
PHOSPHATIDYLSERINE TRANSLOCATION IN MAMMALIAN CELLS
76
PHOSPHATIDYLSERINE TRANSLOCATION IN MAMMALIAN CELLS Liisa Heikinheimo Department of Biosciences, Division of Biochemistry Faculty of Science University of Helsinki, Finland Institute of Biomedicine / Biochemistry Faculty of Medicine University of Helsinki, Finland Academic Dissertation To be presented for public criticism, with the permission of the Faculty of Science of the University of Helsinki, in the lecture hall 2 of Biomedicum (Haartmaninkatu 8, Meilahti, Helsinki), on June 28 th , 2002, at 12 o’clock. Helsinki 2002
Transcript of PHOSPHATIDYLSERINE TRANSLOCATION IN MAMMALIAN CELLS

PHOSPHATIDYLSERINE TRANSLOCATION
IN MAMMALIAN CELLS
Liisa Heikinheimo
Department of Biosciences, Division of Biochemistry
Faculty of Science
University of Helsinki, Finland
Institute of Biomedicine / Biochemistry
Faculty of Medicine
University of Helsinki, Finland
Academic Dissertation
To be presented for public criticism,
with the permission of the Faculty of Science of the University of Helsinki,
in the lecture hall 2 of Biomedicum (Haartmaninkatu 8, Meilahti, Helsinki),
on June 28th, 2002, at 12 o’clock.
Helsinki 2002

2
Supervisor: Docent Pentti Somerharju
Institute of Biomedicine / Biochemistry
University of Helsinki, Finland
Reviewers: Docent Matti Jauhiainen
Department of Molecular Medicine
National Public Health Institute, Finland
Docent Vesa Olkkonen
Department of Molecular Medicine
National Public Health Institute, Finland
Opponent: Professor Günther Daum
Institut für Biochemie
Technische Universität Graz, Austria
ISBN 952-91-4665-5 (nid.)
ISBN 952-10-0545-9 (pdf)
http://ethesis.helsinki.fi
Cosmoprint Oy
Espoo 2002

3
Omistettu suomalaiselle sisulle,
jonka avulla mennään läpi harmaiden
– ja mustienkin – kivien.

4
ORIGINAL PUBLICATIONS
I Heikinheimo Liisa and Somerharju Pentti: “Preferential decarboxylation of
hydrophilic phosphatidylserine species in cultured cells. Implications on the
mechanism of transport to mitochondria and cellular aminophospholipid species
compositions.” Journal of Biological Chemistry 273: 3 327 – 3 335 (1998).
II Heikinheimo Liisa and Somerharju Pentti: “Translocation of phosphatidylthreonine
and phosphatidylserine to mitochondria diminishes exponentially with increasing
molecular hydrophobicity.” Traffic 3: 367 – 377 (2002).
III Heikinheimo Liisa and Somerharju Pentti: “Translocation of pyrene-labeled
phosphatidylserine from the plasma membrane to mitochondria diminishes
systematically with molecular hydrophobicity. Implications on the maintenance of
high phosphatidylserine content in the inner leaflet of the plasma membrane.”
Biochimica et Biophysica Acta, in press.
Publications I-III are reprinted with kind permissions from The American Society for
Biochemistry and Molecular Biology, Munksgaard International Publishers and Elsevier
Science, respectively.

5
TABLE OF CONTENTS
ORIGINAL PUBLICATIONS............................................................................ 4
TABLE OF CONTENTS.................................................................................... 5
ACKNOWLEDGEMENTS ................................................................................ 8
ABBREVIATIONS............................................................................................. 10
INTRODUCTION............................................................................................... 11
REVIEW OF THE LITERATURE..................................................................... 14
1. Phosphatidylserine metabolism.................................................................................... 14
1.1. Phosphatidylserine synthesis............................................................................. 14
1.2. Phosphatidylserine synthesis in yeast ............................................................... 17
1.3. Transbilayer movement of phosphatidylserine in endoplasmic reticulum........ 18
1.4. Phosphatidylserine remodelling........................................................................ 19
1.5. Phosphatidylserine decarboxylation ................................................................. 20
1.6. Phosphatidylserine decarboxylation in yeast.................................................... 22
2. Phosphatidylthreonine.................................................................................................. 23
3. Intracellular lipid translocation mechanisms ............................................................... 23
3.1. Lipid carrier proteins ........................................................................................ 24
3.2. Yeast phosphatidylinositol transfer protein ...................................................... 25
3.3. Non-specific lipid transfer protein .................................................................... 25
3.4. Vesicular transfer .............................................................................................. 26
3.5. Spontaneous diffusion........................................................................................ 27
3.6. Lateral diffusion ................................................................................................ 29
4. Methods for studying intracellular lipid translocation mechanisms ............................ 29
4.1. Radioactive phospholipids ................................................................................ 29
4.2. Fluorescent phospholipids ................................................................................ 30
4.3. Spin-labelled phospholipids .............................................................................. 32
4.4. Stable isotope labelling ..................................................................................... 32
5. Phosphatidylserine translocation from ER to mitochondria ........................................ 32
6. Phosphatidylserine translocation from ER to mitochondria in yeast ........................... 35

6
7. Phosphatidylserine translocation from outer mitochondrial membrane to
inner mitochondrial membrane.............................................................................36
8. Phosphatidylethanolamine translocation from mitochondria to ER
and other destinations ...........................................................................................39
9. Phosphatidylserine translocation from plasma membrane to mitochondria.................40
9.1. Plasma membrane aminophospholipid translocase ..........................................40
AIMS OF THE PRESENT STUDY ................................................................... 42
EXPERIMENTAL PROCEDURES ................................................................... 43
Cell culture .......................................................................................................................43
Cell labelling procedures ..................................................................................................43
Labelling with � 3H�-serine (I) ..................................................................................43
Labelling with deuterium-serine or 13C-threonine (II) .............................................44
Introduction of dipyrene phosphatidylserine species to the plasma
membrane (III) ..................................................................................................44
Control experiments in Triton X-100 micelles.................................................................44
Decarboxylation of � 3H�phosphatidylserine species in Triton X-100-micelles (I) ..44
Decarboxylation of phosphatidylserine in Triton X-100 micelles (II)......................45
Determination of the substrate specificity of phosphatidylserine
decarboxylase (III)............................................................................................45
Lipid extraction and molecular species composition analysis (I) ....................................45
Lipid extraction and analysis (II and III)..........................................................................46
Mass spectrometry and data analysis (II) .........................................................................46
Cell fractionation and enzyme assays (II) ........................................................................47
Measurement of spontaneous intervesicle translocation rates in vitro (III) .....................48
RESULTS............................................................................................................ 49
Translocation of phosphatidylserine molecular species from ER to mitochondria..........49
Phosphatidylethanolamine is more “hydrophilic” than its precursor
phosphatidylserine (I) .......................................................................................49
Preferential translocation of hydrophilic phosphatidylserine species
to mitochondria (I)............................................................................................50

7
The kinetics of phosphatidylserine translocation to mitochondria has
two components (I) ........................................................................................... 50
Mass spectroscopic studies of phosphatidylserine molecular species
translocation to mitochondria (II).................................................................... 51
Translocation of phosphatidylthreonine molecular species from ER to mitochondria.... 53
Identification of phosphatidylisopropanolamine, a decarboxylation product of
phosphatidylthreonine (II)................................................................................ 53
Preferential translocation of hydrophilic phosphatidylthreonine species
to mitochondria (II) .......................................................................................... 54
Phosphatidylthreonine is a poor substrate for phosphatidylserine
decarboxylase (II)............................................................................................. 55
Translocation of phosphatidylserine species from plasma membrane to mitochondria .. 55
Preferential translocation of short-chain dipyrene phosphatidylserine species
from plasma membrane to mitochondria (III).................................................. 55
The spontaneous translocation rate of diPyr12PS is similar to that of
18:0/18:1-PS (III)............................................................................................. 57
ESI-MS and stable isotope -labelled precursors in lipid trafficking studies (II) ............. 58
DISCUSSION ..................................................................................................... 59
Effect of lipid hydrophobicity on its rate of translocation depends on the
mechanism of translocation ..................................................................................... 59
Mechanism of phosphatidylserine translocation from ER to mitochondria .................... 60
Mechanism of phosphatidylthreonine translocation from ER to mitochondria............... 61
Mechanism of phosphatidylserine translocation from plasma membrane to
mitochondria ............................................................................................................ 61
Maintenance of high phosphatidylserine concentration in the inner leaflet of
plasma membrane .................................................................................................... 62
CONCLUSIONS................................................................................................. 65
REFERENCES.................................................................................................... 66

8
ACKNOWLEDGEMENTS
Tämä väitöskirjatyö ei ole yksilösuoritus, vaan siihen on tavalla tai toisella tarvittu teitä
kaikkia. Ilman teitä en olisi jaksanut maaliin. Kiitos kaikille!
Pena on ohjannut työtäni kärsivällisesti kaikki nämä vuodet. Olen aina voinut mennä
kysymään samaa asiaa uudemmankin kerran ja saanut ystävällisen vastauksen.
Biomedicumiin muuton jälkeen en ole käytännössä lainkaan koskenut pipettiin, vaan
olen viimeiset 1,5 vuotta vain kirjoittanut. Se on vaatinut enemmän kärsivällisyyttä
kuin omaan, mutta Pena on jaksanut tukea ja auttaa kaikin mahdollisin tavoin, ja jopa
kehua. Olen oppinut Penalta todella paljon, sekä märkäkemiaa että tieteellistä
kirjoittamista. Kiitos!
Tarja on äidillisesti kasvattanut solut kokeisiini. Hän on kanssani leimannut, uuttanut, ajanut
houplesia ja niin edelleen. Lisäksi Tarja on labran sielu ja kaikkien olkapää. Ilot ja
surut on aina voinut jakaa Tarjan kanssa. Kiitos kaikesta!
Kimmo on tuhannet kerrat auttanut mua tietsikan kanssa. Lisäksi olemme lukemattomat kerrat
pohtineet lipidien metkuja teekupin ääressä. Ja lounaalla ja vapaalla on sitten puhuttu
Elämästä. Kiitos ystävyydestäsi!
Anulle, Mirkalle ja Reijolle kiitokset henkisestä tuesta.
Kaikille kesätyöntekijöillemme kiitos piristävästä ja nuorentavasta lounasseurasta Unicaféssa.
Perttu Haimille kiitos Excel-makrosta, jota ilman en olisi jaksanut prosessoida massadataa.
Kaija Nivalle kiitokset avusta mitokondrioiden kanssa.
Juha Okkerille kiitos avusta skannauksessa ja sekvenssihakujen kanssa.
Kaija Tiilikalle kiitos 21 kappaleesta mustia vahakantisia vihkoja ja muusta tilpehööristä.
Kainuun Annalle kiitokset superhyvästä siivouksesta.
Tertulle, Liisa-kaimalle ja Maaritille kiitokset silyloitujen kimaksien hellävaraisesta
tiskaamisesta.
Nina Katajamäelle kiitokset sadoista artikkeleista.
Leena Karhiselle kiitos hiivan pikakurssista.
Masalle ja Vesalle kiitokset hyvistä kommenteista väitöskirjaani.
Kimmolle, Martinille ja Reijolle kiitos väitöskirjani oikoluvusta.
Sofia Finnilälle suurkiitokset upeasta kansikuvasta.
Ich danke herzlich den Professor Günter Daum dafür, dass er mein Opponent sein wollte.

9
Jukka on elänyt kanssani yli 4 vuotta eli suurimman osan tämän väitöskirjatyön ajasta. Jukka
on opettanut minut relaamaan. Tärkeintä elämässä ei ole tohtorinhattu, vaan irtautua
työhuolista katsomalla toimintaleffaa! Kiitos kun olet kestänyt Känkkäränkkää!
Äiti ja Isä ovat tukeneet minua vielä kauemmin kuin 4 vuotta, kaikin mahdollisin tavoin. He
ovat vastanneet tämän saavutuksen dopingista, sillä peruskuntoni väitöskirjatyön
tekemiseen on saavutettu Villiruusun perunoilla ja mustaherukkamehulla, ja
viimeistely on tapahtunut Villiruusun minttua juoden. Myös rahallisia lahjuksia olen
ottanut vastaan heiltä akateemisen urani alkuvaiheessa. Erikoiskiitos vielä loppu-
metrien kannustuksesta ja avusta karonkkajärjestelyissä.
Avoppivanhempieni Aunen ja Hannun koti Laukaassa on ollut minulle rauhan satama kaukana
kaupungin hälystä. Siellä olen viettänyt monta onnellista viikkoa pääsiäis-, kesä- ja
joululomilla. Aune on laittanut herkullista kotiruokaa ja Hanski on näyttänyt minulle
luonnon ihmeitä ja saanut minut innostumaan linnuista. Kiitos!
Pialle ja Pauliinalle kiitos kun olette lainalapsina piristäneet viikonloppujamme ja
tutustuttaneet minut Harry Potteriin.
Katalle ja Pekalle kiitos yhteisistä opiskeluvuosista Vuorikadulla dinosaurusajalla ennen
Kumpuloita ja Viikkejä.
Anulle kiitos yhteisestä väitöskirjatuskasta, sekä muusta jaetusta Elämästä.
Maukalle, Mikalle ja Hannulle kiitos nyrkkeilyharkoista ja kaikille reenikavereille kiitos että
mäiskitte hikisinä kanssani. Parasta vastapainoa tietokoneelle!
Astangajoogaopettajalleni Gabriellalle kiitos kärsivällisestä opetuksesta ja Eevalle kiitos
kyydistä ja kahvilaseurasta.
Ateneumin Ystävien sihteerille Minna Erwelle kiitos upeista taide-elämyksistä.
Entisille partio-, kuoro-, ja judokavereille kiitos yhteisistä hetkistä ja retkistä.
Katriina Koskelalle kiitos kallonkutistuksesta.
Raija Raanamolle, Leena Larvalle, Nabil Jaserille sekä Anja Juvakselle kiitos hyvästä
hoidosta.
Sonjalle, Sarille ja Irinalle kaunis kiitos kaunistamisestani.
Marja Raitaselle Stokkan pukeutumispalveluun kiitos hyvistä neuvoista.
Kiitokset apurahoista Ella ja Georg Ehrnroothin säätiölle (tuplasti), Wiipurilaisen osakunnan
stipendirahastolle ja Magnus Ehrnroothin säätiölle sekä Suomen Akatemialle ja Sigrid
Juseliuksen säätiölle.

10
ABBREVIATIONS
BHK cells baby hamster kidney cells
BODIPY boron dipyrromethene difluoride
�-CD carboxyethyl-�-cyclodextrin
CHO cells chinese hamster ovary cells
diPyrnPS dipyrene phosphatidylserine (n = number of aliphatic carbons in the pyrenylacyl chain)
DNP 1,4-dinitrophenol
DOPE dioleoyl-phosphatidylethanolamine
DOTMA dioleyloxypropyl trimethylammonium chloride
DTT dithiotreitol
EM electron microscopy
ER endoplasmic reticulum
ESI-MS electrospray ionisation mass spectrometry
ESR electron spin resonance
I-MEM CO2 -independent growth medium
IMM inner mitochondrial membrane
MAM mitochondria-associated membrane
NBD nitrobenzoxadiazole
NL neutral loss
NEM N-ethylmaleimide
nsL-TP nonspecific lipid transfer protein
ODS octadecylsilica
OMM outer mitochondrial membrane
PC phosphatidylcholine
PE phosphatidylethanolamine
POPC 1-palmitoyl-2-oleoyl-phosphatidylcholine
PPA phosphatidylisopropanolamine
PS phosphatidylserine
PSD phosphatidylserine decarboxylase
PT phosphatidylthreonine
RP-HPLC reverse phase high performance liquid chromatography
RRT relative retention time
SM sphingomyelin
TLC thin layer chromatography
TNP- trinitrophenyl-
yeast the yeast Saccharomyces cerevisiae

11
INTRODUCTION
Phosphatidylserine (PS) is an essential phospholipid present in all mammalian cellular
membranes, and it is especially abundant at the plasma membrane inner leaflet. The plasma
membrane of eukaryotic cells is asymmetric, with choline phospholipids phosphatidylcholine
(PC) and sphingomyelin (SM) concentrated in the outer leaflet, and PS and phosphatidyl-
ethanolamine (PE) in the inner leaflet. The asymmetry is most strict for PS, with no PS
exposed on the cell surface under normal conditions. The asymmetry is maintained by the
aminophospholipid translocase that rapidly pumps PS from the outer leaflet to the inner
leaflet (see section 9).
PS exposed on the cell surface is a signal for cell phagocytosis by macrophages. PS is
exposed on apoptotic red blood cells (Schroit et al., 1985) and lymphocytes (Fadok et al.,
1992), and this leads to clearance by macrophages. Also sickled erythrocytes in sickle cell
anaemia expose PS (Lubin et al., 1981) and their lifetime in the circulation is therefore shorter
than that of normal erythrocytes. A PS-receptor on macrophages has been found (Fadok et al.,
2000). PS exposed also on the surface of macrophages is needed for phagocytosis of apoptotic
thymocytes (Callahan et al., 2000). The engulfment process seems to require interactions
between PS-exposing membranes of both the engulfing and target cells. PS exposure is
needed yet for another important event, blood coagulation. Activation of platelets leads to loss
of plasma membrane lipid asymmetry, thus exposing PS, which promotes assembly of
enzyme complexes of the blood coagulation cascade (Bevers et al., 1983, 1996). PS exposure
on cell surface is a result of redistribution of plasma membrane phospholipids, which is
accomplished by concomitant activation of plasma membrane scramblase and inhibition of
the aminophospholipid translocase, both launched by an elevation of intracellular Ca2+
concentration (Bevers et al., 1996; Bratton et al., 1997; Zhao et al., 1998).
It has been proposed that an ATP-binding cassette transporter, ABC-A1, promotes Ca2+-
induced PS translocation into the outer leaflet of plasma membrane (Hamon et al., 2000).
This ABC-A1-induced PS-exposure is suggested to be needed (i) in macrophages to enable
the engulfment of apoptotic cells and (ii) in mouse embryo fibroblasts in the release of
membrane phospholipids and cholesterol to apo-A1, the major apolipoprotein component of
the cholesterol-shuttling high-density lipoprotein particle (Hamon et al., 2000). However, it

12
has been argued that cell surface PS is not sufficient to mediate cellular apo-A1 binding and
lipid efflux (Smith et al., 2002).
In addition, PS is an essential cofactor for the activation of protein kinase C (Nishizuka,
1988), and PS regulates in vitro various enzymes including glucocerebrosidase (Berent and
Radin, 1981), diacylglycerol kinase (Sakane et al., 1991), dynamin GTPase (Tuma et al.,
1993), RAF-1 protein kinase (Ghosh et al., 1994) and nitric oxide synthase (Calderon et al.,
1994).
PE has the important ability to adopt the hexagonal HII phase and other non-bilayer structures,
which is suggested to be important in membrane fusion. PS is also believed to be helpful in
fusion, as it has a tendency to destabilise bilayer structures in the presence of Ca2+ (Cullis and
de Kruijff, 1979). A possible example of this is that when Sindbis virus is internalised into a
CHO cell line defective in PS-synthesis (strain PSA-3), virus production is severely reduced.
The proposed reason is that the internalised virus cannot release its nucleocapsid because it
cannot fuse with the host endosome membranes having a diminished amount of PS (and/or PE
derived from PS) (Kuge et al., 1989).
PS is thus involved in many important cellular events. It is synthesised only in the ER (and a
specialised region of ER called MAM, mitochondria-associated membrane), but it is needed
in all cellular membranes. Therefore, translocation of PS from the site of synthesis to other
locations in the cell must take place. In addition, the concentration of PS varies among
cellular organelles, being highest in the plasma membrane. Also the distribution of PS
molecular species differs between organelles (Schneiter et al., 1999). How is intracellular PS
translocation mediated without perturbing the distinct lipid contents of organelles? Possible
lipid translocation mechanisms include transfer mediated by lipid transfer proteins, vesicular
transfer, spontaneous diffusion via cytoplasm and lateral diffusion via membrane contacts
(see section 3). How is the translocation regulated? These questions have not yet been
thoroughly answered.
In this thesis and the original publications, results are presented indicating that spontaneous
diffusion has an important role in intracellular PS translocation. It was shown that from PS
synthesised in the ER/MAM, the less hydrophobic species are rapidly translocated to
mitochondria and decarboxylated therein to PE, while more hydrophobic ones are

13
translocated much more slowly (I, II). As a result, the PS species remaining in the ER/MAM
are, on the average, more hydrophobic than those initially synthesised. Many of these
obviously incorporate to exocytic vesicles and are transported to the plasma membrane. It was
also shown that PS translocation from the plasma membrane to mitochondria is much slower
for the more hydrophobic PS species (III). Therefore the hydrophobic PS species, once
arrived into the plasma membrane, may not be prone to efflux from the membrane, which
could be crucial for the maintenance of a high concentration of PS in the inner leaflet of the
plasma membrane.

14
REVIEW OF THE LITERATURE
This thesis is focused on mammalian cells, but for comparative purposes data from the yeast
Saccharomyces cerevisiae is included. This eukaryotic organism is well known, and a lot of
essential information on PS metabolism and translocation has been obtained from yeast
studies.
1. Phosphatidylserine metabolism
1.1. Phosphatidylserine synthesis
In mammalian cells, PC and PE are synthesised de novo by the Kennedy-pathway from
diacylglycerol and CDP-choline or CDP-ethanolamine (Kennedy and Weiss, 1956). PS is
synthesised from existing PC and PE molecules by a base-exchange reaction, in which the
choline or ethanolamine is replaced by serine (Kanfer, 1980). It is important to note that the
base-exchange reaction is not a reversal of phospholipase D reaction. Phospholipase D
purified from rat brain hydrolyses phospholipids to phosphatidic acid, but it has no base-
exchange activity (Taki and Kanfer, 1978).
PS synthesis occurs in the ER, particularly in a specialised region of the ER called MAM for
mitochondria-associated membrane. The base-exchange reaction needs no energy, but Ca2+ is
required (Borkenhagen et al., 1961; Hübscher, 1962; Kanfer, 1980). Several agents with
activity to empty intracellular Ca2+ stores inhibit PS synthesis in glioma cells. These include
the neurotransmitters glutamate and acetylcholine which bind to cell surface receptors and
induce increase in intracellular Ca2+ concentration, thapsigargin which inhibits the ER Ca2+ -
ATPase, and the ionophore A23187 which renders cell membranes permeable to Ca2+ (Czarny
et al., 1992). Also in Jurkat T cells PS synthesis is inhibited by EGTA, Ca2+ -ionophores and
inhibitors of the Ca2+ -ATPase (Pelassy et al., 1992). Moreover, PS synthesis is decreased
markedly in activated T cells, where the intracellular Ca2+ stores have been depleted (Pelassy
et al., 1992).
It has long been argued whether the PS synthesis occurs in the cytoplasmic or luminal leaflet
of ER membranes. The requirement for Ca2+ and the inhibition of synthesis upon emptying
intracellular Ca2+ stores point to the luminal side. The PS synthase has maximal activity at 10
mM Ca2+ (Taki and Kanfer, 1978). For comparison, the cytosolic calcium concentration is

15
only 0.1-0.2 �M in resting cells (Carafoli, 1987), whereas in the ER lumen the concentration
can rise to millimolar range (Baranska, 1989). Another proof pointing to the luminal side
hypothesis is that in rat brain microsomes, the serine (and ethanolamine) base-exchange
activities were found to be trypsin-sensitive, in contrast to choline base-exchange activity
(Buchanan and Kanfer, 1980). Yet another possibility might be that the active site (and/or the
end-product inhibition site) of the enzyme is on the cytosolic side, but the Ca2+ activation site
is luminal.
There are two enzymes responsible for PS synthesis, phosphatidylserine synthase (PSS) I and
II. In vitro PSS I is capable of exchanging choline, ethanolamine and serine and PSS II is
capable of exchanging ethanolamine and serine but not choline (Kuge et al., 1986a). In vivo
PSS I catalyses the PC to PS conversion (Kuge and Nishijima, 1997), and PSS II the PE to PS
conversion (Saito et al., 1998).
PC is the major precursor for PS synthesis in CHO cells (Miller and Kent, 1986; Voelker and
Frazier, 1986; Stone and Vance, 1999) and resting keratinocytes (Arthur and Lu, 1993). Since
PS in turn is the major precursor for PE by PS decarboxylation in mitochondria (see section
1.5), this leads to the following scheme (Figure 1). As can be seen, choline and ethanolamine
released by the PS synthase I and II are recycled to PC and PE synthesis via the Kennedy-
pathway. This means that no PC, PE, choline or ethanolamine is consumed in the synthesis of
PS and PE.
PC PS PSPE
Ser Cho Ser EtCO2
PSS I (ER) PSS II (ER)PSD (mito)
Recycling via Kennedy-pathway
Recycling via Kennedy-pathway
Figure 1. Biosynthetic pathways for phosphatidylserine and phosphatidylethanolamine in mammalian cells. Theabbreviations used are: PSS, phosphatidylserine synthase; PSD, phosphatidylserine decarboxylase; mito,mitochondrial; PC, phosphatidylcholine; PS, phosphatidylserine; PE, phosphatidylethanolamine; Ser, serine;Cho, choline, Et, ethanolamine.

