
NMR-spektroskopische Untersuchungen an cis- und trans-N-Methyl-N-benzyl-thioformamid. Zuordnung der...
8
1966 W. Waiter. G. Maerten und H. Rose 25 NMR-spektroskopische Untersuchungen an cis- und trans-N- Methyl-N-benzyl-thioformamid. Zuordnung der Konfigurationen und Kinetik der Urnwandlung von Wolfgang Walter, Gerhard Maerten und Heinrich Rose Aus dem Chemischen Staatsinstitut, Institut fur Organische Chemie der Universitat Hamburg Eingegangen am 4. Juni 1965 Aus einem Gemisch der cis-trans-Isomeren von N-Methyl-N-benzyl-thioformamid konnten bei Raumtemperatur das stabile Isomere A und ein angereichertes labiles Isomeres B abge- trennt werden. Aufgrund von NMR-Spektren wurde A die trans- und B die cis-Konfiguration zugeordnet. Die Kinetik der Isomerisierung wurde zwischen 50 und 60" untersucht und die Aktivierungsenergie zu 25.1 kcal/Mol bestimmt. Durch Thioformylierung von Aminen mit Dichlorcarben und Natriumhydrogen- sulfid konnten wir eine Anzahl von N-substituierten Thioformamiden darstellen 1). Hierbei erhielten wir das N-Methyl-N-benzyl-thioformamid (1) als Gemisch von zwei Isomeren. Das Isomere A (Schmp. 67.5-68") konnte rein isoliert werden. Das bei Raumtemperatur fliissige Isomere B konnte nur bis zu ca. 75 % angereichert werden. Es isomerisiert langsam und lien sich nur bei -15" unverandert aufbewahren. Wir deuteten dieses Verhalten, insbesondere aus spektroskopischen Befunden 1) als cis-trans-Isomerie um die partielle Doppelbindung der zentralen C -N-Amidbindung und wahlten die Bezeichnung ,,trans" fur die Konfiguration, in der der Thiocarbonyl- gruppe die Benzylgruppe gegeniibersteht *). A ,,trans" 1 B ,,cis" Eine nach dem chemischen und spektroskopischen Verhalten von vornherein un- wahrscheinliche Tautomerie unter Verschiebung der CH3-Gruppe zwischen Stickstoff *) Die Konstitution der Verbindung ist bezuglich der Substituenten hinreichend von der Peptidbindung abgesetzt, so daD Verwechslungen mit den Definitionen fur eine dieser Strukturen trans R\C-N/H) nicht zu erwarten sind. i OF \R 1) W. Walter und G. Maerten, Liebigs Ann. Chem. 669, 66 (1963).
-
Upload
wolfgang-walter -
Category
Documents
-
view
212 -
download
0
Transcript of NMR-spektroskopische Untersuchungen an cis- und trans-N-Methyl-N-benzyl-thioformamid. Zuordnung der...

1966 W. Waiter. G . Maerten und H. Rose 25
NMR-spektroskopische Untersuchungen an cis- und trans-N- Methyl-N-benzyl-thioformamid. Zuordnung der Konfigurationen und Kinetik der Urnwandlung
von Wolfgang Walter, Gerhard Maerten und Heinrich Rose
Aus dem Chemischen Staatsinstitut, Institut fur Organische Chemie der Universitat Hamburg
Eingegangen am 4. Juni 1965
Aus einem Gemisch der cis-trans-Isomeren von N-Methyl-N-benzyl-thioformamid konnten bei Raumtemperatur das stabile Isomere A und ein angereichertes labiles Isomeres B abge- trennt werden. Aufgrund von NMR-Spektren wurde A die trans- und B die cis-Konfiguration zugeordnet. Die Kinetik der Isomerisierung wurde zwischen 50 und 60" untersucht und die Aktivierungsenergie zu 25.1 kcal/Mol bestimmt.
Durch Thioformylierung von Aminen mit Dichlorcarben und Natriumhydrogen- sulfid konnten wir eine Anzahl von N-substituierten Thioformamiden darstellen 1).
