
Jürgen Thomas · Thomas Gemming Analytische Transmissions- · 10.17 Potential in elektrostatischen...
368
123 Analytische Transmissions- elektronenmikroskopie Eine Einführung für den Praktiker Jürgen Thomas · Thomas Gemming
-
Upload
duongthuan -
Category
Documents
-
view
214 -
download
0
Transcript of Jürgen Thomas · Thomas Gemming Analytische Transmissions- · 10.17 Potential in elektrostatischen...

123
AnalytischeTransmissions- elektronenmikroskopieEine Einführung für den Praktiker
Jürgen Thomas · Thomas Gemming

AnalytischeTransmissionselektronenmikroskopie

Jürgen Thomas � Thomas Gemming
Analytische Transmissions-elektronenmikroskopie
Eine Einführung für den Praktiker

Jürgen ThomasMikro- und NanostrukturenIFW DresdenDresden, Deutschland
ISBN 978-3-7091-1439-1 ISBN 978-3-7091-1440-7 (eBook)DOI 10.1007/978-3-7091-1440-7
Die Deutsche Nationalbibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie;detaillierte bibliografische Daten sind im Internet über http://dnb.d-nb.de abrufbar.
© Springer-Verlag Wien 2013Dieses Werk einschließlich aller seiner Teile ist urheberrechtlich geschützt. Jede Verwertung, die nichtausdrücklich vom Urheberrechtsgesetz zugelassen ist, bedarf der vorherigen Zustimmung des Verlags.Das gilt insbesondere für Vervielfältigungen, Bearbeitungen, Übersetzungen, Mikroverfilmungen unddie Einspeicherung und Verarbeitung in elektronischen Systemen.
Die Wiedergabe von Gebrauchsnamen, Handelsnamen, Warenbezeichnungen usw. in diesem Werk be-rechtigt auch ohne besondere Kennzeichnung nicht zu der Annahme, dass solche Namen im Sinne derWarenzeichen- und Markenschutz-Gesetzgebung als frei zu betrachten wären und daher von jedermannbenutzt werden dürften.
Gedruckt auf säurefreiem und chlorfrei gebleichtem Papier.
Springer ist Teil der Fachverlagsgruppe Springer Science+Business Mediawww.springer.com
Thomas GemmingMikro- und NanostrukturenIFW DresdenDresden, Deutschland
Springer Wien Heidelberg New York Dordrecht London

Vorwort Wozu brauchen wir eigentlich ein Durchstrahlungselektronenmikroskop (englisch: Transmission Electron Microscope, „eingedeutscht“: Transmissionselektronenmi-kroskop)? Es ist teuer in der Anschaffung, verursacht hohe Betriebskosten, liefert teilweise schwer verständliche Daten, die auch noch falsch gedeutet werden kön-nen, und erfordert womöglich Spezialisten und - damit verbunden - zusätzliche Lohnkosten.
Andererseits ist es ein Mikroskop, d.h. es liefert als Ergebnis vergrößerte Bil-der, und Bilder braucht man nur anzusehen, da gibt es doch scheinbar keine Ver-ständnisprobleme. Wozu also auch noch ein neues Buch zu diesem Thema?
Goethe: „Mikroskope und Fernrohre verwirren eigentlich den reinen Menschensinn.“ [0.1]
Die analytische Transmissionselektronenmikroskopie umfasst aber nicht allein die mikroskopische Abbildung. Elektronenbeugung und chemische Analyse mit speziellen Spektrometern für Röntgenstrahlung und Energieverluste der Elektro-nen gehören ebenfalls dazu. Das analytische Transmissionselektronenmikroskop vereint vier anspruchsvolle Methoden: Elektronenmikroskopische Abbildung, Elektronenbeugung, Analyse charakteristischer Röntgenstrahlung und Elektronen-energieverlust-Analyse. Für jede dieser Methoden gibt es Spezialisten, trotzdem sollte der Experimentator einen Überblick über alle Möglichkeiten der analyti-schen Transmissionselektronenmikroskopie haben. Er sollte sie sowohl hinsicht-lich der Gerätebedienung beherrschen als auch mit den Grundsätzen der Inter-pretation der Messergebnisse vertraut sein.
Diesen Überblick soll dieses Buch verschaffen. Die Idee dazu ist uns während der Arbeit in unserem elektronenmikroskopischen Labor des Leibniz-Institutes für Festkörper- und Werkstoffforschung (IFW) Dresden gekommen. Bei der Unter-richtung von Studenten der Werkstoffwissenschaft sowohl in Vorlesungen als auch in Praktika zur analytischen Transmissionselektronenmikroskopie sowie beim Anlernen von Diplomanden, Doktoranden und Technikern am Elektronen-mikroskop haben wir Erfahrungen gesammelt, einerseits hinsichtlich häufig ge-stellter Fragen und besonderer Probleme des Anfängers, andererseits aber auch hinsichtlich der didaktischen Vorgehensweise beim Erklären der Funktionsweise des Transmissionselektronenmikroskops und der praktischen Arbeit daran.
Diese Erfahrungen sollen in dieses Buch einfließen. Es wendet sich insbeson-dere an Personen, die an einem Transmissionselektronenmikroskop arbeiten wol-len oder müssen, aber noch keine ausgebildeten Elektronenmikroskopiker sind. Servicetechnikern soll es helfen, den Überblick über die Grundlagen der von ihnen gewarteten Geräte zu behalten. Das Buch soll aber auch unterhaltsam sein, um möglicherweise auch bei Außenstehenden das Interesse an der Elektronenmikro-skopie zu wecken.

Vorwort 2
Schwerpunkte werden Erklärungen anhand einfacher Modellvorstellungen und Hinweise zur praktischen elektronenmikroskopischen Arbeit sein. Bereits die Ka-pitelüberschriften deuten auf dieses Anliegen hin. Dies unterscheidet dieses Buch von anderen Einführungen in die Elektronenmikroskopie. Wir versuchen, auf Er-klärungen, die allein auf mathematischen Formalismen beruhen, weitgehend zu verzichten.
In diesem Zusammenhang sei eine allgemeine Bemerkung zu Modellvorstel-lungen gestattet: Im Buch sprechen wir manchmal von Elektronen als Teilchen, manchmal als Wellen. Oder die Position der Probe im Elektronenmikroskop: Manchmal zeichnen wir die Probe außerhalb des Objektivs, manchmal inmitten des magnetischen Feldes des Objektivs. Der eine oder andere Leser wird darin einen Widerspruch sehen. Es ist aber kein Widerspruch sondern die Eigenart von Modellen, die dazu dienen, spezielle Sachverhalte zu erklären. Je nach experimen-teller Anordnung beobachten wir einerseits den Teilchen- und andererseits den Wellencharakter der Elektronen. Oder: Zur Erklärung der mehrstufigen Abbildung im Elektronenmikroskop zeichnen wir die Probe außerhalb des Magnetfeldes, weil die genaue Elektronenbahn innerhalb der Linse in diesem Fall keine Rolle spielt. Es ist nur wichtig, wie die Bahn außerhalb der Linse verläuft. Wenn wir aber dis-kutieren, welche Probleme magnetische Proben bereiten, spielt natürlich die di-rekte Wechselwirkung mit dem Magnetfeld des Objektivs eine Rolle, und unser Modell muss anders angelegt sein. Die Modelle sollen so einfach wie möglich und nur so kompliziert wie unbedingt nötig sein.
Ungeachtet des Versuchs plausibler Erklärungen lassen sich manche Zusam-menhänge mit Hilfe der Mathematik besser verstehen. Für quantitative Angaben werden Formeln benötigt. Besonders das letzte Kapitel 10 nimmt darauf Rück-sicht. Dort sind einige Grundlagen noch einmal näher erklärt, gegebenenfalls wird im laufenden Text auf solche vertiefenden Abschnitte hingewiesen. Vereinzelt tauchen auch in den vorhergehenden Kapiteln bereits Gleichungen auf, die beispielsweise Elemente der Infinitesimalrechnung, wie Differentiale und Integra-le, enthalten. Bei Definitionen und physikalischen Grundlagen ist dies mitunter notwendig. Dies sollte den Leser nicht davon abhalten weiterzulesen, selbst wenn ihm die eine oder andere Gleichung unverständlich erscheint.
Für den Spezialisten kann das Kapitel 10 den Griff zu einem Fachbuch über die Spezialgebiete der Elektronenmikroskopie allerdings nicht ersetzen. Einige Vor-schläge für solche Bücher haben wir in den Literaturhinweisen am Ende dieses Buches aufgeschrieben.
Schließlich möchten wir uns bedanken: Bei unseren Lehrern, Freunden und Kollegen, die uns an die Elektronenmikroskopie herangeführt oder später die Ar-beit an modernen Geräten ermöglicht haben. Einige Namen möchten wir nennen: Prof. Alfred Recknagel, Dr. Hans-Dietrich Bauer und Prof. Klaus Wetzig in Dresden sowie Prof. Manfred Rühle, Prof. Frank Ernst, Dr. Günter Möbus und Prof. Joachim Mayer, die zur fraglichen Zeit am Max-Planck-Institut für Metall-forschung in Stuttgart tätig waren.

Vorwort 3
Elektronenmikroskopische Untersuchungen sind nur möglich mit geeignet prä-parierten dünnen Proben. Viele der im Buch gezeigten elektronenmikroskopischen Bilder wären deshalb ohne die sorgfältige Präparation durch Dipl.-Ing. (FH) Birgit Arnold und Dina Lohse nicht vorhanden gewesen.
Mit Prof. Josef Zweck aus Regensburg und Prof. Klaus Wetzig aus Dresden ha-ben wir bereits in einem frühen Stadium über unser Buchprojekt gesprochen. Sie haben uns in unserem Vorhaben bestärkt und durch ihre Fürsprache beim Sprin-ger-Verlag maßgeblich dazu beigetragen, dass dieses Buch, insbesondere auch in deutscher Sprache, herausgegeben wurde.
Dr. Alois Sillaber und B.S. Stephen Soehnlen waren unsere Verhandlungspart-ner beim Springer-Verlag in Wien. Ohne ihr Wohlwollen wäre dieses Buch nicht erschienen.
M.A. Nora Thomas hat mit ihren Kenntnissen bei der Auswahl von Zitaten ge-holfen.
Bei allen genannten Personen sowie bei denjenigen Dresdner Elektronenmikro-skopikern, die das Buchmanuskript oder Teile davon gelesen und hilfreiche Kor-rekturhinweise gegeben haben, aber auch bei Freunden, Kollegen und Studenten, die uns durch Fragen und Kommentare zum Nachdenken über Sachverhalte anreg-ten, die eigentlich „vollkommen klar“ sind, bedanken wir uns.
Goethe: „Alles Gescheite ist schon mal gedacht worden, man muß nur versuchen, es noch einmal zu denken.“ [0.2]

Inhalt
Vorwort 1
Inhalt 5
1 Wozu dieser Aufwand? 9
1.1 Das Problem mit der Vergrößerung 9 1.2 Das Auflösungsvermögen 10 1.3 Elektronenwellen 14 1.4 Die Bedeutung der Vergrößerung 16
2 Was wir über Elektronenoptik und den Aufbau eines Elektronenmikroskops wissen sollten 19
2.1 Das Prinzip der mehrstufigen Abbildung 19 2.2 Rotationssymmetrische magnetische Felder als Elektronenlinsen 20 2.3 Abbildungsfehler 24 2.4 Auflösungsvermögen mit Berücksichtigung des Öffnungsfehlers 28 2.5 Die Elektronenkanone 29 2.6 Der Richtstrahlwert 33 2.7 Wir bauen ein Elektronenmikroskop 36 2.7.1 Das Beleuchtungssystem 37 2.7.2 Das Abbildungssystem 38 2.7.3 Die Probenbühne 39 2.7.4 Die Registrierung des Bildes 41 2.7.5 Das Vakuumsystem 43 2.7.6 Sonstiges 47
3 Wir präparieren elektronentransparente Proben 49
3.1 Wo liegt das Problem? 49 3.2 „Klassische“ Methoden 51 3.3 Schneiden, Schleifen und Ionendünnen 55 3.4 Focused Ion Beam („FIB“) Techniken 60
4 Wir beginnen mit der praktischen Arbeit 65
4.1 Was wir „am Rande“ benötigen 66 4.2 Wir bauen die Probe in den Halter und schleusen diesen ins Mikroskop 67 4.3 Wir überprüfen den (Justage)-Zustand des Mikroskops 69 4.4 Scharfstellen des Bildes – Schärfe und Kontrast 77 4.5 Kontamination und Objektschädigung 78

Inhalt 6
5 Wir schalten um auf Elektronenbeugung 83
5.1 Wieso Beugungsreflexe? 83 5.2 Kristallgitter und Netzebenen 86 5.3 Feinbereichs- und Feinstrahlbeugung 94 5.4 Was können wir aus Feinbereichs-Beugungsmustern lernen? 99 5.4.1 Radien in Ringdiagrammen 100 5.4.2 Auslöschungsregeln 102 5.4.3 Intensitäten der Beugungsreflexe 107 5.4.4 Positionen der Beugungsreflexe in Punktdiagrammen 108 5.4.5 Indizierung der Beugungsreflexe 110 5.5 Kikuchi- und HOLZ-Linien 114 5.6 Amorphe Proben 118
6 Warum sehen wir Kontraste im Bild? 121
6.1 Elastische Streuung der Elektronen in der Probe 121 6.2 Streuabsorptions- und Beugungskontrast 123 6.3 Hell- und Dunkelfeldabbildung 126 6.4 Biegekonturen, Versetzungen und semikohärente Ausscheidungen 129 6.5 Dickenkonturen, Stapelfehler und Zwillinge 135 6.6 Moiré-Muster 139 6.7 Magnetische Domänen: Lorentzmikroskopie 140
7 Wir erhöhen die Vergrößerung 145
7.1 Abbildung von Atomsäulen in Kristallgittern: Phasenkontrast 145 7.2 Kontrastübertragung durch die Objektivlinse 150 7.3 Wellenoptische Deutung des Auflösungsvermögens 153 7.4 Periodische Helligkeitsverteilung in Bildern: Fourieranalyse 155 7.5 Streuabsorptions- und Phasenkontrast 158 7.6 Kontrast bei amorphen Proben 160 7.7 Korrektur des Astigmatismus 163 7.8 Messung des Auflösungsvermögens 164 7.9 Korrektur von Öffnungs- und Farbfehler 166 7.10 Interpretation hochaufgelöster Bilder 169
8 Wir schalten um auf Rastertransmissionselektronenmikroskopie 171
8.1 Was ändert sich elektronenoptisch? 171 8.2 Auflösungsvermögen oder: Wie klein kann die Elektronensonde werden? 173 8.3 Kontrast im rastertransmissionselektronenmikroskopischen Bild 178 8.4 Spezialfall: Weitwinkel-Dunkelfeld-Ringdetektor 181

Inhalt 7
9 Wir nutzen die analytischen Möglichkeiten 183
9.1 Analytische Signale als Folge inelastischer Wechselwirkung 183 9.1.1 Emission von Röntgenstrahlung 183 9.1.2 Energieverluste der Primärelektronen 188 9.2 Energiedispersive Spektroskopie charakteristischer Röntgenstrah-
lung („EDXS“) 192 9.2.1 Röntgenspektrometer und Röntgenspektren 192 9.2.2 Qualitative Interpretation der Röntgenspektren 197 9.2.3 Quantifizierung von Röntgenspektren 201 9.2.4 Linienprofile und Elementverteilungsbilder 209 9.3 Elektronenenergieverlust-Spektroskopie („EELS“) 211 9.3.1 Elektronenenergie-Spektrometer 211 9.3.2 Low-Loss und Core-Loss Bereich der Spektren 212 9.3.3 Qualitative Elementanalyse 215 9.3.4 Untergrund und Vielfachstreuung: Anforderungen an die Probe 217 9.3.5 Messung der Probendicke 219 9.3.6 Kantenfeinstruktur: Bindungsanalyse 222 9.3.7 Quantifizierung von Energieverlust-Spektren 225 9.4 Energiegefilterte Abbildung 227 9.5 Vergleich zwischen EDXS und EELS 231
10 Grundlagen genauer erklärt (etwas mehr Mathematik) 233
10.1 Beugung an einer Kante (Huygenssches Prinzip) 233 10.2 Wellenfunktion für Elektronen 234 10.3 Elektronenwellenlänge relativistisch berechnet 239 10.4 Elektronenbahnen in rotationssymmetrischen magnetischen Feldern 241 10.5 Auflösungsvermögen mit Berücksichtigung des Öffnungsfehlers 251 10.6 Schottky-Effekt 252 10.7 Elektrisches Potential in rotationssymmetrischen Elektrodenanordnungen 254 10.8 Laue-Gleichungen und reziprokes Gitter, Ewald-Konstruktion 258 10.9 Kinematisches Modell: Gitterfaktor und Strukturfaktor 270 10.10 Debye-Streuung 278 10.11 Elektronen im Feld einer Zentralkraft 282 10.12 Mittlere freie Weglänge für elastische Streuung 288 10.13 Abstände in Moiré-Mustern 290 10.14 Kontrastübertragungsfunktion 293 10.15 Scherzer-Fokus 302 10.16 Delokalisation 306 10.17 Potential in elektrostatischen Multipolen 310 10.18 Elektronensonde und Abbildungsfehler 313 10.19 Klassischer inelastischer Stoß 319

Inhalt 8
10.20 Effizienz von energiedispersiven Röntgendetektoren 321 10.21 Berechnung von Cliff-Lorimer-k-Faktoren 327 10.22 Absorptionskorrektur bei EDXS 332 10.23 Prismen für Elektronen 335 10.24 Faltung von Funktionen 339
Resümee und Ausblick 343
Physikalische Konstanten 345
Abkürzungsverzeichnis 347
Literaturhinweise 351
Quellenverzeichnis 355
Sachwortverzeichnis 363

1 Wozu dieser Aufwand?
Ziel: In der modernen Werkstoffforschung spielen sehr kleine, sogenannte „Na-nostrukturen“ eine immer bedeutsamere Rolle: Kobalt-Kupfer-Wechselschichten mit Einzeldicken um 1 nm führen zu unerwartet starken Änderungen des elektri-schen Widerstandes bei Änderung des Magnetfeldes und ermöglichen das Ausle-sen der Information von Computer-Festplatten („Giant Magnetoresistance“), Na-noteilchen auf Oberflächen verhindern den direkten Kontakt von Wasser mit der Oberfläche: die Wassertropfen perlen ab („Lotuseffekt“), Nanoteilchen ermög-lichen durch ihre große spezifische Oberfläche verbesserte Katalysatorwirkung usw. Die spannende Frage ist: „Wie können solche ‚Nanostrukturen’ untersucht und charakterisiert werden?“
Nun, wenn wir an der Straßenbahnhaltestelle stehen und die Nummer der heran-nahenden Bahn nicht erkennen, warten wir bis diese näher herangekommen ist: Und siehe da, es ist die „7“. Die Erfahrung lehrt: Um kleinere Details zu erkennen, müssen wir näher herangehen. Oder anders ausgedrückt: Der Sehwinkel (s. Bild 1-1) muss möglichst groß sein.
Bild 1-1. Definition des Sehwinkels σ.
1.1 Das Problem mit der Vergrößerung Es ist allerdings unmöglich, sich dem betrachteten Gegenstand beliebig weit zu nähern, die Physiker gehen von einem optimalen Betrachtungsabstand von S = 25 cm („deutliche Sehweite“) aus. Mit dieser Randbedingung ist der Sehwinkel σ vorgegeben durch die Größe y des Gegenstandes:
tany
Sσ = bzw. bei kleinen Winkeln (σ << 1):
y
S=σ . (1.1)
Für Strukturen in der Größenordnung von Nanometern folgt daraus ein Sehwinkel von weniger als 10-5 mrad ≈ 0,0000006°. Das wäre so, als wenn man eine Straßen-bahn in 100000 km Entfernung sehen wollte. Was wir brauchen, ist ein (optisches) Gerät, mit dem wir den Sehwinkel vergrößern können, ohne uns dem Gegenstand nähern zu müssen. Im einfachsten Fall wäre das eine Lupe (Bild 1-2).
Damit können wir die Vergrößerung eines optischen Gerätes definieren:
J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie, DOI 10.1007/978-3-7091-1440-7_1, © Springer-Verlag Wien 2013

Kapitel 1 10
Sehwinkel mit optischem Gerät 'Vergrößerung =
Sehwinkel ohne optisches GerätM
σ
σ= . (1.2)
Bild 1-2. Vergrößerung des Sehwinkels mit Hilfe einer Lupe.
Der Weg zur Charakterisierung von Nanostrukturen scheint geklärt: Wir müssen nur viele Lupen hintereinander schalten, um einen hinreichend großen Sehwinkel zu bekommen, und dabei einige optische Gesetzmäßigkeiten berücksichtigen, da-mit wir auch ein scharfes Bild erhalten. Dies ist leider ein Trugschluss! Warum dies ein Trugschluss ist, erfahren wir im nächsten Abschnitt „Auflösungsvermö-gen“.
1.2 Das Auflösungsvermögen Bisher haben wir die Natur des Lichtes außer Acht gelassen. Wir wissen, dass Licht sowohl Teilchen- als auch Wellencharakter hat. Betrachten wir das Licht als Welle, stellen uns vor, eine solche Lichtwelle trifft auf eine Lochblende und über-legen, wie sich die Welle hinter der Blende ausbreitet. Die naheliegende Vermu-tung ist eine scharfe seitliche Begrenzung (vgl. Bild 1-3a).
Bild 1-3. Ausbreitung einer Welle hinter einer Blende. a) Naive Vermutung. b) Realität (Huygenssches Prinzip). Abschnitt 10.1
Diese Vermutung ist allerdings falsch. Schauen wir beispielsweise durch ein klei-nes Loch in einer schwarzen Pappe auf eine helle Lichtquelle, so werden wir den Lochrand nur unscharf wahrnehmen. Der Grund ist, dass die Welle hinter der

Wozu dieser Aufwand? 11
Blende eben nicht scharf begrenzt ist sondern um das Hindernis „herumgebeugt“ wird. Die physikalische Grundlage liefert das Huygenssche Prinzip1 für die Wel-lenausbreitung: Jeder Punkt einer Wellenfront ist Ausgangspunkt einer Elementar-welle. Die Überlagerung der Elementarwellen bildet die neue Wellenfront. Damit muss das obige Bild korrigiert werden (vgl. Bild 1-3b).
Stellen wir uns weiter vor, wir hätten ein zweites Loch in der Pappe mit sehr geringem Abstand vom ersten. Wegen des Beugungseffektes der Lichtwellen wür-den wir die zwei Löcher nicht mehr getrennt wahrnehmen können, selbst wenn sie sich nicht unmittelbar berühren. Wir können die beiden Löcher nicht mehr „auf-lösen“.
Wir wollen nun versuchen, diesen Sachverhalt für die lichtmikroskopische Ab-bildung mathematisch zu beschreiben, d. h. eine Formel zu finden, mit der das Auflösungsvermögen eines Lichtmikroskops berechnet werden kann. Die Idee dazu entwickelte Ernst Abbe2, der wissenschaftliche Mitarbeiter von Carl Zeiss3, etwa im Jahre 1870. In der geometrischen Optik wird für den Abbildungsprozess gefordert, dass alle Strahlen, die von einem Dingpunkt ausgehen, in einem Bild-punkt vereinigt werden. Beschrieben wird dies durch die Abbildungsgleichung (hier für dünne Linsen), welche die Brennweite f, die Dingweite g (auch Gegen-standsweite oder Objektweite genannt) und die Bildweite b miteinander verknüpft:
1 1 1
f g b= + ( 1.3)
Daraus folgen Merksätze, wie beispielsweise dieser: „Parallelstrahlen schneiden sich im bildseitigen Brennpunkt.“ Parallelstrahlen verlaufen nämlich im Dingraum parallel zur optischen Achse, d. h. ihr Dingabstand g ist unendlich. Damit folgt für den Bildabstand b = f wie im Merksatz gesagt.
Wie sollen wir uns aber nun den Abbildungsprozess vorstellen, wenn wir an den Wellencharakter des Lichtes denken? Warum entsteht das Bild gerade in einem bestimmten Abstand von der Linse? Die Erklärung liegt auf der Hand: Das Bild ist das Ergebnis der Interferenz aller Wellen, die am Objekt gestreut und durch die Linse wieder vereinigt werden. Damit die Überlagerung „phasenrichtig“ (d. h. ohne Phasenschiebung innerhalb des Abbildungssystems) erfolgen kann, müssen die optischen Weglängen sopt für alle diese Wellen gleich sein. Die optische Weglänge ist das Produkt aus geometrischer Weglänge s und Brechzahl n des Mediums, in dem sich das Licht ausbreitet. Da n nicht auf dem gesamten Weg s gleich bleiben muss, gilt allgemein:
1 Christiaan Huygens, niederländischer Physiker , 1629 – 1695 2 Ernst Abbe, deutscher Physiker, 1840 - 1905 3 Carl Zeiss, deutscher Mechaniker und Unternehmer, 1816 - 1888

Kapitel 1 12
( )0
0
s
opts n s ds= ⋅ . (1.4)
Die Forderung nach Gleichheit der optischen Weglängen stellt eine andere Formu-lierung der Abbildungsgleichung dar. Der Sachverhalt sei anhand der folgenden Skizze 1-4 veranschaulicht: Es ist klar, dass die geometrischen Weglängen für die beiden (durch ihre Ausbreitungsrichtung gekennzeichneten) Wellen unterschied-lich sind. Sie legen aber auch unterschiedliche Wege innerhalb der Linse zurück: Die auf der optischen Achse verlaufende, ungebeugte Welle legt infolge der Linsenform einen längeren Weg in der Linse zurück als die gebeugte Welle. Da-mit kann die Forderung nach gleicher optischer Weglänge erfüllt werden (vgl. Bild 1-4).
Bild 1-4. Weglängen beim Abbildungsprozess.
Wir nehmen an, die Linsenumgebung habe die Brechzahl 1 und das Linsenma-terial die Brechzahl n. Dann muss für die fehlerfreie Abbildung gelten:
1 2 3 1 2 3n ' n ' 's s s s s s+ ⋅ + = + ⋅ + . (1.5)
Es ist klar, dass dies spezielle Anforderungen an die Form der Linse stellt. Wenn aber das Bild eine Folge der Wellenüberlagerung (Interferenz) ist, so
müssen mindestens zwei Wellen durch die Linse gehen: die ungebeugte und eine gebeugte Welle. Die nächste Frage lautet, wie groß denn der Beugungswinkel α ist. Zur Beantwortung dieser Frage schauen wir auf Bild 1-5.
Eine Welle fällt senkrecht auf einen Doppelspalt (Spaltabstand δ). Hinter dem Doppelspalt breiten sich Elementarwellen aus, die sich überlagern. Verstärkung tritt dann ein, wenn die (optischen) Gangunterschiede Δs ein ganzzahliges Viel-faches der Wellenlänge λ betragen. Aus der Skizze folgt
sins δ αΔ = ⋅ (1.6)
bzw. mit n als Brechzahl im Gebiet hinter dem Doppelspalt (rechte Seite):

Wozu dieser Aufwand? 13
n sins δ αΔ = ⋅ ⋅ . (1.7)
Bild 1-5. Interferenz an einem Doppelspalt.
Für den Fall, dass der Gangunterschied gerade gleich der Wellenlänge ist, gilt
n sin bzw. =n sin
sλ
λ δ α δα
= Δ = ⋅ ⋅⋅
. (1.8)
Die letzte Formel beschreibt, wie weit die beiden Spalte bei vorgegebener Wellen-länge λ und „numerischer Apertur“ n⋅sinα bei einer Abbildung voneinander ent-fernt sein dürfen oder allgemeiner, welchen Abstand zwei Struktureinzelheiten ha-ben müssen, damit sie bei der Abbildung getrennt gesehen, d.h. „aufgelöst“ wer-den können. Damit haben wir eine Formel für das Auflösungsvermögen gefunden. Eine genauere Betrachtung der Intensitätsverteilungen in den Beugungsscheibchen einschließlich der Berücksichtigung des Rayleigh4-Kriteriums (Der Abstand der beiden Intensitätsmaxima muss mindestens so groß sein wie der Abstand zwischen Maximum und erstem Intensitätsminimum.) liefert für das Auflösungsvermögen eines Lichtmikroskops:
B0,61
= n sin
λδ δ
α
⋅=
⋅ . (1.9)
Den Index B ergänzen wir, um darauf hinzuweisen, dass die Ursache der Begren-zung in der Beugung liegt.
Wir wollen abschätzen, welche minimalen Strukturgrößen mit einem Lichtmi-kroskop aufgelöst, d.h. untersucht werden können: Die Wellenlänge des sichtbaren Lichtes beträgt etwa 0,5 μm, die Brechzahl n von Luft, was im Allgemeinen das Umgebungsmedium ist, ist gleich 1 (mit speziellen Immersionsölen kann bis 1,4
4 Lord John W. S. Rayleigh, englischer Physiker, 1842 – 1919, Nobelpreis für Physik 1904

Kapitel 1 14
erreicht werden) und sinα kann höchstens 1 werden. Mit diesen Werten folgt für das Auflösungsvermögen eines Lichtmikroskops ca. 0,2 μm.
Es ist demzufolge auf diese Weise physikalisch unmöglich, Nanostrukturen aufzulösen, wie in der Einleitung gefordert. Da hilft auch keine noch so hohe Ver-größerung. Wir müssen das Auflösungsvermögen des optischen Instruments ver-bessern. Der Weg dahin erscheint klar: Wenn wir die beschriebene „klassische“ optische Abbildung5 beibehalten wollen, benötigen wir anstelle von Licht ein Me-dium mit kürzerer Wellenlänge.
1.3 Elektronenwellen Dank Louis de Broglie6 ist seit 1924 bekannt, dass auch Teilchen mit einer Ruhe-masse größer als Null (beispielsweise Elektronen) Wellencharakter besitzen. Für deren Wellenlänge λ gilt ( Abschnitt 10.2):
h =
pλ (1.10)
mit dem Planckschen7 Wirkungsquantum h und dem Impuls p. Andererseits war bekannt, dass Elektronen in elektrischen und magnetischen
Feldern abgelenkt werden können. Hans Busch8 schlug 1926 vor, mit solchen Fel-dern Linsen zu bauen, die auf Elektronen ähnlich wirken wie Glaslinsen auf Licht. Damit war die Elektronenoptik geboren und eine Möglichkeit eröffnet, das Auf-lösungsvermögen eines solchen „Übermikroskops“9 gegenüber dem des Lichtmi-kroskops zu verbessern.
Um dies zu verstehen, müssen wir zunächst die für die Elektronen erreichbare Wellenlänge berechnen. Ausgangspunkt ist die o.g. de Broglie-Formel. Für den Impuls des Elektrons gilt:
0mp v= ⋅ , (1.11)
5 Das Auflösungsvermögen kann auch verbessert werden, wenn nicht der gesamte abzubildende Probenbereich beleuchtet wird, sondern nur ein extrem kleiner Spot für Anregung und Informa-tionsgewinnung genutzt wird („nichtklassische“ Methoden, z.B. Rasterverfahren mit Licht). 6 Louis de Broglie, französischer Physiker, 1892 – 1987, Nobelpreis für Physik 1929 7 Max Planck, deutscher Physiker, 1858 – 1947, Nobelpreis für Physik 1918, gilt als Begründer der Quantenphysik 8 Hans Busch, deutscher Physiker, 1884 – 1973, gilt als Begründer der Elektronenoptik 9 „Übermikroskop“ war der Name des ersten kommerziellen Durchstrahlungselektronenmikro-skops der Firma Siemens (Markteinführung 1939). Maßgebliche Entwickler waren die deutschen Elektrotechniker Ernst Ruska (1906 – 1988, Nobelpreis für Physik 1986), Bodo von Borries (1905 – 1955) und Max Knoll (1897- 1969). Vor 1939 und später gab es Laborgeräte, anhand derer u.a. Baueinheiten entwickelt und getestet wurden, die die Geräteentwicklung beeinflusst haben, z.B. im Institut von Manfred von Ardenne (1907 – 1997) in Berlin-Lichterfelde.

Wozu dieser Aufwand? 15
d.h. Produkt aus Ruhemasse m0 und Geschwindigkeit v. Die Geschwindigkeit folgt aus dem Energiesatz, d.h. die aufgewendete Beschleunigungsarbeit WB wird in kinetische Energie umgewandelt. Die Arbeit ist definiert als
B SW F ds= ⋅ (1.12)
(FS: Kraftkomponente in Richtung des Wegelementes ds). Eine Elektronenladung –e (negative Elementarladung) erfährt im elektrischen Feld der Feldstärke E die Kraft
eSF E= − ⋅ . (1.13)
Vereinfachend setzen wir zwischen Kathode und Anode (Abstand d) ein homoge-nes Feld voraus. Dann gilt mit Berücksichtigung der Feldrichtung von Anode zu Kathode:
BUE
d= − (1.14)
(UB: Beschleunigungsspannung zwischen Kathode und Anode), d.h.
0
e ed
BB B
UW ds U
d= ⋅ ⋅ = ⋅ . (1.15)
Damit lautet der Energiesatz in klassischer Rechnung:
20me
2 Bv U= ⋅ (1.16)
mit der Elementarladung e und der Elektronenruhemasse m0. Demnach gilt:
0
h
2 e m BUλ =
⋅ ⋅ ⋅. (1.17)
Bei Beschleunigungsspannungen über 50 kV sind relativistische Effekte nicht mehr zu vernachlässigen. Die Wellenlänge berechnet sich dann nach der Formel ( Abschnitt 10.3):
0 2
h
ee 2 m
cB
BU
U
λ =⋅
⋅ ⋅ +
, (1.18)

Kapitel 1 16
in der die Lichtgeschwindigkeit c im Vakuum als weitere Konstante auftaucht. Für die Wellenlänge von Elektronen, die mit 300 kV beschleunigt wurden,
erhalten wir in nichtrelativistischer Näherung 2,24 pm, relativistisch gerechnet 1,97 pm ≈ 0,002 nm (s. Bild 1-6). Die Wellenlänge nach klassischer Rechnung wäre bei einer Beschleunigungsspannung von 300 kV um 14% falsch!
Bild 1-6. Elektronenwellen-länge in Abhängigkeit von der Beschleunigungsspan-nung in der Elektronen-kanone.
Bei Übernahme der Formel (1.9) für das Auflösungsvermögen des Lichtmi-kroskops betrüge das Auflösungsvermögen des Elektronenmikroskops etwa 1,6 pm = 0,0016 nm. In der Praxis werden derzeit ca. 0,1 nm erreicht. Wir werden später (Abschnitt 2.4) sehen, warum dies so ist.
1.4 Die Bedeutung der Vergrößerung Wir haben erkannt, dass das Auflösungsvermögen das entscheidende Qualitäts-merkmal eines optischen Gerätes ist. Doch welche Bedeutung hat nun die Ver-größerung?
Das Auflösungsvermögen ist eine Länge, die sich auf das Objekt bezieht. Für das Transmissionselektronenmikroskop war ein Wert von etwa 0,1 nm angegeben worden. Selbstverständlich muss diese Länge soweit vergrößert werden, dass sie mit dem Auge, auf einem Film oder mit einer Kamera auch erkannt werden kann, d.h. auch das Registrierinstrument hat ein Auflösungsvermögen. Wir wollen die-ses mit δReg bezeichnen. Für das menschliche Auge gilt beispielsweise δReg ≈ 0,1 mm.
„Mit dem Auge erkennen“ bedeutet, dass die mit dem optischen Instrument erreichbare Auflösungsgrenze bis auf 0,1 mm vergrößert werden muss. Allgemein gilt für diese förderliche Vergrößerung:
RegfördM
δ
δ= . (1.18)
Höhere Vergrößerungen als Mförd führen nicht zu besserer Detailerkennbarkeit, sondern im Gegenteil: Sie liefern unscharfe Bilder.

Wozu dieser Aufwand? 17
Benutzen wir das Auge zur Registrierung, so erhalten wir für das Lichtmikro-skop (δ ≈ 0,2 μm) eine förderliche Vergrößerung von ca. 500, beim Transmis-sionselektronenmikroskop (δ ≈ 0,1 nm) hingegen eine förderliche Vergrößerung von ca. 1 Million.
Heutzutage werden in der Elektronenmikroskopie häufig sogenannte „CCD-Kameras“ (CCD: Charge Coupled Device – vgl. Abschnitt 2.7.4) verwendet, um die Bilder zu registrieren. Die digitalen Bilder bestehen aus einzelnen Pixeln, deren Standardgröße für die zurzeit gebräuchlichen Kameras bei 15 μm - 25 μm liegt. Die elektronenoptisch förderliche Vergrößerung reduziert sich für das Transmissionselektronenmikroskop damit auf 150000 bis 250000. Die Pixelgröße auf dem Computerbildschirm hängt von dessen Größe und Auflösung ab, sie liegt typischerweise zwischen 0,2 mm und 0,3 mm, d.h. oberhalb der Auflösungsgrenze des Auges.

2 Was wir über den Aufbau eines Elektronen-mikroskops und Elektronenoptik wissen sollten
Ziel: Wenn in diesem Buch vom Elektronenmikroskop geschrieben wird, dann ist damit immer das Transmissionselektronenmikroskop gemeint und nicht das Ra-sterelektronenmikroskop. Damit geht es um ein Mikroskop im engeren Sinn, d.h. um ein Gerät, in dem ein Gegenstand, d.h. die zu untersuchende Probe, durch-strahlt und optisch abgebildet wird. Ungeachtet dessen kann in diesem Mikroskop der Elektronenstrahl auch auf die Probe fokussiert und gerastert werden („STEM“ – s. Kapitel 8). Es bleibt aber bei der Durchstrahlung der Probe. Das Transmis-sionselektronenmikroskop ist im Prinzip wie ein (Durchstrahlungs-) Lichtmikro-skop aufgebaut. Davon ausgehend werden wir uns in diesem Kapitel mit einigen elektronenoptischen Gesetzmäßigkeiten, Abbildungsfehlern und wichtigen Bau-gruppen des Elektronenmikroskops befassen.
2.1 Das Prinzip der mehrstufigen Abbildung Die förderliche Vergrößerung wird beim Lichtmikroskop (und erst recht beim Elektronenmikroskop) durch eine mehrstufige Abbildung erreicht. Bild 2-1 zeigt dies für die zweistufige Abbildung des Lichtmikroskops.
Bild 2-1. Zweistufige Abbil-dung beim Lichtmikroskop. Das Licht der (unten ange-ordneten) Lampe fällt durch eine Kondensorlinse mit Blende, durch die die Be-leuchtungsstärke auf der Pro-be eingestellt wird. Das Ob-jektiv erzeugt ein reelles Zwi-schenbild, welches mit einer Projektivlinse zum Endbild vergrößert wird. Dieses kann beispielsweise auf einer Foto-platte festgehalten werden. Oft ist das Projektiv durch ein Okular ersetzt, durch welches das reelle Zwischenbild mit dem Auge wie durch eine Lupe vergrößert wahrge-nommen wird.
DOI 10.1007/978-3-7091-1440-7_2, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 2 20
Das Elektronenmikroskop ist im Prinzip genauso aufgebaut. Allerdings ist es in den allermeisten Fällen auf den „Kopf gestellt“, d.h. der Kondensor ist die oberste Linse.
Für ein System aus zwei (dünnen) Linsen mit den Brennweiten f1 und f2 sowie dem Linsenabstand t gilt für die resultierende Brennweite f
1 2 1 2
1 1 1 t
f f f f f= + −
⋅
. (2.1)
Im Elektronenmikroskop ist der Bildabstand b durch die feste Position der Linsen und des Beobachtungsschirms bzw. der Kamera vorgegeben. Bei Veränderung der Brennweite wird es möglich, unterschiedlich entfernte (Ding)-Ebenen auf dem Beobachtungsschirm abzubilden (vgl. Abbildungsgleichung 1.3). Wir werden spä-ter sehen, welche Möglichkeiten dadurch im Elektronenmikroskop eröffnet wer-den.
Für die Gesamtvergrößerung M einer mehrstufigen Abbildung mit den einzel-nen Vergrößerungsstufen M1, M2, ... Mn gilt:
1 2 ... nM M M M= ⋅ ⋅ ⋅ . (2.2)
Im allgemeinen Sprachgebrauch wird die Vergrößerung oft mit dem Abbil-dungsmaßstab gleichgesetzt, obwohl dies strenggenommen zwei unterschiedliche Dinge sind: Die Vergrößerung ist der Quotient aus den Sehwinkeln mit und ohne optische Vorrichtung (1.2); der Abbildungsmaßstab AM ist der Quotient aus Bild- und Dinggröße y’ bzw. y. Eine einfache geometrische Überlegung (Strahlensatz) mit Vorzeichenberücksichtigung zeigt, dass AM gleich dem negativen Quotienten aus Bild- und Dingweite b bzw. g ist. Schließlich kann b mit der Abbildungs-gleichung (1.3) aus g und Brennweite f berechnet werden:
My b f
Ay g f g
′= = − =
− (2.3)
Am gravierendsten äußert sich der Unterschied zur Vergrößerung darin, dass der Abbildungsmaßstab im Gegensatz zur Vergrößerung eine vorzeichenbehaftete Größe ist: Ein negativer Abbildungsmaßstab bedeutet, dass das Bild auf dem Kopf steht. Nimmt man aber nur den Betrag des Abbildungsmaßstabes, so stimmt der Zahlenwert mit der Vergrößerung überein.
2.2 Rotationssymmetrische magnetische Felder als Elektronenlinsen Wir wollen nun überlegen, wieso die üblicherweise in Elektronenmikroskopen eingesetzten, rotationssymmetrischen magnetischen Felder als Elektronenlinsen

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 21
wirken, d.h. auf Elektronen den gleichen Einfluss ausüben wie Glaslinsen auf Licht. Der schematische Aufbau einer solchen Elektronenlinse ist in Bild 2-2 dar-gestellt.
Bild 2-2. Schematische Darstellung einer magne-tischen Elektronenlinse. Der Spulenstrom kann bis zu 30 A betragen. Eine Wasser-kühlung sorgt für konstante Temperatur in der Linse. Der Polschuh formt und konzen-triert das Magnetfeld.
Damit sich die gewünschte Linsenwirkung einstellt, müssen alle Elektronen, die sich nicht entlang der Mittelachse („optische Achse“) bewegen, eine Kraft erfah-ren, die sie zur Achse hinzieht. Wir wissen, dass auf Elektronen, die sich mit der Geschwindigkeit v in einem Magnetfeld der Stärke (genauer: der magnetischen In-duktion) B bewegen, die Lorentzkraft10
e= − ⋅ ×F v B (2.4)
wirkt. Darin geht die Elementarladung e ein. Außerdem ist berücksichtigt, dass die Kraft F, die Geschwindigkeit v und die magnetische Induktion B Vektoren sind, d.h. sie sind gekennzeichnet durch Betrag (Zahlenwert) und Richtung. Es ist üb-lich, solche Vektoren durch Pfeile darzustellen, deren Länge den Betrag veran-schaulicht und deren Winkel gegen eine Bezugsachse die Richtung. In den Bildern kennzeichnen wir Vektorgrößen durch einen kleinen Pfeil über dem Buchstaben, in Formeln und Text durch Fettdruck.
Die Frage ist nun, wie man die Richtung der Lorentzkraft bei dem in Formel (2.4) dargestellten „Kreuzprodukt“ ermittelt. Mathematisch handelt es sich bei v, B, F um ein „Rechtssystem“, d.h. die Kraft F steht senkrecht auf der von v und B aufgespannten Ebene (vgl. Bild 2-3). Bei der Festlegung der Kraftrichtung dürfen wir das negative Vorzeichen in Formel (2.4) nicht vergessen. Praktisch bedient man sich bei der Richtungsfestlegung beispielsweise der „rechte Hand Regel“: Man spreize Daumen, Zeige- und Mittelfinger der rechten Hand so, dass sie jeweils einen rechten Winkel miteinander bilden. Dann drehe man die Hand so, dass der Daumen in Richtung der Geschwindigkeit und der Zeigefinger in
10 Hendrik Antoon Lorentz, niederländischer Mathematiker und Physiker, 1853 – 1928, Nobelpreis für Physik 1902

Kapitel 2 22
Richtung der Induktion zeigt. Der Mittelfinger gibt dann die Richtung des Kreuzproduktes (hier die Kraft) an.
Bild 2-3. Richtung der Lo-rentzkraft F auf ein Elektron mit der Ladung –e, welches sich mit der Geschwindig-keit v in einem Magnetfeld der Induktion B bewegt.
Als nächstes müssen wir überlegen, wie groß der Betrag F der Lorentzkraft ist. Er entspricht beim Kreuzprodukt der Fläche der durch die beiden Vektoren v und B aufgespannten Ebene:
( )e sin ,F v B= ⋅ ⋅ ⋅ v B . (2.5)
Wir sehen, dass bei paralleler Richtung von Geschwindigkeit und Magnetfeld kei-ne Kraft auf das Elektron ausgeübt wird. Mit anderen Worten: Ein homogenes Magnetfeld in Richtung der optischen Achse hat keinen Einfluss auf Elektronen, die parallel zur optischen Achse in das Magnetfeld einfallen (in der geometrischen Optik sind das „Parallelstrahlen“). Wir benötigen für die Linsenwirkung also eine Komponente des Magnetfeldes senkrecht zur Bewegungsrichtung der Elektronen, d.h. wir benötigen ein inhomogenes Magnetfeld.
Für die weitere Diskussion ist es zweckmäßig, ein Koordinatensystem in die magnetische Linse zu legen. Wegen der Rotationssymmetrie wählen wir Zylinder-koordinaten r, ϕ und z (vgl. Bild 2-4).
Bild 2-4. Zylinderkoordina-tensystem im Bereich des Polschuhspaltes mit ein-gezeichneten Feldlinien der magnetischen Induktion B.
Wir erkennen, dass die Feldlinien keine Komponente in azimutaler (d.h. ϕ -) Rich-tung, sondern nur solche in r- (Br) und z-Richtung (Bz) haben. Die Konsequenzen für unseren o.g. Parallelstrahl sollen anhand von Bild 2-5 erläutert werden. Daraus

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 23
ist ersichtlich, dass unser Elektron, welches den Parallelstrahl symbolisiert, tatsächlich zur optischen Achse gelenkt wird ( Abschnitt 10.4).
Bild 2-5. Bewegung eines Elektrons im inhomogenen Magnetfeld der Linse. a) Das Elektron hat nur eine Geschwindigkeitskomponente vz („Parallelstrahl“). Es er-fährt durch Vermittlung der Radialkomponente Br der magnetischen Induktion eine Lorentzkraft Fϕ in azimutaler Richtung (d.h. in die Zeichen-ebene hinein), wird in diese Richtung beschleunigt und erhält demzufolge eine Ge-schwindigkeitskomponente vϕ in azimutaler Richtung. b) Infolge der nunmehr vor-handenen Geschwindigkeits-komponente vϕ in azimutaler Richtung erfährt das Elektron durch Vermittlung der axialen Komponente Bz der magneti-schen Induktion eine Kraft zur optischen Achse hin, wie wir das für eine Linse erwar-ten.
Wir erkennen gleichzeitig zwei grundlegende Eigenschaften der rotationssym-metrischen magnetischen Elektronenlinse:
1. Die Elektronen bewegen sich wegen der azimutalen Geschwindigkeitskom-ponente auf Schraubenbahnen mit veränderlichem Abstand von der Mittelachse durch die Linse, d.h. das Bild ist gegenüber der Probe gedreht. In den folgenden Bildern mit Strahlengängen ist immer die r(z)-Ebene dargestellt.
2. Der Grad der Feldinhomogenität, d.h. das Verhältnis Br / Bz an jeder Stelle innerhalb des Linsenfeldes (Form der Feldlinien), bestimmt maßgeblich die Bre-chungseigenschaften. Später (Abschnitt 2.7.3) werden wir sehen, dass die Probe im Magnetfeld der Objektivlinse angeordnet ist. Wenn es sich dabei um magneti-sches Material handelt, wird die Feldinhomogenität verändert und dies auch noch in Abhängigkeit vom genauen Probenort. Probleme bei der Abbildung derartiger Proben sind also zu erwarten.

Kapitel 2 24
2.3 Abbildungsfehler Die nächste Frage ist nunmehr, ob wir denn den „richtigen“ Grad der Feldinho-mogenität einstellen können, um nicht nur prinzipiell eine Linsenwirkung er-reichen sondern auch eine fehlerfreie optische Abbildung erzielen zu können. Oder auf die Lichtoptik übertragen: Was passiert, wenn wir die Forderung nach gleichen optischen Weglängen für alle zwischen Ding- und Bildpunkt denkbaren Wellen nicht erfüllen können (vgl. Abschnitt 1.2), weil unsere Glaslinse falsch ge-schliffen ist?
In der Tat ist dies für die besprochenen rotationssymmetrischen Elektronenlin-sen ein Problem. Es ist unter Beibehaltung der Rotationssymmetrie unmöglich, die Feldinhomogenität nach Bedarf „zurechtzubiegen“. Dieses Problem wurde bereits 1936 von Otto Scherzer11 erkannt [2.1]. Die Folge ist, dass in der Elektronenoptik Abbildungsfehler eine weit stärkere Rolle spielen als in der Lichtoptik. Wir wollen uns hier mit drei wesentlichen Fehlern befassen: dem Öffnungsfehler (auch sphä-rische Aberration genannt), dem Farbfehler (auch chromatische Aberration ge-nannt) und dem Astigmatismus.
- Öffnungsfehler (sphärische Aberration):
Der Name sagt es bereits: Dieser Fehler tritt bei stärker geöffneten Strahlenbün-deln auf. Der zweite Name „sphärische Aberration“ liefert einen Hinweis auf die Ursache: Die Linsenoberfläche hat nicht die erforderliche Form, sie ist „nicht rich-tig“ geschliffen oder auf die magnetischen Elektronenlinsen übertragen: Die Feld-form („Feldinhomogenität“) entspricht nicht den Forderungen für eine fehlerfreie Abbildung (→ Abschnitt 10.4). Die Abweichungen zwischen Forderung und Rea-lität sind umso größer, je schwächer das rotationssymmetrische magnetische Feld ist. Langbrennweitige magnetische Linsen haben deshalb einen größeren Öff-nungsfehler als kurzbrennweitige.
Die Folge der „falschen“ Feldform ist in Bild 2-6 dargestellt.
Bild 2-6. Auswirkung des Öffnungsfehlers. Das wenig geöffnete Strahlenbündel A erzeugt den Bildpunkt in der Gaußschen Bildebene. Dem-gegenüber wird das stärker (Apertur α) geöffnete Bün-del B stärker gebrochen. Als Folge entsteht in der Gauß-schen Bildebene ein Zer-streuungsscheibchen mit dem Radius rS.
11 Otto Scherzer, deutscher Physiker und Elektronenoptiker, 1909 - 1982

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 25
Die Größe des Fehlerscheibchens hängt von der Apertur α ab. In der Gaußschen12 Bildebene gilt:
3SCSr M α= ⋅ ⋅ (2.6)
mit CS als Öffnungsfehlerkonstante und M als Vergrößerung bei der Abbildung. Die Öffnungsfehlerkonstante kann als ein Qualitätsparameter der Linse angesehen werden. Bei der rotationssymmetrischen magnetischen Linse hat sie etwa den Wert der Brennweite.
Üblicherweise werden Fehlerscheibchen auf die Dingebene bezogen. Dort gilt:
3SCSδ α= ⋅ . (2.7)
Dies ist unabhängig von der Vergrößerung und relevant für den Öffnungsfehler. Wegen der Abhängigkeit der Größe des Fehlerscheibchens von der dritten Potenz der Apertur wird der Öffnungsfehler auch als Abbildungsfehler dritter Ordnung bezeichnet.
- Farbfehler (chromatische Aberration):
Aus der Formel (2.4) für die Lorentzkraft geht hervor, dass die Kraft auf das Elek-tron von der magnetischen Induktion und von der Geschwindigkeit abhängt. An-dererseits wissen wir inzwischen, dass die Elektronenwellenlänge λ durch die Elektronenenergie und damit durch die Geschwindigkeit bestimmt ist (Formeln (1.15) bis (1.17)). Unterschiedliche Wellenlängen, aber natürlich auch unter-schiedliche Linsenfeldstärken, haben unterschiedliche Brechkräfte der magneti-schen Linse und damit einen Abbildungsfehler zur Folge. Beim sichtbaren Licht repräsentieren unterschiedliche Wellenlängen verschiedene Farben. Analog dazu wird der hier beschriebene Abbildungsfehler als Farbfehler bezeichnet. Ähnlich wie beim Öffnungsfehler entsteht in der Gaußschen Bildebene ein Zerstreu-ungsscheibchen, dessen Radius δC (auf die Dingebene bezogen) vom Öffnungs-winkel α des Strahlenbündels, der relativen Schwankung der Brechkraft der Linse ΔS/S und der Farbfehlerkonstante CC der Linse abhängt. Es gilt:
CCCS
Sδ α
Δ= ⋅ ⋅ (2.8)
Die Schwankung der Brechkraft kann zwei Ursachen haben: Änderung der Elek-tronengeschwindigkeit (bzw. Wellenlänge) und Änderung des Magnetfeldes. Für eine Plausibilitätserklärung betrachten wir die Quadrate von Geschwindigkeit und magnetischer Induktion. Das Quadrat der Elektronengeschwindigkeit ist propor-tional zur Elektronenenergie e⋅U (vgl. Formel (1.16)), und die magnetische Induk-
12 Carl Friedrich Gauß, deutscher Mathematiker und Physiker, 1777 - 1855

Kapitel 2 26
tion ist proportional zum Spulenstrom I (Amperesches13 Gesetz, auch Durchflu-tungsgesetz genannt). Da die Brechkraft S der Linse mit stärker werdendem Mag-netfeld und für kleinere Elektronenenergie wächst, können wir schreiben:
2
e
IS
U⋅ . (2.9)
Für kleine Änderungen ΔS folgt daraus:
2S I U
S I U
Δ Δ Δ= ⋅ + . (2.10)
Die für die Qualität der Abbildung maßgebliche Linse ist das Objektiv. Wir müs-sen bezüglich des Farbfehlers demzufolge in erster Linie auf die Stabilität des Ob-jektivlinsenstromes achten. Die Schwankung der Elektronenenergie kann ver-schiedene Ursachen haben: Schwankungen der Beschleunigungsspannung, endli-che Energiebreite der aus der Kathode austretenden Elektronen, aber auch inela-stische Wechselwirkungen zwischen den Elektronen und der Probe, d.h. Wechsel-wirkungen, die mit Energieverlusten verbunden sind.
Nehmen wir weiter an, dass der Farbfehler im Vergleich zum Öffnungsfehler keine Rolle spielen soll, so bedeutet das:
C Sδ δ bzw. 3C SC 2 C
I U
I Uα α
Δ Δ⋅ ⋅ + ⋅ ⋅ . (2.11)
Die Fehlerkonstanten CS und CC sind von gleicher Größenordnung, die Apertur α liegt bei etwa 10 mrad, die Forderung lautet also
42 10I U
I U−Δ Δ
⋅ + . (2.12)
Das bedeutet eine erhebliche Anforderung an die Stabilität der Strom- und Spannungsversorgung eines Elektronenmikroskops. Bei den derzeit höchstauflö-senden Geräten mit Korrektoren für Öffnungs- und Farbfehler (vgl. Abschnitt 7.9) wird sogar die Forderung < 10-8 realisiert [2.2].
- (axialer) Astigmatismus
Im Gedankenexperiment greifen wir uns aus einem Elektronenbündel, das durch eine Linse fällt, zwei Ebenen heraus (in Bild 2-7 rot und grün gezeichnet). Astig-
13 André-Marie Ampére, französischer Physiker, 1775 - 1836

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 27
matismus bedeutet, dass die Brennweiten der Linse in diesen zwei (oder mehr) Ebenen unterschiedlich sind.
Die Ursache sind Abweichungen von der Rotationssymmetrie, möglicherweise bedingt durch „Unrundheit“ der Polschuhbohrung, durch kleinste Materialinhomo-genitäten, durch verschmutzte Blenden oder auch durch im Linsenfeld befind-liches magnetisches Material. Wir beschränken uns hier auf den zweizähligen Astigmatismus, wie er in Bild 2-7 skizziert ist.
Bild 2-7. Schematische Dar-stellung einer Linse mit zweizäh-ligem axialen Astigmatismus.
Die zwei Ebenen stehen senkrecht aufeinander und haben die astigmatische Brennweitendifferenz ΔfA. Für den Radius des (blau gezeichneten) Kreises der kleinsten Verwirrung gilt offensichtlich:
min1 1
'2 2
r b bM
αα= ⋅Δ ⋅ = ⋅Δ ⋅ (2.13)
mit M als Vergrößerung. Bei hohen Vergrößerungen (Dingweite g ≈ Brennweite f) folgt für (kleine) Bildweitendifferenzen der Zusammenhang (vgl. Formeln (10.14.5) und 10.14.6))
2Ab f MΔ = Δ ⋅ (2.14)
mit der astigmatischen Brennweitendifferenz ΔfA. Bezieht man die Größe des Feh-lerscheibchens wie üblich auf die Dingseite, so ist der Radius durch M zu dividie-ren und wir erhalten:
min 1
2A Ar
fM
δ α= = ⋅Δ ⋅ . (2.15)
Kapitel 2 28
2.4 Auflösungsvermögen mit Berücksichtigung des Öffnungsfehlers Am Ende des Abschnitts 1.3 hatten wir versprochen, nochmals auf das Auflö-sungsvermögen zurückzukommen. Zur Erinnerung: Mit Elektronenwellen hatten wir ein Auflösungsvermögen von ca. 2 pm erwartet, erreicht werden allerdings nur etwa 0,1 nm (d.h. 100 pm).
Wir haben nunmehr eine Ursache für diese Diskrepanz erkannt: Unsere rota-tionssymmetrischen magnetischen Linsen haben im Gegensatz zu Glaslinsen einen Öffnungsfehler, der zusätzlich zum (wellenspezifischen) Beugungsfehler das Auf-lösungsvermögen des Elektronenmikroskops begrenzt.
Wir wissen bereits, wie die Radien δB und δS der beiden Fehlerscheibchen in der Objektebene vom Öffnungswinkel α abhängen (Formeln (1.9) und (2.7)). Un-ter Berücksichtigung sehr kleiner Öffnungswinkel (sinα ≈ α) und von n = 1 (Va-kuum) lauten diese:
0,61B
λδ
α
⋅= und 3
SCSδ α= ⋅ (2.16)
Wir erkennen eine gegenläufige Abhängigkeit der Größe der Zerstreuungs-scheibchen vom Öffnungswinkel α (vgl. Bild 2-8). Damit existiert eine optimale Apertur αopt, bei der die Größe δ des Gesamtfehlerscheibchens minimal ist. Für die Abschätzung von δ benutzen wir das Fehlerfortpflanzungsgesetz:
2 2S Bδ δ δ= + . (2.17)
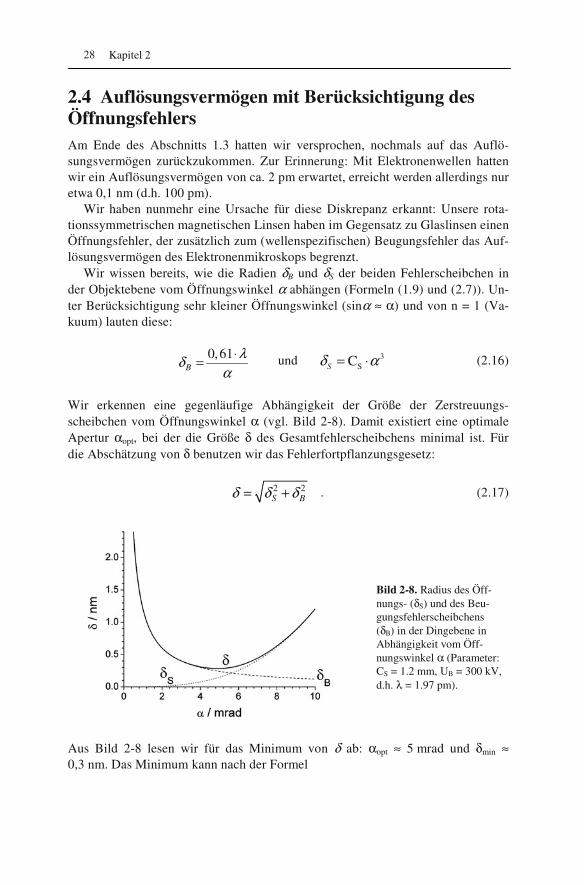
Bild 2-8. Radius des Öff-nungs- (δS) und des Beu-gungsfehlerscheibchens (δB) in der Dingebene in Abhängigkeit vom Öff-nungswinkel α (Parameter: CS = 1.2 mm, UB = 300 kV, d.h. λ = 1.97 pm).
Aus Bild 2-8 lesen wir für das Minimum von δ ab: αopt ≈ 5 mrad und δmin ≈ 0,3 nm. Das Minimum kann nach der Formel

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 29
34min S0,9 C δ λ= ⋅ (2.18)
berechnet werden ( Abschnitt 10.5). Damit sind wir dem praktisch erreichten Auflösungsvermögen sehr nahe gekommen, allerdings ist der errechnete Wert nunmehr etwas zu groß. Es ist demzufolge notwendig, später (Abschnitt 7.3) noch einmal auf das Auflösungsvermögen zurückzukommen.
2.5 Die Elektronenkanone Nun ist es an der Zeit zu überlegen, wie die „freien“ Elektronen, die mit der Probe wechselwirken und durch Linsen zur Formung einer optischen Abbildung veran-lasst werden sollen, überhaupt erzeugt werden können.
Die Frage, die wir uns dazu am Anfang stellen müssen, ist die: „Warum fließen die Elektronen normalerweise in einem Draht und treten nicht von allein aus die-sem heraus?“ Die Antwort findet man im mittleren positiven Potential innerhalb des Drahtes, erzeugt durch die Atomkerne. Dieses Potential hält die Elektronen im Draht, ähnlich wie ein Topf das Wasser in seinem Inneren hält. Mit diesem ein-fachen Modell wird auch klar, wie die Elektronen aus dem Draht befreit werden können: Durch Erhitzen. Stellen wir den Topf, gefüllt mit Wasser, auf eine heiße Kochplatte. Wir werden sehen, dass das heiße Wasser zu wallen beginnt und schließlich über den Topfrand sprudelt. Genauso können wir uns die thermische Emission von Elektronen vorstellen. Anstelle des Kochtopfes verwenden wir ein „Potentialtopf-Modell“ (s. Bild 2-9). Die Energiezustände der Elektronen sind durch Quantenzahlen beschrieben. Jeder Zustand kann nur durch ein Elektron besetzt werden (Pauli14-Prinzip). Beim absoluten Nullpunkt der Temperatur (0 K = -273,16 °C) füllen die Elektronen die minimal möglichen Energiezustände auf. Die dabei erreichte energetische Obergrenze wird als Fermi15-Energie bezeichnet. Damit Elektronen den Draht verlassen können, müssen sie mindestens auf das Po-tential in Drahtumgebung („Vakuumniveau“) angehoben werden. Die dazu zu ver-richtende Arbeit wird als Austrittsarbeit WA bezeichnet.
Bild 2-9. Potentialtopfmodell für Temperatur T = 0 K (a) und T > 0 K (b).
14 Wolfgang Pauli, österreichischer Physiker, 1900 – 1958, Nobelpreis für Physik 1945 15 Enrico Fermi, italienisch/amerikanischer Physiker, 1901 – 1954, Nobelpreis für Physik 1938
Kapitel 2 30
Bei der thermischen Emission wird diese Arbeit in Form von Wärme zugeführt; dabei erhitzt sich der Draht. Es ist klar, dass die notwendige Wärme und damit die Drahttemperatur umso größer sein müssen, je größer die Austrittsarbeit des Ma-terials ist. Außerdem darf das Material nicht schmelzen. In der Praxis hat sich Wolfram (Schmelztemperatur 3695 K = 3422 °C) bewährt. Im einfachsten Fall ist die Kathode ein dünner (Durchmesser ca. 0,5 mm) Wolframdraht, der wie eine Haarnadel gebogen ist („Wolfram-Haarnadelkathode“). Die Austrittsarbeit von Wolfram beträgt etwa 4,6 eV, zur Anregung einer ausreichenden thermischen Emission sind Temperaturen von mehr als 2500 °C erforderlich. Die dabei emit-tierte Elektronenstromdichte j kann nach der Richardson16-Gleichung
2 kA eAW
Tj T−
⋅= ⋅ ⋅ (2.19)
(A: Richardson-Konstante, T: absolute Temperatur, k: Boltzmann17-Konstante) be-rechnet werden.
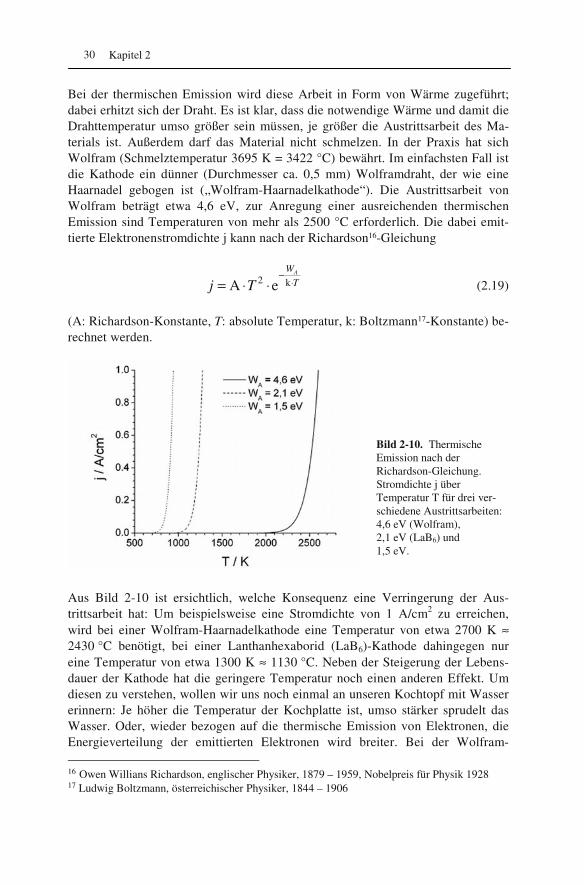
Bild 2-10. Thermische Emission nach der Richardson-Gleichung. Stromdichte j über Temperatur T für drei ver-schiedene Austrittsarbeiten: 4,6 eV (Wolfram), 2,1 eV (LaB6) und 1,5 eV.
Aus Bild 2-10 ist ersichtlich, welche Konsequenz eine Verringerung der Aus-trittsarbeit hat: Um beispielsweise eine Stromdichte von 1 A/cm2 zu erreichen, wird bei einer Wolfram-Haarnadelkathode eine Temperatur von etwa 2700 K ≈ 2430 °C benötigt, bei einer Lanthanhexaborid (LaB6)-Kathode dahingegen nur eine Temperatur von etwa 1300 K ≈ 1130 °C. Neben der Steigerung der Lebens-dauer der Kathode hat die geringere Temperatur noch einen anderen Effekt. Um diesen zu verstehen, wollen wir uns noch einmal an unseren Kochtopf mit Wasser erinnern: Je höher die Temperatur der Kochplatte ist, umso stärker sprudelt das Wasser. Oder, wieder bezogen auf die thermische Emission von Elektronen, die Energieverteilung der emittierten Elektronen wird breiter. Bei der Wolfram- 16 Owen Willians Richardson, englischer Physiker, 1879 – 1959, Nobelpreis für Physik 1928 17 Ludwig Boltzmann, österreichischer Physiker, 1844 – 1906
Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 31
Haarnadelkathode beträgt sie etwa 5 eV - 6 eV, für die LaB6-Kathode reduziert sie sich auf etwa 2 eV – 3 eV.
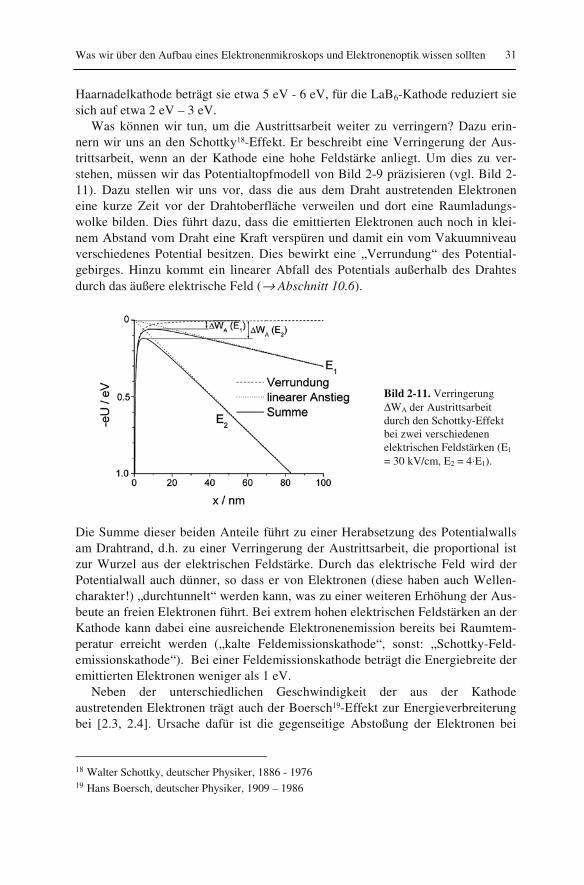
Was können wir tun, um die Austrittsarbeit weiter zu verringern? Dazu erin-nern wir uns an den Schottky18-Effekt. Er beschreibt eine Verringerung der Aus-trittsarbeit, wenn an der Kathode eine hohe Feldstärke anliegt. Um dies zu ver-stehen, müssen wir das Potentialtopfmodell von Bild 2-9 präzisieren (vgl. Bild 2-11). Dazu stellen wir uns vor, dass die aus dem Draht austretenden Elektronen eine kurze Zeit vor der Drahtoberfläche verweilen und dort eine Raumladungs-wolke bilden. Dies führt dazu, dass die emittierten Elektronen auch noch in klei-nem Abstand vom Draht eine Kraft verspüren und damit ein vom Vakuumniveau verschiedenes Potential besitzen. Dies bewirkt eine „Verrundung“ des Potential-gebirges. Hinzu kommt ein linearer Abfall des Potentials außerhalb des Drahtes durch das äußere elektrische Feld (→ Abschnitt 10.6).
Bild 2-11. Verringerung ΔWA der Austrittsarbeit durch den Schottky-Effekt bei zwei verschiedenen elektrischen Feldstärken (E1 = 30 kV/cm, E2 = 4⋅E1).
Die Summe dieser beiden Anteile führt zu einer Herabsetzung des Potentialwalls am Drahtrand, d.h. zu einer Verringerung der Austrittsarbeit, die proportional ist zur Wurzel aus der elektrischen Feldstärke. Durch das elektrische Feld wird der Potentialwall auch dünner, so dass er von Elektronen (diese haben auch Wellen-charakter!) „durchtunnelt“ werden kann, was zu einer weiteren Erhöhung der Aus-beute an freien Elektronen führt. Bei extrem hohen elektrischen Feldstärken an der Kathode kann dabei eine ausreichende Elektronenemission bereits bei Raumtem-peratur erreicht werden („kalte Feldemissionskathode“, sonst: „Schottky-Feld-emissionskathode“). Bei einer Feldemissionskathode beträgt die Energiebreite der emittierten Elektronen weniger als 1 eV.
Neben der unterschiedlichen Geschwindigkeit der aus der Kathode austretenden Elektronen trägt auch der Boersch19-Effekt zur Energieverbreiterung bei [2.3, 2.4]. Ursache dafür ist die gegenseitige Abstoßung der Elektronen bei
18 Walter Schottky, deutscher Physiker, 1886 - 1976 19 Hans Boersch, deutscher Physiker, 1909 – 1986

Kapitel 2 32
hohen Stromdichten, wie sie bei Fokussierung der Elektronen innerhalb der Kanone im engsten Strahlquerschnitt auftreten können. Dieser Anteil liegt bei wenigen 0,1 eV und ist bei unseren Zahlenangaben zur Energiebreite berück-sichtigt.
An einer Kugeloberfläche ist die elektrische Feldstärke Er umgekehrt pro-portional zum Kugelradius r:
1rE
r , (2.20)
d.h. bei sehr kleinem Krümmungsradius der Kathodenspitze wird die Feldstärke dort sehr groß. Feldemissionskathoden sind deshalb extrem spitz (Krümmungs-radius ca. 1 μm) und ihr Emissionsverhalten hängt dramatisch von der Spitzen-form ab. Ein Beschuss der Spitze mit Restgas-Ionen würde die Spitze verrunden und muss deshalb vermieden werden. Die Konsequenz ist, dass der Restgasdruck im Kathodenraum extrem niedrig sein muss, wir benötigen Ultrahochvakuum (Druck < 10-8 Pa).
Um die für ein gutes Auflösungsvermögen notwendigen kurzen Wellenlängen zu erreichen, müssen die aus der Kathode ausgetretenen freien Elektronen be-schleunigt werden. Dies geschieht im elektrischen Feld zwischen Kathode (Spitze) und Anode (Platte mit kleinem Loch) (s. auch Formeln (1.13) bis (1.15)). Zu-sätzlich soll der aus der Anode austretende Elektronenstrom verändert werden können. Es hängt von der Kathodenart ab, wie dies organisiert wird.
Bild 2-12. Elektroden in der Elektronenkanone. a) Triodensystem für Haarnadel- und LaB6-Kathoden. b) Elektrodensystem in einer Elektronenkanone mit Feldemissionskathode (Bezeichnung der Elektroden und Potentiale).
Bei Elektronenkanonen mit Wolfram-Haarnadel- oder LaB6-Kathode wird ein Triodensystem benutzt, das ähnlich wie eine Trioden-Elektronenröhre arbeitet (vgl. Bild 2-12a). Die geheizte Kathode liegt auf negativem Hochspannungspoten-tial und emittiert den Strahlstrom I. Dieser verursacht am Widerstand R einen

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 33
Spannungsabfall, d.h. der Fußpunkt des Widerstandes ist negativer (bis zu weni-gen 100 V) als die Kathode selbst. Dieses Potential liegt an der Wehnelt20-Elektro-de, die als Steuerelektrode für den Strahlstrom I arbeitet: Je negativer diese Elek-trode, desto kleiner der Strahlstrom. Die Schaltung stabilisiert gleichzeitig den Strahlstrom: Bei kleiner werdendem Strahlstrom verringert sich der Spannungs-abfall an R, die Wehnelt-Elektrode wird etwas positiver und erhöht damit den Strahlstrom. Das Potential ist so geformt, dass die Elektronen bereits in der Kano-ne fokussiert werden (→ Abschnitt 10.7). Der dabei etwa in Höhe der Anode entstehende engste Strahlquerschnitt wird als „cross over“ bezeichnet. Dieser stellt elektronenoptisch die eigentliche Quelle dar.
Etwas anders sieht die Elektronenkanone bei Verwendung einer Feldemissions-kathode aus (vgl. Bild 2-12b). Im Interesse einer hohen Feldstärke vor der Katho-denspitze ist der Extraktor positiv gegenüber der Kathode. Die gegen Kathoden-potential leicht negative Suppressor-Kappe verhindert, dass Elektronen, die nicht an der Kathodenspitze ausgelöst werden, in den Elektronenstrahl gelangen. Die Linsenelektrode ermöglicht eine Fokussierung des Elektronenstrahls, d.h. die For-mung eines cross over, der durch Veränderung der Potentiale an den Elektroden (UE, UL) auf der Mittelachse verschoben werden kann. Bei bestimmten Verhältnissen von UL/UE kann es passieren, dass die Elektronen am Durchtritt durch die Linse gehindert werden, d.h. es kommt kein Strahlstrom zustande.
2.6 Der Richtstrahlwert Die Frage ist nun: Gibt es einen Parameter für die Elektronenkanone, der die Qua-lität dieser Kanone hinsichtlich ihrer Funktion im Elektronenmikroskop be-schreibt? Ja, den gibt es: Es ist der Richtstrahlwert R21. Er ist definiert als Quotient aus der die Kathode verlassende Elektronenstromdichte j und dem Raumwinkel Ω, in den die Elektronen emittiert werden:
jR =
Ω . (2.21)
Im rotationssymmetrischen Fall gilt bei kleinen Winkeln
2αΩ = ⋅ , (2.22)
wobei α der halbe Öffnungswinkel ist. Wir wollen nun überlegen, wovon α be-einflusst wird. Dazu verwenden wir ein stark vereinfachtes Modell der Elektro-nenkanone, in dem nur Kathode und Anode vorhanden sind (s. Bild 2-13).
20 Arthur Wehnelt, deutscher Physiker, 1871 - 1944 21 1939 eingeführt von E. Ruska und B. von Borries

Kapitel 2 34
Bild 2-13. Skizze zur Berechnung des Winkels α für den Richtstrahlwert (v: Geschwindigkeit der Elek-tronen, vT: Komponente durch thermische Emission, vB: Komponente durch Beschleunigungsfeld).
Den größten Winkel α erreichen die Elektronen, die mit der thermischen Energie
2me
2 T Tv U⋅ = ⋅ (2.23)
senkrecht zur Achse aus der Kathode austreten. Aus Bild 2-13 folgt für kleine Winkel (α << 1) mit dem Energieerhaltungssatz (1.16)
e
eT T T
B B B
v U U
v U Uα
⋅= = =
⋅ . (2.24)
Für die thermische Emission können wir die Stromdichte aus dem Richardson-schen Gesetz (2.19) berechnen und erhalten somit als Abschätzung für den Richt-strahlwert
2 kA eAW
TB
T
T UR
U
−⋅⋅ ⋅ ⋅
=⋅
. (2.25)
Für die Wolfram-Haarnadelkathode (WA ≈ 4,6 eV, T ≈ 2700 K, e⋅UT ≈ 5 eV) folgt daraus bei einer Beschleunigungsspannung von 200 kV ein Richtstrahlwert von R ≈ 3⋅104 A/cm2, für die LaB6-Kathode (WA ≈ 2,1 eV, T ≈ 1800 K, e⋅UT ≈ 3 eV) R ≈ 107 A/cm2. Bei Schottky-Feldemissionskathoden wird eine mit Zirkonium- oder Thoriumoxid dotierte Wolframspitze benutzt und damit die Austrittsarbeit auf Werte verringert, die etwa der des LaB6 entsprechen. Hinzu kommt die weitere Reduzierung der Austrittsarbeit durch den Schottky-Effekt. Bei einer Extraktor-spannung von 4 kV und einem Spitzenradius von 1 μm beträgt die elektrische Feldstärke an der Kathode etwa 4⋅104 kV/cm. Dadurch wird die Austrittsarbeit um etwa 0,8 eV erniedrigt. Mit WA ≈ 1,3 eV, T ≈ 1500 K, e⋅UT ≈ 1 eV erhalten wir al-lein für die thermische Emission einen Richtstrahlwert von ca. 7⋅108 A/cm2. In Wirklichkeit wird die Stromdichte durch den Tunneleffekt größer sein, so dass bei der Schottky-Kathode mit einem Richtstrahlwert in der Größenordnung 109 A/cm2 zu rechnen ist. Bei der kalten Feldemissionskathode ist der Krümmungsradius der Kathodenspitze im Interesse einer extrem hohen Feldstärke kleiner als bei der

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 35
Schottky-Kathode und die Energiebreite der emittierten Elektronen beträgt nur etwa 0,3 eV. Das Resultat ist ein Richtstrahlwert höher als 1010 A/cm2.
Die Frage ist nun, welche praktischen Konsequenzen ein höherer Richtstrahl-wert hat. Dazu überlegen wir, was mit ihm entlang des Abbildungsstrahlenganges passiert. Der Strahlstrom IS soll aus einer Kathodenfläche AK in den Raumwinkel
2K KΩ α= ⋅ (2.26)
emittiert werden. Für den auf die Kathode bezogenen Richtstrahlwert gilt demzu-folge
2S
KK K
IR
A α=
⋅ ⋅ . (2.27)
Durch die Kondensorlinse wird die Fläche AK in eine Fläche AB abgebildet, für die mit der Vergrößerung M
2B KA A M= ⋅ . (2.28)
gilt. Dabei wird der (halbe) Öffnungswinkel α in
KB M
αα = (2.29)
geändert. Wenn keine Blenden beteiligt sind, ändert sich der Strahlstrom IS nicht. Für den auf AB bezogenen Richtstrahlwert RB gilt demnach:
2
2 2 2S S
B KB B K K
I I MR R
A A Mα α
⋅= = =
⋅ ⋅ ⋅ ⋅ ⋅ (2.30)
d. h. der Richtstrahlwert ändert sich durch die Linsen nicht. Die Gleichung (2.30) gilt also auch für die Probenebene. Wollen wir den Elektronenstrahl in dieser Ebe-ne auf einen möglichst kleinen kreisförmigen Fleck mit dem Durchmesser dS fo-kussieren und dabei einen Strahlstrom IS erreichen, so gilt:
2 2 2 2
4 S
S
IjR
dα α
⋅= =
⋅ ⋅ ⋅
(2.31)
bzw.
2 2 2
4S
Sd
I Rα⋅ ⋅
= ⋅ , (2.32)

Kapitel 2 36
d.h. der (längs des Strahlenganges konstante) Strahlstrom hängt u. a. vom Fleck-durchmesser dS ab. Der Richtstrahlwert R ist der Parameter, der beschreibt, welcher Strom maximal in einer kleinen Elektronensonde vorhanden sein kann. Wir werden später sehen, dass ein hoher Richtstrahlwert noch weitere Konsequen-zen für die Elektronenmikroskopie hat.
2.7 Wir bauen ein Elektronenmikroskop Nachdem wir die Funktion von Elektronenkanone und Elektronenlinsen verstan-den haben, können wir daraus und mit einigen Ergänzungen gedanklich ein Elek-tronenmikroskop zusammenstellen. Wir nehmen uns dazu das Lichtmikroskop zum Vorbild, drehen es aber so, dass der Kondensor die oberste Linse ist (s. Bild 2-14). Selbstverständlich ist die Lampe durch eine Elektronenkanone ersetzt, und die Linsen sind elektrische Spulen mit Polschuhen, die rotationssymmetrische magnetische Felder erzeugen.
Darüber hinaus werden für Kondensor und Projektiv keine einzelnen Linsen sondern Linsensysteme benutzt, die im Folgenden genauer beschrieben werden sollen. Außerdem ist eine besondere Probenbühne notwendig, mit der die Probe hinreichend stabil gehalten und sehr präzise bewegt werden kann.
Bild 2-14. Vergleich zwischen Lichtmikroskop (rechts) und Transmis-sionselektronenmikroskop (links).
Da wir kein Sinnesorgan für Elektronen besitzen (was auch gut ist), brauchen wir spezielle Sensoren, um das Bild in der Endbildebene zu beobachten. Schließlich benötigen wir ein Vakuumsystem, um mit den freien Elektronen arbeiten zu können. Da bei modernen Elektronenmikroskopen die Bedienelemente in eng-lischer Sprache bezeichnet sind, werden wir bei den folgenden Beschreibungen auch die englischen Begriffe erwähnen.

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 37
2.7.1 Das Beleuchtungssystem
Das Beleuchtungssystem umfasst die Elektronenkanone und das Kondensorsy-stem, d.h. salopp gesagt: „Alles, was sich im Strahlengang vor der Probe befin-det.“ Es hat die Aufgabe, die Elektronenstromdichte auf der Probe und unter-schiedliche Bestrahlungsbedingungen für die Probe zu realisieren und damit u.a. die Bildhelligkeit einzustellen. In diesem Zusammenhang spielt die Beleuchtungs-apertur β eine wichtige Rolle (s. Bild 2-16). Der Abstand zwischen Kondensor und Probe beträgt einige Zentimeter und ist damit für elektronenoptische Verhält-nisse groß. Um den im Elektronenstrahler erzeugten cross over auf die Probe ab-zubilden, würde eine schwache Linse mit großer Brennweite benötigt. Solche Lin-sen weisen jedoch einen großen Öffnungsfehler auf und sind deshalb ungeeignet. Der Ausweg besteht in der Benutzung von zwei (oder drei) Kondensorlinsen. Am gebräuchlichsten ist der sogenannte „Doppelkondensor“ (s. Bild 2-15).
Bild 2-15. Doppelkondensor mit verschiedenen Erregungen der Kondensorlinse 2: a) Schwache Erregung. b) Parallele Beleuchtung. c) Konvergente Beleuchtung. d) Starke Erregung. Durch Verkürzung der Brennweite der Kondensorlinse 1 wird der cross over stärker verkleinert, d.h. es wird ein kleinerer Fleck auf der Probe erreicht. Deshalb wird diese Einstellung als „Spot Size“ bezeichnet. Wir sollten uns hier allerdings an den Abschnitt 2.6 über den Richtstrahlwert erinnern: Ein kleinerer Durch-messer des Elektronenstrahls auf der Probe bedeutet auch weniger Strahlstrom.
Bild 2-16. Einstellung der Bestrahlungsapertur β durch eine Blende in Hö-he der Kondensorlinse 2 („C2-Blende“).

Kapitel 2 38
Durch Veränderung der Brennweite der Kondensorlinse 2 wird die Stromdichte auf der Probe und damit die Bildhelligkeit eingestellt (deshalb „Intensity“). Wich-tige Grenzfälle sind die parallele und die konvergente Beleuchtung. Manchmal ist es notwendig, die Bestrahlungsapertur β bei konvergenter Beleuchtung zu ver-ändern (s. Kapitel 5 Elektronenbeugung). Dazu dient eine Blende nahe der Kon-densorlinse 2 (vgl. Bild 2-16). Es ist klar, dass durch eine kleinere Kondensor-2-Blende eine größere Anzahl von Elektronen aus dem Strahlengang entfernt wird: Der Strahlstrom und auch der Richtstrahlwert werden kleiner.
2.7.2 Das Abbildungssystem
Das Abbildungssystem erzeugt ein optisches Bild der durchleuchteten Probe und umfasst damit alle Linsen, die sich unterhalb der Probe befinden.
Bild 2-17. Strahlengang in einem Mikroskop bei drei-stufiger Abbildung (zwei Projektivlinsen). Ohne Be-rücksichtigung der Bilddre-hung in magnetischen Linsen ist in diesem Fall das Endbild gegenüber dem Objekt umgekehrt.
Für die mehrstufige Abbildung ist eine genaue Abstimmung von Abständen und Brennweiten innerhalb des Abbildungssystems notwendig. Dazu gehört auch die

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 39
Position der Probe, d. h. des Objektes. Bei Veränderung der Vergrößerungseinstel-lung („Magnification“) am Mikroskop werden die Brennweiten der einzelnen Projektivlinsen verändert, die Brennweite des Objektivs bleibt konstant. Da die Endbildebene unveränderlich ist, werden bei Variation der Brennweite fProj des Gesamtprojektivlinsensystems unterschiedliche Ebenen in die Endbildebene abge-bildet (vgl. Abbildungsgleichung (1.3)). Im Abbildungsstrahlengang betrifft das die erste reelle Zwischenbildebene.
Die reellen Zwischenbildebenen sind ortsselektive Ebenen: Wählt man bei-spielsweise mit einer kleinen Blende einen Bereich in einer dieser Ebenen aus, so entspricht dies einem bestimmten Ortsbereich auf dem Objekt. In diesem Zusam-menhang ist eine weitere Ebene im Abbildungssystem hervorhebenswert: Die bildseitige Brennebene des Objektivs. Aus der Abbildungsgleichung (1.3) folgt, dass für alle parallel zueinander in das Objektiv einfallenden Strahlen (Dingweite g ∞) die Bildweite b gleich der Brennweite f ist, d. h. diese Strahlen treffen sich in der bildseitigen Brennebene. Verlaufen sie auch noch parallel zur optischen Achse („Parallelstrahlen“), dann schneiden sie sich bekanntermaßen im bildseiti-gen Brennpunkt. Sind die parallelen Strahlen gegen die optische Achse geneigt, so wandert der Schnittpunkt aus dem Brennpunkt heraus, verbleibt bei kleinen Nei-gungswinkeln aber in der Brennebene (s. Bild 2-18).
Bild 2-18. Bildseitige Brenn-ebene als winkelselektive Ebene: Ein paralleles Strah-lenbündel, das um den Win-kel α gegen die optische Achse geneigt ist, schneidet sich in der bildseitigen Brennebene im Abstand α⋅f von der optischen Achse (α<<1).
Setzen wir unsere kleine Blende in die bildseitige Brennebene und wählen damit einen kleinen Bereich aus, so entspricht das auf der Dingseite der Linse einem Winkelbereich für die einfallenden Strahlen. Die bildseitige Brennebene ist folge-richtig eine winkelselektive Ebene.
2.7.3 Die Probenbühne
Auf den ersten Blick erscheint es vielleicht verwunderlich, dass wir der Proben-bühne für das Elektronenmikroskop einen besonderen Abschnitt widmen. Wenn wir aber daran denken, dass die förderliche Vergrößerung des Elektronenmikro-skops bei etwa 106 liegt, dann wird die besondere Bedeutung der Probenbühne klar: Will man ein Bild mit 106-facher Vergrößerung registrieren und die Probe

Kapitel 2 40
bewegt sich während der Registrierung, die beispielsweise 1 s dauert, nur um 0,1 nm, dann sieht man im Bild eine Unschärfe von 0,1 mm, was etwa dem Auf-lösungsvermögen des Auges entspricht. Mit anderen Worten: das Bild ist un-scharf!
Damit ist eine außerordentliche Forderung an die Stabilität der Probenhalterung formuliert: Die Probe darf sich in einer Sekunde nur um weniger als ein Zehn-millionstel Millimeter bewegen. Wir wissen, dass der Wärmeausdehnungskoef-fizient für Metalle in der Größenordnung von 10-5 K-1 liegt. Das heißt, bei einer Temperaturschwankung von nur 1 K würde sich eine Halterung von nur 1 mm Länge bereits um 10 nm verlängern oder verkürzen. Die Probenhalterung muss also nicht nur eine außerordentlich gute mechanische Stabilität besitzen, auch die Umgebungstemperatur sollte auf besser als 0,1 K konstant gehalten werden.
Aber nicht nur das: Zum Aufsuchen von Details auf der Probe muss es möglich sein, die Probe definiert zu bewegen, zunächst in zwei senkrechten Richtungen (x und y) in der Probenebene. Aus den Überlegungen zum Abbildungssystem (Ab-schnitt 2.7.2) wissen wir aber auch, dass es eine Objektebene gibt, die zumindest näherungsweise eingehalten werden muss. Andernfalls funktioniert die Abstim-mung der Brennweiten des Projektivlinsensystems nicht. Das bedeutet, die Probe muss auch in Richtung der Objektivachse („optische Achse“, z-Richtung) definiert verschiebbar sein.
Schließlich (und das werden wir später besser verstehen) ist es für einige Un-tersuchungen an kristallinem Material erforderlich, die Probe um zwei Achsen zu kippen, um sie geeignet zum Elektronenstrahl orientieren zu können.
Als nächstes wollen wir überlegen, an welcher Stelle des Objektivlinsenfeldes sich eigentlich die Objektebene (Dingweite des Objektivs) befindet. Um den Öff-nungsfehler klein zu halten, benutzen wir als Objektiv eine kurzbrennweitige Linse; ihre Brennweite liegt bei 1 mm bis 2 mm. Angenommen, wir wünschen eine Vergrößerung durch das Objektiv von 100, dann beträgt die Differenz zwi-schen dingseitigem Brennpunkt und Objektebene nur 1% der Brennweite (vgl. Formel (2.3)). Die Probe ist also nahezu in Linsenmitte, d. h. innerhalb des Pol-schuhs des Objektivs anzuordnen (s. Bild 2-19).
Bild 2-19. Anordnung der Probe innerhalb des Pol-schuhs der Objektivlinse.

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 41
Die Probe soll in drei Raumrichtungen verschoben und mindestens um die Pro-benhalterachse gekippt werden. Dazu wird der Halter in eine Goniometer genann-te Vorrichtung eingeschoben, die neben den Bewegungsmöglichkeiten auch die Vakuumdichtheit gewährleistet.
2.7.4 Die Registrierung des Bildes
Üblicherweise wird das Endbild zunächst mit den Augen betrachtet. Elektronen müssen dazu in sichtbares Licht umgewandelt werden. Dafür wird ein Effekt aus-genutzt, der als „Kathodolumineszenz“ bezeichnet wird. Durch die einfallenden Strahlelektronen werden die Elektronen eines Festkörperatoms auf höhere Bahnen gehoben (in höhere Energiezustände gebracht). Dieser „angeregte“ Zustand ist nicht stabil, die „angehobenen“ Elektronen kehren wieder in ihren Grundzustand zurück und das Atom emittiert dabei elektromagnetische Strahlung (vgl. Abschnitt 9.1.1). Die Wellenlänge der emittierten Strahlung hängt vom Energieunterschied zwischen angeregtem und Grundzustand und damit vom Material („Leuchtstoff“) ab. Sichtbares Licht mit Wellenlängen von etwa 500 nm (grünes Licht) entsteht bei einem Energieunterschied von ca. 2,5 eV (z.B. Cadmiumsulfid, Zinkselenid). Der Beobachtungsschirm in der Endbildebene ist ein Metallblech, welches mit einer dünnen Leuchtstoffschicht bedeckt ist. Er wird durch ein Bleiglasfenster von außen betrachtet.
In manchen Fällen (Elektronenmikroskop durch ein Gehäuse komplett nach außen abgeschirmt oder Endbildebene besonders tief gelegen) ist die direkte Be-trachtung des Bildschirms unmöglich. Dann hilft eine auf den Bildschirm gerich-tete Fernsehkamera.
Der zweite Schritt ist die Dokumentation der elektronenmikroskopischen Bil-der. Bis in die 1990-er Jahre erfolgte dies hauptsächlich fotografisch mit speziel-len elektronenempfindlichen Emulsionen. Die bei modernen Elektronenmikro-skopen immer noch vorhandene Anzeige der Belichtungszeit („Exposure Time“) erinnert an diese Zeit. Vorteilhaft an diesem Verfahren ist das vergleichsweise große erfasste Gesichtsfeld, nachteilig die Tatsache, dass die Filme oder Foto-platten in das Vakuum des Mikroskops gebracht werden müssen. Da die Emul-sionen Wasser enthalten, ist trotz vorheriger Vakuumtrocknung eine Beeinträch-tigung des Vakuums im Elektronenmikroskop nicht auszuschließen. Und schließ-lich sind die Bilder nicht sofort verfügbar: Wir benötigen Zeit, Chemikalien (Entwickler, Fixiersalz), eine Dunkelkammer und nicht zuletzt eine Person mit Erfahrung.
Bild 2-20. Schematische Darstellung einer CCD-Kamera.

Kapitel 2 42
Gegenwärtig werden die Bilder hauptsächlich mit einer „CCD-Kamera“ (CCD: Charge-Coupled Device, d.i. „Ladungsgekoppelte Vorrichtung“) registriert. Sie besteht im Wesentlichen aus einem Durchsichtsleuchtschirm, einer Faseroptik und einem CCD-Array (s. Bild 2-20).
Der Leuchtschirm soll eine hohe laterale Auflösung haben, deshalb wird ein dünner Yttrium-Aluminium-Granat („YAG“) Kristall benutzt. Er wandelt das Sig-nal der einfallenden Elektronen in ein Lichtsignal um, welches über Glasfasern zu den CCD-Elementen weitergeleitet wird. Die Dicke des Leuchtschirms beeinflusst sowohl die Lichtausbeute (ein dickerer Schirm ist empfindlicher) als auch die Verteilung des Lichts in benachbarte CCD-Elemente (ein dickerer Schirm verteilt die Information über einen größeren Bereich). Hier ist ein Kompromiss für die üblicherweise im Elektronenmikroskop benutzte Beschleunigungsspannung not-wendig.
Ein CCD-Element besteht aus Silizium, das mit einer dünnen Silizium-oxidschicht und einer (lichtdurchlässigen) Metallelektrode bedeckt ist („MOS-Kondensator“). Beim Auftreffen von Licht entstehen Elektron-Loch-Paare, die zur Aufladung des kleinen Kondensators führen. Die Größe dieser Aufladung hängt von der Lichtintensität und von der Belichtungszeit ab, die zur Aufladung zur Verfügung steht („Exposure Time“). Nach Ablauf der Aufladungszeit werden die Aufladungen ausgelesen und das CCD-Element in seinen Ausgangszustand zurückgesetzt. Die Kamera ist für die nächste Bildaufnahme bereit.
Wegen der thermischen Bewegung der Ladungsträger entstehen auch ohne Einfall von Licht geringe Aufladungen. Dieser „Dunkelstrom“ (Dark current) hängt von der Aufladungszeit ab und wird üblicherweise vor Beginn der eigentli-chen Bildaufnahme gemessen. Das Bild kann dann später im Rechner damit korri-giert werden. Der Dunkelstrom wird durch Peltier22-Kühlung des CCD-Arrays verringert. Ein weiterer Effekt, der korrigiert werden kann, ist die unterschiedliche Effizienz der einzelnen CCD-Elemente („Pixel“), d.h. die Unterschiede im Aus-gangssignal bei gleicher Lichtintensität. Um dies zu korrigieren („Gain Cor-rection“ oder „Flat Field Correction“) wird ein Referenzbild benötigt, das bei gleichmäßiger Beleuchtung des gesamten YAG-Leuchtschirms aufgenommen wurde. Kameras mit größerem Gesichtsfeld sind aus vier Einzelarrays zusammen-gesetzt („Quadranten“), deren unterschiedliche Effizienz gleichfalls auf die be-schriebene Weise korrigiert wird.
Wird die Zahl der Elektron-Loch-Paare zu groß, so fließen Ladungen auf be-nachbarte CCD-Elemente über („Übersprechen“, „Blooming“). In diesem Fall muss die Elektronenintensität oder die Aufladungszeit verringert werden. Bei län-ger andauernder Überladung kann das CCD-Element dauerhaft geschädigt wer-den. Bei geringer Elektronenintensität (Bildhelligkeit) oder erwünschten kurzen Aufladungszeiten können mehrere Pixel zusammengeschaltet werden („Binning“). Binning 2 bedeutet beispielsweise, dass 2 x 2 = 4 Elemente verbunden sind. Die
22 Jean Peltier, französischer Physiker, 1785 – 1845

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 43
Empfindlichkeit wird damit (allerdings auf Kosten der lateralen Auflösung durch die damit verbundene Vergrößerung der effektiven Pixelgröße) auf das Vierfache erhöht.
Besonders bei sehr geringen Elektronenintensitäten mit langen Aufladungszei-ten kann es passieren, dass einzelne Pixel im Bild der CCD-Kamera extrem hell erscheinen. Die Ursache sind von Elektronen ausgelöste Röntgenquanten, die wegen ihrer höheren Energie in dem von ihnen getroffenen CCD-Element zu einer großen Ausbeute von Elektronen-Loch-Paaren führen.
Ein Nachteil der CCD-Kamera, speziell bei Übersichtsbildern, ist das ver-gleichsweise kleine erfasste Gesichtsfeld. Bei einer (üblichen) Pixelgröße von 24 μm auf dem YAG-Leuchtschirm und einer sogenannten 1Kx1K-Kamera (das sind 1024 x 1024 Pixel) ist das Gesichtsfeld nur 24,6 mm x 24,6 mm groß. Kameras mit größerer Pixelzahl haben in der Regel eine kleinere Pixelgröße und damit ein besseres Auflösungsvermögen, so dass damit das Gesichtsfeld nicht ent-scheidend vergrößert wird. Abhilfe schafft die Fotomontage von Einzelbildern.
2.7.5 Das Vakuumsystem
Vakuum ist aus mehreren Gründen im Elektronenmikroskop erforderlich: • Die Elektronen sollen sich frei bewegen können und nicht mit Luftmole-
külen zusammenstoßen. • Der Heizdraht der Kathode soll nicht durchbrennen. • Die Kathodenspitze soll nicht durch Ionenbeschuss verrundet werden. • Die Elektronenkanone soll spannungsfest sein, d.h. zwischen Kathode
und Anode sollen elektrische Überschläge vermieden werden. • Die Probe soll nicht verschmutzt werden.
Daraus lassen sich Forderungen für den maximal zulässigen Druck ableiten. Aus der Zustandsgleichung
Rp V m T⋅ = ⋅ ⋅ (2.33)
(p: Gasdruck, V: Gasvolumen, m: Gasmasse, R: Gaskonstante, T: absolute Tem-peratur) für ideale Gase folgt für die Teilchendichte
AA
NN
R k
pm pn
V T T
⋅= ⋅ = =
⋅ ⋅ (2.34)
(NA: Avogadro23-Konstante, k: Boltzmann-Konstante). Wenn sich die Elektronen ohne Zusammenstoß mit Luftmolekülen im Elektro-
nenmikroskop bewegen sollen, muss die mittlere freie Weglänge Λ zwischen zwei
23 Amedeo Avogadro, italienischer Mathematiker und Physiker, 1776 - 1856

Kapitel 2 44
Stößen sehr groß gegen die Höhe hM des Mikroskops (ca. 2 m) sein. Für die mitt-lere freie Weglänge gilt näherungsweise
2 2
1 k
M M
T
n r p r
⋅Λ ≈ =
⋅ ⋅ ⋅ ⋅ (2.35)
mit rM als Molekülradius. Da Λ >> hM ist, gilt
2
k
M M
Tp
h r
⋅<<
⋅ ⋅ . (2.36)
Für Sauerstoff und Stickstoff ist rM ≈ 150 pm, d.h. bei Raumtemperatur (293 K) muss der Druck p in der Mikroskopsäule deutlich kleiner als 3⋅10-2 Pa sein. Über-setzen wir „deutlich kleiner“ mit „etwa zehnfach kleiner“, so folgt daraus ein Druck p < 10-3 Pa.
Interessant ist in diesem Zusammenhang ein Blick auf die Teilchendichte n. Sie beträgt bei 10-3 Pa immer noch ca. 3⋅108 Moleküle pro mm3. Aus der Gaskinetik (Maxwellsche24 Geschwindigkeitsverteilung der Moleküle) folgt für den Teilchen-strom aus dem Halbraum auf eine Wand der Fläche A
AN8 RR
4 k 2 2 k
dN n A p A TT p A
dt T T
⋅ ⋅ ⋅= ⋅ ⋅ ⋅ = ⋅ = ⋅ ⋅
⋅ ⋅ ⋅ ⋅ ⋅ . (2.37)
Für Luft (1 mol = 29 g) und eine Fläche von 3 μm2 (Kathodenspitze mit Krüm-mungsradius 1 μm) folgt daraus ein Teilchenstrom von ca. 107 Teilchen pro Se-kunde, der auf die Kathodenspitze trifft. Selbst wenn nur jedes millionste Teilchen die nötige Energie für eine Veränderung der Spitze hat, sind das immer noch 10 kritische Ereignisse pro Sekunde. Daraus ist ersichtlich, wie wichtig ein niedriger Druck im Kathodenraum ist. Bei Ultrahochvakuum (p < 10-8 Pa) reduziert sich nach unserer Abschätzung die Zahl der kritischen Ereignisse auf weniger als 1 pro Stunde.
Wir erkennen, dass die Anforderungen an das Vakuum im Elektronenmikro-skop nicht überall gleich sind. In der Elektronenkanone mit Feldemissionskathode sollte der Druck kleiner als 10-8 Pa sein, in der restlichen Mikroskopsäule kleiner als 10-3 Pa. Dieser Druckunterschied ist möglich, weil sich zwischen Kanone und Säule sehr kleine Blenden befinden.
Um diese Vakua zu erzeugen und aufrecht zu erhalten, benötigen wir Pumpen. Wir unterscheiden zwischen Transportpumpen und Speicherpumpen. Transport-pumpen arbeiten mit veränderlichen Volumina, in denen das Gas aus dem zu eva-kuierenden Behälter wechselweise angesaugt und dann verdichtet an die Umge-bung abgegeben wird (Verdrängerpumpen: Drehschieberpumpe, Membranpumpe, Rootspumpe). Bei der anderen Variante der Transportpumpe wird der Brown-
24 James Clerk Maxwell, schottischer Physiker, 1831 – 1879

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 45
schen25 Bewegung der Gasmolekeln eine Vorzugsrichtung erteilt. Dies kann durch Berühren mit dem Rotor einer Turbomolekularpumpe oder durch Zusammenstoß mit einem gerichteten Strahl von Ölmolekülen (Öldiffusionspumpe) erfolgen. Die-se Pumpenart kann nicht gegen den äußeren Luftdruck arbeiten, sie benötigt ein Vorvakuum, das durch eine Transportpumpe der ersten Art geschaffen wird.
Speicherpumpen speichern das gepumpte Gas in ihrem Innern. Die gebräuch-lichste Variante für den Hoch- und Ultrahochvakuum-Bereich ist die Ionengetter-pumpe. Im einfachsten Fall besteht sie aus einem geerdeten Edelstahlgehäuse mit einem Gitter aus Titan im Innern (vgl. Bild 2-21). Das Ganze ist im Magnetfeld eines Permanentmagneten untergebracht. Zwischen Gitter und Gehäuse liegt eine Spannung von etwa 5 kV. Unterhalb eines Drucks von einigen zehn Pa brennt eine Gasentladung, die durch Stoßionisation befördert wird, Titan vom Gitter abstäubt und als dünne Schicht auf der Gehäusewand deponiert. Diese Titanschicht bindet Stickstoff und Sauerstoff chemisch und wird durch den Sputterprozess ständig erneuert. Edelgase können nach Physisorption nur von den neuen Schichten „zugeweht“ werden, das Saugvermögen der Getterpumpe ist für Edelgase dem-entsprechend schlecht.
Bild 2-21. Ionengetterpumpe (Gehäuse aufgesägt, ohne Permanentmagnet). In der Mitte sind Interferenzfarben dünner Schichten zu sehen.
Zum Zünden benötigt auch die Getterpumpe ein Vorvakuum, im „normalen“ Be-trieb ist keine Vorvakuumpumpe erforderlich. Das Magnetfeld verlängert die Ionenbahnen und erhöht damit die Ionisationswahrscheinlichkeit der Restgasmole-keln. Die Pumpe regelt sich selbst: Bei höherem Druck verstärkt sich die Gasent-ladung, bei niedrigem Druck schwächt sie sich ab. Der Pumpenstrom ist also gleichzeitig ein Maß für den Druck. Läuft die Pumpe allerdings längere Zeit bei Drücken im Vorvakuumbereich, so erhitzt sie sich und die Titanschichten können das gebundene Gas teilweise wieder abgeben. Dann hilft nur, die Pumpe auszu-schalten und zunächst mit einer anderen Pumpe zu arbeiten bis die Ionenget-terpumpe wieder abgekühlt ist. Ein Vorteil der Pumpe ist, dass sie völlig ohne
25 Robert Brown, schottischer Botaniker, 1773 - 1858

Kapitel 2 46
mechanisch bewegte Teile auskommt, was für die Stabilität der Mikroskopsäule sehr hilfreich ist.
Schließlich muss der Druck kontrolliert, d.h. gemessen werden. Beim Elek-tronenmikroskop werden dazu im Allgemeinen zwei verschiedene Messverfahren benutzt, die in unterschiedlichen Druckbereichen arbeiten.
Das erste Verfahren basiert auf der Wärmeleitung in Gasen. Sie wird durch die Bewegung der Gasmolekeln (Konvektion) bewerkstelligt. Die Messröhre besteht aus einem stromdurchflossenen Draht in einem Metallzylinder. Der Draht erhitzt sich beim Stromdurchfluss, wird aber gleichzeitig gekühlt durch die Konvektion, deren Intensität durch den Gasdruck bestimmt ist. Andererseits hängt der elektri-sche Widerstand des Drahtes von dessen Temperatur ab, d.h. der elektrische Wi-derstand wird durch den Gasdruck bestimmt. Die Messröhre wird mit „Pirani“26
bezeichnet und arbeitet vorzugsweise bei Drücken zwischen 1 Pa und 100 Pa. Das zweite Messverfahren („Penning“27) arbeitet nach dem Gasentladungsprin-
zip, welches bereits bei der Beschreibung der Ionengetterpumpe erläutert wurde. Bei der Druckmessröhre wird allerdings auf das Titangitter verzichtet, es ist durch einen steifen Draht ersetzt. Schließlich wollen wir messen und nicht pumpen! Es wird bei Drücken unterhalb von 1 Pa eingesetzt und ergänzt sich mit dem Pirani-Manometer.
Bild 2-22. Schema des Vakuumsystems für ein Elektronenmikroskop mit Schottky-Feldemissions-kathode.
In Bild 2-22 ist das Prinzip des Vakuumsystems für ein Elektronenmikroskop mit Schottky-Feldemissionskathode dargestellt. Im Fall der LaB6-Kathode sind die Anforderungen an das Vakuum in der Elektronenkanone geringer, so dass dort im Allgemeinen keine zusätzlichen Pumpen erforderlich sind und lediglich eine äußere Rohrverbindung mit geringem Strömungswiderstand zur Pumpe an der Mi-kroskopsäule besteht. 26 Marcello Pirani, deutscher Physiker, 1880 - 1968 27 Frans Michel Penning, niederländischer Physiker, 1894 - 1953

Was wir über den Aufbau eines Elektronenmikroskops und Elektronenoptik wissen sollten 47
2.7.6 Sonstiges
Prinzipiell ist unser Elektronenmikroskop nunmehr fertig: Wir haben ein Beleuch-tungssystem, die Probenbühne, das Abbildungssystem, eine Möglichkeit zur Re-gistrierung des Bildes und wir erzeugen das notwendige Vakuum. Allerdings fehlen noch einige wichtige Details, die hier aufgezählt werden sollen.
- Strahlführung: Trotz größter Sorgfalt beim Zusammenbau des Elektronenmikroskops ist die
mechanische Präzision für elektronenoptische Erfordernisse ungenügend. Es muss deshalb möglich sein, Elektronenstrahl und Linsen während des Betriebes zueinander zu justieren. In den ersten Geräten konnten dazu Elektronenkanone und Linsen von außen mechanisch gekippt und verschoben werden. Dies geht auf Kosten der mechanischen Stabilität und ist bei modernen Geräten nicht mehr vorgesehen. Stattdessen wird der Weg des Elektronenstrahls durch eine Vielzahl von Ablenkelementen den geometrischen Verhältnissen angepasst. Der Elektro-nenstrahl wird bis zur Beobachtungskammer in einem evakuierten Rohr mit einem Durchmesser von etwa 1 cm geführt, in das die von außen justierbaren Blenden vakuumgedichtet hineinragen.
- Blenden: Die Funktion der Kondensor-2-Blende wurde bereits beschrieben (Abschnitt
2.7.1). Wir werden später sehen, dass mindestens noch zwei weitere Blenden not-wendig sind („Objektivblende“ und „Feinbereichsblende“). Diese Blenden müssen von außen justierbar sein.
- Abschirmung der ionisierenden Strahlung: Wo immer energiereiche Elektronen auf Festkörper treffen, wird Röntgen-
strahlung ausgelöst. Diese gefährdet den Experimentator und muss deshalb am Austritt aus dem Mikroskop gehindert werden. Die Mikroskopsäule und die Beobachtungskammer sind deshalb bleiummantelt, das Einblickfenster ist aus Bleiglas.
-Objektraumkühlung: Um die Verschmutzung („Kontamination“) der Probe zu minimieren, kann der
Objektbereich mit Kupferblechen umgeben sein, die von außen mit flüssigem Stickstoff gekühlt werden. Dadurch wird der Partialdruck der Kohlenwasserstoffe und des Wasserdampfes in Objektumgebung reduziert (vgl. Abschnitt 4.5). Was-serdampf und andere „hochsiedende“ Gase frieren an den gekühlten Blechen an und werden nach Entfernen des Flüssig-Stickstoff-Behälters wieder frei. Die Get-terpumpen speichern dabei in kurzer Zeit vergleichsweise viel Gas. Dies wird durch Starten eines „Kryo-Zyklus“ vermieden: Nach Entfernen des Flüssig-Stick-stoff-Behälters werden die für den Objektraum zuständigen Getterpumpen für etwa eine Stunde ausgeschaltet. In dieser Zeit wird der Objektraum mit einer anderen Pumpe (im Vakuumsystem von Bild 2-22 mit der Turbomolekularpumpe)

Kapitel 2 48
evakuiert. Danach sind die Kühlbleche aufgewärmt und der starke Gasanfall ist zu Ende. Die Getterpumpen werden wieder eingeschaltet.
- Abschirmung von äußeren Einflüssen: Geringe Erschütterungen, wie sie beispielsweise beim Vorbeilaufen von Perso-
nen entstehen, müssen von der Mikroskopsäule ferngehalten werden. Dazu ist die Säule auf Luftpolstern gelagert. Darüber hinaus stören bei hohen Vergrößerungen auch (laute) Geräusche, Luftverwirbelungen und magnetische Wechselfelder. Ja, selbst der Experimentator kann mit seiner Wärmeentwicklung stören. Moderne Elektronenmikroskope werden deshalb häufig in speziellen schalldichten Kabinen aufgebaut, deren Wände aus Metall mit hoher Permeabilität („μ-Metall“) be-stehen. Oft ist noch die Möglichkeit der Magnetfeldkompensation eingebaut. Der Experimentator sitzt außerhalb der Kabine und steuert das Mikroskop fern.
- Strom- und Spannungsversorgung: Zum Betrieb wird eine Vielzahl von Versorgungsspannungen und –strömen
benötigt. Besondere Stabilitätsanforderungen werden an die Beschleunigungs-spannung (zwischen Kathode und Anode) und an die Linsenströme gestellt (vgl. Abschnitt 2.3). Gleiches gilt für die Versorgung der Ablenkelemente. Da die Ver-sorgungsgeräte viel Wärme entwickeln, werden sie in einem separaten Raum un-tergebracht.
- Klimaanlage und Wasserkühler mit Thermostaten: Die Wärme, die von Linsenströmen, Elektronik, Experimentator usw. erzeugt
wird, muss zuverlässig abgeführt werden. Im Probenraum soll die Temperatur-schwankung weniger als 0,1 K betragen (vgl. Abschnitt 2.7.3), was nur durch di-rekte Wasserkühlung der Linsen zu erreichen ist. Innerhalb der Rohrleitungen und Behälter für die Wasserkühlung dürfen keine Luftblasen vorhanden sein. Deren „Blubbern“ würde schon wieder unzulässige Erschütterungen hervorrufen. Der Mikroskopraum muss klimatisiert (20°C) sein, wobei die Klimaanlage verwirbe-lungsfrei arbeiten muss. Die Mikroskopsäule darf nicht direkt von Luft angeblasen werden.
- Computer: Schließlich ist ein leistungsfähiger Computer notwendig, der das Mikroskop
mit seinem Zubehör und Messprogrammen steuert, wichtige Funktionen über-wacht, die Aufzeichnung von Daten erlaubt und den Experimentator beim Mikro-skopieren unterstützt.

