
Management von Risiken in der Lieferketten - dqs.de · Prozesses auf und integriert diese in ihr...
21
3 Armin F. Schwolgin / Egon H. Trump / Frank O. Bayer (Hrsg.) Management von Risiken in der Lieferketten April 2014 Duale Hochschule Baden-Württemberg Lörrach Studiengang BWL-Spedition, Transport und Logistik
Transcript of Management von Risiken in der Lieferketten - dqs.de · Prozesses auf und integriert diese in ihr...

3
Armin F. Schwolgin / Egon H. Trump / Frank O. Bayer (Hrsg.)
Management von Risiken in der Lieferketten
April 2014
Duale Hochschule Baden-Württemberg Lörrach Studiengang BWL-Spedition, Transport und Logistik

Inhaltsverzeichnis
1 Einleitung ........................................................................................................... 29
2 Ansatz der Parenteral Drug Association zum Risikomanagement .................... 30
2.1 Risk Assessment ........................................................................................ 33
2.2 Risk Control ................................................................................................. 38
2.3 Risk Review ................................................................................................. 41
2.3.1 Behandlung von Abweichungen ............................................................ 43
2.3.2 Beschwerdemanagement ..................................................................... 43
2.3.3 Audit ...................................................................................................... 44
2.3.4 Trendanalyse ........................................................................................ 44
2.4 Re-Assessment of Risk ............................................................................... 46
3 Ausblick ............................................................................................................. 46
Literaturverzeichnis .................................................................................................. 47
28

Anforderungen an ein Risikomanagement in der
Pharmalogistik
Dr. Nicola Spiggelkötter
Geschäftsführerin / Inhaberin
Knowledge & Support
1 Einleitung
Pharmalogistiker sind Grenzgänger, denn die Pharmalogistik ist in zwei Welten
beheimatet: in der Pharmaindustrie und in der Logistik. Sie wird geprägt durch die
Bedingungen und Anforderungen, aber auch Praktiken beider Branchen. Gerade auf
der Seite der Logistik ist das oft herausfordernd: der pharmazeutische Hersteller ist
seiner GMP-/GDP-Compliance verpflichtet und der Transportdienstleister hat diese
umzusetzen und zu erfüllen. Die Etablierung und Aufrechterhaltung eines Qualitäts-
risikomanagementsystems, kurz Risikomanagements, ist dabei nur ein Aspekt, wenn
auch ein sehr gewichtiger, den wir nachfolgend betrachten.
Risikomanagement wird in vielen Wirtschaftszweigen und Branchen betrieben, stellt
doch das Abwägen von Risiken eine grundlegende unternehmerische Tätigkeit dar
(1). Jeder Unternehmer wägt Chancen und Risiken ab, bevor er eine Investition
tätigt, ein neues Projekt beginnt, oder eine Kundenbeziehung eingeht. Der Begriff
des Risikomanagements fand seine erste Anwendung in der Finanz- und
Versicherungswirtschaft. Eine systematische Übertragung und Anwendung auf das
pharmazeutische Umfeld erfolgte im Jahr 2005 durch die International Conference on
Harmonisation (ICH) mit der Veröffentlichung der ICH Leitlinie Q9 zum Qualitäts-
risikomanagement. Diese Leitlinie wurde von der Europäischen Kommission im März
2008 als Anhang 20 in den EU-GMP-Leitfaden (Good Manufacturing Practices)
aufgenommen. Als Grundsätze eines pharmazeutischen Qualitätsrisikomanagements
wird im Anhang 20 des EG-Leitfadens zur guten Herstellpraxis folgendes formuliert:
„Die Bewertung der Qualitätsrisiken sollte auf wissenschaftlichen Erkenntnissen
beruhen und vor dem Hintergrund des Patientenschutzes gesehen werden und der
29

Grad des Aufwands, der Formalitäten und der Dokumentation des Verfahrens zum
Qualitäts-Risikomanagement sollte der Risikostufe angemessen sein.“ (2).
Die Anforderungen an ein Risikomanagement in der Pharmalogistik können auch klar
aus der revidierten EU Good Distribution Guideline (GDP) abgeleitet werden: „Quality
risk management should ensure that the evaluation of risk to quality is based on
scientific knowledge and experience with the process […].“ (3). Nachfolgend stellen
wir einen Ansatz der Parenteral Drug Association (PDA) vor, der diesen Anfor-
derungen gerecht wird (4). Die PDA greift dabei Kernelemente eines ICH Q 9 -
Prozesses auf und integriert diese in ihr Konzept (5).
2 Ansatz der Parenteral Drug Association zum
Risikomanagement
Allgemein wird anerkannt, dass ein Risiko als eine Kombination aus der
Eintrittswahrscheinlichkeit eines Schadens und dem Ausmaß des Schadens definiert
ist (siehe Abbildung 1). Das auf die Qualität von Arzneimitteln bezogene Risiko-
management hat das unumstößliche Ziel, einen substantiellen Beitrag zur
Arzneimittelsicherheit zu leisten, indem die Risiken von Arzneimitteln für den
Patienten verringert werden und zwar über die gesamte Supply Chain.
Abbildung 1: Bestimmungsgrößen der Risikoprioritätszahl (eigene Darstellung)
30