Hierbei erhielten wir das N-Methyl-N-benzyl-thioformamid (1) als Gemisch von zwei Isomeren. Das Isomere A (Schmp. 67.5-68") konnte rein isoliert werden. Das bei Raumtemperatur fliissige Isomere B konnte nur bis zu ca. 75 % angereichert werden. Es isomerisiert langsam und lien sich nur bei -15" unverandert aufbewahren.
Wir deuteten dieses Verhalten, insbesondere aus spektroskopischen Befunden 1) als cis-trans-Isomerie um die partielle Doppelbindung der zentralen C -N-Amidbindung und wahlten die Bezeichnung ,,trans" fur die Konfiguration, in der der Thiocarbonyl- gruppe die Benzylgruppe gegeniibersteht *).
A ,,trans" 1 B ,,cis"
Eine nach dem chemischen und spektroskopischen Verhalten von vornherein un- wahrscheinliche Tautomerie unter Verschiebung der CH3-Gruppe zwischen Stickstoff
* ) Die Konstitution der Verbindung ist bezuglich der Substituenten hinreichend von der Peptidbindung abgesetzt, so daD Verwechslungen mit den Definitionen fur eine dieser
Strukturen trans R\C-N/H) nicht zu erwarten sind. i OF \ R 1) W. Walter und G . Maerten, Liebigs Ann. Chem. 669, 66 (1963).
26 W. Walter, C. Maerten und H . Rose Bd. 691
und Schwefel konnten wir durch Darstellung des N-Benzyl-thioformimidomethyl- esters (2) mit Sicherheit ausschlienen.
S-CH3
H c~H~-cH,-N=c: 2
2 ist eine Flussigkeit, die sich durch ihren niedrigeren Siedepunkt, ihr IR-Spektrum und ihre Zersetzlichkeit (schon bei Raumtemperatur) eindeutig von 1 unterscheidet.
Zuordnung der Konfigurationen von A und B
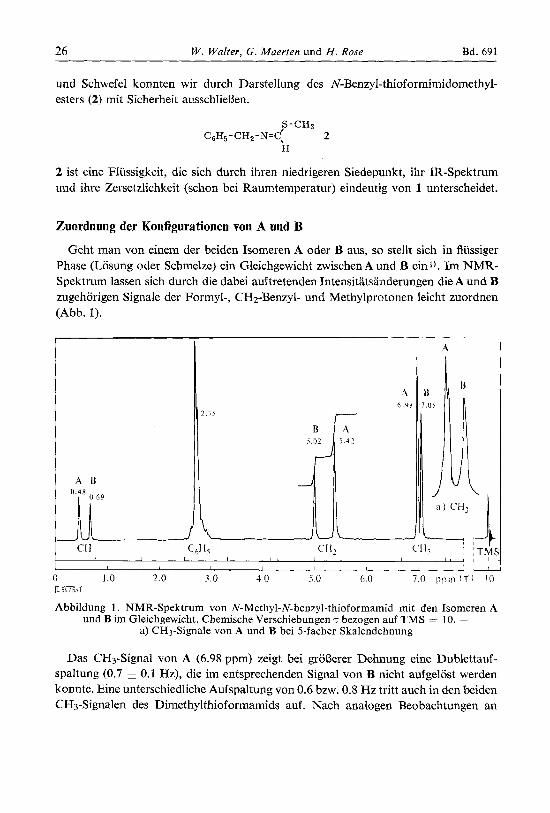
Geht man von einem der beiden Isomeren A oder B aus, so stellt sich in flussiger Phase (Losung oder Schmelze) ein Gleichgewicht zwischen A und B einl). Im NMR- Spektrum lassen sich durch die dabei auftretenden Tntensitatsanderungen die A und B zugehorigen Signale der Formyl-, CHz-Benzyl- und Methylprotonen leicht zuordnen (Abb. 1).
