
Pharmakokinetische und pharmakodynamische Untersuchungen ... · Pharmakodynamik von Naproxen...
123
Pharmakokinetische und pharmakodynamische Untersuchungen zu selektiven und nicht selektiven Cyclooxygenasehemmern Den naturwissenschaftlichen Fakultäten der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades vorgelegt von Olga Cheremina aus Kysyl / Russland
-
Upload
dangkhuong -
Category
Documents
-
view
231 -
download
0
Transcript of Pharmakokinetische und pharmakodynamische Untersuchungen ... · Pharmakodynamik von Naproxen...

Pharmakokinetische und pharmakodynamische
Untersuchungen zu selektiven und nicht selektiven
Cyclooxygenasehemmern
Den naturwissenschaftlichen Fakultäten
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
vorgelegt von
Olga Cheremina
aus Kysyl / Russland

Als Dissertation genehmigt von den Naturwissenschaftlichen Fakultäten der Universität
Erlangen-Nürnberg
Tag der mündlichen Prüfung: 19.06.09
Vorsitzender
der Promotionskommission: Professor Dr. E. Bänsch
Erstberichterstatter: Professor Dr. R. Troschütz
Zweitberichterstatter: Professor Dr. Dr. K. Brune

Für meinen Sohn

Inhaltverzeichnis 1
Inhaltverzeichnis
Inhaltverzeichnis.......................................................................................................................1
Abkürzungsverzeichnis............................................................................................................6
1 Zusammenfassung ...........................................................................................................7
2 Summary .........................................................................................................................10
3 Einleitung.........................................................................................................................13
3.1 Cyclooxygenase ........................................................................................................14
3.2 Isoformen der COX ...................................................................................................15
3.3 Produkte des Cyclooxygenaseweges: Prostaglandine und Thromboxane.................16
3.3.1 Prostaglandin E2 ................................................................................................16
3.3.2 Thromboxan B2..................................................................................................17
3.4 Hemmung der Prostaglandinsynthese als Wirkungsmechanismus antipyretischer
Analgetika.............................................................................................................................17
3.4.1 Saure Antipyretika .............................................................................................17
3.4.2 Nicht saure Antipyretika.....................................................................................18
3.4.2.1 Wirkungmechanismus von Metamizol ........................................................19
3.4.2.2 Wirkungmechanismus von Paracetamol ....................................................22
3.5 Cyclooxygenasehemmer ...........................................................................................24
3.5.1 Nicht selektive Cyclooxygenasehemmer............................................................25
3.5.1.1 Thrombozytenhemmende und potenziell kardioprotektive Wirkung von
Naproxen ..................................................................................................................26
3.5.2 Selektive Cyclooxygenasehemmer ....................................................................27
3.5.2.1 Lumiracoxib ein hochselektiver COX-2-Hemmer........................................29
4 Ziele..................................................................................................................................31
4.1 Untersuchungen zum Wirkungmechanismus von Metamizol .....................................31
4.2 Untersuchungen zur Wirkungsmechanismus von Paracetamol .................................32
4.3 Entwicklung und Validierung einer analytischen Methode zur Bestimmung von
Lumiracoxib in Plasma von menschlichen Probanden ..........................................................32
4.4 Untersuchungen zur COX-2-Hemmung durch Lumiracoxib in verschiedenen
Dosierungen .........................................................................................................................32
4.5 Untersuchung zur thrombozytenhemmenden und potenziell kardioprotektiven Wirkung
von Naproxen .......................................................................................................................32
5 Experimenteller Teil ........................................................................................................34
5.1 Materialien.................................................................................................................34
5.1.1 Lösungen...........................................................................................................34
5.1.2 Chemikalien.......................................................................................................35

Inhaltverzeichnis 2
5.1.3 Verbrauchsmaterialien.......................................................................................36
5.1.4 Geräte ...............................................................................................................37
5.1.5 Software ............................................................................................................38
5.1.6 Medikamente .....................................................................................................38
5.2 Methoden ..................................................................................................................39
5.2.1 Pharmakokinetische Untersuchungen................................................................39
5.2.2 Pharmakodynamische Untersuchungen ............................................................39
5.2.2.1 COX-1-Aktivität im menschlichen Vollblut ..................................................40
5.2.2.2 COX-2-Aktivität im menschlichen Vollblut ..................................................40
5.2.2.3 Quantifizierung der Konzentration von Prostanoid im Vollblut mittels ELISA ..
..................................................................................................................40
5.2.2.3.1 Messung von TXB2 .................................................................................40
5.2.2.3.2 Messung von PGE2 ................................................................................41
5.2.3 Korrelationsanalyse zwischen Pharmakodynamik und Pharmakokinetik............41
5.2.3.1 Pharmakodynamische Analyse ..................................................................41
5.2.3.2 Pharmakokinetische Analyse .....................................................................42
5.2.3.3 Berechnung der Korrelation zwischen Pharmakodynamik und
Pharmakokinetik............................................................................................................42
5.2.4 Validierung einer analytischen Methode ............................................................43
5.2.4.1 Spezifität und Selektivität ...........................................................................43
5.2.4.2 Linearität ....................................................................................................43
5.2.4.3 Nachweis- und Bestimmungsgrenze ..........................................................44
5.2.4.4 Richtigkeit ..................................................................................................44
5.2.4.5 Präzision ....................................................................................................44
5.2.4.6 Wiederfindung (Recovery)..........................................................................45
5.2.4.7 Stabilität .....................................................................................................45
5.3 Studien......................................................................................................................46
5.3.1 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Metamizol.....................................................................................46
5.3.1.1 Studienziel .................................................................................................46
5.3.1.2 Studiendesign ............................................................................................46
5.3.1.3 Probandenpopulation .................................................................................46
5.3.1.4 Studienmedikation .....................................................................................46
5.3.1.5 Studienplan................................................................................................47
5.3.1.6 In-vitro Untersuchungen.............................................................................47
5.3.1.6.1 Wirkungen von Metamizol Metaboliten auf die COX-1 und COX-2-Aktivität
im menschlichen Vollblut ...........................................................................................47

Inhaltverzeichnis 3
5.3.1.7 Ex-vivo Untersuchungen ............................................................................48
5.3.1.7.1 Hemmung der COX-Aktivitäten in Blut nach einmaliger oraler Gabe von
Metamizol ...............................................................................................................48
5.3.1.7.2 Bestimmung von 4-MAA und 4-AA im menschlichen Plasma .................48
5.3.2 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Paracetamol .................................................................................49
5.3.2.1 Studienziel .................................................................................................49
5.3.2.2 Studiendesign ............................................................................................49
5.3.2.3 Probandenpopulation .................................................................................49
5.3.2.4 Studienmedikation .....................................................................................49
5.3.2.5 Studienplan................................................................................................49
5.3.2.6 In vitro Untersuchungen .............................................................................50
5.3.2.6.1 Wirkungen von Paracetamol auf COX-1 und COX-2-Aktivität im
menschlichen Vollblut................................................................................................50
5.3.2.6.2 Effekt von Paracetamol auf COX-2-Aktivität im Human rekombinante
COX-2 ...............................................................................................................50
5.3.2.6.3 Effekt von Paracetamol auf COX-2-Aktivität in humanen Blutmonocyten51
5.3.2.6.4 Effekt von Paracetamol auf die COX-2-Expression in humanen
Blutmonocyten mittels Westernblot-Analyse..............................................................51
5.3.2.7 Ex-vivo Untersuchungen ............................................................................52
5.3.2.7.1 Hemmung der COX-Aktivitäten nach einmaliger oraler Gabe von
Paracetamol 1000 mg ...............................................................................................52
5.3.2.7.2 Bestimmung von Paracetamol in menschlichem Plasma ........................53
5.3.3 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Lumiracoxib..................................................................................54
5.3.3.1 Studienziel .................................................................................................54
5.3.3.2 Studiendesign ............................................................................................54
5.3.3.3 Probandenpopulation .................................................................................54
5.3.3.4 Studienmedikation .....................................................................................54
5.3.3.5 Studienplan................................................................................................54
5.3.3.6 Hemmung der COX-2-Aktivität durch Lumiracoxib ex-vivo .........................55
5.3.3.7 Bestimmung von Lumiracoxib in menschlichem Plasma. ...........................55
5.3.3.8 Statistik ......................................................................................................55
5.3.4 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Naproxen .....................................................................................57
5.3.4.1 Studienziel .................................................................................................57
5.3.4.2 Studiendesign ............................................................................................57

Inhaltverzeichnis 4
5.3.4.3 Probandenpopulation .................................................................................57
5.3.4.4 Studienmedikation .....................................................................................57
5.3.4.5 Studienplan................................................................................................57
5.3.4.6 Hemmung der COX-Aktivitäten durch Naproxen 200mg zweimal am Tag für
7 Tage bei menschlichen Probanden. ...........................................................................58
5.3.4.7 Bestimmung von Naproxen in menschlichem Plasma................................58
6 Ergebnisse.......................................................................................................................60
6.1 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Metamizol ........................................................................................60
6.1.1 In-vitro Wirkungen von 4-MAA und 4-AA auf die COX-1 und COX-2-Aktivität in
menschlichem Vollblut ......................................................................................................60
6.1.2 Ex-vivo Hemmung der COX-1 und COX-2-Aktivität in menschlichem Vollblut
nach einmaliger Einnahme von Metamizol ........................................................................60
6.1.3 Pharmakokinetik von Metamizolmetaboliten nach oraler Gabe von Metamizol in
den Dosierungen von 500 mg und 1000 mg......................................................................62
6.1.4 Korrelation zwischen Plasmakonzentration von 4-MAA und ex-vivo-Hemmung
der COX-Isoformen...........................................................................................................64
6.2 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Paracetamol.....................................................................................66
6.2.1 In-vitro Wirkung von Paracetamol auf die COX-1- und COX-2-Aktivität in
menschlichem Vollblut ......................................................................................................66
6.2.2 Pharmakokinetik von Paracetamol nach einmaliger oraler Gabe von 1000 mg
Paracetamol......................................................................................................................66
6.2.3 Ex-vivo-Hemmung der COX-1 und COX-2-Aktivität in menschlichem Vollblut
nach einmaliger Einnahme von 1000 mg Paracetamol......................................................68
6.2.4 Korrelation zwischen den Plasmakonzentrationen von Paracetamol und den ex-
vivo-Hemmungen von COX-1 und COX-2.........................................................................69
6.2.5 In-vitro-Wirkung von Paracetamol auf die COX-2-Aktivität von humanem
rekombinanten Enzym und Blutmonozyten .......................................................................71
6.2.6 In-vitro-Wirkung von Paracetamol auf die COX-2-Expression ............................71
6.3 Entwicklung und Validierung einer HPLC-UV Methode zur Bestimmung der
Konzentrationen von Lumiracoxib in menschlichem Plasma .................................................73
6.3.1 Methodenentwicklung ........................................................................................73
6.3.1.1 Standardlösungen......................................................................................73
6.3.1.2 Probenaufarbeitung und Messung .............................................................74
6.3.2 Validierung.........................................................................................................75
6.3.2.1 Spezifität, Linearität und Bestimmungsgrenze............................................75

Inhaltverzeichnis 5
6.3.2.2 Repräsentative Chromatogramme .............................................................76
6.3.2.3 Intra-day und - Inter-day-Präzision .............................................................77
6.3.2.4 Wiederfindung............................................................................................78
6.3.2.5 Stabilität .....................................................................................................79
6.4 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Lumiracoxib......................................................................................80
6.4.1 Pharmakokinetik von Lumiracoxib nach oraler Gabe .........................................80
6.4.2 Ex-vivo-Hemmung der COX-2-Aktivität nach oraler Gabe von Lumiracoxib .......82
6.4.3 Korrelation zwischen den Lumiracoxibplasmakonzentrationen und der ex-vivo
COX-2-Hemmung .............................................................................................................84
6.5 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik und
Pharmakodynamik von Naproxen .........................................................................................85
6.5.1 Pharmakokinetik von Naproxen und ex-vivo-Hemmung der COX-1- und COX-2-
Aktivität nach einmaliger oraler Gabe von 220 mg Naproxen Natrium bei gesunden
Probanden ........................................................................................................................85
6.5.2 Pharmakokinetik von Naproxen und ex-vivo-Hemmung der COX-1 und COX-2-
Aktivität nach oraler Gabe von 220 mg Naproxen Natrium im Steady-state.......................86
6.5.3 Korrelation zwischen den Naproxenplasmakonzentrationen und ex-vivo-
Hemmungen der COX-Isoformen......................................................................................88
7 Diskussion.......................................................................................................................90
7.1 Untersuchungen zum Wirkungmechanismus von Metamizol .....................................90
7.2 Untersuchungen zum Wirkungsmechanismus von Paracetamol ...............................94
7.3 Entwicklung und Validierung einer analytischen Methode zur Bestimmung von
Lumiracoxib in Plasma von menschlichen Probanden ..........................................................98
7.4 Untersuchungen zur COX-2-Hemmung durch Lumiracoxib in verschiedenen
Dosierungen .........................................................................................................................99
7.5 Untersuchung zur thrombozytenhemmende und potenziell kardioprotektive Wirkung
von Naproxen .....................................................................................................................101
8 Schlussfolgerungen......................................................................................................103
9 Literaturverzeichnis ......................................................................................................104
10 Lebenslauf .....................................................................................................................116
11 Veröffentlichungen .......................................................................................................118
Danksagung ..........................................................................................................................120
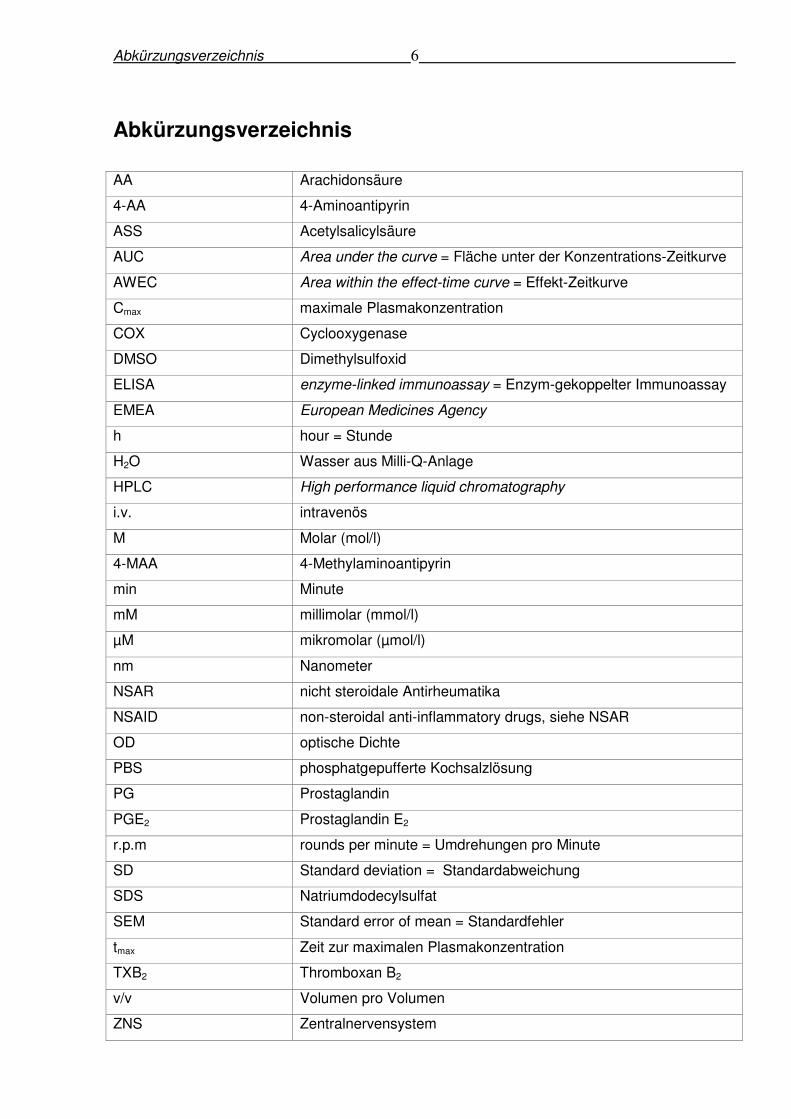
Abkürzungsverzeichnis 6
Abkürzungsverzeichnis
AA Arachidonsäure
4-AA 4-Aminoantipyrin
ASS Acetylsalicylsäure
AUC Area under the curve = Fläche unter der Konzentrations-Zeitkurve
AWEC Area within the effect-time curve = Effekt-Zeitkurve
Cmax maximale Plasmakonzentration
COX Cyclooxygenase
DMSO Dimethylsulfoxid
ELISA enzyme-linked immunoassay = Enzym-gekoppelter Immunoassay
EMEA European Medicines Agency
h hour = Stunde
H2O Wasser aus Milli-Q-Anlage
HPLC High performance liquid chromatography
i.v. intravenös
M Molar (mol/l)
4-MAA 4-Methylaminoantipyrin
min Minute
mM millimolar (mmol/l)
µM mikromolar (µmol/l)
nm Nanometer
NSAR nicht steroidale Antirheumatika
NSAID non-steroidal anti-inflammatory drugs, siehe NSAR
OD optische Dichte
PBS phosphatgepufferte Kochsalzlösung
PG Prostaglandin
PGE2 Prostaglandin E2
r.p.m rounds per minute = Umdrehungen pro Minute
SD Standard deviation = Standardabweichung
SDS Natriumdodecylsulfat
SEM Standard error of mean = Standardfehler
tmax Zeit zur maximalen Plasmakonzentration
TXB2 Thromboxan B2
v/v Volumen pro Volumen
ZNS Zentralnervensystem

Zusammenfassung 7
1 Zusammenfassung
Die nichtsteroidalen Antirheumatika (NSAR) wurden schon viele Jahre therapeutisch eingesetzt,
bevor ihr Wirkmechanismus erkannt wurde. Ihre Wirkung beruht im Wesentlichen auf einer
unselektiven Hemmung der beiden bekannten Cyclooxygenaseisoformen (COX-1 und COX-2).
Dadurch wird die Synthese von Prostaglandinen und Thromboxanen gehemmt. Diese
Substanzen stellen wichtige Mediatoren für Schmerz- und Entzündungsreaktionen dar. Sie
erfüllen darüber hinaus Schutzfunktionen in zahlreichen Organen des menschlichen Körpers,
z.B in der Magen- Darmschleimhaut, den Nieren und den Blutgefäßen. In den 90-er Jahren
wurden selektiven COX-2-Hemmer (Coxibe), als Analgetika mit niedrigerer gastrointestinalen
Toxizität im Vergleich zu den traditionellen NSARs, entwickelt.
Der Schwerpunkt dieser Dissertation wurde auf die Hemmung der peripheren Cyclooxygenasen
in menschlichen Blutzellen durch die Vertreter von selektiven und nichtselektiven COX-
Inhibitoren gelegt. Die in dieser Dissertation verwendete Methode erlaubt aus der gleichen
Blutprobe (ex-vivo) die Wirkstoffkonzentration (Pharmakokinetik) sowie die durch diese
Konzentration ausgelöste Hemmung beider Cyclooxygenasen (Pharmakodynamik) zu
bestimmen. Die Hemmung der COX-Aktivitäten wurde mittels enzymgekoppelten
Immunadsorptionstest (ELISA) bestimmt, wobei das koagulationsinduzierte Thromboxan B2
(TXB2) und das Lipopolysaccharid-induzierte Prostaglandin E2 (PGE2) ex-vivo und in-vitro im
menschlichen Vollblut als Marker der COX-1 und COX-2 Aktivität herangezogen wurden.
Aufgrund der Analyse von Ausmaß und Dauer der COX-2-Hemmung konnte die kleinste
effektive analgetische Dosis und das entsprechende Dosisintervall bestimmt werden. Durch
diese Erkenntnisse könnte man Überdosierungen und damit verbundene Nebenwirkungen
vermeiden. Hinsichtlich der Analyse der COX-1-Hemmung diente eine relevante Hemmung der
Thrombozytenaggregation (über 95%) als erhöhtes Risiko für Blutungen sowie als Prognose zur
Kardioprotektion eines Medikamentes.
Die gleichzeitige Analyse der Pharmakokinetik und Pharmakodynamik den untersuchten
Wirkstoffen nach der oralen Einnahme von Medikamenten ermöglichte verschiedene
Fragestellungen zu untersuchen.
Zunächst wurden die Untersuchungen zum Wirkungsmechanismus von Metamizol und
Paracetamol als Vertreter der nicht sauren Analgetika durchgeführt. Diese Medikamente üben
analgetische und antipyretische Effekte aus, weisen jedoch keine nennenswerte
entzündungshemmende Wirkkomponente sowie nur geringe gastrointestinale Nebenwirkungen
auf. Dagegen besitzen saure Analgetika potente analgetische, antipyretische und
antiphlogistische Eigenschaften und führen z.T. zu schweren gastrointestinalen und renalen
Nebenwirkungen.

Zusammenfassung 8
Metamizol ist ein Prodrug und wird im Organismus zu 4-Methylaminoantipyrin (4-MAA)
hydrolisiert, das weiter zu 4-Aminoantipyrin (4-AA) demethyliert wird. Die vorliegende Studie mit
Metamizol ist die erste, die ex-vivo beim Menschen die Selektivität von Metamizol zu den
peripheren COX-Enzymen sowie die Bedeutung der entsprechenden Metamizolmetaboliten (4-
MAA und 4-AA) untersuchte. Die Pharmakokinetiken der Metaboliten und die ex-vivo COX-
Hemmungen wurden bei fünf Probanden bestimmt, die einzelne orale Dosen von 500 oder
1000 mg Metamizol erhielten. Es zeigte sich, dass Metamizol in therapeutischer Dosierung eine
anhaltende Hemmung der beiden COX-Isoformen durch 4-MAA verursacht. Die COX-2-
Hemmung über 80% wurde bereits mit einer Dosierung von 500 mg erreicht. Nach Dosierung
von 1000 mg wurde eine über 95%-ige TXB2-Hemmung bei einigen Probanden gemessen.
Höhere Einzeldosen erscheinen aufgrund dieses Befundes nicht effektiver sein, erhöhen aber
das Risiko von unerwünschten Effekten.
Die Tatsache, dass Paracetamol funktionell einem selektiven COX-2 Hemmer ähnelt, führte zur
Überprüfung der Hypothese, dass Paracetamol bevorzugt über eine Hemmung von COX-2
analgetisch wirkt. Dafür wurden die Pharmakokinetik von Paracetamol und die ex-vivo COX-
Hemmung bei fünf Probanden bestimmt, die 1000 mg als einmalige orale Dosis erhielten.
Zusätzlich wurden in-vitro-Versuche durchgeführt, um festzustellen, ob die Expression oder
Aktivität von COX-2 in Blutmonozyten nach oraler Paracetamolgabe beeinflusst wird. In-vitro
sowie ex-vivo zeigte Paracetamol eine 4-fach höhere Selektivität für COX-2-Hemmung. Nach
der oralen Gabe des Medikamentes (1000 mg) wurde COX-2 (83%) im Vergleich zu COX-1
(56%) deutlich effektiver gehemmt. Mittels weiterer in-vitro Versuche konnte nachgewiesen
werden, dass Paracetamol die COX-2-Aktivität beeinflußt jedoch nicht die Expression von COX-
2 in Blutmonozyten.
Weiterhin wurde die COX-2-Hemmung durch den selektiven COX-2-Inhibitor Lumiracoxib
untersucht. Unter den im Jahr 2007 verfügbaren COX-2-Inhibitoren war Lumiracoxib der einzige
Arzneistoff für den in einer großen Patientenstudie Vorteile hinsichtlich gastrointestinaler
Nebenwirkungen bei Patienten mit Osteoarthritis demonstriert werden konnten. Dennoch wurde
im August 2007 von Fällen berichtet, in denen sich schwerwiegende lebertoxische
Nebenwirkung nach Lumiracoxibeinnahme in höheren Dosierungen (200 mg, 400 mg pro Tag)
zeigte. In der vorliegenden Studie wurde untersucht, ob eine ausreichende schmerzlindernde
COX-2-Hemmung (80%) auch nach oraler Einnahme von 50 mg erreicht werden kann. Um
dieses Ziel zu erreichen wurde eine Methode zur Bestimmung von Lumiracoxib im
menschlichen Plasma entwickelt und validiert. Anschließend wurde eine randomisierte, dreifach
cross-over Studie mit vier Probanden, die einmalig 50 mg, 100 mg oder 200 mg Lumiracoxib
eingenommen hatten, durchgeführt. Es konnte gezeigt werden, dass Lumiracoxib innerhalb
eines Zeitraums von 1 bis 1,5 Stunden nach Applikation zur kompletten Hemmung der COX-2-
Aktivität bei allen untersuchten Dosierungen führte. Die schmerzlindernde Hemmung der COX-

Zusammenfassung 9
2 (80%) wurde bis zu 6 Stunden bereits bei der kleinsten untersuchten Dosierung festgestellt,
bis zu 8 Stunden bei 100 mg und bis zu 10 Stunden bei 200 mg. Daraus folgt, dass Lumiracoxib
in der Dosierung von 50 mg eine vollständige und anhaltende COX-2-Hemmung hervorruft.
Sinnvolle therapeutische Effekte müssten aufgrund dieser Befunde bereits in der Dosierung von
100 mg erreichbar sein.
Anschließend wurde eine Untersuchung mit dem nicht selektiven COX-Hemmer Naproxen
durchgeführt um die immer wieder postulierte kardioprotektive Wirkung von Naproxen zu
untersuchen. Es ist bekannt, dass Naproxen beide Enzyme effektiv hemmt, er hat dazu eine
lange Halbwertzeit, deswegen könnte eine langandauernde Hemmung von TXB2 Synthese über
95% ein möglicher Mechanismus für die kardioprotektive Wirkung sein. Dafür wurde eine Studie
mit vier Probanden durchgeführt, die 220 mg Naproxen Natrium zweimal täglich für 7 Tage
einnahmen. Die stärkste Hemmung der COX-1 ergab sich mit 94% nach der ersten
Tabletteneinnahme und mit 93% im Steady-state. Stärkere Hemmung als 95% wurde bei zwei
von vier Probanden nach der Einzeldosis und bei einem von vier Probanden im Steady - state
beobachtet. Daraus folgt, dass Naproxen in OTC (rezeptfreie) Dosierung mögliche aber keine
zuverlässige kardioprotektive Wirkung hat.

Summary 10
2 Summary
The nonsteroidal antiinflammatory drugs (NSAIDs) had been in use for years before their mode
of action became known. Their activity is based largely on the non-selective inhibition of the two
known cyclooxygenases (COX-1 and COX-2), which results in a reduction of prostaglandins
and thromboxanes, both of which are important mediators of pain and inflammation. In addition,
they play a protective role in many organs of the human body - for example in gastrointestinal
mucosa, in the kidney and blood vessels. In the 1990s, selective inhibitors of the COX-2
enzyme (coxibs) were developed as analgesics with a lower gastrointestinal toxicity than
traditional NSAIDs.
The major focus of the present dissertation was the investigation of the inhibition of the
peripheral cyclooxygenases in human blood cells by members of selective and nonselective
COX-inhibitors. The method employed in this study makes it possible to determine, in one and
the same blood sample, both the concentration of the active agent (pharmacokinetics) and the
inhibition of the two cyclooxygenases at that concentration (pharmacodynamics). The inhibition
of the COX activities was determined by the enzyme-linked immunosorbent assay (ELISA),
using the coagulation-induced thromboxane B2 (TXB2) and the lipopolysaccharide-induced
prostaglandin E2 (PGE2), measured ex vivo and in vitro in human whole blood, as indices of
COX-1 and COX-2 activity.
An analysis of the extent and duration of COX-2 inhibition made it possible to determine the
minimal analgesic dose and the dosing interval. This knowledge might help to avoid overdosing
related side effects. In the case of COX-1 inhibition a clinically relevant inhibition of platelet
aggregation was used as an indication of whether a drug may be the cause of an elevated risk
of bleeding or provide cardioprotection.
The simultaneous analysis of the pharmacokinetics and pharmacodynamics of the substances
investigated following oral administration of the drugs made it possible to study a number of
questions.
First, the mode of action of metamizol (dipyrone) and paracetamol (acetaminophen) as
representatives of the non-acidic analgesics was investigated. These drugs have analgesic and
antipyretic activities, but no appreciable anti-inflammatory effects and only mild gastrointestinal
side effects. In contrast, acidic analgesics have potent analgesic, antipyretic and anti-
inflammatory properties and in some cases may cause severe gastrointestinal and renal side
effects.
Metamizol is a prodrug and is hydrolysed in the organism to 4-methylamnioantipyrine (4-MAA),
which is then demethylated to 4-aminoantipyrine (4-AA). The present study is the first to

Summary 11
investigate, ex-vivo in humans, the selectivity of metamizol for the peripheral COX enzymes and
the significance of the corresponding metabolites (4-MAA und 4-AA). The pharmacokinetics of
the metabolites and ex vivo COX inhibition were investigated in volunteers receiving metamizol
at single oral doses of 500 or 1000 mg. It was found that metamizol in therapeutic doses brings
about sustained inhibition of both COX isoforms via 4-MAA. Even the lower dose (500 mg)
results in a COX-2 inhibition of more than 80%. At a dose of 1000 mg a greater than 95%
inhibition of TXB2 was seen in some volunteers. Higher single doses would thus not appear to
be more effective, but would increase the risk of undesirable side effects.
The fact that paracetamol (acetaminophen) has an action similar to that of a selective COX-
inhibitor, prompted us to investigate the hypothesis that its analgesic activity might be mediated
by the inhibition of COX-2. To this end ex vivo COX inhibition and the pharmacokinetics of
paracetamol were assessed in five volunteers receiving single oral 1000 mg dose. In addition, in
vitro experiments were done to establish whether the expression or activity of COX-2 in blood
monocytes is influenced by oral paracetamol. Paracetamol showed a 4-fold greater selectivity
towards COX-2 inhibition, both in vitro and ex vivo. After administration of the drug the activity of
COX-2 was inhibited considerably more strongly (83%) than that of COX-1 (56%). Further in
vitro experiments showed that while paracetamol influenced COX-2 activity it had no influence
on the expression of COX-2 in blood monocytes.
COX-2 inhibition by the selective COX-2 inhibitor lumiracoxib was also investigated. In a study
involving a large number of patients, lumiracoxib showed to have advantages in terms of
gastrointestinal side effects in patients with osteoarthritis. Nevertheless, in August 2007 a
number of patients were reported to have developed severe liver problems following high daily
doses of lumiracoxib (200 mg, 400 mg). The present study investigated the question whether an
oral dose of 50 mg could also achieve adequate COX-2 inhibition (80%). For this purpose a
method for the determination of lumiracoxib in human plasma was developed and validated. A
randomised cross-over study was then done on four volunteers who had taken a single dose of
50 mg, 100 mg or 200 mg lumiracoxib. It was shown that all three doses resulted in complete
inhibition of COX-2 activity within a period of 1 to 1.5 hours. The analgesic effect of COX-2
inhibition (over 80%) persisted for 6 hours at the smallest analysed dose investigated, for 8
hours at a dose of 100 mg, and for 10 hours at 200 mg. This implies that Lumiracoxib at a dose
of 50 mg produces complete and sustained COX-2 inhibition. On the basis of these data, a dose
of 100 mg should suffice to achieve an acceptable therapeutic effect.
A further investigation was carried out with a non-selective COX-inhibitor – naproxen, to study
its potential cardioprotective action. It is known that naproxen effectively inhibits both COX-
enzyme isoforms and has a long half life. Hence, a possible mechanism of its cardioprotective
action might be prolonged inhibition of TXB2 synthesis above 95%. To investigate this, four

Summary 12
subjects were given a twice daily dose of 220 mg naproxen sodium for 7 days. The maximum
COX-1 inhibition after the first dose was 94% and at steady state 93%. A greater than 95%
inhibition was observed in two of four volunteers at time of maximal plasma concentration after
the first dose and in one of four volunteers throughout the 12-hour dose interval at steady-state.
Thus, a cardioprotective effect of over-the-counter doses of naproxen is possible but not reliably
certain.

Summary 13
The results may be summarised as follows:
• Via its metabolite 4-MAA, metamizol (dipyrone) dose-dependently inhibits the two COX
enzymes to an appreciable extent in vitro and ex vivo in the blood of volunteers. The
rapid and profound inhibition of COX-2 in human blood monocytes supports the
assumption that the analgesic action of metamizol is mediated by peripheral
mechanisms. In this connection no COX-1/COX-2 selectivity was observed. With regard
to the observed COX-1 inhibition it is assumed that physicochemical factors (lack of
acidity) are responsible for the superior gastrointestinal tolerability of metamizol as
compared with acidic COX inhibitors. The good tissue penetration properties of
metamizol suggest that, in the CNS, too, it inhibits COX-2 sufficiently to produce an
analgesic effect. Since a single dose of 1 g metamizol has a prolonged effect, larger
doses would appear unnecessary.
• Paracetamol (acetaminophen) inhibited COX-2 by more than 80%, i.e. to a degree
comparable with that seen with non-steroidal anti-inflammatory drugs (NSAIDs) and
selective COX-2 inhibitors. On the other hand, a relevant COX-1 inhibition (at least
95%), as is necessary for the suppression of platelet function, was not observed. In vitro
and ex vivo paracetamol revealed an approximately 4-fold higher selectivity for COX-2.
In view of its substantial COX-2 inhibition, recently defined cardiovascular warnings
against the use of COX-2 inhibitors should also be considered for this substance.
• Lumiracoxib at a dose of 50 mg elicits complete and prolonged COX-2 inhibition. It
would appear meaningful to consider using lumiracoxib for pain therapy at therapeutic
doses of not more than 100 mg.
• The administration of naproxen sodium at OTC doses was associated with a profound
inhibition of both COX enzymes. Although in some volunteer an anticoagulant, i.e.
cardioprotective effect of COX-1 inhibition was observed already at OTC dose, reliable
cardioprotection probably requires a dose of ca. 1 g a day.
In the light of the results presented here, a critical review of the “usual” doses of
cyclooxygenase inhibitors would appear justified.

Einleitung 14
3 Einleitung
3.1 Cyclooxygenase
Die Cyclooxygenase (COX), auch Prostaglandin-H-Synthase (PGHS) genannt, ist ein Enzym,
das die Umsetzung der Arachidonsäure (AA) zu wichtigen Botenstoffen katalysiert. Der Name
der Arachidonsäure leitet sich von der Erdnuss (Arachis hypogaea) her, aus der die AA zuerst
isoliert wurde. Arachidonsäure (all-cis-5,8,11,14-Eicosatetraensäure) wird aus den
Membranphospholipiden durch die Phospholipase (PLA2) freigesetzt und kann über die
Cyclooxygenase und Lipoxygenase weiter verstoffwechselt werden. Durch die Katalyse der
Lipoxygenase entstehen Leukotriene, durch die Cyclooxygenase werden Prostaglandine und
Thromboxane synthetisiert. Neben Prostaglandinen werden aus Arachidonsäure auch andere
Fettsäureprodukte, wie z.B. Epoxyeicosatriensäuren (EET), Isoprostane (IP),
Endocannabinoide, Lipoxine (LX) und Hepoxiline (HX) gebildet. Sie alle werden unter dem
Begriff Eicosanoide zusammengefasst, da sie alle von der Fettsäure Prostansäure, die aus 20
(griech: eikosi) Kohlenstoffatomen aufgebaut ist, abgeleitet sind.
Abbildung 1: Schematische Darstellung der Arachidonsäurekaskade. Die Cyclooxygenasen COX-1 und COX-2 verstoffwechseln die Arachidonsäure zu Prostaglandinen, Prostazyklin und Thromboxan in zwei aufeinander folgenden Reaktionen. Dabei wird zuerst die AA durch die Endoperoxidsynthase-Reaktion zu Prostaglandin G2 (PGG2) umgewandelt. Dann vermittelt die Peroxidase-Reaktion die Umwandlung zu Prostaglandin H2. Inhibitoren der Cyclooxygenase hemmen dabei die Endoperoxidsynthase-Reaktion.
Die Cyclooxygenase ist ein bifunktionelles Enzym und besitzt daher zwei aktive Zentren
(Hawkey, 1999). Als Endoperoxid-Synthase (Cyclooxygenase-Reaktion) oxidiert und zyklisiert
PGH2
Arachidonsäure
Endoperoxidsynthase-Reaktion
PGG2
Peroxidase-Reaktion
PGF2α
PGD2
PGE2
PGI2
TXA2
COX

Einleitung 15
sie die AA zum Prostaglandin G2 (PGG2), wobei der charakteristische Cyclopentanring der
Prostaglandine entsteht. Ein zweites katalytisches Zentrum der COX, die Peroxidase, reduziert
die Peroxidgruppe am C-15 zum Alkohol. Das dadurch entstandene Prostaglandin H2 (PGH2)
wird durch gewebespezifische Enzyme weiter zu den verschiedenen Substanzen, wie
Prostaglandinen (PGE, PGF und PGD), Prostazyklin (PGI), und Thromboxan (TXA)
transformiert (Abbildung 1).
3.2 Isoformen der COX
Zurzeit sind zwei COX-Isoformen, die COX-1 und COX-2 bekannt. Beide Isoenzyme
unterscheiden sich in ihrer Gewebeverteilung und der Regulation ihrer Expression. Die COX-1-
Isoform wird in fast allen Geweben einschließlich Gastrointestinaltrakt, Thrombozyten,
Endothelzellen und Nieren konstitutiv exprimiert (Crofford, 1997) und ist vor allem an
physiologischen Funktionen, wie z.B. die Protektion der Magenschleimhaut, die Kontrolle des
renalen Blutflusses und die Plättchenaggregation beteiligt. Im Gegensatz zu der überall
vorkommenden COX-1 findet man COX-2 unter physiologischen Bedingungen nur in wenigen
Geweben wie ZNS (Beiche et al., 1996), Niere (Harris et al., 1994; Catella-Lawson et al., 1999;
McAdam et al., 1999), weibliche Reproduktionsgewebe (Kniss, 1999) und Magen (Zimmermann
et al., 1998). Alle anderen Gewebe produzieren das Enzym lediglich bei Bedarf, so nach
unterschiedliche Stimuli wie z.B. proinflammatorische Zytokine (Mitchell et al., 1994),
Wachstumsfaktoren (DeWitt et al., 1993), Endotoxine und Tumorpromotoren (Lee and Ip, 1992),
sowie als Antwort auf Stress durch chemische Reizung, Verletzung, UV-Bestrahlung, bakterielle
Infektion usw. Da es bei solchen Stressreaktionen meist zu Entzündungen kommt, gilt COX-2
als „proinflammatorisches Notfallenzym“. Zum anderen spielt die COX-2 eine wichtige Rolle bei
der Wund- bzw. Ulkusheilung und bei der Regulierung des Augeninnendruckes (Hinz and Brune,
2002).
Beide Isoformen zeigen eine ca. 60 %-ige Homologie in ihrer Aminosäuresequenz (Vane et al.,
1998). Sie bestehen aus ca. 600 Aminosäuren (Smith et al., 1996), wobei die COX-1 576
Aminosäuren, und die COX-2 587 Aminosäuren enthält. Beide Isoformen besitzen ähnliche
aktive Zentren, einen langen und engen, tunnelartigen, hydrophoben Kanal an dessen Ende die
Enzymreaktionen katalysiert werden (Garavito, 1996). Der wichtigste Unterschied liegt in der
Tertiärstruktur der katalytischen Zentren beider Enzyme. Durch den Austausch von Isoleucin
(Ile) gegen das kleinere Valin (Val) bei der COX-2 an der Position 523 wird die Bindungstasche
der COX-2 gegenüber der COX-1 um 25 % größer (Smith et al., 2000; Garavito et al., 2002;
Gupta et al., 2004). Die größere Bindungstasche der COX-2 wurde bei der Entwicklung von
selektiven COX-2-Inhibitoren (siehe Abschnitt 3.5.2) ausgenutzt (Hinz and Brune, 2002).

Einleitung 16
Zusätzlich zu diesen gut charakterisierten Isoformen wurden mehrere Splicevarianten von
COX-1 und COX-2 in verschiedenen Geweben nachgewiesen (Simmons et al., 2004). Für eine
katalytisch aktive COX-1-Variante, die in Hunden entdeckt wurde und durch Paracetamol
gehemmt wurde (Chandrasekharan et al., 2002), konnten in weiteren genetischen
Untersuchungen jedoch gezeigt werden, dass diese Variante beim Menschen nur ein Protein
mit geringer COX-Homologie und ohne COX-Aktivität darstellt (Kis et al., 2005). Daher ist
dieses Protein beim Menschen vermutlich nicht an der Prostaglandin-Biosynthese beteiligt.
3.3 Produkte des Cyclooxygenaseweges: Prostaglandine und
Thromboxane
Die Produkte, die unter der katalytischen Wirkung der COX-Enzyme gebildet werden, sind unter
anderen die Prostaglandine und Thromboxane. Sie werden auch unter dem Begriff Prostanoide
zusammengefasst. Die Prostanoide sind wegen ihrer raschen metabolischen Inaktivierung nur
für wenige Sekunden bis Minuten am Ort ihrer Bildung wirksam und müssen also ständig neu
synthetisiert werden.
3.3.1 Prostaglandin E2
Das Prostaglandin E2 (PGE2) ist ein primäres Prostaglandin, das aus PGH2 durch die PGE-
Synthase entsteht. Zurzeit sind drei PGE-Synthasen, die Membran-gebundene (mikrosomale)
PGE-Synthase-1 (mPGES-1) (Jakobsson et al., 1999), die Membran-gebundene (mikrosomale)
PGE-Synthase-2 (mPGES-2) (Murakami et al., 2003) und die cytosolische PGE-Synthase
(cPGES) bekannt (Tanioka et al., 2000). mPGES-1 ist funktionell an COX-2 gekoppelt und kann
genauso wie COX-2 durch proinflammatorische Stimuli (IL-1β) hochreguliert werden
(Stichtenoth et al., 2001; Korotkova et al., 2005). Die hauptsächlich im Gehirn vorkommende
zytostolische PGE-Synthase (cPGES) ist dagegen funktionell an COX-1 gebunden und wird wie
diese konstitutiv exprimiert (Tanioka et al., 2000). Die mPGES-2 kann zusammen sowohl mit
COX-1 als auch mit COX-2 vorkommen (Murakami et al., 2003). Es wird vermutet, dass die
physiologische PGE2-Funktionen vorwiegend durch cPGES / mPGES-2 / COX-1 und die
pathophysiologische PGE2-Funktionen durch das mPGES-1 / COX-2-System vermittelt werden
(Helliwell et al., 2004).
PGE2 kann viele und teilweise gegensätzlichen Wirkungen, wie z.B. eine Kontraktion oder
Relaxation von glatten Muskeln verursachen, eine Stimulation oder Suprimierung der Sekretion
von Hormonen hervorrufen. Diese sind auf die PGE-Rezeptor-Subtypen: EP1, EP2, EP3 und
EP4 zurückzuführen. (Bos et al., 2004). Eine Aktivierung des EP1-Rezeptors führt zur
Kontraktion glatter Muskelzellen in den Bronchien und dem Gastrointestinaltrakt (Narumiya et
al., 1999). Eine Aktivierung des EP2-Rezeptors ruft dagegen eine Relaxation von glatten

Einleitung 17
Muskelzellen in den Bronchien, im Gastrointestinaltrakt und in Blutgefäßen hervor. Eine
Aktivierung des EP3-Rezeptors führt zur Hemmung der Magensäureproduktion, einer
verstärkten Uteruskontraktion in der Schwangerschaft (Mutschler E., 2001). Darüber hinaus ist
der EP3-Rezeptor im Hypothalamus an der Induktion des Fiebers beteiligt (Nakamura et al.,
1999). Der EP4-Rezeptor vermittelt eine vermehrte Schleimsekretion des Magens und hält den
Ductus arteriosus Botalli offen (Mutschler E., 2001). Im Bereich des Intestinaltraktes ist u.a. das
PGE2 von zytoproliferativer Bedeutung. Es wurde berichtet, dass exogenes Prostaglandin E2 die
Proliferation von Stammzellen intestinaler Krypten stimuliert (Cohn et al., 1997).
3.3.2 Thromboxan B2
Das Thromboxan B2 (TXB2) ist ein stabiler Metabolit des Thromboxan A2 (TXA2), das in
physiologischer Umgebung eine äußerst instabile Verbindung ist. Das Thromboxan A2 wird von
der Thromboxansynthase, das zu den Cytochrom P450 Isoenzymen gehört (Wang and
Kulmacz, 2002), in den Thrombozyten gebildet. TXA2 vermittelt die Plättchenaggregation und
die Kontraktion glatter Muskelzellen in den Blutgefäßen und den Bronchien über den TXA2-
Rezeptor. Mit einer Halbwertzeit von 30 Sekunden zerfällt es nicht enzymatisch zu dem stabilen,
aber inaktiven TXB2 (Hamberg et al., 1975; Needleman et al., 1976), das man nach
Blutkoagulation messen kann. Im Urin wurden zahlreiche Metabolite des TXB2 nachgewiesen,
vor allem jedoch 2,3-Dinor-TXB2 and 11-Dehydro-TXB2.
3.4 Hemmung der Prostaglandinsynthese als
Wirkungsmechanismus antipyretischer Analgetika
Antipyretische Analgetika sind chemisch heterogene Medikamente, die sich aus
pharmakologischer und physikochemischer Sicht in zwei Gruppen einteilen lassen: saure und
nicht saure Antipyretika.
3.4.1 Saure Antipyretika
Zu den sauren Analgetika gehören die Pharmaka, die neben analgetischen und antipyretischen
Effekten in höherer Dosierung auch durch eine ausgeprägte antiphlogistische Wirkung
charakterisiert sind. Nach der Entdeckung der antiinflammatorischen Wirkung der
Glucocorticoide im Jahre 1949 werden die antiphlogistisch wirkenden sauren antipyretischen
Analgetika auch kollektiv als nicht steroidale Antiphlogistika bezeichnet ("nonsteroidal
antiinflammatory drugs", NSAIDs, NSAR). Die entsprechenden Substanzen sind schwache
Säuren (pKA-Wert 3-5,5) wie z.B. Acetylsalicylsäure, 2-Arylpropionsäuren (Profen),
Arylessigsäuren und heterozyklische Ketoenolsäuren. Sie weisen eine ausgeprägte Polarität
sowie eine hochgradige Bindung an Plasmaproteine (>90%) auf (Hinz and Brune, 2007).

