
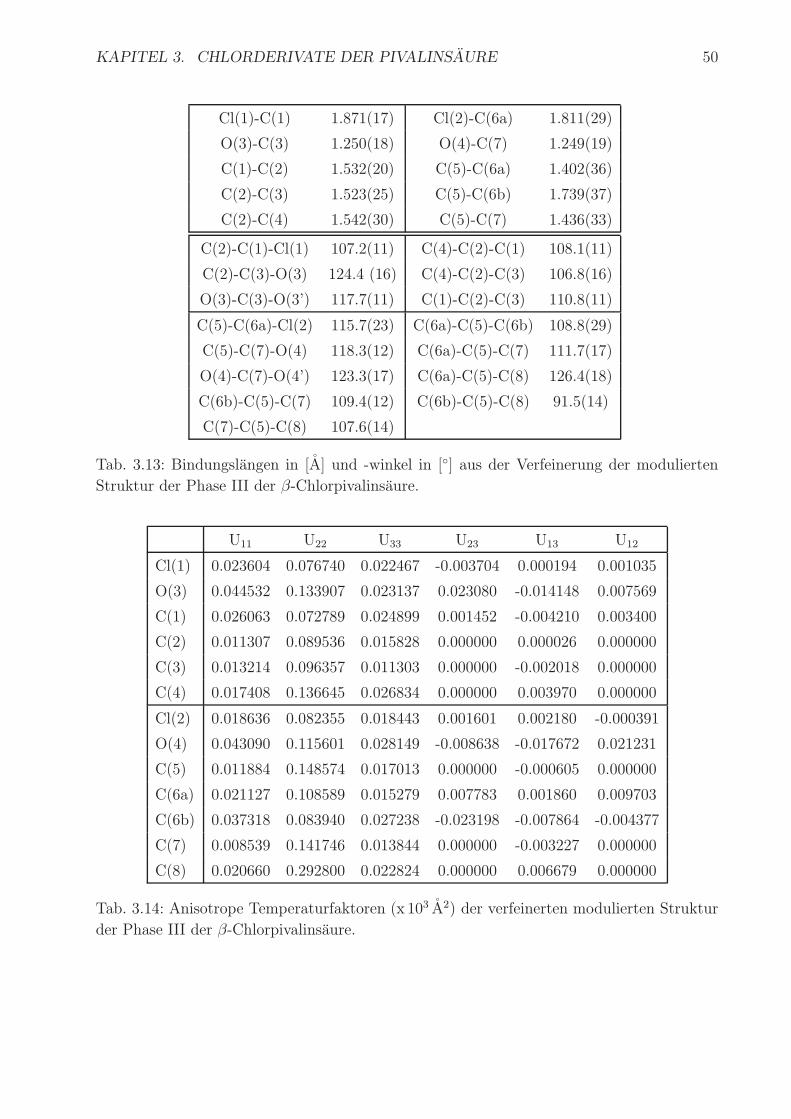
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE¨ 50 Cl(1)-C(1) 1...
30
KAPITEL 3. CHLORDERIVATE DER PIVALINS ¨ AURE 50 Cl(1)-C(1) 1.871(17) Cl(2)-C(6a) 1.811(29) O(3)-C(3) 1.250(18) O(4)-C(7) 1.249(19) C(1)-C(2) 1.532(20) C(5)-C(6a) 1.402(36) C(2)-C(3) 1.523(25) C(5)-C(6b) 1.739(37) C(2)-C(4) 1.542(30) C(5)-C(7) 1.436(33) C(2)-C(1)-Cl(1) 107.2(11) C(4)-C(2)-C(1) 108.1(11) C(2)-C(3)-O(3) 124.4 (16) C(4)-C(2)-C(3) 106.8(16) O(3)-C(3)-O(3’) 117.7(11) C(1)-C(2)-C(3) 110.8(11) C(5)-C(6a)-Cl(2) 115.7(23) C(6a)-C(5)-C(6b) 108.8(29) C(5)-C(7)-O(4) 118.3(12) C(6a)-C(5)-C(7) 111.7(17) O(4)-C(7)-O(4’) 123.3(17) C(6a)-C(5)-C(8) 126.4(18) C(6b)-C(5)-C(7) 109.4(12) C(6b)-C(5)-C(8) 91.5(14) C(7)-C(5)-C(8) 107.6(14) Tab. 3.13: Bindungsl¨ angen in [ ˚ A] und -winkel in [ ◦ ] aus der Verfeinerung der modulierten Struktur der Phase III der β -Chlorpivalins¨ aure. U 11 U 22 U 33 U 23 U 13 U 12 Cl(1) 0.023604 0.076740 0.022467 -0.003704 0.000194 0.001035 O(3) 0.044532 0.133907 0.023137 0.023080 -0.014148 0.007569 C(1) 0.026063 0.072789 0.024899 0.001452 -0.004210 0.003400 C(2) 0.011307 0.089536 0.015828 0.000000 0.000026 0.000000 C(3) 0.013214 0.096357 0.011303 0.000000 -0.002018 0.000000 C(4) 0.017408 0.136645 0.026834 0.000000 0.003970 0.000000 Cl(2) 0.018636 0.082355 0.018443 0.001601 0.002180 -0.000391 O(4) 0.043090 0.115601 0.028149 -0.008638 -0.017672 0.021231 C(5) 0.011884 0.148574 0.017013 0.000000 -0.000605 0.000000 C(6a) 0.021127 0.108589 0.015279 0.007783 0.001860 0.009703 C(6b) 0.037318 0.083940 0.027238 -0.023198 -0.007864 -0.004377 C(7) 0.008539 0.141746 0.013844 0.000000 -0.003227 0.000000 C(8) 0.020660 0.292800 0.022824 0.000000 0.006679 0.000000 Tab. 3.14: Anisotrope Temperaturfaktoren (x10 3 ˚ A 2 ) der verfeinerten modulierten Struktur der Phase III der β -Chlorpivalins¨ aure.
Transcript of KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE¨ 50 Cl(1)-C(1) 1...
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 50
Cl(1)-C(1) 1.871(17) Cl(2)-C(6a) 1.811(29)
O(3)-C(3) 1.250(18) O(4)-C(7) 1.249(19)
C(1)-C(2) 1.532(20) C(5)-C(6a) 1.402(36)
C(2)-C(3) 1.523(25) C(5)-C(6b) 1.739(37)
C(2)-C(4) 1.542(30) C(5)-C(7) 1.436(33)
C(2)-C(1)-Cl(1) 107.2(11) C(4)-C(2)-C(1) 108.1(11)
C(2)-C(3)-O(3) 124.4 (16) C(4)-C(2)-C(3) 106.8(16)
O(3)-C(3)-O(3’) 117.7(11) C(1)-C(2)-C(3) 110.8(11)
C(5)-C(6a)-Cl(2) 115.7(23) C(6a)-C(5)-C(6b) 108.8(29)
C(5)-C(7)-O(4) 118.3(12) C(6a)-C(5)-C(7) 111.7(17)
O(4)-C(7)-O(4’) 123.3(17) C(6a)-C(5)-C(8) 126.4(18)
C(6b)-C(5)-C(7) 109.4(12) C(6b)-C(5)-C(8) 91.5(14)
C(7)-C(5)-C(8) 107.6(14)
Tab. 3.13: Bindungslangen in [A] und -winkel in [◦] aus der Verfeinerung der modulierten
Struktur der Phase III der β-Chlorpivalinsaure.
U11 U22 U33 U23 U13 U12
Cl(1) 0.023604 0.076740 0.022467 -0.003704 0.000194 0.001035
O(3) 0.044532 0.133907 0.023137 0.023080 -0.014148 0.007569
C(1) 0.026063 0.072789 0.024899 0.001452 -0.004210 0.003400
C(2) 0.011307 0.089536 0.015828 0.000000 0.000026 0.000000
C(3) 0.013214 0.096357 0.011303 0.000000 -0.002018 0.000000
C(4) 0.017408 0.136645 0.026834 0.000000 0.003970 0.000000
Cl(2) 0.018636 0.082355 0.018443 0.001601 0.002180 -0.000391
O(4) 0.043090 0.115601 0.028149 -0.008638 -0.017672 0.021231
C(5) 0.011884 0.148574 0.017013 0.000000 -0.000605 0.000000
C(6a) 0.021127 0.108589 0.015279 0.007783 0.001860 0.009703
C(6b) 0.037318 0.083940 0.027238 -0.023198 -0.007864 -0.004377
C(7) 0.008539 0.141746 0.013844 0.000000 -0.003227 0.000000
C(8) 0.020660 0.292800 0.022824 0.000000 0.006679 0.000000
Tab. 3.14: Anisotrope Temperaturfaktoren (x 103 A2) der verfeinerten modulierten Struktur
der Phase III der β-Chlorpivalinsaure.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 51
Abstande im Bereich von 3.5-4.0 A vergroßert und dadurch der dreidimensionale Aufbau im
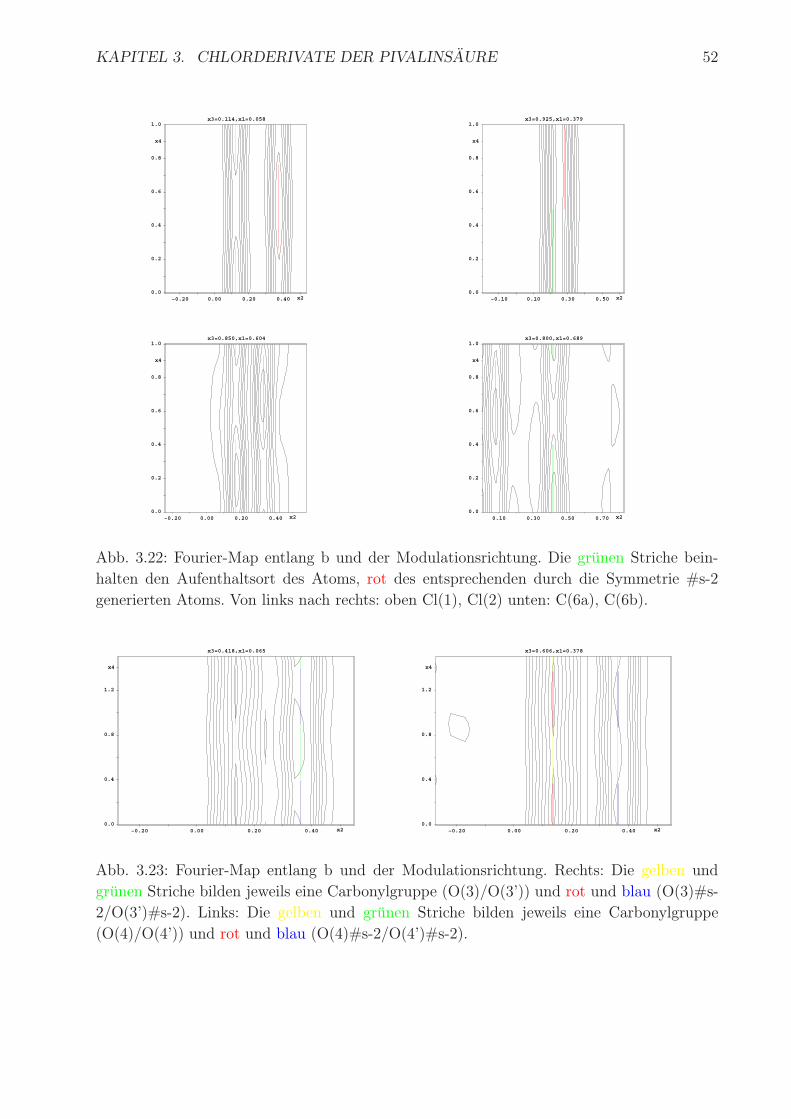
Kristall gefestigt wird. In Tab. 3.15 sind die Chlorabstande aufgelistet. In Abb. 3.22 ist an-
schaulich die Modulation der Atome Cl(1), Cl(2), C(6a) und C(6b) gezeigt. Man erkennt
deutlich den Sprung von einer Besetzung zur anderen, die bei den Atomen Cl(1), C(6a) und
C(6b) am großten ist. Bei Cl(1) ist die Sprungweite gering, da nur eine alternative Position
fur das Atom in der Basisstruktur gefunden werden konnte. In Abb. 3.23 sind die Sauerstoff-
positionen gezeigt. So bilden O(3)/O(3’) und O(4)/O(4’) jeweils eine Carboxyleinheit, die in
einer anderen Elementarzelle durch O(3)#s-2/O(3’)#s-2 und O(4)#s-2/O(4’)#s-2 gebildet
wird. #s-2 bezeichnet die Translation entlang der Modulation. Im Vergleich zu den Fourier-
Maps aus den gemessenen Daten zeigen Abb. 3.24 und Abb. 3.25 die Simulation des zuvor
beschriebenen Modells mit einer maximalen Auflosung von sinθ/λ=0.7 und maximalem In-
dex von m=±1. Der Vergleich zeigt, daß die verfeinerte Struktur in guter Ubereinstimmung
mit den simulierten Modell ist.