Abbildung 1 . NMR-Spektrum von N-Methyl-N-benzyl-thioformamid mit den Isomeren A und B im Gleichgewicht. Chemische Verschiebungen T bezogen auf TMS = 10. -
a) CH3-Signale von A und B bei 5-facher Skalendehnung
Das CH3-Signal von A (6.98 ypm) zeigt bei grol3erer Dehnung eine Dublettauf- spaltung (0.7 k 0.1 Hz), die im entsprechenden Signal von B nicht aufgelost werden konnte. Eine unterschiedliche Aufspaltung von 0.6 bzw. 0.8 Hz tritt auch in den beiden CH3-Signalen des Dimethylthioformamids auf. Nach analogen Beobachtungen an
1966 NMR-Untersuchungen an N-Methyl-N-benzyl-thioformamid 27
substituierten Formarniden2.3) mulj die groljere Aufspaltung einer trans-Spinkopplung der N-Methylprotonen rnit dem Formylproton zugeschrieben werden.
Eine andere Methode der Zuordnung beruht auf der unterschiedlichen Verschiebung der Kernresonanzsignale von Amiden bei Verdiinnung rnit Benzol2). Generell ver- schieben sich alle Signale mit zunehmender Verdunnung nach hoheren Feldstarken. Nach Hutton und Richards2) tritt jedoch das aromatische x-Elektronensystem rnit dem durch Mesomerie positivierten Stickstoff des Amids bevorzugt an der dem negati- vierten Sauerstoff abgewandten Seite in Wechselwirkung.
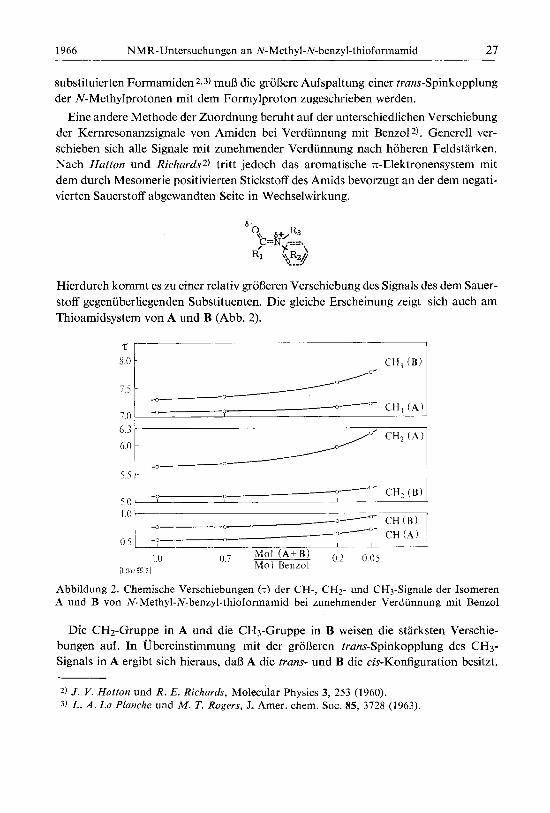
Hierdurch kommt es zu einer relativ groljeren Verschiebung des Signals des dem Sauer- stoff gegeniiberliegenden Substituenten. Die gleiche Erscheinung zeigt sich auch am Thioamidsystern von A und B (Abb. 2).
p- o------- CH2 ( B ) 5.0 ,
o-oA- CH ( B ) o-L--o- CH ( A )
0 . S ~ ~ -9 , I I
0.7 Mo' ( A + B ) 0.2 0.05 Mol Beiizol 1.0
1194/6521
Abbildung 2. Chemische Verschiebungen (T) der CH-, CH2- und CH3-Signale der Isomeren A und B von N-Methyl-N-benzyl-thioformamid bei zunehmender Verdiinnung rnit Benzol
Die CH2-Gruppe in A und die CH3-Gruppe in B weisen die starksten Verschie- bungen auf. In Ubereinstimmung mit der groljeren trans-Spinkopplung des CH3- Signals in A ergibt sich hieraus, dal3 A die trans- und B die cis-Konfiguration besitzt.
2) J. V. Hatton und R. E. Richards, Molecular Physics 3, 253 (1960). 3) L. A. La Plunche und M. T. Rogers, J. Amer. chem. SOC. 85, 3728 (1963).

