
Physikalische Chemie I-B: Kinetik - physik.leech.it · 1 EINFUHRUNG 7 Die zeitliche Analyse der Sto...
105
Physikalische Chemie I-B: Kinetik Marcus Elstner, Patrick Weis 11. Februar 2011 1
Transcript of Physikalische Chemie I-B: Kinetik - physik.leech.it · 1 EINFUHRUNG 7 Die zeitliche Analyse der Sto...

Physikalische Chemie I-B: Kinetik
Marcus Elstner, Patrick Weis
11. Februar 2011
1

INHALTSVERZEICHNIS 2
Inhaltsverzeichnis
1 Einfuhrung 61.1 Thermodynamik vs. Kinetik . . . . . . . . . . . . . . . . . . . 61.2 Zeitskalen in der (Bio-)Chemie . . . . . . . . . . . . . . . . . . 71.3 Literatur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2 Grundbegriffe 82.1 Definition der Rate (Reaktionsgeschwindigkeit) . . . . . . . . 82.2 Ratengleichung, Ratenkonstanten und Reaktionsordnung . . . 92.3 Elementarreaktionen . . . . . . . . . . . . . . . . . . . . . . . 11
3 Zeitgesetze einfacher Reaktionen 113.1 Reaktion 0. Ordnung: m=0 . . . . . . . . . . . . . . . . . . . . 123.2 Reaktion 1. Ordnung: m=1 . . . . . . . . . . . . . . . . . . . . 133.3 Halbwertszeit und Zeitkonstante . . . . . . . . . . . . . . . . . 153.4 Reaktion 2. Ordnung: m=2 . . . . . . . . . . . . . . . . . . . . 163.5 Reaktion 3. Ordnung: m=3 . . . . . . . . . . . . . . . . . . . . 193.6 Methoden zur Bestimmung der Reaktionsordnung . . . . . . . 19
4 Erweiterungen und wichtige Konzepte 224.1 Temperaturabhangigkeit: Arrhenius Gleichung . . . . . . . . . 224.2 Reaktion 1. Ordnung mit Ruckreaktion . . . . . . . . . . . . . 234.3 Parallelreaktionen . . . . . . . . . . . . . . . . . . . . . . . . . 25
4.3.1 Reaktion erster Ordnung ohne Ruckreaktion . . . . . . 254.3.2 Reaktion erster Ordnung mit Ruckreaktion: kinetische
vs. thermodynamische Kontrolle . . . . . . . . . . . . . 274.4 Folgereaktionen . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.4.1 Folgereaktionen erster Ordnung . . . . . . . . . . . . . 294.4.2 Quasistationaritatsprinzip . . . . . . . . . . . . . . . . 314.4.3 Der geschwindigkeitsbestimmende Schritt . . . . . . . . 324.4.4 Beispiel . . . . . . . . . . . . . . . . . . . . . . . . . . 334.4.5 Vorgelagertes Gleichgewicht . . . . . . . . . . . . . . . 34
4.5 Komplexe Temperaturabhangigkeiten . . . . . . . . . . . . . . 364.6 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . . . . 36
5 Komplexe Kinetiken 385.1 Unimolekulare Reaktionen: Lindeman-Hinshelwood Mechanis-
mus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 385.2 Assoziationsreaktion: Radikal-Radikal Rekombination . . . . . 405.3 Kettenreaktionen . . . . . . . . . . . . . . . . . . . . . . . . . 41

INHALTSVERZEICHNIS 3
5.3.1 Kettenreaktion ohne Verzweigung . . . . . . . . . . . . 425.3.2 Radikalische Polymerisation . . . . . . . . . . . . . . . 445.3.3 Kettenreaktion mit Verzweigung: Explosion . . . . . . 45
5.4 Enzymatische Katalyse: Michaelis-Menten . . . . . . . . . . . 485.5 Photochemie . . . . . . . . . . . . . . . . . . . . . . . . . . . . 535.6 Heterogene Katalyse: Reaktionen an Oberflachen . . . . . . . 55
5.6.1 Langmuir Adsorptionsisotherme . . . . . . . . . . . . . 555.6.2 Oberflachenreaktionen: Heterogene Katalyse . . . . . . 56
6 Das mikroskopische Bild: kinetische Gastheorie 596.1 Stoßzahl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 606.2 Geschwindigkeitsverteilung-1 . . . . . . . . . . . . . . . . . . . 626.3 Mikrosopischer Druck und Gleichverteilungssatz . . . . . . . . 646.4 Geschwindigkeitsverteilung-2 . . . . . . . . . . . . . . . . . . . 656.5 3-D Geschwindigkeitsverteilung von Maxwell und Boltzmann . 666.6 Mittelwerte der 3-D Maxwell-Boltzmann Geschwindigkeitsver-
teilung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 686.6.1 Wahrscheinlichste Geschwindigkeit . . . . . . . . . . . 686.6.2 Mittlere Geschwindigkeit . . . . . . . . . . . . . . . . . 696.6.3 Mittleres Geschwindigkeitsquadrat . . . . . . . . . . . 696.6.4 Mittlere Relativgeschwindigkeit . . . . . . . . . . . . . 70
6.7 Arrhenius 2: Mindestenergie . . . . . . . . . . . . . . . . . . . 736.8 Arrhenius 3: Energieabhangiger Stoßparameter . . . . . . . . . 74
7 Theorie des Ubergangszustandes 777.1 Potentialhyperflachen: der Ubergangszustand . . . . . . . . . . 777.2 Aktivierter Komplex . . . . . . . . . . . . . . . . . . . . . . . 807.3 Energie eines Molekuls . . . . . . . . . . . . . . . . . . . . . . 81
7.3.1 Molekulschwingungen . . . . . . . . . . . . . . . . . . . 837.4 Statistische Mechanik . . . . . . . . . . . . . . . . . . . . . . . 84
7.4.1 Molekulschwingungen: Harmonischer Oszilator . . . . . 857.4.2 Elektronische Energie . . . . . . . . . . . . . . . . . . . 857.4.3 Mittlere (Innere) Energie . . . . . . . . . . . . . . . . . 857.4.4 Entropie . . . . . . . . . . . . . . . . . . . . . . . . . 867.4.5 Freie Enthalpie . . . . . . . . . . . . . . . . . . . . . . 87
7.5 Freie Enthalpie und Gleichgewichtskonstante . . . . . . . . . . 897.6 ’Imaginare’ Schwingungsfrequenzen am Ubergangszustand . . 917.7 Eyring Gleichung . . . . . . . . . . . . . . . . . . . . . . . . . 937.8 Verbindung zu Arrhenius . . . . . . . . . . . . . . . . . . . . . 94

INHALTSVERZEICHNIS 4
8 Experimentelle Techniken 958.1 Langsame Reaktionen . . . . . . . . . . . . . . . . . . . . . . 958.2 ’stopped flow’ Technik . . . . . . . . . . . . . . . . . . . . . . 958.3 Relaxationsmethoden . . . . . . . . . . . . . . . . . . . . . . . 958.4 Blitzlichtphotolyse . . . . . . . . . . . . . . . . . . . . . . . . 978.5 Geschwindigkeitsfilter . . . . . . . . . . . . . . . . . . . . . . . 97
9 Reaktionen in Flussigkeiten: Diffusion 999.1 Diffusion (stationar) . . . . . . . . . . . . . . . . . . . . . . . 999.2 Diffusion als zeitabhangiger Transport . . . . . . . . . . . . . 1009.3 Berechnung des Diffusionskoeffizienten D aus der Gastheorie . 1029.4 Thermodynamische Behandlung der Diffusion . . . . . . . . . 104

INHALTSVERZEICHNIS 5
’If the Lord Almighty had consulted me before embarking upon thecreation, I should have recommended something simpler’(Alphonso X, the Wise of Spain (1223-1284))
Dieses Skript folgt in der inhaltlichen Auswahl und Darstellung im Wesent-lichen den Vorlesungen von Prof. Kappes und Prof. Freyland. Dank gebuhrtauch Kai Welke und Michael Messer fur eine kritische Durchsicht.

1 EINFUHRUNG 6
1 Einfuhrung
1.1 Thermodynamik vs. Kinetik
Die (Gleichgewichts-) Thermodynamik (wie bisher besprochen) machteine fundamentale Aussage uber die Energetik von chemischen Reaktionenund bestimmt damit ihre Richtung: die Konzentration der Reaktanden wirdzu kleinerer freier Enthalpie G verschoben. Es wird aber keine Ausssage uberAktivierungsenergie oder Reaktionsgeschwindigkeit gemacht.
Desweiteren bestimmt sie das thermodynamische Gleichgewicht, dassich im Grenzfall langer Reaktionszeiten einstellt. Betrachten wir dazu fol-gende Reaktion:
A B
Das Verhaltnis der Konzentrationen [A] und [B] ist im Gleichgewicht durchdie Gleichgewichtskonstante K gegeben, und diese ist ist mit ∆G0 ver-knupft:
∆G0 = −RTlnKDie Thermodynamik gibt also Auskunft uber das Ergebnis einer Reaktion(wenn man sehr lange wartet), sagt aber nichts uber den Verlauf, die Ge-schwindigkeit oder den Mechanismus aus. Da z.B. ein Katalysator nichtsan der Energetik der Reaktion andert, macht die Thermodynamik identischeAussagen uber katalysierte und unkatalysierte Reaktionen.
Die Kinetik dagegen untersucht die Prozesse im Nichtgleichgewicht.
• Z.B. wird das obige System durch Reduktion von [B] stark aus demGleichgewicht ausgelenkt und dann der Stoffumsatz von A betrachtet.Welche Gesetzmassigkeiten gelten fur die Reaktion, d.h. wie sieht derzeitliche Ablauf aus?
• Lassen sich unterschiedliche Reaktionen charakterisieren?
• Woraus resultieren unterschiedlichen Reaktionsgeschwindigkeiten?
Was beeinflusst nun die Reaktionsgeschwindigkeit?
• Mechanismus: Welche Einzelschritte laufen bei der Stoffumwandlungab?
• Die Geschwindigkeit, mit der jeder der Einzelschritte ablauft, be-stimmt die Geschwindigkeit, mit der das Gleichgewicht erreicht wird.

1 EINFUHRUNG 7
Die zeitliche Analyse der Stoffumwandlungen erlaubt nun Aufschlusse uberden Mechanismus und die Zeitskala, auf der die Reaktion ablauft. Diesist das Ziel der chemischen Kinetik.
Eine Analyse von Reaktionen in Begriffen der Thermodynamik (∆G, K)und Kinetik (Mechanismus, Reaktionsrate) ist zentral fur das Verstand-nis von chemischen Reaktionen. Dies gilt genauso fur die Biochemie. DieFunktionsweise von Proteinen wird oft uberhaut erst durch eine solche Ana-lyse moglich. Es ist die Frage nach der Effizienz: Wie gelingt es Proteinen,bestimmte Prozesse so effizient umzusetzen?
1.2 Zeitskalen in der (Bio-)Chemie
Abbildung 1: (Bio-)Physikalische Zeitskalen, aus: D. Zhong, Ultrafast cataly-tic processes in enzymes, Current Opinion in Chemical Biology 2007, 11:174
1.3 Literatur
• P. Atkins, Physical Chemistry, Oxford University Press.
• J. P. Allen, Biophyiscal Chemistry, Wiley-Blackwell.
• G. Wedler, Lehrbuch der Physikalischen Chemie, VCH Weinheim.