16
CHO cell mutants lacking functional PSS I have been isolated. They have lowered PS and PE
levels, which confirms that PS is indeed a key precursor for PE in these cultured cells.
Without proper supplementation the mutants die. Mutant M.9.1.1 is rescued by addition of
PS, PE, lyso-PE or ethanolamine (Voelker and Frazier, 1986). Mutant PSA-3 is rescued by
addition of either PS or PE, but not by ethanolamine (Kuge et al., 1986a), indicating that this
mutant must be somehow defective also in the Kennedy-pathway.
The cDNAs for the PSS enzymes were first cloned from CHO cells. pssA encodes PSS I
(Kuge et al., 1991) and pssB encodes PSS II (Kuge et al., 1997). The predicted proteins
contain 471 and 474 residues, respectively, with a 32 % sequence identity. Hydrophobicity
analysis revealed that PSS I and II exhibit very similar hydrophobicity profiles. They are
highly hydrophobic proteins containing several potential membrane spanning domains (Kuge
and Nishijima, 1997). Both PSS enzymes have an ER-targeting sequence: PSS I has a C-
terminal double lysine motif (Gly-Val-Gly-Lys-Lys), and PSS II has an N-terminal double
arginine motif (Met-Arg-Arg-Ala-Glu) (Kuge and Nishijima, 1997).
Later, both PSS I and II have been cloned also from mouse. The open reading frame for both
enzymes contains 473 residues, again with only about 30 % sequence identity (Stone and
Vance, 1999). Mouse PSS I has five putative transmembrane domains, it contains the C-
terminal double lysine motif, and it is > 90 % identical to that of CHO cells and a human
myeloblast cell line (Stone et al., 1998). Mouse PSS II has six putative transmembrane
domains, it contains the N-terminal double arginine motif, and it is almost identical to the PSS
II of CHO cells (Stone and Vance, 1999).
PS synthesis is stimulated by amphiphilic cations such as oleylamine and sphingosine, and
inhibited by amphiphilic anions such as cholesterol sulfate in rat brain (Kanfer and
McCartney, 1993), rat liver and glioma C6 cell microsomes (Wiktorek-Wojcik et al., 1997).
Sphingosine is known to release Ca2+ from intracellular stores, but it is still stimulatory, at
least in microsomes. It has been speculated that sphingosine could be stimulatory in intact
cells as well, as it does not cause the Ca2+ stores to empty totally, in contrast to thapsigargin
and Ca2+ -ionophores (Sabala et al., 1996). Both the stimulators and inhibitors of PSS affect
only the Vmax without changing the Km for serine, suggesting that the mechanism of action is
related to interactions of PSS with the membrane phospholipids. The C-terminus of PSS I
carrying the double lysine motif and the N-terminus of PSS II carrying the double arginine

17
motif are presumably located on the cytosolic side of the ER. These positively charged
sequences of the enzyme might interact with the negatively charged phosphates of the
membrane phospholipids in the cytosolic leaflet (Wiktorek-Wojcik et al., 1997). Another
possibility is that these positive sequences of the enzyme would interact with the negatively
charged product, i.e. PS, leading to end-product inhibition of the enzyme. The mechanism of
stimulation for PS synthesis by amphiphilic cations could then be that the positively charged
amphiphile interacts with the negatively charged PS, thus reducing the end-product inhibition
(Wiktorek-Wojcik et al., 1997).
Exogenous PS can be efficiently incorporated into CHO cells, and it suppresses endogenous
PS synthesis, with no reduction in the amount of the base-exchange enzyme (Nishijima et al.,
1986). PS inhibits the serine base-exchange activity of both PSS I and II in vitro (Hasegawa
et al., 1989). A CHO cell mutant (mutant 29) is highly resistant to this inhibition by
exogenous PS, resulting in overproduction of PS and PE (Hasegawa et al., 1989). Mutant 29
has a point mutation in the pssA gene encoding PSS I, resulting in replacement of Arg-95 by a
lysine residue (Kuge et al., 1998). Thus Arg-95 seems to be critical for PSS I regulation by PS
in CHO cells (Kuge et al., 1998). Analogously, Arg-97 seems to be critical for PSS II
regulation by PS in CHO cells (Kuge et al., 1999). Together, the putative cytosolic location of
the C-terminus of PSS I and the N-terminus of PSS II (Wiktorek-Wojcik et al., 1997) and the
predicted location of the transmembrane domains (Stone et al., 1998; Stone and Vance, 1999),
suggest that Arg-95 and Arg-97 are also located on the cytosolic side. In mouse liver, it seems
that PSS II, but not PSS I, is inhibited by PS (Stone et al., 1998; Stone and Vance, 1999).
However, also mouse liver PSS I and II contain Arg-95 and Arg-97, respectively (Stone et al.,
1998; Stone and Vance, 1999). Therefore, the exact mechanism of PS synthesis regulation
remains unresolved.
1.2. Phosphatidylserine synthesis in yeast
In the yeast Saccharomyces cerevisiae PS synthesis is not a base-exchange reaction as in
higher eukaryotes, but it occurs by de novo synthesis from CDP-diglyceride and serine as in
Escherichia coli (PS synthase EC 2.7.8.8) (Henry et al., 1984). PS is the precursor for both
PE and PC in yeast. PS is first decarboxylated to PE, which is then methylated to PC (Letts et
al., 1983). However, PS itself is not vital for yeast, as a mutant (cho1) unable to synthesise PS
is viable when supplemented with either choline, ethanolamine, mono- or dimethylethanol-

18
amine (Atkinson et al., 1980). This mutant synthesises PC and PE via the Kennedy-pathway
(Kennedy and Weiss, 1956) and is thus rescued. The defect in the cho1-mutant is the total lack
of CHO1 gene, which has been shown to encode PS-synthase (Letts et al., 1983). Even
though the cho1-mutant grows well in supplemented growth medium, it has mitochondrial
abnormalities and diploid homozygotes are defective in sporulation (Atkinson et al., 1980).
The yeast PS synthase is not Ca2+-dependent and does not directly use ATP, but the
enzymatic reaction is energy-dependent owing to the requirement for CDP-diacylglycerol as
substrate (Daum and Vance, 1997). The expression of yeast PS synthase is transcriptionally
repressed by myo-inositol and choline in a coordinate manner with other phospholipid
synthesising enzymes. This theme is beyond the scope of this thesis and has been thoroughly
reviewed (Yamashita and Nikawa, 1997; Carman and Henry, 1999).
Yeast PS synthase is located in ER and MAM (Zinser et al., 1991; Gaigg et al., 1995), like
the two PS synthases of mammals. Even though the PS synthesis occurs via totally distinct
pathways in mammals and yeast, the site of synthesis and subsequent translocation to
mitochondria to yield PE are similar.
1.3. Transbilayer movement of phosphatidylserine in endoplasmic reticulum
It is still controversial whether the synthesis of PS occurs at the cytosolic or luminal surface
of the ER/MAM membrane. In any case, the newly-synthesised PS has to be translocated to
the other leaflet, in order to prevent one leaflet from expanding disproportionately. The same
is true for other phospholipids as well. It has been shown that transbilayer movement (flip-
flop) of phospholipids in microsomal vesicles is rapid, occuring in the order of seconds to
minutes (Zilversmit and Hughes, 1977; Bishop and Bell, 1985; Menon, 1995; Buton et al.,
1996). In contrast, flip-flop in pure lipid vesicles (or vesicles containing membrane proteins
from irrelevant sources, i.e. membrane proteins from biomembranes that do not synthesise
significant quantities of lipids) is very slow, in the order of hours to days (Kornberg and
McConnell, 1971a; Bishop and Bell, 1985; Menon, 1995). These large rate differences
suggest that specific membrane proteins may catalyse the rapid movement of phospholipids in
the ER membrane. Indeed, such proteins have been found and termed flippases. They are
ATP-independent, stereospecific, sensitive to proteolysis and sulfhydryl-modifying reagents,
and catalyse a bidirectional process with no apparent selectivity towards the phospholipid

19
headgroup or glycerol vs. ceramide backbone (Bishop and Bell, 1985; Herrmann et al., 1990;
Menon, 1995; Menon et al., 2000). The flippase protein is required to overcome the high
energy barrier associated with taking a charged phospholipid headgroup through the
hydrophobic core of the membrane (Bishop and Bell, 1985; Menon, 1995). It has been
suggested that PE present in microsomes would facilitate the flip-flop of phospholipids due to
its ability to destabilise bilayer structures by adopting the hexagonal HII phase (van Duijn et
al., 1986), but also contradicting views exist (Buton et al., 1996).
It is possible that PS synthase itself acts as a PS flippase, since it has been suggested that lipid
synthesising enzymes having multiple transmembrane domains might act as flippases of their
product molecules, thereby integrating phospholipid synthesis and transmembrane transport
(Hjelmstad and Bell, 1991).
1.4. Phosphatidylserine remodelling
Theoretically, the acyl chains of all phospholipids, including PS, can be exchanged by
deacylation-reacylation (MacDonald and Sprecher, 1991). Phospholipase A1 and A2 can
remove the acyl chain from the sn-1 and sn-2 position, respectively, yielding a lysophospho-
lipid. This lysolipid can then be reacylated via one of two enzymes, either acyl-CoA-
lysophosphatide acyltransferase (acyl-CoA + lysolipid) or transacylase (intact phospholipid +
lysolipid) (MacDonald and Sprecher, 1991). The deacylation and reacylation reactions most
probably occur in the ER (Schmid et al., 1991).
Rat liver microsomes can carry out in vitro acylation of both 2-acyl-sn-glycero-3-
phosphoserine at the sn-1 position (unsaturated fatty acids preferred, particularly 18:0,
(Thompson and Belina, 1986)) and of 1-acyl-sn-glycero-3-phosphoserine at the sn-2 position
(unsaturated fatty acids preferred, (Holub, 1980)). However, the deacylation-reacylation
pathway seems to play a very limited role in determining the fatty acid composition of
hepatocyte PS (Bjerve, 1985). In rat liver microsomes the pattern of PS molecular species
(mostly stearoyl-unsaturated species) is markedly different from both its precursors, PC and
PE. It has been shown that this is not achieved by post-synthetic deacylation-reacylation, but
rather via direct preference by PS synthase for the 18:0/20:4 and 18:0/22:6 PC and PE species
(Ellingson and Seenaiah, 1994).

20
All of the data available on PS remodelling is obtained from studies with rat liver, which is a
very specialised organ. Thus extensive conclusions applying to other types of cells or tissues
can not be drawn.
1.5. Phosphatidylserine decarboxylation
Phosphatidylethanolamine (PE) can be synthesised in three different ways in mammalian
cells: (1) by de novo –synthesis via the CDP-ethanolamine pathway (Kennedy-pathway,
(Kennedy and Weiss, 1956)), (2) by decarboxylation of PS (Borkenhagen et al., 1961) or (3)
by a base-exchange reaction. The contribution of the last, however, has been found negligible,
at least in rat liver (Bjerve, 1973). In contrast, both the Kennedy-pathway and PS
decarboxylation are important, and their contributions to total PE synthesis vary according to
cell type and nutrition.
PS decarboxylation occurs in mitochondria (Dennis and Kennedy, 1972) and is mediated by
the enzyme PS decarboxylase (PSD), which is present only in the mitochondrial inner
membrane (van Golde et al., 1974; Percy et al., 1983) with its active site facing the
intermembrane space (Zborowski et al., 1983). It is not clear if PS has to translocate to the
inner mitochondrial membrane to be decarboxylated, or if the enzyme can reach PS present in
the inner leaflet of the mitochondrial outer membrane (see section 7). PSD is not selective
towards the fatty acid composition of natural PS, at least in rat hepatocytes (Bjerve, 1985).
Voelker has shown that PS decarboxylation provides the majority of PE in BHK and CHO
cells, even in the presence of ethanolamine, since addition of ethanolamine had only a slight
inhibitory effect upon incorporation of [3H]serine to PE (Voelker, 1984). In the case of CHO
cells, there are many studies using mutant cell lines confirming that PE is mostly derived from
PS (Kuge et al., 1986a, 1986b; Miller and Kent, 1986; Voelker and Frazier, 1986). PS
decarboxylation is also the major quantitative contributor to rat hepatocyte PE (Vance, 1988),
but in the presence of ethanolamine the Kennedy-pathway in enhanced (Tijburg et al., 1989).
The same is true for human keratinocytes (Arthur and Lu, 1993), and thus there seems to be
coordination of the two pathways to prevent excess production of PE in hepatocytes and
keratinocytes.

21
The physiological ethanolamine concentration in serum is 20-25 �M (rat serum, Sundler and
Åkesson, 1975). This implies that in vivo cells are exposed to a constant supply of
ethanolamine. Common cell culture media do not contain ethanolamine, but they are
supplemented with fetal bovine serum that contains approximately 23 �M ethanolamine
(Lipton et al., 1990). Despite the presence of ethanolamine the clear majority of PE is formed
via PS decarboxylation in cultured BHK and CHO cells (Voelker, 1984; Miller and Kent,
1986).
Why are there two different functional pathways for PE synthesis, is an intriguing question.
Most cultured mammalian cells do not have the need for exogenous ethanolamine, so it seems
likely that they have the capacity to derive PE exclusively by decarboxylation of PS. If this
indeed is the case, what is the physiological significance of the Kennedy-pathway? It has been
suggested that the Kennedy-pathway is required for synthesis of ethanolamine plasmalogen
species (alkenyl/acyl species) in neuronal and cardiac cells, in which ethanolamine
plasmalogens account for about half of total ethanolamine phosphoglycerides (Yorek et al.,
1985). It has been shown that in retinoblastoma Y79 cells serine is rapidly incorporated to
diacyl PE, but not to ethanolamine plasmalogen (Yorek et al., 1985). Heart tissue contains
more ethanolamine plasmalogens than liver or kidney, and the decarboxylation pathway is
less active in this tissue (Arthur and Page, 1991). There are contradicting results suggesting
that PS might act as a precursor for both diacyl and ether ethanolamine phosphoglycerides in
cultured rat brain cells (Yavin and Zeigler, 1977) and in cultured glioma cells (Xu et al.,
1991). These data might, however, be biased by metabolism of the labelled serine precursor to
compounds like acetate, formate and glycine (Xu et al., 1991), which incorporate to the
glycerol and fatty acid moieties of phospholipids (Voelker, 1984; Vance and Vance, 1986; Xu
et al., 1991).
According to Vance, in rat hepatocytes, where PE is methylated to PC, the two PE
synthesising pathways seem to have specific roles. PS-derived PE and PC are used mainly for
the synthesis of secreted lipoproteins (as well as CDP-choline-derived PC), whereas CDP-
ethanolamine-derived PE and PC are used by the cell itself (Vance and Vance, 1986; Vance,
1988).

22
1.6. Phosphatidylserine decarboxylation in yeast
As in mammals, yeast PSD has been localised to mitochondria (Kuchler et al., 1986; Zinser et
al., 1991). It was cloned in 1993 (Clancey et al., 1993; Trotter et al., 1993). The gene was
disrupted in a haploid strain, resulting in loss of detectable PSD activity, but unexpectedly the
gene was found unessential for growth as its disruption did not lead to ethanolamine
auxotrophy, PS accumulation or elimination of PE (Clancey et al., 1993; Trotter et al., 1993).
This was very surprising, since mutation of the PS synthase is lethal in absence of
ethanolamine or choline (Atkinson et al., 1980). It was suggested that a second PSD exists.
Indeed, a non-mitochondrial PSD2 was identified and cloned two years later, and it was
exclusively present in the Golgi and vacuoles (Trotter and Voelker, 1995). PSD2 shows
surprisingly low homology to PSD1 (Voelker, 1997). PSD2 accounts for only 2-5 % of wild
type PSD activity, but this small activity is still sufficient to support growth without
ethanolamine (Trotter and Voelker, 1995). Even though a mutant lacking PSD1 is viable and
has a normal phospholipid composition, it has unstable mitochondria with a markedly reduced
content of PE, indicating that PE formed by PSD2 is not efficiently transported to
mitochondria (Trotter and Voelker, 1995). A double mutant psd1� psd2� is auxotrophic for
ethanolamine or choline, and has only 30 % of PE compared to wild type cells (Trotter and
Voelker, 1995).
The function and synthesis of PE via three different pathways in yeast has been studied
extensively. Even though it was first believed that only PC is essential for growth, it now
seems that also a minimal amount of PE is needed for cell viability (Birner et al., 2001;
Storey et al., 2001). Small amounts of PE can be produced in psd1� psd2� mutants in the
absence of ethanolamine supplementation by sphingolipid breakdown. This yields
ethanolamine-phosphate (by dihydrosphingosine phosphate lyase 1, Dpl1, (Gottlieb et al.,
1999)), which is incorporated to PE via the Kennedy-pathway. The possible role of this
minimal amount of PE needed for viability might be providing PE for GPI-anchored proteins
(Birner et al., 2001; Storey et al., 2001). It has been shown that PE is the terminal
phosphoethanolamine group donor for GPI-anchored proteins (Menon et al., 1993).

23
2. Phosphatidylthreonine
A structural analogue of PS, phosphatidylthreonine (PT) was first found in egg yolk (Rhodes
and Lea, 1957) and tuna fish muscle (Igarashi et al., 1958). Later, PT was detected in BHK-
21 cells and polyoma virus transformed hamster embryo fibroblasts (Mark-Malchoff et al.,
1978). Only a trace amount of PT occurs in normal cells and tissues of hamster and mouse
embryos and adults (Mark-Malchoff et al., 1978). BHK cells can be considered transformed,
since this cell line was selected for continual growth in tissue culture, and BHK cells have
been shown to produce tumors upon inoculation into adult hamsters (Defendi et al., 1963).
Thus it seems that PT is a lipid that is produced in continuously growing or transformed
hamster cells in culture, but hardly in normal tissues (Mark-Malchoff et al., 1978).
Hippocampal astroglial cells release significant amounts of L-serine and this amino acid is
trophic for hippocampal neurons (Mitoma et al., 1998a). When hippocampal neurons are
maintained in culture without astroglial cells, the levels of sphingolipids and PS decrease
markedly in the absence of external serine or glycine, and PT appears (Mitoma et al., 1998b).
Thus PT seems to be a phospholipid appearing upon serine depletion in cultured cells. PT is
not detected in hippocampal regions of normal rat brain tissues, suggesting that serine is not
depleted in normal brain (Mitoma et al., 1998b). Indeed, it has been shown that extracellular
free L-serine is present in the central nervous system (Shimada et al., 1993).
The rat brain microsomal base-exchange enzyme synthesising PS has a 150-fold lower
affinity for L-threonine as compared to L-serine (Mitoma et al., 1998b). Thus PT synthesis is
inhibited strongly already at very low concentrations of L-serine (Mitoma et al., 1998b).
These figures provide a plausible explanation for the fact that PT is synthesised only upon L-
serine deprivation.
3. Intracellular lipid translocation mechanisms
With a few exceptions, phospholipids are synthesised in the ER, but they are needed in all
cellular membranes. Accordingly, extensive intracellular lipid translocation is needed. The
possible lipid transfer mechanisms (Figure 2) will be briefly discussed below.

24
Lateral diffusion via membrane(hemi)fusion sites
Lipid carrier protein-mediated diffusion
Spontaneous diffusion via cytoplasm
Vesicular transfer
Figure 2. Intracellular lipid translocation mechanisms.
3.1. Lipid carrier proteins
Three specific phospholipid carrier proteins have been characterised from mammalian
sources: the phosphatidylcholine transfer protein (PC-TP) and two phosphatidylinositol
transfer proteins (PI-TP and SM/PI-TP, also called PI-TP� and PI-TP���respectively). PC-TP
binds and transfers only PC, PI-TP� binds and transfers both PI and PC, but shows an
approximately 16-fold preference for PI (van Paridon et al., 1987). Both PC-TP and PI-TP�
have three separate binding sites: one for the polar headgroup, second for the sn-1 acyl chain
and third for the sn-2 acyl chain (Somerharju et al., 1987; van Paridon et al., 1988). They
exhibit clear selectivity for the hydrophobicity and steric properties of the acyl chains
(Kasurinen et al., 1990). The structure of PI-TP� complexed with one molecule of PC has
been determined by x-ray diffraction techniques. A single �-sheet and several long �-helices
define an enclosed internal cavity in which a single phospholipid molecule is accommodated
with its polar headgroup in the centre of the protein and fatty acyl chains projected towards
the surface (Yoder et al., 2001).
PI-TP��also binds and transfers PI (and PC with lower affinity), but prefers sphingomyelin (de
Vries et al., 1995; Westerman et al., 1995). Recently also PI-TP� was shown to bind and
transfer sphingomyelin (Li et al., 2002). PI-TP� is predominantly present in the cytoplasm

25
and nucleus, PI-TP� is preferentially associated with the Golgi region (de Vries et al., 1995).
PI-TP� is highly conserved among species (Wirtz, 1997). The amino acid sequence is 77 %
identical and 94 % similar to PI-TP� (Tanaka and Hosaka, 1994), but shows no homology to
PC-TP (Wirtz and Gadella, 1990).
Functionally the two mammalian PI-TPs are thought not to perform actual net transfer of lipid
in vivo. Instead, they seem to act as regulators of the secretory pathway, e.g. in the budding of
transport vesicles from trans-Golgi network and in the fusion of the vesicles with the plasma
membrane (reviewed in Wirtz, 1997). No physiologically relevant functions for PC-TP have
been found so far even though the issue has been actively investigated e.g. in a knock-out
mouse model (van Helvoort et al., 1999).
3.2. Yeast phosphatidylinositol transfer protein
A PI-TP with dual specificity for PI and PC has been found also in yeast, again with a higher
affinity for PI than PC (Szolderits et al., 1989). It was found essential for cell viability (Wirtz,
1997). The yeast PI-TP gene is identical to the SEC14 gene encoding a protein required for
transport of secretory proteins from the Golgi membranes (Wirtz, 1997). Interestingly both
mammalian PI-TPs restore yeast sec14 mutants, even though neither has any sequence
homology with Sec14p (reviewed in Wirtz, 1997). Bankaitis and co-workers have shown that
yeast PI-TP is associated with the Golgi, and proposed that the role of PI-TP is to maintain the
PI/PC-ratio of yeast Golgi membranes (McGee et al., 1994; Skinner et al., 1995).
3.3. Non-specific lipid transfer protein
In addition to the specific lipid carrier proteins mentioned above, a non-specific lipid transfer
protein (nsL-TP, also called sterol carrier protein 2, SCP2) exists. It is very conserved among
species (Ossendorp and Wirtz, 1993) and transfers all common phospholipids, glycolipids,
cholesterol and oxysterols (Wirtz and Gadella, 1990). Its transfer rate in vitro correlates
inversely with lipid hydrophobicity (Nichols and Pagano, 1983; van Amerongen et al., 1989).
It has been suggested that nsL-TP does not actually carry lipids, but lowers the energy barrier
for lipid monomer dissociation and efflux from a membrane and thus enhances
intermembrane lipid transfer (Wirtz and Gadella, 1990). In all tissues except liver, the bulk of
nsL-TP is membrane-associated (Ossendorp and Wirtz, 1993).