3 Wir präparieren elektronentransparente Proben
Ziel: Transmissionselektronenmikroskopie wird in der Regel gleichgesetzt mit moderner Gerätetechnik, Elektronenoptik auf dem neuesten Stand, Interpretation neuartiger Ergebnisse; auch über Sinn und Zweck des Ganzen wird diskutiert. Bei Neueinrichtung eines elektronenmikroskopischen Labors ist aber unbedingt zu beachten, dass für die Transmissionselektronenmikroskopie geeignete dünne Pro-ben benötigt werden und die Präparation solcher Proben alles andere als einfach und selbstverständlich ist. Dieses Kapitel gibt einen Überblick über geeignete Prä-parationsmethoden.
Die Qualität elektronenmikroskopischer Ergebnisse ist nur so gut wie die Qualität der elektronenmikroskopischen Präparation!
3.1 Wo liegt das Problem? Beim Durchgang von Elektronen durch Festkörper wechselwirken diese mit den Atomkernen, d.h. sie erfahren eine starke Coulomb28-Kraft. Diese Wechselwir-kung ist wesentlich stärker als beispielsweise diejenige von Röntgenstrahlung mit Materie. Dies hat zur Folge, dass Elektronen nur sehr dünne Folien durchdringen können. Ohne die Durchstrahlung der Probe funktioniert aber unser „Durchstrah-lungsmikroskop“ nicht. Bei den im Transmissionselektronenmikroskop üblichen Elektronenenergien von 60 keV bis 300 keV sollten die Probendicken bei etwa 100 nm liegen. Wir werden später genauer sehen, dass dies nur eine grobe Orientierung ist. In der Praxis hängt die erforderliche Probendicke von der Frage-stellung ab. Für die atomare Abbildung benötigt man beispielsweise extrem dünne Proben (Dicken um 5 nm ... 20 nm), für Messungen von Elektronenenergie-verlusten sind Dicken um 30 nm bis 50 nm optimal. Demgegenüber sollten die Proben bei Realstrukturuntersuchungen, z.B. bei der Abbildung von Versetzun-gen, dicker als 150 nm sein.
Die Problematik soll an einem einfachen Beispiel veranschaulicht werden. Stel-len wir uns doch einmal vor, wir wollen eine Keramik mit dem Elektronenmikro-skop untersuchen. Solche Keramiken können beispielsweise Hochtemperatur-Supraleiter29 sein. Porzellan ist eine geläufigere Keramiksorte. In unserem kleinen 28 Charles Augustin de Coulomb, französischer Physiker, 1736 – 1806 29 Supraleiter leiten den elektrischen Strom unterhalb einer „Sprungtemperatur” ohne elektri-schen Widerstand. Bei klassischen Supraleitern (Metallen) liegt diese Temperatur bei wenigen K. Bei Hochtemperatur-Supraleitern liegt sie deutlich höher, z.B. oberhalb von 77 K = -196 °C (Temperatur von flüssigem Stickstoff).
DOI 10.1007/978-3-7091-1440-7_3, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 3 50
Gedankenexperiment nehmen wir einen Teller aus dem Küchenschrank, schneiden eine kleine Scheibe mit 3 mm Durchmesser aus dem Tellerboden heraus und bringen diese Scheibe in der Mitte auf eine Restdicke von 50 nm. (Ein mensch-liches Haar ist übrigens etwa 50000 nm dick!) Zeitaufwändig und nicht ganz un-problematisch, oder?
Natürlich gibt es für diese Präparation erprobte Methoden, von denen wir im Folgenden einige erläutern wollen. Dabei beschränken wir uns auf solche, die uns für die Materialwissenschaft besonders wichtig erscheinen und mit denen wir selbst praktische Erfahrungen gesammelt haben. Für Präparationsmethoden von biologischen Proben und Sonderfälle verweisen wir auf die Literatur (z.B. [3.1], [3.2]). Eine Herausforderung bleibt die elektronenmikroskopische Probenpräpara-tion allerdings in (fast) jedem Fall.
Und wenn wir endlich ein geeignetes Präparat hergestellt und untersucht haben, tauchen neue Fragen auf:
- Inwieweit ist das Material durch die Präparation verändert worden?
- Wie repräsentativ ist denn eine so winzige Menge für die gesamte Probe?
Wir wollen zunächst die zweite Frage beantworten. Streng genommen müssen wir einen Stichprobenplan aufstellen, wofür in der mathematischen Statistik Vor-schriften existieren. Je größer die Zahl der Stichproben ist, desto größer ist die sta-tistische Sicherheit des Ergebnisses. Soweit die Theorie. In der elektronenmikro-skopischen Praxis steht dem häufig der Zeitaufwand bei der Präparation einer Pro-be entgegen, so dass wir die Probenzahl auf das Notwendigste beschränken müs-sen. Um die Frage zu entschärfen, stellen wir uns vor, wir hätten eine Kiste mit 100 Schrauben und nur eine davon hat ein falsches Gewinde. Die Wahrschein-lichkeit dafür, dass wir beim Griff in die Kiste ausgerechnet die falsche, untypi-sche Schraube fassen, ist 0,01, und das ist schon ziemlich unwahrscheinlich. Aber unmöglich ist es nicht.
Schwieriger ist die Beantwortung der ersten Frage. Es hängt extrem vom Mate-rial und vom gewählten Präparationsverfahren ab, inwieweit die Probe durch die Präparation verändert wird. Möglicherweise stellt die Probenpräparation bereits eine echte wissenschaftliche Herausforderung dar, insbesondere wenn bei neu-artigen Materialien keine Erfahrungswerte vorliegen. Auf solche Erfahrungswerte werden wir bei der Beschreibung der Präparationsmethoden hinweisen. In der Regel gibt es für die einzelnen Präparationsschritte verschiedene Verfahren: Ent-weder strapazieren sie das Material stark, sind aber schnell; oder sie strapazieren die Probe weniger, dauern dafür aber länger. Durch systematische Variation der Verfahren kann man feststellen, inwieweit die Präparation die Probe verändert.
Schließlich noch eine grundsätzliche Bemerkung. „Wir benötigen einige trans-missionselektronenmikroskopische Bilder von unserer Probe“, ist ein häufig ge-äußerter Wunsch von Auftraggebern für das elektronenmikroskopische Labor. Elektronenmikroskopiker bevorzugen eine konkrete Fragestellung, z.B.: Wie groß sind die Partikel? Wie dick sind die Schichten? Welche Morphologie und welche

Wir präparieren elektronentransparente Proben
51
Phasen liegen vor? In der Regel ist es zweckmäßig, bereits bei der Präparation diese Fragestellung zu beachten oder anders gesagt: Wenn transmissionselektro-nenmikroskopische Untersuchungen an einem Material geplant sind, so sollte die notwendige elektronenmikroskopische Präparation frühzeitig in die Überlegungen einbezogen werden. Oft kann die Probengeometrie den Präparationserfordernissen angepasst werden. Oder eine Schicht kann auf einem für die spätere Präparation geeigneteren Substrat abgeschieden werden usw.
3.2 „Klassische“ Methoden Das Problem der dünnen Proben begleitet die Elektronenmikroskopie seit ihren Anfängen. Es verwundert nicht, dass die ersten wissenschaftlich relevanten Ergeb-nisse an biologischen Proben erhalten wurden. Mikrotom-Schnitte waren in den 1930-er Jahren bekannt; die Hauptbestandteile Wasserstoff, Stickstoff und Sauer-stoff in biologischen Proben lenken Elektronen wegen ihrer niedrigen Kern-ladungszahl nur wenig ab, deshalb dürfen diese Proben vergleichsweise dick sein. Helmut Ruska30 hat 1939 erste Ergebnisse veröffentlicht und darin Zaponlack als Trägerfilm für elektronenmikroskopische Präparate sowie die „Metallimprägna-tion“ und ein anderes gezieltes Beeinflussen einzelner Zellbestandteile zur Ver-änderung von Dichte und/oder Dicke und damit zur Kontrastverstärkung vorge-schlagen [3.3]. Eine wichtige Rolle spielte zu dieser Zeit die Sichtbarmachung von Viren im Elektronenmikroskop, die bis dahin wegen des begrenzten Auflösungs-vermögens des Lichtmikroskops als „ultravisibel“ galten.
Damit sind wir bei der ersten „klassischen“ Methode für die elektronenmikro-skopischen Probenpräparation angelangt, dem
- Aufbringen kleiner Teilchen auf Trägerfilme
Diese Methode ist geeignet, um Form- und Größenverteilungen von Partikeln im Submikrometerbereich zu messen. Als Trägerfilme werden Kohle- oder Kunst-stofffilme benutzt, die auf einem Stützgitter aus Kupfer oder einem anderen Mate-rial aufgebracht sind (vgl. Bild 3-1).
Bild 3-1. Trägernetz für die Transmissionselektronen-mikroskopie (Dicke: 10 μm ... 30 μm, Maschenweite: 20 μm ... 400 μm, Drahtdicke: 10 μm ... 15 μm, Material: Kupfer, Molybdän, Edelstahl, Nickel, Gold u.a.).
30 Helmut Ruska, deutscher Mediziner, 1908 – 1973, Bruder von Ernst Ruska

Kapitel 3 52
Anstelle des Netzes sind auch andere Öffnungen üblich: kreisrunde Löcher, Schlitze und Kombinationen von Schlitzen.
Als Träger sind auch Siliziumnitrid-Fenster verfügbar, bei denen in ein 200 μm bis 300 μm dickes Si3N4-Scheibchen (Außendurchmesser 3 mm) mehrere dünne Fenster mit einer Größe von etwa 100 μm x 100 μm hineingeätzt worden sind. Die Restdicke in den Fenstern liegt zwischen 30 nm und 100 nm.
Die Art des Aufbringens der Teilchen hängt von deren Konsistenz ab. Liegen sie als Rauch vor, so wird ein befilmtes Trägernetz am Rand mit einer Pinzette ge-fasst und kurzzeitig in den Rauch gehalten. Mit Pulvern kann eine wässrige oder alkoholische Suspension hergestellt werden, von der ein kleiner Tropfen mit einem dünnen Glasstab auf das befilmte Trägernetz gegeben wird, der dann in staubfreier Umgebung eintrocknet. Beim Betropfen wird das Netz mit einer Pinzette niedergehalten. Es gehört etwas Übung dazu, denn der Tropfen soll nicht auf der Pinzette landen und mit ihr weggezogen werden. Der Anfänger fertigt oft Proben mit viel zu großer Teilchendichte an. Es genügt eine kleine Pinzettenspitze Pulver auf ein Reagenzglas mit Flüssigkeit, also so wenig, dass man glauben möchte, es käme kaum etwas auf das Netz. Wenn das Pulver zur Zusammenbal-lung neigt, empfiehlt sich vor dem Auftropfen eine Behandlung der Suspension in einem Ultraschallbad.
Sind die Teilchen elektronentransparent, so sind an derartig hergestellten Prä-paraten auch weitere TEM-Untersuchungen möglich: Phasenbestimmung mittels Elektronenbeugung, Hochauflösungsabbildung und Elementanalysen. Größere Teilchen können unter Umständen im Mörser zerrieben werden, um die Elektro-nentransparenz zu erreichen.
- Ankleben an Trägernetze
Eine Präparationsmethode, die sich vor allem bei Filzen aus nanoskaligen Fasern (z.B. Kohlenstoff-Nanoröhren) bewährt hat, ist das Ankleben kleiner Filzstück-chen an das Trägernetz. Dazu wird das Trägernetz mit wenig Klebstoff bestrichen, indem ein Klebestift vorsichtig über das mit einer Pinzette gehaltene Netz geführt wird. Anschließend wird mit diesem Netz über den Filz gestrichen. Auch hier gilt: Je weniger desto besser. Der Vorteil dieser Methode ist, dass die Fasern frei liegen und der Trägerfilm nicht den Bildkontrast beeinträchtigt. Der Nachteil ist, dass sich einzelne Fasern auch leicht bewegen, so dass die Gefahr der Bewegungsun-schärfe während der elektronenmikroskopischen Aufnahme besteht. Oft muss ein Kompromiss gefunden werden, indem Faserbereiche aufgesucht werden, die beid-seitig von dichterem Filz gestützt sind.
- Aufbringen dünner Filme auf Trägernetze
Liegt die Probe in Form von freitragenden dünnen Filmen vor, so ist es möglich diese Filme direkt auf ein Trägernetz zu bringen. In der Regel schwimmen die Filme in kleinen (ca. 3 mm x 3 mm) Stücken auf destilliertem Wasser. Das Trä-gernetz wird mit einer Pinzette gefasst, neben einem Filmflitterchen unter den

Wir präparieren elektronentransparente Proben
53
Wasserspiegel getaucht, unter das Flitterchen geführt und dann mit dem Flitter-chen herausgehoben. Auch hier ist einige Übung erforderlich, um die richtige Geschwindigkeit beim Herausheben zu finden. Das Flitterchen soll auf dem Netz bleiben und nicht heruntergespült werden. Das anschließende Trocknen sollte auf sauberem und wenig saugfähigem Papier erfolgen, zu starke Saugfähigkeit könnte den Film vom Netz herunterziehen.
Die Frage ist nun, wie wir zu den freitragenden Filmen kommen. Eine Mög-lichkeit ist das Deponieren einer dünnen (einige 10 nm dick) Schicht auf einem Kochsalz-Einkristall. Die Schicht wird mit einem Skalpell oder einer Rasierklinge in kleine Quadrate (ca. 3 mm x 3 mm) eingeritzt, der Kristall mit einer Pinzette gefasst und mit der Schicht nach oben vorsichtig in destilliertes Wasser einge-schoben. Das Wasser dringt zwischen Schicht und Kochsalz ein und löst die Schicht vom Kristall ab. Die Filme werden dann aufgefischt (s. oben). Bei kri-stallinen Proben können auf diese Weise die Kornmorphologie, der Phasengehalt, evtl. Ver- oder Entmischungen und die Eigenschaften von Korngrenzen analysiert werden. Bei amorphen Proben können Nahordnungen ermittelt werden. Werden hitzebeständige Trägernetze (z. B. solche aus Molybdän) benutzt, ist es bei Ver-wendung geeigneter Probenhalter im Mikroskop möglich, die Filme in situ zu heizen und so Korngrößenwachstum und Phasenumwandlungen zu verfolgen.
Es ist auf diese Weise auch möglich, Wachstumsprozesse auf dem NaCl-Kristall direkt zu untersuchen [3.4]. Dazu werden evtl. Wachstumsstufen (d.h. an der Oberfläche auftretende Kristallbaufehler) dekoriert, indem die (geheizte) Kri-stalloberfläche so dünn mit Gold bedampft wird, dass keine zusammenhängende Goldschicht entsteht. Die Goldatome bewegen sich bevorzugt zu Stufen oder an-deren energetisch bevorzugten Stellen auf der Kristalloberfläche (Dekoration). Anschließend wird eine dünne (10 nm ... 20 nm) Kohlenstoffschicht aufgedampft, die Gold-Kohlenstoff-Schicht in destilliertem Wasser abgelöst und s.o.
Ähnlich funktioniert die Herstellung von Extraktionsreplica. Dabei geht es um die Untersuchung von Ausscheidungen an der Oberfläche von Festkörpern. Um die Ausscheidungen freizulegen, muss die Oberfläche evtl. angeätzt werden. Dann wird eine dünne Schicht Kohlenstoff aufgedampft, die mit einem Lack verstärkt und anschließend vorsichtig abgezogen wird. Der Lack muss nun wieder entfernt werden. Alternativ kann anstelle der Lackverstärkung eine Chemikalie benutzt werden, die die Oberfläche, nicht aber die zu untersuchenden Ausscheidungen an-löst. Dadurch kann der Film wie bei NaCl mit Wasser abgelöst werden. Wir erhal-ten einen Kohlenstofffilm, in dem die Ausscheidungen eingebettet sind und hin-sichtlich Morphologie, Phase und chemischer Zusammensetzung analysiert wer-den können. Insbesondere die Auswahl geeigneter Ätzmittel und -bedingungen ist wesentlich für den Erfolg der Präparation und erfordert erheblichen Aufwand.
Bis in die 1970-er Jahre war die Herstellung von Oberflächenabdrücken eine weit verbreitete Präparationsmethode. Ziel ist die Analyse von Oberflächentopo-grafien im Submikrometerbereich. Dazu wird auf die zu untersuchende Oberfläche eine Matrize aufgedrückt oder aufgedampft. Beispielsweise kann dazu ein ca. 0,5 mm dickes und ca. 1 cm2 großes Stück Plexiglas benutzt werden. Das Plexi-

Kapitel 3 54
glas wird mit einem Tropfen Chloroform angelöst und aufgedrückt. Es nimmt die Topografie der Oberfläche an und härtet nach dem Verdunsten des Chloroforms aus. Nach dem Ablösen der Matrize wird deren Oberfläche zunächst schräg mit einer dünnen (ca. 10 nm) Schwermetallschicht (z.B. Chrom) und anschließend aus unterschiedlichen Richtungen mit ca. 20 nm Kohlenstoff bedampft (vgl. Bild 3-2).
Bild 3-2. Herstellung eines Oberflächenabdruckes mit Plexiglasmatrize.
Infolge der Schrägbedampfung spiegelt sich die Topografie in der „Licht-Schat-ten-Verteilung“ der Chromschicht wider. Wegen der guten Beweglichkeit der Kohlenstoffatome während des Bedampfens umhüllt die Kohlenstoffschicht die Cr-Partikel als zusammenhängende dünne Schicht. Die Plexiglasmatrize wird in kleine Quadrate (ca. 3 mm x 3 mm) zerteilt, die auf Trägernetze aufgelegt werden. Nun muss noch das Plexiglas langsam in einer Chloroformatmosphäre (selbstver-ständlich unter einem Abzug!) weggelöst werden.
Mit der Verbreitung der Rasterelektronenmikroskope ist die Bedeutung der Oberflächenabdruckmethode stark zurückgegangen; die Oberflächentopografie kann mit dem Rasterelektronenmikroskop ebenfalls im Submikrometerbereich er-fasst werden und der Präparationsaufwand ist weitaus geringer.
- Elektrolytisches Dünnen
Für elektronenmikroskopische Untersuchungen zur Realstruktur (Gitterbaufehler) von Metallen ist seit den 1950-er Jahren das elektrolytische Polieren etabliert. Es wird eingesetzt bei elektrisch leitenden, homogenen Proben. Auf die als Anode geschaltete Probe (Scheibchen mit 3 mm Durchmesser, ca. 0,5 mm dick) wird aus einer als Kathode geschalteter Düse ein Elektrolytstrahl gerichtet (s. Bild 3-3).
Bild 3-3. Prinzip des elektrolytischen Dünnens.

Wir präparieren elektronentransparente Proben
55
Gedünnt wird bis in der Mitte der Probe ein kleines Loch entsteht. Am Lochrand steigt die Probendicke keilförmig an, so dass in diesem Bereich für die elektronen-mikroskopische Untersuchung geeignete Dicken gefunden werden können. Der Keil hat den Vorteil, dass die für die Lösung unterschiedlicher Fragestellungen optimale Dicke zur Verfügung steht.
Die Probleme beim elektrolytischen Dünnen bestehen in der Auswahl des geeigneten Elektrolyten und seiner Temperatur. Die Strom-Spannungs-Kennlinie muss gemessen werden, um die Kenndaten für den Polierbereich zu bestimmen. Andernfalls wird geätzt und es entstehen raue Oberflächen, d. h. stark fluktuieren-de Dicken. Vor Präparationsbeginn ist ein Literaturstudium besonders wichtig, um evtl. Erfahrungen anderer Autoren mit dem Material nutzen zu können.
- Ultramikrotom
Bevorzugt an organischen Proben sind auch Dünnschnitte wie in der Licht-mikroskopie möglich [3.5]. Sie dürfen allerdings nur etwa 0,1 μm dick sein, wes-halb von Ultramikrotom-Schnitten gesprochen wird. In der Regel wird die Probe vor dem Schneiden in Epoxidharz eingebettet. Vereinzelt werden solche Schnitte auch an hartem, anorganischem Material ausgeführt. Das Besondere an den Dia-mantmessern ist der vergleichsweise große Schneidwinkel von teilweise mehr als 35°. Das Schneiden erinnert an das Hobeln in der Tischlerei, nur ist alles sehr viel kleiner. Die Schnitte sind nur einige 10 Mikrometer groß und werden aus einem Wasserbad auf befilmte Trägernetze aufgefischt.
- Mechanisches Spalten („Cleavage“)
In Sonderfällen (z.B. Silizium) ist es möglich, dünne Scheiben entlang von Kristallebenen zu brechen und dadurch kleine Keile mit Keilwinkeln von etwa 20° bei atomar glatten Bruchflächen zu erhalten [3.6]. Derartige Keile sind an der Spitze für die transmissionselektronenmikroskopische Untersuchung hinreichend dünn.
3.3 Schneiden, Schleifen und Ionendünnen Die zurzeit gebräuchlichste Methode der elektronenmikroskopischen Präparation in der Werkstoffwissenschaft kann mit Schneiden, Schleifen und Ionendünnen beschrieben werden. Diese Methode ist für die meisten Fragestellungen geeignet und ermöglicht auch die Präparation inhomogener Proben, wie z.B. von Schicht-systemen. Das Ziel ist eine Probengeometrie, wie sie bereits beim elektrolytischen Dünnen erreicht worden ist: Eine Scheibe mit 3 mm Durchmesser, deren Dicke vom Rand zur Mitte hin abnimmt und die in der Mitte ein kleines Loch (weniger als 1 μm im Durchmesser) hat (vgl. Bild 3-4).

Kapitel 3 56
Bild 3-4. Geometrie für abgedünnte TEM-Proben. Der Durchmesser von 3,0 mm muss auf ± 0,1 mm genau eingehalten werden. An-derenfalls besteht die Gefahr, dass die Probe nicht in den Halter passt.
Es gibt zwei verschiedene Möglichkeiten der Präparation. Besteht das Ziel in der Untersuchung eines kompakten Materials oder einer dünnen Oberflächenschicht, so wird die Probe in einer „Draufsicht“ (englisch: plan-view) präpariert, d.h. das dünne Präparat wird parallel zur Probenoberfläche herausgearbeitet. Dies ermög-licht die Analyse der Kornmorphologie, die Phasenbestimmung, die Bestimmung der chemischen Zusammensetzung und der chemischen Bindung; bei amorphen Proben auch die Bestimmung von Nahordnungszuständen. Die andere Mög-lichkeit ist die Querschnittspräparation (englisch: cross-section), bei der das Prä-parat senkrecht zur Probenoberfläche gerichtet ist. Dies ist besonders wün-schenswert für die Analyse von Schichtsystemen (Einzelschichtdicken, Grenz-schichten, Gradienten in der Kornmorphologie, im Phasengehalt, in der chemi-schen Zusammensetzung usw.).
- „Draufsicht“-Proben
Als erstes wird ein Scheibchen mit einem Durchmesser von 3 mm benötigt. Für dessen Dicke sind 0,3 mm bis 0,5 mm optimal. Auch quadratische Stücke mit einer Diagonale von 3 mm sind zulässig. Je nach Material kann dieses Scheibchen durch Stanzen, Kernlochbohren, Brechen oder Sägen herausgearbeitet werden. Duktiles Material kann gestanzt werden, Plättchen aus Silizium u. Ä. werden mit einem Ultraschall-Kernlochbohrer bearbeitet. Das Werkzeug ist dabei ein dünnes Stahlrohr mit 3 mm Innendurchmesser und einer gehärteten Schneide, welches mit hoher (Ultraschall-) Frequenz auf die Probe gestoßen wird (s. Bild 3-5).
Bild 3-5. Prinzip des Kernlochbohrens.
Schneide und Probenoberfläche werden mit einer Schleifmittelsuspension benetzt. Das Ergebnis ist ein kreisrundes Scheibchen mit 3 mm Durchmesser. Wir sehen, dass eine plattenförmige Probengeometrie als Ausgangszustand vorteilhaft ist.

Wir präparieren elektronentransparente Proben
57
Sollte dies nicht der Fall sein, muss im ersten Schritt mit einer Diamantsäge oder Fadensäge ein Plättchen aus dem Probenblock herausgesägt werden. Die Diamant-säge ist eine kleine Kreissäge, deren Sägeblatt mit Diamantsplittern belegt ist. Die Fadensäge erinnert an eine Bügelsäge, bei der ein dünner, mit Schleifmittel-suspension benetzter Wolframdraht (ca. 0,5 mm dick) an der Schnittstelle hin und her bewegt wird und das Material langsam durchschleift. Ähnlich wie das Farb-band bei einer mechanischen Schreibmaschine wird der Wolframdraht ständig von einer Rolle nachgeführt. Beim Sägen sind normalerweise zwei Schnitte notwen-dig: Zuerst wird eine Bezugsfläche gesägt, dann mit einer (eingebauten) Mikro-meterschraube definiert (z.B. 0,5 mm) nachgestellt und der zweite Schnitt ausge-führt. Natürlich muss dabei die Sägeblatt- bzw. Drahtdicke berücksichtigt werden. Unterschiede zwischen den beiden Sägemethoden bestehen einmal im Grad der Strapazierung des Materials und zum anderen in der notwendigen Zeit. Das Diamantsägen ist schneller, strapaziert aber mehr als das Fadensägen. Überhaupt gilt für viele Präparationsmethoden: Je schneller die Prozedur, desto strapaziöser ist sie für die Probe, d.h. desto eher sind Veränderungen durch die Präparation zu erwarten!
Sollte das Scheibchen zu dick sein, so wird es mit den aus der Metallografie be-kannten Schleifmaschinen und zunehmend feineren Schleifpapieren und –pasten dünner geschliffen.
Der nächste Schritt ist das Konkavschleifen (Dimpeln). Darunter wird das Hi-neinschleifen einer Kugelkalotte in das dünne Probenscheibchen verstanden, so dass die in Bild 3-4 gezeigte Probengeometrie entsteht. Auch hierbei existieren zwei verschiedene Möglichkeiten. Das erste Gerät müssen wir uns wie eine Art Plattenspieler vorstellen, bei dem der Plattenteller aus Stahl am Rand eine Rille hat, in der eine Stahlkugel läuft. Diese Stahlkugel ist mit Schleifmittel (z.B. Dia-mantpaste unterschiedlicher Körnung) benetzt und reibt gegen die schräg gestellte Probe (s. Bild 3-6).
Bild 3-6. Prinzip des Dimpelns mit Kugel.
Die zweite Variante arbeitet mit einem Schleifrad und sich drehender Probe (vgl. Bild 3-7). Auch hier entsteht eine kalottenförmige Vertiefung. Der Vorteil gegen-über der Dimpelkugel ist, dass die Andrückkraft des Schleifrades als zusätzlicher Parameter zur Körnung dosiert werden kann, was das Dimpeln unter Umständen beschleunigt. Allerdings gilt auch hier das Gleiche wie beim Sägen: Je schneller desto strapaziöser.

Kapitel 3 58
Bild 3-7. Prinzip des Dimpelns mit Schleifrad.
Anstelle des Dimpelns kann die Probe mit einer speziellen Vorrichtung („Tripod“ [3.7]) zum Einstellen flacher (wenige Grad) Winkel auch keilförmig geschliffen werden.
Bedauerlicherweise wird die Probenoberfläche beim mechanischen Schleifen trotz feinster Körnung bis in eine Tiefe von bis zu einigen Mikrometern verändert: Sie wird aufgeraut, Kristallgefüge werden zerstört. Diese gestörte Schicht muss beseitigt werden. Deshalb wird nicht bis zum Durchbruch der Probe im Zentrum der Kalotte gedimpelt, sondern es bleibt ein Rest von ca. 10 μm bis 20 μm stehen. Dieser Rest wird durch Sputtern mit Argon-Ionen entfernt („Ionendünnen“ - vgl. Bild 3-8). Dazu benutzt man eine Argon-Ionenquelle mit Ionenenergien zwischen einigen 100 eV und 5 keV.
Bild 3-8. Letzter Schritt der Präparation: Ionendünnen, d.h. Sputtern mit Ar+-Ionen (Ionenenergie: 200 eV ... 5 keV, Beschusswinkel gegen Probenteller: 5° ... 15°).
Zu beachten ist, dass sich das gesputterte Material in der Probenumgebung nieder-schlägt („Redeposit“) und dass beim Sputtern auch die Probenbühne erfasst wer-den kann. Sie sollte deshalb aus einem schwer sputterbaren Material (Tantal, Gra-phit) bestehen oder damit bedeckt sein. Bei inhomogenen Proben können Proben-teile bevorzugt gesputtert werden und am Ende im elektronentransparenten Be-reich fehlen. Dem wird durch die Probenrotation entgegengewirkt. Da die Sputter-rate auch vom Einfallswinkel der Ionen abhängt (senkrechter Einfall – geringe Sputterrate), werden Rauheiten der Oberfläche bei gleichbleibendem Ioneneinfall verstärkt. Auch diesem Effekt wirkt die Probenrotation entgegen. Zur Glättung der Oberfläche und zur Beseitigung von Verunreinigungen sollte stets auf beiden

Wir präparieren elektronentransparente Proben
59
Seiten ionengedünnt werden. Bei der Untersuchung auf Substrat befindlicher dün-ner Schichten wird von der Substratseite aus gedimpelt und gesputtert, die Schichtseite wird zum Abschluss nur „zart“, d.h. mit Ionen geringer Energie gereinigt.
- Querschnittsproben
Stellen wir uns nun vor, wir wollen auf den Querschnitt beispielsweise eines Schichtstapels schauen. Dazu muss der Stapel aufgesägt werden wie in Bild 3-9 gezeigt, so dass wir eine neue Draufsicht erhalten. Der Rest geht vom Prinzip her wie beschrieben. In der Praxis werden solche Querschnitte von Schichtstapeln et-was anders hergestellt. Wir müssen nämlich dafür sorgen, dass interessierende De-tails, wie beispielsweise Übergänge (Grenzschichten) zwischen einzelnen Schich-ten auch tatsächlich elektronentransparent werden.
Bild 3-9. Zum Verständnis der Querschnittspräparation: Durch Aufsägen eines Schichtstapels wird eine neue Draufsicht geschaffen.
Nehmen wir an, wir hätten eine dünne (z.B. 50 nm dicke) Schicht auf einem Sub-strat mit einer Dicke von 0,5 mm. Davon werden zwei etwa 1 mm breite Streifen abgetrennt (Anritzen und Brechen oder Sägen), die dann Schicht an Schicht (eng-lisch: face-to-face) mit Epoxidharz zusammengeklebt werden (Bild 3-10). Dabei sind dünnflüssiges Epoxidharz und eine Vorrichtung zum Zusammenpressen der beiden Streifen empfehlenswert, um den Klebespalt möglichst schmal zu halten.
Bild 3-10. Querschnittspräparation. a) Abtrennen von zwei Streifen. b) Zusammenkleben Schicht an Schicht. c) Einkleben in ein Keramikröhrchen (Außendurchmesser 3 mm, Wandstärke ca. 0,5 mm).
Das Schichtpaket wird in ein dünnes Keramikröhrchen (in der Regel Al2O3) ein-geklebt. Nach dem Aushärten des Epoxidharzes wird mit zwei Schnitten (glatte Bezugsseite und eigentliches Abschneiden) ein 0,3 mm ... 0,5 mm dickes Scheib-chen abgesägt, das dann mit planparallelem Schleifen, Dimpeln und Ionendünnen wie bei der Draufsicht-Probe weiterbehandelt wird. Es entsteht ein TEM-Präparat, bei dem der Übergang zwischen Substrat und Schicht an vier Stellen betrachtet werden kann (vgl. Bild 3-11).

Kapitel 3 60
Bild 3-11. Ergebnis der Schicht-an-Schicht (face-to-face) Querschnittspräparation. a) Schematische Darstellung. b) TEM-Bild.
3.4 Focused Ion Beam („FIB“) Techniken Um Focused-Ion-Beam (d.i. Fokussierter Ionenstrahl)-Techniken nutzen zu kön-nen, ist erst einmal eine größere Investition erforderlich: Wir brauchen ein neues Gerät, ein „Rasterionenmikroskop“. Dieses funktioniert wie ein Rasterelektro-nenmikroskop, arbeitet allerdings mit einer Ionensonde anstelle der Elektronen-sonde. Eine Ionenoptik erzeugt die sehr feine Ionensonde mit einem Durchmesser von wenigen Nanometern, mit der die Probenoberfläche Punkt für Punkt abgera-stert wird und die dabei ausgelösten Sekundärelektronen zur Helligkeitssteuerung der entsprechenden Bildpunkte auf dem Monitor genutzt werden. Das Bild wird demzufolge seriell, d.h. Pixel für Pixel, zusammengesetzt. Es handelt sich dabei nicht um eine optische Abbildung wie im Transmissionselektronenmikroskop.
Es ist vorteilhaft, ein Gerät mit zwei Säulen zu verwenden. Die erste erzeugt einen feinfokussierten Elektronenstrahl zur rasterelektronenmikroskopischen Ab-bildung, die zweite einen feinfokussierten Gallium-Ionenstrahl. Sie bildet das o.g. Rasterionenmikroskop. Gallium ist bereits bei 30°C flüssig, es ist deshalb ver-gleichsweise einfach, eine Gallium-Ionenquelle zu bauen. Mit diesem fokussierten Ionenstrahl ist es möglich, die Probe wie mit einem Schaftfräser zu bearbeiten, nur dass der Fräser einen Durchmesser von lediglich einigen Millionstel Millimetern hat! Und das Ganze erfolgt unter rasterelektronenmikroskopischer Kontrolle, dazu dient die erste Säule. Die Benutzung von Elektronen für die Abbildung vermeidet, dass die Probe bereits bei der Beobachtung „gefräst“ wird.
Um das Fräsen in vertretbarer Zeit (1 – 2 Stunden für eine Lamelle) erledigen zu können, wird standardmäßig mit einer vergleichsweise hohen Ionenenergie von 30 keV gearbeitet. Dies ist verbunden mit zwei nachteiligen Effekten: Einer Amorphisierung der Oberfläche bei kristallinen Proben und einer Implantation von Gallium-Ionen in der Probe. Besonders kritisch ist dies bei senkrechtem Ionenaufprall. Um die Oberfläche der herauszuarbeitenden Lamelle davor zu schützen, wird vor Beginn des Fräsens ein schmaler Schutzriegel auf die Oberfläche aufgebracht (vgl. Bild 3-12).

Wir präparieren elektronentransparente Proben
61
Bild 3-12. Schutzstreifen auf der Oberfläche der späteren FIB-Lamelle (rasterelektro-nenmikroskopisches Bild).
Dazu wird mittels eines lokalen Gasinjektionssystems bei geringem Druck ein schwermetallhaltiges (Platin, Wolfram) Kohlenwasserstoffgas auf die Probenober-fläche geleitet und der zu schützende Bereich mit dem Elektronen- oder Ionen-strahl abgerastert. Durch die damit verbundene Energiezufuhr vernetzen die Kohlenwasserstoffe und bilden an der Auftreffstelle von Elektronen- oder Ionen-strahl eine feste, schwermetallhaltige Schutzschicht (vgl. Abschnitt 4.5).
Wir wollen nun sehen, wie uns das „Nanofräswerkzeug“ bei der Präparation von dünnen Querschnittslamellen für die Transmissionselektronenmikroskopie helfen kann. Drei Varianten dazu sollen beschrieben werden.
- H-Balken-Methode
Die H-Balken-Methode beginnt wie die „klassische“ Querschnittspräparation: Es muss ein schmaler Streifen abgetrennt werden, der dann allerdings auf einen Kupfer-Halbring geklebt wird (s. Bild 3-13). Im Unterschied zur klassischen Prä-paration muss dieser Streifen sehr schmal sein, möglichst nicht breiter als 100 μm.
Bild 3-13. Veranschaulichung der FIB-H-Balken-Methode. a) Aufgeklebter Probenstreifen. b) Geometrie der „herausgefrästen“ Lamelle.

Kapitel 3 62
Der Vorteil dieser Methode ist die stabile Befestigung der TEM-Probe. Es ist möglich, bei Bedarf die Lamelle „nachzuschneiden“, das heißt, wiederholt im FIB-Gerät nachzudünnen. Nachteilig ist das kleine Lamellenfenster, verbunden mit hohen Kanten am Fensterrand. Besonders bei Untersuchungen der Kristall-struktur (Beugung) kann dieser Rand den Kippwinkel der Probe zum Elektronen-strahl begrenzen. Kritisch kann auch das vergleichsweise große Materialvolumen in Fensternähe sein. Speziell bei der Röntgenspektrometrie (EDXS - vgl. Ab-schnitt 9.2) führt dies zu Artefakten. Wir werden später auf solche Probleme zu-rückkommen, wenn wir die elektronenmikroskopische Praxis beschreiben.
- Herausschneiden und Ablegen der Lamelle auf Trägerfilm
Um die o.g. Nachteile zu vermeiden, wird die Lamelle komplett aus der Probe he-rausgeschnitten. Dabei erübrigt es sich, vorher einen schmalen Streifen herzustel-len. Wir nehmen die Probe und bringen wie oben einen Schutzriegel auf die Ober-fläche. Die weitere Verfahrensweise ist aus dem Schema in Bild 3-14 ersichtlich.
Bild 3-14. Herausschneiden einer FIB-Lamelle und Ablegen auf einem Trägernetz a) Aufbrin-gen des Schutzriegels. b) Freischneiden der Lamelle. c) Heraustrennen der Lamelle d) Heraus-heben der Lamelle mit einer Glasnadel. e) Ablegen der Lamelle auf einem befilmten Trägernetz (Film mit Löchern). f) Transmissionselektronenmikroskopisches Bild einer Lamelle auf Träger-netz.
Nach dem Ablegen haftet die Lamelle durch Adhäsionskräfte fest auf dem Träger. Haftprobleme treten mitunter auf, wenn die Lamelle unter mechanischer Span-nung steht und sich deshalb durchbiegt. Im Bild 3-14f sehen wir, dass die Lamelle nicht gleichmäßig dick ist sondern zwei dickere „Balken“ enthält. Dazu müssen wir uns noch einmal vergegenwärtigen, dass unsere Lamelle weniger als 0,1 μm dick ist (d.h. etwa 500-mal dünner als ein menschliches Haar!). Obwohl sie auch nur etwa 10 μm x 10 μm groß ist, neigt sie zur Durchbiegung, d.h. sie muss mechanisch stabilisiert werden. Dazu sind die Balken da. Weiterhin erkennen wir, dass der Trägerfilm eher einem Spinnennetz gleicht als einem zusammen-hängenden Film. Auch das ist Absicht, erhöht doch der Trägerfilm die Proben-

Wir präparieren elektronentransparente Proben
63
dicke. Wir werden unsere elektronenmikroskopische Untersuchung also möglichst in einem Lamellenbereich ausführen, der über einem Loch im Trägerfilm liegt. Dazu gehört ein bisschen Glück, wie manchmal beim Experimentieren. Nicht nur auf das Glück kann man sich beim Herausheben der Lamelle verlassen. Es erfolgt unter lichtmikroskopischer Kontrolle mit Hilfe einer Glasnadel, die an einem Mikromanipulator befestigt ist. Erinnern wir uns an das Auflösungsvermögen des Lichtmikroskops. Es beträgt etwa 0,2 μm. Ohne Erfahrung wird man die Lamelle wahrscheinlich gar nicht im Lichtmikroskop erkennen!
Leider hat auch diese Methode einen gravierenden Nachteil: Wenn sich bei der TEM-Untersuchung herausstellt, dass die Lamelle zu dick ist, muss die gesamte Prozedur wiederholt werden. Ein Nachschneiden ist nicht möglich.
- Herausschneiden und Anschweißen der Lamelle an einem Träger
Der Kritikpunkt ist das Ablegen der Lamelle auf dem Trägernetz. Die Lamelle selbst ist dann nicht mehr einzeln handhabbar. Um dies zu vermeiden, wird die Lamelle unter rasterelektronenmikroskopischer Kontrolle an einem Träger ange-schweißt. Sie glauben nicht, dass das funktioniert? Es funktioniert!
Bild 3-15. Anschweißen einer FIB-Lamelle. a) Schema. b) – d) Rasterelektronenmikroskopi-sche Bilder: b) Freigeschnittene Lamelle. c) Träger mit angeschweißter Lamelle. d) Ange-schweißte Lamelle nach dem finalen Dünnen.
Bis zum Herausheben ist die Prozedur die Gleiche wie in Bild 3-14 beschrieben. Allerdings bleibt die Lamelle etwa 1 μm dick. Der Mikromanipulator befindet sich nunmehr in der FIB-Kammer und ist mit einer kleinen Metallspitze ausge-

Kapitel 3 64
rüstet. Die Lamelle wird vorsichtig herausgebrochen und dann zu einem speziellen Träger geführt, angehalten und angeschweißt. Dabei ist das Anschweißen eher ein Ankleben. Benutzt wird der gleiche Mechanismus wie beim Aufbringen des Schutzriegels: Das schwermetallhaltige Kohlenwasserstoffgas wird mit dem Gas-injektionssystem in die Nähe der Schweißstelle geleitet, die dann wiederholt mit dem Elektronenstrahl abgerastert wird. Durch die Kohlenwasserstoffvernetzung entsteht eine feste Verbindung zwischen Lamelle und Träger. In diesem Zustand erfolgt das finale Dünnen der Lamelle mit dem Ionenstrahl.
Auch dabei werden zu deren mechanischer Stabilisierung einige Balken dicker belassen (vgl. Bild 3-15).
- Verbesserung der Qualität der FIB-Lamellen
Für viele elektronenmikroskopische Fragestellungen reicht die Qualität der Lamel-le vollkommen aus: Für die Analyse von Korngrößen und Phasen, für Realstruk-turuntersuchungen, auch für viele analytische Experimente, d.h. Bestimmungen der chemischen Zusammensetzung mit hoher lateraler Auflösung. In anderen Fäl-len stören die amorphen Schichten, oder die Lamellen sind einfach noch zu dick: für Aufnahmen mit atomarer Auflösung oder für Elektronenenergieverlust-Analy-sen der Kantenfeinstruktur (s. Abschnitt 9.3.6).
In diesen Fällen sollte beim finalen Dünnen die Energie der Gallium-Ionen re-duziert und die Lamelle um wenige Grad aus ihrer senkrechten Position verkippt werden. Im Allgemeinen kann bei den FIB-Geräten mit Ionenenergien bis he-runter zu 5 keV gearbeitet werden. Bei weiterer Verringerung beeinflussen häufig Aufladungen und die höhere Störanfälligkeit der langsameren Ionen gegenüber äußeren Einflüssen die Qualität der Ionensonde. Das führt dazu, dass der Sonden-querschnitt so groß und instabil wird, dass ein gezieltes Abdünnen nicht mehr möglich ist.
Die Alternative bei den angeschweißten Lamellen ist ein Nachdünnen in einer anderen Anlage, z.B. mit Argon-Ionen einer Energie zwischen 200 eV und 1 keV. Dabei ist zu beachten, dass der Träger nicht von Ionen getroffen wird, d.h. diese Anlage muss die Fokussierung des Argon-Ionenstrahls auf einen Fleck von etwa 1 μm und eine Kontrolle des Lamellenortes gestatten. Anderenfalls wird die Probe durch Redeposit vom massiven Träger verschmutzt.
Wir sehen, dass das Präparieren untrennbar mit der Transmissionselektronen-
mikroskopie verbunden ist und dazu auch ein erheblicher apparativer Aufwand betrieben werden muss. Unersetzbar ist der Erfahrungsschatz der Mitarbeiter, die diese Präparationsarbeiten ausführen.

4 Wir beginnen mit der praktischen Arbeit
Ziel: In den Kapiteln 1 bis 3 haben wir in groben Zügen beschrieben, was sich elektronenoptisch im Mikroskop abspielt, wir wissen, wie die Proben vorbereitet werden müssen und sitzen nun gedanklich vor einem Transmissionselektronen-mikroskop. Zeit, sich mit seinem Äußeren vertraut zu machen. Natürlich sieht jeder Mikroskoptyp etwas anders aus, das hängt von der Herstellungsfirma aber auch von der Baureihe ab. Es ist aber wie mit dem Auto: Es gibt auch Gemein-samkeiten und wir wollen versuchen, uns auf solche zu beschränken. Schließlich erwerben wir den Führerschein auch nicht für einen bestimmten Autotyp. Zu Beginn ist der Justagezustand des Mikroskops zu überprüfen und gegebenenfalls zu korrigieren. Auf evtl. Veränderungen der Probe während der Bestrahlung mit Elektronen wird hingewiesen.
In Bild 4-1 ist ein Transmissionselektronenmikroskop schematisch dargestellt. Die wichtigsten Teile sind bezeichnet, damit wir wissen, wovon wir später sprechen.
Bild 4-1. Schematische Darstellung eines Trans-missionselektronen-mikroskops. a) Ansicht von rechts. b) Vorderansicht.
Der erste Arbeitsschritt am TEM ist die Kontrolle des Justagezustandes. Die be-sten Bilder werden mit achsennahen Strahlen, sogenannten „Paraxialstrahlen“, er-halten. In dem mehrstufigen Abbildungssystem müssen demzufolge die einzelnen Linsen hinreichend gut zueinander zentriert sein. Die Frage ist, was bedeutet „hinreichend gut“? Wir wollen wieder den Vergleich mit dem Lichtmikroskop he-ranziehen. Dort reicht die mechanische Zentrierung bei der Herstellung aus. Er-innern wir uns an die förderliche Vergrößerung: Sie beträgt beim Lichtmikroskop etwa 500 ... 1000, beim Elektronenmikroskop aber ca. 1 Million. Grob gesagt, muss also die Zentrierung beim Elektronenmikroskop 1000-mal besser sein als
DOI 10.1007/978-3-7091-1440-7_4, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 4 66
beim Lichtmikroskop. Im Allgemeinen ist dies allein durch Sorgfalt bei der Herstellung und dem Zusammenbau nicht zu erreichen. Die Linsen und die Blen-den müssen während des Betriebs nachjustiert werden können. Bis in die 1970-er Jahre wurde dies zumindest teilweise durch mechanische Verschiebemöglich-keiten der Linsenkörper während des Betriebes realisiert. Mit Verbesserung des Auflösungsvermögens der Geräte erlangt allerdings die mechanische Stabilität der Mikroskopsäule immer größere Bedeutung. Die genannten Verschiebemöglich-keiten verhindern eine feste Verbindung der einzelnen Säulenteile und beein-trächtigen damit die mechanische Stabilität. In modernen Elektronenmikroskopen werden deshalb nicht die Linsen zueinander ausgerichtet, sondern der Elektronen-strahl wird so „gebogen“, dass er jeweils optimal durch die Linsenfelder tritt. Bei nicht-rotationssymmetrischen Einheiten (z.B. Stigmator – vgl. Abschnitte 7.7 und → 10.17) können die Linsenfelder elektrisch bzw. magnetisch geschoben und damit dem Strahlverlauf angepasst werden. Nur die Blenden werden weiterhin mechanisch zentriert. Deshalb gehören zu jeder Blende zwei senkrecht zueinander angeordnete Triebe, die eben diese Feinzentrierung erlauben. Einer der Triebe hat außerdem eine grobe Verschiebemöglichkeit, mit der eine andere Blendengröße in den Strahlengang gebracht werden kann, die danach wieder feinjustiert wird. Inzwischen sind diese Blendentriebe häufig motorisiert, und die Stellmotoren reproduzieren die Blendenpositionen mit großer Zuverlässigkeit. Diese Positionen können im Computer gespeichert und bei Bedarf zurückgerufen werden, was die Arbeit des Experimentators erleichtert. Bevor wir jedoch mit der Justage be-ginnen, müssen wir einige andere Dinge organisieren und erledigen.
4.1 Was wir „am Rande“ benötigen Selbstverständlich ist hier an erster Stelle die elektronentransparente Probe zu nennen. Wir hatten in Kapitel 3 erfahren, dass zu ihrer Herstellung neben aller Er-fahrung u. a. auch ein erheblicher apparativer Aufwand betrieben wird. Die Aus-wahl der Präparationsmaschinen hängt von den Aufgabenstellungen ab, die bei Neueinrichtung eines elektronenmikroskopischen Labors oder Änderung der For-schungsrichtung bekannt sein müssen. In der Werkstoffwissenschaft werden i. A. Schleifmaschinen benötigt, wie sie in der Metallografie üblich sind. Hinzu kom-men Pinzetten, Skalpelle, Messwerkzeuge, Lichtmikroskope, Heizplatten, Sägen und Dimpler sowie Maschinen zum Ionendünnen. Unter Umständen ist auch die Anschaffung einer Ausrüstung zum elektrolytischen Dünnen oder eines Ultra-mikrotoms sinnvoll. Zu all diesen Maschinen gehören Verbrauchsmaterialien, wie Schleifpapiere, Schleifpasten und –sprays, Epoxidharze und andere Kleber, Gase (Argon) usw. Dies alles ist auch bei der jährlichen Finanzplanung zu berück-sichtigen.
Die Probe muss mit aller Vorsicht in einen Probenhalter eingebaut und fest-gespannt werden. Dies geschieht mit Pinzette oder Vakuumpinzette; das ist eine feine Kanüle, die mit einer kleinen Vakuumpumpe verbunden ist und mit der das kleine Probenscheibchen angesaugt und an der richtigen Stelle im Probenhalter

Wir beginnen mit der praktischen Arbeit 67
wieder losgelassen werden kann. Oft muss die kleine Probe in einer bestimmten Lage in den Halter eingesetzt und ihre Fixierung kontrolliert werden. Das ist nur mit Hilfe eines Lichtmikroskops möglich. Bewährt hat sich dazu ein Stereo-Auflichtmikroskop mit veränderlicher Vergrößerung zwischen 5- und 100-fach und einem Arbeitsabstand von mindesten 3 cm.
Wir werden später erläutern, dass die Verunreinigung der Probenoberfläche im Elektronenmikroskop während der Bestrahlung mit Elektronen durch Kohlenwas-serstoffkontamination stören kann. Deshalb ist es zweckmäßig, die Probe unmit-telbar vor dem Einschleusen in das Mikroskop in einem Plasmacleaner zu reini-gen. Dabei werden die kohlenstoffhaltigen Oberflächenbedeckungen der Probe in einem niederenergetischen (< ca. 100 eV) Plasma verbrannt, d.h. es ist ein sauer-stoffhaltiges Inertgas erforderlich. Oft wird Argon mit 20% Sauerstoff empfohlen, „normale“ Luft ist aber ebenso geeignet.
Zum gleichen Zweck, nämlich der Verringerung der Kohlenwasserstoffkonta-mination, ist die Probe im Transmissionselektronenmikroskop von gekühlten Kup-ferblechen umgeben. Die Kühlung der Bleche erfolgt über einen Kupferbolzen, der vakuumdicht in die Mikroskopsäule ragt und außen mit flüssigem Stickstoff gekühlt wird. Wir benötigen also flüssigen Stickstoff; wenn das Elektronenmikro-skop mit einem energiedispersiven Röntgendetektor ausgerüstet ist, evtl. auch für die Kühlung des Detektorkristalls.
Turbopumpen werden nach dem Schließen der Ventile zur Pumpe und dem Abschalten „von außen“ belüftet. Dies erfolgt mit reinem Stickstoff (99,999%), da-mit keine Luftfeuchtigkeit eingebracht wird. Auch zur evtl. notwendigen Belüf-tung der Mikroskopsäule wird dieser Stickstoff benötigt. Eine entsprechende Gas-flasche ist anzuschaffen. Und da wir gerade bei Gasen sind: Der Tank für die Er-zeugung der Hochspannung und der Behälter für die Elektronik an der Elektronen-kanone sind mit einigen bar Schwefelhexafluorid (SF6) gefüllt, um Überschläge zu vermeiden. Die entsprechende Gasflasche ist ebenfalls vorrätig zu halten.
4.2 Wir bauen die Probe in den Halter und schleusen diesen ins Mikroskop Nun wird es wirklich konkret: Wir wollen die Probe in den Probenhalter einbauen. Zuerst wollen wir uns mit einem typischen Probenhalter für das Transmissions-elektronenmikroskop vertraut machen (s. Bild 4-2).
Bild 4-2. Probenhalter für das Transmissionselektro-nenmikroskop.
Die Probe wird unter lichtmikroskopischer Kontrolle an der dafür vorgesehenen Position (im Bild rechts) in den Probenhalter eingelegt und fixiert. Je nach Pro-

Kapitel 4 68
benhalter erfolgt dies in unterschiedlicher Weise: Beispielsweise mit einer feder-belasteten Klappe oder durch einen Gewinde- bzw. Sprengring. Spezialproben-halter haben häufig auch spezielle Vorrichtungen, um die Probe zu fixieren.
Die Probenschleuse ist ein Rohr, welches am mikroskopseitigen Ende mit einem Ventil verschlossen ist. In dieses Rohr wird der Halter eingeführt, wobei darauf zu achten ist, dass der Stift am Halter in eine dafür vorgesehene Nut im In-neren des Schleusenrohres gleitet. Der Dichtring des Halters dichtet nunmehr das äußere Ende des Schleusenrohres ab, so dass zwischen innerem Ventil und dem Dichtring ein abgeschlossener Raum entsteht, der mit einer Vakuumpumpe evaku-iert wird. Durch Drehen des Halters am Griff öffnet der Stift das innere Ventil, und der Halter gleitet in die Arbeitsposition. Es ist nicht notwendig, den Halter mit Muskelkraft zu schieben, der äußere Luftdruck sorgt schon für die Bewegung. Im Gegenteil, es ist besser, sich auf das Bremsen der Halterbewegung einzustellen.
Für die Transmissionselektronenmikroskopie gibt es kommerziell eine Vielzahl von Probenhaltern: Einfach- und Doppelkipphalter, Rotationskipphalter, „Low-Background“-Halter, Halter mit Heiz- oder Kühleinrichtung, Halter für Zugver-suche und Halter, in denen eine Spitze gegenüber der Probe angeordnet wird, die piezoelektrisch bewegt werden kann („Tunnelmikroskop“). Darüber hinaus exi-stieren Spezialanfertigungen für besondere Untersuchungen, z.B. in magnetischen Feldern.
Wir wollen uns auf die Halter beschränken, die für die in diesem Buch ge-schilderten elektronenmikroskopischen Untersuchungen notwendig sind. Mit an-deren Worten: Vor Halterauswahl muss klar sein, welche Eigenschaften der Probe mit welchen Methoden gemessen werden sollen. Für Korngrößen und Phasen in polykristallinem Material und für Nahordnungen in amorphem Schichten genügt der Einfachkipphalter. Für Beugungs- und Hochauflösungsuntersuchungen an ein-kristallinen oder epitaktisch gewachsenen Proben benötigen wir den Doppelkipp-halter, um geeignete Kristallrichtungen auswählen zu können. Bei diesem Halter wird die Probe in der Regel in ein kleines Körbchen eingelegt, dessen Innen-durchmesser nur wenig größer als 3 mm ist. Ist das Probenscheibchen größer, kann dieses nicht benutzt werden, und der Präparationsaufwand war vergeblich. Sollen auch Analysen mittels energiedispersiver Röntgenspektroskopie („EDXS“) erfolgen, so ist ein Low-Background-Halter (Probenaufnahme aus Beryllium, vgl. Abschnitt 9.2.2) auszuwählen. Es lohnt sich auf jeden Fall, vor dem Einbau der Probe über diese Dinge nachzudenken. Jeder Probenein- und –ausbau birgt die Gefahr in sich, dass die Probe zerbricht oder auf andere Art verloren geht!
Bei allen Maßnahmen ist auf Sauberkeit des Halters und der Probe zu achten. Der probenseitige Teil des Halters sollte ab Dichtring nicht mit bloßen Fingern be-rührt werden, das Anfassen der Probe mit bloßen Händen verbietet sich schon we-gen der Kleinheit der Proben. Wenn möglich, sollte die eingebaute Probe zusam-men mit dem Halter vor dem Einschleusen ins Mikroskop in einem Plasmacleaner gereinigt werden. Doch Vorsicht, beim Plasmareinigen handelt es sich im Wesent-lichen um ein plasmagestütztes Verbrennen von Kohlenstoff in sauerstoffhaltiger Atmosphäre. Präparate mit Kohlenstoff als Stützfilm oder mit Materialien, die zur

Wir beginnen mit der praktischen Arbeit 69
Oxidation neigen (z.B. Kupfer), sollten also nicht im Plasmacleaner behandelt werden.
Wir sollten nun auch die Objektraumkühlung in Betrieb nehmen, falls sie es nicht bereits ist, d.h. wir füllen flüssigen Stickstoff in das betreffende Dewar-gefäß31.
Schließlich noch eine Bemerkung zu ferromagnetischen Proben: Aus Kapitel 2 wissen wir, dass sich die Probe inmitten des starken Objektivlinsen-Magnetfeldes befindet, d.h. auf die Probe wirkt eine magnetische Kraft. Bei unzureichender Be-festigung der Probe im Halter (z.B. nur durch die federbelastete Klappe) wird die Probe u. U. im Mikroskop aus dem Halter herausgezogen und klebt dann am Pol-schuh. Im günstigeren Fall erhöht das lediglich den Astigmatismus, im ungün-stigen Fall behindert die Probe den Strahlengang und die Mikroskopsäule muss demontiert werden, um die Probe zu entfernen. Magnetische Proben sollten also hinreichend fest im Halter fixiert werden. Beim Einschleusen solcher Proben sollte der Objektivlinsenstrom ausgeschaltet oder zumindest stark reduziert wer-den (Umschalten in den „Low-Magnification“-Bereich).
4.3 Wir überprüfen den (Justage-)Zustand des Mikroskops Nun ist es soweit: Die Probe ist eingeschleust, ein Blick auf die Druckanzeige in der Säule zeigt uns nach einigen Minuten Wartezeit, dass das Vakuum in der Mikroskopsäule hinreichend gut ist (p < 10-3 Pa), um das Ventil zwischen Elek-tronenkanone und Säule öffnen zu können. Wegen der guten Stabilität moderner Transmissionselektronenmikroskope ist eine komplette und zeitaufwändige Justa-ge im Allgemeinen nur nach Änderungen und Demontagen der Mikroskopsäule erforderlich. Es ist jedoch empfehlenswert, zu Beginn der elektronenmikroskopi-schen Untersuchung einige Merkmale der Gerätejustage zu überprüfen.
Bild 4-3. Modell des Elek-tronenmikroskops zum Ver-ständnis der Justageschritte. Die Ablenksysteme sind in der x-z-Ebene gezeichnet. Weitere, identische Einheiten sind in der dazu senkrechten y-z-Ebene angeordnet, um den Elektronenstrahl in alle x-y-Richtungen ablenken zu können.
31 Sir James Dewar, schottischer Physiker, 1842 - 1923

Kapitel 4 70
Um das Prinzip der Justage zu verstehen, benutzen wir ein vereinfachtes Modell des Elektronenmikroskops, welches nur aus der Elektronenkanone, einer Konden-sorlinse, der Objektivlinse und einer Projektivlinse besteht. Zwischen diesen Ein-heiten sind Ablenksysteme angebracht, die ein Verschieben und Kippen des Elek-tronenstrahls in zwei zueinander senkrechten Richtungen ermöglichen. Unser Modell enthält elektrostatische Ablenksysteme, weil die Skizzen damit leichter zu verstehen sind. In der Praxis werden meistens magnetische Systeme eingesetzt; auf das Funktionsprinzip hat dies keine Auswirkung. Zusätzlich besitzt unser Mo-dell eine Blende: die verstellbare Kondensorblende in Höhe der Kondensorlinse, die oft auch als Kondensor-2-(C2)-Blende bezeichnet wird (vgl. Bild 4-3). Die Prüfung der Justage erfolgt in fünf Schritten.
1. Justage der Blende im 2. Kondensor („C2-Blende“):
Die Kondensorblende beeinflusst die Ausleuchtung des Beobachtungsfeldes und muss deshalb zuerst zentriert werden. Die einzelnen Justierschritte sollen anhand der Bilder 4-4 und 4-5 erläutert werden. Darin sind nur die für diese Erläuterung wichtigen Teile unseres Modells gezeichnet.
Bild 4-4. Zur Justage der Kondensorblende, Strahlengänge. a)-c) Dezentrierte Blende. d)-f) Zentrierte Blende. Weitere Erläuterungen im Text.
Wir gehen von einer schiefen Beleuchtung der Kondensorlinse aus (dies wird erst später korrigiert). Bei „moderater“ Vergrößerung (< 10000) wird die Kondensor-brennweite („Intensity“) so eingestellt, dass auf dem Bildschirm ein möglichst kleiner Leuchtfleck entsteht (Bilder 4-4a und 4-4d). Dieser kleine Fleck wird mit dem Ablenksystem 2 („Beam Shift“) in das Zentrum des Bildschirms geschoben (Bilder 4-4b und 4-4e). Dabei spielt die evtl. Dezentrierung der Kondensorblende keine Rolle. Bei Veränderung der Kondensorbrennweite vergrößert sich der Fleck auf dem Bildschirm und wir bemerken einen Einfluss der Blendenzentrierung: Bei
Wir beginnen mit der praktischen Arbeit 71
dezentrierter Blende verschiebt sich die Mitte des größer werdenden Scheibchens (Bild 4-4c bzw. Bild 4-5a), bei zentrierter Blende öffnet sich der Fleck konzen-trisch (Bild 4-4f bzw. Bild 4-5b).
Bild 4-5. Zur Justage der Kondensorblende, Ansicht auf dem Bildschirm bei Variation der Kondensor-brennweite. a) Dezentrierte Blende. b) Zentrierte Blende.
Beim „Spielen“ mit der Kondensorbrennweite stellen wir fest, dass sich aus-gehend vom fokussierten Spot der beleuchtete Bereich öffnet, unabhängig davon in welche Richtung wir den Intensity-Knopf drehen. Leser, die davon überrascht sind, sehen bitte noch einmal im Abschnitt 2.7.1 (Beleuchtungssystem) nach.
Die Blende ist bei Bedarf solange zu verschieben, bis der Fall von Bild 4-5b eintritt. Wenn der Fleck nicht kreisrund sondern elliptisch ist und sich die Rich-tung der langen Ellipsenachse beim Durchgang durch die „kleinste-Fleck-Posi-tion“ um ca. 90° ändert, liegt ein zweizähliger Kondensorastigmatismus vor. Die-ser wird mit dem Kondensor-Stigmator korrigiert.
2. Justage der Elektronenkanone:
Ziel dieses Justageschrittes ist ein möglichst helles und gleichmäßig ausge-leuchtetes Bild. Auch hier wollen wir uns am vereinfachten Modell Strahlengang und Erscheinungsbild auf dem Schirm überlegen. Dabei müssen wir das Intensi-tätsprofil im Strahlquerschnitt berücksichtigen. Es ist nicht kastenförmig sondern hat in der Mitte sein Maximum und fällt dann langsam zum Rand hin ab. Mathe-matisch ist es recht gut mit einer Gauß-Funktion zu beschreiben (s. Bild 4-6).
Bild 4-6. Stromdichte im Strahlquerschnitt. a) Seitenansicht und Draufsicht. b) Kastenprofil (unzutreffend). c) Gaußprofil (gute Näherung für den justierten Strahl).
Mit dieser Kenntnis können wir die Forderung nach dem „gleichmäßig ausge-leuchteten Bild“ präzisieren: Das Strahlprofil auf dem Schirm soll symmetrisch bzgl. des Mittelpunktes sein, wie es in Bild 4-6a auch gezeichnet ist. Anhand von

Kapitel 4 72
Bild 4-7 wollen wir überlegen, wie eine schiefe Beleuchtung aufgrund einer ge-ringfügigen Dezentrierung der Elektronenkanone mit Hilfe der Ablenksysteme korrigiert werden kann.
Im Strahlenbündel ist der intensitätsreiche Kern (Maximum der Stromdichte) hell eingezeichnet. Wir gehen vom dezentrierten Zustand aus, d.h. die Elektronen-kanone ist verdreht und verschoben (Bild 4-7a). Der Elektronenstrahl trifft teil-weise auf die Kondensorblende, die Intensität (d.h. der Strom) auf dem Bildschirm ist reduziert. Zuerst fokussieren wir den Strahl bei kleiner Vergrößerung (ca. 5000) mit dem Kondensor („Intensity“), so dass auf dem Schirm ein kleiner heller Spot entsteht. Bei Bedarf kann dieser mit dem Ablenksystem 2 („Beam Shift“) ins Zentrum des Schirms geschoben werden. Der Strahl wird nun mit dem Ablenk-system 1 gekippt („Gun Tilt“) bis der Spot seine maximale Helligkeit erreicht hat. Wenn der Spot aus dem Zentrum wandert, kann dies jederzeit mit „Beam Shift“ korrigiert werden.
Bild 4-7. Zur Justage der Elektronenkanone. a) Strahlengang im dejustierten Zustand. b) Nach Strahlkippung mit fokussiertem Strahl. c) Nach Strahlkippung mit „aufgezogenem“ Strahl. d) Nach Strahlkippung und –verschiebung mit aufgezogenem Strahl. Ansicht auf dem Bild-schirm bei (e) ungenügender Strahlverschiebung und (f) justiertem Zustand.
Es ist nicht einfach, die Helligkeit des Spots visuell zu kontrollieren. Deshalb be-nutzen wir als Hilfsmittel die angezeigte Belichtungszeit. Dies ist eine Referenz an die Zeiten, in denen das elektronenmikroskopische Bild auf Film oder Fotoplatten aufgenommen wurde. Um die Belichtungszeit einstellen zu können, wurde der Elektronenstrom, der auf den Bildschirm gelangt, gemessen und daraus nach Kali-brierung für das benutzte Fotomaterial die Belichtungszeit festgelegt. Obwohl heute im Elektronenmikroskop kaum noch fotografiert wird, sind die Messung des Stroms auf dem Schirm und die Anzeige der Belichtungszeit erhalten geblieben.

Wir beginnen mit der praktischen Arbeit 73
Je größer der Strom ist, desto kürzer wird die Belichtungszeit. Dies nutzen wir aus, um die Helligkeit im Spot zu kontrollieren. Wir ändern also „Gun Tilt“ so-lange, bis die angezeigte Belichtungszeit minimal wird (Bild 4-7b).
Nun ändern wir die Brennweite der Kondensorlinse („Intensity“) und erhöhen die Vergrößerung bis die in Bild 4-7e skizzierte Figur auf dem Schirm zu sehen ist: Innerhalb eines kreisrunden Flecks ist ein heller Spot zu sehen. Bei mecha-nisch ordnungsgemäß vorzentrierten Feldemissionskathoden sollte dies problem-los möglich sein, bei LaB6-Kathoden muss evtl. der Heizstrom etwas verringert werden; auch ist die äußere Umrandung der Figur dabei kein Kreis. Durch zusätzliche Verschiebung des Strahls mit dem Ablenksystem 1 („Gun Shift“) wird der helle Spot ins Zentrum des kreisrunden Flecks gebracht (Bilder 4-7d und f). Bei LaB6-Kathoden ist eine hohe Symmetrie der Figur auf dem Leuchtschirm das Ziel. Damit ist das Bild gleichmäßig ausgeleuchtet. Wie bereits bei der Justage der Kondensorblende wird ein evtl. auftretender Kondensorastigmatismus zwischen-durch mit dem Kondensor-Stigmator korrigiert. Bei großer Dejustage muss die Korrektur unter Umständen in mehreren kleinen Schritten erfolgen.
3. Einstellen der euzentrischen Höhe:
Bei einer optischen Abbildung ist das Bild genau dann scharfgestellt, wenn die Abbildungsgleichung
1 1 1
f g b= + (1.3)
(f: Brennweite, g: Dingweite, b: Bildweite) erfüllt ist. Dazu gibt es offensichtlich drei verschiedene Möglichkeiten: Dingweite wird Brenn- und Bildweite angepasst (Beispiel: Lichtmikroskop), Bildweite wird Brenn- und Dingweite angepasst (Fo-toapparat) oder Brennweite wird Ding- und Bildweite angepasst (Auge). Der Vorteil der letzten Methode bzgl. eines Elektronenmikroskops ist, dass die Brenn-weite der magnetischen Linsen einfach, feinfühlig und mit sehr guter Genauigkeit durch Änderung des Linsenstromes eingestellt werden kann und die Fokussierung ohne mechanische Verschiebung auskommt. Aus Abschnitt 2.1 wissen wir aber, dass sich bei Brennweitenänderung auch der Abbildungsmaßstab ändert, was die Nutzung des Mikroskops als Messmaschine für kleinste Längen unmöglich macht. Bei einer Kombination mehrerer Linsen sind deren Brennweiten aufeinander ab-gestimmt. Wird eine der Brennweiten der Linsen stark geändert, beeinflusst dies die Abstimmung, und andere Brennweiten müssen nachgeführt werden. Um dies zu vermeiden, wird beim Transmissionselektronenmikroskop in zwei Stufen scharfgestellt: Eine Grobeinstellung wird wie beim Lichtmikroskop mit einer An-passung der Dingweite vorgenommen, die Feinfokussierung erfolgt dann durch Veränderung der Objektivbrennweite.
Wir wollen uns Kriterien überlegen, nach denen wir die Güte der Grobein-stellung beurteilen können. Naheliegend ist es, in gleicher Weise wie beim Licht-mikroskop vorzugehen: Wir stellen durch Verschieben der Probenbühne in z-

Kapitel 4 74
Richtung („optische Achse“) scharf. Für die Grobeinstellung ist das feinfühlig ge-nug. Im Unterschied zum Lichtmikroskop ist allerdings im Elektronenmikroskop auch die Brennweite variabel, so dass wir vorher eine Bezugsbrennweite festlegen müssen. Das geschieht durch Drücken eines entsprechenden Knopfes auf dem Bedienpult (Bezeichnung z.B. „Eucentric Focus“). Damit wird ein vorgegebener Objektivlinsenstrom und damit die Bezugsbrennweite eingestellt.
Die Frage ist: „Wie können wir diese Bezugsbrennweite kontrollieren?“ Oder anders: „Gibt es ein anderes Kriterium zur Beurteilung der ‚richtigen’ Höhenein-stellung der Probe?“ Erinnern wir uns: Der Probenhalter wird in ein Goniometer eingeführt und kann um die Goniometerachse gedreht werden. Die Goniometer-achse ist geeignet, als Bezugshöhe ohne Rücksicht auf die Objektivbrennweite zu gelten. In Bild 4-8 ist skizziert, was wir bei Abweichungen und Übereinstimmung der Probenhöhe mit der Goniometerachse zu erwarten haben.
Bild 4-8. Zur Einstellung der euzentrischen Höhe. a) Probe unterhalb der Goniometerachse. b) Probe in Höhe der Goniometerachse („euzentrische Höhe“).
Auf dem Bildschirm des Mikroskops sehen wir die (vergrößerte) Projektion der Probe. Wird das Goniometer mit der Probe gedreht, verzerrt sich deren Bild. Be-findet sich die Probe unterhalb der Goniometerachse (Bild 4-8a), so verschieben sich die im Bild der Probe sichtbaren Details zur Seite und zwar systematisch mit der Drehrichtung. Bei gleicher Höhe von Probe und Goniometerachse bleibt es bei der Verzerrung, eine Verschiebung findet nicht statt (Bild 4-8b). Damit haben wir das gesuchte brennweitenunabhängige Kriterium für die Einstellung der euzentri-schen Höhe gefunden. Da die mechanische Verschiebung vergleichsweise „unsen-sibel“ ist, sollte die Vergrößerung bei diesem Schritt nicht zu groß gewählt werden (< 10000). Bei modernen Geräten übernimmt ein Computer die Steuerung der pe-riodischen Drehbewegung („Wobbeln“).
4. Einstellen der Umlenkpunkte für die Strahlkippung:
In der Transmissionselektronenmikroskopie gibt es Kontrastphänomene, die von der Orientierung der Probe zum Elektronenstrahl abhängen. Wir werden dies im Kapitel 6 genauer erläutern. Um solche Kontraste genauer zu analysieren, wird die Orientierung geändert, d.h. entweder wird die Probe gekippt oder der Strahl. Wie schon mehrfach betont, sind mechanische Bewegungen im Elektronenmikroskop problematisch, und eine elektrische (bzw. magnetische) Strahlkippung ist durch-

Wir beginnen mit der praktischen Arbeit 75
aus eine wünschenswerte Alternative, die mit Hilfe der Ablenksysteme 2 und 3 (vgl. Bild 4-3) auch realisiert werden kann. Natürlich muss bei der Strahlkippung der gleiche Probenbereich beleuchtet bleiben, d.h. der Kipppunkt (englisch: „Pivot Point“) muss exakt in der Probenebene liegen. Die Probenebene ist in Bild 4-3 nicht eingezeichnet, sie ist aber hier für das Verständnis erforderlich und wird deshalb in Bild 4-9 ergänzt. In diesem Bild ist zu sehen, dass der Strahl durch Nutzen beider Etagen des Ablenksystems 2 in der gewünschten Weise gekippt werden kann. Der Umlenkpunkt der Kippung (Kipppunkt) wird durch Einstellen des „richtigen“ Verhältnisses der Umlenkwinkel beider Etagen in die Probenebene geschoben. Wir sehen, dass der Fokuspunkt des gekippten Strahls unterhalb der Objektivlinse von der optischen Achse weggeschoben wird. Das Ablenksystem 3 sorgt dafür, dass das Strahlenbündel auch bei gekipptem Strahl optimal in die Projektivlinse eintritt.
Bild 4-9. Strahlengänge bei Kippung des Strahlenbündels. a) Ungekippt. b) Kippung nach rechts. c) Kippung nach links.
Das Ziel dieses Justageschrittes ist es, den Kipppunkt genau in die Probenebene zu schieben. Die praktische Vorgehensweise sei anhand von Bild 4-10 erläutert.
Bild 4-10. Bilder auf dem Beobach-tungsschirm bei Einstellung des Kipp-punktes („Wobbeln“). a) Dejustiert mit falscher Fokussierung. b) Dejustiert mit richtiger Fokussierung. c) Justiert.
Voraussetzung ist natürlich, dass wir exakt die Probenebene auf dem Beobach-tungsschirm abbilden, d.h. die Probe muss exakt scharfgestellt sein. Der Strahl wird nun „gewobbelt“, d.h. er wird periodisch hin- und hergekippt. Wir konzen-trieren uns auf ein leicht zu beobachtendes Detail im Bild der Probe und können daran erkennen, ob wirklich exakt scharfgestellt wurde. Falls dies nicht der Fall ist, sehen wir ein Doppelbild (Bild 4-10a). Wichtig ist, dass wir uns auf das Detail konzentrieren und nicht auf den Rand des Strahlenbündels. Das Doppelbild wird

Kapitel 4 76
durch Anpassen der Objektivbrennweite („Focus“) beseitigt. Nun konzentrieren wir uns auf den Rand des Strahlenbündels. Im dejustierten Zustand (Bild 4-10b) ist er doppelt zu sehen. Dies wird durch Verschiebung des Kipppunktes längs der optischen Achse korrigiert (Bild 4-10c - Einstellmöglichkeit in der Regel mit „Pivot Point X“ bzw. „Pivot Point Y“ bezeichnet). Es gibt zwei Einstellmöglich-keiten, weil in zwei zueinander senkrechten Richtungen (x,y) gekippt werden kann.
5. Justage des Rotationszentrums:
Die Objektivlinse hat den stärksten Einfluss auf die Abbildungsqualität. Deshalb ist es wichtig, dass das Elektronenbündel bei ungekipptem Strahl symmetrisch zur optischen Achse des Objektivs verläuft. Wie können wir das kontrollieren?
Auch hier wollen wir uns zunächst den Strahlengang überlegen (vgl. Bild 4-11). Die Abweichung zwischen der Symmetrieachse des Strahlenbündels und der Mittelachse (d.i. die optische Achse) der Objektivlinse ist durch Kippung der Ob-jektivlinse veranschaulicht. Das Bild sei scharfgestellt, die Bildverschiebung auf dem Beobachtungsschirm durch das Ablenksystem 3 korrigiert (Bild 4-11a). Bei Veränderung der Objektivbrennweite wird das Bild unscharf und ändert seine Größe, bei magnetischen Linsen dreht es sich zusätzlich um einen kleinen Winkel. Bei unveränderter Einstellung des Ablenksystems 3 verschiebt sich das Zentrum des Bildes (Bild 4-11b). Die Justage erfolgt durch Zusammenspiel der Ab-lenksysteme 2 und 3: Mit System 2 wird der Strahl in die Mittelachse des Ob-jektivs gekippt, mit System 3 die daraus folgende Bildverschiebung korrigiert (Bild 4-11c). Im Unterschied zum dejustierten Zustand bleibt jetzt das Zentrum des Bildes auf dem Beobachtungsschirm an Ort und Stelle (Bild 4-11d).
Bild 4-11. Strahlengänge zur Veranschaulichung der Abweichungen des Strahlenbündels von der optischen Achse des Objektivs. a) Dejustierter Zustand, fokussiertes Bild. b) Dejustierter Zustand nach Änderung der Objektivbrennweite. c) Justierter Zustand, fokussiertes Bild. d) Justierter Zustand nach Änderung der Objektivbrennweite.
Nun zur praktischen Vorgehensweise (vgl. Bild 4-12): Zunächst verschieben wir die Probe so, dass sich ein gut zu sehendes Detail im Zentrum des Beobach-tungsschirmes befindet. Günstig ist eine Spitze wie in Bild 4-12 angenommen. Nun schalten wir den Wobbler für die Objektivbrennweite („Rotation Center“) ein und sehen, dass sich die Spitze periodisch aus dem Zentrum wegbewegt.

Wir beginnen mit der praktischen Arbeit 77
Außerdem rotiert das Bild etwas (Bild 4-12a). Die Korrektur ist erfolgreich, wenn die Spitze unverändert im Zentrum bleibt und sich das Bild um diese Spitze dreht.
Bild 4-12. Bilder auf dem Beobachtungsschirm bei Wobbeln der Objektivbrennweite zur Justage des Rotationszentrums. a) Dejustiert. b) Justiert.
Daher kommt auch der Name dieses Justageschrittes: Das Zentrum der Bildrota-tion ist ortsfest.
Alternativ kann für diese Justage anstelle des Objektivlinsenstromes auch die Beschleunigungsspannung periodisch geändert werden (Hochspannungszentrum). Die Objektivbrennweite wird in diesem Fall durch die sich verändernde Elek-tronenwellenlänge gewobbelt (Einzelheiten s. [4.1]).
4.4 Scharfstellen des Bildes – Schärfe und Kontrast Im vorhergehenden Abschnitt haben wir wiederholt vom „Scharfstellen des Bil-des“ geschrieben. Wann ist aber nun ein elektronenmikroskopisches Bild scharf? Es passiert dem Anfänger häufig, dass er Schärfe mit Kontrast verwechselt. So er-scheint ein unscharfes aber kontrastreiches Bild in der Regel schärfer als ein scharfes aber kontrastarmes Bild. Das hat eine gewisse Berechtigung, denn was nützt ein scharfes Bild, wenn darin kein Kontrast ist, wir also nichts sehen. Ande-rerseits brauchen wir ein scharfes Bild, wenn wir kleinste Strukturen auflösen wollen.
Zur Erklärung dieses Widerspruchs zwischen Schärfe und Kontrast im Trans-missionselektronenmikroskop blicken wir auf Bild 4-13.
Bild 4-13. TEM-Bild vom Rand eines Kohlenstofffilms. a) Unterfokus. b) Fokus. c) Überfokus.

Kapitel 4 78
Es zeigt ein elektronenmikroskopisches Bild vom Rand eines dünnen Kohlenstofffilms, auf dem sich einige Goldpartikel befinden. Das fokussierte Bild 4-13b zeigt im Vergleich zu den beiden anderen Bildern nur schwachen Kontrast. Vakuum und C-Film haben fast die gleiche Graufärbung. Demgegenüber verläuft bei den defokussierten Bildern parallel zum Kohlenstoffrand ein heller oder dunkler Saum, der den Rand kontrastreicher macht. Dieser Saum ist eine Folge des Wellencharakters der Elektronen. Hinter dem Kohlenstoffrand ist die Intensität nicht gleichmäßig verteilt sondern besitzt infolge der Interferenz der Elementarwellen Maxima und Minima (→ Abschnitt 10.1). Diese Maxima und Minima sind nur zu sehen, wenn eben gerade nicht exakt die Kohlenstofffilm-Ebene abgebildet wird. Wir sehen den kontrastverstärkenden Saum also nur im defokussierten Bild. Das Bild ist also gerade dann optimal scharfgestellt, wenn der Kontrast minimal ist.
Wir erkennen aber auch, dass die Abbildung beispielsweise von Kohlenstoff-strukturen im Elektronenmikroskop wegen des geringen Kontrastes problematisch ist. In diesem Fall muss häufig ein Kompromiss eingegangen werden: Wir defokussieren ein wenig, um überhaupt etwas zu sehen.
4.5 Kontamination und Objektschädigung „... Die Entwicklung der heutigen Elektronenmikroskope war im Wesentlichen ein Kampf gegen die unerwünschten Folgen derselben Eigenschaften von Elektronen-strahlen, die die sublichtmikroskopische Auflösung erst ermöglicht haben. So ist z. B. die kurze Materiewelle - die Voraussetzung der guten Auflösung - an die we-gen der Objektbelastung nicht erwünschte hohe Elektronenenergie gekoppelt. ...“ [4.2].
Dieses Zitat von Ernst Ruska zeigt, dass wir neben den erwünschten, kontrast-formenden Wechselwirkungen zwischen den Elektronen und der Probe (auf die wir in Kapitel 6 eingehen werden) auch unerwünschte Ergebnisse dieser Wechsel-wirkung beachten müssen. Wir wollen uns mit zwei derartigen Problemen näher beschäftigen.
- Kontamination:
Die Kontamination, genauer die Kohlenwasserstoffkontamination, ist ein Prozess, der in allen Elektronenstrahlgeräten auftritt, in der Transmissionselektronenmi-kroskopie aber besonders stört. Obwohl in der Probenumgebung ein „gutes“ Hochvakuum (Druck < 10-3 Pa) herrscht, gibt es dort eine große Anzahl von Gas-molekeln (Teilchendichte ca. 3⋅108 mm-³ bei Raumtemperatur und 10-3 Pa). Selbst wenn nur 0,01 % davon Kohlenwasserstoffmoleküle sind, wären das immer noch 30000 pro mm3. Da die Gasmolekeln auf die Wände sowie auf die Proben-oberfläche treffen und auch für eine kurze Zeit dort verweilen, bildet sich eine Gasbedeckung, an der auch die Kohlenwasserstoffmoleküle teilnehmen. Zwischen ankommenden und wegfliegenden Molekeln besteht ein Gleichgewicht, das

Wir beginnen mit der praktischen Arbeit 79
druck- und temperaturabhängig ist. Bei höherer Wand- (bzw. Proben-) Temperatur wird die Verweilzeit der Molekeln auf der Oberfläche kürzer, die Dicke der Be-deckungsschicht sinkt. Bei höherem Druck im Gasraum wächst der Molekelstrom auf die Oberfläche, die Dicke der Bedeckungsschicht nimmt zu.
Wir stellen uns vor, dass wir die Probe „großflächig“ mit Elektronen bestrahlen (großflächig bedeutet im Elektronenmikroskop einen Fleck mit einem Durchmes-ser von wenigen Mikrometern). Dies wird in Bild 4-14 vorausgesetzt.
Wir sehen das dynamische Gleichgewicht der auf die Probe auftreffenden und von ihr wegfliegenden gasförmigen Kohlenwasserstoffmoleküle, die auch von Elektronen getroffen werden. Wenn allerdings zufällig zwei Moleküle auf der Pro-benoberfläche nahe beieinander liegen, führt der Elektronenbeschuss zu einer Ver-netzung, und es bilden sich feste Kohlenwasserstoffe, die Kontaminationsschicht.
Bild 4-14. Kontamination bei großflächiger Proben-bestrahlung mit Elektronen.
Bei Abschätzung des Dickenwachstums durch Kontamination ist zu beachten, dass die Kontaminationsschicht im TEM sowohl auf der Oberseite als auch auf der Unterseite der Probe aufwächst. Dies kann durchaus mit einigen Nanometern pro Sekunde geschehen. Um die Kontamination zu verringern, muss entweder die mittlere Verweilzeit der gasförmigen Kohlenwasserstoffmoleküle auf der Proben-oberfläche oder/und der Kohlenwasserstoff-Partialdruck in der Probenumgebung verringert werden. Die Verweilzeit hängt von der Oberflächentemperatur ab, eine Möglichkeit wäre, die Probe zu heizen. Wegen der Empfindlichkeit der Proben (z.B. Einbettung in Epoxidharz) und der Notwendigkeit stabiler Temperaturver-hältnisse bei hoher Vergrößerung (Vermeidung von thermischer Probendrift) ist dies nur eingeschränkt möglich. Stattdessen wird üblicherweise der Kohlenwas-serstoff-Partialdruck gesenkt, indem in der (sowieso schon sehr engen) Probenum-gebung Kupferbleche angeordnet werden, die von außen gekühlt sind [4.3]. Dazu dient das mit flüssigem Stickstoff gefüllte Dewargefäß, welches in Bild 4-1 mit „Objektraumkühlung“ bezeichnet ist. Natürlich sollte die Probenoberfläche vor dem Einschleusen in das Mikroskop sauber sein, beispielsweise durch Reinigen im Plasmacleaner.
Etwas anders ist der Kontaminationsmechanismus bei Bestrahlung der Probe mit einer kleinen Elektronensonde, wie dies für das Rasterelektronenmikroskop oder im Rastermodus des Transmissionselektronenmikroskops typisch ist. In die-

Kapitel 4 80
sem Fall besteht eine weitere Möglichkeit für die Nachlieferung von Kohlen-wasserstoffmolekülen zum Kontaminationsort: Diffusion über die Probenober-fläche (vgl. Bild 4-15).
Bild 4-15. Kontamination bei sondenartiger Probenbestrah-lung mit Elektronen. Die mit Ausrufezeichen gekennzeich-neten Moleküle bewegen sich über die Oberfläche zum Kontaminationsort.
Die treibende Kraft für die Oberflächendiffusion ist der Konzentrationsunterschied zwischen den festen Kohlenwasserstoffen am Sondenort und den beweglichen, gasförmigen Kohlenwasserstoffmolekülen. Dieser Mechanismus ist sehr effektiv, weil die gesamte Probenoberfläche gewissermaßen als „Antenne“ für den Emp-fang der Moleküle wirkt. Die Kontaminationsschicht am Sondenort wächst oft schneller als bei großflächiger Bestrahlung.
- Objektschädigung:
Was passiert, wenn energiereiche Elektronen auf das um seine Ruhelage schwin-gende Atom eines Festkörpers treffen? Prinzipiell sind zwei Effekte möglich: Das Atom selbst kann verändert werden, oder/und das Atom wird „angestoßen“ und führt die Schwingung mit größerer Amplitude aus oder wird sogar von seinem Platz entfernt.
Bei den im Transmissionselektronenmikroskop üblicherweise benutzten Elek-tronenenergien zwischen 60 keV und 300 keV kann die Änderung des Atoms nur in dessen Elektronenhülle erfolgen. Wir sprechen nun von zwei verschiedenen Sorten von Elektronen: Zum einen von den Elektronen der Atomhülle und zum anderen von den Elektronen im Strahlenbündel. Zur Unterscheidung bezeichnen wir die Strahlelektronen auch als Primärelektronen. Elektronen der Hülle können aus ihren angestammten Energiezuständen (Bahnen) in höhere Energiezustände gehoben oder völlig aus der Hülle entfernt werden. Man spricht von Ionisation. Diesen Vorgang werden wir später in Kapitel 9 genauer erläutern. Die Wahr-scheinlichkeit für solche Ionisationsprozesse sinkt mit wachsender Energie der Primärelektronen.
Um die Vorgänge beim „Anstoßen“ des Atoms zu verstehen, müssen wir zu-nächst überlegen, warum sich ein Atom überhaupt in einem bestimmten Abstand von den anderen Atomen aufhält. Vereinfachend stellen wir uns vor, wir hätten nur zwei Atome, die je nach Abstand Abstoßungs- oder Anziehungskräfte auf-einander ausüben, so als ob sie mit einer Feder verbunden wären. Die Atome werden also den Abstand voneinander haben, in dem beide Kräfte gleich groß

Wir beginnen mit der praktischen Arbeit 81
sind. Um die Schwingung zu verstehen, ist es nützlich, anstelle der Kräfte die damit verbundene potentielle Energie zu betrachten. Bild 4-16 zeigt ein verein-fachtes Potentialmodell. Es basiert auf der Überlagerung der abstandsabhängigen Abstoßungs- und Anziehungskräfte.
Bei einer Temperatur von 0 K schwingen die Atome nicht, ihr Abstand ent-spricht dem Ort des Potentialminimums. Bei höherer Temperatur kann sich unser herausgegriffenes Atom in dem durch die Potentialmulde vorgegebenen Abstands-bereich bewegen: es schwingt um seine Mittellage. Je höher die Temperatur ist, desto breiter ist die Potentialmulde, d.h. desto größer ist die Schwingungsam-plitude. In diesem Zusammenhang ist es erwähnenswert, dass uns dieses Poten-tialmodell auch eine Erklärung für die Wärmeausdehnung liefert. Es liegt an der asymmetrischen Form der Potentialmulde. Die Mittellage des Bewegungsberei-ches verschiebt sich bei höherer Energie zu größeren Abständen hin.
Bild 4-16. Vereinfachtes Potentialmodell zur Erklärung des Abstandes und des Schwingungsverhaltens der Atome.
Wir stellen uns nun vor, dass die Energie nicht als Wärme zugeführt wird sondern durch Zusammenstöße zwischen den Primärelektronen und den Atomen. Obwohl die Massen von Atomkern und Elektron extrem unterschiedlich sind, kann Energie von den Elektronen auf die Atome übertragen werden, was zu deren stärkerer Bewegung führt und zwar umso eher, je leichter die Atome sind. Organische Materialien bestehen im Wesentlichen aus Wasserstoff, Stickstoff, Kohlenstoff und Sauerstoff, weshalb sie in dieser Hinsicht besonders gefährdet sind. Wenn es viele Atome betrifft, wäre dies als Temperaturerhöhung messbar. Es ist schwer abzuschätzen, wie hoch die Temperatur werden kann, weil dabei nicht nur die Energiezufuhr und die Probenmasse eine Rolle spielen sondern auch die schwer vorhersehbare Wärmeableitung (abhängig von Probengeometrie, Wärmekontakten innerhalb der Probe und zum Halter, Wärmestrahlung). Bei wärmeempfindlichen Proben ist die Möglichkeit der Temperaturerhöhung zu berücksichtigen, unter Umständen hilft die Benutzung eines Kühlhalters.
Schließlich wollen wir den Fall betrachten, dass die durch die Elektronen zuge-führte Energie größer als die in Bild 4-16 eingezeichnete „Ablöseenergie“ ist. In diesem Fall wird ein Potentialniveau erreicht, auf dem sich das Atom frei bewegen kann. Wenn das eine Vielzahl von Atomen betrifft, schmilzt die Probe. Es kann al-

Kapitel 4 82
lerdings auch nur auf einzelne Atome zutreffen, die dann von ihrem angestammten Platz entfernt werden. Im realen Gitter gibt es solche Gitterleerstellen, deren An-zahl als Teil des thermodynamischen Gleichgewichts mit steigender Temperatur wächst. Stöße mit Elektronen können zusätzliche Leerstellen hervorrufen oder be-stimmte Atome aus dem Gitterverband herauslösen. Häufig betrifft dies Sauerstoff oder Halogene wie Fluor oder Chlor. Es ist leicht einzusehen, dass solche Vor-gänge eine materialabhängige Mindestenergie erfordern (auch als Bindungs-energie bezeichnet). Andererseits benötigt eine solche Wechselwirkung eine (wenn auch sehr kurze) Zeit, d.h. bei sehr hohen Elektronenenergien (und damit –geschwindigkeiten) kann die Wahrscheinlichkeit derartiger Probenschädigungen auch wieder sinken.
Aufgrund der Beweglichkeit der Atome im Kristallgitter können elektronen-strahlinduzierte Defekte auch wieder ausheilen, so dass eine konkrete Vorhersage möglicher Schädigungen schwierig ist.
Amorphe Festkörper („metallische Gläser“) sind häufig thermodynamisch me-tastabil, und eine geringe Energiezufuhr durch den Elektronenstrahl kann zur be-grenzten Kristallisation führen.

5 Wir schalten um auf Elektronenbeugung
Ziel: Ein großer Vorteil des Transmissionselektronenmikroskops ist es, dass auf einfache Weise zwischen der Abbildung von sehr kleinen Strukturen in einer dün-nen Probe und dem Beugungsmuster von den gleichen Strukturen umgeschaltet werden kann. Wir wollen deshalb erklären, wieso überhaupt Elektronenbeugungs-muster entstehen und was an den Linsen innerhalb des Elektronenmikroskops ge-ändert werden muss, damit wir die Beugungsmuster auch sehen können. Schließ-lich wollen wir erläutern, welche materialwissenschaftlichen Erkenntnisse aus den Beugungsmustern erhalten werden können. Dazu ist es notwendig, einige Grund-kenntnisse über den Aufbau der Kristalle zu vermitteln.
5.1 Wieso Beugungsreflexe? Zunächst wollen wir einen kleinen Ausflug in die Wissenschaftsgeschichte unter-nehmen. Bis etwa 1910 waren zwei wichtige Fragen der Physik ungeklärt:
1. Welcher Natur sind die 1895 von Wilhelm Conrad Röntgen32 entdeckten „X-Strahlen“? 2. Sind die Atome eines Festkörpers periodisch in Kristallgittern angeordnet?
Basierend auf Überlegungen von A. Sommerfeld33 hatte Max von Laue34 die Idee, beide Fragen durch ein Experiment zu beantworten: Wenn die Röntgenstrah-len Wellencharakter haben und die Atome in Abständen in der Größenordnung der angenommenen Röntgen-Wellenlänge periodisch angeordnet sind, dann sind bei Durchstrahlung des Kristalls Beugungsreflexe zu erwarten. Und tatsächlich fanden W. Friedrich35 und P. Knipping36 nach dem Experiment keine gleichmäßig ge-schwärzte Fotoplatte sondern einzelne, besonders schwarze Flecken, die Beu-gungsreflexe [5.1, 5.2, 5.3]. Das Experiment wurde 1927 von C. Davisson37 und L.H. Germer38 mit Elektronen an einem Nickelkristall wiederholt [5.4].
Wie kam M. von Laue auf seine Idee? Bild 5-1 soll uns das veranschaulichen. In Bild 5-1a sind acht Atome gezeichnet, die periodisch, d. h. gitterartig angeord-net sind. Das Gitter wird durch die Verbindungslinien hervorgehoben. Davon wählen wir zwei parallele Ebenen aus (Bild 5-1b). Derartige Gitterebenen werden
32 Wilhelm Conrad Röntgen, deutscher Physiker, 1845 – 1923, Nobelpreis für Physik 1901 (erster Physik-Nobelpreis) 33 Arnold Sommerfeld, deutscher Physiker, 1868 - 1951 34 Max von Laue, deutscher Physiker, 1879 – 1960, Nobelpreis für Physik 1914 35 Walter Friedrich, deutscher Biophysiker, 1883 – 1968 36 Paul Knipping, deutscher Physiker, 1883 – 1935 37 Clint Davisson, amerikanischer Physiker, 1881 - 1958 38 Lester Germer, amerikanischer Physiker, 1896 - 1971
DOI 10.1007/978-3-7091-1440-7_5, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 5 84
als Netzebenen bezeichnet. Wir stellen uns diese Ebenen als teilweise durchlässige Spiegel vor, auf die unter flachem Winkel eine Welle (hier: Elektronenwelle) fällt. Die Welle wird teilweise an der vorderen, teilweise an der hinteren Netzebene reflektiert, und wir haben in Reflexionsrichtung zwei Wellen, die sich überlagern, die miteinander interferieren (Bild 5-1c).
Bild 5-1. Beugungsexperiment an Kristallen. a) Periodische Anordnung von Atomen im Kristallgitter. b) Ausgewählte Kristallebenen („Netzebenen“). c) Netzebenen wirken wie halb-durchlässige Spiegel: Die Welle wird teilweise reflektiert.
Auch diese Überlagerung wollen wir uns veranschaulichen, und zwar in Bild 5-2. Die Bilder sind Momentaufnahmen der an den Netzebenen teilweise reflektierten Wellen bei zwei unterschiedlichen Wellenlängen. Die Teilbilder b) und e) zeigen den Moment, in dem ein Wellental die rechte Netzebene erreicht und teilweise reflektiert wird. Der Rest der Welle durchdringt diese Netzebene und wird an der linken reflektiert. Dabei legt sie einen zusätzlichen Weg zurück, trifft dann wieder auf die erste Teilwelle und interferiert mit ihr.
Bild 5-2. Interferenz von Elektronenwellen nach Streuung an Kristallebenen. a) – c) Destruktive Interferenz (Auslöschung). d) – f) Kon-struktive Interferenz bei veränderter Wellenlänge (Verstärkung). Die Bilder sind Momentauf-nahmen der fortschreitenden Wellen. Bei b) bzw. e) hat gerade ein Wellental die rechte Netzebene erreicht und wird reflektiert.
Im Allgemeinen sind die beiden Teilwellen infolge des zusätzlich zurückgelegten Weges der zweiten Teilwelle („Gangunterschied“) gegeneinander phasenverscho-ben, d. h. Wellental trifft nicht auf Wellental. Das Ergebnis ist eine Verminderung der Amplitude der resultierenden Welle. Wir können durch Verändern der Wellen-länge allerdings erreichen, dass doch Wellental auf Wellental trifft, d. h. eine Ver-

Wir schalten um auf Elektronenbeugung 85
stärkung der resultierenden Welle eintritt. Offenbar muss der Gangunterschied gerade ein ganzzahliges Vielfaches der Wellenlänge sein.
Wir wollen nun anhand von Bild 5-3 überlegen, wovon der Gangunterschied zwischen den beiden Teilwellen abhängt. Wir abstrahieren: Die Wellen charakteri-sieren wir durch die Vektoren k0 und k (Wellenzahlvektoren), die in die Aus-breitungsrichtung der einfallenden bzw. der reflektierten Welle zeigen. Den Ab-stand der beiden reflektierenden Netzebenen bezeichnen wir mit d, den Winkel zwischen dem Wellenzahlvektor der einfallenden Welle und der Netzebene mit θ/2. Nach dem Reflexionsgesetz ist er gleich dem Winkel zwischen der Netzebene und dem Wellenzahlvektor der reflektierten Welle.
Bild 5-3. Zur Berechnung des Gangunterschiedes bei Beugung einer Welle an zwei Netzebenen innerhalb eines Kristalls.
Wie bereits in Bild 5-2 dargestellt, wird das eingezeichnete Wellental auch in Bild 5-3 direkt an der rechten Netzebene reflektiert. Im durch diese Netzebene hin-durchgehenden Teil der Welle, der dann an der linken Netzebene reflektiert wird, muss dieses Wellental zweimal die Distanz x zusätzlich zurücklegen. Der Gang-unterschied ist also
2s xΔ = ⋅ . (5.1)
Wir wissen bereits, dass konstruktive Interferenz mit maximaler Verstärkung auf-tritt, wenn dieser Gangunterschied ein ganzzahliges Vielfaches der Wellenlänge λ ist. Mit n als ganzer Zahl gilt damit als Bedingung für ein Intensitätsmaximum:
n 2 xλ⋅ = ⋅ . (5.2)
Da Winkel, deren Schenkel paarweise senkrecht aufeinander stehen, gleich groß sind, ist auch der Winkel zwischen dem eingezeichneten Wellental und der Ab-standsgeraden d zwischen den Netzebenen gleich θ/2. Das Wellental erstreckt sich senkrecht zur Ausbreitungsrichtung. Damit lesen wir aus Bild 5-3 ab:

Kapitel 5 86
sin2
x dθ
= ⋅ (5.3)
bzw.
n 2 sin2
dθ
λ⋅ = ⋅ ⋅ (5.4)
Diese Gleichung ist fundamental für die Beugung und ist als Braggsches39 Gesetz bekannt. Das Modell mit den teilweise reflektierenden Netzebenen stammt von Vater und Sohn Bragg [5.5, 5.6]. Das Braggsche Gesetz gibt an, unter welchen Winkeln Beugungsmaxima („Beugungsreflexe“) zu erwarten sind. In der Regel ist die Wellenlänge bekannt, so dass aus den gemessenen Winkeln die Netzebenenab-stände d berechnet werden können, die ein maßgebliches Kennzeichen der Kristal-le sind.
Welche Größenordnung ist für diese Winkel bei der Elektronenbeugung im Transmissionselektronenmikroskop zu erwarten? In Bild 1-6 war die Wellenlänge in Abhängigkeit von der Beschleunigungsspannung aufgetragen. Für die im Trans-missionselektronenmikroskop üblichen Beschleunigungsspannungen liegt die Wellenlänge bei wenigen Pikometern, sagen wir bei typischerweise 3 pm. Die Netzebenenabstände hängen selbstverständlich vom Material ab, nehmen wir für unsere Abschätzung 0,3 nm an. Mit diesen Werten erhalten wir für das erste Beugungsmaximum (n = 1) einen Winkel von etwa 0,01 rad = 10 mrad ≈ 0,6°. Im Unterschied zur Röntgenbeugung können wir bei derartig kleinen Winkeln
sin2 2
θ θ= (5.5)
setzen und erhalten als „Grundgleichung für die Elektronenbeugung“
n dλ θ⋅ = ⋅ . (5.6)
5.2 Kristallgitter und Netzebenen Wir wollen uns nun der Frage widmen, welche Netzebenenabstände im Kristall zu erwarten sind. Die Elemente und Verbindungen bilden unterschiedliche Kristalle, die Werkstoffspezialisten sprechen von unterschiedlichen „Phasen“. Vorsicht! Diese „Werkstoffphasen“ dürfen wir nicht mit der Phase einer Welle verwechseln. Die Unterschiede zwischen den Kristallen können sich in unterschiedlichen Kri-stallformen und in unterschiedlichen Atomabständen äußern. Zur Kennzeichnung
39 William Hennry Bragg und William Lawrence Bragg: australisch/englische Physiker (Vater und Sohn), 1862 – 1942 bzw. 1890 – 1971, Nobelpreis für Physik 1915

Wir schalten um auf Elektronenbeugung 87
wird eine Elementarzelle eingeführt, deren periodische Aneinanderreihung das gesamte Kristallgitter ergibt. Diese Elementarzelle wird in Form und Größe durch drei Achsen und drei Winkel beschrieben (s. Bild 5-4).
Bild 5-4. Bezeichnung von Achsen und Winkeln in einer Elementarzelle.
Wir wollen die drei Achsen mit a1, a2 und a3 bezeichnen, in der Literatur sind auch a, b und c dafür gebräuchlich. Der Winkel α zwischen a2 und a3 liegt gegenüber der Achse a1 in der a2-a3-Ebene, der Winkel β gegenüber der Achse a2 in der a1-a3-Ebene und der Winkel γ gegenüber der Achse a3 in der a1-a2-Ebene. Aus den Achsen- und Winkelrelationen ergeben sich sieben Kristallsysteme (s. Tabelle 5-1). Das kubische System hat die höchste Symmetrie, das trikline die niedrigste.
Nr. Kristallsystem Achsenlängen Achsenwinkel
1 kubisch a1 = a2 = a3 α = β = γ = 90°
2 tetragonal a1 = a2 ≠ a3 α = β = γ = 90°
3 orthorhombisch a1 ≠ a2 ≠ a3 α = β = γ = 90°
4 hexagonal (auch: trigonal mit
hexagonalen Achsen) a1 = a2 ≠ a3 α = β = 90°, γ = 120°
5 rhomboedrisch (trigonal mit rhomboedrischen Achsen)
a1 = a2 = a3 α = β = γ ≠ 90°
6 monoklin a1 ≠ a2 ≠ a3 α = γ = 90°, β beliebig
7 triklin a1 ≠ a2 ≠ a3 α ≠ β ≠ γ, beliebig
Tabelle 5-1. Definition der sieben Kristallsysteme [5.7].
Die Symmetrieeigenschaften der Kristallgitter werden im Allgemeinen durch Raumgruppen beschrieben (vgl. Tabelle 5-2).