2 GRUNDBEGRIFFE 8
2 Grundbegriffe
2.1 Definition der Rate (Reaktionsgeschwindigkeit)
Die ’Geschwindigkeit’ einer Reaktion gibt an, wie schnell ein Stoff umgesetztwird. Bei der Reaktion
A→ B
wird man z. B. messen, wie schnell sich die Konzentration der Substanz A,[A], andert.
∆[A]
∆t→ d[A]
dt= v
v ist damit ein Maß fur die Reaktionsgeschwindigkeit.
Betrachten wir aber die Reaktion:
A+ 2B → 4C +D
Aus der Stochiometrie folgt nun fur die Anderungen der Konzentrationen:
−d[A]
dt= −1
2
d[B]
dt=d[D]
dt=
1
4
d[C]
dt
• die Konzentrationen von A und B nehmen ab, d.h. die Ableitungensind negativ: daher das ’-’ vor den Ableitungen.
• Die Konzentration von C nimmt vier mal schneller zu als die von D:daher der Faktor 1/4.
Die Geschwindigkeiten der Bildung bzw. des Abbaus der Produkte bzw.Edukte unterscheiden sich offensichtlich durch die stochiometrischen Ko-effizienten νi ( νA = 1, νB = 2, νC = 4, νD = 1).
Um eine eindeutige Beschreibung der Reaktionsgeschwindigkeit zu erhalten,wird daher die sogenannte Reaktionslaufzahl ξ eingefuhrt:
ξ =niνi
(2.1)
ni gibt die Stoffmenge der chemischen Spezies in ’mol’ an. Die Anderung derReaktionslaufzahl wahrend einer Reaktion ist dann:
dξ =dniνi
=dnjνj
= ... (2.2)

2 GRUNDBEGRIFFE 9
Am obigen Beispiel A+ 2B → 4C +D:
dξ =dnC
4=dnD
1= −dnA
1= −dnB
2
Damit sieht man sofort den Sinn der Reaktionslaufzahl: sie charakterisiertdie Reaktion als solche, ist also unabhangig davon, welchen Reaktanden manbetrachtet! Damit ist also eine eindeutige Beschreibung einer Reaktionsge-schwindigkeit erst ermoglicht.
Nun wollen wir diese Gleichungen nochmals umschreiben, um mit Konzen-trationen arbeiten zu konnen. Die Konzentation ci = [i] ist gegeben durch:
ci =niV
(V: Volumen), in Gl. 2.2 eingesetzt:
1
Vdξ =
dciνi
=dcjνj
= ...
Jetzt teilen wir noch durch ’dt’ und erhalten die Reaktionsgeschwindig-keit rv
rv =1
V
dξ
dt=
1
νi
d[i]
dt=
1
νj
d[j]
dt= ... (2.3)
Bitte beachten Sie, dass rv auf ein Volumen V bezogen ist. Eine alternativeDefinition der Reaktionsgeschwindigkeit bezieht sich nur auf die Molzahlen,rξ = dξ/dt.
2.2 Ratengleichung, Ratenkonstanten und Reaktions-ordnung
In vielen Fallen hangt die Reaktionsrate von der Konzentration der Reaktan-den ab. Diese Abhangigkeit kann sehr komplex sein, i.A. kann man schreiben(A+B → P):
rv = f([A], [B]) (2.4)

2 GRUNDBEGRIFFE 10
Wenn Ruckreaktionen auftreten, so gehen hier auch die Konzentrationen derProdukte ein. Die genaue Form der Kinetik gibt Aufschluss uber den Me-chanismus. In einem einfachen Fall einer solchen Ratengleichung waref([A], [B]) beispielsweise durch ein Produkt gegeben:
rv = k[A][B], (2.5)
wobei k als Ratenkonstante (Geschwindigkeitskonstante) bezeichnetwird.
Einfache Ratengleichungen (Geschwindigkeitsgesetze), wie sie im nachstenKapitel diskutiert werden sollen, haben die folgende Form (A,B,C: Reaktan-den):
rv = k[A]m1 [B]m2 [C]m3 (2.6)
• m = m1 + m2 + m3 wird die (Gesamt-) Reaktionsordnung genannt.
• die mi sind oft ganze Zahlen: Achtung, sie haben in der Regel nichtsmit der Stochiometrie νi zu tun!
• rv ist selbst zeitabhangig, d.h. man muss rv(t) schreiben. Die Reakti-onsgeschwindigkeit kann sich im Laufe der Reaktion andern.
• Die Dimension der Zeitkonstante ist unterschiedlich fur die verschiede-nen Reaktionsordnungen! (s.u.)
• in diesen einfachen Ratengleichungen tauchen zunachst nur die Kon-zentrationen der Reaktanden auf, Ruckreaktionen werden spater be-trachtet.
Beispiele:
• 2N2O5 → 4NO2 + O2.Exp. Befund:
rv =d[N2O5]
−2dt=d[NO2]
4dt= k[N2O5]1
Dies ist eine Reaktion erster Ordnung, wobei der stochiometrische Ko-effizient 2 ist.

3 ZEITGESETZE EINFACHER REAKTIONEN 11
• 2NO + O2 = 2NO2
Exp. Befund:
rv =d[NO2]
−2dt= k[NO]2[O2]1
Dies ist eine Reaktion dritter Ordnung, 2ter Ordnung in NO, ersterOrdnung in O2. Dass die stoch. Koeff. gleich der Reaktionsordnungsind, ist eher die Ausnahme.
• H2+ Br2 → 2 HBrExp. Befund:zunachst
rv =d[HBr]
2dt= k[H2][Br]1/2
gebrochene Reaktionsordnung, d.h. komplizierter Mechanismusspater:
rv =d[HBr]
2dt=k[H2][Br]1/2
1 + k‘[HBr]
ist viel HBr gebildet, wird die Kinetik noch komplizierter, man kannkeine Reaktionsordnung mehr angeben
2.3 Elementarreaktionen
Oft resultiert die Gesamtreaktion aus mehreren Elementarreaktionen. DieAufklarung des Mechanismus der Reaktion fuhrt zu einer Identifizierung derElementarreaktionen. Diese unterscheidet man anhand ihrer Molekularitat:
• monomolekular: z.B. Dissoziation I2 → 2I oder Isomerisierung.
• dimolekular: ein ’Zweierstoss’ fuhrt zur Reaktion, z.B. NO + O3 →NO2 +O2
• trimolekular: Existenz unklar.
3 Zeitgesetze einfacher Reaktionen
Die obigen Zeitgesetze haben die mathematische Form von Differential-gleichungen (DGL), d.h., sie geben die Anderung der Konzentration inAbhangigkeit der Konzentrationen der Reaktanden an. Denken Sie dabei z.B.an das radioaktive Zerfallsgesetz. Hier haben wir auch die Zahl der Zerfallepro Zeit, N in Abhangigkeit von der Teilchenzahl N ausgedruckt:
N = −λN

3 ZEITGESETZE EINFACHER REAKTIONEN 12
Diese Gesetzmassigkeit beschreibt die zeitliche Anderung. Was uns aber i.A.interessiert ist, wie sich N zeitlich andert. Dies erhalt man durch Integrationder Differentialgleichung:
N(t) = N0e−λt
Im Folgenden werden wir die einfachen Zeitgesetze integrieren (d.h. die Diffe-rentialgleichungen losen, siehe PC0), um den zeitlichen Verlauf der Konzen-trationen zu erhalten. Damit erhalt man aus den experimentell bestimmtenKonzentrationsverlaufen c(t)=[i](t) Aufschlusse uber das Geschwindigkeits-gesetz, die Ordnung und damit den Mechanismus.
3.1 Reaktion 0. Ordnung: m=0
Dies sind Reaktionen, bei denen die Reaktionsgeschwindingkeiten unabhangigvon der Anfangskonzentration sind.Beispiele:
• Verdunsten von Wasser aus einem Glas. Wieviel Wasser pro Stundeverdunstet, hangt von der Oberflache, Druck, Temperatur etc. ab, abernicht von der Wassermenge im Glas.
• eher ungewohnlich in der Chemie, treten insbes. bei heterogenen Re-aktionen (Oberflachen) auf.
• Enzymatische Reaktionen, in denen ein Uberschuss an Substrat vorliegt
z.B.:A→ B
Reaktionsgeschwindigkeit ist unabhangig von der Konzentration [A].
rv = −d[A]
dt= k0 (3.1)
Die Ableitung ist negativ, die Konstante k0 aber positiv, daher wird die linkeSeite mit (-1) multipliziert (wir schreiben im Folgenden [A](t) = [A]t = [A]).Umformen:
−d[A] = k0dt (3.2)
Integration:
−∫ [A]t1
[A]t0
d[A] =
∫ t1
t0
k0dt (3.3)

3 ZEITGESETZE EINFACHER REAKTIONEN 13
Stammfunktion:
−([A]t1 − [A]t0) = k0(t1 − t0) (3.4)
oder (sei t0 =0, [A]t0 = [A]0):
[A]t1 = [A]0 − k0t1 (3.5)
Wir konnen auch schreiben (wir ersetzen t1 durch t):
[A]t = [A]0 − k0t (3.6)
Die Konzentration nimmt also linear mit der Zeit ab, wie aus Abb. 2 ersicht-lich.
Abbildung 2: Zeitabhangigkeit der Konzentration fur m=0. Gezeigt ist hierder Verlauf mit [A]0 = 3 und k = 0.5
Die Dimension der Reaktionskonstante ist
dim(k0) =mol
ls
(Eigentlich schreibt man fur die Dimension [k0], hier besteht aber Verwechs-lungsgefahr mit der Konzentration).
3.2 Reaktion 1. Ordnung: m=1
Dieses Geschwindigkeitsgesetz gilt fur Reaktionen, bei denen die Reaktions-geschwindigkeit proportional zur Konzentration eines der Reaktanden ist.Man findet es z.B. fur:
• radioaktiven Zerfall
• viele Dissoziationsreaktionen, zB.: 2N2O5 → 4NO2 +O2
• viele Isomerisierungsreaktionen, z.B. Cyclopropan → Propen

3 ZEITGESETZE EINFACHER REAKTIONEN 14
Die genannten Reaktionen sind monomolekular:
A→ B
rv = −d[A]
dt= k1[A] (3.7)
Trennung der Variablen:
d[A]
[A]= −k1dt (3.8)
Stammfunktion (∫
(1/x)dx = ln(x)):
ln([A])− ln([A]0) = −k1t (3.9)
oder
ln[A]
[A]0= −k1t (3.10)
Losung:
[A] = [A]0e−kt (3.11)
Wie beim radioaktiven Zerfall sinkt die Anfangskonzentration exponentiell.Graphisch ist es oft gunstiger Gl. 3.10 statt Gl. 3.11 aufzutragen. Durch Auf-tragung von ln [A]
[A]0uber t kann man -k1 direkt aus der Steigung der Geraden
ablesen. Die Dimension der Reaktionskonstante ist (kt ist dimensionslos):
dim(k1) =1
s.
Wie sieht nun die Bildung von [B] aus? Wie oben angenommen, gibt es keineRuckreaktion, wir haben daher ([B]0 = 0):
[B] = [A]0 − [A]
Mit [A] = [A]0e−kt erhalten wir:
[B] = [A]0 − [A] = [A]0 − [A]0e−kt = [A]0(1− e−kt) (3.12)
Damit ist die zeitliche Entwicklung von [B] wie in Abb. 4 gezeigt.

3 ZEITGESETZE EINFACHER REAKTIONEN 15
Abbildung 3: Zeitabhangigkeit der Konzentration fur m=1. Gezeigt ist hierder Verlauf nach Gl. 3.11 und Gl. 3.10 mit [A]0 = 1000 und k = 10
3.3 Halbwertszeit und Zeitkonstante
Die Halbwertszeit t1/2 ist definiert als die Zeit, in der die Anfangskonzen-tration um die Halfte abnimmt, also:
[A]
[A]0=
1
2.
Mit Gl. 3.10 gilt dann:
ln(1
2) = −k1t1/2 (3.13)
d.h.:
t1/2 =ln2
k1
(3.14)
Bei Reaktionen erster Ordnung ist also die Halbwertszeit unabhangig vonder Konzentration.
Eine weitere wichtige Grosse ist die Zeitkonstante τ . Sie ist definiert alsdie Zeit, in der die Konzentration of 1/e abgefallen ist, d.h.
[A]
[A]0=
1
e.
Analog zur Halbwertszeit erhalt man
τ =1
k1
. (3.15)
τ ist eine charakteristische Grosse der Dynamik.
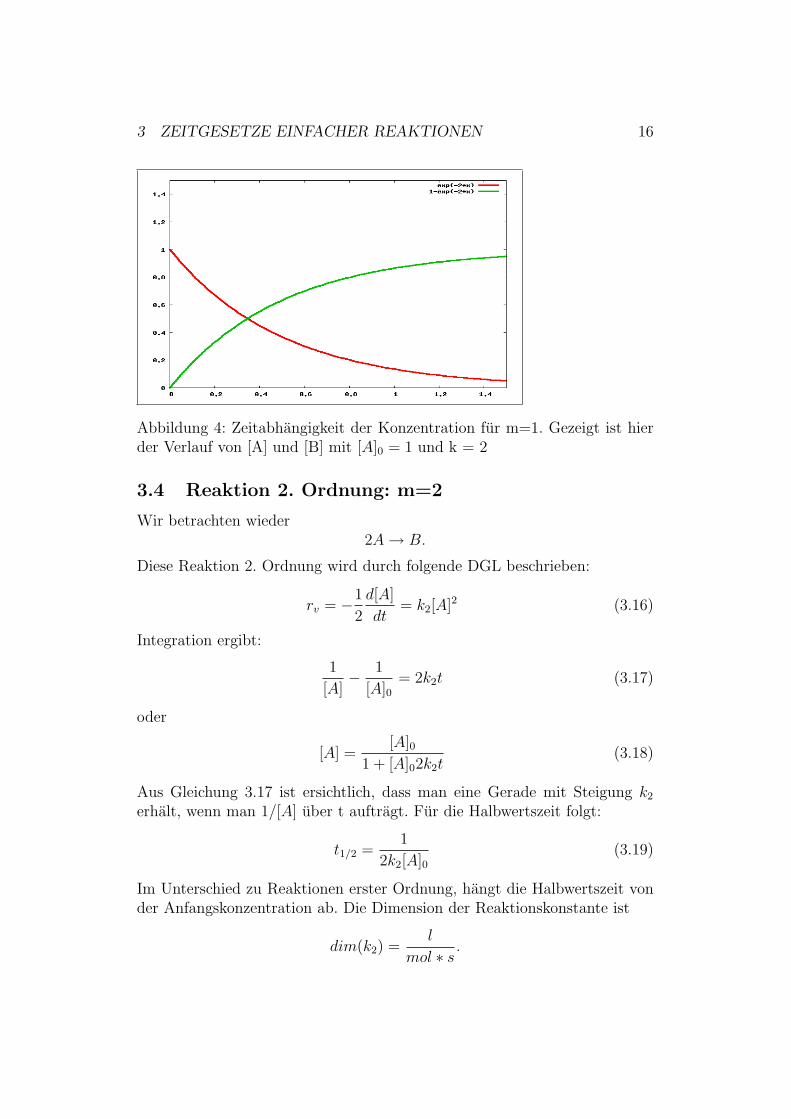
3 ZEITGESETZE EINFACHER REAKTIONEN 16
Abbildung 4: Zeitabhangigkeit der Konzentration fur m=1. Gezeigt ist hierder Verlauf von [A] und [B] mit [A]0 = 1 und k = 2
3.4 Reaktion 2. Ordnung: m=2
Wir betrachten wieder2A→ B.
Diese Reaktion 2. Ordnung wird durch folgende DGL beschrieben:
rv = −1
2
d[A]
dt= k2[A]2 (3.16)
Integration ergibt:
1
[A]− 1
[A]0= 2k2t (3.17)
oder
[A] =[A]0
1 + [A]02k2t(3.18)
Aus Gleichung 3.17 ist ersichtlich, dass man eine Gerade mit Steigung k2
erhalt, wenn man 1/[A] uber t auftragt. Fur die Halbwertszeit folgt:
t1/2 =1
2k2[A]0(3.19)
Im Unterschied zu Reaktionen erster Ordnung, hangt die Halbwertszeit vonder Anfangskonzentration ab. Die Dimension der Reaktionskonstante ist
dim(k2) =l
mol ∗ s.

