
Kurs “ Allgemeine und systematische Pharmakologie und ... II_3.pdf · Ihr Referat sollte folgende...
45
Kurs “ Allgemeine und systematische Pharmakologie und Toxikologie” Wintersemester 2018/19 Seminarthema II: Antiphlogistische und antipyretische Analgetika Der Inhalt bzw. die Gliederung der Referate ist frühzeitig mit der/dem zuständigen Dozentin/en abzusprechen. Alle Referate sollten ca. 20 Minuten dauern und den Einsatz von Hilfsmitteln (Powerpoint-Präsentation) umfassen. Bei Wiederverwendung von Powerpoint-Präsentationen von Kolleginnen/en voran- gegangener Seminare werden keine Jokerpunkte (siehe Link "Creditsystem") vergeben. Prof. Dr. Barbara Möpps, N 26-5208, Tel. 500-65505 Referat I: Prothrombotische, antithrombotische und cardiovaskuläre Effekte von NSAIDs und Coxiben - Grosser, T., Fries, S., and FitzGerald, G.A. (2006): Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 116: 4-15 - Patrono, C. (2016): Cardiovscular effects of cyclooxygenase-2 inhibitors: a mechanistic and clinical perspective. Fanelli, A., Ghisi, D., Aprile, P.L., and Lapi, F. (2017): Cardiovascular and cerebrovascular risk with nosteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: latest evidence and clinical implications. Ther. Adv. Drug saf. 8: 173-182. Ihr Referat sollte folgende Punkte umfassen: - Strukturelle Unterschiede und Physiologie der COX1- und COX2-Enzyme - Vorstellung COX-2-selektiver Hemmer, Wirkungen und unerwünschten Wirkungen - prothrombotische, antithrombotische und kardiovaskuläre Effekte der NSAIDs und Coxibe Referat II: Pharmakotherapie der Osteoarthrose/Osteoarthritis (OA). Paracetamol als Analgetikum. - Toussaint, K. et al. (2010). What do we (not) know about how paracetamol (acetaminophen) works? J. Clin. Pharm. Ther. 35: 617-638. - Nambiar, N.J. (2012): Management of paracetamol poisoning: the old and the new. J. Clin. Diagnost. Res. 6: 1101-1104. Sharma, C.V., Metha, V. (2014): Paracetamol; mechanisms and updates. BJA education, Continuing Education in Anaesthesia Critical Care & Pain 14: 155 Ihr Referat sollte folgende Punkte umfassen: - Darstellung der möglichen molekularen Wirkmechanismen von Paracetamol - Einsatz von Paracetamol - Nebenwirkungen von Paracetamol und toxische Wirkung bei Überdosierung. Referat III: Neuropathischer Schmerz: therapeutische Ansätze - Cohen, S.P., and Mao, J. (2014): Neuropathic pain: mechanisms and their clinical implications. BMJ, 384: f7656. - Sommer, C. (2013): Neuropathische Schmerzen. Pathophysiologie, Diagnostik und Therapie. Schmerz 27: 619-634. - Magrinelli, F., Zanette, G., and Tamburin, S. (2013): Neuropathic pain: diagnosis and treatment. Prac. Neurol. 13: 292-307. - Leitlinien für Diagnostik und Therapie in der Neurologie: http://www.awmf.org/leitlinien/detail/ll/030-114.html Ihr Referat sollte folgende Punkte umfassen: - Evidenzbasierte Therapie des neuropathischen Schmerzes - Welche Rolle spielen NSAIDs in der medikamentösen Therapie?
Transcript of Kurs “ Allgemeine und systematische Pharmakologie und ... II_3.pdf · Ihr Referat sollte folgende...

Kurs “ Allgemeine und systematische Pharmakologie und Toxikologie” Wintersemester 2018/19
Seminarthema II: Antiphlogistische und antipyretische Analgetika Der Inhalt bzw. die Gliederung der Referate ist frühzeitig mit der/dem zuständigen Dozentin/en abzusprechen. Alle Referate sollten ca. 20 Minuten dauern und den Einsatz von Hilfsmitteln (Powerpoint-Präsentation) umfassen. Bei Wiederverwendung von Powerpoint-Präsentationen von Kolleginnen/en voran-gegangener Seminare werden keine Jokerpunkte (siehe Link "Creditsystem") vergeben. Prof. Dr. Barbara Möpps, N 26-5208, Tel. 500-65505
Referat I: Prothrombotische, antithrombotische und cardiovaskuläre Effekte von NSAIDs und Coxiben - Grosser, T., Fries, S., and FitzGerald, G.A. (2006): Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 116: 4-15 - Patrono, C. (2016): Cardiovscular effects of cyclooxygenase-2 inhibitors: a mechanistic and clinical perspective. Fanelli, A., Ghisi, D., Aprile, P.L., and Lapi, F. (2017): Cardiovascular and cerebrovascular risk with nosteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: latest evidence and clinical implications. Ther. Adv. Drug saf. 8: 173-182.
Ihr Referat sollte folgende Punkte umfassen: - Strukturelle Unterschiede und Physiologie der COX1- und COX2-Enzyme - Vorstellung COX-2-selektiver Hemmer, Wirkungen und unerwünschten Wirkungen - prothrombotische, antithrombotische und kardiovaskuläre Effekte der NSAIDs und Coxibe
Referat II: Pharmakotherapie der Osteoarthrose/Osteoarthritis (OA). Paracetamol als
Analgetikum. - Toussaint, K. et al. (2010). What do we (not) know about how paracetamol (acetaminophen) works? J. Clin. Pharm. Ther. 35: 617-638. - Nambiar, N.J. (2012): Management of paracetamol poisoning: the old and the new. J. Clin. Diagnost. Res. 6: 1101-1104. Sharma, C.V., Metha, V. (2014): Paracetamol; mechanisms and updates. BJA education, Continuing Education in Anaesthesia Critical Care & Pain 14: 155
Ihr Referat sollte folgende Punkte umfassen: - Darstellung der möglichen molekularen Wirkmechanismen von Paracetamol - Einsatz von Paracetamol - Nebenwirkungen von Paracetamol und toxische Wirkung bei Überdosierung.
Referat III: Neuropathischer Schmerz: therapeutische Ansätze - Cohen, S.P., and Mao, J. (2014): Neuropathic pain: mechanisms and their clinical implications. BMJ, 384: f7656. - Sommer, C. (2013): Neuropathische Schmerzen. Pathophysiologie, Diagnostik und Therapie. Schmerz 27: 619-634. - Magrinelli, F., Zanette, G., and Tamburin, S. (2013): Neuropathic pain: diagnosis and treatment. Prac. Neurol. 13: 292-307. - Leitlinien für Diagnostik und Therapie in der Neurologie: http://www.awmf.org/leitlinien/detail/ll/030-114.html
Ihr Referat sollte folgende Punkte umfassen:
- Evidenzbasierte Therapie des neuropathischen Schmerzes - Welche Rolle spielen NSAIDs in der medikamentösen Therapie?

1 of 12For personal use only
Rationale for mechanism based treatmentOne reason for the high prevalence rate of chronic pain, and neuropathic pain in particular, is the absence of effective treatments. Unlike opioids and non-steroidal anti-inflam-matory drugs, which form the cornerstone of drug treatment for nociceptive pain, the adjuvants used to treat neuropathic pain tend to have only a modest effect and in a minority of patients. The main reason for this is the inability to target underlying mechanisms precisely; this is why syndromes (such as fibromyalgia), which lack distinct pathophysiologi-cal mechanisms, tend to be associated with lower treatment success rates than diseases.5
Generally, mechanism based treatments, which target specific pain mechanisms, are superior to disease based or cause based treatments, which target less proximate causes. This may be one reason why so many drugs that are success-ful in preclinical studies fail in clinical trials.6 As a general rule, painful conditions such as inflammatory arthritis, in which the mechanisms have been clearly identified, have more effective treatments.7 But in clinical practice, eluci-dating the pain mechanisms responsible for neuropathic symptoms can be difficult. One method for identifying mechanisms and predicting treatment outcomes is the use of intravenous infusion tests, such as intravenous ketamine to predict response to dextromethorphan or other NMDA (N-methyl-D-aspartate) receptor antagonists. However, studies that have evaluated these treatments have been methodologically flawed and usually have reported only modest predictive value.8
In the past decade, several reviews have been written on the mechanisms of neuropathic pain, most of which
IntroductionPain is a survival mechanism that serves as a warning sign of ongoing or impending tissue damage. According to an Institute of Medicine report released in 2011, one in three Americans experiences chronic pain—more than the total number affected by heart disease, cancer, and diabetes combined.1 In Europe, the prevalence of chronic pain is 25-30%.2 About a fifth of people who report chronic pain are thought to have predominantly neuropathic pain.3 4
SUMMARY POINTSAs more accurate instruments have been developed to identify neuropathic pain, estimates of its prevalence and socioeconomic impact have increasedThe development of neuropathic pain requires a plethora of different mechanisms that extend from the periphery to the central nervous system where they involve the spinal cord, brain, and descending modulation systemsAlthough conceptually appealing, the mechanism based treatment of pain is challenging to implementMany drugs shown to be effective in preclinical models of neuropathic pain fail in clinical studies, mostly because animal models tend to emphasize evoked, rather than spontaneous, pain and do not account for the emotional aspects of painIn view of the large degree of overlap between neuropathic and nociceptive pain in terms of mechanisms and treatment response, many experts view them as different points on a chronic pain continuum, rather than distinct entities
Neuropathic pain: mechanisms and their clinical implicationsSteven P Cohen,1 2 Jianren Mao3
ABSTRACT Neuropathic pain can develop after nerve injury, when deleterious changes occur in injured neurons and along nociceptive and descending modulatory pathways in the central nervous system. The myriad neurotransmitters and other substances involved in the development and maintenance of neuropathic pain also play a part in other neurobiological disorders. This might partly explain the high comorbidity rates for chronic pain, sleep disorders, and psychological conditions such as depression, and why drugs that are effective for one condition may benefit others. Neuropathic pain can be distinguished from non-neuropathic pain by two factors. Firstly, in neuropathic pain there is no transduction (conversion of a nociceptive stimulus into an electrical impulse). Secondly, the prognosis is worse: injury to major nerves is more likely than injury to non-nervous tissue to result in chronic pain. In addition, neuropathic pain tends to be more refractory than non-neuropathic pain to conventional analgesics, such as non-steroidal anti-inflammatory drugs and opioids. However, because of the considerable overlap between neuropathic and nociceptive pain in terms of mechanisms and treatment modalities, it might be more constructive to view these entities as different points on the same continuum. This review focuses on the mechanisms of neuropathic pain, with special emphasis on clinical implications.
1Departments of Anesthesiology and Critical Care Medicine and Physical Medicine and Rehabilitation, Johns Hopkins School of Medicine, Baltimore, MD 21029, USA2Uniformed Services University of the Health Sciences, Bethesda, MD, USA3Massachusetts General Hospital, Harvard Medical School, Boston, MA, USACorrespondence to: S P Cohen [email protected] this as: BMJ 2014;348:f7656doi: 10.1136/bmj.f7656
STATE OF THE ART REVIEW

2 of 12
are directed at neuroscientists. Yet it is essential that clini-cians understand the mechanisms too, because such an understanding can steer future research and guide clinical practice.
Search methodsIn September 2013, we searched the databases on Medline via PubMed and Ovid, Embase, and CINAHL Plus using the keywords “neuropathic pain”, “sensitization”, “neuroplas-ticity”, “mechanisms”, “reorganization”, “sympathetically maintained”, “antinociceptive”, and “descending modu-lation”, with no date restrictions. For individual sections, keywords relating to specific topics and mechanisms were identified from the initial search (for example, “ion channel expression”, “cytokine”, “glial cell”) and searched using the same databases. Additional articles and prime references were obtained by cross referencing all search terms with “review article” and searching through reference lists. We considered animal studies, experimental and clinical trials, and review articles published in English.
Physiology and classificationThe generation of pain in response to tissue injury involves four basic elements:• Transduction: a function of nociceptors that converts
noxious stimulation to nociceptive signals• Transmission: a process that sends nociceptive signals
along nerve fibers from the site of injury to the central nervous system (CNS)
• Transformation or plasticity: a mechanism that modulates nociceptive signals at synaptic sites and at the level of the CNS through ascending, descending, or regional facilitation and inhibition
• Perception: a key component of the clinical pain experience that integrates cognitive and affective (emotional) responses. In evolutionary terms the activation of high threshold
mechanical nociceptors or other types of specialized nocic-eptor served a protective role, acting as a warning system for dangerous stimuli. But whereas inflammatory pain is adap-tive, evolution has failed to account for our enhanced ability to survive trauma, disease, or iatrogenic trauma intended to prolong or enhance quality of life (such as surgery). In these contexts pain no longer serves a useful function but becomes the disease itself.
Although it is easy to conceptualize pain as a homoge-neous entity, this is overly simplistic. In reality there are several different types, each with distinct neurobiological and pathophysiological mechanisms. The most common categorization divides pain into two main types: neuro-pathic and nociceptive pain (table 1). This distinction is important because it not only reflects the cause of pain but also informs treatment.
Nociceptive pain can be classified as somatic (for exam-ple, muscles, joints) or less often visceral (internal organs). Because of the high concentration of nociceptors in somatic tissues, chronic somatic pain is typically well localized and often results from degenerative processes (such as arthri-tis). By contrast, internal organs are usually unresponsive to classic painful stimuli, such as cutting and burning, but respond to ischemia (for example, angina), inflammation
Allodynia: Painful response to a normally innocuous stimulusCentral pain: A subset of neuropathic pain caused by a lesion or disease of the central somatosensory nervous systemCentral sensitization: Increased responsiveness of nociceptive neurons in the central nervous system to normal or subthreshold sensory inputDeafferentation pain: Pathological pain condition associated with a partial or complete loss of sensory input from a part of the body after lesions in somatosensory pathways, often as a result of reorganization in the central nervous system. Common examples include phantom limb pain and brachial plexopathy Descending modulation: The process by which pathways that descend from the brain to the spinal cord modify incoming somatosensory information so that the perception of and reactions to somatosensory stimuli are altered, resulting in increased or decreased painEctopic discharge: Trains of ongoing electrical nerve impulses that occur spontaneously without stimulation or originate at sites other than the normal location (or both). This phenomenon typically occurs after nerve injuryEphaptic transmission: The phenomenon by which two independent nerves communicate with each other through an artificial synapse, which often develops after injury to the insulating myelin sheath that normally prevents crosstalk between parallel nervesHyperalgesia: Increased pain response to a normally painful stimulusNeuropathic pain: Pain caused by a lesion or disease of the somatosensory nervous systemNeuroplasticity: Changes in neural pathways and synapses that result from bodily injury or changes in behavior, the environment, or neural processes. This is consistent with the concept that the brain is a dynamic organ that constantly changes in response to internal and outside events throughout lifeNociception: The neural responses of encoding and processing noxious stimuliNociceptive pain: Pain that arises from the activation of peripheral nerve endings (nociceptors) that respond to noxious stimulation. Nociceptive pain arises from actual or potential damage to non-neural tissue and can be categorized as visceral or somaticNoxious stimulus: A stimulus that damages or threatens to damage normal tissuesPeripheral sensitization: A lowering of the stimulus (pain) threshold for nociceptor activation and an increased frequency of nerve impulse firing in response to stimulation (hyperexcitability). Peripheral sensitization is often found at the site of tissue damage or inflammationSympathetically maintained pain: Pain that is enhanced or maintained by a functional abnormality of the sympathetic nervous system, such as functional sympathetic afferent coupling or increased expression of adrenergic receptors at the peripheral terminals of nociceptive afferent fibersWindup: Progressive increase in the frequency and magnitude of firing of dorsal horn neurons produced by repetitive activation of C fibers above a critical threshold, leading to a perceived increase in pain intensity
DEFINITIONS
Table 1 | Classification of neuropathic and nociceptive painClinical characteristic
Neuropathic pain Nociceptive pain
Cause Injury to the nervous system, often accompanied by maladaptive changes in the nervous system
Damage or potential damage to tissues
Descriptors Lancinating, shooting, electric-like, stabbing pain Throbbing, aching, pressure-like pain
Sensory deficits Common—for example, numbness, tingling, pricking Uncommon; if present they have a non-dermatomal or non-nerve distribution
Motor deficits Neurological weakness may be present if a motor nerve is affected; dystonia or spasticity may be associated with central nervous system lesions and sometimes peripheral lesions (such as complex regional pain syndrome)
May have pain induced weakness
Hypersensitivity Pain often evoked by non-painful (allodynia) or painful (exaggerated response) stimuli
Uncommon except for hypersensitivity in the immediate area of an acute injury
Character Distal radiation common Distal radiation less common; proximal radiation more common
Paroxysms Exacerbations common and unpredictable Exacerbations less common and often associated with activity
Autonomic signs
Color changes, temperature changes, swelling, or sudomotor (sweating) activity occur in a third to half of patients
Uncommon
STATE OF THE ART REVIEW
For personal use only

3 of 12
differences between individuals, the degree of pathology tends to correlate poorly with the intensity of pain for con-ditions such as back pain.13 To illustrate, conditions such as fibromyalgia have high reported pain scores despite the absence of overt disease.
Secondary order neurons arising in the spinal cord trans-mit nociceptive input to the thalamus through ascending pathways such as the spinothalamic tract, which functions as a relay station to higher cortical centers. These centers include:• The anterior cingulate cortex, which is involved in
anxiety, anticipation of pain, attention to pain, and motor responses
• The insular cortex, which may play a role in the sensory discriminative and affective aspects of pain that contribute to the negative emotional responses and behaviors associated with painful stimuli14
• The prefrontal cortex, which is important for sensory integration, decision making, memory retrieval, and attention processing in relation to pain15
• The primary and secondary somatosensory cortices that localize and interpret noxious stimuli16
• The nucleus accumbens, which is involved in placebo analgesia17
• The amygdala, hippocampus, and other parts of the limbic system, which are involved in the formation and storage of memories associated with emotional events, affect, arousal, and attention to pain and learning. The limbic system may also be partially responsible for the fear that accompanies pain.18 Because pain is multidimensional experience, it is not
surprising that psychosocial factors such as depression, somatization, poor coping skills, social stressors, and nega-tive job satisfaction can predict the development of chronic pain after an acute episode.19 20 In addition, the context in which a painful stimulus occurs affects how we perceive it. This is why an injury that occurs during a football game may be less painful than a similar injury that occurs while walk-ing to school, and why acute pain, which we anticipate will get better, is better tolerated than chronic pain.
Peripheral mechanismsPeripheral sensitizationOnce injury occurs, inflammation and reparatory processes ensue, leading to a hyperexcitable state known as periph-eral sensitization. In most patients, this state resolves as healing occurs and inflammation subsides. However, when nociception persists because of repeated stimulation from ongoing injury or disease (for example, in diabetes), the changes in primary afferent neurons may persist.
Several factors can contribute to peripheral sensitization. Inflammatory mediators such as calcitonin gene related peptide and substance P, which are released from nocic-eptive terminals, increase vascular permeability, leading to localized edema and the escape of the byproducts of injury, such as prostaglandins, bradykinin, growth factors, and cytokines. These substances can sensitize as well as excite nociceptors, resulting in lowered firing thresholds and ectopic discharges. The fact that multiple substances can sensitize nociceptors may partly explain why no drug is universally effective and there is a ceiling effect for antago-
(appendicitis), or occlusion of flow that results in capsular distension (bowel obstruction).
Neuropathic pain is defined as pain resulting from injury to, or dysfunction of, the somatosensory system.9 In neuro-pathic pain, tissue damage directly affects the nervous sys-tem, resulting in the generation of ectopic discharges that bypass transduction.10 One subtype of neuropathic pain is central pain (for example, as a result of spinal cord injury), which manifests as a constellation of signs and symptoms that follows an insult to the CNS as a necessary, but not always sufficient, inciting event. Although many forms of nociceptive pain, and some forms of neuropathic pain, may confer evolutionary benefits, chronic neuropathic pain is always maladaptive.
Compared with previous studies, estimates of the preva-lence of neuropathic pain have significantly increased over the past decade since the development of instruments designed to identify such pain.11 Around 15-25% of people with chronic pain are currently thought to have neuropathic pain.3 4 However, the prevalence of neuropathic pain may belie its socioeconomic impact, because studies have found that it is associated with a greater negative impact on qual-ity of life than nociceptive pain.12
Emotional versus physiological aspectsA common misconception is that pain is purely a physi-ological phenomenon. In fact, “pain” represents a final integrative package, the components of which consist of neurophysiological processes as well as contextual, psy-chological, and sociocultural factors. This is one reason for the discrepancies between preclinical studies (which measure increased tolerance to painful stimuli in animals (anti-nociception)), clinical studies (which assess efficacy), and clinical practice, which measures effectiveness (table 2). Partly because of these factors and the neurophysiological
Table 2 | Comparison of pain in animal models and clinical painVariable Animal models Clinical painStudy methods Deficiencies in blinding, randomization,
and power calculation (low numbers) are common
Greater attention to methodological quality in large scale clinical trials
Time course of development
Minutes to hours to days; largely coincides with the time course of tissue damage
Weeks to months to years; usually outlasts the time course of tissue damage
Hallmark Increased neuronal excitability, decreased nociceptive threshold, and expanded receptive fields
Altered pain quality with or without neurological deficits (such as numbness, weakness)
Pattern Correlations between tissue damage and cellular responses; near uniformity in response patterns; sustainability of neuronal responses not confirmed
Considerable individual variations in pain experience; dynamic changes in pain intensity, timing, location, modality, and quality
Presentation Mainly hyperalgesia and allodynia; rarely contralateral behavioral changes
Usually spontaneous pain; often extends beyond a single nerve or dermatome distribution; contralateral responses may be present; behavioral responses common
Affective component Absent or unknown Powerful contributor to the pain experience; has a strong influence on treatment outcome
Intervention Behavioral changes can be prevented or reversed (or both) by blocking key cellular elements
Similar approaches (blocking key cellular elements) have had mostly negative outcomes in clinical trials; long interval between positive results and implementation of intervention in practice
Outcomes Considered over the course of days or weeks; treatment response rates are higher than in clinical trials
Considered over the course of weeks or months for clinical trials, and months or years in practice; success rates are lower than in animal models, especially in clinical practice
STATE OF THE ART REVIEW
For personal use only