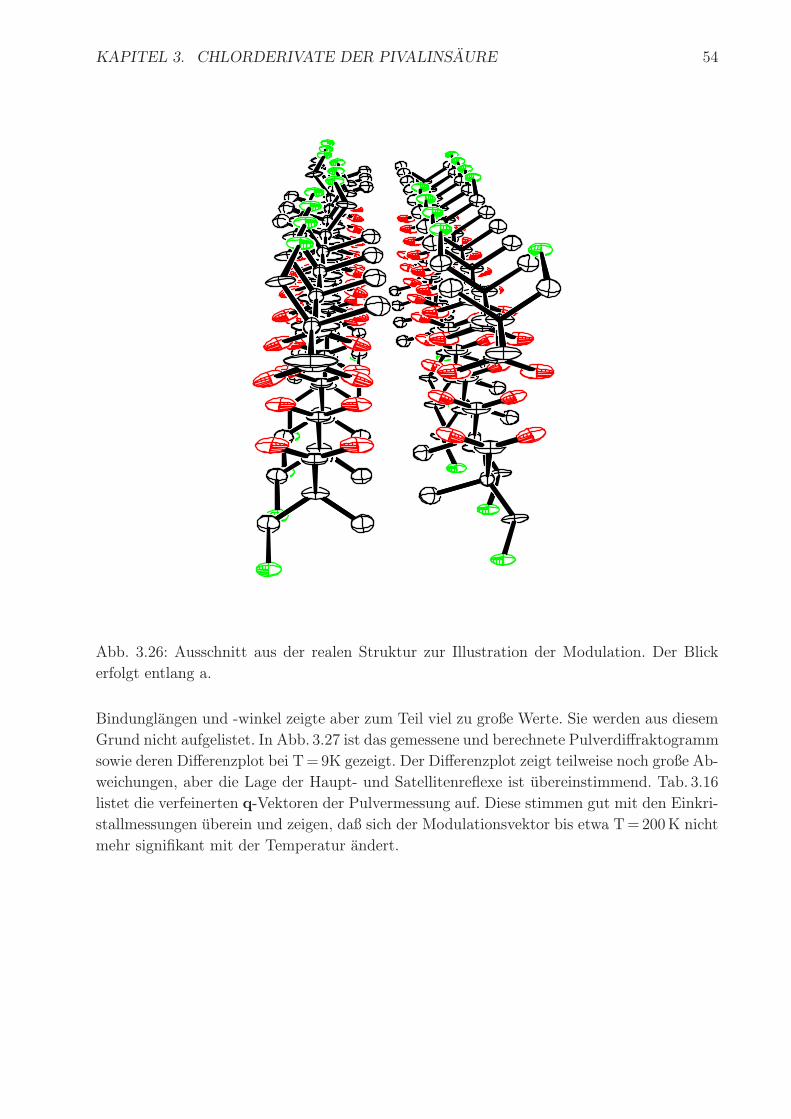
In Abb. 3.26 ist ein Ausschnitt aus dem realen dreidimensionalen Aufbau der Struktur
gezeigt. Die Modulation ist am deutlichsten an der Position des Cl(1)-Atoms zu erkennen. So
erkennt man in diesem Ausschnitt, daß - von vorne nach hinten gesehen - erst ein Molekul mit
Position Cl(1), anschließend 4 Molekule mit Position Cl(1)#s-2 und dann erneut 3 Molekule
mit Position Cl(1) aufgereiht sind. Fur das Chloratom findet man die Abfolge 4,3,3. Der
Ausschnitt aus dem realen Aufbau ist willkurlich und die Wiederholung dieser Stapelfolge
nicht anschließend gegeben.
Zur Beschreibung der dreidimensionalen Struktur dient am Besten ein Modell der Stape-
lung. Es sind 4 verschiedene Dimere in der Struktur moglich, die sich aus den Molekulpaaren
AC, AD, BC und BD zusammensetzen (siehe Newman-Projektion in Abb. 3.16). Die Anord-
nung dieser Dimeren ist entlang der a-Achse im gewissen Sinne zufallig, d.h. es existiert eine
Stapelfolge, die ungleich der Translationssymmetrie der Elementarzelle ist. Gleiches gilt fur
die Stapelung in c-Richtung: Die Stapelung dieser Reihen von Dimeren ist ebenso zufallig.
Ebenfalls mit dem Programm JANA konnten die gemessenen Pulverdiffraktogramme bei
T=9, 155K und 220K analysiert werden. Der fur diese Analyse notwendige Teil des Pro-
grammpakets ist noch im Entwicklungsstadium, daher ist die Profilanpassung nicht gut.
Diese reichte nicht aus, um eine stabile Verfeinerung der modulierten Struktur zu erhalten.
Die Verfeinerung der Gitterkonstanten und des q-Vektors sind aber ohne Probleme mog-
lich. Die Temperaturfaktoren wurden nur isotrop verfeinert. Der omsin-Parameter fiel aber
auch hier bei unterschiedlichen Startwerten auf den gleichen Wert zuruck. Die Analyse der
Cl(1)-Cl(1) 1 3.50(1) Cl(1)-Cl(2) 2 3.22(1)
Cl(1)-Cl(2) 3 3.46(2)
Tab. 3.15: Intermolekulare Chlorabstande in [A] in der modulierten Struktur der Phase III
der β-Chlorpivalinsaure. Symmetrieoperatoren 1: -x, -y, -z#s-1, 2: x, y, -1+z#t0,0,-1, 3: x,
1/2-y, -1+z#s-2t0,0,-1.
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 52
x3=0.114,x1=0.058
−0.20 0.00 0.20 0.40 x20.0
0.2
0.4
0.6
0.8
1.0
x4
x3=0.925,x1=0.379
−0.10 0.10 0.30 0.50 x20.0
0.2
0.4
0.6
0.8
1.0
x4
x3=0.850,x1=0.604
−0.20 0.00 0.20 0.40 x20.0
0.2
0.4
0.6
0.8
1.0
x4
x3=0.800,x1=0.689
0.10 0.30 0.50 0.70 x20.0
0.2
0.4
0.6
0.8
1.0
x4
Abb. 3.22: Fourier-Map entlang b und der Modulationsrichtung. Die grunen Striche bein-
halten den Aufenthaltsort des Atoms, rot des entsprechenden durch die Symmetrie #s-2
generierten Atoms. Von links nach rechts: oben Cl(1), Cl(2) unten: C(6a), C(6b).
x3=0.418,x1=0.065
−0.20 0.00 0.20 0.40 x20.0
0.4
0.8
1.2
x4
x3=0.606,x1=0.378
−0.20 0.00 0.20 0.40 x20.0
0.4
0.8
1.2
x4
Abb. 3.23: Fourier-Map entlang b und der Modulationsrichtung. Rechts: Die gelben und
grunen Striche bilden jeweils eine Carbonylgruppe (O(3)/O(3’)) und rot und blau (O(3)#s-
2/O(3’)#s-2). Links: Die gelben und grunen Striche bilden jeweils eine Carbonylgruppe
(O(4)/O(4’)) und rot und blau (O(4)#s-2/O(4’)#s-2).

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 53
x3=0.114,x1=0.058
−0.20 0.00 0.20 0.40 x20.0
0.2
0.4
0.6
0.8
1.0
x4
x3=0.925,x1=0.379
−0.10 0.10 0.30 0.50 x20.0
0.2
0.4
0.6
0.8
1.0
x4
x3=0.850,x1=0.604
−0.20 0.00 0.20 0.40 x20.0
0.2
0.4
0.6
0.8
1.0
x4
x3=0.800,x1=0.689
0.10 0.30 0.50 0.70 x20.0
0.2
0.4
0.6
0.8
1.0
x4
Abb. 3.24: Simulierte Fourier-Map entlang b und der Modulationsrichtung. Die grunen Stri-
che beinhalten den Aufenthaltsort des Atoms, rot des entsprechenden durch die Symmetrie
#s-2 generierten Atoms. Von links nach rechts: oben Cl(1), Cl(2) unten: C(6a), C(6b).
x3=0.418,x1=0.065
−0.20 0.00 0.20 0.40 x20.0
0.4
0.8
1.2
x4
x3=0.606,x1=0.378
−0.20 0.00 0.20 0.40 x20.0
0.4
0.8
1.2
x4
Abb. 3.25: Simulierte Fourier-Map entlang b und der Modulationsrichtung. Rechts: Die gel-
ben und grunen Striche bilden jeweils eine Carbonylgruppe (O(3)/O(3’)) und rot und blau
(O(3)#s-2/O(3’)#s-2). Links: Die gelben und grunen Striche bilden jeweils eine Carbonyl-
gruppe (O(4)/O(4’)) und rot und blau (O(4)#s-2/O(4’)#s-2).
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 54
Abb. 3.26: Ausschnitt aus der realen Struktur zur Illustration der Modulation. Der Blick
erfolgt entlang a.
Bindunglangen und -winkel zeigte aber zum Teil viel zu große Werte. Sie werden aus diesem
Grund nicht aufgelistet. In Abb. 3.27 ist das gemessene und berechnete Pulverdiffraktogramm
sowie deren Differenzplot bei T=9K gezeigt. Der Differenzplot zeigt teilweise noch große Ab-
weichungen, aber die Lage der Haupt- und Satellitenreflexe ist ubereinstimmend. Tab. 3.16
listet die verfeinerten q-Vektoren der Pulvermessung auf. Diese stimmen gut mit den Einkri-
stallmessungen uberein und zeigen, daß sich der Modulationsvektor bis etwa T=200K nicht
mehr signifikant mit der Temperatur andert.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 55
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
4069 COUNTS(o) 3525 COUNTS(c)
0.0
0.1
0.2
0.0
6.0 8.0 10.0 12.0 14.0 16.0 18.0 20.0
Inte
nsitä
t [a.
u.]
2Θ [°]
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.0
0.1
0.2
0.3
0.4
0.0
20.0 22.0 24.0 26.0 28.0 30.0 32.0 34.0 36.0 38.0 40.0
Inte
nsitä
t [a.
u.]
2Θ [°]
Abb. 3.27: Gemessenes und berechnetes Pulverspektrum, sowie der Differenzplot der Phase
III bei T=9K der β-Chlorpivalinsaure. Die grunen Marker zeigen die Position der Satelli-
tenreflexe an.
