
Peptid-Funktionalisierte Gold (III)-Komplexe als … · fe beim Anfertigen der Verbindungen danken...
173
Peptid-Funktionalisierte Gold(III)-Komplexe als Substrat für Matrix-Metalloprotease 2 Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Chemie und Biochemie der Ruhr-Universität Bochum vorgelegt von M. Sc. Jan Christoph Dittrich aus Meerbusch Bochum Oktober 2013
Transcript of Peptid-Funktionalisierte Gold (III)-Komplexe als … · fe beim Anfertigen der Verbindungen danken...

Peptid-Funktionalisierte
Gold(III)-Komplexe
als Substrat für
Matrix-Metalloprotease 2
Dissertation
zur Erlangung des Grades eines
Doktors der Naturwissenschaften
der Fakultät für Chemie und Biochemie
der Ruhr-Universität Bochum
vorgelegt von
M. Sc. Jan Christoph Dittrich
aus Meerbusch
Bochum Oktober 2013

Diese Arbeit wurde zwischen Oktober 2009 bis zum Oktober 2013 unter Anleitung von Herrn
Prof. Dr. Nils Metzler-Nolte im Lehrstuhl für Anorganische Chemie I − Bioanorganische
Chemie in der Fakultät für Chemie und Biochemie der Ruhr-Universität Bochum angefertigt.
Referent: Prof. Dr. Nils Metzler-Nolte
Koreferent: Prof. Dr. Ingo Ott
Tag der mündlichen Prüfung: 29.November 2013

Meiner Familie

Danksagung
Danksagung
Herrn Prof. Dr. Nils Metzler-Nolte danke ich herzlich für das Thema, das mir entgegenge-
brachte Vertrauen, die anregenden fachlichen Diskussionen und alle großartigen Gespräche.
Herrn Prof. Dr. Ingo Ott für seine Bereitschaft bei meiner Dissertation als Koreferent zur Ver-
fügung zu stehen und alle die gute Zeit die ich mit ihm und seiner Arbeitsgruppe auf Konfe-
renzen verbringen durfte.
Meinen Praktikanten Frau Barbara Hoffknecht, Frau Maike Wiemann, Herrn Martin Strack,
Frau Janine Parthier, Herrn Florian Seidel und Herrn Dennis Pache möchte ich für ihre Mithil-
fe beim Anfertigen der Verbindungen danken und die gute Zeit die wir im Labor verbracht
haben.
Frau Prof. Dr. Chiara Chabrele und insbesondere Frau Saskia Neukirchen danke ich für die
Möglichkeit mit dem Syro Multiple Peptide Synthesizer der Firma Syro 2000 zu arbeiten und
mir bei dem Gerät mit Rat und Tat zur Seite zu stehen.
Frau Prof. Dr. Martina Havenith-Newen und ganz besonders Frau Dr. Meike Mischo danke
ich für das Anfertigen und Auswerten der Ramanspektren. Herrn Dr. Pablo Nieto danke ich
für die Berechnungen der Schwingungsbande, welche eine Auswertung erst möglich gemacht
haben.
Herrn Dr. Klaus Merz danke ich für die Messung der Röntgenstrukturdaten und seine Hilfe
bei der Auswertung.
Frau Nicole Ray danke ich für ihre Hilfe bei allen administrativen Problemen.
Herrn Dr. Bauke Albada danke ich für seine Hilfe mit den HPLC-Anlagen und guten fachli-
chen Gespräche.
Frau Andrea Ewald danke ich für ihren Einsatz rund um das ESI-Massenspektrometer, für die
Messungen die sie für mich angefertigt hat und ganz besonders für ihren schnellen Reparatu-
ren, wenn doch mal etwas defekt war.

Danksagung
Frau Anna Sosniak und Frau Barbara Hoffknecht danke ich für alle angefertigten MALDI-
Spektren.
Herrn Jochen Lügger danke ich ganz herzlich für seinen Einsatz bei allen technischen Prob-
lemen im Labor und den Erklärungen die ich zusätzlich dazu erhalten habe.
Frau Annegret Knüfer möchte ich meinen Dank für ihre Anstrengungen aussprechen, für alle
Reparaturen am Liberty Mikrowellen Peptid Synthesizer
Dr. Christiane Klare danke ich für ihr Engagement für mehr Sicherheit im Labor.
Herrn Gregor Barchan und Herrn Martin Gartmann danke ich für ihren Einsatz bei allen
NMR-Spektrometern.
Frau Sabine Bendix danke ich für die Messungen der Elementaranalyse.
Den Mitgliedern des Lehrstuhles für Anorganische Chemie I danke ich für die angenehme
Arbeitsatmosphäre mit vielen spannenden wissenschaftlichen Diskussionen und ganz beson-
ders danke ich Herrn Dr. Nicola Alzakhem, Frau Anna Sosniak und Frau Jessica Wahsner, die
mich beim anfertigen des schriftlichen Teils der Arbeit besonders unterstützt haben.
Und nicht zuletzt danke ich meiner Familie und meinen Freunden, die mich sehr unterstützt
haben.

Inhaltsverzeichnis
I
Inhaltsverzeichnis
1. Einleitung ........................................................................................................................... 1 2. Aufgabenstellung ............................................................................................................. 12 3. Ergebnisse ........................................................................................................................ 14
Der Gold(III)-Komplex ........................................................................................................ 14
Mögliche Modifizierung zur Kopplung von Peptiden ......................................................... 15
Funktionalisierung des Phenylringes ................................................................................... 16
Einfügung einer Alkylkette .............................................................................................. 18
Methylester ....................................................................................................................... 20
Funktionalisierung des Pyridinringes ................................................................................... 21
Transmetallierung ............................................................................................................. 23
Verwendung von Bipyridin als Ligand ............................................................................ 23
Peptide ...................................................................................................................................... 26
Bindung der Peptide direkt zum Gold(III)-Zentrum ........................................................ 26
HSAB Prinzip ....................................................................................................................... 26
Austausch der Chloridliganden ............................................................................................ 26
Suche nach einem Peptidbindungsmotive ........................................................................ 27
Zusammenfassung der Suche nach einem Peptidbindungsmotive ................................... 29
Stabilitätstest in der Abspaltlösung ...................................................................................... 29
Ergänzung des Bindungsmotives um einen geladen Peptidteil ............................................ 30
Orthogonal geschützt Glutaminsäure ................................................................................... 32
Kupferkatalysierte [3+2] Cycloaddition ............................................................................... 32
[3+2] Cycloaddition der Teilsequenzen ........................................................................... 34
Zusammenfassung [3+2] Cycloaddition der Teilsequenzen ............................................ 36
[3+2] Cycloaddition der kompletten Sequenz .................................................................. 36
Zusammenfassung [3+2] Cycloaddition der kompletten Sequenz ................................... 38
Bessere Löslichkeit in Wasser .............................................................................................. 38
Zusammenfassung Bessere Löslichkeit in Wasser ........................................................... 41
300 potentielle Bindungsmotive ........................................................................................... 41
Bindenden Aminosäuren .................................................................................................. 42
Abstandsaminosäuren ....................................................................................................... 43
Aufbau der Sequenz ......................................................................................................... 43
Synthese ........................................................................................................................... 44
Analyse ............................................................................................................................. 45

Inhaltsverzeichnis
II
Farbe der Harzkugeln ....................................................................................................... 45
Farbe im Fluoreszenzmikroskop ...................................................................................... 45
Ramanspektroskopie ........................................................................................................ 46
Auswertung des 300 Peptide Projekts .................................................................................. 49
Auswertung der Farbe ...................................................................................................... 49
Auswertung der Fluoreszenzmikroskopie ........................................................................ 50
Auswertung der Ramanspektren ...................................................................................... 51
Diskussion zu dem 300 Peptideprojekt ............................................................................ 52
Stabilität in Lösung .............................................................................................................. 53
Zusammenfassung des 300 Peptideprojekts ..................................................................... 58
Mehr Abstandsaminosäuren ................................................................................................. 58
Bindung über eine α-Helix ............................................................................................... 64
Erhöhung der Löslichkeit in Wasser ................................................................................ 66
Zusammenfassung der Kettenverlängerung ..................................................................... 68
16 Peptide ............................................................................................................................. 69
[3+2] Cycloaddition mit dem stabilen Bindungsmotiv ........................................................ 72
Andere Bindungsmotive in der [3+2] Cycloaddition ....................................................... 74
Zusammenfassung der [3+2] Cycloaddition mit dem stabilen Bindungsmotiv ............... 79
[3+2] Cycloadditionen ohne Kupferkatalysator ................................................................... 79
Orthogonale Bindung über ein Imin ..................................................................................... 83
Direkter Weg ........................................................................................................................ 85
4. Zusammenfassung ............................................................................................................ 90 5. Experimentalteil ............................................................................................................... 94
Material ................................................................................................................................ 94
Methoden .............................................................................................................................. 96
Synthesen ............................................................................................................................. 98
Synthese von 2-Phenylpyridin-1-gold(III)trichlorid 1 ..................................................... 98
Synthese von 2-Phenylpyridin-1,1´-gold(III)dichlorid 2 ................................................. 99
Synthese von 2-Phenylpyridin-3´-säure 3 ...................................................................... 100
Synthese von 2-Phenylpyridin-3´-säure-1´-gold(III)trichlorid 4 ................................... 101
Synthese von 2-Phenylpyridin-3´-acrylsäure 6 .............................................................. 102
Synthese von 2-Phenylpyridin-3´-propansäure 7 ........................................................... 103
Synthese von 2-Phenylpyridin-3´-propansäure-1-gold(III)trichorid 8 ........................... 104
Synthese von 2-Phenylpyridin-3´-acrylat 10 .................................................................. 105
Synthese von 2-Phenylpyridin-3´-propansäuremethylester 11 ...................................... 106

Inhaltsverzeichnis
III
Synthese von 2-Phenylpyridin-3´-propansäuremethylester-1-gold(III)trichorid 12 ...... 107
Synthese von 4-Brom-2-Phenylpyridin 14 ..................................................................... 108
Synthese von 4-Brom-2-Phenylpyridin-1-gold(III)trichorid 15 .................................... 110
Synthese von 2-Phenylquinolin-4-methylcarboxylat-1´-chloromercury 17................... 111
Synthese von Methyl-6,6´-dimethyl-[2,2´-bipyridin]-4-carboxylat-1-gold(III)trichloride
19 .................................................................................................................................... 112
Synthese von 2-Phenylpyridin-1,1´-golddiaceatat 20 .................................................... 113
Synthese von 2-Phenylpyridin-1-gold(III)tribromid 650 ............................................... 114
Synthese von 2-Phenylpyridin-1,1´gold(II)dibromid 651 .............................................. 115
Synthese von Tetraethylammoniumtetrachloroaurat(III) ............................................... 116
Synthese von Tetraethylammoniumtetrabromoaurat(III) ............................................... 117
Synthese von Tetraethylammoniumphenyltrichloroaurat(III) 652................................. 118
Synthese von Tetraethylammoniumphenyltribromoaurat(III) 653 ................................ 119
Synthese von Pyridin-1-gold(III)trichlorid 654 ............................................................. 120
Synthese von Pyridin-1-gold(III)tribromid 655 ............................................................. 121
Synthese von Bipyridin-1,1´gold(III)dichlorid tetrachloroaurat(III) 656 ...................... 122
Synthese von Bipyridin-1,1´gold(III)dibromid tetrabromoaurat(III) 657 ...................... 123
Synthese von 2-Phenylpyridin-1,1´gold(III)ethanditihol 658 ........................................ 124
Peptide: ............................................................................................................................... 125
Festphasen Peptidsynthese ............................................................................................. 126
Kopplung von AuPhPyCl2 mit Peptiden ............................................................................ 134
Vor dem 300 Peptidprojekt ............................................................................................ 134
[3+2] Cycloaddition ....................................................................................................... 135
300 Peptidprojekt ........................................................................................................... 136
Nach dem 300 Peptidprojekt .......................................................................................... 137
Literatur .................................................................................................................................. 140 Anhang ................................................................................................................................... 146
Nomenklatur ................................................................................................................... 146
300 Peptidprojekt ........................................................................................................... 147

Abkürzungen
IV
Abkürzungen
A, Ala Alanin
Ac Acetat
AcO2 Acetanhydrid
ACN Acetonitril
AuPhPyCl2 2-Phenylpyridin-1,1´-gold(III)dichlorid
AS Aminosäure
B, Pal3 3-(3-Pyridin)-alanin
Boc tert-Butylcarbamat
C, Cys Cystein
°C Grad Celsius
cm-1
reziproke Zentimeter
CPP cell-penetrating peptides
d Duplett
D, Asp Asparaginsäure
δ chemische Verschiebung
Δ, Aib 2-Aminoisobutansäure
DC Dünnschichtchromatographie
DCM Dichlormethan
dd Duplett vom Duplett
DET Diethylether
DFT Dichtefunktionaltheorie
DIBAL Diisobutylaluminiumhydrid
DIPEA N,N-Diisopropylethylamin
Dmab 4-((1-4,4-Dimethyl-2,6-dioxocyclohexyliden)-3-methybutyl)amino)-benzyl
DMF N,N-Dimethylformamid
DMSO Dimethylsulfoxid
DMSO-d6 deuteriertes Dimethylsulfoxid
DNS Desoxyribonukleinsäure
E, Glu Glutaminsäure
ECM Extrazellularmatrix
EDT 1,2-Ethandithiol
ESI Elektrosprayionisation
EtOH Ethanol
F, Phe Phenylalanin
FDA Food and Drug Administration
Fmoc 9-Fluorenylmethyl
g Gram
G, Gly Glycin
Γ Methyladipoylchlorid
Gew.-% Massenprozent
h Stunde
H, His Histidin
HBTU
2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate
HIV Humane Immundefizienz-Virus
H2O Wasser

Abkürzungen
V
HOBT 1-Hydroxybenzotriazole
HP homing peptide
HPLC high performance liquid chromatography
HSAB hard and soft acids and bases
Hz Hertz
J Kopplungskonstante
J, Pal4 3-(4-Pyridin)-alanin
K, Lys Lysin
KHMDS Kaliumhexamethyldisilizan
l Liter
L, Leu Leucin
LC liquid chromatography
m Multiplett
mW Megawatt
M molar
M, Met Methionin
MALDI matrix assisted laser desorption ionization
MB Macro Beads
mbar Millibar
MeOH Methanol
mg Milligram
MHz Megahertz
min Minuten
ml Milliliter
mm Millimeter
µl Mikroliter
µm Mikromter
mmol Millimol
MMP Matrix-Metalloproteasen
mol Mol
MS Massenspektroskopie
m/z Masse geteilt durch die Ladung
nm Nanometer
NMR nuclear magnetic resonance
NMP N-Methyl-2-pyrrolidon
O Azidoessigsäure
OtBu tertiäre-Butylgruppe
P, Pro Prolin
Π, Pen 4-Pentinsäure
Pbf 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
PDA photo diode array
PhPy 2-Phenylpyridin
ppm parts per million
% Prozent
q Quartett
R, Arg Arginin
RNS Ribonukleinsäure
RT Raumtemperatur
s strong

Abkürzungen
VI
Die Peptide der Gold(III)-Biokonjugate werden im Einbuchstabencode und die gesamte Ver-
bindung wird ohne Ladung aus Gründen der Übersichtlichkeits wiedergegeben.
s Singulett
SPPS solid phase peptide synthesis
t Triplett
TBTA Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amin)
TBTU O-(Benzotriazol-1-yl)-N,N,N′,N′- tetramethyluronium tetrafluoroborat
t-Butanol 2-Methyl-2-propanol
td Triplett vom Duplett
TFA trifluoroacetic acid
THF Tetrahydrofuran
TIS Triisopropylsilan
TOF time of flight
Trt Triphenylmethyl
UV Ultraviolett
Vol.-% Volumenprozent
w weak
Y, Tyr Thyrosin
Z, Dap 2,3-Diaminopropionsäure

Einleitung
Seite | 1
1. Einleitung
In Deutschland wird traditionell zwischen der organischen und der anorganischen Chemie
unterschieden. Die organische Chemie ist demnach die Chemie der belebten, die anorganische
Chemie, die der unbelebten Materie. 1828 zeigte Friedrich Wöhler jedoch schon, dass sich der
organische Harnstoff aus den beiden anorganischen Edukten Ammoniumsulfat und
Kaliumcyanat darstellen lässt.1
Viele Metalle, die der anorganischen Chemie zugerechtet werden, werden von den Lebewesen
auf diesem Planeten genutzt. Zum einen sind dies die Alkali und Erdalkali Elemente Natrium,
Magnesium, Kalium und Calcium, die unter anderem als Elektrolyte eine entscheidende Rol-
le spielen und letzteres natürlich entscheidend für den Knochenaufbau ist.2-4
Zum anderen
sind dies Übergangsmetalle wie Vanadium, Chrom, Mangan, Eisen, Kobalt, Kupfer, Zink und
Molybdän. Diese machen nur einen geringen Prozentsatz der Masse eines Lebewesens aus,
sind jedoch von entscheidender Bedeutung für das jeweilige Lebewesen. Das jeweilige Metall
besitzt besondere Eigenschaften, welche so von keinem anderen Element geboten werden.5-8
Als Beispiel kann hier das Eisen angeführt werden. 4-5 g enthält der menschliche Körper
durchschnittlich. Es gibt diverse eisenhaltige Enzyme wie Peroxidasen, Zytochrome und die
Katalase, es kommt in Eisentransportern wie dem Transferrin vor, es liegt gespeichert im
Ferritin und Hämosiderin vor, und Eisen ist zur temporären Sauerstoffbindung im Myoglobin
und Hämoglobin vorhanden.9
Die menschlichen Hämoglobintypen haben sich zum Beispiel im Verlauf der Evolution zu
einem hervorragenden Sauerstofftransporter entwickelt. Es bindet Sauerstoff bei einem hohen
Sauerstoffpartialdruck und einem relativ hohen pH-Wert. Sinkt der Partialdruck und der pH-
Wert, dann wird das vorher gebundene Sauerstoffmolekül wieder abgeben. So kann der Sau-
erstoff von der Lunge mit hohem Sauerstoffpartialdruck in Bereiche mit geringem Partial-
druck, wie den Muskeln, transportiert werden. Mit einem Anstieg der Kohlenstoffdioxidkon-
zentration im Blut sinkt der pH-Wert, was eine Sauerstoffabgabe durch das Hämoglobin wei-
ter begünstigt.10
Das Hämoglobin besteht aus jeweils zwei verschiedenen Untereinheiten. Die eine Unterein-
heit wird als α, die andere als β bezeichnet. Jeweils zwei verschiedene Untereinheiten bilden
ein Dimer, zwei Dimere bilden das Hämoglobin. Jede Untereinheit wird aus 141 bis 146
Einleitung
Seite | 2
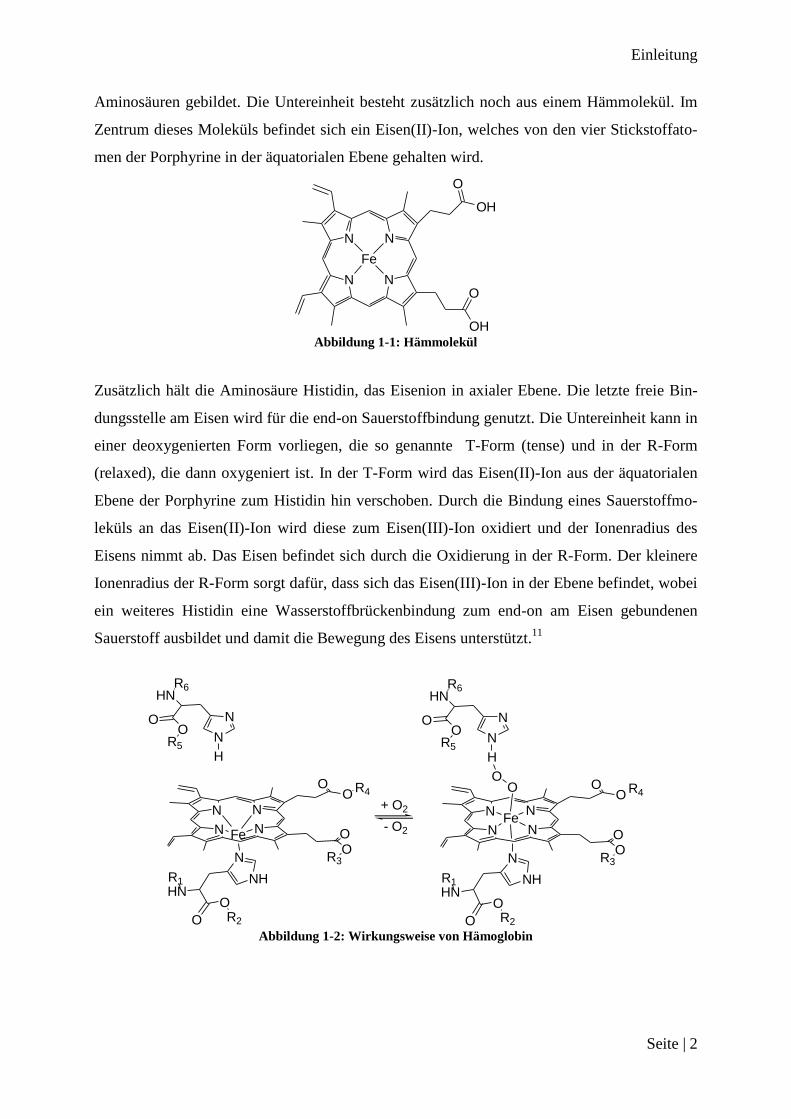
Aminosäuren gebildet. Die Untereinheit besteht zusätzlich noch aus einem Hämmolekül. Im
Zentrum dieses Moleküls befindet sich ein Eisen(II)-Ion, welches von den vier Stickstoffato-
men der Porphyrine in der äquatorialen Ebene gehalten wird.
N
NN
N
Fe
OH
O
OH
O
Abbildung 1-1: Hämmolekül
Zusätzlich hält die Aminosäure Histidin, das Eisenion in axialer Ebene. Die letzte freie Bin-
dungsstelle am Eisen wird für die end-on Sauerstoffbindung genutzt. Die Untereinheit kann in
einer deoxygenierten Form vorliegen, die so genannte T-Form (tense) und in der R-Form
(relaxed), die dann oxygeniert ist. In der T-Form wird das Eisen(II)-Ion aus der äquatorialen
Ebene der Porphyrine zum Histidin hin verschoben. Durch die Bindung eines Sauerstoffmo-
leküls an das Eisen(II)-Ion wird diese zum Eisen(III)-Ion oxidiert und der Ionenradius des
Eisens nimmt ab. Das Eisen befindet sich durch die Oxidierung in der R-Form. Der kleinere
Ionenradius der R-Form sorgt dafür, dass sich das Eisen(III)-Ion in der Ebene befindet, wobei
ein weiteres Histidin eine Wasserstoffbrückenbindung zum end-on am Eisen gebundenen
Sauerstoff ausbildet und damit die Bewegung des Eisens unterstützt.11
+ O2
- O2 N
NN
NFe
OO
OO R4
R3
HNOR2
NH
N
O
R1
HN
OR5
N
NO
R6
OO
H
N
NN
N FeOO
OO R4
R3
HNOR2
NH
N
O
R1
HN
OR5
N
NO
R6
H
Abbildung 1-2: Wirkungsweise von Hämoglobin

Einleitung
Seite | 3
Durch Veränderungen der Umgebung, wie dem pH-Wert und der Sauerstoffkonzentration,
wird die quartäre Struktur des Proteins beeinflusst und eine Bindung in Bereichen mit hoher
Sauerstoffkonzentration nach dem oben beschriebenen Prinzip begünstigt.
Dieses Beispiel veranschaulicht, dass schon während der Evolution die einzigartigen Eigen-
schaften der Metalle ihre lebensnotwendige Anwendung gefunden haben. Es ist deswegen
nahe liegend, diese Eigenschaften auch in der Medizin zu verwenden.
Dies wurde auch schon in vorchristlicher Zeit getan, wo zum Beispiel schon vor 2500 Jahren
Kupferverbindungen zur Desinfektion von Wasser verwendet wurde.12
Seit dem 6. Jahrhundert vor Christus werden Quecksilberverbindungen in der traditonellen
chinesischen und indischen Medizin verwendet. 13-15
Seit dem 18. Jahrhundert werden Amalgane in der Zahnmedizin verwendet, wo sie wegen
ihrer Härte geschätzt werden, jedoch geht ihre Verwendung inzwischen aufgrund von mögli-
chen toxischen Nebeneffekten wieder zurück.16, 17
1859 wurde beschrieben, dass die Gabe von Lithiumionen bei rheumatischen Erscheinungen
helfen kann. Seit 1949 wird Lithiumbromid als erstes modernes Antikonvulsivum bei Epilep-
sie eingesetzt.18
Manisch-depressiv erkrankte Menschen lassen sich sehr gut mit Lithiumsalz
behandeln. Nicht nur helfen die Mittel bei akuten Phasen der Krankheit, sondern sie helfen
auch langfristig.19, 20
Neue technische Möglichkeiten können zur Entwicklung von neuen Medikamenten führen. So
zog die Entwicklung der Röntgengeräte einen Bedarf nach Kontrastmitteln nach sich. Rönt-
genstrahlung wird von besonders elektronenreichen Verbindungen gebremst. Es war deswe-
gen nahe liegend, Metall mit hohen Ordnungszahlen mit in Betracht zu ziehen. Heute werden
unter anderem bariumhaltige Verbindungen als Kontrastmittel bei Röntgenuntersuchungen
verwendet.21
Die Entdeckung der Radioaktivität öffnete die Möglichkeit, diese zu Therapie- und Diagnose-
zwecken einzusetzen.22
Das häufigste in der Diagnostik eingesetzte Isotop ist das
99mTechnetium.
23 Es können jedoch auch Isotope des Galliums, des Indiums, des Kupfers und
des Yttriums verwendet werden.24
In der Therapie können Isotope des Strontiums, Zinns,
Einleitung
Seite | 4
Rheniums, Samariums und Holmiums Anwendung finde.25
Die Entwicklung moderner che-
mischer Synthesen führte dazu, dass nicht nur radioaktive Substanzen für die Therapie entwi-
ckelt wurden.
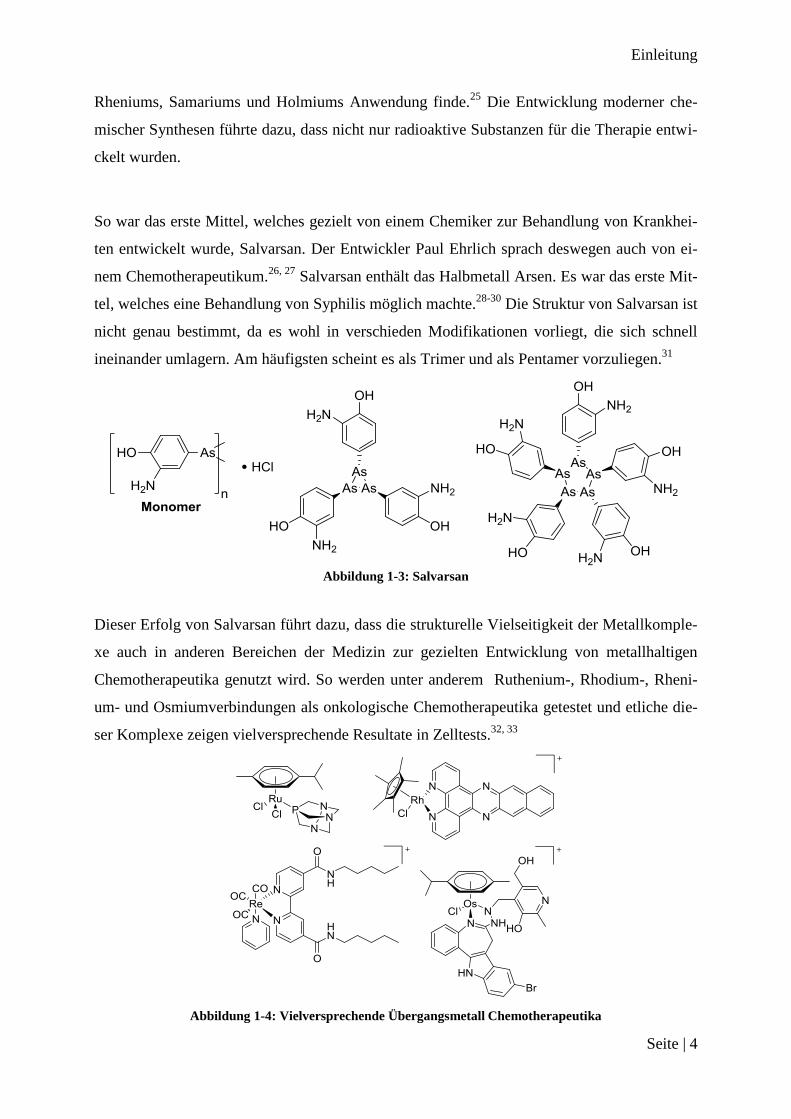
So war das erste Mittel, welches gezielt von einem Chemiker zur Behandlung von Krankhei-
ten entwickelt wurde, Salvarsan. Der Entwickler Paul Ehrlich sprach deswegen auch von ei-
nem Chemotherapeutikum.26, 27
Salvarsan enthält das Halbmetall Arsen. Es war das erste Mit-
tel, welches eine Behandlung von Syphilis möglich machte.28-30
Die Struktur von Salvarsan ist
nicht genau bestimmt, da es wohl in verschieden Modifikationen vorliegt, die sich schnell
ineinander umlagern. Am häufigsten scheint es als Trimer und als Pentamer vorzuliegen.31
Abbildung 1-3: Salvarsan
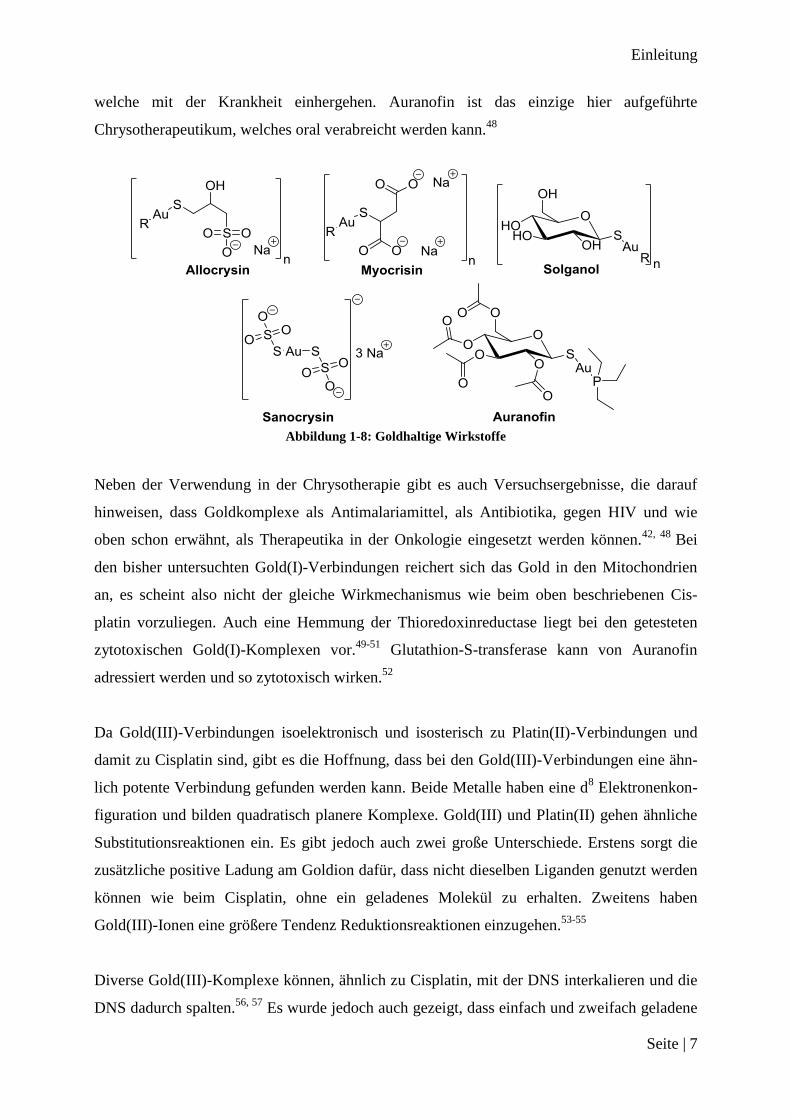
Dieser Erfolg von Salvarsan führt dazu, dass die strukturelle Vielseitigkeit der Metallkomple-
xe auch in anderen Bereichen der Medizin zur gezielten Entwicklung von metallhaltigen
Chemotherapeutika genutzt wird. So werden unter anderem Ruthenium-, Rhodium-, Rheni-
um- und Osmiumverbindungen als onkologische Chemotherapeutika getestet und etliche die-
ser Komplexe zeigen vielversprechende Resultate in Zelltests.32, 33
Abbildung 1-4: Vielversprechende Übergangsmetall Chemotherapeutika

Einleitung
Seite | 5
Einige dieser Verbindungen wurden auch schon in klinischen Studien getestet, was die Frage
nach dem Wirkmechanismus dieser Verbindungen aufwirft. Die Zytostatika wirken nicht alle
auf dieselben Zellbestanteile des malignomen Gewebes und der Mechanismus ist in allen Fäl-
len schwer aufzuklären. Einige der Verbindungen scheinen gegen Proteine im Zytosol zu wir-
ken, andere Verbindungen wirken gegen Zellorganellen, wie zum Beispiel den Mitochondrien
und bei einigen Verbindungen wirkt die Freisetzung von Kohlenstoffmonoxid. Sehr häufig
lässt sich eine Interkalation der Verbindungen mit der DNS beobachten, welche dann eine
Duplikation der DNS verhindert. Diese Hinderung stört den Stoffwechsel der Zelle und es
kommt Final zur Apoptose.34-36
Cisplatin gehört zu den anorganischen Zytostatika, die mit der DNS interkalieren und so die
Apoptose einleitet. Es eines der gebräuchlichsten anorganische Zytostatika und hat seine Zu-
lassung schon 1978 von der FDA (Food and Drug Administration) erhalten.37
Abbildung 1-5: Cisplatin
Es dringt bis in den Zellkern vor, die Chloridliganden werden ausgetauscht und das Platin
bindet an zwei Purine, die zu benachbarten Guanosinen der DNS gehört. Dies stört die Fal-
tung, Transkription und Duplikation der DNS und führt am Ende zur Apoptose.38
Der große Erfolg von Cisplatin führte zur Entwicklung weiterer Platin basierter Zytostatika,
von den Oxaliplatin39
und Carboplatin40
eine weltweite Zulassung erhalten haben. Bei beiden
Verbindungen ist der Wirkmechanismus anders als bei Cisplatin, mit diesen beiden Verbin-
dungen können karzinogene Gewebe behandelt werden, welche nicht auf Cisplatin reagieren.
Abbildung 1-6: links Oxaliplatin und rechts Carboplatin
Es gibt jedoch weitere Gewebe, welche auf bisher keine Therapie ansprechen. Gegen diese
Tumore werden weitere Cisplatinderivate entwickelt. Zu diesen Neuentwicklungen gehören
unter anderem das Pyriplatin und das Phenanthriplatin. Beide Verbindungen werden gerade
auf ihre Wirksamkeit und Verträglichkeit getestet.41

Einleitung
Seite | 6
Pt
H3N NH3
Cl N
Pt
H3N NH3
Cl N
Abbildung 1-7: links Pyriplatin und rechts Phenanthriplatin
Eine weitere Behandlungsmöglichkeit von resistentem karzinogenem Gewebe ist es, das zyto-
toxische wirksame Metall zu wechseln. Zu den potentiellen Metallen gehört das Gold, wel-
ches mit der Ordnungsnummer 79 nur eine Ordnungsnummer weiter im Periodensystem steht
als das Platin. Gold wird schon seit der Antike eine heilende Wirkung nachgesagt. Diese
nachgesagte Wirksamkeit zieht sich durch alle Kulturen, jedoch lässt sich kein wissenschaftli-
cher Effekt bei der Anwendung von gediegenem Gold gegen irgendeine Krankheit nachwei-
sen.42
Gold Legierungen werden lediglich für Prothesen wie zum Beispiel in der Zahnmedizin
verwendet.43
Für eine Anwendung als Chemotherapeutikum kommt vornämlich in Komplexen gebundenes
Gold in Frage. Die stabilen Oxidationsstufen des Goldes sind (I) und (III). Gold(I)-Verbin-
dungen haben in der Regel zwei linear gebundene Liganden, Gold(III)-Komplexe besitzt zu-
meist vier quadratisch planar gebundene Liganden. Als Liganden kommen häufig Halogenide,
Phosphine, Thiole, Thioether, Alkohole, das Stickstoffatom von N-heterocyclen und verschie-
den Bindungen zu Kohlenstoffatomen vor. Zu den Kohlenstoffbindungen gehört die Bindung
an ein Kohlenstoffatom eines konjugierten Systems, an Carbene sowie end-on und side-on an
Alkine.44, 45, 46
Das erste von Robert Koch entdeckte auf Gold basierende Chemotherapeutikum, ist eine klas-
sische anorganische Verbindung. Koch konnte zeigen, dass Kaliumdicyanoaurat(I) eine bakte-
riostatische Wirkung hat.47
Als Chemotherapeutikum werden Goldverbindungen gegen rheumatische Arthritis eingesetzt.
Zu den verwendeten Verbindungen gehört Aurothioglucose (Solganol), Aurothiomalat
(Myocrisin), Aurothiosulfat (Sanocrysin), Aurothiopropanolsulfonat (Allocrysin) und
(3,4,5,6-Tetra-O-acetyl-1-thio-β-D-glucopyranosato)(trieethylphosphan)gold(I) (Auranofin).
Rheumatische Arthritis kann zurzeit nicht geheilt werden, die antirheumatischen Goldkom-
plexe verlangsamen jedoch den Krankheitsverlauf und senken Knochen- und Knorpelschäden,
Einleitung
Seite | 7
welche mit der Krankheit einhergehen. Auranofin ist das einzige hier aufgeführte
Chrysotherapeutikum, welches oral verabreicht werden kann.48
Abbildung 1-8: Goldhaltige Wirkstoffe
Neben der Verwendung in der Chrysotherapie gibt es auch Versuchsergebnisse, die darauf
hinweisen, dass Goldkomplexe als Antimalariamittel, als Antibiotika, gegen HIV und wie
oben schon erwähnt, als Therapeutika in der Onkologie eingesetzt werden können.42, 48
Bei
den bisher untersuchten Gold(I)-Verbindungen reichert sich das Gold in den Mitochondrien
an, es scheint also nicht der gleiche Wirkmechanismus wie beim oben beschriebenen Cis-
platin vorzuliegen. Auch eine Hemmung der Thioredoxinreductase liegt bei den getesteten
zytotoxischen Gold(I)-Komplexen vor.49-51
Glutathion-S-transferase kann von Auranofin
adressiert werden und so zytotoxisch wirken.52
Da Gold(III)-Verbindungen isoelektronisch und isosterisch zu Platin(II)-Verbindungen und
damit zu Cisplatin sind, gibt es die Hoffnung, dass bei den Gold(III)-Verbindungen eine ähn-
lich potente Verbindung gefunden werden kann. Beide Metalle haben eine d8 Elektronenkon-
figuration und bilden quadratisch planere Komplexe. Gold(III) und Platin(II) gehen ähnliche
Substitutionsreaktionen ein. Es gibt jedoch auch zwei große Unterschiede. Erstens sorgt die
zusätzliche positive Ladung am Goldion dafür, dass nicht dieselben Liganden genutzt werden
können wie beim Cisplatin, ohne ein geladenes Molekül zu erhalten. Zweitens haben
Gold(III)-Ionen eine größere Tendenz Reduktionsreaktionen einzugehen.53-55
Diverse Gold(III)-Komplexe können, ähnlich zu Cisplatin, mit der DNS interkalieren und die
DNS dadurch spalten.56, 57
Es wurde jedoch auch gezeigt, dass einfach und zweifach geladene

Einleitung
Seite | 8
Gold(III)-Verbindungen an nucleophilen Heteroatome von L-Histidin, Inosin, Inosin-5′-
monophosphat und Guanosin-5′-monophosphat binden können. Moleküle wie diese kommen
häufig in der Zelle vor und verhindern eine Migration der Gold(III)-Komplexe zum Zell-
kern.58
Auch ein Wirken auf Selenoproteine, thiolhaltige Proteine, die Thioredoxinreductase
und Mitochondrien werden als möglicher Wirkort für Gold(III)-Verbindungen diskutiert. Dies
wäre analog zu Gold(I)-Komplexen.42, 48, 59
Möglich ist auch eine Wirkung auf das Proteasom
oder die Veränderung verschiedener Kinasen.60
Aktuell werden Gold(III)-Komplexe mit N-
heterocyclischen-, S-heterocyclischen-, Carben-, Phosphin- und Chloridliganden als antiproli-
ferative Wirkstoffe getestet, wobei die Häufigkeit der N-heterocyclischen Liganden besonders
auffällt.33, 61-66
Dieses besonders gute Bindungsverhalten wurde genutzt um das N-heterocyclische, häufig in
der Biologie verwendete, Porphyrin mit einem Gold(III)-Komplexe zu einem Biokonjugaten
umzusetzen und so einen weiteren potenten Tumorwirkstoff zu erzeugen.67
Dabei sind Bio-
konjugate Verbindungen aus mindestens zwei Molekülen, die die Eigenschaft der beiden ein-
zelnen Moleküle in der neuen Verbindung vereinigen. Eins der Moleküle muss eine biologi-
sche Funktion innehaben.68
Ein weiteres biologisch aktives Moleküle können Aminosäureketten sein, die mit dem
Gold(III)-Komplexe verbunden werden können, um ein neues Biokonjugat zu erzeugen. Zur
Gewinnung dieser Aminosäureketten gibt es zwei Möglichkeiten:
Erstens gibt es die heute übliche Technik zur Gewinnung von Aminosäureketten durch re-
kombinante Proteine. Diese ermöglicht über Genveränderungen gezielt alle denkbaren Protei-
ne zu erzeugen, bei welchen die Peptide schon zu einem Protein gefalten sind. Jedoch ist die
Darstellung von Proteinen auf diese Weise teuer, aufwendig und zeitintensiv.69-71
Die zweite vorher entwickelte, günstigere und verhältnismäßig schnelle Möglichkeit zeigte
Merrifield 1963 auf und erhielt 1984 hierfür den Nobelpreis.72
Merrifield entwickelte die
Möglichkeit kurze Peptide an einem, in den verwendeten Lösungsmitteln, unlöslichem Harz
darzustellen.73
Die Sequenz wird Baustein für Baustein aufgebaut. Um ein Peptid mittels der
Festphasenpeptidsynthese (SPPS) darzustellen, wird das verwendete Harz in dem für die Syn-
these verwendete Lösungsmittel quellen gelassen. Es folgenden die Schritte Entschützung,
Waschung, Kopplung und wieder Waschung für jeden zu koppelnden Baustein. Sobald die

Einleitung
Seite | 9
gewünschte Kettenlänge erreicht ist, folgen Entschützung, Waschung, Abspaltung und das
Aufreinigen.74, 75
quellen des Harzes
entschützen
waschen
koppeln
waschen
entschützen
nächster Baustein
waschen
abspalten
aufreinigen Abbildung 1-9: Schritte in der SPPS
Von großer Bedeutung für die Festphasenpeptidsynthese sind orthogonale Reaktionssysteme.
Eine Orthogonalität ist dann gegeben, wenn es zwei oder mehr voneinander unabhängigen
Funktionalitäten gibt, an der Reaktionen durchgeführt werden können. Die Reaktionen müs-
sen unabhängig und in beliebiger Reihenfolge voneinander durchgeführt werden können. Bei
der Festphasenpeptidsynthese werden häufig Säure/Base Reaktionen genutzt. Das Entschüt-
zen wird dann mittels einer Base durchgeführt, das Abspalten mittels einer Säure. Es kann
auch genau anders herum vorgegangen werden. Die Seitenketten sind mit Schutzgruppen ge-
schützt, welche erst unter den Abspaltbedingungen gespalten werden oder sogar orthogonal
zum Entschützen und Abspalten geschützt sind. Die Seitenschutzgruppen sind während des
gesamten Peptidaufbaus permanent am Harz geschützt, im Gegensatz zum temporär geschütz-
ten Peptidende, an dem die Schutzgruppe vor jedem Kopplungsschritt abgespalten wird.76-78
Mit der SPPS können Peptide aufgebaut werden, die die Fähigkeit haben, die Plasmamembran
von Zellen zu durchdringen. Diese Zellmembran durchdringenden Peptide (CPPs) sind fast
alle positiv geladen, um mit negativ geladen Glykosaminoglykanen und vor allem den
Phospholipiden in der Zellmembran wechselwirken zu können. Durch diese Wechselwirkung
haben die Peptide die Eigenschaft in die Zelle zu gelangen, wobei der Mechanismus nicht
genau aufgeklärt ist. Es sind eine Vielzahl von Sequenzen mit dieser Eigenschaft bekannt,
dabei sind alle Sequenzen zwischen 5-30 Aminosäuren lang, zum Beispiel ist eine der ein-

Einleitung
Seite | 10
fachsten CPPs das Polyarginin79
. Die Fähigkeit der Sequenzen in die Zelle zu gelangen und
daneben noch weitere Stoffe mit in die Zelle zu transportieren, wird heute genutzt Plasmid-
DNS, Oligonucleotide, RNS-Fragmente, Proteine, Peptide, Kontrastmittel, Wirkstoffe und
sogar Nanopartikel, welche sich in Mizellen befinden, in die Zelle zu befördern. Die Sequen-
zen selber haben keinen Effekt auf die Zelle, sie sind nicht selektiv, sondern wandern durch
jede Zellmembran.80-83
Um bei den CPPs eine Selektivität zu erhalten, werden diesem Peptid weitere Aminosäuren
angehängt. Diese neuen Peptidabschnitte sind spezifisch für die Zielzelle. Bei diesem heute
gängigen Vorgehen, ist also ein Abschnitt des Peptides für die Erkennung der Zielzelle zu-
ständig, der andere Abschnitt für den Transport in die Zelle. An das gesamte Peptid können
dann die oben schon erwähnten Substanzen gebunden werden und in die Zelle transportiert
werden.84, 85
Der CPP-Abschnitt sorgt jedoch weiterhin für einen Transport auch in andere
Zellen.86
Eine Erweiterung dieser Möglichkeit mit CPPs selektiv karzinogene Zellen zu markieren,
haben Jiang et al. 2004 vorgestellt, als sie ein Fluorophoren mit einem Peptid verknüpften.
Das Peptid bestand aus einem positiv geladen CPP-Abschnitt, dann einem durch die Matrix-
Metalloprotease 2 (MMP2) schneidbaren Stück und einem negativ geladenem Abschnitt, um
den CPP-Abschnitt zu inaktivieren. Um das CPP zu aktivieren, musste der negativ geladene
Abschnitt durch Schneiden bei der MMP-Schnittsequenz von diesem CPP-Abschnitt getrennt
werden.87
Bei der gerade beschrieben Arbeit wurde genutzt, dass MMPs häufig bei karzinogenen Zellen
überexprimiert werden. Gebärmutter-, Lungen-, Prostata- und Brusttumore werden von Zellen
ausgelöst, welche MMP2 (Gelatinase-A) und MMP9 (Gelatinase-B) überexprimieren.88-91
Diese beiden MMPs scheinen von entscheidender Bedeutung für die Metastasenbildung zu
sein. Gelatinasen sind für die finale Degeneration der Fasern des Kollagens verantwortlich.92
MMP2 ist die am häufigsten überexprimierte MMP bei allen untersuchten carcinogen Zel-
len.93
Sequenzen, die von MMP2 gespalten werden, können mit sechs Aminosäuren recht
kurz sein.94
MMPs gehören zu einer Familie von strukturverwandten, Zink enthaltenden
Endopeptidasen. Die Gesamtheit aller MMPs ist dadurch in der Lage nahezu alle Verbindun-
gen der Extrazellularmatrix (ECM) abzubauen. MMPs spielen bei gesunden Zellen eine wich-
tige Rolle im physologischen Abbau der ECM, sind an der Gewebemorphogenese, der Gewe-

Einleitung
Seite | 11
bereparatur und der Angiogenese maßgeblich beteiligt.95
MMPs sind jedoch wie oben schon
angedeutet bei pathologischen Veränderungen, die mit einem massiven Abbau der ECM im
Zusammenhang stehen, von großer Bedeutung. Hier zu zählen: rheumatische Arthritis, Arth-
rose, Lungenemphysem, Atherosklerose, Multiple Sklerose, Parodontitis, autoimmunen Bla-
senbildung der Epidermis und bei der Tumorausbreitung und der Metastasenbildung. 95-98
Es gibt aktuell viele Versuche das Wissen über die Beteiligung von MMPs bei der Krank-
heitsentstehung zu nutzen. So werden Inhibitoren für MMPs entwickelt, die sowohl bei Ent-
zündungen als auch bei Tumoren in klinischen Studien getestet werden.99-102
Weniger häufig
werden Moleküle, die von MMPs geschnitten werden sollen, eingesetzt um einen Wirkstoff
freizusetzen.103
So wird der Artikel von Jiang et al.87
bis heute als Beispiel angegeben, wie
eine größere Selektivität von CPPs erreicht werden kann.104, 105
Von Goun et al. wurde das
Konzept adaptiert und CPPs für den Wirkstofftransport genutzt, jedoch wurden keine durch
eine MMP schneidbare Sequenz gewählt, sondern dies sollte durch ein Prostata spezifisches
zelluläres Antigen erfolgen.106
Das von Jiang et al.87
vorgestellte Konzept wurde nur von
Nguyen et al. Wieder aufgegriffen und als Möglichkeit der Bildgebung bei Tumoroperationen
vorgestellt.107
Somit ist es eine interessante neue Idee das Konzept von Jiang et al.87
wieder
aufzugreifen und die Peptidsequenz nicht mit einer Verbindung zur Bildgebung auszustatten,
sondern das Peptid mit einem Gold(III)-Metallkomplex zu einem Biokonjugat zu verknüpfen
und somit die Möglichkeit zu schaffen, den Metallkomplex gezielt gegen karzinogenes Ge-
webe einzusetzen.
Aufgabenstellung
Seite | 12
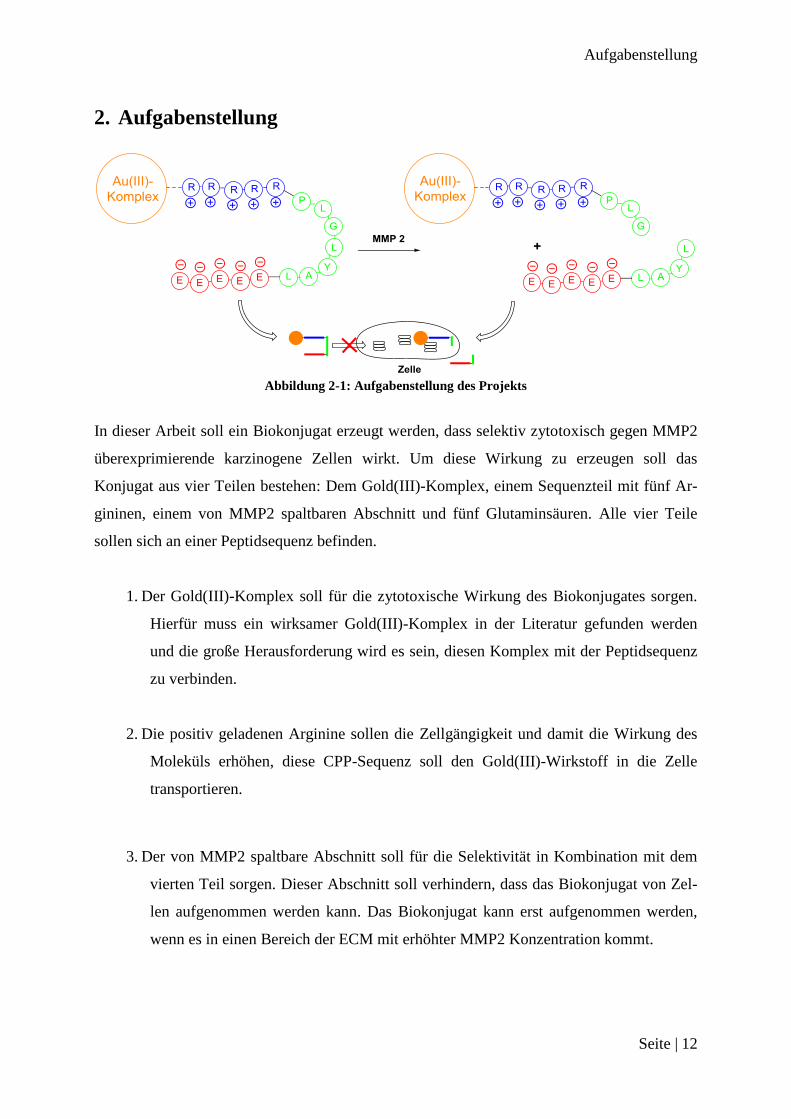
2. Aufgabenstellung
Abbildung 2-1: Aufgabenstellung des Projekts
In dieser Arbeit soll ein Biokonjugat erzeugt werden, dass selektiv zytotoxisch gegen MMP2
überexprimierende karzinogene Zellen wirkt. Um diese Wirkung zu erzeugen soll das
Konjugat aus vier Teilen bestehen: Dem Gold(III)-Komplex, einem Sequenzteil mit fünf Ar-
gininen, einem von MMP2 spaltbaren Abschnitt und fünf Glutaminsäuren. Alle vier Teile
sollen sich an einer Peptidsequenz befinden.
1. Der Gold(III)-Komplex soll für die zytotoxische Wirkung des Biokonjugates sorgen.
Hierfür muss ein wirksamer Gold(III)-Komplex in der Literatur gefunden werden
und die große Herausforderung wird es sein, diesen Komplex mit der Peptidsequenz
zu verbinden.
2. Die positiv geladenen Arginine sollen die Zellgängigkeit und damit die Wirkung des
Moleküls erhöhen, diese CPP-Sequenz soll den Gold(III)-Wirkstoff in die Zelle
transportieren.
3. Der von MMP2 spaltbare Abschnitt soll für die Selektivität in Kombination mit dem
vierten Teil sorgen. Dieser Abschnitt soll verhindern, dass das Biokonjugat von Zel-
len aufgenommen werden kann. Das Biokonjugat kann erst aufgenommen werden,
wenn es in einen Bereich der ECM mit erhöhter MMP2 Konzentration kommt.

Aufgabenstellung
Seite | 13
4. Die negativ geladenen Glutaminsäuren sollen sich an die positiv geladenen Arginine
anlagern. Durch die Anlagerung soll die gesamte Verbindung neutral geladen sein.
Erst wenn der dritte Teil der Verbindung von MMP2 gespalten worden ist, trennt
sich der Sequenzteil mit den Glutaminsäuren vom Rest des Biokonjugats. Der Rest
kann dann von Zellen aufgenommen werden.

Ergebnisse
Seite | 14
3. Ergebnisse
Der Gold(III)-Komplex
Analog zu dem bekanntesten anorganischen Chemotherapeutikum Cisplatin,108
wurde nach
einer zytotoxischen Gold(III)-Verbindung gesucht, welche in Tests schon ihr Potential gegen
karzinogene Zelllinien gezeigt hatte.109
Für die Zelltests muss die Verbindung stabil gegen-
über Wasser als Lösungsmittel des Organismus sein, der Komplex darf nicht leicht oxidier-
oder reduzierbar sein, stabil gegenüber Sauerstoff sein und die Verbindung sollte einen gut
modifizierbaren Liganden besitzen.110, 111
Abbildung 3-1: Umsetzung von Hydrogentetrachloroaurat(III)hydrat mit 2-Phenylpyridin zu
2-Phenylpyridin-1-gold(III)trichlorid
Von Constable und Leese wurde 1989 die Synthese von der Verbindung 2-Phenylpyridin-
1,1´-gold(III)dichlorid 2 [AuPhPyCl2] beschrieben.112
Der Komplex 2 hat die oben geforder-
ten Eigenschaften.
Für die Synthese dieser Verbindung 2 wurde Hydrogentetrachloroaurat(III)hydrat und 2-
Phenylpyridin in Wasser und Acetonitril oder Ethanol zusammen gegeben, so dass sich die
Gold-Stickstoff Bindung ausbildete und 2-Phenylpyridin-1-gold(III)trichlorid 1 erhalten wur-
de. Verbindung 1 wurde für die Cyclometallierung in Wasser und Acetonitril in der Siedehit-
ze umgesetzt.
Ergebnisse
Seite | 15
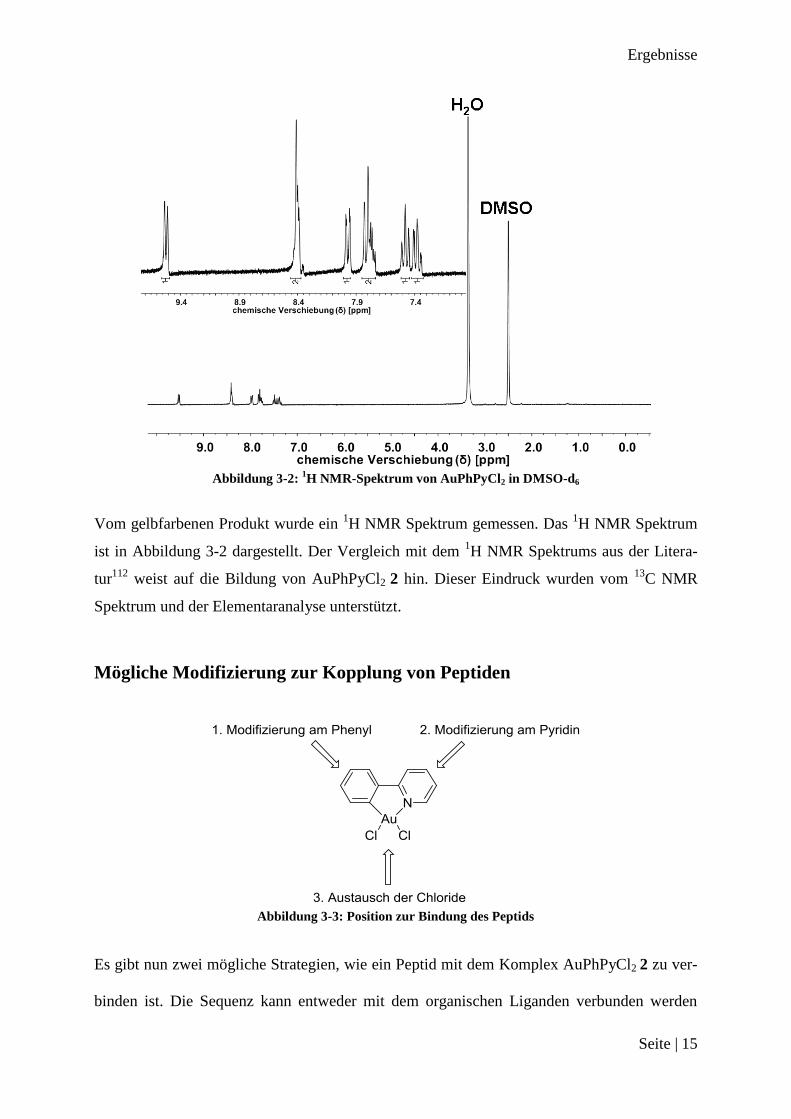
Abbildung 3-2:
1H NMR-Spektrum von AuPhPyCl2 in DMSO-d6
Vom gelbfarbenen Produkt wurde ein 1H NMR Spektrum gemessen. Das
1H NMR Spektrum
ist in Abbildung 3-2 dargestellt. Der Vergleich mit dem 1H NMR Spektrums aus der Litera-
tur112
weist auf die Bildung von AuPhPyCl2 2 hin. Dieser Eindruck wurden vom 13
C NMR
Spektrum und der Elementaranalyse unterstützt.
Mögliche Modifizierung zur Kopplung von Peptiden
Abbildung 3-3: Position zur Bindung des Peptids
Es gibt nun zwei mögliche Strategien, wie ein Peptid mit dem Komplex AuPhPyCl2 2 zu ver-
binden ist. Die Sequenz kann entweder mit dem organischen Liganden verbunden werden

Ergebnisse
Seite | 16
oder durch Austausch der Chloridliganden direkt mit dem Gold(III)-Ion gekoppelt werden. Da
eine zu Cisplatin analoge Verbindung generiert werden sollte und es bei diesem Chemothera-
peutikum zwei gebundene Chloride am Metallion gibt, die für dessen Wirksamkeit wichtig
sind, sollte zuerst der Ligand modifiziert werden. Der Ligand kann sowohl am Phenylring, als
auch am Pyridinring modifiziert werden.
Funktionalisierung des Phenylringes
Zuerst sollte der Phenylring des Liganden von AuPhPyCl2 2 modifiziert werden. Für diese
Modifizierung des Liganden steht eine ganze Reihe von organischen Synthesen zur Verfü-
gung.113-115
Zum Beispiel kann eine Carbonsäure am Phenylring eingefügt werden.
Abbildung 3-4: Syntheseroute zu 2-Phenylpyridin-3´-säure-1,1´gold(III)dichlorid
Zur Darstellung der modifizierten Verbindung wurde eine Suzuki-Kupplung zwischen 2-
Brompyridin und Carboxybenzolboronsäure durchgeführt.116
Das so erhaltene Produkt 3 wur-
de in Ethanol gelöst und mit Hydrogentetrachloroaurat(III)-Lösung zusammen gegeben. Von
dem aus dieser Reaktion erhalten gelbfarbenen Rückstand wurde ein 1H NMR Spektrum auf-
genommen.
Ergebnisse
Seite | 17
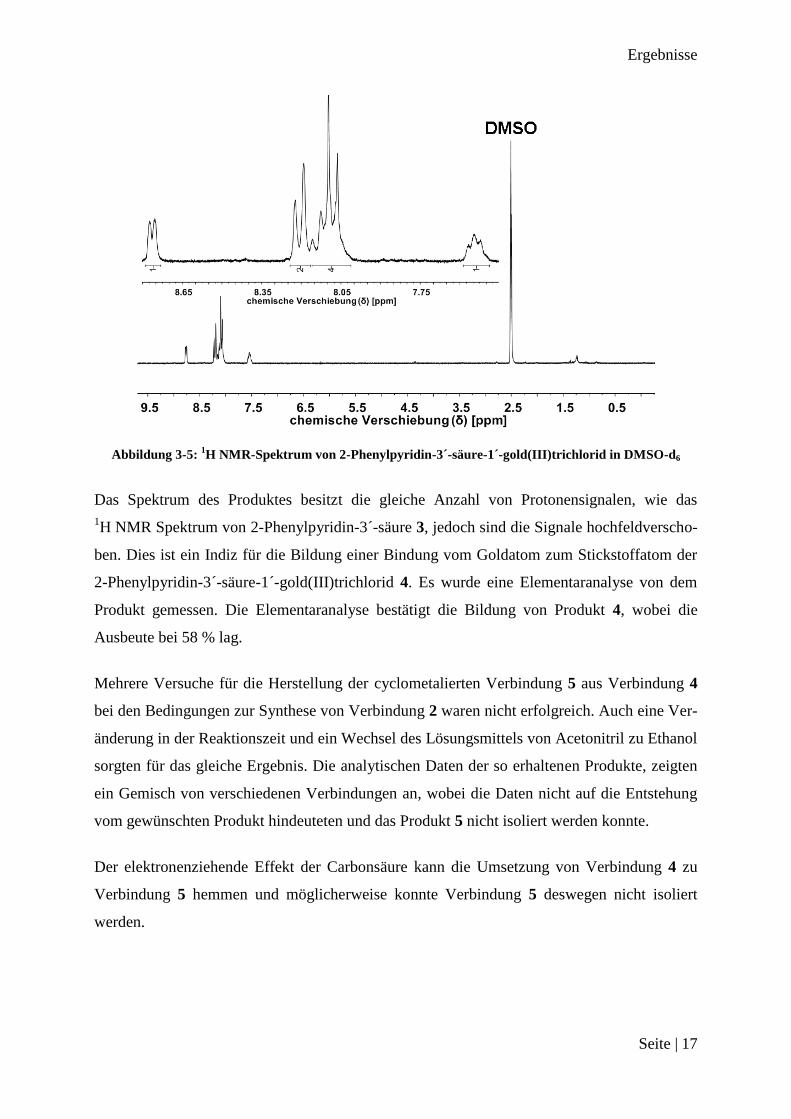
Abbildung 3-5: 1H NMR-Spektrum von 2-Phenylpyridin-3´-säure-1´-gold(III)trichlorid in DMSO-d6
Das Spektrum des Produktes besitzt die gleiche Anzahl von Protonensignalen, wie das
1H NMR Spektrum von 2-Phenylpyridin-3´-säure 3, jedoch sind die Signale hochfeldverscho-
ben. Dies ist ein Indiz für die Bildung einer Bindung vom Goldatom zum Stickstoffatom der
2-Phenylpyridin-3´-säure-1´-gold(III)trichlorid 4. Es wurde eine Elementaranalyse von dem
Produkt gemessen. Die Elementaranalyse bestätigt die Bildung von Produkt 4, wobei die
Ausbeute bei 58 % lag.
Mehrere Versuche für die Herstellung der cyclometalierten Verbindung 5 aus Verbindung 4
bei den Bedingungen zur Synthese von Verbindung 2 waren nicht erfolgreich. Auch eine Ver-
änderung in der Reaktionszeit und ein Wechsel des Lösungsmittels von Acetonitril zu Ethanol
sorgten für das gleiche Ergebnis. Die analytischen Daten der so erhaltenen Produkte, zeigten
ein Gemisch von verschiedenen Verbindungen an, wobei die Daten nicht auf die Entstehung
vom gewünschten Produkt hindeuteten und das Produkt 5 nicht isoliert werden konnte.
Der elektronenziehende Effekt der Carbonsäure kann die Umsetzung von Verbindung 4 zu
Verbindung 5 hemmen und möglicherweise konnte Verbindung 5 deswegen nicht isoliert
werden.

