. (1991). High-cell-density cultivation of Escherichia coli. Curr Opin Biotechnol 2: 380-4.
Riesenberg D., and Guthke R. (1999). High-cell-density cultivation of microorganisms. Appl Microbiol Biotechnol 51: 422-30.
Riesenberg D., Schulz V., Knorre W. A., Pohl H. D., Korz D., Sanders E. A., Ross A., and Deckwer W. D. (1991). High cell density cultivation of Escherichia coli at controlled specific growth rate. J Biotechnol 20: 17-27.
Ruzheinikov S. N., Burke J., Sedelnikova S., Baker P. J., Taylor R., Bullough P. A., Muir N. M., Gore M. G., and Rice D. W. (2001). Glycerol dehydrogenase. structure, specificity, and mechanism of a family III polyol dehydrogenase. Structure (Camb) 9: 789-802.
Sanden A. M., Shokri A., and Larsson G. (2002). Growth rate-dependent changes in Escherichia coli membrane structure and protein leakage. Appl Microbiol Biotechnol 58: 386-92.

Literaturverzeichnis
138
Sanger F., Air G. M., Barrell B. G., Brown N. L., Coulson A. R., Fiddes C. A., Hutchison C. A., Slocombe P. M., and Smith M. (1977). Nucliotide sequence of bacteriophage phi X174 DNA. Nature 265: 687-95.
Schütte H., Flossdorf J., Sahm H., and Kula M. R. (1976). Purification and properties of formaldehyde dehydrogenase and formate dehydrogenase from Candida boidinii. Eur J Biochem 62: 151-60.
Sekimoto T., Fukui T., and Tanizawa K. (1994). Involvement of conserved lysine 68 of Bacillus stearothermophilus leucine dehydrogenase in substrate binding. J Biol Chem 269: 7262-6.
Shaked S., and Whitesides G. (1980). Enzyme-catalyzed Organic Synthesis: NADH Regeneration by Using Formate Dehydrogenase. J. Am. Chem. Soc. 102: 7105-7107.
Sharp P. M., Cowe E., Higgins D. G., Shields D. C., and Wolfe K. H. W., F. (1988). Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within species diversity. Nucl. Acids Res. 16: 8207-8211.
Shin C. S., Hong M. S., Bae C. S., and Lee J. (1997). Enhanced production of human mini-proinsulin in fed-batch cultures at high cell density of Escherichia coli BL21(DE3)[pET-3aT2M2]. Biotechnol Prog 13: 249-57.
Slusarczyk H., Felber S., Kula M. R., and Pohl M. (2000). Stabilization of NAD-dependent formate dehydrogenase from Candida boidinii by site-directed mutagenesis of cysteine residues. Eur J Biochem 267: 1280-9.
Solovjeva O. N., and Kochetov G. A. (1999). Inhibition of transketolase by p-hydroxyphenylpyruvate. FEBS Lett 462: 246-8.
Stader J. (1995). Gene expression in recombinant Escherichia coli. Bioprocess Technol 22: 1-51.
Stemmer W. P. (1994). Rapid evolution of a protein in vitro by DNA shuffling. Nature 370: 389-91.
Stols L., and Donnelly M. I. (1997). Production of succinic acid through overexpression of NAD(+)-dependent malic enzyme in an Escherichia coli mutant. Appl Environ Microbiol 63: 2695-701.
Takada H., Yoshimura T., Ohshima T., Esaki N., and Soda K. (1991). Thermostable phenylalanine dehydrogenase of Thermoactinomyces intermedius: cloning, expression, and sequencing of its gene. J Biochem (Tokyo) 109: 371-6.
Tedin K., Resch A., and Blasi U. (1997). Requirements for ribosomal protein S1 for translation initiation of mRNAs with and without a 5' leader sequence. Mol Microbiol 25: 189-99.
Thein S. L., and Wallace R. B. (1988). The use of synthetic oligonucleotides as a specific hybridization probes in the diagnosis of genetic disorders. Human genetic diseases: a practical approach (Davis, K.E., ed.) IRL Press, Herndon, Virginia.
Uhlig H. (1991). Enzyme arbeiten für uns. Technische Enzyme und ihre Anwendung. Carll Hanser Verlag, München.
Vacca J., Christen P., Malashkevich V. N., Jansonius J. N., and Sandmeier E. (1995). Substitution of apolar residues in the active site of aspartate aminotransferase by histidine-effects on reaction and substrate specificity. Biochem. 227: 481-487.