T [K]
9 -0.141(1) 0.0 0.403(1)
155 -0.141(1) 0.0 0.401(1)
220 -0.142(1) 0.0 0.403(1)
Tab. 3.16: Temperaturabhangiger Propagationsvektor q der modulierten Phase III der β-
Chlorpivalinsaure. Der q-Vektor wurde aus der Profilanpassung der jeweiligen Synchrotron-
messung erhalten.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 56
3.2.5 IR-Spektroskopie
Die IR-Messungen wurden an einem FT-IR Gerat Perkin-Elmer 1750 durchgefuhrt. Es wur-
de der mittlere Infrarotbereich von 4000 cm−1 bis 400 cm−1 untersucht. Jedes Spektrum be-
steht aus 20 Einzelscans mit einer Auflosung von 2 cm−1. Die jeweilige Temperatur wurde
durch eine Kuhlzelle der Firma Graseby-Specac mit Flussigstickstoff als Kuhlmedium er-
reicht und gegen geheizt. Die Temperatur wurde uber einen Eurothermregler eingestellt. Der
abgeschatzte Fehler in der Temperatur liegt bei etwa 2K, da das Thermoelement an den
Halterungen fur die KBr Fenster angebracht ist und nicht direkt an der Probe aufliegt. Die
Spektren zeigen im relevanten Fingerprintbereich keine deutlichen Anderungen der Schwin-
gungsbanden zwischen 77K und Zimmertemperatur (siehe Abb. 3.28). Zusatzlich ist das
1800 1600 1400 1200 1000
5
10
15
20
25
30
Tra
nsm
issi
on [
%]
Wellenzahl ν [cm-1]
1000 900 800 700 600 500 400
5
10
15
20
25
30
Tra
nsm
issi
on
[%
]
Wellenzahl ν [cm-1]
Abb. 3.28: IR-Spektrum der β-Chlorpivalinsaure bei verschiedenen Temperaturen. : flussi-
ge Phase, : Raumtemperatur, : 273K, 223K, : 153K, : 77K. Die Spektren sind
in der Reihenfolge steigender Temperatur aufgenommen.
Spektrum der Flussigkeit gezeigt. Es kann nicht mit Sicherheit festgelegt werden, ob wah-
rend der Aufnahme des IR-Spektrums die plastische Phase des Systems vorlag, obwohl mit
großer Sorgfalt die Spektren in dem relevanten Bereich untersucht wurden. Es ist bekannt,
daß sich die Spektren der plastischen Phasen nicht von denen der flussigen unterscheiden.
Der Unterschied in der flussigen Phase zu den festen Phasen besteht hauptsachlich in einer
Verbreiterung der Banden. Dies wird durch die großere Rotationsfreiheit hervorgerufen.
Im Spektrum der niedrigsten Temperatur (77K, gelbe Linie) ist bei ca. 670 cm−1 eine
deutliche Bande zu erkennen, die bei hoheren Temperaturen drastisch an Intensitat verliert,
und in Phase II nicht mehr zu erkennen ist. Dies weist auf eine zusatzliche Torsionsschwin-
gung hin, die bei hoheren Temperaturen nicht mehr auftritt. Ebenso erkennt man bei ge-
nauerer Analyse bei den schwachen Banden oftmals Intensitatsverschiebungen, die darauf
hinweisen, daß Gerustschwingungen der tert.-Butylgruppe in Phase III zunehmen.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 57
3.2.6 35Cl NQR-Spektroskopie
Eine frisch sublimierte Probe wurde in eine Glasampulle mit 5mm Durchmesser und 1.5 cm
Lange gefullt und vorsichtig zugeschmolzen, damit sich kein Schmelzkuchen bilden konnte.
Obwohl mit mehreren Kuhlraten und bis zu 48 h tempern in Flussigstickstoff die Probe un-
terschiedlich behandelt wurde, konnte kein Signal im abgescannten Bereich (25 - 45MHz) ge-
funden werden. Dies kann auf Unreinheiten der Probe zuruckzufuhren sein, wobei ein Einfluß
durch die Praparation ausgeschlossen ist. Es ist eher anzunehmen, daß durch die Modula-
tion der Phase III keine Kristallfeldaufspaltung mit einer nachweisbaren Intensitat auftritt.
Um jedes Chloratom der Molekulpaare A,B und C,D sind leicht verschiedene Umgebungen
vorhanden, die zu einer unterschiedlichen Kristallfeldaufspaltung fuhren.
3.2.7 1H NMR-Spektroskopie
1HNMR Messungen an der β-Chlorpivalinsaure wurden an der physikalischen Fakultat der
Universitat Leipzig bei 100MHz an einem Bruker FT MSL-100 durchgefuhrt. Es wurde ein1H NMR Probenkopf mit Stickstoffkuhlung verwendet. Die Probe wurde in eine Glasampulle
mit 5mm Durchmesser und einer Lange von 15mm eingeschlossen und auf einen 1H NMR
Tieftemperaturprobenkopf eingespannt. Die Kuhlung erfolgt uber einen Kryostaten mit Flus-
sigstickstoff als Kuhlmedium und der π/2-Puls wurde genau extrahiert. Die T1-Messungen
wurden wie in Kapitel 2.7.7 auf Seite 25 beschrieben mit der Standard-Inversion Recove-
ry Methode [T - 180 ◦ - τ - 90 ◦ - FID] mit typischerweise 12 Wiederholungen von τ und einer
Verzogerungszeit von T≥T1 durchgefuhrt. Da sich die Spin-Gitter-Relaxationszeit mit der
Temperatur stark andert, wurde auch dementsprechend das Meßprogramm angepaßt. Fur al-
le Temperaturen konnte ein exponentieller Verlauf innerhalb der Fehler gefunden werden und
aus dem exponentiellen Verlauf uber den Marquardt-Levenberg-Algorithmus T1 extrahiert
werden.
Linienbreite und 2.Moment
Die Extraktion der Linienbreite wird aus dem Meßzyklus der T1-Inversion-Recovery-Methode
mit der langsten Haltezeit (τ12) durchgefuhrt. Dadurch ist gewahrleistet, daß sich das System
im Gleichgewicht mit seiner Umgebung befindet (Breitband Methode). Aus dem gemessenen
Spektrum wurde das Signal einer Gauß-Kurve angepaßt und die Linienbreite ermittelt. Das
2.Moment wurde ebenfalls aus diesem Spektrum nach Gleichung 2.28 ermittelt. Bei der
Auswertung mußte streng auf die Signalbreite geachtet werden, wie in Abb. 3.29 angedeutet.
Nimmt man das gesamte Spektrum, so erhalt man, wie in Kapitel 2.7.2 auf Seite 16 erlautert,
immer den gleichen Wert fur das 2.Moment.
In Abb. 3.30 sind die Linienbreite und das 2.Moment in Abhangigkeit von der Temperatur
gezeigt. Sowohl das 2.Moment als auch die Linienbreite zeigen den gleichen Gang mit der
Temperatur. Es ist ein deutlicher Abfall zwischen 123K und 160K erkennbar, der zweite
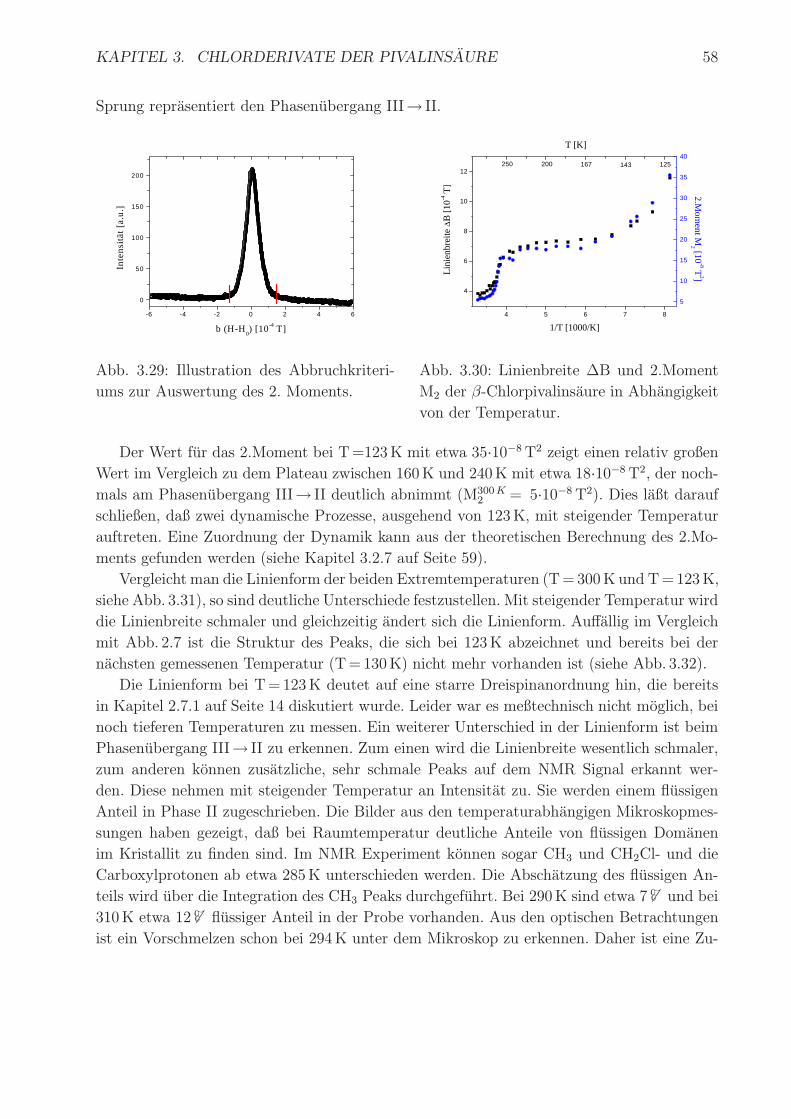
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 58
Sprung reprasentiert den Phasenubergang III→ II.
-6 -4 -2 0 2 4 6
0
50
100
150
200
Inte
nsi
tät
[a.u
.]
b (H-H0) [10-4 T]
Abb. 3.29: Illustration des Abbruchkriteri-
ums zur Auswertung des 2. Moments.
4 5 6 7 8
4
6
8
10
12200 143 125167250
T [K]
1/T [1000/K]
Lin
ienb
reite
∆B
[10
-4 T
]
5
10
15
20
25
30
35
40
2.Mom
ent M2 [10
-8 T2]
Abb. 3.30: Linienbreite ∆B und 2.Moment
M2 der β-Chlorpivalinsaure in Abhangigkeit
von der Temperatur.
Der Wert fur das 2.Moment bei T=123K mit etwa 35·10−8T2 zeigt einen relativ großen
Wert im Vergleich zu dem Plateau zwischen 160K und 240K mit etwa 18·10−8T2, der noch-
mals am Phasenubergang III→ II deutlich abnimmt (M300K2 = 5·10−8T2). Dies laßt darauf
schließen, daß zwei dynamische Prozesse, ausgehend von 123K, mit steigender Temperatur
auftreten. Eine Zuordnung der Dynamik kann aus der theoretischen Berechnung des 2.Mo-
ments gefunden werden (siehe Kapitel 3.2.7 auf Seite 59).
Vergleicht man die Linienform der beiden Extremtemperaturen (T=300K und T=123K,
siehe Abb. 3.31), so sind deutliche Unterschiede festzustellen. Mit steigender Temperatur wird
die Linienbreite schmaler und gleichzeitig andert sich die Linienform. Auffallig im Vergleich
mit Abb. 2.7 ist die Struktur des Peaks, die sich bei 123K abzeichnet und bereits bei der
nachsten gemessenen Temperatur (T=130K) nicht mehr vorhanden ist (siehe Abb. 3.32).
