
Maximale Flüssigkeitsaufnahmekapazität von dispersen ... · η Dynamische Viskosität γ...
167
Lehrstuhl für Maschinen- und Apparatekunde Technische Universität München Maximale Flüssigkeitsaufnahmekapazität von dispersen Trägerstoffen beim Beladen und Tränken Heidi Lankes Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktor-Ingenieurs (Dr.-Ing.) genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr.-Ing. Horst Weisser Prüfer der Dissertation: 1. Univ.-Prof. Dr.-Ing. Karl Sommer 2. Univ.-Prof. Dr. rer. nat., Dr. agr. habil. Harun Parlar Die Dissertation wurde am 03.05.2002 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 10.06.2002 angenommen.
Transcript of Maximale Flüssigkeitsaufnahmekapazität von dispersen ... · η Dynamische Viskosität γ...

Lehrstuhl für Maschinen- und Apparatekunde
Technische Universität München
Maximale Flüssigkeitsaufnahmekapazität von dispersen
Trägerstoffen beim Beladen und Tränken
Heidi Lankes
Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan
für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur
Erlangung des akademischen Grades eines
Doktor-Ingenieurs
(Dr.-Ing.)
genehmigten Dissertation.
Vorsitzender: Univ.-Prof. Dr.-Ing. Horst Weisser
Prüfer der Dissertation: 1. Univ.-Prof. Dr.-Ing. Karl Sommer
2. Univ.-Prof. Dr. rer. nat., Dr. agr. habil. Harun Parlar
Die Dissertation wurde am 03.05.2002 bei der Technischen Universität München
eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für
Ernährung, Landnutzung und Umwelt am 10.06.2002 angenommen.

Die vorliegende Arbeit wurde in der Zeit von Mai 1999 bis April 2002 an der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München am Lehrstuhl für Maschinen- und Apparatekunde in Zusammenarbeit mit dem Adalbert-Raps-Zentrum für Arznei- und Gewürzpflanzen-forschung durchgeführt. Herrn Prof. K. Sommer, dem Lehrstuhlinhaber, möchte ich für die Möglichkeit danken, eine Doktorarbeit an seinem Institut durchführen zu können. Auch dem zweiten Prüfer Herrn Prof. H. Parlar und dem Prüfungsvorsitzenden Herrn Prof. H. Weisser danke ich für die Übernahme des jeweiligen Amtes. Während der Zeit am Lehrstuhl für Maschinen- und Apparatekunde, an dem ich schon während des Studiums als Hilfswissenschaftlerin, dann als Diplomandin und letztendlich als Doktorandin tätig war, habe ich Dank der vielseitigen Interessen von Herrn Prof. K. Sommer, nicht nur im Bereich der mechanischen Verfahrenstechnik, sondern auch in vielen anderen Gebieten mein Wissen bereichern können. Dafür, dass Herr Prof. K. Sommer sein Möglichstes gibt, uns Wissenschaftlern nicht zu „Fachidioten“, sondern viel mehr zu offenen, kritischen, sozial kompetenten und gut ausgebildeten Menschen zu erziehen, die bereit sind, Verantwortung in vielen Bereichen des Lebens zu übernehmen, möchte ich mich ganz besonders bei ihm bedanken. Auch allen anderen Mitarbeitern des Lehrstuhles gilt mein besonderer Dank. Das hervorragende Arbeitsklima sowie die vorhandene Hilfsbereitschaft und Fach-kompetenz, sowohl unter den Wissenschaftlern, als auch unter allen anderen Mitarbeitern und in der Werkstatt, ermöglichten mir ein angenehmes und effektives Arbeiten am Institut. Herr S. Palzer hat es mir während meines Studiums ermöglicht, als Hilfswissen-schaftlerin bei ihm zu arbeiten. So bin ich durch ihn an den Lehrstuhl für Maschinen- und Apparatekunde gekommen und von ihm habe ich sehr viel gelernt, was das wissenschaftliche Arbeiten betrifft. Dafür möchte ich mich an dieser Stelle bedanken. Dank Herrn V. Kehlenbeck sind viele Fehler aus meinen Veröffentlichungen, Vorträgen und auch aus der vorliegenden Arbeit verschwunden, da er wissen-schaftliche Texte mit einer Geduld und einer Genauigkeit zu korrigieren und diskutieren vermag, für die ich mich an dieser Stelle noch einmal bedanken möchte. Auch Herr G. Niederreiter hat entscheidend zum Gelingen dieser Arbeit beigetragen. Nicht nur durch die zahlreichen fachlichen Diskussionen und durch die Hilfe mit Mathcad, sondern auch durch die positive Stimmung, für die er als Bürokollege fast immer sorgte.

Bei der Adalbert-Raps-Stiftung möchte ich mich für die Finanzierung des Forschungsvorhabens bedanken. Ohne die Zusammenarbeit mit dem Adalbert-Raps-Zentrum, wäre es nicht möglich gewesen, diese Arbeit nach drei Jahren erfolgreich zu beenden. Namentlich erwähnen möchte ich in diesem Zusammenhang Herrn B. Weinreich, Frau S. Grüner und Herrn F. Otto. Zu Dank verpflichtet bin ich weiterhin den vielen studentischen Mitarbeitern, die einen wesentlichen Beitrag zu der vorliegenden Arbeit geleistet haben. Zu danken habe ich allen, die als Hilfswissenschaftler für mich gearbeitet oder eine Semesterarbeit bei mir angefertigt haben. Allen voran möchte ich mich bei den Diplomanden Herrn M. Päs, Frau D. Herold und Herrn M. Leutner für ihre hervorragende geleistete Arbeit bedanken. In diesem Zusammenhang möchte ich auch noch Herrn J. Götz meinen Dank aussprechen, für die ausgezeichnete Zusammenarbeit im Bereich der NMR-Messungen, die alle unter seiner Anleitung durchgeführt wurden und deren Ergebnisse wir stets gemeinsam diskutiert und weiterverarbeitet haben. Zu guter Letzt möchte ich meinem Freund A. Förg danken, der mich nicht nur privat sondern auch fachlich in allen erdenklichen Lagen zu unterstützen wusste. Ohne dessen Hilfe wäre vieles in meinem Leben nicht so gut gelungen. Auch meine Schwester S. Lankes, deren Freund M. Weinzierl, meine Eltern und meine Freunde haben viel dazu beigetragen, dass ich die Zeit als Studentin und Doktorandin in Weihenstephan für immer in sehr guter Erinnerung behalten werde.

Inhaltsverzeichnis I
Inhal tsverzeichnis Formelzeichen und Abkürzungen IV
1 Einleitung...................................................................................................... 1 1.1 Vorwort........................................................................................................... 1
1.2 Problemstellung ............................................................................................. 1
2 Theoretische Grundlagen und Kenntnisstand .......................................... 3 2.1 Partikelherstellung.......................................................................................... 3
2.2 Partikelcharakterisierung................................................................................ 6
2.2.1 Partikelgröße und Partikelform....................................................................... 6
2.2.2 Oberfläche und Porosität................................................................................ 8
2.2.3 Feststoffdichte, Schüttdichte und Stampfdichte ........................................... 15
2.3 Eigenschaften von Flüssigkeiten.................................................................. 17
2.3.1 Viskosität...................................................................................................... 17
2.3.2 Oberflächenspannung.................................................................................. 18
2.3.3 Eigenschaften von überkritischem CO2........................................................ 19
2.4 Wechselwirkungen zwischen Flüssigkeiten und Partikeln............................ 20
2.4.1 Benetzung und Kapillarität ........................................................................... 20
2.4.2 Bindungsmechanismen zwischen Partikeln in einem System mit
Flüssigkeit .................................................................................................... 23
2.4.3 Flüssigkeitsverteilung in einem dispersen Feststoffsystem.......................... 26
2.4.4 NMR-Messung zur Bestimmung der Beweglichkeit und der
Bindungszustände von Wasser an Feststoffpartikeln................................... 31
2.5 Verfahren zum Herstellen von flüssigkeitsbeladenen Pulvern...................... 36
2.5.1 Wurfmischer mit Eindüsvorrichtung.............................................................. 38
2.5.2 CPF-Sprühverfahren .................................................................................... 39
3 Material und Methoden .............................................................................. 43 3.1 Messmethoden............................................................................................. 43
3.1.1 Messmethoden zur Partikelcharakterisierung .............................................. 43
3.1.2 Messmethoden zur Produktbeurteilung........................................................ 45
3.2 Rohstoffe...................................................................................................... 47
3.2.1 Trägerstoffe.................................................................................................. 47
3.2.2 Flüssigkeiten ................................................................................................ 63
3.3 Herstellung der Modellpartikeln.................................................................... 64

Inhaltsverzeichnis II
3.3.1 Eingesetzte Materialien zur Herstellung von Modellpartikeln ....................... 64
3.3.2 JetCutter zur Erzeugung von Modellpartikeln............................................... 66
3.4 Verfahren zum Beladen der Trägerstoffe ..................................................... 68
3.4.1 CPF-Sprühverfahren .................................................................................... 68
3.4.2 Pflugscharmischer mit Eindüsvorrichtung .................................................... 71
4 Ergebnisse und Diskussion ...................................................................... 73 4.1 Bestimmung der maximalen Aufnahmekapazität ......................................... 74
4.1.1 Optische Beurteilung der beladenen Pulver ................................................. 74
4.1.2 Veränderung der Partikelgrößenverteilung mit zunehmender
Beladung...................................................................................................... 75
4.1.3 Veränderung von Schütt- und Stampfdichte mit zunehmender
Beladung...................................................................................................... 77
4.1.4 Bestimmung des Agglomeratanteils............................................................. 81
4.1.5 Bestimmung der freien Flüssigkeit ............................................................... 82
4.2 Einfluss des Trägerstoffes auf die maximale
Flüssigkeitsaufnahmekapazität .................................................................... 84
4.2.1 Maximale Beladungskapazität verschiedener Trägerstoffe .......................... 84
4.2.2 Vergleich verschiedener Trägerstoffe hinsichtlich Oberfläche, DVS-
Messungen und maximal möglicher Beladung............................................. 89
4.2.3 Einfluss der Stampfdichte auf die maximale Aufnahmekapazität ................. 96
4.2.4 Modellpartikeln aus dem Strahlschneiderverfahren ................................... 100
4.2.5 Veränderung von Trägerstoffpartikeln, um die maximale
Aufnahmekapazität zu erhöhen.................................................................. 112
4.3 Einfluss der Flüssigkeit auf die maximale Flüssigkeitsauf-
nahmekapazität.......................................................................................... 117
4.3.1 Kieselsäure Aerosil R972 mit Speiseöl, Triacetin und Wasser................... 117
4.3.2 Superabsorber HySorb mit Speiseöl und Wasser ...................................... 121
4.3.3 Maltodextrin Paselli MD6 mit Speiseöl und Triacetin ................................ 122
4.4 Einfluss des Verfahrens auf die maximale Flüssigkeits-
aufnahmekapazität..................................................................................... 124
4.4.1 Versuche im Pflugscharmischer mit Einstoffdüse ...................................... 124
4.4.2 Versuche im Pflugscharmischer mit Zweistoffdüse .................................... 127
4.4.3 Homogenität der Sprühprodukte aus dem CPF-Verfahren und aus
dem Mischer............................................................................................... 129

Inhaltsverzeichnis III
4.5 Flüssigkeitsverteilung in den Produkten..................................................... 130
4.5.1 Modellvorstellung der verschieden fest gebundenen
Flüssigkeitsanteilen.................................................................................... 130
4.5.2 Herstellung und Charakterisierung der Sprühprodukte .............................. 133
4.5.3 Ergebnisse der NMR-Messungen .............................................................. 135
5 Zusammenfassung................................................................................... 142
6 Literaturverzeichnis ................................................................................. 144
7 Anhang...................................................................................................... 148 7.1 Berechnung einer Flüssigkeitsbrücke ........................................................ 148
7.2 Wassergehalt der einzelnen Stichproben aus der Mischertrommel ........... 149
7.3 Berechnung der Kurvenverläufe der unterschiedlich fest gebundenen
Wasseranteile ............................................................................................ 151
7.3.1 Berechnung des Kurvenverlaufs für zwei unterschiedlich fest
gebundene Anteile ..................................................................................... 151
7.3.2 Berechnung des Kurvenverlaufs für drei unterschiedlich fest
gebundene Anteile ..................................................................................... 151
7.4 Zahlenwerte aller Ergebnisse der NMR-Messungen.................................. 152
Abbildungsverzeichnis i
Tabellenverzeichnis v

Formelzeichen und Abkürzungen IV
Formelzeichen und Abkürzungen Lateinisches Alphabet
0B�
Äußeres Magnetfeld
M�
Magnetisierung
∆E Energiedifferenz a Abstand A Fläche c Proportionalitätskonstante d Durchmesser D Schergefälle E Einwaage F Feuchte Fr Froudezahl g Erdbeschleunigung H Höhe h Plancksches Wirkungsquantum HK Haftkraft L Leitfähigkeit m Masse n Drehzahl p Druck P Kernimpuls, Eigendrehimpuls R, r Radius S Sättigung s Schichtdicke T Temperatur t Zeit u Geschwindigkeit U Spannung V Volumen z Anzahl

Formelzeichen und Abkürzungen V
Griechisches Alphabet
ρ Dichte
ϕ Dimensionsloses Volumen einer Flüssigkeitsbrücke
η Dynamische Viskosität
γ Grenzflächenspannung, Oberflächenspannung
ν Kinematische Viskosität
ν0 Larmorfrequenz
µ Magnetisches Moment
ε Porosität
δ Kontaktwinkel, Randwinkel, Benetzungswinkel
τ Schubspannung
λ Wellenlänge
β Winkel einer Flüssigkeitsbrücke zwischen zwei Partikeln („half-filling“)
ω Winkelgeschwindigkeit
∆p Druckdifferenz
Ι Drehimpulszahl, Kernspinquantenzahl Indices A Agglomerat B Bulk g Gas (gas) ges gesamt gP Geschlossene Pore im immobil k Kapillare kr Krümmung krit. kritisch l Flüssigkeit (liquid) lg Flüssigkeit / Gas (liquid / gas) lm weniger mobil (less mobile) mo mobil oP Offene Pore P Partikel R Rohstoff s Feststoff (solid)

Formelzeichen und Abkürzungen VI
Indices (Fortsetzung) sg Feststoff / Gas (solid / gas) sl Feststoff / Flüssigkeit (solid / liquid) Sch Schüttung St Stampfdichte t Tropfen Abkürzungen CPMG Carr-Purcell-Meiboom-Gill-Impulsfolge FG Fettgehalt HF Hochfrequenz REM Rasterelektronenmikroskop NMR Kernresonaz-Spektroskopie (Nuclear Magnetic Resonance) RH Relative Feuchte (relative humidity) WP Wendepunkt

Einleitung 1
1 Einleitung 1.1 Vorwort Die Verbraucher wünschen heutzutage immer mehr natürliche und naturbelassene Lebensmittel. Dies spiegelt sich auch in den Leitsätzen des Deutschen Lebensmittel-buches wieder, wo es heißt, dass Gewürze nicht mehr als technisch notwendig bearbeitete Teile (Wurzeln, Wurzelstöcke, Zwiebeln, Rinden, Blätter, Kräuter, Blüten, Früchte, Samen oder Teile davon) einer bestimmten Pflanzenart sind, die wegen ihres natürlichen Gehalts an Geschmacks- und Geruchsstoffen als würzende oder geschmacksgebende Zutaten zu Lebensmitteln bestimmt sind. Auf der anderen Seite ist Convenience ein Ausdruck, der immer häufiger in Zusammenhang mit Lebensmitteln fällt. Convenience heißt übersetzt Verbraucher-freundlichkeit, und es soll für die Kunden gewährleistet werden, dass Lebensmittel eine gleichbleibende Qualität aufweisen sowie lange haltbar und einfach zuzubereiten sind. Aus diesen beiden Ansprüchen kann für die Produktion von Lebensmitteln ein gewisser Widerspruch entstehen: Beispielsweise unterliegen Gewürze naturgemäß gewissen Schwankungen hinsichtlich Farbe, Geruch und Geschmack. Das liegt am wechselnden Gehalt der wirksamen, d. h. geschmacks- oder farbgebenden, Komponenten in den Pflanzenteilen in Abhängigkeit von der Bodenbeschaffenheit, den Witterungsbedingungen, vom Erntezeitpunkt und von anderen Faktoren. Wird daher ein naturbelassenes Gewürz angeboten, das traditionell durch Trocknen und Vermahlen hergestellt wird, kann keine gleich-bleibende Qualität garantiert werden. Dadurch treten bei der Weiterverarbeitung von Gewürzen häufig Probleme auf. Bei der industriellen Fertigung von Lebensmitteln wird idealerweise zu den Produkten immer dieselbe Gewürzmenge gegeben. Dies setzt voraus, dass die Würzkraft der eingesetzten Zutaten konstant ist. Um dieses Ziel zu erreichen, wurden in den letzten Jahren vermehrt Gewürzextrakte unter Einsatz von überkritischem CO2 als Lösungsmittel hergestellt. Dieses Verfahren ist sehr produktschonend und es erlaubt das Erzeugen von Gewürzextrakten aus Naturstoffen, die standardisierte Inhaltsstoffgehalte aufweisen und frei von mikrobiellen Verunreinigungen sind. Allerdings sind diese Extrakte meist zähflüssig und lassen sich somit nicht einfach in pulverförmige Gewürzmischungen umwandeln. So entstand ein Anwendungsbereich, in dem aus flüssigen und zum Teil hoch-viskosen Stoffen pulverförmige Substanzen formuliert werden müssen.
1.2 Problemstellung In der Lebensmittelindustrie gibt es zahlreiche Anwendungen, bei denen Flüssigkeiten und pulverförmige Substanzen miteinander gemischt bzw. Flüssigkeiten auf disperse Feststoffe aufgebracht werden. Im Allgemeinen sollen die fertigen Produkte hochkonzentriert und streufähig sein. Um dies zu erreichen, ist die maximal mögliche Beladung der Partikeln von Bedeutung. Als Trägerstoffe kommen

Einleitung 2
in der Lebensmittelindustrie verschiedene Rohstoffe in Frage. Beispiele sind Kochsalz, Stärke, Maltodextrin oder Kieselsäure. Da es notwendig ist, die Zusammenhänge zwischen den Partikeleigenschaften und der maximalen Beladung zu verstehen, um mögliche Trägerstoffe zu optimieren, sollen diese Zusammen-hänge in der vorliegenden Dissertation bearbeitet werden. Die erste Fragestellung war, ob und wie sich die Aufnahmekapazität eines Feststoffes messen und bestimmen lässt. Dazu wurde zunächst nach einem Kriterium zur Bestimmung der maximalen Flüssigkeitsaufnahmekapazität von Pulvern gesucht. In Frage kam hier zum Beispiel die Fließfähigkeit, der Agglomerationsgrad oder die Menge der freien Flüssigkeit in der Schüttung beladener Pulver. Weiterhin erfolgte eine Analyse der Einflussgrößen auf die maximale Flüssigkeitsaufnahmekapazität. Hauptsächlich sollten die Eigenschaften der Trägerstoffe hinsichtlich der Auswirkung auf die maximale Flüssigkeitsaufnahme-kapazität untersucht werden. Auch um mögliche Trägerstoffe gegebenenfalls zu optimieren, ist es notwendig, die Zusammenhänge zwischen Pulvereigenschaften und maximaler Aufnahmekapazität zu verstehen. Aber auch andere Einflussgrößen, wie die Eigenschaften der verwendeten Flüssigkeiten oder das angewendete Verfahren, wurden mit berücksichtigt. Neben dem umfangreichen experimentellen und anwendungsorientierten Teil der Arbeit, sind auch die theoretischen Grundlagen und Hintergründe einzelner Prozessschritte diskutiert worden. Von Interesse dabei war, wie die Flüssigkeit in einem beladenen Haufwerk verteilt ist, wie die Flüssigkeit an die Feststoffpartikeln gebunden ist und vor allem welche Menge an Flüssigkeit in verschiedenen dispersen Feststoffen gebunden werden kann. Zusammenfassend sind folgende Fragestellungen im Rahmen dieser Arbeit behandelt worden:
� Wie viel Flüssigkeit können verschiedene Trägerstoffe aufnehmen, und von welchen Eigenschaften der Trägerstoffe hängt die maximale Aufnahmekapazität ab?
� Welchen Einfluss haben die Eigenschaften der Flüssigkeiten auf die maximale Aufnahmekapazität?
� Ist die maximale Beladungskapazität verschiedener Trägerstoffe abhängig vom angewendeten Verfahren?
� Wie ist die Flüssigkeit im beladenen Produkt verteilt, und wie wird sie an die Feststoffpartikeln gebunden?
� Wie kann die Flüssigkeitsverteilung im Feststoff und die Bindung an den Feststoff gemessen werden?
Die vorliegende Arbeit zeigt die Veränderung des Schüttgutes mit zunehmendem Flüssigkeitsanteil, erörtert Möglichkeiten zur Beurteilung flüssigkeitsbeladener Produkte und erläutert die wichtigsten Einflussparameter auf die maximal mögliche Beladung von dispersen Trägerstoffen mit Flüssigkeiten. Zum Beladen kamen zwei verschiedene Verfahren zum Einsatz, die miteinander verglichen werden.

Theoretische Grundlagen und Kenntnisstand 3
2 Theoretische Grundlagen und Kenntnisstand 2.1 Partikelherstellung Pulver sind disperse Systeme (fest in gasförmig), wobei die disperse Phase so hoch konzentriert ist, dass die einzelnen Partikeln sich gegenseitig berühren [1]. Um Partikeln zu gewinnen, gibt es zwei grundsätzlich verschiedene Methoden. Entweder können Feststoffe zerkleinert werden oder es können sich Moleküle durch Kristallisation bzw. Partikeln durch Agglomeration zu größeren Gebilden zusammen-lagern. Die so entstehenden Produkte können sich in den Eigenschaften Partikel-größenverteilung und Partikelform stark voneinander unterscheiden. Durch Auswahl der geeigneten Methode und Variation der entsprechenden Prozessparameter können diese Eigenschaften aber auch gezielt beeinflusst werden, um Partikeln in gewünschter Form und Größe zu produzieren. Beim mechanischen Zerkleinern werden die Partikeln je nach Material in dazu geeigneten Mühlen oder Brechern Kräften ausgesetzt. Dabei entstehen Spannungen in den Partikeln, die bei hinreichender Stärke zum Bruch führen. [36] Der Zerkleinerungsvorgang lässt sich durch entsprechende Betriebsbedingungen steuern. Die Eigenschaften des Mahlgutes und der danach ausgewählte Mühlentyp bestimmen die Qualität des Produktes, da die verschiedenen Mühlen hinsichtlich Beanspruchungsart, Beanspruchungsintensität und Beanspruchungshäufigkeit variieren. Nach Rumpf [43] werden vier Beanspruchungsarten unterschieden, die in Abbildung 1 dargestellt sind.
Abbildung 1: Beanspruchungsarten nach Rumpf [43]
a) Druck-Schub-Beanspruchung: Es sind mindesten zwei Kontaktstellen zwischen
den Partikeln und dem Mahlwerkzeug oder zwischen den Partikeln untereinander vorhanden.
b) Prallbeanspruchung: Es tritt eine Kontaktstelle zwischen dem Partikel und der Wand auf.
c) Beanspruchung durch das umgebende Medium: Diese Art der Zerkleinerung reicht meist nur zur Desagglomeration aus.
d) Beanspruchung durch nichtmechanische Energiezufuhr: Möglich ist eine elektrische, thermische oder chemische Energiezufuhr.
a) b) d)c)

Theoretische Grundlagen und Kenntnisstand 4
Die verschiedenen Mühlen können hinsichtlich ihrer Bauform in drei große Gruppen eingeteilt werden [36]:
� Prallmühlen: Die gasgetragenen Partikeln werden mit hohen Stoß-geschwindigkeiten (ca. 100 m/s) belastet, indem sie entweder gegen sich bewegende Mahlorgane (zum Beispiel Stifte oder Klötze) oder in Strahlmühlen durch Druckluft beschleunigt und aufeinander geschossen werden.
� Mahlkörpermühlen: Mahlkugeln werden durch Rotation der Mahlkammer oder durch Rührer (Rührwerkskugelmühlen) bewegt, und das Mahlgut wird zwischen den Kugeln zermahlen. Rührwerkskugelmühlen werden unter Zugabe von Flüssigkeit betrieben.
� Maschinen zur Druckzerkleinerung: Das Mahlgut wird zwischen einem sich drehenden Mahlteller und darauf abrollenden Kollern (Walzenschüsselmühlen) oder zwischen zwei Walzen, die gegeneinander gepresst werden (Gutbett-walzenmühlen), beansprucht. Beide Mühlentypen liefern hohe Durchsätze und sind energetisch günstig.
Durch den Einsatz der verschiedenen Zerkleinerungsmaschinen lässt sich die Partikelgröße eines Gutes senken. Die Partikelgrößenverteilung kann allerdings mit einfachen Mühlen nur unzureichend beeinflusst werden. Um enge Partikel-größenverteilungen zu erhalten, ist es notwendig, Mühlen mit internen oder externen Sichtern zu verwenden. Eine weitere Möglichkeit, einen Feststoff in kleine Partikeln zu teilen, stellen verschiedene Vertropfungsverfahren dar. Aus einer flüssigen Phase werden einheitliche Tropfen gebildet, welche dann in einer geeigneten Flüssigkeit oder in der Gasphase aufgrund der Oberflächenspannung zu monodispersen Kugeln erstarren. Die einzelnen Technologien unterscheiden sich in erster Linie im Zerteilungsvorgang.
� Beim pulsierten Hüllstrahlprillen tritt die disperse Phase, die eine Schmelze oder Suspension darstellt, als laminarer Strahl aus der Düse aus und wird durch eine überlagerte Schwingung in Tropfen zerteilt [18].
� Auch die Technik des BRACE-Mikrokugelverfahrens basiert auf dem Prinzip des laminaren Strahlzerfalls. Durch eine von außen eingebrachte Störung, z. B. eine Schwingungsanregung der Düse in Flussrichtung des Flüssigkeitsstrahls, lassen sich monodisperse Tropfen erzeugen. [4]
� Bei einem von der Firma Messer Griesheim entwickelten Verfahren wird flüssiger Stickstoff mit –196 °C als laminarer Strom durch eine Pelletierrinne gepumpt, über deren Anfang eine Tropfvorrichtung angebracht ist. Aus dieser wird das flüssige Produkt (z. B. chemische Schmelze) in den Stickstoff eingetropft und erstarrt dort innerhalb von wenigen Sekunden zu Kugeln, die mit dem Stickstoffstrom auf ein engmaschiges Förderband transportiert werden. [20]

Theoretische Grundlagen und Kenntnisstand 5
� Das von der Firma geniaLab entwickelte Strahlschneider-Verfahren (JetCutter) beruht auf der mechanischen Trennung eines Flüssigkeitsstrahls. Das zu verarbeitende Material, das z. B. eine Polymerlösung, ein anorganisches Sol, ein Wachs oder eine andere Schmelze sein kann, wird unter Druck durch eine Düse gepresst, wobei sich ein Flüssigkeitsvollstrahl bildet. Dieser wird unmittelbar nach der Düse durch ein rotierendes Schneidwerkzeug, das aus speichenradförmig angeordneten Drähten besteht, in gleichmäßige Zylindersegmente geschnitten. Aufgrund der Oberflächenspannung formen sich diese Segmente während des Fallens zu sphärischen Partikeln, die in einem geeigneten Medium aufgefangen und ausgehärtet werden, falls sie beim Auftreffen noch nicht fest sind. Das Volumen der entstehenden Kugeln ist abhängig von der Anzahl der Schneid-drähte z, der Drehzahl n des Schneidwerkzeuges und dem Volumenstrom der Flüssigkeit durch die Düse, der wiederum vom Düsendurchmesser dDüse und der Geschwindigkeit der Flüssigkeit ul abhängt [41]:
zn4u)(dV l
2Düse
⋅⋅⋅⋅
=π (Gleichung 1)
Neben der Herstellung von Partikeln mittels mechanischer Verfahren kann auch ein thermisches Verfahren angewendet werden, nämlich die Kristallisation. Eine Suspension aus Kristallen befindet sich nur dann im Gleichgewicht , wenn sich Druck und Temperatur zwischen Dampfphase, flüssiger Phase und Kristallen ausgeglichen haben. Dann hat sich ein Gleichgewicht sowohl zwischen der Gasphase und der Flüssigkeit als auch zwischen der Flüssigkeit und den Kristallen eingestellt. Befindet sich eine Lösung nicht in diesem Zustand, sondern ist die Konzentration an kristallisierender Substanz höher als der Gleichgewichtswert, so tritt eine Übersättigung auf, welche die treibende Kraft für die Keimbildung und das Wachstum darstellt. [23] Ein weitere Möglichkeit, Partikeln herzustellen, bietet ein Verfahren in dem die zu mikronisierenden Feststoffe eingangs in überkritischen Fluiden gelöst werden. Durch eine schnelle Zustandsänderung kommt es zu spontanen Phasenübergängen und zum Ausfällen der Feststoffe. [53] Mit dem RESS-Verfahren (Rapid Expansion of Supercritical Solutions) lassen sich beispielsweise kleinste kugelige Partikeln mit einer relativ engen Partikelgrößenverteilung herstellen. Bei Partikeln in dieser Größenordnung übertreffen die Haftkräfte die Gewichtskraft um ein Vielfaches, so dass sich die runden Primärpartikeln unmittelbar nach der Mikronisierung zu größeren Agglomeraten zusammenlagern. Um diese Agglomeratbildung zu vermeiden, müssen die gebildeten Partikeln z. B. in wässrigen Schutzkolloidlösungen stabilisiert werden. [45] Verschiedene Arbeitsgruppen beschäftigen sich neben dem RESS Verfahren mit der Entwicklung unterschiedlicher Techniken, wie z. B. GAS (Gas Antisolvent Crystallization) oder PCA (Particles with a Compressed Fluid Antisolvent). [54]

Theoretische Grundlagen und Kenntnisstand 6
Partikeln werden meistens für bestimmte Anwendungen hergestellt und sollen deswegen definierten Anforderungen genügen. Dazu müssen verschiedene Eigenschaften, wie Partikelgröße, Partikelform oder Porosität definiert werden. Zur Kontrolle der Produkteigenschaften dient die Partikelcharakterisierung.
2.2 Partikelcharakterisierung Da Pulver disperse Systeme sind, die aus einer festen dispersen Phase und einer gasförmigen Phase bestehen [1], lassen sich daraus zwei grundsätzlich verschiedene Ansätze ableiten, wie sich Pulver charakterisieren lassen. Zum einen gibt es die Eigenschaften des gesamten Schüttgutes, wie die Partikelgrößen-verteilung, die Schütt- und Stampfdichte, die Porosität und Durchströmbarkeit der Schüttung, die Fließfähigkeit sowie die Benetzungseigenschaften. Auch die Farbe, der Geruch oder der Geschmack eines Pulvers können in der Lebensmittelindustrie eine wichtige Rolle spielen. Auf der anderen Seite existieren die Eigenschaften der Einzelpartikeln. Hier kann unterschieden werden zwischen den geometrischen Merkmalen, wie z. B. der Größe oder der Form, die zum Teil schwer zu messen sind, und den physikalischen Merkmalen, wie der Sinkgeschwindigkeit oder der Lichtstreuung. Ist der Zusammenhang zwischen den physikalischen und den geometrischen Merkmalen bekannt, können die erst genannten gemessen und daraus die zweiten berechnet werden.
2.2.1 Partikelgröße und Partikelform
Bei runden, monodispersen Partikeln ist es sehr einfach, Aussagen über die Partikelgröße zu machen. Ist der Durchmesser d der monodispersen Kugeln angegeben, ist die Größe eindeutig definiert. In der Realität sind Pulverschüttungen meistens weder monodispers noch sind die Einzelpartikeln kugelförmig, was die Messung und die Aussagen über Partikelgrößen und Partikelformen erheblich erschwert. Es können verschiedene Partikelmerkmale verwendet werden, um Angaben über die Partikelgröße zu machen. Beispiele sind die Sehnenlänge, die Oberfläche, die Masse, das Volumen, die Projektionsfläche, die Sinkgeschwindigkeit oder Feld-störungen, die von Partikeln ausgelöst werden. Um Partikelgrößen anzugeben, werden Äquivalentdurchmesser genutzt. Ein Äquivalentdurchmesser ist der Durch-messer einer Kugel, die dieselben physikalischen Eigenschaften aufweist wie das gemessene, unregelmäßig geformte Partikel. Wird als Messgröße zum Beispiel die Sinkgeschwindigkeit verwendet, erhält ein unregelmäßig geformtes Partikel den Durchmesser, den ein kugelförmiges Partikel hätte, das dieselbe Sinkge-schwindigkeit aufweist. Um die Partikelgrößenverteilung eines Haufwerkes zu messen, gibt es sehr unterschiedliche Messverfahren. Beispiele sind Siebanalysen, Sedimentations-verfahren und optische Verfahren. Bei den letzteren wird die Störung eines

Theoretische Grundlagen und Kenntnisstand 7
Lichtstrahls als Partikelmerkmal verwendet und die Streuung sowie die Absorption von Licht an kleinen Partikeln gemessen. Wenn die Partikeln sehr klein gegenüber der Wellenlänge der Strahlung sind, tritt die sogenannte Rayleigh-Streuung auf. Das Streumuster dieser Rayleigh-Streuung ist eine Dipolstreuung. Sehr viel komplizierter ist es, wenn die Partikelgröße nicht mehr vernachlässigbar klein gegenüber der Wellenlänge ist. Diese Art von Streuung wird als Mie-Streuung bezeichnet. G. Mie hat im Jahre 1908 auf der Grundlage der Maxwellschen Gleichung für elektrische und magnetische Felder eine vollständige Theorie für kugelförmige Partikeln beschrieben. [27] Um Partikelgrößen zu bestimmen, können Beugungserscheinungen als Partikel-merkmal dienen. Wird ein kugelförmiges Partikel (d >> λ), eine Kreisscheibe oder eine kreisförmige Öffnung in einer wie in Abbildung 2 dargestellten optischen Anordnung von monochromatischem Licht beleuchtet, so erscheint in der Brenn-ebene ein Frauenhofersches Beugungsbild. Dieses entsteht durch Interferenz (Über-lagerungen von Wellensystemen gleicher Wellenlänge) der an der Partikel-umrandung gebeugten Lichtwellen und es besteht aus abwechselnd hellen und dunklen Zonen. Der Radius r des ersten dunklen Ringes lässt sich berechnen. Er ist umso kleiner, je größer der Partikeldurchmesser d ist. Bei vorgegebener Brennweite lässt sich aus der Lage von r die Größe von d bestimmen. [27] Setzt sich die Probe aus Partikeln mit unterschiedlichen Durchmessern zusammen, können Anzahl und Größe der jeweiligen Partikeln aus der Intensitätsverteilung berechnet werden.
Abbildung 2: Prinzip der Beugungsspektrenanalyse
Nach der Messung der Partikelgrößenverteilung ist es wichtig, dass auch deren korrekte Darstellung erfolgt. Eine Möglichkeit, Partikelgrößenverteilungen darzustellen, ist die Summenhäufigkeit (Abbildung 3), die folgendermaßen definiert ist:
Laser
Strahlaufweitung
Messzone
Partikeln
Linse
Detektor

Theoretische Grundlagen und Kenntnisstand 8
PartikelnallerMengexxPartikelnallerMenge(x)Q i
r≤
= (Gleichung 2)
Dabei werden verschiedene Mengenarten unterschieden, wie z. B. die Anzahl (r = 0) oder das Volumen (r = 3). In seltenen Fällen kann auch die Länge (r = 1) oder die Fläche (r = 2) verwendet werden. [50]
Abbildung 3: Summenhäufigkeit Qr(x)
Alle beschriebenen Messverfahren gehen von runden Partikeln aus, oder es werden die Äquivalentdurchmesser als Partikelgrößenverteilungen dargestellt. Deswegen ist es nicht möglich, Partikelgrößenverteilungen, die mit unterschiedlichen Mess-verfahren aufgenommen wurden, direkt miteinander zu vergleichen. Um Pulver bzw. Partikeln genauer zu beschreiben, spielt neben der Partikelgröße auch die Partikelform eine wichtige Rolle. Partikelformen können unter anderem durch folgende Ausdrücke beschrieben werden: Kugelförmig, rund, oval, nadel-förmig, glatt, faserförmig, eckig usw. Um Formunterschiede zwischen einer Kugel und einem von der Kugelform abweichenden realen Partikel zahlenmäßig angeben zu können, gibt es Formfaktoren. Beispiele sind Verhältnisse von Äquivalentdurch-messern oder anderen charakteristischen Partikelabmessungen. [27]
2.2.2 Oberfläche und Porosität
Ein weiteres wichtiges Merkmal von Partikeln und Pulverschüttungen ist die Oberfläche und die Porosität der Einzelpartikeln. Beide Größen sind eng miteinander verknüpft und beeinflussen sich gegenseitig sehr stark. Bei großen geometrischen Körpern mit glatten Oberflächen ist es verhältnismäßig einfach, die Oberfläche, das Porenvolumen oder die Porenradien zu bestimmen. Bei Pulvern und Partikeln handelt es sich nicht immer um eine eindeutig definierte
xmin
WP
Qr (x)1,0
0,5
xh,r xmax x

Theoretische Grundlagen und Kenntnisstand 9
Größe, da sich reale Oberflächen durch mehr oder weniger ausgeprägte Rauhigkeiten oder Poren von glatten Oberflächen unterscheiden. [25] Das Porenvolumen besteht aus der Summe der Volumina aller Kanalporen sowie der offenen und geschlossenen Poren eines Körpers (Abbildung 4).
Die Partikelporosität εp ist das Verhältnis des Porenvolumens (geschlossen: VgP, offen: VoP) zum Gesamtvolumen Vges:
ges
oPgPP V
VVε
+= (Gleichung 3)
Abbildung 4: Porosität eines festen Körpers
Wenn die geschlossenen Poren nicht messbar oder nicht von Einfluss sind, muss die Partikelporosität εP durch die scheinbare Partikelporosität εscheinbar ersetzt werden:
ges
oPscheinbar V
V=ε (Gleichung 4)
Die durch die Poren gebildete Oberfläche bezeichnet man als innere Oberfläche, die zusammen mit der äußeren Oberfläche die gesamte Oberfläche eines Partikels bildet. Für Beurteilungen kann die spezifische Oberfläche, entweder volumen-bezogen oder massebezogen, angegeben werden. Bei Berechnung der spezifischen Oberfläche aus der Teilchengrößenverteilung, wird ausschließlich der Anteil der äußeren Oberfläche ermittelt [25]. Umgekehrt kann aus der äußeren Oberfläche auf einen entsprechenden Mittelwert der Teilchengröße geschlossen werden. Werden die Partikeln kleiner, wird es immer diffiziler, einen absoluten Wert für diese Größen anzugeben. Die an einem realen Körper gemessene Oberfläche hängt davon ab, ob und bis zu welcher Feinheit Rauhigkeiten und Poren mit zum Messwert beitragen. Die gemessene Oberfläche ist daher vom jeweiligen Messverfahren abhängig.
Geschlossene Pore
Kanalpore
Weite Pore mit enger Öffnung("Inkbottle Pore")
Offene Pore

Theoretische Grundlagen und Kenntnisstand 10
Es gibt verschiedene Messmethoden, für die Bestimmung der Partikelporosität. Ob Partikeln porös oder nicht porös sind, lässt sich grundsätzlich z. B. mit Hilfe einer rasterelektronenmikroskopischen Aufnahme (vgl. Abbildung 5) der Probe feststellen.
40 µm 10 µm
Abbildung 5: Stärkepartikel, links mit Poren, rechts ohne Poren
Die zwei bekanntesten Messmethoden zur Bestimmung der Porosität sind das Quecksilberintrusionsverfahren und die Gassorption. Daneben kommt z. B. für die Bestimmung von Metalloberflächen üblicherweise eine selektive Chemisorption zum Einsatz, wie die Chemisorption von N2O und H2 zur Oberflächenbestimmung von Kupfer-Katalysatoren [19]. 2.2.2.1 Quecksilberintrusionsverfahren Das Verfahren beruht auf der Messung des Quecksilbervolumens, das in einen porösen Feststoff in Abhängigkeit von dem aufgebrachten Druck hineingepresst wird. Der Feststoff darf von Quecksilber nicht benetzt werden, was durch den Kontaktwinkel gegeben ist. Zudem darf das Quecksilber nicht mit dem Feststoff reagieren. Es werden keine geschlossenen Poren erfasst und bei den offenen Poren nur diejenigen, in die Quecksilber eindringen kann.
In Abhängigkeit der Druckdifferenz ∆ p dringt Quecksilber mit der Oberflächen-spannung γ und dem Kontaktwinkel δ entsprechend der Young-Laplace-Gleichung in die Poren mit den Radien r ein [38]:
rcos2p∆ δγ ⋅⋅= (Gleichung 5)
Ein Problem dieser Messmethode ist der sogenannte Inkbottle Effekt. Große Poren, die durch kleine Kanäle zugänglich sind, werden dem Mengenanteil der kleinen Poren zugerechnet (siehe auch Abbildung 4) [38]. Weiterhin kann das zu messende

Theoretische Grundlagen und Kenntnisstand 11
Material durch die hohen aufzuwendenden Drücke teilweise verformt oder zerstört werden, was das Messergebnis verfälscht. Für den Kontaktwinkel von Quecksilber wird grundsätzlich ein Wert angenommen. Die sich tatsächlich einstellenden Intrusions- und Extrusionswinkel werden nicht exakt bestimmt. 2.2.2.2 Sorptionsversuche Eine Möglichkeit, Aussagen über die Beschaffenheit und Größe der Oberfläche und der Porosität von Feststoffen zu treffen, ist die Adsorption von Gasen. Wenn ein Feststoff in einem geschlossenen Raum einem Gas oder einem Dampf ausgesetzt ist, beginnt der Feststoff das Gas zu adsorbieren. Dabei nimmt das Gewicht des Feststoffes zu und der Druck des Gases sinkt. Nach einiger Zeit stellt sich ein konstanter Druck p ein und das Gewicht des Feststoffes steigt nicht weiter an. Es wird dabei zwischen physikalischer und chemischer Adsorption unterschieden. Bei der physikalischen Adsorption wirken van-der-Waals-Kräfte, während bei der chemischen Adsorption Reaktionen auftreten und chemische Bindungen geschlossen werden. Die vom Feststoff aufgenommene Gasmenge n hängt dabei von der Masse ms des Feststoffes, von der Temperatur T, vom Druck p, vom Feststoff und dem umgebenden Gas ab und wird in Mol pro Gramm Feststoff angegeben: [14]
)Gas Feststoff,p, T,,(mfn s= (Gleichung 6)
„Sorbere“ bedeutet „zu sich nehmen, einschlürfen“. Das Wort Sorption wurde von McBain 1909 für den Vorgang vorgeschlagen, bei dem ein Festkörper oder eine Flüssigkeit Moleküle aus dem umgebenden Gasraum aufnimmt [13]. Ein Spezialfall ist die sogenannte Oberflächensorption oder die Sorption ohne Strukturänderung, bei der die Moleküle nur an der Oberfläche eines kompakten Festkörpers gebunden sind. Unterschieden wird zwischen Adsorption (Aufnahme) und Desorption (Abgabe) der Moleküle. Brunauer, Emmet und Teller haben folgenden bei der Gasadsorption geltenden Zusammenhang beschrieben: Zwischen der adsorbierten Gasmenge V an einer Oberfläche von 1 g Substanz, der Gasmenge Vm, die zu einer monomolekularen Bedeckung der Oberfläche erforderlich ist, dem Druck p, dem Sättigungsdampfdruck p0 des betreffenden Gases und der Proportionalitätskonstanten C gilt folgende Beziehung, die als BET-Gleichung bekannt ist [1]:
0mm0 pCVp1)(C
CV1
p)(pVp
⋅⋅⋅−+
⋅=
−⋅ (Gleichung 7)

Theoretische Grundlagen und Kenntnisstand 12
Ist diese Gleichung gültig, ergibt die Auftragung von p/(V(p0-p)) gegen p/p0 eine Gerade, deren Steigung und Ordinatenabschnitt die Größen C und Vm bestimmen. Wenn das zur Ausbildung einer monomolekularen Schicht erforderliche Volumen Vm des Gases bekannt ist, kann man mittels der Fläche Am, die ein Mol des Gases bei monomolekularer Ausbreitung bedeckt, die Oberfläche einer Probe Om berechnen [1]:
M
mAmm V
VNAO ⋅⋅= (Gleichung 8)
NA: Avogadrosche Zahl (6,02 ⋅ 10²³ Moleküle pro Mol) VM: Molvolumen des Gases Im Allgemeinen wird für die Messmethode ein Inertgas wie Stickstoff oder Helium verwendet. Es kann aber z. B. auch mit Wasserdampf gemessen werden. Dabei hängt das Ergebnis stark vom verwendeten Medium ab, da die Moleküle ein unterschiedliches Eindringvermögen in die Poren aufweisen. Grundsätzlich kann die Mehrheit der Isothermen, die bei physikalischer Adsorption aufgenommen werden, in fünf Klassen eingeteilt werden. [14] In Abbildung 6 sind diese fünf Klassen und zusätzlich eine stufenweise Isotherme dargestellt.