3 ZEITGESETZE EINFACHER REAKTIONEN 17
Abbildung 5: Zeitabhangigkeit der Konzentration fur m=2. Gezeigt ist hierder Verlauf nach Gl. 3.18 und Gl. 3.17 mit [A]0 = 2 und k = 1
Reaktion 2. Ordnung: Zwei Reaktanden
Ein weiteres Beispiel fur eine Reaktion 2. Ordnung ist
A+B → P.
Wenn die Reaktion jeweils erster Ordnung in den einzelnen Reaktanden ist,erhalt man:
−d[A]
dt= k2[A][B] (3.20)
Diese DGL kann nun nicht ohne Weiteres integriert werden, man benotigteine Bedingung, die die Konzentration von B mit der von A in Verbindungsetzt.
Nach einer gewissen Zeit ist die Konzentration von A um den Wert x ge-sunken, d.h.
[A] = [A]0 − xAufgrund der Stochiometrie gilt daher auch fur B:
[B] = [B]0 − x,
man kann also schreiben (d[A]/dt = −dx/dt):
dx
dt= k2([A]0 − x)([B]0 − x). (3.21)

3 ZEITGESETZE EINFACHER REAKTIONEN 18
Integration ergibt (siehe Ubungsblatt):
ln
([B]/[B]0[A]/[A]0
)= ([B]0 − [A]0)kt (3.22)
Durch Auftragen der rechten Seite vs. t kann wiederum k bestimmt werden.
Spezialfalle:
• [A]0 = [B]0: diese Bedingung in Gl. 3.21 eingesetzt ergibt:
1
[A]− 1
[A]0= k2t
• [B]0 � [A]0: hier kann man annehmen, dass sich [B] wahrend derReaktion kaum andert, damit erhalt man eine Reaktion 1. Ordnungin[A].

3 ZEITGESETZE EINFACHER REAKTIONEN 19
3.5 Reaktion 3. Ordnung: m=3
Reaktionen dritter Ordung sind bereits sehr selten. Ein Beispiel ist
2NO +O2 → 2NO2.
Um diese zu analysieren ist es zweckmassig, die Anfangskonzentrationen imstochiometrischen Verhaltnis zu wahlen, d.h.
[A]0νA
=[B]0νB
=[C]0νC
= a.
Damit erhalt man analog zur Reaktion 2.Ordnung ([A] = [A]0 − νAx, ...):
dx
dt= k3([A]0 − νax)([B]0 − νBx)([C]0 − νCx). (3.23)
oder
dx
dt= k3νAνBνC(a− x)3 (3.24)
Dies kann man durch Trennung der Variablen wieder elementar integrieren(Details, siehe Wedler).
Der Speziallfall3A→ P
fuhrt mit νA = 3 auf die einfache DGL
−1
3
d[A]
dt= k3[A]3, (3.25)
die elementar zu integrieren ist
1
[A]2=
1
[A]20+ 6k3t. (3.26)
3.6 Methoden zur Bestimmung der Reaktionsordnung
Wenn man fur eine Reaktion
• 0. Ordnung [A]− [A]0
• 1. Ordnung ln [A][A]0
• 2. Ordnung 1[A]

3 ZEITGESETZE EINFACHER REAKTIONEN 20
• 3. Ordnung 1[A]2
uber der Zeit t auftragt, erhalt man eine Gerade mit der Steigung k. Damitkann man also sowohl den Grad der Reaktion als auch die Reaktionskonstanteidentifizieren. Die verschiedenen Auftragungsarten ermoglichen meist einebessere Differenzierung als die direkte Zeitabhangigkeit, wie in Abb. 6 gezeigt.Man kann die Reaktionsordnung daher direkt durch diese Auftragungsarten
Abbildung 6: Vergleich der Zeitabhangigkeit der Konzentrationen furm=0,1,2. Gezeigt ist hier der Verlauf mit [A]0 = 1 und k = 1 und [A]0 = 2und k = 1
bestimmen. Eine weitere Methode zur Bestimmung der Reaktionsordnungfur Reaktionen des Typs
mA→ P
ist das Verfahren von Van’t Hoff. Hier muss man die Reaktionsgeschwin-digkeit als Funktion der Zeit messen.
− 1
m
d[A]
dt= k[A]m, (3.27)
−d[A]
dt= mk[A]m
ln
(−d[A]
dt
)= ln(mk[A]m) = ln(mk) +mln[A]
Wenn man also ln(−d[A]dt
) vs. ln[A] auftragt, findet man als Steigung die Re-aktionsordnung m.
Eine andere Moglichkeit ist das Halbwertszeitverfahren. Fur m=1 habenwir gefunden:
t1/2 =ln2
k1
, ln(t1/2) = const.

3 ZEITGESETZE EINFACHER REAKTIONEN 21
Allgemein (m 6= 1) kann man zeigen:
t1/2 =2m−1 − 1
k(m− 1)[A]m−10
ln(t1/2) = ln
(2m−1 − 1
k(m− 1)
)− (m− 1)ln[A]0
D.h., wenn die Halbwertszeit fur verschiedene Anfangskonzentrationen ge-messen wird, kann man durch Auftragen von t1/2 vs [A]0 direkt (m-1) alsSteigung ablesen.
Beispiele:
• m=0: wie oben erwahnt, findet man diese Ordnung z.B. bei enzymkata-lysierten Reaktionen mit Substratuberschuss. In diesem Fall spielt dieKonzentration im Zeitgesetz keine Rolle, das Produkt wird mit kon-stanter Rate gebildet, die nur von der Reaktionsgeschwindigkeit undder Enzymkonzentration abhangt. Die Konzentration des Reaktandengeht nicht mit ein.
• m=1: Dieses Verhalten kann am Beispiel von monomolekularen Re-aktionen verstanden werden. Jedes Molekul reagiert (zerfallt) mit einerbestimmten Wahrscheinlichkeit, die unabhangig von der Gesamtzahlder vorhandenen Molekule ist. Wieviele Molekule pro Zeiteinheit dannumgesetzt werden, hangt von der Gesamtzahl ab. Damit erhalt mandas aus der Radioaktivitat bekannte exponentielle Verhalten.
• m=2: Hier geht die Konzentration im Quadrat ein. Dies kann manmit Hilfe der bimolekularen Reaktion A + A → P verstehen (z.B.2NO2 → 2NO+O2). Jedes Molekul benotigt, im Gegensatz zum ’Zer-fall’, einen Partner fur die Reaktion. Damit hangt die Reaktion auchdavon ab, dass zwei Molekule zusammenstossen. Die Zahl der Zusam-menstosse hangt jedoch von der Konzentration des einen Partners Aund des anderen Partners A ab, damit von dem Produkt der Konzen-trationen (allgemein: A + B → P ). Wenn eine Reaktion aus einemelementaren bimolekularen Prozess besteht, dann findet man eine Ki-netik 2. Ordnung. Der Umkehrschluss muss jedoch nicht gelten.
Die Reaktion A+ B → P , die eine Reaktion 2. Ordnung ist, kann jedoch ineine Reaktion erster Ordnung ubergehen, wenn z.B. einer der Reaktionspart-ner im Uberschuss vorhanden ist. Dann findet z.B. jedes A sofort ein B, unddie Reaktionsgeschwindigkeit hangt wieder nur von [A] ab.

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 22
4 Erweiterungen und wichtige Konzepte
Die bisher besprochenen Kinetiken stellen Spezialfalle dar. Es wurde derBeginn einer einfachen Reaktion beschrieben. Wenn sich die Reaktion demGleichgewicht nahert wird i.A. eine Ruckreaktion stattfinden. Des Weiterenkonnen Verzweigungen oder Folgereaktionen auftreten. Diese Falle sollen imFolgenden besprochen werden. Zunachst betrachten wir jedoch die Tempera-turabhangigkeit einfacher Reaktionen. Diese erlaubt eine mikroskopischeInterpretation der Geschwindigkeitskonstante.
4.1 Temperaturabhangigkeit: Arrhenius Gleichung
Im Allgemeinen ist die Geschwindigkeitskonstante abhangig von der Tempe-ratur, d.h. es gilt:
kn = k(T )
In umfangreichen Experimenten hat Arrhenius (1889) folgende Temperatu-rabhangigkeit gefunden (A, C: Konstanten):
kn = Ae−C/T
Mit εa := Ck und Ea = NAεa erhalten wir die beruhmte Arrhenius Glei-chung:
kn = Ae−εa/kT = Ae−Ea/RT (4.1)
kT hat die Einheit der Energie (kT = 0.593 kcal/mol bei Raumtermpera-tur), daher stellt εa eine Energie dar. Wie in der Thermodynamik gezeigt,gibt e−εa/kT den Bruchteil der Molekule an, die eine Energie grosser odergleich kT besitzen. Die Rate ist damit also abhangig von der Anzahl der Mo-lekule, die eine Energie grosser kT haben. Dies legt ein einfaches Bild nahe,bei dem zwar oft Stosse zwischen zwei Molekulen vorkommen, jedoch nursolche Stosse zu einer Reaktion fuhren, bei der die Partner die Mindestener-gie εa > kT mitbringen. Im Allgemeinen bezieht man sich jedoch auf molareGrossen, d.h. auf Ea = NAεa. Man nennt Ea die Aktivierungsenergie.
Dies lasst sich anschaulich darstellen (Abb. 7). Bei der Hinreaktion muss eineBarriere der Energie Ea, bei der Ruckreaktion der Energie E ′a, uberwundenwerden. Die Differenz der Aktivierungsenergien
∆H = Ea − E ′aist die Reaktionsenthalpie ∆H. Die Aktivierungsenergie kann ermittelt wer-den, wenn die Geschwindigkeitskonstante fur verschiedene Temperaturen ge-messen wird. Man kann ln(k) vs 1/T auftragen,

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 23
Abbildung 7: Linke Seite: Reaktionsbarriere fur Hin- (Ea) und Ruckreaktion(Ea’). Rechte Seite: ln(k) vs.1/T
ln(k) = lnA− EaRT
(4.2)
und aus der Steigung der Geraden die Aktivierungsenergie ablesen. Die Be-deutung der Konstante A wird weiter unten ausfuhrlich diskutiert. A ist einMass fur die Anzahl der Stosse, die aber nur mit der Wahrscheinlichkeite−Ea/RT genugend Energie aufbringen, die Barriere zu uberwinden.
4.2 Reaktion 1. Ordnung mit Ruckreaktion
Betrachten wir folgende Reaktion erster Ordnung mit Ruckreaktion ersterOrdnung
A B,
wobei die Geschwindigkeitskonstanten fur die Hinreaktion mit k1 und fur dieRuckreaktion mit k−1 bezeichnet werden.
−d[A]
dt= k1[A]− k−1[B] (4.3)
Des Weiteren gilt fur die Bildung von B:
d[B]
dt= k1[A]− k−1[B] (4.4)
Dies ist ein System gekoppelter DGL, da die Entwicklung von [A] auchvon [B] abhangt. Um die erste DGL zu Losen, benotigt man [B], das man