4 of 12
nists that work at only one receptor (such as non-steroidal anti-inflammatory drugs (NSAIDs)).
Ectopic discharges can give rise to spontaneous pain and may originate from the dorsal root ganglion, other points along an injured nerve, or even uninjured adjacent fibers.21 The process by which adjacent uninjured nerve fib-ers become excited as a result of non-synaptic “cross talk” is known as ephaptic transmission. Allodynia refers to pain produced by a normally non-painful stimulus, and it may result from decreased stimulation thresholds. Allodynia can be classified as mechanical (pain in response to light touch) or thermal, and it can readily be detected on physical exam-ination. An example is a patient with diabetic neuropathy whose feet are sensitive to putting on socks.
Hyperalgesia refers to exaggerated pain perception as a result of damaged peripheral pain fibers, and it can be categorized as primary or secondary. Primary hyperalge-sia occurs in injured tissue as a result of sensitization of peripheral nociceptors (for example, tenderness after a cut), whereas secondary hyperalgesia is seen in adjacent undamaged tissue owing to sensitization within the CNS and can be assessed with a sharp object. In part, this may be caused by ephaptic transmission or the expansion of recep-tive fields of injured nerves (or both). A clinical example of hyperalgesia might be an amputee who is unable to use a prosthesis because of tenderness overlying the stump. Both allodynia and hyperalgesia are forms of evoked, or stimulus dependent, pain. Although spontaneous neuropathic pain is often more common and distressing than evoked pain in clinical practice, preclinical studies usually measure evoked pain (fig 1).22 It is still not clear whether animals that develop evoked pain incited by models of peripheral nerve injury experience spontaneous pain.
Expression of ion channelsOne contributor to spontaneous firing of nerve fibers after injury is the increased expression of sodium channels in dorsal root ganglia and around the terminal injury site (neuroma) of injured axons.23 Since this discovery, further preclinical studies have shown that a variety of sodium channels are involved in pain. After nerve injury, the
expression of some of these channels increases de novo, the expression of others diminishes, and some translocate into different cellular compartments.24 The proliferation of heterotopic sodium channels, such as Nav1.3, Nav1.7, and Nav1.8, may lower the stimulation threshold and provoke ectopic discharge, resulting in spontaneous pain. In addi-tion, the spread of sodium channels may trigger central sensitization, leading to allodynia. Several adjuvant drugs, such as carbamazepine, act through the blockade of sodium channels. Yet, because none of these drugs is selective for channel subtypes involved in pain, all have low therapeutic indices and many side effects.
Certain types of calcium channels (N-type, T-type, and L-type), and to a lesser extent potassium channels (hyper-polarization activated cyclic nucleotide gated channels), also play a role in neuropathic pain. After nerve injury, the expression of α2δ calcium channels increases in and around the dorsal root ganglia, increasing excitability.25 These voltage gated calcium channels are the primary site of action for gabapentinoids, a first-line treatment for neu-ropathic pain,26 which have been shown in preclinical stud-ies to reduce hyperalgesia and spontaneous pain (table 3).27
Phenotypic switchDifferentiated neurons have different properties from undif-ferentiated ones, which enable them to perform specific functions (Aδ and C fibers transmit pain). After nerve injury, hundreds of genes that affect nerve function are upregulated or downregulated, and this can affect excitability, as well as transduction and transmission properties. Because gene expression affects cellular characteristics, this can result in a change in the phenotype of the nerve fiber, such that neu-romodulators usually expressed in C fibers (such as calci-tonin gene related peptide, substance P) are now expressed in other fibers.28 This may theoretically result in stimuli that are usually innocuous being perceived as painful.
Sensory denervation and sprouting of collateral nerve fibersAfter injury to a sensory nerve, atrophic changes (wallerian degeneration) cause a decrease in the size of the cell body and the axon diameter, and eventually neuronal death. This leads to a decreased density of intraepidermal nociceptors. Depending on the type of nerve injury, this may cause loss of sensation or, paradoxically, hyperalgesia and increased pain (deafferentation pain).29 Severing the link between a nerve and its end organ also deprives the nerve of nerve growth factor and other neurotrophins, which are essential for growth and maintenance and serve as signaling mole-cules. One example of deafferentation pain is phantom limb pain after amputation. Although electrodiagnostic studies may be normal in people with a loss of small pain transmit-ting nerve fibers, a decreased density of C fibers can be seen on skin biopsy. In response to local release of nerve growth factor, collateral sprouting may follow neuronal loss.
Sympathetically maintained painSympathetically maintained pain is pain that is enhanced or maintained by an abnormality in the sympathetic nervous system. Functional coupling between the sym-pathetic nervous system and somatosensory nerves after
Fig 1 | Diagram depicting normally perceived pain, as well as allodynia and hyperalgesia after injury
T0
T1
Pai
n in
ten
sity
Stimulus intensity
Injury Normal Pain
AllodyniaHyperalgesiaNon-injured pain response curveAmplified pain response curve
T0 = Pre-injury pain threshold
T1 = Post-injury pain threshold
BMJ 2014;348:f7656
STATE OF THE ART REVIEW
For personal use only
5 of 12
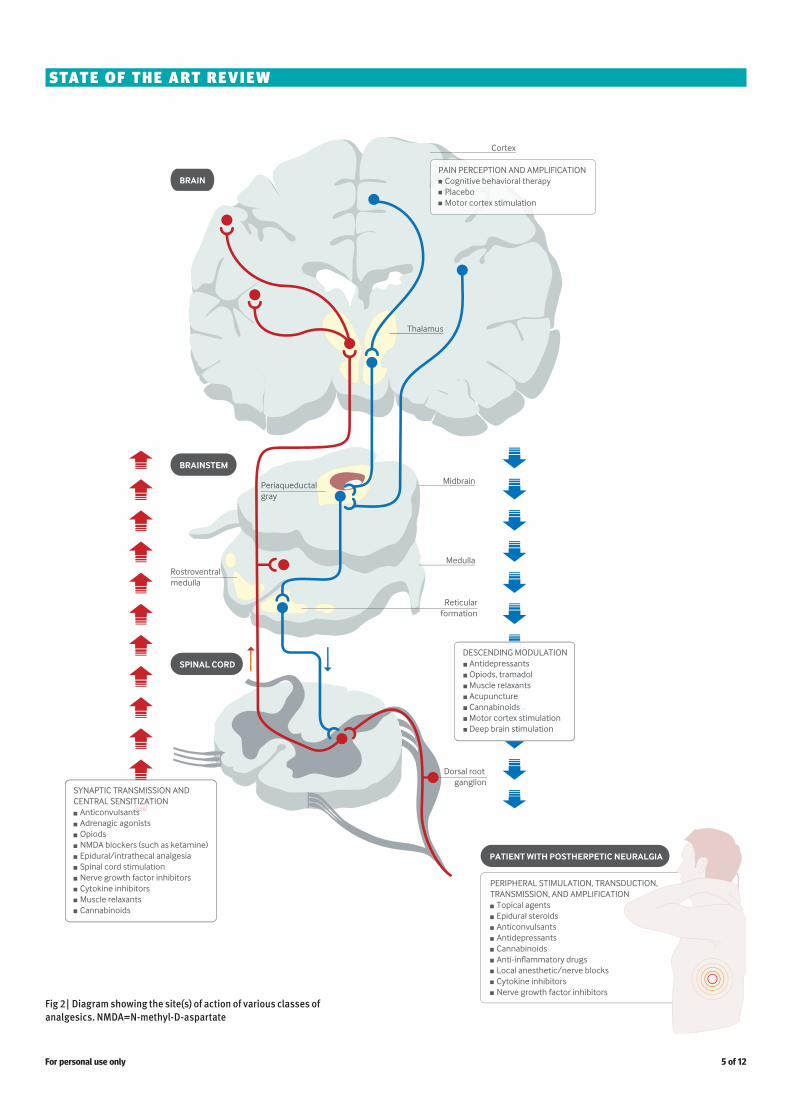
Fig 2 | Diagram showing the site(s) of action of various classes of analgesics. NMDA=N-methyl-D-aspartate
STATE OF THE ART REVIEW
For personal use only
6 of 12
and spatial summation (increased neuronal responses to repeated noxious stimulation in a time and region depend-ent manner).32 Other components include expanded recep-tive fields of nociceptors and second order neurons,33 and increased neuronal excitability of ascending nociceptive pathways that send pain signals to supraspinal regions.34 These neuroplastic changes take place along nociceptive pathways in the spinal cord and in multiple brain regions.35
In the neural circuit, nociceptive signals generated by nerve damage are modulated by supraspinal descending inhibition or facilitation that converges onto dorsal horn neurons (or both).36 At the cellular level, transmission of nociceptive signals within the central nervous system is regulated by cellular and intracellular elements that include37 38:• Ion (Na+, Ca++, K+) channels • Ionotropic and metabotropic receptors such as
glutamatergic, GABA (γ-aminobutyric acid)ergic, serotoninergic, adrenergic, neurokinin, and vanilloid receptors
• Inflammatory cytokines released from activated glial cells
nerve injury has been noted since the American civil war. Although the concept of sympathetically maintained pain is most commonly linked to complex regional pain syndrome, the same principles apply to other pain conditions, such as postherpetic neuralgia.8 The interaction between the ana-tomically distinct autonomic and somatosensory systems is complex but probably includes the expression of α adreno-ceptors on primary afferent sensory fibers, sympathetic sprouting into dorsal root ganglia, and impaired oxygena-tion and nutrition in response to sympathetically mediated vasoconstriction.30 Clinically, sympathetically maintained pain may manifest as temperature or color changes (or both) in an affected extremity, swelling or atrophy, and pain worsened by cold weather or stress, which enhances sym-pathetic outflow. Among the various diagnostic tests used to detect sympathetically maintained pain, clinical studies have found that sympathetic blocks are more sensitive but less specific than intravenous infusion of phentolamine.31
Spinal mechanismsAn important spinal component of neuropathic pain mechanisms is synaptic plasticity in the form of temporal
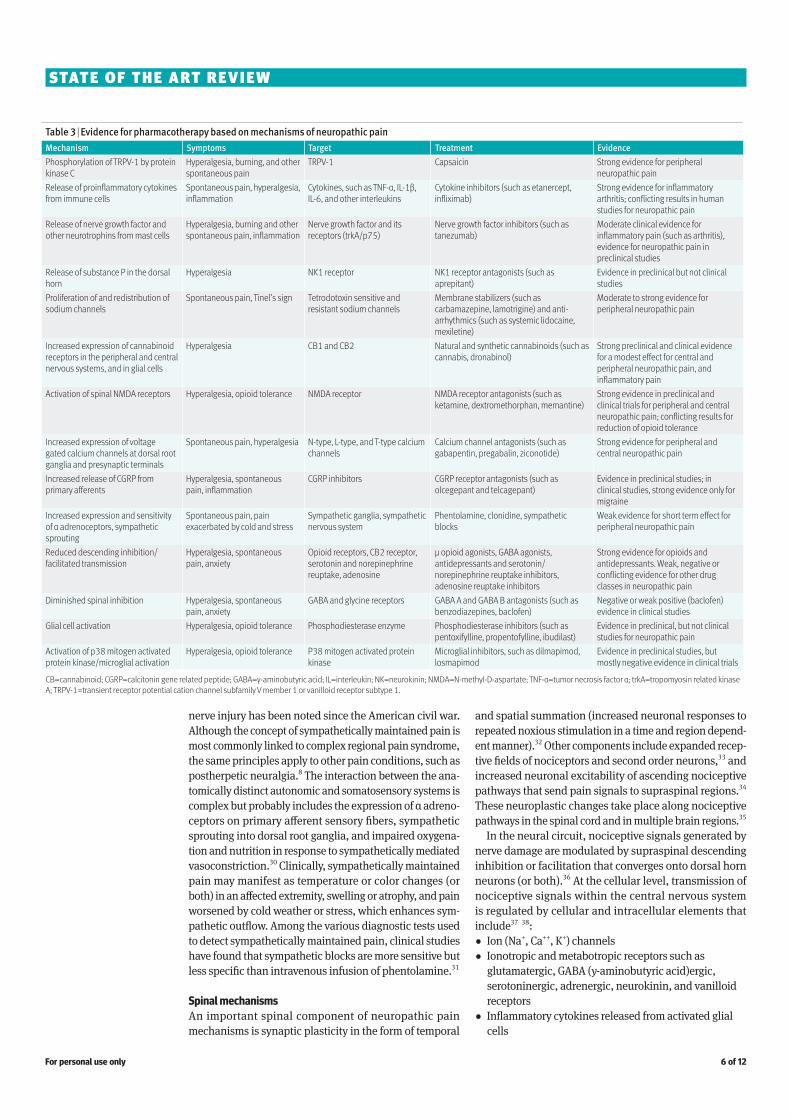
Table 3 | Evidence for pharmacotherapy based on mechanisms of neuropathic painMechanism Symptoms Target Treatment EvidencePhosphorylation of TRPV-1 by protein kinase C
Hyperalgesia, burning, and other spontaneous pain
TRPV-1 Capsaicin Strong evidence for peripheral neuropathic pain
Release of proinflammatory cytokines from immune cells
Spontaneous pain, hyperalgesia, inflammation
Cytokines, such as TNF-α, IL-1β, IL-6, and other interleukins
Cytokine inhibitors (such as etanercept, infliximab)
Strong evidence for inflammatory arthritis; conflicting results in human studies for neuropathic pain
Release of nerve growth factor and other neurotrophins from mast cells
Hyperalgesia, burning and other spontaneous pain, inflammation
Nerve growth factor and its receptors (trkA/p75)
Nerve growth factor inhibitors (such as tanezumab)
Moderate clinical evidence for inflammatory pain (such as arthritis), evidence for neuropathic pain in preclinical studies
Release of substance P in the dorsal horn
Hyperalgesia NK1 receptor NK1 receptor antagonists (such as aprepitant)
Evidence in preclinical but not clinical studies
Proliferation of and redistribution of sodium channels
Spontaneous pain, Tinel’s sign Tetrodotoxin sensitive and resistant sodium channels
Membrane stabilizers (such as carbamazepine, lamotrigine) and anti-arrhythmics (such as systemic lidocaine, mexiletine)
Moderate to strong evidence for peripheral neuropathic pain
Increased expression of cannabinoid receptors in the peripheral and central nervous systems, and in glial cells
Hyperalgesia CB1 and CB2 Natural and synthetic cannabinoids (such as cannabis, dronabinol)
Strong preclinical and clinical evidence for a modest effect for central and peripheral neuropathic pain, and inflammatory pain
Activation of spinal NMDA receptors Hyperalgesia, opioid tolerance NMDA receptor NMDA receptor antagonists (such as ketamine, dextromethorphan, memantine)
Strong evidence in preclinical and clinical trials for peripheral and central neuropathic pain; conflicting results for reduction of opioid tolerance
Increased expression of voltage gated calcium channels at dorsal root ganglia and presynaptic terminals
Spontaneous pain, hyperalgesia N-type, L-type, and T-type calcium channels
Calcium channel antagonists (such as gabapentin, pregabalin, ziconotide)
Strong evidence for peripheral and central neuropathic pain
Increased release of CGRP from primary afferents
Hyperalgesia, spontaneous pain, inflammation
CGRP inhibitors CGRP receptor antagonists (such as olcegepant and telcagepant)
Evidence in preclinical studies; in clinical studies, strong evidence only for migraine
Increased expression and sensitivity of α adrenoceptors, sympathetic sprouting
Spontaneous pain, pain exacerbated by cold and stress
Sympathetic ganglia, sympathetic nervous system
Phentolamine, clonidine, sympathetic blocks
Weak evidence for short term effect for peripheral neuropathic pain
Reduced descending inhibition/facilitated transmission
Hyperalgesia, spontaneous pain, anxiety
Opioid receptors, CB2 receptor, serotonin and norepinephrine reuptake, adenosine
µ opioid agonists, GABA agonists, antidepressants and serotonin/norepinephrine reuptake inhibitors, adenosine reuptake inhibitors
Strong evidence for opioids and antidepressants. Weak, negative or conflicting evidence for other drug classes in neuropathic pain
Diminished spinal inhibition Hyperalgesia, spontaneous pain, anxiety
GABA and glycine receptors GABA A and GABA B antagonists (such as benzodiazepines, baclofen)
Negative or weak positive (baclofen) evidence in clinical studies
Glial cell activation Hyperalgesia, opioid tolerance Phosphodiesterase enzyme Phosphodiesterase inhibitors (such as pentoxifylline, propentofylline, ibudilast)
Evidence in preclinical, but not clinical studies for neuropathic pain
Activation of p38 mitogen activated protein kinase/microglial activation
Hyperalgesia, opioid tolerance P38 mitogen activated protein kinase
Microglial inhibitors, such as dilmapimod, losmapimod
Evidence in preclinical studies, but mostly negative evidence in clinical trials
CB=cannabinoid; CGRP=calcitonin gene related peptide; GABA=γ-aminobutyric acid; IL=interleukin; NK=neurokinin; NMDA=N-methyl-D-aspartate; TNF-α=tumor necrosis factor α; trkA=tropomyosin related kinase A; TRPV-1=transient receptor potential cation channel subfamily V member 1 or vanilloid receptor subtype 1.
STATE OF THE ART REVIEW
For personal use only
7 of 12
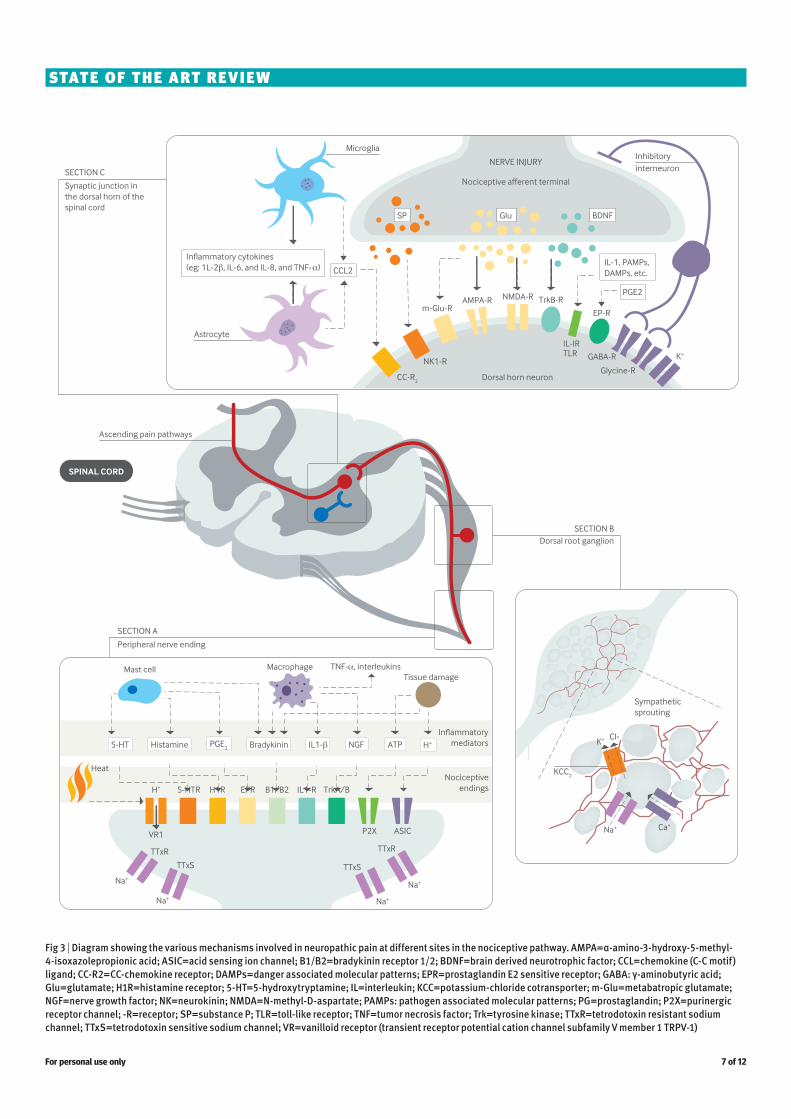
Fig 3 | Diagram showing the various mechanisms involved in neuropathic pain at different sites in the nociceptive pathway. AMPA=α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ASIC=acid sensing ion channel; B1/B2=bradykinin receptor 1/2; BDNF=brain derived neurotrophic factor; CCL=chemokine (C-C motif) ligand; CC-R2=CC-chemokine receptor; DAMPs=danger associated molecular patterns; EPR=prostaglandin E2 sensitive receptor; GABA: γ-aminobutyric acid; Glu=glutamate; H1R=histamine receptor; 5-HT=5-hydroxytryptamine; IL=interleukin; KCC=potassium-chloride cotransporter; m-Glu=metabatropic glutamate; NGF=nerve growth factor; NK=neurokinin; NMDA=N-methyl-D-aspartate; PAMPs: pathogen associated molecular patterns; PG=prostaglandin; P2X=purinergic receptor channel; -R=receptor; SP=substance P; TLR=toll-like receptor; TNF=tumor necrosis factor; Trk=tyrosine kinase; TTxR=tetrodotoxin resistant sodium channel; TTxS=tetrodotoxin sensitive sodium channel; VR=vanilloid receptor (transient receptor potential cation channel subfamily V member 1 TRPV-1)
STATE OF THE ART REVIEW
For personal use only

8 of 12
and neuroma formation.49-51 In animal models, adminis-tration of cytokine inhibitors before nerve injury reduces neuropathology and pain-related behaviors.52 53 However, in controlled clinical trials, most of which were performed in patients with radiculopathy, the use of systemic and neuraxial cytokine inhibitors has been largely disappoint-ing.54 55
Glial cells comprise about 70% of the central nervous system and play an important role in maintenance and homeostasis. Microglia are activated within 24 hours of nerve injury, and astrocytes follow shortly thereafter, with activation persisting for up to 12 weeks. Glial cells undergo structural and functional transformation after injury, with astrocytes releasing a host of different pronociceptive fac-tors, such as prostaglandins, excitatory amino acids, and cytokines.56
Microglial cells comprise less than 20% of spinal glial cells under normal conditions but proliferate rapidly at the dorsal root ganglia and spinal cord after nerve injury.56 57 On activation, microglial cells stimulate the complement component of the immune system and release cytokines, chemokines, and cytotoxic substances such as nitric oxide and free radicals.56 58 59 This proinflammatory milieu begins at synaptic sites in the brain stem and the site of nerve injury but spreads to more distant sites. The ensu-ing release of cytokines from astrocytes and microglia induces an array of cellular responses such as upregula-tion of glucocorticoid and glutamate receptors, leading to spinal excitation and neuroplastic changes.60 IL-1β also enhances conditioned “fear memory” (conditioned fear related memories associated with behavioral responses) through glucocorticoids,61 suggesting that proinflamma-tory cytokines may participate in the affective experience of pain. Drugs that modulate microglia, such as minocycline, pentoxifylline, and propentofylline, have shown some effi-cacy in preclinical models of neuropathic pain but have not proved effective in a clinical context (table 3; fig 2).62
Supraspinal mechanismsNociceptive signals can also be altered at supraspinal lev-els. The brains of patients with chronic pain are different from those without pain, with variations in metabolism and regional concentrations of neurotransmitters occurring in areas such as the thalamus and cingulate cortex. These dif-ferences vary according to the type of pain experienced (for example, acute pain or allodynia).63 In patients with neu-ropathic pain, cortical reorganization occurs after injury, and the extent of the changes seems to correlate with the degree of pain. For example, in upper extremity amputees with phantom limb pain, because of the close proximity of their somatotopic representations, the area of the brain responsible for moving the lips transgresses into the hand movement area of the motor cortex; this phenomenon does not occur in amputees without phantom limb pain.64 The observation that these changes occur after injury suggests that disinhibition may not only be a consequence of nerve injury, but may render patients susceptible to chronic pain.65
Preclinical studies demonstrating changes in gene expression after nerve injury have provided insight into how changes in signal transduction and neuroprotection/
• Nerve growth factors • Intracellular regulators such as protein kinases (for
example, protein kinase C) and transcriptional factors (such as nuclear factor-κB).
Spinal glutamatergic regulationPeripheral nerve injury increases neuronal excitability in the spinal cord by activating excitatory glutamate recep-tors.39 Nerve injury also induces downregulation of spinal glutamate transporters responsible for maintaining synap-tic glutamate homeostasis. Increased regional glutamate availability secondary to loss of glutamate transporters can result in persistent and enhanced activation of both iono-tropic (for example, NMDA and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)) and metabotropic glutamate receptors (such as metabotropic glutamate recep-tor 2), leading to lower activation thresholds and increased neuronal excitability and neurotoxicity.40 41
The term “windup” refers to the progressive increase in the frequency and magnitude of firing of dorsal horn neurons produced by repetitive activation of C fibers, a phenomenon that requires glutamatergic NMDA receptor activity. Spinal glutamatergic activity can in turn initiate intracellular signaling cascades, including activation of protein kinase C, that result in long-lasting neuroplastic changes in the spinal cord.42 Similar to the role of central glutamatergic mechanisms in the pathogenesis of other neurological disorders such as epilepsy and Alzheimer’s disease, glutamate receptors are integral to the develop-ment of central sensitization, and blockade of both NMDA and non-NMDA receptors has been shown to attenuate neuropathic pain in animal models.43 Because of its pri-mary role in neuroplasticity and excitotoxicity, the NMDA receptor has been implicated in such diverse areas as memory, opioid tolerance, and opioid induced hyperalge-sia—the phenomenon whereby opioid use paradoxically increases pain sensitivity.44 In clinical practice, the use of NMDA receptor antagonists to prevent opioid tolerance and hyperalgesia has been disappointing.45 The long-term use of these drugs to treat chronic neuropathic pain has also had mixed results, and their use may be limited by side effects, particularly psychomimetic ones, which seem to increase in proportion to potency. The use of ketamine infusions as a treatment for refractory neuropathic pain has generated intense interest, although studies are limited by methodo-logical flaws and lack of long term follow-up (table 3).46 The rationale behind these infusions is that high doses may “reset” the nervous system back to its pre-injury state, in essence reversing central sensitization.
Glial activation and proinflammatory cytokinesThe role of glial activation and cytokines in neuropathic pain has been extensively studied. Proinflammatory cytokines including interleukin 1β (IL-1β), IL-6, and tumor necrosis factor α (TNF-α) are produced peripherally and centrally in response to nerve injury.47 These proin-flammatory cytokines play a crucial role in inflammatory responses after nerve injury through intracellular mediators such as protein kinase C and 3′,5′-cAMP.48 Proinflamma-tory cytokines also play an important role in sensitization of the CNS and may contribute to allodynia, hyperalgesia,
STATE OF THE ART REVIEW
For personal use only

9 of 12
trials provide evidence for a small effect size for ketamine in neuropathic pain, although the high doses given make it difficult to identify the precise mechanism responsible for analgesia.8 46 There is some evidence to support the use of the GABA-B agonist baclofen for trigeminal neuralgia, but most of the evidence in favor of benzodiazepines as anal-gesics is anecdotal (table 3).79 80
Descending inhibition plays an important role in deter-mining how people experience pain. Recently, it has been shown that descending modulation can be both inhibitory and facilitatory, with conflicting signals often arising from the same regions.81 The balance between inhibition and amplification is dynamic and influenced by context, behav-ior, emotions, expectations, timing, and pathology. After injury, there is an initial spike mediated by changes in the activation and gene expression of NMDA and AMPA excita-tory glutaminergic receptors, and a subsequent decrease in the excitability of neurons in the rostral ventromedial medulla, which lead to facilitation and inhibition, respec-tively.82 The evolutionary advantage of these changes is that the initial stimulus is reinforced to ensure that it is given priority, but once this occurs the brain seeks to mitigate the consequences.
Expectations and context also play a role in descending modulation. In one randomized study,83 20 healthy sub-jects were subjected to painful electrical stimulation of the sural nerve after immersion of an arm in cold water. Half the subjects were told that the immersion would decrease the pain, whereas the other half were told that it would exac-erbate the pain. Normally, exposure to a spatially distinct noxious stimulus should decrease the response to pain, a concept known as “descending (or diffuse) noxious inhibi-tory control.” The study found that the analgesia expec-tancy group experienced a 77% decrease in pain intensity during immersion compared with no significant reduction in pain in the group that anticipated hyperalgesia. Moreo-ver, corresponding changes in activity levels were noted in cortical areas involved in descending inhibition and pla-cebo analgesia.84 These findings agree with other studies that have found that a host of psychosocial factors such as emotions, expectations, and attention affect our intrinsic ability to inhibit pain.84 85 This may explain why positive expectations tend to result in better treatment outcomes and a higher placebo response rate, and why we are less likely to perceive pain when an injury occurs while we are preoccupied (for example, during a sports game rather than at bedtime).86
Supraspinal levelDescending pathways that modulate transmission of noci-ceptive signals originate in the periaqueductal gray, locus coeruleus, anterior cingulate gyrus, amygdala, and hypo-thalamus, and are relayed through brainstem nuclei in the periaqueductal gray and medulla to the spinal cord. The inhibitory transmitters involved in these pathways include norepinephrine (noradrenaline), 5-hydroxytryptamine, dopamine, and endogenous opioids. After nerve injury, several processes take place that mitigate the normal pain attenuating pathways. These include a diminution in tonic noradrenergic inhibition and a shift from a predominantly inhibitory role to a facilitative function for descending
apoptosis contribute to neuropathic pain.66 67 Changes that occur in supraspinal regions may explain the strong associ-ation between neuropathic pain and mood disorders. Inves-tigators recently found that altered corticotropin releasing factor signaling in the limbic system, an area involved in emotions, may play a role in the development of neuro-pathic pain.68 Patients with chronic pain have also been shown to have reduced gray matter compared with control patients, and this can be partially reversed by treatment.69
DisinhibitionSpinal cord levelOnce a nociceptive stimulus is transmitted to higher cor-tical centers, a series of events occurs that results in the activation of inhibitory neurons that attenuate pain. At the spinal cord level, there is increased release of GABA and glycine from primary afferent terminals, and enhanced activity in inhibitory GABAergic and glycinergic dorsal horn interneurons. These spinal interneurons synapse with cen-tral terminals of primary afferent neurons, thereby reduc-ing their activity, and also regulate activity in ascending secondary order neurons. Spinal inhibitory systems may exert a greater effect on the development of mechanical hyperalgesia than on thermal hyperalgesia.70 71
After nerve injury, a loss of inhibitory currents occurs as a result of dysfunctional GABA production and release mech-anisms; impaired intracellular homeostasis from reduced activity of K+Cl− cotransporter or increased activity of Na+K−Cl− cotransporter (or both), leading to increased Cl− levels; and apoptosis of spinal inhibitory interneurons.72 73 Loss of inhibitory control has been shown to provoke tactile allodynia and hyperalgesia,74 and to facilitate structural changes that increase transmission from Aβ fibers that nor-mally transmit non-painful stimuli to nociceptive specific secondary order neurons in the dorsal horn.75
After nerve injury, dorsal root ganglia exhibit decreased expression of µ opioid receptors and secondary spinal neurons become less responsive to opioids.76 By contrast, inflammation may result in an increase in the number and affinity of opioid receptors, thereby enhancing the efficacy of opioids.77 This may explain why patients with chronic neuropathic pain require higher doses of opioids than those with acute and chronic nociceptive pain.78 In preclinical studies, the administration of NMDA receptor antagonists, protein kinase Cγ inhibitors, and GABA-A agonists has been shown to reverse allodynia and hyperalgesia.75 79 Clinical
KEY RESEARCH QUESTIONSIs it possible to devise a valid animal model that accounts for the “affective-motivational” (emotional) aspect of pain as well as the “sensory-discriminative” (physio-anatomical) aspect?Are there any measures that can be taken before (pre-emptive analgesia) or during the early phase after nerve injury that can prevent the transition to chronic neuropathic pain?Is neuropathic pain represented differently in the brain than other chronic pain conditions?Can we develop better animal models to reflect spontaneous pain, rather than those that emphasize stimulus dependent pain (for example, allodynia), which may be less relevant in clinical practice?Although the concept of mechanism based pain treatment is intellectually enticing, can this be routinely incorporated into clinical practice?
STATE OF THE ART REVIEW
For personal use only