Ergebnisse
Seite | 18
Einfügung einer Alkylkette
Zwei Wege wurden probiert, um den elektronenziehenden Effekt der Carbonsäure auf den
Aromaten zu senken. In beiden Wegen sollten einige Kohlenstoffatome zwischen dem Aro-
maten und der Carbonsäure eingefügt werden. Im ersten Weg sollte sich eine freie Carbonsäu-
re am Liganden befinden, beim zweiten Weg sollte die Carbonsäure methylgeschützt sein.
Der Vergleich zwischen geschützter und nicht geschützter Carbonsäure sollte den Einfluss der
Säure auf die Cyclometallierung zeigen.
Abbildung 3-6: Syntheseweg für 2-Phenylpyridin-3´-propansäure-1,1´-gold(III)dichorid
Für den ersten Weg wurden 4-(2-Pyridin)phenyl-3-aldehyd und Malonsäure in Pyridin gelöst
und Piperidin als Base zugegeben, um 2-Phenylpyridin-3´-acrylsäure 6 zu erhalten.117
Die
Reduktion der Doppelbindung der Alkylkette mit Wasserstoff und Palladium auf Aktivkohle,
sollte das konjugierte System von Verbindung 6, welches über den ganzen Liganden bis zur
Carbonsäure reichte, unterbrechen. Die Carbonsäure hätte durch dieses konjugierte System
einen Einfluss auf die Ausbildung der Gold-Kohlenstoffbindung haben können. Die so erhal-
tene 2-Phenylpyridin-3´-propansäure 7 sollte nun mit dem Stickstoffatom zum Gold(III)-Ion
binden und wurde in Acetonitril gelöst, mit Hydrogentetrachloroaurat(III) in Wasser gelöst,
zusammen gegeben. Das erhaltene Rohprodukt wurde säulenchromatographisch getrennt.
Von dem gelbfarbenen Produkt wurde ein 1H NMR Spektrum gemessen.
Ergebnisse
Seite | 19
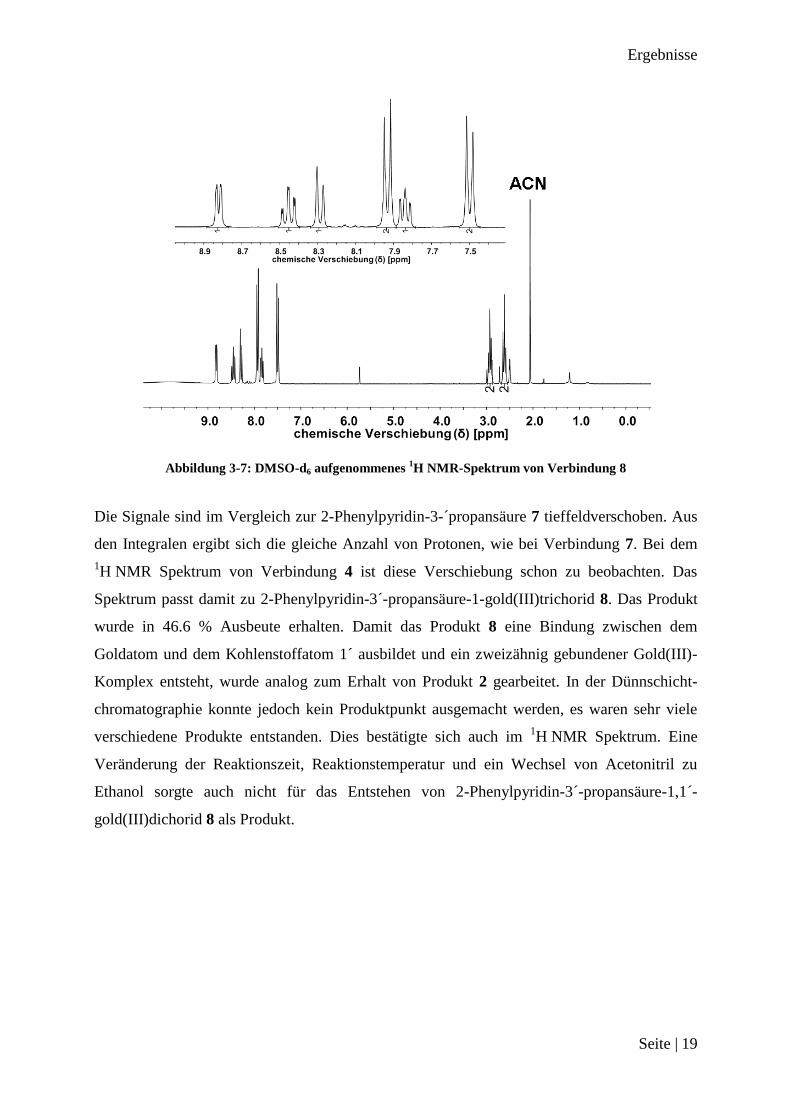
Abbildung 3-7: DMSO-d6 aufgenommenes 1H NMR-Spektrum von Verbindung 8
Die Signale sind im Vergleich zur 2-Phenylpyridin-3-´propansäure 7 tieffeldverschoben. Aus
den Integralen ergibt sich die gleiche Anzahl von Protonen, wie bei Verbindung 7. Bei dem
1H NMR Spektrum von Verbindung 4 ist diese Verschiebung schon zu beobachten. Das
Spektrum passt damit zu 2-Phenylpyridin-3´-propansäure-1-gold(III)trichorid 8. Das Produkt
wurde in 46.6 % Ausbeute erhalten. Damit das Produkt 8 eine Bindung zwischen dem
Goldatom und dem Kohlenstoffatom 1´ ausbildet und ein zweizähnig gebundener Gold(III)-
Komplex entsteht, wurde analog zum Erhalt von Produkt 2 gearbeitet. In der Dünnschicht-
chromatographie konnte jedoch kein Produktpunkt ausgemacht werden, es waren sehr viele
verschiedene Produkte entstanden. Dies bestätigte sich auch im 1H NMR Spektrum. Eine
Veränderung der Reaktionszeit, Reaktionstemperatur und ein Wechsel von Acetonitril zu
Ethanol sorgte auch nicht für das Entstehen von 2-Phenylpyridin-3´-propansäure-1,1´-
gold(III)dichorid 8 als Produkt.

Ergebnisse
Seite | 20
Methylester
Abbildung 3-8: Schema für 2-Phenylpyridin-3´-propansäuremethylester-1,1´-gold(III)trichorid
Für den zweiten Weg wurde mit einer Wittig-Reaktion in Tetrahydrofuran (Essigsäureme-
thylester)triphenylphosphoniumchlorid und 4-(2-Pyridin)phenyl-3-aldehyd mit Hilfe von
Kaliumhexamethyldisilizan umgesetzt. Die orangefarbene Reaktionssuspension wurde säulen-
chromatographisch aufgetrennt und Phenylpyridin-3´-acrylat 10 erhalten. Das konjugierte
System von Verbindung 10 wurde analog zum Erhalt von Verbindung 7 in einer Wasserstoff-
atmosphäre zusammen mit Palladium auf Aktivkohle in Ethanol gerührt und 2-Phenylpyridin-
3´-propansäuremethylester 11 erhalten. Dieser Methylester 11 wurde mit Hydrogentetra-
chloroaurat(III)hydrat umgesetzt. Nach der Reinigung der Reaktionsmischung wurde ein Pro-
dukt erhalten. Von diesem Produkt wurde ein 1H NMR Spektrum gemessen.
Ergebnisse
Seite | 21
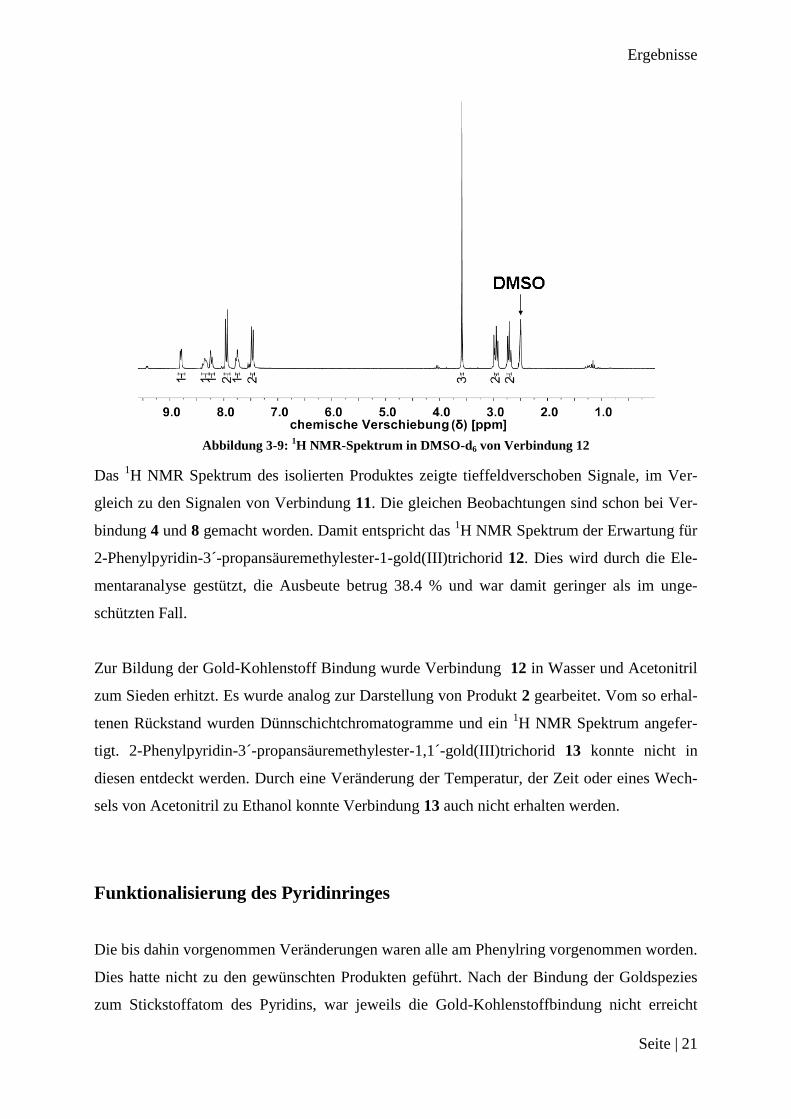
Abbildung 3-9:
1H NMR-Spektrum in DMSO-d6 von Verbindung 12
Das 1H NMR Spektrum des isolierten Produktes zeigte tieffeldverschoben Signale, im Ver-
gleich zu den Signalen von Verbindung 11. Die gleichen Beobachtungen sind schon bei Ver-
bindung 4 und 8 gemacht worden. Damit entspricht das 1H NMR Spektrum der Erwartung für
2-Phenylpyridin-3´-propansäuremethylester-1-gold(III)trichorid 12. Dies wird durch die Ele-
mentaranalyse gestützt, die Ausbeute betrug 38.4 % und war damit geringer als im unge-
schützten Fall.
Zur Bildung der Gold-Kohlenstoff Bindung wurde Verbindung 12 in Wasser und Acetonitril
zum Sieden erhitzt. Es wurde analog zur Darstellung von Produkt 2 gearbeitet. Vom so erhal-
tenen Rückstand wurden Dünnschichtchromatogramme und ein 1H NMR Spektrum angefer-
tigt. 2-Phenylpyridin-3´-propansäuremethylester-1,1´-gold(III)trichorid 13 konnte nicht in
diesen entdeckt werden. Durch eine Veränderung der Temperatur, der Zeit oder eines Wech-
sels von Acetonitril zu Ethanol konnte Verbindung 13 auch nicht erhalten werden.
Funktionalisierung des Pyridinringes
Die bis dahin vorgenommen Veränderungen waren alle am Phenylring vorgenommen worden.
Dies hatte nicht zu den gewünschten Produkten geführt. Nach der Bindung der Goldspezies
zum Stickstoffatom des Pyridins, war jeweils die Gold-Kohlenstoffbindung nicht erreicht

Ergebnisse
Seite | 22
worden, wenn der Phenylring des Kohlenstoffatoms einen weiteren Liganden trug. Eine wei-
tere Möglichkeit war die Modifizierung des Pyridinringes. Es sollte so geprüft werden, ob
eine Veränderung am Pyridinring einen weniger starken Einfluss auf die Eigenschaften des
Phenylringes hatte. Aus diesem Grund sollte das Phenylpyridin mit einem Bromatom an der
vierten Position des Pyridinring dargestellt werden. Dieser Ligand sollte dann mit Hydrogen-
tetrachloroaurat(III) umgesetzt werden. Analog zu Verbindung 2 sollte dann 4-Brom-2-Phe-
nylpyridin-1,1´gold(III)dichlorid 16 erhalten werden. Das Bromatom ist eine gute Abgangs-
gruppe und kann für eine Vielzahl von weiteren Reaktionen genutzt werden. Das Bromatom
kann zum Beispiel in einer nucleophilen bimolekularen Substitution durch ein Azid ausge-
tauscht werden.118, 119
Das Azid könnte dann in einer [3+2] Cycloaddition mit einem Peptid
mit terminalem Alkin gekoppelt werden.120
Das Bromatom eignet sich ebenfalls für eine me-
tallorganische C-C Verknüpfungsreaktion.121, 122
Abbildung 3-10: Syntheseroute zu 4-Brom-2-Phenylpyridin-1,1´-gold(III)dichorid
Für die Darstellung des neuen Liganden wurde ein Grignardreagenz aus Magnesiumspänen
und Brombenzol in Tetrahydrofuran hergestellt. Diese Mischung wurde langsam in eine Mi-
schung aus 4-Brompyridin in Tetrahydrofuran gegeben. Nach der Aufarbeitung wurde 4-
Brom-2-Phenylpyridin 14 erhalten.
Ligand 14 wurde in Acetonitril gelöst und mit Hydrogentetrachloroaurat(III)hydrat umge-
setzt. Durch Aufarbeitung der Reaktion wurde ein Feststoff erhalten. Von diesem wurde ein
1H NMR-Spektrum aufgenommen. Die chemischen Verschiebungen der
1H NMR-Signale
sind wie bei den Verbindungen 4, 8 und 11. Dies lässt vermuten, dass sich 4-Brom-2-Phenyl-
pyridin-1-gold(III)trichorid 15 in 27.6 % Ausbeute gebildet hatte.
Verbindung 15 wurde dann in einer Mischung aus 50 Vol.-% Wasser und 50 Vol.-% Aceto-
nitril bis zum Sieden erhitzt. Auf diese Weise sollte eine Bindung zwischen dem Goldatom
und Kohlenstoffatom 1´ entstehen. Jedoch konnte Verbindung 16 nicht isoliert werden. Ana-
lytische Daten zeigten, dass nur wieder Verbindung 15 isoliert werden konnte und kein Um-
satz bei der Reaktion erfolgt war.

Ergebnisse
Seite | 23
Transmetallierung
Bei den bisherigen Versuchen war kein Ligand erhalten worden, der das Gold(III)-Ion cyclo-
metalliert gebunden hatte. Pritchard et al. hat 2000 einen sehr ähnlichen Verbindung zu
AuPhPyCl2 2 veröffentlicht.123
Die dort veröffentlichte Gold(III)-Verbindung hat jedoch an-
stelle des Pyridinringes ein Quinolinsystem mit einer methylgeschützten Carbonsäure. Es
wird beschrieben, dass die so erhaltene Gold(III)-Verbindung zusätzlich noch eine erhöhte
Löslichkeit in Wasser aufweisen soll, was eine Anforderung an das hier verwendete System
war. Mit Hilfe einer Transmetallierung wurde diese Verbindung erhalten. Bei dieser
Transmetallierung ist das Kohlenstoffatom, welches die Bindung zu Goldatom eingehen soll,
an Quecksilber gebunden und so für die Reaktion aktiviert.123
Abbildung 3-11: 4-(4-(methoxycarbonyl)quinolin-2-yl)benzen-1-ylium-1,1´-gold(III)dichorid
Zur Darstellung dieser Verbindung wurde Quecksilber(II)acetonat zusammen mit 4-(4-(me-
thoxycarbonyl)quinolin-2-yl)benzen-1-ylium in Ethanol über Nacht in der Siedehitze gerührt.
Nach der Behandlung mit Lithiumchlorid und der Aufarbeitung der Reaktionslösung wurde
das Produkt mittels 1H und
13C NMR-Spektroskopie untersucht. Der Vergleich der Spektren
mit den Literaturspektren123
zeigt den Erhalt von Verbindung 17. Verbindung 17 in
Dichlormethan wurde zusammen mit einer äquimolaren Menge von Hydrogentetrachloro-
aurat(III)hydrat in Acetetonitril gerührt. Das erhalten Rohprodukt wurde mittels NMR-
Spektroskopie untersucht. Die Auswertung des 1H NMR-Spektrums ergab, dass es sich um
Verbindung 17 handelte. Verbindung 18 konnte nicht erhalten werden.
Verwendung von Bipyridin als Ligand
Da der Weg den Pyridinring zu modifizieren ebenfalls zu keiner für das Projekt nutzbare Ver-
bindung geführt hatte, wurde nach einer weiteren Alternative gesucht, den Liganden zu ver-
ändern. Die bisherigen Zielverbindungen waren nicht entstanden, da sich die Gold-
Ergebnisse
Seite | 24
Kohlenstoff-bindung nicht ausbildete. Um diese Widrigkeit nun zu umgehen, wurde ein
Bipyridinderivat genutzt. Dieses Bipyridinderivat besitzt einen Methylester, bei welchem
schon gezeigt werden konnte, dass er nach Verseifung an Peptide gekoppelt werden kann.124,
125
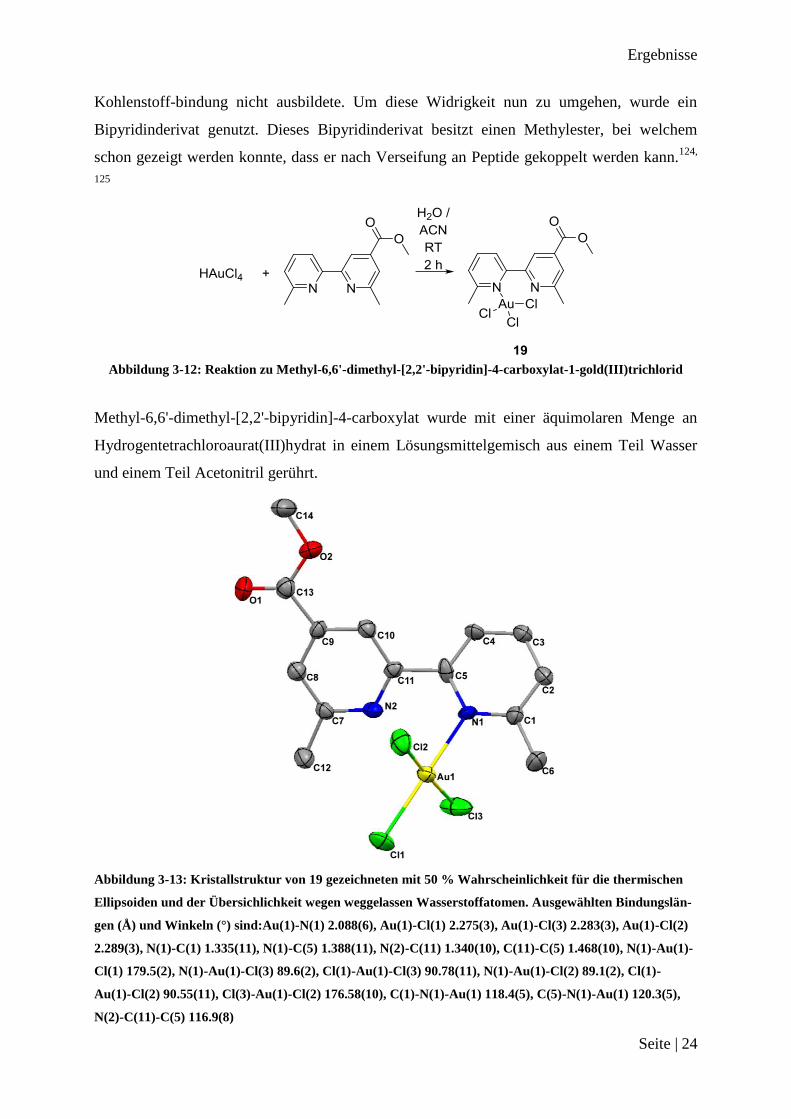
Abbildung 3-12: Reaktion zu Methyl-6,6'-dimethyl-[2,2'-bipyridin]-4-carboxylat-1-gold(III)trichlorid
Methyl-6,6'-dimethyl-[2,2'-bipyridin]-4-carboxylat wurde mit einer äquimolaren Menge an
Hydrogentetrachloroaurat(III)hydrat in einem Lösungsmittelgemisch aus einem Teil Wasser
und einem Teil Acetonitril gerührt.
Abbildung 3-13: Kristallstruktur von 19 gezeichneten mit 50 % Wahrscheinlichkeit für die thermischen
Ellipsoiden und der Übersichlichkeit wegen weggelassen Wasserstoffatomen. Ausgewählten Bindungslän-
gen (Å) und Winkeln (°) sind:Au(1)-N(1) 2.088(6), Au(1)-Cl(1) 2.275(3), Au(1)-Cl(3) 2.283(3), Au(1)-Cl(2)
2.289(3), N(1)-C(1) 1.335(11), N(1)-C(5) 1.388(11), N(2)-C(11) 1.340(10), C(11)-C(5) 1.468(10), N(1)-Au(1)-
Cl(1) 179.5(2), N(1)-Au(1)-Cl(3) 89.6(2), Cl(1)-Au(1)-Cl(3) 90.78(11), N(1)-Au(1)-Cl(2) 89.1(2), Cl(1)-
Au(1)-Cl(2) 90.55(11), Cl(3)-Au(1)-Cl(2) 176.58(10), C(1)-N(1)-Au(1) 118.4(5), C(5)-N(1)-Au(1) 120.3(5),
N(2)-C(11)-C(5) 116.9(8)

Ergebnisse
Seite | 25
Das gelbfarbenen Produkt wurde mittels 1H NMR-Spektroskopie und kristallographisch un-
tersucht. Die 1H NMR-Spektroskopie Untersuchungen ergibt eine ähnliche chemische Ver-
schiebung, wie sie schon bei 4, 8, 11 und 15 zu beobachten waren. Dies deutet auf den Erhalt
von Methyl-6,6'-dimethyl-[2,2'-bipyridin]-4-carboxylat-1-gold(III)trichlorid 19 hin. Die Kris-
tallstruktur bestätigt diesen Eindruck und zeigt keine Entstehung einer Bindung vom
Goldatom zum Stickstoffatom des substituierten Aromaten. Auch in diesem Fall verhindert
der Methylester die Reaktion am Aromaten.

Ergebnisse
Seite | 26
Peptide
Bindung der Peptide direkt zum Gold(III)-Zentrum
Nachdem die Modifizierung des Liganden nicht zu einer nutzbaren Verbindung geführt hatte,
gibt es nun die oben beschriebene Möglichkeit, die Peptide direkt an das an das Gold(III)-
Zentrum zu binden.
HSAB Prinzip
Es wurde nun eine Aminosäure gesucht, welche eine Seitenfunktionalität besitzt, die nach
dem HSAB Prinzip126
eine stabile Bindung zum Gold(III)-Ion ausbilden kann. Im HSAB
Prinzip werden Ionen nach ihrer Härte eingeteilt. Je nachdem wie groß der Ionenradius ist,
wie hoch die Ladung jeweils ist und wie gut das Ion polarisierbar ist, wird es zu den harten
oder weichen Ionen gezählt. Wenn ein Ion positiv geladen ist, wird es zu den Säuren gezählt.
Negativ geladene Ionen werden als Base betrachtet. Es wird dann angenommen, dass weiche
Säuren eher mit weichen Basen reagieren und harte Säuren mit harten Basen. Das Gold(III)-
Ion hat zwar eine höhere Ladung und es hat eine geringere Polarisierbarkeit als zum Beispiel
ein Gold(I)-Ion, aber es besitzt immer noch einen großen Ionenradius, da es zu den späten
Übergangsmetallen gehört. Es wird deswegen noch zu den weichen Säuren gezählt. Es wurde
also nach einer Aminosäure als Bindungspartner gesucht, die in ihrer Seitenkette eine weiche
Base als Bindungspartner besitzt. Es fielen deswegen alle Aminosäuren mit einer harten Sei-
tenfunktionalität wie zum Beispiel Lysin und Asparaginsäure als Bindungspartner weg. Bei
Thiolen ist bekannt, dass sie als weiche Basen sehr gut zum Gold binden.127
Cystein trägt in
der Seitengruppe diese Funktionalität. Um diese Aminosäure sollte deswegen eine Sequenz
dargestellt werden. Da die Peptidsequenz zweizähnig binden sollte, wurden jeweils immer
zwei Cysteine verwendet.
Austausch der Chloridliganden
Da es nicht sicher zu sein schien, dass die Chloridionen eine ausreichend gute Abgangsgrupe
waren, wurde das 2-Phenylpyridin-1,1´gold(III)acetonat 20 aus Silberacetonat und
AuPhPyCl2 2 erzeugt. Die Chloridionen erwiesen sich im Laufe der Versuche jedoch als aus-

Ergebnisse
Seite | 27
reichend gute Abgangsgruppe und auf die Erzeugung des Acetonats 20 wurde verzichtet. Die
Verknüpfung des AuPhPyCl2 2 mit Peptiden ist durch den Austausch der Chloridliganden und
der Bindung von Seitengruppen der Peptide direkt zum Gold(III)-Ion in guter Ausbeute zu
erreichen. Es schien wichtig, dass auch das Peptid den Goldkomplex zwei zähnig band. Da-
durch sollte eine in Lösung stabile Verbindung erzeugt werden.
Suche nach einem Peptidbindungsmotiv
Die erste synthetisierte Sequenz war Ac-CC-NH2 21. Diese Sequenz wurde mit AuPhPyCl2 2
in Wasser und Acetonitril umgesetzt und eine analytische HPLC Messung wurde vom Pro-
dukt angefertigt. Das nach der analytischen HPLC erhalten Chromatogramm gab eine Viel-
zahl von Produkten wieder. Es wurde eine Kombination von analytischer HPLC und Massen-
spektroskopie durchgeführt. Eine HPLC-Anlage wurde vor das Massenspektrometer geschal-
tet. Keines der so erhalten Massenspektren hatte die erwartete Massen für das Produkt
Ac-CC-NH2AuPhPy 22.
Eine mögliche Erklärung, dass sich Ac-CC-NH2AuPhPy 22 nicht gebildet hatte, war der Ab-
stand zwischen den beiden Cysteinen. Der beiden Cysteine waren möglicherweise zu nah bei-
sammen, die Bindungswinkel waren deswegen ungünstig. Um den Abstand zwischen den
beiden Cysteinen zu vergrößern, wurden Glycin, Prolin und Phenylalanin zwischen den bei-
den Cysteinen in der Sequenz ergänzt. Glycin ist die einfachste achirale Aminosäure ohne
funktionelle Seitenkette und ist damit gut als Abstandsaminosäure geeignet. Prolin ist die ein-
zige Aminosäure, die mit einem sekundären Amin im Peptidrückgrat bindet. Dies macht das
Rückgrat weniger flexibel und zwingt die Sequenz in einen bestimmten Winkel. Um das Pep-
tid bei einer Wellenlänge von 254 nm besser in der HPLC detektieren zu können, wurde als
Abstandsaminosäure Phenylalanin in die Sequenz eingebaut, es konnte so auch heraus gefun-
den werden, ob eine sterisch anspruchsvoller natürliche Aminosäure eine Reaktion von
Gold(III)-Komplex 2 verhindert. Es wurden somit folgende drei Sequenzen synthetisiert:
Nummer Name
23 Ac-CGC-NH2
24 Ac-CPC-NH2
25 Ac-CFC-NH2 Tabelle 3-1
Diese Sequenzen wurden mit AuPhPyCl2 2 umgesetzt, um die Produkte aus Abbildung 3-14
zu erhalten.
Ergebnisse
Seite | 28
Abbildung 3-14: Ac-CGC-NH2AuPhPy (links), Ac-CPC-NH2AuPhPy (mitte)
und Ac-CFC-NH2AuPhPy (rechts)
Die erhalten Produkte wurden mit der analytischen HPLC und dem ESI-MS untersucht.
Exemplarisch ist das Chromatogramm von Verbindung 26 in Abbildung 3-15 wiedergegeben.
Abbildung 3-15: HPLChromatogramm vom Rohprodukt Ac-CGC-NH2AuPhPy 26
Das Chromatogramm zeigt einen Peak bei einer Retentionszeit 17.1 min. Dies ließ auf die
Entstehung von nur einem Produkt schließen, welches in allen drei Fällen fast quantitativ er-
halten worden ist. Der intensive Peak zu Beginn der Messung kommt durch die Verwendung
von DMSO zum Lösen des Produktes zustande. Das Massenspektrum des Produktes ist in
Abbildung 3-16 gezeigt.
Ergebnisse
Seite | 29
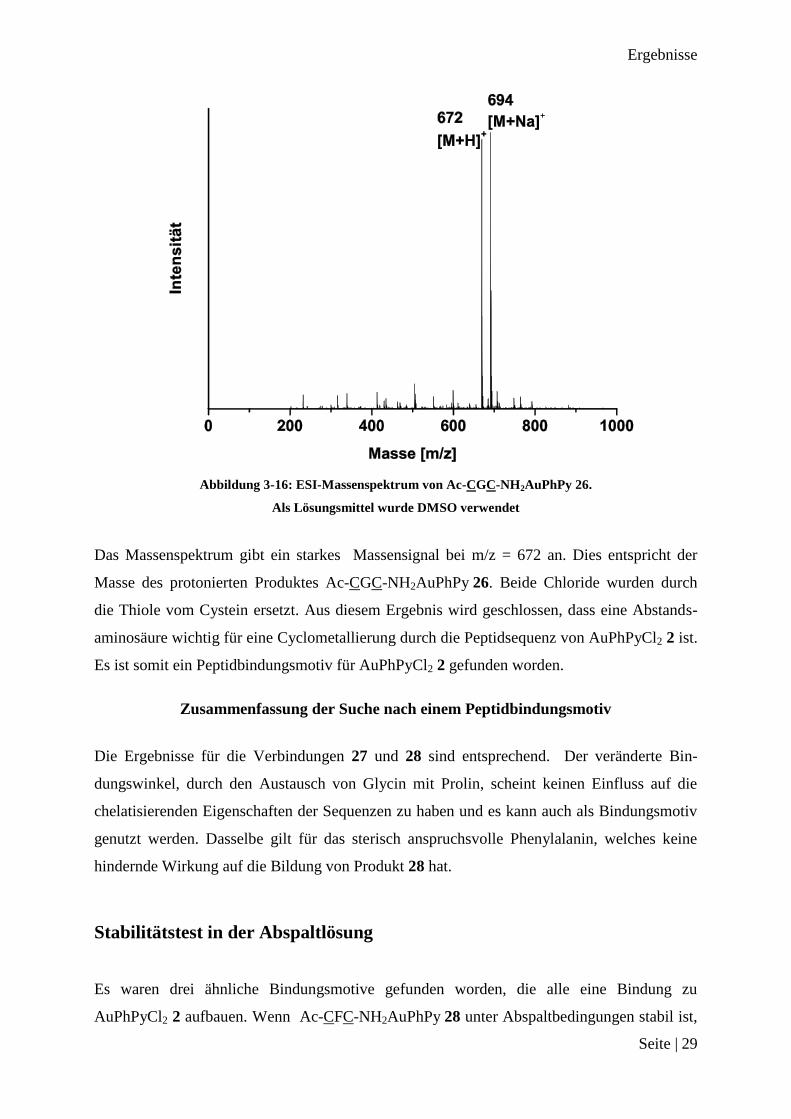
Abbildung 3-16: ESI-Massenspektrum von Ac-CGC-NH2AuPhPy 26.
Als Lösungsmittel wurde DMSO verwendet
Das Massenspektrum gibt ein starkes Massensignal bei m/z = 672 an. Dies entspricht der
Masse des protonierten Produktes Ac-CGC-NH2AuPhPy 26. Beide Chloride wurden durch
die Thiole vom Cystein ersetzt. Aus diesem Ergebnis wird geschlossen, dass eine Abstands-
aminosäure wichtig für eine Cyclometallierung durch die Peptidsequenz von AuPhPyCl2 2 ist.
Es ist somit ein Peptidbindungsmotiv für AuPhPyCl2 2 gefunden worden.
Zusammenfassung der Suche nach einem Peptidbindungsmotiv
Die Ergebnisse für die Verbindungen 27 und 28 sind entsprechend. Der veränderte Bin-
dungswinkel, durch den Austausch von Glycin mit Prolin, scheint keinen Einfluss auf die
chelatisierenden Eigenschaften der Sequenzen zu haben und es kann auch als Bindungsmotiv
genutzt werden. Dasselbe gilt für das sterisch anspruchsvolle Phenylalanin, welches keine
hindernde Wirkung auf die Bildung von Produkt 28 hat.
Stabilitätstest in der Abspaltlösung
Es waren drei ähnliche Bindungsmotive gefunden worden, die alle eine Bindung zu
AuPhPyCl2 2 aufbauen. Wenn Ac-CFC-NH2AuPhPy 28 unter Abspaltbedingungen stabil ist,

Ergebnisse
Seite | 30
dann kann die komplette Sequenz an fester Phase synthetisiert werden. Cystein könnte dann
mit orthogonalen Schutzgruppen in der Sequenz synthetisiert werden. Diese orthogonalen
Schutzgruppen könnten an fester Phase abgespalten werden und AuPhPyCl2 2 könnte gekop-
pelt werden. Danach könnte das Peptid mit dem gebunden Gold(III)-Komplex 2 abgespalten
werden und das gewünschte Biokonjugat würde erhalten.
Als Test für diese Möglichkeit wurde Konjugat 28 in die Abspaltlösung gegeben und für
sechs Stunden geschüttelt. In die Lösung wurde - 70 °C Diethylether zum Fällen des Produk-
tes gegeben. Von dem Rückstand wurde eine analytische HPLC-Messung durchgeführt. Der
Peak bei 22.9 min ist nicht mehr vorhanden. Im Chromatogramm sind viele, zum Teil sehr
wenig intensive neue Peaks zu beobachten. Von dem Rückstand wurde ein ESI-
Massenspektrum aufgenommen. Keines der Signale passt zum Ac-CFC-NH2AuPhPy 28. Es
ist nötig AuPhPyCl2 2 in Lösung an die Peptide zu koppeln, um die gewünschten Biokonjuga-
te zu erhalten.
Ergänzung des Bindungsmotives um einen geladen Peptidteil
Das kurze Bindungsmotiv CFC sollte nun direkt mit den beiden geladen Teilen der langen
Sequenz synthetisiert werden. Hierdurch sollte herausgefunden werden, ob der Gold(III)-
Komplex 2 nicht an irgendwelche Funktionalitäten der langen Peptidsequenz binden kann.
Zum einen wurde das Bindungsmotiv mit fünf Argininen, zum anderen das Bindungsmotiv
mit fünf Glutaminsäuren synthetisiert.
Abbildung 3-17: Ac-CFCRRRRR-NH2AuPhPy
Ergebnisse
Seite | 31
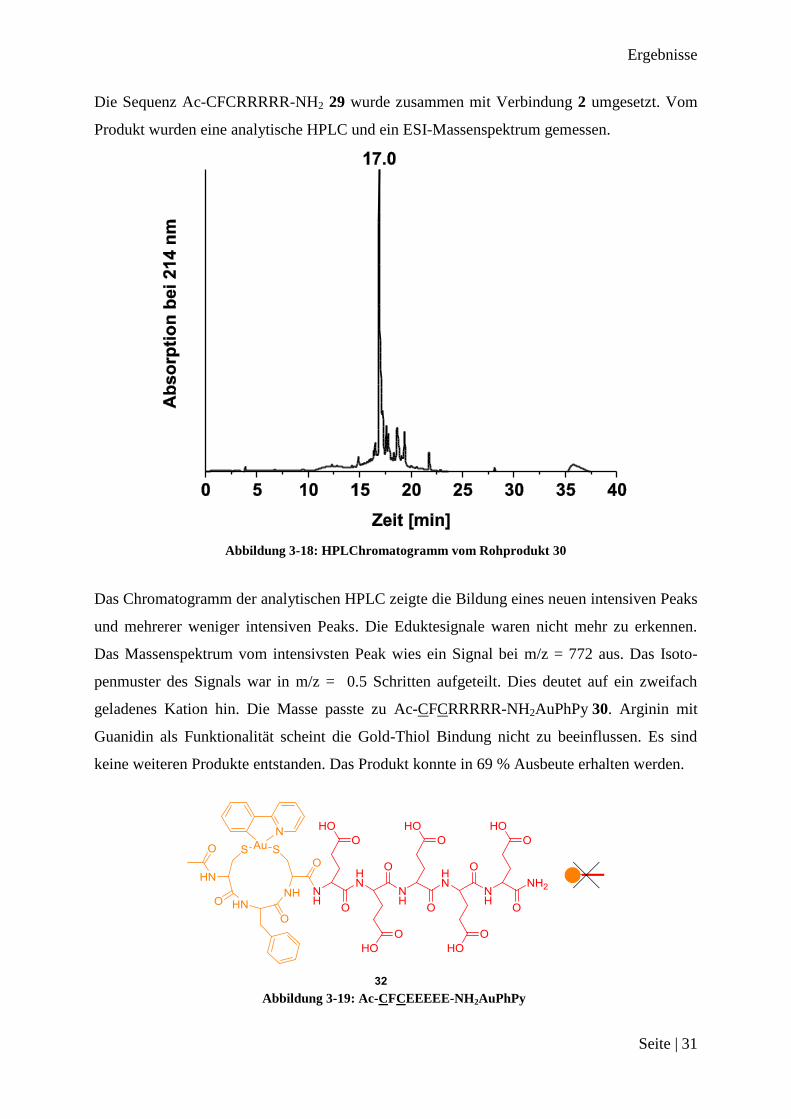
Die Sequenz Ac-CFCRRRRR-NH2 29 wurde zusammen mit Verbindung 2 umgesetzt. Vom
Produkt wurden eine analytische HPLC und ein ESI-Massenspektrum gemessen.
Abbildung 3-18: HPLChromatogramm vom Rohprodukt 30
Das Chromatogramm der analytischen HPLC zeigte die Bildung eines neuen intensiven Peaks
und mehrerer weniger intensiven Peaks. Die Eduktesignale waren nicht mehr zu erkennen.
Das Massenspektrum vom intensivsten Peak wies ein Signal bei m/z = 772 aus. Das Isoto-
penmuster des Signals war in m/z = 0.5 Schritten aufgeteilt. Dies deutet auf ein zweifach
geladenes Kation hin. Die Masse passte zu Ac-CFCRRRRR-NH2AuPhPy 30. Arginin mit
Guanidin als Funktionalität scheint die Gold-Thiol Bindung nicht zu beeinflussen. Es sind
keine weiteren Produkte entstanden. Das Produkt konnte in 69 % Ausbeute erhalten werden.
Abbildung 3-19: Ac-CFCEEEEE-NH2AuPhPy

Ergebnisse
Seite | 32
Es wurde nun Ac-CFCEEEEE-NH2 31 mit AuPhPyCl2 2 zur Reaktion gebracht. Mit dem
Rückstand wurde ein analytischer HPLC Lauf gemessen. Es wurde auch ein Massenspektrum
aufgenommen. Das Chromatogramm hatte sehr viele Peaks, die teilweise ineinander übergin-
gen. Die Bestimmung der genauen Anzahl war damit nicht möglich. Im Massenspektrum wa-
ren sehr viele verschiedene Massen zu erkennen, keine der Massen konnte
Ac-CFCEEEEE-NH2AuPhPy 32 zugeordnet werden. Das Produkt 32 konnte nicht hergestellt
worden. Die Carbonsäure der Glutaminsäure scheint ebenfalls eine bindende Funktionalität
für AuPhPyCl2 2 zu sein. Es musste eine andere Syntheseroute gefunden werden.
Orthogonal geschützt Glutaminsäure
Die Carbonsäure der Glutaminsäure sollte nun orthogonal geschützt werden. So könnte die
ganze Sequenz am Harz synthetisiert werden und dann abgespalten werden, so dass nur noch
die Seitenketten der Glutaminsäuren geschützt sind. Diese Sequenz könnte dann mit
AuPhPyCl2 2 umgesetzt werden und die Seitenketten der Glutaminsäuren selektiv entschützt
werden. Als Schutzgruppe für die orthogonale Schützung der Seitenketten der Glutaminsäu-
ren kann 4-((1-4,4-Dimethyl-2,6-dioxocyclohexyliden)-3-methybutyl)amino)-benzyl (Dmab)
genutzt werden. Diese Schutzgruppe ist sowohl Säure stabil als auch Base stabil. Zum Ab-
spalten der Schutzgruppe wird eine Hydrazinlösung verwendet.128, 129
Versuche mit der Verbindung Ac-CFCE(ODmab)E(ODmab)-NH2 33 ergaben jedoch, dass
die Goldbindung zum Schwefel unter den reduktiven Abspaltebedingungen mit 2 Gew.-%
Hydrazinlösung für die orthogonale Schutzgruppe nicht stabil ist.
Kupferkatalysierte [3+2] Cycloaddition
Eine weitere Möglichkeit ist die [3+2] Cycloaddition zwischen einem Azid und einem termi-
nalen Alkin. Dies ist eine orthogonale Reaktion zu allen Funktionalitäten der natürlichen
Aminosäuren.76
Die Reaktion kann in Anwesenheit aller natürlichen Aminosäuren ablaufen,
ohne von den Seitengruppen der Aminosäuren beeinflusst zu werden oder sogar mit diesen zu
reagieren. Das als Katalysator bei dieser Reaktion verwendete zytotoxische Kupfer, muss vom
Produkt abgetrennt werden.130, 131
Ergebnisse
Seite | 33
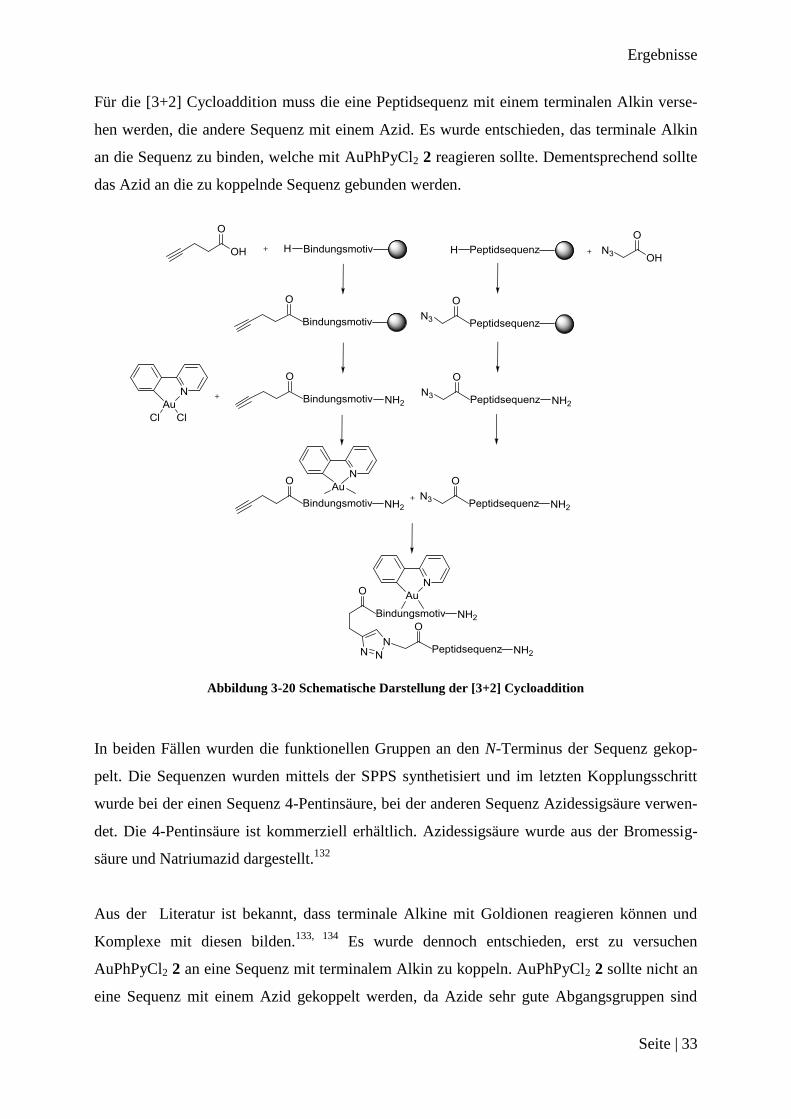
Für die [3+2] Cycloaddition muss die eine Peptidsequenz mit einem terminalen Alkin verse-
hen werden, die andere Sequenz mit einem Azid. Es wurde entschieden, das terminale Alkin
an die Sequenz zu binden, welche mit AuPhPyCl2 2 reagieren sollte. Dementsprechend sollte
das Azid an die zu koppelnde Sequenz gebunden werden.
Abbildung 3-20 Schematische Darstellung der [3+2] Cycloaddition
In beiden Fällen wurden die funktionellen Gruppen an den N-Terminus der Sequenz gekop-
pelt. Die Sequenzen wurden mittels der SPPS synthetisiert und im letzten Kopplungsschritt
wurde bei der einen Sequenz 4-Pentinsäure, bei der anderen Sequenz Azidessigsäure verwen-
det. Die 4-Pentinsäure ist kommerziell erhältlich. Azidessigsäure wurde aus der Bromessig-
säure und Natriumazid dargestellt.132
Aus der Literatur ist bekannt, dass terminale Alkine mit Goldionen reagieren können und
Komplexe mit diesen bilden.133, 134
Es wurde dennoch entschieden, erst zu versuchen
AuPhPyCl2 2 an eine Sequenz mit terminalem Alkin zu koppeln. AuPhPyCl2 2 sollte nicht an
eine Sequenz mit einem Azid gekoppelt werden, da Azide sehr gute Abgangsgruppen sind

Ergebnisse
Seite | 34
und es so zu unerwünschten Nebenreaktionen hätte kommen können. Eine mögliche Reaktion
wäre der Austausch eines Chloridliganden von AuPhPyCl2 2 durch das Azid.135, 136
Abbildung 3-21: ΠCFC-NH2AuPhPy
Für den Test, ob das terminale Alkin am Ende der Sequenz mit dem Gold(III)-Komplex rea-
giert, wurden äquimolare Mengen ΠCFC-NH2 34 und AuPhPyCl2 2 umgesetzt. Das erhalten
gelbfarbene Produkt wurde mit der analytischen HPLC und dem ESI-Massenspektrometer
analysiert. Das Chromatogramm hat einen Peak bei 20.9 min, im Massenspektrum sind ein
Signal bei m/z = 822 und ein Signal bei m/z = 800 zu beobachten. Das Ergebnis der analyti-
schen HPLC deutet auf die Entstehung von nur einem Produkt hin, welches quantitativ erhal-
ten wird. Die erste gemessene Masse entspricht [35+Na]+, die zweite Masse entspricht dem
protonierten ΠCFC-NH2AuPhPy 35. Das terminale Alkin am Ende der Peptidsequenz beein-
flusst die Entstehung vom Produkt 35 nicht, es reagiert nicht mit AuPhPyCl2 2. Da Produkt 35
ohne großen Aufwand zu erhalten war und die Darstellung der Sequenzen mit dem Azid am
N-Terminus auch ohne Widrigkeiten gelang, konnten beiden Teile nun in einer [3+2]
Cycloaddition mit einander zur Reaktion gebracht werden.
[3+2] Cycloaddition der Teilsequenzen
Als erstes sollte die [3+2] Cycloreaktion zwischen ΠCFC-NH2AuPhPy 35 und den einzelnen
Abschnitten der kompletten Sequenz des Projektes probiert werden. Es wurden deswegen
folgende Peptide dargestellt:
Nummer Name
36 ORRRRR-NH2
37 OPLGLYAL-NH2
38 OEEEEE-NH2 Tabelle 3-2
Ergebnisse
Seite | 35
Durch die Tests mit den einzelnen Abschnitten sollten synthetische Herausforderungen leich-
ter lokalisierbar sein.
Beim positiv geladenen Abschnitt konnten die Guanidine der Argininreste an den Kupferkata-
lysator binden und so eine Reaktion verhindern. Das Kupfer(II)sulfat, als Katalysatorquelle
wurde deswegen im dreifachen Überschuss zugegeben.
Der durch MMP2 spaltbare Abschnitt hat außer Tyrosin keine koordinativen Funktionen,
sondern nur aliphatische Gruppen in den Seitenketten, jedoch wurde aus Gründen der Ver-
gleichbarkeit derselbe Überschuss an Katalysator zugegeben.
Als drittes wurde der Abschnitt mit den Glutaminsäuren in der orthogonalen [3+2] Cyclo-
addition umgesetzt. Diese Sequenz mit den Glutaminsäuren hatte bei der direkten Umsetzung
keine spezifische Bindung zum Gold(III)-Komplex gezeigt und die [3+2] Cycloaddition soll
nun zeigen, ob so eine Bindung zwischen Peptid und Gold(III)-Komplex möglich ist.
Da das Bindungsmotiv mit dem Gold(III)-Komplex kaum in den üblichen Lösungsmittelen
der kupferkatalysierten [3+2] Cycloaddition, wie zum Beispiel Wasser, THF oder Aceton
löslich ist, wurden neben Wasser bei dieser Reaktion auch DMSO verwendet.
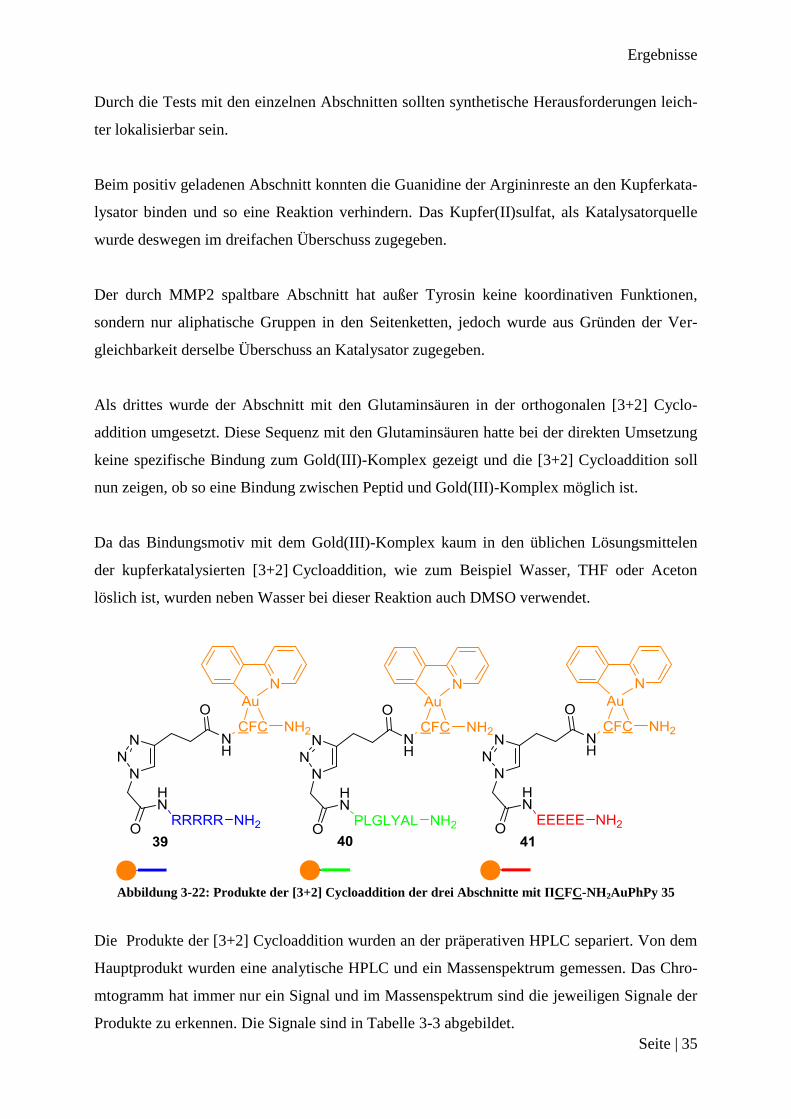
Abbildung 3-22: Produkte der [3+2] Cycloaddition der drei Abschnitte mit ΠCFC-NH2AuPhPy 35
Die Produkte der [3+2] Cycloaddition wurden an der präperativen HPLC separiert. Von dem
Hauptprodukt wurden eine analytische HPLC und ein Massenspektrum gemessen. Das Chro-
mtogramm hat immer nur ein Signal und im Massenspektrum sind die jeweiligen Signale der
Produkte zu erkennen. Die Signale sind in Tabelle 3-3 abgebildet.

Ergebnisse
Seite | 36
Nummer Signal bei m/z
39 1680, 841
40 1628, 815, 545
41 1545, 774 Tabelle 3-3
Zusammenfassung [3+2] Cycloaddition der Teilsequenzen
Es konnten alle drei Abschnitte erfolgreich in knapp 15 % Ausbeute umgesetzt werden, ohne
das die Funktionalitäten in den Seitenketten zu Schwierigkeiten bei der Umsetzung geführt
haben. Es konnte nun versucht werden mit der [3+2] Cycloaddition alle drei Abschnitte zu-
sammen mit ΠCFC-NH2AuPhPy 35 zu verbinden.
[3+2] Cycloaddition der kompletten Sequenz
Die erfolgreich verlaufene [3+2] Cycloaddition zwischen ΠCFC-NH2AuPhPy 35 und den drei
Abschnitten 36, 37 und 38 zeigt, dass es keine Probleme mit den Funktionalitäten in den Se-
quenzen gibt. Es sollten nun probiert werden, die einzelnen Abschnitte zusammen als kom-
plette Sequenz 42 mit ΠCFC-NH2AuPhPy 35 zu verbinden. Neben Sequenz 42 sollten noch
zwei weitere Sequenzen 43 und 44 in einer [3+2] Cycloaddition verbunden werden. Alle drei
Peptidsequenzen unterscheiden sich nur in dem durch MMP2 spaltbaren Bereich und es sollte
ermittelt werden, ob ein Unterschied in der Reaktivität besteht.
Abbildung 3-23: Produkte der [3+2] Cycloaddition von drei kompletten Sequenzen mit
ΠCFC-NH2AuPhPy 35
Es wurden dieselben Reaktionsbedingungen für die [3+2] Cycloaddition, wie schon bei den
einzelnen Abschnitten gewählt. Von den drei Ansätzen wurde mit der präperativen HPLC
jeweils der größte Peak separiert und eine analytische HPLC-Messungen und ein ESI-
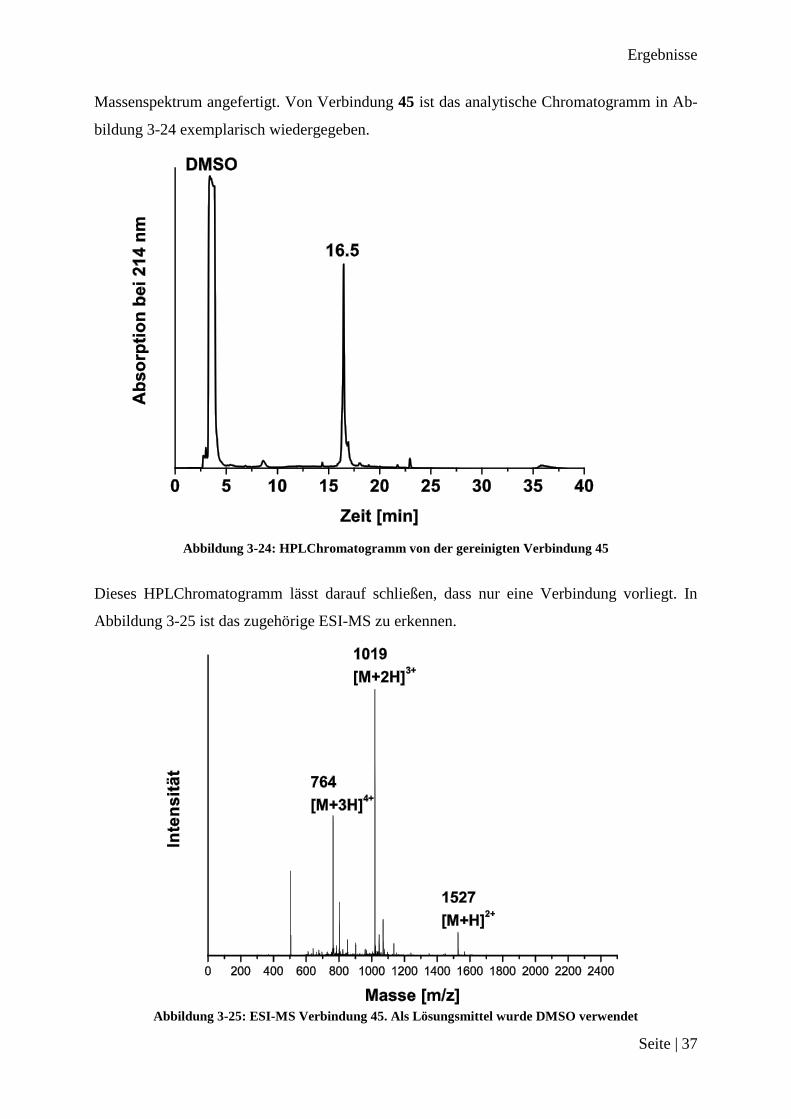
Ergebnisse
Seite | 37
Massenspektrum angefertigt. Von Verbindung 45 ist das analytische Chromatogramm in Ab-
bildung 3-24 exemplarisch wiedergegeben.
Abbildung 3-24: HPLChromatogramm von der gereinigten Verbindung 45
Dieses HPLChromatogramm lässt darauf schließen, dass nur eine Verbindung vorliegt. In
Abbildung 3-25 ist das zugehörige ESI-MS zu erkennen.
Abbildung 3-25: ESI-MS Verbindung 45. Als Lösungsmittel wurde DMSO verwendet

Ergebnisse
Seite | 38
Im Massenspektrum sind drei Signale hervorgehoben. Das erste Signal von einer zweifach
geladenen Spezies liegt bei m/z = 1527, das zweite einer dreifach geladenen Verbindung bei
m/z = 1019 und das dritte Signal einer vierfach geladenen Verbindung bei m/z = 765. Alle
drei Signale können der Zielverbindung 45 zugeordnet werden. Die Daten von Verbindung 46
und 47 sind entsprechend. Aus den Daten konnte gefolgert werden, dass alle drei Sequenzen
erfolgreich mit ΠCFC-NH2AuPhPy 37 umgesetzt werden konnten und es wurden jeweils um
die 10 % Ausbeute erhalten.
Zusammenfassung [3+2] Cycloaddition der kompletten Sequenz
Damit sind gleich drei Verbindungen synthetisiert worden, die der Zielverbindung des Projek-
tes entsprechen. In allen drei Verbindungen ist ein Gold(III)-Komplex für die zytotoxische
Wirkung vorhanden, der an eine längere Peptidsequenz gebunden ist. Die längere
Peptidsequenz besteht aus dem positiv geladen, einem durch MMP2 spaltbaren und dem ne-
gativ geladen Abschnitt.
Bessere Löslichkeit in Wasser
Die über die [3+2] Cycloaddition dargestellten Verbindungen 43, 46 und 47 sind die ersten
Biokonjugate, welche die Zielsetzung dieser Arbeit erfüllten. Bisher sind die Verbindungen
jedoch so schlecht in Wasser löslich, dass sie immer erst in DMSO gelöst werden mussten.
Um diese Verbindungen besser in Wasser lösen zu können und so HPLC-Anwendungen und
Zelltests zu vereinfachen, sollte der besonders schlecht lösliche Teil um das Edukt 35 modifi-
ziert werden. Zwei Teile von Verbindung 35 konnten verändert werden, um eine höhere Lös-
lichkeit in Wasser zu erreichen. Zum einen der Ligand am Gold(III)-Ion und zum anderen das
Bindungsmotiv. Es hatte sich schon herausgestellt, dass es aufwendiger war, den Liganden als
das Bindungsmotiv zu verändern. Aus diesem Grund sollte die Abstandsaminosäure des Bin-
dungsmotivs erneut verändert werden. In diesem Fall sollte sie verändert werden, um die Lös-
lichkeit in Wasser zu erhöhen. Es wurde nun eine neue Abstandsaminosäure benötigt, die
nicht an den Gold(III)-Komplex koordiniert und eine hohe Löslichkeit der neuen Verbindung
in Wasser bewirkte. Aus den Versuchen mit Verbindung 30 ist die hohe Löslichkeit von Ar-
ginin in Wasser bekannt. Die Versuche haben auch ergeben, dass die Guanidingruppe nicht an
AuPhPyCl2 2 bindet.