Kapitel 5 88
Kristallsystem Raumgruppen PEARSON-Symbol
kubisch Nr. 195 bis 230 (Anzahl: 36)
cP, cI,cF
tetragonal Nr. 75 bis 142 (Anzahl: 68)
tP, tI
orthorhombisch Nr. 16 bis 74 (Anzahl: 59)
oP, oI, oF, oC, oA, oB
hexagonal
Nr. 168 bis 194 (Anzahl: 27)
hP, hR
rhomboedrisch (auch: trigonal mit hexagonalen oder
rhomboedrischen Achsen)
Nr. 143 bis 167 (Anzahl: 25)
hP, hR
monoklin Nr. 3 bis 15 (Anzahl: 13)
mP, mC, mA, mI
triklin Nr. 1 und 2 (Anzahl: 2)
aP
Tabelle 5-2. Zuordnung der Raumgruppen zu den Kristallsystemen (das Pearson40-Symbol ist eine andere Bezeichnungsweise, aus der das Kristallsystem sofort ersichtlich ist - siehe [5.8]).
Richtungen im Kristall werden durch eine Linearkombination der drei Vektoren a1, a2, a3 beschrieben und durch ein Zahlentripel in eckigen Klammern bezeichnet:
[h k l] entspricht der Richtung h k l⋅ + ⋅ + ⋅1 2 3a a a .
Die Kristallachsen tragen demzufolge die Bezeichnungen [100], [010] und [001]. Außer an den Ecken der Elementarzelle befinden sich im Allgemeinen auch
Atome im Inneren derselben. Zu deren Beschreibung muss entweder zu dem Kri-stallsystem eine zusätzliche Information über die Packungsart gegeben werden (s. Tabelle 5-3), oder die Positionen der inneren Atome werden unter Kenntnis der für die Raumgruppen bekannten Symmetrieeigenschaften aus wenigen Grundposi-tionen berechnet. Die Ermittlung der Raumgruppe und dieser Grundpositionen sind Ziel der Bestimmung der Kristallstruktur einer unbekannten Phase.
In der elektronenmikroskopischen Beugungspraxis ist die Zuordnung eines gemessenen Beugungsmusters zu einer bekannten Phase die weitaus häufigste Aufgabenstellung. Wir wollen an einem einfachen Beispiel erklären, wie die für die Berechnung eines Beugungsmusters notwendigen kristallografischen Struktur-daten (Abmessungen der Elementarzelle und Atompositionen innerhalb derselben) aus den in der Literatur üblichen Angaben gewonnen werden können. Als Beispiel benutzen wir Natriumchlorid (NaCl). Zunächst brauchen wir eine Datenbasis oder eine Literaturstelle, die Informationen zur angenommenen Phase NaCl enthält. Wir können im „Pearson“, einem Buch mit Zusammenstellung der kristallografi-
40 Frederic Pearson Treadwell, amerikanisch-schweizer Chemiker, 1857 - 1918

Wir schalten um auf Elektronenbeugung 89
schen Daten bekannter Phasen [5.8] oder im Internet in der „Inorganic Crystal Structures Database“ [5.9] nachschauen, wozu eine Lizenz erforderlich ist. Bei neuen, d. h. erst vor kurzer Zeit entdeckten Phasen, hilft oft ein Blick in die aktuellen wissenschaftlichen Publikationen weiter.
Nr. Packungsart Atom-zahl
Atompositionen Schema der
Elementarzelle
1 primitiv
(Raumgruppen P…) 1 0;0;0
2 raumzentriert
Raumgruppen I…) 2
0;0;0 1/2; 1/2; 1/2
3 flächenzentriert
(allseitig)
Raumgruppen F…) 4
0;0;0 1/2; 1/2; 0
1/2; 0; 1/2 0; 1/2; 1/2
4
basisflächenzentriert (Basisfläche C =
Grundfläche)
Raumgruppen C…)
2 0;0;0 1/2; 1/2; 0
5 Diamantgitter 8
0;0;0 1/2; 1/2; 0
1/2; 0; 1/2 0;1/2; 1/2
1/4; 1/4; 1/4 1/4; 3/4; 1/4
3/4; 1/4; 3/4 1/4; 3/4; 3/4
6 hexagonal dichteste
Kugelpackung
Raumgruppen R…)
2
2/3; 1/3;1/4 1/3; 2/3;3/4
Tabelle 5-3. Zahl und Anordnung der Atome in der Elementarzelle bei verschiedenen Packungsarten.
Für NaCl finden wir folgende Daten: Zellparameter: 5,64 Å; 5,64 Å; 5,64 Å; 90.0°; 90,0°; 90,0°
Volumen: 179,41 Å3
Raumgruppe: Fm-3m (225); Pearson Symbol: cF8

Kapitel 5 90
Element Wyckoff41 Symbol x y z
Na 4a 0,000 0,000 0,000
Cl 4b 0,500 0,500 0,500
Mitunter werden die Maßeinheiten auch weggelassen. In der ersten Zeile finden wir als Zellparameter die Längen der Achsen a1, a2, a3 (auch als Gitterkonstanten bezeichnet) und die Winkel α, β und γ zwischen den Achsen. In der zweiten Zeile steht das Volumen der Elementarzelle, das aus den Zellparametern folgt. Die dritte Zeile beinhaltet die Raumgruppe (F: Flächenzentrierung) mit Nummer und das Pearson-Symbol, welches aussagt, dass es sich um ein kubisch flächenzentriertes System mit acht Atomen in der Elementarzelle handelt.
Wie errechnen wir aber nun die Positionen der acht Atome? Dabei helfen uns die Angaben in den beiden unteren Zeilen in Verbindung mit Kenntnis der Raum-gruppe. Für den Praktiker ist es am einfachsten, wenn er dafür ein geeignetes Computerprogramm zur Verfügung hat. Er kann aber auch die „International Tables for Crystallography” [5.7] benutzen. Dort sind die Berechnungsvorschrif-ten für die Atompositionen für alle Raumgruppen aufgelistet. In Bild 5-5 ist als Beispiel eine der Seiten für die Raumgruppe 225 gezeigt.
Die mit A bezeichnete Zeile beschreibt, dass es sich um eine flächenzentrierte Elementarzelle handelt. Zu jeder der später folgenden Positionen sind die Tripel (0,0,0), (0,½,½), (½,0, ½) bzw. (½,½,0) zu addieren, d. h. aus einer Positions-angabe werden vier Atompositionen ausgerechnet. Für die Positionsangaben be-nötigen wir die zwei letzten Zeilen im Datensatz. Für Natrium gilt: x = 0, y = 0, z = 0. Diese Werte könnten wir in die in Bild 5-5 unter A stehenden Vervielfälti-gungsvorschriften (1) bis (48) einsetzen und die Dopplungen streichen ( x x= − ). Wir sehen aber, dass in unserem Fall (0,0,0) alle Vervielfältigungen immer nur auf das Tripel (0,0,0) führen, d. h. nur auf ein Na-Atom. Mit Berücksichtigung der Flächenzentrierung erhalten wir für die Na-Positionen:
(0,0,0) + (0,0,0) = (0,0,0), (0,½,½) + (0,0,0) = (0,½,½), (½,0, ½) + (0,0,0) = (½,0, ½) und (½, ½,0) + (0,0,0) = (½, ½,0).
Wir können uns das Ganze etwas vereinfachen, indem wir die Wykoff-Sym-bole beachten. Für Na gilt 4a. 4 ist die Zahl der Na-Atompositionen und a ist eine Angabe zur Symmetrie. Wir finden das Symbol am linken Rand der unteren Zeile von Block B in Bild 5-5. Dort ist sofort die eine Positionsangabe (0,0,0) ab-zulesen. Analog folgt für das Chlor-Atom aus der oberen Zeile von Block B: (½,½,½) und für die vier Cl-Positionen:
(0,0,0) + (½,½,½)=(½,½,½), (0,½,½) + (½,½,½)= (½,0,0),
41 Ralph Walter Graystone Wyckoff, amerikanischer Kristallograph, 1897 - 1994
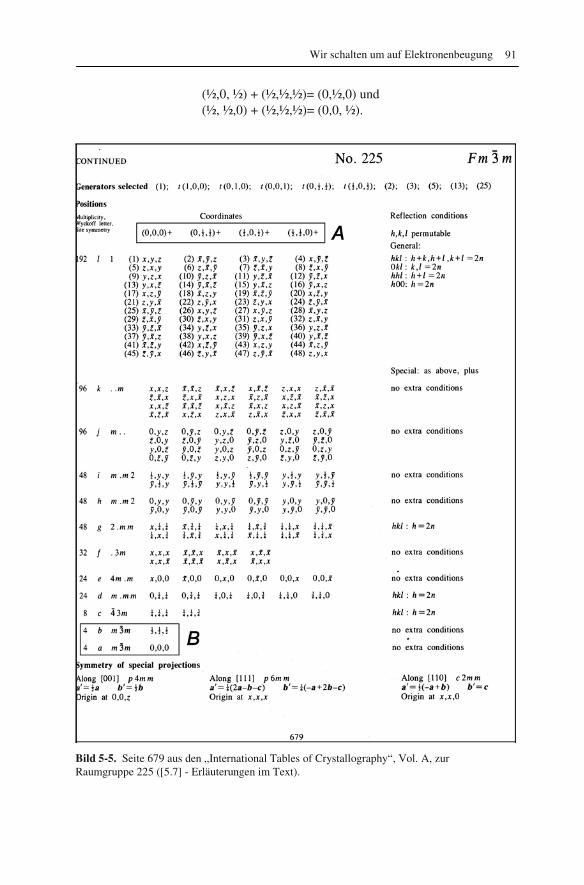
Wir schalten um auf Elektronenbeugung 91
(½,0, ½) + (½,½,½)= (0,½,0) und (½, ½,0) + (½,½,½)= (0,0, ½).
Bild 5-5. Seite 679 aus den „International Tables of Crystallography“, Vol. A, zur Raumgruppe 225 ([5.7] - Erläuterungen im Text).

Kapitel 5 92
Hier verwundert, dass für die Summe ½ + ½ = 0 geschrieben wird. Um dies zu verstehen, müssen wir beachten, was beispielweise mit der Angabe x = ½ gemeint ist. Dies ist bezogen auf die Achsenlänge a1, d. h. exakt müsste x = ½ ⋅ a1 ge-schrieben werden. Wegen der Translationseigenschaft des Gitters wiederholt sich die Elementarzelle; das Atom an Position (1,0,0) ist dasselbe wie das an Position (0,0,0). Dies gilt analog für die beiden anderen Achsen, d. h. die Koordinaten der Atompositionen beziehen sich immer auf die Achsenlängen und sind deshalb Zah-len zwischen 0 und <1.
Ein weitverbreiteter Standard zur Speicherung und zum Austausch von Kri-stallstrukturdaten ist das CIF-Format. Diese ASCII-Textdateien können mit jedem Texteditor gelesen werden und erklären sich mit dem in diesem Abschnitt erlang-ten Wissen „von selbst”.
Zur Beschreibung der Netzebenen gehen wir von einer beliebigen Elementar-zelle aus. Eine Ebene darin wird durch ein Zahlentripel (hkl) beschrieben, wobei 1/h, 1/k und 1/l die Schnittpunkte der Ebene mit den Achsen a1, a2 bzw. a3 der Zelle sind. Verläuft eine Ebene parallel zu einer Achse, so liegt der Schnittpunkt beider im Unendlichen. Der Kehrwert ist Null. Beispielsweise ist also die (100)-Ebene die a2-a3-Ebene. In Bild 5-6 sind Beispiele für Netzebenen im kubischen, tetragonalen und hexagonalen Kristallsystem gezeichnet.
Bild 5-6. Verschiedene Netzebenen im a) kubischen, b) tetragonalen und c) hexagonalen Kristallsystem.
Die Indizes h, k, l werden auch als Millersche42 Indizes bezeichnet. Manchmal wird behauptet, dass die Richtung [hkl] senkrecht auf der Netzebene (hkl) steht. Doch Vorsicht, allgemein gilt das nur im kubischen System.
Für die Beugungswinkel sind die Netzebenenabstände interessant. Wir berück-sichtigen nunmehr die Indizierung der Netzebenen und präzisieren die Gleichung (5.6):
n hkl hkldλ θ⋅ = ⋅ , (5.7)
d. h. zu jedem Netzebenenabstand gehört ein Beugungswinkel.
42 William Hallowes Miller, britischer Kristallograph, 1801 - 1880

Wir schalten um auf Elektronenbeugung 93
kubisch: a1 = a2
= a3 1
2 2 2hkla
dh k l
=+ +
tetra-gonal: a1 = a2
2 2 2
2 21 3
1hkld
h k l
a a
=+
+
ortho-rhom-bisch:
2 2 2
2 2 21 2 3
1hkld
h k l
a a a
=
+ +
hexago-nal: a1 = a2 ( )
22 2
2 21 3
1
4
3
hkldl
h h k ka a
=
+ ⋅ + +⋅
rhombo-edrisch:
a1 = a2 = a3
α = β = γ
( )
( )
2 22 2
2 21 1 11 1
2 22 2 2
1
cotsin sin
mit
cos cot 2 1 cos
und
cos coscot cos cos cos
sin
hkldh h k A l
a a aa a
A h k l
α
α ε
ε α α
ε αα ε α α
α
=
⋅+ − + + +
⋅ ⋅
= ⋅ ⋅ −
−= ⋅ ⋅ − − − ⋅ +
mono-klin:
22 2
2 21 31 2
1
cotsin
hkldh k h l
a aa a
β
β
=
⋅+ + − +
⋅
triklin:
22 2
21
2
1 2
2 2 2 22
1 2 3
2 2
1
mit
cot
sin
und
cot cos cos cos cos cos
sin sin sin sin
sowie
cos 2 cos cos cos coscos
sin
hkl
hk hkl
hk
hkl
dh
C Ca
h kC
a a
h k lC
a a a
γ
γ
γ ε β β ε β
ε γ ε ε
α α β γ βε
γ
=
+ +
⋅= − +
⋅
⋅ ⋅ − − ⋅ −= − +
⋅ ⋅ ⋅ ⋅
− ⋅ ⋅ ⋅ +=
Tabelle 5-4. Berechnungsformeln für die Netzebenenabstände dhkl in den sieben Kristall-systemen.

Kapitel 5 94
Bei der Berechnung der Netzebenenabstände dhkl muss die Form der Elemen-tarzelle berücksichtigt werden, d. h. es müssen für die verschiedenen Kristallsyste-me unterschiedliche Formeln benutzt werden. In Tabelle 5-4 sind diese Formeln zusammengestellt, die Grundlagen dafür sind in Abschnitt 10.8 erläutert.
Für das hexagonale System gibt es hinsichtlich der Indizierung eine Besonder-heit: Ebenen und Richtungen werden häufig mit vier Indizes gekennzeichnet. Dies ist nicht zwingend notwendig, erleichtert aber das Erkennen kristallografisch gleichartiger Richtungen im hexagonalen System. Um das zu verstehen, stellen wir uns zunächst ein kubisches System vor. Die Würfelachsen [1 0 0], [0 1 0], [0 0 1] sind kristallografisch gleichartig. Das ist sofort zu sehen: Die Indizierun-gen enthalten die gleichen Zahlenkombinationen. Anders sieht es bei der drei-zähligen Indizierung [uvw] im hexagonalen System aus. Gleichberechtigt mit den beiden Achsen [1 0 0] und [0 1 0] ist die Richtung [1 1 0]. Wird eine vierzählige Indizierung [qrst] nach der Vorschrift
( ) ( ) ( )1 1 1
2 , r 2 , - , 3 3 3
q u v v u s u v t w= ⋅ − = ⋅ − = + = (5.8)
eingeführt, so erhalten wir anstelle von [1 0 0] nunmehr [2/3 -1/3 -
1/3 0] bzw. nach Erweiterung mit drei: [2 -1 -1 0]. Die Richtung [0 1 0] wird zu [-1 2 -1 0] und die Richtung [1 1 0] zu [1 1 -2 0]. Wir sehen, vom Vorzeichen abgesehen treten die gleichen Zahlen auf. Weitere Details der Indizierung hexagonaler Systeme werden u. a. in [5.10] und [5.11] diskutiert.
5.3 Feinbereichs- und Feinstrahlbeugung Wir wollen uns nun wieder der Praxis am Elektronenmikroskop zuwenden. Zu-nächst soll erläutert werden, was an den Linsen geändert werden muss, wenn wir das Beugungsmuster einer kristallinen Probe auf dem Beobachtungsschirm sehen wollen. Bisher sahen wir ein Bild der Probe, erzeugt durch eine mehrstufige Ab-bildung. Die Brennweiten der Projektivlinsen waren so abgestimmt, dass die reelle Zwischenbildebene des Objektivs auf dem Schirm zu sehen ist.
Aus Abschnitt 5.1 wissen wir, dass Beugungsmaxima unter ganz bestimmten Winkeln zu beobachten sind. Wenn wir die Ausbreitungsrichtung der Welle für eine geometrisch-optische Betrachtung als „Strahl“ interpretieren, wird ein Beu-gungsmaximum durch ein Bündel von Strahlen repräsentiert, die parallel zuein-ander in die Objektivlinse einfallen. Derartige parallele Strahlen werden in der bildseitigen Brennebene des Objektivs vereinigt (vgl. Abschnitt 2.7.2), erzeugen dort also einen hellen Beugungsreflex. Bild 5-7 soll das verdeutlichen.

Wir schalten um auf Elektronenbeugung 95
Bild 5-7. Objektivumgebung bei Feinbereichsbeugung mit paralleler Beleuchtung. a) Schnitt entlang der optischen Achse. b) Perspektivische Darstellung.
Um das Beugungsbild zu sehen, muss die Brennweite des Projektivlinsensystems so eingestellt werden, dass die bildseitige Objektivbrennebene auf dem Beobach-tungsschirm abgebildet wird. Oft erfolgt dieses Umstellen durch Hinzuschalten einer zusätzlichen Beugungslinse, der Experimentator hat lediglich den entspre-chenden Beugungsknopf (englisch: Diffraction) auf dem Bedienfeld zu drücken.
Zwei Fragen sind noch zu beantworten: Welche Voraussetzungen müssen er-füllt sein, damit wir „scharfe“ Reflexe sehen, und wie wird aus dem Abstand eines Beugungsreflexes vom Zentrum (d. h. vom Reflex der ungebeugten Welle) der Beugungswinkel ermittelt?
Die Voraussetzung für scharfe Reflexe haben wir stillschweigend bereits in Bild 5-7 eingezeichnet: Die kristalline Probe wird mit einer ebenen Welle be-strahlt, d. h. wir beleuchten parallel. Diese parallele Beleuchtung wird durch geeignete Erregung der Kondensor-2-Linse (d. h. durch Betätigen des „Intensity“-Knopfes – vgl. Abschnitt 2.7.1) eingestellt.
Für die Ermittlung der Netzebenenabstände nach dem Braggschen Gesetz ist der Beugungswinkel θ maßgebend. Fällt ein paralleles Strahlenbündel unter einem Winkel θ zur optischen Achse in das Objektiv mit der Brennweite fObj ein, so schneiden sich die Strahlen in der bildseitigen Brennebene im Abstand rf = θ ⋅ fObj von der optischen Achse (vgl. Bild 2-18). Das Projektivlinsensystem vergrößert diesen Abstand zusätzlich um MPro:
Obj ProObj Pro
bzw. r
r f Mf M
θ θ= ⋅ ⋅ =⋅
. (5.9)
Den Abstand r vom zentralen Reflex (der die Position der ungebeugten Welle angibt) messen wir im Beugungsbild auf dem Schirm, der Fotoplatte oder dem Kamerabild auf dem Monitor. Wenn wir sehr präzise messen, finden wir, dass wegen des Öffnungsfehlers der Objektivlinse die Messwerte systematisch nach unten von den theoretisch erwarteten abweichen und zwar umso mehr, je größer der Beugungswinkel ist.
In der Praxis wird das Produkt aus Objektivbrennweite und Vergrößerung durch die Projektivlinsen als Kameralänge L bezeichnet:

Kapitel 5 96
r
Lθ = . (5.10)
bzw. für zusammengehörige Reflexabstände und Beugungswinkel:
hklhkl
r
Lθ = . (5.11)
Befänden sich keine Linsen zwischen Probe und Beobachtungsschirm, wäre L der Abstand zwischen den beiden (vgl. Bild 5-8).
Bild 5-8. Erläuterung des Begriffs „Kameralänge“ L. Bei der Elektronenbeugung im TEM ist θ << 1.
Wenn Objektivbrennweite und Projektivvergrößerung bekannt sind, kann daraus die Kameralänge berechnet werden. Sie wird auch auf dem Mikroskopmonitor nach Umschalten auf Beugung angezeigt. Wir sehen aber auch, dass die Kamera-länge von der Brennweiteneinstellung des Objektivs abhängt und somit u. U. vari-ieren kann. Der Praktiker geht deshalb einen anderen Weg: Er misst die Kamera-länge anhand des Beugungsdiagramms einer bekannten Substanz. In diesem Zu-sammenhang lohnt sich ein Blick zurück auf die präzisierte „Grundgleichung für die Elektronenbeugung“ (5.7). Wir setzen n = 1 (1. Beugungsmaximum) und be-rücksichtigen (5.11):
bzw. hklhkl hkl hkl
rd L d r
Lλ λ= ⋅ ⋅ = ⋅ . (5.12)
Wichtig ist also nicht die Kameralänge allein, sondern das Produkt aus Kamera-länge und Wellenlänge. Natürlich könnten wir die Wellenlänge aus der Beschleu-nigungsspannung berechnen, aber wenn wir sowieso an einem Standard messen, können wir auch gleich das Produkt bestimmen und damit evtl. Unsicherheiten der Beschleunigungsspannungsangabe ausschließen. Das Produkt λ⋅L wird auch als Gerätekonstante bezeichnet und oft in Å⋅mm angegeben. Auf die praktische Aus-

Wir schalten um auf Elektronenbeugung 97
führung einer solchen Kalibrierungsmessung kommen wir später im Abschnitt 5.4 zurück.
Bisher haben wir von der „parallelen Beleuchtung“ der Probe gesprochen. Ein Vorteil der Elektronenbeugung im Transmissionselektronenmikroskop ist aber ge-rade, dass wir „besonders kleine“ Bereiche der Probe auswählen können. Wie geht das? Am einfachsten vorstellbar ist es, nur einen kleinen Teil der Probe zu be-strahlen. Dazu müssen wir das Strahlenbündel mit dem Kondensor 2 „zusammen-ziehen“, d. h. die Probe wird nicht mehr parallel beleuchtet, die Reflexe werden unscharf. Wollen wir bei paralleler Beleuchtung nur einen Teil der Probe be-strahlen, so brauchen wir entweder eine dritte Kondensorlinse oder (und das ist zurzeit der üblichere Weg) eine Gesichtsfeldblende, mit der wir die Größe des be-leuchteten Probenbereichs begrenzen. Naheliegend wäre es, den Halter für diese Blende unmittelbar über der Probe anzuordnen. Die Blendenbohrung sollte kleiner als 1 μm sein, um solch kleine Bereiche auch auswählen zu können. Beides ist in der Praxis nicht oder nur schwer zu realisieren. Zur Einstellung der euzentrischen Höhe wird die Probe längs der optischen Achse bewegt, der Blendenhalter müsste mitbewegt werden. Auch Bohrungen von 1 μm Durchmesser sind schwer zu be-herrschen, denken wir nur an evtl. Verschmutzungen. Der bessere Weg ist, die Blende in der ersten reellen Zwischenbildebene anzuordnen. Auch hier wirkt sie als Gesichtsfeldblende, es ist mehr Platz und die Blendenbohrung wird mit dem Objektiv „rückwärts“ auf die Probe verkleinert. Bei einer angenommenen Objek-tivvergrößerung von 100 genügt also ein Lochdurchmesser von 0,1 mm, um De-tails von 1 μm Größe auszuwählen. Von dieser Auswahl eines „feinen Bereichs“ rührt auch die Bezeichnung dieser Methode her: Feinbereichsbeugung (englisch: Selected Area Electron Diffraction – SAED). Sie wurde bereits 1936 von H. Boersch [5.12] vorgeschlagen.
Leider sinkt mit abnehmendem Blendendurchmesser auch die Gesamtintensität der Elektronen in der Beobachtungsebene, das Beugungsmuster ist dann schwer erkennbar und eine gezielte Orientierung einer bestimmten kristallografischen Richtung zum Elektronenstrahl fast unmöglich. Hinzu kommt, dass aufgrund der Abbildungsfehler und der Toleranzen bei der Justage der elektronenoptischen Ein-heiten zueinander das Gesichtsfeld auf der Probe beim Umschalten zwischen Ab-bildung und Beugung um mehr als 10 nm verschoben sein kann. Bei Auswahl von Bereichen unter ca. 50 nm Größe wäre also die Zuordnung des beugenden Be-reiches zum Bild der Probe nicht mehr sicher. Aus diesen Gründen liegt im All-gemeinen die untere Grenze des auswählbaren Bereiches bei der Feinbereichs-beugung bei etwa 50 nm.
Feinbereichsbeugung ist sowohl an polykristallinem Gefüge (Ringdiagramm) als auch an einzelnen Kristallen (Punktdiagramm) möglich. Punktdiagramme sind aber nur zu beobachten, wenn der Kristall größer als der mit der Blende ausge-wählte Bereich ist und durch die gesamte Probendicke reicht, d. h. wenn nur ein einzelner Kristall erfasst wird. Es gibt auch Zwischenstadien, deren Aussehen von dem Verhältnis aus der Größe des erfassten Probenbereiches und der Kristal-litgröße, d. h. der Anzahl n der zum Beugungsbild beitragenden Kristallite ab-

Kapitel 5 98
hängt. Ist diese Zahl groß (n >> 10) und sind alle Kristallorientierungen statistisch gleichverteilt, so entstehen Ringdiagramme mit in sich geschlossenen Ringen. Symmetrisch verteilte Intensitätsfluktuationen auf einzelnen Ringen weisen auf kristallografische Vorzugsorientierungen (Texturen) hin (vgl. Abschnitt 6.3). Die Ringe sind umso schärfer, je größer die einzelnen Kristallite sind. Bei Verrin-gerung von n sind die Ringe nicht mehr geschlossen und lösen sich in Einzel-reflexe auf. Liegt n zwischen 1 und 10, so ist mit mehreren überlagerten Punktdia-grammen zu rechnen, was die Auswertung erschweren kann.
Was können wir tun, um auch „echte Nanostrukturen“ mit Hilfe der Elektro-nenbeugung untersuchen zu können?
Wir können auf die parallele Beleuchtung verzichten und das Strahlenbündel auf der Probe zu einem Spot zusammenziehen (Feinstrahlbeugung). Die Größe dieses Spots kann durch die Brennweite der Kondensorlinse 1 beeinflusst werden („Spot Size“). Anstelle der „scharfen“ Beugungsreflexe erhalten wir Beugungs-scheibchen, deren Durchmesser von der Bestrahlungsapertur (Konvergenzwinkel) abhängt. Der Konvergenzwinkel wird durch die Kondensor-2-Blende bestimmt (s. Bild 2-16). Wir stellen uns zunächst einen sehr kleinen Konvergenzwinkel von nur wenigen Millirad vor (s. Bild 5-9).
Bild 5-9. Objektivumgebung bei Feinstrahlbeugung mit kleinem Konvergenzwinkel. a) Schnitt entlang der optischen Achse. b) Perspektivische Darstellung.
In diesem Fall sind die Abweichungen der Einfallsrichtungen vom Braggwinkel kleiner als der Anregungsfehler bei der Elektronenbeugung (darauf kommen wir später zurück), d. h. wenn der Konvergenzwinkel hinreichend klein ist, sind die Positionen der Beugungsreflexe durch die Zentren der Beugungsscheibchen gege-ben und das Beugungsbild wird wie ein Punktdiagramm bei der Feinbereichs-beugung ausgewertet. Das Verfahren wird als auch als Nanobeugung (englisch: Nano Beam Electron Diffraction – NBED) bezeichnet. Ringdiagramme wären bei dieser Methode nicht sinnvoll, da durch die konvergente Beleuchtung gerade nur ein Kristallit getroffen werden soll. Wenn die Kristallite kleiner als die Proben-dicke sind gibt es ein Problem mit übereinanderliegenden Kristalliten: In der Pro-jektionsebene treffen wir scheinbar nur einen Kristall, in Wirklichkeit aber meh-rere mit unterschiedlicher Kristallorientierung oder/und Phasen. Wir erhalten also mehrere überlagerte Punktdiagramme, was die Auswertung des Beugungsmusters sehr erschweren kann.

Wir schalten um auf Elektronenbeugung 99
Was passiert, wenn wir größere Konvergenzwinkel benutzen, d. h. eine größere Kondensor-2-Blende einsetzen? Zur Beantwortung dieser Frage schauen wir auf Bild 5-10.
Zunächst beobachten wir ein sehr viel größeres zentrales Beugungsscheibchen. Das Besondere ist, dass dieses Beugungsscheibchen nunmehr eine „innere Struk-tur“ aufweist. Der Grund ist folgender: Wir bieten nunmehr dem Kristallgitter einen Kegel sehr unterschiedlicher Elektroneneinfallsrichtungen an. Eine davon (sie ist in Bild 5-10a mit * gekennzeichnet) erfüllt für eine Netzebenenrichtung ge-rade die Bragg-Gleichung, d. h. es entsteht ein intensitätsreiches Beugungsmaxi-mum.
Bild 5-10. Objektiv-umgebung bei Feinstrahl-beugung mit großem Konvergenzwinkel. a) Schnitt entlang der optischen Achse. b) Perspektivische Darstellung.
Die mit * gekennzeichnete Gerade repräsentiert eine Ebene, die senkrecht auf der Papierebene in Bild 5-10a steht. Diese Gerade ist die Schnittlinie der Zei-chenebene mit der Ebene, in der alle die Elektronenstrahlen liegen, die das Bragg-sche Gesetz erfüllen (vgl. Bild 5-10b). Demzufolge sehen wir auf dem Beobach-tungsschirm ein Linienpaar: Eine dunkle Linie in der geradlinigen Fortsetzung der „Bragg-Richtung“ und eine helle Linie in einem Abstand, der dem Bragg-Winkel θ entspricht. Diese Linien werden als Kikuchi43-Linien (oder -bänder) bezeichnet [5.13].
5.4 Was können wir aus den Feinbereichs-Beugungs-mustern lernen? Nun ist es an der Zeit zu überlegen, welche werkstoffwissenschaftlich relevanten Ergebnisse die Elektronenbeugung liefert. Dazu kommen wir zur Feinbereichs-beugung zurück und behandeln zunächst die Ringdiagramme, die bei Elektronen-beugung an polykristallinen Gefügen entstehen, wenn sehr viele kleine Kristallite an der Beugung teilnehmen und deren Orientierungen statistisch gleichverteilt sind.
43 Seishi Kikuchi, japanischer Physiker, 1902 – 1974, entdeckte und erklärte 1928 die beschriebenen Bänder

Kapitel 5 100
5.4.1 Radien in Ringdiagrammen
Jede Werkstoffphase besitzt in der Aufeinanderfolge der möglichen Netzebenen-abstände eine Art „Fingerabdruck“, der durch die Kristallstruktur vorgegeben ist und nach den Gleichungen in Tabelle 5-4 berechnet werden kann. Nach dem Braggschen Gesetz (5.12) können wir aus den Ringradien rhkl die Netzebenenab-stände dhkl und damit die Fingerabdrücke der Phasen berechnen, wenn die Geräte-konstante λ⋅L bekannt ist.
Wir wollen uns deshalb zunächst mit der Bestimmung der Gerätekonstante be-fassen. Notwendig ist ein elektronentransparentes Präparat einer bekannten Sub-stanz, deren Kristallgitter auch in diesem extrem dünnen Zustand nicht oder zu-mindest nur sehr wenig verformt ist. Oft wird eine Probe benutzt, die kleine, etwa 10 nm bis 20 nm große Goldinseln auf einem dünnen Kohlenstofffilm enthält. Derartige Präparate können als Testobjekte für die Elektronenmikroskopie von Lieferfirmen für elektronenmikroskopisches Zubehör gekauft werden. Bild 5-11 zeigt das Elektronenbeugungsdiagramm einer solchen Probe.
Auf Fotos können die Ringradien mit einem Lineal oder fotometrisch gemessen werden. Beim Messen von Abständen in vergrößerten Bildern muss die Vergröße-rung bekannt sein, um aus den Messwerten die Abstände in der Probe berechnen zu können. Die Gerätekonstante kann als Maß für die Vergrößerung bei Beu-gungsbildern verstanden werden, d. h. ihr Wert ist nur für eine Einstellung ein-schließlich evtl. fotografischer Nachvergrößerung gültig.
Bild 5-11. Elektronenbeu-gungsdiagramm von einer feinkristallinen Goldprobe mit überlagertem Polarkoordina-tensystem. Der zentrale (000)-Reflex ist oft so hell, dass er bei der Aufnahme alles über-strahlt, evtl. auch die CCD-Kamera beschädigt. Deshalb wird vor der Aufnahme des Bildes oberhalb des Be-obachtungsschirms bzw. der Kamera eine kleine Spitze („Beam Stopper“) einge-fahren, die diesen hellen Spot abdeckt. Diese Spitze ist im Bild von rechts eingeschoben worden.
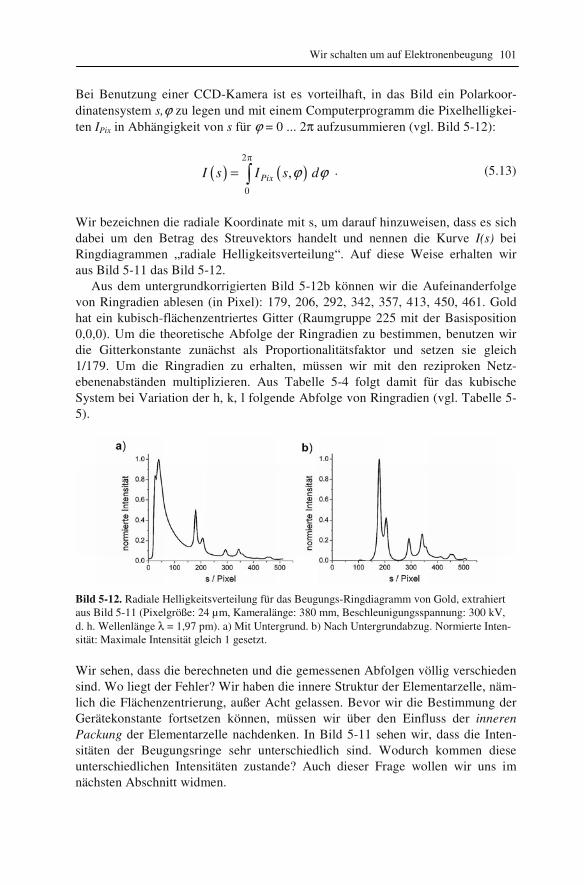
Wir schalten um auf Elektronenbeugung 101
Bei Benutzung einer CCD-Kamera ist es vorteilhaft, in das Bild ein Polarkoor-dinatensystem s,ϕ zu legen und mit einem Computerprogramm die Pixelhelligkei-ten IPix in Abhängigkeit von s für ϕ = 0 ... 2π aufzusummieren (vgl. Bild 5-12):
( ) ( )2
0
, PixI s I s dϕ ϕ= . (5.13)
Wir bezeichnen die radiale Koordinate mit s, um darauf hinzuweisen, dass es sich dabei um den Betrag des Streuvektors handelt und nennen die Kurve I(s) bei Ringdiagrammen „radiale Helligkeitsverteilung“. Auf diese Weise erhalten wir aus Bild 5-11 das Bild 5-12.
Aus dem untergrundkorrigierten Bild 5-12b können wir die Aufeinanderfolge von Ringradien ablesen (in Pixel): 179, 206, 292, 342, 357, 413, 450, 461. Gold hat ein kubisch-flächenzentriertes Gitter (Raumgruppe 225 mit der Basisposition 0,0,0). Um die theoretische Abfolge der Ringradien zu bestimmen, benutzen wir die Gitterkonstante zunächst als Proportionalitätsfaktor und setzen sie gleich 1/179. Um die Ringradien zu erhalten, müssen wir mit den reziproken Netz-ebenenabständen multiplizieren. Aus Tabelle 5-4 folgt damit für das kubische System bei Variation der h, k, l folgende Abfolge von Ringradien (vgl. Tabelle 5-5).
Bild 5-12. Radiale Helligkeitsverteilung für das Beugungs-Ringdiagramm von Gold, extrahiert aus Bild 5-11 (Pixelgröße: 24 μm, Kameralänge: 380 mm, Beschleunigungsspannung: 300 kV, d. h. Wellenlänge λ = 1,97 pm). a) Mit Untergrund. b) Nach Untergrundabzug. Normierte Inten-sität: Maximale Intensität gleich 1 gesetzt. Wir sehen, dass die berechneten und die gemessenen Abfolgen völlig verschieden sind. Wo liegt der Fehler? Wir haben die innere Struktur der Elementarzelle, näm-lich die Flächenzentrierung, außer Acht gelassen. Bevor wir die Bestimmung der Gerätekonstante fortsetzen können, müssen wir über den Einfluss der inneren Packung der Elementarzelle nachdenken. In Bild 5-11 sehen wir, dass die Inten-sitäten der Beugungsringe sehr unterschiedlich sind. Wodurch kommen diese unterschiedlichen Intensitäten zustande? Auch dieser Frage wollen wir uns im nächsten Abschnitt widmen.

Kapitel 5 102
h k l 2 2 2h k l+ + 2 2 2179 h k l⋅ + + Gemessenes s / Pixel
1 0 0 1 179 179
1 1 0 2 253 206
1 1 1 3 310 292
2 0 0 4 358 342
2 1 0 5 400 357
2 1 1 6 438 413
2 2 0 8 506 450
2 2 1 9 537 461
Tabelle 5-5. Gegenüberstellung der gemessenen „Fingerabdrücke“ für die Abfolge der Ringradien in Bild 5-11 mit den Erwartungen für eine kubisch primitive Elementarzelle.
5.4.2 Auslöschungsregeln
In Bild 5-13 sind zwei kubische Elementarzellen dargestellt. In der primitiven Zel-le (Teilbild a) sind nur die Ecken des Würfels von Atomen besetzt, d. h. die Ele-mentarzelle enthält nur ein Atom. (Die restlichen sieben gezeichneten Atome ge-hören jeweils zu einer anderen, angrenzenden Elementarzelle.)
Für das 1. Beugungsmaximum (n = 1, kleinster möglicher Beugungswinkel) gilt mit dem Netzebenenabstand d der primitiven Elementarzelle nach dem Bragg-schen Gesetz (5.6)
prim d
λθ = . (5.14)
Bild 5-13. Beugung an einer a) primitiven b) flächenzentrierten Elementarzelle.
In der flächenzentrierten Elementarzelle (Bild 5-13b) sind zusätzliche Atome im Schnittpunkt der Flächendiagonalen eingeschoben, der Abstand der reflektieren-den Netzebenen verkürzt sich auf die Hälfte. Für den kleinsten Beugungswinkel gilt

Wir schalten um auf Elektronenbeugung 103
2
/ 2fz d d
λ λθ
⋅= = , (5.15)
d. h. der erste Beugungsreflex erscheint, verglichen mit dem der primitiven Zelle, in doppeltem Abstand. Der erste Reflex der primitiven Zelle ist „ausgelöscht“.
Wie können wir solche Auslöschungsregeln allgemein berechnen? Dazu müs-sen wir uns überlegen, wodurch die Intensitäten der Beugungsreflexe bestimmt werden. Bisher haben wir nur die Beugungswinkel berechnet. Wir müssen unser Modell von den reflektierenden Netzebenen präzisieren. Wir haben nicht nur zwei sondern eine Vielzahl von Wellen, die an den Gitteratomen gestreut werden und sich überlagern (→ Abschnitt 10.9).
Die Intensität der Beugungsreflexe wird durch das Streuvermögen der einzel-nen Atome in den Netzebenen und die atomare Besetzungsdichte der Netzebenen bestimmt. Diese Einflüsse werden im Strukturfaktor zusammengefasst. Er ist netz-ebenenspezifisch und hat deshalb den Index hkl, der die für den Beugungseffekt verantwortliche Netzebenenschar hkl kennzeichnet. In → Abschnitt 10.9 wird ge-zeigt, dass für den Strukturfaktor
( )2 ie j j jx h y k z l
hkl jj
F f− ⋅ ⋅ + ⋅ + ⋅
= ⋅ (5.16)
(fj: Atomformamplitude für Atom j, i: imaginäre Einheit, xj, yj, zj, Koordinaten des Atoms j innerhalb der Elementarzelle) gilt. Wir kommen zurück auf das Problem aus Abschnitt 5.4.1 und wollen überlegen, welche Strukturfaktoren für die flä-chenzentrierte Gold-Zelle gelten. Darin gibt es vier Goldatome mit der Atomform-amplitude fAu auf den Plätzen
j xj yj zj j xj yj zj
1 0 0 0 3 0,5 0 0,5
2 0 0,5 0,5 4 0,5 0,5 0
Mit diesen Atompositionen folgt aus (5.16):
( )
l l k2 i( ) 2 i( ) 2 i( )0 2 2 2 2 2 2
i( ) i( ) i( )
e e e
1 e e e
k h h
hkl Au
k l h l h khkl Au
F f e
F f
− ⋅ + − ⋅ + − ⋅ +
− ⋅ + − ⋅ + − ⋅ +
= ⋅ + + +
= ⋅ + + +
(5.17)
Für die weitere Auswertung berücksichtigen wir die Eulersche44 Formel
44 Leonhard Euler, schweizer Mathematiker, 1707 - 1783

Kapitel 5 104
ie cos i sinϕ ϕ ϕ− ⋅ = − ⋅ (5.18)
sowie
( ) ( )
( )( ) ( )( )1 12 2
cos n 2 1, sin n 2 0 und
cos n 2 1, sin n 2 0
⋅ = ⋅ =
+ ⋅ = − + ⋅ =
(5.19)
(n: ganze Zahl). Die Summen (k+l), (h+l) und (h+k) müssen gerade Zahlen ergeben, damit der Strukturfaktor von Null verschieden ist. Diese Forderung ist erfüllt, wenn die h, k, l alle gerade oder alle ungerade sind. Dies ist die Auslöschungsregel für das (all-seitig) flächenzentrierte Gitter mit nur einer Atomsorte.
h k l e-π⋅i(k+l) e-π⋅i(h+l) e-π⋅i(h+k) Fhkl / fAu
1 0 0 1 -1 -1 1+1-1-1=0
1 1 0 -1 -1 1 1-1-1+1=0
1 1 1 1 1 1 1+1+1+1=4
2 0 0 1 1 1 1+1+1+1=4
2 1 0 -1 1 -1 1-1+1-1=0
2 1 1 1 -1 -1 1+1-1-1=0
2 2 0 1 1 1 1+1+1+1=4
2 2 1 -1 -1 1 1-1-1+1=0
2 2 2 1 1 1 1+1+1+1=4
Tabelle 5-6. Strukturfaktoren für die flächenzentrierte Elementarzelle (h,k,l = 0...2). Mit diesem Wissen können wir die Tabelle 5-5 korrigieren:
h k l 2 2 2h k l+ + 2 2 2179
h k l3⋅ + +
Gemessen/Pixel
1 1 1 3 179 179
2 0 0 4 207 206
2 2 0 8 292 292
3 1 1 11 343 342
2 2 2 12 358 357
4 0 0 16 413 413
3 3 1 19 451 450
4 2 0 20 462 461
Tabelle 5-7. Gegenüberstellung der gemessenen „Fingerabdrücke“ für die Abfolge der Ring- radien in Bild 5-11 mit den Erwartungen für eine kubisch-flächenzentrierte Elementarzelle.

Wir schalten um auf Elektronenbeugung 105
Wir sehen eine sehr gute Übereinstimmung von gemessenem und berechnetem Muster. Damit können wir mit Hilfe der Gitterkonstante von Gold (d = 0,4078 nm = 4,078 Å) die Gerätekonstante bestimmen. Gut geeignet ist dafür eine grafische Lösung, bei der wir die reziproken Netzebenenabstände über den zugehörigen Pixelabständen auftragen (s. Bild 5-14).
Für den Anstieg m der Kurve gilt (vgl. (5.12)):
1/hkl
hkl hklhkl
rm d r L
dλ= = ⋅ = ⋅ , (5.20)
der zunächst die ungebräuchliche Maßeinheit Pixel⋅nm hat. Für eine „echte“ Län-geneinheit muss die Pixelgröße berücksichtigt werden.
Bild 5-14. Gemessene Ring-radien (Pixel) über den rezi-proken Netzebenenabständen. Die für die Messpunkte ver-antwortlichen Netzebenen sind in Form ihrer Miller-schen Indizes (hkl) ange-tragen. Die Ausgleichs- gerade hat einen Anstieg m = 42,09 Pixel⋅nm.
Die Pixelgröße war 24 μm, d. h. λ⋅L = 1010 μm⋅nm, umgewandelt in eine ge-bräuchlichere Maßeinheit: λ⋅L = 10,10 Å⋅mm. Auf dem Computerbildschirm sind die Pixel größer, beispielsweise 0,26 mm. Die Gerätekonstante wäre dann 109,4 Å⋅mm. Wir sehen das Dilemma: Die Gerätekonstante hängt von der Pixel-größe ab. Ähnlich wie bei vergrößerten Bildern wäre ein Maßstabsbalken auf dem Beugungsbild vorteilhaft. Seine Länge ändert sich mit der Pixelgröße und er bleibt korrekt. Da die Abstände der Beugungsreflexe proportional zu den rezipro-ken Netzebenenabständen sind, steht auch der Maßstabsbalken für eine reziproke Längeneinheit, hat aber selbst natürlich eine reale Länge (Pixelanzahl). Wir geben einen reziproken Abstand 1/d vor und berechnen daraus mit Kenntnis von λ⋅L die zugehörige Länge r des Maßstabsbalkens in Pixeln:
1r m
d= ⋅ . (5.21)

Kapitel 5 106
In unserem Fall wäre ein Maßstabsbalken, der einen reziproken Abstand von 5 nm-1 repräsentiert, 210,4 Pixel lang. Damit können reziproke Längen direkt im Beugungsbild gemessen werden (s. Bild 5-15).
Zum Abschluss dieses Abschnitts stellen wir in Tabelle 5-8 die Auslöschungs-regeln für die in Tabelle 5-3 aufgelisteten Packungsarten zusammen.
Bild 5-15. Elektronenbeugungsdia-gramm von einer feinkri-stallinen Goldprobe mit eingezeichnetem Maßstabs-balken. Die Maßzahl ist 5, die Maßeinheit nm-1, d. h. die Größe ist 5 nm-1.
Nr. Packungsart Atom-zahl
Atompositionen Auslöschungsregel
1 primitiv
(Raumgruppen P…) 1 0;0;0 keine Auslöschung
2 raumzentriert
Raumgruppen I…) 2 0;0;0 1/2;
1/2; 1/2 Summe h+k+l ungeradzahlig
3 flächenzentriert
(allseitig) Raumgruppen F…)
4 0;0;0 1/2;
1/2; 0
1/2; 0; 1/2 0; 1/2;
1/2 h,k,l gemischt geradzahlig und
ungeradzahlig
4
basisflächenzentriert (Basisfläche C =
Grundfläche) Raumgruppen C…)
2 0;0;0 1/2; 1/2;
0 Summe h+k ungeradzahlig
5 Diamantgitter
(Raumgruppen F…) 8
0;0;0 1/2; 1/2;
0 1/2;
0; 1/2 0;1/2;
1/2
1/4; 1/4;
1/4 3/4;
3/4; 1/4
3/4; 1/4;
3/4 1/4;
3/4; 3/4
wie bei Nr 3, zusätzliche Auslöschung, wenn:
(h+k+l)/2, (3h+3k+l)/2, (3h+k+3l)/2, (h+3k+3l)/2 alle
ungeradzahlig
6 hexagonal dichteste
Kugelpackung Raumgruppen R…)
2
2/3; 1/3;
1/4 1/3;
2/3;3/4 h=k, l ungeradzahlig
Tabelle 5-8. Auslöschungsregeln.

Wir schalten um auf Elektronenbeugung 107
5.4.3 Intensitäten der Beugungsreflexe
Im kinematischen Modell (→ Abschnitt 10.9) wird die Intensität der Beugungs-reflexe durch zwei Einflussfaktoren bestimmt, den bereits aus dem vorhergehen-den Abschnitt bekannten Strukturfaktor Fhkl und den Gitterfaktor G. Im Struktur-faktor sind die Auswirkungen der Atomanordnung innerhalb der Elementarzelle auf die Beugungsreflexe erfasst, im Gitterfaktor diejenigen des gesamten Kristalls. Die Intensität I der Elektronenwellen ist proportional zum Produkt aus den Be-tragsquadraten von Struktur- und Gitterfaktor (vgl. auch Gleichungen (10.9.11), (10.9.15) und (10.9.30) → Abschnitt 10.9):
( )
( )
( )
( )
( )
( )
( )
( )
2 2
22 i2
2 2 21 1 2 2 3 32
2 2 2 21 1 2 2 3 3
mit
e und
sin sin sin1
j j j
hkl
h x k y l zhkl j
j
EZ
I F G
F f
u M u M u MG
V M u M u M u
− ⋅ ⋅ ⋅ + ⋅ + ⋅
⋅
= ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅= ⋅ ⋅ ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅
k
(5.22)
mit h, k, l als Millersche Indizes der beugenden Netzebenen, xj, yj, zj als Koordi-naten des Atoms j innerhalb der Elementarzelle sowie VEZ als Volumen der Ele-mentarzelle. M1, M2, M3 kennzeichnen als Zahl der Elementarzellen in a1-, a2- und a3-Richtung zusammen mit VEZ die Kristallgröße. u1, u2 und u3 sind die Kompo-nenten des Anregungsfehlers
1 2 3u u u= ⋅ + ⋅ + ⋅1 2 3u b b b , (5.23)
für die
1 2 31 2 3
1 1 1, , u u u
M M M= = = (5.24)
gilt.
Bild 5-16. Atomform-amplituden für Silizium und Silber in Abhängig- keit vom Betrag des Streuvektors s = |k – k0| nach [5.14].

Kapitel 5 108
Wir sehen, dass die Atomformamplituden nicht nur elementspezifisch sind son-dern auch vom Betrag des Streuvektors abhängen. Das bedeutet, dass die Intensi-tät der Beugungsreflexe mit wachsendem Beugungswinkel abnimmt.
Der Gitterfaktor bestimmt die Form der Beugungsreflexe. Ist der Kristall in einer kristallografischen Richtung besonders dünn (d. h. plättchenförmig), dann sind strichförmig verzerrte Reflexe zu erwarten. Bei Ringdiagrammen, d. h. einer Vielzahl von Kristallen mit unterschiedlichen kristallografischen Orientierungen im Beugungsbereich, bestimmt die Kristallgröße die Schärfe der Ringe: Je größer die Kristalle, umso schärfer die Ringe.
Schließlich gibt es Gefüge, bei denen viele Kristalle zum Beugungsbild bei-tragen, deren Orientierungen aber nicht statistisch gleichverteilt sind sondern eine Vorzugsorientierung aufweisen. Dies wird als Textur bezeichnet. Je nach Lage der Vorzugsorientierung zur Elektronenstrahlrichtung äußert sich eine Textur ent-weder als periodische Helligkeitsschwankung auf einzelnen Ringen oder Ringe, die nach den diesbezüglichen Regeln nicht ausgelöscht sein sollten, fehlen.
5.4.4 Positionen der Beugungsreflexe in Punktdiagrammen
Nach Einführung des reziproken Gitters ist mit der Ewald-Konstruktion (→ Abschnitt 10.8) die Vorschrift für die Berechnung der Positionen der Beugungs-reflexe von einem Einkristall gegeben. Wir wollen versuchen, diese Positionen auch ohne das Modell des reziproken Gitters zu erklären. Die Abstände der Re-flexe vom Zentrum lassen sich mit dem Braggschen Gesetz (z.B. (5.12)) aus den Netzebenenabständen berechnen. Die Formeln dafür sind in Tabelle 5-4 zusam-mengestellt. Aus Bild 5-1 geht bereits hervor, dass die Beugungsreflexe stets senkrecht auf den für sie verantwortlichen Netzebenen stehen. Die Winkel ξ zwi-schen den Beugungsreflexen sind also dieselben wie diejenigen zwischen den betreffenden Netzebenen (h1, k1, l1) und (h2, k2, l2). Für das kubische System ist die Rechnung einfach, weil die Netzebenennormalen die gleiche Indizierung (hkl) wie die Netzebenen selbst haben. Für die anderen Kristallsysteme bedienen wir uns wieder der Transformationen in ein orthogonales System (→ Abschnitt 10.8).
Wir benötigen einen Bezugsreflex mit der Indizierung (h1,k1,l1) und können mit Hilfe der Formeln in Tabelle 5-9 bei Kenntnis der Elementarzelle (Achsenlängen und Achsenwinkel) den Winkel ξ zu jedem anderen Reflex berechnen. In einem Polarkoordinatensystem sind mit Abstand und Winkel die Positionen der Beugungsreflexe bestimmt.
kubisch: a1 = a2 = a3
α = β = γ = 90°
ε = 90°
( ) ( )1 2 1 2 1 2
2 2 2 2 2 21 1 1 2 2 2
cosh h k k l l
h k l h k lξ
⋅ + ⋅ + ⋅=
+ + ⋅ + +

Wir schalten um auf Elektronenbeugung 109
tetragonal: a1 = a2 a3
α = β = γ = 90°
ε = 90°
2
11 2 1 2 1 2
3
2 22 2 2 2 2 21 11 1 1 2 2 2
3 3
cos
ah h k k l l
a
a ah k l h k l
a a
ξ
⋅ + ⋅ + ⋅
=
+ + ⋅ + +
ortho-rhombisch:
a1 a2 a3
α = β = γ = 90°
ε = 90°
22
1 11 2 1 2 1 2
2 3
2 22 22 2 2 2 2 21 1 1 11 1 1 2 2 2
2 3 2 3
cos
a ah h k k l l
a a
a a a ah k l h k l
a a a a
ξ
⋅ + ⋅ ⋅ + ⋅ ⋅
=
+ ⋅ + ⋅ + ⋅ +
hexagonal: a1 = a2 a3
α = β = 90°
γ = 120°
ε = 90°
( )2
11 2 1 2 2 1 1 2 1 2
3
2 22 2 2 2 2 21 11 1 1 1 1 2 2 2 2 2
3 3
13
2cos
3 3
ah h h k h k k k l l
a
a ah h k k l h h k k l
a a
ξ
⋅ + ⋅ + ⋅ + ⋅ + ⋅ ⋅ ⋅
=
+ ⋅ + + ⋅ ⋅ + ⋅ + + ⋅
rhombo-edrisch:
a1 = a2 = a3
α = β = γ 90°
( ) ( )
( ) ( )( ) ( ) ( )( )
( ) ( )
1 2 1 1 2 1 2 2 1 2 1 2 1 2
2 21 3 1 1 4 1 1 1 2 3 2 2 4 2 2 2
2 1 21 1 2 1 2 1 2 1 2 2 1 2
, , , , , , , ,cos
, , , , , ,
mit
cot, , , cot
sin sin
h h F h h k k F h h k k l l
h F h k F h k l h F h k F h k l
k kF h h k k h h h k h k
ξ
αα
α α
⋅ + +=
+ + ⋅ + +
⋅= ⋅ ⋅ − ⋅ + ⋅ ⋅ +
( )
( )
( )
( ) ( )
2 1 2 1 2 1 2
22
1 2 1 2 1 2222
21 2 2 1
2
1 2 2 1 1 2 2 1 22
, , , , ,
1 coscot tan
2 cossin
cos cot tan +
2 cos
cos cot tan
2 cos
F h h k k l l
h h k k l l
h k h k
h l h l k l k l
αα
α
α
α
α
αα
ε
α α
αα
=
= ⋅ ⋅ + ⋅ ⋅ ⋅ + ⋅ +⋅
⋅ ⋅⋅ + ⋅ ⋅ −
⋅
− ⋅ + ⋅ ⋅ − ⋅ + ⋅ ⋅ ⋅⋅
( )2
2 23 2
cot, cot 2
sin sin
kF h k h h k
αα
α α= ⋅ − ⋅ ⋅ ⋅ +
( ) ( )2
22 2 24 22
2
2
2
22
1 cos, , cot tan
2 cossin
cos cot tan
cos
cos 2 cot tan
cos
F h k l h k l
h k
h l k l
αα
α
α
αα
αα
ε
α α
αα
= ⋅ + ⋅ ⋅ + +⋅
⋅ ⋅+ ⋅ ⋅ −
− ⋅ ⋅ − ⋅ ⋅ ⋅ ⋅
sowie ( )cos cot 2 1 cosε α α= ⋅ ⋅ −

Kapitel 5 110
monoklin: a1 a2 a3
α = γ = 90°
β beliebig
ε = β
( )
( ) ( )
( )
1 2 2 11 2 1 2 1 22 2 2 2 2 21 2 1 3 3
5 1 1 1 5 2 2 2
2 2 2
5 2 2 2 2 2 21 2 1 3 3
cos
sin sin sincos
, , , ,
mit
2 cos, ,
sin sin sin
h l h lh h k k l l
a a a a a
F h k l F h k l
h k h l lF h k l
a a a a a
β
β β βξ
β
β β β
⋅ + ⋅ ⋅⋅ ⋅ ⋅+ − +
⋅ ⋅ ⋅ ⋅=
⋅
⋅ ⋅ ⋅= + − +
⋅ ⋅ ⋅ ⋅
triklin:
a1 a2 a3
α β γ
( )cos ,⋅
=⋅
1 21 2
1 2
B BB B
B B
( ) ( )( ) ( )
21 2 1 1 1 1 2 2 1 2 2
1 1 1 2 1 3 2 1 2 2 2 3
= h
x y y y y
z z z z z z
h b h b k b h b k b
h b k b l b h b k b l b
⋅ + + ⋅ +
+ + + ⋅ + +
1 2B B
( ) ( ) ( )
( ) ( ) ( )
22 21 1 1 1 1 2 1 1 1 2 1 3
22 22 1 2 1 2 2 2 1 2 2 2 3
= h
h
x y y z z z
x y y z z z
b h b k b h b k b l b
b h b k b h b k b l b
⋅ + + + + +
⋅ + + + + +
1 2B B
2 2
1 1 11 1 1
2 2
2 22 2
33
2 2
cot cos cos cos1 cot, ,
sin
cos cos1,
sin sin sin
1
a sin
cos 2 cos cos cos cossowie cos
sin
x y z
y z
z
b b ba a a
b ba a
b
γ ε β βγ
ε
ε β
γ γ ε
ε
α α β γ βε
γ
⋅ − −−= = =
⋅
− −= =
⋅ ⋅ ⋅
=⋅
− ⋅ ⋅ ⋅ +=
Tabelle 5-9. Formeln zur Berechnung der Winkel ξ zwischen zwei Punktreflexen (h1,k1,l1) und (h2,k2,l2) im Beugungsbild.
5.4.5 Indizierung der Beugungsreflexe
Mit Kenntnis der vorhergehenden Abschnitte des Kapitels 5 ist es einfach, die Frage nach der Indizierung der Beugungsreflexe zu beantworten: Die Beugungs-reflexe haben die gleiche Indizierung wie die sie hervorrufenden Netzebenen (vgl. Bild 5-17).

Wir schalten um auf Elektronenbeugung 111
Bild 5-17. Indizierung eines punktförmigen Beugungs-musters: Die Indizes der Beugungsreflexe sind diejenigen der beugenden Netzebenen. Die Schnittlinie aller beugenden Netzebenen wird als Zonenachse bezeichnet.
Wir wollen an einem Beispiel demonstrieren, wie die Indizierung eines Punkt-diagramms praktisch vorgenommen werden kann. Wir benutzen dafür ein Beu-gungsmuster von Silizium (Bild 5-18).
Bild 5-18. Punkt- diagramm von Silizium (gemessenes Beugungsmuster). Reflexabstände: s1 = s4 = 5,13 nm-1, s2 = s5 = 5,11 nm-1, s3 = s6 = 7,21 nm-1, Winkel zwischen s1 und s2: 89,9°, Winkel zwischen s2 und s3: 45,0°.
Zuerst ist zu überlegen, welche Beugungsreflexe von Silizium zu den Abständen s1, s2 und s3 passen. Silizium ist kubisch mit Diamantstruktur, d. h. ein Reflex (hkl) erscheint nur, wenn die h, k, l entweder alle gerade oder alle ungerade sind. Aus dieser Menge von Indizes scheiden zusätzlich diejenigen aus, für die (h+k+l)/2, (3h+3k+l)/2, (3h+k+3l)/2 und (h+3k+3l)/2 alle ganzzahlig und ungerade sind (vgl. Tabelle 5-8).
Die Achsenlänge in der Elementarzelle (Gitterkonstante) ist für Silizium a1 = 0,543 nm. Aus Tabelle 5-4 nehmen wir die Formel
2 2 2
1
1
hkl
h k l
d a
+ += (5.25)

Kapitel 5 112
für das kubische Kristallsystem. Damit sind u. a. die in Tabelle 5-10 aufgelisteten reziproken Netzebenenabstände möglich.
Die gemessenen reziproken Abstände weichen um weniger als 2 % von den be-rechneten für (220) und (400) ab. Dies ist eine zulässige Toleranz bei der Feinbe-reichsbeugung. Wir müssen noch die Winkel zwischen verschiedenen Reflexen berechnen und solche Indizierungen auswählen, für die 90° bzw. 45° gilt. Die not-wendige Formel für die Winkelberechnung entnehmen wir der Tabelle 5-9:
( ) ( )1 2 1 2 1 2
2 2 2 2 2 21 1 1 2 2 2
cosh h k k l l
h k l h k lξ
⋅ + ⋅ + ⋅=
+ + ⋅ + +
. (5.26)
Für einen Winkel ξ = 90° gilt cos∠(s1,s2) = 0, d. h. h1⋅h2 + k1⋅k2 + l1⋅l2 = 0.
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )111 11 1 1 11 111 1 1 1 11 1 1 11 1 1 1, , , , , , , 113 19
hkl
, nmd
−=
( ) ( ) ( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) ( ) ( )
220 202 022 220 220 220
202 202 202 022 022 022
, , , , , ,
, , , , ,
115 21
hkl
, nmd
−=
( ) ( ) ( ) ( ) ( ) ( )400 040 004 400 040 004, , , , , 117 37
hkl
, nmd
−=
Tabelle 5-10. Reziproke Netzebenenabstände in Silizium.
Da s1 und s2 beide etwa gleich 5,21 nm-1 sind, müssen wir die Indizes aus der zweiten Reihe von Tabelle 5-10 nutzen. Ein Index ist jeweils Null, um obige For-derung zu erfüllen, muss gelten:
1 2 1 2 1 2 1 2 1 2 1 2 oder oder h h k k h h l l k k l l⋅ = − ⋅ ⋅ = − ⋅ ⋅ = − ⋅ .
Dies ist erfüllt für die Reflexpaare
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
220 bzw. 220 und 220 bzw. 220 ,
202 bzw. 202 und 202 bzw. 202 ,
022 bzw. 022 und 022 bzw. 022
Wir benutzen (willkürlich) das erste Paar und indizieren s1 mit ( )220 und s2 mit
( )220 . Der Winkel zu s3 beträgt 45°, d. h.
( )( )
( ) ( ) ( )2 2 2 2
2 2 2 22 2 2 2
2 1cos ,
24 4 2
h k h k
h k h k
⋅ − + − += = =
+ ⋅ + ⋅ +2 3s s . (5.27)

Wir schalten um auf Elektronenbeugung 113
Für h2 = -4 und k2 = 0 ist diese Forderung offensichtlich erfüllt. Damit sind die drei ausgewählten Reflexe indiziert, die gegenüberliegenden sind Spiegelungen davon. Es bietet sich an, für solche Auswertungen ein Computerprogramm zu benutzen, mit dem reziproke Netzebenenabstände und Winkel zwischen Reflexen berechnet werden können (z. B. ELDISCA [5.15]). Das Ergebnis ist ein vollstän-dig indiziertes Beugungsdiagramm (s. Bild 5-19).
Das Punktmuster hängt von der Zonenachse ab. Wir wollen abschließend die Zonenachse für die in den Bildern 5-18 und 5-19 gezeigten Muster bestimmen. Wir kennen die Indizierung von mindestens zwei Beugungsreflexen: Beispiels-weise ( )220 und ( )220 .
Die Komponenten der Zonenachse im Kristallachsensystem sind (→ Abschnitt 10.8)
( ) ( ) ( )1 2 2 1 2 1 1 2 1 2 2 1, und k l k l h l h l h k h k⋅ − ⋅ ⋅ − ⋅ ⋅ − ⋅ , (5.28)
d. h. in unserem Fall [008] bzw. nach Kürzen der gemeinsamen Vielfachen aller Indizes [001].
Bild 5-19. Berechnetes und vollständig indizier- tes Beugungsdiagramm von Silizium, Zonenachse [001].
Nützliche Übersichten über indizierte Beugungsdiagramme gebräuchlicher Zonen-achsen für die dichtest gepackten Kristallsysteme sind beispielsweise in [5.16] dargestellt.

Kapitel 5 114
5.5 Kikuchi- und HOLZ-Linien Erinnern wir uns: Zur Scharfstellung des Beugungsbildes wird bei der Feinbe-reichsbeugung eine parallele Beleuchtung der Probe eingestellt, d. h. dem Kristall-gitter wird nur eine einzige Einfallsrichtung angeboten. Wir wollen nun auf die Feinstrahlbeugung mit großem Konvergenzwinkel zurückkommen, d. h. wir bieten dem Kristallgitter eine Vielfalt unterschiedlicher Einfallsrichtungen an. Wir nehmen an, dass die Symmetrieachse des Strahlenkegels parallel zur Netzebene (hkl) im Kristall verläuft (vgl. Bild 5-20).
Die Netzebenen (hkl) suchen sich aus der Richtungsvielfalt diejenigen Rich-tungen aus, für die das Braggsche Gesetz erfüllt ist. Es sind Ebenen, die innerhalb des Strahlenkegels parallel zu den Netzebenen (hkl) verlaufen. Sie sind in Bild 5-20 schwarz eingezeichnet. Nach Reflexion an der Netzebene (hkl) haben diese Ebenen den Winkelabstand θ/2 (θ: Braggwinkel) von der Symmetrieachse des Strahlenkegels. Die reziproken Gittervektoren stehen senkrecht auf den gleich-indizierten Netzebenen. Ihre Länge charakterisiert den zugehörigen Braggwinkel, d. h. ihre halbe Länge den halben Braggwinkel. Damit haben wir eine einfache Möglichkeit gefunden, zu einem Punktdiagramm das erwartete Kikuchi-Muster zu konstruieren. Als Beispiel benutzen wir das aus Bild 5-19 bekannte Si-Punkt-diagramm mit der Zonenachse [001] und beschränken uns dabei auf die inneren, intensitätsreichen Reflexe.
Bild 5-20. Schema zur Erklärung der Kikuchi-Linien.
In Bild 5-21a ist die Verfahrensweise anhand des (040)-Reflexes demonstriert: Die (040)-Kikuchi-Linie ist die Mittelsenkrechte der Verbindungslinie zwischen dem (000)- und dem (040)-Reflex. Dementsprechend sind die weiteren Kikuchi-Linien in Bild 5-21b ergänzt worden. Die Intensitäten der Kikuchi-Linien entsprechen denen der zu ihrer Konstruktion dienenden Punktreflexe.

Wir schalten um auf Elektronenbeugung 115
Bild 5-21. Konstruktion eines Kikuchi-Linien-Musters für Silizium, Zonenachse [001]. a) Konstruktion der (040)-Linie aus dem (040)-Reflex. b) Komplettes Kikuchi-Muster für die inneren Reflexe.
Die Kikuchi-Linien in dieser Form entstehen nur dann, wenn die durch den Bragg-winkel geforderten Einfallsrichtungen innerhalb des Strahlenkegels auch zur Ver-fügung stehen, d. h. der halbe Braggwinkel muss kleiner sein als der halbe Öff-nungswinkel des Strahlenkegels (Bestrahlungsapertur bzw. Konvergenzwinkel β): θ/2 < β.
Was passiert aber, wenn die Netzebenen (hkl) gegen die Symmetrieachse des Strahlenkegels geneigt sind (s. Bild 5-22). Praktisch bedeutet dies, dass wir den Kristall um einen Winkel τ (wenige mrad) aus seiner voreingestellten Zonenachse (z.B. [001]) herauskippen.
Bild 5-22. Veränderung des Kikuchi-Linien-Musters bei geringer Kippung des Kristalls.
Wir erkennen, dass dann der Symmetriepunkt des Kikuchi-Musters (Pol) aus-wandert. Die Richtung der Wanderung hängt davon ab, um welche kristallogra-fische Achse gekippt wird. Für unser Beispiel in Bild 5-21 verschiebt sich das Mu-

Kapitel 5 116
ster horizontal, wenn um die Kristallachse a1 und vertikal, wenn um die Kristall-achse a2 gekippt wird. Die Größe der Verschiebung ist proportional zum Winkel τ. Wird der weiter erhöht, so sind zwei Fälle möglich:
1. Der halbe Braggwinkel liegt nur noch „einseitig“ innerhalb des Strahlen-kegels. In der geraden Fortsetzung der Bragg-orientierten Ebene fehlt die ge-beugte Intensität und es entsteht eine dunkle Linie parallel zur hellen Linie im Winkelabstand θ von ihr. Dieser Umstand wurde im Abschnitt 5.3 als allgemeiner Fall bei Feinstrahlbeugung mit großem Konvergenzwinkel geschildert.
2. Eine andere („niedrig indizierte“) Netzebenenschar gelangt in die Nähe der Bragg-Position. Es entsteht ein neuer Pol, der zunächst nicht im Zentrum liegt, aber durch weiteres Kippen um die „richtigen“ Kristallachsen dorthin geschoben werden kann. Das bedeutet andererseits, dass es durch Zentrierung des Kikuchi-Musters möglich ist, im Elektronenmikroskop eine niedrig indizierte Zonenachse sehr genau einzustellen (vgl. Bild 7-6b).
Wir wollen uns diesen Sachverhalt mit dem reziproken Gittermodell veran-schaulichen (s. Bild 5-23).
Bild 5-23. Konvergente Beleuchtung und reziprokes Gitter (ebener Schnitt).
In der Schnittdarstellung sind vom Strahlenkegel die grenzwertigen Einfalls-richtungen mit den zugehörigen Ewaldkugeln gezeichnet. Wir sehen, dass zwi-schen den beiden Kugeln ein Bereich existiert, der Beugungsreflexe („Bragg-Po-sitionen“ von Netzebenen) einschließt, die zum Linienmuster beitragen können. Insbesondere sehen wir auch, dass sich dieser Bereich über mehrere Ebenen des reziproken Gitters erstreckt. Diese Ebenen werden „Laue-Zonen genannt. Im Englischen heißt die 0. Laue-Zone: Zero Order Laue Zone („ZOLZ“), die 1. Laue-Zone: First Order Laue Zone („FOLZ“) und allgemein die Laue-Zone höherer Ordnung: High Order Laue Zone („HOLZ“). Wir ersehen aus Bild 5-23 auch, dass die HOLZ-Linien bei vergleichsweise großen Beugungswinkeln erscheinen, die evtl. außerhalb des Beobachtungsbereiches liegen. Wir wissen aber, dass das Beugungsmuster durch geringfügiges Kippen der Probe auf dem Beobachtungs-schirm verschoben werden kann. In der Praxis wird die Probe um wenige mrad aus der niedrigindizierten Achse herausgekippt, d. h. es wird eine höher indizierte Zonenachse eingestellt. In der Regel sind dann keine Linienpaare wie bei den Kikuchi-Mustern, sondern nur einzelne Linien zu sehen (vgl. Bild 5-24).

Wir schalten um auf Elektronenbeugung 117
Bild 5-24. HOLZ-Linien-Muster von a) Aluminium b) Kupfer. Zonenachse: nahe [136]. Insbesondere bei Kupfer sind Linienver-biegungen zu sehen (rechts oben), die mit dem kine-matischen Modell nicht erklärt werden können. (Aufnahmen: M. Hofmann)
Da die Beugungswinkel für die HOLZ-Linien vergleichsweise groß sind, haben selbst geringfügige Änderungen der Gitterkonstanten einen messbaren Einfluss auf die Linienmuster. So ist es prinzipiell möglich, anhand solcher Muster die Git-terkonstanten mit einer Genauigkeit von besser als 0,01 % in Bereichen von weni-gen Nanometern zu bestimmen. Allerdings sind wir dabei auf einkristalline Be-reiche mit kleinen mechanischen Spannungsgradienten angewiesen. Auch die Auswertung der Messungen kann problematisch sein. Wir sehen in Bild 5-24b an Kreuzungspunkten mehrerer Linien Linienverbiegungen, die durch die Wechsel-wirkung der Elektronenwellen untereinander zustande kommen. Solche Wechsel-wirkungen bleiben im kinematischen Modell unberücksichtigt, ebenso die Wech-selwirkungen der gebeugten Wellen mit dem Kristallgitter. Folglich ist das kine-matische Modell insbesondere bei Gittern mit schwereren Elementen zu unpräzise, um die o.g. Genauigkeit bei der Auswertung erreichen zu können. Wir benötigen ein Modell, welches die gegenseitige Beeinflussung der gebeugten Wellen und des Kristallgitters berücksichtigt: Das dynamische Modell, das aber ungleich rechen-intensiver und schwerer verständlich ist. Es erfordert die Lösung der Schrödinger-Gleichung45 (das ist die Grundgleichung für Materiewellen → Abschnitt 10.2) im Kristallpotential [5.17]. Neben der bereits erwähnten HOLZ-Linienverbiegung führt dies zu weiteren Konsequenzen:
Die Elektronenwellenlänge hängt vom Kristallpotential und damit auch von der Richtung im Kristallgitter ab. Ein positives Kristallpotential verkürzt die Wellen-länge. Die Ewald-Konstruktion (→ Abschnitt 10.8) ist dann keine Kugel mit konstantem Radius mehr, sondern ihr Radius ist kristallrichtungsabhängig. Die exakten Reflexpositionen sind damit gegenüber der kinematischen Näherung ge-ringfügig verschoben. Bei der Auswertung von Feinbereichs-Beugungsdiagram-men spielt dies i. A. keine Rolle, wohl aber bei der genauen Gitterkonstanten-messung mit Hilfe von HOLZ-Linienmustern.
Vielfachstreuung der Elektronenwellen innerhalb des Kristallgitters ist mög-lich. Die Intensität oszilliert zwischen gebeugten und nichtgebeugten Wellen wäh-rend ihres Durchgangs durch den Kristall. Damit hängt die Reflexintensität von der Probendicke ab.
45 Erwin Schrödinger, österreichischer Physiker, 1887 – 1961, Nobelpreis für Physik 1933

Kapitel 5 118
Es ist möglich, den Einfluss dynamischer Effekte und denjenigen des Anre-gungsfehlers auf das Beugungsbild zu reduzieren, indem die Ewald-Kugel nicht festgehalten wird sondern „taumelt“. Damit wird eine Vielzahl reziproker Gitter-punkte im zeitlichen Mittel in gleicher Weise getroffen, was gleichbedeutend mit identischen Anregungsbedingungen für alle Beugungsreflexe ist. Praktisch wird dies durch eine Präzessionsbewegung des Elektronenstrahls vor der Probe bewerk-stelligt, die nach der Probe wieder ausgeglichen werden muss („Precessing Electron Diffraction“ – PED [5.18]).
Das (genauere) dynamische Modell ermöglicht weitere Schlussfolgerungen: Die (hier nicht näher erläuterte) Intensitätsverteilung in den Beugungsscheibchen bei der konvergenten Beugung (englisch: Convergent Beam Electron Diffraction – CBED) wird maßgeblich durch die Streueigenschaften des Gitters beeinflusst. Da-mit hat man prinzipiell Zugriff auf die Messung der elektronischen Struktur, z.B. der Elektronendichte, des Festkörpers [5.19].
5.6 Amorphe Proben Wir kehren zurück zur Feinbereichsbeugung, d. h. zur parallelen Beleuchtung. Das beobachtete Beugungsdiagramm hängt in starkem Maße von der Größe der Kri-stallite ab. Bei Kristalliten, die der Größe des ausgewählten Bereiches entspre-chen, werden Punktdiagramme beobachtet. Bei Verkleinerung der Kristallite ent-stehen Ringdiagramme, deren scharfe Maxima schließlich in diffuse Ringe über-gehen.
Wir wollen uns hier dem letzten Fall genauer widmen und damit Charakterisie-rungsmöglichkeiten für sogenannte amorphe Substanzen aufzeigen. Dabei ist zu beachten, dass vom Standpunkt der Charakterisierung her der Übergang zwischen dem feinkristallinen und dem amorphen Zustand fließend ist und das Mess-ergebnis stark vom Verfahren abhängt. Der Begriff „röntgenamorph“ für Materialien mit Kristallitgrößen unter 5 nm deutet auf diesen Umstand hin.
Das Messergebnis, nämlich die Abhängigkeit der Intensität vom Streuwinkel, soll hierbei als Streukurve bezeichnet werden (entspricht der radialen Helligkeits-verteilung bei Ringdiagrammen – vgl. Abschnitt 5.4.1). Die Atome sind die Streu-zentren für die einfallenden Strahlelektronen. Entscheidend für das Aussehen der Streukurve sind demzufolge die Atomart und die Atomanordnung. In unserer Mo-dellvorstellung wollen wir uns auf den Einfluss der Atomanordnung beschränken.
Einkristalle zeichnen sich durch die periodische Anordnung der Atome aus. Die Folge ist, dass nur diskrete Abstände auftreten (wenn man von Wärmebewegun-gen absieht) und die Abstände richtungsabhängig sind (vgl. Bild 5-25a).

Wir schalten um auf Elektronenbeugung 119
Bild 5-25. Atomanordnung in kristallinem (a) und amorphem (b) Festkörper.
Die Verteilung der Atome ρ hängt von Abstand r und den Winkeln δ und η ab:
( ), ,rρ ρ δ η= , (5.29)
sie ist richtungsanisotrop. Wird die periodische Anordnung aufgelöst, was durch Temperaturerhöhung
über den Schmelzpunkt, durch anders geartete Kraftfelder in der Atomumgebung oder auch durch besondere Bedingungen beim Erstarren des Festkörpers erreicht werden kann, so verschwindet neben den scharfen Abständen in der Vertei-lungsfunktion insbesondere auch deren Richtungsanisotropie (s. Bild 5-25b). Nun-mehr gilt:
( )rρ ρ= . (5.30)
Trotzdem existieren selbstverständlich nach wie vor zwischenatomare entfer-nungsabhängige Kräfte, so dass Vorzugsabstände der Atome und damit Modula-tionen der Dichteverteilung auch nach Auflösung des Kristallgitters zu erwarten sind. Die im Kristall vorhandene Fernordnung geht verloren und wird durch eine Nahordnung ersetzt.
Bild 5-26 zeigt ein Beugungsbild von einer dünnen amorphen Probe.
Bild 5-26. Beugungsbild von einem amorphem Film und daraus extrahierte radiale Helligkeitsverteilung nach Untergrundabzug (Streukurve).
Um dieses Messergebnis auswerten zu können, müssen wir verstehen, wie es zu-stande kommt. Die Streukurve I(s) ist das Resultat der Überlagerung aller an der Atomanordnung gestreuten Wellen. Für die Intensität gilt
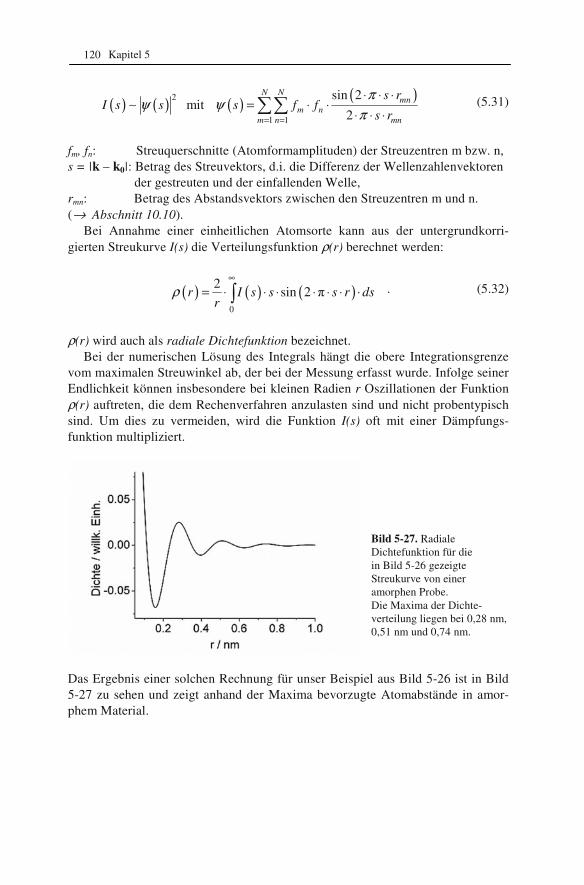
Kapitel 5 120
( ) ( ) ( )( )2
1 1
sin 2 mit
2
N Nmn
m nm n mn
s rI s s s f f
s r
πψ ψ
π= =
⋅ ⋅ ⋅= ⋅ ⋅
⋅ ⋅ ⋅ (5.31)
fm, fn: Streuquerschnitte (Atomformamplituden) der Streuzentren m bzw. n, s = |k – k0|: Betrag des Streuvektors, d.i. die Differenz der Wellenzahlenvektoren der gestreuten und der einfallenden Welle, rmn: Betrag des Abstandsvektors zwischen den Streuzentren m und n. (→ Abschnitt 10.10).
Bei Annahme einer einheitlichen Atomsorte kann aus der untergrundkorri-gierten Streukurve I(s) die Verteilungsfunktion ρ(r) berechnet werden:
( ) ( ) ( )0
2sin 2r I s s s r ds
rρ
∞
= ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ . (5.32)
ρ(r) wird auch als radiale Dichtefunktion bezeichnet. Bei der numerischen Lösung des Integrals hängt die obere Integrationsgrenze
vom maximalen Streuwinkel ab, der bei der Messung erfasst wurde. Infolge seiner Endlichkeit können insbesondere bei kleinen Radien r Oszillationen der Funktion ρ(r) auftreten, die dem Rechenverfahren anzulasten sind und nicht probentypisch sind. Um dies zu vermeiden, wird die Funktion I(s) oft mit einer Dämpfungs-funktion multipliziert.
Bild 5-27. Radiale Dichtefunktion für die in Bild 5-26 gezeigte Streukurve von einer amorphen Probe. Die Maxima der Dichte-verteilung liegen bei 0,28 nm, 0,51 nm und 0,74 nm.
Das Ergebnis einer solchen Rechnung für unser Beispiel aus Bild 5-26 ist in Bild 5-27 zu sehen und zeigt anhand der Maxima bevorzugte Atomabstände in amor-phem Material.

6 Warum sehen wir Kontraste im Bild?
Ziel: Wenn wir im Gedankenexperiment mit einem weißen Stift zwei Punkte auf ein weißes Blatt Papier zeichnen, werden wir diese Punkte nicht sehen können. Der weiße Stift liefert keinen Kontrast. Auch im Elektronenmikroskop benötigen wir nicht nur ein hohes Auflösungsvermögen sondern auch einen Bildkontrast. Dieser ist das Ergebnis der Wechselwirkung zwischen den Strahlelektronen (Pri-märelektronen) und der durchstrahlten Probe. Anders als in der Lichtmikroskopie, wo die Schwächung der Amplitude der Lichtwelle eine wesentliche Kontrast-ursache ist, spielt die Absorption von Elektronen in der Probe (d. h. die Verringe-rung der Amplitude der Elektronenwelle) in der Transmissionselektronenmikro-skopie nur eine untergeordnete Rolle. Hier ist die unterschiedliche Streuung (Ab-lenkung) der Elektronen in verschiedenen Probenbereichen die dominierende Ur-sache für den Kontrast. Später werden wir sehen, dass insbesondere bei sehr hohen Vergrößerungen auch Phasenschiebungen der Elektronenwelle für den Kontrast verantwortlich sein können.
6.1 Elastische Streuung der Elektronen in der Probe Wir stellen uns vor, dass die Strahlelektronen beim Durchgang durch die Probe nur mit den Atomkernen wechselwirken. Wegen der extrem unterschiedlichen Massen von Atomkern und Elektron (die Masse eines Protons ist 1836 mal größer als die Masse eines Elektrons) wird die Lage des Kerns nicht verändert, ähnlich wie beim Stoß einer Billardkugel mit dem Rand des Billardtisches. Wir sprechen demzufolge von einer elastischen Wechselwirkung, der Betrag der Geschwindig-keit und damit die Energie des Elektrons ändern sich dabei nicht. Für die Wahr-scheinlichkeit, mit der die Elektronen durch Coulomb-Wechselwirkung mit einem Atomkern um den Winkel θ abgelenkt und in das durch θ gekennzeichnete Raumwinkelsegment dΩ gestreut werden, gilt (vgl. Bild 6-1 – differentieller Rutherfordscher46 Streuquerschnitt → Abschnitt 10.11):
22
40 0
1 e 1
4 4 sin2
d Z
d E
σ
θε
⋅= ⋅ ⋅
Ω ⋅ ⋅ (6.1)
(ε0: Influenzkonstante, Z: Ordnungszahl, e: Elementarladung, E0: Primärelektro-nenenergie). Dabei vernachlässigen wir die teilweise Abschirmung des elektri- 46 Ernest Rutherford, neuseeländisch/englischer Physiker, 1871 – 1937, Nobelpreis für Chemie 1908
DOI 10.1007/978-3-7091-1440-7_6, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 6 122
schen Potentials des Atomkerns durch die Elektronenhülle. Die Gesamtstreuung der Elektronen wird auch durch die Dichte der Atome in der Probe beeinflusst. Für die Anzahl N von Atomen im Volumen V gilt:
ANN Vρ= ⋅ ⋅ (6.2)
(NA: Avogadro-Konstante, ρ: Dichte).
Bild 6-1. Zur Erläuterung des differentiellen Wirkungsquerschnitts.
Das Streuvermögen eines einzelnen Atoms wird durch den Streuquerschnitt be-schrieben. Wir interessieren uns für alle Elektronen, deren Streuwinkel größer als ein angenommener Akzeptanzwinkel α ist. Aus (6.1) folgt nach Integration (→ Abschnitt 10.12):
( )22
20 0 2
e 1
8 tan
Z
E ασ α
⋅= ⋅
⋅ ⋅ ⋅ . (6.3)
Selbstverständlich hängt die Stärke der Streuung auch von der Probendicke ab. Wie in → Abschnitt 10.12 gezeigt wird, können wir für die Zahl NE von Elek-tronen, die nach Zurücklegen einer Strecke s innerhalb der Probe um weniger als den Winkel α abgelenkt werden, schreiben:
,0 e elsE EN N − Λ= ⋅ . (6.4)
NE,0 ist die Zahl der in die Probe einfallenden Elektronen, Λel ist eine statistische Größe, die als mittlere freie Weglänge für die elastische Streuung bezeichnet wird. Für sie gilt mit unserem einfachen Modell:
2 20 0 2
2A
tan8
e Nel
E
Z
α
ρ
⋅⋅ ⋅Λ = ⋅
⋅ ⋅ . (6.5)

Warum sehen wir Kontraste im Bild? 123
6.2 Streuabsorptions- und Beugungskontrast Um das unterschiedliche Streuvermögen in einen Bildkontrast umzusetzen, müs-sen die stärker gestreuten Elektronen aus dem Strahlengang entfernt werden. Dies geschieht mit einer Blende, deren Radius den Akzeptanzwinkel bestimmen soll. Diese Blende muss demzufolge in einer winkelselektiven Ebene, d. h. in der bild-seitigen Brennebene der Objektivlinse angeordnet werden (Objektivblende - vgl. Abschnitt 2.7.2 und Bild 6-2). Wegen ihrer kontrastverstärkenden Wirkung wird sie auch als Kontrastblende bezeichnet.
Bild 6-2. Anordnung und Wirkungsweise der Kontrastblende.
Ihre Wirkungsweise ist aus Bild 6-2 ersichtlich: Schwach gestreute Elektronen be-wegen sich nahezu parallel zur optischen Achse. Sie treffen sich in der bildseiti-gen Brennebene nahe dem Brennpunkt und werden durch die Kontrastblende hin-durchgelassen. Stärker gestreute Elektronen treffen auf die Blende, werden damit aus dem Strahlengang entfernt und tragen nicht zur Bildhelligkeit bei. Die stärker streuenden Probenbereiche erscheinen im Bild dunkel.
Wir wollen nun mit Hilfe der Formeln (6.4) und (6.5) abschätzen, welcher Kontrast im Falle von kleinen, wenige 10 nm großen Goldinseln, auf einer Kohle-folie zu erwarten ist. Wir definieren den Kontrast K1-2 zwischen zwei Bildbe-reichen 1 und 2 durch die Helligkeiten H1 und H2 in diesen Bildbereichen:
1 21 2
1 2
2H H
KH H−
−= ⋅
+ . (6.6)
Für die Bildhelligkeit ist die Anzahl der pro Zeit und Fläche einfallenden Elek-tronen verantwortlich. Mit (6.4) können wir schreiben:
,
,
1 ,0
2 ,0
e
e
Au el Au
C el C
sAu E
sC E
H H N
H H N
− Λ
− Λ
= = ⋅
= = ⋅ (6.7)
bzw.

Kapitel 6 124
, ,
, ,
e e2
e e
Au el Au C el C
Au el Au C el C
s s
Au C s sK− Λ − Λ
− − Λ − Λ
−= ⋅
+ . (6.8)
Zur Vereinfachung setzen wir für die Probendicken sAu = sC = s und berechnen den Kontrast KAu-C in Abhängigkeit von der Probendicke s bei zwei Kontrastblen-dendurchmessern d (s. Bild 6-3). Für den Akzeptanzwinkel α gilt (vgl. Bild 2-18):
2 2 mm
d d
fα = ≈
⋅ . (6.9)
Bild 6-3. Erwarteter Kontrast im elektronen-mikroskopischen Bild von Goldinseln auf Kohlefolie bei einer Primärenergie von 200 keV und zwei verschiedenen Kontrast-blendendurchmessern d in Abhängigkeit von der Probendicke s.
Für einen Vergleich mit dem experimentellen Ergebnis ziehen wir das elektronen-mikroskopische Bild 6-4 von Goldinseln auf Kohlefolie heran. Der daraus er-mittelte Kontrast zwischen Kohlefolie und Gold von 0,72 wird nach Bild 6-3 bei einer Probendicke von 30 nm erreicht, was durchaus plausibel erscheint.
Bild 6-4. Transmissions-elektronenmikroskopi-sches Bild von einer Kohlefolie mit Gold-inseln (E0 = 200 keV, Kontrastblendendurch-messer: 100 μm). In den zwei weiß gerahmten Rechtecken wurde die mittlere Bildhelligkeit ge-messen: Sie beträgt im oberen Rechteck (Gold) 82 und im unteren (Kohlenstoff) 175. Daraus folgt nach (6.6) ein Kontrast von 0,72.

Warum sehen wir Kontraste im Bild? 125
Diese Übereinstimmung zeigt, dass unser einfaches Modell zur Erklärung dieses Kontrastes geeignet ist. Wir können daraus ableiten, welche Maßnahmen zur Kon-trastverstärkung ohne Veränderung der Probe ergriffen werden können: Verrin-gerung der Beschleunigungsspannung (d. h. der Primärelektronenenergie E0) und Verkleinerung der Kontrastblende.
Der Name für den beschriebenen Kontrastmechanismus fasst dessen zwei physikalische Ursachen zusammen: unterschiedliche Streuung der Elektronen in der Probe und Absorption der stark gestreuten Elektronen an der Kontrastblende: Streuabsorptionskontrast. Im Englischen ist der Name „Mass thickness contrast“ (Massendickekontrast) gebräuchlich.
Bei kristallinen Proben kann eine unterschiedliche Ablenkung der Elektronen auch infolge Beugung bewirkt werden (vgl. Kapitel 5). Wir stellen uns dazu eine polykristalline Probe vor, bei der die Kristallite (Körner) unterschiedlich orientiert sind und deren Kristallgitter damit verschiedene Winkel mit der Elektronenein-fallsrichtung bilden. Erfüllt der Winkel zwischen einer Netzebenenschar und der Elektroneneinfallsrichtung gerade die Bragg-Bedingung (5.6), so entstehen inten-sitätsreiche Beugungsmaxima. Die dahin gestreuten Elektronen werden von der Kontrastblende aus dem Strahlengang entfernt, die in „Bragg-Lage“ befindlichen Kristallite erscheinen im Bild dunkler (s. Bild 6-5). Wir sprechen vom Beu-gungskontrast. Er ermöglicht die Unterscheidung einzelner Kristallite im elektro-nenmikroskopischen Bild.
Bild 6-5. Transmissions-elektronenmikroskopisches Bild von einer dünnen polykristallinen Ni/NiO-Schicht. Die Körner (Kristallite) sind aufgrund des Beugungskontrastes deutlich zu unterscheiden.
Wegen des bei der Elektronenbeugung zulässigen Anregungsfehlers muss die Bragg-Bedingung nicht exakt erfüllt sein, um Beugungsreflexe zu erhalten (vgl. Abschnitt 5.4.3 sowie → Abschnitt 10.9). Somit existiert auch für die Bragg-Lage eine Toleranz, die von der Korngröße abhängt.
Im Unterschied zum Streuabsorptionskontrast ist der Beugungskontrast orien-tierungsabhängig, d. h. er ändert sich beim Kippen der Probe. Damit ist es für den Experimentator möglich, zwischen beiden Kontrastarten zu unterscheiden: Er

Kapitel 6 126
muss die Probenbühne kippen (dies ist bei allen Transmissionselektronen-mikroskopen zumindest um eine Achse möglich). Ändert sich dabei der Kontrast, so handelt es sich um Beugungskontrast und damit um eine kristalline Probe.
6.3 Hell- und Dunkelfeldabbildung Bisher sind wir davon ausgegangen, dass die Kontrastblende mittig zur optischen Achse zentriert ist, das heißt, dass sie die nicht oder wenig gestreuten Elektronen hindurchlässt. Dieser Fall wird als Hellfeldabbildung bezeichnet. Die Bilder 6-4 und 6-5 sind solche Hellfeldbilder.
Was geschieht aber, wenn wir die Blende zur Seite schieben und auf diese Wei-se die wenig gestreuten Elektronen aus dem Strahlengang entfernen. Dafür pas-siert zumindest ein Teil der stärker gestreuten Elektronen die Blende (Dunkelfeld-abbildung). Bei einer nichtkristallinen Probe erhalten wir den inversen Kontrast und somit keinen zusätzlichen Erkenntnisgewinn. Anders ist der Sachverhalt bei kristallinen Proben. Wir nehmen zunächst an, dass in einer dünnen Schicht alle Kornorientierungen mit gleicher Wahrscheinlichkeit auftreten, oder mit anderen Worten: Es gibt keine Vorzugsorientierung. In Bild 6-6a ist das zu erwartende Hellfeldbild schematisch dargestellt. Verschieben wir in unserem Rechenmodell die Kontrastblende an zwei unterschiedliche Stellen, so entstehen die Dunkel-feldbilder 6-6b und 6-6c.
Bild 6-6. Berechnete Hell- und Dunkelfeldbilder einer polykristallinen Schicht ohne Vorzugs-orientierung der Körner. a) Hellfeldbild, mittlere Helligkeit: 165. b) Dunkelfeldbild 1, mittlere Helligkeit: 126, c) Dunkelfeldbild 2, mittlere Helligkeit: 124.
Wie erwartet ist die mittlere Helligkeit in den Dunkelfeldbildern signifikant niedriger als im Hellfeldbild; sie unterscheidet sich kaum in den beiden Dunkel-feldbildern. Das ändert sich, wenn die Körner eine Vorzugsorientierung (Textur) aufweisen.
Unter dieser Voraussetzung wurden die Bilder in 6-7 berechnet. Die Kontrast-blendeneinstellung bei den Dunkelfeldbildern ist die gleiche wie in Bild 6-6. Im Gegensatz zu Bild 6-6 unterscheiden sich hier die mittleren Helligkeiten der bei-den Dunkelfeldbilder deutlich.

Warum sehen wir Kontraste im Bild? 127
Bild 6-7. Berechnete Hell- und Dunkelfeldbilder einer polykristallinen Schicht mit Vorzugs-orientierung der Körner. a) Hellfeldbild, mittlere Helligkeit: 160. b) Dunkelfeldbild 1, mittlere Helligkeit: 149. c) Dunkelfeldbild 2, mittlere Helligkeit: 99. Mit der Kontrastblende können im Beugungsbild auch einzelne Reflexe ausge-wählt werden. Im zugehörigen Dunkelfeldbild erscheinen dann diejenigen Körner hell, die für den ausgewählten Reflex verantwortlich sind. Bei statistisch gleich-verteilten Orientierungen sind im Mittel etwa gleich viele Körner hell, unabhängig vom ausgewählten Reflex (s. Bilder 6-6b und c). Anders ist es bei texturierten Schichten. Wird hier ein Reflex ausgewählt, für den die vorzugsorientierten Kör-ner verantwortlich sind, so wird im Dunkelfeldbild eine größere Anzahl Körner hell erscheinen als wenn ein Reflex gewählt wird, der zu Kornorientierungen ge-hört, die mit geringer Wahrscheinlichkeit auftreten (vgl. Bilder 6-7b und c). Die Texturen wirken sich auch direkt auf das Beugungsbild aus, wie das einfache Beispiel in Bild 6-8 zeigt.
Bild 6-8. Schicht mit <001> - Vorzugsorientierung. a) Skizze einer texturierten Schicht. b) Lage der beugenden Netzebenen. c) Beugungsbild dieser Schicht (schematisch).
In diesem Beispiel setzen wir kubische Kristalle voraus, deren [001]-Achsen aus-nahmslos nach oben zeigen und die geringfügig um diese Achse pendeln (Bild 6-8a). Die Beugungsintensitäten sind demzufolge nicht gleichmäßig auf den Ringen verteilt sondern auf symmetrisch angeordnete azimutale Bereiche konzentriert (Bild 6-8c). Wird die Kontrastblende zur Dunkelfeldabbildung auf eine Position mit hoher Intensität im Beugungsbild gesetzt, so werden besonders viele Körner hell erscheinen. Bei systematischer Verschiebung der Kontrastblende innerhalb des Beugungsbildes können in polykristallinen Gefügen mit der Dunkelfeldab-

Kapitel 6 128
bildung Texturen erkannt und charakterisiert werden. Es ist allerdings experimen-tell schwierig, die Blende mechanisch mit der nötigen Genauigkeit und Reprodu-zierbarkeit an die vorgesehenen Stellen zu schieben.
Allerdings kann der gleiche Effekt wie beim Verschieben der Kontrastblende auch durch Kippen des Elektronenstrahls erreicht werden (vgl. Bild 6-9). Es ist wichtig, dass der Punkt, um den der Elektronenstrahl gekippt wird, exakt in der Probenebene (Objektebene) liegt. Anderenfalls wird infolge des Kippens eine an-dere Probenstelle beleuchtet. Wir erinnern uns: Die Höheneinstellung dieses Kipp-punktes („Pivot Point“) war Teil der Kontrolle des Justagezustandes des Mikro-skops, wie es im Abschnitt 4.3 beschrieben ist. Die Strahlkippung erfolgt mit Hilfe elektromagnetischer Ablenksysteme.
Bild 6-9. Zwei Möglichkeiten, den ( -1.)-Beugungsreflex zur Dunkelfeldabbildung zu verwen-den. a) Verschieben der Kontrastblende. b) Kippen des Elektronenstrahls.
Neben der höheren Präzision bei der Einstellung hat dieses Verfahren noch zwei weitere Vorteile:
1. Wie aus Bild 6-9b ersichtlich ist, bleibt die Kontrastblende bei der Kipp-Me-thode weiterhin konzentrisch zur optischen Achse justiert, d. h. auch für die Dun-kelfeldabbildung werden achsennahe Strahlen benutzt, die Abbildungsfehler damit minimiert.
2. Durch geeignete Ansteuerung der Strahlkippungssysteme ist es möglich, den Elektronenstrahl auf einem Kegelmantel zu führen, wobei die Kegelspitze in der Objektebene liegt (Conical Darkfield). Dies ist der gleiche Effekt, wie wenn die Kontrastblende auf einem Ring geführt wird (was mechanisch kaum möglich ist). Der Ringradius ist durch den Kegelwinkel der Strahlführung bestimmt.
Damit eröffnet sich eine weitere Möglichkeit der Phasenanalyse. Wir nehmen an, dass sich die beiden Werkstoffphasen eines Phasengemisches kristallografisch derart unterscheiden, dass einzelne Ringe des Beugungsdiagramms nur einer Pha-se zugeordnet und mit der Kontrastblende selektiert werden können. Dies ist zum einen eine Frage der kristallografischen Struktur und zum anderen eine Frage der Größe der Kontrastblende.

Warum sehen wir Kontraste im Bild? 129
Bild 6-10. Phasenanalyse von Ni / NiO durch Dunkelfeldabbildung (Conical Darkfield); Kontrastblende: 50 μm. a) Hellfeldbild. b) Dunkelfeldbild mit eingesetztem Beugungsbild (c). Für ein Gemisch von Nickel und Nickeloxid ist das möglich. Beide bilden kubische Phasen mit Gitterkonstanten von 0,35 nm bzw. 0,42 nm. Damit kann der innerste Ring des Beugungsdiagramms dem NiO mit der größeren Gitterkonstante zugeordnet werden (vgl. Bild 6-10). Durch Einstellung eines Kegelwinkels von 8 mrad wurde dieser Ring mit der Kontrastblende selektiert und ein Dunkelfeld-bild bei konischer Strahlführung erzeugt (Bild 6-10b). In diesem Dunkelfeldbild erscheinen die NiO-Kristallite hell.
6.4 Biegekonturen, Versetzungen und semikohärente Ausscheidungen Wir haben gesehen, wie wichtig das Orientierungsverhältnis zwischen Netzebenen und einfallendem Elektronenstrahl für den Beugungskontrast ist. Dieser Beu-gungskontrast ist die Grundlage für viele elektronenmikroskopische Untersuchun-gen zur Gitterstruktur der Probe. Mit anderen Worten: Alle Abweichungen (Kri-stallbaufehler), die zu lokalen Veränderungen von Netzebenenorientierungen füh-ren, rufen Beugungskontraste hervor. Mit einigen solcher Abweichungen von der idealen Gitterstruktur und ihren Auswirkungen auf das elektronenmikroskopische Bild wollen wir uns im Folgenden beschäftigen.
Als Erstes stellen wir uns einen auf eine Dicke von weniger als 100 nm präpa-rierten Einkristall als Probe vor. Es ist nicht verwunderlich, wenn dieser wahrhaft „hauchdünne“ Kristall verbogen ist (vgl. Bild 6-11).
Die Netzebenen, von denen in Bild 6-11 nur diejenigen nahe der Bragg-Lage skizziert sind, folgen dieser Verbiegung des Kristalls, d. h. ihre Orientierung schwankt etwas. Folglich gibt es kleine Probenbereiche mit Netzebenen, deren Winkel zum Elektronenstrahl exakt dem Bragg-Winkel entspricht. Der Teil des Elektronenstrahls, der diesen Bereich trifft, wird demzufolge mit höherer Inten-sität in die durch den Bragg-Winkel vorgegebene Richtung gestreut, seine Intensi-tät in Durchgangsrichtung ist reduziert.

Kapitel 6 130
Bild 6-11. Ursache der Biegekonturen: Gekrümmte Probe mit unterschiedlichen Winkeln zwischen Netzebenen und einfallendem Elektro-nenstrahl. Ergebnis: Dunkle Linie in der Bildebene.
Die gestreute Intensität wird durch die Kontrastblende aus dem Strahlengang ent-fernt, die in Bragg-Lage befindlichen Probenbereiche erscheinen im Bild dunkel. In Bild 6-11 ist dies durch die dunkle Linie in der Bildebene illustriert. Wie bereits im Abschnitt 6.2 erwähnt, muss in Wirklichkeit die Bragg-Bedingung wegen des zulässigen Anregungsfehlers bei der Elektronenbeugung nur näherungsweise erfüllt sein. Deshalb beobachten wir keine scharfen sondern „verwaschene“ Biegekonturen (englisch: Bending contours). Außerdem ist nicht zu erwarten, dass die durch die elektronenmikroskopische Präparation eingebrachten mechanischen Spannungen lediglich zu einer Krümmung des Kristalls in einer Ebene führen. Wir müssen vielmehr mit Verdrehungen des Kristalls rechnen, die gekrümmte Biegekonturen zur Folge haben.
Bild 6-12. Biegekonturen in einem gedünnten Silizium-Kristall. Vor Aufnahme von Bild b) wurde der Kristall um 2° gegenüber dem Zustand in Bild a) gekippt. Der Kristall ist gerissen, die Risslinie (Pfeil) verschiebt sich bei Kippung nicht.

Warum sehen wir Kontraste im Bild? 131
Werden Probe oder Elektronenstrahl gekippt, so geraten andere Probenbereiche in die Bragg-Lage. Im Bild „wandern“ die Biegekonturen über die Probe (s. Bild 6-12). Der im Hellfeldmodus abgebildete Silizium-Kristall wurde gedimpelt und mit Argon-Ionen bis zum Durchbruch ionengedünnt (vgl. Abschnitt 3.3). Links unten ist in den Bildern ein Stück des Lochrandes zu sehen (Streuabsorptionskontrast). An dessen Form ist zu erkennen, dass es sich tatsächlich um die gleiche Proben-stelle handelt, nur die Biegekonturen haben sich infolge der Kippung innerhalb der Probe verschoben.
Versetzungen sind eine andere Möglichkeit, lokale Gitterverzerrungen hervor-zurufen. Bei einer Stufenversetzung ist eine zusätzliche Netzebene in das Gitter eingeschoben (vgl. Bild 6-13). In der Umgebung des Versetzungskerns (hier be-ginnt die eingeschobene Netzebene) relaxieren die Atome, was zu einer Krüm-mung der Netzebenen führt. Es passiert das Gleiche wie bei den Biegekonturen: Bei geeigneter Elektroneneinstrahlrichtung gerät ein Teil der gekrümmten Netz-ebenen in die Bragg-Lage, die an dieser Stelle einfallenden Elektronen werden verstärkt gestreut, und im Bild entsteht eine dunkle Linie, die die gleiche Richtung wie die eingeschobene Netzebene hat.
Bild 6-13. Entstehung des Beugungskontrastes an einer Stufenversetzung. In der Skizze ist nur der Probenbereich dargestellt, die Anordnung von Objektiv und Kontrastblende sind wie in Bild 6-11 dargestellt.
Im Unterschied zu den Biegekonturen ist die Gitterverzerrung aber auf den Be-reich um den Versetzungskern beschränkt, d. h. beim Kippen der Probe wandert die dunkle Linie nicht, sondern sie verschwindet bei hinreichend großem Kipp-winkel. Dies ermöglicht es dem Experimentator, zwischen Versetzungslinie und Biegekontur zu unterscheiden und unterstreicht, wie wichtig es ist, dass die Probe beim Mikroskopieren gekippt werden kann.
Bei der Stufenversetzung tritt die Gitterverzerrung nur längs einer Linie auf und beschränkt sich auf eine Netzebenenschar. Dies führt einerseits dazu, dass die Sichtbarkeit der Versetzungslinie von der Beobachtungsrichtung abhängt, ermög-licht aber andererseits die Lagebestimmung der Versetzung. Wir wollen uns dies anhand der Skizze in Bild 6-14 veranschaulichen.