Einleitung 18
1971 klärte John R. Vane den Wirkmechanismus von NSAR auf und teilte sich dafür 1982 den
Nobelpreis für Physiologie und Medizin mit den Drs. Sune Bergstrom, Bengt Samuelson. Vane
zeigte, dass die sauren antiphlogistischen Analgetika durch Hemmung der Cyclooxygenase zur
Reduktion proinflammatorischer Prostaglandine führen (Vane, 1971). Die Prostaglandine
erhöhen im traumatisierten Gewebe die Empfindlichkeit von feiner Nervenendigungen
(Nozizeptoren) auf andere Stimuli, indem sie normalerweise nicht erregbare polymodale
Rezeptoren („silent nociceptors“) in einen Zustand leichter Erregbarkeit überführen. Die NSAR
normalisieren die erhöhte Empfindlichkeit Nozizeptoren im geschädigten Gewebe. Die
entsprechenden analgetischen Substanzen üben insofern im strengen Sinne keinen
analgetischen Effekt aus, sondern wirken durch Modulierung der Sensitivität polymodaler
Nozizeptoren „antihyperalgetisch“ (Schaible and Grubb, 1993).
Die Arbeitsgruppe um Brune zeigte anhand radiographischer Studien, dass sich die sauren
Analgetika besonders im Blut, in Leber, Milz und Knochenmark, aber auch in Kompartimenten
mit saurem extrazellulärem pH-Wert, insbesondere in den entzündeten Geweben, in der Wand
des oberen Gastrointestinaltrakts und in den Sammelrohren der Nieren, anreichern (Brune,
1974; Brune et al., 1976; Rainsford et al., 1981). Die Akkumulation saurer Analgetika im
entzündeten Gewebe wird als entscheidender Faktor ihrer antiinflammatorischen Effekte
angesehen. Im entzündeten Gewebe verhindern die sauren NSAR die pathologische
Überproduktion von Prostaglandinen und damit die vor allem von PGE2 ausgelösten
entzündlichen Reaktionen (Brune and Zeilhofer, 1999).
3.4.2 Nicht saure Antipyretika
Nicht saure Analgetika (Paracetamol, Pyrazolinone) üben analgetische und antipyretische
Effekte aus, besitzen jedoch keine nennenswerte antiphlogistische Wirkkomponente. Ihre
Vertreter sind gering polar, neutral (Paracetamol) oder schwach alkalisch (Pyrazolinone) und
nur geringgradig an Plasmaproteine gebunden. Sie verteilen sich schnell und homogen im
Organismus und penetrieren auch die Blut-Hirn-Schranke (Brune et al., 1980). Ebenso weisen
sie nicht die für saure Analgetika typischen Nebenwirkungen im Magen-Darm-Trakt, in der
Niere und bei der Blutgerinnung auf.
Die genaueren Beschreibungen zum Wirkmechanismus von nicht sauren Analgetika erfolgen in
den Abschnitten 3.4.2.1 und 3.4.2.2. Jedoch ist der genaue analgetische
Wirkungsmechanismus dieser Substanzklasse bisher noch nicht vollständig geklärt.

Einleitung 19
3.4.2.1 Wirkungmechanismus von Metamizol
Chemische Bezeichnung: Natrium-[(1,5-dimethyl-3-oxo-
2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-
N-methylamino]methansulfonat
Synonym: Dipyron, Noramidopyrin, Novaminsulfon
Summenformel: C13H16O4N3NaS
Molekulargewicht: 351,4 g/mol
Abbildung 2: Strukturformel von Metamizol Natrium.
Metamizol ist ein häufig verschriebenes, potentes spasmolytisches Analgetikum und
Antipyretikum (Carlsson et al., 1986; Vlahov et al., 1990). Ein antiphlogistischer Effekt tritt
jedoch erst bei höheren Dosierungen auf (Gelgor et al., 1992). Indikationen für dieses
Medikament sind starke akute und chronische Schmerzen, sowie Tumorschmerzen (Aventis,
2003). Auf Grund der krampflösenden Wirkung wird es zudem häufig bei kolikartigen
Schmerzen im Gastrointestinaltrakt und Urogenitaltrakt eingesetzt. Die Anwendung ist
beschränkt auf Schmerzzustände, bei denen eine Therapie mit NSAR nicht ausreichend ist.
Diese Anwendungsbeschränkung und die Einführung der Verschreibungspflicht im Jahre 1987
sind Folgen des in den 70er Jahren entdeckten Zusammenhangs zwischen der Einnahme von
Metamizol und dem Auftreten einer Agranulozytose. Als Folge der daraus entstandenen
kontroversen Diskussion über das Nutzen-Risiko-Potential von Metamizol ist der Arzneistoff in
vielen Ländern gänzlich vom Markt verschwunden. Währenddessen kommt dieser Arzneistoff
besonders in Entwicklungsländern zur Anwendung und ist dort ohne Verschreibung erhältlich.
Die Inzidenz dieser unerwünschten Effekte ist dennoch seltener als bei den NSAR (Maj and
Centkowski, 2004; Ibanez et al., 2005). Somit ist Metamizol ein weitgehend sicheres
Medikament, dessen analgetische Potenz mit schwachen Opiaten vergleichbar ist.
Metamizol ist in oralen und rektalen Darreichungsformen verfügbar (Aventis, 2003). Durch
Einführung einer Sulfonsäurestruktur und Bildung des Natriumsalzes konnte eine waserlösliche
Formulierung erzielt werden. Dies erlaubt auch intravenöse Applikationen dieses Medikamentes.
Da nach schneller intravenöser Injektion von Metamizol Schockreaktionen mit tödlichem
Ausgang beschrieben worden sind, sollte die Substanz langsam injeziert und unter strenger
Indikationsstellung i.v. eingesetzt werden.
Der Metabolismus von Metamizol im Körper führt über zahlreiche Biotransformationsprodukte.
Nach der oralen Verabreichung von Metamizol-Natriumsalz (Prodrug von Metamizol)
(Abbildung 2) erfolgt eine schnelle nicht enzymatische Hydrolyse im Gastrointestinaltrakt zu 4-
NN
NCH3
CH3 O
CH2SO3Na
CH3

Einleitung 20
Methylaminoantipyrin (4-MAA) (Abbildung 3, links), das nahezu vollständig resorbiert wird.
(Brune, 1988). Anschließend wird 4-Methylaminoantipyrin zu 4-Formylaminoantipyrin (4-FAAP)
enzymatisch oxidiert (Noda et al., 1976), sowie zu 4-Aminoantipyrin (4-AA) (Abbildung 3,
rechts) demethyliert, das durch eine polymorphe N-Acetyl-Transferase in der Leber zum
4-Acetylaminoantipyrin (4-AAA) acetyliert wird. Das Ausmaß dieses Metabolisierungsschrittes
ist vom Acetylierungsphänotyp des Patienten abhängig (Zylber-Katz et al., 1992). Diese vier
Hauptmetaboliten (4-MAA, 4-AA, 4-FAAP, 4-AAA) von Metamizol stellen ungefähr 65% der
verabreichten Dosis dar (Volz and Kellner, 1980). In sehr geringem Maße wird 4-Aminoantipyrin
auch zum atypischen Metaboliten 4-Hydroxyantipyrin umgesetzt (Reinhardt et al., 2006). Nach
hohen Dosen kann im Urin der rot gefärbte, jedoch harmlose Metabolit Rubazonsäure
nachgewiesen werden (Reinhardt et al., 2006).
Abbildung 3: Strukturformeln der untersuchten Metamizolmetaboliten: 4-Methylaminoantipyrin (links) und 4-Aminoantipyrin (rechts).
4-MAA und sein demethyliertes Derivat 4-AA werden als pharmakologisch wichtigste Metabolite
diskutiert (Weithmann and Alpermann, 1985). Sie sind, chemisch gesehen, schwach basische
Amine. Aus diesem Grund findet eine Anreicherung im leicht sauren, entzündeten Gewebe wohl
kaum statt. (Frolich et al., 1986). Daher kann man wahrscheinlich nur bei Dosierungen, die
oberhalb des therapeutischen Bereichs liegen, eine antiphlogistische Wirkung beobachten.
Weiterhin sind 4-MAA und 4-AA hydrophil und verteilen sich gleichmäßig in allen wässrigen
Kompartimenten. Sie werden bis zu 58 % bzw. 48 % an Plasmaproteine gebunden. Ihre
Plasmahalbwertzeiten variieren interindividuell sehr stark. Sie ist im Falle des 4-AA vom
Acetylierungsphänotyp abhängig und liegt deshalb im Bereich von 3,7 bis 8,1 Stunden. Die von
enzymatischen Reaktionen unabhängige Plasmahalbwertzeit des 4-MAA ist mit ca 2,5 Stunden
dagegen relativ kurz (Zylber-Katz et al., 1992).
Bis jetzt wurde angenommen, dass die antinozizeptive Wirkung von Metamizol wenigsten zum
Teil über einen Wirkmechanismus im zentralen Nervensytem vermittelt wird. (Carlsson et al.,
1986; Gelgor et al., 1992; Neugebauer et al., 1994; Shimada et al., 1994; Tortorici and Vanegas,
1995; Beirith et al., 1998). Da 4-MAA als auch 4-AA die Blut-Hirn-Schranke sehr gut
überwinden können, werden nach oraler Einnahme von Metamizol (1000 mg) maximale
Konzentrationen von 5-30 µg/ml (4-MAA) bzw. 1-7 µg/ml (4-AA) in der cerebrospinalen
NN
NCH3
CH3 O
CH3
H
NN
NCH3
CH3 O
H
H

Einleitung 21
Flüssigkeit erreicht. (Cohen et al., 1998). In Übereinstimmung damit wurde eine zeitabhängige
Senkung der Thromboxan B2-Konzentrationen in der cerebrospinalen Flüssigkeit bei Patienten,
die Metamizol erhalten hatten, nachgewiesen (Levy et al., 1998). Das unterstützt ebenfalls
einen zentral vermittelten Wirkungsmechanismus von Metamizol, der erstmalig von Tomek
bereits 1955 vermutet wurde. (Tomek, 1955). Die Hemmung der Prostaglandinsynthese wurde
zusätzlich dazu auch an anderen Orten des zentralen Nervensystems festgestellt. (Dembinska-
Kiec, 1976; Yaksh et al., 2001). Eine Bindung von 4-MAA, 4-AA bzw. Metamizol an Opioid-
Rezeptoren ist dagegen eher unwahrscheinlich. (Masuko et al., 1979; Goldenberg and De Boni,
1983).
Es wurde jedoch gezeigt, dass Metamizol auch die periphere Synthese von Eicosanoiden
einschließlich TXB2 in menschlichen Blutplättchen inhibiert. (Eldor et al., 1984; Weithmann and
Alpermann, 1985; Frolich et al., 1986; Geisslinger et al., 1996).
Eine periphere Wirkungsweise wurde 1985 von Lorenzetti postuliert, da er eine durch Carageen,
PGE2, Isoprenalin und Calciumchlorid ausgelöste Hyperalgesie durch Metamizol unterdrückten
konnte. Dabei wurde hauptsächlich PGE2 die Hauptrolle für die Hyperalgesie zugeschrieben.
(Lorenzetti and Ferreira, 1985). Weitere Studien konnten eine Hemmung der PGE2-Synthese in
menschlicher Magenschleimhaut (Peskar et al., 1986) und eine Absenkung der PGE2-
Konzentration im Magensaft nachweisen. (Frolich et al., 1986). Untersuchungen an Astrozyten
und Makrophagen zeigten, dass 4-MAA ein vergleichbares Hemmpotential der
Prostaglandinsynthese in beiden Zelltypen aufwies. (Lanz et al., 1986). In Übereinstimmung mit
diesen früheren Erkentnissen verhindert Metamizol, wie vor kurzem gezeigt wurde, die
enzymatischen Aktivitäten der beiden Cyclooxygenasen-Isoformen sowohl in gereinigten
Enzymen, als auch in verschiedenen peripheren Zellen (Campos et al., 1999).

Einleitung 22
3.4.2.2 Wirkungmechanismus von Paracetamol
Chemische Bezeichnung: 4´-Hydroxyacetanilid
Synonym: Acetaminophen
Summenformel: C8H9NO2
Molekulargewicht: 151,2 g/mol
Abbildung 4: Strukturformel von Paracetamol
Paracetamol, ist eines der weltweit am meisten verwendeten freiverkäuflichen, antipyretischen
und analgetischen Arzneimittel. Es wird als Therapeutikum erster Wahl gegen den
Osteoarthrose-assoziierten Schmerz (Schnitzer, 2002) empfohlen. Paracetamol gehört zu den
Aniliden und ist ein p-Aminophenol-Derivat. Es kann oral, rektal und intravenös appliziert
werden. Nach oraler Gabe erfolgt eine schnelle und vollständige Resorption sowie
gleichmäßige Verteilung in alle Gewebe. Die Inaktivierung erfolgt in der Leber durch die schnell
sättigbare Konjugation an Glucuron- oder Schwefelsäure. Ein geringerer Teil wird über das
Cytochrom P-450 Enzyme zum giftigen N-Acetyl-p-benzochinonimin metabolisiert, das über
Glutathion entgiftet und an Cystein gebunden ausgeschieden wird. Bei Überdosierung steht
nach gewisser Zeit nicht genügend Cystein für die Entgiftung zur Verfügung. Die Elimination
erfolgt mit einer HWZ von ca. zwei Stunden hauptsächlich renal. Paracetamol besitzt bei
therapeutischer Dosierung (4 g pro Tag bei Erwachsenen, 15 mg pro kg Körpergewicht am Tag
bei Kindern) keine nennenswerten Nebenwirkungen. Bei akuter Überdosierung (bei Dosen über
10 g pro Tag) tritt Hepatotoxizität auf, die zu schweren, meist tödlich verlaufenden
Leberzellnekrosen führt (Prescott et al., 1971). Trotz der Tatsache, dass Paracetamol schon vor
100 Jahren entdeckt wurde sowie auch seit ungefähr 50 Jahren vielfältig verwendet wird, ist der
komplette Wirkungsmechanismus immer noch unklar. Es wird allgemein angenommen, dass
Paracetamol zentral wirkt und bestenfalls ein schwacher Inhibitor der durch COX-1 und COX-2
vermittelten Prostaglandinsynthese ist (Botting, 2000). Dieses Konzept beruht auf der frühen
Arbeit von Flower und Vane, die zeigten, dass Paracetamol die Prostaglandin-Synthese im
Gehirn zehnfach stärker hemmt als in der Milz (Flower et al., 1972). Jedoch wurde diese
Entdeckung durch spätere Studien (Lanz et al., 1986; Warner et al., 2004) nicht bestätigt.
Außerdem stellte es sich heraus, dass Paracetamol keine messbare Inhibition der Bildung von
Prostaglandinen in aufgeschlossenen Zellpräparationen jedoch aber eine deutliche Hemmung
in intakten Zellen hervorruft (Graham and Scott, 2005).
Lucas et al. konnten zeigen, dass die Reduktion der nervalen Prostaglandin-Synthese durch
Paracetamol nicht auf transkriptioneller Ebene sondern auf Proteinebene reguliert wird (Lucas
et al., 2005). Als weitere Einflussgröße konnte die zelluläre Konzentration an Peroxiden
identifiziert werden. Bei niedriger Peroxidkonzentration ist das COX-Inhibitionspotential des

Einleitung 23
Paracetamols deutlich größer, da hohe Peroxidkonzentration die reduzierenden Eigenschaften
von Paracetamol antagonisieren und gleichzeitig die COX in einen höheren Oxidationszustand
überführen (Lucas et al., 2005). Auf Grundlage der pH-Abhängigkeit wurde ebenfalls
vorgeschlagen, dass Paracetamol keine Affinität zum aktiven Zentrum der COX habe, sondern
die COX durch Reduktion der aktiven oxidierten Form in die inaktive Form überführe (Lucas et
al., 2005). Da das Nervensystem über einen niedrigen Oxidationsstatus verfügt, könnte das
eine plausible Erklärung für die Wirkung im zentralen Nervensystem sein.
Auch wurde die Annahme, dass die Wirkung von Paracetamol auf die Hemmung von COX-3
zurückzuführen ist (Chandrasekharan et al., 2002), aus verschiedenen Gründen (Kis et al.,
2005) verworfen. Dies erfolgte vor allem aufgrund der Tatsache, dass die Existenz einer
funktionellen menschlichen COX-3 in Frage gestellt wurde. Im Falle der COX-3, ein Produkt des
COX-1 Genes, führt der Verbleib des Intron 1 in der COX-3 mRNA zu einem kürzeren, nicht
funktionellen Protein (Dinchuk et al., 2003; Schwab et al., 2003a).
Qin et al. berichteten über eine niedrige Expression von drei Splice-Varianten der COX-1 in
menschlichen Geweben (Qin et al., 2005). Es konnte aber kein relevanter Unterschied
zwischen der Paracetamol-vermittelten Hemmung von COX-1 und der Intron 1-enthaltenden
Splicevariante nachgewiesen werden. Obwohl die Aktivität der caninen COX-3 vollständig durch
Paracetamol inhibierbar ist, trifft diese Erklärung für die pharmakologischen Wirkungen des
Paracetamols beim Menschen nicht zu (Schwab et al., 2003b; Lucas et al., 2005). Deshalb wird
die Forschung auf dem Gebiet der Identifizierung der Wirkmechanismen von Paracetamol
weiter fortgesetzt.

Einleitung 24
3.5 Cyclooxygenasehemmer
Die fibersenkend wirkenden Analgetika werden als Cyclooxygenaseinhibitoren (COX-Inhibitoren,
COX-Hemmer) bezeichnet, weil sie ihre Wirkung über eine periphere und / oder zentrale
Hemmung der Cyclooxygenase entfalten. Je nach den bevorzugt gehemmten Isoenzymen
werden nicht selektive und selektive Inhibitoren unterschieden.
Die analgetischen, antipyretischen und antiphlogistischen Wirkungen nicht steroidaler
Antirheumatika (NSAR) beruhen im Wesentlichen auf einer unselektiven Hemmung der beiden
bekannten Cyclooxygenaseisoformen COX-1 und COX-2. Deswegen werden diese
Medikamente auch als nicht selektive Cyclooxygenase-Inhibitoren bezeichnet. Seit einiger Zeit
steht neben den traditionellen nicht steroidalen antiinflammatorischen Substanzen die Klasse
der selektiven COX-2-Hemmer (Coxibe) zur Verfügung. Eine Substanz wird als ein selektiver
COX-2-Inhibitor betrachtet, wenn sie die COX-2 hemmt, aber keine klinisch relevante COX-1-
Hemmung in therapeutischen Dosen verursacht.
Die 3D-Strukturen der COX-1 und COX-2, die mit Hilfe von Röntgenstrukturanalysen ermittelt
wurden, ermöglichten Einblicke in den molekularen Wirkmechanismus von NSAR. Diese
Erkenntnisse führten auch zu einem verbesserten Verständnis darüber wie die Selektivität von
Hemmstoffen erreicht wird. Die Daten zeigten, dass NSAR den hydrophoben Enzymkanal der
COX-1 in der Mitte blockieren. Dies wird über Wasserstoffbrückenbindungen zwischen dem
Arzneistoff und dem polarem Arginin an der Position 120 vermittelt. Arginin ist an dieser
Position sowohl in COX-1 als auch in COX-2 vorzufinden. Kritisch für die Selektivität von COX-
Hemmer ist die Aminosäureposition 523. In COX-1 befindet sich an dieser Stelle ein Isoleucin
wogegen in COX-2 ein Valin vorzufinden ist. Der Unterschied an dieser Position führt dazu,
dass die COX-2 selektiv durch Coxibe gehemmt werden kann (Hawkey, 1999).
Für die Bestimmung der COX-1 / COX-2-Selektivität hat sich der von Patrono und Mitarbeitern
beschriebene „Vollblutassay“ als vorteilhaft erwiesen. (Patrignani et al., 1994). Mit diesem
Assay (siehe Abschnitt 5.2.2) wird die COX-1- und COX-2-Aktivität eines Hemmstoffes an
klinisch relevanten humanen Blutzellen bestimmt (COX-1: Thrombozyten bei der Blutgerinnung,
COX-2: Monozyten nach Lipopolysaccharidstimulation). Dieser Test ermöglicht die
Untersuchung der COX-Selektivität unter physiologischen Bedingungen bei denen die
Plasmaproteinbindung berücksichtigt wird. (Patrignani et al., 1997).

Einleitung 25
3.5.1 Nicht selektive Cyclooxygenasehemmer
Untersuchungen mit dem in Abschnitt 5.2.2 beschriebenen Versuchsystem zeigten, dass keines
der derzeit therapeutisch eingesetzten NSAR die COX-2 selektiv hemmt (Patrignani et al.,
1997). Einige Substanzen (Diclofenac, Aceclofenac, Meloxicam) weisen eine bevorzugte
Hemmung des COX-2-Enzyms auf und können deshalb als präferentielle Hemmer der COX-2
betrachtet werden (Tabelle 1) (Hinz and Brune, 1999).
Tabelle 1: Relative Potenz einiger analgetischer, antipyretischer, antiphlogistischer Wirkstoffe hinsichtlich ihrer COX-1 und COX-2- Hemmungen.
Substanz COX-1 / COX-2 IC50-Ratio
Ibuprofen 0,50
Naproxen 0,56
S-Ketoprofen 0,61
Flurbiprofen 1,00
Indomethacin 1,90
Piroxicam 3,10
Meloxicam 11,16
Nimesulid 17,70
Diclofenac 18,90
Die IC50-Werte wurden ex-vivo mittels des humanen Vollblutassays (siehe Abschnitt 3.5) bestimmt. Die Daten stammen aus der Arbeit von Patrignani (Patrignani et al., 1997). Für die aufgeführten Substanzen ist das Verhältnis der COX-1- zur COX-2-Hemmung angegeben (Quotient IC50 COX-1 bzw. IC50 COX-2). Ist der Quotient größer als 1, dann ist diese Substanz durch eine vergleichsweise stärkeren COX-2-Hemmung gekennzeichnet.
Als unerwünschte Begleiterscheinungen der nicht selektiven COX-Inhibitoren, die sich
besonders im Bereich des oberen Gastrointestinaltraktes manifestieren, treten
Schleimhautschädigungen, petechiale Blutungen, Erosionen sowie Ulzerationen auf (Graham
and Smith, 1988; Langman, 1989; McCarthy, 1989; Cheatum et al., 1999).

Einleitung 26
3.5.1.1 Thrombozytenhemmende und potenziell kardioprotektive Wirkung
von Naproxen
Chemische Bezeichnung: (S)-2-(6'-Methoxy-2'-naphthyl)-propionsäure
Synonym: Naproxenum
Summenformel: C14H14O3
Molekulargewicht: 230,26 g/mol
Abbildung 5: Strukturformel von Naproxen
Das Arylpropionsäurederivat Naproxen ist ein gut verträgliches NSAR mittlerer Wirkstärke. Es
wird nach oraler Gabe des Natriumsalzes rasch und gut resorbiert, maximale
Plasmakonzentration wird bereits nach einer Stunde erreicht. Infolge der langen
Serumhalbwertzeit von 13-18 Stunden braucht Naproxen auch bei chronischen entzündlichen
Schmerzen nur ein- bis zweimal täglich eingenommen zu werden.
Im Laufe der letzten Jahre entwickelten sich Bedenken hinsichtlich der kardiovaskulären
Sicherheit von selektiven Cyclooxygenase-2-Hemmern und nicht steroidalen Antirheumatika bei
höheren Dosierungen (FitzGerald and Patrono, 2001; Tegeder and Geisslinger, 2006; Hinz and
Brune, 2007). Tatsächlich konnte in gut geplanten, Plazebo-kontrollierten Studien gezeigt
werden, dass die langfristige Einnahme von COX-2-Hemmern bei Patienten, die anamnestisch
ein kolorektales Adenom aufwiesen, mit erhöhten kardiovaskulären Nebenwirkungen in
Verbindung gebracht wurden (Bresalier et al., 2005; Solomon et al., 2005). Außerdem konnte
bei Patienten mit einem erhöhten kardiovaskulären Risiko die Kurzzeitbehandlung mit
Valdecoxib oder Parecoxib mit einer höheren Anzahl von starken thromboembolischen
Ereignissen assoziiert werden (Nussmeier et al., 2005).
Obwohl keine plazebokontrollierten, randomisierten Studien durchgeführt wurden, um das
kardiovaskuläre Risiko von traditionellen NSAR zu untersuchen, zeigten bevölkerungsbasierte
Fall-Kontroll-Studien deutlich, dass der gegenwärtige Gebrauch von COX-2 Hemmstoffen und
traditionellen NSAR mit einer erhöhten Anzahl von Myokardinfarkten assoziiert war (Hippisley-
Cox and Coupland, 2005). Außerdem demonstrierte die kürzlich veröffentlichte MEDAL-Studie,
dass NSAR (Diclofenac) und COX-2-Hemmer (Etoricoxib) ein vergleichbares kardiovaskuläres
Risikopotential aufwiesen (Cannon et al., 2006). Deshalb wurde vorgeschlagen, dass keine
Unterscheidung zwischen diesen Gruppen von Medikamenten hinsichtlich der kardiovaskulären
Toxizität notwendig ist. Die ursprüngliche Annahme, dass NSAR durch die Hemmung der
prothrombotischen COX-1 zur Senkung des kardiovaskulären Risikos führen sollte, konnte nicht
bestätig werden, weil die COX-1-Aktivität in Thrombozyten über 95% unterdrückt werden muss,
um die Hemmung der Thrombozytenaggregation hervorzurufen (Reilly and FitzGerald, 1987).

Einleitung 27
Jedoch wurde in einer Meta-Analyse gezeigt, dass das Auftreten von ernsten vaskulären
Ereignissen von Naproxen mit Placebo vergleichbar ist (Kearney et al., 2006).
3.5.2 Selektive Cyclooxygenasehemmer
Viele unerwünschte Effekte der NSAR wie z.B. Magen- und Darm-Ulzerationen, Blutungen und
Thrombozytenfunktionsstörungen wurden mit einer Hemmung der COX-1-abhängigen
protektiven Prostaglandine assoziiert, wohingegen die erwünschten therapeutischen,
antientzündlichen, schmerzlindernden und fiebersenkenden Effekte dieser Verbindungen mit
einer Hemmung der COX-2-abhängigen proinflammatorischen Prostaglandine in Verbindung
gebracht wurden. Als Folge entstand die Hypothese, dass die selektive Hemmung der COX-2
die traditionellen therapeutischen Wirkungen der NSAR ohne deren unerwünschten
Nebenwirkungen hervorrufen könnte. Diese Idee führte letztlich zur Entwicklung von selektiven
COX-2-Hemmstoffen.
Die selektiven COX-2-Inhibitoren, die sich zurzeit in klinischer Anwendung befinden, sind
Celecoxib (Sulfonamid), Etoricoxib (Methylsulfon) und Parecoxib, ein intravenös applizierbares
Prodrug von Valdecoxib (Sulfonamid).
Tabelle 2: Relative Potenz der selektiven COX-Inhibitoren hinsichtlich ihrer COX-1- und COX-2- Hemmungen.
Substanz COX-1 / COX-2 IC50-Ratio
Celecoxib 30
Etoricoxib 344
Lumiracoxib 515
Die Daten wurden ex-vivo mit dem humanen Vollblutassay generiert (Brune and Hinz, 2004). Lumiracoxib weist dabei die höchste COX-2-Selektivität auf, gefolgt von Etoricoxib und Celecoxib.
Die nicht saure COX-2-Hemmer Celecoxib und Etoricoxib verteilen sich homogen im Körper.
Aufgrund seiner hohen Lipophilie wird Celecoxib relativ langsam und unvollständig resorbiert.
Etoricoxib wird dagegen schnell resorbiert, jedoch langsam aus dem Körper eliminiert (t1/2: ca.
20–26 h). (Hinz et al., 2006). Die unterschiedlichen Pharmakokinetiken der Coxibe sind
Grundlage für die verschiedenen Indikationen der entsprechenden COX-2-Hemmstoffe. So ist
z.B. Celecoxib für die Behandlung von akuten Schmerzen aufgrund seiner langsamen
Resorption und des variablen First-pass-Metabolismus ungeeignet. Hingegen erscheint
Etoricoxib aufgrund seiner langen Plasmahalbwertszeit besonders für Indikationen, bei denen
eine verlängerte Wirkdauer (akute Gichtarthritis, rheumatische Arthritis) erforderlich ist, sehr

Einleitung 28
vielversprechend. Parecoxib, als ein intravenös applizierbares Coxib ist für die Behandlung des
postoperativen Schmerzens gut geeignet. Ein anderer hochselektiver Cyclooxygenase-2-
Hemmer, der für die symptomatische Behandlung von Osteoarthritis und akutem Schmerz
verwendet wurde ist das Phenylacetylsäurederivat Lumiracoxib (Bannwarth and Berenbaum,
2007). Lumiracoxib wurde in Januar 2007 in Deutschland zugelassen, jedoch bereits im
Dezember 2007 wieder vom Markt zurückgenommen.

Einleitung 29
F Cl
NH
COOH
H3C
3.5.2.1 Lumiracoxib ein hochselektiver COX-2-Hemmer
Chemische Bezeichnung: 2-((2-chloro-6-fluorophenyl)amino)-
5-methyl- Benzenessigsäure
Synonym: COX189, Prexige®
Summenformel: C15H13ClFNO2
Molekulargewicht: 293,7 g/mol
Abbildung 6: Strukturformel von Lumiracoxib
Lumiracoxib unterscheidet sich von anderen existierenden selektiven COX-2-Hemmer durch
das Fehlen eines Schwefelanteils und durch eine Carboxylgruppe wodurch es leicht saure
Eigenschaften (pKa 4,7) aufweist. Als ein chemisches Analogon zu Diclofenac besitzt
Lumiracoxib im Vergleich zu den anderen selektiven COX-2-Inhibitoren (Abschnitt 3.5.2)
einzigartige pharmakokinetische Eigenschaften. Es wird rasch resorbiert (tmax 2 h), besitzt eine
relativ kurze Plasmahalbwertzeit (Brune and Hinz, 2004) und weist anhaltend (12 bis 24 h nach
Einnahme) hohe Konzentrationen in der Synovial-Flüssigkeit bei Patienten mit rheumatoider
Arthritis auf (Scott et al., 2004). Lumiracoxib wird durch die Oxidation von Cytochrom P450 2C9
nahezu vollständig metabolisiert (Mangold et al., 2004). Die Hauptmetaboliten von Lumiracoxib
im Plasma, die durch die Oxidation der 5-Methyl-Gruppe, oder Hydroxylierung seines
dihaloaromatischen Rings entstehen, sind der 5-Carboxy, 4'-Hydroxy und 4'-Hydroxy-5-Carboxy
Derivat (Esser et al., 2005).
Unter den verfügbaren COX-2-Hemmstoffen ist Lumiracoxib der einzige Arzneistoff für den in
einer großen Patientenstudie Vorteile hinsichtlich gastrointestinaler Nebenwirkungen bei
Patienten mit Osteoarthritis demonstriert werden konnten. Lumiracoxib senkte dabei im
Vergleich zu Ibuprofen und Naproxen die Häufigkeit von Ulkuskomplikationen im oberen
Bereich des Gastrointestinaltraktes um mehr als 70% (Schnitzer et al., 2004b).
In mehreren Ländern wurde Lumiracoxib mit den Dosierungen von 200 mg und 400 mg zur
Behandlung der Osteoarthritis bzw. des akuten Schmerzens zugelassen. Im Falle beider
Dosierungen wurde die für die COX-2-Hemmung notwendige Plasmakonzentration deutlich
überschritten (Cheremina et al., 2006; Hinz et al., 2007). Diese Beobachtung stimmt mit einer
bereits veröffentlichten klinischen Studie überein. Dabei wurden Patienten mit einer Knie- oder
Hüftosteoarthritis hinsichtlich der Lumiracoxibdosis randomisiert. Die Patienten nahmen
Lumiracoxib von 50 mg, 100 mg oder 200 mg zweimal täglich oder 400 mg einmal täglich und
dabei wurde eine vergleichbare Schmerzlinderung beobachtet (Schnitzer et al., 2004a).
Bedauerlicherweise fehlt eine systematische Analyse bezüglich der dosisabhängigen
therapeutischen Effekte von Lumiracoxib.

Einleitung 30
Im August 2007 hat Australia´s Therapeutic Goods Administration die Zulassung von
Lumiracoxib auf Grund von acht Fällen von Lebertoxizität, einschließlich zweier Todesfälle
zurückgenommen. Diese Patienten hatten Lumiracoxib in täglichen Dosen von ≥ 200 mg
eingenommen (Therapeutic Goods Administration, 13. August 2007; Burton, 2007). Obwohl die
Daten des Herstellers (Novartis, 2007) darauf schließen lassen, dass die in Europa
zugelassene Dosierung von einmal täglich 100 mg Lumiracoxib im Vergleich zu anderen NSAR
nicht mit einem erhöhten hepatischen Risiko assoziiert ist (Rubenstein and Laine, 2004), hat die
European Medicines Agency (EMEA) ebenfalls wegen einiger nicht tödlicher Fälle von
Lebertoxizitäten die Rücknahme von Lumiracoxib im Dezember 2007 empfohlen (European
edicines gency, 13 Dezember 2007).

Ziele 31
4 Ziele
Korrelationsanalysen zwischen pharmakokinetischen und pharmakodynamischen Parametern
aus menschlichen Blutproben ermöglichen es verschiedene Zielsetzungen zu untersuchen. Das
primäre Bestreben war es, einen Beitrag zur Aufklärung der COX-inhibierenden Eigenschaften
der nicht sauren Antipyretika Metamizol und Paracetamol nach oraler Gabe und somit zu ihrem
Wirkungsmechanismus zu leisten. Ein weiteres Ziel war die Untersuchung des Einflusses der
verschiedenen Dosierungen von Lumiracoxib auf die Dauer und das Ausmaß der COX-2-
Hemmung nach einmaliger oraler Einnahme, um die kleinste effektive Dosis für die
analgetische Wirkung zu bestimmen. Zuvor musste deshalb eine analytische Methode zur
Bestimmung von Lumiracoxibplasmakonzentration entwickelt und validiert werden. Als letzte
Fragestellung wurde die thrombozytenhemmende und potenziell kardioprotektive Wirkung des
nicht selektiven sauren Antipyretikums Naproxen, untersucht. Genauere Beschreibung der
Zielsetzungen erfolgt im Weiteren.
4.1 Untersuchungen zum Wirkungmechanismus von Metamizol
Auf der Basis von IC50-Werten, die in in-vitro-Studien von Campos (Campos et al., 1999)
ermittelt wurden, kann man vermuten, dass Metamizol auch in-vivo eine selektive COX-2-
Hemmung hervorrufen könnte - eine potentiell wichtige Frage, die das reduzierte Risiko
gastrointestinaler Blutungen nach Verabreichung von Metamizol im Vergleich zu
Acetylsalicylsäure und anderen NSAR erklären könnte (Laporte and Carne, 1987; Laporte et al.,
1991). Jedoch fehlte bisher der Nachweis aus in-vivo Studien. Weiterhin bestand Unklarheit
bezüglich der Hemmung peripherer COX-Enzyme durch Metamizol. Dementsprechend fehlt
immer noch eine ausführliche vergleichende Analyse, die sich mit der Selektivität,
Dosisabhängigkeit und Dauer der Hemmung peripherer COX-Enzyme in Probanden nach oraler
Verabreichung von Metamizol beschäftigt. Überdies, trotz der wohlbekannten hemmenden
Wirkung des Metaboliten 4-Methylaminoantipyrin (4-MAA) sowie der Inaktivität der Metaboliten
4-Acetylaminoantipyrin und 4-Formylaminoantipyrin auf die Produktion von Prostanoiden
(Weithmann and Alpermann, 1985) bleibt die Frage unbeantwortet, ob 4-Aminoantipyrin (4-AA)
nach Einnahme von Metamizol zur Hemmung der Eicosanoiden-Bildung beiträgt (Levy et al.,
1995). Aus all diesen Gründen untersuchte die vorliegende Studie den Beitrag der
Hauptmetaboliten (4-MAA und 4-AA) zum Wirkungsmechanismus von Metamizol hinsichtlich
der Hemmung der Prostanoid-Synthese.

Ziele 32
4.2 Untersuchungen zur Wirkungsmechanismus von Paracetamol
Das pharmakologische Profil von Paracetamol ist dem der selektiven COX-2 Hemmer (Coxibe)
sehr ähnlich. Wie Coxibe, ruft Paracetamol bei oralen empfohlenen Einzeldosen keine toxische
Wirkung im Gastrointestinaltrakt hervor (Garcia Rodriguez and Hernandez-Diaz, 2001), hemmt
nicht die Thrombozytenaggregation (Mielke, 1981; Catella-Lawson et al., 2001) und provoziert
kaum Bronchokonstriktionen in Aspirin-empfindlichen Asthmatikern (Jenkins et al., 2004). Die
Tatsache, dass Paracetamol funktionell den Coxiben ähnlich ist, führte zur Untersuchung der
Hypothese, ob Paracetamol durch selektive COX-2 Hemmung wirkt.
4.3 Entwicklung und Validierung einer analytischen Methode zur
Bestimmung von Lumiracoxib in Plasma von menschlichen
Probanden
Um die Plasmakonzentrationen von Lumiracoxib in weitere Studie zu bestimmen, wurde es
nötig eine passende dafür Methode zu entwickeln und zu validieren.
4.4 Untersuchungen zur COX-2-Hemmung durch Lumiracoxib in
verschiedenen Dosierungen
Berichte über eine dosisabhängige Lebertoxizität von Lumiracoxib führten in jüngster Zeit zu
einem gesteigerten Interesse an einer Behandlung mit niedrig dosiertem Lumiracoxib.
In Anbetracht des gastrointestinalen Vorteiles von Lumiracoxib, seiner offenbar
dosisabhängigen hepatischen Toxizität (Schnitzer et al., 2004b) sowie einer möglichen
Überdosierung in einigen Ländern bei langfristigem Gebrauch, wurden Dosierungen unter
100 mg für die Schmerztherapie in Betracht gezogen. Die deshalb durchgeführte vorliegende
Studie ist eine Analyse, die die Dauer und das Ausmaß der COX-2-Hemmung in Abhängigkeit
von der Lumiracoxibdosis (50 mg, 100 mg bzw 200 mg) untersuchte.
4.5 Untersuchung zur thrombozytenhemmenden und potenziell
kardioprotektiven Wirkung von Naproxen
Während der letzten Jahre, wurde eine potenzielle kardioprotektive Wirkung für die 2-
Arylpropionsäure Naproxen diskutiert. Eine Meta-Analyse von 138 veröffentlichten und
unveröffentlichten, randomisierten Untersuchungen (Kearney et al., 2006) deutete daraufhin,
dass das Auftreten von ernsten vaskulären Ereignissen zwischen einem COX-2-Hemmer und

Ziele 33
jedem NSAR außer Naproxen ähnlich ist, und dass das Risiko von Naproxen mit Placebo
vergleichbar ist. Da in publizierten Studien bereits vorgeschlagen wurde, dass die 95%-ige
Hemmung der TXB2-Synthese im Serum die Thrombozytenfunktion maßgeblich beeinflußt
(Reilly and FitzGerald, 1987), könnte ein möglicher Mechanismus für die potenzielle
kardioprotektive Wirkung von Naproxen die ausgeprägte und lang andauernde Hemmung der
COX-1 sein.
Die vorliegende Studie untersuchte, ob Dosierungen von freiverkäuflichem (OTC) Naproxen
Natrium eine ausreichende COX-1-Hemmung nach einmaliger Gabe und im Steady-state
erzeugen können. Parallel dazu wurde auch die COX-2-Hemmung untersucht.