Theoretische Grundlagen und Kenntnisstand 13
Abbildung 6: Adsorptionsisothermen (Typ I bis V) und eine stufenweise Isotherme (VI) [14]
Die Adsorptionsisothermen Typ IV und Typ V enthalten eine Hysterese. Dabei stellt der untere Ast jeweils die Messung dar, die bei Zugabe von Gas zum System und der obere Ast die Messung, die bei der Entnahme von Gas entsteht. Auch bei allen anderen Typen können Hysteresen auftreten. Neben diesen Typen von Isothermen können auch Grenzfälle zwischen zwei oder mehreren Typen auftreten, und es existieren weitere Kurven, die sich zu keinem der Typen eindeutig zuordnen lassen. [14] Bei Gregg und Singh [14] werden die einzelnen Typen der Isothermen behandelt, deren detaillierte Interpretation aber umstritten ist. Zusammenfassend wird festgestellt, dass Typ II Isothermen auf nicht poröse Feststoffe hinweisen, während Typ IV Isothermen charakteristisch für ein mikroporöses System sind. Isothermen vom Typ I werden bei Festsstoffen mit Mikroporen gemessen und Isothermen vom Typ III und Typ V scheinen aufzutreten, wenn die Bindungen zwischen Gas und Feststoff sehr schwach sind. Diese beiden Typen von Isothermen treten weniger häufig auf als die drei anderen.
I II
III IV
VIV
Druck p / Sättigungsdampfdruck p0
Men
ge d
es a
dsor
bier
ten
Gas
es

Theoretische Grundlagen und Kenntnisstand 14
Ein Problem, das bei der Durchführung von Sorptionsversuchen an porösen Systemen auftreten kann, ist die Überlagerung der Oberflächenadsorption durch die Kapillarkondensation. In zylindrischen Poren mit den Radien r kann der bei genügend großem relativen Druck p/p0 auftretende Zusammenhang der Kapillarkondensation mit der Kelvin-Gleichung beschrieben werden:
rcos2
TRV)(p/pln 0
δγ ⋅⋅⋅⋅
−= (Gleichung 9)
R: Allgemeine Gaskonstante Poren können daher in drei Größenklassen eingeteilt werden [25]: Mikroporen, die für die Anwendung der Kelvin-Gleichung zu klein sind, Meso- oder Übergangsporen, die typisch für die Kapillarkondensation sind und Makroporen, die für die Messung der Kapillarkondensation zu groß sind. Bei Gregg und Sing [14] wird folgende Klassifizierung genannt: Tabelle 1: Klassifizierung der Poren anhand der Porenweite [14]
Bezeichnung Porenweite Mikroporen Weniger als ~ 20 Å (2 nm)
Mesoporen Zwischen ~ 20 Å und ~ 500 Å (2 und 50 nm)
Makroporen Mehr als ~ 500 Å (50 nm)
In der Praxis kann die Adsorption von Gasen an Feststoffen also dazu dienen, Informationen über deren Oberflächen und Poren (Struktur und Größe) zu erhalten. Meist wird dazu Stickstoff bei dessen Siedepunkt von 77 K verwendet. Neben den Stickstoffisothermen besteht die Möglichkeit, die Wasserdampf-sorptionsisothermen von Pulvern aufzunehmen. Kommt das Wasser mit dem dispersen Feststoff in Kontakt, können unterschiedliche Mechanismen auftreten:
� Absorption in der Schüttung, � Oberflächen-Adsorption, � Kapillarkondensation, � Chemische Reaktion, � Komplexierung, � Lösungsprozesse etc.

Theoretische Grundlagen und Kenntnisstand 15
Tritt eine reine Oberflächenadsorption auf, können über die molekularen Sorptionskonstanten des Wassers Aussagen über die Oberflächenbeschaffenheit des Feststoffes getroffen werden. Tabelle 2: Molekulare Sorptionskonstanten des Wassers [13]
Volumenbedarf pro Molekül im Wasser ca. 30 ų
Belegte Fläche pro Wassermolekül in unimolekularer Schicht 10,6 Ų
Flächenäquivalent von 1 g Wasser in unimolekularer Schicht 35 m²
Allerdings hängt die Wasserdampfsorption von festen Sorbentien von der chemischen Zusammensetzung und der physikalischen Struktur ab, so dass eine exakte theoretische Auswertung der Wasserdampfsorptionsisothermen nur in seltenen einfachen Fällen möglich ist. [13]
2.2.3 Feststoffdichte, Schüttdichte und Stampfdichte
Allgemein wird die Dichte ρ als Quotient aus der Masse m durch das Volumen V definiert.
Vm=ρ (Gleichung 10)
Bei dispersen Systemen ist zusätzlich zwischen der wahren Feststoffdichte, der Schüttdichte einer losen Schüttung und der Stampfdichte einer verdichteten Schüttung zu unterscheiden (Abbildung 7).
Abbildung 7: Schematische Darstellung verschiedener Dichten eines dispersen Systems
Feststoffdichte ρρρρs
Schüttdichte Stampfdichte
geschlossene Pore VgP
Hohlraumvolumen VH
offene Pore VoP

Theoretische Grundlagen und Kenntnisstand 16
Auch hier spielen die Hohlräume in den Partikeln, die von außen nicht zugänglich sind, eine große Rolle. Die wahre Dichte des Feststoffes ρs entspricht dann folgender Gleichung:
s
ss V
m=ρ (Gleichung 11)
Eine sogenannte Rohdichte oder scheinbare Dichte wird ermittelt, wenn geschlossene Poren in den einzelnen Partikeln vorhanden sind. Dies kann gegebenenfalls vermieden werden, indem die Partikeln so weit zerkleinert werden, bis alle Poren von außen zugänglich sind. Allerdings ist auch die Rohdichte eine entscheidende Größe, z. B. für das Bewegungsverhalten eines Partikels, und deshalb oft von verfahrenstechnischem Interesse. [1], [27] Die Schütt- und die Stampfdichte lassen sich auf die gleiche Art und Weise berechnen, wobei das Hohlraumvolumen VH zwischen den Partikeln bei der Schüttdichte größer ist als bei der Stampfdichte:
HoPs
s
VVVm
ρ++
= (Gleichung 12)
Schütt- und Stampfdichte werden als Bulkdichten bezeichnet und sind von großer technologischer Bedeutung. Beispiele für Anwendungen aus der Pharmazie, bei denen die Bulkdichten eine große Rolle spielen, sind das Tablettieren und das Befüllen von Kapseln. Um die Bulkdichten reproduzierbar messen zu können, muss beachtet werden, dass die Messbedingungen konstant gehalten werden. Das bedeutet für die Schüttdichte, dass die Pulverproben aus einem einheitlichem System in einen Behälter mit bekanntem Volumen gegeben werden und die Masse dieser Schüttung bestimmt wird. Um die Stampfdichte zu bestimmen, wird die Schüttung durch definierte Stöße solange verdichtet, bis sich das Volumen der Schüttung nicht mehr verringert. Um Feststoffdichten zu bestimmen, können Flüssigkeits- oder Gaspyknometer verwendet werden. Die Masse der Probe wird dabei jeweils gewogen. Wird ein Gaspyknometer verwendet, gilt bei konstanter Temperatur nach Boyle-Mariotte:
.constVpVp 2211 =⋅=⋅ (Gleichung 13)
Sind beide Drücke und ein Volumen bekannt, lässt sich das zweite Volumen berechnen.

Theoretische Grundlagen und Kenntnisstand 17
Abbildung 8: Prinzipbild eines Gaspyknometers [27]
V1 wird definiert als das Leervolumen des Gefäßes (1) mit dem Verbindungsrohr (2) und dem Kompressionsrohr (3) mit der Kolbenoberkante des Kolbens (4) in Position z1. In der Kolbenposition z2 wird das Volumen V1 um ∆V1 verringert, und es herrscht der Druck p1 + ∆p1. Man misst am Manometer (5) p1 und ∆p1. Ist in den Behälter ein bestimmtes Feststoffvolumen VS gefüllt, gilt folgende allgemein gültige Gleichung für Gaspyknometer [27]:
+⋅−
+⋅= 1
∆pp∆V1
∆pp∆VV
2
22
1
11S (Gleichung 14)
2.3 Eigenschaften von Flüssigkeiten Charakteristische Eigenschaften von Flüssigkeiten sind die Dichte, die Wärme-leitfähigkeit, die spezifische Wärmekapazität, die Schmelz- und Siedetemperatur, die Viskosität sowie die Oberflächenspannung. Beim Benetzen und Tränken von Fest-stoffpartikeln spielen vor allem die Viskosität und die Oberflächenspannung eine Rolle. Deshalb werden die theoretischen Grundlagen dieser Eigenschaften kurz zusammengefasst. Da in die Flüssigkeiten beim Beladen zum Teil überkritisches CO2 gelöst wird, werden auch die Eigenschaften von überkritischem CO2 beschrieben.
2.3.1 Viskosität
Rheologie bezeichnet die Lehre von den Fließeigenschaften von Stoffen und Stoffsystemen. Darunter fallen auch die Gesetzmäßigkeiten des Fließens von Flüssigkeiten. Um diese zu beschreiben, spielt die Viskosität eine wichtige Rolle.
P
P1bzw.
P2
z
z2
z1
z3
(1)
(2) (3)
(4)
(5)

Theoretische Grundlagen und Kenntnisstand 18
Unterschieden wird zwischen der dynamischen Viskosität (Zähigkeit, innere Reibung) η und der kinematischen Viskosität ν, die der Quotient aus der dynamischen Viskosität η und der Dichte der Flüssigkeit ρl ist.
lρην = (Gleichung 15)
Nach Newton kann die Viskosität einer idealviskosen Flüssigkeit durch Gleichung 16 beschrieben werden [1]:
Ddxdvη ⋅=⋅= ητ (Gleichung 16)
Dabei ist τ die Schubspannung und D das Schergefälle. Für newtonsche Flüssigkeiten ist das Schergefälle in Abhängigkeit von der Schubspannung durch eine Gerade gekennzeichnet. Um Viskositäten zu messen, können Kapillar-viskosimeter, Kugelfallviskosimeter oder Rotationsviskosimeter eingesetzt werden.
2.3.2 Oberflächenspannung
Die Oberflächenspannung γ stellt die Grenzflächenspannung von Festkörpern und Flüssigkeiten gegenüber der Dampfphase bzw. gegenüber Luft dar. Die Einheit der Oberflächenspannung ist Kraft pro Längeneinheit. In Flüssigkeiten sind die Moleküle frei beweglich, und es treten mehr oder weniger starke intermolekulare Wechselwirkungen zwischen den Molekülen auf. Während innerhalb der Flüssigkeit (a) auf die Moleküle gleiche zwischenmolekulare Kräfte aus allen Richtungen wirken, sind diese Kräfte an der Flüssigkeit/Dampf-Grenzfläche (b) nicht ausgeglichen (siehe Abbildung 9).
Flüssigkeit
FlüssigkeitsoberflächeDampf
a
b
Abbildung 9: Zwischenmolekulare Kräfte [8]
Eine Flüssigkeit ist bestrebt, ihre Oberfläche zu verkleinern. Deshalb versuchen Tröpfchen die energetisch günstigste Form, die Kugelform, anzunehmen. Mit

Theoretische Grundlagen und Kenntnisstand 19
folgenden Methoden lassen sich die Oberflächenspannungen messen: Meniskus-Bildung und Steighöhe in Kapillaren, Blasendruck einer in der Flüssigkeit austretenden Gasblase, Ring-Methode, Wilhelmy-Methode oder die optische Messung der Gestalt liegender oder hängender Tropfen. [8]
2.3.3 Eigenschaften von überkritischem CO2
Da die Flüssigkeiten im Rahmen der Untersuchungen mit Hilfe des CPF-Sprühverfahrens [57], das in Kapitel 2.5.2 ausführlich erklärt wird, auf die Trägerstoffe aufgesprüht werden, wird CO2 in die verwendeten Flüssigkeiten gelöst. In diesem Zusammenhang wird erläutert, was der sogenannte überkritische Zustand eines Gases bedeutet. Zudem wird kurz über Löslichkeit von CO2 in Flüssigkeiten berichtet. Im überkritischen Bereich löst sich die Phasengrenze zwischen Gas und Flüssigkeit auf und eine Unterscheidung ist nicht mehr möglich. Die kinetische Energie der Flüssigkeitsteilchen ist hier so groß geworden, dass die van-der-Waals Kräfte nicht mehr ausreichen, um die Moleküle zusammenzuhalten. Das Fluid hat den überkritischen Bereich erreicht. Folgt man der Dampfdruckkurve eines Stoffes aufwärts, es liegt also ein Zweiphasengemisch vor, so erreicht man den kritischen Zustand im kritischen Punkt, dem Endpunkt der Dampfdruckkurve (siehe auch Abbildung 10).
überkritischerBereich
flüssig
fest
gasförmig
Temperatur
Dru
ckp k
rit.
Tkrit.
krit. Punk
Abbildung 10: Phasendiagramm [22]
Im Rahmen dieser Dissertation sowie in vielen Anwendungen in der Lebensmittel-industrie wird als Gas CO2 verwendet, da es ungiftig, nicht brennbar, nicht korrosiv, billig und in unbegrenzter Menge sowie in ausreichender Reinheit verfügbar und

Theoretische Grundlagen und Kenntnisstand 20
herstellbar ist. Außerdem ist CO2 ein natürlicher Bestandteil vieler Lebensmittel sowie der Luft. CO2 hat günstige kritische Daten, was sich positiv auf Investitions- und Prozesskosten auswirkt. Des weiteren hemmt es durch die Verdrängung von Sauerstoff das Wachstum vieler Mikroorganismen und die Oxidation von Inhaltsstoffen. [22], [51] Die kritische Temperatur von CO2 beträgt 31,3 °C und der kritische Druck 72,9 bar [51]. Im CPF-Verfahren wird CO2, unter Bedingungen bei denen es überkritisch vorliegt (beispielsweise 40 °C und 80 bis 100 bar), in einen statischen Mischer gefördert und dort mit der Flüssigkeit, die auf die Trägerstoffe aufgesprüht wird, vermischt. Welche Menge an überkritischem CO2 sich in einem bestimmten Stoff löst, hängt von der Flüssigkeit, dem Druck und der Temperatur ab. In dieser Mischung liegen im Gleichgewichtszustand maximal drei Phasen vor: CO2 mit gelöster Flüssigkeit, Flüssigkeit mit gelöstem CO2 und eine reine Phase aus Flüssigkeit oder CO2.
2.4 Wechselwirkungen zwischen Flüssigkeiten und Partikeln Um Trägerstoffpartikeln zu tränken, müssen diese zunächst mit der Flüssigkeit benetzt werden. Das Benetzen kann in zwei Vorgänge unterteilt werden: Das Spreiten, d. h. das Verteilen der Flüssigkeit auf der Oberfläche und das Eindringen von Flüssigkeiten in Kapillaren. Über diese beiden Phänomene hat Palzer [35] eingehende Untersuchungen vorgenommen, die am Anfang dieses Kapitels kurz zusammengefasst werden, da sie auch für dieses Vorhaben von großer Bedeutung sind. Im Rahmen dieser Arbeit wurde die Verteilung der Flüssigkeit im Produkt weiterführend theoretisch diskutiert und praktisch untersucht.
2.4.1 Benetzung und Kapillarität
2.4.1.1 Grundlagen des Benetzens von Partikeln Der Begriff Benetzung bezeichnet die Fähigkeit von Flüssigkeiten mit Festkörpern eine neue gemeinsame Grenzfläche auszubilden. Benetzungsvorgänge und deren Folgen können sowohl in der Natur als auch in Bereichen der Technik von großem Interesse sein. Beispielsweise ist schlechtes Benetzen beim Abperlen von Wasser an den Blättern der Lotuspflanze oder beim wasserabweisenden Federkleid eines Vogels erwünscht. In der Technik ist die gute Benetzbarkeit eines Feststoffes durch eine Flüssigkeit in zahlreichen Bereichen, wie z. B. beim Dispergieren, Nassmahlen, bei der Herstellung von Suspensionen, sowie bei Flotations- und Waschprozessen eine wichtige Voraussetzung. Die Benetzbarkeit ist definiert als Vermögen einer Flüssigkeit, sich auf der Oberfläche eines Festkörpers auszubreiten. Sie ist eine physikochemische Eigenschaft von festen Körpern. Ein Maß der Benetzbarkeit ist der Kontaktwinkel (auch Benetzungswinkel, Randwinkel) δ (siehe Abbildung 11). Dieser Winkel stellt sich zwischen den drei Phasen Flüssigkeit, Feststoff und umgebendes Gas ein und

Theoretische Grundlagen und Kenntnisstand 21
hängt von den Grenzflächenspannungen zwischen dem Festkörper und der Flüssigkeit, dem Festkörper und dem Gas sowie der Flüssigkeit und dem Gas ab. [9]
Abbildung 11: Schematische Darstellung eines auf einer Feststoffoberfläche liegenden Tropfens und dem sich einstellenden Kontaktwinkel
Zwischen den Grenzflächenspannungen stellt sich am Tropfen ein Gleichgewicht ein, das durch die Youngsche Gleichung beschrieben werden kann [9]:
δγγγ coslgslsg ⋅+= (Gleichung 17)
Die tatsächlich vorliegenden Feststoffoberflächen sind nicht immer glatt, sondern können rau oder porös sein, was Auswirkungen auf das Benetzen und den Kontaktwinkel hat. Palzer [35] unterscheidet in seiner Arbeit daher sowohl statische und dynamische, als auch mikroskopische und makroskopische Kontaktwinkel. Der durch die Youngsche Gleichung beschriebene Kontaktwinkel wird als mikroskopisch bezeichnet und ist material-, druck- und temperaturabhängig. Der makroskopische Kontaktwinkel hingegen hängt von der Geometrie der Oberfläche ab. 2.4.1.2 Kapillarität Nach dem Verteilen der Flüssigkeit über einen bestimmten Bereich folgt das Eindringen der Flüssigkeit in die Kapillaren und Poren. Dabei muss bei dispersen Feststoffen, wie sie in dieser Arbeit verwendet wurden, zwischen Kapillaren und Poren in den einzelnen Partikeln und zwischen Hohlräumen, die sich zwischen den Partikeln einstellen, unterschieden werden. Kapillarität ist ein Sammelbegriff für alle physikalischen Erscheinungen, die infolge der Grenzflächenspannung von Flüssigkeiten an engen Hohlräumen von Fest-körpern, d. h. in Kapillaren, Spalten und bei Porosität auftreten. Hierzu gehören unter anderem das Aufsteigen bzw. Absinken einer Flüssigkeit in einer Kapillare und die Kapillarkondensation. [8] Wenn eine Flüssigkeit mit einer Kapillare in Kontakt tritt, stellt sich an der Innenwand der Kapillare der Kontaktwinkel δ ein. Daraus folgt eine Krümmung der Grenzfläche zwischen Flüssigkeit und Gas, deren Krümmungsradius rkr aus den Größen Radius
γγγγsg γγγγsl
γγγγlg
ΘΘΘΘm
Flüssigkeit
Feststoff
Gas
δδδδ

Theoretische Grundlagen und Kenntnisstand 22
der Kapillare rk und dem Cosinus des Randwinkels cos δ berechnet werden kann [35]:
δ cosrr k
kr = (Gleichung 18)
Weiterhin kann der sogenannte Kapillardruck pk berechnet werden:
k
lgk r
cos2p
δγ ⋅⋅= (Gleichung 19)
Durch den Kapillardruck dringt die Flüssigkeit in Kapillaren bis zu einer Steighöhe h ein, sofern der Kontaktwinkel δ kleiner als 90° ist und ein kapillares Benetzen erfolgt. Bei einem Kontaktwinkel δ größer als 90° ergibt sich ein negativer Kapillardruck, man spricht dann von kapillarer Depression (Abbildung 12).
hδ < 90° δ = 90° δ > 90°
Abbildung 12: Beispiele von unterschiedlich benetzenden Flüssigkeiten in zylindrischen Kapillaren
Die Flüssigkeit liegt in diesem Beispiel als Kontinuum vor. Eine weitere Möglichkeit ist, dass die Flüssigkeit als Form eines Tropfens am Ende einer Kapillare vorliegt. Palzer konnte zeigen, dass dann auch Flüssigkeiten, die den Feststoff der Kapillare nicht benetzen, in diese eindringen können. Dabei stellt der Innendruck des Tropfens, der sogenannte Laplacedruck die maßgebliche Größe dar, denn die Summe aus Kapillardruck und Laplacedruck muss positiv sein, damit eine nicht benetzende Flüssigkeit in die Kapillare eindringt [35]:
0r
2r
cos2
t
lg
k
lg >⋅
+⋅⋅ γδγ
(Gleichung 20)

Theoretische Grundlagen und Kenntnisstand 23
a b c Abbildung 13: Eindringen von nicht benetzenden Flüssigkeiten in Einzelkapillaren [35]
Wie in Abbildung 13b dargestellt, kann also auch eine nicht benetzende Flüssigkeit in eine Kapillare eindringen, wenn der Durchmesser des Tropfens nur klein genug gegenüber dem Durchmesser der Kapillare ist. Bei großen Tropfen (vgl. Abbildung 13c) oder einem Flüssigkeitskontinuum (vgl. Abbildung 13a) dagegen, dringt eine nicht benetzende Flüssigkeit nicht in die Kapillare ein. Dieses Phänomen konnte Palzer [35] auch empirisch nachvollziehen, indem er kleine Wassertropfen in hydrophobierte Glaskapillaren eindringen ließ. Für die Praxis des Beladens und Tränkens von dispersen Feststoffen bedeutet dies, dass auch nicht benetzbare Flüssigkeiten in Kapillaren oder zwischen den Poren eingelagert werden können, wenn die Tröpfchen, die zum Beladen verwendet werden, klein genug sind.
2.4.2 Bindungsmechanismen zwischen Partikeln in einem System mit Flüssigkeit
Grundsätzlich können zwischen einzelnen Feststoffpartikeln verschiedene Arten von Bindungen wirken: Haftkräfte mit Festkörperbrücken, Haftkräfte ohne stoffliche Verbindung, wie van-der-Waals-Kräfte oder elektrostatische Kräfte, formschlüssige Bindungen und Flüssigkeitsbrücken [42]. Letztere sind beim Beladen und Tränken von Partikelschüttungen von Interesse, wenn zwischen der Flüssigkeit und dem Feststoff keine chemische Reaktion auftritt. Das ist z. B. beim Beladen mit Speiseölen, Fetten oder Gewürzextrakten der Fall. Grundsätzlich können, wie in Abbildung 14 dargestellt, drei Arten von Bindungen zwischen Flüssigkeit und Partikel unterschieden werden.

Theoretische Grundlagen und Kenntnisstand 24
Adsorptionsschicht
Flüssigkeitsbrücke
Porenflüssigkeit
Abbildung 14: Bindungsmechanismen zwischen Partikeln und Flüssigkeit
Die Flüssigkeit kann entweder als dünne Adsorptionsschicht um die einzelnen Partikeln liegen, in den Poren der Einzelpartikeln gespeichert sein oder als Flüssigkeitsbrücke zwischen zwei Partikeln vorliegen. In der Praxis kommen alle drei Bindungsarten parallel vor. Die Flüssigkeitsbrücken können sich zu flüssigkeits-gefüllten Bereichen ausweiten, wie im folgenden Kapitel über die Flüssigkeits-verteilung in dispersen Feststoffsystemen dargestellt wird. Eine weitere Möglichkeit besteht darin, dass die Feststoffpartikeln mit der Flüssigkeit reagieren, d. h. dass intermolekulare Wechselwirkungen auftreten und beispielsweise eine Vernetzung oder eine Gelbildung auftritt. Wenn Partikeln keine Poren aufweisen oder wenn alle Poren mit Flüssigkeit gesättigt sind, lagert sich die Flüssigkeit in Form von Flüssigkeitsbrücken zwischen zwei Partikeln an. Es existieren zahlreiche Untersuchungen über das Volumen einer Flüssigkeitsbrücke zwischen zwei Partikeln. [28], [34], [37], [49], [55] Diese sind vor allem im Zusammenhang mit der Zugfestigkeit von Agglomeraten von Interesse. Im Rahmen dieser Dissertation sollen verschiedene theoretische Ansätze mit einem Versuchsergebnis verglichen werden. Bei zwei sphärischen, starren und gleich großen Partikeln, die durch eine Flüssig-keitsbrücke verbunden sind, lassen sich Haftkraft und Flüssigkeitsvolumen der Flüssigkeitsbrücke berechnen. Allerdings handelt es sich dabei um sehr komplizierte Gleichungssysteme, so dass für die numerische Auswertung vereinfachende Annahmen getroffen werden [37]: Die Projektionsfläche einer Flüssigkeitsbrücke zwischen zwei gleich großen Partikeln beschreibt eine Linie, die sich mit hinreichender Genauigkeit durch einen Kreisbogen mit dem Radius R1 beschreiben lässt (vgl. Abbildung 15).

Theoretische Grundlagen und Kenntnisstand 25
ββββ
a
dPartikel
δδδδ
R2
R1
Abbildung 15: Flüssigkeitsbrücke zwischen zwei Partikeln [55]
Es lassen sich dann folgende geometrische Beziehungen angeben [37]:
( )( )δβ
β+⋅
+−⋅=
cos2acos1d
R p1 (Gleichung 21)
und
( )( )1sinR2sindR 1
P2 −+⋅+
⋅= δββ (Gleichung 22)
und außerdem gilt 21 RRb += .
Auch die Haftkraft, die zwischen zwei gleich großen Kugeln wirkt, kann mit den gegebenen Größen berechnet werden. Sie setzt sich aus zwei Anteilen zusammen, zum einen aus dem Haftkraftanteil (HK1) als Folge des kapillaren Unterdrucks und zum anderen aus dem Haftkraftanteil (HK2) als Folge der Oberflächenspannung der Flüssigkeit [37].
21 HKHKHK += (Gleichung 23)
βπγ 22P
211 sind
4R1
R1HK ⋅⋅⋅
−⋅= (Gleichung 24)
)(sinsindHK P2 δββπγ +⋅⋅⋅⋅= (Gleichung 25)

Theoretische Grundlagen und Kenntnisstand 26
Um das Volumen der Flüssigkeitsbrücke aus den vorhanden Größen zu berechnen,
kann folgende Gleichung herangezogen werden [37]:
( )
−⋅+⋅−
−−++⋅+⋅⋅
−+⋅
−+⋅⋅+⋅⋅=
24)²cos(1cos2d)R)(sin)(cosRb
3)(cosR)(cosR)b(R
V 3P2
12
1
31
122
1
Fl ββδβπδβδβ
δβδβπ
2(
2
3
(Gleichung 26)
Neben diesem Ansatz von Pietsch und Rumpf existieren zahlreiche weitere Ansätze, um das Volumen der Flüssigkeitsbrücke zu berechnen. Diese wurden zum Teil von Weigert und Ripperger [55] im Jahr 1999 zusammengestellt. Von den Autoren Weigert und Rippberger [55] selbst wird ein empirischer Zusammenhang zwischen dem Flüssigkeitsvolumen VFl und den Größen Partikeldurchmesser dp und halber Zentriwinkel β vorgeschlagen, der wesentlich einfacher ist als viele in der Literatur genannte Gleichungen und dennoch die Realität genau beschreibt. Ca und Cδ stellen Korrekturfunktionen dar. Ca für den Abstand a und Cδ für den Kontaktwinkel δ.
δCCβsind0,12V a43
PFl ⋅⋅⋅⋅= (Gleichung 27)
Pa d
a61C ⋅+= (Gleichung 28)
δδ sin1,11C ⋅+= (Gleichung 29)
2.4.3 Flüssigkeitsverteilung in einem dispersen Feststoffsystem
Wenn disperse Feststoffe mit Flüssigkeiten beladen und getränkt werden, spielen die Vorgänge des Benetzens, des Spreitens und des Eindringens in Kapillaren und Poren eine große Rolle. Am Ende liegt ein Produkt vor, das aus einem dispersen Feststoff besteht, an dessen Einzelpartikeln die Flüssigkeit auf irgend eine Art und Weise gebunden ist. Das Ziel vieler Anwendungen ist es, die Flüssigkeit möglichst homogen auf den Feststoff zu verteilen. Dabei kann es sich entweder um sehr kleine Mengen handeln, die homogen in der Schüttung verteilt werden sollen oder es kann möglichst große Mengen Flüssigkeit betreffen, die gleichmäßig im Produkt verteilt

Theoretische Grundlagen und Kenntnisstand 27
vorliegen sollen, ohne dass Inhomogenitäten, wie zum Beispiel feuchte Agglomerate, auftreten. Anwendungen, bei denen Flüssigkeiten in disperse Systeme eingebracht werden, sind beispielsweise die Agglomeration und die Granulation mit flüssigen Bindemitteln. Allerdings muss beachtet werden, dass diese Prozesse nicht in allen Schritten mit den Vorgängen beim Beladen und Tränken, wie es in diesem Vorhaben untersucht wurde, verglichen werden können. Eine Voraussetzung für das Beladen und Tränken ist, dass die Flüssigkeit auf den Partikeln bleibt und nicht abgetrocknet wird. Die Flüssigkeit soll auch nicht mit den Trägerstoffen reagieren, sondern sowohl die Flüssigkeit als auch der Trägerstoff sollen chemisch im Ausgangszustand verbleiben. Bei der Agglomeration ist es dagegen oft so, dass das Bindemittel den Feststoff löst, die flüssige Komponente in einem weiteren Prozessschritt abgetrocknet wird, und dadurch Feststoffbrücken zwischen den Partikeln hergestellt werden. Laut Sartor und Schubert [44] bezeichnet der Begriff Agglomerieren das Zusammenfügen disperser Stoffe zu größeren Stoffgebilden. Der Begriff des Granulierens ist umfassender, er bezieht sich nur auf die Form des Endproduktes und gibt keinerlei Auskünfte über die unterschiedlichen verfahrenstechnischen Prozesse, die zum Granulat führen. So umfasst dieser Begriff neben dem Agglomerieren zum Beispiel auch die Granulatbildung durch Zerkleinern. Wird eine Flüssigkeit zu einem dispersen Stoff gegeben und setzt man voraus, dass die Flüssigkeit den Feststoff benetzt (Randwinkel Θ < 90°), lässt sich nach Schubert [49] je nach Mengenanteil der Flüssigkeit die in Abbildung 16 dargestellte Einteilung treffen.
Abbildung 16: Flüssigkeitsverteilung in einem dispersen System [49]
Bei einer kleinen Flüssigkeitsmenge bilden sich einzelne Flüssigkeitsbrücken zwischen den Teilchen (Abbildung 16a). Dieser Bereich wird als Brückenbereich bezeichnet. Durch eine Steigerung des Flüssigkeitsanteils ergibt sich zunächst ein Übergangsbereich (Abbildung 16b), bei dem sowohl Flüssigkeitsbrücken als auch mit Flüssigkeit ausgefüllte Bereiche nebeneinander existieren und anschließend ein flüssigkeitserfüllter Bereich bzw. Kapillarbereich (Abbildung 16c). Wird der
Theoretische Grundlagen und Kenntnisstand 28
Flüssigkeitsanteil weiter erhöht, bildet sich ein Tropfen, in dessen Inneren oder an dessen Oberfläche sich Teilchen befinden können (Abbildung 16d). Auch Newitt und Conway-Jones [34] unterscheiden drei Zustände kugelförmiger Partikeln in Abhängigkeit vom Wassergehalt:
a) Wassergehalt niedrig: Das Wasser wird als diskrete Wasserbrücken mit punktförmiger Verbindung zu den Feststoffteilchen in der Schüttung gehalten.
b) Wassergehalt mittel: Die Flüssigkeit bildet ein zusammenhängendes Netz. c) Wassergehalt hoch: Die Flüssigkeit füllt die Kapillaren zwischen den Partikeln
vollständig aus.
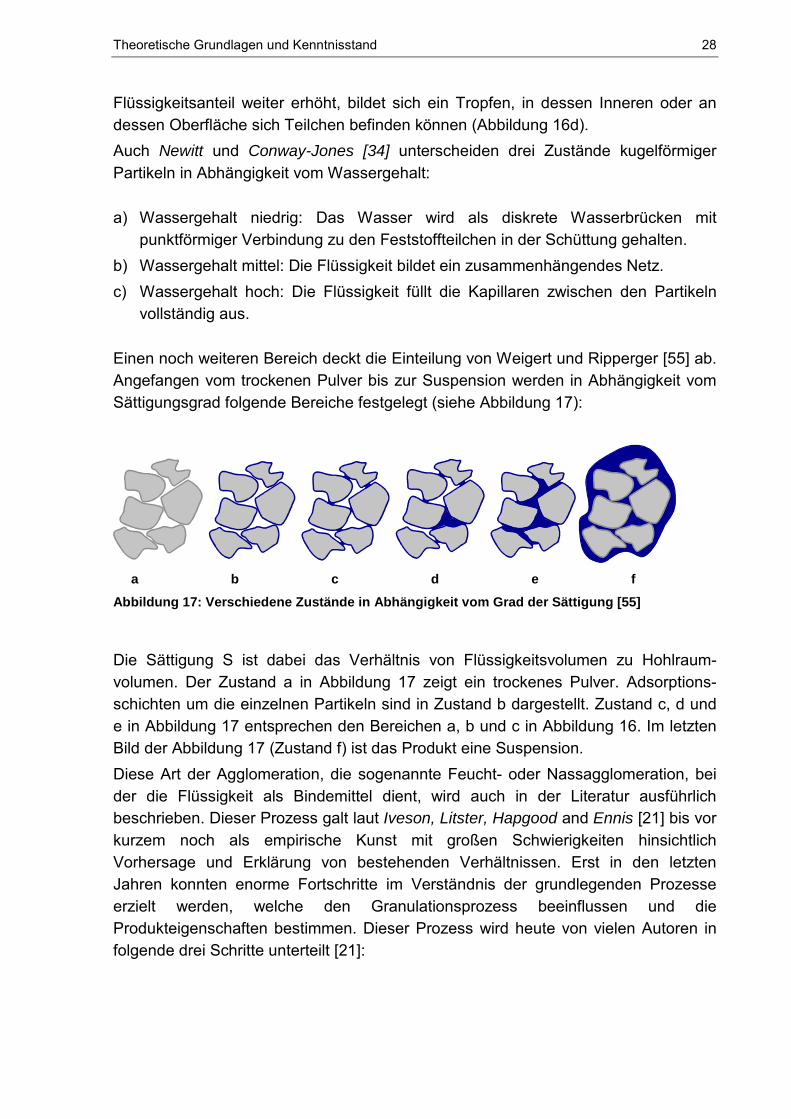
Einen noch weiteren Bereich deckt die Einteilung von Weigert und Ripperger [55] ab. Angefangen vom trockenen Pulver bis zur Suspension werden in Abhängigkeit vom Sättigungsgrad folgende Bereiche festgelegt (siehe Abbildung 17):
a b c d e f Abbildung 17: Verschiedene Zustände in Abhängigkeit vom Grad der Sättigung [55]
Die Sättigung S ist dabei das Verhältnis von Flüssigkeitsvolumen zu Hohlraum-volumen. Der Zustand a in Abbildung 17 zeigt ein trockenes Pulver. Adsorptions-schichten um die einzelnen Partikeln sind in Zustand b dargestellt. Zustand c, d und e in Abbildung 17 entsprechen den Bereichen a, b und c in Abbildung 16. Im letzten Bild der Abbildung 17 (Zustand f) ist das Produkt eine Suspension. Diese Art der Agglomeration, die sogenannte Feucht- oder Nassagglomeration, bei der die Flüssigkeit als Bindemittel dient, wird auch in der Literatur ausführlich beschrieben. Dieser Prozess galt laut Iveson, Litster, Hapgood and Ennis [21] bis vor kurzem noch als empirische Kunst mit großen Schwierigkeiten hinsichtlich Vorhersage und Erklärung von bestehenden Verhältnissen. Erst in den letzten Jahren konnten enorme Fortschritte im Verständnis der grundlegenden Prozesse erzielt werden, welche den Granulationsprozess beeinflussen und die Produkteigenschaften bestimmen. Dieser Prozess wird heute von vielen Autoren in folgende drei Schritte unterteilt [21]:

Theoretische Grundlagen und Kenntnisstand 29
(1) Benetzung und Keimbildung: Zunächst wird die als Bindemittel dienende Flüssigkeit mit dem trockenen Pulver in Kontakt gebracht und in der Schüttung verteilt, so dass eine Verteilung von gebildeten Keimen auftritt.
(2) Vereinigung und Wachstum: Hier führen die Zusammenstöße von gebildeten Agglomeraten untereinander, Agglomeraten mit Ausgangsprodukt und Agglomeraten mit Anlageteilen zur Kompaktierung und zum Wachstum.
(3) Abrieb und Bruch: In diesem Stadium zerbrechen die gebildeten Agglomerate auf Grund von Stößen, Abnutzung oder nachfolgender Produktbehandlung.
Wie beim Beladen und Tränken von Trägerstoffen ist es das Ziel des ersten Agglomerationsschrittes, die Flüssigkeit möglichst gleichmäßig auf den Partikeln zu verteilen. Auch dieser Schritt kann weiter unterteilt werden in die Keimbildung, die abhängig ist von der Thermodynamik und der Kinetik des Benetzens, und in die Flüssigkeitsverteilung, die eine Funktion der Prozessparameter darstellt. Bei einer schlechte Kombination aus Pulver und Bindemittel, beispielsweise mit einem großen Kontaktwinkel ist sowohl das Produkt als auch die Produktion sehr schwer zu kontrollieren. Gleiches gilt bei unzureichender Dispergierung des flüssigen Binde-mittels auf Grund eines zu hohen Massenstromes oder einer schlechten Sprühvorrichtung. [21] Außer der bisher diskutierten Verteilung der Flüssigkeit im Haufwerk kann noch ein weiterer Fall auftreten. Schæfer und Mathiesen [46] stellen bei der Schmelzagglomeration zwei verschiedene Mechanismen der Keimbildung in Abhängigkeit vom Verhältnis der Tropfengröße des Bindemittels zur Größe der Einzelpartikeln vor, die in Abbildung 18 dargestellt sind.
Feststoff Bindemittel Verteilung Vereinigung
Feststoff Bindemittel Immersion
Abbildung 18: Die Mechanismen der Keimbildung bei der Agglomeration in Abhängigkeit des Verhältnisses von Tropfengröße zu Einzelpartikel [46]
Im oberen Teil der Abbildung 18 wird ein Bindemittel zu dem Feststoff gegeben, welches sich dann gleichmäßig auf die Partikeln verteilt und diese dadurch, wie bereits beschrieben, agglomerieren. Im unteren Teil dagegen sind die

Theoretische Grundlagen und Kenntnisstand 30
Flüssigkeitstropfen größer als die Feststoffteilchen und stabil. Die Feststoffteilchen lagern sich in diesem Fall an den Tropfen an. Palzer [35] zeigt als mögliche Bindungsmechanismen für Flüssigkeiten in Pulverschüttungen die in Abbildung 19 dargestellten Varianten auf. Die Fälle a, b und d sind bereits ausführlich diskutiert worden. Der Fall c ist die oben angesprochene Variante, die laut Palzer [35] vorkommt, wenn eine nicht benetzende, erstarrende oder viskose Flüssigkeit in Form von Tropfen, die größer als die Einzelpartikeln der Schüttung sind, in diese eingebracht wird. In diesem Fall wird ein stabiler Tropfen mit anhaftenden Feststoffteilchen überzogen.
a b c d
Abbildung 19: Bindungsmechanismen für Flüssigkeiten in Pulverschüttungen [35]
Um die verschiedenen Bindungsvarianten nicht nur theoretisch zu diskutieren, sondern auch durch Messungen zu belegen, wurde die NMR-Spektroskopie eingesetzt, um Produkte aus Kieselsäure und Wasser zu untersuchen. Von Interesse ist nicht nur, wie die Flüssigkeit im Haufwerk verteilt ist, sondern vor allem die Menge an Flüssigkeit, die von einer Schüttung aufgenommen werden kann, ohne dass Inhomogenitäten wie feuchte Agglomerate auftreten. Kristensen [26] zeigte eine Methode, wie die sogenannte Überfeuchtung demonstriert werden kann. Er untersuchte den Sättigungsgrad von Agglomeraten während des Granulationsprozesses von Kalziumhydrogenphosphat (x50 ≈ 10 µm) mit einer wässrigen Lösung in einem Mischer (Fielder PMAT 25 high shear mixer).
Theoretische Grundlagen und Kenntnisstand 31
Feuchtegehalt [%]
Mitt
lere
Agg
lom
erat
größ
e x
[µm
]
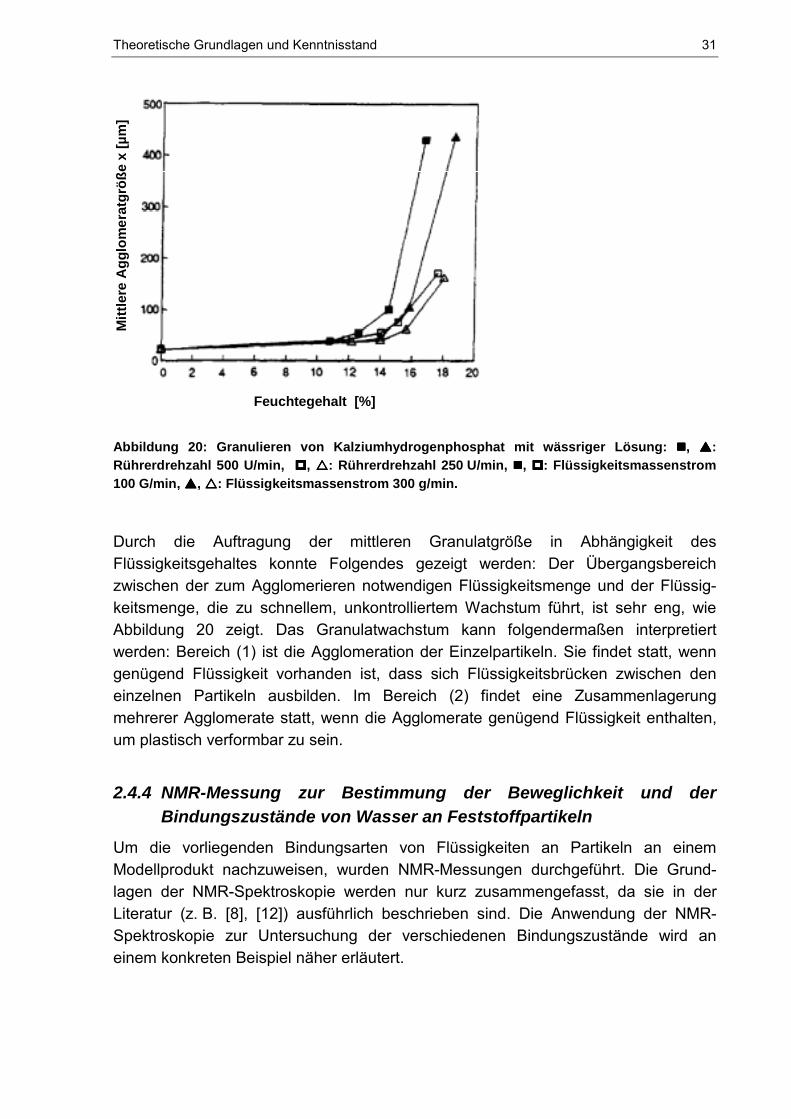
Abbildung 20: Granulieren von Kalziumhydrogenphosphat mit wässriger Lösung: ����, ����: Rührerdrehzahl 500 U/min, ����, ����: Rührerdrehzahl 250 U/min, ����, ����: Flüssigkeitsmassenstrom 100 G/min, ����, ����: Flüssigkeitsmassenstrom 300 g/min.
Durch die Auftragung der mittleren Granulatgröße in Abhängigkeit des Flüssigkeitsgehaltes konnte Folgendes gezeigt werden: Der Übergangsbereich zwischen der zum Agglomerieren notwendigen Flüssigkeitsmenge und der Flüssig-keitsmenge, die zu schnellem, unkontrolliertem Wachstum führt, ist sehr eng, wie Abbildung 20 zeigt. Das Granulatwachstum kann folgendermaßen interpretiert werden: Bereich (1) ist die Agglomeration der Einzelpartikeln. Sie findet statt, wenn genügend Flüssigkeit vorhanden ist, dass sich Flüssigkeitsbrücken zwischen den einzelnen Partikeln ausbilden. Im Bereich (2) findet eine Zusammenlagerung mehrerer Agglomerate statt, wenn die Agglomerate genügend Flüssigkeit enthalten, um plastisch verformbar zu sein.
2.4.4 NMR-Messung zur Bestimmung der Beweglichkeit und der Bindungszustände von Wasser an Feststoffpartikeln
Um die vorliegenden Bindungsarten von Flüssigkeiten an Partikeln an einem Modellprodukt nachzuweisen, wurden NMR-Messungen durchgeführt. Die Grund-lagen der NMR-Spektroskopie werden nur kurz zusammengefasst, da sie in der Literatur (z. B. [8], [12]) ausführlich beschrieben sind. Die Anwendung der NMR-Spektroskopie zur Untersuchung der verschiedenen Bindungszustände wird an einem konkreten Beispiel näher erläutert.