Ergebnisse
Seite | 39
Abbildung 3-26: ΠCRC-NH2AuPhPy
Für die Darstellung dieser besser wasserlöslichen Verbindung wurde das neue Bindungsmotiv
ΠCRC-NH2 48 zusammen mit einer äquimolaren Menge an AuPhPyCl2 2 umgesetzt. Von der
Reaktionslösung wurde nach einer Stunde eine analytische HPLC durchgeführt. Das Chroma-
togramm der analytischen HPLC hat einen Peak bei 17.9 min. Es ist sonst kein Peak zu er-
kennen und es wurde ein Produkt quantitativ erhalten. Es ist nur eine Verbindung aus der Re-
aktion hervorgegangen. Es wurde ein ESI-MS durchgeführt. Das Spektrum hat zwei Signale.
Das eine Signal ist bei m/z = 809 und das andere bei m/z = 406. Diese beiden Signale ent-
sprechen dem Produkt 49 mit einfacher Ladung und mit zweifacher Ladung.
Es ist versucht worden ΠCRC-NH2AuPhPy 49 direkt mit der Verbindung ORRRRRPL-
GLYALEEEEE-NH2 42 in einer [3+2] Cycloaddition umzusetzen. Die Reaktion wurde unter
den oben genutzten Bedingungen mehrfach durchgeführt. Mit der analytischen HPLC wurden
Reaktionskontrollen durchgeführt. In den Chromatogrammen ist der Peak von ORRRRR-
PLGLYALEEEEE-NH2 42 weiter vorhanden, während der Peak von ΠCRC-NH2AuPhPy 49
an Intensität verliert, entstehen viele neue wenig intensive Peaks. Keine dieser Reaktionen
führte zu dem erwarteten Produkt der [3+2] Cycloaddition. Durch die Beobachtungen in der
analytischen HPLC wurde vermutet, ΠCRC-NH2AuPhPy 49 könne in Lösung nicht stabil
sein. Zur Überprüfung dieses Verdachtes wurde etwas ΠCRC-NH2AuPhPy 49 in Wasser ge-
löst. Von dieser Lösung wurden in Zeitabständen analytische HPLC Läufe gemessen.
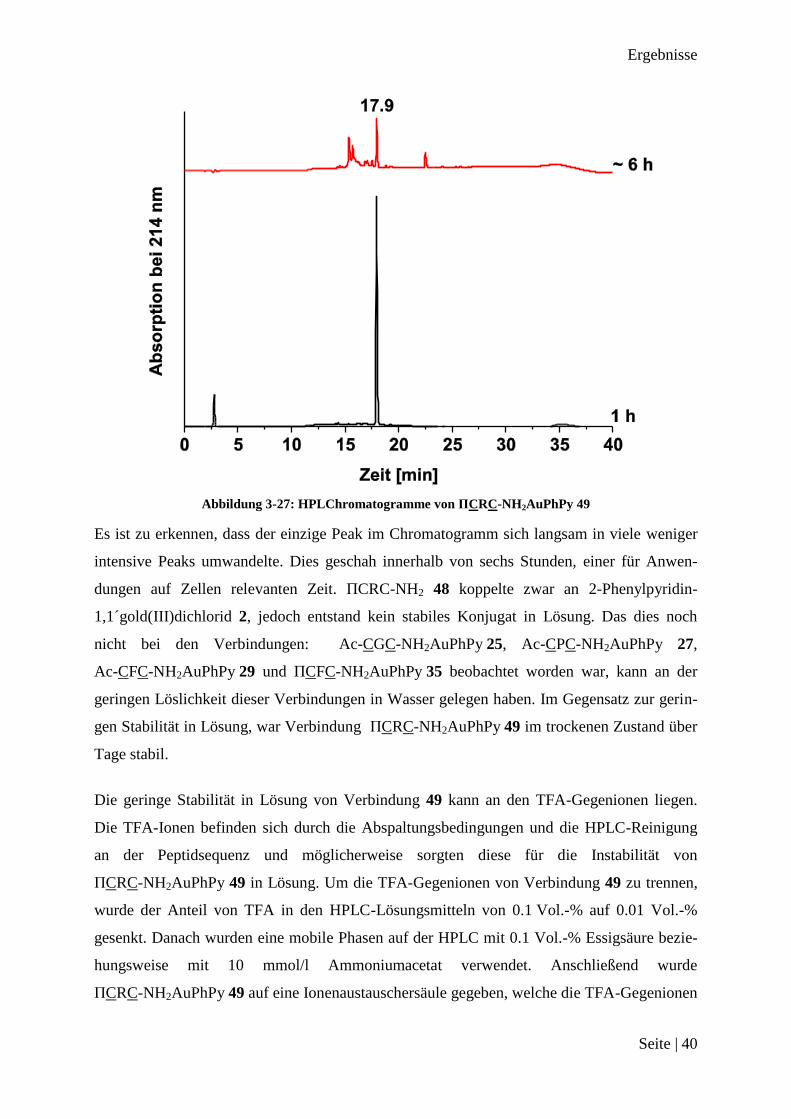
Ergebnisse
Seite | 40
Abbildung 3-27: HPLChromatogramme von ΠCRC-NH2AuPhPy 49
Es ist zu erkennen, dass der einzige Peak im Chromatogramm sich langsam in viele weniger
intensive Peaks umwandelte. Dies geschah innerhalb von sechs Stunden, einer für Anwen-
dungen auf Zellen relevanten Zeit. ΠCRC-NH2 48 koppelte zwar an 2-Phenylpyridin-
1,1´gold(III)dichlorid 2, jedoch entstand kein stabiles Konjugat in Lösung. Das dies noch
nicht bei den Verbindungen: Ac-CGC-NH2AuPhPy 25, Ac-CPC-NH2AuPhPy 27,
Ac-CFC-NH2AuPhPy 29 und ΠCFC-NH2AuPhPy 35 beobachtet worden war, kann an der
geringen Löslichkeit dieser Verbindungen in Wasser gelegen haben. Im Gegensatz zur gerin-
gen Stabilität in Lösung, war Verbindung ΠCRC-NH2AuPhPy 49 im trockenen Zustand über
Tage stabil.
Die geringe Stabilität in Lösung von Verbindung 49 kann an den TFA-Gegenionen liegen.
Die TFA-Ionen befinden sich durch die Abspaltungsbedingungen und die HPLC-Reinigung
an der Peptidsequenz und möglicherweise sorgten diese für die Instabilität von
ΠCRC-NH2AuPhPy 49 in Lösung. Um die TFA-Gegenionen von Verbindung 49 zu trennen,
wurde der Anteil von TFA in den HPLC-Lösungsmitteln von 0.1 Vol.-% auf 0.01 Vol.-%
gesenkt. Danach wurden eine mobile Phasen auf der HPLC mit 0.1 Vol.-% Essigsäure bezie-
hungsweise mit 10 mmol/l Ammoniumacetat verwendet. Anschließend wurde
ΠCRC-NH2AuPhPy 49 auf eine Ionenaustauschersäule gegeben, welche die TFA-Gegenionen

Ergebnisse
Seite | 41
binden sollte. Keines dieser Verfahren sorgte für eine Erhöhung der Stabilität von dem Kom-
plex ΠCRC-NH2AuPhPy 49.
Zusammenfassung Bessere Löslichkeit in Wasser
Da das Bindungsmotiv von zwei Cysteinen mit einer Abstandsaminosäure zu keiner in Lö-
sung stabilen Verbindung in Kombination mit dem 2-Phenylpyridin-1,1´gold(III)-Komplex
führt, wurde beschlossen ein neues Bindungsmotiv zu suchen.
300 potentielle Bindungsmotive
Um eine Peptidsequenz zu finden, die einen stabilen Komplex mit AuPhPyCl2 2 bildet, sollten
mehrere Sequenzen in einem kombinatorischen Projekt ausprobiert werden.137
Die neuen Se-
quenzen sollten wieder aus den drei oben schon verwendeten Komponenten, bindenden Ami-
nosäuren, Abstandsaminosäure zwischen den beiden bindenden Aminosäuren und am C-
Terminus die 4-Pentinsäure, aufgebaut werden. Es ergibt sich folgender allgemeiner Aufbau:
Π bindende AS1 Abstands AS2 bindende AS3 Harz
Die 4-Pentinsäure soll bei den über das Projekt gefundenen stabilen Bindungsmotiven wieder
für die [3+2] Cycloaddition genutzt werden. In Abbildung 3-28 sind alle Monomere für das
Kombinatorische Projekt dargestellt.

Ergebnisse
Seite | 42
Abbildung 3-28: Valenzstrichformel mit Einbuchstabencode und Namen der Monomere
Bindende Aminosäuren
Dass Cystein zu AuPhPyCl2 2 binden kann, ist durch die bisher durchgeführten Versuche be-
kannt. In der Kombination mit einer anderen bindenden Aminosäure sollte ein stabiler neuer
Gold(III)-Komplex erzeugt werden. Im nachfolgenden werden die verwendeten bindenden
Aminosäuren aufgeführt.
Methionin ist die einzige andere natürliche Aminosäure mit einem Schwefelatom in der Sei-
tenkette. Das Schwefelatom befindet sich in einem Thioether und dient damit nur über die
freien Elektronenpaare als potentieller Bindungspartner für das Gold(III)-Zentrum.138-140
Die beiden unnatürlichen Aminosäuren 3-(3-Pyridin)-alanin und 3-(4-Pyridin)-alanin wurden
wegen des Pyridins in der Seitenfunktion gewählt. Das Pyridin ist bekannt für seine Fähigkeit
an Gold(III)-Komplexe zu binden.141, 142
Sie unterscheiden sich in der Stellung des Stickstoff-
atoms im Pyridinring. Dadurch wird der Bindungswinkel beeinflusst, was einen Einfluss auf
das Bindungsmotiv hat.

Ergebnisse
Seite | 43
Histidin ist eine natürliche Aminosäure mit einem N-heterocyclischen Aromaten und ähnelt
damit den Pyridinen der zuletzt genannten unnatürlichen Aminosäuren.
Das primäre Amin in der Seitenkette des Lysin und die unnatürlichen 2,3-Diaminopropion-
säure sollen eine Bindung zum Gold(III)-Ion ausbilden.143, 144
Die beiden Aminosäuren unter-
scheiden sich in der Länge der Alkylkette. Dies sollte einen Einfluss auf den Bindungswinkel
haben.
Asparaginsäure und Glutaminsäure sollten durch ihre Carbonsäure in der Seitengruppe eine
Bindung zum Goldatom vom AuPhPyCl2 2 ausbilden können.145
Nach dem HSAB Prinzip
sollte dies zwar nicht wahrscheinlich sein,126
jedoch hatten die Versuche bei Verbindung 32
gezeigt, dass die Carbonsäuren sehr wohl eine Bindung zum Gold(III)-Komplex aufbauen
konnten. Die unterschiedliche lange Alkylkette sollte, analog zu dem oben genannten Fall mit
den primären Aminen, zu unterschiedlichen Bindungsverhältnissen führen und damit beein-
flussen, ob eine Bindung zu Goldatom ausgebildet werden kann.
Alanin wurde als Blindprobe ausgewählt, welche keine Bindung durch ihre Funktionalität
ausbilden kann.
Abstandsaminosäuren
Es wurden drei verschiedene Abstandsaminosäuren ausgewählt. Die erste Abstandsaminosäu-
re ist Glycin. Glycin wurde als einfachste Aminosäure, welches lediglich ein Wasserstoffatom
in der Seitengruppe besitzt, verwendet. Die Zweite ist Prolin, die einzige natürliche Amino-
säure mit sekundärem Amin, wurde verwendet um die Bindungswinkel zu verändern. Analog
zu Verbindung 26 sollte hiermit herausgefunden werden, ob auch der Winkel einen Einfluss
auf die Bindung von der Peptidsequenz zum AuPhPyCl2 2 hat. Als dritte Abstandsaminosäure
wurde Leucin verwendet, da diese Aminosäure durch die Isopropylfunktion als Seitengruppe
einen gewissen sterischen Anspruch hat.
Aufbau der Sequenz
In der Tabelle 3-4 ist der Aufbau der Sequenzen wiedergegeben. Die Sequenzen sollten an
einem Harz ohne Linker dargestellt werden.

Ergebnisse
Seite | 44
Π-AS1-AS2-AS3-Harz
Π AS1 AS2 AS3
4-Pentinsäure A,B,C,D,E,H,J,K,M,Z G,P,L A,B,C,D,E,H,J,K,M,Z Tabelle 3-4: Schematischer Aufbau der Sequenzen am Harz im Einbuchstabencode
Die Peptide sind von N-Terminus beginnend wie folgt aufgebaut: Die 4-Pentinsäure, es folgt
eine der bindende Aminosäure, dann kommt eine Abstandsaminosäure, wieder eine bindende
Aminosäure und am C-Terminus befindet sich das Harz. In diesem Projekt wurden jede mög-
liche Kombinationen synthetisiert, dafür wurden alle zehn Möglichkeiten von AS1 verwendet,
während AS2 und AS3 nicht verändert wurden. Dann wurde eine neue AS3, bei gleichbleiben-
der AS2, verwendet und wieder alle zehn Möglichkeiten von AS1 in dieser Kombination dar-
gestellt. Nachdem alle 100 Kombinationen von AS1 und AS3 synthetisiert waren, wurde AS2
gewechselt und alle Möglichkeiten in dieser Zusammensetzung dargestellt. Das ganze wurde
dann für die dritte Abstandsaminosäure AS2 wiederholt und alle 100 hierbei möglichen Kom-
bination synthetisiert.
Synthese
Die Synthese der Peptidsequenzen wurden an einem Syro Multiple Peptid Synthesizer der
Firma Syro 2000 ausgeführt, wobei an einem Tentagal Macro Beads Harz gearbeitet wurde.
Jede einzelne Harzkugel hatte eine Partikelgröße zwischen 280 µm und 320 µm und eine Be-
ladung von 0.24 mmol/g. Die Lösungen mit den natürlichen Aminosäuren wurden in dem
Gerät in ihre vorgesehen Postion gesetellt, die unnatürlichen Aminosäuren, zu den auch die 4-
Pentinsäure gezählt wurde, wurden an Positionen gestellt, die vorher im Programm des Syn-
thesizer für sie eingestellt worden waren. Die Kupplungen wurden im fünffachen Überschuss
durchgeführt. Die Kupplungen jeder Aminosäuren wurden doppelt durchgeführt, damit ge-
währleistet war, dass jedes Peptid in maximaler Ausbeute am Harz vorhanden war. Dies ist
entscheidend, da eine Überprüfung der Beladung und der Reinheit der Sequenzen am Harz
nicht möglich ist. Nach der Synthese der Peptidsequenzen am Synthesizer wurde manuell
weiter gearbeitet. Die Seitenschutzgruppen der einzelnen Aminosäure wurden, wenn die Se-
quenz mindestens ein Methionin oder ein Cystein beinhaltete, mit einer Abspaltlösung mit
EDT behandelt, welches die Bildung von Disulfidbrücken unterbinden soll. Bei allen anderen
Sequenzen wurde der Abspaltlösung kein EDT zugegeben. Da das Harz keinen Linker besitzt,
blieben die Sequenzen am Harz gebunden und nur die Seitenschutzgruppen von den einzelnen

Ergebnisse
Seite | 45
Aminosäuren wurden abgespalten. Das Harz wurde gewaschen. Anschließend wurde die
Hälfte des Harzes aus den einzelnen Spritzen heraus genommen und in eine zweite 2 ml
Spritze mit Polypropylenfilter gegeben. In diese zweite Spritze wurde AuPhPyCl2 2 gegeben
und mit jeweils beiden Spritzen parallel weiter gearbeitet.
Analyse
Bei der Synthese war ein Harz ohne Linker verwendet worden. Dies sollte die Reinigung der
einzelnen Sequenzen und vor allem die Analytik beschleunigen. Eine schnelle Analytik ist bei
kombinatorischen Projekten häufig die größte Herausforderung.146, 147
Zur analytischen Auf-
klärung wurde von allen Harzkugeln die Farbe bestimmt, aus allen Spritzen wurden einige
Harzkugeln unter dem Fluoreszenzmikroskop untersucht und jede Sequenzen mit Glycin als
Abstandsaminosäure und Behandlung mit AuPhPyCl2 2 wurden unter dem Ramanspektro-
meter untersucht.148-150
Farbe der Harzkugeln
Die Farbe jeder Harzkugel wurde visuell und damit qualitativ bestimmt. Die Farbe der Kugel
mit und ohne Behandlung von AuPhPyCl2 2 konnten verglichen werden. Auf diese Weise
wurde ein Indiz erhalten, ob der Gold(III)-Komplex gebunden worden war, da AuPhPyCl2 2
gelbfarben ist. Violettfarbene sind ein Hinweis darauf, dass sich kolloidales Gold gebildet
hat.151
Dieses kann sowohl die Raman wie auch die Fluoreszenzmikroskopie beeinflussen.
Der Vergleich ist innerhalb von wenigen Augenblicken möglich.
Farbe im Fluoreszenzmikroskop
Die Untersuchungen am Fluoreszenzmikroskop sind bei vorbereiteten Proben innerhalb von
einer Minute möglich und damit stellte die Fluoreszenzmikroskopie ein sehr schnelles Verfah-
ren dar. Im UV-Licht des Fluoreszenzmikroskops wurde die Farbe der Harzkugeln qualitativ
bestimmt. Es wurden Kugeln beobachtet, welche blaufarben, blaufarben mit
gold/gelbfarbenen Rand, blaufarben mit schwach gold/gelbfarbenen Rand und farblos sind.
Beispiele für alle Fälle sind in den nächsten Abbildungen 3-29 und 3-30 gegeben.

Ergebnisse
Seite | 46
Abbildung 3-29: Fluoreszenzmikroskopaufnahme von ΠCGC-MB 50 auf der linken Seite und
ΠCGC-MBAuPhPy 350 auf der rechten Seite
Abbildung 3-30 Fluoreszenzmikroskopaufnahme von ΠCGD-MBAuPhPy 390 auf der linken Seite und
ΠZGE-MBAuPhPy 406 auf der rechten Seite
Da es sich nur um einen qualitativen Vergleich mit dem Fluoreszenzmikroskop bei den ge-
bundenen Gold(III)-Komplexen handelt, wurde ein quantitatives Verfahren bei einer aussage-
kräftigen großen Gruppe von Harzkugeln benötigt.
Ramanspektroskopie
Die Ramanspektroskopie stellt ein quantitatives Verfahren zur Bestimmung der Schwin-
gungsbande von Molekülen dar.152, 153
Es wurden die Schwingung der ersten 100 mit

Ergebnisse
Seite | 47
AuPhPyCl2 2 behandelten Sequenzen von Meike Mischo aus der Physikalischen Chemie II
der Ruhr Universität Bochum untersucht und damit alle Bindungsmotive mit Glycin als Ab-
standsaminosäure und alle Kombinationen der bindenden Aminosäuren. Somit waren alle
Bindungsmotive ohne einen starren Bindungswinkel und ohne sterischen Anspruch abgedeckt
und sollten so bei der Bewertung der aus der Fluoreszenzmikroskopie erhaltenen Daten unter-
stützend wirken. Durch die im Ramanspektrometer erhalten Daten ist es möglich, Aussagen
über die Bindungsverhältnisse am der Sequenz am Harz zu tätigen. Für die Zuordnung der
einzelnen Banden wurden folgende Vergleichsverbindungen dargestellt:
Nummer Name
1 2-Phenylpyridin-1-gold(III)trichlorid
2 2-Phenylpyridin-1,1´gold(III)dichlorid
650 2-Phenylpyridin-1-gold(III)tribromid
651 2-Phenylpyridin-1,1´gold(III)dibromid
652 Tetraethylammoniumphenyltrichloroaurat(III)
653 Tetraethylammoniumphenyltribromoaurat(III)
654 Pyridin-1-gold(III)trichlorid
655 Pyridin-1-gold(III)tribromid
656 Bipyridin-1,1´gold(III)dichlorid tetrachloroaurat(III)
657 Bipyridin-1,1´gold(III)dibromid tetrabromoaurat(III)
658 2-Phenylpyridin-1,1´gold(III)ethandtihiol Tabelle 3-5: Verbindung zur Aufklärung der Ramanbanden
Die Verbindungen 1, 2, 652, 654 und 656 sind gemessen worden, um die Schwingungen des
2-Phenylpyridins zuordnen zu können und die Schwingung der Bindungen zum Goldatom.
Zur Bestimmung der Schwingungen der am Gold gebunden Chloride wurden die analogen
Verbindungen mit Bromid Liganden dargestellt. Verbindung 658 soll die Verschiebungen der
Schwingung bei einer Bindung zu einer Peptidsequenz wiederspiegeln.
Zur Aufklärung der Schwingungsbanden wurden folgende Verbindungen noch zusätzlich von
Dr. Pablo Nieto aus der Physikalischen Chemie II der Ruhr Universität Bochum
quantenmechanisch berechnet, um die Spektren simulieren zu können: 2-Phenylpyridin-
1,1´gold(III)dichlorid 2, 2-Phenylpyridin-1,1´gold(III)dibromid 651 und 2-Phenylpyridin-
1,1´gold(III)ethandtihiol 658. Die Rechnungen wurden von in der Dichtefunktionaltheorie
(DFT) durchgeführt. Sie wurden in einem Gaussian09 Code, mit der Berichtigung C.01 unter
Benutzung des Hybridaustauschfunktionals B3LYP ausgeführt. Alle Elektronen der Hetero-
atome und der Chloratome wurden mit den Basissätzen 6-31G(d`) und VTZ berechnet. Mit
dem gleichen Basissätzen wurden die Elektronen der Bromatome berechnet, wobei sie mit
Pseudopotentialen behandelt wurden, um die Validität der Ergebnisse zu erhöhen. Die Elekt-
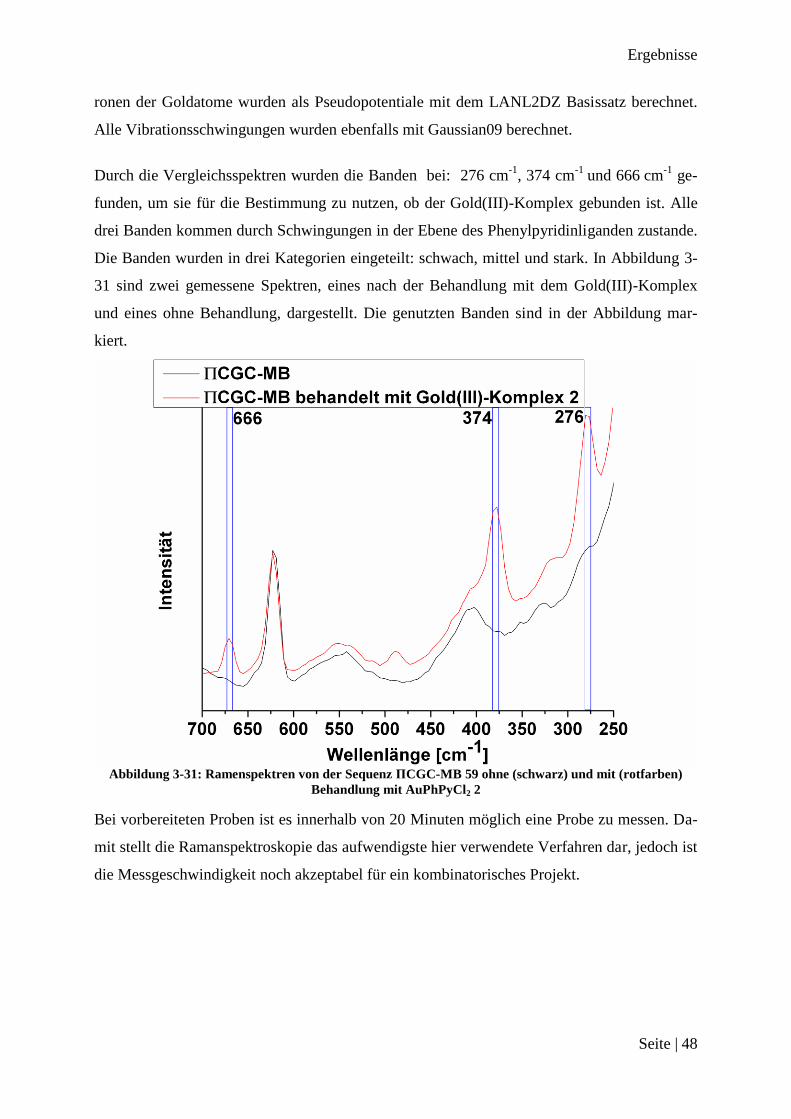
Ergebnisse
Seite | 48
ronen der Goldatome wurden als Pseudopotentiale mit dem LANL2DZ Basissatz berechnet.
Alle Vibrationsschwingungen wurden ebenfalls mit Gaussian09 berechnet.
Durch die Vergleichsspektren wurden die Banden bei: 276 cm-1
, 374 cm-1
und 666 cm-1
ge-
funden, um sie für die Bestimmung zu nutzen, ob der Gold(III)-Komplex gebunden ist. Alle
drei Banden kommen durch Schwingungen in der Ebene des Phenylpyridinliganden zustande.
Die Banden wurden in drei Kategorien eingeteilt: schwach, mittel und stark. In Abbildung 3-
31 sind zwei gemessene Spektren, eines nach der Behandlung mit dem Gold(III)-Komplex
und eines ohne Behandlung, dargestellt. Die genutzten Banden sind in der Abbildung mar-
kiert.
Abbildung 3-31: Ramenspektren von der Sequenz ΠCGC-MB 59 ohne (schwarz) und mit (rotfarben)
Behandlung mit AuPhPyCl2 2
Bei vorbereiteten Proben ist es innerhalb von 20 Minuten möglich eine Probe zu messen. Da-
mit stellt die Ramanspektroskopie das aufwendigste hier verwendete Verfahren dar, jedoch ist
die Messgeschwindigkeit noch akzeptabel für ein kombinatorisches Projekt.

Ergebnisse
Seite | 49
Auswertung des 300 Peptide Projekts
Über die Auswertung der Daten des kombinatorischen Projektes sollten nun ein stabiles Bin-
dungsmotiv ermittelt werden. Hierfür wurde die Bestimmung der Farbe der Harzkugel ge-
nutzt, die Daten des Fluoreszenzmikroskops und die Ramanspektren.
Auswertung der Farbe
Die meisten Harzkugeln sind farblos, wenige schwach gelbfarben, nach der Behandlung ohne
AuPhPyCl2 2. Mit AuPhPyCl2 2 behandelte Harzkugeln konnten farblos, gelbfarben oder vio-
lettfarben sein.
Farblose Harzkugeln sind ein Anzeichen für die Abwesenheit des Gold(III)-Komplexes, wenn
die Kugeln gelbfarben sind, dann ist dies ein Indiz für seine Anwesenheit. Jedoch konnten die
Kugeln auch gelbfarben ohne die Behandlung mit AuPhPyCl2 2 sein. Um dies zu klären wur-
den die beiden anderen Messverfahren eingesetzt. Die beiden anderen Messverfahren konnten
jedoch von den violettfarbenen Harzkugeln behindert werden, da diese Färbung auf kolloida-
les Gold hindeutet und die Fluoreszenz von Goldnanopartikeln bekannt ist.151
Bei den Ab-
standsaminosäuren Glycin und Prolin sind jeweils 13 Peptisequenzen violettfarben im sicht-
baren Wellenlängenbereich. Bei Leucin ist es eine Sequenz mehr. In Tabelle 3-6 ist die Häu-
figkeit von bindenden Aminosäuren in violettfarbenen Peptidsequenzen aufgelistet.
Bindende
AS
Vorkommen in
violetten Sequenzen
Z 19
A 17
K 15
D 11
E 6
J 6
B 3
H 2 Tabelle 3-6: Vorkommen von Bindendenaminosäuren in violettfarbenen Sequenzen
Es fällt auf, dass bei keiner der Sequenzen mit Methionin und Cystein sich die violette Farbe
ausgebildet hat. Wie häufig sich die Färbung bei welchen bindenden Aminosäuren gebildet
hat, ist ein für die Stabilität der Bindung der funktionellen Gruppe zum Gold(III)-Komplex

Ergebnisse
Seite | 50
wichtiges Indiz. Umso häufiger sich kolloidalles Gold bei einer Aminosäure gebildet hat, um-
so ungünstiger ist diese als Bindungspartner. Nach dieser Theorie sind die beiden, mit primä-
ren Aminen in der Seitengruppe ausgestatteten Aminosäuren 2,3-Diaminopropionsäure und
Lysin die ungünstigsten. Alanin ist fast ebenso ungünstig. Bei dieser Aminosäure ist nicht die
Funktionalität in der Seitenkette für die Reduktion verantwortlich. Möglicherweise liegt es
daran, dass aus der Peptidsequenz, welche einen zweizähnigen Liganden bilden sollte, ein
einzähniger Ligand geworden ist. Dieser Ligand stabilisierte den Gold(III)-Komplex 2 dann
nicht mehr in ausreichendem Maße und es bildete sich kolloidales Gold. Die nächsten beiden
Aminosäuren, die bei Verwendung zur Bildung der violetten Farbe führen können, sind Aspa-
raginsäure und Glutaminsäure. Die Carbonsäure in der Seitengruppe, scheint in manchen Fäl-
len ebenfalls zur Bildung von elementarem Gold beigetragen zu haben. Schwach scheint der
Effekt bei den drei aromatischen Aminosäuren 3-(4-Pyridin)-alanin, 3-(3-Pyridin)-alanin und
Histidin ausgeprägt gewesen zu sein. Cystein und Methionin scheinen demnach den
Gold(III)-Komplex 2 zu stabilisieren. Demnach ergibt sich folgende Reihenfolge für die Sta-
bilität:
Z ≈ A ≈ K < D < E ≈ J < B ≈ H < C ≈ M
instabil stabil
Auswertung der Fluoreszenzmikroskopie
Die im visuellen getätigte Negativkontrolle für die Stabilität der Gold(III)-Peptidkomplexe
sollte mit der Fluoreszenzmikroskopie durch eine Positivkontrolle bestätigt werden. Es wur-
den folgende Kugeln im UV-Licht beobachtet: blaufarbene Kugeln, blaufarbene Kugeln mit
gelbfarbenem Rand, farblose Kugeln, gelbfarbene Kugel und blaufarbene Kugeln mit
schwach gelbfarbenen Rand.
Im UV-Licht sind alle Kugeln ohne Behandlung mit AuPhPyCl2 2 blaufarben, daraus wird
geschlossen, dass auch die blaufarbenen Harzkugeln nach AuPhPyCl2 2 Behandlung nicht den
Komplex gebunden haben. Dieser Eindruck wird durch die Farbe im sichtbaren Bereich ge-
stützt, denn diese Kugeln sind fast immer farblos, mal leicht gelbfarben.

Ergebnisse
Seite | 51
Harzkugeln, die blaufarben mit einem gelbfarbenen Rand im UV-Licht des Fluoreszenzmik-
roskops sind, haben fast immer eine gelbe Farbe im visuellen Bereich. Vermutlich binden
diese Kugeln den Gold(III)-Komplex.
Beim Vergleich der Sequenzen fällt auf, dass es zu meist keinen Unterschied gibt, welche
Abstandsaminosäure verwendet worden ist, bei gleichen bindende Aminosäuren ist im Prinzip
das Ergebnis sowohl im visuellen-, als auch im UV-Bereich dasselbe. Dieses Prinzip ist für
Prolin nicht so stark ausgeprägt. Während 18 Sequenzen mit Prolin nach den Fluoreszenzda-
ten den Gold(III)-Komplex 2 nicht binden, kam es zur Bindung bei Glycin oder Leucin. Für
Glycin und Leucin war dies nur bei jeweils drei Sequenzen der Fall. Eine Erklärung hierfür ist
der veränderte Winkel in der Sequenz durch das Prolin.
Zweimal binden Peptidsequenzen mit Glycin als Abstandsaminosäure, wenn die gleichen Se-
quenzen mit Prolin oder Leucin nicht koppelten. Zwei Sequenzen mit Prolin taten das selbige.
Bei Leucin sind es fünf Sequenzen, welche nach den Fluoreszenzdaten den Gold(III)-
Komplex 2 zu binden schienen, während die gleichen Sequenzen mit Glycin oder Prolin dies
nicht beobachten lassen. Eine mögliche Erklärung für diese etwas höhere Bindefähigkeit des
Leucins ist der durch die Isopropylgruppe leicht veränderte Bindungswinkel.
Auswertung der Ramanspektren
Da die Auswertung der Fluoreszenzspektren nur qualitativ und nicht quantitativ ist und eine
Zuordnung der selten vorkommenden farblosen Kugeln und gelbfarbene Kugel sowie der
häufiger vorkommenden blaufarbene Kugeln mit schwach gelbfarbenen Rand nicht möglich
ist, wurden von den ersten 100 Sequenzen, welche mit AuPhPyCl2 2 behandelt worden waren,
Ramanspektren gemessen.15
Die quantitativen Ramanspektren zeigen, dass nur die Kugeln
blaufarbene Kugeln mit gelbfarbenen Rand bei allen drei Banden eine mittlere oder eine star-
ke Ausprägung der Banden haben. Diese Kugeln haben den Gold(III)-Komplex gebunden.
Dies trifft auf folgende Verbindungen zu:
Nummer Name
350 ΠCGC-MBAuPhPy
351 ΠMGC-MBAuPhPy
354 ΠDGC-MBAuPhPy
355 ΠEGC-MBAuPhPy
356 ΠZGC-MBAuPhPy

Ergebnisse
Seite | 52
357 ΠBGC-MBAuPhPy
360 ΠCGM-MBAuPhPy
361 ΠMGM-MBAuPhPy
362 ΠHGM-MBAuPhPy
365 ΠEGM-MBAuPhPy
366 ΠZGM-MBAuPhPy
367 ΠBGM-MBAuPhPy
369 ΠKGM-MBAuPhPy
370 ΠCGH-MBAuPhPy
371 ΠMGH-MBAuPhPy
380 ΠCGA-MBAuPhPy
381 ΠMGA-MBAuPhPy
391 ΠMGD-MBAuPhPy
400 ΠCGE-MBAuPhPy
401 ΠMGE-MBAuPhPy
410 ΠCGZ-MBAuPhPy
411 ΠMGZ-MBAuPhPy
431 ΠMGJ-MBAuPhPy
441 ΠMGK-MBAuPhPy Tabelle 3-7: Verbindungen die den Gold(III)-Komplex binden
Wird das Auftreten der einzelnen bindenden Aminosäuren gezählt, dann kann folgende Liste
für die Stabilität der bindenden Aminosäuren daraus abgeleitet werden:
A ≈ J < D ≈ K ≈ B < E ≈ Z ≈ H < C < M
instabil stabil
Diskussion zu dem 300 Peptideprojekt
Die beiden in dem Projekt erstellten Stabilitätslisten zeigen, dass Cystein und Methionin die
beiden günstigsten Aminosäuren sind, um AuPhPyCl2 2 zu binden. Sequenzen mit günstigen
Aminosäuren sind in Tabelle 3-7 abgebildet. Die Tabelle 3-7 ist aus den Daten der Fluores-
zenzspektren und vor allem aus den Ramanspektren erstellt worden. Es ergeben sich aus den
Daten somit viele Verbindungen, die den Gold(III)-Komplex gebunden haben. Aus diesen
Verbindungen wurde die Stabilitätsliste erstellt. Bei der Zuordnung zur zweiten Liste, wurden
einige Verbindungen nicht berücksichtigt. Die Verbindungen ΠDGM-MBAuPhPy 373,
ΠCGB-MBAuPhPy 429 und ΠMGB-MBAuPhPy 430 haben einen gold/gelbfarbenen Rand
bei der Betrachtung im UV-Licht des Fluoreszenzmikroskops. Im Raman Spektrum sind bei
den Verbindungen die ersten beiden Banden mittel ausgeprägt, jedoch ist bei ihnen die Bande
bei 666 cm-1
nur schwach ausgeprägt. ΠAGM-MBAuPhPy 372 zeigt, trotz des Randes, nur
bei 374 cm-1
eine mittlere Bande. Die anderen Banden sind schwach ausgeprägt. Schwach

Ergebnisse
Seite | 53
sind alle Banden, trotz eines gold/gelbfarbenen Randes, bei den Verbindungen
ΠHGC-MBAuPhPy 361, ΠAGC-MBAuPhPy 362, ΠJGM-MBAuPhPy 377,
ΠCGD-MBAuPhPy 399 und ΠCGJ-MBAuPhPy 439. Die Verbindungen ΠJGE-MBAuPhPy
417, ΠDGJ-MBAuPhPy 443, ΠEGJ-MBAuPhPy 444, ΠBGJ-MBAuPhPy 446 und
ΠJGJ-MBAuPhPy 447 sind im UV-Licht des Fluoreszenzmikroskops blaufarben oder blau-
farben mit schwachem gold/gelbfarbenen Rand, haben jedoch neben den beiden schwachen
Banden 276 cm-1
und 374 cm-1
, auch eine mittlere Bande bei 666 cm-1
. ΠHGH-MBAuPhPy
381 und ΠEGH-MBAuPhPy 384 sind blaufarben beziehungsweise blaufarben mit schwach
gold/gelbfarbenen Rand, haben jedoch eine mittlere Bande bei 276 cm-1
. Das blaufarbene
ΠAGZ-MBAuPhPy 422 hat eine mittlere Band bei 374 cm-1
im Spektrum. ΠJGC-MBAuPhPy
367 und ΠCGE-MBAuPhPy 409 mit schwach gold/gelbfarbenen Rand hat mittlere Banden
bei allen drei Referenzwerten. Es kommen besonders häufig bei den Abweichungen die bei-
den Aminosäuren 3-(4-Pyridin)-alanin und Cystein vor. Jeweils eine als besonders günstige
und eine nicht besonders stabil eingeordnete Aminosäure. Lysin ist bei keiner abweichenden
Sequenz vorhanden, 2,3-Diaminopropionsäure befindet sich nur in einer. Die anderen Amino-
säuren sind gleich häufig in den Sequenzen vorhanden.
Bei den Sequenzen gibt es einen gemeinsamen Trend in allen Messmethoden, jedoch entzie-
hen sich einige Sequenzen diesem Trend. Dies kann dadurch zustande gekommen sein, dass
einige Banden als schwach einsortiert werden, aber doch schon fast die Größe einer mittleren
Bande erreicht haben. Dies erklärt zum Teil die oben abweichenden Werte zwischen Fluores-
zenzmikroskopie und Ramanspektroskopie. Eine weitere Erklärung für die Abweichung sind
störende Teilchen, wie zum Beispiel die violettfarbenen Goldnanopartikel. Die Bildung von
anderen Spezies ist daneben denkbar, die dann eine Bande bei 276 cm-1
, 374 cm-1
oder
666 cm-1
haben.
Stabilität in Lösung
Bei der Synthese der 300 Peptidsequenzen am Harz ohne Linker sind sehr viele potentielle
Bindungsmotive entdeckt worden. Um diese Entdeckungen unter dem Fluoreszenzmikroskop
und im Ramanspektrometer zu stützen, wurden einige der potentiell bindenden Peptidse-
quenzen an einem Harz mit Linker dargestellt. Diese potentiell bindenden Peptidsequenzen
müssen mit AuPhPyCl2 2 in Lösung getestet werden, um die langfristige Stabilität des entste-

Ergebnisse
Seite | 54
henden Adduktes zu klären und eine in der Praxis verwendbare Sequenz zu finden. Wichtig
für die Praxis ist ein Bindungsmotiv, welches über mehrere Tage in Lösung stabil ist.
Zur Einschränkung der Auswahl der möglichen stabilen Bindungsmotive, wurden nur Se-
quenzen mit einem Glycin als Abstandsaminosäure AS2 gewählt. Es wurde also nicht aus al-
len 300 Peptiden ausgewählt, sondern nur aus den ersten 100 Peptiden. Von diesen 100 Se-
quenzen wurden dann solche Peptide ausgewählt, die intensive Hinweise zur Bindung des
Gold(III)-Komplexes 2 im Fluoreszenzmikroskop oder im Ramanspektrometer geliefert ha-
ben. Zwei Sequenzen wurden ausgewählt, da sich bei ihnen das Harz violett gefärbt hatte. Es
ergaben sich folgende Sequenzen:
Nummer Name
659 ΠCGC-NH2
660 ΠMGC-NH2
661 ΠEGC-NH2
662 ΠZGC-NH2
663 ΠBGC-NH2
664 ΠCGM-NH2
665 ΠZGM-NH2
666 ΠJGM-NH2
667 ΠKGM-NH2
668 ΠCGH-NH2
669 ΠMGH-NH2
670 ΠCGA-NH2
671 ΠMGA-NH2
672 ΠZGD-NH2
673 ΠKGD-NH2
674 ΠCGZ-NH2
675 ΠDGZ-NH2
676 ΠZGZ-NH2
677 ΠCGB-NH2
678 ΠBGB-NH2
679 ΠJGB-NH2
680 ΠCGJ-NH2
681 ΠMGK-NH2 Tabelle 3-8 Sequenzen für Tests in Lösung
Diese Sequenzen wurden mittels der Festphasenpeptidsynthese dargestellt, vom Harz abge-
spalten und in Lösung mit AuPhPyCl2 2 umgesetzt. Es wurde immer nach einer Stunde und
nach fünf Stunden eine analytische HPLC-Messung von der Reaktionslösung durchgeführt.
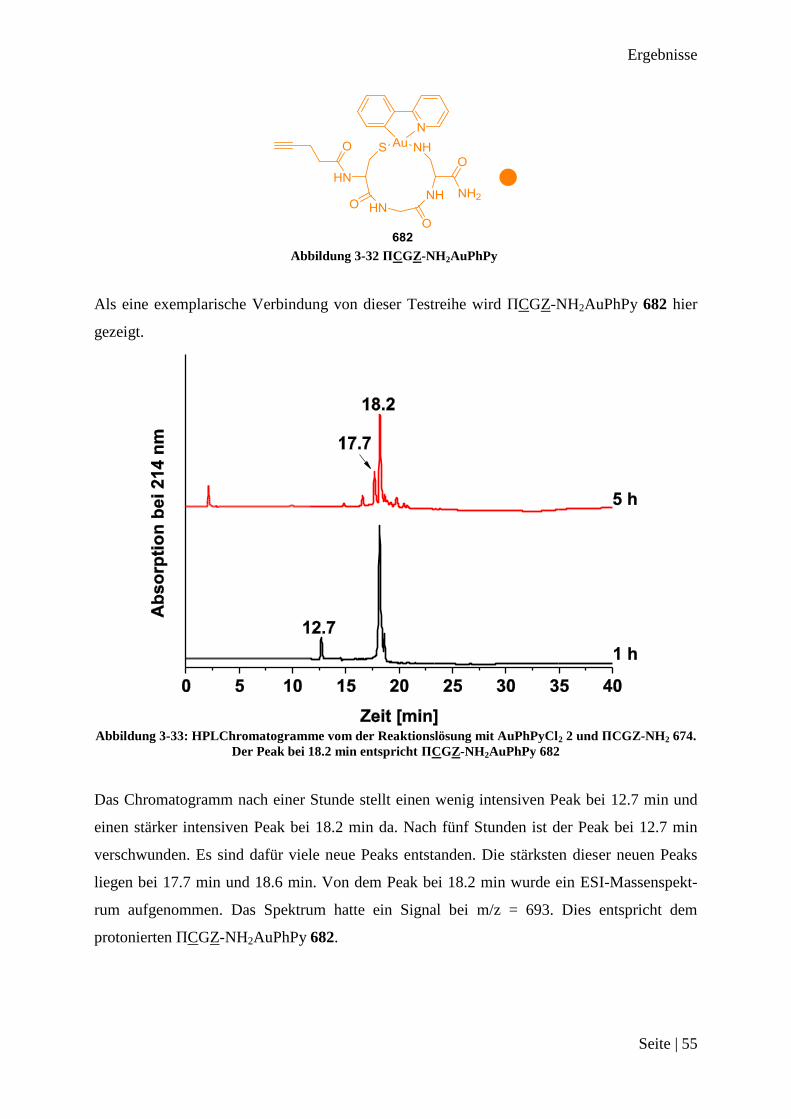
Ergebnisse
Seite | 55
Abbildung 3-32 ΠCGZ-NH2AuPhPy
Als eine exemplarische Verbindung von dieser Testreihe wird ΠCGZ-NH2AuPhPy 682 hier
gezeigt.
Abbildung 3-33: HPLChromatogramme vom der Reaktionslösung mit AuPhPyCl2 2 und ΠCGZ-NH2 674.
Der Peak bei 18.2 min entspricht ΠCGZ-NH2AuPhPy 682
Das Chromatogramm nach einer Stunde stellt einen wenig intensiven Peak bei 12.7 min und
einen stärker intensiven Peak bei 18.2 min da. Nach fünf Stunden ist der Peak bei 12.7 min
verschwunden. Es sind dafür viele neue Peaks entstanden. Die stärksten dieser neuen Peaks
liegen bei 17.7 min und 18.6 min. Von dem Peak bei 18.2 min wurde ein ESI-Massenspekt-
rum aufgenommen. Das Spektrum hatte ein Signal bei m/z = 693. Dies entspricht dem
protonierten ΠCGZ-NH2AuPhPy 682.
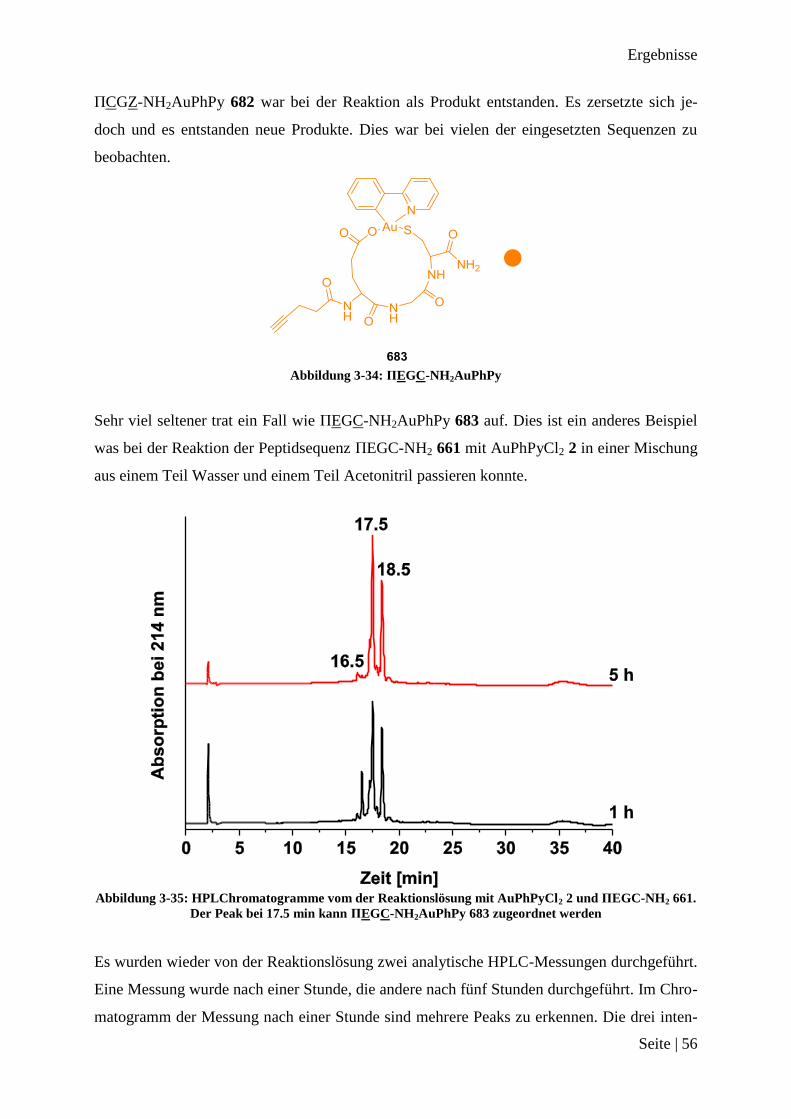
Ergebnisse
Seite | 56
ΠCGZ-NH2AuPhPy 682 war bei der Reaktion als Produkt entstanden. Es zersetzte sich je-
doch und es entstanden neue Produkte. Dies war bei vielen der eingesetzten Sequenzen zu
beobachten.
Abbildung 3-34: ΠEGC-NH2AuPhPy
Sehr viel seltener trat ein Fall wie ΠEGC-NH2AuPhPy 683 auf. Dies ist ein anderes Beispiel
was bei der Reaktion der Peptidsequenz ΠEGC-NH2 661 mit AuPhPyCl2 2 in einer Mischung
aus einem Teil Wasser und einem Teil Acetonitril passieren konnte.
Abbildung 3-35: HPLChromatogramme vom der Reaktionslösung mit AuPhPyCl2 2 und ΠEGC-NH2 661.
Der Peak bei 17.5 min kann ΠEGC-NH2AuPhPy 683 zugeordnet werden
Es wurden wieder von der Reaktionslösung zwei analytische HPLC-Messungen durchgeführt.
Eine Messung wurde nach einer Stunde, die andere nach fünf Stunden durchgeführt. Im Chro-
matogramm der Messung nach einer Stunde sind mehrere Peaks zu erkennen. Die drei inten-

Ergebnisse
Seite | 57
sivsten Peaks waren nach 16.5 min, 17.5 min und 18.5 min. Nach fünf Stunden ist das Signal
bei 16.5 min gänzlich verschwunden und die beiden anderen Signale sind noch zu erkennen.
Der Peak nach 17.5 min wurde isoliert und mittels eines ESI-MS charakterisiert. Es ist ein
Signal bei m/z = 735 zu beobachten. Dieses Signal entspricht der Masse von
ΠEGC-NH2AuPhPy 683. Die Verbindung 683 wurde in Wasser gelöst und eine analytische
HPLC wurde nach einer halben Stunde von der Lösung gemessen. Das Chromatogramm ent-
spricht dem oben gezeigten der Reaktionslösungsmessung nach fünf Stunden. In Lösung ist
ΠEGC-NH2AuPhPy 683 somit nicht stabil.
Durch Zugabe von AuPhPyCl2 2 zu den oben gezeigten Peptiden und nach der Charakterisie-
rung mittels der analytischen HPLC und der ESI-MS konnten folgende Verbindungen erhal-
ten werden:
Nummer Name
684 ΠCGC-NH2AuPhPy
685 ΠMGC-NH2AuPhPy
683 ΠEGC-NH2AuPhPy
686 ΠZGC-NH2AuPhPy
687 ΠBGC-NH2AuPhPy
688 ΠCGM-NH2AuPhPy
689 ΠZGM-NH2AuPhPy
690 ΠJGM-NH2AuPhPy
691 ΠKGM-NH2AuPhPy
692 ΠCGH-NH2AuPhPy
693 ΠMGH-NH2AuPhPy
694 ΠCGA-NH2AuPhPy
695 ΠMGA-NH2AuPhPy
696 ΠKGD-NH2AuPhPy
682 ΠCGZ-NH2AuPhPy
697 ΠDGZ-NH2AuPhPy
698 ΠZGZ-NH2AuPhPy
699 ΠCGB-NH2AuPhPy
700 ΠJGB-NH2AuPhPy
701 ΠCGJ-NH2AuPhPy
702 ΠMGK-NH2AuPhPy Tabelle 3-9: Sequenzen die den Gold(III)-Komplex binden
Der Reaktionsverlauf zur Synthese dieser Verbindungen wurden immer nach einer und nach
fünf Stunden mit der analytischen HPLC kontrolliert. Die meisten der Kontrollen zur Synthe-
se dieser Verbindungen verhielten sich, analog zum oben beschrieben Fall für
ΠCGZ-NH2AuPhPy 682. Das Produkt bildete sich am Anfang und zersetzte sich dann in Ne-
benprodukte. Ein sehr viel seltenerer Fall verlief analog zu ΠEGC-NH2AuPhPy 683. Es sind

Ergebnisse
Seite | 58
im Chromatogramm nach einer Stunde viele Peaks zu erkennen, deren Anzahl nach fünf
Stunden abgenommen hat.
Zusammenfassung des 300 Peptideprojekts
Von den Peptidsequenzen in Lösung haben fast alle AuPhPyCl2 2 gebunden. Keine der
Peptidsequenzen bindet jedoch den Gold(III)-Komplex 2 über längere Zeit stabil. Es ist durch
das Kombinatorische Projekt herausgefunden worden, dass von den eingesetzten bindenden
Aminosäuren: Alanin, Asparaginsäure, 2,3-Diaminopropionsäure, Cystein, Glutaminsäure,
Histidin, Lysin, Methionin, 3-(3-Pyridin)-alanin und 3-(4-Pyridin)-alanin zum AuPhPyCl2 2
koordinierten und lediglich das als Blindprobe eingesetzte Alanin überhaupt nicht gebunden
hat. Die beiden mit Carbonsäurefunktion eingesetzten Aminosäuren Glutaminsäure und Aspa-
raginsäure bauten die instabilste Bindung zum AuPhPyCl2 2 auf. Die über die Stickstoffatome
koordinierenden Aminosäuren Lysin, Histidin, 3-(3-Pyridin)-alanin, 3-(4-Pyridin)-alanin und
2,3-Diaminopropionsäure binden etwa gleich stabil zum Gold(III)-Komplex 2. Es fällt jedoch
tendenziell ein Unterschied zwischen Lysin und 2,3-Diaminopropionsäure auf. Beide tragen
ein primäres Amin als Bindungspartner und unterschieden sich nur in der Länge der Alkylket-
te. Trotzdem schien 2,3-Diaminopropionsäure etwas besser zu binden. Dies kann an den güns-
tigeren Bindungswinkeln durch die kürzere Alkylkette gelegen haben. Histidin, 3-(3-Pyridin)-
alanin und 3-(4-Pyridin)-alanin wichen kaum bei den Untersuchungen voneinander ab. Me-
thionin und Cystein erwiesen sich als die Aminosäuren, welche die stabilste Bindung zum
AuPhPyCl2 2 aufgebaut haben. Cystein erwies sich hierbei als noch etwas stärker in seiner
Bindefähigkeit.
A < E ≈ D < K ≈ Z < H ≈ B ≈ J < M < C
instabil stabil
Mehr Abstandsaminosäuren
Das Projekt der 300 Peptidsequenzen hat eine Vielzahl von neuen Bindungsmotiven hervor-
gebracht. Jedoch bildete keines der Bindungsmotive einen über fünf Stunden stabilen Kom-
plex mit AuPhPyCl2 2, aber es wurden gezeigt wie gut die bindenden Aminosäuren zum
Gold(III)-Komplex binden. Dadurch hat sich bei den bisher getätigten Versuche Cystein als
die beste bindende Aminosäure herausgestellt. Eine mögliche Ursache, warum
ΠCGC-NH2AuPhPy 684 dennoch keinen stabilen Komplex hervorgebracht hat, kann eine zu
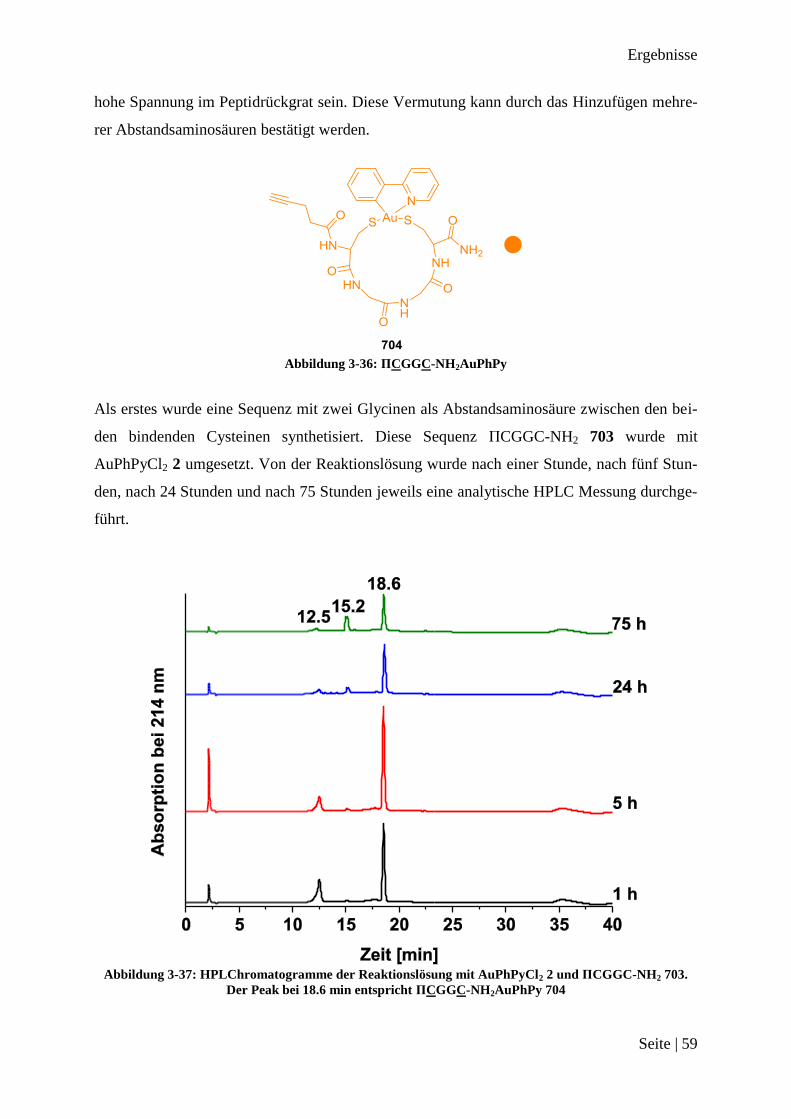
Ergebnisse
Seite | 59
hohe Spannung im Peptidrückgrat sein. Diese Vermutung kann durch das Hinzufügen mehre-
rer Abstandsaminosäuren bestätigt werden.
Abbildung 3-36: ΠCGGC-NH2AuPhPy
Als erstes wurde eine Sequenz mit zwei Glycinen als Abstandsaminosäure zwischen den bei-
den bindenden Cysteinen synthetisiert. Diese Sequenz ΠCGGC-NH2 703 wurde mit
AuPhPyCl2 2 umgesetzt. Von der Reaktionslösung wurde nach einer Stunde, nach fünf Stun-
den, nach 24 Stunden und nach 75 Stunden jeweils eine analytische HPLC Messung durchge-
führt.
Abbildung 3-37: HPLChromatogramme der Reaktionslösung mit AuPhPyCl2 2 und ΠCGGC-NH2 703.
Der Peak bei 18.6 min entspricht ΠCGGC-NH2AuPhPy 704