Die Linienform bei T=123K deutet auf eine starre Dreispinanordnung hin, die bereits
in Kapitel 2.7.1 auf Seite 14 diskutiert wurde. Leider war es meßtechnisch nicht moglich, bei
noch tieferen Temperaturen zu messen. Ein weiterer Unterschied in der Linienform ist beim
Phasenubergang III→ II zu erkennen. Zum einen wird die Linienbreite wesentlich schmaler,
zum anderen konnen zusatzliche, sehr schmale Peaks auf dem NMR Signal erkannt wer-
den. Diese nehmen mit steigender Temperatur an Intensitat zu. Sie werden einem flussigen
Anteil in Phase II zugeschrieben. Die Bilder aus den temperaturabhangigen Mikroskopmes-
sungen haben gezeigt, daß bei Raumtemperatur deutliche Anteile von flussigen Domanen
im Kristallit zu finden sind. Im NMR Experiment konnen sogar CH3 und CH2Cl- und die
Carboxylprotonen ab etwa 285K unterschieden werden. Die Abschatzung des flussigen An-
teils wird uber die Integration des CH3 Peaks durchgefuhrt. Bei 290K sind etwa 7% und bei
310K etwa 12% flussiger Anteil in der Probe vorhanden. Aus den optischen Betrachtungen
ist ein Vorschmelzen schon bei 294K unter dem Mikroskop zu erkennen. Daher ist eine Zu-

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 59
-2 -1 0 1 2
0
50
100
150
200
250
300
Inte
nsitä
t [a.
u.]
b (H-H0) [10-4 T]
Abb. 3.31: Vergleich der Absorptionslinien
im 1H NMR Spektrum der beiden Extrem-
temperaturen der β-Chlorpivalinsaure. :
T= 123K, : T= 300K.
-2 -1 0 1 2
0
50
100
150
200
250
Inte
nsitä
t [a.
u.]
b (H-H0) [10-4 T]
Abb. 3.32: Vergleich der Absorptionslinien
im 1H NMR Spektrum der β-Chlorpivalin-
saure. : T= 123K, : T= 130K.
ordnung zur ODIC Phase I ausgeschlossen. Ebenfalls mußte der ODIC Reflex der Phase I
im Pulverdiffraktogramm zu erkennen sein. Dies konnte nicht beobachtet werden.
Theoretische Berechnung des 2.Moments
Zur Berechnung des 2.Moments wurden nur intramolekulare Wechselwirkungen angenom-
men. Testrechnungen haben gezeigt, daß ein intermolekularer Anteil bis zu einem Radius
von 5 A nur ca. 0.2 · 10−8T2 fur eine starre Struktur ausmachen. Die H-H- und die H-Cl-
Abstande wurden aus den jeweiligen Einkristallstrukturdaten entnommen. Da die H-Atome
meist geometrisch positioniert wurden und die C-H Bindungslange entsprechend der gemes-
senen Temperatur angefittet wurde, liegt hier kein realer Zustand vor. Fur die Raumtempe-
raturstruktur wurde das 2.Moment unter Berucksichtigung der dimeren Einheit berechnet.
Bei der Tieftemperatur wurden alle 4 Konformere berucksichtigt und von zwei Dimeren-
paaren ausgegangen. Dadurch ist gewahrleistet, daß der Einfluß der Carboxylgruppe in die
Berechnung miteinbezogen ist.
T=300K a) starres Molekul
i)∑
j>k r−6jk = 1.07 · 1060m−6 mit N=18 folgt
MH−H2 = 42.50 · 10−8 T 2
ii)∑
f,j r−6fj = 2.99 · 1058m−6 mit N=18 folgt
MH−Cl2 = 0.01 · 10−8T 2
=⇒Mstarr2 =42.5·10−8T2

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 60
b) Methylgruppenrotation
i)∑
j>k r−6jk = 3.51 · 1059m−6 mit N=18 folgt
MH−H2 = 13.95 · 10−8 T 2
ii)∑
f,j r−6fj = 2.61 · 1058m−6 mit N=18 folgt
MH−Cl2 = 0.01 · 10−8T 2
=⇒Mstarr2 =13.96·10−8T2
c) Methyl/Chlormethyl-Rotation
i)∑
j>k r−6jk = 2.66 · 1059m−6 mit N=18 folgt
MH−H2 = 10.59 · 10−8 T 2
ii)∑
f,j r−6fj = 6.53 · 1057m−6 mit N=18 folgt
MH−Cl2 = 0.002 · 10−8T 2
=⇒Mstarr2 =10.59·10−8T2
T=200K a) starres Molekul
i)∑
j>k r−6jk = 1.87 · 1060m−6 mit N=36 folgt
MH−H2 = 37.17 · 10−8 T 2
ii)∑
f,j r−6fj = 5.22 · 1058m−6 mit N=36 folgt
MH−Cl2 = 0.02 · 10−8T 2
=⇒Mstarr2 =37.19·10−8T2
b) Methylgruppenrotation
i)∑
j>k r−6jk = 8.74 · 1059m−6 mit N=36 folgt
MH−H2 = 17.38 · 10−8 T 2
ii)∑
f,j r−6fj = 5.22 · 1058m−6 mit N=36 folgt
MH−Cl2 = 0.02 · 10−8T 2

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 61
=⇒Mstarr2 =17.40·10−8T2
c) Methyl/Chlormethyl-Rotation
i)∑
j>k r−6jk = 4.51 · 1059m−6 mit N=36 folgt
MH−H2 = 8.96 · 10−8 T 2
ii)∑
f,j r−6fj = 1.31 · 1058m−6 mit N=36 folgt
MH−Cl2 = 0.002 · 10−8T 2
=⇒Mstarr2 =8.96·10−8T2
T=107K a) starres Molekul
i)∑
j>k r−6jk = 1.88 · 1060m−6 mit N=36 folgt
MH−H2 = 37.43 · 10−8 T 2
ii)∑
f,j r−6fj = 5.22 · 1058m−6 mit N=36 folgt
MH−Cl2 = 0.02 · 10−8T 2
=⇒Mstarr2 =37.45·10−8T2
b) Methylgruppenrotation
i)∑
j>k r−6jk = 8.83 · 1059m−6 mit N=36 folgt
MH−H2 = 17.55 · 10−8 T 2
ii)∑
f,j r−6fj = 5.22 · 1058m−6 mit N=36 folgt
MH−Cl2 = 0.02 · 10−8T 2
=⇒Mstarr2 =17.57·10−8T2

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 62
c) Methyl/Methylchlorid-Rotation
i)∑
j>k r−6jk = 4.57 · 1059m−6 mit N=36 folgt
MH−H2 = 9.08 · 10−8 T 2
ii)∑
f,j r−6fj = 1.31 · 1058m−6 mit N=36 folgt
MH−Cl2 = 0.002 · 10−8T 2
=⇒Mstarr2 =9.08·10−8T2
Isotrope Reorientierung Bei der Berechnung des 2.Moments fur die plastische Phase
wird nach Boden [28] die Gittersumme des fcc-Gitters bestimmt (siehe Gleichung 2.36).
Fur ein kubisch flachenzentriertes Gitter ergibt sich mit Z=4 und a0=8.84
∑j>k r
−6jk = 2.45 · 1056m−6
=⇒M2=0.4 ·10−8T2
Der Vergleich der berechneten 2.Momente zeigt, daß der Unterschied bei verschiedenen
Temperaturen relativ gering ist. Hierbei ist noch zu erwahnen, daß bei der Berechnung der
Diffusionsanteil unberucksichtigt bleibt. Der berechnete Wert nimmt demzufolge immer einen
kleineren Wert an, als das gemessene 2.Moment.
In Tab. 3.17 sind die berechneten 2.Momente mit den entsprechenden gemessenen Werten
gegenuber gestellt. Hierbei ergibt sich folgender Zusammenhang:
Bei der tiefsten gemessenen Temperatur T=123K ist der Rotationsprozeß noch nicht ab-
geschlossen. Es liegt nahe, daß bei noch tieferen Temperaturen eine vollig starre Struk-
tur erreicht wird. Zwischen 140K und dem Phasenubergang III→ II findet Methylgruppen-
Rotation statt. Am Phasenubergang kommt zusatzlich zur Methylgruppen-Rotation min-
destens ein weiterer dynamischer Prozeß hinzu. Der berechnete Wert fur eine kombinierte
starres Molekul CH3- CH3/CH2Cl- Molekul- isotrope
Rotation Rotation Reorientierung Reorientierung
berechnet 37 17 10 0.4
gemessen 35 18 5
Zuordnung Phase III Phase III Phase II Phase I
T< 140K T> 140K
Tab. 3.17: Zusammenfassung der theoretisch berechneten und gemessenen 2.Momente
(x 10−8T2) fur die β-Chlorpivalinsaure.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 63
Methyl- und Chlormethyl-Rotation ist aber viel zu groß im Vergleich zu dem gemessenen
Wert von 5·10−8T2. Es liegt nahe, daß in der Phase II das gesamte Molekul eine Rotation
ausfuhren muß, die aber nicht mit der fur plastische Phasen typischen isotropen Reorientie-
rungen gleichgesetzt werden darf. Der berechnete Wert ist wesentlich zu klein. Es ist aber
anzunehmen, daß in der plastischen Phase isotrope Reorientierung vorherrscht.
T1-Messungen
In Abb. 3.33 sind die experimentellen Ergebnisse der Inversion-Recovery Methode tempe-
raturabhangig abgebildet. Man erkennt ein fur Molekulkristalle typisches Minimum. Die
Spin-Gitter-Relaxationszeit ist ein Maß fur die Starke der Wechselwirkung zwischen Spinsy-
stem und Gitter. Auf der Hochtemperaturseite steigt T1 nahezu linear mit der Temperatur
an. Es gilt ω0τc � 1, d.h. die relaxationsbestimmende Bewegung ist zu schnell, um effektiv
zur Relaxation beizutragen. Auf der Tieftemperaturseite findet man ebenfalls einen linearen
Verlauf, der aber bei Temperaturen ab etwa 140K von der Linearitat abweicht und abflacht.
Es gilt ω0τc � 1. Hier ist die relaxationsbestimmende Bewegung viel zu langsam, um effek-
tiv zur Relaxation beizutragen. Wie in Kapitel 2.7.6 auf Seite 24 beschrieben, kann man die
T1-Kurve mathematisch beschreiben und daraus die Korrelationszeit τc0 bestimmen. Hier-
bei wird angenommen, daß sich die Relaxationszeit aus den Bewegungen der Methyl- und
Chlormethylgruppen zusammensetzt.
1
T1
=1
TCH31
+1
TCH2Cl1
(3.2)
Die experimentelle Kurve wurde mit dem Marquardt-Levenberg-Algorithmus nach Glei-
chung 3.3 angefittet.
1
T1
= ACH3
(τCH3
1 + ω2τ 2CH3
+4τCH3
1 + 4ω2τ 2CH3
)
+ ACH2Cl
(τCH2Cl
1 + ω2τ 2CH2Cl
+4τCH2Cl
1 + 4ω2τ 2CH2Cl
) (3.3)
mit Ai = 920
ni
Nγ4�2
r6iund τi = τ0 exp{ Ei
a
RT}.
ni: Anzahl der Protonen, N: Gesamtzahl der Protonen, ri: Proton-Proton Abstand innerhalb
der Gruppe.