10 of 12
and the functional and practical classification. Consider-ing the large overlap between neuropathic and nociceptive pain, similar to the classification of other neurological dis-orders (such as tension-type and migraine headaches) that share pathophysiological mechanisms and overlap in their response to treatment,99 the different types of chronic pain might best be viewed as points on the same continuum.
Emerging treatmentsIt is anticipated that translational pain research will play an important role in understanding pain mechanisms, formu-lating treatment and research paradigms, and developing new analgesics in the next decade. To facilitate this, several emerging developments must unfold.
Firstly, new animal models should account for the influ-ence of clinical comorbidities such as depression on nocicep-tive behaviors.100 This will be challenging, because animal models for emotional outcomes tend to be less studied than those for physiological parameters.
Secondly, behavioral assessment tools should be capable of measuring the various dimensions of pain experiences, such as the use of conditional place preference or aversion (forms of pavlovian conditioning used to measure the moti-vational effects of positive and negative experiences) and behavioral coding in preclinical studies.101 102 For exam-ple, because the relief of pain is a reward in itself, analgesic agents that are not rewarding in the absence of pain should become rewarding only in the presence of pain.
Thirdly, the association between brain reorganization seen on advanced imaging and the chronicity of pain should be further explored, with emphasis on how changes on imaging relate to pain behaviors and response to treatment.103
Lastly, the identification of biomarkers and the genotyping or phenotyping of pain characteristics may provide tools that enable us to understand better the heterogeneity of clinical pain and formulate individualized treatment regimens.104 105
These research advances, together with the development of newer drugs tailored to individual patients and specific pain mechanisms, will probably improve the treatment of neuropathic pain in the coming years.
ConclusionsInjury to the peripheral or central nervous system results in maladaptive changes in neurons along the nociceptive pathway that can cause neuropathic pain. Unlike acute pain, chronic neuropathic pain confers no individual or evolutionary advantage and is often considered to be a disease in itself. The myriad mechanisms involved in neu-ropathic pain overlap considerably with non-neuropathic pain and other neurological conditions. Although treatment based on the mechanism(s) of pain is widely accepted to be theoretically better than treatment based on the cause of pain, or empirical treatment, this paradigm can be difficult to implement in clinical practice. The multitude of different mechanisms, and the affective-motivational component of chronic pain that distinguishes “human pain” from nocic-eption tested in preclinical pain models, make neuropathic pain notoriously refractory to treatment. This in turn has resulted in chronic pain being considered not only a medi-cal problem but also a socioeconomic concern that requires urgent attention.
serotonergic modulation.87 The manifold roles of these neurotransmitters to affect pain, mood, and sleep may partially explain the high comorbidity rates between pain, depression, anxiety, and sleep disturbances.88 Monoamine reuptake inhibitors such as tricyclic antidepressants are not only effective for neuropathic pain and depression but also alleviate anxiety and improve sleep (fig 3).89
Neuropathic versus nociceptive pain: different entities or part of the same continuum?It is generally acknowledged that neuropathic and non-neuropathic pain are distinct entities, but some experts dispute this assertion, considering it part of our natural tendency to categorize things. There are two main factors that distinguish neuropathic pain from nociceptive pain:• Nociceptive pain requires transduction to convert a
non-electrical signal (for example, mechanical) to an electrochemical one, whereas neuropathic pain involves direct nerve stimulation
• Different prognosis: most people with nociceptive pain (for example, after surgery) recover, but injury to a major nerve (for example, plexopathy or limb amputation) often results in persistent pain.90
Even the requirement for “nerve injury” in neuropathic pain is contentious. After a nociceptive stimulus, we feel pain because microscopic nerve fibers are embedded in the injured tissue. The difference between neuropathic and non-neuropathic pain might therefore be considered one of scope (large v small nerve injury), although many forms of neuropathic pain, such as small fiber neuropathy, also do not involve discrete nerve injury.
Neuroscientists use distinct models for non-neuropathic (for example, Carrageenan) and neuropathic pain, and even different models (>40) of neuropathic pain to reflect myriad causes (for example, chronic constriction injury, spared nerve injury models).91 Yet, the same neurotransmitters, neuropeptides, cytokines, and enzymes are implicated in both types of pain, with a large degree of overlap. NMDA receptor antagonists are often considered to be effective for neuropathic pain only, being intricately involved in the process of central sensitization, but preclinical and clinical studies have shown that they alleviate nociceptive pain too.46 92 93 Similarly, the voltage gated calcium channel subunit α-2δ-1 is upregulated in injured dorsal root gan-glion neurons but not in inflammatory pain.25 However, drugs that block these channels, such as gabapentin, are effective in both preclinical models of nociceptive pain94 and in preventing chronic postsurgical pain when given pre-emptively.95 Conversely, drugs widely acknowledged to be effective only for nociceptive pain may also alleviate neuropathic pain. NSAIDs are so widely viewed as being ineffective for neuropathic pain that no major guidelines even mention them in their algorithm.26 But preclinical and clinical studies have demonstrated efficacy for NSAIDs in neuropathic pain states,96 97 and they are commonly pre-scribed for neuropathic pain (tables 2 and 3).98
It is important to note that ascending spinal path-ways, supraspinal regions that process these signals, and descending modulation pathways are essentially the same for neuropathic and non-neuropathic pain. This creates a difference between the taxonomic classification of pain
STATE OF THE ART REVIEW
For personal use only

11 of 12
26 Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 2010;17:1113-e88.
27 Field MJ, McCleary S, Hughes J, Singh L. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain 1999;80:391-8.
28 Ueda H. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Ther 2006;109:57-77.
29 Schüning J, Scherens A, Haussleiter IS, Schwenkreis P, Krumova EK, Richter H, et al. Sensory changes and loss of intraepidermal nerve fibers in painful unilateral nerve injury. Clin J Pain 2009;25:683-90.
30 Nickel FT, Seifert F, Lanz S, Maihofner C. Mechanisms of neuropathic pain. Eur J Neuropsychopharmacol 2012;22:81-91.
31 Wehnert Y, Muller B, Larsen B, Kohn D: Sympathetically maintained pain (SMP): phentolamine test vs sympathetic nerve blockade. Comparison of two diagnostic methods [in German]. Orthopade 2002;31:1076-83.
32 Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science 2000;288:1769-72.
33 Willis WD Jr. Role of neurotransmitters in sensitization of pain responses. Ann NY Acad Sci 2001:933:142-56.
34 Dougherty PM, Willis WD. Enhanced responses of spinothalamic tract neurons to excitatory amino acids accompany capsaicin-induced sensitization in the monkey. J Neurosci 1992;12:883-94.
35 Zhuo M. Glutamate receptors and persistent pain: targeting forebrain NR2B subunits. Drug Discov Today 2002;7:259-67.
36 Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev 2004;27:729-37.
37 Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, et al. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci U S A 1999;96:7640-4.
38 Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov 2003;2:973-85.
39 Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, et al. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci 2002;22:6208-17.
40 Miller KE, Hoffman EM, Sutharshan M, Schechter R. Glutamate pharmacology and metabolism in peripheral primary afferents: physiological and pathophysiological mechanisms. Pharmacol Ther 2011;130:283-309.
41 Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci 2003;23:2899-910.
42 Malmberg AB, Chen C, Tonegawa S, Basbaum AI. Preserved acute pain and reduced neuropathic pain in mice lacking PKC gamma. Science 1997;278:279-83.
43 Mao J, Mayer DJ, Hayes RL, Lu J, Price DD. Differential roles of NMDA and non-NMDA receptor activation in induction and maintenance of thermal hyperalgesia in rats with painful peripheral mononeuropathy. Brain Res 1992;598:271-8.
44 Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312-23.
45 Liu Y, Zheng Y, Gu X, Ma Z. The efficacy of NMDA receptor antagonists for preventing remifentanil-induced increase in postoperative pain and analgesic requirement: a meta-analysis. Minerva Anesthesiol 2012;78:653-67.
46 Cohen SP, Liao W, Gupta A, Plunkett A. Ketamine in pain management. Adv Psychosom Med 2011;30:139-61.
47 Vallejo R, Tilley DM, Vogel L, Benyamin R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract 2010;10:167-84.
48 Barkhudaryan N, Dunn AJ. Molecular mechanisms of actions of interleukin-6 on the brain, with special reference to serotonin and the hypothalamo-pituitary-adrenocortical axis. Neurochem Res 1999;24:1169-80.
49 Sorkin LS, Doom CM. Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst 2000;5:96-100.
50 Leung L, Cahill CM. TNF-alpha and neuropathic pain—a review. J Neuroinflammation 2010;7:27.
51 Lu G, Beuerman RW, Zhao S, Sun G, Nguyen DH, Ma S, et al. Tumor necrosis factor-alpha and interleukin-1 induce activation of MAP kinase in human neuroma fibroblasts. Neurochem Int 1997;30:401-10.
52 Olmarker K, Rydevik B. Selective inhibition of tumor necrosis factor-alpha prevents nucleus pulposus-induced thrombus formation, intraneural edema, and reduction of nerve conduction velocity. Possible implications for future pharmacologic treatment strategies of sciatica. Spine 2001;26:863-9.
53 Quintao NL, Balz D, Santos AR, Campos MM, Calixto JB. Long-lasting neuropathic pain induced by brachial plexus injury in mice: role triggered by the pro-inflammatory cytokine, tumour necrosis factor alpha. Neuropharmacology 2006;50:614-20.
Thanks to Srinivasa Raja and Tony Yaksh for their help.Contributors: SPC conceived, designed, partly wrote, and reviewed the article and tables, and helped with the figures. JM wrote part of the article and tables and critically reviewed the article.Funding: Funded in part by the Centers for Rehabilitation Sciences Research, Uniformed Services University of the Health Sciences, Bethesda, MD, USA.Competing interests: We have read and understood the BMJ Group policy on declaration of interests and declare the following interests: None.The opinions or assertions contained herein are the private views of the authors and must not be construed as official or as reflecting the views of the US Department of the Army or the Department of Defense.Provenance and peer review: Commissioned; externally peer reviewed.1 Institute of Medicine Report from the Committee on Advancing Pain
Research, Care, and Education. Relieving pain in America. A blueprint for transforming prevention, care, education and research. National Academies Press, 2011. http://books.nap.edu/openbook.php?record_id=13172&page=1.
2 Leadley RM, Armstrong N, Lee YC, Allen A, Kleijnen J. Chronic diseases in the European Union: the prevalence and health cost implications of chronic pain. J Pain Palliat Care Pharmacother 2012;26:310-25.
3 Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. J Pain 2006;7:281-9.
4 Bouhassira D, Lantéri-Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136:380-7.
5 Henningsen P, Zipfel S, Herzog W. Management of functional somatic syndromes. Lancet 2007;369:946-55.
6 Woolf CJ, Max MB. Mechanism-based pain diagnosis: issues for analgesic drug development. Anesthesiology 2001;95:241-9.
7 Walsh DA, McWilliams DF. Pain in rheumatoid arthritis. Curr Pain Headache Rep 2012;16:509-17.
8 Cohen SP, Kapoor SG, Rathmell JP. Intravenous infusion tests have limited utility for selecting long-term drug therapy in patients with chronic pain: a systematic review. Anesthesiology 2009;111:416-31.
9 Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: Redefinition and a grading system for clinical and research purposes. Neurology 2008;70:1630-5.
10 Devor M. Neuropathic pain and injured nerve: peripheral mechanisms, Br Med Bull 1991;47:619-30.
11 Freynhagen R, Baron R, Gockel U, Tolle TR. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin 2006;22:1911-20.
12 Smith BH, Torrance N, Bennett MI, Lee AJ. Health and quality of life associated with chronic pain of predominantly neuropathic origin in the community. Clin J Pain 2007;23:143-9.
13 Jarvik JG, Deyo RA. Diagnostic evaluation of low back pain with emphasis on imaging. Ann Intern Med 2002;137:586-97.
14 Giesecke T, Gracely RH, Grant MA, Nachemson A, Petzke F, Williams DA, et al. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum 2004;50:613-23.
15 Bornhovd K, Quante M, Glauche V, Bromm B, Weiller C, Buchel C. Painful stimuli evoked different stimulus-reponse functions in the amygdala, prefrontal, insula and somatosensory cortex: a single-trial fMRI study. Brain 2002;125:1326-36.
16 Chen JI, Ha B, Bushnell MC, Pike B, Duncan GH. Differentiating noxious- and innocuous-related activation of human somatosensory cortices using temporal analysis of fMRI. J Neurophysiol 2002;88:464-74.
17 Zubieta JK, Stohler CS. Neurobiological mechanisms of placebo response. Ann NY Acad Sci 2009;1156:198-210.
18 Jaggi AS, Singh N. Role of different brain areas in peripheral nerve injury-induced neuropathic pain. Brain Res 2011;1381:187-201.
19 Pincus T, Burton A, Vogel S, Field AP. A systematic review of psychosocial factors as predictors of chronicity/disability in prospective cohorts of low back pain. Spine 2002;27:E109-120.
20 Shipton EA. The transition from acute to chronic post surgical pain. Anaesth Intensive Care 2011;39:824-36.
21 Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 1983;17:321-39.
22 Rasmussen PV, Sindrup SH, Jensen TS, Bach FW. Symptoms and signs in patients with suspected neuropathic pain. Pain 2004;110:461-9.
23 Devor M, Keller CH, Deerinck TJ, Levinson SR, Ellisman MH. Na+ channel accumulation on axolemma of afferent endings in nerve end neuromas in Apteronotus. Neurosci Lett 1989;102:149-54.
24 Levinson SR, Luo S, Henry MA. The role of sodium channels in chronic pain. Muscle Nerve 2012;46:155-65.
25 Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci 2001;21:1868-75.
STATE OF THE ART REVIEW
For personal use only

12 of 12
78 Benedetti F, Vighetti S, Amanzio M, Casadio C, Oliaro A, Bergamasco B, et al. Dose-response relationship of opioids in nociceptive and neuropathic postoperative pain. Pain 1998;74:205-11.
79 Zeilhofer HU, Benke D, Yevenes GE. Chronic pain states: pharmacological strategies to restore diminished inhibitory spinal pain control. Annu Rev Pharmacol Toxicol 2012;52:111-33.
80 Zakrzewska JM, McMillan R. Trigeminal neuralgia: the diagnosis and management of this excruciating and poorly understood facial pain. Postgrad Med J 2011;87:410-6.
81 Zhuo M, Gebhart GF. Biphasic modulation of spinal nociceptive transmission from the medullary raphe nuclei in the rat. J Neurophysiol 1997;78:746-58.
82 Dubner R. The neurobiology of persistent pain and its clinical implications. Suppl Clin Neurophysiol 2004;57:3-7.
83 Goffaux P, Redmond WJ, Rainville P, Marchand S. Descending analgesia—when the spine echoes what the brain expects. Pain 2007;130:137-43.
84 Wager TD, Rilling JK, Smith EE, Sokolik A, Casey KL, Davidson RJ, et al. Placebo-induced changes in fMRI in the anticipation and experience of pain. Science 2004;303:1162-7.
85 Valet M, Sprenger T, Boecker H, Willoch F, Rummeny E, Conrad B, et al. Distraction modulates connectivity of the cingulo-frontal cortex and the midbrain during pain—an fMRI analysis. Pain 2004;109:399-408.
86 Enck P, Benedetti F, Schedlowski M. New insights into the placebo and nocebo responses. Neuron 2008;59:195-206.
87 Wei F, Dubner R, Zou S, Ren K, Bai G, Wei D, et al. Molecular depletion of descending serotonin unmasks its novel facilitatory role in the development of persistent pain. J Neurosci 2010;30:8624-36.
88 Bannister K, Bee LA, Dickenson AH. Preclinical and early clinical investigations related to monoaminergic pain modulation. Neurotherapeutics 2009;6:703-12.
89 McCleane G. Antidepressants as analgesics. CNS Drugs 2008;22:139-56.90 Ciaramitaro P, Mondelli M, Logullo F, Grimaldi S, Battiston B, Sard A, et al;
Italian Network for Traumatic Neuropathies. Traumatic peripheral nerve injuries: epidemiological findings, neuropathic pain and quality of life in 158 patients. J Peripher Nerv Syst 2010;15:120-7.
91 Barrot M. Tests and models of nociception and pain in rodents. Neuroscience 2012;211:39-50.
92 Quintero GC, Herrera J, Bethancourt J. Cortical NR2B NMDA subunit antagonism reduces inflammatory pain in male and female rats. J Pain Res 2011;4:301-8.
93 Siu A, Drachtman R. Dextromethorphan: a review of N-methyl-d-aspartate receptor antagonist in the management of pain. CNS Drug Rev 2007;13:96-106.
94 Yoon MH, Yaksh TL. The effect of intrathecal gabapentin on pain behavior and hemodynamics on the formalin test in the rat. Anesth Analg 1999;89:434-9.
95 Clarke H, Bonin RP, Orser BA, Englesakis M, Wijeysundera DN, Katz J. The prevention of chronic postsurgical pain using gabapentin and pregabalin: a combined systematic review and meta-analysis. Anesth Analg 2012;115:428-42.
96 Vo T, Rice AS, Dworkin RH. Non-steroidal anti-inflammatory drugs for neuropathic pain: how do we explain continued widespread use? Pain 2009;143:169-71.
97 Cohen KL, Harris S. Efficacy and safety of nonsteroidal anti-inflammatory drugs in the therapy of diabetic neuropathy. Arch Int Med 1987;147:1442-4.
98 Dieleman JP, Kerklaan J, Huygen FJ, Bouma PA, Sturkenboom CJ. Incidence rates and treatment of neuropathic pain conditions in the general population. Pain 2008;137:681-8.
99 Schulman EA. Overview of tension-type headache. Curr Pain Headache Rep 2001;5:454-62.
100 Seminowicz DA, Laferriere AL, Millecamps M, Yu JS, Coderre TJ, Bushnell MC. MRI structural brain changes associated with sensory and emotional function in a rat model of long-term neuropathic pain. Neuroimage 2009;47:1007-14.
101 King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, et al. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci 2009;12:1364-6.
102 Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, et al. Coding of facial expressions of pain in the laboratory mouse. Nat Methods 2010;7:447-9.
103 Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, et al. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci 2012;15:1117-9.
104 Goodsaid F. Challenges of biomarkers in drug discovery and development. Expert Opin Drug Discov 2012;7:457-61.
105 Lariviere WR, Mogil JS. The genetics of pain and analgesia in laboratory animals. Methods Mol Biol 2010;617:261-78.
54 Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Bowman C, et al. The treatment of disc-herniation-induced sciatica with infliximab: one-year follow-up results of FIRST II, a randomized controlled trial. Spine (Phila Pa 1976) 2006;31:2759-66.
55 Cohen SP, White RL, Kurihara C, Larkin TM, Chang A, Griffith SR, et al. Epidural steroids, etanercept, or saline in subacute sciatica: a multicenter randomized trial. Ann Intern Med 2012;156:551-9.
56 Mika J, Zychowska M, Popiolek-Barczyk K, Rojewska E, Przewlocka B. Importance of glial activation in neuropathic pain. Eur J Pharmacol 2013; published online 13 Mar.
57 Mika J, Rojewska E, Makuch W, Przewlocka B. Minocycline reduces the injury-induced expression of prodynorphin and pronociceptin in DRG in a rat model of neuropathic pain. Neuroscience 2010;165:1420-8.
58 Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci 2004;24:7353-65.
59 Opree A, Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci 2000;20:6289-93.
60 Wang S, Lim G, Zeng Q, Yang L, Sung B, Mao J. Central glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J Neurosci 2005;25:488-95.
61 Song C, Phillips AG, Leonard B. Interleukin 1 beta enhances conditioned fear memory in rats: possible involvement of glucocorticoids. Eur J Neurosci 2003;18:1739-43.
62 Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol 2012;234:340-50.
63 Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol 2009;87:81-97.
64 Lotze M, Grodd W, Birbaumer N, Erb M, Huse E, Flor H. Does use of a myoelectric prosthesis prevent cortical reorganization and phantom limb pain? Nat Neurosci 1999;2:501-2.
65 Moseley GL, Flor H. Targeting cortical representations in the treatment of chronic pain: a review. Neurorehabil Neural Repair 2012;26:646-52.
66 Tang Y, Chu GY, He HX, Yu CP, An JX, Guo XY. Screening of differentially expressed genes in the hypothalamus of a rat neuropathic pain model following sciatic nerve injury. Chin Med J (Engl) 2009;122:2893-7.
67 Kõks S, Fernandes C, Kurrikoff K, Vasar E, Schalkwyk LC. Gene expression profiling reveals upregulation of Tlr4 receptors in Cckb receptor deficient mice. Behav Brain Res 2008;188:62-70.
68 Rouwette T, Vanelderen P, Roubos EW, Kozicz T, Vissers K. The amygdala, a relay station for switching on and off pain. Eur J Pain 2012;16:782-92.
69 Seminowicz DA, Wideman TH, Naso L, Hatami-Khoroushahi Z, Fallatah S, Ware MA, et al. Effective treatment of chronic low back pain in humans reverses abnormal brain anatomy and function. J Neurosci 2011;31:7540-50.
70 Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 1989;37:111-23.
71 Polgár E, Hughes DI, Riddell JS, Maxwell DJ, Puskár Z, Todd AJ. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain 2003;104:229-39.
72 Janssen SP, Truin M, Van Kleef M, Joosten EA. Differential GABAergic disinhibition during the development of painful peripheral neuropathy. Neuroscience 2011;184:183-94.
73 Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 2002;22:6724-31.
74 Drew GM, Siddall PJ, Duggan AW. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain 2004;109:379-88.
75 Miraucourt LS, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through PKC gamma interneurons. PLoS One 2007;2:e1116.
76 Kohno T, Ji RR, Ito N, Allchorne AJ, Befort K, Karchewski LA, et al. Peripheral axonal injury results in reduced mu opioid receptor pre- and post-synaptic action in the spinal cord. Pain 2005;117:77-87.
77 Przewłocki R, Przewłocka B. Opioids in chronic pain. Eur J Pharmacol 2001;429:79-91.
STATE OF THE ART REVIEW
For personal use only

Punkte sammeln auf...
springermedizin.de/eAkademieTeilnahmemöglichkeitenDiese Fortbildungseinheit steht Ihnen als e.CME und e.Tutorial in der Springer Medizin e.Akademie zur Verfügung. – e.CME: kostenfreie Teilnahme im
Rahmen des jeweiligen Zeitschriften-abonnements
– e.Tutorial: Teilnahme im Rahmen des e.Med-Abonnements
ZertifizierungDiese Fortbildungseinheit ist mit 3 CME-Punkten zertifiziert von der Landesärzte-kammer Hessen und der Nord rheinischen Akademie für Ärztliche Fort- und Weiter-bildung und damit auch für andere Ärzte-kammern anerkennungsfähig.
Hinweis für Leser aus ÖsterreichGemäß dem Diplom-Fortbildungs-Pro-gramm (DFP) der Österreichischen Ärzte-kammer werden die in der e.Akademieerworbenen CME-Punkte hierfür 1:1 alsfachspezifische Fortbildung anerkannt.
Kontakt und weitere InformationenSpringerVerlag GmbHSpringer Medizin KundenserviceTel. 0800 77 80 777E-Mail: [email protected]
Schmerz 2013 · 27:619–634DOI 10.1007/s00482-013-1344-8Online publiziert: 13. November 2013© Deutsche Schmerzgesellschaft e.V. Published by Springer-Verlag Berlin Heidelberg - all rights reserved 2013
C. SommerNeurologische Klinik, Universitätsklinikum Würzburg
Neuropathische SchmerzenPathophysiologie, Diagnostik und Therapie
ZusammenfassungNeuropathische Schmerzen sind bedingt durch Läsionen im somatosensorischen System. Charakteristische, aber nicht exklusive Merkmale sind spontane Brennschmerzen, elektrisie-rende und einschießende Schmerzen, Hyperalgesie und Allodynie. Das Grundkonzept der Pathophysiologie neuropathischer Schmerzen besteht in der Kombination aus peripherer und zentraler Sensibilisierung. Die Kenntnisse über molekulare Mechanismen sind in den ver-gangenen Jahren exponentiell angewachsen. Problematisch sind die Gewichtung der einzel-nen Mechanismen sowie die Erstellung eines integrierten Gesamtkonzepts. Fortschritte gab es auch in der Diagnostik, z. B. wurden die Methoden zur Erfassung von Funktionsstörun-gen der Nozizeptoren deutlich verbessert. Für die Therapie stehen heute weit mehr Optio-nen zur Verfügung als noch vor 15 Jahren. Unter den verfügbaren Medikamenten sind Anti-depressiva, Antikonvulsiva, Opiate und Lokaltherapeutika. Daten aus kontrollierten Studien und Empfehlungen aus Leitlinien liegen vor.
SchlüsselwörterPeriphere Sensibilisierung · Zentrale Sensibilisierung · Transkutane elektrische Nervenstimu-lation · Gabapentin · Pregabalin
CME Zertifizierte Fortbildung
Teile dieses Beitrags wurden bereits in Akt Neurol 2010; 37(9):447–453, DOI: 10.1055/s-0030-1265966, veröffentlicht.
© K
laus
Rüs
chho
ff, Sp
ringe
r Med
izin
RedaktionH. Göbel, KielR. Sabatowski, Dresden
619Der Schmerz 6 · 2013 |

CME
Lernziele
Nach Lektüre dieses Beitrags über neuropathische SchmerzenF kennen Sie die aktuelle Definition und die wichtigsten Merkmale neuropathischer
Schmerzen.F kennen Sie die Grundzüge und einige neue Aspekte zur Pathophysiologie.F wissen Sie, welche Diagnostik bei Verdacht auf neuropathische Schmerzen einzuleiten
ist.F kennen Sie die aktuellen Therapieprinzipien.
Definition und Unterformen
Laut Definition der International Association for the Study of Pain (IASP) liegt bei neuropathischen Schmerzen eine Läsion oder Erkrankung des somatosensorischen Systems, also in den meisten Fäl-len des schmerzleitenden Systems selbst vor [1]. Die physiologische Warnfunktion geht beim neuro-pathischen Schmerz verloren.
Ursachen für neuropathische Schmerzen können auf jeder Ebene des schmerzleitenden oder schmerzverarbeitenden Systems liegen. Häufig entstehen neuropathische Schmerzen durch Erkran-kungen des peripheren Nervensystems, z. B. bei Engpasssyndromen, Nervenverletzungen und Po-lyneuropathien. Auf der Ebene des Plexus brachialis oder lumbalis können infektiöse oder autoim-mune Plexusneuritiden, Kompressionen durch Tumoren oder Lymphknoten sowie Plexusavulsio-nen als Folge eines Traumas zu neuropathischen Schmerzen führen. Auf radikulärer Ebene kom-men Wurzelkompressionssyndrome und Wurzelabrisse vor. Nach Herpes zoster kann es radikulär zu einer postherpetischen Neuralgie kommen. An den Hirnnerven ist die Trigeminusneuralgie ein Prototyp neuropathischer Schmerzen. Zentral bedingte neuropathische Schmerzen können nach Rü-ckenmarksverletzungen, bei Syringomyelie, spinalen Angiomen, nach ischämischen Infarkten oder bei Tumoren im zentralen Nervensystem (ZNS) auftreten, zudem bei entzündlichen ZNS-Erkran-kungen wie der multiplen Sklerose (. Tab. 1).
Pathophysiologie
Die Pathophysiologie neuropathischer Schmerzen beinhaltet komplexe Zusammenhänge, die nur langsam verstanden werden. Es gibt periphere und zentrale Adaptations- und Maladaptationsme-chanismen sowohl in pro- als auch antinozizeptiven Systemen. Die molekularen Veränderungen sind vielfältig.
Beim neuropathischen Schmerz geht die physiologische Warnfunktion verloren
Häufig entstehen neuropathische Schmerzen durch Erkrankungen des peripheren Nervensystems
An den Hirnnerven ist die Trigeminusneuralgie ein Prototyp neuropathischer Schmerzen
Neuropathic pain · Pathophysiology, assessment, and therapy
AbstractNeuropathic pain is caused by lesions in the somatosensory system. Characteristic but not exclusive features are spontaneous burning pain, electrifying and shooting pain, hyperalgesia, and allodynia. The basic concept of the pathophysiology of neuropathic pain is the combination of peripheral and central sensitization. Knowledge on the molecular mechanisms has grown exponentially in recent years. The problem lies in identifying the individual mechanisms and in determining a comprehen-sive concept. Progress has also been made in assessment, e.g., methods for detecting dysfunction of nociceptors have significantly improved. In addition, there are many more therapeutic options avail-able than 15 years ago. The drugs available include antidepressants, anticonvulsants, opioids, and topical medications. Data from controlled trials and recommendations from guidelines are available.
KeywordsPeripheral sensitization · Central sensitization · Transcutaneous electric nerve stimulation · Gabapentin · Pregabalin
620 | Der Schmerz 6 · 2013
CME
Spontanaktivität
Die ektope Impulsgeneration im peripheren Nervensystem ist das Korrelat von Spontanschmerzen und paroxysmalen einschießenden Schmerzen. Ektope Aktivität wurde auf verschiedenen Ebenen des Nervensystems in zahlreichen Tiermodellen gezeigt, auch mithilfe neurophysiologischer Ein-zelfaserableitungen an Patienten und gesunden Versuchspersonen [2]. Vor allem die Untergrup-pe der mechanoinsensitiven C-Fasern scheint für die Spontanaktivität verantwortlich zu sein [3].
Die Ursache der Spontanaktivität liegt in einem veränderten Gleichgewicht der Ionenkanalakti-vität. Die Depolarisierung der Axonmembran erfolgt durch einen Einwärtsstrom positiv geladener Natriumionen, vermittelt durch die Öffnung spannungsabhängiger Natriumkanäle. Das negative Ru-hepotenzial wird später nach Inaktivierung der Natriumkanäle bzw. Aktivierung spannungsabhän-giger Kaliumkanäle wieder erreicht. Somit kann eine vermehrte Erregbarkeit der Membran durch mangelnde Inaktivierung von Natriumkanälen oder durch unzureichende Aktivierung von Kalium-kanälen bedingt sein.
Die Gene SCN1A bis SCN11A codieren verschiedene spannungsabhängige Natriumkanäle. Die Proteine werden Nav1,1–1,9 genannt (v = spannungsabhängig; [4]). Mit der Entdeckung von Mu-tationen im Gen für Nav1,7 bei der Erythromelalgie, einer Erkrankung mit sehr starken akralen Brennschmerzen bei Erwärmung und Belastung, und bei der Erkrankung mit paroxysmalen extre-men Schmerzen [“paroxysmal extreme pain disorder“ (PEPD)] wurden erstmals Natriumkanalmu-tationen als Ursache erblicher Schmerzerkrankungen identifiziert [5]. Auch bei einigen Patienten mit idiopathischer schmerzhafter Small-fiber-Neuropathie wurden Nav1,7- und kürzlich auch Nav1,8-Mutationen gefunden [5]. Eine erbliche Ursache für neuropathische Schmerzen ist somit wahrschein-lich häufiger als bislang angenommen.
Ektope Aktivität wurde auf verschiedenen Ebenen des Nervensystems von Tiermodellen und Menschen gezeigt
Natriumkanalmutationen wurden als eine Ursache erblicher Schmerzerkrankungen identifiziert
Tab. 1 Häufige Ursachen neuropathischer Schmerzen
Lokalisation Diagnose
Peripher Mononeuropathien:– traumatisch– Engpasssyndrome– diabetisch– Postmastektomieschmerz, Postthorakotomieschmerz, Narbenschmerzen
Polyneuropathien und Small-fiber-Neuropathien:– Diabetes mellitus, Alkohol, Hypothyreose– akute inflammatorische Polyradikuloneuropathie (Guillain-Barré-Syndrom), HIV-Neuropathie,
chronische Polyneuritis– antiretrovirale Substanzen, Cisplatin, Oxaliplatin, Disulfiram, Ethambutol, Isoniazid, Nitrofuran-
toin, Thalidomid, Thiouracil, Vincristin, Chloramphenicol, Metronidazol, Taxoide, Gold– Amyloidose, Morbus Fabry, Morbus Charcot-Marie-Tooth Typ 2B und 5, hereditäre sensibel-
autonome Neuropathien Typ 1 und 1B, hereditäre Neuropathie mit MPZ-Mutation– Erythromelalgie
Plexusläsionen:– traumatisch– Tumorinfiltration
Komplexes regionales Schmerzsyndrom I und II
Hirnnerven Neuralgien (Trigeminusneuralgie, Glossopharyngeusneuralgie)
Neuropathien
Radikulär Wurzelkompressionssyndrome, Postdiskektomiesyndrom
Radikulitis, Radikuloneuritis, z. B. Borreliose
Ganglionitis
Akuter Herpes zoster, postherpetische Neuralgie
Spinal Trauma
Angiom
Syringomyelie
Zerebral Ischämie, insbesondere Thalamus, Hirnstamm
Tumor
Multiple Sklerose
Phantomschmerz (mit peripheren Anteilen)
621Der Schmerz 6 · 2013 |