Experimenteller Teil 34
5 Experimenteller Teil
5.1 Materialien
5.1.1 Lösungen
Natriumacetat Puffer 30 mM 2,46 g Natriumacetat in 1 l H2O, mit Essigsäure auf pH 5,6
einstellen
Phosphatpuffer 20 mM 3,12 g Natriumdihydrogenphosphat Dihydrat in 1 l H2O, mit
Phosphorsäure auf pH 2,5 einstellen
PBS pH=7,4 NaCl 40 g
KCl 1 g
KH2PO4 1 g
Na2HPO4 × 12H2O 14,5 g
In 1 l H2O, autoklavieren
SDS-Polyacrylamidgel 10% Acrylamid 30 % / Bis 37,5 : 1 10,0 ml
1,5 M Tris-HCl, pH 8,8 1,5 ml
SDS 10 % 300 µl
Tetramethylethylendiamin 12,0 µl
Milliporwasser 11,9 ml
Ammoniumperoxodisulfat 10 % 300 µl
Solubilisierungspuffer HEPES pH 7,4 50 mM
NaCl 150 mM
EDTA 1 mM
Triton® X-100 1 % (v/v)
Glyzerin 10 % (v/v)
Phenylmethylsulfonyl Fluorid 1 mM
Leupeptin 1 µg/ml
Aprotinin 10 µg/ml
Magermilchlösung Milchpulver 5 % in PBS
Trichloressigsäure 0,05 % Trichloressigsäure 0,5 g in 1 l H2O
Experimenteller Teil 35
5.1.2 Chemikalien
Acetaminophen Sigma, Steinheim, Deutschland
Acetonitril (HPLC Grade) Merk, Darmstadt, Deutschland
Antipyrin Sigma, Steinheim, Deutschland
Aspirin Sigma, Steinheim, Deutschland
ß-actin Calbiochem, Bad Soden, Deutschland
COX Inhibitor Screening Assay Cayman Chemicals, Ann Arbor,
Massachussets, USA
COX-2-Antikörper BD Biosciences, Heidelberg, Deutschland
Diethylether (für LC) Merk, Darmstaft, Deutschland
Dimethylsulfoxid Merk, Darmstaft, Deutschland
Dinatriumhydrogenphosphat-Dihydrat Merk, Darmstaft, Deutschland
Essigsäure Merk, Darmstadt, Deutschland
Fab-spezifische Antimaus IgG Sigma, Steinheim, Deutschland
Flurbiprofen Sigma, Steinheim, Deutschland
Kaliumdihydrogensphosphat Merk, Darmstaft, Deutschland
Lumiracoxib Novartis, East Hanover, USA
Lipopolysaccharid von Escherichia coli
(Serotyp 026:B6) Sigma, Steinheim, Deutschland
Methanol Merk, Darmstadt, Deutschland
Methylaminoantipyrin Sanofi-Aventis, Frankfurt/Main, Deutschland
Natriumazetat Sigma, Steinheim, Deutschland
Natriumdihydrogensphosphat Dihydrat Merk, Darmstaft, Deutschland
Natriumhydroxid Merk, Darmstadt, Deutschland
Nifluminsäure Sigma, Deisenhofen, Deutschland
n-Hexan Merk, Darmstadt, Deutschland
Perchlorsäure Merk, Darmstadt, Deutschland
PGE2 Enzyme Immunoassay Kit Cayman Chemicals, Ann Arbor,
Massachussets, USA
RPMI-1640 Medium Invitrogen, Karlsruhe, Deutschland
Salzsäure Merk, Darmstadt, Deutschland
Trichloressigsäure Fluka, Steinheim, Deutschland
TXB2-Enzyme Immunoassay Kit Cayman Chemicals, Ann Arbor,
Massachussets, USA
Wasser aus Milli-Q-Anlage Millipore, Neu-Isenburg
Westernblot-Detektionsreagenzien Amersham Pharmacia Biotech, Deutschland
Experimenteller Teil 36
5.1.3 Verbrauchsmaterialien
Adapter Combitips plus 50 ml Eppendorf AG, Hamburg
Bechergläser SCHOT Duran
Blut-Monovetten mit Heparin Saarstedt, Nümbrecht
Blut-Monovetten mit EDTA, Saarstedt, Nümbrecht
Blut-Monovetten neutral Saarstedt, Nümbrecht
Braunüle B.Braun, Melsungen
Cutasept® F BODE CHEMIE, Hamburg
Einmalspritze 2 ml B.Braun, Melsungen
Eppendorf Pipetten Eppendorf AG, Hamburg
Falkon Tube 14 ml Falcon Becton Dickinson and
Company Labware, Heidelberg
Injektionsnadel: grün, gelb B.Braun, Melsungen
Injektionspflaster Paul Hartmann AG, Heidenheim
Multiadapter Sarstedt, Nümbrecht
Multipette® plus Eppendorf AG, Hamburg
Multipetteaufsätze: Combitips plus 0,5/ 5/ 10 /50 ml Eppendorf AG, Hamburg
Obturator 20GA 1,1*30 mm Beckton Dickinson, Spanien
Pasteur-Pipetten Glas 15 mm VWR International
Pipettenspitze 10 µl, 200 µl, Sarstedt, Nümbrecht
Pipettenspitze 1000 µl Eppendorf AG, Hamburg
Reagenzgläser Soda, gl. Rand, rd. Boden VWR International
Reagiergefäß 1,5 ml Sarstedt, Nümbrecht
Röhre mit Verschluß 101×16,5 mm Sarstedt, Nümbrecht
Tupfer Paul Hartmann AG, Heidenheim
Venenkatheterverband Beckton Dickinson, Spanien
Venenwerweilkanüle 20GA 1,1*30 mm Beckton Dickinson, Spanien

Experimenteller Teil 37
5.1.4 Geräte
Autosampler, Model 231 Gilson, Abimed
Blotkammer Protean®, BIO-RAD
Dilutor, Model 401 Gilson, Abimed
Elektrophoresekammer Sigma-Aldrich, Steinheim, Deutschland
Elektronische Analysenwaage AE 260
Delterange® Mettler Waage GmbH, Schweiz
HPLC-Pumpe Spectra System P 4000 Finnigan MAT
HPLC-System (Entgaser, Säulenofen) Finnigan MAT
HPLC-Säule CC125/4 Nucleosil 120-3 C8 Macherey-Nagel, Düren, Deutschland
HPLC-Säule CC125/4 Nucleodur C18 Pyramid,
3 µmol/l Macherey-Nagel, Düren, Deutschland
HPLC-Säule CC125/4 Nucleosil 120-3 C18 Macherey-Nagel, Düren, Deutschland
Laborwaage PE 3600 Mettler Waage GmbH, Schweiz
optischen Scanner Gel Doc 2000; Bio-Rad-Laboratories,
Hercules, CA
Photometer für 96-well Platten DIAS, Dynatech Laboratories
pH Meter Model pH 211 HANNA Instruments, Portugal
SHAKER DRS-12 Sky-Line ELMI LTF LabortechnikGmbH & CO.KG,
Wasserburg, Deutschland
Ultraschalbad Bioblock Scientific, Deutschland
UV-Detektor Spectra 100 Spectra-Physics, San Jose, CA, USA
Überkopfschüttler GFL 3025 GmbH für Labortechnik, Burkwedel,
Deutschland
Vorsäule CC 8 / 4 Nucleodur C8 3µ Macherey-Nagel, Düren, Deutschland
Vorsäule CC 8 / 4 Nucleosil C8 3µ Macherey-Nagel, Düren, Deutschland
Vorsäule CC 8 / 4 Nucleosil C18 3µ Macherey-Nagel, Düren, Deutschland
Wasseraufbereitungssystem TKA TKA GmbH Niederelbert
Wasserbad und Thermostat GmbH für Labortechnik, Burkwedel,
Deutschland
Zentrifuge 5810R Eppendorf AG, Hamburg
Experimenteller Teil 38
5.1.5 Software
Borwin Version 1,5 Jasco, Groß-Umstadt
Exel Microsoft, Unterschleißheim
Multi-Analyst Programm, Version 1.1 Bio-Rad-Laboratories, Hercules, CA
PRISM Version 3.0 GraphPad, San Diego, CA, USA
WinNonlin Version 3.3 Pharsight, Gebirgssicht, CA, USA
Word Microsoft, Unterschleißheim
5.1.6 Medikamente
Arzneistoff Wirkstoff Hersteller
Novalgin® Filmtabletten 500 mg Metamizol Sanofi-Aventis, Frankfurt am
Main, Deutschland
Paracetamol-ratiopharm® 500 mg Paracetamol Ratiopharm GmbH, Ulm,
Deutschland
Prexige® 100mg Filmtabletten Lumiracoxib Novartis Pharma GmbH,
Nürnberg, Deutschland
Aleve® 200 mg Naproxen Bayer Vital GmbH,
Leverkusen, Deutschland

Experimenteller Teil 39
5.2 Methoden
5.2.1 Pharmakokinetische Untersuchungen
Die Quantifizierung von Arzneistoffen und deren Metabolite in Plasma erfolgte in allen in dieser
Dissertation beschriebenen Studien mit Hilfe von Hochleistungsflüssigkeitschromatography mit
ultravioletem Detektor (HPLC-UV-Methoden).
HPLC stellt ein Trennverfahren dar, bei dem die Probenflüssigkeit mittels einer flüssigen Phase
(Eluent) unter hohem Druck über die stationäre Phase (Trennsäule) transportiert wird. Wenn die
stationäre Phase unpolar ist, geht es um umgekehrt (engl.: "reversed") Phase (RP). Die RP-
HPLC ist in der Praxis die gängigste Methode. 70 % aller analytischen HPLC-Trennungen und
alle Methoden in dieser Arbeit sind RP-Trennungen. Als mobile Phase werden meist
Mischungen aus Wasser oder Puffer und Acetonitril oder Methanol eingesetzt. In allen
vorliegenden Untersuchungen wurde isokratische Trennung verwendet, bei der die
Zusammensetzung der mobilen Phase während der gesamten Zeit gleich bleibt. Bei
Gradiententrennungen wird dagegen die Polarität des Fließmittelgemisches während der
Analyse verändert. Elutionskraft bei RP-HPLC steigt mit senkender Polarität des Eluentes, weil
da der Eluent in grösserer Konkurrenz zur festen Phase ist. Besondere Anwendung findet die
RP-HPLC bei der Auftrennung von polaren Analyten, die auf Normalphasen zu hohe
Retentionszeiten aufweisen würden. Die Darstellung des Ergebnisses der Stofftrennung erfolgt
in Form eines Chromatogrammes, einer Elutionskurve. Sie stellt die Abhängigkeit für die Menge
(Konzentration) der eluierten Substanzen von der Zeit dar.
Zur Detektion wurde UV / VIS-Spektrometer verwendet. Die UV / Vis-Spektroskopie ist eine
Spektroskopie, die elektromagnetische Wellen des ultravioletten (UV: 1-380 nm) und des
sichtbaren (VIS: 380-780 nm) Lichts nutzt.
5.2.2 Pharmakodynamische Untersuchungen
In dieser Arbeit wurde der Einflüss von selektiven und nicht selektiven COX-Inhibitoren auf die
Aktivität der Cyclooxygenasen in Vollblut, das das periphere Kompartiment im menschlichen
Körper repräsentiert, untersucht.
COX-1 und COX-2 Aktivitäten im Vollblut wurden nach der Methode von Patrignani et al.
bestimmt (Patrignani et al., 1994). Diese Methode gibt einen direkten Aufschluss über das
inhibitorische Potential einer Substanz bezüglich COX-1 indem die Hemmung der
Thromboxansynthese von aktivierten Thrombozyten während der Blutgerinnung untersucht wird.
Das Hemmpotential einer Substanz bezüglich COX-2 wird durch die Hemmung der
Prostaglandin E2-Synthese in Lipopolysaccharid-induzierten Monozyten bestimmt. Dieser Test

Experimenteller Teil 40
ermöglicht die Untersuchung der COX-Aktivität unter physiologischen Bedingungen bei denen
u.a. die Plasmaproteinbindung berücksichtigt wird.
Das Blut wurde durch Venenpunktion von gesunden Freiwilligen gewonnen, die zwei Wochen
vor der Blutentnahme keine NSAR genommen hatten.
5.2.2.1 COX-1-Aktivität im menschlichen Vollblut
Aliquoten des Vollblutes ohne Antikoagulans (500 µl) wurden unmittelbar nach der
Blutentnahme in die Reagenzgläser übertragen. Für die in-vitro-Versuche wurden die
Reagensgläser mit Testagent oder PBS (als Kontrolle) versetzt. Blutproben sind im Wasserbad
eine Stunde bei 37°C geronnen. Serum wurde durch Zentrifugation bei 1500×g, 4°C und 5
Minuten getrennt und Thromboxan B2-Konzentrationen wurden gemessen.
5.2.2.2 COX-2-Aktivität im menschlichen Vollblut
Für die in-vitro-Versuche wurden die Reagensgläser mit Testagent oder PBS (als Kontrolle)
versetzt. Blut von Probanden wurde in Heparinbeschichteten Röhrchen gesammelt und in die
Reagenzgläser übertragen. Weiterhin wurden die Blutproben mit LPS (10 µg/ml) und ASS
(10 µg/ml) 24 Stunden bei 37°C im Wasserbad inkubiert. Anschließend wurde das Plasma
durch Zentrifugation bei 1500×g, 4°C und 5 Minuten abgetrennt und PGE2-Konzentration wurde
bestimmt.
5.2.2.3 Quantifizierung der Konzentration von Prostanoid im Vollblut mittels
ELISA
ELISA ist ein Assay, der auf Immunfixation der gesuchten Substanz an mit spezifischem,
monoklonalen Antikörper gegen diese Substanz beschichteter Oberfläche einer Kunststoffplatte
basiert. Nicht fixierte Substanzen werden abgewaschen. Ein Enzym-markierter
Sekundärantikörper wird in einem Sandwichverfahren an das gebundene Substrat gebunden.
Das markierende Enzym ist in der Lage, zugegebenes Substrat unter Farbumschwung
umzusetzen. Die Intensität des Farbumschlages kann photometrisch bestimmt werden und
korreliert mit der Konzentration der zu bestimmenden Substanz.
5.2.2.3.1 Messung von TXB2
Es wurde ein kommerziell erhältliches Fertigkit von Cayman Chemicals (Ann Arbor,
Massachussets, USA) verwendet.
Die Messplatte wurde gebrauchsfertig geliefert. Proben- und Waschpuffer wurden als
Trockensubstanz geliefert und mussten nach Herstellervorgabe in H2O gelöst werden. Tracer

Experimenteller Teil 41
(Konjugat) und Antikörper wurden ebenfalls nach Herstellervorgabe im Probenpuffer gelöst.
Eine Verdünnungsreihe des Standards von 1000 bis 7,8 pg/ml wurde nach Herstellervorgabe
mit dem Assaypuffer angesetzt. Die 96-Well-Platte wurde mit Nullwert, 50 µl des Standards und
50 µl Probe / Well als Dreifachbestimmung befüllt, die Proben ggf. vorher nach Bedarf mit
Probenpuffer verdünnt. Nach Zugabe von 50 µl Antikörper und 50 µl Konjugat / Well wurde die
Platte mit Plastikfilm zugedeckt und 18 Stunden bei Raumtemperatur inkubiert. Anschließend
wurde die Flüssigkeit abgekippt, die Platte fünf Mal mit Waschpuffer gewaschen, getrocknet, mit
200 µl des Ellmann´s Reagent / Well befüllt und zugedeckt bei Raumtemperatur auf dem
Schüttler 60 bis 90 Minuten inkubiert (entwickelt). Dann wurde die Platte im Bereich von 405 bis
420 nm abgelesen. Die Berechnung der endgültigen Konzentrationen erfolgte mit Hilfe
spezieller Exel-Tabelle.
5.2.2.3.2 Messung von PGE2
Es wurde ein kommerziell erhältliches Fertigkit von Cayman Chemicals (Ann Arbor,
Massachussets, USA) verwendet.
Die Messplatte wurde gebrauchsfertig geliefert. Proben- und Waschpuffer wurden als
Trockensubstanz geliefert und mussten nach Herstellervorgabe in H2O gelöst werden. Tracer
(Konjugat) und Antikörper wurden ebenfalls nach Herstellervorgabe im Probenpuffer gelöst.
Eine Verdünnungsreihe des Standards von 1000 bis 7,8 pg/ml wurde nach Herstellervorgabe
mit dem Assaypuffer angesetzt. Die 96-Well-Platte wurde nach Plan mit Nullwert, 50 µl des
Standards und 50 µl Probe / Well als Dreifachbestimmung befüllt. Die Proben wurden vorher
nach Bedarf mit Probenpuffer verdünnt. Nach Zugabe von 50 µl Antikörper und 50 µl Konjugat /
Well wurde die Platte mit Plastikfilm zugedeckt und 18 Stunden bei 4°C im Kühlschrank
inkubiert. Anschließend wurde die Flüssigkeit abgekippt, die Platte fünf Mal mit Waschpuffer
gewaschen, getrocknet, mit 200 µl des Ellmann´s Reagent / Well befüllt und zugedeckt (im
Dunklen) bei Raumtemperatur auf dem Schüttler 60 bis 90 Minuten inkubiert (entwickelt). Dann
wurde die Platte im Bereich von 405 bis 420 nm abgelesen. Die Berechnung der endgültigen
Konzentrationen erfolgte mit Hilfe spezieller Exel-Tabelle.
5.2.3 Korrelationsanalyse zwischen Pharmakodynamik und
Pharmakokinetik
5.2.3.1 Pharmakodynamische Analyse
Das Ausmaß der COX-1 oder COX-2-Hemmung wurde als die Prozentänderung der
Eicosanoidkonzentration im Blut (COX-1: TXB2; COX-2: PGE2) in verschiedenen Zeitpunkte
nach der Medikamentengabe im Vergleich zu Eicosanoidkonzentrationen vor der
Medikamentengabe berechnet. Im Fall von COX-2 wurden die PGE2-Werte, die ohne LPS-

Experimenteller Teil 42
Induktion gemessen wurden, von LPS-induzierten PGE2-Werten substrahiert. Die maximale
Hemmung der COX Isoformen und Zeit, um dies zu erreichen, wurde direkt von den Wirkungs-
Zeitkurven erhalten. Die Fläche unter der Wirkungs-Zeitkurven (AWECs) von 0 bis bestimmten
Zeitpunkt nach Dosisverabreichung wurde mit Hilfe der linearen trapezoidal-Regel berechnet.
5.2.3.2 Pharmakokinetische Analyse
Plasmakonzentration-Zeitkurven von untersuchten Medikamenten und deren Metabolite (Studie
mit Metamizol) wurden durch nicht kompartimentale Analyse bestimmt, wozu die WinNonlin
Version 3,3 (Pharsight, Gebirgssicht, CA, USA,) benutzt wurde. Die maximale
Plasmakonzentrationen (Cmax) und die Zeiten bis zu Cmax (tmax) wurden direkt von der
individuellen Plasmakonzentration gegen Zeitkurven ermittelt. Die Fläche unter der
Plasmakonzentration-Zeitkurve bis zur letzten quantitativen Plasmakonzentration (AUCt) wurde
mittels der linearen trapezoidalen Methode errechnet.
5.2.3.3 Berechnung der Korrelation zwischen Pharmakodynamik und
Pharmakokinetik
Für das Bewerten der Korrelation zwischen Plasmakonzentrationen von Medikamenten und
Änderungen in COX-1 oder COX-2-Hemmung wurden Plasmakonzentrations-Aktivitätskurven
mittels einer sigmoidalen Regression mit variablem Enstieg errechnet. Die IC50-Werte für die
COX-1 bzw. COX-2-Hemmung wurden mit Hilfe der PRISM® Version 3,0 (GraphPad, San Diego,
CA, USA) bestimmt.

Experimenteller Teil 43
5.2.4 Validierung einer analytischen Methode
Unter Validierung versteht man den Nachweis und die Dokumentation der Zuverlässigkeit einer
Methode für die Bestimmung eines Analyten in einer definierten Matrix. Die
Methodenvalidierung in der Bioanalytik muss zeigen, dass eine bestimmte Methode zur
Quantifizierung eines Analyten in einer biologischen Matrix, wie beispielsweise Plasma bzw.
Serum verlässlich und reproduzierbar ist.
In dieser Arbeit wurde die Validierung einer neu entwickelten Methode auf der Basis der von
Shah (2000) zusammengefassten Richtlinien der Konferenz „Analytical methods validation:
Bioavailability, bioequivalence and pharmacokinetic studies“ (Shah et al., 2000) durchgeführt.
Versuchsreihen zur Spezifität, Linearität, Präzision und Richtigkeit sind für bioanalytische
Methoden von essentieller Bedeutung.
5.2.4.1 Spezifität und Selektivität
Spezifität ist die Fähigkeit einer Methode, eine bestimmte Substanz ohne Verfälschung durch
andere in der Probe vorhandene Komponenten zu erfassen und sie somit eindeutig zu
identifizieren.
Selektivität ist die Fähigkeit einer Methode, verschiedene nebeneinander zu bestimmende
Analyten ohne gegenseitige Störungen oder Störungen durch andere endogene oder exogene
Substanzen (Verunreinigungen, Abbauprodukte, Matrix) zu erfassen.
Spezifität und Selektivität wurde nachgewiesen, indem man Leerplasmaproben von sechs
verschiedenen Probanden untersucht hat.
5.2.4.2 Linearität
Die Linearität beschreibt die Fähigkeit einer Methode, über einen festgelegten
Konzentrationsbereich Ergebnisse zu erbringen, die eine direkte Proportionalität zur
Analytkonzentration aufweisen. Der Nachweis der Linearität erfolgt über eine graphische
Auswertung mit Angabe der Regressionsfunktion und des Korrelationskoeffizienten.
Bei einer linearen Regression hängen die Werte linear voneinander ab. Daraus lässt sich
folgende Regressionsgleichung ermitteln:
y = a * x + b
a = Steigung
b = Ordinatenabschnitt
Zur Bestimmung der Linearität der Eichgeraden (kleine, mittlere, große) wurden jeweils fünf
Eichkonzentrationen pro Gerade analysiert. Die einzelnen Eichkonzentrationen wurden jeweils
dreifach bestimmt.

Experimenteller Teil 44
5.2.4.3 Nachweis- und Bestimmungsgrenze
Die Nachweisgrenze (Limit of Detection, LOD) ist die kleinste erfaßbare Menge des Analyten in
einer Probe. Zur Ermittlung der Nachweisgrenze muß das Signal-Rausch-Verhältnis bestimmt
werden. An der Nachweisgrenze soll das Signal größer als das dreifache Grundrauschen sein.
Die Bestimmungsgrenze (Limit of Quantification, LOQ), auch Quantifizierungsgrenze genannt,
ist die kleinste quantifizierbare Konzentration, die noch mit hinreichender Präzision und
Richtigkeit ermittelt werden kann. In der chromatographischen Praxis gilt das neunfache
Rauschen als Bestimmungsgrenze, wonach also folgendes gilt: Bestimmungsgrenze = 3 x
Nachweisgrenze. Signale, die diese Bedingung nicht erfüllen, können somit nicht korrekt
quantifiziert werden und müssen bei der Auswertung entsprechend gekennzeichnet werden.
Die Präzision, berechnet als Variationskoeffizient innerhalb Proben einer Konzentration, die als
Bestimmungsgrenze dient, sollte nicht größer als 20% sein, die Richtigkeit um nicht mehr als ±
20% vom Sollwert abweichen.
5.2.4.4 Richtigkeit
Unter Richtigkeit versteht man das Maß der Übereinstimmung zwischen dem gemessenen Wert
und einem als richtig angesehenen Wert (Sollwert). Zur Bestimmung der Richtigkeit wurden
sieben verschiedenen Lumiracoxibkonzentrationen (10, 50, 100, 500, 1000, 5000 und 10000
ng/ml) jeweils 5-fach extrahiert und gemessen. Aus den Ergebnissen der Untersuchungen, die
an drei unterschiedlichen Analysentagen wiederholt worden sind, wurde die relative Fehler
([Mittelwert der Analysenergebnisse x 100 / eingesetzte Konzentration] - 100) in Prozent
errechnet.
5.2.4.5 Präzision
Die Präzision (Reproduzierbarkeit der Methode) beschreibt die Streuung der einzelnen Werte
um den Mittelwert, wobei zufällige Fehler erfasst werden. Das Maß für die Präzision wird als
eine relative Standardabweichung (Variationskoeffizient) der Meßergebnisse in % angegeben.
Es wird zwischen einer “Intra-day” Präzision und einer “Inter-day” Präzision unterschieden. Mit
der Intra-day-Präzision soll die Streuung der Meßergebnisse, die durch Aufarbeitung und
Messung mehrerer aus einem Pool stammender Aliquote an einem Tag resultiert, festgestellt
werden. Damit soll ermittelt werden, ob die Aufarbeitung einer Serie von Proben zu gleichen
Ergebnissen führt.
Die Inter-day-Präzision zeigt die Streuung der Meßergebnisse von Tag zu Tag. Dadurch soll die
Streuung der Meßergebnisse mehrerer aus einem Pool stammender Aliquote an
unterschiedlichen Tagen ermittelt werden. Diese Streuung zeigt, ob die Art der Aufarbeitung

Experimenteller Teil 45
von Proben an unterschiedlichen Tagen divergiert und inwieweit das Gerät, das zur Messung
der Proben verwendet wird, von Tag zu Tag Schwankungen zeigt.
Zur Bestimmung der Intra-day-Präzision und -Richtigkeit des Messverfahrens wurden jeweils
fünf Proben bei sieben verschiedenen Lumiracoxibkonzentrationen über den gesamten
Messbereich analysiert. Dafür wurden mit 50%-igem wässrigen Methanol Verdünnungen der
Stammlösungen von Lumiracoxib hergestellt, mit denen Leerplasmaproben versetzt wurden, so
dass Plasmakonzentrationen von 10, 50, 100, 500, 1000, 5000 und 10000 ng/ml Lumiracoxib
resultierten. Diese Proben wurden wie unten beschrieben (Abschnitt 6.3.1.2) aufgearbeitet und
in einer Serie mit dem HPLC - UV System vermessen. Zur Bestimmung der Inter-day-Präzision
und -Richtigkeit wurde dieses Verfahren an zwei weiteren Tagen wiederholt. Ergebnisse der
Untersuchungen, die an drei unterschiedlichen Analysentagen wiederholt worden sind, wurde
die relative Standardabweichung (Variationskoeffizient = Standardabweichung x 100 / Mittelwert
der Analysenergebnisse) ermittelt. Die Präzision der Konzentrationsmessungen im
Konzentrationsbereich außerhalb der Bestimmungsgrenze sollte nicht mehr als 15% betragen,
die Richtigkeit sollte nicht um mehr als ± 15% vom Sollwert abweichen.
5.2.4.6 Wiederfindung (Recovery)
Zur Bestimmung der Wiederfindung wurde der Prozentsatz der absoluten Peakhöhen von
Lumiracoxib der aufgearbeiteten Proben der Validierung von den jeweiligen Peakhöhen nach
Einspritzen von Standardlösungen der entsprechenden Konzentrationen berechnet.
5.2.4.7 Stabilität
Die Stabilität einer Substanz in biologischem Material ist von den Lagerungsbedingungen, den
chemischen Eigenschaften, sowie der Probenmatrix abhängig. Müssen Proben über einen
längeren Zeitraum gelagert werden, bevor sie analysiert werden können, sollte die Stabilität der
zu bestimmenden Substanzen gesichert sein. Die Stabilität von Lumiracoxib wurde bei sieben
verschiedenen Konzentrationen (je Dreifachbestimmung) im Plasma nach 5 Wochen bei -70°C
gemessen.

Experimenteller Teil 46
5.3 Studien
5.3.1 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik
und Pharmakodynamik von Metamizol
5.3.1.1 Studienziel
Die vorliegende Studie erforschte den Beitrag von Metamizolmetaboliten (4-MAA und 4-AA)
zum Wirkungsmechanismus von Metamizol hinsichtlich der Prostanoid-Synthese in Vollblut.
5.3.1.2 Studiendesign
Zuerst wurde eine in-vitro-Analyse beider Verbindungen bezüglich der Aktivität der COX-
Enzyme in menschlichem Vollblut durchgeführt. Als Weiteres wurde den Vergleich der Kinetik
von 4-MAA und 4-AA und die begleitende ex-vivo-Hemmung der COX-Enzyme bei gesunden
männlichen Probanden gemacht, die klinisch empfohlene orale Dosen von entweder 500 mg
oder 1000 mg Metamizol erhielten. Dabei handelte es sich um eine zweifach Cross-over nicht
Plazebokontrolierte Probandenstudie. Die Studie wurde am Lehrstuhl für Pharmakologie und
Toxikologie der Universität Erlangen-Nürnberg im Februar-März 2006 durchgeführt.
5.3.1.3 Probandenpopulation
An der Studie nahmen 5 männliche Probanden im Alter von 39 bis 65 (durchschnittlicher Alter:
44,8 Jahre) mit einer Körpermasse 70,2 ± 3,0 kg (mean ± SEM) teil. Alle Probanden waren die
approbierten Ärzte am Institut für Experimentelle und Klinische Pharmakologie und Toxikologie.
Voraussetzung für die Teilnahme war das Einverständnis des Probanden und keine Einnahme
von NSAR zwei Wochen vor der ersten Blutentnahme. Die Ausschlusskriterien entsprachen den
in der Fachinformation für Novalgin® genannten Gegenanzeigen.
5.3.1.4 Studienmedikation
Als Studienmedikation wurde Novalgin® Filmtabletten (Sanofi-Aventis, Frankfurt am Main,
Deutschland) verwendet, das von einer öffentlichen Apotheke bezogen wurde. 1 Tablette
enthält 500 mg Metamizol. Die Probanden nahmen klinisch empfohlene Dosen von 500 mg
oder 1000 mg Metamizol ein.

Experimenteller Teil 47
5.3.1.5 Studienplan
Die Probanden erhielten im Abstand von Minimum 1 Woche einmalig 1 Tablette oder 2
Tabletten von Novalgin® Filmtabletten. Die Prüfmedikation wurde nüchtern mit 150 ml Wasser
zwischen 8:00 und 9:00 Uhr verabreicht. Eine Stunde später wurde Frühstück gereicht. Venöse
Blutproben wurden mit einer Verweilkanüle unmittelbar vor der Tabletteeinnahme (Zeitpunkt 0)
und 0,25; 0,75; 1,5; 3; 5; 8 und 12 Stunden nach der Medikation abgenommen. Für die
Bestimmung von Metamizolmetaboliten wurden die heparinisierten Blutproben zentrifugiert und
die Aliquote vom Plasma wurden bei -20°C bis zu zwei Wochen gelagert.
5.3.1.6 In-vitro Untersuchungen
5.3.1.6.1 Wirkungen von Metamizol Metaboliten auf die COX-1 und COX-2-Aktivität im
menschlichen Vollblut
COX-1: Blut wurde von gesunden Blutspendern entnommen, die zwei Wochen lang vor
Blutentnahme keinen NSAR eingenommen hatten. Aliquote von Vollblut (ohne Antikoagulans)
wurden sofort in Reagenzgläser mit Testsubstanzen (4-MAA bzw 4-AA: 0,01; 0,1; 0,3; 1; 3; 10;
30; 100 oder 300 µmol/l) oder Kontrolle (PBS mit DMSO) pipettiert. Die Endkonzentrationen von
DMSO in Reaktionsgemisch waren 0,1% (v/v) [für Konzentrationen von 0,01-100 µmol/l und
seine Kontrolle] und 0,3% (v/v) [für 300 µmol/l bzw. Kontrolle]. Das Blut wurde für 1 Stunde bei
37°C inkubiert. Das Serum wurde durch Zentrifugation abgetrennt und die Serum-TXB2-
Konzentrationen wurden bestimmt.
COX-2: Aliquote von heparinisiertem Blut von gesunden Blutspendern wurden mit LPS (10
µg/ml) plus Testsubstanzen (4-MAA bzw 4-AA: 0,01; 0,1; 0,3; 1; 3; 10; 30; 100 oder 300 µmol/l)
oder Kontrolle (PBS mit DMSO) für 24 Stunden bei 37°C inkubiert. Die Aktivität der
Blutplättchen-COX-1 wurde durch Zusatz von Aspirin (10 µg/ml) am Anfang der Inkubation
unterdrückt. Das Plasma wurde bei Zentrifugation abgetrennt und anschließend wurde PGE2-
Konzentrationen in Plasma gemessen.
Für beide COX-Assays wurden Konzentrations-Aktivitätskurven durch eine sigmoidale
Regression mit dem variablen Anstieg errechnet. Die IC50-Werte wurden durch den Gebrauch
der PRISM Version 3.0 (GraphPad, San Diego, CA) bestimmt. Die COX-1- bzw. COX-2-
Hemmung wurde als die prozentualle Änderung von Plasmaeicosanoiden (COX-1: TXB2;
COX-2: PGE2) in Gegenwart von Metamizol berechnet. Im Fall vom COX-2 wurden basale
PGE2-Werte von jedem PGE2-Wert in der LPS- und LPS / Metamizol-behandelten Gruppen vor
der Berechnung der Prozentänderung abgezogen.

Experimenteller Teil 48
5.3.1.7 Ex-vivo Untersuchungen
5.3.1.7.1 Hemmung der COX-Aktivitäten in Blut nach einmaliger oraler Gabe von
Metamizol
COX-1: Sofort nach den Blutentnahmen wurden Vollblutproben ohne Antikoagulans für 1
Stunde bei 37°C inkubiert, anschließend zentrifugiert, und die Serum-TXB2-Konzentrationen
bestimmt.
COX-2: Sofort nach den Blutentnahmen wurden heparinisierte Vollblutproben mit 10 µg/ml LPS
für 24 Stunden bei 37° inkubiert. Die Blutplättchen-COX-1-Aktivität wurde durch Zusatz von
Aspirin (10 µg/ml) am Beginn der Inkubation unterdrückt. Das Plasma wurde durch
Zentrifugation abgetrennt und die PGE2-Konzentrationen wurden bestimmt.
5.3.1.7.2 Bestimmung von 4-MAA und 4-AA im menschlichen Plasma
Die Metamizolmetaboliten wurden mit einer modifizierten Methode (Agundez et al., 1994)
analysiert. Für die Probenaufarbeitung wurden 0,2 ml 1 M Natrium Hydroxid und 0,1 ml interner
Standard (10 µg Antipyrin / ml destilliertes Wasser) zu 0,5 ml Plasma hinzufügt. Als
Extraktionsmittel wurde 4 ml Chloroform verwendet. Die Reagenzgläser wurden verschlossen
und die Proben wurden in einem Shaker für 5 min über Kopf geschüttelt und anschließend in
einer Kühlzentrifuge (Eppendorf 5810R) bei 4000 r.p.m. für eine Dauer von 5 Minuten bei 4°C
zentrifugiert. Die organische Phase wurde in einen Reagenzglas überführt und wurde bei
Zimmertemperatur unter einem Strom des Stickstoffes evaporiert. Der Rückstand wurde in
150 µl FM aufgelöst und 100 µl wurde für HPLC-Analyse benutzt. Die Substanzen wurden
mittels RP-Säule 125/4 Nucleodur C18 Pyramid, 3 µmol/l (Macherey-Nagel, Düren,
Deutschland) und eine C18 Vorsäule (3 µmol/l) getrennt. Die mobile Phase bestand aus einer
87:13 (v/v) Mischung von 30 mM Natriumacetat (pH-Wert = 5,6) und Acetonitril. Der Fluss war
1 ml/min. UV-Detektion wurde auf 257 nm gesetzt. 4-AA und 4-MAA wurden eluiert bei 9 min
bzw. 12 min. Die Retentionszeit von Antipyrin (interner Standard) war 7,6 min. Linearität der
Standardkurven wurde über einen Konzentrationsbereich von 50 bis 20 000 ng/ml bestimmt.
Der Messbereich wurde in zwei kleinere Messbereiche (50 - 1000 und 1000 - 20 000 ng/ml)
aufgeteilt.

Experimenteller Teil 49
5.3.2 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik
und Pharmakodynamik von Paracetamol
5.3.2.1 Studienziel
Untersuchungen zum Wirkungsmechanismus von Paracetamol hinsichtlich der Prostanoid-
Synthese in Vollblut.
5.3.2.2 Studiendesign
Zuerst wurde eine in-vitro-Wirkung von Paracetamol auf die Aktivität der COX-Enzyme in
menschlichem Vollblut durchgeführt. Dann wurde ex-vivo-Hemmung der COX-Enzyme und die
begleitende Kinetik von Paracetamol bei gesunden männlichen Probanden, die klinisch
empfohlene orale Dosen von 1000 mg Paracetamol erhielten, analysiert. Eine nicht Plazebo-
kontrolierte Probandenstudie wurde am Lehrstuhl für Pharmakologie und Toxikologie der
Universität Erlangen-Nürnberg im November-Dezember 2006 durchgeführt.
5.3.2.3 Probandenpopulation
In die Studie wurden 5 männliche Probanden im Alter von 39 bis 65 Jahren (durchschnittliches
Alter: 44,8 Jahre) mit einer Körpermasse 69 ± 2,9 kg (mean ± SEM) eingeschlossen. Alle
Probanden waren die approbierten Ärzte am Institut für Experimentelle und Klinische
Pharmakologie und Toxikologie. Voraussetzung für die Teilnahme war das Einverständnis des
Probanden und keine Einnahme von NSAR zwei Wochen vor der ersten Blutentnahme. Die
Ausschlusskriterien entsprachen den in der Fachinformation für Paracetamol-ratiopharm®
genannten Gegenanzeigen.
5.3.2.4 Studienmedikation
Als Studienmedikation wurde Paracetamol-ratiopharm® 500 (ratiopharm GmbH, Ulm,
Deutschland) verwendet, das von einer öffentlichen Apotheke bezogen wurde. 1 Tablette
enthält 500 mg Paracetamol. Die Probanden nahmen 1000 mg Paracetamol (2 Tabletten).
5.3.2.5 Studienplan
Die Prüfmedikation wurde nüchtern mit 150 ml Wasser zwischen 8:00 und 8:30 Uhr verabreicht.
Eine Stunde später wurde ein Frühstück gereicht. Venöse Blutproben wurden mit einer
Verweilkanüle unmittelbar vor der Tabletteeinnahme (Zeitpunkt 0) und 0,25; 0,75; 1,5; 3; 5; 8;

Experimenteller Teil 50
10 und 24 Stunden nach der Medikamentgabe abgenommen. Für die Paracetamolbestimmung
wurden die heparinisierten Blutproben zentrifugiert und die Aliquote vom Serum wurden bei
-20°C bis zu einer Woche gelagert.
5.3.2.6 In vitro Untersuchungen
5.3.2.6.1 Wirkungen von Paracetamol auf COX-1 und COX-2-Aktivität im menschlichen
Vollblut
COX-1: Blut wurde von gesunden Blutspendern, die zwei Wochen lang vor Blutentnahme
keinen NSAR genommen hatten, entnommen. Aliquote von Vollblut (ohne Antikoagulans)
wurden sofort in Reagenzgläser mit Testsubstanz (Paracetamol 0,1; 1; 10; 30; 100; 300 oder
1000 µmol/l) oder Kontrolle (PBS mit DMSO) überführt. Endkonzentrationen von DMSO im
Reaktionsgemisch waren 0,1% (v/v). Dann wurde Blut für 1 Stunde bei 37°C inkubiert.
Anschließend wurde Serum bei Zentrifugation getrennt um Serum-TXB2-Konzentrationen zu
bestimmen.
COX-2: Aliquoten von heparinisiertem Blut von gesunden Blutspendern wurden mit LPS (10
µg/ml) plus Testsubstanz (Paracetamol 0,1; 1; 10; 30; 100; 300 oder 1000 µmol/l) oder Kontrolle
(PBS mit DMSO) für 24 Stunden bei 37°C inkubiert. Die Aktivität der Blutplättchen-COX-1
wurde durch Zusatz von Aspirin (10 µg/ml) am Anfang der Inkubation unterdrückt. Plasma
wurde durch Zentrifugation getrennt und anschließend wurde zur quantitativen PGE2-
Bestimmung verwendet.
Für beide COX-Assays wurden Konzentrations-Aktivitätskurven durch eine sigmoidale
Regression mit dem variablen Anstieg errechnet. Die IC50-Werte wurden durch den Gebrauch
der PRISM Version 3.0 (GraphPad, San Diego, CA, die USA) bestimmt. Die COX-1 oder COX-2
Hemmung wurde als die prozentualle Änderung von Plasmaeicosanoiden (COX-1: TXB2; COX-
2: PGE2) in Gegenwart von Paracetamol berechnet. Im Fall vom COX-2 wurden basale PGE2-
Werte von jedem PGE2-Wert in der LPS- und LPS / Paracetamol-behandelten Gruppen vor der
Berechnung der Prozentänderung abgezogen.
5.3.2.6.2 Effekt von Paracetamol auf COX-2-Aktivität im Human rekombinante COX-2
Die hemmende Wirkung von Paracetamol auf die humane rekombinante COX-2 wurde mittels
des COX-Inhibitor-Screening-Assay from Cayman gemessen. Die Versuche wurden gemäß den

Experimenteller Teil 51
Angaben des Herstellers durchgeführt, wobei eine 12-minutige Inkubationszeit des Enzyms mit
Paracetamol verwendet wurde. Der Assay bestimmt direkt PGF2α, das durch die Zinnchlorid-
Reduktion des COX-2 gebildeten PGH2 produziert wird.
5.3.2.6.3 Effekt von Paracetamol auf COX-2-Aktivität in humanen Blutmonocyten
Mononuklearen Zellen wurden aus heparinisiertem Vollblut von gesunden Blutspendern durch
Dichtegradientenzentrifugation mit Histopaque-1077 isoliert. (Hinz et al., 2000).
In einer 48-Well Zellkulturplatten wurden 1 x 106 Zellen pro Well ausgesät und bei
Raumtemperatur für 3 Stunden inkubiert. Nichtadhärente Zellen wurden durch Waschen
entfernt. Die adhärenten Monocyten wurden in serumfreiem RPMI-1640 Medium bei 37°C und
5% CO2 kultiviert. Um die Wirkung von Paracetamol auf die COX-2-Aktivität zu bewerten,
wurden die Monocyten mit Acetylsalicylsäure (250 µmol/l) für 2,5 Stunden inkubiert, um die
endogene COX-Aktivität zu hemmen. Danach wurden Zellen gründlich gewaschen und
anschließend mit dem Vehicle (Gruppe 1) oder 10 µg/ml LPS (Gruppen 2 und 3) für 18 Stunden
inkubiert. Nach dem Mediumwechsel wurden Kontrolle (Gruppen 1 und 2) oder 100 µmol/l
Paracetamol (Gruppe 3) zu den Zellen hinzugefügt. Nach einer 90-minutigen Inkubationszeit
wurden die Zellkulturüberstände entnommen und die PGE2-Konzentrationen bestimmt. Die
COX-2-Hemmung wurde als die prozentualle Änderung der PGE2-Konzentration in LPS /
Paracetamol-behandelten Zellen im Vergleich zu Kontrollzellen, die nur mit LPS behandelt
wurden, berechnet. Zu diesem Zweck wurden die basalen PGE2-Werte (Gruppe 1) von jedem
Wert in den Gruppen 2 und 3 abgezogen.
5.3.2.6.4 Effekt von Paracetamol auf die COX-2-Expression in humanen Blutmonocyten
mittels Westernblot-Analyse
In einer 6-Well Platte wurden 5 x 106 mononukleare Zellen pro Well ausgesät und bei
Raumtemperatur für 3 Stunden inkubiert. Nach dem Abwaschen von nicht adhärenten Zellen,
wurden adhärente Monocyten entweder mit Kontrolle, Paracetamol (100 µmol/l), LPS (10 µg/ml)
oder LPS plus Paracetamol für 24 Stunden inkubiert. Später wurden Zellen gewaschen,
geerntet und durch Zentrifugation pelletiert. Die Zellen wurden mittels Ultraschall im
Solubilisierungspuffer lysiert und bei 10 000 r.p.m. für 5 Minuten zentrifugiert. Die Überstände
wurden für die Westernblot-Analyse verwendet. Um Proteine mittels der SDS-
Polyacrylamidgelelektrophorese (SDS-PAGE) bzw. Westernblot zu analysieren, werden die
Proben vorher einer proteindenaturienden Behandlung unterzogen. Dabei werden die Proteine
durch SDS-Behandlung denaturiert und bekommen im Verhältnis zu ihrem Molekulargewicht
eine konstante negative Ladung.

Experimenteller Teil 52
Die Proteine werden durch die Polyacrylamid-Gelelektrophorese aufgrund ihrer
unterschiedlichen Molekulargewichte aufgetrennt. Dabei migrieren Proteine mit geringerem
Molekulargewicht nach Anlegen einer Spannung schneller zur Anode als Proteine mit höherem
Molekulargewicht.
Die Auftragelösungen wurden in die Probentaschen des Gels pipettiert und durch Anlegen einer
konstanten Spannung im Bereich von 35 V bis 200 V auf einem 10%-igen SDS-Gel aufgetrennt.
Als “Blotten” wird der Transfer von Proteinen von einem SDS-Polyacrylamidgel auf eine
Nitrocellulose- oder Polyvinylidendifluorid (PVDF)-Membran unter Anlegen einer Spannung
oder einer Stromstärke bezeichnet. Der Transfer der Proteine erfolgte auf Nitrocellulose in der
einer Miniblotkammer (Protean®, Biorad). Darauf folgte das Blockieren der Membranen mit 5 %
Milchpulver in PBS. Zur Detektion des zu untersuchenden Proteins wird ein spezifischer
Primärantikörper verwendet, an den sich wiederum ein sekundärer Peroxidase-gekoppelter
Antikörper anlagert. Als Primärantikörper wurden ein spezifischer COX-2-Antikörper oder ein
Antikörper gegen ß-actin, das als interne Kontrolle diente, verwendet. Als Sekundärantikörper
diente ein Meerrettich-Peroxidase-konjugierter Fab-spezifische Antimaus IgG. Zum Abschluss
wurden die Membranen mit dem Westernblot-Detektionsreagenzien inkubiert. Die COX-2 wurde
mittels Chemilumineszenz auf einem Film visualisiert. Die densitometrische Analyse der COX-2
Band-Intensitäten (normalisiert zu ß-actin) wurden durch einen optischen Scanner (Gel Doc
2000; Bio-Rad-Laboratories, Hercules, CA) und das Multi-Analyst Programm, Version 1.1 (Bio-
Rad-Laboratories, Hercules, CA) durchgeführt.
5.3.2.7 Ex-vivo Untersuchungen
5.3.2.7.1 Hemmung der COX-Aktivitäten nach einmaliger oraler Gabe von Paracetamol
1000 mg
COX-1: Sofort nach Blutentnahme wurden Vollblutproben ohne Antikoagulantien für 1 Stunde
bei 37°C inkubiert, anschließend zentrifugiert, und die Serum-TXB2-Konzentrationen wurden
bestimmt.
COX-2: Sofort nach den Blutentnahmen wurden heparinisierte Vollblutproben mit 10 µg/ml LPS
für 24 Stunden bei 37° inkubiert. Die Blutplättchen COX-1-Aktivität wurde mit Zusatz von Aspirin
(10 µg/ml) am Anfang von Inkubation unterdrückt. Das Plasma wurde durch Zentrifugation
abgetrennt und PGE2- Konzentrationen wurden bestimmt.