Als Beispiel für die Anwendung des geforderten risikobasierten Ansatzes dient uns
ein besonders kritisches Segment der Pharmalogistik - der Transport von
temperaturempfindlichen Arzneimitteln. Diese Auswahl erfolgt aus verschiedenen
Gründen: zum einen wird der Anteil an temperaturempfindlichen Arzneimitteln künftig
stark zunehmen, allein schon durch den wachsenden Anteil an Biosimilars und bio-
technologischen Produkten und zum anderen, weil gerade hier Abweichungen, ein
„Nicht-Einhalten“ von Temperaturkorridoren, fatale Konsequenzen haben kann. So
sind Insuline, die einmal gefroren waren, wirkungslos, was der Patient und Anwender
jedoch durch eine visuelle Kontrolle nicht feststellen kann. Hier ist es daher mehr als
angebracht, Risiken zu identifizieren, zu analysieren und zu bewerten, um diese letzt-
endlich zu verringern - somit eine fundierte Risikoanalyse im Rahmen des Qualitäts-
risikomanagements durchzuführen. Damit komplexe Prozesse wie Transportabläufe
analysiert werden können, erfolgt die Aufgliederung in vier Phasen:
Phase 1: Risk Assessment (Requirements)
Phase 2: Risk Control (Design, Qualification and Quality Management)
Phase 3: Risk Review (CAPA Management)
Phase 4: Re-Assessment of Risk (Change Control)
Abbildung 2: Qualitätsrisikomanagement und temperaturgeführte Logistik (eigene Darstellung)
Diese vier Phasen werden in dem Technical Report mit einem Risikomanagement-
prozess verknüpft (siehe Abbildung 2), der in Anlehnung an die ICH Q9 (Abbildung
3) erfolgt. Die grauen, oberen Felder skizzieren den ICH Q9 Prozess. Die Risiko-
beurteilung (risk assessment) findet hierbei in den 3 Stufen Risikoidentifizierung,
31

Risikoanalyse und Risikobeurteilung statt. Die Risikosteuerung (risk control) besteht
aus den Teilschritten Risikoreduzierung und Risikoakzeptanz. Darauf erfolgt die
Risikoüberwachung (risk review) und ggf. eine erneute Risikobeurteilung. Im
Folgenden werden nun die vier oben benannten Teilschritte näher erläutert.
Abbildung 3: Typischer Ablauf eines Risikoprozesses nach ICH Q9 (5)
32

2.1 Risk Assessment
In der Phase des Risk Assessments erfolgt die Ausarbeitung und Festlegung der
Eckdaten und Erfordernisse für den temperaturgeführten Versand. Aus den
individuellen Produktanforderungen mit Lager-/Transportbedingungen, Haltbarkeit
und vorliegenden Stabilitätsdaten ergeben sich die Grundanforderungen. Diese
legen unumstößlich die Eckpunkte für den Transport fest. Bei den Lager- und
Transportbedingungen hat sich eine Gleichsetzung der Lager- mit der Transport-
temperatur durchgesetzt, die man auf den Nenner bringen kann: wie gelagert, so zu
transportieren. Abweichungen von dieser Grundregel kann der pharmazeutische
Versender definieren und zwar auf Grundlage der ihm hierzu vorliegenden
Stabilitätsdaten.
Danach wird der Frage nach den regulatorischen Anforderungen nachgegangen:
Was wird von Seiten der Good Distribution Practice, der Good Importation Practice
und weiteren Sicherheitsauflagen wie Gefahrgutverordnung und/oder TAPA Freight
Security Requirements (FSR) eingefordert (6)? Was besagen hausinterne Standards
zum Versand? Welche nationalen regulatorischen Bestimmungen müssen bedacht
werden (im Land des Versenders und des Empfängers)? Sobald produktspezifische
sowie regulatorische Erfordernisse ermittelt worden sind, können die eigentlichen
Anforderungen an den Transport abgeleitet werden. Dabei gilt es, einen um-
fänglichen Fragenkatalog zu berücksichtigen. Exemplarisch sind wesentliche Leit-
fragen in Tabelle 1 aufgeführt.
Diese Phase der Datenerhebung ist wesentlich, da sie letztendlich auch den Umfang
und die Tiefe der nachfolgenden Schritte bestimmt. Dies entspricht dem Risk
Assessment im Rahmen eines Risikomanagements in Anlehnung an ICH Q9: “Risk
assessment consists of the identification of hazards and the analysis and evaluation
of risks associated with exposure to those hazards (as defined below).” (7).
Die Phasen der Risikoidentifizierung, Risikoanalyse und Risikobewertung lassen sich
durch die folgenden Schlüsselfragen abbilden:
Welche Gefahren oder Risiken könnten eintreten?
Wie hoch ist die Wahrscheinlichkeit, dass ein Schaden eintritt?
Was sind dann die Folgen davon?
Wie steht es um die Erkennbarkeit?
33