CME
Periphere Sensibilisierung
Neben spontanen Schmerzen und Parästhesien treten bei Patienten mit neuropathischen Schmer-zen häufig evozierte Schmerzen auf. Dazu gehört die verstärkte Wahrnehmung schmerzhafter Reize ( Hyperalgesie) oder die Wahrnehmung von Schmerz bei normalerweise nichtschmerzhaften Reizen (Allodynie). Diese Phänomene erklärt man z. T. durch eine Sensibilisierung der afferenten Neurone. Sensibilisierung bedeutet, dass eine geringere Depolarisation nötig ist, um ein Aktionspotenzial zu initiieren, und somit die Schwelle für die Auslösung eines Aktionspotenzials herabgesetzt ist. Zudem können überschwellige Reize eine vermehrte Antwort hervorrufen, d. h. die Reiz-Antwort-Funktion ist nach links verschoben.
Zu den Substanzen, die eine Sensibilisierung afferenter Neurone bewirken können, gehören Ent-zündungsmediatoren und Nervenwachstumsfaktoren. Diese aktivieren membrangebundene Rezep-toren, welche über eine Signaltransduktionkaskade Kinasen wie Proteinkinase A (PKA) und Protein-kinase C (PKC) aktivieren. Diese Kinasen können Natriumkanäle wie Nav1,8 und Nav1,9 phosphory-lieren und so den Natriumstrom durch die Kanäle fördern [6].
Proinflammatorische Zytokine steigen im Nerv nach einer Läsion rasch an [7]. Die klinische Bedeutung dieser tierexperimentellen Daten wird durch die Befunde gestützt, dass Patienten mit schmerzhafter Polyneuropathie im Vergleich zu Patienten mit Neuropathien ohne Schmerzen ver-mehrt proinflammatorische Zytokinprofile im peripheren Blut exprimieren [8] und dass bei be-stimmten Formen der Small-fiber-Neuropathie Zytokine in der betroffenen schmerzhaften Haut vermehrt sind [9].
Nach Nervenläsion kommt es zunächst zu einer Reduktion des normalerweise von den Endor-ganen zum Nerv transportierten Nervenwachstumsfaktors [“nerve growth factor“ (NGF)], dann je-doch wird NGF vermehrt in Schwann-Zellen produziert und kann die Expression von Ionenkanä-len und Rezeptoren steigern. Der neutralisierende NGF-Antikörper Tanezumab ist nach ersten Stu-diendaten ein vielversprechendes Schmerzmedikament [10].
Ebenso an der Sensibilisierung beteiligt sind Rezeptoren der Transient-receptor-potential-vanil-loid(TRPV)-Familie; am besten erforscht ist der Capsaicinrezeptor TRPV-1 [11]. Bei der schmerz-haften diabetischen Neuropathie wurde im letzten Jahr das Stoffwechselprodukt Methylglyoxal als zentraler Verursacher von Schmerzen identifiziert [12], wobei ein möglicher Wirkmechanismus die Sensibilisierung von Nav1,8-Kanälen einschließt. Viele der Mechanismen, die zu neuropathischen Schmerzen beitragen, werden durch die Degeneration von Axonen und die damit einhergehenden Abläufe ausgelöst (Waller-Degeneration; [13]). Substanzen, die zugleich neuroprotektive bzw. rege-nerationsfördernde wie auch analgetische Eigenschaften haben, wären somit für die Therapie neuro-pathischer Schmerzen besonders geeignet.
Zentrale Sensibilisierung
Auch in zentralen Neuronen ist eine Sensibilisierung möglich [14]. Dabei kann die periphere Akti-vität die zentralen Vorgänge dynamisch unterhalten. Eine wichtige Rolle bei der zentralen Sensibi-lisierung spielen Glutamatrezeptoren. Glutamat vermittelt seine Wirkungen über 3 Typen von Re-zeptoren, die ionotropen N-Methyl-D-Aspartat(NMDA)- und Nicht-NMDA-Rezeptoren, sog. α-Amino-3-Hydroxy-5-Methylisoxazol-4-Proprionsäure(AMPA)/Kainat-Rezeptoren, sowie die me-tabotropen Rezeptoren.
Neben den Neuronen spielt die spinale Mikroglia eine wichtige Rolle. Diese wird nach Nerven-läsion über die Chemokine Fraktalkin und CCL2 sowie über die Toll-like-Rezeptoren (TRL) akti-viert [15]. Aktivierte Mikrogliazellen produzieren vermehrt Entzündungsmediatoren, wodurch die Schmerzkaskade potenziert wird. Proinflammatorische Zytokine fördern im Rückenmark das An-sprechen der Neurone auf exzitatorische Neurotransmitter; antiinflammatorische Zytokine verstär-ken die Wirkung inhibitorischer Neurotransmitter [16]. Die Inhibition der Mikrogliaaktivierung, z. B. durch Minozyklin, bewirkt in vielen Schmerzmodellen eine Analgesie; die Übertragung der Be-funde auf die klinische Praxis ist jedoch noch nicht gelungen.
Ein wichtiger inhibitorischer Transmitter im Rückenmark mit prä- und postsynaptischer Wirkung ist die γ-Aminobuttersäure (GABA). Die Freisetzung des Neurotrophins „brain-derived neurotro-phic factor“ (BDNF) aus Mikroglia kann die Wirkung von GABA im Hinterhorn von einer inhibi-
Bei Patienten mit neuropathischen Schmerzen tritt häufig eine Hyperalgesie oder Allodynie auf
Entzündungsmediatoren und Nervenwachstumsfaktoren können eine Sensibilisierung afferenter Neurone bewirken
Zugleich neuroprotektive und analgetische Substanzen wären für die Therapie neuropathischer Schmerzen besonders geeignet
Bei der zentralen Sensibilisierung spielen Glutamatrezeptoren eine wichtige Rolle
Proinflammatorische Zytokine fördern im Rückenmark das Ansprechen der Neurone auf exzitatorische Neurotransmitter
622 | Der Schmerz 6 · 2013
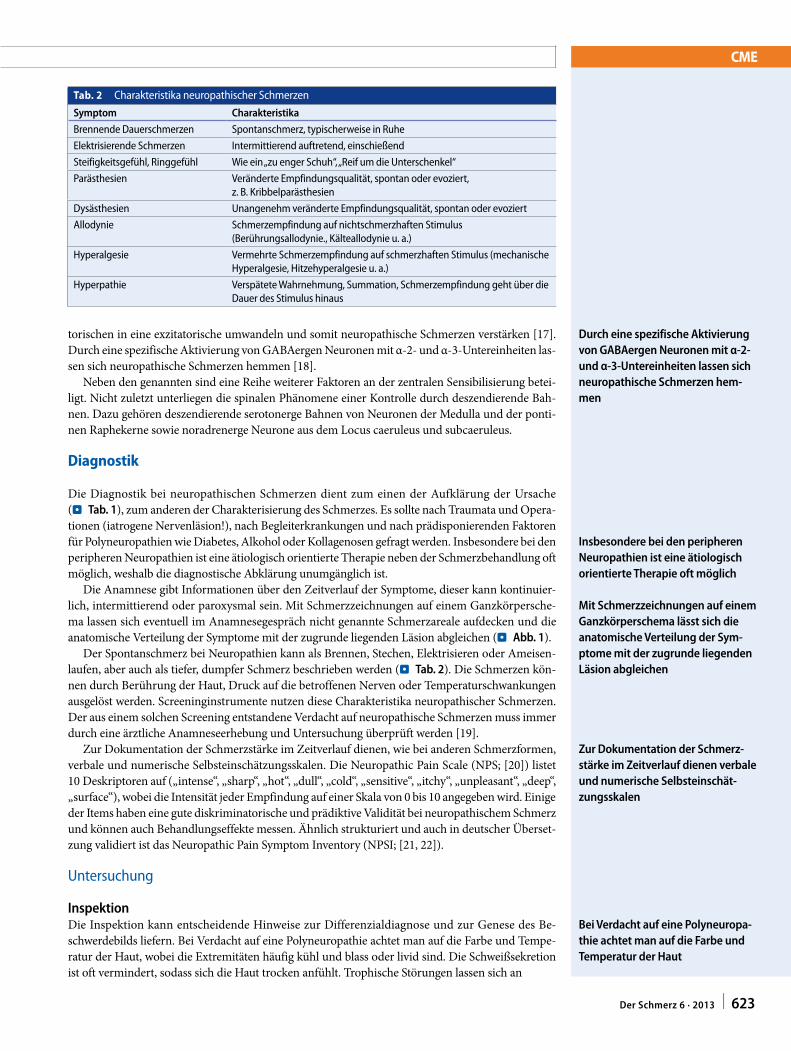
CME
torischen in eine exzitatorische umwandeln und somit neuropathische Schmerzen verstärken [17]. Durch eine spezifische Aktivierung von GABAergen Neuronen mit α-2- und α-3-Untereinheiten las-sen sich neuropathische Schmerzen hemmen [18].
Neben den genannten sind eine Reihe weiterer Faktoren an der zentralen Sensibilisierung betei-ligt. Nicht zuletzt unterliegen die spinalen Phänomene einer Kontrolle durch deszendierende Bah-nen. Dazu gehören deszendierende serotonerge Bahnen von Neuronen der Medulla und der ponti-nen Raphekerne sowie noradrenerge Neurone aus dem Locus caeruleus und subcaeruleus.
Diagnostik
Die Diagnostik bei neuropathischen Schmerzen dient zum einen der Aufklärung der Ursache (. Tab. 1), zum anderen der Charakterisierung des Schmerzes. Es sollte nach Traumata und Opera-tionen (iatrogene Nervenläsion!), nach Begleiterkrankungen und nach prädisponierenden Faktoren für Polyneuropathien wie Diabetes, Alkohol oder Kollagenosen gefragt werden. Insbesondere bei den peripheren Neuropathien ist eine ätiologisch orientierte Therapie neben der Schmerzbehandlung oft möglich, weshalb die diagnostische Abklärung unumgänglich ist.
Die Anamnese gibt Informationen über den Zeitverlauf der Symptome, dieser kann kontinuier-lich, intermittierend oder paroxysmal sein. Mit Schmerzzeichnungen auf einem Ganzkörpersche-ma lassen sich eventuell im Anamnesegespräch nicht genannte Schmerzareale aufdecken und die anatomische Verteilung der Symptome mit der zugrunde liegenden Läsion abgleichen (. Abb. 1).
Der Spontanschmerz bei Neuropathien kann als Brennen, Stechen, Elektrisieren oder Ameisen-laufen, aber auch als tiefer, dumpfer Schmerz beschrieben werden (. Tab. 2). Die Schmerzen kön-nen durch Berührung der Haut, Druck auf die betroffenen Nerven oder Temperaturschwankungen ausgelöst werden. Screeninginstrumente nutzen diese Charakteristika neuropathischer Schmerzen. Der aus einem solchen Screening entstandene Verdacht auf neuropathische Schmerzen muss immer durch eine ärztliche Anamneseerhebung und Untersuchung überprüft werden [19].
Zur Dokumentation der Schmerzstärke im Zeitverlauf dienen, wie bei anderen Schmerzformen, verbale und numerische Selbsteinschätzungsskalen. Die Neuropathic Pain Scale (NPS; [20]) listet 10 Deskriptoren auf („intense“, „sharp“, „hot“, „dull“, „cold“, „sensitive“, „itchy“, „unpleasant“, „deep“, „surface“), wobei die Intensität jeder Empfindung auf einer Skala von 0 bis 10 angegeben wird. Einige der Items haben eine gute diskriminatorische und prädiktive Validität bei neuropathischem Schmerz und können auch Behandlungseffekte messen. Ähnlich strukturiert und auch in deutscher Überset-zung validiert ist das Neuropathic Pain Symptom Inventory (NPSI; [21, 22]).
Untersuchung
InspektionDie Inspektion kann entscheidende Hinweise zur Differenzialdiagnose und zur Genese des Be-schwerdebilds liefern. Bei Verdacht auf eine Polyneuropathie achtet man auf die Farbe und Tempe-ratur der Haut, wobei die Extremitäten häufig kühl und blass oder livid sind. Die Schweißsekretion ist oft vermindert, sodass sich die Haut trocken anfühlt. Trophische Störungen lassen sich an
Durch eine spezifische Aktivierung von GABAergen Neuronen mit α2 und α3Untereinheiten lassen sich neuropathische Schmerzen hemmen
Insbesondere bei den peripheren Neuropathien ist eine ätiologisch orientierte Therapie oft möglich
Mit Schmerzzeichnungen auf einem Ganzkörperschema lässt sich die anatomische Verteilung der Sym ptome mit der zugrunde liegenden Läsion abgleichen
Zur Dokumentation der Schmerzstärke im Zeitverlauf dienen verbale und numerische Selbsteinschätzungsskalen
Bei Verdacht auf eine Polyneuropathie achtet man auf die Farbe und Temperatur der Haut
Tab. 2 Charakteristika neuropathischer Schmerzen
Symptom Charakteristika
Brennende Dauerschmerzen Spontanschmerz, typischerweise in Ruhe
Elektrisierende Schmerzen Intermittierend auftretend, einschießend
Steifigkeitsgefühl, Ringgefühl Wie ein „zu enger Schuh“, „Reif um die Unterschenkel“
Parästhesien Veränderte Empfindungsqualität, spontan oder evoziert, z. B. Kribbelparästhesien
Dysästhesien Unangenehm veränderte Empfindungsqualität, spontan oder evoziert
Allodynie Schmerzempfindung auf nichtschmerzhaften Stimulus (Berührungsallodynie., Kälteallodynie u. a.)
Hyperalgesie Vermehrte Schmerzempfindung auf schmerzhaften Stimulus (mechanische Hyperalgesie, Hitzehyperalgesie u. a.)
Hyperpathie Verspätete Wahrnehmung, Summation, Schmerzempfindung geht über die Dauer des Stimulus hinaus
623Der Schmerz 6 · 2013 |
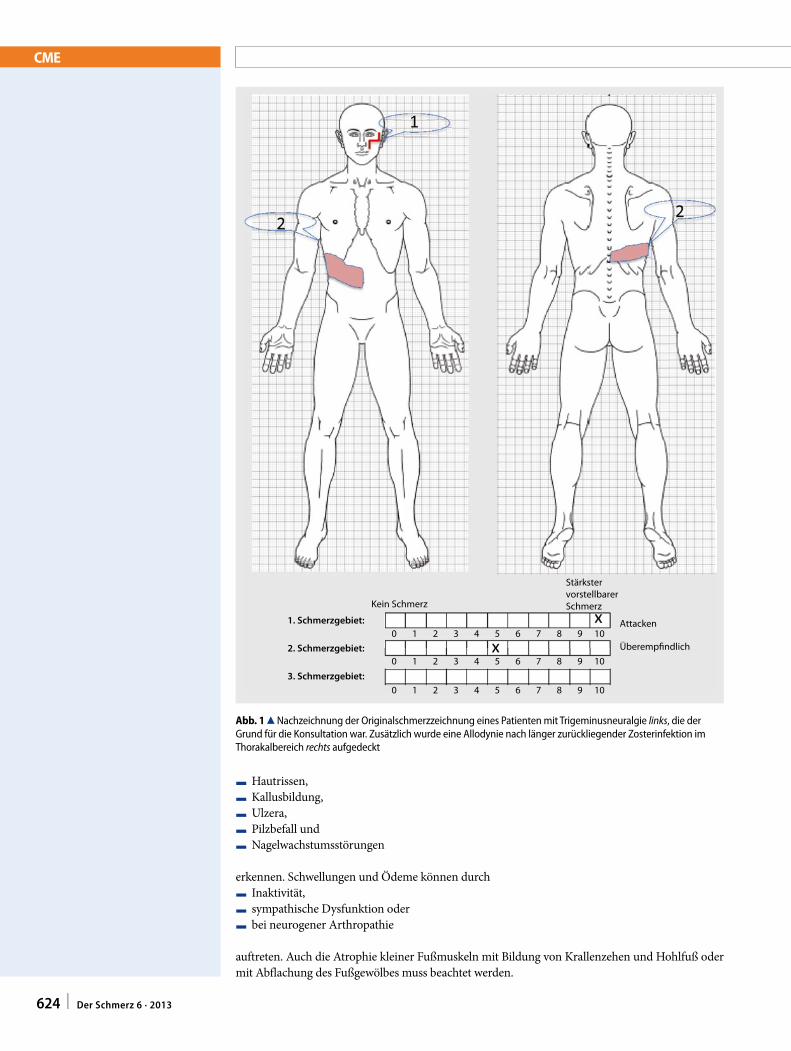
CME
F Hautrissen,F Kallusbildung,F Ulzera,F Pilzbefall undF Nagelwachstumsstörungen
erkennen. Schwellungen und Ödeme können durchF Inaktivität,F sympathische Dysfunktion oderF bei neurogener Arthropathie
auftreten. Auch die Atrophie kleiner Fußmuskeln mit Bildung von Krallenzehen und Hohlfuß oder mit Abflachung des Fußgewölbes muss beachtet werden.
Attacken
StärkstervorstellbarerSchmerzKein Schmerz
1. Schmerzgebiet:
2. Schmerzgebiet:
3. Schmerzgebiet:
0 1 2 3 4 5 6 7 8 9 10
0 1 2 3 4 5 6 7 8 9 10
0 1 2 3 4 5 6 7 8 9 10
Überemp�ndlich
Abb. 1 8 Nachzeichnung der Originalschmerzzeichnung eines Patienten mit Trigeminusneuralgie links, die der Grund für die Konsultation war. Zusätzlich wurde eine Allodynie nach länger zurückliegender Zosterinfektion im Thorakalbereich rechts aufgedeckt
624 | Der Schmerz 6 · 2013

CME
Bei Wurzelläsionen kann man eine Schonhaltung von Rumpf oder Extremitäten und einen seg-mentalen Muskelhartspann beobachten. Beim Postamputationsschmerz ist die Inspektion und Pal-pation des Narbenbereichs wichtig, um Wundheilungsstörungen und eine Neurombildung als even-tuelle Schmerzursache zu identifizieren. Bei Schmerzen nach zentralen Läsionen (zerebraler Infarkt, Tumor) hilft die Inspektion, eine Spastik oder Myoklonien als Schmerzursache festzustellen sowie Fehlhaltungen, Immobilität, Ödembildung und trophische Störungen zu erkennen. Damit lassen sich zentrale und periphere Ursachen der Schmerzgenese identifizieren.
Sensibles SystemDie Untersuchung des sensiblen Systems dient dazu, sensible Ausfälle („Minussymptome“) und Reiz-symptome („Plussymptome“) nachzuweisen. Man untersucht hierzu mit einem geeigneten Instru-ment die verschiedenen sensiblen Qualitäten:F OberflächensensibilitätF SchmerzempfindungF TemperaturempfindungF VibrationsempfindenF LageempfindenF ZweipunktdiskriminationF Stereognosie
Bereiche veränderter Sensibilität werden auf einem Ganzkörperschema notiert (. Tab. 3). Die Area-le der verschiedenen Ausfälle bzw. Reizsymptome können mit farbigen Stiften auf der Haut des Pa-tienten eingezeichnet und für Verlaufsuntersuchungen fotographisch dokumentiert werden.
MotorikDie Untersuchung der Motorik beinhaltet die Inspektion auf Muskelatrophien, die Palpation be-züglich der Schmerzhaftigkeit, die Prüfung des Muskeltonus, der Muskeleigenreflexe und patholo-gischen Reflexe sowie die Prüfung der Kraft und Feinmotorik. Ziel dieser Untersuchung bei Patien-ten mit Verdacht auf neuropathischen Schmerz ist zum einen der mögliche Nachweis einer zentra-len oder peripheren Läsion, zum anderen die Abgrenzung zu anderen Schmerzursachen, z. B. arth-rogenen oder myogenen Ursachen.
Beim Postamputationsschmerz ist die Inspektion und Palpation des Narbenbereichs wichtig
Ziele der motorischen Untersuchung sind der Nachweis einer zent ralen oder peripheren Läsion sowie die Abgrenzung zu anderen Schmerzursachen
healthy subjects (n=1080 test sites)
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
polyneuropathy (n=343)
postherpetic neuralgia (n=72)
peripheral nerve injury (n=154)
CRPS (n=403)
trigeminal neuralgia (n=92)
central pain (n=51)
other neuropathy (n=121)
all (n=1236)
not any pathologyonly loss
only gaingain and loss
Abb. 2 8 Die Z-Profile (standardnormalverteilte Daten nach Z-Transformation in Bezug auf Mittelwert und Stan-dardabweichung) der quantitativen sensorischen Testung zeigen Unterschiede in der Verteilung von Plus- und Minussymptomen zwischen diagnostischen Gruppen, aber auch innerhalb derselben. [Aus [28], mit freundl. Geneh-migung der International Association for the Study of Pain® (IASP). Die Abbildung darf nicht ohne Genehmigung für andere Zwecke weiterverwendet werden]
625Der Schmerz 6 · 2013 |
CME
Autonome FunktionsstörungenDie Untersuchung autonomer Funktionsstörungen beschränkt sich klinisch auf die Pupillomoto-rik, die Beobachtung der Schweißsekretion und einfache Tests der kardiovaskulären Funktion, z. B. den Schellong-Test. Aufwendigere apparative Verfahren stehen je nach Indikationsstellung zur Ver-fügung.
Apparative Untersuchungsverfahren
NeurographieIn der diagnostischen Abklärung von Neuropathien ist die Elektroneurographie die wichtigste neuro-physiologische Untersuchungsmethode. Die konventionelle Neurographie erfasst nur die nichtnozi-zeptiven bemarkten Fasern, allerdings lassen sich oft indirekte Schlüsse auf eine wahrscheinliche Mit-beteiligung der nozizeptiven Fasern ziehen. Auch die somatosensibel evozierten Potenziale (SSEP) erfassen die markhaltigen nichtnozizeptiven Bahnen, sind jedoch in Bezug auf den Nachweis einer Mitbeteiligung der zentralen Afferenzen wertvoll.
Die direkte mikroneurographische Ableitung unbemarkter Nervenfasern ist bislang wenigen Speziallabors vorbehalten [3]. Aδ-Nozizeptoren lassen sich mithilfe laserevozierter Potenziale (LEP; [24]), hitzeevozierter Potenziale [“contact heat evoked potentials“ (CHEPS); [25]] oder schmerzevo-zierter Potenziale [“pain-related evoked potentials“ (PREP); [26]] untersuchen.
Quantitative sensorische TestungDie quantitative sensorische Testung (QST) ist eine Ergänzung und Vertiefung der sensiblen Unter-suchung. Hierbei werden mit einer computergestützten Apparatur definierte thermische und mecha-nische Stimuli in aufsteigender Intensität nach einem standardisierten Algorithmus appliziert [27]. Der Proband gibt die Empfindungs- und Schmerzschwellen für den jeweiligen Reiz an; das standar-disierte QST-Protokoll des Deutschen Forschungsverbunds Neuropathischer Schmerz (DFNS) er-fasst zusätzlich auch Phänomene wie Allodynie und Hyperalgesie [27]. Interessant ist die Verteilung von Plus- und Minussymptomen, auch im Hinblick auf einen eventuellen prädiktiven Wert bezüg-lich des Ansprechens auf eine Therapie. Eine Datenbankauswertung zeigte deutliche Unterschiede in der Verteilung von Plus- und Minussymptomen zwischen diagnostischen Gruppen, aber auch in-nerhalb derselben (. Abb. 2, [28]).
Die konventionelle Neurographie erfasst nur die nichtnozizeptiven bemarkten Fasern
SSEP sind in Bezug auf den Nachweis einer Mitbeteiligung der zentralen Afferenzen wertvoll
Tab. 3 Negative und positive sensible Symptome bei neuropathischen Schmerzen. (Modifiziert nach [23])
Symptom,Befund
Definition Untersuchung,BedsideTest
Erwartete Antwort
Neg
ativ
sym
ptom
e
Hypästhesie Reduzierte Empfin-dung nichtschmerz-hafter Reize
Bestreichen der Haut mit Fingern, Pinsel oder Watte-träger
Reduzierte Empfindung, Taubheitsgefühl
Pallhypästhesie Reduzierte Empfin-dung eines Vibrations-reizes
Applikation der Stimm-gabel über Knochen oder Gelenk
Verlust oder rasches Verschwinden des Vibra-tionsempfindens
Hypalgesie Reduzierte Empfin-dung schmerzhafter Reize
Berühren der Haut mit spit-zem Gegenstand (z. B. mit Zahnstocher oder steifem von-Frey-Haar)
Empfindung nur als Druck, nicht als Schmerz
Therm- hypästhesie
Reduzierte Empfin-dung eines Warm- oder Kaltreizes
Berührung der Haut mit kalten Gegenständen (z. B. 10°C; Metallrolle, Wasser-glas, Acetonspray);Berührung der Haut mit warmen Gegenständen (z. B. 45°C; Metallrolle, Wasserglas)
Reduzierte Wahrneh-mung, bei Schädigung der Kaltfasern auch paradoxe Hitzeempfin-dung
626 | Der Schmerz 6 · 2013
CME
HautbiopsieDer Nachweis intraepidermaler Nervenfasern in Hautbiopsien kann bei sehr weit in der Peripherie lokalisierten Erkrankungen wie der Small-fiber-Neuropathie, bei denen die konventionelle Neuro-physiologie unauffällige Befunde erbringt, diagnostisch hilfreich sein [29]. Kürzlich wurde eine ver-minderte Hautinnervation auch bei Patienten mit Fibromyalgiesyndrom nachgewiesen [30], was auf eine mögliche Beteiligung des peripheren Nervensystems bei diesem Beschwerdebild hinweist.
Messung des „axon flare“Eine weitere potenziell sehr nützliche Methode ist die Messung des „axon flare“ nach Stimulation. Die Flare-Antwort (Hautrötung) entsteht durch Freisetzung von Neuropeptiden aus C-Fasern, die im Gewebe durch Vasodilatation und Verstärkung der Plasmaextravasation aus Gefäßen ein Ödem und Erythem hervorrufen. Die Ausdehnung des Flare-Areals korreliert relativ gut mit der Dichte in-traepidermaler Nervenfasern [31].
Der Nachweis intraepidermaler Nervenfasern in Hautbiopsien kann bei sehr weit in der Peripherie lokalisierten Erkrankungen diagnostisch hilfreich sein
Die Ausdehnung des FlareAreals korreliert relativ gut mit der Dichte intraepidermaler Nervenfasern
Tab. 3 Negative und positive sensible Symptome bei neuropathischen Schmerzen. (Modifiziert nach [23]) (Fortsetzung)
Symptom,Befund
Definition Untersuchung,BedsideTest
Erwartete Antwort
Posi
tivsy
mpt
ome
Spon
tane
Em
pfin
dung
; Sp
onta
nsch
mer
z
Parästhesie Nichtschmerzhafte, anhaltende krib-belnde Empfindung (Ameisenlaufen)
Fragen nach Intensität (z. B. NRS)
–
Dysästhesie Unangenehme Miss-empfindung
Fragen nach Intensität (z. B. NRS)
–
Einschießende Schmerz- attacke
Elektrisierende Schocks von Sekun-dendauer
Fragen nach Anzahl pro Zeit und Intensität (z. B. NRS);Fragen nach auslösenden Faktoren
–
Oberflächlicher Schmerz
Anhaltende schmerz-hafte Empfindung, oft brennend
Fragen nach Intensität (z. B. NRS)
–
Evoz
iert
er S
chm
erz
Mechanisch-dynamische Allodynie
Normalerweise nicht-schmerzhafter leich-ter Reiz auf der Haut löst Schmerz aus
Bestreichen der Haut mit Pinsel oder Watteträger;Größe der Fläche in cm2
Brennender, stechender Schmerz in der primär betroffenen Zone und darüber hinaus (sekun-däre Zone)
Mechanisch statische Allo-dynie
Normalerweise nichtschmerzhafter leichter statischer Druck auf der Haut löst Schmerz aus
Leichter Druck mit einem Watteträger auf der Haut;Größe der Fläche in cm2
Dumpfer Schmerz in der primär betroffenen Zone
Mechanische Pinprick-Hyper-algesie
Normalerweise leicht stechender Reiz auf der Haut löst einen stärkeren Schmerz aus
Berühren der Haut mit spit-zem Gegenstand (z. B. mit Zahnstocher oder steifem von-Frey-Haar);Größe der Fläche in cm2
Stechender Schmerz in der primär betroffenen Zone und darüber hin-aus (sekundäre Zone)
Kälteallodynie Normalerweise nichtschmerzhafter Kaltreiz auf der Haut löst einen (stärkeren) Schmerz aus
Berührung der Haut mit kalten Gegenständen (z. B. 10°C; Metallrolle, Wasser-glas, Acetonspray)
Schmerzhaft-brennende Temperaturmissemp-findungen in der primär betroffenen Zone, para-doxe Hitzeempfindung
Hitzeallodynie (Hyperalgesie)
Normalerweise nicht-schmerzhafter (leicht schmerzhafter) Warm-reiz auf der Haut löst einen (stärkeren) Schmerz aus
Berührung der Haut mit warmen Gegenständen (z. B. 40°C; Metallrolle, Wasserglas)
Schmerzhaft-brennende Temperaturmissemp-findungen in der primär betroffenen Zone
NRS Numerische Ratingskala, bei der dem Wert 0 die Aussage „Symptom nicht vorhanden“ und dem Wert 10 die Aussage „maximal vorstellbare Ausprägung des Symptoms“ (z. B. Parästhesien oder Brennschmerzen) zugeordnet wird.
627Der Schmerz 6 · 2013 |