28 W. Walter, G . Muerten und H . Rose Bd. 691
Die aus den NMR-Spektren gezogenen Schlusse konnten durch eine Rontgen- strukturanalyse von A eindeutig bestatigt werden4) (Abb. 3).
Abbildung 3 (entnommen aus Lit.4)). Bindungslangen (k0.025 -0.035 A) und Bindungs- winkel (k2-3") in trans-N-Methyl-N-benzyl-thioformamid. Die durch starke Linien verbun- denen Atome liegen (innerhalb der Fehlergrenzen) in einer Ebene. Die Lage des Phenylrings
wird durch Drehung in Pfeilrichtung mit den angegebenen Winkeln erhalten.
Kinetik der cis-tuans-Isomerisierung Die Aktivierungsenergien der behinderten Rotation wurden an einer Anzahl von
N-substituierten Amiden aus der Temperaturabhangigkeit der aufgespaltenen NMR- Signale ermittelts). Die mit der Teniperatur zunehmende Rotation macht sich dabei in einer Austauschverbreiterung und schlieRlich in einer Verschmelzung der getrennten Signale bemerkbar. Fur verschiedene Amide wurden Aktivierungsenergien von 6 bis 12 kcaI/Mol, fur Formamids) und Dimethylformamid7) von ca. 18 kcal/Mol gefunden. Wesentlich hohere Werte von 28 bzw. 32 kcal/Mol wurden nach dieser Methode fur Dimethyl- und Diisopropylthioformamid bestimmt 8).
NMR-Spektren des Gleichgewichtsgemisches von A und B zeigten bis zu einer Temperatur von 180" keine wesentliche Veranderung durch Austauschverbreiterung. Die Trennung der Isomeren ermoglichte hier jedoch eine unmittelbare kinetische
Verfolgung der Gleichgewichtseinstellung A -A B. Fur diesen Vorgang gilt 9 ) kz
ki
Ig (CA(G)-CA) = - ___- (kl + kz) t + const. 2.303
4) A . M. Piuzzesi, R . Bardi, M. Mammi und W. Walter, Ricerca sci. 34 (11-A), 173 (1964)
5 ) A. Loewenstein und T. M . Connor, Z . Elektrochem., Ber. Bunsenges. physik. Chem. 67,
6 ) B. Sunners. L. H. Piette und W. G. Schneider, Canad. J. Chem. 38, 681 (1960). 7) T. M. Rogers und J . C. Woodbrey, J. physic. Chem. 66, 540 (1962). 8) A. Loewenstein, A . Melera, P. Rigny und W. Walter, J. physic. Chem. 68, 1597 (1964). 9) A. A. Frost und R. G. Pearson, Kinetik und Mechanismen homogener chemischer Reak-
tionen, Verlag Chemie GrnbH, Weinheim/Bergstr. 1964, S. 173.
[C. A. 63, 2878 (1965)l.
280 (1963).
1966 NMR- Untersuchungen an N-Methyl-N-benzyl-thioformamid 29
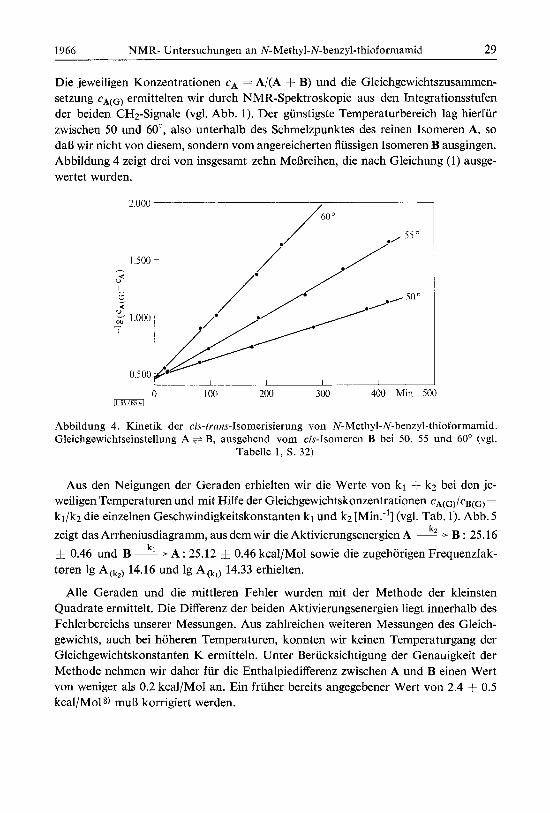
Die jeweiligen Konzentrationen cA = A/(A + B) und die Gleichgewichtszusammen- setzung cA(G) ermittelten wir durch NMR-Spektroskopie aus den Integrationsstufen der beiden CH2-Signale (vgl. Abb. 1). Der gunstigste Temperaturbereich lag hierfur zwischen 50 und 60°, also unterhalb des Schmelzpunktes des reinen Isomeren A, so da13 wir nicht von diesem, sondern vom angereicherten flussigen Isomeren B ausgingen. Abbildung 4 zeigt drei von insgesamt zehn NfeBreihen, die nach Gleichung (1) ausge- wertet wurden.