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 24
erst durch die Losung der zweiten DGL erhalt. Zur Vereinfachung werdenwir die DGL entkoppeln. Dazu verwendet man die Stoffbilanz:
[A] + [B] = [A]0 + [B]0 = [A]∞ + [B]∞ (4.5)
nach [B] auflosen und in Gl. 4.3 einsetzen:
−d[A]
dt= k1[A]− k−1([A]0 + [B]0 − [A]) = (4.6)
= (k1 + k−1)[A]− k−1([A]0 + [B]0) =
= (k1 + k−1)
([A]− k−1
k1 + k−1
([A]0 + [B]0)
)=
=: (k1 + k−1) ([A]− g)
Mit x = [A]− g ergibt sich(d[A]dt
= dxdt
):
−dxdt
= (k1 + k−1)x (4.7)
Losung:
ln(x) = −(k1 + k−1)t+ c (4.8)
oder (c’ = ec)
x = c′ ∗ e−(k1+k−1)t, (4.9)
d.h.
[A] = c′ ∗ e−(k1+k−1)t + g, (4.10)
Bestimme c aus Anfangsbedingungen, t=0 ([A]t=0 = [A]0):
c′ = [A]0 − g (4.11)
Jetzt c’ einsetzen:
[A] = ([A]0 − g) ∗ e−(k1+k−1)t + g, (4.12)
Fur t→∞ erhalt man (Thermo. GG):
[A]∞ = g, (4.13)
also
[A] = [A]∞ + ([A]0 − [A]∞) ∗ e−(k1+k−1)t (4.14)

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 25
Die Konzentration fallt also, wie bei der Reaktion 1.Ordnung, exponentiellab. Allerdings ist der Grenzwert nicht gleich ’0’ sondern gleich der Konzen-tration im thermodynamischen Gleichgewicht [A]∞. Aufgrund der StoffbilanzGl. 4.5 gilt:
[B] = [B]∞ + ([B]0 − [B]∞) ∗ e−(k1+k−1)t (4.15)
Die DGL 4.3 ergibt fur t→∞
0 = −d[A]
dt= k1[A]∞ − k−1[B]∞ (4.16)
und man erhalt eine Beziehung zwischen der Kinetik und der Thermodyna-mik:
k1
k−1
=[B]∞[A]∞
= Keq (4.17)
Im Grenzfall ’langer’ Reaktionszeiten erreicht die Reaktion das Thermo-dynamische Geichgewicht. Abb. 8 zeigt den Verlauf von [A] und [B] .
Abbildung 8: [A] und [B] mit A0 =1 und B0 =0, k1=1, k−1=0.5 (links) undmit A0 =1 und B0 =0, k1=2, k−1=1 (rechts)
Die Gleichgewichtskonzentrationen sind durch das Verhaltnis der Geschwin-digkeitskonstanten gegeben. Eine proportionale Anderung der Geschwindig-keitskonstanten (z.b. auf 2 und 1) beschleunigt oder verzogert das Erreichendes Gleichgewichts.
4.3 Parallelreaktionen
4.3.1 Reaktion erster Ordnung ohne Ruckreaktion
Das Edukt A reagiert zu zwei verschiedenen Produkten B und C mit denRatenkonstanten kB und kC :
AkB−→ B

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 26
AkC−→ C
Seien[B]0 = [C]0 = 0, dann gilt die Massenbilanz:
[A] + [B] + [C] = [A]0 (4.18)
Ratengleichung fur A:
−d[A]
dt= kB[A] + kC [A] = (kB + kC)[A] (4.19)
Losung (analog m=1):
[A] = [A]0e−(kB+kC)t (4.20)
Ratengleichung fur B:
d[B]
dt= kB[A] = kB[A]0e
−(kB+kC)t (4.21)
Losung:
[B]− [B]0 = kB[A]0
∫ t
0
e−(kB+kC)tdt (4.22)
mit [B]0 = 0:
[B] =kB
kB + kC[A]0
(1− e−(kB+kC)t
)(4.23)
Analog fur C:
[C] =kC
kB + kC[A]0
(1− e−(kB+kC)t
)(4.24)
d.h.
[B]
[C]=kBkC
(4.25)
Das Verhaltnis der Produkte ist also durch das Verhaltnis der Ratenkonstan-ten gegeben.

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 27
4.3.2 Reaktion erster Ordnung mit Ruckreaktion: kinetische vs.thermodynamische Kontrolle
A B
A C
Wir verwenden wieder [B]0 = [C]0 = 0 und die Massenbilanz:
[A] + [B] + [C] = [A]0 (4.26)
Ratengleichungen:
−d[A]
dt= kB[A] + kC [A]− k−B[B]− k−C [C] (4.27)
d[B]
dt= kB[A]− k−B[B]
d[C]
dt= kC [A]− k−C [C]
Im Prinzip kann man diese gekoppelten DGL losen. Zur Verdeutlichung desPrinzips der kinetischen vs. thermodynamischen Kontrolle betrachtenwir jedoch nur zwei Grenzfalle:
a) kurze Zeiten t:Hier sind die Konzentrationen [B] und [C] noch klein, die Ruckreaktionenspielen keine Rolle, d.h. die letzen Terme in der obigen DGL werden ver-nachlassigt. Die Losung ist dann wie oben (ohne Ruckreaktion):
[B]
[C]=kBkC
Kinetische Kontrolle: Die Produktkonzentrationen werden durch die Ge-schwindigkeitskonstanten bestimmt. Wie oben ausgefuhrt, hangen diese mitder Aktivierungsenergie Ea zusammen. Kinetische Kontrolle bedeutet da-her, dass die Aktivierungsenergie und NICHT die Reaktionsenergie (Ent-halpie) ∆G0 fur die Reaktion bestimmend ist.
b) lange Zeiten t: Thermodynamisches Gleichgewicht
d[i]
dt= 0
Damit erhalt man aus der 2. und 3. DGL von Gl. 4.27:
0 =d[B]
dt= kB[A]∞ − k−B[B]∞ (4.28)

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 28
0 =d[C]
dt= kC [A]∞ − k−C [C]∞ (4.29)
d.h.
[B]∞[A]∞
=kBk−B
= KBeq (4.30)
[C]∞[A]∞
=kCk−C
= KCeq (4.31)
und:
[B]∞[C]∞
=kBk−Ck−BkC
=KBeq
KCeq
(4.32)
Nun kann man die sich einstellenden Konzentrationen direkt ausrechnen. Ausder Stoffbilanz
[A]0 = [A]∞ + [B]∞ + [C]∞
erhalt man sofort:
[A]∞ =[A]0
1 +KBeq +KC
eq
, [B]∞ =[A]0K
Beq
1 +KBeq +KC
eq
, [C]∞ =[A]0K
Ceq
1 +KBeq +KC
eq
Thermodynamische Kontrolle: Die Produktkonzentationen werden durchdie Gleichgewichtskonstanten Keq bestimmt. Wie oben ausgefuhrt, hangendiese mit der Reaktionsenthalpie ∆G0 zusammen. ThermodynamischeKontrolle bedeutet daher, dass die Reaktionsenthalpie und NICHT die Ak-tivierungsenergie fur die Reaktion bestimmend ist.
Am Anfang der Reaktion ist das Produktverhaltnis demnach durch das Verhalt-nis der Geschwindigkeitskonstanten bestimmt, am Ende durch das Verhaltnisder Gleichgewichtskonstanten.
Dies kann man anhand eines Beispiels illustrieren. Mit kB = 1s−1, kC =0.1s−1, k−B = 0.01s−1, k−C = 0.0001s−1 erhalt man fur den Beginn der Re-aktion
[B]Anfang[C]Anfang
=kBkC
= 10
(kinetische Kontrolle) und fur lange Zeiten das thermodynamische Gleichge-wicht
[B]∞[C]∞
=kBk−Ck−BkC
= 0.1.
(thermodynamische Kontrolle). Die beiden Falle sind in Abb. 9 dargestellt.

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 29
Abbildung 9: Anfang der Reaktion (links) und Einstellen des thermodyna-mischen Gleichgewichts (rechts). Abbildungen von Dr. P. Weis
4.4 Folgereaktionen
4.4.1 Folgereaktionen erster Ordnung
Wir betrachten folgende Reaktion
AkB−→ B
kC−→ C
mit den Geschwindigkeitskonstanten kB und kC . Die DGL’s sind folglich:
−d[A]
dt= kB[A] (4.33)
d[B]
dt= kB[A]− kC [B]
d[C]
dt= kC [B]
Als Anfangsbedingungen wahlen wir [B]0 = [C]0 = 0. Fur [A] ist die Losungwieder trivial
[A] = [A]0e−kBt, (4.34)
aber fur [B] erhalten wir mit dieser Losung eine etwas ungewohnliche Form:
d[B]
dt+ kC [B] = kB[A]0e
−kBt

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 30
Dies ist eine DGL der Form ([B] = y, t=x):
dy
dx+ f(x)y = g(x)
Allgemein lost man diese mit der Methode der Integralfaktoren. Man multipliziert beideSeiten mit:
eRf(x)dx = e
RkCdt = ekCt
und erhalt:
ekCtd[B]
dt+ ekCtkC [B] = kB[A]0e
−kBtekCt
Mitd([B]ekCt
)dt
= ekCtd[B]
dt+ ekCtkC [B]
ergibt sich:d([B]ekCt
)dt
= kB[A]0e(kC−kB)t
Integration: ∫d([B]ekCt
)= kB[A]0
∫e(kC−kB)tdt
[B]ekCt − [B]0e0 =
kB[A]0kC − kB
(e(kC−kB)t − 1)
Mit der Anfangsbed. [B]0 = 0 erhalten wir die Losung:
[B] =kB[A]0kC − kB
(e−kBt − e−kCt
)(4.35)
Fur [C] mussen wir nicht die DGL losen, wir konnen auch einfach die Rand-bedingungen und die Stoffbilanz verwenden:
[C] = [A]0 − [A]− [B]
[C] = [A]0
(1− e−kBt − kB
kC − kB(e−kBt − e−kCt
))oder
[C] = [A]0
(1 +
kBe−kCt − kCe−kBt
kC − kB
)(4.36)
Die Gl. 4.34,4.35,4.36 sind die Losungen des Systems von linearen gekoppel-ten DGL 4.33, der Verlauf der Graphen ist in Abb. 11 dargestellt.

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 31
Abbildung 10: Verlauf von [A], [B] und [C] mit den Parameterwerten (a)kB = 1.0, kC = 1.2, (b) kB = 20.0, kC = 1.0, (c) kB = 1.0, kC = 20.0 und (d)kB = 1.0, kC = 20.0 mit Quasistationaritatsprinzip (s.u.)
4.4.2 Quasistationaritatsprinzip
Dieses Prinzip stellt eine Naherung dar die es erlaubt, die DGLn zu verein-fachen. Wir haben bisher nur sehr einfache DGLn integriert, und man kannsich schon vorstellen, dass komplexere Systeme analytisch oft nicht losbarsind. Oft verwendet man daher Mathematikprogramme zu deren numeri-scher Losung. In vielen Fallen kann man jedoch Naherungen einfuhren,die eine approximative analytische Losung erlauben. Beim Quasistationa-ritatsprinzip (QS) nimmt man nun an, dass sich die Konzentrationen derIntermediate [I] nach einer Anfangsphase uber weite Bereiche der Reakti-on nur langsam andern, d.h. die Zeitableitungen ihrer Konzentrationen ver-nachlassigbar sind.
d[I]
dt≈ 0 (4.37)

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 32
Dies ist beim obigen Beispiel
A→ B → C
etwa dann der Fall, wenn gilt:
kB << kC , d.h. [B] << [A], [C].
Dann wird aus dem DGL System 4.33
−d[A]
dt= kB[A] (4.38)
0 = kB[A]− kC [B]
d[C]
dt= kC [B]
Die quasistationare Konzentration von B, [B]QS ist allerdings nicht konstant,sondern folgt der Konzentration von A:
[B]QS =kBkC
[A] =kBkC
[A]0e−kBt (4.39)
d[C]
dt= kB[A]0e
−kBt
Integration:
[C]QS = [A]0(1− e−kBt) (4.40)
Abb. 11 (d) zeigt den Kurvenverlauf bei Anwendung des QS. Offensichtlichist die Anderung von [B] nur klein, wenn kB/kC klein ist, d.h. nur dann istdie Konzentration von B annahernd konstant und das QS kann angewendetwerden.
4.4.3 Der geschwindigkeitsbestimmende Schritt
Wenn ein Reaktionsschritt wesentlich langsamer ablauft als alle anderen, soist dieser der geschwindigkeitsbestimmende Schritt der Gesamtreakti-on. Die gesamte Reaktionszeit ist durch diesen Schritt bestimmt, die anderenSchritte spielen dann kaum eine Rolle. Dies gilt allerdings nur, wenn die-ser Reaktionschritt nicht etwa durch eine Parallelreaktion umgangen werdenkann. Im letzten Beispiel war dies der Schritt
A→ B.
Der Schritt B → C ist dagegen so schnell, dass B sofort umgesetzt wird, alsoB nicht stark akkumuliert wird.