CME
BildgebungUnter den bildgebenden Verfahren sind die kranielle und spinale Computertomographie (CT) und Magnetresonanztomographie (MRT) Standarduntersuchungen zur Evaluation zentraler Schmerz-ursachen. Während sich etwa ein Thalamusinfarkt, der zentrale neuropathische Schmerzen verur-sacht, leicht nachweisen lässt, sind für den Nachweis zentraler Veränderungen bei anderen neuro-genen Schmerzsyndromen, z. B. beim Phantomschmerz, Methoden der funktionellen Bildgebung (funktionelle MRT, PET) erforderlich. Diese haben allerdings noch nicht den Weg in die diagnosti-sche Routine gefunden [19]. Läsionen peripherer Nerven lassen sich heute teilweise schon mithilfe der hochauflösenden MRT oder sonographisch nachweisen.
Therapie
Nichtmedikamentöse Therapie
Die Evidenz zur Wirksamkeit der nichtmedikamentösen Therapieverfahren ist schwach, sodass rein empirisch vorgegangen wird. Manche Patienten profitieren von warmen Fußbädern oder milder Inf-rarotstrahlung. Bei anderen ist Kälte schmerzlindernd, es können Eisbeutel oder Gefrierelemente (für nicht mehr als 10–30 min) verwendet werden. Bei Parästhesien der Füße können Zweizellenbäder oder die transkutane elektrische Nervenstimulation (TENS) hilfreich sein. Eine Cochrane-Analy-se schloss 25 Studien mit fast 1300 Teilnehmern zur TENS bei chronischen Schmerzen ein, inklusive neuropathischer Schmerzen [32]. In 13 von 22 Studien war die TENS wirksamer als Placebo. Daten zur Akupunktur bei neuropathischem Schmerz sind rar. In einer Metaanalyse aller Studien mit Aku-punktur bei chronischem Schmerz fand sich eine schwache Evidenz für die Wirksamkeit [33].
Physiotherapie und Ergotherapie sind Bestandteil der interdisziplinären Schmerztherapie, dies gilt auch für neuropathische Schmerzen [34]. Neben der Schmerzlinderung wird hierdurch angestrebt, Fehlhaltungen zu beseitigen, pathologische Bewegungsabläufe zu kompensieren und eine adäquate Funktion zu erhalten. Fußpflege, adäquates Schuhwerk und ggf. Orthesen müssen bei Patienten mit Polyneuropathien bedacht werden.
Medikamentöse Therapie
Generelle PrinzipienEine kausale Therapie bzw. Therapie der Grunderkrankung sollte angestrebt werden, wo immer mög-lich, z. B. beim Karpaltunnelsyndrom oder bei der diabetischen Neuropathie. Die Dosierung muss in-dividuell angepasst werden. Bei den meisten Substanzen kann erst nach einer Gabe über 2–4 Wochen die Wirksamkeit eingeschätzt werden. Unwirksame Medikamente sollten wieder abgesetzt werden.
Verschiedene Fachgesellschaften haben Leitlinien zur medikamentösen Behandlung neuropa-thischer Schmerzen erstellt. Zu den aktuellsten gehören die Leitlinie der Deutschen Gesellschaft für Neurologie (DGN; [34]), der European Federation of Neurological Societies (EFNS; [35]), der Neu-ropathic Pain Special Interest Group (NeuPSIG) der IASP [36] und des britischen National Institu-te for Health and Clinical Excellence (NICE; [37]). Die Leitlinien gehen von der jeweils aktuellen Datenlage randomisierter klinischer Studien aus, unterscheiden sich jedoch in der Interpretation und Gewichtung, wobei auch nationale Aspekte der Zulassung und Verfügbarkeit zu beachten sind.
Eine breite Zulassung für die Therapie neuropathischer Schmerzen haben in Deutschland Gaba-pentin und Pregabalin, Antikonvulsiva mit Wirkung auf neuronale Kalziumkanäle. Für die Schmerz-therapie im Allgemeinen zugelassen sind Amitriptylin, Clomipramin und Imipramin als Vertreter der trizyklischen Antidepressiva. Der Serotonin-Noradrenalin-Wiederaufnahmehemmer (SNRI) Duloxetin hat eine Zulassung für die schmerzhafte diabetische Neuropathie. Eine Indikation für mit-telstarke und starke Opiate besteht bei starken, anderweitig nicht beherrschbaren Schmerzzustän-den, wovon neuropathische Schmerzen nicht ausgenommen sind. Als topische Therapeutika gibt es das Lidocainpflaster Versatis® für die postherpetische Neuralgie und das hoch dosierte 8%-Cap-saicinpflaster (Qutenza®) für peripher bedingte neuropathische Schmerzen mit Ausnahme der dia-betischen Neuropathie (. Tab. 4).
Verfahren der funktionellen Bildgebung haben noch nicht den Weg in die diagnostische Routine gefunden
Nichtmedikamentöse Therapieverfahren werden bei neuropathischen Schmerzen rein empirisch angewendet
Physiotherapie und Ergotherapie sind auch bei neuropathischen Schmerzen Bestandteil der interdisziplinären Schmerztherapie
Bei den meisten Substanzen kann erst nach einer Gabe über 2–4 Wochen die Wirksamkeit eingeschätzt werden
Eine breite Zulassung für die Therapie neuropathischer Schmerzen haben in Deutschland Gabapentin und Pregabalin
628 | Der Schmerz 6 · 2013
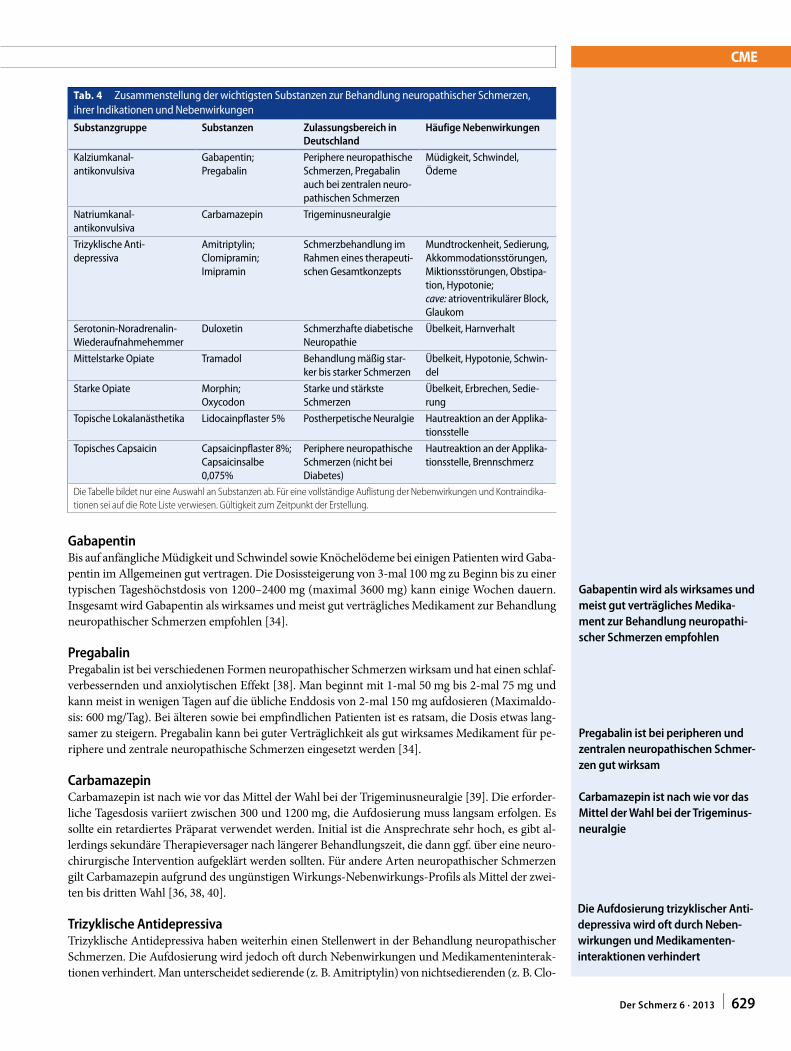
CME
GabapentinBis auf anfängliche Müdigkeit und Schwindel sowie Knöchelödeme bei einigen Patienten wird Gaba-pentin im Allgemeinen gut vertragen. Die Dosissteigerung von 3-mal 100 mg zu Beginn bis zu einer typischen Tageshöchstdosis von 1200–2400 mg (maximal 3600 mg) kann einige Wochen dauern. Insgesamt wird Gabapentin als wirksames und meist gut verträgliches Medikament zur Behandlung neuropathischer Schmerzen empfohlen [34].
PregabalinPregabalin ist bei verschiedenen Formen neuropathischer Schmerzen wirksam und hat einen schlaf-verbessernden und anxiolytischen Effekt [38]. Man beginnt mit 1-mal 50 mg bis 2-mal 75 mg und kann meist in wenigen Tagen auf die übliche Enddosis von 2-mal 150 mg aufdosieren (Maximaldo-sis: 600 mg/Tag). Bei älteren sowie bei empfindlichen Patienten ist es ratsam, die Dosis etwas lang-samer zu steigern. Pregabalin kann bei guter Verträglichkeit als gut wirksames Medikament für pe-riphere und zentrale neuropathische Schmerzen eingesetzt werden [34].
CarbamazepinCarbamazepin ist nach wie vor das Mittel der Wahl bei der Trigeminusneuralgie [39]. Die erforder-liche Tagesdosis variiert zwischen 300 und 1200 mg, die Aufdosierung muss langsam erfolgen. Es sollte ein retardiertes Präparat verwendet werden. Initial ist die Ansprechrate sehr hoch, es gibt al-lerdings sekundäre Therapieversager nach längerer Behandlungszeit, die dann ggf. über eine neuro-chirurgische Intervention aufgeklärt werden sollten. Für andere Arten neuropathischer Schmerzen gilt Carbamazepin aufgrund des ungünstigen Wirkungs-Nebenwirkungs-Profils als Mittel der zwei-ten bis dritten Wahl [36, 38, 40].
Trizyklische AntidepressivaTrizyklische Antidepressiva haben weiterhin einen Stellenwert in der Behandlung neuropathischer Schmerzen. Die Aufdosierung wird jedoch oft durch Nebenwirkungen und Medikamenteninterak-tionen verhindert. Man unterscheidet sedierende (z. B. Amitriptylin) von nichtsedierenden (z. B. Clo-
Gabapentin wird als wirksames und meist gut verträgliches Medikament zur Behandlung neuropathischer Schmerzen empfohlen
Pregabalin ist bei peripheren und zentralen neuropathischen Schmerzen gut wirksam
Carbamazepin ist nach wie vor das Mittel der Wahl bei der Trigeminusneuralgie
Die Aufdosierung trizyklischer Anti depressiva wird oft durch Nebenwirkungen und Medikamenteninteraktionen verhindert
Tab. 4 Zusammenstellung der wichtigsten Substanzen zur Behandlung neuropathischer Schmerzen, ihrer Indikationen und Nebenwirkungen
Substanzgruppe Substanzen Zulassungsbereich in Deutschland
Häufige Nebenwirkungen
Kalziumkanal- antikonvulsiva
Gabapentin;Pregabalin
Periphere neuropathische Schmerzen, Pregabalin auch bei zentralen neuro-pathischen Schmerzen
Müdigkeit, Schwindel, Ödeme
Natriumkanal- antikonvulsiva
Carbamazepin Trigeminusneuralgie
Trizyklische Anti- depressiva
Amitriptylin;Clomipramin;Imipramin
Schmerzbehandlung im Rahmen eines therapeuti-schen Gesamtkonzepts
Mundtrockenheit, Sedierung, Akkommodationsstörungen, Miktionsstörungen, Obstipa-tion, Hypotonie;cave: atrioventrikulärer Block, Glaukom
Serotonin-Noradrenalin-Wiederaufnahmehemmer
Duloxetin Schmerzhafte diabetische Neuropathie
Übelkeit, Harnverhalt
Mittelstarke Opiate Tramadol Behandlung mäßig star-ker bis starker Schmerzen
Übelkeit, Hypotonie, Schwin-del
Starke Opiate Morphin;Oxycodon
Starke und stärkste Schmerzen
Übelkeit, Erbrechen, Sedie-rung
Topische Lokalanästhetika Lidocainpflaster 5% Postherpetische Neuralgie Hautreaktion an der Applika-tionsstelle
Topisches Capsaicin Capsaicinpflaster 8%;Capsaicinsalbe 0,075%
Periphere neuropathische Schmerzen (nicht bei Diabetes)
Hautreaktion an der Applika-tionsstelle, Brennschmerz
Die Tabelle bildet nur eine Auswahl an Substanzen ab. Für eine vollständige Auflistung der Nebenwirkungen und Kontraindika-tionen sei auf die Rote Liste verwiesen. Gültigkeit zum Zeitpunkt der Erstellung.
629Der Schmerz 6 · 2013 |

CME
mipramin) trizyklischen Antidepressiva und kann diese entsprechend der gewünschten Wirkung ver-ordnen, so etwa Amitriptylin retard zur Nacht bei zusätzlichen Schlafstörungen [34]. Die wirksame Dosis variiert interindividuell sehr stark, sodass Tagesdosen zwischen 25 und 150 mg möglich sind. In jedem Fall ist eine langsame Aufdosierung erforderlich.
DuloxetinDer selektive SNRI Duloxetin ist bei der schmerzhaften diabetischen Neuropathie wirksam. Das NICE wertet Duloxetin in der Dosierung von 60 mg/Tag als Medikament mit der besten Kosteneffi-zienz und dem höchsten zu erwartenden therapeutischen Nutzen bei der Behandlung der schmerz-haften diabetischen Polyneuropathie [41].
OpioidanalgetikaOpioidanalgetika sind wirksam in der Behandlung neuropathischer Schmerzen [42], jedoch gelten sie als Mittel der zweiten bis dritten Wahl. Nebenwirkungen und eine Toleranzentwicklung können die Anwendung in der Praxis limitieren [34]. Eine langfristige Therapiekontrolle mit Schmerztage-büchern und Dokumentation der therapeutischen Auswirkungen auf alle Lebensbereiche wird emp-fohlen [34].
Topische TherapeutikaDas 5%-Lidocainpflaster kann zur Mono- oder Kombinationstherapie bei der postherpetischen Neu-ralgie eingesetzt werden [43]. Die Pflaster werden für 12 h auf das schmerzhafte Areal aufgeklebt, wo-rauf ein mindestens 12-stündiges applikationsfreies Intervall folgen muss. Das hoch dosierte Cap-saicinpflaster (8%) kann bei nichtdiabetischen Patienten mit peripheren neuropathischen Schmer-zen angewendet werden. Der Therapieerfolg einer Anwendung kann bis zu 3 Monate anhalten. Die Patienten müssen allerdings gewarnt werden, dass an den ersten Tagen verstärkte Brennschmerzen auftreten können. Für diesen Fall sollten sie eine Bedarfsmedikation erhalten.
Medikamentöse KombinationstherapieIntuitiv erscheint es sinnvoll, Medikamente mit unterschiedlichen Angriffspunkten zu kombinieren, um bei niedrigeren Dosen und geringeren Nebenwirkungen eine optimale Wirkung zu erzielen. Be-sonders plausibel erscheint dies für die Kombination von lokalen und systemischen Präparaten, z. B. Lidocainpflaster und Pregabalin bei der postherpetischen Neuralgie. Die Datenlage für einen Vor-teil der Kombinationstherapie ist jedoch schwach. Die zusätzlichen Effekte sind gering [44, 45] bzw. werden durch Addition von Nebenwirkungen aufgehoben [46]. Dabei handelt es sich insbesonde-re um ZNS-Nebenwirkungen. Zudem hatten die Gruppen mit Kombinationstherapie in Studien die höhere Abbruchrate [47]. In der Praxis muss das wirksame Medikament bzw. die wirksame Kombi-nation individuell durch Erprobung gefunden werden; dabei sind das Beschwerdebild, die Neben-wirkungen und Kontraindikationen zu berücksichtigen [34].
Invasive Therapieverfahren
Eine differenzierte Darstellung der invasiven Verfahren zur Behandlung neuropathischer Schmer-zen sprengt den Rahmen dieser Darstellung. Es wird auf aktuelle Übersichtsartikel und Leitli-nien verwiesen [48, 49]. Zur Verfügung stehen verschiedene Neurostimulationsverfahren sowie Opiatpumpen. Neuroablative Verfahren werden mit wenigen Ausnahmen sehr kritisch gesehen. Es besteht Konsens darüber, dass die invasiven Maßnahmen Patienten mit ansonsten refraktären neuro-pathischen Schmerzen vorbehalten sind. Etwas niedriger liegt die Schwelle für interventionelle Ver-fahren bei der klassischen Trigeminusneuralgie [50].
Fazit für die Praxis
F Wenn die IASPDefinition von Schmerzen mit Läsion oder Erkrankung des somatosensorischen Systems beachtet wird, kann die Diagnose neuropathischer Schmerzen anhand der Anamnese und Untersuchung mit wenigen Zusatzuntersuchungen gestellt werden.
F Das aktuelle pathophysiologische Konzept geht von einer Kombination aus peripherer und zentraler Sensibilisierung aus.
Opioidanalgetika gelten als Mittel der zweiten bis dritten Wahl
Hoch dosierte Capsaicinpflaster können bei nichtdiabetischen Patienten mit peripheren neuropathischen Schmerzen angewendet werden
Die Datenlage für einen Vorteil der Kombinationstherapie ist schwach
In der Praxis muss das wirksame Medikament bzw. die wirksame Kombination individuell durch Erprobung gefunden werden
Invasive Maßnahmen sind Patien ten mit ansonsten refraktären neuropathischen Schmerzen vorbehalten
630 | Der Schmerz 6 · 2013

CME
F Medikamente und andere Verfahren zur Behandlung neuropathischer Schmerzen stehen zur Verfügung. Evidenz und Empfehlungen sind in mehreren Leitlinien zusammengefasst worden.
F Die Therapieentscheidung muss individuell getroffen werden.
Korrespondenzadresse
Prof. Dr. C. SommerNeurologische Klinik, Universitätsklinikum WürzburgJosef-Schneider-Str. 11, 97080 Wü[email protected]
Einhaltung ethischer Richtlinien
Interessenkonflikt. C. Sommer ist Mitglied in wissenschaftlichen Beiräten der Firmen Astellas, Lilly sowie Pfizer und hat Vorträge im Auftrag dieser Firmen gehalten.
Dieser Beitrag beinhaltet keine Studien an Menschen oder Tieren.
Literatur 1. Treede RD, Jensen TS, Campbell JN
et al (2008) Neuropathic pain: redefi-nition and a grading system for clini-cal and research purposes. Neurolo-gy 70:1630–1635
2. Ochoa JL, Campero M, Serra J, Bos-tock H (2005) Hyperexcitable poly-modal and insensitive nociceptors in painful human neuropathy. Muscle Nerve 32:459–472
3. Serra J, Bostock H, Sola R et al (2012) Microneurographic identification of spontaneous activity in C-nocicep-tors in neuropathic pain states in hu-mans and rats. Pain 153:42–55
4. Goldin AL, Barchi RL, Caldwell JH et al (2000) Nomenclature of volta-ge-gated sodium channels. Neuron 28:365–368
5. Dib-Hajj SD, Yang Y, Black JA, Wax-man SG (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14:49–62
6. Jin X, Gereau RWT (2006) Acute p38-mediated modulation of tetrodo-toxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci 26:246–255
7. Üçeyler N, Sommer C (2008) Cyto-kine regulation in animal models of neuropathic pain and in human di-seases. Neurosci Lett 437:194–198
8. Üçeyler N, Rogausch JP, Toyka KV, Sommer C (2007) Differential expres-sion of cytokines in painful and pain-less neuropathies. Neurology 69:42–49
9. Üçeyler N, Kafke W, Riediger N et al (2010) Elevated proinflammatory cy-tokine expression in affected skin in small fiber neuropathy. Neurology 74:1806–1813
10. Katz N, Borenstein DG, Birbara C et al (2011) Efficacy and safety of tanezu-mab in the treatment of chronic low back pain. Pain 152:2248–2258
11. Palazzo E, Luongo L, Novellis V de et al (2012) Transient receptor poten-tial vanilloid type 1 and pain deve-lopment. Curr Opin Pharmacol 12:9–17
12. Bierhaus A, Fleming T, Stoyanov S et al (2012) Methylglyoxal modifica-tion of Nav1.8 facilitates nociceptive neuron firing and causes hyperalge-sia in diabetic neuropathy. Nat Med 18:926–933
13. Dubovy P (2011) Wallerian degene-ration and peripheral nerve condi-tions for both axonal regeneration and neuropathic pain induction. Ann Anat 193:267–275
14. Woolf CJ (2011) Central sensitization: implications for the diagnosis and treatment of pain. Pain 152:S2–S15
15. Scholz J, Woolf CJ (2007) The neuro-pathic pain triad: neurons, immune cells and glia. Nat Neurosci 10:1361–1368
16. Kawasaki Y, Zhang L, Cheng JK, Ji RR (2008) Cytokine mechanisms of cen-tral sensitization: distinct and over-lapping role of interleukin-1beta, in-terleukin-6, and tumor necrosis fac-tor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 28:5189–5194
17. Coull JA, Beggs S, Boudreau D et al (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017–1021
18. Knabl J, Witschi R, Hosl K et al (2008) Reversal of pathological pain through specific spinal GABAA re-ceptor subtypes. Nature 451:330–334
19. Haanpää M, Attal N, Backonja M et al (2011) NeuPSIG guidelines on neuropathic pain assessment. Pain 152:14–27
20. Galer BS, Jensen MP (1997) Develop-ment and preliminary validation of a pain measure specific to neuropat-hic pain: the Neuropathic Pain Scale. Neurology 48:332–338
21. Bouhassira D, Attal N, Fermanian J et al (2004) Development and valida-tion of the Neuropathic Pain Symp-tom Inventory. Pain 108:248–257
22. Sommer C, Richter H, Rogausch JP et al (2011) A modified score to iden-tify and discriminate neuropathic pain: a study on the German version of the neuropathic pain symptom in-ventory (NPSI). BMC Neurol 11:104
23. Baron R, Binder A, Birklein F et al (2012) Diagnostik neuropathischer Schmerzen. In: Diener H-C, Weimar C (Hrsg) Leitlinien für Diagnostik und Therapie in der Neurologie, 5. Aufl. Thieme, Stuttgart
24. Treede RD, Lorenz J, Baumgartner U (2003) Clinical usefulness of laser-evoked potentials. Neurophysiol Clin 33:303–314
25. Chen AC, Niddam DM, Arendt-Niel-sen L (2001) Contact heat evoked potentials as a valid means to study nociceptive pathways in human sub-jects. Neurosci Lett 316:79–82
26. Hansen N, Obermann M, Üçey-ler N et al (2012) Klinische Anwen-dung schmerzevozierter Potenziale. Schmerz 26:8–15
27. Rolke R, Baron R, Maier C et al (2006) Quantitative sensory testing in the German Research Network on Neu-ropathic Pain (DFNS): standardized protocol and reference values. Pain 123:231–243
28. Maier C, Baron R, Tolle TR et al (2010) Quantitative sensory testing in the German Research Network on Neu-ropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndro-mes. Pain 150:439–450
631Der Schmerz 6 · 2013 |