Experimenteller Teil 53
5.3.2.7.2 Bestimmung von Paracetamol in menschlichem Plasma
Paracetamol wurde nach einer vorher veröffentlichte Methode (Lau and Critchley, 1994)
analysiert. Für die Kalibrierungsstandard und Probenaufarbeitung wurden 0,2 ml 10% wässrige
Perchlosäure zu 0,2 ml Plasma hinzugefügd, vortexed für 10 sec. und anschließend in einer
Kühlzentrifuge (Eppendorf 5810R) bei 4000 r.p.m. für eine Dauer von 5 Minuten bei 5°C
zentrifugiert. Klare Überstände (50 µl jeder) wurden direkt ins HPLC System mit einer RP-Säule
CC 125/4 Nucleosil 120-3 C18 (Machery-Nagel, Düren, Deutschland) und eine Vorsäule CC 8/4
Nucleosil C18 injeziert. Das Fliesmittel bestand aus 96:4 (v/v) Mischung von 20 mM
Natriumdihydrogenphosphat Dihydrat (pH = 2,5) und Acetonitril. Der Fluss war 0,9 ml/min. UV-
Detektion wurde auf 254 nm gesetzt. Retentionszeit von Paracetamol war 7,0 Minuten.
Paracetamol-Konzentrationen wurden als Equivalent von den Peakflächen berechnet. Die
Linearität der Standardkurven wurde von 0,2 bis 40 µg/ml mit Regressionskoeffizienten größer
als 0,999 bewiesen.

Experimenteller Teil 54
5.3.3 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik
und Pharmakodynamik von Lumiracoxib
5.3.3.1 Studienziel
Die vorliegende Studie liefert eine vergleichende Analyse zu Pharmakokinetiken, zur
Dosisabhängigkeit und Dauer der COX-2-Hemmung bei Probanden nach der einmaligen oralen
Verabreichung von Lumiracoxib in Dosen von 50, 100 oder 200 mg.
5.3.3.2 Studiendesign
Es handelte sich um eine dreifache Cross-over Probandenstudie, die am Lehrstuhl für
Pharmakologie und Toxikologie der Universität Erlangen-Nürnberg im September-November
2007 durchgeführt wurde.
5.3.3.3 Probandenpopulation
In die Studie wurden 4 Probanden (drei Männer und eine Frau) im Alter von 27 bis 41 Jahren
(durchschnittliches Alter: 36,8 Jahre) mit einer Körpermasse 70,0 ± 3,9 kg (mean ± SEM)
eingeschlossen. Alle Probanden waren die approbierten Ärzte am Institut für Experimentelle
und Klinische Pharmakologie und Toxikologie. Voraussetzung für die Teilnahme war das
Einverständnis des Probanden und keine Einnahme von NSAR zwei Wochen vor der ersten
Blutentnahme. Die Ausschlusskriterien entsprachen den in der Fachinformation für Prexige®
genannten Gegenanzeigen.
5.3.3.4 Studienmedikation
Als Studienmedikation wurde Lumiracoxib (Prexige, Novartis Pharma, Nürnberg, Deutschland)
verwendet, das von einer öffentlichen Apotheke bezogen wurde. 1 Tablette enthält 100 mg
Lumiracoxib. Die Probanden nahmen Lumiracoxib in Dosierungen von 50 mg (die Hälfte der
100 mg Tablette), 100 mg oder 200 mg (zwei 100 mg Tabletten).
5.3.3.5 Studienplan
Die Probanden erhielten im Abstand von zwei Wochen einmalig Prüfdosierungen (50 mg,
100 mg bzw. 200 mg) von Lumiracoxib. Die Prüfmedikation wurde nüchtern mit 150 ml Wasser
zwischen 8:00 und 9:00 Uhr verabreicht. Venöse Blutproben wurden mit einer Verweilkanüle
unmittelbar vor der Tabletteeinnahme (Zeitpunkt 0) und 0,25; 0,75; 1,5; 3; 5; 8, 10 und 24

Experimenteller Teil 55
Stunden nach der Medikation abgenommen. Für die Bestimmung von Lumiracoxib wurden die
heparinisierten Blutproben zentrifugiert und die Aliquote vom Plasma wurden bei -20°C bis zu
einer Woche gelagert.
5.3.3.6 Hemmung der COX-2-Aktivität durch Lumiracoxib ex-vivo
Sofort nach den Blutentnahmen wurden heparinisierte Vollblutproben mit 10 µg/ml LPS für 24
Stunden bei 37° inkubiert. Die Blutplättchen COX-1-Aktivität wurde mit Zusatz von Aspirin (10
µg/ml) am Anfang von Inkubation unterdrückt. Das Plasma wurde durch Zentrifugation
abgetrennt und PGE2- Konzentrationen wurden bestimmt.
5.3.3.7 Bestimmung von Lumiracoxib in menschlichem Plasma.
Lumiracoxib wurde nach einer neu entwickelten und validierten Methode mittels HPLC–UV
(Cheremina et al., 2006) analysiert (Abschnitt 6.3).
Für die Probenaufarbeitung und Kalibrierungsstandard wurden 0,5 ml 0,1 M Salzsäure und 0,1
ml interner Standard (100 µg/ml Nifluminsäure in Methanol) zu 1 ml Plasma hinzugefügt.
Anschließend wurde 4 ml Mischung n-Hexan:Diethylether 70:30 (v/v) gegeben. Die
Reagenzgläser mit der Mischung wurden in einem Shaker für 10 min über Kopf geschüttelt und
anschließend in einer Kühlzentrifuge (Eppendorf 5810R) bei 4000 r.p.m. für eine Dauer von 10
Minuten bei 4°C zentrifugiert. Die organische Phase wurde in einen Reagenzglas überführt und
wurde bei Raumtemperatur unter einem Strom des Stickstoffes evaporiert. Der Rückstand
wurde in 150 µl Methanol:Wasser 50:50 (v:v) aufgelöst, vortext und 100 µl wurde in HPLC-
Anlage injeziert. Die Substanzen wurden mittels RP-Säule CC 125/4 Nucleosil 120-3 C8
(Machery-Nagel, Düren, Germany) und eine Vorsäule CC 8/4 Nucleosil C8 3µ getrennt. Die
mobile Phase bestand aus einer Mischung vom Acetonitril und 0,05 % Trichloressigsäure 35:65
(v/v). Der Fluss war 1,0 ml/min. UV-Detektion wurde auf 270 nm gesetzt. Lumiracoxib wurde bei
16,9 min und Nifluminsäure bei 10,4 min eluiert. Linearität der Standardkurven wurde über
einen Konzentrationsbereich von 10 bis 10 000 ng/ml, mit Regressionskoeffizienten größer als
0,997 bewiesen.
5.3.3.8 Statistik
Potenziale Unterschiede in pharmakodynamischen Parametern in verschiedenen Zeitintervalen
(0,25 bis 5, 8, 12 oder 24 Stunden nach Medikamentverabreichung für durchschnittliche
Hemmungen; 0 bis 5, 8, 12 oder 24 Stunden nach der Medikamentverabreichung für AWEC-
Werte) der drei Gruppen (50 mg gegen 100 mg; 50 mg gegen 200 mg; 100 mg gegen 200 mg)

Experimenteller Teil 56
wurden vom Student´s paired t-Test bewertet. A P-Wert weniger als 0,05 wurde als signifikant
betrachtet. Statistische Analysen wurden mit PRISM® Version 3.0 (GraphPad, San Diego, CA,
USA) durchgeführt.

Experimenteller Teil 57
5.3.4 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik
und Pharmakodynamik von Naproxen
5.3.4.1 Studienziel
Die vorliegende Studie untersuchte, ob Dosierungen von freiverkäuflichem (OTC) Naproxen
Natrium eine für Thrombozytenaggregation ausreichende TXB2-Hemmung nach einmaliger
Gabe und im Steady-state (200 mg Naproxen zweimal am Tag für 7 Tage) und dadurch
mögliche kardioprotektive Wirkung erzeugen können.
5.3.4.2 Studiendesign
Es handelte sich um eine mehrfachdosierte nichtverblindete Probandenstudie, die am Lehrstuhl
für Pharmakologie und Toxikologie der Universität Erlangen-Nürnberg im Dezember 2006 - Mai
2007 durchgeführt wurde.
5.3.4.3 Probandenpopulation
In die Studie wurden 4 Probanden (drei Männer und eine Frau) im Alter von 26 bis 66 Jahren
(durchschnittliches Alter: 44,8 Jahre) mit einer Körpermasse 75,8 ± 7,1 kg (mean ± SEM)
eingeschlossen. Alle Probanden waren die approbierten Ärzte am Institut für Experimentelle
und Klinische Pharmakologie und Toxikologie. Voraussetzung für die Teilnahme war das
Einverständnis des Probanden und keine Einnahme von NSAR zwei Wochen vor der ersten
Blutentnahme. Die Ausschlusskriterien entsprachen den in der Fachinformation für Aleve®
genannten Gegenanzeigen.
5.3.4.4 Studienmedikation
Als Studienmedikation wurde Naproxen Natrium 220 mg (entspricht den 200 mg Naproxen,
Aleve®, Bayer, Germany), das von einer öffentlichen Apotheke bezogen wurde. Die Probanden
nahmen 1 Tablette zweimal täglich für 7 Tage ein.
5.3.4.5 Studienplan
Am 1. Tag wurde die Prüfmedikation nüchtern zwischen 8:00 und 8:30 Uhr verabreicht. Venöse
Blutproben wurden mit einer Verweilkanüle unmittelbar vor der Tabletteeinnahme (Zeitpunkt 0)
und 1, 3, 6 und 12 Stunden nach der Medikamentgabe abgenommen. In den Tagen 2 bis 5
wurde Blut sofort vor der Einnahme der Morgendosis, d.h. 12 Stunden nach der Abenddosis
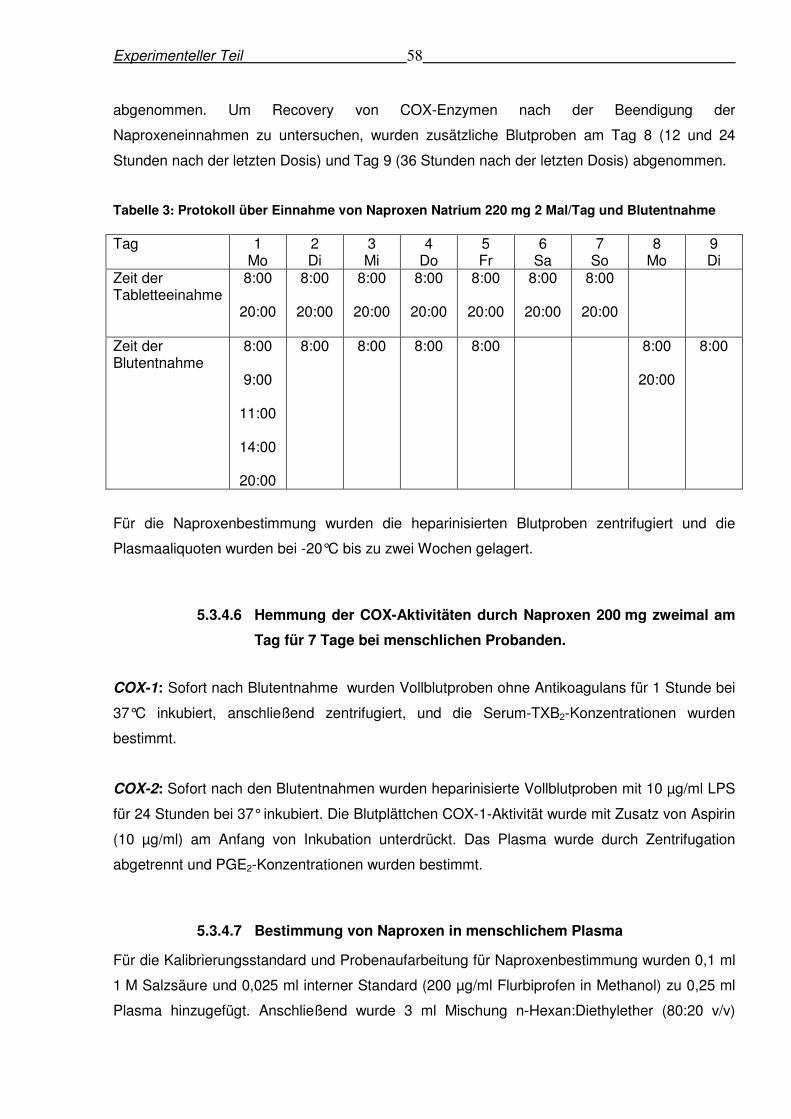
Experimenteller Teil 58
abgenommen. Um Recovery von COX-Enzymen nach der Beendigung der
Naproxeneinnahmen zu untersuchen, wurden zusätzliche Blutproben am Tag 8 (12 und 24
Stunden nach der letzten Dosis) und Tag 9 (36 Stunden nach der letzten Dosis) abgenommen.
Tabelle 3: Protokoll über Einnahme von Naproxen Natrium 220 mg 2 Mal/Tag und Blutentnahme
Tag 1 Mo
2 Di
3 Mi
4 Do
5 Fr
6 Sa
7 So
8 Mo
9 Di
Zeit der Tabletteeinahme
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
Zeit der Blutentnahme
8:00
9:00
11:00
14:00
20:00
8:00
8:00
8:00
8:00
8:00
20:00
8:00
Für die Naproxenbestimmung wurden die heparinisierten Blutproben zentrifugiert und die
Plasmaaliquoten wurden bei -20°C bis zu zwei Wochen gelagert.
5.3.4.6 Hemmung der COX-Aktivitäten durch Naproxen 200 mg zweimal am
Tag für 7 Tage bei menschlichen Probanden.
COX-1: Sofort nach Blutentnahme wurden Vollblutproben ohne Antikoagulans für 1 Stunde bei
37°C inkubiert, anschließend zentrifugiert, und die Serum-TXB2-Konzentrationen wurden
bestimmt.
COX-2: Sofort nach den Blutentnahmen wurden heparinisierte Vollblutproben mit 10 µg/ml LPS
für 24 Stunden bei 37° inkubiert. Die Blutplättchen COX-1-Aktivität wurde mit Zusatz von Aspirin
(10 µg/ml) am Anfang von Inkubation unterdrückt. Das Plasma wurde durch Zentrifugation
abgetrennt und PGE2-Konzentrationen wurden bestimmt.
5.3.4.7 Bestimmung von Naproxen in menschlichem Plasma
Für die Kalibrierungsstandard und Probenaufarbeitung für Naproxenbestimmung wurden 0,1 ml
1 M Salzsäure und 0,025 ml interner Standard (200 µg/ml Flurbiprofen in Methanol) zu 0,25 ml
Plasma hinzugefügt. Anschließend wurde 3 ml Mischung n-Hexan:Diethylether (80:20 v/v)

Experimenteller Teil 59
gegeben, die Reagenzgläser mit der Mischung in einem Shaker für 5 min über Kopf geschüttelt
und anschließend in einer Kühlzentrifuge (Eppendorf 5810R) bei 4000 r.p.m. für eine Dauer von
5 Minuten bei 4°C zentrifugiert. Die organische Phase wurde in einen Reagenzglas überführt
und wurde bei Raumtemperatur unter einem Strom des Stickstoffes evaporiert. Der Rückstand
wurde in 200 µl Fließmittel aufgelöst und 50 µl wurde für HPLC-Analyse benutzt. Die
Substanzen wurden mittels RP-Säule CC 125/4 Nucleosil 120-3 C8 (Machery-Nagel, Düren,
Germany) und eine Vorsäule CC 8/4 Nucleosil C8 3µ getrennt. Die mobile Phase bestand aus
einer Mischung vom Methanol und Wasser 60:40 (v/v) (pH = 3,1). Der Fluss war 0,7 ml/min.
UV-Detektion wurde auf 230 nm gesetzt. Naproxen wurde bei 4,5 min und Ibuprofen bei 6,5 min
eluiert. Linearität der Standardkurven wurde über einen Konzentrationsbereich von 1 bis 50
µg/ml mit Regressionskoeffizienten größer als 0,999 bewiesen.
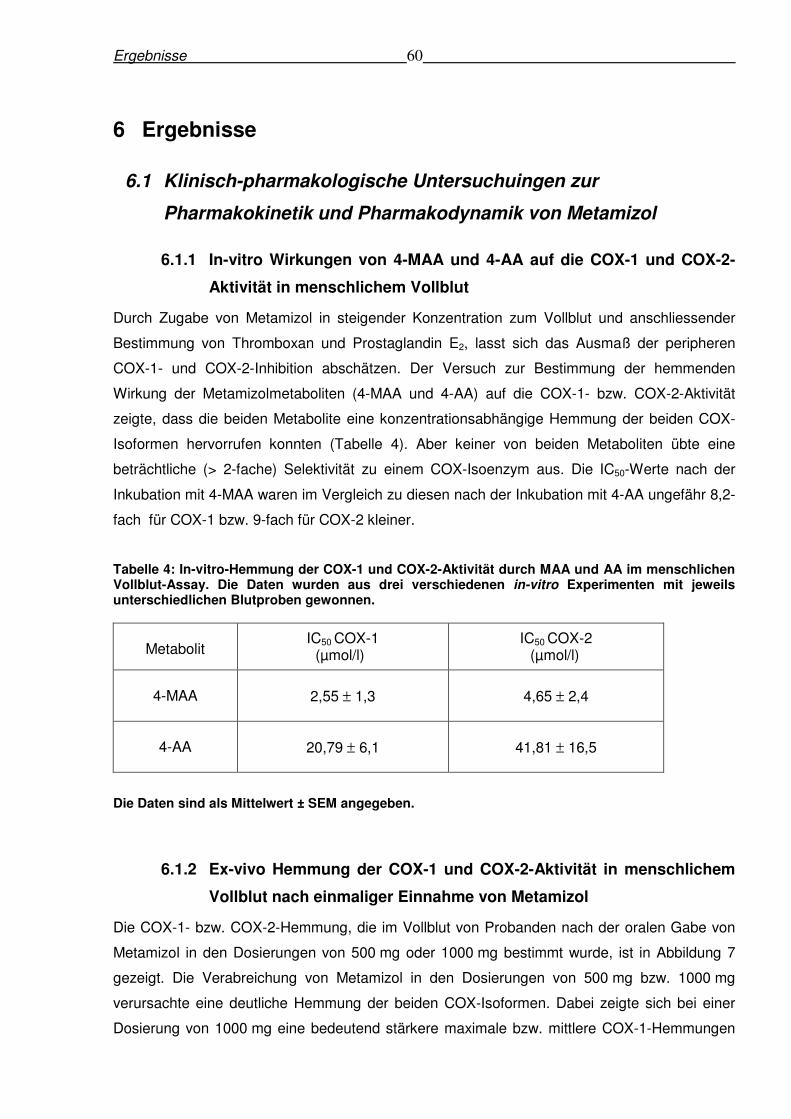
Ergebnisse 60
6 Ergebnisse
6.1 Klinisch-pharmakologische Untersuchuingen zur
Pharmakokinetik und Pharmakodynamik von Metamizol
6.1.1 In-vitro Wirkungen von 4-MAA und 4-AA auf die COX-1 und COX-2-
Aktivität in menschlichem Vollblut
Durch Zugabe von Metamizol in steigender Konzentration zum Vollblut und anschliessender
Bestimmung von Thromboxan und Prostaglandin E2, lasst sich das Ausmaß der peripheren
COX-1- und COX-2-Inhibition abschätzen. Der Versuch zur Bestimmung der hemmenden
Wirkung der Metamizolmetaboliten (4-MAA und 4-AA) auf die COX-1- bzw. COX-2-Aktivität
zeigte, dass die beiden Metabolite eine konzentrationsabhängige Hemmung der beiden COX-
Isoformen hervorrufen konnten (Tabelle 4). Aber keiner von beiden Metaboliten übte eine
beträchtliche (> 2-fache) Selektivität zu einem COX-Isoenzym aus. Die IC50-Werte nach der
Inkubation mit 4-MAA waren im Vergleich zu diesen nach der Inkubation mit 4-AA ungefähr 8,2-
fach für COX-1 bzw. 9-fach für COX-2 kleiner.
Tabelle 4: In-vitro-Hemmung der COX-1 und COX-2-Aktivität durch MAA und AA im menschlichen Vollblut-Assay. Die Daten wurden aus drei verschiedenen in-vitro Experimenten mit jeweils unterschiedlichen Blutproben gewonnen.
Metabolit
IC50 COX-1 (µmol/l)
IC50 COX-2 (µmol/l)
4-MAA
2,55 ± 1,3 4,65 ± 2,4
4-AA
20,79 ± 6,1 41,81 ± 16,5
Die Daten sind als Mittelwert ± SEM angegeben.
6.1.2 Ex-vivo Hemmung der COX-1 und COX-2-Aktivität in menschlichem
Vollblut nach einmaliger Einnahme von Metamizol
Die COX-1- bzw. COX-2-Hemmung, die im Vollblut von Probanden nach der oralen Gabe von
Metamizol in den Dosierungen von 500 mg oder 1000 mg bestimmt wurde, ist in Abbildung 7
gezeigt. Die Verabreichung von Metamizol in den Dosierungen von 500 mg bzw. 1000 mg
verursachte eine deutliche Hemmung der beiden COX-Isoformen. Dabei zeigte sich bei einer
Dosierung von 1000 mg eine bedeutend stärkere maximale bzw. mittlere COX-1-Hemmungen
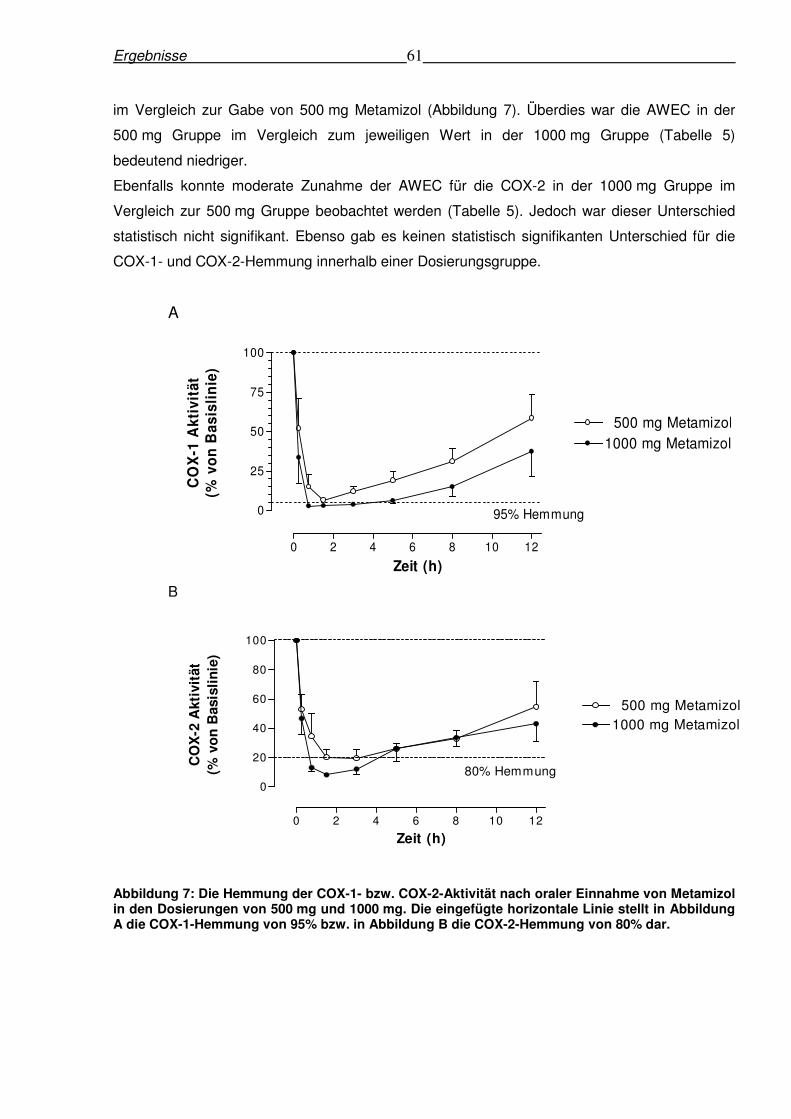
Ergebnisse 61
0 2 4 6 8 10 12
0
25
50
75
100
500 mg Metamizol1000 mg Metamizol
Zeit (h)
CO
X-1
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
95% Hemmung
im Vergleich zur Gabe von 500 mg Metamizol (Abbildung 7). Überdies war die AWEC in der
500 mg Gruppe im Vergleich zum jeweiligen Wert in der 1000 mg Gruppe (Tabelle 5)
bedeutend niedriger.
Ebenfalls konnte moderate Zunahme der AWEC für die COX-2 in der 1000 mg Gruppe im
Vergleich zur 500 mg Gruppe beobachtet werden (Tabelle 5). Jedoch war dieser Unterschied
statistisch nicht signifikant. Ebenso gab es keinen statistisch signifikanten Unterschied für die
COX-1- und COX-2-Hemmung innerhalb einer Dosierungsgruppe.
A
B
Abbildung 7: Die Hemmung der COX-1- bzw. COX-2-Aktivität nach oraler Einnahme von Metamizol in den Dosierungen von 500 mg und 1000 mg. Die eingefügte horizontale Linie stellt in Abbildung A die COX-1-Hemmung von 95% bzw. in Abbildung B die COX-2-Hemmung von 80% dar.
0 2 4 6 8 10 12
0
20
40
60
80
100
500 mg Metamizol1000 mg Metamizol
80% Hemmung
Zeit (h)
CO
X-2
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
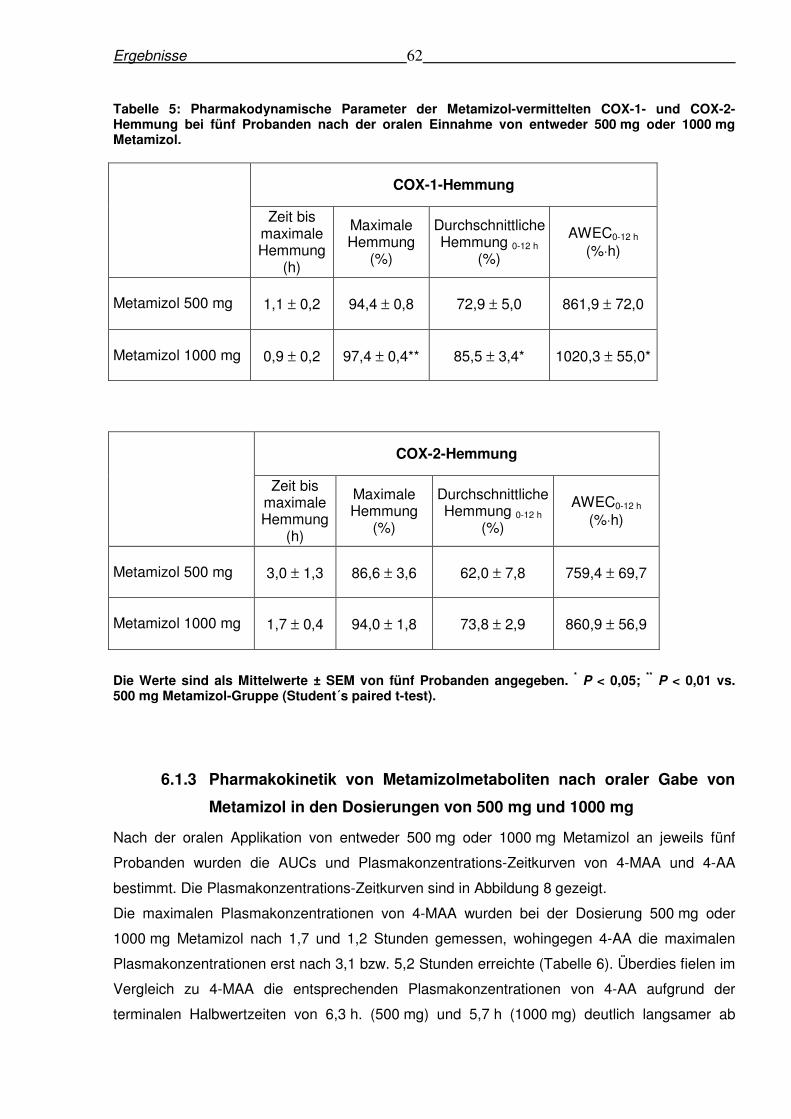
Ergebnisse 62
Tabelle 5: Pharmakodynamische Parameter der Metamizol-vermittelten COX-1- und COX-2-Hemmung bei fünf Probanden nach der oralen Einnahme von entweder 500 mg oder 1000 mg Metamizol.
COX-1-Hemmung
Zeit bis maximale Hemmung
(h)
Maximale Hemmung
(%)
Durchschnittliche Hemmung 0-12 h
(%)
AWEC0-12 h (%⋅h)
Metamizol 500 mg
1,1 ± 0,2
94,4 ± 0,8
72,9 ± 5,0
861,9 ± 72,0
Metamizol 1000 mg
0,9 ± 0,2
97,4 ± 0,4**
85,5 ± 3,4*
1020,3 ± 55,0*
COX-2-Hemmung
Zeit bis maximale Hemmung
(h)
Maximale Hemmung
(%)
Durchschnittliche Hemmung 0-12 h
(%)
AWEC0-12 h (%⋅h)
Metamizol 500 mg
3,0 ± 1,3
86,6 ± 3,6
62,0 ± 7,8
759,4 ± 69,7
Metamizol 1000 mg
1,7 ± 0,4
94,0 ± 1,8
73,8 ± 2,9
860,9 ± 56,9
Die Werte sind als Mittelwerte ± SEM von fünf Probanden angegeben. * P < 0,05; ** P < 0,01 vs. 500 mg Metamizol-Gruppe (Student´s paired t-test).
6.1.3 Pharmakokinetik von Metamizolmetaboliten nach oraler Gabe von
Metamizol in den Dosierungen von 500 mg und 1000 mg
Nach der oralen Applikation von entweder 500 mg oder 1000 mg Metamizol an jeweils fünf
Probanden wurden die AUCs und Plasmakonzentrations-Zeitkurven von 4-MAA und 4-AA
bestimmt. Die Plasmakonzentrations-Zeitkurven sind in Abbildung 8 gezeigt.
Die maximalen Plasmakonzentrationen von 4-MAA wurden bei der Dosierung 500 mg oder
1000 mg Metamizol nach 1,7 und 1,2 Stunden gemessen, wohingegen 4-AA die maximalen
Plasmakonzentrationen erst nach 3,1 bzw. 5,2 Stunden erreichte (Tabelle 6). Überdies fielen im
Vergleich zu 4-MAA die entsprechenden Plasmakonzentrationen von 4-AA aufgrund der
terminalen Halbwertzeiten von 6,3 h. (500 mg) und 5,7 h (1000 mg) deutlich langsamer ab
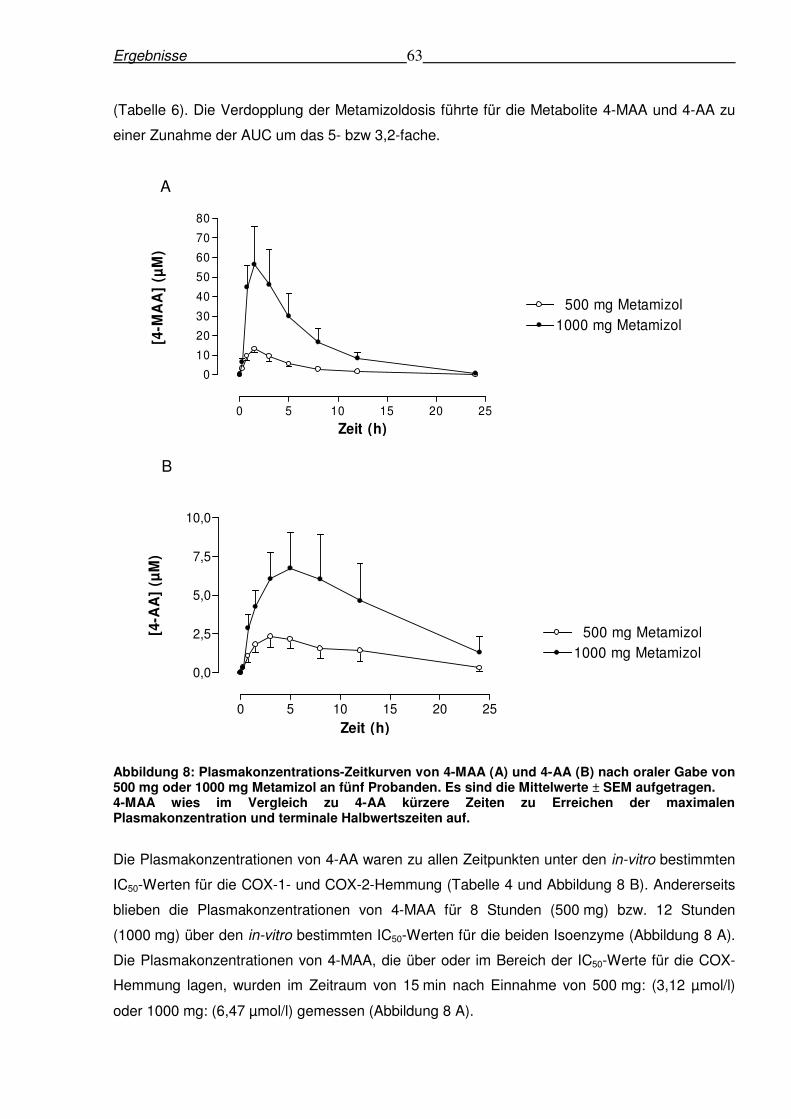
Ergebnisse 63
(Tabelle 6). Die Verdopplung der Metamizoldosis führte für die Metabolite 4-MAA und 4-AA zu
einer Zunahme der AUC um das 5- bzw 3,2-fache.
A
B
Abbildung 8: Plasmakonzentrations-Zeitkurven von 4-MAA (A) und 4-AA (B) nach oraler Gabe von 500 mg oder 1000 mg Metamizol an fünf Probanden. Es sind die Mittelwerte ± SEM aufgetragen. 4-MAA wies im Vergleich zu 4-AA kürzere Zeiten zu Erreichen der maximalen Plasmakonzentration und terminale Halbwertszeiten auf.
Die Plasmakonzentrationen von 4-AA waren zu allen Zeitpunkten unter den in-vitro bestimmten
IC50-Werten für die COX-1- und COX-2-Hemmung (Tabelle 4 und Abbildung 8 B). Andererseits
blieben die Plasmakonzentrationen von 4-MAA für 8 Stunden (500 mg) bzw. 12 Stunden
(1000 mg) über den in-vitro bestimmten IC50-Werten für die beiden Isoenzyme (Abbildung 8 A).
Die Plasmakonzentrationen von 4-MAA, die über oder im Bereich der IC50-Werte für die COX-
Hemmung lagen, wurden im Zeitraum von 15 min nach Einnahme von 500 mg: (3,12 µmol/l)
oder 1000 mg: (6,47 µmol/l) gemessen (Abbildung 8 A).
0 5 10 15 20 25
0
10
20
30
40
50
60
70
80
500 mg Metamizol1000 mg Metamizol
Zeit (h)
[4-M
AA
] (µ
M)
0 5 10 15 20 25
0,0
2,5
5,0
7,5
10,0
500 mg Metamizol1000 mg Metamizol
Zeit (h)
[4-A
A]
(µM
)
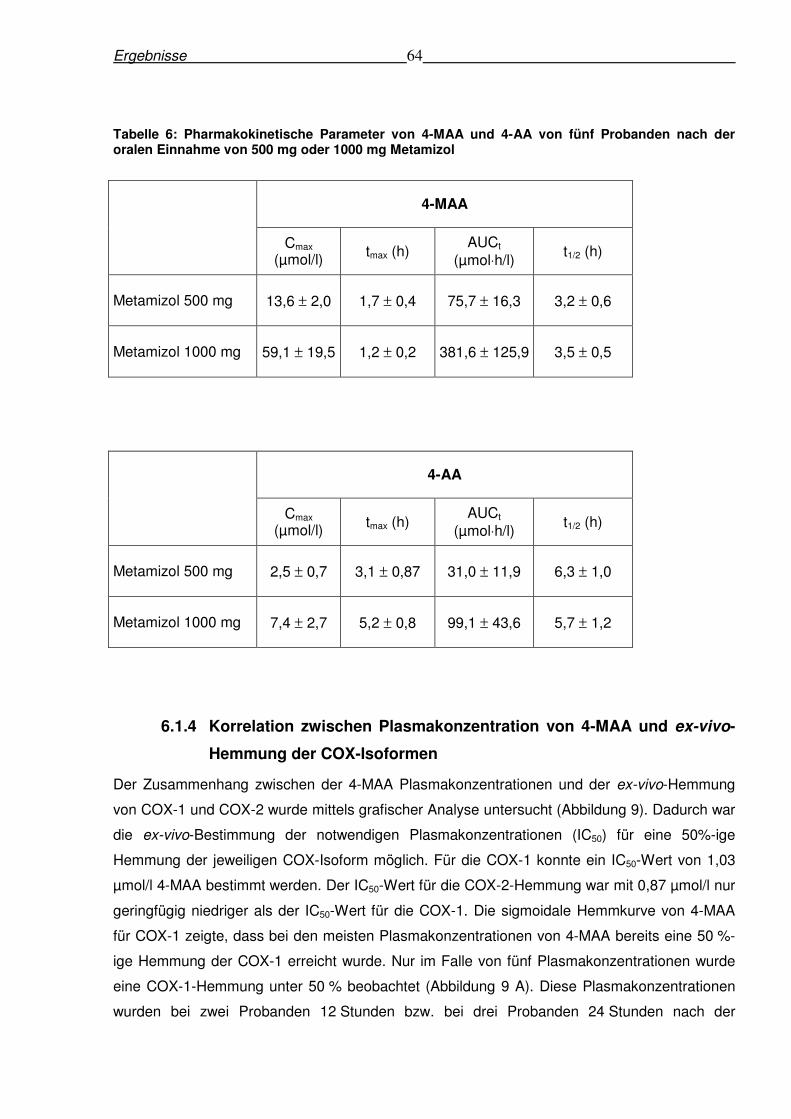
Ergebnisse 64
Tabelle 6: Pharmakokinetische Parameter von 4-MAA und 4-AA von fünf Probanden nach der oralen Einnahme von 500 mg oder 1000 mg Metamizol
4-MAA
Cmax (µmol/l) tmax (h)
AUCt (µmol⋅h/l)
t1/2 (h)
Metamizol 500 mg
13,6 ± 2,0 1,7 ± 0,4 75,7 ± 16,3 3,2 ± 0,6
Metamizol 1000 mg
59,1 ± 19,5 1,2 ± 0,2 381,6 ± 125,9 3,5 ± 0,5
4-AA
Cmax (µmol/l) tmax (h)
AUCt (µmol⋅h/l)
t1/2 (h)
Metamizol 500 mg
2,5 ± 0,7 3,1 ± 0,87 31,0 ± 11,9 6,3 ± 1,0
Metamizol 1000 mg
7,4 ± 2,7 5,2 ± 0,8 99,1 ± 43,6 5,7 ± 1,2
6.1.4 Korrelation zwischen Plasmakonzentration von 4-MAA und ex-vivo-
Hemmung der COX-Isoformen
Der Zusammenhang zwischen der 4-MAA Plasmakonzentrationen und der ex-vivo-Hemmung
von COX-1 und COX-2 wurde mittels grafischer Analyse untersucht (Abbildung 9). Dadurch war
die ex-vivo-Bestimmung der notwendigen Plasmakonzentrationen (IC50) für eine 50%-ige
Hemmung der jeweiligen COX-Isoform möglich. Für die COX-1 konnte ein IC50-Wert von 1,03
µmol/l 4-MAA bestimmt werden. Der IC50-Wert für die COX-2-Hemmung war mit 0,87 µmol/l nur
geringfügig niedriger als der IC50-Wert für die COX-1. Die sigmoidale Hemmkurve von 4-MAA
für COX-1 zeigte, dass bei den meisten Plasmakonzentrationen von 4-MAA bereits eine 50 %-
ige Hemmung der COX-1 erreicht wurde. Nur im Falle von fünf Plasmakonzentrationen wurde
eine COX-1-Hemmung unter 50 % beobachtet (Abbildung 9 A). Diese Plasmakonzentrationen
wurden bei zwei Probanden 12 Stunden bzw. bei drei Probanden 24 Stunden nach der
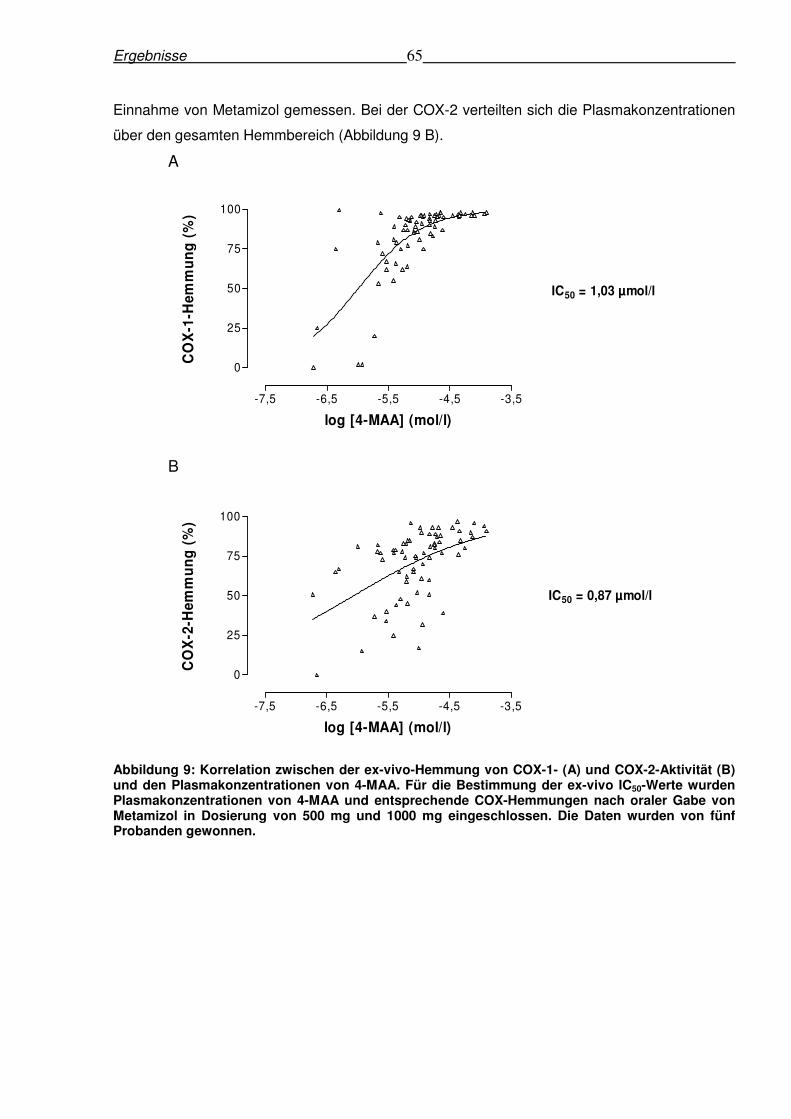
Ergebnisse 65
Einnahme von Metamizol gemessen. Bei der COX-2 verteilten sich die Plasmakonzentrationen
über den gesamten Hemmbereich (Abbildung 9 B).
A
B
Abbildung 9: Korrelation zwischen der ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) und den Plasmakonzentrationen von 4-MAA. Für die Bestimmung der ex-vivo IC50-Werte wurden Plasmakonzentrationen von 4-MAA und entsprechende COX-Hemmungen nach oraler Gabe von Metamizol in Dosierung von 500 mg und 1000 mg eingeschlossen. Die Daten wurden von fünf Probanden gewonnen.
-7,5 -6,5 -5,5 -4,5 -3,5
0
25
50
75
100
IC50 = 1,03 µmol/l
log [4-MAA] (mol/l)
CO
X-1
-Hem
mu
ng
(%
)
-7,5 -6,5 -5,5 -4,5 -3,5
0
25
50
75
100
IC50 = 0,87 µmol/l
log [4-MAA] (mol/l)
CO
X-2
-Hem
mu
ng
(%
)