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 33
4.4.4 Beispiel
Zerfall von H2O2: Quasistationaritatsprinzip
2H2O2k−→ 2H2O +O2
(2Ak−→ 2B + C)
Mechanismus/Elementarreaktionen:(OH• ≡ X, HO2• ≡ Y)
H2O2k1,slow−→ 2OH • A
k1,slow−→ 2X (4.41)
H2O2 +OH• k2−→ HO2 •+H2O A+Xk2−→ Y +B
HO2 •+OH• k3−→ O2 +H2O X + Yk3−→ B + C
HO2 •+HO2•k4−→ O2 +H2O2 Y + Y
k4−→ A+ C
Dieses Reaktionsschema fuhrt zu einem recht komplexen System von DGLn:
a) H2O2 : −d[A]dt
= k1[A] + k2[A][X]− k4[Y ]2
b) OH• : −d[X]dt
= k2[A][X] + k3[X][Y ]− 2k1[A]
c) HO2• : −d[Y ]dt
= k3[X][Y ] + 2k4[Y ]2 − k2[A][X]
d) H2O : d[B]dt
= k2[A][X] + k3[X][Y ]
e) O2 : d[C]dt
= k3[X][Y ] + k4[Y ]2
Beim Aufstellen von (a) haben wir Folgendes bedacht:
• A ’zerfallt’ mit k1 in 2X (deswegen ’-’ Zeichen!)
• es gibt eine Parallelreaktion mit k2, die von [A][X] abhangt, d.h. ein-fache Kinetik 2. Ordnung.
• es gibt eine Ruckreaktion mit k4, die von [Y ]2 abhangt.
Aufstellen von (b):
• X reagiert mit A (k2) in Reaktion 2. Ordnung

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 34
• es gibt eine Parallelreaktion mit k3, die von [Y ][X] abhangt, d.h. ein-fache Kinetik 2. Ordnung.
• X wird gebildet mit k1, hangt von 1/2[A] ab.
etc. ..
Wir suchen nun die Losung von (a), d.h. [H2O2](t). (a) ist allerdings ge-koppelt an die Losung der DGL (b) und (c), wir benotigen ja die Konzen-trationsverlaufe von X und Y! Hier kann man das Quasistationaritatsprinzipausnutzen. Da OH• und HO2• sehr reaktive Intermediate sind, sind die Ge-schwindigkeitskonstanten k2 und k3 sehr gross, d.h. wir konnen annehmen(X und Y reagieren ’sofort’ weiter, sobald sie gebildet wurden):
d[X]
dt≈ d[Y ]
dt≈ 0
Zur Losung ziehen wir Gl. (c) von Gl. (b) ab:
k2[A][X] + k3[X][Y ]− 2k1[A] = k3[X][Y ] + 2k4[Y ]2 − k2[A][X]
k2[A][X]− 2k1[A] = +2k4[Y ]2 − k2[A][X]
d.h.k1[A] = k2[A][X]− k4[Y ]2
Einsetzen in (a):
−d[A]
dt= k1[A] + k2[A][X]− k4[Y ]2 = 2k1[A]
d.h.[H2O2] = [H2O2]0e
−2k1t
H2O2 zerfallt also formell nach Kinetik erster Ordnung, obwohl der Mecha-nismus wesentlich komplexer ist.
4.4.5 Vorgelagertes Gleichgewicht
Betrachen wir nun die folgende Reaktion
A+Bk1
Ik2→ P,

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 35
bei der gilt: k1, k−1 >> k2. D.h. es kann sich ein intermediares Gleichge-wicht zwischen I und den Edukten einstellen, das erst langsam in RichtungP abgebaut wird. Dann gilt:
K =[I]
[A][B]=
k1
k−1
Dabei nehmen wir an, dass die Folgereaktion zu langsam ist, um das Gleich-gewicht zwischen I und A+B zu storen und es gilt:
d[P ]
dt= k2[I] = k2K[A][B]
Diese Reaktion hat damit die Form einer Reaktion 2. Ordnung mit der ef-fektiven Ratenkonstante:
k = k2K =k1k2
k−1
(4.42)
Naherung mit QS
Wir wollen nun nicht vernachlassigen, dass I abgebaut wird. Damit erhal-ten wir die DGL:
d[P ]
dt= k2[I] (4.43)
d[I]
dt= k1[A][B]− k−1[I]− k2[I]
Quasistationaritatsprinzip:d[I]
dt≈ 0
Es wird nicht angenommen, dass [I] klein ist, sondern dass es sich nur sehrlangsam verandert. Damit folgt:
[I] ≈ k1[A][B]
k−1 + k2
,
d.h. die effektive Ratenkonstante ist:
k =k1
k−1 + k2
welche identisch mit der oben gefundenen ist wenn gilt: k2 << k−1 Fur Perhalten wir damit:
[P ] ≈ k1k2
k−1 + k2
[A][B] (4.44)
Dieser Typ von Reaktionen ist typisch fur die enzymatische Katalyse.

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 36
Abbildung 11: Vorgelagertes Gleichgewicht (links) und Losung mitQS(rechts). Grafiken von P. Weis.
4.5 Komplexe Temperaturabhangigkeiten
Wir hatten gesehen, dass komplexe Reaktionen eine einfache Gestalt anneh-men konnen, so fanden wir fur die Reaktion mit vorgelagertem Gleichge-wicht eine effektive Reaktion 2. Ordnung mit Gesamtgeschwindigkeitskon-stante (Gl. 4.42)
k =k1k2
k−1
Mit den Arrhenius Parametern (A1, A−1, A2) und Einsetzen der ArrheniusGleichung erhalten wir:
k =A1A2e
−E1/RT−E2/RT
A−1e−E−1/RT=A1A2
A−1
e−(E1+E2−E−1)/RT
Wir erhalten damit eine effektive Aktivierungsenergie
Ea = E1 + E2 − E−1
Diese kann jedoch, je nach Hohe der einzelnen Barrieren, positiv oder ne-gativ sein. Fur Ea > 0 erhalt man das erwartete Arrhenius Verhalten, dieRate steigt mit der Temeratur, fur Ea < 0 erhalt man das Gegenteil: dieRate sinkt mit der Temperatur. Dies bedeutet, dass die Ruckreaktion sehrsensitiv gegenuber einer Temperaturerhohung ist und die Konzentration vonI vermindert, was die Gesamtrate verkleinert.
4.6 Zusammenfassung
Bei komplexen Reaktionen geht man wie folgt vor:

4 ERWEITERUNGEN UND WICHTIGE KONZEPTE 37
Abbildung 12: Reaktion mit zwei Reaktionsbarrieren:
1. Elementarreaktionen identifizieren.
2. Aufstellung der DGL fur jeden Stoff, Annahme:
• unimolekularer Schritt: 1.Ordnung
• bimolekularer Schritt: 2.Ordnung
3. DGLn prufen: Massenbilanz, Stochiometrie ± 1νi
d[i]dt
4. Entweder Naherung (Quasistationaritat) oder exakte Losung, eventu-ell numerisch (Mathematika, Maple etc.) unter Berucksichtigung derRandbedingungen (A0 etc.)

5 KOMPLEXE KINETIKEN 38
5 Komplexe Kinetiken
5.1 Unimolekulare Reaktionen: Lindeman-HinshelwoodMechanismus
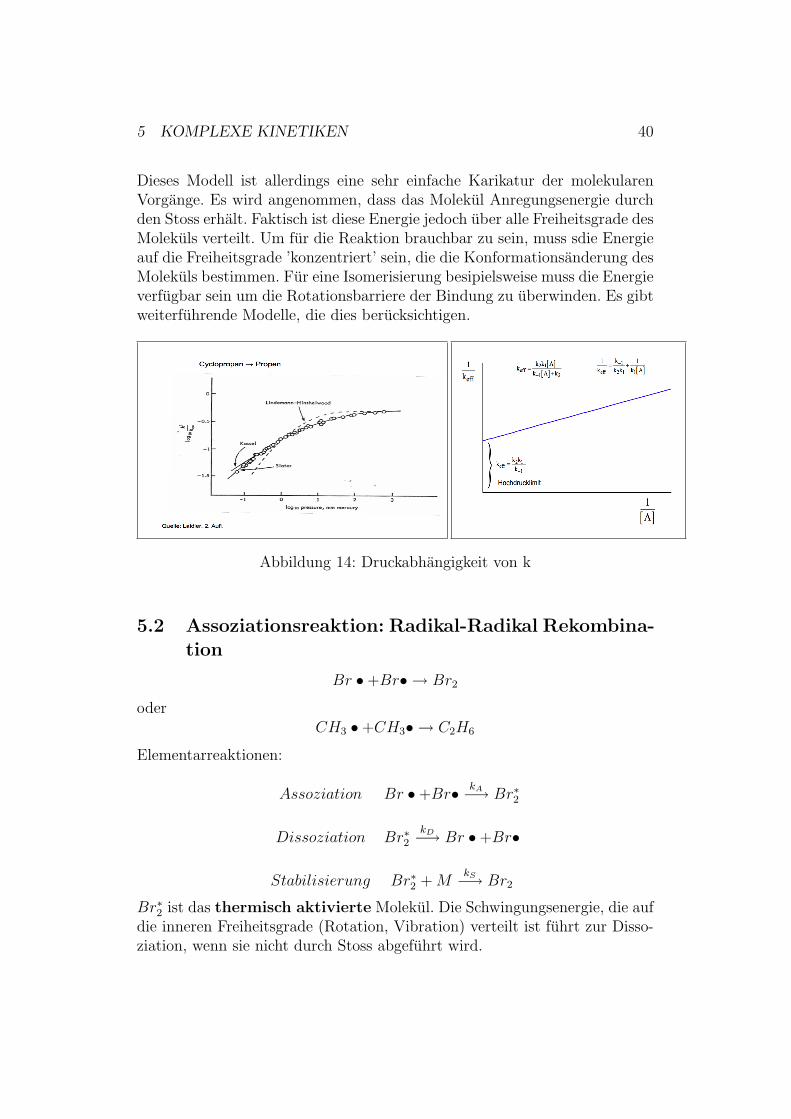
Bei unimolekularen Reaktionen stellt man i.A. eine Druckabhangigkeit derReaktionsordnung fest, was zunachst sehr eigenartig erscheint. Wie oben dis-
Abbildung 13: Druckabhangigkeit von k
kutiert erwartet man, dass diese Reaktionen 1.Ordnung sind. So andert sichbei der Isomerisierung
cycloC3H6 → CH3CH = CH2
nur die Struktur des Molekuls, es muss kein weiteres Molekul angelagertwerden, was eine Reaktion 2. Ordnung bedingen wurde. Dennoch ist auchhier die Situation komplexer. Es ist namlich moglich, dass ein Molekul diezur Umlagerung notige Energie durch den Stoss mit einem anderen Molekulerhalt (Aktivierung). Dann wurde auch eine unimolekulare Reaktion einenReaktionsschritt 2. Ordnung enthalten.Der Lindemann-Hinshelwood Mechanismus nimmt eine Aktivierungdes Molekuls A durch Stoss mit einem Partner an, das aktivierte Molekulwird mit A∗ bezeichnet. Molekule sind nicht starre Korper, sie haben vie-le Schwingungsfreiheitsgrade. Ein Stoss kann solche Schwingungen anregen,wobei bestimmte Schwingungen zum ’Bruch’ des Molekuls oder dessen Iso-merisierung fuhren konnen.
A+ AkA−→ A∗ + A