26
NsL-TP is highly concentrated in peroxisomes, but also found to a lesser extent in
mitochondria, ER and cytosol (Tsuneoka et al., 1988; Keller et al., 1989). NsL-TP is not
essential for cell viability, as a CHO cell line deficient in peroxisomes lacks nsL-TP, but is
still viable and shows no alteration in phospholipid compositions (van Heusden et al., 1990).
NsL-TP is synthesised as a precursor with a 20 amino acid signal peptide, and peroxisomes
are thought to play a role in its conversion to the mature 14 kDa protein (Ossendorp and
Wirtz, 1993). NsL-TP contains a C-terminal tripeptide Ala-Lys-Leu, which is a potential
peroxisomal targeting sequence (Ossendorp and Wirtz, 1993). Peroxisomes play an important
role in lipid metabolism, including �-oxidation of very-long-chain and branched-chain fatty
acids and the synthesis of cholesterol, bile acids, ether-linked phospholipids and dolichol
(reviewed in van den Bosch et al., 1992). It is thought nsL-TP could be involved in these
processes. NsL-TP stimulates various steps of cholesterol metabolism in vitro (Ossendorp and
Wirtz, 1993) and it can bind fatty acids and long-chain fatty-acyl-CoAs (Wirtz, 1997). A
mouse lacking the gene for nsL-TP was created. No differences were observed in oxidation of
very long-chain fatty acids and palmitic acid, but the sera of these mice were 10-fold enriched
in phytanic acid, indicating that metabolism of branched-chain fatty acids was affected (Wirtz
et al., 1998). NsL-TP could also have a role in cholesterol transport to mitochondria (Daum
and Vance, 1997; Gallegos et al., 2000) and to and from the plasma membrane (Baum et al.,
1997; Atshaves et al., 2000; Gallegos et al., 2001).
3.4. Vesicular transfer
The major routes of vesicular traffic in living cells are (i) the biosynthetic pathway
responsible for the transport of proteins synthesised in the ER to the extracellular space
(secretion) or to other membrane compartments and (ii) the endocytic pathway responsible for
the uptake of compounds from the extracellular milieu to be used in cellular metabolism. The
outgoing and incoming pathways communicate through the exchange of material between the
Golgi apparatus and the endosomal elements (Olkkonen and Ikonen, 2000). Many of these
vesicular traffic steps are microtubulus-dependent (Kelly, 1990). The pathways that do not
involve microtubules are of short range, for example transfer between Golgi cisternae (Kelly,
1990).
Pagano and co-workers have studied vesicular transport in chinese hamster lung fibroblasts
(V79 fibroblasts) and CHO cells with NBD-labelled lipid analogues and radioactive lipids.

27
Sphingolipids, i.e. sphingomyelin and glycolipids, travel through Golgi en route to the plasma
membrane. Upon monensin-treatment (Lipsky and Pagano, 1985) or during mitosis
(Kobayashi and Pagano, 1989), which both inhibit protein secretion, they are arrested within
the Golgi membrane, which indicates an important role of vesicular transport in trafficking of
sphingolipids. In contrast, PE and PC seem to reach the cell surface much more rapidly, and
their transport is not affected by inhibitors of protein secretion (Sleight and Pagano, 1983;
Kaplan and Simoni, 1985): (i) brefeldin A which causes disassembly of Golgi (Vance et al.,
1991), (ii) mitosis (Kobayashi and Pagano, 1989), or (iii) low temperature (Kaplan and
Simoni, 1985). However, in these transport studies only a few percent (around 2 %) of the
labelled PE and PC did reach the plasma membrane rapidly. Thus it could be an artefact
resulting from generation of labelled lysolipids which can rapidly diffuse to the plasma
membrane and be reacylated therein. Therefore the interpretation that PE and PC travel to
plasma membrane by other means than vesicular transfer could be erroneous.
3.5. Spontaneous diffusion
One of the possible mechanisms of intracellular lipid trafficking is spontaneous diffusion of
lipid monomers via the cytosol. However, this mode of transfer has not been considered
quantitatively significant in total lipid flux for two reasons. First, it has been argued that
various cell membranes are known to have different lipid compositions and this would not be
possible if spontaneous lipid transfer occurred at a significant rate. Second, the spontaneous
transfer of long-chain natural phospholipids between vesicles is relatively slow (e.g. POPC t½
= 48-50 h at 37 °C (McLean and Phillips, 1981; Jones and Thompson, 1989)). However,
these reasons no longer appear to be sufficient to rule out some kind of role for spontaneous
lipid transfer in membrane biogenesis. First, it is now known that donor membrane
composition plays a critical role in regulating lipid exchange rate. The properties of the donor
membrane that affect the efflux rate include the physical state, packing defects, membrane
curvature and presence of membrane proteins. In addition, temperature, pH and ionic strength
of the aqueous medium affect the process (reviewed in Brown, 1992). All these factors
together regulate spontaneous lipid transfer and thus could have a major role in maintaining
compositional differences in cellular membranes. As for the second argument, it has been
shown that transfer rates of long-chain PCs increase significantly when the vesicle
concentration is high (Jones and Thompson, 1989, see below), and it is reasonable to assume

28
that inside the cell the cytoplasm is highly packed with various intracellular membranes and
thus the “membrane concentration” in vivo is likely to be high.
In the lipid concentration range below 2 mM transfer processes are best described by a first-
order kinetics, independent of vesicle concentration. However, at acceptor lipid
concentrations above 2 mM, a second-order term proportional to acceptor concentration must
be added (Jones and Thompson, 1990). The rate of transfer is limited by the formation of the
“activated complex” (see Figure 3) illustrated as a lipid monomer which has moved
perpendicularly to the plane of the bilayer with only the terminal carbons of its acyl chains
remaining in the bilayer. The rate of monomer desorption is enhanced by interaction with a
closely apposed membrane. The two membranes in this model are not in direct contact, but
the optimal separation distance is assumed to be approximately 15 Å, at which equal repulsive
and attractive forces are present (Jones and Thompson, 1989).
Rel
a tiv
ef r
eee n
ergy
,G
bilayer water
Position of terminal carbon
�G++
A
B++
Figure 3. Energy diagram for formation of the activated complex for monomer transfer. Panel A: Modelmembrane studies have shown that the rate-limiting step in spontaneous diffusion is the efflux of a lipidmonomer from the donor membrane (arrow). Panel B: The free energy of an effluxing lipid molecule as afunction of its transversal position relative to the membrane. ‡ indicates the transition state.

29
The rate-limiting step in spontaneous transfer is the desorption of the lipid monomer from the
donor membrane (McLean and Phillips, 1981; Lund-Katz et al., 1982; Massey et al., 1982a).
The efflux rate increases with decreasing chain length and increasing unsaturation (Massey et
al., 1982a; Massey et al., 1984; Ferrell et al., 1985; Massey et al., 1985; Silvius and Leventis,
1993). Also the polar headgroup influences the rate of spontaneous transfer, due to differences
in charge and hydrogen bonding properties (Massey et al., 1982b).
3.6. Lateral diffusion
It is possible that lipids could be transferred from one membrane to another by lateral
diffusion via membrane fusion/hemifusion sites. Lipidic bridges between organelles have
been postulated to exist between MAM and mitochondria (Vance, 1990) and between
mitochondrial inner and outer membranes (van Venetië and Verkleij, 1982).
Lateral diffusion of lipids is very rapid. It has been calculated that a membrane lipid would
diffuse from one end of a bacterium to the other (~ 1 �M) in much less than 5 minutes, if not
obstructed by membrane proteins (Kornberg and McConnell, 1971b). Theoretically lateral
diffusion should be similar for all lipids in the same matrix. Indeed, lateral diffusion in
liposomal membranes is relatively insensitive to the nature of lipid or the number or length of
the acyl chains (Derzko and Jacobson, 1980). Small amounts of cholesterol (less than 10
mole%) increase lipid diffusion, while higher concentrations decrease it (Kuo and Wade,
1979).
4. Methods for studying intracellular lipid translocation mechanisms
Intracellular protein transport can be studied using specific antibodies, but unfortunately
useful antibodies against phospholipids are not available. Intracellular lipid trafficking has
been studied with various phospholipid analogues including radioactive, fluorescent and spin-
labelled ones.
4.1. Radioactive phospholipids
Cultured cells can be labelled with [3H]-, [14C]- or [32P]precursors that incorporate to cellular
phospholipids. For example, [3H]serine yields [3H]PS in ER/MAM by the base-exchange
reaction, and it is then converted to [3H]PE by PS decarboxylase in mitochondria. Using

30
pulse-chase experiments, information on [3H]PS transport to mitochondria can be obtained
(Voelker, 1985).
The draw-back in using radiolabelled precursors is the need for many control experiments.
For example, when measuring PE labelling from [3H]serine, it is necessary to inhibit the
incorporation of [3H]serine to sphingolipids, because sphingolipid breakdown yields
phosphoethanolamine that is used for PE synthesis via the Kennedy-pathway (Hanada et al.,
1992). Another complicating issue is that serine is rapidly metabolised to acetate, formate and
glycine (Xu et al., 1991), which then incorporate to the glycerol and fatty acid moieties of
phospholipids (Voelker, 1984; Vance and Vance, 1986; Xu et al., 1991).
4.2. Fluorescent phospholipids
The advantage of using fluorescent phospholipid analogues is that they can be visualised by
microscopy, thereby avoiding laborious cell fractionations.
The first fluorescent lipid analogues used were nitrobenzoxadiazole (NBD) derivatives. The
fluorescent NBD-moiety can be attached to either the polar headgroup or at the end of one of
the acyl chains, usually at the sn-2 position. Due to their water solubility NBD-analogues are
easily incorporated to the plasma membrane from vesicles (in contrast to natural
phospholipids). However, the transport data obtained with these analogues might be
misleading, as spontaneous diffusion rates of these water soluble analogues are much higher
than those of their natural counterparts (Pagano and Sleight, 1985). In addition, acyl-NBD-
analogues are degraded rapidly in cells, with an approximate half-time of 2 hours (Pagano et
al., 1983; Sleight and Pagano, 1984, 1985; Kasurinen and Somerharju, 1995). Furthermore,
the NBD-moiety is not fully embedded in the bilayer but loops towards the membrane
interface (Chattopadhyay and London, 1987; Wolf et al., 1992; Huster et al., 2001).
Boron dipyrromethene difluoride (BODIPY) labelled lipids (reviewed in Pagano and Chen,
1998) have also been used in lipid trafficking studies. The BODIPY-fluorophore has a higher
fluorescence yield and it is more photostable than NBD. BODIPY-labelled lipids exhibit a
shift in their fluorescence emission maximum from green to red wavelengths with increasing
concentration in membranes. However, BODIPY-labelled lipids are also more hydrophilic

31
than their natural counterparts (Tanhuanpää and Somerharju, 1999) and therefore diffuse
spontaneously between all accessible membrane surfaces.
Pyrene-labelled phospholipids provide a probe in which the fluorophore is suggested to
minimally affect the properties of the labelled lipid (reviewed in Pownall and Smith, 1989;
Somerharju, 2002). The pyrene-moiety can be incorporated to either sn-1 or sn-2 acyl chain
(monopyrene lipid) or both (dipyrene lipid), see Figure 4. The pyrene-moiety is hydrophobic
and thus (i) the spontaneous diffusion rates of long-chain pyrene lipids are similar to natural
lipids, (ii) the pyrene-moiety is embedded in the membrane (Somerharju, 2002), (iii) pyrene
lipids are transferred by the lipid transfer proteins PC-TP (Somerharju et al., 1987), PI-TP
(van Paridon et al., 1988) and nsL-TP (Huuskonen et al., 1998). Additional advantages are
that (iv) pyrene lipids are much more stable with a half-time of degradation over 24 hours
(Kasurinen and Somerharju, 1995), (v) pyrene lipids can be synthesised with varying acyl
chain lengths, which gives useful information on intracellular lipid transfer mechanism, and
(vi) dipyrene lipids exhibit excimer fluorescence (480 nm) in addition to monomer
fluorescence (395 nm), and thus selective monitoring of intact dipyrene lipids is possible
(Kasurinen and Somerharju, 1995).
O
O
O
O
HCH2C CH2
O
O
O
O
CHH2C CH2
O
P O
O
OH3N
HH
HO OO
P O
O
O
HH
HO OO
P O
O
OH3N
HH
HO OO
P O
O
O
HH
HO O
Figure 4. Pyrene-labelled phosphatidylserine analogues. On the left a monopyrene phosphatidylserine(14:0/Pyr10-PS) and on the right a dipyrene phosphatidylserine (DiPyr10PS) are shown. The subscript numberafter Pyr denotes the number of aliphatic carbons in the pyrenylacyl chain.

32
4.3. Spin-labelled phospholipids
Spin-labelled lipids have a nitroxide group attached to the polar headgroup or to one of the
acyl chains. The nitroxide group is a stable radical, whose unpaired electron enables
monitoring with electron spin resonance (ESR) spectrometer. Kornberg and McConnell
devised an experimental set-up to study phospholipid flip-flop using spin-labelled
phospholipids (Kornberg and McConnell, 1971a). The spin-labelled molecules in the outer
leaflet of a vesicle can be reduced with ascorbate, resulting in loss of ESR-signal. Ascorbate
does not cross the membrane, and thus the inner leaflet spin-labelled lipids remain intact.
Another possibility for asymmetry determination is to extract the spin-labelled species from
the outer leaflet with bovine serum albumin (Morrot et al., 1989). Spin-labelled PS and PE
analogues contributed to the discovery of the plasma membrane aminophospholipid
translocase (Seigneuret and Devaux, 1984).
4.4. Stable isotope labelling
Electrospray mass spectrometry in combination with stable isotope -labelled precursors is a
potential method for studying intracellular lipid trafficking. Deuterium-labelled ethanolamine
and choline have been used to study the contributions of the Kennedy-pathway and PE
methylation pathway in PC synthesis in rat hepatocytes (DeLong et al., 1999). The use of
headgroup-specific scanning modes allows one to selectively detect and quantify both labelled
and unlabelled lipid molecular species conveniently and with high sensitivity (Han and
Gross, 1995).
5. Phosphatidylserine translocation from ER to mitochondria
Mitochondria are able to perform de novo synthesis of cardiolipin (Hostetler et al., 1971) and
to decarboxylate imported PS to PE (Dennis and Kennedy, 1972). The remainder of lipids
must be imported from the main site of cellular lipid synthesis, the ER. How is PS
translocated to mitochondria? The arrival of PS to mitochondria can be measured by
monitoring the conversion of PS to PE (Voelker, 1985; Vance and Shiao, 1996), assuming
that transfer to mitochondria rather than decarboxylation itself is rate-limiting (Voelker,
1989a). The translocation is rather slow, with a half-time of 5-7 hours (Voelker, 1985; Vance
and Vance, 1988). This seems inconsistent with the existence of a permanent membrane

33
continuum between ER/MAM and mitochondrial membranes, which would allow fast lateral
diffusion of PS to mitochondria.
Vesicle-mediated transfer is not likely either since (i) no vesicle transport to mitochondria has
been observed, (ii) 45-fold dilution of the cytosol in permeabilised CHO cells did not affect
PS translocation to mitochondria (Voelker, 1990) and (iii) in permeabilised and sheared CHO
cells PS translocation was restricted to autologous mitochondria, i.e. mitochondria of the
same cell (Voelker, 1993).
The involvement of cytosolic proteins seems also unlikely, since both in cell-free systems of
reconstituted microsomal and mitochondrial membranes (Voelker, 1989a; Vance, 1991) and
in permeabilised CHO cells (Voelker, 1989b, 1990) translocation of PS to mitochondria was
independent of cytosolic proteins. Specifically, nsL-TP is not involved, since PS transport to
mitochondria is normal in CHO mutant cells lacking this protein (van Heusden et al., 1990).
Inhibitors of cytoskeleton do not affect PS translocation to mitochondria in intact CHO cells
(Voelker, 1989b). There seems to be no other cofactors needed than ATP, Ca2+ and Mg2+
(Voelker, 1990). Energy is needed for PS translocation to mitochondria in intact and
permeabilised cells (Voelker, 1985, 1989b, 1990). However, no ATP is needed in cell-free
systems of reconstituted microsomal and mitochondrial membranes (Voelker, 1989a). This
lead to the conclusion that PS translocation to mitochondria is a two-step process, the first
step being ATP-dependent and the second one being ATP-independent (Voelker, 1989b). It is
not clear for what ATP is needed. One possibility could be because mammalian PS synthesis
requires it (Daum and Vance, 1997).
It seems that MAM has a major role in PS translocation to mitochondria. First, both PS
synthases are enriched in MAM (Vance, 1990; Stone and Vance, 2000). Second, newly-
synthesised PS is preferentially translocated to mitochondria in isolated organelles of rat liver
(Vance, 1991) and brain (Corazzi and Arienti, 1992; Corazzi et al., 1993) and in intact CHO
cells (Shiao et al., 1995). Third, it has been shown that newly-synthesised PS traverses MAM
en route to mitochondria (Shiao et al., 1995). The ER/MAM has been shown to be closely
associated with mitochondria in thin section electron microscopy of tissues or isolated
mitochondria and microsomes (Franke and Kartenbeck, 1971; Morré et al., 1971; Lewis and
Tata, 1973; Meier et al., 1981). Even though there is apparently no membrane continuity,

34
there might be some kind of channelling of PS from ER/MAM to mitochondria, as PS is
translocated only to autologous mitochondria but not to those derived from a different cell
(Voelker, 1993), and as extensive dilution of permeabilised cells had no effect on the
translocation of newly-synthesised PS from ER to mitochondria (Voelker, 1990).
The most recent data suggest that some membrane-associated proteins are involved in PS
translocation to mitochondria. A mitochondrial fusogenic glycoprotein has been proposed to
promote PS transfer from donor liposomes to isolated rat brain mitochondria (Camici and
Corazzi, 1997). Treatment of mitochondria with pronase or with a carboxyl modifying reagent
partly inhibited the putative fusion, and the purified protein markedly restored the “fusion”
capability of pronase-treated mitochondria (Camici and Corazzi, 1997). Analogously, in a rat
liver cell-free system of isolated microsomes and mitochondria, proteolysis of mitochondria
with trypsin or treatment with sulfhydryl-modifying reagents inhibited PS translocation to
mitochondria, whereas proteolysis of MAM had no effect (Shiao et al., 1998). Thus a
mitochondrial protein appears to promote PS translocation to mitochondria. However,
Voelker found that pronase and trypsin treatment had no effect in isolated rat liver organelles
(Voelker, 1989a). A possible explanation (Shiao et al., 1998) for this discrepancy could be
that Voelker’s mitochondria contained associated MAM, which could have protected them
from proteolysis.
Recently, it was discovered that a soluble protein from bovine brain cytosol enhances PS
translocation to mitochondria in permeabilised CHO cells (Kuge et al., 2001). The partial
sequence of the protein was determined and found identical to an EF-hand calcium-binding
protein, S100B (Kuge et al., 2001). Also this finding is in contrast with previous work by
Voelker where cytosol was slightly inhibitory in permeabilised CHO cells (Voelker, 1990). A
possible explanation (Kuge et al., 2001) could be that different assay conditions were used.
Voelker examined the effect of cytosol in the presence of EGTA which chelates Ca2+ and
arrests PS synthesis, whereas the assay of Kuge et al. contained Ca2+ allowing PS synthesis to
continue.
In conclusion, the exact mechanism of PS transfer from ER/MAM to mitochondria is still
unresolved. It seems likely that vesicular transport, nsL-TP or lateral diffusion via permanent
membrane continuity are not involved. The remaining possibilities are spontaneous diffusion
and protein-mediated translocation. Possibly PS-binding proteins on mitochondrial surface

35
preferentially bind regions of MAM enriched in PS, thereby enhancing PS translocation from
MAM to mitochondria (Shiao et al., 1998).
6. Phosphatidylserine translocation from ER to mitochondria in yeast
In yeast, as in mammalian cells, PS synthase is enriched in MAM (Gaigg et al., 1995), which
is closely associated with mitochondria as it co-sediments with them (Zinser et al., 1991).
Also electron microscopy data support this close association (Achleitner et al., 1999). The
translocation of PS to mitochondria is independent of ATP, cytosol, bivalent cations and
ongoing PS synthesis (Simbeni et al., 1993; Achleitner et al., 1995; Achleitner et al., 1999).
The cytoskeleton is probably not involved either (Gnamusch et al., 1992). Proteinase
treatment of mitochondria diminished PS translocation by 30 %, and treatment of both MAM
and mitochondria diminished PS translocation by 50 % (Achleitner et al., 1999). Thus it
appears that mitochondrial proteins, and maybe MAM proteins, are involved in the
translocation event.
Voelker and co-workers have examined the translocation of PS to the sites of PSD1
(mitochondria) and PSD2 (Golgi/vacuoles) using several yeast mutants. Using a mutant
lacking PSD1, it was found that PE formed by PSD2 is not efficiently transported to
mitochondria, as indicated by a markedly reduced content of PE in mitochondria (Trotter and
Voelker, 1995). Another mutant lacking PSD1 (pstB1) accumulates PS and shows diminished
PE formation because of a defect in translocating PS to PSD2 in Golgi/vacuoles (Trotter et
al., 1998). This mutation is complemented by a gene encoding a PI-4-kinase (Trotter et al.,
1998). Another PS transport mutant (pstB2) is complemented by a gene encoding a protein
that is homologous to Sec14, i.e. the yeast PI-TP (Wu et al., 2000). PS translocation to the site
of PSD2 seems to require Mn2+ and to be inhibited by EGTA in permeabilised yeast cells (Wu
and Voelker, 2001).
In summary, PS is probably translocated to mitochondria from MAM, which is tightly
associated with mitochondria. In electron microscopy (EM) pictures of yeast thin sections the
MAM and mitochondrial membranes were clearly distinct with no fusion sites detected, and a
distance less than 30 nm was considered association (Achleitner et al., 1999). Putative
proteins are thought to enhance the association. The mechanism by which the lipids are
translocated between the two apposed membranes is still unclear.

36
7. Phosphatidylserine translocation from outer mitochondrial membrane to inner
mitochondrial membrane
PSD is present only in the inner mitochondrial membrane (IMM) with its active site facing
the intermembrane space (Zborowski et al., 1983). It is, however, not clear if PS has to really
move into IMM to be decarboxylated (Voelker, 1989a; Jasinska et al., 1993; Trotter and
Voelker, 1994).
Three alternative models for intramitochondrial transfer of PS and PE have been proposed by
Jasinska et al. (1993) based on data from the authors themselves and others (Figure 5). Model
A proposes that contact sites between the IMM and outer mitochondrial membrane (OMM)
could form a closed system (Fig. 5a). This would explain why newly-formed PE is not mixed
with the IMM-PE in rat liver (Hovius et al., 1992) and yeast mitochondria (Simbeni et al.,
1990; Simbeni et al., 1993). (See section 8. for contradicting data regarding the fate of newly-
formed PE.)
In Model B (Fig. 5b) PS transfer from OMM to IMM takes place by spontaneous or protein-
mediated diffusion, which both are highly dependent on lipid hydrophobicity (see Figure 13
in Discussion). PS decarboxylation in isolated rat liver mitochondria was studied using
pyrene-labelled PS (PyrnPS). Various PyrnPS species were decarboxylated at a similar rate
irrespective of their hydrophobicity (Jasinska et al., 1993), thus contradicting Model B. In
addition, no evidence so far for a lipid transfer protein that would mediate the transfer
between OMM and IMM has been found in the intermembrane space of rat liver (Blok et al.,
1971) or yeast mitochondria (Simbeni et al., 1990).
It is possible that PS does not necessarily have to move into IMM to be decarboxylated, but
that PSD can reach to PS present at the inner leaflet of OMM, depicted by Model C (Fig. 5c).
It was concluded that both model A and C are probable, as both involve lateral diffusion,
which is quick and not dependent on lipid hydrophobicity (Jasinska et al., 1993). In both
models, PS has to first move to the inner leaflet of OMM, which is suggested to take place via
lateral diffusion at membrane pore sites in OMM (Jasinska et al., 1993). No evidence for the
presence of a flippase in OMM has been found (Sperka-Gottlieb et al., 1988).