Ergebnisse 66
6.2 Klinisch-pharmakologische Untersuchuingen zur
Pharmakokinetik und Pharmakodynamik von Paracetamol
6.2.1 In-vitro Wirkung von Paracetamol auf die COX-1- und COX-2-Aktivität
in menschlichem Vollblut
Analog zur Aktivitätsbestimmung der peripheren COX im Vollblut nach Metamizolzugabe wurde
die Suppression der COX-1- und COX-2-Aktivität durch die Zugabe von Paracetamol in
verschiedenen Konzentrationen zu Vollblutproben untersucht (siehe Abschnitt 5.3.2.6.1). Als
Aktivitätsmaß für die COX-1 wurde Thromboxan B2 bzw. für die COX-2 Prostaglandin E2
bestimmt. Der in-vitro-Versuch zeigte, dass Paracetamol eine konzentrationsabhängige
Hemmung der beiden COX-Isoformen hervorruft (Tabelle 7). Paracetamol beeinflusste die
COX-2 jedoch deutlich effektiver als die COX-1. Auf Grundlage des Quotienten aus den IC50-
Werten, dem sogenannten Selektivitätsquotient, zeigte Paracetamol eine 4,4-fach höhere
Selektivität zu COX-2.
Tabelle 7: In-vitro-Hemmung der COX-1 und COX-2-Aktivität durch Paracetamol im menschlichen Vollblut-Assay. Die Daten wurden aus vier verschiedenen in-vitro Experimenten mit jeweils unterschiedlichen Blutproben gewonnen.
COX-1 IC50 (µmol/l) 113,7 ± 17,2
COX-2 IC50 (µmol/l) 25,8 ± 1,8
Selektivitätsquotient (COX-1/COX-2) 4,4
Die Daten sind als Mittelwerte ± SEM angegeben.
6.2.2 Pharmakokinetik von Paracetamol nach einmaliger oraler Gabe von
1000 mg Paracetamol
Nach der oralen Applikation von 1000 mg Paracetamol an jeweils fünf Probanden wurden die
Plasmakonzentrations-Zeitkurven von Paracetamol bestimmt (Abbildung 10).
Die durchschnittlichen Plasmakonzentrationen von Paracetamol blieben unter den in-vitro
bestimmten IC50-Wert für die COX-1-Hemmung innerhalb des gesamten Zeitraums der Studie.
Jedoch lagen die Plasmakonzentrationen von Paracetamol für mindestens 5 Stunden nach der
Medikamenteneinnahme über dem in-vitro bestimmten IC50-Wert für die COX-2-Hemmung
(Abbildung 10).
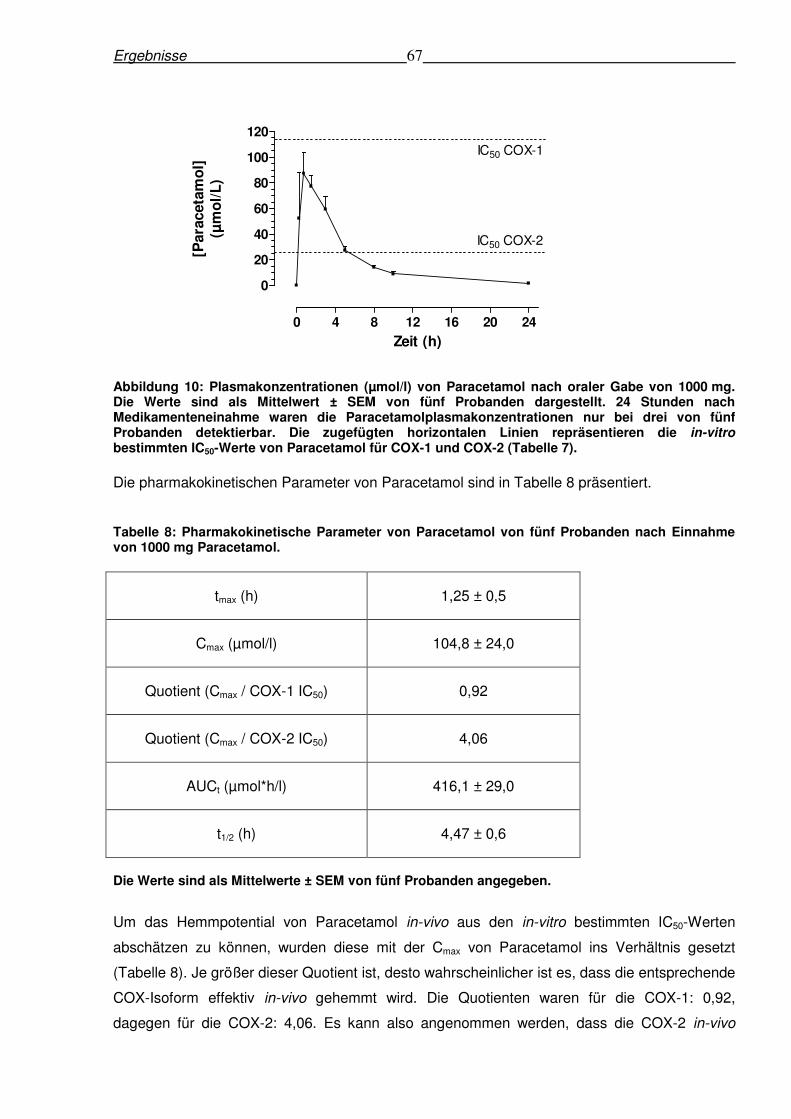
Ergebnisse 67
Abbildung 10: Plasmakonzentrationen (µmol/l) von Paracetamol nach oraler Gabe von 1000 mg. Die Werte sind als Mittelwert ± SEM von fünf Probanden dargestellt. 24 Stunden nach Medikamenteneinahme waren die Paracetamolplasmakonzentrationen nur bei drei von fünf Probanden detektierbar. Die zugefügten horizontalen Linien repräsentieren die in-vitro bestimmten IC50-Werte von Paracetamol für COX-1 und COX-2 (Tabelle 7).
Die pharmakokinetischen Parameter von Paracetamol sind in Tabelle 8 präsentiert.
Tabelle 8: Pharmakokinetische Parameter von Paracetamol von fünf Probanden nach Einnahme von 1000 mg Paracetamol.
tmax (h) 1,25 ± 0,5
Cmax (µmol/l) 104,8 ± 24,0
Quotient (Cmax / COX-1 IC50) 0,92
Quotient (Cmax / COX-2 IC50) 4,06
AUCt (µmol*h/l) 416,1 ± 29,0
t1/2 (h) 4,47 ± 0,6
Die Werte sind als Mittelwerte ± SEM von fünf Probanden angegeben.
Um das Hemmpotential von Paracetamol in-vivo aus den in-vitro bestimmten IC50-Werten
abschätzen zu können, wurden diese mit der Cmax von Paracetamol ins Verhältnis gesetzt
(Tabelle 8). Je größer dieser Quotient ist, desto wahrscheinlicher ist es, dass die entsprechende
COX-Isoform effektiv in-vivo gehemmt wird. Die Quotienten waren für die COX-1: 0,92,
dagegen für die COX-2: 4,06. Es kann also angenommen werden, dass die COX-2 in-vivo
0 4 8 12 16 20 24
0
20
40
60
80
100
120IC50 COX-1
IC50 COX-2
Zeit (h)
[Par
acet
amo
l](µ
mo
l/L
)
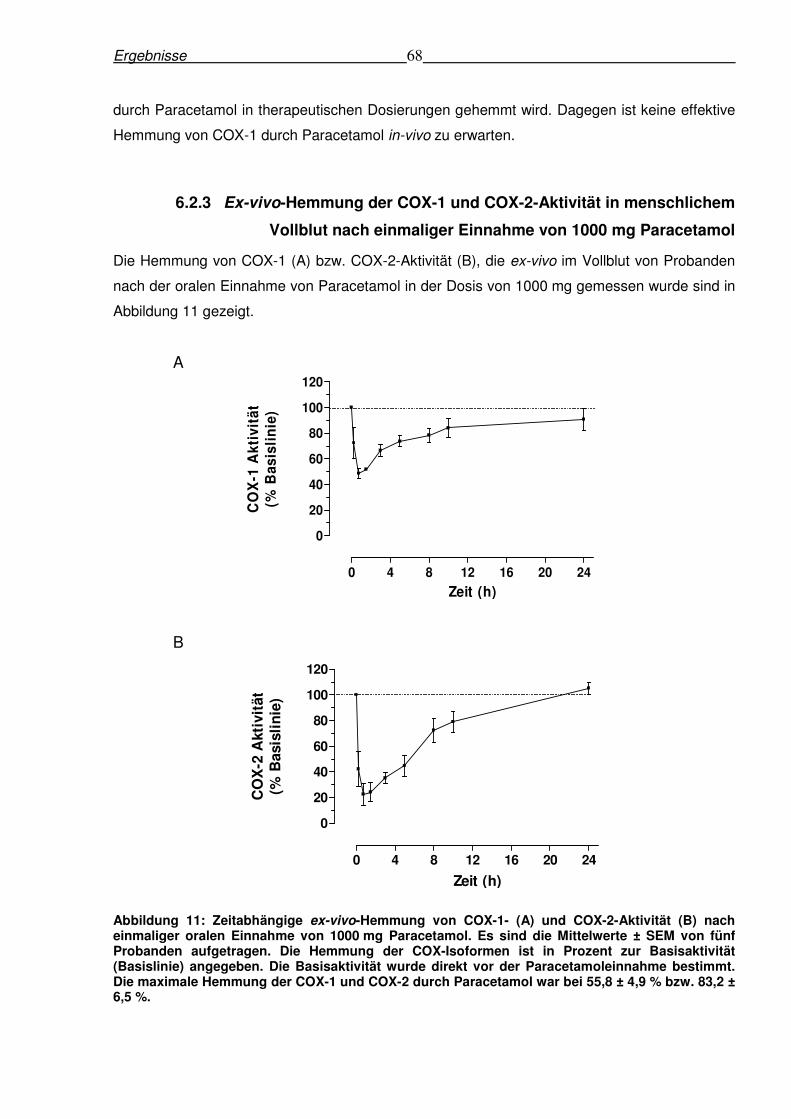
Ergebnisse 68
durch Paracetamol in therapeutischen Dosierungen gehemmt wird. Dagegen ist keine effektive
Hemmung von COX-1 durch Paracetamol in-vivo zu erwarten.
6.2.3 Ex-vivo-Hemmung der COX-1 und COX-2-Aktivität in menschlichem
Vollblut nach einmaliger Einnahme von 1000 mg Paracetamol
Die Hemmung von COX-1 (A) bzw. COX-2-Aktivität (B), die ex-vivo im Vollblut von Probanden
nach der oralen Einnahme von Paracetamol in der Dosis von 1000 mg gemessen wurde sind in
Abbildung 11 gezeigt.
A
B
Abbildung 11: Zeitabhängige ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) nach einmaliger oralen Einnahme von 1000 mg Paracetamol. Es sind die Mittelwerte ± SEM von fünf Probanden aufgetragen. Die Hemmung der COX-Isoformen ist in Prozent zur Basisaktivität (Basislinie) angegeben. Die Basisaktivität wurde direkt vor der Paracetamoleinnahme bestimmt. Die maximale Hemmung der COX-1 und COX-2 durch Paracetamol war bei 55,8 ± 4,9 % bzw. 83,2 ± 6,5 %.
0 4 8 12 16 20 24
0
20
40
60
80
100
120
Zeit (h)
CO
X-1
Akt
ivit
ät(%
Bas
isli
nie
)
0 4 8 12 16 20 24
0
20
40
60
80
100
120
Zeit (h)
CO
X-2
Akt
ivit
ät(%
Bas
isli
nie
)
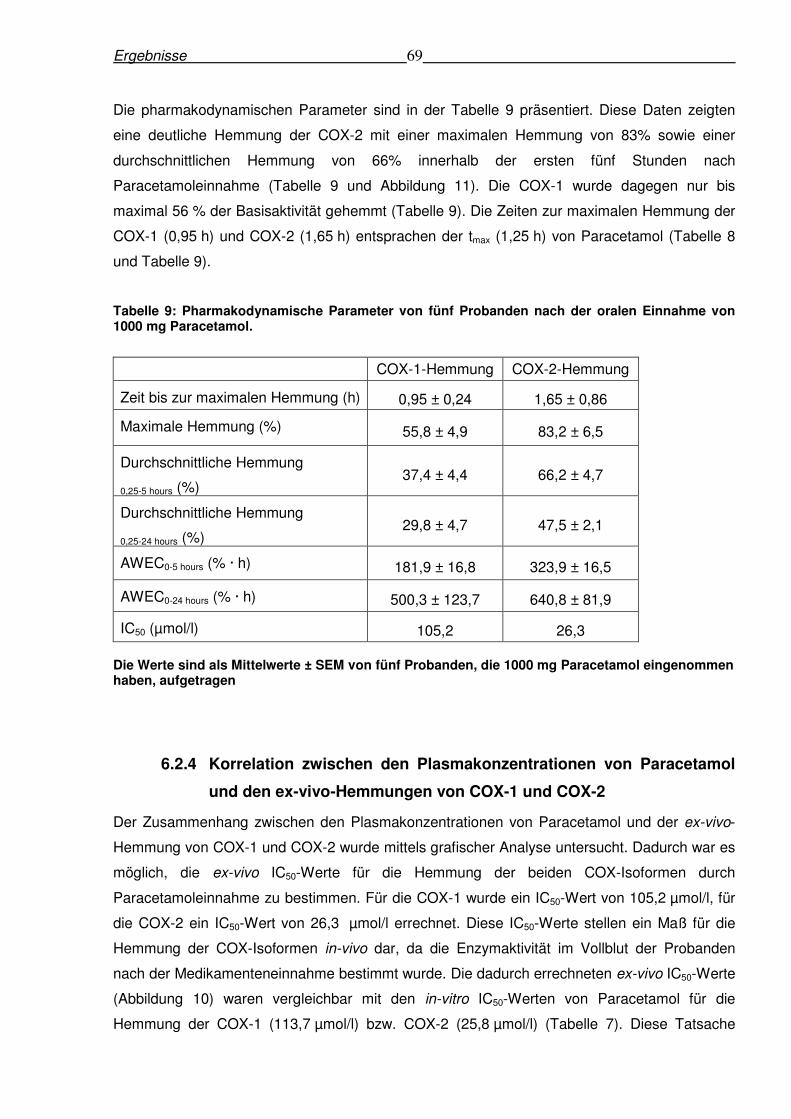
Ergebnisse 69
Die pharmakodynamischen Parameter sind in der Tabelle 9 präsentiert. Diese Daten zeigten
eine deutliche Hemmung der COX-2 mit einer maximalen Hemmung von 83% sowie einer
durchschnittlichen Hemmung von 66% innerhalb der ersten fünf Stunden nach
Paracetamoleinnahme (Tabelle 9 und Abbildung 11). Die COX-1 wurde dagegen nur bis
maximal 56 % der Basisaktivität gehemmt (Tabelle 9). Die Zeiten zur maximalen Hemmung der
COX-1 (0,95 h) und COX-2 (1,65 h) entsprachen der tmax (1,25 h) von Paracetamol (Tabelle 8
und Tabelle 9).
Tabelle 9: Pharmakodynamische Parameter von fünf Probanden nach der oralen Einnahme von 1000 mg Paracetamol.
COX-1-Hemmung COX-2-Hemmung
Zeit bis zur maximalen Hemmung (h) 0,95 ± 0,24 1,65 ± 0,86
Maximale Hemmung (%) 55,8 ± 4,9 83,2 ± 6,5
Durchschnittliche Hemmung
0,25-5 hours (%) 37,4 ± 4,4 66,2 ± 4,7
Durchschnittliche Hemmung
0,25-24 hours (%) 29,8 ± 4,7 47,5 ± 2,1
AWEC0-5 hours (% · h) 181,9 ± 16,8 323,9 ± 16,5
AWEC0-24 hours (% · h) 500,3 ± 123,7 640,8 ± 81,9
IC50 (µmol/l) 105,2 26,3 Die Werte sind als Mittelwerte ± SEM von fünf Probanden, die 1000 mg Paracetamol eingenommen haben, aufgetragen
6.2.4 Korrelation zwischen den Plasmakonzentrationen von Paracetamol
und den ex-vivo-Hemmungen von COX-1 und COX-2
Der Zusammenhang zwischen den Plasmakonzentrationen von Paracetamol und der ex-vivo-
Hemmung von COX-1 und COX-2 wurde mittels grafischer Analyse untersucht. Dadurch war es
möglich, die ex-vivo IC50-Werte für die Hemmung der beiden COX-Isoformen durch
Paracetamoleinnahme zu bestimmen. Für die COX-1 wurde ein IC50-Wert von 105,2 µmol/l, für
die COX-2 ein IC50-Wert von 26,3 µmol/l errechnet. Diese IC50-Werte stellen ein Maß für die
Hemmung der COX-Isoformen in-vivo dar, da die Enzymaktivität im Vollblut der Probanden
nach der Medikamenteneinnahme bestimmt wurde. Die dadurch errechneten ex-vivo IC50-Werte
(Abbildung 10) waren vergleichbar mit den in-vitro IC50-Werten von Paracetamol für die
Hemmung der COX-1 (113,7 µmol/l) bzw. COX-2 (25,8 µmol/l) (Tabelle 7). Diese Tatsache
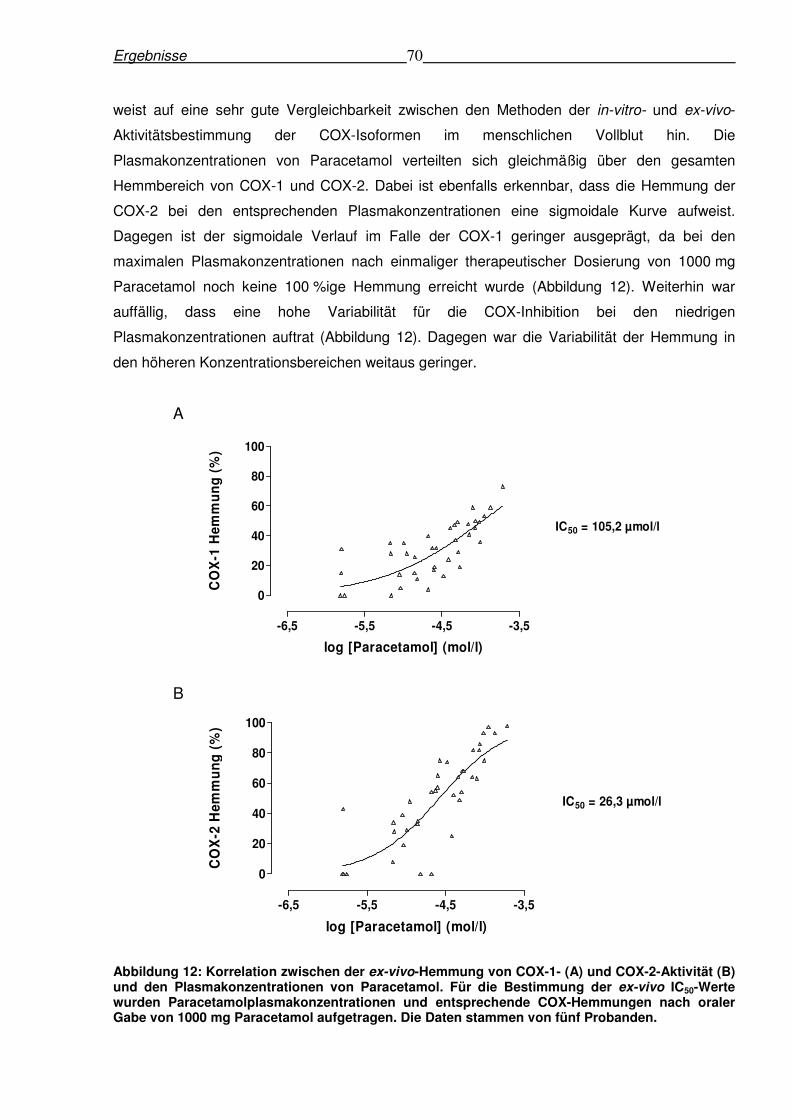
Ergebnisse 70
weist auf eine sehr gute Vergleichbarkeit zwischen den Methoden der in-vitro- und ex-vivo-
Aktivitätsbestimmung der COX-Isoformen im menschlichen Vollblut hin. Die
Plasmakonzentrationen von Paracetamol verteilten sich gleichmäßig über den gesamten
Hemmbereich von COX-1 und COX-2. Dabei ist ebenfalls erkennbar, dass die Hemmung der
COX-2 bei den entsprechenden Plasmakonzentrationen eine sigmoidale Kurve aufweist.
Dagegen ist der sigmoidale Verlauf im Falle der COX-1 geringer ausgeprägt, da bei den
maximalen Plasmakonzentrationen nach einmaliger therapeutischer Dosierung von 1000 mg
Paracetamol noch keine 100 %ige Hemmung erreicht wurde (Abbildung 12). Weiterhin war
auffällig, dass eine hohe Variabilität für die COX-Inhibition bei den niedrigen
Plasmakonzentrationen auftrat (Abbildung 12). Dagegen war die Variabilität der Hemmung in
den höheren Konzentrationsbereichen weitaus geringer.
A
B
Abbildung 12: Korrelation zwischen der ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) und den Plasmakonzentrationen von Paracetamol. Für die Bestimmung der ex-vivo IC50-Werte wurden Paracetamolplasmakonzentrationen und entsprechende COX-Hemmungen nach oraler Gabe von 1000 mg Paracetamol aufgetragen. Die Daten stammen von fünf Probanden.
-6,5 -5,5 -4,5 -3,5
0
20
40
60
80
100
IC50 = 105,2 µmol/l
log [Paracetamol] (mol/l)
CO
X-1
Hem
mu
ng
(%
)
-6,5 -5,5 -4,5 -3,5
0
20
40
60
80
100
IC50 = 26,3 µmol/l
log [Paracetamol] (mol/l)
CO
X-2
Hem
mu
ng
(%
)
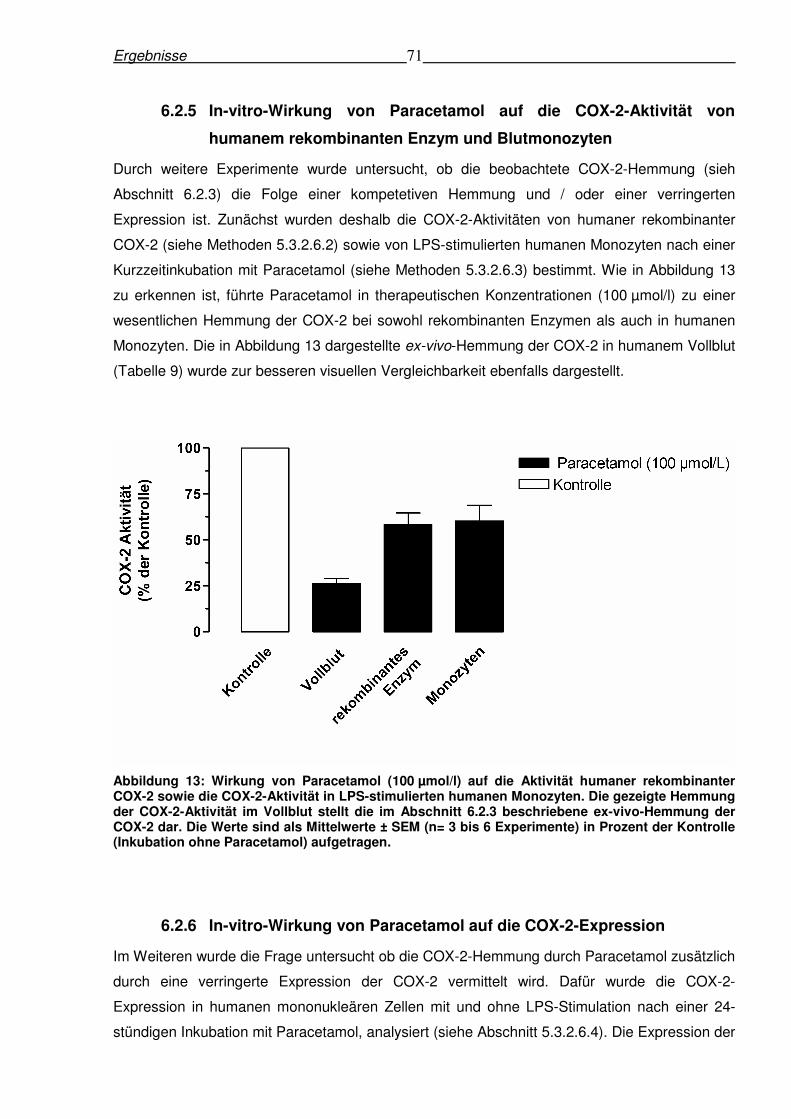
Ergebnisse 71
6.2.5 In-vitro-Wirkung von Paracetamol auf die COX-2-Aktivität von
humanem rekombinanten Enzym und Blutmonozyten
Durch weitere Experimente wurde untersucht, ob die beobachtete COX-2-Hemmung (sieh
Abschnitt 6.2.3) die Folge einer kompetetiven Hemmung und / oder einer verringerten
Expression ist. Zunächst wurden deshalb die COX-2-Aktivitäten von humaner rekombinanter
COX-2 (siehe Methoden 5.3.2.6.2) sowie von LPS-stimulierten humanen Monozyten nach einer
Kurzzeitinkubation mit Paracetamol (siehe Methoden 5.3.2.6.3) bestimmt. Wie in Abbildung 13
zu erkennen ist, führte Paracetamol in therapeutischen Konzentrationen (100 µmol/l) zu einer
wesentlichen Hemmung der COX-2 bei sowohl rekombinanten Enzymen als auch in humanen
Monozyten. Die in Abbildung 13 dargestellte ex-vivo-Hemmung der COX-2 in humanem Vollblut
(Tabelle 9) wurde zur besseren visuellen Vergleichbarkeit ebenfalls dargestellt.
Abbildung 13: Wirkung von Paracetamol (100 µmol/l) auf die Aktivität humaner rekombinanter COX-2 sowie die COX-2-Aktivität in LPS-stimulierten humanen Monozyten. Die gezeigte Hemmung der COX-2-Aktivität im Vollblut stellt die im Abschnitt 6.2.3 beschriebene ex-vivo-Hemmung der COX-2 dar. Die Werte sind als Mittelwerte ± SEM (n= 3 bis 6 Experimente) in Prozent der Kontrolle (Inkubation ohne Paracetamol) aufgetragen.
6.2.6 In-vitro-Wirkung von Paracetamol auf die COX-2-Expression
Im Weiteren wurde die Frage untersucht ob die COX-2-Hemmung durch Paracetamol zusätzlich
durch eine verringerte Expression der COX-2 vermittelt wird. Dafür wurde die COX-2-
Expression in humanen mononukleären Zellen mit und ohne LPS-Stimulation nach einer 24-
stündigen Inkubation mit Paracetamol, analysiert (siehe Abschnitt 5.3.2.6.4). Die Expression der

Ergebnisse 72
COX-2 wurde dann mittels Immunoblotanalyse visualisiert und ausgewertet. Es konnte gezeigt
werden, dass sowohl in LPS-stimulierten Zellen als auch in nicht LPS-stimulierten Zellen sich
die COX-2-Expression nicht durch Paracetamol änderte (Abbildung 14). Somit kann
geschlussfolgert werden, dass Paracetamol nur einen Effekt auf die COX-2-Aktivität hat.
Abbildung 14: Repräsentativer Immunoblot zum Einfluss von Paracetamol auf die Expression von COX-2 in humanen mononukleären Zellen. Die Immunoblotanalyse zeigt, dass die COX-2-Expression durch Paracetamol nicht beeinflusst wird. Als interner Standard wurde β-Aktin nach dem „Stripping“ nachgewiesen. Über den Blots sind die densitometrischen bestimmten Expressionsdaten der COX-2 in Prozent der Kontrolle gezeigt (n=4 Immunoblots). Innerhalb der nicht LPS-stimulierten Zellen bzw. der LPS-stimulierten Zellen zeigte sich keine Änderung der COX-2-Expression durch Paracetamol. Die LPS-Stimulation der Zellen führte zu einer Zunahme der COX-2-Expression um durchschnittlich 160 %.

Ergebnisse 73
6.3 Entwicklung und Validierung einer HPLC-UV Methode zur
Bestimmung der Konzentrationen von Lumiracoxib in
menschlichem Plasma
6.3.1 Methodenentwicklung
Zur Bestimmung der Plasmakonzentrationen von Lumiracoxib war es notwendig, eine Methode
zu entwickeln. Die zu entwickelnde Methode müsste für laborübliche Analysengeräte
zugeschnitten sein. Deswegen wurde die Analyse mittels HPLC und UV-Detektion durchgeführt.
Nach der Spektroskopie hat es sich zwei Maximume ergeben. Deswegen wurde Messung von
Lumiracoxib bei Wellenlänge: 210, 212, 270, 280 nm durchgetestet. Dabei wurden bei niedrigen
Wellenlängen zweimal größere Peakflächen von Lumiracoxib, als bei höheren Wellenlängen
gemessen. Jedoch wurden mehrere Störpeaks mitgemessen. Deshalb wurde es für
Wellenlänge 270 nm entschieden. Die Lumiracoxib-Plasmakonzentrationen wurden mithilfe
einer Internen-Standard-Methode bestimmt. Als interner Standard wurde Nifluminsäure und
Diclofenac untersucht. Es wurde für Nifluminsäure entschieden, weil Diclofenac unmittelbar vor
dem Lumiracoxibpeak rausgegangen ist, wodurch schlechtere Auflösung erzielt wurde.
Als mobile Phase für die Trennung von Analyten wurde Gemisch aus 0,05% Trichloressigsäure
und Acetonitril im verschiedenen Verhältnisse überprüft. Bei Mischungsverhältnis von 0,05%
Trichloressigsäure und Acetonitril 35:65 (v/v) war die Trennung von Lumiracoxib und Internen
Standard optimal, ohne dass Interferenzen mit den übrigen Plasmabestandteilen auftraten.
Es hat sich angeboten, ein zweiphasiges Lösungsmittelgemisch bestehend aus einem polaren,
und einem weniger polaren, leicht verdampfbaren Lösungsmittel zu verwenden. Nach diesen
Vorüberlegungen wurden zweiphasige Lösungsmittelsysteme, so wie Pentan:Chloroform und
Hexan:Diethylether auf ihre Eignung als Extraktionsmittel untersucht. Ein Gemisch aus Hexan
und Diethylether stellte sich als das effektivste Extraktionsmittel heraus. Dabei wurden größere
Peakflächen bei der Verhältnis 70:30 (v/v) von Hexan:Diethylether gemessen.
Die Methode ermöglichte die Bestimmung von Lumiracoxib im Konzentrationsbereich von 10 –
10 000 ng/ml.
6.3.1.1 Standardlösungen
4,5 mg Lumiracoxib wurden in 4,5 ml Methanol gelöst so dass eine Standardlösung von 1
mg/ml Lumiracoxib erhalten wurde. Diese Lösung wurde weiter 1:10 mit 50%-igem (v/v)
wässrigen MeOH verdünnt, so dass die eine Standardlösung von 100 µg/ml erhalten wurde.

Ergebnisse 74
Die Standardlösung des internen Standards Nifluminsäure mit einer Konzentration von 10 µg/ml
wurde durch Lösen von 1 mg Nifluminsäure in 100 ml MeOH erhalten. Alle Standardlösungen
wurden bei -20°C aufbewahrt.
6.3.1.2 Probenaufarbeitung und Messung
Zu 1000 µl Plasma wurden 100 µl interner Standard (Nifluminsäure 10 µg/ml Methanol), 500 µl
1M HCl sowie 4 ml Extraktionsmittel (Hexan:Diethylether 70:30) gegeben. Die Proben wurden
10 Minuten über Kopf geschüttelt und anschließend in einer Kühlzentrifuge (Eppendorf 5810R)
bei 4000 r.p.m. für eine Dauer von 10 Minuten bei 4°C zentrifugiert. Organische Phase wurde
abgehoben und unter Stickstoff evaporiert.
Als mobile Phase für die HPLC-Analyse diente ein Gemisch aus 0,05% Trichloressigsäure und
Acetonitril im Verhältnis 35:65 (v/v). Die Fließgeschwindigkeit lag bei 1,0 ml/min. Temperatur
des Säulenofens wurde auf 28°C gestellt. Das Fließmittel wurde vor der Verwendung in
Ultraschalbad gestellt, um gelöste Gase zu beseitigen, die die HPLC-Analytik stören könnten.
Vor Beginn der Messungen wurde die HPLC-Apparatur mindestens eine halbe Stunde lang mit
der mobilen Phase äquilibriert. Die Leitungen des automatischen Probengebers und des
Injektionssystems wurden vor und nach jeder Messung mit entgastem 50%-igem wässrigen
Acetonitril gespült. Die Detektion erfolgte mithilfe eines Ultraviolettdetektors bei einer
Wellenlänge von 270 nm. Die Retentionszeit für Nifluminsäure (IS) lag bei 10,4 Minuten und für
Lumiracoxib bei 16,9 Minuten. Bei jeder Messung während der Lumiracoxibanalytik wurde
Qualitätskontrollproben (10, 50, 100, 500, 1000, 5000 und 10 000 ng/ml) mitgemessen. Die
Schwankungen des Peakhöhenverhältnisses von Lumiracoxib zu Nifluminsäure in den
Kontrollproben, berechnet als Variationskoeffizient, lagen im Lauf der Bestimmungen bei
weniger als 15%, bei Lumiracoxibkonzentration von 10 ng/ml bei weniger als 20%. Das HPLC-
System lief unter Verwendung der Software Borwin (Jasco-Borwin Version 1.50).