CME
29. Sommer C, Lauria G (2007) Skin bi-opsy in the management of periphe-ral neuropathy. Lancet Neurol 6:632–642
30. Üçeyler N, Zeller D, Kahn AK et al (2013) Small fibre pathology in pa-tients with fibromyalgia syndrome. Brain 136(Pt 6):1857–1867
31. Bickel A, Heyer G, Senger C et al (2009) C-fiber axon reflex flare size correlates with epidermal nerve fiber density in human skin biopsies. J Pe-ripher Nerv Syst 14:294–299
32. Nnoaham KE, Kumbang J (2008) Transcutaneous electrical ner-ve stimulation (TENS) for chro-nic pain. Cochrane Database Syst Rev:CD003222
33. Ezzo J, Berman B, Hadhazy VA et al (2000) Is acupuncture effective for the treatment of chronic pain? A sys-tematic review. Pain 86:217–225
34. Baron R, Binder A, Birklein F et al (2012) Pharmakologische nicht-in-terventionelle Therapie chronisch neuropathischer Schmerzen. In: Die-ner HC, Weimar C (Hrsg) Leitlinien für Diagnostik und Therapie in der Neurologie, 5. Aufl. Thieme, Stuttgart
35. Attal N, Cruccu B, Baron R et al (2009) EFNS guidelines on the pharmacolo-gical treatment of neuropathic pain: 2009 revision. Eur J Neurol 17:1010–1018
36. Dworkin RH, O’Connor AB, Audette J et al (2010) Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc 85:S3–S14
37. National Institute for Health and Cli-nical Excellence. http://www.nice.org.uk/guidance/CG96
38. Attal N, Cruccu G, Baron R et al (2010) EFNS guidelines on the phar-macological treatment of neuropat-hic pain: 2010 revision. Eur J Neurol 17:1113–e88
39. Zakrzewska JM, McMillan R (2011) Trigeminal neuralgia: the diagno-sis and management of this excru-ciating and poorly understood facial pain. Postgrad Med J 87:410–416
40. Finnerup NB, Sindrup SH, Jensen TS (2010) The evidence for pharmacolo-gical treatment of neuropathic pain. Pain 150:573–581
41. Hoitsma E, Marziniak M, Faber CG et al (2002) Small fibre neuropathy in sarcoidosis. Lancet 359:2085–2086
42. Eisenberg E, McNicol E, Carr DB (2006) Opioids for neuropat-hic pain. Cochrane Database Syst Rev 3:CD006146
43. Khaliq W, Alam S, Puri N (2007) To-pical lidocaine for the treatment of postherpetic neuralgia. Cochrane Database Syst Rev:CD004846
44. Gilron I, Bailey JM, Tu D et al (2009) Nortriptyline and gabapentin, alo-ne and in combination for neuropat-hic pain: a double-blind, randomi-sed controlled crossover trial. Lancet 374:1252–1261
45. Gilron I, Bailey JM, Tu D et al (2005) Morphine, gabapentin, or their com-bination for neuropathic pain. N Engl J Med 352:1324–1334
46. Gatti A, Sabato AF, Occhioni R et al (2009) Controlled-release oxycodo-ne and pregabalin in the treatment of neuropathic pain: results of a mul-ticenter Italian study. Eur Neurol 61:129–137
47. Wiffen PJ (2012) Combination phar-macotherapy for the treatment of neuropathic pain in adults. J Pain Palliat Care Pharmacother 26:380
48. Cruccu G, Aziz TZ, Garcia-Larrea L et al (2007) EFNS guidelines on neu-rostimulation therapy for neuropat-hic pain. Eur J Neurol 14:952–970
49. Nizard J, Raoul S, Nguyen JP, Lefau-cheur JP (2012) Invasive stimulation therapies for the treatment of refrac-tory pain. Discov Med 14:237–246
50. Zakrzewska JM, Coakham HB (2012) Microvascular decompression for tri-geminal neuralgia: update. Curr Opin Neurol 25:296–301
Springer-Preis für den besten CME-Beitrag 2013
2013 wird der Springer-Medizin-Verlag erstmalig einen Buchpreis für den besten Beitrag aus der Rubrik CME Zertifizierte Fortbildung vergeben, der in der Zeitschrift Der Schmerz veröffentlicht wurde.
Der Buchpreis wird jeweils an Autoren und Autorinnen verliehen, die einen didaktisch besonders wertvollen CME-Beitrag verfasst haben.
Als Auswahlgremium fungieren die Rubrikherausgeber von Der Schmerz, Herr Prof. Göbel und Herr Prof. Sabatowski sowie die Redaktion des Springer-Verlags.
Der Gewinner hat die Auswahl zwischen drei Büchern des Springer- Verlags:
Die KopfschmerzenUrsachen, Mechanismen, Diagnostik und Therapie in der Praxis Göbel, Hartmut (Hrsg.)
Praktische SchmerztherapieInterdisziplinäre Diagnostik - multimodale TherapieR. Baron et al. (Hrsg.)
und
Die AnästhesiologieAllgemeine und spezielle Anästhesiologie, Schmerztherapie und Intensivmedizin Rossaint, Rolf; Werner, Christian; Zwißler, Bernhard (Hrsg.)
632 | Der Schmerz 6 · 2013

633Der Schmerz 6 · 2013 |
springermedizin.de/eAkademie
?Was ist eine typische Symptomkonstellation bei neuropathischen Schmerzen?
Hyperalgesie und Arthralgien. Analgesie und Hyperästhesie. Kältehypästhesie und Wärmeallodynie. Spontaner Brennschmerz und Allodynie. Schmerz an „tender points“ und Kribbel-
parästhesien.
?Welches ist die aktuell akzeptierte Definition neuropathischer Schmerzen?
Neuropathische Schmerzen sind durch eine Läsion deszendierender schmerz-modulierender Bahnen bedingt.
Neuropathische Schmerzen sind durch eine Läsion oder Erkrankung des somato-sensorischen Systems bedingt.
Neuropathische Schmerzen sind durch eine Störung von Nozizeptoren bedingt.
Neuropathische Schmerzen sind durch eine periphere Nervenschädigung be-dingt.
Neuropathische Schmerzen sind durch einen Thalamusinfarkt bedingt.
?Welche Aussage zur Spontanaktivität peripherer Nerven ist zutreffend?
Sie wurde bisher nur im Tierversuch beob-achtet.
Sie schließt eine zentrale Schmerzursache aus.
Sie wird durch einen Generator in spina-len Neuronen verursacht.
Sie ist das pathophysiologische Korrelat spontaner Brennschmerzen und paroxys-maler einschießender Schmerzen.
Sie wird v. a. in Aδ-Fasern generiert.
?Welche Aussage zu Sensibilisierungsprozessen bei neuropathischen Schmerzen ist korrekt?
Eine periphere und zentrale Sensibilisie-rung schließen sich gegenseitig aus.
Eine spezifische Aktivierung von GABAer-gen Neuronen mit α-2- und α-3-Unterei-nheiten trägt zur zentralen Sensibilisie-rung bei.
Proinflammatorische Zytokine fördern im Rückenmark das Ansprechen von Neuro-nen auf exzitatorische Neurotransmitter.
NMDA-Rezeptoren spielen eine herausra-gende Rolle bei der peripheren Sensibili-sierung.
Aktivierte Mikrogliazellen hemmen die zentrale Sensibilisierung.
?Welche Aussage zur Diagnostik neuropathischer Schmerzen ist falsch?
Die Diagnostik bei neuropathischen Schmerzen dient der Aufklärung der Ursache und der Charakterisierung des Schmerzes.
Die Anamnese gibt Informationen über den Zeitverlauf der Symptome.
Bei Schmerzen nach zentralen Läsionen hilft die Inspektion, eine Spastik oder Myoklonien als Schmerzursache zu iden-tifizieren.
Die Untersuchung des sensiblen Systems dient dazu, sensible Ausfälle (“Minus-symptome”) und Reizsymptome (“Plus-symptome”) nachzuweisen.
Die Untersuchung der Motorik spielt bei neuropathischen Schmerzen keine Rolle.
?Welche Aussage zur Zusatzdiagnostik bei neuropathischen Schmerzen ist richtig?
Die quantitative sensorische Testung ist ein objektives, nicht von der Kooperation des Probanden abhängiges Messverfah-ren.
Die Routineelektroneurographie ermög-licht Aussagen über die Funktion von C-Fasern.
Läsionen peripherer Nerven lassen sich heute z. T. mit der hochauflösenden MRT oder mit dem Ultraschall nachweisen.
Die funktionelle MRT-Bildgebung sollte bei allen neuropathischen Schmerzsyn-dromen angewendet werden.
Laserevozierte Potenziale messen die Funktion von Aβ-Fasern.
?Welche Aussage zur Behandlung neuropathischer Schmerzen ist korrekt?
Es gibt gute Evidenz aus kontrollierten Studien zur Wirkung von milder Infrarot-strahlung.
In einer Metaanalyse aller Studien mit Akupunktur bei chronischem Schmerz fand sich eine schwache Evidenz für die Wirksamkeit.
Physiotherapie und Ergotherapie sind bei neuropathischen Schmerzen nicht indi-ziert.
Bei den meisten Medikamenten zur Be-handlung neuropathischer Schmerzen kann nach 2–3 Tagen über die Wirksam-keit entschieden werden.
Eine kausale Therapie bzw. Therapie der Grunderkrankung ist bei neuropathischen Schmerzen grundsätzlich nicht möglich, da die Läsion im somatosensorischen Sys-tem liegt.
CME-FragebogenBitte beachten Sie: • Teilnahme nur online unter: springermedizin.de/eAkademie• Die Frage-Antwort-Kombinationen werden online individuell zusammengestellt.• Es ist immer nur eine Antwort möglich.
Für Zeitschriftenabonnenten ist die Teilnahme am e.CME kostenfreiD
633Der Schmerz 6 · 2013 |

CME-Fragebogen
634 | Der Schmerz 6 · 2013
?Welche Aussage zur medikamentösen Behandlung neuropathischer Schmerzen ist falsch?
Trizyklische Antidepressiva haben in Deutschland keine Zulassung zur Schmerztherapie.
Pregabalin ist bei verschiedenen Formen neuropathischer Schmerzen wirksam und hat einen schlafverbessernden und anxio-lytischen Effekt.
Opioidanalgetika gelten nach Aussage der meisten Leitlinien als Mittel der zweiten bis dritten Wahl.
Carbamazepin ist nach wie vor das Mittel der Wahl bei der Trigeminusneuralgie.
Duloxetin ist wirksam bei schmerzhafter diabetischer Neuropathie.
?Welche Aussage zur medikamentösen Kombinationstherapie bei neuropathischen Schmerzen ist korrekt?
Die Kombination mehrerer Präparate mit gleichem Wirkmechanismus ist sinnvoll.
Eine Kombinationstherapie hat prinzipiell weniger Nebenwirkungen als die Maxi-maldosis eines einzelnen Medikaments.
Die Datenlage belegt den eindeutigen Vorteil der Kombinationstherapie gegen-über Monotherapien.
Die Kombination von topischen und syste-mischen Präparaten ist unbedingt zu ver-meiden.
Kombinationstherapien können sinnvoll sein, jedoch muss auf die Aufaddierung insbesondere von ZNS-Nebenwirkungen geachtet werden.
?Welche Dosisempfehlung in der Behandlung neuropathischer Schmerzen ist zutreffend?
Lidocainpflaster werden mit 20%iger Konzentration verwendet.
Bei Trizyklika werden Tagesdosen von 10–20 mg empfohlen.
Die aktuell verfügbaren Capsaicinpflaster enthalten 5% Capsaicin.
Die maximale Tagesdosis von Pregabalin beträgt 600 mg.
Die Tageshöchstdosis von Gabapentin be-trägt 300 mg.
Diese zertifizierte Fortbildung ist 12 Monate auf springermedizin.de/ eAkademie verfügbar.Dort erfahren Sie auch den genauen Teilnahmeschluss. Nach Ablauf des Zertifizierungszeitraums können Sie diese Fortbildung und den Fragebogen weitere 24 Monate nutzen.
Mit dem e.Med-Komplettpaket können Sie neben den bis-herigen e.CMEs (Beitrags-PDF plus CME-Fragebogen) auch die neuen e.Learningformate e.Tutorial und e.Tutorial.plus nutzen.
D Das e.Tutorial ist speziell für die Online-Fortbildung konzi-piert und didaktisch optimiert. Klar gegliederte Lernabschnitte, besondere Hervorhebung von Merksätzen, zoomfähige Abbil-dungen und Tabellen sowie verlinkte Literatur erleichtern das Lernen und den Erwerb von CME-Punkten.
D Das e.Tutorial.plus bietet multimedialen Zusatznutzen in Form von Audio- und Videobeiträgen, 3D-Animationen, Experteninterviews und weiterführende Informationen. CME-Fragen und Multiple-Choice-Fragen innerhalb der einzelnen Lernabschnitte ermöglichen die Lernerfolgskontrolle.
D Wissenscheck: Kurse, deren Zertifizierungszeitraum ab-gelaufen ist, können weiterhin für Ihre Fortbildung und Ihren persönlichen Wissenscheck genutzt werden.
Im e.Med-Komplettpaket ist der uneingeschränkte Zugang zur e.Akademie enthalten. Hier stehen Ihnen alle Kurse der Fachzeitschriften von Springer Medizin zur Verfügung.
Teilnehmen und weitere Informationen unter: springermedizin.de/eAkademie
Unser Tipp: Testen Sie e.Med gratis und unverbindlich unter springermedizin.de/eMed
Exklusiv mit e.Med – neue Kursformate für Ihre Online-Fortbildung
634 | Der Schmerz 6 · 2013

Neuropathic pain: diagnosisand treatment
Francesca Magrinelli,1 Giampietro Zanette,2 Stefano Tamburin1
▸ Additional material ispublished online only. To viewplease visit the journal online(http://dx.doi.org/10.1136/practneurol-2013-000536).
1Department of Neurological,Neuropsychological,Morphological and MovementSciences, University of Verona,Verona, Italy2Section of Neurology, PederzoliHospital, Peschiera del Garda,Verona, Italy
Correspondence toDr Stefano Tamburin,Department of Neurological,Neuropsychological,Morphological and MovementSciences, University of Verona,Piazzale Scuro 10,Verona 37134, Italy;[email protected],[email protected]
Published Online First16 April 2013
To cite: Magrinelli F,Zanette G, Tamburin S. PractNeurol 2013;13:292–307.
ABSTRACTNeuropathic pain (NP) develops as aconsequence of a lesion or disease affecting thesomatosensory pathways in the peripheral orcentral nervous system, and occurs in manyneurological diseases (eg, peripheral neuropathy,radiculopathy, spinal cord injury, stroke andmultiple sclerosis). It affects 6%–8% of thegeneral population and its impact on quality oflife, mood and sleep exceeds the burden of itscausative pathology. A peculiar feature of NP isthe coexistence of negative and positivesymptoms and signs, reflecting loss-of-functionand gain-of-function of the somatosensorysystem, respectively. NP has long beenconsidered a difficult clinical issue because of thelack of a diagnostic gold standard and theunsatisfactory response to treatment. In recentyears, a redefinition, diagnostic algorithm, andsome guidelines on diagnosis and treatment ofNP have been published. This review offers anupdated overview on the definition,pathophysiology, clinical evaluation, diagnosisand treatment of NP and focuses on some of themost frequent NP conditions. We intend to helpovercome uncertainties on NP and bridge thegap between evidence based medicine and thereal clinical world.
INTRODUCTIONNeuropathic pain (NP) affects 6%–8% ofthe general population and has a greatimpact on the patients’ quality of life anddisability.1 2 While these epidemiologicalfigures suggest that NP is endemic, with aprevalence similar to that of diabetes mel-litus and asthma, NP is still considered dif-ficult to diagnose and treat by the generalpractitioners and by pain and neurologyspecialists. The recent redefinition anddiagnostic algorithm and the guidelineson diagnosis and treatment of NP mayhelp overcome these uncertainties andbridge the gap between evidence basedmedicine and the real world (table 1).The International Association for the
Study of Pain (IASP) stipulated that NP is
initiated or caused by a primary lesion ordysfunction in the nervous system.3
However, this classical definition lackedpathophysiological and anatomical speci-ficity because it did not specify what fellunder the umbrella term dysfunction, andincluded patients with pain secondary tomotor disorders (eg, spasticity, dystonia).To overcome these limitations, theNeuropathic Pain Special Interest Groupof the IASP (NeuPSIG) recently redefinedNP as arising as a direct consequence of alesion or disease affecting the somatosen-sory system.4 According to this newdefinition, some conditions (eg, fibro-myalgia, complex regional pain syndrometype 1) which were traditionally definedas putative NP according to the classicalIASP definition should not be consideredas such (table 2).We favour the NeuPSIG redefinition
because it is more specific than the IASPone and offers a diagnostic gradingsystem (see below), which is similar tothose used for other neurological diseases(eg, Alzheimer’s disease, Parkinson’sdisease, multiple sclerosis, motor neuronedisease), but there is no consensus onwhich NP definition is better.5
NP syndromes can be divided intothose that are peripheral or central, basedon the anatomical location of the causa-tive lesion or disease (box 1).The pathophysiology of NP involves
both the peripheral and the centralnervous systems. The term peripheral sen-sitisation indicates changes in the excit-ability of the peripheral nerve and thedorsal root ganglion, and central sensi-tisation includes changes in the spinalcord neurones, the descending pain-controlling systems and abnormal brainplasticity (table 3).6
CLINICAL PRESENTATIONThe clinical presentation of NP is similardespite different aetiologies. Common
REVIEW
292 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536

features include burning, cold and shock-like qualitiesof spontaneous pain, paraesthesia, numbness and painevoked to various stimuli, but some patients find diffi-culties in describing their pain. Studies exploringwhether any pain quality or combination of qualitieshelps to separate NP from other types of pain couldnot identify a specific pattern, as they found consider-able overlap between definite/possible and unlikelyNP.7 8 NP patients usually show both negative andpositive symptoms and signs (figure 1): this combin-ation strongly suggests NP. However, this is still notspecific for NP because patients with nociceptive painmay also have negative signs.9 A pain distribution thatsuggests a lesion or disease to a specific peripheral orcentral nervous system site (ie, a plausible neuroana-tomical distribution) would be more specific for NP.4
Asking the patient to draw the distribution of painand related symptoms on a body diagram often helps(figure 2). The frequent extraterritorial spread of
NP10 11 should be considered as it may make it diffi-cult to define to what extent a pain distribution isneuroanatomically plausible or not.12
CLINICAL DIAGNOSISAt present, no single diagnostic procedure allows a def-inite diagnosis of NP. There are some validated screen-ing tools to enable a quick and easy identification ofpatients with NP (table 4).13–19 They include questionson pain qualities, itemising the most common verbalpain descriptors and a simplified examination of thepatient. Their sensitivity and specificity are high, whenexplored by their developers in expert pain centres,but there is no information on their predictive value innon-expert settings (general practitioners, primaryneurological centres), where they would representimportant tools. Screening tools are for operationalguidance to give more robust diagnostic evaluation andnever replace clinical judgement and a global
Table 2 NP syndromes according to the IASP classical definition and the NeuPSIG revised one
IASP definition3 NeuPSIG definition4
Pain initiated or caused by a primary lesion or dysfunction in thenervous system
Pain arising as a direct consequence of a lesion or disease of thesomatosensory system
Asymmetrical (focal and multifocal) lesions in the PNS including CRPS-2*Symmetrical lesions of the peripheral nervous system (painful polyneuropathy)Lesions in the CNS
Pain in movement disorders (Parkinson’s disease, dystonia)
Pain secondary to spasticity
Frozen shoulder in patients with previous stroke
Fibromyalgia
CRPS-1*
Miscellaneous nociceptive pain conditions with central sensitisationphenomena
*CRPS is a very painful condition, which is often limited to one limb and arises usually after trauma. It is characterised by sensory, autonomic (vasomotor/sudomotor), motor and trophic changes limited to one limb. A distinction is made between CRPS-1, where a nerve lesion cannot be identified, andCRPS-2, where it can.CNS, central nervous system; CRPS, complex regional pain syndrome; IASP, International Association for the Study of Pain; NeuPSIG, Neuropathic PainSpecial Interest Group of IASP; NP, neuropathic pain; PNS, peripheral nervous system.
Table 1 Pain terminology
Pain Unpleasant sensory and emotional experience associated with actual or potential tissue damage,or described in terms of such damage
Nociceptive pain Pain that arises from actual or potential damage to non-neural tissue and is due to the physiologicalactivation of nociceptors
Neuropathic pain Pain arising as a direct consequence of a lesion or disease of the somatosensory system
Hyperaesthesia Increased sensitivity to somatic non-nociceptive stimulation
Hyperalgesia Increased pain from a stimulus that normally provokes pain
Allodynia Pain due to a stimulus that does not normally provoke pain
Hypaesthesia Decreased sensitivity to somatic non-nociceptive stimulation
Hypopallaesthesia Decreased sensitivity to vibration
Hypalgesia Diminished pain in response to a painful stimulus
Analgesia Absence of pain in response to stimulation that normally provokes pain
Painful anaesthesia Spontaneous pain in a body area or region which is anaesthetised
Temporal summation Increased pain intensity over time in response to a painful stimulus, which is delivered repeatedly above a critical rate
Paraesthesia Abnormal somatic sensation, whether spontaneous or evoked
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 293

Box 1 NP syndromes according to the site of damage of the somatosensory system
▸ Asymmetrical (focal and multifocal) lesions in the peripheral nervous system– Entrapment mononeuropathies (carpal tunnel syndrome, ulnar nerve entrapment at the elbow, meralgia paraesthe-
tica due to injury to lateral femoral cutaneous nerve, peroneal nerve entrapment at the fibular head)– Post-traumatic and postsurgical mononeuropathies– Phantom limb pain– Cervical, thoracic and lumbosacral radiculopathies– Trigeminal neuralgia– Diabetic monoradiculopathies and mononeuropathies– Postherpetic neuralgia– Brachial and lumbosacral plexopathies (inflammatory, traumatic, brachial plexus avulsion, neoplastic, radiotherapy,
diabetic lumbosacral radiculoplexus neuropathies)– Vasculitic multineuropathies
▸ Symmetrical lesions of the peripheral nervous system (painful polyneuropathies)– Diabetic distal symmetrical and small fibre polyneuropathies– Metabolic (alcohol-related, secondary to vitamin deficiency) neuropathies– Malignancy-associated neuropathies– Immune-mediated polyneuropathies– Neuropathy secondary to chemotherapy– HIV/AIDS-related polyneuropathy– Hereditary sensory neuropathies, amyloid neuropathies– Neuropathy in Fabry’s disease
▸ Lesions in the central nervous system– Spinal cord lesions (injury, infarction, inflammatory, spondylotic)– Central poststroke pain– Multiple sclerosis-related NP
NP, neuropathic pain.
Table 3 Main pathophysiological mechanisms of NP
Level of the nervous system Pathophysiological mechanisms
PNS
Peripheral nerve ▸ Release of pain-related mediators (BK, PG, TNFα, ILs, His, ATP and potassium ions)▸ Upregulation of TRP proteins in uninjured C fibres▸ Dysregulation of the synthesis or the functioning of voltage-gated sodium channels▸ Dysregulation of the synthesis or the functioning of potassium channels
Dorsal root ganglion ▸ Increased activity in dorsal root ganglions▸ Dorsal root ganglion infiltration by activated macrophages▸ Increased synthesis of proinflammatory cytokines in dorsal root ganglions
CNS
Spinal cord neurones ▸ Functional reorganisation (neuroplasticity) of dorsal horn nociceptive neurones▸ Increased release of glutamate and substance P▸ Increased expression of Nav1.3 in dorsal horn second-order neurones▸ Increased activity in voltage-gated calcium channels▸ Selective apoptotic loss of GABA-releasing interneurones▸ Reduction of KCC2 in lamina I neurone▸ Intracellular changes induced by the activation of NMDA receptors or other receptors (ie, glutamatemetabotropic receptors) by excitatory amino acids released by primary afferents
▸ Microglial activation
Brainstem (descendingpain-controlling systems)
▸ Loss of function in descending inhibitory opioidergic, serotoninergic and noradrenergic pathways▸ Changes in the modulatory control of nociceptive pathways
Brain ▸ Functional reorganisation (neuroplasticity) of thalamic and cortical (prefrontal and somatosensory) nociceptiveneurones
ATP, adenosine-5'-triphosphate; BK, bradykinin; CNS, central nervous system; GABA, γ-aminobutyric acid; His, histamine; IL, interleukin; KCC2, potassiumchloride co-transporter 2; Nav1.3, voltage-gated sodium channel 1.3; NMDA, N-methyl-D-aspartate; NP, neuropathic pain; PG, prostaglandin; PNS,peripheral nervous system; TNFα, tumour necrosis factor α; TRP, transient potential receptor.
REVIEW
294 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536