Leitfragen Checkpunkte
Welche Anforderungen stellt das Transportgut?
Konfektionierung & Versandvorbereitung
Versandkarton (Abmessung/Gewicht)
Palettierung (Abmessung/Gewicht)
Sondervorschriften für Stoss & Vibration
Feuchte / Temperatur
Welche Anforderungen stellt der Empfänger?
Haftungsfragen / Gefahrenübergang / INCOTERMS
Zeiten Warenannahme
Dokumentation / Nachweise / Informationsfluss
Warenannahmebeschränkungen (Auslastung Personal, Auslastung Laderampen)
Welche Anforderungen hat der Verlader zu erfüllen?
Transporttemperaturen (zu erwartende)
Transportdauer inkl. Versandvorbereitung, Verladung, Zwischenlagerung, Umschlag
Auswahl Transporteur (Qualität, Performance, Pharmaverständnis/-schulung)
Verlade-/Transportvolumen
Bedingungen Zwischenlagerung/Umschlag
Temperaturüberwachungssystem
Kommunikationsmanagement
Notfallmanagement
Technische Spezifikation Thermotruck (Qualifizierungsstatus, GPS, ATP)
Qualitätsvereinbarung
Servicevertrag
Bei passiven Thermoversandgebinden zusätzlich
• Spezifikation Thermogebinde
• „green logistics“
Tabelle 1: Leitfragen zur Erarbeitung eines Anforderungsprofils (eigene Darstellung)
34

Beim Transport von temperaturempfindlichen Produkten kommen beispielsweise die
folgenden Risiken ins Spiel:
Produktstabilität
Umgebungstemperatur während der Transporte (Sommer, Winter,
historische Daten wie zu erwartende Temperaturen)
Transportart (Luft, Land, See, Teilladung, Vollladung, Stückgut,
Kurierdienst)
Transportdauer
Primär-/Sekundär-/Umverpackung
Temperaturüberwachung
Qualität des Transportdienstleisters
Auswirkung von Produktschaden/-verlust und von Qualitätseinbußen
auf Patient
Diese Aufzählung ist keineswegs vollständig, weitere Risiken, die die Produkt-
temperatur und/oder die Produktintegrität betreffen, kommen hinzu. Unter letztere
fällt die Untermischung von mutmaßlich gefälschten Arzneimitteln in die legale
Vertriebskette, ein Gesichtspunkt, der immer mehr an Bedeutung gewinnt.
Die Diversität der Risiken kann nur durch einen systemischen Ansatz identifiziert,
analysiert und evaluiert werden. In den nationalen wie internationalen Regelwerken
ist hier keine besondere Vorgehensweise fest vorgeschrieben. Die einmal gewählte
Vorgehensweise muss allerdings verstanden, geschult und dokumentiert sein.
Durchgesetzt hat sich dabei weitgehend die Failure Mode and Effects Analysis
(FMEA) (8). Die FMEA ist eine Methode, bei der identifizierte Fehler und deren
potenzielle Ursachen quantitativ beurteilt werden hinsichtlich der Faktoren:
Eintrittswahrscheinlichkeit (A)
Bedeutung des Fehlers (B)
Entdeckungswahrscheinlichkeit (E)
Besonders beim Beschwerdemanagement erfolgt die Analyse der Beschwerden seit
geraumer Zeit durch FMEA-Prozesse, wobei die Root-Cause-Analyse im Vorder-
35