Theoretische Grundlagen und Kenntnisstand 32
2.4.4.1 Grundlagen der NMR-Spektroskopie Die NMR (Nuclear Magnetic Resonance) - Spektroskopie ist heute ein unentbehrliches Werkzeug für Chemiker, Biochemiker, Biologen, Physiker und Mediziner [12]. Das Hauptanwendungsgebiet der NMR-Spektroskopie ist die Strukturaufklärung von Molekülen. Aber auch in der Lebensmittelindustrie gibt es zahlreiche Anwendungsmöglichkeiten, bei denen Produkteigenschaften, wie Wassergehalt, Proteingehalt, Öl- bzw. Fettgehalt oder der Festfettanteil in Fetten, bestimmt werden können [58]. Ein konkretes Beispiel ist die Untersuchung der Wasserbindung in Lebensmitteln. Das Verfahren, das von F. Bloch und E. M. Purcell 1946 entwickelt wurde, beruht auf der Resonanz-Wechselwirkung zwischen einem hochfrequenten magnetischen Wechselfeld und bestimmten Atomkernen [8]. Die Voraussetzung für die Anwendung der NMR sind die Atome mit magnetischen Dipolmomenten in der zu untersuchenden Probe [17]. Viele Kerne haben einen Kern- oder Eigendrehimpuls P, der mit Gleichung 30 beschrieben werden kann [12]:
�⋅+⋅= 1)(ΙΙP (Gleichung 30)
mit 2πh=� und das Plancksche Wirkungsquantum h = 6,6256 ⋅ 10-34 J ⋅ s;
Ι = Drehimpuls- oder Kernspinquantenzahl: Ι = 0, ½, 1, 3/2, 2,...,6; Mit dem Drehimpuls P ist das magnetische Moment µ verknüpft: Pc ⋅=µ , wobei das gryomagnetische Verhältnis c eine Proportionalitätskonstante darstellt, die für jedes Isotop der verschiedenen Elemente eine charakteristische Konstante ist. Aus den oben genannten Gleichungen ergibt sich folgender Zusammenhang [12]:
�⋅+⋅⋅= 1)(ΙΙcµ (Gleichung 31)
Aus dieser Gleichung folgt, dass Kerne mit dem Kernspin Ι = 0 kein magnetisches Kernmoment besitzen [12]. Kerne, wie zum Beispiel das Wasserstoffisotop 1H, besitzen ein magnetisches Moment und richten sich, ähnlich wie eine Kompass-nadel, in einem äußeren Magnetfeld (0,0,1)BB 00 =
�
parallel oder antiparallel zu diesem aus [6]. Kerne mit Drehimpuls kann man sich vereinfacht als Stabmagneten (vgl. Abbildung 21) vorstellen, die ein um ihre Drehachse rotationssymmetrisches Magnetfeld ausbilden [17].

Theoretische Grundlagen und Kenntnisstand 33
Abbildung 21: Schematische Darstellung eines Protons als Stabmagnet [17]
Die zufällig angeordneten magnetischen Momente richten sich bei Anbringen eines äußeren, homogenen Magnetfeldes 0B
�
entweder parallel oder antiparallel zu diesem aus. Die parallele Ausrichtung des Dipolmomentes ist energetisch günstiger und wird deshalb häufiger eingenommen [12]. Atomkerne mit magnetischem Moment führen in einem äußeren Magnetfeld die sogenannte Präzessionsbewegung mit einer bestimmten Frequenz, der sogenannten Larmor-Frequenz, aus. Wird der Raum als kartesisches Koordinatensystem dargestellt, so stellt sich wie in Abbildung 22 veranschaulicht, die Präzession der Kernmagnetisierung M
�
um 0B�
ein.
Abbildung 22: (a) Kernmagnetisierung M�
ist parallel zu 0B�
; (b) M präzediert um 0B�
mit
Larmorfrequenz 0ν ; (c) Wird M�
in eine zu 0B�
senkrecht liegende Ebene ausgelenkt, wird ein
maximales Amplitudensignal gemessen [7]
Wird ein magnetisches Wechselfeld 1B�
mit der Frequenz ν senkrecht zu 0B�
angelegt, so können Kerne bei Übereinstimmung von ν mit 0ν in ein höheres Energieniveau eintreten. Wirkt auf ein Spinsystem ein Hochfrequenz-Impuls, so dreht sich die Magnetisierung M
�
aus der Richtung des statischen Magnetfeldes 0B�
. Es wird eine x,y-Magnetisierung induziert. In Abbildung 23 wird ein solcher Impuls dargestellt.

Theoretische Grundlagen und Kenntnisstand 34
Abbildung 23: Erzeugung der x,y-Magnetisierung durch den Impuls B1 [7]
Ist die Zeit des Impulses genügend lang, um ein Kippen der Magnetisierung um 90° zu erzielen, so wird von einem 90 °-Impuls gesprochen, bei 180 ° entsprechend von einem 180 °-Impuls. Die Magnetisierung M
�
strebt anschließend wieder der Ausgangsorientierung zu, die energetisch günstiger ist. Dieser Vorgang wird durch die Relaxationszeiten T1 und T2 beschrieben. T1 charakterisiert die Relaxation der z-Komponente der Magnetisierung in ihr Gleichgewicht. Sie wird auch als longitudinale Relaxationszeit bezeichnet. Die Relaxation der Quermagnetisierung beschreibt hingegen T2. Aus der longitudinalen Relaxationszeit (T1) und der transversalen Relaxationszeit (T2) lassen sich Rückschlüsse über die Bindungsart des Wassers ziehen [7][29]. T1- und T2-Messungen sind ein weit verbreitetes Instrument zur zerstörungsfreien Analyse von Feststoffen und Flüssigkeiten. Dies beruht darauf, dass bei der Protonen-Relaxation freies Wasser wesentlich längere Relaxationszeiten aufweist als chemisch gebundenes oder physikalisch immobilisiertes Wasser. [58][59]
2.4.4.2 Anwendung der NMR Buggisch und Götz [6] stellten fest, dass man mittels NMR zeigen kann, welcher Mengenanteil an destilliertem Wasser in einem Feststoff- und Flüssigkeitsgemisch an den Feststoff gebunden bzw. immobilisiert und welcher Anteil frei vorliegt. Die Kernresonanz-Methode liefert nicht nur die Protonendichte und damit die Feuchte, sondern auch Informationen über die Art der Bindung des Wassers an den Feststoff. Es kann zwischen Kristallwasser, gebundenem und freiem Wasser unterschieden werden und deren Anteile an der Gesamtfeuchte ermittelt werden. Dabei ist grundsätzlich zu beachten, dass auf verschiedene Eigenschaften des Wassers beruhende Auslegungen von gebundenem Wasser existieren. Bei Lebens-mitteln können zum Beispiel folgende Definitionen hinsichtlich der thermo-dynamischen Eigenschaften von gebundenem Wasser unterschieden werden [59]: � Wasser, das bei einer bestimmten relativen Luftfeuchte und Temperatur an
das Lebensmittel gebunden ist, oder
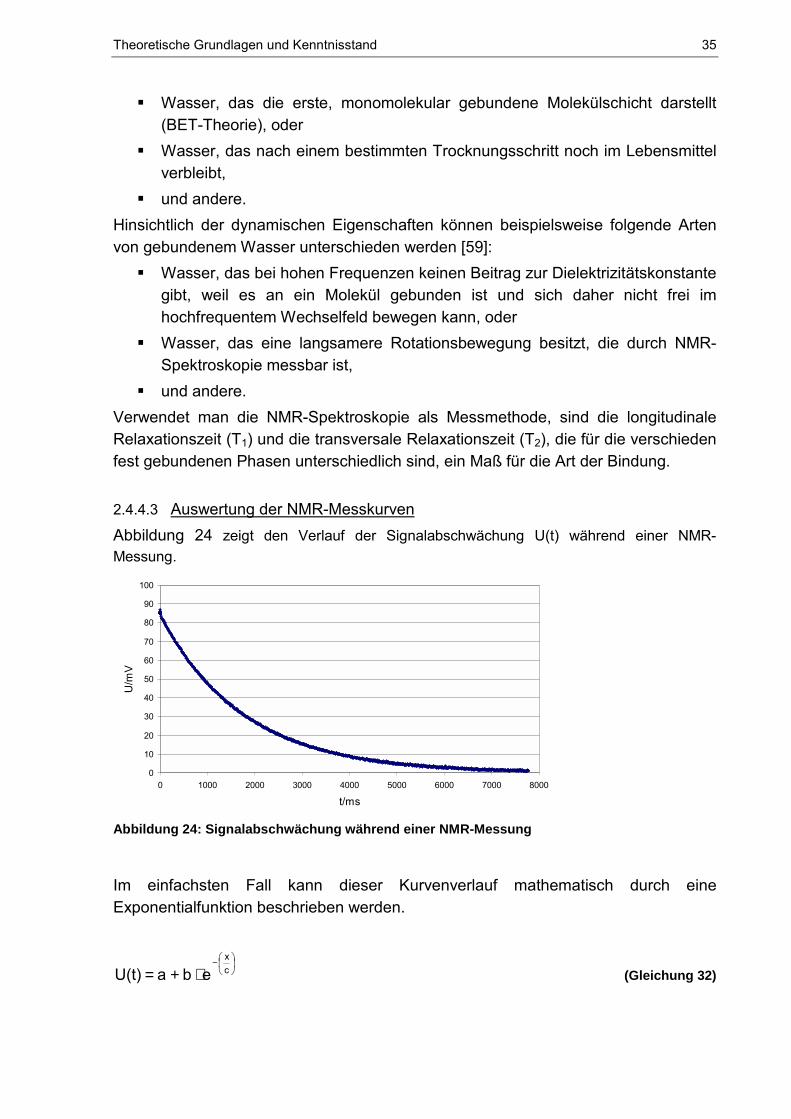
Theoretische Grundlagen und Kenntnisstand 35
� Wasser, das die erste, monomolekular gebundene Molekülschicht darstellt (BET-Theorie), oder
� Wasser, das nach einem bestimmten Trocknungsschritt noch im Lebensmittel verbleibt,
� und andere. Hinsichtlich der dynamischen Eigenschaften können beispielsweise folgende Arten von gebundenem Wasser unterschieden werden [59]: � Wasser, das bei hohen Frequenzen keinen Beitrag zur Dielektrizitätskonstante
gibt, weil es an ein Molekül gebunden ist und sich daher nicht frei im hochfrequentem Wechselfeld bewegen kann, oder
� Wasser, das eine langsamere Rotationsbewegung besitzt, die durch NMR-Spektroskopie messbar ist,
� und andere. Verwendet man die NMR-Spektroskopie als Messmethode, sind die longitudinale Relaxationszeit (T1) und die transversale Relaxationszeit (T2), die für die verschieden fest gebundenen Phasen unterschiedlich sind, ein Maß für die Art der Bindung. 2.4.4.3 Auswertung der NMR-Messkurven
Abbildung 24 zeigt den Verlauf der Signalabschwächung U(t) während einer NMR-Messung.
0
10
20
30
40
50
60
70
80
90
100
0 1000 2000 3000 4000 5000 6000 7000 8000
t/ms
U/m
V
Abbildung 24: Signalabschwächung während einer NMR-Messung
Im einfachsten Fall kann dieser Kurvenverlauf mathematisch durch eine Exponentialfunktion beschrieben werden.
−⋅+= c
x
ebaU(t) (Gleichung 32)

Theoretische Grundlagen und Kenntnisstand 36
Dabei stellt der Parameter c die T2-Zeit der untersuchten Probe dar. Diese Gleichung trifft allerdings nur bei absolut reinen, einphasigen Flüssigkeiten zu. Wenn mehr als eine Phase präsent sind, können die Anteile und die T2 Zeiten der einzelnen Phasen bestimmt werden. Dazu wird eine Kurve an die Messkurve mittles einer Summe aus Exponentialgleichungen oder aus Gausfunktionen angepasst. Ob Exponential- oder Gaußfunktionen verwendet werden, hängt davon ab, wie groß die Relaxationszeiten sind.
−
⋅−
⋅+⋅+= ∑i2,
2
2,1 Tt
i iT2t
1 ededxU(t) (Gleichung 33)
mit 1di i =∑
T2,1 < T2,2 < T2,3 < ... x: Achsenabschnitt d1, T2,1: Anteil und durchschnittliche T2 Zeit der immobilsten Phase
di, T2,i: Anteile und durchschnittliche T2 Zeiten der mobileren Phasen z Anzahl der unterscheidbaren Phasen
Können beispielsweise vier verschiedene Anteile unterschieden werden, gehören zu den T2-Zeiten T2,1, T2,2, T2,3 und T2,4 die entsprechenden Werte der Parameter d1, d2, d3 und d4. Mit diesen Werten können die Anteile a der auftretenden Wasser-bindungsarten an der Gesamtwassermasse berechnet werden, wie am Beispiel der immobilsten Phase mit der folgenden Gleichung gezeigt wird:
4321
11 d dd d
da+++
= (Gleichung 34)
2.5 Verfahren zum Herstellen von flüssigkeitsbeladenen Pulvern Für das Herstellen von flüssigkeitsbeladenen Pulverprodukten kommen verschie-dene Verfahren in Frage, die sich alle aus den in Abbildung 25 dargestellten Einzelschritten zusammensetzen. [35] Das Ziel dieser Verfahren ist es immer, die Flüssigkeit vollständig und homogen auf den Partikeln der Schüttung zu verteilen. Dabei kommt es in den meisten Fällen zu einer Agglomeration, d. h. mehrere Partikeln schließen sich durch Flüssigkeits-brücken zu größeren Gebilden zusammen. Diese Agglomeration kann durchaus erwünscht sein, wird aber ab einem gewissen Grad unerwünscht: Wenn sich feuchte Agglomerate bilden und ein großer Teil der Flüssigkeit in diesen Agglomeraten gebunden wird, so dass kein homogenes Produkt mehr vorliegt. Deswegen wird zwischen einer homogenen Agglomeration der Schüttung infolge der Flüssigkeits-

Theoretische Grundlagen und Kenntnisstand 37
zugabe und einer partiellen Agglomeration einer kleinen Teilmenge des Partikelkollektives unterschieden. [35] Um solche großen Agglomerate in der Schüttung wieder zu zerstören, können in Mischern zum Beispiel sogenannte Zerhacker eingesetzt werden.
Abbildung 25: Einzelne Prozessschritte beim Anreichern von Pulvern mit Flüssigkeiten
Im Folgenden werden zwei Verfahren beschrieben, die zur Herstellung von flüssigkeitsbeladenen Pulverprodukten verwendet werden können und im Rahmen dieser Arbeit eingesetzt wurden. Neben diesen gibt es noch eine Vielzahl anderer Methoden, um Flüssigkeiten auf Trägerstoffe aufzubringen. Während es beim vorliegenden Projekt das Ziel war, möglichst viel Flüssigkeit auf Trägerstoffen zu fixieren, werden die meisten dieser Verfahren dazu genutzt, Agglomerate zu produzieren, die unter anderem für Instantprodukte eingesetzt werden. Die Flüssigkeit kann dabei in Form von kondensiertem Dampf, von versprühter Flüssigkeit oder als Mischung von beidem aufgebracht werden. Die wichtigsten Verfahren sind in Abbildung 26 dargestellt. [48]
Zerstäuben der Flüssigkeit
Zertropfen der Flüssigkeit
Zugabe als Kontinuum
Fluidisieren des Pulvers Mechanisch oder pneumatisch
Mischen und Benetzen des Pulversmit der Flüssigkeit Mechanische oder pneumatischeDurchmischung, ggf. Einsatz von Messerköpfen oder Schneidmühlen
bewegte Pulverschüttung
Flüssigkeitskontinuum grobe Tropfen
Aerosol
Produkt Fließfähigkeit, Partikelgröße Konzentration der wirksamen KomponenteHomogenität, u. a.

Theoretische Grundlagen und Kenntnisstand 38
A
L
P
P + A
L
G
G
L
P + A
P
A + G
G
L
(Dampf)
P
AA
LLP Trocknen
BrechenKühlen
A
a b c
d e f
Abbildung 26: Feuchtagglomerationsverfahren mit den Teilschritten Befeuchten, Agglo-merieren, Trocknen und evtl. Brechen: (a) Fließbett, (b) Mischer, (c) Teller (Rollagglomeration, (d) Strahl, (e) mittels rotierender Scheibe oder (f) auf einem Band, Trocknen und evtl. Brechen Abkürzungen: A = Agglomerat, G = Gas, L = Flüssigkeit oder Dampf, P = Pulver [48] Während Fließbett-, Misch- und Strahlagglomeration hauptsächlich zur Instanti-sierung eingesetzt werden, ist die Rollagglomeration im Agglomerierteller für diesen Zweck nicht gut geeignet. Hier entstehen beim Abrollen gleichmäßige Agglomerate mit einer engen Porengrößenverteilung, die zum Wiederbefeuchten eher von Nachteil ist. Das Verfahren mittels Bandagglomerierer stellt insofern eine Sonderform dar, als das dies die einzig aufgeführte statische Methode ist. Hier wird das Produkt nach dem Trocknen und Kühlen erst am Ende in die gewünschte Agglomeratgröße gebrochen. [48]
2.5.1 Wurfmischer mit Eindüsvorrichtung
Mischaggregate können anhand verschiedener Kriterien in Gruppen eingeteilt werden. Eine Möglichkeit ist die Einteilung nach Art der Bewegungserzeugung: Mischer mit bewegten Mischbehältern, Mischer mit bewegten Mischwerkzeugen oder Mischer, in denen beides bewegt wird. Weiterhin existieren Schwerkraftmischer, statische oder pneumatische Mischer und mischende Lagerverfahren, wie Misch-betten oder Mischhalden.

Theoretische Grundlagen und Kenntnisstand 39
Bei allen Mischern mit rotierender Bewegung kann die Intensität der Drehbewegung durch das Verhältnis von Zentrifugalbeschleunigung ω zu Erdbeschleunigung g gekennzeichnet werden. Dieses Verhältnis wird Froudezahl genannt.
gωRFr
2⋅= (Gleichung 35)
Diese sogenannte Froudezahl Fr stellt eine dimensionslose Drehfrequenz dar. In ihr kommt keine Stoffeigenschaft vor. Pflugschar-, Schleuder- und Mehrstromfluid-mischer arbeiten im Bereich von Froudezahlen größer als eins, was zur Folge hat, dass zumindest in der Nähe der Werkzeugaussenkante die Zentrifugalkraft größer als die Gewichtskraft wird und die Partikeln geworfen werden. Es findet statt einer Schubbewegung eine Flugbewegung statt. Dabei nehmen bei sehr hohen Froude-zahlen die Scherkräfte auf das Mischgut stark zu und die Prallbeanspruchung ist enorm. Das Produkt heizt sich durch die dissipierte Energie stark auf, wobei die Wärme durch die Reibung zwischen den Mischwerkzeugen und dem Feststoff sowie durch die Reibung der Feststoffpartikeln untereinander entsteht. [56] Solche Mischer mit Froudezahlen größer eins werden auch für die Agglomeration und zur Befeuchtung eingesetzt, denn ein Prozessschritt bei der Herstellung flüssigkeits-beladener Pulver ist das Fluidisieren der Pulverschüttung. Fluidisieren ist das in Schwebe bringen von Feststoffpartikeln. Die Feststoffschüttung beginnt beim Fluidisieren zu fließen, die einzelnen Partikeln berühren sich nicht mehr und sind frei zugänglich für die Flüssigkeit, mit der sie in Kontakt gebracht werden sollen.
2.5.2 CPF-Sprühverfahren Im Kapitel 2.1 wurden neben den klassischen Verfahren zur Partikelherstellung dem Mahlen und dem Kristallisieren sowie dem Strahlschneideverfahren, das in dieser Arbeit zum Einsatz kam, auch Hochdruckverfahren wie RESS (Rapid Expansion of Supercritical Solutions), GAS (Gas-Antisolvent Crystallization) and PCA (Particles with Compressed Fluid Antisolvent) vorgestellt. Ein solches Hochdruckverfahren wurde auch zum Beladen von Trägerstoffen entwickelt, das sogenannte CPF-Verfahren (Concentrated Powder Form) [15], [57]. Bei diesem Verfahren wird die Flüssigkeit, die auf den Trägerstoff aufgebracht werden soll, in einem statischen Mischer mit überkritischem CO2 gemischt und diese gesättigte Lösung über eine Düse entspannt. In den feinen Sprühnebel wird ein pulverförmiger Trägerstoff dosiert, der die feinen Tröpfchen aufnimmt. Das Prinzip des Verfahrens ist in Abbildung 27 dargestellt.

Theoretische Grundlagen und Kenntnisstand 40
12 3
4
5
6
7
7
Abbildung 27: Prinzip des CPF-Sprühturmes: (1) Pulverförmiger Trägerstoff; (2) Flüssigkeit; (3) Überkritisches CO2; (4) Statischer Mischer; (5) Düse; (6) Zyklon; (7) Beladenes Produkt.
Ein großer Vorteil dieses Verfahrens ist es, dass auch hochviskose Flüssigkeiten, wie zum Beispiel Gewürzextrakte, verarbeitet werden können, weil durch das Mischen der Komponenten mit überkritischem Gas eine dünnflüssige Lösung entsteht, die versprüht werden kann. Das beruht auf einer Absenkung der Viskosität und der Oberflächenspannung. Dazu wird das in Abbildung 27 dargestellte Verfahren zusätzlich mit einem Autoklaven ausgestattet, der zur Gleichgewichtseinstellung geschüttelt werden kann. Der hochviskose Rohstoff wird in diesem bei geeigneter Druck- und Temperatureinstellung mit überkritischem Gas gemischt. Dabei können sich maximal drei Phasen ausbilden: Überkritisches Gas in dem Flüssigkeit gelöst ist, Flüssigkeit, in der überkritisches Gas gelöst ist und entweder Flüssigkeit oder überkritisches Gas in reiner Form. Diese Mischung wird von dort zur Düse gefördert. In ihrer Dissertation beschrieb Grüner [15] unter anderem die maximale Aufnahme-kapazität verschiedener Trägerstoffe, wie Kieselsäuren, Maisstärken, Cellulosen, Maltodextrine und Emulgatoren im CPF-Verfahren. Als flüssige Komponenten kamen in dieser Arbeit Gewürzextrakte zum Einsatz. Das Ergebnis war, dass die Belad-barkeit der Trägerstoffe sehr stark von deren Partikelform und deren Stampfdichte abhängt. Dazu wurden die Trägerstoffe zunächst in drei Gruppen unterteilt: Feinkörnige, kugelförmige Trägerstoffpartikeln, grobkörnige, flockige und faserige Trägerstoffpartikeln sowie Emulgatorpartikeln. Von den Trägerstoffen wurde eine sogenannte „Rütteldichte“ bestimmt und die maximale Aufnahmekapazität der Trägerstoffe in Abhängigkeit dieser „Rütteldichte“ aufgetragen. Da diese Ergebnisse hinsichtlich des Einflusses von Trägerstoffmerkmalen auf die maximale

Theoretische Grundlagen und Kenntnisstand 41
Flüssigkeitsaufnahmekapazität in der vorliegenden Arbeit genauer untersucht und modelliert werden sollen, sind die Ergebnisse der Arbeit von Grüner hier noch einmal in Tabelle 3 und Abbildung 28 zusammengestellt. Tabelle 3: Maximaler Extraktgehalt (Thymianextrakt) auf verschiedenen Trägerstoffen [15]
Trägerstoffgruppe Trägerstoff „Rütteldichte“ [kg/m³]
Max. Extraktgehalt [Gew.-%]
Cabosil M5 56 80
Sipernat 22 270 60
Maisstärke 7598 670 25
HI-FLO* 720 20
C*EmCap 06376 850 20
C*EmCap 06377 850 20
Vivapur 105 500 40
Feinkörnige, kugelförmige
Trägerstoffpartikel
C*Dry MD 019R9 530 35
C*Pulp Tex 12930 320 50
C*Gel-Instant 530 35
Aero Myl 33** 110 60
Vitacel L 600-20 500 40
C*Dry MD 01955 560 30
Grobkörnige, flockige, fasrige
Trägerstoffpartikel
C*Dry MD 01997 140 50
Lamegin 750 15
Lamegin pulverisiert 210 40
Dimodan PV* 620 20 Emulgatorpartikel
Dimodan PV pulverisiert 170 40
*Paprikaextrakt, **Muskatextrakt

Theoretische Grundlagen und Kenntnisstand 42
0
20
40
60
80
100
0 200 400 600 800 1000
Rütteldichte des Trägerstoffes in kg/m³
Max
imal
er E
xtra
ktge
halt
in %
Feinkörnig, kugelförmig Grobkörnig, flockig Emulgatoren
Abbildung 28: Maximaler Extraktgehalt in Abhängigkeit von der „Rütteldichte“ der verschiedenen Trägerstoffe [15]
Als flüssige Komponenten wurden 16 Extrakte aus elf verschiedenen Gewürzen (u. a. Ingwer, Oregano, Paprika, Pfeffer, usw.) verwendet. Als Trägerstoff diente unter anderem die Kieselsäure Cabosil M5. Diese Extrakte unterscheiden sich stark hinsichtlich ihrer Viskosität. Die Versuche zeigten, dass der Einfluss der Extrakte auf die maximale Beladbarkeit nur von geringer Bedeutung ist: Alle Extrakte, gleich in welcher Zusammensetzung und von welcher Viskosität, ließen sich versprühen und die Kieselsäure Cabosil M5 ließ sich unabhängig von der Extraktsorte stets mit der gleichen Menge beladen, bis das Maximum erreicht wurde. [15]

Material und Methoden 43
3 Material und Methoden 3.1 Messmethoden
3.1.1 Messmethoden zur Partikelcharakterisierung
3.1.1.1 Messung der Partikelgrößenverteilung Die Partikelgrößenverteilungen der dispersen Feststoffe wurden mittels Laser-beugung aufgenommen. Zum Einsatz kam das Messgerät Helos der Firma Sympatec (Clausthal-Zellerfeld) in Verbindung mit verschiedenen Dispergiereinheiten. Bei der Dispergiereinheit Rhodos werden die zu vermessenden Partikeln über eine Vibrationsrinne in eine luftdurchströmte Düse gefördert, in der sie vereinzelt werden und im Luftstrom durch einen Laserstrahl transportiert werden. Weiterhin steht ein Fallschacht zur Verfügung, bei dem die Partikeln ohne mechanische Beanspruchung durch die Messzone rieseln. Auch eine flüssige Dispergierung zur Messung von Teilchengrößen in Suspensionen ist möglich. Aus der gemessenen Beugung dieses Laserstrahles lässt sich die jeweilige Partikelgröße berechnen. Als Ergebnis wird die Summen- oder Dichteverteilung in Abhängigkeit vom Äquivalentdurchmesser beugungsgleicher Kugeln aufgetragen. 3.1.1.2 Bestimmung von Feststoffdichte, Schütt- und Stampfdichte Die Feststoffdichte wurde nur zum Teil gemessen. Einige Werte konnten aus der Literatur entnommen werden. Zur Messung wurde ein Autopyknometer der Firma Micromeritics Instrument Corporation (Norcross, USA) verwendet. In die Probenkammer des Autopyknometers werden 2-3 g getrocknete Probe eingewogen. Anschließend wird das reine Feststoffvolumen der Probe, wie in Kapitel 2.2.3 erläutert, bestimmt. Das Verhältnis der Masse zum Feststoffvolumen der Probe entspricht der Feststoffdichte. Um die Schüttdichte zu messen, kam eine einfache Vorrichtung zum Einsatz, die in Abbildung 29 links abgebildet ist. Nach dem Öffnen eines Schiebers wird das Pulver auf reproduzierbare Art und Weise aus definierter Höhe in ein zylindrisches Gefäß mit definiertem Volumen gefüllt. Die überschüssige Pulvermasse wird mit einem Schaber abgestrichen und der Quotient aus dem Volumen des Gefäßes und der enthaltenen Masse bestimmt. Um die Stampfdichte zu ermitteln, wurde ein Stampf-Volumeter vom Typ PT-TD (siehe Abbildung 29 rechts) der Firma Pharma Test (Hainburg) verwendet. Etwa 200 cm3 des Probengutes werden in einen Glaszylinder eingewogen und durch Hübe mit einer Hubhöhe von 3 ± 0,1 mm verdichtet. Nach je 1250 Hüben wird das Volumen abgelesen und die Versuchsreihe wird beendet, wenn bei zwei aufeinanderfolgenden Versuchen der Volumenunterschied weniger als 2% beträgt.

Material und Methoden 44
Abbildung 29: Vorrichtungen zur Bestimmung der Schüttdichte (links) und der Stampfdichte (rechts)
3.1.1.3 Trockensubstanzbestimmung Um die Trockenmasse der Trägerstoffe zu ermitteln, wurden diese für etwa 14 Stunden im Trockenschrank bei 105 °C aufbewahrt. Durch Wägung der Massen vor und nach dem Trocknen kann die Trockensubstanz berechnet werden. 3.1.1.4 Messung der Oberfläche durch Stickstoffadsorption nach BET Für die Stickstoffadsorptionsmessungen diente das Messgerät TriStarTM 3000 der Firma Micromeritics Instrument Corporation (Norcross, USA). Dieses vollautomatische Gasadsorptionsgerät nutzt physikalische Adsorptions- und Kapillarkondensationsphänomene, um Aussagen über die Oberfläche und die Porosität der Proben zu machen. Alle Messungen wurden bei 20 °C durchgeführt. 3.1.1.5 Aufnahme der Wasserdampfsorptionsisothermen Von einigen Pulvern wurden die Wasserdampfsorptionsisothermen bei 20 °C aufgenommen. Dazu diente das Gerät DVS (Dynamic Vapour Sorption) der Firma Porotec GmbH (Hofheim). Dieses Gerät ist mit einer sehr präzisen Waage ausgerüstet, und ein Video-Mikroskop erlaubt die Betrachtung einer Probe bis zur 200-fachen Vergrößerung. Für die Versuchsreihen wurde jeweils eine kleine Menge der Probe auf die Waagschale gebracht und zunächst getrocknet. Anschließend wird über die Probe ein Luftstrom mit 20 °C und definierter relativer Feuchte geleitet. Die relative Feuchte wird von 10 % stufenweise auf 90 % erhöht. Dabei wird ein Wert solange konstant gehalten, bis die Probe keine weitere Massenzunahme erfährt, dass heißt, bis die Probe kein weiteres Wasser aus dem Luftstrom mehr aufnimmt. Danach wird die relative Feuchte wieder von 90 % auf 10 % stufenweise absenkt. Aus den gewonnen Daten können dann die Sorptions- und die Desorptionsisotherme ermittelt und die Oberfläche der Probe mittels BET Auswertung berechnet werden.

Material und Methoden 45
3.1.2 Messmethoden zur Produktbeurteilung
3.1.2.1 Schnelltest zur Bestimmung der freien Flüssigkeit Um Sprühprodukte mit unterschiedlichen Flüssigkeitskonzentrationen untereinander vergleichen zu können und die maximale Aufnahmekapazität zu ermitteln, wurde im Rahmen dieser Arbeit ein Schnelltest entwickelt. Von den hergestellten Sprüh-produkten kommt eine Probe von etwa 5 g auf zwei übereinander liegende Rundfilterpapiere (Schleicher & Schuell, 5891, Schwarzband). Die Probe wird auf eine definierte Fläche für eine Minute mit einem Gewicht belastet. Anschließend bestimmt man die Gewichtszunahme der Filter gravimetrisch. Je höher das Pulver beladen ist, desto mehr Flüssigkeit geht von dem Pulver auf den Filter über.
Abbildung 30: Schnelltest zur Bestimmung der "freien" Flüssigkeit“ in der Schüttung
Es muss bei dieser Methode berücksichtigt werden, dass die absoluten Mengen an entzogener Flüssigkeit aus Sprühprodukten mit unterschiedlichen Trägerstoffen nicht direkt miteinander verglichen werden können. Die Konkurrenzverhältnisse zwischen dem Filter und dem Porensystem der Schüttung bestimmen die absolute Menge an Flüssigkeit, die vom Pulver auf den Filter übergeht. Da die Trägerstoffe eine ganz unterschiedliche Porenstruktur besitzen, werden in Abhängigkeit vom Trägerstoff unterschiedliche absolute Mengen an das Porensystem des Filters abgegeben. Aber der Verlauf der Kurven zeigt an, ab welcher Beladung sich zuviel Flüssigkeit auf einem Trägerstoff befindet und somit ist es möglich, verschiedene Trägerstoffe miteinander zu vergleichen. Mit diesem Test wurde ein Werkzeug geschaffen, um die maximale Flüssigkeitsaufnahmekapazität von Trägerstoffen zu ermitteln.
Rundfilter
Gewicht: 500 g
Bodenplatte
Pulverprobe

Material und Methoden 46
3.1.2.2 Bestimmung des Fettgehaltes nach Soxhlet Für die Bestimmung des Fettgehaltes der Sprühprodukte, die mit Speiseöl beladen wurden, wurden je 10 g der Probe in fettfreie Extraktionshülsen (Schleicher & Schüll Nr. 33/90) eingewogen und in einer Extraktionsapparatur nach Soxhlet [30] vier Stunden lang mit Diethylether extrahiert. Am Ende der Versuche wurde das Lösungsmittel abdestilliert und der Kolben nach dem Trocknen und Abkühlen rückgewogen. Der prozentuale Fettanteil FG kann nach folgender Formel berechnet werden [30]:
[ ] 100E
mm%FG 12 ⋅−= % (Gleichung 36)
E: Einwaage m1: Masse des Kolbens nach der Extraktion m2: Masse des Kolbens vor der Extraktion 3.1.2.3 NMR-Spektroskopie Für die NMR-Messungen kamen ein gepulstes, niedrigauflösendes Protonen-Kernresonanzspektrometer zum Einsatz. Das Wort Protonen bedeutet in dieser Bezeichnung, dass Wasserstoffkerne nachgewiesen werden und niedrigauflösend drückt aus, dass keine Protonenspezies, wie CH-, CH2, OH-Protonen unterschieden werden können. Lediglich die Phasen (mobil, immobil), in der sich die Protonen befinden, werden erkannt. Gepulst bedeutet, dass Hochfrequenzimpulse in die Probe eingestrahlt werden [17]. Für die T2-Messungen wurde das niedrigauflösende NMR-Spektrometer minispec NMQ 20 der Firma Bruker Analytik (Rheinstetten) verwendet. Der Permanentmagnet dieses Messgerätes weist eine konstante Temperatur von 40 °C und eine Feldstärke von 0,47 Tesla auf. Daraus ergibt sich eine Frequenz des HF-Feldes von 20 MHz. In einem Zeitraum von 0 bis 0,2 ms nach dem 90°-Impuls wird der FID gemessen. Danach wird die CPMG-Impulsfolge eingesetzt. Insgesamt werden 19.600 Einzel-signale mit einem zeitlichen Abstand von 0,2 ms aufgezeichnet. Das Probenmaterial wird in Probenröhrchen aus protonenfreiem Spezialglas gefüllt. Die optimale Füllhöhe beträgt 11 mm. Die Messungen erfolgten bei einer Probentemperatur von 6 °C, die durch einen temperierbaren Probenkopf konstant gehalten wird. Alle Versuchsergebnisse sind mit dem Programm TableCurve 2D, Version 4.0 der Firma SPSS Science (Chicago) ausgewertet worden. Die mathematischen Grund-lagen dazu sind in Kapitel 2.4.4.3 erläutert.

Material und Methoden 47
3.2 Rohstoffe Viele Größen der Rohstoffe wurden im Rahmen der vorliegenden Dissertation gemessen. Die Ergebnisse sind Mittelwerte aus Dreifachbestimmungen, die mit einem Konfidenzintervall für 95 % Wahrscheinlichkeit angegeben werden. Andere Daten wurden aus Herstellerangaben entnommen uns sind als solche kenntlich gemacht.
3.2.1 Trägerstoffe
Die verwendeten Trägerstoffe dienen dazu, mit verschiedenen Verfahren aufgesprühte Flüssigkeit aufzunehmen. In der Lebensmittel- und Pharmaindustrie kommen dazu verschiedene Stoffe in Frage. Die wichtigsten Stoffgruppen sind Stärken, Maltodextrine, Zellulosen und Kieselsäuren. Im Rahmen des Projektes wurden neben diesen praxisrelevanten Trägerstoffen auch Modellpartikeln eingesetzt. Zum einen waren dies runde monodisperse Zellulosepartikeln und zum anderen wurden selbst Partikeln mittels Strahlschneiderverfahren hergestellt. 3.2.1.1 Stärken Stärken dienen bei Pflanzen als Reservekohlenhydrat und sind als Bestandteil vieler Lebensmittel die wichtigste Kohlenhydratquelle für die menschliche Ernährung [2]. Als Rohstoffe für die Stärkegewinnung werden hauptsächlich Mais, Kartoffel, Maniok und Weizen verwendet. Im Folgenden werden die Stärken, die für die Versuche verwendet werden, vorgestellt. K a r t o f f e l s t ä r k e A e r o M y l 3 3
Die Kartoffelstärke Aero Myl 33 wird von der Firma Südstärke (Schrobenhausen) hergestellt. Aero Myl 33 ist eine physikalisch modifizierte Kartoffelstärke, die in kaltem Wasser löslich ist. Folgende physikalische Eigenschaften sind nach den oben beschriebenen Methoden ermittelt bzw. vom Hersteller angegeben worden.

Material und Methoden 48
Tabelle 4: Physikalische Eigenschaften von Kartoffelstärke Aero Myl 33
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] 1500 ± 10 -
Schüttdichte [kg/m³] 123 ± 2 110
Stampfdichte [kg/m³] 186 ± 5 -
x50 - Wert [µm] 188 ± 25 90% < 1 mm (Siebanalyse)
Trockensubstanzgehalt [%] 95,2 minimal 92 %
Spez. Oberfläche (BET) [m²/g] 0,47 -
Spez. Oberfläche (DVS) [m²/g] 205,6 -
Die Partikelgrößenverteilung ist in Abbildung 31 dargestellt.
Abbildung 31: Partikelgrößenverteilung von Aero Myl 33 (Laserbeugung)
Die Form der Partikeln ist anhand der rasterelektronenmikroskopischen Aufnahmen in Abbildung 32 zu erkennen. Die Partikeln sind sehr unregelmäßig geformt und weisen eine große Anzahl an Poren und Hohlräume auf. Die Struktur erinnert an Blätterteig.
0
20
40
60
80
100
0 100 200 300 400 500 600 700 800
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 49
300 µm 150 µm
Abbildung 32: REM Aufnahmen der Aero Myl 33 Partikeln
3.2.1.2 Maltodextrine Maltodextrine sind durch den enzymatischen Abbau von Stärke gewonnene wasserlösliche Kohlenhydrate mit einer Kettenlänge von 5–10 Zucker-Einheiten und einem hohen Anteil an Maltose. [8] P a s e l l i M D 6
Das Maltodextrin Paselli MD6 wird von der Firma Avebe (Veendam, Holland) hergestellt. Tabelle 5: Physikalische Eigenschaften von Maltodextrin Paselli MD6
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] - -
Schüttdichte [kg/m³] 411 ± 11 300 - 450
Stampfdichte [kg/m³] 533 ± 21 -
x50 - Wert [µm] 59,5 ± 1,2 -
Trockensubstanzgehalt [%] 96,9 ± 0,2 max. 92
In Abbildung 33 ist die Partikelgrößenverteilung und in Abbildung 34 eine REM Aufnahme der Paselli MD6 Partikeln abgebildet. Die Partikelgrößen variieren in einem relativ weiten Bereich und die Partikelform ist sehr unregelmäßig.

Material und Methoden 50
Abbildung 33: Partikelgrößenverteilung von Paselli MD6 (Laserbeugung)
120 µm
Abbildung 34: REM Aufnahme von Paselli MD6
3.2.1.3 Cellulosen Cellulose ist der Hauptbestandteil pflanzlicher Zellwände und wird durch körpereigene Enzyme des menschlichen Intestinaltrakts nicht angegriffen. Cellulose ist Bestandteil des Ballaststoffanteils von Lebensmitteln pflanzlicher Herkunft, der die Darmperistaltik anregen soll. Sie besteht aus β-Glucopyranoseresten, die über 1, 4-Bindungen verknüpft sind. Unterschieden wird zwischen mikrokristalliner Cellulose und verschiedenen Cellulosederivaten. Beispielsweise Alkylcellulose und Hydroxy-alkylcellulose, die durch Umsetzung von Cellulose mit Alkali oder Methylchlorid bzw. Propylenoxid hergestellt werden. Dabei werden Methylgruppen oder Hydroxy-methylgruppen eingefügt, wobei der Substitutionsgrad von den Reaktions-bedingungen abhängig ist. Die Substituenten bewirken eine Störung der regel-mäßigen Packung und je nach Art und Anzahl der Substituenten entstehen unterschiedlich gut quellbare oder wasserlösliche Produkte. [2]
0
20
40
60
80
100
0 100 200 300 400 500 600 700
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 51
P u l v e r z e l l u l o s e P 2 9 0
Die VITACEL Pulverzellulose der Firma J. Rettenmaier & Söhne GmbH + Co (Rosenberg) ist eine gereinigte, mechanisch zerkleinerte Zellulose. Die Charakterisierungsergebnisse sind in Tabelle 6 und Abbildung 35 dargestellt. Tabelle 6: Physikalische Eigenschaften von Pulverzellulose P290
Eigenschaft Ermittelter Wert Herstellerangaben Feststoffdichte [kg/m³] - -
Schüttdichte [kg/m³] 276 ± 2 290 ± 15%
Stampfdichte [kg/m³] 456 ± 22 -
x50 - Wert [µm] 63 ± 1 50
Trockensubstanzgehalt [%] 99,7 ± 0,2 min. 93
Spez. Oberfläche [m²/g] 0,85 -
Die Partikelgrößenverteilung ist in Abbildung 35 dargestellt.
Abbildung 35: Partikelgrößenverteilung von Pulverzellulose P290 (Laserbeugung)
Mittels der REM Aufnahme in Abbildung 36 wird die Partikelform ersichtlich. Die Partikeln liegen als Fasern vor, die eine ungleichmäßige Form aufweisen. Anhand der Bilder kann man keine Poren ausmachen.
0
20
40
60
80
100
0 50 100 150 200 250 300
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 52
60 µm
Abbildung 36: REM Aufnahme von Pulverzellulose P290
C e l l e t s
Weitere Cellulosepartikeln, die verwendet wurden, sind sogenannte Cellets der Firma Syntapharm (Mühlheim-Ruhr). Cellets sind mikrokristalline Cellulose Kugeln, die in verschiedenen Fraktionen geliefert werden können. Die gemessenen Eigenschaften dieser drei Fraktionen sind in Tabelle 7 aufgelistet.
Tabelle 7: Eigenschaften der Cellets
Fraktion (lt. Hersteller) 120 – 200 µm 500 – 700 µm 1000 – 1400 µm Feststoffdichte [kg/m³] - - 1448 ± 10
Schüttdichte [kg/m³] 803 ± 1 773 ± 8 787 ± 7
Stampfdichte [kg/m³] 911 ± 10 842 ± 18 881 ± 11
Trockenmasse [%] 98,0 ± 0,8 94,9 ± 2,7 94,5 ± 0,1
x50-Wert [µm] 172 ± 3 595 ± 70 1096 ± 42
Die REM Aufnahmen sind in Abbildung 37 abgebildet, und die Partikelgrößen-
verteilungen sind in Abbildung 38 dargestellt. Die REM Aufnahmen zeigen, dass es
sich um nahezu runde Partikeln handelt, die keine sichtbaren Poren aufweisen. Auch
die Partikelgrößenverteilungen sind sehr eng, da es sich um nahezu monodisperse
Fraktionen handelt.

Material und Methoden 53
400 µm 400 µm 400 µm
Abbildung 37: REM Aufnahmen der Cellets
Abbildung 38: Partikelgrößenverteilungen der Cellets Fraktionen (Laserbeugung)
3.2.1.4 Kieselsäuren Kieselsäure ist eine Sammelbezeichnung für Verbindungen der allgemeinen Formel SiO2·nH2O. Setzt man ein Siliciumhalogenid (z. B. SiCl4) mit Wasser um, bildet sich primär die Orthokieselsäure (Monokieselsäure)[8]:
SiCl4+4H2O → Si(OH)4+4HCl Die Orthokieselsäure H4SiO4, ist eine sehr schwache Säure, die nur in großer Verdünnung und bei pH-Werten zwischen zwei und drei stabil ist. Bei abweichenden pH-Werten spaltet sie intermolekulares Wasser ab. Als erstes Kondensationsprodukt entsteht Pyrokieselsäure (Dikieselsäure, H6Si2O7). Nach weiterer Kondensation treten kugelförmige Polykieselsäuren auf. Das Endprodukt der Kondensation ist polymeres Siliciumdioxid (SiO2)x, das Anhydrid der Kieselsäure. Bei der Kondensation laufen kettenverlängernde, ringbildende und verzweigende Prozesse nebeneinander ab, so dass die Polykieselsäuren ungeordnet aufgebaut (amorph) sind. Die Si-Atome befinden sich bei allen Ketten im Mittelpunkt von unregelmäßig miteinander verknüpften Tetraedern, an deren vier Eckpunkten sich O-Atome befinden, die gleichzeitig den Nachbartetraedern angehören. [8]
0
20
40
60
80
100
0 200 400 600 800 1000 1200 1400 1600Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%
Fraktion 120 bis 200 µm
Fraktion 500 bis 700 µm
Fraktion 1000 bis 1400 µm

Material und Methoden 54
Abbildung 39: Strukturformel von Siliciumdioxid
Bei der Herstellung unterscheidet man grundsätzlich zwei verschiedene Verfahren: Einmal die Nassverfahren und einmal die pyrogene Erzeugung. Alle synthetischen Kieselsäuren werden aus Sand gewonnen.
Abbildung 40: Herstellung von Kieselsäuren [8]
S i p e r n a t 5 0 u n d S i p e r n a t 5 0 S
Die Kieselsäure Sipernat 50 ist eine sprühgetrocknete Trägerkieselsäure der Degussa AG (Frankfurt). Die physikalischen Eigenschaften sind in Tabelle 8 zusammengestellt.