5 KOMPLEXE KINETIKEN 39
A∗ kann die Energie durch erneuten Stoss wieder verlieren
A+ A∗k−A−→ A+ A,
oder ins Produkt ubergehen:
A∗kP−→ P
Wir erhalten die folgenden DGLn fur die Einzelschritte:
d[A∗]
dt= kA[A]2 − k−A[A][A∗]− kP [A∗] (5.1)
Nun kommt es darauf an, welcher der ratenbestimmende Schritt ist.Wenn es der Letzte ist, erhalt man eine Gesamtreaktion der 1. Ordnung.Dies kann man mit Hilfe des Anwendung des Quasistationaritatsprinzipszeigen:
[A∗] =kA[A]2
kP + k−A[A](5.2)
und fur das Produkt erhalt man:
d[P ]
dt= kP [A∗] =
kPkA[A]
kP + k−A[A][A] (5.3)
Dieses Geschwindigkeitsgesetz ist nun eindeutig nicht 1. Ordnung! Man kanneine effektive Geschwindigkeitskonstante fur die Gesamtreaktion definieren:
keff =kPkA[A]
kP + k−A[A]
1
keff=
k−AkPkA
+1
kA[A](5.4)
Wir wollen nun zur weiteren Analyse zwei Grenzfalle unterscheiden:
• Hochdrucklimit: [A] ist sehr gross, d.h. k−A[A] >> kP :
keff ≈kPkAk−A
==>d[P ]
dt=kPkAk−A
[A]
Effektiv ist dies eine Reaktion 1. Ordnung. Der geschwindigkeitsbestim-mende Schritt ist der Zerfall von A∗.
• Niederdrucklimit: [A] ist sehr klein, d.h. kA[A] << kP :
keff ≈ kA[A] ==>d[P ]
dt= kA[A]2.
Reaktion 2.Ordnung. Der geschwindigkeitsbestimmende Schritt ist diebimolekulare Erzeugung von A∗.
5 KOMPLEXE KINETIKEN 40
Dieses Modell ist allerdings eine sehr einfache Karikatur der molekularenVorgange. Es wird angenommen, dass das Molekul Anregungsenergie durchden Stoss erhalt. Faktisch ist diese Energie jedoch uber alle Freiheitsgrade desMolekuls verteilt. Um fur die Reaktion brauchbar zu sein, muss sdie Energieauf die Freiheitsgrade ’konzentriert’ sein, die die Konformationsanderung desMolekuls bestimmen. Fur eine Isomerisierung besipielsweise muss die Energieverfugbar sein um die Rotationsbarriere der Bindung zu uberwinden. Es gibtweiterfuhrende Modelle, die dies berucksichtigen.
Abbildung 14: Druckabhangigkeit von k
5.2 Assoziationsreaktion: Radikal-Radikal Rekombina-tion
Br •+Br• → Br2
oderCH3 •+CH3• → C2H6
Elementarreaktionen:
Assoziation Br •+Br• kA−→ Br∗2
Dissoziation Br∗2kD−→ Br •+Br•
Stabilisierung Br∗2 +MkS−→ Br2
Br∗2 ist das thermisch aktivierte Molekul. Die Schwingungsenergie, die aufdie inneren Freiheitsgrade (Rotation, Vibration) verteilt ist fuhrt zur Disso-ziation, wenn sie nicht durch Stoss abgefuhrt wird.

5 KOMPLEXE KINETIKEN 41
Aufstellen der Ratengleichungen und Verwendung des Stationaritatsprinzipsfuhrt zu der folgenden DGL (Beweis: Ubung.):
d[Br•]dt
= −2kAkS[M ]
kD + kS[M ][Br•]2 =: −2keff [Br•]2 (5.5)
Es kann also eine effektive Geschwindigkeitskonstante definiert werden. Wirunterscheiden nun 2 Grenzfalle:
• hoher Druck, d.h. [M ] gross: kS[M ] >> kD
d[Br•]dt
≈ −2kA[Br•]2
Geschwindigkeitsbest. Schritt: Assoziation. Reaktion (pseudo) 2. Ord-nung.
• [M ] klein: kS[M ] << kD
d[Br•]dt
≈ −2kAkSkD
[M ][Br•]2
Reaktion (pseudo) 3. Ordnung.
Abbildung 15: Druckabhangigkeit von k, Abb. von P. Weis.
5.3 Kettenreaktionen
Viele Reaktionen in der Gasphase und Polymerisierungsreaktionen in Losungsind sogenannte Kettenreaktionen. Nach Initiierung der Reaktion bilden die

5 KOMPLEXE KINETIKEN 42
Intermediate in einem folgenden Schritt weitere Intermediate, usw., bis ei-ne Inhibierung zum Abbruch der Kettenreaktion fuhrt. Die Intermediate,die diese Kettenreaktion treiben, werden Kettentrager genannt. Bei Kern-reaktionen beispielsweise sind diese Kettentrager Neutronen, in radikalischenKettenreaktionen, wie dem folgenden Beispiel, sind sie Radikale.
5.3.1 Kettenreaktion ohne Verzweigung
Die (Gasphasen-) Reaktion
H2(g) +Br2(g)→ 2HBr(g)
hat eine sehr komplizierte Reaktionsordnung, wie oben angesprochen. Umdiese zu verstehen wurde der folgende Mechanismus vorgeschlagen (Folgevon Elementarreaktionen):
Initiierung Br2 +Mk1−→ Br •+Br •+M
Kettenwachstum Br •+H2k2−→ HBr +H•
H •+Br2k3−→ HBr +Br•
Inhibierung H •+HBrk4−→ H2 +Br•
Abbruch Br •+Br •+Mk5−→ Br2 +M
(5.6)
Im Initiierungsschritt wird Br2 durch einen Stoss mit M (H2 oder Br2)aktiviert. Dadurch erhalt das Molekul kinetische Energie, die zum Zerfallfuhrt (Lindemann-Hinshelwood). Damit ist diese Reaktion, je nach Druck,entweder 1. oder 2. Ordnung. Ebenso muss im Abbruch-Schritt diese Ener-gie wieder abgefuhrt werden. Dies geschieht wieder durch einen Stoss mit M.Diese Reaktion ist daher 2. oder 3. Ordnung, analog zum Initiierungsschritt,abhangig vom Druck. Im Inhibierungsschritt wird H• entzogen, damitwird die Reaktion verlangsamt, aber die Terminierung erfolgt erst durch BrRekomination.Ratengleichung fur Produktformation:
d[HBr]
dt= k2[Br•][H2] + k3[H•][Br2]− k4[H•][HBr] (5.7)

5 KOMPLEXE KINETIKEN 43
Um diese zu losen, benotigen wir die Losung der DGL der Kettentrager, diewir wieder durch Anwendung des Quasistationaritatsprinzips erhalten:
(a) 0 =d[Br•]dt
= 2k1[Br2][M ]− k2[Br•][H2] + k3[H•][Br2] + k4[H•][HBr]− 2k5[Br•]2[M ]
(b) 0 =d[H•]dt
= k2[Br•][H2]− k3[H•][Br2]− k4[H•][HBr]
Gln. (a) und (b) haben gleiche Beitrage (k2, k3 und k4) mit umgekehrtenVorzeichen, das bringt uns auf die Idee, die beiden Gleichungen zu addieren:
0 = 2k1[Br2][M ]− 2k5[Br•]2[M ]
Da [M ] wegfallt, erhalt man:
[Br•] =
(k1
k5
)1/2
[Br2]1/2 (5.8)
Gl. (b) nach [H•] auflosen und Gl. 5.8 einsetzen:
[H•] =k2[Br•][H2]
k3[Br2] + k4[HBr]=k2
(k1
k5
)1/2
[Br2]1/2[H2]
k3[Br2] + k4[HBr](5.9)
Dies konnen wir nun in Gl. 5.7 einsetzen:
d[HBr]
dt= k2
(k1
k5
)1/2
[Br2]1/2[H2] +k3k2
(k1
k5
)1/2
[Br2]1/2[H2][Br2]
k3[Br2] + k4[HBr]−
k4k2
(k1
k5
)1/2
[Br2]1/2[H2][HBr]
k3[Br2] + k4[HBr]
Nun erweitern wir den 1. Term mit (k3[Br2] + k4[HBr]), verwenden die ef-fektiven Geschwindigkeitskonstanten
k := k2
(k1
k5
)1/2
k′ :=k4
k3
und erhalten:
d[HBr]
dt=
2k[H2][Br2]3/2
[Br2] + k′[HBR](5.10)
Man sieht sofort, dass die Reaktion sich mit Bildung von [HBr] verlangsamt.Dies liegt an der Konkurrenzreaktion (Inhibierung), HBr bildet mit H• wiederdas Edukt H2. Die Gleichung kann numerisch integriert werden.

5 KOMPLEXE KINETIKEN 44
5.3.2 Radikalische Polymerisation
Kettenwachstum erfolgt oft uber radikalische Reaktionen, bei denen ein In-itiatormolekul I in zwei Radikale R• zerfallt und dadurch die MonomereM zu einer Kette gefugt werden.
Initiierung Iki−→ 2R•
Kettenwachstum M +R• k1−→ RM•
M +RM• k2−→ RM2•
...
M +RMn−1•kn−→ RMn•
Abbruch RMm •+RMn•ka−→ RMn+mR
(5.11)
Man kann nun annehmen, dass die Geschwindigkeitskonstanten unabhangigvon der Kettenlange sind, d.h.
k := k1 = k2 = ... = kn
Nun definieren wir noch die Gesamtkonzentration der Radikale,
[GR•] := [R•] + [RM•] + [RM2•] + ...+ [RMn•]
und wir konnen die DGL aufstellen:
−d[M ]
dt= k1[R•][M ] + k2[RM•][M ] + k3[RM2•][M ] + ...+ kn[RMn•][M ] =
= k[GR•][M ] (5.12)
Die Gesamtkonzentration der Radikale andert sich nur am Anfang und beimAbbruch, d.h. wir konnen wieder das Quasistationaritatsprinzip anwenden:
−d[GR•]dt
= 2ki[I]− 2ka[GR•]2 = 0 (5.13)
D.h.
[GR•] =√ki/ka
√[I] (5.14)
in Gl. 5.12 eingesetzt ergibt:
−d[M ]
dt= k√ki/ka
√[I][M ] =: keff
√[I][M ] (5.15)
Die Reaktionsgeschwindigkeit ist damit proportional zur Wurzel der Konzen-tration des Initiatormolekuls.

5 KOMPLEXE KINETIKEN 45
5.3.3 Kettenreaktion mit Verzweigung: Explosion
Es gibt zwei Typen von Explosionen, die Thermische, bei der mit jedem(exothermen) Reaktionschritt die Temperatur erhoht wird, was zu einer wei-teren Beschleunigung fuhrt, und die Verzweigungs-Explosion, bei der dieAnzahl der Kettentrager exponentiell wachst.Ein Beispiel ist die Knallgasreaktion,
2H2(g) +O2(g)→ 2H2O(g).
Die Gesamtreaktion sieht sehr einfach aus, der Mechanismus ist jedoch sehrkomplex und noch nicht bis ins letzte Detail geklart. Die Kettenreaktioninvolviert eine Reihe radikalischer Intermediate.
Initiierung H2 +O2 +Mk1−→ 2OH •+M
OH •+H2k2−→ H2O +H•
V erzweigung H •+O2k3−→ •O •+OH•
•O •+H2k4−→ H •+OH•
Abbruch H •+H •+Mk5−→ H2 +M
(5.16)
Hier wird in jedem Reaktionsyklus die Anzahl der Radikale verdoppelt. Lei-der verlauft diese Reaktion auf noch komplexere Weise, z.B. wird auch HO2
gebildet, daher werden wir diese Reaktion nicht im Detail betrachten.Stattdessen wollen wir im Folgenden das stark vereinfachte Modell betrach-ten:
A+B → C
Initiierung Ak1−→ X
V erzweigung B +Xk2−→ C + αX
Abbruch Xk3−→ A
(5.17)
(X: radikalische Intermediate) Dabei wird sowohl der Lindemann-HinshelwoodMechanismus im Initiierungsschritt ignoriert, wie auch der Stosspartner Mbeim Abbruch. α > 1, bei der Reaktion 5.16 gilt α = 2.