grund steht, was wir in unseren Ausführungen zu Phase 3 Risk Review und CAPA
vertiefend darstellen werden.
Bei der FMEA werden Risiken in Form von Zahlenwerten widergespiegelt, die sich
aus der Bewertung der genannten Bestimmungsgrößen ergeben (siehe Abbildung 1).
Die Multiplikation dieser drei Faktoren ergibt die Risikoprioritätszahl (RPZ).
Demgegenüber erfolgt beispielsweise bei der HACCP (Hazard Analysis and Critical
Control Points) die Bewertung des möglichen Risikos eher durch subjektive
Abschätzung, ob sich das Risiko unter Kontrolle befindet oder nicht und deshalb
eventuell zusätzliche Kontrollen und Maßnahmen notwendig werden. Bei der
Klassifizierung von Risiken gibt es unterschiedliche Ansätze, entweder ein
kompaktes System aus einer 3-stufigen Bewertung (tief, mittel, hoch) oder eine
Skalierung von 1 bis 10 oder höher. Je feingliedriger das angesetzte System ist,
umso schwieriger ist es allerdings oft auch, sich im Risikoteam auf eine Bewertung
zu einigen.
Die detaillierte Beschreibung der Transportroute bildet die Basis für die weiteren
Schritte. Dabei werden pro Segment die folgenden Teilschritte durchlaufen, wobei in
Teilschritten die FMEA-Methodik Anwendung findet:
1. Aufteilung der Transportrouten in Segmente und deren Beschreibung
2. Evaluierung der einzelnen Teilsegmente hinsichtlich: Schwere,
Eintrittswahrscheinlichkeit und Erkennbarkeit der Risiken
3. Durchführung der FMEA Analyse der Transporte unter worst case
Bedingungen
4. Berechnung der Risikoprioritätszahl (RPZ)
5. Ausarbeitung und Vorschlag von CAPA-Maßnahmen (korrigierende wie
präventive Maßnahmen)
6. Neuberechnung der Risikoprioritätszahl (RPZ)
Wir greifen exemplarisch den Prozessschritt Laderaumtemperatur bei aktiv tem-
perierten Fahrzeugen auf und veranschaulichen daran diese Schritte (Tabelle 2). Die
unter Punkt 14 berechnete Risikoprioritätszahl sollte unter der in Punkt 09
ausgewiesenen RPZ liegen, anderenfalls haben die getroffenen Maßnahmen, die zu
einer Risikoreduzierung führen sollten, nicht gegriffen und/oder wurden nicht richtig
umgesetzt und praktiziert.
36
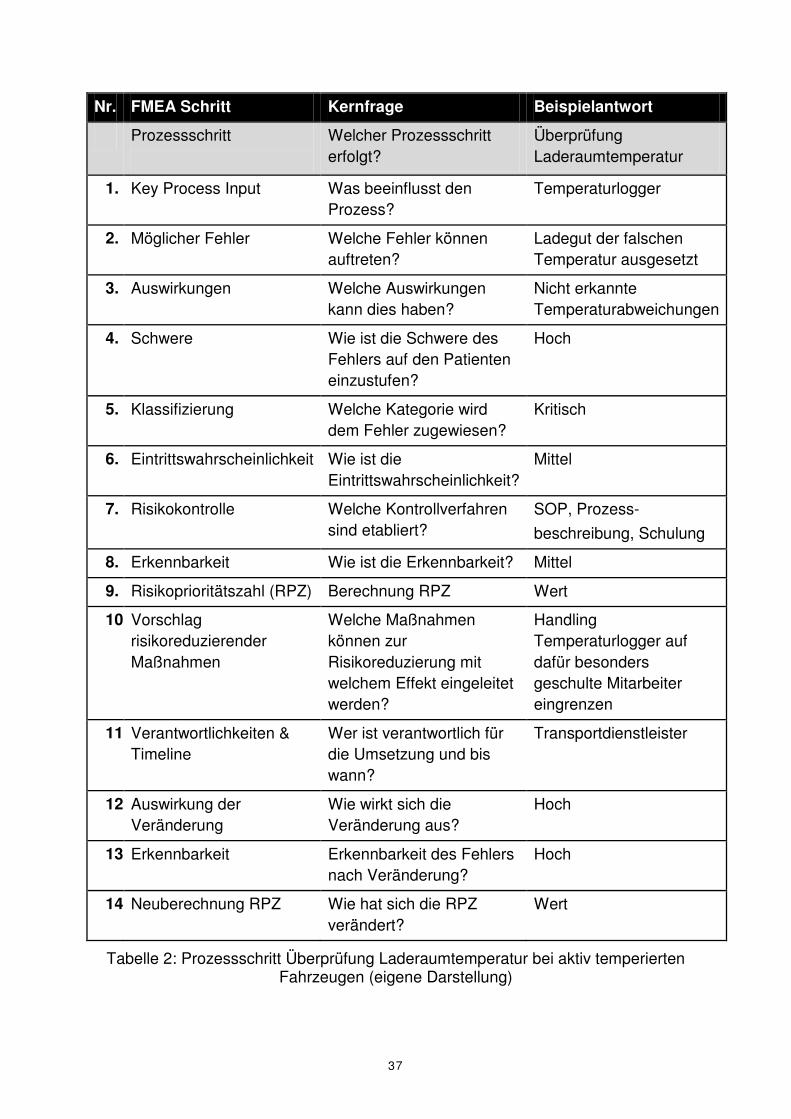
Nr. FMEA Schritt Kernfrage Beispielantwort
Prozessschritt
Welcher Prozessschritt erfolgt?
Überprüfung Laderaumtemperatur
1. Key Process Input Was beeinflusst den Prozess?
Temperaturlogger
2. Möglicher Fehler Welche Fehler können auftreten?
Ladegut der falschen Temperatur ausgesetzt
3. Auswirkungen Welche Auswirkungen kann dies haben?
Nicht erkannte Temperaturabweichungen
4. Schwere Wie ist die Schwere des Fehlers auf den Patienten einzustufen?
Hoch
5. Klassifizierung Welche Kategorie wird dem Fehler zugewiesen?
Kritisch
6. Eintrittswahrscheinlichkeit Wie ist die Eintrittswahrscheinlichkeit?
Mittel
7. Risikokontrolle Welche Kontrollverfahren sind etabliert?
SOP, Prozess-
beschreibung, Schulung
8. Erkennbarkeit Wie ist die Erkennbarkeit? Mittel
9. Risikoprioritätszahl (RPZ) Berechnung RPZ Wert
10 Vorschlag risikoreduzierender Maßnahmen
Welche Maßnahmen können zur Risikoreduzierung mit welchem Effekt eingeleitet werden?
Handling Temperaturlogger auf dafür besonders geschulte Mitarbeiter eingrenzen
11 Verantwortlichkeiten & Timeline
Wer ist verantwortlich für die Umsetzung und bis wann?
Transportdienstleister
12 Auswirkung der Veränderung
Wie wirkt sich die Veränderung aus?
Hoch
13 Erkennbarkeit Erkennbarkeit des Fehlers nach Veränderung?
Hoch
14 Neuberechnung RPZ Wie hat sich die RPZ verändert?
Wert
Tabelle 2: Prozessschritt Überprüfung Laderaumtemperatur bei aktiv temperierten Fahrzeugen (eigene Darstellung)
37