Ergebnisse
Seite | 60
Das Chromatogramm für die Messung nach einer Stunde hat zwei Peaks. Der erste Peak wird
bei 12.5 min beobachtet und der zweite Peak kann bei 18.6 min erkannt werden. Der Peak bei
12.5 min verliert im Vergleich zum Peak bei 18.6 min an Intensität. Nach 24 Stunden ist der
Peak nach 12.5 min gänzlich verschwunden, der Peak bei 18.6 min jedoch immer noch zu
erkennen, dafür sind mehrere Peaks um 15.1 min entstanden. Die Peaks um 15.1 min sind bei
75 Stunden noch intensiver geworden. Vom Peak bei 18.6 min wurde ein ESI- Massenspekt-
rum aufgenommen. Dieses Massenspektrum lässt eine Masse von m/z = 767 beobachten.
Diese Masse passte zur protonierten Verbindung 704.
Die abstandhaltende Glycinkette zu verlängern, schien die Fähigkeit der Sequenz zu erhöhen
einen stabilen Komplex mit AuPhPyCl2 2 auszubilden. ΠCGGC-NH2 703 bildete im Ver-
gleich zu ΠCGC-NH2 659 einen stabileren Komplex aus. Die Chromatogramme von Kom-
plex ΠCGGC-NH2AuPhPy 704 zeigen, dass die Verbindung 704 sich immer noch zersetzt.
Aufgrund dieser Zersetzung wurde die Kette noch um eine weitere Aminosäure verlängert.
Abbildung 3-38: ΠCGGGC-NH2AuPhPy
ΠCGGGC-NH2 705 wurde zusammen mit Verbindung 2 in einer Lösungsmittelmischung aus
Wasser und Acetonitril zusammen gebracht. Es wurden erneut zeitabhängige Messungen auf
der analytischen HPLC durchgeführt.
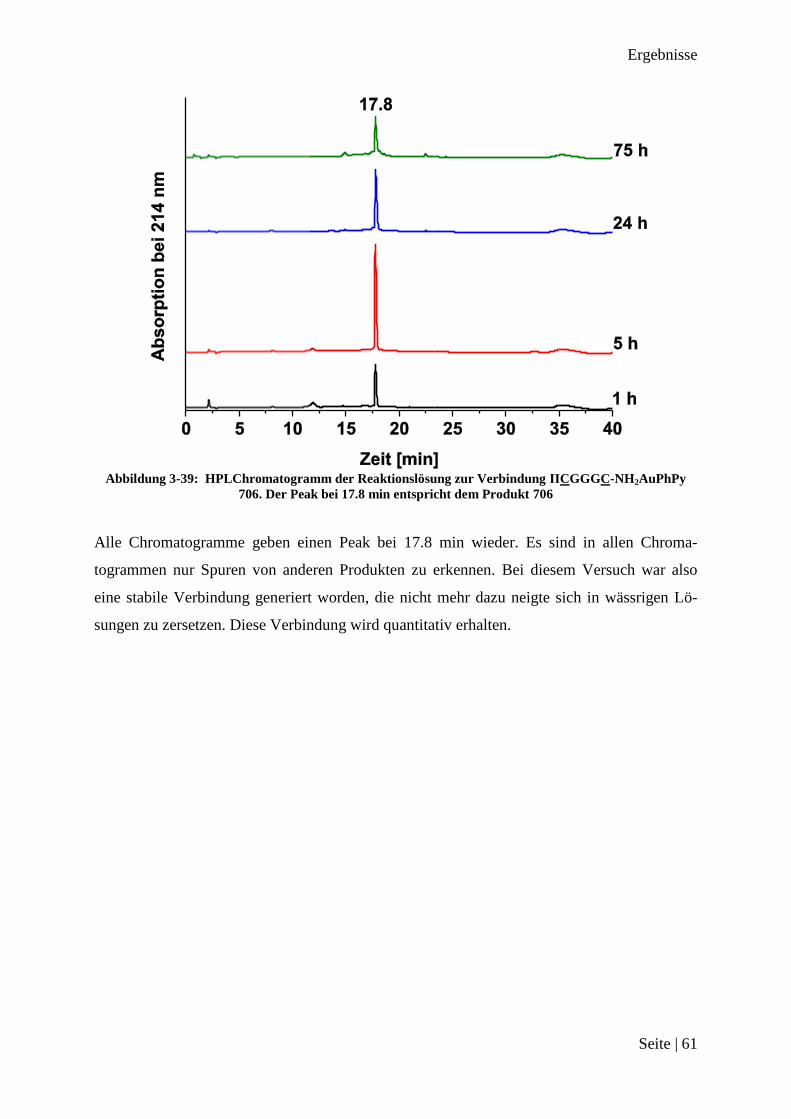
Ergebnisse
Seite | 61
Abbildung 3-39: HPLChromatogramm der Reaktionslösung zur Verbindung ΠCGGGC-NH2AuPhPy
706. Der Peak bei 17.8 min entspricht dem Produkt 706
Alle Chromatogramme geben einen Peak bei 17.8 min wieder. Es sind in allen Chroma-
togrammen nur Spuren von anderen Produkten zu erkennen. Bei diesem Versuch war also
eine stabile Verbindung generiert worden, die nicht mehr dazu neigte sich in wässrigen Lö-
sungen zu zersetzen. Diese Verbindung wird quantitativ erhalten.
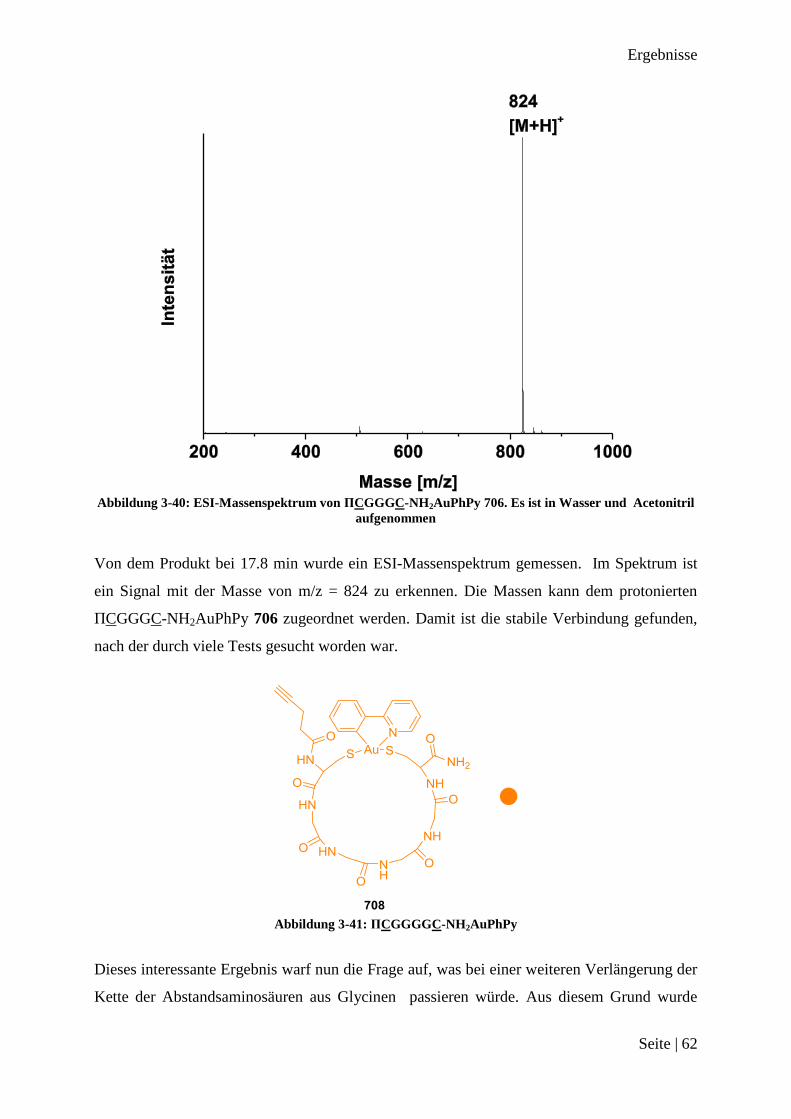
Ergebnisse
Seite | 62
Abbildung 3-40: ESI-Massenspektrum von ΠCGGGC-NH2AuPhPy 706. Es ist in Wasser und Acetonitril
aufgenommen
Von dem Produkt bei 17.8 min wurde ein ESI-Massenspektrum gemessen. Im Spektrum ist
ein Signal mit der Masse von m/z = 824 zu erkennen. Die Massen kann dem protonierten
ΠCGGGC-NH2AuPhPy 706 zugeordnet werden. Damit ist die stabile Verbindung gefunden,
nach der durch viele Tests gesucht worden war.
Abbildung 3-41: ΠCGGGGC-NH2AuPhPy
Dieses interessante Ergebnis warf nun die Frage auf, was bei einer weiteren Verlängerung der
Kette der Abstandsaminosäuren aus Glycinen passieren würde. Aus diesem Grund wurde
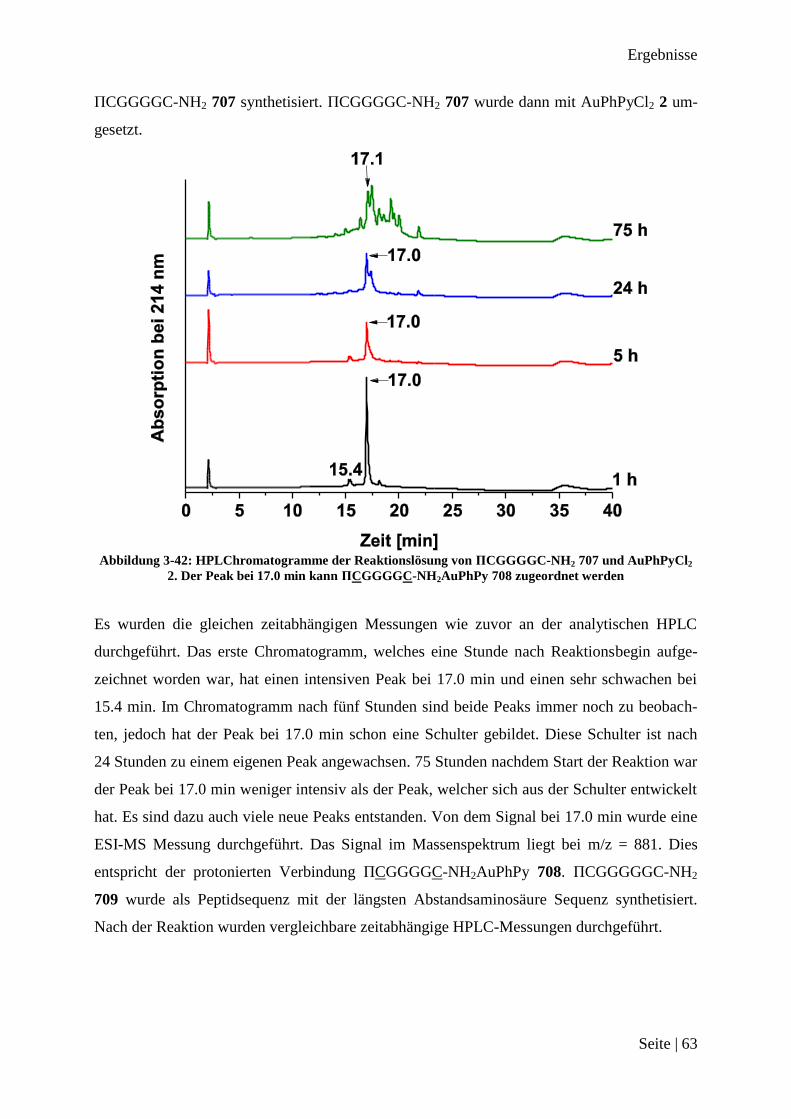
Ergebnisse
Seite | 63
ΠCGGGGC-NH2 707 synthetisiert. ΠCGGGGC-NH2 707 wurde dann mit AuPhPyCl2 2 um-
gesetzt.
Abbildung 3-42: HPLChromatogramme der Reaktionslösung von ΠCGGGGC-NH2 707 und AuPhPyCl2
2. Der Peak bei 17.0 min kann ΠCGGGGC-NH2AuPhPy 708 zugeordnet werden
Es wurden die gleichen zeitabhängigen Messungen wie zuvor an der analytischen HPLC
durchgeführt. Das erste Chromatogramm, welches eine Stunde nach Reaktionsbegin aufge-
zeichnet worden war, hat einen intensiven Peak bei 17.0 min und einen sehr schwachen bei
15.4 min. Im Chromatogramm nach fünf Stunden sind beide Peaks immer noch zu beobach-
ten, jedoch hat der Peak bei 17.0 min schon eine Schulter gebildet. Diese Schulter ist nach
24 Stunden zu einem eigenen Peak angewachsen. 75 Stunden nachdem Start der Reaktion war
der Peak bei 17.0 min weniger intensiv als der Peak, welcher sich aus der Schulter entwickelt
hat. Es sind dazu auch viele neue Peaks entstanden. Von dem Signal bei 17.0 min wurde eine
ESI-MS Messung durchgeführt. Das Signal im Massenspektrum liegt bei m/z = 881. Dies
entspricht der protonierten Verbindung ΠCGGGGC-NH2AuPhPy 708. ΠCGGGGGC-NH2
709 wurde als Peptidsequenz mit der längsten Abstandsaminosäure Sequenz synthetisiert.
Nach der Reaktion wurden vergleichbare zeitabhängige HPLC-Messungen durchgeführt.
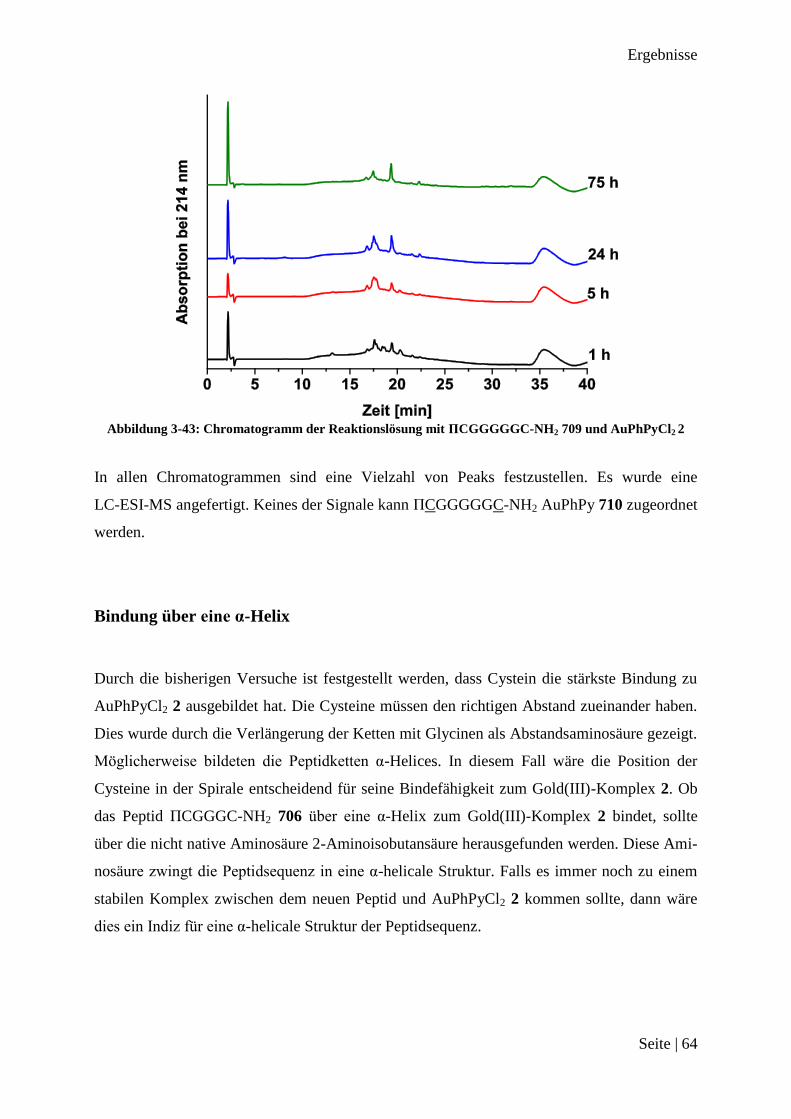
Ergebnisse
Seite | 64
Abbildung 3-43: Chromatogramm der Reaktionslösung mit ΠCGGGGGC-NH2 709 und AuPhPyCl2 2
In allen Chromatogrammen sind eine Vielzahl von Peaks festzustellen. Es wurde eine
LC-ESI-MS angefertigt. Keines der Signale kann ΠCGGGGGC-NH2 AuPhPy 710 zugeordnet
werden.
Bindung über eine α-Helix
Durch die bisherigen Versuche ist festgestellt werden, dass Cystein die stärkste Bindung zu
AuPhPyCl2 2 ausgebildet hat. Die Cysteine müssen den richtigen Abstand zueinander haben.
Dies wurde durch die Verlängerung der Ketten mit Glycinen als Abstandsaminosäure gezeigt.
Möglicherweise bildeten die Peptidketten α-Helices. In diesem Fall wäre die Position der
Cysteine in der Spirale entscheidend für seine Bindefähigkeit zum Gold(III)-Komplex 2. Ob
das Peptid ΠCGGGC-NH2 706 über eine α-Helix zum Gold(III)-Komplex 2 bindet, sollte
über die nicht native Aminosäure 2-Aminoisobutansäure herausgefunden werden. Diese Ami-
nosäure zwingt die Peptidsequenz in eine α-helicale Struktur. Falls es immer noch zu einem
stabilen Komplex zwischen dem neuen Peptid und AuPhPyCl2 2 kommen sollte, dann wäre
dies ein Indiz für eine α-helicale Struktur der Peptidsequenz.
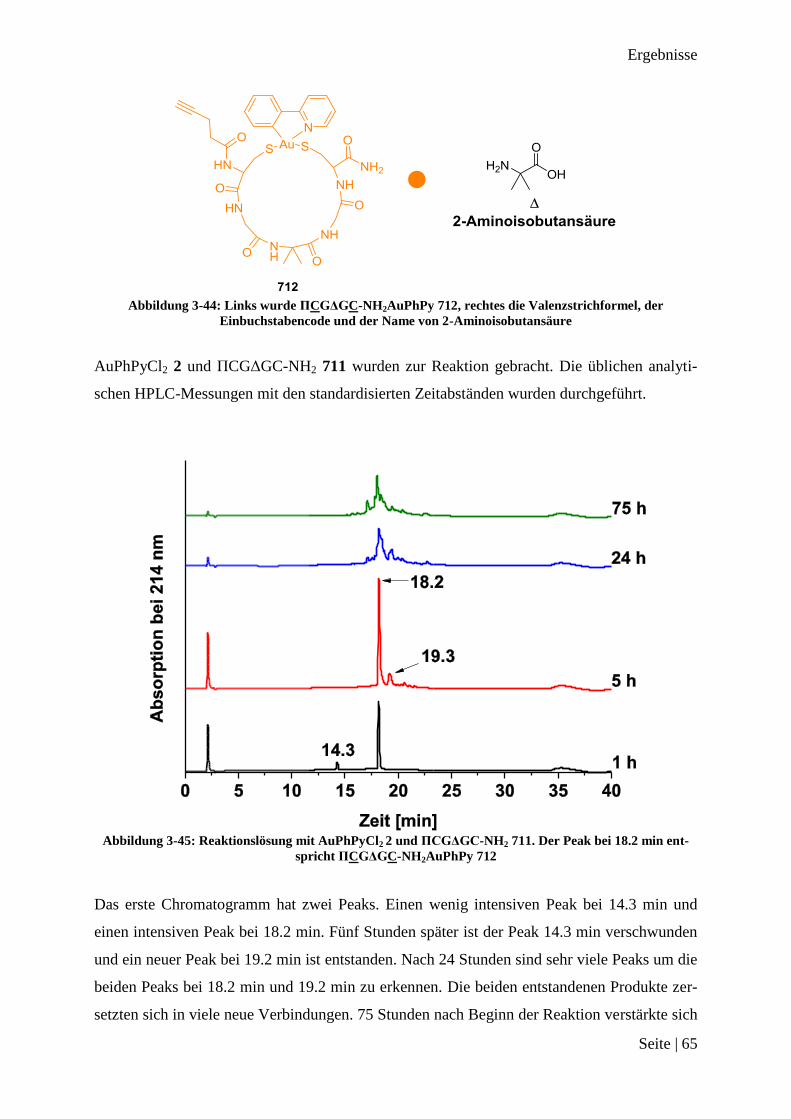
Ergebnisse
Seite | 65
Abbildung 3-44: Links wurde ΠCGΔGC-NH2AuPhPy 712, rechtes die Valenzstrichformel, der
Einbuchstabencode und der Name von 2-Aminoisobutansäure
AuPhPyCl2 2 und ΠCGΔGC-NH2 711 wurden zur Reaktion gebracht. Die üblichen analyti-
schen HPLC-Messungen mit den standardisierten Zeitabständen wurden durchgeführt.
Abbildung 3-45: Reaktionslösung mit AuPhPyCl2 2 und ΠCGΔGC-NH2 711. Der Peak bei 18.2 min ent-
spricht ΠCGΔGC-NH2AuPhPy 712
Das erste Chromatogramm hat zwei Peaks. Einen wenig intensiven Peak bei 14.3 min und
einen intensiven Peak bei 18.2 min. Fünf Stunden später ist der Peak 14.3 min verschwunden
und ein neuer Peak bei 19.2 min ist entstanden. Nach 24 Stunden sind sehr viele Peaks um die
beiden Peaks bei 18.2 min und 19.2 min zu erkennen. Die beiden entstandenen Produkte zer-
setzten sich in viele neue Verbindungen. 75 Stunden nach Beginn der Reaktion verstärkte sich

Ergebnisse
Seite | 66
dieser Effekt noch. Von dem Peak bei 18.2 min war ein ESI-MS aufgenommen worden. Das
Signal liegt bei m/z = 852. Dies entspricht der protonierten Masse von
ΠCGΔGC-NH2AuPhPy 712.
Nach diesen Ergebnissen scheint keine Peptid α-Helix den GoldKomplex 2-Phenylpyridin-
1,1´gold(III) stabil zu binden. Wenn die Sequenz in die Struktur einer α-Helix durch 2-Ami-
noisobutansäure gezwungen wurde, entstand zwar ΠCGΔGC-NH2AuPhPy 712, jedoch ist
dies das kinetisch stabile Produkt, doch nicht das thermodynamisch stabilste.
ΠCGΔGC-NH2AuPhPy 712 zersetzte sich wieder und es entstehen viele neue Produkte. Die
Peptidsequenz ΠCGGGC-NH2 706 scheint nicht in einer α-helicalen Struktur in
ΠCGGGC-NH2AuPhPy 707 vorzuliegen.
Erhöhung der Löslichkeit in Wasser
Nachdem der erste Versuch eine neue Abstandsaminosäure in das Bindungsmotiv 706 nicht
zu einer neuen stabil chelatisierenden Sequenz geführt hatte, sollte das Peptid weiter modifi-
ziert werden. Das Bindungsmotiv sollte so modifiziert werden, dass es besser in Wasser lös-
lich sein sollte. Die Löslichkeit von ΠCGGGC-NH2AuPhPy 707 in Wasser ist nur mäßig,
schon bei Konzentrationen von 1 mol/l fällt es aus Lösung aus. Um höhere Löslichkeit in
Wasser zu erreichen, sollten analog zu Verbindung ΠCRC-NH2AuPhPy 49 die Glycine durch
Arginin ausgetauscht werden.
Abbildung 3-46: ΠCRRRC-NH2AuPhPy
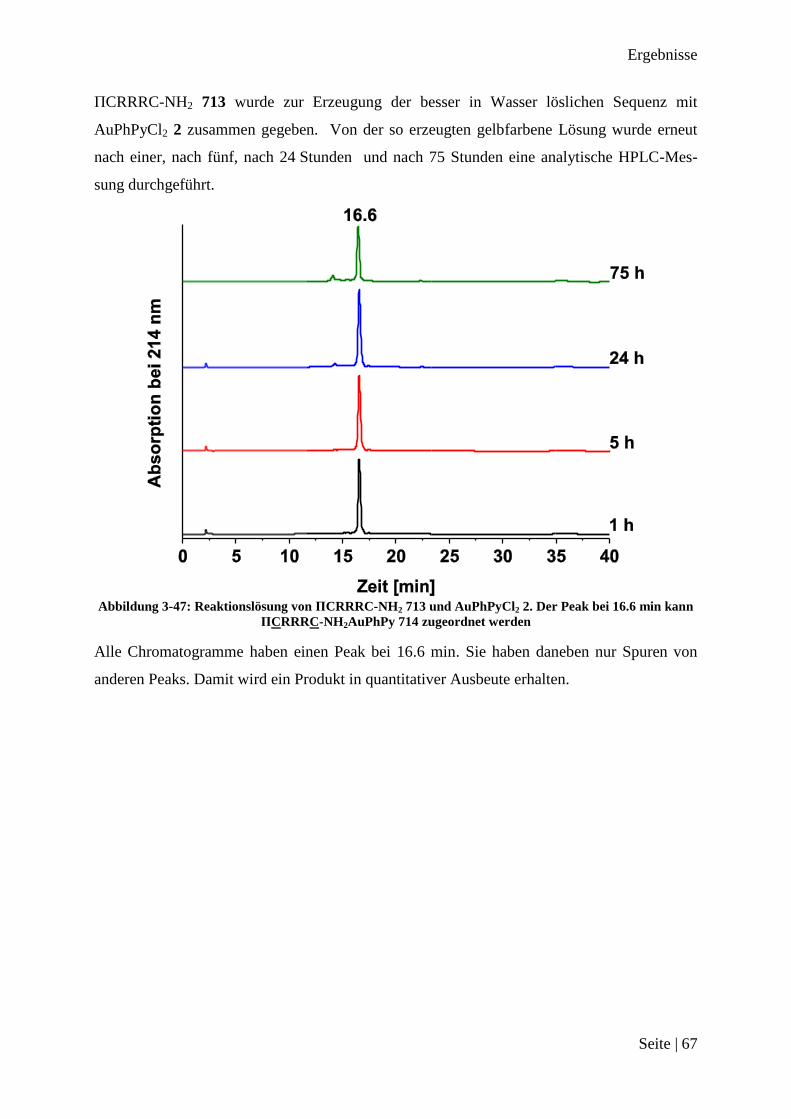
Ergebnisse
Seite | 67
ΠCRRRC-NH2 713 wurde zur Erzeugung der besser in Wasser löslichen Sequenz mit
AuPhPyCl2 2 zusammen gegeben. Von der so erzeugten gelbfarbene Lösung wurde erneut
nach einer, nach fünf, nach 24 Stunden und nach 75 Stunden eine analytische HPLC-Mes-
sung durchgeführt.
Abbildung 3-47: Reaktionslösung von ΠCRRRC-NH2 713 und AuPhPyCl2 2. Der Peak bei 16.6 min kann
ΠCRRRC-NH2AuPhPy 714 zugeordnet werden
Alle Chromatogramme haben einen Peak bei 16.6 min. Sie haben daneben nur Spuren von
anderen Peaks. Damit wird ein Produkt in quantitativer Ausbeute erhalten.

Ergebnisse
Seite | 68
Abbildung 3-48: ESI-Massenspektrum von ΠCRRRC-NH2AuPhPy 714. Gemessen in Wasser und Aceto-
nitril
Es wurde von der neuen Verbindung ein ESI Massenspektrum aufgenommen. Das Spektrum
hat ein Signal bei m/z = 561. Dies entspricht der zweifach geladenen Masse von
ΠCRRRC-NH2AuPhPy 714. Damit ist es gelungen, ein stabiles und gut in Wasser lösliches
Biokonjugat darzustellen.
Zusammenfassung der Kettenverlängerung
Die Kettenverlängerung mit zwei weiteren Abstandsaminosäuren sorgt für eine höhere Stabi-
lität der Verbindungen in Wasser. Erneut instabiler in Waser wird das Konjugat bei einer wei-
teren Verlängerung der Kette, damit scheinen die Cysteine das AuPhPyCl2 2 am besten mit
drei Abstandsaminosäuren binden zu können. Wird die Nomenklatur für α-Helices verwendet,
dann würde die Bezeichnung i+(i+3) für das günstigste Peptid lauten. Hierbei steht das „i“ für
jeweils ein bindendes Cystein und die Zahl für die Anzahl der Abstandsaminosäuren. Die
Versuche mit 2-Aminoisobutansäure deuten darauf hin, dass das bindende Peptid jeweils
nicht über eine α-Helix an das AuPhPyCl2 2 bindet. Es scheint keinen Einfluss auf das
Konjugat zu haben, wenn sterisch anspruchsvolle Gruppen an den Abstandsaminosäuren ge-
bunden sind. Dies war durch die Verwendung von Arginin zu beobachten. Aus allen bisher
herhalten Ergebnissen wird geschlossen, dass die Struktur des Peptides aus Kettenlänge und
Freiheitsgraden des Peptidrückgrats von genauso entscheidender Bedeutung ist, wie die Aus-

Ergebnisse
Seite | 69
wahl der zum Gold(III)-Komplex 2 bindenden Aminosäuren. Wenn ΠCRRRC-NH2AuPhPy
714 in DMSO gelöst wurde, dann konnte die Zersetzung des Produktes in Chromatogrammen
von analytischen HPLC-Messungen beobachtet werden. DMSO war ein ungeeignetes Lö-
sungsmittel für die Verbindung ΠCRRRC-NH2AuPhPy 714. Das Lösungsmittel hat evidenten
Einfluss auf die Stabilität der Verbindungen.
16 Peptide
ΠCRRRC-NH2AuPhPy 714 bindet als stabile und gut in Wasser lösliche Sequenz. Diese Se-
quenz 713 ist über die beiden Cysteine an den Gold(III)-Komplex gebunden. Was passiert
nun, wenn eines der beiden Cysteine durch eine der anderen oben getesteten bindenden Ami-
nosäuren ausgetauscht wird? Wird eine weitere stabile Sequenz gefunden? Um eine Antwort
auf diese Fragen zu erhalten, wurden alle 16 möglichen Sequenzen dargestellt, bei der die
eine bindenden Aminosäure Cystein ist und andere bindende Aminosäure entweder Aspara-
ginsäure, 2,3-Diaminopropionsäure, Glutaminsäure, Histidin, Lysin, Methionin,
3-(3-Pyridin)-alanin oder 3-(4-Pyridin)-alanin sind. Die Blindprobe Alanin wurde nicht ver-
wendet, da sie oben keine stabilisierenden Eigenschaften gezeigt hat. Es wurden folgende
Peptide synthetisiert:
Änderung der C-Terminal
bindenden AS
Änderung der N-Terminal
bindenden AS
ΠCRRRB-NH2 715 ΠBRRRC-NH2 716
ΠCRRRD-NH2 717 ΠDRRRC-NH2 718
ΠCRRRE-NH2 719 ΠERRRC-NH2 720
ΠCRRRH-NH2 721 ΠHRRRC-NH2 722
ΠCRRRJ-NH2 723 ΠJRRRC-NH2 724
ΠCRRRK-NH2 725 ΠKRRRC-NH2 726
ΠCRRRM-NH2 727 ΠMRRRC-NH2 728
ΠCRRRZ-NH2 729 ΠZRRRC-NH2 730 Tabelle 3-10: Die Sequenzen der 16 Sequenzen
Jedes der Peptide wurde in Lösung mit AuPhPyCl2 2 umgesetzt. Von den gelbfarbenen Reak-
tionslösungen wurden zeitabhängige analytische HPLC-Messungen durchgeführt. In den
Chromatogrammen nach einer Stunde ist bei allen Verbindungen, bei welchen Cystein näher
am C-Terminus sitzt, nur ein Peak zu erkennen. Dies ist bei den anderen Sequenzen nur bei
ΠCRRRD-NH2 717 und ΠCRRRH-NH2 721 der Fall. In allen Chromatogrammen ist über die
ersten drei Zeiten ein intensiver Peak zu beobachten. Bei den Sequenzen mit Cystein in der

Ergebnisse
Seite | 70
Nähe des N-Terminus ist nur in Kombination mit Histidin und Lysin der intensivste Peak
nach 75 Stunden immer noch gut auszumachen und es sind jeweils nur zwei neue Peaks zu
erkennen. Wenn Cystein in der Nähe des C-Terminus ist, dann gilt das gerade beschriebene
bei allen Aminosäuren außer bei Asparaginsäure und Glutaminsäure. Zwischen den beiden
Aminosäuren 3-(3-Pyridin)-alanin und 3-(4-Pyridin)-alanin konnte kein Unterschied ausge-
macht werden. In beiden Fällen ist es wieder günstiger, wenn Cystein in der Nähe des C-
Terminus befindlich ist. Methionin in den Sequenzen zeigt bei den Addukten ähnliche Stabili-
tät, mit der gleichen Anzahl und Intensität der Nebenpeaks in den Chromatogrammen, wie die
nicht nativen Aminosäuren 2,3-Diaminopropionsäure, 3-(3-Pyridin)-alanin und 3-(4-Pyridin)-
alanin. Ein ESI-Massenspektrum wurde jeweils vom Hauptpeak, der sich zuerst gebildet hat-
te, aufgenommen. Diese geben jeweils ein starkes Signal für ein zweifach geladenes Ion an.
Diese Signale können in allen Fällen dem Addukt aus Peptid und Gold(III)Komplex 2 zuge-
ordnet werden. Nach 75 Stunden wurden LC-ESI-MS Läufe gemessen. Mit den so erhalten
Signalen wurde festgestellt, dass einige Peaks der Masse der jeweiligen Zielverbindung plus
der Masse von Wasser, 2-Phenylpyridin-1,1´gold(III) oder Peptid zugeordnet werden können.
Änderung der C-Terminal
bindenden AS mit AuPhPy
Änderung der N-Terminal
bindenden AS mit AuPhPy
ΠCRRRB-NH2AuPhPy 731 ΠBRRRC-NH2AuPhPy 732
ΠCRRRD-NH2AuPhPy 733 ΠDRRRC-NH2AuPhPy 734
ΠCRRRE-NH2AuPhPy 735 ΠERRRC-NH2AuPhPy 736
ΠCRRRH-NH2AuPhPy 737 ΠHRRRC-NH2AuPhPy 738
ΠCRRRJ-NH2AuPhPy 739 ΠJRRRC-NH2AuPhPy 740
ΠCRRRK-NH2AuPhPy 741 ΠKRRRC-NH2AuPhPy 742
ΠCRRRM-NH2AuPhPy 743 ΠMRRRC-NH2AuPhPy 744
ΠCRRRZ-NH2AuPhPy 745 ΠZRRRC-NH2AuPhPy 746 Tabelle 3-11: Sequenzen mit 2-Phenylpyridin-1,1´gold(III)-Ligand
Aus den Chromatogrammen ergab sich dann folgende Reihenfolge der AS für die Stabilität
der Verbindungen:
E ≈ D < B ≈ J ≈ Z ≈ M < H < K
instabil stabil
Allerdings spielt für die Stabilität die Position des Cysteins in der Sequenz eine Schlüsselrol-
le. Die Verbindungen mit Cystein in der Nähe des C-Terminus sind stabiler, als die mit Cys-
tein in der Nähe des N-Terminus. Mögliche Erklärungen hierfür sind eine Abschirmung der
N-terminalen Aminosäure durch die 4-Pentinsäure und das benachbarte Arginin oder ein
günstiges herausstehen des C-terminalen Cysteins aus der Sequenz. Keine der neuen Verbin-

Ergebnisse
Seite | 71
dungen ist in den Versuchen so stabil wie ΠCRRRC-NH2AuPhPy 714. Cystein ist bei drei
Abstandsaminosäuren der günstigste Ligand.
Ergebnisse
Seite | 72
[3+2] Cycloaddition mit dem stabilen Bindungsmotiv
Abbildung 3-49: Reaktionsgleichung der [3+2] Cycloreaktion zwischen ΠCRRRC-NH2AuPhPy 714 und
ORRRRRPLGLYALEEEEE-NH2 42
Mit ΠCRRRC-NH2AuPhPy 714 ist eine stabile Verbindung erhalten worden, mit der die
[3+2] Cycloaddition versucht werden konnte. Als Reaktionspartner wurde ORRRRRPLG-
LYALEEEEE-NH2 42 ausgewählt, um die angestrebte Zielverbindung darzustellen. In der
Tabelle 3-12 sind die verwendeten Reaktionsbedingungen wiedergegeben:
Lösungsmittel Kupferquelle Hilfsreagenz Base
H2O/ACN Kupfersulfat Natriumascorbat
H2O/t-Butanol Kupfersulfat Natriumascorbat
H2O/Aceton Kupfersulfat Natriumascorbat
THF Kupfersulfat Natriumascorbat
H2O/ACN Kupfersulfat Natriumascorbat Triethylamin
H2O/t-Butanol Kupfersulfat Natriumascorbat Triethylamin
THF Kupfersulfat Natriumascorbat DIPEA
H2O/t-Butanol Kupferiodid
H2O/t-Butanol Kupferiodid Natriumascorbat
H2O/ACN CuI(Ph3P)3 TBTA DIPEA
DMF CuI(Ph3P)3 TBTA DIPEA

Ergebnisse
Seite | 73
Tabelle 3-12: Verwendeten Reaktionsbedingungen der [3+2] Cycloaddition zwischen
ΠCRRRC-NH2AuPhPy 714 und ORRRRRPLGLYALEEEEE-NH2 42
Da ΠCRRRC-NH2AuPhPy 714 in DMSO nicht stabil ist, konnten nicht genau dieselben
Reaktionsbedingen der [3+2] Cycloaddition zwischen ΠCFC-NH2AuPhPy 34 und Verbin-
dung 42 verwendet werden. Anstelle der vorher verwendeten Lösungsmittelmischung aus
Wasser und DMSO, wurde die Reaktion in Wasser mit Acetonitril, Wasser mit t-Butanol,
Wasser und Aceton und reinem Tetrahydrofuran probiert.154, 155
Alle Ansätze wurden mittels
analytischer HPLC und ESI-MS untersucht. Aus diesen Untersuchungen lässt sich schließen,
dass keine der Reaktionsbedingungen zu dem angestrebten Produkt geführt hat.
Möglicherweise binden die Seitenfunktionalitäten der verwendeten Peptide den eingesetzten
Kupferkatalysator. Es wurden deswegen mehr Äquivalente des Kupferkatalysators in ver-
schiedenen Lösungsmitteln genutzt. Diese Ansätze wurden mit den oben schon beschriebenen
Methoden analysiert und das gleiche Ergebnis wurde beobachtet.
Weil die Verwendung von mehr Äquivalenten des Kupferkatalysators nicht zum gewünschten
Produkt geführt hatte, wurde gehofft durch Beschleunigung der Reaktion in der Mikrowelle
die angestrebte Zielverbindung zu erhalten.156
Deswegen wurde eine Mischung aus den bei-
den Edukten, Natriumascorbat, Kupfer(II)sulfat, Wasser und Acetonitril in der Mikrowelle
umgesetzt. Anschließend wurde eine analytische HPLC-Messung durchgeführt.Das so erhal-
ten Chromatogramm besitzt sehr viele Peaks. Beide Edukte wurden in viele Nebenprodukte in
der Mikrowelle zersetzt. Das angestrebte Produkt wurde nicht erhalten.
Nachdem die Edukte sich in der Mikrowelle zersetzt hatten, sollte die Synthese unter stärker
basischen Reaktionsbedingungen zum Produkt führen.157-159
Als Base wurde dafür
Triethylamin und DIPEA genutzt. In den angefertigten analytischen HPLChromatogrammen
und den ESI-Massenspektren konnte das Produkt nicht ausgemacht werden.
Der Zusatz von Base hatte erneut nicht zu der Zielverbindung geführt, eventuell wurde das
verwendet Kupfer(II)sulfat unter den verwendeten Reaktionsbedingungen nicht zum Kup-
fer(I)-Katalysator reduziert. Es wurde deswegen Kupfer(I)iodid als Kupfer(I)-Quelle verwen-
det.160, 161
Das Kupfer(I)iodid wurde mit beiden Edukten in Wasser und t-Butanol gelöst. Von
der Reaktionsmischung wurden in regelmäßigen Abstanden analytische HPLC-Messungen

Ergebnisse
Seite | 74
durchgeführt. Die Messungen haben kein neues intensives Hauptsignal, welches auf das Ge-
lingen der [3+2] Cycloaddition schließen lässt.
Das Kupfer(I)iodid wurde durch Triethylphosphitkupfer(I)iodid ersetzt. Der Ligand am Kup-
fer sollte verhindern, dass das Kupfer mit den Funktionalitäten der Seitengruppe an der Pep-
tidsequenz reagierte.162
Als Hilfsreagenz wurde TBTA zugegeben. Dieses sollte den gleichen
Effekt wie Natriumascorbat haben und die Oxidation und eine Disproportionierung verhin-
dern. Eine Reaktionskontrolle wurde mit der analytischen HPLC durchgeführt. Es waren die
Edukt Peaks zu erkennen und einige wenig intensive neue Peaks. Es entstand kein neuer in-
tensiver Peak. Es wurden LC-ESI-MS aufgenommen. In keinem der Spektren waren Massen-
signale zu erkennen, welche dem Produkt zugeordnet werden konnten.
Andere Bindungsmotive in der [3+2] Cycloaddition
Da der Versuch mit dem neuen Katalysator ebenfalls nicht zum Produkt geführt hatte, wurde
ein störender Einfluss der Arginine am ΠCRRRC-NH2AuPhPy 714 auf die Reaktion vermu-
tet. Um dies zu überprüfen wurde die Reaktion mit ΠCGGGC-NH2AuPhPy 706 und
ORRRRRPLGLYALEEEEE-NH2 42 durchgeführt. Es wurden erneut verschieden Lösungs-
mittel ausprobiert. Es wurden Reaktionskontrollen mit der analytischen HPLC durchgeführt.
Keines der Chromatogramme zeigt andere Peaks, als die der Eduktpeaks. Es sind keine neuen
Produkte entstanden. Die angefertigten ESI-Massenspektren bestätigen diesen Eindruck.
Die Reaktion wurde mit Triethylphosphitkupfer(I)iodid und TBTA in DMF wiederholt. Es
wurden analytische HPLC-Messungen durchgeführt. Diese geben keinen neuen Hauptpeak
neben den beiden Eduktpeaks in den Chromatogrammen an. Es sind nur viele wenig intensive
Peaks zu erkennen. Es wurde eine LC-ESI-MS-Messung durchgeführt. In dem Spektrum sind
keine der zu erwartenden Massen für das Produkt zu beobachten.
Da es offensichtlich nicht am Arginin lag, dass die Reaktion nicht ablief, wurde überlegt wel-
cher Parameter noch verändert werden könnte, um eine ähnliche Verbindung wie
ΠCFC-NH2AuPhPy 34 darzustellen, mit welchem die [3+2] Cycloaddition abgelaufen war.
Aus diesem Grund wurden drei Phenylalanin als Abstandsaminosäuren gewählt.

Ergebnisse
Seite | 75
Abbildung 3-50: ΠCFFFC-NH2AuPhPy
Es wurde die Peptidsequenz ΠCFFFC-NH2 747 in einer Mischung aus Wasser und Acetonitril
zusammen AuPhPyCl2 2 gekoppelt. Von der Reaktionslösung wurde eine analytische HPLC-
Messung durchgeführt. Diese ergibt einen Peak bei 24.3 min. Von dem Peak wurde ein Mas-
senspektrum angefertigt. Das Massenspektrum hat ein Signal bei m/z = 1094. Dieses Massen-
signal kann dem protonierten ΠCFFFC-NH2AuPhPy 748 zugeordnet werden, damit wurde
das Produkt quantitatv erhalten. ΠCFFFC-NH2AuPhPy 748 war kaum in Wasser, Acetonitril,
Aceton, THF und t-Butanol löslich. Aus diesem Grund wurde die [3+2] Cycloaddition in
DMF mit TBTA und Triethylphosphitkupfer(I)iodid ausgeführt. Eine Reaktionskontrolle
wurde an der analytischen HPLC durchgeführt. Diese zeigte keinen Peak, welcher zum Pro-
dukt passen kann.
Abbildung 3-51: ΠCFRFC-NH2AuPhPy
Da ΠCFFFC-NH2AuPhPy 748 schlecht in vielen Lösungsmitteln löslich war, wurde die Ver-
bindung ΠCFRFC-NH2 749 mit AuPhPyCl2 2 umgesetzt. Das gelbfarbene Produkt wurde mit
der analytischen HPLC und dem ESI-Massenspektrometer untersucht. Das Chromatogramm
hat einen Peak bei 19.6 min, im Massenspektrum ist ein Signal bei m/z = 552 zu beobachtet.
Das Signal gehört zu einem zweifach geladenen Produkt. Das Ergebnis der analytischen
HPLC deutet auf die Entstehung von nur einem Produkt hin. Das Signal im ESI-

Ergebnisse
Seite | 76
Massenspektrum entspricht dem zweifach geladen ΠCFRFC-NH2AuPhPy 750, welches quan-
titativ erhalten worden ist.
ORRRRRPLGLYALEEEEE-NH2 42 wurde nun mit ΠCFRFC-NH2AuPhPy 750 Kupfersulfat
und Natriumascorbat in Wasser und Acetonitril umgesetzt. Es wurden Proben entnommen
und mit Hilfe der analytischen HPLC untersucht. Es waren Peaks bei den Zeiten der Edukte
zu erkennen. Es waren einige neue Peaks zu erkennen. Die neu entstandenen Peaks wurden
im ESI-Massenspektrometer untersucht. Die erwartete Produktmasse konnte nicht in den
Spektren gefunden werden.
Abbildung 3-52: ΠCGRGC-NH2AuPhPy
Das Phenylalanin schien nicht die [3+2] Cycloadditionsreaktion zu unterstützen. Es wurde
deswegen eine neue Sequenz mit einer Mischung der bisher genutzten Abstandsaminosäuren
verwendet. Es sollten zwei Glycin und ein Arginin genutzt werden. Das Glycin war eine ein-
fache Aminosäure, welche bei allen weiteren Synthesen keinen negativen Einfluss auf die
reaktiven Eigenschaften der Sequenz haben sollte und die hydrophoben Eigenschaften der
neuen Verbindung kaum erhöhte. Das Arginin sollte die Löslichkeit der neuen Verbindung in
Wasser erhöhen. Für die Synthese wurde die Sequenz ΠCGRGC-NH2 751 zusammen mit
Verbindung 2 gegeben.
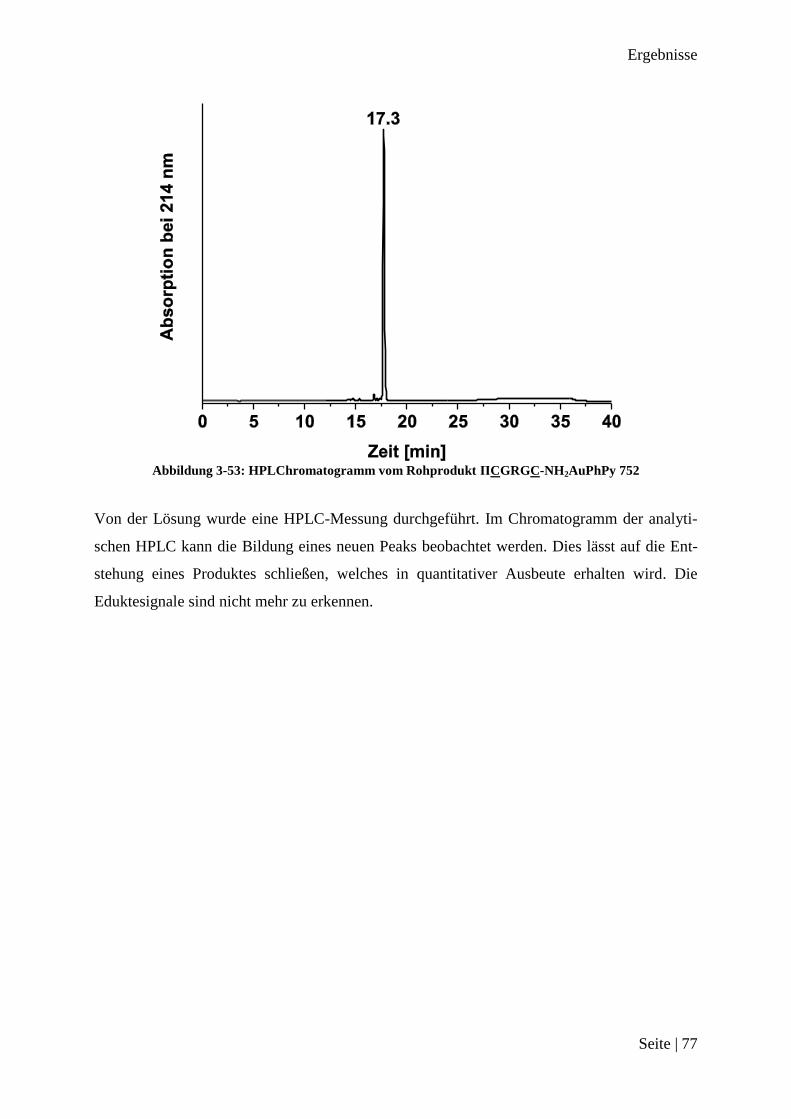
Ergebnisse
Seite | 77
Abbildung 3-53: HPLChromatogramm vom Rohprodukt ΠCGRGC-NH2AuPhPy 752
Von der Lösung wurde eine HPLC-Messung durchgeführt. Im Chromatogramm der analyti-
schen HPLC kann die Bildung eines neuen Peaks beobachtet werden. Dies lässt auf die Ent-
stehung eines Produktes schließen, welches in quantitativer Ausbeute erhalten wird. Die
Eduktesignale sind nicht mehr zu erkennen.
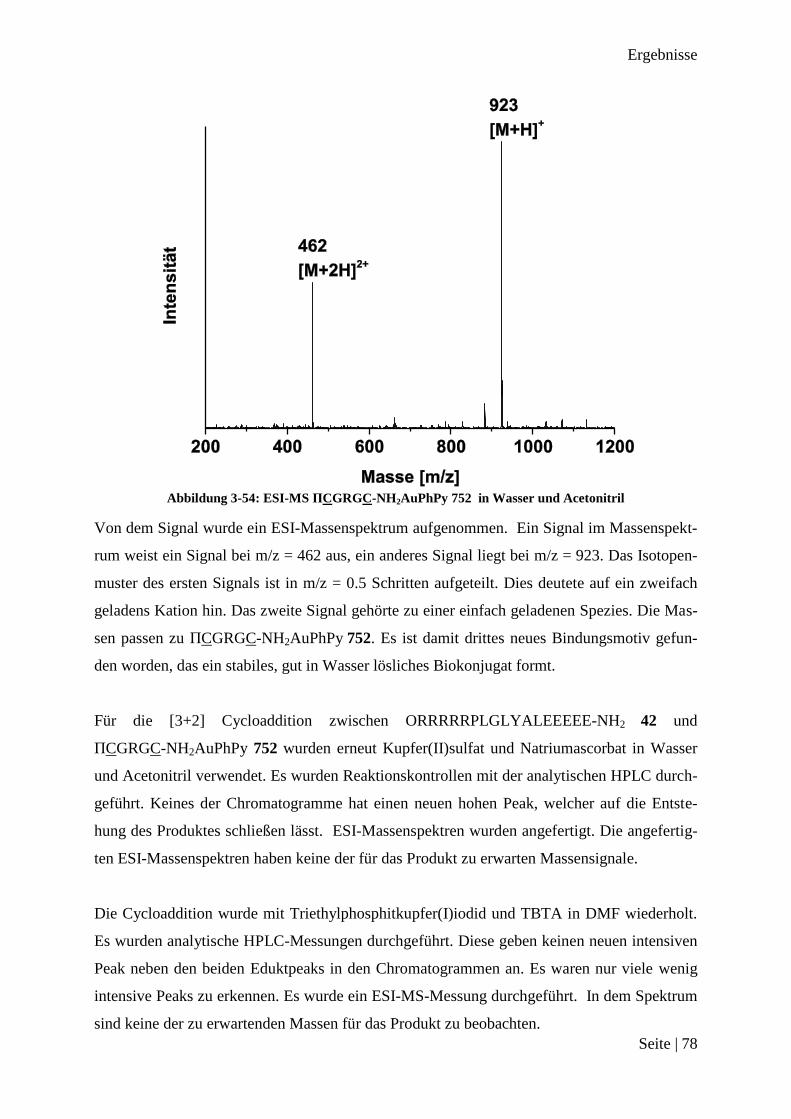
Ergebnisse
Seite | 78
Abbildung 3-54: ESI-MS ΠCGRGC-NH2AuPhPy 752 in Wasser und Acetonitril
Von dem Signal wurde ein ESI-Massenspektrum aufgenommen. Ein Signal im Massenspekt-
rum weist ein Signal bei m/z = 462 aus, ein anderes Signal liegt bei m/z = 923. Das Isotopen-
muster des ersten Signals ist in m/z = 0.5 Schritten aufgeteilt. Dies deutete auf ein zweifach
geladens Kation hin. Das zweite Signal gehörte zu einer einfach geladenen Spezies. Die Mas-
sen passen zu ΠCGRGC-NH2AuPhPy 752. Es ist damit drittes neues Bindungsmotiv gefun-
den worden, das ein stabiles, gut in Wasser lösliches Biokonjugat formt.
Für die [3+2] Cycloaddition zwischen ORRRRRPLGLYALEEEEE-NH2 42 und
ΠCGRGC-NH2AuPhPy 752 wurden erneut Kupfer(II)sulfat und Natriumascorbat in Wasser
und Acetonitril verwendet. Es wurden Reaktionskontrollen mit der analytischen HPLC durch-
geführt. Keines der Chromatogramme hat einen neuen hohen Peak, welcher auf die Entste-
hung des Produktes schließen lässt. ESI-Massenspektren wurden angefertigt. Die angefertig-
ten ESI-Massenspektren haben keine der für das Produkt zu erwarten Massensignale.
Die Cycloaddition wurde mit Triethylphosphitkupfer(I)iodid und TBTA in DMF wiederholt.
Es wurden analytische HPLC-Messungen durchgeführt. Diese geben keinen neuen intensiven
Peak neben den beiden Eduktpeaks in den Chromatogrammen an. Es waren nur viele wenig
intensive Peaks zu erkennen. Es wurde ein ESI-MS-Messung durchgeführt. In dem Spektrum
sind keine der zu erwartenden Massen für das Produkt zu beobachten.

Ergebnisse
Seite | 79
Zusammenfassung der [3+2] Cycloaddition mit dem stabilen Bindungsmotiv
Keine der Kupferkatalysierten [3+2] Cycloadditionen hat zu dem erwarteten Produkt geführt.
Die Variation der Lösungsmittel, der Hilfsreagenzien und der Kupfer(I)quelle führt nicht zum
[3+2] Cycloreaktionsprodukt. Das Edukt mit dem gebundenen Gold(III)-Komplex wurde ver-
ändert, da ein störender Einfluss der Funktionalitäten in den Seitenketten vermutet wurde. So
wurde ebenfalls nicht das angestrebte Produkt synthetisiert. Möglicherweise konnte das Kup-
fer(I) aus sterischen Gründen die Reaktion nicht katalysieren.
[3+2] Cycloadditionen ohne Kupferkatalysator
Die 4-Pentinsäure sollte aus diesem Grund durch ((1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-
yl)methyl (2,5-dioxopyrrolidin-1-yl) carbonat 753 ersetzt werden. Diese Verbindung besteht
aus einem Achtring mit einem Alkin. Durch die Ringspannung geht dieser Achtring [3+2]
Cycloadditionenreaktion ohne Kupferkatalysator ein.163
Abbildung 3-55: ((1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-yl)methyl (2,5-dioxopyrrolidin-1-yl) carbonat
H-CGRGC-Rink AM Harz wurde an SPPS synthetisiert. Die Fortschritte in der Synthese der
Peptidsequenz wurden mittels des Kaisertestes überprüft.164
Anderthalb Äquivalente von Ver-
bindung 753 wurden wie eine Aminosäure an fester Phase an die Sequenz gebunden. Die Se-
quenz wurde vom Harz mit Hilfe einer Trifluoressigsäurelösung abgespalten und mit - 70 °C
Diethylether ausgefällt. Der Rückstand wurde in Wasser gelöst, eingefroren und lyophilisiert.
Von dem schaumartigen Produkt wurden eine analytische HPLC-Lauf und ein ESI-LC-MS
gemessen. Im Chromatogramm ist ein intensiver Peak bei 2.5 min zu erkennen. Im Spektrum
können diesem die Massen m/z = 247 und m/z = 493 zugeordnet werden. Diese entsprechen

Ergebnisse
Seite | 80
dem Peptid ohne das Alkin 753. Das Produkt war nicht auszumachen. Das Alkin scheint unter
den Abspaltebedingungen nicht stabil an das Peptid gebunden worden zu sein.
Aus diesem Grund sollte erst die Sequenz H-CGRGC-NH2 754 synthetisiert werden. Pep-
tidsequenz 754 sollte dann mit AuPhPyCl2 2 in den oben verwendeten Bedingungen zu Ver-
bindung H-CGRGC-NH2AuPhPy 755 umgesetzt werden. Verbindung 755 sollte dann unter
basischen Bedingungen mit Verbindung 753 reagieren.
Abbildung 3-56: Darstellung von Peptid mit Cyclooctaalkin 753
H-CGRGC-NH2 754 wurde zusammen mit einer äquimolaren Menge von AuPhPyCl2 2 in
einem Lösungsmittelgemisch aus einem Teil Wasser und einem Teil Acetonitril zusammen
gegeben. Es wurde eine analytische HPLC-Messung vorgenommen. Das Chromatogramm hat
zwei Peaks. Der erste Peak entspricht der Retentionszeit der Peptidsequenz. Der zweite Peak
wurde in der ESI-Massenspektroskopie untersucht. Das Spektrum zeigt zwei Signale. Das
erste Signal liegt bei m/z = 597 und das zweite Signal bei m/z = 1193. Das erste Signal gehör-
te zu einer zweifach geladen Spezies. Die Massen der beiden Signale passten zu
H-CGRGC-NH2AuPhPy 755 mit einem zusätzlich gebunden AuPhPyCl2 2 ohne die beiden
Chloridliganden. Das Produkt H-CGRGC-NH2AuPhPy 756 wurde nicht beobachtet.

Ergebnisse
Seite | 81
Offensichtlich reagierte das Peptid jeweils mit zwei Gold(III)-Komplexen. Eine mögliche
freie Bindungsstelle war das endständige Amin des Peptidrückgrats. Die Guanidingruppe und
die Amide hatten bisher in keinem Experiment eine besondere Affinität zum Gold(III)-
Zentrum gezeigt.
Zur Umgehung der Nebenreaktion wurde die Fmoc-Schutzgruppe 758 auf der Peptidsequenz
belassen. Die Sequenz mit der Fmoc-Schutzgruppe 758 wurde mit AuPhPyCl2 2 zur Reaktion
gebracht. Es wurden ein analytischer HPLC-Lauf und ein ESI-Massenspektrum gemessen.
Das Chromatogramm hat einen Produkt Peak, im Spektrum sind eine einfach und eine zwei-
fach geladene Spezies zu erkennen, welche beide zur Masse von m/z = 1064 passten. Die Er-
gebnisse stimmen mit den Erwartungen für Verbindung Fmoc-CGRGC-NH2AuPhPy 759
überein.
Die Fmoc-Schutzgruppe sollten vom Fmoc-CGRGC-NH2AuPhPy 759 abgespalten werden.
Hierfür wurde Fmoc-CGRGC-NH2AuPhPy 759 in verschiedene Lösungen mit den Basen
Piperidin, DIPEA, Triethylamin und Kaliumhydroxid gegeben. Von allen Ansätzen wurden
analytische HPLC-Messungen durchgeführt und es wurden von einigen Proben ESI-LC-MS-
Messungen aufgenommen. Es konnte beobachtet werden, dass in den Chromatogrammen die
Anzahl der Peaks mit der Erhöhung der Basenkonzentration zu nahm. Keine der ESI-LC-MS-
Messungen lässt die Masse der Fmoc-entschützen Spezies beobachten.
Erst den Gold(III)-Komplex 2 zu koppeln und dann die Fmoc-Schutzgruppe abzuspalten ist
nicht gelungen. Bei zu niedrigen Basekonzentrationen wurde die Fmoc-Schutzgruppe nicht
abgespalten, bei zu hohen Konzentrationen bilden sich neue Verbindungen. Die freie
Guanidingruppe konnte eine reaktive Stelle für Nebenreaktionen gewesen sein. Nebenreakti-
onen können auch an den Thiolen der Cysteine mit dem vormals gebunden 2-Phenylpyridin-
1,1´gold(III) stattgefunden haben, wenn der Gold(III)-Komplex durch stark basische Bedin-
gungen abgespalten worden war. Das freie 2-Phenylpyridin-1,1´gold(III) würde dann auch
wieder Nebenreaktionen eingegangen sein. Sobald die Fmoc-Schutzgruppe nicht mehr am
Amin gebunden ist, könnte das Amin auch mit diversen reaktiven Spezies in der
Abspaltlösung reagieren.
Ein neuer Weg zur Synthese von Verbindung 757 musste gefunden werden. Das Peptid 754
sollte ohne Fmoc-Schutzgruppe vom Harz abgespalten werden. Das Peptid 754 sollte darauf
so behandelt werden, dass sich zwischen den beiden Thiolen in der Sequenz intramolekulare

Ergebnisse
Seite | 82
Thiolbrücken ausbildeten. Alkin 753 sollte dann unter leicht basischen Bedingungen gekop-
pelt werden. Anschließend würden die Thiolbrücke gebrochen werden, um dann diese Ver-
bindung mit AuPhPyCl2 2 umzusetzen.
Abbildung 3-57: Alternative Route zu Peptid mit Cyclooctaalkin 753
Nach der Synthese und Aufreinigung der Peptidsequenz 754 wurde diese in eine Lösung aus
Wasser und DMSO gelöst. Es wurde in viel Lösungsmittel gearbeitet. Die große Verdünnung
sollte eine intramolekulare Reaktion durch den Luftsauerstoff bevorzugen.165, 166
Von der
Reaktionslösung wurden in regelmäßigen Abständen kleine Proben genommen. Mit diesen
Proben wurde ein Ellmanstest durchgeführt.167
Färbte sich die Probe gelbfarben, ist dies ein
Hinweis auf die Anwesenheit von freien Thiolen. Sobald der Ellmanstest negativ ausfiel,
wurde das Lösungsmittel entfernt. Der Rückstand wurde über die präperative HPLC aufge-
reinigt. Die gesammelte Fraktion wurde einer analytischen HPLC-Messung und einem ESI-
Massenspektrums Aufnahme unterzogen. Da die Ergebnisse des Massenspektrums identisch
zum Peptid ohne Disulfidbrücke sind, wurde erneut ein Ellmanstest durchgeführt. Der Ell-
manstest blieb farblos. Aus dem Test wurde geschlossen, dass keine freien Thiole mehr im

Ergebnisse
Seite | 83
Peptid vorhanden waren. Das cyclisierte Produkt 754 wurde in Lösung mit DIPEA und dem
Alkin 753 zusammen gegeben. Eine analytische HPLC Messung wurde von der Reaktionslö-
sung gemessen. Das Chromatogramm hat mehrere neue Peaks. Bei der Retentionszeit der
Peptidsequenz ist kein Peak mehr erkennbar. Die neuen Peaks wurden mittels ESI-MS unter-
sucht. Einer der Peaks hat ein zweifach geladenes Massesignal von m/z = 350. Dies entspricht
der Masse vom zweifach geladenen, nicht mehr cyclisierten Produkt 760. Ein erneut durchge-
führter Ellmanstest wurde gelbfarben. Es hatten sich freie Thiole gebildet. Die Destabilisie-
rung der Disulfidbrücke und das damit verbundene Auftreten von freien Thiolen, erklärt die
große Anzahl an weiteren Produkten, die im Chromatogramm zu beobachten sind. Hierdurch
wurde nur eine sehr geringe Ausbeute erreicht.
Orthogonale Bindung über ein Imin
Die Syntheseroute zur Darstellung eines Gold(III)-Komplexes an einem Peptid mit der Alkin-
verbindung 753 hatte sich als nicht besonders ergiebig erwiesen. Es musste eine neue Mög-
lichkeit der Kopplung der lange Peptidsequenz ORRRRRPLGLYALEEEEE-NH2 42 gefun-
den werden. Nun wurde überlegt die beiden Sequenzteile mittels eines Imins zu verbinden.
Das Imin sollte sich aus einem Amin der einen Sequenz und dem Aldehyd der anderen Se-
quenz bilden. Für den Erhalt der Sequenz mit dem Amin wurde bei der Synthese der langen
Sequenz auf die Kopplung der Azidoessigsäure verzichtet. So wurde folgende Sequenz erhal-
ten: H-RRRRRPLGLYALEEEEE-NH2 761. Das Aldehyd sollte entsprechend an der Sequenz
mit dem Gold(III)-Komplex 2 gebunden sein. Dafür sollte die Peptidsequenz mit einem Ester
abschließen, welcher dann zum Aldehyd reduziert werden sollte. Diese Sequenz mit Aldehyd
sollte mit AuPhPyCl2 2 zur Reaktion gebracht werden. Das Aldehyd könnte dann mit dem
Amin zum Halbaminal reagieren. Die reversible Reaktion zum Halbaminal könnte in einer
irreversiblen Reaktion zum Imin reduziert werden.