5 KOMPLEXE KINETIKEN 46
Losung I: Quasistationaritat (obwohl bei Explosion fraglich!)
0 =d[X]
dt= k1[A]− k2[B][X] + αk2[B][X]− k3[X] = (5.18)
= k1[A] + (k2(α− 1)[B]− k3)[X] =: k1[A]−λ[X]
[X] =k1[A]
λ(5.19)
Fallunterscheidung:
1. λ > 0, d.h. k3 > k2(α−1)[B]: Abbruch uberwiegt Verzweigung, Losungwie bei Kettenreaktion (HBr) oben.
2. λ < 0, d.h. k3 < k2(α−1)[B]: Verzweigung uberwiegt den Abbruch, [X]wird negativ, QS offensichtlich nicht anwendbar. Bei Explosion steigt[X] schnell an, im Gegensatz zur QS Annahme.
Losung II: Integration ohne QS
Betrachte den Anfang der Reaktion.
[A], [B] >> [X], [A] ≈ [A]0, [B] ≈ [B]0, [X]0 = 0
d.h.−λ = (k2(α− 1)[B]0 − k3)
d[X]
dt= k1[A]0−λ[X] (5.20)
dx
dt= a− bx
∫dx
a− bx= −1
bln(a− bx)
−1
λ[ln(k1[A]0 − λ[X])]
[X][X]0=0 = t
−1
λln(k1[A]0 − λ[X]) +
1
λln(k1[A]0) = t

5 KOMPLEXE KINETIKEN 47
Abbildung 16: Verlauf der Konzentration [X] fur die drei Falle: k=1, [A]0 =5, λ =1,0,-1
Fur kurze Zeiten t erhalten wir (Anfang der Reaktion):
[X]Anfang =k1[A]0λ
(1− e−λt
)(5.21)
Fallunterscheidung:
1. λ > 0, d.h. k3 > k2(α− 1)[B]: Abbruch uberwiegt Verzweigung.[X] konvergiert mit t gegen den QS Wert Gl. 5.19
[X]Anfang =k1[A]
λ
2. λ = 0 : 00
entweder l’ Hospital oder Reihendarstellung der exp Funktion.
exp(x) =∞∑0
xn
n!
[X]Anfang = k1[A]0
(t− λt2
2!+
(λ2t3
3!−)
Fur λ = 0 : erhalten wir:
[X]Anfang = k1[A]0t
3. λ < 0, d.h. k3 < k2(α − 1)[B], Verzweigung uberwiegt den Abbruch:Explosion. mit λ = −|λ|
[X]Anfang =k1[A]0|λ|
(e+|λ|t − 1
)

5 KOMPLEXE KINETIKEN 48
Hier wachst die Anzahl der Kettentrager exponentiell, damit steigt auchder Druck exponentiell. Eine Explosion findet nur fur λ < 0 statt, d.h.es gibt einen Mindestdruck.
k3 > k2(α− 1)[B]→ [B] >k3
k2(α− 1).
Man sollte aber beachten, dass dies ein sehr einfaches Modell fur dieExplosion ist, z.B. wurde die Druckabhangigkeit der Abbruchreaktionvernachlassigt, aber auch die termischen Aspekte: hohere Temperaturvergrossert die k’s, beschleunigt also die Reaktion weiter.
Abbildung 17: Bereiche der Explosionsreaktion: Grafik aus Atkins
5.4 Enzymatische Katalyse: Michaelis-Menten
Ein Katalysator ist ein Stoff, der eine Reaktion beschleunigt ohne dabeiselbst chemisch verbraucht zu werden, er vermindert die AktivierungsenergieEa einer Reaktion, andert aber ∆G nicht. Der Katalysator greift damit in dieKinetik ein, lasst aber die Thermodynamik unverandert. Man unterscheidetdie Homogene Katalyse, wo Katalysator und Reaktanden in der gleichenPhase vorliegen, von der Heterogenen Katalyse, wo diese in unterschiedli-chen Phasen vorliegen, wie z.B. die Metalloberflache, die als Katalysator furGasphasenreaktionen dienen kann. Ein Enzym ist ein biologischer Kataly-sator (Homogene Katalyse), meist ein Protein oder RNA.

5 KOMPLEXE KINETIKEN 49
Beispiel: Die ReaktionH2O2 → 2H2O +O2
ist mit ∆G =-103 kJ/Mol exotherm, doch stark inhibiert durch eine hoheBarriere von Ea =76 kJ/Mol. Zugabe von Iodid Ionen vermindert die Barriereauf EA = 57 kJ/Mol, d.h. die Rate wird etwa um den Faktor 2000 erhoht.Das Enzym Katalase reduziert die Barriere allerdings viel drastischer aufEA = 8 kJ/Mol, d.h.die Rate steigt um den Faktor 1015.Aus dem Substrat S und Enzym E bildet sich in einem ersten Schrittder Enzym-Substrat-Komplex ES , dieser Schritt ist reversibel. Es gibtzwei Modellvorstellung fur diesen Schritt. In dem Schlussel-Schloss Modellwird angenommen, dass das Substrat genau in die Bindungstasche des En-zyms passt, dieses Modell gibt ein suggestives Bild fur die Enzymspezifizitat.Allerdings sind Enzyme strukturell flexibel. Das ’induced fit’ Modell tragtdem Rechnung indem es eine Anpassung der Bindungstasche an das Substratberucksichtigt.In einem zweiten Schritt zerfallt ES in das Enzym E und das Produkt P
E + Sk1,k−1
ESk2−→ E + P
Diese Reaktion haben wir oben schon einmal diskutiert (vorgelagertes Gleich-gewicht). Wir haben die DGLn
d[P ]
dt= k2[ES] (5.22)
d[ES]
dt= k1[E][S]− k−1[ES]− k2[ES]
Anwenden von QS auf die 2. Gleichung ergibt:
[ES] =k1
k−1 + k2
[E][S] =:1
KM
[E][S] (5.23)
mit der Michaelis-Menten Konstante
KM =k−1 + k2
k1
=[E][S]
[ES].
Im Fall k2 << k−1 ist KM identisch mit der Gleichgewichtskonstanten Keq.
Nun verwenden wir die Massenbilanz fur das Enzym:
[E]0 = [ES] + [E]

5 KOMPLEXE KINETIKEN 50
Wenn wir nun [E] = [E]0 − [ES] in die Gleichung 5.23 einsetzen und umfor-men erhalten wir:
[ES] =[E]0[S]
KM + [S](5.24)
Dies in Gl. 5.22 fur [P ] einsetzen ergibt die Michaelis-Menten Gleichung:
d[P ]
dt=k2[E]0[S]
KM + [S](5.25)
Abbildung 18: Michaelis Menten Kinetik, Abb. von P. Weis.
Grenzfalle:
1. [S] << KM , d.h. wenig Substrat in Losung:
d[P ]
dt=k2[E]0KM
[S]
Kinetik 1.Ordnung.
2. [S] >> KM , Sattigungskinetik
d[P ]
dt= k2[E]0
Kinetik 0. Ordnung. Geschwindigkeit nur abhangig von Enzymmenge.Dies ist auch die maximale Geschwindigkeit rmax = k2[E]0 da (sieheGl. 5.25)
[S]
KM + [S]< 1.

5 KOMPLEXE KINETIKEN 51
Die beiden Grenzfalle sind in Abb. 18 dargestellt.
Die Reaktionsgeschwindigkeit r = d[P ]/dt lasst sich durch rmax ausdrucken:
r =rmax[S]
KM + [S](5.26)
Betrachten wir die halbe maximale Geschwindigkeit, rmax/2 = rmax/2, diesich bei der Konzentration [S]max/2 einstellt.
rmax/2 =rmax[S]max/2KM + [S]max/2
(5.27)
oder:[S]max/2 = KM
Man kann also KM direkt aus dem Michaelis-Menten Plot ablesen (Abb. 19links). Sehr nutzlich ist auch die Lineweaver-Burk Darstellung, die Gl. 5.26invertiert.
1
r=KM + [S]
rmax[S]=
1
rmax+KM
rmax
1
[S]
Diese Darstellung hat die Form einer Geradengleichung f(x) = a + bx, d.h.durch Auftragen von
1
rvs
1
[S]
kann man direkt die Steigung b= KMrmax
und a= 1rmax
ablesen (Abb. 19 rechts).
Katalytische Effizienz
Die maximale Geschwindigkeit wird unter Sattigungsbedingungen gemessen,wo die maximale Geschwindigkeit durch rmax = k2[E]0 gegeben ist. Ein Maßfur die Effizienz des Enzyms konnte daher die katalytische Konstante
kcat = k2
sein. kcat wird Wechselzahl genannt (’turnover frequencey’) und gibt die An-zahl der katalytischen Zyklen pro Zeiteinheit an. Enzyme arbeiten jedochunter physiologischen Bedingungen die weit von der Sattigung entfernt sind,die Geschwindigkeit ist daher nur
v = kcat[ES].

5 KOMPLEXE KINETIKEN 52
Abbildung 19: Michaelis Menten Kinetik, Abb. von P. Weis (links) undLineweaver-Burcke Darstellung (rechts, Quelle: www.chemgapedia.de)
Die Bildung von [ES] wird jedoch von KM bestimmt, die katalytische Effizi-enz hangt im Fall [S] << KM (s.o.) wegen
v =kcatKM
[E]0[S]
von dem Quotienten ε = kcat/KM ab, der die enzymatische Umsatzgeschwin-digkeit charakterisiert. Diese kann die Entstehungsrate von [ES] nicht uber-schreiten, die durch die Diffusionsgeschwindigkeit begrenzt ist.
Inhibierung
Die Enzymfunktion kann auf zwei Weisen inhibiert werden, kompetitiv,wenn der Inhibitor I an der gleichen Stelle bindet wie das Substrat, dannist das Enzym blockiert. Oder nicht-kompetitiv, hier bindet I an einer an-deren Stelle als S, deaktiviert aber dann den Komplex ES durch Bildungvon ESI. Um dies zu beschreiben, mussen in der Kinetik noch die folgendenbeiden Gleichgewichte berucksichtigt werden.
EI E + I KI =[E][I]
[EI](5.28)
ESI ES + I K ′I =[ES][I]
[ESI]
(5.29)
Mit den Definitionen:
α = 1 + [I]/KI α′ = 1 + [I]/K ′I

5 KOMPLEXE KINETIKEN 53
erhalt man eine modifizierte Michaelis-Menten Kinetik
d[P ]
dt=
k2[E]0[S]
α′KM + α[S](5.30)
Durch kinetische Analyse kann mit Hilfe der Lineweaver-Burk Darstellungdie Art der Inhibierung festgestellt werden (fur Details, siehe etwa Atkinsoder Allen).
5.5 Photochemie
Die Photochemie beschaftigt sich mit Reaktionen, die durch Licht (Photo-nen) initiiert werden. Um diese formal richtig behandeln zu konnen, benotigtman die Quantenmechanik (QM: PCII). Im Molekul sind die Elektronen imRaum um die positiv geladenen Kerne verteilt, wie genau, das beschreibtdie QM. Die genaue Verteilung ist jedoch wichtig, um die chemische Bin-dung verstehen zu konnen. Die Elektronenanordnung fuhrt zu einer Kom-pensation der Abstossung der positiv geladenen Kerne, und eine effektiveAnziehung/Bindung zwischen den Atomen ensteht. Die genaue Anordnungder Elektronen im Molekul bestimmt damit auch die Details der Geometrieim Molekul, d.h. die Bindungsabstande, Winkel etc.
Abbildung 20: Bohrsches Atommodell (links) und Energieniveaus eines Mo-lekuls
Das Bohrsche Atommodell, das eine Analogie zum Planetenmodell darstellt,ist viel zu einfach um den Sachverhalt beschreiben zu konnen. Eine qua-litativ richtige Einsicht besteht jedoch in der Quantelung der Energie, dieElektronen konnen sich nur auf bestimmten, energetisch determinierten Bah-nen bewegen. So kann Licht nur zu bestimmten Anregungen der Elektronen

5 KOMPLEXE KINETIKEN 54
auf andere ’Bahnen’ (diese Konzept wird in der QM revidiert) fuhren. Manbezeichnet den elektronischen Grundzustand mit S0, den ersten ange-regten Zustand mit S1 etc. Wenn also durch Licht der S1 angeregt wird,verandert sich die Elektronenanordnung im Molekul, dadurch verandert sichauch die Bindung zwischen den Atomen.
Wenn ein Molekul durch Licht (hν) elektronisch angeregt wird, konnenmehrere Prozesse stattfinden:
1. Emission: das Molekul kann das Licht wieder abgeben (Fluoreszenz).
2. Da durch Veranderung der Elektronenverteilung sich andere Bindungs-verhaltnisse einstellen konnen, kann das Molekul im S1 (oder hoherangeregt Zustande) (Abb. 21):
• dissoziieren, wenn es kein Energieminimum im S1 gibt.
• die Struktur relaxieren, wenn sich das Energieminimum im S1
von dem im S0 unterscheidet. Wenn das Molekul dann wiederein Photon abgibt (Fluoreszenz), hat dieses Photon eine geringereEnergie (Rotverschiebung gegenuber der Anregungswellenlange).
• isomerisieren, wenn im S1 eine Rotation von Winkeln favorisertwird.
3. Inter-System-Crossing (ISC): Es kann ein Ubergang von einem Sin-gulett (S1) in einen Triplett (T1) Zustand stattfinden. Wenn der T1
durch Strahlung in den S0 ubergeht, nennt man das Phosphoreszenz(Abb. 20).
Betrachten wir nun eine photoinduzierte Reaktion (z.B: Spaltung von Br2):
A+ hνka,kf A∗
kr→ B
A∗ bezeichne das Molekul im ersten angeregten Zustand.
d[A∗]
dt= ka[A]Φ− kf [A∗]− kr[A∗] (5.31)
d[B]
dt= kr[A
∗]
Die Photonen werden hier wie ein Stoff behandelt, Φ ist die Lichtintensitat(Photonen/Zeit Probenvolumen). Anwenden des QS auf die erste Gleichung:
[A∗] =ka[A]Φ
kf + kr