2.2 Risk Control
Nachdem die Exposition von potentiellen Risiken erfasst ist, stellt sich in Phase 2 die
Frage, wie diese kontrolliert bzw. gesteuert werden können (Risk Control). Die
Risikosteuerung kann grundsätzlich auf vier unterschiedlichen Weisen erfolgen:
1. Risikovermeidung (durch Tourenwahl, Wahl der Transportart, Wahl der
Ausrüstung)
2. Risikominimierung (durch Schulung, Personalauswahl, Auswahl des
Transportdienstleisters)
3. Risikoakzeptanz (sollte allerdings nur für „Restrisiken Gültigkeit haben,
wie Naturgewalten und Aschewolken)
4. Risiken bzw. die Verantwortung darüber weitergeben (Beispiel:
vertragliche Weitergabe an einen Dienstleister, Temperaturdaten
auswerten)
Abzuwägen gilt es dabei stets zwischen den Kosten/Folgen, die durch den Verlust
des Produktes entstehen und den Kosten, die für eine Implementierung und
Aufrechterhaltung eines Qualitätssicherungssystems anfallen.
Risikosteuerung im Rahmen des ICH Q9-Ansatzes zielt auf die Reduzierung der
Risiken auf ein akzeptables Niveau ab. Auch an diesem Punkt spielt die Ange-
messenheit der Mittel eine tragende Rolle: “Risk control includes decision making to
reduce and/or accept risks. The purpose of risk control is to reduce the risk to an
acceptable level. The amount of effort used for risk control should be proportional to
the significance of the risk.” (9).
Das Design und die anschließende Qualifizierung von Einrichtungen (Lägern), sowie
Gegenständen (Fahrzeugen, Versandgebinden) sind neben dem Qualitätssiche-
rungssystem die zentralen Tools der Risikosteuerung. Ein Blick auf die
internationalen Guidelines zu den guten Vertriebspraktiken (GDP) zeigt ganz klar: der
Einsatz von qualifizierten Fahrzeugen ist darin fest verankert. Die Qualifizierung auch
von aktiven Großsystemen ist ebenfalls eine zentrale Anforderung der Arzneimittel-
und Wirkstoffherstellungsverordnung (AMWHV) §7.5: „Die Verfahren für die
38

Lagerung und den Transport sind schriftlich festzulegen. Soweit sie einen Einfluss
auf die Qualität der Ausgangsstoffe und Zwischenprodukte für die Arzneimittel-
herstellung oder für die Arzneimittel haben können, ist die Geeignetheit der
Verfahren nachzuweisen.“ (10).
Diese „Geeignetheit der Verfahren“ bedeutet im Pharmaumfeld nichts anderes als
eine Qualifizierung. Insofern sind Qualifizierungen und Risikokontrolle eng
miteinander verbunden. Die Forderung nach einer Qualifizierung erstreckt sich nicht
ausschließlich darauf, Einrichtungen und Gegenstände einzusetzen, sondern auch
auf Lieferanten und Dienstleister. Da Transportdienstleistungen im Pharmabereich
weitgehend an externe Dienstleister vergeben werden, ist hierbei eine detaillierte
Abstimmung zwischen Auftraggeber und Auftragnehmer erforderlich. Die Quali-
fizierung von Transportdienstleistern ist ein wesentlicher Punkt davon.
Als erste Orientierung zur Qualifizierung von Transporten dienen die in Tabelle 3
formulierten Leitfragen. Durch ein angemessenes Design und die Qualifizierung der
eingesetzten Läger, Thermoverpackungen, Temperaturüberwachung und Transport-
routen, aber auch deren Dienstleister und Transportdienstleister, kann die
Wahrscheinlichkeit von Transportschäden verringert werden. Dies kann allerdings
nur dann greifen, wenn die fraglichen Prozesse in ein umfassendes Qualitäts-
managementsystem eingebettet sind. Das Qualitätsmanagement und seine
Ausgestaltung ist ebenso eine tragende Säule der Risikokontrolle. Kernpunkte eines
so konzipierten Qualitätssicherungssystems sind die Beschreibung aller Prozesse in
Arbeitsanweisungen (Standard Operation Procedures) und deren Schulung,
Kontrolle und Evaluierung durch Schlüsselkennzahlen (Key Performance Indicators).
Die in Tabelle 4 aufgeführten Punkte könnten gleichfalls bei der Lieferanten-
qualifizierung eingesetzt werden, denn auch diese ist Teil der Risikosteuerung.
39

Leitfragen Checkpunkte
Lager / Depot
Abmessungen Lager / Fassungsvermögen
Ausstattung (Hochregale, Kühlzellen, BTM Lager)
Qualifizierungsstatus (Sommer/Winter)
Volllast
Access Control / Pest Control/ Notfallpläne
Temperaturüberwachung/ Alarmierung
Dediziertes Pharmalager
Transportumgebung
(aktive oder passive Systeme)
Spezifikation des eingesetzten Systems
Maximale Beladung (Beladungsgrenzen)
Qualifizierungsstatus (Temperaturprofile)
Temperaturmonitoring (Intervall, Anzahl Temperaturfühler)
Temperaturüberwachung
Temperaturmonitoringstrategie
Kalibrierungsstatus
Temperaturüberwachung Einstellungen (Intervall, Grenzwerte, Alarmgebung)
Positionierung Temperaturfühler
United States Pharmacopeia (USP) 1118 Compliance
Datenbanken
Warenwirtschaftssystem/ Lagerverwaltungsprogramm
Übersicht Temperaturüberwachungssysteme und Temperaturdaten
Daten Temperaturabweichungen / Abweichungen
Zugriffsrechte / Benutzerprofile
Transportrouten
Destinationen/ Umschläge
Transportart (Kühllaster, Reefer, Envirotainer)
Saisonale Aspekte (Winter-/Sommer-Konfigurationen)
Schutzverpackungen Transportgut
Transportdauer
Auditierung / Qualifizierung Transportdienstleister
Qualitätsvereinbarung Transportdienstleister
Servicevereinbarung Kunde (inkl. INCOTERMS)
Tabelle 3: Leitfragen Phase 2 Risk Control (eigene Darstellung)
40