Der Fit zeigt folgende Ergebnisse:
ECH3a =15.3(2) kJmol−1 τCH3
0 =1.88(2) · 10−12s
ECH2Cla =20.7(9) kJmol−1 τCH2Cl
0 =2.08(9) · 10−13s
Die gemessene und simulierte Kurve sowie die Einzelkurven fur den Relaxationsprozeß
der Methylgruppen und der Chlormethylgruppe sind in Abb. 3.34 gezeigt. Den Erwartungen

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 64
0.003 0.004 0.005 0.006 0.007 0.008
0.1
1
10
125143167200250333
T [K]
Rel
axat
ions
zeit
T1 [
1/s]
1/T [1/K]
Abb. 3.33: Auftragung der Spin-Gitter-Relaxationszeiten gegen die Temperatur der β-Chlor-
pivalinsaure.
entsprechend findet man eine hohere Aktivierungsenergie fur die CH2Cl-Gruppe, da diese
durch die Beteiligung des Chlor-Atoms eine wesentlich großere Masse hat. Dadurch ist auch
die Korrelationszeit τc0 großer im Vergleich zur Methylgruppe. Das Abflachen der T1-Kurve
ab etwa 140K ist mit dem Einfrieren der Methylgruppenrotation korreliert, wie es auch
die Ergebnisse der Linienbreitenanalyse und des 2.Moments zeigen. Durch den Wegfall der
Dynamik der Methylgruppe wird die Moglichkeit der Spins zu relaxieren eingeschrankt und
erhoht damit die Relaxationszeit T1.
Vergleich der 1H NMR Untersuchungen mit der Literatur
Es wurden eingehende NMR Untersuchungen an der β-Chlorpivalinsaure von Aksnes et al.
im Temperaturbereich von 350 -160K durchgefuhrt [52]. Das Hauptaugenmerk ihrer Un-
tersuchungen lag dabei auf Diffusionsvorgangen in den festen und der flussigen Phase. Es
wurden T1 und T2-Messungen an 1H und 13C durchgefuhrt sowie 2H-Messungen an der d1-
deuterierten Saure. Aksnes et al. fanden ebenfalls im Temperaturbereich von der Flussigkeit
bis ca. 285K einen breiten Peak, der von sehr schmalen CH3, CH2Cl und COOH Peaks
uberlagert ist. Ihre Annahme, daß diese von Phase I herruhren, konnte hier nicht bestatigt
werden. Allerdings lagen ihnen nicht die umfangreichen thermodynamischen und strukturel-
len Untersuchungen vor. Obwohl sie von einem Phasenubergang II↔ III in ihrer Einfuhrung
ausgehen, wird dieser Aspekt nicht wieder aufgegriffen, da keine markanten Anderungen von
T1 meßbar sind. Ebenfalls fanden sie einen Wert fur das 2.Moment fur die Tieftemperatursei-
te des T1-Minimums von 11 · 10−8T2, daher nahmen sie C3 und C’3-Rotation an. Die hier
prasentierten Ergebnisse zeigen, daß nur Methylgruppenrotation in Phase III stattfindet. Es
zeigt sich, daß bei der Messung und Berechnung des 2.Moments große Sorgfalt bezuglich des
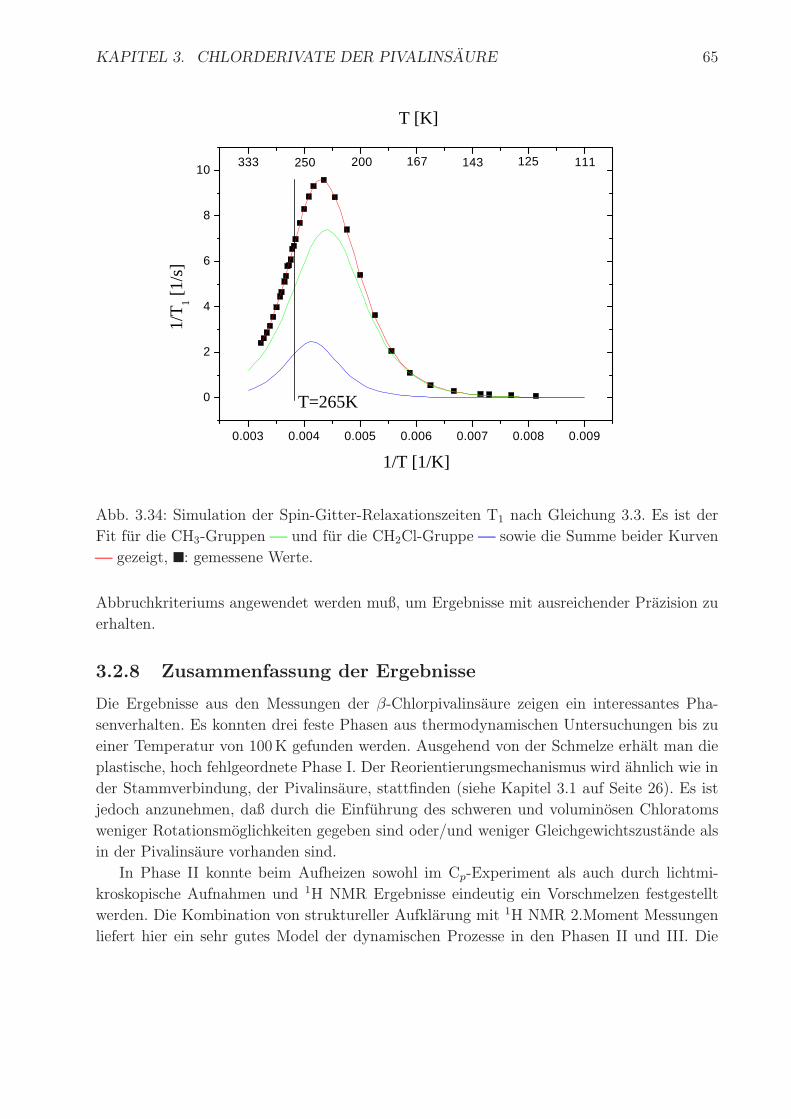
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 65
0.003 0.004 0.005 0.006 0.007 0.008 0.009
0
2
4
6
8
10111125143167250 200333
T [K]
T=265K
1/T
1 [1/
s]
1/T [1/K]
Abb. 3.34: Simulation der Spin-Gitter-Relaxationszeiten T1 nach Gleichung 3.3. Es ist der
Fit fur die CH3-Gruppen und fur die CH2Cl-Gruppe sowie die Summe beider Kurven
gezeigt, ■: gemessene Werte.
Abbruchkriteriums angewendet werden muß, um Ergebnisse mit ausreichender Prazision zu
erhalten.
3.2.8 Zusammenfassung der Ergebnisse
Die Ergebnisse aus den Messungen der β-Chlorpivalinsaure zeigen ein interessantes Pha-
senverhalten. Es konnten drei feste Phasen aus thermodynamischen Untersuchungen bis zu
einer Temperatur von 100K gefunden werden. Ausgehend von der Schmelze erhalt man die
plastische, hoch fehlgeordnete Phase I. Der Reorientierungsmechanismus wird ahnlich wie in
der Stammverbindung, der Pivalinsaure, stattfinden (siehe Kapitel 3.1 auf Seite 26). Es ist
jedoch anzunehmen, daß durch die Einfuhrung des schweren und voluminosen Chloratoms
weniger Rotationsmoglichkeiten gegeben sind oder/und weniger Gleichgewichtszustande als
in der Pivalinsaure vorhanden sind.
In Phase II konnte beim Aufheizen sowohl im Cp-Experiment als auch durch lichtmi-
kroskopische Aufnahmen und 1H NMR Ergebnisse eindeutig ein Vorschmelzen festgestellt
werden. Die Kombination von struktureller Aufklarung mit 1H NMR 2.Moment Messungen
liefert hier ein sehr gutes Model der dynamischen Prozesse in den Phasen II und III. Die

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 66
NMR Ergebnisse zeigen, daß in Phase II das gesamte Molekul eine Drehung ausfuhren muß,
um den geringen berechneten Wert des 2.Moments zu erklaren. Die strukturelle Fehlordnung
kann nun - ausgehend von den spektroskopischen Messungen - als eine dynamische Fehlord-
nung angesehen werden. Das Rotationsmodell ist anhand von Abb. 3.11 leicht zu erklaren.
Um von der Konformation des Molekuls A nach B zu kommen, reicht eine Rotation der
tert.-Butylgruppe von 120◦ entlang der C(3)-C(4)-Verbindungsachse aus. Gleichzeitig ver-
schiebt sich das Chloratom um etwa 1.48(1) A. Es ist ausgeschlossen, daß eine 240◦-Drehungstattfindet, da strukturell gesehen nicht genugend Platz vorhanden ist, um das Chloratom
so weit zu verschieben. Folglich findet sicherlich eine reine Methylgruppen-Rotation in Phase
II statt, die aber an die 120◦-Drehung des β-Chlorpivalinsaure Molekuls gekoppelt ist. Ob
noch zusatzlich eine 180◦-Drehung der Carboxylgruppe stattfindet, kann ausgeschlossen wer-
den. Die theoretische Berechnung einer 180◦-Drehung hat keinen vermindernden Effekt auf
das 2.Moment. Aus Einkristalluntersuchungen konnte die Carbonyl- (C=O) und Hydroxyl-
(C-O) Bindung eindeutig zugeordnet werden. Eine 180◦-Drehung der Carboxylgruppe wur-
de im Rontgenbild eine gemittelte Struktur liefern, in der nicht zwischen Carbonyl- und
Hydroxylbindungen unterschieden werden kann.
Am Phasenubergang II→ III friert die 120◦-Drehung ein, wobei vier unterschiedliche
Konformere aus den Zimmertemperaturkonformeren entstehen. Am Phasenubergang kann
aus Molekul A der Phase II direkt Molekul A der Phase III hervorgehen. Uber eine 30◦-Drehung wird Molekul C erhalten. Theoretisch kann Molekul D durch eine 150◦-Drehungerhalten werden. Molekul B wird, wie oben beschrieben, aus Molekul A der Phase II erhalten.
Gleiches gilt fur die Abbildung von Molekul B aus Phase II zu den vier Konformeren der
Phase III. Das Einfrieren ist ein kooperativer Prozeß, der die Anordnung, d.h. die Stapelung
der moglichen dimeren Einheiten (AC, AD, BC, und BD) wie in Kapitel 3.2.4 auf Seite 46
beschrieben, zulaßt.
In Phase III liegen zwei unterschiedliche dynamische Verhaltnisse vor. Die 2.Moment
Messungen zeigen, daß bis zu einer Temperatur von ca. 140K Methylgruppen-Rotation vor-
handen ist. Unterhalb dieser Temperatur wird die Rotationsgeschwindigkeit langsamer, bis
eine vollig geordnete Struktur erreicht ist.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 67
3.3 Zusammenfassung der experimentellen Ergebnisse
der β,β’-Dichlorpivalinsaure
Dieser Abschnitt dient der kurzen Zusammenfassung der Ergebnisse der β,β’-Dichlorpivalin-
saure. Die Untersuchungen wurden von Strauss im Rahmen seiner Dissertation durchgefuhrt.
Siehe hierzu auch [3, 53].
Alle hier vorgestellten Ergebnisse wurden mit Proben, die durch Sublimation gereinigten
wurden, durchgefuhrt.
Die thermodynamischen Messungen wurden an einem selbstgebauten DTA-DSC-Gerat
[3] in Abhangigkeit der Kuhlrate durchgefuhrt.