0 rn 100 200 300 400 Min. 500
Abbildung 4. Kinetik der cis-trans-Isomerisierung von N-Methyl-N-benzyl-thioformamid. Gleichgewichtseinstellung A + B, ausgehend vom cis-Isomeren B bei 50, 55 und 60" (vgl.
Tabelle 1 , S. 32)
Aus den Neigungen der Geraden erhielten wir die Werte von kl -1- k2 bei den je- weiligen Temperaturen und mit Hilfe der Gleichgewichtskonzentrationen CA(G)/CB(G) =
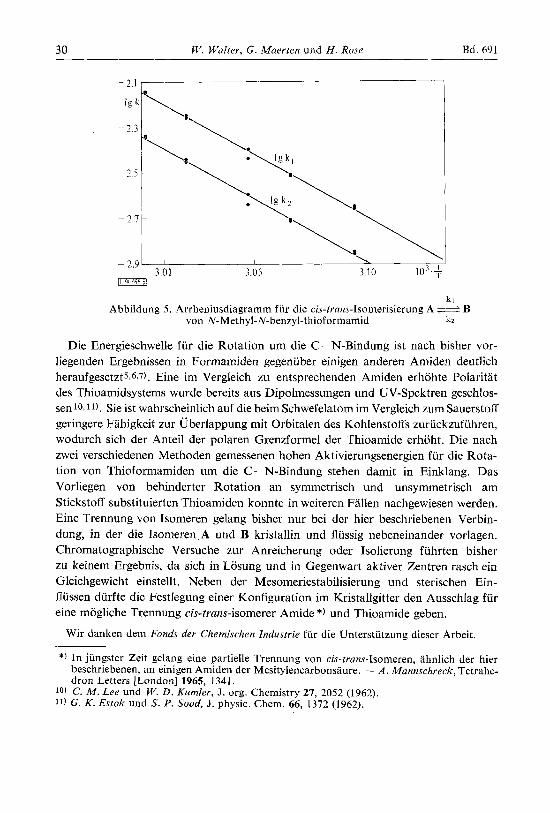
kl/k2 die einzelnen Geschwindigkeitskonstanten kl und k2 [Min.-'] (vgl. Tab. 1). Abb. 5 zeigt das Arrheniusdiagramm, aus dem wir die Aktivierungsenergien A 2j B : 25.16 Ilt 0.46 und B 2 4 A : 25.12 & 0.46 kcal/Mol sowie die zugehorigen Frequenzfak- toren Ig A(kz) 14.16 und Ig A(kl) 14.33 erhielten.