Material und Methoden 55
Tabelle 8: Physikalische Eigenschaften von Sipernat 50
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] - -
Schüttdichte [kg/m³] 132 ± 20 -
Stampfdichte [kg/m³] 179 ± 5 180
x50 - Wert [µm] 26,1 ± 4,1 50*
Trockensubstanzgehalt [%] 94,2 ± 1 94
Spez. Oberfläche (BET) [m²/g] - 450
Spez. Oberfläche (DVS) [m²/g] 185,6 -
* Multisizer, 100 µm Kapillare
Abbildung 41: Partikelgrößenverteilung von Sipernat 50 (Laserbeugung)
Zusätzlich gibt es noch die Kieselsäure Sipernat 50S, die durch Vermahlung von Sipernat 50 gewonnen wird. Dessen physikalische Eigenschaften sind in Tabelle 9 aufgelistet und Abbildung 42 zeigt die Partikelgrößenverteilung.
0
20
40
60
80
100
0 20 40 60 80 100
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 56
Tabelle 9: Physikalische Eigenschaften von Sipernat 50S
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] - -
Schüttdichte [kg/m³] 103 ± 8 -
Stampfdichte [kg/m³] 123 ± 16 90
x50 - Wert [µm] 5,6 ± 0,6 7,5
Trockensubstanzgehalt [%] 97 94
Spez. Oberfläche (BET) [m²/g] - 450
Spez. Oberfläche (DVS) [m²/g] - -
Abbildung 42: Partikelgrößenverteilung von Sipernat 50S (Laserbeugung)
In Abbildung 43 ist eine REM Aufnahme von Sipernat 50S abgebildet.
0
20
40
60
80
100
0 10 20 30 40 50 60 70 80 90 100
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 57
30 µm
Abbildung 43: REM Aufnahme von Sipernat 50S
S i p e r n a t 2 2 0 0
Die Kieselsäure Sipernat 2200 von der Degussa AG (Frankfurt) wird als mikrogranuläre Trägerkieselsäure bezeichnet, die über eine ausgesprochen hohe Saugfähigkeit verfügt. In Tabelle 10 sind die gemessenen Werte und die Herstellerangaben zusammengestellt. Tabelle 10: Physikalische Eigenschaften von Sipernat 2200
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] - 2,0
Schüttdichte [kg/m³] 228 ± 1 -
Stampfdichte [kg/m³] 253 ± 2 260
x50 - Wert [µm] 265 ± 12 230
Trockensubstanzgehalt [%] 95,4 ± 0,6 95
Spez. Oberfläche (N2) m²/g - 185

Material und Methoden 58
Abbildung 44: Partikelgrößenverteilung von Sipernat 2200
Abbildung 45: REM Aufnahme von Sipernat 2200
A e r o s i l 1 3 0
Die Kieselsäure Aerosil 130 von der Degussa AG (Frankfurt) wird in einem thermischen Verfahren erzeugt. Ihre Eigenschaften sind in Tabelle 11 aufgelistet. Die sehr kleinen Primärteilchen lagern sich zu größeren Agglomeraten zusammen, die dann eine hohe Porosität aufweisen.
0
20
40
60
80
100
0 200 400 600 800 1000 1200
Partikelgröße x in µm
Vert
eilu
ngss
umm
e Q
3 in
%

Material und Methoden 59
Tabelle 11: Physikalische Eigenschaften von Aerosil 130
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] - -
Schüttdichte [kg/m³] 51 ± 4 -
Stampfdichte [kg/m³] 55 ± 2 50
x50 - Wert [µm] 779 ± 91 Primärteilchen: x50 = 16 nm
Trockensubstanzgehalt [%] 98,3 ± 2,7 min. 98,5
Spez. Oberfläche (N2) m²/g - 130 ± 25
Abbildung 46: Partikelgrößenverteilung von Aerosil 130 (Laserbeugung)
20 µm
Abbildung 47: REM Aufnahme von Aerosil 130
0
20
40
60
80
100
0 500 1000 1500 2000 2500
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 60
A e r o s i l R 9 7 2
Die Kieselsäure Aerosil R972 der Degussa AG (Frankfurt) weist eine hydrophobierte Oberfläche auf. Sie wird hergestellt, indem hydrophiles Aerosil 130 mit Dimethyldichlorsilan in einem Fließbettreaktor umgesetzt wird, wie in Abbildung 48 dargestellt.
Si SiOH
OH
SiCH3Cl
CH3
Cl
SiO
SiCH3
CH3
SiOSi
CH3
OCH3
H2O
DDCS+ 4 HCl
hydrophiles AEROSIL hydrophobes AEROSIL Abbildung 48: Schematische Darstellung der Reaktion von Dimethyldichlorsilan mit der Kieselsäureoberfläche: Überführung von "hydrophil" in "hydrophob"
Die physikalischen Eigenschaften von Aerosil R972 sind in Tabelle 12 zusammengestellt und die Partikelgrößenverteilung ist in Abbildung 49 dargestellt. Tabelle 12: Physikalische Eigenschaften von Aerosil R972
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] - -
Schüttdichte [kg/m³] 59 -
Stampfdichte [kg/m³] 74 50
x50 - Wert [µm] 779 Primärteilchen: x50 = 16 nm
Trockensubstanzgehalt [%] 99 min. 99,5
Spez. Oberfläche (N2) m²/g - 110 ± 20

Material und Methoden 61
Abbildung 49: Partikelgrößenverteilung von Aerosil R972 (Laserbeugung)
C a b o s i l
Cabosil ist eine pyrogene Kieselsäure mit einer kleinen Partikelgröße und einer großen spezifischen Oberfläche. Geliefert wird dieser Trägerstoff von der Firma Wacker (München) oder der Firma Cabot (Hanau). Tabelle 13: Physikalische Eigenschaften von Cabosil
Eigenschaft Ermittelter Wert Herstellerangabe (Cabot)
Feststoffdichte [kg/m³] - 2,2
Schüttdichte [kg/m³] 48 ± 2 40
Stampfdichte [kg/m³] 51 ± 4 ca. 50
x50 - Wert [µm] 11,63 ± 0,1 12 nm (Primärpartikeln)
Trockensubstanzgehalt [%] 99 ± 1 98,5
Spez. Oberfläche DVS [m²/g] 199,6 -
Spez. Oberfläche (N2) [m²/g] - 200 ± 25
0
20
40
60
80
100
0 500 1000 1500 2000 2500
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%
Material und Methoden 62
Abbildung 50: Partikelgrößenverteilung von Cabosil (Laserbeugung)
3.2.1.5 Sonstige Materialien H y S o r b C 7 0 1 5
HySorb C 7015 ist ein Natriumpolyacrylat, das für Personal Care Produkte wie Babywindeln hergestellt wird. HySorb C 7015 hat laut dem Hersteller BASF ein gutes Adsorptionsvermögen unter Last und ein hervorragendes Rückhaltevermögen im Zentrifugentest. HySorb ist ein sogenannter Superabsorber. So werden vernetzte Polymere bezeichnet, die extrem quellfähig sind und unter Gelbildung dazu in der Lage sind, ein Vielfaches ihres eigenen Gewichtes an Flüssigkeit zu speichern. Die Speicherkapazität nimmt mit steigendem Salzgehalt der flüssigen Komponente ab, bleibt aber auch unter Druck erhalten. Die ermittelten Eigenschaften von HySorb sind in Tabelle 14 zusammengestellt und die Partikelgrößenverteilung ist in Abbildung 51 zu sehen. Tabelle 14: Eigenschaften von HySorb
Eigenschaft Ermittelter Wert Herstellerangabe Feststoffdichte [kg/m³] -
Schüttdichte [kg/m³] 682 ± 20
Stampfdichte [kg/m³] 749 ± 9
„Scheinbare Dichte 600“
x50 - Wert [µm] 420 ± 46 99 % zwischen 106 und 850
Trockensubstanzgehalt [%] 95,4 ± 3,3 min. 94
0
20
40
60
80
100
0 50 100 150 200
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 63
Abbildung 51: Partikelgrößenverteilung von Hy Sorb (Laserbeugung)
3.2.2 Flüssigkeiten
Auf die oben aufgelisteten Trägerstoffe wurden im Rahmen dieser Arbeit verschiedene Flüssigkeiten aufgebracht, die im Folgenden charakterisiert werden. W a s s e r
Eine verwendete Flüssigkeit war Leitungswasser. Die gemessenen Werte für Dichte, Viskosität und Oberflächenspannung sind in Tabelle 15 zusammengestellt. Zum Vergleich sind Werte aus der Literatur für destilliertes Wasser angegeben. Tabelle 15: Eigenschaften von Wasser
Dichte
[g/cm3]
bei 20 °C
Viskosität ηηηη
[[[[mPas] bei 20 °C
Oberflächenspannung σσσσl,g
[N/m*10-3] bei 20 °C
Experimentell
bestimmte Werte 1,0019 ± 0,007 1,11 ± 0,25 68,33 ± 2,9
Literatur [16] 1,0000 1,00 72,80
S p e i s e ö l
Bei dem verwendeten Öl handelt es sich um Delios V, das von der Firma Grünau
(Illertissen) geliefert wird. In Tabelle 16 werden die physikalischen Größen
aufgelistet. Es handelt sich um ein pflanzliches Öl, das ein mittelkettiges Triglycerid
mit einem Caprylsäuregehalt von 60 % und einem Caprinsäuregehalt von 40 % ist.
0
20
40
60
80
100
0 200 400 600 800 1000 1200
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%

Material und Methoden 64
Tabelle 16: Eigenschaften von Speiseöl Delios
Dichte ρρρρ
[[[[g/cm3]
bei 20 °C
Viskosität ηηηη
[[[[mPas] bei 20 °C
Oberflächenspannung
σσσσl,g [N/m*10-3] bei 20 °C
Experimentell
bestimmte Werte 0,9111 ± 0,013 69,27 ± 0,76 33,20 ± 0,04
Literaturwerte unbekannt unbekannt unbekannt
T r i a c e t i n Triacetin ist ein Glycerinacetat und bezeichnet die Essigsäureester des Glycerins, die
durch Erhitzen von Glycerin mit Essigsäure entstehen. Man unterscheidet Mono-, Di-
und Triacetate. Glycerintriacetat (Triacetin, 1,2,3-Triacetoxypropan) ist eine farblose
Flüssigkeit, mischbar mit Alkohol, Ether und Benzol, jedoch schwer in Wasser löslich.
Triacetin wird in der Aromenindustrie als Lösungsmittel verwendet. Die
Glycerinacetate sind synthetische Produkte, die in der Natur nicht vorkommen. [8]
Tabelle 17 zeigt die relevanten Größen.
Tabelle 17: Eigenschaften von Triacetin
Dichte ρρρρ [g/cm]3
bei 20 °C
Viskosität ηηηη
[[[[mPas] bei 20 °C
Oberflächenspannung
σσσσl,g [N/m*10-3] bei 20 °
Experimentell
bestimmte Werte 1,1551 ± 0,020 21,70 ± 1,84 36,15 ± 0,36
Literaturwerte 1,16* 23,00* unbekannt
*Merck Produktspezifikation 108238
3.3 Herstellung der Modellpartikeln
3.3.1 Eingesetzte Materialien zur Herstellung von Modellpartikeln
M o n t a n w a c h s
Zur Herstellung von Modellpartikeln wurde im JetCutter das Montanwachs Licolub WE 4 FL der Firma Clariant GmbH (Gersthofen) eingesetzt. Das in Flocken vorliegende gelbe Wachs wird entweder im Wasserbad oder im Trockenschrank über zwei bis drei Stunden bei 110 °C geschmolzen. Das entstehende flüssige Wachs besitzt laut Hersteller bei einer Temperatur von 120 °C

Material und Methoden 65
eine Dichte von 0,85 g/cm3 und eine dynamisch Viskosität von ca. 60 mPas. Die Dichte des Wachses in fester Form beträgt bei 20 °C ca. 1,01-1,03 g/cm3. Montanwachs weist einen Schmelzbereich von 80–84 °C auf, stellt ein echtes Wachs dar und besteht aus ca. 53 % Estern von C22- bis C34-Fettsäuren (hauptsächlich Hexacosan-, Octacosan- und Triacontansäure) mit C24-, C26- und C28-Wachsalkoholen. Weitere Bestandteile sind freie Fettsäuren (17 %), freie Wachs-alkohole (1–2 %), sowie Montanharze (15–25 %), Ketone und asphaltartiges Material. Die Herstellung erfolgt durch Extraktion aus zerkleinerter Braunkohle mit Toluol. Das braunschwarze Rohmontanwachs kann mit Chromsäure oxidativ zu einem farblosen bis gelben Produkt gereinigt werden [8]. K o c h s a l z Zum Teil wurde in die Wachsschmelze das Siedespeisesalz Mikrozo der Firma Akzo Nobel Salz GmbH (Hamburg) gemischt. Bei dem Kochsalz Microzo handelt es sich um mikrofeines Siede-Speisesalz mit Tricalciumphosphat als Trennmittel. Die mittlere Korngröße beträgt ungefähr 13 µm und alle Partikeln sind kleiner als 87 µm. A l g i n a t , C a l c i u m l ö s u n g u n d T i t a n d i o x d Um poröse Partikeln aus Alginat herzustellen, wurde Natriumalginat der Firma FMC BioPolymer (Drammen, Norwegen) verwendet, das mittels Ultraturax in einer Konzentration von 2 – 3 % in destilliertes Wasser eingerührt wurde. Alginat ist die Bezeichnung für Salze und Ester der Alginsäure (siehe Abbildung 52).
Abbildung 52: Strukturformel von Alginsäure [8]
Von den Salzen hat Natriumalginat dank der Wasserlöslichkeit und der übrigen Eigenschaften die größte Bedeutung als Verdickungsmittel, Emulgator bzw. Emulsionsstabilisator und Gelgrundlage für die Nahrungsmittel-, Pharma- und Kosmetikindustrie. [8] Zum Aushärten tropften die Partikeln in eine 2 %-ige Calciumchloridlösung. Zur Stabilisierung kam Titandioxid der Firma Kronos

Material und Methoden 66
(Leverkusen) zum Einsatz, das in einer Konzentration von 40 % der Lösung zugesetzt wurde.
3.3.2 JetCutter zur Erzeugung von Modellpartikeln
Das von der Firma geniaLab (Braunschweig) entwickelte Strahlschneider-Verfahren (JetCutter), dessen Prinzip in Abbildung 53 dargestellt ist, beruht auf der mechanischen Trennung eines Flüssigkeitsstrahls. [39] Das zu verarbeitende Material, das z. B. eine Polymerlösung, ein anorganisches Sol, ein Wachs oder eine andere Schmelze sein kann, wird aus einem beheizbaren Druckbehälter zu einer Düse gefördert, durch die sich ein Flüssigkeitsvollstrahl ausbildet (siehe Abbildung 53). Dieser wird unmittelbar unter der Düse durch ein rotierendes Schneidwerkzeug (siehe Abbildung 54), das aus speichenradförmig angeordneten Drähten besteht, in gleichmäßige Zylindersegmente geschnitten. Der verwendete JetCutter ist komplett temperierbar durch Heizelemente der Firma Hillesheim (Waghäusel), so dass Temperaturen von 130 °C in allen Anlageteilen bis hin zur Düse realisiert werden können.
Abbildung 53: Prinzip des JetCutters
Motor
Schneid-draht
nFluid-strahl
abgeschnittener Zylinder
Kugel
vDraht
vFluid
dFluid
T
p
Druck-luft
Druck-behälter
Düse
Heiz-mantel

Material und Methoden 67
Abbildung 54: Schneidewerkzeug mit speichenförmig angeordneten Drähten
Aufgrund der Oberflächenspannung formen sich diese Segmente während des Fallens zu sphärischen Partikeln, die, falls sie beim Auftreffen noch nicht fest sind, in einem geeigneten Medium aufgefangen und ausgehärtet werden. Die Größe der entstehenden Kugeln ist abhängig von der Zahl der Schneiddrähte z, der Drehzahl des Schneidwerkzeugs n sowie vom Volumenstrom des Fluids durch die Düse, welcher wiederum vom Düsendurchmesser d und der Fluidgeschwindigkeit uFluid beeinflusst wird, wie auch in Gleichung 1 dargestellt ist. [41] Das auf diese Weise errechnete Volumen V ist allerdings größer als das praktisch ermittelte Volumen, da die beim Schneidvorgang auftretenden Verluste noch nicht berücksichtigt sind. Beim Schnitt durch den Fluidstrahl wird ein zylindrisches Segment von der Höhe des Drahtdurchmessers aus dem Strahl geschlagen, der sogenannte Schnittverlust. Da sich währenddessen der Strahl weiterbewegt, ergibt sich ein schräger Schnitt, und es kommt an den Zylinderhufen zu weiteren Verlusten, dem sogenannten Spritzverlust [41]. Berücksichtigt man auch diesen Verlust, kann der Partikeldurchmesser folgendermaßen berechnet werden:
3 DrahtFluid2
DüseP )dzn
u(d
23d −
⋅⋅⋅= (Gleichung 37)
Um die jeweils notwendigen Anlagenparameter zur Herstellung bestimmter Partikelgrößen zu ermitteln, wurde die Software BeadSim 2.1 der Firma geniaLab (Braunschweig) eingesetzt. Dieses Programm liefert auf der Grundlage von Gleichung 37 den Partikeldurchmesser, der mit den gegebenen Parametern erzeugt wird. Allerdings gilt es dabei zu beachten, dass je nach verwendetem Material auch zusätzliche Faktoren beachtet werden müssen, die zusätzlichen Einfluss auf die Partikelgröße haben. Bei der Erzeugung von Alginatpartikeln aus einer Alginatlösung wird beispielsweise ein Korrekturfaktor von 0,75 eingerechnet, da die Partikeln beim

Material und Methoden 68
Gelieren schrumpfen. Weiterhin werden von der Software die auftretenden Spritzverluste und der Schlankheitsgrad h/D der geschnittenen Zylinder ermittelt.
3.4 Verfahren zum Beladen der Trägerstoffe Um die pulverförmigen Trägerstoffe mit den Flüssigkeiten zu beladen, kamen zwei Verfahren zum Einsatz. Zum einen wurde das CPF-Verfahren [57] angewendet und zum anderen ein Pflugscharmischer, in den die Flüssigkeit mittels Einstoff- oder Zweistoffdüse zudosiert werden kann.
3.4.1 CPF-Sprühverfahren
Der verwendete CPF-Sprühturm wurde von der Firma Raps GmbH & Co. KG (Kulmbach) konstruiert und zur Verfügung gestellt. In Abbildung 55 ist der Aufbau der Anlage dargestellt. Bei allen Versuchen wurde CO2 als überkritisches Gas verwendet. Im statischen Mischer wurde ein Sprühdruck von 100 bar und eine Temperatur von 40 °C eingestellt. Am Ende des Versuches wurde die erreichte Beladung durch folgende Massenbilanz berechnet, wobei ml die Masse der Flüssigkeit und ms die Feststoffmasse darstellt:
% 100mm
mozentGewichtsprinBeladungsl
l ⋅+
= (Gleichung 38)
Alle im Folgenden angegebenen Beladungen sind Massenprozent. Die flüssige Komponente wurde über eine Dreifach-Membrandosierpumpe mit einem mittleren Massenstrom von 200 g/min in den Sprühturm gefördert. Die Zugabe des Trägerstoffes erfolgte per Hand. Dabei wurde darauf geachtet, das Pulver möglichst gleichmäßig zuzugeben, um den Massenstrom während der Dauer des Versuches konstant zu halten.

Material und Methoden 69
10
TIC
TIC
PI
PI
1
3
4
27
5
6
7
98
T
28
2
23
11
12
TIC
24
22
14
19
17 21
16
18
20
1326
PI T
TIC
25
CO230
29
31
15
32
Abbildung 55: Aufbau der CPF-Sprühanlage
Die einzelnen Bauteile sind im Folgenden aufgelistet: (1) Vorlagebehälter, Volumen ca. 3 Liter
(2) Heizmatte der Firma Winkler GmbH, Behälter elektrisch beheizbar bis 50°C

Material und Methoden 70
(3) AE-Ventil der Firma MAXIMATOR
(4) Elektrische Rohrbegleitheizung (beheizbar bis 50°C)
(5) Dreifach-Membrandosierpumpe Typ EKM 3 der Firma LEWA
(6) Elektrische Rohrbegleitheizung (beheizbar bis 50°C)
(7) Absperrventil vor statischem Mischer von der Firma MAXIMATOR
(8) Statischer Mischer der Firma SULZER CHEMTECH
(9) Elektrische Begleitheizung des statischen Mischers (beheizbar bis 50°C, im Mischer-
block integriert)
(10) Düse (Einbau verschiedener Düsen möglich)
(11) AE-Ventil der Firma MAXIMATOR
(12) Betriebs-Druckmessgerät (0 bis 250 bar)
(13) Berstscheibe als Sicherheit gegen Überdruck ab 300 bar
(14) Rohrbegleitheizung elektrisch bis 50°C
(15) AE-Ventil der Firma MAXIMATOR
(16) Membrandosierpumpe Typ ELM der Firma LEWA
(17) Kühlleitung für CO2
(18) Druckanzeige bis 160 bar
(19) AE-Ventil zur CO2-Entspannung
(20) Flexibler Anschluss CO2 (Druckschlauch 100 bar)
(21) Kühlwasserkreislauf der Firma Bosch
(22) Druckluftanschluss mit Reduzierstation (1 bar)
(23) Sprühturm
(24) Feststoffdosierung
(25) Zyklon für Abluftstrom
(26) Naß-/Trockensauger der Firma Kärcher
(27) Spülanschluss mit Ventil / Extraktleitung
(28) Temperaturfühler Sprühturm
(29) Druckanzeige Autoklav
(30) Autoklav mit regelbarer Temperatur
(31) Absperrventil Extraktseite
(32) Anschlussstück für beide Extraktleitungen

Material und Methoden 71
Der verwendete CPF-Sprühturm ohne Autoklav ist in Abbildung 56 zu sehen.
Schaltschrank
Zyklon Pulverzugabe
Membranpumpe für CO2
Mischerblock
Abbildung 56: CPF-Sprühturm ohne Autoklav
3.4.2 Pflugscharmischer mit Eindüsvorrichtung
Der verwendete Pflugscharmischer wurde von der Firma Gebrüder Lödige, Maschinenbau GmbH (Paderborn) zur Verfügung gestellt. Dieser Mischer ist in Abbildung 57 dargestellt. Die technischen Daten sind in Tabelle 18 zusammengefasst.
PflugscharDüse
Zerhacker
Abbildung 57: Lödige Pflugscharmischer

Material und Methoden 72
Tabelle 18: Technische Daten des Pflugscharmischers
Fassungsvolumen 15 l Drehzahl der Welle, elektrisch regelbar 47 – 245 U/min Drehzahl des Zerhackers, elektrisch regelbar 600 – 3000 U/min Leistung Motor, Antrieb der Welle 2,2 kW Leistung Motor, Zerhacker 0,55 kW
Der Mischer war mit einer Einstoffdüse ausgestattet und wurde zusätzlich mit einer Zweistoffdüse ausgerüstet, so dass die Zugabe der Flüssigkeit sowohl über eine Einstoffdüse, als auch über eine Zweistoffdüse erfolgen konnte.

Ergebnisse und Diskussion 73
4 Ergebnisse und Diskussion Wie sich im Laufe der Arbeit herausstellte, kommen beim Beladen von Trägerstoffen mit Flüssigkeiten im Wesentlichen drei große Einflussfaktoren in Frage, die in Abbildung 58 im Schaubild dargestellt sind. Um diese zu untersuchen und deren Einfluss auf die maximal mögliche Beladung zu bestimmen, müssen die hergestellten Produkte miteinander verglichen werden. Dafür wurde eine einfache Messmethode (Filtermethode) entwickelt. Für das theoretische Verständnis ist in diesem Zusammenhang auch die Flüssigkeitsverteilung im Haufwerk von großem Interesse.
- Partikelgröße- Partikelform- Porosität- Schütt- u. Stampfdichte- Feststoffdichteusw.
- Viskosität- Oberflächenspannung- Zusammensetzung.
- Benetzbarkeit- Reaktion zwischen Trägerstoff und Flüssigkeit
- CPF-Sprühverfahren- Wurfmischer mit Düse
- Optische Beurteilung- Partikelgrößenverteilung- Agglomerationsgrad- Freie Flüssigkeit in der Schüttung- Bindungsmechanismen
Maximale Aufnahmekapazitätvon Trägerstoffen
Einfluss der Trägerstoffe Einfluss der Flüssigkeit
Einfluss des verwendetenVerfahren
Beurteilung der beladenen Produkte und derFlüssigkeitsverteilung im Haufwerk
Abbildung 58: Einflussfaktoren auf die maximale Aufnahmekapazität
Zum einen können die Eigenschaften des Trägerstoffes für die maximale Aufnahme-kapazität von Flüssigkeiten eine Rolle spielen. Untersuchungen erfolgten bei den verwendeten Pulvern in Bezug auf Partikelgröße, Partikelform, spezifische Oberfläche, Porosität sowie verschiedene Dichten. Weiterhin wurden die Flüssigkeiten hinsichtlich Viskosität, Oberflächenspannung und Zusammensetzung charakterisiert. Auch die Benetzungseigenschaften zwischen Flüssigkeit und Trägerstoff und mögliche Reaktionen zwischen den beiden Komponenten sind berücksichtigt worden. Es kamen für diese Arbeit zwei Verfahren zum Beladen der Pulver mit Flüssigkeiten zum Einsatz, zum einen das CPF-Sprühverfahren und zum anderen ein Pfugscharmischer mit Eindüsvorrichtung. Um die hergestellten Sprühprodukte zu beurteilen und untereinander zu vergleichen, sind anfangs

Ergebnisse und Diskussion 74
verschiedene Merkmale wie die Partikelgrößenverteilung, der Agglomerationsgrad und die freie Flüssigkeit in der Schüttung als Beurteilungskriterien getestet worden. Um nähere Informationen über die Flüssigkeitsverteilung im Haufwerk und die Bindungsmechanismen zwischen Partikeln und Flüssigkeit zu erforschen, ist ein Modell entwickelt und mittels NMR-Messungen quantifiziert worden. Alle im Folgenden diskutierten Beladungen sind in % angegeben. Das heißt, ein Produkt, das eine Beladung von 30 % aufweist, besteht zu 30 Gew.-% aus Flüssigkeit und 70 Gew.-% aus Trägerstoff.
4.1 Bestimmung der maximalen Aufnahmekapazität Wird ein Trägerstoff mit steigenden Mengen Flüssigkeit beladen, wird irgendwann eine Grenze erreicht und das Pulver kann die eingesprühte Flüssigkeitsmenge nicht mehr aufnehmen. Wird die Grenze überschritten, befindet sich freie Flüssigkeit in der Schüttung oder als Belag an den Bauteilen des verwendeten Apparates. Untersucht wird in dieser Arbeit die Menge, die ein Trägerstoff maximal aufnehmen kann, ohne dass es zu unerwünschter Agglomeration und zu freier Flüssigkeit in der Schüttung kommt. Wird durch Flüssigkeit der Feinanteil eines Pulvers an größere Partikeln gebunden, kann man auch von Agglomeratbildung sprechen, allerdings wird diese nicht als störend empfunden. Um diese beiden Kriterien zu dokumentieren, wurden folgende Untersuchungen vorgenommen. Zunächst fand eine optische Beurteilung der beladenen Pulver statt. Dann wurde zum einen die Partikelgrößenverteilung des Rohstoffes sowie die Verteilungen der unterschiedlich hoch beladenen Sprüh-produkte und die Veränderungen der Schütt- und Stampfdichte in Abhängigkeit von der Beladung gemessen. Außerdem wurde der Agglomeratanteil durch Absieben mittels Handsieb bestimmt. Zum anderen wurde während der Arbeit eine neue Methode entwickelt. Es ist ein Schnelltest, mit dem die Aufnahmekapazität der Trägerstoffe sehr gut ermittelt werden kann.
4.1.1 Optische Beurteilung der beladenen Pulver
Werden in der Praxis beispielsweise flüssige Aromaöle auf Gewürzmischungen aufgesprüht, dann wird optisch beurteilt, ab welcher Menge Flüssigkeit die Mischung zu hoch beladen ist. Dass anhand einer optischen Beurteilung sinnvolle Schlüsse gezogen werden können, sollen die folgenden Abbildungen zeigen. Die Produkte der ersten Versuchsreihe, bei der eine Kieselsäure mit steigenden Mengen Wasser beladen wurde, sind in Abbildung 59 zu sehen. Wie man deutlich erkennen kann, ist eine Beladung bis zu 70 % problemlos möglich, bei einer Beladung mit 80 % Wasser dagegen ist das Produkt kein streu- und fließfähiges Pulver mehr, das aus einzelnen Partikeln besteht, sondern eine hochkonzentrierte Suspension. Anhand dieser Ergebnisse kann man feststellen, dass die maximale

Ergebnisse und Diskussion 75
Aufnahmekapazität der Kieselsäure Sipernat 2200 für Wasser zwischen 70 und 80 % betragen muss.
80%40% 60% 70%
Abbildung 59: Kieselsäure Sipernat 2200 mit verschiedenen Mengen Wasser
Ein weiteres Beispiel ist in Abbildung 60 dargestellt: Das Maltodextrin Paselli MD 6 wurde mit steigenden Mengen Triacetin beladen. Bis zu einer Beladung von 45 % kann das Produkt als feinkörniges Pulver bezeichnet werden. Ab einem Triacetin-gehalt von 50 % besteht das Produkt aus feuchten Agglomeraten und ist somit zu hoch beladen. Die maximale Aufnahmekapazität von Paselli MD 6 für Triacetin ist daher circa 45 %.
40% 45% 50%
Abbildung 60: Maltodextrin Paselli MD 6 mit verschiedenen Mengen Triacetin
Diese beiden Beispiele zeigen, dass eine optische Beurteilung der beladenen Produkte sinnvoll ist und angewendet werden kann. Zusätzlich soll aber noch eine Messgröße herangezogen werden, um die Aufnahmegrenzen zu dokumentieren.
4.1.2 Veränderung der Partikelgrößenverteilung mit zunehmender Beladung
Die ersten Untersuchungen befassten sich mit der Veränderung der Partikelgrößenverteilungen bei steigenden Beladungen. Dazu wurden die Rohstoffe mit zunehmenden Mengen Flüssigkeit beladen, und die Partikelgrößenverteilungen mittels Laserbeugung gemessen. Zur Pulverdispergierung kam der Fallschacht zum

Ergebnisse und Diskussion 76
Einsatz, um vorhandene Agglomerate nicht durch Einwirkung von Scherkräften zu zerstören. Exemplarisch sind in den folgenden Abbildungen die Kurvenverläufe der Kieselsäure Sipernat 50S und der Kartoffelstärke Aero Myl 33 mit Speiseöl dargestellt.
Abbildung 61: Partikelgrößenverteilungen der Kieselsäure Sipernat 50S mit verschiedenen Mengen Speiseöl gemessen mittels Laserbeugung (Fallschacht)
Abbildung 62: Partikelgrößenverteilungen der Kartoffelstärke Aero Myl 33 mit verschiedenen Mengen Speiseöl gemessen mittels Laserbeugung (Fallschacht)
Bei beiden Produkten (Abbildung 61 und Abbildung 62) ist zu sehen, dass schon bei einer Beladung der Rohstoffe mit kleinen Mengen an Speiseöl eine Agglomeration auftritt. Die Partikelgrößenverteilungen verschieben sich signifikant nach rechts zu größeren Partikeldurchmessern, weil sich die einzelnen Primärpartikeln zu Agglomeraten zusammenlagern. Bei weiter ansteigenden Flüssigkeitsmengen verschieben sich die Partikelgrößen zwar weiterhin in Richtung größerer Durch-messer, allerdings nicht mehr signifikant. Dieses Ergebnis zeigt, dass die zusätzlich aufgebrachte Flüssigkeit hauptsächlich vorhandene Hohlräume der bereits gebildeten Agglomerate auffüllt und sich nur vereinzelt weitere Partikeln anlagern,
0102030405060708090
100
0 500 1000 1500 2000 2500
Partikelgröße x in µm
Sum
men
vert
eilu
ng Q
3 in
%
Rohstoff50%60%70%
0
20
40
60
80
100
0 500 1000 1500 2000 2500
Partikelgröße x in µm
Sum
men
vert
eilu
ng Q
3 in
%
Rohstoff10%20%40%55%60%65%

Ergebnisse und Diskussion 77
welche die Agglomeratdurchmesser etwas vergrößern. Dieses Ergebnis stimmt gut mit den theoretischen Betrachtungen von Schubert [49] und Weigert [55] überein, die in Kapitel 2.4.2 diskutiert wurden. Diese Darstellungen der Partikelgrößenverteilungen können indes nicht zur Beurteilung der maximalen Aufnahmekapazität verwendet werden, da beispielsweise die Kartoffelstärke Aero Myl 33 mit höchstens 55 bis 60 % beladen werden kann. Bei einer Beladung von 65 % ist dieser Rohstoff, wie man anhand der optischen Beurteilung erkennen kann, zu hoch beladen und es sind feuchte Agglomerate in der Schüttung sichtbar. Bei den Partikelgrößenverteilungen treten aber zwischen 60 % und 65 % Beladung keine signifikanten Unterschiede auf. Auch für den Rohstoff Sipernat 50S, der mit höchstens 60 bis 70 % beladen werden kann, sind anhand der Partikelgrößenverteilungen keine Aussagen hinsichtlich der maximal Beladungs-kapazität möglich.
4.1.3 Veränderung von Schütt- und Stampfdichte mit zunehmender Beladung
Um die Eigenschaften von beladenen Pulvern weiter zu untersuchen, wurden die Veränderungen der Schütt- und der Stampfdichte (Definition vgl. Kapitel 2.2.3) mit zunehmender Beladung ermittelt. Nachfolgend werden exemplarisch die Ergebnisse der Kartoffelstärke Aero Myl 33 und der Kieselsäure Sipernat 50S mit Speiseöl dargestellt. Die Ergebnisse, die mit anderen Rohstoffen erzielt wurden, entsprechen diesen Resultaten. In Abbildung 63 sind die Schütt- und Stampfdichten der Kartoffelstärke Aero Myl 33 dargestellt.
Abbildung 63: Schütt- und Stampfdichten von Sprühprodukten aus Speiseöl und Aero Myl 33 mit steigenden Speiseölmengen
Für Aero Myl 33 zeigt die Abbildung 63, dass die Bulkdichten bis zu einer Beladung von 30 % annähernd konstant bleiben, obwohl in den Schüttungen mehr und mehr
0
100
200
300
400
500
0 10 20 30 40 50 55Beladung in %
Schü
tt- b
zw. S
tam
pfdi
chte
in
kg/
m³
SchüttdichteStampfdichte

Ergebnisse und Diskussion 78
Flüssigkeit vorhanden ist. Dies bedeutet, dass die Porosität mit zunehmendem Flüssigkeitsanteil zunächst ansteigt. Danach steigen die Bulkdichten signifikant an, was damit zu erklären ist, dass die Porosität der Schüttungen nun konstant bleibt, und die Poren zwischen den Partikeln mit Flüssigkeit gefüllt werden. Dieses Ergebnis stimmt sehr gut mit den Resultaten einer Studie von Feng und Yu [11] überein, welche die Packungsstruktur monodisperser Kugeln, vor allem in Bezug auf die Flüssigkeitszugabe untersuchten. Das Packungsverhalten von feuchten Partikeln unterscheidet sich demnach von demjenigen trockener Partikeln, was anhand von Glaskugeln experimentell bestimmt wurde. Bei konstanten Ausgangsbedingungen durchläuft die ursprüngliche Porosität des trockenen Pulvers bei Zugabe von Flüssigkeit zunächst ein Maximum und bleibt bei weiterer Zugabe danach konstant. Dabei unterscheiden Feng und Yu [11] zwei Abschnitte:
a) Es existiert ein Abschnitt der Benetzung, in dem die Porosität mit steigendem Flüssigkeitsgehalt zunimmt, weil der Einfluss der durch Flüssigkeit hervorgerufener Kräfte zunimmt.
b) Weiterhin findet ein Füllen der Partikelzwischenräume statt, so dass die Bulkdichten zunehmen.
Gleiches gilt für das zweite Beispiel Sipernat 50S. Die Schütt- und Stampfdichten sind in Abbildung 64 zu sehen. Hier ist nur eine niedrige Beladung, nämlich 10 %, vorhanden, bei der die Porosität zugenommen hat und die Bulkdichten daher genauso groß sind wie die des Ausgangsproduktes. Anschließend folgen die Beladungen 50 %, 60 % und 70 %. Bei diesen hohen Beladungen ist schon das Stadion erreicht, in dem die Porosität konstant bleibt und die Partikelzwischenräume mit Flüssigkeit aufgefüllt werden.

Ergebnisse und Diskussion 79
Abbildung 64: Schütt- und Stampfdichten von Sprühprodukten aus Speiseöl auf Sipernat 50S mit verschiedenen Mengen Speiseöl
Wenn die Porosität der trockenen Schüttung (ohne Flüssigkeitsanteil) mit steigendem Flüssigkeitsanteil zunächst ein Maximum erreicht, bedeutet das, dass die Bulkdichten der „trockenen“ Schüttungen ein Minimum erreichen müssen. Dies ist in Abbildung 65 dargestellt.
Abbildung 65: Schütt- und Stampfdichten von Sprühprodukten aus Aero Myl 33 ohne den Flüssigkeitsanteil
Der Verlauf der Schütt- und Stampfdichten des Pulvers Aero Myl 33 ohne Flüssigkeitsanteil zeigt deutlich die Abnahme bis zu einem Minimum. Zwischen Beladungen von 20 und 40 % bleiben die Bulkdichten in Etwa konstant und anschließend steigen sie wieder an. Dieser Anstieg kann dadurch erklärt werden kann, dass die Beladungen nun an die maximale Aufnahmekapazität der Trägerstoffe kommen und sich jetzt auch Flüssigkeitsbrücken zwischen den Agglo-meraten ausbilden können, welche die Porosität der Schüttung wieder verringern.
0
100
200
300
400
500
0 10 50 60 70Beladung in %
Schü
tt- b
zw. S
tam
pfdi
chte
in
kg/
m³
SchüttdichteStampfdichte
0
100
200
300
0 10 20 30 40 50 55
Beladung in %
Schü
tt- b
zw. S
tam
pfdi
chte
in
kg/
m³
SchüttdichteStampfdichte

Ergebnisse und Diskussion 80
Diese Agglomeration ist in der Praxis meist unerwünscht, und bei der optischen Beurteilung wird freie Flüssigkeit in der Schüttung festgestellt und das Produkt beginnt zu kleben und zu klumpen. Ein ähnlicher Verlauf ist in Abbildung 66 zu erkennen, in der die Bulkdichten der trockenen Schüttung (ohne Flüssigkeitsanteil) aus Sipernat 50S dargestellt sind.
Abbildung 66: Schütt- und Stampfdichten von Sprühprodukten aus Speiseöl und Sipernat 50S ohne den Flüssigkeitsanteil
Durch diese Ergebnisse kann gezeigt werden, dass beim Beladen und Tränken der Pulverpartikeln zunächst einzelne Flüssigkeitsbrücken ausgebildet werden, die zu einer Porositätserhöhung der Schüttung führen. Anschließend werden die Poren in und zwischen den Partikeln mit Flüssigkeit gefüllt. Erneut stimmen die Ergebnisse der Untersuchungen sehr gut mit der Modellvorstellung überein, die in den Veröffentlichungen von Schubert [49] und Weigert [55] diskutiert wurde. Veranschaulicht wird das Ganze noch einmal in Abbildung 67.
Geringe Flüssigkeitsmenge Hohe FlüssigkeitsmengeTrockene Schüttung
Porosität nimmt zu Porosität bleibt konstant
Maximale Flüssigkeitsmenge
Porosität nimmt ab
ρρρρSch ρρρρSt ρρρρSch ρρρρSt ρρρρSch ρρρρSt ρρρρSch ρρρρSt
Abbildung 67: Modellvorstellung: Schütt- und Stampfdichten von beladenen Pulvern
0
100
200
0 10 50 60 70Beladung in %
Schü
tt- b
zw. S
tam
pfdi
chte
in
kg/
m³
SchüttdichteStampfdichte

Ergebnisse und Diskussion 81
Allerdings kann anhand dieser Messergebnisse wiederum keine Aussage hinsichtlich der maximal möglichen Flüssigkeitsmenge getroffen werden. Es tritt bei Erreichen der maximalen Flüssigkeitsmenge in der Schüttung keine signifikante Änderung auf, die als Kennzeichen der maximalen Beladung verwendet werden könnte.
4.1.4 Bestimmung des Agglomeratanteils
Ein Kriterium um festzustellen, ab wann ein Trägerstoff zu hoch beladen ist, stellt der Agglomerationsgrad dar. Der Agglomerationsgrad kann ermittelt werden, indem Proben des Sprühproduktes abgesiebt werden und der Masenanteil des Rückstandes gewogen wird. Zum Absieben können verschiedene Siebverfahren, wie Handsieb, Luftstrahlsieb oder Rüttelsieb verwendet werden. Bei den im Folgenden dargestellten Ergebnissen kam ein Handsieb mit einer durchschnittlichen Maschenweite von 1 mm zum Einsatz. Beispielhaft sind die Ergebnisse von zwei Sprühprodukten in Abbildung 68 und Abbildung 69 dargestellt. Auch bei den Messungen anderer, hier nicht explizit dargestellter Produkte steigt der als Rückstand verbleibende Agglomeratanteil mit höherer Beladung an. Allerdings treten bei den meisten Messungen sehr große Schwankungen innerhalb der einzelnen Messungen der Dreifachbestimmung auf, wie vor allem in Abbildung 68, bei Aero Myl 33 mit Speiseöl zu sehen ist. Da alle Werte die auf dem Konfidenzintervall liegen gleichberechtigt sind, lassen sich anhand dieser Ergebnisse keine Aussagen hinsichtlich des Agglomeratanteils in Abhängigkeit von der Beladung treffen.
Abbildung 68: Überkornanteil der Sprühprodukte aus Speiseöl und Aero Myl 33
In Abbildung 69 ist der Agglomeratanteil von Sipernat 50S mit Speiseöl dargestellt. Hier sind die Konfidenzintervalle deutlich kleiner. Vor allem zwischen einer Beladung von 60 % und 70 % ist ein signifikanter Anstieg auszumachen. Anhand dieses Diagramms kann die Aussage getroffen werden, dass ab einer Beladung von 70 % ein signifikant höherer Agglomeratanteil vorliegt und die Beladung dementsprechend
01020304050607080
0 10 20 30 40 50 60 70
Beladung in %
Übe
rkor
nant
eil i
n %
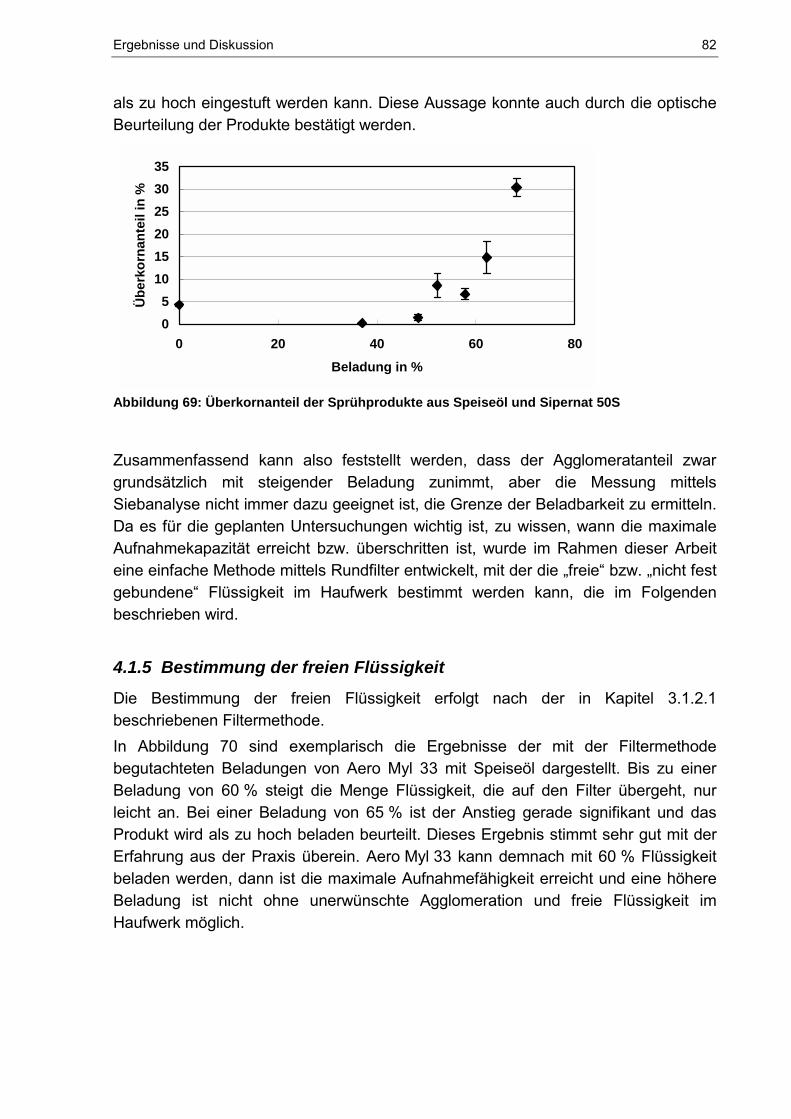
Ergebnisse und Diskussion 82
als zu hoch eingestuft werden kann. Diese Aussage konnte auch durch die optische Beurteilung der Produkte bestätigt werden.
Abbildung 69: Überkornanteil der Sprühprodukte aus Speiseöl und Sipernat 50S
Zusammenfassend kann also feststellt werden, dass der Agglomeratanteil zwar grundsätzlich mit steigender Beladung zunimmt, aber die Messung mittels Siebanalyse nicht immer dazu geeignet ist, die Grenze der Beladbarkeit zu ermitteln. Da es für die geplanten Untersuchungen wichtig ist, zu wissen, wann die maximale Aufnahmekapazität erreicht bzw. überschritten ist, wurde im Rahmen dieser Arbeit eine einfache Methode mittels Rundfilter entwickelt, mit der die „freie“ bzw. „nicht fest gebundene“ Flüssigkeit im Haufwerk bestimmt werden kann, die im Folgenden beschrieben wird.
4.1.5 Bestimmung der freien Flüssigkeit
Die Bestimmung der freien Flüssigkeit erfolgt nach der in Kapitel 3.1.2.1 beschriebenen Filtermethode. In Abbildung 70 sind exemplarisch die Ergebnisse der mit der Filtermethode begutachteten Beladungen von Aero Myl 33 mit Speiseöl dargestellt. Bis zu einer Beladung von 60 % steigt die Menge Flüssigkeit, die auf den Filter übergeht, nur leicht an. Bei einer Beladung von 65 % ist der Anstieg gerade signifikant und das Produkt wird als zu hoch beladen beurteilt. Dieses Ergebnis stimmt sehr gut mit der Erfahrung aus der Praxis überein. Aero Myl 33 kann demnach mit 60 % Flüssigkeit beladen werden, dann ist die maximale Aufnahmefähigkeit erreicht und eine höhere Beladung ist nicht ohne unerwünschte Agglomeration und freie Flüssigkeit im Haufwerk möglich.
05
101520253035
0 20 40 60 80
Beladung in %
Übe
rkor
nant
eil i
n %
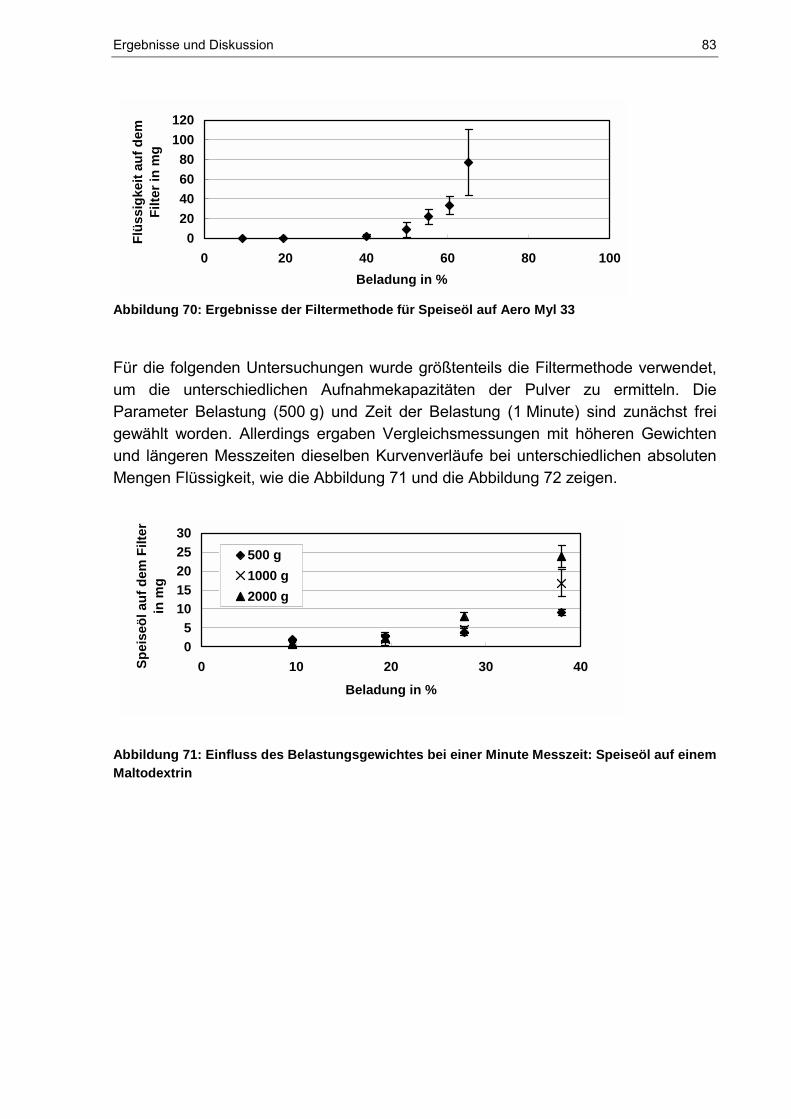
Ergebnisse und Diskussion 83
Abbildung 70: Ergebnisse der Filtermethode für Speiseöl auf Aero Myl 33
Für die folgenden Untersuchungen wurde größtenteils die Filtermethode verwendet, um die unterschiedlichen Aufnahmekapazitäten der Pulver zu ermitteln. Die Parameter Belastung (500 g) und Zeit der Belastung (1 Minute) sind zunächst frei gewählt worden. Allerdings ergaben Vergleichsmessungen mit höheren Gewichten und längeren Messzeiten dieselben Kurvenverläufe bei unterschiedlichen absoluten Mengen Flüssigkeit, wie die Abbildung 71 und die Abbildung 72 zeigen.
Abbildung 71: Einfluss des Belastungsgewichtes bei einer Minute Messzeit: Speiseöl auf einem Maltodextrin
020406080
100120
0 20 40 60 80 100Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg
05
1015202530
0 10 20 30 40
Beladung in %
Spei
seöl
auf
dem
Filt
er
in m
g
500 g1000 g2000 g