Kapitel 6 132
Bild 6-14. Atomanordnung bei einer Stufenversetzung in verschiedenen Projektionen: Richtungen 1 und 2: keine Verzerrung, d. h. keine un-terschiedlichen Netzebenen-orientierungen bezüglich der Einfallsrichtung. Richtung 3: Verschiebungen der Atome in Projektionsrichtung sichtbar, d. h. Variation der Netz-ebenenorientierung.
Wir erkennen, dass die Versetzungslinie im Hellfeldbild bei der Elektronenein-fallsrichtung k03 mit hohem Bildkontrast als dunkle Linie zu sehen sein wird. Für die weitere Diskussion nehmen wir an, es handele sich um einen kubischen Kri-stall und die drei Einfallsrichtungen stimmen (bis auf das Vorzeichen) mit den Kristallrichtungen [100], [010] und [001] überein. In unserem Fall sind die (100)-Ebenen gekrümmt. Zur kristallografischen Beschreibung der Versetzung dient der Burgers47-Vektor.
Bild 6-15. Burgers-Umlauf um den Versetzungskern zur Erläuterung des Burgers-Vektors. Ausgangspunkt ist das schwarz umrandete Atom. Von dort gehen wir fünf Atome nach unten (Pfeile mit u1 – u5 gekennzeichnet). Dann sieben Atome nach links (l1 – l7), acht Atome nach oben (o1 – o8) und wieder sieben Atome nach rechts (r1 – r7). Da wir die gleiche Anzahl Atome nach unten wie nach oben gehen müssen, fehlen drei Schritte nach unten. Dies wird nach-geholt (u6 – u8) und wir landen beim Nachbar des Ausgangsatoms. Die Lücke wird ausgehend vom Aus-gangsatom durch den Bur-gers-Vektor b geschlossen.
Seine Richtung und sein Betrag werden durch einen „Burgers-Umlauf“ bestimmt, wie das für unser Beispiel in Bild 6-15 erläutert ist. Wir schauen dazu auf die Git-
47 Johannes Martinus Burgers, niederländischer Physiker, 1895 - 1981

Warum sehen wir Kontraste im Bild? 133
terprojektion in k02-Richtung und greifen die Stelle um den Versetzungskern heraus.
Wir erkennen, dass der Burgers-Vektor senkrecht auf der Versetzungslinie steht (hier Kristallrichtung [100]). Dies ist das Kennzeichen einer Stufenver-setzung. Wir sehen außerdem, dass für die Skalarprodukte der drei Einfallsrich-tungen mit dem Burgers-Vektor gilt:
( )0, 0, 0, denn cos ,⋅ ≠ ⋅ = ⋅ = ⋅ = ⋅ ⋅0,1 02 03 0,i 0,i 0,ik b k b k b k b k b k b . (6.10)
Nehmen wir Dunkelfeldbilder „im Lichte“ des (010)- bzw. des (001)-Reflexes auf, so bleibt die Versetzungslinie in diesen Bildern unsichtbar. Das Kreuzprodukt aus diesen beiden Reflexrichtungen liefert die Richtung des Burgers-Vektors: [100]. Dies ist die Grundlage für die Bestimmung von Burgers-Vektoren mit Hilfe von transmissionselektronenmikroskopischen Abbildungen. In der Praxis sind dabei drei wichtige Dinge zu beachten:
1. Die Probe muss während der mikroskopischen Beobachtung in geeignete kristallografische Richtungen orientiert werden können. Der Einsatz eines Doppel-kipphalters ist deshalb bei solchen Untersuchungen zwingend erforderlich.
2. Vor dem Einzeichnen des Burgers-Vektors in das elektronenmikroskopische Bild ist eine evtl. Bilddrehung zwischen Beugungs- und Abbildungsmodus mit Hilfe dazu geeigneter Objekte (z.B. orthorhombische α-MoO3-Nadeln, deren Ach-sen die kristallografische [001]-Richtung haben, und die kommerziell verfügbar sind) zu kontrollieren und in der Zeichnung zu korrigieren. Wir erinnern uns: Die Elektronen laufen auf Schraubenbahnen durch die magnetischen Linsen. Beim Umschalten von Abbildung auf Beugung werden die Projektivlinsen unter-schiedlich erregt (Abbildung der Zwischenbildebene bzw. der bildseitigen Brenn-ebene des Objektivs), was zu der erwähnten Bilddrehung führen kann. In moder-nen Elektronenmikroskopen wird diese Bilddrehung in der Regel durch geeignetes Zusammenspiel der Projektivlinsen kompensiert. Doch das sollte man kontrol-lieren!
3. Im Abschnitt 6.3 hatten wir erläutert, dass Dunkelfeldbilder vorteilhafter-weise durch Kippung des Elektronenstrahls erzeugt werden sollten. Dies ist bei Versetzungsuntersuchungen zu überdenken, weil die Einfallsrichtung eine ent-scheidende Rolle spielt und diese beim Kippen verändert wird. Unter Umständen muss für die Dunkelfeldabbildung die Kontrastblende verschoben werden, es sei denn, die Strahlkippung ist beabsichtigt (Weak-Beam-Technik).
Die Stufenversetzung, bei der die Versetzungslinie nahezu parallel zur Proben-oberfläche verläuft, diente uns zur Erläuterung des Prinzips der Kontrastentste-hung. Es gibt weitere Versetzungskonfigurationen: Die Schraubenversetzung, bei der der Burgers-Vektor parallel zur Versetzungslinie verläuft, und Versetzungen, bei denen die Versetzungslinie gegen die Probenoberfläche geneigt ist und innerhalb der Probe endet.

Kapitel 6 134
Der letzte Fall führt zu Krümmungen mehrerer Netzebenenscharen, so dass diese Enden der Versetzungslinien bei mehreren Elektroneneinfallsrichtungen zu sehen sind. Sie dominieren deshalb häufig in Bildern, die ohne Auswahl einer speziellen Elektroneneinfallsrichtung aufgenommen worden sind (vgl. Bild 6-16).
Bild 6-16. Abbildung von Versetzungen im TEM. a) Krümmung der Netzebenen am Ende einer Stufenversetzung. b) TEM-Hellfeldbild von Al: keine Versetzungen sichtbar. c) Gleiche Proben-stelle, aber Probe um 8° gekippt: Versetzungslinien und –enden sichtbar.
Schließlich wollen wir eine dritte Möglichkeit der Entstehung lokaler Gitterver-zerrungen diskutieren: Semikohärente Ausscheidungen. Darunter wollen wir Parti-kel verstehen, die in eine Matrix eingelagert sind und deren kristallografische Struktur und Gitterkonstanten denjenigen der Matrix sehr ähnlich sind. Bild 6-17 zeigt eine Skizze, der die Annahme zugrunde liegt, dass die Gitterkonstante des Partikels kleiner als die der umgebenden Matrix ist.
Bild 6-17. Abbildung semikohärenter Ausscheidungen im TEM. a) Netzebenenanpassung durch Verzerrung und Einschub (Stufenversetzung). b) Perspektivische Darstellung der Verzerrungen. c) TEM-Bild von 20 nm großen Kupfer-Partikeln in einer Blei-Matrix.
Typisch für semikohärente Ausscheidungen ist ein kaffeebohnenartiger Kontrast im elektronenmikroskopischen Bild. Voraussetzung ist selbstverständlich eine ge-eignete Einstrahlrichtung wie bei allen Untersuchungen, die den Beugungskontrast ausnutzen.

Warum sehen wir Kontraste im Bild? 135
6.5 Dickenkonturen, Stapelfehler und Zwillinge Wir wollen uns nun mit Kontrastphänomenen in transmissionselektronenmikro-skopischen Bildern von kristallinen Proben beschäftigen, die nicht allein durch Verzerrungen von Netzebenen und den damit verbundenen lokal begrenzten Bragg-Lagen erklärt werden können. Wir betrachten Bild 6-18. Es zeigt die Korn-struktur innerhalb einer abgedünnten Kupferschicht. Neben zahlreichen Kontra-sten durch lokale Verzerrungen der Netzebenen sehen wir am Rand eines Korns dunkle Streifen, deren Ursache wir verstehen wollen.
Dazu erinnern wir uns an die Beugung der Elektronenwelle im Kristallgitter. Bei den Überlegungen zum Braggschen Gesetz (Abschnitt 5.1) waren wir von einer partiellen Reflexion der Elektronenwelle an der Netzebenenschar ausge-gangen und hatten danach nicht weiter verfolgt, was noch mit dem gebeugten Wellenanteil im Kristall passieren kann.
Bild 6-18. Dickenkonturen am Rand eines Korns in einer dünnen Cu-Schicht (s. Pfeil).
Um die Streifen in Bild 6-18 erklären zu können, müssen wir unser Modell erweitern und betrachten dazu Bild 6-19.
Wir gehen von einem Kristall aus, dessen Dicke am Rand keilförmig ansteigt (Bild 6-19a). Die im Ausschnitt skizzierten Netzebenen bilden mit der Elektronen-einfallswelle den Bragg-Winkel θ, d. h. der Kristall befindet sich in exakter Bragg-Lage. In unserem einfachen Modell soll das Streuvermögen der Netzebenen gleich sein und die gesamte Intensität der Elektronenwelle an einer Netzebene reflektiert werden. Sie erreicht dann die benachbarte Netzebene und wird wieder in die ursprüngliche Richtung reflektiert (vgl. Bild 6-19b). Infolge der Vielfachreflexion pendelt die Intensität periodisch zwischen der durchgehenden und der gestreuten Welle.

Kapitel 6 136
Die Länge der Periode wird als Extinktionslänge Lext bezeichnet. Wir erkennen, dass die Probendicke bestimmt, ob die durchgehende oder die gestreute Welle die intensitätsreichere nach Verlassen der Probe ist. Offenbar ist im Proben-dickenintervall (i = 0, 1, 2, ..)
( )12i iext extL t L⋅ ≤ < + ⋅ (6.11)
die gestreute Welle intensitätsreicher, und im Dickenintervall
( ) ( )12i i 1ext extL t L+ ⋅ ≤ < + ⋅ (6.12)
ist hingegen die durchgehende Welle diejenige mit der größeren Intensität. Die gestreute Intensität wird durch die Kontrastblende aus dem Strahlengang entfernt und wir erhalten im Bild helle und dunkle Streifen, die als Dickenkonturen be-zeichnet werden.
Bild 6-19. Zur Erläuterung von Dickenkonturen. a) Kristalline Probe mit keilförmig ansteigender Dicke in exakter Bragg-Lage: Elektronenwelle pendelt zwischen reflektierenden Netzebenen. b) Bragg-Winkel θ, Netzebenenabstand d, Extinktionslänge Lext und Probendicke t.
Nach Bild 6-19b gilt für kleine Bragg-Winkel θ:
2
2extL d
θ
⋅= (6.13)
und nach dem Braggschen Gesetz (5.6) für n = 1 (erste Beugungsordnung) mit der Elektronenwellenlänge λ

Warum sehen wir Kontraste im Bild? 137
24ext
dL
λ
⋅= . (6.14)
Die Netzebenenabstände sind orientierungsabhängig. Wir schreiben deshalb statt d besser dhkl. Damit ist auch die Extinktionslänge orientierungsabhängig:
2
,4 hkl
ext hkld
Lλ
⋅= . (6.15)
Nach diesem einfachen geometrischen Modell erhalten wir beispielsweise für die (111)-Netzebenenschar von Kupfer (Gitterkonstante: 0,362 nm) und 300 keV-Elektronen (λ = 0,00197 nm) eine Extinktionslänge von 91 nm. In unserem Mo-dell haben wir das Streuvermögen der Netzebenen (Strukturfaktor und Volumen der Elementarzelle) außer Acht gelassen, welches die Extinktionslänge ebenfalls beeinflusst. Formel (6.15) liefert demzufolge nur die Größenordnung der Extink-tionslänge.
Durch Zählen der dunklen Streifen ist es möglich, die Dicke des Kristalls abzuschätzen: Wir multiplizieren die Streifenzahl mit der Extinktionslänge. Da die Elektronenwelle in Wirklichkeit nur teilweise von den Netzebenen reflektiert wird, „verwaschen“ die Dickenkonturen mit zunehmender Probendicke.
In diesem Abschnitt sind wir bis hierher von einem ungestörten Gitter ausge-gangen. Was passiert aber, wenn Unregelmäßigkeiten im Netzebenenverlauf auf-treten und dadurch deren Streuvermögen lokal verändert ist? Stapelfehler sind typische Vertreter solcher Störungen. Zur Erklärung des Stapelfehlers nehmen wir an, dass nur eine Atomsorte vorliegt und die Atome „auf Lücke“ gestapelt sind (vgl. Bild 6-20a). An der mit „Stapelfehler“ gekennzeichneten Stelle ist diese Anordnung gestört, die Atome sind nicht auf Lücke gestapelt. Im dreidimen-sionalen Fall entstehen dadurch Änderungen in der atomaren Besetzung der Netz-ebenen (s. Bild 6-20b). Diese Abweichungen von der regulären Atomanordnung führen zu zusätzlicher Phasenschiebung beim Durchgang der Welle durch das Kristallgitter. Wir veranschaulichen uns dies in dem Modell zur Erklärung der Dickenkonturen durch kleine Lücken in den Netzebenen, die Veränderungen im Reflexionsverhalten zur Folge haben (Bild 6-20c).
Bild 6-20. Kontrastentstehung an einem Stapelfehler. a) Entstehung des Stapelfehlers. b) Perspektivische Darstellung eines Stapelfehlers. c) Auswirkung auf die Elektronenwelle.

Kapitel 6 138
In unserem Modell befindet sich eine Netzebenenschar in Bragg-Lage. Wie bei den Dickenkonturen pendelt die Intensität zwischen durchgehender und gestreuter Welle. Im ungestörten Kristallbereich dominiert die Intensität der durchgehenden Welle, d. h. der Kristall erscheint im elektronenmikroskopischen Hellfeldbild hell. Die Lücken führen dazu, dass im Bereich des Stapelfehlers die Intensität auf die gebrochene Welle umverteilt ist, d. h. der Bereich des Stapelfehlers erscheint im Bild dunkel.
Bis hierher sind wir von einem Stapelfehler ausgegangen, der nahezu senkrecht zur Einfallsrichtung der Elektronenwelle orientiert ist. Bei Neigungen gegen diese Richtung kann der Stapelfehler auch als dünner, dunkler Streifen abgebildet wer-den oder es erscheinen am Rand des Stapelfehlerbildes zusätzliche Dickenkon-turen (helle und dunkle Streifen).
Die mit dem Stapelfehler verbundenen Verschiebungen der Atome bewirken eine Veränderung der Gesamtenergie des Systems; die Differenz wird als Stapel-fehlerenergie bezeichnet. Aufgrund der zwischenatomaren Kräfte sind einige we-nige Stapelfehlerkonfigurationen bevorzugt.
Bild 6-21. Zwei mögliche Stapelungen atomarer Schichten „auf Lücke“.
Wir wollen nun zwei der verschiedenen Möglichkeiten der Stapelung von atomaren Schichten betrachten (s. Bild 6-21). Die beiden Möglichkeiten (1) und (2) unterscheiden sich seitlich um einen halben Atomabstand und in der Höhe um 3
6 Atomabstände. Wenn innerhalb des Kristalls von der Stapelvariante (1) auf die Variante (2) umgeschaltet wird, entstehen gespiegelte Netzebenen.
Bild 6-22. Zwillinge durch Stapelfehler. a) Perspektivische Skizze. b) Erläuterung des resultierenden Beugungskontrastes. c) Elektronenmikroskopisches Hellfeldbild von Zwillingen in einer Bi-Te-Legierung.

Warum sehen wir Kontraste im Bild? 139
Ein Beispiel ist in Bild 6-22a durch die schwarze Linie hervorgehoben. Diese gespiegelten Netzebenen sind typisch für Kristallzwillinge, die Spiegelebenen werden als Zwillingsebenen bezeichnet.
Wir hatten im vorhergehenden Abschnitt diskutiert, welchen Einfluss die Kri-stallorientierung auf den Kontrast des elektronenmikroskopischen Bildes hat. Wenn der Kristall so zur einfallenden Elektronenwelle orientiert wird, dass der mittlere Netzebenenbereich in Bild 6-22b in Bragg-Lage kommt, dann wird nach dem Ausblenden der stark gestreuten Intensitäten dieser Teil des Zwillings im Bild dunkel erscheinen (vgl. Bilder 6-22b und c). Auch hier ändern sich die Kontraste beim Kippen der Probe.
6.6 Moiré-Muster Wir wollen nun den Fall betrachten, dass zwei Kristalle in Elektroneneinfallsrich-tung (wird auch als Projektionsrichtung bezeichnet) übereinanderliegen. Die Netz-ebenen repräsentieren periodische Strukturen, deren Überlagerung zusätzliche Streifen, sogenannte „Moiré-Muster“48 zur Folge haben. In Bild 6-23 sind drei mögliche Varianten skizziert.
Bild 6-23. Moire-Muster durch Überlagerung von Kristallen. a) Verdrehung mit gleichen Netzebenenabständen. b) Ohne Verdrehung mit unterschiedlichen Netzebenenabständen. c) Mit Verdrehung bei unterschiedlichen Netzebenenabständen.
Die Streifenabstände h können mit Hilfe der Formeln
2 sin2
dh
δ=
⋅
(6.16)
aus dem Netzebenenabstand d und dem Drehwinkel δ im Fall von Bild 6-23a,
1 2
1 2
d dh
d d
⋅=
− (6.17)
aus den beiden Netzebenenabständen d1 und d2 im Fall von Bild 6-23b und
48 vom französischen „moirer”: marmorieren

Kapitel 6 140
1 2
2 21 2 1 22 cos
d dh
d d d d δ
⋅=
+ − ⋅ ⋅ ⋅
(6.18)
aus Netzebenenabständen und Drehwinkel im Fall Bild 6-23c berechnet werden (→ Abschnitt 10.13).
Das elektronenmikroskopische Bild 6-24 zeigt ein praktisches Beispiel für die Moiré-Streifen. Sie dürfen nicht mit evtl. größeren Netzebenenabständen verwech-selt werden.
Bild 6-24. Moiré-Streifen (weiße Markierung) durch Überlagerung zweier TiN-Kristalle in Projektions-richtung im transmissions-elektronenmikroskopischen Bild.
Für die Beugung bedeuten die übereinanderliegenden Kristalle, dass starke Beu-gungsintensitäten des ersten Kristalls beim Durchlaufen des zweiten Kristalls zu-sätzliche Reflexe erzeugen können. Prinzipiell ist dies auch innerhalb eines ein-zelnen dickeren Kristalls möglich. Dieses Phänomen wird als Doppelbeugung be-zeichnet; das betreffende Beugungsmuster kann als Linearkombination der einzel-nen Beugungsbilder verstanden werden.
6.7 Magnetische Domänen: Lorentzmikroskopie Nach der Diskussion der Kontrastentstehung durch die Orientierungsabhängigkeit der Elektronenwelleninterferenzen wollen wir zum Teilchenmodell zurückkehren und überlegen, wie die Elektronen in einer ferromagnetischen Probe abgelenkt werden und wie diese Ablenkung für Kontraste im elektronenmikroskopischen Bild sorgen kann. Die ferromagnetische Probe besteht aus Domänen („Weisssche

Warum sehen wir Kontraste im Bild? 141
Bezirke“49), innerhalb derer die magnetischen Momente der Atome (Elementar-magnete) gleichgerichtet sind. Die einzelnen Domänen sind durch Bloch50-Wände voneinander getrennt. Innerhalb einer Blochwand dreht die Magnetisierungsrich-tung. Benachbarte Domänen haben häufig antiparallel ausgerichtete Magnetisie-rungen.
Ein Ausschnitt aus einer solchen ferromagnetischen Probe mit antiparallel aus-gerichteten Domänen ist in Bild 6-25 gezeichnet. Die Elektronen folgen beim Durchgang durch die Probe der Lorentzkraft
e= − ⋅ ×F v B (2.4)
mit der Elementarladung e, der Elektronengeschwindigkeit v und der magneti-schen Induktion B. Diese Formel war uns bereits im Kapitel 2.2. begegnet, wo die Funktion rotationssymmetrischer Magnetfelder als Elektronenlinsen erläutert wur-de. Von dort ist uns auch bekannt, welche Richtung die Lorentzkraft hat. Die mag-netische Induktion hat die gleiche Richtung wie die Magnetisierung. Dies ist in Bild 6-25 berücksichtigt. Die Ablenkung der Elektronen in der Probe hat in der Ebene 1 unterhalb der Probe eine Modulation der Elektronendichte zur Folge: Im Bereich der Blochwände, die Domänen mit der Magnetisierung nach hinten auf der linken Seite und solche mit der Magnetisierung nach vorn auf der rechten Sei-te trennen, ist die Elektronendichte geringer. Dies ist durch schwarze Streifen in Ebene 1 gekennzeichnet. Blochwände zwischen Domänen mit nach vorn gerichte-ter Magnetisierung links und nach hinten gerichteter Magnetisierung rechts haben eine erhöhte Elektronendichte zur Folge, was durch helle Streifen symbolisiert wird. In der Probenebene 0 tritt diese Modulation der Elektronendichte nicht auf.
Bild 6-25. Elektronendichte in verschiedenen Ebenen nach Ablenkung in antiparallel ausgerich-teten magnetischen Domänen innerhalb einer ferromagnetischen Probe. Die schwarzen Pfeile kennzeichnen die Magnetisierungsrichtung.
Wir wissen bereits, dass im Elektronenmikroskop durch Veränderung der Linsen-brennweite verschiedene Ebenen auf dem Leuchtschirm abgebildet werden kön-nen. So ist es möglich, sowohl die Probenebene 0 (was der exakten Fokussierung entspricht) als auch die Ebene 1 (was als Überfokussierung bezeichnet wird) auf 49 Pierre-Ernest Weiss, französischer Physiker, 1865 - 1940 50 Felix Bloch, schweizer Physiker, 1905 – 1983, Nobelpreis für Physik 1952

Kapitel 6 142
dem Leuchtschirm abzubilden. Im ersten Fall sind die Domänengrenzen nicht sichtbar, im zweiten Fall erzeugen die unterschiedlichen Elektronendichten einen Kontrast, die Domänengrenzen heben sich im Bild als dunkle und helle Linien heraus.
Die Variation der Linsenbrennweite hat eine weitere Konsequenz. Wir können auch die Ebene 2 oberhalb der Probe abbilden. Die rückwärtigen Verlängerungen der Elektronen-Ablenkungsrichtungen führen zu einer Umkehr des Kontrastes in dieser Ebene, d. h. bei Erhöhung des Linsenstromes vom Unterfokus zum Überfokus sehen wir zunächst die Domänenwände als helle und dunkle Linien, im scharfen Bild werden sie unsichtbar und tauchen schließlich mit umgekehrtem Kontrast als dunkle und helle Linien wieder auf.
Bei der praktischen Ausführung dieser Lorentzmikroskopie gibt es ein schwer-wiegendes Problem: Normalerweise befindet sich die Probe inmitten des sehr starken Objektiv-Magnetfeldes. Die Magnetisierungen richten sich nach dem äußeren Magnetfeld aus, die Blochwände verschwinden. Um dies zu vermeiden, ist eine spezielle Objektivlinse (Lorentzlinse) notwendig, bei der das Magnetfeld in Probenumgebung deutlich abgeschwächt ist. Unglücklicherweise wird dadurch der Öffnungsfehler vergrößert, so dass dies bei rotationssymmetrischen Linsen zur Verschlechterung des Auflösungsvermögens führt. Eine andere Möglichkeit be-steht darin, die konventionelle Objektivlinse vollkommen auszuschalten oder zu-mindest mit drastisch reduziertem Spulenstrom zu betreiben. Dies ist einfach zu bewerkstelligen, indem im geringen Vergrößerungsbereich (englisch: Low Magnification) gearbeitet wird. Die (geringere) Vergrößerung wird dann im We-sentlichen mit den Projektivlinsen erreicht, was auch dramatische Konsequenzen für das Auflösungsvermögen hat. Der Vorteil ist, dass man sich den Kauf der teu-ren Lorentzlinse und den Geräteumbau erspart.
Die elektronenmikroskopischen Bilder 6-26 von magnetischen Domänen in einem nanoskaligen Kobalt-Balken wurden im „Low-Mag“-Betrieb des Objektivs aufgenommen [6.1]. Die Probengeometrie ist Folge der Querschnittspräparation eines Co/Cu/Co-Schichtstapels auf Silizium-Substrat.
Bild 6-26. Lorentzmikroskopie an einem Querschnittspräparat von einem Co/Cu/Co-Schicht-stapel. a) Unterfokus. b) Fokus. c) Überfokus. Der Pfeil kennzeichnet die gleiche Probenstelle.
Die obere Co-Schicht (Co2) ist teilweise oxidiert und nicht ferromagnetisch. Die Co-Schicht auf dem Substrat (Co1) zeigt das erwartete Verhalten: Die Domänen-

Warum sehen wir Kontraste im Bild? 143
grenzen sind im fokussierten Bild 6-26b unsichtbar. Beim Übergang von Unterfo-kus zu Überfokus kehrt sich der Kontrast um.
Bild 6-27. Erklärung des Bildkontrastes in Bild 6-26. a) Überlagerung der grün (Unterfokus) und rot (Überfokus) kolorierten Bilder 6-26a und c. b) Skizze zur Erklärung der Magnetisie-rungsrichtungen in den Domänen.
Unter Zugrundelegung des in Bild 6-25 skizzierten Sachverhaltes können aus den elektronenmikroskopischen Bildern die Magnetisierungsrichtungen festgelegt werden, die zu den beobachteten Bildkontrasten führen (s. Bild 6-27).

7 Wir erhöhen die Vergrößerung
Ziel: Wir wollen uns nun mit der Abbildung von Strukturen beschäftigen, deren Größen nahe der Auflösungsgrenze des Transmissionselektronenmikroskops lie-gen. Die Atomabstände in den Kristallgittern sind in dieser Größenordnung. Auch die Untersuchung von Korngrenzen und anderen Grenzflächen auf atomarer Skale gehört dazu. Wir wollen aber auch die Effekte bedenken, die bei der hochver-größerten und hochaufgelösten Abbildung amorpher Materialien auftreten.
7.1 Abbildung von Atomsäulen in Kristallgittern: Phasenkontrast Wir wollen mit einem Beispiel beginnen. Dazu betrachten wir die Elementarzelle eines Siliziumkristalls aus verschiedenen Richtungen. Silizium bildet ein Dia-mantgitter, welches durch die Raumgruppe 227 ( 3Fdm ) mit den Basis-Atom-positionen 0,25; 0,25; 0,25 beschrieben wird. Wir stellen uns dazu zwei kubisch flächenzentrierte Gitter vor, wobei das zweite um ein Viertel auf der Raum-diagonale des ersten verschoben ist (vgl. Bild 7-1a). Die Gitterkonstante beträgt 0,543 nm.
Bild 7-1. Kristallstruktur von Silizium (Diamant-gitter). a) Eine Elementar-zelle. b) Nach Vervielfäl-tigung der Elementarzelle in beliebiger Blickrichtung auf den Kristall.
Bei der beliebigen Blickrichtung auf das Kristallgitter in Bild 7-1b können wir keinerlei Ordnung erkennen. Die Atome überlappen sich in Projektionsrichtung, einzelne Atomsäulen sind nicht zu erkennen. Dies ändert sich, wenn wir den Kri-stall drehen und beispielsweise in Richtung einer Würfelkante der Elementarzelle auf den Kristall schauen, wie dies in Bild 7-2a gezeigt ist.
Das elektronenmikroskopische Bild (HRTEM-Bild, aus dem Englischen: High Resolution TEM, d.h. Hochauflösungs-TEM) in Bild 7-2b entspricht genau der Gitterprojektion entlang der Würfelachse der Si-Elementarzelle (vgl. Bild 7-2c).
DOI 10.1007/978-3-7091-1440-7_7, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 7 146
Aus Kapitel 5 wissen wir, dass die Würfelachsen kristallografisch mit [100], [010] oder [001] bezeichnet werden. Im kubischen Kristall sind diese Achsen gleichbe-rechtigt, zusammenfassend schreibt man dafür <100>. Wir sehen, dass die Indizes dieser Richtung klein sind. Solche niedrig indizierten Richtungen sind für die Ab-bildung vorteilhaft, weil in diesem Fall die projizierten Atomabstände vergleichs-weise groß sind.
Bild 7-2. Interpretation eines hochaufgelösten elektronenmikroskopischen Bildes von Silizium. a) Siliziumgitter in [100]-Richtung. b) HRTEM-Bild von Si. c) HRTEM-Bild mit überlagerter Elementarzelle von Si in [100]-Projektion.
Wir wollen nun überlegen, wieso wir die Atomsäulen im transmissionselektronen-mikroskopischen Bild sehen können, welcher Kontrastmechanismus dafür verant-wortlich ist. Aus Kapitel 6 sind uns zwei Kontrastarten bekannt: Streuabsorptions- und Beugungskontrast. Streuabsorptionskontrast erfordert unterschiedliche Mas-sendicken, Beugungskontrast unterschiedliche Orientierungen des Kristallgitters gegen die Richtung des einfallenden Elektronenstrahls. Bei der Abbildung des Si-liziumkristalls ist keine der beiden Voraussetzungen erfüllt. Es muss also noch einen weiteren Kontrastmechanismus geben, der insbesondere bei der Hochauf-lösungsabbildung im Transmissionselektronenmikroskop von Bedeutung ist.
Um diesen Kontrastmechanismus zu verstehen, erinnern wir uns an den Wel-lencharakter der Elektronen und bedenken die Wechselwirkung der Elektronen-welle mit den periodisch angeordneten Atomen im Kristallgitter. Aus der Schrö-dinger-Gleichung (→ Abschnitt 10.2) folgt, dass die Wellenlänge λKr der Elektro-nenwelle innerhalb eines Potentials Φ nach
2
h
2 m eKr
pλ =
+ ⋅ ⋅ ⋅Φ
(7.1)
(h: Plancksches Wirkungsquantum, p: Impuls des Elektrons, m: Elektronenmasse, e: Elementarladung) berechnet werden kann. In nichtrelativistischer Näherung gilt für den Zusammenhang zwischen Impuls p, Energie E und Beschleunigungsspan-nung UB:

Wir erhöhen die Vergrößerung 147
2 2 m 2 m e Bp E U= ⋅ ⋅ = ⋅ ⋅ ⋅ , (7.2)
d.h. mit Berücksichtigung der Ortsabhängigkeit des Potentials im periodischen Kristallgitter (vgl. Bild 7-3):
( )( )
h
2 m e , ,Kr
BU x y zλ =
⋅ ⋅ ⋅ + Φ
. (7.3)
Bild 7-3. Veranschauli- chung des Gitterpotentials. a) Festlegung des Koordinatensystems. b) Schema des Kristall-potentials in der x-y-Ebene.
Außerhalb des Kristalls (Φ = 0) beträgt die Wellenlänge
h
2 m e BUλ =
⋅ ⋅ ⋅ . (7.4)
Im Kristall (positives Potential) verkürzt sich demnach die Wellenlänge, was in-nerhalb einer Kristallschicht der Dicke dz eine Phasenschiebung dφ zur Folge hat:
2 2 1Kr Kr
dz dz dzd
λφ
λ λ λ λ= ⋅ ⋅ − = ⋅ ⋅ ⋅ − (7.5)
Mit (7.3) und (7.4) folgt daraus
( )( )
( )
2 m e , ,2 1
2 m e
, ,2 1 1
B
B
B
U x y zdzd
U
x y zdz
U
φλ
λ
⋅ ⋅ ⋅ + Φ= ⋅ ⋅ ⋅ −
⋅ ⋅ ⋅
Φ= ⋅ ⋅ ⋅ + −
. (7.6)

Kapitel 7 148
Bei den gebräuchlichen Beschleunigungsspannungen im Transmissionselektro-nenmikroskop ist UB >> Φ(x,y,z) und damit
( ), ,1
B
x y z
U
Φ (7.7)
Wir entwickeln den Wurzelausdruck in (7.6) in eine Taylor-Reihe bei
( ), ,0
B
x y z
U
Φ= , (7.8)
brechen diese nach dem zweiten Glied ab und erhalten
( )( )
, ,12 1 1 , ,
2 B B
x y zdzd x y z dz
U Uφ
λ λ
Φ= ⋅ ⋅ + − = ⋅Φ ⋅
⋅ . (7.9)
Die Phasenschiebung φ bei einer Probe der Dicke t erhalten wir durch Integration:
( )0
, ,t
B
x y z dzU
φλ
= ⋅ Φ ⋅⋅
. (7.10)
Unter der Voraussetzung, dass das Potential nur von x und y abhängt und längs z konstant bleibt, gilt
( ) ( ), ,B
tx y x y
Uφ
λ
⋅= ⋅Φ
⋅ , (7.11)
Bild 7-4. Phasenmodulation durch das Kristallpotential beim Durchgang einer einfallenden ebenen Elek-tronenwelle durch ein Kristallgitter.
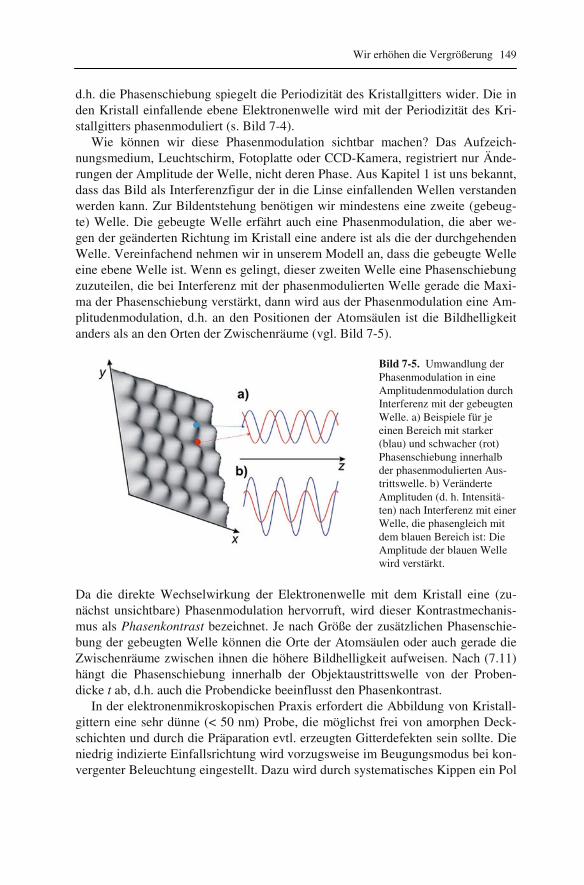
Wir erhöhen die Vergrößerung 149
d.h. die Phasenschiebung spiegelt die Periodizität des Kristallgitters wider. Die in den Kristall einfallende ebene Elektronenwelle wird mit der Periodizität des Kri-stallgitters phasenmoduliert (s. Bild 7-4).
Wie können wir diese Phasenmodulation sichtbar machen? Das Aufzeich-nungsmedium, Leuchtschirm, Fotoplatte oder CCD-Kamera, registriert nur Ände-rungen der Amplitude der Welle, nicht deren Phase. Aus Kapitel 1 ist uns bekannt, dass das Bild als Interferenzfigur der in die Linse einfallenden Wellen verstanden werden kann. Zur Bildentstehung benötigen wir mindestens eine zweite (gebeug-te) Welle. Die gebeugte Welle erfährt auch eine Phasenmodulation, die aber we-gen der geänderten Richtung im Kristall eine andere ist als die der durchgehenden Welle. Vereinfachend nehmen wir in unserem Modell an, dass die gebeugte Welle eine ebene Welle ist. Wenn es gelingt, dieser zweiten Welle eine Phasenschiebung zuzuteilen, die bei Interferenz mit der phasenmodulierten Welle gerade die Maxi-ma der Phasenschiebung verstärkt, dann wird aus der Phasenmodulation eine Am-plitudenmodulation, d.h. an den Positionen der Atomsäulen ist die Bildhelligkeit anders als an den Orten der Zwischenräume (vgl. Bild 7-5).
Bild 7-5. Umwandlung der Phasenmodulation in eine Amplitudenmodulation durch Interferenz mit der gebeugten Welle. a) Beispiele für je einen Bereich mit starker (blau) und schwacher (rot) Phasenschiebung innerhalb der phasenmodulierten Aus-trittswelle. b) Veränderte Amplituden (d. h. Intensitä-ten) nach Interferenz mit einer Welle, die phasengleich mit dem blauen Bereich ist: Die Amplitude der blauen Welle wird verstärkt.
Da die direkte Wechselwirkung der Elektronenwelle mit dem Kristall eine (zu-nächst unsichtbare) Phasenmodulation hervorruft, wird dieser Kontrastmechanis-mus als Phasenkontrast bezeichnet. Je nach Größe der zusätzlichen Phasenschie-bung der gebeugten Welle können die Orte der Atomsäulen oder auch gerade die Zwischenräume zwischen ihnen die höhere Bildhelligkeit aufweisen. Nach (7.11) hängt die Phasenschiebung innerhalb der Objektaustrittswelle von der Proben-dicke t ab, d.h. auch die Probendicke beeinflusst den Phasenkontrast.
In der elektronenmikroskopischen Praxis erfordert die Abbildung von Kristall-gittern eine sehr dünne (< 50 nm) Probe, die möglichst frei von amorphen Deck-schichten und durch die Präparation evtl. erzeugten Gitterdefekten sein sollte. Die niedrig indizierte Einfallsrichtung wird vorzugsweise im Beugungsmodus bei kon-vergenter Beleuchtung eingestellt. Dazu wird durch systematisches Kippen ein Pol
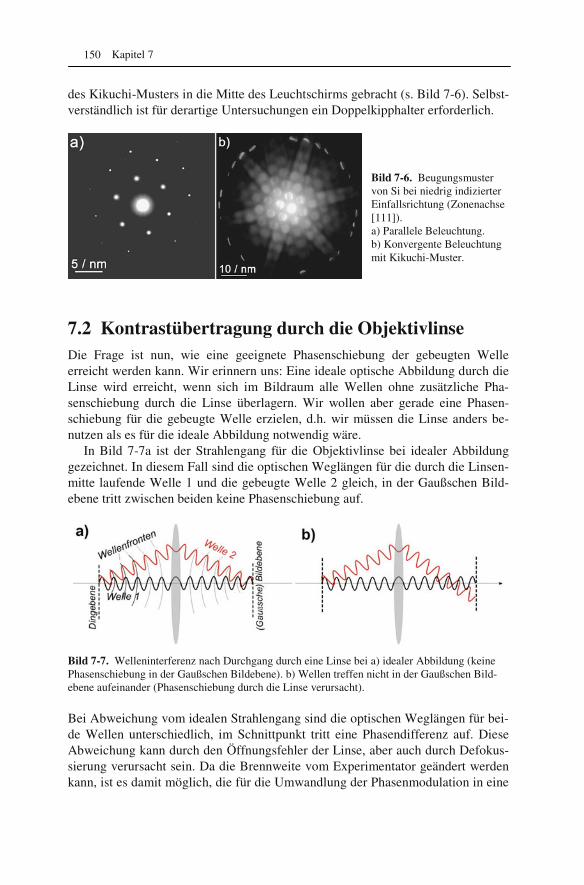
Kapitel 7 150
des Kikuchi-Musters in die Mitte des Leuchtschirms gebracht (s. Bild 7-6). Selbst-verständlich ist für derartige Untersuchungen ein Doppelkipphalter erforderlich.
Bild 7-6. Beugungsmuster von Si bei niedrig indizierter Einfallsrichtung (Zonenachse [111]). a) Parallele Beleuchtung. b) Konvergente Beleuchtung mit Kikuchi-Muster.
7.2 Kontrastübertragung durch die Objektivlinse Die Frage ist nun, wie eine geeignete Phasenschiebung der gebeugten Welle erreicht werden kann. Wir erinnern uns: Eine ideale optische Abbildung durch die Linse wird erreicht, wenn sich im Bildraum alle Wellen ohne zusätzliche Pha-senschiebung durch die Linse überlagern. Wir wollen aber gerade eine Phasen-schiebung für die gebeugte Welle erzielen, d.h. wir müssen die Linse anders be-nutzen als es für die ideale Abbildung notwendig wäre.
In Bild 7-7a ist der Strahlengang für die Objektivlinse bei idealer Abbildung gezeichnet. In diesem Fall sind die optischen Weglängen für die durch die Linsen-mitte laufende Welle 1 und die gebeugte Welle 2 gleich, in der Gaußschen Bild-ebene tritt zwischen beiden keine Phasenschiebung auf.
Bild 7-7. Welleninterferenz nach Durchgang durch eine Linse bei a) idealer Abbildung (keine Phasenschiebung in der Gaußschen Bildebene). b) Wellen treffen nicht in der Gaußschen Bild-ebene aufeinander (Phasenschiebung durch die Linse verursacht).
Bei Abweichung vom idealen Strahlengang sind die optischen Weglängen für bei-de Wellen unterschiedlich, im Schnittpunkt tritt eine Phasendifferenz auf. Diese Abweichung kann durch den Öffnungsfehler der Linse, aber auch durch Defokus-sierung verursacht sein. Da die Brennweite vom Experimentator geändert werden kann, ist es damit möglich, die für die Umwandlung der Phasenmodulation in eine

Wir erhöhen die Vergrößerung 151
Amplitudenmodulation notwendige Phasenschiebung zwischen durchgehender und gebeugter Welle einzustellen und das Kristallgitter sichtbar zu machen.
Wir wollen diese Möglichkeit mit Hilfe einer Bildsimulation unter Benutzung der für diese Zwecke sehr gebräuchlichen Software JEMS von P. Stadelmann [7.1] demonstrieren (vgl. Bild 7-8). Dazu dient ein Kupfer-Kristall, den wir genau senkrecht von oben (kristallografische [001]-Richtung) betrachten.
Das projizierte Potential (Bild 7-8b) spiegelt exakt die Positionen der Kupfer-atome wider. Bei einer Defokussierung von 79 nm erscheinen genau diese Positio-nen im berechneten Hochauflösungsbild hell (Bild 7-8c), bei einer Defokussierung von 99 nm demgegenüber dunkel (Bild 7-8d).
Bild 7-8. Einfluss der Fokussierung auf den Phasenkontrast. a) Cu-Gitter in [001]-Projektion (4x4 Elementarzellen). b) Projiziertes Potential. c) Berechnetes HRTEM-Bild (UB = 300 kV, CS = 1,2 mm, CC =2 mm, ΔE = 1 eV, t = 11 nm, Defokus Δf = 79 nm) d) wie c, aber Δf = 99 nm.
Neben der einstellbaren Linsenbrennweite (Defokus Δf, d.h. Abweichung der eingestellten Brennweite von derjenigen für Fokussierung in der Gaußschen Bild-ebene) und dem Öffnungsfehler (gekennzeichnet durch die Öffnungsfehlerkon-stante CS) hat allerdings auch der Winkel θ der gebeugten Welle gegen die opti-sche Achse einen Einfluss auf die Phasenschiebung φ durch die Linse:
( ), ,SC fφ φ θ= Δ . (7.12)
Im → Abschnitt 10.14 wird dies mathematisch abgehandelt. Wir wollen hier den Sachverhalt plausibel erklären.
Wird für einen bestimmten Beugungswinkel θ die Phasenschiebung φ so einge-stellt, dass im Bild der Gangunterschied zwischen durchgehender und gebeugter Welle ein ganzzahliges Vielfaches der Elektronenwellenlänge λ beträgt, tritt Ver-stärkung, d.h. hohe Bildhelligkeit, ein. Bei einem Gangunterschied gleich dem un-geradzahligen Vielfachen von λ/2 erfolgt Auslöschung, d.h. geringe Bildhellig-keit. Aus dem Braggschen Gesetz (5.6) folgt, dass der Winkel θ umgekehrt pro-portional zum Netzebenenabstand d ist. Diese Aussage verallgemeinern wir: Der Winkel θ ist umgekehrt proportional zum Abstand d zweier abzubildender Objekt-details:

Kapitel 7 152
1
dθ . (7.13)
Der Term 1/d (reziproke Länge) wird auch als Raumfrequenz q bezeichnet (mit-unter wird dafür auch 2π/d benutzt). Die Phasenschiebung φ hängt demzufolge von der Raumfrequenz ab:
( ), ,Sq C fφ φ= Δ . (7.14)
Welche Konsequenzen hat das für die Abbildung? Wir nehmen an, dass für einen herausgegriffenen Detailabstand d1 bei gegebenem Öffnungsfehler ein Fokus ge-wählt wird, der diese beiden Details im Bild infolge konstruktiver Interferenz von durchgehender und gebeugter Welle mit großer Helligkeit, d.h. mit gutem Kon-trast, darstellt. Bei anderem Detailabstand d2 ändert sich der Beugungswinkel θ und damit auch die Phasenschiebung φ. Die Bedingung für maximale Verstärkung bei der Interferenz ist nicht mehr erfüllt, der Kontrast für diese Details ist geringer. Das andere Extrem ist eine abstandsbedingte Phasenschiebung, die bei Interferenz zur Auslöschung führt. Mit anderen Worten: Der Bildkontrast hängt von der Größe der abzubildenden Strukturen ab. Diese dramatische Konsequenz ist in Bild 7-9 veranschaulicht.
Bild 7-9. Kontrastübertra-gung in Abhängigkeit vom Detailabstand: a) Bei Phasenschiebung, die zu maximaler Verstärkung führt. b) Zwischenstadium. c) Bei Phasenschiebung, die zur Auslöschung führt.
Variierende Abstände zwischen den Bilddetails führen zu vollkommen unter-schiedlichen Kontrastverhältnissen. Ohne genaues Verständnis dieser Zusammen-hänge wird die Bildinterpretation zweifelhaft:
„Glaube erst was du siehst, wenn du verstanden hast, warum du es siehst!“

Wir erhöhen die Vergrößerung 153
7.3 Wellenoptische Deutung des Auflösungsvermögens Die im vorherigen Abschnitt beschriebene Abhängigkeit des Bildkontrastes vom abgebildeten Abstand (d. h. Raumfrequenz) wird für die anschauliche Erläuterung in einer oft guten Näherung durch die lineare Phasenkontrastübertragungsfunktion CTF beschrieben. Mit Kenntnis der Kontrastübertragungsfunktion (→ Abschnitt 10.14)
( ) ( )
22 2 2 4
03 4 2sin 2 e2
CE
C qE
SCTF q C q f qλ
λ λ
Δ− ⋅ ⋅ ⋅
= ⋅ ⋅ − ⋅Δ ⋅ ⋅ ⋅ (7.15)
(q: Raumfrequenz, CS: Öffnungsfehlerkonstante, λ: Elektronenwellenlänge, Δf: Defokus, d.h. Differenz der aktuellen Brennweite zu derjenigen für Fokussierung in der Gaußschen Bildebene, CC: Farbfehlerkonstante, ΔE: Energiebreite der Elek-tronen, E0: Primärelektronenenergie) wollen wir uns erneut mit der Definition des Auflösungsvermögens beschäftigen. Das Argument der Sinusfunktion zeigt, dass der Einfluss des Öffnungsfehlers durch geeignete Defokussierung gemindert werden kann. Die „beste“ Kontrastübertragungseigenschaft, d.h. die höchste Raumfrequenz, bis zu der die Bilder einfach zu interpretieren sind, wird bei einer geringfügigen Defokussierung erhalten. Diese Überlegung führt zum Scherzer-Fokus (→ Abschnitt 10.15)
1, 2Sch Sf C λΔ = ⋅ ⋅ . (7.16)
Ein Beispiel für die Kontrastübertragungsfunktion ist in Bild 7-10 zu sehen.
Bild 7-10. Kontrastüber-tragungsfunktion für E0 = 300 keV, ΔE = 0,7 eV (Schottky-Feldemissions-kathode), CS = 1,2 mm, CC = 1,5 mm, Δf = 58 nm (Scherzer-Fokus).
Raumfrequenzen bis qδ werden ohne Kontrastumkehr übertragen, bis dahin treten keine Probleme bei der Bildinterpretation wegen der oszillierenden Kontrastüber-tragungsfunktion auf. Allerdings werden auch höhere Raumfrequenzen über-tragen, wenn auch mit variierendem Kontrast. Es gibt deshalb zwei unter-schiedliche Raumfrequenzen, die als Grenzen definiert werden.

Kapitel 7 154
Der Kehrwert von qδ wird als Punktauflösung bezeichnet:
1P qδ
δ = . (7.17)
Nach (7.15) gilt für die erste Nullstelle:
3 4 2
2
2
2
S
S
C q f q
fq
C
δ δ
δ
λ λ
λ
⋅ ⋅ = ⋅Δ ⋅ ⋅
⋅Δ=
⋅
(7.18)
und bei Benutzen des Scherzer-Fokus (7.16):
2 2 4 34
2 1, 2 2, 42, 4S S
S S S
C Cq
C C Cδ
λ λ
λ λ λ
⋅ ⋅ ⋅ ⋅= = ⋅ =
⋅ ⋅ ⋅
(7.19)
d.h. für das Punktauflösungsvermögen
340,645P SCδ λ= ⋅ ⋅ . (7.20)
Der Radikand stimmt mit dem aus Formel (2.18) überein, der Faktor vor der Wurzel ist bei der wellenoptischen Betrachtungsweise kleiner (0,645 statt 0,9). Für die in Bild 7-9 genannten Parameter folgt eine Punktauflösung von 0,20 nm.
Der zweite Grenzwert ist diejenige Raumfrequenz, die unabhängig von den Oszillationen noch übertragen wird. Dafür ist die Dämpfung verantwortlich. Bei der Festlegung dieses Grenzwertes herrscht eine gewisse Willkür. Wie üblich be-nutzen wir dafür die Raumfrequenz, bei der die Amplitude der Kontrastüber-tragungsfunktion auf 1/e2 = 0,135 gesunken ist. Der Kehrwert dieser Raumfre-quenz wird als Informationslimit bezeichnet und berechnet nach
lim0
1,49 CE
CE
δ λΔ
= ⋅ ⋅ ⋅ (7.21)
(→ Abschnitt 10.14). Das Informationslimit für den Parametersatz von Bild 7-10 beträgt demnach 0,12 nm.
Die hier beschriebene Näherung wird für eine korrekte Beschreibung um die wechselseitige Überlagerung aller mit unterschiedlichem Wellenvektor auftreten-den Wellen, die durch einen Transmissions-Kreuzkoeffizienten beschrieben wird,

Wir erhöhen die Vergrößerung 155
ergänzt. Rechenprogramme zur HRTEM-Bildsimulation können dies berücksich-tigen, eine anschauliche Erläuterung ist jedoch nicht möglich.
7.4 Periodische Helligkeitsverteilung in Bildern: Fourieranalyse Bei der Abbildung sehr kleiner Strukturen im Bereich des Auflösungsvermögens des Transmissionselektronenmikroskops spielt offenbar die Raumfrequenz eine bedeutende Rolle für den Bildkontrast. Der Begriff „Frequenz“ erinnert an die harmonische Analyse von Schwingungsvorgängen, d.h. die Darstellung beliebiger periodischer Zeitfunktionen mit einer Schwingungsdauer T durch eine Summe von Sinus- und Kosinusfunktionen mit unterschiedlichen Vorfaktoren Ak und Bk sowie Vielfachen einer Grundfrequenz ω = 2π/T im Argument der trigonometrischen Funktionen:
( ) ( ) ( )01 1
cos sinn n
k kk k
f t A A k t B k tω ω= =
= + ⋅ ⋅ ⋅ + ⋅ ⋅ ⋅ . (7.22)
Die Vorfaktoren werden als Fourierkoeffizienten51 bezeichnet. Sie wichten die Si-nus- und Kosinusfunktionen für unterschiedliche Frequenzen und vermitteln damit Informationen über bevorzugt auftretende Periodizitäten. Wenn wir die Zeit t durch den Ort x und die Frequenz ω durch die Raumfrequenz q = 1/d ersetzen, erhalten wir
( ) ( ) ( )01 1
cos 2 sin 2n n
k kk k
f x A A k q x B k q x= =
= + ⋅ ⋅ ⋅ ⋅ + ⋅ ⋅ ⋅ ⋅ , (7.23)
d.h. die Fourierkoeffizienten geben an, welche periodischen Abstände (gekenn-zeichnet durch Vielfache einer Basisraumfrequenz) dominieren.
Mit anderen Worten: Die Fourieranalyse (bzw. –transformation) liefert Infor-mationen über auftretende Periodizitäten, in einem Bild sind dies Helligkeits-schwankungen, z. B. in Form von parallelen Streifen oder Gittern.
Bei digitalisierten Bildern wird eine numerische Methode benutzt, mit deren Hilfe ermittelt wird, welche sinus- bzw. kosinusförmige Schablone unterschiedli-cher Periodenlänge die im Bild vorhandene Helligkeitsmodulation am besten be-schreibt. Wir wollen diese Methode zunächst an einer einzelnen Bildzeile erklä-ren.
51 Joseph Fourier, französischer Mathematiker, 1768 - 1830

Kapitel 7 156
Bild 7-11. Beschreibung einer periodischen Helligkeitsmodulation einer Bildzeile durch Kosinusfunktionen unterschiedlicher Periodenlänge (bzw. Raumfrequenz) mit zugehörigem „Übereinstimmungsparameter“ S. Die beste Übereinstimmung wird in Bildteil c) erreicht. In Bild 7-11 ist die periodische Helligkeitsmodulation einer Bildzeile durch das Quadrat einer Kosinusfunktion dargestellt. Darüber sind als „Schablonen“ Kosi-nusfunktionen mit unterschiedlichen Periodenlängen gelegt. Offenbar stimmt die Periodenlänge der Variante in Bild 7-11c mit derjenigen der Helligkeitsmodu-lation überein. Der „Übereinstimmungsparameter“ S für eine Raumfrequenz q wird nach
( ) ( ) ( )cos 2k kk
S q I x q x= ⋅ ⋅ ⋅ (7.24)
errechnet. Für die Raumfrequenz mit optimaler Übereinstimmung erreicht S ein Maximum. Damit ist die numerische Fourieranalyse möglich. Das Ergebnis wird in einem Bild mit S als Helligkeitswert dargestellt (s. Bild 7-12). Zur besseren Sichtbarkeit ist die Bildzeile in senkrechter Richtung auseinander gezogen.
Bild 7-12. Bildzeilen mit Helligkeitsmodulation unter-schiedlicher Periodenlänge mit zugehörigen Fourier-transformierten (FT).

Wir erhöhen die Vergrößerung 157
Wir erkennen die umgekehrte Proportionalität: Je größer die Periodenlänge in der Bildzeile, desto kleiner der Abstand der Helligkeitsmaxima in der Fouriertransfor-mierten.
Wir übertragen dies auf ein (zweidimensionales) Bild. Das Bild habe Nx Pixel in horizontaler und Ny Pixel in senkrechter Richtung. Die Helligkeit des Pixels an der Stelle (x,y) sei I(x,y). Die Helligkeit des Pixels (qx, qy) im fouriertransformier-ten Bild kann dann nach
( ) ( ) ( ) ( )11
0 0
, , cos 2 cos 2yx
NN
x y x yx y
I q q I x y q x q y−−
= =
= ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ (7.25)
berechnet werden. qx und qy sind Raumfrequenzen, d.h. reziproke Längen. Anstelle der Kosinus- kann auch die Sinusfunktion oder eine Kombination aus
beiden in Form einer komplexen Funktion
( ) ( )2 ie cos 2 i sin 2q x q x q x⋅ ⋅ ⋅ = ⋅ ⋅ + ⋅ ⋅ ⋅ (7.26)
benutzt werden. Im letzteren Fall sind die Ergebnisse der Fouriertransformation komplexe Zahlen, im Bild wird deren Betrag als Helligkeit angezeigt. Dieses Bild wird auch als Powerspektrum bezeichnet.
Bild 7-13 zeigt das Ergebnis der Transformation eines Gitterbildes mit quadra-tischen Maschen nach Formel (7.24).
Bild 7-13. Fouriertrans-formation eines Bildes mit quadratischem Gitter. a) Originalbild. b) Fouriertransformiertes Bild.
Zurück zum Elektronenmikroskop: Periodische Atomanordnungen in kristallinen Proben haben Beugungsmuster zur Folge, die in der bildseitigen Brennebene der Objektivlinse beobachtet werden können. Nach dem Braggschen Gesetz ist der Abstand eines Beugungsreflexes vom Zentrum bei kleinen Beugungswinkeln um-gekehrt proportional zum Netzebenenabstand, d. h. direkt proportional zur Raum-frequenz. Das Beugungsbild kann demzufolge auch als Analyse der in der Probe auftretenden periodischen Netzebenenabstände interpretiert werden. Die Fourier-transformation des abbildungsfehlerfreien elektronenmikroskopischen Netzebe-nenbildes einer kristallinen Probe liefert also die gleiche Information wie das Beu-gungsbild dieser Probe.

Kapitel 7 158
7.5 Streuabsorptions- und Phasenkontrast Mit der Fouriertransformation und ihrer Inversion haben wir eine Methode ge-funden, um die Auswirkungen der Kontrastblende auf das elektronenmikrosko-pische Bild zu studieren. Die Kontrastblende ist in Höhe der bildseitigen Brenn-ebene des Objektivs angeordnet (vgl. Abschnitt 6.2), in der auch das Beugungsbild zu beobachten ist. Wir erkennen die Analogie: Ein mathematisches Filter im fouriertransformierten Bild hat die gleiche Wirkung wie die Kontrastblende. Nach Anwendung dieses Filters und anschließender Rücktransformation in den Ortsraum erhalten wir das Analogon zum elektronenmikroskopischen Bild (s. Bild 7-14).
Bild 7-14a ist das Ausgangsbild, im Elektronenmikroskop entspräche das der Probe. Es könnte sich dabei um vier unregelmäßig geformte Kristallite auf einer amorphen Trägerfolie handeln. In den „Kristalliten“ ist die periodische Atom-anordnung modelliert. Die Fouriertransformierte dieses Bildes (7-14b) zeigt die strenge Periodizität der Helligkeitsmodulation innerhalb der „Kristallite“. Nach Filterung im Fourierraum gemäß Bild 7-14d und Rücktransformation sehen wir in 7-14c keine „Atomsäulen“ mehr sondern nur noch die Form der „Kristallite“. Auf das Elektronenmikroskop übertragen bedeutet dies, dass die an den „Atomsäulen“ gebeugten Wellen durch die Kontrastblende ausgeblendet werden, eine Interferenz mit ihnen wird unmöglich, die Information über die kleinen Strukturen in der Probe geht verloren. Nur die kleinen Raumfrequenzen werden übertragen, Streu-absorptionskontrast ist sichtbar.
Bild 7-14. Einfluss der Kontrastblende auf Streuabsorptions- und Phasenkontrast (Erläuterungen im Text).
Bei größerer Kontrastblende werden auch gebeugte Wellen durch die Kontrast-blende hindurchgelassen, Interferenz ist möglich und im Bild sind sowohl die Um-risse der „Kristallite“ als auch die „Atomsäulen“ im Innern zu sehen (Bilder 7-14e
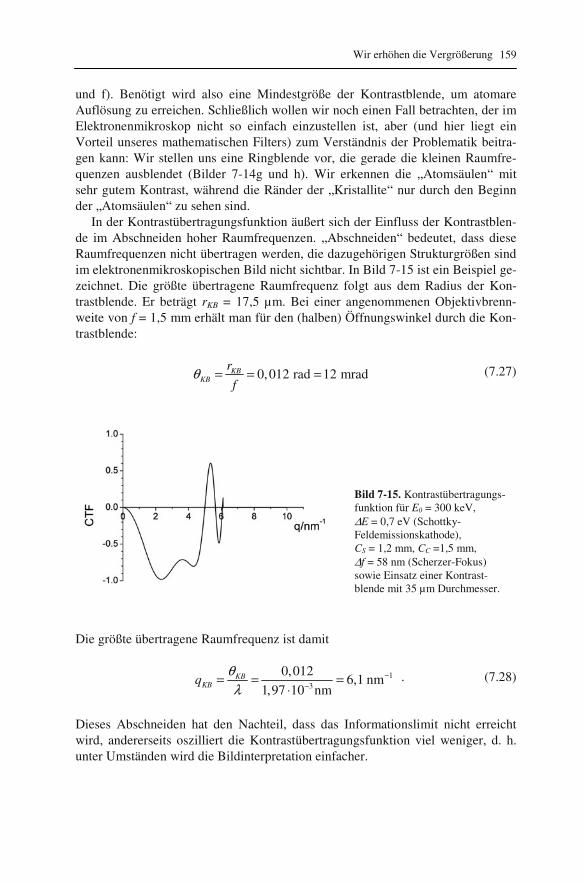
Wir erhöhen die Vergrößerung 159
und f). Benötigt wird also eine Mindestgröße der Kontrastblende, um atomare Auflösung zu erreichen. Schließlich wollen wir noch einen Fall betrachten, der im Elektronenmikroskop nicht so einfach einzustellen ist, aber (und hier liegt ein Vorteil unseres mathematischen Filters) zum Verständnis der Problematik beitra-gen kann: Wir stellen uns eine Ringblende vor, die gerade die kleinen Raumfre-quenzen ausblendet (Bilder 7-14g und h). Wir erkennen die „Atomsäulen“ mit sehr gutem Kontrast, während die Ränder der „Kristallite“ nur durch den Beginn der „Atomsäulen“ zu sehen sind.
In der Kontrastübertragungsfunktion äußert sich der Einfluss der Kontrastblen-de im Abschneiden hoher Raumfrequenzen. „Abschneiden“ bedeutet, dass diese Raumfrequenzen nicht übertragen werden, die dazugehörigen Strukturgrößen sind im elektronenmikroskopischen Bild nicht sichtbar. In Bild 7-15 ist ein Beispiel ge-zeichnet. Die größte übertragene Raumfrequenz folgt aus dem Radius der Kon-trastblende. Er beträgt rKB = 17,5 μm. Bei einer angenommenen Objektivbrenn-weite von f = 1,5 mm erhält man für den (halben) Öffnungswinkel durch die Kon-trastblende:
0,012 rad 12 mradKBKB
r
fθ = = = (7.27)
Bild 7-15. Kontrastübertragungs-funktion für E0 = 300 keV, ΔE = 0,7 eV (Schottky-Feldemissionskathode), CS = 1,2 mm, CC =1,5 mm, Δf = 58 nm (Scherzer-Fokus) sowie Einsatz einer Kontrast-blende mit 35 μm Durchmesser.
Die größte übertragene Raumfrequenz ist damit
13
0,0126,1 nm
1,97 10 nmKB
KBqθ
λ
−
−= = =
⋅ . (7.28)
Dieses Abschneiden hat den Nachteil, dass das Informationslimit nicht erreicht wird, andererseits oszilliert die Kontrastübertragungsfunktion viel weniger, d. h. unter Umständen wird die Bildinterpretation einfacher.

Kapitel 7 160
Da die gebeugten Wellen von kleinen Strukturen, z. B. den Netzebenen eines Kristalls, größere Winkel mit der optischen Achse des Objektivs einschließen als diejenigen von gröberen Strukturen, die sich durch Streuabsorptionskontrast dar-stellen, sind die gebeugten Wellen dem Einfluss des Öffnungsfehlers stärker un-terworfen. Dies hat eine Verschiebung des Netzebenenbildes gegenüber dem Streuabsorptionsbild, eine sogenannte Delokalisation zur Folge (→ Abschnitt 10.16). Diese Delokalisation δD hängt vom Öffnungsfehler (Öffnungsfehler-konstante CS), der Defokussierung Δf, der Elektronenwellenlänge λ und der Raumfrequenz q ab:
( )2 2D Sq C q fδ λ λ= ⋅ ⋅ ⋅ ⋅ + Δ . (7.29)
Sie kann einige Nanometer betragen und erschwert es beispielsweise, im Bild die Grenze zwischen amorphen und kristallinen Probenbereichen festzulegen.
7.6 Kontrast bei amorphen Proben Im Abschnitt 5.6 hatten wir beschrieben, dass die Atome im amorphen Material nicht gitterartig angeordnet sind, wie das von Kristallen bekannt ist. Es gibt keine Fernordnung und auch nicht die damit verbundene Richtungsanisotropie. Trotz-dem ist die Anordnung der Atome im amorphen Material nicht vollkommen zufäl-lig: Es existiert eine Nahordnung in Form wahrscheinlichster nächster Nachbar-abstände. Aus Abschnitt 5.6 wissen wir auch, dass diese Nahordnung durch Ent-faltung von Streukurven, die im Beugungsmodus mit dem Transmissionselektro-nenmikroskop gemessen werden, charakterisiert werden kann.
Bild 7-16. Elektronenmikroskopische Bilder von einer dünnen amorphen Siliziumschicht (Parameter: E0 = 300 keV, CS = 1,2 mm, CC = 1,2 mm, ΔE ≈ 1 eV (Schottky-Feldemissions-kathode). a) Fokussiert. b) Defokussiert.

Wir erhöhen die Vergrößerung 161
Leichtsinnigerweise könnte man annehmen, dass diese Vorzugsabstände in einem Transmissionselektronenmikroskop mit hohem Auflösungsvermögen bei hoher Vergrößerung auch abgebildet werden können. Wir wollen diskutieren, warum diese Vermutung falsch ist. Dazu betrachten wir Bild 7-16. In beiden Teilbildern ist dieselbe Probenstelle zu sehen. Dennoch sind die Bildeindrücke vollkommen verschieden: Im rechten Bild 7-16b sind die Strukturen scheinbar größer als im linken Bild 7-16a, obwohl zwischen beiden Bildern nur die Fokussierung geändert wurde. Offenbar sehen wir keine Strukturen der Probe sondern die Eigenschaft des Abbildungssystems. Um dies genauer zu verstehen, überlegen wir, welche Bilder von Strukturen in der amorphen Schicht überhaupt zu erwarten sind, und wir erinnern uns an die Kontrastübertragung durch die Objektivlinse.
Wenn wir im Gedankenexperiment durch die dünne amorphe Schicht schauen, sehen wir lediglich die Projektion der räumlichen Atompositionen in der Abbil-dungsebene. Im Raum bewirken die Vorzugsabstände periodische Atomdichte-schwankungen, in der Projektion mitteln sich diese Dichteschwankungen heraus. Wir erwarten keinen Kontrast im elektronenmikroskopischen Bild. Für die Kon-trastübertragungsfunktion bedeutet dies, dass eine breite Vielfalt von Raum-frequenzen anfällt, die durch die Objektivlinse in unterschiedlicher Weise übertra-gen werden (vgl. Bild 7-17).
Die Bilderserie 7-17 wurde gänzlich ohne ein Elektronenmikroskop geschaffen. Mit Hilfe eines Bildbearbeitungsprogramms wurde ein Bild mit statistischem („weißem“) Rauschen (Bild 7-16a) erzeugt. Das Bild zeigt keinerlei Kontraste, die von Strukturen in der amorphen Schicht herrühren könnten und entspricht damit vollkommen unserer Erwartungshaltung an ein elektronenmikroskopisches Bild von einer solchen Schicht. Da keine periodischen Kontraste in diesem Bild vor-handen sind, gibt es in seiner Fouriertransformierten 7-17b keine markanten Hel-ligkeitsmaxima. In Bild 7-17c ist die Kontrastübertragungsfunktion für die in Bild 7-16 angegebenen Mikroskopparameter bei eingestelltem Scherzer-Fokus gezeich-net. Darin sind die ersten drei mit gleichem (negativem) Kontrast übertragenen Raumfrequenzbereiche grau unterlegt. Im fouriertransformierten Bild entspricht der Abstand von der Bildmitte der Raumfrequenz. Die Oszillation der Kontrast-übertragungsfunktion kann demnach als Folge von ringförmigen mathematischen Filtern im fouriertransformierten Bild realisiert werden. In den Bildern 7-17d (für Δf = 58 nm) und g (für Δf = 20 nm) sind drei dieser Ringfilter auf das fourier-transformierte Bild angewendet worden. Die Rücktransformation in den Ortsraum liefert das zu erwartende elektronenmikroskopische Bild.
Wir erkennen, dass das defokussierte Bild 7-17h tatsächlich gröbere (Schein)-strukturen zeigt als das Bild 7-17e im Scherzer-Fokus. Die Ursache liegt in der Filterung durch die Kontrastübertragungsfunktion. Ähnlich wie im verrauschten Ausgangsbild treten in der amorphen Probe alle möglichen Atomabstände in Pro-jektionsrichtung auf. Aus dieser Vielfalt filtert die Objektivlinse einige heraus und stellt sie im Bild kontrastreich dar. Beschrieben wird dieses Verhalten durch die Kontrastübertragungsfunktion. Im elektronenmikroskopischen Bild von amorphen

Kapitel 7 162
Proben sehen wir also keine „amorphen Strukturen“ sondern ein Abbild der Kon-trastübertragungsfunktion.
Bild 7-17. Simulation hochvergrößerter Bilder von dünnen amorphen Schichten zur Demonstra-tion des Einflusses der Kontrastübertragungsfunktion. a) Mit Computer erzeugtes, verrauschtes Bild. b) Fouriertransformation von Bild a. c) Kontrastübertragungsfunktion für die in Bild 7-15 angegebenen Parameter bei Scherzer-Fokus (Δf = 58 nm). d) Anwendung der in der Kontrast-übertragungsfunktion grau eingezeichneten Fenster („Filter“) auf das fouriertransformierte Bild b. e) Rücktransformation der gefilterten Fouriertransformierten in den Ortsraum. f) – h) Wiederholung für Defokus Δf = 20 nm.
Oft werden elektronenmikroskopische Hochauflösungsbilder zur Rauschminde-rung „fouriergefiltert“. Dabei muss streng darauf geachtet werden, dass nicht un-typische Bilddetails als typisch herausgehoben werden.

Wir erhöhen die Vergrößerung 163
7.7 Korrektur des Astigmatismus Die Fouriertransformation eines elektronenmikroskopischen Bildes von einer dün-nen amorphen Folie spiegelt nach den Überlegungen des vorhergehenden Ab-schnitts die Kontrastübertragungsfunktion wider. Diesen Sachverhalt wollen wir anhand von Bild 7-18 genauer betrachten. Infolge der Oszillationen der Kontrast-übertragungsfunktionen werden die Raumfrequenzen mit unterschiedlicher Effi-zienz übertragen. Die Intensität interpretieren wir als Quadrat der Kontrastüber-tragungsfunktion. Außerdem lassen wir die Funktion „um die optische Achse rotieren“, d. h. wir erhalten im Powerspektrum zusätzlich eine Information über die Kontrastübertragungsfunktion bei verschiedenen Azimutwinkeln.
Was passiert aber, wenn beispielsweise die Kontrastübertragungsfunktion in senkrechter Richtung (Azimut 90°) eine andere ist als in waagerechter Richtung (Azimut 0°), weil zwischen beiden Schnittebenen eine Brennweitendifferenz besteht? Wir erinnern uns: Dies ist das Kennzeichen des Astigmatismus (vgl. Ab-schnitt 2.3). Die Helligkeitsmaxima im Powerspektrum haben dann unterschied-liche Abstände vom Zentrum, die Ringe werden verzerrt. Damit können bizarre Muster entstehen, bei geeignetem Defokus sind es nahezu Ellipsen (s. Bild 7-19).
Bild 7-18. Kontrastübertragungs-funktionen und Power-Spektren für Defokus Δf = 0 (a und b) sowie Δf = 58 nm (Scherzer-Fokus für UB = 300 kV und CS = 1,2 mm – c und d).
Zur Astigmatismus-Korrektur wird eine dünne amorphe Folie als Probe benutzt. Oft genügt auch eine schmaler amorpher Rand oder Bereich auf dem zu unter-suchenden Objekt.
Bei geeignetem Defokus (einige Ringe müssen zu sehen sein) wird das elek-tronenmikroskopische Bild mit einer CCD-Kamera aufgenommen und synchron zur Bildaufnahme fouriertransformiert.

Kapitel 7 164
Bild 7-19. Kontrastübertragungs-funktionen und Power-Spektren bei Astigmatismus. a) Kontrastübertragungs-funktion im waagerechten Schnitt (Δf = 140 nm). b) Kontrastübertragungs-funktion im senkrechten Schnitt (Δf = 0 nm): c) Resultierendes, berech-netes Powerspektrum. d) Fouriertransformiertes gemessenes Bild einer amorphen Si-Folie mit Astigmatismus. e) Wie d) nach Astigmatis-mus-Korrektur.
Der (Objektiv-)Stigmator (Korrektiv für den Astigmatismus) wird so eingestellt, dass die Ringe kreisförmig sind (s. Bild 7-19e). Zweckmäßigerweise beginnt man die Korrektur im starken Unterfokus (viele Ringe) und verfeinert dann unter Annäherung an den Fokus (s. z. B. auch [7.2]). Wichtigstes Element eines Stigmators ist der Oktupol. Sein Prinzip und seine Wirkungsweise sind in → Abschnitt 10.17 beschrieben.
7.8 Messung des Auflösungsvermögens Bis in die 1970-er Jahre wurden zur Messung des Auflösungsvermögens spezielle Präparate benutzt, die Subnanometer große Schwermetall-Partikel enthielten und somit einen für die fotografische Registrierung ausreichenden Streuabsorptions-kontrast lieferten. Da die Vergrößerung der damaligen Transmissionselektronen-mikroskope auf etwa 100000-fach begrenzt war, mussten die Bilder fotografisch bis zu 10-fach nachvergrößert werden. Um auszuschließen, dass die „dunklen Punkte“ im vergrößerten Bild vom Plattenkorn herrühren, wurden zwei Bilder un-ter gleichen Bedingungen (Defokus) auf zwei verschiedenen Fotoplatten aufge-nommen. Kleine Abstände zwischen „dunklen Punkten“ galten nur dann als signi-fikant für das Auflösungsvermögen, wenn sie auf beiden Platten nachgewiesen werden konnten. Zudem mussten die Abstände in verschiedenen Richtungen auf-treten, um den Einfluss des Astigmatismus auszuschließen.
Später wurden Netzebenenabbildungen, beispielsweise von kleinen Gold-Kri-stallen, zum Nachweis des Auflösungsvermögens genutzt. Damit waren nur dis-krete Werte nachweisbar, nämlich die ausgewählten Netzebenenabstände.

Wir erhöhen die Vergrößerung 165
Heute wird das Auflösungsvermögen (genauer: das Informationslimit) durch Auswertung von Youngschen Interferenzmustern („Young52 fringes“) experimen-tell bestimmt. Als Probe dient eine dünne amorphe Folie. Während der Bildauf-nahme wird die Probe um wenige Nanometer in der Probenebene verschoben, was in der Regel durch geeignete Ablenkung des Elektronenstrahls simuliert wird („Image Shift“). Alternativ können auch zwei Bilder aufgenommen und an-schließend überlagert (addiert) werden, wobei die Probenposition zwischen den beiden Bildern geändert wird.
Dieses resultierende Bild wird fouriertransformiert. Wir erkennen Streifen, deren Kontrast zum Bildrand hin abnimmt (Bild 7-20).
Die Bildverschiebung erzeugt eine zusätzliche periodische Helligkeitsmodula-tion in Verschiebungsrichtung, die in der Fouriertransformierten durch die Streifen dokumentiert ist. Alle Abstände, die im Bild vorhanden sind, werden mit verscho-ben, allerdings sinkt ihr Kontrast aufgrund der Dämpfung der Kontrastübertra-gungsfunktion zu kleineren Abständen (d. h. zu größeren Raumfrequenzen) hin. Abstände, die nicht mehr im Bild dargestellt sind, rufen auch keine Helligkeits-modulation hervor, für die zugehörigen Raumfrequenzen treten keine Interferenz-muster mehr auf.
Bild 7-20. Youngsches Interferenzmuster (amor-pher Rand einer abgedünn-ten Si-Probe) zur Demon-stration der Messung des Informationslimits. Der Kreis markiert eine Raum-frequenz von 6,7 nm-1. Der Si(220)-Reflex liegt bei 5,2 nm-1.
Als Informationslimit gilt der Kehrwert der Raumfrequenz, bis zu der die Young-schen Interferenzmuster reichen. In Bild 7-20 ist diese Grenze durch einen weißen Ring markiert. Das Informationslimit liegt nach dieser Messung bei 0,15 nm.
Diese Methode ist einfach in ihrer Anwendung und wird beispielsweise beim Nachweis der Qualitätsparameter eines Mikroskops benutzt. Kritikpunkt ist, dass dabei nicht zwischen verschiedenen Einflussfaktoren auf den Kontrast der Inter-
52 Thomas Young, englischer Universalgelehrter, 1773 - 1829

Kapitel 7 166
ferenzmuster unterschieden wird. Neben dem eigentlichen Informationslimit wir-ken sich darauf beispielsweise Probendrift, Probendicke und elektronische In-stabilitäten aus [7.3].
7.9 Korrektur von Öffnungs- und Farbfehler Astigmatismus, Öffnungs- und Farbfehler haben eines gemeinsam: Elektronen, die außeraxial durch das Linsenfeld treten, werden „falsch“ abgelenkt. Beim zwei-zähligen Astigmatismus (vgl. Abschnitt 2.3), dessen Korrektur im Abschnitt 7.7 besprochen wurde, betrifft dies die Elektronen in zwei senkrechten Schnitten. Die Korrektur kann prinzipiell mit einem Quadrupol erfolgen, der die Elektronen in einer Ebene (nämlich der mit der kürzeren Brennweite) nach außen zieht und in der Ebene mit der längeren Brennweite nach innen drückt. Um das Feld innerhalb des Korrektors ohne mechanische Verschiebungen zu drehen und zu zentrieren, wird im Allgemeinen ein Oktupol benutzt (→ Abschnitt 10.17).
Der Öffnungsfehler äußert sich dadurch, dass Elektronen in außeraxialen Be-reichen durch das rotationssymmetrische Linsenfeld zu stark abgelenkt werden (vgl. Abschnitt 2.3). Zur Korrektur ist ein Multipol erforderlich, der die Elek-tronen ähnlich wie bei der Korrektur des Astigmatismus in der kurzbrennweitigen Ebene nach außen zieht. Allerdings muss dies für alle Azimute geschehen. Nach H. Rose [7.4] ist dazu ein Hexapol geeignet, der allerdings astigmatische Zwi-schenbilder erzeugt. Zu deren Korrektur dient ein weiterer Hexapol. Grundsätzlich bestehen Korrektoren für den Öffnungsfehler („CS-Korrektoren“ [7.5]) aus Multi-polpaaren und Übertragungslinsen, die dafür sorgen, dass die Zwischenbildebenen „an den richtigen Stellen“ entstehen.
Der Farbfehler entsteht dadurch, dass die Brechkraft der Linse von der Ge-schwindigkeit, d. h. der Wellenlänge der Elektronen, abhängt. Um die Möglichkeit der Korrektur zu verstehen, benutzen wir ein stark vereinfachtes Modell einer magnetischen Elektronenlinse: Wir überlegen, welche Bahnkurve Elektronen der Geschwindigkeit vz0 in einem homogenen Magnetfeld der Induktion By (s. Bild 7-21a) beschreiben.
Bild 7-21. Bewegung von Elektronen in (homogenen) Feldern. a) Magnetfeld. b) Gekreuztes magnetisches und elektrisches Feld (Wien53-Filter).
53 Wilhelm Wien: deutscher Physiker, 1864 – 1928, Nobelpreis für Physik 1911

Wir erhöhen die Vergrößerung 167
Auf das in z-Richtung in das Magnetfeld einfallende Elektron wirkt die Lorentz-kraft
e= − ⋅ ×LF v B (7.30)
bzw. unter Berücksichtigung der in Bild 7-21 gekennzeichneten Richtungen:
0 0e ez y z y xLv B v B F= − ⋅ ⋅ ⋅ × = ⋅ ⋅ ⋅ = ⋅L z y x xF e e e e . (7.31)
Eingesetzt in die Bewegungsgleichung folgt damit für die x-Komponente:
00 0
0 0 00 0
2 20 0 0
0 0
e
m m
e e
m m
1 e 1 e
2 m 2 m
xLx z y
x x z y x z y
x z y z y
Fa v B
v a dt v B t v v B t
x v dt v B t x v B t
= = ⋅ ⋅
= ⋅ = ⋅ ⋅ ⋅ + = ⋅ ⋅ ⋅
= ⋅ = ⋅ ⋅ ⋅ ⋅ + = ⋅ ⋅ ⋅ ⋅
(7.32)
(e/m0: spezifische Ladung des Elektrons). Für die z-Komponente gilt:
0
0
0z
z z
z
a
v v
z v t
=
=
= ⋅
. (7.33)
Dies eingesetzt in (7.32) liefert die gesuchte Gleichung x(z) für die Bahnkurve
2
0 0
1 e
2 my
z
Bx z
v= ⋅ ⋅ ⋅ . (7.34)
Daraus folgt für den Ablenkwinkel:
0 0
etan
my
z
Bdxz
dz vα = = ⋅ ⋅ , (7.35)
d. h. langsamere Elektronen (längere Wellenlänge) werden stärker abgelenkt als schnellere Elektronen.
Zur Korrektur dieser geschwindigkeitsabhängigen Ablenkung benötigen wir ein zweites Magnetfeld in entgegengesetzter Richtung. Dies hätte allerdings zur Folge, das jegliche Ablenkung aufgehoben würde. Ziel ist aber nur eine Korrektur der „falschen Ablenkung“ von Elektronen anderer Geschwindigkeiten als vz0. Wir

Kapitel 7 168
brauchen eine Gegenkraft zur Lorentzkraft. Diese Gegenkraft wird durch ein elek-trisches Feld Ex erzeugt (vgl. Bild 7-21b). Damit erhalten wir für die Gesamtkraft, die das Elektron erfährt:
( )
( )
0
0
e e
e
z y x
z y x x
v B E
v B E F
= + = − ⋅ ⋅ ⋅ × − + ⋅ ⋅
= ⋅ − ⋅ + ⋅ = ⋅
L E z y x
x x
F F F e e e
e e (7.36)
bzw. für die Bahnkurve
22
0 0 0
1 e
2 my x
z z
B Ex z
v v= ⋅ ⋅ − + ⋅ (7.37)
und für den Ablenkwinkel
20 0 0
etan
my x
z z
B Ez
v vα = ⋅ − + ⋅ . (7.38)
Durch das gekreuzte elektrische und magnetische Feld werden Elektronen der Ge-schwindigkeit vz0 nicht abgelenkt, wenn
0
xy
z
EB
v= (7.39)
eingestellt wird. Sind die Elektronen schneller (vz > vz0), so erhalten wir
20 0
etan 0
mx x
z z z
E Ez
v v vα = ⋅ − + ⋅ <
⋅ , (7.40)
sind sie dagegen langsamer (vz < vz0), so gilt
tan 0α > . (7.41)
Mit gekreuzten magnetischen und elektrischen Feldern (Wien-Filter) ist es also möglich, die unterschiedliche Brechung von Elektronen unterschiedlicher Wellen-länge zu korrigieren. In Wirklichkeit ist der Korrektor für den Farbfehler wesent-lich komplizierter aufgebaut. Das Grundprinzip der gekreuzten Felder ist erhalten, sie werden in Form von alternierend angeordneten elektrischen und magnetischen Dipolen in komplexen Quadrupol- und Oktupolsystemen realisiert [7.4]. Die elektronische Stabilität muss extremen Anforderungen genügen [2.2].

Wir erhöhen die Vergrößerung 169
Aus den Abschnitten 7.3, 7.6 und 10.14 wissen wir, dass sowohl Öffnungs-fehler als auch Farbfehler Einfluss auf die Kontrastübertragungsfunktion haben. Prinzipiell bildet sich diese Funktion im Powerspektrum (d. h. nach Fouriertrans-formation) hochaufgelöster Bilder einer dünnen amorphen Folie ab. Ähnlich wie bei der Korrektur des Astigmatismus können auch Öffnungs- und Farbfehler mit Hilfe solcher Powerspektren gemessen werden. Ein einzelnes reicht dafür aller-dings nicht aus, sondern es werden mehrere Bilder unter verschiedenen Kipp- und Azimutwinkeln (wie bei der konischen Dunkelfeldabbildung ohne Benutzung der Kontrastblende – vgl. Abschnitt 6.3) aufgenommen und fouriertransformiert („Zemlin-Tableau“ [7.6]). Daraus werden die Fehlerkonstanten berechnet. Wegen des damit verbundenen großen Rechenaufwandes sind dazu leistungsfähige Com-puter erforderlich. Dies ist einer der Gründe, weshalb derartige Korrektoren erst ab etwa dem Jahre 2000 kommerziell erhältlich sind.
7.10 Interpretation hochaufgelöster Bilder Was kann man tun, um hochaufgelöste Bilder mit Raumfrequenzen im oszil-lierenden Bereich der Kontrastübertragungsfunktion zu interpretieren, wenn kein öffnungsfehlerkorrigiertes Mikroskop zur Verfügung steht, welches bei geeigneter Fokussierung diesen kritischen Bereich nach höheren Raumfrequenzen ver-schiebt?
Dazu gibt es drei Möglichkeiten: 1. Man geht von einer Atomanordnung aus, die aus thermodynamischen oder anderen Überlegungen resultiert, erzeugt daraus eine „Superzelle” mit periodi-schen Anschlüssen (d. h. die Atomanordnungen an den Rändern der Superzelle stimmen überein) und berechnet unter Berücksichtigung der Kontrastüber-tragungsfunktion das resultierende Bild. Leider gibt es dabei mindestens zwei Parameter, die gewöhnlich nicht hinreichend genau bekannt sind: Die Probendicke und die Defokussierung. Deshalb werden diese beiden Parameter variiert und eine Matrix von Bildern berechnet [7.7]. Der Vergleich mit dem gemessenen Bild zeigt, ob die angenommene Atomanordnung geeignet ist, das elektronenmikro-skopische Bild zu erklären. Evtl. muss die Superzelle verändert werden, um ans Ziel zu gelangen. Dieses „trial-and-error“-Verfahren ist u. U. sehr zeitaufwändig.
2. Im Mikroskop wird eine Serie von Bildern bei schrittweise geänderter Fokussierung aufgenommen. Daraus kann die Kontrastübertragungsfunktion und damit die Austrittswellenfunktion rekonstruiert werden („Fokusserienrekonstruk-tion“, „Exit wave reconstruction“ [7.8], [7.9]). Das Problem dabei liegt in der mangelnden Stabilität der Probe. Deren Drift in der Probenebene kann rechnerisch korrigiert werden, nicht aber deren evtl. Verformung. Unkontrollierte Proben-bewegung in z-Richtung (optische Achse) verändert den Defokus-Wert, so dass die Defokus-Schrittweite oft nicht hinreichend genau bekannt ist.
3. Man rechnet „rückwärts“, d. h. man rekonstruiert aus dem Bild die Elektro-nenaustrittswelle aus der Probe (dem Objekt). Dabei wird der Einfluss der Abbil-dungsfehler rechnerisch korrigiert. Da die Welle durch Amplitude und Phase ge-

Kapitel 7 170
kennzeichnet ist, im Bild aber normalerweise nur die Amplitudeninformation auf-gezeichnet ist, muss die Phaseninformation zusätzlich registriert werden. Dies geschieht durch Interferenz der Objektwelle mit einer Referenzwelle und Auf-zeichnung des Interferenzmusters. Die Referenzwelle wird durch Elektronen gebildet, die nicht durch die Probe gehen (z. B. durch das Loch in einer abgedünn-ten Probe oder am Rand einer FIB-Lamelle vorbei). In Höhe der Feinbereichs-blende befindet sich ein dünner Draht auf positivem elektrischen Potential, der Objektwelle und Referenzwelle zueinander ablenkt und überlagert („Holografie“ [7.10]) Mit Hilfe der Holografie können nicht nur Hochauflösungsbilder inter-pretiert, sondern beispielsweise auch elektrische und magnetische Felder in der Probe oder am Rand derselben gemessen werden.

8 Wir schalten um auf Rastertransmissions-elektronenmikroskopie
Ziel: Bei analytischen Untersuchungen im Transmissionselektronenmikroskop ist im Interesse einer hohen räumlichen Auflösung ein nanoskaliges Anregungsgebiet erwünscht. Die Elektronenoptik des Beleuchtungssystems eines TEM (Kondensor-system und Teil des Objektivfeldes vor dem Objekt) ist in der Lage, den Elek-tronenstrahl sehr fein zu fokussieren und damit Elektronensondendurchmesser von weniger als 0,1 nm in der Probenebene zu erzeugen. Ablenksysteme ermöglichen das Rastern dieser feinen Elektronensonde auf der Probe, analog zu dem aus der konventionellen Rasterelektronenmikroskopie bekannten Verfahren. Ähnlich wie beim Namen „Transmission Electron Microscope oder Microscopy – TEM“ hat sich auch für diese Methode ein Kürzel eingebürgert, dessen Ursprung in der englischen Bezeichnung liegt: „STEM“. Es steht für „Scanning Transmission Electron Microscope oder Microscopy“.
Die Unterschiede zur konventionellen Rasterelektronenmikroskopie liegen zum einen in der erreichbaren Kleinheit der Sonde: Aufgrund der günstigen elektro-nenoptischen Bedingungen im TEM (sehr kleiner Arbeitsabstand) gelingt es, die o.g. kleinen Sondendurchmesser im Ångström-Bereich zu erzielen. Zum anderen beruht aufgrund der Durchstrahlbarkeit der Probe der Kontrastmechanismus auf den gleichen Prinzipien wie bei der konventionellen („Ruhebild“-) Transmis-sionselektronenmikroskopie: Wenig gestreute Elektronen gelangen in den Detek-tor, stark gestreute Elektronen nicht („STEM-Hellfeldbild“) oder umgekehrt („STEM-Dunkelfeldbild“). Die elektronentransparenten Proben haben einen ent-scheidenden Vorteil: Im Gegensatz zu den kompakten Objekten im konven-tionellen Rastermikroskop bildet sich in den dünnen Folien kein größeres Anre-gungsgebiet (sogenannte „Anregungsbirne“) aus. Damit gelingt es, auch im STEM-Verfahren eine Auflösung von besser als 0,1 nm zu erreichen.
8.1 Was ändert sich elektronenoptisch? Auf den ersten Blick erscheint es sehr einfach, den Elektronenstrahl auf die Probe zu fokussieren und zeilenweise über die Probe zu führen: Wir stellen die Brennweite des Kondensors 2 (im Falle eines Doppelkondensors) auf konvergente Beleuchtung ein, so wie wir es bereits bei der Feinstrahlbeugung (vgl. Abschnitte 2.7.1 und 5.3) kennengelernt haben. Zum Rastern schwenkt ein Ablenksystem den Elektronenstrahl in zwei senkrechten Richtungen. Bei dieser Verfahrensweise tre-ten zwei Probleme auf:
1. Die Kondensor-2-Linse ist eine langbrennweitige Linse. Ihr Öffnungsfehler ist dementsprechend groß (vgl. Abschnitt 2.3). Wir werden später sehen (Ab-
DOI 10.1007/978-3-7091-1440-7_8, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 8 172
schnitt 8.2), dass es damit nicht gelingt, eine Elektronensonde von weniger als einigen Nanometer im Durchmesser zu erreichen.
2. Der konvergente Elektronenstrahl muss unabhängig von der Position auf der Probe die gleiche Einstrahlrichtung haben (s. Bild 8-1). Einfaches Ablenken in zwei senkrechten Richtungen erfüllt diese Forderung nicht.
Um den Ausweg aufzuzeigen, müssen wir unser Modell von der Objektivlinse verändern. Bisher sind wir von einer einzigen Spule zur Erzeugung des Objektiv-Magnetfeldes ausgegangen. Wir wissen, dass sich die Probe innerhalb des Pol-schuhs befindet. Mit mehreren Spulen kann der Feldteil vor (d.h. oberhalb) der Probe getrennt von dem nach (d.h. unterhalb) der Probe eingestellt werden. Nach unserem neuen Modell besteht die Objektivlinse aus zwei Teilen: einem „Objek-tiv-Vorfeld“ und einem „Objektiv-Nachfeld“. Die Probe befindet sich zwischen beiden, d.h. unmittelbar am Vorfeld. Damit können beide Probleme beseitigt werden.
Bild 8-1. Parallele Einstrahlrichtung des konvergenten Elektro-nenbündels als Voraus-setzung für den STEM-Betrieb des Transmissions-elektronenmikroskops.
Wir benutzen das Vorfeld als zusätzliche Kondensorlinse, was in der Regel als „Nanoprobe-Mode“ bezeichnet wird. Wegen des sehr geringen Arbeitsabstandes (Distanz zwischen letzter Kondensorlinse und Probe) hat das Vorfeld eine kurze Brennweite und damit einen geringen Öffnungsfehler.
Bild 8-2. Schematische Darstellung des Strahlen-ganges im Beleuchtungs-system einschließlich Objektiv-Vorfeld beim STEM-Betrieb.
Die parallele Verschiebung des konvergenten Elektronenstrahls beim Abrastern der Probe wird möglich, wenn der Kipppunkt des Ablenksystems exakt im ding-

Wir schalten um auf Rastertransmissionselektronenmikroskopie 173
seitigen Brennpunkt des Objektiv-Vorfeldes liegt (vgl. Bild 8-2). Wir erinnern uns: Brennpunktstrahlen verlaufen im Bildraum parallel zur optischen Achse. Da der Kontrast hauptsächlich durch unterschiedliche Streuung der Elektronen innerhalb der Probe entsteht, muss dafür gesorgt werden, dass wenig und stark gestreute Elektronen nach Passieren der Probe räumlich voneinander getrennt sind, um deren Selektion mit einem elektronenempfindlichen Detektor zu ermög-lichen. Dies ist in der bildseitigen Brennebene des Objektivs, nach unserem Mo-dell vom zweigeteilten Objektiv in der bildseitigen Brennebene des Objektiv-Nachfeldes, der Fall (vgl. Abschnitt 2.7.2). In der Regel befindet sich der Detektor für das STEM-Signal in Höhe des Beobachtungsschirms. Um die bildseitige Brennebene des Objektivs in diese Höhe abzubilden, ist auf den Beugungsmodus umzuschalten. Damit sind die elektronenoptischen Voraussetzungen für STEM-Abbildungen gegeben.
Da es sich bei der rastertransmissionselektronenmikroskopischen Abbildung um keine „echte“ optische Abbildung handelt sondern um eine elektronische Sig-nalverarbeitung, spielt das Signal-Rausch-Verhältnis eine wesentliche Rolle. Bei der Vorbereitung des Mikroskops auf den STEM-Betrieb ist deshalb großer Wert auf gute Justage der Elektronenkanone zu legen (vgl. Abschnitt 4.3). Es ist zweckmäßig, deren Justagezustand im Nanoprobe-Mode vor Umschalten auf „STEM“ zu prüfen und gegebenenfalls zu verbessern.
8.2 Auflösungsvermögen oder: Wie klein kann die Elektronensonde werden? Die im STEM-Betrieb notwendige kleine Elektronensonde wird durch mehrstufige Verkleinerung des cross over der Elektronenkanone erzeugt. Unabhängig von der konkreten Art und Anordnung der Verkleinerungslinsen sind einige grundsätzliche Dinge zu beachten, mit denen wir uns hier beschäftigen wollen. Das Auflösungs-vermögen im STEM-Betrieb kann nicht besser sein als der Durchmesser der Elektronensonde auf der Probe.
Angenommen, der verkleinerte Durchmesser des cross over in der Probenebene sei dco. Wegen der Erhaltung des Richtstrahlwertes
jR =
Ω (8.1)
(j: Stromdichte, Ω: Raumwinkel, in den emittiert wird) längs des Strahlenganges ohne Blenden (vgl. Abschnitt 2.6) gilt unter Annahme eines kreisförmigen Son-denquerschnitts und dem Strahlstrom IP in der Sondenebene die Beziehung
2 2
4 1P
co
IR
d α
⋅= ⋅
⋅ ⋅ (8.2)

Kapitel 8 174
(α: Strahlapertur, d.h. halber Öffnungswinkel). Daraus folgt für den Sondendurch-messer
2 2
4 2P Pco
I Id
RR αα
⋅= = ⋅
⋅⋅ ⋅ . (8.3)
Diese Formel hat eine weitreichende Bedeutung: Es gibt einen Zusammenhang zwischen dem Sondendurchmesser und dem Strahlstrom, d.h. je kleiner der Strahlstrom ist, desto kleiner kann auch die Sonde werden (s. Bild 8-3a). Um ein ausreichendes Signal-Rausch-Verhältnis zu erreichen, ist ein Mindeststrahlstrom notwendig, der dann häufig die Kleinheit der Sonde begrenzt (Bild 8-3b).
Bild 8-3. Auswirkungen des Richtstrahlwertes auf Strahlstrom und minimalen Sondendurchmes-ser im STEM-Modus, berechnet für α = 10 mrad. a) Strahlstrom und Sondendurchmesser für drei Richtstrahlwerte, die typisch für die angegebenen Kathoden sind. b) Richtstrahlwert und Sondendurchmesser für drei verschiedene Sondenströme.
Wir erkennen die dramatischen Unterschiede zwischen den Kathodenarten. Ange-nommen, zur STEM-Abbildung ist ein Strahlstrom von 10 pA erforderlich, dann ist mit einer Schottky-Feldemissionskathode ein Sondendurchmesser von etwa 0,1 nm möglich, bei einer LaB6-Kathode sind es ca. 1 nm und bei einer einfachen Wolfram-Haarnadelkathode 10 nm. Für analytische Messungen sind höhere Strahlströme nötig. 0,5 nA werden bei der Schottky-Feldemissionskathode bei einem Sondendurchmesser von 0,5 nm erreicht, bei der LaB6-Kathode sind es 4,7 nm und bei der Wolfram-Haarnadelkathode 86 nm. Analytik im Bereich weni-ger Nanometer („echter Nanometerbereich“) ist nur mit einem Elektronen-mikroskop möglich, das mit einer Feldemissionskathode ausgerüstet ist.
So wie das Auflösungsvermögen des Transmissionselektronenmikroskops wird auch die elektronenoptische Verkleinerung durch Abbildungsfehler beeinträchtigt. Wir wollen den Einfluss von Beugungs-, Öffnungs- und Farbfehler sowie Astig-matismus betrachten (vgl. auch Abschnitt 2.3).

Wir schalten um auf Rastertransmissionselektronenmikroskopie 175
Für den Durchmesser des Beugungsfehlerscheibchens gilt mit λ als Elektronen-wellenlänge unter der Voraussetzung sehr kleiner Aperturen α:
1, 22Bdλ
α= ⋅ (8.4)
und für den Durchmesser des Öffnungsfehlerscheibchens bei Fokussierung auf die Gaußsche Bildebene mit CS als Öffnungsfehlerkonstante
32S Sd C α= ⋅ ⋅ . (8.5)
Die Gaußsche Bildebene ist allerdings nicht die optimale Einstellung für den kleinsten Sondendurchmesser. Wie im → Abschnitt 10.18 gezeigt wird, gilt für den Radius in der Ebene der kleinsten Verwirrung
3 31 1, d.h.
4 2S S S Sr C d Cα α= ⋅ ⋅ = ⋅ ⋅ (8.6)
Analog gilt für den Sondendurchmesser in der Ebene der kleinsten Verwirrung bei Sondenverbreiterung durch den Farbfehler:
1, d.h.
2C C C CE E
r C d CE E
α αΔ Δ
= ⋅ ⋅ ⋅ = ⋅ ⋅ (8.7)
(ΔE: Energiebreite der Elektronen, E: Primärelektronenenergie). Für den Astigma-tismus gilt
1 , d.h.
2A A A Ar f d fα α= ⋅Δ ⋅ = Δ ⋅ (8.8)
mit ΔfA als astigmatischer Brennweitendifferenz. Für die Abschätzung der erreichbaren Sondengröße d addieren wir die Quadra-
te der Einflussfaktoren. Dabei vernachlässigen wir, dass die Ebenen der kleinsten Verwirrung für die einzelnen Fehler an unterschiedlichen Positionen liegen kön-nen:
2 2 2 2 2 2co B S C Ad d d d d d= + + + + . (8.9)
Unter Berücksichtigung der Gleichungen (8.4) bis (8.8) folgt:

Kapitel 8 176
( )22 6
22 2 2 22 2
411,5
4SP
C ACI E
d C fER
αλ α
α
⋅⋅ Δ= ⋅ + ⋅ + + ⋅ + Δ ⋅
⋅ . (8.10)
Ohne Öffnungs-, Farbfehler und Astigmatismus könnte demnach der Sonden-durchmesser durch Vergrößerung der Apertur α theoretisch unbegrenzt verkleinert werden. In der Praxis steht dem allerdings die Tatsache entgegen, dass die Lin-senfelder und Ablenksysteme nur in Nähe der optischen Achse „funktionieren“, d.h. aus diesen Gründen eine Begrenzung der Apertur auf 50 mrad ...100 mrad notwendig ist. Mit den drei Fehlern erkennen wir in Gleichung (8.10) eine gegensätzliche Abhängigkeit der Einzelterme von der Apertur: Im ersten Term steht α im Nenner, im zweiten und dritten im Zähler. Daraus folgt eine optimale Apertur, bei der der Sondendurchmesser minimal wird (→ Abschnitt 10.18):
( )
22
4 2
222
1224,5
3
CA CAPopt
S S S
CA C A
K KI
C C CR
Emit K C f
E
α λ⋅
= + + ⋅ −⋅ ⋅
Δ= ⋅ + Δ
. (8.11)
Wenn durch hinreichend kleine Energiebreite der Elektronen (Feldemissions-kathode) und Korrektur des Astigmatismus der Öffnungsfehler dominiert, verein-facht sich Gleichung (8.11) zu
2
82 2 2
16 2
3P
optS S
I
C R C
λα
⋅ ⋅= +
⋅ ⋅ ⋅ (8.12)
und der minimale Sondendurchmesser beträgt
32 28
2
162
3P
SI
d CR
λ⋅
= ⋅ + ⋅⋅ ⋅
. (8.13)
In modernen Geräten ist eine Korrektur des Öffnungsfehlers durch Multipole auch im Kondensorsystem möglich (vgl. Abschnitt 7.9). Bei dominierendem Farbfehler und Astigmatismus entfällt der Term mit der sechsten Potenz der Apertur in (8.10). Die optimale Apertur ist in diesem Fall:
24
2
411,5P
optCA
I
K Rα λ
⋅= ⋅ + ⋅
⋅ (8.14)

Wir schalten um auf Rastertransmissionselektronenmikroskopie 177
und der minimale Sondendurchmesser
242
44 1,5P
CAI
d KR
λ⋅
= ⋅ ⋅ + ⋅⋅
. (8.15)
In Bild 8-4 sind diese Zusammenhänge an einem Beispiel veranschaulicht. Da Nanoanalytik nur mit einer Feldemissionskathode möglich ist, beschränken wir uns dabei auf die Schottky-Kathode. Wir erkennen den Vorteil der Öffnungsfehlerkorrektur auch für die Analytik: Bei einem angenommenen Mindeststrahlstrom von 0,5 nA beträgt bei einer Öffnungsfehlerkonstanten von 1,6 mm der minimale Sondendurchmesser 0,76 nm, mit Öffnungsfehlerkorrektur (hier: CS = 10 μm) hingegen 0,23 nm.
Bild 8-4. Zusammenhang zwischen minimalem Sondendurchmesser dmin und Strahlstrom bei verschiedenen Öffnungsfehlerkonstanten CS. Parameter: E = 300 keV, ΔE = 0,7 eV, CC = 1,5 mm, R = 109 A/cm2, ΔfA = 0. a) Überblick. b) Für hochauflösende Abbildung und Nano-analytik interessanter Ausschnitt.
Die Verkleinerung des cross over wird durch die Stärke des Kondensors 1 be-stimmt („Spot Size“). Dessen Wahl richtet sich zunächst nach der Größe der abzu-bildenden Strukturen. Will man atomare Auflösung erreichen, so muss die Elek-tronensonde kleiner sein als die Atomabstände (vorausgesetzt, die Abbildungs-fehler lassen dies überhaupt zu). Wir wissen inzwischen, dass mit kleiner werden-der Sonde der Strahlstrom sinkt, sich damit also das Signal-Rausch-Verhältnis verschlechtert. Insofern ist die Wahl der „Spot-Größe“ die Suche nach einem gu-ten Kompromiss zwischen der Kleinheit der Elektronensonde und einem aus-reichenden Signal-Rausch-Verhältnis. Selbstverständlich kann man das Signal-Rausch-Verhältnis auch durch Verlängerung der Messzeit, d.h. durch eine längere Verweilzeit des Elektronensonde auf einem Rasterpunkt, verbessern. Damit dauert der gesamte Bildaufbau länger. Bei Nanostrukturen kann dies wegen der Proben-drift zum Problem werden, folglich müssen wir bei unserem Kompromiss auch noch die Stabilität der Probe berücksichtigen.

Kapitel 8 178
Ziel des Scharfstellens im STEM-Betrieb ist es, die Elektronensonde durch ge-eignete Einstellung der Brennweiten von Kondensorlinsen und Objektiv-Vorfeld so klein wie möglich zu machen, d.h. den cross over in die Probenebene abzu-bilden. Im Allgemeinen ist die Güte dieser Einstellung an der Schärfe des Raster-bildes zu erkennen.
Die Bildvergrößerung ist gleich dem Verhältnis aus der Größe des Rasterfeldes auf dem Monitor und derjenigen des Rasterfeldes auf der Probe. Eine Ver-größerungsänderung erfordert lediglich eine andere Ablenkamplitude der Elektro-nensonde auf der Probe, die elektronenoptische Verkleinerung der Sonde bleibt dieselbe. Es empfiehlt sich deshalb, bei möglichst hohen Vergrößerungen scharf zu stellen. Doch Vorsicht: Erinnern wir uns an die förderliche Vergrößerung (s. Abschnitt 1.4). Bei einer (mit Spot Size eingestellten) Sondengröße von 1 nm be-trägt die förderliche Vergrößerung ca. 100000. Bei höheren Vergrößerungen ist es ohne Verkleinerung der Sonde unmöglich, ein wirklich scharfes Bild zu erhalten!
Zur Korrektur des Astigmatismus wird die Brennweite des Objektiv-Vorfeldes periodisch verändert. Astigmatismus äußert sich dabei als Vorzugsrichtung im Bild, die bei der periodischen Änderung um 90° (zweizähliger Astigmatismus) umklappt.
Bei kohärenter Beleuchtung, d.h. im Falle einer Feldemissionskathode, gibt es noch ein anderes Kriterium zur Beurteilung der Güte der Scharfstellung: das Ronchigramm. Dieses ist das vergrößerte Beugungsscheibchen nullter Ordnung. Wenn die Elektronensonde eine amorphe Probenstelle durchstrahlt, ähnelt dieses Ronchigramm dem Powerspektrum eines hochvergrößerten TEM-Bildes von einer amorphen Folie. Bei exakter Scharfstellung erreichen die im Ronchigramm sicht-baren Ringe ihren maximalen Durchmesser, bei astigmatischer Abbildung sind sie elliptisch verzerrt.
Schließlich müssen wir noch beachten, dass das Auflösungsvermögen eines di-gitalen Bildes durch die Zahl der Pixel, d.h. durch die Zahl der Rasterpunkte im abgebildeten Bereich, bestimmt wird. Die optimale Pixelzahl ist erreicht, wenn die Schrittweite der Elektronensonde auf der Probe gleich ihrem Durchmesser d ist.
8.3 Kontrast im rastertransmissionselektronen-mikroskopischen Bild Grundlage für den Bildkontrast ist wie bei der transmissionselektronenmikro-skopischen Abbildung die Wechselwirkung der Strahlelektronen mit der Probe. Anders als beim konventionellen Rasterelektronenmikroskop ist unsere Probe elektronentransparent. Wir hatten bereits früher (s. Abschnitt 6.1) die mittlere freie Weglänge Λel für die elastische Streuung als statistische Größe eingeführt, die das Streuvermögen eines Materials mit der Dichte ρ und der Ordnungszahl Z bei einer Primärelektronenenergie E0 in Abhängigkeit vom Streuwinkel α kennzeichnet. Bei kleinen Streuwinkeln α << 1 gilt:
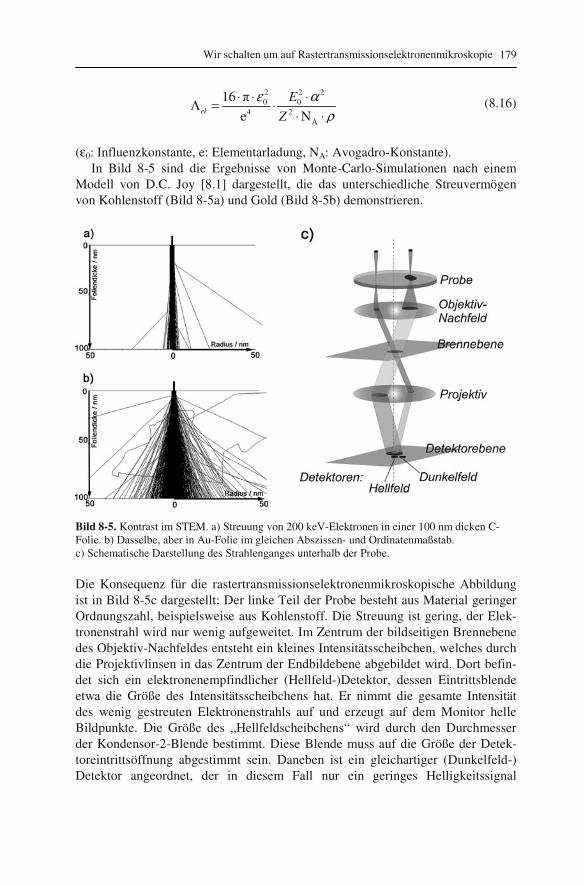
Wir schalten um auf Rastertransmissionselektronenmikroskopie 179
2 2 20 0
4 2A
16
e Nel
E
Z
ε α
ρ
⋅ ⋅ ⋅Λ = ⋅
⋅ ⋅ (8.16)
(ε0: Influenzkonstante, e: Elementarladung, NA: Avogadro-Konstante). In Bild 8-5 sind die Ergebnisse von Monte-Carlo-Simulationen nach einem
Modell von D.C. Joy [8.1] dargestellt, die das unterschiedliche Streuvermögen von Kohlenstoff (Bild 8-5a) und Gold (Bild 8-5b) demonstrieren.
Bild 8-5. Kontrast im STEM. a) Streuung von 200 keV-Elektronen in einer 100 nm dicken C-Folie. b) Dasselbe, aber in Au-Folie im gleichen Abszissen- und Ordinatenmaßstab. c) Schematische Darstellung des Strahlenganges unterhalb der Probe.
Die Konsequenz für die rastertransmissionselektronenmikroskopische Abbildung ist in Bild 8-5c dargestellt: Der linke Teil der Probe besteht aus Material geringer Ordnungszahl, beispielsweise aus Kohlenstoff. Die Streuung ist gering, der Elek-tronenstrahl wird nur wenig aufgeweitet. Im Zentrum der bildseitigen Brennebene des Objektiv-Nachfeldes entsteht ein kleines Intensitätsscheibchen, welches durch die Projektivlinsen in das Zentrum der Endbildebene abgebildet wird. Dort befin-det sich ein elektronenempfindlicher (Hellfeld-)Detektor, dessen Eintrittsblende etwa die Größe des Intensitätsscheibchens hat. Er nimmt die gesamte Intensität des wenig gestreuten Elektronenstrahls auf und erzeugt auf dem Monitor helle Bildpunkte. Die Größe des „Hellfeldscheibchens“ wird durch den Durchmesser der Kondensor-2-Blende bestimmt. Diese Blende muss auf die Größe der Detek-toreintrittsöffnung abgestimmt sein. Daneben ist ein gleichartiger (Dunkelfeld-) Detektor angeordnet, der in diesem Fall nur ein geringes Helligkeitssignal

Kapitel 8 180
empfängt. Im rechten Teil der Probe befindet sich ein Material hoher Ordnungs-zahl, beispielsweise Gold. Es streut mehr, der Elektronenstrahl wird deutlich stär-ker aufgeweitet. Im Zentrum der bildseitigen Brennebene entsteht ein größeres In-tensitätsscheibchen, welches wiederum in die Endbildebene abgebildet wird. Der Hellfelddetektor erfasst nun allerdings nur einen Teil der Intensität, die Bildpunkte von diesem Probenbereich sind dunkler als vom wenig streuenden Teil der Probe. Demgegenüber erreicht das größere Intensitätsscheibchen auch den Dunkelfeld-detektor, er liefert ein höheres Signal als vorher.
Wir wollen diesen Sachverhalt quantitativ untermauern. Gemäß der Definition der mittleren freien Weglänge Λel für die elastische Streuung gilt für das Verhält-nis der in einen Winkel kleiner als αG abgelenkten Elektronen (vgl. (6.4)) bei einer Probendicke s und zwei verschiedenen Materialien 1 und 2:
,1,2 ,1
,2
1 1
,1
,2
ee
e
el
el el
el
s sE
sE
N
N
− Λ ⋅ −Λ Λ
− Λ= = (8.17)
und unter Berücksichtigung von (8.16):
( )4
2 22 A 2 2 1 A1 12 2 2
0 0
e 1N N
,1 16
,2
es
Z ZE E
E
N
N
ρ ρε α
⋅⋅ ⋅ ⋅ ⋅ − ⋅ ⋅
⋅ ⋅ ⋅= . (8.18)
Daraus folgt die zugeschnittene Größengleichung
( )
( ) ( )-3 -32 12 2
2 122,2 ,10
/ g cm / g cm/ nm0,00392 Z Z
M M/ keV,1
,2
er rG
s
EE
E
N
N
ρ ρ
α
⋅ ⋅⋅ ⋅ ⋅ − ⋅
⋅
= . (8.19)
Das Verhältnis hängt demnach von folgenden Parametern ab: Probendicke s, Akzeptanzwinkel αG, sowie Materialeigenschaften, die durch die Ordnungszahl Z, die Dichte ρ und Atom- bzw. Molekulargewicht Mr repräsentiert sind. Für unseren bereits diskutierten Fall Kohlenstoff (Material 1) und Gold (Material 2) ist das Ergebnis in Bild 8-6a dargestellt. Es repräsentiert die Benutzung des Hellfelddetektors. Das Verhältnis der Zahl der an Kohlenstoff zu derjenigen der an Gold gestreuten Elektronen steigt mit kleiner werdendem Akzeptanzwinkel α an, d.h. der Kontrast wird umso besser, je kleiner die Detektoreintrittsblende ist. Gleichzeitig sinkt allerdings auch die Intensität, so dass der Verkleinerung dieser Blende Grenzen gesetzt sind. In der Praxis liegt der Akzeptanzwinkel des Hell-felddetektors bei 5 ... 10 mrad.