Schwerpunkt Evaluierungspunkte
SOP
Abgrenzung der Verantwortlichkeiten und Aufgaben (Stellenbeschreibungen)
Warenannahme (Prüfpunkte, Prioritäten, Zurückweisungen)
Lagerung
Datenfluss /-verwaltung
Versandvorbereitung (pick & pack, Sommer-/Winterkonfiguration, Vortemperierung)
Umgang mit der Ware während des Transportes
Verhalten bei Abweichungen (CAPA)
Change Control
Versanddokumentation /-papiere
Schulungen Fahrerschulungen
Versandmitarbeiter
Disponenten und weitere am Prozess Beteiligte
Wartung
Kalibrierung der Messsysteme
Präventive Wartung der Gegenstände
Requalifizierungen
Management der Wartungen (Zeitinterval, Ergebnisse, Nachverfolgung der Reparaturen)
Key Performance Indicators
Temperaturüber-/unterschreitungen
Zurückgewiesene Ware
Fehlerhafte Exportdokumentation
Lieferpünktlichkeit
Beschwerden
Tabelle 4: Checkpunkte Qualitätsmanagement eines Transportdienstleisters (eigene Darstellung)
2.3 Risk Review
In der Phase 3 erfolgt die Überprüfung der eingeleiteten Maßnahmen. Dieser
Prozess wird folgendermaßen definiert: “Review or monitoring of output / results of
the risk management process considering (if appropriate) new knowledge and
experience about the risk.” (11).
41

Eine regelmäßige Überprüfung der Prozesse, Annahmen und Qualifizierungen ist
eine Voraussetzung für ein greifendes Qualitätsrisikomanagement. Dieser Review-
prozess kann anlassbezogen eingeleitet werden, bei Transportvorfällen, Kunden-
beschwerden, festgestellten Abweichungen bei internen wie externen Audits sowie
bei Ergebnissen von Trendanalysen. Gerade aus realen Vorkommnissen im Rahmen
von Transporten können wichtige Erkenntnisse für die Anpassung der Prozesse und
der Implementierung von erforderlichen Präventivmaßnahmen generiert werden.
Hierin findet dann das grundlegende Prinzip der ICH Q9 aus Daten und
Informationen, Wissen und Erkenntnis zu generieren, seine erneute Bestätigung.
Diese Daten können reaktiv und proaktiv generiert werden. Tabelle 5 zeigt mögliche
Informationsquellen.
Proaktiv Evaluierungspunkte
• Key Performance Indicators
Benchmarking
Trendanalysen
Realdaten (Temperatur, GPS)
Reaktiv Evaluierungspunkte
• Auditberichte /Auditabweichungen / Management Reviews
Abweichungen
Beschwerden
CAPA-Maßnahmen
Risikoprioritätszahl (ursprüngliche)
Risikoprioritätszahl (revidierte nach Abschluss der eingeleiteten CAPA-Maßnahmen)
Transportvorfälle (inkl. Near Miss Events)
Tabelle 5: Informationsquellen (eigene Darstellung)
Ein CAPA-System (Corrective And Preventive Actions) ist längst integraler
Bestandteil eines jeden Qualitätssicherungssystems; dies gilt insbesondere für die
stark regulierte Pharmaindustrie (12). Der Fokus von CAPA liegt auf der syste-
matischen Untersuchung von Diskrepanzen (z.B. Ausfall und/oder Abweichungen)
und auf dem Versuch, das wiederholte Auftreten zu verhindern (corrective action),
oder das Auftreten bereits im Vorfeld zu vermeiden (preventive action). Zielführend
42

erweist sich dabei, die erforderlichen CAPA-Maßnahmen strukturiert vier
Themenkomplexen zu zuordnen:
Behandlung von Abweichungen
Beschwerdemanagement
Ergebnisse von internen und externen Audits und
Trendanalysen
Ein CAPA-Verfahren ist einzuleiten, wenn Abweichungen festgestellt wurden,
Beschwerden eingegangen sind, Audits Abweichungen von den Sollvorgaben
aufgedeckt haben, oder Rückrufe eingeleitet werden mussten, um nur einige
exemplarisch anzuführen. Nun muss die Frage beantwortet werden, was exakt
darunter zu verstehen ist.
2.3.1 Behandlung von Abweichungen
Der Umgang mit Abweichungen erfolgt nach der klaren Ermittlung der Gründe, die
hierzu geführt haben. Diese Root-Cause-Analysis bringt immer wieder die Gefahr
von Oberflächlichkeit mit sich. Bei so manchen diesbezüglichen Analysen verbleibt
man leider an der Oberfläche oder erfasst eben nicht alle Faktoren und Umstände,
die für die Ermittlung einer Abweichung herangezogen werden sollten. Um dieser
Tendenz entgegen zu wirken, erweist es sich als hilfreich, auf systemische Tools der
Risikoanalyse zurückzugreifen (Fishbone, Fault Tree, FMEA). Der Einsatz entsprech-
ender Analysetools ist unverzichtbar, um der Anforderung nach Nachvollziehbarkeit
gerecht zu werden. Dies sind wichtige Aspekte, die von behördlichen aber auch
Kundeninspektionen eingefordert werden. Inspektionssicherheit in Bezug auf
Risikoanalysen bedeutet auch, dass die Risikoanalysen für Außenstehende
nachvollziehbar sein müssen.
2.3.2 Beschwerdemanagement
In der Behandlung von Beschwerden wurden CAPA-Verfahren zuerst eingesetzt. Die
Abarbeitung der unterschiedlichen Prozessschritte erfolgt dabei meist auf Basis eines
Flussdiagrammes (siehe Abbildung 4). Im Beschwerdemanagement wird beispiels-
weise unterschieden nach Beschwerden aufgrund von Temperaturabweichungen,
43