Ergebnisse und Diskussion 84
Abbildung 72: Einfluss der Messzeit bei 500 g Belastung: Speiseöl auf einem Maltodextrin
Anhand der optischen Beurteilung und aufgrund von Erfahrungen aus der Praxis ist bekannt, dass das verwendete Maltodextrin bis ca. 30 % beladen werden kann. Dies kann mit allen dargestellten Zeit- und Gewichtskombinationen nachgewiesen werden, da bei einer Beladung von 38 % jeweils ein signifikanter Sprung auftritt. Der signifikante Anstieg bei dieser Beladung konnte mit einer längeren Messzeit oder einem höheren Gewicht zwar deutlicher dargestellt werden, allerdings kann es bei großen Mengen Flüssigkeit, die auf den Filter übergehen, dazu kommen, dass dieser übersättigt wird, und so die Messungen verfälscht werden. Daher wird für die Messungen im Rahmen dieser Dissertation eine Messzeit von einer Minute und ein Gewicht von 500 g gewählt.
4.2 Einfluss des Trägerstoffes auf die maximale Flüssigkeits-aufnahmekapazität
Die wichtigste Einflussgröße auf die Flüssigkeitsmenge, die auf einen Trägerstoff aufgesprüht werden kann, ist der Trägerstoff selbst. Im Rahmen der vorliegenden Dissertation wurden sehr viele verschiedene Pulver mit Flüssigkeiten beladen und getränkt. Zunächst werden die Ergebnisse der einzelnen Versuchsreihen dargestellt und anschließend in Hinblick auf verschiedene Eigenschaften der Trägerstoffe diskutiert.
4.2.1 Maximale Beladungskapazität verschiedener Trägerstoffe
Verschiedene Trägerstoffe (Stärken, Maltodextrine, Zellulosen und Kieselsäuren) sind im CPF-Sprühverfahren mit Speiseöl beladen und die Sprühprodukte mittels optischer Beurteilung und Filtermethode beurteilt worden. Bei allen Versuchen herrschte im statischen Mischer eine Temperatur von 40 °C und der Sprühdruck betrug 80 bis 100 bar. Die Beladung der Produkte wurde aus der jeweiligen Massenbilanz des Sprühversuches ermittelt. Bei einzelnen Versuchen wurde die
0
510
15
2025
30
0 10 20 30 40Beladung in %
Spei
seöl
auf
dem
Filte
r in
mg
1 Minute
3 Minuten 5 Minuten

Ergebnisse und Diskussion 85
Konzentration des Speiseöls analytisch durch die Fettbestimmung nach Soxhlet (vgl. Kapitel 3.1.2.2) ermittelt, um die Gültigkeit der Massenbilanz zu überprüfen. K a r t o f f e l s t ä r k e A e r o M y l 3 3 Das Ergebnis der Versuche mit Aero Myl 33 ist in Abbildung 70 dargestellt. In dem Diagramm kann man erkennen, dass maximal eine Beladung von 60 % Speiseöl erreicht werden kann. Auch die optische Beurteilung ergab dieses Ergebnis: Belädt man die Kartoffelstärke Aero Myl 33 mit mehr als 60 % Speiseöl, so enthält die Schüttung viele feuchte Agglomerate und es befindet sich freies Öl im Produkt, so dass nicht mehr von einem frei fließfähigen, feinkörnigen Pulver gesprochen werden kann. P a s e l l i M D 6 Das Maltodextrin Paselli MD 6 dagegen kann nur mit 20 % Speiseöl beladen werden, wie in Abbildung 73 ersichtlich wird. Bis zu einer Beladung mit 20 % erhält man ein homogenes Produkt ohne feuchte Agglomerate. Bei höheren Beladungen geht signifikant mehr Flüssigkeit auf den Filter über. Auch optisch beurteilt ist das Produkt zu hoch beladen.
Abbildung 73: Ergebnisse der Filtermethode für Maltodextrin Paselli MD6 mit Speiseöl
P u l v e r z e l l u l o s e P 2 9 0 Das nächste Beispiel ist die Pulverzellulose P290, die im CPF-Verfahren mit Speiseöl beladen wurde. Bei Beladungen von 10 % und 20 % geht so gut wie keine Flüssigkeit auf das Filterpapier über. Allerdings ist eine Beladung über 35 % zu hoch und die Menge Speiseöl, die auf den Filter übergeht, steigt signifikant an. Auch hier stimmt das Ergebnis wieder gut mit Erfahrungen aus der Praxis und der optischen Beurteilung überein: Diese Zellulose kann maximal 30 bis 35 % Flüssigkeit aufnehmen.
0
5
10
15
20
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g

Ergebnisse und Diskussion 86
Abbildung 74: Ergebnisse der Filtermethode für Pulverzellulose P290 mit Speiseöl
Nach der Kartoffelstärke, dem Maltodextrin und der Zellulose wurden eine Reihe von Kieselsäuren beladen. Bei allen Kieselsäuren konnte gezeigt werden, dass sie im Vergleich zu den oben genannten Stoffen verhältnismäßig viel Flüssigkeit auf-nehmen können, wie die folgenden Ergebnisse zeigen. S i p e r n a t 2 2 0 0 Als erste Kieselsäure wurde Sipernat 2200 mit Speiseöl beladen. Anhand Abbildung 75 kann man erkennen, dass Beladungen bis 60 % möglich sind und erst bei einer Beladung von 70 % ein sprunghafter Anstieg der Flüssigkeitsmenge auf dem Filter detektiert wurde.
Abbildung 75: Ergebnisse der Filtermethode für Sipernat 2200 mit Speiseöl
S i p e r n a t 5 0 S Ein weiteres Sipernat ist das Sipernat 50S, das sich in einigen Eigenschaften von Sipernat 2200 unterscheidet. Auf diese Kieselsäure konnten bis zu 70 % Speiseöl aufgebracht werden, ohne dass sich die Menge, die auf den Filter übergeht, verändert (siehe Abbildung 76). Bei einer Beladung von 80 % war das Produkt
0
10
20
30
40
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em F
ilter
in
mg
0
5
10
15
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g

Ergebnisse und Diskussion 87
allerdings kein Pulver mehr, sondern eine Suspension, so dass kein Filtertest mehr durchgeführt werden konnte.
Abbildung 76: Ergebnisse der Filtermethode für Sipernat 50S mit Speiseöl
Eine ebenso hohe Beladung konnte auf der folgenden Kieselsäure erzielt werden. A e r o s i l 1 3 0 Die Kieselsäure Aerosil 130 kann mit 70 % Speiseöl beladen werden. In Abbildung 77 sieht man, dass ab einer Beladung von 80 % die Menge an Speiseöl, die auf den Filter übergeht, signifikant ansteigt.
Abbildung 77: Ergebnisse der Filtermethode für Speiseöl auf Aerosil 130
Das selbe Ergebnis erzielt man mit der optischen Beurteilung (siehe Abbildung 78): Bei einer Beladung mit 79 % Speiseöl treten deutlich sichtbare Agglomerate in der Schüttung auf. Das Produkt kann nicht mehr als feinkörnig und frei fließfähig beschrieben werden.
0
5
10
15
20
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg
02468
101214
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em F
ilter
in
mg

Ergebnisse und Diskussion 88
57% 69% 79%
Agglomerat
Abbildung 78: Aerosil 130 mit steigenden Beladungen Speiseöl
Zusätzlich gibt es auch noch Aerosil R972, die hydrophobierte Variante des Aerosil 130, das auch mit Speiseöl beladen wurde. A e r o s i l R 9 7 2 Die Ergebnisse der Versuche mit Aerosil R972 und Speiseöl entsprechen den Ergebnissen von Aerosil 130 und Speiseöl (vgl. Abbildung 79).
Abbildung 79: Ergebnisse der Filtermethode für Speiseöl auf Aerosil R972
C a b o s i l Als letztes Beispiel wurde die Kieselsäure Cabosil mit Speiseöl beladen. Auch dieser Trägerstoff kann relativ hoch beladen werden. Erst Beladungen über 60 % führen zu signifikant höheren Mengen Speiseöl auf dem Filter (siehe Abbildung 80).
0
5
10
15
20
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g

Ergebnisse und Diskussion 89
Abbildung 80: Ergebnisse der Filtermethode für Speiseöl auf der Kieselsäure Cabosil
Zusammengefasst sind die Ergebnisse der Beladungsversuche in Tabelle 19. Tabelle 19: Maximal mögliche Beladung verschiedener Trägerstoffe mit Speiseöl
Trägerstoff Stoffklasse Maximale Beladung Aero Myl 33 Kartoffelstärke 60 % Paselli ND6 Maltodextrin 20 % Pulverzellulose P290 Zellulose 30 % Sipernat 2200 60 %
Sipernat 50S 70 % Aerosil 130 70 % Aerosil R972 70 % Cabosil
Kieselsäure
60 %
4.2.2 Vergleich verschiedener Trägerstoffe hinsichtlich Oberfläche, DVS-Messungen und maximal möglicher Beladung
Von einigen Trägerstoffen wurde die Oberfläche mittels Wasserdampf- oder Stickstoffadsorption oder mittels beider Verfahren gemessen. Diskutiert werden die Messergebnisse, die Form der Sorptionsisothermen und die nach BET berechneten Oberflächen im Zusammenhang mit der maximalen Flüssigkeitsaufnahmekapazität der jeweiligen Trägerstoffe.
0
5
10
15
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g

Ergebnisse und Diskussion 90
A e r o M y l 3 3 Von der Kartoffelstärke Aero Myl 33 wurde die spezifische Oberfläche sowohl mittels Stickstoffadsorption als auch anhand einer Wasserdampfsorptionsisotherme ermittelt. Die Auswertung erfolgte jeweils nach der BET-Methode. Die Wasserdampfsorptionsisotherme wurde bei 20°C aufgenommen. In Abbildung 81 ist sowohl die relative Feuchte des Luftstromes, der über die Probe geleitet wird, als auch die Massenzunahme der Probe bei der jeweiligen Feuchte aufgetragen. Zu Beginn wird die Probe solange getrocknet, bis keine Massenabnahme mehr stattfindet. Danach wird die relative Feuchte (RH) zunächst stufenweise von 10 % auf 90 % angehoben, um dann wieder bis auf 10 % abgesenkt zu werden. Die Masse der Probe steigt bei Erhöhung der Feuchte jeweils solange an, bis sie im Gleichgewicht wieder einen konstanten Wert erreicht. Wird die umgebende Feuchte wieder abgesenkt, nimmt auch die Masse der Probe wieder ab.
Abbildung 81: Messkurve der DVS Messung von Aero Myl 33
Da nun zu jeder relativen Feuchte RH sowohl die relative Massenzunahme bei der Sorption als auch die relative Massenabnahme bei der Desorption bekannt sind, kann aus der Messkurve in Abbildung 81 die Sorptionsisotherme berechnet werden.
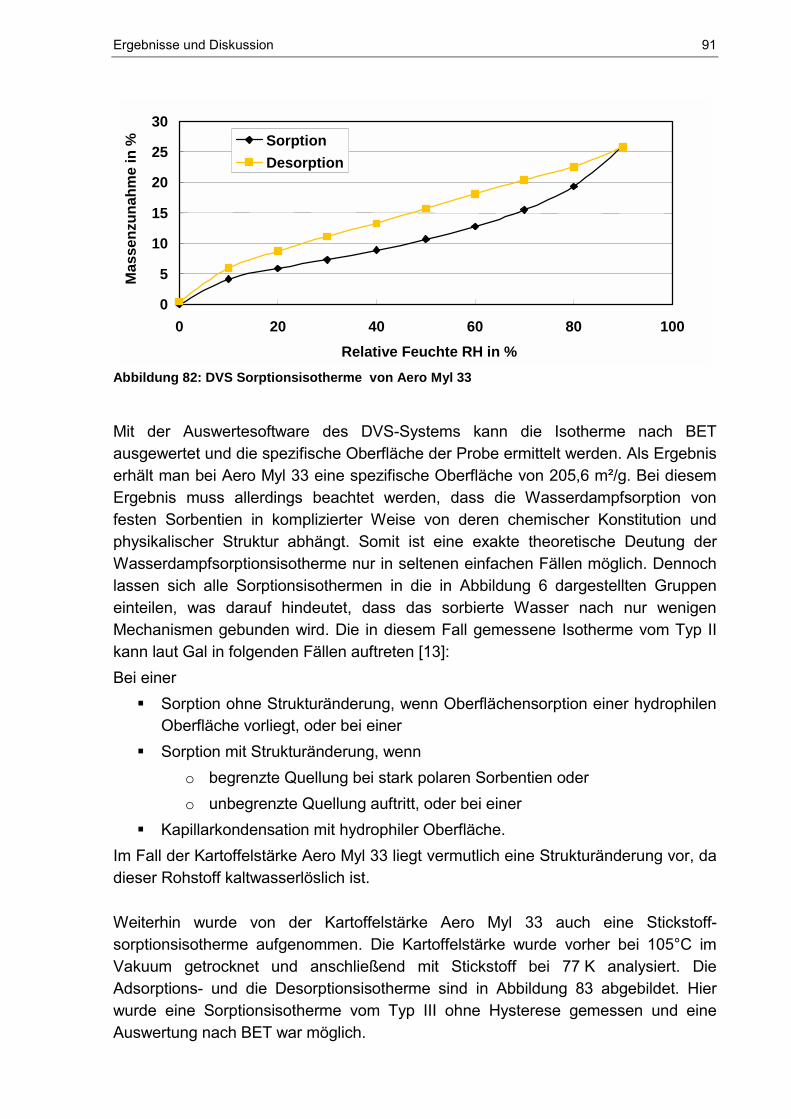
Ergebnisse und Diskussion 91
Abbildung 82: DVS Sorptionsisotherme von Aero Myl 33
Mit der Auswertesoftware des DVS-Systems kann die Isotherme nach BET ausgewertet und die spezifische Oberfläche der Probe ermittelt werden. Als Ergebnis erhält man bei Aero Myl 33 eine spezifische Oberfläche von 205,6 m²/g. Bei diesem Ergebnis muss allerdings beachtet werden, dass die Wasserdampfsorption von festen Sorbentien in komplizierter Weise von deren chemischer Konstitution und physikalischer Struktur abhängt. Somit ist eine exakte theoretische Deutung der Wasserdampfsorptionsisotherme nur in seltenen einfachen Fällen möglich. Dennoch lassen sich alle Sorptionsisothermen in die in Abbildung 6 dargestellten Gruppen einteilen, was darauf hindeutet, dass das sorbierte Wasser nach nur wenigen Mechanismen gebunden wird. Die in diesem Fall gemessene Isotherme vom Typ II kann laut Gal in folgenden Fällen auftreten [13]: Bei einer � Sorption ohne Strukturänderung, wenn Oberflächensorption einer hydrophilen
Oberfläche vorliegt, oder bei einer � Sorption mit Strukturänderung, wenn
o begrenzte Quellung bei stark polaren Sorbentien oder o unbegrenzte Quellung auftritt, oder bei einer
� Kapillarkondensation mit hydrophiler Oberfläche. Im Fall der Kartoffelstärke Aero Myl 33 liegt vermutlich eine Strukturänderung vor, da dieser Rohstoff kaltwasserlöslich ist.
Weiterhin wurde von der Kartoffelstärke Aero Myl 33 auch eine Stickstoff-sorptionsisotherme aufgenommen. Die Kartoffelstärke wurde vorher bei 105°C im Vakuum getrocknet und anschließend mit Stickstoff bei 77 K analysiert. Die Adsorptions- und die Desorptionsisotherme sind in Abbildung 83 abgebildet. Hier wurde eine Sorptionsisotherme vom Typ III ohne Hysterese gemessen und eine Auswertung nach BET war möglich.
0
5
10
15
20
25
30
0 20 40 60 80 100Relative Feuchte RH in %
Mas
senz
unah
me
in %
Sorption Desorption

Ergebnisse und Diskussion 92
Abbildung 83: Stickstoffsorptionsisothermen von Aero Myl 33
Die dabei ermittelte spezifische Oberfläche wies einen Wert von 0,4657 ± 0,0007 m²/g auf. Mit den beiden unterschiedlichen Messverfahren Stickstoffadsorption und Wasser-dampfsorption sind vollkommen unterschiedliche Werte für die spezifische Oberfläche erzielt worden. Dies kann dadurch erklärt werden, dass sich zwischen Wasser und Kartoffelstärke andere Bindungsmechanismen einstellen, als zwischen Stickstoff und der Stärke. Dennoch ist vor allem die kleine spezifische Oberfläche, die mittels Stickstoffadsorptionsmessung ermittelt wurde, fragwürdig. Anhand der REM Aufnahme ist zu erkennen, dass die Partikeln eine poröse Struktur aufweisen, die eine große spezifische Oberfläche zur Folge haben muss. Man kann daraus schließen, dass die Ergebnisse und Auswertungen solcher Messungen grundsätzlich hinterfragt und diskutiert werden müssen, um daraus auf Partikelmerkmale zu schließen. Weitere Versuche wurden mit Kieselsäuren durchgeführt. S i p e r n a t 5 0 Von der Kieselsäure Sipernat 50 wurde die Wasserdampfsorptionsisotherme aufgenommen. Die relative Feuchte (RH) wurde von 0 % stufenweise auf 90 % erhöht und im Anschluss daran wieder abgesenkt. In Abbildung 84 ist die gemessene Kurve abgebildet. Die Auswertung nach BET ergibt eine spezifische Oberfläche von 185,6 m²/g.
0
1
1
2
2
3
0 0,2 0,4 0,6 0,8 1
Relativdruck p/p0 in -
Ads
orbi
erte
s Vo
ulm
en in
cm
3 /g
SorptionDesorption

Ergebnisse und Diskussion 93
Abbildung 84: Messkurve und Sorptionsisotherme der DVS Messung von Sipernat 50
Schon anhand der Messkurve, die in Abbildung 84 dargestellt ist, ist zu erkennen, dass die Kieselsäure im Verlauf der DVS-Messung deutlich mehr Wasser aufnimmt, als die oben untersuchte Stärke. Die Kieselsäure nimmt im Verlauf der Messung nahezu 70 % Wasser aus der Luft auf, was auch den Ergebnissen im CPF-Sprühturm entspricht.
Abbildung 85: DVS Sorptionsisotherme von Sipernat 50
Die Isotherme in Abbildung 85 weist eine Form von Typ II auf, wobei bei dieser Kieselsäure keine Reaktion mit Wasser und somit keine Strukturänderung stattfindet, sodass hier Oberflächenadsorption oder Kapillarkondensation mit hydrophiler Oberfläche vermutet werden kann. Allerdings weist folgendes Anzeichen auf eine poröse Struktur hin: Bei der eingestellten maximalen Feuchte von 90 % wird während
0 10
20 30
40 50
60 70
0 20 40 60 80 100Relative Feuchte RH in %
Mas
senz
unah
me
in %
Sorption Desorption

Ergebnisse und Diskussion 94
der Messzeit kein Gleichgewichtszustand erreicht, was auf eine Kapillarkondensation schließen lässt. Die Struktur des Sorbens hat einen großen Einfluss auf den zeitlichen Ablauf der Sorptionsvorgänge, welche bei reiner Oberflächensorption am schnellsten vonstatten gehen und bei vorhandenen Poren und Kapillaren entsprechend mehr Zeit beanspruchen. [13] Bei einer weiteren Kieselsäure, bei Cabosil, wurde dagegen ein anderes Ergebnis erzielt, was im Folgenden beschrieben wird. C a b o s i l Auch von der Kieselsäure Cabosil wurde die Wasserdampfsorptionsisotherme unter denselben oben beschriebenen Bedingungen aufgenommen. Im Sprühturm kann auch dieser Rohstoff mit bis zu 60 % beladen werden. Während der DVS-Messung dagegen nahm das Pulver nur sehr wenig Wasser auf, wie in Abbildung 86 und Abbildung 87 zu sehen ist.
Abbildung 86: Messkurve der DVS Messung von Cabosil

Ergebnisse und Diskussion 95
Abbildung 87: DVS Sorptionsisotherme von Cabosil
Die Auswertung nach BET ergibt eine spezifische Oberfläche von 199,6 m²/g. Dieses Ergebnis stimmt gut mit der Herstellerangabe überein, wonach diese Kieselsäure nach Stickstoffsorptionsmessung eine Oberfläche von 200 m²/g aufweist. Zusammengefasst sind die Ergebnisse in Tabelle 20. Anhand dieser Zusammen-stellung ist zu erkennen, dass die Ergebnisse der Oberflächenmessung in jedem Fall hinterfragt und diskutiert werden müssen und dass aus diesen Messergebnissen keine Rückschlüsse auf eine mögliche Beladbarkeit gezogen werden können. Tabelle 20: BET-Oberflächen, aufgenommene Wassermenge und Flüssigkeitsaufnahme-kapazität der drei Trägerstoffe
Träg
erst
off
BE
T-O
berfl
äche
D
VS
-Mes
sung
Auf
geno
mm
ene
Was
serm
enge
(D
VS
-Mes
sung
)
BE
T-O
berfl
äche
S
ticks
toffa
dsor
btio
n
Max
. Flü
ssig
keits
-au
fnah
mek
apaz
ität
Aero Myl 33 205,6 m²/g ca. 25 % 0,47 m²/g 55 – 60 % Sipernat 50S 185,6 m²/g ca. 65 % 450 m²/g 70 % Cabosil 199,6 m²/g ca. 6 % 200 m²/g 60 %
0
2
4
6
8
0 20 40 60 80 100Relative Feuchte RH in %
Mas
senz
unah
me
in %
Sorption Desorption

Ergebnisse und Diskussion 96
4.2.3 Einfluss der Stampfdichte auf die maximale Aufnahmekapazität
Wie schon in Kapitel 3.4.1 über das CPF-Verfahren erwähnt wurde, fand Grüner [15] einen Zusammenhang zwischen der Stampfdichte und der maximalen Beladung verschiedener Trägerstoffe. Dieser Zusammenhang konnte im Rahmen der vorliegenden Dissertation bestätigt werden, da die Ergebnisse aller durchgeführten Versuche sehr gut mit der Aussage übereinstimmten, dass die maximal mögliche Beladung umso höher ist, je geringer die Stampfdichte der Trägerstoffe. In der Abbildung 88 sind sowohl die Ergebnisse von Grüner [15], als auch die im Rahmen dieser Arbeit entstanden Wertepaare, die auch in Tabelle 21 aufgeführt sind, zusammengestellt.
0
20
40
60
80
100
0 100 200 300 400 500 600 700 800 900
Stampfdichte des Trägerstoffes in kg/m³
Max
imal
e B
elad
ung
in %
Feinkörnig, kugelförmigGrobkörnig, flockigMesswerte
Abbildung 88: Maximaler Extraktgehalt in Abhängigkeit von der Stampfdichte: Vergleich Grüner [15] und den Messwerten der vorliegenden Dissertation
Tabelle 21: Maximale Flüssigkeitsmenge (Speiseöl) auf verschiedenen Trägerstoffen
Trägerstoff Stampfdichte [kg/m³]
Max. Flüssigkeitsmenge [Gew.-%]
Cabosil Kieselsäure 51 80 ± 5
Sipernat 50S 123 70 ± 5
Kartoffelstärke Aero Myl 33 177 57,5 ± 5
Maisstärke C-Pulp Tex 315 42,5 ± 5
Vitacel Pulvercellulose 449 35 ± 5
Maltodextrin C-Pur 616 25 ± 5
Bei den eigenen Messwerten sind dabei Fehlerbalken von ± 5 Gew.-% aufgetragen, da die maximal mögliche Beladung nicht genauer ermittelt werden kann. Eine Unterscheidung in feinkörnig und kugelförmig bzw. grobkörnig und flockig ist dann

Ergebnisse und Diskussion 97
nicht mehr notwendig. Die Ergebnisse liegen dennoch im gleichen Bereich wie die Resultate von Grüner [15]. Daher ist es von Interesse, von welchen Größen die Stampfdichte der Trägerstoffe abhängt. In Abbildung 7 sind die einzelnen Einflussparameter gekennzeichnet. Es handelt sich dabei um � die Feststoffdichte der Partikeln, � das Hohlraumvolumen zwischen den Partikeln, � das Volumen der offenen Poren und � das Volumen der geschlossenen Poren.
Die Partikelporosität εP wird bestimmt durch das Feststoffvolumen Vs und das Partikelvolumen Vp:
P
sP V
V)(1 =− ε (Gleichung 39)
Analog dazu kann auch die Porosität eines Agglomerates εA, das sich aus mehreren Einzelpartikeln zusammensetzt, berechnet werden:
A
PA V
V)(1 =− ε (Gleichung 40)
Letztendlich kann auch die Porosität einer Schüttung εB so beschrieben werden:
B
AB V
V)(1 =− ε (Gleichung 41)
Für die Bulkdichte ρB gilt folgender Zusammenhang:
)(1)-(1)-(1V
V)(1)-(1)-(1V
)(1VV
)(1VVm
BPASB
BBPAS
B
PAAS
B
PPS
B
sB
εεερεεερ
εερερρ
−⋅⋅⋅=⋅−⋅⋅⋅
=
−⋅⋅−⋅=
−⋅⋅==
)1(
(Gleichung 42)
Die Schüttungsporosität εB darf nicht mit Flüssigkeit gefüllt werden, da sonst das Sprühprodukt zu hoch beladen wäre, was mittels optischer Beurteilung und Filtermethode angezeigt wird. Unter der Voraussetzung, dass die Feststoffdichte ρS
immer im selben Größenbereich liegt und sich bei den Stampfdichtemessungen immer dieselbe Schüttungsporosität einstellt, was im Folgenden noch näher erläutert

Ergebnisse und Diskussion 98
wird, ist die Bulkdichte eine Funktion der Agglomeratporosität und der Partikelporosität :
( ) ( ) ( )PABs
B 11 εεερ
ρ−⋅−=
−⋅ 1 (Gleichung 43)
Daher kann die gemessene Stampfdichte als Maß für die maximal mögliche Beladung herangezogen werden. Sowohl die Poren in den Partikeln als auch die Poren in den Agglomeraten werden bei der Beladung mit Flüssigkeit gefüllt. Handelt es sich bei den Trägerstoffpartikeln um runde, monodisperse Partikeln, so hat das Hohlraumvolumen zwischen den Partikeln keinen Einfluss mehr, wie das folgende Beispiel verdeutlicht. Gleichgroße Kugeln können sich in verschiedenen Packungen zusammenlagern. Ein Beispiel ist die kubische Packung, wie sie in Abbildung 89 dargestellt ist.
d
Abbildung 89: Kubische Packung gleich großer Kugeln
Diese Packung kann aus Elementarzellen aufgebaut werden, die jeweils einen, die Kugel umhüllenden Würfel, darstellen. Die Koordinationszahl dieser Packung k = 6 und die Porosität der Schüttung εSch kann durch folgende Formel berechnet werden [32].
0,477ε6d³d³)ε(1 SchSch =⇒
⋅=⋅− π (Gleichung 44)
Aus der Gleichung ergibt sich, dass die Porosität der kubischen Packung vom Partikeldurchmesser unabhängig ist. Eine andere Möglichkeit, wie sich Kugeln zusammenlagern können, ist die sogenannte dichteste Kugelpackung, wobei die Koordinationszahl k = 12 ist.

Ergebnisse und Diskussion 99
Abbildung 90: Aufbau einer dichtesten Kugelpackung
Die Porosität der dichtesten Kugelpackung ist auch vom Partikeldurchmesser unabhängig und entspricht εSch = 0,259. Die Stampfdichten der verschiedenen Celletsfraktionen müssten demzufolge gleich groß sein. Die Werte 911 ± 10 kg/m³, 842 ± 18 kg/m³ und 881 ± 11 kg/m³ unterscheiden sich dennoch um etwa 10 % voneinander, was damit erklärt werden kann, dass die Partikeln nicht absolut monodispers und rund sind. Unter der Voraussetzung, dass die mittlere Stampfdichte der Cellets ρSt = 880 kg/m³ beträgt und die Partikeln nicht porös sind, d.h. die Rohdichte gleich der Feststoffdichte ρS = 1447 kg/m³ ist, kann man die Porosität der Schüttung εSch, die sich bei der Stampfdichtemessung der Cellets einstellt, berechnen:
0,392m³kg1447m³kg8801
ρρ1ε
S
StSch =−=−= (Gleichung 45)
Diese Porosität liegt zwischen den Porositäten der kubischen und der dichtesten Kugelpackung, was in der Realität zu erwarten ist. Auch in der Literatur werde Werte um die 0,4 angegeben, für Porositäten von Zufallsschüttungen. [52][33] Geht man davon aus, dass sich beim Messen der Stampfdichte von Kieselsäure Sipernat 2200, die auch aus annähernd runden und monodispersen Partikeln besteht, dieselbe Schüttungsporosität einstellt, kann man die Porosität der Kieselsäurepartikel εPa abschätzen.
0,392)ε(1ρ
ρ1ρρ1ε
PS
St
R
StSch =
−⋅−=−=
0,68m³kg20000,392
m³kg2531ρ0,392
ρ1εS
StPa =
⋅−=
⋅−=⇒ (Gleichung 46)
Allerdings bezieht sich diese Porosität der Partikel sowohl auf offene, als auch auf geschlossene Poren. Zum Beladen mit Flüssigkeiten stehen allerdings nur die offenen Poren zur Verfügung.

Ergebnisse und Diskussion 100
Beide Pulver wurden im CPF-Verfahren beladen. Bei Sipernat 2200 sind Beladungen bis 60 % möglich und erst bei einer Beladung von 70 % war ein sprunghafter Anstieg der Flüssigkeitsmenge auf dem Filter detektiert worden (Kapitel 4.2.1). Dies zeigt, dass nicht nur die Poren in den Partikeln, sondern auch Hohlräume zwischen den Partikeln gefüllt worden sind. Werden alle Poren mit Flüssigkeit gefüllt (d. h. es existieren keine geschlossenen Poren, die von außen nicht zugänglich sind), würde lediglich eine Beladung von 52 % erreicht werden, wie folgende Berechnung zeigt:
[ ]
[ ] %52%1000,90,682,00,68)(1
0,90,68
%100ρερε)(1
ρε%inBeladungls
l
=⋅
⋅+⋅−⋅=
⋅
⋅+⋅−⋅
= (Gleichung 47)
Da die erzielte Beladung im Sprühturm höher als dieser berechnet Wert liegt und vermutlich von außen nicht zugängliche Poren vorhanden sind, werden auch Poren in den Agglomeraten, also zwischen einzelnen Partikeln gefüllt, wie im Kapitel Grundlagen und Kenntnisstand beschrieben wurde. Die Cellets konnten dagegen nur mit 5 % Flüssigkeit beladen werden und schon bei dieser geringen Beladung befand sich freie Flüssigkeit in der Schüttung. Die Partikeln neigten zur Agglomeration. Höhere Beladungen waren nicht mehr durchführbar, sodass auch die Filtermethode nicht angewendet wurde. Um solche Beladungen auf monodispersen runden Partikeln genauer zu untersuchen, wurden im nächsten Abschnitt der Arbeit Modellpartikeln mit einem Strahlschneiderverfahren hergestellt.
4.2.4 Modellpartikeln aus dem Strahlschneiderverfahren Um noch weitere Untersuchungen an Modellpartikeln vornehmen zu können, sind Trägerstoffpartikeln mittels JetCutter produziert worden. Zunächst erfolgte die Herstellung monodisperser Kugeln aus Montanwachs in drei verschiedenen Größen. Anschließend wurde versucht, poröse Montanwachspartikeln mittels Kochsalz herzustellen und in einer weiteren Versuchsreihe wurden Partikeln aus Titandioxid erzeugt, die eine hohe Feststoffdichte und eine poröse Struktur aufweisen. 4.2.4.1 Monodisperse, runde Partikeln aus Montanwachs Für die Herstellung von Partikeln ohne Poren, wurden Wachse aus Estern der Montansäure mit der Handelsbezeichnung Licolub WE4 verwendet. Zur Formung und Verfestigung der flüssigen Wachskugeln wurde über die gesamte zur Verfügung stehende Fallstrecke von ca. 12 Metern vertropft. Die festen Partikeln konnten mit einer Plastikwanne unbeschadet aufgefangen werden.

Ergebnisse und Diskussion 101
Ziel dieser ersten Versuchsreihe war es, monodisperse runde Partikeln in drei Fraktionen herzustellen. Dabei sollte die größte Fraktion einen Partikeldurchmesser von 1000 µm nicht überschreiten. Die dazu notwendigen Einstellungsparameter wurden mit Hilfe der Software BeadSim 2.1 ermittelt. Tabelle 22: Erste Versuchsreihe: Partikeln aus Montanwachs aus dem JetCutter
Versuch 1 2 3 4 5 6
dDüse [µm] 200 400 200 400 600 600
pDruckehälter [bar] 2,2 1,5 4,2 2,7 1,1 0,8
TDruckbehälter [°C] 120 130 125 125 125 125
m� Flüssigkeit [g/s] 0,2 0,934 0,3 1,72 2,4 1,97
nSchneidwerkzeug [1/min] 10000 5000 11030 11058 6600 6000
x50 / berechnet [µm] 373 783 392 699 938 948
x50 / Laserbeugung [µm] 416 ± 3 979 ± 78 410 ± 3 665 ± 5 - 804 ± 6
Anteil Partikeln größer 1000 µm
[%] 1,5 51,49 0,35 10,5 - 11,5
Eine Herstellung von Partikeln mit Durchmessern über 800 µm war zunächst mit der gegebenen Anlage nicht möglich, da die vorhandene Fallhöhe zur notwendigen Partikelverfestigung nicht ausreichte. Da die Leitung zur Düse hin nicht komplett beheizbar war, mussten bei den restlichen Anlageteilen Temperaturen eingestellt werden, die sehr weit über dem Schmelzpunkt des Wachses lagen. Deswegen konnten im Versuch 5 keine Partikeln hergestellt werden. Durch den Einbau einer zusätzlichen Heizelementes konnten diese Probleme beseitigt werden und der Versuch 6 mit niedrigeren Temperaturen durchgeführt werden. Die erzeugten Partikeln wiesen zunächst nicht die gewünschten engen Partikel-größenverteilungen auf, wie Abbildung 91 zeigt.

Ergebnisse und Diskussion 102
Abbildung 91: Partikelgrößenverteilungen (Laserbeugung) der mittels JetCutter hergestellten Montanwachspartikeln
Wie in Abbildung 91 zu erkennen ist, sind die Partikelgrößenverteilungen der Versuche 1 und 3 am engsten. Dies ist der Fall, wenn kleine Partikeln herstellt werden und dazu eine hohe Drehzahl des Schneidwerkzeuges mit einem kleinen Massenstrom kombiniert wird. Bei den Versuchen 4 und 6 zeigen die Partikelgrößen-verteilungen zwei durch ein Plateau getrennte Bereiche. Die kleinere „Fraktion“ entseht hier aus dem Schnittverlust, der in Kapitel 3.3.2 erwähnt wurde. Bei der Herstellung der größten Partikeln entstand die breiteste Partikelgrößenverteilung, da hier mit der geringsten Drehzahl gearbeitet wurde. Um die gewünschten drei Fraktionen zu erhalten, wurden die Partikeln folgender-maßen zusammengemischt und fraktioniert: � Feinfraktion (300 µm - 500 µm) aus den Versuchen 1 und 3 � Mittelfraktion (500 µm - 800 µm) aus den Versuchen 2 und 4 � Grobfraktion (800 µm - 1000 µm) aus den Versuchen 2, 4 und 6 Die resultierenden Partikelgrößenverteilungen sind in Abbildung 92 dargestellt. Die Schütt- und Stampfdichten sind in Tabelle 23 zusammengestellt. Tabelle 23: Schütt- und Stampfdichten der Montanwachspartikeln
Feinfraktion Mittelfraktion Grobfraktion Schüttdichte [kg/m³] 584 ± 7 576 ± 10 583 ± 1
Stampfdichte [kg/m³] 631 ± 52 606 ± 7 609 ± 13
0
20
40
60
80
100
0 500 1000 1500 2000 2500Partikelgröße x in µm
Q3 i
n %
Versuch 1Versuch 2Versuch 3Versuch 4Versuch 6
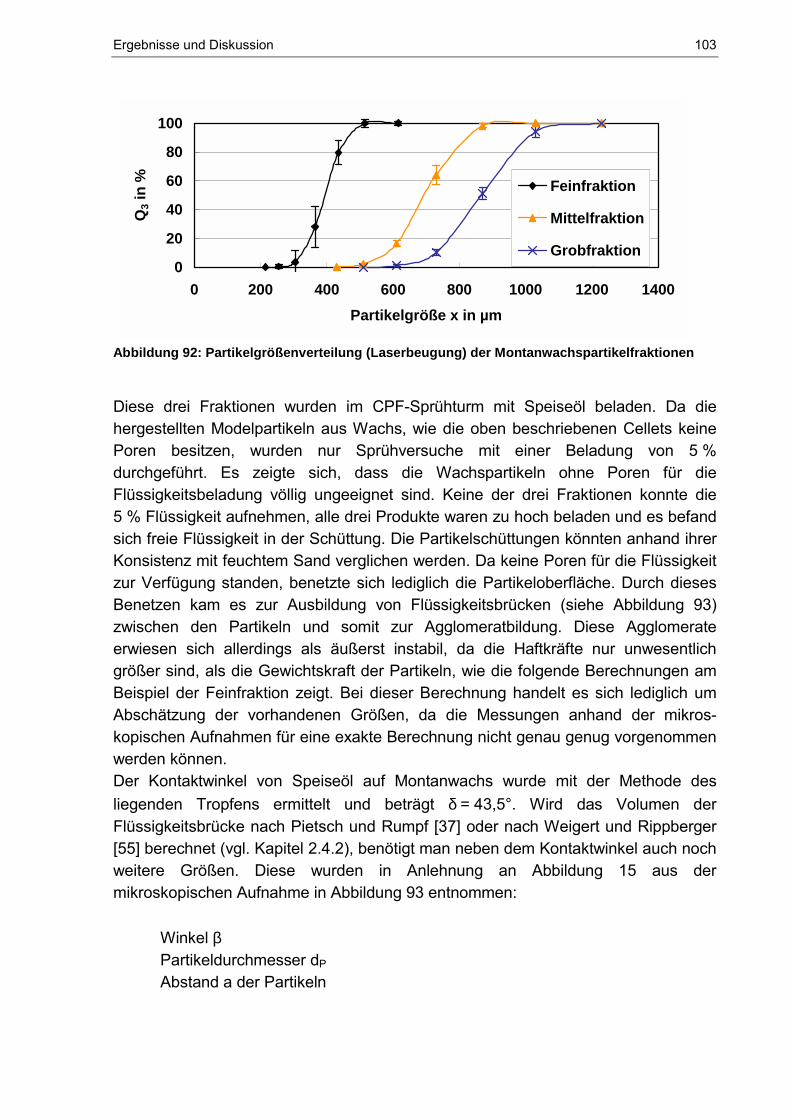
Ergebnisse und Diskussion 103
Abbildung 92: Partikelgrößenverteilung (Laserbeugung) der Montanwachspartikelfraktionen
Diese drei Fraktionen wurden im CPF-Sprühturm mit Speiseöl beladen. Da die hergestellten Modelpartikeln aus Wachs, wie die oben beschriebenen Cellets keine Poren besitzen, wurden nur Sprühversuche mit einer Beladung von 5 % durchgeführt. Es zeigte sich, dass die Wachspartikeln ohne Poren für die Flüssigkeitsbeladung völlig ungeeignet sind. Keine der drei Fraktionen konnte die 5 % Flüssigkeit aufnehmen, alle drei Produkte waren zu hoch beladen und es befand sich freie Flüssigkeit in der Schüttung. Die Partikelschüttungen könnten anhand ihrer Konsistenz mit feuchtem Sand verglichen werden. Da keine Poren für die Flüssigkeit zur Verfügung standen, benetzte sich lediglich die Partikeloberfläche. Durch dieses Benetzen kam es zur Ausbildung von Flüssigkeitsbrücken (siehe Abbildung 93) zwischen den Partikeln und somit zur Agglomeratbildung. Diese Agglomerate erwiesen sich allerdings als äußerst instabil, da die Haftkräfte nur unwesentlich größer sind, als die Gewichtskraft der Partikeln, wie die folgende Berechnungen am Beispiel der Feinfraktion zeigt. Bei dieser Berechnung handelt es sich lediglich um Abschätzung der vorhandenen Größen, da die Messungen anhand der mikros-kopischen Aufnahmen für eine exakte Berechnung nicht genau genug vorgenommen werden können. Der Kontaktwinkel von Speiseöl auf Montanwachs wurde mit der Methode des liegenden Tropfens ermittelt und beträgt δ = 43,5°. Wird das Volumen der Flüssigkeitsbrücke nach Pietsch und Rumpf [37] oder nach Weigert und Rippberger [55] berechnet (vgl. Kapitel 2.4.2), benötigt man neben dem Kontaktwinkel auch noch weitere Größen. Diese wurden in Anlehnung an Abbildung 15 aus der mikroskopischen Aufnahme in Abbildung 93 entnommen:
Winkel β Partikeldurchmesser dP Abstand a der Partikeln
0
20
40
60
80
100
0 200 400 600 800 1000 1200 1400Partikelgröße x in µm
Q3 i
n % Feinfraktion
Mittelfraktion
Grobfraktion

Ergebnisse und Diskussion 104
Abbildung 93: Zwei Montanwachspartikeln (Feinfraktion) mit Flüssigkeitsbrücke aus Speiseöl
Die für die Berechnung verwendeten Zahlenwerte sind in Tabelle 24 und die Ergebnisse der Berechnungen in Tabelle 25 zusammengestellt. Die Berechnung ist im Anhang ausführlich dargestellt. Tabelle 24: Zahlenwerte für die Berechnung einer Flüssigkeitsbrücke der Feinfraktion
dP [µm] R1 [µm] R2 [µm] a [µm] ββββ [°] 400 21 40 3,2 17
Tabelle 25: Ergebnisse der Berechnungen einer Flüssigkeitsbrücke der Feinfraktion
Volumen der Flüssigkeitsbrücke [m3]
Nach Pietsch und Rumpf [37]
Nach Weigert und Rippberger [55]
Interpartikuläre Kraft [N] Gewichtskraft [N]
15 ⋅ 10-15 10 ⋅ 10-14 35 ⋅ 10-6 34 ⋅ 10-8
In Tabelle 25 ist zu sehen, dass die Abschätzung nach Weigert und Rippberger ein Volumen der Flüssigkeitsbrücke ergibt, das um eine Zehnerpotenz größer ist, als das nach Pietsch und Rumpf berechnete Volumen. Die interpartikuläre Kraft ist um zwei Zehnerpotenzen höher, als die Gewichtskraft der Montanwachspartikeln. Daher bildeten sich Flüssigkeitsbrücken aus. Diese waren allerdings nur in Ruhe stabil. Sobald das Produkt in Bewegung gebracht wird, fallen die Agglomerate auseinander und es entsteht freie Flüssigkeit in der Schüttung.