Wir schalten um auf Rastertransmissionselektronenmikroskopie 181
Bild 8-6. Verhältnis der Elektronenzahlen innerhalb (a) und außerhalb (b) eines Streuwinkels α bei Streuung in Kohlenstoff und Gold. Parameter: Probendicke s = 100 nm, Kohlenstoff: Z = 6, Mr = 12, ρ = 2,26 g/cm3; Gold: Z = 79, Mr = 197, ρ = 19,3 g/cm3.
Für die Dunkelfeldabbildung werden die stärker gestreuten Elektronen als Hellig-keitssignal genutzt. Für das Verhältnis der in Material 1 bzw. 2 gestreuten Elektro-nen mit einem Streuwinkel größer als αG gilt:
,1
,2
,1
,2
1 e
1 e
el
el
sE
sE
N
N
− Λ
− Λ
−=
− . (8.20)
In diesem Fall steigt der Kontrast mit wachsendem Winkel (s. Bild 8-6b).
8.4 Spezialfall: Weitwinkel-Dunkelfeld-Ringdetektor Der bisher beschriebene Dunkelfelddetektor hat einen gravierenden Nachteil: Er erfasst nur einen kleinen azimutalen Anteil der stark gestreuten Elektronen. Besser ist ein ringförmiger Detektor, der in der Regel oberhalb des Beobachtungsleucht-schirms angebracht ist. Die englische Bezeichnung für diesen Detektor lautet: High Angle Annular Darkfield Detector. Daraus leitet sich die übliche Bezeich-nung HAADF-Detektor ab. Mit diesem können auch stark gestreute Elektronen mit guter Effizienz nachgewiesen werden. Deren Winkelbereich wird durch die Kameralänge L (vgl. Abschnitt 5.3) beeinflusst (s. Bild 8-7).
Wir ersehen daraus, dass mit dem Ringdetektor Winkel von über 200 mrad erfasst werden können, was für den Kontrast im STEM-Dunkelfeldbild vorteilhaft ist. Durch geeignete Abstimmung von Kameralänge und Bestrahlungsapertur (Kondensor-2-Blende) ist das Intensitätsscheibchen der nicht gestreuten Elektro-nen so klein zu halten, dass es den Ringdetektor nicht erreicht.
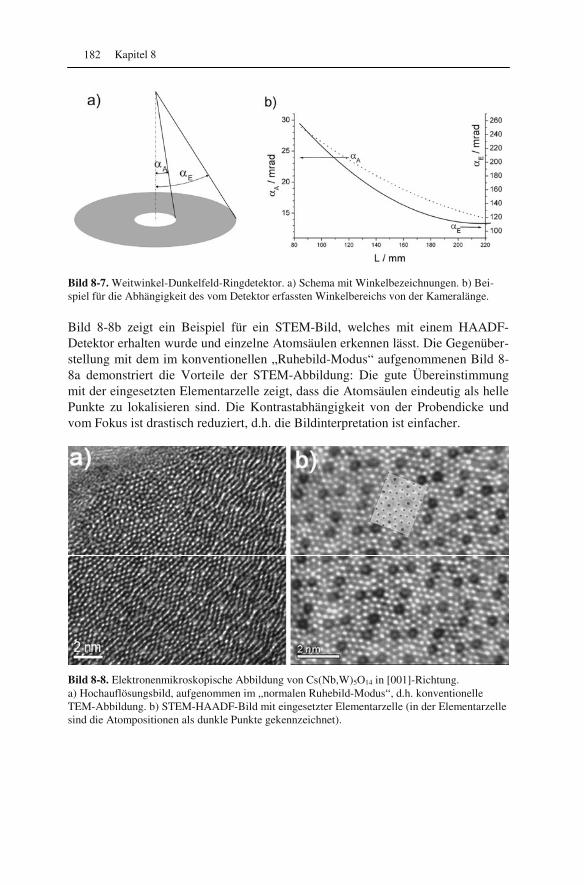
Kapitel 8 182
Bild 8-7. Weitwinkel-Dunkelfeld-Ringdetektor. a) Schema mit Winkelbezeichnungen. b) Bei-spiel für die Abhängigkeit des vom Detektor erfassten Winkelbereichs von der Kameralänge.
Bild 8-8b zeigt ein Beispiel für ein STEM-Bild, welches mit einem HAADF-Detektor erhalten wurde und einzelne Atomsäulen erkennen lässt. Die Gegenüber-stellung mit dem im konventionellen „Ruhebild-Modus“ aufgenommenen Bild 8-8a demonstriert die Vorteile der STEM-Abbildung: Die gute Übereinstimmung mit der eingesetzten Elementarzelle zeigt, dass die Atomsäulen eindeutig als helle Punkte zu lokalisieren sind. Die Kontrastabhängigkeit von der Probendicke und vom Fokus ist drastisch reduziert, d.h. die Bildinterpretation ist einfacher.
Bild 8-8. Elektronenmikroskopische Abbildung von Cs(Nb,W)5O14 in [001]-Richtung. a) Hochauflösungsbild, aufgenommen im „normalen Ruhebild-Modus“, d.h. konventionelle TEM-Abbildung. b) STEM-HAADF-Bild mit eingesetzter Elementarzelle (in der Elementarzelle sind die Atompositionen als dunkle Punkte gekennzeichnet).

9 Wir nutzen die analytischen Möglichkeiten
Ziel: Bei allen bisher beschriebenen Abbildungsvarianten und Kontrastphäno-menen wurde im Transmissionselektronenmikroskop die elastische Streuung der Elektronen im Festkörper, d. h. die Wechselwirkung ohne Energieverlust, ausge-nutzt. Inelastische Streuprozesse, d. h. solche, bei denen die Strahlelektronen Energie verlieren, waren unerwünscht. Sie führen zu Änderungen der Elektronen-wellenlänge und damit zu Auflösungsverlust durch chromatische Aberration und zu stärkerem Untergrund in den Beugungsbildern. Andererseits sind inelastische Wechselwirkungen häufig elementspezifisch, d. h. ihre Ergebnisse hängen davon ab, welchem Element das wechselwirkende Atom angehört. Damit wird eine chemische Analyse im Nanometerbereich mög-lich. Wir werden zunächst den inelastischen Streuprozess erläutern, die zu dessen praktischer Nutzung erforderlichen Spektrometer und Verfahren beschreiben und schließlich einige Beispiele anführen.
9.1 Analytische Signale als Folge inelastischer Wechselwirkung Bisher haben wir lediglich die Coulombsche Wechselwirkung zwischen den Atomkernen der Probe und den Strahlelektronen berücksichtigt, wobei kein Ener-gieübertrag vom Strahlelektron an das Atom stattfindet (elastische Streuung). Um zu verstehen, welche Möglichkeiten des Energieübertrags an die Elektronenhülle bestehen (inelastische Streuung), benötigen wir ein Modell, welches die Eigen-schaften der Elektronenhülle erfasst. Für das Strahlelektron ist die Konsequenz klar: Es verliert Energie. Die Elektronenhülle verändert ihren energetischen Zu-stand und emittiert u. a. Röntgenstrahlung. Elektronenenergie-Verluste und die Energie der charakteristischen Röntgenstrahlung werden im analytischen Trans-missionselektronenmikroskop gemessen und zur chemischen Analyse ausgenutzt. Deshalb beschränken wir uns auf diese beiden Sachverhalte. Zur plausiblen Er-klärung legen wir unterschiedliche Atommodelle zugrunde. Für die Röntgenemis-sion genügt das Rutherford-Bohrsche54 Modell. Für die Erörterung der Energie-verluste müssen wir dieses Modell um bindungsspezifische Details erweitern.
9.1.1 Emission von Röntgenstrahlung
Aus der Elektrodynamik (Maxwellsche Gleichungen) wissen wir, dass beschleu-nigte elektrische Ladungen veränderliche Magnetfelder erzeugen und diese wiede-
54 Niels Bohr, dänischer Physiker, 1885 – 1962, Nobelpreis für Physik 1922
DOI 10.1007/978-3-7091-1440-7_9, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 9 184
rum veränderliche elektrische Felder. Diese periodisch wechselnden Felder sind bekannt als elektromagnetische Wellen, welche bei der Beschleunigung von Elektronen entstehen. Wir erinnern uns an die Bewegung von Elektronen im Coulombschen Kraftfeld eines Atomkerns (→ Abschnitt 10.11): Die Elektronen werden abgelenkt, d. h. ihre Geschwindigkeitsrichtung ändert sich. Jede Ge-schwindigkeitsänderung erfordert eine Beschleunigung; auch diese Richtungsän-derung, so dass die Ablenkung der Elektronen mit der Emission von elektro-magnetischer Strahlung verbunden ist. Diese Strahlung breitet sich mit Licht-geschwindigkeit c aus, ihre Energie E bestimmt die Wellenlänge λ (→ Abschnitt 10.2):
ch h
2E
ω
λ= ⋅ = ⋅
⋅ (9.1)
(h: Plancksches Wirkumsquantum). Liegt die Wellenlänge zwischen 0,05 nm und 10 nm, wird die elektromagnetische Strahlung als Röntgenstrahlung (bzw. –welle) bezeichnet. Die darin enthaltene Energie wird dem abgelenkten Elektron entzo-gen, d. h. das Elektron wird abgebremst. Man spricht deshalb von Bremsstrahlung.
Die Wellenlängen dieser Bremsstrahlung bilden ein Kontinuum, ihre Energie kann allerdings höchstens gleich der Primärenergie E0 der Strahlelektronen sein. Wegen der umgekehrten Proportionalität von Energie und Wellenlänge gibt es demzufolge eine untere, kurzwellige Grenze λmin dieser Bremsstrahlung:
min0
h c
Eλ
⋅= . (9.2)
Die Wahrscheinlichkeit für die Entstehung der Bremsstrahlung („Streuquer-schnitt“) ist energieabhängig. Ihre Intensität wird näherungsweise durch die Kra-merssche55 Formel [9.1]
( ) 0Z 1BrE
I E CE
= ⋅ ⋅ − (9.3)
beschrieben (C: Konstante, die u. a. den Primärelektronenstrom und die Proben-dicke berücksichtigt, Z: Ordnungszahl). Wir sehen, dass die Intensität der Brems-strahlung mit der Ordnungszahl zunimmt und mit wachsender Energie hyperbo-lisch abnimmt. Bei der Messung kommt der Wirkungsgrad (Effizienz) des De-tektors hinzu, der ebenfalls energieabhängig ist und bei kleinen Röntgenenergien sinkt.
Die Bremsstrahlung liefert keine elementspezifischen Signale, sie beeinflusst aber den Untergrund im Röntgenspektrum (d.i. die Intensität der Röntgenstrahlung 55 Hendrik Anthony Kramers, niederländischer Physiker, 1894 – 1952

Wir nutzen die analytischen Möglichkeiten 185
in Abhängigkeit von ihrer Energie bzw. Wellenlänge) und kann die quantitative Auswertung der Spektren vor allem im niederenergetischen Bereich stören.
Die inelastische Wechselwirkung zwischen Strahlelektronen und den Atomen der Probe kann allerdings noch eine andere Konsequenz haben: Veränderungen in der Elektronenhülle des Atoms. Zu deren Verständnis müssen wir uns etwas ge-nauer mit dieser Hülle befassen und benutzen dazu das Rutherford-Bohrsche Mo-dell. Ausgangspunkt ist die Vorstellung, dass die (einfach) negativ geladenen Elektronen den positiv geladenen Kern umkreisen, wobei die Zahl der Elektronen mit der Zahl der (einfach) positiv geladenen Protonen im Kern übereinstimmt und der Ordnungszahl im Periodensystem der Elemente entspricht. Insgesamt ist das Atom elektrisch neutral. Die Coulomb-Kraft wirkt als Radialkraft und hält die Elektronen auf ihrer Bahn. Diese Vorstellung ist mit zwei Problemen verbunden:
1. Wieso treibt die Coulomb-Kraft die positiv geladenen Protonen nicht aus-einander?
2. Eingangs hatten wir darauf hingewiesen, dass beschleunigte Ladungsträger elektromagnetische Strahlung aussenden. Elektronen auf einer Kreisbahn sind aber beschleunigte Ladungsträger. Sie müssten demzufolge abgebremst werden und schließlich in den Kern stürzen.
Das erste Problem wird mit der Vorstellung gelöst, dass die Protonen nicht nur die positive Ladung mitbringen, sondern auch „Gluonen“, d. h. eine Art Kitt, der die Protonen zusammenhält. Mit wachsender Ordnungszahl wächst die elektrische Abstoßungskraft, es werden mehr Gluonen benötigt als die Protonen allein liefern können. Dieses Dilemma wird beseitigt, indem zusätzliche Teilchen ohne Ladung aber mit Gluonen, die Neutronen, in den Kern eingebaut werden.
Zur Lösung des zweiten Problems erinnern wir uns an den Wellencharakter der Elektronen. Wir nehmen an, dass sich das Elektron auf einer Kreisbahn um den Kern bewegt. Wegen seines Wellencharakters muss der Umfang der Kreisbahn ein ganzzahliges Vielfaches der Wellenlänge λ des Elektrons betragen, andernfalls löscht sich die Welle und damit das Elektron durch Interferenz selbst aus:
2 nr λ⋅ = ⋅ (9.4)
(r: Bahnradius, n: ganze Zahl). Die Radialkraft ist gleich der Coulomb-Kraft (v: Geschwindigkeit, m0: Elektronenmasse, e: Elementarladung, ε0 : Influenzkonstan-te, Z: Ordnungszahl):
2 2
0 20
1 Z em
4
v
r rε
⋅⋅ = ⋅
⋅ . (9.5)
Mit 20m
2E v= ⋅ (9.6)

Kapitel 9 186
als (kinetische) Elektronenenergie folgt daraus
2
0
1 Z e
8E
r
⋅= ⋅
⋅ (9.7)
und mit Berücksichtigung der de Broglie-Formel (1.10) in nichtrelativistischer Nä-herung
0
h
2 m Eλ =
⋅ ⋅ (9.8)
(h: Plancksches Wirkumsquantum) sowie (9.4):
220
20
m1 Z e
8 hnnE
⋅= ⋅ ⋅
⋅ . (9.9)
Da n eine ganze Zahl ist, sind die durch (9.9) vorgegebenen Energiewerte (bzw. -niveaus) diskret. Den Index n ergänzen wir, um auf diesen Umstand hinzuweisen.
Um Missverständnissen vorzubeugen, möchten wir betonen, dass es sich bei En um die kinetische Energie des Elektrons handelt. Diese ist umso größer, je kleiner der Bahnradius ist. Für die potentielle Energie (das „Potential“) gilt im elektro-statischen Kraftfeld:
( )2 2
20 0
Z e Z e 1
4 8e
Per
drW R
r rε ε
∞⋅ ⋅
= − ⋅ = − ⋅⋅ ⋅ ⋅ ⋅
, (9.10)
d. h. wegen des negativen Vorzeichens wird sie mit sinkendem Radius re kleiner. Die später folgende Erörterung einzelner Energiezustände bzw. Energieniveaus bezieht sich auf die potentielle Energie.
Der Wellencharakter des Elektrons hat eine weitere Konsequenz: Zwei identi-sche Elektronen, d. h. zwei Elektronen mit exakt gleicher Wellenfunktion würden miteinander interferieren, d. h. sich gegenseitig beeinflussen. Es gibt Möglich-keiten, sich ungleiche, aber sehr ähnliche Bahnen (und damit Wellenfunktionen) vorzustellen: Die Bahnen können elliptisch sein, sie können in verschiedenen Richtungen durchlaufen werden, und zusätzlich können sich die Elektronen selbst links- oder rechtsherum drehen (Spin). Die ähnlichen Energieniveaus werden zu Schalen zusammengefasst. Die verschiedenen Möglichkeiten werden durch vier Quantenzahlen beschrieben: Die Hauptquantenzahl n kennzeichnet den Bahnra-dius (bzw. die große Halbachse einer elliptischen Bahn) und damit die Schale (n = 1: K-Schale, n = 2: L-Schale, n = 3: M-Schale usw.); die Nebenquantenzahl l kennzeichnet den Bahndrehimpuls (l = 0, ... , n-1; l = 0: s-Elektronen, l = 1: p-

Wir nutzen die analytischen Möglichkeiten 187
Elektronen, l = 2: d-Elektronen, l = 3: f-Elektronen usw.), die magnetische Quantenzahl m die z-Komponente des Drehimpulses (m = -l ... +l) und die Spinquantenzahl s den Eigendrehimpuls des Elektrons (s = ± 1/2). Die Elektronen müssen sich in mindestens einer dieser Quantenzahlen unterscheiden (Pauli-Prinzip).
Daraus folgt für die Maximalzahl Nn an Elektronen, die eine Schale aufnehmen kann (vgl. auch Tabelle 9-1):
22nN n= ⋅ . (9.11)
Bezeichnung der Schale
n l Bezeichnung des Elektrons
m s
K-Schale (2 Elektronen)
1 1
0 0
1s 1s
0 0
+½ -½
L-Schale (8 Elektronen)
2 2 2 2 2 2 2 2
0 0 1 1 1 1 1 1
2s 2s 2p 2p 2p 2p 2p 2p
0 0 -1 -1 0 0 1 1
+½ -½ +½ -½ +½ -½ +½ -½
Tabelle 9-1. Quantenzahlen für die Besetzung der K- und L-Schale.
Die Gesamtdrehimpuls-Quantenzahl ist j = l + s. Strahlungsübergänge sind quantenmechanisch nur erlaubt, wenn Δl = ± 1 und Δj = 0, ± 1 gilt.
Bild 9-1. Energieniveaus (Termschema) für die ersten drei Schalen K, L und M mit möglichen Strahlungs-übergängen.

Kapitel 9 188
Die möglichen Elektronenenergiezustände für die inneren drei Schalen und die zwischen ihnen erlaubten Strahlungsübergänge sind schematisch in Bild 9-1 dar-gestellt. Wegen des Pauli-Prinzips sind solche mit der Emission von elektromag-netischer Strahlung verbundenen Übergänge nur möglich, wenn vorher ein Ener-gieniveau auf einer unteren Schale frei geworden ist. Hier kommen wir zur inela-stischen Wechselwirkung der Strahlelektronen mit dem Atom zurück: Das Strahl-elektron kann durch entsprechenden Energieübertrag dafür sorgen, dass ein sol-ches „inneres Energieniveau“ frei wird, es kann eine innere Schale ionisieren.
Wir nehmen an, dass ein Platz auf der K-Schale (n = 1) frei wird und dieser beim anschließenden Strahlungsübergang mit einem Elektron aus der L-Schale (n = 2) aufgefüllt wird, vernachlässigen die Unterschiede der Energieniveaus inner-halb einer Schale und benutzen Formel (9.9), um die damit verbundene Energie-differenz abzuschätzen:
220
1, 2 2 20 1 2
m Z e 1 1
8 hn nEn n
⋅Δ = ⋅ ⋅ −
⋅ . (9.12)
Für Chrom (Z = 24) folgt daraus beispielsweise eine Energiedifferenz von 9,413⋅10-16 Nm bzw. 5876 eV. Nach (9.2) entspricht das einer Wellenlänge von 0,211 nm, d. h. die emittierte Strahlung ist Röntgenstrahlung. Da die Differenzen der Energieniveaus elementabhängig sind, ist auch die Energie der auf diese Weise entstehenden Röntgenstrahlung elementabhängig. Sie heißt deshalb im Un-terschied zur Bremsstrahlung charakteristische Röntgenstrahlung.
9.1.2 Energieverluste der Elektronen
Nachdem wir die charakteristische Röntgenemission als Resultat der Veränderun-gen in der Elektronenhülle infolge der Wechselwirkung von Primärelektronen mit den Atomen der Probe erläutert haben, wollen wir uns nun der Veränderung der Energie der Primärelektronen als Folge dieses Prozesses widmen. Auf den ersten Blick erscheint dies simpel; die auf die Röntgenquanten übertragene Energie wird dem Strahlelektron entzogen. Für die Bremsstrahlung trifft dies tatsächlich zu. Bei der Emission von charakteristischer Röntgenstrahlung ist diese Aussage allerdings falsch. In Wirklichkeit wird hierbei nur ein Teil des Elektronenenergie-Verlustes auf die Röntgenquanten übertragen (vgl. Bild 9-2).
Dem Primärelektron werde eine Energie ΔE1 entzogen. Diese Energie wird benutzt, um die K-Schale zu ionisieren, d. h. ein Elektron von der K-Schale auf den ersten freien oder einen höheren Energiezustand zu befördern oder auch voll-kommen aus dem Kraftfeld des Atoms zu entfernen und eine zusätzliche kineti-sche Energie zu erteilen. Das emittierte Röntgenquant hat demgegenüber nur die Energie ΔE2, was der Differenz eines bereits besetzten Zustandes und der Energie der K-Schale entspricht. Sie ist kleiner als ΔE1. Für die Ionisation der K-Schale

Wir nutzen die analytischen Möglichkeiten 189
von Sauerstoff ist beispielsweise mindestens eine Energie von 532 eV notwendig, die O-K-Röntgenlinie hat demgegenüber nur eine Energie von 523 eV.
Bild 9-2. Ablauf der Emission von charakteristischer Röntgenstrahlung. a) Das Strahlelektron (Primärelektron) ionisiert die K-Schale. Das vorher dort befindliche Elektron wird mindestens auf den ersten freien Zustand gehoben (Energiedifferenz ΔE1). b) Die K-Schale ist ionisiert, d. h. ein Zustand ist frei. c) Ein Elektron der L-Schale füllt den ionisierten Zustand auf (Strahlungsübergang mit Energiedifferenz ΔE2).
Die Energieverluste der Strahlelektronen hängen von den Energieniveaus in der Elektronenhülle der Atome ab. Dafür gibt es weitere als die bis hierher beschrie-benen Möglichkeiten. Bisher haben wir immer nur einzelne Atome betrachtet. In unserer Probe sind aber viele Atome enthalten, bei kristallinen Materialien sind sie periodisch und nahe beieinander angeordnet. Dies hat Konsequenzen für die Elek-tronenhüllen, d. h. wir müssen unser Atommodell ergänzen. In diesem Zusam-menhang stellt sich auch die Frage nach den Ursachen der chemischen Bindung. Was hält die Atome zusammen? Eigentlich erwarten wir eine Coulombsche Ab-stoßung, wenn sich die Elektronenhüllen einander annähern und damit ein Aus-einanderdriften der Atome.
Dies wird allerdings verhindert, wenn das Überlappen der Elektronenhüllen mit einem Energiegewinn verbunden ist, d. h. wenn die Gesamtenergie des verbunde-nen Zustandes kleiner ist als die Gesamtenergie im getrennten Zustand. Es ist wie mit einem Schlitten am Rodelhang: Die Hangabtriebskraft zieht ihn nach unten, und seine potentielle Energie ist im Tal kleiner als auf dem Berg.
Alle Bindungsmöglichkeiten haben dieses Überlappen der Elektronenhüllen gemeinsam. Eine Variante setzt voraus, dass die äußere Schale eines Atoms nur schwach besetzt ist, ein typisches Beispiel ist Natrium mit der Ordnungszahl 11. Seine K-Schale enthält zwei s-Elektronen (Schreibweise: 1s2), die L-Schale zwei s-Elektronen und sechs p-Elektronen (2s2 2p6) und die M-Schale ein s-Elektron (3s1 bzw. 3s). Für die Kennzeichnung der gesamten Elektronenkonfiguration ist dafür die Schreibweise 1s2 2s2 2p6 3s üblich. Chlor mit der Ordnungszahl 17 hat demgegenüber die Elektronenkonfiguration 1s2 2s2 2p6 3s2 3p5. Zur energetisch günstigeren Edelgaskonfiguration fehlt lediglich ein 3p-Elektron. Dieses wird vom Natrium gestiftet und im Ergebnis entstehen zwei Ionen mit gegensätzlicher La-dung, die sich anziehen (Ionenbindung). Wichtig ist, dass sich dabei die Ge-samtenergie vermindert, d. h. es entsteht ein veränderter Energiezustand, der cha-rakteristisch für diese Bindung ist.

Kapitel 9 190
Bei einer anderen Überlappungsmöglichkeit gehören einige der Elektronen bei-den Atomen an, z. B. beim Wasserstoffmolekül. Ein Wasserstoffatom hat die Elektronenkonfiguration 1s, im Molekül ist sie 1s2, was energetisch günstiger ist als zweimal 1s (kovalente Bindung).
Schließlich können wir uns noch eine Konfiguration vorstellen, bei der die äußeren Elektronen nicht mehr lokal gebunden sind, d. h. nicht mehr zu einem einzelnen Atompaar gehören sondern sich frei im Kristallgitter bewegen können (metallische Bindung). Die quantenmechanische Rechnung (Lösung der Schrö-dinger-Gleichung im periodischen Kristallpotential) zeigt, dass in diesem Fall „Energiebänder“ existieren; die darin befindlichen Elektronen sind nicht an einzel-ne Atomkerne gebunden.
Für eine grafische Darstellung erinnern wir uns daran, dass die Quantenzahlen Energiezustände beschreiben, die u. a. durch unterschiedliche Bahnformen reprä-sentiert sind. Der Bahndrehimpuls l = 0 (s-Elektronen) impliziert nur eine Bahn-form (m = 0) und wird deshalb durch eine Kugel dargestellt, deren Orientierung im Raum keine Rolle spielt. Bei l = 1 (p-Elektronen) existieren drei unterschied-liche Bahnformen (m = -1, 0, 1), die durch Ellipsen symbolisiert werden, die sich in die drei Raumrichtungen erstrecken (s. Bild 9-3).
Bild 9-3. Grafische Dar-stellung der s- und p-Elektronen. Die skizzierten Bereiche werden als Orte hoher Aufenthaltswahr-scheinlichkeit interpretiert und als Orbitale bezeichnet.
In dieser Darstellungsweise lassen sich drei Grenzfälle von Überlappungen kon-struieren, die speziellen Bindungsarten entsprechen (s. Bild 9-4).
Bild 9-4. Grenzfälle von Bindungen. a) s-s σ-Bindung. b) p-p σ-Bindung. c) p-p π-Bindung. (vgl. auch [9.2]).

Wir nutzen die analytischen Möglichkeiten 191
Es sind auch Mischformen dieser Bindungen denkbar, die als Hybridorbitale be-zeichnet werden. Alle diese Bindungen produzieren zusätzliche Energiezustände, in die im Falle unvollständiger Besetzung bei inelastischen Stoßprozessen Elektro-nen befördert werden können.
In den Energiebändern können sich die Elektronen ähnlich wie ein Gas bewe-gen, man spricht deshalb auch vom „Elektronengas“. Dieses Gas schwingt bei äußerer Anregung, wie sie bei inelastischer Wechselwirkung stattfindet.
Für eine Plausibilitätserklärung stellen wir uns vor, dass die quasifreien Elek-tronen im Elektronengas von einem Primärelektron angestoßen und dadurch ver-schoben werden. Infolge der Ladungsverschiebung entsteht ein elektrisches Feld E und damit eine rücktreibende Kraft
eF E= − ⋅ (9.13)
(e: Elementarladung). Wir stellen uns die Ladungsverschiebung entlang einer Achse u vor und entnehmen aus den Maxwellschen Gleichungen:
0
E uρ
ε= ⋅
⋅ (9.14)
(ρ: Ladungsdichte, 0: Influenzkonstante, : Dielektrizitätszahl) und erhalten mit
ee
Q n Vn
V Vρ
⋅ ⋅= = = ⋅ (9.15)
(Q: Ladung, n: Zahl der Elektronen im Volumen V) die Bewegungsgleichung
2
0
e0
m
nu u
ε
⋅+ ⋅ =
⋅ ⋅ (9.16)
(m: Elektronenmasse), d. h. die Gleichung einer harmonischen Schwingung mit der Eigenfrequenz
0
emP
nω
ε= ⋅
⋅ ⋅ , (9.17)
was einer Energie
h
2P PE ω= ⋅ (9.18)

Kapitel 9 192
(h: Plancksches Wirkumsquantum) und damit zusätzlichen Möglichkeiten für Energieverluste entspricht. Diese Eigenschwingungen des Elektronengases wer-den als Plasmonen bezeichnet. Die Ladungsverteilung ist an der Oberfläche des Festkörpers anders als im Volumen, deshalb wird zwischen Oberflächen- und Volumenplasmonen unterschieden.
Im Festkörpermodell werden die Bindungen zwischen den Atomen manchmal durch mechanische Federn symbolisiert. Trotz der extrem unterschiedlichen Mas-sen von Elektronen und Atomkernen ist es möglich, dass Strahlelektronen Energie auf diese „Federkonstruktion“ übertragen und diese zu schwingen beginnt. Die Eigenfrequenzen dieser Schwingungen werden als Phononen bezeichnet.
9.2 Energiedispersive Spektroskopie charakteri-stischer Röntgenstrahlung („EDXS“) Aus Abschnitt 9.1.1 wissen wir, dass Atome bei Strahlungsübergängen zwischen inneren Schalen charakteristische Röntgenstrahlung aussenden, deren Energie von der Ordnungszahl, d. h. vom Element, abhängt. Wir wollen uns nun der Praxis zu-wenden und erläutern, wie die Energie dieser Röntgenstrahlung gemessen wird, wie die Spektren aussehen, welche Artefakte auftreten können und welche Schlussfolgerungen aus den Röntgenspektren gezogen werden können.
9.2.1 Röntgenspektrometer und Röntgenspektren
Da Energie und Wellenlänge von Röntgenstrahlung umgekehrt proportional sind (vgl. (9.1)), kann die Röntgenenergie über deren Wellenlänge gemessen werden. Aus Kapitel 5 wissen wir, dass der Bragg-Winkel von der Wellenlänge abhängt. Mit einem Kristall bekannter Gitterkonstante ist es möglich, die Wellenlänge durch Messung des Bragg-Winkels zu bestimmen (wellenlängendispersives Spek-trometer). Allerdings müssen bei der Messung der Einfallswinkel der Röntgen-strahlung in den Kristall und der Ausfallswinkel aus dem Kristall gleich groß sein (die Summe aus beiden ist der Bragg-Winkel), so dass der Kristall zur Messung auf einer Kreisbahn bewegt werden muss. Die dazu notwendige Vorrichtung passt geometrisch kaum an ein Transmissionselektronenmikroskop, so dass derartige Spektrometer in der Praxis der analytischen Transmissionselektronenmikroskopie keine Rolle spielen.
Es gibt aber noch eine andere Möglichkeit der Energiemessung. Um diese zu verstehen, drehen wir in Gedanken den Prozess der Röntgenemission um. Wir stellen uns vor, dass ein Röntgenquant ein Elektron von einem besetzten Energie-zustand in einen unbesetzten Zustand befördert, ähnlich wie dies als Voraus-setzung für die Röntgenemission durch das Strahlelektron geschieht. Bei Halb-leitern erfolgt die Verschiebung der Elektronen vorzugsweise zwischen Energie-bändern. Die beiden oberen Bänder werden als Valenz- und Leitungsband bezeichnet. Bei einem undotierten Halbleiter ist das Valenzband bei sehr niedrigen

Wir nutzen die analytischen Möglichkeiten 193
Temperaturen vollständig besetzt, das Leitungsband hingegen unbesetzt (vgl. Bild 9-5). Die Elektronen können sich deshalb nicht frei bewegen, der elektrische Widerstand ist sehr hoch. Die Energielücke zwischen Valenz- und Leitungsband ist bei Halbleitern vergleichsweise klein, bei Silizium beispielsweise beträgt sie etwa 1,2 eV. Bei höheren Temperaturen gelangen Elektronen in das Leitungsband, deshalb sinkt bei Halbleitern der elektrische Widerstand mit wachsender Tempe-ratur.
Bild 9-5. Schematische Darstellung der Bandstruktur eines Halbleiters.
Auch ein Röntgenquant mit der Energie ERQ kann Elektronen in das Leitungsband befördern („innerer Photoeffekt“). Dazu muss pro Elektron eine Energie EL aufge-bracht werden, die mindestens der Größe der Bandlücke entspricht, d. h. die Höchstzahl Nmax der „Leitfähigkeitselektronen“ ist
maxRQ
L
EN
E= . (9.19)
Im Halbleiterkristall wird mit zwei Elektroden ein elektrisches Feld erzeugt; nach dem Eindringen eines Röntgenquants fließt kurzzeitig ein Strom, dessen Stärke von der Zahl der Leitfähigkeitselektronen, d. h. von der Energie des Röntgenquants abhängt. Damit wird es möglich, die Röntgenenergie direkt zu messen (energiedispersive Röntgenspektroskopie, englisch: Energy Dispersive X-ray Spectroscopy – EDXS).
Allerdings ist der Energieunterschied EL der Bandlücke nur ein Mindestwert. Die pro Leitfähigkeitselektron benötigte Energie kann variieren, weil innerhalb der Energiebänder unterschiedliche Energiezustände besetzt werden können. Die Besetzung unterliegt einer Wahrscheinlichkeitsverteilung, d. h. es gibt Energie-zustände, die bevorzugt eingenommen werden. Damit folgt auch die Zahl der Leitfähigkeitselektronen und die gemessene Energie dieser Wahrscheinlich-keitsverteilung. Eine diskrete Röntgenenergie wird im Spektrum nicht als Linie sondern als schmale Gauß-ähnliche Kurve („Röntgen-Peaks“) dargestellt. Die Halbwertsbreite dieser Gauß-Kurve wird als energetisches Auflösungsvermögen des Spektrometers bezeichnet. Es ist begrenzt und hängt von der Röntgenenergie ab. Bei einer Röntgenenergie von 5,9 keV beträgt es etwa 120 eV ... 130 eV.
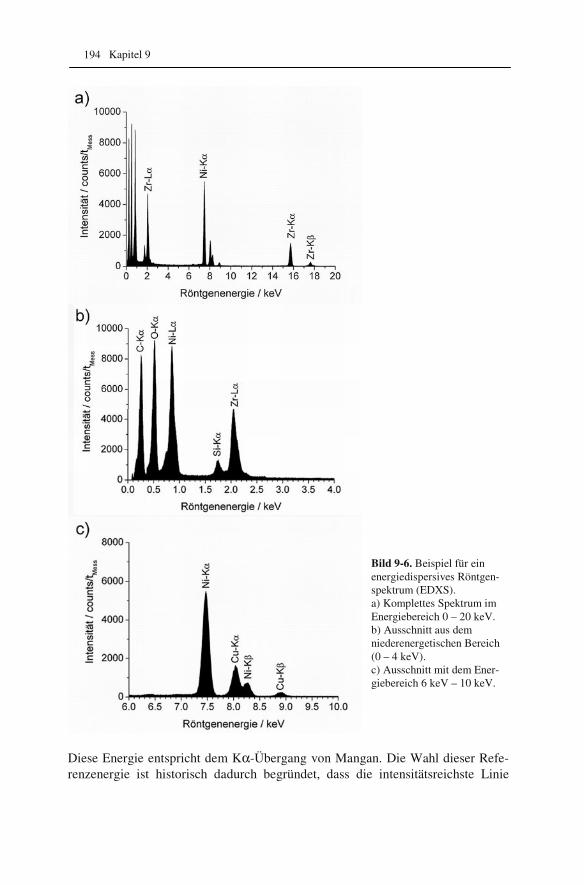
Kapitel 9 194
Bild 9-6. Beispiel für ein energiedispersives Röntgen-spektrum (EDXS). a) Komplettes Spektrum im Energiebereich 0 – 20 keV. b) Ausschnitt aus dem niederenergetischen Bereich (0 – 4 keV). c) Ausschnitt mit dem Ener-giebereich 6 keV – 10 keV.
Diese Energie entspricht dem Kα-Übergang von Mangan. Die Wahl dieser Refe-renzenergie ist historisch dadurch begründet, dass die intensitätsreichste Linie

Wir nutzen die analytischen Möglichkeiten 195
beim radioaktiven Zerfall des Fe55-Isotops genau diese Energie hat. Es ist deshalb möglich, ein EDX-Spektrometer mit einer Fe55-Quelle ohne Elektronenmikro-skop zu testen. Bei niedrigeren Röntgenenergien verbessert sich die Energieauf-lösung und erreicht für Kohlenstoff (Kα-Linie bei 277 eV) einen Wert von etwa 60 eV.
In Bild 9-6 ist ein EDX-Spektrum gezeigt. Die Bezeichnung der Röntgen-Peaks im Spektrum leitet sich aus dem Termschema (Bild 9-1) ab: Der Großbuchstabe (z. B. K) gibt die durch die Strahlelektronen ionisierte Schale an, die durch einen Strahlungsübergang aufgefüllt wird. Der griechische Buchstabe gibt an, aus wel-cher Schale das Auffüllelektron stammt. α bedeutet Auffüllelektron aus der ener-getisch nächst höheren Schale, d. h. Kα heißt Auffüllen aus der L-Schale, Kβ aus der M-Schale usw.
Wir sehen, dass der Untergrund im niederenergetischen Bereich höher ist als im hochenergetischen Bereich. Hier macht sich die Bremsstrahlung bemerkbar.
Bereits bei Raumtemperatur ist die Wärmeenergie so groß, dass einige Elek-tronen in das Leitungsband gehoben werden können, was zu einem geringen Strom zwischen den Detektorelektroden führt und damit den Rauschuntergrund vergrößert. Verunreinigungen des Halbleiters wirken als Dotierungen und erhöhen ebenfalls den Rauschuntergrund. Der Detektorkristall muss deshalb extrem rein sein und wird mit flüssigem Stickstoff oder mit Peltier-Elementen gekühlt. In Bild 9-7 ist der Aufbau eines EDX-Detektors schematisch dargestellt (Detektoreffi-zienz → Abschnitt 10.20).
Bild 9-7. Aufbau eines EDX-Detektors mit Flüssig-Stickstoff-Kühlung. a) Außenansicht. b) Detail: Rohr mit Detektor-Kristall.
In den Halbleiter-Kristall (in der Regel Silizium) wird Lithium eindiffundiert, wel-ches durch evtl. Verunreinigungen entstehende Ladungsträger kompensiert, so dass eine möglichst breite ladungsträgerfreie Zone entsteht, in der bewegliche La-dungen nur durch eindringende Röntgenquanten erzeugt werden. Das elektrische Feld im Kristall wird durch Goldelektroden erzeugt. Der Si-Kristall ist etwa 3 mm bis 5 mm dick bei einer Querschnittsfläche von 10 mm2 bis 30 mm2. Die Detektor-einheit steckt in einem Rohr, welches in den meisten Fällen zur Röntgenquelle mit einem dünnen Kunststofffenster druckdicht verschlossen ist und einschließlich eines Vorverstärkers gekühlt wird. Zur Röntgenanalyse wird das Rohr mit dem

Kapitel 9 196
Kristall in den Polschuh der Objektivlinse hineingeschoben, im zurückgezogenen Zustand verschließt eine kleine Klappe das Rohrende und verhindert, dass ener-giereiche Rückstreuelektronen das dünne Kunststofffenster vor dem Kristall zer-stören. Im „Normalbetrieb“ des Mikroskops, d. h. bei kurzbrennweitigem Objektiv, ist diese Gefahr gering, weil die Rückstreuelektronen vom Linsenfeld gebündelt werden und nicht zum Detektor gelangen. Kritisch wird es, wenn im niedrigen Vergrößerungsbereich mit ausgeschaltetem bzw. stark geschwächtem Objektiv gearbeitet wird. In diesem Fall ist der Detektor vorher unbedingt in seine Ruhestellung außerhalb des Polschuhs zu bringen.
Die Vakua in der Detektorumgebung und in der Mikroskopsäule werden durch das Fenster voneinander getrennt. Damit wird verhindert, dass der gekühlte Detek-tor bei Belüftung der Mikroskopsäule vereist. Doch Vorsicht, der dünne Kunst-stofffilm wird auf der Detektorseite von einem Aluminiumgitter gestützt, um eine Druckdifferenz von etwas mehr als 1 bar aufzunehmen. Druck von 1 bar in der Mikroskopsäule ist möglich, Vakuum in der Mikroskopsäule und ein Druck von 1 bar in der Detektorumgebung zerstören das Fenster! Das Vakuum in Detektor-umgebung wird durch ein Sorptionsmittel aufrecht erhalten, welches ebenfalls durch den flüssigen Stickstoff im Dewar-Gefäß gekühlt wird. Nach mehrjährigem Betrieb hat das Sorptionsmittel soviel Gas gespeichert, dass beim Erwärmen ein Druck von mehr als 1 bar in der Detektorumgebung entstehen kann. Wird dann das Nachfüllen von flüssigem Stickstoff vergessen, kann dies zur Zerstörung des Fensters führen. Es ist deshalb günstig, wenn im Rahmen der Wartungsarbeiten der Detektorraum alle 2 ... 3 Jahre kontrolliert erwärmt und mit einer separaten Pumpe evakuiert wird.
Eine schnell arbeitende Elektronik („Pulsprozessor“) sorgt dafür, dass die durch Röntgenquanten erzeugten Ladungsträgerwolken abgesaugt, getrennt wahr-genommen und ihrer Stärke folgend in Energiekanäle einsortiert werden. In der Zeit, die für das „Abarbeiten“ der Ladungsträgerwolke gebraucht wird, ist der Detektor nicht arbeitsfähig (Totzeit). Bei hoher Röntgenintensität folgen die Rönt-genquanten u.U. so schnell aufeinander, dass deren Trennung unmöglich wird. Die Totzeit steigt an und kann den Detektor vollständig sperren. In diesem Fall muss die Röntgenintensität verringert oder die Elektronik beschleunigt werden. Im zweiten Fall wird die „Abarbeitungszeit“ verkürzt und die Ladungsträgerwolken können dann nicht mehr vollständig „abgearbeitet“ werden. Die Röntgenpeaks werden breiter, d. h. die Energieauflösung schlechter, und die Energieskale des Spektrums muss neu kalibriert werden. Der Pulsprozessor ist rechnergesteuert, und die „Abarbeitungszeiten“ werden mit der Energiekalibrierung in Kalibrie-rungsdateien niedergelegt und gespeichert.
Gegenwärtig werden die beschriebenen „klassischen“ Detektoren mehr und mehr durch Silizium-Drift-Detektoren (SDD – [9.3, 9.4]) ersetzt, bei denen der Siliziumkristall mit ringförmigen Elektroden versehen ist. Damit werden größere Eintrittsflächen bei gleichzeitigem schnelleren Abführen der Ladungsträgerwolken möglich. Diese Detektoren sind empfindlicher und können höhere Röntgeninten-sitäten verarbeiten. Im Allgemeinen genügt eine Peltier-Kühlung, so dass dem Ex-
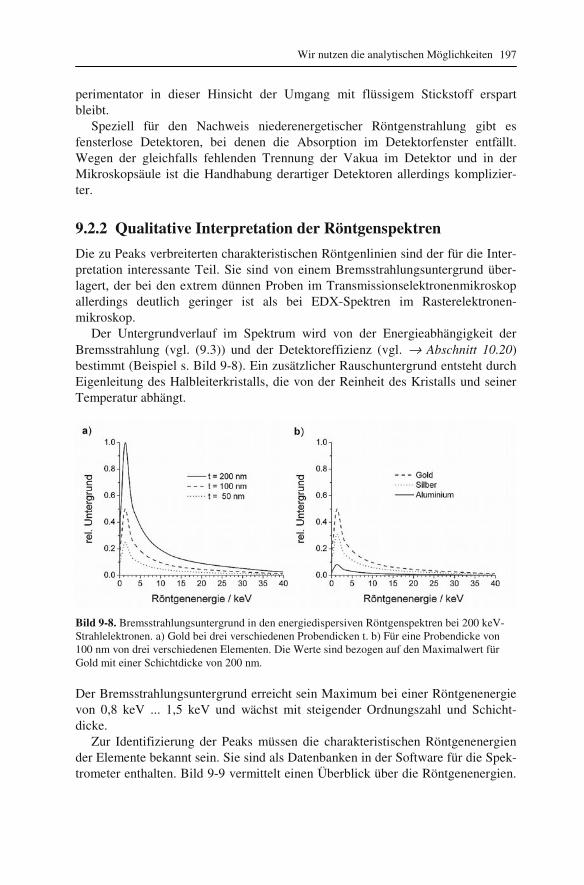
Wir nutzen die analytischen Möglichkeiten 197
perimentator in dieser Hinsicht der Umgang mit flüssigem Stickstoff erspart bleibt.
Speziell für den Nachweis niederenergetischer Röntgenstrahlung gibt es fensterlose Detektoren, bei denen die Absorption im Detektorfenster entfällt. Wegen der gleichfalls fehlenden Trennung der Vakua im Detektor und in der Mikroskopsäule ist die Handhabung derartiger Detektoren allerdings komplizier-ter.
9.2.2 Qualitative Interpretation der Röntgenspektren
Die zu Peaks verbreiterten charakteristischen Röntgenlinien sind der für die Inter-pretation interessante Teil. Sie sind von einem Bremsstrahlungsuntergrund über-lagert, der bei den extrem dünnen Proben im Transmissionselektronenmikroskop allerdings deutlich geringer ist als bei EDX-Spektren im Rasterelektronen-mikroskop.
Der Untergrundverlauf im Spektrum wird von der Energieabhängigkeit der Bremsstrahlung (vgl. (9.3)) und der Detektoreffizienz (vgl. → Abschnitt 10.20) bestimmt (Beispiel s. Bild 9-8). Ein zusätzlicher Rauschuntergrund entsteht durch Eigenleitung des Halbleiterkristalls, die von der Reinheit des Kristalls und seiner Temperatur abhängt.
Bild 9-8. Bremsstrahlungsuntergrund in den energiedispersiven Röntgenspektren bei 200 keV-Strahlelektronen. a) Gold bei drei verschiedenen Probendicken t. b) Für eine Probendicke von 100 nm von drei verschiedenen Elementen. Die Werte sind bezogen auf den Maximalwert für Gold mit einer Schichtdicke von 200 nm.
Der Bremsstrahlungsuntergrund erreicht sein Maximum bei einer Röntgenenergie von 0,8 keV ... 1,5 keV und wächst mit steigender Ordnungszahl und Schicht-dicke.
Zur Identifizierung der Peaks müssen die charakteristischen Röntgenenergien der Elemente bekannt sein. Sie sind als Datenbanken in der Software für die Spek-trometer enthalten. Bild 9-9 vermittelt einen Überblick über die Röntgenenergien.
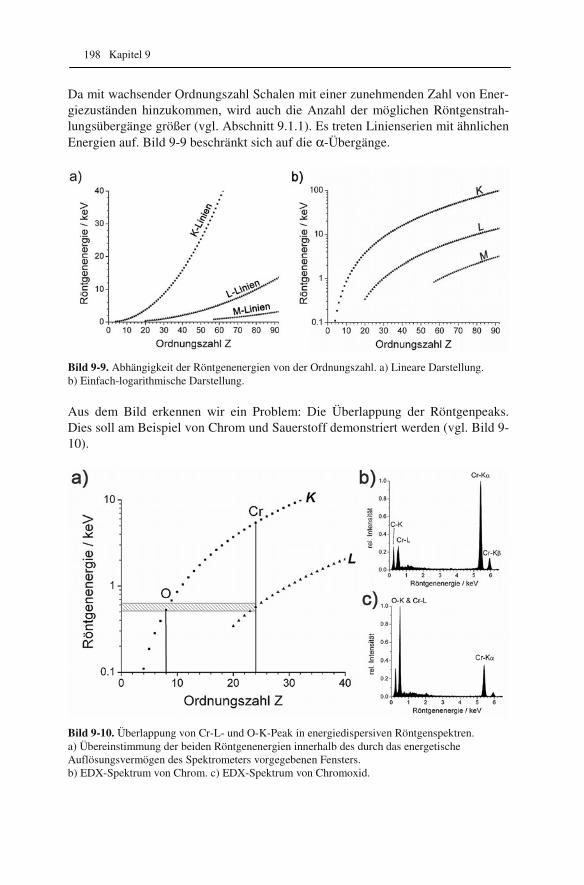
Kapitel 9 198
Da mit wachsender Ordnungszahl Schalen mit einer zunehmenden Zahl von Ener-giezuständen hinzukommen, wird auch die Anzahl der möglichen Röntgenstrah-lungsübergänge größer (vgl. Abschnitt 9.1.1). Es treten Linienserien mit ähnlichen Energien auf. Bild 9-9 beschränkt sich auf die α-Übergänge.
Bild 9-9. Abhängigkeit der Röntgenenergien von der Ordnungszahl. a) Lineare Darstellung. b) Einfach-logarithmische Darstellung.
Aus dem Bild erkennen wir ein Problem: Die Überlappung der Röntgenpeaks. Dies soll am Beispiel von Chrom und Sauerstoff demonstriert werden (vgl. Bild 9-10).
Bild 9-10. Überlappung von Cr-L- und O-K-Peak in energiedispersiven Röntgenspektren. a) Übereinstimmung der beiden Röntgenenergien innerhalb des durch das energetische Auflösungsvermögen des Spektrometers vorgegebenen Fensters. b) EDX-Spektrum von Chrom. c) EDX-Spektrum von Chromoxid.

Wir nutzen die analytischen Möglichkeiten 199
Chrom (Z = 24) hat eine K-Linie bei 5,41 keV und eine L-Linie bei 0,57 keV. Die Sauerstoff-K-Linie liegt bei 0,523 keV, d. h. unmittelbar neben der Cr-L-Linie. Das energetische Auflösungsvermögen des Röntgenspektrometers beträgt bei dieser Energie 70 eV - 80 eV, d. h. der L-Peak von Chrom und der K-Peak von Sauerstoff können nicht getrennt werden.
Der Vergleich der Linienintensitäten zeigt, dass im Spektrum 9-10c Sauerstoff beteiligt ist. Wenn das Vergleichsspektrum von reinem Cr unter gleichen Be-dingungen aufgenommen worden ist, ist diese Schlussfolgerung zwingend. Liegt allerdings kein Vergleichsspektrum vor, ist Vorsicht angeraten: Das Verhältnis der Intensitäten von Cr-K- und Cr-L-Peak hängt u. a. von der Spektrometereffizienz ab. Diese kann sich ändern, beispielsweise durch Verschmutzung des Detektorfen-sters. Das Intensitätsverhältnis wird auch durch die Probendicke beeinflusst, wie wir in Abschnitt 9.2.3 noch sehen werden.
Außer dieser Peak-Überlappung gibt es zwei Artefakte in den EDX-Spektren, die zu falschen Schüssen bzgl. der angezeigten Elemente führen können: Escape- und Summen-Peaks.
Zur Erklärung des Escape-Peaks erinnern wir uns an das Geschehen in der ladungsträgerfreien Zone des Detektorkristalls: Das eindringende Röntgenquant erzeugt freie Ladungsträger und baut dabei seine Energie ab. Es kann aber auch die K-Schale des Siliziums ionisieren (wir nehmen an, dass es sich um einen Si-liziumkristall handelt). Das Elektron von der K-Schale wird zum freien La-dungsträger, allerdings ist die dazu verbrauchte Energie gleich der Energie der Absorptionskante von Silizium (1,84 keV), d. h. wesentlich größer als die Band-lücke (1,2 eV). Das Auffüllen des freien Platzes in der K-Schale ist mit der Emis-sion eines Röntgenquants (Energie 1,74 keV) verbunden, dessen Energie im Kri-stall durch Erzeugung von Ladungsträgern weiter abgebaut wird. Dies wird ver-hindert, wenn letzteres Röntgenquant den Kristall verlässt (deshalb „Escape“). Dann fehlt diese Energie und es entsteht ein zusätzlicher Peak im Spektrum, des-sen Energie um 1,74 keV kleiner ist als der Bezugspeak. Beispielsweise wird der Cr-Kα-Peak (5,41 keV) begleitet von einem Escape-Peak bei 3,67 keV. Dies ist aber gerade die Kα-Energie von Kalzium (3,69 keV). Unvorsichtigerweise könnte also auf geringe Mengen Kalzium in der Probe geschlossen werden; tatsächlich handelt es sich aber um den Escape-Peak von Chrom. Die Wahrscheinlichkeit für derartige Escape-Peaks hängt u. a. von der Größe des Detektorkristalls ab, sie sollte aber bei der Analyse der Spektren nicht außer Acht gelassen werden.
Summen-Peaks kommen zustande, wenn zwei Röntgenquanten vom gleichen Element zeitgleich in den Kristall eindringen. Zeitgleich heißt, dass sie vom Pulsprozessor nicht getrennt wahrgenommen werden können. In diesem Fall ent-steht ein Peak mit der doppelten Energie des Bezugspeaks. Die Wahrscheinlich-keit für das Auftreten von Summen-Peaks wächst mit der Intensität der Röntgen-strahlung. In modernen EDX-Spektrometern ist sie gering, weil der Pulsprozessor sehr schnell arbeitet.
Wir wollen noch auf einen Umstand hinweisen, der häufig unterschätzt wird. Trotz rastertransmissionselektronenmikroskopischer Arbeitsweise mit subnano-
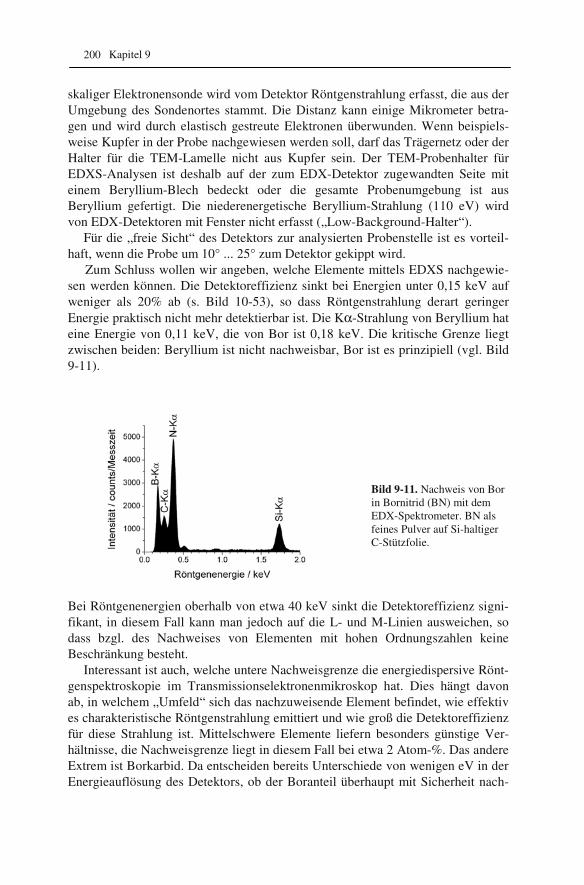
Kapitel 9 200
skaliger Elektronensonde wird vom Detektor Röntgenstrahlung erfasst, die aus der Umgebung des Sondenortes stammt. Die Distanz kann einige Mikrometer betra-gen und wird durch elastisch gestreute Elektronen überwunden. Wenn beispiels-weise Kupfer in der Probe nachgewiesen werden soll, darf das Trägernetz oder der Halter für die TEM-Lamelle nicht aus Kupfer sein. Der TEM-Probenhalter für EDXS-Analysen ist deshalb auf der zum EDX-Detektor zugewandten Seite mit einem Beryllium-Blech bedeckt oder die gesamte Probenumgebung ist aus Beryllium gefertigt. Die niederenergetische Beryllium-Strahlung (110 eV) wird von EDX-Detektoren mit Fenster nicht erfasst („Low-Background-Halter“).
Für die „freie Sicht“ des Detektors zur analysierten Probenstelle ist es vorteil-haft, wenn die Probe um 10° ... 25° zum Detektor gekippt wird.
Zum Schluss wollen wir angeben, welche Elemente mittels EDXS nachgewie-sen werden können. Die Detektoreffizienz sinkt bei Energien unter 0,15 keV auf weniger als 20% ab (s. Bild 10-53), so dass Röntgenstrahlung derart geringer Energie praktisch nicht mehr detektierbar ist. Die Kα-Strahlung von Beryllium hat eine Energie von 0,11 keV, die von Bor ist 0,18 keV. Die kritische Grenze liegt zwischen beiden: Beryllium ist nicht nachweisbar, Bor ist es prinzipiell (vgl. Bild 9-11).
Bild 9-11. Nachweis von Bor in Bornitrid (BN) mit dem EDX-Spektrometer. BN als feines Pulver auf Si-haltiger C-Stützfolie.
Bei Röntgenenergien oberhalb von etwa 40 keV sinkt die Detektoreffizienz signi-fikant, in diesem Fall kann man jedoch auf die L- und M-Linien ausweichen, so dass bzgl. des Nachweises von Elementen mit hohen Ordnungszahlen keine Beschränkung besteht.
Interessant ist auch, welche untere Nachweisgrenze die energiedispersive Rönt-genspektroskopie im Transmissionselektronenmikroskop hat. Dies hängt davon ab, in welchem „Umfeld“ sich das nachzuweisende Element befindet, wie effektiv es charakteristische Röntgenstrahlung emittiert und wie groß die Detektoreffizienz für diese Strahlung ist. Mittelschwere Elemente liefern besonders günstige Ver-hältnisse, die Nachweisgrenze liegt in diesem Fall bei etwa 2 Atom-%. Das andere Extrem ist Borkarbid. Da entscheiden bereits Unterschiede von wenigen eV in der Energieauflösung des Detektors, ob der Boranteil überhaupt mit Sicherheit nach-

Wir nutzen die analytischen Möglichkeiten 201
gewiesen werden kann. Vorsicht ist geboten bei Benutzung der häufig in der Software eingebauten Möglichkeit der automatischen Peakidentifizierung. „Was man im Spektrum nicht mit bloßem Auge sieht, ist auch nicht vorhanden!“ Das Signal sollte dreimal höher sein als der Untergrund (Rose-Kriterium [9.5]).
9.2.3 Quantifizierung von Röntgenspektren
Die Energie der Röntgenquanten spiegelt sich in der Zahl der im Detektorkristall erzeugten freien Ladungsträger und damit in der Höhe des Strompulses wider. Die Intensität der Röntgenstrahlung richtet sich nach der Zahl der Atome, die angeregt werden können, und bestimmt die Zahl der Strompulse („counts“) während der Messzeit und damit die Größe der Peaks. Das Verhältnis der Peakgrößen IA und IB zweier Elemente A und B ist proportional zum Verhältnis der Zahl der Atome NA und NB, die angeregt werden. Den Proportionalitätsfaktor nennen wir kAB:
A AAB
B B
I Nk
I N= ⋅ . (9.20)
Die Elementkonzentration cA ist gleich dem Quotienten aus der Anzahl der Atome der Sorte A und der Gesamtzahl der Atome. Analoges gilt für die Sorte B:
, A BA B
A B A B
N Nc c
N N N N= =
+ + . (9.21)
Im Allgemeinen werden Konzentrationen in Prozent angegeben. Im o.g. Fall sind dies Atomprozent. Auch die Angabe in Gewichts- bzw. Masseprozent ist üblich. Dazu muss die Atomzahl mit der Atommasse M multipliziert werden. Für die Konzentrationsverhältnisse gilt damit:
, A
, B
M bzw.
MA MA A A
B B B M B
cc N N
c N c N
⋅= =
⋅ (9.22)
(MA,B: Atomgewichte der Elemente A und B). Wir erhalten demzufolge das Ver-hältnis der Konzentration von zwei Elementen in der Probe aus dem Verhältnis der Intensitäten der betreffenden Röntgenpeaks:
, B
, A
M 1
MA MA A
B B M AB B
cc I
c c k I= ⋅ = ⋅ . (9.23)
Diese Gleichung wird als „Cliff-Lorimer-Gleichung“ bezeichnet, die Proportiona-litätsfaktoren sind auch als „Cliff-Lorimer-k-Faktoren“ bekannt [9.6]. Mit der Normierung

Kapitel 9 202
1A Bc c+ = (9.24)
folgt aus dem Konzentrationsverhältnis für die einzelnen Konzentrationen:
A B
A B A B
c c 1 und
1+ c c 1+ c cA Bc c= = . (9.25)
Sind nicht nur zwei sondern n Elemente beteiligt, wird ein Bezugselement B aus-gewählt und die Konzentrationsverhältnisse zu diesem Element werden nach (9.23) berechnet:
3 11 2, , , .... , n
B B B B
c cc c
c c c c− . (9.26)
Die Normierungsbedingung lautet nun:
1
1
1n
B ii
c c−
=
+ = . (9.27)
Damit wird als erstes die Konzentration des Bezugselementes ausgerechnet:
1
1
1
1+B n
i Bi
c
c c−
=
= . (9.28)
Die Bestimmung der restlichen Konzentrationen ist trivial:
ii B
B
cc c
c= ⋅ . (9.29)
Wir sehen zwei grundsätzliche Aufgaben, die zur Quantifizierung der Röntgen-spektren gelöst werden müssen: Die Bestimmung der Peakintensitäten und die Ermittlung des Proportionalitätsfaktors 1/kAB. - Bestimmung der Peakintensitäten:
Die Lösung dieser Aufgabe erscheint einfach: Wir bestimmen die Peakflächen. Allerdings müssen wir davon den energieabhängigen Untergrund U(E) abziehen, der durch Bremsstrahlung und Rauschen infolge Eigenleitung im Detektorkristall entsteht. Wir bezeichnen das Spektrum als Funktion S(E). Die Spektrenfunktion liegt in Form einer Wertetabelle vor, d. h. als Zahl (counts) in einem Energie-

Wir nutzen die analytischen Möglichkeiten 203
bereich von E-ΔE/2 bis E+ΔE/2 für die Energie E („Kanal“ - vgl. Bild 9-12a). Die Grenzen iA und iE markieren die Kanäle am Beginn und am Ende des Peaks von Element A:
( )E
A
i
A i ii i
I S U=
= − . (9.30)
Mit Berücksichtigung der Tatsache, dass die Proben extrem dünn sind, ändert sich die Bremsstrahlung bei höheren Energien nahezu linear mit der Energie (vgl. Bild 9-8). Der Untergrundverlauf kann durch eine Gerade angenähert werden, deren Gleichung aus den Mittelwerten der Intensitäten an zwei Stützstellen U1 und U2 außerhalb des Röntgenpeaks bestimmt wird. .
Bild 9-12. Peakflächenbestimmung und Untergrundapproximation in EDX-Spektren. a) Summation der Kanalinhalte und Untergrundverlauf im höherenergetischen Teil des Spektrums. b) Untergrund im niederenergetischen Teil des Spektrums.
Problematischer ist die Untergrundapproximation im niederenergetischen Teil des Spektrums. Hier ändern sich sowohl Bremsstrahlung als auch Spektrometer-effizienz dramatisch mit der Energie (s. Bild 9-8). Bei Vernachlässigung des Absorptionskanteneffektes in der Spektrometereffizienz (vgl. → Abschnitt 10.20) kann der Untergrundverlauf durch eine Funktion der Form
( ) ( )201 1 1 e c EE
U E cE
− ⋅= ⋅ − ⋅ − (9.31)
angenähert werden. Bisher sind wir davon ausgegangen, dass die Röntgenpeaks voneinander ge-
trennt sind. Dies ist nicht immer der Fall, wie Bild 9-10 zeigt. Bei derartigen

Kapitel 9 204
Überlagerungen versagt die einfache Summation der Kanalintensitäten. Die Zu-ordnung von Intensitäten zu den beteiligten Peaks wird dann durch eine Model-lierung der gemessenen Peaks durch Gauß- (oder ähnlich geformte) Kurven vor-genommen. Die Maxima der Gaußkurven werden auf die Nennenergien der betei-ligten Peaks gesetzt, für die Halbwertsbreite wird das Energieauflösungsvermögen benutzt. Die Gesamtfläche unter den Gauß-Kurven wird der aufsummierten Fläche im Spektrum durch Variation der Maximalwerte der Gauß-Kurven angepasst. Die einzelnen Peakintensitäten entsprechen dann den Flächen unter den einzelnen Gauß-Kurven. Mitunter sieht die Spektrometer-Software auch eine experimentelle Ermittlung der Peakform vor. Bei der Installation müssen dann Musterspektren an Standards aufgenommen werden, anhand derer das passendste mathematische Peakform-Modell ausgewählt wird. - Ermittlung der Proportionalitätsfaktoren 1/kAB:
Die beste Methode ist es, die Proportionalitätsfaktoren durch Messung an Stan-dards bekannter Zusammensetzung experimentell zu bestimmen. Das Verhältnis cA/cB vom Standard ist bekannt, das Verhältnis der Peakintensitäten IA/IB wird aus dem Spektrum ermittelt und der Proportionalitätsfaktor nach Gleichung (9.19) be-rechnet. Diese Methode hat den Vorteil, dass alle individuellen Merkmale des Spektrometers und der Spektrenauswertung berücksichtigt sind. Sie hat aber auch gravierende Nachteile: Wir benötigen Standards und wir müssen von diesen Stan-dards Proben für das Transmissionselektronenmikroskop präparieren. Dieser Auf-wand lohnt sich nur, wenn man sich über längere Zeit mit einem speziellen Ma-terialsystem beschäftigt.
Für die „schnelle“ Quantifizierung von Röntgenspektren wird deshalb eine an-dere Methode bevorzugt, die ohne Standards auskommt: Die Berechnung der Pro-portionalitätsfaktoren. Das Ganze heißt dann standardlose Quantifizierung.
Der Berechnung liegt ein Modell zugrunde, welches die Wahrscheinlichkeiten, mit der die einzelnen Schritte der Röntgenemission und ihres Nachweises eintre-ten, miteinander multipliziert. Die einzelnen Schritte sind:
- die Ionisation einer inneren Schale (Ionisationswahrscheinlichkeit Q), - das Auffüllen dieser Schale unter Aussendung eines Röntgenquants
(Fluoreszenzausbeute ω) und - der Nachweis des Röntgenquants im Detektor (Detektoreffizienz Deff).
Für eine bessere Übersicht ist es zweckmäßig, den Proportionalitätsfaktor in der Form
1 A
AB B
k
k k= (9.32)
zu schreiben. Unter Berücksichtigung der o.g. Schritte und des Kehrwertes erhal-ten wir

Wir nutzen die analytischen Möglichkeiten 205
,
,
B B eff BA
B A A eff A
Q Dk
k Q D
ω
ω
⋅ ⋅=
⋅ ⋅ . (9.33)
In der Literatur werden diese Faktoren in der Regel auf Masseprozente bezogen, so dass das Verhältnis der Atomgewichte zu berücksichtigen ist:
, B
, A
M
MB B eff BA
B A A eff AM
Q Dk
k Q D
ω
ω
⋅ ⋅ ⋅=
⋅ ⋅ ⋅ . (9.34)
Die Röntgenstrahlung wird gleichmäßig (isotrop) in den gesamten Raum emittiert. Der Anteil, der davon vom Detektor erfasst wird, hängt vom Raumwinkel ab, den das Detektoreintrittsfenster überdeckt, d. h. von der Geometrie des Detektors und seinem Abstand von der Probe. Auch der Winkel zwischen Probenoberfläche und Detektorachse („Abnahmewinkel“) spielt eine Rolle. Diese Einflüsse sind aber für alle Elemente gleich und wirken sich deshalb im Verhältnis zweier Konzentratio-nen nicht aus. Die Detektoreffizienz wird in → Abschnitt 10.20 genauer behandelt. Für die Ionisationswahrscheinlichkeit und die Fluoreszenzausbeute gibt es ver-schiedene Modelle, die unterschiedliche Näherungen bedeuten (vgl. → Abschnitt 10.21).
Bild 9-13. Berechnete Cliff-Lorimer-k-Faktoren, jeweils bezogen auf die Si-Kα-Linie für 300 keV-Elektronen (Rechnung → Abschnitt 10.21).
Insofern ist die standardlose Quantifizierung mit systematischen Fehlern belastet, die von der Güte der Modelle abhängen. Häufig gelten die Modelle nur für einen eingeschränkten Bereich von Röntgenenergien. Bei der Quantifizierung mittel-schwerer Elemente (Ordnungszahlen 20 ... 40) unter Verwendung der K-Linien sind die kleinsten Fehler zu erwarten (relativ etwa 5%). Die Fehler sind deutlich größer beim Vergleich von K- mit L- bzw. M-Linien. Die in Bild 9-13 darge-stellten k-Faktoren sind auf Masseprozente und die Referenz Si-Kα bezogen, wie es in der Literatur üblich ist.

Kapitel 9 206
Bild 9-13 zeigt auch, dass sich die k-Faktoren der einzelnen Elemente sehr stark unterscheiden. Es ist deshalb falsch, durch bloßen Vergleich der Peak-intensitäten auf Konzentrationsverhältnisse zu schließen!
Je geringer die Effizienz bei Röntgenemission und –nachweis ist, desto größer muss der zugehörige k-Faktor sein, um diese Unterschiede bei der Umrechnung von Intensitäts- in Konzentrationsverhältnisse auszugleichen. Das bedeutet aber auch: Je größer der k-Faktor desto geringer das Messsignal. Wir sehen aus Bild 9-13, dass Elemente mit Ordnungszahlen bis etwa 40 am effizientesten anhand der K-Linien zu messen sind. Bei Elementen mit Ordnungszahlen über 40 sind die L-Linienpaare zu bevorzugen. In der Regel sind bei großen Unterschieden in den k-Faktoren größere systematische Fehler bei der standardlosen Quantifizierung zu erwarten.
- Absorptionskorrektur:
Für leichte Elemente kann die Absorption der niederenergetischen Röntgen-strahlung in der dünnen Probe das Ergebnis zusätzlich verfälschen (→ Abschnitt 10.22). Die Schichtdicke t, bei welcher der systematische Fehler infolge unter-schiedlicher Absorption 10% nicht überschreitet, kann nach
( ) ( )
0, 2 sinEA EB
tδ
ρ μ ρ μ ρ
⋅<
⋅ −
(9.35)
(δ: Abnahmewinkel der Röntgenstrahlung, ρ: Probendichte, EA(μ/ρ) bzw. EB(μ/ρ): Schwächungskoeffizienten der Probe für Röntgenenergien EA bzw. EB – vgl. auch (10.22.14)) berechnet werden. Wir wollen zwei Beispiele für die Grenz-schichtdicke ausrechnen: CrSi und CrO.
Die Konzentrationen (in Atomprozent) sind für CrSi: cCr = 0,5 und cSi = 0,5. Wir nehmen näherungsweise an, dass sich Dichte und Schwächungskoeffizienten der Probe aus den Werten für Chrom und Silizium mit den Konzentrationen als Wichtungsfaktoren berechnen lassen:
3 3 30,5 7,19 g/cm 0,5 2,33 g/cm 4,76 g/cm
Cr Cr Si Sic cρ ρ ρ= ⋅ + ⋅
= ⋅ + ⋅ = . (9.36)
Für die Schwächungskoeffizienten benötigen wir eine mittlere Ordnungszahl Z, um nach der Formel
( )2 -1
12,396
cm g / keV
E
Z C ZE
α
βμ ρ
= ⋅ ⋅⋅
(10.20.3)
mit den Parameterwerten aus Tabelle 10-4 rechnen zu können:

Wir nutzen die analytischen Möglichkeiten 207
0,5 24 0,5 14 19Cr Cr Si SiZ c Z c Z= ⋅ + ⋅ = ⋅ + ⋅ = . (9.37)
Für die Schwächungskoeffizienten erhalten wir 980 cm2/g für die Si-Kα- und 50 cm2/g für die Cr-Kα-Strahlung. Bei einem Abnahmewinkel von 20° folgt da-raus eine Grenzschichtdicke von ca. 150 nm.
Für CrO gilt: cCr = 0,5 und cO = 0,5. Nach analoger Rechnung wie bei CrSi mit ρ = 3,58 g/cm3, Z = 16, OK(μ/ρ) ≈ 13850 cm2/g, CrK(μ/ρ) ≈ 23 cm2/g) erhalten wir eine Grenzschichtdicke von etwa 15 nm.
Bei Probendicken von typischerweise 100 nm im Transmissionselektronen-mikroskop spielt die Absorption bei der Quantifizierung der Röntgenspektren im Fall von Chrom und Silizium keine Rolle; bei Chrom und Sauerstoff muss demgegenüber mit Fehlern deutlich größer als 10% gerechnet werden.
Wie kann man diesen Absorptionseinfluss korrigieren? Natürlich durch eine Rechnung (→ Abschnitt 10.22). Allerdings müssen dazu Probendicke, Proben-dichte und Schwächungskoeffizienten hinreichend genau bekannt sein. Bei unse-ren Beispielen hatten wir uns mit Näherungen für Dichte und Schwächungs-koeffizienten begnügt. Die Messung der Probendicke ist problematisch, wenn dies routinemäßig durchgeführt werden soll. Wir wollen deshalb auf ein Verfahren von Z. Horita [9.7] zur Absorptionskorrektur hinweisen, welches ohne Kenntnis dieser Werte auskommt.
Bild 9-14. Absorptions-korrektur bei EDXS-Messung des Silizium-Chrom-Verhältnisses nach dem Verfahren von Horita [9.7].
Voraussetzung sind eine Probe mit kontinuierlich veränderlicher Dicke, wie dies bei abgedünnten Proben am keilförmigen Rand in der Regel gegeben ist, und ein Material, das mindestens eine hochenergetische Röntgenlinie emittiert. Mehrere Spektren werden an Stellen unterschiedlicher Probendicke unter sonst identischen Bedingungen aufgenommen (Strahlstrom, Abnahmewinkel, Messzeit) und ohne Absorptionsberücksichtigung quantifiziert. Die Quantifizierungsergebnisse wer-den in einem Diagramm als Ordinatenwerte über den Intensitäten einer hochener-getischen Röntgenlinie (hängt linear von der Probendicke ab) aufgetragen und ihr Verlauf zum Abszissenwert Null (d. h. Dicke Null) extrapoliert. Der zugehörige Ordinatenwert ist das absorptionskorrigierte Quantifizierungsergebnis (vgl. Bild 9-

Kapitel 9 208
14). Das Bild zeigt die Korrektur am Beispiel einer gesputterten Silizium-Chrom-Mischschicht. Das Target enthielt 74 Atom-% Silizium und 26 Atom-% Chrom. Die Spektren wurden an drei verschieden dicken Stellen der Probe aufgenommen (Dicken etwa 50 nm, 100 nm und 150 nm), die drei Gruppen von cSi/cCr-Ver-hältnissen ergeben: Bei Cr-Kα-Intensitätswerten von etwa 1500 Counts/Messzeit diejenigen von der dünnsten Stelle mit einem Mittelwert von 2,9, die bei etwa 3000 Counts/Messzeit mit einem Mittelwert von 2,65 und die von der dicksten Stelle bei 4000 bis 5000 Counts/Messzeit mit einem Mittelwert von 2,5. Die Ex-trapolation liefert einen Wert von 3,04 für das cSi/cCr-Verhältnis. Das entspricht einem Siliziumanteil von 75,2 Atom-% und kommt damit dem Targetwert sehr nahe, wie das in diesem Fall erwartet wurde. Das an der 150 nm dicken Proben-stelle gemessene Konzentrationsverhältnis weicht demgegenüber um ca. 18% vom Sollwert ab. Diese Dicke ist gerade die oben berechnete Grenzschichtdicke für 10% Abweichung, allerdings berechnet für ein anderes Si/Cr-Verhältnis. Die größenordnungsmäßige Übereinstimmung zeigt aber, dass unser Modell zur Ab-schätzung des Absorptionseinflusses geeignet ist.
- zufällige Fehler:
Schließlich wollen wir auf einige Fehler bei EDXS-Messungen hinweisen, die vom Experimentator beeinflusst werden können. Zunächst erinnern wir daran, dass sowohl die Röntgenemission als auch der Nachweis statistische Prozesse sind. Bei einer Zahl N von statistischen Ereignissen ist der relative Fehler
1 N (9.38)
oder anders ausgedrückt: Soll der statistische Fehler bei Auswertung von zwei Röntgenpeaks mit den counts (nachgewiesene Ereignisse) N1 = N und N2 = a⋅N kleiner als 1% sein, so muss die Zahl N der counts im Bezugspeak
( )210000
1+N aa
> ⋅ (9.39)
sein, bei zwei gleich großen Peaks also je 40000 counts. Ein zweites Problem sind Hindernisse, die die Röntgenstrahlung auf ihrem
Weg zum Detektor überwinden muss („Abschattung“). Dies kann der Steg eines Objektträgernetzes sein oder ein überstehendes Probendetail oder auch der Rand des Probenhalters. Im EDX-Spektrum äußert sich dies in fehlendem oder redu-ziertem Signal bei niedrigen Energien einschließlich des Bremsstrahlungsunter-grundes. Kippen der Probe in Richtung des Röntgendetektors kann die Lage ver-bessern. Bereits beim Einbau der Probe in den Halter muss darauf geachtet werden, dass die Röntgenstrahlung ungehindert den Detektor erreichen kann. Trägernetze sind mit der Schicht nach oben einzubauen. (Achtung, oft sind die Halter in Einbaustellung 180° um ihre Achse gegen die Arbeitsstellung im Mikro-

Wir nutzen die analytischen Möglichkeiten 209
skop gedreht!) Halter mit angeschweißten FIB-Lamellen (vgl. Abschnitt 3.4) sollten so eingelegt werden, dass die offene Seite zum Detektor zeigt. Es lohnt sich, vor dem Probeneinbau über diese Dinge nachzudenken, denn jeder Proben-ein- und –ausbau birgt die Gefahr der Beschädigung der Probe in sich.
9.2.4 Linienprofile und Elementverteilungsbilder
In rastertransmissionselektronenmikroskopischer Arbeitsweise ist es möglich, den Elektronenstrahl schrittweise auf einer im STEM-Bild vorgegebenen Linie zu führen und an jedem Haltepunkt ein EDX-Spektrum aufzunehmen. Diese Verfah-rensweise wird auch als „Linescan“ bezeichnet. In den Spektren wird dann ein charakteristischer Röntgenpeak ausgewählt, ein Fenster mit einer Weite von 100 eV ... 200 eV über diesen Peak gelegt und die Intensitäten in diesem Fenster aus allen Spektren extrahiert. Das Ergebnis ist ein Profil des Anteils des zum aus-gewählten Peak gehörenden Elements entlang der vorgegebenen Linie (s. Bild 9-15).
Bild 9-15. EDXS-Linescan-Messung an eisengefüllten und mit Tantal ALD56-beschichteten Kohlenstoff-Nanoröhren. a) EDX-Spektrum als Summe aller Einzelspektren. Die Energiefenster sind grau eingefärbt und erfassen den Ta-M-Peak und den Fe-Kα-Peak. b) STEM-HAADF-Bild mit eingezeichneter Linie (Position) entlang derer die Einzelspektren (70 Stück) aufgenommen wurden. c) Profil der Röntgenintensitäten des Ta-M- und des Fe-Kα-Peaks entlang der Positions-linie. Die kleinen Ta-Peaks am Rande des Fe-Profils deuten auf eine Ta-Hülle der Eisenfüllung hin. Die Probe wurde freundlicherweise von S. Menzel zur Verfügung gestellt.
Das Intensitätsprofil der Ta-M-Linie in Bild 9-15c zeigt den typischen Verlauf für eine Rohrwandbeschichtung [9.8].
An querschnittspräparierten Proben (vgl. Abschnitte 3.3 und 3.4) ist es mit die-ser Linescan-Methode möglich, Diffusionsprofile aufzunehmen. Dabei interessiert die Ortsauflösung des Verfahrens. Wir wissen bereits von Bild 8-5, dass der Elek-tronenstrahl durch elastische Streuung innerhalb der Probe aufgeweitet wird. In Bild 9-16a ist dies schematisch dargestellt. Die Durchmesseraufweitung b kann nach J.I. Goldstein [9.9] mit einer Näherungsformel
56 ALD: Atomic Layer Deposition
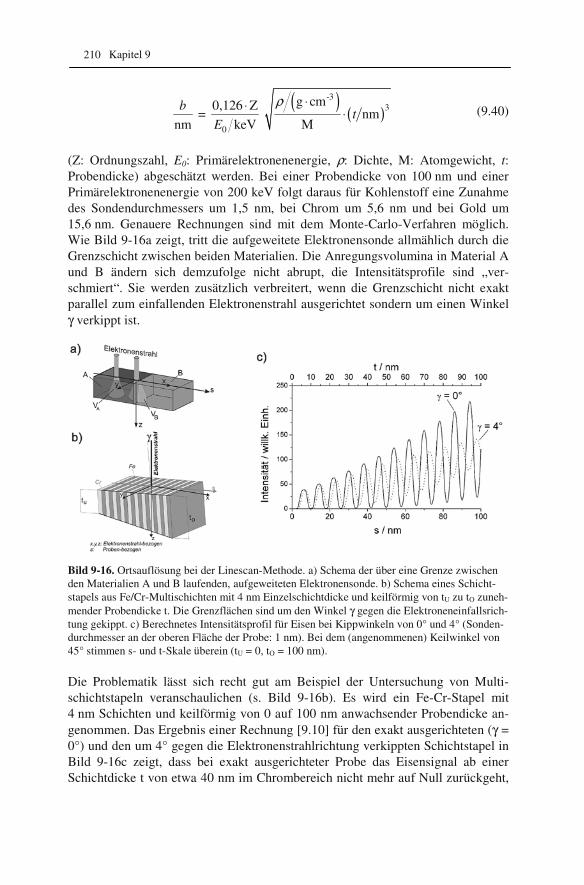
Kapitel 9 210
( )( )
-33
0
g cm0,126 Z = nm
nm keV M
bt
E
ρ ⋅⋅⋅ (9.40)
(Z: Ordnungszahl, E0: Primärelektronenenergie, ρ: Dichte, M: Atomgewicht, t: Probendicke) abgeschätzt werden. Bei einer Probendicke von 100 nm und einer Primärelektronenenergie von 200 keV folgt daraus für Kohlenstoff eine Zunahme des Sondendurchmessers um 1,5 nm, bei Chrom um 5,6 nm und bei Gold um 15,6 nm. Genauere Rechnungen sind mit dem Monte-Carlo-Verfahren möglich. Wie Bild 9-16a zeigt, tritt die aufgeweitete Elektronensonde allmählich durch die Grenzschicht zwischen beiden Materialien. Die Anregungsvolumina in Material A und B ändern sich demzufolge nicht abrupt, die Intensitätsprofile sind „ver-schmiert“. Sie werden zusätzlich verbreitert, wenn die Grenzschicht nicht exakt parallel zum einfallenden Elektronenstrahl ausgerichtet sondern um einen Winkel γ verkippt ist.
Bild 9-16. Ortsauflösung bei der Linescan-Methode. a) Schema der über eine Grenze zwischen den Materialien A und B laufenden, aufgeweiteten Elektronensonde. b) Schema eines Schicht-stapels aus Fe/Cr-Multischichten mit 4 nm Einzelschichtdicke und keilförmig von tU zu tO zuneh-mender Probendicke t. Die Grenzflächen sind um den Winkel γ gegen die Elektroneneinfallsrich-tung gekippt. c) Berechnetes Intensitätsprofil für Eisen bei Kippwinkeln von 0° und 4° (Sonden-durchmesser an der oberen Fläche der Probe: 1 nm). Bei dem (angenommenen) Keilwinkel von 45° stimmen s- und t-Skale überein (tU = 0, tO = 100 nm).
Die Problematik lässt sich recht gut am Beispiel der Untersuchung von Multi-schichtstapeln veranschaulichen (s. Bild 9-16b). Es wird ein Fe-Cr-Stapel mit 4 nm Schichten und keilförmig von 0 auf 100 nm anwachsender Probendicke an-genommen. Das Ergebnis einer Rechnung [9.10] für den exakt ausgerichteten (γ = 0°) und den um 4° gegen die Elektronenstrahlrichtung verkippten Schichtstapel in Bild 9-16c zeigt, dass bei exakt ausgerichteter Probe das Eisensignal ab einer Schichtdicke t von etwa 40 nm im Chrombereich nicht mehr auf Null zurückgeht,

Wir nutzen die analytischen Möglichkeiten 211
seine Modulationshöhe aber weiter ansteigt. Bei der verkippten Probe sinkt die Modulationshöhe demgegenüber dramatisch, so dass ab einer Probendicke von ca. 150 nm kein Schichtstapel mehr erkennbar wäre.
Wenn die Elektronensonde nicht nur eine Linie sondern eine Fläche abrastert, erhält man ein zweidimensionales Elementverteilungsbild. Oft werden derartige Bilder für verschiedene Elemente aufgenommen, unterschiedlich eingefärbt und überlagert. Bild 9-17 zeigt das Ergebnis von einer Cr/Fe-Multischicht wie sie be-schrieben wurde. Der Bildeindruck stimmt mit der Erwartung überein: Mit wach-sender Probendicke verschmieren die Farben zunehmend. Am rechten Rand ist nur noch gelb als Mischfarbe zu sehen.
Bild 9-17. In Rasterarbeits-weise mit EDXS aufgenom-mene Elementverteilungsbil-der für Chrom und Eisen, koloriert (Cr: grün, Fe: rot) und überlagert. Der Schicht-stapel beginnt links mit einer 15 nm dicken Cr-Schicht, die folgenden Schichtdicken betragen (4 ... 5) nm. Die Probendicke steigt nach rechts keilförmig an.
9.3 Elektronenenergieverlust-Spektroskopie („EELS“) Nachdem wir uns mit den Röntgenstrahlungsübergängen innerhalb der Elektro-nenhüllen der Atome und ihrer Messung befasst haben, wollen wir uns wieder den Primärelektronen zuwenden. Aus der Beschleunigungsspannung zwischen Katho-de und Anode können wir deren Energie vor dem Eindringen in die Probe be-rechnen. Wenn wir ihre Energie nach Verlassen der Probe messen, kennen wir den Energieverlust, den diese Elektronen in der Probe erlitten haben. Aus Abschnitt 9.1.2 wissen wir, dass diese Energieverluste charakteristisch für die Elemente und für die Bindungen zwischen den Atomen im Festkörper sind. Wir wollen uns nun den praktischen Gesichtspunkten dieser Elektronenenergieverlust-Spektroskopie (englisch: Electron Energy Loss Spectroscopy – EELS) widmen: Wie können Elektronenenergien gemessen werden? Wie sehen die Spektren aus, und was können wir daraus lernen? Welche Möglichkeiten in Verbindung mit dem Transmissionselektronenmikroskop gibt es?
9.3.1 Elektronenenergie-Spektrometer
Aufgabe des Spektrometers ist es, die einfallenden Elektronen nach ihrer Energie zu trennen, d. h. Elektronen unterschiedlicher Energie sollen das Spektrometer an verschiedenen Orten bzw. unter verschiedenen Bahnneigungen verlassen. In der

Kapitel 9 212
Lichtoptik erledigt dies ein Glasprisma. In → Abschnitt 10.23 wird gezeigt, dass magnetische oder elektrische Felder diese Aufgabe für Elektronen übernehmen können. Das magnetische oder elektrostatische Prisma muss durch elektronenopti-sche Bauelemente ergänzt werden, die u. a. dafür sorgen, dass die Elektronen weitgehend parallel in das Prisma einfallen, und die Änderung der Dispersion er-möglichen (vgl. Bild 9-18).
Im Zusammenspiel mit den elektronenoptischen Einheiten (Multipole) erzeugt das Prisma eine energieselektive Ebene, in der sich Elektronen gleicher Energie am selben Ort treffen. Die Entfernung Δs zwischen den Auftrefforten von Elektronen mit den Energien E1 und E2 ist durch die Dispersion (Empfindlichkeit)
1 2
sD
E E
Δ=
− . (9.41)
Bild 9-18. Schematische Darstellung eines Elektro-nenenergie-Spektrometers.
des Spektrometers bestimmt. Die Dispersion wird auch durch die Primärenergie beeinflusst. Je kleiner diese ist, desto höher wird die Dispersion (→ Abschnitt 10.23). Prinzipiell ist es möglich, mit Hilfe sogenannter „Immersionslinsen“ die Elektronen insgesamt zu verzögern, um die Dispersion zu vergrößern.
Die energieselektive Ebene wird auf eine CCD-Kamera (vgl. Abschnitt 2.7.4) abgebildet und die Helligkeitswerte ausgelesen. Die Justage des Spektrometers umfasst die Kontrolle und evtl. Korrektur des senkrechten Elektroneneinfalls in das Prisma und der „Fokussierung“ von Elektronen gleicher Energie in die ener-gieselektive Ebene. Bei modernen Spektrometern wird diese Justage von einer Software übernommen.
9.3.2 Low-Loss- und Core-Loss-Bereich der Spektren
Wir wollen uns nun ein Elektronenenergieverlust-Spektrum ansehen, wie es ty-pisch für die im Transmissionselektronenmikroskop aufgenommenen Spektren ist (s. Bild 9-19).

Wir nutzen die analytischen Möglichkeiten 213
Die Spektren sind normiert, d. h. die höchste Intensität im ausgewählten Energiebereich ist gleich Eins gesetzt. Auf der Abszisse ist der Energieverlust, d. h. die Differenz aus der mit dem Spektrometer gemessenen Elektronenenergie und der durch die Beschleunigungsspannung zwischen Kathode und Anode be-stimmten ursprünglichen Energie, aufgetragen. Die Intensität hängt außer vom Energieverlust auch vom Strahlstrom und der Dispersion des Spektrometers ab. Die Pixelgröße der CCD-Kamera bestimmt ein Energiefenster, innerhalb dessen die Elektronen hinsichtlich ihrer Energie nicht unterschieden werden. Die Breite dieses Energiefensters sinkt mit wachsender Dispersion, d. h. die Zahl der pro Pixel registrierten Elektronen wird kleiner.
Bild 9-19. Elektronenenergieverlust-Spektrum von Nickeloxid. a) Komplettes Spektrum mit Kennzeichnung der drei wesentlichen Abschnitte. b) Core-Loss-Abschnitt, separat aufgenommen mit höherer Intensität und längerer Messzeit als (a). c) Bereich der O-K- und der Ni-L-Kante von (b) nach weiterer Verstärkung und Untergrundabzug.
Bei hinreichend dünnen Proben rührt die weitaus größte Intensität (> 90%) von elastisch gestreuten Elektronen her (Bild 9-19a). Zur Erinnerung: Elastische Streu-ung erfolgt ohne Energieverlust. Im EEL-Spektrum sind englische Bezeichnungen üblich: Zero-Loss-Peak. Eigentlich sollte dies eine einzelne Linie sein. Dies wird zum einen durch die Unterschiede in der Energie der aus der Kathode austretenden Elektronen (Energiebreite – vgl. Abschnitt 2.5) und zum anderen durch das be-grenzte energetische Auflösungsvermögen des Spektrometers (Dispersion und Ka-mera-Pixelgröße) verhindert. Das Spektrometer wird so eingestellt, dass der Ener-gieverlust Null auf das Maximum des Zero-Loss-Peaks fällt. Die Intensität im negativen Teil der Abszisse kommt also nicht durch beschleunigte Elektronen zustande sondern durch die endliche Breite dieses Peaks.
An den Peak elastisch gestreuter Elektronen schließt sich der Low-Loss-Bereich an. Derartig geringe Energieverluste entstehen, wenn durch die Primärelektronen Übergänge zwischen eng benachbarten Energiezuständen, beispielsweise Über-gänge innerhalb des Leitungsbandes, oder Schwingungen des Elektronengases als Ganzes (Plasmonen) bzw. des Verbandes der Festkörperatome (Phononen mit Energien unter 0,1 eV) angeregt werden. Phononen spielen in der EELS-Praxis keine Rolle.
Größere Energieverluste entstehen bei Ionisation innerer Schalen (Core-Loss-Bereich). Die Intensität wird so gering, dass in der Regel zur Messung in diesem
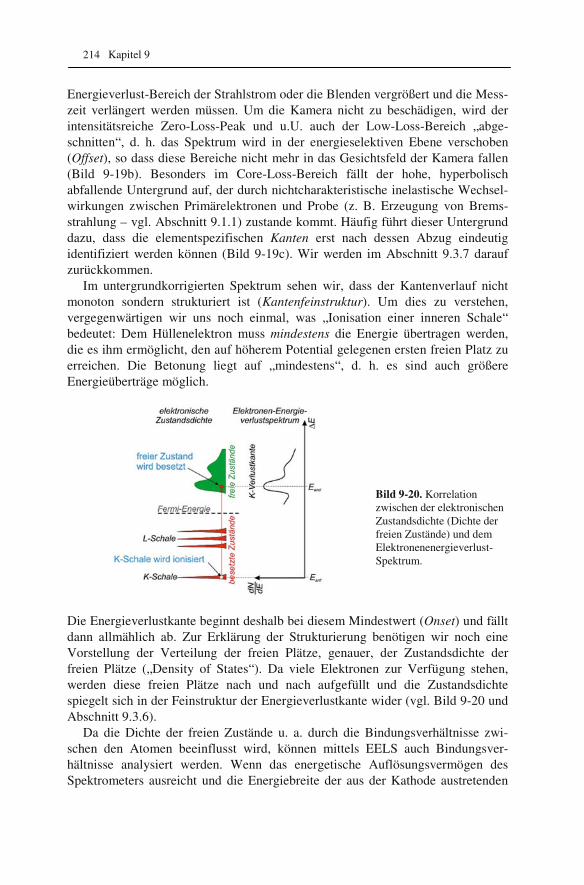
Kapitel 9 214
Energieverlust-Bereich der Strahlstrom oder die Blenden vergrößert und die Mess-zeit verlängert werden müssen. Um die Kamera nicht zu beschädigen, wird der intensitätsreiche Zero-Loss-Peak und u.U. auch der Low-Loss-Bereich „abge-schnitten“, d. h. das Spektrum wird in der energieselektiven Ebene verschoben (Offset), so dass diese Bereiche nicht mehr in das Gesichtsfeld der Kamera fallen (Bild 9-19b). Besonders im Core-Loss-Bereich fällt der hohe, hyperbolisch abfallende Untergrund auf, der durch nichtcharakteristische inelastische Wechsel-wirkungen zwischen Primärelektronen und Probe (z. B. Erzeugung von Brems-strahlung – vgl. Abschnitt 9.1.1) zustande kommt. Häufig führt dieser Untergrund dazu, dass die elementspezifischen Kanten erst nach dessen Abzug eindeutig identifiziert werden können (Bild 9-19c). Wir werden im Abschnitt 9.3.7 darauf zurückkommen.
Im untergrundkorrigierten Spektrum sehen wir, dass der Kantenverlauf nicht monoton sondern strukturiert ist (Kantenfeinstruktur). Um dies zu verstehen, vergegenwärtigen wir uns noch einmal, was „Ionisation einer inneren Schale“ bedeutet: Dem Hüllenelektron muss mindestens die Energie übertragen werden, die es ihm ermöglicht, den auf höherem Potential gelegenen ersten freien Platz zu erreichen. Die Betonung liegt auf „mindestens“, d. h. es sind auch größere Energieüberträge möglich.
Bild 9-20. Korrelation zwischen der elektronischen Zustandsdichte (Dichte der freien Zustände) und dem Elektronenenergieverlust-Spektrum.
Die Energieverlustkante beginnt deshalb bei diesem Mindestwert (Onset) und fällt dann allmählich ab. Zur Erklärung der Strukturierung benötigen wir noch eine Vorstellung der Verteilung der freien Plätze, genauer, der Zustandsdichte der freien Plätze („Density of States“). Da viele Elektronen zur Verfügung stehen, werden diese freien Plätze nach und nach aufgefüllt und die Zustandsdichte spiegelt sich in der Feinstruktur der Energieverlustkante wider (vgl. Bild 9-20 und Abschnitt 9.3.6).
Da die Dichte der freien Zustände u. a. durch die Bindungsverhältnisse zwi-schen den Atomen beeinflusst wird, können mittels EELS auch Bindungsver-hältnisse analysiert werden. Wenn das energetische Auflösungsvermögen des Spektrometers ausreicht und die Energiebreite der aus der Kathode austretenden
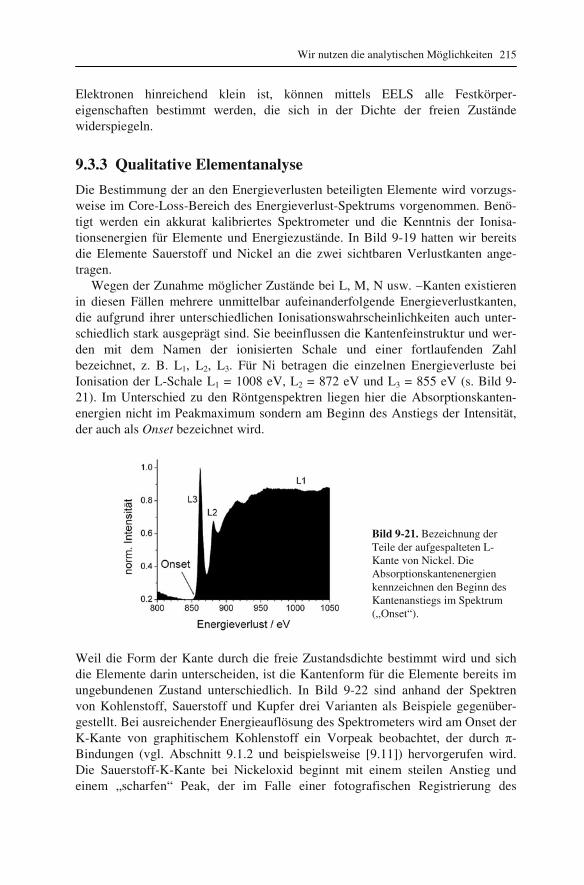
Wir nutzen die analytischen Möglichkeiten 215
Elektronen hinreichend klein ist, können mittels EELS alle Festkörper-eigenschaften bestimmt werden, die sich in der Dichte der freien Zustände widerspiegeln.
9.3.3 Qualitative Elementanalyse
Die Bestimmung der an den Energieverlusten beteiligten Elemente wird vorzugs-weise im Core-Loss-Bereich des Energieverlust-Spektrums vorgenommen. Benö-tigt werden ein akkurat kalibriertes Spektrometer und die Kenntnis der Ionisa-tionsenergien für Elemente und Energiezustände. In Bild 9-19 hatten wir bereits die Elemente Sauerstoff und Nickel an die zwei sichtbaren Verlustkanten ange-tragen.
Wegen der Zunahme möglicher Zustände bei L, M, N usw. –Kanten existieren in diesen Fällen mehrere unmittelbar aufeinanderfolgende Energieverlustkanten, die aufgrund ihrer unterschiedlichen Ionisationswahrscheinlichkeiten auch unter-schiedlich stark ausgeprägt sind. Sie beeinflussen die Kantenfeinstruktur und wer-den mit dem Namen der ionisierten Schale und einer fortlaufenden Zahl bezeichnet, z. B. L1, L2, L3. Für Ni betragen die einzelnen Energieverluste bei Ionisation der L-Schale L1 = 1008 eV, L2 = 872 eV und L3 = 855 eV (s. Bild 9-21). Im Unterschied zu den Röntgenspektren liegen hier die Absorptionskanten-energien nicht im Peakmaximum sondern am Beginn des Anstiegs der Intensität, der auch als Onset bezeichnet wird.
Bild 9-21. Bezeichnung der Teile der aufgespalteten L-Kante von Nickel. Die Absorptionskantenenergien kennzeichnen den Beginn des Kantenanstiegs im Spektrum („Onset“).
Weil die Form der Kante durch die freie Zustandsdichte bestimmt wird und sich die Elemente darin unterscheiden, ist die Kantenform für die Elemente bereits im ungebundenen Zustand unterschiedlich. In Bild 9-22 sind anhand der Spektren von Kohlenstoff, Sauerstoff und Kupfer drei Varianten als Beispiele gegenüber-gestellt. Bei ausreichender Energieauflösung des Spektrometers wird am Onset der K-Kante von graphitischem Kohlenstoff ein Vorpeak beobachtet, der durch -Bindungen (vgl. Abschnitt 9.1.2 und beispielsweise [9.11]) hervorgerufen wird. Die Sauerstoff-K-Kante bei Nickeloxid beginnt mit einem steilen Anstieg und einem „scharfen“ Peak, der im Falle einer fotografischen Registrierung des
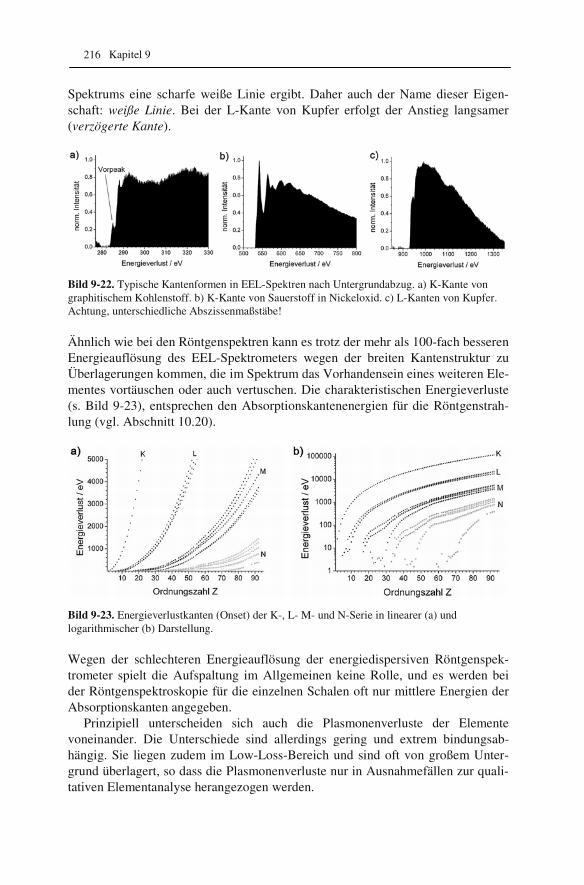
Kapitel 9 216
Spektrums eine scharfe weiße Linie ergibt. Daher auch der Name dieser Eigen-schaft: weiße Linie. Bei der L-Kante von Kupfer erfolgt der Anstieg langsamer (verzögerte Kante).
Bild 9-22. Typische Kantenformen in EEL-Spektren nach Untergrundabzug. a) K-Kante von graphitischem Kohlenstoff. b) K-Kante von Sauerstoff in Nickeloxid. c) L-Kanten von Kupfer. Achtung, unterschiedliche Abszissenmaßstäbe!
Ähnlich wie bei den Röntgenspektren kann es trotz der mehr als 100-fach besseren Energieauflösung des EEL-Spektrometers wegen der breiten Kantenstruktur zu Überlagerungen kommen, die im Spektrum das Vorhandensein eines weiteren Ele-mentes vortäuschen oder auch vertuschen. Die charakteristischen Energieverluste (s. Bild 9-23), entsprechen den Absorptionskantenenergien für die Röntgenstrah-lung (vgl. Abschnitt 10.20).
Bild 9-23. Energieverlustkanten (Onset) der K-, L- M- und N-Serie in linearer (a) und logarithmischer (b) Darstellung.
Wegen der schlechteren Energieauflösung der energiedispersiven Röntgenspek-trometer spielt die Aufspaltung im Allgemeinen keine Rolle, und es werden bei der Röntgenspektroskopie für die einzelnen Schalen oft nur mittlere Energien der Absorptionskanten angegeben.
Prinzipiell unterscheiden sich auch die Plasmonenverluste der Elemente voneinander. Die Unterschiede sind allerdings gering und extrem bindungsab-hängig. Sie liegen zudem im Low-Loss-Bereich und sind oft von großem Unter-grund überlagert, so dass die Plasmonenverluste nur in Ausnahmefällen zur quali-tativen Elementanalyse herangezogen werden.