nicht termingerechter Belieferung, Lieferung in nicht abgemessener Anzahl,
Lieferung in nicht entsprechender Qualität (qualitätsbezogene Reklamationen), Bruch
der Ware, aber auch mutmaßlich gefälschter Ware. All diese Beschwerdekategorien
verlangen nach unterschiedlichen Vorgehensweisen. Dabei ist ein entsprechender
Entscheidungsbaum für die Vorgehensweise und Abarbeitung, aber auch für die
Nachweiserbringungspflicht sowie Dokumentation hilfreich.
2.3.3 Audit
Die Abarbeitung von Inspektionsmängeln erfolgt ebenso nach CAPA-Vorgaben. Dies
ist nicht nur im Pharmabereich übliche Praxis, auch bei IFS (International Food
Standard) Food Logistik Inspektionen oder ISO Selbstinspektionen gibt CAPA die
Struktur vor, insbesondere für die Überwachung der terminnahen Beseitigung der
entdeckten Mängel.
2.3.4 Trendanalyse
Trendanalysen geben wichtige Informationen über zentrale Leistungsparameter.
Diese werden nicht nur kontinuierlich erhoben, sondern werden auch hinsichtlich
ihrer Entwicklung ausgewertet. Das ist ein zentrales Tool zur Vermeidung von
Abweichungen und Fehlern. Hierbei geht man beispielsweise der folgenden Frage
nach: Wie wirken sich die Jahreszeiten auf die Häufigkeit und die Schwere von
Temperaturabweichungen aus? Sind im Winter mehr Vorfälle mit Temperatur-
unterschreitungen zu verzeichnen. Sollte dies der Fall sein, wäre beispielsweise die
Winterkonfiguration der passiven Thermogebinde zu verändern (Menge Kühlenergie
und/oder Konditionierung der Kühlelemente) oder bei Kühlfahrzeugen müsste die
Einstellung der Kühlmaschine verändert werden (Setpoint).
44
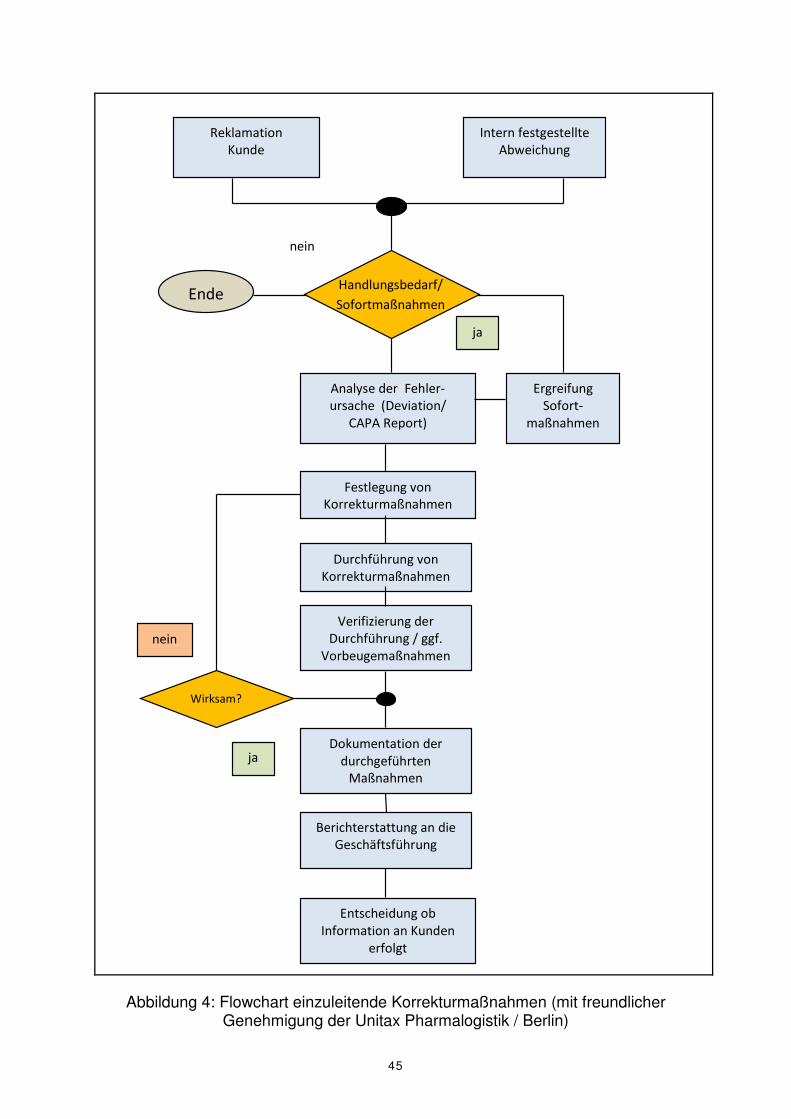
Intern festgestellte
Abweichung
Reklamation
Kunde
nein
ja
Ende
Analyse der Fehler-
ursache (Deviation/
CAPA Report)
Ergreifung
Sofort-
maßnahmen
Dokumentation der
durchgeführten
Maßnahmen
Berichterstattung an die
Geschäftsführung
Festlegung von
Korrekturmaßnahmen
Durchführung von
Korrekturmaßnahmen
Verifizierung der
Durchführung / ggf.
Vorbeugemaßnahmen
nein
ja
Wirksam?
Entscheidung ob
Information an Kunden
erfolgt
Handlungsbedarf/
Sofortmaßnahmen
Abbildung 4: Flowchart einzuleitende Korrekturmaßnahmen (mit freundlicher Genehmigung der Unitax Pharmalogistik / Berlin)
45