37
Figure 5. Models for the intramitochondrial transfer of PS and PE. According to Model A, lipids move betweenOMM and IMM by lateral diffusion through so called hemi-fusion sites bridging the two membranes. In Model
B this movement is accounted by either lipid transfer proteins (LTP) or by spontaneous diffusion through theintermembrane space. Model C implies that the decarboxylation takes place while PS remains in the inner leafletof the outer membrane. The pores drawn to OMM may correspond those known to allow the pass-through ofvarious kinds of small molecules. OM, Outer membrane; IM, inner membrane; DC, phosphatidylserinedecarboxylase; LTP, lipid transfer protein. Reprinted with permission from (Jasinska et al., 1993).
Contact sites (reviewed in Moynagh, 1995) are areas where OMM and IMM are closely
associated, possibly the membranes transiently hemi-fused. They were first observed by
Hackenbrock in EM images of rat liver mitochondria (Hackenbrock, 1968). The existence of
contact sites is supported by the atypical behaviour of the fracture plane in freeze-fractured
mitochondria, which is characterised by frequent jumping of the fracture plane between the
adjacent membranes. Contact sites are enriched in PE and cardiolipin both in mouse liver
(Ardail et al., 1990) and yeast mitochondria (Simbeni et al., 1991). These lipids favour a
hexagonal non-bilayer structure (HII phase), which has been proposed to be involved in the
formation of hemi-fusion sites between OMM and IMM (van Venetië and Verkleij, 1982).
Contact sites seem to be involved in intramitochondrial translocation of PS in mouse liver
(Ardail et al., 1991) and yeast mitochondria (Simbeni et al., 1990). Contact sites are closely
associated with ER/MAM as seen in EM pictures of both thin sections of whole mouse liver

38
cells and of isolated mitochondria (Ardail et al., 1993). Dinitrophenol (DNP) inhibits PS
decarboxylation by decreasing the amount of contact sites (Knoll and Brdiczka, 1983;
Biermans et al., 1990) rather than by inhibiting PSD itself (Hovius et al., 1992). It has been
postulated that upon incorporation of the negatively charged DNP into the mitochondrial
membranes the intermembrane space is increased due to electrostatic repulsion between the
membranes (Bücheler et al., 1991), thus pulling the contact sites apart.
There is one report of PS accumulating into contact sites of mouse liver mitochondria when
PS decarboxylase was inhibited with hydroxylamine (Ardail et al., 1991). However, in rat
liver mitochondria treated with hydroxylamine, no PS segregation in OMM was found
(Jasinska et al., 1993). Neither was there found PS enrichment to contact sites in yeast
mitochondria treated with hydroxylamine (Simbeni et al., 1993). Even though PSD is found in
contact sites, it is not enriched as compared to IMM (Simbeni et al., 1991; Hovius et al.,
1992).
One study reported that adriamycin, which inhibits import of mitochondrial precursor proteins
to mitochondria, inhibits PS translocation to mitochondria in permeabilised CHO cells
(Voelker, 1991). This finding could not be reproduced with isolated rat liver mitochondria
(Jasinska et al., 1993) or yeast mitochondria (Simbeni et al., 1993). Therefore adriamycin
could actually inhibit PS translocation from ER/MAM to OMM rather than intramito-
chondrial PS translocation (Jasinska et al., 1993).
Recently, a CHO cell mutant with decreased cellular PE level was isolated, in which neither
PS synthesis nor decarboxylase activities were affected (Emoto et al., 1999). The
translocation-dependent decarboxylation in isolated mitochondria was defective, whereas
normal decarboxylation was observed in mechanically disrupted mitochondria. This suggests
that the intramitochondrial infrastructure was disturbed in this mutant and thus PS
translocation was defective (Emoto et al., 1999). This mutant could therefore be useful in
further studies of intramitochondrial PS translocation.

39
8. Phosphatidylethanolamine translocation from mitochondria to ER and other
destinations
What are the sites where the newly-formed PE will proceed from mitochondria? As IMM is
enriched in PE (Daum, 1985), it would be logical to assume that this PE would be derived
from PS. This seems to be the case in CHO cells, where the majority of mitochondrial PE is
derived from PS in situ, even when ethanolamine is supplied in the culture medium (Shiao et
al., 1995). Also in rat brain mitochondria IMM-PE derives from PS (Carlini et al., 1993;
Camici and Corazzi, 1995). However, it appears that newly-formed PE does not mix with pre-
existing PE of IMM but translocates rapidly to OMM in rat liver mitochondria (Hovius et al.,
1992) and in yeast (Simbeni et al., 1990; Simbeni et al., 1993). Also in mouse liver
mitochondria newly-formed PE is exported very rapidly to mitochondrial surface via contact
sites (Ardail et al., 1991).
Newly-made PE has been shown to be translocated to ER in isolated rat liver organelles
(Vance, 1991) and to the cell surface in rat hepatocytes (Vance et al., 1991). In BHK cells,
about half of the PE pulse-labelled from [3H]serine had moved from mitochondria to
microsomes during the 7.5 hour chase period (Voelker, 1985).
How could one explain these contradicting results regarding the intracellular translocation of
newly-synthesised PS-derived PE? It is possible that the fate of PE depends on the cell type
and on the contribution of the decarboxylation pathway to total PE synthesis. If most of the
PE is formed via decarboxylation, then PS-derived PE has to account for both mitochondrial
PE as well as that in other organelle membranes. If only a minority of PE is formed via
decarboxylation, as in rat brain (Butler and Morell, 1983), only IMM-PE is derived from
decarboxylation and OMM-PE derives from the Kennedy-pathway (Camici and Corazzi,
1995). It is likely that also the growth status of cells (actively growing/dividing vs.
resting/confluent cells) could affect the fate of PE, since an actively dividing cell obviously
needs more phospholipids than one in a stationary state.

40
9. Phosphatidylserine translocation from plasma membrane to mitochondria
Exogenous PS can be incorporated into CHO cells, as deduced from the facts that it
suppresses PS synthesis (Nishijima et al., 1986) and supports growth of mutant CHO cells
defective in PS synthesis (Kuge et al., 1986a; Voelker and Frazier, 1986). Part of the
exogenous PS is further converted to PE, implying that it is translocated to mitochondria
(Voelker and Frazier, 1986). It is unclear how hydrophobic PS molecules have entered the
cell in these studies where sonicated PS donor vesicles were incubated with cells. It is
possible that only the lyso-PS present in the PS preparation had moved to the cells.
Exogenously added fluorescent NBD12PS has been shown to incorporate to the plasma
membrane and then move to mitochondria and the Golgi apparatus. It was suggested that a
transfer protein would be responsible for this transfer (Kobayashi and Arakawa, 1991). This
conclusion was, however, based only on lack of effect with various transport inhibitors
(Kobayashi and Arakawa, 1991). Later, evidence was provided that NBD12PS is much less
hydrophobic than the typical natural PS species and can thus diffuse quite rapidly between
membranes without the assistance of transfer proteins (Tanhuanpää and Somerharju, 1999).
Also short-chain fluorescent dipyrene PS species rapidly distribute to intracellular membranes
including mitochondria (Kasurinen and Somerharju, 1995; Tanhuanpää and Somerharju,
1999). In contrast, a long-chain dipyrene PS species seemed to stay in the plasma membrane
as well as in the connected endosomal membranes in fluorescence imaging studies
(Tanhuanpää and Somerharju, 1999).
9.1. Plasma membrane aminophospholipid translocase
The plasma membrane of eukaryotic cells is asymmetric in terms of phospholipid
composition. Choline phospholipids (PC and SM) are enriched in the outer leaflet and the
aminophospholipids (PS and PE) in the inner leaflet (Devaux, 1991). The asymmetric
distribution of aminophospholipids is maintained by the plasma membrane
aminophospholipid translocase (flippase), that was first found by Seigneuret and Devaux
(1984) from erythrocytes. Thereafter this activity was detected in platelets, lymphocytes,
reticulocytes, endothelial cells, human fibroblasts, sperm cells, and yeast cells, as well as in
chromaffin granules (reviewed in Dolis et al., 1997). The flippase prefers PS, while PE is
translocated at a slower rate (Zachowski et al., 1986).

41
Evidence for a protein being responsible for aminophospholipid asymmetry comes from the
(i) translocation being inhibited by sulfhydryl modifying reagents (Daleke and Huestis, 1985;
Zachowski et al., 1986) and from selectivity for the substrate: (ii) Although both the L-serine
and D-serine analogues of PS are transported equally well, inversion of the stereochemistry of
the glycerol group abolishes translocation (Daleke and Lyles, 2000). (iii) Replacing the
diacylglycerol moiety of PS with ceramide reduces translocation 100-fold (Morrot et al.,
1989). (iv) In contrast, the flippase is relatively insensitive to the fatty acid composition, but
the presence of two fatty acids is required, as lyso-PS is translocated at a negligible rate
(Morrot et al., 1989).
CHO cell mutants defective in endocytosis-independent uptake of PS have been isolated, and
the defect has been postulated to be in the aminophospholipid translocase (Hanada and
Pagano, 1995; Endo et al., 1996). The cDNA encoding a Mg2+-ATPase was cloned from
bovine chromaffin granules (Tang et al., 1996). A highly homologous DRS2 gene was found
in yeast, and a drs2 null mutant was shown to be defective in aminophospholipid transport
(Tang et al., 1996). The potential role of the Drs2 protein in aminophospholipid transport was
however questioned because (i) no change in the uptake or distribution of NBD-PS and NBD-
PE was observed, and (ii) the distribution of endogenous PE in the plasma membrane was not
affected in the drs2 null mutant (Siegmund et al., 1998).

42
AIMS OF THE PRESENT STUDY
The overall goal was to study by which mechanism phosphatidyl-
serine translocates from ER and from plasma membrane to
mitochondria. To approach this goal we studied:
� The effect of hydrophobicity of phosphatidylserine on its
translocation from ER to mitochondria
� The effect of hydrophobicity of phosphatidylthreonine on its
translocation from ER to mitochondria
� The effect of hydrophobicity of phosphatidylserine on its
translocation from plasma membrane to mitochondria
In addition, the suitability of electrospray ionisation mass
spectroscopy in combination with stable isotope precursor labelling
for intracellular lipid trafficking studies was explored.

43
EXPERIMENTAL PROCEDURES
Sources of lipids and other reagents can be found in the original papers (I-III).
Cell culture
BHK-21 cells (C-13, CCL-10, from the American Type Culture Collection, Manassas, VA)
were cultured on plastic tissue culture dishes or flasks (Nunc, Roskilde, Denmark) in DMEM
(Gibco 41965) supplemented with 10 % fetal bovine serum, 2 mM glutamine, penicillin (200
units / ml) and streptomycin (200 �g / ml) under 5 % CO2 at 37 �C. Wild type CHO-K1 cells
and the CHO-82 mutant cells lacking the non-specific lipid transfer protein (van Heusden et
al., 1990) were provided by Dr. R. A. Zoeller (Boston University School of Medicine,
Boston, MA), and were grown in Ham’s F-12 medium (Gibco 21765) with the supplements
listed above. All cell culture media and supplements were from Gibco BRL (Life
Technologies Inc., San Diego, CA, USA). For labelling, cells were plated either on 5-cm
(diPyrnPS and [3H]serine labellings) or 14-cm (stable isotope labellings) dishes 2-3 days prior
to the experiment. A CO2-independent medium (I-MEM, Gibco 18045) was used when the
labelling and the chase were performed outside the CO2-incubator. All labelling and chase
media were serum-free. To deplete ATP, the cells were maintained in a glucose-free medium
(Gibco 11966) containing NaN3 and deoxyglucose.
Cell labelling procedures
Labelling with � 3H�-serine (I)
Confluent monolayers of BHK or CHO cells were preincubated in a serum-free medium for
12 hours, washed twice with PBS, and then labelled for 15 min at 37 �C in serine-free MEM
(Gibco 21090) containing 15 �Ci of �3H�-serine. In some experiments, �-chloroalanine (1
mM), an inhibitor of serine palmitoyltransferase (Medlock and Merrill, 1988; Chen et al.,
1993) was added in the medium two hours prior to the pulse. After the pulse, the cells were
washed twice with PBS and chased at 37 �C in serum-free medium containing serine (1 mM)
and ethanolamine (1 mM) for up to 22 h. They were then washed twice with PBS and scraped
into PBS.

44
Labelling with deuterium-serine or 13C-threonine (II)
Half-confluent BHK cells were incubated for 1 hour with 0.64 mM D3-L-serine in 10 ml
serine-free MEM (Gibco 21090), washed twice with PBS, and chased at +37°C in normal
medium without serum for 1-2 days. Alternatively, the cells were incubated in 10 ml serine-
free MEM containing 5.1 mM 13C4-L-threonine for up to 4 days. After these incubations the
cells were washed twice with PBS and once with 0.25 M sucrose and scraped into ice-cold
0.25 M sucrose.
Introduction of dipyrene phosphatidylserine species to the plasma membrane (III)
Two alternative methods were used to introduce diPyrnPS to the plasma membrane of BHK
cells. First, carboxyethyl-�-cyclodextrin (�-CD) was used to transfer the fluorescent PS to cells
at +37 °C (Tanhuanpää and Somerharju, 1999). The donor vesicles consisted of diPyrnPS and
POPC (1/9 molar ratio) plus a trace amount of [3H]cholesteryl ether (1,000,000 cpm) as a
nonexchangeable vesicle marker. The labelling time and the concentrations of diPyrnPS and �-
CD were adjusted depending on the diPyrnPS used as specified in Table 1 of III. The second
labelling method employed fusion of cationic donor vesicles with cells (Felgner et al., 1987;
Düzgünes et al., 1989). The donor vesicles consisted of diPyrnPS, DOPE and DOTMA (5 / 15
/ 20 nmol) and [3H]cholesterol ester (200,000 cpm). The labelling was performed at +8 °C for
1 h.
Control experiments in Triton X-100 micelles
PS decarboxylase (PSD) from rat liver (Dygas and Zborowski, 1989) was used because it was
impractical to obtain sufficient amounts of enzyme from BHK cells. It is very unlikely that
the specificities of the rat and hamster enzymes would differ significantly since mammalian
PS decarboxylase is highly conserved (see II and III). The buffer used in these micelle
experiments was 0.1 M phosphate (pH 7.4) containing 1.5 mM Triton X-100 and 1 mM
EDTA.
Decarboxylation of � 3H�phosphatidylserine species in Triton X-100-micelles (I)
�3H�-PS (approx. 450 nmol) was blown to dryness, phosphate buffer was added and the
mixture was sonicated for 4 min on a bath sonicator to assure complete dispersion of the lipid.

45
After addition of crude rat liver PSD (0.9 mg) solubilised in the same buffer, the reaction
mixture was incubated in a shaking water-bath at 37 �C and aliquots were removed at 5, 10,
15 and 20 min of incubation and mixed with chloroform-methanol (1:2). Lipids were then
extracted and �3H�-PS and �3H�-PE were separated with TLC as described below, except that
the TLC plate was first developed with acetone to remove �-mercaptoethanol (originating
from the PSD preparation) which interferes with the subsequent TNP-derivatisation step.
Decarboxylation of phosphatidylserine in Triton X-100 micelles (II)
The “PS” fraction was isolated from BHK cells by HPLC and pipetted to a silylated glass
tube. After removal of the solvents, 100 �l phosphate buffer was added and the suspension
was bath-sonicated for 4 minutes at 50 °C. Then 450 �g of crude rat liver PSD was added and
the reaction mixture was incubated at 37 °C. After various incubation times, aliquots were
taken to silylated glass tubes containing 1 ml of methanol, the lipids were extracted and the
“PE” and “PS” fractions were isolated by HPLC and then analysed by ESI-MS.
Determination of the substrate specificity of phosphatidylserine decarboxylase (III)
To test if PSD is sensitive to the chain length of diPyrnPS, the species (0.5 nmol) were
pipetted either separately or together into silylated glass tubes containing bovine brain PS
carrier (30 nmol). After removal of the solvents, 100 �l phosphate buffer was added and the
suspension was bath-sonicated for 4 minutes at 50 °C. Then 80 �g of crude rat liver PSD was
added and the reaction mixture was incubated at 37 °C. After various incubation times,
aliquots were taken to silylated glass tubes containing 1 ml of methanol and the lipids were
extracted and analysed by HPLC as detailed below.
Lipid extraction and molecular species composition analysis (I)
Bovine brain PS carrier (50 nmol/dish) was added to the cell pellet, and lipids were extracted
according to Bligh and Dyer (1959) using 2 % NaCl / 5 % acetic acid instead of water. PS and
PE were purified by TLC using chloroform / methanol / acetic acid / water (25:15:4:1) as
solvent, and the lipids were eluted from silica with chloroform / methanol / 0.2 M acetic acid
(1:1:0.2). To analyse the �3H�-labelling of PS and PE molecular species, these lipids were
converted to trinitrophenyl- (TNP-) derivatives (Hullin et al., 1989). The TNP-derivatives
were purified by TLC using chloroform / acetone / methanol / acetic acid / water

46
(10:4:2:2:0.5) as solvent and extracted from silica as above. TNP-PS and TNP-PE molecular
species were then separated on an Ultrasphere 4.6 * 250 mm ODS (5 �m) column (Beckman
Inc., CA, USA) using 30 mM choline chloride in methanol / 25 mm KH2PO4 / acetonitrile /
acetic acid (906:62:25:8) (Patton et al., 1982) as solvent. Fractions were collected and their
radioactivity was measured by liquid scintillation counting. The identity of the cellular PS and
PE molecular species was determined by comparing their relative retention times with TNP-
PE and -PS standards prepared from phosphatidylcholine species by phospholipase D -media-
ted transphosphatidylation (Comfurius et al., 1990). The identification was confirmed using
various other methods (see I).
Lipid extraction and analysis (II and III)
Lipids were extracted according to Folch et al. (1957). All extractions were performed in
silylated glass tubes using 0.58 % NaCl / 0.1 M HCl instead of water. This protocol was
found necessary to obtain a good recovery of PS. Incubation in the acidic solvent also
hydrolysed the alkenylether species of PE, which simplifies the MS spectra. For mass
spectroscopy, the extracts were subjected to HPLC fractionation on a Lichrosphere 100 Diol
column (Alltech, 250 x 4.6 mm, 5 µm particle size) equipped with a light scattering detector
(Silversand and Haux, 1997) and “PE” and “PS” fractions were collected into silylated screw
cap vials (Alltech, Deerfield, IL). For pyrene PS studies, the decarboxylation of diPyrnPS to
diPyrnPE was determined by HPLC using the same Diol column but with on-line fluorescence
detection.
Mass spectrometry and data analysis (II)
Mass-spectrometry was carried out with a PE Sciex API 300 or API 3000 triple quadrupole
instrument as described previously (Koivusalo et al., 2001). The different phospholipid
classes were selectively detected by using headgroup-specific neutral loss scanning (Brügger
et al., 1997). PE and PPA give the characteristic neutral losses of 141 and 155, respectively,
in the positive ion mode, while PS and PT were selectively detected using the neutral losses
of 87 and 101, respectively, in the negative ion mode. PS species labelled from D3-serine
were detected using neutral loss of 90 and 13C4-threonine labelled PT using neutral loss of
105. PPA derived from 13C4-PT gives the neutral-loss of 158, rather than 159 because one of

47
the four 13C-atoms of the threonine headgroup is lost upon decarboxylation. Isotope peak and
sodium adduct subtractions were made when necessary (see II).
The hydrophobicity (effective carbon number) of the different molecular species was
determined from their retention times on a reverse-phase column (cf. Patton et al., 1982).
This method indicated that addition of one, two, three, four, five and six double bonds to the
acyl chain decreases the effective carbon number of an acyl chain by 1.8, 3.1, 4.4, 5.7, 6.9 and
8.2 units, respectively.
Lipid compositions of the subcellular membranes were determined by quantitative ESI-MS
using internal standards. PC and SM were detected by scanning for the precursors of 184 in
the positive ion mode, PI by scanning for the precursors of 241 in the negative ion mode, and
PE and PS by scanning for the neutral loss 141 and 185, respectively in the positive ion mode
(Brügger et al., 1997).
Cell fractionation and enzyme assays (II)
Unlabeled confluent BHK cells (thirty 14-cm dishes) were fractionated essentially as
described (Shiao et al., 1995). Mitochondria were further fractionated into inner and outer
membranes as described (Hovius et al., 1990), except that the digitonin concentration was
increased to 1.35 mg / mg mitochondrial protein to maximally remove the outer membrane
and thus ensure a high purity of the inner membrane fraction.
Membrane fractions were assayed for the following marker enzymes using published
procedures: monoamine oxidase (EC 1.4.3.4) (Weissbach et al., 1960), cytochrome c oxidase
(EC 1.9.3.1) (Storrie and Madden, 1990), NADPH cytochrome c reductase (EC 1.6.2.5)
(Roberts et al., 1991) and �-glucosidase (EC 3.2.1.21) (van Hoof and Hers, 1968).
Phosphatidylserine synthase activity was measured as described previously (Rakowska et al.,
1997), except that the extraction was performed at room temperature in the presence of MgCl2
(Trotter et al., 1998).

48
Measurement of spontaneous intervesicle translocation rates in vitro (III)
The spontaneous intervesicle translocation rates of diPyr12PS and 18:0/18:1-PS were
determined by using a previously described assay (Jones and Thompson, 1989). Briefly,
negatively charged donor vesicles consisting of POPC, POPG, diPyr12PS and [3H]18:0/18:1-
PS (850 / 150 / 1 / 10 nmol) and 50,000 cpm [14C]-cholesterol oleate, and uncharged acceptor
vesicles consisting of POPC only were prepared in 10 mM Hepes – 25 mM KCl – 0.5 mM
EDTA, pH 7.0 by probe sonication. Donor and acceptor vesicles (1 and 10 �mol total lipid,
respectively) were coincubated at 37 °C in the dark, aliquots were withdrawn at various time
points and applied to DEAE-Sephacel (Amersham Pharmacia Biotech, Uppsala, Sweden)
minicolumn to trap the negatively charged donors. The eluent was then analysed for
diPyr12PS and [3H]18:0/18:1-PS content by measuring the pyrene excimer fluorescence
intensity or radioactivity, respectively. The leakage of donor vesicles from the column, as
estimated by the amount of [14C]-radioactivity in the eluent, was minor (1.8 ± 1.4 %).