Ergebnisse und Diskussion 105
4.2.4.2 Herstellung poröser Montanwachspartikeln durch Zusatz von Kochsalz Da in Kapitel 4.2.3 der maßgebliche Einfluss der Partikelporosität auf die maximal mögliche Beladung nachgewiesen wurde, sollten Modellpartikeln erzeugt werden, die sowohl monodispers als auch rund sind und bestimmte Partikelporositäten aufweisen. Dazu wurde Kochsalz unter einer Wachsschmelze aus Montanwachs dispergiert und diese Mischung im JetCutter vertropft. Aus den hergestellten Partikeln sollte das Kochsalz anschließend wieder herausgelöst werden, um die Porosität zu erzeugen. Wie viel sich von dem eingerührtem Kochsalz aus den Partikeln tatsächlich wieder löste, wurde mittels Leitfähigkeitsmessung bestimmt. Das zu Grunde liegende Arbeitsschema ist in Abbildung 94 dargestellt.
Abbildung 94: Modellvorstellung: Herstellung und Analyse der Monatnwachspartikeln mit Kochsalz im JetCutter
Für die Partikelherstellung im JetCutter wurde in drei Versuchen Kochsalz in einer Konzentration von 33 % zu einer Wachsschmelze aus Montanwachs Licolub WE4 hinzugefügt. Das Salz konnte ohne Probleme untergerührt werden, und es entstand eine homogene Masse, die sich ebenso gut wie die Wachsschmelze im JetCutter vertropfen ließ. Zum Einsatz kam dabei eine Düse mit einem Durchmesser von 400 µm, so dass auch die Salzpartikeln, die einen mittleren Durchmesser x50 von 13 µm aufweisen und deren Partikeln alle kleiner als 87 µm sind, durch die Düse gefördert werden konnten. Der Massenstrom betrug bei diesen Versuchen 0,97 g/s Die in Tabelle 26 aufgelisteten Produkte sind in drei Versuchen erzeugt worden.
H2O
Leitfähigkeitsmessung
Kochsalz geht in Lösung
Wachsschmelze mit Kochsalz
(a) (b) (c)

Ergebnisse und Diskussion 106
Tabelle 26: Montanwachspartikeln mit Kochsalz
Versuch Schüttdichte
[kg/m³]
Partikelgröße x50 Laserbeugung
[µm]
Partikelgröße x50
Software BeadSim 2.1 [µm]
1 650 829 ± 46 693 2 664 831 ± 22 737 3 600 958 ± 190 795
Nach der Partikelherstellung, die sich problemlos realisieren ließ, stellte sich die Frage, wie die Partikeln nach dem Herauslösen des Kochsalzes aussahen (siehe auch Abbildung 94 (a) – (c)):
(a) Das Kochsalz ist komplett durch das Wachs bedeckt und kann nicht bzw. nur von der Oberfläche gelöst werden.
(b) Das Kochsalz, das sich in der Nähe der Oberfläche befindet, kann durch das umgebende Wasser herausgelöst werden und es entstehen einzelne Hohlräume an der Partikeloberfläche.
(c) Die Kochsalzpartikeln liegen so dicht beieinander, dass sich durch das Herauslösen des Kochsalzes durchgängige Poren durch die Partikeln ausbilden.
Die Menge Kochsalz, die aus den Partikeln herausgelöst wurde, konnte mittels Leitfähigkeitsmessung bestimmt werden. Dazu wurde zunächst eine Eichgerade für die Leitfähigkeit erstellt, die in Abbildung 95 dargestellt ist.
Abbildung 95: Eichgerade für die Leitfähigkeitsmessung
Die salzhaltigen Partikeln wurden in destilliertes Wasser eingerührt und unmittelbar danach wurde die Leitfähigkeit L0 gemessen. Die Ansätze wurden drei Tage stehen
y = 15,316x
0
4
8
12
16
20
0 0,2 0,4 0,6 0,8 1
Konzentration der Kochsalzlösung in g/100 ml
Leitf
ähig
keit
in m
S/cm
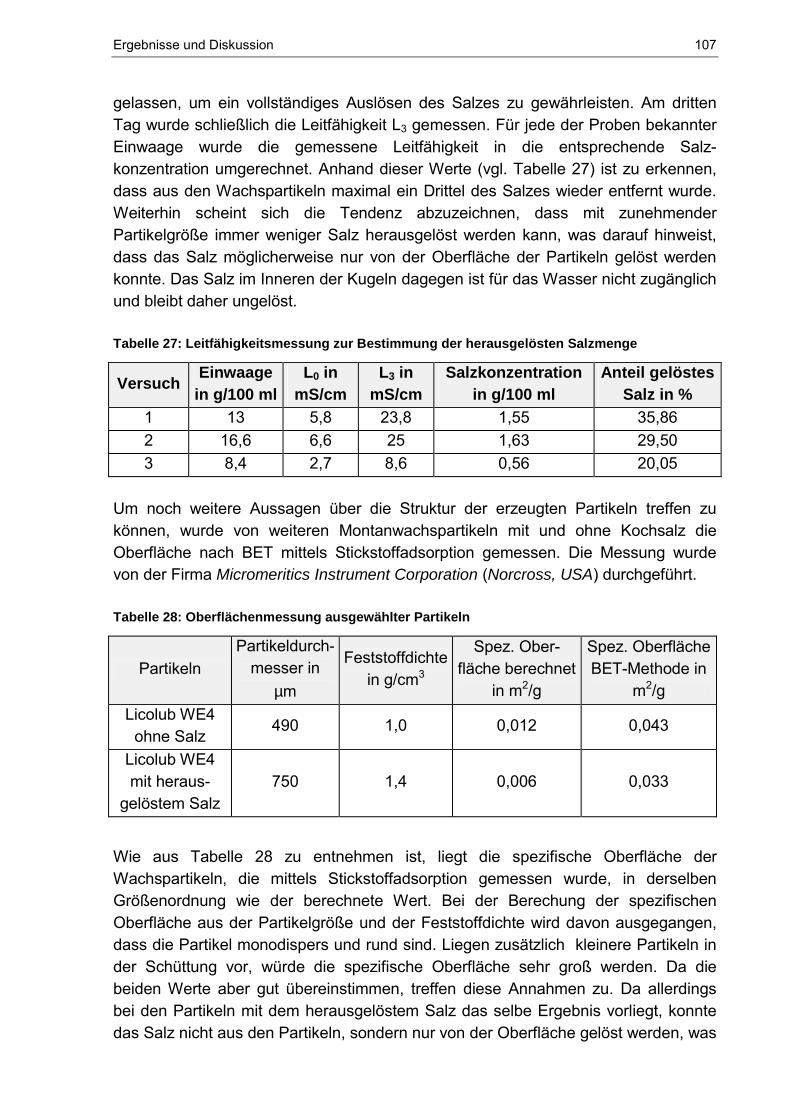
Ergebnisse und Diskussion 107
gelassen, um ein vollständiges Auslösen des Salzes zu gewährleisten. Am dritten Tag wurde schließlich die Leitfähigkeit L3 gemessen. Für jede der Proben bekannter Einwaage wurde die gemessene Leitfähigkeit in die entsprechende Salz-konzentration umgerechnet. Anhand dieser Werte (vgl. Tabelle 27) ist zu erkennen, dass aus den Wachspartikeln maximal ein Drittel des Salzes wieder entfernt wurde. Weiterhin scheint sich die Tendenz abzuzeichnen, dass mit zunehmender Partikelgröße immer weniger Salz herausgelöst werden kann, was darauf hinweist, dass das Salz möglicherweise nur von der Oberfläche der Partikeln gelöst werden konnte. Das Salz im Inneren der Kugeln dagegen ist für das Wasser nicht zugänglich und bleibt daher ungelöst. Tabelle 27: Leitfähigkeitsmessung zur Bestimmung der herausgelösten Salzmenge
Versuch Einwaage in g/100 ml
L0 in mS/cm
L3 in mS/cm
Salzkonzentration in g/100 ml
Anteil gelöstes Salz in %
1 13 5,8 23,8 1,55 35,86 2 16,6 6,6 25 1,63 29,50 3 8,4 2,7 8,6 0,56 20,05
Um noch weitere Aussagen über die Struktur der erzeugten Partikeln treffen zu können, wurde von weiteren Montanwachspartikeln mit und ohne Kochsalz die Oberfläche nach BET mittels Stickstoffadsorption gemessen. Die Messung wurde von der Firma Micromeritics Instrument Corporation (Norcross, USA) durchgeführt. Tabelle 28: Oberflächenmessung ausgewählter Partikeln
Partikeln Partikeldurch-
messer in µm
Feststoffdichte in g/cm3
Spez. Ober-fläche berechnet
in m2/g
Spez. OberflächeBET-Methode in
m2/g Licolub WE4
ohne Salz 490 1,0 0,012 0,043
Licolub WE4 mit heraus-
gelöstem Salz 750 1,4 0,006 0,033
Wie aus Tabelle 28 zu entnehmen ist, liegt die spezifische Oberfläche der Wachspartikeln, die mittels Stickstoffadsorption gemessen wurde, in derselben Größenordnung wie der berechnete Wert. Bei der Berechung der spezifischen Oberfläche aus der Partikelgröße und der Feststoffdichte wird davon ausgegangen, dass die Partikel monodispers und rund sind. Liegen zusätzlich kleinere Partikeln in der Schüttung vor, würde die spezifische Oberfläche sehr groß werden. Da die beiden Werte aber gut übereinstimmen, treffen diese Annahmen zu. Da allerdings bei den Partikeln mit dem herausgelöstem Salz das selbe Ergebnis vorliegt, konnte das Salz nicht aus den Partikeln, sondern nur von der Oberfläche gelöst werden, was

Ergebnisse und Diskussion 108
schon die Ergebnisse der Leitfähigkeitsmessung zeigten. Daher ist es auf diesem Wege nicht möglich, poröse Partikeln zu erzeugen und ein neuer Versuch mit Modellpartikeln aus Natriumalginat wurde unternommen. 4.2.4.3 Modellpartikeln aus Natriumalginat mit Zusatz von Titandioxid Um poröse Partikeln zum Beladen zur Verfügung zu stellen, wurden mit dem JetCutter Partikeln aus einer 2%-igen Natriumalginatlösung hergestellt, die in Calciumchloridlösung eingetropft wurden. Bei der verwendeten Methode wird die Gelierung der Alginatgele mit Calcium-Ionen ausgenutzt (vgl. Abbildung 96). [24] Beim Eintropfen eines Natriumalginattropfens in eine Lösung mit Ca2+-Ionen wird zunächst an der Grenzfläche zwischen Tropfen und Lösung eine dünne, ionen-durchlässige Membran aus Calciumalginat gebildet. Dabei werden benachbarte Polymergruppen des Alginats mit Ca2+-Ionen vernetzt. Durch weitere Diffusion der Ca2+-Ionen in den Natriumalginattropfen geliert dieser vollständig. [40]
Abbildung 96: Schematische Darstellung der Bildung von Ca-Alginat-Hydrogelen durch ionotrope Gelierung von außen nach innen, nach [24]
Dass solche Partikeln eine hohe Porosität aufweisen, wird von Mofidi u. a. [31] beschrieben, die Alginatkugeln mit einem anderen Verfahren herstellten. Dabei wurde eine 2 – 4 %-ige Alginatlösung in einem Reaktor in eine nicht wässrige Flüssigkeit eingerührt, sodass eine homogene Suspension aus kleinen Alginat-tröpfchen entstand. Unter Rühren wurde eine entsprechende Menge Koagulant zugegeben, so dass die Alginatpartikeln ausfielen. Die so erzeugten Kugeln wiesen einen mittleren Partikeldurchmesser von 500 µm und eine durchschnittliche Porengröße von 0,36 µm auf. Diese bereits in zahlreichen Versuchen erfolgreich als Verkapselungsmatrix eingesetzte Lösung [41], [31] ließ sich im JetCutter zu gleichmäßigen Partikeln im Bereich 300 - 1000 µm vertropfen. Die erzielten Natriumalginatkugeln, die in CaCl2 oder Wasser suspendiert zunächst eine runde Form aufweisen, flachen erfahrungs-gemäß beim Trocknen deutlich ab. Um diesen Formverlust während des Trocknungsprozesses zu verhindern, wurde durch Zugabe von Titandioxid der Feststoffanteil der Lösung erhöht. Die Tropfen gelierten nun so, dass die entstandenen Partikeln rund und gleichmäßig waren (siehe Abbildung 97).

Ergebnisse und Diskussion 109
~800 µm Abbildung 97: Partikeln aus 1,5 % Alginat mit 40 % TiO2
Um zu überprüfen, ob diese Partikeln tatsächlich Poren aufweisen, wurde von einer ersten Probe die BET-Oberfläche mittels Stickstoffadsorbtion gemessen und mit der aus dem Partikeldurchmesser berechneten Oberfläche einer Kugel verglichen. Tabelle 29: Spezifische Oberfläche von Natriumalginatpartikeln
Partikeldurchmesser in µm
Feststoffdichte in g/cm3
Spez. Oberfläche berechnet in m2/g
Spez. Oberfläche BET-Methode in m2/g
415 4 0,0036 11,88
Aus den Werten der Tabelle 29 wird deutlich, dass die mittels Stickstoffadsorption bestimmte Oberfläche um mehrere Zehnerpotenzen höher liegt als der aus dem Durchmesser abgeschätzte Wert. Man kann also davon ausgehen, dass die mittels JetCutter produzierten Partikeln eine Porosität aufweisen. Für die endgültige Partikelherstellung ist eine Mischung aus folgender Zusammen-setzung verwendet worden: Destilliertes Wasser: 58,8 % Natriumalginat: 1,2 % Titandioxid: 40 % Am JetCutter wurden Einstellungen gewählt: Druck im Vorlagebehälter: 5 bar Temperatur im Vorlagebehälter: Raumtemperatur Massenstrom: 1,0 bis 1,5 g/s Durchmesser der Düse: 400 µm Drehzahl des Schneidwerkzeuges: 10.000 min -1

Ergebnisse und Diskussion 110
Die entstandenen Partikeln wurden in zwei Fraktionen getrennt, deren Partikelgrößen in Abbildung 98 dargestellt und deren physikalische Eigenschaften in Tabelle 30 zusammengestellt sind. Tabelle 30: Physikalische Eigenschaften der erzeugten Alginatpartikeln
Eigenschaft Feinere Fraktion Größere Fraktion Feststoffdichte [kg/m³] 4,3 ± 0,2 -
Schüttdichte [kg/m³] 1300 ± 2 1224 ± 1
Stampfdichte [kg/m³] 1428 ± 16 1405 ± 56
x50 - Wert [µm] 452 ± 7 678 ± 13
Trockensubstanzgehalt [%] 97,9 ± 1,0 98,4 ± 0,9
Abbildung 98: Partikelgrößenverteilung der Alginatpartikeln (Laserbeugung)
Anschließend wurden diese Partikeln im CPF-Verfahren mit Speiseöl in steigenden Konzentrationen beladen. Dabei wurde im Mischer eine Temperatur von 40 °C und ein Sprühdruck von 100 bar eingestellt. Der Massenstrom des Speiseöls betrug in etwa 0,5 g/s. Obwohl diese Partikeln eine hohe Stampfdichte aufweisen und gemäß Abbildung 28 und Abbildung 88 kaum Flüssigkeit aufnehmen können, konnten sie dennoch mit bis zu 20 % beladen werden. Die Erklärung dafür ist die abweichende Feststoffdichte von 4,3 kg/m³. Bei den bisher untersuchten Trägerstoffen lag die Feststoffdichte immer zwischen 1,5 und 2,5 kg/m³. Die Alginatpartikeln weisen einen hohen Anteil an Titandioxid auf und haben deswegen eine sehr viel höhere Feststoffdichte, obwohl die Partikeln porös sind. Die Ergebnisse der Filtermethode sind in Abbildung 100 und Abbildung 101 dargestellt und eine Zusammenstellung der Versuche ist in Tabelle 31 gegeben. Beladungen bis 20 % sind bei beiden Fraktionen
0
20
40
60
80
100
0 500 1000 1500
Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%
Größere Fraktion Feinere Frakiton

Ergebnisse und Diskussion 111
problemlos möglich, erst bei Beladungen über 20 % tauchen Agglomerate und freie Flüssigkeit in der Schüttung auf, wie auch in Abbildung 99 zu sehen ist. Tabelle 31: Im CPF-Verfahren mit Speiseöl beladene Alginatpartikeln
Trägerstoff Speiseöl in % Optische Beurteilung 8,3 10,0 17,4
Feinkörnig, ohne Agglomerate Feinere Fraktion
24,2 Ölig, Agglomerate, zu hoch beladen
13,0 20,0
Feinkörnig, ohne Agglomerate Gröbere Fraktion
26,5 Ölig, Agglomerate, zu hoch beladen
17,4 % 24,2 %
Abbildung 99: Feine Fraktion der Alginatpartikeln mit Speiseöl
Abbildung 100: Ergebnisse der Filtermethode für Speiseöl auf der feinere Fraktion der Alginatpartikeln
012345
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg

Ergebnisse und Diskussion 112
Abbildung 101: Ergebnisse der Filtermethode für Speiseöl auf der gröberen Fraktion der Alginatpartikeln
Anhand dieser Untersuchung konnte gezeigt werden, dass die Abhängigkeit der maximal möglichen Beladung von der Stampfdichte nur dann gegeben ist, wenn die Feststoffdichte konstant bleibt. Dies bedeutet, dass die maximal mögliche Beladung vor allem von der Partikelporosität abhängt, wie auch schon in Kapitel 4.2.3 diskutiert wurde.
4.2.5 Veränderung von Trägerstoffpartikeln, um die maximale Aufnahme-kapazität zu erhöhen
Da in der Praxis oft eine möglichst hohe Aufnahmekapazität erwünscht ist, stellte sich die Frage, wie gegebene Trägerstoffpartikeln verändert werden können, um die maximale Aufnahmekapazität zu erhöhen. Dazu wurde ein Versuch mit Kochsalz durchgeführt, das in seiner ursprünglichen Form nur mit sehr geringen Mengen Flüssigkeit beladen werden kann. In dieser Form weist das Kochsalz folgende, in Tabelle 32 zusammengestellte, Eigenschaften auf. Tabelle 32: Physikalische Eigenschaften von Kochsalz
Eigenschaft Ermittelter Wert Herstellerangaben Feststoffdichte [kg/m³] 2,2 -
Schüttdichte [kg/m³] 1254 ± 12 1213
Stampfdichte [kg/m³] 1380 ± 28 -
Partikelgröße [µm] x50 = 180 ± 16* 5 % > 355 und 10 % < 63**
Trockensubstanzgehalt [%] 99,95 99,85 *Laserbeugung, ** gesiebt nach ISO 3310
0
1
2
3
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg

Ergebnisse und Diskussion 113
Dieses Kochsalz wurde sprühgetrocknet, um die Partikelgröße und die Partikelform zu verändern. Dazu wurden 8,1 kg Salz in 33,0 kg destilliertem Wasser gelöst, um eine 20 %-ige Lösung herzustellen. Diese gesättigte Kochsalzlösung wurde in einem Sprühturm (Niro Atomizer Ltd., Typ Minor Production) am Institut für Lebensmittel-verfahrenstechnik und Molkereitechnologie an der Technischen Universität München, Weihenstephan, unter folgenden Bedingungen getrocknet:
Massenstrom der Trocknungsluft: 425 kg/h Temperatur am Eintritt: 200 –210 °C Temperatur am Austritt: 80 °C Rotationsdüse Drehzahl: 2400 U/min
Umfangsgeschwindigkeit: 150 m/s Bohrungsdurchmesser: 2 mm
Versuchsdauer: 2 Stunden Masse verdampftes Wasser: 33 kg Resultierende Salzmenge: 6,4 kg
Die Eigenschaften des sprühgetrockneten Kochsalzes sind in Tabelle 33 aufgeführt. Tabelle 33: Physikalische Eigenschaften von sprühgetrocknetem Kochsalz
Eigenschaft Ermittelter Wert Feststoffdichte [kg/m³] 2200
Schüttdichte [kg/m³] 566 ± 42
Stampfdichte [kg/m³] 797 ± 20
x50 - Wert (Laserbeugung) [µm] 16,2 ± 2,7
Trockensubstanzgehalt [%] 99,85
In Abbildung 102 sind die Partikelgrößenverteilungen und in Abbildung 103 die mittels Rasterelektronenmikroskop angefertigten Aufnahmen des Kochsalzes vor und nach dem Sprühtrocknen abgebildet.

Ergebnisse und Diskussion 114
Abbildung 102: Partikelgrößenverteilungen von Kochsalz original und sprühgetrocknet (Laserbeugung)
Anhand der Partikelgrößenverteilung und der REM Aufnahmen lässt sich deutlich die Abnahme der Partikelgröße erkennen. Auch die Zusammenlagerung der kleinen Partikeln zu größeren Gebilden ist in Abbildung 103 gut ersichtlich.
100 µm 100 µm
Abbildung 103: REM Aufnahmen von Kochsalz: Links original, rechts sprühgetrocknet
Wie in Kapitel 4.2.3 diskutiert worden ist, ist die Porosität von monodispersen, runden Partikeln unabhängig von der Partikelgröße. Dies gilt allerdings nicht für sehr feine Partikel. Wie auch Feng und Yu [11] feststellten, nimmt die Porosität von monodispersen runden Partikeln die kleiner als 200 µm sind, mit abnehmender Partikelgröße zu, weil die Haftkräfte wie Van-der-Waals-Kräfte oder elektrostatische Kräfte zwischen den Partikeln im Verhältnis zu der Gewichtskraft der Partikeln immer stärkeren Einfluss haben, wie die Abbildung 104 zeigt.
0
20
40
60
80
100
0 100 200 300 400 500Partikelgröße x in µm
Sum
men
häuf
igke
it Q
3 in
%
Kochsalz original
Kochsalz sprühgetrocknet

Ergebnisse und Diskussion 115
0 0,01 0,02 0,03 0,050,040,35
0,40
0,45
0,50
0,55
0,60
Durchmesser der Glaskugeln [cm]
Poro
sitä
t εε εε
[−]
[−]
[−]
[−]
Abbildung 104: Abhängigkeit der Porosität vom Durchmesser der Glaskugeln [11]
Auch wenn die Salzpartikeln nicht rund und monodispers sind, spiegelt sich dieses Phänomen in den Werten der Schütt- und Stampfdichten der Kochsalzschüttungen wieder: Werden die Eigenschaften des Salzes vor und nach dem Sprühtrocknen verglichen, ist ersichtlich, dass die Schüttungsporosität der kleinen, sprüh-getrockneten Partikeln deutlich höher ist als die Schüttungsporosität der größeren Salzpartikeln. Diese geringere Stampfdichte weist auch darauf hin, dass die sprühgetrockneten Salzpartikeln im Sprühturm höher mit Flüssigkeiten beladen werden können als das ursprüngliche Kochsalz. Beide Arten Kochsalz wurden im CPF-Sprühturm mit steigenden Mengen Speiseöl beladen, bis die maximale Beladungskapazität laut optischer Beurteilung erreicht war und die Produkte anschließend mittels Filtermethode untersucht. Die durchgeführten Sprühversuche inklusive optischer Beurteilung sind in Tabelle 34 zusammengefasst die Ergebnisse der Filtermethode sind in Abbildung 105 und Abbildung 106 dargestellt.

Ergebnisse und Diskussion 116
Tabelle 34: Sprühversuche mit Kochsalz und Speiseöl im CPF-Verfahren
Trägerstoff Beladung [%] Optische Beurteilung 2,5 Keine Agglomerate sichtbar, wirkt feucht 6,7 Produkt beginnt zu agglomerieren, am Maximum Kochsalz
9,4 Produkt zu hoch beladen 7,1
7,9 Produkt feinkörnig, ohne Agglomeration
11,0 Pulver wirkt feucht, neigt zum agglomerieren Kochsalz sprühgetrocket
16,0 Maximal beladen, Beginn der Agglomeration
Abbildung 105: Ergebnisse der Filtermethode für Speiseöl auf Kochsalz
Abbildung 106: Ergebnisse der Filtermethode für Speiseöl auf sprühgetrocknetem Kochsalz
Während beim ursprünglichen Kochsalz die maximale Beladungskapazität schon bei einer Beladung von 9 % überschritten war, konnte das sprühgetrocknete Kochsalz mit 11 % noch beladen werden und die maximale Kapazität wurde erst bei 16 % erreicht. Anhand dieses Beispieles kann gezeigt werden, dass die Beladungs-
05
1015202530
0 10 20 30 40 50Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g
02468
1012
0 10 20 30 40 50Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g

Ergebnisse und Diskussion 117
kapazität eines Trägerstoffes gezielt verbessert werden kann. Allerdings muss berücksichtigt werden, dass durch die Sprühtrocknung ein Produkt entsteht, das in der Praxis nicht mehr zu verarbeiten ist, da die Schüttung stark hygroskopisch ist und an der Luft sofort zu kompaktieren beginnt. Das Produkt ist nur unter absolutem Luftausschluss lagerfähig, sonst entsteht aus dem freifließendem Schüttgut ein zusammengebackener Block.
4.3 Einfluss der Flüssigkeit auf die maximale Flüssigkeitsauf-nahmekapazität
Im dritten Abschnitt des Ergebnisteils wird der Einfluss der Flüssigkeit auf die maximale Beladbarkeit diskutiert. Palzer [35] erläutert in seiner Arbeit den Einfluss der Flüssigkeit auf die Anreicherung und das Benetzen von Pulvern mit Flüssigkeiten und kommt zu dem Ergebnis, dass bei hochporösen Einzelpartikeln und einer ausreichend feinen Zerstäubung die Flüssigkeit in die Partikeln penetriert und sich deswegen die Fließfähigkeit und die Partikelgröße des Pulvers bis zur Sättigung nur unwesentlich ändert. Er stellte fest, das dies bei agglomerierenden Pulverprodukten anders ist und die Flüssigkeit hier einen erheblichen Einfluss auf die pulvertypischen Eigenschaften hat, da sie auf der Partikeloberfläche vorliegt. Grüner [15] hat in ihrer Arbeit Trägerstoffe im CPF-Verfahren mit den unterschiedlichsten Gewürzextrakten beladen und kommt zu dem Ergebnis, dass von Seiten der Extrakte nur ein geringer Einfluss auf die Anwendung der CPF-Technologie zu verzeichnen ist. Alle von ihr verwendeten Gewürzextrakte (u.a. dünnflüssiger Ingwerextrakt, stichfester Basilikumextrakt und zäher Oreganoextrakt) ließen sich pulverisieren und die unterschiedlichen Extraktzusammensetzungen und Viskositäten spielten keine bzw. nur eine untergeordnete Rolle hinsichtlich der Beladbarkeit. Die Kieselsäure Cabosil M5 ließ sich beispielsweise unabhängig von der Extraktsorte stets mit annähernd gleicher Extraktmenge beladen. Auch im Rahmen der vorliegenden Arbeit wurden für einige Trägerstoffe unterschiedliche Flüssigkeiten zum Beladen ausgewählt, und die Ergebnisse miteinander verglichen. Als erstes Beispiel werden die Ergebnisse des Beladens von Kieselsäure Aerosil R972 mit Speiseöl, Triacetin und Wasser diskutiert.
4.3.1 Kieselsäure Aerosil R972 mit Speiseöl, Triacetin und Wasser
Diese Kieselsäure kann mit Speiseöl bis zu 60 % beladen werden, bei 80 % ist die maximale Aufnahmekapazität deutlich überschritten, wie in Abbildung 79 in Kapitel 4.2.1 zu erkennen ist. Dieselbe Versuchsreihe wurde mit Triacetin als flüssige Komponente wiederholt.

Ergebnisse und Diskussion 118
0
5
10
15
20
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g
Abbildung 107: Ergebnisse der Filtermethode für Aerosil R972 mit Triacetin
Wie in Abbildung 107 zu sehen ist, kann kein großer Unterschied zwischen der maximal möglichen Beladung mit Speiseöl und mit Triacetin festgestellt. Bei beiden Flüssigkeiten liegt das Maximum zwischen 60 und 70 %. Eine weitere Versuchsreihe wurde mit Wasser durchgeführt. Wie in Abbildung 108 zu erkennen ist, dringt ein Wassertropfen, der auf eine Schüttung aus Aerosil R972 gegeben wird, nicht in diese ein, sondern bleibt als Tropfen liegen. Dies liegt an der hydrophobierten Oberfläche der Kieselsäure, zwischen der und Wasser sich ein Kontaktwinkel > 90 ° einstellt.
Abbildung 108: Wassertropfen auf einer Schüttung aus Aerosil R972
Dennoch war es möglich, die Kieselsäure im CPF-Verfahren mit Wasser zu beladen und als Produkt wurde ein frei fließfähiges Pulver erzeugt. Die Ergebnisse der Filtermethode sind in Abbildung 109 dargestellt und es wird deutlich, dass auch bei einer Beladung über 70 % ein stabiles Produkt vorliegt und kaum mehr Wasser als bei niedrigeren Beladungen auf den Filter übergeht. Der Anstieg beginnt erst bei über 80%.
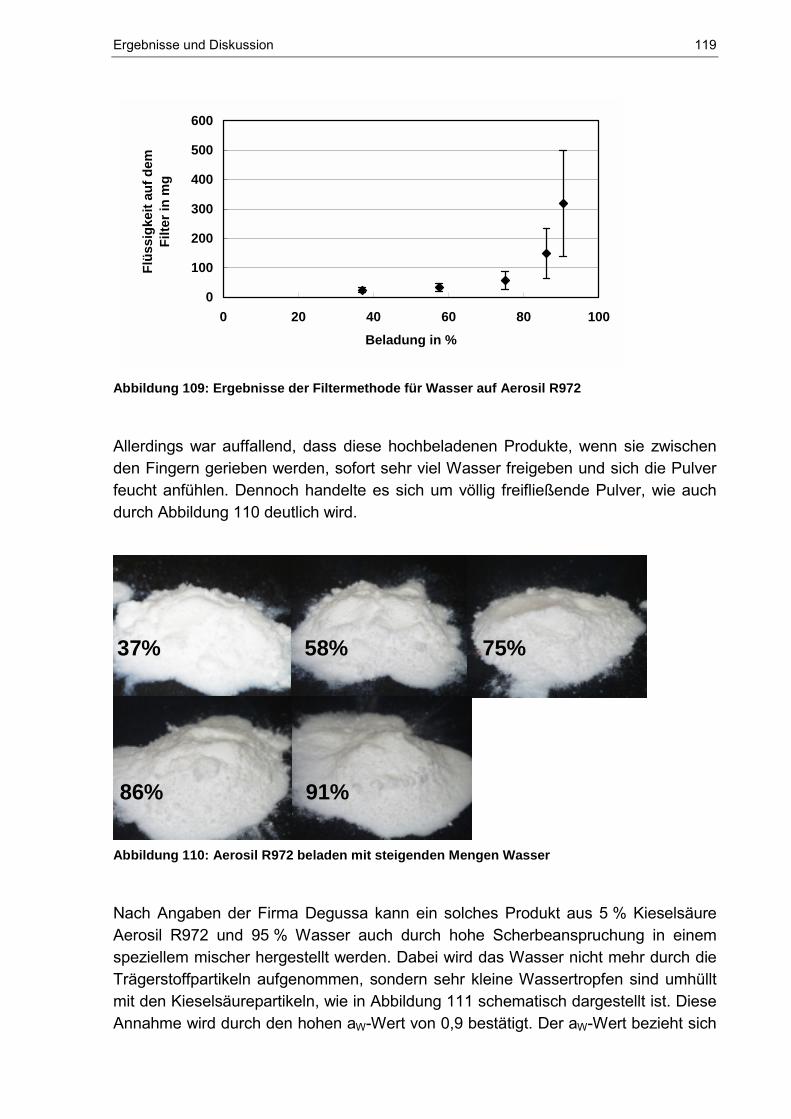
Ergebnisse und Diskussion 119
0
100
200
300
400
500
600
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g
Abbildung 109: Ergebnisse der Filtermethode für Wasser auf Aerosil R972
Allerdings war auffallend, dass diese hochbeladenen Produkte, wenn sie zwischen den Fingern gerieben werden, sofort sehr viel Wasser freigeben und sich die Pulver feucht anfühlen. Dennoch handelte es sich um völlig freifließende Pulver, wie auch durch Abbildung 110 deutlich wird.
37% 58% 75%
86% 91%
Abbildung 110: Aerosil R972 beladen mit steigenden Mengen Wasser
Nach Angaben der Firma Degussa kann ein solches Produkt aus 5 % Kieselsäure Aerosil R972 und 95 % Wasser auch durch hohe Scherbeanspruchung in einem speziellem mischer hergestellt werden. Dabei wird das Wasser nicht mehr durch die Trägerstoffpartikeln aufgenommen, sondern sehr kleine Wassertropfen sind umhüllt mit den Kieselsäurepartikeln, wie in Abbildung 111 schematisch dargestellt ist. Diese Annahme wird durch den hohen aW-Wert von 0,9 bestätigt. Der aW-Wert bezieht sich

Ergebnisse und Diskussion 120
auf die Dampfphase, die sich mit dem festen Material oder einer Lösung im Gleichgewicht befindet. Er gibt den Quotienten aus der Konzentration an Wasser in der Dampfphase, d. h. im Luftraum über dem Material und der Wasserkonzentration im Luftraum über reinem Wasser bei einer bestimmten Temperatur an [47]. Eine solche Produktstruktur, tritt laut Palzer [35] auf, wenn ein Pulver mit einer nicht benetzenden, einer erstarrenden oder einer hoch viskosen Flüssigkeit gemischt wird und die Tropfen größer als die zu beladenen Pulverpartikeln sind. Im Fall der Kieselsäure Aerosil R972 ist Wasser eine nicht benetzende Flüssigkeit und die Primärpartikelchen sind mit einem mittleren Durchmesser von 16 nm sehr klein.
Abbildung 111: Schematische Darstellung eines Wassertropfens umhüllt von Feststoffpartikeln
Eine mikroskopische Aufnahme des Produktes mit einem Anteil von 91 % Wasser ist in Abbildung 112 abgebildet. Man kann auch hier die vorhanden Wassertropfen erkennen. Durch den im Bild daneben abgebildeten Maßstab, kann die Größe der Wassertropfen abgeschätzt werden, da beide Bilder dieselbe Vergrößerung aufweisen. Demnach sind die Wassertropfen, die im CPF Sprühturm erzeugt wurden bis zu 0,09 mm groß.
Abbildung 112: Aerosil R972 mit 91% Wasser unter dem Mikroskop
Als Ergebnis kann festgehalten werden, dass in diesem Fall eine hydrophobe Kieselsäure mit Speiseöl und Triacetin gleich hoch beladen werden kann, mit Wasser dagegen eine höhere Beladung erzielt werden kann. Die Erklärung dafür ist, dass diese hydrophobe Kieselsäure mit Wasser nicht benetzbar ist, und aus diesem Grund eine grundlegend andere Produktstruktur entsteht. In so einem Fall spielen die
1 mmWassertropfen

Ergebnisse und Diskussion 121
Eigenschaften der Flüssigkeiten für die maximale Beladung eines Trägerstoffes eine entscheidende Rolle. Um den Einfluss der Flüssigkeiten weiter zu untersuchen, wurde ein Superabsorber, das sogenannte HySorb der Firma BASF, beladen.
4.3.2 Superabsorber HySorb mit Speiseöl und Wasser
Als Flüssigkeiten kamen bei diesem Trägerstoff Speiseöl und Wasser zum Einsatz. Die Ergebnisse der Filtermethode sind in Abbildung 113 und Abbildung 114 dargestellt.
Abbildung 113: Ergebnisse der Filtermethode für HySorb mit Speiseöl
Abbildung 114: Ergebnisse der Filtermethode für HySorb mit Wasser
Es ist deutlich zu erkennen, dass mit Speiseöl schon bei 20 % die maximale Aufnahmekapazität überschritten ist, bei Wasser dagegen sehr hohe Beladungen möglich sind. Der Grund dafür sind Vernetzungen, die sich bei der Reaktion zwischen HySorb und Wasser ausbilden. In Abbildung 115 ist ein Polymernetzwerk, wie es für Superabsorber typisch ist, schematisch dargestellt. Die Punkte stellen die Quervernetzungen zwischen den spiralförmigen Polymeren dar.
0
5
10
15
20
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g
02468
101214
0 20 40 60 80 100Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g
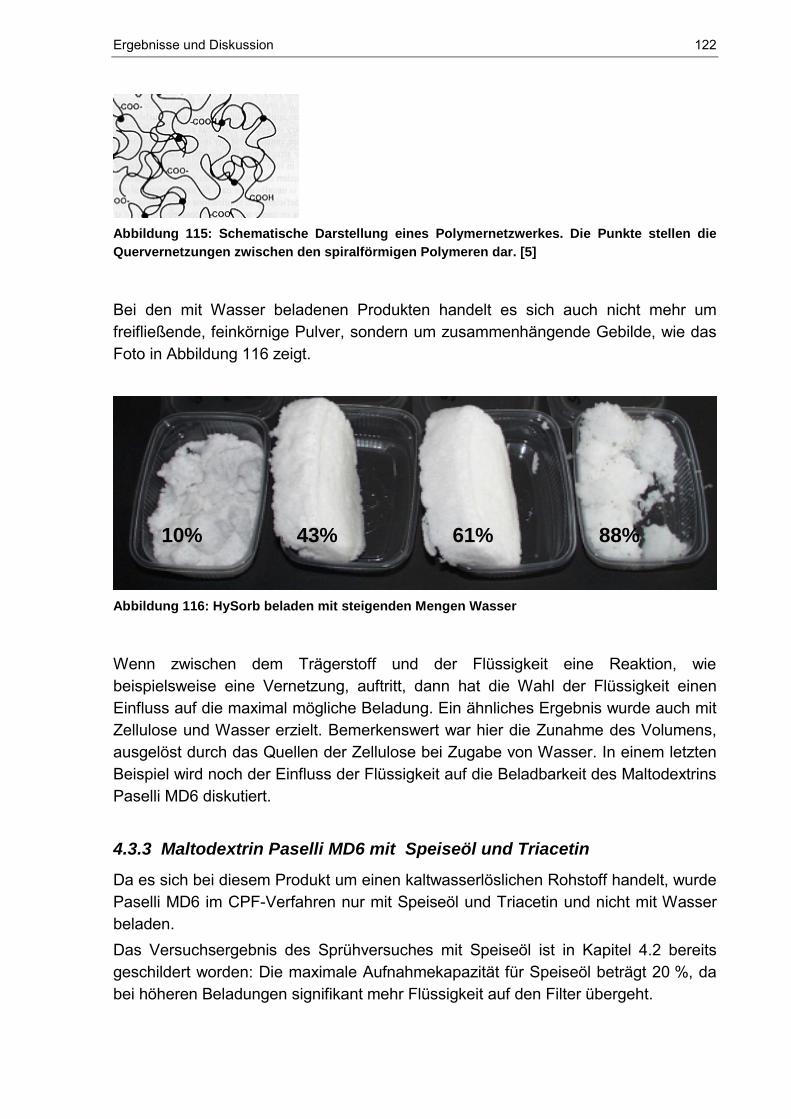
Ergebnisse und Diskussion 122
Abbildung 115: Schematische Darstellung eines Polymernetzwerkes. Die Punkte stellen die Quervernetzungen zwischen den spiralförmigen Polymeren dar. [5]
Bei den mit Wasser beladenen Produkten handelt es sich auch nicht mehr um freifließende, feinkörnige Pulver, sondern um zusammenhängende Gebilde, wie das Foto in Abbildung 116 zeigt.
10% 43% 88%61%
Abbildung 116: HySorb beladen mit steigenden Mengen Wasser
Wenn zwischen dem Trägerstoff und der Flüssigkeit eine Reaktion, wie beispielsweise eine Vernetzung, auftritt, dann hat die Wahl der Flüssigkeit einen Einfluss auf die maximal mögliche Beladung. Ein ähnliches Ergebnis wurde auch mit Zellulose und Wasser erzielt. Bemerkenswert war hier die Zunahme des Volumens, ausgelöst durch das Quellen der Zellulose bei Zugabe von Wasser. In einem letzten Beispiel wird noch der Einfluss der Flüssigkeit auf die Beladbarkeit des Maltodextrins Paselli MD6 diskutiert.
4.3.3 Maltodextrin Paselli MD6 mit Speiseöl und Triacetin
Da es sich bei diesem Produkt um einen kaltwasserlöslichen Rohstoff handelt, wurde Paselli MD6 im CPF-Verfahren nur mit Speiseöl und Triacetin und nicht mit Wasser beladen. Das Versuchsergebnis des Sprühversuches mit Speiseöl ist in Kapitel 4.2 bereits geschildert worden: Die maximale Aufnahmekapazität für Speiseöl beträgt 20 %, da bei höheren Beladungen signifikant mehr Flüssigkeit auf den Filter übergeht.
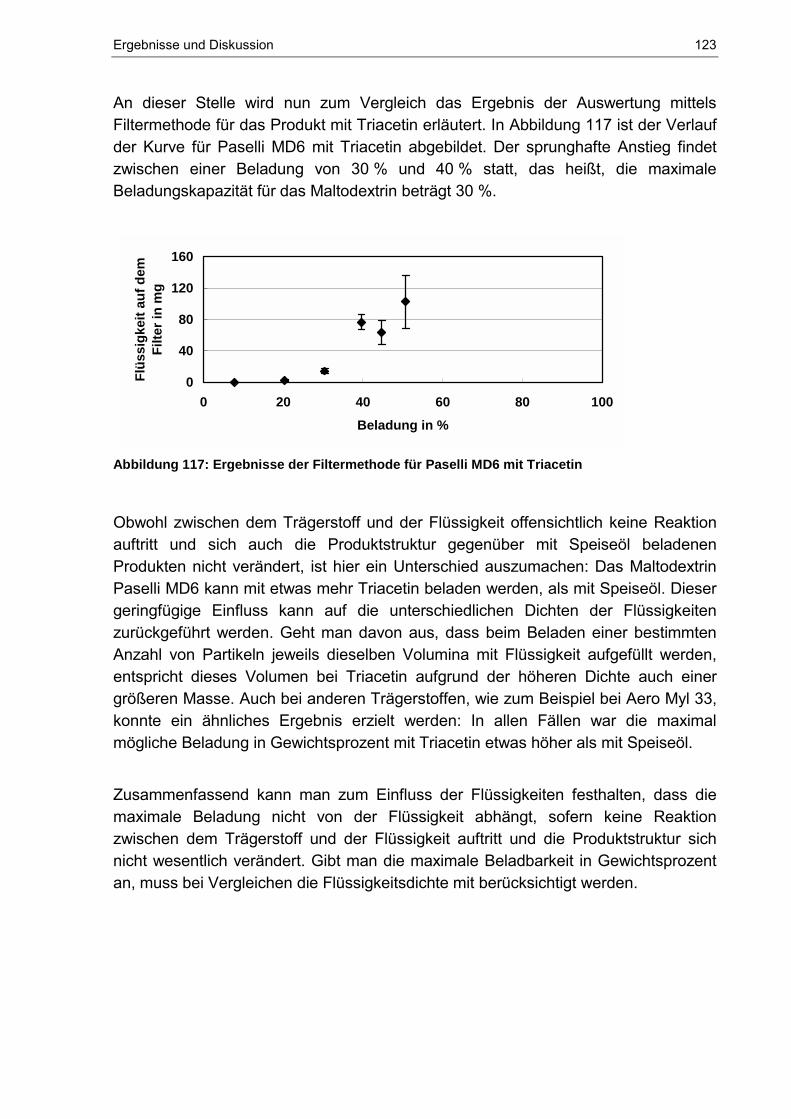
Ergebnisse und Diskussion 123
An dieser Stelle wird nun zum Vergleich das Ergebnis der Auswertung mittels Filtermethode für das Produkt mit Triacetin erläutert. In Abbildung 117 ist der Verlauf der Kurve für Paselli MD6 mit Triacetin abgebildet. Der sprunghafte Anstieg findet zwischen einer Beladung von 30 % und 40 % statt, das heißt, die maximale Beladungskapazität für das Maltodextrin beträgt 30 %.
0
40
80
120
160
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
emFi
lter i
n m
g
Abbildung 117: Ergebnisse der Filtermethode für Paselli MD6 mit Triacetin
Obwohl zwischen dem Trägerstoff und der Flüssigkeit offensichtlich keine Reaktion auftritt und sich auch die Produktstruktur gegenüber mit Speiseöl beladenen Produkten nicht verändert, ist hier ein Unterschied auszumachen: Das Maltodextrin Paselli MD6 kann mit etwas mehr Triacetin beladen werden, als mit Speiseöl. Dieser geringfügige Einfluss kann auf die unterschiedlichen Dichten der Flüssigkeiten zurückgeführt werden. Geht man davon aus, dass beim Beladen einer bestimmten Anzahl von Partikeln jeweils dieselben Volumina mit Flüssigkeit aufgefüllt werden, entspricht dieses Volumen bei Triacetin aufgrund der höheren Dichte auch einer größeren Masse. Auch bei anderen Trägerstoffen, wie zum Beispiel bei Aero Myl 33, konnte ein ähnliches Ergebnis erzielt werden: In allen Fällen war die maximal mögliche Beladung in Gewichtsprozent mit Triacetin etwas höher als mit Speiseöl. Zusammenfassend kann man zum Einfluss der Flüssigkeiten festhalten, dass die maximale Beladung nicht von der Flüssigkeit abhängt, sofern keine Reaktion zwischen dem Trägerstoff und der Flüssigkeit auftritt und die Produktstruktur sich nicht wesentlich verändert. Gibt man die maximale Beladbarkeit in Gewichtsprozent an, muss bei Vergleichen die Flüssigkeitsdichte mit berücksichtigt werden.