Ergebnisse 75
6.3.2 Validierung
In dieser Arbeit wurde die Validierung auf der Basis der von Shah (2000) zusammengefassten
Richtlinien der Konferenz „Analytical methods validation: Bioavailability, bioequivalence and
pharmacokinetic studies“ (Shah et al., 2000) durchgeführt.
6.3.2.1 Spezifität, Linearität und Bestimmungsgrenze
Spezifität (auch Selektivität) wurde nachgewiesen, indem man Leerplasmaproben von sechs
verschiedenen Probanden untersucht hat und keine Störpeaks an der Stelle der Analytes
festgestellt hat.
Linearität der Kalibriergerade für Lumiracoxib ist in Messbereich von 10 ng/ml bis 10000 ng/ml
vorhanden. Diesen Bereich wurde in 3 kleinere Messbereiche aufgeteilt: kleinen 10-100 ng/ml,
mittleren 100-1000 ng/ml und großen 1000-10 000 ng/ml und für jedes Bereich separat validiert.
Die Linearität der Messung wurde durch die Analyse von jeden Messbereich bestehend aus
jeweils fünf Konzentrationen ermittelt. Die Korrelationskoeffiziente waren größer als 0,997. Die
Bestimmungsgrenze lag bei 10 ng/ml.
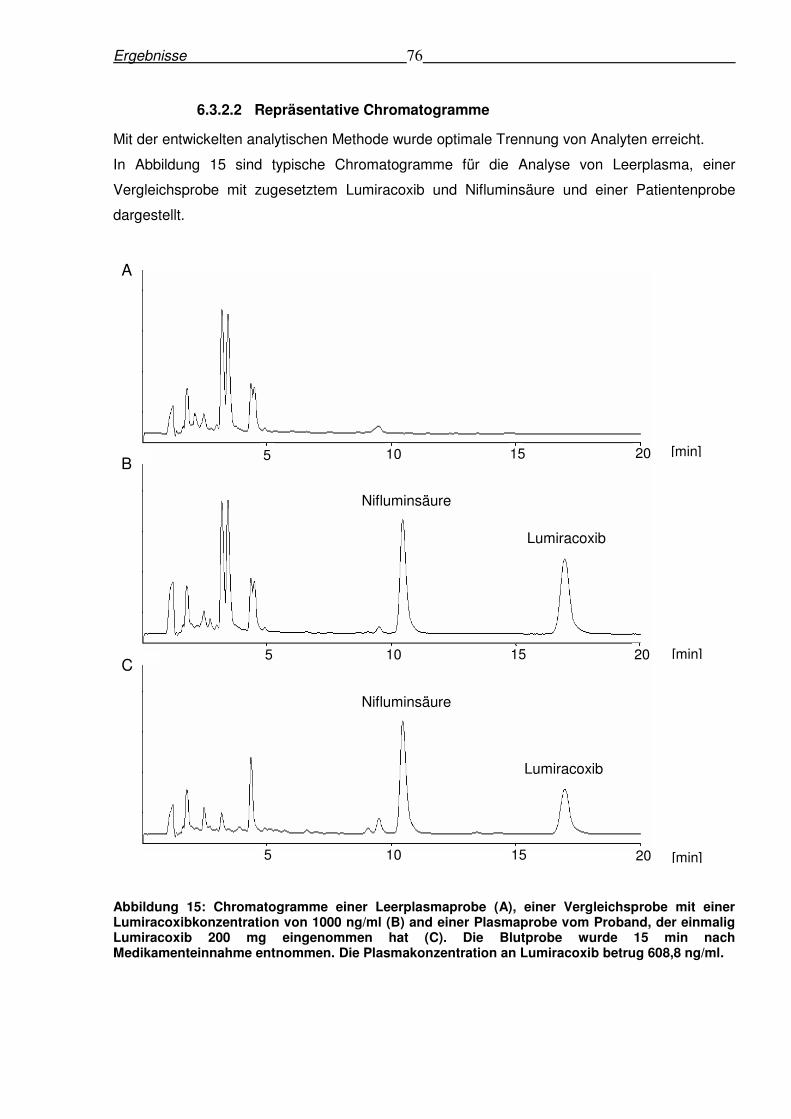
Ergebnisse 76
6.3.2.2 Repräsentative Chromatogramme
Mit der entwickelten analytischen Methode wurde optimale Trennung von Analyten erreicht.
In Abbildung 15 sind typische Chromatogramme für die Analyse von Leerplasma, einer
Vergleichsprobe mit zugesetztem Lumiracoxib und Nifluminsäure und einer Patientenprobe
dargestellt.
Abbildung 15: Chromatogramme einer Leerplasmaprobe (A), einer Vergleichsprobe mit einer Lumiracoxibkonzentration von 1000 ng/ml (B) and einer Plasmaprobe vom Proband, der einmalig Lumiracoxib 200 mg eingenommen hat (C). Die Blutprobe wurde 15 min nach Medikamenteinnahme entnommen. Die Plasmakonzentration an Lumiracoxib betrug 608,8 ng/ml.
A
B
C
5
5
5
10
10
10 15
15
15 20
20
20
Lumiracoxib
Lumiracoxib
Nifluminsäure
Nifluminsäure
[min]
[min]
[min]
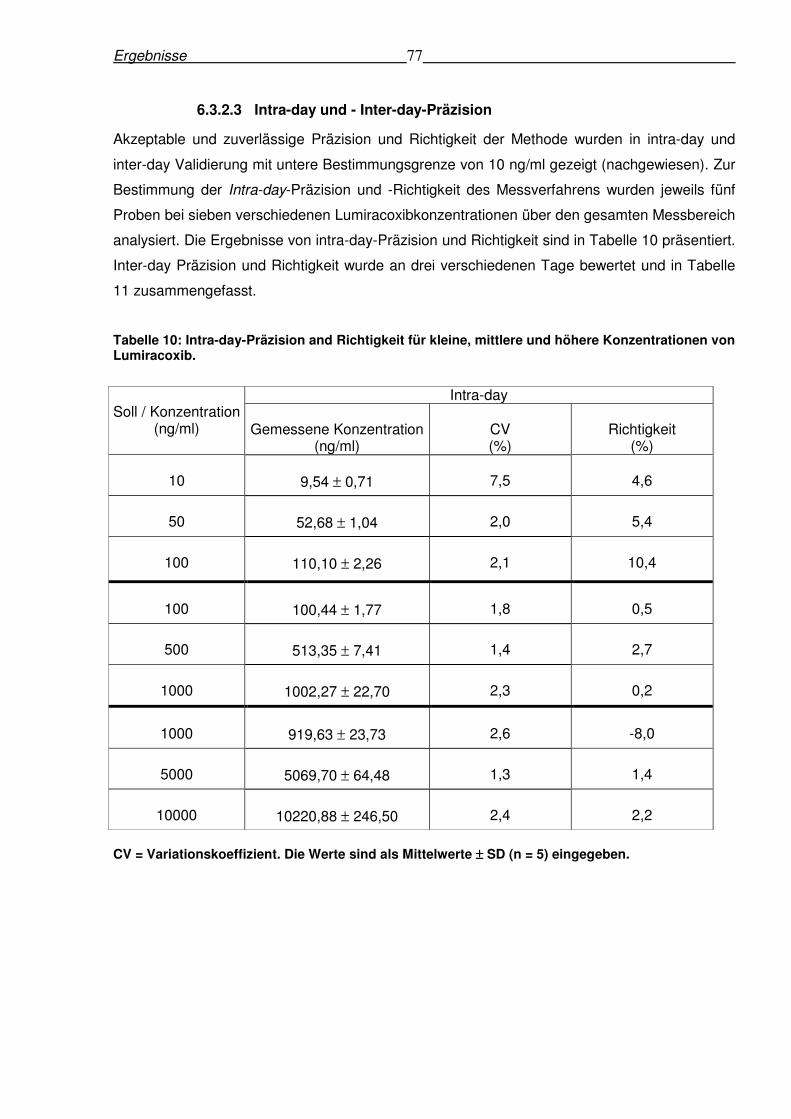
Ergebnisse 77
6.3.2.3 Intra-day und - Inter-day-Präzision
Akzeptable und zuverlässige Präzision und Richtigkeit der Methode wurden in intra-day und
inter-day Validierung mit untere Bestimmungsgrenze von 10 ng/ml gezeigt (nachgewiesen). Zur
Bestimmung der Intra-day-Präzision und -Richtigkeit des Messverfahrens wurden jeweils fünf
Proben bei sieben verschiedenen Lumiracoxibkonzentrationen über den gesamten Messbereich
analysiert. Die Ergebnisse von intra-day-Präzision und Richtigkeit sind in Tabelle 10 präsentiert.
Inter-day Präzision und Richtigkeit wurde an drei verschiedenen Tage bewertet und in Tabelle
11 zusammengefasst.
Tabelle 10: Intra-day-Präzision and Richtigkeit für kleine, mittlere und höhere Konzentrationen von Lumiracoxib.
Intra-day Soll / Konzentration
(ng/ml)
Gemessene Konzentration (ng/ml)
CV (%)
Richtigkeit
(%)
10
9,54 ± 0,71
7,5
4,6
50
52,68 ± 1,04
2,0
5,4
100
110,10 ± 2,26
2,1
10,4
100
100,44 ± 1,77
1,8
0,5
500
513,35 ± 7,41
1,4
2,7
1000
1002,27 ± 22,70
2,3
0,2
1000
919,63 ± 23,73
2,6
-8,0
5000
5069,70 ± 64,48
1,3
1,4
10000
10220,88 ± 246,50
2,4
2,2
CV = Variationskoeffizient. Die Werte sind als Mittelwerte ±±±± SD (n = 5) eingegeben.
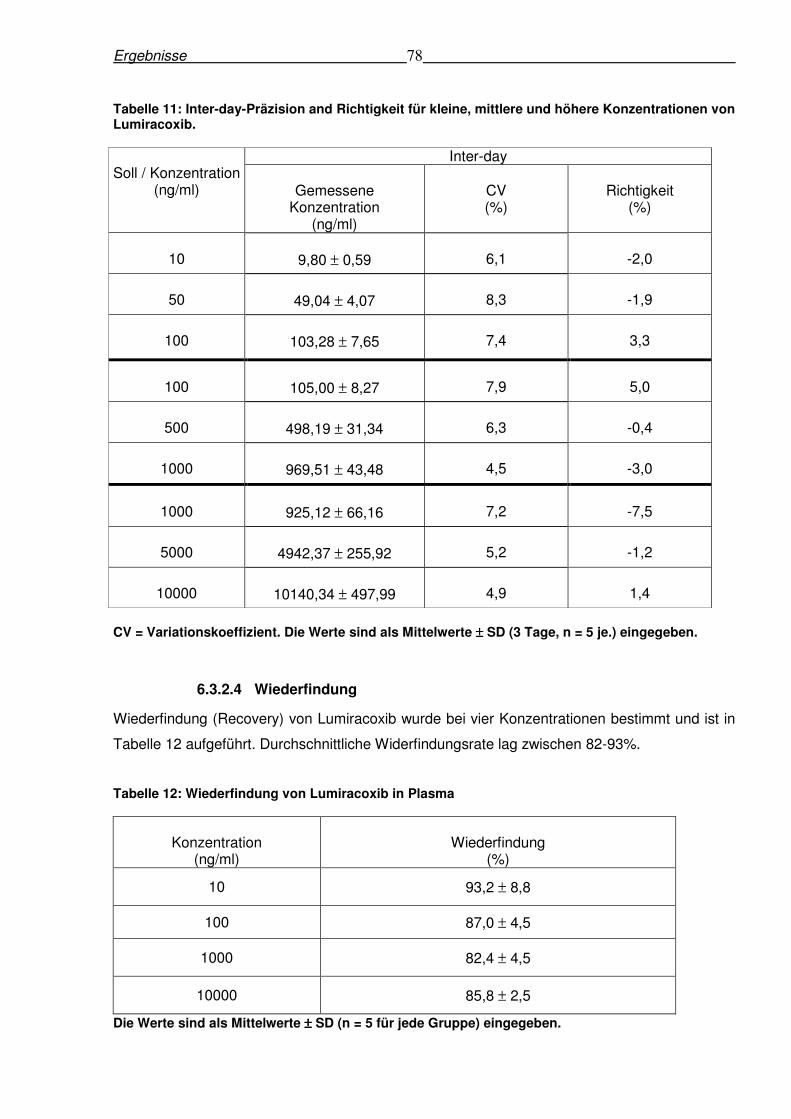
Ergebnisse 78
Tabelle 11: Inter-day-Präzision and Richtigkeit für kleine, mittlere und höhere Konzentrationen von Lumiracoxib.
Inter-day Soll / Konzentration
(ng/ml)
Gemessene Konzentration
(ng/ml)
CV (%)
Richtigkeit
(%)
10
9,80 ± 0,59
6,1
-2,0
50
49,04 ± 4,07
8,3
-1,9
100
103,28 ± 7,65
7,4
3,3
100
105,00 ± 8,27
7,9
5,0
500
498,19 ± 31,34
6,3
-0,4
1000
969,51 ± 43,48
4,5
-3,0
1000
925,12 ± 66,16
7,2
-7,5
5000
4942,37 ± 255,92
5,2
-1,2
10000
10140,34 ± 497,99
4,9
1,4
CV = Variationskoeffizient. Die Werte sind als Mittelwerte ±±±± SD (3 Tage, n = 5 je.) eingegeben.
6.3.2.4 Wiederfindung
Wiederfindung (Recovery) von Lumiracoxib wurde bei vier Konzentrationen bestimmt und ist in
Tabelle 12 aufgeführt. Durchschnittliche Widerfindungsrate lag zwischen 82-93%.
Tabelle 12: Wiederfindung von Lumiracoxib in Plasma
Konzentration
(ng/ml)
Wiederfindung
(%)
10 93,2 ± 8,8
100 87,0 ± 4,5
1000 82,4 ± 4,5
10000 85,8 ± 2,5
Die Werte sind als Mittelwerte ±±±± SD (n = 5 für jede Gruppe) eingegeben.
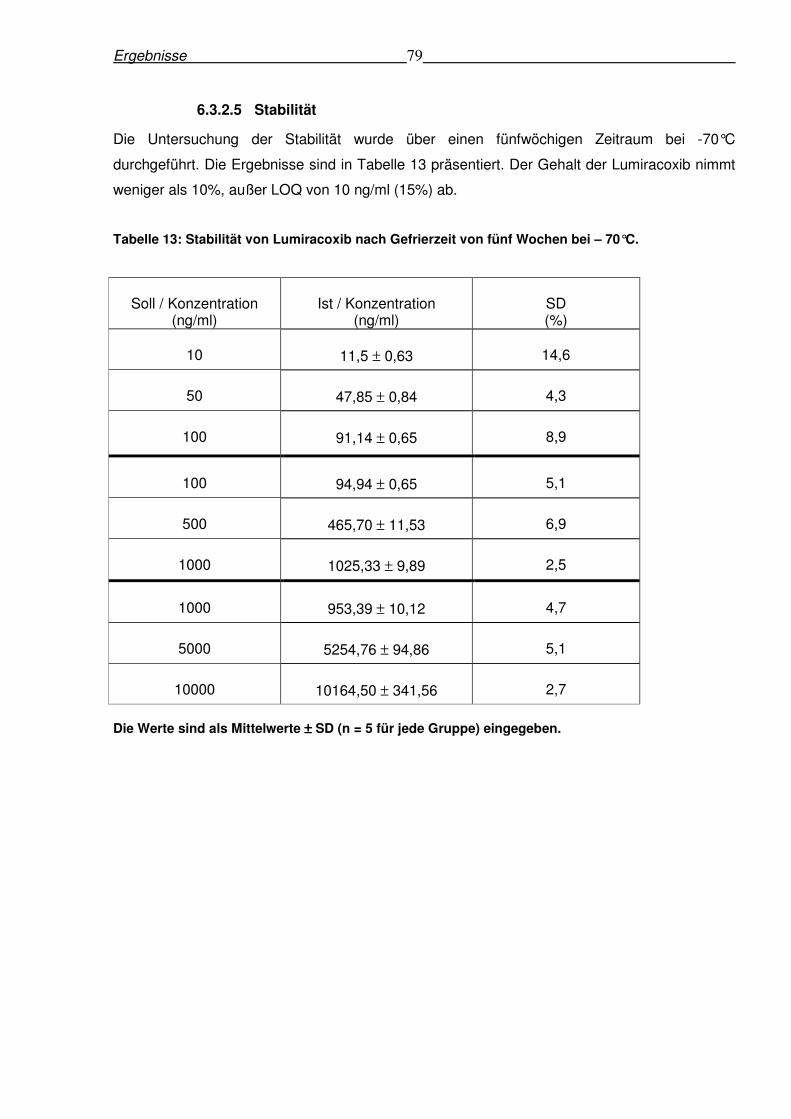
Ergebnisse 79
6.3.2.5 Stabilität
Die Untersuchung der Stabilität wurde über einen fünfwöchigen Zeitraum bei -70°C
durchgeführt. Die Ergebnisse sind in Tabelle 13 präsentiert. Der Gehalt der Lumiracoxib nimmt
weniger als 10%, außer LOQ von 10 ng/ml (15%) ab.
Tabelle 13: Stabilität von Lumiracoxib nach Gefrierzeit von fünf Wochen bei – 70°C.
Soll / Konzentration
(ng/ml)
Ist / Konzentration
(ng/ml)
SD (%)
10
11,5 ± 0,63
14,6
50
47,85 ± 0,84
4,3
100
91,14 ± 0,65
8,9
100
94,94 ± 0,65
5,1
500
465,70 ± 11,53
6,9
1000
1025,33 ± 9,89
2,5
1000
953,39 ± 10,12
4,7
5000
5254,76 ± 94,86
5,1
10000
10164,50 ± 341,56
2,7
Die Werte sind als Mittelwerte ±±±± SD (n = 5 für jede Gruppe) eingegeben.
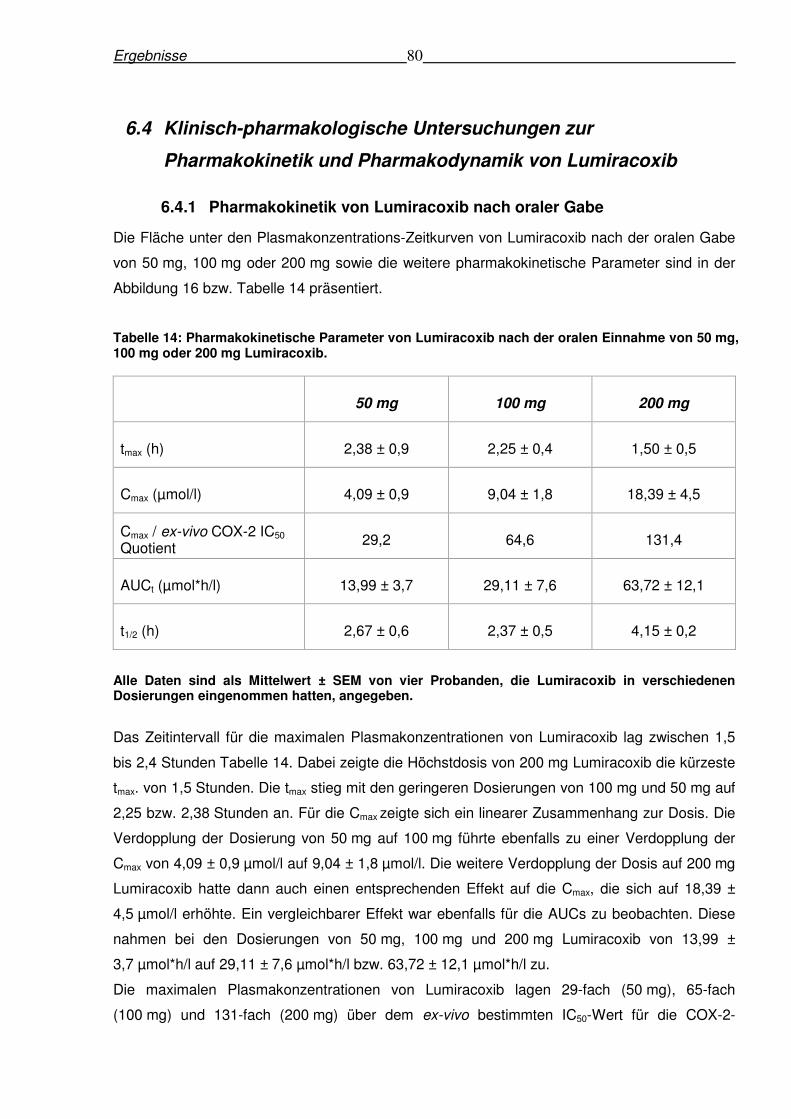
Ergebnisse 80
6.4 Klinisch-pharmakologische Untersuchungen zur
Pharmakokinetik und Pharmakodynamik von Lumiracoxib
6.4.1 Pharmakokinetik von Lumiracoxib nach oraler Gabe
Die Fläche unter den Plasmakonzentrations-Zeitkurven von Lumiracoxib nach der oralen Gabe
von 50 mg, 100 mg oder 200 mg sowie die weitere pharmakokinetische Parameter sind in der
Abbildung 16 bzw. Tabelle 14 präsentiert.
Tabelle 14: Pharmakokinetische Parameter von Lumiracoxib nach der oralen Einnahme von 50 mg, 100 mg oder 200 mg Lumiracoxib.
50 mg 100 mg 200 mg
tmax (h) 2,38 ± 0,9 2,25 ± 0,4 1,50 ± 0,5
Cmax (µmol/l) 4,09 ± 0,9 9,04 ± 1,8 18,39 ± 4,5
Cmax / ex-vivo COX-2 IC50
Quotient 29,2 64,6 131,4
AUCt (µmol*h/l) 13,99 ± 3,7 29,11 ± 7,6 63,72 ± 12,1
t1/2 (h) 2,67 ± 0,6 2,37 ± 0,5 4,15 ± 0,2
Alle Daten sind als Mittelwert ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen eingenommen hatten, angegeben.
Das Zeitintervall für die maximalen Plasmakonzentrationen von Lumiracoxib lag zwischen 1,5
bis 2,4 Stunden Tabelle 14. Dabei zeigte die Höchstdosis von 200 mg Lumiracoxib die kürzeste
tmax. von 1,5 Stunden. Die tmax stieg mit den geringeren Dosierungen von 100 mg und 50 mg auf
2,25 bzw. 2,38 Stunden an. Für die Cmax zeigte sich ein linearer Zusammenhang zur Dosis. Die
Verdopplung der Dosierung von 50 mg auf 100 mg führte ebenfalls zu einer Verdopplung der
Cmax von 4,09 ± 0,9 µmol/l auf 9,04 ± 1,8 µmol/l. Die weitere Verdopplung der Dosis auf 200 mg
Lumiracoxib hatte dann auch einen entsprechenden Effekt auf die Cmax, die sich auf 18,39 ±
4,5 µmol/l erhöhte. Ein vergleichbarer Effekt war ebenfalls für die AUCs zu beobachten. Diese
nahmen bei den Dosierungen von 50 mg, 100 mg und 200 mg Lumiracoxib von 13,99 ±
3,7 µmol*h/l auf 29,11 ± 7,6 µmol*h/l bzw. 63,72 ± 12,1 µmol*h/l zu.
Die maximalen Plasmakonzentrationen von Lumiracoxib lagen 29-fach (50 mg), 65-fach
(100 mg) und 131-fach (200 mg) über dem ex-vivo bestimmten IC50-Wert für die COX-2-
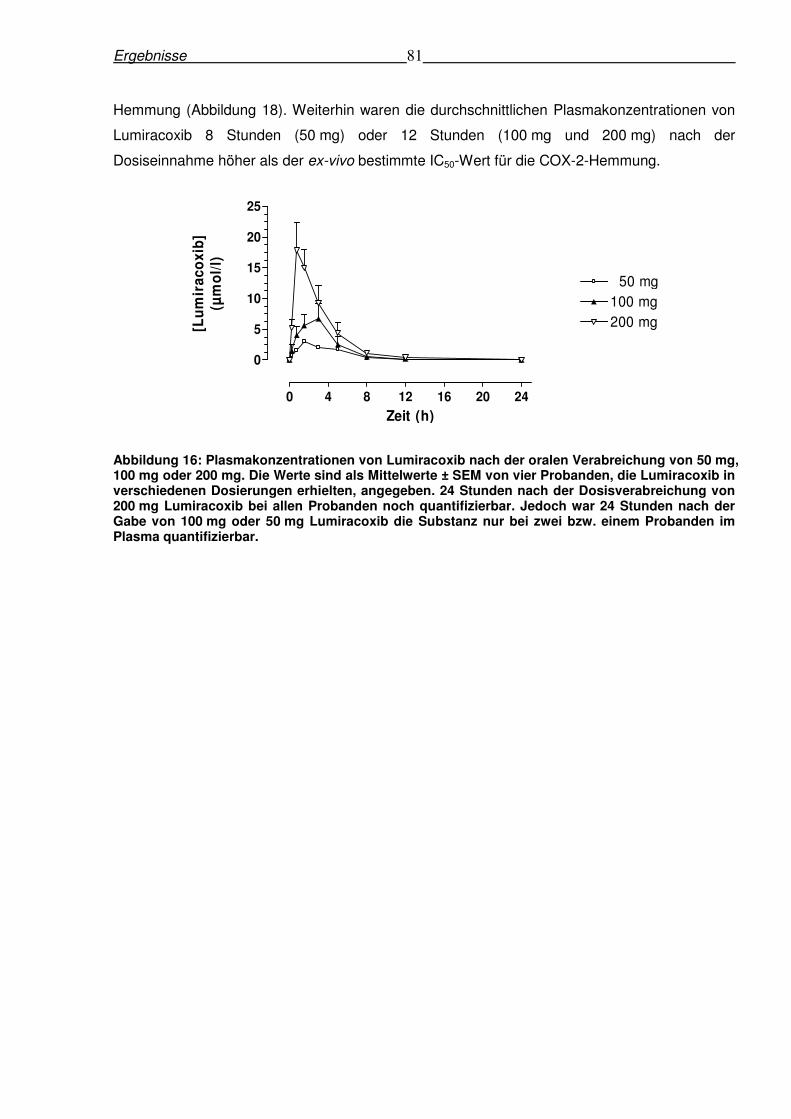
Ergebnisse 81
0 4 8 12 16 20 24
0
5
10
15
20
25
100 mg200 mg
50 mg
Zeit (h)
[Lu
mir
aco
xib
](µ
mo
l/l)
Hemmung (Abbildung 18). Weiterhin waren die durchschnittlichen Plasmakonzentrationen von
Lumiracoxib 8 Stunden (50 mg) oder 12 Stunden (100 mg und 200 mg) nach der
Dosiseinnahme höher als der ex-vivo bestimmte IC50-Wert für die COX-2-Hemmung.
Abbildung 16: Plasmakonzentrationen von Lumiracoxib nach der oralen Verabreichung von 50 mg, 100 mg oder 200 mg. Die Werte sind als Mittelwerte ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen erhielten, angegeben. 24 Stunden nach der Dosisverabreichung von 200 mg Lumiracoxib bei allen Probanden noch quantifizierbar. Jedoch war 24 Stunden nach der Gabe von 100 mg oder 50 mg Lumiracoxib die Substanz nur bei zwei bzw. einem Probanden im Plasma quantifizierbar.
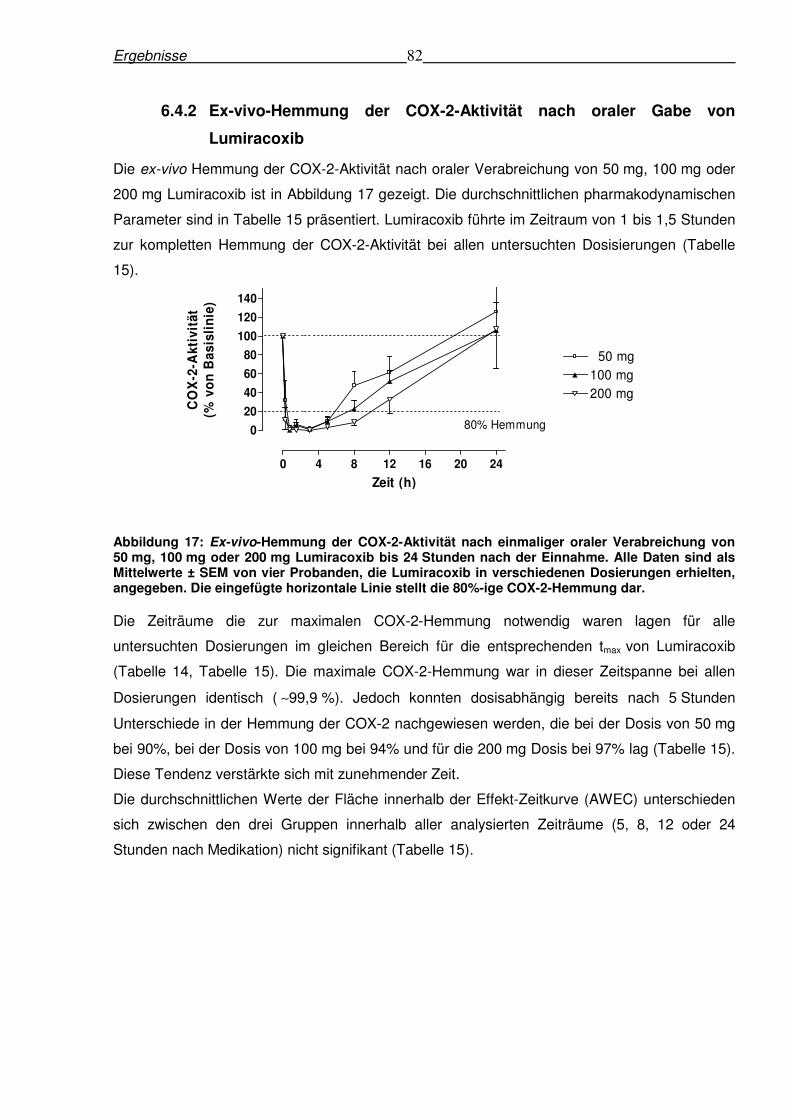
Ergebnisse 82
0 4 8 12 16 20 24
0
20
40
60
80
100
120
140
100 mg200 mg
50 mg
Zeit (h)
CO
X-2
-Akt
ivit
ät(%
vo
n B
asis
lin
ie)
80% Hemmung
6.4.2 Ex-vivo-Hemmung der COX-2-Aktivität nach oraler Gabe von
Lumiracoxib
Die ex-vivo Hemmung der COX-2-Aktivität nach oraler Verabreichung von 50 mg, 100 mg oder
200 mg Lumiracoxib ist in Abbildung 17 gezeigt. Die durchschnittlichen pharmakodynamischen
Parameter sind in Tabelle 15 präsentiert. Lumiracoxib führte im Zeitraum von 1 bis 1,5 Stunden
zur kompletten Hemmung der COX-2-Aktivität bei allen untersuchten Dosisierungen (Tabelle
15).
Abbildung 17: Ex-vivo-Hemmung der COX-2-Aktivität nach einmaliger oraler Verabreichung von 50 mg, 100 mg oder 200 mg Lumiracoxib bis 24 Stunden nach der Einnahme. Alle Daten sind als Mittelwerte ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen erhielten, angegeben. Die eingefügte horizontale Linie stellt die 80%-ige COX-2-Hemmung dar. Die Zeiträume die zur maximalen COX-2-Hemmung notwendig waren lagen für alle
untersuchten Dosierungen im gleichen Bereich für die entsprechenden tmax von Lumiracoxib
(Tabelle 14, Tabelle 15). Die maximale COX-2-Hemmung war in dieser Zeitspanne bei allen
Dosierungen identisch ( ∼99,9 %). Jedoch konnten dosisabhängig bereits nach 5 Stunden
Unterschiede in der Hemmung der COX-2 nachgewiesen werden, die bei der Dosis von 50 mg
bei 90%, bei der Dosis von 100 mg bei 94% und für die 200 mg Dosis bei 97% lag (Tabelle 15).
Diese Tendenz verstärkte sich mit zunehmender Zeit.
Die durchschnittlichen Werte der Fläche innerhalb der Effekt-Zeitkurve (AWEC) unterschieden
sich zwischen den drei Gruppen innerhalb aller analysierten Zeiträume (5, 8, 12 oder 24
Stunden nach Medikation) nicht signifikant (Tabelle 15).
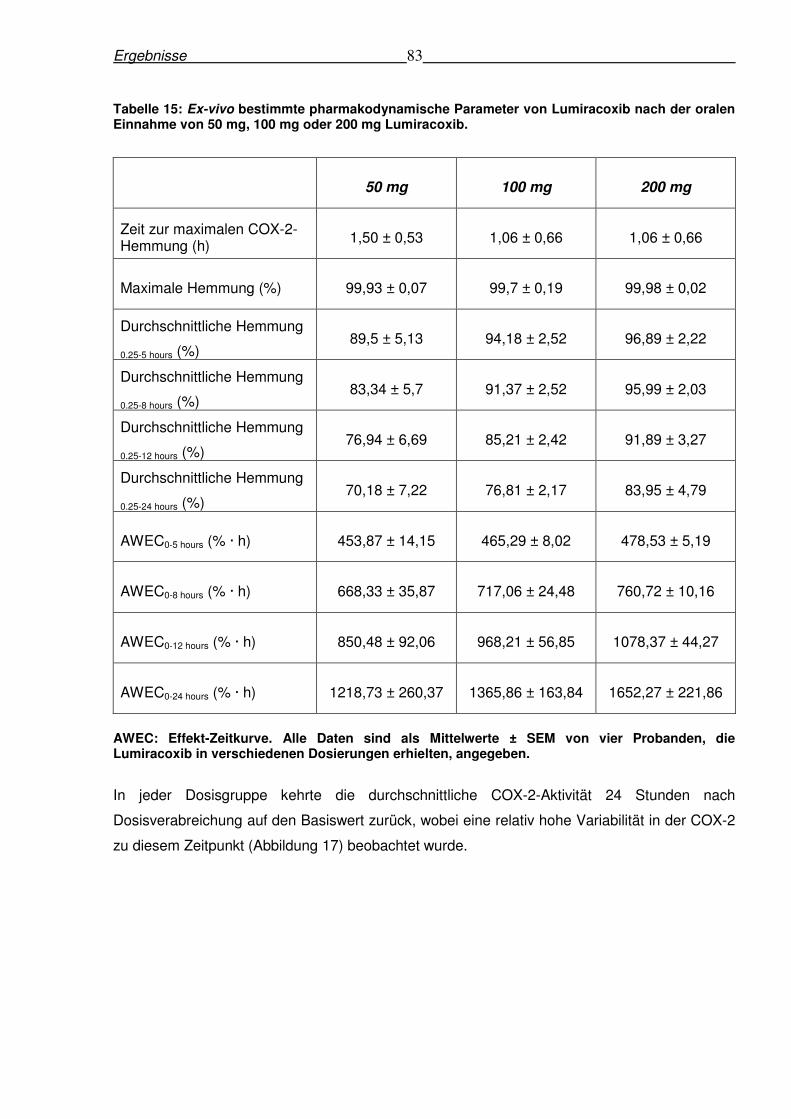
Ergebnisse 83
Tabelle 15: Ex-vivo bestimmte pharmakodynamische Parameter von Lumiracoxib nach der oralen Einnahme von 50 mg, 100 mg oder 200 mg Lumiracoxib.
50 mg 100 mg 200 mg
Zeit zur maximalen COX-2-Hemmung (h) 1,50 ± 0,53 1,06 ± 0,66 1,06 ± 0,66
Maximale Hemmung (%) 99,93 ± 0,07 99,7 ± 0,19 99,98 ± 0,02
Durchschnittliche Hemmung
0.25-5 hours (%) 89,5 ± 5,13 94,18 ± 2,52 96,89 ± 2,22
Durchschnittliche Hemmung
0.25-8 hours (%) 83,34 ± 5,7 91,37 ± 2,52 95,99 ± 2,03
Durchschnittliche Hemmung
0.25-12 hours (%) 76,94 ± 6,69 85,21 ± 2,42 91,89 ± 3,27
Durchschnittliche Hemmung
0.25-24 hours (%) 70,18 ± 7,22 76,81 ± 2,17 83,95 ± 4,79
AWEC0-5 hours (% · h) 453,87 ± 14,15 465,29 ± 8,02 478,53 ± 5,19
AWEC0-8 hours (% · h) 668,33 ± 35,87 717,06 ± 24,48 760,72 ± 10,16
AWEC0-12 hours (% · h) 850,48 ± 92,06 968,21 ± 56,85 1078,37 ± 44,27
AWEC0-24 hours (% · h) 1218,73 ± 260,37 1365,86 ± 163,84 1652,27 ± 221,86
AWEC: Effekt-Zeitkurve. Alle Daten sind als Mittelwerte ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen erhielten, angegeben.
In jeder Dosisgruppe kehrte die durchschnittliche COX-2-Aktivität 24 Stunden nach
Dosisverabreichung auf den Basiswert zurück, wobei eine relativ hohe Variabilität in der COX-2
zu diesem Zeitpunkt (Abbildung 17) beobachtet wurde.
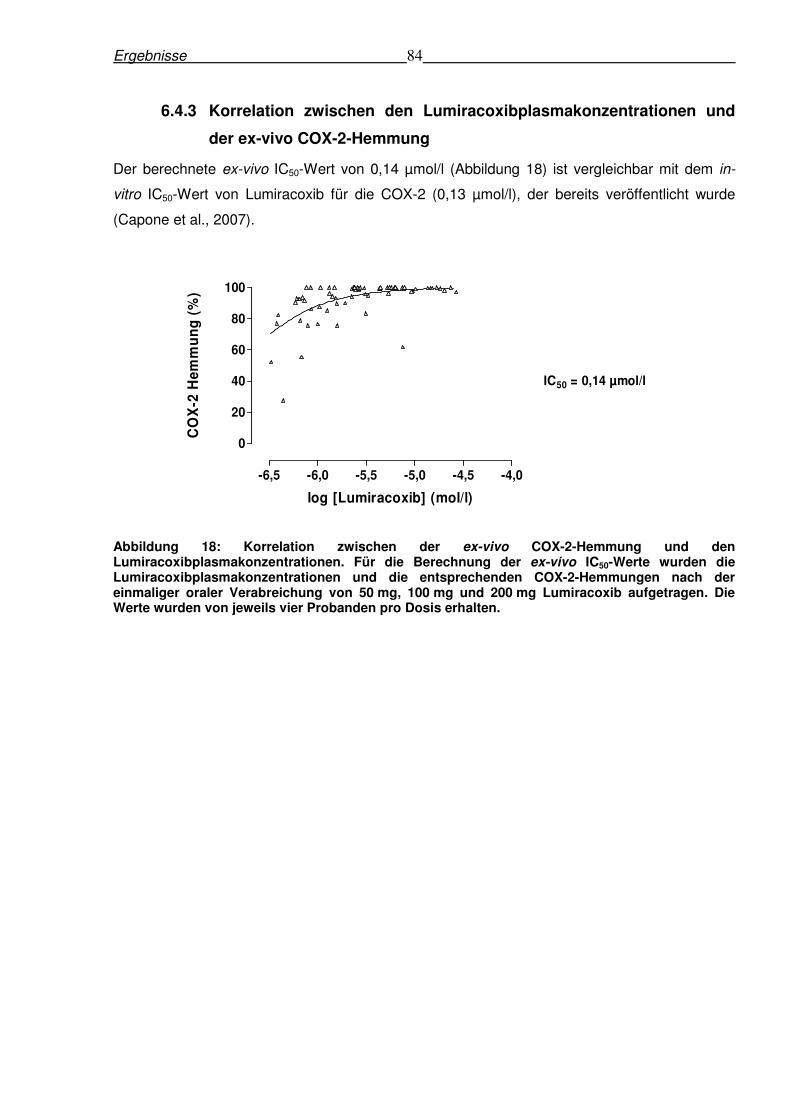
Ergebnisse 84
-6,5 -6,0 -5,5 -5,0 -4,5 -4,0
0
20
40
60
80
100
IC50 = 0,14 µmol/l
log [Lumiracoxib] (mol/l)
CO
X-2
Hem
mu
ng
(%
)
6.4.3 Korrelation zwischen den Lumiracoxibplasmakonzentrationen und
der ex-vivo COX-2-Hemmung
Der berechnete ex-vivo IC50-Wert von 0,14 µmol/l (Abbildung 18) ist vergleichbar mit dem in-
vitro IC50-Wert von Lumiracoxib für die COX-2 (0,13 µmol/l), der bereits veröffentlicht wurde
(Capone et al., 2007).
Abbildung 18: Korrelation zwischen der ex-vivo COX-2-Hemmung und den Lumiracoxibplasmakonzentrationen. Für die Berechnung der ex-vivo IC50-Werte wurden die Lumiracoxibplasmakonzentrationen und die entsprechenden COX-2-Hemmungen nach der einmaliger oraler Verabreichung von 50 mg, 100 mg und 200 mg Lumiracoxib aufgetragen. Die Werte wurden von jeweils vier Probanden pro Dosis erhalten.
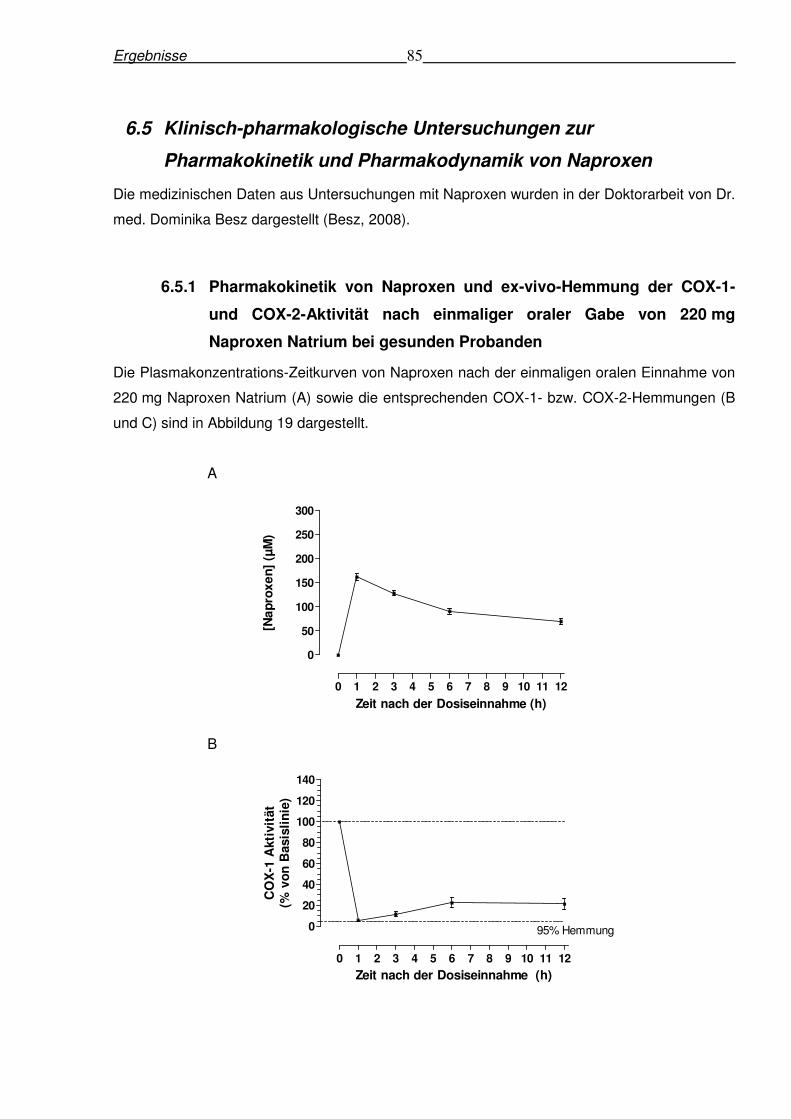
Ergebnisse 85
6.5 Klinisch-pharmakologische Untersuchungen zur
Pharmakokinetik und Pharmakodynamik von Naproxen
Die medizinischen Daten aus Untersuchungen mit Naproxen wurden in der Doktorarbeit von Dr.
med. Dominika Besz dargestellt (Besz, 2008).
6.5.1 Pharmakokinetik von Naproxen und ex-vivo-Hemmung der COX-1-
und COX-2-Aktivität nach einmaliger oraler Gabe von 220 mg
Naproxen Natrium bei gesunden Probanden
Die Plasmakonzentrations-Zeitkurven von Naproxen nach der einmaligen oralen Einnahme von
220 mg Naproxen Natrium (A) sowie die entsprechenden COX-1- bzw. COX-2-Hemmungen (B
und C) sind in Abbildung 19 dargestellt.
A
B
0 1 2 3 4 5 6 7 8 9 10 11 12
0
20
40
60
80
100
120
140
95% Hemmung
Zeit nach der Dosiseinnahme (h)
CO
X-1
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
0 1 2 3 4 5 6 7 8 9 10 11 12
0
50
100
150
200
250
300
Zeit nach der Dosiseinnahme (h)
[Na
pro
xe
n]
(µM
)
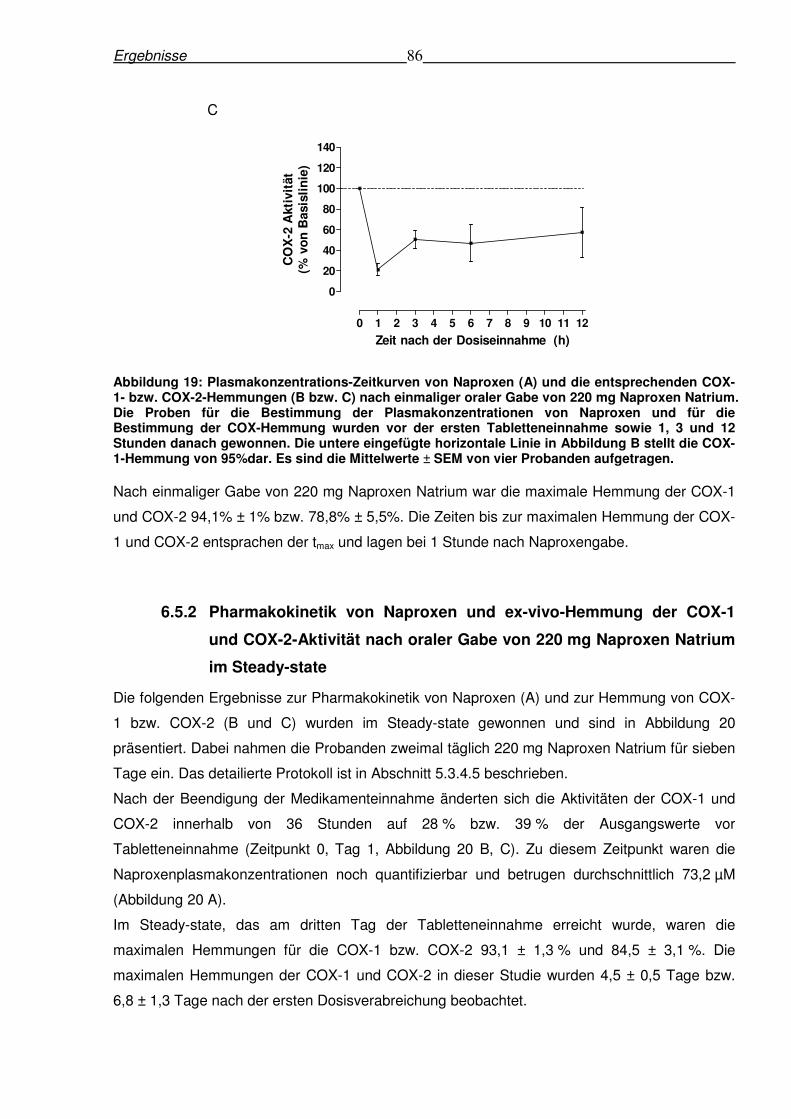
Ergebnisse 86
C
Abbildung 19: Plasmakonzentrations-Zeitkurven von Naproxen (A) und die entsprechenden COX-1- bzw. COX-2-Hemmungen (B bzw. C) nach einmaliger oraler Gabe von 220 mg Naproxen Natrium. Die Proben für die Bestimmung der Plasmakonzentrationen von Naproxen und für die Bestimmung der COX-Hemmung wurden vor der ersten Tabletteneinnahme sowie 1, 3 und 12 Stunden danach gewonnen. Die untere eingefügte horizontale Linie in Abbildung B stellt die COX-1-Hemmung von 95%dar. Es sind die Mittelwerte ± SEM von vier Probanden aufgetragen. Nach einmaliger Gabe von 220 mg Naproxen Natrium war die maximale Hemmung der COX-1
und COX-2 94,1% ± 1% bzw. 78,8% ± 5,5%. Die Zeiten bis zur maximalen Hemmung der COX-
1 und COX-2 entsprachen der tmax und lagen bei 1 Stunde nach Naproxengabe.
6.5.2 Pharmakokinetik von Naproxen und ex-vivo-Hemmung der COX-1
und COX-2-Aktivität nach oraler Gabe von 220 mg Naproxen Natrium
im Steady-state
Die folgenden Ergebnisse zur Pharmakokinetik von Naproxen (A) und zur Hemmung von COX-
1 bzw. COX-2 (B und C) wurden im Steady-state gewonnen und sind in Abbildung 20
präsentiert. Dabei nahmen die Probanden zweimal täglich 220 mg Naproxen Natrium für sieben
Tage ein. Das detailierte Protokoll ist in Abschnitt 5.3.4.5 beschrieben.
Nach der Beendigung der Medikamenteinnahme änderten sich die Aktivitäten der COX-1 und
COX-2 innerhalb von 36 Stunden auf 28 % bzw. 39 % der Ausgangswerte vor
Tabletteneinnahme (Zeitpunkt 0, Tag 1, Abbildung 20 B, C). Zu diesem Zeitpunkt waren die
Naproxenplasmakonzentrationen noch quantifizierbar und betrugen durchschnittlich 73,2 µM
(Abbildung 20 A).
Im Steady-state, das am dritten Tag der Tabletteneinnahme erreicht wurde, waren die
maximalen Hemmungen für die COX-1 bzw. COX-2 93,1 ± 1,3 % und 84,5 ± 3,1 %. Die
maximalen Hemmungen der COX-1 und COX-2 in dieser Studie wurden 4,5 ± 0,5 Tage bzw.
6,8 ± 1,3 Tage nach der ersten Dosisverabreichung beobachtet.
0 1 2 3 4 5 6 7 8 9 10 11 12
0
20
40
60
80
100
120
140
Zeit nach der Dosiseinnahme (h)
CO
X-2
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
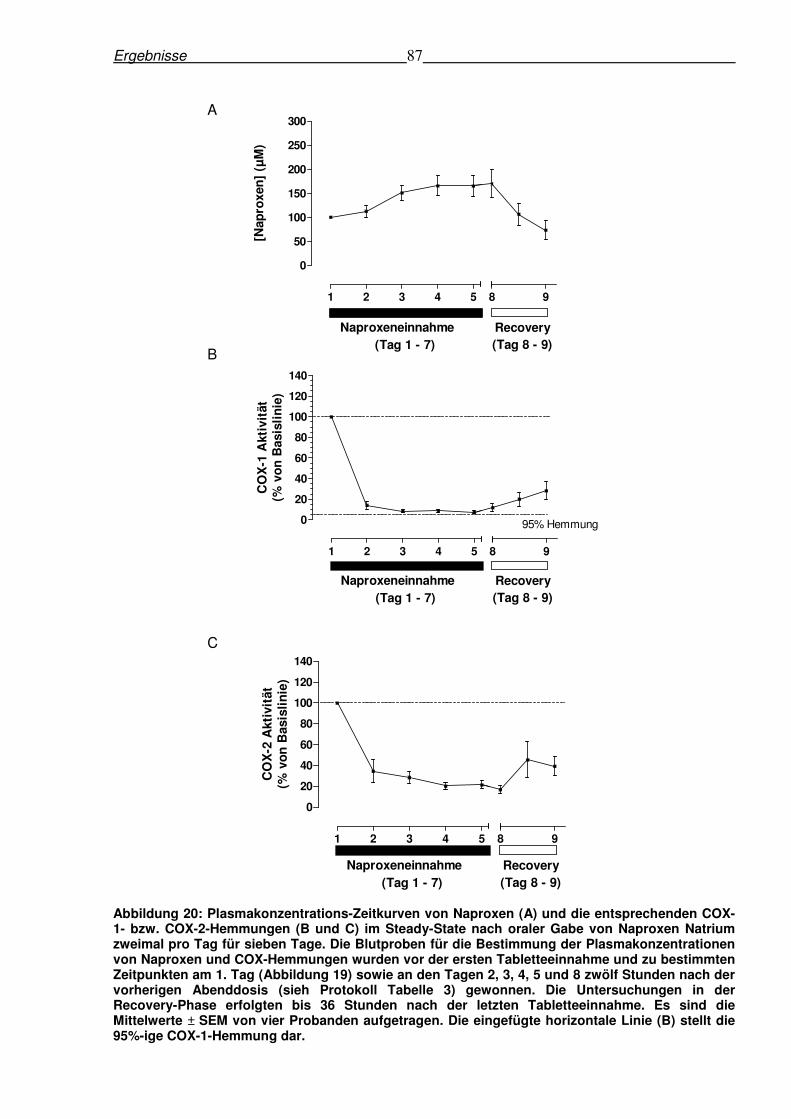
Ergebnisse 87
1 2 3 4 5
0
50
100
150
200
250
300
8 9
Naproxeneinnahme Recovery(Tag 8 - 9)(Tag 1 - 7)
[Nap
roxe
n]
(µM
)
A
B
C
Abbildung 20: Plasmakonzentrations-Zeitkurven von Naproxen (A) und die entsprechenden COX-1- bzw. COX-2-Hemmungen (B und C) im Steady-State nach oraler Gabe von Naproxen Natrium zweimal pro Tag für sieben Tage. Die Blutproben für die Bestimmung der Plasmakonzentrationen von Naproxen und COX-Hemmungen wurden vor der ersten Tabletteeinnahme und zu bestimmten Zeitpunkten am 1. Tag (Abbildung 19) sowie an den Tagen 2, 3, 4, 5 und 8 zwölf Stunden nach der vorherigen Abenddosis (sieh Protokoll Tabelle 3) gewonnen. Die Untersuchungen in der Recovery-Phase erfolgten bis 36 Stunden nach der letzten Tabletteeinnahme. Es sind die Mittelwerte ± SEM von vier Probanden aufgetragen. Die eingefügte horizontale Linie (B) stellt die 95%-ige COX-1-Hemmung dar.
1 2 3 4 5
0
20
40
60
80
100
120
140
8 9
Naproxeneinnahme Recovery(Tag 8 - 9)(Tag 1 - 7)
95% Hemmung
CO
X-1
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
1 2 3 4 5
0
20
40
60
80
100
120
140
8 9
Naproxeneinnahme Recovery(Tag 8 - 9)(Tag 1 - 7)
CO
X-2
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
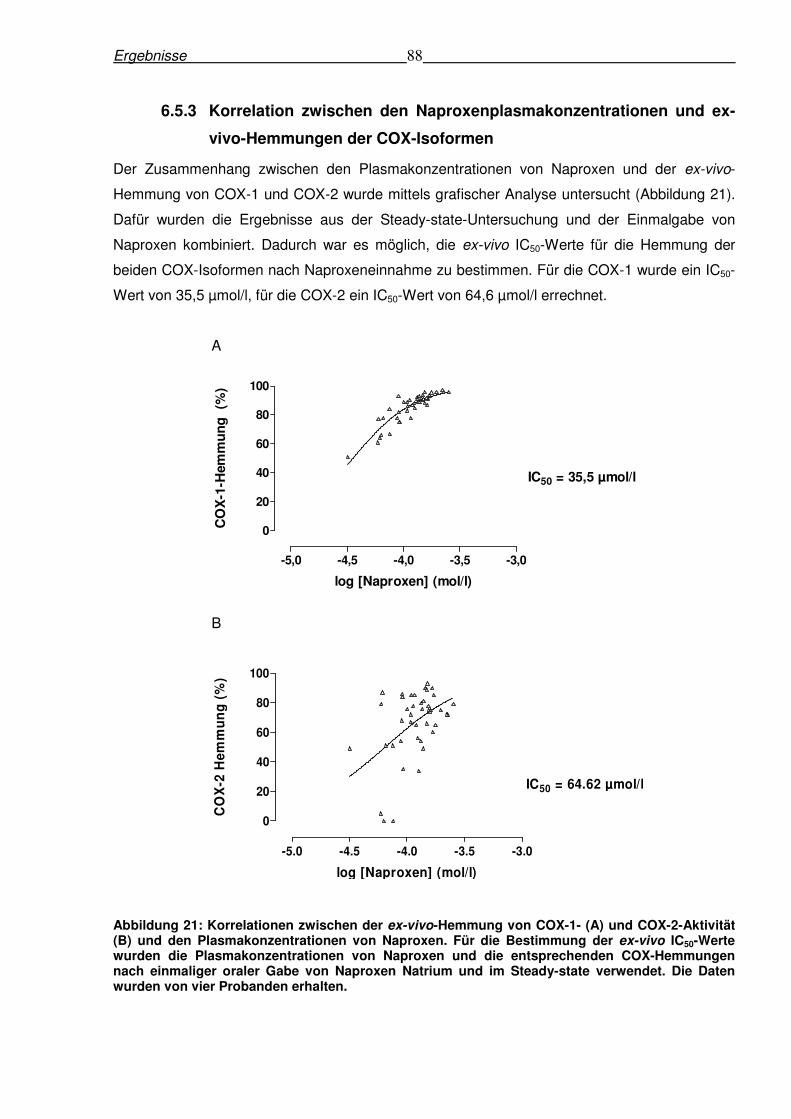
Ergebnisse 88
6.5.3 Korrelation zwischen den Naproxenplasmakonzentrationen und ex-
vivo-Hemmungen der COX-Isoformen
Der Zusammenhang zwischen den Plasmakonzentrationen von Naproxen und der ex-vivo-
Hemmung von COX-1 und COX-2 wurde mittels grafischer Analyse untersucht (Abbildung 21).
Dafür wurden die Ergebnisse aus der Steady-state-Untersuchung und der Einmalgabe von
Naproxen kombiniert. Dadurch war es möglich, die ex-vivo IC50-Werte für die Hemmung der
beiden COX-Isoformen nach Naproxeneinnahme zu bestimmen. Für die COX-1 wurde ein IC50-
Wert von 35,5 µmol/l, für die COX-2 ein IC50-Wert von 64,6 µmol/l errechnet.
A
B
Abbildung 21: Korrelationen zwischen der ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) und den Plasmakonzentrationen von Naproxen. Für die Bestimmung der ex-vivo IC50-Werte wurden die Plasmakonzentrationen von Naproxen und die entsprechenden COX-Hemmungen nach einmaliger oraler Gabe von Naproxen Natrium und im Steady-state verwendet. Die Daten wurden von vier Probanden erhalten.
-5,0 -4,5 -4,0 -3,5 -3,0
0
20
40
60
80
100
IC50 = 35,5 µmol/l
log [Naproxen] (mol/l)
CO
X-1
-Hem
mu
ng
(%
)
-5.0 -4.5 -4.0 -3.5 -3.0
0
20
40
60
80
100
IC50 = 64.62 µmol/l
log [Naproxen] (mol/l)
CO
X-2
Hem
mu
ng
(%
)

Ergebnisse 89
Der sigmoidale Verlauf ist im Falle der COX-1 deutlicher ausgeprägt, da bei den maximalen
Plasmakonzentrationen nach einmaliger therapeutischer Dosierung von 220 mg Naproxen
Natrium eine 100 %ige Hemmung erreicht wurde (Abbildung 21). Weiterhin war auffällig, dass
für die COX-2 der sigmoidale Kurvenverlauf weniger deutlich ausgeprägt war als für die COX-1.
Dies ist auf die hohe Variabilität für die COX-Inhibition über den gesamten Bereich der
Plasmakonzentrationen zurückzuführen (Abbildung 21).