49
RESULTS
Translocation of phosphatidylserine molecular species from ER to mitochondria
Phosphatidylethanolamine is more “hydrophilic” than its precursor phosphatidylserine (I)
Most, or even all PE is derived from PS decarboxylation in BHK and CHO cells (see section
1.5). Thus it is striking to find that the molecular species composition of PE is very different
from that of PS. In BHK-PS the major species are 18:0/18:1 (peak 15), 18:1/18:1 (peak 8) and
16:0/18:1 + 16:1/18:0 (peak 7) which constitute about 43, 20 and 17 % of all PS species,
respectively (Fig. 6, upper panel). Notably, there are only minor amounts of the rapidly
eluting species (peaks 1-6). In BHK-PE those rapidly eluting species are relatively much more
abundant than in PS, while the reverse is true for the slowly eluting ones (Fig. 6, bottom
panel). Analogous differences in mass distribution of PS and PE were found in CHO cells
(not shown). Below, the rapidly eluting species will be referred as ”hydrophilic” and to the
slowly eluting species as ”hydrophobic” for convenience.
Figure 6. Reverse phase HPLC analysis of PS and PE molecular species from BHK cells. BHK cells were grownto confluence in a complete medium and harvested into PBS with a cell scraper. The lipids were extracted, PSand PE fractions isolated with TLC, converted to TNP-derivatives and separated into molecular species on a C18reverse phase column. The detection was at 340 nm. The retention times were normalised using the 18:0/18:1peak as the reference. RRT = relative retention time. The major molecular species eluting in each peak are listedin Table 1 of (I). Upper panel: PS molecular species. Bottom panel: PE molecular species. Plasmalogens aremarked with an asterix. (Figure 1 in I)

50
Preferential translocation of hydrophilic phosphatidylserine species to mitochondria (I)
Pulse-chase labelling studies with [3H]serine showed that the distribution of radioactivity in
both PS and PE molecular species is similar to the distribution of molecular species in natural
lipids (Figure 2 in I). Extensive control studies were performed to exclude the possibility that
the differences in the [3H]PS and [3H]PE profiles would be due to (i) incorporation of
[3H]label to PE via the sphingosine-phosphoethanolamine pathway (Figure 3 in I), (ii)
extensive incorporation of [3H]label to the diglycerol moiety of PS and PE (Figure 4 in I),
(iii) differences in degradation or remodelling among species (Figure 5 in I), or (iv) selectivity
of the PS decarboxylase enzyme towards specific PS species (Figure 7 in I). Thus the
differences in the [3H]PS and [3H]PE profiles indeed appear to be caused by faster
decarboxylation of the hydrophilic PS species. This, in turn, suggests that hydrophilic species
are translocated from ER/MAM to mitochondria more rapidly than the hydrophobic ones,
since it has been shown that transfer to mitochondria rather than decarboxylation itself is rate-
limiting (Voelker, 1989a).
The kinetics of phosphatidylserine translocation to mitochondria has two components (I)
Figure 7 shows the decay of radioactivity of different �3H�PS peaks during a 7.5-hour chase.
Note that some peaks that are minor or not well separated have been grouped together. The
plots indicate that, in each case, there are apparently two components of decay, a fast and a
slow one. However, the fractional contribution of these components varies significantly. For
instance, the fast component accounts for 20, 12 and 2.5% of the total decay in peak groups 1-
4, 7-9 and 14-15, respectively. Also the half-time of the slow component varies significantly
being approx. 10, 18 and 32 h, respectively. It was concluded that there are two, kinetically
distinct pools of PS molecules in BHK cells: one which consists predominantly of the more
hydrophilic PS species and is characterised by a rapid conversion to PE, and another that
consists mostly of the more hydrophobic species which are converted to PE much more
slowly. For the total PS pool, the half-times of the fast and slow components were determined
to be 22 minutes and 28 hours, respectively, and approximately 12 % of the total pool of the
newly-synthesised PS molecules was estimated to be transported to mitochondria via the fast
process in BHK cells. These data are consistent with previous studies suggesting, in a more
qualitative manner, the presence of two pools of PS with different decarboxylation kinetics in
cultured cells (Bjerve, 1985; Vance, 1991; Shiao et al., 1995).

51
2 4 6
0.6
0.7
0.8
0.9
peaks 14-15
T1 T2 - 32.1 hF1 F22.5 % 97.5 %
[3 H]-
PS
/ (
[3 H]-
PS
+ [
3 H]-
PE
)
CHASE TIME (h)
0.6
0.7
0.8
0.9 peaks 10-11 T1 T2 - 23.7 hF1 F29 % 91 %
0.5
0.6
0.7
0.8
peaks 7-9T1 T219 min 18.3 hF1 F212 % 88 %
0.4
0.5
0.6
0.7 peaks 1-4 T1 T225 min 10.3 hF1 F220 % 80 %
Figure 7. Analysis of the decarboxylation kineticsof PS species. BHK cells were labelled 15 minuteswith �3H�serine and chased for up to 7.5 hours.Since all peaks were not adequately separated, theywere pooled as indicated. The fraction of�
3H�radioactivity in PS is plotted as a function ofchase time. The data was analysed by employingthe two-exponential decay model, as it gavesignificantly better fits than the single exponentialmodel. The fractional contributions and half-timesobtained for the fast (F1, T1) and slow (F2, T2)decay components are shown in the panels. Forsome peaks the half-time of the fast componentcould not be determined due to paucity of relevantdata points. (Figure 6 in I)
Mass spectroscopic studies of phosphatidylserine molecular species translocation to
mitochondria (II)
Electrospray ionisation mass spectroscopy (ESI-MS) allows a more accurate analysis on the
effect of hydrophobicity than the reverse phase HPLC method used above. By use of
headgroup specific scanning modes nearly all individual molecular species can be quantified.

52
BHK cells labelled with D3-serine showed an inverse exponential correlation between the D3-
PE/D3-PS ratio and the calculated molecular hydrophobicity, i.e. the effective carbon number
(Fig 8a, inset). The more hydrophilic species are far more prominent in D3-PE as compared to
its precursor D3-PS. The same is true for unlabelled species as well (Fig. 8b). As PE is derived
from PS by decarboxylation in mitochondria, these results suggest that translocation of PS to
mitochondria is inversely correlated with molecular hydrophobicity.
10
20
30 A
D3-PS
D3-PE30 32 34 36
0.5
1.0
1.5
2.0
D3-P
E /
D3-P
S
Effective CN
38
:6
33
:2
40
:7
37
:5
35
:3
32
:1
36
:4
34
:2
38
:5
36
:3
33
:1
37
:4
40
:6
35
:2
32
:0
34
:1
38
:4
36
:2
37
:3
33
:0
40
:5
35
:1
42
:6
38
:3
37
:2
34
:0
36
:1
40
:4
38
:2
35
:0
37
:1
40
:3
36
:0
38
:1
40
:2
40
:1
42
:2
42
:1
10
20
30
40
B
Molecular species in order of increasing hydrophobicity
PS
PE30 32 34 36
5
10
15
20
PE
/ P
S
Effective CN
Figure 8. The PE/PS ratio decreases as a function of lipid hydrophobicity in BHK cells. Molecular speciesprofiles are shown for D3-PS and D3-PE (Panel A), and unlabelled PS and PE (Panel B). The D3-PE/D3-PS-ratiois plotted in the inset of panel A as function of increasing molecular hydrophobicity, i.e., the effective carbonnumber (CN); see Experimental Procedures. The PE/PS ratio is similarly plotted in the inset of panel B. (Part ofFigure 4 in II)
Also the lack of selectivity of the PSD enzyme towards different PS molecular species was
confirmed by ESI-MS studies. The BHK cell PS fraction was incubated with rat liver PSD in
Triton X-100 micelles. After a 50-min incubation more than 60 % of the total PS pool was
decarboxylated. The relative concentration of nearly all PS species remained essentially
constant at all time points (Figure 6 in II), indicating that the decarboxylase is not sensitive to
the acyl chain structure of PS.
% o
f m
ole
cu
lar
sp
ecie
s s
um

53
Translocation of phosphatidylthreonine molecular species from ER to mitochondria
Identification of phosphatidylisopropanolamine, a decarboxylation product of phosphatidyl-
threonine (II)
In accordance with previous data (Mark-Malchoff et al., 1978), significant amounts of phos-
phatidylthreonine (PT) were found in BHK cells by use of ESI-MS and head group -specific
neutral loss scanning. The decarboxylation product of PT, phosphatidylisopropanolamine
(PPA), has not been described so far as an intrinsic component of biological membranes. To
study whether PPA could be produced from PT in BHK cells, a head group -specific neutral
loss specific for isopropanolamine was used to detect PPA. A small amount of PPA was
found in BHK cells (Fig. 9b). The identification was confirmed by presence of 13C3-PPA in13C4-threonine-labelled cells (Fig. 9c). Thus BHK cells indeed contain PPA, the decarboxyla-
tion product of PT. The concentration of PT can reach or even exceed that of PS in BHK
cells. In contrast, the concentration of PPA in growing cells is low, only about 1 % of that of
diacyl PE. The amount of both PT and PPA increases when the cultures age or if the cells are
kept in serine-free medium, indicating that their production increases upon serine deprivation.
700 725 750 775 800
714 739 764 789 814
PEA
PPAB
717 742 767 792 817
13C-PPAC
717 742 767 792 817
ControlD
Figure 9. ESI-MS spectra obtained from BHK “PE” fraction. BHK cells were labelled with 5.1 mM 13C4-threonine for 3 days, lipids were extracted and the “PE” fraction was isolated with HPLC and then subjected toscans specific for PE (NL 141, panel A), PPA (NL 155, panel B) and 13C3-PPA (NL 158, panel C). Panel Dshows the spectrum (NL 158) for cells incubated identically but in the absence of 13C4-threonine. The y-axismaxima are 200,000 (panel A); 2,500 (panel B) and 3,500 cps (panels C and D). Note that the mass axis scaleshave been adjusted to ease comparison. (Figure 2 in II)
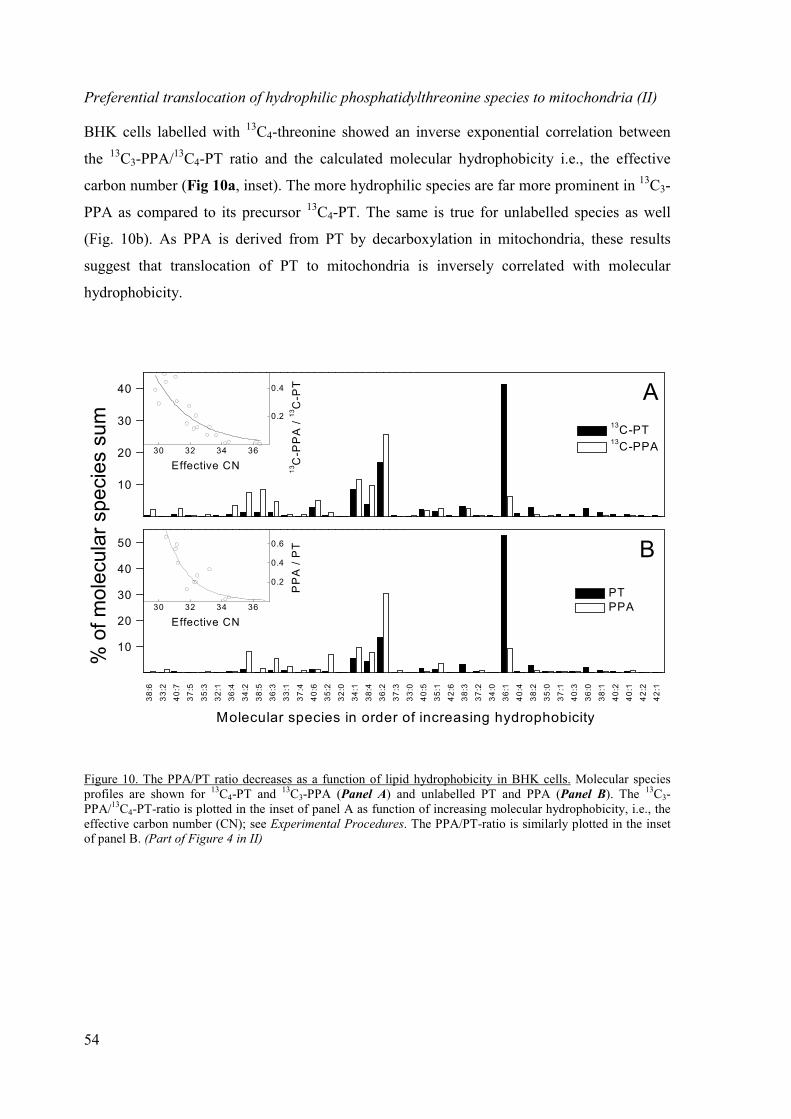
54
Preferential translocation of hydrophilic phosphatidylthreonine species to mitochondria (II)
BHK cells labelled with 13C4-threonine showed an inverse exponential correlation between
the 13C3-PPA/13C4-PT ratio and the calculated molecular hydrophobicity i.e., the effective
carbon number (Fig 10a, inset). The more hydrophilic species are far more prominent in 13C3-
PPA as compared to its precursor 13C4-PT. The same is true for unlabelled species as well
(Fig. 10b). As PPA is derived from PT by decarboxylation in mitochondria, these results
suggest that translocation of PT to mitochondria is inversely correlated with molecular
hydrophobicity.
10
20
30
40 A
13C-PT
13C-PPA30 32 34 36
0.2
0.4
13C
-PP
A /
13C
-PT
Effective CN
38
:6
33
:2
40
:7
37
:5
35
:3
32
:1
36
:4
34
:2
38
:5
36
:3
33
:1
37
:4
40
:6
35
:2
32
:0
34
:1
38
:4
36
:2
37
:3
33
:0
40
:5
35
:1
42
:6
38
:3
37
:2
34
:0
36
:1
40
:4
38
:2
35
:0
37
:1
40
:3
36
:0
38
:1
40
:2
40
:1
42
:2
42
:110
20
30
40
50
Molecular species in order of increasing hydrophobicity
B
PT
PPA30 32 34 36
0.2
0.4
0.6
PP
A /
PT
Effective CN
Figure 10. The PPA/PT ratio decreases as a function of lipid hydrophobicity in BHK cells. Molecular speciesprofiles are shown for 13C4-PT and 13C3-PPA (Panel A) and unlabelled PT and PPA (Panel B). The 13C3-PPA/13C4-PT-ratio is plotted in the inset of panel A as function of increasing molecular hydrophobicity, i.e., theeffective carbon number (CN); see Experimental Procedures. The PPA/PT-ratio is similarly plotted in the insetof panel B. (Part of Figure 4 in II)
% o
f m
ole
cu
lar
sp
ecie
s s
um

55
Phosphatidylthreonine is a poor substrate for phosphatidylserine decarboxylase (II)
The concentration of PPA in BHK cells remained low even when the concentration of its
precursor, PT, exceeded that of PS. This was shown to be due to PT being a poor substrate for
the decarboxylase as compared to PS. In Triton X-100 micelles containing PS decarboxylase
the PS/PT-ratio fell to 1/20 during the 180 min-incubation (Figure 5 in II). Although PT was
a poor substrate for PS decarboxylase, it did not accumulate to IMM in large amounts. The
PT/PS-ratio in IMM was approximately double as compared to the PT/PS-ratio in the
postnuclear supernatant and other organelles, but the amounts of both PS and PT were
extremely low, 0.7 % and 1.2 % of total phospholipids of IMM, respectively (Table 1 in II).
Translocation of phosphatidylserine species from plasma membrane to mitochondria
Preferential translocation of short-chain dipyrene phosphatidylserine species from plasma
membrane to mitochondria (III)
Different dipyrene PS (diPyrnPS, n = 4-14) species were introduced to the plasma membrane
of BHK cells. The rate of diPyrnPS decarboxylation decreased strongly with increasing acyl
chain length, irrespective of whether vesicle fusion or �-CD-mediated transfer was used to
introduce the labelled lipids to cells (Fig. 11a,b). The deviating behaviour of diPyr4PS is
probably due to that its translocation to mitochondria is so fast (> 5 % decarboxylated already
during the 1 minute labelling period) that decarboxylation becomes rate-limiting. This is
unlikely to occur with the more hydrophobic species because for them the rate of
decarboxylation exceeds the rate of translocation. As seen in Figure 11c, diPyr4PS is a poor
substrate for PS decarboxylase. This unnaturally short PS species was decarboxylated about 9
times slower than DiPyr14PS in Triton micelles. Although it is not possible to accurately
correct the transfer rate obtained for diPyr4PS based on the data in Fig. 11c, it is likely that
such a correction would place the diPyr4PS data point on the dashed line drawn in Fig. 11a,b.
In conclusion, it seems that the rate of diPyrnPS transport from the plasma membrane to
mitochondria decreases in a nearly logarithmic manner with increasing acyl chain length.
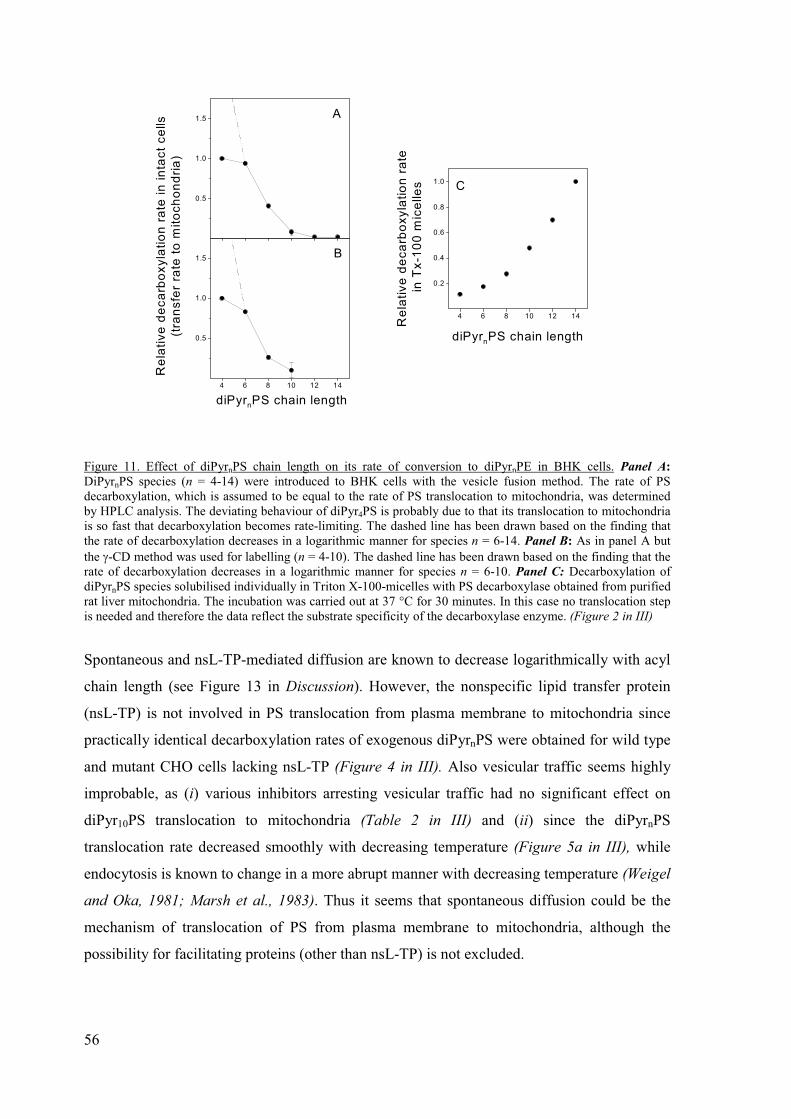
56
4 6 8 10 12 14
0.5
1.0
1.5 B
diPyrnPS chain length
Re
lati
ve
de
ca
rbo
xyla
tio
n r
ate
in
in
tac
t c
ells
(tra
nsfe
r ra
te t
o m
ito
ch
on
dri
a)
0.5
1.0
1.5 A
4 6 8 10 12 14
0.2
0.4
0.6
0.8
1.0
Re
lativ
e d
eca
rbo
xy
latio
n r
ate
in T
x-1
00
mic
elle
s C
diPyrnPS chain length
Figure 11. Effect of diPyrnPS chain length on its rate of conversion to diPyrnPE in BHK cells. Panel A:
DiPyrnPS species (n = 4-14) were introduced to BHK cells with the vesicle fusion method. The rate of PSdecarboxylation, which is assumed to be equal to the rate of PS translocation to mitochondria, was determinedby HPLC analysis. The deviating behaviour of diPyr4PS is probably due to that its translocation to mitochondriais so fast that decarboxylation becomes rate-limiting. The dashed line has been drawn based on the finding thatthe rate of decarboxylation decreases in a logarithmic manner for species n = 6-14. Panel B: As in panel A butthe �-CD method was used for labelling (n = 4-10). The dashed line has been drawn based on the finding that therate of decarboxylation decreases in a logarithmic manner for species n = 6-10. Panel C: Decarboxylation ofdiPyrnPS species solubilised individually in Triton X-100-micelles with PS decarboxylase obtained from purifiedrat liver mitochondria. The incubation was carried out at 37 °C for 30 minutes. In this case no translocation stepis needed and therefore the data reflect the substrate specificity of the decarboxylase enzyme. (Figure 2 in III)
Spontaneous and nsL-TP-mediated diffusion are known to decrease logarithmically with acyl
chain length (see Figure 13 in Discussion). However, the nonspecific lipid transfer protein
(nsL-TP) is not involved in PS translocation from plasma membrane to mitochondria since
practically identical decarboxylation rates of exogenous diPyrnPS were obtained for wild type
and mutant CHO cells lacking nsL-TP (Figure 4 in III). Also vesicular traffic seems highly
improbable, as (i) various inhibitors arresting vesicular traffic had no significant effect on
diPyr10PS translocation to mitochondria (Table 2 in III) and (ii) since the diPyrnPS
translocation rate decreased smoothly with decreasing temperature (Figure 5a in III), while
endocytosis is known to change in a more abrupt manner with decreasing temperature (Weigel
and Oka, 1981; Marsh et al., 1983). Thus it seems that spontaneous diffusion could be the
mechanism of translocation of PS from plasma membrane to mitochondria, although the
possibility for facilitating proteins (other than nsL-TP) is not excluded.

57
The spontaneous translocation rate of diPyr12PS is similar to that of 18:0/18:1-PS (III)
To act as good probes, fluorescent phospholipid analogues should have similar hydro-
phobicity and spontaneous diffusion rates as their natural counterparts. The hydrophobicity of
diPyr12PS and that of the most abundant species of natural PS, 18:0/18:1-PS, are similar as
estimated from reverse phase HPLC retention times (Tanhuanpää and Somerharju, 1999). It
was further investigated whether also their spontaneous diffusion rates would be similar, and
the spontaneous intervesicle translocation rate of diPyr12PS was found to be similar to that of
18:0/18:1-PS (Figure 12) as was shown by using an in vitro transfer assay (Jones and
Thompson, 1989). Both PS species were incorporated to the same donor vesicles to exclude
effects due to possible variation of the vesicle structure. The spontaneous translocation rate of
diPyr12PS is virtually identical to that of 18:0/18:1-PS (half-times 349 ± 22 and 323 ± 8 hours,
respectively). Thus the long-chain diPyrnPS species should report correctly on trafficking
properties of the natural PS species.
10 20 30 40 50
0.02
0.04
0.06
0.08
0.10
diP
yr 1
2P
S o
r [
3H
]-P
S in
ac
ce
pto
rs
Time (h)
Figure 12. Spontaneous intervesicle translocation rate of diPyr12PS and 18:0/18:1-PS. Donor vesicles containingboth diPyr12PS (open symbols) and [3H]-18:0/18:1-PS (closed symbols) were incubated with unlabelled acceptorvesicles for the indicated times, after which the acceptors were isolated using an anion-exchange column andtheir diPyr12PS and [3H]-18:0/18:1-PS contents were analysed based on pyrene excimer fluorescence intensityand radioactivity, respectively. The y-axis indicates the fraction of labelled PS species found in the acceptors.Contamination of the acceptor fraction by donor vesicles was 1.85 ± 1.47 % as estimated based on the content of[14C]-cholesterol oleate, the non-exchangeable donor marker. The lines represent fits of a single exponentialequation to the diPyr12PS data (dashed line) and [3H]-18:0/18:1-PS data (solid line). It was assumed that 67 % ofboth PS species was available for transfer, i.e. present in the outer leaflet of the donor vesicles (cf. Wimley andThompson, 1990). (Figure 3 in III)

58
ESI-MS and stable isotope -labelled precursors in lipid trafficking studies (II)
Our study has also important methodological relevance. Stable isotope -labelling in combina-
tion with ESI-MS has not been used for studying intracellular lipid trafficking before. This
approach has many advantages over conventional chromatographic methods. First of all, it is
possible to determine both the unlabelled and labelled molecular species simply by using
specific neutral loss values. Second, the specific labelling can be determined with a 3-5 orders
of magnitude higher sensitivity than with radioactive labelling. Third, quantification of the
molecular species as well as their labelling is far more rapid and convenient with ESI-MS as
compared to chromatographic methods. Fourth, nonspecific labelling due to metabolism of
the precursor does not hamper analysis, since the neutral loss scanning detects only the
molecules which have incorporated the labelled precursor to their head group. Finally, the
high specificity of the ESI-MS detection allows the use of multiple precursors simultaneously.
The drawback of the ESI-method is the requirement for an expensive instrument.