Ergebnisse und Diskussion 124
4.4 Einfluss des Verfahrens auf die maximale Flüssigkeits-aufnahmekapazität
Alle bisher diskutierten Sprühprodukte entstanden im CPF-Sprühturm. Neben diesem Verfahren wurde auch ein Pflugscharmischer zum Beladen der dispersen Feststoffe mit Flüssigkeit verwendet. Das Ziel dieser Versuchsreihe war es, mögliche Unterschiede in der maximalen Beladungskapazität herauszufinden. Zwei Versuchsreihen wurden mit den Trägerstoffen Kartoffelstärke Aero Myl 33, Pulverzellulose P290 und Maltodextrin Paselli MD6 durchgeführt. In der ersten Versuchsreihe wurden die Trägerstoffe mittels Einstoffdüse mit Speiseöl beladen und in der zweiten Versuchsreihe wurde dafür eine Zweistoffdüse eingesetzt. Die im Mischer erzielten Beladungen wurden aus den eingesetzten Massen berechnet. Um diese Massenbilanzen zu überprüfen, wurden Stichproben genommen und der Fettgehalt nach Soxhlet bestimmt.
4.4.1 Versuche im Pflugscharmischer mit Einstoffdüse
In Abbildung 118 sind die Ergebnisse der ersten Versuchsreihe mit Aero Myl 33 im Pflugscharmischer mit den Ergebnissen im CPF-Sprühverfahren in einem Diagramm zusammengestellt. Es kann kein Unterschied zwischen den Produkten aus den beiden Verfahren festgestellt werden. Der Kurvenverlauf ist für beide Versuchsreihen gleich. Dies bestätigt auch die optische Beurteilung der Produkte. In beiden Verfahren beträgt die maximale Aufnahmekapazität von Aero Myl 33 für Speiseöl 60 %.
Abbildung 118: Ergebnisse der Filtermethode für Aero Myl 33 mit Speiseöl im Pflugscharmischer und im CPF-Verfahren
Bei höheren Beladungen ist zusätzlich zu den feuchten Agglomeraten und Inhomogenitäten im Produkt im Mischerinnenraum eine vermehrte Belagbildung zu
0
40
80
120
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg CPF
Mischer
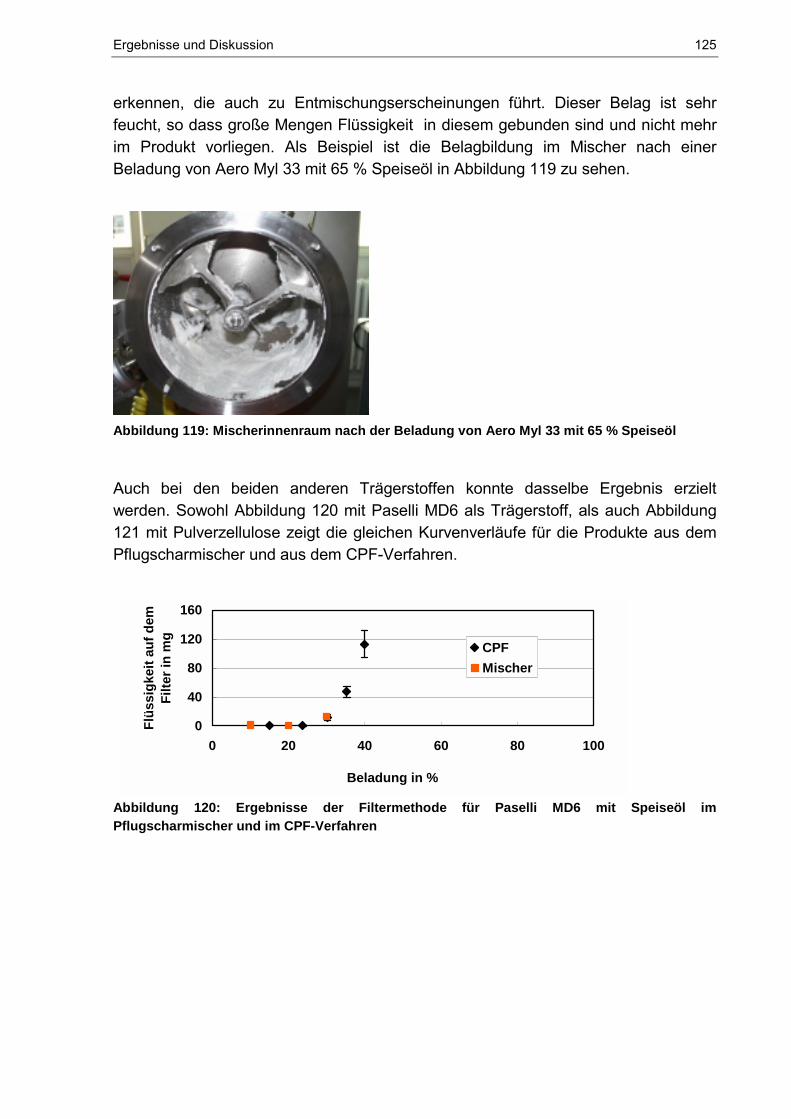
Ergebnisse und Diskussion 125
erkennen, die auch zu Entmischungserscheinungen führt. Dieser Belag ist sehr feucht, so dass große Mengen Flüssigkeit in diesem gebunden sind und nicht mehr im Produkt vorliegen. Als Beispiel ist die Belagbildung im Mischer nach einer Beladung von Aero Myl 33 mit 65 % Speiseöl in Abbildung 119 zu sehen.
Abbildung 119: Mischerinnenraum nach der Beladung von Aero Myl 33 mit 65 % Speiseöl
Auch bei den beiden anderen Trägerstoffen konnte dasselbe Ergebnis erzielt werden. Sowohl Abbildung 120 mit Paselli MD6 als Trägerstoff, als auch Abbildung 121 mit Pulverzellulose zeigt die gleichen Kurvenverläufe für die Produkte aus dem Pflugscharmischer und aus dem CPF-Verfahren.
Abbildung 120: Ergebnisse der Filtermethode für Paselli MD6 mit Speiseöl im Pflugscharmischer und im CPF-Verfahren
0
40
80
120
160
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg
CPFMischer

Ergebnisse und Diskussion 126
Abbildung 121: Ergebnisse der Filtermethode für Zellulose mit Speiseöl im Pflugscharmischer und im CPF-Verfahren
Ferner wurde die Flüssigkeit Triacetin zusätzlich zum CPF-Verfahren auch im Pflugscharmischer auf einige Trägerstoffe aufgebracht. Exemplarisch ist das Ergebnis eines Versuches mit Triacetin und Aero Myl 33 in Abbildung 122 dargestellt. Auch hier ist kein Unterschied zwischen den beiden Verfahren zu erkennen, und mit Triacetin kann auf Grund der höheren Dichte eine etwas höhere Beladung erzielt werden.
Abbildung 122: Ergebnisse der Filtermethode für Triacetin auf Aero Myl 33 im Pflugscharmischer und im CPF-Verfahren
Neben dieser Versuchsreihe wurde in einer weiteren Versuchsreihe die Einstoffdüse durch eine Zweistoffdüse ersetzt. Um auch hier die berechneten Beladungen zu überprüfen, wurde bei Stichproben der Fettgehalt nach Soxhlet ermittelt.
0
20
40
60
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg
CPFMischer
0
100
200
300
400
500
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg CPF
Mischer

Ergebnisse und Diskussion 127
Tabelle 35: Fettgehalte der Produkte aus dem Mischer
Trägerstoff Beladung in % nach
Massenbilanz Fettgehalt in % nach
Soxhlet
Aero Myl 33 40 41,37
Aero Myl 33 60 60,61
Aero Myl 33 60 58,06
Aero Myl 33 65 61,63
Cellulose 30 30,74
Cellulose 40 40,05
Paselli 30 30,02
Die Ergebnisse nach Soxhlet stimmen in fast allen Fällen sehr genau mit den angegebenen Beladungen nach der Massenbilanz überein. Die einzige Ausnahme stellt die Beladung von 65 % laut Massenbilanz da: Mittels Soxhlet-Extraktion konnte nut ein Fettgehalt von 62 % ermittelt werden. Die Begründung dafür liegt darin, dass hier die maximale Beladung der Kartoffelstärke gerade überschritten wurde und nicht mehr die gesamte Flüssigkeit vom Trägerstoff aufgenommen werden konnte. Ein Teil des eingesprühten Speiseöles ist zum Beispiel in den Belägen an der Mischerinnen-wand gebunden (vgl. Abbildung 119).
4.4.2 Versuche im Pflugscharmischer mit Zweistoffdüse
Durch die Zweistoffdüse lässt sich ein feinerer Sprühkegel erzeugen als durch die Einstoffdüse (vgl. Abbildung 123). Dennoch konnte keine höhere Beladung erzielt werden als in den bisherigen Versuchen.
Abbildung 123: Sprühkegel der Einstoffdüse (links) im Vergleich zur Zweistoffdüse (rechts)
Exemplarisch sind zwei Ergebnisse mit Aero Myl 33 und Speiseöl in Abbildung 124 und mit Triacetin in Abbildung 125 dargestellt.

Ergebnisse und Diskussion 128
Abbildung 124: Ergebnisse der Filtermethode für Aero Myl 33 mit Speiseöl im Mischer mit Einstoff- und Zweistoffdüse
Abbildung 125: Ergebnisse der Filtermethode für Aero Myl 33 mit Triacetin im Mischer mit Einstoff- und Zweistoffdüse
Bei den verwendeten Rohstoffen konnte hinsichtlich der Qualität der Sprühprodukte insgesamt weder ein Unterschied zwischen Einstoff- und Zweistoffdüse noch zwischen CPF-Verfahren und Pflugscharmischer festgestellt werden. Zu ersterem sei hinzugefügt, dass es sich bei allen flüssigen Komponenten, die für diese Versuche zum Einsatz kamen, um niedrig viskose sowie leicht zu fördernde und zu versprühende Substanzen handelt. Sollen hochviskose Medien, wie beispielsweise zähflüssige Gewürzextrakte, auf Trägerstoffe aufgesprüht werden, können mit der Zweistoffdüse bessere Ergebnisse erzielt werden als mit der Einstoffdüse [35]. Können die flüssigen Komponenten nicht mehr zur Düse gefördert werden, bietet das CPF-Verfahren den Vorteil, dass die zähflüssige Masse mit überkritischem CO2 ins Gleichgewicht gebracht werden kann und diese gasgesättigte Lösung versprüht werden kann [15]. Ein weiterer Nachteil des Beladens im Pflugscharmischer ist die Erwärmung der Produkte während des Mischvorganges durch den hohen Energie-eintrag. Im CPF-Verfahren dagegen kühlt sich der Sprühturm durch die Entspannung des überkritischen Gases stark ab, so dass die Temperaturen durchschnittlich
0
40
80
120
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg Einstoffdüse
Zweistoffdüse
0
100
200
300
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em
Filte
r in
mg Einstoffdüse
Zweistoffdüse

Ergebnisse und Diskussion 129
zwischen – 10 °C und + 5 °C liegen. Des weiteren ist es von großem Interesse, wie homogen die Beladung der Trägerstoffe in den beiden Verfahren erfolgt.
4.4.3 Homogenität der Sprühprodukte aus dem CPF-Verfahren und aus dem Mischer
Um die Homogenität der Produkte aus dem CPF-Verfahren abzuschätzen, wurden aus den mit Wasser beladenen Produkten kleine Proben gezogen und auf ihren Wassergehalt hin untersuchen. Die produzierte Menge pro Sprühversuch variierte zwischen 500 g und 2000 g. Daraus wurden je drei Stichproben in der Größen-ordnung von ein bis zwei Gramm gezogen und deren Wassergehalt im Trockenschrank gemessen. Tabelle 36: Wassergehalte der Sprühprodukte aus Kieselsäure und Wasser
Trägerstoff Wassergehalt in % nach Massenbilanz
Wassergehalt in % nach Trockenschrankmethode
38,1 39,24 ± 0,59 59,0 59,54 ± 0,07 78,9 79,99 ± 0,63 89,6 89,36 ± 0,23
Aerosil R972
94,8 95,07 ± 0,24 59,2 58,61 ± 0,83 68,8 68,35 ± 0,37 Aerosil 130 79,4 78,36 ± 0,04
Die Werte in Tabelle 36 zeigen, wie viele andere Versuche auch, dass im Sprühturm eine sehr homogene Beadlung stattfindet, da bei allen Dreifachbestimmungen die Konfidenzintervalle der Wassergehalte sehr klein sind. Es treten also innerhalb des Produktes in dieser Größenordnung der Proben keine Schwankungen hinsichtlich des Wassergehaltes auf. Welche Vorgänge bei der Entspannung der Mischung aus Flüssigkeit und überkritischem CO2 genau ablaufen, ist wissenschaftlich noch nicht im Detail geklärt. Aber damit könnte es möglicherweise zusammenhängen, dass die einzelnen Trägerstoffpartikeln jeweils mit derselben Menge Flüssigkeit beladen werden. Hier besteht in Zukunft noch Forschungsbedarf. Für die Anwendung in der Praxis ist es jedoch schon jetzt von großem Vorteil, dass Produkte mit einer so großen Homogenität hergestellt werden können. Um im Vergleich dazu auch die Homogenität der beladenen Produkte aus dem Pflugscharmischer zu untersuchen, ist folgender Versuch durchgeführt worden: Zur Überprüfung der Homogenität des Produktes innerhalb der Mischertrommel wurden bei dem Versuch mit Pulvercellulose P290 und Wasser mit 40%-iger Beladung 30 Proben an unterschiedlichen Stellen der Trommel entnommen und zur

Ergebnisse und Diskussion 130
Bestimmung des Wassergehaltes für 24 Stunden bei 105°C in den Trockenschrank gestellt. Die Werte schwanken dabei um einen Mittelwert von 38,34 %, wobei als maximale Feuchte der Wert von 40,03 % und als minimale Feuchte der Wert von 37,22 % auftritt. Die einzelnen Messwerte sind im Anhang in Tabelle 40 aufgelistet. Insgesamt ist also die Beladung im Mischer auch ziemlich homogen, auch wenn geringe Schwankungen nachgewiesen werden konnten. Allerdings treten bei höheren Beladungen dennoch Entmischungserscheinungen auf, wie die Belag-bildung in Abbildung 119 erkennen lässt: An den Mischerbauteilen im Innenraum bilden sich Beläge aus, die im Durchschnitt einen höheren Flüssigkeitsanteil aufweisen, als das bewegte Mischgut im Inneren.
4.5 Flüssigkeitsverteilung in den Produkten Um die Flüssigkeitsverteilung in beladenen Schüttungen zu untersuchen, wurde zunächst ein Modell aufgestellt, in dem zwischen verschieden fest gebundenen Anteilen Flüssigkeit unterschieden wird. Diese Modell wird dann mittels NMR-Messungen überprüft. Als Modellsubstanz dient die Kieselsäure Sipernat 2200, die mit steigenden Mengen destilliertem Wasser beladen wurde.
4.5.1 Modellvorstellung der verschieden fest gebundenen Flüssigkeits-anteilen
In der Literatur werden verschiedene Bindungsarten von Flüssigkeiten an disperse Feststoffe diskutiert. Zum Teil werden unterschiedliche Mechanismen in Abhängig-keit der Flüssigkeitsmenge unterschieden, wie in Kapitel 2.4 beschrieben wird. In den nachfolgenden Überlegungen wird von Wasser als benetzender Flüssigkeit ausgegangen. Bisher wurde die Beladung, die aus der Massenbilanz nach Gleichung 38 berechnet wird, in Prozent angegeben. Nun wird die Feuchte Fges als Verhältnis der Masse der Flüssigkeit zur Gesamtmasse definiert.
sl
lges mm
mF+
= (Gleichung 48)
Es wird in dieser Versuchsreihe davon ausgegangen, dass die am Partikel gebildete Hydrathülle stärker gebunden ist, als die sich darüber befindenden Schichten. Je weiter sich die Wassermoleküle vom Partikel entfernt befinden, desto schwächer sind sie an ihn gebunden. Der Übergang von stark gebundenem bis hin zu freiem Wasser erfolgt dabei kontinuierlich. Ab einer bestimmten Beladung sind alle Plätze, an denen das Wasser fest gebunden ist, belegt. Deswegen sinkt bei steigender Beladung der Anteil an fest gebundenem Wasser gegenüber dem weniger fest gebundenem Wasser. Anhand von zwei Modellvorstellungen, mit zwei bzw. drei unterschiedlich

Ergebnisse und Diskussion 131
fest gebundenen Wasseranteilen, soll der Verlauf der Anteile in Abhängigkeit der Beladung diskutiert werden. Z w e i u n t e r s c h i e d l i c h f e s t g e b u n d e n e F l ü s s i g k e i t s a n t e i l e
Es werden zwei verschieden fest gebundenen Flüssigkeitsanteile unterschieden: Ein stark gebundener immobilisierter Anteil aim und ein weniger bis überhaupt nicht gebundener, mobiler Anteil amo. Wird der disperse Feststoff mit Flüssigkeit beladen, wird die Flüssigkeit zunächst fest an die Partikeln gebunden. Dies geschieht an der Oberfläche der Partikeln und in den kleinen Kapillaren. Der Anteil an immobiler Flüssigkeit ist in diesem Bereich gleich eins. Wenn alle Stellen, an denen die Flüssigkeit fest gebunden werden kann, besetzt sind, werden die Bereiche zwischen den Partikeln mit Flüssigkeit gefüllt. Hier ist das Wasser nicht mehr so stark gebunden. Der Anteil des stärker gebundenen, immobilen Wassers sinkt und der Anteil des mobilen Wassers steigt. Für einen Sättigungszustand des immobilen Anteils aim bei einer willkürlich bestimmten Feuchte Fo ergibt sich rechnerisch folgender Zusammenhang:
l
imim m
mWassersgesamtendesMasse
WassersgebundenenfestdesMassea == (Gleichung 49)
Aus Gleichung 48 kann ml berechnet werden:
ges
sgesl F-1
mFm
⋅= (Gleichung 50)
Da bei einer Feuchte F0 alle Stellen, an denen das Wasser fest gebunden ist, belegt sind, ergibt sich für die Masse mim des immobilen Wassers folgender Zusammenhang:
0
s0im F-1
mFm ⋅= (Gleichung 51)
Daraus ergibt sich für aim als Funktion von Fges:
Wenn 0 ≤ Fges ≤ F0: aim = 1 und amo = 1 – aim
Wenn F0 < Fges ≤ 1: )F(1F)F(1F
mm)(Fa
0ges
ges0
l
imgim −⋅
−⋅== und amo = 1 – aim
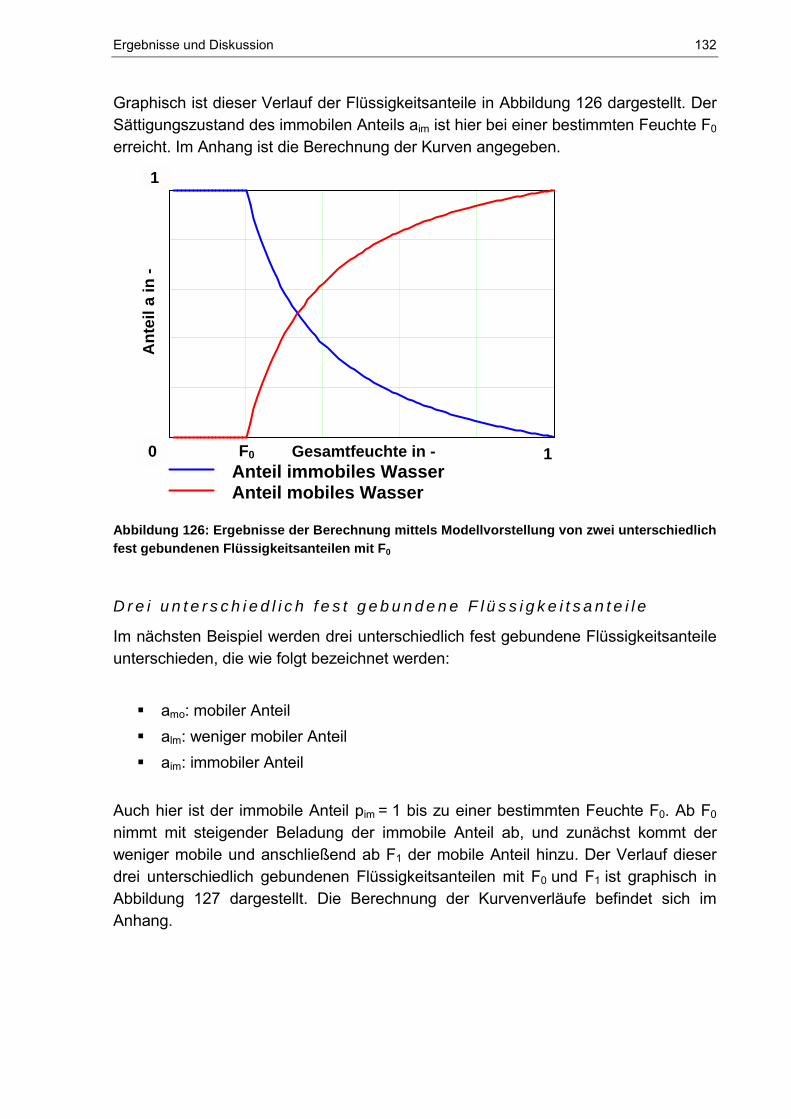
Ergebnisse und Diskussion 132
Graphisch ist dieser Verlauf der Flüssigkeitsanteile in Abbildung 126 dargestellt. Der Sättigungszustand des immobilen Anteils aim ist hier bei einer bestimmten Feuchte F0 erreicht. Im Anhang ist die Berechnung der Kurven angegeben.
Anteil immobiles WasserAnteil mobiles Wasser
Gesamtfeuchte in -
Ant
eil a
in -
Abbildung 126: Ergebnisse der Berechnung mittels Modellvorstellung von zwei unterschiedlich fest gebundenen Flüssigkeitsanteilen mit F0
D r e i u n t e r s c h i e d l i c h f e s t g e b u n d e n e F l ü s s i g k e i t s a n t e i l e
Im nächsten Beispiel werden drei unterschiedlich fest gebundene Flüssigkeitsanteile unterschieden, die wie folgt bezeichnet werden: � amo: mobiler Anteil � alm: weniger mobiler Anteil � aim: immobiler Anteil
Auch hier ist der immobile Anteil pim = 1 bis zu einer bestimmten Feuchte F0. Ab F0 nimmt mit steigender Beladung der immobile Anteil ab, und zunächst kommt der weniger mobile und anschließend ab F1 der mobile Anteil hinzu. Der Verlauf dieser drei unterschiedlich gebundenen Flüssigkeitsanteilen mit F0 und F1 ist graphisch in Abbildung 127 dargestellt. Die Berechnung der Kurvenverläufe befindet sich im Anhang.
F0 10
1

Ergebnisse und Diskussion 133
Anteil an immobilem WasserAnteil an weniger mobilem WasserAnteil an mobilem Wasser
Gesamtfeuchte in -
Ant
eil a
in -
Abbildung 127: Ergebnisse der Berechnung mittels Modelvorstellung der Anteile an immobilem, weniger mobilem und mobilem Wasser mit F0 und F1
4.5.2 Herstellung und Charakterisierung der Sprühprodukte
Für die Überprüfung dieser Modellvorstellungen wurde die Kieselsäure Sipernat 2200 im CPF-Sprühturm mit steigenden Mengen destilliertem Wasser beladen. In Tabelle 37 sind die Sprühprodukte zusammengestellt. Die Beladung Fges wurde mittels Massenbilanz nach dem Sprühversuch ermittelt und im Trockenschrank (14 Stunden, 105 °C) überprüft. Da auch der Rohstoff eine Feuchte von Fges = 0,043 aufweist, sind die durch Trocknung erhaltenen Wassergehalte etwas höher als die Werte aus den Massenbilanzen. Tabelle 37: Sprühprodukte aus Sipernat 2200 und Wasser
Beladung Fges laut Massenbilanz
Beladung Fges laut Trockenschrank
Optische Beurteilung
0 (Rohmaterial) 0,043 frei fließend, staubend 0,035 0,104
0,060 0,122
0,113 0,162
keine Agglomeration, frei fließend, staubend
1
1
0 F0 F1

Ergebnisse und Diskussion 134
Beladung Fges laut Massenbilanz
Beladung Fges laut Trockenschrank
Optische Beurteilung
0,178 0,224
0,231 0,264
0,277 0,316
0,338 0,384
0,363 0,411
0,431 0,456
keine Agglomeration, frei fließend
0,482 0,511
0,539 0,564
0,578 0,604
einzelne, lockere Agglomerate
0,645 0,665 einzelne, große Agglomerate
0,692 0,708 feuchte Agglomerate, Beladung am Maximum
0,743 0,760 zu hoch beladen, feucht
0,791 0,797 kein Pulver mehr, Suspension
Rohstoff Fges = 0 Fges = 0,34 Fges = 0,69
Fges = 0,74 Fges = 0,79
Agglomerate
Abbildung 128: Sipernat 2200 mit verschiedenen Feuchten Fges
Einige Produkte sind in Abbildung 128 abgebildet. Der Rohstoff und das Produkt mit einer Feuchte von Fges = 0,34 sind feinkörnige Pulver ohne Agglomerate. Bei einer

Ergebnisse und Diskussion 135
Feuchte Fges = 0,69 sind dagegen die ersten Agglomerate deutlich sichtbar. Das Produkt mit Fges = 0,74 ist kein fließfähiges Pulver mehr und eine Beladung mit Fges = 0,79 ergibt eine Suspension. Mittels Filtermethode wurden die Produkte hinsichtlich ihrer maximalen Aufnahme-kapazität untersucht. Der Kurvenverlauf ist in Abbildung 129 dargestellt. Es ist deutlich zu sehen, dass sich bei Beladungen bis 70 % die auf den Filter übergegangene Flüssigkeit wenig ändert und, dass zwischen 70 % und 75 % ein signifikanter Anstieg zu verzeichnen ist. Das Produkt mit 79 % Wasser (entspricht F = 0,79) war kein feuchtes Schüttgut, sondern eine Suspension, so dass eine Anwendung der Filtermethode weder sinnvoll noch möglich war.
Abbildung 129: Ergebnisse der Filtermethode für destilliertes Wasser auf Sipernat 2200
4.5.3 Ergebnisse der NMR-Messungen
Als Voraussetzung für die Diskussion der Messergebnisse gilt, dass Wasser-moleküle, die fest an Feststoffpartikeln gebunden sind, kaum beweglich sind, d. h. immobil vorliegen und kleine T2 Relaxationszeiten aufweisen. Die T2 Zeiten werden um so größer, je freier sich die Moleküle bewegen können. Mobiles Wasser zeigt die größten Relaxationszeiten. Dazwischen können sich beliebig viele Abstufungen befinden. Mittels NMR-Spektroskopie sind die in Abbildung 130 aufgetragenen T2 Zeiten in Abhängigkeit von der Gesamtfeuchte gemessen worden. Dabei wurde auch eine Einfachmessung mit reinem Wasser durchgeführt. Das Ergebnis ohne Konfidenzintervall stellt den Messpunkt bei einer Gesamtfeuchte Fges = 1 dar. In Abbildung 130 ist zunächst auffällig, dass bei einer Gesamtfeuchte Fges = 0,8 eine starke Veränderung der Messergebnisse auftritt. Wie auch schon mittels optischer Beurteilung und Filtermethode festgestellt wurde, findet hier der Umschlag von einem beladenen Pulver zur Suspension statt. Vor allem bei den beiden immobileren Anteilen schwankt die T2 Zeit plötzlich in einem großem Intervall und steigt beim weniger immobilen Anteil signifikant an, im Vergleich zu den Werten bei geringeren Gesamtfeuchten Fges. Der vierte, mobile Anteil tritt bei dieser Feuchte zum ersten Mal
0
100
200
300
400
0 20 40 60 80 100
Beladung in %
Flüs
sigk
eit a
uf d
em F
ilter
in
mg

Ergebnisse und Diskussion 136
auf, was dadurch zu erklären ist, dass das Produkt jetzt als Suspension vorliegt, und nun auch freies mobiles Wasser vorhanden ist.
0
0,02
0,04
0,06
0,08
0 0,2 0,4 0,6 0,8 1Fges in -
T 2,1
in m
s
Immobiler Anteil
0,0
2,0
4,0
6,0
8,0
0 0,2 0,4 0,6 0,8 1Fges in -
T 2,2
in m
s
Weniger immobilerAnteil
0
20
40
60
0 0,2 0,4 0,6 0,8 1Fges in -
T 2,3
in m
s
Moderat mobiler Anteil
0200400600800
100012001400160018002000
0 0,2 0,4 0,6 0,8 1Fges in -
T 2,4
in m
sMobiler Anteil
Abbildung 130: Ergebnisse der T2 Zeit Messungen der vier unterscheidbaren Fraktionen: T2,1 und d1: Immobil, T2,2 und d2: weniger immobil, T2,3 und d3: moderat mobil, T2,4 und d4: mobil
Deutlich zu erkennen sind auch die vier unterschiedlichen Größenordnungen der Relaxationszeiten T2 der vier Phasen (immobil, weniger immobil und moderat mobil und mobil) die in Tabelle 38 für die Feuchten von 0,43 bis 0,76 noch einmal zusammengestellt sind. Bei der immobilen und der weniger immobilen Phase bleiben die T2 Zeiten bis zu der kritischen Feuchte von 0,76 annähernd konstant und verändern sich erst ab einer Feuchte von 0,8. Daraus folgt, dass es sich hier um Wasser handelt, dass an der Oberfläche der Partikeln gebunden ist. Das gebundene Wasser kann man sich hier als Schichtenmodell vorstellen. Unabhängig davon, welcher Teil der Oberfläche belegt ist, das Wasser ist immer gleich stark gebunden. Die T2 Zeiten der moderat mobilen Phase dagegen treten zum ersten Mal bei einer Feuchte von 0,12 auf, und steigen dann kontinuierlich an. Dies bedeutet, dass diese Phase um so mobiler wird, je höher die Beladung wird, weil die moderat mobile Phase von Wasser herrührt, das in den Poren gebunden ist. Je mehr von diesem Wasser vorliegt, desto größere Poren werden gefüllt. Zuletzt tritt noch mobiles Wasser auf, dass sich zwischen den Partikeln befindet.

Ergebnisse und Diskussion 137
Tabelle 38: T2 Zeiten der verschieden fest gebundenen Phasen
Phase Minimale T2-Zeit in ms
Maximale T2-Zeit in ms
Mittlere T2-Zeit in ms Kurvenverlauf
Immobil 0,004 0,009 0,006 ± 0,001 Keine Steigung Weniger immobil 0,026 0,271 0,125 ± 0,040 Keine Steigung Moderat mobil 0,938 33,988 9,050 ± 4,799 Steigung
Die Ergebnisse der ersten Messreihe sind in Abbildung 131 noch einmal logarithmisch und ohne Konfidenzintervalle in Abhängigkeit der Beladung in % aufgetragen. Alle dazugehörigen Zahlenwerte sind im Anhang aufgelistet.
Abbildung 131: Logarithmische Auftragung der T2-Relaxationszeiten: T2,1 und d1: Immobil, T2,2 und d2: weniger immobil, T2,3 und d3: moderat mobil, T2,4 und d4: mobil
Als nächstes wurden aus den T2-Zeiten die Anteile der Phasen berechnet (vgl. Kapitel 2.4.4.3). Die so berechneten Anteile sind in den Kurven in Abbildung 132 dargestellt.
1E-03
1E-02
1E-01
1E+00
1E+01
1E+02
1E+03
1E+04
0 20 40 60 80 100
Beladung in %
T 2-R
elax
atio
nsze
it in
ms
immobilweniger immobilmoderat mobilmobil

Ergebnisse und Diskussion 138
0,0
0,2
0,4
0,6
0,8
1,0
0,0 0,2 0,4 0,6 0,8 1,0Fges in -
Ant
eil a
n F g
es in
- Immobil
0,0
0,2
0,4
0,6
0,8
1,0
0,0 0,2 0,4 0,6 0,8 1,0Fges in -
Ant
eil a
n F g
es in
-
Weniger immobil
0,0
0,2
0,4
0,6
0,8
1,0
0,0 0,2 0,4 0,6 0,8 1,0Fges in -
Ant
eil a
n F g
es in
-
Moderat mobil0,0
0,2
0,4
0,6
0,8
1,0
0,0 0,2 0,4 0,6 0,8 1,0Fges in -
Ant
eil a
n F g
es in
- Mobil
Abbildung 132: Anteile a der verschiedenen Phasen aus T2-Zeiten berechnet: T2,1 und d1: Immobil, T2,2 und d2: weniger immobil, T2,3 und d3: moderat mobil, T2,4 und d4: mobil
Es ist in den Diagrammen der T2 Zeiten zu sehen, dass bei den ersten beiden Proben, die den geringsten Wasseranteil aufweisen, nur zwei verschieden fest gebundenen Wasserfraktionen unterschieden werden können. Ab einer Beladung von 12 % kann in der Probe ein zusätzlicher, etwas mobilerer Anteil nachgewiesen werden. Erst bei einer Beladung von 80 % kommt eine vierte Wasserfraktion hinzu, der sogenannte mobile Anteil. In Abbildung 132 kann man erkennen, dass der immobile Anteil, wie in dem Modell in Kapitel 4.5.1 beschrieben, mit zunehmender Gesamtfeuchte abnimmt. Allerdings sind in dieser Messreihe die Konfidenzintervalle zum Teil sehr hoch, so dass der genaue Kurvenverlauf nicht ersichtlich wird. Auch der weniger immobile Anteil nimmt mit zunehmender Gesamtfeuchte ab. Hier kann auf Grund der großen Konfidenz-intervalle der exakte Kurvenverlauf ebenfalls nicht gezeigt werden. Der moderat mobile Anteil steigt zunächst an bis eine Gesamtfeuchte von 0,76 erreicht ist, dann taucht der vierte, mobile Anteil auf, und der moderat mobile Anteil fällt signifikant ab. Um die Messwerte mit dem in Kapitel 4.5.1 vorgestelltem Modell zu vergleichen, werden für die nächsten Überlegungen die immobile und die weniger immobile Phase zu einer neuen immobilen Phase zusammengefasst. Außerdem wurden die Messwerte bei F = 0,316 und F = 0,456 für den Vergleich verworfen, da diese Werte nicht dem erwarteten Kurvenverlauf entsprechen. Es besteht die Annahme, dass es sich bei diesen beiden Messwerten um Mess- oder Auswertefehler bei den NMR-Experimenten handelt. Um hier sicher festzustellen, ob das tatsächlich der Fall war, müsste die Messreihe wiederholt werden, um einen Irrtum auszuschließen.

Ergebnisse und Diskussion 139
Das Ergebnis zeigen die in Abbildung 133 dargestellten Kurvenverläufe: Im Diagramm verlaufen die aus den T2 Zeiten ermittelten Anteile entlang des berechneten Modells und es ist eine qualitativ gute Übereinstimmung zu erkennen.
0 0.2 0.4 0.6 0.8 10
0.2
0.4
0.6
0.8
1
Anteil an immobilem WasserAnteil an moderat mobilem WasserAnteil an mobilem WasserAnteil an immobilem Wasser / NMR ErgebnisseAnteil an moderat mobilem Wasser / NMR ErgebnisseAnteil an mobilem Wasser / NMR Ergebnisse
Gesamtfeuchte in -
Ant
eil a
in -
Abbildung 133: Messwerte und Modellvorstellung für drei Anteile mit F0 = 0,12 und F1 = 0,8
Die Probe mit einer Gesamtfeuchte Fges = 1 besteht aus reinem Wasser, dennoch kann ein kleiner Anteil festgestellt werden, der etwas immobiler ist. Eine mögliche Erklärung dafür ist, dass ein Teil der Wassermoleküle durch die Glaswand des Probengefäßes beeinflusst wird und daher etwas immobiler erscheint, als das restliche Wasser. In diesem Zusammenhang stellt sich auch die Frage, welche Schichtdicke die Wasserschichten aufweisen, die auf den Kieselsäurepartikeln gebunden sind. Für diese Überlegung werden die beiden immobilen Schichten zu einer neuen immobilen Schicht zusammengefasst. Für den unbeladenen Rohstoff Sipernat 2200, der laut Trockenschrankmethode (105 °C für 14 Stunden) eine Feuchte von 0,043 aufweist, ergibt sich dann folgender Zusammenhang: 1 g Produkt besteht aus 0,957 g Feststoff. Diese Menge Feststoff verfügt über eine Fläche von 177 m² (die mittels BET-Stickstoffadsorption gemessenen spezifischen Oberfläche beträgt 185 m²/g). Auf dieser Fläche sind 0,043 g Wasser immobil gebunden, was bei einer Dichte von

Ergebnisse und Diskussion 140
998000 g/m³ einem Volumen von 0,043⋅10-6 m³ entspricht. Daraus ergibt sich eine Schichtdicke der Wassermoleküle von 2,43⋅10-10 m. Ein Wassermolekül hat, wie in Abbildung 134 zu sehen, einen definierten Platzbedarf. Nimmt man als „Schichtdicke“ eines Wassermoleküls dH2O = 0,152 nm, kann die Anzahl der Wassermoleküle in der belegten Schicht abgeschätzt werden:
1,5m100,152m102,43
dsz 9
10
H2O
=⋅
⋅== −
−
(Gleichung 52)
Abbildung 134: Wassermolekül [8]
Bei diesem Ergebnis muss allerdings beachtet werden, dass die mittels Stickstoff-adsorption gemessene Fläche nicht unbedingt komplett für die Wassermoleküle zugänglich sein muss. Bei diesen Partikeln handelt es außerdem sich um hochporöse Substanzen und möglicherweise ist ein Teil des Wassers auch in kleinen Kapillaren sehr fest gebunden. Dennoch ist die Größenordnung von ein bis zwei Wassermolekülen in der immobilen Schicht durchaus realistisch. Auch für alle anderen Feuchten ist die Schichtdicke berechnet worden. Die Ergebnisse sind in Tabelle 39 zusammengestellt. Tabelle 39: Schichtdicken und Anzahl der Wassermoleküle der immobilen Phase
F Fläche in m²/g
Volumen H2O in m³/g
s immobil in m
Anzahl H2O Moleküle in der Schicht
0,043 177,138 4,2585 ⋅10-8 2,4041⋅10-10 1,6 0,104 165,742 1,0431 ⋅10-7 6,2935 ⋅10-10 4,1 0,122 162,430 1,2224 ⋅10-7 5,4116 ⋅10-10 3,5 0,162 155,086 1,6202 ⋅10-7 6,5481 ⋅10-10 4,3 0,224 143,653 2,2395 ⋅10-7 6,6253 ⋅10-10 4,3 0,264 136,086 2,6493 ⋅10-7 8,4589 ⋅10-10 5,5 0,384 113,997 3,8457 ⋅10-7 8,6973 ⋅10-10 5,7 0,411 108,947 4,1192 ⋅10-7 1,2436 ⋅10-9 8,1 0,511 90,558 5,1152 ⋅10-7 1,6112 ⋅10-9 10,5 0,564 80,586 5,6553 ⋅10-7 1,5653 ⋅10-9 10,2

Ergebnisse und Diskussion 141
F Fläche in m²/g
Volumen H2O in m³/g
s immobil in m
Anzahl H2O Moleküle in der Schicht
0,604 73,316 6,0491 ⋅10-7 9,8254 ⋅10-10 6,4 0,665 61,938 6,6653 ⋅10-7 1,4238 ⋅10-9 9,3 0,708 53,946 7,0982 ⋅10-7 1,1816 ⋅10-9 7,7 0,760 44,474 7,6112 ⋅10-7 2,3618 ⋅10-9 15,4 0,797 37,608 7,9831 ⋅10-7 2,4657 ⋅10-9 16,1
Aus den Werten in Tabelle 39 wird ersichtlich, dass auch die immobile Schicht, die am stärksten an den Partikeln gebunden ist, bei steigender Beladung weiter anwächst. Bei dem Rohstoff mit einer Feuchte F = 0,043 beträgt die Schichtdicke ein bis zwei Wassermoleküle. Bei einer Beladung von knapp 80 % liegen durchschnittlich 16 Wassermoleküle übereinander als Hydrathülle an der Feststoffoberfläche. Aufgrund der Ergebnisse der NMR-Messung der mit Wasser beladenen Kieselsäure-partikeln kann man sagen, dass sehr interessante Ergebnisse und Einblicke in die Verteilung des Wassers im feuchten Haufwerk gewonnen werden konnten. Die NMR-Messung stellt auf diese Weise in Zukunft eine Möglichkeit dar, mit Wasser beladenen Pulver zu untersuchen und zu charakterisieren.

Zusammenfassung 142
5 Zusammenfassung Werden Flüssigkeiten und pulverförmige Substanzen miteinander gemischt bzw. Flüssigkeiten auf disperse Feststoffe aufgebracht, wie es beispielsweise in der Lebensmittelindustrie häufig der Fall ist, entstehen Produkte mit sehr unter-schiedlichen Flüssigkeitsgehalten und Produkteigenschaften. Oft sollen dabei große Mengen Flüssigkeit auf die Trägerstoffe aufgebracht werden, ohne dass das Produkt feuchte Agglomerate aufweist. Um dies zu erreichen, ist die maximal mögliche Beladung der Trägerstoffpartikeln von Bedeutung. Im Rahmen dieser Dissertation wurden Trägerstoffe in zwei verschiedenen Verfahren mit unterschiedlichen Flüssigkeiten beladen. Sowohl die Rohstoffe als auch die beladenen Produkte sind mit verschiedenen Messmethoden untersucht worden. Dabei stellte sich heraus, dass der Trägerstoff, der mit der Flüssigkeit beladen wird, den größten Einfluss auf die maximale Beladbarkeit hat. Weniger bedeutend sind die Eigenschaften der Flüssigkeit oder das angewendete Verfahren. Die Eigenschaften der Flüssigkeit spielen nur dann eine Rolle, wenn zwischen dem Trägerstoff und der Flüssigkeit Reaktionen ablaufen und dadurch die Struktur des Produktes beeinflusst wird. Anhand eines Beispieles konnte gezeigt werden, dass bei einer nicht benetzenden Flüssigkeit in Verbindung mit sehr kleinen Trägerstoff-partikeln, ein Produkt gebildet wird, das aus Flüssigkeitströpfchen, die von Trägerstoffpartikeln umgeben sind, besteht. Tritt zwischen den Partikeln und der Flüssigkeit eine Vernetzung auf, kann in den Zwischenräumen des Netzwerkes eine wesentlich größere Menge Flüssigkeit aufgenommen werden, als in den Poren und Zwischenräumen der Partikeln ohne Vernetzung. Wird eine niedrig viskose Flüssigkeit verwendet, die sich leicht zur Düse fördern und durch sie verdüsen lässt, können Trägerstoffe auf herkömmliche Art und Weise in einem Pflugscharmischer mittels Ein- oder Zweistoffdüse genauso hoch und homogen mit dieser Flüssigkeit beladen werden wie mit dem neueren CPF-Verfahren (Concentrated Powder Form). Bei diesem Verfahren wird die Flüssigkeit mit überkritischem CO2 gemischt und dann durch die Entspannung auf Raumtemperatur und Normaldruck fein zerstäubt. Die Anwendung dieses Verfahrens hat den Vorteil, dass bei hochviskosen Flüssigkeiten, wie zum Beispiel Gewürzextrakten, durch die Mischung mit überkritischem CO2 die Viskosität herabgesetzt wird, so dass sich diese Stoffe fördern, verdüsen und auf Trägerstoffe aufbringen lassen. Ein weiterer Vorteil sind, dass die Temperaturen, die sich durch die Entspannung des überkritischen Gases im Sprühturm einstellen, sehr niedrig sind, so dass das Verfahren auch für hitzeempfindliche Stoffe geeignet ist. Bemerkenswert ist weiterhin, dass alle mit dem CPF-Verfahren hergestellten Produkte, ob mit geringer oder sehr hoher Beladung, eine sehr hohe Homogenität aufweisen. Die genauen Vorgänge bei der Zustandsänderung im CPF-Sprühturm, um dieses Phänomen zu erklären, sind wissenschaftlich noch nicht eindeutig beschrieben.