Diskussion 90
7 Diskussion
7.1 Untersuchungen zum Wirkungsmechanismus von Metamizol
Der Wirkungsmechanismus von Metamizol konnte lange nicht aufgeklärt werden. Zuerst wurde
das Medikament mit einer vorwiegenden Hemmung der PGE-Synthese im Gehirngewebe in
Zusammenhang gebracht (Dembinska-Kiec, 1976). Jedoch widersprach eine spätere Studie
einer zellspezifischen Modulation, da eine vergleichbare hemmende Wirkung von dem
Hauptmetabolit von Metmizol (4-MAA) auf die Prostanoid-Freisetzung in Astrozyten und
Makrophagen gezeigt werden konnte (Lanz et al., 1986). Darüberhinaus wurde vor kurzem
behauptet, dass Metamizol eine Variante des COX-1-Genes, die sogenannte COX-3, selektiv
hemmt (Chandrasekharan et al., 2002). Diese Splicevariante ist durch den Verbleib des Intron-1
charakterisiert. Jedoch wurde in dieser Studie die COX-3 von Hunden und COX-1 bzw. COX-2
von Mäusen mit jeweils unterschiedlichen Aktivitäten verglichen. Metamizol zeigte dabei nur
eine schwache COX-3-Hemmung (IC50 = 52 µmol/l). Ein weiteres Problem ist die Tatsache,
dass die Induktion und die Aufrechterhaltung der Hyperalgesie eher durch Prostaglandine, die
durch die zentrale COX-2 gebildet wurden, als durch die COX-1 assozierten PGs vermittelt wird.
Zusätzlich dazu wurde kürzlich die Existenz einer humanen COX-3 in Frage gestellt, da die
Retention des Intron-1 beim Menschen zur Verschiebung des Leserahmens (frame-shift), zum
frühzeitigem Translationsstop und unvollständigem COX-3 Protein führte (Patrono, 2001; Hinz
et al., 2006).
Qin et al. gelang es, die geringe Expression von drei Splicevarianten der COX-1 im humanen
Gewebe nachzuweisen (Qin et al., 2005). Jedoch konnte kein Unterschied hinsichtlich der
Hemmung durch Metamizol zwischen der Wildtypvariante der COX-1 und einer Variante mit
Intron-1 festgestellt werden. Aufgrund der erwähnten Gründe mussten die Versuche, die
pharmakologische Wirkung von nichtsauren Analgetika mit der Hemmung der zentralen COX-3
zu erklären, verworfen werden. Obwohl weitere ausgezeichnete Arbeiten veröffentlicht wurden,
die die zentralen Wirkmechanismen des antinozizeptiven Effekts von Metamizol unterstützen
(Carlsson et al., 1986; Gelgor et al., 1992; Neugebauer et al., 1994; Shimada et al., 1994),
wurde eine periphere Wirkung in vergangenen Jahren weitgehend vernachlässigt.
Die vorliegende Arbeit ist die erste, die ex-vivo beim Menschen die Selektivität von Metamizol
zu den peripheren COX-Enzymen sowie die Bedeutung der entsprechenden
Metamizolmetabolite untersuchte. Hier wurde gezeigt, dass der Hauptmetabolit von Metamizol,
Methylaminoantipyrin (4-MAA), in-vitro und ex-vivo im Blut von Probanden zu einer deutlichen
Hemmung von COX-1 und COX-2 führt. In-vitro hemmte 4-MAA die COX-1 und COX-2
praktisch in gleichem Maße. Nach oraler Gabe von Metamizol waren die maximalen
Plasmakonzentrationen von 4-MAA bis zu 23,2- und 12,7-fach höher (1000 mg Dosis) als die

Diskussion 91
jeweiligen in-vitro bestimmten IC50-Werte für COX-1 bzw. COX-2. Aufgrund eines schnellen
Anflutens von 4-MAA innerhalb von 15 Minuten nach Tabletteneinnahme kam es bei beiden
Dosierungen (500 md und 1000 mg) zu einer unmittelbaren Hemmung der zwei Isoformen. Die
Plasmakonzentrationen von 4-MAA blieben nach Verabreichung für 8 Stunden (500 mg Dosis)
bzw. 12 Stunden (1000 mg Dosis) über den für jedes Isoenzym entsprechenden in-vitro
bestimmten IC50-Werten. Die ex-vivo Hemmung der COX-Enzyme korrelierte mit der
Plasmakonzentration von 4-MAA, wobei die ex-vivo-bestimmten IC50-Werte 2,5-fach für COX-1
und 5,3-fach für COX-2 niedriger als die jeweiligen in-vitro Werte waren. Im Einklang mit bereits
veröffentlichten Daten über andere COX-2-Inhibitoren (Hinz et al., 2006) wurde eine
beträchtliche Variabilität zwischen Probanden bezüglich der Plasmakonzentrationen von 4-MAA
und der ex-vivo-Inhibition von COX-2 beobachtet. Diese Variabilität könnte auch für den oben
erwähnten, obgleich sehr kleinen Unterschied zwischen den in-vitro und ex-vivo bestimmten
IC50-Werten mitverantwortlich sein.
Entsprechend wurden signifikante Unterschiede zwischen den Parametern der COX-Hemmung
(maximale und mittlere Hemmung, AWEC-Werte) für beide Metamizol-Dosen für COX-1
ermittelt. Im Gegensatz dazu, zeigten sich keine Unterschiede für die COX-2. Die Gründe dafür
sind unklar, aber genetische und interindividuelle Variabilitäten in der COX-2 oder in der
Pharmakokinetik sind möglicherweise dafür verantwortlich. (Patrono, 2001).
Im Gegensatz zu 4-MAA waren die maximalen Plasmakonzentrationen vom anderen
Metamizolmetabolit 4-Aminoantipyrin (4-AA) nach der 1000 mg Metamizol Dosis 2,8- und 5,7-
fach unter den in-vitro IC50-Werten für COX-1 bzw. COX-2. So erscheint ein signifikanter Beitrag
von 4-AA nach einmaliger Einnahme von Metamizol zur Hemmung beider Enzyme
unwahrscheinlich.
Aufgrund der schnellen Hydrolyse von Metamizol erreichte 4-MAA in kurzer Zeit die maximale
Plasmakonzentration. Dieses pharmakokinetische Profil steht im Einklang mit dem klinisch
erforderlichen schnellen Wirkungseintritt und einer viermaligen täglichen Einnahme. Bei
Verdoppelung der Dosis kann man eine mehr als zweifache Zunahme in den AUC-Werten
beider Metaboliten feststellen. Dies unterstützt die Hypothese einer nicht linearen
Pharmakokinetik von Metamizol (Levy et al., 1995). Die Zunahme nach Dosisverdoppelung war
deutlicher bei 4-MAA. Im Vergleich zu diesem Metabolit erreicht 4-AA die maximale
Plasmakonzentration später und wird auch mit mittleren terminalen Halbwertzeiten von 6,3
Stunden (500 mg Dosis) bzw. 5,7 Stunden (1000 mg Dosis) langsamer und variabler eliminiert.
Agranulozytose wird als schwerwiegende Nebenwirkung in Zusammenhang mit Metamizol
gebracht. Obwohl hier ein statistisch signifikanter Zusammenhang zu bestehen scheint, ist das
Auftreten von Agranulozytose äußerst selten (1 Fall pro Millionen Behandlungsperioden)
(Anonymous, 1986; Kaufman DW, 1991; Ibanez et al., 2005). Diese Tatsache verhinderte

Diskussion 92
ausführliche klinische Untersuchungen mit diesem Medikament. Aus diesem Grund gibt es auch
keine klinischen Studien zur Wirkung von Metamizol bei chronischen Schmerzen (Hackenthal,
1997). Dementsprechend sind tierische Studien, die auf eine im Vergleich zu NSAR geringere
entzündungshemmende Wirkung von Metamizol hinwiesen (Brune and Alpermann, 1983;
Tatsuo et al., 1994), nicht auf Menschen angewandt worden.
In vorliegende Untersuchungen rief Metamizol eine beträchtliche Hemmung der COX-2-Aktivität
hervor, die eindeutig über der COX-2-Hemmung anderer nicht saurer Wirkstoffe wie z.B.
Celecoxib und Rofecoxib nach einmaliger Dosierung von klinisch empfohlenen Dosen von
200 mg bzw. 25 mg lag (Hinz et al., 2006). Obwohl noch nicht nachgewiesen, weisen die
gegenwärtigen Daten daraufhin, dass die empfohlene 4-mal tägliche Gabe von 500 mg oder
1000 mg Metamizol (Levy et al., 1995) mit konstanten 70-90%-igen Hemmung von COX-2
assoziiert wird.
Hinsichtlich der nicht selektiven Hemmung der COX-Enzyme sind vorliegende Daten in
Einklang mit den Ergebnissen von Campos et al. (Campos et al., 1999), die gereinigte Enzyme
für ihre Untersuchungen benutzten. Dabei wurden jedoch verhältnismäßig hohe IC50-Werte (426
µmol/l für COX-1 und COX-2) beobachtet. Einer der Gründe für diese Tatsache liegt
möglicherweise in der kurzzeitigen (10 Minuten) Verwendung des inaktiven Prodrug Metamizol
in einem enzymatischen Assay. In der gleichen Veröffentlichung wurden weiterhin IC50-Werte
für die COX-1-Hemmung in Endothelialzellen der Rinderaorta und frischen menschlichen
Blutplättchen beschrieben, die weit über den notwendigen pharmakologischen Konzentrationen
lagen. Die Autoren schlossen daraus, dass Metamizol eine selektive COX-2-Hemmung in-vivo
hervorrufen könnte (Campos et al., 1999). In der Tat könnte diese Hypothese das reduzierte
Risiko für gastrointestinale Blutungen von Metamizol im Vergleich zu ASS und anderen NSAR
erklären (Laporte and Carne, 1987; Laporte et al., 1991). Trotz alledem konnten wir im Vollblut,
das eher die in-vivo Situation repräsentiert, eine signifikante Hemmung der COX-1-vermittelten
TXB2-Synthese beobachten. Bisherige Daten weisen darauf hin, dass im Serum nur eine
Hemmung der TXB2-Synthese von über 95% die Thrombozytenfunktion maßgeblich beeinflusst
(Reilly and FitzGerald, 1987). In vorliegenden Untersuchungen lag die Hemmung der COX-1-
vermittelten TXB2-Bildung in der 500 mg Gruppe unter 95%. Eine Inhibition der TXB2-Synthese
über 95% konnte jedoch in der 1000 mg Gruppe im Zeitraum zwischen 0,75 und 3 Stunden
nach Metamizoleinmahme beobachtet werden. Im Vergleich zu einer Einmaldosis von 100 mg
Aspirin, die zu einer Reduktion des thrombozytären TXB2 von über 95% für 24 Stunden führt
(Patrignani et al., 1982), hat Metamizol nur einen sehr kurzzeitigen thrombozytenhemmenden
Effekt. Andererseits war das Ausmaß der COX-1-Hemmung durch Metamizol stärker als bei
einer 75 mg Retardform von Diclofenac (Hinz et al., 2003). Aufgrund der geringen
gastrointestinalen Nebenwirkungen von Metamizol in epidemiologischen Studien und dem
widersprüchlichen Einfluss auf COX-1, wird vermutet, dass physikochemische Faktoren (z.B.

Diskussion 93
die nicht vorhandenen Säureeigenschaften) für die bessere gastrointestinale Verträglichkeit im
Vergleich zu den NSAR verantwortlich sind. Tatsächlich sind zwei Mechanismen hauptsächlich
für den ulcerativen Effekt von NSAR verantwortlich. Dabei handelt es sich zum einen um die
lokale Reizung der Mukosaschleimhaut und zum anderen um die Hemmung der
Prostaglandinsynthese. Die lokale Reizung der Schleimhaut ist dabei nur auf die NSAR
beschränkt, da diese sich aufgrund ihres sauren Charakters im sauren Milieu („Ionenfalle“)
akkumulieren (Brune et al., 1980). Weitere Studien weisen darauf hin, dass nicht nur die
alleinige Inhibition der Prostaglandinsynthese für die gastrointestinalen Komplikationen
verantwortlich ist (Frolich et al., 1986). So konnte gezeigt werden, dass eine dreimal tägliche
Gabe von 1000 mg Metamizol für 3 Tage trotz einer substantiellen Verringerung der PGE2-
Konzentrationen im Magensaft nicht zu schweren gastrointestinalen Beeinträchtigungen führte.
Zusammengefasst weißt die schnelle und deutliche Hemmung von COX-2 durch therapeutische
Dosierungen von Metamizol in menschlichen Blutmonozyten daraufhin, dass ein signifikanter
Anteil der analgetischen Wirkung von Metamizol wahrscheinlich durch periphere Mechanismen
vermittelt wird. Metamizol in Dosierung von 1000 mg ruft eine COX-2-Hemmung über 80 %, d.h.
noch höhere Dosierung (z.B. 2,5 mg) kann nur noch wenig zum Effekt beitragen, dagegen aber
die COX-1 vermittelten unerwünschten Effekte potenzieren.
Im Hinblick auf zu NSAR vergleichbare COX-1-Hemmung wird vermutet, dass eher
physikochemische Faktoren (z.B. die nicht vorhandenen Säureeigenschaften) für die bessere
gastrointestinale Verträglichkeit im Vergleich zu den sauren NSAR verantwortlich sind.
Aufgrund der ausgeprägten analgetischen Wirkung von Metamizol, seiner guten
gastrointestinalen Verträglichkeit und der freiverkäuflichen Anwendung in vielen Ländern
(insbesondere Lateinamerika, Asien, Ost- und Mitteleuropa) sind weitere klinische Studien zur
Aufklärung der peripheren analgetischen und antientzündlichen Mechanismen sinnvoll.

Diskussion 94
7.2 Untersuchungen zum Wirkungsmechanismus von Paracetamol
Die Wirkungsweise von Paracetamol gehört auch wie Metamizol noch immer zu den
pharmakologischen Rätseln in der Schmerz-Forschung. Während das Arzneimittel zuerst mit
einer vorwiegenden Unterdrückung der PG-Synthese im Hirngewebe (Flower and Vane, 1972)
in Verbindung gebracht wurde, stellten nachfolgende Studien eine zellspezifische Modulation in
Frage (Lanz et al., 1986; Swierkosz et al., 2002; Warner et al., 2004). Einer der Gründe für
diese kontroversen Ergebnisse könnte an der Tatsache liegen, dass Paracetamol keine
messbaren Inhibitionen der PG-Bildung in aufgeschlossenen Zell-Präparationen, aber eine
profunde Unterdrückung in intakten Zellen auslöst (Graham and Scott, 2005). Trotz dieser
Ergebnisse wird Paracetamol als ein schwacher Inhibitor von peripheren COX-1 und COX-2
Enzymen angesehen. Auf der anderen Seite tauchen erste Anzeichen auf, die andeuten, dass
Paracetamol viele Eigenschaften mit selektiven COX-2 Inhibitoren teilt, da es keine toxischen
Effekte auf den Gastrointestinaltrakt auslöst (Garcia Rodriguez and Hernandez-Diaz, 2001),
keine Thrombozytenfunktion hemmt (Mielke, 1981; Catella-Lawson et al., 2001) und kaum
bronchiale Verengungen bei Aspirin-sensitiven Asthmatikern provoziert (Jenkins et al., 2004).
Basierend auf diesen klinischen Beobachtungen wurde im Rahmen dieser Arbeit die Hypothese
einer selektiven COX-2- Inhibition durch Paracetamol untersucht, wobei menschliche Vollblut-
Proben benutzt wurden (Patrignani et al., 1994). Dadurch war es möglich den aussagekräftigen
Selektivitäts-Index zu bestimmen, mit dem die COX-Inhibition in einem physiologischen Milieu
gemessen wird und dadurch intakte zelluläre Interaktionen, die für den
Arachidonsäuremetabolismus essentiell sind, sowie die Arzneistoffbindung an Plasmaproteine,
berücksichtigt wird.
Die vorliegende Studie demonstriert, dass Paracetamol eine vierfach höhere Selektivität für die
Inhibition von COX-2 in-vitro und ex-vivo zeigt. Die Verabreichung einer Standarddosis des
Arzneistoffes bewirkte eine beinahe komplette Inhibition von COX-2 in menschlichen
Probanden, wohingegen nur eine moderate Inhibition von COX-1 beobachtet wurde. In
vorliegenden Studie war die Inhibition der COX-2-Aktivität durch Paracetamol sogar höher als
das entsprechend bei Einzeldosierung von Celecoxib und Rofecoxib bei klinisch empfohlenen
Dosierungen von 200 mg und 25 mg (Hinz et al., 2006). Im Gegensatz zu den letzteren
Verbindungen wurde keine substanzielle Variabiliät zwischen den Patienten bezüglich der
Plasmakonzentration von Paracetamol und dem entsprechendem Grad der ex-vivo COX-2
Inhibition beobachtet. Obwohl es noch nicht untersucht ist, wird es durch die bestehenden
Daten offensichtlich, dass die empfohlene viermalige Dosierung von 1000 mg Paracetamol am
Tag mit einer permanenten 60-80%-igen Inhibition von COX-2 verbunden sein könnte. Frühere
Daten (Catella-Lawson et al., 2001), die eine viel langsamere und unselektivere ex-vivo

Diskussion 95
Inhibition von COX-2 durch Paracetamol andeuteten, entstanden wahrscheinlich aufgrund der
Auswertung innerhalb eines begrenzten Zeitrahmens.
In Anbetracht des ähnlichen Verhaltens von Paracetamol und Coxiben auf die COX-2
vermittelten PGE2-Konzentration im menschlichen Vollblut, deuten vorliegende Daten an, dass
Paracetamol eine periphere COX-2 während einer Entzündung, ähnlich wie Rofecoxib und
Celecoxib, beeinflusst. Obwohl eine solche Annahme gerechtfertigt erscheint, wurde von
Paracetamol angenommen, keine entzündungshemmende Aktivität zu besitzen. In der Tat
basiert dieser Befund auf eine frühere Arbeit, die zeigt, dass Paracetamol keine Entzündungen,
die mit Gelenkrheumatismus assoziiert sind, hemmt (Boardman and Hart, 1967; Ring et al.,
1974). Dieses könnte durch die hohe extrazelluläre Konzentration der Arachidonsäure und
Peroxid im entzündeten Gewebe erklärt werden; beides schwächt den Effekt von Paracetamol
auf die PG-Synthese (Ouellet and Percival, 2001; Boutaud et al., 2002; Graham and Scott,
2005). Auf der anderen Seite mindert Paracetamol die Gewebe-Anschwellung nach einer
Kiefer-Operation bei Menschen, mit einer Wirksamkeit, sehr ähnlich der von Ibuprofen
(Skjelbred and Lokken, 1979; Bjornsson et al., 2003). Eine periphere entzündungshemmende
Wirkung wird ferner gestützt durch Ergebnisse, die Paracetamol als ein Inhibitor von
Nozizeption und Ödemen im “rat carrageenan footpad model” zeigen (Ferreira et al., 1978;
Honore et al., 1995), einem inflammatorischen Zustand, der sehr abhängig von COX-2-
vermittelten PGs ist (Smith et al., 1998).
Vorliegende Ergebnisse zeigen ferner, dass die COX-2-Blockade durch Paracetamol in
menschlichen Gefäßen, Beachtung auslösen sollte. Tatsächlich ist eine permanente Blockade
von COX-2-abhängigen PGs, einschließlich Prostacyclin und PGE2 die zurzeit plausibelste
Erklärung für die kardiovaskuläre Gefährdung, die von selektiven und nicht selektiven COX-2
Inhibitoren ausgeht (Cannon et al., 2006; Hinz et al., 2006). In diesem Zusammenhang wurde
ein anhaltender Anstieg des Blutdruckes nach langfristiger Anwendung als Hauptursache für die
kardiovaskulären Nebenwirkungen vorgeschlagen.
Bezüglich der Auswirkungen von Paracetamol auf den Blutdruck, fand eine große
Gruppenstudie heraus, dass die geregelte Anwendung von Paracetamol im Vergleich zur
Nichtanwendung durch ein signifikant höheres Risiko für die Entwicklung von Hypertonie
begleitet wurde (Forman et al., 2005). Bemerkenswerterweise war das relative Risiko von
Paracetamol ähnlich wie das der NSAR. In Übereinstimmung mit dieser Erkenntnis, zeigte sich
in einer großen, prospektiven, kürzlich veröffentlichten Studie, dass die Anwendung von
Paracetamol bei mehr als 15 Tabletten pro Woche nahezu das gleiche Risiko für
kardiovaskuläre Ereignisse, wie bei traditionellen NSAR mit sich bringt (Chan et al., 2006).
In der vorliegenden Arbeit reduzierte Paracetamol die COX-2-Proteinmenge in menschlichen
Monozyten weder in der Abwesenheit noch in der Anwesenheit von LPS. Dies schließt einen

Diskussion 96
signifikanten Beitrag zu einer verminderten COX-2-Expression durch eine verringerte PGE2-
Konzentraton in menschlichem Blut aus. Ferner wurde die Hemmung der COX-2 Aktivität durch
Paracetamol in Kurzzeit-Experimenten, in denen sowohl menschliche rekombinante COX-2
Enzyme als auch menschliche Monozyten als Matrix von COX-2 eingesetzt wurden, bestätigt.
Wie früher schon diskutiert wurde, hängt die Fähigkeit von Paracetamol als COX-2-Inhibitor
stark vom oxidierenden / antioxidierenden Status des umgebenden Systemes ab (Ouellet and
Percival, 2001; Graham and Scott, 2005). Daher kann die Anwesenheit von verschiedenen
enzymatischen und nichtenzymatischen antioxidierenden Komponenten im menschlichen
Plasma (Halliwell and Gutteridge, 1990) erklären, warum Paracetamol die ausgeprägteste
COX-2-Inhibition im menschlichen Vollblut im Vergleich mit den beiden anderen
experimentellen Systemen auslöst.
Die nur moderate Inhibition von COX-1 durch Paracetamol bei Probanden spiegelt sich durch
dessen schwache antithrombozytenaggregatorische Aktivität und der guten gastrointestinalen
Verträglichkeit wieder. Tatsächlich deuten frühere Daten an, dass nur eine Steigung über 95%
der Inhibition des TXB2 im Serum signifikant die Thrombozytenfunktion beeinflusst (Reilly and
FitzGerald, 1987). In Übereinstimmung mit unseren Daten hemmt Paracetamol bei einzelnen
oralen Dosierungen von 1000 mg (Mielke, 1981) oder von bis zu 1950 mg (Catella-Lawson et
al., 2001) nicht die Thrombozytenfunktion. Klinische Studien, die von einer Inhibition der
Thrombozytenfunktion durch Paracetamol berichten, benutzten hohe parenterale Dosierungen
des Arzneimittels (Niemi et al., 2000; Munsterhjelm et al., 2005). Dieselbe Abhängigkeit von der
Dosierung gilt auch für die gastrointestinale Verträglichkeit von Paracetamol. Demnach führen
höhere Dosierungen von Paracetamol, im Vergleich mit geringeren Dosierungen, zu höheren
Raten von gastrointestinalen Nebenwirkungen (z.B. Dyspepsie), wahrscheinlich aufgrund der
ausgeprägteren COX-1-Inhibition. Im Gegensatz zu mehreren, kurzzeitigen, randomisierten
Studien, deuten Ergebnisse von epidemiologischen Studien an, dass Paracetamol, bei
täglichen Dosierungen von >2 oder >2,6 g, das Risiko von stärkeren gastrointestinalen
Nebenwirkungen, einschließlich gastrointestinaler Blutungen oder Perforation, erhöht (Mielke,
1981; Rahme et al., 2002). Allerdings kann aufgrund dieser Daten nicht geschlussfolgert
werden, dass Paracetamol zu erhöhten gastrointestinalen Nebenwirkungen führt, da oftmals
Patienten mit einem gastrointestinalem Risiko bzw. Komplikationen mit Paracetamol behandelt
werden (Abramson, 2002). Demnach wird Paracetamol immer noch als frei von größerer
gastrointestinaler Toxizität angesehen (Bannwarth, 2004; Graham et al., 2005). Dennoch sollten
großangelegte, randomisierte, gastrointestinale Ausgangsstudien an Patienten durchgeführt
werden, die langfristig Paracetamol einehmen.
Mit Bezug auf die gastrointestinale Sicherheit von Paracetamol, erscheint eine andere Tatsache
bemerkenswert. Neben seiner signifikant geringeren Auswirkung auf COX-1 im Vergleich zu
den sauren NSAR, können physikochemische Faktoren, so wie das Fehlen der Säurefunktion

Diskussion 97
für die günstige Magenverträglichkeit sowohl von Paracetamol, als auch von Metamizol
verantwortlich sein. Folglich, abgesehen von der Fähigkeit, die PG-Synthese zu unterdrücken,
wird die ulzerogene Wirkung von NSAR im Magen von einem topisch reizenden Effekt auf das
Epithel, aufgrund des Phänomens „ ion traping“, d.h. aufgrund der Akkumulation von saueren
NSAR in gastrischen Epithelzellen begleitet (McCormack and Brune, 1987; Somasundaram et
al., 1997; Wallace, 1997). In diesem Zusammenhang wurde behauptet, dass die sauren NSAR
Schleimhautverletzungen produzieren, indem oxidative mitochondrielle Phosphorylierungen in
Epithelzellen ausgelöst werden, die eine verminderte zelluläre ATP-Produktion zur Folge haben,
sowie zelluläre Toxizität und eine gesteigerte mukosale Permeabilität (Somasundaram et al.,
1997). Ein COX-unabhängiger Mechanismus für die gastrointestinale Toxizität von NSAR wird
desweiteren gestützt durch Befunde in COX-1 Knock-out Mäusen, die noch gastrale Reaktionen
als Antwort auf die orale Verabreichung von Indomethacin entwickelten (Langenbach et al.,
1995).
Letztendlich begründet die Frage der nachgewiesenen selektiven ex-vivo COX-2-Inhibition in
Erwachsenen einen generellen Ablauf, der zu ermitteln bleibt. Im Falle des Geschlechtes
jedenfalls ist die früher nachgewiesene 22-prozentige Clearance (Ausscheidung) von
Paracetamol bei Männern im Vergleich zu Frauen (Miners et al., 1983) nicht als klinisch wichtig
betrachtet worden.
In Bezug auf Kinder ist die zu erzielende Plasmakonzentration von Paracetamol für die
Antipyrese 10-20 µg/ml (z.B., 66-133 µmol/l) (Rumack, 1978) was im Bereich der
Paracetamolkonzentrationen, die zu einer starken COX-2 Inhibition in menschlichem Vollblut
von Erwachsenen führen, liegt. Obwohl die ideale Plasmakonzentration für die Analgesie
unbestimmt bleibt, wird angenommen, dass sie innerhalb desselben Bereiches liegt (Mantzke
and Brambrink, 2002). Auf der anderen Seite empfehlen aktuelle Produkte eine rektale
Paracetamoldosis von 10-15 mg/kg für Kinder, die maximale Plasmakonzentrationen erbringt,
die geringer sind als die zitierte pharmakologische Konzentration, weshalb die Frage
aufgeworfen wird, ob diese Dosis für eine effektive post-operative Schmerztherapie geeignet ist
(Mantzke and Brambrink, 2002). In diesem Zusammenhang hat eine frühere Studie gezeigt,
dass in Kindern eine anfängliche rektale Paracetamoldosis von 40 mg/kg gefolgt von einer 20
mg/kg Dosis alle sechs Stunden benötigt wird, um geeignete antipyretische
Plamakonzentrationen zu erreichen (Birmingham et al., 2001). Hinsichtlich älterer Patienten
zeigen frühere Daten keine altersbezogenen Veränderungen in der Rate und im Ausmaß der
Resorption sowie Plasmakonzentrationen von Paracetamol (Triggs and Nation, 1975).

Diskussion 98
7.3 Entwicklung und Validierung einer analytischen Methode zur
Bestimmung von Lumiracoxib in Plasma von menschlichen
Probanden
Eine einfache, sensitive analytische Methode zur Bestimmung von Lumiracoxib in Plasma
mittels Flüssig-flüssig-Extraktion und HPLC-UV-Detektion wurde entwickelt und validiert.
Die Spezifität der Methode wurde dadurch nachgewiesen, dass in sechs untersuchten Leer-
plasmen keine Störpeaks an der Stelle der Analyten festgestellt wurden. Eine
Zusammensetzung der mobilen Phase aus Acetonitril:0.05 % Trichloressigsäure 35:65 (v/v)
erlaubt eine effektive Trennung der Analyten von einander und die Trennung des internen
Standardes vom Störpeak vor ihm.
Der Messbereich von 10 ng/ml bis 10000 ng/ml wurde in drei kleinere Messbereiche aufgeteilt.
Die Kalibrationsgerade jedes Bereiches bestand aus fünf Kalibrationspunkten. Die Linearität der
Kalibrationsgerade wurde in jedem Messbereich nachgewiesen und die
Lumiracoxibkonzentration von 10 ng/ml wurde als LOQ festgelegt.
Die Untersuchung der Präzision und Richtigkeit wurde für sieben verschiedene Konzentrationen
mit jeweils fünf Proben (Messungen) an einem Tag durchgeführt. Bei allen Konzentrationen
wurden eine niedrige Standardabweichung und ein Variationskoeffizient bis zu maximal 7,5 %
ermittelt, woraus sich eine sehr geringe Streuung der Analysenergebnisse und damit eine hohe
Präzision der Methode ergab. Auch die Bestimmung der Inter-day-Präzision ergab mit
Variationskoeffizienten von 4,5 % bzw. 8,3 % gute Ergebnisse, wodurch die Gleichmäßigkeit
und Qualität von Aufarbeitung und Messung auch an unterschiedlichen Tagen bestätigt wurde.
Diese analytische Methode wurde für die Bestimmung von Lumiracoxib in Plasma bei gesunden
Probanden in der nachstehenden Studie verwendet. Alle gemessenen Konzentrationen lagen
im Messbereich dieser Methode.

Diskussion 99
7.4 Untersuchungen zur COX-2-Hemmung durch Lumiracoxib in
verschiedenen Dosierungen
Die vorliegende Arbeit demonstrierte, dass die orale Gabe von 50 mg Lumiracoxib bei
gesunden Probanden die COX-2 in Blutmonozyten in einem vergleichbaren Ausmaß wie
100 mg oder 200 mg Lumiracoxib hemmt. Die maximale Plasmakonzentrationen von
Lumiracoxib waren 29- (50 mg), 65- (100 mg) und 131-fach (200 mg) über der Konzentration
(IC50), die die Hälfte der maximalen COX-2-Hemmung ex-vivo bewirkt. Das spiegelt eine
schnelle und vollständige Hemmung dieses Enzymes durch jede Dosis wider.
Die maximalen Plasmakonzentrationen des Medikamentes wurden innerhalb eines Zeitraums
von 1,5 bis zu 2,4 Stunden nach der Verabreichung beobachtet, was mit dem schnellen
Einsetzen der Schmerzlinderung durch das Medikament übereinstimmt (Bannwarth and
Berenbaum, 2007). Im Einklang mit der kurzen Plasmahalbwertzeit von Lumiracoxib kehrten die
durchschnittlichen COX-2-Aktivitätswerte nach 24 Stunden auf das Niveau vor der
Tabletteneinnahme zurück. Trotz seiner schnellen Elimination scheinen klinische Studien nur
die einmalige tägliche Dosierung von Lumiracoxib zu empfehlen (Bannwarth and Berenbaum,
2007). Ein möglicher Grund für diese offenbare Diskrepanz kann die Tatsache sein, dass der
saure Arzneistoff in das entzündete Gewebe abgesondert wird und dadurch die Hemmung der
Prostaglandinsynthese an den entzündeten Stellen über das ganze Dosisintervall gewährleistet.
Dementsprechend hatten die Patienten mit rheumatischer Arthritis, die mit Lumiracoxib 400 mg
einmal täglich für 7 Tage behandelt wurden, ca. 3-fach höhere Steady-state-Konzentrationen
von Lumiracoxib in der Synovialflüssigkeit als im Plasma (Scott et al., 2004). Um diesen Befund
zu verallgemeinern, sind jedoch mehr Daten, insbesondere von Patienten, die einmal täglich mit
niedrigeren Dosen von Lumiracoxib behandelt wurden, notwendig.
Es wird angenommen, dass die Hemmung der COX-2 ex-vivo ein Marker der antientzündlichen
und schmerzlindernden Wirkung von COX-Inhibitoren darstellt. Blutproben passen in diesem
Zusammenhang gut, da die Messungen in einem physiologischen Milieu durchgeführt werden,
wodurch die variable Bindung des Medikamentes zu Plasmaproteinen berücksichtigt wird. Die
Tatsache, dass Lumiracoxib in den Dosen von 50 mg, 100 mg und 200 mg eine ähnliche COX-
2 hemmende Wirkung in diesem System hervorruft, wirft die Frage auf, ob die Dosierungen
kleiner als 100 mg für die Schmerztherapie genügend sind. In der Tat zeigte eine klinische
Studie, dass Patienten mit Osteoarthritis, die randomisiert Lumiracoxib (50, 100 oder 200 mg
zweimal täglich oder 400 mg einmal täglich) oder Diclofenac (75 mg zweimal täglich) erhielten,
eine vergleichbare Schmerzlinderung hatten (Schnitzer et al., 2004a).
Im Hinblick auf die offensichtlich dosisabhängige Lebertoxizität von Lumiracoxib (Schnitzer et
al., 2004b), könnten niedrigere Dosen eventuell Leberschäden vermeiden, wodurch die Vorteile

Diskussion 100
dieses Arzneistoffes wieder genutzt werden könnten. Laut einer jüngsten Behauptung des
Herstellers (Novartis, 2007) wurde Lumiracoxib in der Dosis von 100 mg mit nur neun starken
hepatischen Ereignissen weltweit in Verbindung gebracht, die alle nach einer Behandlung von
länger als einem Monat aufgetreten sind. Das entspricht einer Rate von 5,2 Ereignissen pro
100 000 Patientenjahren, die innerhalb der erwarteten Rate für NSAR liegt (Rubenstein and
Laine, 2004).
Außerdem deutet ein neuer Hinweis (Li et al., 2008) darauf hin, dass sich die Lebertoxizität
durch Lumiracoxib einen gemeinsamen Mechanismus mit dem strukturell ähnlichen Diclofenac
teilt, indem beide Substanzen eine CYP2C9-vermittelte Biotransformation zu chemisch
reaktiven Metaboliten (elektrophile Chinon Imine Arten) erfahren. Tatsächlich geht der
Gebrauch von Diclofenac mit einer ähnlichen Lebertoxizität einher, aber im Hinblick auf seine
therapeutischen Vorteile wird diese Gefahr als akzeptabel betrachtet.
Insgesamt untersucht die vorliegende Studie die Notwendigkeit, Lumiracoxib in den Dosen über
100 mg zu verabreichen, und ob die Dosen bis zu 100 mg für die COX-2-Hemmung und
Schmerzlinderung ausreichen können. Im Hinblick auf eine anhaltende praktisch 100%-ige
Hemmung der COX-2 bei den untersuchten Dosierungen (50 mg, 100 mg, 200 mg) erscheinen
die hohen Dosen von 200 und 400 mg sinnlos. In Anbetracht der eindrucksvollen
gastrointestinalen Verträglichkeit von Lumiracoxib (Schnitzer et al., 2004b) könnte
vorgeschlagen werden, die niedrigeren Dosierungen von Lumiracoxib für die Schmerztherapie
noch einmal zu überlegen. Es könnte durchaus sein, dass Lumiracoxib ein verbessertes
Analogon (z.B. weniger gastrointestinale Toxizität, kein Einfluss auf Blutkoagulation) des
weltweit bevorzugten nichtselektiven NSAR Diclofenac ist.

Diskussion 101
7.5 Untersuchung zur thrombozytenhemmende und potenziell
kardioprotektive Wirkung von Naproxen
Der Einfluss von Naproxen auf das kardiovaskuläre System konnte bisher nur schwer
interpretiert werden. Einerseits weisen neue Daten der vorzeitig abgebrochenen Alzheimer`s
Disease Antiinflammatory Prevention (ADAPT)-Studie mit Celecoxib, Naproxen und Placebo bei
Patienten mit Alzheimer auf ein erhöhtes kardiovaskuläres und cerebrovaskuläres Risiko von
Naproxen hin (ADAPT Reserach Group, 2006). Jedoch sind diese Daten keineswegs endgültig,
da diese Studie nicht geplant wurde um Unterschiede im kardiovaskulären und
cerebrovaskulären Risiko von COX-Inhibitoren nachzuweisen und da die Ergebnisse
retrospektiv zusammengefasst wurden. Außerdem stehen diese Ergebnisse im Gegensatz zu
einer Meta-Analyse von 138 veröffentlichten und unveröffentlichten, randomisierten
Untersuchungen (Kearney et al., 2006) die zeigten, dass das Auftreten von ernsten vaskulären
Ereignissen zwischen einem COX-2-Hemmer und jedem NSAR außer Naproxen ähnlich ist,
und dass das Risiko von Naproxen mit Placebo vergleichbar ist. Die Ereignisrate von
vaskulären Nebenwirkungen war in den untersuchten Studien im Vergleich zum Placebo 0,92
für Naproxen, 1,51 für Ibuprofen und 1,63 für Diclofenac. Auf der anderen Seite konnte
nachgewiesen werden, dass die andauernde und regelmäßige Einnahme von Naproxen
(500 mg zweimal täglich) die COX-1-Aktivität in Thrombozyten und die dadurch vermittelte
Plättchenaggregation während des gesamten Dosierungsintervals in einigen, aber nicht in allen
Probanden beeinflussen konnte (Capone et al., 2004). Auf Grund der publizierten Studien
besteht Konsens, dass die 95 %-ige Hemmung der TXB2-Synthese im Serum die
Thrombozytenfunktion klinisch relevant beeinflusst (Reilly and FitzGerald, 1987). Deshalb
könnte eine ausgeprägte und lang andauernde Hemmung der COX-1 durch Naproxen ein
möglicher Mechanismus für die potentiell kardioprotektive Wirkung sein.
Die vorliegende Studie untersuchte, ob auch OTC Dosierungen von freiverkäuflichem Naproxen
Natrium (220 mg zweimal täglich) eine ausreichende COX-1-Hemmung nach einer einzelnen
Dosisverabreichung und im Steady-State erzeugen können. Es konnte festgestellt werden, dass
die Hemmung der COX-1-vermittelten TXB2-Synthese nach einer einzelnen Naproxendosis
mehr als 95 % in zwei von vier Probanden zum Zeitpunkt der maximalen Plasmakonzentration
(tmax) war. Jedoch war im Vergleich zur Einmalgabe von 100 mg Aspirin®, die eine anhaltende
Hemmung des Thrombozyten-TXB2 für 24 h (Reilly and FitzGerald, 1987) verursacht, die
Wirkung von 220 mg Naproxen Natrium auf TXB2 nur kurzzeitig. Weiterhin wurde festgestellt,
dass bei einem von vier Probanden die COX-1 im Steady-State über 95 % innerhalb des
gesamten Dosierungsintervalls gehemmt wurde. Auf der Grundlage dieser Daten ist es
vorstellbar, dass die dauernde Behandlung mit Naproxen Natrium in Dosen von 220 mg für die
Kardioprotektion in allen Patienten nicht ausreichend ist.

Diskussion 102
Die Analyse der COX-2-Aktivität in Probanden, die mit Naproxen behandelt wurden, zeigte
maximale Hemmungen von 79 % (einzelne Dosis) und von 80 % (Steady-state). Im Hinblick auf
vorherige Daten, die eine direkte Korrelation in-vitro COX-2 IC80-Werte zu analgetischen
Plasmakonzentrationen der verschiedenen COX-2-Hemmer (Huntjens et al., 2005) zeigte,
könnte man diese Hemmung als ausreichend für die effiziente Schmerzlinderung in OTC
Dosierungen betrachten. Tatsächlich lag die gemessene COX-2-Hemmung durch niedrige
Dosierungen von Naproxen nicht unter der COX-2-Hemmung, die durch analgetische
Standarddosierungen von selektiven COX-2-Hemmer erreicht wird. Entsprechend rief Celecoxib
(200 mg zweimal täglich) und Rofecoxib (25 mg einmal täglich) eine 81%- oder 72%-ige COX-
2-Hemmung im Steady-state hervor (Hinz et al., 2006). Beachtenswerterweise wurde in dieser
Arbeit eine wesentliche Variabilität zwischen Probanden bezüglich der Plasmakonzentrationen
von Naproxen und dem entsprechenden Grad der COX-2-Hemmung beobachtet. Diese
Beobachtung stimmt mit vorherigen Daten überein, die mit den selektiven COX-2-Hemmern
(Hinz et al., 2006) erreicht wurden. Der Grund für dieses Verhalten ist unklar. Aber mehrere
Faktoren, einschließlich genetischer Variabilitäten im Zielprotein, in arzneimittel-
metabolisierenden Enzymen oder die Variabilität in der Pharmakokinetik zwischen den
Probanden scheinen mögliche Gründe zu sein (Patrono, 2001).
In einer Veröffentlichung wurde berichtet, dass die in-vitro IC50-Werte von Naproxen in
menschlichen Vollblut innerhalb der pharmakologischen Konzentrationsreihe des NSAR, d. h.
bei ca. 16 µM für COX-1 und bei 28 µM für COX-2 lagen (Patrignani et al., 1997). In
Übereinstimmung mit diesen Daten zeigt die vorliegende Analyse der Korrelation von
Naproxenplasmakonzentrationen mit der ex-vivo Hemmungen von COX-1 und COX-2 eine
bevorzugte Blockade der COX-1. Die berechneten ex-vivo IC50-Werte waren 35,5 µM für COX-1
bzw. 64,6 µM für den COX-2. Diese Daten erklären auch die langsame Wiederherstellung
(Recovery) von beiden Enzymen nach Naproxeneinnahme. Dementsprechend waren die
durchschnittliche Naproxenplasmakonzentrationen, die 36 Stunden nach der Beendigung der
Naproxeneinnahme gemessen wurden, noch über oder leicht unter den ex-vivo IC50-Werten für
die COX-1 bzw. COX-2. Deswegen wurden die COX-1 und COX-2-Aktivitäten nur zu 28% bzw.
39% im Bezug auf die jewelige Aktivität vor Naproxeneinahme an diesem Zeitpunkt
wiederhergestellt.
Zusammengefasst wird die verlängerte Behandlung von gesunden Probanden mit Naproxen
Natrium mit OTC Dosen mit einer größeren als 90%-igen Hemmung der COX-1 und einer
ausreichenden bis zu 80%-igen Hemmung der COX-2 begleitet. Naproxen-Natrium kann
funktionell relevante antiaggregatorische Wirkung auf Grund einer fast ganzen und anhaltenden
Hemmung des COX-1 in einer beschränkten Zahl von Patienten hervorrufen, aber das muss in
weiteren Analysen bestätigt werden.