assessment of the patient.20 The use of screening toolsshould be governed by the specific clinical applicationsfor which they have been validated (eg, peripheral and/or central NP, neuropathic low back pain).
The NeuPSIG revised definition comes with a diag-nostic algorithm, defining five levels of certainty(unlikely, possible, probable, definite and unconfirmedNP; figure 3).4 The first two questions of the
Figure 1 Clinical features of NP. NP syndromes are characterised by the coexistence of negative symptoms and signs, which areexpression of loss-of-function of the somatosensory system, and positive symptoms and signs, which indicate gain-of-function of thesomatosensory system. Figure lists symptoms and signs of NP syndromes and their underlying pathophysiology. NP, neuropathic pain.
Figure 2 Body diagram showing pain distribution in a representative patient. Example of a diabetic patient with long-lasting andcomplex NP scenario that was difficult to understand when collecting clinical history. Asking the patient to draw the distribution ofpain indicates the presence of different types of NP, which turned out to depend upon the coexistence of three types of DMneuropathy (ie, distal symmetrical neuropathy, bilateral diabetic lumbosacral radiculoplexus neuropathy and thoracic radiculopathy;see section on Diabetic NP). DM, diabetes mellitus; NP, neuropathic pain; NRS, Numerical Rating Scale.
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 295
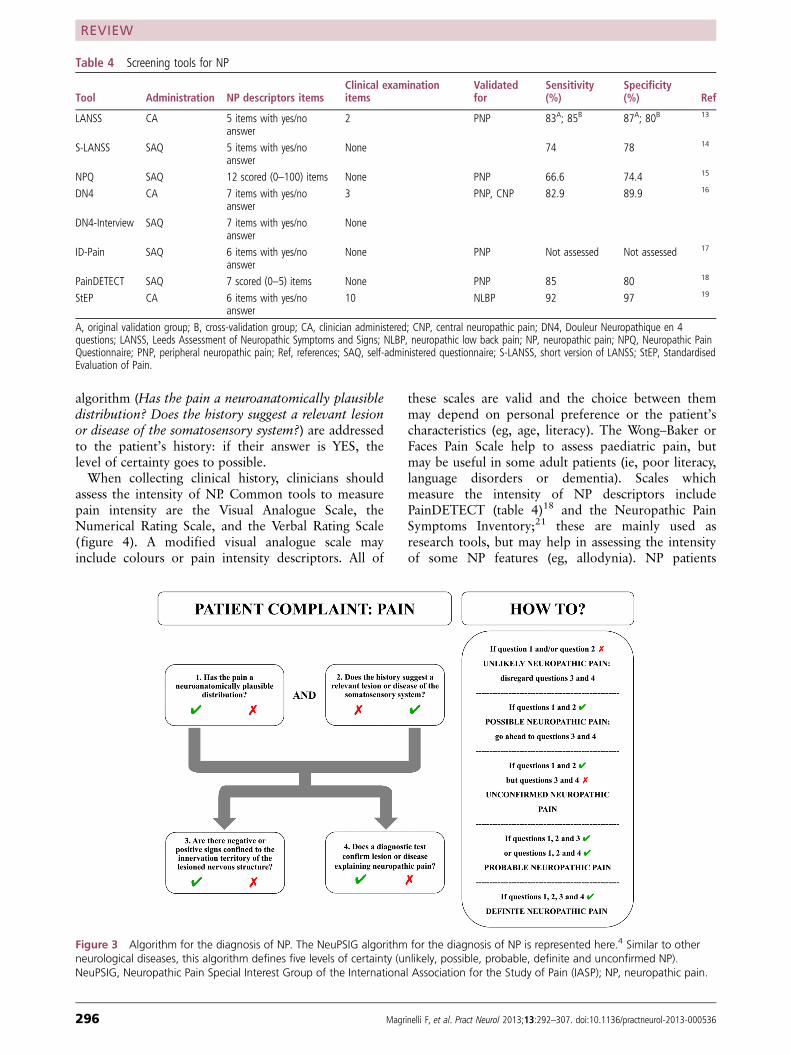
algorithm (Has the pain a neuroanatomically plausibledistribution? Does the history suggest a relevant lesionor disease of the somatosensory system?) are addressedto the patient’s history: if their answer is YES, thelevel of certainty goes to possible.When collecting clinical history, clinicians should
assess the intensity of NP. Common tools to measurepain intensity are the Visual Analogue Scale, theNumerical Rating Scale, and the Verbal Rating Scale(figure 4). A modified visual analogue scale mayinclude colours or pain intensity descriptors. All of
these scales are valid and the choice between themmay depend on personal preference or the patient’scharacteristics (eg, age, literacy). The Wong–Baker orFaces Pain Scale help to assess paediatric pain, butmay be useful in some adult patients (ie, poor literacy,language disorders or dementia). Scales whichmeasure the intensity of NP descriptors includePainDETECT (table 4)18 and the Neuropathic PainSymptoms Inventory;21 these are mainly used asresearch tools, but may help in assessing the intensityof some NP features (eg, allodynia). NP patients
Figure 3 Algorithm for the diagnosis of NP. The NeuPSIG algorithm for the diagnosis of NP is represented here.4 Similar to otherneurological diseases, this algorithm defines five levels of certainty (unlikely, possible, probable, definite and unconfirmed NP).NeuPSIG, Neuropathic Pain Special Interest Group of the International Association for the Study of Pain (IASP); NP, neuropathic pain.
Table 4 Screening tools for NP
Tool Administration NP descriptors itemsClinical examinationitems
Validatedfor
Sensitivity(%)
Specificity(%) Ref
LANSS CA 5 items with yes/noanswer
2 PNP 83A; 85B 87A; 80B 13
S-LANSS SAQ 5 items with yes/noanswer
None 74 78 14
NPQ SAQ 12 scored (0–100) items None PNP 66.6 74.4 15
DN4 CA 7 items with yes/noanswer
3 PNP, CNP 82.9 89.9 16
DN4-Interview SAQ 7 items with yes/noanswer
None
ID-Pain SAQ 6 items with yes/noanswer
None PNP Not assessed Not assessed 17
PainDETECT SAQ 7 scored (0–5) items None PNP 85 80 18
StEP CA 6 items with yes/noanswer
10 NLBP 92 97 19
A, original validation group; B, cross-validation group; CA, clinician administered; CNP, central neuropathic pain; DN4, Douleur Neuropathique en 4questions; LANSS, Leeds Assessment of Neuropathic Symptoms and Signs; NLBP, neuropathic low back pain; NP, neuropathic pain; NPQ, Neuropathic PainQuestionnaire; PNP, peripheral neuropathic pain; Ref, references; SAQ, self-administered questionnaire; S-LANSS, short version of LANSS; StEP, StandardisedEvaluation of Pain.
REVIEW
296 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536
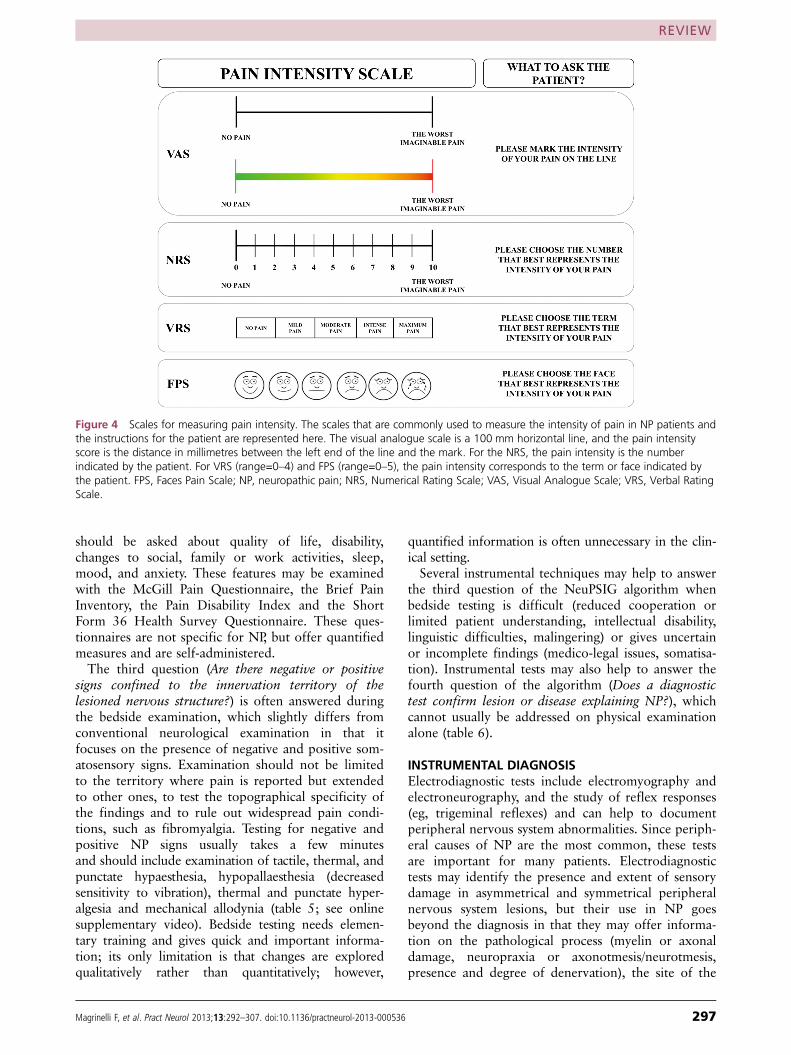
should be asked about quality of life, disability,changes to social, family or work activities, sleep,mood, and anxiety. These features may be examinedwith the McGill Pain Questionnaire, the Brief PainInventory, the Pain Disability Index and the ShortForm 36 Health Survey Questionnaire. These ques-tionnaires are not specific for NP, but offer quantifiedmeasures and are self-administered.The third question (Are there negative or positive
signs confined to the innervation territory of thelesioned nervous structure?) is often answered duringthe bedside examination, which slightly differs fromconventional neurological examination in that itfocuses on the presence of negative and positive som-atosensory signs. Examination should not be limitedto the territory where pain is reported but extendedto other ones, to test the topographical specificity ofthe findings and to rule out widespread pain condi-tions, such as fibromyalgia. Testing for negative andpositive NP signs usually takes a few minutesand should include examination of tactile, thermal, andpunctate hypaesthesia, hypopallaesthesia (decreasedsensitivity to vibration), thermal and punctate hyper-algesia and mechanical allodynia (table 5; see onlinesupplementary video). Bedside testing needs elemen-tary training and gives quick and important informa-tion; its only limitation is that changes are exploredqualitatively rather than quantitatively; however,
quantified information is often unnecessary in the clin-ical setting.Several instrumental techniques may help to answer
the third question of the NeuPSIG algorithm whenbedside testing is difficult (reduced cooperation orlimited patient understanding, intellectual disability,linguistic difficulties, malingering) or gives uncertainor incomplete findings (medico-legal issues, somatisa-tion). Instrumental tests may also help to answer thefourth question of the algorithm (Does a diagnostictest confirm lesion or disease explaining NP?), whichcannot usually be addressed on physical examinationalone (table 6).
INSTRUMENTAL DIAGNOSISElectrodiagnostic tests include electromyography andelectroneurography, and the study of reflex responses(eg, trigeminal reflexes) and can help to documentperipheral nervous system abnormalities. Since periph-eral causes of NP are the most common, these testsare important for many patients. Electrodiagnostictests may identify the presence and extent of sensorydamage in asymmetrical and symmetrical peripheralnervous system lesions, but their use in NP goesbeyond the diagnosis in that they may offer informa-tion on the pathological process (myelin or axonaldamage, neuropraxia or axonotmesis/neurotmesis,presence and degree of denervation), the site of the
Figure 4 Scales for measuring pain intensity. The scales that are commonly used to measure the intensity of pain in NP patients andthe instructions for the patient are represented here. The visual analogue scale is a 100 mm horizontal line, and the pain intensityscore is the distance in millimetres between the left end of the line and the mark. For the NRS, the pain intensity is the numberindicated by the patient. For VRS (range=0–4) and FPS (range=0–5), the pain intensity corresponds to the term or face indicated bythe patient. FPS, Faces Pain Scale; NP, neuropathic pain; NRS, Numerical Rating Scale; VAS, Visual Analogue Scale; VRS, Verbal RatingScale.
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 297

lesion (root, plexus, nerve trunk), the extent ofdamage (sensory or sensorimotor, involvement ofother peripheral nervous sites, even subclinical) andthe prognosis (timing and degree of reinnervation).Somatosensory evoked potentials may document a
lesion in the central sensory pathways in patients withsuspected central NP, and give information on thepathological process (demyelination, degenerative),the site of the lesion (spinal cord, brain) and the pres-ence of subclinical involvement at other sites.Electrodiagnostic tests and somatosensory evoked
potentials explore large myelinated nerve fibres andlemniscal pathways, which do not convey nociceptiveafferents, but these tests are important in the diagnosisof NP because they are available in many neurologicalcentres. They have diagnostic value, because most dis-eases causing NP do not result in damage or lesionlimited to the nociceptive system, but extend to the
whole peripheral nerve or involve non-nociceptivepathways.In patients with selective damage to either small
nerve fibres (eg, diabetic small fibre neuropathy) orcentral nociceptive pathways (eg, Wallenberg’s syn-drome), electrodiagnostic tests and somatosensoryevoked potentials may be normal; instrumental tests,which selectively explore the nociceptive system, maybe necessary to reach a definite diagnosis of NP. Theyinclude quantitative sensory testing, laser evokedpotentials, skin biopsy, autonomic tests, microneuro-graphy and functional neuroimaging; these tests areusually available only in tertiary or specialisedcentres.22 23
Quantitative sensory testing is non-invasive andmeasures a patient’s response to thermal and mechan-ical stimuli of standardised intensity. It gives quanti-fied information on detection and pain thresholds and
Table 5 How to test negative and positive NP signs during bedside examination (see also online supplementary video)
Tactile sensationApply a gentle touch with fingers, cotton wisp, fine hairbrush or von Frey hairs to the skin of the body regionwhich you want to examine. Ask the patient to close his/her eyes and say ‘yes’ when touched. Compare sensationin different body regions▸ Patients with tactile hypaesthesia report reduced sensation in the affected body region compared with thecorresponding normal one
▸ Patients with mechanical allodynia report pain in response to light touch (static mechanical allodynia) or gentlebrushing (dynamic mechanical allodynia)
Thermal sensationTubes or vials of warm and cold water can be used but this is usually not much practical. The Lindblomthermoroller is not widely present. Use the handle of the reflex hammer or the tuning fork, which is normallyperceived as cold, and the palm of your hand, which is normally perceived as warm, to test thermal sensation. Askthe patient to close his/her eyes and identify when touched with warm or cold stimuli. Compare sensation indifferent body regions▸ Patients with thermal hypaesthesia report reduced cold and/or warm sensation▸ Patients with thermal hyperalgesia report that the thermal stimulus is discomforting or painful
Sensation to punctate stimuliApply a punctate stimulus with a pin or with the sharp end of a broken wooden cotton tip. Ask the patient toidentify ‘sharp or dull’ with his/her eyes closed▸ Patients with punctate hypaesthesia report non-punctate touch-like sensation in response to the punctatestimulus
▸ Patients with punctate hyperalgesia report sharp pain in response to the punctate stimulus
Vibratory sensation (pallaesthesia)Place a vibrating low-frequency (128 Hz) tuning fork over a bone salience. Ask the patient to report whether he/she feels vibration and then to report when it stops. You can measure the minimal threshold for vibrationsensation, in case the tuning fork is graduated, or compare different body regions▸ Patients with hypopallaesthesia report decreased vibration sensation
Temporal summation of pain (wind up) (optional)Apply first a single punctate stimulus and then a series of 5/10 of the same punctate stimuli. Ask the patient torate pain/discomfort intensity to the single stimulus and to the series of stimuli; compare two body regions▸ Patients with enhanced wind-up report increased ratio between the series of stimuli and the single stimulus
NP, neuropathic pain.
REVIEW
298 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536
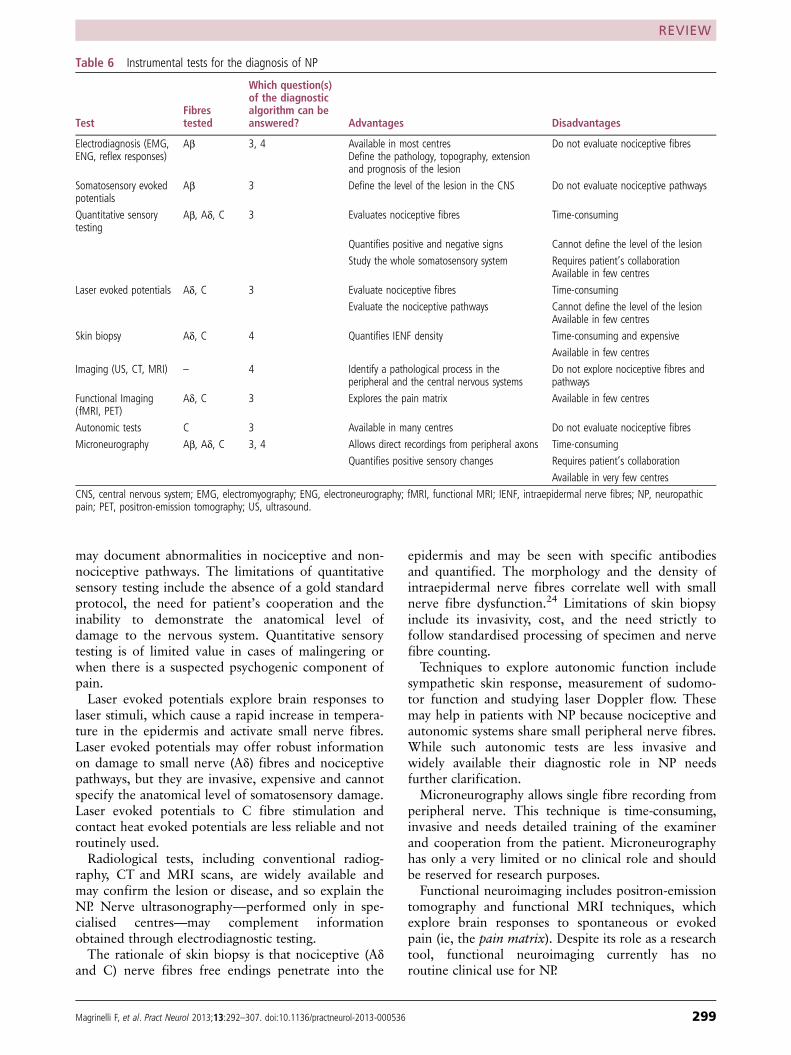
may document abnormalities in nociceptive and non-nociceptive pathways. The limitations of quantitativesensory testing include the absence of a gold standardprotocol, the need for patient’s cooperation and theinability to demonstrate the anatomical level ofdamage to the nervous system. Quantitative sensorytesting is of limited value in cases of malingering orwhen there is a suspected psychogenic component ofpain.Laser evoked potentials explore brain responses to
laser stimuli, which cause a rapid increase in tempera-ture in the epidermis and activate small nerve fibres.Laser evoked potentials may offer robust informationon damage to small nerve (Aδ) fibres and nociceptivepathways, but they are invasive, expensive and cannotspecify the anatomical level of somatosensory damage.Laser evoked potentials to C fibre stimulation andcontact heat evoked potentials are less reliable and notroutinely used.Radiological tests, including conventional radiog-
raphy, CT and MRI scans, are widely available andmay confirm the lesion or disease, and so explain theNP. Nerve ultrasonography—performed only in spe-cialised centres—may complement informationobtained through electrodiagnostic testing.The rationale of skin biopsy is that nociceptive (Aδ
and C) nerve fibres free endings penetrate into the
epidermis and may be seen with specific antibodiesand quantified. The morphology and the density ofintraepidermal nerve fibres correlate well with smallnerve fibre dysfunction.24 Limitations of skin biopsyinclude its invasivity, cost, and the need strictly tofollow standardised processing of specimen and nervefibre counting.Techniques to explore autonomic function include
sympathetic skin response, measurement of sudomo-tor function and studying laser Doppler flow. Thesemay help in patients with NP because nociceptive andautonomic systems share small peripheral nerve fibres.While such autonomic tests are less invasive andwidely available their diagnostic role in NP needsfurther clarification.Microneurography allows single fibre recording from
peripheral nerve. This technique is time-consuming,invasive and needs detailed training of the examinerand cooperation from the patient. Microneurographyhas only a very limited or no clinical role and shouldbe reserved for research purposes.Functional neuroimaging includes positron-emission
tomography and functional MRI techniques, whichexplore brain responses to spontaneous or evokedpain (ie, the pain matrix). Despite its role as a researchtool, functional neuroimaging currently has noroutine clinical use for NP.
Table 6 Instrumental tests for the diagnosis of NP
TestFibrestested
Which question(s)of the diagnosticalgorithm can beanswered? Advantages Disadvantages
Electrodiagnosis (EMG,ENG, reflex responses)
Aβ 3, 4 Available in most centresDefine the pathology, topography, extensionand prognosis of the lesion
Do not evaluate nociceptive fibres
Somatosensory evokedpotentials
Aβ 3 Define the level of the lesion in the CNS Do not evaluate nociceptive pathways
Quantitative sensorytesting
Aβ, Aδ, C 3 Evaluates nociceptive fibres Time-consuming
Quantifies positive and negative signs Cannot define the level of the lesion
Study the whole somatosensory system Requires patient’s collaborationAvailable in few centres
Laser evoked potentials Aδ, C 3 Evaluate nociceptive fibres Time-consuming
Evaluate the nociceptive pathways Cannot define the level of the lesionAvailable in few centres
Skin biopsy Aδ, C 4 Quantifies IENF density Time-consuming and expensive
Available in few centres
Imaging (US, CT, MRI) – 4 Identify a pathological process in theperipheral and the central nervous systems
Do not explore nociceptive fibres andpathways
Functional Imaging(fMRI, PET)
Aδ, C 3 Explores the pain matrix Available in few centres
Autonomic tests C 3 Available in many centres Do not evaluate nociceptive fibres
Microneurography Aβ, Aδ, C 3, 4 Allows direct recordings from peripheral axons Time-consuming
Quantifies positive sensory changes Requires patient’s collaboration
Available in very few centres
CNS, central nervous system; EMG, electromyography; ENG, electroneurography; fMRI, functional MRI; IENF, intraepidermal nerve fibres; NP, neuropathicpain; PET, positron-emission tomography; US, ultrasound.
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 299
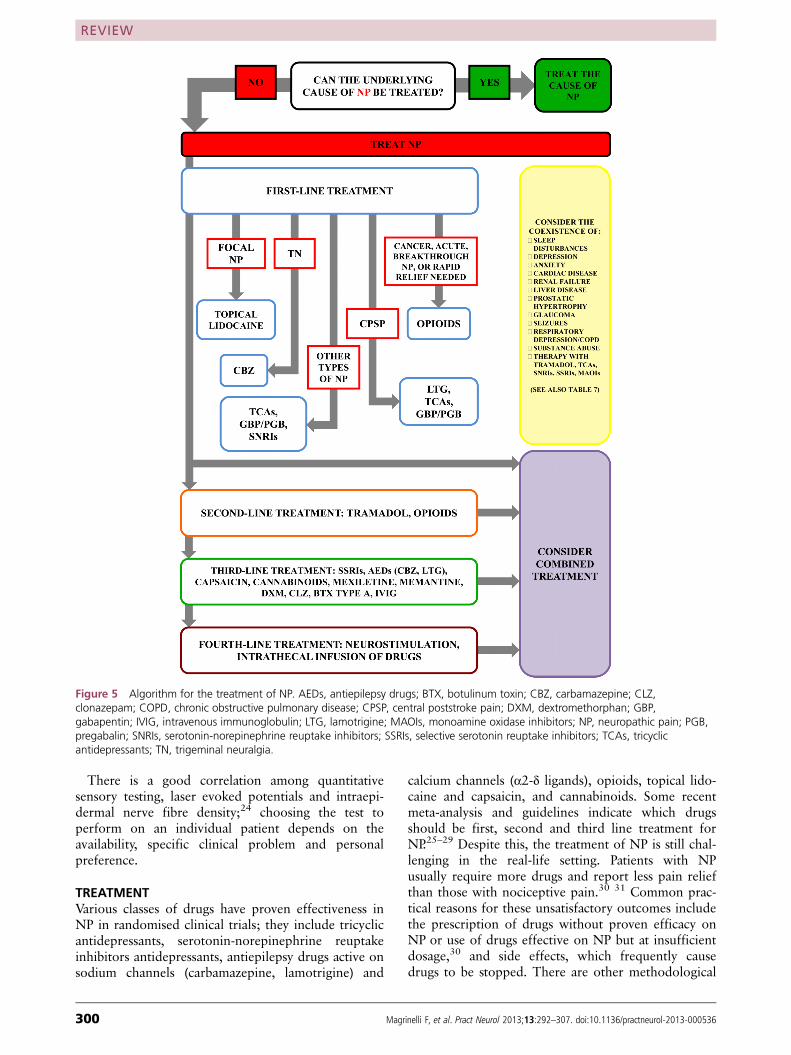
There is a good correlation among quantitativesensory testing, laser evoked potentials and intraepi-dermal nerve fibre density;24 choosing the test toperform on an individual patient depends on theavailability, specific clinical problem and personalpreference.
TREATMENTVarious classes of drugs have proven effectiveness inNP in randomised clinical trials; they include tricyclicantidepressants, serotonin-norepinephrine reuptakeinhibitors antidepressants, antiepilepsy drugs active onsodium channels (carbamazepine, lamotrigine) and
calcium channels (α2-δ ligands), opioids, topical lido-caine and capsaicin, and cannabinoids. Some recentmeta-analysis and guidelines indicate which drugsshould be first, second and third line treatment forNP.25–29 Despite this, the treatment of NP is still chal-lenging in the real-life setting. Patients with NPusually require more drugs and report less pain reliefthan those with nociceptive pain.30 31 Common prac-tical reasons for these unsatisfactory outcomes includethe prescription of drugs without proven efficacy onNP or use of drugs effective on NP but at insufficientdosage,30 and side effects, which frequently causedrugs to be stopped. There are other methodological
Figure 5 Algorithm for the treatment of NP. AEDs, antiepilepsy drugs; BTX, botulinum toxin; CBZ, carbamazepine; CLZ,clonazepam; COPD, chronic obstructive pulmonary disease; CPSP, central poststroke pain; DXM, dextromethorphan; GBP,gabapentin; IVIG, intravenous immunoglobulin; LTG, lamotrigine; MAOIs, monoamine oxidase inhibitors; NP, neuropathic pain; PGB,pregabalin; SNRIs, serotonin-norepinephrine reuptake inhibitors; SSRIs, selective serotonin reuptake inhibitors; TCAs, tricyclicantidepressants; TN, trigeminal neuralgia.
REVIEW
300 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536
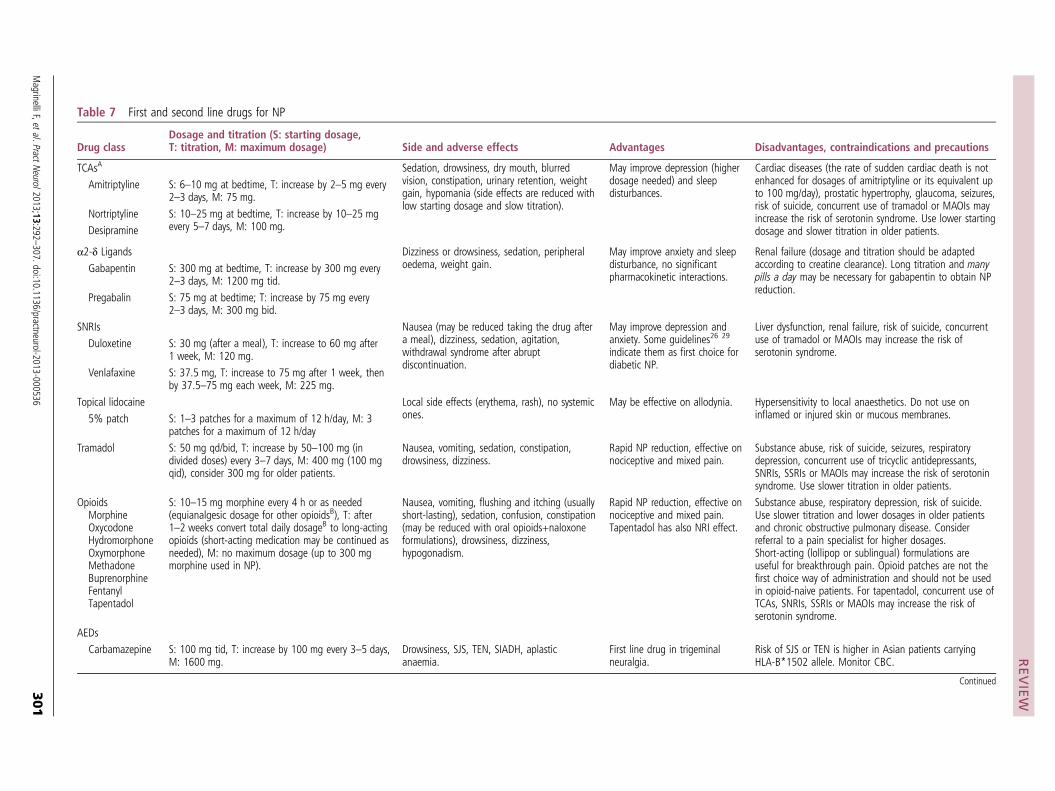
Table 7 First and second line drugs for NP
Drug classDosage and titration (S: starting dosage,T: titration, M: maximum dosage) Side and adverse effects Advantages Disadvantages, contraindications and precautions
TCAsA Sedation, drowsiness, dry mouth, blurredvision, constipation, urinary retention, weightgain, hypomania (side effects are reduced withlow starting dosage and slow titration).
May improve depression (higherdosage needed) and sleepdisturbances.
Cardiac diseases (the rate of sudden cardiac death is notenhanced for dosages of amitriptyline or its equivalent upto 100 mg/day), prostatic hypertrophy, glaucoma, seizures,risk of suicide, concurrent use of tramadol or MAOIs mayincrease the risk of serotonin syndrome. Use lower startingdosage and slower titration in older patients.
Amitriptyline S: 6–10 mg at bedtime, T: increase by 2–5 mg every2–3 days, M: 75 mg.
Nortriptyline S: 10–25 mg at bedtime, T: increase by 10–25 mgevery 5–7 days, M: 100 mg.Desipramine
α2-δ Ligands Dizziness or drowsiness, sedation, peripheraloedema, weight gain.
May improve anxiety and sleepdisturbance, no significantpharmacokinetic interactions.
Renal failure (dosage and titration should be adaptedaccording to creatine clearance). Long titration and manypills a day may be necessary for gabapentin to obtain NPreduction.
Gabapentin S: 300 mg at bedtime, T: increase by 300 mg every2–3 days, M: 1200 mg tid.
Pregabalin S: 75 mg at bedtime; T: increase by 75 mg every2–3 days, M: 300 mg bid.
SNRIs Nausea (may be reduced taking the drug aftera meal), dizziness, sedation, agitation,withdrawal syndrome after abruptdiscontinuation.
May improve depression andanxiety. Some guidelines26 29
indicate them as first choice fordiabetic NP.
Liver dysfunction, renal failure, risk of suicide, concurrentuse of tramadol or MAOIs may increase the risk ofserotonin syndrome.
Duloxetine S: 30 mg (after a meal), T: increase to 60 mg after1 week, M: 120 mg.
Venlafaxine S: 37.5 mg, T: increase to 75 mg after 1 week, thenby 37.5–75 mg each week, M: 225 mg.
Topical lidocaine Local side effects (erythema, rash), no systemicones.
May be effective on allodynia. Hypersensitivity to local anaesthetics. Do not use oninflamed or injured skin or mucous membranes.5% patch S: 1–3 patches for a maximum of 12 h/day, M: 3
patches for a maximum of 12 h/day
Tramadol S: 50 mg qd/bid, T: increase by 50–100 mg (individed doses) every 3–7 days, M: 400 mg (100 mgqid), consider 300 mg for older patients.
Nausea, vomiting, sedation, constipation,drowsiness, dizziness.
Rapid NP reduction, effective onnociceptive and mixed pain.
Substance abuse, risk of suicide, seizures, respiratorydepression, concurrent use of tricyclic antidepressants,SNRIs, SSRIs or MAOIs may increase the risk of serotoninsyndrome. Use slower titration in older patients.
OpioidsMorphineOxycodoneHydromorphoneOxymorphoneMethadoneBuprenorphineFentanylTapentadol
S: 10–15 mg morphine every 4 h or as needed(equianalgesic dosage for other opioidsB), T: after1–2 weeks convert total daily dosageB to long-actingopioids (short-acting medication may be continued asneeded), M: no maximum dosage (up to 300 mgmorphine used in NP).
Nausea, vomiting, flushing and itching (usuallyshort-lasting), sedation, confusion, constipation(may be reduced with oral opioids+naloxoneformulations), drowsiness, dizziness,hypogonadism.
Rapid NP reduction, effective onnociceptive and mixed pain.Tapentadol has also NRI effect.
Substance abuse, respiratory depression, risk of suicide.Use slower titration and lower dosages in older patientsand chronic obstructive pulmonary disease. Considerreferral to a pain specialist for higher dosages.Short-acting (lollipop or sublingual) formulations areuseful for breakthrough pain. Opioid patches are not thefirst choice way of administration and should not be usedin opioid-naive patients. For tapentadol, concurrent use ofTCAs, SNRIs, SSRIs or MAOIs may increase the risk ofserotonin syndrome.
AEDs
Carbamazepine S: 100 mg tid, T: increase by 100 mg every 3–5 days,M: 1600 mg.
Drowsiness, SJS, TEN, SIADH, aplasticanaemia.
First line drug in trigeminalneuralgia.
Risk of SJS or TEN is higher in Asian patients carryingHLA-B*1502 allele. Monitor CBC.
Continued
REVIEW
MagrinelliF,etal.PractN
eurol2013;13:292–307.doi:10.1136/practneurol-2013-000536
301