Zusammenfassung 143
Von den Trägerstoffen hängt es ab, wie viel Flüssigkeit aufgebracht werden kann. Liegen poröse Partikeln vor, können diese mit deutlich mehr Flüssigkeit beladen werden, als nicht poröse Partikeln. Lagern sich viele kleine Partikeln zu stabilen Agglomeraten zusammen, ist die Flüssigkeit auch zwischen den Partikeln gebunden. Die Stampfdichte der Schüttung, die als Trägerstoffe verwendet werden soll, kann als Maß für die maximale Beladbarkeit dienen. Je geringer die Stampfdichte der Schüttung, desto höher die maximal mögliche Beladung. Dies gilt, weil sich bei der Messung der Stampfdichte immer die gleiche Schüttungsporosität einstellt und die Stampfdichte deswegen bei gleichbleibender Feststoffdichte eine Funktion der Partikel- und der Agglomeratporosität ist. Die Poren in den Partikeln und in den Agglomeraten werden beim Beladen mit Flüssigkeit gefüllt. Zur Untersuchung der beladenen Produkte wurde neben einer optischen Beurteilung ein dafür entwickelter Schnelltest mittels Filter herangezogen und die Flüssigkeits-verteilung und Flüssigkeitsbindung in einem dispersen System mittels NMR- Messungen untersucht. Der Schnelltest misst die Flüssigkeitsmenge, die vom Produkt unter einer bestimmten Belastung an den Filter abgegeben wird. Diese bleibt zunächst mit steigender Beladung nahezu konstant und steigt ab einer bestimmten Beladung, der sogenannten maximalen Beladung, sprunghaft an. Dieser Test kann für alle Produkte angewendet werden, ist schnell und einfach auszuführen und liefert zuverlässige Ergebnisse über die maximal mögliche Beladung verschiedener Pulver-Flüssigkeitssysteme. Die NMR Messung ist wesentlich aufwendiger und wurde bereits erfolgreich für die Untersuchung einer Kieselsäure, die mit unterschiedlichen Wassermengen beladen war, angewendet. Durch die Bestimmung der T2 Relaxationszeiten konnten verschiedenen Phasen des Wassers im System unter-schieden werden, die unterschiedlich stark an die Partikeln gebunden sind. Die erzielten Ergebnisse stimmten sowohl mit der optischen Beurteilung als auch mit den Ergebnissen der Filtermethode überein. Zusätzlich konnte eine qualitativ gute Übereinstimmung mit einem Modell erzielt werden, das sich zum Berechnen der unterschiedlichen Anteile heranziehen lässt.

Literaturverzeichnis 144
6 Literaturverzeichnis [1] BAUER, K. H.; FRÖMMING; K.-H., FÜLLER, C.: Pharmazeutische
Technologie. 5. Aufl. Stuttgart: Gustav Fischer, 1997
[2] BELITZ, H.-D.; GROSCH, W.: Lehrbuch der Lebensmittelchemie. 4. Aufl. Berlin: Springer, 1992
[3] BORHO, K.; POLKE, R.; WINTERMANTEL, K.; SCHUBERT, H.; SOMMER, K.: Produkteigenschaften und Verfahrenstechnik. Chem.-Ing.-Tech. 63 (1991), Nr. 8, S. 792-808
[4] BRANDAU, TH.: Herstellung von Mikrokugeln in einer Vertropfungsanlage. Fraunhofer UMSICHT-Tage, Beitrag der BRACE GmbH, 2000
[5] BUCHHOLZ, F. L.; GRAHAM, A. T.: Modern Superabsorbent Polymer Technology. New York: Wiley-VCH, 1998, S. 6
[6] BUGGISCH, H.; GÖTZ, J.: Scherinduzierte Strukturänderungen in Pasten verbunden mit der Relativbewegung des Fluids. Abschlußbericht über das DFG-Vorhaben BU 485/11, 1993
[7] CANET, D.: NMR – Konzepte und Methoden. Berlin: Springer, 1994
[8] CD RÖMPP; 9. Aufl. des Römpp Chemie Lexikons auf CD-ROM, Version 1.0, Herausgeber: Jürgen Falbe und Manfred Regnitz, Stuttgart. New York: Thieme, 1995
[9] DÖRFLER, H.: Grenzflächengleichgewichte – Grundlagen zur Messung der Ober- und Grenzflächenspannungen von Flüssigkeiten. In Dörfler, H. (Hrsg.): Grenzflächen- und Kolloidchemie. Weinheim: Wiley-VCH, 1994
[10] EVERETT, D. H.: Manual of symbols and terminology for physicochemical quantities and units. Appendix 2, Definitions Terminology Symbols in Colloid and Surface Chemistry, Part 1. London: Butterworths, 1971
[11] FENG; C. L.; YU, A. B.: Effect of liquid addition on the packing of mono-sized coarse spheres. Powder Technol. 99 (1998), Nr. 1, S. 22-28.
[12] FRIEBOLIN, H.: Ein- und zweidimensionale NMR-Spektroskopie, Eine Einführung. 2. Aufl., Weinheim: VCH, 1992
[13] GAL, S.: Die Methodik der Wasserdampf-Sorptionsmessungen. Berlin: Springer, 1967
[14] GREGG, S. J.; SING, K. S. W.: Adsorption, Surface Area and Porosity. London: Academic Press, 1982
[15] GRÜNER, S.: Entwicklung eines Hochdrucksprühverfahrens zur Herstellung hochkonzentrierter flüssigkeitsbeladener Pulver. Erlangen, Friedrich-Alexander-Universität, Diss., 1999

Literaturverzeichnis 145
[16] HAMMER, A.; HAMMER, K.: Physikalische Formeln und Tabellen. 4. Aufl. München: Lindauer, 1985
[17] HARZ, H.-P.; WEISSER, H.: Einsatz von Kernresonanz-Spektrometern in der Lebensmittelindustrie, Vortrag, gehalten anlässlich des INATEC-Congresses in Köln, 1986. ZFL 37 (1986), Nr. 4, S. 278-281
[18] HEINZEN, CH.; WIDMER, F.: Herstellung von monodispersen Mikrokugeln mit pulsiertem Hüllstrahlprillen. Chem.-Ing.-Tech. 69 (1997), Nr. 5, S. 667-670
[19] HINRICHSEN, O.; GENGER, T.; MUHLER, M.: Die Chemiesorption von N2O und H2 zur Oberflächenbestimmung von Kupfer-Katalysatoren. Chem.-Ing.-Tech. 72 (2000), Nr. 1+2, S. 94-98
[20] HOFFMANNS, W.; KOSOCK, ST.; MOSER, F.: Kugeln in Top-Form. gas aktuell 57, Info-Prospekt der Messer Griesheim GmbH, 2001
[21] IVESON, S. M.; LITSTER, J. D.; HAPGOOD, K.; ENNIS, B. J.: Nucleation, growth and breakage phenomena in agitated wet granulation processes: a review. Powder Technol.117 (2001), Nr. 1, S. 3-39.
[22] KESSLER, H. G.: Lebensmittel- und Bioverfahrenstechnik – Molkerei-technologie. 4. Aufl. Freising: A. Kessler, 1996
[23] KIND, M.: Kristallieren und Fällen. Vortrag auf dem Hochschulkurs „Pulverförmige Formulierungen“. Technische Hochschule Karlsruhe, 2001
[24] KLEIN, J.; VORLOP, K.-D.: Immobilization techniques: cells, in Comprehensive Biotechnology, Vol. 2 (Hrsg.: Moo-Young, M.), Oxford: Pergamon Press, 1985, S. 203-224
[25] KOGLIN, B.; LESCHONSKI, K.; ALEX, W.: Teilchengrößenanalyse. Chem.-Ing.-Tech. 46 (1974), Nr. 23, S. 984-987
[26] KRISTENSEN, H. G.: Particle agglomeration in high shear mixers. Powder Technol. 88 (1996), S. 197-202
[27] LESCHONSKI, K.: Grundlagen und moderne Verfahren der Partikelmesstechnik. 21. Clausthaler Kursus; 18. bis 22. Oktober 1999
[28] LIAN, G.; THORTON, C.; ADAMS, M. J.: A theoretical study of the liquid bridge forces between two rigid spherical bodies. J. Colloid Interface Sci.161, (1993), Nr. 1, S. 138-147
[29] LIEBENSPACHER, F.: Untersuchungen über Wasserdampfsorption und Wasserdampftransport in Packstoffen auf Zellulosebasis. München, Technische Universität, Lehrstuhl für Brauereianlagen und Lebensmittel-Verpackungstechnik, Diss., 1991
[30] MATISSEK, R.; SCHNEPEL, F.-M.; STEINER, G.: Lebensmittelanalytik, Grundzüge, Methoden, Anwendungen. Berlin: Springer, 1992, S. 32-35

Literaturverzeichnis 146
[31] MOFIDI, N.; AGHAI-MOGHADAM, M.; SARBOLOUKI, M. N.: Mass preparation and charakterization of alginat microspheres. Process Biochemistry 35 (2000), Nr. 9, S. 885-888
[32] MOLERUS, O.; WIRTH, K.-E.: 2. Grundlagen der Mechanischen Verfahrenstechnik, Vorlesungsskript der Universität Erlangen, Lehrstuhl für Mechanische Verfahrenstechnik, 1992
[33] NEESSE, T.; FAHLAND, M.: Model Calculation for Porosity, Pore Size Distribution and Flow Resistance in Filter Cakes. Aufbereitungs-Techn. 35 (1994), Nr. 3, S. 117-124
[34] NEWITT, D. M.; CONWAY-JONES, J. M.: A contribution to the theory and practice of granulation. Trans. Instn Chem. Engrs. 36 (1958), S. 422-440
[35] PALZER, S.: Anreichern und Benetzen von pulverförmigen Lebensmitteln mit Flüssigkeiten in diskontinuierlichen Mischaggregaten. München, Technische Universität, Lehrstuhl für Maschinen- und Apparatekunde, Diss., 2000
[36] PEUKERT, W.; VOGEL, L.: Zerkleinern. Vortrag auf dem Hochschulkurs „Pulverförmige Formulierungen“. Technische Hochschule Karlsruhe, 2001
[37] PIETSCH, W.; RUMPF, H.: Haftkraft, Kapillardruck, Flüssigkeitsvolumen und Grenzwinkel einer Flüssigkeitsbrücke zwischen zwei Kugeln, Chem.-Ing.-Tech. 39 (1967), Nr. 15, S. 885-893
[38] POLKE, R.; HERRMANN, W.; SOMMER, K.: Charakterisierung von Agglomeraten. Chem.-Ing.-Tech. 51 (1979), Nr. 4, S. 283-288
[39] PRÜßE, U., DALLUHN, J., BREFORD, J., VORLOP, K.-D.: Herstellung sphärischer Partikel mit dem Strahlschneider-Verfahren. Chem.-Ing.-Tech. 72 (2000), S. 852-858.
[40] PRÜßE, U.: Entwicklung, Charakterisierung und Einsatz von Edelmetall-katalysatoren zur Nitratreduktion mit Wasserstoff und Ameisensäure sowie des Strahlschneider-Verfahrens zur Herstellung Polyvinylalkohol-verkapselter Katalysatoren. Wissenschaftliche Mitteilungen der Bundesforschungsanstalt für Landwirtschaft (FAL), Braunschweig, 2000, S. 30-31
[41] PRÜßE, U.; BRUSKE, F.; BREFORD, J.; VORLOP, K.-D.: Verbesserung des Strahlschneiderverfahrens zur Herstellung kugelförmiger Partikel aus viskosen Polymerlösungen. Chem.-Ing.-Tech. 70 (1998), Nr. 5, S. 556-560
[42] RUMPF, H.: Grundlagen und Methoden des Granulierens, Chem.-Ing.-Techn. 30 (1958), Nr. 3, S. 144-158
[43] RUMPF, H.: Mechanische Verfahrenstechnik. München: Carl Hanser, 1975
[44] SARTOR, K.-H.; SCHUBERT, H.: Agglomerieren fester Stoffe: Grundlagen und Verfahren. MM Würzburg, 84 (1978), Nr. 70, S. 1365-1369

Literaturverzeichnis 147
[45] SCHABER, K.; TÜRK, M.: Erzeugen von Nanopartikeln mit überkritischen Fluiden. Vortrag auf dem Hochschulkurs „Pulverförmige Formulierungen“. Technische Hochschule Karlsruhe, 2001
[46] SCHÆFER, T.; MATHIESEN, C.: Melt pelletization in a high shear mixer: IX. Effects of binder particle size. Int. J. Pharm. 139 (1996) Nr. 1+2, S. 139-148
[47] SCHLEGEL, H.G.: Allgemeine Mikrobiologie, 7. Aufl., Stuttgart: Georg Thieme, 1992, S. 196
[48] SCHUBERT, H.: Instantisieren pulverförmiger Lebensmittel. Chem.-Ing.-Tech. 62 (1990), Nr. 11, S. 892-906
[49] SCHUBERT, H.: Kapillardruck und Zugfestigkeit von feuchten Haufwerken aus körnigen Stoffen. Chem.-Ing.-Tech. 45 (1973), Nr. 6, S. 396-401
[50] SOMMER, K.: 40 Jahre Darstellung von Partikelgrößenverteilungen – und immer noch falsch? Chem.-Ing.-Tech. 72 (2000), Nr. 8, S. 809-812
[51] STAHL, E.; QUIRIN, K. W., GERAD, D.: Verdichtete Gase zur Extraktion und Raffination, Berlin: Springer, 1987
[52] STIESS, M.: Mechanische Verfahrenstechnik 1. Berlin: Springer, 1992
[53] TOM, J. W.; DEBENEDETTI, P. G.: Particle formation with supercritical fluids – a review. Journal Aerosol Science 22 (1991) Nr. 5, S. 555-584
[54] TSCHERNJAEW, J., BERGER, T., WEBER, A., KÜMMEL, R.: Herstellung von Mikropartikeln durch Kristallisation mittels hochkomprimierter Gase. Chem.-Ing.-Tech. 69 (1997), Nr. 5, S. 670-674
[55] WEIGERT, T.; RIPPERGER S.: Calculation of the Liquid Bridge Volume and Bulk Saturation from the Half-filling Angle. Part. Part. Syst. Charact. 16 (1999), S. 238-242
[56] WEINEKÖTTER, R.; GERICKE, H.: Mischen von Feststoffen, Prinzipien, Verfahren, Mischer. Berlin: Springer, 1995
[57] WEINREICH, B.; WEIDNER, E.; DIRSCHERL, J.; STEINER, R.: Method for producing a powder product from a liquid substance or mixture of substances, European Patent EP 1021241 A, 2000
[58] WEISSER, H.: Anwendungen der Kernresonanz-Spektroskopie in der Lebensmittelverfahrenstechnik. ZFL 28 (1977), Nr. 3, S. 97-101
[59] WEISSER, H.: Untersuchungen zum gebundenen Wasser mittels Kernresonanz-Spektroskopie. ZFL 31 (1980), Nr. 3, S. 143-147

Anhang 148
7 Anhang
7.1 Berechnung einer Flüssigkeitsbrücke Gegebene Werte:
δ 43.5Grad:= a 3.24 10 6−⋅ m⋅:= γ 0.032Nm 1−:=
β 17Grad:= dp 0.4mm:= ρs 1.02kg l 1−:=
Berechnung der Radien R1 und R2:
R1dp 1 cos β( )−( )⋅ a+
2 cos β δ+( )⋅:= R1 2.104 10 5−× m=
R2 dpsin β( )
2⋅
R1 sin β δ+( ) 1−⋅( )⋅+:= R2 4.016 10 5−× m=
Berechnung der Haftkraft F zwischen Partikeln nach Pietsch und Rumpf:
pk γ1
R11
R2+
⋅:=
F1
π dp2( )⋅ sin β( )( )2⋅ pk⋅4
:=
F2 γ π⋅ dp⋅ sin β( )⋅ sin β δ+( )⋅:=
F1 2.49 10 5−× kg ms -2=
F2 1.023 10 5−× kg ms-2=
F F1 F2+:=
F 3.513 10 5−× kg ms -2=

Anhang 149
Volumen der Flüssigkeitsbrücke nach Pietsch und Rumpf:
b R1 R2+:=
b 6.12 10 5−× m=
T1 R12 b2+( ) R1⋅ cos β δ+( )⋅R13 cos β δ+( )( )3⋅
3−:=
T2 b R12 cos β δ+( )⋅ sin β δ+( )⋅ R12 π2
β− δ−
⋅+
⋅:=
T31 dp3⋅ 2 cos β( )+( )⋅ 1 cos β( )−( )2⋅
24:=
Vbr 2 π⋅ T1 T2− T3−( ):=
Vbr 1.514 10 14−× m3=
Volumen der Flüssigkeitsbrücke nach Weigert und Rippberger:
Ca 1 6adp
⋅+
:= Ca 1.049=
Cb 1 1.1 sin δ( )⋅+:= Cb 1.757=
Vbr 0.12 dp3⋅ sin β( )( )4⋅ Ca⋅ Cb⋅:= Vbr 1.034 10 13−× m3=
Gewichtskraft Partikeln Fg:
Fgπ ρs⋅ g⋅ dp3⋅
6:=
Fg 3.352 10 7−× kg ms-2=
7.2 Wassergehalt der einzelnen Stichproben aus der Mischertrommel
Tabelle 40: Wassergehalte der Proben aus dem Mischer aus Zellulose und Wasser
Probe Wassergehalt in % nach Trockenschrankmethode (Wassergehalt-Mittelwert)²
1 38,71 0,076 2 38,80 0,139 3 38,73 0,091

Anhang 150
Probe Wassergehalt in % nach Trockenschrankmethode (Wassergehalt-Mittelwert)²
4 37,68 0,567 5 38,84 0,168 6 39,05 0,385 7 37,83 0,354 8 39,03 0,361 9 38,05 0,143 10 38,11 0,105 11 38,41 0,001 12 38,90 0,216 13 38,06 0,138 14 38,72 0,086 15 38,49 0,004 16 39,02 0,343 17 38,29 0,021 18 38,97 0,291 19 38,03 0,158 20 38,66 0,053 21 38,20 0,055 22 38,81 0,147 23 40,03 (Maximum) 2,547 24 38,87 0,191 25 38,51 0,006 26 37,63 0,643 27 37,86 0,322 28 37,77 0,429 29 37,63 0,638 30 37,22 (Minimum) 1,457
Mittelwert 38,43

Anhang 151
7.3 Berechnung der Kurvenverläufe der unterschiedlich fest gebundenen Wasseranteile
7.3.1 Berechnung des Kurvenverlaufs für zwei unterschiedlich fest gebundene Anteile
Die Sättigungsfeuchte ist F0 0.1:=
Die Gesamtfeuchte Fges 0 0.01, 1..:=
aim Fges( ) 1 0 Fges< F0≤if
F0 1 Fges−( )⋅
Fges 1 F0−( )⋅
F0 Fges< 1≤if
:=
amo Fges( ) 0 0 Fges< F0≤if
1F0 1 Fges−( )⋅
Fges 1 F0−( )⋅−
F0 Fges< 1≤if
:=
7.3.2 Berechnung des Kurvenverlaufs für drei unterschiedlich fest gebundene Anteile
Sättigungsfeuchte F0 0.1:=
Fges 0 0.01, 1..:=
F1 0.3:=
aim Fges( ) 1 0 Fges< F0≤if
F0 1 Fges−( )⋅
Fges 1 F0−( )⋅
F0 Fges< F1≤if
F0 1 Fges−( )⋅
Fges 1 F0−( )⋅
F1 Fges< 1≤if
:=

Anhang 152
amo Fges( ) 0 0 Fges< F0≤if
0 F0 Fges< 1≤if
1F1 1 Fges−( )⋅
Fges 1 F1−( )⋅
− F1 Fges< 1≤if
:=
almo Fges( ) 0 0 Fges< F0≤if
1F0 1 Fges−( )⋅
Fges 1 F0−( )⋅−
F0 Fges< F1≤if
1 aim Fges( )− amo Fges( )− F1 Fges< 1≤if
:=
7.4 Zahlenwerte aller Ergebnisse der NMR-Messungen Die T2-Messungen sind Dreifachbestimmungen und das Ergebnis wird als Mittelwert inklusive einem Konfidenzintervall für 95 %-ige Wahrscheinlichkeit angegeben. Tabelle 41: Ergebnisse der T2-Messungen
Fges c ±±±± e ±±±± g ±±±± i ±±±± 0,0425* 0,0088 0,0040 0,2714 0,0749 - - - - 0,1041 0,0061 0,0039 0,0743 0,1959 - - - - 0,1220 0,0055 0,0030 0,0589 0,1159 0,9380 0,3843 - - 0,1617 0,0051 0,0036 0,0264 0,0106 1,3505 0,1038 - - 0,2235 0,0081 0,0029 0,0768 0,0630 2,0343 0,1434 - - 0,2644 0,0074 0,0047 0,0419 0,0294 2,6936 0,1023 - - 0,3160 0,0054 0,0083 0,0444 0,0456 3,5592 0,7429 - - 0,3838 0,0084 0,0053 0,1555 0,0378 5,3772 0,2098 - - 0,4111 0,0058 0,0045 0,0640 0,0569 5,9023 0,1087 - - 0,4560 0,0040 0,0037 0,1873 0,0724 6,8775 0,2744 - - 0,5105 0,0057 0,0056 0,1936 0,0955 8,3792 0,2592 - - 0,5644 0,0055 0,0061 0,2201 0,1434 10,1584 0,0500 - - 0,6037 0,0066 0,0063 0,1560 0,0995 11,9620 0,2097 - - 0,6652 0,0082 0,0019 0,1603 0,0973 15,4074 0,2783 - - 0,7084 0,0087 0,0022 0,1878 0,1241 18,0729 0,6936 - - 0,7596 0,0035 0,0003 0,0777 0,0949 33,9884 8,3730 - - 0,7967 0,0316 0,0395 5,1873 1,6099 37,7016 16,3122 278,9594 107,5040
1,0000** - - 0,1968 - - - 1764,0412 - *Rohstoff, **reines Wasser

Anhang 153
Tabelle 42: Anteile berechnet aus den T2-Messungen
Fges c ±±±± e ±±±± g ±±±± i ±±±± 0,0425* 0,6097 0,1623 0,3903 0,1623 - - - - 0,1041 0,6432 0,2269 0,3568 0,1896 - - - - 0,1220 0,5184 0,3837 0,2006 0,3243 0,2809 0,1442 - - 0,1617 0,3934 0,7966 0,2334 0,3055 0,3732 0,4910 - - 0,2235 0,3132 0,0664 0,1118 0,0778 0,5750 0,0372 - - 0,2644 0,2835 0,1102 0,1510 0,0653 0,5655 0,1756 - - 0,3160 0,5000 0,2954 0,1014 0,1272 0,3986 0,3462 - - 0,3838 0,1841 0,1492 0,0737 0,0258 0,7422 0,1739 - - 0,4111 0,2520 0,1856 0,0769 0,0241 0,6711 0,2089 - - 0,4560 0,4997 0,6779 0,0287 0,0426 0,4385 0,6360 - - 0,5105 0,2473 0,4027 0,0379 0,0252 0,6856 0,3824 - - 0,5644 0,1922 0,2607 0,0309 0,0110 0,7510 0,2563 - - 0,6037 0,1027 0,6124 0,0164 0,0054 0,8566 0,6169 - - 0,6652 0,0957 0,0018 0,0366 0,0145 0,8677 0,0133 - - 0,7084 0,0614 0,0034 0,0284 0,0141 0,9102 0,0131 - - 0,7596 0,1115 0,0775 0,0265 0,0099 0,8620 0,0683 - - 0,7967 0,0277 0,0084 0,0885 0,0648 0,5256 0,0426 0,3582 0,2612
1,0000** - - 0,0245 - - - 0,9755 - *Rohstoff, **reines Wasser

Abbildungsverzeichnis i
Abbi ldungsverzeichnis Abbildung 1: Beanspruchungsarten nach Rumpf [43]............................................................................. 3 Abbildung 2: Prinzip der Beugungsspektrenanalyse............................................................................... 7 Abbildung 3: Summenhäufigkeit Qr(x) ..................................................................................................... 8 Abbildung 4: Porosität eines festen Körpers ........................................................................................... 9 Abbildung 5: Stärkepartikel, links mit Poren, rechts ohne Poren .......................................................... 10 Abbildung 6: Adsorptionsisothermen (Typ I bis V) und eine stufenweise Isotherme (VI) [14] .............. 13 Abbildung 7: Schematische Darstellung verschiedener Dichten eines dispersen Systems ................. 15 Abbildung 8: Prinzipbild eines Gaspyknometers [27] ............................................................................ 17 Abbildung 9: Zwischenmolekulare Kräfte [8] ......................................................................................... 18 Abbildung 10: Phasendiagramm [22] .................................................................................................... 19 Abbildung 11: Schematische Darstellung eines auf einer Feststoffoberfläche liegenden Tropfens
und dem sich einstellenden Kontaktwinkel ..................................................................... 21 Abbildung 12: Beispiele von unterschiedlich benetzenden Flüssigkeiten in zylindrischen
Kapillaren ........................................................................................................................ 22 Abbildung 13: Eindringen von nicht benetzenden Flüssigkeiten in Einzelkapillaren [35]...................... 23 Abbildung 14: Bindungsmechanismen zwischen Partikeln und Flüssigkeit .......................................... 24 Abbildung 15: Flüssigkeitsbrücke zwischen zwei Partikeln [55]............................................................ 25 Abbildung 16: Flüssigkeitsverteilung in einem dispersen System [49].................................................. 27 Abbildung 17: Verschiedene Zustände in Abhängigkeit vom Grad der Sättigung [55] ......................... 28 Abbildung 18: Die Mechanismen der Keimbildung bei der Agglomeration in Abhängigkeit des
Verhältnisses von Tropfengröße zu Einzelpartikel [46] .................................................. 29 Abbildung 19: Bindungsmechanismen für Flüssigkeiten in Pulverschüttungen [35] ............................. 30 Abbildung 20: Granulieren von Kalziumhydrogenphosphat mit wässriger Lösung: �, �:
Rührerdrehzahl 500 U/min, �, �: Rührerdrehzahl 250 U/min, �, �: Flüssigkeitsmassenstrom 100 G/min, �, �: Flüssigkeitsmassenstrom 300 g/min. ...... 31
Abbildung 21: Schematische Darstellung eines Protons als Stabmagnet [17] ..................................... 33 Abbildung 22: (a) Kernmagnetisierung M
�
ist parallel zu 0B�
; (b) M präzediert um 0B�
mit
Larmorfrequenz 0ν ; (c) Wird M�
in eine zu 0B�
senkrecht liegende Ebene
ausgelenkt, wird ein maximales Amplitudensignal gemessen [7]................................... 33 Abbildung 23: Erzeugung der x,y-Magnetisierung durch den Impuls B1 [7] .......................................... 34 Abbildung 24: Signalabschwächung während einer NMR-Messung .................................................... 35 Abbildung 25: Einzelne Prozessschritte beim Anreichern von Pulvern mit Flüssigkeiten..................... 37 Abbildung 26: Feuchtagglomerationsverfahren mit den Teilschritten Befeuchten, Agglomerieren,
Trocknen und evtl. Brechen: (a) Fließbett, (b) Mischer, (c) Teller (Rollagglomeration, (d) Strahl, (e) mittels rotierender Scheibe oder (f) auf einem Band, Trocknen und evtl. Brechen.................................................................................. 38
Abbildung 27: Prinzip des CPF-Sprühturmes: (1) Pulverförmiger Trägerstoff; (2) Flüssigkeit; (3) Überkritisches CO2; (4) Statischer Mischer; (5) Düse; (6) Zyklon; (7) Beladenes Produkt. ........................................................................................................................... 40
Abbildung 28: Maximaler Extraktgehalt in Abhängigkeit von der „Rütteldichte“ der verschiedenen Trägerstoffe [15].............................................................................................................. 42
Abbildung 29: Vorrichtungen zur Bestimmung der Schüttdichte (links) und der Stampfdichte (rechts) ............................................................................................................................ 44
Abbildung 30: Schnelltest zur Bestimmung der "freien" Flüssigkeit“ in der Schüttung ......................... 45 Abbildung 31: Partikelgrößenverteilung von Aero Myl 33 (Laserbeugung)........................................... 48 Abbildung 32: REM Aufnahmen der Aero Myl 33 Partikeln................................................................... 49 Abbildung 33: Partikelgrößenverteilung von Paselli MD6 (Laserbeugung)........................................... 50 Abbildung 34: REM Aufnahme von Paselli MD6 ................................................................................... 50

Abbildungsverzeichnis ii
Abbildung 35: Partikelgrößenverteilung von Pulverzellulose P290 (Laserbeugung) ............................ 51 Abbildung 36: REM Aufnahme von Pulverzellulose P290..................................................................... 52 Abbildung 37: REM Aufnahmen der Cellets .......................................................................................... 53 Abbildung 38: Partikelgrößenverteilungen der Cellets Fraktionen (Laserbeugung).............................. 53 Abbildung 39: Strukturformel von Siliciumdioxid ................................................................................... 54 Abbildung 40: Herstellung von Kieselsäuren [8].................................................................................... 54 Abbildung 41: Partikelgrößenverteilung von Sipernat 50 (Laserbeugung)............................................ 55 Abbildung 42: Partikelgrößenverteilung von Sipernat 50S (Laserbeugung) ......................................... 56 Abbildung 43: REM Aufnahme von Sipernat 50S.................................................................................. 57 Abbildung 44: Partikelgrößenverteilung von Sipernat 2200 .................................................................. 58 Abbildung 45: REM Aufnahme von Sipernat 2200................................................................................ 58 Abbildung 46: Partikelgrößenverteilung von Aerosil 130 (Laserbeugung) ............................................ 59 Abbildung 47: REM Aufnahme von Aerosil 130 .................................................................................... 59 Abbildung 48: Schematische Darstellung der Reaktion von Dimethyldichlorsilan mit der
Kieselsäureoberfläche: Überführung von "hydrophil" in "hydrophob"............................. 60 Abbildung 49: Partikelgrößenverteilung von Aerosil R972 (Laserbeugung) ......................................... 61 Abbildung 50: Partikelgrößenverteilung von Cabosil (Laserbeugung) .................................................. 62 Abbildung 51: Partikelgrößenverteilung von Hy Sorb (Laserbeugung) ................................................. 63 Abbildung 52: Strukturformel von Alginsäure [8] ................................................................................... 65 Abbildung 53: Prinzip des JetCutters .................................................................................................... 66 Abbildung 54: Schneidewerkzeug mit speichenförmig angeordneten Drähten..................................... 67 Abbildung 55: Aufbau der CPF-Sprühanlage ........................................................................................ 69 Abbildung 56: CPF-Sprühturm ohne Autoklav....................................................................................... 71 Abbildung 57: Lödige Pflugscharmischer .............................................................................................. 71 Abbildung 58: Einflussfaktoren auf die maximale Aufnahmekapazität.................................................. 73 Abbildung 59: Kieselsäure Sipernat 2200 mit verschiedenen Mengen Wasser.................................... 75 Abbildung 60: Maltodextrin Paselli MD 6 mit verschiedenen Mengen Triacetin ................................... 75 Abbildung 61: Partikelgrößenverteilungen der Kieselsäure Sipernat 50S mit verschiedenen
Mengen Speiseöl gemessen mittels Laserbeugung (Fallschacht) ................................. 76 Abbildung 62: Partikelgrößenverteilungen der Kartoffelstärke Aero Myl 33 mit verschiedenen
Mengen Speiseöl gemessen mittels Laserbeugung (Fallschacht) ................................. 76 Abbildung 63: Schütt- und Stampfdichten von Sprühprodukten aus Speiseöl und Aero Myl 33 mit
steigenden Speiseölmengen........................................................................................... 77 Abbildung 64: Schütt- und Stampfdichten von Sprühprodukten aus Speiseöl auf Sipernat 50S mit
verschiedenen Mengen Speiseöl .................................................................................... 79 Abbildung 65: Schütt- und Stampfdichten von Sprühprodukten aus Aero Myl 33 ohne den
Flüssigkeitsanteil ............................................................................................................. 79 Abbildung 66: Schütt- und Stampfdichten von Sprühprodukten aus Speiseöl und Sipernat 50S
ohne den Flüssigkeitsanteil............................................................................................. 80 Abbildung 67: Modellvorstellung: Schütt- und Stampfdichten von beladenen Pulvern......................... 80 Abbildung 68: Überkornanteil der Sprühprodukte aus Speiseöl und Aero Myl 33 ................................ 81 Abbildung 69: Überkornanteil der Sprühprodukte aus Speiseöl und Sipernat 50S............................... 82 Abbildung 70: Ergebnisse der Filtermethode für Speiseöl auf Aero Myl 33.......................................... 83 Abbildung 71: Einfluss des Belastungsgewichtes bei einer Minute Messzeit: Speiseöl auf einem
Maltodextrin..................................................................................................................... 83 Abbildung 72: Einfluss der Messzeit bei 500 g Belastung: Speiseöl auf einem Maltodextrin ............... 84 Abbildung 73: Ergebnisse der Filtermethode für Maltodextrin Paselli MD6 mit Speiseöl ..................... 85 Abbildung 74: Ergebnisse der Filtermethode für Pulverzellulose P290 mit Speiseöl............................ 86 Abbildung 75: Ergebnisse der Filtermethode für Sipernat 2200 mit Speiseöl....................................... 86 Abbildung 76: Ergebnisse der Filtermethode für Sipernat 50S mit Speiseöl ........................................ 87 Abbildung 77: Ergebnisse der Filtermethode für Speiseöl auf Aerosil 130 ........................................... 87

Abbildungsverzeichnis iii
Abbildung 78: Aerosil 130 mit steigenden Beladungen Speiseöl .......................................................... 88 Abbildung 79: Ergebnisse der Filtermethode für Speiseöl auf Aerosil R972 ........................................ 88 Abbildung 80: Ergebnisse der Filtermethode für Speiseöl auf der Kieselsäure Cabosil ....................... 89 Abbildung 81: Messkurve der DVS Messung von Aero Myl 33............................................................. 90 Abbildung 82: DVS Sorptionsisotherme von Aero Myl 33 .................................................................... 91 Abbildung 83: Stickstoffsorptionsisothermen von Aero Myl 33 ............................................................. 92 Abbildung 84: Messkurve und Sorptionsisotherme der DVS Messung von Sipernat 50 ...................... 93 Abbildung 85: DVS Sorptionsisotherme von Sipernat 50...................................................................... 93 Abbildung 86: Messkurve der DVS Messung von Cabosil .................................................................... 94 Abbildung 87: DVS Sorptionsisotherme von Cabosil ............................................................................ 95 Abbildung 88: Maximaler Extraktgehalt in Abhängigkeit von der Stampfdichte: Vergleich Grüner
[15] und den Messwerten der vorliegenden Dissertation................................................ 96 Abbildung 89: Kubische Packung gleich großer Kugeln ....................................................................... 98 Abbildung 90: Aufbau einer dichtesten Kugelpackung.......................................................................... 99 Abbildung 91: Partikelgrößenverteilungen (Laserbeugung) der mittels JetCutter hergestellten
Montanwachspartikeln .................................................................................................. 102 Abbildung 92: Partikelgrößenverteilung (Laserbeugung) der Montanwachspartikelfraktionen........... 103 Abbildung 93: Zwei Montanwachspartikeln (Feinfraktion) mit Flüssigkeitsbrücke aus Speiseöl ........ 104 Abbildung 94: Modellvorstellung: Herstellung und Analyse der Monatnwachspartikeln mit
Kochsalz im JetCutter ................................................................................................... 105 Abbildung 95: Eichgerade für die Leitfähigkeitsmessung.................................................................... 106 Abbildung 96: Schematische Darstellung der Bildung von Ca-Alginat-Hydrogelen durch ionotrope
Gelierung von außen nach innen, nach [24] ................................................................. 108 Abbildung 97: Partikeln aus 1,5 % Alginat mit 40 % TiO2 ................................................................... 109 Abbildung 98: Partikelgrößenverteilung der Alginatpartikeln (Laserbeugung) .................................... 110 Abbildung 99: Feine Fraktion der Alginatpartikeln mit Speiseöl .......................................................... 111 Abbildung 100: Ergebnisse der Filtermethode für Speiseöl auf der feinere Fraktion der
Alginatpartikeln.............................................................................................................. 111 Abbildung 101: Ergebnisse der Filtermethode für Speiseöl auf der gröberen Fraktion der
Alginatpartikeln.............................................................................................................. 112 Abbildung 102: Partikelgrößenverteilungen von Kochsalz original und sprühgetrocknet
(Laserbeugung)............................................................................................................. 114 Abbildung 103: REM Aufnahmen von Kochsalz: Links original, rechts sprühgetrocknet.................... 114 Abbildung 104: Abhängigkeit der Porosität vom Durchmesser der Glaskugeln [11]........................... 115 Abbildung 105: Ergebnisse der Filtermethode für Speiseöl auf Kochsalz .......................................... 116 Abbildung 106: Ergebnisse der Filtermethode für Speiseöl auf sprühgetrocknetem Kochsalz .......... 116 Abbildung 107: Ergebnisse der Filtermethode für Aerosil R972 mit Triacetin..................................... 118 Abbildung 108: Wassertropfen auf einer Schüttung aus Aerosil R972 ............................................... 118 Abbildung 109: Ergebnisse der Filtermethode für Wasser auf Aerosil R972 ...................................... 119 Abbildung 110: Aerosil R972 beladen mit steigenden Mengen Wasser ............................................. 119 Abbildung 111: Schematische Darstellung eines Wassertropfens umhüllt von Feststoffpartikeln...... 120 Abbildung 112: Aerosil R972 mit 91% Wasser unter dem Mikroskop................................................. 120 Abbildung 113: Ergebnisse der Filtermethode für HySorb mit Speiseöl ............................................. 121 Abbildung 114: Ergebnisse der Filtermethode für HySorb mit Wasser............................................... 121 Abbildung 115: Schematische Darstellung eines Polymernetzwerkes. Die Punkte stellen die
Quervernetzungen zwischen den spiralförmigen Polymeren dar. [5] ........................... 122 Abbildung 116: HySorb beladen mit steigenden Mengen Wasser ...................................................... 122 Abbildung 117: Ergebnisse der Filtermethode für Paselli MD6 mit Triacetin ...................................... 123 Abbildung 118: Ergebnisse der Filtermethode für Aero Myl 33 mit Speiseöl im Pflugscharmischer
und im CPF-Verfahren .................................................................................................. 124 Abbildung 119: Mischerinnenraum nach der Beladung von Aero Myl 33 mit 65 % Speiseöl ............. 125

Abbildungsverzeichnis iv
Abbildung 120: Ergebnisse der Filtermethode für Paselli MD6 mit Speiseöl im Pflugscharmischer und im CPF-Verfahren .................................................................................................. 125
Abbildung 121: Ergebnisse der Filtermethode für Zellulose mit Speiseöl im Pflugscharmischer und im CPF-Verfahren .................................................................................................. 126
Abbildung 122: Ergebnisse der Filtermethode für Triacetin auf Aero Myl 33 im Pflugscharmischer und im CPF-Verfahren .................................................................................................. 126
Abbildung 123: Sprühkegel der Einstoffdüse (links) im Vergleich zur Zweistoffdüse (rechts) ............ 127 Abbildung 124: Ergebnisse der Filtermethode für Aero Myl 33 mit Speiseöl im Mischer mit
Einstoff- und Zweistoffdüse........................................................................................... 128 Abbildung 125: Ergebnisse der Filtermethode für Aero Myl 33 mit Triacetin im Mischer mit
Einstoff- und Zweistoffdüse........................................................................................... 128 Abbildung 126: Ergebnisse der Berechnung mittels Modellvorstellung von zwei unterschiedlich
fest gebundenen Flüssigkeitsanteilen mit F0 ................................................................ 132 Abbildung 127: Ergebnisse der Berechnung mittels Modelvorstellung der Anteile an immobilem,
weniger mobilem und mobilem Wasser mit F0 und F1 .................................................. 133 Abbildung 128: Sipernat 2200 mit verschiedenen Feuchten Fges........................................................ 134 Abbildung 129: Ergebnisse der Filtermethode für destilliertes Wasser auf Sipernat 2200 ................. 135 Abbildung 130: Ergebnisse der T2 Zeit Messungen der vier unterscheidbaren Fraktionen: T2,1 und
d1: Immobil, T2,2 und d2: weniger immobil, T2,3 und d3: moderat mobil, T2,4 und d4: mobil.............................................................................................................................. 136
Abbildung 131: Logarithmische Auftragung der T2-Relaxationszeiten: T2,1 und d1: Immobil, T2,2 und d2: weniger immobil, T2,3 und d3: moderat mobil, T2,4 und d4: mobil ....................... 137
Abbildung 132: Anteile a der verschiedenen Phasen aus T2-Zeiten berechnet: T2,1 und d1: Immobil, T2,2 und d2: weniger immobil, T2,3 und d3: moderat mobil, T2,4 und d4: mobil.. 138
Abbildung 133: Messwerte und Modellvorstellung für drei Anteile mit F0 = 0,12 und F1 = 0,8 ........... 139 Abbildung 134: Wassermolekül [8] ...................................................................................................... 140

Tabellenverzeichnis v
Tabel lenverzeichnis Tabelle 1: Klassifizierung der Poren anhand der Porenweite [14] ........................................................ 14 Tabelle 2: Molekulare Sorptionskonstanten des Wassers [13] ............................................................. 15 Tabelle 3: Maximaler Extraktgehalt (Thymianextrakt) auf verschiedenen Trägerstoffen [15]............... 41 Tabelle 4: Physikalische Eigenschaften von Kartoffelstärke Aero Myl 33............................................. 48 Tabelle 5: Physikalische Eigenschaften von Maltodextrin Paselli MD6 ................................................ 49 Tabelle 6: Physikalische Eigenschaften von Pulverzellulose P290....................................................... 51 Tabelle 7: Eigenschaften der Cellets..................................................................................................... 52 Tabelle 8: Physikalische Eigenschaften von Sipernat 50...................................................................... 55 Tabelle 9: Physikalische Eigenschaften von Sipernat 50S ................................................................... 56 Tabelle 10: Physikalische Eigenschaften von Sipernat 2200................................................................ 57 Tabelle 11: Physikalische Eigenschaften von Aerosil 130 .................................................................... 59 Tabelle 12: Physikalische Eigenschaften von Aerosil R972.................................................................. 60 Tabelle 13: Physikalische Eigenschaften von Cabosil .......................................................................... 61 Tabelle 14: Eigenschaften von HySorb ................................................................................................. 62 Tabelle 15: Eigenschaften von Wasser ................................................................................................. 63 Tabelle 16: Eigenschaften von Speiseöl Delios .................................................................................... 64 Tabelle 17: Eigenschaften von Triacetin ............................................................................................... 64 Tabelle 18: Technische Daten des Pflugscharmischers ....................................................................... 72 Tabelle 19: Maximal mögliche Beladung verschiedener Trägerstoffe mit Speiseöl.............................. 89 Tabelle 20: BET-Oberflächen, aufgenommene Wassermenge und Flüssigkeitsaufnahmekapazität
der drei Trägerstoffe........................................................................................................ 95 Tabelle 21: Maximale Flüssigkeitsmenge (Speiseöl) auf verschiedenen Trägerstoffen ....................... 96 Tabelle 22: Erste Versuchsreihe: Partikeln aus Montanwachs aus dem JetCutter............................. 101 Tabelle 23: Schütt- und Stampfdichten der Montanwachspartikeln .................................................... 102 Tabelle 24: Zahlenwerte für die Berechnung einer Flüssigkeitsbrücke der Feinfraktion .................... 104 Tabelle 25: Ergebnisse der Berechnungen einer Flüssigkeitsbrücke der Feinfraktion ....................... 104 Tabelle 26: Montanwachspartikeln mit Kochsalz................................................................................. 106 Tabelle 27: Leitfähigkeitsmessung zur Bestimmung der herausgelösten Salzmenge........................ 107 Tabelle 28: Oberflächenmessung ausgewählter Partikeln .................................................................. 107 Tabelle 29: Spezifische Oberfläche von Natriumalginatpartikeln ........................................................ 109 Tabelle 30: Physikalische Eigenschaften der erzeugten Alginatpartikeln ........................................... 110 Tabelle 31: Im CPF-Verfahren mit Speiseöl beladene Alginatpartikeln .............................................. 111 Tabelle 32: Physikalische Eigenschaften von Kochsalz...................................................................... 112 Tabelle 33: Physikalische Eigenschaften von sprühgetrocknetem Kochsalz...................................... 113 Tabelle 34: Sprühversuche mit Kochsalz und Speiseöl im CPF-Verfahren ........................................ 116 Tabelle 35: Fettgehalte der Produkte aus dem Mischer...................................................................... 127 Tabelle 36: Wassergehalte der Sprühprodukte aus Kieselsäure und Wasser.................................... 129 Tabelle 37: Sprühprodukte aus Sipernat 2200 und Wasser................................................................ 133 Tabelle 38: T2 Zeiten der verschieden fest gebundenen Phasen ....................................................... 137 Tabelle 39: Schichtdicken und Anzahl der Wassermoleküle der immobilen Phase............................ 140 Tabelle 40: Wassergehalte der Proben aus dem Mischer aus Zellulose und Wasser........................ 149 Tabelle 41: Ergebnisse der T2-Messungen ......................................................................................... 152 Tabelle 42: Anteile berechnet aus den T2-Messungen........................................................................ 153


