Ergebnisse
Seite | 84
Abbildung 3-58: Der Aldehyd haltige Gold(III)-Komplex
Die Sequenz H-CGRGC-Rink AM wurde dargestellt und mit Methyladipoylchlorid und
DIPEA am Harz behandelt. Nach dem Waschen des Harzes, wurde eine DIBAL-Lösung in n-
Hexan zum Harz gegeben.168
Erneut wurde das Harz gewaschen und dann wurde die
Peptidsequenz abgespalten. Von dem so erhaltenen Produkt wurde eine analytische HPLC-
Messung aufgenommen. Das Chromatogramm hat mehrere Peaks, wobei ein besonders inten-
siver Peak hervorsticht. Dieser Peak wurde isoliert und mit dem ESI-Massenspektrum analy-
siert. Das Produkt hat eine Masse von m/z = 636. Dies entspricht der Verbindung
ΓCGRGC-NH2 762. Von dem Rohprodukt wurde ein ESI-LC-MS gemessen. Die Massen der
Sequenz, bei der der Ester von ΓCGRGC-NH2 762 zum Aldehyd reduziert war, konnte dabei
nicht entdeckt werden. Die Kopplung von Methyladipoylchlorid hatte gut funktioniert, nicht
jedoch die Reduktion des Esters zum Aldehyd. Der Versuch der Reduktion des Esters zum
Aldehyd am Harz wurde deswegen mit DIBAL in Toluol wiederholt. Es führte zum gleichen
Ergebnis. Der Wechsel zu Dichlormethan brachte ebenso keine Veränderung. Die dreifache
Behandlung mit DIBAL in Toluol führte ebenfalls nicht zum reduzierten Aldehyd Produkt.
Eine Behandlung von abgespaltenen ΓCGRGC-NH2 762 mit DIBAL in Toluol führte eben-
falls nicht zu dem erwartet Produkt mit Aldehyd.

Ergebnisse
Seite | 85
Möglicherweise war die Reduktion aus sterischen Gründen nicht am Harz möglich, deswegen
wurde ΓCGRGC-NH2 762 mit AuPhPyCl2 2 umgesetzt. Von der Reaktionslösung wurden
eine analytische HPLC-Messung und ein ESI-Massenspektrum aufgenommen. In der HPLC-
Messung war ein Peak zu erkennen. Dieser Peak hatte im ESI-MS ein zweifach geladenes
Massesignal von m/z = 493. Dieses Massensignal entsprach dem zweifach geladenen Produkt
ΓCGRGC-NH2AuPhPy 763, welches in guter Ausbeute erhalten werden konnte.
ΓCGRGC-NH2AuPhPy 763 wurde DIBAL behandelt. Von der Reaktionslösung wurde ein
ESI-LC-MS gemessen. Das Chromatogramm gibt einen intensiven Hauptpeak wieder. Das
Massensignal dieses Peaks liegt bei m/z = 493. Dies entspricht dem Edukt
ΓCGRGC-NH2AuPhPy 763. Keine der anderen Spezies entspricht dem aus dem Methylester
entstanden Aldehyd Produkt. Es war nicht gelungen ein Aldehyd am Bindungsmotiv zu er-
zeugen und damit die Zielverbindung über eine Iminkopplung darzustellen.
Direkter Weg
Da auch über die Iminkopplung nicht die Zielverbindung dargestellt werden konnte, sollte das
Produkt über die Behandlung der ganzen Sequenz mit AuPhPyCl2 2 erhalten werden. Von der
ganzen Sequenz hatte es sich gezeigt, dass nur die Carbonsäuren der Seitenketten der Gluta-
minsäuren zum AuPhPyCl2 2 binden können. Falls dies passieren sollte, dann müssten die
Carbonsäuren orthogonal geschützt werden. Jedoch sollte zuerst die einfachste Möglichkeit
probiert werden und alle Sequenzen mit ungeschützten Aminosäuren verwendet werden. Erst
sollten der positiv geladene Sequenzteil mit den Argininen und der von MMP2 spaltbare Se-
quenzteil jeweils inklusive des Cysteinbindungsmotives dargestellt werden. Es sollte so her-
ausgefunden werden, ob bei den dort verwendeten Aminosäuren neue synthetische Probleme
auftraten.

Ergebnisse
Seite | 86
Abbildung 3-59: Oben Ac-CGRGCRRRRR-NH2AuPhPy und unten
Ac-CGRGCRRRRRCPLGLYAL-NH2AuPhPy
Die Sequenzen Ac-CGRGCRRRRR-NH2 764 und Ac-CGRGCRRRRRCPLGLYAL-NH2 765
wurden dargestellt und gereinigt. Die Peptidsequenzen wurden dann jeweils zusammen mit
AuPhPyCl2 2 umgesetzt. Nach einer Stunde wurden analytische HPLC-Messungen durchge-
führt. In beiden Chromatogrammen war jeweils ein intensiver Peak zu erkennen. Beim Ansatz
mit der Sequenz Ac-CGRGCRRRRR-NH2 764 liegt der Peak bei 14.1 min. Beim zweiten
Ansatz mit Ac-CGRGCRRRRRCPLGLYAL-NH2 765 ist der Peak bei 17.0 min zu erkennen.
Die beiden Peaks wurden an der präperativen HPLC von allen sehr weniger intensiven Peaks
getrennt. Von den erhaltenen Produkten wurden MALDI-Massenspektren aufgenommen.
Beim Ansatz mit Ac-CGRGCRRRRR-NH2 764 sind drei Massensignale zu erkennen. Das
erste Signal liegt bei m/z = 1315, das zweite Signal bei m/z = 1512 und das dritte Signal bei
m/z = 1665. Das dritte Signal kann der Verbindung Ac-CGRGCRRRRR-NH2AuPhPy 766
zugeordnet werden. Das zweite Signal entspricht dieser Verbindung ohne 2-Phenylpyridin
und das erste Signal stimmt mit der Zielverbindung 766 ohne 2-Phenylpyridin-1,1´gold(III)
überein. Das Spektrum von dem Ansatz mit Ac-CGRGCRRRRRCPLGLYAL-NH2 765 hat
drei Signale. Diese sind bei m/z = 2043, m/z = 2238 und m/z = 2393. Das Signal bei
m/z = 2393 entspricht Ac-CGRGCRRRRRCPLGLYAL-NH2AuPhPy 767. Die anderen bei-
den Signale entsprechen analog zu Verbindung 766 diesem Produkt ohne 2-Phenylpyridin-

Ergebnisse
Seite | 87
1,1´gold(III) beziehungsweise ohne 2-Phenylpyridin. Das jeweils die Produktmassen ohne
2-Phenylpyridin-1,1´gold(III) und ohne 2-Phenylpyridinzu erkennen gewesen sind, hat etwas
mit der Ionisierungsart des MALDI-Massenspektrometers zu tun. Das Metallionen und Li-
ganden von Metallionen abgespalten werden, ist eine Eigenart dieser Methode.169, 170
Die Carbonsäuren der Glutaminsäuren konnten ebenfalls eine Bindung zum Gold(III)-
Komplex 2 aufbauen. Es war aus diesem Grund nicht gelungen Verbindung 32 zu synthetisie-
ren. Jedoch war das Bindungsmotiv im Laufe dieser Arbeit optimiert. Die optimierte
Peptidsequenz bot AuPhPyCl2 2 eine bessere Bindungsgeometrie. Durch diese verbesserte
Bindungsgeometrie war es potentielle Möglich, dass freie Carbonsäuren in der Sequenz vor-
handen waren und AuPhPyCl2 2 dennoch an der vorgesehen Bindungsstelle zur
Peptidsequenz gebunden wurde.
Abbildung 3-60: Ac-CGRGCRRRRRCPLGLYALEEEEE-NH2AuPhPy
Zur Überprüfung dieser Theorie wurde die Sequenz Ac-CGRGCRRRRRCPLGLYAL-
EEEEE-NH2 768 zusammen gegeben mit AuPhPyCl2 2. Von der Lösung wurde eine HPLC-
Messung durchgeführt. Das Chromatogramm der HPLC gibt die Bildung mehrerer neuer
Peaks wieder. Einer dieser neuen Peaks sticht durch sein besonders großflächiges Integral
hervor. Das analytische HPLChromatogramm nach der Reinigung auf der preparativen HPLC
ist in Abbildung 3-61 dargestellt.
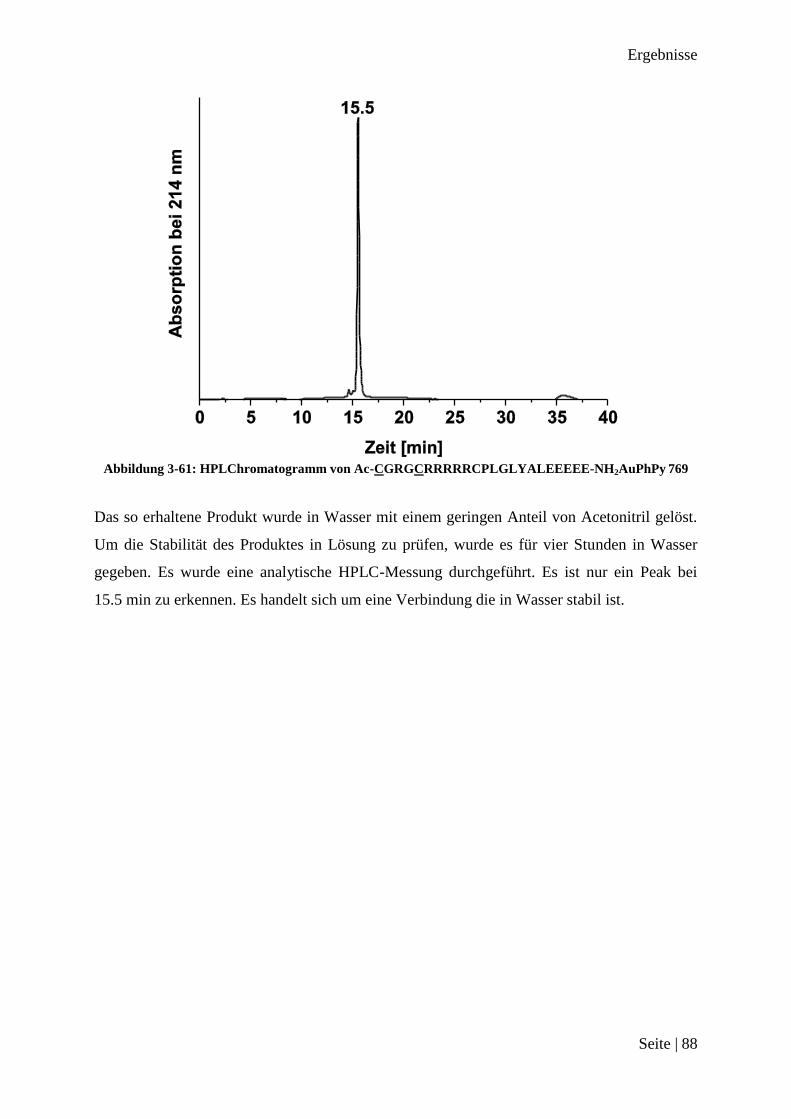
Ergebnisse
Seite | 88
Abbildung 3-61: HPLChromatogramm von Ac-CGRGCRRRRRCPLGLYALEEEEE-NH2AuPhPy 769
Das so erhaltene Produkt wurde in Wasser mit einem geringen Anteil von Acetonitril gelöst.
Um die Stabilität des Produktes in Lösung zu prüfen, wurde es für vier Stunden in Wasser
gegeben. Es wurde eine analytische HPLC-Messung durchgeführt. Es ist nur ein Peak bei
15.5 min zu erkennen. Es handelt sich um eine Verbindung die in Wasser stabil ist.
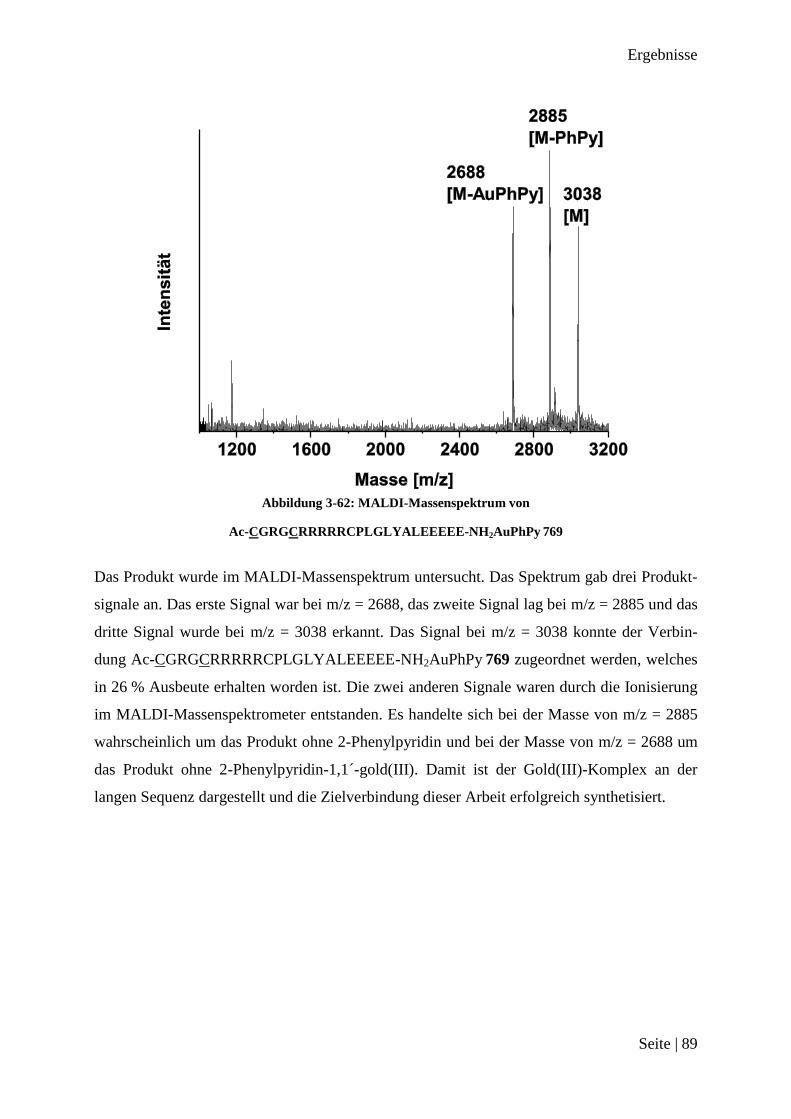
Ergebnisse
Seite | 89
Abbildung 3-62: MALDI-Massenspektrum von
Ac-CGRGCRRRRRCPLGLYALEEEEE-NH2AuPhPy 769
Das Produkt wurde im MALDI-Massenspektrum untersucht. Das Spektrum gab drei Produkt-
signale an. Das erste Signal war bei m/z = 2688, das zweite Signal lag bei m/z = 2885 und das
dritte Signal wurde bei m/z = 3038 erkannt. Das Signal bei m/z = 3038 konnte der Verbin-
dung Ac-CGRGCRRRRRCPLGLYALEEEEE-NH2AuPhPy 769 zugeordnet werden, welches
in 26 % Ausbeute erhalten worden ist. Die zwei anderen Signale waren durch die Ionisierung
im MALDI-Massenspektrometer entstanden. Es handelte sich bei der Masse von m/z = 2885
wahrscheinlich um das Produkt ohne 2-Phenylpyridin und bei der Masse von m/z = 2688 um
das Produkt ohne 2-Phenylpyridin-1,1´-gold(III). Damit ist der Gold(III)-Komplex an der
langen Sequenz dargestellt und die Zielverbindung dieser Arbeit erfolgreich synthetisiert.

Zusammenfassung
Seite | 90
4. Zusammenfassung
Diese Arbeit beschäftigt sich mit dem Komplex AuPhPyCl2 2 und seiner Funktionalisierung
mit einem Peptid. Das Peptid kann entweder über den Liganden oder direkt über das Metall-
atom selber mit dem Gold(III)-Komplex 2 verknüpft werden. Im Laufe der Untersuchungen
zeigte es sich, dass ein Bindungsmotiv aus zwei Cysteinen und einer Abstandsaminosäure für
die Verknüpfung zum Gold(III)-Ion günstig ist. Das AuPhPyCl2 2 konnte über dieses Bin-
dungsmotiv erfolgreich zu folgenden Verbindungen umgesetzt werden:
Nummer Name
26 Ac-CGC-NH2AuPhPy
27 Ac-CPC-NH2AuPhPy
28 Ac-CFC-NH2AuPhPy
30 Ac-CFCRRRRR-NH2AuPhPy Tabelle 4-1: Sequenzen mit Gold(III)-Komplex
Bei Sequenzen mit dem Bindungsmotiv und freien Carbonsäuren band der Gold(III)-Komplex
jedoch unspezifisch an die Sequenz, so dass kein reines Biokonjugat erhalten wurde. Dieses
Biokonjugat konnte dann über die orthogonale Kupferkatalysierte [3+2] Cycloaddition aus
dem Edukt ΠCFC-NH2AuPhPy 35 mit terminalem Alkin und den in folgender Tabelle ange-
geben Sequenzen mit einem Azid erhalten werden:
Nummer Name
36 ORRRRR-NH2
37 OPLGLYAL-NH2
38 OEEEEE-NH2
42 ORRRRRPLGLYALEEEEE-NH2
43 ORRRRRPLGLLEEEEE-NH2
44 ORRRRRLGLYLEEEEE-NH2 Tabelle 4-2: Sequenzen mit terminalem Azid
Als Test Verbindungen wurden 39, 40 und 41 mittels der [3+2] Cycloaddition aus den Ab-
schnitten 36, 37 und 38 erzeugt und die kompletten Sequenzen führten zu den Biokonjugate
45, 46 und 47 aus Abbildung 4-1. Diese letzten drei Biokonjugate entsprechen der in der Auf-
gabenstellung gesuchten Verbindung. Damit schien das Projekt zu einem erfolgreichen Ab-
schluss gebracht worden zu sein und lediglich ihre Löslichkeit in Wasser sollte erhöht wer-
den.

Zusammenfassung
Seite | 91
Abbildung 4-1: [3+2] Cycloadditionprodukte dreier kompletter Sequenzen mit ΠCFC-NH2AuPhPy 35
Um die Löslichkeit der Sequenzen in Wasser zu erhöhen, wurde ΠCRC-NH2AuPhPy 49 mit
terminalem Alkin synthetisiert. Diese Verbindung 49 war gut in Wasser löslich, es zeigte sich
aber eine geringe Stabilität in Lösung bei dieser Art von Bindungsmotiv.
Zur Verbesserung der Stabilität wurden viele neue Kombinationen von bindenden Aminosäu-
ren getestet. Es wurden dafür 300 verschieden Peptide an einem Harz ohne Linker syntheti-
siert und mit AuPhPyCl2 2 umgesetzt. Die vielversprechensten Peptide hiervon wurden an
einem Harz mit Linker synthetisiert, in Lösung mit AuPhPyCl2 2 umgesetzt und dabei wurden
unter anderen folgenden Verbindungen dargestellt:
Nummer Name
682 ΠCGZ-NH2AuPhPy
683 ΠEGC-NH2AuPhPy
684 ΠCGC-NH2AuPhPy
685 ΠMGC-NH2AuPhPy
686 ΠZGC-NH2AuPhPy
687 ΠBGC-NH2AuPhPy
688 ΠCGM-NH2AuPhPy
689 ΠZGM-NH2AuPhPy
690 ΠJGM-NH2AuPhPy
691 ΠKGM-NH2AuPhPy
692 ΠCGH-NH2AuPhPy
693 ΠMGH-NH2AuPhPy
694 ΠCGA-NH2AuPhPy
695 ΠMGA-NH2AuPhPy
696 ΠKGD-NH2AuPhPy
697 ΠDGZ-NH2AuPhPy
698 ΠZGZ-NH2AuPhPy
699 ΠCGB-NH2AuPhPy

Zusammenfassung
Seite | 92
700 ΠJGB-NH2AuPhPy
701 ΠCGJ-NH2AuPhPy
702 ΠMGK-NH2AuPhPy Tabelle 4-3: Gold(III)-Sequenzen aus dem 300 Peptidprojekt
Die Stabilität blieb bei diesen neuen Verbindungen unverändert. Um eine verbesserte Stabili-
tät herbei zu führen, wurden erneut viele Verbindungen getestet. Bei diesen Verbindungen
wurde im Bindungsmotiv die Anzahl der Abstandsaminosäuren erhöht und es wurden erneut
viele verschiedene Kombinationen von bindenden Aminosäuren ausprobiert. Durch diese
Modifizierungen wurden viele neue Substanzen mit höherer Stabilität synthetisiert:
Nummer Name
704 ΠCGGC-NH2AuPhPy
708 ΠCGGGGC-NH2AuPhPy
712 ΠCGΔGC-NH2AuPhPy
731, 733,
735, 737,
739, 741,
743, 745
ΠCRRRX1-NH2AuPhPy
732, 734,
736, 738,
740, 742,
744, 746
ΠX1RRRC-NH2AuPhPy
X1 = B, D, E, H, J, K, M oder Z
Tabelle 4-4: Optimierte Gold(III)-Sequenzen
Diese Substanzen waren zwar stabiler in Lösung, aber nicht über 75 Stunden. Dieses Kriteri-
um erfüllten folgende Verbindungen jedoch erfolgreich:
Nummer Name
706 ΠCGGGC-NH2AuPhPy
714 ΠCRRRC-NH2AuPhPy
748 ΠCFFFC-NH2AuPhPy
750 ΠCFRFC-NH2AuPhPy
752 ΠCGRGC-NH2AuPhPy
763 ΓCGRGC-NH2AuPhPy Tabelle 4-5: Stabilste Gold(III)-Sequenzen
Als stabilstes Bindungsmotiv haben sich in dieser Arbeit Sequenzen gezeigt, die aus zwei
bindenden Cysteinen und drei flexiblen Abstandsaminosäuren bestehen. Die Abstandsamino-
säuren können mit allen aliphatischen Aminosäuren frei kombiniert werden und verleihen den
Biokonjugat eine große Variabilität im Bindungsmotiv. Mit diesem neuen Typ von Bin-
dungsmotiv wurden zahlreiche orthogonale Reaktionen durchgeführt. Zum Schluss zeigte

Zusammenfassung
Seite | 93
sich, dass das neue Bindungsmotiv auch in Anwesenheit von Carbonsäuren selektiv ohne
[3+2] Cycloaddition zum Gold(III)-Komplex bindet und folgende neue Biokonjugate konnten
erfolgreich synthetisiert werden:
Nummer Name
766 Ac-CGRGCRRRRR-NH2AuPhPy
767 Ac-CGRGCRRRRRCPLGLYAL-NH2AuPhPy
769 Ac-CGRGCRRRRRCPLGLYALEEEEE-NH2AuPhPy Tabelle 4-6: Gold(III)-Biokonjugate
Das Biokonjugat 769 erfüllt dabei die Vorgabe einen Gold(III)-Komplex mit einer
Peptidsequenz, aus einem positiv geladenen Arginin, einem durch MMP2 spaltbaren und ei-
nem negativ geladenen Glutaminsäure Teil zu funktionalisieren.
Abbildung 4-2: Zielverbindung Ac-CGRGCRRRRRCPLGLYALEEEEE-NH2AuPhPy
Das Projekt ist damit zu einem synthetisch erfolgreichen Ende gebracht worden und die neuen
Biokonjugate können pharmakologisch getestet werden.
Experimentalteil
Seite | 94
5. Experimentalteil
Material
Name Lieferant CAS-Nummer
Ammoniumacetat J.T.Baker [631-61-8]
Ammoniumchlorid J.T.Baker [12125-02-9]
((1R,8S,9S)-Bicyclo[6.1.0]non-4-yn-9-
yl)methyl(2,5-dioxopyrrolidin-1-yl)carbonat
SynAffix
2,2´-Bipyridin ABCR [366-18-7]
Brombenzol Merck [108-95-4]
2-Brompyridin Aldrich [109-04-6]
4-Brompyridin Hydrochlorid Aldrich [19524-06-2]
4-Carboxybenzolboronsäure ChemPur [14047-29-1]
o-Chloranilin Aldrich [2435-53-2]
DIBAL in Hexan Aldrich [1191-15-7]
DIBAL in Toluol Alfa Aesar [1191-15-7]
Essigsäure (49-51%) HPLC Sigma-Aldrich [64-19-7]
(Essigsäuremethylester)triphenylphosphoniumchlorid ABCR [2181-97-7]
Hydrogentetrachloroaurat(III)hydrat ChemPur [16961-25-4]
Kaliumhexamethyldisilizan 0.5 M in Toluoln Aldrich [40949-94-8]
Kaliumhydroxid J.T.Baker [1310-58-3]
Kaliumtetrabromoaurat(III) ChemPur [14323-32-1]
Kupfer(I)iodid Aldrich [7681-65-4]
Kupfer(II)sulfat Pentahydrat Aldrich [7758-99-8]
Lithiumchlorid Merck [7447-41-8]
Magnesiumspäne Riedele de Haën [7439-95-4]
Magnesiumsulfat Aldrich [7487-88-9]
Malonsäure ABCR [141-82-2]
Methyl-2-phenylquinolin-4-carboxylat Aldrich [4546-48-9]
Mercaptoethanol Aldrich [60-24-2]
Natriumacetat Trihydrat Aldrich [6131-90-4]
Natriumascorbat Across [134-03-2]
Natriumcarbonat J.T.Baker [24551-51-7]
Natriumhydroxid J.T.Baker [1310-73-2]
Palladium auf Aktivkohle Fluka [7440-05-3]
Palladium(0)tetrakis(triphenylphosphin) ABCR [14221-01-3]
Phenylhydraziniumchlorid Aldrich [71-91-0]
2-Phenylpyridin Aldrich [1008-89-5]
Piperidin ABCR/biosolve [141-82-2]
Pyridin J.T.Baker [110-86-1]
4-(2-Pyridin)phenyl-3-aldehyd Aldrich [127406-56-8]
Quecksilber(II)acetat Acros [1600-27-7]
Silberacetat Fluka [563-63-3]
Sinapinsäure Bruker [530-59-6]
Tetraethylammoniumbromid Aldrich [71-91-0]
Tetraethylammoniumchlorid Aldrich [56-34-8]
Triethylamin J.T.Baker [121-44-8]
Experimentalteil
Seite | 95
Triethylphosphitkupfer(I)iodid Lehrstuhl [51717-23-8]
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amin) Lehrstuhl [510758-28-8]
Wasserstoff Air Liquid [1333-74-0] Tabelle 5-1
Name Abkürzung Code Lieferant CAS-Nummer
Acetanhydrid Ac2O Aldrich [108-24-7]
1,2-Ethandithiol EDT Alfa Aesar [540-63-6]
Fmoc-Aib-OH Aib Δ Iris [62-57-7]
Fmoc-Gly-OH Gly G Iris [29022-11-5]
Fmoc-L-Ala-OH Ala A novabiochem [04-12-1006]
Fmoc-L-Asp(OtBu)-OH Asp D Iris [71989-14-5]
Fmoc-L-Arg(Pbf)-OH Arg R Iris/Biosolve [154445-77-9]
Fmoc-L-Cys(Trt)-OH Cys C Iris [103213-32-7]
Fmoc-L-Dap(Boc)-OH Dap Z Iris [162558-25-0]
Fmoc-L-Glu(ODmab)-OH Glu E Iris [268730-86-5]
Fmoc-L-Glu(OtBu)-OH Glu E Iris [71989-18-9]
Fmoc-L-His(Trt)-OH His H novabiochem [109425-51-6]
Fmoc-L-Leu-OH Leu L Iris/Biosolve [35661-60-0]
Fmoc-L-Lys(Boc)-OH Lys K Iris [71989-26-9]
Fmoc-L-Pal3-OH Pal3 B Iris [175453-07-3]
Fmoc-L-Pal4-OH Pal4 J Iris [169555-95-7]
Fmoc-L-Phe-OH Phe F Iris [35661-40-6]
Fmoc-L-Pro-OH Pro P Iris [71989-31-6]
Fmoc-L-Met-OH Met M novabiochem [71989-28-1]
2-(1H-Benzotriazole-1-yl)-1,1,3,3-
tetramethyl-
uronium hexafluorophosphate HBTU Biosolve [94790-37-1]
1-Hydroxybenzotriazole HOBT molekula [2592-95-2]
Methyladipoylchlorid Γ ABCR [35444-44-1]
N,N-Diisopropylethylamin DIPEA Aldrich [7087-68-5]
O-(Benzotriazol-1-yl)-N,N,N′,N′-
tetramethyluronium tetrafluoroborat TBTU Iris [125700-67-6]
4-Pentinsäure Pen Π Aldrich/ABCR [6089-09-04]
Triisopropylsilan TIS Aldrich [6485-79-6] Tabelle 5-2
Lösungsmittel Abkürzung Lieferant
Aceton VWR
Acetonitril ACN J.T.Baker/ Biosolve
Dichlormethan DCM Fisher
Diethylether DET J.T.Baker
N,N-Dimethylformamid DMF Roth/ biosolve
Dimethylsulfoxid DMSO J.T.Baker
Essigsäure VWR
Ethanol EtOH Aldrich
n-Hexen VWR
Methanol MeOH J.T.Baker

Experimentalteil
Seite | 96
N-Methyl-2-pyrrolidon NMP Roth/ Biosolve
Salzsäurelösung 37 % J.T.Baker/ VWR
Tetrahydrofuran THF VWR
Trifluoressigsäure TFA Acros/ Alfa Aesar/ Biosolve Tabelle 5-3
Methoden
Jede kommerziell erhalten Chemikalien wurden ohne weitere Reinigung verwendet, außer es
ist in der Vorschrift anders vermerkt. Bei jeder Luft empfindlichen Reaktion unter Schutzgas
wurden standard Schlenktechniken verwendet.
Die NMR-Messungen wurden auf folgenden Geräten der Firma Bruker durchgeführt: DPX
200, DPX 250 und DRX 400. Die Messungen erfolgten bei Raumtemperatur. Die chemische
Verschiebung (δ) wird in ppm bezogen auf TMS und das das nicht vollständig deuterierte
Lösungsmittel angegeben. Die Multiplizitäten sind wie folgt abgekürzt: Singulett = s, Duplett
= d, Triplett = t, Quartett = q, Duplett vom Duplett = dd , Triplett vom Duplett = td, Multiplett
= m.
Alle Elementaranalysen wurden auf einer Vario EL Elementar Hanau im C,H,N,S-Modus
durchgeführt.
Als Zentrifuge wurde eine Universal 320 R von Hettich Zentrifugen genutzt.
An einem automatisierten Bruker Autoflex MALDI-TOF Massenspektrometer wurden die
MALDI Massenspektren gemessen. Als Matrix wurde Sinapinsäure verwendet, die Spektren
wurden im positiven Modus, in der Reflektoreinstellung gemessen.
Die ESI Massenspektren wurden auf einem Bruker Esquire 6000 Massenspektrometer gemes-
sen. Die Proben wurden Manuel aufgegeben. Es wurde meist im positiven (ESI+), manchmal
auch im negativen (ESI-) gemessen. Das Verhältnis von Masse geteilt durch die Ladung (m/z)
wird als dimensionslose Zahl angegeben.
Die HPLC-MS Versuche wurden an einem Agilent 1100, welches am Bruker Esquire 6000
Massenspektrometer angeschlossen war, durchgeführt. Die Agilent 1100 beinhaltete ein Pro-
ben Automation. Als Säule wurde ein Agilent Zorbax SB-C18 2.7 µm (2.1 x 5.0 mm) ver-
wendet. Die Chromatogramme wurden bei 220 nm aufgenommen. Das Laufmittel bestand aus
Wasser mit 0.05 % Trifluoressigsäure und Acetonitril. Der Gradient begann bei 100 % Was-

Experimentalteil
Seite | 97
ser mit TFA und nach 10 min wurden 100 % Acetonitril verwendet. Es wurden für 2 min
100 % Acetonitril verwendet, um dann innerhalb von 6 min wieder auf 100 % Wasser mit
TFA umzuschalten.
Die analytischen HPLC Versuche wurden an einem Knauer HPLC Instrument durchgeführt.
Es wurden zwei verschieden Lösungsmittelgemische verwendet. Das erste Lösungsmittelge-
misch A besteht aus 95 % Wasser, 5 % Acetonitril und 0.1 % Trifluressigsäure. Lösungsmit-
telgemisch B besteht aus 95 % Acetonitril, 5 % Wasser und 0.1 % Trifluoressigsäure. Alle
analytischen HPLC Versuche wurden im Gradientenverfahren durchgeführt. Jede Messung
dauerte 40 min. Die ersten 5 min wurde 100 % von Gemisch A verwendet, dann startete der
Gradient und nach 25 min wurden für 5 min 100 % von Lösungsmittelgemisch B genutzt, um
dann nach 35 min wieder für 5 min100 % von Lösungsmittelgemisch A zu nutzen. Es wurde
bei einem Fluss von 1 ml/min gearbeitet. Die Chromatogramme wurden bei 214 nm und bei
254 nm aufgenommen. Als Säulenmaterial wurde eine Dr. Maisch Reprosil-C18-AQ (250 x
4.6 mm), Knauer Eurospher II 100-5 C18A (250 x 4 mm), und eine Dr. Maisch ReproSil-Pur
C8 5 µm (250 x 4.6 mm) genutzt.
Die semipräperative HPLC Aufreinigungen wurden auf dem Knauer HPLC Instrument
durchgeführt. Es wurde mit einer Merck LiChrospher® 100 RP-18e 10 µm (250 x 10 mm) bei
einer Flussrate von 5 ml/min gearbeitet. Beim Gradienten wurde zuerst mit 100 % von Ge-
misch A für 5 min gearbeitet, dann wurde innerhalb von 20 min der Gradient auf 100 % von
Lösungsmittelgemisch B umgestellt. Für 10 min wurde dann mit 100 % von Gemisch B gear-
beitet. Innerhalb von 5 min wurde die mobile Phase wieder auf 100 % Gemisch A umgestellt
und so für die letzten 5 min der Aufreinigung belassen. Die Detektion erfolgte bei 214 nm und
bei 254 nm.
Präperative HPLC Reinigung wurden auf einer Varian ProStar 210 mit PDA Detektor durch-
geführt. Die Versuche wuden bei einer Flussrate von 20 ml/min durchgeführt. Als stationäre
Phase wurde eine HIBAR Lichrospher 100 RP-18e (250 x 25 mm) verwendet. In den ersten 5
min wurde eine mobile Phase von 100 % Lösungsmittelgemisch A genutzt. Nach 5 min wurde
der Anteil von Lösungsmittelgemisch B sukzessive erhöht, bis er in Minute 25 des Laufes
99 % von Lösungsmittelgemisch B erreicht hatte. Es wurde dann für 5 min 99 % von Ge-
misch B gepumpt, um danach innerhalb von 5 min wieder 100 % von Gemisch A zu pumpen.
Es wurde dann für die letzten 5 min der Reinigung bei 100 % von Gemisch A gepumpt.

Experimentalteil
Seite | 98
Das genutzte Fluoreszenzmikroskop ist ein IX-81 Olympus. Die Anregung erfolgte bei
330 nm – 385 nm, die Emission erfolgte bei > 420 nm.
Mikrowellereaktionen wurden in einem CEM Discover Mikrowellengerät ausgeführt.
Es wurde ein WITec Alpha300 R/A/S Ramanspektrometer verwendet. Alle Messungen wur-
den mit einem Laser bei 785 nm bei 200 mW durchgeführt. Die jeweilige Bande kann
schwach ausgeprägt sein, dann ist die Bande mit einem „w“ gekennzeichnet. Falls die Bande
mittel ausgeprägt ist, ist sie mit einem „m“ gekennzeichnet. Alle mit einem „s“ gekennzeich-
neten Banden, sind stark ausgeprägt.
Synthesen
Synthese von 2-Phenylpyridin-1-gold(III)trichlorid 1
250 mg (0.7353 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 20 ml Wasser gelöst.
Es bildete sich eine gelbfarbene Lösung. 0.13 ml (0.8834 mmol) 2-Phenylpyridin wurden in
20 ml Ethanol gelöst und langsam in die erste Lösung getropft. An der Eintropfstelle bildete
sich umgehend ein gelbfarbener Niederschlag. Die Suspension wurde für zwei Stunden ge-
rührt. Der Feststoff wurde von der Lösung getrennt und getrocknet.
Ausbeute: 232.5 mg (0.5088 mmol) bezogen auf das eingesetzte Hydrogentetrachloroaurat-
(III)hydrat: 69.2 %
1H NMR (250 MHz, DMSO-d6) δ 9.30 (d, J = 5.2 Hz, 1H), 8.48 (td, J = 7.8, 1.2 Hz 1H),
8.12 (d, J = 7.4 Hz, 1H), 7.99 (t, J = 7.6 Hz, 1H), 7.91 – 7.80 (m, 4H), 7.76 (t, J = 6.5 Hz,
1H).

Experimentalteil
Seite | 99
Synthese von 2-Phenylpyridin-1,1´-gold(III)dichlorid 2
232.5 mg (0.5088 mmol) 2-Phenylpyridin-1-gold(III)trichlorid 1 wurden in einer Mischung
aus 20 ml Wasser und 20 ml Acetonitril suspendiert. Die Suspension wurde refluxiert, wobei
eine gelbfarbene Lösung entstandt. Die Lösung wurde für vier Stunden in der Siedehitze ge-
rührt. Langsam wurde die Lösung auf Raumtemperatur abgekühlt. Es entstand ein
gelbfarbener Niederschlag. Der Feststoff wurde von der Lösung getrennt und getrocknet.
Ausbeute: 94.6 mg (0.2247 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-1-
gold(III)trichlorid 1: 44.2 %
1H NMR (250 MHz, DMSO-d6) δ 9.52 (d, J = 5.9 Hz, 1H), 8.46 – 8.37 (m, 2H), 7.97 (d, J =
7.8 Hz, 1H), 7.85 – 7.73 (m, 2H), 7.48 (t, J = 7.3 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 183.5, 168.2, 149.3, 129.1, 127.5, 127.0, 126.1, 125.5,
124.9, 124.2, 122.9
Elementaranalyse (kalkuliert in Klammern): C 31.26 (31.30), N 3.26 (3.32), H 1.96 (1.91)

Experimentalteil
Seite | 100
Synthese von 2-Phenylpyridin-3´-säure 3 116
2-Brompyridin wurde destilliert. 939.5 µl (9.64 mmol) der Fraktion, welche bei einer Bad-
temperatur von 125 °C, einer Kopftemperatur von 74 °C und einem Druck von 16 mbar erhal-
ten wurden, wurden zusammen mit 1.6 g (9.64 mmol) Carboxybenzolboronsäure und
578.4 mg (0.482 mmol) Palladium(0)tetrakis(triphenylphosphin) in 100 ml Acetonitril und
100 ml 0.4 M Natriumcarbonatlösung suspendiert. Die Lösung wurde 20 min mit Stickstoff
durchblasen. Die Lösung wurde für 20 Stunden refluxiert. Die Suspension wurde heiß filtriert.
Es blieb ein gelbfarbener Rückstand im Filter. Die Lösung war leicht gelbfarben. Sie wurde
bis zur Hälfte eingeengt, wobei sich ein farbloser Schaum bildete. Die Lösung wurde dreimal
mit etwa 200 ml Dichlormethan gewaschen. Die wässrige Phase war gelbfarben, ihr wurden
3 ml konzentrierte Salzsäurelösung zugegeben. Es bildete sich eine Trübung der Lösung. Die
Lösung wurde über Nacht bei – 20 °C gelagert. Die Suspension wurde aufgetaut und der farb-
lose Niederschlag wurde von der Lösung getrennt und getrocknet.
Ausbeute: 1.75 g (8.81 mmol) bezogen auf das eingesetzte 2-Brompyridin: 91.4 %
1H NMR (250 MHz, DMSO-d6) δ 8.82 (d, J = 4.9 Hz, 1H), 8.31-8.19 (m, 4H), 8.10 (d, J = 8.4
Hz, 2H), 7.73 (td, 6.4 Hz, 2.2 Hz, 1H).

Experimentalteil
Seite | 101
Synthese von 2-Phenylpyridin-3´-säure-1´-gold(III)trichlorid 4
500 mg (2,51 mmol) 2-Phenylpyridin-3´-säure 3 wurden in 30 ml Ethanol gelöst. 675.7 mg
(2 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 30 ml Wasser gelöst und zur ersten
Lösung gegeben. Es entstand eine gelbfarbenen Suspension. Die Suspension wurde über
Nacht gerührt. Die Suspension wurde zentrifugiert, der Rückstand wurde getrocknet.
Ausbeute: 581.5 mg (1.1608 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-3´-säure 3:
58 %
1H NMR (250 MHz, DMSO-d6) δ 8.76 (d, J = 4.7 Hz, 1H), 8.21 (d, J = 7.9 Hz, 2H), 8.14 –
8.09 (m, 4H), 7.55 (t, J = 5.6 Hz, 1H).
Elementaranalyse (kalkuliert in Klammern): C 28.56 (28.68), N 2.73 (2.79), H 1.73 (1.81)
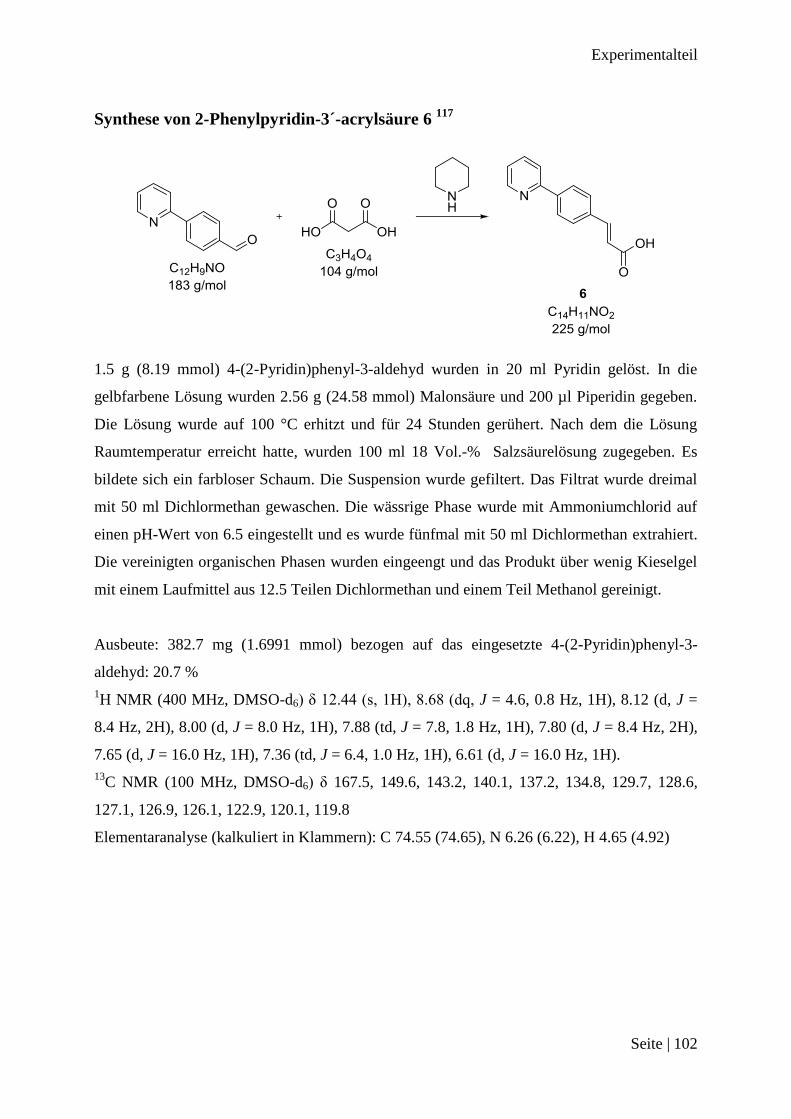
Experimentalteil
Seite | 102
Synthese von 2-Phenylpyridin-3´-acrylsäure 6 117
1.5 g (8.19 mmol) 4-(2-Pyridin)phenyl-3-aldehyd wurden in 20 ml Pyridin gelöst. In die
gelbfarbene Lösung wurden 2.56 g (24.58 mmol) Malonsäure und 200 µl Piperidin gegeben.
Die Lösung wurde auf 100 °C erhitzt und für 24 Stunden gerühert. Nach dem die Lösung
Raumtemperatur erreicht hatte, wurden 100 ml 18 Vol.-% Salzsäurelösung zugegeben. Es
bildete sich ein farbloser Schaum. Die Suspension wurde gefiltert. Das Filtrat wurde dreimal
mit 50 ml Dichlormethan gewaschen. Die wässrige Phase wurde mit Ammoniumchlorid auf
einen pH-Wert von 6.5 eingestellt und es wurde fünfmal mit 50 ml Dichlormethan extrahiert.
Die vereinigten organischen Phasen wurden eingeengt und das Produkt über wenig Kieselgel
mit einem Laufmittel aus 12.5 Teilen Dichlormethan und einem Teil Methanol gereinigt.
Ausbeute: 382.7 mg (1.6991 mmol) bezogen auf das eingesetzte 4-(2-Pyridin)phenyl-3-
aldehyd: 20.7 %
1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 8.68 (dq, J = 4.6, 0.8 Hz, 1H), 8.12 (d, J =
8.4 Hz, 2H), 8.00 (d, J = 8.0 Hz, 1H), 7.88 (td, J = 7.8, 1.8 Hz, 1H), 7.80 (d, J = 8.4 Hz, 2H),
7.65 (d, J = 16.0 Hz, 1H), 7.36 (td, J = 6.4, 1.0 Hz, 1H), 6.61 (d, J = 16.0 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 167.5, 149.6, 143.2, 140.1, 137.2, 134.8, 129.7, 128.6,
127.1, 126.9, 126.1, 122.9, 120.1, 119.8
Elementaranalyse (kalkuliert in Klammern): C 74.55 (74.65), N 6.26 (6.22), H 4.65 (4.92)

Experimentalteil
Seite | 103
Synthese von 2-Phenylpyridin-3´-propansäure 7
In einen Schlenkkolben wurden 150 mg (0.666 mmol) 2-Phenylpyridin-3´-acrylsäure 6 gege-
ben und in 20 ml Ethanol suspendiert. In die Suspension wurde für 10 min Stickstoff eingelei-
tet. Es wurden 17 mg einer Mischung aus einem Teil Palladium und neun Teilen Aktivkohle
in die Suspension gegeben. Mittels einer Wasserstrahlpumpe wurde der Druck im Kolben
vermindert. Nun wurde an den Schlenkkolben Wasserstoffgas angeschlossen und der Kolben
mit dem Gas geflutet. Nach 40 Stunden unter Wasserstoffatmosphäre wurde etwas Kieselgel
zugegeben und das Lösungsmittel entfernt. Das Rohprodukt wurde über wenig Kieselgel mit
einem Laufmittel aus neun Teilen Dichlormethan und einem Teil Methanol gereinigt.
Ausbeute: 116.5 mg (0.513 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-3´-acrylsäure
6: 77 %
1H NMR (250 MHz, DMSO-d6) δ 8.64 (dt, J = 4.8, 0.8 Hz 1H), 7.99 (d, J = 8.2 Hz, 2H), 7.94
– 7.81 (m, 2H), 7.40 – 7.26 (m, 3H), 2.88 (t, J = 7.5 Hz, 2H), 2.58 (t, J = 7.5 Hz, 2H).
N
O
OH
6C14H11NO2
225 g/mol
H2
H2
2 g/mol
PalladiumAktivkohle N
O
OH
7C14H13NO2
227 g/mol
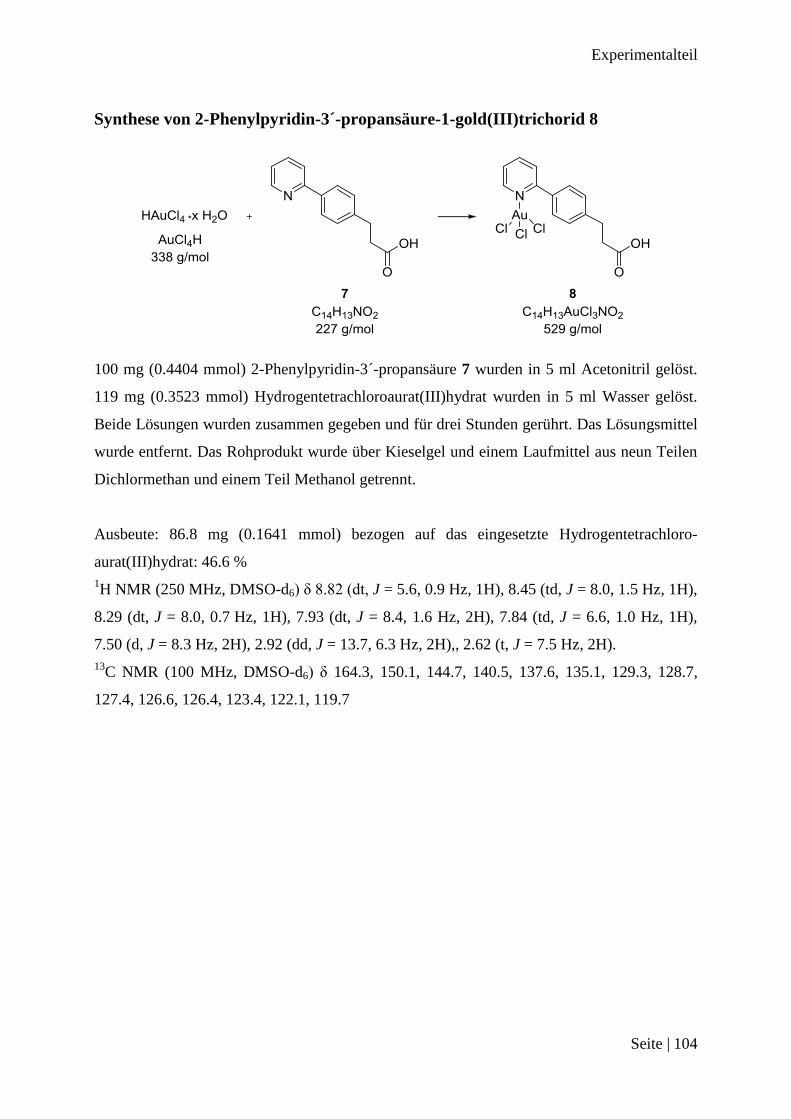
Experimentalteil
Seite | 104
Synthese von 2-Phenylpyridin-3´-propansäure-1-gold(III)trichorid 8
100 mg (0.4404 mmol) 2-Phenylpyridin-3´-propansäure 7 wurden in 5 ml Acetonitril gelöst.
119 mg (0.3523 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 5 ml Wasser gelöst.
Beide Lösungen wurden zusammen gegeben und für drei Stunden gerührt. Das Lösungsmittel
wurde entfernt. Das Rohprodukt wurde über Kieselgel und einem Laufmittel aus neun Teilen
Dichlormethan und einem Teil Methanol getrennt.
Ausbeute: 86.8 mg (0.1641 mmol) bezogen auf das eingesetzte Hydrogentetrachloro-
aurat(III)hydrat: 46.6 %
1H NMR (250 MHz, DMSO-d6) δ 8.82 (dt, J = 5.6, 0.9 Hz, 1H), 8.45 (td, J = 8.0, 1.5 Hz, 1H),
8.29 (dt, J = 8.0, 0.7 Hz, 1H), 7.93 (dt, J = 8.4, 1.6 Hz, 2H), 7.84 (td, J = 6.6, 1.0 Hz, 1H),
7.50 (d, J = 8.3 Hz, 2H), 2.92 (dd, J = 13.7, 6.3 Hz, 2H),, 2.62 (t, J = 7.5 Hz, 2H).
13C NMR (100 MHz, DMSO-d6) δ 164.3, 150.1, 144.7, 140.5, 137.6, 135.1, 129.3, 128.7,
127.4, 126.6, 126.4, 123.4, 122.1, 119.7
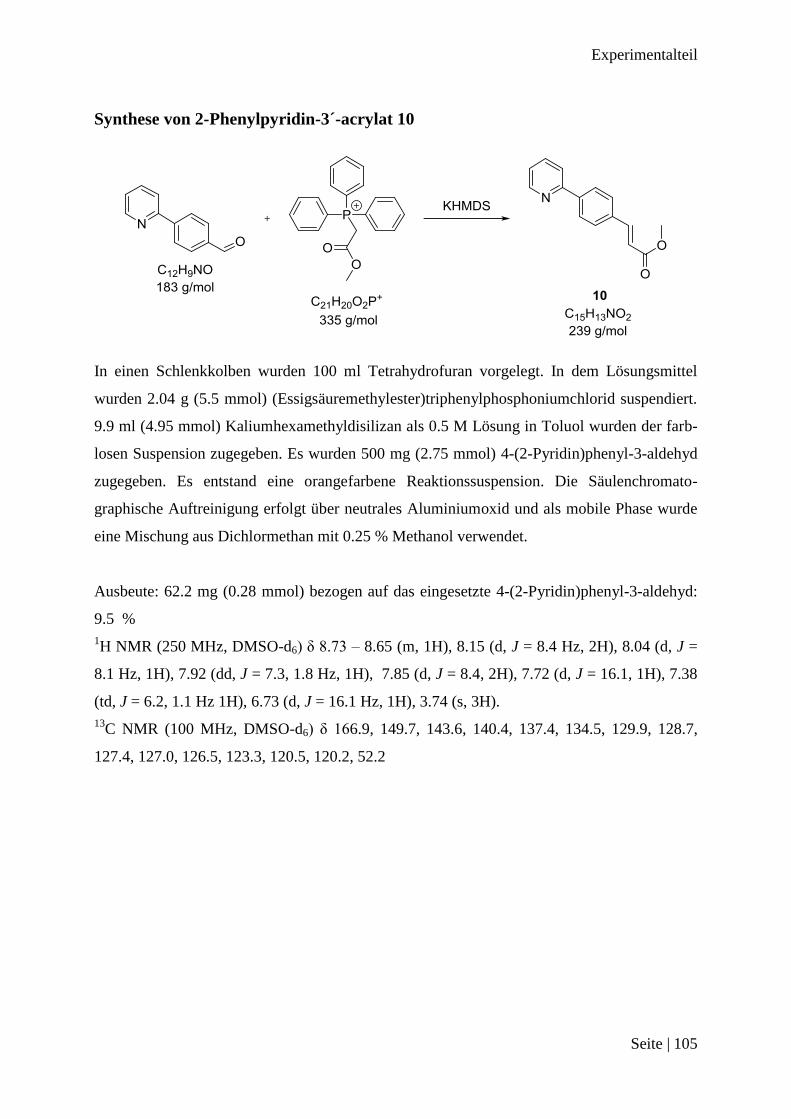
Experimentalteil
Seite | 105
Synthese von 2-Phenylpyridin-3´-acrylat 10
In einen Schlenkkolben wurden 100 ml Tetrahydrofuran vorgelegt. In dem Lösungsmittel
wurden 2.04 g (5.5 mmol) (Essigsäuremethylester)triphenylphosphoniumchlorid suspendiert.
9.9 ml (4.95 mmol) Kaliumhexamethyldisilizan als 0.5 M Lösung in Toluol wurden der farb-
losen Suspension zugegeben. Es wurden 500 mg (2.75 mmol) 4-(2-Pyridin)phenyl-3-aldehyd
zugegeben. Es entstand eine orangefarbene Reaktionssuspension. Die Säulenchromato-
graphische Auftreinigung erfolgt über neutrales Aluminiumoxid und als mobile Phase wurde
eine Mischung aus Dichlormethan mit 0.25 % Methanol verwendet.
Ausbeute: 62.2 mg (0.28 mmol) bezogen auf das eingesetzte 4-(2-Pyridin)phenyl-3-aldehyd:
9.5 %
1H NMR (250 MHz, DMSO-d6) δ 8.73 – 8.65 (m, 1H), 8.15 (d, J = 8.4 Hz, 2H), 8.04 (d, J =
8.1 Hz, 1H), 7.92 (dd, J = 7.3, 1.8 Hz, 1H), 7.85 (d, J = 8.4, 2H), 7.72 (d, J = 16.1, 1H), 7.38
(td, J = 6.2, 1.1 Hz 1H), 6.73 (d, J = 16.1 Hz, 1H), 3.74 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 166.9, 149.7, 143.6, 140.4, 137.4, 134.5, 129.9, 128.7,
127.4, 127.0, 126.5, 123.3, 120.5, 120.2, 52.2
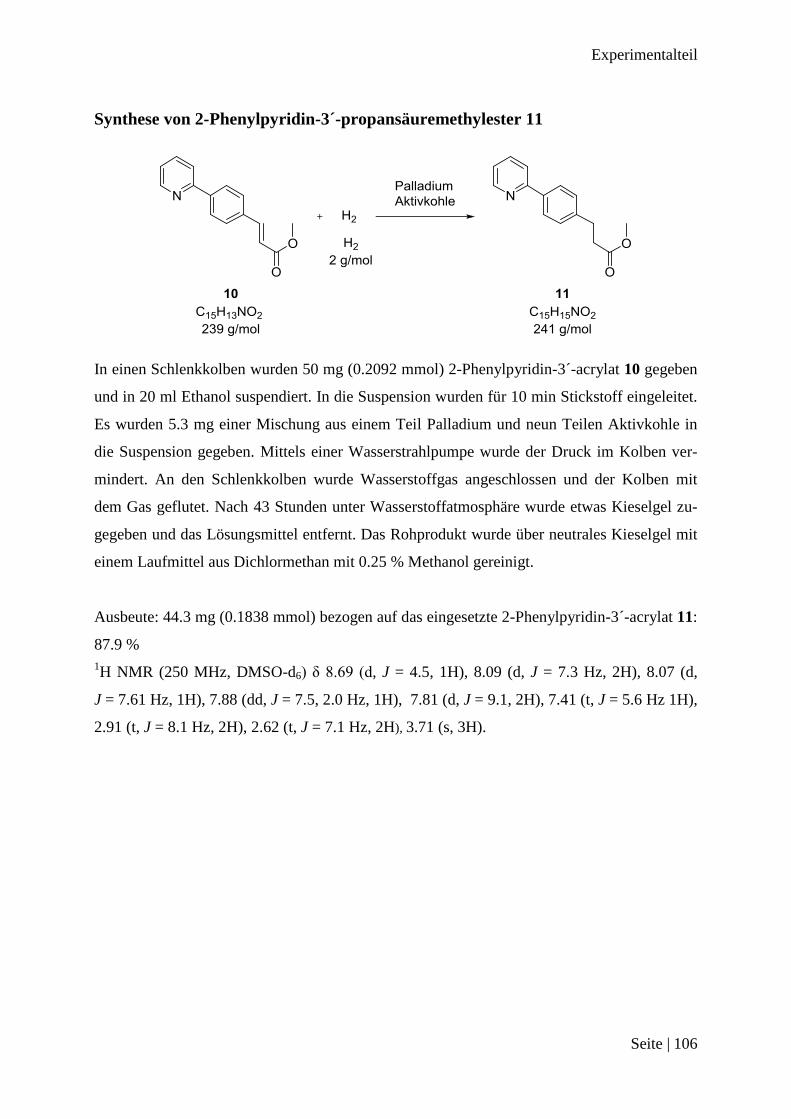
Experimentalteil
Seite | 106
Synthese von 2-Phenylpyridin-3´-propansäuremethylester 11
In einen Schlenkkolben wurden 50 mg (0.2092 mmol) 2-Phenylpyridin-3´-acrylat 10 gegeben
und in 20 ml Ethanol suspendiert. In die Suspension wurden für 10 min Stickstoff eingeleitet.
Es wurden 5.3 mg einer Mischung aus einem Teil Palladium und neun Teilen Aktivkohle in
die Suspension gegeben. Mittels einer Wasserstrahlpumpe wurde der Druck im Kolben ver-
mindert. An den Schlenkkolben wurde Wasserstoffgas angeschlossen und der Kolben mit
dem Gas geflutet. Nach 43 Stunden unter Wasserstoffatmosphäre wurde etwas Kieselgel zu-
gegeben und das Lösungsmittel entfernt. Das Rohprodukt wurde über neutrales Kieselgel mit
einem Laufmittel aus Dichlormethan mit 0.25 % Methanol gereinigt.
Ausbeute: 44.3 mg (0.1838 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-3´-acrylat 11:
87.9 %
1H NMR (250 MHz, DMSO-d6) δ 8.69 (d, J = 4.5, 1H), 8.09 (d, J = 7.3 Hz, 2H), 8.07 (d,
J = 7.61 Hz, 1H), 7.88 (dd, J = 7.5, 2.0 Hz, 1H), 7.81 (d, J = 9.1, 2H), 7.41 (t, J = 5.6 Hz 1H),
2.91 (t, J = 8.1 Hz, 2H), 2.62 (t, J = 7.1 Hz, 2H), 3.71 (s, 3H).

Experimentalteil
Seite | 107
Synthese von 2-Phenylpyridin-3´-propansäuremethylester-1-gold(III)trichorid 12
32.4 mg (0.1344 mmol) 2-Phenylpyridin-3´-propansäuremethylester 11 wurden in 3 ml Ace-
tonitril gelöst. 36.5 mg (0.1075 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 3 ml
Wasser gelöst. Beide Lösungen wuden zusammen gegeben und für drei Stunden gerührt. Das
Lösungsmittel wurde entfernt. Das Rohprodukt wurde über neutrales Aluminiumoxid und
einem Laufmittel aus hundert Teilen Dichlormethan und einem Teil Methanol getrennt.
Ausbeute: 22.4 mg (0.0413 mmol) bezogen auf das eingesetzte Hydrogentetrachloro-
aurat(III)hydrat: 38.4 %
1H NMR (250 MHz, DMSO-d6) δ 8.85 (dt, J = 4.7, 1.2 Hz, 1H), 8.46 (td, J = 8.5, 1.1 Hz, 1H),
8.25 (dt, J = 6.3, 0.3 Hz, 1H), 7.99 (dt, J = 8.2, 1.0 Hz, 2H), 7.86 (td, J = 5.6, 1.9 Hz, 1H),
7.63 (d, J = 7.7 Hz, 2H), 2.88 (t, J = 7.8 Hz, 2H), 2.55 (t, J = 6.9 Hz, 2H), 3.78 (s, 3H).
Elementaranalyse (kalkuliert in Klammern): C 33.16 (33.08), N 2.46 (2.57), H 2.69 (2.78)

Experimentalteil
Seite | 108
Synthese von 4-Brom-2-Phenylpyridin 14
2.1067 g (11.7 mmol) Magnesiumspäne wurden in einen Schlenkkolben in 4 ml Tetrahydro-
furan suspendiert. Der Mischung wurden langsam 1.22 ml (11.7 mmol) Brombenzol zuge-
tropft. Die Lösung wurde für eine Stunde refluxiert.
In einem zweiten Schlenkkolben wurden 750 mg (4.78 mmol) 4-Brompyridin Hydrochlorid in
50 ml Tetrahydrofuran suspendiert. Der Kolben wurde auf -78 °C gekühlt und es wurde für
eine halbe Stunde gerührt. Die erste nun graufarbene Lösung wurde langsam in die Suspensi-
on getropft. Es wurde eine halbe Stunde nach dem zu tropfen weiter bei -78 °C gerührt. Es
werden 705 µl (5.58 mmol) Phenylchlorformiat in 5 ml Tetrahydrofuran zugegeben. Die
gelbfarbene Lösung wurde eine halbe Stunde bei Raumtemperatur gerührt. Die Lösung wurde
in einen Kolben mit 20 Gew.-% Ammoniumchloridlösung bei 0 °C gegeben. Es wurde drei-
mal mit 30 ml Diethylether extrahiert. Die vereinigten Diethyletherphasen wurden mit 50 ml
Wasser gewaschen, mit 50 ml 10 Vol.-% Salzsäurelösung und wieder mit 50 ml Wasser ge-
waschen. Die organische Phase wurde über Magnesiumsulfat getrocknet. Das Lösungsmittel
wurde evaporiert und das zurückbleibende gelbfarbene Öl wurde für 20 Stunden getrocknet.
Das Öl wurde in einem Schlenkkolben in 30 ml Toluol gelöst. Der Lösung wurden 1.451 g
(5.95 mmol) o-Chloranilin in 20 ml Eisessig zugegeben. Die rotfarbene Lösung wurde für
22 Stunden gerührt. Es wurden 40 ml einer 10 Gew.-% Natriumhydroxidlösung zugegeben.
Die Lösung färbte sich tief dunkel rotfarben und es bildete sich ein schwarzfarbener Nieder-
schlag. Die Suspension erwärmt sich. Sie wurde durch Celite gefiltert. Das Filtrat wurde
dreimal mit 50 ml Wasser gewaschen. Die organische Phase wurde dreimal mit 10 Vol.-%
Salzsäurelösung extrahiert. Die vereinigten wässrigen Phasen wurden dreimal mit 50 ml
Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Magnesiumsulfat
getrocknet, gefiltert und das Lösungsmittel bei vermindertem Druck entfernt. Der orangefar-
bene Rückstand wurde getrocknet.