reasons for the gap between guidelines/meta-analysisand real life. The randomised controlled trialsenrolled patients mostly with diabetic NP and posther-petic neuralgia, and it is unclear whether their conclu-sions can be extrapolated to other NP conditions.More recent trials have had more robust designs andexplored measures of quality of life, sleep, anxietyand depression in addition to NP intensity (which wasthe only outcome in older studies). As a result, theevidence for older drugs (eg, tricyclic antidepressants)may be less robust and less complete than for newerones. There are very few head-to-head randomisedcontrolled trials; comparisons between drugs arebased on their respective number needed-to-treat andnumber needed-to-harm, which may be influenced bydifferent patient populations and differences betweenplacebo effects between trials.There is a good agreement between guidelines that
tricyclic antidepressants, α2-δ ligands, serotonin-norepinephrine reuptake inhibitors, carbamazepine(for trigeminal neuralgia) and topical lidocaine (forlocalised peripheral NP) are the first line drugs, andtramadol and opioids are second line drugs (figure 5;table 7). Concerns about side effects, long-term safety,opioids hyperalgesia and addiction have made opioidssecond line treatment, except in cancer NP, acute orbreakthrough (ie, with transitory worsening) NP, orwhen rapid pain relief is needed while titrating otherdrugs.When first and second line treatments are ineffect-
ive or not tolerated, there are other possibilities.They include selective serotonin reuptake inhibitors,other antidepressants (bupropion, citalopram, parox-etine), antiepileptic medications (carbamazepine,oxcarbazepine, lamotrigine, phenytoin, topiramate,valproate), high-concentration capsaicin patches, can-nabinoids (for central NP, especially multiple scler-osis), mexiletine, memantine, dextromethorphan,clonazepam, botulinum toxin type A and intravenousimmunoglobulin.32
Few randomised controlled trials have examinedcombination therapy in NP, but they converged onthere being a more marked pain reduction and fewerside effects when using two drugs together (α2-δligands+opioids; α2-δ ligands+tricyclic antidepres-sants) than each drug alone.33 The combination of NPdrugs may also target different NP mechanisms in thesame patient.Mixed pain (ie, a combination of NP and nocicep-
tive pain) is present in many conditions, includingneuropathic low back pain, nerve/root compressionand cancer pain. Evidence based medicine does notoffer any information for these patients, but combin-ing NP drugs and analgesics for nociceptive painseems reasonable.Invasive treatments (neurostimulation, intrathecal
infusion of opioids, local anaesthetics, baclofen andziconotide) are commonly used for patients whose NPTa
ble7
Continued
Drugclass
Dosag
ean
dtitratio
n(S:startingdo
sage
,T:
titratio
n,M:m
axim
umdo
sage
)Side
andad
verseeffects
Adv
antage
sDisad
vantag
es,con
traind
ications
andprecau
tions
Oxcarbazepine
S:300mg,T:increase
by600mgeveryweek,
M:2
400mg.
Drow
siness,dizziness,headache,SIADH
.Firstlinedrug
intrigeminal
neuralgia.
Bettertolerated
than
carbam
azepine.Uselowerstarting
dosage
andslowertitrationinolderp
atients.
Lamotrigine
S:25
mg,T:increase
25mgevery2weeks,M
:400mg.
Drow
siness,blurredvision,
SJS,TEN,D
RESS.
Firstlinedrug
incentral
poststroke
pain.
Adversereactions
arereducedwith
slowtitration.
Itmay
take
manyweeks
toreacheffective
dosage.
A=TCAs
dosageseffective
onNParegenerally
lowerthan
thoseused
ford
epression.Do
sageshigherthan
thosereportedhereareusually
poorlytoleratedinolderp
atients,butm
aybe
used
inyoungero
nes(sideeffects
andECGabnormalities
shouldbe
monitored).B
=Equianalgesic
opioidchartsareavailableas
pocket-sizedcardsandopioidscalcu
lator/converterappsareavailableforiPhoneandAn
droidmobile
devices.
AEDs,antiepilepsydrugs;bid,twice
daily;C
BC,com
pletebloodcount;DR
ESS,drug
reactionwith
eosinophilia
andsystem
icsymptom
s;MAO
Is,monoamineoxidaseinhibitors;N
P,neuropathicpain;N
RI,norepinephrine
reuptake
inhibitor;qd,d
aily;
qid,four
times
aday;SIAD
H,syndromeof
inappropriateantidiuretic
hormone;SJS,Stevens-Johnsonsyndrome;SNRIs,serotonin-norepinephrinereuptake
inhibitors;SSRIs,
selective
serotonin
reuptake
inhibitors;TCA
s,tricyclicantidepressants;TEN
,toxicepidermalnecrosis;
tid,three
times
aday.
REVIEW
302 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536

is refractory to other therapies. There is only a weakquality of evidence base for these invasiveapproaches,34 largely from open case series without acontrol group. Spinal cord stimulation is a reasonablechoice in refractory neuropathic low back pain, andmotor cortex stimulation may be the last resort inrefractory central poststroke pain.Despite the availability of several drugs, no more
than 30%–50% of NP patients achieve a satisfactoryresponse. Randomised controlled trials recruit patientsaccording to the NP aetiology, and this fails to capturethe complex relationship among causes, pathophysi-ology and clinical manifestations of NP.35 Furthermore,most trials do not assess pain quality, although somedrugs might be effective in subgroups of patients withspecific clinical phenotypes or with some NP features.For these reasons, the definition of sensory profilesthrough symptoms (NP questionnaires) and signs(bedside assessment and quantitative sensory testing)may better stratify patients in randomised controlledtrials and personalise treatment of NP. This perspectiveis tempting but its clinical utility has not yet beendemonstrated.Patients with NP often receive non-pharmacological
treatments, including physical exercise, physical ther-apies (eg, transcutaneous electrical nerve stimulation,graded motor imagery), cognitive behavioural therapyor supportive psychotherapy. There is only limitedevidence supporting these treatments, but they play arole in the comprehensive and multidisciplinary man-agement of a complex clinical problem.The delivery of care for NP may vary between
countries, but it is usually performed in an outpatientsetting. General practitioners play an important rolein first line treatment, given the high prevalence ofNP, while consultations with specialised neurology orpain centres are limited to refractory cases. Specialisedcentres should include medical and paramedical spe-cialties to offer a multidisciplinary approach.
SPECIFIC NP CLINICAL PICTURESWe will briefly review some NP syndromes, which arefrequent and present specific diagnostic and/or treat-ment problems.
Diabetic NPPeripheral neuropathy affects 30%–50% of patientswith diabetes mellitus and includes different syn-dromes, divided into the frequent symmetric poly-neuropathies (distal sensorimotor, autonomic, smallfibre polyneuropathy) and the less common focal/multifocal neuropathies (cranial neuropathy, thoracic/lumbar radiculopathy, lumbosacral radiculoplexusneuropathy). About half of diabetic patients withneuropathy (ie, 10%–20% of diabetic patients) haveNP, which is usually chronic.36 Acute painful neur-opathy is a separate condition precipitated by strictglycaemic control. Diabetic NP correlates with age,
body mass index, waist circumference, physical activ-ity, diabetic duration, nephropathy, peripheral arterialdisease and severity of the neuropathy.The diagnosis of NP in diabetes mellitus should
conform to the NeuPSIG diagnostic algorithm (figure 3)4
and is easily made from the patient’s history and bedsidetesting. Some validated questionnaires or checklists canscore NP symptoms and signs. Electrodiagnostic testsmay help in special cases (eg, for research purposes, atyp-ical or asymmetrical presentations).The treatment of diabetic NP follows the treatment
algorithm (figure 5). Better glycaemic control,improving other metabolic markers and reducing car-diovascular risk factors are recommended but theireffect on NP is still unclear. Intravenous α-lipoic acidmay reduce NP, but there is insufficient evidence onthe efficacy of the oral preparation. Tricyclic antide-pressants, α2-δ ligands and serotonin-norepinephrinereuptake inhibitors are the first line drugs, while tra-madol and opioids are second line treatments.
Postherpetic neuralgiaPostherpetic neuralgia is defined as severe pain in thearea of distribution of a herpes zoster eruption, persist-ing more than 30 days after the onset of rash or aftercutaneous healing. In all, 10%–15% of patients withzoster develop postherpetic neuralgia, the risk factorsincluding age >50 years, immunodeficiency and pro-dromal sensory symptoms. The pain is usuallydescribed as continuous deep aching, burning, stabbingand shooting; allodynia is frequent. The condition isself-limiting, resolving within 2 months in half of thepatients, but may persist longer—up to years—withconsiderable impact on functional status andhealth-related quality of life. Although randomisedcontrolled trials give contradictory results on theeffectiveness of antiviral agents in preventing posther-petic neuralgia, we consider that patients at high riskshould receive these drugs during the acute phase of azoster eruption. Postherpetic neuralgia is usuallydifficult to treat and first line treatments includeantidepressants (tricyclics, serotonin-norepinephrinereuptake inhibitors), α2-δ ligands (gabapentin, prega-balin), opioids or their combination. Topical applica-tion of capsaicin or lidocaine patches may bringtemporary relief. High-concentration capsaicin patchesor invasive treatments may help in refractory cases.
Trigeminal neuralgiaTrigeminal neuralgia and persistent idiopathic facialpain are the commonest causes of facial pain (figure 6).Trigeminal neuralgia is a chronic NP condition,
affecting one or more branches of the trigeminalnerve. It is characterised by unilateral, sudden, shock-like and brief (fractions of a second to minutes)painful attacks, which follow the distribution of tri-geminal nerve branches, and with no other sensori-motor or autonomic signs and symptoms. Simple
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 303

activities (washing the face or teeth, eating, talking)and the stimulation of trigger zones may unleashattacks. Trigeminal neuralgia presents spontaneousremissions, even without treatment. Bilateral symp-toms suggest multiple sclerosis or other secondarycauses. MRI and neurophysiological investigationsmay exclude secondary trigeminal neuralgia (eg,lesions of the posterior fossa, multiple sclerosis).Carbamazepine and oxcarbazepine are first choicesfor trigeminal neuralgia and reduce symptoms innearly 70% of cases. α2-δ Ligands and baclofen aresecond line drugs. Invasive treatments (eg, micro-vascular decompression, Gasserian ganglion radiofre-quency) may help refractory trigeminal neuralgia.Persistent idiopathic facial pain persists throughout
the whole day, or most of it. It usually starts in a smallarea of the face (eg, the nasolabial fold), and then canspread to the jaw or larger areas of the face and neck,irrespective of nerve branches distribution. The pain isdeep and poorly localised, and there are no sensori-motor signs or symptoms. Dental procedures oftenprecede its onset. Secondary causes of facial painshould be ruled out. Radiological and neurophysio-logical investigations are normal. Persistent idiopathicfacial pain often responds to non-steroidal anti-inflammatory drugs (especially indometacin) and tri-cyclic antidepressants.
Low back painLow back pain has a lifetime prevalence of 70% andan important burden, especially when chronic. Thereis often an NP component, giving mixed pain, in25%–30% of patients.37 Various spinal osteoarticularstructures are responsible for nociceptive low back
pain. Frequent causes of neuropathic low back paininclude herniated disc with root compression, lumbaror foraminal stenosis, and scar tissue from previousspinal surgery.Separating nociceptive from neuropathic low back
pain is difficult. Radicular NP radiates to the lowerlimb according to the involved root (crural pain: L4,sciatic pain: L5, S1), but nociceptive low back painmay have pseudoradicular distribution because ofreferred pain phenomena.History taking, including a pain diagram and
bedside examination, is important in patients withlow back pain. Red flags (weight loss, fever, pain atrest and during the night, no relief after 6–8 weeks oftreatment, distal numbness and weakness, saddleanaesthesia, loss of bowel and bladder control)suggest a potentially serious cause and should alwaysbe sought. Yellow flags (poor compliance with exer-cise, poor work history, catastrophising, depression,feeling useless) are associated with poor prognosis.37
Standardised Evaluation of Pain is a screening toolwith high sensitivity and specificity for separatingneuropathic from nociceptive low back pain.19 Thestraight leg raising (Lasègue’s sign), the cross–straightleg raising and the femoral nerve stretch tests arehighly sensitive (88%–90%) but poorly specific(26%–29%) for root compression. Motor and sensorynegative signs and reduced reflexes suggest neuro-pathic low back pain.Spinal MRI and CT scan are often inconclusive in
patients with low back pain, with poor correlationbetween radiological and clinical features, but shouldbe performed in the presence of red flags.Electrodiagnostic tests and somatosensory evoked
Figure 6 Trigeminal neuralgia and persistent idiopathic facial pain. Clinical and treatment differences between the two mostfrequent causes of facial pain are presented here. NSAIDs, non-steroidal anti-inflammatory drugs; TCAs, tricyclic antidepressants.
REVIEW
304 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536
potentials are usually negative in patients with lowback pain who have a normal neurological examin-ation and no red flags.Treatment of neuropathic low back pain should adhere
to the NP treatment algorithm (figure 5). Nociceptive ormixed low back pain should be treated with non-steroidal anti-inflammatory drugs and analgesics.
Pain in patients with strokePain is a frequent complaint in chronic stroke, involv-ing up to half of the patients. The reasons for pain
after stroke include pre-existing pain conditions(30%–40%), shoulder pain (30%–40%), painful spas-ticity (7%–10%), headache (5%–10%) and centralpoststroke pain (6%–8%).38 Among these, onlycentral poststroke pain represents true NP, and shouldbe diagnosed if developing after a stroke in a bodyregion affected by the associated sensory abnormal-ities.4 There are proposed diagnostic criteria forcentral poststroke pain;38 applying them is straightfor-ward for some patients, particularly after strokesinvolving the lateral medulla, the ventroposterior
Figure 7 Diagnostic algorithm for pain in stroke patients. CPSP, central poststroke pain; GBP, gabapentin; LTG, lamotrigine; MCS,motor cortex stimulation; PGB, pregabalin; rTMS, repetitive transcranial magnetic stimulation; SNRIs, serotonin-norepinephrinereuptake inhibitors; TCAs, tricyclic antidepressants.
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 305

thalamus and the insula. However, diagnosing centralpoststroke pain may be a hard row to hoe in manystroke patients. The reasons for this difficulty includecognitive or language problems (making it difficult todemonstrate sensory signs), the presence of sensorydeficits not related to NP (not all stroke patients withsensory deficits have central poststroke pain) and thecoexistence of other types of pain.Pain descriptors and thermal allodynia may help but
are insufficient to make the diagnosis of central post-stroke pain; other causes of pain (ie, nociceptive painand peripheral NP) should be excluded (figure 7).Brain CT and MRI scans may show the stroke loca-tion. Quantitative sensory testing and laser evokedpotentials may to help document the sensory deficits,but are not routine.The treatment of central poststroke pain is disap-
pointing (figure 7). The few randomised controlledtrials on small populations of patients have given posi-tive results for oral tricyclic antidepressants (amitrip-tyline 75 mg), pregabalin (300–600 mg), lamotrigine(200 mg), and intravenous lidocaine and propofol.38
Lamotrigine titration may take up to 3 months toavoid severe cutaneous reactions. Serotonin-norepinephrine reuptake inhibitors, opioids and psy-chotherapy help in other types of NP and could beconsidered in central poststroke pain.38 Case seriesreport positive results for repetitive transcranial mag-netic stimulation of the motor cortex, motor cortexstimulation and deep brain stimulation of the thal-amus and the periacqueductal grey matter.
Pain in multiple sclerosisPain affects 57%–65% of multiple sclerosis patientsand includes NP (ongoing extremity pain, 12%–28%of patients; Lhermitte’s phenomenon, 15%; trigem-inal neuralgia, 2%–5%, often bilateral), musculoskel-etal pain (pain related to spasticity, <50%; painfultonic spasms, 6%–11%; back pain, 10%–16%), head-ache (21%–34%) and painful optic neuritis (8%).39 40
Extremity pain is continuous and burning, affects legsand feet bilaterally and usually worsens at night andduring physical activity. Lhermitte’s phenomenon isan electric shock sensation involving the neck, backand occasionally the limbs after neck flexion. Painfultonic spasms are stereotyped and short-lasting(<2 min), but may occur several times a day and betriggered by movement, sensory stimuli or emotions.MRI may show the location of demyelinating
lesions responsible for NP. Quantitative sensory testingand laser evoked potentials may document sensorydeficits, but are not largely diffused.There is debate over the treatment of multiple
sclerosis-related pain.39 Carbamazepine and oxcarba-zepine are the first line drugs for trigeminal neuralgia.Tricyclic antidepressants, α2-δ ligands and lamotriginemight help because they are useful in other types ofcentral NP. It would also be reasonable to use drugs
effective on peripheral NP (serotonin-norepinephrinereuptake inhibitors, tramadol, opioids). Despite somepositive randomised controlled trials, cannabinoidshave important side effects (psychosis, risk of addic-tion) and are not routinely recommended for NP orspasticity in multiple sclerosis. Botulinum toxin, intra-thecal baclofen and, to a lesser extent, four oral drugs(baclofen, dantrolene, diazepam, tizanidine) mayreduce spasticity and improve function, but theireffect on pain has not been specifically studied.
Key messages
▪ Neuropathic pain (NP) arises as a direct consequenceof a lesion or disease of the somatosensory system; itaffects 6%–8% of the general population and iscaused by a range of conditions, including peripheralneuropathy, radiculopathy, spinal cord injury, strokeand multiple sclerosis.
▪ A common feature of NP syndromes is the coexist-ence of negative and positive symptoms and signs,which reflect, respectively, loss-of-function andgain-of-function of the somatosensory system.
▪ As with other neurological diseases, there is agrading system to define different degrees of diagnos-tic certainty for NP.
▪ Although the diagnosis of NP has long been consid-ered difficult, validated screening tools and a thor-ough bedside examination remain important toidentify NP and to guide further investigations.
▪ In randomised clinical trials, the best medications forNP achieve satisfactory pain relief in only 30%–50%of patients; side effects are a common reason forwithdrawal of drugs.
Contributors FM designed the article, collected andinterpreted the data, drafted the manuscript andrevised it. GZ and ST designed the article, collectedand interpreted the data, and revised the manuscriptfor important intellectual content. All authorsapproved the final version of the article. FM and STtake full responsibility for the content of this review.
Competing interests FM received travel grants fromGrifols and Astellas for participation in scientificconferences and meetings. GZ received travel grantsfrom Grifols and Pfizer for participation in scientificconferences and meetings. ST received travel grantsfrom Grifols, Astellas and Pfizer for participation inscientific conferences and meetings. The Institution ofST received money from Pfizer for educationalactivities on neuropathic pain. The institution of STreceived EFIC Grünenthal Grant 2010.
Patient consent Obtained.
REVIEW
306 Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536

Provenance and peer review Commissioned; externallypeer reviewed. This paper was reviewed by LionelGinsberg, London, UK.
REFERENCES1 Torrance N, Smith BH, Bennett MI, et al. The epidemiology of
chronic pain of predominantly neuropathic origin. Results froma general population survey. J Pain 2006;7:281–9.
2 Bouhassira D, Lantéri-Minet M, Attal N, et al. Prevalence ofchronic pain with neuropathic characteristics in the generalpopulation. Pain 2008;136:380–7.
3 Merskey H, Bogduk N. Classification of chronic pain:descriptions of chronic pain syndromes and definitions of painterms. 2nd edn. Seattle, WA: IASP Press, 1994.
4 Treede RD, Jensen TS, Campbell JN, et al. Neuropathic pain:redefinition and a grading system for clinical and researchpurposes. Neurology 2008;70:1630–5.
5 Lynch ME, Clark AJ, Moulin DE, et al. Modifications aresuggested for the Special Interest Group (SIG) on NeuropathicPain proposed definition and guidelines for neuropathic pain.Pain 2011;152:1682; author reply 1683–4.
6 Baron R. Mechanisms of disease: neuropathic pain—a clinicalperspective. Nat Clin Pract Neurol 2006;2:95–106.
7 Rasmussen PV, Sindrup SH, Jensen TS, et al. Symptoms andsigns in patients with suspected neuropathic pain. Pain2004;110:461–9.
8 Lin CP, Kupper AE, Gammaitoni AR, et al. Frequency ofchronic pain descriptors: implications for assessment of painquality. Eur J Pain 2011;15:628–33.
9 Geber C, Magerl W, Fondel R, et al. Numbness in clinical andexperimental pain—a cross sectional study exploring themechanisms of reduced tactile function. Pain 2008;139:73–81.
10 Zanette G, Marani S, Tamburin S. Extra-median spread ofsensory symptoms in carpal tunnel syndrome suggests thepresence of pain-related mechanisms. Pain 2006;122:264–70.
11 Zanette G, Cacciatori C, Tamburin S. Central sensitization incarpal tunnel syndrome with extraterritorial spread of sensorysymptoms. Pain 2010;148:227–36.
12 Geber C, Baumgärtner U, Schwab R, et al. Revised definition ofneuropathic pain and its grading system: an open case seriesillustrating its use in clinical practice. Am J Med 2009;122:S3–12.
13 Bennett M. The LANSS Pain Scale: the Leeds assessment ofneuropathic symptoms and signs. Pain 2001;92:147–57.
14 Bennett MI, Smith BH, Torrance N, et al. The S-LANSS scorefor identifying pain of predominantly neuropathic origin:validation for use in clinical and postal research. J Pain2005;6:149–58.
15 Krause SJ, Backonja MM. Development of a neuropathic painquestionnaire. Clin J Pain 2003;19:306–14.
16 Bouhassira D, Attal N, Alchaar H, et al. Comparison of painsyndromes associated with nervous or somatic lesions anddevelopment of a new neuropathic pain diagnosticquestionnaire (DN4). Pain 2005;114:29–36.
17 Portenoy R. Development and testing of a neuropathic painscreening questionnaire: ID Pain. Curr Med Res Opin2006;22:1555–65.
18 Freynhagen R, Baron R, Gockel U, et al. painDETECT: a newscreening questionnaire to identify neuropathic components inpatients with back pain. Curr Med Res Opin 2006;22:1911–20.
19 Scholz J, Mannion RJ, Hord DE, et al. A novel tool for theassessment of pain: validation in low back pain. PLoS Med2009;6:e1000047.
20 Bouhassira D, Attal N. Diagnosis and assessment of neuropathicpain: the saga of clinical tools. Pain 2011;152:S74–83.
21 Bouhassira D, Attal N, Fermanian J, et al. Development andvalidation of the Neuropathic Pain Symptom Inventory. Pain2004;108:248–57.
22 Cruccu G, Sommer C, Anand P, et al. EFNS guidelines onneuropathic pain assessment: revised 2009. Eur J Neurol2010;17:1010–18.
23 Haanpää M, Attal N, Backonja M, et al. NeuPSIG guidelineson neuropathic pain assessment. Pain 2011;152:14–27.
24 Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria forsmall fibre neuropathy: from symptoms to neuropathology.Brain 2008;131:1912–25.
25 Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologicmanagement of neuropathic pain: evidence-basedrecommendations. Pain 2007;132:237–51.
26 Attal N, Cruccu G, Baron R, et al. EFNS guidelines on thepharmacological treatment of neuropathic pain: 2010 revision.Eur J Neurol 2010;17:1113–23, e67–e88.
27 Moulin DE, Clark AJ, Gilron I, et al. Pharmacologicalmanagement of chronic neuropathic pain—consensusstatement and guidelines from the Canadian Pain Society. PainRes Manag 2007;12:13–21.
28 Finnerup NB, Sindrup SH, Jensen TS. The evidence forpharmacological treatment of neuropathic pain. Pain2010;150:573–81.
29 National Institute for Health and Clinical Excellence.Neuropathic pain: the pharmacological management ofneuropathic pain in adults in non-specialist settings. London:National Institute for Health and Clinical Excellence, 2010.http://www.nice.org.uk/guidance/CG96 (accessed 16 Jan 2013).
30 Torrance N, Smith BH, Watson MC, et al. Medication andtreatment use in primary care patients with chronic pain ofpredominantly neuropathic origin. Fam Pract 2007;24:481–5.
31 O’Connor AB, Dworkin RH. Treatment of neuropathic pain:an overview of recent guidelines. Am J Med 2009;122:S22–32.
32 Tamburin S, Zanette G. Intravenous immunoglobulin for thetreatment of diabetic lumbosacral radiculoplexus neuropathy.Pain Med 2009;10:1476–80.
33 Gilron I, Bailey JM, Tu D, et al. Nortriptyline and gabapentin,alone and in combination for neuropathic pain: a double-blind,randomised controlled crossover trial. Lancet 2009;374:1252–61.
34 Cruccu G, Aziz TZ, Garcia-Larrea L, et al. EFNS guidelines onneurostimulation therapy for neuropathic pain. Eur J Neurol2007;14:952–70.
35 Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis,pathophysiological mechanisms, and treatment. Lancet Neurol2010;9:807–19.
36 Callaghan BC, Cheng HT, Stables CL, et al. Diabeticneuropathy: clinical manifestations and current treatments.Lancet Neurol 2012;11:521–34.
37 Freynhagen R, Baron R. The evaluation of neuropathiccomponents in low back pain. Curr Pain Headache Rep2009;13:185–90.
38 Klit H, Finnerup NB, Jensen TS. Central post-stroke pain:clinical characteristics, pathophysiology, and management.Lancet Neurol 2009;8:857–68.
39 O’Connor AB, Schwid SR, Herrmann DN, et al. Painassociated with multiple sclerosis: systematic review andproposed classification. Pain 2008;137:96–111.
40 Truini A, Barbanti P, Pozzilli C, et al. A mechanism-basedclassification of pain in multiple sclerosis. J Neurol2013;260:351–67.
REVIEW
Magrinelli F, et al. Pract Neurol 2013;13:292–307. doi:10.1136/practneurol-2013-000536 307


