59
DISCUSSION
Effect of lipid hydrophobicity on its rate of translocation depends on the mechanism of
translocation
In theory, any of the following mechanisms could be involved in the translocation of PS from
other organelles to mitochondria: (i) vesicular transport, (ii) spontaneous or (iii) protein-
assisted diffusion via the cytoplasm or (iv) lateral diffusion via membrane fusion (or
hemifusion) sites. One possibility to distinguish between these alternative mechanisms is to
determine the effect of lipid hydrophobicity on the rate of translocation (Figure 13). First, it is
very likely that vesicular transport does not depend on the hydrophobicity of the lipid to be
transferred. Also transport relying on lateral diffusion via membrane (hemi)fusion sites should
not depend significantly on lipid hydrophobicity (Derzko and Jacobson, 1980). In contrast, an
optimum regarding the hydrophobicity of the lipid to be transferred has been observed for the
mammalian phospholipid carrier proteins (Somerharju et al., 1987; van Paridon et al., 1988;
Kasurinen et al., 1990). On the other hand, spontaneous diffusion via an aqueous phase is
known to decrease logarithmically with acyl chain length because the efflux of the lipid from
the donor membrane is the rate-limiting step (McLean and Phillips, 1981; Massey et al.,
1982a; Massey et al., 1984; Silvius and Leventis, 1993). The ability of the nonspecific lipid
transfer protein (nsL-TP) to enhance (phospho)lipid transfer also decreases logarithmically
with lipid hydrophobicity (Nichols and Pagano, 1983; van Amerongen et al., 1989).
carrier protein
spontaneous or
nsL-TP mediated
diffusion
tra
nslo
cation r
ate
vesicle transport
or lateral diffusion
lipid hydrophobicity
Figure 13. Effect of lipid hydrophobicity on the different mechanisms of intermembrane lipid transfer. Thecurves are based either on theoretical predictions or experimental data (see text). It is important to note that ifseveral mechanisms operate simultaneously, the observed hydrophobicity dependency can be a combination ofthe curves shown here, depending on the relative importance of the different mechanisms. However, spontaneoustranslocation of the less hydrophobic species will always occur and will thus contribute to the observedhydrophobicity dependence. (Figure 6 in III)

60
The effect of PS hydrophobicity on its translocation rate was used in these studies to find out
by which mechanism PS is translocated from ER/MAM and plasma membrane to
mitochondria.
Mechanism of phosphatidylserine translocation from ER to mitochondria
It was found that translocation of PS from ER to mitochondria is strongly dependent on
molecular hydrophobicity. The hydrophilic PS species are rapidly translocated to
mitochondria where they are converted to PE, whereas the hydrophobic PS species are
translocated in much slower rate (I, II). This suggests that the rate-limiting step in the overall
translocation process is the desorption of the PS molecule from the ER/MAM membrane,
which in turn indicates that spontaneous translocation via cytoplasm is the translocation
mechanism. This result may seem unexpected since translocation of natural phospholipids by
spontaneous efflux is generally considered as a very slow process (half-time tens of hours).
However, the rate of spontaneous translocation can increase remarkably at higher membrane
concentrations. It has been shown that the half-time for transfer of palmitoyl-oleoyl-PC
between lipid vesicles decreased from 46 to 7 hours when the acceptor vesicle concentration
was increased from 2.5 to 40 mM (Jones and Thompson, 1990). The latter half-time is similar
to that for the transport of newly-synthesised PS from ER/MAM to mitochondria (Voelker,
1985; Vance and Vance, 1988, article I). The enhancement of transfer at higher membrane
concentrations has been attributed to stabilisation of the transition-state monomers by their
interaction with the acceptor membrane (Jones and Thompson, 1990). According to
morphological (Franke and Kartenbeck, 1971; Morré et al., 1971; Lewis and Tata, 1973;
Meier et al., 1981) and biochemical studies (Vance, 1990; Gaigg et al., 1995) the ER/MAM
and the outer mitochondrial membranes are closely associated. This means that the local
concentration of the donor and acceptor membranes is very high, which should favour
collision-enhanced intermembrane monomer diffusion.
The results indicate that PS transfer from ER/MAM to mitochondria would occur by
spontaneous diffusion. However, mitochondrial (Camici and Corazzi, 1997; Shiao et al.,
1998; Achleitner et al., 1999) and soluble proteins (Kuge et al., 2001) have been shown to
participate in PS translocation to mitochondria. The involvement of proteins does not by any
means rule out spontaneous diffusion, or vice versa. The role of protein(s) could be that PS-

61
binding protein(s) present on mitochondrial surface preferentially bind regions of MAM
enriched in PS (Shiao et al., 1998).
Mechanism of phosphatidylthreonine translocation from ER to mitochondria
The concentration of PPA in BHK cells remained low even when the concentration of its
precursor, PT, exceeded that of PS (II). The reason for this seems to be that that PT is a poor
substrate for the PS decarboxylase (Figure 5 in II). Alternatively, PT might not translocate
efficiently to mitochondria. This latter possibility seems unlikely since the PT content of IMM
was higher than that of PS (Table 1 in II). On the other hand, if PT indeed moves efficiently
to mitochondria, why does it not, being a poor substrate for the decarboxylase, accumulate in
IMM? A feasible explanation is that the high negative charge of the IMM bilayer, due to the
presence of high concentrations of cardiolipin in this membrane (Daum, 1985), repels PT
which is also negatively charged. Thus, even if PT would readily enter IMM, its steady-state
concentration therein would remain low due to its fast exit back to ER.
In conclusion, phosphatidylthreonine is translocated to mitochondria but is poorly decarboxy-
lated to phosphatidylisopropanolamine. The preference of PS decarboxylase for PS over PT is
reminiscent of that of PS synthase (Mitoma et al., 1998b).
Mechanism of phosphatidylserine translocation from plasma membrane to mitochondria
Like translocation from ER/MAM to mitochondria, PS translocation from the plasma
membrane to mitochondria is also strongly dependent on its hydrophobicity (III), indicating
that the desorption from a membrane surface is the rate-limiting step of translocation.
Therefore, it seems that this translocation occurs by spontaneous diffusion. This putative
mechanism is supported by the high value obtained for diPyr8PS activation energy (85
kJ/mol), typical for desorption of both natural (McLean and Phillips, 1984; Wimley and
Thompson, 1990) as well as pyrene (Massey et al., 1982a; Tanhuanpää et al., 2001) lipids
from a bilayer.
There are several reasons to believe that the data obtained with diPyrnPS is relevant for
natural PS species as well. First, even though the pyrene moiety is relatively bulky, it actually
only slightly increases the effective surface area of the labelled lipid molecule (Somerharju et

62
al., 1985). Second, pyrene does not significantly distort the conformation of the acyl chain or
the whole molecule (Eklund et al., 1992; Sassaroli et al., 1995). Third, pyrene lipids are
metabolised similarly to the natural ones (Naylor et al., 1991; Kasurinen and Somerharju,
1992, 1995). Fourth, pyrene-labelled lipids are good substrates for phospholipid carrier
proteins (Somerharju et al., 1987; van Paridon et al., 1988) as well as for nsL-TP (van
Amerongen et al., 1989). Finally, and most importantly, the spontaneous intermembrane
diffusion rates of the long-chain diPyrnPS species (n = 8-14) appear to be very similar to that
of typical natural species. This was confirmed by showing that the spontaneous
intermembrane translocation rate of diPyr12PS is virtually identical to that of 18:0/18:1-PS,
the most common PS species in BHK cells (Figure 12). One can estimate that the range of
spontaneous translocation rates of diPyr8PS - diPyr14PS species covers well that of the typical
natural species (cf. Tanhuanpää and Somerharju, 1999).
The results suggest that PS transfer from plasma membrane to mitochondria is likely to occur
by spontaneous diffusion. However, there could be proteins bringing the plasma membrane
and mitochondria temporarily to close proximity, thereby enhancing PS efflux from the
former to the latter. Again, the possible involvement of proteins would not rule out
spontaneous diffusion.
Maintenance of high phosphatidylserine concentration in the inner leaflet of plasma
membrane
It has not been clear how the cell achieves and maintains the high PS content, up to one third
of all phospholipids, in the inner leaflet of its plasma membrane. The following tentative
model that could explain this is proposed (Figure 14). PS is synthesised in ER/MAM, from
where a fraction of the newly-synthesised PS molecules rapidly moves to mitochondria (thick
black arrow) where it is decarboxylated to PE. Notably, of the newly-synthesised PS species
the more hydrophilic (i.e., species having shorter and/or more unsaturated acyl chains) ones
move to mitochondria much more rapidly than the hydrophobic ones. As a result, the PS
species remaining in the ER/MAM are, on the average, more hydrophobic than those initially
synthesised. Many of these PS molecules obviously incorporate to exocytic vesicles and are
thus transported to the plasma membrane. During this transport and after their arrival to the
plasma membrane the still remaining hydrophilic PS species tend to diffuse to mitochondria
(thin arrows) either directly or via other organelles, such as endosomes and ER. Supporting
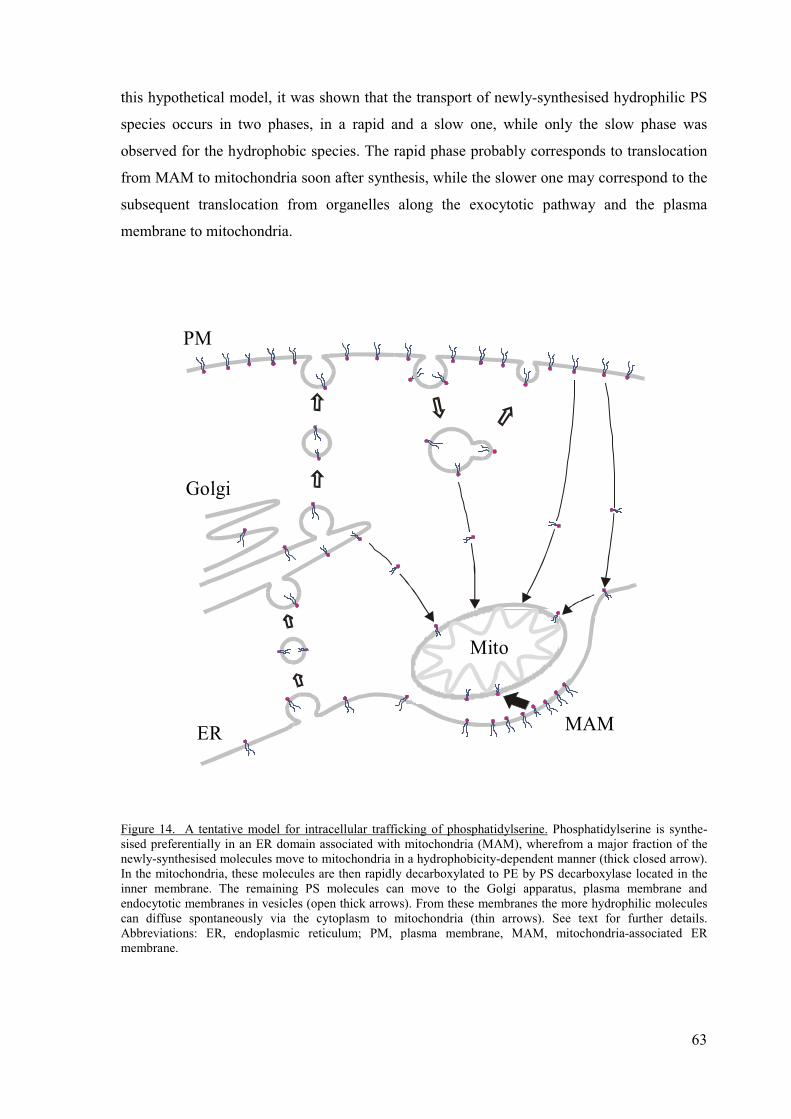
63
this hypothetical model, it was shown that the transport of newly-synthesised hydrophilic PS
species occurs in two phases, in a rapid and a slow one, while only the slow phase was
observed for the hydrophobic species. The rapid phase probably corresponds to translocation
from MAM to mitochondria soon after synthesis, while the slower one may correspond to the
subsequent translocation from organelles along the exocytotic pathway and the plasma
membrane to mitochondria.
PM
Golgi
ER
Mito
MAM
Figure 14. A tentative model for intracellular trafficking of phosphatidylserine. Phosphatidylserine is synthe-sised preferentially in an ER domain associated with mitochondria (MAM), wherefrom a major fraction of thenewly-synthesised molecules move to mitochondria in a hydrophobicity-dependent manner (thick closed arrow).In the mitochondria, these molecules are then rapidly decarboxylated to PE by PS decarboxylase located in theinner membrane. The remaining PS molecules can move to the Golgi apparatus, plasma membrane andendocytotic membranes in vesicles (open thick arrows). From these membranes the more hydrophilic moleculescan diffuse spontaneously via the cytoplasm to mitochondria (thin arrows). See text for further details.Abbreviations: ER, endoplasmic reticulum; PM, plasma membrane, MAM, mitochondria-associated ERmembrane.

64
The depletion of more hydrophilic PS species due to their easy translocation to mitochondria
after their synthesis in the ER/MAM could be crucial for the maintenance of a high
concentration of PS in the inner leaflet of the plasma membrane, because the remaining
hydrophobic PS molecules would have little tendency to efflux from this membrane. Would
the cell synthesise only relatively hydrophilic PS species, it would be confronted with
frustrating efforts of maintaining adequate levels of PS in the plasma membrane as well as
other membranes. Notably, the model also predicts that if the rate of PS synthesis is relatively
slow compared to the rate of its desorption from membranes, the PS molecular species
compositions of different cellular organelle membranes should be similar. In this context, it is
relevant to mention that the PS molecular species composition of the influenza and vesicular
stomatitis viruses budding from the plasma membrane of human skin fibroblasts are closely
similar to that of the whole cell (Blom et al., 2001).

65
CONCLUSIONS
The translocation of phosphatidylserine from ER and plasma
membrane to mitochondria was studied. The following main
conclusions were drawn:
� Desorption from the donor membrane seems to be the rate-limiting
step of phosphatidylserine translocation from ER and plasma
membrane to mitochondria. This suggests that spontaneous
diffusion is the translocation mechanism. However, the
involvement of (unknown) proteins cannot be excluded.
� Preferential synthesis of hydrophobic phosphatidylserine species
and rapid depletion of the hydrophilic ones (due to their rapid
translocation to mitochondria) is likely to be important for the
maintenance of a high concentration of phosphatidylserine in the
inner leaflet of the plasma membrane.
� Spontaneous monomer diffusion via the cytoplasm could play an
important role in regulating the lipid composition of organelle
membranes.
� Electrospray ionisation mass spectroscopy in combination with
stable isotope precursor labelling is a very promising tool in
intracellular lipid trafficking studies.

66
REFERENCES
Achleitner, G., Zweytick, D., Trotter, P.J., Voelker, D.R. and Daum, G. (1995). Synthesis and intracellulartransport of aminoglycerophospholipids in permeabilized cells of the yeast, Saccharomyces cerevisiae.J Biol Chem 270: 29836-42.
Achleitner, G., Gaigg, B., Krasser, A., Kainersdorfer, E., Kohlwein, S.D., Perktold, A., Zellnig, G. and Daum, G.(1999). Association between the endoplasmic reticulum and mitochondria of yeast facilitatesinterorganelle transport of phospholipids through membrane contact. Eur J Biochem 264: 545-53.
Ardail, D., Privat, J.P., Egret-Charlier, M., Levrat, C., Lerme, F. and Louisot, P. (1990). Mitochondrial contactsites. Lipid composition and dynamics. J Biol Chem 265: 18797-802.
Ardail, D., Lerme, F. and Louisot, P. (1991). Involvement of contact sites in phosphatidylserine import into livermitochondria. J Biol Chem 266: 7978-81.
Ardail, D., Gasnier, F., Lerme, F., Simonot, C., Louisot, P. and Gateau-Roesch, O. (1993). Involvement ofmitochondrial contact sites in the subcellular compartmentalization of phospholipid biosyntheticenzymes. J Biol Chem 268: 25985-92.
Arthur, G. and Page, L. (1991). Synthesis of phosphatidylethanolamine and ethanolamine plasmalogen by theCDP-ethanolamine and decarboxylase pathways in rat heart, kidney and liver. Biochem J 273: 121-5.
Arthur, G. and Lu, X. (1993). The ethanolamine requirement of keratinocytes for growth is not due to defectivesynthesis of ethanolamine phosphoacylglycerols by the decarboxylation pathway. Biochem J 293: 125-30.
Atkinson, K.D., Jensen, B., Kolat, A.I., Storm, E.M., Henry, S.A. and Fogel, S. (1980). Yeast mutantsauxotrophic for choline or ethanolamine. J Bacteriol 141: 558-64.
Atshaves, B.P., Starodub, O., McIntosh, A., Petrescu, A., Roths, J.B., Kier, A.B. and Schroeder, F. (2000). Sterolcarrier protein-2 alters high density lipoprotein-mediated cholesterol efflux. J Biol Chem 275: 36852-61.
Baranska, J. (1989). Mechanism of the ATP-dependent phosphatidylserine synthesis in liver subcellularfractions. FEBS Lett 256: 33-7.
Baum, C.L., Reschly, E.J., Gayen, A.K., Groh, M.E. and Schadick, K. (1997). Sterol carrier protein-2overexpression enhances sterol cycling and inhibits cholesterol ester synthesis and high densitylipoprotein cholesterol secretion. J Biol Chem 272: 6490-8.
Berent, S.L. and Radin, N.S. (1981). Mechanism of activation of glucocerebrosidase by co-beta-glucosidase(glucosidase activator protein). Biochim Biophys Acta 664: 572-82.
Bevers, E.M., Comfurius, P. and Zwaal, R.F. (1983). Changes in membrane phospholipid distribution duringplatelet activation. Biochim Biophys Acta 736: 57-66.
Bevers, E.M., Comfurius, P. and Zwaal, R.F. (1996). Regulatory mechanisms in maintenance and modulation oftransmembrane lipid asymmetry: pathophysiological implications. Lupus 5: 480-7.
Biermans, W., Bakker, A. and Jacob, W. (1990). Contact site between inner and outer mitochondrial membrane:a dynamic microcompartment for creatine kinase activity. Biochim Biophys Acta 1018: 225-8.
Birner, R., Bürgermeister, M., Schneiter, R. and Daum, G. (2001). Roles of phosphatidylethanolamine and of itsseveral biosynthetic pathways in Saccharomyces cerevisiae. Mol Biol Cell 12: 997-1007.
Bishop, W.R. and Bell, R.M. (1985). Assembly of the endoplasmic reticulum phospholipid bilayer: thephosphatidylcholine transporter. Cell 42: 51-60.
Bjerve, K.S. (1973). The Ca2+-dependent biosynthesis of lecithin, phosphatidylethanolamine andphosphatidylserine in rat liver subcellular particles. Biochim Biophys Acta 296: 549-562.
Bjerve, K.S. (1985). The biosynthesis of phosphatidylserine and phosphatidylethanolamine from L-[3-14C]serine in isolated rat hepatocytes. Biochim Biophys Acta 833: 396-405.
Blok, M.C., Wirtz, K.W. and Scherphof, G.L. (1971). Exchange of phospholipids between microsomes and innerand outer mitochondrial membranes of rat liver. Biochim Biophys Acta 233: 61-75.
Blom, T.S., Koivusalo, M., Kuismanen, E., Kostiainen, R., Somerharju, P. and Ikonen, E. (2001). Massspectrometric analysis reveals an increase in plasma membrane polyunsaturated phospholipid speciesupon cellular cholesterol loading. Biochemistry 40: 14635-14644.
Borkenhagen, L.F., Kennedy, E.P. and Fielding, L. (1961). Enzymatic decarboxylation of phosphatidylserine. JBiol Chem 236: PC28-PC30.
Bratton, D.L., Fadok, V.A., Richter, D.A., Kailey, J.M., Guthrie, L.A. and Henson, P.M. (1997). Appearance ofphosphatidylserine on apoptotic cells requires calcium- mediated nonspecific flip-flop and is enhancedby loss of the aminophospholipid translocase. J Biol Chem 272: 26159-65.
Brown, R.E. (1992). Spontaneous lipid transfer between organized lipid assemblies. Biochim Biophys Acta 1113:375-89.

67
Brügger, B., Erben, G., Sandhoff, R., Wieland, F.T. and Lehmann, W.D. (1997). Quantitative analysis ofbiological membrane lipids at the low picomole level by nano-electrospray ionization tandem massspectrometry. Proc Natl Acad Sci U S A 94: 2339-44.
Buchanan, A.G. and Kanfer, J.N. (1980). Topographical distribution of base exchange activities in rat brainsubcellular fractions. J Neurochem 34: 720-5.
Butler, M. and Morell, P. (1983). The role of phosphatidylserine decarboxylase in brain phospholipidmetabolism. J Neurochem 41: 1445-54.
Buton, X., Morrot, G., Fellmann, P. and Seigneuret, M. (1996). Ultrafast glycerophospholipid-selectivetransbilayer motion mediated by a protein in the endoplasmic reticulum membrane. J Biol Chem 271:6651-7.
Bücheler, K., Adams, V. and Brdiczka, D. (1991). Localization of the ATP/ADP translocator in the innermembrane and regulation of contact sites between mitochondrial envelope membranes by ADP. Astudy on freeze-fractured isolated liver mitochondria. Biochim Biophys Acta 1056: 233-42.
Calderon, C., Huang, Z.H., Gage, D.A., Sotomayor, E.M. and Lopez, D.M. (1994). Isolation of a nitric oxideinhibitor from mammary tumor cells and its characterization as phosphatidyl serine. J Exp Med 180:945-58.
Callahan, M.K., Williamson, P. and Schlegel, R.A. (2000). Surface expression of phosphatidylserine onmacrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ 7: 645-53.
Camici, O. and Corazzi, L. (1995). Import of phosphatidylethanolamine for the assembly of rat brainmitochondrial membranes. J Membr Biol 148: 169-76.
Camici, O. and Corazzi, L. (1997). Phosphatidylserine translocation into brain mitochondria: involvement of afusogenic protein associated with mitochondrial membranes. Mol Cell Biochem 175: 71-80.
Carafoli, E. (1987). Intracellular calcium homeostasis. Annu Rev Biochem 56: 395-433.Carlini, E., Corazzi, L., Camici, O. and Arienti, G. (1993). The fate of phosphatidylethanolamine formed by
decarboxylation in rat brain mitochondria. Biochem Mol Biol Int 29: 821-9.Carman, G.M. and Henry, S.A. (1999). Phospholipid biosynthesis in the yeast Saccharomyces cerevisiae and
interrelationship with other metabolic processes. Prog Lipid Res 38: 361-99.Chattopadhyay, A. and London, E. (1987). Parallax method for direct measurement of membrane penetration
depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26: 39-45.Chen, H., Born, E., Mathur, S.N. and Field, F.J. (1993). Cholesterol and sphingomyelin syntheses are regulated
independently in cultured human intestinal cells, CaCo-2: role of membrane cholesterol andsphingomyelin content. J Lipid Res 34: 2159-67.
Clancey, C.J., Chang, S.C. and Dowhan, W. (1993). Cloning of a gene (PSD1) encoding phosphatidylserinedecarboxylase from Saccharomyces cerevisiae by complementation of an Escherichia coli mutant. JBiol Chem 268: 24580-90.
Comfurius, P., Bevers, E.M. and Zwaal, R.F. (1990). Enzymatic synthesis of phosphatidylserine on small scaleby use of a one-phase system. J Lipid Res 31: 1719-21.
Corazzi, L. and Arienti, G. (1992). The transfer of phosphatidylserine from rat brain microsomes tomitochondria is regulated by microsomal lipid pattern. Biochem Int 27: 853-60.
Corazzi, L., Pistolesi, R., Carlini, E. and Arienti, G. (1993). Transport of phosphatidylserine from microsomes tothe inner mitochondrial membrane in brain tissue. J Neurochem 60: 50-6.
Cullis, P.R. and de Kruijff, B. (1979). Lipid polymorphism and the functional roles of lipids in biologicalmembranes. Biochim Biophys Acta 559: 399-420.
Czarny, M., Sabala, P., Ucieklak, A., Kaczmarek, L. and Baranska, J. (1992). Inhibition of phosphatidylserinesynthesis by glutamate, acetylcholine, thapsigargin and ionophore A23187 in glioma C6 cells. BiochemBiophys Res Commun 186: 1582-7.
Daleke, D.L. and Huestis, W.H. (1985). Incorporation and translocation of aminophospholipids in humanerythrocytes. Biochemistry 24: 5406-16.
Daleke, D.L. and Lyles, J.V. (2000). Identification and purification of aminophospholipid flippases. BiochimBiophys Acta 1486: 108-27.
Daum, G. (1985). Lipids of mitochondria. Biochim Biophys Acta 822: 1-42.Daum, G. and Vance, J.E. (1997). Import of lipids into mitochondria. Prog Lipid Res 36: 103-30.de Vries, K.J., Heinrichs, A.A., Cunningham, E., Brunink, F., Westerman, J., Somerharju, P.J., Cockcroft, S.,
Wirtz, K.W. and Snoek, G.T. (1995). An isoform of the phosphatidylinositol-transfer protein transferssphingomyelin and is associated with the Golgi system. Biochem J 310: 643-9.
Defendi, V., Lehman, J. and Kraemer, P. (1963). "Morfologically normal" hamster cells with malignantproperties. Virology 19: 592-598.
DeLong, C.J., Shen, Y.J., Thomas, M.J. and Cui, Z. (1999). Molecular distinction of phosphatidylcholinesynthesis between the CDP- choline pathway and phosphatidylethanolamine methylation pathway. JBiol Chem 274: 29683-8.