Wir nutzen die analytischen Möglichkeiten 217
9.3.4 Untergrund und Vielfachstreuung: Anforderungen an die Probe
In Bild 9-19b hatten wir gesehen, dass die elementspezifischen Kanten nur wenig aus dem hyperbolisch abfallenden Untergrund herausragen und oft erst nach Ab-zug dieses Untergrundes genauer zu identifizieren sind. Wir wollen nun überlegen, woher dieser Untergrund kommt, wie wir ihn mathematisch modellieren können und welche Möglichkeiten es zu seiner Reduzierung gibt.
Ursachen für den Untergrund sind nichtcharakteristische Energieverluste der Primärelektronen: Die Erzeugung von Bremsstrahlung, Elektronen, die vollkom-men aus den Atomhüllen entfernt und irgendeine kinetische Energie ins Vakuum mitnehmen (z. B. Sekundärelektronen), Übergänge zwischen Energiezuständen im Valenz- und Leitungsband (Interbandübergänge) sind einige der Möglichkeiten. Schließlich kann es passieren, dass ein Primärelektron zunächst einen charak-teristischen Energieverlust erleidet, anschließend aber noch vor Verlassen der Probe einen der aufgezählten nichtcharakteristischen Verluste erfährt und damit wiederum zum Untergrund beiträgt. Für ein hohes Nettokantensignal ist offensichtlich eine Optimierung der Probendicke möglich: Die Wahrscheinlichkeit für einen charakteristischen Energieverlust steigt mit wachsender Probendicke. Andererseits steigt damit auch die Wahrscheinlichkeit für einen anschließenden nichtcharakteristischen Energieverlust, so dass dieses Elektron nicht zur Netto-kantenintensität beiträgt. Die Wahrscheinlichkeit für diesen zweiten Energiever-lust ist durch die Form des Spektrums selbst gegeben. Besonders hohe Intensitäten (und damit Wahrscheinlichkeiten) treten im Zero-Loss- und im Low-Loss-Bereich auf (vgl. Bild 9-19). Mathematisch gesehen ist jede Energieverlust-Kante mit dem Zero-Loss- und Low-Loss-Bereich des Spektrums gefaltet (→ Abschnitt 10.24). Die Entfernung der Vielfachstreu-Anteile aus dem Spektrum erfolgt deshalb durch Entfaltung des Spektrums. Natürlich müssen dazu auch der Zero-Loss- und Low-Loss-Bereich gemessen worden sein.
Um den Untergrund quantitativ zu erfassen, müssen wir einen Formalismus für den Untergrundverlauf u(ΔE) finden. Wir verzichten auf den Versuch, dies exakt physikalisch zu begründen, sondern beschränken uns auf eine mathematische An-passung. Das funktioniert zumindest in kleineren Energieverlustintervallen von (200 ... 300) eV recht gut mit der Funktion
( ) 11
aE
u E uE
−Δ
Δ = ⋅Δ
(9.42)
(u1: Untergrundhöhe bei Energieverlust ΔE1, a: Anpassungsparameter). In Bild 9-24 ist dies am Beispiel einer Ni-Mn-Schicht demonstriert. Zur Bestimmung von a benötigen wir zwei Stützstellen u1(ΔE1) und u2(ΔE2). Da die Messkurven ver-rauscht sind, werden für diese Stützstellen schmale Fenster benutzt und die Unter-

Kapitel 9 218
grundhöhen u1 und u2 über diese Fenster gemittelt (vgl. Bild 9-24a). Für a erhalten wir aus (9.42) mit den beiden Stützstellen:
1 2
2 1
ln lnu E
au E
Δ=
Δ (9.43)
Wir sehen, dass damit der Untergrund für die jeweilige Energieverlust-Kante gut modelliert wird. Wir erkennen aber auch, dass sich Messkurve und modellierter Untergrund innerhalb des Energieverlustintervalls bis 1200 eV nicht angleichen. Bei der Bestimmung der Nettokantenintensität (s. Bilder 9-24b und c)) spielt dem-zufolge die Breite des Energieverlustfensters, innerhalb dessen die Kanteninten-sität ermittelt wird, eine Rolle.
Bild 9-24. Untergrundapproximation und –subtraktion im EEL-Spektrum von einer Mn-Ni-Schicht. a) Originalspektrum mit eingezeichneten Untergrund für die Mn-L- und die Ni-L-Kante. b) Profil der Mn-L-Kante nach Untergrundabzug. c) Profil der Ni-L-Kante nach Untergrund-abzug. Wir kommen nun auf die Optimierung der Probendicke zurück und wollen dies am Beispiel eines Nickeloxid-Kristalls mit variierender Dicke demonstrieren (vgl. Bild 9-25).
Im STEM-HAADF-Bild (vgl. Abschnitt 8.4) sehen wir im Wesentlichen einen Massendickekontrast, d. h. bei konstanter chemischer Zusammensetzung einen Dickekontrast: Je dicker die Probenstelle desto heller das Bild davon. Entlang der in Bild 9-25a eingezeichneten weißen Linie wurden 18 EDX- und EEL-Spektren aufgenommen, außerdem die Helligkeit im STEM-HAADF-Detektor registriert.
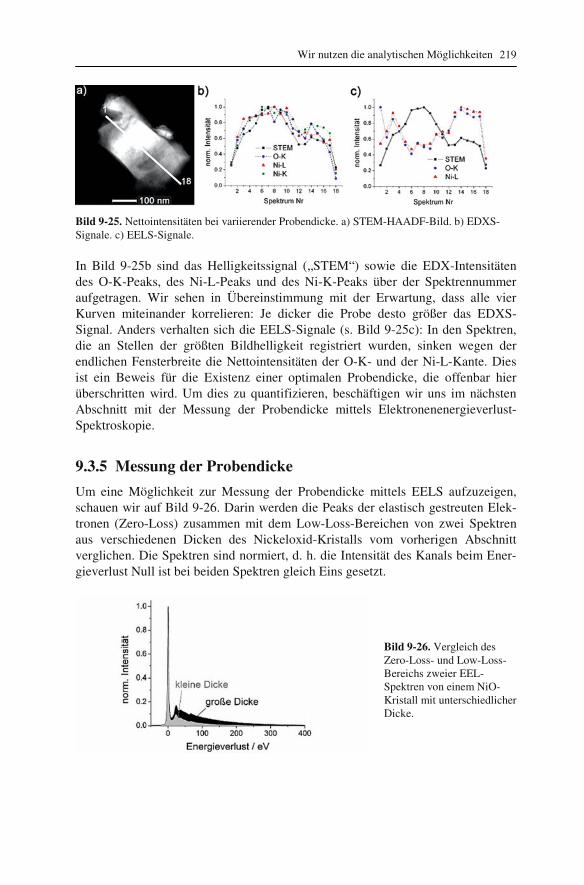
Wir nutzen die analytischen Möglichkeiten 219
Bild 9-25. Nettointensitäten bei variierender Probendicke. a) STEM-HAADF-Bild. b) EDXS-Signale. c) EELS-Signale.
In Bild 9-25b sind das Helligkeitssignal („STEM“) sowie die EDX-Intensitäten des O-K-Peaks, des Ni-L-Peaks und des Ni-K-Peaks über der Spektrennummer aufgetragen. Wir sehen in Übereinstimmung mit der Erwartung, dass alle vier Kurven miteinander korrelieren: Je dicker die Probe desto größer das EDXS-Signal. Anders verhalten sich die EELS-Signale (s. Bild 9-25c): In den Spektren, die an Stellen der größten Bildhelligkeit registriert wurden, sinken wegen der endlichen Fensterbreite die Nettointensitäten der O-K- und der Ni-L-Kante. Dies ist ein Beweis für die Existenz einer optimalen Probendicke, die offenbar hier überschritten wird. Um dies zu quantifizieren, beschäftigen wir uns im nächsten Abschnitt mit der Messung der Probendicke mittels Elektronenenergieverlust-Spektroskopie.
9.3.5 Messung der Probendicke
Um eine Möglichkeit zur Messung der Probendicke mittels EELS aufzuzeigen, schauen wir auf Bild 9-26. Darin werden die Peaks der elastisch gestreuten Elek-tronen (Zero-Loss) zusammen mit dem Low-Loss-Bereichen von zwei Spektren aus verschiedenen Dicken des Nickeloxid-Kristalls vom vorherigen Abschnitt verglichen. Die Spektren sind normiert, d. h. die Intensität des Kanals beim Ener-gieverlust Null ist bei beiden Spektren gleich Eins gesetzt.
Bild 9-26. Vergleich des Zero-Loss- und Low-Loss-Bereichs zweier EEL-Spektren von einem NiO-Kristall mit unterschiedlicher Dicke.

Kapitel 9 220
Wir sehen, dass im Low-Loss-Bereich der Untergrund im Vergleich zum elastischen Zero-Loss-Peak bei größerer Probendicke größer ist als bei geringerer Dicke. Das ist zu erwarten, steigt doch bei zunehmender Probendicke die Wahrscheinlichkeit für inelastische Wechselwirkungen an.
Dieser Effekt wird zur Messung der Probendicke ausgenutzt. Benötigt wird ein EEL-Spektrum wie in Bild 9-26, d. h. ein Spektrum, das den Peak der elastisch gestreuten Elektronen enthält (s. Bild 9-27). Die Anteile der elastisch und der inelastisch gestreuten Elektronen werden mathematisch voneinander getrennt (Flächen unter der Spektrenkurve), wie dies in Bild 9-27 angedeutet ist.
Bild 9-27. Unterscheidung der Intensität Iel im elasti-schen Peak von der Intensität Iinel der inelastisch gestreuten Elektronen.
Ähnlich wie bei der elastischen Streuung (vgl. Abschnitt 6.1) definieren wir eine mittlere freie Weglänge für die inelastische Streuung Λinel, innerhalb derer der Anteil der elastisch gestreuten Elektronen an der Gesamtintensität I0 auf 1/e (e = 2,71828..., natürliche Zahl) abgesunken ist:
( )0 e einel inelt tel el inelI I I I− Λ − Λ= ⋅ = + ⋅ (9.44)
bzw.
ln el inel
inel el
I It
I
+=
Λ (9.45)
(t: Probendicke). Damit erhalten wir die Probendicke als Vielfaches der mittleren freien Weglänge für die elastische Streuung. Für diese Weglänge existieren Näherungsformeln, beispielsweise gilt nach R.F. Egerton [9.12]:
0
0
106 keV
nm keV2ln
mrad
inel
mm
F E
EE
Eβ
Λ ⋅ ⋅=
⋅⋅ ⋅
(9.46)

Wir nutzen die analytischen Möglichkeiten 221
(β: Akzeptanzwinkel, d. h. maximaler Streuwinkel der inelastisch gestreuten Elek-tronen, die vom Spektrometer erfasst werden) mit dem relativistischen Korrektur-faktor
( )0
20
1+ E 1022 keV
1+ E 511 keVF = (9.47)
und der ordnungszahlabhängigen Größe Em
0,367,6 ZmE ≈ ⋅ (9.48)
mit Z als gewichteter Ordungszahl der beteiligten Elemente. Andere Autoren [9.13] schlagen anstelle der Ordnungszahlabhängigkeit eine (plausiblere) Abhän-gigkeit von der Materialdichte ρ vor. Danach lautet der (etwas vereinfachte) For-malismus:
( )( )
02
0,33 00,33
18, 2 keV
nmkeV mrad1
g cm ln2 7,8 g cm
F E
F E
λ
βρ
ρ
−
−
⋅ ⋅≈
⋅ ⋅⋅ ⋅ +
⋅ ⋅
(9.49)
Aus Bild 9-28 erkennen wir die Größenordnung der mittleren freien Weglänge für die inelastische Streuung: Für die im Transmissionselektronenmikroskop üblichen Parameter und Probenmaterialien liegt sie zwischen 50 und 300 nm.
Bild 9-28. Mittlere freie Weglänge für die inelastische Streuung in Abhängigkeit von der Ord-nungszahl für drei Primärenergien und einen Akzeptanzwinkel von 10 mrad. a) Nach Formel (9.48). b) Nach Formel (9.49).
Mit Formel (9.44) können wir die Spektren von Bild 9-25 quantifizieren und so eine optimale Dicke für EELS-Untersuchungen angeben (s. Bild 9-29).
Aus Bild 9-25 ist ersichtlich, dass die maximale Nettokantenintensität in den Spektren 14 bis 19 erreicht wird und deren Einbruch bei den Spektren 4 bis 10 auftritt.

Kapitel 9 222
Bild 9-29. Verhältnis der Probendicke t zur mittleren freien Weglänge Λinel für die inelastische Streuung in den EEL-Spektren von NiO (vgl. Bild 9-25). Zum Vergleich ist das (auf 1 normierte) STEM-Signal aus Bild 9-25 zusätzlich eingezeichnet.
Aus Bild 9-29 lesen wir für die Spektren 4 bis 10 ein Verhältnis t/Λinel > 0,9 und für die Spektren 14 bis 18 ein t/Λinel zwischen 0,2 und 0,8 ab. Die Praxis lehrt, dass eine Probendicke t zwischen (0,3 und 0,4)⋅ Λinel für EELS optimal ist.
9.3.6 Kantenfeinstruktur: Bindungsanalyse
Aus Abschnitt 9.3.2 wissen wir, dass sich in der Kantenfeinstruktur die Dichte der freien Zustände und damit die Art der Bindung zwischen den Atomen abbildet. Allerdings betrifft dies nur den Teil der Kantenfeinstruktur, der in unmittelbarer Nähe (d. h. in einer energetischen Entfernung von bis zu (40 ...50) eV) vom Onset der Ionisationskante auftritt. Die englische Bezeichnung ist „Energy Loss Near Edge Fine Structure“, woraus die Abkürzung ELNES für die Bindungsanalyse anhand von EEL-Spektren abgeleitet ist.
Bild 9-30. Bereiche und Unterscheidungsmerkmale der EELS-Kantenfein-struktur.
Eine weitere Kantenfeinstruktur tritt in einer energetischen Entfernung bis zu mehr als 100 eV auf („Extended Energy Loss Fine Structure“ – EXELFS). Dafür sind Interferenzen der am Atomverband gestreuten Elektronenwellen verant-wortlich (vgl. Bild 9-30).
Wir wollen uns zuerst mit der kantennahen Feinstruktur beschäftigen. Zur Be-rechnung der Bandstruktur und damit der Zustandsdichte ist die Lösung der Schrödinger-Gleichung im Festkörper unter Berücksichtigung der von Atomker-
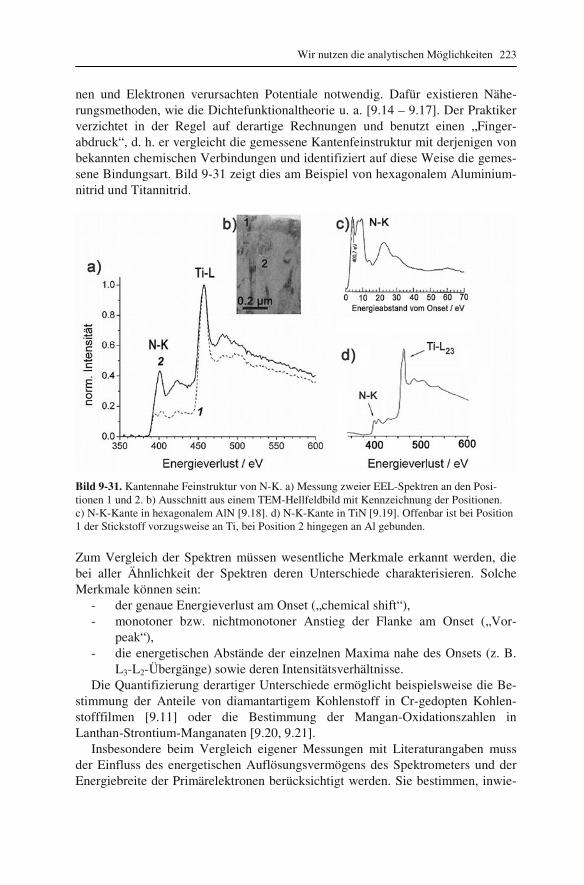
Wir nutzen die analytischen Möglichkeiten 223
nen und Elektronen verursachten Potentiale notwendig. Dafür existieren Nähe-rungsmethoden, wie die Dichtefunktionaltheorie u. a. [9.14 – 9.17]. Der Praktiker verzichtet in der Regel auf derartige Rechnungen und benutzt einen „Finger-abdruck“, d. h. er vergleicht die gemessene Kantenfeinstruktur mit derjenigen von bekannten chemischen Verbindungen und identifiziert auf diese Weise die gemes-sene Bindungsart. Bild 9-31 zeigt dies am Beispiel von hexagonalem Aluminium-nitrid und Titannitrid.
Bild 9-31. Kantennahe Feinstruktur von N-K. a) Messung zweier EEL-Spektren an den Posi-tionen 1 und 2. b) Ausschnitt aus einem TEM-Hellfeldbild mit Kennzeichnung der Positionen. c) N-K-Kante in hexagonalem AlN [9.18]. d) N-K-Kante in TiN [9.19]. Offenbar ist bei Position 1 der Stickstoff vorzugsweise an Ti, bei Position 2 hingegen an Al gebunden.
Zum Vergleich der Spektren müssen wesentliche Merkmale erkannt werden, die bei aller Ähnlichkeit der Spektren deren Unterschiede charakterisieren. Solche Merkmale können sein:
- der genaue Energieverlust am Onset („chemical shift“), - monotoner bzw. nichtmonotoner Anstieg der Flanke am Onset („Vor-
peak“), - die energetischen Abstände der einzelnen Maxima nahe des Onsets (z. B.
L3-L2-Übergänge) sowie deren Intensitätsverhältnisse. Die Quantifizierung derartiger Unterschiede ermöglicht beispielsweise die Be-
stimmung der Anteile von diamantartigem Kohlenstoff in Cr-gedopten Kohlen-stofffilmen [9.11] oder die Bestimmung der Mangan-Oxidationszahlen in Lanthan-Strontium-Manganaten [9.20, 9.21].
Insbesondere beim Vergleich eigener Messungen mit Literaturangaben muss der Einfluss des energetischen Auflösungsvermögens des Spektrometers und der Energiebreite der Primärelektronen berücksichtigt werden. Sie bestimmen, inwie-
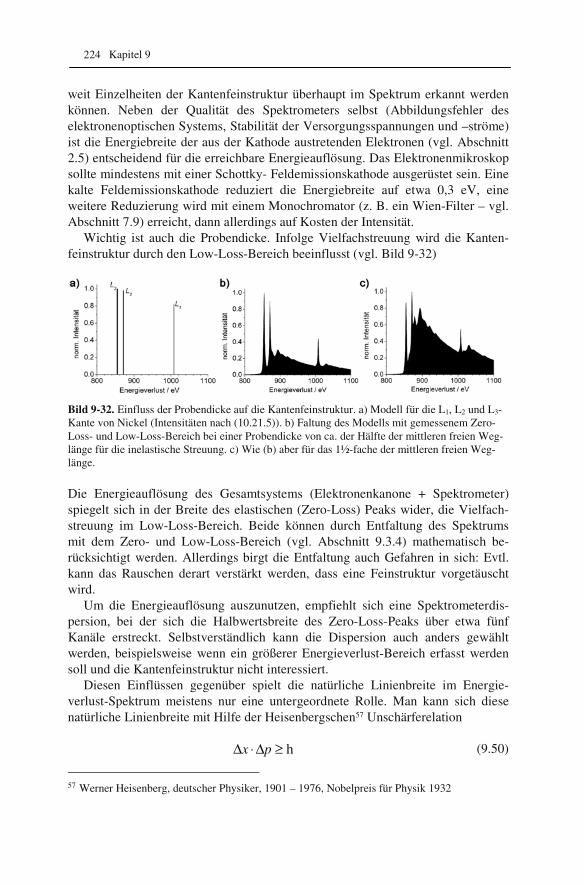
Kapitel 9 224
weit Einzelheiten der Kantenfeinstruktur überhaupt im Spektrum erkannt werden können. Neben der Qualität des Spektrometers selbst (Abbildungsfehler des elektronenoptischen Systems, Stabilität der Versorgungsspannungen und –ströme) ist die Energiebreite der aus der Kathode austretenden Elektronen (vgl. Abschnitt 2.5) entscheidend für die erreichbare Energieauflösung. Das Elektronenmikroskop sollte mindestens mit einer Schottky- Feldemissionskathode ausgerüstet sein. Eine kalte Feldemissionskathode reduziert die Energiebreite auf etwa 0,3 eV, eine weitere Reduzierung wird mit einem Monochromator (z. B. ein Wien-Filter – vgl. Abschnitt 7.9) erreicht, dann allerdings auf Kosten der Intensität.
Wichtig ist auch die Probendicke. Infolge Vielfachstreuung wird die Kanten-feinstruktur durch den Low-Loss-Bereich beeinflusst (vgl. Bild 9-32)
Bild 9-32. Einfluss der Probendicke auf die Kantenfeinstruktur. a) Modell für die L1, L2 und L3-Kante von Nickel (Intensitäten nach (10.21.5)). b) Faltung des Modells mit gemessenem Zero-Loss- und Low-Loss-Bereich bei einer Probendicke von ca. der Hälfte der mittleren freien Weg-länge für die inelastische Streuung. c) Wie (b) aber für das 1½-fache der mittleren freien Weg-länge.
Die Energieauflösung des Gesamtsystems (Elektronenkanone + Spektrometer) spiegelt sich in der Breite des elastischen (Zero-Loss) Peaks wider, die Vielfach-streuung im Low-Loss-Bereich. Beide können durch Entfaltung des Spektrums mit dem Zero- und Low-Loss-Bereich (vgl. Abschnitt 9.3.4) mathematisch be-rücksichtigt werden. Allerdings birgt die Entfaltung auch Gefahren in sich: Evtl. kann das Rauschen derart verstärkt werden, dass eine Feinstruktur vorgetäuscht wird.
Um die Energieauflösung auszunutzen, empfiehlt sich eine Spektrometerdis-persion, bei der sich die Halbwertsbreite des Zero-Loss-Peaks über etwa fünf Kanäle erstreckt. Selbstverständlich kann die Dispersion auch anders gewählt werden, beispielsweise wenn ein größerer Energieverlust-Bereich erfasst werden soll und die Kantenfeinstruktur nicht interessiert.
Diesen Einflüssen gegenüber spielt die natürliche Linienbreite im Energie-verlust-Spektrum meistens nur eine untergeordnete Rolle. Man kann sich diese natürliche Linienbreite mit Hilfe der Heisenbergschen57 Unschärferelation
hx pΔ ⋅Δ ≥ (9.50)
57 Werner Heisenberg, deutscher Physiker, 1901 – 1976, Nobelpreis für Physik 1932

Wir nutzen die analytischen Möglichkeiten 225
(Δx: Ortsunsicherheit, Δp: Impulsunsicherheit, h: Plancksches Wirkungsquantum) veranschaulichen. Nach Ersetzen der Differenzen durch Differentiale erhält man
hdx dp⋅ ≥ . (9.51)
In nichtrelativistischer Näherung können wir dafür schreiben:
m hv dt dv⋅ ⋅ ⋅ ≥ (9.52)
(v: Geschwindigkeit, m: Masse). Mit v⋅dv = d(v2/2) folgt daraus
2mh
2d v dt dE dt⋅ = ⋅ ≥ , (9.53)
d. h. die Linienbreite dE wird durch die Lebensdauer dt der am Elektronenüber-gang beteiligten Zustände bestimmt. Die Größenordnung dieser Lebensdauer für die K-Serie wird beispielsweise von G. Wentzel [10.15] mit 1/(109
⋅Z4) s (Z: Ord-nungszahl) abgeschätzt. Damit folgen aus (9.53) Linienbreiten zwischen 0,003 eV und 3 eV für Elemente mit Ordnungszahlen zwischen 5 (Bor) und 30 (Zink)
Die kantenferne Feinstruktur entsteht durch den Einfluss der nächsten Nach-baratome auf den inelastischen Streuprozess. Die Welle des bei der Ionisierung einer inneren Schale entfernten Elektrons wird an den Nachbaratomen gestreut (reflektiert) und interferiert mit dieser gestreuten Welle. Die Wellenlänge hängt von der durch das Primärelektron beim Ionisationsprozess übertragenen Energie, d. h. von dessen Energieverlust ab. Der Abstand der nächsten Nachbaratome wiederum bestimmt den Gangunterschied der miteinander interferierenden Wellen und damit diejenigen Energieverluste, bei denen Verstärkung und Abschwächung auftreten. Ähnlich wie bei der Analyse von Beugungsbildern amorpher Materia-lien lassen sich aus der kantenfernen Feinstruktur nächste Nachbarabstände be-stimmen. Da die Modulationen hinter einer elementspezifischen Ionisationskante auftreten, steht in diesem Fall fest, welchem Element das Bezugsatom angehört. Allerdings sind die Modulationen oft nur schwach ausgeprägt, so dass das Signalrauschen zu falschen Schlüssen führen kann.
9.3.7. Quantifizierung von Energieverlust-Spektren
Prinzipiell läuft die Quantifizierung von EEL-Spektren wie diejenige von EDX-Spektren (vgl. Abschnitt 9.2.3) ab: Das Verhältnis zweier Elementkonzentrationen cA und cB ist proportional zum Verhältnis der Nettointensitäten IA und IB der beiden elementspezifischen Verlustkanten. Als Proportionalitätsfaktor ist hierbei das reziproke Verhältnis der beiden Ionisationsquerschnitte (Anregungswahr-scheinlichkeiten für die Verlustkanten A und B) QA und QB einzusetzen:

Kapitel 9 226
A B A
B A B
c Q I
c Q I= ⋅ (9.54)
Im Vergleich zu den EDX-Spektren gibt es allerdings vier gravierende Unter-schiede, die die Quantifizierung der EEL-Spektren erschweren: 1. Der Untergrund ist im EEL-Spektrum im Allgemeinen wesentlich höher als im EDX-Spektrum und variiert stark mit der Probendicke sowie durch die Nähe weiterer Verlustkanten, d. h. die Untergrundmodellierung hat wesentlich größere Bedeutung bei der Bestimmung der Nettokantenintensitäten. 2. Im Allgemeinen erreicht das Spektrum auch in größerem Abstand von der Kan-te nicht wieder das Untergrundniveau (vgl. Bild 9-24), so dass die Nettokanten-intensität auch durch die Breite des Energiefensters für die Messung beeinflusst wird. 3. Die Röntgenemission erfolgt unabhängig von der Röntgenenergie isotrop in den gesamten Raumwinkel. Geringe Abhängigkeiten von der Kristallorientierung [9.22] spielen bei der Quantifizierung keine Rolle. Bei den inelastisch gestreuten Elektronen hängen demgegenüber Streuwinkel θinel und Energieverlust ΔE von-einander ab. Nach den klassischen Stoßgesetzen (→ Abschnitt 10.19) besteht zwischen beiden der Zusammenhang
02inelE
Eθ
Δ=
⋅ (9.55)
(E0: Energie der Primärelektronen). Bei den Ionisationsquerschnitten in (9.54) muss demzufolge neben der Energie- auch die Winkelabhängigkeit berücksichtigt werden, da nur ein kleiner aber veränderbarer Winkelbereich erfasst wird:
( ), inelQ Q E θ= Δ . (9.56)
Die Wahrscheinlichkeit, mit der ein Elektron inelastisch mit dem Energieverlust ΔE in einen bestimmten Winkel gestreut wird, wird auch als Generalisierte Oszillatorstärke (GOS) bzw. als Bethe58-Oberfläche bezeichnet [9.23, 9.24].
Das Quantifizierungsergebnis hängt demzufolge auch vom Akzeptanzwinkel des Spektrometers ab, der durch die Geometrie, die Linseneinstellungen des Elek-tronenmikroskops (Abbildungs- bzw. Beugungsmodus) sowie Kontrast- bzw. Spektrometereintrittsblendendurchmesser bestimmt ist. In der Regel findet man in der Bedienungsanleitung des Mikroskops Hinweise zur Berechnung des Akzep-tanzwinkels. Größenordnungsmäßig beträgt er 5 mrad … 20 mrad, er sollte auf je-den Fall größer sein als θinel. Bei konvergenter Beleuchtung (z. B. STEM) sollte
58 Hans Bethe, deutsch/amerikanischer Physiker, 1906 – 2005, Nobelpreis für Physik 1967

Wir nutzen die analytischen Möglichkeiten 227
der Akzeptanzwinkel auch größer als der Konvergenzwinkel der Beleuchtung sein [9.25].
Für die Berechnung der Ionisationsquerschnitte (Lösung der Schrödinger-Gleichung) existieren unterschiedliche Modelle (vgl. z. B. [9.26]), die in der Quantifizierungssoftware enthalten sind. Wegen der Vielzahl an Parametern ist ein Vergleich mit Messungen an Standards bei der EELS-Quantifizierung besonders wichtig [9.27]. 4. Die Kantenprofile sind mit dem Low-Loss-Bereich des Spektrums gefaltet, d. h. eine Entfaltung ist trotz der damit verbundenen Probleme (vgl. Abschnitt 9.3.6) wünschenswert.
9.4. Energiegefilterte Abbildung Wie bei der energiedispersiven Röntgenspektroskopie beschrieben (vgl. Abschnitt 9.2.4) können auch mittels EELS Linescan-Profile und zweidimensionale Vertei-lungsbilder der Nettokantenintensitäten im STEM-Modus des Elektronenmikro-skops gemessen werden, indem an jedem Pixelhalt des fokussierten Elektronen-strahls ein EEL-Spektrum aufgezeichnet wird. Die Auswertung ist etwas aufwän-diger als bei EDXS, weil die Verlustspektren zur Extraktion der Nettokanten-intensitäten in jedem Fall untergrundkorrigiert werden müssen. Bei hinreichend deutlichen Unterschieden in der Kantenfeinstruktur ist auch ein bindungsspezi-fisches Verteilungsbild möglich.
Für die Elektronenenergieverlust-Spektroskopie gibt es jedoch noch eine andere Möglichkeit zur Aufnahme von Elementverteilungsbildern: Die energiege-filterte Abbildung (englisch: Energy Filtered Transmission Electron Microscopy – EFTEM). Voraussetzung dafür ist ein „abbildendes Energiefilter“, d. h. ein elektronenoptisches Transfersystem, welches ein reelles Zwischenbild in ein anderes überführt, gleichzeitig aber ein Elektronenprisma enthält. Prinzipiell kann dieses System in das Projektivlinsensystem integriert („In-Column-Filter“) oder der Endbildebene nachgeschaltet („Post-Column-Filter“) werden. Die wesentliche Eigenschaft dieses abbildenden Energiefilters ist die Erzeugung einer energie-selektiven Ebene, d. h. einer Ebene, in der sich alle Elektronen gleicher Energie an einer Stelle (bzw. in einer Linie) treffen (vgl. Abschnitt 9.3.1).
Wir erinnern uns: Derartige ausgezeichnete Ebenen hatten wir bereits kennen-gelernt (vgl. Abschnitt 2.7.2): Die bildseitige Brennebene des Objektivs ist eine winkelselektive Ebene. In ihr entsteht das Beugungsbild. Mit der in dieser Ebene gelegenen Kontrastblende können wir stark gestreute Elektronen aus dem Strah-lengang entfernen und somit für Streuabsorptions- und Beugungskontrast sorgen. Mit einer Blende in der ortsselektiven Zwischenbildebene wählen wir ein Ge-sichtsfeld und damit den Bereich der Probe aus, aus dem die Information im Beu-gungsbild stammt (Feinbereichsbeugung).
Die energieselektive Ebene ermöglicht es uns, mit einer Blende Elektronen einer bestimmten Energie, genauer eines bestimmten Energiebereichs (wegen der endlichen Blendengröße), auszuwählen. Nur diese Elektronen verbleiben im

Kapitel 9 228
Strahlengang und tragen zum Bild bei. Wegen der speziellen elektronenoptischen Eigenschaften des Elektronenprismas treffen sich Elektronen gleicher Energie nicht in einem Punkt sondern in einer Linie, als Blende genügt deshalb ein Schlitz.
Übertragen auf ein EEL-Spektrum wirkt dieser Schlitz wie ein Energiefenster. Setzt man dieses Energiefenster auf einen elementspezifischen Energieverlust, beispielsweise auf 532 eV (Sauerstoff – s. Bild 9-33), so werden alle sauerstoff-reichen Probenbereiche im Bild hell erscheinen.
Bild 9-33. Energiegefilterte Abbildung einer Ta-Mg-O-Schicht ohne Untergrundsubtraktion. a) TEM-Hellfeldbild (Zero-Loss-Peak mit 10 eV-Energiefenster). b) EEL-Spektrum von der Probe mit O-K-Kante und eingezeichnetem 25 eV-Energiefenster für Aufnahme von Bild c).
Dies ist in Bild 9-33c nicht der Fall. Wir wissen, dass im Hellfeldbild 9-33a die Tantal-Körner wegen der großen Ordnungszahl von Tantal (73) infolge des Streu-absorptionskontrastes dunkel erscheinen. Im Bild 9-33c, das „im Lichte“ der Elek-tronen mit einem Energieverlust zwischen 532 eV und 557 eV aufgenommen wor-den ist, erscheinen diese Körner heller als ihre Umgebung. Die naheliegende Ver-mutung, dass es sich um Tantaloxid handelt, ist falsch! Wir haben den Untergrund nicht berücksichtigt. Die stärker streuenden Ta-Körner erhöhen den Untergrund, so dass an ihren Positionen auch ein höheres Sauerstoff-Signal vorgetäuscht wird. Wir sehen an diesem Beispiel, wie wichtig die Untergrundsubtraktion bei EFTEM ist.
Im Wesentlichen werden zwei Methoden der Untergrundbehandlung ange-wendet: Die Zwei- und die Drei-Fenster Methode. Bei der Zwei-Fenster-Methode wird neben dem bereits in Bild 9-33 gesetzten Energiefenster auf (bzw. unmittel-bar nach) der Verlustkante ein zweites unmittelbar vor der Kante gesetzt und ein „Vorkantenbild“ aufgenommen. Die Helligkeitswerte im Vor- und im „Nach-kantenbild“ werden für jedes Bild durcheinander dividiert und in einem dritten Bild als Helligkeitswerte dargestellt (Jump-Ratio-Methode). Diese Helligkeiten sind damit ein Maß dafür, wie weit sich die Verlustkante aus dem unmittelbar vor ihr gemessenen Untergrund heraushebt.
Wenn die beiden Fenster im Zero- und Low-Loss-Bereich gesetzt werden, lässt sich auf diese Weise auch die Probendickeverteilung aufnehmen. Ein Nachteil dieser Methode ist, dass damit keine „echten“ Nettokantenintensitäten erfasst werden.

Wir nutzen die analytischen Möglichkeiten 229
Dieser Nachteil wird mit der Drei-Fenster-Methode vermieden (vgl. Bild 9-34). Bei dieser Methode werden drei Bilder mit verschiedenen Energieverlustfenstern aufgenommen: Zwei vor der Verlustkante und eins auf bzw. unmittelbar hinter der Kante. Aus den zwei Vorkantenbildern wird für jedes Pixel die Untergrundhellig-keit approximiert (vgl. Abschnitt 9.3.4, Formeln (9.41) und (9.42)) und dieser Wert vom gleichen Pixel im Nachkantenbild subtrahiert. Auf diese Weise wird das Verteilungsbild (hier für Sauerstoff, s. Bild 9-34f) berechnet. Wir erkennen, dass im Gegensatz zur Vermutung aus Bild 9-33c in den Tantalbereichen kein Sauerstoff auftritt. Die drei Bilder müssen exakt von der gleichen Probenstelle stammen, was wegen der Probendrift und Aufnahmezeiten von einigen Sekunden bis zu wenigen Minuten pro Bild durchaus nicht selbstverständlich ist. In der Regel wird deshalb vor der Untergrundberechnung und –subtraktion eine Driftkorrektur an den Einzelbildern vorgenommen.
Bild 9-34. Energiegefilterte Abbildung einer Ta-Mg-O-Schicht nach der Drei-Fenster-Methode. a) TEM-Hellfeldbild (Zero-Loss-Peak mit 10 eV-Energiefenster). b) EEL-Spektrum mit Kenn-zeichnung der Energiefenster für die drei Bilder. c) Vorkantenbild 1 (Energieverlustbereich 475 eV – 500 eV). d) Vorkantenbild 2 (Energieverlustbereich 503 eV – 528 eV). e) Nachkanten-bild (Energieverlustbereich 532 eV – 557 eV). f) Sauerstoff-Verteilungsbild, berechnet aus den Bildern c), d) und e).
Besonders kritisch wird es, wenn sich die Probe während der Aufnahme der Bilder verformt. Insbesondere bei abrupten Änderungen der Elementkonzentrationen (z. B. Schichtgrenzen) können infolge der damit verbundenen abrupten Änderung des Untergrundes Elementkonzentrationen an der Schichtgrenze vorgetäuscht werden. Dies ist umso kritischer, je kleiner die zu analysierenden Strukturen sind. Es empfiehlt sich, die Helligkeitswerte in den berechneten Bildern zu kontrollieren.

Kapitel 9 230
Eine Anhäufung von negativen Helligkeitswerten deutet auf Probleme bei der Untergrundmodellierung hin. In diesen Fällen ist es hilfreich, wenn nicht nur die berechneten Verteilungsbilder, sondern auch alle Einzelbilder gespeichert wurden. Man kann dann versuchen, durch erneute Driftkorrektur oder durch Mittelung über eine Zahl von Pixeln eine Verbesserung zu erreichen.
Der Praktiker versucht dieses Problem zu entschärfen, indem er die Auf-nahmezeiten möglichst kurz hält. Dies erfordert eine Strahlerzeugereinstellung, die einen vergleichsweise hohen Strahlstrom garantiert, und möglichst breite Ener-giefenster.
Zum Schluss wollen wir uns einigen elektronenoptischen Gesichtspunkten bei der energiegefilterten Abbildung widmen. Wenn wir mit dem Schlitz in der energieselektiven Ebene Elektronen auswählen, die einen Energieverlust ΔE erlitten haben, benutzen wir diese Elektronen auch zur Abbildung. Fokussiert wird im „normalen“ TEM-Bild d. h. im Wesentlichen mit elastisch gestreuten Elektro-nen (ΔE = 0). Beim Umschalten auf EFTEM verändert sich der Fokus infolge der veränderten Elektronenenergie. Das Problem wird reduziert, indem nicht der Schlitz in der energieselektiven Ebene verschoben, sondern die Beschleunigungs-spannung um ΔUB = ΔE/e (e: Elementarladung) erhöht wird. Nach Energieverlust in der Probe haben dann die ausgewählten Elektronen wieder die Energie, für die Linsen und Justageelemente eingestellt sind. Dies gilt allerdings nicht für das Kondensorsystem. Es befindet sich vor der Probe und arbeitet im Wesentlichen mit Elektronen der höheren Energie, d. h. Brennweiten und Justage des Konden-sorsystems müssen bei Veränderung der Energiefenster geeignet nachgestellt wer-den.
Das Ortsauflösungsvermögen wird durch die gleichen Einflussfaktoren wie die „normale“ TEM-Abbildung beeinflusst. Die dabei benutzten elastisch gestreuten Elektronen haben eine Energiebreite von etwa 1 eV (Schottky-Feldemissions-kathode). Bei EFTEM benutzen wir Energiefensterbreiten von mehr als 10 eV, so dass der Farbfehler wesentlichen Einfluss gewinnt. Der Radius δC des Farbfehler-scheibchens hängt von der Energiebreite δE ab (vgl. Abschnitt 2.3):
0C C
EC
E
δδ α= ⋅ ⋅ (9.57)
(α: Objektivapertur, CC: Farbfehlerkonstante, E0: Primärelektronenenergie). Wenn wir diesen Radius als Ortsauflösungsvermögen benutzen, erhalten wir beispiels-weise bei einer Apertur α = 10 mrad, einer Farbfehlerkonstante CC = 1,5 mm und einer Primärelektronenenergie von E0 = 200 keV den Zusammenhang
nm =0,075 eVC Eδ δ⋅ , (9.58)
d. h. bei einer Energiefensterbreite von 20 eV eine Ortsauflösung von 1,5 nm. Eine Verbesserung ist durch Verkleinerung des Energiefensters möglich, aller-

Wir nutzen die analytischen Möglichkeiten 231
dings sinkt dann auch die Intensität. Um signifikant über dem Rauschuntergrund zu bleiben, muss die Messzeit verlängert werden, verbunden mit den o.g. Pro-blemen.
In der Praxis erfordert EFTEM einen Kompromiss, der durch die Struktur-größe, die Probendicke, die Form der Verlustkante, die energetische Lage der Verlustkante und benachbarter Kanten sowie den Richtstrahlwert bestimmt wird.
9.5 Vergleich zwischen EDXS und EELS Für den Praktiker läuft dieser Vergleich auf die Frage hinaus, die er sich zu Be-ginn analytischer Messungen im Transmissionselektronenmikroskop stellt: „Wo-mit fange ich an?“
Zunächst spricht viel für die energiedispersive Röntgenspektroskopie: Die Spektren sind übersichtlich mit geringem Untergrund. Die Probendicke hat den erwarteten Einfluss: Je dicker die Probe, umso höher das Signal. Die Quantifi-zierung der Spektren im Rahmen der erläuterten Fehlergrenzen ist mit der zu den Spektrometern gelieferten Software „auf Knopfdruck“ möglich.
Demgegenüber erfordert die Elektronenenergieverlust-Spektroskopie mehr Hintergrundwissen: Der Untergrund ist hoch, mitunter können die elementspe-zifischen Kanten erst nach Untergrundabzug eindeutig identifiziert werden. Der Untergrund wird durch die Probendicke stark beeinflusst, die Nettokanten-intensität wächst zunächst mit steigender Probendicke, sinkt dann aber wieder. Die Proben müssen in einer geeigneten Dicke präpariert werden. Die Form der Verlustkanten ist unterschiedlich (weiße Linien, verzögerte Kanten), damit sind die Kanten unterschiedlich deutlich über dem Untergrund erkennbar. Eine erwar-tete, aber nicht sichtbare Verlustkante bedeutet nicht, dass das betreffende Ele-ment auch nicht in der Probe enthalten ist. Die Quantifizierung der Spektren er-fordert die Eingabe von Parametern, wie Akzeptanzwinkel des Spektrometers oder Energiefensterbreite zur Bestimmung des Untergrundes und der Nettokanteninten-sität, die den unerfahrenen Experimentator durchaus abschrecken kann.
Es gibt allerdings Fälle, wo EDXS nicht weiterhilft. Das energetische Auflö-sungsvermögen der energiedispersiven Röntgenspektrometer liegt bei etwa 130 eV. Peaküberlagerungen sind nicht selten, beispielsweise Titan und Sauerstoff sind deshalb im Röntgenspektrum kaum zu unterscheiden. Energieverlust-Spek-trometer haben demgegenüber ein Energieauflösungsvermögen von 1 eV oder besser. Titan und Sauerstoff können getrennt nachgewiesen und Bindungsver-hältnisse können analysiert werden, d. h. wir können feststellen, ob beide als Titanoxid vorliegen.
In der Regel liegt die Achse des Röntgenspektrometers wenige Millimeter oberhalb der Probe. Hindernisse im Weg der Röntgenstrahlung zum Detektor beeinflussen insbesondere die niederenergetische Strahlung leichter Elemente. Oft muss deshalb die Probe zum Detektor hin gekippt werden. Bei der Untersuchung von Grenzschichten und Schichtstapeln schränkt dies die Möglichkeiten zur Aus-

Kapitel 9 232
richtung der Grenzflächen zum Elektronenstrahl unzulässig ein. Diese Nachteile gibt es bei EELS nicht.
Unterschiede gibt es in der Nachweiseffizienz: Bei geeigneten Probendicken lassen sich leichte Elemente effizienter mit EELS nachweisen, bei Elementen mit Ordnungszahlen zwischen etwa 11 und 30 gibt es kaum Unterschiede, für schwerere Elemente ist EDXS besser geeignet.
Schließlich noch einige Worte zur Ortsauflösung im STEM-Betrieb. Elastische Streuung erfolgt in größere Winkel als inelastische Streuung. Röntgenstrahlung wird von allen Probenbereichen emittiert, die von Elektronen erreicht werden, also aus dem gesamten, durch die elastische Streuung erreichten Bereich. Elastisch gestreute Elektronen spielen aber bei EELS keine Rolle. Der Winkel- und damit Anregungsbereich ist kleiner und wird zusätzlich durch die Spektrometer-Ein-trittsblende begrenzt. Die Ortsauflösung im STEM-Betrieb ist bei EELS daher besser als bei EDXS.

10 Grundlagen genauer erklärt (etwas mehr Mathematik)
Ziel: Können die Intensitätsmodulationen beim Auftreffen einer Welle auf eine Kante wirklich durch das Huygenssche Prinzip erklärt werden? Besitzen rotations-symmetrische magnetische Felder für Elektronen tatsächlich Linseneigenschaften? Wie kommt man auf die Formeln für die Berechnung von Beugungsreflexab-ständen und Winkeln zwischen Beugungsreflexen, wenn es sich nicht um ein ku-bisches Gitter handelt? Wir wollen versuchen, diese und ähnliche Fragen in die-sem Kapitel zu beantworten. Dabei lässt es sich nicht vermeiden, dass die Mathe-matik eine weitaus größere Rolle spielt als vorher. In einigen Fällen haben wir kleine Computerprogramme geschrieben, um aus den Modellen quantitative Aus-sagen zu gewinnen.
10.1 Beugung an einer Kante (Huygenssches Prinzip) Das Huygenssche Prinzip ist geeignet, um Beugungseffekte bei der Wellenaus-breitung zu beschreiben. Es besagt, dass jeder Punkt einer Wellenfront Ausgangs-punkt einer Elementarwelle ist und die Überlagerung der Elementarwellen die neue Wellenfront bildet.
Wir stellen uns vor, dass eine ebene (Elektronen-)Welle von oben auf eine Kante trifft, wie dies beispielsweise im Transmissionselektronenmikroskop der Fall ist, wenn wir eine dünne Folie mit Loch abbilden. Im Bild 10-1a ist sche-matisch dargestellt, was sich nach dem Huygensschen Prinzip abspielt. Für eine quantitative Betrachtung ist diese schematische Darstellung allerdings zu grob. Die Elementarwellen sind keine diskreten Kreise sondern haben eine kosinus-förmige Intensitätsverteilung, die sich in unserer ebenen Ansicht kreisförmig ausbreitet (Kugelwellen im Raum). Außerdem müssen wir uns die Kugelwellen sehr eng beieinander denken („Jeder Punkt einer Wellenfront ...“). Wenn wir diese Voraussetzungen berücksichtigen und die Einzelintensitäten aller Wellen an jedem Punkt summieren, erhalten wir das Bild 10-1b.
Bild 10-1. Beugung einer ebenen Welle an einer Kante (Hyugenssches Prinzip). a) Schema. b) Ergebnis der Rechnung.
DOI 10.1007/978-3-7091-1440-7_10, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Kapitel 10 234
Wir finden den bekannten Sachverhalt bestätigt, dass die Welle um die Kante „herumgebeugt“ wird und dort Minima und Maxima („Interferenzmuster“) auf-weist. Als nächstes wollen wir herausfinden, wie die Winkelverteilung der Intensi-täten hinter der Kante ist. Dazu betrachten wir Bild 10-2.
Bild 10-2. Intensitätsver-teilung einer gebeugten Welle hinter einer Kante in Abhängigkeit vom Winkel α bei verschiedenen Wellen-längen.
Wir sehen, dass das Maximum der Intensität nicht bei 0° (gedachte Verlängerung der Kante) auftritt, sondern um einige Grad dazu verschoben ist. Die Verschiebung wächst mit größer werdender Wellenlänge. Das bedeutet, dass in Ebenen, die nicht mit der Kantenebene übereinstimmen, helle Streifen, sogenannte „Beugungssäume“, auftreten.
10.2 Wellenfunktion für Elektronen Das Wort „Elektronen“ impliziert, dass es sich dabei um Teilchen handelt. Viele Beobachtungsergebnisse lassen sich auch gut mit dem Teilchenmodell der Elek-tronen erklären: Elektronen werden im elektrischen Feld beschleunigt, sie erfahren Kräfte in elektrischen und magnetischen Feldern und werden abgelenkt, sie erzeu-gen Defekte im Festkörper usw.
Es gibt allerdings auch Erscheinungen, die sich mit dem Teilchenbild nicht ver-einbaren lassen: Elektronen zeigen Interferenzen, die nur mit dem Wellencharak-ter erklärbar sind.
Für eine tiefergehende, mathematische Behandlung der mit dem Wellencharak-ter verbundenen Phänomene ist die Kenntnis der orts- und zeitabhängigen Wellen-funktion ψ(r,t) von grundlegender Bedeutung, mit der wir uns deshalb hier be-fassen wollen.
Die Größe ψ sagt etwas aus über den Zustand innerhalb der Welle in Abhängigkeit von Ort und Zeit. Am einfachsten kann man sich das am Beispiel einer mechanischen Transversalwelle veranschaulichen (s. Bild 10-3).

Grundlagen genauer erklärt (etwas mehr Mathematik) 235
Bild 10-3. Schema einer mechanischen Transversal-welle. a) Momentaufnahmen zu den Zeiten t0 und t1 > t0. b) Zeitfunktion für zwei Oszillatoren an den Orten x0 und x1 > x0.
Voraussetzung ist eine Anordnung von Oszillatoren (in Bild 10-3a grün bzw. rot gezeichnet), die miteinander gekoppelt sein müssen. Zur Zeit t = t0 = 0 seien die Oszillatoren derart ausgelenkt, dass ihre Verbindungslinie die grüne Kurve ergibt. Jeder einzelne Oszillator bewegt sich nach einer Zeitfunktion wie im rechten Teilbild dargestellt, d. h. die ersten Oszillatoren nahe x = 0 haben sich etwas nach unten bewegt, diejenigen hinter dem ersten (grünen) Wellenberg etwas nach oben usw. Die Verbindungslinie der Oszillatoren zur Zeit t = t1 ist rot eingezeichnet. Durch diese Bewegung der Oszillatoren hat sich der Wellenberg („Phase“) etwas nach rechts verschoben, ohne dass sich die Oszillatoren nach rechts verschoben haben.
Zur mathematischen Behandlung gehen wir von der harmonischen Schwin-gungsgleichung aus:
( )( )
22
20
d tt
dt
ψω ψ+ ⋅ = (10.2.1)
mit ω = 2π/T (T: Schwingungsdauer) und der Lösung
( ) ( )cost A tψ ω φ= ⋅ ⋅ + (10.2.2)
(A: Amplitude = const). Für die im Bild 10-3b blau gezeichnete Zeitfunktion für den Oszillator an der Stelle x0 gilt für die Phasenschiebung beispielsweise φ = π/2. Wir sehen auch, dass die Zeitfunktion für den Oszillator an der Stelle x1 lediglich einen anderen Wert für φ erfordert. Wir können also die Ortsabhängigkeit in die Wellenfunktion einbinden, indem wir eine ortsabhängige Phasenschiebung be-rücksichtigen:
( ) ( )( )0, cosx t A t xψ ω φ φ= ⋅ ⋅ + + . (10.2.3)
Wir setzen eine konstante Geschwindigkeit für die Bewegung des Wellenberges (allgemein: „der Phase“ – Phasengeschwindigkeit) voraus. Offensichtlich bewegt sich der Wellenberg in der Zeit, in der ein Oszillator gerade eine volle Schwin-gung (Dauer T) vollführt, gerade um eine Wellenlänge λ weiter. Für die Phasen-geschwindigkeit gilt demzufolge

Kapitel 10 236
cT
λλ ν= = ⋅ (10.2.4)
(ν: Frequenz). Für den Wellenberg gilt
( )( ) ( )0 0 0, d.h. cos t+ x 1 bzw. t+ x 0Aψ ψ ω φ φ ω φ φ= = ⋅ + = ⋅ + = . (10.2.5)
Damit erhalten wir den Zusammenhang
( ) 0x tφ ω φ= − ⋅ − . (10.2.6)
Für die Bewegung der Welle (Phase) in x-Richtung gilt
( ) 0 0 02
und damit 2x T x
x c t x xc T
πφ ω φ φ π φ
λ λ= ⋅ = − ⋅ − = − ⋅ ⋅ − = − ⋅ − . (10.2.7)
Der Term
1k
λ= (10.2.8)
wird als Wellenzahl bezeichnet. Für die Wellenfunktion folgt damit:
( ) ( ), cos 2x t A t k xψ ν= ⋅ ⋅ − ⋅ . (10.2.9)
Prinzipiell (aber schwerer verständlich) ist es auch möglich, die Ortsabhängigkeit der Wellenfunktion direkt in die Differentialgleichung für die harmonische Schwingung einzufügen. Dazu berücksichtigen wir die Gleichwertigkeit der Orts- (-k⋅x) und der Zeitabhängigkeit (ν⋅t) innerhalb des Argumentes, gehen über zu partiellen Ableitungen und erhalten:
( ) ( ) ( ) ( )2 2 2 2
2 2 2 2 2 2 2
, , , ,1 1 10 bzw. 0
x t x t x t x t
t k x c t x
ψ ψ ψ ψ
ν
∂ ∂ ∂ ∂⋅ − ⋅ = ⋅ − =
∂ ∂ ∂ ∂. (10.2.10)
Durch zweimaliges partielles Differenzieren und Einsetzen lässt sich zeigen, dass unsere Wellenfunktion
( ) ( ), cos 2x t A t k xψ ν= ⋅ ⋅ − ⋅ (10.2.11)
eine Lösung dieser partiellen Differentialgleichung ist:

Grundlagen genauer erklärt (etwas mehr Mathematik) 237
( )( ) ( )
( )( ) ( )
22
2
22
2
,2 cos 2
,2 cos 2
x tA t k x
t
x tA k t k x
x
ψν ν
ψν
∂= − ⋅ ⋅ ⋅ ⋅ − ⋅
∂
∂= − ⋅ ⋅ ⋅ ⋅ − ⋅
∂
. (10.2.12)
Die allgemeinere Lösung der partiellen Differentialgleichung für die Wellen-funktion lautet
( ) ( )2 i, e t k xx t A νψ
− ⋅ − ⋅= ⋅ , (10.2.13)
wovon man sich durch zweimalige Differentiation und Einsetzen überzeugen kann.
Die Grundgleichung für Materiewellen ist die Schrödinger-Gleichung. In ihrer allgemeinen Form lautet sie
( ) ( ) ( )2
i , ,2 m
t V ttψ ψ
∂ −⋅ ⋅ = Δ +
∂ ⋅r r r (10.2.14)
(i2 = -1, h / 2= , h: Plancksches Wirkungsquantum, Δ: Laplace59-Operator, V: potentielle Energie). Wir setzen V zur Vereinfachung zunächst gleich Null, d. h. wir betrachten die Wellenfunktion freier Elektronen, die sich in x-Richtung bewegen sollen. Damit gilt für die Schrödinger-Gleichung:
( ) ( )2
2
, ,i
2 m
x t x t
t x
ψ ψ∂ ∂−⋅ = ⋅
∂ ⋅ ∂ . (10.2.15)
Wir wollen versuchen, ob die oben angegebene, allgemeinere Lösung (10.2.13) auch Lösung der vereinfachten Schrödinger-Gleichung ist. Dazu differenzieren wir partiell:
( )
( ) ( )
2 i
22 2 i
2
2 i e
2 e
t k x
t k x
At
k Ax
ν
ν
ψν
ψ
− ⋅ − ⋅
− ⋅ − ⋅
∂= − ⋅ ⋅ ⋅
∂
∂= − ⋅ ⋅ ⋅
∂
(10.2.16)
und setzen in die Schrödinger-Gleichung ein:
59 Pierre-Simon Laplace, französischer Mathematiker, 1749 – 1827

Kapitel 10 238
( ) ( ) ( )2 i 2 i2 2 2
2 2
i 2 e 4 e2 m
h2
2 m 2 m
t k x t k xA k A
k k
ν νν
ν
− ⋅ − ⋅ − ⋅ − ⋅−− ⋅ ⋅ ⋅ ⋅ = ⋅ − ⋅ ⋅ ⋅
⋅
= ⋅ ⋅ = ⋅⋅ ⋅
. (10.2.17)
Für die Energie gilt (p: Impuls)
2 2
h d.h. 2 m =2 m h
p pE ν
ν= ⋅ = ⋅
⋅ ⋅ . (10.2.18)
Damit folgt
2 2
2
h h h, d.h. h
2 m
k kp k
p
νν
⋅ ⋅ ⋅ ⋅= = = ⋅
⋅ . (10.2.19)
Mit
1 h folgt k p
λ λ= = (10.2.20)
und damit die bekannte de Broglie-Formel
h
pλ = . (10.2.21)
Damit haben wir auch die Wellenfunktion für die Elektronen im potentialfreien Raum gefunden.
Wir wollen nun überlegen, welche Änderungen sich ergeben, wenn sich die Elektronen in einem konstanten Potential Φ0 ungleich Null bewegen, beispiels-weise in einem Kristall mit einem mittleren konstanten Potential Φ0. Es gilt
0eV = − ⋅Φ (10.2.22)
mit e als Elementarladung. Die Elektronen sollen sich weiterhin in x-Richtung bewegen. Die Schrödinger-
Gleichung für diesen Fall lautet:
( ) ( )( )
22
02
, ,i e ,
2
x t x tx t
t m x
ψ ψψ
∂ ∂−⋅ ⋅ = ⋅ − ⋅Φ ⋅
∂ ∂ . (10.2.23)
Unter der Voraussetzung, dass die gleiche Funktion wie oben die Lösung der par-tiellen Differentialgleichung ist, erhalten wir

Grundlagen genauer erklärt (etwas mehr Mathematik) 239
( )
( ) ( ) ( )
2 i
22 2 i 2
0
22
0
2 22
0
2 2 20
2 e
2 e e e2 m
hh e
2 m
he
2 m 2 m
h 2 m e
t k x
t k x i t k x
A
k A A
k
pk
p k
ν
ν π ν
ν
ν
− ⋅ − ⋅
− ⋅ − ⋅ − ⋅ − ⋅
⋅ ⋅ ⋅ ⋅ =
⋅ ⋅ ⋅ ⋅ − ⋅Φ ⋅ ⋅⋅
⋅ = ⋅ − ⋅Φ⋅
= ⋅ − ⋅Φ⋅ ⋅
= ⋅ − ⋅ ⋅ ⋅Φ
(10.2.24)
Daraus folgt schließlich für die Wellenlänge im Potential Φ0:
202 m e
h
pλ =
+ ⋅ ⋅ ⋅Φ
. (10.2.25)
Ein positives Potential verkürzt demzufolge die Wellenlänge, im potentialfreien Raum erhalten wir die de Broglie-Formel.
Schließlich fehlt noch eine anschauliche Deutung der Elektronen-Wellenfunk-tion. In der Quantenmechanik wird das Produkt aus der komplexen Wellenfunk-tion ψ und ihrer Konjugierten ψ* als Aufenthaltswahrscheinlichkeit interpretiert. Für das Produkt gilt:
( ) ( ) ( ) ( ) ( )22 i 2 i 2, * , e e ,t k x t k xx t x t A A A x tν ν
ψ ψ ψ− ⋅ − ⋅ ⋅ − ⋅
⋅ = ⋅ ⋅ ⋅ = = . (10.2.26)
Da die Elektronen eine Ladung transportieren, kann das Betragsquadrat der Elek-tronen-Wellenfunktion als Ladungsdichte angesehen werden.
Im Allgemeinen werden in der Elektronenmikroskopie stationäre Zustände be-rechnet, d. h. die Zeitabhängigkeit wird außer Acht gelassen. Damit und mit Über-gang von der Koordinate x zum Vektor r im Raum erhalten wir für die Wellen-funktion:
( ) 2 ieAψ − ⋅ ⋅= ⋅ k rr . (10.2.27)
10.3 Elektronenwellenlänge relativistisch berechnet Wir gehen von der de Broglie-Beziehung aus (h: Plancksches Wirkungsquantum) :
h
m vλ =
⋅ . (10.3.1)

Kapitel 10 240
Zur Berechnung der beiden Unbekannten Masse m und Geschwindigkeit v be-nutzen wir den relativistischen Energiesatz (c: Lichtgeschwindigkeit im Vakuum, e: Elementarladung, UB: Beschleunigungsspannung, m0: Ruhemasse des Elek-trons)
( ) 20m c e Bm U− ⋅ = ⋅ (10.3.2)
und die relativistische Masse-Geschwindigkeits-Relation:
( )
0
2
c
m
1 v
m =
−. (10.3.3)
Daraus folgen
0 2
em
cBU
m⋅
= + (10.3.4)
und
( )0
2
220
mc 1
m e / cB
vU
= −+ ⋅
. (10.3.5)
Daraus lässt sich die Wellenlänge zu
0 2
h
ee 2 m
cB
B
UU
λ =⋅
⋅ ⋅ +
(10.3.6)
berechnen. Für die Handhabung von Zahlenwerten z. B. in Computerprogrammen wird
häufig eine andere Formel benutzt. Aus (10.3.2) und (10.3.3) folgt
( )22
0c
e11
511,06 kVm c1
B B
v
U U⋅− = =
⋅−
(10.3.7)
und daraus
( )2
c
511,06 kV1
+511,06 kVv
relB
KU
= − = (10.3.8)

Grundlagen genauer erklärt (etwas mehr Mathematik) 241
als relativistische Korrektur. Aus (10.3.1) wird dann
2 20
h2,4263 pm
m c 1- 1-
rel rel
rel rel
K K
K Kλ = ⋅ = ⋅
⋅ . (10.3.9)
10.4 Elektronenbahnen in rotationssymmetrischen magnetischen Feldern Die Elektronenbahnen im Magnetfeld der Induktion B werden durch die Lorentz-kraft
e= − ⋅ ×F v B (10.4.1)
(e: Elementarladung) bestimmt. Zur Berechnung der Elektronenbahnen müssen ein Anfangswert der Elektronengeschwindigkeit v (Betrag und Richtung) sowie die Feldverteilung B (ebenfalls Betrag und Richtung) bekannt sein. Die Lorentz-kaft ist in die Bewegungsgleichung einzusetzen und aus der daraus folgenden Beschleunigung kann der Ort-Zeit-Verlauf für die Elektronen mit Hilfe der Anfangsbedingungen berechnet werden.
Im Folgenden wollen wir uns auf einen gebräuchlichen Spezialfall be-schränken: die rotationssymmetrische Polschuhlinse. Sie ist die in Elektronenmi-kroskopen am häufigsten benutzte Linse. Wegen der Rotationssymmetrie benut-zen wir Zylinderkoordinaten r, ϕ, z (vgl. Bild 10-4).
Bild 10-4. Zylinderkoordinaten für die rotationssymmetrische magnetische Linse.
Wir schreiben die Bewegungsgleichung in der Form
( )d
mdt
= ⋅F v . (10.4.2)

Kapitel 10 242
Bei konstanter Masse (d. h. in nichtrelativistischer Näherung) gilt
md
dt= ⋅
vF . (10.4.3)
Als Nächstes ermitteln wir den Geschwindigkeitsvektor in Zylinderkoordinaten. Für den Ort s gilt (bitte nicht mit dem Polschuhspalt verwechseln):
r z= ⋅ + ⋅r zs e e . (10.4.4)
Daraus folgt für die Geschwindigkeit:
dd dr dzr
dt dt dt dt= = ⋅ + ⋅ + ⋅r
r zes
s e e . (10.4.5)
Für die zeitliche Ableitung des Einheitsvektors in r-Richtung nehmen wir eine An-leihe in kartesischen Koordinaten:
cos sin
sin cosd dd d
dt dt d dt
ϕ ϕ
ϕ ϕϕ ϕ ϕ
ϕ
= ⋅ + ⋅
= ⋅ = ⋅ − ⋅ + ⋅ = ⋅
r x y
r rx y
e e e
e ee e e
, (10.4.6)
d. h.
r r zϕ= ⋅ + ⋅ ⋅ + ⋅r zs e e e . (10.4.7)
Damit lautet die Formel für die Lorentzkraft (10.4.1) in Zylinderkoordinaten:
( ) ( )
e
0
e e e
r z
z r z r
r r z
B B
r B z B r B r B
ϕ
ϕ ϕ
= − ⋅ ⋅
= − ⋅ ⋅ ⋅ ⋅ − ⋅ ⋅ − ⋅ − ⋅ ⋅ − ⋅ ⋅
r z
r z
e e e
F
e e e
(10.4.8)
bzw. für die Komponenten der Lorentzkraft:
e , e e , er z z r z rF r B F r B z B F r Bϕϕ ϕ= − ⋅ ⋅ ⋅ = ⋅ ⋅ − ⋅ ⋅ = ⋅ ⋅ ⋅ . (10.4.9)
Damit und mit der Bewegungsgleichung kann der zeitliche Verlauf der Elek-tronenbewegung numerisch berechnet werden:

Grundlagen genauer erklärt (etwas mehr Mathematik) 243
( ) ( ) ( ) ( ) ( ) ( )
( ) ( ) ( )
( ) ( ) ( ) ( ) ( )( )
( ) ( ) ( )
( ) ( ) ( ) ( )
( ) ( ) ( )
e,
m m
e, ,
m m
e,
m m
rz
z r
zr
Fr t t r t t r t r t t B r z t
r t t r t r t t
Ft t t t t r B r z z B r z t
t t t t t
Fz t t z t t z t r B r z t
z t t z t z t t
ϕ
ϕ
ϕ ϕ ϕ
ϕ ϕ ϕ
ϕ
+ Δ = + ⋅Δ = − ⋅ ⋅ ⋅ ⋅Δ
+ Δ = + ⋅Δ
+ Δ = + ⋅Δ = + ⋅ ⋅ − ⋅ ⋅Δ
+ Δ = + ⋅Δ
+ Δ = + ⋅Δ = + ⋅ ⋅ ⋅ ⋅Δ
+ Δ = + ⋅Δ
(10.4.10)
mit der spezifische Elektronenladung e/m. Zur weiteren Rechnung benötigen wir die Komponenten Br und Bz der magnetischen Induktion. Nach den Maxwellschen Gleichungen gilt für das magnetische Potential Ψ im stromlosen Raum die Laplace-Gleichung
0ΔΨ = . (10.4.11)
In kartesischen Koordinaten heißt das
( ) ( ) ( )2 2 2
2 2 2
, , , , , ,0
x y z x y z x y z
x y z
∂ Ψ ∂ Ψ ∂ Ψ+ + =
∂ ∂ ∂ . (10.4.12)
Die Komponenten Bx, By und Bz der magnetischen Induktion B hängen mit dem magnetischen Potential wie folgt zusammen:
, , x y zB B Bx y z
∂Ψ ∂Ψ ∂Ψ= − = − = −
∂ ∂ ∂ . (10.4.13)
Die Laplace-Gleichung in Zylinderkoordinaten lautet:
( ) ( ) ( ) ( )2 2 2
2 2 2 2
, , , , , , , ,1 10
r z r z r z r z
r rr z r
ϕ ϕ ϕ ϕ
ϕ
∂ Ψ ∂ Ψ ∂Ψ ∂ Ψ+ + ⋅ + ⋅ =
∂∂ ∂ ∂ . (10.4.14)
Im rotationssymmetrischen Feld hängt das Potential nicht von ϕ ab, daher ist
( )2
2
, ,0
r zϕ
ϕ
∂ Ψ=
∂ (10.4.15)
und die Laplace-Gleichung vereinfacht sich zu

Kapitel 10 244
( ) ( ) ( )2 2
2 2
, , ,10
r z r z r z
r rr z
∂ Ψ ∂ Ψ ∂Ψ+ + ⋅ =
∂∂ ∂ . (10.4.16)
Die Komponenten der magnetischen Induktion sind dann
, r zB Br z
∂Ψ ∂Ψ= − = −
∂ ∂ . (10.4.17)
Eine Möglichkeit zur Lösung der Differentialgleichung
( ) ( ) ( )2 2
2 2
, , ,10
r z r z r z
r rr z
∂ Ψ ∂Ψ ∂ Ψ+ ⋅ + =
∂∂ ∂ (10.4.18)
ist ein Reihenansatz. Dabei berücksichtigen wir die Rotationssymmetrie, d. h.
( ) ( ), ,r z r zΨ = Ψ − . (10.4.19)
Damit sind in der Reihe nur geradzahlige Potenzen von r möglich:
( ) ( ) ( ) ( ) ( ) ( )2 4 6 80 2 4 6 8, ...r z K z K z r K z r K z r K z rΨ = + ⋅ + ⋅ + ⋅ + ⋅ + . (10.4.20)
Für r = 0 muss
( ) ( )00, z K zΨ = (10.4.21)
gelten. Die Terme der Laplace-Gleichung lauten damit:
( )( ) ( ) ( ) ( )
( )( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) ( )
( )
22 4 6
2 4 6 82
2 4 62 4 6 8
2 2 2 2 22 4 62 4 6
2 2 2 2 2
28 82
,2 12 30 56 ...
,12 4 6 8 ...
, 0,
...
r zK z K z r K z r K z r
rr z
K z K z r K z r K z rr r
r z z K z K z K zr r r
z z z z z
K zr
z
∂ Ψ= ⋅ + ⋅ ⋅ + ⋅ ⋅ + ⋅ ⋅ +
∂
∂Ψ⋅ = ⋅ + ⋅ ⋅ + ⋅ ⋅ + ⋅ ⋅ +
∂
∂ Ψ ∂ Ψ ∂ ∂ ∂= + ⋅ + ⋅ + ⋅ +
∂ ∂ ∂ ∂ ∂
∂+ ⋅ +
∂
(10.4.22)
Einsetzen in die Laplace-Gleichung und Vergleich der Koeffizienten vor gleichen Potenzen von r liefert:

Grundlagen genauer erklärt (etwas mehr Mathematik) 245
( ) ( )( )
( )( )2 2
02 2 22 2
0, 0,1: 2 2 0, d.h.
4
z zr K z K z K z
z z
∂ Ψ ∂ Ψ⋅ + ⋅ + = = − ⋅
∂ ∂
( ) ( )
( )
( )( ) ( )
222
4 4 2
2 42
4 2 4
: 12 4 0,
0,1 1 d.h.
16 64
K zr K z K z
z
K z zK z
z z
∂⋅ + ⋅ + =
∂
∂ ∂ Ψ= − ⋅ = ⋅
∂ ∂
( ) ( )
( )
( )( ) ( )
244
6 6 2
2 64
6 2 6
: 30 6 0,
0,1 1 d.h.
36 2304
K zr K z K z
z
K z zK z
z z
∂⋅ + ⋅ + =
∂
∂ ∂ Ψ= − ⋅ = − ⋅
∂ ∂
( ) ( )
( )
( )( ) ( )
266
8 8 2
2 86
8 2 8
: 56 8 0,
0,1 1 d.h.
64 147456
K zr K z K z
z
K z zK z
z z
∂⋅ + ⋅ + =
∂
∂ ∂ Ψ= − ⋅ = ⋅
∂ ∂
.
Die Reihe für das magnetische Potential lautet damit:
( ) ( )( ) ( )
( ) ( )
2 42 4
2 4
6 86 8
6 8
0, 0,, 0,
4 64
0, 0,...
2304 147456
z zr rr z z
z z
z zr r
z z
∂ Ψ ∂ ΨΨ = Ψ − ⋅ + ⋅ −
∂ ∂
∂ Ψ ∂ Ψ− ⋅ + ⋅ − +
∂ ∂
. (10.4.23)
Offenbar lässt sich diese Reihe allgemein als Summe (n: Nummer des Gliedes, beginnend bei n = 0)
( )( )
( )
( )2 2
2 2
1 0,,
4 !
n n n
nnn
r zr z
zn
⋅− ⋅ ∂ ΨΨ = ⋅
∂⋅
(10.4.24)
schreiben. Wir sehen, dass mit Kenntnis des Potentialverlaufs auf der Achse das Magnetfeld vollständig beschrieben werden kann.
Bild 10-5. Induktionsspule zur Messung von magneti-schen Linsenfeldern.

Kapitel 10 246
Zur Bestimmung des Magnetfeldes einer rotationssymmetrischen Linse wird im Allgemeinen die magnetische Induktion Bz(0,z) auf der optischen Achse (Mit-telachse) der Linse mit einer kleinen Induktionsspule (s. Bild 10-5) gemessen. Für die gesättigte magnetische Polschuhlinse kann sie typischerweise durch die Glasersche60 Glockenkurve [10.1]
( ) 02
0,
1
zz
BB z
z
d
=
+
(10.4.25)
beschrieben werden (s. Bild 10-6), wobei d die Breite der Glockenkurve charakte-risiert. Mit Kenntnis dieses Verlaufs können wir das magnetische Potential auf der Linsenmittelachse (r = 0) berechnen:
( ) ( ), d.h. 0, 0,z zB z B z dzz
∂Ψ= − Ψ = − ⋅
∂ (10.4.26)
Bild 10-6. Glasersche Glockenkurven der z-Komponente der magne-tischen Induktion B auf der Linsenmittelachse für verschiedene Breiten d.
Wir setzen die Funktion (10.4.25) für die Glasersche Glockenkurve ein und er-halten
( ) 2002 2 2
20
0,
1
arctan
zz
z
B dz dzz B d
d zz
d
B d zC
d d
⋅Ψ = − = − ⋅
+−
⋅= − ⋅ +
. (10.4.27)
60 Walter Glaser, österreichischer Physiker und Elektronenoptiker, 1906 –1960

Grundlagen genauer erklärt (etwas mehr Mathematik) 247
Analog zum elektrischen Potential in einem Plattenkondensator fordern wir für die Mitte der Linse (z = 0) ein magnetisches Potential Ψ(0,0) = 0 und erhalten damit (vgl. Bild 10-7):
( ) 00, arctanzz
z B dd
Ψ = − ⋅ ⋅ . (10.4.28)
Bild 10-7. Verlauf des magnetischen Potentials auf der Linsenmittelachse für verschiedene Glasersche Glockenkurven der z-Komponente der magne-tischen Induktion B.
Für achsennahe Gebiete („Paraxialstrahlen“) können wir die Reihe nach dem 3. Glied abbrechen:
( ) ( )( ) ( )2 42 4
2 4
0, 0,, 0,
4 64
z zr rr z z
z z
∂ Ψ ∂ ΨΨ = Ψ − ⋅ + ⋅
∂ ∂ . (10.4.29)
Für die Ableitungen gilt bei Benutzung des Glaserschen Glockenfeldes
( )( )2
02 2
0,0, z
z
zB dB z
zd z
∂Ψ⋅= = −
∂+ (10.4.30)
( )
( )
2 20
2 2 2
22 0
0 2 2 22 2
0,
21
z
zz
z d B
zz d z
d B zd B
z d z d z
∂ Ψ ⋅∂= −
∂∂ +
⋅ ⋅ ⋅∂= − ⋅ ⋅ =
∂ + +
(10.4.31)

Kapitel 10 248
( )
( )
( ) ( )
3 20
3 22 2
2 22 2
0 02 32 2 2 2
0, 2
2 2
z
z z
z d B z
zz d z
z z dd B d B
z d z d z
∂ Ψ ⋅ ⋅ ⋅∂= −
∂∂ +
∂ −= − ⋅ ⋅ ⋅ = ⋅ ⋅ ⋅
∂ + +
(10.4.32)
( )
( )
( )
( )
( )
( )
4 2 22
04 32 2
2 22
0 42 2
2 22
0 42 2
0,2
4 22
28
z
z
z
z z dd B
zz d z
z d zd B
d z
d zz d B
d z
∂ Ψ ∂ −= − ⋅ ⋅ ⋅
∂∂ +
⋅ ⋅ −= − ⋅ ⋅ ⋅
+
⋅ −= − ⋅ ⋅ ⋅ ⋅
+
(10.4.33)
( )
( )
( )
5 2 32
05 42 2
4 2 2 42
0 52 2
0, 28
2 58
z
z
z d z zd B
zz d z
d d z zd B
d z
∂ Ψ ∂ ⋅ ⋅ −= − ⋅ ⋅ ⋅
∂∂ +
⋅ − ⋅ ⋅ −= − ⋅ ⋅ ⋅
+
(10.4.34)
und damit
( ) ( )( )
( )
( )
( ) ( )( )
( )
( )
2 222 420
02 42 2 2 2
2 2 2
2 20 2 42 2 2 2
2, 0,
2 8
21, 0,
2 8
zz
z
d zd B zr rr z z z d B
d z d z
r d zr z z d B r z
d z d z
⋅ −⋅ ⋅Ψ = Ψ − ⋅ − ⋅ ⋅ ⋅ ⋅
+ +
⋅ ⋅ −Ψ = Ψ − ⋅ ⋅ ⋅ ⋅ +
⋅ + ⋅ +
.(10.4.35)

Grundlagen genauer erklärt (etwas mehr Mathematik) 249
Wir haben außerdem die Möglichkeit, die beiden für die Lorentzkraft wichtigen Komponenten Bz(r,z) sowie Br(r,z) der magnetischen Induktion analytisch zu berechnen:
( ) ( ) ( ) ( )
( ) ( )
2 42 4
2 4
2 43
2 4
, 0, 0, 0,
4 64
0, 0,
2 16
r
r
r z z z zr rB
r r r rz z
z zr rB
z z
∂Ψ ∂Ψ ∂Ψ ∂Ψ∂ ∂= − = − + ⋅ − ⋅
∂ ∂ ∂ ∂∂ ∂
∂Ψ ∂Ψ= ⋅ − ⋅
∂ ∂
( )
( )
( )
2 2 22
0 2 42 2 2 2
21
2r z
r d zB r z d B
d z d z
⋅ ⋅ −= ⋅ ⋅ ⋅ ⋅ +
+ ⋅ +
(10.4.36)
bzw.
( ) ( ) ( ) ( )
( )( )
( )
( )
3 52 4
3 5
2 2 22
0 32 2
4 4 2 2 42
0 52 2
20
2 2
2 2 2 2 4 2 22
0 32 2
, 0, 0, 0,
4 64
0,2
2 5
8
2 5
2 4
z
z z z
z
zz
z
r z z z zr rB
z z z z
r z dB B z d B
d z
r d d z zd B
d z
B dB
d z
r z d r d d zd B
d z
∂Ψ ∂Ψ ∂ Ψ ∂ Ψ= − = − + ⋅ − ⋅
∂ ∂ ∂ ∂
−= + ⋅ ⋅ ⋅ −
+
⋅ − ⋅ ⋅ −− ⋅ ⋅ ⋅
+
⋅= +
+
− ⋅ − ⋅ ⋅ −+ ⋅ ⋅ ⋅ − ⋅
+ ( )
4
52 2
z
d z+
. (10.4.37)
Für den Praktiker ist eine Information über die Breite d der Glaserschen Glocken-kurve notwendig. Aus einer Grafik von Glaser [10.2], in der der Quotient d/b über s/b (b: Durchmesser der Polschuhbohrung, s: Polschuhspalt) für die gesättigte Polschuhlinse (Feldstärke so hoch, dass Polschuhmaterial magnetisch gesättigt ist) dargestellt ist, lässt sich für einen Bereich s/b = 0,2 ... 1 näherungsweise ein Zusammenhang
0,35 0,25d b s≈ ⋅ + ⋅ (10.4.38)

Kapitel 10 250
erkennen. Realistische Werte für Polschuhspalt und Polschuhbohrung sind s = 5,4 mm und b = 2,2 mm. Daraus folgt nach (10.4.38) eine Glockenfeldbreite d = 2,1 mm. In Bild 10-8 sind die daraus resultierenden Feldverläufe dargestellt.
Im Transmissionselektronenmikroskop ist die Probe etwa in der Polschuhmitte (z = 0) angeordnet. Aus Bild 10-8 ist ersichtlich, dass die Radialkomponente des magnetischen Feldes in radialer Richtung von der Mitte aus anwächst. Beim Ein-schleusen von magnetischen Proben kann dies zum Verschieben der Probe im Halter führen.
Bild 10-8. Radiale (Br - a) und axiale (Bz - b) Komponente der magnetischen Induktion in einer gesättigten Polschuhlinse.
Bild 10-9a vermittelt einen zweidimensionalen Eindruck vom magnetischen Po-tential in einer Polschuhlinse. Daraus sind nach den Formeln (10.4.17) und (10.4.10) die gleichfalls in Bild 10-9 dargestellten Elektronenbahnen berechnet worden.
Bild 10-9. Magnetisches Potential in einer Polschuhlinse (a) und daraus berechnete Elektronen-bahnen in der (verdrillten) r-z-Ebene (b) und (c). Die Elektronen bewegen sich auf Schrauben-bahnen durch das magnetische Feld.
Sie zeigen das typische Verhalten einer optischen Linse: Parallel in die Linse ein-fallende Strahlen werden im Brennpunkt vereinigt (Bild 10-9b) und von einem Dingpunkt ausgehende Strahlen treffen sich im Bildpunkt (Bild 10-9c). Bei

Grundlagen genauer erklärt (etwas mehr Mathematik) 251
genauerem Hinsehen erkennen wir aber auch die Schwachstelle der rotations-symmetrischen magnetischen Linse: Die Linsenaußenzonen brechen stärker als die Linsenmittenzone. Dieser Sachverhalt wurde bereits in Abschnitt 2.3 beschrie-ben und wird als Öffnungsfehler bezeichnet.
10.5 Auflösungsvermögen mit Berücksichtigung des Öffnungsfehlers Das Auflösungsvermögen δ des TEM resultiert aus der Überlagerung von Beu-gungs- und Öffnungsfehlerscheibchen. Wir benutzen dafür vereinfachend die Wurzel aus der Summe aus den Quadraten der beiden Fehlerscheibchen, setzen die Brechzahl in der Objektumgebung n = 1 und berücksichtigen die aufgrund des Öffnungsfehlers notwendige Begrenzung der Apertur auf Werte α << 1:
( )2
22 2 30 6B S S
, C
⋅= + = + ⋅ . (10.5.1)
Wegen der gegenläufigen Abhängigkeit der Fehlerscheibchen von α existiert eine optimale Apertur αopt, bei der δ seinen minimalen Wert annimmt. Die Differentia-tion ergibt:
( )
22 5
3
223
0,362 6
0 62
S
S
Cd
d ,
C
λα
αα
⋅− ⋅ + ⋅ ⋅
=
⋅+ ⋅
. (10.5.2)
Mit der Extremwertbedingung
| 0opt
d
d αα
= , (10.5.3)
d. h.
22 5
3
0,362 6 0S opt
opt
Cλ
αα
⋅− ⋅ + ⋅ ⋅ = (10.5.4)
erhält man
4opt 0,77 SC
λα = (10.5.5)

Kapitel 10 252
und
( )2
23 34min S
0 6 0,9 CS opt
opt
, C
⋅= + ⋅ = ⋅ . (10.5.6)
Bei 300 kV Beschleunigungsspannung (λ = 1,97 pm) folgt daraus mit CS = 1,2 mm: αopt ≈ 4,9 mrad und δmin ≈ 2,8 Å, was gut mit den bereits früher (Ab-schnitt 2.4) aus Bild 2.8 abgelesenen Werten übereinstimmt.
10.6 Schottky-Effekt Wir stellen uns vor, dass sich die Elektronen bei der thermischen Emission eine kurze Zeit in der Nähe der Kathodenoberfläche aufhalten und dort eine Raumla-dung bilden. Das elektrische Feld zwischen einem Elektron und der Drahtober-fläche ist identisch mit einem Feld zwischen dem erwähnten Elektron und einer positiven Ladung im gleichen Abstand hinter der Drahtoberfläche („Bildladung“ – vgl. Bild 10-10).
Bild 10-10. Elektrisches Feld an einer Metalloberfläche (Bildladung).
Die Potentialkraft F für das Elektron mit der Ladung –e im Abstand x vor der Drahtoberfläche ist die Coulomb-Kraft zwischen den Ladungen e und –e im Abstand 2⋅x:
( )2 2
2 20 0
1 e e 1
4 164RF x
x x
− −= ⋅ = ⋅
⋅ ⋅ ⋅ ⋅⋅ (10.6.1)
(Elementarladung e und Influenzkonstante ε0). Liegt ein zusätzliches elektrisches Feld E an der Kathode an, so erfährt das
Elektron durch dieses Feld die Kraft (Kathode ist negativ!)
eEF E= ⋅ (10.6.2)
Für die potentielle Energie W unter Einfluss dieser beiden Kräfte gilt dann:

Grundlagen genauer erklärt (etwas mehr Mathematik) 253
( ) ( )( )2
00
ee
16E RW x F F x dx W E xx
= + ⋅ = + ⋅ ⋅ +⋅ ⋅ ⋅
(10.6.3)
Wir wollen nun die Auswirkungen auf das Potentialtopf-Modell betrachten. Wir müssen berücksichtigen, dass in der grafischen Darstellung dieses Modells das Potential nach oben hin negativer wird. Außerdem müssen wir einen Bezugspunkt („Nullpunkt“) festlegen. Wir wollen dazu die Energie des Vakuumniveaus be-nutzen (vgl. Bild 10-11). In diesem Fall ist W0 = 0.
Es gilt: ( ) E RW x W W= + für 0x ≥
und ( ) FermiW x W= für 0x < .
Für die Austrittsarbeit gilt:
( ) ( )( )0 0A Fermi E RW W W x W x= − = + = (10.6.4)
Bild 10-11. Erniedrigung des Potentialwalls durch den Schottky-Effekt (Parameter: E = 20 kV/cm).
Aus Bild 10-11 ist ersichtlich, dass damit die Austrittsarbeit verringert wird. Zur Abschätzung dieser Verringerung bestimmen wir das Maximum der Funktion
( ) ( )( ) E RW x W x W x= + :
( )2
0
ee
16W x E x
x= ⋅ ⋅ +
⋅ ⋅ ⋅ (10.6.5)
max
2
20 max
e| e 0
16x x
dWE
dx x= = ⋅ − =
⋅ ⋅ ⋅ (10.6.6)
max0
e
16x
E=
⋅ ⋅ ⋅ (10.6.7)

Kapitel 10 254
Diesen Wert setzen wir in W(x) ein und erhalten für das Maximum:
( )2
max max max0 max
2
00
0
ee
16
e ee
16 e16
16
W W x E xx
EE
E
= = ⋅ ⋅ +⋅ ⋅ ⋅
= ⋅ ⋅ +⋅ ⋅ ⋅
⋅ ⋅ ⋅⋅ ⋅ ⋅
( )
2 2 4 3
max2
0 00
0
e e e e2
e16 161616
E EW
EEε
⋅ ⋅ ⋅= + = ⋅
⋅ ⋅ ⋅ ⋅ ⋅⋅ ⋅ ⋅
⋅ ⋅ ⋅
(10.6.8)
35
max -10
e3,8 10 eV
4 V m
E EW −⋅
= = ⋅ ⋅⋅ ⋅ ⋅
. (10.6.9)
Die Verringerung der Austrittsarbeit durch den Schottky-Effekt ist demzufolge proportional zur Wurzel aus der an der Kathode anliegenden elektrischen Feldstär-ke. Bei Benutzung gebräuchlicher Maßeinheiten folgt:
, -10,0038 eV
kV cmA Schottky
EWΔ = ⋅
⋅ . (10.6.10)
10.7 Elektrisches Potential in rotationssymmetrischen Elektrodenanordnungen Um die Arbeitsweise einer Elektronenkanone (bzw. eines Strahlerzeugers) zu ver-stehen, ist es notwendig, die Potentialverteilung innerhalb des Strahlerzeugers zu kennen. Wir wollen diese deshalb berechnen. Wir gehen dabei von einer vorgege-benen Elektrodenanordnung mit bekannten Potentialen aus. Im Falle eines Triodensystems sind dies die geometrische Anordnung von Kathode, Wehnelt-Elektrode und Anode sowie die Potentiale von Kathode und Wehnelt-Elektrode. Das Potential der Anode ist normalerweise Null, d. h. die Anode ist geerdet.
Uns interessiert die Potentialverteilung zwischen den Elektroden. In diesem Bereich gibt es keine Ladungen, es gilt für das Potential U die Laplace-Gleichung
0UΔ = . (10.7.1)
In kartesischen Koordinaten x,y,z heißt das:

Grundlagen genauer erklärt (etwas mehr Mathematik) 255
2 2 2
2 2 2
U U UU
x y z
∂ ∂ ∂Δ = + +
∂ ∂ ∂ . (10.7.2)
Für rotationssymmetrische Anordnungen benutzen wir das Zylinderkoordinaten-system r, ϕ, z (vgl. Bild 10-4):
( ) ( ) ( ) ( )2 2 2
2 2 2 2
, , , , , , , ,1 10
U r z U r z U r z U r z
r rr z r
ϕ ϕ ϕ ϕ
ϕ
∂ ∂ ∂ ∂+ + ⋅ + ⋅ =
∂∂ ∂ ∂ . (10.7.3)
Wegen der Rotationssymmetrie ist
( )2
2
, ,0
U r zϕ
ϕ
∂=
∂ (10.7.4)
und die Laplace-Gleichung vereinfacht sich zu
( ) ( ) ( )2 2
2 2
, , ,10
U r z U r z U r z
r rr z
∂ ∂ ∂+ + ⋅ =
∂∂ ∂ . (10.7.5)
Diese partielle Differentialgleichung wollen wir numerisch lösen. Dazu wandeln wir die Differentialquotienten in Differenzenquotienten um:
( ) ( )( )( )
( ) ( ) ( )
( )
( ) ( )( )
( )
( ) ( ) ( )
( )
( ) ( ) ( ) ( )
2
2 2 2
2
2 2 2
,, , 2 , ,
,, , 2 , ,
, , , ,
2
U r zU r z U r r z U r z U r r z
r r r
U r zU r z U r z z U r z U r z z
z z z
U r z U r z U r r z U r r z
r r r
Δ Δ∂ + Δ − ⋅ + −Δ→ =
∂ Δ Δ
Δ Δ∂ + Δ − ⋅ + −Δ→ =
∂ Δ Δ
∂ Δ + Δ − −Δ→ =
∂ Δ ⋅Δ
. (10.7.6)
Zur Vereinfachung benutzen wir ein Netz mit gleichen Maschenweiten Δr = Δz = Δ und bezeichnen die Masche U(r,z) unter Benutzung von r = i⋅Δ sowie z = j⋅Δ mit U(i,j). Damit gilt für die Laplace-Gleichung (10.7.5):
( ) ( ) ( )
( ) ( ) ( ) ( ) ( )
, 2 , ,
, 2 , , , ,0
2
U r z U r z U r z
U r z U r z U r z U r z U r z
r
+ Δ − ⋅ + − Δ+
Δ
+ Δ − ⋅ + − Δ + Δ − − Δ+ + =
Δ ⋅
(10.7.7)

Kapitel 10 256
bzw.
( ) ( ) ( )
( ) ( )
4 , 1 , 1 ,2 2
, ,
U r z U r z U r zr r
U r z U r z
Δ Δ⋅ = + ⋅ + Δ + − ⋅ − Δ +
⋅ ⋅
+ + Δ + − Δ
(10.7.8)
( ) ( ) ( )
( ) ( )}
1 1 1, 1 1, 1 1,
4 2 2
, 1 , 1
U i j U i j U i ji i
U i j U i j
= + ⋅ + + − ⋅ − +⋅ ⋅
+ + + −
. (10.7.9)
Dies ist eine Rekursionsformel, nach der das Ergebnis des k-ten Schrittes aus dem (k-1)-ten Schritt berechnet werden kann. Die Verteilung des nullten Schrittes ist durch die Randbedingungen (Potentiale der Elektroden) gegeben. Die Rekursions-formel lautet damit:
( ) ( ) ( )
( ) ( )}
1 1
1 1
1 1 1, 1 1, 1 1,
4 2 2
, 1 , 1
k k k
k k
U i j U i j U i ji i
U i j U i j
− −
− −
= + ⋅ + + − ⋅ − +⋅ ⋅
+ + + −
. (10.7.10)
Da i im Nenner auftaucht, gibt es ein Problem für i = 0, welches wir separat lösen müssen. Dafür benutzen wir die Differenzengleichung in kartesischen Koordina-ten:
( ) ( ) ( )
( )
( ) ( ) ( )
( )
( ) ( ) ( )
( )
2
2
2
, , 2 , , , ,
, , 2 , , , ,
, , 2 , , , ,0
U x x y z U x y z U x x y z
x
U x y y z U x y z U x y y z
y
U x y z z U x y z U x y z z
z
+ Δ − ⋅ + −Δ+
Δ
+ Δ − ⋅ + −Δ+ +
Δ
+ Δ − ⋅ + −Δ+ =
Δ
. (10.7.11)
Für r = 0 ist x = 0 und y = 0. Außerdem setzen wir Δx = Δy =Δz = Δ:
( ) ( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
,0, 2 0,0, ,0, 0, , 2 0,0,
0, , 0,0, 2 0,0, 0,0, 0
U z U z U z U z U z
U z U z U z U z
Δ − ⋅ + −Δ + Δ − ⋅ +
+ −Δ + +Δ − ⋅ + −Δ = . (10.7.12)
Nun erinnern wir uns wieder an die Rotationssymmetrie und sehen:

Grundlagen genauer erklärt (etwas mehr Mathematik) 257
( ) ( )
( ) ( ) ( )
( ) ( ) ( )
0, 0, 0,
, 0, 0, , ,
, 0, 0, , ,
U x y z U r z
U x y z U x y z U r z
U x y z U x y z U r z
= = = =
= Δ = = = = Δ = = Δ
= −Δ = = = = −Δ = = Δ
(10.7.13)
Damit folgt aus Gleichung (10.7.12):
( ) ( ) ( ) ( ) ( ) ( )
( ) ( ) ( )
, 2 0, , , 2 0, ,
0, 2 0, 0, 0
U z U z U z U z U z U z
U z U z U z
Δ − ⋅ + Δ + Δ − ⋅ + Δ +
+ + Δ − ⋅ + − Δ = (10.7.14)
bzw.
( ) ( ) ( ) ( )6 0, 4 , 0, 0,U z U z U z U z⋅ = ⋅ Δ + + Δ + − Δ (10.7.15)
und damit als Rekursionsformel für die Berechnung des Potentials auf der z-Achse (r = 0):
( ) ( ) ( ) ( )1 1 12 1
0, 1, 0, 1 0, 13 6k k k kU j U j U j U j− − −= ⋅ + + + − . (10.7.16)
Damit ist es möglich, die Potentialverteilung zu berechnen: Die Ausgangsvertei-lung ist durch die Randbedingungen gegeben. Daraus wird schrittweise zunächst nach (10.7.16) die Verteilung auf der z-Achse und anschließend nach (10.7.10) die restliche Verteilung berechnet. Dies wird iterativ wiederholt, bis die Veränderung zwischen den Schritten vernachlässigbar klein wird.
Bild 10-12. Verteilung des elektrischen Potentials in rotationssymmetrischen Elektronenkanonen. a) Triodensystem. b) Schottky-Feldemis-sionskathode. (Gleiche Farben bedeuten gleiche Potentiale.)
Nach diesem Verfahren sind die Potentialverteilungen in Bild 10-12 berechnet worden. Aus der Form der Potentiallinien ist die fokussierende Wirkung beider Systeme ersichtlich (Kraftvektoren stehen senkrecht auf den Potentiallinien).

Kapitel 10 258
10.8 Laue-Gleichungen und reziprokes Gitter, Ewald-Konstruktion Die Braggsche Gleichung verknüpft Gittereigenschaften in Form der Netzebenen-abstände mit den Beugungswinkeln. Ein bestimmter Netzebenenabstand erzeugt einen Beugungsreflex, im Transmissionselektronenmikroskop kann das ein heller Spot sein. Was ist aber nun, wenn wir zwei Netzebenenscharen haben, die gegen-einander verdreht sind, beispielsweise (1 0 0)- und (0 1 0)-Ebenen, die in orthogo-nalen Systemen (alle Achsenwinkel gleich 90°) senkrecht aufeinander stehen. Wir erwarten zwei Beugungsreflexe, deren Verbindungslinien zum Zentrum gleichfalls senkrecht aufeinander stehen. Wie können wir diese Reflexpositionen berechnen?
Wir gehen vom Braggschen Gesetz
n dλ θ⋅ = ⋅ (5.6)
(n: ganze Zahl, λ: Wellenlänge, d: Netzebenenabstand, θ: Beugungswinkel) aus und betrachten dazu Bild 10-13. Der Wellenzahlvektor k0 der einfallenden Welle schließt mit dem Wellenzahlvektor k der reflektierten Welle den Winkel θ ein. Bei den in der Elektronenbeugung kleinen Beugungswinkeln können wir für den Betrag der Differenz der beiden Wellenzahlvektoren („Streuvektor“) unter Be-rücksichtigung des Braggschen Gesetzes (5.6) schreiben:
n
d
λθ
⋅− = ⋅ = ⋅0k k k k . (10.8.1)
Bild 10-13. Wellenzahl- vektoren bei Reflexion einer Elektronenwelle an Netzebenen.
Wir setzen elastische Streuung voraus, d. h. die Beträge der beiden Wellen-zahlvektoren sind gleich:
1
λ= =0k k , (10.8.2)
d. h.

Grundlagen genauer erklärt (etwas mehr Mathematik) 259
nd ⋅ − =0k k . (10.8.3)
Wir ersetzen d durch a1 und n durch die ganze Zahl h, berücksichtigen, dass a1 und k – k0 parallel sind und erhalten
( ) h⋅ − =1 0a k k . (10.8.4a)
Analog gilt für die beiden anderen Raumrichtungen
( )
( )
k
l
⋅ − =
⋅ − =
2 0
3 0
a k k
a k k . (10.8.4b)
Die Gleichungen (10.8.4) werden als Laue-Gleichungen bezeichnet. Aus Gleichung (10.8.1) ist ersichtlich, dass die Länge des Streuvektors und da-
mit der Beugungswinkel θ proportional zum reziproken Wert des Netzebenenab-standes d sind. Es ist deshalb zweckmäßig bei Berechnungen zur Beugung zur Be-schreibung der Atomanordnung im Kristall ein „reziprokes Gitter“ zu benutzen. Seine Basisvektoren b1, b2 und b3 sind definiert durch
1 wenn (i, j = 1,2,3)
0
i j
i j
=⋅ =
≠i ja b . (10.8.5)
Damit können wir schreiben:
( )
( )
( )
2 3
2 3
2 3
h k l h
h k l k
h k l l
⋅ ⋅ + ⋅ + ⋅ =
⋅ ⋅ + ⋅ + ⋅ =
⋅ ⋅ + ⋅ + ⋅ =
1 1
2 1
3 1
a b b b
a b b b
a b b b
(10.8.6)
Nach Vergleich mit den Laue-Gleichungen (10.8.4) folgt
( )2 3h k l⋅ + ⋅ + ⋅ = −1 0b b b k k , (10.8.7)
d. h. der Streuvektor muss ein reziproker Gittervektor sein, wenn ein Beugungs-maximum entstehen soll. Der durch (hkl) charakterisierte reziproke Gittervektor steht damit stets senkrecht auf den Netzebenen (hkl). Wir wollen nun ein Gedan-kenexperiment ausführen: Wir verschieben den Vektor k0 parallel bis seine Spitze einen reziproken Gitterpunkt trifft und überlegen, welche Richtung der Vektor k mit gleicher Länge haben muss, damit er ebenfalls einen (anderen) reziproken Gitterpunkt trifft. Die geometrische Lösung dieses Problems ist eine Kugelschale,

Kapitel 10 260
deren Mittelpunkt der Ursprung des Vektors k0 ist (vgl. Bild 10-14). Das Verfah-ren heißt Ewald61-Konstruktion.
Bild 10-14. Ewald-Konstruktion. a) Perspektivische Darstellung der Kugelschale. b) Ansicht der Ebene, die die Wellenzahlvektoren k und k0 enthält.
Wir haben damit eine Vorschrift zur Konstruktion des Beugungsbildes:
Wir zeichnen den Wellenzahlvektor k0 der einfallenden Welle (Länge: 1/λ) so, dass dessen Ende (Pfeil) auf einen reziproken Gitterpunkt trifft. Wenn die Kugel um den Ursprung dieses Vektors mit dem Radius 1/λ einen reziproken Gitterpunkt trifft, kennzeichnet dies einen Beugungsreflex.
Bei der Elektronenbeugung im Transmissionselektronenmikroskop liegen die Wellenlängen in der Größenordnung 3 pm, 1/λ beträgt dann etwa 0,3 pm-1. Die Atomabstände im Kristallgitter liegen typischerweise bei etwa 0,3 nm, der Kehr-wert ist dann ungefähr 3 nm-1. Das Verhältnis vom Radius der Ewald-Kugel zu den Abständen im reziproken Gitter beträgt etwa 100, d. h. wir können den Teil der Kugeloberfläche um den Auftreffpunkt des Vektors k0 auf das reziproke Gitter als Ebene annähern.
Wir sehen, dass das reziproke Gitter von grundlegender Bedeutung für die In-terpretation von Beugungsmustern ist. Wie können wir es aus dem „realen“ Gitter berechnen?
Ausgangspunkt ist die Definition (10.8.5) des reziproken Gitters. Das Skalar-produkt zweier Vektoren ist Null, wenn beide senkrecht aufeinander stehen und wird maximal, wenn beide in die gleiche Richtung zeigen. Wir erkennen, dass
2 2 3 3|| , || und ||1 1b a b a b a (10.8.8)
sowie
61 Paul Peter Ewald, deutscher Physiker, 1888 – 1985

Grundlagen genauer erklärt (etwas mehr Mathematik) 261
, , , , , ⊥ ⊥ ⊥ ⊥ ⊥ ⊥1 2 1 3 2 1 2 3 3 1 3 2b a b a b a b a b a b a (10.8.9)
gelten muss. Das Kreuzprodukt (auch: Vektorprodukt) zweier Vektoren steht senkrecht auf der von den beiden Vektoren aufgespannten Ebene. Damit lassen sich die Forderungen (10.8.9) erfüllen; wir müssen noch Normierungsgrößen V1, V2 und V3 berücksichtigen, damit sich beim Skalarprodukt maximal 1 ergibt wie in der Definition des reziproken Gitters gefordert:
( )
( )
( )
1
2
3
1
1
1
V
V
V
= ⋅
= ⋅
= ⋅
1 2 3
2 3 1
3 1 2
b a ×a
b a ×a
b a ×a
. (10.8.10)
Zur Berechnung von V1, V2 und V3 setzen wir die Gleichungen (10.8.10) in die Definitionsgleichung (10.8.5) ein und erhalten:
( )( )
( )( )
( )( )
11
22
33
1
1
1
VV
VV
VV
⋅⋅ = = = ⋅
⋅⋅ = = = ⋅
⋅⋅ = = = ⋅
1 2 31 1 1 2 3
2 3 12 2 2 3 1
3 1 23 3 3 1 2
a a ×aa b a a ×a
a a ×aa b a a ×a
a a ×aa b a a ×a
. (10.8.11)
Da a1, a2, a3 nach Voraussetzung ein Rechtssystem bilden, ändert die zyklische Vertauschung der Vektoren in (10.8.11) nichts am Ergebnis, d. h. es ist V1 = V2 = V3 = V. V ist das Volumen der durch die drei Vektoren a1, a2 und a3 aufgespannten Elementarzelle.
Im rechtwinkligen (orthogonalen) Koordinatensystem ist die Rechnung trivial, wir erhalten V = a1⋅a2⋅a3, das Produkt der Beträge der drei Vektoren a1, a2 und a3. Die Basisvektoren des reziproken Gitters sind in diesem Fall
1 2 3
, und a a a
= = = 31 21 2 3
aa ab b b (10.8.12)
Im nicht-orthogonalen Fall wird es etwas komplizierter. Wir wollen ein beliebiges Koordinatensystem a1, a2, a3 (triklines Gitter) in ein orthogonales mit den Ein-heitsvektoren ex, ey, ez transferieren und die Rechnung damit wieder auf den einfa-chen Fall zurückführen.

Kapitel 10 262
Bild 10-15. Triklines und kartesisches Achsenkreuz. Die x-Achse stimmt mit der a1-Achse überein, die x-y-Ebene liegt in der a1-a2-Ebene. a) Kartesische Koordi-naten der a2-Achse. b) Karte-sische Koordinaten der a3-Achse.
Aus Bild 10-15a lesen wir ab:
1a= ⋅1 xa e (10.8.13)
und
( )2 cos sina γ γ= ⋅ ⋅ + ⋅2 x ya e e . (10.8.14)
Zur Berechnung der kartesischen Koordinaten der a3-Achse führen wir die beiden Winkel ρ und ε ein (vgl. Bild 10-15b). Die Achsen ez und a3 liegen in der Ebene OBCD. In dieser Ebene wird auch der Winkel ε zwischen der x-y-Ebene und der a3-Achse gemessen. Die Schnittline der OBCD-Ebene mit der x-y-Ebene ist um den Winkel ρ gegen die x-Achse gedreht. Für die Strecke OA (x-Komponente von a3) im rechtwinkligen Dreieck OAC gilt
3 cosOA a β= ⋅ . (10.8.15)
Im Dreieck OAB gilt für die Strecke AB (y-Komponente von a3):
3 cos sinAB a ε ρ= ⋅ ⋅ . (10.8.16)
Die z-Komponente von a3 ist die Strecke BC . Im Dreieck OBC lesen wir
3 sinBC a ε= ⋅ (10.8.17)
ab. Wir müssen noch die beiden Winkel ε und ρ bestimmen. Dazu nutzen wir aus, dass wir die Strecke OA sowohl im Dreieck OAC – wie in (10.8.15) geschehen – als auch im Dreieck OAB berechnen können:
3 cos cosOA a ε ρ= ⋅ ⋅ , (10.8.18)
d. h.