2.4 Re-Assessment of Risk
Mit dem letzten und vierten Schritt Re-Assessment of Risk (Change Control) bzw.
Änderungsmanagement wird die abschließende Risk Assessment Phase eingeleitet.
Änderungen, die sich aus Schritt 3 ergaben, werden hier realisiert im Rahmen eines
Änderungsmanagements. Aber zuerst bedarf es einer genaueren Abgrenzung des
Begriffes Change Control. Allgemein versteht man darunter den Prozess, der das
Einführen von Änderungen steuert. Darunter ist aber ein formaler Prozess zu
verstehen, der im GxP-Umfeld dafür sorgt, dass alle Änderungen an Prozessen und
Gegenständen sowie Einrichtungen in der gesamten Prozesslandschaft des
Qualitätsmanagementsystems kommuniziert und umgesetzt werden. Letztendlich
haben alle GxP-relevanten Änderungen aufgrund eines formalen Freigabeprozesses
der zuständigen Quality Unit zu erfolgen. Auf logistische Belange übertragen wäre
beispielsweise der Einsatz eines anderen Transportdienstleisters eine GxP-relevante
Änderung. Der neue Dienstleister kann erst eingesetzt werden, wenn er den Status
„freigegebener“ Lieferant/Dienstleister erlangt hat, also nach einer erfolgreich
durchlaufenen Lieferantenqualifizierung.
3 Ausblick
Eine Risikoanalyse alleine ist noch kein funktionierendes Qualitätsrisikomanagement-
System. Zu einem Risikomanagement-Prozess wird die Risikoanalyse erst, wenn sie
mit den weiteren Elementen wie der Risikokommunikation verbunden wird. Die
Risikokommunikation findet periodisch oder aperiodisch, meist Anlass bezogen, statt.
Bei dem Umgang mit Risiken, deren Identifizierung, Analyse und Evaluierung, darf
der Einfluss von psychologischen Effekten nicht außer Acht gelassen werden:
Verdrängen und Ausblenden von Risiken. Dieses Verhalten tritt besonders dann auf,
wenn die Gefahr und die hiermit assoziierten Risiken über einen längeren Zeitraum
nicht in Form des auftretenden Negativereignisses wahrgenommen werden konnten.
Weiterhin wird vergessen, dass das Nichtauftreten eines Ereignisses auch ein
Ergebnis der statistischen Verteilung sein kann.
46

Literaturverzeichnis
(1) Vgl. dazu den Beitrag von Schwolgin in diesem Band
(2) O.V.: Anlage 3 zur Bekanntmachung des Bundesministeriums für Gesundheit zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherstellungsverord-nung – AMWHV – vom 18. Juli 2008 (BAnz. S. 2798, Anhang 20 zum EG-Leitfaden der Guten Herstellungspraxis, Qualitäts-Risikomanagement, S. 4
(3) Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use, 2013/C 343/01, §1.5 Quality Risk Management, S.3
(4) Vgl. o.V.: Parenteral Drug Association, PDA, Technical Report 58, Risk Management for Temperature Controlled Distribution, September 2012
(5) Vgl. o.V.: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Harmonised Tripartite Quality Guideline, Quality Risk Management (Q9), Current Step 4 version dated 9 November 2005
(6) Vgl. dazu o.V.: Transported Asset and Protection Association: Standards, zitiert nach: http://www.tapaonline.org/standards
(7) O.V.: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Harmonised Tripartite Quality Guideline, Quality Risk Management (Q9), Current Step 4 version dated 9 November 2005, S. 2
(8) Vgl. dazu z.B. Eberhardt, O.: Risikobeurteilung mit FMEA, Renningen 2012 sowie Mathe, R.: FMEA für das Supply Chain Management. Prozessrisiken frühzeitig erkennen und wirksam vermeiden mit Matrix-FMEA, Düsseldorf 2012
(9) O.V.: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Harmonised Tripartite Quality Guideline, Quality Risk Management (Q9), Current Step 4 version dated 9 November 2005, S. 4
(10) O.V.: Verordnung über die Anwendung der Guten Herstellungspraxis bei der Herstellung von Arzneimitteln und Wirkstoffen und über die Anwendung der Guten fachlichen Praxis bei der Herstellung von Produkten menschlicher Herkunft (Arzneimittel- und Wirkstoffherstellungsverordnung - AMWHV), § 7.5, S. 8
(11)
O.V.: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Harmonised Tripartite Quality Guideline, Quality Risk Management (Q9), Current Step 4 version dated 9 November 2005, S. 9
(12) Vgl. dazu Rodriguez-Pérez, J.: CAPA for the FDA-Regulated Industry, Milwaukee 2011
47


