68
Dennis, E.A. and Kennedy, E.P. (1972). Intracellular sites of lipid synthesis and the biogenesis of mitochondria.J Lipid Res 13: 263-7.
Derzko, Z. and Jacobson, K. (1980). Comparative lateral diffusion of fluorescent lipid analogues in phospholipidmultibilayers. Biochemistry 19: 6050-57.
Devaux, P.F. (1991). Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30: 1163-73.Dolis, D., Moreau, C., Zachowski, A. and Devaux, P.F. (1997). Aminophospholipid translocase and proteins
involved in transmembrane phospholipid traffic. Biophys Chem 68: 221-31.Dygas, A. and Zborowski, J. (1989). Effect of Triton X-100 on the activity and solubilization of rat liver
mitochondrial phosphatidylserine decarboxylase. Acta Biochim Pol 36: 131-41.Düzgünes, N., Goldstein, J.A., Friend, D.S. and Felgner, P.L. (1989). Fusion of liposomes containing a novel
cationic lipid, N-[2,3- (dioleyloxy)propyl]-N,N,N-trimethylammonium: induction by multivalent anionsand asymmetric fusion with acidic phospholipid vesicles. Biochemistry 28: 9179-84.
Eklund, K.K., Virtanen, J.A., Kinnunen, P.K., Kasurinen, J. and Somerharju, P.J. (1992). Conformation ofphosphatidylcholine in neat and cholesterol-containing liquid-crystalline bilayers. Application of anovel method. Biochemistry 31: 8560-5.
Ellingson, J.S. and Seenaiah, B. (1994). The selective use of stearoyl-polyunsaturated molecular species ofphosphatidylcholine and phosphatidylethanolamine for the synthesis of phosphatidylserine. BiochimBiophys Acta 1213: 113-7.
Emoto, K., Kuge, O., Nishijima, M. and Umeda, M. (1999). Isolation of a Chinese hamster ovary cell mutantdefective in intramitochondrial transport of phosphatidylserine. Proc Natl Acad Sci U S A 96: 12400-5.
Endo, T.A., Kobayashi, T. and Ohki, K. (1996). A Chinese hamster ovary cell mutant resistant tophosphatidylserine is defective in transbilayer movement of cell surface phosphatidylserine. Exp CellRes 228: 341-6.
Fadok, V.A., Voelker, D.R., Campbell, P.A., Cohen, J.J., Bratton, D.L. and Henson, P.M. (1992). Exposure ofphosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removalby macrophages. Journal of Immunology 148: 2207-16.
Fadok, V.A., Bratton, D.L., Rose, D.M., Pearson, A., Ezekewitz, R.A. and Henson, P.M. (2000). A receptor forphosphatidylserine-specific clearance of apoptotic cells. Nature 405: 85-90.
Felgner, P.L., Gadek, T.R., Holm, M., Roman, R., Chan, H.W., Wenz, M., Northrop, J.P., Ringold, G.M. andDanielsen, M. (1987). Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. ProcNatl Acad Sci U S A 84: 7413-7.
Ferrell, J.E., Jr., Lee, K.J. and Huestis, W.H. (1985). Lipid transfer between phosphatidylcholine vesicles andhuman erythrocytes: exponential decrease in rate with increasing acyl chain length. Biochemistry 24:2857-64.
Franke, W.W. and Kartenbeck, J. (1971). Outer mitochondrial membrane continuous with endoplasmicreticulum. Protoplasma 73: 35-41.
Gaigg, B., Simbeni, R., Hrastnik, C., Paltauf, F. and Daum, G. (1995). Characterization of a microsomalsubfraction associated with mitochondria of the yeast, Saccharomyces cerevisiae. Involvement insynthesis and import of phospholipids into mitochondria. Biochim Biophys Acta 1234: 214-20.
Gallegos, A.M., Schoer, J.K., Starodub, O., Kier, A.B., Billheimer, J.T. and Schroeder, F. (2000). A potentialrole for sterol carrier protein-2 in cholesterol transfer to mitochondria. Chem Phys Lipids 105: 9-29.
Gallegos, A.M., Atshaves, B.P., Storey, S.M., McIntosh, A.L., Petrescu, A.D. and Schroeder, F. (2001). Sterolcarrier protein-2 expression alters plasma membrane lipid distribution and cholesterol dynamics.Biochemistry 40: 6493-506.
Ghosh, S., Xie, W.Q., Quest, A.F., Mabrouk, G.M., Strum, J.C. and Bell, R.M. (1994). The cysteine-rich regionof raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras.J Biol Chem 269: 10000-7.
Gnamusch, E., Kalaus, C., Hrastnik, C., Paltauf, F. and Daum, G. (1992). Transport of phospholipids betweensubcellular membranes of wild-type yeast cells and of the phosphatidylinositol transfer protein-deficientstrain Saccharomyces cerevisiae sec 14. Biochim Biophys Acta 1111: 120-6.
Gottlieb, D., Heideman, W. and Saba, J.D. (1999). The DPL1 gene is involved in mediating the response tonutrient deprivation in Saccharomyces cerevisiae. Mol Cell Biol Res Commun 1: 66-71.
Hackenbrock, C.R. (1968). Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc Natl Acad Sci U S A 61: 598-605.
Hamon, Y., Broccardo, C., Chambenoit, O., Luciani, M.F., Toti, F., Chaslin, S., Freyssinet, J.M., Devaux, P.F.,McNeish, J., Marguet, D. and Chimini, G. (2000). ABC1 promotes engulfment of apoptotic cells andtransbilayer redistribution of phosphatidylserine. Nat Cell Biol 2: 399-406.
Han, X. and Gross, R.W. (1995). Structural determination of picomole amounts of phospholipids viaelectrospray ionization tandem mass spectrometry. J Am Soc Mass Spectrom 6: 1202-10.

69
Hanada, K., Nishijima, M., Kiso, M., Hasegawa, A., Fujita, S., Ogawa, T. and Akamatsu, Y. (1992).Sphingolipids are essential for the growth of Chinese hamster ovary cells. Restoration of the growth ofa mutant defective in sphingoid base biosynthesis by exogenous sphingolipids. J Biol Chem 267:23527-33.
Hanada, K. and Pagano, R.E. (1995). A Chinese hamster ovary cell mutant defective in the non-endocytic uptakeof fluorescent analogs of phosphatidylserine: isolation using a cytosol acidification protocol. J Cell Biol128: 793-804.
Hasegawa, K., Kuge, O., Nishijima, M. and Akamatsu, Y. (1989). Isolation and characterization of a Chinesehamster ovary cell mutant with altered regulation of phosphatidylserine biosynthesis. J Biol Chem 264:19887-92.
Henry, S.A., Klig, L.S. and Loewy, B.S. (1984). The genetic regulation and coordination of biosyntheticpathways in yeast: amino acid and phospholipid synthesis. Annu Rev Genet 18: 207-31.
Herrmann, A., Zachowski, A. and Devaux, P.F. (1990). Protein-mediated phospholipid translocation in theendoplasmic reticulum with a low lipid specificity. Biochemistry 29: 2023-7.
Hjelmstad, R.H. and Bell, R.M. (1991). Molecular insights into enzymes of membrane bilayer assembly.Biochemistry 30: 1731-40.
Holub, B.J. (1980). The biosynthesis of phosphatidylserines by acylation of 1-acyl-sn- glycero-3-phosphoserinein rat liver. Biochim Biophys Acta 618: 255-62.
Hostetler, K.Y., Van den Bosch, H. and Van Deenen, L.L. (1971). Biosynthesis of cardiolipin in livermitochondria. Biochim Biophys Acta 239: 113-9.
Hovius, R., Lambrechts, H., Nicolay, K. and de Kruijff, B. (1990). Improved methods to isolate andsubfractionate rat liver mitochondria. Lipid composition of the inner and outer membrane. BiochimBiophys Acta 1021: 217-26.
Hovius, R., Faber, B., Brigot, B., Nicolay, K. and de Kruijff, B. (1992). On the mechanism of the mitochondrialdecarboxylation of phosphatidylserine. J Biol Chem 267: 16790-5.
Hullin, F., Kim, H.Y. and Salem, N., Jr. (1989). Analysis of aminophospholipid molecular species by highperformance liquid chromatography. J Lipid Res 30: 1963-75.
Huster, D., Müller, P., Arnold, K. and Herrmann, A. (2001). Dynamics of Membrane Penetration of theFluorescent 7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl (NBD) Group Attached to an Acyl Chain ofPhosphatidylcholine. Biophys J 80: 822-831.
Huuskonen, J., Olkkonen, V.M., Jauhiainen, M., Sareneva, T., Somerharju, P. and Ehnholm, C. (1998).Oxidative modification of HDL3 in vitro and its effect on PLTP-mediated phospholipid transfer.Biochim Biophys Acta 1391: 181-92.
Hübscher, G. (1962). Metabolism of phospholipids. VI. Effect of metal ions on the incorporation of L-serine intophosphatidylserine. Biochim Biophys Acta 57: 555-561.
Igarashi, H., Zama, K. and Katada, M. (1958). Presence of a threonine-containig phospholipid in tunny muscle.Nature 181: 1282-83.
Jasinska, R., Zborowski, J. and Somerharju, P. (1993). Intramitochondrial distribution and transport ofphosphatidylserine and its decarboxylation product, phosphatidylethanolamine. Application of pyrene-labeled species. Biochim Biophys Acta 1152: 161-70.
Jones, J.D. and Thompson, T.E. (1989). Spontaneous phosphatidylcholine transfer by collision between vesiclesat high lipid concentration. Biochemistry 28: 129-34.
Jones, J.D. and Thompson, T.E. (1990). Mechanism of spontaneous, concentration-dependent phospholipidtransfer between bilayers [published errata appear in Biochemistry 1990 May 15;29(19):4746 and 1991Nov 5;30(44):10818]. Biochemistry 29: 1593-600.
Kanfer, J.N. (1980). The base exchange enzymes and phospholipase D of mammalian tissue. Can J Biochem 58:1370-80.
Kanfer, J.N. and McCartney, D.G. (1993). Modulation of the serine base exchange enzyme activity of rat brainmembranes by amphiphilic cations and amphiphilic anions. J Neurochem 60: 1228-35.
Kaplan, M.R. and Simoni, R.D. (1985). Intracellular transport of phosphatidylcholine to the plasma membrane. JCell Biol 101: 441-5.
Kasurinen, J., van Paridon, P.A., Wirtz, K.W. and Somerharju, P. (1990). Affinity of phosphatidylcholinemolecular species for the bovine phosphatidylcholine and phosphatidylinositol transfer proteins.Properties of the sn-1 and sn-2 acyl binding sites. Biochemistry 29: 8548-54.
Kasurinen, J. and Somerharju, P. (1992). Metabolism of pyrenyl fatty acids in baby hamster kidney fibroblasts.Effect of the acyl chain length. J Biol Chem 267: 6563-9.
Kasurinen, J. and Somerharju, P. (1995). Metabolism and distribution of intramolecular excimer-formingdipyrenebutanoyl glycerophospholipids in human fibroblasts. Marked resistance to metabolicdegradation. Biochemistry 34: 2049-57.

70
Keller, G.A., Scallen, T.J., Clarke, D., Maher, P.A., Krisans, S.K. and Singer, S.J. (1989). Subcellularlocalization of sterol carrier protein-2 in rat hepatocytes: its primary localization to peroxisomes. J CellBiol 108: 1353-61.
Kelly, R.B. (1990). Microtubules, membrane traffic, and cell organization. Cell 61: 5-7.Kennedy, E.P. and Weiss, S.B. (1956). The function of cytidine coenzymes in the biosynthesis od
phospholipides. J Biol Chem 222: 193-214.Knoll, G. and Brdiczka, D. (1983). Changes in freeze-fractured mitochondrial membranes correlated to their
energetic state. Dynamic interactions of the boundary membranes. Biochim Biophys Acta 733: 102-10.Kobayashi, T. and Pagano, R.E. (1989). Lipid transport during mitosis. Alternative pathways for delivery of
newly synthesized lipids to the cell surface. J Biol Chem 264: 5966-73.Kobayashi, T. and Arakawa, Y. (1991). Transport of exogenous fluorescent phosphatidylserine analogue to the
Golgi apparatus in cultured fibroblasts. J Cell Biol 113: 235-44.Koivusalo, M., Haimi, P., Heikinheimo, L., Kostiainen, R. and Somerharju, P. (2001). Quantitative
determination of phospholipid compositions by ESI-MS: effects of acyl chain length, unsaturation, andlipid concentration on instrument response. J Lipid Res 42: 663-72.
Kornberg, R.D. and McConnell, H.M. (1971a). Inside-outside transitions of phospholipids in vesicle membranes.Biochemistry 10: 1111-20.
Kornberg, R.D. and McConnell, H.M. (1971b). Lateral diffusion of phospholipids in a vesicle membrane. ProcNatl Acad Sci U S A 68: 2564-8.
Kuchler, K., Daum, G. and Paltauf, F. (1986). Subcellular and submitochondrial localization of phospholipid-synthesizing enzymes in Saccharomyces cerevisiae. J Bacteriol 165: 901-10.
Kuge, O., Nishijima, M. and Akamatsu, Y. (1986a). Phosphatidylserine biosynthesis in cultured Chinese hamsterovary cells. II. Isolation and characterization of phosphatidylserine auxotrophs. J Biol Chem 261: 5790-4.
Kuge, O., Nishijima, M. and Akamatsu, Y. (1986b). Phosphatidylserine biosynthesis in cultured Chinese hamsterovary cells. III. Genetic evidence for utilization of phosphatidylcholine and phosphatidylethanolamineas precursors. J Biol Chem 261: 5795-8.
Kuge, O., Akamatsu, Y. and Nishijima, M. (1989). Abortive infection with Sindbis virus of a Chinese hamsterovary cell mutant defective in phosphatidylserine and phosphatidylethanolamine biosynthesis. BiochimBiophys Acta 986: 61-9.
Kuge, O., Nishijima, M. and Akamatsu, Y. (1991). A Chinese hamster cDNA encoding a protein essential forphosphatidylserine synthase I activity. J Biol Chem 266: 24184-9.
Kuge, O. and Nishijima, M. (1997). Phosphatidylserine synthase I and II of mammalian cells. Biochim BiophysActa 1348: 151-6.
Kuge, O., Saito, K. and Nishijima, M. (1997). Cloning of a Chinese hamster ovary (CHO) cDNA encodingphosphatidylserine synthase (PSS) II, overexpression of which suppresses the phosphatidylserinebiosynthetic defect of a PSS I- lacking mutant of CHO-K1 cells. J Biol Chem 272: 19133-9.
Kuge, O., Hasegawa, K., Saito, K. and Nishijima, M. (1998). Control of phosphatidylserine biosynthesis throughphosphatidylserine- mediated inhibition of phosphatidylserine synthase I in Chinese hamster ovarycells. Proc Natl Acad Sci U S A 95: 4199-203.
Kuge, O., Saito, K. and Nishijima, M. (1999). Control of phosphatidylserine synthase II activity in Chinesehamster ovary cells. J Biol Chem 274: 23844-9.
Kuge, O., Yamakawa, Y. and Nishijima, M. (2001). Enhancement of transport-dependent decarboxylation ofphosphatidylserine by S100B protein in permeabilized Chinese hamster ovary cells. J Biol Chem 276:23700-6.
Kuo, A.L. and Wade, C.G. (1979). Lipid lateral diffusion by pulsed nuclear magnetic resonance. Biochemistry18: 2300-8.
Letts, V.A., Klig, L.S., Bae-Lee, M., Carman, G.M. and Henry, S.A. (1983). Isolation of the yeast structural genefor the membrane-associated enzyme phosphatidylserine synthase. Proc Natl Acad Sci U S A 80: 7279-83.
Lewis, J.A. and Tata, J.R. (1973). A rapidly sedimenting fraction of rat liver endoplasmic reticulum. J Cell Sci13: 447-59.
Li, H., Tremblay, J.M., Yarbrough, L.R. and Helmkamp, G.M., Jr. (2002). Both isoforms of mammalianphosphatidylinositol transfer protein are capable of binding and transporting sphingomyelin. BiochimBiophys Acta 1580: 67-76.
Lipsky, N.G. and Pagano, R.E. (1985). Intracellular translocation of fluorescent sphingolipids in culturedfibroblasts: endogenously synthesized sphingomyelin and glucocerebroside analogues pass through theGolgi apparatus en route to the plasma membrane. J Cell Biol 100: 27-34.
Lipton, B.A., Davidson, E.P., Ginsberg, B.H. and Yorek, M.A. (1990). Ethanolamine metabolism in culturedbovine aortic endothelial cells. J Biol Chem 265: 7195-201.

71
Lubin, B., Chiu, D., Bastacky, J., Roelofsen, B. and Van Deenen, L.L. (1981). Abnormalities in membranephospholipid organization in sickled erythrocytes. J Clin Invest 67: 1643-9.
Lund-Katz, S., Hammerschlag, B. and Phillips, M.C. (1982). Kinetics and mechanism of free cholesterolexchange between human serum high- and low-density lipoproteins. Biochemistry 21: 2964-9.
MacDonald, J.I. and Sprecher, H. (1991). Phospholipid fatty acid remodeling in mammalian cells. BiochimBiophys Acta 1084: 105-21.
Mark-Malchoff, D., Marinetti, G.V., Hare, G.D. and Meisler, A. (1978). Characterization ofphosphatidylthreonine in polyoma virus transformed fibroblasts. Biochemistry 17: 2684-8.
Marsh, M., Bolzau, E. and Helenius, A. (1983). Penetration of Semliki Forest virus from acidic prelysosomalvacuoles. Cell 32: 931-40.
Massey, J.B., Gotto, A.M., Jr. and Pownall, H.J. (1982a). Kinetics and mechanism of the spontaneous transfer offluorescent phosphatidylcholines between apolipoprotein-phospholipid recombinants. Biochemistry 21:3630-6.
Massey, J.B., Gotto, A.M., Jr. and Pownall, H.J. (1982b). Kinetics and mechanism of the spontaneous transfer offluorescent phospholipids between apolipoprotein-phospholipid recombinants. Effect of the polarheadgroup. J Biol Chem 257: 5444-8.
Massey, J.B., Hickson, D., She, H.S., Sparrow, J.T., Via, D.P., Gotto, A.M. and Pownall, H.J. (1984).Measurement and prediction of the rates of spontaneous transfer of phospholipids between plasmalipoproteins. Biochim Biophys Acta 794: 274-80.
Massey, J.B., Hickson-Bick, D., Via, D.P., Gotto, A.M., Jr. and Pownall, H.J. (1985). Fluorescence assay of thespecificity of human plasma and bovine liver phospholipid transfer proteins. Biochim Biophys Acta 835:124-31.
McGee, T.P., Skinner, H.B., Whitters, E.A., Henry, S.A. and Bankaitis, V.A. (1994). A phosphatidylinositoltransfer protein controls the phosphatidylcholine content of yeast Golgi membranes. J Cell Biol 124:273-87.
McLean, L.R. and Phillips, M.C. (1981). Mechanism of cholesterol and phosphatidylcholine exchange ortransfer between unilamellar vesicles. Biochemistry 20: 2893-900.
McLean, L.R. and Phillips, M.C. (1984). Kinetics of phosphatidylcholine and lysophosphatidylcholine exchangebetween unilamellar vesicles. Biochemistry 23: 4624-30.
Medlock, K.A. and Merrill, A.H., Jr. (1988). Inhibition of serine palmitoyltransferase in vitro and long-chainbase biosynthesis in intact Chinese hamster ovary cells by beta- chloroalanine. Biochemistry 27: 7079-84.
Meier, P.J., Spycher, M.A. and Meyer, U.A. (1981). Isolation and characterization of rough endoplasmicreticulum associated with mitochondria from normal rat liver. Biochim Biophys Acta 646: 283-97.
Menon, A.K., Eppinger, M., Mayor, S. and Schwarz, R.T. (1993). Phosphatidylethanolamine is the donor of theterminal phosphoethanolamine group in trypanosome glycosylphosphatidylinositols. Embo J 12: 1907-14.
Menon, A.K. (1995). Flippases. Trends in cell biology 5: 355-360.Menon, A.K., Watkins, W.E.r. and Hrafnsdottir, S. (2000). Specific proteins are required to translocate
phosphatidylcholine bidirectionally across the endoplasmic reticulum. Curr Biol 10: 241-52.Miller, M.A. and Kent, C. (1986). Characterization of the pathways for phosphatidylethanolamine biosynthesis
in Chinese hamster ovary mutant and parental cell lines. J Biol Chem 261: 9753-61.Mitoma, J., Furuya, S. and Hirabayashi, Y. (1998a). A novel metabolic communication between neurons and
astrocytes: non- essential amino acid L-serine released from astrocytes is essential for developinghippocampal neurons. Neurosci Res 30: 195-9.
Mitoma, J., Kasama, T., Furuya, S. and Hirabayashi, Y. (1998b). Occurrence of an unusual phospholipid,phosphatidyl-L-threonine, in cultured hippocampal neurons. Exogenous L-serine is required for thesynthesis of neuronal phosphatidyl-L-serine and sphingolipids. J Biol Chem 273: 19363-6.
Morré, D.J., Merritt, W.D. and Lembi, C.A. (1971). Connections between mitochondria and endoplasmicreticulum in rat liver and onion stem. Protoplasma 73: 43-9.
Morrot, G., Herve, P., Zachowski, A., Fellmann, P. and Devaux, P.F. (1989). Aminophospholipid translocase ofhuman erythrocytes: phospholipid substrate specificity and effect of cholesterol. Biochemistry 28: 3456-62.
Moynagh, P.N. (1995). Contact sites and transport in mitochondria. Essays Biochem 30: 1-14.Naylor, B.L., Picardo, M., Homan, R. and Pownall, H.J. (1991). Effects of fluorophore structure and
hydrophobicity on the uptake and metabolism of fluorescent lipid analogs. Chem Phys Lipids 58: 111-9.Nichols, J.W. and Pagano, R.E. (1983). Resonance energy transfer assay of protein-mediated lipid transfer
between vesicles. J Biol Chem 258: 5368-71.