Grundlagen genauer erklärt (etwas mehr Mathematik) 263
cos cos cosβ ε ρ= ⋅ . (10.8.19)
Bezugsachse war a1. Wenn wir in gleicher Weise mit Bezug auf a2 verfahren, müssen wir den Winkel β (zwischen a1- und a3-Achse) durch den Winkel α (zwi-schen a2- und a3-Achse) sowie den Winkel ρ (zwischen a1-Achse und Schnittlinie von OBCD- und x-y-Ebene) durch den Winkel γ-ρ (zwischen a2-Achse und Schnittlinie von OBCD- und x-y-Ebene) ersetzen, d. h.
( )cos cos cosα ε γ ρ= ⋅ − . (10.8.20)
Damit haben wir zwei Bestimmungsgleichungen für die Winkel ε und ρ. Mit
( )cos cos cos sin sinγ ρ γ ρ γ ρ− = ⋅ + ⋅ (10.8.21)
und (10.8.20) gilt:
( )cos cos cos cos cos sin sincos cos
cos cos cos
cos cos cos cos sin tan
γ ρ β γ ρ β γ ρα β
ρ ρ ρ
α β γ β γ ρ
− ⋅ ⋅ ⋅ ⋅= ⋅ = +
= ⋅ + ⋅ ⋅
(10.8.22)
bzw. cos
coscos
tansin
αγ
βρ
γ
−
= . (10.8.23)
Unter Berücksichtigung von
2
1cos
1 tanρ
ρ=
+
(10.8.24)
folgt mit (10.8.19):
2
2
22 2
2
coscos
coscoscos cos 1
cos sin
cos cos coscos sin 2 cos cos
sin coscos
αγ
ββε β
ρ γ
β α αε γ γ γ
γ ββ
−
= = +
= ⋅ + − ⋅ ⋅ +
(10.8.25)
sowie

Kapitel 10 264
( )2 2sin cos 1γ γ+ = :
2 2cos 2cos cos cos coscos =
sin
α α β γ βε
γ
− ⋅ ⋅ + . (10.8.26)
Außerdem gilt
( )2 2 2 2cos sin = cos 1 cos = cos cosε ρ ε ρ ε β⋅ ⋅ − − . (10.8.27)
Wir können die Vorschrift für die Transformation in das orthogonale System zu-sammenfassen:
( )
( )
1
2
2 23
2 2
= a
= a cos + sin
= a cos + cos cos + sin
cos 2cos cos cos cos mit cos =
sin
γ γ
β ε β ε
α α β γ βε
γ
⋅
⋅ ⋅ ⋅
⋅ ⋅ ⋅ − ⋅
− ⋅ ⋅ +
1 x
2 x y
3 x y z
a e
a e e
a e e e (10.8.28)
und haben damit die Voraussetzungen für die Berechnung der reziproken Gitter-basisvektoren b1, b2, b3 nach Gleichung (10.8.10) geschaffen. Für die Kreuzpro-dukte gilt nunmehr:
( )
2 2
2 23 3 3
2 3 2 3
2 22 3
cos sin 0
cos cos cos sin
sin sin cos sin
cos cos cos sin cos
a a
a a a
a a a a
a a
γ γ
β ε β ε
γ ε γ ε
γ ε β γ β
= ⋅ ⋅
⋅ ⋅ − ⋅
= ⋅ ⋅ ⋅ ⋅ − ⋅ ⋅ ⋅ ⋅
+ ⋅ ⋅ ⋅ − − ⋅
x y z
2 3
2 3 x y
z
e e e
a ×a
a ×a e e
e
, (10.8.29)
2 23 3 3
1
cos cos cos sin
0 0
a a a
a
β ε β ε= ⋅ ⋅ − ⋅
x y z
3 1
e e e
a ×a

Grundlagen genauer erklärt (etwas mehr Mathematik) 265
2 21 3 1 3sin cos cosa a a aε ε β= ⋅ ⋅ ⋅ − ⋅ ⋅ ⋅ −3 1 y za ×a e e (10.8.30)
1
2 2
1 2
0 0
cos sin 0
sin
a
a a
a a
γ γ
γ
=
⋅ ⋅
= ⋅ ⋅ ⋅
x y z
1 2
1 2 z
e e e
a ×a
a ×a e
. (10.8.31)
Für das Volumen der Elementarzelle folgt damit
1 2 3=a a a sin sinV ε γ⋅ ⋅ ⋅ ⋅ (10.8.32)
und für die reziproken Gitterbasisvektoren:
2 2
1
cot cos cos cos1 = - cot
a sin
γ ε β βγ
ε
⋅ − −⋅ ⋅ + ⋅1 x y zb e e e (10.8.33)
2 2
2
3
cos cos1 -
sin sin
= a sin
a
ε β
γ ε
ε
−= ⋅ ⋅
⋅
⋅
2 y z
z3
b e e
eb
. (10.8.33a)
Diesen Formalismus fassen wir in einer Transformationsmatrix zusammen:
2 2
1 1 1
2 2
2 2
3
cot cos cos cos1 cot
sin
cos cos10
sin sin sin
10 0
a sin
a a a
a a
γ ε β βγ
ε
ε β
γ γ ε
ε
⋅ − −−
⋅
− −= ⋅
⋅ ⋅ ⋅
⋅
1 x
2 y
3 z
b e
b e
b e
(10.8.33b)

Kapitel 10 266
bzw.
1 1 1
2 2
3
0
0 0
x y z
y z
z
b b b
b b
b
= ⋅
1 x
2 y
3 z
b e
b e
b e
. (10.8.33c)
Damit können wir beliebige reziproke Gittervektoren ausrechnen. Wir erinnern uns an die Gleichungen (10.8.1) und (10.8.2), setzen n = 1 und erhalten
1 1 bzw. d
d− = =
−0
0
k kk k
. (10.8.1a)
Mit (10.8.7) folgt daraus
hkl2 3
1
h k ld =
⋅ + ⋅ + ⋅1b b b , (10.8.34)
eine allgemeine Formel zur Berechnung von Netzebenenabständen. Aufgrund der Achsen- und Winkelvorgaben bei den verschiedenen Kristallsystemen vereinfa-chen sich die Elemente b1x, b2x, b3x, b2y, b2z, b3z der Transformationsmatrix in un-terschiedlicher Weise (vgl. Tabelle 10-1). Die Ergebnisse für die Netzebenenab-stände sind in der Tabelle 5-4 im Kapitel 5 aufgelistet.
kubisch: a1 = a2 = a3
α = β = γ = 90°
ε = 90°
1
1
1
10 0
10 0
10 0
a
a
a
tetragonal: a1 = a2 ≠ a3
α = β = γ = 90°
ε = 90°
1
1
3
10 0
10 0
10 0
a
a
a
ortho- rhombisch:
a1 ≠ a2 ≠ a3
α = β = γ = 90°
ε = 90°
1
2
3
10 0
10 0
10 0
a
a
a

Grundlagen genauer erklärt (etwas mehr Mathematik) 267
hexagonal: a1 = a2 ≠ a3
α = β = 90°
γ = 120°
ε = 90°
1 1
1
3
1 10
3
20 0
3
10 0
a
a a
a
⋅
⋅
rhombo- edrisch:
a1 = a2 = a3
α = β = γ ≠ 90° 1 1 1 2
2
1 1
1
1 cot cos
2 cos sin
cot tan10
sin sin
10 0
a sin
a a a
a a
α
α
α α
ε
α
α ε
ε
− −
⋅ ⋅ ⋅
− ⋅
⋅ ⋅
⋅
mit ( )cos cot 2 1 cosε α α= ⋅ ⋅ −
monoklin: a1 ≠ a2 ≠ a3
α = γ = 90°
β beliebig
ε = β
1 1
2
3
1 cot0
10 0
10 0
a sin
a a
a
β
β
−
⋅
triklin: a1 ≠ a2 ≠ a3
α ≠ β ≠ γ
2 2
1 1 1
2 2
2 2
3
2 2
cot cos cos cos1 cot
sin
cos cos10
sin sin sin
10 0
a sin
cos 2 cos cos cos cossowie cos
sin
a a a
a a
γ ε β βγ
ε
ε β
γ γ ε
ε
α α β γ βε
γ
⋅ − −−
⋅
− −
⋅ ⋅ ⋅
⋅
− ⋅ ⋅ ⋅ +=
Tabelle 10-1. Transformationsmatrizen für die Berechnung von orthogonalen reziproken Gitter-vektoren.
Der Winkel zwischen zwei punktförmigen Beugungsreflexen (d. h. zwischen zwei reziproken Gittervektoren B1, B2) ist gleich dem Winkel zwischen den für die bei-den Beugungsreflexe verantwortlichen Netzebenenscharen. Wir können diesen Winkel mit Hilfe des Skalarproduktes berechnen:
( )cos ,⋅
=⋅
1 21 2
1 2
B BB B
B B . (10.8.35)

Kapitel 10 268
Für die beiden reziproken Gittervektoren gilt:
1 1 1
2 2 2
h k l
h k l
= ⋅ + ⋅ + ⋅
= ⋅ + ⋅ + ⋅
1 1,1 2,1 3,1
2 1,2 2,2 3,2
B b b b
B b b b . (10.8.36)
Für die Berechnung des Skalarproduktes und der Beträge der beiden reziproken Gittervektoren benutzen wir die Transformation (10.8.33) in das orthogonale Sy-stem. Allgemein gilt:
( ) ( )( ) ( )
21 2 1 1 1 1 2 2 1 2 2
1 1 1 2 1 3 2 1 2 2 2 3
= h h h k h k
h k l h k l
x y y y y
z z z z z z
b b b b b
b b b b b b
⋅ + + ⋅ +
+ + + ⋅ + +
1 2B B (10.8.37)
sowie
( ) ( ) ( )
( ) ( ) ( )
22 21 1 1 1 1 2 1 1 1 2 1 3
22 22 1 2 1 2 2 2 1 2 2 2 3
= h h k h k l
h h k h k l
x y y z z z
x y y z z z
b b b b b b
b b b b b b
⋅ + + + + +
⋅ + + + + +
1 2B B . (10.8.38)
Wegen der festen Vorgaben bzgl. Achsen und Winkeln vereinfachen sich die For-meln teilweise (vgl. Tabelle 10-1). Diese vereinfachten Formeln sind in Tabelle 5-9 zusammengestellt.
Das Punktmuster von einem einzelnen Kristall hängt nicht nur von der Kristall-struktur ab sondern auch von der Einfallsrichtung der Elektronenwellen in das Kristallgitter. Wegen der sehr kleinen Beugungswinkel bei der Elektronenbeugung im Transmissionselektronenmikroskop stimmen Einfallsrichtung und Zonenachse (d.i. die Schnittlinie der beugenden Netzebenen) faktisch überein. Wir wollen ab-schließend überlegen, wie wir die Zonenachse bei Kenntnis von zwei indizierten Reflexen berechnen können. Die reziproken Gittervektoren stehen senkrecht auf den beugenden Netzebenen, d. h. das Kreuzprodukt von zwei reziproken Gitter-vektoren liegt in beiden verantwortlichen Netzebenen, ist also die Schnittlinie beider Ebenen (vgl. Bild 10-16).
Für den Vektor Z, der die Richtung der Zonenachse angibt, gilt demzufolge
= ×1 2Z B B . (10.8.39)
Wir benutzen wieder die Transformation (10.8.33) in das orthogonale Achsensy-stem und erhalten mit

Grundlagen genauer erklärt (etwas mehr Mathematik) 269
Bild 10-16. Skizze zur Erläuterung der Berech- nung der Zonenachse.
( ) ( )
( ) ( )
1 1 1 1 1 2 1 1 1 2 1 3
2 1 2 1 2 2 2 1 2 2 2 3
= h + h k + h k l
= h + h k + h k l
x y y z z z
x y y z z z
b b b b b b
b b b b b b
⋅ ⋅ ⋅ + ⋅ ⋅ + ⋅ + ⋅
⋅ ⋅ ⋅ + ⋅ ⋅ + ⋅ + ⋅
1 x y z
2 x y z
B e e e
B e e e . (10.8.40)
unter Berücksichtigung von
1
1 2
1 2 3
=
=
=
x
y y
z z z
b
b b
b b b
⋅
⋅ + ⋅
⋅ + ⋅ + ⋅
x 1
y 1 2
z 1 2 3
e a
e a a
e a a a
(10.8.41)
für das Kreuzprodukt
( ) ( )
( ) ( )
( ) ( )
( ) ( )
( )
1 1y 2 1 2 3
1 1 1 1 1 2 1 1 1 2 1 3
2 1 2 1 2 2 2 1 2 2 2 3
1 2 2 1 2 1 1 21 2 3
1 2 2 1
b +
= h h k h k l
h h k h k l
k l k l h l h l =
h k h k
x y z z z
x y y z z z
x y y z z z
x y z
b b b b b
b b b b b b
b b b b b b
b b b
+ +
× + + +
+ + +
⋅ − ⋅ ⋅ + ⋅ − ⋅ ⋅ +
+ ⋅ − ⋅ ⋅
1 2 2 1 2 3
1 2
1 2
3
a a a a a a
B B
a a
a
. (10.8.42)
Wir geben die Zonenachse als Richtung im Kristallachsensystem a1, a2, a3 an. Gemeinsame Vielfache der Komponenten, wie das Produkt b1x·b2y·b3z, werden nicht berücksichtigt:
( ) ( ) ( )1 2 2 1 2 1 1 2 1 2 2 1k l k l h l h l h k h k= ⋅ − ⋅ ⋅ + ⋅ − ⋅ ⋅ + ⋅ − ⋅ ⋅1 2 3Z a a a . (10.8.43)

Kapitel 10 270
10.9 Kinematisches Modell: Gitterfaktor und Strukturfaktor Nach dem Braggschen Gesetz können wir mit Hilfe des reziproken Gittermodells die Positionen der Beugungsreflexe ermitteln („geometrische Theorie“). Zur Berechnung der Reflexintensitäten müssen wir das Modell erweitern: Wir berück-sichtigen nunmehr die Überlagerung aller am Kristallgitter gestreuten Wellen, ver-nachlässigen aber weiterhin Wechselwirkungen zwischen den Elektronenwellen und dem Kristallgitter, die über den einfachen Streuprozess hinausgehen (z. B. Veränderungen der Wellenlänge im Kristall) und Wechselwirkungen der gestreu-ten Welle mit der ungestreuten Welle. Die Intensitäten der Streuwellen sind klein gegen die Intensität der einfallenden Welle, deren Intensitätsänderung wird ver-nachlässigt. Diese Vorstellung wird als „kinematisches Modell“ bezeichnet. In der Praxis gilt diese Näherung nur für sehr dünne Proben (Dicke: wenige 10 nm).
Im Ergebnis der Überlagerung erhalten wir eine Summe von Elektronenwellen (Amplituden fj), die gegenüber der einfallenden Welle (Amplitude A0) um χj phasenverschoben sind:
( )i 2 i2 i0 j 0 j
j j
e e ej jA f A fχ χ− ⋅ ⋅ + − ⋅− ⋅ ⋅
Ψ = ⋅ ⋅ = ⋅ ⋅ ⋅0 0k r k r . (10.9.1)
Die Phasenverschiebung rührt vom Gangunterschied Δs her, den die gestreuten Elektronenwellen erfahren haben. Ein Gangunterschied von λ entspricht einer Phasenschiebung von 2π. Allgemein gilt:
j j2
sχλ
⋅= ⋅Δ (10.9.2)
Bei kleinen Streuvektoren k – k0 gilt nach (5.1), (5.4), (5.5) und (10.8.4):
( )js λΔ = ⋅ − ⋅gj 0r k k (10.9.3)
bzw. mit (10.9.2):
( )j 2χ = ⋅ ⋅ −gj 0r k k (10.9.4)
und mit (10.9.1):
( )2 i2 i0 j
j
e eA f− ⋅ ⋅ −− ⋅ ⋅
Ψ = ⋅ ⋅ ⋅ gj 00r k kk r (10.9.5)

Grundlagen genauer erklärt (etwas mehr Mathematik) 271
Da wir im kinematischen Modell die Wechselwirkung mit der einfallenden Welle vernachlässigen, können wir den ersten Term normieren und gleich 1 setzen:
( )2 ij
j
ef− ⋅ ⋅ −
Ψ = ⋅ gj 0r k k . (10.9.6)
Den Abstandsvektor rgj zwischen den Streuzentren teilen wir in zwei Summanden auf: In einen Translationsvektor rg, der die Position der Elementarzelle im Gitter angibt, und in einen Vektor, der vom Ursprung (0,0,0) der Elementarzelle zum Atom j mit den Koordinaten xj, yj, zj innerhalb der Elementarzelle zeigt:
j j jx y z= ⋅ + ⋅ + ⋅j 1 2 3r a a a . (10.9.7)
Unter diesen Voraussetzungen wird aus (10.9.6):
( ) ( ) ( ) ( )2 i 2 i 2 ij j
j j
e e ef f− ⋅ ⋅ + ⋅ − − ⋅ ⋅ ⋅ − − ⋅ ⋅ ⋅ −
Ψ = ⋅ = ⋅ ⋅g j 0 g 0 j 0r r k k r k k r k k (10.9.8)
Die erste Term wird als Gitterfaktor bezeichnet. Wir müssen noch berücksichti-gen, dass jede Elementarzelle ihren Beitrag zum Gitterfaktor leistet. Mit M als Zahl der zum Beugungsmuster beitragenden Elementarzellen gilt:
( ) ( )2 i
M
eG − ⋅ ⋅ ⋅ −− = g 0r k k
0k k . (10.9.9)
Der zweite Term heißt Strukturfaktor:
( ) ( )2 ij
j
eF f − ⋅ ⋅ ⋅ −− = ⋅ j 0r k k
0k k . (10.9.10)
Das Produkt ihrer Quadrate beschreibt die Intensitätsverteilung I(k-k0) im Beu-gungsmuster:
( ) ( ) ( ) ( ) ( )2 22 2I F G F G− − ⋅ − = − ⋅ −0 0 0 0 0k k k k k k k k k k . (10.9.11)
Im Strukturfaktor wird berücksichtigt, welchen Einfluss die Atomanordnung in-nerhalb der Elementarzelle auf die Intensität der Beugungsreflexe hat. Ist er gleich Null, so spricht man von „ausgelöschten Reflexen“. Da für einen Beugungsreflex der Streuvektor k-k0 ein Vektor des reziproken Gitters ist, folgt aus den Glei-chungen (10.8.7) und (10.9.7):

Kapitel 10 272
( ) ( ) ( )j j j 2 3
j j j
h k l
h k l
x y z
x y z
⋅ − = ⋅ + ⋅ + ⋅ ⋅ ⋅ + ⋅ + ⋅
= ⋅ + ⋅ + ⋅
j 0 1 2 3 1r k k a a a b b b (10.9.12)
und für den Strukturfaktor
( ) ( )j j j2 i h k lhkl j
j
ex y z
F F f− ⋅ ⋅ ⋅ + ⋅ + ⋅
− = = ⋅0k k . (10.9.13)
In Abschnitt 5.2 hatten wir die Atomanordnung bei Kochsalz (NaCl) erläutert. Sie ist in Tabelle 10-2 zusammengestellt. Insgesamt enthält die Elementarzelle acht Atome.
Na-Atome:
(0,0,0), (0,½,½) (½,0, ½) (½, ½,0)
Cl-Atome:
(½,½,½), (½,0,0), (0,½,0)
(0,0, ½).
Tabelle 10-2. Anordnung der Atome in einer NaCl-Elementarzelle. Für den Strukturfaktor gilt:
( ) ( )
( ) ( )
( ) ( )
( ) ( )
1 12 2
1 1 1 12 2 2 2
1 1 1 12 2 2 2
1 12 2
2 i h 0 k l2 i h 0 k 0 l 0
2 i h k 0 l 2 i h k l 0
2 i h k l 2 i h k 0 l 0
2 i h 0 k l 0 2 i h 0 k 0 l
e e
e e
e e
e e
hkl Na
Cl
F f
f
− ⋅ ⋅ ⋅ + ⋅ + ⋅− ⋅ ⋅ ⋅ + ⋅ + ⋅
− ⋅ ⋅ ⋅ + ⋅ + ⋅ − ⋅ ⋅ ⋅ + ⋅ + ⋅
− ⋅ ⋅ ⋅ + ⋅ + ⋅ − ⋅ ⋅ ⋅ + ⋅ + ⋅
− ⋅ ⋅ ⋅ + ⋅ + ⋅ − ⋅ ⋅ ⋅ + ⋅ + ⋅
+ += ⋅
+ +
+ ++ ⋅
+ +
(10.9.14)
( ) ( ) ( )( )( )( )
i k l i h l i h k
i h k l i h i k i l
1 e e
e e e e
hkl Na
Cl
F f e
f
− ⋅ ⋅ + − ⋅ ⋅ + − ⋅ ⋅ +
− ⋅ ⋅ + + − ⋅ ⋅ − ⋅ ⋅ − ⋅ ⋅
= ⋅ + + +
+ ⋅ + + +
(10.9.15)
Für den Na-Anteil beziehen wir uns auf den Abschnitt 5.4.2, wo die Auslö-schungsregeln für das flächenzentrierte Gitter am Beispiel von Gold abgeleitet wurden. Danach müssen die h, k, l alle entweder geradzahlig oder ungeradzahlig

Grundlagen genauer erklärt (etwas mehr Mathematik) 273
sein, damit der Reflex nicht ausgelöscht wird. Wir ergänzen den Cl-Einfluss und erhalten Tabelle 10-3.
h k l Na-Anteil Cl-Anteil Summe
(10.9.15)
geradzahlig geradzahlig geradzahlig 1+1+1+1 1+1+1+1 4⋅(fNa+ fCl)
geradzahlig geradzahlig ungeradzahlig 1-1-1+1 -1+1+1-1 0
geradzahlig ungeradzahlig geradzahlig 1-1+1-1 -1+1-1+1 0
ungeradzahlig geradzahlig geradzahlig 1+1-1-1 -1-1+1+1 0
geradzahlig ungeradzahlig ungeradzahlig 1+1-1-1 1+1-1-1 0
ungeradzahlig geradzahlig ungeradzahlig 1-1-1+1 1-1+1-1 0
ungeradzahlig ungeradzahlig geradzahlig 1-1-1+1 1-1-1+1 0
ungeradzahlig ungeradzahlig ungeradzahlig 1+1+1+1 -1-1-1-1 4⋅(fNa- fCl)
Tabelle 10-3. Strukturfaktoren für NaCl.
Im Vergleich zum einfachen flächenzentrierten Gitter (d. h. nur eine Atomsorte) erkennen wir einen zusätzlichen Unterschied im Strukturfaktor für Reflexe mit geradzahligen und ungeradzahligen Indizes. Voraussetzung für die Berechnung der Beugungsintensitäten ist die Kenntnis des Strukturfaktors, der von der Atom-sorte, der Packung der Atome innerhalb der Elementarzelle und von der beugen-den Netzebenenschar (h, k, l) abhängt.
Als Nächstes wollen wir uns mit dem Gitterfaktor befassen. Die Ewald-Kon-struktion besagt, dass für einen Beugungsreflex der Streuvektor k-k0 ein Vektor b des reziproken Gitters ist. Wir fragen uns nun, welche Toleranz u dabei zulässig ist. Dazu stellen wir uns vor, dass das reziproke Gitter kein „Punktgitter“ ist, son-dern die Gitterpunkte zu kleinen Ellipsoiden entartet sind (vgl. Bild 10-17). Damit wäre der Betrag u der Toleranz gleich der Höhe eines solchen Ellipsoids.
Bild 10-17. „Entartetes“ reziprokes Gitter mit Kennzeichnung der Toleranz u für die Beugungsbedingung für ein Beugungsmaximum.
Aus Bild 10-17 ist ersichtlich:
− = +0k k b u , (10.9.16)

Kapitel 10 274
wobei b ein Vektor im reziproken Gitter ist. Für den Gitterfaktor (10.9.9) gilt damit
( )2 i 2 i 2 i
M M
e e eG − ⋅ ⋅ ⋅ + − ⋅ ⋅ ⋅ − ⋅ ⋅ ⋅= = ⋅g g gr b u r b r u
. (10.9.17)
Da rg ein Vektor des realen Gitters und b ein Vektor des reziproken Gitters ist, er-gibt das Skalarprodukt aus beiden eine ganze Zahl. Damit wird
2 ie 1− ⋅ ⋅ ⋅=gr b
(10.9.18)
und (10.9.17) zu
2 i
M
eG− ⋅ ⋅ ⋅
= gr u. (10.9.19)
Wir berücksichtigen die drei Raumrichtungen und nennen die Zahl der Elementar-zellen in den drei Richtungen M1, M2 und M3. Die Gesamtzahl ist dann M = M1⋅M2⋅M3 und
1 2 3
2 i
M M M
eG − ⋅ ⋅ ⋅= gr u
. (10.9.20)
Da der Vektor u im reziproken Gitter auftritt, können wir ihn als Linearkom-bination der reziproken Gitterbasisvektoren darstellen:
1 2 3u u u= ⋅ + ⋅ + ⋅1 2 3u b b b . (10.9.21)
Für den Vektor im realen Gitter gilt analog:
m n o= ⋅ + ⋅ + ⋅g 1 2 3r a a a (10.9.22)
und für den Gitterfaktor (10.9.20) mit Festlegung des Bezugspunktes in der Mitte des Kristalls:
( )31 2
1 2 3
1 2 3
M /2M /2 M /22 i
M /2 M /2 M /2
e m u n u o uG − ⋅ ⋅ ⋅ + ⋅ + ⋅
− − −
= . (10.9.23)
Zur analytischen Ausführung dieser Summation stellen wir uns eine einzelne Ele-mentarzelle infinitesimal klein im Vergleich zum gesamten Kristallgitter vor. Eine

Grundlagen genauer erklärt (etwas mehr Mathematik) 275
einzelne Zelle mit dem Volumen dV erbringt dann einen Beitrag dG zum gesam-ten Gitterfaktor, wobei der durch m, n und o charakterisierte Ort der Elementarzel-le im Kristall zu berücksichtigen ist. Mit VEZ als Volumen einer Elementarzelle gilt dann:
( )
( )
1 2 3
1 2 3
2 i
2 i
1 2 3
e
eM M M
m u n u o u
Kristall
m u n u o u
EZ
dVdG
V
dm dn do
V
− ⋅ ⋅ ⋅ + ⋅ + ⋅
− ⋅ ⋅ ⋅ + ⋅ + ⋅
= ⋅
⋅ ⋅= ⋅
⋅ ⋅ ⋅
. (10.9.24)
Unter diesen Voraussetzungen wird die Summe (10.9.23) zu
( )31 2
1 2 3
1 2 3
M / 2M / 2 M / 22 i
M / 2 M / 2 M / 2
1 2 3
e
M M M
m u n u o u
EZ
dm dn do
GV
− ⋅ ⋅ ⋅ + ⋅ + ⋅
− − −
⋅ ⋅ ⋅
=⋅ ⋅ ⋅
. (10.9.25)
Wir schreiben die Exponentialfunktion in anderer Weise und trennen dann die Variablen:
( )
( )
( )
1
1
1
2
2
2
3
3
3
M / 22 i
1 M / 2
M / 22 i
2 M / 2
M / 22 i
3 M / 2
1 1e
M
1e
M
1e
M
m u
EZ
n u
o u
G dmV
dn
do
− ⋅ ⋅ ⋅
−
− ⋅ ⋅ ⋅
−
− ⋅ ⋅ ⋅
−
= ⋅ ⋅
⋅ ⋅
⋅
. (10.9.26)
Zur Vereinfachung betrachten wir eine orthogonale Elementarzelle mit VEZ = a1⋅a2⋅a3 und schreiben für (10.9.26)
1 2 3 G G G G= ⋅ ⋅ (10.9.27)
mit

Kapitel 10 276
( )1
1
1
M / 22 i
11 1 M / 2
1e
Mm uG dm
a− ⋅ ⋅ ⋅
−
=⋅
, (10.9.27a)
( )2
2
2
M / 22 i
22 2 M / 2
1e
Mn uG dn
a− ⋅ ⋅ ⋅
−
=⋅
, (10.9.27b)
( )3
3
3
M / 22 i
33 3 M / 2
1e
Mo uG do
a− ⋅ ⋅ ⋅
−
=⋅
. (10.9.27c)
Die Auswertung des Integrals für G1 liefert:
1
1
1
1
1
1
1 1 1 1
M / 22 i
11 1 M / 2
M2 i 2
M1 1 12
i M i M1
1 1 1
1e
M
1 e
M 2 i
1e e
2 i M
u m
u m
u u
G dma
a u
Ga u
− ⋅ ⋅ ⋅
−
− ⋅ ⋅ ⋅
−
− ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
=⋅
=⋅ − ⋅ ⋅
= − +⋅ ⋅ ⋅ ⋅
. (10.9.28)
Wir erinnern uns an die Eulersche Formel (5.18):
( ) ( )
( ) ( )
( )
1 1 1 1 11 1 1
1 1 1 1
1 11
1 1 1
1cos M i sin M
2 i M
cos M i sin M
sin M
M
G u ua u
u u
uG
a u
π
= − ⋅ ⋅ + ⋅ ⋅ ⋅ +⋅ ⋅ ⋅ ⋅
+ ⋅ ⋅ + ⋅ ⋅ ⋅
⋅ ⋅=
⋅ ⋅ ⋅
. (10.9.29)
Analog gilt:

Grundlagen genauer erklärt (etwas mehr Mathematik) 277
( )
( )
2 22
2 2 2
3 33
3 3 3
sin M
M
sin M
M
uG
a u
uG
a u
⋅ ⋅=
⋅ ⋅ ⋅
⋅ ⋅=
⋅ ⋅ ⋅
(10.9.30)
Welche Konsequenz hat dieses Ergebnis für die Beugungspraxis? Wir erinnern uns: Die Intensität I des Beugungsreflexes ist proportional zum Quadrat des Git-terfaktors, d. h.
( )
( )
( )
( )
( )
( )
( )
( )
( )
( )
( )
( )
2 2 21 2 3
2 2 21 1 2 2 3 3
2 2 2 2 2 21 2 3 1 1 2 2 3 3
2 2 21 1 2 2 3 3
2 2 2 21 1 2 2 3 3
sin M sin M sin M1
M M M
sin M sin M sin M1
M M MEZ
I G G G
u u uI
a a a u u u
u u uI
V u u u
⋅ ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅⋅ ⋅ ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅⋅ ⋅ ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅
. (10.9.31)
Da u1, u2 und u3 Komponenten im reziproken Raum sind, beschreibt (10.9.31) die Intensitätsverteilung im Beugungsbild. Wir sehen, dass dabei eine Funktion der Form
( )2
sin xF x
x= (10.9.32)
eine wesentliche Rolle spielt und wollen uns den Graphen dieser Funktion in Bild 10-18 anschauen.
Bild 10-18. Graph der Funktion
( )2
sin xF x
x=
zur Diskussion der Intensitätsverteilung der Beugungsreflexe.

Kapitel 10 278
Die dem Hauptmaximum benachbarten Nullstellen dieser Funktion liegen offen-bar bei x = ±π. Wenn wir die Ausdehnungen der Ellipsoide unseres entarteten re-ziproken Gitters durch diese Nullstellen definieren, sind die Komponenten unserer Toleranz u damit:
1 2 31 2 3
1 1 1, und
M M Mu u u= = = , (10.9.33)
d. h. die Ellipsoide sind umso kleiner, je größer die Zahl der Elementarzellen ist bzw. je größer der Kristall ist. Die Toleranz wird auch als Anregungsfehler bei der Beugung bezeichnet.
Ist die Zahl der Elementarzellen in einer Raumrichtung besonders klein (bei-spielsweise bei plättchenförmigen Kristallen), so sind die Ellipsoide in dieser Richtung besonders lang, d. h. auch die Beugungsreflexe sind strichartig („streaks“). Aus der Form der Beugungsreflexe lassen sich u .U. also Rückschlüs-se auf die Kristallform ziehen.
10.10 Debye-Streuung Um Elektronenstreukurven von amorphen Substanzen auszuwerten, wird in der Regel ein Formalismus benutzt, der auf P. Debye62 zurückgeht. Das ursprüngliche Ziel war die Auswertung von Streukurven nach Durchgang von Licht durch kol-loidale Lösungen [10.3]. Von Debye-Streuung wird gesprochen, wenn die Teil-chen größer als 1/20 der Wellenlänge der gestreuten Wellen sind. Dies ist für Ato-me und Elektronenwellen im Transmissionselektronenmikroskop der Fall.
Die Streukurven sind das Ergebnis der Überlagerung von Elektronenwellen, die an einer Atomversammlung bzw. an einer Ansammlung anderer N Streuzentren gestreut werden. Für die Intensität I in Abhängigkeit von der Streurichtung (reprä-sentiert durch den Wellenzahlvektor k) gilt:
( ) ( ) ( )N N2 2 i
m nm 1 n 1
mit eI f fψ ψ− − ⋅
= =
= ⋅ ⋅ 0 mnk k rk k (10.10.1)
(fm, fn: Streuquerschnitte (Atomformamplituden) der Streuzentren m bzw. n, k, k0: Wellenzahlvektoren der gestreuten bzw. der einfallenden Welle rmn: Abstandsvektor zwischen den Streuzentren m und n (vgl. Bild 10-19)).
Für den Betrag des Streuvektors s gilt (θ << 1 vorausgesetzt):
s kθ
θ θλ
= = ⋅ = ⋅ =s k . (10.10.2)
62 Peter Debye, niederländischer Physiker, 1884 – 1966, Nobelpreis für Chemie 1936

Grundlagen genauer erklärt (etwas mehr Mathematik) 279
Bild 10-19. Streuung einer Welle an zwei Streuzentren m und n.
Für die Richtungen der rmn wird Isotropie vorausgesetzt, d. h. alle Richtungen treten mit gleicher Wahrscheinlichkeit auf. Demnach befindet sich das Atom n mit gleicher Wahrscheinlichkeit am Ort (1) oder (2) oder an jedem anderen Ort auf der Kugeloberfläche mit dem Radius rmn (vgl. Bild 10-20a). Daraus lesen wir ab:
cosmns r γ⋅ = ⋅ ⋅mns r . (10.10.3)
Wir überlegen nun, welche Radiusrichtungen den gleichen Winkel γ mit einem (bzgl. Länge und Richtung) festgehaltenem s haben. Diese erbringen den gleichen Beitrag zum Skalarprodukt (10.10.3). Diese gleichberechtigten Richtungen bilden den Raumwinkel dΩ (s. Bild 10-20b):
Bild 10-20. Skizzen zur Erläuterung der Rechnung. a) Isotrope Richtungs-verteilung. b) Gleichberechtigte Radiusrichtungen.
mn mn2 2
mn mn
2 sin2 sin
r r ddfd d
r r
γ γγ γ
⋅ ⋅ ⋅ ⋅ ⋅Ω = = = ⋅ ⋅ ⋅ . (10.10.4)
Die Atome im Raumwinkelsegment dΩ mit dem Abstand rmn vom Bezugsatom im Zentrum erbringen den Beitrag
( ) mn2 i cosmn m n, e s rd
d r s f f γψ − ⋅ ⋅ ⋅ ⋅Ω= ⋅ ⋅ ⋅
Ω (10.10.5)
zur Streuwellenfunktion, d. h. für den Beitrag aller Atome im Abstand rmn zur Streuwelle gilt unter Berücksichtigung von (10.10.4):

Kapitel 10 280
( ) mn2 i cosmn m n
0
2, e sins rr s f f d
πγψ γ γ− ⋅ ⋅ ⋅ ⋅⋅
= ⋅ ⋅ ⋅ ⋅ ⋅Ω
. (10.10.6)
Zur Lösung dieses Integrals substituieren wir
mn
mn
mn
2 i cos
2 i sin
2 i sin
x s r
dxs r
d
dxd
s r
γ
γγ
γγ
= − ⋅ ⋅ ⋅ ⋅ ⋅
= ⋅ ⋅ ⋅ ⋅ ⋅
=⋅ ⋅ ⋅ ⋅ ⋅
(10.10.7)
und erhalten mit Berücksichtigung von Ω = 4π:
( )1
0
m nmn
mn
, e4 i
xx
x
f fr s dx
s rψ
⋅= ⋅ ⋅
⋅ ⋅ ⋅ ⋅ . (10.10.8)
Die Integrationsgrenzen lauten:
( )
( )
0 mn
1 mn
0 2 i ,
2 i .
x x s r
x x s r
γ
γ π
= = = − ⋅ ⋅ ⋅ ⋅
= = = ⋅ ⋅ ⋅ ⋅ (10.10.9)
Aus (10.10.8) wird damit
( )
( ) ( )
( ) ( )
( )( )
mn mn2 i 2 im nmn
mn
m nmn mn
mn
mn mn
mnmn m n
mn
, e e4 i
cos 2 i sin 24 i
cos 2 i sin 2
sin 2, .
2
s r s rf fr s
s r
f fs r s r
s r
s r s r
s rr s f f
s r
ψ
ψ
⋅ ⋅ ⋅ ⋅ − ⋅ ⋅ ⋅ ⋅⋅= ⋅ −
⋅ ⋅ ⋅ ⋅
⋅= ⋅ ⋅ ⋅ ⋅ + ⋅ ⋅ ⋅ ⋅ −
⋅ ⋅ ⋅ ⋅
− ⋅ ⋅ ⋅ + ⋅ ⋅ ⋅ ⋅
⋅ ⋅ ⋅= ⋅ ⋅
⋅ ⋅ ⋅
(10.10.10)
Um die gesamte Streuwelle zu erhalten, müssen wir noch über alle Abstände rmn summieren:

Grundlagen genauer erklärt (etwas mehr Mathematik) 281
( )( )mn
m nm 1 n 1 mn
sin 2
2
N N s rs f f
s rψ
= =
⋅ ⋅ ⋅= ⋅ ⋅
⋅ ⋅ ⋅ . (10.10.11)
Die Intensität der Streukurve ist gleich dem Quadrat der Streuwellenfunktion. Sie kann mit (10.10.11) bei Kenntnis der Abstandsverteilung rmn berechnet werden. Bei praktischen Messungen wollen wir allerdings die Abstandsverteilung aus der gemessenen Streukurve berechnen.
Nach unserem Modell vom amorphen Zustand sind die Atome richtungsisotrop angeordnet. Ihre (gesuchte) radiale Dichteverteilung sei ρ(r). In einer Kugelschale mit dem Radius r und der Dicke dr sind damit
( ) ( )24N r r r drρ= ⋅ ⋅ ⋅ ⋅ (10.10.12)
Atome enthalten. Wir vereinfachen die Gleichung (10.10.11), indem wir den Ein-fluss der Atomformamplituden in den Untergrundanteil der Streukurve schieben. Die Strukturinformation steckt in dem Quotienten mit dem Sinus-Term. Des Weiteren betrachten wir als Intensität der Streukurve den einfachen Betrag der Summe aller Streuwellen. Damit folgt für die Streukurve:
( ) ( )( )
0
sin 22
s rI s r r dr
sρ
∞ ⋅ ⋅ ⋅= ⋅ ⋅ ⋅ ⋅ . (10.10.13)
In der Streukurve sind die Streuwellenbeiträge aller Atomabstände überlagert. Mathematisch ist demzufolge eine Entfaltung erforderlich. Dazu nutzen wir das Fourier-Integral für ungerade Funktionen:
( ) ( ) ( ) ( )0 0
2sin sinf x u x du f t u t dt
∞ ∞
= ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ . (10.10.14)
Darin setzen wir für die Variablen
und 2x t r u s= = = ⋅ ⋅ (10.10.15)
sowie für die Funktionen
( ) ( ) ( )2f x f t r rρ= = ⋅ ⋅ (10.10.16)
und erhalten damit aus (10.10.14)

Kapitel 10 282
( ) ( ) ( ) ( )
( ) ( ) ( )( )
0 0
0 0
22 sin 2 2 2 sin 2
sin 22 sin 2 2
r r s r ds r r s r dr
s rr r s r ds s r r dr
s
ρ ρπ
ρ ρ
∞ ∞
∞ ∞
⋅ ⋅ = ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
⋅ ⋅ ⋅⋅ = ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
(10.10.17)
sowie nach Vergleich mit (10.10.13)
( ) ( ) ( )0
2 sin 2r r I s s s r dsρ∞
⋅ = ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ (10.10.18)
bzw.
( ) ( ) ( )0
2sin 2r I s s s r ds
rρ
∞
= ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ . (10.10.19)
Damit ist es möglich, aus einer gemessenen, untergrundkorrigierten Streukurve die (isotrope) Verteilung der Atomabstände zu bestimmen, wie dies an einem Beispiel in Abschnitt 5.6 gezeigt wurde.
10.11 Elektronen im Feld einer Zentralkraft Wir wollen überlegen, wovon die Ablenkung der Elektronen im Kraftfeld eines Atomkerns abhängt. Benachbarte Atome lassen wir unberücksichtigt. Dabei gehen wir von einer elastischen Wechselwirkung aus, d. h. der Betrag der Geschwindig-keit und damit die Energie des Elektrons ändern sich dabei nicht. Wir wollen uns diese elastische Wechselwirkung anhand von Bild 10-21a veranschaulichen.
Das Elektron bewegt sich auf einer geradlinigen Bahn, die den (senkrechten) Abstand a vom Kernmittelpunkt hat, auf den Atomkern zu. Der aktuelle Abstand zwischen Elektron und Kern sei r, der Winkel zwischen dem Vektor r und der Verbindungsgeraden zwischen Kernmittelpunkt und Scheitelpunkt der Bahnkurve des Elektrons sei ϕ. Elektron und Atomkern sind elektrisch geladen, das Elektron trägt die (negative) Elementarladung -e, der Kern die (positive) Ladung Z⋅e mit Z als Ordnungszahl im Periodensystem der Elemente. Beide ziehen sich mit der Coulomb-Kraft
2
20
1 Z e
4 r
⋅= ⋅
⋅CF (10.11.1)

Grundlagen genauer erklärt (etwas mehr Mathematik) 283
(ε0: Influenzkonstante) an. Wir vernachlässigen dabei die partielle Abschirmung der Kernladung durch die Elektronenhülle. Für die Ablenkung ist die Komponente FS verantwortlich:
Bild 10-21. a) Elastische Ablenkung eines Elektrons im Kraftfeld eines Atomkerns. b) Veränderung des Impulses bei elastischer Wechselwirkung zwischen Elektron und Atomkern.
2
20
1 Z ecos cos
4 rϕ ϕ
⋅= ⋅ = ⋅ ⋅
⋅S CF F (10.11.2)
Diese Kraftkomponente führt zu einer Richtungsumkehr der Komponente pS des Impulses des Elektrons (vgl. Bild 10-21b). Die Kraftkomponente FW kehrt sich nach Durchgang des Elektrons durch den Scheitelpunkt um; in der Summe hebt sich ihre Wirkung zwischen den Elektronenpositionen 1 und 2 auf. Der Betrag der Impulsänderung von 1 nach 2 ist:
2 2 sin 2 sin2 2
m vθ θ
= ⋅ = ⋅ ⋅ = ⋅ ⋅ ⋅Sp p p (10.11.3)
(m: Elektronenmasse, v: Elektronengeschwindigkeit). Für infinitesimal kleine Än-derungen gilt:

Kapitel 10 284
d dm m d= ⋅ + ⋅p v v (10.11.4)
bzw. in nichtrelativistischer Näherung (Elektronenmasse konstant):
d m d= ⋅p v . (10.11.5)
Diese Impulsänderung wird durch die Kraft F bewirkt:
bzw. d
m d dtdt m
= ⋅ = ⋅v F
F v . (10.11.6)
Für die Impulsänderung gilt damit:
2
1
bzw. d dt dt= ⋅ Δ = ⋅p F p F (10.11.7)
und für das Elektron:
2
20 0
Z e cos2 sin
2 4m v dt
r
θ ϕ∞
⋅⋅ ⋅ ⋅ = ⋅
⋅ . (10.11.8)
Bei der Bewegung im Zentralkraftfeld treten keine äußeren Drehmomente auf, es gilt der Drehimpulssatz:
const= × =L r p . (10.11.9)
Der Betrag des Drehimpulses ist
2 dm r
dt
ϕ= ⋅ ⋅L (10.11.10)
und, verglichen mit dem des Elektrons am Bahnanfang,
2 da m v m r
dt
ϕ⋅ ⋅ = ⋅ ⋅ . (10.11.11)
Damit können wir in (10.11.8) dt eliminieren:
2rdt d
a vϕ= ⋅
⋅ (10.11.12)

Grundlagen genauer erklärt (etwas mehr Mathematik) 285
und erhalten
( )
( ) 22
0 2
Z e cossin
2 8m v d
a v
θ
θ
θ ϕϕ
+
− +
⋅⋅ ⋅ = ⋅
⋅ ⋅ . (10.11.13)
Integration und Umformung liefert:
( )( ) ( )( )
( )( )
2
20
2
20
2
20
Z esin sin 2 sin 2
2 8
Z esin 2 sin 2
2 8
Z esin cos
2 24
m v a
m v a
m v a
θθ θ
θθ
θ θ
⋅= + − − +
⋅ ⋅ ⋅ ⋅
⋅= ⋅ ⋅ +
⋅ ⋅ ⋅ ⋅
⋅= ⋅
⋅ ⋅ ⋅ ⋅
. (10.11.14)
Damit folgt für den Ablenkwinkel θ:
2
20
Z etan
2 4 m v a
θ ⋅=
⋅ ⋅ ⋅ ⋅ . (10.11.15)
Problematisch ist die Abhängigkeit vom Parameter a, dem Bahnabstand vom Kern. Die Elektronenbahnen haben beliebige Abstände. Wir greifen deshalb einen Abstand a heraus und stellen uns dazu einen kreisförmigen Ring mit dem Radius a und der Breite da vor (s. Bild 10-22).
Bild 10-22. Elektronen bewegen sich durch einen infinitesimal dünnen Ring im Abstand a von der Achse, auf der das Atom liegt, auf das streuende Atom zu.
Die Fläche dieses Ringes ist

Kapitel 10 286
2d a daσ = ⋅ ⋅ ⋅ . (10.11.16)
Elektronen, die durch diese Fläche treten, werden in das Raumwinkelintervall dΩ abgelenkt. Dieses Raumwinkelintervall wird aus dem Ablenkwinkel θ berechnet (vgl. 10.10.4):
2 sind dθ θΩ = ⋅ ⋅ ⋅ (10.11.17)
und wir erhalten nach Division:
sin
d a da
d d
σ
θ θ= ⋅
Ω , (10.11.18)
was als differentieller Wirkungsquerschnitt der Streuung bezeichnet wird. Er be-schreibt die „Stärke der Streuung“ und ist ein Maß für die Wahrscheinlichkeit, mit der ein Elektron in θ-Richtung gestreut wird.
Wir berücksichtigen (10.11.15):
2
20
Z e
4 tan sin2
d da
d dm v
σ
θ θθ
⋅= ⋅
Ω⋅ ⋅ ⋅ ⋅ ⋅
, (10.11.19)
2 2
2 220 0
Z e 1 Z e 1
4 4tan 2 sin2 2
da d
d dm v m vθ θθ θ
⋅ ⋅ −= ⋅ = ⋅
⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
. (10.11.20)
Für den Betrag gilt:
22
220
22
230
Z e 1
4 tan sin 2 sin2 2
cosZ e 24 2 sin sin
2
d
d m v
m v
σ
θ θθ
θ
θθ
⋅= ⋅
Ω ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
⋅= ⋅
⋅ ⋅ ⋅ ⋅ ⋅
. (10.11.21)
Mit der bekannten trigonometrischen Relation
( )sin 2 2 sin cosα α α⋅ = ⋅ ⋅ (10.11.22)
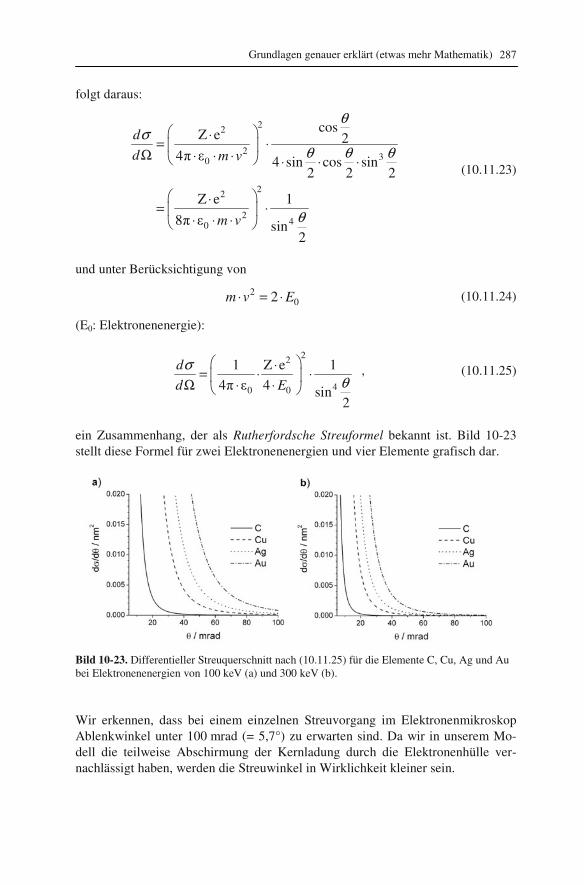
Grundlagen genauer erklärt (etwas mehr Mathematik) 287
folgt daraus:
22
230
22
240
cosZ e 24 4 sin cos sin
2 2 2
Z e 1
8 sin2
d
d m v
m v
θσ
θ θ θ
θ
⋅= ⋅
Ω ⋅ ⋅ ⋅ ⋅ ⋅ ⋅
⋅= ⋅
⋅ ⋅ ⋅
(10.11.23)
und unter Berücksichtigung von
202m v E⋅ = ⋅ (10.11.24)
(E0: Elektronenenergie):
22
40 0
1 Z e 1
4 4 sin2
d
d E
σ
θ
⋅= ⋅ ⋅
Ω ⋅ ⋅ , (10.11.25)
ein Zusammenhang, der als Rutherfordsche Streuformel bekannt ist. Bild 10-23 stellt diese Formel für zwei Elektronenenergien und vier Elemente grafisch dar.
Bild 10-23. Differentieller Streuquerschnitt nach (10.11.25) für die Elemente C, Cu, Ag und Au bei Elektronenenergien von 100 keV (a) und 300 keV (b).
Wir erkennen, dass bei einem einzelnen Streuvorgang im Elektronenmikroskop Ablenkwinkel unter 100 mrad (= 5,7°) zu erwarten sind. Da wir in unserem Mo-dell die teilweise Abschirmung der Kernladung durch die Elektronenhülle ver-nachlässigt haben, werden die Streuwinkel in Wirklichkeit kleiner sein.

Kapitel 10 288
10.12 Mittlere freie Weglänge für elastische Streuung Nachdem wir wissen, mit welcher Wahrscheinlichkeit ein Elektron in ein be-stimmtes Raumwinkelelement gestreut wird, wollen wir ausrechnen, wie groß die Wahrscheinlichkeit σ(α) dafür ist, dass ein Elektron in einen beliebigen Winkel θ ≥ α gestreut wird. Dazu berücksichtigen wir (10.11.25) und integrieren:
( )22
40 0
Z e 2 sin
16 sin2
dE
α
θσ α θ
θ
⋅ ⋅ ⋅= ⋅ ⋅
⋅ ⋅ ⋅ (10.12.1)
(Z: Ordnungszahl, e: Elementarladung, ε0: Influenzkonstante, E0: Elektronenener-gie). Mit (10.11.22) folgt:
( )22
2 24
0 0 2
222
30 0 2
22
20 0 2
22
20 0 2
22
0 0
2 sin cosZ e2
16 sin
cosZ e
8 sin
Z e 1
8 sin
Z e 11
8 sin
1Z e
8
dE
dE
E
E
E
θ θ
θα
θ
θα
θ
α
α
σ α θ
θ
⋅ ⋅⋅= ⋅ ⋅ ⋅ ⋅
⋅ ⋅ ⋅
⋅= ⋅ ⋅ ⋅
⋅ ⋅ ⋅
⋅= ⋅ ⋅ −
⋅ ⋅ ⋅
⋅= ⋅ ⋅ − +
⋅ ⋅ ⋅
⋅= ⋅
⋅ ⋅
22
22
sin
sin
α
α
−
⋅
(10.12.2)
bzw.
( )22
20 0 2
Z e 1
8 tanE ασ α
⋅= ⋅
⋅ ⋅ ⋅ . (10.12.3)
Was passiert, wenn die Elektronen auf mehrere Atome treffen? Wir stellen uns eine dünne Schicht mit der Querschnittsfläche A und der Dicke ds vor. Diese Schicht enthält N Atome:
ANN A dsρ= ⋅ ⋅ ⋅ (10.12.4)

Grundlagen genauer erklärt (etwas mehr Mathematik) 289
(NA: Avogadro-Konstante, ρ: Dichte). In diese Schicht fallen pro Zeit und Flä-cheneinheit NE Elektronen ein. Davon werden beim Durchgang durch die Schicht dNE Elektronen in einen Winkel ≥ α (Akzeptanzwinkel) gestreut (s. Bild 10-24).
Bild 10-24. Bilanz der Elektronenzahl bei Streuung in einer dünnen Schicht.
Dadurch ändert sich die Zahl der in einen Winkel < α gestreuten Elektronen um
( ) ( )Ad NE E E
NN N N ds
Aσ α ρ σ α= − ⋅ ⋅ = − ⋅ ⋅ ⋅ ⋅ . (10.12.5)
Diese Differentialgleichung wird durch Trennung der Variablen gelöst:
( )
( )A
A
N1 2
dN
e
E
E
sE
Nds
N
N C C ρ σ α
ρ σ α
− ⋅ ⋅ ⋅
= − ⋅ ⋅ ⋅
= + ⋅
. (10.12.6)
Die Integrationskonstanten C1 und C2 bestimmen wir aus den Randbedingungen:
( ) ( ),00 und 0E E EN s N N s= = →∞ = (10.12.7)
und erhalten
( )AN,0 e s
E EN N ρ σ α− ⋅ ⋅ ⋅= ⋅ . (10.12.8)
Wir setzen
2 20 0 2
2A
tan8
Z e Nel
E α
ρ
⋅⋅ ⋅Λ = ⋅
⋅ ⋅ (10.12.9)
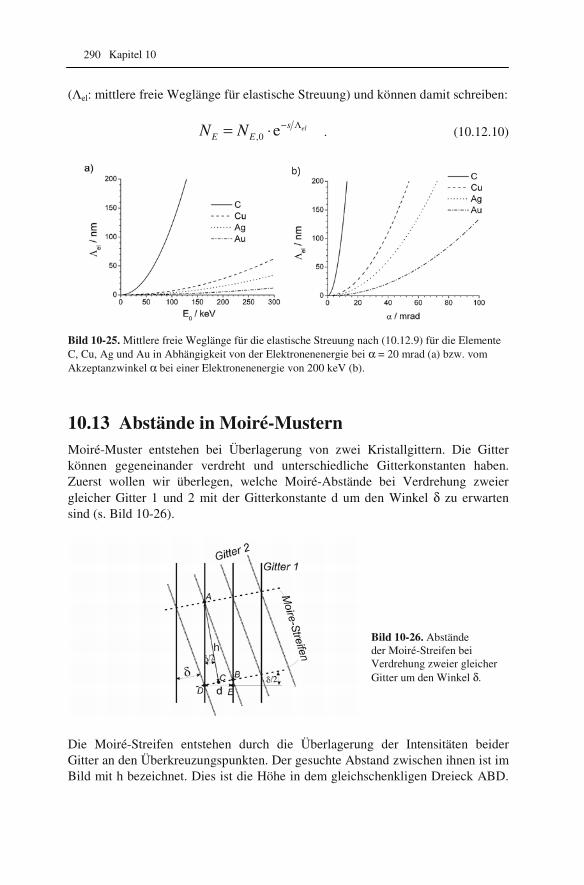
Kapitel 10 290
(Λel: mittlere freie Weglänge für elastische Streuung) und können damit schreiben:
,0 e elsE EN N − Λ= ⋅ . (10.12.10)
Bild 10-25. Mittlere freie Weglänge für die elastische Streuung nach (10.12.9) für die Elemente C, Cu, Ag und Au in Abhängigkeit von der Elektronenenergie bei α = 20 mrad (a) bzw. vom Akzeptanzwinkel α bei einer Elektronenenergie von 200 keV (b).
10.13 Abstände in Moiré-Mustern Moiré-Muster entstehen bei Überlagerung von zwei Kristallgittern. Die Gitter können gegeneinander verdreht und unterschiedliche Gitterkonstanten haben. Zuerst wollen wir überlegen, welche Moiré-Abstände bei Verdrehung zweier gleicher Gitter 1 und 2 mit der Gitterkonstante d um den Winkel δ zu erwarten sind (s. Bild 10-26).
Bild 10-26. Abstände der Moiré-Streifen bei Verdrehung zweier gleicher Gitter um den Winkel δ.
Die Moiré-Streifen entstehen durch die Überlagerung der Intensitäten beider Gitter an den Überkreuzungspunkten. Der gesuchte Abstand zwischen ihnen ist im Bild mit h bezeichnet. Dies ist die Höhe in dem gleichschenkligen Dreieck ABD.

Grundlagen genauer erklärt (etwas mehr Mathematik) 291
Diese Höhe ist die Ankathete im rechtwinkligen Dreieck ACD, die Strecke von C nach D ist die Gegenkathete:
( )2
CD
tanh
δ= . (10.13.1)
Andererseits gilt im Dreieck BDE für die Strecke zwischen B und D:
( )2
BD 2 CDcos
dδ
= ⋅ = (10.13.2)
und damit für den gesuchten Abstand h:
( ) ( ) ( )2 2 2
d
2 tan cos 2 sin
dh
δ δ δ= =
⋅ ⋅ ⋅ . (10.13.3)
Für kleine Verdrehungen (δ << 1) gilt demnach
dh
δ= . (10.13.4)
Als Nächstes überlegen wir, welcher Streifenabstand sich bei unverdrehter Über-lagerung zweier Gitter mit unterschiedlichen Gitterkonstanten d1 bzw. d2 ergibt. Maßgebend ist die Dichte der Linien in der Überlagerung, deshalb ist die Lo-kalisierung des Streifenabstandes nicht so einfach möglich wie bei den verdrehten Gittern. Zur Berechnung der Liniendichte nehmen wir eine sinusförmige Hellig-keitsverteilung in den Gitterbildern an. Die Gitterkonstante gibt die Periode der Sinusfunktion vor, wir schreiben für die Helligkeitsverteilung der beiden Gitter mit der auf eins normierten Amplitude:
1 21 2
sin 2 bzw. sin 2x x
H Hd d
= ⋅ ⋅ = ⋅ ⋅ . (10.13.5)
Die reziproken Werte der Gitterkonstanten haben die Bedeutung von Raumfre-quenzen, ein Begriff, den wir in Kapitel 7 kennengelernt hatten. Für die Über-lagerung gilt:
1 21 2
sin 2 sin 2x x
H H Hd d
= + = ⋅ ⋅ + ⋅ ⋅ . (10.13.6)

Kapitel 10 292
Unter Berücksichtigung des Additionstheorems für die Summe zweier Sinus-funktionen gilt:
1 2 1 2
1 1 1 12 sin cosH x x
d d d d= ⋅ ⋅ ⋅ + ⋅ ⋅ ⋅ − , (10.13.7)
d. h. wir erhalten eine Überlagerung von einer hohen und einer niedrigen Raum-frequenz (vgl. Bild 10-27). Dieser Sachverhalt wird in der Elektrotechnik als Schwebung bezeichnet. Die niedrige Raumfrequenz in der Kosinusfunktion kenn-zeichnet den Abstand h der Moiré-Streifen:
1 2
1 2 2 1
1 1 1 bzw.
d dh
h d d d d
⋅= − =
− . (10.13.8)
Im Bildbeispiel sind zwei Gitter mit den Konstanten d1 = 1/11 nm ≈ 0,091 nm und d2 = 1/10 nm = 0,1 nm überlagert. Das Ergebnis ist eine Helligkeitsmodulation (Moiré-Streifen) im Abstand von 1 nm.
Bild 10-27. Moiré-Streifen bei verdrehungsfreier Überlagerung zweier Gitter mit den Gitterkon-stanten d1 = 1/11 nm und d2 = 1/10 nm.
Zur Behandlung des allgemeinen Falls mit gedrehten Gittern und unterschied-lichen Gitterkonstanten ergänzen wir unser „Raumfrequenz-Modell“ um die Rich-tung, d. h. wir betrachten die beiden Raumfrequenzen, die die Periodizitäten der beiden Gitter kennzeichnen, als Vektoren r1 und r2. Die Raumfrequenz, die den Abstand der Moiré-Streifen beschreibt, sei rh. Offenbar gilt allgemein:
= −h 1 2r r r . (10.13.9)
Der Abstand der Moiré-Streifen ist dann
1h =
hr . (10.13.10)

Grundlagen genauer erklärt (etwas mehr Mathematik) 293
Bild 10-28. Bilanz der Raumfrequenzen bei verdrehten Gittern mit unterschiedlichen Gitterkonstanten.
Aus der Skizze in Bild 10-28 folgt unter Verwendung des Kosinussatzes für den Betrag des Vektors rh:
2 2 2
2
2 2 21 21 2
12 cos
1 1 1 2 cosh
d dh d d
δ
δ
= = + − ⋅ ⋅ ⋅
⋅= + −
⋅
h 1 2 1 2r r r r r . (10.13.11)
Daraus folgt für den Abstand der Moiré-Streifen:
1 2
2 21 2 1 22 cos
d dh
d d d d δ
⋅=
+ − ⋅ ⋅ ⋅
. (10.13.12)
Die beiden vorher hergeleiteten Formeln sind Spezialfälle von (10.13.12). Für die Verdrehung zweier gleicher Gitter ist d1 = d2 = d und damit
( ) ( )
2
2 122 1 cos2 1 cos 2 sin
2
d d dh
d δδδ= = =
⋅ −⋅ − ⋅
, (10.13.13)
d. h. identisch mit (10.13.3). Für die verdrehungsfreie Überlagerung zweier ungleicher Gitter ist δ = 0 und damit
( )
1 2 1 2 1 2
2 2 21 21 2 1 2 1 2
2
d d d d d dh
d dd d d d d d
⋅ ⋅ ⋅= = =
−+ − ⋅ ⋅ −
, (10.13.14)
d. h. identisch mit (10.13.8).
10.14 Kontrastübertragungsfunktion Im Abschnitt 7.2 hatten wir durch qualitative Überlegungen gezeigt, dass es mög-lich sein muss, die für die Umwandlung des Phasenkontrastes in einen Amplitu-denkontrast notwendige Phasenschiebung durch die Objektivlinse zu erreichen.

Kapitel 10 294
Wir erinnern uns: Die Phasenschiebung φ sollte demnach abhängen von der Struk-turgröße d, der Öffnungsfehlerkonstanten CS und einer im Vergleich zur exakten Fokussierung in der Gaußschen Bildebene vorgenommenen Brennweitenänderung Δf:
1, ,SC f
dφ φ= Δ . (7.14)
Im Folgenden wollen wir versuchen, diesen Zusammenhang quantitativ zu ver-stehen. Bild 10-29 soll die Rolle von Öffnungsfehler und Defokussierung ver-anschaulichen.
Bild 10-29. Einfluss von Öffnungsfehler (a) und Defokussierung (b) auf den Strahlengang (s. auch [10.4]).
Die Abkürzungen in Bild 10-29 haben folgende Bedeutung: P: Dingpunkt, P’ Bildpunkt bei fehlerfreier Abbildung, P’’ Bildpunkt bei fehlerbehafteter Abbil-dung, θ: Öffnungswinkel des in die Linse einfallenden Strahls, θ’: Öffnungswin-kel des gebrochenen Strahls, R: Abstand des Strahls von der optischen Achse beim Einfall in die Linse, F: dingseitiger Brennpunkt, f: Brennweite, Δf: Änderung der

Grundlagen genauer erklärt (etwas mehr Mathematik) 295
Brennweite bei Defokussierung, g: Dingweite, b: Bildweite, Δb: Änderung der Bildweite bei Defokussierung, CS: Öffnungsfehlerkonstante und M: Vergrößerung.
Die Skizze 10-29a zeigt, dass der Öffnungsfehler zu einer Abweichung εö des Öffnungswinkels der gebrochenen Strahlen führt. Es gilt:
3S
Ö
C Mr
b b
θε
⋅ ⋅Δ= = (10.14.1)
sowie mit
, (bei hoher Vergrößerung) und R b
g f Mg g
θ = ≈ = : (10.14.2)
3 3
3 4S
SÖ
C R b RC
g g b fε
⋅ ⋅= =
⋅ ⋅ . (10.14.3)
Aus der Skizze 10-29b folgt für den Einfluss einer Defokussierung (Brennweiten-änderung Δf) ohne Berücksichtigung der Richtung der Winkelveränderung:
'
2f
b b R b Rx
b b b b b
θε
Δ ⋅ Δ ⋅ Δ ⋅Δ= = = =
⋅ . (10.14.4)
Aus der Abbildungsgleichung
1 1 1 bzw.
1 1 11 ... 1 ...
f f g b b
f b
f f g b b
= ++ Δ + Δ
Δ Δ− + = + − +
(10.14.5)
folgt
2 2
2 2
1 1 1
1 1 1bzw. mit :
f b
f f g b b
f b
f g b f b
Δ Δ− = + −
Δ Δ= + =
(10.14.6)
sowie für die Winkeländerung
2f
f R
fε
Δ ⋅= . (10.14.7)

Kapitel 10 296
Betrachten wir nun die Auswirkungen der Gesamtwinkeländerung
fÖε ε ε= −
auf die Phasenschiebung
( ) 2s
φ θλ
Δ= ⋅ (10.14.8)
mit Δs als durch die fehlerhafte Abbildung hervorgerufenen Gangunterschied zwi-schen den Wellenfronten und λ als Elektronenwellenlänge. Zur Berechnung von Δs schauen wir auf Bild 10-30.
Bild 10-30. Zur Herleitung des Gangunterschiedes bei fehlerbehafteter Abbildung.
Die Änderung des Öffnungswinkels θ um einen infinitesimal kleinen Betrag dθ führt zu einem Gangunterschied
ds dRε= ⋅ . (10.14.9)
Der gesamte Gangunterschied Δs wird durch Integration über alle ds berechnet:
( )0 0
R R
fÖs ds dR dRε ε εΔ = = ⋅ = − ⋅ . (10.14.10)
Nach Einsetzen der o.g. Zusammenhänge (10.14.3), (10.14.7) und (10.14.8) folgt damit für die durch die Linse verursachte Phasendifferenz zwischen durchgehen-der und gebeugter Welle:

Grundlagen genauer erklärt (etwas mehr Mathematik) 297
3
4 20
4 2
4 2
2( ) 2 ( )
2
R
S
S
s R RR C f dR
f f
C R Rf
f f
φλ λ
λ
⋅
Δ= ⋅ = −Δ ⋅ ⋅
= ⋅ − Δ ⋅
. (10.14.11)
Mit Berücksichtigung von
und R R
qg f d
λθ θ λ= ≈ = = ⋅ (Braggsches Gesetz)
folgt daraus:
4 2( )2
SCfφ θ θ θ
λ= ⋅ − Δ ⋅ (10.14.12)
bzw.
( )3 4 2( ) 22 Sq C q f qφ λ λ= ⋅ ⋅ − ⋅Δ ⋅ ⋅ . (10.14.13)
Dies ist die gesuchte Funktion (7.13). Die Einfallswelle in das Objekt sei eine ebene Welle der Form
( ) 2 ieEin Aψ − ⋅ ⋅= ⋅ k rr . (10.2.27)
Beim Durchgang durch das Kristallgitter erfährt sie eine ortsabhängige Phasen-schiebung (vgl. Abschnitt 7.1)
( ) ( ), ,B
tx y x y
Uϕ
λ
⋅= ⋅Φ
⋅ , (7.11)
d. h. die Objektaustrittswelle hat die Form
( ) ( )( ) ( )i 2 . i ,2 ie e ex y x y
Aus A Aϕ ϕ
ψ− ⋅ ⋅ + − ⋅− ⋅ ⋅= ⋅ = ⋅ ⋅
k r k rr . (10.14.14)
Vorausgesetzt, dass die Phasenmodulation durch das Kristallgitter klein gegen 1 ist, gilt:

Kapitel 10 298
( ) ( )i ,e 1 i ,x y x yϕϕ
− ⋅≈ − ⋅ (10.14.15)
und damit
( ) ( )( )2 ie 1 i ,Aus A x yψ ϕ− ⋅ ⋅= ⋅ ⋅ − ⋅k rr . (10.14.16)
Diese Welle wird mit einer ebenen Welle überlagert, die durch die Linse um φ(q) (s. Formel (10.14.13)) phasenverschoben ist. Im Bild entsteht die Wellenfunktion
( ) ( ) ( )( )i 2e q
Bild Aus Aφ
ψ ψ− ⋅ ⋅ +
= + ⋅k rr r (10.14.17)
und mit (10.14.16)
( ) ( )( ) ( )
( ) ( ) ( ) ( )( )
i2 i 2 i
2 i
e 1 i , e e
e 1 cos i , sin
qBild
Bild
A x y A
A q x y q
φψ ϕ
ψ φ ϕ φ
− ⋅− ⋅ ⋅ − ⋅ ⋅
− ⋅ ⋅
= ⋅ ⋅ − ⋅ + ⋅ ⋅
= ⋅ ⋅ + − ⋅ +
k r k r
k r
r
r . (10.14.18)
Die Intensität I(x,y) im Bild ist gleich der Wellenfunktion, multipliziert mit ihrer Konjugierten:
( )
( ) ( ) ( ) ( )( )
( ) ( ) ( )( )
*
i
i
.
. e 1 cos i , sin
e 1 cos i , sin
Bild BildI x y
I x y A q x y q
A q x y q
ψ ψ
φ ϕ φ
φ ϕ φ
− ⋅ ⋅
+ ⋅ ⋅
= ⋅
= ⋅ ⋅ + − ⋅ + ⋅
⋅ ⋅ ⋅ + + ⋅ +
k r
k r
(10.14.19a)
( ) ( )( ) ( )( ) ( ) ( )( )
( )( ) ( ) ( )( ) ( ) ( )( )
( ) ( )( ) ( ) ( )( )
22
2
2 22
. 1 cos 1 cos i , sin
1 cos i , sin , sin
. 1 cos , sin
I x y A q q x y q
q x y q x y q
I x y A q x y q
φ φ ϕ φ
φ ϕ φ ϕ φ
φ ϕ φ
= ⋅ + + + ⋅ ⋅ + −
− + ⋅ ⋅ + + +
= ⋅ + + +
(10.14.19b)
Im quadratischen Term mit der Kosinusfunktion von φ(q) wird die konstante Zahl 1 addiert, d. h. dieser Teil trägt nicht zur Helligkeitsmodulation im Bild bei, er ist Bestandteil des (konstanten) Bilduntergrundes. Demgegenüber ist im zweiten qua-dratischen Term die ortsabhängige Phasenmodulation ϕ(x,y) mit der Sinusfunk-tion von φ(q) kombiniert, d. h. die Funktion

Grundlagen genauer erklärt (etwas mehr Mathematik) 299
( )sinCTF qφ= (10.14.20)
ist maßgebend für die Phasenkontrastübertragung, sie wird deshalb als Kontrast-übertragungsfunktion (englisch: Contrast Transfer Funktion – CTF), genauer: Pha-senkontrastübertragungsfunktion, bezeichnet und hängt von der Raumfrequenz, dem Öffnungsfehler, der Wellenlänge und der Defokussierung ab (10.14.13):
( ) ( )3 4 2, , , sin 22S SCTF q C f C q f qλ λ λΔ = ⋅ ⋅ − ⋅Δ ⋅ ⋅ (10.14.21)
(s. Bild 10-31).
Bild 10-31. Beispiele für Kontrastübertragungsfunktionen für UB = 300 kV, Δf = 60 nm und zwei verschiedene Öffnungsfehlerkonstanten: a) CS = 1 mm. b) CS = 2 mm.
Die Kontrastübertragungsfunktion beginnt bei (0,0). Bis zum nächsten Nulldurch-gang schließt sind ein „Band“ von Raumfrequenzen an, bei denen der Kontrast zwar stärker und schwächer sein kann, sein Vorzeichen sich aber nicht ändert: der Kontrast invertiert nicht. In diesem Raumfrequenzband ist die Bildinterpretation einfach. Bild 10-31 zeigt, dass dieser Nulldurchgang bei kleinerem Öffnungsfehler zu höheren Raumfrequenzen, d. h. zu kürzeren Abständen, verschoben ist. Die Verringerung des Öffnungsfehlers erhöht nicht nur das Auflösungsvermögen son-dern vereinfacht auch die Bildinterpretation.
Wir sehen auch, dass Kontrast auch für höhere Raumfrequenzen übertragen wird, allerdings nicht monoton. Die Übertragungsfunktion oszilliert, d. h. manche Raumfrequenzen werden mit „positivem“, andere mit „negativem“ Kontrast und wieder andere gar nicht übertragen. Eine Bildinterpretation ist dann nur mit genauer Kenntnis der Kontrastübertragungsfunktion möglich.
Es fällt auf, dass die Kontrastübertragungsfunktionen in Bild 10-31 ungedämpft sind. Kontrastübertragung wäre demnach bis zu unendlich hohen Raumfrequenzen möglich, wenn auch mit einigen Lücken. Dies widerspricht der Erfahrung.
In Wirklichkeit ist die Übertragungsfunktion gedämpft, d. h. die Amplitude nimmt mit steigender Raumfrequenz ab. Dies hat verschiedene Ursachen: Mecha-nische Vibrationen der Probe „verwackeln“ die kleinen Abstände, d. h. die großen

Kapitel 10 300
Raumfrequenzen. Sie sind deshalb im Bild nicht mehr sichtbar. Dies hängt aller-dings nicht unmittelbar mit der Übertragungseigenschaft der Linse zusammen. Wir wollen uns mit einer anderen Ursache für die Dämpfung näher beschäftigen, die durch die Linse direkt beeinflusst wird: dem Farbfehler der Linse.
Bisher sind wir davon ausgegangen, dass alle Elektronen eine einheitliche Wel-lenlänge λ haben. Dies ist aber nicht der Fall. Abhängig vom Kathodentyp haben die von der Kathode emittierten Elektronen eine Energiebreite von bis zu einigen eV (s. Abschnitt 2.5). Eine Schwankung der Wellenlänge und ein Farbfehler (vgl. Abschnitt 2.3) sind die Folge. Dieser Farbfehler wird ähnlich wie der Öff-nungsfehler durch ein Farbfehlerscheibchen mit dem (auf das Bild bezogenen) Radius
0C C
Er C M
Eθ
ΔΔ = ⋅ ⋅ ⋅ (10.14.22)
(ΔE: Energiebreite der Elektronen, E0: Primärelektronenenergie, CC: Farbfehler-konstante, M: Vergrößerung) berücksichtigt. Analog zur Überlegung zum Einfluss des Öffnungsfehlers lässt sich daraus für die Phasenschiebung durch den Farb-fehler die Beziehung
2
0
( )C CE
q C qE
φ λΔ
= ⋅ ⋅ ⋅ (10.14.23)
herleiten. Dies ist allerdings die maximal mögliche Phasenschiebung. Die aktuelle Energie E eines Elektrons liegt zwischen E0-½ΔE und E0+½ΔE. Die daraus resul-tierenden unterschiedlichen Phasenschiebungen führen zu „verschmierten“ Inter-ferenzen und damit zu einer Abnahme der Amplitude der resultierenden Welle. Dämpfungen werden im Allgemeinen durch Exponentialfunktionen beschrieben. Wir benutzen deshalb eine Dämpfungsfunktion der Form
( )( )
2
2 2 2 42
0( ) e eC
C
EC q
q ED qλ
φ
Δ− ⋅ ⋅ ⋅
−= = (10.14.24)
und erhalten für die gedämpfte Kontrastübertragungsfunktion
( )
22 2 2 4
0
0
3 4 2
, , , , ,
sin 2 e2
C
S C
EC q
ES
ECTF q C f C
E
C q f qλ
λ
λ λ
Δ− ⋅ ⋅ ⋅
ΔΔ =
⋅ ⋅ − ⋅Δ ⋅ ⋅ ⋅
. (10.14.25)

Grundlagen genauer erklärt (etwas mehr Mathematik) 301
Da der Richtstrahlwert und somit die Energiebreite ΔE vom Kathodentyp abhän-gen, hat auch der Kathodentyp einen Einfluss auf die Kontrastübertragung (s. Bild 10-32).
Bei der Schottky-Kathode liegt der erste Nulldurchgang der Kontrastüber-tragungsfunktion bei einer Raumfrequenz von 5,5 nm-1, d. h. bei Strukturgrößen von 0,18 nm, weitere Raumfrequenzen werden oszillierend übertragen. Bei glei-cher Objektivlinse, aber LaB6-Kathode gibt es keinen Nulldurchgang. Die Dämp-fung ist so stark, dass sich die Kontrastübertragungsfunktion der Abszisse asymptotisch nähert und diese bei etwa 4,5 nm-1 berührt, was einer Strukturgröße von 0,22 nm entspricht. Oszillationen treten nicht auf. Das hat einerseits den Vor-teil der einfacheren Bildinterpretation, andererseits aber den Nachteil, dass das Auflösungsvermögen kleiner wird.
Bild 10-32. Einfluss der Kathode auf die Kontrastübertragung (UB = 300 kV, Δf = 60 nm, CS = 1 mm, CC = 1,5 mm): a) ΔE = 0.7 eV (Schottky-Feldemissionskathode), b) ΔE = 3 eV (LaB6-Kathode) gestrichelte Linien: Dämpfungsfunktion.
Offenbar beeinflusst die Dämpfung die maximal übertragbare Raumfrequenz. Es ist zweckmäßig, dafür eine Größe anzugeben, die als Informationslimit bezeichnet wird. In unserem Modell legen wir für diese Größe diejenige Raumfrequenz zu-grunde, bei der die gedämpfte Amplitude auf 1/e2 = 0,135 (e = 2,7182..., natür-liche Zahl) gesunken ist:
( )
2
2 2 2 4lim
02lim
22 2 2 4
lim0
lim
0
e e
2
2
C
EC q
E
C
C
D q
EC q
E
qE
CE
λ
λ
λ
Δ− ⋅ ⋅ ⋅
−= =
Δ= ⋅ ⋅ ⋅
=Δ
⋅ ⋅ ⋅
. (10.14.26)
Für das Informationslimit im Ortsraum gilt damit:

Kapitel 10 302
lim0
1,49 CE
CE
δ λΔ
= ⋅ ⋅ ⋅ . (10.14.27)
Für die Mikroskope, gekennzeichnet durch die in Bild 10-32 genannten Parameter, beträgt das Informationslimit bei Verwendung einer LaB6-Kathode 0,26 nm, bei Einsatz einer Schottky-Kathode demgegenüber 0,12 nm, d. h. eine Verbesserung um mehr als den Faktor 2.
Schließlich wollen wir auf die Bedeutung des ersten Nulldurchganges der Kon-trastübertragungsfunktion hinweisen: Raumfrequenzen bis zu diesem Wert werden ohne Kontrast-Oszillation übertragen. Damit eröffnet sich eine Möglichkeit, das Auflösungsvermögen wellenoptisch zu interpretieren. Der Kehrwert der Raum-frequenz des ersten Nulldurchgangs wird im Unterschied zum Informationslimit als Punktauflösung bezeichnet (s. Abschnitt 7.3).
10.15 Scherzer-Fokus Aus dem Argument
( )3 4 2( ) 22 Sq C q f qφ λ λ= ⋅ ⋅ − ⋅Δ ⋅ ⋅ . (10.14.13)
der Kontrastübertragungsfunktion folgt, dass der Einfluss des Öffnungsfehlers durch einen geeigneten Defokus Δf ausgeglichen werden kann. Allerdings ist der Ausgleich raumfrequenzabhängig.
In Bild 10-33 ist die ungedämpfte Phasenkontrastübertragungsfunktion für drei verschiedene Defoki bei sonst gleichen Parametern dargestellt. Bei der Funktion von Bild 10-33b liegt der erste Nulldurchgang bei der größten Raumfrequenz, im Raumfrequenzbereich bis dahin gibt es keine Kontrastinversion. Wir wollen den-jenigen Defokuswert Δfr bestimmen, bei dem der erste Nulldurchgang am weite-sten nach rechts verschoben ist.
Für die Nullstellen q0,n der Kontrastübertragungsfunktion gilt:
( ) ( )0, 0,sin 0 bzw. n mit n = 0, 1, 2, 3, ...n nq qφ φ= = ± ⋅ (10.15.1)
und unter Berücksichtigung von (10.14.13):
3 4 22 m mit m = 0, 2, 4, ... geradzahligSC q f qλ λ⋅ ⋅ − ⋅Δ ⋅ ⋅ = ± . (10.15.2)
Die Nullstelle für n = 0 liegt bei q = 0. Dies kann allerdings auch für Raumfre-quenzen q > 0 erreicht werden unter der Bedingung
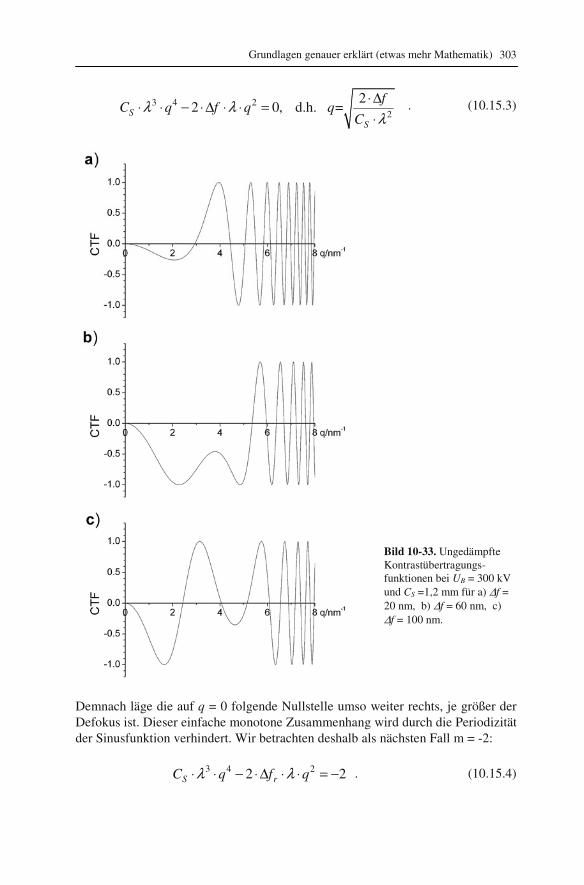
Grundlagen genauer erklärt (etwas mehr Mathematik) 303
3 4 22
22 0, d.h. =S
S
fC q f q q
Cλ λ
λ
⋅Δ⋅ ⋅ − ⋅Δ ⋅ ⋅ =
⋅ . (10.15.3)
Bild 10-33. Ungedämpfte Kontrastübertragungs-funktionen bei UB = 300 kV und CS =1,2 mm für a) Δf = 20 nm, b) Δf = 60 nm, c) Δf = 100 nm.
Demnach läge die auf q = 0 folgende Nullstelle umso weiter rechts, je größer der Defokus ist. Dieser einfache monotone Zusammenhang wird durch die Periodizität der Sinusfunktion verhindert. Wir betrachten deshalb als nächsten Fall m = -2:
3 4 22 2S rC q f qλ λ⋅ ⋅ − ⋅Δ ⋅ ⋅ = − . (10.15.4)
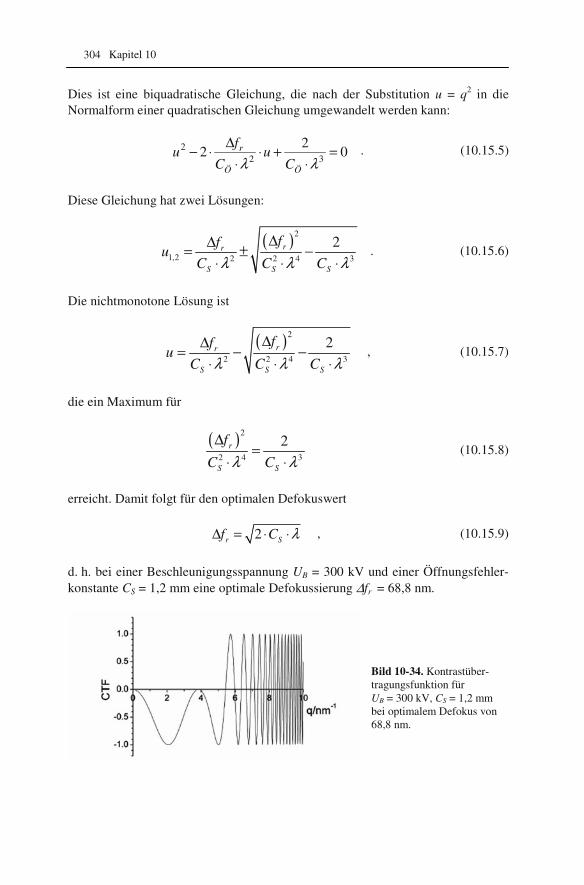
Kapitel 10 304
Dies ist eine biquadratische Gleichung, die nach der Substitution u = q2 in die Normalform einer quadratischen Gleichung umgewandelt werden kann:
22 3
22 0r
Ö Ö
fu u
C Cλ λ
Δ− ⋅ ⋅ + =
⋅ ⋅ . (10.15.5)
Diese Gleichung hat zwei Lösungen:
( )2
1,2 2 2 4 3
2rr
S S S
ffu
C C Cλ λ λ
ΔΔ= ± −
⋅ ⋅ ⋅ . (10.15.6)
Die nichtmonotone Lösung ist
( )2
2 2 4 3
2rr
S S S
ffu
C C Cλ λ λ
ΔΔ= − −
⋅ ⋅ ⋅ , (10.15.7)
die ein Maximum für
( )2
2 4 3
2r
S S
f
C Cλ λ
Δ=
⋅ ⋅ (10.15.8)
erreicht. Damit folgt für den optimalen Defokuswert
2r Sf C λΔ = ⋅ ⋅ , (10.15.9)
d. h. bei einer Beschleunigungsspannung UB = 300 kV und einer Öffnungsfehler-konstante CS = 1,2 mm eine optimale Defokussierung Δfr = 68,8 nm.
Bild 10-34. Kontrastüber-tragungsfunktion für UB = 300 kV, CS = 1,2 mm bei optimalem Defokus von 68,8 nm.

Grundlagen genauer erklärt (etwas mehr Mathematik) 305
In Bild 10-34 ist die Kontrastübertragungsfunktion für diese optimale Defokussie-rung gezeichnet. Problematisch ist die dramatische Kontrastverringerung auf Null bei q ≈ 3,8 nm-1, die vermieden werden muss. Deshalb benutzen wir einen anderen Algorithmus.
Wir fordern, dass die Kontrastübertragungsfunktion bis zum Abfall unmittelbar vor der ersten Inversion sich der Abszisse höchstens bis auf 80 % nähert:
sin ( ) 0,8qφ ≥ . (10.15.10)
Bild 10-35. Sinusfunktion mit Veranschaulichung eines Mindest-Maximalwertes.
Dieser Sachverhalt ist in Bild 10-35 veranschaulicht. Wir wählen den gekenn-zeichneten, größtmöglichen Abszissenwert aus, d. h.
max( ) 2, 214qφ = . (10.15.11)
Für die Raumfrequenz qmax, bei der dieser Extremwert erreicht wird, gilt:
( )max
3 3max max
2max 2
4 4 02 S
q
S
dC q f q
dq
fq
C
φλ λ
λ
= ⋅ ⋅ ⋅ − ⋅Δ ⋅ ⋅ =
Δ=
⋅
, (10.15.12)
eingesetzt in Gleichung (10.14.13), liefert:
2
3max 2 2
( ) 2 2, 2142 S
S S
f fq C f
C Cφ λ λ
λ λ
Δ Δ= ⋅ ⋅ ⋅ − ⋅Δ ⋅ ⋅ =
⋅ ⋅ (10.15.13)
bzw.

Kapitel 10 306
( ) ( ) ( )2 2 2
2 1, 41S S S
f f f
C C Cλ λ λ
Δ Δ Δ− ⋅ = =
⋅ ⋅ ⋅ . (10.15.14)
Bei Einstellung der daraus berechneten Brennweitenänderung
1,41 1, 2Sch S Sf C Cλ λΔ = ⋅ ⋅ = ⋅ ⋅ (10.15.15)
spricht man vom Scherzer-Fokus. In Bild 10-36 ist die Kontrastübertragungs-funktion für diese Defokussierung dargestellt.
Bild 10-36. Kontrastübertragungs-funktion für UB = 300 kV, CS = 1,2 mm bei optimalem Defokus von 58,3 nm.
Wie erwartet wird bis zum ersten Nulldurchgang der Funktion ein Abstand von 0,8 von der Abszisse nicht unterschritten. Dieser Abstand von 0,8 wurde willkür-lich festgelegt. Insofern ist auch der Faktor 1,2 in Formel (10.15.15) einer Willkür unterworfen. Er stimmt aber mit dem in der Literatur üblichen Wert überein.
10.16 Delokalisation Aus den Kapiteln 5 und 7 wissen wir, dass bei der Phasenkontrastabbildung sehr kleiner Strukturen die für die Interferenz benötigten gebeugten Elektronenwellen Beugungswinkel θ von mehr als 10 mrad haben können. Sie müssen durch das Objektiv übertragen werden, wobei der Einfluss des Öffnungsfehlers infolge des größeren Winkels größer ist als bei der Abbildung gröberer Strukturen. Die Skiz-zen in Bild 10-37 sollen diese Problematik veranschaulichen.
Bei der Abbildung mit achsennahen Elektronenwellen bzw. –strahlen, wie dies beispielsweise für den Streuabsorptionskontrast typisch ist, spielt der Öffnungs-fehler keine Rolle (vgl. Bild 10-37a). Die Gaußsche Bildebene ist die Fokusebene. Dies ändert sich bei Verwendung stärker geneigter Wellen, wie sie in den Beu-gungsmaxima gegeben sind. In diesem Fall bewirkt der Öffnungsfehler eine stär-kere Brechung der Strahlen, diese schneiden sich bereits vor der Gaußschen Bild-ebene in einem Punkt, der außerdem senkrecht zur optischen Achse z verschoben ist (s. Bild 10-37b). Diese Verschiebung der Bilder bei Abbildung mit achsen-nahen und achsenfernen Strahlen wird Delokalisation genannt.
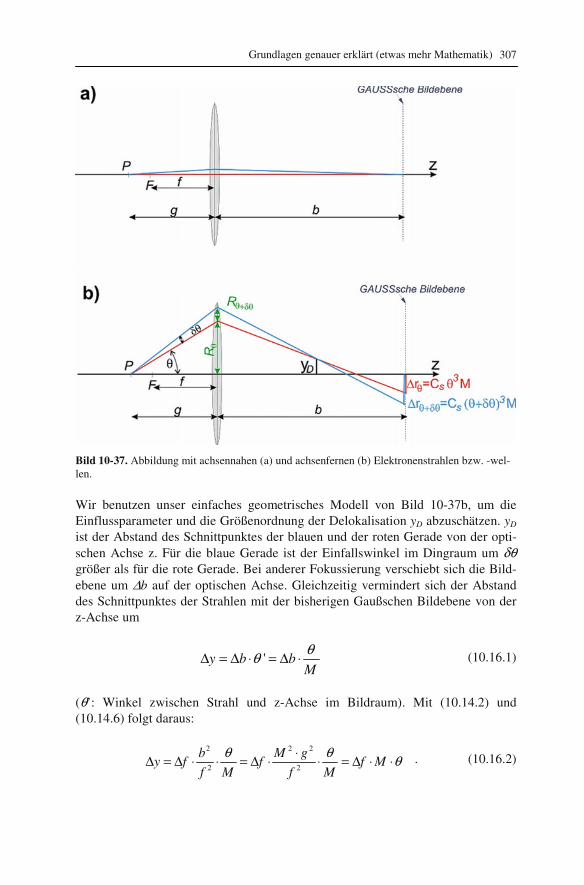
Grundlagen genauer erklärt (etwas mehr Mathematik) 307
Bild 10-37. Abbildung mit achsennahen (a) und achsenfernen (b) Elektronenstrahlen bzw. -wel-len.
Wir benutzen unser einfaches geometrisches Modell von Bild 10-37b, um die Einflussparameter und die Größenordnung der Delokalisation yD abzuschätzen. yD ist der Abstand des Schnittpunktes der blauen und der roten Gerade von der opti-schen Achse z. Für die blaue Gerade ist der Einfallswinkel im Dingraum um δθ größer als für die rote Gerade. Bei anderer Fokussierung verschiebt sich die Bild-ebene um Δb auf der optischen Achse. Gleichzeitig vermindert sich der Abstand des Schnittpunktes der Strahlen mit der bisherigen Gaußschen Bildebene von der z-Achse um
'y b bM
θθΔ = Δ ⋅ = Δ ⋅ (10.16.1)
(θ’: Winkel zwischen Strahl und z-Achse im Bildraum). Mit (10.14.2) und (10.14.6) folgt daraus:
2 2 2
2 2
b M gy f f f M
f M f M
θ θθ
⋅Δ = Δ ⋅ ⋅ = Δ ⋅ ⋅ = Δ ⋅ ⋅ . (10.16.2)

Kapitel 10 308
Die beiden Geradengleichungen lauten:
3
rot
S
r y Ry R z
b
M C f M gg z
b
θ θθ
θ θ θθ
Δ −Δ += − ⋅
⋅ ⋅ − Δ ⋅ ⋅ + ⋅= ⋅ − ⋅
(10.16.3)
und
( )( ) ( )
3
blau
S
r y Ry R z
b
M C f M gg z
b
θ δθ θ δθθ δθ
θ δθ θ θ δθθ δθ
+ ++
Δ −Δ += − ⋅
⋅ ⋅ + − Δ ⋅ ⋅ + ⋅ += ⋅ + − ⋅
. (10.16.4)
Für die z-Koordinate des Schnittpunktes beider Geraden gilt:
( )( ) ( )
3
3
SS
SS
M C f M gg z
b
M C f M gg z
b
θ θ θθ
θ δθ θ θ δθθ δθ
⋅ ⋅ − Δ ⋅ ⋅ + ⋅⋅ − ⋅ =
⋅ ⋅ + − Δ ⋅ ⋅ + ⋅ +⋅ + − ⋅
(10.16.5)
und daraus nach Beschränkung auf lineare Terme von δθ:
23SS
g bz
M C gθ
⋅=
⋅ ⋅ ⋅ + . (10.16.6)
Einsetzen in die Geradengleichung (10.16.3) liefert:
3
2
2
2
3
13
SD
S
SD
S
M C f M g g by g
b M C g
M C f M gy g
M C g
θ θ θθ
θ
θθ
θ
⋅ ⋅ − Δ ⋅ ⋅ + ⋅ ⋅= ⋅ − ⋅
⋅ ⋅ ⋅ +
⋅ ⋅ − Δ ⋅ += ⋅ −
⋅ ⋅ ⋅ +
(10.16.7)
bzw.
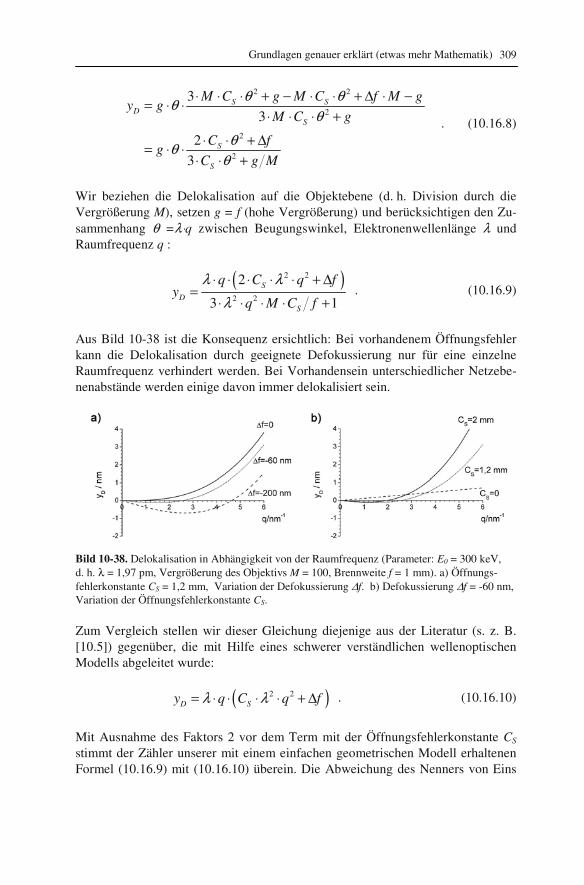
Grundlagen genauer erklärt (etwas mehr Mathematik) 309
2 2
2
2
2
3
3
2
3
S SD
S
S
S
M C g M C f M gy g
M C g
C fg
C g M
θ θθ
θ
θθ
θ
⋅ ⋅ ⋅ + − ⋅ ⋅ + Δ ⋅ −= ⋅ ⋅
⋅ ⋅ ⋅ +
⋅ ⋅ + Δ= ⋅ ⋅
⋅ ⋅ +
. (10.16.8)
Wir beziehen die Delokalisation auf die Objektebene (d. h. Division durch die Vergrößerung M), setzen g = f (hohe Vergrößerung) und berücksichtigen den Zu-sammenhang θ =λ⋅q zwischen Beugungswinkel, Elektronenwellenlänge λ und Raumfrequenz q :
( )2 2
2 2
2
3 1S
DS
q C q fy
q M C f
λ λ
λ
⋅ ⋅ ⋅ ⋅ ⋅ + Δ=
⋅ ⋅ ⋅ ⋅ + . (10.16.9)
Aus Bild 10-38 ist die Konsequenz ersichtlich: Bei vorhandenem Öffnungsfehler kann die Delokalisation durch geeignete Defokussierung nur für eine einzelne Raumfrequenz verhindert werden. Bei Vorhandensein unterschiedlicher Netzebe-nenabstände werden einige davon immer delokalisiert sein.
Bild 10-38. Delokalisation in Abhängigkeit von der Raumfrequenz (Parameter: E0 = 300 keV, d. h. λ = 1,97 pm, Vergrößerung des Objektivs M = 100, Brennweite f = 1 mm). a) Öffnungs-fehlerkonstante CS = 1,2 mm, Variation der Defokussierung Δf. b) Defokussierung Δf = -60 nm, Variation der Öffnungsfehlerkonstante CS.
Zum Vergleich stellen wir dieser Gleichung diejenige aus der Literatur (s. z. B. [10.5]) gegenüber, die mit Hilfe eines schwerer verständlichen wellenoptischen Modells abgeleitet wurde:
( )2 2D Sy q C q fλ λ= ⋅ ⋅ ⋅ ⋅ + Δ . (10.16.10)
Mit Ausnahme des Faktors 2 vor dem Term mit der Öffnungsfehlerkonstante CS stimmt der Zähler unserer mit einem einfachen geometrischen Modell erhaltenen Formel (10.16.9) mit (10.16.10) überein. Die Abweichung des Nenners von Eins

Kapitel 10 310
bleibt bei typischen Parametern im Transmissionselektronenmikroskop (CS ≈ f, M ≈ 100, E0 = 300 keV, q < 6 nm-1) kleiner als 0,015.
10.17 Potential in elektrostatischen Multipolen Für Stigmatoren und Korrektoren für sphärische und chromatische Aberrationen sind nicht-rotationssymmetrische elektronenoptische Einheiten, sogenannte Multi-pole, erforderlich. Dies ist seit Erscheinen der Arbeiten von Otto Scherzer im Jahre 1936 bekannt. Darauf basierend realisierte O. Rang 1949 einen Stigmator [10.6]. Ein (wenn auch mit geringem Auflösungsvermögen) nur aus Quadrupolen bestehendes Objektiv konstruierte und erprobte H.-D. Bauer im Jahre 1965 [10.7].
Für das Verständnis der Eigenschaften eines Multipols werden wir die Poten-tialverteilung in einem Quadrupol und einem Oktupol berechnen und grafisch dar-stellen. Aus Abschnitt 10.7 ist die Verfahrensweise bekannt: Wir müssen die Laplace-Gleichung
2 2 2
2 2 20
U U UU
x y z
∂ ∂ ∂Δ = + + =
∂ ∂ ∂ , (10.17.1)
die hier in kartesischen Koordinaten aufgeschrieben ist, lösen. Vereinfachend ge-hen wir von einem in z-Richtung unendlich ausgedehnten Multipol aus, d. h. die Potentialverteilung ist in z-Richtung konstant. Damit vereinfacht sich Gleichung (10.17.1) zu
2 2
2 20
U U
x y
∂ ∂+ =
∂ ∂ . (10.17.2)
Zur numerischen Lösung wandeln wir diese Differentialgleichung nach der Vor-schrift
( ) ( )( )
( )
( ) ( ) ( )
( )
( ) ( )( )
( )
( ) ( ) ( )
( )
2
2 2 2
2
2 2 2
,, , 2 , ,
,, , 2 , ,
U x yU x z U x x y U x y U x x y
x x x
U x yU x y U x y y U x y U x y y
y y y
Δ Δ∂ + Δ − ⋅ + − Δ→ =
∂ Δ Δ
Δ Δ∂ + Δ − ⋅ + − Δ→ =
∂ Δ Δ
(10.17.3)
in eine Differenzengleichung um. Bei gleichen Differenzen Δx und Δy (= Δ) sowie
bzw. x i y j= ⋅Δ = ⋅Δ (10.17.4)

Grundlagen genauer erklärt (etwas mehr Mathematik) 311
folgt
( ) ( ) ( ) ( )
( ) ( )
1, 2 , 1, , 1
2 , , 1 0
U i j U i j U i j U i j
U i j U i j
+ − ⋅ + − + + −
− ⋅ + − = (10.17.5)
und daraus die Rekursionsformel
( ) ( ) ( ) ( ) ( )1
, 1, 1, , 1 , 14
U i j U i j U i j U i j U i j= + + − + + + − (10.17.6)
zur Berechnung der Potentialverteilung aus den Randbedingungen. Die nicht durch die Randbedingungen abgedeckten Randpotentiale werden aus den Formeln
( ) ( ) ( )
( ) ( ) ( )
1,0 1,0 1,0 für i>0
21
0, 0, 1 0, 1 für j>02
U i U i U i
U j U j U j
= + + −
= + + −
(10.17.7)
erhalten. Für die Festlegung der Randbedingungen wählen wir als Multipolele-mente Stäbe mit kreisförmigem Querschnitt, die in der Mitte der Seiten eines Qua-drates bzw. eines regelmäßigen Achtecks angeordnet sind (vgl. Bild 10-39). Prin-zipiell ist ein Quadrupol als Stigmator geeignet. Um dies zu verstehen, schauen wir auf Bild 10-40. Dort ist die Potentialverteilung auf zwei verschiedene Arten dargestellt: In Bild 10-40a farbkodiert und in Bild 10-40b als Potentialgebirge.
Bild 10-39. Schematische Darstellung eines Quadrupols (a) und eines Oktupols (b) mit möglichen Potentialen.
Bild 10-40b sieht aus wie ein Sattel. Der Punkt, in dem die Elektronen keine Ablenkungskraft erfahren (Potential = 0) heißt deshalb auch „Sattelpunkt“. Wir können ihn auch als Durchstoßpunkt der optischen Achse durch die gezeichnete Multipolebene verstehen.
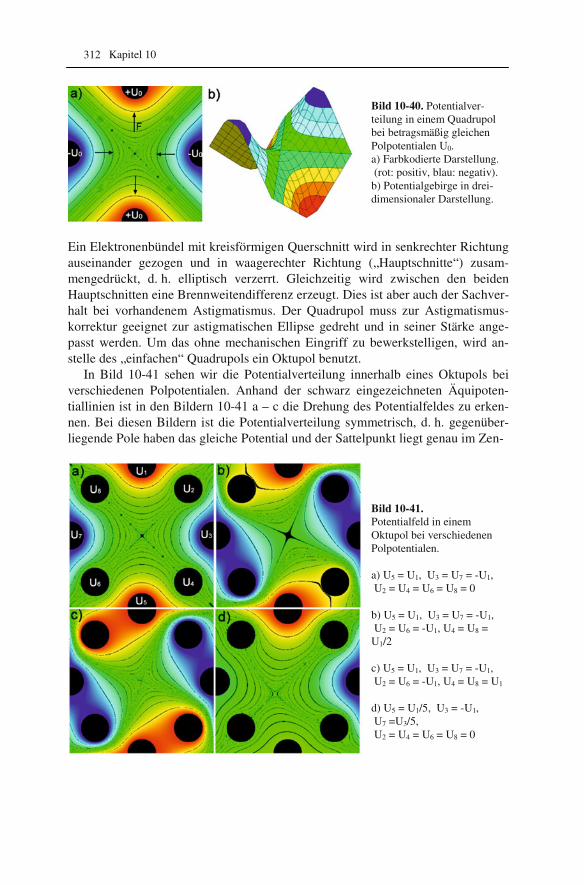
Kapitel 10 312
Bild 10-40. Potentialver-teilung in einem Quadrupol bei betragsmäßig gleichen Polpotentialen U0. a) Farbkodierte Darstellung. (rot: positiv, blau: negativ). b) Potentialgebirge in drei-dimensionaler Darstellung.
Ein Elektronenbündel mit kreisförmigen Querschnitt wird in senkrechter Richtung auseinander gezogen und in waagerechter Richtung („Hauptschnitte“) zusam-mengedrückt, d. h. elliptisch verzerrt. Gleichzeitig wird zwischen den beiden Hauptschnitten eine Brennweitendifferenz erzeugt. Dies ist aber auch der Sachver-halt bei vorhandenem Astigmatismus. Der Quadrupol muss zur Astigmatismus-korrektur geeignet zur astigmatischen Ellipse gedreht und in seiner Stärke ange-passt werden. Um das ohne mechanischen Eingriff zu bewerkstelligen, wird an-stelle des „einfachen“ Quadrupols ein Oktupol benutzt.
In Bild 10-41 sehen wir die Potentialverteilung innerhalb eines Oktupols bei verschiedenen Polpotentialen. Anhand der schwarz eingezeichneten Äquipoten-tiallinien ist in den Bildern 10-41 a – c die Drehung des Potentialfeldes zu erken-nen. Bei diesen Bildern ist die Potentialverteilung symmetrisch, d. h. gegenüber-liegende Pole haben das gleiche Potential und der Sattelpunkt liegt genau im Zen-
Bild 10-41. Potentialfeld in einem Oktupol bei verschiedenen Polpotentialen. a) U5 = U1, U3 = U7 = -U1, U2 = U4 = U6 = U8 = 0 b) U5 = U1, U3 = U7 = -U1, U2 = U6 = -U1, U4 = U8 = U1/2 c) U5 = U1, U3 = U7 = -U1, U2 = U6 = -U1, U4 = U8 = U1
d) U5 = U1/5, U3 = -U1, U7 =U3/5, U2 = U4 = U6 = U8 = 0

Grundlagen genauer erklärt (etwas mehr Mathematik) 313
trum. In Bild 10-41d haben gegenüberliegende Pole ungleiches Potential, der Sattelpunkt ist nach links unten verschoben. Damit ist es möglich, den Oktupol zur optischen Achse der astigmatischen Linse zu zentrieren.
10.18 Elektronensonde und Abbildungsfehler Als Ergänzung zu Abschnitt 8.2 wollen wir hier einige der dort getroffenen Aus-sagen und Gleichungen begründen. Im Unterschied zur vergrößernden Abbildung soll der Sondenquerschnitt verkleinert werden. Die Fehlerscheibchen begrenzen in diesem Fall direkt die minimale Sondengröße, ihre optische Verkleinerung ist un-möglich. In den Gleichungen für die Fehlerscheibchengröße taucht die Vergröße-rung deshalb hier nicht mehr auf.
- Öffnungsfehlerscheibchen in der Ebene der kleinsten Verwirrung
Die Gaußsche Bildebene ist nicht die optimale Einstellung für den kleinsten Son-dendurchmesser (vgl. Bild 10-42).
Bild 10-42. Zur Berech- nung des Sondenradius in der Ebene der kleinsten Verwirrung bei vorhan- denem Öffnungsfehler. In der Linsenmitte ist z = 0.
Wir sehen, dass für den Öffnungswinkel α der Radius y(β) des Zerstreuungs-scheibchens signifikant kleiner ist als CS⋅α
3 in der Gaußschen Bildebene. Die Ebe-ne mit dem kleinsten Radius wird als „Ebene der kleinsten Verwirrung“ bezeich-net. Wir wollen nun geometrisch-optisch abschätzen, wie klein der minimale Son-dendurchmesser (bzw. –radius) werden kann. Dazu bestimmen wir zunächst die beiden Geradengleichungen y1(z) und y2(z):
( ) ( ) ( )
( )
31 1 1 1 1
3
1
, 0 , S
S
y z m z n y g y b C
C gy z z g
b
α α
α αα
= ⋅ + = − ⋅ = ⋅
⋅ + ⋅= ⋅ − ⋅
(10.18.1)
sowie

Kapitel 10 314
( ) ( ) ( )
( )
32 2 2 2 2
3
2
, 0 , S
S
y z m z n y g y b C
C gy z z g
b
β β
β ββ
= ⋅ + = ⋅ = − ⋅
⋅ + ⋅= − ⋅ + ⋅
(10.18.2)
Offensichtlich hängt der Radius y im Schnittpunkt der beiden Geraden vom Win-kel β ab, d. h. y = y(β). Ziel ist nun, den Winkel β mit maximalem y zu finden. Dies ist der minimale Sondenradius. Zunächst wird die Funktion y(β) ermittelt. Im Schnittpunkt der beiden Geraden bei zs gilt:
( ) ( )
( ) ( )
1 2
3 3
3 3
s s
S Ss s
sS S
y z y z
C g C gz g z g
b bz
C g C g gb
α α β βα β
α α β β α β
=
⋅ + ⋅ ⋅ + ⋅⋅ − ⋅ = − ⋅ + ⋅
⋅ + ⋅ + ⋅ + ⋅ = ⋅ +
(10.18.3)
bzw.
( )
( ) ( )3 3s
S
b gz
C g
α β
α β α β
⋅ ⋅ +=
⋅ + + ⋅ + . (10.18.4)
Dies wird eingesetzt in die Geradengleichung y1(z):
( )( )
( ) ( )
3
1 3 3S
S
b gC gy z g
b C g
α βα αα
α β α β
⋅ ⋅ +⋅ + ⋅= ⋅ − ⋅
⋅ + + ⋅ + , (10.18.5)
und wir erhalten nach Umformung die gesuchte Funktion
( )( )
( ) ( )
2 2
3 3
S
S
g Cy
C g
α β α ββ
α β α β
⋅ ⋅ ⋅ −=
⋅ + + ⋅ + . (10.18.6)
Vom Dingpunkt geht ein Kegel mit dem halben Öffnungswinkel α aus, d. h. für β gilt
0 β α≤ ≤ .
Die minimale Sondengröße in der Ebene der kleinsten Verwirrung entspricht dem Maximum der Funktion y(β):

Grundlagen genauer erklärt (etwas mehr Mathematik) 315
( ) ( ) ( )
( ) ( )
( ) ( )
2 3 2 3
23 3
2 3 2 3
3 2 3
3 2| 0
0 3 2
3 2
S
Max S
S
S
C gyg C
C g
C g
α α α β ββα
β α β α β
α α α β β
α α β β
⋅ + ⋅ − ⋅ ⋅ − ⋅∂= = ⋅ ⋅ ⋅
∂⋅ + + ⋅ +
= ⋅ + ⋅ − ⋅ ⋅ − ⋅
= ⋅ ⋅ + ⋅
. (10.18.7)
Diese Gleichung ist offensichtlich für β = α/2 erfüllt. Dieses Ergebnis setzen wir in Gleichung 10.18.6) ein und erhalten für den Radius des minimalen Sondenquer-schnitts:
2 22
3
min 3 23
2 4 3
3 9 128 2
SS
SS
g CC
rC
C g g
α αα
α
α αα α
⋅ ⋅ ⋅ −⋅ ⋅
= =
⋅ ⋅ +⋅ + + ⋅
. (10.18.8)
Für α <<1 und der bei Verkleinerungslinsen gültigen Bedingung g > CS gilt für den Nenner:
29 12SC
gα⋅ ⋅ << (10.18.9)
und damit
33
min3 1
12 4S
SC
r Cα
α⋅ ⋅
= = ⋅ , (10.18.10)
d. h. in der Ebene der kleinsten Verwirrung ist das Zerstreuungsscheibchen nur ein Viertel so groß wie in der Gaußschen Bildebene.
- Farbfehlerscheibchen in der Ebene der kleinsten Verwirrung
Die gleiche Überlegung wiederholen wir für das Farbfehlerscheibchen. Dazu betrachten wir Bild 10-43.
In der Gaußschen Bildebene liege der Bildpunkt für Elektronen mit der Energie E, d. h. ΔE = 0. Der betreffende Strahlengang ist grün gezeichnet und im Bildraum mit y2(z) bezeichnet. Elektronen der Energie E-ΔE (y1(z): rot) werden aufgrund des Farbfehlers stärker gebrochen und erreichen die Gaußsche Bildebene im Abstand CC⋅(ΔE/E)⋅α von der Achse (CC: Farbfehlerkonstante). Für die beiden Geradengleichungen liest man aus der Skizze ab:

Kapitel 10 316
Bild 10-43. Zur Berech- nung des Sondenradius in der Ebene der kleinsten Verwirrung bei vorhan- denem Farbfehler.
( )
( )
1
2und
CE
y z C g z gE b
gy z z g
b
αα
αα
Δ= ⋅ + ⋅ ⋅ − ⋅
⋅= − ⋅ + ⋅
. (10.18.11)
Bei z = zs gilt:
( ) ( )1 2S Sy z y z= (10.18.12)
und
2
2
C S S
SC
E gC g z g z g
E b b
bz
C Eg E
α αα α
Δ ⋅⋅ + ⋅ ⋅ − ⋅ = − ⋅ + ⋅
⋅=
Δ⋅ +
. (10.18.13)
Dies wird eingesetzt in die Geradengleichung für y2:
( ) ( )22
2
21
2
S SC
C
g by z y z g
C Ebg E
gC E
g E
αα
α
⋅ ⋅= = − ⋅ + ⋅
Δ⋅ +
= ⋅ ⋅ −Δ
⋅ +
(10.18.14)

Grundlagen genauer erklärt (etwas mehr Mathematik) 317
und wir erhalten für den minimalen Abstand von der z-Achse:
min
2
C
C
EC
Er
C E
g E
αΔ
⋅ ⋅
=Δ
⋅ +
. (10.18.15)
Auch hier gilt g > CC und damit
2CC E
g E
Δ⋅ << (10.18.16)
bzw.
min1
2 CE
r CE
αΔ
= ⋅ ⋅ ⋅ , (10.18.17)
d. h. das Farbfehlerscheibchen ist in der Ebene der kleinsten Verwirrung halb so groß wie in der Gaußschen Bildebene.
- Lösung der Extremwertaufgabe zur Bestimmung der optimalen Apertur:
Bei Berücksichtigung von Astigmatismus (astigmatische Brennweitendifferenz ΔfA), Öffnungs- und Farbfehler (Fehlerkonstanten CS bzw. CC) hängt der Son-dendurchmesser d vom Strahlstrom IP, vom Richtstrahlwert R, von der relativen Energiebreite ΔE/E und der Wellenlänge λ der Elektronen ab (vgl. Abschnitt 8.2, Formel (8.10):
22 6 2
2 4IR S
CAK C
d Kλ α αα
= + ⋅ + ⋅ (10.18.18)
mit
( )2
22 22
41,5 und P
IR CA C AI E
K K C fER
λ λπ
⋅ Δ= + ⋅ = ⋅ + Δ
⋅ . (10.18.19)
Zur Extremwertberechnung wird nach α differenziert und die erste Ableitung gleich Null gesetzt:

Kapitel 10 318
( )2
2 53
2 53
8 42 2
2 32
22 3
22
4 40
3 3
IRS CA
IRS opt CA opt
opt
CA IRopt opt
S S
d KC K
KC K
K K
C C
λ
λ
λ
α αα α
α αα
α α
∂ ⋅= − + ⋅ ⋅ + ⋅ ⋅
∂
⋅= ⋅ ⋅ + ⋅ ⋅
= + ⋅ ⋅ − ⋅
. (10.18.20)
Wir substituieren
4opt xα = (10.18.21)
und erhalten die quadratische Gleichung
22 2
4 40
3 3CA IR
S S
K Kx x
C Cλ= + ⋅ ⋅ − ⋅ . (10.18.22)
Deren Lösung lautet
22 2
2
1,2 2 2 2
4 40
3 3
2 4 4
3 9 3
CA IR
S S
CA CA IR
S S S
K Kx x
C C
K K Kx
C C C
λ
λ
= + ⋅ ⋅ − ⋅
= − ⋅ ± ⋅ + ⋅
. (10.18.23)
Die Lösung muss positiv sein, deshalb gilt das positive Vorzeichen vor der Wur-zel:
2
2 2 2
2 4 4
3 9 3CA CA IR
S S S
K K Kx
C C Cλ= − ⋅ + ⋅ + ⋅ (10.18.24)
2
2
23
3CA CA
IRS S S
K Kx K
C C C λ= ⋅ − + + ⋅⋅
. (10.18.25)
Mit Berücksichtigung von (10.18.21) erhalten wir für die optimale Apertur:

Grundlagen genauer erklärt (etwas mehr Mathematik) 319
2
42
23
3CA CA
opt IRS S S
K KK
C C C λα = ⋅ − + + ⋅⋅
. (10.18.26)
Wird der Öffnungsfehler auf Null korrigiert, folgt aus (10.18.18) mit CS = 0 nach Extremwertbestimmung:
4IR
optCA
K
Kλα = . (10.18.27)
In Abschnitt 8.2 wird diskutiert, welche Konsequenzen dieser Formalismus für das rastertransmissionselektronenmikroskopische Auflösungsvermögen hat.
10.19 Klassischer inelastischer Stoß Bei inelastischen Wechselwirkungen dient ein Teil der Energie der Strahlelektro-nen dazu, Veränderungen in der Elektronenhülle der Atome hervorzurufen. Wir wollen die Konsequenzen zunächst ohne Berücksichtigung der Abläufe in der Elektronenhülle diskutieren und dazu als Modell das des klassischen inelastischen Stoßes benutzen. In Bild 10-44 sind die Impulsbilanzen für den elastischen und den inelastischen Stoß für den Fall kleiner Streuwinkel (θ << 1) gegenübergestellt (vgl. auch [10.8]).
Bild 10-44. Impulsbilanzen für den elastischen (a) und den inelastischen (b) Stoß.
Beim elastischen Stoß bleiben Impuls und Energie erhalten, d. h.
0 1
p p
p pθ
Δ Δ Δ= = =
0
p
p . (10.19.1)

Kapitel 10 320
Demgegenüber verbleibt beim inelastischen Stoß ein Teil ΔE der Elektronen-energie „im Atom“, so dass für die Energiebilanz in nichtrelativistischer Näherung
2 20 1
2 2
p pE
m mΔ = −
⋅ ⋅ (10.19.2)
(m: Elektronenmasse) und für die Impulsbilanz
= −Δ1 0p p p (10.19.3)
gilt. Nach Quadrieren und Ausmultiplizieren der Skalarprodukte folgt daraus
( ) ( ) ( )2 2 22 2
1 0 0 bzw. 2 cosp p p p pη= −Δ = − ⋅ ⋅Δ ⋅ + Δ1 0p p p (10.19.4)
und unter Berücksichtigung von (10.19.2) sowie Δp << p0:
0 cosp p
Em
η⋅Δ
Δ = ⋅ . (10.19.5)
Aus Bild 10-44b liest man im kleinen rechtwinkligen Dreieck
( ) ( )2 22 2 2 2
0 cosp x y p pθ ηΔ = + ≈ ⋅ + Δ ⋅ (10.19.6)
ab. Der erste Term p0⋅θ kennzeichnet nach (10.19.1) die Richtungsänderung des Impulses beim elastischen Stoß, der zweite Term kann dementsprechend als Rich-tungsänderung infolge des inelastischen Stoßes interpretiert werden. Dessen cha-rakteristischer Winkel sei θie. Damit gilt:
( ) ( )2 2 2 20 iep p θ θΔ = ⋅ + (10.19.7)
bzw. unter Berücksichtigung von (10.19.5) und
20
0 2
pE
m=
⋅ (10.19.8)
02 2
0 00 0
coscos
2iep pp E m E
p Ep p
ηηθ
⋅Δ ⋅Δ ⋅ Δ ⋅ Δ= = = =
⋅ . (10.19.9)
Bei einer Primärelektronenenergie E0 von 200 keV und einem Energieverlust von 100 eV beträgt der charakteristische Ablenkwinkel für die inelastische Streuung

Grundlagen genauer erklärt (etwas mehr Mathematik) 321
0,25 mrad und ist damit deutlich kleiner als derjenige für die elastische Streuung (vgl. Abschnitt 10.11).
10.20 Effizienz von energiedispersiven Röntgen-detektoren Das Kernstück eines EDX-Detektors ist ein zylindrischer Halbleiterkristall (mei-stens Silizium), der sich in einem elektrischen Feld befindet.
Bild 10-45. Schema eines Halbleiterdetektors.
Wir stellen uns die beiden Deckflächen des Zylinders als schmale p- bzw. n-leitende Halbleiterbereiche vor. Dazwischen ist eine vergleichsweise breite, la-dungsträgerfreie Zone (s. Bild 10-45). Um die Effizienz (Wirkungsgrad) des De-tektors zu berechnen, müssen wir die Wahrscheinlichkeiten bestimmen, mit denen das Röntgenquant die in Bild 10-45 dargestellten Hürden überwindet.
- Transparenz des Fensters
Zur Mikroskopsäule hin ist das Detektorrohr mit einem ultradünnen Polymer-fenster (Dicke ca. 0,3 μm) abgeschlossen. Die erste Hürde, die die Röntgenstrah-lung überwinden muss, ist dieses Fenster. Seine Transparenz hängt von der Rönt-genenergie ab und wird als wesentliches Merkmal vom Hersteller offengelegt (s. Bild 10-46).
Bild 10-46. Transparenz TF verschiedener ultradünner Fenster in Abhängigkeit von der Energie der Röntgen-strahlung ER (Quelle: [10.9]).