Alle Geraden und die mittleren Fehler wurden mit der Methode der kleinsten Quadrate ermittelt. Die Differenz der beiden Aktivierungsenergien liegt innerhalb des Fehlerbereichs unserer Messungen. Aus zahlreichen weiteren Messungen des Gleich- gewichts, auch bei hoheren Temperaturen, konnten wir keinen Temperaturgang der Gleichgewichtskonstanten K ermitteln. Unter Berucksichtigung der Genauigkeit der Methode nehmen wir daher fur die Enthalpiedifferenz zwischen A und B einen Wert von weniger als 0.2 kcal/Mol an. Ein fruher bereits angegebener Wert von 2.4 i 0.5 kcal/Molg) mu13 korrigiert werden.
k
k
30 W. Walter, G. Maerten und H. Rose Bd. 691
ki
von N-Methyl-N-benzyl-thioformamid k2
Abbildung 5. Arrheniusdiagramm fur die cis-trans-Isomerisierung A B
Die Energieschwelle fur die Rotation um die C-N-Bindung ist nach bisher vor- liegenden Ergebnissen in Formamiden gegenuber einigen anderen Amiden deutlich heraufge~etzt~~6~7). Eine im Vergleich zu entsprechenden Amiden erhohte Polaritat des Thioamidsystems wurde bereits aus Dipolniessungen und UV-Spektren geschlos- sen 10,11). Sie ist wahrscheinlich auf die beim Schwefelatom im Vergleich zum Sauerstoff geringere Fahigkeit zur Uberlappung mit Orbitalen des Kohlenstoffs zuruckzufuhren, wodurch sich der Anteil der polaren Grenzformel der Thioamide erhoht. Die nach zwei verschiedenen Methoden gemessenen hohen Aktivierungsenergien fur die Rota- tion von Thioformamiden um die C-N-Bindung stehen damit in Einklang. Das Vorliegen von behinderter Rotation an symmetrisch und unsymmetrisch am Stickstoff substituierten Thioaniiden konnte in weiteren Fallen nachgewiesen werden. Eine Trennung von Isomeren gelang bisher nur bei der hier beschriebenen Verbin- dung, in der die Isomeren A und B kristallin und flussig nebeneinander vorlagen. Chromatographische Versuche zur Anreicherung oder Isolierung f uhrten bisher zu keinem Ergebnis, da sich in Losung und in Gegenwart aktiver Zeritren rasch ein Gleichgewicht einstellt. Neben der Mesomeriestabilisierung und sterischen Ein- flussen durfte die Festlegung einer Konfiguration im Kristallgitter den Ausschlag fur eine mogliche Trennung cis-trans-isomerer Amide *) und Thioamide geben.
Wir danken dem Fonds der Chemischen lndustrie fur die Unterstiitzung dieser Arbeit.
*) In jungster Zeit gelang eine partielle Trennung von cis-trans-Isomeren, ahnlich der hier beschriebenen, an einigen Amiden der Mesitylencarbonsiiure. - A. Mannschreck, Tetrahe- dron Letters [London] 1965, 1341.
10) C. M. Lee und W. D. Kumler, J. org. Chemistry 27, 2052 (1962). 11) G . K. Esfok und S. P . Sood, J. physic. Chem. 66, 1372 (1962).

1966 NMR-Untersuchungen an N-Methyl-N-benzyl-thioformamid 31
Beschreibung der Versuche N-Benzyl-thioformimidomethylester(2). - Die Darstellung erfolgte nach Bottcher und
Buuer12). 6 g N-Benzyl-thioformamid und 5 g Methyljodid wurden in 50 ccm trockenem Ace- ton 1 Stde. unter RuckfluR erhitzt. Nach dem Abdampfen wurde der olige Ruckstand unter Eiskiihlung mit 7 g KOH in 25 ccm Wasser geschuttelt. Es wurde sofort mehrmals rnit Ather extrahiert, rnit Natriumsulfat getrocknet und destilliert : hellgelbe Flussigkeit vom Sdp.o.03 64". Ausbeute 2.5 g (38 %).
C ~ H I I N S (165.3) Ber. C 65.41 H 6.71 N 8.48 S 19.40 Gef. 65.29 6.76 8.89 19.47
Die Verbindung hat einen widerwartigen Geruch und zersetzt sich schon bei Raumtempera- tur unter Abspaltung von Methylmercaptan. Das IR-Spektrum weist charakteristische Unter- schiede gegenuber dem Spektrum des N-Methyl-N-benzyl-thioformamids auf. Die C=N- Valenzschwingung liegt bei 1612 cm-1, wahrend die entsprechende C-N-Schwingung mit hohem Doppelbindungsanteil im Thioamid bei 1515 cm-1 auftritt.