72
Nishijima, M., Kuge, O. and Akamatsu, Y. (1986). Phosphatidylserine biosynthesis in cultured Chinese hamsterovary cells. I. Inhibition of de novo phosphatidylserine biosynthesis by exogenous phosphatidylserineand its efficient incorporation. J Biol Chem 261: 5784-9.
Nishizuka, Y. (1988). The molecular heterogeneity of protein kinase C and its implications for cellularregulation. Nature 334: 661-5.
Olkkonen, V.M. and Ikonen, E. (2000). Genetic defects of intracellular-membrane transport. N Engl J Med 343:1095-104.
Ossendorp, B.C. and Wirtz, K.W. (1993). The non-specific lipid-transfer protein (sterol carrier protein 2) and itsrelationship to peroxisomes. Biochimie 75: 191-200.
Pagano, R.E., Longmuir, K.J. and Martin, O.C. (1983). Intracellular translocation and metabolism of afluorescent phosphatidic acid analogue in cultured fibroblasts. J Biol Chem 258: 2034-40.
Pagano, R.E. and Sleight, R.G. (1985). Defining lipid transport pathways in animal cells. Science 229: 1051-7.Pagano, R.E. and Chen, C.S. (1998). Use of BODIPY-labeled sphingolipids to study membrane traffic along the
endocytic pathway. Ann N Y Acad Sci 845: 152-60.Patton, G.M., Fasulo, J.M. and Robins, S.J. (1982). Separation of phospholipids and individual molecular
species of phospholipids by high-performance liquid chromatography. J Lipid Res 23: 190-6.Pelassy, C., Breittmayer, J.P. and Aussel, C. (1992). Agonist-induced inhibition of phosphatidylserine synthesis
is secondary to the emptying of intracellular Ca2+ stores in Jurkat T-cells. Biochem J 288: 785-9.Percy, A.K., Moore, J.F., Carson, M.A. and Waechter, C.J. (1983). Characterization of brain phosphatidylserine
decarboxylase: localization in the mitochondrial inner membrane. Arch Biochem Biophys 223: 484-94.Pownall, H.J. and Smith, L.C. (1989). Pyrene-labeled lipids: versatile probes of membrane dynamics in vitro and
in living cells. Chem Phys Lipids 50: 191-211.Rakowska, M., Jasinska, R., Lenart, J., Komanska, I., Makowski, P., Dygas, A. and Pikula, S. (1997). Membrane
integrity and phospholipid movement influence the base exchange reaction in rat liver microsomes. MolCell Biochem 168: 163-76.
Rhodes, D.N. and Lea, C.H. (1957). Phospholipids. 4. On the composition of hen's egg phospholipids. Biochem J65: 526-533.
Roberts, C.J., Raymond, C.K., Yamashiro, C.T. and Stevens, T.H. (1991). Methods for studying the yeastvacuole. Methods Enzymol 194: 644-61.
Sabala, P., Wiktorek, M., Czarny, M., Chaban, V. and Baranska, J. (1996). Sphingosine stimulates calciummobilization and modulates calcium signals evoked by thapsigargin in glioma C6 cells. Acta NeurobiolExp 56: 507-13.
Saito, K., Nishijima, M. and Kuge, O. (1998). Genetic evidence that phosphatidylserine synthase II catalyzes theconversion of phosphatidylethanolamine to phosphatidylserine in Chinese hamster ovary cells. J BiolChem 273: 17199-205.
Sakane, F., Yamada, K., Imai, S. and Kanoh, H. (1991). Porcine 80-kDa diacylglycerol kinase is a calcium-binding and calcium/phospholipid-dependent enzyme and undergoes calcium-dependent translocation. JBiol Chem 266: 7096-100.
Sassaroli, M., Ruonala, M., Virtanen, J., Vauhkonen, M. and Somerharju, P. (1995). Transversal distribution ofacyl-linked pyrene moieties in liquid- crystalline phosphatidylcholine bilayers. A fluorescencequenching study. Biochemistry 34: 8843-51.
Schmid, P.C., Johnson, S.B. and Schmid, H.H. (1991). Remodeling of rat hepatocyte phospholipids by selectiveacyl turnover. J Biol Chem 266: 13690-7.
Schneiter, R., Brügger, B., Sandhoff, R., Zellnig, G., Leber, A., Lampl, M., Athenstaedt, K., Hrastnik, C., Eder,S., Daum, G., Paltauf, F., Wieland, F.T. and Kohlwein, S.D. (1999). Electrospray ionization tandemmass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeastsubcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species enroute to the plasma membrane. J Cell Biol 146: 741-54.
Schroit, A.J., Madsen, J.W. and Tanaka, Y. (1985). In vivo recognition and clearance of red blood cellscontaining phosphatidylserine in their plasma membranes. J Biol Chem 260: 5131-8.
Seigneuret, M. and Devaux, P.F. (1984). ATP-dependent asymmetric distribution of spin-labeled phospholipidsin the erythrocyte membrane: relation to shape changes. Proc Natl Acad Sci U S A 81: 3751-5.
Shiao, Y.J., Lupo, G. and Vance, J.E. (1995). Evidence that phosphatidylserine is imported into mitochondria viaa mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamineis derived from decarboxylation of phosphatidylserine. J Biol Chem 270: 11190-8.
Shiao, Y.J., Balcerzak, B. and Vance, J.E. (1998). A mitochondrial membrane protein is required fortranslocation of phosphatidylserine from mitochondria-associated membranes to mitochondria. BiochemJ 331: 217-23.
Shimada, N., Graf, R., Rosner, G. and Heiss, W.D. (1993). Ischemia-induced accumulation of extracellularamino acids in cerebral cortex, white matter, and cerebrospinal fluid. J Neurochem 60: 66-71.

73
Siegmund, A., Grant, A., Angeletti, C., Malone, L., Nichols, J.W. and Rudolph, H.K. (1998). Loss of Drs2p doesnot abolish transfer of fluorescence-labeled phospholipids across the plasma membrane ofSaccharomyces cerevisiae. J Biol Chem 273: 34399-405.
Silversand, C. and Haux, C. (1997). Improved high-performance liquid chromatographic method for theseparation and quantification of lipid classes: application to fish lipids. J Chromatogr B Biomed SciAppl 703: 7-14.
Silvius, J.R. and Leventis, R. (1993). Spontaneous interbilayer transfer of phospholipids: dependence on acylchain composition. Biochemistry 32: 13318-26.
Simbeni, R., Paltauf, F. and Daum, G. (1990). Intramitochondrial transfer of phospholipids in the yeast,Saccharomyces cerevisiae. J Biol Chem 265: 281-5.
Simbeni, R., Pon, L., Zinser, E., Paltauf, F. and Daum, G. (1991). Mitochondrial membrane contact sites ofyeast. Characterization of lipid components and possible involvement in intramitochondrialtranslocation of phospholipids. J Biol Chem 266: 10047-9.
Simbeni, R., Tangemann, K., Schmidt, M., Ceolotto, C., Paltauf, F. and Daum, G. (1993). Import ofphosphatidylserine into isolated yeast mitochondria. Biochim Biophys Acta 1145: 1-7.
Skinner, H.B., McGee, T.P., McMaster, C.R., Fry, M.R., Bell, R.M. and Bankaitis, V.A. (1995). TheSaccharomyces cerevisiae phosphatidylinositol-transfer protein effects a ligand-dependent inhibition ofcholine-phosphate cytidylyltransferase activity. Proc Natl Acad Sci U S A 92: 112-6.
Sleight, R.G. and Pagano, R.E. (1983). Rapid appearance of newly synthesized phosphatidylethanolamine at theplasma membrane. J Biol Chem 258: 9050-8.
Sleight, R.G. and Pagano, R.E. (1984). Transport of a fluorescent phosphatidylcholine analog from the plasmamembrane to the Golgi apparatus. J Cell Biol 99: 742-51.
Sleight, R.G. and Pagano, R.E. (1985). Transbilayer movement of a fluorescent phosphatidylethanolamineanalogue across the plasma membranes of cultured mammalian cells. J Biol Chem 260: 1146-54.
Smith, J.D., Waelde, C., Horwitz, A. and Zheng, P. (2002). Evaluation of the Role of PhosphatidylserineTranslocase Activity in ABCA1-mediated Lipid Efflux. J Biol Chem 277: 17797-803.
Somerharju, P. (2002). Pyrene-labeled lipids as tools in membrane biophysics and cell biology. Chem PhysLipids in press.
Somerharju, P.J., Virtanen, J.A., Eklund, K.K., Vainio, P. and Kinnunen, P.K. (1985). 1-Palmitoyl-2-pyrenedecanoyl glycerophospholipids as membrane probes: evidence for regular distribution in liquid-crystalline phosphatidylcholine bilayers. Biochemistry 24: 2773-81.
Somerharju, P.J., van Loon, D. and Wirtz, K.W. (1987). Determination of the acyl chain specificity of the bovineliver phosphatidylcholine transfer protein. Application of pyrene-labeled phosphatidylcholine species.Biochemistry 26: 7193-9.
Sperka-Gottlieb, C.D., Hermetter, A., Paltauf, F. and Daum, G. (1988). Lipid topology and physical properties ofthe outer mitochondrial membrane of the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta 946:227-34.
Stone, S.J., Cui, Z. and Vance, J.E. (1998). Cloning and expression of mouse liver phosphatidylserine synthase-1cDNA. Overexpression in rat hepatoma cells inhibits the CDP- ethanolamine pathway forphosphatidylethanolamine biosynthesis. J Biol Chem 273: 7293-302.
Stone, S.J. and Vance, J.E. (1999). Cloning and expression of murine liver phosphatidylserine synthase (PSS)-2:differential regulation of phospholipid metabolism by PSS1 and PSS2. Biochem J 342: 57-64.
Stone, S.J. and Vance, J.E. (2000). Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem 275: 34534-40.
Storey, M.K., Clay, K.L., Kutateladze, T., Murphy, R.C., Overduin, M. and Voelker, D.R. (2001).Phosphatidylethanolamine has an essential role in Saccharomyces cerevisiae that is independent of itsability to form hexagonal phase structures. J Biol Chem 276: 48539-48548.
Storrie, B. and Madden, E.A. (1990). Isolation of subcellular organelles. Methods Enzymol 182: 203-25.Sundler, R. and Åkesson, B. (1975). Regulation of phospholipid biosynthesis in isolated rat hepatocytes. Effect
of different substrates. J Biol Chem 250: 3359-67.Szolderits, G., Hermetter, A., Paltauf, F. and Daum, G. (1989). Membrane properties modulate the activity of a
phosphatidylinositol transfer protein from the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta986: 301-9.
Taki, T. and Kanfer, J.N. (1978). A phospholipid serine base exchange enzyme. Biochim Biophys Acta 528: 309-17.
Tanaka, S. and Hosaka, K. (1994). Cloning of a cDNA encoding a second phosphatidylinositol transfer proteinof rat brain by complementation of the yeast sec14 mutation. J Biochem (Tokyo) 115: 981-4.
Tang, X., Halleck, M.S., Schlegel, R.A. and Williamson, P. (1996). A subfamily of P-type ATPases withaminophospholipid transporting activity. Science 272: 1495-7.

74
Tanhuanpää, K. and Somerharju, P. (1999). gamma-cyclodextrins greatly enhance translocation of hydrophobicfluorescent phospholipids from vesicles to cells in culture. Importance of molecular hydrophobicity inphospholipid trafficking studies. J Biol Chem 274: 35359-66.
Tanhuanpää, K., Cheng, K.H., Anttonen, K., Virtanen, J.A. and Somerharju, P. (2001). Characteristics of pyrenephospholipid/gamma-cyclodextrin complex. Biophys J 81: 1501-10.
Thompson, W. and Belina, H. (1986). Rapid accumulation of diacyl lipid in rat liver microsomes by selectiveacylation of 2-acyl-sn-glycero-3-phosphorylserine. Biochim Biophys Acta 876: 379-86.
Tijburg, L.B., Geelen, M.J. and Van Golde, L.M. (1989). Biosynthesis of phosphatidylethanolamine via theCDP-ethanolamine route is an important pathway in isolated rat hepatocytes. Biochem Biophys ResCommun 160: 1275-80.
Trotter, P.J., Pedretti, J. and Voelker, D.R. (1993). Phosphatidylserine decarboxylase from Saccharomycescerevisiae. Isolation of mutants, cloning of the gene, and creation of a null allele. J Biol Chem 268:21416-24.
Trotter, P.J. and Voelker, D.R. (1994). Lipid transport processes in eukaryotic cells. Biochim Biophys Acta 1213:241-62.
Trotter, P.J. and Voelker, D.R. (1995). Identification of a non-mitochondrial phosphatidylserine decarboxylaseactivity (PSD2) in the yeast Saccharomyces cerevisiae. J Biol Chem 270: 6062-70.
Trotter, P.J., Wu, W.I., Pedretti, J., Yates, R. and Voelker, D.R. (1998). A genetic screen for aminophospholipidtransport mutants identifies the phosphatidylinositol 4-kinase, STT4p, as an essential component inphosphatidylserine metabolism. J Biol Chem 273: 13189-96.
Tsuneoka, M., Yamamoto, A., Fujiki, Y. and Tashiro, Y. (1988). Nonspecific lipid transfer protein (sterol carrierprotein-2) is located in rat liver peroxisomes. J Biochem (Tokyo) 104: 560-4.
Tuma, P.L., Stachniak, M.C. and Collins, C.A. (1993). Activation of dynamin GTPase by acidic phospholipidsand endogenous rat brain vesicles. J Biol Chem 268: 17240-6.
van Amerongen, A., Demel, R.A., Westerman, J. and Wirtz, K.W. (1989). Transfer of cholesterol and oxysterolderivatives by the nonspecific lipid transfer protein (sterol carrier protein 2): a study on its mode ofaction. Biochim Biophys Acta 1004: 36-43.
van den Bosch, H., Schutgens, R.B., Wanders, R.J. and Tager, J.M. (1992). Biochemistry of peroxisomes. AnnuRev Biochem 61: 157-97.
van Duijn, G., Luiken, J., Verkleij, A.J. and de Kruijff, B. (1986). Relation between lipid polymorphism andtransbilayer movement of lipids in rat liver microsomes. Biochim Biophys Acta 863: 193-204.
van Golde, L.M., Raben, J., Batenburg, J.J., Fleischer, B., Zambrano, F. and Fleischer, S. (1974). Biosynthesis oflipids in Golgi complex and other subcellular fractions from rat liver. Biochim Biophys Acta 360: 179-92.
van Helvoort, A., de Brouwer, A., Ottenhoff, R., Brouwers, J.F., Wijnholds, J., Beijnen, J.H., Rijneveld, A., vander Poll, T., van der Valk, M.A., Majoor, D., Voorhout, W., Wirtz, K.W., Elferink, R.P. and Borst, P.(1999). Mice without phosphatidylcholine transfer protein have no defects in the secretion ofphosphatidylcholine into bile or into lung airspaces. Proc Natl Acad Sci U S A 96: 11501-6.
van Heusden, G.P., Bos, K., Raetz, C.R. and Wirtz, K.W. (1990). Chinese hamster ovary cells deficient inperoxisomes lack the nonspecific lipid transfer protein (sterol carrier protein 2). J Biol Chem 265: 4105-10.
van Hoof, F. and Hers, H.G. (1968). The abnormalities of lysosomal enzymes in mucopolysacc- haridoses. Eur JBiochem 7: 34-44.
van Paridon, P.A., Gadella, T.W., Jr., Somerharju, P.J. and Wirtz, K.W. (1987). On the relationship between thedual specificity of the bovine brain phosphatidylinositol transfer protein and membranephosphatidylinositol levels. Biochim Biophys Acta 903: 68-77.
van Paridon, P.A., Gadella, T.W., Jr., Somerharju, P.J. and Wirtz, K.W. (1988). Properties of the binding sitesfor the sn-1 and sn-2 acyl chains on the phosphatidylinositol transfer protein from bovine brain.Biochemistry 27: 6208-14.
van Venetië, R. and Verkleij, A.J. (1982). Possible role of non-bilayer lipids in the structure of mitochondria. Afreeze-fracture electron microscopy study. Biochim Biophys Acta 692: 397-405.
Vance, J.E. and Vance, D.E. (1986). Specific pools of phospholipids are used for lipoprotein secretion bycultured rat hepatocytes. J Biol Chem 261: 4486-91.
Vance, J.E. (1988). Compartmentalization of phospholipids for lipoprotein assembly on the basis of molecularspecies and biosynthetic origin. Biochim Biophys Acta 963: 70-81.
Vance, J.E. and Vance, D.E. (1988). Does rat liver Golgi have the capacity to synthesize phospholipids forlipoprotein secretion? J Biol Chem 263: 5898-909.
Vance, J.E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem265: 7248-56.

75
Vance, J.E. (1991). Newly made phosphatidylserine and phosphatidylethanolamine are preferentiallytranslocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem 266: 89-97.
Vance, J.E., Aasman, E.J. and Szarka, R. (1991). Brefeldin A does not inhibit the movement ofphosphatidylethanolamine from its sites for synthesis to the cell surface. J Biol Chem 266: 8241-7.
Vance, J.E. and Shiao, Y.J. (1996). Intracellular trafficking of phospholipids: import of phosphatidylserine intomitochondria. Anticancer Res 16: 1333-9.
Weigel, P.H. and Oka, J.A. (1981). Temperature dependence of endocytosis mediated by the asialoglycoproteinreceptor in isolated rat hepatocytes. Evidence for two potentially rate-limiting steps. J Biol Chem 256:2615-7.
Weissbach, H., Smith, T.E., Daly, J.W., Witkop, B. and Udenfriend, S. (1960). A rapid spectrometric assay ofmonoamine oxidase based on the rate of disappearance of kynuramine. J Biol Chem 235: 1160-3.
Westerman, J., de Vries, K.J., Somerharju, P., Timmermans-Hereijgers, J.L., Snoek, G.T. and Wirtz, K.W.(1995). A sphingomyelin-transferring protein from chicken liver. Use of pyrene- labeled phospholipid.J Biol Chem 270: 14263-6.
Wiktorek-Wojcik, M., Banasiak, M., Czarny, M., Stepkowski, D. and Baranska, J. (1997). Serine base exchangeenzyme activity is modulated by sphingosine and other amphiphilic compounds: possible role ofpositive charge in increasing the synthesis of phosphatidylserine. Biochem Biophys Res Commun 241:73-8.
Wimley, W.C. and Thompson, T.E. (1990). Exchange and flip-flop of dimyristoylphosphatidylcholine in liquid-crystalline, gel, and two-component, two-phase large unilamellar vesicles. Biochemistry 29: 1296-303.
Wirtz, K.W. and Gadella, T.W., Jr. (1990). Properties and modes of action of specific and non-specificphospholipid transfer proteins. Experientia 46: 592-9.
Wirtz, K.W., Wouters, F.S., Bastiaens, P.H., Wanders, R.J., Seedorf, U. and Jovin, T.M. (1998). The non-specific lipid transfer protein (sterol carrier protein 2) acts as a peroxisomal fatty acyl-CoA bindingprotein. Biochem Soc Trans 26: 374-8.
Wirtz, K.W.A. (1997). Phospholipid transfer proteins revisited. Biochemical Journal 324: 353-360.Voelker, D.R. (1984). Phosphatidylserine functions as the major precursor of phosphatidylethanolamine in
cultured BHK-21 cells. Proc Natl Acad Sci U S A 81: 2669-73.Voelker, D.R. (1985). Disruption of phosphatidylserine translocation to the mitochondria in baby hamster kidney
cells. J Biol Chem 260: 14671-6.Voelker, D.R. and Frazier, J.L. (1986). Isolation and characterization of a Chinese hamster ovary cell line
requiring ethanolamine or phosphatidylserine for growth and exhibiting defective phosphatidylserinesynthase activity. J Biol Chem 261: 1002-8.
Voelker, D.R. (1989a). Reconstitution of phosphatidylserine import into rat liver mitochondria. J Biol Chem 264:8019-25.
Voelker, D.R. (1989b). Phosphatidylserine translocation to the mitochondrion is an ATP- dependent process inpermeabilized animal cells. Proc Natl Acad Sci U S A 86: 9921-5.
Voelker, D.R. (1990). Characterization of phosphatidylserine synthesis and translocation in permeabilizedanimal cells. J Biol Chem 265: 14340-6.
Voelker, D.R. (1991). Adriamycin disrupts phosphatidylserine import into the mitochondria of permeabilizedCHO-K1 cells. J Biol Chem 266: 12185-8.
Voelker, D.R. (1993). The ATP-dependent translocation of phosphatidylserine to the mitochondria is a processthat is restricted to the autologous organelle. J Biol Chem 268: 7069-74.
Voelker, D.R. (1997). Phosphatidylserine decarboxylase. Biochim Biophys Acta 1348: 236-44.Wolf, D.E., Winiski, A.P., Ting, A.E., Bocian, K.M. and Pagano, R.E. (1992). Determination of the transbilayer
distribution of fluorescent lipid analogues by nonradiative fluorescence resonance energy transfer.Biochemistry 31: 2865-73.
Wu, W.I., Routt, S., Bankaitis, V.A. and Voelker, D.R. (2000). A new gene involved in the transport-dependentmetabolism of phosphatidylserine, PSTB2/PDR17, shares sequence similarity with the gene encodingthe phosphatidylinositol/phosphatidylcholine transfer protein, SEC14. J Biol Chem 275: 14446-56.
Wu, W.I. and Voelker, D.R. (2001). Characterization of phosphatidylserine transport to the locus ofphosphatidylserine decarboxylase 2 in permeabilized yeast. J Biol Chem 276: 7114-21.
Xu, Z.L., Byers, D.M., Palmer, F.B., Spence, M.W. and Cook, H.W. (1991). Serine utilization as a precursor ofphosphatidylserine and alkenyl- (plasmenyl)-, alkyl-, and acylethanolamine phosphoglycerides incultured glioma cells. J Biol Chem 266: 2143-50.
Yamashita, S. and Nikawa, J. (1997). Phosphatidylserine synthase from yeast. Biochim Biophys Acta 1348: 228-35.
Yavin, E. and Zeigler, B.P. (1977). Regulation of phospholipid metabolism in differentiating cells from rat braincerebral hemispheres in culture. Serine incorporation into serine phosphoglycerides: base exchange anddecarboxylation patterns. J Biol Chem 252: 260-7.

76
Yoder, M.D., Thomas, L.M., Tremblay, J.M., Oliver, R.L., Yarbrough, L.R. and Helmkamp, G.M., Jr. (2001).Structure of a multifunctional protein. Mammalian phosphatidylinositol transfer protein complexed withphosphatidylcholine. J Biol Chem 276: 9246-52.
Yorek, M.A., Rosario, R.T., Dudley, D.T. and Spector, A.A. (1985). The utilization of ethanolamine and serinefor ethanolamine phosphoglyceride synthesis by human Y79 retinoblastoma cells. J Biol Chem 260:2930-6.
Zachowski, A., Favre, E., Cribier, S., Herve, P. and Devaux, P.F. (1986). Outside-inside translocation ofaminophospholipids in the human erythrocyte membrane is mediated by a specific enzyme.Biochemistry 25: 2585-90.
Zborowski, J., Dygas, A. and Wojtczak, L. (1983). Phosphatidylserine decarboxylase is located on the externalside of the inner mitochondrial membrane. FEBS Lett 157: 179-82.
Zhao, J., Zhou, Q., Wiedmer, T. and Sims, P.J. (1998). Level of expression of phospholipid scramblase regulatesinduced movement of phosphatidylserine to the cell surface. J Biol Chem 273: 6603-6.
Zilversmit, D.B. and Hughes, M.E. (1977). Extensive exchange of rat liver microsomal phospholipids. BiochimBiophys Acta 469: 99-110.
Zinser, E., Sperka-Gottlieb, C.D., Fasch, E.V., Kohlwein, S.D., Paltauf, F. and Daum, G. (1991). Phospholipidsynthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomycescerevisiae. J Bacteriol 173: 2026-34.


