Experimentalteil
Seite | 109
Ausbeute: 607.3 mg (2.6067 mmol) bezogen auf das eingesetzte 4-Brompyridin: 54.5 %
1H NMR (200 MHz, CD2Cl2) δ 8.08 (d, J = 4.8 Hz, 1H), 7.63 – 7.54 (m, 2H), 7.51 (dd, J =
1.6, 0.4 Hz 1H), 7.06 (d, J = 1.9 Hz, 1H), 7.03 (t, J = 1.8 Hz, 2H), 6.99 (d, J = 1.8 Hz, 1H).
13C NMR (100 MHz, CD2Cl2) δ 156.3, 151.6, 138.7, 133.9, 131.2, 129.6, 128.7, 127.9, 127.6,
127.3, 123.5
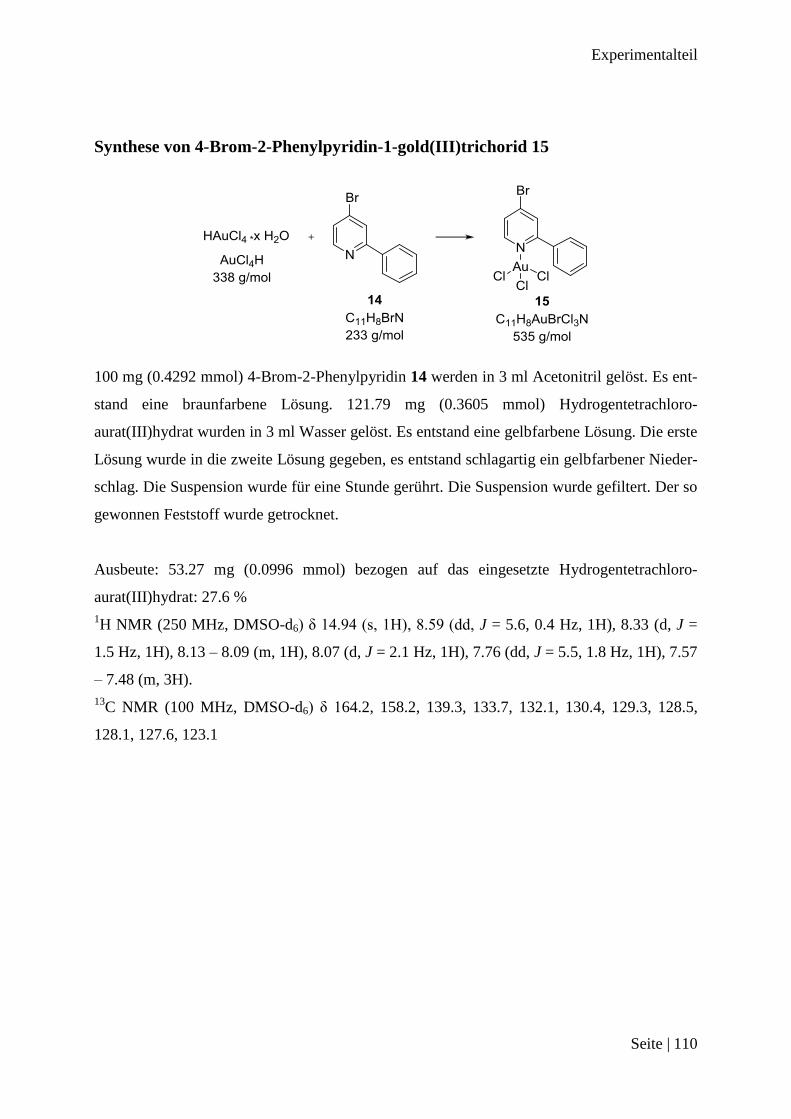
Experimentalteil
Seite | 110
Synthese von 4-Brom-2-Phenylpyridin-1-gold(III)trichorid 15
100 mg (0.4292 mmol) 4-Brom-2-Phenylpyridin 14 werden in 3 ml Acetonitril gelöst. Es ent-
stand eine braunfarbene Lösung. 121.79 mg (0.3605 mmol) Hydrogentetrachloro-
aurat(III)hydrat wurden in 3 ml Wasser gelöst. Es entstand eine gelbfarbene Lösung. Die erste
Lösung wurde in die zweite Lösung gegeben, es entstand schlagartig ein gelbfarbener Nieder-
schlag. Die Suspension wurde für eine Stunde gerührt. Die Suspension wurde gefiltert. Der so
gewonnen Feststoff wurde getrocknet.
Ausbeute: 53.27 mg (0.0996 mmol) bezogen auf das eingesetzte Hydrogentetrachloro-
aurat(III)hydrat: 27.6 %
1H NMR (250 MHz, DMSO-d6) δ 14.94 (s, 1H), 8.59 (dd, J = 5.6, 0.4 Hz, 1H), 8.33 (d, J =
1.5 Hz, 1H), 8.13 – 8.09 (m, 1H), 8.07 (d, J = 2.1 Hz, 1H), 7.76 (dd, J = 5.5, 1.8 Hz, 1H), 7.57
– 7.48 (m, 3H).
13C NMR (100 MHz, DMSO-d6) δ 164.2, 158.2, 139.3, 133.7, 132.1, 130.4, 129.3, 128.5,
128.1, 127.6, 123.1

Experimentalteil
Seite | 111
Synthese von 2-Phenylquinolin-4-methylcarboxylat-1´-chloromercury 17 123
In einen 250 ml Kolben wurden 11 g (0.0344 mol) Quecksilber(II)acetat in 40 ml Ethanol
suspendiert. Es wurden 9.044 g (0.0344 mol) Methyl-2-phenylquinolin-4-carboxylat in 40 ml
Ethanol suspendiert und in den 250 ml Kolben gegeben. Das Gefäß in dem das Methyl-2-
phenylquinolin-4-carboxylat suspendiert war, wurde dreimal mit 20 ml Ethanol gespült. Die
Spüllösungen wurde in den 250 ml Kolben gegeben. Die leicht gelbfarbene Suspension wurde
für 12 Stunden unter Rückfluss zum Sieden erhitzt. Es wurden 80 ml Ethanol zur Suspension
zugegeben. Die Partikel in der Suspension waren farblos, das Lösungsmittel war gelbfarben.
Die heiße Suspension wurde durch einen Faltenfilter in eine Methanollösung aus 50 ml Me-
thanol und 2.0218 g (0.04816 mol) Lithiumchlorid gefiltert. Es entsteht ein farbloser Nieder-
schlag, die Lösung war leicht gelbfarben. Der farblose Feststoff wurde von der Lösung ge-
trennt, dreimal mit 50 ml Wasser und dreimal mit 50 ml Ethanol gewaschen. Der Feststoff
wurde getrocknet.
Ausbeute: 9.7103 g (0.0196 mol) bezogen auf das eingesetzte Methyl-2-phenylquinolin-4-
carboxylat: 56.6 %
1H NMR (200 MHz, CDCl3) δ 8.59 (s, 1H), 8.43 (t, J = 2.1 Hz 1H), 8.37 (t, J = 2.2 Hz, 1H),
8.01 (t, J = 1.9 Hz, 1H), 7.54 (d, J = 1.8 Hz, 1H), 7.42 – 7.38 (m, 3H), 7.04 (d, J = 1.7 Hz,
1H), 4.01 (s, 3H).
13C NMR (100 MHz, CDCl3) δ 148.3, 140.9, 128.3, 129.3, 128.6, 138.3, 155.0, 110.9, 124.9,
125.6, 131.1, 128.2, 130.9, 136.7, 147.9, 166.5, 53.1
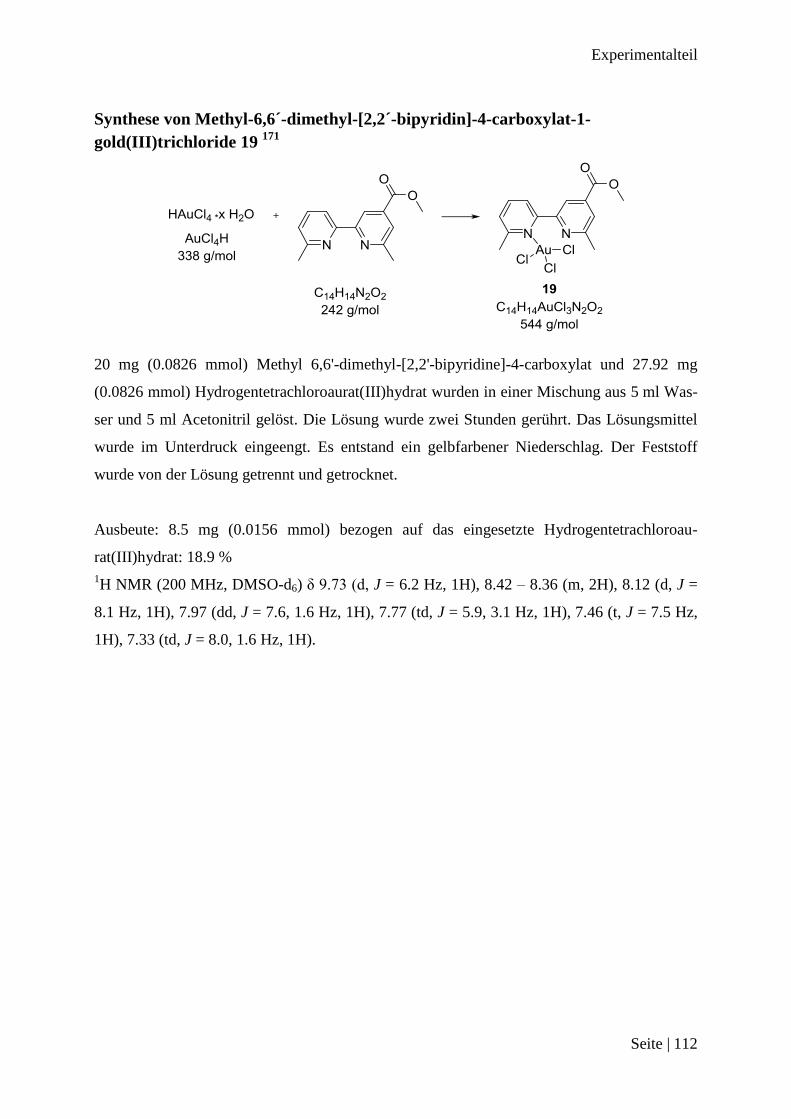
Experimentalteil
Seite | 112
Synthese von Methyl-6,6´-dimethyl-[2,2´-bipyridin]-4-carboxylat-1-
gold(III)trichloride 19 171
20 mg (0.0826 mmol) Methyl 6,6'-dimethyl-[2,2'-bipyridine]-4-carboxylat und 27.92 mg
(0.0826 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in einer Mischung aus 5 ml Was-
ser und 5 ml Acetonitril gelöst. Die Lösung wurde zwei Stunden gerührt. Das Lösungsmittel
wurde im Unterdruck eingeengt. Es entstand ein gelbfarbener Niederschlag. Der Feststoff
wurde von der Lösung getrennt und getrocknet.
Ausbeute: 8.5 mg (0.0156 mmol) bezogen auf das eingesetzte Hydrogentetrachloroau-
rat(III)hydrat: 18.9 %
1H NMR (200 MHz, DMSO-d6) δ 9.73 (d, J = 6.2 Hz, 1H), 8.42 – 8.36 (m, 2H), 8.12 (d, J =
8.1 Hz, 1H), 7.97 (dd, J = 7.6, 1.6 Hz, 1H), 7.77 (td, J = 5.9, 3.1 Hz, 1H), 7.46 (t, J = 7.5 Hz,
1H), 7.33 (td, J = 8.0, 1.6 Hz, 1H).

Experimentalteil
Seite | 113
Synthese von 2-Phenylpyridin-1,1´-golddiaceatat 20
100 mg (0.23 mmol) 2-Phenylpyridin-1,1´-gold(III)dichlorid 2 wurden in 500 ml Aceton ge-
löst. Durch die farblose Lösung wurden 10 min Stickstoff geblasen. Es wurden 86.7 mg
(0.5077 mmol) Silberacetat zugegeben. Die Suspension wurde filtriert. Das Filtrat wurde ein-
geengt, bis ein graufarbener Niederschlag entstanden war. Dieser Niederschlag wurde von der
Lösung getrennt und getrocknet.
Ausbeute: 21.1 mg (0.045 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-1,1´-
gold(III)dichlorid 2: 19.6 %
1H NMR (250 MHz, DMSO-d6) δ 8.55 (d, J = 5.9 Hz, 1H), 8.41 (m, 2H), 7.93 (d, J = 6.7 Hz,
1H), 7.72 (td, J = 7.8, 3.3 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.36 (t, J = 7.3 Hz, 1H), 7.12 (d, J
= 7.5 Hz, 1H), 2.12 (s, 3H), 2.99 (s, 3H).

Experimentalteil
Seite | 114
Synthese von 2-Phenylpyridin-1-gold(III)tribromid 650
100 mg (0.1813 mmol) Kaliumtetrabromoaurat(III) wurden in 5 ml Wasser gelöst. 31.1 µl
(0.2175 mmol)) 2-Phenylpyridin wurden in 5 ml Ethanol gelöst und langsam in die erste
dunkelrotfarbene Lösung getropft. Es entstand ein gelbfarbener Schaum. Die Lösung wurde
für drei Stunden gerührt. Es hatte sich ein rotfarbener Niederschlag gebildet, dieser wurde
vom Überstand getrennt und getrocknet.
Ausbeute: 76.4 mg (0.1296 mmol) bezogen auf das eingesetzte Kaliumtetrabromoaurat(III):
71.6 %
1H NMR (200 MHz, DMSO-d6) δ 9.42 (d, J = 6.1 Hz, 1H), 8.62 (t, J = 8.1, 1H),
8.25 (dt, J = 7.4, 1.2 Hz 1H), 8.11 (t, J = 8.2 Hz, 1H), 8.01 – 7.90 (m, 4H), 7.85 (t, J = 7.2 Hz,
1H).

Experimentalteil
Seite | 115
Synthese von 2-Phenylpyridin-1,1´gold(II)dibromid 651
76.4 mg (0.1296 mmol) 2-Phenylpyridin-1-gold(III)tribromid 650 wurden in einer Mischung
aus 15 ml Wasser und 15 ml Acetonitril suspendiert. Die Suspension wurde sechs Stunden
refluxiert, wobei eine rotfarbene Lösung entstand. Langsam wurde die Lösung auf Raumtem-
peratur abgekühlt. Es entstand ein gelbfarbener Niederschlag. Der Feststoff wurde von der
Lösung getrennt und getrocknet.
Ausbeute: 17.26 mg (0.0339 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-1-
gold(III)tribromid 650: 26 %
1H NMR (200 MHz, DMSO-d6) δ 9.73 (d, J = 6.2 Hz, 1H), 8.42 – 8.36 (m, 2H), 8.12 (d, J =
8.1 Hz, 1H), 7.97 (dd, J = 7.6, 1.6 Hz, 1H), 7.77 (td, J = 5.9, 3.1 Hz, 1H), 7.46 (t, J = 7.5 Hz,
1H), 7.33 (td, J = 8.0, 1.6 Hz, 1H). 13
C NMR (100 MHz, DMSO-d6) δ 185.1, 155.7, 145.2, 132.4, 127.9, 127.5, 126.1, 125.9,
125.6, 124.9, 124.3

Experimentalteil
Seite | 116
Synthese von Tetraethylammoniumtetrachloroaurat(III) 172
0.2 g (0.51 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 30 ml Ethanol gelöst. Zur
gelbfarbenen Lösung wurden 84.28 mg (0.51 mmol) Tetraethylammoniumchlorid zugegeben.
Es fiel ein gelbfarbener Feststoff aus, welcher von der Lösung getrennt wurde und aus Etha-
nol umkristallisiert wurde. Der Feststoff wurde getrocknet.
Ausbeute: 147 mg (0.31 mmol) bezogen auf das eingesetzte Hydrogentetrachloroau-
rat(III)hydrat: 62 %

Experimentalteil
Seite | 117
Synthese von Tetraethylammoniumtetrabromoaurat(III) 172
Bei einer Temperatur von 55 °C wurden zu einer Lösung von 0.2 g (0.51 mmol)
Hydrogentetrachloroaurat(III)hydrat in 30 ml Ethanol, 1.0671 g (0.51 mmol)
Tetraethylammoniumbromid in 20 ml Ethanol gegeben. Die vorher gelbfarbene Lösung färbte
sich bei der Zugabe schlagartig dunkel rotfarben. Die Suspension wurde bei 55 °C für zwei
Stunden gerührt. Der violettfarbene Feststoff wurde von der Lösung getrennt, in Ethanol
umkristallisiert und getrocknet.
Ausbeute: 253 mg (0.3936 mmol) bezogen auf das eingesetzte Hydrogentetrachloroau-
rat(III)hydrat: 77.1 %

Experimentalteil
Seite | 118
Synthese von Tetraethylammoniumphenyltrichloroaurat(III) 652 173
In einen 50 ml Kolben werden 144.61 (0.3097 mmol) Tetraethylammoniumtetrachloroau-
rat(III) in 20 ml Ethanol suspendiert. Die leicht gelbfarbene Suspension wurde auf 50 °C er-
hitzt. In einem zweiten 50 ml Kolben wurde eine Suspension aus 44.83 mg (0.3097 mmol)
Phenylhydraziniumchlorid in 20 ml Ethanol angesetzt. Diese Mischung wurde in dem ersten
Kolben mit beigegeben. Die so entstandene Suspension wurde für drei Stunden gerührt und
heiß gefiltert. Die Lösung wurde für 12 Stunden bei -20 °C gelagert. Es hatte sich ein
gelbfarbener Feststoff gebildet, welcher von der Lösung getrennt und getrocknet wurde.
Ausbeute: 8.72 mg (0.0171 mmol) bezogen auf das eingesetzte Tetraethylammoniumtetra-
chloroaurat(III): 5.5 %
1H NMR (200 MHz, DMSO-d6) δ 7.62 – 7.49 (m, 5H), 3.27 (q, J = 8.4 Hz, 8H), 1.25 (t, J =
6.8 Hz, 12H).

Experimentalteil
Seite | 119
Synthese von Tetraethylammoniumphenyltribromoaurat(III) 653 173
120 mg (0.1866 mmol) Tetraethylammoniumtetrabromoaurat(III) wurden in 5 ml Ethanol
suspendiert. Die Suspension wurde auf 45 °C erhitzt. Es wuden zu dieser Mischung 30.01 mg
(0.2084 mmol) Phenylhydraziniumchlorid in 5 ml Ethanol gegeben. Die rotfarbene Reakti-
onssuspension wurde gelbfarben während den fünf Stunden bei der sie bei 45 °C erhitzt wur-
de. Die Mischung wurde warm gefiltert und für drei Tage bei – 18 °C gelagert. Es war ein
gelbfarbener Niederschlag entstanden. Dieser wurde von der Lösung getrennt und getrocknet.
Ausbeute: 12.56 mg (0.0196 mmol) bezogen auf das eingesetzte Tetraethylammoniumtetra-
bromoaurat(III): 10.5 %
1H NMR (200 MHz, DMSO-d6) δ 7.94 – 7.78 (m, 5H), 3.26 (q, J = 9.2 Hz, 8H), 1.23 (t, J =
7.3 Hz, 12H).

Experimentalteil
Seite | 120
Synthese von Pyridin-1-gold(III)trichlorid 654 174, 175
10.4 mg (0.0306 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 0.5 ml Wasser gelöst.
Zur gelbfarbenen Lösung wurden 0.5 ml Acetonitril und 100 µl Pyridin gegeben. Die Lösung
wurde für acht Stunden gerührt. Teile des Lösungsmittels wurden im Unterdruck entfernt. Die
Lösung wurde für drei Tage bei -18 °C gelagert. Der entstanden Niederschlag wurde von der
Lösung getrennt und vorsichtig mit Wasser und Acetonitril gewaschen.
Ausbeute: 3.2 mg (0.0084 mmol) bezogen auf das eingesetzte Hydrogentetrachloro-
aurat(III)hydrat: 27.4 %
1H NMR (200 MHz, DMSO-d6) δ 9.25 (d, J = 5.2 Hz, 2H), 8.61 – 8.48 (m, 3H)

Experimentalteil
Seite | 121
Synthese von Pyridin-1-gold(III)tribromid 655
7.6 mg (0.0138 mmol) Kaliumtetrabromoaurat(III) wurden in 0.5 ml Wasser gelöst. Zur
rotfarbenen Lösung wurden 100 µl Pyridin und 0.5 ml Acetonitril gegeben. Die Lösung wur-
de vier Stunden gerührt. Die Lösung wurde für sieben Tage bei -18 °C gelagert. Der Nieder-
schlag wurde von der Lösung getrennt, mit Wasser und Acetonitril mit Bedacht gewaschen
und anschließend getrocknet.
Ausbeute: 2.7 mg (0.0053 mmol) bezogen auf das eingesetzte Kaliumtetrabromoaurat(III):
38.4 %
1H NMR (200 MHz, DMSO-d6) δ 9.34 (d, J = 6.2 Hz, 2H), 8.74 – 8.56 (m, 3H)

Experimentalteil
Seite | 122
Synthese von Bipyridin-1,1´gold(III)dichlorid tetrachloroaurat(III) 656 112, 176
32 mg (0.2042 mmol) Bipyridin wurden in 3 ml Acetonitril gelöst und 53.4 mg
(0.1571 mmol) Hydrogentetrachloroaurat(III)hydrat wurden in 3 ml Wasser gelöst. Die zweite
Lösung wurde der ersten Lösung zugegeben. Es bildete sich ein gelbfarbener Niederschlag.
Dieser wurde von der Lösung getrennt. Der Feststoff wurde mit Wasser und Acetonitril gewa-
schen und danach getrocknet.
Ausbeute: 94,4 mg (0.1241 mmol) bezogen auf das eingesetzte Hydrogentetrachloroaurat-
(III)hydrat: 79.1 %
1H NMR (200 MHz, DMSO-d6) δ 9.42 (d, J = 5.4 Hz, 1H), 8.92 (t, J = 4.8, 1H),
8.74 (d, J = 7.9 Hz, 1H), 8.13 (dd, J = 7.6, 5.3 Hz, 1H)
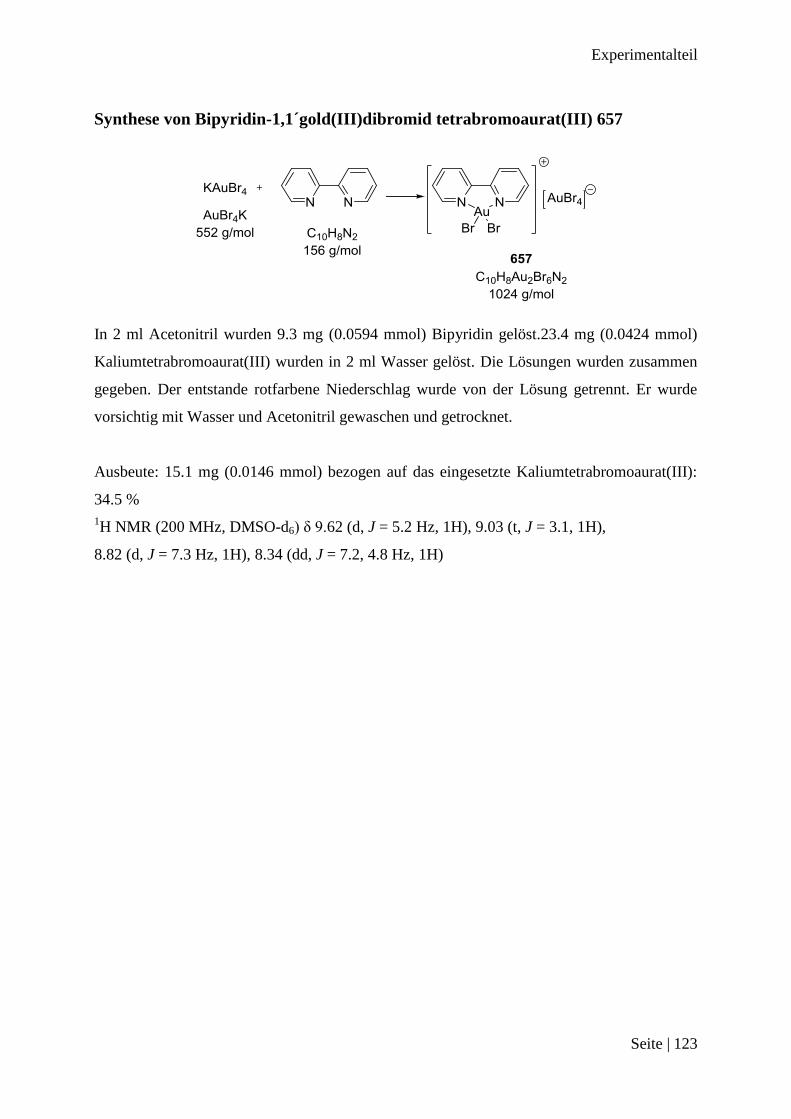
Experimentalteil
Seite | 123
Synthese von Bipyridin-1,1´gold(III)dibromid tetrabromoaurat(III) 657
In 2 ml Acetonitril wurden 9.3 mg (0.0594 mmol) Bipyridin gelöst.23.4 mg (0.0424 mmol)
Kaliumtetrabromoaurat(III) wurden in 2 ml Wasser gelöst. Die Lösungen wurden zusammen
gegeben. Der entstande rotfarbene Niederschlag wurde von der Lösung getrennt. Er wurde
vorsichtig mit Wasser und Acetonitril gewaschen und getrocknet.
Ausbeute: 15.1 mg (0.0146 mmol) bezogen auf das eingesetzte Kaliumtetrabromoaurat(III):
34.5 %
1H NMR (200 MHz, DMSO-d6) δ 9.62 (d, J = 5.2 Hz, 1H), 9.03 (t, J = 3.1, 1H),
8.82 (d, J = 7.3 Hz, 1H), 8.34 (dd, J = 7.2, 4.8 Hz, 1H)

Experimentalteil
Seite | 124
Synthese von 2-Phenylpyridin-1,1´gold(III)ethanditihol 658 171, 177
15.6 mg (0.0371 mmol) 2-Phenylpyridin-1,1´-gold(III)dichlorid (2) wurden in 3 ml einer Mi-
schung aus 50 Vol.-% Wasser und 50 Vol.-% Acetonitril suspendiert. Es wurden 100 µl 1,2-
Ethandithiol zugeben. Die vorher leicht gelbfarbene Suspension wurde intensiver gelbfarben.
Die Mischung wurde sechs Stunden gerührt. Der Feststoff wurde von der Lösung getrennt
und mit Wasser und Acetonitril gewaschen, bis der typische Geruch von 1,2-Ethandithiol
nicht mehr wahr zu nehmen war. Der Feststoff wurde getrocknet.
Ausbeute: 8.4 mg (0.019 mmol) bezogen auf das eingesetzte 2-Phenylpyridin-1,1´-
gold(III)dichlorid 2: 51.1 %
1H NMR (200 MHz, DMSO) δ 9.52 (d, J = 6.4 Hz, 1H), 8.44 – 8.35 (m, 2H), 7.95 (d, J = 7.6
Hz, 1H), 7.82 – 7.69 (m, 2H), 7.43 (t, J = 6.2 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H) 3.01 – 2.94 (m,
4H).
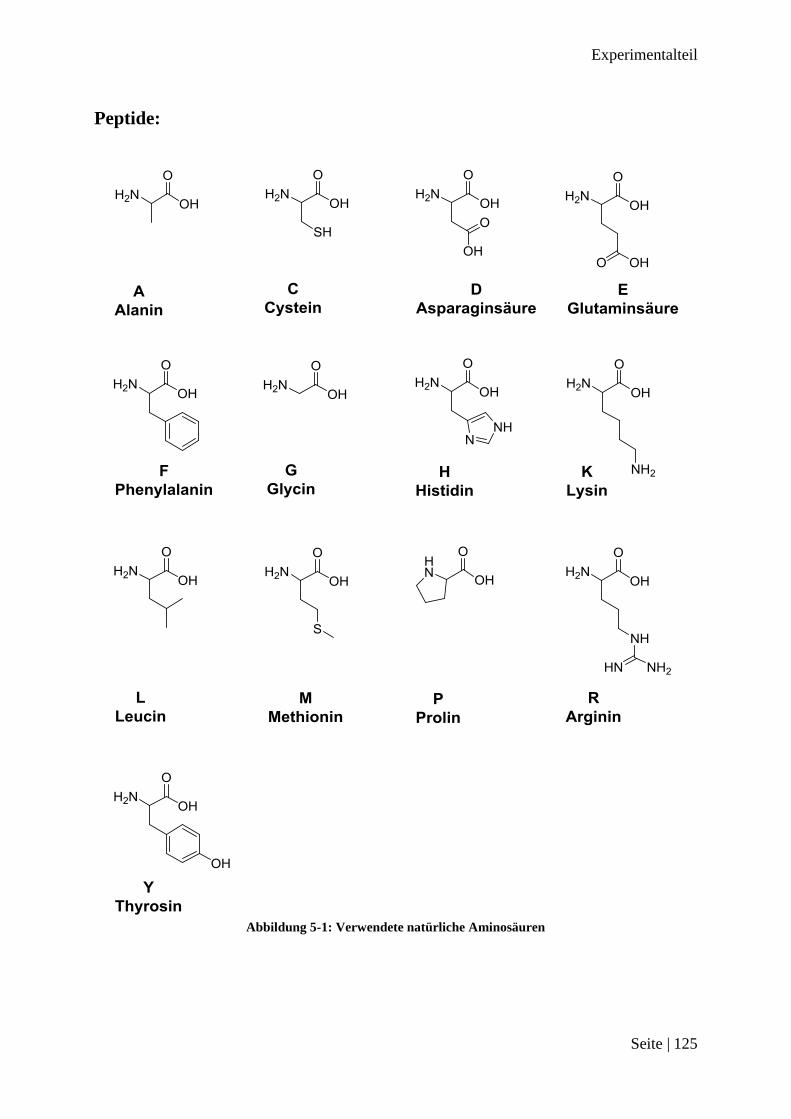
Experimentalteil
Seite | 125
Peptide:
Abbildung 5-1: Verwendete natürliche Aminosäuren
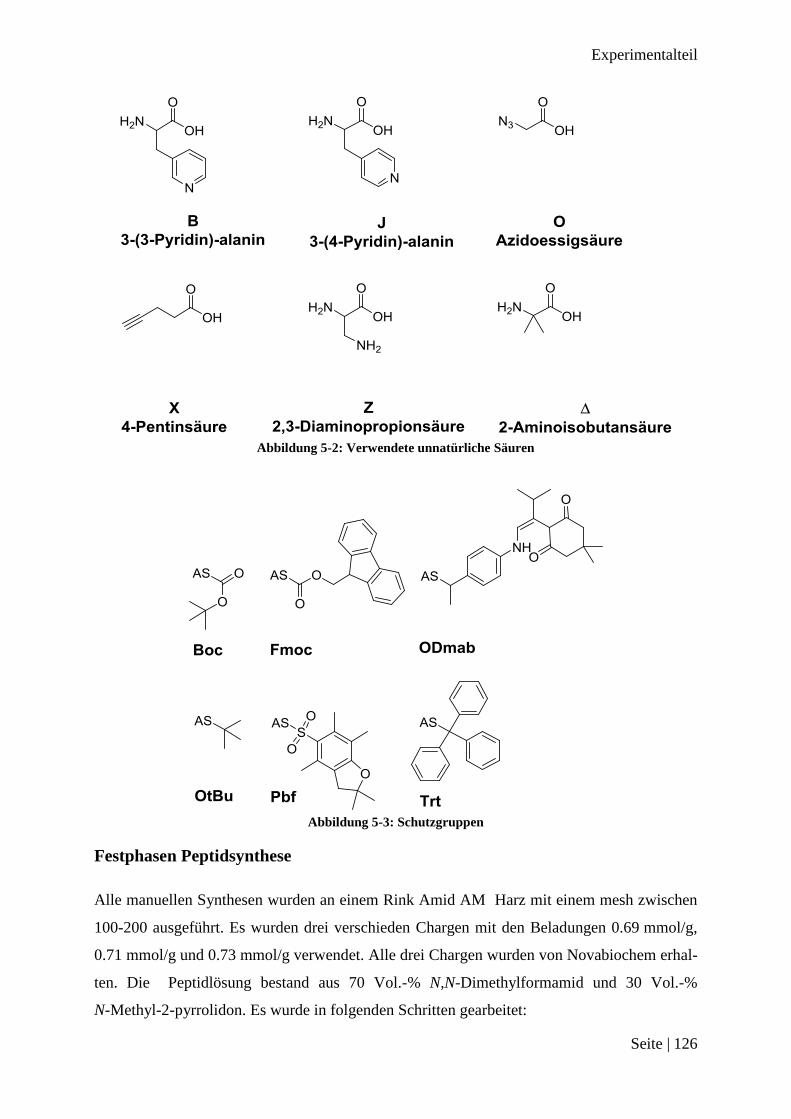
Experimentalteil
Seite | 126
Abbildung 5-2: Verwendete unnatürliche Säuren
Abbildung 5-3: Schutzgruppen
Festphasen Peptidsynthese
Alle manuellen Synthesen wurden an einem Rink Amid AM Harz mit einem mesh zwischen
100-200 ausgeführt. Es wurden drei verschieden Chargen mit den Beladungen 0.69 mmol/g,
0.71 mmol/g und 0.73 mmol/g verwendet. Alle drei Chargen wurden von Novabiochem erhal-
ten. Die Peptidlösung bestand aus 70 Vol.-% N,N-Dimethylformamid und 30 Vol.-%
N-Methyl-2-pyrrolidon. Es wurde in folgenden Schritten gearbeitet:

Experimentalteil
Seite | 127
1. Das Harz wurde in der Peptidlösung für eine halbe Stunde quellen gelassen.
2. Mit einer Lösung aus 20 Vol.-% Piperidin in der Peptidlösung wurde das Harz für
10 min geschüttelt.
3. In die Spritze mit dem Harz wurde dreimal zum Waschen die Peptidlösung gezogen.
4. Die Spritze wurde erneut für 10 min mit der Lösung aus 20 Vol.-% Piperidin in der
Peptidlösung gewaschen.
5. Fünfmal wurde das Harz mit der Peptidlösung gewaschen.
6. In ein verschließbares Gefäß wurden vier Äquivalente der zu koppelnde Aminosäure,
HOBT und TBTU gegeben. Es wurde wenig der Peptidlösung zugegeben, niemals so
viel, dass das Volumen der verwendeten Spritze überschritten wurde. Sobald sich eine
Lösung gebildet hatte, wurden vier Äquivalente DIPEA zugegeben und der Inhalt so-
fort in die Spritze gezogen. Die Spritze wurde für 40 min bis 60 min geschüttelt.
7. Fünfmal wurde das Harz mit der Peptidlösung gewaschen.
8. Die Schritte 2. bis 8. wurden wiederholt, bis die gewünschte Kettenlänge erreicht war
und die gewünschte Sequenz synthetisiert war.
9. Das Harz wurde fünfmal mit DCM gewaschen. Das Abspalten erfolgte für mindestens
sechs Stunden in einer Lösung aus 92 Vol.-% TFA, 2.5 Vol.-% Wasser und
5.5 Vol.-% TIS, bei allen Peptide welche mindestens ein Methionin oder ein Cystein
enthalten, wurde eine Mischung aus 90 Vol.-% TFA, 2.5 Vol.-% EDT, 2.5 Vol.-%
Wasser und 5 Vol.-% TIS verwendet. Wenn sich das Harz und die Abspaltlösung bei
der Zugabe gelbfärbten, dann wurde wenig TIS zugegeben. Die Lösung wurde in ein
Plastikgefäß mit Deckel gegeben. In die Spritze wurde TFA gezogen und ebenfalls in
das Gefäß gegeben. In das Gefäß wurde Diethylether mit einer Temperatur von
- 70 °C gegeben. Es fiel ein farbloses Pulver aus, welches von der Lösung, in einer
Zentrifuge Rotina 46 von Hettich mit getrennt wurde. Abschließend wurde die Lösung
dekantiert und der feste Rückstand wurde zweimal mit DET bei - 70 °C gewaschen
und mit Zentrifugieren und Dekantieren von dem Lösungsmittel getrennt. Das Pulver
wurde in einer Mischung aus Wasser und ACN gelöst und in flüssigem Stickstoff ein-
gefroren. Die eingefrorene Lösung wurde lyophilisiert. Es wurde ein farbloses schau-
martiges Pulver erhalten.
Bei Kettenlängen über fünf Aminosäuren wurden doppelte Kopplungen durchgeführt. Das
bedeutet nach Schritt 7. wurde Schritt 6. mit der gleichen Aminosäure wiederholt. Es folgt
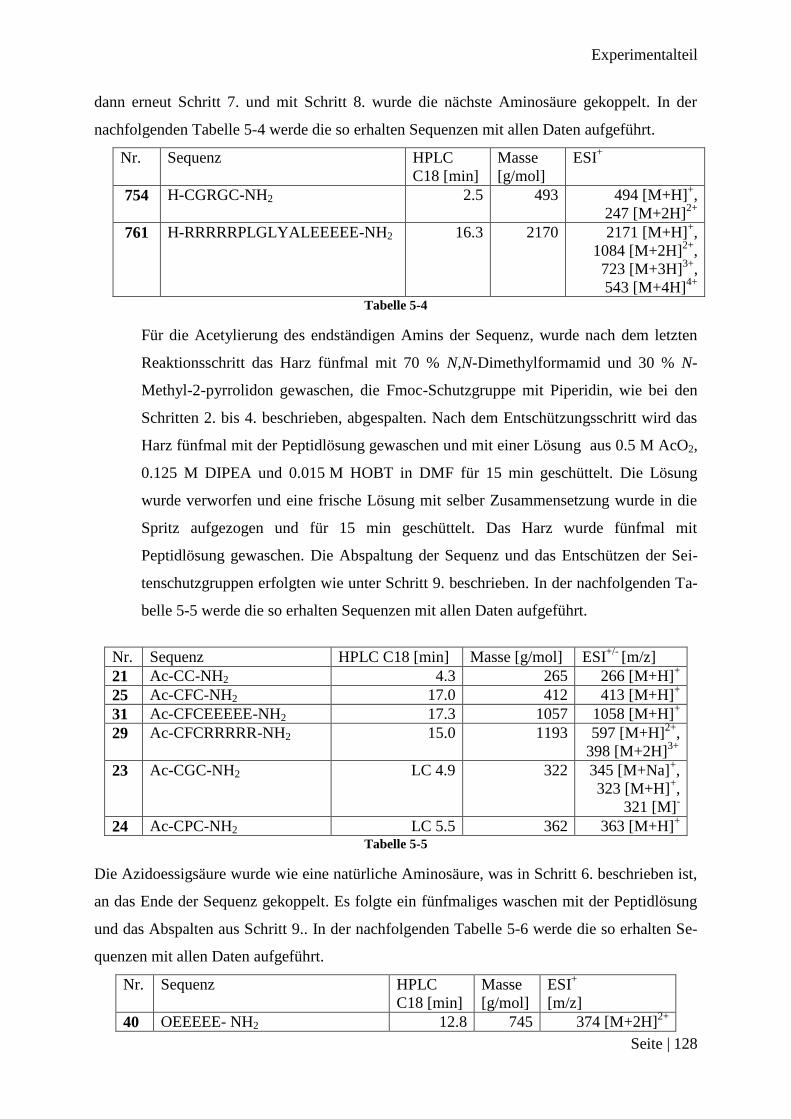
Experimentalteil
Seite | 128
dann erneut Schritt 7. und mit Schritt 8. wurde die nächste Aminosäure gekoppelt. In der
nachfolgenden Tabelle 5-4 werde die so erhalten Sequenzen mit allen Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
754 H-CGRGC-NH2 2.5 493 494 [M+H]+,
247 [M+2H]2+
761 H-RRRRRPLGLYALEEEEE-NH2 16.3 2170 2171 [M+H]+,
1084 [M+2H]2+
,
723 [M+3H]3+
,
543 [M+4H]4+
Tabelle 5-4
Für die Acetylierung des endständigen Amins der Sequenz, wurde nach dem letzten
Reaktionsschritt das Harz fünfmal mit 70 % N,N-Dimethylformamid und 30 % N-
Methyl-2-pyrrolidon gewaschen, die Fmoc-Schutzgruppe mit Piperidin, wie bei den
Schritten 2. bis 4. beschrieben, abgespalten. Nach dem Entschützungsschritt wird das
Harz fünfmal mit der Peptidlösung gewaschen und mit einer Lösung aus 0.5 M AcO2,
0.125 M DIPEA und 0.015 M HOBT in DMF für 15 min geschüttelt. Die Lösung
wurde verworfen und eine frische Lösung mit selber Zusammensetzung wurde in die
Spritz aufgezogen und für 15 min geschüttelt. Das Harz wurde fünfmal mit
Peptidlösung gewaschen. Die Abspaltung der Sequenz und das Entschützen der Sei-
tenschutzgruppen erfolgten wie unter Schritt 9. beschrieben. In der nachfolgenden Ta-
belle 5-5 werde die so erhalten Sequenzen mit allen Daten aufgeführt.
Nr. Sequenz HPLC C18 [min] Masse [g/mol] ESI+/-
[m/z]
21 Ac-CC-NH2 4.3 265 266 [M+H]+
25 Ac-CFC-NH2 17.0 412 413 [M+H]+
31 Ac-CFCEEEEE-NH2 17.3 1057 1058 [M+H]+
29 Ac-CFCRRRRR-NH2 15.0 1193 597 [M+H]2+
,
398 [M+2H]3+
23 Ac-CGC-NH2 LC 4.9 322 345 [M+Na]+,
323 [M+H]+,
321 [M]-
24 Ac-CPC-NH2 LC 5.5 362 363 [M+H]+
Tabelle 5-5
Die Azidoessigsäure wurde wie eine natürliche Aminosäure, was in Schritt 6. beschrieben ist,
an das Ende der Sequenz gekoppelt. Es folgte ein fünfmaliges waschen mit der Peptidlösung
und das Abspalten aus Schritt 9.. In der nachfolgenden Tabelle 5-6 werde die so erhalten Se-
quenzen mit allen Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
40 OEEEEE- NH2 12.8 745 374 [M+2H]2+
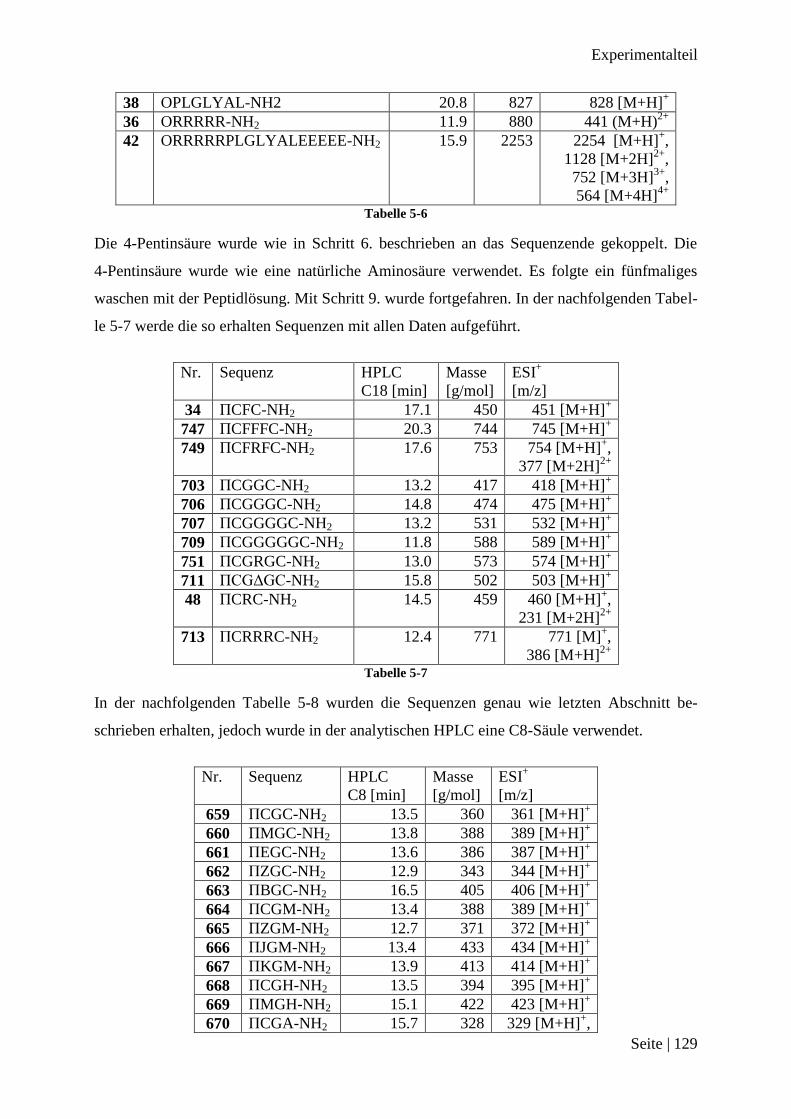
Experimentalteil
Seite | 129
38 OPLGLYAL-NH2 20.8 827 828 [M+H]+
36 ORRRRR-NH2 11.9 880 441 (M+H)2+
42 ORRRRRPLGLYALEEEEE-NH2 15.9 2253 2254 [M+H]+,
1128 [M+2H]2+
,
752 [M+3H]3+
,
564 [M+4H]4+
Tabelle 5-6
Die 4-Pentinsäure wurde wie in Schritt 6. beschrieben an das Sequenzende gekoppelt. Die
4-Pentinsäure wurde wie eine natürliche Aminosäure verwendet. Es folgte ein fünfmaliges
waschen mit der Peptidlösung. Mit Schritt 9. wurde fortgefahren. In der nachfolgenden Tabel-
le 5-7 werde die so erhalten Sequenzen mit allen Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
34 ΠCFC-NH2 17.1 450 451 [M+H]+
747 ΠCFFFC-NH2 20.3 744 745 [M+H]+
749 ΠCFRFC-NH2 17.6 753 754 [M+H]+,
377 [M+2H]2+
703 ΠCGGC-NH2 13.2 417 418 [M+H]+
706 ΠCGGGC-NH2 14.8 474 475 [M+H]+
707 ΠCGGGGC-NH2 13.2 531 532 [M+H]+
709 ΠCGGGGGC-NH2 11.8 588 589 [M+H]+
751 ΠCGRGC-NH2 13.0 573 574 [M+H]+
711 ΠCGΔGC-NH2 15.8 502 503 [M+H]+
48 ΠCRC-NH2 14.5 459 460 [M+H]+,
231 [M+2H]2+
713 ΠCRRRC-NH2 12.4 771 771 [M]+,
386 [M+H]2+
Tabelle 5-7
In der nachfolgenden Tabelle 5-8 wurden die Sequenzen genau wie letzten Abschnitt be-
schrieben erhalten, jedoch wurde in der analytischen HPLC eine C8-Säule verwendet.
Nr. Sequenz HPLC
C8 [min]
Masse
[g/mol]
ESI+
[m/z]
659 ΠCGC-NH2 13.5 360 361 [M+H]+
660 ΠMGC-NH2 13.8 388 389 [M+H]+
661 ΠEGC-NH2 13.6 386 387 [M+H]+
662 ΠZGC-NH2 12.9 343 344 [M+H]+
663 ΠBGC-NH2 16.5 405 406 [M+H]+
664 ΠCGM-NH2 13.4 388 389 [M+H]+
665 ΠZGM-NH2 12.7 371 372 [M+H]+
666 ΠJGM-NH2 13.4 433 434 [M+H]+
667 ΠKGM-NH2 13.9 413 414 [M+H]+
668 ΠCGH-NH2 13.5 394 395 [M+H]+
669 ΠMGH-NH2 15.1 422 423 [M+H]+
670 ΠCGA-NH2 15.7 328 329 [M+H]+,
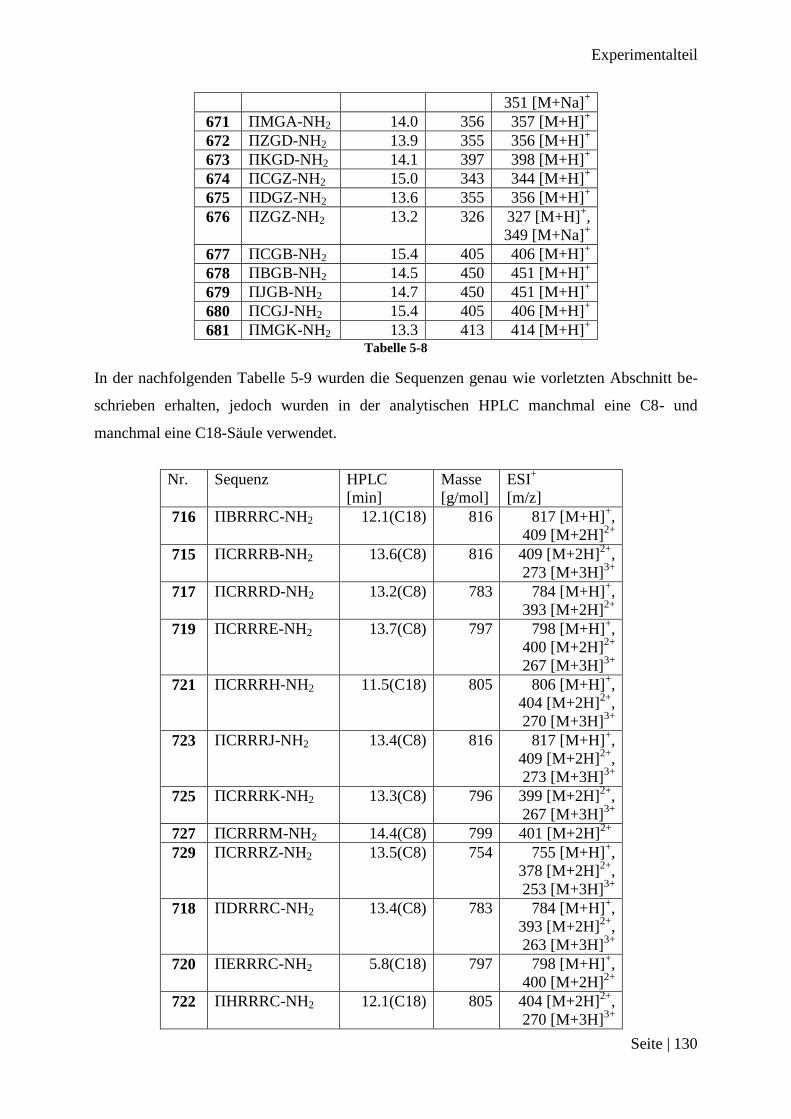
Experimentalteil
Seite | 130
351 [M+Na]+
671 ΠMGA-NH2 14.0 356 357 [M+H]+
672 ΠZGD-NH2 13.9 355 356 [M+H]+
673 ΠKGD-NH2 14.1 397 398 [M+H]+
674 ΠCGZ-NH2 15.0 343 344 [M+H]+
675 ΠDGZ-NH2 13.6 355 356 [M+H]+
676 ΠZGZ-NH2 13.2 326 327 [M+H]+,
349 [M+Na]+
677 ΠCGB-NH2 15.4 405 406 [M+H]+
678 ΠBGB-NH2 14.5 450 451 [M+H]+
679 ΠJGB-NH2 14.7 450 451 [M+H]+
680 ΠCGJ-NH2 15.4 405 406 [M+H]+
681 ΠMGK-NH2 13.3 413 414 [M+H]+
Tabelle 5-8
In der nachfolgenden Tabelle 5-9 wurden die Sequenzen genau wie vorletzten Abschnitt be-
schrieben erhalten, jedoch wurden in der analytischen HPLC manchmal eine C8- und
manchmal eine C18-Säule verwendet.
Nr. Sequenz HPLC
[min]
Masse
[g/mol]
ESI+
[m/z]
716 ΠBRRRC-NH2 12.1(C18) 816 817 [M+H]+,
409 [M+2H]2+
715 ΠCRRRB-NH2 13.6(C8) 816 409 [M+2H]2+
,
273 [M+3H]3+
717 ΠCRRRD-NH2 13.2(C8) 783 784 [M+H]+,
393 [M+2H]2+
719 ΠCRRRE-NH2 13.7(C8) 797 798 [M+H]+,
400 [M+2H]2+
267 [M+3H]3+
721 ΠCRRRH-NH2 11.5(C18) 805 806 [M+H]+,
404 [M+2H]2+
,
270 [M+3H]3+
723 ΠCRRRJ-NH2 13.4(C8) 816 817 [M+H]+,
409 [M+2H]2+
,
273 [M+3H]3+
725 ΠCRRRK-NH2 13.3(C8) 796 399 [M+2H]2+
,
267 [M+3H]3+
727 ΠCRRRM-NH2 14.4(C8) 799 401 [M+2H]2+
729 ΠCRRRZ-NH2 13.5(C8) 754 755 [M+H]+,
378 [M+2H]2+
,
253 [M+3H]3+
718 ΠDRRRC-NH2 13.4(C8) 783 784 [M+H]+,
393 [M+2H]2+
,
263 [M+3H]3+
720 ΠERRRC-NH2 5.8(C18) 797 798 [M+H]+,
400 [M+2H]2+
722 ΠHRRRC-NH2 12.1(C18) 805 404 [M+2H]2+
,
270 [M+3H]3+

Experimentalteil
Seite | 131
724 ΠJRRRC-NH2 11.1(C18) 816 817 [M+H]+,
408 [M+2H]2+
,
273 [M+3H]3+
726 ΠKRRRC-NH2 6.3(C18) 796 797 [M+H]+,
399 [M+2H]2+
,
267 [M+3H]3+
728 ΠMRRRC-NH2 12.9(C18) 799 800 [M+H]+,
401 [M+2H]2+
730 ΠZRRRC-NH2 2.7(C18) 754 755 [M+H]+,
378 [M+2H]2+
,
253 [M+3H]3+
Tabelle 5-9
Die automatische Peptidsynthese erfolgte an einem CEM Liberty Mikrowellen Peptid Synthe-
sizer. An dem Gerät können zwei Ansatzgrößen synthetisiert werden: 0.1 mmol und
0.25 mmol. Bei allen Synthesen wurde ein Rink Amid AM Harz mit einem mesh zwischen
100-200 verwendet. Es wurden drei verschieden Chargen mit den Beladungen 0.69 mmol/g,
0.71 mmol/g und 0.73 mmol/g verwendet. Alle drei Chargen werden von Novabiochem erhal-
ten. Alle benötigten Massen und Volumen wurden von der Software PepDriver berechnet.
Das Harz wurde für die Synthese in ein Gefäß eingewogen und in N,N-Dimethylformamid für
eine halbe Stunde quellen gelassen. Die berechneten Massen an Aminosäuren wurden in spe-
zielle Gefäße eingewogen und in den vorgegebenen Mengen einer Lösung aus 70 Vol.-%
N,N-Dimethylformamid und 30 Vol.-% N-Methyl-2-pyrrolidon gelöst. Sobald die Aminosäu-
ren gelöst waren, wurden sie an die vorgesehen Halterungen geschraubt. In eine Schraubde-
ckelflasche aus Glass wurde die Lösung zur Fmoc-Entschützung aus 20 Vol.-% Piperidin in
DMF gegeben und an das Gerät angeschlossen. Die vorgegebene Menge an Aktivatorlösung
aus HOBT und TBTU in DMF wurden mittels einer Schraubdeckelflasche an das Gerät ange-
schlossen. Dasselbe wurde mit der Aktivatorbase DIPEA in NMP gemacht. Nachdem alle
Lösungen angeschlossen waren, konnte das Gerät gestartet werden. Das fertige Peptid am
Harz befand sich in demselben Gefäß an dem auch schon das Harz selber angeschlossen wor-
den war. Das mit Peptid beladene Harz wurde in 5 ml Polypropylenspritzen mit einem Filter
bei 0.1 mmol Ansätzen und bei 0.25 mmol Ansätzen in eine 10 ml Spritze gegeben. Es gab
vier Möglichkeiten weiter fortzufahren.
1) Die Fmoc-Entschütze Sequenz wurde zweimal mit einer Lösung aus 0.5 M AcO2,
0.125 M DIPEA und 0.015 HOBT in DMF für 15 min behandelt. Zwischen dem ers-
ten und dem zweiten Anwenden wurde das Harz fünfmal mit der Peptidlösung gewa-
schen. In der nachfolgenden Tabelle 5-10 werde die so erhalten Sequenzen mit allen
Daten aufgeführt.
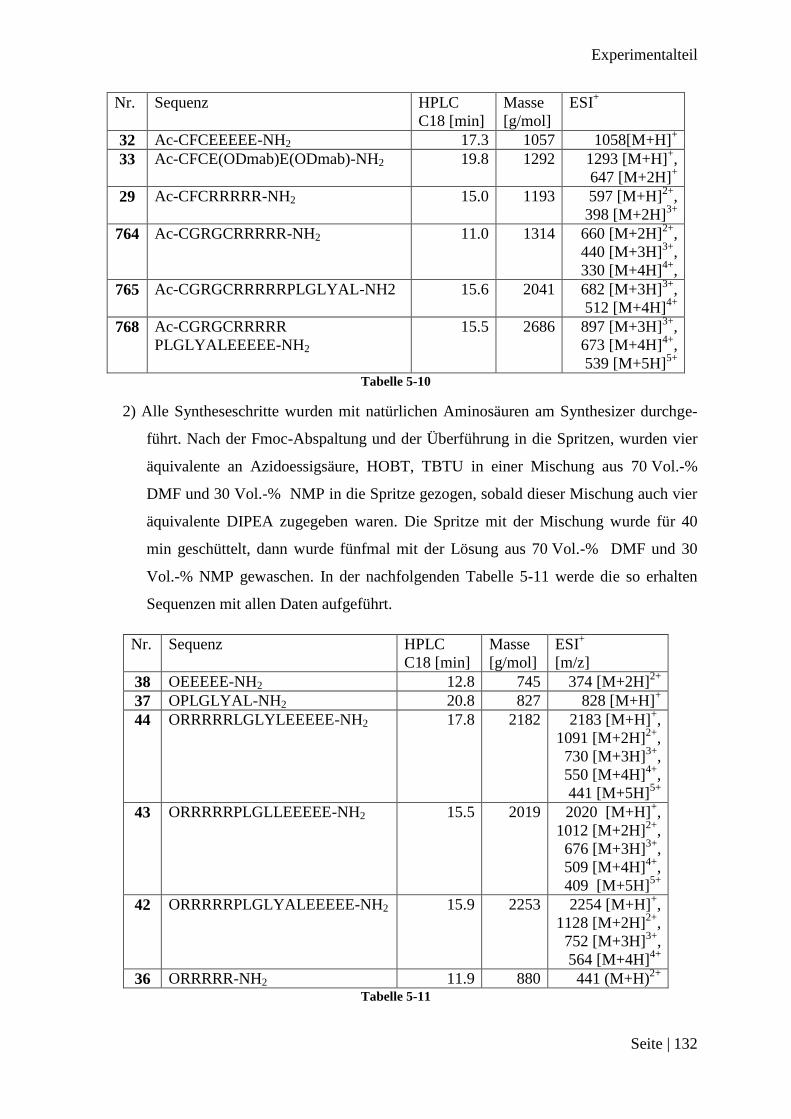
Experimentalteil
Seite | 132
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
32 Ac-CFCEEEEE-NH2 17.3 1057 1058[M+H]+
33 Ac-CFCE(ODmab)E(ODmab)-NH2 19.8 1292 1293 [M+H]+,
647 [M+2H]+
29 Ac-CFCRRRRR-NH2 15.0 1193 597 [M+H]2+
,
398 [M+2H]3+
764 Ac-CGRGCRRRRR-NH2 11.0 1314 660 [M+2H]2+
,
440 [M+3H]3+
,
330 [M+4H]4+
,
765 Ac-CGRGCRRRRRPLGLYAL-NH2 15.6 2041 682 [M+3H]3+
,
512 [M+4H]4+
768 Ac-CGRGCRRRRR
PLGLYALEEEEE-NH2
15.5 2686 897 [M+3H]3+
,
673 [M+4H]4+
,
539 [M+5H]5+
Tabelle 5-10
2) Alle Syntheseschritte wurden mit natürlichen Aminosäuren am Synthesizer durchge-
führt. Nach der Fmoc-Abspaltung und der Überführung in die Spritzen, wurden vier
äquivalente an Azidoessigsäure, HOBT, TBTU in einer Mischung aus 70 Vol.-%
DMF und 30 Vol.-% NMP in die Spritze gezogen, sobald dieser Mischung auch vier
äquivalente DIPEA zugegeben waren. Die Spritze mit der Mischung wurde für 40
min geschüttelt, dann wurde fünfmal mit der Lösung aus 70 Vol.-% DMF und 30
Vol.-% NMP gewaschen. In der nachfolgenden Tabelle 5-11 werde die so erhalten
Sequenzen mit allen Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
38 OEEEEE-NH2 12.8 745 374 [M+2H]2+
37 OPLGLYAL-NH2 20.8 827 828 [M+H]+
44 ORRRRRLGLYLEEEEE-NH2 17.8 2182 2183 [M+H]+,
1091 [M+2H]2+
,
730 [M+3H]3+
,
550 [M+4H]4+
,
441 [M+5H]5+
43 ORRRRRPLGLLEEEEE-NH2 15.5 2019 2020 [M+H]+,
1012 [M+2H]2+
,
676 [M+3H]3+
,
509 [M+4H]4+
,
409 [M+5H]5+
42 ORRRRRPLGLYALEEEEE-NH2 15.9 2253 2254 [M+H]+,
1128 [M+2H]2+
,
752 [M+3H]3+
,
564 [M+4H]4+
36 ORRRRR-NH2 11.9 880 441 (M+H)2+
Tabelle 5-11
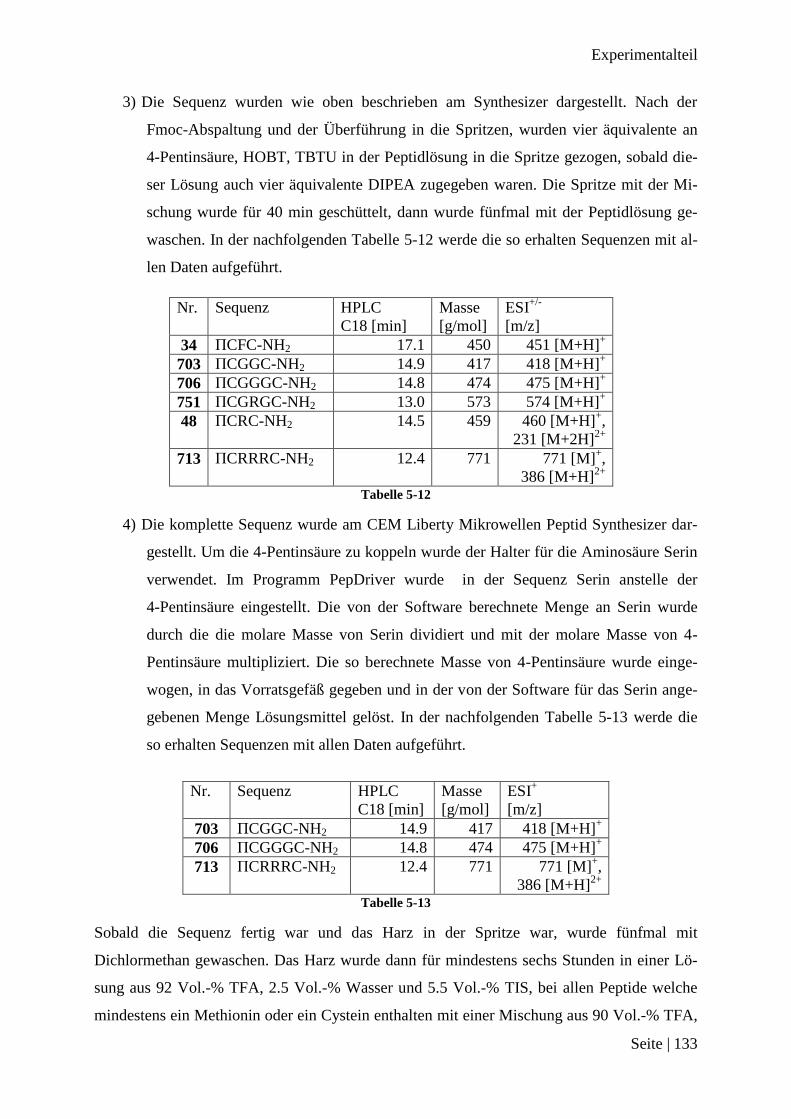
Experimentalteil
Seite | 133
3) Die Sequenz wurden wie oben beschrieben am Synthesizer dargestellt. Nach der
Fmoc-Abspaltung und der Überführung in die Spritzen, wurden vier äquivalente an
4-Pentinsäure, HOBT, TBTU in der Peptidlösung in die Spritze gezogen, sobald die-
ser Lösung auch vier äquivalente DIPEA zugegeben waren. Die Spritze mit der Mi-
schung wurde für 40 min geschüttelt, dann wurde fünfmal mit der Peptidlösung ge-
waschen. In der nachfolgenden Tabelle 5-12 werde die so erhalten Sequenzen mit al-
len Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+/-
[m/z]
34 ΠCFC-NH2 17.1 450 451 [M+H]+
703 ΠCGGC-NH2 14.9 417 418 [M+H]+
706 ΠCGGGC-NH2 14.8 474 475 [M+H]+
751 ΠCGRGC-NH2 13.0 573 574 [M+H]+
48 ΠCRC-NH2 14.5 459 460 [M+H]+,
231 [M+2H]2+
713 ΠCRRRC-NH2 12.4 771 771 [M]+,
386 [M+H]2+
Tabelle 5-12
4) Die komplette Sequenz wurde am CEM Liberty Mikrowellen Peptid Synthesizer dar-
gestellt. Um die 4-Pentinsäure zu koppeln wurde der Halter für die Aminosäure Serin
verwendet. Im Programm PepDriver wurde in der Sequenz Serin anstelle der
4-Pentinsäure eingestellt. Die von der Software berechnete Menge an Serin wurde
durch die die molare Masse von Serin dividiert und mit der molare Masse von 4-
Pentinsäure multipliziert. Die so berechnete Masse von 4-Pentinsäure wurde einge-
wogen, in das Vorratsgefäß gegeben und in der von der Software für das Serin ange-
gebenen Menge Lösungsmittel gelöst. In der nachfolgenden Tabelle 5-13 werde die
so erhalten Sequenzen mit allen Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
703 ΠCGGC-NH2 14.9 417 418 [M+H]+
706 ΠCGGGC-NH2 14.8 474 475 [M+H]+
713 ΠCRRRC-NH2 12.4 771 771 [M]+,
386 [M+H]2+
Tabelle 5-13
Sobald die Sequenz fertig war und das Harz in der Spritze war, wurde fünfmal mit
Dichlormethan gewaschen. Das Harz wurde dann für mindestens sechs Stunden in einer Lö-
sung aus 92 Vol.-% TFA, 2.5 Vol.-% Wasser und 5.5 Vol.-% TIS, bei allen Peptide welche
mindestens ein Methionin oder ein Cystein enthalten mit einer Mischung aus 90 Vol.-% TFA,