Literaturverzeichnis
139
van den Bogaard P. T., Kleerebezem M., Kuipers O. P., and de Vos W. M. (2000). Control of lactose transport, beta-galactosidase activity, and glycolysis by CcpA in Streptococcus thermophilus: evidence for carbon catabolite repression by a non-phosphoenolpyruvate-dependent phosphotransferase system sugar. J Bacteriol 182: 5982-9.
van Soolingen D., Hermans P. W., de Haas P. E., Soll D. R., and van Embden J. D. (1991). Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence- dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J Clin Microbiol 29: 2578-86.
Vanhooke J. L., Thoden J. B., Brunhuber N. M., Blanchard J. S., and Holden H. M. (1999). Phenylalanine dehydrogenase from Rhodococcus sp. M4: high-resolution X-ray analyses of inhibitory ternary complexes reveal key features in the oxidative deamination mechanism. Biochemistry 38: 2326-39.
Vellanoweth R. L., and Rabinowitz J. C. (1992). The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol 6: 1105-14.
Watts J. L., Lowery D. E., Teel J. F., Ditto C., Horng J. S., and Rossbach S. (2001). Phylogenetic studies on Corynebacterium bovis isolated from bovine mammary glands. J Dairy Sci 84: 2419-23.
Wendel U., Koppelkamm M., and Hummel W. (1991). Enzymatic phenylalanine estimation for the management of patients with phenylketonuria. Clin Chim Acta 201: 95-8.
Wendel U., Koppelkamm M., Hummel W., Sander J., and Langenbeck U. (1990). A new approach to the newborn screening for hyperphenylalaninemias: use of L-phenylalanine dehydrogenase and microtiter plates. Clin Chim Acta 192: 165-70.
Wichmann R., Wandrey C., Bückmann A. F., and Kula M. R. (1981). Continuous enzymatic transformation in an enzyme membrane reactor with simultaneous NAD(H) regeneration. Biotechnol. Bioeng. 23: 2789-2802.
Wilkinson A. J., Fersht A. R., Blow D. M., Carter P., and Winter G. (1984). A large increase in enzyme-substrate affinity by protein engineering. Nature 307: 187-8.
Willeford K. O., and Wedding R. T. (1987). Evidence for a multiple subunit composition of plant NAD malic enzyme. J Biol Chem 262: 8423-9.
Winberry L. K., Morris S. M., Jr., Fisch J. E., Glynias M. J., Jenik R. A., and Goodridge A. G. (1983). Molecular cloning of cDNA sequences for avian malic enzyme. Nutritional and hormonal regulation of malic enzyme mRNA levels in avian liver cells in vivo and in culture. J Biol Chem 258: 1337-42.
Wong C., and Whitesides G. (1981). Enzyme-catalyzed Organic Synthesis: NAD(P)H Cofactor Regeneration by using Glucose-6-Phosphate and the Glucose-6-Phosphate Dehydrogenase from Leuconoctoc mesenteroides. J. Am. Chem. Soc. 103: 4890-4899.
Xu Y., Shen M., Zhao Q., and Zhang H. (1994). Construction and expression of high efficiency expressing plasmid of transforming growth factor alpha-Pseudomonas aeruginosa exotoxin fusion protein TGF alpha-PE40. Chin J Biotechnol 10: 233-9.
Yang Q. H., Wu C. L., Lin K., and Li L. (1997). Low concentration of inducer favors production of active form of 6- phosphofructo-2-kinase/fructose-2,6-bisphosphatase in Escherichia coli. Protein Expr Purif 10: 320-4.

Literaturverzeichnis
140
Yee L., and Blanch H. W. (1992). Recombinant protein expression in high cell density fed-batch cultures of Escherichia coli. Biotechnology (N Y) 10: 1550-6.
You L., and Arnold F. H. (1996). Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide [published erratum appears in Protein Eng 1996 Aug;9(8):719]. Protein Eng 9: 77-83.
Zaks A. (2001). Industrial biocatalysis. Curr Opin Chem Biol 5: 130-6.
Zhang Z., Schofield C. J., Baldwin J. E., Thomas P., and John P. (1995). Expression, purification and characterization of 1-aminocyclopropane-1- carboxylate oxidase from tomato in Escherichia coli. Biochem J 307: 77-85.
Zhao H., and Arnold F. H. (1999). Directed evolution converts subtilisin E into a functional equivalent of thermitase. In "Protein Eng", pp. 47-53.
Zhao H., Giver L., Shao Z., Affholter J. A., and Arnold F. H. (1998). Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat Biotechnol 16: 258-61.
Zhou L., Zhao Y., and Bi R. (1996). Thermal stable subtilisin with oxidation-resistant property. Enzyme Engineering VIII. Ann. N.Y. Acad. Sci. 799: 82-84.
Zurita M., Bolivar F., and Soberon X. (1984). Construction and characterization of new cloning vehicles. VII. Construction of plasmid pBR327par, a completely sequenced, stable derivative of pBR327 containing the par locus of pSC101. Gene 28: 119-22.

141
An dieser Stelle möchte ich mich bei allen recht herzlich bedanken, die durch Rat und Tat zum Gelingen dieser Arbeit beigetragen haben, insbesondere danke ich Herrn Priv.- Doz. Dr. Werner Hummel für die Überlassung des Themas, seine vielfältigen konstruktiven Anregungen, seine stete Diskussionsbereitschaft und für die kritische Durchsicht der Arbeit, Herrn Prof. Dr. Cornelis P. Hollenberg für die Übernahme des Korreferats, Frau Prof. Dr. M. –R. Kula für die Möglichkeit, diese Dissertation am Institut für Enzymtechnologie durchführen zu können und für die sehr guten Arbeitsbedingungen an ihrem Institut, Herrn Dipl. Chem. Frank Schneider für die kritische Durchsicht der Arbeit, allen Mitgliedern meiner Arbeitsgruppe und allen anderen Mitarbeiterinnen und Mitarbeitern des Instituts, die mir mit fachlichen Ratschlägen zur Seite standen.


