Kapitel 10 322
Die Transparenz TF(ER) ist die Wahrscheinlichkeit, mit der ein Röntgenquant der Energie ER das Fenster durchdringt. In den Kurven von Bild 10-46 sind vier Abweichungen vom monotonen Verlauf bei folgenden Energien zu sehen: 283 eV, 401 eV, 532 eV und 1560 eV. Diese Energien sind notwendig, um ein Elektron der K-Schale von Kohlenstoff, Stickstoff, Sauerstoff und Aluminium auf den ersten freien Energiezustand zu befördern. Diese Energien werden als Absorp-tionskanten bezeichnet und sind etwas größer als die Energie der Röntgen-K-Strahlung (vgl. Abschnitt 9.1.2).
Bisher waren wir davon ausgegangen, dass diese Energien von Strahlelektro-nen aufgebracht werden, sie können allerdings auch von Röntgenstrahlung genü-gend hoher Energie stammen („Fluoreszenz“).
Offenbar besteht das Polymerfenster aus den Elementen C, N und O, was für organische Polymere nicht verwunderlich ist. Die Al-Absorptionskante bei 1560 eV rührt vermutlich von dem Al-Stützgitter her, was auch dafür sorgt, dass die Transparenz selbst bei hohen Röntgenenergien auf ca. 80% beschränkt bleibt.
- Durchlässigkeit der Goldelektrode
Die zweite Hürde ist die dünne Goldelektrode. Allgemein wird die Schwächung von Röntgenstrahlung in Abhängigkeit von Material und Energie durch „Schwä-chungskoeffizienten“ beschrieben.
Bild 10-47. Skizze zur Erläuterung des Schwä-chungsgesetzes für Röntgenstrahlung.
Beim Durchgang von Röntgenstrahlung durch eine Materialscheibe mit der Dicke ds, gekennzeichnet durch Dichte ρ und energieabhängigen Schwächungskoeffi-zient
( )E
Zμ ρ ,
verringert sich die Intensität um dI:
( )E
ZdI I dsμ ρ ρ= − ⋅ ⋅ ⋅ . (10.20.1)
Daraus folgt für die Intensität:

Grundlagen genauer erklärt (etwas mehr Mathematik) 323
( )0 e
EZ
sI I μ ρ ρ− ⋅ ⋅= ⋅ (10.20.2)
mit I0 als Anfangsintensität und der Gesamtmaterialdicke s. Der Schwächungs-koeffizient kann mit Hilfe der Formel
( )2 -1
12,396
cm g / keV
E
Z C ZE
α
βμ ρ
= ⋅ ⋅⋅
(10.20.3)
(E: Röntgenenergie, Z: mittlere Ordnungszahl des schwächenden Materials) mit den Parametern C, α und β aus Tabelle 10-4 berechnet werden.
E ≥ EK EK > E ≥ EL EL >E ≥ EM
3 ≤ Z ≤ 10
C
α
β
5,4⋅10-3
2,92
3,07
11 ≤ Z ≤ 18
C
α
β
1,38⋅10-2
2,79
2,73
5,33⋅10-4
2,74
3,03
19 ≤ Z ≤ 36
C
α
β
3,12⋅10-2
2,66
2,47
9,59⋅10-4
2,70
2,90
2,73⋅10-5
2,44
3,47
37 ≤ Z ≤ 54
C
α
β
1,03⋅10-3
2,70
2,88
2,73⋅10-5
2,44
3,47
55 ≤ Z ≤ 71
C
α
β
1,24⋅10-3
2,70
2,83
1,58⋅10-4
2,50
2,98
72 ≤ Z ≤ 86
C
α
β
1,03⋅10-4
2,50
3,38
9,39⋅10-5
2,55
3,09
87 ≤ Z ≤ 92
C
α
β
5,76⋅10-7
2,63
4,26
Tabelle 10-4. Parameter zur Berechnung der Schwächungskoeffizienten (nach [10.10]). Kommen wir zurück zur Goldelektrode. Für Gold (Z = 79) können die Schwä-chungskoeffizienten mit den Werten aus Tabelle 10-4 für Energien bis herunter zur M-Kante (2,2 keV) ermittelt werden. Für niedrigere Energien findet man Angaben des „National Institute of Standards and Technology (NIST)“ der USA

Kapitel 10 324
im Internet [10.11]. Im Energiebereich bis zur M-Kante lassen sich diese Angaben für den Schwächungskoeffizienten KSchw durch eine Funktion
2,2 0,5keV
2 -1800 10000 e
cm g
E
SchwK − ⋅ −
= + ⋅⋅
(10.20.4)
Bild 10-48. Schwächungskoeffizienten für Gold in Abhängigkeit von der Röntgenenergie. a) Nach Formeln (10.20.3) und (10.20.4). b) Vergleich mit Messwerten nach NISTIR 5632 im Niedrigenergiebereich.
anfitten (vgl. Bild 10-48). Für die energieabhängige Transparenz (vgl. Bild 10-49) der Goldelektrode mit der Dicke s folgt damit aus Gleichung (10.20.2):
( )79
0
eE
Au s
Au
IT
Iμ ρ ρ− ⋅ ⋅
= = (10.20.5)
(Dichte von Gold: ρAu = 19,32 g/cm3).
Bild 10-49. Transparenz TAu der Goldelektrode für Röntgenstrahlung unter-schiedlicher Energie für drei verschiedene Gold-schichtdicken s.
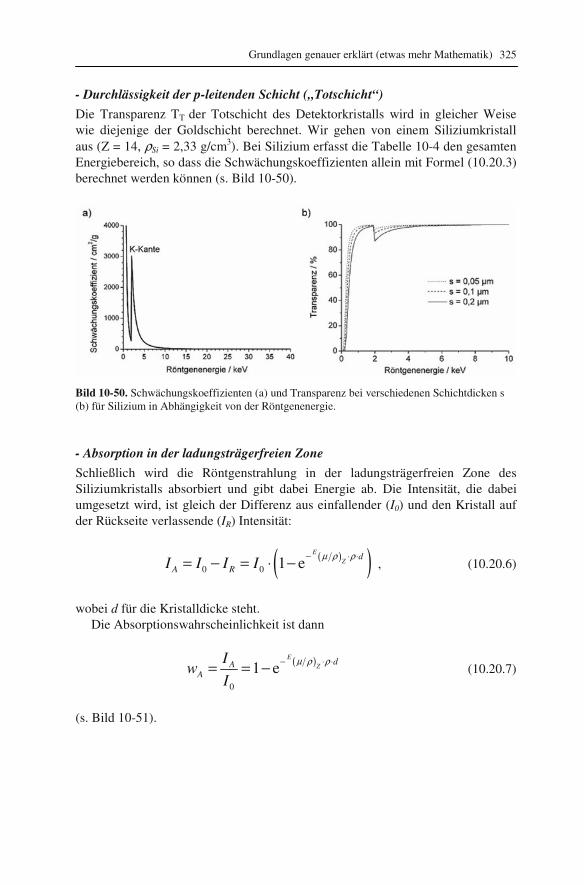
Grundlagen genauer erklärt (etwas mehr Mathematik) 325
- Durchlässigkeit der p-leitenden Schicht („Totschicht“)
Die Transparenz TT der Totschicht des Detektorkristalls wird in gleicher Weise wie diejenige der Goldschicht berechnet. Wir gehen von einem Siliziumkristall aus (Z = 14, ρSi = 2,33 g/cm3). Bei Silizium erfasst die Tabelle 10-4 den gesamten Energiebereich, so dass die Schwächungskoeffizienten allein mit Formel (10.20.3) berechnet werden können (s. Bild 10-50).
Bild 10-50. Schwächungskoeffizienten (a) und Transparenz bei verschiedenen Schichtdicken s (b) für Silizium in Abhängigkeit von der Röntgenenergie.
- Absorption in der ladungsträgerfreien Zone
Schließlich wird die Röntgenstrahlung in der ladungsträgerfreien Zone des Siliziumkristalls absorbiert und gibt dabei Energie ab. Die Intensität, die dabei umgesetzt wird, ist gleich der Differenz aus einfallender (I0) und den Kristall auf der Rückseite verlassende (IR) Intensität:
( )( )0 0 1 eE
Zd
A RI I I Iμ ρ ρ− ⋅ ⋅
= − = ⋅ − , (10.20.6)
wobei d für die Kristalldicke steht. Die Absorptionswahrscheinlichkeit ist dann
( )
0
1 eE
ZdA
A
Iw
Iμ ρ ρ− ⋅ ⋅
= = − (10.20.7)
(s. Bild 10-51).
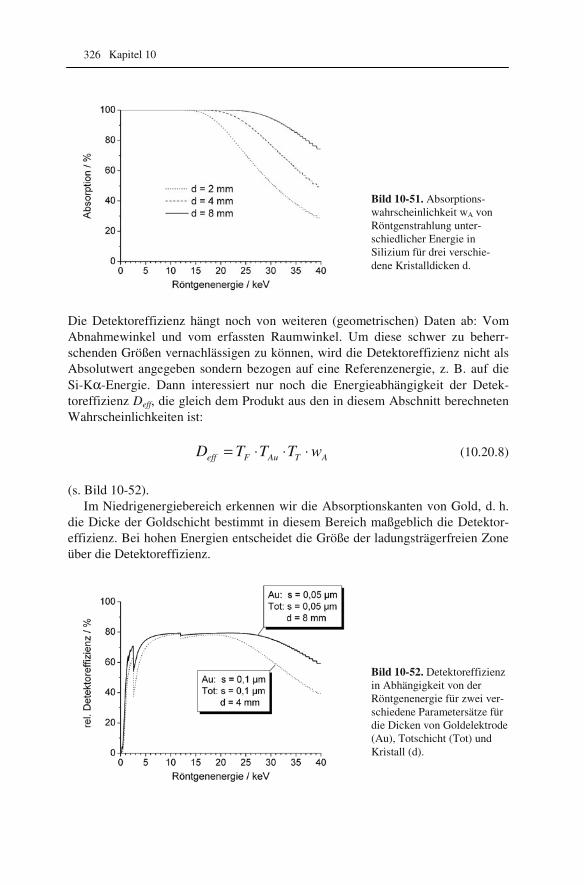
Kapitel 10 326
Bild 10-51. Absorptions-wahrscheinlichkeit wA von Röntgenstrahlung unter-schiedlicher Energie in Silizium für drei verschie- dene Kristalldicken d.
Die Detektoreffizienz hängt noch von weiteren (geometrischen) Daten ab: Vom Abnahmewinkel und vom erfassten Raumwinkel. Um diese schwer zu beherr-schenden Größen vernachlässigen zu können, wird die Detektoreffizienz nicht als Absolutwert angegeben sondern bezogen auf eine Referenzenergie, z. B. auf die Si-Kα-Energie. Dann interessiert nur noch die Energieabhängigkeit der Detek-toreffizienz Deff, die gleich dem Produkt aus den in diesem Abschnitt berechneten Wahrscheinlichkeiten ist:
eff F Au T AD T T T w= ⋅ ⋅ ⋅ (10.20.8)
(s. Bild 10-52). Im Niedrigenergiebereich erkennen wir die Absorptionskanten von Gold, d. h.
die Dicke der Goldschicht bestimmt in diesem Bereich maßgeblich die Detektor-effizienz. Bei hohen Energien entscheidet die Größe der ladungsträgerfreien Zone über die Detektoreffizienz.
Bild 10-52. Detektoreffizienz in Abhängigkeit von der Röntgenenergie für zwei ver-schiedene Parametersätze für die Dicken von Goldelektrode (Au), Totschicht (Tot) und Kristall (d).

Grundlagen genauer erklärt (etwas mehr Mathematik) 327
10.21 Berechnung von Cliff-Lorimer-k-Faktoren Aus Abschnitt 9.2.3 wissen wir, dass die Einflussgrößen für die Empfindlichkeits-faktoren durch den Vorgang der Röntgenemission bei Elektronenanregung gege-ben sind: Die Ionisation des angeregten Atoms erfolgt mit einer Wahrscheinlich-keit Q; die Wahrscheinlichkeit dafür, dass dieses ionisierte Atom ein Röntgen-quant emittiert, sei ω (Fluoreszenzausbeute); schließlich wird dieses Quant mit einer Wahrscheinlichkeit Deff (Detektoreffizienz) vom Detektor nachgewiesen.
Diese Größen sind für beide beteiligten Elemente A und B zu berechnen. Außerdem gehen der Anteil a der berücksichtigten Linie (z. B. Kα) an der Inten-sität der gesamten Serie (z. B. K-Serie) und bei Angaben in Masse-% das Atom-gewicht M ein. Für das Verhältnis der Empfindlichkeitsfaktoren zweier Elemente A und B gilt damit:
, B
, AM
M
MB B B eff BA
B A A A eff A
Q a Dk
k Q a D
ω
ω
⋅ ⋅ ⋅ ⋅=
⋅ ⋅ ⋅ ⋅ . (10.21.1)
Dies ist die Formel (9.34), ergänzt durch den Anteil a der Kα-Linien an der K-Serie. Bei L- und M-Serien liegen die Peaks im Allgemeinen so nahe beieinander, dass eine Unterscheidung wegen der begrenzten Energieauflösung der Spektro-meter unmöglich ist und die Anteile a entfallen. Die Detektoreffizienz hatten wir bereits im vorherigen Abschnitt 10.20. besprochen, mit den drei restlichen Ein-flüssen werden wir uns hier befassen und dabei auf Literaturangaben zurück-greifen.
- Ionisationswahrscheinlichkeit (bzw. –querschnitt) Q
Zur Berechnung des Ionisationsquerschnittes Qn,l für ein Elektron in Schale n mit der Nebenquantenzahl l wird gewöhnlich eine Formel benutzt, die 1930 von H. Bethe publiziert wurde [10.12]:
4 2n ,
, 2,,
2 e Z 2lnn l
n ln ln l
b m vQ
Em v E
⋅ ⋅ ⋅ ⋅ ⋅= ⋅
⋅ ⋅ (10.21.2)
(e: Elementarladung, m: Elektronen-Ruhemasse, v: Geschwindigkeit der Strahl-elektronen, En,l: Ionisierungsenergie, d. h. Energie der Absorptionskante, für das Elektron n,l, Zn: Zahl der Elektronen in Schale n, bn,l: quantenmechanischer Para-meter, der von Bethe unter Benutzung der Elektronen-Eigenfunktionen von Was-serstoff zu 0,2 ... 0,6 berechnet wurde). In nichtrelativistischer Näherung ist
20e
2 Bm
v U E⋅ = ⋅ = (10.21.3)

Kapitel 10 328
(UB: Beschleunigungsspannung, E0: Energie der Strahlelektronen), so dass wir für (10.21.2)
4n , 0
,0 , ,
e Z 4lnn l
n ln l n l
b EQ
E E E
⋅ ⋅ ⋅ ⋅= ⋅
⋅ (10.21.4)
schreiben können. In der Praxis wird die Formel (10.21.4) modifiziert: Die bn,l und
die Ionisierungsenergien werden nur hinsichtlich der Hauptquantenzahl unter-schieden und die 4 im Logarithmus wird durch eine weitere Konstante cn ersetzt:
4n 0
0
e Zlnn n
nn n
b c EQ
E E E
⋅ ⋅ ⋅ ⋅= ⋅
⋅ . (10.21.5)
Die Konstanten bn und cn werden durch Anfitten an Messergebnisse bestimmt. Mit dem „Überspannungsverhältnis“ UÜ = E0/En wird aus Gleichung (10.21.5)
( )4
n2
e Zln
n
nn n Üd
n Ü
bQ c U
E U
⋅ ⋅ ⋅= ⋅ ⋅
⋅ . (10.21.6)
Der Exponent dn am Überspannungsverhältnis wird nach Schreiber und Wims [10.13] hinzugefügt, um eine bessere Übereinstimmung mit Messwerten zu errei-chen. In Tabelle 10-5 sind die Anpassungsparameter nach [10.13] und Powell [10.14] zusammengestellt (Z: Ordnungszahl).
Powell [10.14] Schreiber und Wims [10.13] (Z ist hier Ordnungszahl)
K-Schale
(n = 1,
Z1 = 2)
cK = 0,94
bK = 0,64
dK = 1
cK = 1
bK= 8,874-8,158⋅lnZ+2.9055⋅(lnZ)2 -0,35778⋅(lnZ)3 für Z≤30
bK= 0,661 für Z>30
dK = 1,0667-0,00476⋅Z
L-Schale
(n = 2,
Z2 = 8)
cL = 0,59
bL = 0,63
dL = 1
cL = 1
bL = 0,2704+0,00726⋅(lnZ)3
dL = 1
M-Schale
(n = 3,
Z3 = 18)
cM = 1
bM = 11,33-2,43⋅lnZ
cM = 1
Tabelle 10-5. Anpassungsparameter c, b und d zur Berechnung von Ionisationsquerschnitten.
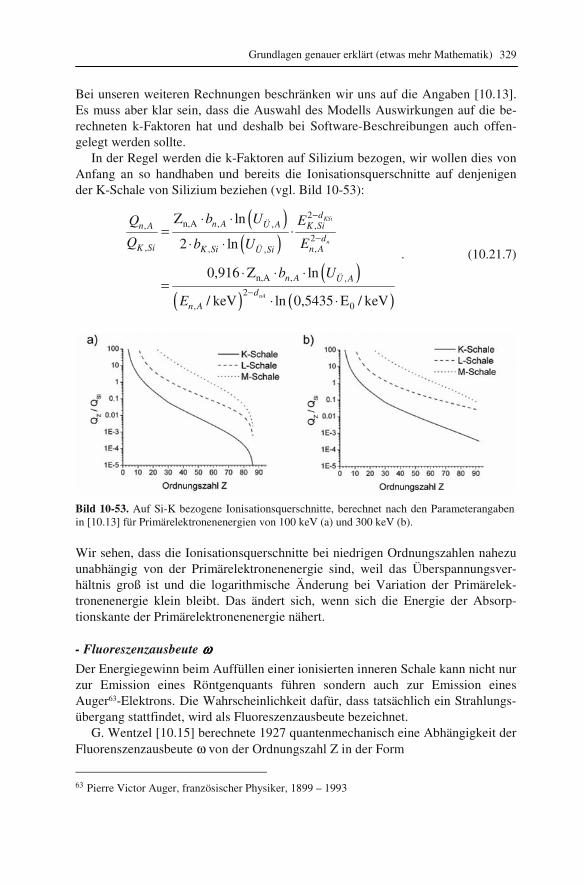
Grundlagen genauer erklärt (etwas mehr Mathematik) 329
Bei unseren weiteren Rechnungen beschränken wir uns auf die Angaben [10.13]. Es muss aber klar sein, dass die Auswahl des Modells Auswirkungen auf die be-rechneten k-Faktoren hat und deshalb bei Software-Beschreibungen auch offen-gelegt werden sollte.
In der Regel werden die k-Faktoren auf Silizium bezogen, wir wollen dies von Anfang an so handhaben und bereits die Ionisationsquerschnitte auf denjenigen der K-Schale von Silizium beziehen (vgl. Bild 10-53):
( )( )
( )
( ) ( )
2n,A , ,, ,
2, ,, ,
n,A , ,
2
, 0
Z ln
2 ln
0,916 Z ln
/ keV ln 0,5435 E / keV
KSi
n
nA
dn A Ü An A K Si
dK Si n AK Si Ü Si
n A Ü A
d
n A
b UQ E
Q Eb U
b U
E
−
−
−
⋅ ⋅= ⋅
⋅ ⋅
⋅ ⋅ ⋅=
⋅ ⋅
. (10.21.7)
Bild 10-53. Auf Si-K bezogene Ionisationsquerschnitte, berechnet nach den Parameterangaben in [10.13] für Primärelektronenenergien von 100 keV (a) und 300 keV (b).
Wir sehen, dass die Ionisationsquerschnitte bei niedrigen Ordnungszahlen nahezu unabhängig von der Primärelektronenenergie sind, weil das Überspannungsver-hältnis groß ist und die logarithmische Änderung bei Variation der Primärelek-tronenenergie klein bleibt. Das ändert sich, wenn sich die Energie der Absorp-tionskante der Primärelektronenenergie nähert.
- Fluoreszenzausbeute ωωωω
Der Energiegewinn beim Auffüllen einer ionisierten inneren Schale kann nicht nur zur Emission eines Röntgenquants führen sondern auch zur Emission eines Auger63-Elektrons. Die Wahrscheinlichkeit dafür, dass tatsächlich ein Strahlungs-übergang stattfindet, wird als Fluoreszenzausbeute bezeichnet.
G. Wentzel [10.15] berechnete 1927 quantenmechanisch eine Abhängigkeit der Fluorenszenzausbeute ω von der Ordnungszahl Z in der Form
63 Pierre Victor Auger, französischer Physiker, 1899 – 1993

Kapitel 10 330
4
4
Z
ZAω =
+ (10.21.8)
(A ≈ 106), die Grundlage für alle weiteren Untersuchungen zu dieser Problematik ist. Ähnlich wie beim Ionisationsquerschnitt wurde diese Formel mit weiteren Parametern versehen, um eine bessere Übereinstimmung mit Messwerten zu er-reichen (Zusammenstellung in [10.16]). Eine Variante nach E.H.S. Burhop [10.17] basiert auf der Formel
( )
( )
43
43
Z Z
1 Z Z
A B C
A B Cω
+ ⋅ + ⋅=
+ + ⋅ + ⋅
(10.21.9)
Die Parameter A, B und C unterscheiden sich für die Schalen und sind in Tabelle 10-6 zusammengestellt.
A B C
K-Schale -0,03795 0,03426 -1,163⋅10-6
L-Schale -0,11107 0,01368 -2,177⋅10-7
M-Schale -0,00036 0,00368 2,010⋅10-7
Tabelle 10-6. Parameter A, B und C zur Berechnung von Fluoreszenzausbeuten nach Gleichung (10.21.9) – s. [10.17].
Wir beziehen auch die Fluoreszenzausbeuten auf diejenige der Si-K-Schale und erhalten dafür die Formel
( )
( )
43n n,
43,n n
Z Z28,05
1+ Z Z
n n nA n
Si Kn n n
A B C
A B C
ω
ω
+ ⋅ + ⋅= ⋅
+ ⋅ + ⋅
(10.21.10)
(grafische Darstellung s. Bild 10-54).
Bild 10-54. Auf Si-K bezogene Fluoreszenz-ausbeuten, berechnet nach den Parameterangaben in Tabelle 10-6.

Grundlagen genauer erklärt (etwas mehr Mathematik) 331
- Kαααα-Anteil a an der K-Serie:
Nach Ionisation der K-Schale kann das Auffüllen sowohl aus der L-Schale (Kα-Strahlung) als auch (falls es sie gibt) aus der M-Schale (Kβ-Strahlung) erfolgen. Bei Silizium liegt die Kα-Linie bei 1,74 keV, die Kβ-Linie bei 1,83 keV. Beide Linien rücken bei Elementen mit höheren Ordnungszahlen auseinander, d. h. sie sind trotz des begrenzten Energie-Auflösungsvermögens des Detektors von etwa 130 eV im Spektrum getrennt. Wir benötigen das Intensitätsverhältnis Kβ/Kα. Ein Polynom der Form
2 3Z Z ZK
K
IA B C D
Iβ
α
= + ⋅ + ⋅ + ⋅ (10.21.11)
(A, B, C, D s. Tabelle 10-7) gibt die Werte von Scofield [10.18] recht gut wieder (s. Bild 10-55).
Z A B C D
11 ≤ Z ≤ 35 -0,9834 0,1282 -0,00486 6,134⋅10-5
Z > 35 -0,065 0,0102 -1,12 4,8⋅10-7
Tabelle 10-7. Parameter zur Berechnung des Kβ/Kα-Intensitätsverhältnisses.
Für den Anteil a gilt damit:
,
, , , ,
1
+ 1+K A
AK A K A K A K A
Ia
I I I Iα
α β β α
= = . (10.21.12)
Bild 10-55. Verhältnis der Kβ- zur Kα-Intensität in Abhängigkeit von der Ordnungszahl. a) Vergleich von Messwerten mit Polynom (10.21.11). b) Anteil a bezogen auf Si.

Kapitel 10 332
Für Silizium ist aSi = 0,9736. Damit sind alle Voraussetzungen für die Berechnung der k-Faktoren gegeben. Wir multiplizieren die auf Silizium bezogenen Einfluss-faktoren (Ergebnis s. Bild 10-56):
,, , A
, , ,
M bzw.
28,09eff SiK Si Si K SiA A A
Si n A A n A eff A Si SiM
DQ ak k k
k Q a D k k
ω
ω= ⋅ ⋅ ⋅ = ⋅ . (10.21.13)
Bild 10-56. Auf Si bezogene Cliff-Lorimer-k-Faktoren für Atomprozent (a) und Masseprozent (b) für 300 keV-Elektronen. Die zugrunde liegenden Modelle sind im Text erläutert.
In den Kurvenverläufen fallen Sprünge auf. Sie liegen bei Röntgenenergien, für die die Detektoreffizienz infolge der Absorptionskanten von Fenster- und Elektro-denmaterial nicht-monoton ist.
10.22 Absorptionskorrektur bei EDXS Obwohl die Proben für die Transmissionselektronenmikroskopie sehr dünn sind, kann niederenergetische Röntgenstrahlung beim Durchgang durch die Probe merklich geschwächt werden und damit bei der Quantifizierung zu einer Unter-bewertung der leichten Elemente führen. Wir wollen in diesem Abschnitt ab-schätzen, unter welchen Umständen dieser Effekt eine Rolle spielt.
Bild 10-57 veranschaulicht das Problem. Längs des Weges z der Elektronen durch die Probe entsteht in dem Wegelement dz Röntgenstrahlung der Intensität dI0, insgesamt auf dem gesamten Weg von z = 0 bis z = z0 die Intensität I0. Bei konstanter Anregung auf dem gesamten Weg gilt:
00
0
IdI dz
z= ⋅ . (10.22.1)

Grundlagen genauer erklärt (etwas mehr Mathematik) 333
Bild 10-57. Skizze zur Erläuterung der Absorption von Röntgenstrahlung in einer dünnen Probe der Dicke t.
Auf dem Weg zum Detektor muss die Röntgenstrahlung die Strecke s(z) innerhalb der Probe zurücklegen, wobei sie teilweise absorbiert wird. In Detektorrichtung tritt aus der Probe an der Stelle z nur noch der Anteil dI aus (vgl. Abschnitt 10.20):
( ) ( ) ( )0 e
Es zdI z dI μ ρ ρ− ⋅ ⋅
= ⋅ (10.22.2)
(E(μ/ρ): Massenschwächungskoeffizient des Probenmaterials für Röntgenstrahlung der Energie E, ρ: Dichte des Probenmaterials). Aus Bild 10-57 lesen wir
( ) cots z z δ= ⋅ (10.22.3)
ab. Den Detektor erreicht die Gesamtintensität
( )0
cot0
0 0
eE
zzI
I dzz
μ ρ ρ δ− ⋅ ⋅ ⋅= ⋅ ⋅ , (10.22.4)
d. h. nach Auflösen des Integrals:
( )
( )
0cot0
00
e
cot
E zz
E
II
z
μ ρ ρ δ
μ ρ ρ δ
− ⋅ ⋅ ⋅−
= ⋅⋅ ⋅
(10.22.5)
und Einsetzen der Integrationsgrenzen sowie Berücksichtigung von

Kapitel 10 334
0 cos cot sin
t tz
δ δ δ= =
⋅ (10.22.6)
erhalten wir
( )
( ) sin0 sin1 e
Et
E
II
t
μ ρ ρ δδ
μ ρ ρ
− ⋅ ⋅⋅= ⋅ −
⋅ ⋅ . (10.22.7)
Den Quotienten aus der am Detektor ankommenden Röntgenintensität I und der ursprünglich in der Probe ausgelösten Intensität I0 interpretieren wir als Absorp-tionskorrektur
( )
( ) sin
0
sin1 e
Et
Absk E
IF
I t
μ ρ ρ δδ
μ ρ ρ
− ⋅ ⋅= = ⋅ −
⋅ ⋅ . (10.22.8)
Für die Spektren-Quantifizierung ist der Vergleich von Elementen mit unter-schiedlichen charakteristischen Röntgenenergien wichtig. Allerdings hängen Schwächungskoeffizient und Dichte von der Materialzusammensetzung in der Probe ab, und es ist nicht sicher, ob sich beide in sehr dünnen Proben genauso ver-halten wie in kompaktem Material. Eine Näherung geht davon aus, dass dies so ist, und sowohl Schwächungskoeffizienten als auch Dichte mit der Konzentration gewichtete Linearkombinationen der Werte für die elementaren Bestandteile der Probe sind. Bei einer Probe, die aus zwei Elementen A und B mit den Masse-Konzentrationen cA.M und cB,M besteht, bedeutet dies:
( ) ( ) ( ), ,E E E
A M B MA Bc cμ ρ μ ρ μ ρ= ⋅ + ⋅ (10.22.9)
bzw.
, ,A M A B M Bc cρ ρ ρ= ⋅ + ⋅ . (10.22.10)
Für das Verhältnis der Absorptionskorrekturen für die Elemente A und B mit den Röntgenenergien EA und EB folgt:
( )
( )
( )
( )
sin,
sin,
1 e
1 e
EA
EB
EB tAbsk A
EA tAbsk B
F
F
μ ρ ρ δ
μ ρ ρ δ
μ ρ
μ ρ
− ⋅ ⋅
− ⋅ ⋅
−= ⋅
− . (10.22.11)
Da die Schichtdicke in der Größenordnung 0,1 μm liegt, entwickeln wir die Expo-nentialfunktionen in Reihen und brechen diese nach dem quadratischen Glied ab:

Grundlagen genauer erklärt (etwas mehr Mathematik) 335
( )
( )
( ) ( )
( ) ( )
( )
( )
2 2 2 21, 2
2 2 2 21, 2
12
12
sin sin
sin sin
1 sin
1 sin
EB EA EAAbsk A
EA EB EBAbsk B
EA
EB
F t t
F t t
t
t
μ ρ μ ρ ρ δ μ ρ ρ δ
μ ρ μ ρ ρ δ μ ρ ρ δ
μ ρ ρ δ
μ ρ ρ δ
⋅ ⋅ − ⋅ ⋅ ⋅= ⋅
⋅ ⋅ − ⋅ ⋅ ⋅
− ⋅ ⋅ ⋅=
− ⋅ ⋅ ⋅
.
(10.22.12)
Diese Funktion entwickeln wir wieder in eine Reihe und brechen nach dem zweiten Glied ab:
( ) ( )( ),
,
11
2 sinEB EAAbsk A
Absk B
F t
F
ρμ ρ μ ρ
δ
⋅= + − ⋅ . (10.22.13)
Soll der Fehler infolge Absorption kleiner als beispielweise 10% gehalten werden, muss
( ) ( )( )10,1
2 sinEB EAtρ
μ ρ μ ρδ
⋅⋅ ⋅ − < . (10.22.14)
gelten. Bis auf den Faktor 1/sinδ stimmt diese Formel mit dem „Dünnschicht-kriterium“ von Goldstein et al. [10.19] überein. Der „Abnahmewinkel“ δ besteht aus zwei Anteilen: Dem Kippwinkel der Probe wie in Bild 10-57 dargestellt und einem Beitrag, der entsteht, wenn die Detektorachse höher als die Kippachse der Probe liegt.
10.23 Prismen für Elektronen Um Elektronenenergieverlust-Spektren erzeugen zu können, benötigen wir ein Dispersionselement für Elektronen, ähnlich einem Glasprisma für Licht. Das Glas-prisma zerlegt weißes Licht in seine Farben, d. h. unterschiedliche Wellenlängen verlassen das Prisma unter verschiedenen Winkeln. Wir wissen, dass Elektronen in elektrischen und magnetischen Feldern abgelenkt werden und wollen überlegen, unter welchen Winkeln Elektronen mit unterschiedlicher Geschwindigkeit der-artige Felder verlassen.
Wir stellen uns ein magnetisches Feld vor, dessen Feldlinien senkrecht aus der Zeichenebene herausragen (s. Bild 10-58a). Die magnetische Induktion B hat die gleiche Richtung. Elektronen mit der ursprünglichen Energie, d. h. solche, die keinen Energieverlust erlitten haben, fallen senkrecht mit der Geschwindigkeit v0 in dieses Magnetfeld ein und erfahren die Lorentzkraft

Kapitel 10 336
e= − ⋅ ×L 0F v B . (7.30)
Der Geschwindigkeitsvektor liegt in der Zeichenebene, die magnetische Induktion steht senkrecht auf ihr, d. h. Geschwindigkeit und Induktion bilden einen rechten Winkel. Der Betrag der Lorentzkraft ist damit gleich dem Produkt der Elementar-ladung e mit den Beträgen von Geschwindigkeit v0 und magnetischer Induktion B:
0eLF v B= ⋅ ⋅ . (10.23.1)
Bild 10-58. Magnetisches (a) und elektrostatisches (b) Prisma für Elektronen (Erläuterungen im Text).
Gemäß (7.30) steht diese Kraft senkrecht auf der von v0 und B aufgespannten Ebe-ne, d. h. immer senkrecht auf der Bahn des Elektrons. Dies ist Kennzeichen einer Radialkraft
20
0r
vF m
r= ⋅ (10.23.2)
bei der Kreisbewegung mit dem Radius r0 (m: Elektronenmasse):
20
00
ev
m v Br
⋅ = ⋅ ⋅ , (10.23.3)
d. h. das Elektron mit der Geschwindigkeit v0 beschreibt im Magnetfeld eine Kreisbahn mit dem Radius
00 e
m vr
B
⋅=
⋅ . (10.23.4)

Grundlagen genauer erklärt (etwas mehr Mathematik) 337
Ein Elektron, das beim Durchgang durch die Probe einen Energieverlust erlitten hat, besitzt eine kleinere Geschwindigkeit v und beschreibt eine Kreisbahn mit dem Radius
e
m vr
B
⋅=
⋅ . (10.23.5)
Aus Bild 10-58a lesen wir für den Winkel α ab:
( )0 0 0sin 1 1r r r vr
r r r vα
−Δ= = = − = − . (10.23.6)
In nichtrelativistischer Näherung gilt für den Zusammenhang zwischen Geschwin-digkeit v und Energie E:
2v E
m= ⋅ , (10.23.7)
d. h.
0 0
0
sin 1 1E E
E E Eα = − = −
−Δ . (10.23.8)
Für ΔE << E0 ist sinα ≈ α und nach Reihenentwicklung von (10.23.8) und Ab-bruch nach dem linearen Term folgt
02
E
Eα
Δ=
⋅ , (10.23.9)
d. h. ein linearer Zusammenhang zwischen Ablenkwinkel und Energieverlust. Wir wollen uns einer zweiten Möglichkeit zuwenden: dem elektrostatischen
Prisma, wie es in Bild 10-58b skizziert ist. Voraussetzung sind zwei kreisförmig gekrümmte Platten im Abstand d, an denen eine elektrische Spannung U anliegt. Diese Spannung ruft unter der Voraussetzung, dass d sehr klein gegen den mittleren Krümmungsradius rM ist, zwischen den Platten näherungsweise eine elektrische Feldstärke
Feld
UE
d= (10.23.10)
hervor. Wir stellen uns vor, dass die elektrostatische Kraft durch geeignete Wahl einer Plattenspannung UM die Elektronen mit der Geschwindigkeit v0 gerade auf

Kapitel 10 338
eine Kreisbahn mit dem Radius rM zwingt. Im Koordinatensystem, das mit dem Elektron mitbewegt wird, herrscht zwischen elektrischer und Zentrifugalkraft in diesem Fall Gleichgewicht:
20 e M
M
v Um
r d⋅ = ⋅ . (10.23.11)
Die nach innen gerichtete Kraft F, die auf ein Elektron mit der Geschwindigkeit v wirkt, ist gleich der Differenz aus elektrostatischer und Zentrifugalkraft:
2
e MU vF m
d r= ⋅ − ⋅ . (10.23.12)
Für die nach innen gerichtete Beschleunigung a gilt damit:
2e MUF va s
m m d r= = = ⋅ − (10.23.13)
und nach zweimaliger Integration über die Zeit t für den Weg:
2 2e
2MU v t
sm d r
= ⋅ − ⋅ . (10.23.14)
Andererseits legt das Elektron in der Zeit t im gekrümmten Prisma die Strecke
l v t= ⋅ (10.23.15)
zurück. Aus (10.23.14) und (10.23.15) folgt für die Bahnkurve:
( ) 22
1 e 1
2MU
s l lm d v r
= ⋅ − ⋅⋅
. (10.23.16)
Mit r ≈ rM folgt für die Winkelabweichung von der Kreisbahn mit dem Radius rM (erste Ableitung s’(l)) unter Berücksichtigung von (10.23.11):
( )20
2M M
v 1s' l l
r v r= − ⋅
⋅ . (10.23.17)
An der Stelle l = π⋅rM/2 (Austritt aus dem Prisma) gilt damit:

Grundlagen genauer erklärt (etwas mehr Mathematik) 339
( )201
2 2' tan 1
2M
vs r
vα⋅ = = − (10.23.18)
bzw.
0tan 12 2
E E
E Eα
Δ= − = ⋅ (10.23.19)
Für ΔE << E0 und kleine Winkel gilt hier:
02
E
Eα
Δ= ⋅ , (10.23.20)
also ebenfalls ein linearer Zusammenhang. Für die technische Realisierung werden magnetische Prismen als Ablenkein-
heiten bevorzugt, da unter typischen TEM-Bedingungen Magnetfelder der Stärke 5 mT den gleichen Ablenkradius wie elektrische Felder der Stärke 10 kV/cm erreichen.
10.24 Faltung von Funktionen Die Faltung von Funktionen ist eine Prozedur, die dazu dient, Überlagerungen verschiedener Einflüsse, die zu „Verwaschungen“ von Messergebnissen führen, zu berechnen. Die Überlagerung verschiedener Fehlerscheibchen bei der Berechnung des optischen Auflösungsvermögens und die Verbreiterung von Peaks in energie-dispersiven Röntgenspektren oder Elektronenenergieverlust-Spektren infolge des begrenzten energetischen Auflösungsvermögens sind Beispiele dafür.
Wir wollen das Prinzip der Faltung von Funktionen an einem einfachen Bei-spiel erläutern: Wir stellen uns vor, dass bei einem Fertigungsprozess Stopfen in das Ende eines Rohres eingepresst werden und die Rohrenden dadurch etwas aufgeweitet werden. Diese Aufweitung soll gleich der Differenz aus Stopfen-durchmesser und Rohrinnendurchmesser sein. Die Rohrwandstärke sei bei allen Rohren gleich, die Häufigkeitsverteilungen r(d) der Rohrinnen- und der Rohr-außendurchmesser vor dem Einpressen der Stopfen sind damit auch gleich. Diese Häufigkeitsverteilung sowie diejenige der Stopfendurchmesser s(d) seien bekannt. Gesucht wird die Häufigkeitsverteilung R(d) der Rohrdurchmesser nach dem Ein-pressen der Stopfen. Zur Messung der Häufigkeitsverteilungen werden die Durch-messer in Intervalle der einheitlichen Breite Δd aufgeteilt. Jeder Durchmesser kann damit als Produkt aus einer ganzen Zahl i und der Intervallbreite Δd dar-gestellt werden:
iid d= ⋅Δ . (10.24.1)

Kapitel 10 340
Die Rohrdurchmesser vor dem Einpressen sollen von dr1 bis dr2 variieren, die Durchmesser der Stopfen von ds1 bis ds2. Die zugehörigen ganzen Zahlen seien ir1, ir2, is1 und is2. Durch Normierung nach den Vorschriften
( ) r2
r1
i
ri=i
mit A =ir ii i
r r
r d rw r
A A= = (10.24.2)
und
( ) s2
s1
i
si=i
mit A =is ii i
s s
s d sw s
A A= = (10.24.3)
erhalten wir daraus die Wahrscheinlichkeiten rwi und swi. Der kleinste Durchmesser Rmin nach dem Einpressen entsteht, wenn der kleinste
Rohrdurchmesser rmin = ir1⋅Δd mit dem kleinsten Stopfendurchmesser smin = is1⋅Δd kombiniert wird:
( )min min min r1 s1i iR r s d= + = + ⋅Δ . (10.24.4)
Die Wahrscheinlichkeit Rwmin für das Auftreten von Rmin ist gleich dem Produkt aus den Wahrscheinlichkeiten von r0 und s0:
min ir1 is1R r sw w w= ⋅ . (10.24.5)
Wir können die Zählung der Durchmesser verallgemeinern und generell bei Null beginnen. Nicht vorhandene Durchmesserwerte haben die Wahrscheinlichkeit Null:
( )0 0 0 0 0R r s d= + = + ⋅Δ (10.24.6)
mit
0 0 0R r sw w w= ⋅ . (10.24.7)
Der nächstgrößere Durchmesserwert kann durch zwei Kombinationen erreicht werden:
( ) ( )1 1 0 0 11 0 0 1R r s d r s d= + = + ⋅Δ = + = + ⋅Δ (10.24.8)
mit

Grundlagen genauer erklärt (etwas mehr Mathematik) 341
1 1 0 0 1R r s r sw w w w w= ⋅ + ⋅ . (10.24.9)
Dies lässt sich fortsetzen bis zum größten möglichen Wert
( )max max max r2 s2i iR r s d= + = + ⋅Δ (10.24.10)
mit
max ir2 is2R r sw w w= ⋅ . (10.24.11)
Allgemein gilt offenbar für die Wahrscheinlichkeit, mit der ein Durchmesserwert Ri auftritt:
0
iR r s
i k i kk
w w w −
=
= ⋅ . (10.24.12)
Nach Einsetzen von zwei Funktionen f(x) und g(x) anstelle der Wahrscheinlich-keiten w und dem Übergang zu infinitesimal kleinen Intervallbreiten dx erhalten wir für das Resultat F(ξ) die bekannte Formel
( ) ( ) ( )F f x g x dxξ ξ∞
−∞
= ⋅ − ⋅ (10.24.13)
für Faltungsoperationen. Zwei Auswirkungen dieser Faltung wollen wir anhand von Bild 10-59 demon-
strieren. In Bild 10-59b ist zu sehen, wie zwei scharfe Linien verbreitert werden. Zwischen beiden Linien erreicht die Funktion nach Faltung nicht mehr den Ordinatenwert Null. In Bild 10-59c wird die Ausgangsfunktion durch zwei Gauß-Funktionen mit unterschiedlicher Halbwertsbreite gebildet, deren Maxima gegen-einander verschoben sind..
Bild 10-59. Faltung mit einer Gauß-Kurve. a) Gauß-Kurve, mit der gefaltet wurde. b) Faltung zweier scharfer δ-Funktionen mit der Gauß-Kurve (a). c) Faltung zweier Gauß-Kurven mit gleicher maximaler Höhe aber unterschiedlichen Halbwertsbreiten mit der Gauß-Kurve (a).

Kapitel 10 342
Im Ergebnis der Faltung entsteht neben der Verbreiterung auch ein Unterschied in der Höhe der beiden Maxima.
Die Umkehrung, d. h. die Entfaltung von Messkurven ist schwieriger. Theore-tisch erscheint eine numerische Entfaltung möglich, versagt aber in der Praxis, weil die Wahrscheinlichkeiten vom Rand beginnend berechnet werden müssen und die Folgewerte jeweils aufeinander aufbauen. Die kleinen Randwerte sind aber nicht zuletzt wegen des Rauschens extrem unsicher. Prinzipiell gibt es zwei Möglichkeiten, trotzdem eine Entfaltung zu versuchen: Die Vorgabe einer ver-muteten Funktion mit anschließender Faltung und Vergleich mit dem Messer-gebnis oder die Rechnung im Fourierraum. Eine Faltungsoperation entspricht einer Multiplikation der Fouriertransformierten mit anschließender Rücktrans-formation in den Ortsraum. Eine Entfaltung wäre dann entsprechend eine Division der Fouriertransformierten. Allerdings täuscht auch hierbei das Rauschen oft Frequenzen und damit Fourierkoeffizienten vor, die in Wirklichkeit nicht vor-handen sind. Die entfaltete Messkurve wird unter Umständen wellig und bekommt eine vorgetäuschte Feinstruktur.
Die Faltung von Funktionen hat prinzipielle Bedeutung für die Interpretation von Messergebnissen. Sie ist die mathematische Grundlage für die Berücksich-tigung der Überlagerung mehrerer Einflüsse auf das Messergebnis, beispielsweise durch das begrenzte Auflösungsvermögen eines Spektrometers, oder durch inela-stische Mehrfachstreuung bei EELS, wie das in Abschnitt 9.6.3 gezeigt wird.

Resümee und Ausblick In den Fußnoten der zehn Kapitel dieses Buches stehen die Namen von 62 be-kannten Wissenschaftlern und Technikern, deren Arbeiten zum Fundament und zur Entwicklung der Transmissionselektronenmikroskopie beigetragen haben. 20 davon sind Nobelpreisträger: „Die Elektronenmikroskopie steht auf den Schultern von Giganten.“
Hans-Dietrich Bauer, der sich bereits in den 1960-er Jahren im Institut von Al-fred Recknagel an der Technischen Universität Dresden mit den experimentellen Problemen beim Einsatz von Vierpollinsen im Elektronenmikroskop beschäftigt hatte, begann damals seine Vorlesung über Durchstrahlungselektronenmikrosko-pie mit zwei (nicht ganz ernst gemeinten) Hauptsätzen der Elektronenmikrosko-pie:
1. Es ist unmöglich, kein Bild zu erhalten! 2. Es ist unmöglich, ein scharfes Bild zu erhalten! Der Hintergrund für den „2. Hauptsatz“ ist die Tatsache, dass in rotationssym-
metrischen und raumladungsfreien Feldern, die zeitlich konstant sind, der Öff-nungsfehler unvermeidlich ist. Der Ausweg war eigentlich klar: Die Verwendung von Multipolen anstelle rotationssymmetrischer Linsen. Trotzdem hat es fast bis zum Jahre 2000 gedauert, bis solche Multipolelemente zur Öffnungsfehlerkor-rektur in kommerzielle Geräte eingebaut werden konnten. Dazu bedurfte es zum einen der Ideen von Harald Rose, Max Haider und Knut Urban und zum anderen schneller Computer. Schließlich muss man in der Lage sein, den Öffnungsfehler in vertretbarer Zeit zu messen, wenn man ihn korrigieren will. Dazu ist hohe Rech-nerleistung erforderlich.
Nach der Korrektur des Öffnungsfehlers erreicht das Auflösungsvermögen das Informationslimit. Damit rückt die Farbfehlerkorrektur in den Mittelpunkt des In-teresses. Auch dies ist mit Multipoleinheiten möglich, in denen elektrostatische und magnetische Dipole kombiniert sind. Damit wird das Informationslimit zu hö-heren Raumfrequenzen, d.h. zu kleineren Abständen verschoben.
Das Informationslimit wird aber nicht allein vom Farbfehler bestimmt. Me-chanische Erschütterungen, Temperaturschwankungen und äußere magnetische Wechselfelder beeinflussen es ebenfalls. Mit der Verbesserung der Elektronenlin-sen richtet sich das Augenmerk auf diese Umgebungseinflüsse: Moderne Höchst-leistungsmikroskope werden in einem vollständig geschlossenen Gehäuse aufge-stellt. Als Labore werden Häuser mit speziellen Fundamenten fernab von ver-kehrsreichen Innenstädten genutzt. Solche Häuser werden dann nur für das Elek-tronenmikroskop gebaut.
Eine Folge dieser Entwicklung ist es, dass nicht mehr allein das Auflösungs-vermögen, das heißt, das Vermögen kleinste Abstände getrennt wahrzunehmen, betrachtet wird, sondern auch die Genauigkeit, mit der solche kleinsten Abstände gemessen werden können. Hier ist man inzwischen im Pikometer-Bereich ange-langt und hat damit die Möglichkeit, Abweichungen einzelner Atompositionen im Kristallgitter zu bestimmen.
DOI 10.1007/978-3-7091-1440-7, © Springer-Verlag Wien 2013 J. Thomas, T. Gemming, Analytische Transmissionselektronenmikroskopie,

Resümee und Ausblick 344
In der analytischen Transmissionselektronenmikroskopie ist die hohe Orts-auflösung allerdings nur ein Aspekt. Sie ist zu kombinieren mit Röntgen- und Elektronenenergieverlust-Spektroskopie. Für die Messung von Bindungszuständen ist eine hohe Energieauflösung des Energieverlustspektrometers im Bereich von wenigen 0,1 eV wünschenswert. Bei Strukturen im Subnanometerbereich ist die Messzeit durch (geringste) mechanische Drift begrenzt. Für ein ausreichendes Signal-Rausch-Verhältnis ist es wichtig, den Strahlstrom besonders in der raster-transmissionselektronenmikroskopischen Arbeitsweise zu erhöhen und die Effi-zienz der Detektoren zu verbessern. Für den hohen Strahlstrom werden dazu ein Strahlerzeuger mit hohem Richtstrahlwert (auserlesene Feldemissionskathoden) und ein Öffnungsfehlerkorrektor für das Kondensorsystem benötigt. Für die Rönt-genspektroskopie wird die Detektoreffizienz durch Vergrößerung des erfassten Raumwinkels, beispielsweise durch Einsatz mehrerer Detektoren, verbessert.
Es kostet viel Geld, die idealen Laborbedingungen zu schaffen, ganz zu schweigen von den Kosten für ein Höchstleistungsgerät. In Biologie und Werk-stoffforschung ist es oft auch gar nicht notwendig, ein solches Spitzengerät einzu-setzen. Elektronenbeugungsmethoden zur Phasenanalyse, Beugungskontrastunter-suchungen zur Bestimmung der realen Gitterstruktur und Abbildung von Zell-strukturen sind Beispiele, bei denen ein „normales“ Transmissionselektronen-mikroskop ausreicht. Wichtiger als Leistung an der Grenze des Machbaren ist in diesen Fällen, dass das Gerät „vor Ort“ steht, d. h. in unmittelbarer Nachbarschaft zu anderen Laboratorien. Schließlich dürfen wir in diesem Zusammenhang auch nicht vergessen, dass für die elektronenmikroskopische Untersuchung ultradünne Proben benötigt werden: Je anspruchsvoller die Elektronenmikroskopie ist, desto höher sind die Anforderungen an die Probenqualität.
Es gibt auch „gemischte“ Fälle, beispielsweise in der Halbleiterindustrie. Bei Halbleiterbauelementen können Dicken von Zwischenschichten im Nanometer-bereich mit der erforderlichen Genauigkeit nur mit dem Transmissionselektronen-mikroskop gemessen werden, welches zur Vermeidung von Ungenauigkeiten durch Delokalisation auch mit Öffnungsfehlerkorrektor ausgerüstet sein sollte. Das Gerät muss „vor Ort“ sein. Hieraus ergibt sich ein Wunsch an die Hersteller von Elektronenmikroskopen: Verringerung der Empfindlichkeit gegenüber Umge-bungseinflüssen.
Wenn der Leser nach dem Studium dieses Buches zu der Überzeugung ge-kommen ist, dass zur Bedienung eines Transmissionselektronenmikroskops und zur fundierten Interpretation elektronenmikroskopischer Ergebnisse mehr Wissen notwendig ist als lediglich „für einen bestimmten Zweck an einem bestimmten Knopf zu drehen“, dann haben wir als Autoren unser Hauptziel erreicht. Wir wün-schen allen gegenwärtigen und zukünftigen Elektronenmikroskopikern ein „glück-liches Händchen“, und denken Sie beim Interpretieren elektronenmikroskopischer Bilder bitte immer daran:
Glaube erst was du siehst, wenn du verstanden hast, warum du es siehst!

Physikalische Konstanten
Werte der physikalischen Konstanten, auf die im Text Bezug genommen wird:
Boltzmannsche Konstante: k = 1,381⋅10-23 J/K
Elementarladung: e = 1,602⋅10-19 A⋅s
Gaskonstante: R = 8,315 J/( mol⋅K)
Influenzkonstante: ε0 = 8,854⋅10-12 A⋅s/(V⋅m)
Lichtgeschwindigkeit im Vakuum: c = 2,998⋅108 m/s
Avogadro-Konstante: NA = 6,022⋅1023 /mol
Plancksches Wirkungsquantum: h = 6,626⋅10-34 J⋅s
Richardson-Konstante: A = 120 A⋅cm-2⋅K-2
Ruhemasse des Elektrons: m0 = 9,109⋅10-31 kg
Spezifische Ladung des Elektrons: e/m0 = 1,759⋅1011 A⋅s/kg
Kombinationen von Konstanten, die oft benötigt werden:
h c
e
⋅ = 1,24⋅10-6 V⋅m
20m c
e
⋅ = 511059 V
0
h
m c⋅ = 2,4263⋅10-12 m = 2,4263 pm
2
1v
c−
= 511,06 kV
511,06 kVBU + (UB: Beschleunigungsspannung)
0
1
4π ε⋅ = 8,99⋅109 V⋅m/(A⋅s)
Umrechnungen:
1 N = 1 kg⋅m/s2 1 J = 1 N⋅m = 1 V⋅A⋅s = 1 W⋅s = 6,242⋅1018 eV
1 Pa = 1 N/m2 1 Torr = 133 Pa 1 bar = 105 Pa
R = k⋅NA

Abkürzungsverzeichnis
ΔE Energiebreite
Δf Defokus
ΔfA astigmatische Brennweitendifferenz
Λ mittlere freie Weglänge
α Beugungswinkel, Apertur, Gitterachsenwinkel
β Bestrahlungsapertur, Gitterachsenwinkel
γ Gitterachsenwinkel
δ Auflösungsvermögen, Spaltabstand
δC Radius des Farbfehlerscheibchens
δD Delokalisation
δS Radius des Öffnungsfehlerscheibchens
θ Bragg-Winkel
λ Wellenlänge
ν Frequenz
Φ (Kristall-) Potential
φ Phase, Phasenschiebung
ρ Dichte
σ Sehwinkel, Streuquerschnitt
ϕ Winkel, Azimut bei Polar- und Zylinderkoordinaten
Ψ magnetisches Potential
ψ Wellenfunktion
Ω Raumwinkel
ω Kreisfrequenz, Fluoreszenzausbeute
a1,a2,a3 Gittervektoren
b1,b2,b3 reziproke Gittervektoren
B magnetische Induktion
b Burgers-Vektor
b Bildweite
CC Farbfehlerkonstante
CS Öffnungsfehlerkonstante
CTF Kontrastübertragungsfunktion
c Konzentration
D Dispersion
Deff Detektoreffizienz
d, dhkl Netzebenenabstand, allgemein: Abstand
E Energie, elektrische Feldstärke

Abkürzungsverzeichnis 348
E0 Primärelektronenenergie
EDXS Energy Dispersive X-ray Spectroscopy (energiedispersive Röntgenspektroskopie)
EELS Electron Energy Loss Spectroscopy (Elektronenenergieverlust-Spektroskopie)
ELNES Energy Loss Near Edge Fine Structure (kantennahe Feinstruktur)
EXELFS Extented Energy Loss Fine Structure (kantenferne Feinstruktur)
f Brennweite
F Kraft
Fhkl Strukturfaktor
G Gitterfaktor
g Gegenstandsweite
I Stromstärke, Intensität
H Helligkeit
hkl Millersche Indizes
i imaginäre Einheit, ganze Zahl
j Stromdichte, Gesamtdrehimpuls-Quantenzahl, ganze Zahl
k Wellenzahlvektor
kAB Cliff-Lorimer-k-Faktor
L Kameralänge
l Nebenquantenzahl
M Vergrößerung, ganze Zahl
Mr Atom- bzw. Molekulargewicht
m magnetische Quantenzahl
m, m0 Masse
N ganze Zahl
n Brechzahl, Hauptquantenzahl, ganze Zahl
p Impuls, Druck
Q Ionisationsquerschnitt (-wahrscheinlichkeit), elektrische Ladung
q Raumfrequenz
R Richtstrahlwert
r Radius (auch bei Polar- und Zylinderkoordinaten)
S, S deutliche Sehweite, Brechkraft
s Streuvektor, Spinquantenzahl
s, sopt Weg, Weglänge, optische Weglänge
STEM Scanning Transmission Electron Microscopy (Rastertransmissionselektronenmikroskopie)
T Schwingungsdauer, absolute Temperatur
TEM Transmission Electron Microscopy (Transmissionselektronenmikroskopie)
U elektrisches Potential, Untergrund, Spannung
UB Beschleunigungsspannung
v Geschwindigkeit

Abkürzungsverzeichnis 349
W Arbeit, potentielle Energie
WA Austrittsarbeit
WP potentielle Energie
x Ortskoordinate
y Dinggröße, Ortskoordinate
yD Delokalisation
y’ Bildgröße
Z Ordnungszahl
z Ortskoordinate (auch bei Zylinderkoordinaten), optische Achse
Hinweis: In den Bildern kennzeichnen wir Vektorgrößen durch einen kleinen Pfeil über dem Buchstaben, in Formeln und Text durch Fettdruck.

Literaturhinweise
Analytische Transmissionselektronenmikroskopie:
Picht, J., Heydenreich, J.: Einführung in die Elektronenmikroskopie, Technik-Verlag, Berlin (1966)
Reimer, L.: Elektronenmikroskopische Untersuchungs- und Präparations-methoden, Springer-Verlag, Berlin (1967)
v. Heimendahl, M.: Einführung in die Elektronenmikroskopie: Verfahren zur Untersuchung von Werkstoffen und anderen Festkörpern, Vieweg-Verlag, Braunschweig (1970)
Bethge, H., Heydenreich, J.: Elektronenmikroskopie in der Festkörperphysik, Deutscher Verlag der Wissenschaften, Berlin (1982)
Bauer, H.-D.: Analytische Transmissionselektronenmikroskopie, Beiträge zur Forschungstechnologie, Heft 13, Akademie-Verlag, Berlin (1986)
Alexander, H.: Physikalische Grundlagen der Elektronenmikroskopie, Teubner-Verlag, Stuttgart (1997)
Colliex, C.: Elektronenmikroskopie – Eine anwendungsbezogene Einführung, übersetzt und bearbeitet von H. Kohl, Wissenschaftliche Verlagsgesellschaft Stuttgart (2008)
Hirsch, P. B.: Electron Microscopy of Thin Crystals, Krieger Publishing Company (1977)
Hirsch, P. B.: Topics in Electron Diffraction and Microscopy of Materials, IOP Publishing Ltd. London (1999)
Ernst, F., Rühle, M. (Eds.): High-Resolution Imaging and Spectrometry of Materi-als, Springer-Verlag, Berlin (2003)
Sigle, W.: Analytical Transmission Electron Microscopy, Annual Rev. Mater. Res. 35, 239-314 (2005)
Reimer, L., Kohl, H.: Transmission Electron Microscopy, Springer-Verlag, Berlin (2008)

Literaturhinweise 352
Williams, D. B., Carter, C. B.: Transmission Electron Microscopy, Springer-Verlag, New York (2009)
Elektronenoptik:
Glaser, W.: Grundlagen der Elektronenoptik, Springer-Verlag, Wien (1952)
Hawkes, P. W., Kasper, E.: Principles of Electron Optics, Academic Press, Lon-don (1989)
Rose, H.: Geometrical Charged-Particle Optics, Springer-Verlag, Heidelberg (2009)
Elektronenbeugung:
Hahn, T. (Ed.): International Tables for Crystallography, Vol. A, Space-Group Symmetry, Kluwer Academic Publishers, Dordrecht, Boston, London (1996)
Champness, P. E.: Electron Diffraction in the Transmission Electron Microscope, BIOS Scientific Publishers Ltd, Oxford (2001)
Spence, J.C.H., Zuo, J. M.: Electron Microdiffraction, Plenum Press, New York (1992)
Hochauflösungs-Transmissionselektronenmikroskopie:
Hillebrand, R., Scheerschmidt, K., Neumann, W., Werner, P., Pippel, A.: Bildinterpretation in der Hochauflösungs-Elektronenmikroskopie, Beiträge zur Forschungstechnologie, Heft 11, Akademie-Verlag Berlin (1984)
Rastertransmissionselektronenmikroskopie:
Pennycook, S. J., Nellist, P. D. (Eds.): Scanning Transmission Electron Microscopy, Springer-Verlag, New York, Dordrecht, Heidelberg, London (2011)

Literaturhinweise 353
Röntgen- und Elektronenenergieverlust-Spektroskopie:
Eggert, F.: Standardfreie Elektronenstrahl-Mikroanalyse mit dem EDX im Rasterelektronenmikroskop – Ein Handbuch für die Praxis, Books on Demand GmbH, Norderstedt (2005)
Reimer, L. (Ed.): Energy-Filtering Transmission Electron Microscopy, Springer-Verlag, Berlin (1995)
Egerton, R. F.: Electron Energy-Loss Spectroscopy in the Electron Microscope, Plenum Press, New York, London (1996)
Elektronenmikroskopische Präparation:
Müller, H.: Präparation von technisch-physikalischen Objekten für die elektronenmikroskopische Untersuchung, Akademische Verl.-Gesellschaft Geest & Portig, Leipzig (1962)
Petzow, G.: Metallografisches, keramografisches, plastografisches Ätzen, Gebr. Bornträger, Berlin, Stuttgart (1994)
Ayache, J., Beaunier, L., Boumendil, J., Ehret, G., Laub, D.: Sample Preparation Handbook for Transmission Electron Microscopy - Methodology, Springer-Verlag, New York, Dordrecht, Heidelberg, London (2010)
Ayache, J., Beaunier, L., Boumendil, J., Ehret, G., Laub, D.: Sample Preparation Handbook for Transmission Electron Microscopy - Techniques, Springer-Verlag, New York, Dordrecht, Heidelberg, London (2010)
Im Internet existiert eine Vielzahl von Links zu Seiten über analytische Transmis-sionselektronenmikroskopie. Doch Vorsicht, nicht alles, was im Internet geschrie-ben wird, ist richtig!

Quellenverzeichnis
Bilder 1-1, 1-2: Gezeichnet unter Verwendung von CorelDraw-Cliparts der Corel Corporation
Biografische Daten: Wikipedia - die freie Enzyklopädie, http://de.wikipedia.org/wiki/Wikipedia Fouriertransformationen von Bildern teilweise unter Verwendung der Software Digital Micrograph, Version 3.11.0, Gatan Inc. Pleasanton, USA
[0.1] von Goethe, J. W.: Wilhelm Meisters Wanderjahre, ed. Erich Trunz, Goethes Werke – Hamburger Ausgabe Bd. 8, Romane und Novellen III, 12. Aufl. München, II/Betrachtungen im Sinne der Wanderer (1989), S. 293
[0.2] von Goethe, J. W.: ebenda, S. 283
[2.1] Scherzer, O.: Über einige Fehler von Elektronenlinsen, Zeitschrift f. Physik 101, 593-603 (1936)
[2.2] Haider, M., Müller, H., Uhlemann, S., Zach, J., Loebau, U., Hoeschen, R.: Prerequisites for a Cc/Cs-corrected ultrahigh-resolution TEM, Ultramicroscopy 108, 167-178 (2008)
[2.3] Boersch, H.: Experimentelle Bestimmung der Energieverteilung in thermisch ausgelösten Elektronenstrahlen, Zeitschr. f. Physik 139, 115-146 (1954)
[2.4] Bronsgeest, M. S., Barth, J. E., Schwind, G. A., Swanson, L. W., Kruit, P.: Extracting the Boersch effect contribution from experimental energy spread measurements for Schottky electron emitters, J. Vac. Sci. Technol. B25(6), 2049-2054 (2007)
[3.1] Lang, G.: Histotechnik – Praxislehrbuch für die biomedizinische Analytik, Springer-Verlag, Wien (2006)
[3.2] Allen, T. D. (Ed.): Introduction to Electron Microscopy for Biologists, Academic Press, Elsevier Inc. (2008)
[3.3] Ruska, H., v. Borries, B., Ruska, E.: Die Bedeutung der Übermikroskopie für die Virusforschung, Arch. ges. Virusforsch., 1, 155-169 (1939), DOI: 10.1007/BF01243399

Quellenverzeichnis 356
[3.4] Bethge, H.: Oberflächenstrukturen und Kristallbaufehler im elektronenmikroskopischen Bild, untersucht am NaCl, Physica status solidi (b) 2, 3-27 und 775-820 (1962)
[3.5] Galetzka, W., Gnägi, H., Godehardt, R., Lebek, W., Michler, G. H., Vastenhout, B.: Ultramikrotomie in der Materialforschung, Hanser-Verlag, München (2004)
[3.6] McCaffrey, J. P.: Small-angle cleavage of semiconductors for transmission electron microscopy, Ultramicroscopy 38, 149-157 (1991)
[3.7] Benedict, J., Anderson, R., Klepeis, S. J.: Recent Developments in the Use of the Tripod Polisher for TEM Specimen Preparation, Specimen Preparation for Transmission Electron Microscopy of Materials-III, MRS Symposium Proceedings 254, 121-140 (1992), doi:10.1557/PROC-254-121
[4.1] Ishizuka, K., Shirota, K.: Lens-field center alignment for high resolution electron microscopy, Ultramicroscopy 65, 71–79 (1996)
[4.2] Ruska, E.: Das Entstehen des Elektronenmikroskops und der Elektronen-mikroskopie, Nobelvortrag am 08. 12. 1986, Stockholm, veröffentlicht in: Phys. Bl. 43, 271ff (1987), s. auch: http://ernst.ruska.de/daten_d/bibliothek/dokumente/ 999.nobelvortrag/vortrag.html
[4.3] Heide, H. G.: Die Objektverschmutzung im Elektronenmikroskop und das Problem der Strahlenschädigung durch Kohlenstoffabbau, Zeitschr. angew. Physik 15, 116–128 (1963)
[5.1] Ewald, P. P.: Die Entdeckung der Röntgeninterferenzen vor zwanzig Jahren und zu Sir William Braggs siebzigstem Geburtstag, Naturwissenschaften 29, 527-530 (1932)
[5.2] Friedrich, W: Erinnerungen an die Entdeckung der Interferenzerscheinungen bei Röntgenstrahlen, Naturwissenschaften 36, 354-356 (1949)
[5.3] Schulze, G. E. R.: M. v. Laue und die Geschichte der Röntgenfeinstruktur-untersuchung, Krist. Techn. 8, 527-543 (1973)
[5.4] Davisson, C., Germer, L. H.: The Scattering of Electrons by a Single Crystal of Nickel, Nature 119, 558-560 (1927)
[5.5] Bragg, W. L.: The Specular Reflection of X-rays, Nature 90, 410 (1912)
[5.6] Bragg, W. H., Bragg, W. L.: The Reflections of X-rays by Crystals, Proc. Royal Soc. London, 88, 428-438 (1913)

Quellenverzeichnis 357
[5.7] Hahn, T. (Ed.): International Tables for Crystallography, Vol. A, Space-Group Symmetry, Kluwer Academic Publishers, Dordrecht, Boston, London (1996)
[5.8] Villars, P., Calvert, L. D.: Pearson's Handbook of Cryst. Data for Intermetallic Phases, ASM Intern. (1991)
[5.9] http://icsd.fiz-karlsruhe.de/icsd/
[5.10] Otte, H. M., Crocker, A. G.: Crystallographic Formulae for Hexagonal Lattices, phys. stat. sol. 9, 441-450 (1965)
[5.11] Nicholas, J.: The Simplicity of Miller-Bravais Indexing, Acta Crystallogr. 21, 880 – 881 (1966)
[5.12] Boersch, H.: Über das primäre und sekundäre Bild im Elektronen-mikroskop, Ann. d. Phys. 26, 631-644 (1936) und 27, 75-80 (1936)
[5.13] Kikuchi, S.: Japan. J. Phys. 5 (1928), 83 und Phys. Z. 31 (1930), 777, zitiert in: Möllenstedt, G.: My Early Work on Convergent-Beam Electron Diffraction, phys. stat. sol. (a) 116, 13-22 (1989)
[5.14] Doyle, P. A., Turner, P. S.: Relativistic Hartree-Fock X-ray and Electron Scattering Factors, Acta Crystallogr. A24, 390-397 (1968)
[5.15] Thomas, J., Gemming, T.: ELDISCA C# - a new version of the program for identifying electron diffraction patterns, in: Luysberg, M., Tillmann, K., Weirich, T. (Eds.), EMC 2008, Aachen, Vol. 1, Springer-Verlag, Berlin, Heidelberg, 231-232 (2008)
[5.16] Williams, D. B., Carter, C. B.: Transmission Electron Microscopy, Springer-Verlag, New York (2009), S. 299ff.
[5.17] Laue, M. von: Materiewellen und ihre Interferenzen, Akad. Verl.-Ges. Geest & Portig, Leipzig (1948)
[5.18] Vincent, R., Midgley, P. A.: Double conical beam-rocking system for measurement of integrated electron diffraction intensities, Ultramicroscopy 53, 271 – 282 (1994)
[5.19] Deininger, C., Mayer, J., Rühle, M.: Determination of the charge-density distribution of crystals by energy-filtered CBED, Optik 99, 135-140 (1995)
[6.1] Brückner, W., Thomas, J., Hertel, R., Schäfer, R., Schneider, C. M.: Magnetic domains in a textured Co nanowire, Journ. Magnetism and Magnetic Mat. 283, 82–88 (2004)

Quellenverzeichnis 358
[7.1] Stadelmann, P.: EMS – a software package for electron diffraction analysis and HREM image simulation in materials science, Ultramicroscopy 21, 131-146 (1987), benutzte Java-EMS-Version: 6.6201U2011
[7.2] Möbus, G., Phillipp, F., Gemming, T., Schweinfest, R., Rühle, M.: Quantita-tive diffractometry at 0.1 nm resolution for testing lenses and recording media of a high-voltage atomic resolution microscope, Journ. Electron Microsc. 46 (5), 381–395 (1997)
[7.3] Barthel, J., Thust, A.: Quantification of the Information Limit of Tranmission Electron Microscopes, Phys. Rev. Letters 101 , 200801-1 - 200801-4 (2008)
[7.4] Rose, H.: Correction of aberrations, a promising means for improving the spatial and energy resolution of energy-filtering electron microscopes, Ultramicroscopy 56, 11-25 (1994)
[7.5] Haider, M., Uhlemann, S., Schwan, E., Rose, H., Kabius, B., Urban, K.: Electron microscopy image enhanced, Nature 392, 768-769 (1998)
[7.6] Zemlin, F., Weiss, K., Schiske, P., Kunath, W., Herrmann, K.-H.: Coma-free alignment of high resolution electron microscopes with the aid of optical diffractograms, Ultramicroscopy 3, 49-60 (1978)
[7.7] Möbus, G., Schweinfest, R., Gemming, T., Wagner, T., Rühle, M.: Iterative structure retrieval techniques in HREM: a comparative study and a modular program package, Journ. of Microscopy 190 (1/2), 109-130 (1998)
[7.8] Thust, A., Lentzen, M., Urban, K.: Non-linear reconstruction of the exit plane wave function from periodic high-resolution electron microscopy images, Ultramicroscopy 53, 101-120 (1994)
[7.9] Thust, A., Coene, W. M. J., Op de Beek, M., Van Dyck, D.: Focal-series reconstruction in HRTEM: simulation studies on non-periodic objects, Ultramicroscopy 64, 211-230 (1996)
[7.10] Lichte, H., Lehmann, M.: Electron holography – basics and applications, Rep. Prog. Phys 71 1-46 (2008)
[8.1] Joy, D. C.: Monte Carlo Modeling for Electron Microscopy and Microanalysis, Oxford University Press, New York, Oxford (1995) [9.1] Kramers, H. A.: On the theory of X-ray absorption and of the continuous X-ray spectrum, Phil. Mag. 46, 836-871 (1923)
[9.2] Kreher, K.: Festkörperphysik, Akademie-Verlag, Berlin (1976), 25ff.

Quellenverzeichnis 359
[9.3] Gatti, E., Rehak, P.: Semiconductor drift chamber – an application of a novel charge transport scheme, Nuclear Instr. and Methods in Physics Research 225, 608-614 (1984)
[9.4] Lechner, P., Eckbauer, S., Hartmann, R., Krisch, S., Hauff, D., Richter, R., Soltau, H., Struder, L., Fiorini, C., Gatti, E., Longoni, A., Sampietro, M.: Silicon drift detectors for high resolution room temperature X-ray spectroscopy, Nuclear Instr. and Methods in Physics Research A 377, 346-351 (1996)
[9.5] Rose, A.: The Sensitivity Performance of the Human Eye on an Absolute Scale, J. Opt. Soc. America. 38, 196-208 (1948)
[9.6] Cliff, G., Lorimer, G. W.: The quantitative Analysis of thin specimens, Journ. of Microscopy 103, 203-207 (1975)
[9.7] Horita, Z., Sano, T., Nemoto, M.: Determination of the Absorption-Free kANi Factors for Quantitative Microanalysis of Nickel Base Alloys, J. Electron Microsc. 35, 324-334 (1986)
[9.8] Thomas, J., Gemming, T.: Shells on nanowires detected by analytical TEM, Appl. Surf. Science 252, 245-251 (2005)
[9.9] Goldstein, J. I.: Principles of Thin Film X-ray Microanalysis, in: Introduction to Analytical Electron Microscopy, Eds. Hren, J. J., Goldstein, J. I., Joy, D. C., Plenum Press, New York, S. 101 (1979)
[9.10] Thomas, J., Rennekamp, R., van Loyen, L.: Characterization of multilayers by means of EDXS in the analytical TEM, Fres. J. Anal. Chem. 361, 633-636 (1998)
[9.11] Fan, X., Dickey, E. C., Pennycook, S. J., Sunkara, M. K.: Z-contrast imaging and electron energy-loss spectroscopy analysis of chromium doped diamond-like carbon films, Appl. Phys. Let. 75, 2740-2742 (1999)
[9.12] Egerton, R. F.: Electron Energy-Loss Spectroscopy in the Electron Microscope, 2nd Edition, Plenum Press, New York, London (1996), S. 305
[9.13] Iakoubovskii, K., Mitsuishi, K., Nakayama, Y., Furuya, K.: Thickness Measurements with Electron Energy Loss Spectroscopy, Microsc. Research Techn. 71, 626-631 (2008)
[9.14] Hohenberg, P., Kohn, W.: Inhomogeneous Electron Gas, Phys. Rev. 136, B864-B8871 (1964)
[9.15] Kohn, W., Sham, L. J.: Self-consistent Equations including Exchange and Correlation Effects, Phys. Rev. 140, 1133-1138 (1965)
[9.16] Rez, P., Alvarez, J. R., Pickard, C.: Calculation of near edge structure“, Ultramicroscopy 78, 175-183 (1999)

Quellenverzeichnis 360
[9.17] Hérbert, C., Luitz, J., Schattschneider, P.: Improvement of energy loss near edge structure calculation using Wien2k, Micron 34, 219-225 (2003)
[9.18] Serin, V., Colliex, C., Brydson, R., Matar, S., Boucher, F.: EELS investigation of the electron conduction-band states in wurtzite AlN and oxygen-doped AlN(O), Phys. Rev. B 58, 5106-5115 (1998)
[9.19] Contreras, O., Duarte-Moller, A., Hirata, G. A., Avalos-Borja, M.: EELS characterization of TiN by the DC sputtering technique, Journ. Electron Spec. and Rel. Phenom. 105, 129-133 (1999)
[9.20] Riedl, T., Gemming, T., Wetzig, K.: Extraction of EELS white-line intensities of manganese compounds: Methods, accuracy, and valence sensitivity, Ultramicroscopy 106, 284-291 (2006)
[9.21] Riedl, T., Gemming, T., Gruner, W., Acker, J., Wetzig, K.: Determination of manganese valency in La1-xSrxMnO3, Micron 38, 224-230 (2007)
[9.22] Spence, J. C. H., Taftø, J: ALCHEMI: a new technique for locating atoms in small crystals, Journ. of Microscopy, 130, 147-154 (1983)
[9.23] Reimer, L. (Ed.): Energy-Filtering Transmission Electron Microscopy, Springer-Verlag, Berlin (1995) , S. 9
[9.24] Hofer, F.: Inner-Shell Ionization, ebenda, S. 225
[9.25] Egerton, R. F.: Electron Energy-Loss Spectroscopy in the Electron Microscope, 2nd Edition, Plenum Press, New York, London (1996), S. 334ff
[9.26] Rez, P.: Electron Ionisation Cross sections for Atomic Subshells, Microsc. Microanal. 9, 42-53 (2003)
[9.27] Hofer, F.: Determination of inner-shell cross-sections for EELS-quantification, Microsc. Microanal. Microstruct. 2, 215-230 (1991)
[10.1] Glaser, W.: Grundlagen der Elektronenoptik, Springer-Verlag, Wien (1952), S. 113
[10.2] Glaser, W.: ebenda, S. 297
[10.3] Debye, P., Bueche, A. M.: Scattering by an inhomogenous solid, J. Appl. Phys. 20, 518-525 (1949)
[10.4] Reimer, L.: Transmission Electron Microscopy, Physics of Image Formation and Microanalysis, Springer-Verlag, Berlin (1997), S. 71ff
[10.5] Williams, D. B., Carter, C. B.: Transmission Electron Microscopy, Springer-Verlag, New York (2009), S. 498
[10.6] Rang, O: Der elektronenoptische Stigmator, ein Korrektiv für astigmatische Elektronenlinsen, Optik 5, 518-530 (1949)

Quellenverzeichnis 361
[10.7] Bauer, H.-D.: Elektronenoptische Eigenschaften einer elektrostatischen Objektivlinse mit Vierpolsymmetrie, Optik 23, 596-609 (1965/66)
[10.8] Reimer, L.: Transmission Electron Microscopy, Physics of Image Formation and Microanalysis, Springer-Verlag, Berlin (1997), S. 160
[10.9] http://www.moxtek.com/x-ray-windows/ap3-ultra-thin-polymer-windows.html
[10.10] Theisen, R., Vollath, D.: Tabellen der Massenschwächungskoeffizienten von Röntgenstrahlen, Verlag Stahleisen mbH, Düsseldorf (1967), S. 11
[10.11] National Institute of Standards and Technology: NISTIR 5632: http://physics.nist.gov/PhysRefData/XrayMassCoef/ElemTab/z79.html
[10.12] Bethe, H.: Zur Theorie des Duchrchgangs schneller Korpuskularstrahlung durch Materie, Annalen der Physik 5, 325-400 (1930)
[10.13] Schreiber, T. P., Wims, A. M.: A quantitative X-ray microanalysis thin film method using K- L- and M-lines, Ultramicroscopy 6, 323-334 (1981)
[10.14] Powell, C. J.: Cross sections for ionization of inner shell electrons by electrons, Rev. Mod. Phys. 48, 33-47 (1976)
[10.15] Wentzel, G.: Über strahlungslose Quantensprünge, Zeitschrift für Physik, 43, 524-530 (1927)
[10.16] Klaar, H. J., Schwaab, P.: Röntgenmikroanalyse im Elektronenmikroskop, Teil 1: Grundlagen und Messtechnik, Prakt. Metallogr. 27, 319-331, Teil 2: Quantitative Analyse, 373-384, Teil 3: Vergleich mit anderen Verfahren, 429-438 (1990)
[10.17] Burhop, E. H. S.: Le rendement de fluorescence, J. Phys. Radium 16, 625-629 (1955), zitiert in [10.16]
[10.18] Scofield, J. H.: Radiative Transitions in: Atomic inner shell processes, Vol. I (Ed. B. Crasemann), Academic Press, New York (1975), S. 265-292
[10.19] Goldstein, J. I., Costley, J. L., Lorimer, G. W., Reed, S. J. B.: Quantitative X-ray analysis in the electron microscope, Scanning Electron Microscopy, ed. O. Johari, Chicago ITTRI 1, 315-324 (1977), zitiert in [10.16]

Sachwortverzeichnis
A Abbe, Ernst, 11 Abbildung
Dunkelfeld-, 126, 133 energiegefilterte, 227 Hellfeld-, 126 Hochauflösungs-, 145 mehrstufige, 19 optische, 29 -sgleichung, 11, 12, 20, 73 -smaßstab, 20 -ssystem, 38 von Atomsäulen, 145
Abbildungsfehler, 24 Astigmatismus, 27, 163, 176, 312, 317 Beugungsfehler, 175 Farbfehler, 25, 166, 176, 300, 310, 315,
317 Korrektur, 165, 310 Öffnungsfehler, 24, 40, 150, 165, 175,
176, 251, 294, 299, 306, 310, 313, 317
-scheibchen, 25, 27, 28, 251, 313, 315 Aberration
chromatische. s. Farbfehler sphärische. s. Öffnungsfehler
Ablenkwinkel bei inelastischer Streuung, 320
Abnahmewinkel, 205, 335 Abschirmung
äußerer Einflüsse, 48 des elektrischen Potentials, 121 ionisierender Strahlung, 47
Absorptionskorrektur, 332 Achse
optische, 12, 21, 22 23 40, 76, 123, 151, 306
Akzeptanzwinkel, 122, 124, 226, 289 Amorphisierung, 60 Ampére, Andre-Marie, 26 Amplitude
-nmodulation, 150 Ankleben, 52 Anode, 15, 32, 254 Anregungsfehler, 98, 107, 278 Apertur, 176
Beleuchtungs-, 37, 98, 115 numerische, 13 optimale, 28, 176, 317, 318
Arbeit, 15
Beschleunigungs-, 15 Ardenne, Manfred von, 14 Atomanordnung, 118 Atomformamplitude, 103, 108, 278, 281 Atomkern, 121, 184, 282 Atompositionen, 90 Auffischen
dünner Filme, 52 Auflösungsvermögen
Auge, 16, 40 bei EFTEM, 230 energetisches, 193, 199, 213 Lichtmikroskop, 11, 13 Messung, 164 mit Öffnungsfehler, 28, 251 Punkt-, 153, 302 STEM, 173 welloptische Deutung, 153
Aufrauchen, 52 Auftropfen, 52 Auger, Pierre Victor, 329
Elektron, 329 Auslöschungsregeln, 102, 105 Ausscheidungen, 53
semikohärente, 134 Austrittsarbeit, 29, 31, 34, 254
B
Basisvektoren Berechnung, 261 des reziproken Gitters, 261
Beam Shift, 72 Beleuchtung
konvergent, 38, 171 parallel, 38
Beleuchtungssystem, 37 Belichtungszeit, 42 Bending contours. s. Biegekonturen Beschleunigungsspannung, 15, 240, 328 Bestrahlungsapertur. s.
Beleuchtungsapertur Bethe, Hans, 226, 327
Oberfläche, 226 Beugung, 11, 13
Doppel-, 140 Elektronen-, 83 Feinbereichs-, 94, 97 Feinstrahl-, 94, 98 konvergente, 118

Sachwortverzeichnis 364
Röntgen-, 83, 86 -sreflexe, 86 -ssaum, 234 -sscheibchen, 13 -swinkel, 12, 86, 95
Beugungsmodus, 173 Beugungsreflex
Intensität, 102, 107 Position, 108
Biegekonturen, 130 Bewegungsgleichung, 241 Bildabstand. s. Bildweite Bilddrehung, 23, 133 Bildkontrast, 77 Bildladung, 252 Bildregistrierung
CCD-Kamera, 42 fotografisch, 41
Bildschärfe, 77 bildseitige Brennebene, 94 Bildsimulation, 151, 169 Bildweite, 11, 73 Bindung, 191
chemische, 189 Ionen-, 189 kovalente, 190 metallische, 190
Binning, 42 Blende, 47, 66
Feinbereichs-, 47 Gesichtsfeld-, 97 Kondensor2, 38 Kontrast-, 123, 127, 128 Objektiv-, 47, 123, 125, 127
Bloch, Felix, 141 -Wände, 141
Blooming, 42 Boersch, Hans
Effekt, 31 Bohr, Niels, 183 Boltzmann, Ludwig
Konstante, 43 Borries, Bodo von, 15 Br, 22 Bragg
-Lage, 125, 129 -sches Gesetz, 86, 95, 108, 114, 136,
258 William Henry, 86 William Lawrence, 86
Brechkraft, 25 Brechzahl, 12, 13 Bremsstrahlung, 184, 197, 217
-suntergrund, 185 Brennpunktstrahlen, 173 Brennweite, 11, 20, 24, 37, 39, 73
astigmatische -ndifferenz, 27, 175 Bezugs-, 74 Linsensystem, 20 Objektiv, 96
Broglie, Louis de, 14 Formel, 238
Brown, Robert, 44 -sche Bewegung, 44
Burgers, Johannes Martinus, 132 Vektor, 132
Busch, Hans, 14 Bz, 22
C
CBED. s. konvergente Beugung CCD-Kamera, 17, 42, 100, 149, 164, 212 chemical shift, 223 CIF-Dateien, 92 Cleavage, 55 Cliff-Lorimer-Faktoren, 201, 332
Berechnung der, 327 Bestimmung der, 204
Conical Darkfield, 128 Core-Loss-Bereich, 213 Coulomb, Charles Augustin de, 49
Kraft, 49, 252, 282 cross over, 33, 37, 173, 177 cross-section. s. Querschnitt CS-Korrektor, 166
D
Dämpfung, 164 der Kontrastübertragungsfunktion, 300
Dämpfungsfunktion, 120, 300 Davisson, Clint, 83 Debye, Peter, 278
Streuung, 278 Defokus, 302 Defokussierung, 150, 153, 162, 294, 299,
304 Delokalisation, 159, 306 Detektor
Effizienz, 184, 200, 204, 321, 326, 327 -fenster, 196, 321
Detektor-Wirkungsgrad. s. Detektor-Effizienz
Dichtefunktionaltheorie, 223 Dickenkonturen, 135, 136 Differentialgleichung

Sachwortverzeichnis 365
harmonische Schwingung, 236 numerische Lösung, 255, 310 Reihenansatz, 244 Wellenfunktion, 237
Differenzengleichung, 310 Differenzenquotienten, 255 Dimpeln, 57 Dingweite, 11, 40, 73 Dispersion, 212, 224 Doppelkondensor, 37 Doppelspalt, 12 Drehimpulssatz, 284 Drei-Fenster-Methode, 228 Dunkelstrom, 42 Dünnen
elektrolytisch, 54 mit Ionen, 55, 58
Dünnschichtkriterium, 335
E Ebene
bildseitige Brenn-, 39 der kleinsten Verwirrung, 175, 313 energieselektive, 212, 227 Objekt-, 40 ortsselektive, 39, 212, 227 Proben-, 40 winkelselektive, 39, 227 Zwischenbild-, 39
EDX Detektor, 195, 321 Spektrometer, 193 Spektrum, 195
EDXS, 68, 192, 193 Abschattung, 208 Absorptionskorrektur, 206, 332 Bornachweis, 200 Elementverteilungsbild, 211 Fehler bei Quantifizierung, 208 Linescan, 209 Quantifizierung, 201 Untergrundapproximation, 203
EEL Spektrometer, 211 Spektrum, 212
EELS, 211, 342 Messung der Probendicke, 219 optimale Probendicke, 219
EELS-Kanten Bezeichnung der, 215 Form der, 215 Überlagerung von, 216
EFTEM, 227 Eigenfunktion, 327 Elektronen
-Bahnen, 241, 250 d-, 187 -energieverlust, 211 -energieverluste, 188 -gas, 191 -hülle, 183, 189 -konfiguration, 189 p-, 187, 190 -prisma, 212, 227, 335 s-, 186, 190 Wellenfunktion, 238
Elektronenenergieverlust-Spektroskopie. s. EELS
Elektronenkanone, 254 Elektronenruhemasse, 15, 240 Elektronensonde, 172, 313
Aufweitung, 209 Elementarladung, 15, 21, 121, 141, 179,
191, 238, 240, 241, 252, 288, 327
Elementarzelle, 87, 88, 90, 271 Volumen der, 261, 275
ELNES. s. Kantenfeinstruktur Emission
thermische, 30 Energie
-band, 190, 192 -bilanz, 320 -breite, 26, 30, 153, 175, 224, 300, 317 -fenster, 227 Ionisierungs-, 327 kinetische, 15, 186 potentielle, 186 -verlust, 26 -zustand, 186, 188, 190
energiedispersive Röntgenspektroskopie. s. EDXS
Energieniveaus. s. Energiezustand Energiesatz, 34
klassisch, 15 relativistisch, 240
Eucentric Focus, 74 Euler, Leonhard, 103
-sche Formel, 103, 276 Ewald, Paul Peter, 260
Konstruktion, 108, 117, 260 Kugel, 116
EXELFS. s. Kantenfeinstruktur Exposure Time, 41 Extinktionslänge, 136

Sachwortverzeichnis 366
Extraktionsreplica, 53 Extraktor, 33
F
Faltung von Funktionen, 217, 339 Faltungsoperation, 341 Farbfehlerkonstante, 25, 153, 217, 315 Feld
elektrisches, 14, 15, 32, 167, 252, 321, 337
inhomogenes, 22, 24 -linien, 23 magnetisches, 14, 20, 166, 335 rotationssymmetrisches, 20, 243
Fermi, Enrico, 29 Energie, 29
Fernordnung, 119 FIB, 60
-Lamelle, 209 Flat Field Correction, 42 Fluoreszenzausbeute, 204, 327, 329 Fokus, 77
Über-, 77, 141 Unter-, 77, 141 Fokusserien-Rekonstruktion, 169 Fotoplatte, 164 Friedrich, Walter, 83 Fourier, Joseph, 154
Analyse, 154 Integral, 281 Koeffizienten, 155 -Raum, 342 Transformation, 157, 164 G
Gain Correction, 42 Gallium-Implantation, 60 Gallium-Ionen, 60 Gangunterschied, 13, 84, 151, 270, 296 Gauß, Carl Friedrich, 25
Funktion, 341 -Kurve, 204 -sche Bildebene, 25, 150, 175, 294,
306, 313, 315 -sche Kurve, 193
Gegenstandsweite. s. Dingweite generalisierte Oszillatorstärke (GOS), 226 Gerätekonstante, 96
Kalibrierung, 100, 104 Germer, Lester, 83 Gitterfaktor, 107 Gitter
-faktor, 107, 271, 274 -konstanten, 90 -leerstellen, 82 reziprokes, 259, 261 von Gold, 101 von NaCl, 88 von Silizium, 145
Glaser, Walter, 246 -sche Glockenkurve, 246
Goldelektrode, 322 Goniometer, 74 Gun Shift, 73 Gun Tilt, 72
H
HAADF. s. STEM-Detektor harmonische Schwingung, 235 H-Balken-Methode, 61 Heisenberg, Werner, 224
-sche Unschärferelation, 224 Hochspannungszentrum, 77 Holografie, 169 HOLZ-Linien, 116 Horita-Verfahren, 207 HRTEM, 145 Huygens, Christiaan, 11
Prinzip, 11, 233 I
Image Shift, 164 Impuls, 14
-bilanz, 320 Indizierung
Beugungsreflexe, 110 vierzählig hexagonal, 94
inelastischer Stoß, 320 Influenzkonstante, 121, 179, 191, 252,
283, 288 Informationslimit, 154, 165, 301 Intensität, 298 Intensity, 38, 71, 72 Interbandübergänge, 213, 217 Interferenz, 11, 84, 149, 152
konstruktive, 85 -muster, 234
Ionisation, 80, 188 der K-Schale, 188 innerer Schalen, 213 -squerschnitt, 225, 327, 329 -swahrscheinlichkeit, 204, 327
Isotropie, 279

Sachwortverzeichnis 367
J
JEMS (von P. Stadelmann), 151 Jump-Ratio-Methode, 228 Justage, 65, 70
Elektronenkanone, 71, 173 euzentrische Höhe, 73 Kipppunkte, 74, 128 Kondensorblende, 70 Rotationszentrum, 76 von EEL-Spektrometern, 212
K
Kα-Anteil, 331 Kameralänge, 95 Kantenfeinstruktur, 214, 222 Kathode, 15, 26, 32, 254
kalte Feldemissions-, 31, 34, 224 Lanthanhexaborid, 30, 34, 174, 301 Schottky, 31, 34, 174, 177, 224, 301 Wolfram-Haarnadel, 30, 34, 174
Kathodolumineszenz, 41 Kernlochbohren, 56 Kikuchi, Seishi, 99
Linien, 99, 113, 115 Muster, 150
Kipppunkt, 172 Knipping, Paul, 83 Kohlenwasserstoffmoleküle, 78 Kondensorastigmatismus, 73 Konkavschleifen. s. Dimpeln Kontamination, 47, 67, 78
-smechanismus, 79 Kontrast, 121, 123
Amplituden-, 293 bei amorphen Proben, 160 bei STEM, 178 Beugungs-, 125 -Inversion, 302 orientierungsabhängiger, 125 Phasen-, 149, 157, 293 Streuabsorptions-, 123, 157 -Übertragung, 150, 160 -Übertragungsfunktion, 153, 158, 162,
164, 168, 293, 299, 302 Kontrastblende. s. auch Objektivblende Konvergenzwinkel. s. Beleuchtungsapertur Konzentration, 201 Kornorientierung, 126 Knoll, Max, 14 Kramers, Hendrik Anthony, 184
-sche Formel, 184
Kreuzprodukt, 21, 261 Kristall, 86
-achsen, 87 -achsenwinkel, 87 Gitter, 83, 87, 149, 297 -größe, 97, 108 Potential, 146 Richtung im, 88 -system, 87 Verbiegung, 129 -Zwillinge, 139
Kristallite, 125 Kristallsysteme
hexagonal, 87 kubisch, 87 monoklin, 87 orthorhombisch, 87 Packungsart, 88 rhomboedrisch, 87 tetragonal, 87 triklin, 87
Krümmungsradius, 32 Kryo-Zyklus, 47 kurzwellige Grenze, 184
L
ladungsträgerfreie Zone, 195, 325 Lamelle
Anschweißen, 63 auf Trägerfilm, 62
Laplace, Pierre-Simon, 237 Gleichung, 243, 254, 310 Operator, 237
Laue, Max von, 83 Gleichungen, 259 Zonen, 116
Lebensdauer eines Energiezustandes, 225 Leuchtstoff, 41 Licht
Teilchen, 10 Welle, 10, 11
Lichtgeschwindigkeit, 16, 184, 240 Linienbreite, 224 Linse, 66
Beugungs-, 95 Elektronen-, 21, 23 Kondensor, 36, 95 langbrennweitige, 171 Objektiv, 23, 26, 39, 76, 150, 160 Projektiv, 36, 95 rotationssymmetrische Polschuh-, 241
Lorentz, Hendrik Antoon, 21

Sachwortverzeichnis 368
Kraft, 21, 22, 25, 141, 166, 241, 335 Kraft in Zylinderkoordinaten, 242 Linse, 142 Mikroskopie, 142
Loschmidt, Josef, 43 Konstante, 43, 179, 289
Low-Magnification, 69, 142 Low-Loss-Bereich, 213 Lupe, 9
M
magnetische Domänen, 140 -Wände, 142
magnetische Induktion, 21, 241, 243 Magnetisierung, 141 Magnification, 39 Massendickekontrast. s.
Streuabsorptionskontrast Massenschwächungskoeffizient. s. auch
Schwächungskoeffizient Materiewellen, 117, 237 Maxwell, James Clerk, 44
-sche Geschwindigkeitsverteilung, 44 -sche Gleichung, 19
Miller, William Hallowes, 92 -sche Indizes, 92
mittlere freie Weglänge elastische, 122, 178, 288 inelastische, 220, 221
Modell geometrisches, 270 dynamisches, 117 kinematisches, 117, 270 Potentialtopf-, 29, 253
Moiré-Muster, 139 Abstände, 290
Monochromator, 224 Monte-Carlo-Simulation, 179 Multipol, 176, 310
Hexapol, 166 Oktupol, 164, 310 Quadrupol, 168, 310
Mörser, 52
N Nachdünnen, 64 Nachkantenbild, 228 Nahordnung, 119, 160 Nanoanalytik, 177 Nanobeugung, 98 nanokristallin, 118 Nanometerbereich, 174
Nanostrukturen, 9, 14, 97 nBED. s. Nanobeugung Netzebene, 84, 114, 135
Kennzeichnung, 92 Krümmung, 131 -nabstand, 92, 164, 258 -nschar, 103 Winkel zwischen -n, 267 O
Oberflächenabdruck, 53 Oberflächendiffusion, 80 Objektiv
Nachfeld, 172, 179 Vorfeld, 172
Objektraumkühlung, 47, 69, 79 Objektschädigung, 80 Objektweite. s. Dingweite Öffnungsfehlerkonstante, 25, 151, 153,
175, 309 Öffnungswinkel. s. Apertur Offset, 214 Onset, 215 Optik
Elektronen-, 14, 24 geometrisch, 11, 22
Orbitale, 191 Ortsauflösung
bei EDXS, 209 Oszillator, 235
P
Packungsart, 101 Diamantgitter, 89 flächenzentriert, 89 hexagonal dichteste Kugelpackung, 89 primitiv, 89 raumzentriert, 89
Parallelstrahlen, 11, 22, 39, 94 Paraxialstrahlen, 65, 247 Pauli, Wolfgang, 29
Prinzip, 29, 187 Pearson, Frederic Treadwell, 87 PED, 118 Phase, 236
-ngeschwindigkeit, 235 -nmodulation, 149, 150 -nschiebung, 11, 137, 147, 151, 270,
294, 300 Phasenanalyse, 88, 128 Phononen, 192, 213 Pivot Point, 75

Sachwortverzeichnis 369
Pixel, 17, 42, 178 Planck, Max, 14
-sches Wirkumsquantum, 14, 146, 184, 186, 192, 224, 237, 239
plan-view. s. Draufsicht Plasmacleaner, 67, 68, 79 Plasmonen, 192, 213 Pol, 115, 116 Polarkoordinaten, 100 Polschuh, 21, 40, 195
-bohrung, 27, 241, 249 -spalt, 241, 249
Potential, 29, 81 im Strahlerzeuger, 254 in elektrostatischen Multipolen, 310 konstantes, 238 Kristall-, 117, 147 magnetisches, 243, 245, 250 -minimum, 81 -mulde, 81 Oktupol, 312 projiziertes, 151
Potentialverteilung mit Schottky-Kathode, 257 Triodensystem, 257
Powerspektrum, 157, 162, 168 Probe
amorphe, 82, 118, 160, 281 Draufsicht, 56 elektronentranparent, 49 magnetische, 23, 69, 250 -nbühne, 39 -ndicke, 49 -npräparation, 50 -nschleuse, 68 Querschnitt, 59
Probenhalter, 67 Doppelkipp-, 68, 133 Einfachkipp-, 68 Kühl-, 81 Low-Background, 68, 200
Pulsprozessor, 196 Punktdiagramm, 97
Q
Quantenzahlen, 29, 186, 190 Gesamtdrehimpuls-Quantenzahl, 187 Hauptquantenzahl, 186, 328 magnetische Quantenzahl, 187 Nebenquantenzahl, 186, 327 Spin, 187
Quantifizierung
standardlose, 204 von EDX-Spektren, 201 von EEL-Spektren, 225 R
radiale Dichtefunktion, 120, 281 radiale Helligkeitsverteilung, 100 Radialkraft, 185, 336 Rastermikroskopie, 171 Raumfrequenz, 152, 153, 154, 158, 161,
164, 292, 299, 305 Raumgruppe, 87 Raumladung, 31 Raumwinkel, 33, 173, 286 Rauschminderung, 162 Rayleigh, Lord John W.S., 13
Kriterium, 13 rechte Hand Regel, 21 Rekursionsformel, 256, 311 Restgas
Druck, 32 Ionen, 32
reziprokes Gitter, 108 reziproke Gittervektoren. s. auch
Basisvektoren Berechnung, 266
Richardson, Owens Willians, 30 Gleichung, 30, 34 Konstante, 30
Richtstrahlwert, 33, 35, 37, 173, 301 Richtung
niedrig indiziert, 146, 149 richtungsanisotrop, 119 richtungsisotrop, 119 Ringdiagramm, 97
Radien, 99 Ronchigramm, 178 Röntgen, Wilhelm Conrad, 83 Röntgen
-energien, 197 Röntgenpeaks
Überlappung von, 198 Bezeichnung der, 195 Escape-, 199 Summen-, 199 Intensität der, 202
Röntgenstrahlung charakteristische, 188
Rose-Kriterium, 201 Ruhebildmikroskopie, 171 Ruska, Ernst, 14, 78

Sachwortverzeichnis 370
Ruska, Helmut, 51 Rutherford, Ernest, 121
Streuformel, 287 Streuquerschnitt, 121
Rutherford-Bohrsches Atommodell, 183 Elektronen, 185 Gluonen, 185 Neutronen, 185 Protonen, 185
S
SAED. s. Feinbereichsbeugung Säge
Diamant-, 57 Faden-, 57
Sattelpunkt, 311 Schale, 186 Scherzer, Otto, 24, 310
Fokus, 153, 306 Schottky, Walter, 31
Effekt, 31, 252 Kathode, 31
Schrägbedampfung, 54 Schraubenbahnen, 23 Schrödinger, Erwin, 117
Gleichung, 117, 237 Schutzriegel, 60 Schwächungskoeffizient, 206, 322, 323,
333 Schwefelhexafluorid, 67 Sehweite deutliche, 9 Sehwinkel, 9 Sekundärelektronen, 217 Signal-Rausch-Verhältnis, 177 Silizium-Drift-Detektor, 196 Skalarprodukt, 267, 320 Sommerfeld, Arnold, 83 Sonden
-durchmesser, 174 -querschnitt, 173
Spalten. s. Cleavage Spektrometer
wellenlängendispersives, 192 Spot Size, 37, 98, 177 Stabilität
elektronische, 168 Mikroskopsäule, 66 Strom und Spannung, 26, 48 Temperatur, 40, 48
Standards, 204 Stapelfehler, 137
-Energie, 138 STEM, 19, 171, 218
Abbildung von Atomsäulen, 182 STEM-Detektor
Dunkelfeld, 180 HAADF, 181 Hellfeld, 179
Stereomikroskop, 67 Stickstoff
flüssiger, 67, 196 gasförmiger, 67
Stigmator, 310 Kondensor, 71, 73 Objektiv, 164
Strahlerzeuger s. Elektronenkanone, 254
Strahlführung, 47 Strahlstrom, 33, 174, 177 Strahlungsübergang, 187, 329 streaks, 278 Streukurve, 118, 281, 282 Streuquerschnitt, 122 Streuung
differentieller Wirkungsquerschnitt, 286
elastische, 121, 183, 220 inelastische, 183, 220 Vielfach-, 217
Streuquerschnitt, 184 Streuvektor, 119 Streuwinkel, 178, 226 Stromdichte, 38 Strukturfaktor, 103, 107, 271
von NaCl, 272 Superzelle, 169 Suppressor, 33
T Totschicht, 325 Trägerfilm, 51
Teilchen auf, 51 Trägernetz, 52 Transparenz, 321 Teilchendichte, 43, 44 Termschema, 188 Textur, 108, 127 Totzeit, 196 Triodensystem, 32, 254
U Übermikroskop, 14 Überspannungsverhältnis, 328

Sachwortverzeichnis 371
Untergrund bei EFTEM, 228 in EDX-Spektren, 197 in EEL-Spektren, 214, 217
Ultramikrotom, 55
V Vakuum
Hoch-, 45 -pinzette, 66 -system, 43, 46 Ultrahoch-, 32, 44
Vakuummessung Penning, Frans Michel, 46 Pirani, Marcello, 46
Vakuumniveau, 29, 253 Vakuumpumpen
Drehschieber-, 44 Ionengetter-, 45 Membran-, 44 Öldiffusions-, 45 Speicher-, 44 Transport-, 44 Turbomolekular-, 45 Vor-, 45
Vektoren, 21 Vergrößerung, 9, 14, 27, 39
bei STEM, 178 förderliche, 16, 19, 65 Gesamt-, 20 Projektiv, 96
Versetzung, 131 Schrauben-, 133 -skern, 131 -slinie, 132 Stufen-, 131
verzögerte Kante, 216 Vorkantenbild, 228 Vorpeak, 223 Vorzugsorientierung. s. Textur
W Wärmeausdehnung, 40, 81 Wechselwirkung
Coulomb-, 121, 183 elastische, 121, 282 inelastische, 26, 183, 319
Weglänge geometrische, 11 optische, 11, 24, 150
Wehnelt, Arthur, 33 Elektrode, 33, 254
weiße Linie, 216 Weiss, Pierre-Ernest, 141
-sche Bezirke, 141 Welle
ebene, 233, 298 Einfalls-, 297 elektromagnetische, 184 Elektronen-, 28, 84, 270 Elementar-, 11, 233 -namplitude, 149 -nfront, 11, 149, 233 -nfunktion, 186, 234, 238, 298 Objektaustritts-, 297
Wellenlänge, 84, 278 Elektronen, 14, 15, 25, 86, 117, 151 Licht, 12 relativistisch, 16, 240
Wellenüberlagerung. s. Interferenz Wellenzahlvektor, 85, 258, 278 Werkstoffphase, 86 Wien, Wilhelm, 166
Filter, 166, 224 Winkel zwischen Beugungsreflexen, 108 Wykoff, Ralph Walter Graystone, 90
Symbol, 90
Y YAG-Kristall, 42 Young fringes. s. Youngsche
Interferenzmuster Young, Thomas, 164
-sches Interferenzmuster, 164 Z
Zeiss, Carl, 11 Zero-Loss-Peak, 213 Zemlin-Tableau, 168 Zonenachse, 113, 268
niedrig indiziert, 116 Richtung der, 268
Zubehör, 66 Zustandsdichte, 214 Zylinderkoordinaten, 22, 241, 255


