Experimentalteil
Seite | 134
2.5 Vol.-% EDT, 2.5 Vol.-% Wasser und 5 Vol.-% TIS behandelt. Wenn sich das Harz und
die Lösung bei der Zugabe gelbfärbten, dann wurde wenig TIS zugegeben. Die Lösung wurde
in ein Plastikgefäß mit Deckel gegeben. In die Spritze wurde TFA gezogen und ebenfalls in
das Gefäß gegeben. In das Gefäß wurde Diethylether mit einer Temperatur von - 70 °C gege-
ben. Es fiel ein farbloses Pulver aus, welches von der Lösung, in einer Zentrifuge mit ab-
schließendem dekantieren, getrennt wird. Das Pulver wurde zweimal mit DET bei - 70 °C
gewaschen und mit Zentrifugieren und Dekantieren von dem Lösungsmittel getrennt. Das
Pulver wurde in einer Mischung aus Wasser und ACN gelöst und in flüssigem Stickstoff ein-
gefroren. Die eingefrorene Lösung wurde lyophilisiert. Es wurde ein farbloses schaumartiges
Pulver erhalten.
Kopplung von AuPhPyCl2 mit Peptiden
Vor dem 300 Peptidprojekt
Äquimolare Mengen von AuPhPyCl2 (2) wurden in einem Kolben mit jeweils einer der fol-
genden Verbindungen: Ac-CFC-NH2 (27), Ac-CFCRRRRR-NH2 (29), Ac-CGC-NH2 (23),
Ac-CPC-NH2 (25), ΠCFC NH2 (33) und ΠCRC-NH2 (48) zusammen gegeben. In den Kolben
wurden zwischen 2 ml und 15 ml einer Lösungsmittelmischung aus 50 Vol.-% Wasser und
50 Vol.-% Acetonitril gegeben. Die entstehende Suspension wurde für mindestens eine Stun-
de gerührt. In den Kolben mit den Verbindungen 29 und 48 entstand nach kurzer Zeit eine
gelbfarbene Lösung, in allen andern Kolben war eine gelbfarbene Suspension zu beobachten.
Das Lösungsmittel wurde am Lyophilisator entfernt. Das erhalten gelbfarbene Pulver wurde
an der präperativen HPLC gereinigt. In der nachfolgenden Tabelle 5-14 werde die so erhalten
Verbindung mit allen Daten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
Ausbeute
[%]
28 Ac-CFC-NH2AuPhPy 22.9 761 762 [M+H]+,
381 [M+H]2+
82
30 Ac-CFCRRRRR-NH2
AuPhPy
17.0 1542 772 [M+H]2+
69
26 Ac-CGC-NH2AuPhPy 18.7 671 672 [M+H]+,
629 [M-Ac]+
84
27 Ac-CPC-NH2AuPhPy 19.2 711 712 [M+H]+,
669 [M-Ac]+
80
34 ΠCFC-NH2AuPhPy 20.9 799 822 [M+Na]+, 86
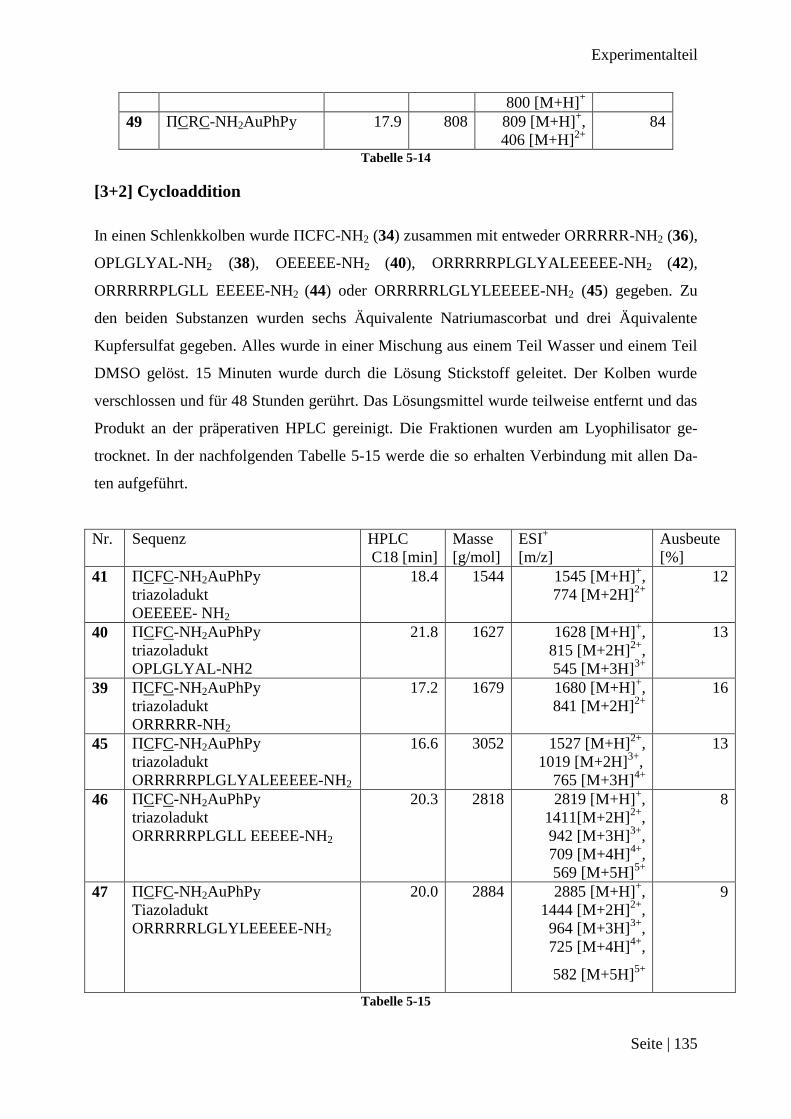
Experimentalteil
Seite | 135
800 [M+H]+
49 ΠCRC-NH2AuPhPy 17.9 808 809 [M+H]+,
406 [M+H]2+
84
Tabelle 5-14
[3+2] Cycloaddition
In einen Schlenkkolben wurde ΠCFC-NH2 (34) zusammen mit entweder ORRRRR-NH2 (36),
OPLGLYAL-NH2 (38), OEEEEE-NH2 (40), ORRRRRPLGLYALEEEEE-NH2 (42),
ORRRRRPLGLL EEEEE-NH2 (44) oder ORRRRRLGLYLEEEEE-NH2 (45) gegeben. Zu
den beiden Substanzen wurden sechs Äquivalente Natriumascorbat und drei Äquivalente
Kupfersulfat gegeben. Alles wurde in einer Mischung aus einem Teil Wasser und einem Teil
DMSO gelöst. 15 Minuten wurde durch die Lösung Stickstoff geleitet. Der Kolben wurde
verschlossen und für 48 Stunden gerührt. Das Lösungsmittel wurde teilweise entfernt und das
Produkt an der präperativen HPLC gereinigt. Die Fraktionen wurden am Lyophilisator ge-
trocknet. In der nachfolgenden Tabelle 5-15 werde die so erhalten Verbindung mit allen Da-
ten aufgeführt.
Nr. Sequenz
HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
Ausbeute
[%]
41 ΠCFC-NH2AuPhPy
triazoladukt
OEEEEE- NH2
18.4 1544 1545 [M+H]+,
774 [M+2H]2+
12
40 ΠCFC-NH2AuPhPy
triazoladukt
OPLGLYAL-NH2
21.8 1627 1628 [M+H]+,
815 [M+2H]2+
,
545 [M+3H]3+
13
39 ΠCFC-NH2AuPhPy
triazoladukt
ORRRRR-NH2
17.2 1679 1680 [M+H]+,
841 [M+2H]2+
16
45 ΠCFC-NH2AuPhPy
triazoladukt
ORRRRRPLGLYALEEEEE-NH2
16.6 3052 1527 [M+H]2+
,
1019 [M+2H]3+
,
765 [M+3H]4+
13
46 ΠCFC-NH2AuPhPy
triazoladukt
ORRRRRPLGLL EEEEE-NH2
20.3 2818 2819 [M+H]+,
1411[M+2H]2+
,
942 [M+3H]3+
,
709 [M+4H]4+
,
569 [M+5H]5+
8
47 ΠCFC-NH2AuPhPy
Tiazoladukt
ORRRRRLGLYLEEEEE-NH2
20.0 2884 2885 [M+H]+,
1444 [M+2H]2+
,
964 [M+3H]3+
,
725 [M+4H]4+
,
582 [M+5H]5+
9
Tabelle 5-15

Experimentalteil
Seite | 136
300 Peptidprojekt
Die Synthesen für die kombinatorischen Untersuchungen wurden an Tenta Gel Macrobeads
mit einer Beladung von 0.24 mmol/g durchgeführt. Die Synthese der Peptide erfolgte an ei-
nem Syro Multiple Peptide Synthesizer der Firma Syro 2000 in 2 ml Spritzen mit einem
Polypropylenfilter. Für die Synthese wurden die Aminosäuren in einer Lösung aus 1-
HydroΠybenzotriazole und 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyl-uronium hexa-
fluorophosphate in 70 Vol.-% N,N-Dimethylformamid und 30 Vol.-% N-Methyl-2-pyrrolidon
gelöst. Die Aminosäure, 1-HydroΠybenzotriazole und 2-(1H-Benzotriazole-1-yl)-1,1,3,3-
tetramethyl-uronium hexafluorophosphate waren in äquimolaren Konzentration in der Lösung
vorhanden. Die Lösungen mit den natürlichen Aminosäuren wurden an vorgesehen Postionen
gestellt. Die unnatürlichen Aminosäuren, zu den auch die 4-Pentinsäure gezählt wird, wurden
an Positionen gestellt die vorher im Programm des Synthesizer für sie eingestellt worden wa-
ren. N,N-Diisopropylethylamine wurde in N-Methyl-2-pyrrolidon gelöst und an seine Positi-
on gestellt. Piperidin wurde in 70 Vol.-% N,N-Dimethylformamid und 30 Vol.-% N-Methyl-
2-pyrrolidon gelöst und positioniert. Das Harz wurde in 2 ml Spritzen mit Filter eingewogene,
die Spritzen wurden ohne Stempel mit der Spitze nach unten in den Synthesizer gestellt. Die
Platte auf der bis zu 24 Spritzen gleichzeitig positioniert werden konnten, hatte ein Schlauch
System, über welchen der Inhalt aus den Spritzen abgesaugt und direkt verworfen werden
konnte. Diese Platte konnte auch vom Synthesizer geschüttelt werden für eine bessere
Durchmischung in den Spritzen. Alle vorher positionierten Lösungen wurden automatisch
vom Synthesizer in einem Saugrüssel aufgenommen und von oben, wo normalerweise der
Stempel sitzen würde, in die Spritze gegeben. Der Saugrüssel wurde vor der der Aufnahme
einer neuen Substanz mit N,N-Dimethylformamid gewaschen. Alle Waschschritte in den
Spritzen wurden ebenfalls in reinem N,N-Dimethylformamid durchgeführt. Die Kupplungen
jeder Aminosäuren wurden doppelt durchgeführt. Die Kopplungen wurden im fünffachen
Überschuss durchgeführt. Die Fmoc-Entschützung erfolgte mittels einer 40 Vol.-%
Piperidinlösung in der Peptidlösung. Die Waschschritte zwischen den einzelnen Synthese-
schritten wurden in reinem DMF durchgeführt. Nach dem letzten Kopplungsschritt mit der 4-
Pentinsäure wurden die Spritzen aus dem Synthesizer entnommen und das Harz wurde fünf-
mal mit Dichlormethan gewaschen. Alle Peptide, welche mindestens ein Methionin oder ein
Cystein enthalten, wurden für zehn Stunden in einer Mischung aus 94 Vol.-% TFA,
2.5 Vol.-% EDT, 2.5 Vol.-% Wasser und 1 Vol.-% TIS, alle anderen in einer Mischung aus
95 Vol.-% TFA, 2.5 Vol.-% Wasser und 2.5 Vol.-% TIS behandelt. Wenn die Lösung bei der
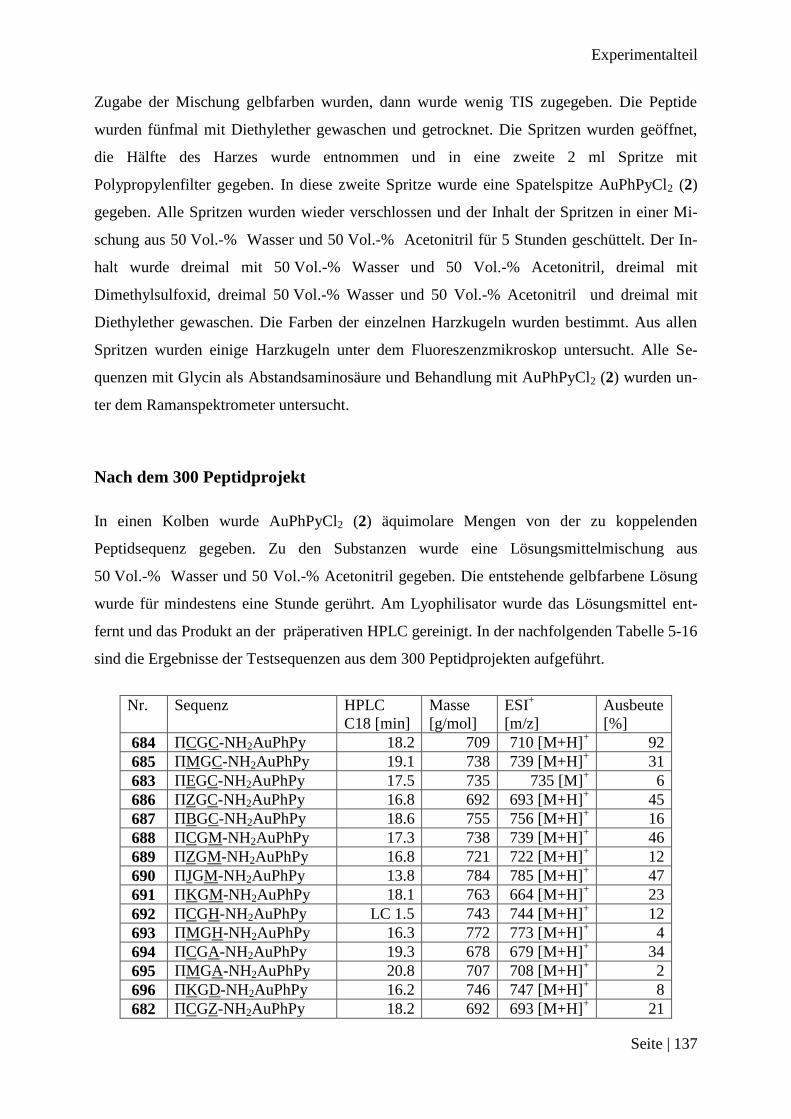
Experimentalteil
Seite | 137
Zugabe der Mischung gelbfarben wurden, dann wurde wenig TIS zugegeben. Die Peptide
wurden fünfmal mit Diethylether gewaschen und getrocknet. Die Spritzen wurden geöffnet,
die Hälfte des Harzes wurde entnommen und in eine zweite 2 ml Spritze mit
Polypropylenfilter gegeben. In diese zweite Spritze wurde eine Spatelspitze AuPhPyCl2 (2)
gegeben. Alle Spritzen wurden wieder verschlossen und der Inhalt der Spritzen in einer Mi-
schung aus 50 Vol.-% Wasser und 50 Vol.-% Acetonitril für 5 Stunden geschüttelt. Der In-
halt wurde dreimal mit 50 Vol.-% Wasser und 50 Vol.-% Acetonitril, dreimal mit
Dimethylsulfoxid, dreimal 50 Vol.-% Wasser und 50 Vol.-% Acetonitril und dreimal mit
Diethylether gewaschen. Die Farben der einzelnen Harzkugeln wurden bestimmt. Aus allen
Spritzen wurden einige Harzkugeln unter dem Fluoreszenzmikroskop untersucht. Alle Se-
quenzen mit Glycin als Abstandsaminosäure und Behandlung mit AuPhPyCl2 (2) wurden un-
ter dem Ramanspektrometer untersucht.
Nach dem 300 Peptidprojekt
In einen Kolben wurde AuPhPyCl2 (2) äquimolare Mengen von der zu koppelenden
Peptidsequenz gegeben. Zu den Substanzen wurde eine Lösungsmittelmischung aus
50 Vol.-% Wasser und 50 Vol.-% Acetonitril gegeben. Die entstehende gelbfarbene Lösung
wurde für mindestens eine Stunde gerührt. Am Lyophilisator wurde das Lösungsmittel ent-
fernt und das Produkt an der präperativen HPLC gereinigt. In der nachfolgenden Tabelle 5-16
sind die Ergebnisse der Testsequenzen aus dem 300 Peptidprojekten aufgeführt.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
Ausbeute
[%]
684 ΠCGC-NH2AuPhPy 18.2 709 710 [M+H]+ 92
685 ΠMGC-NH2AuPhPy 19.1 738 739 [M+H]+ 31
683 ΠEGC-NH2AuPhPy 17.5 735 735 [M]+ 6
686 ΠZGC-NH2AuPhPy 16.8 692 693 [M+H]+ 45
687 ΠBGC-NH2AuPhPy 18.6 755 756 [M+H]+ 16
688 ΠCGM-NH2AuPhPy 17.3 738 739 [M+H]+ 46
689 ΠZGM-NH2AuPhPy 16.8 721 722 [M+H]+ 12
690 ΠJGM-NH2AuPhPy 13.8 784 785 [M+H]+ 47
691 ΠKGM-NH2AuPhPy 18.1 763 664 [M+H]+ 23
692 ΠCGH-NH2AuPhPy LC 1.5 743 744 [M+H]+ 12
693 ΠMGH-NH2AuPhPy 16.3 772 773 [M+H]+ 4
694 ΠCGA-NH2AuPhPy 19.3 678 679 [M+H]+ 34
695 ΠMGA-NH2AuPhPy 20.8 707 708 [M+H]+ 2
696 ΠKGD-NH2AuPhPy 16.2 746 747 [M+H]+ 8
682 ΠCGZ-NH2AuPhPy 18.2 692 693 [M+H]+ 21
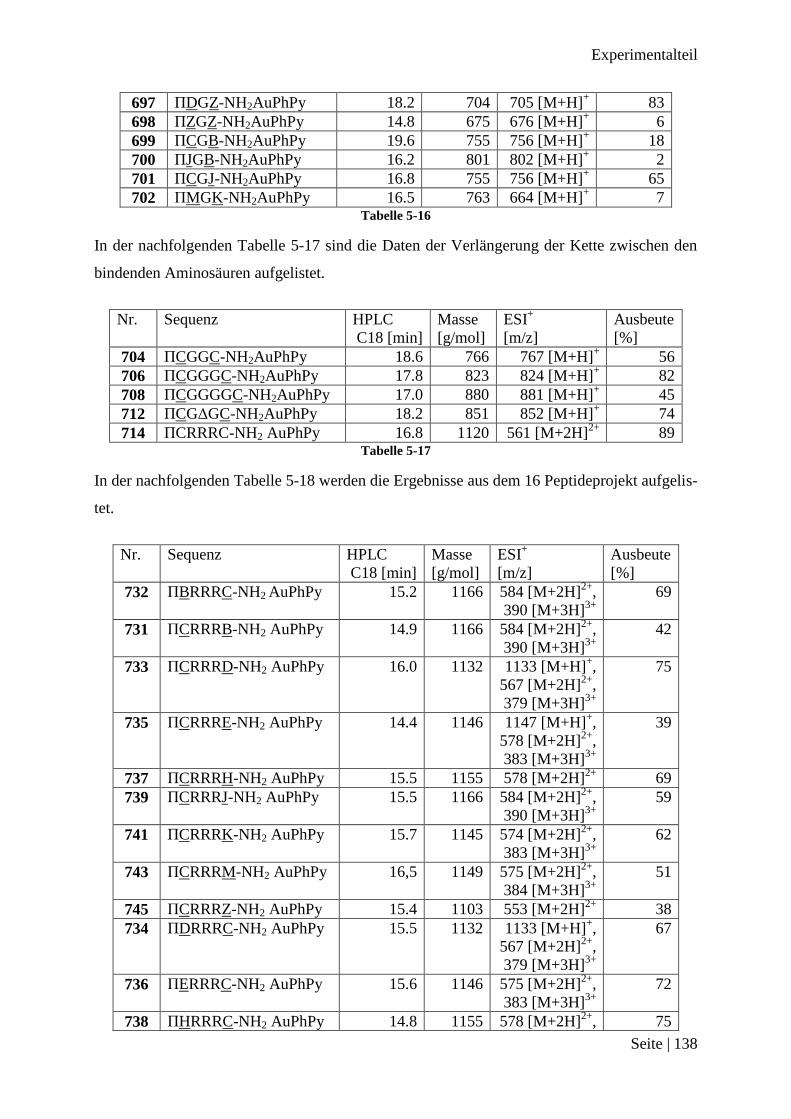
Experimentalteil
Seite | 138
697 ΠDGZ-NH2AuPhPy 18.2 704 705 [M+H]+ 83
698 ΠZGZ-NH2AuPhPy 14.8 675 676 [M+H]+ 6
699 ΠCGB-NH2AuPhPy 19.6 755 756 [M+H]+ 18
700 ΠJGB-NH2AuPhPy 16.2 801 802 [M+H]+ 2
701 ΠCGJ-NH2AuPhPy 16.8 755 756 [M+H]+ 65
702 ΠMGK-NH2AuPhPy 16.5 763 664 [M+H]+ 7
Tabelle 5-16
In der nachfolgenden Tabelle 5-17 sind die Daten der Verlängerung der Kette zwischen den
bindenden Aminosäuren aufgelistet.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
Ausbeute
[%]
704 ΠCGGC-NH2AuPhPy 18.6 766 767 [M+H]+ 56
706 ΠCGGGC-NH2AuPhPy 17.8 823 824 [M+H]+ 82
708 ΠCGGGGC-NH2AuPhPy 17.0 880 881 [M+H]+ 45
712 ΠCGΔGC-NH2AuPhPy 18.2 851 852 [M+H]+ 74
714 ΠCRRRC-NH2 AuPhPy 16.8 1120 561 [M+2H]2+
89 Tabelle 5-17
In der nachfolgenden Tabelle 5-18 werden die Ergebnisse aus dem 16 Peptideprojekt aufgelis-
tet.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
Ausbeute
[%]
732 ΠBRRRC-NH2 AuPhPy 15.2 1166 584 [M+2H]2+
,
390 [M+3H]3+
69
731 ΠCRRRB-NH2 AuPhPy 14.9 1166 584 [M+2H]2+
,
390 [M+3H]3+
42
733 ΠCRRRD-NH2 AuPhPy 16.0 1132 1133 [M+H]+,
567 [M+2H]2+
,
379 [M+3H]3+
75
735 ΠCRRRE-NH2 AuPhPy 14.4 1146 1147 [M+H]+,
578 [M+2H]2+
,
383 [M+3H]3+
39
737 ΠCRRRH-NH2 AuPhPy 15.5 1155 578 [M+2H]2+
69
739 ΠCRRRJ-NH2 AuPhPy 15.5 1166 584 [M+2H]2+
,
390 [M+3H]3+
59
741 ΠCRRRK-NH2 AuPhPy 15.7 1145 574 [M+2H]2+
,
383 [M+3H]3+
62
743 ΠCRRRM-NH2 AuPhPy 16,5 1149 575 [M+2H]2+
,
384 [M+3H]3+
51
745 ΠCRRRZ-NH2 AuPhPy 15.4 1103 553 [M+2H]2+
38
734 ΠDRRRC-NH2 AuPhPy 15.5 1132 1133 [M+H]+,
567 [M+2H]2+
,
379 [M+3H]3+
67
736 ΠERRRC-NH2 AuPhPy 15.6 1146 575 [M+2H]2+
,
383 [M+3H]3+
72
738 ΠHRRRC-NH2 AuPhPy 14.8 1155 578 [M+2H]2+
, 75
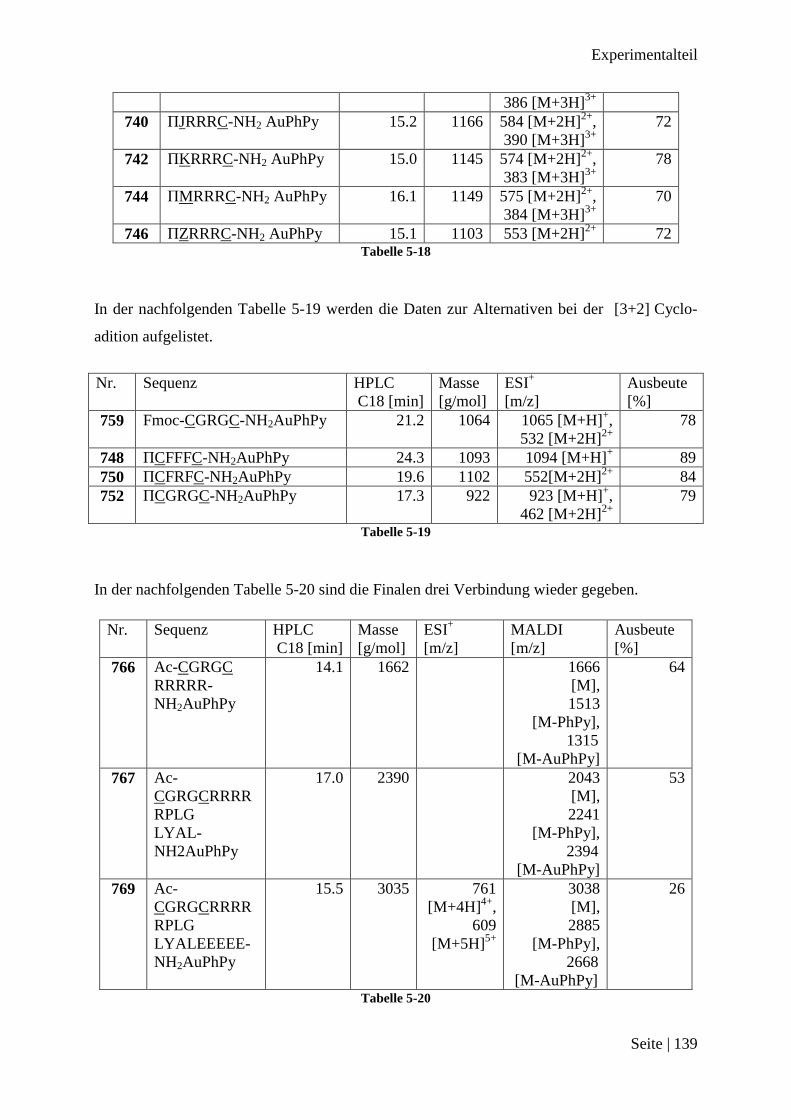
Experimentalteil
Seite | 139
386 [M+3H]3+
740 ΠJRRRC-NH2 AuPhPy 15.2 1166 584 [M+2H]2+
,
390 [M+3H]3+
72
742 ΠKRRRC-NH2 AuPhPy 15.0 1145 574 [M+2H]2+
,
383 [M+3H]3+
78
744 ΠMRRRC-NH2 AuPhPy 16.1 1149 575 [M+2H]2+
,
384 [M+3H]3+
70
746 ΠZRRRC-NH2 AuPhPy 15.1 1103 553 [M+2H]2+
72 Tabelle 5-18
In der nachfolgenden Tabelle 5-19 werden die Daten zur Alternativen bei der [3+2] Cyclo-
adition aufgelistet.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
Ausbeute
[%]
759 Fmoc-CGRGC-NH2AuPhPy 21.2 1064 1065 [M+H]+,
532 [M+2H]2+
78
748 ΠCFFFC-NH2AuPhPy 24.3 1093 1094 [M+H]+ 89
750 ΠCFRFC-NH2AuPhPy 19.6 1102 552[M+2H]2+
84
752 ΠCGRGC-NH2AuPhPy 17.3 922 923 [M+H]+,
462 [M+2H]2+
79
Tabelle 5-19
In der nachfolgenden Tabelle 5-20 sind die Finalen drei Verbindung wieder gegeben.
Nr. Sequenz HPLC
C18 [min]
Masse
[g/mol]
ESI+
[m/z]
MALDI
[m/z]
Ausbeute
[%]
766 Ac-CGRGC
RRRRR-
NH2AuPhPy
14.1 1662 1666
[M],
1513
[M-PhPy],
1315
[M-AuPhPy]
64
767 Ac-
CGRGCRRRR
RPLG
LYAL-
NH2AuPhPy
17.0 2390 2043
[M],
2241
[M-PhPy],
2394
[M-AuPhPy]
53
769 Ac-
CGRGCRRRR
RPLG
LYALEEEEE-
NH2AuPhPy
15.5 3035 761
[M+4H]4+
,
609
[M+5H]5+
3038
[M],
2885
[M-PhPy],
2668
[M-AuPhPy]
26
Tabelle 5-20

Literatur
Seite | 140
Literatur
1. Mortimer, Mueller, Chemie: Das Basiswissen der Chemie Thieme, Stuttgart: 2003.
2. L.D. Quarles, J. Clin. Invest. 2008, 118, 3820-3828.
3. J.P. Ceron-Carrasco, D. Jacquemin, ChemPhysChem 2011, 12, 2615-2623.
4. J.S. Becker, M. Zoriy, A. Matusch, B. Wu, D. Salber, C. Palm, J.S. Becker, Mass
Spectrom. Rev. 2009, 29, 156-175.
5. D.G. Mendoza-Cozatl, T.O. Jobe, F. Hauser, J.I. Schroeder, Curr. Opin. Plant Biol.
2011, 14, 554-562.
6. A.J. Guerra, D.P. Giedroc, Arch. Biochem. Biophys. 2011, 519, 210-222.
7. M. Murakami, T. Hirano, Cancer Sci. 2008, 99, 1515-1522.
8. G. Grass, L. Rensing, C. Rensing, Metallomics 2011, 3, 1095-1097.
9. M.U. Muckenthaler, B. Galy, M.W. Hentze, Annu. Rev. Nutr. 2008, 28, 197-213, 194
10. R.E. Weber, K.L. Campbell, Acta Physiol. 2011, 202, 549-562.
11. M.K. Safo, M.H. Ahmed, M.S. Ghatge, T. Boyiri, Biochim. Biophys. Acta, Proteins
Proteomics 2011, 1814, 797-809.
12. C. Orvig, M.J. Abrams, Chem. Rev. 1999, 99, 2201-2203.
13. N. Singh, A. Chaudhary, Int. J. Res. Ayurveda Pharm. 2012, 3, 5-9.
14. H. Sakurai, J. Health Sci. 2010, 56, 129-143.
15. K. Kneipp, H. Kneipp, I. Itzkan, R.R. Dasari, M.S. Feld, Chem. Rev. 1999, 99, 2957-
2975.
16. A. Tillberg, B. Jaervholm, A. Berglund, Dent. Mater. 2008, 24, 940-943.
17. Y. Ucar, W.A. Brantley, Int. J. Dent. 2011, 981595, 981597 pp.
18. N.J. Birch, Chem. Rev. 1999, 99, 2659-2682.
19. J. Deberitz, G. Boche, Chem. Unserer Zeit 2003, 37, 258-266.
20. M.P. Freeman, S.A. Freeman, Am. J. Med. 2006, 119, 478-481.
21. S.-B. Yu, A.D. Watson, Chem. Rev. 1999, 99, 2353-2377.
22. V. Sharma, D. Piwnica-Worms, Chem. Rev. 1999, 99, 2545-2560.
23. S. Liu, D.S. Edwards, Chem. Rev. 1999, 99, 2235-2268.
24. C.J. Anderson, M.J. Welch, Chem. Rev. 1999, 99, 2219-2234.
25. W.A. Volkert, T.J. Hoffman, Chem. Rev. 1999, 99, 2269-2292.
26. K.A. Sepkowitz, N. Engl. J. Med. 2011, 365, 291-293.
27. K. Tripathi, Asian J. Res. Chem. 2009, 2, 14-18.
28. N. Metzler-Nolte, Nachr. Chem. 2006, 54, 966-970.
29. K.H. Antman, Oncologist 2001, 6, 1-2.
30. K.A. Sepkowitz, N. Engl. J. Med. 2011, 365, 291-293.
31. N.C. Lloyd, H.W. Morgan, B.K. Nicholson, R.S. Ronimus, Angew. Chem., Int. Ed.
2005, 44, 941-944.
32. N. Farrell, Coord. Chem. Rev. 2002, 232, 1-4.
33. G. Gasser, I. Ott, N. Metzler-Nolte, J. Med. Chem. 2011, 54, 3-25.
34. W. Liu, R. Gust, Chem. Soc. Rev. 2013, 42, 755-773.
35. C.G. Hartinger, N. Metzler-Nolte, P.J. Dyson, Organometallics 2012, 31, 5677-5685.
36. C.G. Hartinger, P.J. Dyson, Chem. Soc. Rev. 2009, 38, 391-401.
37. T.C. Johnstone, J.J. Wilson, S.J. Lippard, Inorg. Chem. 2013.
38. E.R. Jamieson, S.J. Lippard, Chem. Rev. 1999, 99, 2467-2498.
39. E. Steinnes, Environ. Pollut. 2013, 22, 411-428.
40. I. Zanellato, I. Bonarrigo, E. Gabano, M. Ravera, N. Margiotta, P.-G. Betta, D. Osella,
Inorg. Chim. Acta 2012, 393, 64-74.
41. G.Y. Park, J.J. Wilson, Y. Song, S.J. Lippard, Proc. Natl. Acad. Sci. U. S. A. 2012,
109, 11987-11992, S11987/11981-S11987/11924.

Literatur
Seite | 141
42. S.J. Berners-Price, A. Filipovska, Metallomics 2011, 3, 863-873.
43. D. Raducanu, A. Cimpean, E. Vasilescu, V.D. Cojocaru, I. Cinca, P. Drob, S.
Ivanescu, Mater. Corros. 2010, 61, 775-782.
44. R.V. Parish, Gold Bull. 1997, 30, 3-12.
45. R.V. Parish, Gold Bull. 1997, 30, 55-62.
46. S.J. Berners-Price, M.J. DiMartino, D.T. Hill, R. Kuroda, M.A. Mazid, P.J. Sadler,
Inorg. Chem. 1985, 24, 3425-3434.
47. C.F. Shaw, III, Chem. Rev. 1999, 99, 2589-2600.
48. I. Ott, Coord. Chem. Rev. 2009, 253, 1670-1681.
49. J.L. Hickey, R.A. Ruhayel, P.J. Barnard, M.V. Baker, S.J. Berners-Price, A.
Filipovska, J. Am. Chem. Soc. 2008, 130, 12570-12571.
50. S.J. Berners-Price, A. Filipovska, Aust. J. Chem. 2008, 61, 661-668.
51. I. Ott, X. Qian, Y. Xu, D.H.W. Vlecken, I.J. Marques, D. Kubutat, J. Will, W.S. Shel-
drick, P. Jesse, A. Prokop, C.P. Bagowski, J. Med. Chem. 2009, 52, 763-770.
52. A. De Luca, C.G. Hartinger, P.J. Dyson, M. Lo Bello, A. Casini, J. Inorg. Biochem.
2013, 119, 38-42.
53. A.P. Shaw, M. Tilset, R.H. Heyn, S. Jakobsen, J. Coord. Chem. 2011, 64, 38-47.
54. V. Milacic, D. Chen, L. Ronconi, R. Landis-Piwowar Kristin, D. Fregona, Q.P. Dou,
Cancer Res 2006, 66, 10478-10486.
55. B.D. Glisic, U. Rychlewska, M.I. Djuran, Dalton Trans. 2012, 41, 6887-6901.
56. M.N. Patel, B.S. Bhatt, P.A. Dosi, Spectrochim. Acta, Part A 2013, 110, 20-27.
57. M.N. Patel, B.S. Bhatt, P.A. Dosi, Inorg. Chem. Commun. 2013, 29, 190-193.
58. A. Djekovic, B. Petrovic, Z.D. Bugarcic, R. Puchta, R. van Eldik, Dalton Trans. 2012,
41, 3633-3641.
59. M.J. McKeage, L. Maharaj, S.J. Berners-Price, Coord. Chem. Rev. 2002, 232, 127-
135.
60. W. Liu, R. Gust, Chem. Soc. Rev. 2013, 42, 755-773.
61. C. Gabbiani, M.A. Cinellu, L. Maiore, L. Massai, F. Scaletti, L. Messori, Inorg. Chim.
Acta 2012, 393, 115-124.
62. T. Zou, C.T. Lum, S.S.-Y. Chui, C.-M. Che, Angew. Chem., Int. Ed. 2013, 52, 2930-
2933.
63. D. Sanghvi Chinar, M. Olsen Pauline, C. Elix, B. Peng Shifang, D. Wang, G. Chen
Zhuo, M. Shin Dong, I. Hardcastle Kenneth, E. Macbeth Cora, F. Eichler Jack, J.
Inorg. Biochem. 2013, 128C, 68-76.
64. N. Lease, V. Vasilevski, M. Carreira, A. de Almeida, M. Sanau, P. Hirva, A. Casini,
M. Contel, J. Med. Chem. 2013, 56, 5806-5818.
65. R.W.-Y. Sun, C.-N. Lok, T.T.-H. Fong, C.K.-L. Li, Z.F. Yang, T. Zou, A.F.-M. Siu,
C.-M. Che, Chem. Sci. 2013, 4, 1979-1988.
66. B. Notash, V. Amani, N. Safari, S.N. Ostad, A. Abedi, M.Z. Dehnavi, Dalton Trans.
2013, 42, 6852-6858.
67. C.T. Lum, A.S.-T. Wong, M.C.M. Lin, C.-M. Che, R.W.-Y. Sun, Chem. Commun.
(Cambridge, U. K.) 2013, 49, 4364-4366.
68. G.T. Hermanson, Bioconjugate Techniques. Academic Press: 1996.
69. N. Rajendran, M.A. Marahiel, Compr. Nat. Prod. Chem. 1999, 4, 195-220.
70. P.G. Arnison, M.J. Bibb, G. Bierbaum, A.A. Bowers, T.S. Bugni, G. Bulaj, J.A.
Camarero, D.J. Campopiano, G.L. Challis, J. Clardy, P.D. Cotter, D.J. Craik, M. Daw-
son, E. Dittmann, S. Donadio, P.C. Dorrestein, K.-D. Entian, M.A. Fischbach, J.S.
Garavelli, U. Goeransson, C.W. Gruber, D.H. Haft, T.K. Hemscheidt, C. Hertweck, C.
Hill, A.R. Horswill, M. Jaspars, W.L. Kelly, J.P. Klinman, O.P. Kuipers, A.J. Link, W.
Liu, M.A. Marahiel, D.A. Mitchell, G.N. Moll, B.S. Moore, R. Mueller, S.K. Nair, I.F.
Nes, G.E. Norris, B.M. Olivera, H. Onaka, M.L. Patchett, J. Piel, M.J.T. Reaney, S.

Literatur
Seite | 142
Rebuffat, R.P. Ross, H.-G. Sahl, E.W. Schmidt, M.E. Selsted, K. Severinov, B. Shen,
K. Sivonen, L. Smith, T. Stein, R.D. Suessmuth, J.R. Tagg, G.-L. Tang, A.W. Truman,
J.C. Vederas, C.T. Walsh, J.D. Walton, S.C. Wenzel, J.M. Willey, W.A. van der Donk,
Nat. Prod. Rep. 2013, 30, 108-160.
71. R. Hubert, L. Thomas, Der Experimentator: Proteinbiochemie/Proteomics Spektrum
Akademischer Verlag, Heidelberg 2009.
72. B. Merrifield, Prix Nobel 1984, 127-153.
73. R.B. Merrifield, J. Am. Chem. Soc. 1963, 85, 2149-2154.
74. H.P. Hemantha, N. Narendra, V.V. Sureshbabu, Tetrahedron 2012, 68, 9491-9537.
75. A. Meister, Advances in Enzymology and Related Areas of Molecular Biology. John
Wiley & Sons: 1969.
76. C.-H. Wong, S.C. Zimmerman, Chem. Commun. 2013, 49, 1679-1695.
77. W.C. Chan, P.D. White, Editors, Fmoc Solid Phase Peptide Synthesis: A Practical
Approach. Oxford University Press: 2000.
78. M. Gongora-Benitez, J. Tulla-Puche, F. Albericio, ACS Comb. Sci. 2013, 15, 217-228.
79. D.J. Mitchell, D.T. Kim, L. Steinman, C.G. Fathman, J.B. Rothbard, J. Pept. Res.
2000, 56, 318-325.
80. C. Bechara, S. Sagan, FEBS Lett. 2013, 587, 1693-1702.
81. J. Regberg, A. Srimanee, U. Langel, Pharmaceuticals 2012, 5, 991-1007.
82. E. Koren, V.P. Torchilin, Trends Mol. Med. 2012, 18, 385-393.
83. K. Park, J. Controlled Release 2012, 159, 153.
84. N. Svensen, J.G.A. Walton, M. Bradley, Trends Pharmacol. Sci. 2012, 33, 186-192.
85. B. Fang, L. Jiang, M. Zhang, F.Z. Ren, Biochimie 2013, 95, 251-257.
86. E. Vives, J. Controlled Release 2005, 109, 77-85.
87. T. Jiang, E.S. Olson, Q.T. Nguyen, M. Roy, P.A. Jennings, R.Y. Tsien, Proc. Natl.
Acad. Sci. U. S. A. 2004, 101, 17867-17872.
88. J.R. Tauro, R.A. Gemeinhart, Bioconjugate Chem. 2005, 16, 1133-1139.
89. M. Mansour Ahmed, J. Drevs, N. Esser, M. Hamada Farid, A. Badary Osama, C.
Unger, I. Fichtner, F. Kratz, Cancer Res 2003, 63, 4062-4066.
90. R.A. Dean, C.M. Overall, Mol. Cell. Proteomics 2007, 6, 611-623.
91. C.F. Albright, N. Graciani, W. Han, E. Yue, R. Stein, Z. Lai, M. Diamond, R.
Dowling, L. Grimminger, S.-Y. Zhang, D. Behrens, A. Musselman, R. Bruckner, M.
Zhang, X. Jiang, D. Hu, A. Higley, S. DiMeo, M. Rafalski, S. Mandlekar, B. Car, S.
Yeleswaram, A. Stern, R.A. Copeland, A. Combs, S.P. Seitz, G.L. Trainor, R. Taub, P.
Huang, A. Oliff, Mol. Cancer Ther. 2005, 4, 751-760.
92. J.L. Mauriz, J. Gonzalez-Gallego, J. Pharm. Sci. 2008, 97, 4129-4154.
93. N. Tandler, B. Mosch, J. Pietzsch, Amino Acids 2012, 43, 2203-2230.
94. Z. Hu, X. Jiang, C.F. Albright, N. Graciani, E. Yue, M. Zhang, S.-Y. Zhang, R.
Bruckner, M. Diamond, R. Dowling, M. Rafalski, S. Yeleswaram, G.L. Trainor, S.P.
Seitz, W. Han, Bioorg. Med. Chem. Lett. 2010, 20, 853-856.
95. P. Vihinen, V.-M. Kahari, Int. J. Cancer 2002, 99, 157-166.
96. J.R. Tauro, B.-S. Lee, S.S. Lateef, R.A. Gemeinhart, Peptides 2008, 29, 1965-1973.
97. A. Talvensaari-Mattila, P. Paeaekkoe, T. Turpeenniemi-Hujanen, Br. J. Cancer 2003,
89, 1270-1275.
98. A. Cobos-Correa, J.B. Trojanek, S. Diemer, M.A. Mall, C. Schultz, Nat. Chem. Biol.
2009, 5, 628-630.
99. R.A.A. Muzzarelli, Carbohydr. Polym. 2009, 76, 167-182.
100. V. Lagente, C. Le Quement, E. Boichot, Expert Opin Ther Targets 2009, 13, 287-295.
101. J. MacFadyen Robert, Curr Opin Pharmacol 2007, 7, 171-178.
102. C. Gialeli, D. Theocharis Achilleas, K. Karamanos Nikos, Febs J 2010, 278, 16-27.
103. G. Vartak Deepali, A. Gemeinhart Richard, J. Drug. Target. 2007, 15, 1-20.

Literatur
Seite | 143
104. P.A. Wender, Tetrahedron 2013, 69, 7529-7550.
105. I. Martin, M. Teixido, E. Giralt, Pharmaceuticals 2010, 3, 1456-1490.
106. E.A. Goun, R. Shinde, K.W. Dehnert, A. Adams-Bond, P.A. Wender, C.H. Contag,
B.L. Franc, Bioconjugate Chem. 2006, 17, 787-796.
107. Q.T. Nguyen, E.S. Olson, T.A. Aguilera, T. Jiang, M. Scadeng, L.G. Ellies, R.Y.
Tsien, Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4317-4322.
108. S. Waissbluth, S.J. Daniel, Hear. Res. 2013, 299, 37-45.
109. C.A. Puckett, R.J. Ernst, J.K. Barton, Dalton Trans. 2010, 39, 1159-1170.
110. D. Fan, C.-T. Yang, J.D. Ranford, P.F. Lee, J.J. Vittal, Dalton Trans. 2003, 2680-
2685.
111. R.V. Parish, Met.-Based Drugs 1999, 6, 271-276.
112. E.C. Constable, T.A. Leese, J. Organomet. Chem. 1989, 363, 419-424.
113. H.G.O. Becker, R. Mayer, W. Berger, K. Müller, G. Domschkem, D. Pavel, E. Fang-
hänel, H. Schmidt, J. Faust, K. Schollberg, M. Fischer, K. Schwetlick, F. Gentz, E.
Seiler, K. Gewald, G. Zeppenfeld, R. Gluch, Organikum, Organisch-chemisches
Grundpraktikum. Wiley-VCH Weinheim: 2004.
114. M. Zaidlewicz, A. Wolan, J. Organomet. Chem. 2002, 657, 129-135.
115. K.P.C. Vollhardt, N.E. Schore, Organische Chemie. Wiley-VCH Weinheim: 2000.
116. Y. Gong, H.W. Pauls, Synlett 2000, 829-831.
117. S.J. Pridmore, J.M.J. Williams, Tetrahedron Lett. 2008, 49, 7413-7415.
118. E. Uggerud, Pure Appl. Chem. 2009, 81, 709-717.
119. H. Jin, B. Zhou, Z. Wu, Y. Shen, Y. Wang, Tetrahedron 2011, 67, 1178-1182.
120. M. Meldal, C.W. Tornoe, Chem. Rev. 2008, 108, 2952-3015.
121. L. Ackermann, A. Althammer, Chem. Unserer Zeit 2009, 43, 74-83.
122. D. Seyferth, Organometallics 2009, 28, 1598-1605.
123. R.V. Parish, J.P. Wright, R.G. Pritchard, J. Organomet. Chem. 2000, 596, 165-176.
124. C. Bischof, Building blocks for near-IR luminescent lanthanide complexes Doktorar-
beit, Ruhr-Universität Bochum 2013.
125. N. Alsakhem, Stable lanthanide cryptates as building block for solid phase peptide
synthesis - synthesis, functionalisation and photophysical properties. Doktorarbeit,
Ruhr-Universität Bochum 2012.
126. R.G. Pearson, J. Am. Chem. Soc. 1963, 85, 3533-3539.
127. D. Fan, C.-T. Yang, J.D. Ranford, J.J. Vittal, P.F. Lee, Dalton Trans. 2003, 3376-
3381.
128. J. Ruczynski, B. Lewandowska, P. Mucha, P. Rekowski, J. Pept. Sci. 2008, 14, 335-
341.
129. W.C. Chan, B.W. Bycroft, D.J. Evans, P.D. White, J. Chem. Soc., Chem. Commun.
1995, 2209-2210.
130. J.E. Macdonald, J.A. Kelly, J.G.C. Veinot, Langmuir 2007, 23, 9543-9545.
131. S.J. Hwang, S.H. Cho, S. Chang, Pure Appl. Chem. 2008, 80, 873-879.
132. H. Pfeiffer, A. Rojas, J. Niesel, U. Schatzschneider, Dalton Trans. 2009, 4292-4298.
133. K.M.-C. Wong, X. Zhu, L.-L. Hung, N. Zhu, V.W.-W. Yam, H.-S. Kwok, Chem.
Commun. 2005, 2906-2908.
134. V.W.-W. Yam, K.M.-C. Wong, L.-L. Hung, N. Zhu, Angew. Chem., Int. Ed. 2005, 44,
3107-3110.
135. S.D. Köster, H. Alborzinia, S. Can, I. Kitanovic, S. Wölfl, R. Rubbiani, I. Ott, P.
Riesterer, A. Prokop, K. Merz, N. Metzler-Nolte, Chem. Sci. 2013, 3, 2062-2072.
136. T.J. Robilotto, N. Deligonul, J.B. Updegraff, III, T.G. Gray, Inorg. Chem. 2013, 52,
9659-9668.
137. F. Collin, I. Sasaki, H. Eury, P. Faller, C. Hureau, Chem. Commun. 2013, 49, 2130-
2132.

Literatur
Seite | 144
138. B.D. Glisic, S. Rajkovic, Z.D. Stanic, M.I. Djuran, Gold Bull. 2011, 44, 91-98.
139. J.P. Hermes, F. Sander, T. Peterle, R. Urbani, T. Pfohl, D. Thompson, M. Mayor,
Chem. - Eur. J. 2011, 17, 13473-13481, S13473/13471-S13473/13421.
140. P. Angelova, E. Solel, G. Parvari, A. Turchanin, M. Botoshansky, A. Goelzhaeuser, E.
Keinan, Langmuir 2013, 29, 2217-2223.
141. M.A. Ivanov, M.V. Puzyk, Russ. J. Gen. Chem. 2001, 71, 1660-1661.
142. C.M. Harris, J. Chem. Soc. 1959, 682-687.
143. S.Y. Quek, L. Venkataraman, H.J. Choi, S.G. Louie, M.S. Hybertsen, J.B. Neaton,
Nano Lett. 2007, 7, 3477-3482.
144. A.H. Pakiari, Z. Jamshidi, J. Phys. Chem. A 2007, 111, 4391-4396.
145. J.M. Hull, M.R. Provorse, C.M. Aikens, J. Phys. Chem. A 2012, 116, 5445-5452.
146. H. De Muynck, A. Madder, N. Farcy, P.J. De Clercq, M.N. Perez-Payan, L.M.
Ohberg, A.P. Davis, Angew. Chem., Int. Ed. 2000, 39, 145-148.
147. A. Clouet, T. Darbre, J.-L. Reymond, Angew. Chem., Int. Ed. 2004, 43, 4612-4615.
148. J. Ryttersgaard, B.D. Larsen, A. Holm, D.H. Christensen, O.F. Nielsen, Spectrochim.
Acta, Part A 1997, 53A, 91-98.
149. J.L. Stair, J.A. Holcombe, Anal. Chem. 2007, 79, 1999-2006.
150. A.A. McConnell, D.H. Brown, W.E. Smith, Spectrochim. Acta, Part A 1982, 38A,
265-270.
151. C.-A.J. Lin, T.-Y. Yang, C.-H. Lee, S.H. Huang, R.A. Sperling, M. Zanella, J.K. Li,
J.-L. Shen, H.-H. Wang, H.-I. Yeh, W.J. Parak, W.H. Chang, ACS Nano 2009, 3, 395-
401.
152. P.L. Goggin, J. Mink, J. Chem. Soc., Dalton Trans. 1974, 1479-1483.
153. G.C. Stocco, R.S. Tobias, J. Amer. Chem. Soc. 1971, 93, 5057-5065.
154. M. Aldhoun, A. Massi, A. Dondoni, J. Org. Chem. 2008, 73, 9565-9575.
155. C. Tang, N. Jiao, J. Am. Chem. Soc. 2012, 134, 18924-18927.
156. H. Bauke Albada, C.J. Arnusch, H.M. Branderhorst, A.-M. Verel, W.T.M. Janssen, E.
Breukink, B. de Kruijff, R.J. Pieters, R.M.J. Liskamp, Bioorg. Med. Chem. Lett. 2009,
19, 3721-3724.
157. E. Moses John, D. Moorhouse Adam, Chem Soc Rev 2007, 36, 1249-1262.
158. W.H. Binder, R. Sachsenhofer, Macromol. Rapid Commun. 2008, 29, 952-981.
159. J.E. Hein, V.V. Fokin, Chem. Soc. Rev. 2010, 39, 1302-1315.
160. S. Punna, J. Kuzelka, Q. Wang, M.G. Finn, Angew. Chem., Int. Ed. 2005, 44, 2215-
2220.
161. J. Choi Won, Z.-D. Shi, M. Worthy Karen, L. Bindu, G. Karki Rajeshri, C. Nicklaus
Marc, J. Fisher Robert, R. Burke Terrence, Jr., Bioorg. Med. Chem. Lett. 2006, 16,
5265-5269.
162. B.W.T. Gruijters, M.A.C. Broeren, F.L. Van Delft, R.P. Sijbesma, P.H.H. Hermkens,
F.P.J.T. Rutjes, Org. Lett. 2006, 8, 3163-3166.
163. J. Dommerholt, S. Schmidt, R. Temming, L.J.A. Hendriks, F.P.J.T. Rutjes, J.C.M. van
Hest, D.J. Lefeber, P. Friedl, F.L. van Delft, Angew. Chem., Int. Ed. 2010, 49, 9422-
9425.
164. E. Kaiser, R.L. Colescott, C.D. Bossinger, P.I. Cook, Anal. Biochem. 1970, 34, 595-
598.
165. A. Esposito, E. Delort, D. Lagnoux, F. Djojo, J.-L. Reymond, Angew. Chem., Int. Ed.
2003, 42, 1381-1383.
166. D.R.W. Hodgson, J.M. Sanderson, Chem. Soc. Rev. 2004, 33, 422-430.
167. G.L. Ellman, Arch. Biochem. Biophys. 1958, 74, 443-450.
168. N. Catozzi, M.G. Edwards, S.A. Raw, P. Wasnaire, R.J.K. Taylor, J. Org. Chem.
2009, 74, 8343-8354.

Literatur
Seite | 145
169. G. Petroselli, M.K. Mandal, L.C. Chen, G.T. Ruiz, E. Wolcan, K. Hiraoka, H.
Nonami, R. Erra-Balsells, J. Mass Spectrom. 2012, 47, 313-321.
170. S. Mounicou, J. Szpunar, R. Lobinski, Chem. Soc. Rev. 2009, 38, 1119-1138.
171. W. Paw, S.D. Cummings, M.A. Mansour, W.B. Connick, D.K. Geiger, R. Eisenberg,
Coord. Chem. Rev. 1998, 171, 125-150.
172. P. Braunstein, R.J.H. Clark, J. Chem. Soc., Dalton Trans. 1973, 1845-1848.
173. P. Braunstein, R.J.H. Clark, Inorg. Chem. 1974, 13, 2224-2229.
174. H.W. Chen, C. Paparizos, J.P. Fackler, Jr., Inorg. Chim. Acta 1985, 96, 137-149.
175. J. Li, J. Hu, G. Li, Catal. Commun. 2011, 12, 1401-1404.
176. C.M. Harris, T.N. Lockyer, J. Chem. Soc. 1959, 3083-3085.
177. M.A. Mansour, R.J. Lachicotte, H.J. Gysling, R. Eisenberg, Inorg. Chem. 1998, 37,
4625-4632.