Kernresonannersuche: Alle NMR-Messungen wurden rnit dem Spektrometer Varian A 60 rnit eingebautem elektronischen Integrator bei 60 MHz ausgefuhrt. Die chemischen Ver- schiebungen beziehen sich auf Tetamethylsilan als inneren Standard mit T = 10.
Fur die Verdiinnungsmessungen (vgl. Abb. 2) wurde aus den Kristallen A durch 1 stdg. Er- hitzen das Isomerengemisch erzeugt und die Losungen durch Einwaage der Substanz und Zu- waage von Benzol hergestellt.
Fur die kinetischen Messungen wurde von dem auf ca. 15 % B angereicherten Isomeren- gemisch ausgegangen (Schmp. ca. 7"). Eine Probe der bei - 15" aufbewahrten Substanz wurde nach dem Aufschmelzen in das NMR-MeRrohrchen gefiillt und dieses bei der Anfangszeit 0 (Min.) in einen Ultrathermostaten rnit der gewahlten Versuchstemperatur gegeben. Zur Messung des Konzentrationsverhaltnisses C A = A/(A f- B) bei der jeweiligen Zeit t wurde die Probe in Abstanden von 30-70 Min. in den vorher auf die Versuchstemperatur einge- stellten Probenkopf des Spektrometers gebracht (Varian-Temperaturregulator V 6040). Es wurden nur die CHz-Signale von A und B und iiber diesen 6mal rasch hintereinander die Integrationsstufen aufgenommen. Die den Konzentrationen proportionalen beiden Stufen- hohen wurden an einem Digitalvoltmeter (Hewlett-Packard 405 CR) direkt abgelesen und die Zeit t notiert. Bei einer MeRdauer von ca. 2 Min. betrugen die moglichen Zeitablesefehler hochstens i 1 Min. Ebenso konnten bei der kurzen Verweilzeit der Probe im Spektro- meter die dort moglichen Temperaturschwankungen von & 2" vernachlassigt werden. Die 6mal aufgenommenen Konzentrationsverhaltnisse wurden zu dem Wert C A bei der Zeit t gemittelt. Es wurden so 5-7 Messungen von CA bis zur Einstellung der Gleichgewichts- konzentration C A (G) durchgefuhrt und diese durch drei weitere Messungen genau bestimmt. Wir werteten die MeRergebnisse nach der Gleichung (1) fur eine einfache reversible Reaktion 1. Ordnung aus. Die Linearitat (vgl. Abb. 4) war gut erfullt, wenn bei guter Stabilitat und Homogenitat des Magnetfeldes besonders Phasenlage und Detektornull an den Integralen sorgfaltig eingestellt wurden13) (Aufnahme der Integrale bei RF 0.1 mG, Filterbandweite 1 , Sweepgeschwindigkeit 100 Sek./500 Hz).
12) B. Bottcher und F. Buuer, Liebigs Ann. Chem. 568, 218 (1950). 13) J. L. Jungnickel und J. W. Forbes, Analytic. Chem. 35, 938 (1963).