In Tab. 3.18 sind die Phasenubergangstemperaturen, -enthalpien und entropien zusam-
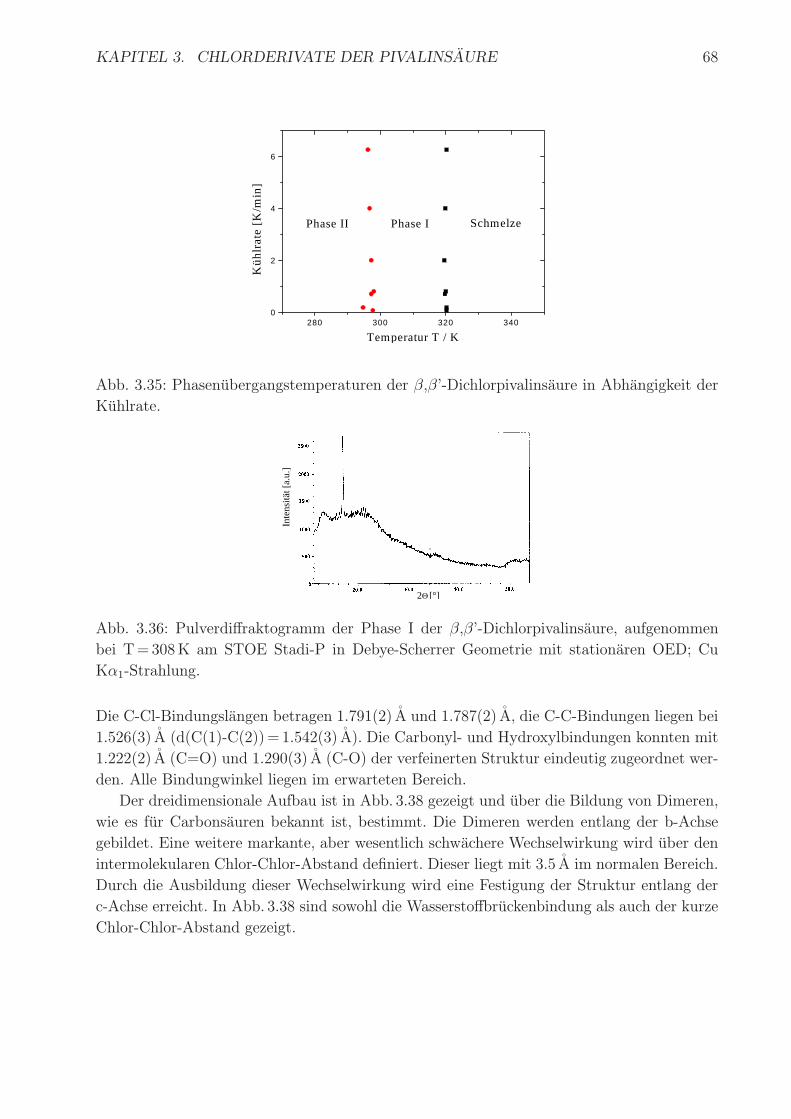
mengefaßt. Abb. 3.35 zeigt die Phasenubergangstemperaturen in Abhangigkeit von der Heiz-
/Kuhlrate. Beim Abkuhlen aus der Schmelze erhalt man die plastische Phase I, die eine
II→m m→ I I→ II
T [K] 343.8 319.7 297.2
∆H [kJmol−1] 21.89 3.44 19.39
∆S [Jmol−1K−1] 62.90 10.77 65.79
∆S/R 7.57 1.30 7.91
Tab. 3.18: Phasenubergangstemperaturen, -enthalpien und -entropien der β,β’-Dichlor-
pivalinsaure.
relativ große Phasenbreite von 23K aufweist. Der geringe Energieinhalt des Erstarrungspro-
zesses ist typisch fur plastische Phasen. Bei 297K findet der Ubergang in Phase II statt.
Die Summe der Phasenubergangsentropien ∆Sm→ I +∆SI→ II ist mit 76 kJmol−1K−1 sehr
hoch und laßt auf eine geordnete Struktur der Phase II schließen. Es konnte kein weiterer
Phasenubergang bis 77K gefunden werden. Beim Aufheizen gelangt man direkt, wie bei der
β-Chlorpivalinsaure, in die Schmelze. Die Hysterese von ca. 24K zwischen Schmelz- und
Erstarrungsprozeß ist nicht unublich.
Im Rontgenpulverdiffraktogramm findet man, wie schon in der β-Chlorpivalinsaure nur
einen Reflex im Bereich vom 11≤ 2Θ≤ 54 ◦. Die Indizierung mit (111) ergibt ein fcc-Gitter
mit Z=4, a= 9.11(1) A fur T=308K. Geht man von der hochst moglichen Symmetrie aus,
so gelangt man auch bei der β,β’-Dichlorpivalinsaure zur Raumgruppe Fm3m. Das Pulver-
diffraktogramm ist in Abb. 3.36 gezeigt.
Einkristallstrukturuntersuchungen bei Zimmertemperatur zeigen eine orthorhombische
Struktur. Meßbedingungen und die Guteparameter der Verfeinerung sind in Tab. 3.19 auf-
gelistet. In Abb. 3.37 ist die asymmetrische Einheit der Phase II gezeigt. Atomkoordinaten,
Bindungslangen und -winkel sowie die anisotropen Temperaturfaktoren sind im Anhang A2
zusammengefaßt. Der intramolekulare Aufbau zeigt keine ungewohnlichen Bindungslangen.
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 68
280 300 320 3400
2
4
6
Phase II Phase I Schmelze
Kü
hlr
ate
[K/m
in]
Temperatur T / K
Abb. 3.35: Phasenubergangstemperaturen der β,β’-Dichlorpivalinsaure in Abhangigkeit der
Kuhlrate.
Inte
nsitä
t [a.
u.]
Θ2 [°]
Abb. 3.36: Pulverdiffraktogramm der Phase I der β,β’-Dichlorpivalinsaure, aufgenommen
bei T=308K am STOE Stadi-P in Debye-Scherrer Geometrie mit stationaren OED; Cu
Kα1-Strahlung.
Die C-Cl-Bindungslangen betragen 1.791(2) A und 1.787(2) A, die C-C-Bindungen liegen bei
1.526(3) A (d(C(1)-C(2))= 1.542(3) A). Die Carbonyl- und Hydroxylbindungen konnten mit
1.222(2) A (C=O) und 1.290(3) A (C-O) der verfeinerten Struktur eindeutig zugeordnet wer-
den. Alle Bindungwinkel liegen im erwarteten Bereich.
Der dreidimensionale Aufbau ist in Abb. 3.38 gezeigt und uber die Bildung von Dimeren,
wie es fur Carbonsauren bekannt ist, bestimmt. Die Dimeren werden entlang der b-Achse
gebildet. Eine weitere markante, aber wesentlich schwachere Wechselwirkung wird uber den
intermolekularen Chlor-Chlor-Abstand definiert. Dieser liegt mit 3.5 A im normalen Bereich.
Durch die Ausbildung dieser Wechselwirkung wird eine Festigung der Struktur entlang der
c-Achse erreicht. In Abb. 3.38 sind sowohl die Wasserstoffbruckenbindung als auch der kurze
Chlor-Chlor-Abstand gezeigt.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 69
Cl1
C3
Cl2 C4
C2
C1 C5
O2
O1
Abb. 3.37: Asymmetrische Einheit der
Phase II der β,β’-Dichlorpivalinsaure. Es
ist die Numerierung der Atome gegeben,
wie sie auch im Text verwendet wird. Die
thermischen Ellipsoide zeigen eine Auf-
enthaltswahrscheinlichkeit von 50%.
Abb. 3.38: Dreidimensionaler Aufbau der
Phase II der β,β’-Dichlorpivalinsaure. Ge-
zeigt sind die Wasserstoffbruckenbindun-
gen (Ausbildung der dimeren Einheiten)
und der kurze Cl-Cl-Abstand von 3.50 A.
50 100 150 200 250 300 350
33.0
33.5
34.0
34.5
35.0
ν (35
Cl)
[M
Hz]
T [K]
Abb. 3.39: 35Cl NQR-Frequenzen [MHz] in Abhangigkeit der Temperatur der β,β’-Dichlor-
pivalinsaure. ■: ν1, ●: ν2.
35Cl NQR Messungen zeigen ein Duplett im Temperaturbereich von 77K bis zur Schmel-
ze, wie aus den kristallographisch inaquivalenten Chlorpositionen zu erwarten war. Die Ab-
hangigkeit der NQR-Frequenz von der Temperatur ist in Abb. 3.39 gezeigt. Beide Resonanz-
linien verschieben sich zu geringeren Frequenzen mit steigender Temperatur.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 70
STOE Stadi-4 T=297K
Kristallsystem orthorhombisch
Raumgruppe Pbca
Gitterkonstanten
a [A] 10.197(3)
b [A] 11.464(3)
c [A] 12.938(3)
Volumen [A3] 1512.3(7)
Z 8
ber. Dichte [Mgm−3] 1.502
Absorptionskoeffizient [mm−1] 0.785
F(000) 704
Kristallgroße [mm3] 0.80 x 0.42 x 0.300
θ Bereich fur Datensammlung [◦] 3.10 bis 25.00
Indizierungsbereich 0≤ h≤ 12
-13≤ k≤ 13
0≤ l≤ 15
Anzahl Reflexe 2858
davon unabhangige 1333 (Rint=0.03)
Absorptionskorrektur empirisch
Verfeinerungsmethode ’full-matrix least-squares on F2’
Daten / Restraints / Parameter 1333 / 0 / 84
GooF [I>σ(I)] 1.06
R-Faktoren [I> 2σ(I)] R1=0.0352, ωR2=0.0913
R-Faktoren (alle Daten) R1=0.0407 ωR2=0.0973
Großte Diff. zw. Berg und Tal [e A−3] 0.319 und -0.335
Tab. 3.19: Meßbedingungen und Guteparameter der Strukturverfeinerung der Phase II der
β,β’-Dichlorpivalinsaure.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 71
3.4 β,β’,β”-Trichlorpivalinsaure
In der Reihe der Chlorderivate der Pivalinsaure fehlt zur Vollstandigkeit noch die β,β’,β”-
Trichlorpivalinsaure mit x= 3. Die Untersuchungen wurden von Frau Goetze im Rahmen
ihrer Diplomarbeit durchgefuhrt [54, 55].
3.4.1 Synthese
Pentaerythrit C(CH2OH)4) wurde nach einer modifizierten Anleitung von Lynch [56] zur
β,β’,β”-Trichlorpivalinsaure umgesetzt. Das Edukt wurde in Pyridin gelost und auf 340K
erhitzt. Thionylchlorid (SO2Cl) wurde unter kraftigem Ruhren langsam zugegeben. Die Lo-
sung wurde fur 12 h bei 390 - 400K unter Ruckfluß erhitzt und kontinuierlich geruhrt. Nach
komplettem Umsatz wurde erst Eiswasser, anschließend Toluol hinzugefugt. Das Produkt
Pentraerythritol Trichlorhydrin (CH2Cl)3CCH2OH wird aus der organischen Phase des To-
luols mit Nitriersaure oxidiert. Nach Extraktion und Einengung konnte β,β’,β”-Trichlor-
pivalinsaure erhalten werden. Nach Umkristallisation in Hexan wurden feine weiße Nadel
erhalten.
3.4.2 Thermische Analyse
DSC-Messungen wurden an einem Setaram DSC 121 im Temperaturbereich von 400K bis
150K durchgefuhrt. Es konnte im DSC-Experiment nur der Schmelz-/Erstarrungsprozess ge-
funden werden. In Tab. 3.20 sind die Phasenubergangstemperaturen, -enthalpien und entro-
pien zusammengefasst. Der hohe Wert fur die Schmelzentropie zeigt, daß keine orientierungs-
ungeordnete Phase auftritt.
Phasenubergang T [K] ∆H [kJmol−1] ∆S [Jmol−1K−1] ∆S/R
krist.→m 383.9 20.9 54.2 6.5
m→ krist. 380.9 21.4 56.3 6.8
Tab. 3.20: Thermodynamische Daten der β,β’,β”-Trichlorpivalinsaure im Temperaturbereich
von 400 - 150K.
3.4.3 Strukturanalyse
Es wurden sowohl Einkristalluntersuchungen am Nonius Enraf CAD4 als auch Rontgenpul-
verbeugungsmessungen am STOE-Stadi-P in Debye Scherrer Geometrie durchgefuhrt. Ein-
kristalle der β,β’,β”-Trichlorpivalinsaure wurden durch Umkristallisation in Hexan erhalten.