Anhang
Anhang
Nomenklatur
Die Peptidsequenzen werden mit dem Einbuchstabencode benannt.
Name Einbuchstabencode
Alanin A
2-Aminoisobutansäure Δ
Arginin R
Asparaginsäure D
Azidoessigsäure O
Cystein C
2,3-Diaminopropionsäure Z
Glutaminsäure E
Glycin G
Histidin H
Leucin L
Lysin K
Methionin M
Methyladipoylchlorid Γ
4-Pentinsäure Π
Phenylalanin F
Prolin P
3-(3-Pyridin)-alanin B
3-(4-Pyridin)-alanin J
Thyrosin Y Tab.: Angabe der Namen und des Einbuchstabencodes der verwendeten natürlichen und unnatürlichen Amino-
säuren.
Die Peptide werden immer von der linken Seite beim N-Terminus nach rechts zum
C-Terminus aufgeschrieben. Bei einem acetylierten N-Terminus wird ein „Ac-“ an die linke
Seite zu Beginn geschrieben, bei einem Amid am C-Terminus werden der Endung „-NH2“
angehängt und ein „-MB“ wenn sich die Sequenz nach am Harz befindet. Wenn es sich um
eine Peptidsequenz handelt, die mit AuPhPyCl2 (2) zur Reaktion gebracht worden ist, dann
werden die Aminosäuren, deren Seitengruppen den Komplex gebunden haben sollte, unter-
strichen. Der Sequenz wird ein „AuPhPy“ angehängt. Pentinsäure wird in den Sequenzen mit
einem „Π“ abgekürzt. Azidessigsäure wird mit einem „O“ abgekürzt. Geladen Gold(III)-
Peptidbiokonjugate werden ohne Ladung wiedergegeben.
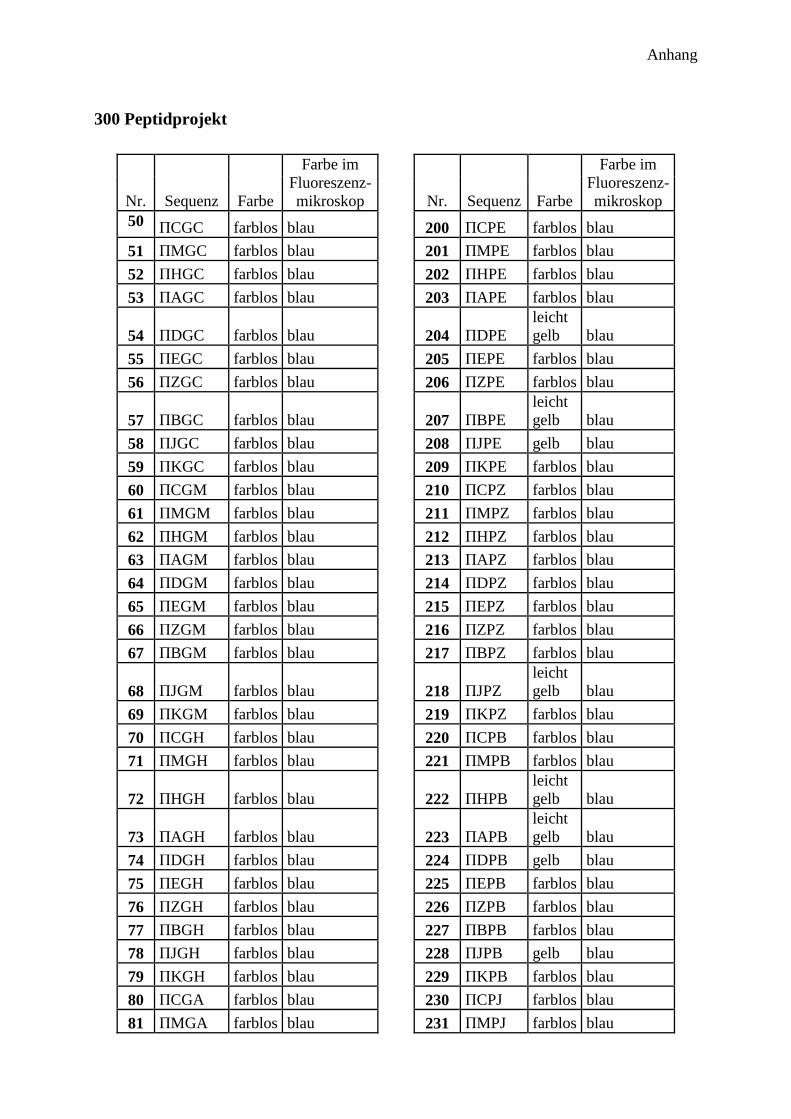
Anhang
300 Peptidprojekt
Nr. Sequenz Farbe
Farbe im
Fluoreszenz-
mikroskop
Nr. Sequenz Farbe
Farbe im
Fluoreszenz-
mikroskop
50 ΠCGC farblos blau 200 ΠCPE farblos blau
51 ΠMGC farblos blau 201 ΠMPE farblos blau
52 ΠHGC farblos blau 202 ΠHPE farblos blau
53 ΠAGC farblos blau 203 ΠAPE farblos blau
54 ΠDGC farblos blau 204 ΠDPE
leicht
gelb blau
55 ΠEGC farblos blau 205 ΠEPE farblos blau
56 ΠZGC farblos blau 206 ΠZPE farblos blau
57 ΠBGC farblos blau 207 ΠBPE
leicht
gelb blau
58 ΠJGC farblos blau 208 ΠJPE gelb blau
59 ΠKGC farblos blau 209 ΠKPE farblos blau
60 ΠCGM farblos blau 210 ΠCPZ farblos blau
61 ΠMGM farblos blau 211 ΠMPZ farblos blau
62 ΠHGM farblos blau 212 ΠHPZ farblos blau
63 ΠAGM farblos blau 213 ΠAPZ farblos blau
64 ΠDGM farblos blau 214 ΠDPZ farblos blau
65 ΠEGM farblos blau 215 ΠEPZ farblos blau
66 ΠZGM farblos blau 216 ΠZPZ farblos blau
67 ΠBGM farblos blau 217 ΠBPZ farblos blau
68 ΠJGM farblos blau 218 ΠJPZ
leicht
gelb blau
69 ΠKGM farblos blau 219 ΠKPZ farblos blau
70 ΠCGH farblos blau 220 ΠCPB farblos blau
71 ΠMGH farblos blau 221 ΠMPB farblos blau
72 ΠHGH farblos blau 222 ΠHPB
leicht
gelb blau
73 ΠAGH farblos blau 223 ΠAPB
leicht
gelb blau
74 ΠDGH farblos blau 224 ΠDPB gelb blau
75 ΠEGH farblos blau 225 ΠEPB farblos blau
76 ΠZGH farblos blau 226 ΠZPB farblos blau
77 ΠBGH farblos blau 227 ΠBPB farblos blau
78 ΠJGH farblos blau 228 ΠJPB gelb blau
79 ΠKGH farblos blau 229 ΠKPB farblos blau
80 ΠCGA farblos blau 230 ΠCPJ farblos blau
81 ΠMGA farblos blau 231 ΠMPJ farblos blau
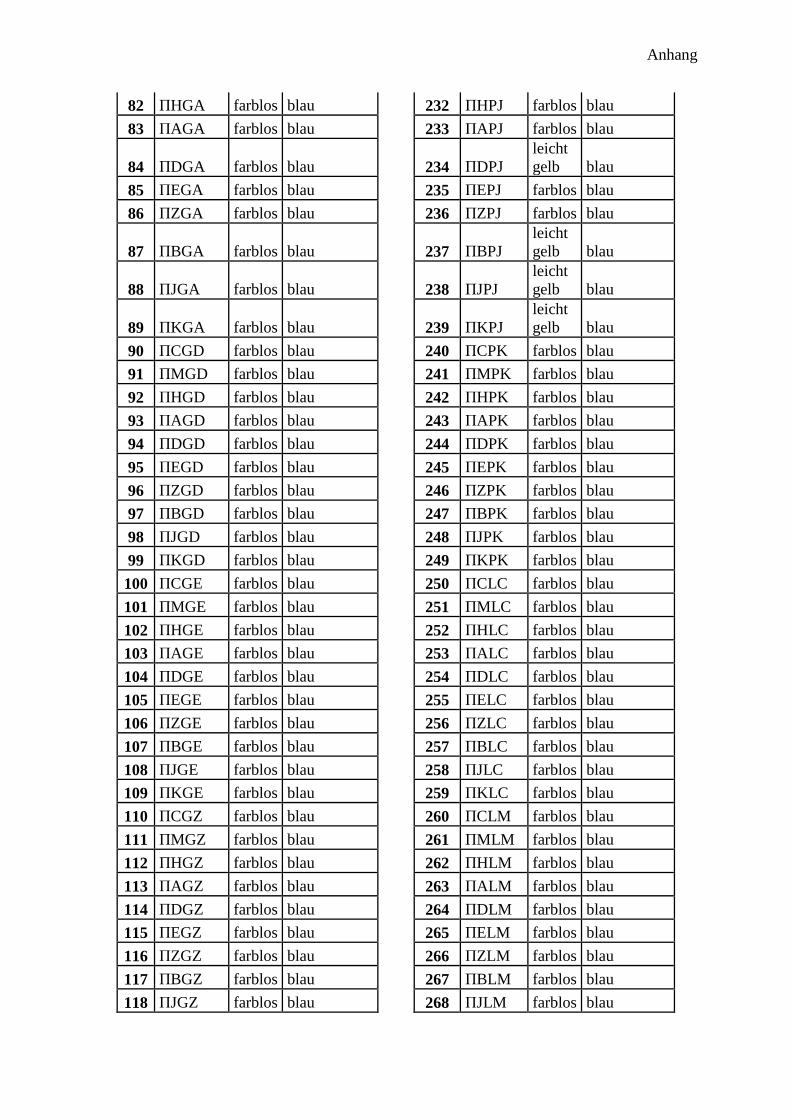
Anhang
82 ΠHGA farblos blau 232 ΠHPJ farblos blau
83 ΠAGA farblos blau 233 ΠAPJ farblos blau
84 ΠDGA farblos blau 234 ΠDPJ
leicht
gelb blau
85 ΠEGA farblos blau 235 ΠEPJ farblos blau
86 ΠZGA farblos blau 236 ΠZPJ farblos blau
87 ΠBGA farblos blau 237 ΠBPJ
leicht
gelb blau
88 ΠJGA farblos blau 238 ΠJPJ
leicht
gelb blau
89 ΠKGA farblos blau 239 ΠKPJ
leicht
gelb blau
90 ΠCGD farblos blau 240 ΠCPK farblos blau
91 ΠMGD farblos blau 241 ΠMPK farblos blau
92 ΠHGD farblos blau 242 ΠHPK farblos blau
93 ΠAGD farblos blau 243 ΠAPK farblos blau
94 ΠDGD farblos blau 244 ΠDPK farblos blau
95 ΠEGD farblos blau 245 ΠEPK farblos blau
96 ΠZGD farblos blau 246 ΠZPK farblos blau
97 ΠBGD farblos blau 247 ΠBPK farblos blau
98 ΠJGD farblos blau 248 ΠJPK farblos blau
99 ΠKGD farblos blau 249 ΠKPK farblos blau
100 ΠCGE farblos blau 250 ΠCLC farblos blau
101 ΠMGE farblos blau 251 ΠMLC farblos blau
102 ΠHGE farblos blau 252 ΠHLC farblos blau
103 ΠAGE farblos blau 253 ΠALC farblos blau
104 ΠDGE farblos blau 254 ΠDLC farblos blau
105 ΠEGE farblos blau 255 ΠELC farblos blau
106 ΠZGE farblos blau 256 ΠZLC farblos blau
107 ΠBGE farblos blau 257 ΠBLC farblos blau
108 ΠJGE farblos blau 258 ΠJLC farblos blau
109 ΠKGE farblos blau 259 ΠKLC farblos blau
110 ΠCGZ farblos blau 260 ΠCLM farblos blau
111 ΠMGZ farblos blau 261 ΠMLM farblos blau
112 ΠHGZ farblos blau 262 ΠHLM farblos blau
113 ΠAGZ farblos blau 263 ΠALM farblos blau
114 ΠDGZ farblos blau 264 ΠDLM farblos blau
115 ΠEGZ farblos blau 265 ΠELM farblos blau
116 ΠZGZ farblos blau 266 ΠZLM farblos blau
117 ΠBGZ farblos blau 267 ΠBLM farblos blau
118 ΠJGZ farblos blau 268 ΠJLM farblos blau
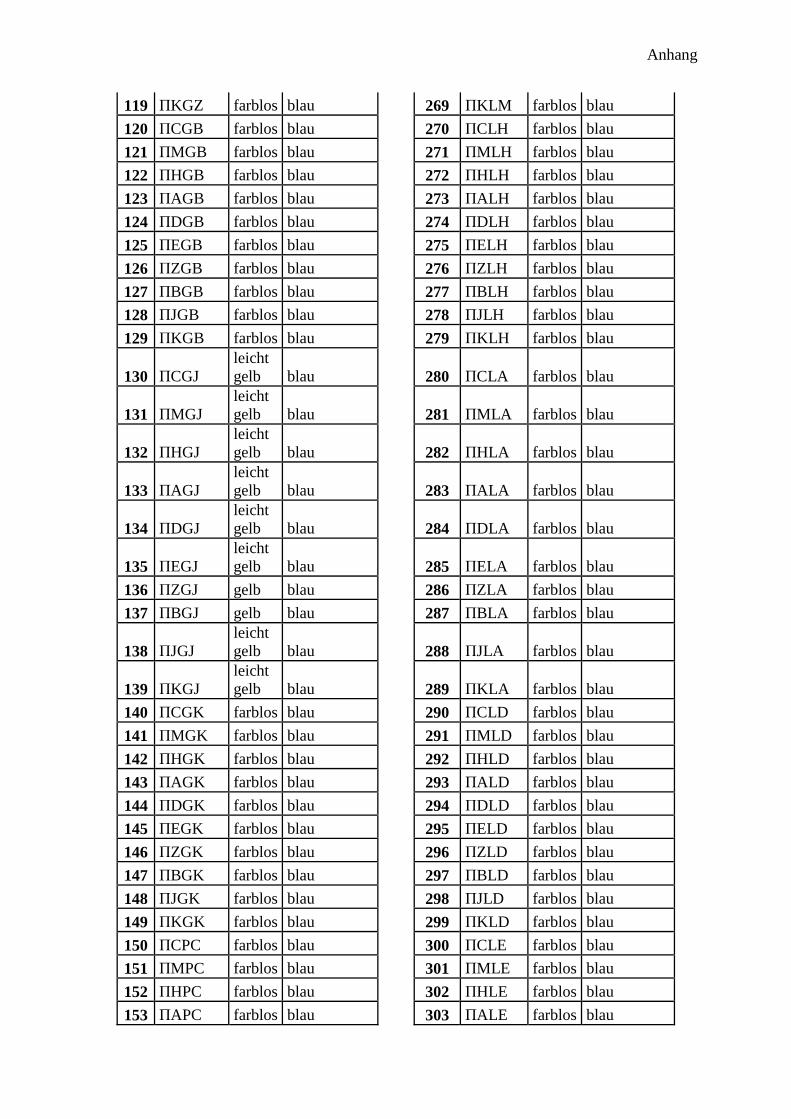
Anhang
119 ΠKGZ farblos blau 269 ΠKLM farblos blau
120 ΠCGB farblos blau 270 ΠCLH farblos blau
121 ΠMGB farblos blau 271 ΠMLH farblos blau
122 ΠHGB farblos blau 272 ΠHLH farblos blau
123 ΠAGB farblos blau 273 ΠALH farblos blau
124 ΠDGB farblos blau 274 ΠDLH farblos blau
125 ΠEGB farblos blau 275 ΠELH farblos blau
126 ΠZGB farblos blau 276 ΠZLH farblos blau
127 ΠBGB farblos blau 277 ΠBLH farblos blau
128 ΠJGB farblos blau 278 ΠJLH farblos blau
129 ΠKGB farblos blau 279 ΠKLH farblos blau
130 ΠCGJ
leicht
gelb blau 280 ΠCLA farblos blau
131 ΠMGJ
leicht
gelb blau 281 ΠMLA farblos blau
132 ΠHGJ
leicht
gelb blau 282 ΠHLA farblos blau
133 ΠAGJ
leicht
gelb blau 283 ΠALA farblos blau
134 ΠDGJ
leicht
gelb blau 284 ΠDLA farblos blau
135 ΠEGJ
leicht
gelb blau 285 ΠELA farblos blau
136 ΠZGJ gelb blau 286 ΠZLA farblos blau
137 ΠBGJ gelb blau 287 ΠBLA farblos blau
138 ΠJGJ
leicht
gelb blau 288 ΠJLA farblos blau
139 ΠKGJ
leicht
gelb blau 289 ΠKLA farblos blau
140 ΠCGK farblos blau 290 ΠCLD farblos blau
141 ΠMGK farblos blau 291 ΠMLD farblos blau
142 ΠHGK farblos blau 292 ΠHLD farblos blau
143 ΠAGK farblos blau 293 ΠALD farblos blau
144 ΠDGK farblos blau 294 ΠDLD farblos blau
145 ΠEGK farblos blau 295 ΠELD farblos blau
146 ΠZGK farblos blau 296 ΠZLD farblos blau
147 ΠBGK farblos blau 297 ΠBLD farblos blau
148 ΠJGK farblos blau 298 ΠJLD farblos blau
149 ΠKGK farblos blau 299 ΠKLD farblos blau
150 ΠCPC farblos blau 300 ΠCLE farblos blau
151 ΠMPC farblos blau 301 ΠMLE farblos blau
152 ΠHPC farblos blau 302 ΠHLE farblos blau
153 ΠAPC farblos blau 303 ΠALE farblos blau
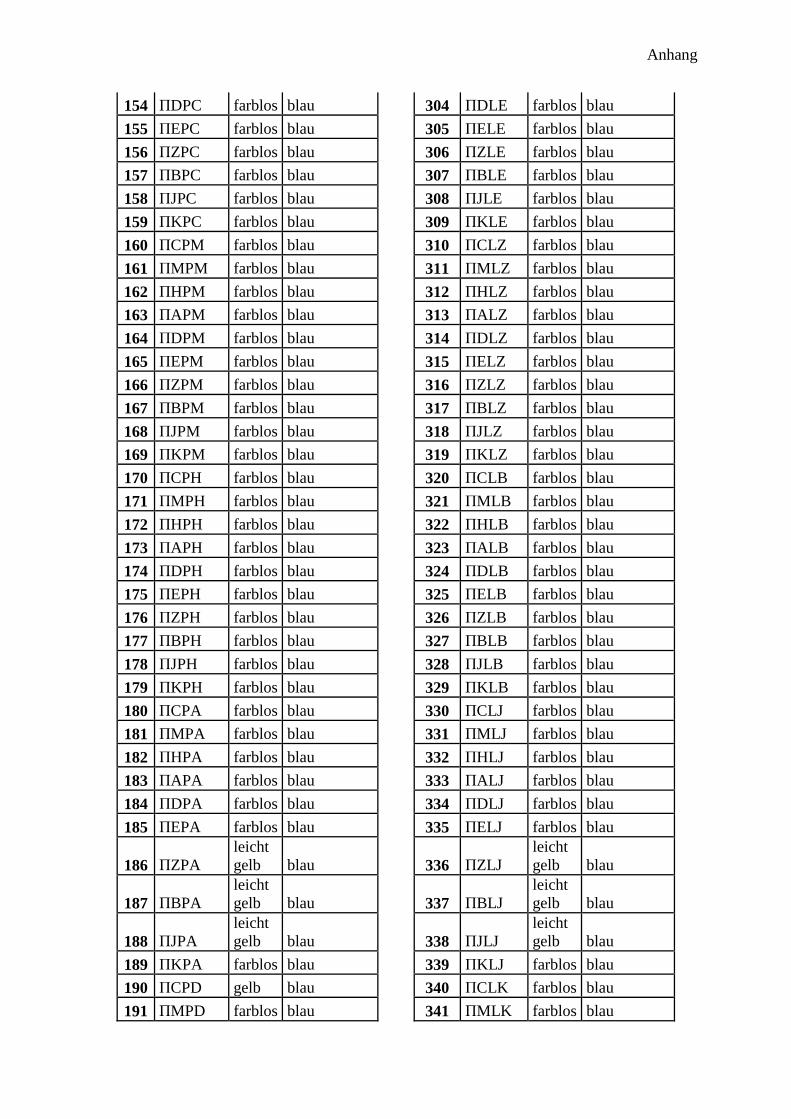
Anhang
154 ΠDPC farblos blau 304 ΠDLE farblos blau
155 ΠEPC farblos blau 305 ΠELE farblos blau
156 ΠZPC farblos blau 306 ΠZLE farblos blau
157 ΠBPC farblos blau 307 ΠBLE farblos blau
158 ΠJPC farblos blau 308 ΠJLE farblos blau
159 ΠKPC farblos blau 309 ΠKLE farblos blau
160 ΠCPM farblos blau 310 ΠCLZ farblos blau
161 ΠMPM farblos blau 311 ΠMLZ farblos blau
162 ΠHPM farblos blau 312 ΠHLZ farblos blau
163 ΠAPM farblos blau 313 ΠALZ farblos blau
164 ΠDPM farblos blau 314 ΠDLZ farblos blau
165 ΠEPM farblos blau 315 ΠELZ farblos blau
166 ΠZPM farblos blau 316 ΠZLZ farblos blau
167 ΠBPM farblos blau 317 ΠBLZ farblos blau
168 ΠJPM farblos blau 318 ΠJLZ farblos blau
169 ΠKPM farblos blau 319 ΠKLZ farblos blau
170 ΠCPH farblos blau 320 ΠCLB farblos blau
171 ΠMPH farblos blau 321 ΠMLB farblos blau
172 ΠHPH farblos blau 322 ΠHLB farblos blau
173 ΠAPH farblos blau 323 ΠALB farblos blau
174 ΠDPH farblos blau 324 ΠDLB farblos blau
175 ΠEPH farblos blau 325 ΠELB farblos blau
176 ΠZPH farblos blau 326 ΠZLB farblos blau
177 ΠBPH farblos blau 327 ΠBLB farblos blau
178 ΠJPH farblos blau 328 ΠJLB farblos blau
179 ΠKPH farblos blau 329 ΠKLB farblos blau
180 ΠCPA farblos blau 330 ΠCLJ farblos blau
181 ΠMPA farblos blau 331 ΠMLJ farblos blau
182 ΠHPA farblos blau 332 ΠHLJ farblos blau
183 ΠAPA farblos blau 333 ΠALJ farblos blau
184 ΠDPA farblos blau 334 ΠDLJ farblos blau
185 ΠEPA farblos blau 335 ΠELJ farblos blau
186 ΠZPA
leicht
gelb blau 336 ΠZLJ
leicht
gelb blau
187 ΠBPA
leicht
gelb blau 337 ΠBLJ
leicht
gelb blau
188 ΠJPA
leicht
gelb blau 338 ΠJLJ
leicht
gelb blau
189 ΠKPA farblos blau 339 ΠKLJ farblos blau
190 ΠCPD gelb blau 340 ΠCLK farblos blau
191 ΠMPD farblos blau 341 ΠMLK farblos blau
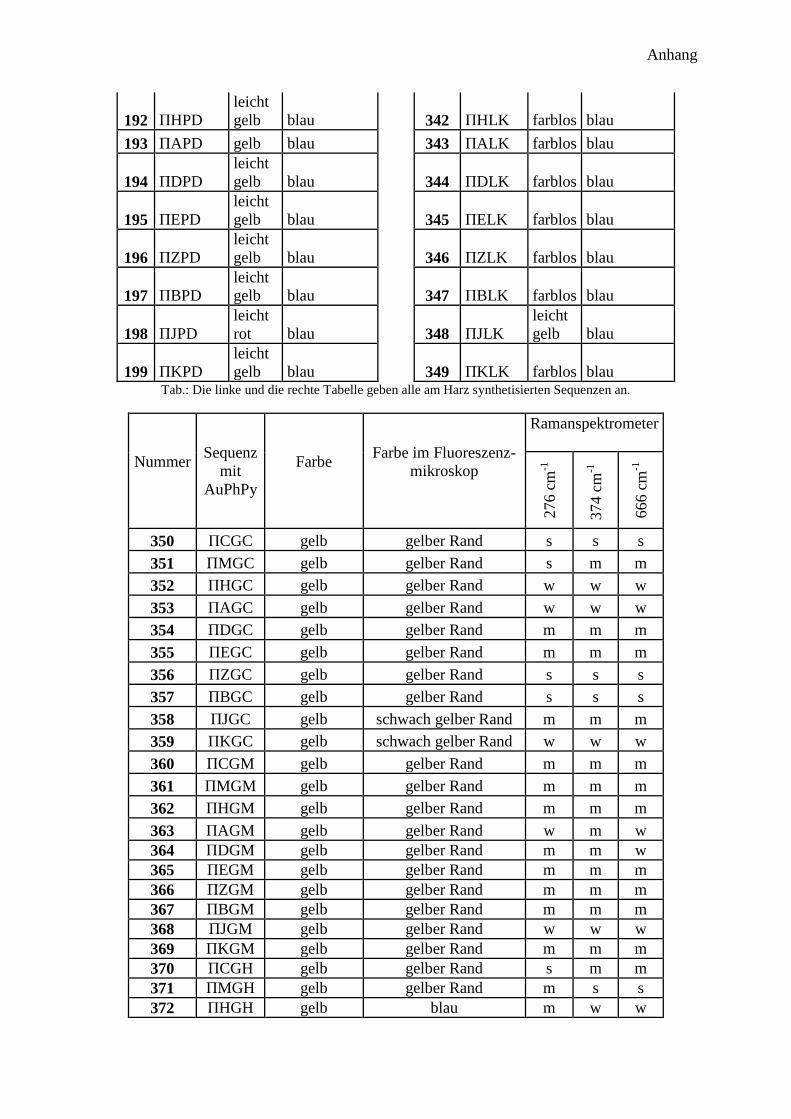
Anhang
192 ΠHPD
leicht
gelb blau 342 ΠHLK farblos blau
193 ΠAPD gelb blau 343 ΠALK farblos blau
194 ΠDPD
leicht
gelb blau 344 ΠDLK farblos blau
195 ΠEPD
leicht
gelb blau 345 ΠELK farblos blau
196 ΠZPD
leicht
gelb blau 346 ΠZLK farblos blau
197 ΠBPD
leicht
gelb blau 347 ΠBLK farblos blau
198 ΠJPD
leicht
rot blau 348 ΠJLK
leicht
gelb blau
199 ΠKPD
leicht
gelb blau 349 ΠKLK farblos blau Tab.: Die linke und die rechte Tabelle geben alle am Harz synthetisierten Sequenzen an.
Nummer
Sequenz
mit
AuPhPy
Farbe
Farbe im Fluoreszenz-
mikroskop
Ramanspektrometer
276 c
m-1
374 c
m-1
666 c
m-1
350 ΠCGC gelb gelber Rand s s s
351 ΠMGC gelb gelber Rand s m m
352 ΠHGC gelb gelber Rand w w w
353 ΠAGC gelb gelber Rand w w w
354 ΠDGC gelb gelber Rand m m m
355 ΠEGC gelb gelber Rand m m m
356 ΠZGC gelb gelber Rand s s s
357 ΠBGC gelb gelber Rand s s s
358 ΠJGC gelb schwach gelber Rand m m m
359 ΠKGC gelb schwach gelber Rand w w w
360 ΠCGM gelb gelber Rand m m m
361 ΠMGM gelb gelber Rand m m m
362 ΠHGM gelb gelber Rand m m m
363 ΠAGM gelb gelber Rand w m w
364 ΠDGM gelb gelber Rand m m w
365 ΠEGM gelb gelber Rand m m m
366 ΠZGM gelb gelber Rand m m m
367 ΠBGM gelb gelber Rand m m m
368 ΠJGM gelb gelber Rand w w w
369 ΠKGM gelb gelber Rand m m m
370 ΠCGH gelb gelber Rand s m m
371 ΠMGH gelb gelber Rand m s s
372 ΠHGH gelb blau m w w
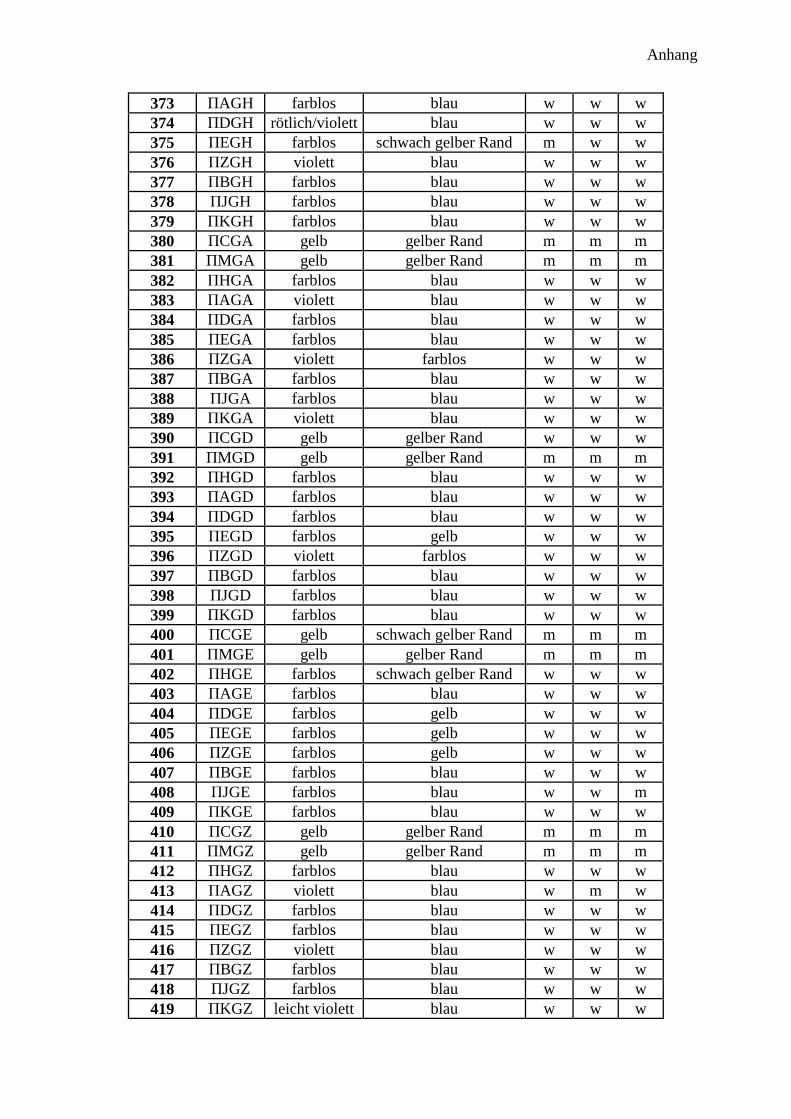
Anhang
373 ΠAGH farblos blau w w w
374 ΠDGH rötlich/violett blau w w w
375 ΠEGH farblos schwach gelber Rand m w w
376 ΠZGH violett blau w w w
377 ΠBGH farblos blau w w w
378 ΠJGH farblos blau w w w
379 ΠKGH farblos blau w w w
380 ΠCGA gelb gelber Rand m m m
381 ΠMGA gelb gelber Rand m m m
382 ΠHGA farblos blau w w w
383 ΠAGA violett blau w w w
384 ΠDGA farblos blau w w w
385 ΠEGA farblos blau w w w
386 ΠZGA violett farblos w w w
387 ΠBGA farblos blau w w w
388 ΠJGA farblos blau w w w
389 ΠKGA violett blau w w w
390 ΠCGD gelb gelber Rand w w w
391 ΠMGD gelb gelber Rand m m m
392 ΠHGD farblos blau w w w
393 ΠAGD farblos blau w w w
394 ΠDGD farblos blau w w w
395 ΠEGD farblos gelb w w w
396 ΠZGD violett farblos w w w
397 ΠBGD farblos blau w w w
398 ΠJGD farblos blau w w w
399 ΠKGD farblos blau w w w
400 ΠCGE gelb schwach gelber Rand m m m
401 ΠMGE gelb gelber Rand m m m
402 ΠHGE farblos schwach gelber Rand w w w
403 ΠAGE farblos blau w w w
404 ΠDGE farblos gelb w w w
405 ΠEGE farblos gelb w w w
406 ΠZGE farblos gelb w w w
407 ΠBGE farblos blau w w w
408 ΠJGE farblos blau w w m
409 ΠKGE farblos blau w w w
410 ΠCGZ gelb gelber Rand m m m
411 ΠMGZ gelb gelber Rand m m m
412 ΠHGZ farblos blau w w w
413 ΠAGZ violett blau w m w
414 ΠDGZ farblos blau w w w
415 ΠEGZ farblos blau w w w
416 ΠZGZ violett blau w w w
417 ΠBGZ farblos blau w w w
418 ΠJGZ farblos blau w w w
419 ΠKGZ leicht violett blau w w w
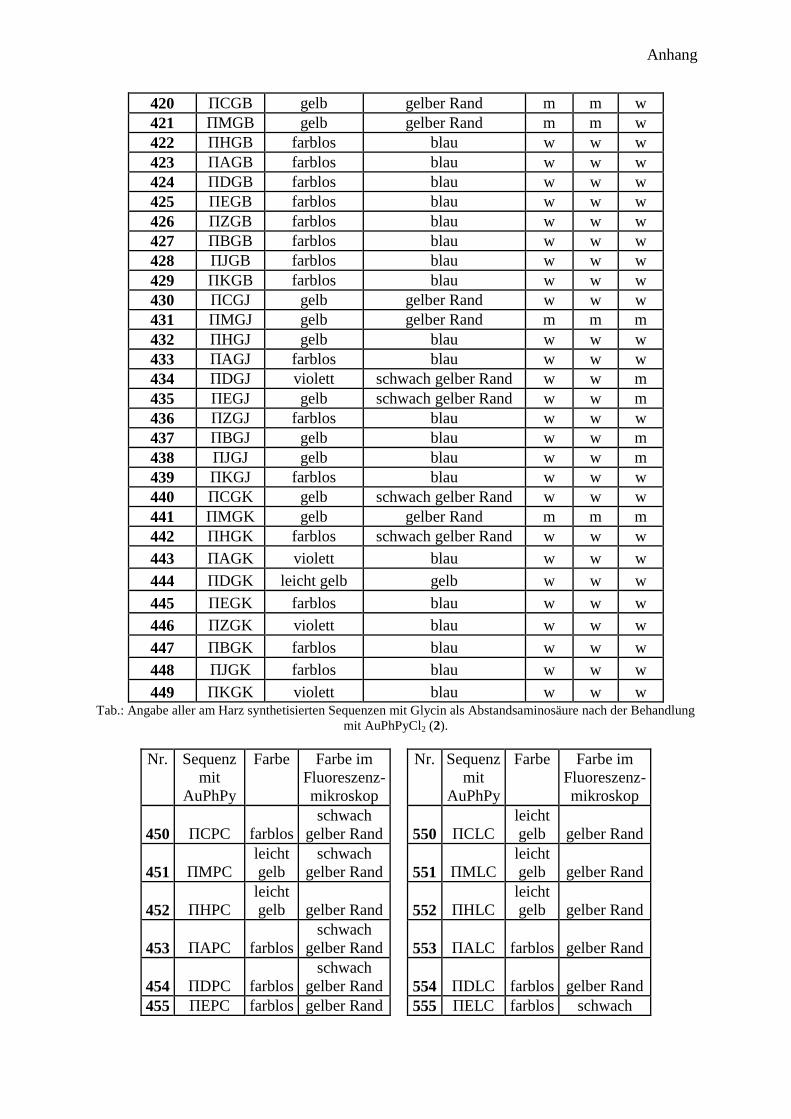
Anhang
420 ΠCGB gelb gelber Rand m m w
421 ΠMGB gelb gelber Rand m m w
422 ΠHGB farblos blau w w w
423 ΠAGB farblos blau w w w
424 ΠDGB farblos blau w w w
425 ΠEGB farblos blau w w w
426 ΠZGB farblos blau w w w
427 ΠBGB farblos blau w w w
428 ΠJGB farblos blau w w w
429 ΠKGB farblos blau w w w
430 ΠCGJ gelb gelber Rand w w w
431 ΠMGJ gelb gelber Rand m m m
432 ΠHGJ gelb blau w w w
433 ΠAGJ farblos blau w w w
434 ΠDGJ violett schwach gelber Rand w w m
435 ΠEGJ gelb schwach gelber Rand w w m
436 ΠZGJ farblos blau w w w
437 ΠBGJ gelb blau w w m
438 ΠJGJ gelb blau w w m
439 ΠKGJ farblos blau w w w
440 ΠCGK gelb schwach gelber Rand w w w
441 ΠMGK gelb gelber Rand m m m
442 ΠHGK farblos schwach gelber Rand w w w
443 ΠAGK violett blau w w w
444 ΠDGK leicht gelb gelb w w w
445 ΠEGK farblos blau w w w
446 ΠZGK violett blau w w w
447 ΠBGK farblos blau w w w
448 ΠJGK farblos blau w w w
449 ΠKGK violett blau w w w Tab.: Angabe aller am Harz synthetisierten Sequenzen mit Glycin als Abstandsaminosäure nach der Behandlung
mit AuPhPyCl2 (2).
Nr. Sequenz
mit
AuPhPy
Farbe Farbe im
Fluoreszenz-
mikroskop
Nr. Sequenz
mit
AuPhPy
Farbe Farbe im
Fluoreszenz-
mikroskop
450 ΠCPC farblos
schwach
gelber Rand 550 ΠCLC
leicht
gelb gelber Rand
451 ΠMPC
leicht
gelb
schwach
gelber Rand 551 ΠMLC
leicht
gelb gelber Rand
452 ΠHPC
leicht
gelb gelber Rand 552 ΠHLC
leicht
gelb gelber Rand
453 ΠAPC farblos
schwach
gelber Rand 553 ΠALC farblos gelber Rand
454 ΠDPC farblos
schwach
gelber Rand 554 ΠDLC farblos gelber Rand
455 ΠEPC farblos gelber Rand 555 ΠELC farblos schwach
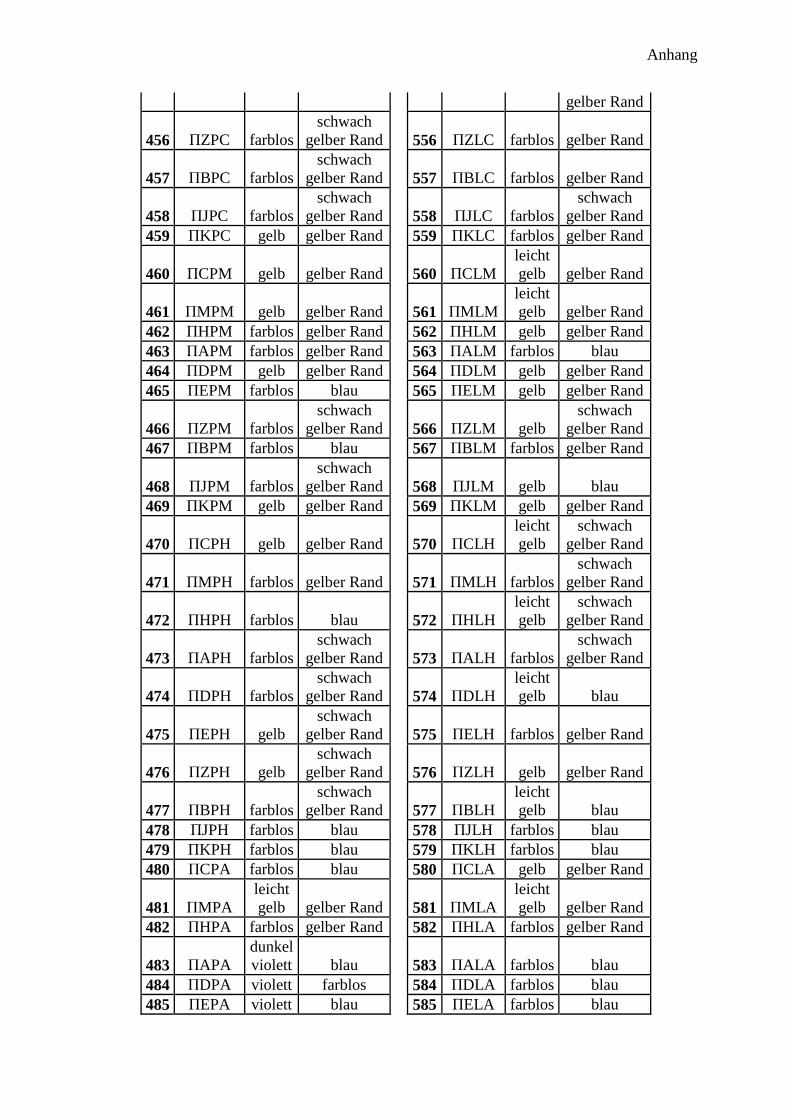
Anhang
gelber Rand
456 ΠZPC farblos
schwach
gelber Rand 556 ΠZLC farblos gelber Rand
457 ΠBPC farblos
schwach
gelber Rand 557 ΠBLC farblos gelber Rand
458 ΠJPC farblos
schwach
gelber Rand 558 ΠJLC farblos
schwach
gelber Rand
459 ΠKPC gelb gelber Rand 559 ΠKLC farblos gelber Rand
460 ΠCPM gelb gelber Rand 560 ΠCLM
leicht
gelb gelber Rand
461 ΠMPM gelb gelber Rand 561 ΠMLM
leicht
gelb gelber Rand
462 ΠHPM farblos gelber Rand 562 ΠHLM gelb gelber Rand
463 ΠAPM farblos gelber Rand 563 ΠALM farblos blau
464 ΠDPM gelb gelber Rand 564 ΠDLM gelb gelber Rand
465 ΠEPM farblos blau 565 ΠELM gelb gelber Rand
466 ΠZPM farblos
schwach
gelber Rand 566 ΠZLM gelb
schwach
gelber Rand
467 ΠBPM farblos blau 567 ΠBLM farblos gelber Rand
468 ΠJPM farblos
schwach
gelber Rand 568 ΠJLM gelb blau
469 ΠKPM gelb gelber Rand 569 ΠKLM gelb gelber Rand
470 ΠCPH gelb gelber Rand 570 ΠCLH
leicht
gelb
schwach
gelber Rand
471 ΠMPH farblos gelber Rand 571 ΠMLH farblos
schwach
gelber Rand
472 ΠHPH farblos blau 572 ΠHLH
leicht
gelb
schwach
gelber Rand
473 ΠAPH farblos
schwach
gelber Rand 573 ΠALH farblos
schwach
gelber Rand
474 ΠDPH farblos
schwach
gelber Rand 574 ΠDLH
leicht
gelb blau
475 ΠEPH gelb
schwach
gelber Rand 575 ΠELH farblos gelber Rand
476 ΠZPH gelb
schwach
gelber Rand 576 ΠZLH gelb gelber Rand
477 ΠBPH farblos
schwach
gelber Rand 577 ΠBLH
leicht
gelb blau
478 ΠJPH farblos blau 578 ΠJLH farblos blau
479 ΠKPH farblos blau 579 ΠKLH farblos blau
480 ΠCPA farblos blau 580 ΠCLA gelb gelber Rand
481 ΠMPA
leicht
gelb gelber Rand 581 ΠMLA
leicht
gelb gelber Rand
482 ΠHPA farblos gelber Rand 582 ΠHLA farblos gelber Rand
483 ΠAPA
dunkel
violett blau 583 ΠALA farblos blau
484 ΠDPA violett farblos 584 ΠDLA farblos blau
485 ΠEPA violett blau 585 ΠELA farblos blau
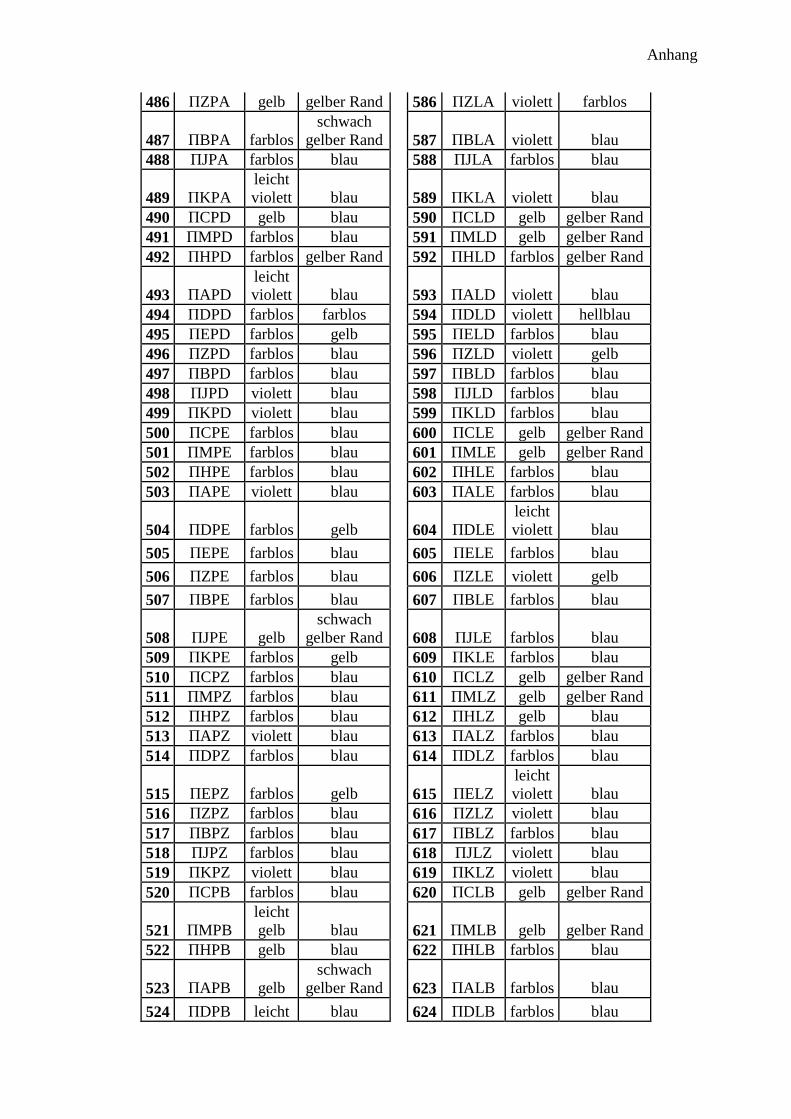
Anhang
486 ΠZPA gelb gelber Rand 586 ΠZLA violett farblos
487 ΠBPA farblos
schwach
gelber Rand 587 ΠBLA violett blau
488 ΠJPA farblos blau 588 ΠJLA farblos blau
489 ΠKPA
leicht
violett blau 589 ΠKLA violett blau
490 ΠCPD gelb blau 590 ΠCLD gelb gelber Rand
491 ΠMPD farblos blau 591 ΠMLD gelb gelber Rand
492 ΠHPD farblos gelber Rand 592 ΠHLD farblos gelber Rand
493 ΠAPD
leicht
violett blau 593 ΠALD violett blau
494 ΠDPD farblos farblos 594 ΠDLD violett hellblau
495 ΠEPD farblos gelb 595 ΠELD farblos blau
496 ΠZPD farblos blau 596 ΠZLD violett gelb
497 ΠBPD farblos blau 597 ΠBLD farblos blau
498 ΠJPD violett blau 598 ΠJLD farblos blau
499 ΠKPD violett blau 599 ΠKLD farblos blau
500 ΠCPE farblos blau 600 ΠCLE gelb gelber Rand
501 ΠMPE farblos blau 601 ΠMLE gelb gelber Rand
502 ΠHPE farblos blau 602 ΠHLE farblos blau
503 ΠAPE violett blau 603 ΠALE farblos blau
504 ΠDPE farblos gelb 604 ΠDLE
leicht
violett blau
505 ΠEPE farblos blau 605 ΠELE farblos blau
506 ΠZPE farblos blau 606 ΠZLE violett gelb
507 ΠBPE farblos blau 607 ΠBLE farblos blau
508 ΠJPE gelb
schwach
gelber Rand 608 ΠJLE farblos blau
509 ΠKPE farblos gelb 609 ΠKLE farblos blau
510 ΠCPZ farblos blau 610 ΠCLZ gelb gelber Rand
511 ΠMPZ farblos blau 611 ΠMLZ gelb gelber Rand
512 ΠHPZ farblos blau 612 ΠHLZ gelb blau
513 ΠAPZ violett blau 613 ΠALZ farblos blau
514 ΠDPZ farblos blau 614 ΠDLZ farblos blau
515 ΠEPZ farblos gelb 615 ΠELZ
leicht
violett blau
516 ΠZPZ farblos blau 616 ΠZLZ violett blau
517 ΠBPZ farblos blau 617 ΠBLZ farblos blau
518 ΠJPZ farblos blau 618 ΠJLZ violett blau
519 ΠKPZ violett blau 619 ΠKLZ violett blau
520 ΠCPB farblos blau 620 ΠCLB gelb gelber Rand
521 ΠMPB
leicht
gelb blau 621 ΠMLB gelb gelber Rand
522 ΠHPB gelb blau 622 ΠHLB farblos blau
523 ΠAPB gelb
schwach
gelber Rand 623 ΠALB farblos blau
524 ΠDPB leicht blau 624 ΠDLB farblos blau
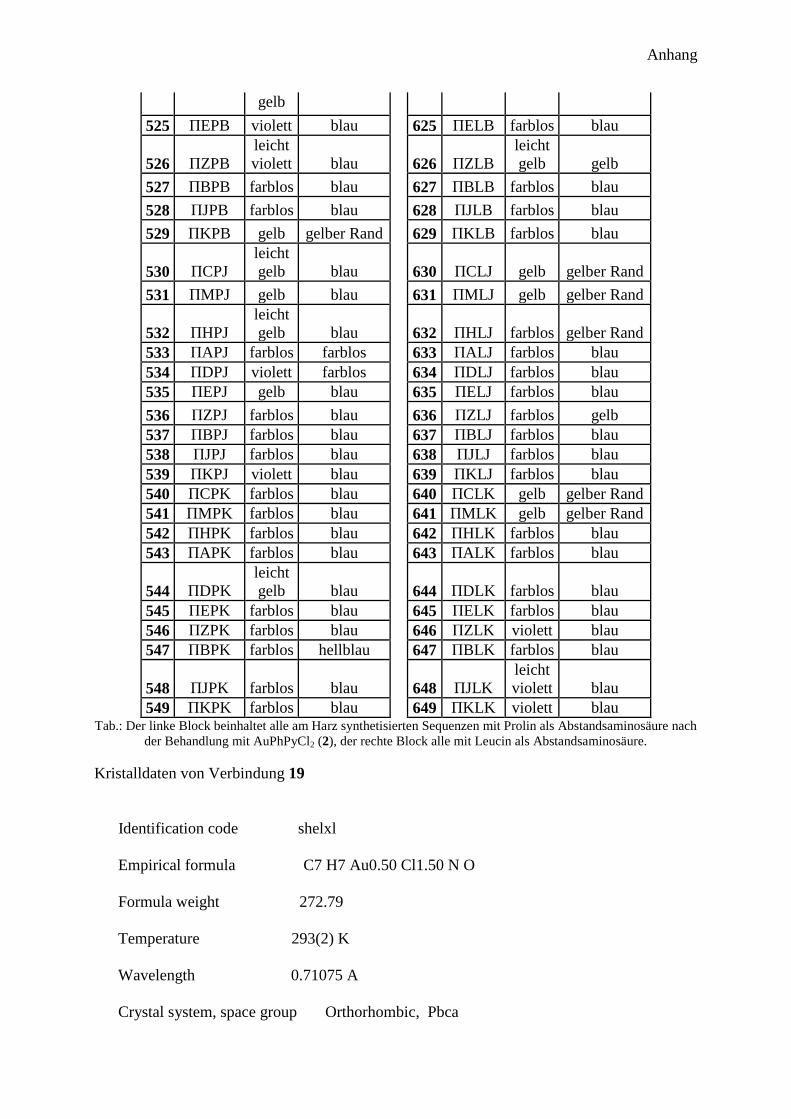
Anhang
gelb
525 ΠEPB violett blau 625 ΠELB farblos blau
526 ΠZPB
leicht
violett blau 626 ΠZLB
leicht
gelb gelb
527 ΠBPB farblos blau 627 ΠBLB farblos blau
528 ΠJPB farblos blau 628 ΠJLB farblos blau
529 ΠKPB gelb gelber Rand 629 ΠKLB farblos blau
530 ΠCPJ
leicht
gelb blau 630 ΠCLJ gelb gelber Rand
531 ΠMPJ gelb blau 631 ΠMLJ gelb gelber Rand
532 ΠHPJ
leicht
gelb blau 632 ΠHLJ farblos gelber Rand
533 ΠAPJ farblos farblos 633 ΠALJ farblos blau
534 ΠDPJ violett farblos 634 ΠDLJ farblos blau
535 ΠEPJ gelb blau 635 ΠELJ farblos blau
536 ΠZPJ farblos blau 636 ΠZLJ farblos gelb
537 ΠBPJ farblos blau 637 ΠBLJ farblos blau
538 ΠJPJ farblos blau 638 ΠJLJ farblos blau
539 ΠKPJ violett blau 639 ΠKLJ farblos blau
540 ΠCPK farblos blau 640 ΠCLK gelb gelber Rand
541 ΠMPK farblos blau 641 ΠMLK gelb gelber Rand
542 ΠHPK farblos blau 642 ΠHLK farblos blau
543 ΠAPK farblos blau 643 ΠALK farblos blau
544 ΠDPK
leicht
gelb blau 644 ΠDLK farblos blau
545 ΠEPK farblos blau 645 ΠELK farblos blau
546 ΠZPK farblos blau 646 ΠZLK violett blau
547 ΠBPK farblos hellblau 647 ΠBLK farblos blau
548 ΠJPK farblos blau 648 ΠJLK
leicht
violett blau
549 ΠKPK farblos blau 649 ΠKLK violett blau Tab.: Der linke Block beinhaltet alle am Harz synthetisierten Sequenzen mit Prolin als Abstandsaminosäure nach
der Behandlung mit AuPhPyCl2 (2), der rechte Block alle mit Leucin als Abstandsaminosäure.
Kristalldaten von Verbindung 19
Identification code shelxl
Empirical formula C7 H7 Au0.50 Cl1.50 N O
Formula weight 272.79
Temperature 293(2) K
Wavelength 0.71075 A
Crystal system, space group Orthorhombic, Pbca

Anhang
Unit cell dimensions a = 7.436(6) A alpha = 90 deg.
b = 19.681(15) A beta = 90 deg.
c = 23.366(17) A gamma = 90 deg.
Volume 3420(5) A^3
Z, Calculated density 16, 2.120 Mg/m^3
Absorption coefficient 9.079 mm^-1
F(000) 2064
Crystal size 0.15 x 0.12 x 0.05 mm
Theta range for data collection 2.71 to 25.00 deg.
Limiting indices -8<=h<=8, -23<=k<=23, -27<=l<=27
Reflections collected / unique 27239 / 3002 [R(int) = 0.0910]
Completeness to theta = 25.00 99.8 %
Absorption correction Empirical
Max. and min. transmission 0.6596 and 0.3429
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 3002 / 0 / 200
Goodness-of-fit on F^2 1.129
Final R indices [I>2sigma(I)] R1 = 0.0418, wR2 = 0.0863
R indices (all data) R1 = 0.0722, wR2 = 0.1010
Extinction coefficient 0.00007(4)
Largest diff. peak and hole 0.965 and -1.135 e.A^-3
Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic
displacement parameters (A^2 x 10^3) for shelxl.
U(eq) is defined as one third of the trace of the orthogonalized
Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________

Anhang
Au(1) 7502(1) 3295(1) 773(1) 39(1)
Cl(1) 7540(4) 2259(1) 1205(1) 65(1)
Cl(2) 10117(3) 3588(2) 1227(1) 58(1)
Cl(3) 4985(4) 3004(1) 270(1) 73(1)
O(1) 6460(12) 5601(4) 3195(3) 73(2)
O(2) 7627(11) 6232(3) 2499(3) 61(2)
N(1) 7491(10) 4245(3) 376(3) 34(2)
N(23) 5647(9) 4176(3) 1438(3) 39(2)
C(14) 8024(17) 6756(5) 2897(5) 75(4)
C(2) 7942(11) 4900(5) -462(4) 41(2)
C(4) 6944(12) 5425(5) 396(4) 41(2)
C(11) 6484(11) 4751(4) 1285(3) 34(2)
C(1) 7988(12) 4283(5) -172(4) 42(2)
C(6) 8642(16) 3652(5) -474(4) 60(3)
C(9) 6375(11) 5171(5) 2239(3) 36(2)
C(8) 5523(11) 4578(5) 2398(4) 41(2)
C(13) 6805(13) 5679(5) 2708(4) 45(2)
C(3) 7426(12) 5478(4) -165(3) 43(2)
C(5) 6986(11) 4821(5) 680(3) 40(2)
C(10) 6874(12) 5259(5) 1679(4) 40(2)
C(12) 4272(13) 3420(5) 2130(4) 51(3)
C(7) 5173(12) 4087(4) 1991(3) 37(2)
________________________________________________________________
Table 3. Bond lengths [A] and angles [deg] for shelxl.
_____________________________________________________________
Au(1)-N(1) 2.088(6)
Au(1)-Cl(1) 2.275(3)
Au(1)-Cl(3) 2.283(3)
Au(1)-Cl(2) 2.289(3)
O(1)-C(13) 1.178(10)
O(2)-C(13) 1.339(11)
O(2)-C(14) 1.419(12)
N(1)-C(1) 1.335(11)
N(1)-C(5) 1.388(11)
N(23)-C(11) 1.340(10)
N(23)-C(7) 1.352(10)
C(14)-H(14A) 0.9600
C(14)-H(14B) 0.9600
C(14)-H(14C) 0.9600
C(2)-C(3) 1.388(12)
C(2)-C(1) 1.391(12)
C(2)-H(2A) 0.9300
C(4)-C(5) 1.363(12)
C(4)-C(3) 1.364(11)
C(4)-H(4A) 0.9300
C(11)-C(10) 1.388(11)
C(11)-C(5) 1.468(10)
C(1)-C(6) 1.510(12)
C(6)-H(6A) 0.9600

Anhang
C(6)-H(6B) 0.9600
C(6)-H(6C) 0.9600
C(9)-C(10) 1.371(11)
C(9)-C(8) 1.379(12)
C(9)-C(13) 1.517(12)
C(8)-C(7) 1.379(12)
C(8)-H(8A) 0.9300
C(3)-H(3A) 0.9300
C(10)-H(10A) 0.9300
C(12)-C(7) 1.510(12)
C(12)-H(12A) 0.9600
C(12)-H(12B) 0.9600
C(12)-H(12C) 0.9600
N(1)-Au(1)-Cl(1) 179.5(2)
N(1)-Au(1)-Cl(3) 89.6(2)
Cl(1)-Au(1)-Cl(3) 90.78(11)
N(1)-Au(1)-Cl(2) 89.1(2)
Cl(1)-Au(1)-Cl(2) 90.55(11)
Cl(3)-Au(1)-Cl(2) 176.58(10)
C(13)-O(2)-C(14) 116.5(8)
C(1)-N(1)-C(5) 121.3(7)
C(1)-N(1)-Au(1) 118.4(5)
C(5)-N(1)-Au(1) 120.3(5)
C(11)-N(23)-C(7) 119.0(7)
O(2)-C(14)-H(14A) 109.5
O(2)-C(14)-H(14B) 109.5
H(14A)-C(14)-H(14B) 109.5
O(2)-C(14)-H(14C) 109.5
H(14A)-C(14)-H(14C) 109.5
H(14B)-C(14)-H(14C) 109.5
C(3)-C(2)-C(1) 118.6(8)
C(3)-C(2)-H(2A) 120.7
C(1)-C(2)-H(2A) 120.7
C(5)-C(4)-C(3) 122.0(9)
C(5)-C(4)-H(4A) 119.0
C(3)-C(4)-H(4A) 119.0
N(23)-C(11)-C(10) 121.9(8)
N(23)-C(11)-C(5) 116.9(8)
C(10)-C(11)-C(5) 121.2(8)
N(1)-C(1)-C(2) 120.7(8)
N(1)-C(1)-C(6) 119.5(8)
C(2)-C(1)-C(6) 119.8(8)
C(1)-C(6)-H(6A) 109.5
C(1)-C(6)-H(6B) 109.5
H(6A)-C(6)-H(6B) 109.5
C(1)-C(6)-H(6C) 109.5
H(6A)-C(6)-H(6C) 109.5
H(6B)-C(6)-H(6C) 109.5
C(10)-C(9)-C(8) 119.2(8)
C(10)-C(9)-C(13) 123.3(9)
Anhang
C(8)-C(9)-C(13) 117.4(8)
C(9)-C(8)-C(7) 119.6(8)
C(9)-C(8)-H(8A) 120.2
C(7)-C(8)-H(8A) 120.2
O(1)-C(13)-O(2) 123.9(9)
O(1)-C(13)-C(9) 124.5(10)
O(2)-C(13)-C(9) 111.7(8)
C(4)-C(3)-C(2) 119.4(8)
C(4)-C(3)-H(3A) 120.3
C(2)-C(3)-H(3A) 120.3
C(4)-C(5)-N(1) 118.0(7)
C(4)-C(5)-C(11) 122.9(9)
N(1)-C(5)-C(11) 119.1(8)
C(9)-C(10)-C(11) 119.0(8)
C(9)-C(10)-H(10A) 120.5
C(11)-C(10)-H(10A) 120.5
C(7)-C(12)-H(12A) 109.5
C(7)-C(12)-H(12B) 109.5
H(12A)-C(12)-H(12B) 109.5
C(7)-C(12)-H(12C) 109.5
H(12A)-C(12)-H(12C) 109.5
H(12B)-C(12)-H(12C) 109.5
N(23)-C(7)-C(8) 121.3(8)
N(23)-C(7)-C(12) 115.7(7)
C(8)-C(7)-C(12) 123.0(8)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
Table 4. Anisotropic displacement parameters (A^2 x 10^3) for shelxl.
The anisotropic displacement factor exponent takes the form:
-2 pi^2 [ h^2 a*^2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
Au(1) 53(1) 26(1) 38(1) 2(1) -5(1) 3(1)
Cl(1) 93(2) 31(1) 71(2) 15(1) -5(2) 10(2)
Cl(2) 57(2) 72(2) 46(1) 8(1) -14(1) -5(1)
Cl(3) 80(2) 36(1) 103(2) -2(2) -42(2) -7(2)
O(1) 124(7) 64(5) 30(4) -3(3) 5(4) -14(5)
O(2) 93(5) 35(4) 54(4) -12(3) 9(4) -5(5)
N(1) 50(4) 19(3) 34(4) -5(3) -11(4) 9(4)
N(23) 45(4) 26(4) 45(4) -2(3) 2(4) -7(3)
C(14) 110(10) 36(6) 78(8) -8(5) 29(7) -11(6)
C(2) 50(6) 35(5) 37(5) 7(4) 0(4) -3(4)
C(4) 56(6) 24(5) 42(5) 5(4) 6(4) 10(4)
C(11) 36(5) 30(5) 35(5) -4(4) -4(4) 7(4)
C(1) 60(6) 36(5) 29(5) -2(4) -5(4) 6(4)
Anhang
C(6) 92(8) 46(7) 42(6) -7(5) 10(6) 13(6)
C(9) 42(5) 38(5) 30(4) -5(4) 6(4) 7(4)
C(8) 42(5) 40(6) 41(5) 3(4) 3(4) 2(4)
C(13) 52(5) 47(6) 36(5) 1(4) -5(4) 8(5)
C(3) 58(6) 30(5) 40(5) 5(4) -4(5) 3(5)
C(5) 36(5) 55(7) 29(4) 14(4) 0(3) 15(4)
C(10) 43(5) 35(6) 41(5) 4(4) 8(4) -2(4)
C(12) 60(6) 51(6) 42(5) 4(4) 3(5) -13(5)
C(7) 42(5) 30(5) 37(5) 8(4) -1(4) 4(4)
_______________________________________________________________________
Anhang
Table 5. Hydrogen coordinates ( x 10^4) and isotropic
displacement parameters (A^2 x 10^3) for shelxl.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(14A) 8608 7124 2703 112
H(14B) 8804 6582 3190 112
H(14C) 6928 6915 3068 112
H(2A) 8250 4923 -847 49
H(4A) 6575 5813 591 49
H(6A) 8591 3273 -216 90
H(6B) 9860 3718 -598 90
H(6C) 7891 3563 -800 90
H(8A) 5187 4509 2777 49
H(3A) 7409 5899 -348 51
H(10A) 7466 5652 1564 47
H(12A) 4158 3155 1788 76
H(12B) 3100 3505 2287 76
H(12C) 4985 3177 2405 76
________________________________________________________________


