32 W. Walter, C. Maerten und H. Rose Bd. 691
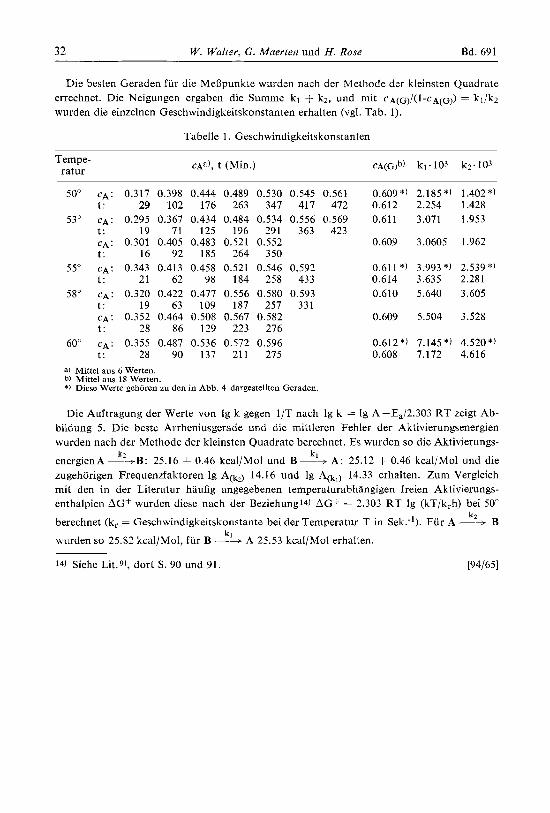
Die besten Geraden fur die MeBpunkte wurden nach der Methode der kleinsten Quadrate errechnet. Die Neigungen ergaben die Summe kl + kZ, und mit CA(G)/(~-CA(G$ = k1/k2 wurden die einzelnen Geschwindigkeitskonstanten erhalten (vgl. Tab. 1).
Tabelle 1. Geschwindigkeitskonstanten
Tempe- ratur
50" CA: 0.317 0.398 0.444 0.489 0.530 0.545 0.561 t: 29 102 176 263 341 411 412
53" CA: 0.295 0.367 0.434 0.484 0.534 0.556 0.569 t : 19 71 125 196 291 363 423 CA: 0.301 0.405 0.483 0.521 0.552 t : 16 92 185 264 350
55" CA: 0.343 0.413 0.458 0.521 0.546 0.592 t : 21 62 98 184 258 433
58" CA: 0.320 0.422 0.471 0.556 0.580 0.593 t: 19 63 109 187 257 331 CA: 0.352 0.464 0.508 0.567 0.582 t : 28 86 129 223 276
60" CA: 0.355 0.487 0.536 0.572 0.596 t : 28 90 137 211 275
a) Mittel aus 6 Werten. b) Mittel aus 18 Werten. *) Diese Werte gehoren zu den in Abb. 4 dargestellten Geraden.
0.609 *) 0.612 0.61 1
0.609
0.611 * )
0.614 0.610
0.609
0.612 * )
0.608
2.185 *)
2.254 3.07 1
3.0605
3.993 *)
3.635 5.640
5.504
7.145 * ) 7.172
1.402 *)
1.428 1.953
1.962
2.539 *)
2.281 3.605
3.528
4.520 *) 4.616
Die Auftragung der Werte von lg k gegen 1/T nach lg k = lg A-Ea/2.303 RT zeigt Ab- bildung 5. Die beste Arrheniusgerade und die mittleren FehIer der Aktivierungsenergien wurden nach der Methode der kleinsten Quadrate berechnet. Es wurden so die Aklivierungs-
energienA-+B: 25.16 0.46 kcal/Mol und B + A : 25.12 f 0.46 kcal/Mol und die zugehorigen Frequenzfaktoren Ig A(k2) 14.16 und Ig A(kl) 14.33 erhalten. Zum Vergleich mit den in der Literatur haufig angegebenen temperaturabhangigen freien Aktivierungs- enthalpien AG* wurden diese nach der Beziehungl4) AG* = 2.303 RT Ig (kT/k,h) bei 50"
berechnet (k, = Geschwindigkeitskonstante bei der Temperatur T in Sek.-'). Fur A -+ B
wurden so 25.82 kcal/Mol, fur B -L A 25.53 kcal/Mol erhalten.
kz ki
kz
k
14) Siehe Lit.g), dort S. 90 und 91. [94/651


![3. Fluoreszenz- und UV/VIS-spektroskopische ......49 3. Fluoreszenz- und UV/VIS-spektroskopische Untersuchungen der dargestellten Imidazo[4,5-c]carbazole, Carbazole und Phenazine 3.1.](https://static.fdokument.com/doc/165x107/60b7520dfe96d2539945bd31/3-fluoreszenz-und-uvvis-spektroskopische-49-3-fluoreszenz-und-uvvis-spektroskopische.jpg)