Schlussfolgerungen 103
8 Schlussfolgerungen
Die Ergebnisse können wie folgt zusammengefasst werden:
• Metamizol führt zu einer wesentlichen dosisabhängigen Hemmung der beiden COX-
Enzymen in-vitro und ex-vivo im Blut von Probanden durch seinen Metabolit 4-MAA. Die
schnelle und deutliche Hemmung von COX-2 in menschlichen Blutmonozyten
unterstützt die Annahme, dass ein signifikanter Anteil der analgetischen Wirkung von
Metamizol wahrscheinlich durch periphere Mechanismen vermittelt wird. Dabei wurde
keine COX-1/COX-2-Selektivität festgestellt. Es wird vermutet, dass physikochemische
Faktoren (z.B. die nicht vorhandenen Säureeigenschaften) für die bessere
gastrointestinale Verträglichkeit im Vergleich zu den sauren NSAR verantwortlich sind.
Auf Grund der guten Gewebepenetration von Metamizol lässt sich vermuten, dass auch
im ZNS eine analgetisch ausreichende COX-2-Hemmung geschieht. Der nachhaltige
Effekt von 1000 mg Metamizol lässt höhere Einzeldosen sinnlos erscheinen.
• Nach einmaliger Einnahme von 1000 mg Paracetamol betrug die Hemmung von COX-2
in-vitro und ex-vivo mehr als 80%, in etwa vergleichbar mit NSAR und COX-2 Inhibitoren.
Andererseits, fehlte eine relevante COX-1-Blockade (mind. 95%), die für die
Unterdrückung der Thrombozytenfunktion notwendig ist. In-vitro und ex-vivo zeigte
Paracetamol eine ca. 4-fach höhere Selektivität für COX-2. Auf Basis der
nachgewiesenen COX-2-Hemmung sollte Paracetamol einer kritischen Analyse
hinsichtlich seines kardiovaskulären Risikopotentials unterzogen werden.
• Lumiracoxib ruft in der Dosierung von 50 mg eine vollständige und anhaltende COX-2-
Hemmung hervor. Es erscheint sinnvoll eine Anwendung von Lumiracoxib in den Dosen
bis zu 100 mg für Schmerztherapie zu überlegen.
• Die Gabe von 220 mg Naproxen Natrium zweimal täglich ruft eine starke Hemmung von
beiden Enzymen hervor. Obwohl eine antikoagulatorische und damit potenziell
kardioprotektive Hemmung der COX-1 bei einzelnen Probanden bereits mit OTC
(rezeptfreie) Dosierung beobachtet wurde, dürfte eine zuverlässige kardioprotektive
Wirkung erst bei Dosen um 1g / Tag erreicht werden.
Fazit: Auf Grund der vorgelegten Untersuchungen erscheint eine kritische Analyse der
„üblichen“ Dosierungen von Cyclooxygenasehemmern geboten.

Literaturverzeichnis 104
Literaturverzeichnis
Abramson SB (2002) Et tu, acetaminophen? Arthritis Rheum 46:2831-2835.
ADAPTReserachGroup (2006) Cardiovascular and cerebrovascular events in the randomized,
controlled Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin
Trials 1:e33.
Agundez JA, Martinez C, Martin R and Benitez J (1994) Determination of aminopyrine, dipyrone
and its metabolites in urine by high-performance liquid chromatography. Ther Drug Monit
16:316-322.
Anonymous (1986) Risks of agranulocytosis and aplastic anemia. A first report of their relation
to drug use with special reference to analgesics. The International Agranulocytosis and
Aplastic Anemia Study. Jama 256:1749-1757.
Aventis P (2003) Fachinformation Novalgin, Aventis Pharma, in.
Bannwarth B (2004) Gastrointestinal safety of paracetamol: is there any cause for concern?
Expert Opin Drug Saf 3:269-272.
Bannwarth B and Berenbaum F (2007) Lumiracoxib in the management of osteoarthritis and
acute pain. Expert Opin Pharmacother 8:1551-1564.
Beiche F, Scheuerer S, Brune K, Geisslinger G and Goppelt-Struebe M (1996) Up-regulation of
cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS
Lett 390:165-169.
Beirith A, Santos AR, Rodrigues AL, Creczynski-Pasa TB and Calixto JB (1998) Spinal and
supraspinal antinociceptive action of dipyrone in formalin, capsaicin and glutamate tests.
Study of the mechanism of action. Eur J Pharmacol 345:233-245.
Besz D (2008) Untersuchung der Zyklooxygenasehemmwirkungen von Naproxen. Dissertation.
Universitat Erlangen-Nürnberg.
Birmingham PK, Tobin MJ, Fisher DM, Henthorn TK, Hall SC and Cote CJ (2001) Initial and
subsequent dosing of rectal acetaminophen in children: a 24-hour pharmacokinetic
study of new dose recommendations. Anesthesiology 94:385-389.
Bjornsson GA, Haanaes HR and Skoglund LA (2003) A randomized, double-blind crossover trial
of paracetamol 1000 mg four times daily vs ibuprofen 600 mg: effect on swelling and
other postoperative events after third molar surgery. Br J Clin Pharmacol 55:405-412.
Boardman PL and Hart FD (1967) Clinical measurement of the anti-inflammatory effects of
salicylates in rheumatoid arthritis. Br Med J 4:264-268.
Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP and Versteeg HH (2004) Prostanoids and
prostanoid receptors in signal transduction. Int J Biochem Cell Biol 36:1187-1205.
Botting RM (2000) Mechanism of action of acetaminophen: is there a cyclooxygenase 3? Clin
Infect Dis 31 Suppl 5:S202-210.

Literaturverzeichnis 105
Boutaud O, Aronoff DM, Richardson JH, Marnett LJ and Oates JA (2002) Determinants of the
cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases.
Proc Natl Acad Sci U S A 99:7130-7135.
Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R,
Morton D, Lanas A, Konstam MA and Baron JA (2005) Cardiovascular events
associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med
352:1092-1102.
Brune K (1974) How aspirin might work: a pharmacokinetic approach. Agents Actions 4:230-
232.
Brune K (1988) The pharmacological profile of non-opioid (OTC) analgesics: aspirin,
paracetamol (acetaminophen), ibuprofen, and phenazones. Agents Actions Suppl 25:9-
19.
Brune K and Alpermann H (1983) Non-acidic pyrazoles: inhibition of prostaglandin production,
carrageenan oedema and yeast fever. Agents Actions 13:360-363.
Brune K, Graf P and Glatt M (1976) Inhibition of prostaglandin synthesis in vivo by nonsteroid
anti-inflammatory drugs: evidence for the importance of pharmacokinetics. Agents
Actions 6:159-164.
Brune K and Hinz B (2004) Selective cyclooxygenase-2 inhibitors: similarities and differences.
Scand J Rheumatol 33:1-6.
Brune K, Rainsford KD and Schweitzer A (1980) Biodistribution of mild analgesics. Br J Clin
Pharmacol 10 Suppl 2:279S-284S.
Brune K and Zeilhofer HU (1999) Antipyretic (non-narcotic) analgesics. In:Wall PD, Melzack R
(eds).Textbook of pain., in pp 1139-1154, Churchil Livingston, Edinburgh.
Burton B (2007) Australian drugs regulator cancels registration of COX 2 inhibitor. Bmj 335:363.
Campos C, de Gregorio R, Garcia-Nieto R, Gago F, Ortiz P and Alemany S (1999) Regulation
of cyclooxygenase activity by metamizol. Eur J Pharmacol 378:339-347.
Cannon CP, Curtis SP, FitzGerald GA, Krum H, Kaur A, Bolognese JA, Reicin AS, Bombardier
C, Weinblatt ME, van der Heijde D, Erdmann E and Laine L (2006) Cardiovascular
outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid
arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL)
programme: a randomised comparison. Lancet 368:1771-1781.
Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG and Patrignani P (2007)
Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid
Mediat 82:85-94.
Capone ML, Tacconelli S, Sciulli MG, Grana M, Ricciotti E, Minuz P, Di Gregorio P, Merciaro G,
Patrono C and Patrignani P (2004) Clinical pharmacology of platelet, monocyte, and

Literaturverzeichnis 106
vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy
subjects. Circulation 109:1468-1471.
Carlsson KH, Helmreich J and Jurna I (1986) Activation of inhibition from the periaqueductal
grey matter mediates central analgesic effect of metamizol (dipyrone). Pain 27:373-390.
Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan
H, Gertz BJ and FitzGerald GA (1999) Effects of specific inhibition of cyclooxygenase-2
on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther
289:735-741.
Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, Vyas SN and
FitzGerald GA (2001) Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N
Engl J Med 345:1809-1817.
Chan AT, Manson JE, Albert CM, Chae CU, Rexrode KM, Curhan GC, Rimm EB, Willett WC
and Fuchs CS (2006) Nonsteroidal antiinflammatory drugs, acetaminophen, and the risk
of cardiovascular events. Circulation 113:1578-1587.
Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS and Simmons DL
(2002) COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other
analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A
99:13926-13931.
Cheatum DE, Arvanitakis C, Gumpel M, Stead H and Geis GS (1999) An endoscopic study of
gastroduodenal lesions induced by nonsteroidal anti-inflammatory drugs. Clin Ther
21:992-1003.
Cheremina O, Brune K and Hinz B (2006) A validated high-performance liquid chromatographic
assay for determination of lumiracoxib in human plasma. Biomed Chromatogr 20:1033-
1037.
Cohen O, Zylber-Katz E, Caraco Y, Granit L and Levy M (1998) Cerebrospinal fluid and plasma
concentrations of dipyrone metabolites after a single oral dose of dipyrone. Eur J Clin
Pharmacol 54:549-553.
Cohn SM, Schloemann S, Tessner T, Seibert K and Stenson WF (1997) Crypt stem cell survival
in the mouse intestinal epithelium is regulated by prostaglandins synthesized through
cyclooxygenase-1. J Clin Invest 99:1367-1379.
Crofford LJ (1997) COX-1 and COX-2 tissue expression: implications and predictions. J
Rheumatol Suppl 49:15-19.
Dembinska-Kiec Z, Krupinska. (1976) Inhibition of prostaglandin synthetase by aspirin-like
drugs in different microsomal preparations. Adv Prostaglandin Thtomboxane Res 1:99-
103.
DeWitt DL, Meade EA and Smith WL (1993) PGH synthase isoenzyme selectivity: the potential
for safer nonsteroidal antiinflammatory drugs. Am J Med 95:40S-44S.

Literaturverzeichnis 107
Dinchuk JE, Liu RQ and Trzaskos JM (2003) COX-3: in the wrong frame in mind. Immunol Lett
86:121.
Eldor A, Zylber-Katz E and Levy M (1984) The effect of oral administration of dipyrone on the
capacity of blood platelets to synthesize thromboxane A2 in man. Eur J Clin Pharmacol
26:171-176.
Esser R, Berry C, Du Z, Dawson J, Fox A, Fujimoto RA, Haston W, Kimble EF, Koehler J,
Peppard J, Quadros E, Quintavalla J, Toscano K, Urban L, van Duzer J, Zhang X, Zhou
S and Marshall PJ (2005) Preclinical pharmacology of lumiracoxib: a novel selective
inhibitor of cyclooxygenase-2. Br J Pharmacol 144:538-550.
EuropeanMedicinesAgency L (13 Dezember 2007) Questions and answers on the
recommendation to withdraw the marketing authorisation for lumiracoxib-containing
medicines. www.emea.europa.eu.
Ferreira SH, Lorenzetti BB and Correa FM (1978) Blockade of central and peripheral generation
of prostaglandins explains the antialgic effect of aspirin like drugs. Pol J Pharmacol
Pharm 30:133-140.
FitzGerald GA and Patrono C (2001) The coxibs, selective inhibitors of cyclooxygenase-2. N
Engl J Med 345:433-442.
Flower R, Gryglewski R, Herbaczynska-Cedro K and Vane JR (1972) Effects of anti-
inflammatory drugs on prostaglandin biosynthesis. Nat New Biol 238:104-106.
Flower RJ and Vane JR (1972) Inhibition of prostaglandin synthetase in brain explains the anti-
pyretic activity of paracetamol (4-acetamidophenol). Nature 240:410-411.
Forman JP, Stampfer MJ and Curhan GC (2005) Non-narcotic analgesic dose and risk of
incident hypertension in US women. Hypertension 46:500-507.
Frolich JC, Rupp WA, Zapf RM and Badian MJ (1986) The effects of metamizol on
prostaglandin synthesis in man. Agents Actions Suppl 19:155-166.
Garavito RM (1996) The cyclooxygenase-2 structure: new drugs for an old target? Nat Struct
Biol 3:897-901.
Garavito RM, Malkowski MG and DeWitt DL (2002) The structures of prostaglandin
endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat 68-69:129-152.
Garcia Rodriguez LA and Hernandez-Diaz S (2001) Relative risk of upper gastrointestinal
complications among users of acetaminophen and nonsteroidal anti-inflammatory drugs.
Epidemiology 12:570-576.
Geisslinger G, Peskar BA, Pallapies D, Sittl R, Levy M and Brune K (1996) The effects on
platelet aggregation and prostanoid biosynthesis of two parenteral analgesics: ketorolac
tromethamine and dipyrone. Thromb Haemost 76:592-597.

Literaturverzeichnis 108
Gelgor L, Cartmell S and Mitchell D (1992) Intracerebroventricular micro-injections of non-
steroidal anti-inflammatory drugs abolish reperfusion hyperalgesia in the rat's tail. Pain
50:323-329.
Goldenberg SS and De Boni U (1983) Pure population of viable neurons from rabbit dorsal root
ganglia, using gradients of Percoll. J Neurobiol 14:195-206.
Graham DY and Smith JL (1988) Gastroduodenal complications of chronic NSAID therapy. Am
J Gastroenterol 83:1081-1084.
Graham GG and Scott KF (2005) Mechanism of action of paracetamol. Am J Ther 12:46-55.
Graham GG, Scott KF and Day RO (2005) Tolerability of paracetamol. Drug Saf 28:227-240.
Gupta K, Selinsky BS, Kaub CJ, Katz AK and Loll PJ (2004) The 2.0 A resolution crystal
structure of prostaglandin H2 synthase-1: structural insights into an unusual peroxidase.
J Mol Biol 335:503-518.
Hackenthal E (1997) Paracetamol and metamizol in the treatment of chronic pain syndromes.
Schmerz 11:269-275.
Halliwell B and Gutteridge JM (1990) The antioxidants of human extracellular fluids. Arch
Biochem Biophys 280:1-8.
Hamberg M, Svensson J and Samuelsson B (1975) Thromboxanes: a new group of biologically
active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A
72:2994-2998.
Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN and Breyer MD (1994)
Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with
salt restriction. J Clin Invest 94:2504-2510.
Hawkey CJ (1999) COX-2 inhibitors. Lancet 353:307-314.
Helliwell RJ, Adams LF and Mitchell MD (2004) Prostaglandin synthases: recent developments
and a novel hypothesis. Prostaglandins Leukot Essent Fatty Acids 70:101-113.
Hinz B and Brune K (1999) [Specific COX-2 inhibitors: prospects of therapy with new analgesic
and anti-inflammatory substances]. Wien Klin Wochenschr 111:103-112.
Hinz B and Brune K (2002) Cyclooxygenase-2--10 years later. J Pharmacol Exp Ther 300:367-
375.
Hinz B and Brune K (2007) Antipyretic analgesics: nonsteroidal antiinflammatory drugs,
selective COX-2 inhibitors, paracetamol and pyrazolinones. Handb Exp Pharmacol:65-
93.
Hinz B, Brune K and Pahl A (2000) Cyclooxygenase-2 expression in lipopolysaccharide-
stimulated human monocytes is modulated by cyclic AMP, prostaglandin E(2), and
nonsteroidal anti-inflammatory drugs. Biochem Biophys Res Commun 278:790-796.

Literaturverzeichnis 109
Hinz B, Dormann H and Brune K (2006) More pronounced inhibition of cyclooxygenase 2,
increase in blood pressure, and reduction of heart rate by treatment with diclofenac
compared with celecoxib and rofecoxib. Arthritis Rheum 54:282-291.
Hinz B, Rau T, Auge D, Werner U, Ramer R, Rietbrock S and Brune K (2003) Aceclofenac
spares cyclooxygenase 1 as a result of limited but sustained biotransformation to
diclofenac. Clin Pharmacol Ther 74:222-235.
Hinz B, Renner B and Brune K (2007) Drug insight: cyclo-oxygenase-2 inhibitors--a critical
appraisal. Nat Clin Pract Rheumatol 3:552-560; quiz 551 p following 589.
Hippisley-Cox J and Coupland C (2005) Risk of myocardial infarction in patients taking cyclo-
oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population
based nested case-control analysis. Bmj 330:1366.
Honore P, Buritova J and Besson JM (1995) Aspirin and acetaminophen reduced both Fos
expression in rat lumbar spinal cord and inflammatory signs produced by carrageenin
inflammation. Pain 63:365-375.
Huntjens DR, Danhof M and Della Pasqua OE (2005) Pharmacokinetic-pharmacodynamic
correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology
(Oxford) 44:846-859.
Ibanez L, Vidal X, Ballarin E and Laporte JR (2005) Agranulocytosis associated with dipyrone
(metamizol). Eur J Clin Pharmacol 60:821-829.
Jakobsson PJ, Thoren S, Morgenstern R and Samuelsson B (1999) Identification of human
prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme,
constituting a potential novel drug target. Proc Natl Acad Sci U S A 96:7220-7225.
Jenkins C, Costello J and Hodge L (2004) Systematic review of prevalence of aspirin induced
asthma and its implications for clinical practice. Bmj 328:434.
Kaufman DW KJ, Levy M, Shapiro S. (1991) The drug etiology of agranulocytosis an aplastic
anemia. Monographs in Epidemiology and Biostatistics. 18.
Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR and Patrono C (2006) Do selective
cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs
increase the risk of atherothrombosis? Meta-analysis of randomised trials. Bmj
332:1302-1308.
Kis B, Snipes JA and Busija DW (2005) Acetaminophen and the cyclooxygenase-3 puzzle:
sorting out facts, fictions, and uncertainties. J Pharmacol Exp Ther 315:1-7.
Kniss DA (1999) Cyclooxygenases in reproductive medicine and biology. J Soc Gynecol
Investig 6:285-292.
Korotkova M, Westman M, Gheorghe KR, af Klint E, Trollmo C, Ulfgren AK, Klareskog L and
Jakobsson PJ (2005) Effects of antirheumatic treatments on the prostaglandin E2
biosynthetic pathway. Arthritis Rheum 52:3439-3447.

Literaturverzeichnis 110
Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee
CA, Goulding EH, Kluckman KD, Kim HS and Smithies O (1995) Prostaglandin synthase
1 gene disruption in mice reduces arachidonic acid-induced inflammation and
indomethacin-induced gastric ulceration. Cell 83:483-492.
Langman MJ (1989) Treating ulcers in patients receiving anti-arthritic drugs. Q J Med 73:1089-
1091.
Lanz R, Polster P and Brune K (1986) Antipyretic analgesics inhibit prostaglandin release from
astrocytes and macrophages similarly. Eur J Pharmacol 130:105-109.
Laporte JR and Carne X (1987) Blood dyscrasias and the relative safety of non-narcotic
analgesics. Lancet 1:809.
Laporte JR, Carne X, Vidal X, Moreno V and Juan J (1991) Upper gastrointestinal bleeding in
relation to previous use of analgesics and non-steroidal anti-inflammatory drugs. Catalan
Countries Study on Upper Gastrointestinal Bleeding. Lancet 337:85-89.
Lau GS and Critchley JA (1994) The estimation of paracetamol and its major metabolites in
both plasma and urine by a single high-performance liquid chromatography assay. J
Pharm Biomed Anal 12:1563-1572.
Lee PP and Ip MM (1992) Regulation of proliferation of rat mammary tumor cells by inhibitors of
cyclooxygenase and lipoxygenase. Prostaglandins Leukot Essent Fatty Acids 45:21-31.
Levy M, Brune K, Zylber-Katz E, Cohen O, Caraco Y and Geisslinger G (1998) Cerebrospinal
fluid prostaglandins after systemic dipyrone intake. Clin Pharmacol Ther 64:117-122.
Levy M, Zylber-Katz E and Rosenkranz B (1995) Clinical pharmacokinetics of dipyrone and its
metabolites. Clin Pharmacokinet 28:216-234.
Li Y, Slatter JG, Zhang Z, Doss GA, Braun MP, Stearns RA, Dean DC, Baillie TA and Tang W
(2008) In vitro metabolic activation of lumiracoxib in rat and human liver preparations.
Drug Metab Dispos 36:469-473.
Lorenzetti BB and Ferreira SH (1985) Mode of analgesic action of dipyrone: direct antagonism
of inflammatory hyperalgesia. Eur J Pharmacol 114:375-381.
Lucas R, Warner TD, Vojnovic I and Mitchell JA (2005) Cellular mechanisms of acetaminophen:
role of cyclo-oxygenase. Faseb J 19:635-637.
Maj S and Centkowski P (2004) A prospective study of the incidence of agranulocytosis and
aplastic anemia associated with the oral use of metamizole sodium in Poland. Med Sci
Monit 10:PI93-95.
Mangold JB, Gu H, Rodriguez LC, Bonner J, Dickson J and Rordorf C (2004) Pharmacokinetics
and metabolism of lumiracoxib in healthy male subjects. Drug Metab Dispos 32:566-571.
Mantzke US and Brambrink AM (2002) [Paracetamol in childhood. Current state of knowledge
and indications for a rational approach to postoperative analgesia]. Anaesthesist 51:735-
746.

Literaturverzeichnis 111
Masuko S, Kuromi H and Shimada Y (1979) Isolation and culture of motoneurons from
embryonic chicken spinal cords. Proc Natl Acad Sci U S A 76:3537-3541.
McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA and FitzGerald GA (1999)
Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human
pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A 96:272-277.
McCarthy DM (1989) Nonsteroidal antiinflammatory drug-induced ulcers: management by
traditional therapies. Gastroenterology 96:662-674.
McCormack K and Brune K (1987) Classical absorption theory and the development of gastric
mucosal damage associated with the non-steroidal anti-inflammatory drugs. Arch Toxicol
60:261-269.
Mielke CH, Jr. (1981) Comparative effects of aspirin and acetaminophen on hemostasis. Arch
Intern Med 141:305-310.
Miners JO, Attwood J and Birkett DJ (1983) Influence of sex and oral contraceptive steroids on
paracetamol metabolism. Br J Clin Pharmacol 16:503-509.
Mitchell JA, Belvisi MG, Akarasereenont P, Robbins RA, Kwon OJ, Croxtall J, Barnes PJ and
Vane JR (1994) Induction of cyclo-oxygenase-2 by cytokines in human pulmonary
epithelial cells: regulation by dexamethasone. Br J Pharmacol 113:1008-1014.
Munsterhjelm E, Munsterhjelm NM, Niemi TT, Ylikorkala O, Neuvonen PJ and Rosenberg PH
(2005) Dose-dependent inhibition of platelet function by acetaminophen in healthy
volunteers. Anesthesiology 103:712-717.
Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, Ohmiya Y, Watanabe K
and Kudo I (2003) Cellular prostaglandin E2 production by membrane-bound
prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem 278:37937-
37947.
Mutschler E. GG, Kroemer H.K. and Schäfer-Korting M (2001) Mutschler Arzneimittelwirkungen.
Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Ichikawa A and Negishi M (1999)
Immunocytochemical localization of prostaglandin EP3 receptor in the rat hypothalamus.
Neurosci Lett 260:117-120.
Narumiya S, Sugimoto Y and Ushikubi F (1999) Prostanoid receptors: structures, properties,
and functions. Physiol Rev 79:1193-1226.
Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M and Samuelsson B (1976)
Identification of an enzyme in platelet microsomes which generates thromboxane A2
from prostaglandin endoperoxides. Nature 261:558-560.
Neugebauer V, Schaible HG, He X, Lucke T, Gundling P and Schmidt RF (1994)
Electrophysiological evidence for a spinal antinociceptive action of dipyrone. Agents
Actions 41:62-70.

Literaturverzeichnis 112
Niemi TT, Backman JT, Syrjala MT, Viinikka LU and Rosenberg PH (2000) Platelet dysfunction
after intravenous ketorolac or propacetamol. Acta Anaesthesiol Scand 44:69-74.
Noda A, Tsubone N, Mihara M, Goromaru T and Iguchi S (1976) Formation of 4-
formylaminoantipyrine as a new metabolite of aminopyrine. II. Enzymatic demethylation
and oxidation of aminopyrine and 4-monomethylaminoantipyrine. Chem Pharm Bull
(Tokyo) 24:3229-3231.
Novartis (2007) United Kingdom and Germany suspend marketing and sale of Prexige pending
outcome of European regulatory review, in MediaReleases.
Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, Boyce SW and
Verburg KM (2005) Complications of the COX-2 inhibitors parecoxib and valdecoxib
after cardiac surgery. N Engl J Med 352:1081-1091.
Ouellet M and Percival MD (2001) Mechanism of acetaminophen inhibition of cyclooxygenase
isoforms. Arch Biochem Biophys 387:273-280.
Patrignani P, Filabozzi P and Patrono C (1982) Selective cumulative inhibition of platelet
thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest 69:1366-
1372.
Patrignani P, Panara MR, Greco A, Fusco O, Natoli C, Iacobelli S, Cipollone F, Ganci A,
Creminon C, Maclouf J and et al. (1994) Biochemical and pharmacological
characterization of the cyclooxygenase activity of human blood prostaglandin
endoperoxide synthases. J Pharmacol Exp Ther 271:1705-1712.
Patrignani P, Panara MR, Sciulli MG, Santini G, Renda G and Patrono C (1997) Differential
inhibition of human prostaglandin endoperoxide synthase-1 and -2 by nonsteroidal anti-
inflammatory drugs. J Physiol Pharmacol 48:623-631.
Patrono C (2001) Measurement of cyclooxygenase isozyme inhibition in humans: exploring the
clinical relevance of biochemical selectivity. Clin Exp Rheumatol 19:S45-50.
Peskar BM, Kleine A, Pyras F and Muller MK (1986) Gastrointestinal toxicity. Role of
prostaglandins and leukotrienes. Med Toxicol 1 Suppl 1:39-43.
Prescott LF, Roscoe P, Wright N and Brown SS (1971) Plasma-paracetamol half-life and
hepatic necrosis in patients with paracetamol overdosage. Lancet 1:519-522.
Qin N, Zhang SP, Reitz TL, Mei JM and Flores CM (2005) Cloning, expression, and functional
characterization of human cyclooxygenase-1 splicing variants: evidence for intron 1
retention. J Pharmacol Exp Ther 315:1298-1305.
Rahme E, Pettitt D and LeLorier J (2002) Determinants and sequelae associated with utilization
of acetaminophen versus traditional nonsteroidal antiinflammatory drugs in an elderly
population. Arthritis Rheum 46:3046-3054.

Literaturverzeichnis 113
Rainsford KD, Schweitzer A and Brune K (1981) Autoradiographic and biochemical
observations on the distribution of non-steroid anti-inflammatory drugs. Arch Int
Pharmacodyn Ther 250:180-194.
Reilly IA and FitzGerald GA (1987) Inhibition of thromboxane formation in vivo and ex vivo:
implications for therapy with platelet inhibitory drugs. Blood 69:180-186.
Reinhardt N, Jantos R, Sinning C and Imming P (2006) Metamizol: Renaissance eines
Analgetikums. Pharmazeutische Zeitung online 32.
Ring EF, Collins AJ, Bacon PA and Cosh JA (1974) Quantitation of thermography in arthritis
using multi-isothermal analysis. II. Effect of nonsteroidal anti-inflammatory therapy on
the thermographic index. Ann Rheum Dis 33:353-356.
Rubenstein JH and Laine L (2004) Systematic review: the hepatotoxicity of non-steroidal anti-
inflammatory drugs. Aliment Pharmacol Ther 20:373-380.
Rumack BH (1978) Aspirin versus acetaminophen: a comparative view. Pediatrics 62:943-946.
Schaible HG and Grubb BD (1993) Afferent and spinal mechanisms of joint pain. Pain 55:5-54.
Schnitzer TJ (2002) Update of ACR guidelines for osteoarthritis: role of the coxibs. J Pain
Symptom Manage 23:S24-30; discussion S31-24.
Schnitzer TJ, Beier J, Geusens P, Hasler P, Patel SK, Senftleber I, Gitton X, Moore A, Sloan VS
and Poor G (2004a) Efficacy and safety of four doses of lumiracoxib versus diclofenac in
patients with knee or hip primary osteoarthritis: a phase II, four-week, multicenter,
randomized, double-blind, placebo-controlled trial. Arthritis Rheum 51:549-557.
Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M, Ehrsam E, Gitton X,
Krammer G, Mellein B, Matchaba P, Gimona A and Hawkey CJ (2004b) Comparison of
lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and
Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised
controlled trial. Lancet 364:665-674.
Schwab JM, Beiter T, Linder JU, Laufer S, Schulz JE, Meyermann R and Schluesener HJ
(2003a) COX-3--a virtual pain target in humans? Faseb J 17:2174-2175.
Schwab JM, Schluesener HJ and Laufer S (2003b) COX-3: just another COX or the solitary
elusive target of paracetamol? Lancet 361:981-982.
Scott G, Rordorf C, Reynolds C, Kalbag J, Looby M, Milosavljev S, Weaver M, Huff JP and Ruff
DA (2004) Pharmacokinetics of lumiracoxib in plasma and synovial fluid. Clin
Pharmacokinet 43:467-478.
Shah VP, Midha KK, Findlay JW, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik
RN, Powell ML, Tonelli A, Viswanathan CT and Yacobi A (2000) Bioanalytical method
validation--a revisit with a decade of progress. Pharm Res 17:1551-1557.
Shimada SG, Otterness IG and Stitt JT (1994) A study of the mechanism of action of the mild
analgesic dipyrone. Agents Actions 41:188-192.

Literaturverzeichnis 114
Simmons DL, Botting RM and Hla T (2004) Cyclooxygenase isozymes: the biology of
prostaglandin synthesis and inhibition. Pharmacol Rev 56:387-437.
Skjelbred P and Lokken P (1979) Paracetamol versus placebo: effects on post-operative
course. Eur J Clin Pharmacol 15:27-33.
Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL,
Seibert K and Isakson PC (1998) Pharmacological analysis of cyclooxygenase-1 in
inflammation. Proc Natl Acad Sci U S A 95:13313-13318.
Smith WL, DeWitt DL and Garavito RM (2000) Cyclooxygenases: structural, cellular, and
molecular biology. Annu Rev Biochem 69:145-182.
Smith WL, Garavito RM and DeWitt DL (1996) Prostaglandin endoperoxide H synthases
(cyclooxygenases)-1 and -2. J Biol Chem 271:33157-33160.
Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A,
Hawk E and Bertagnolli M (2005) Cardiovascular risk associated with celecoxib in a
clinical trial for colorectal adenoma prevention. N Engl J Med 352:1071-1080.
Somasundaram S, Rafi S, Hayllar J, Sigthorsson G, Jacob M, Price AB, Macpherson A,
Mahmod T, Scott D, Wrigglesworth JM and Bjarnason I (1997) Mitochondrial damage: a
possible mechanism of the "topical" phase of NSAID induced injury to the rat intestine.
Gut 41:344-353.
Stichtenoth DO, Thoren S, Bian H, Peters-Golden M, Jakobsson PJ and Crofford LJ (2001)
Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and
glucocorticoids in primary rheumatoid synovial cells. J Immunol 167:469-474.
Swierkosz TA, Jordan L, McBride M, McGough K, Devlin J and Botting RM (2002) Actions of
paracetamol on cyclooxygenases in tissue and cell homogenates of mouse and rabbit.
Med Sci Monit 8:BR496-503.
Tanioka T, Nakatani Y, Semmyo N, Murakami M and Kudo I (2000) Molecular identification of
cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1
in immediate prostaglandin E2 biosynthesis. J Biol Chem 275:32775-32782.
Tatsuo MA, Carvalho WM, Silva CV, Miranda AE, Ferreira SH and Francischi JN (1994)
Analgesic and antiinflammatory effects of dipyrone in rat adjuvant arthritis model.
Inflammation 18:399-405.
Tegeder I and Geisslinger G (2006) Cardiovascular risk with cyclooxygenase inhibitors: general
problem with substance specific differences? Naunyn Schmiedebergs Arch Pharmacol
373:1-17.
TherapeuticGoodsAdministration A (13. August 2007) Urgent advice regarding management of
patients taking lumiracoxib (Prexige). Safety alert. www.tga.gov.au/alerts/prexige.htm.
Tomek S (1955) New consideration on mechanism of action of pyrasolone derivatives.
Arzneimittelforschung. 5:53-60.

Literaturverzeichnis 115
Tortorici V and Vanegas H (1995) Anti-nociception induced by systemic or PAG-microinjected
lysine-acetylsalicylate in rats. Effects on tail-flick related activity of medullary off- and on-
cells. Eur J Neurosci 7:1857-1865.
Triggs EJ and Nation RL (1975) Pharmacokinetics in the aged: a review. J Pharmacokinet
Biopharm 3:387-418.
Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like
drugs. Nat New Biol 231:232-235.
Vane JR, Bakhle YS and Botting RM (1998) Cyclooxygenases 1 and 2. Annu Rev Pharmacol
Toxicol 38:97-120.
Vlahov V, Badian M, Verho M and Bacracheva N (1990) Pharmacokinetics of metamizol
metabolites in healthy subjects after a single oral dose of metamizol sodium. Eur J Clin
Pharmacol 38:61-65.
Volz M and Kellner HM (1980) Kinetics and metabolism of pyrazolones (propyphenazone,
aminopyrine and dipyrone). Br J Clin Pharmacol 10 Suppl 2:299S-308S.
Wallace JL (1997) Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second
hundred years. Gastroenterology 112:1000-1016.
Wang LH and Kulmacz RJ (2002) Thromboxane synthase: structure and function of protein and
gene. Prostaglandins Other Lipid Mediat 68-69:409-422.
Warner TD, Vojnovic I, Giuliano F, Jimenez R, Bishop-Bailey D and Mitchell JA (2004)
Cyclooxygenases 1, 2, and 3 and the production of prostaglandin I2: investigating the
activities of acetaminophen and cyclooxygenase-2-selective inhibitors in rat tissues. J
Pharmacol Exp Ther 310:642-647.
Weithmann KU and Alpermann HG (1985) Biochemical and pharmacological effects of dipyrone
and its metabolites in model systems related to arachidonic acid cascade.
Arzneimittelforschung 35:947-952.
Yaksh TL, Dirig DM, Conway CM, Svensson C, Luo ZD and Isakson PC (2001) The acute
antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal
prostaglandin E2 is mediated by the inhibition of constitutive spinal cyclooxygenase-2
(COX-2) but not COX-1. J Neurosci 21:5847-5853.
Zimmermann KC, Sarbia M, Schror K and Weber AA (1998) Constitutive cyclooxygenase-2
expression in healthy human and rabbit gastric mucosa. Mol Pharmacol 54:536-540.
Zylber-Katz E, Granit L and Levy M (1992) Formation and excretion of dipyrone metabolites in
man. Eur J Clin Pharmacol 42:187-191.

Lebenslauf 116
9 Lebenslauf
Name Olga Cheremina
Geburtsdatum 28. Februar 1970
Geburtsort Kysyl / Russland
Staatsangehörigkeit Russisch
e-mail [email protected]
Familienstand verheiratet, 1 Kind (10 Jahre alt)
Beruflicher Werdegang
09/1977 – 07/1987 Allgemeinbildende Schule in Kysyl / Russland
Abschluss: Hochschulzugangsberechtigung
09/1987 – 06/1992 Polytechnische Universität, Tomsk
Studium: Chemische Technologie der biologisch
aktiven Verbindungen
Abschluss: Diplom-Ingenieur-Chemietechnologe
09/1992 – 02/1996 Chemiebetrieb „Synthesekautschuk“, Toljatti
Schichtchemikerin in analytischem Labor
03/1996 – 07/1998 Medizinische Fachschule, Omsk
Umzug und Ausbildung zur Krankenschwester
Abschluss: Diplom-Krankenschwester
08/1998 – 12/1999 Familienzeit
01/2000 – 06/2000 Regionale Pharmaziegesellschaft „Pharmamarket“ GmbH Moskau
Außendienstmitarbeiterin in Omsk
26.07.2000 Einreise in die Bundesrepublik Deutschland
08/2000 – 05/2001 Familienzeit
06/2001 – 06/2003 Sprach- und Integrationskurse in Bundesrepublik Deutschland

Lebenslauf 117
07/2003 – 12/2003 Friedrich-Alexander-Universität Erlangen-Nürnberg
Institut für Experimentelle und Klinische Pharmakologie und
Toxikologie, Arbeitsgruppe Arzneistoffanalytik
Praktikum als Stipendiatin der Otto Benecke Stiftung
01/2004 – 12/2005 Friedrich-Alexander-Universität Erlangen-Nürnberg
Institut für Experimentelle und Klinische Pharmakologie und
Toxikologie, Arbeitsgruppe Arzneistoffanalytik
Wissenschaftliche Hilfskraft
01/2006 – 06/2009 Friedrich-Alexander-Universität Erlangen-Nürnberg
Institut für Experimentelle und Klinische Pharmakologie und
Toxikologie, Arbeitsgruppe Arzneistoffanalytik
Wissenschaftliche Angestellte und Promotionsstudium unter
Betreuung von Prof. Dr. med. Dr. h.c. K. Brune und Prof. Dr. rer.
nat. B. Hinz

Veröffentlichungen 118
10 Veröffentlichungen
Aus dieser Dissertation sind die folgenden Publikationen hervorgegangen: Originalarbeiten
Dezember 2005 Cheremina O., Bachmakov J., Neubert A., Brune K., Fromm M.F., Hinz
B., (2005). Simultaneous determination of oxycodone und its major
metabolite, noroxycodone in human plasma by high-performance
liquid chromatography. Biomed Chromatogr., 19:777-82
Oktober 2006 Cheremina O., Brune K., Hinz B., (2006). A validated high-performance
liquid chromatographic assay for determination of lumiracoxib in
human plasma. Biomed Chromatogr., 20:1033-7
August 2007 Hinz B., Cheremina O., Bachmakov J., Renner B., Zolk O., Fromm M. F.,
Brune K. (2007). Dipyrone elicits substantial inhibition of peripheral
cyclooxygenases: new insights into the pharmacology of an old
analgesic. FASEB J., 21:2343-51
Februar 2008 Hinz B., Cheremina O., Brune K. (2008). Acetaminophen (Paracetamol)
is a selective cyclooxygenase-2 inhibitor in man. FASEB J., 22: 383-
90.
Sertürner-Preis 2007
April 2008 Hinz B, Cheremina O, Besz D, Slotnick S, Brune K. (2008). Impact of
naproxen sodium at over-the-counter doses on cyclooxygenase
isoforms in human volunteers. Int J Clin Pharmacol., 46: 180-6
Februar 2009 Hinz B, Renner B, Cheremina O, Besz D, Zolk O, Brune K. (2009).
Lumiracoxib inhibits cyclo-oxygenase 2 completely at the 50 mg
dose: is liver toxicity avoidable by adequate dosing? Ann Rheum
Dis., 68: 289-91

Veröffentlichungen 119
Posterpräsentationen
März 2007 Cheremina O., Bachmakov J., Brune K., Hinz B. Metamizol elicits
substantial inhibition of peripheral cyclooxygenases in human
volunteers.
(48. Frühjahrstagung der Deutsche Gesellschaft für Experimentelle und
Klinische Pharmakologie und Toxikologie in Mainz).
Oktober 2007 Cheremina O., Brune K., Hinz B. Acetaminophen (Paracetamol) is a
selective Cyclooxygenase-2 inhibitor in man.
(8. Jahreskongress der Deutsche pharmazeutischen Gesellschaft in
Erlangen).
Oktober 2007 Cheremina O., Bachmakov J., Brune K., Hinz B. Metamizol elicits
substantial inhibition of peripheral cyclooxygenases in human
volunteers.
(8. Jahreskongress der Deutsche pharmazeutischen Gesellschaft in
Erlangen).
März 2008 Cheremina O., Besz D, Renner B., Brune K., Hinz B. The 50-mg dose of
lumiracoxib is sufficient to elicit complete cyclooxygenase-2
inhibition in man.
(49. Frühjahrstagung der Deutsche Gesellschaft für Experimentelle und
Klinische Pharmakologie und Toxikologie in Mainz).
März 2008 Cheremina O., Brune K., Hinz B. Acetaminophen (Paracetamol) is a
selective Cyclooxygenase-2 inhibitor in man.
(49. Frühjahrstagung der Deutsche Gesellschaft für Experimentelle und
Klinische Pharmakologie und Toxikologie in Mainz).

Danksagung 120
Danksagung
Sehr herzlich bedanken möchte ich mich bei Herrn Professor Dr. Dr. Kay Brune, dass er mir die
Durchführung dieser Arbeit am Institut für Experimentelle und Klinische Pharmakologie und
Toxikologie der Friedrich-Alexander-Universität Erlangen-Nürnberg ermöglicht hat. Mit großer
Bewunderung habe ich Herrn Professor Brune gesehen, wie er sich selbstlos für die
Wissenschaft als Proband einsetzt. Ich schätze mich sehr glücklich, dass ich mit ihm diese
Versuche durchführen konnte. Ich möchte an dieser Stelle nochmals meinen besonderen Dank
zum Ausdruck bringen.
Mein großer Dank geht auch an Herrn Professor Dr. Burkhard Hinz für die Übertragung der
interessanten Themen, für seine Unterstützung und sein stetes Interesse am Fortgang der
Arbeit. Seine wissenschaftliche und methodische Fragestellungen, sowie die konstruktive Kritik
trugen entscheidend zur Realisierung meines Promotionsvorhabens bei.
Herrn Professor Dr. Reinhard Troschütz möchte ich danken, dass er meine Arbeit seitens der
Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität betreute, sowie Herrn
Professor Geoffrey Lee, dass er sich als Vorsitzender bei Promotionsprüfung zur Verfügung
gestellt hat.
Ich bedanke mich sehr bei Ulrike Weinzierl für die Durchführung der ELISA Messungen und die
Westernblot-Analyse.
Auch gilt mein großer Dank den Ärzten unseres Institutes, die sich als Probanden für
Selbstversuche zur Verfügung gestellt haben. Besonderes möchte ich mich bei dem Dr. Bertold
Renner bedanken.
Mein besonderer Dank geht an Dr. Hartmut Gläser, weil er immer für Fragen und Diskussionen
zur Verfügung stand und da ich immer auf seine geduldige Unterstützung zählen konnte.
Auch gilt mein großer Dank Martin Gillich und Daniel Auge für die technische Unterstützung in
Fragen der Arzneistoffanalytik.
Bei dieser Gelegenheit möchte ich nicht versäumen, meinen Dank an allen Kollegen des
Lehrstuhls für Klinische Pharmakologie und Klinische Toxikologie für die sehr angenehme
Arbeitsatmosphäre und für das nette Beisammsein im Sozialraum auszurichten.











![5.1 Thorax.ppt [Kompatibilitätsmodus] - cox-radiology.orgcox-radiology.org/content/e1653/e1541/e1863/e1864/e1866/5.1_Thorax.pdf · 6. linke A. pulmonalis 7. rechte A. pulmonalis](https://static.fdokument.com/doc/165x107/5cf1031088c993d72c8d1629/51-kompatibilitaetsmodus-cox-radiologyorgcox-radiologyorgcontente1653e1541e1863e1864e186651thoraxpdf.jpg)