Da die Verbindung hygroskopisch ist, wurde der Kristall in eine dunnwandige Kapillare ein-
geschlossen und auf dem Diffraktometer justiert. Die Meßbedingungen und Guteparameter
der Verfeinerung sind in Tab. 3.21 zusammengefaßt.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 72
Enraf Nonius CAD 4 T=304K
Kristallsystem monoklin
Raumgruppe P 21
Gitterkonstanten
a [A] 6.961(1)
b [A] 9.979(2)
c [A] 12.059(3)
β 94.01(2) ◦
Volumen [A3] 835.6(3)
Z 4
ber. Dichte [Mgm−3] 1.633
Absorptionskoeffizient [mm−1] 1.034
F(000) 416
Kristallgroße [mm3] 0.50 x 0.08 x 0.08
θ Bereich fur Datensammlung [◦] 1.69 bis 25.95
Indizierungsbereich -8≤ h≤ 1
-12≤ k≤ 0
-14≤ l≤ 14
Anzahl Reflexe 1899
davon unabhangige 1726 (Rint=0.03)
Absorptionskorrektur empirisch
Verfeinerungsmethode ’full-matrix least-squares on F2’
Daten / Restraints / Parameter 1735 / 1 / 187
GooF [I>σ(I)] 1.02
R-Faktoren [I> 2σ(I)] R1=0.0403, ωR2=0.0862
R-Faktoren (alle Daten) R1=0.1100 ωR2=0.1073
Absoluter Strukturparameter 0.1(2)
Großte Diff. zw. Berg und Tal [e A−3] 0.322 und -0.281
Tab. 3.21: Meßbedingungen und Guteparameter der Verfeinerung der kristallinen Phase der
β,β’,β”-Trichlorpivalinsaure.
Die Struktur wurde in der monoklinen Raumgruppe P 21 gelost mit Z=4, a= 6.961(1) A,
b= 9.979(2) A, c= 12.059(3) A und β=94.01(2) ◦, V=835.6(3) A3. Alle nicht Wasserstoff-
atome wurden uber Differenzfouriersynthese gefunden und nach der Methode der kleinsten
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 73
Fehlerquadrate verfeinert. Das Wasserstoffatom zwischen O(2) und O(3) wurde uber Dif-
ferenzfourieranalyse lokalisiert und verfeinert. Alle anderen Wasserstoffpositionen wurden
geometrisch generiert und nicht verfeinert. Die asymmetrische Einheit ist in Abb. 3.40 mit
der Numerierung der Atome gezeigt.
Cl4
Cl6
C10C8
C7
O3
C6
O4Cl5
C9Cl3 Cl1
C4
C3
C5
Cl2
O2
C1O1
C2
Abb. 3.40: Asymmetrische Einheit der kristallinen Phase der β,β’,β”-Trichlorpivalinsaure.
Es ist die Numerierung der Atome angegeben, wie sie auch im Text verwendet wird.
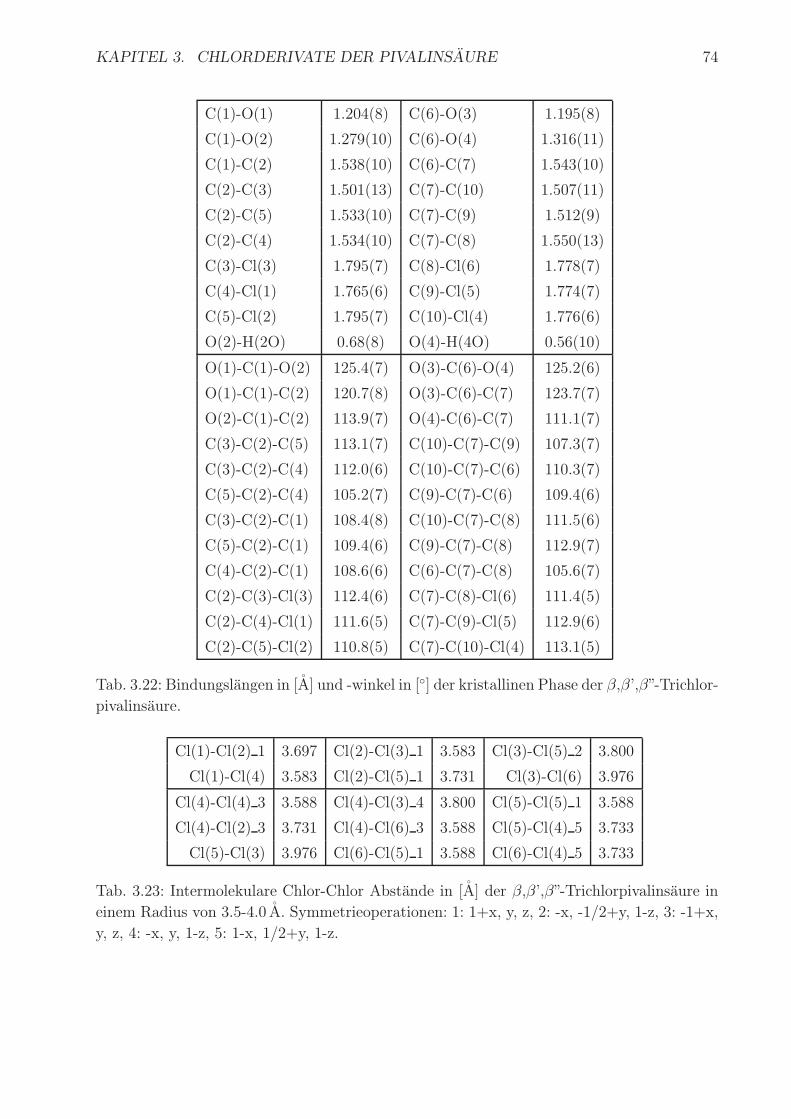
Bindungslangen und -winkel sind in Tab. 3.22 aufgelistet. Atomkoordinaten und anisotro-
pe Temperaturfaktoren befinden sich im Anhang A3. Bindungslangen und -winkel der bei-
den Molekule sind im erwarteten Bereich. Die C-Cl-Bindungslangen liegen zwischen 1.765 A
und 1.795 A, die C-C-Cl Winkel im Bereich von 110.3 ◦ und 112.9 ◦. Die Bindungslangen
der C-O-Bindungen sind ein geeignetes Kriterium der Zuordnung zur Hydroxyl- und Car-
bonylgruppe. Die Wasserstoffbruckenbindung ist mit 2.671(1) A im erwarteten Bereich. Die
dreidimensionale Anordnung ist in Abb. 3.41 gezeigt. Sie zeigt die Ausbildung der Wasser-
stoffbruckenbindung als charakteristisches Element in der Struktur.
Betrachtet man den strukturellen Aufbau entlang [101] (Bilddiagonale von links unten
nach rechts oben), so kann man den Aufbau folgendermaßen beschreiben. Es sind immer
zwei Dimereinheiten direkt ubereinander gestapelt, die dritte und vierte Dimereinheit ist um
eine halbe Dimereinheit (ein Molekul) entlang [-101] verschoben.
Die intermolekularen Cl-Cl Abstande sind in Tab. 3.23 aufgefuhrt. Man kann deutlich
erkennen, daß durch die oben beschriebene dreidimenionale Anordnung die Cl-Cl-Abstande
zwischen 3.5 A und 4.0 A liegen. Dies ist ein optimaler Abstand zur Ausbildung von Wech-
selwirkungen zwischen zwei Chloratomen [5].
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 74
C(1)-O(1) 1.204(8) C(6)-O(3) 1.195(8)
C(1)-O(2) 1.279(10) C(6)-O(4) 1.316(11)
C(1)-C(2) 1.538(10) C(6)-C(7) 1.543(10)
C(2)-C(3) 1.501(13) C(7)-C(10) 1.507(11)
C(2)-C(5) 1.533(10) C(7)-C(9) 1.512(9)
C(2)-C(4) 1.534(10) C(7)-C(8) 1.550(13)
C(3)-Cl(3) 1.795(7) C(8)-Cl(6) 1.778(7)
C(4)-Cl(1) 1.765(6) C(9)-Cl(5) 1.774(7)
C(5)-Cl(2) 1.795(7) C(10)-Cl(4) 1.776(6)
O(2)-H(2O) 0.68(8) O(4)-H(4O) 0.56(10)
O(1)-C(1)-O(2) 125.4(7) O(3)-C(6)-O(4) 125.2(6)
O(1)-C(1)-C(2) 120.7(8) O(3)-C(6)-C(7) 123.7(7)
O(2)-C(1)-C(2) 113.9(7) O(4)-C(6)-C(7) 111.1(7)
C(3)-C(2)-C(5) 113.1(7) C(10)-C(7)-C(9) 107.3(7)
C(3)-C(2)-C(4) 112.0(6) C(10)-C(7)-C(6) 110.3(7)
C(5)-C(2)-C(4) 105.2(7) C(9)-C(7)-C(6) 109.4(6)
C(3)-C(2)-C(1) 108.4(8) C(10)-C(7)-C(8) 111.5(6)
C(5)-C(2)-C(1) 109.4(6) C(9)-C(7)-C(8) 112.9(7)
C(4)-C(2)-C(1) 108.6(6) C(6)-C(7)-C(8) 105.6(7)
C(2)-C(3)-Cl(3) 112.4(6) C(7)-C(8)-Cl(6) 111.4(5)
C(2)-C(4)-Cl(1) 111.6(5) C(7)-C(9)-Cl(5) 112.9(6)
C(2)-C(5)-Cl(2) 110.8(5) C(7)-C(10)-Cl(4) 113.1(5)
Tab. 3.22: Bindungslangen in [A] und -winkel in [◦] der kristallinen Phase der β,β’,β”-Trichlor-
pivalinsaure.
Cl(1)-Cl(2) 1 3.697 Cl(2)-Cl(3) 1 3.583 Cl(3)-Cl(5) 2 3.800
Cl(1)-Cl(4) 3.583 Cl(2)-Cl(5) 1 3.731 Cl(3)-Cl(6) 3.976
Cl(4)-Cl(4) 3 3.588 Cl(4)-Cl(3) 4 3.800 Cl(5)-Cl(5) 1 3.588
Cl(4)-Cl(2) 3 3.731 Cl(4)-Cl(6) 3 3.588 Cl(5)-Cl(4) 5 3.733
Cl(5)-Cl(3) 3.976 Cl(6)-Cl(5) 1 3.588 Cl(6)-Cl(4) 5 3.733
Tab. 3.23: Intermolekulare Chlor-Chlor Abstande in [A] der β,β’,β”-Trichlorpivalinsaure in
einem Radius von 3.5-4.0 A. Symmetrieoperationen: 1: 1+x, y, z, 2: -x, -1/2+y, 1-z, 3: -1+x,
y, z, 4: -x, y, 1-z, 5: 1-x, 1/2+y, 1-z.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 75
Abb. 3.41: Dreidimensionale Anordnung der kristallinen Phase der β,β’,β”-Trichlorpivalin-
saure, Blick entlang [010].
Es wurde ebenfalls ein Rontgenpulverdiffraktogramm aufgenommen und mit den Ein-
kristalldaten verglichen. Das Ergebnis der Rietveldverfeinerung ist in Abb. 3.42 dargestellt.
Der Differenzplot zeigt gute Ubereinstimmmung zwischen beobachteten und berechneten
Profilen. Die verfeinerten Strukturparameter stehen im Einklang mit den Einkristallstruk-
turdaten. Die resultierenden Atomkoordinaten sind im Anhang A3 zusammengefaßt.
Inte
nsitä
t [a.
u.]
2Θ [°]
Abb. 3.42: Gemessenes und berechnetes Pulverdiffraktogramm der kristallinen Phase der
β,β’,β”-Trichlorpivalinsaure. Der Differenzplot ist ebenfalls gezeigt.

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 76
3.4.4 Zusammenfassung
In der Reihe der Chlorderivate der Pivalinsaure zeigt, wie zu erwarten, die Verbindung mit
x= 3, β,β’,β”-Trichlorpivalinsaure, keine orientierungsungeordnete Phase. Man gelangt di-
rekt von der geordneten Phase in die Schmelze. Durch den Einbau von drei Chloratomen
und die Ausbildung der Wasserstoffbruckenbindung wird die Moglichkeit des Reorientie-
rungsprozesses gehemmt. Der dreidimensionale Aufbau der kristallinen Phase ist durch die
Ausbildung der Wasserstoffbruckenbindung und durch Chlor-Chlor-Wechselwirkungen, deren
Abstande idealerweise zwischen 3.5-4.0 A liegen sollten [5], gekennzeichnet.
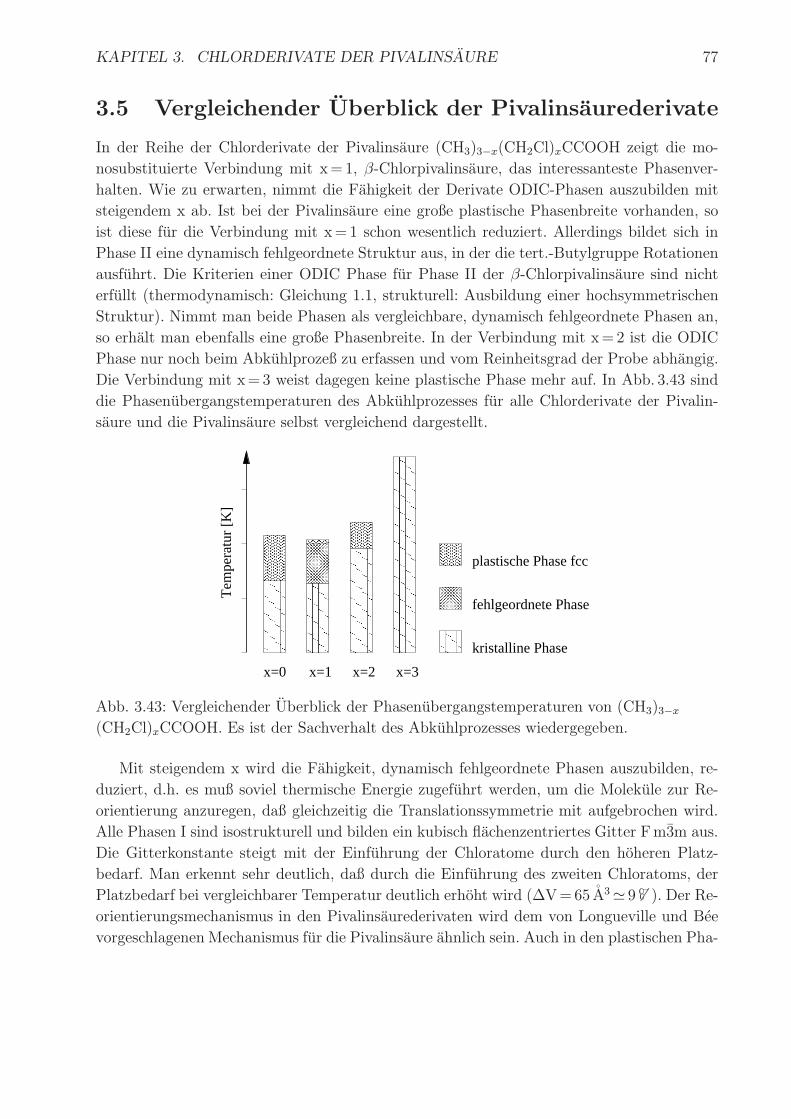
KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 77
3.5 Vergleichender Uberblick der Pivalinsaurederivate
In der Reihe der Chlorderivate der Pivalinsaure (CH3)3−x(CH2Cl)xCCOOH zeigt die mo-
nosubstituierte Verbindung mit x= 1, β-Chlorpivalinsaure, das interessanteste Phasenver-
halten. Wie zu erwarten, nimmt die Fahigkeit der Derivate ODIC-Phasen auszubilden mit
steigendem x ab. Ist bei der Pivalinsaure eine große plastische Phasenbreite vorhanden, so
ist diese fur die Verbindung mit x= 1 schon wesentlich reduziert. Allerdings bildet sich in
Phase II eine dynamisch fehlgeordnete Struktur aus, in der die tert.-Butylgruppe Rotationen
ausfuhrt. Die Kriterien einer ODIC Phase fur Phase II der β-Chlorpivalinsaure sind nicht
erfullt (thermodynamisch: Gleichung 1.1, strukturell: Ausbildung einer hochsymmetrischen
Struktur). Nimmt man beide Phasen als vergleichbare, dynamisch fehlgeordnete Phasen an,
so erhalt man ebenfalls eine große Phasenbreite. In der Verbindung mit x= 2 ist die ODIC
Phase nur noch beim Abkuhlprozeß zu erfassen und vom Reinheitsgrad der Probe abhangig.
Die Verbindung mit x= 3 weist dagegen keine plastische Phase mehr auf. In Abb. 3.43 sind
die Phasenubergangstemperaturen des Abkuhlprozesses fur alle Chlorderivate der Pivalin-
saure und die Pivalinsaure selbst vergleichend dargestellt.
��
��
��
��������
��������
������������
������������
������������������������������������
������������������������������������
����������
����������
��������
������������
������������
����������������������������
����������������
��������
������
������
��������
��������
Tem
pera
tur
[K]
x=0 x=1 x=2 x=3
kristalline Phase
fehlgeordnete Phase
plastische Phase fcc
Abb. 3.43: Vergleichender Uberblick der Phasenubergangstemperaturen von (CH3)3−x
(CH2Cl)xCCOOH. Es ist der Sachverhalt des Abkuhlprozesses wiedergegeben.
Mit steigendem x wird die Fahigkeit, dynamisch fehlgeordnete Phasen auszubilden, re-
duziert, d.h. es muß soviel thermische Energie zugefuhrt werden, um die Molekule zur Re-
orientierung anzuregen, daß gleichzeitig die Translationssymmetrie mit aufgebrochen wird.
Alle Phasen I sind isostrukturell und bilden ein kubisch flachenzentriertes Gitter Fm3m aus.
Die Gitterkonstante steigt mit der Einfuhrung der Chloratome durch den hoheren Platz-
bedarf. Man erkennt sehr deutlich, daß durch die Einfuhrung des zweiten Chloratoms, der
Platzbedarf bei vergleichbarer Temperatur deutlich erhoht wird (∆V=65 A3� 9%). Der Re-
orientierungsmechanismus in den Pivalinsaurederivaten wird dem von Longueville und Bee
vorgeschlagenen Mechanismus fur die Pivalinsaure ahnlich sein. Auch in den plastischen Pha-

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 78
x 0 1 2
a [A] 8.87 8.84 9.11
T [K] 293 301 308
Tab. 3.24: Zusammenstellung der Gitterkonstante a der kubischen Phase I der Chlorderivate
der Pivalinsaure (CH3)3−x(CH2Cl)xCCOOH mit x= 0, 1, 2.
sen der Chlorderivate sollte die Reorientierung stark von der Bildung der Dimeren beeinflußt
sein. Die schweren und voluminosen Chloratome werden die Rotation der tert.-Butylgruppe
beeinflussen und die Reorientierung des gesamten Molekuls verlangsamen.
Der Vergleich der geordneten Chlorderivate zeigt, daß die Ausbildung der Wasserstoffbrucken-
bindung das strukturelle Hauptmerkmal ist. Ein zweiter Aspekt ist die Ausbildung von van
der Waals Wechselwirkungen, deren Bildungsmoglichkeit uber intermolekulare Chlor-Chlor-
Abstande definiert wird.
Sowohl die Trichlor- als auch die Dichlorverbindung zeigen einen sehr ahnlichen Aufbau. In
Abb. 3.44 ist der strukturelle Aufbau der β,β’-Dichlorpivalinsaure und der β,β’,β”-Trichlor-
pivalinsaure mit gleicher Ansicht gezeigt. Jeweils senkrecht zur Dimerenverbindungsachse
sind die Molekule so angeordnet, daß nach zwei Molekulen die Dimereneinheit um 1/2 (nach
unten) verschoben ist. Fur β,β’,β”-Trichlorpivalinsaure ist in jeder Reihe noch eine zusatz-
liche Verschiebung entlang b (nach unten) vorhanden. Mit dieser Anordnung werden Chlor-
Chlor-Abstande im Bereich von 3.5-4.0 A erreicht, die den dreidimensionalen Verband zusatz-
lich starken. Die Chlor-Chlor-Abstande sind in Tab. 3.25 aufgelistet. Im Gegensatz dazu ist
β,β’-Dichlorpivalinsaure
Cl(1)-Cl(2) 3.794 Cl(1)-Cl(2) 1 3.496
β,β’,β”-Trichlorpivalinsaure
Cl(1)-Cl(2) 2 3.697 Cl(1)-Cl(4) 3 3.583
Cl(2)-Cl(3) 4 3.583 Cl(3)-Cl(2) 3.910
Cl(2)-Cl(5) 4 3.731 Cl(3)-Cl(5) 5 3.800
Cl(3)-Cl(6) 3.976 Cl(4)-Cl(5) 4 3.711
Cl(4)-Cl(6) 6 3.733 Cl(5)-Cl(6) 3.766
Cl(5)-Cl(6) 2 3.588 Cl(6)-Cl(5) 4 3.588
Tab. 3.25: Intermolekulare Chlor-Chlor-Abstande der Verbindungen mit x= 2 und x=3,
(CH3)3−x(CH2Cl)xCCOOH. Die Fehler liegen bei 0.002 A. Symmetrieoperatoren: 1: 1/2-x,
1/2+y, z, 2: -1+x, y, z, 3: x, y, -1+z, 4: 1+x, y, z, 5: -x, -1/2+y, 1-z, 6: 1-x, -1/2+y, 1-z.
der dreidimensionale Verband der β-Chlorpivalinsaure aus Reihen von Dimeren aufgebaut.
Durch die versetzte Anordnung der Chloratome, wie in Kapitel 3.2.4 auf Seite 46 beschrie-

KAPITEL 3. CHLORDERIVATE DER PIVALINSAURE 79
Abb. 3.44: Vergleich der dreidimensionalen Struktur der Verbindung mit x= 2 (links, Blick
entlang [100]) und x=3 (rechts, Blick entlang [100]). Es ist eine vergleichbare strukturelle
Ansicht gewahlt.
ben, konnen sich fur diese geometrische Anordnung optimale Chlor-Chlor-Wechselwirkungen
ausbilden (siehe Tab. 3.15).
Zusammenfassend ist zu bemerken, daß in der Reihe der Chlorderivate der Pivalinsaure,
das Verhalten der Verbindung mit x= 1, β-Chlorpivalinsaure außergewohnlich ist.
Die Analyse der dreidimensionalen Struktur der geordneten Phasen zeigt einen sehr ahnli-
chen Aufbau der β,β’-Dichlorpivalinsaure und β,β’,β”-Trichlorpivalinsaure, die die gleichen
strukturellen Merkmale besitzen.













![Roth, Gabrielle - Enseñanzas de una chamán urbananewfield.education/...1475086195-04Ensennanzasdeunachamanurbana.pdf · 56/&)$+3-3$"74#(!!!!! &3#"&0)! [1'! 3'! 4'%\#! f]x&(! ,-.?9k.68!](https://static.fdokument.com/doc/165x107/5e12c1a54f52f07167614c94/roth-gabrielle-enseanzas-de-una-chamn-563-374-30.jpg)