5 KOMPLEXE KINETIKEN 55
Abbildung 21: Dissoziation, Relaxation und Isomerisierung im ersten ange-regten Zustand
und in die Zweite einsetzen:
d[B]
dt=krka[A]Φ
kf + kr(5.32)
5.6 Heterogene Katalyse: Reaktionen an Oberflachen
5.6.1 Langmuir Adsorptionsisotherme
Reaktionen, die von Festkorperoberflachen katalysiert werden setzen in ei-nem ersten Schritt die Adsorption der Molekule auf der Oberflache (OF)voraus. Man unterscheidet zwischen Physisorption, wo die Molekule eineVdW (Dispersion) Bindung mit der OF eingehen und der Chemisorption,deren Bindungscharakter eher der chemischen Bindung entspricht.
Eine Oberflache weist nun eine bestimmte Anzahl an Bindungsstellen Oauf. Z.B. kann jedes Oberflachenatom ein ungepaartes Elektron haben dasprinzipiell in der Lage ist, eine Bindung mit einem anderen Atom/MolekulA einzugehen (Abb. 22). Da dies reversibel ist, betrachten wir folgendenMechanismus
A+Ok1,k−1
OA
mit der Ratengleichung:
d[OA]
dt= k1[O][A]− k−1[OA] (5.33)
Die Oberflache ist 2-dimensional, die Konzentrationen [O] und [OA] sinddaher als (Anzahl Teilchen)/Flache gegeben. Nun hat die Oberflache eine

5 KOMPLEXE KINETIKEN 56
maximale Anzahl an Bindungsstellen O, d.h. die Konzentration von OA er-reicht ihr Maximum [OA]max, wenn alle Bindungsstellen belegt sind. Die Zahl(0≤ θ ≤1)
θ =[OA]
[OA]max
gibt die Oberflachenbedeckung an und wir konnen die Ratengleichung damitauch schreiben als ([O] + [OA] = [OA]max):
dθ
dt= k1(1− θ)[A]− k−1θ (5.34)
Mit pV = NkT sehen wir sofort, dass bei V,T = konst. der Druck propor-
Abbildung 22: Obeflachenbedeckung mit θ = 0, θ = 0.5 und θ = 1. Abb. vonP. Weis
tional zur Anzahl der Teilchen im Volumen V ist, p ist also proportional zu[A]. Damit konnen wir sofort schreiben:
dθ
dt= k1(1− θ)p− k−1θ (5.35)
Im Gleichgewicht (dθ/dt = 0)erhalten wir mit K = k1
k−1damit die Langmuir-
Adsorptionsisotherme (Abb. 23):
θ =Kp
1 +Kp(5.36)
Abweichungen von dieser Isotherme treten bei Multilagen-Adsorption undbei unterschiedlich stark bindenden Adsorptionsplatzen auf.
5.6.2 Oberflachenreaktionen: Heterogene Katalyse
Beispiele:
• Ammoniaksynthese: 3H2 +N2 → 2NH3

5 KOMPLEXE KINETIKEN 57
Abbildung 23: Langmuir Isotherme (rechts) und Abweichung bei Multilagen-Adsorption. (Abb. von P. Weis)
• Abgaskatalysator: NOx zu N2, CO zu CO2.
Diese Reaktionen vom TypA+B → C
wollen wir naherungsweise als vorgelagertes Gleichgewicht betrachten.
A+Ok1,k−1
OA
B +Ok2,k−2
OB
OA+OBk3,slow−→ OC
OCk4,fast−→ O + C
Wir verwenden nun Gl. 5.35 fur A und B an und beachten, dass fur dieHinreaktion die Anzahl der freien Bindungsplatze durch (1−θA−θB) gegebenist. Dies fuhrt auf die DGLn:
(a)dθAdt
= k1(1− θA − θB)pA − k−1θA = 0 (5.37)
(b)dθBdt
= k2(1− θA − θB)pB − k−2θB = 0
(c)dθCdt
= k3θAθB − k4θC = 0
(d)d[C]
dt= k4θC
Fur Gl. (a) und (b) nehmen wir thermodynamisches Gleichgewicht an (wieoben), fur (c) wenden wir QS an. Mit QS fur (c), k3θAθB = k4θC , erhalten

5 KOMPLEXE KINETIKEN 58
wir fur (d):d[C]
dt= k4θC = k3θAθB
Wir benotigen also θA und θB. Dazu teilen wir (a) durch (b) und erhalten:
KApAKBpB
=θAθB
Dies nach θB auflosen und in (a) einsetzen:
θA =KApA
1 +KApA +KBpB
Man erhalt ein analoges Ergebnis fur B und erhalt fur (d):
d[C]
dt= k4θC = k3θAθB = k3
KApAKBpB(1 +KApA +KBpB)2
(5.38)
Diese Gleichung geht von dem sehr einfachen Modell aus, jeder Schritt kannkomplizierter sein. So kann insb. auch der Desorptionsschritt (d) langsamsein, dann gabe es eine Inhibierung durch das Produkt. OA und OB konnenauf der Oberflache diffundieren, k3 ist also auch durch die Diffusion auf derOberfache limitiert.

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 59
6 Das mikroskopische Bild: kinetische Gas-
theorie
’Fur den Physiker ist die Kuh ne Kugel’ (alte Volksweisheit)
Wir haben die Kinetik bisher phanomenologisch betrieben, d.h. wir haben eineReaktion
A+Bk−→ C +D
betrachtet und die Reaktionsordnungen und Ratenkonstanten empirisch ermittelt.Wir haben bisher keinen Bezug auf die Molekule, ihre relativen Geschwindigkeitenund die molekularen Details der Reaktionen genommen. Bei der Diskussion derArrhenius Gleichungen hatten wir einen ersten Ansatz einer mikroskopischenSicht: Nur solche Molekule, die
• einen Stoß ausfuhren und
• durch diesen Stoß genugend kinetische Energie aufbringen, die Aktivierungs-barriere zu uberwinden,
werden zum Produkt fuhren. Um zu einer mikroskopischen Interpretation derArrheniusparameter A und EA zu kommen, mussen wir also mikroskopischabschatzen, wie oft Stoße zwischen Molekulen vorkommen und wie oft dabei dieEnergie zur Reaktion ausreicht. Dies fuhrt uns zur Stoßzahl und zur Geschwin-digkeitsverteilung von Maxwell und Boltzmann.
Wikipedia zur kinetischen Gastheorie:(http://de.wikipedia.org/wiki/Kinetische Gastheorie)Die wichtigsten Aussagen der Theorie sind:
1. Die Teilchen eines Gases (Atome, Molekule) sind standig in ungeordneter,nur statistisch fassbarer Bewegung.
2. Zwischen ihren Zusammenstoßen bewegen sie sich gleichformig und unabhangigvoneinander, ohne Bevorzugung einer Richtung.
3. Die Teilchen uben keine Krafte aufeinander aus, solange sie sich nicht ge-genseitig beruhren.
4. Zusammenstoße der Teilchen mit der Gefaßwand gehorchen dem Gesetz deselastischen Stoßes und verursachen den Gasdruck.

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 60
6.1 Stoßzahl
Zuerst ermitteln wir, wie haufig Stoße zwischen Molekulen vorkommen. Dazu wer-den wir diese als (harte) Kugeln beschreiben. Betrachten wir zwei Spezies mit denRadien rA und rB. Um die Stoßzahl, d.h. die Anzahl der Stoße pro Zeiteinheit∆t abzuschatzen, ist ihre Relativgeschwindigkeit vr von Bedeutung. Um diemittlere Anzahl der Stoße z zu berechnen, benotigen wir die mittlere Rela-tivgeschwindigkeit < vr >. Eine Kugel A wird mit anderen Kugeln B zusam-menstoßen, wenn deren Mittelpunkt innerhalb eines Zylinders mit der Oberflacheσ (Stoßquerschnitt)
σ = r2ABπ rAB = (rA + rB)
liegt, wie in Abb. 24 (links) skizziert. Faktisch wird die Kugel A bei jedem Stoßeine Richtungsanderung durchfuhren, d.h. der Zylinder bekommt einen ’Knick’(Abb. 24 (rechts)). Diese ’Knicke’ wollen wir vernachlassigen und die Lange des
Abbildung 24:
Zylinders, der in der Zeit ∆t durchlaufen wird, mit L =< vr > ∆t bezeichnen,d.h. wir konnen ein Zylindervolumen berechnen als:
VZ = σL = σ < vr > ∆t
Mit wie vielen Kugeln B kollidiert A nun in der Zeit ∆t? Das hangt an der AnzahlnB von B im Zylinder VZ ,
nB = VZ ∗ [B]NA = σ < vr > ∆tNA[B]
(Avogadrozahl NA = 6.022 1023mol−1). Die Stoßzahl z ist die Anzahl der Stoßeder Kugel A pro Zeit, d.h.
z =nB∆t
= σ < vr > NA[B] (6.1)

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 61
Die Gesamtzahl der Stoße ZAB pro Zeit ∆t und pro Volumen V hangt von derKonzentration der Kugeln A (nA) ab,
nA = [A]NA,
d.h.
ZAB = nAz = σ < vr > N2A[A][B] (6.2)
Bitte beachten:- VZ ist das Volumen des ’Stoßzylinders’.- [A] ist eine Konzentration, d.h. gibt die Anzahl Mol/(Volumen V) an. NA[A] gibtdemnach die (Anzahl der Teilchen)/(Volumen V) an.- ZAB ist die Anzahl der Stoße pro Zeit und pro Volumen V, ist sozusagen dieDichte der Stoße pro Zeit. ZABV ist dann die Gesamtzahl der Stoße pro Zeit imVolumen V.
Betrachten wir nun die Reaktion
A+B → P
Jetzt nehmen wir an, jeder Stoß fuhrt zu einer Reaktion, d.h. zu einer Abnahmevon A: Wir haben:
• die Anzahl der Atome A pro Volumen V:
[A]NA
• die Anderung der Anzahl der Atome A pro Volumen V durch die Reaktion:
−d([A]NA)dt
• Dies ist gleich der Gesamtzahl der Stoße von A und B pro Volumen und proZeit:
ZAB
D.h. wir erhalten:
−d([A]NA)dt
= ZAB (6.3)
oder
−d[A]dt
=ZABNA
= σ < vr > NA[A][B] (6.4)
Vergleichen wir das mit der DGL fur die obige Reaktion,
−d[A]dt
= k[A][B]

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 62
so konnen wir die Geschwindigkeitskonstante mikroskopisch wie folgt definieren:
k = σ < vr > NA (6.5)
Vergleich mit Arrhenius:k = Ae−EA/kT
Daher muss in der mittleren Geschwindigkeit die exponentielle Abhangigkeit vonder Temperatur ’versteckt’ sein. Dies wollen wir nun im Folgenden genauer unter-suchen.
6.2 Geschwindigkeitsverteilung-1
In der kinetischen Gastheorie nahern wir Molekule als Punkte ohne innere Frei-heitsgrade. Die Molekule haben nicht alle die gleiche Geschwindigkeit, d.h. es wirdeine Geschwindigkeitsverteilung geben die angibt, mit welcher Wahrscheinlich-keit ein Molekul eine Geschwindigkeit im Intervall v+dv besitzt.
Fur Molekule benotigen wir nun eine kontinuierliche Verteilungsfunktion der Ge-schwindigkeiten F(−→v ), die Geschwindigkeit ist ein Vektor mit den karthesischenKomponenten
~v = (vx, vy, vz) v = |~v| =√v2x + v2
y + v2z
d.h. man kann auch schreiben:
F (~v)d3~v = F (vx, vy, vz)dvxdvydvz
Dies gibt die Wahrscheinlichkeit an, ein Molekul mit der Geschwindigkeit im In-tervall zwischen ~v und ~v + d~v zu finden bzw. die Wahrscheinlichkeit, ein Molekulmit den Geschwindigkeitskomponenten zwischen (vx, vy, vz) und (vx + dvx, vy +dvy, vz + dvz) zu finden. Wir nehmen nun an, dass die Verteilung isotrop ist, d.h.die Geschwindigkeit z.B. in x-Richung nicht von der Geschwindigkeit in y-Richtungabhangt, d.h. die Verteilungen der Komponenten sind unabhangig voneinander.Dann konnen wir schreiben:
F (~v)d~v = f(vx)f(vy)f(vz)dvxdvydvz (6.6)
Diese Funktionen sind normiert, die Wahrscheinlichkeit, dass das Teichen irgend-eine Geschwindigkeit hat ist:
1 =∫ ∞−∞
F (~v)d3~v =∫ ∞−∞
∫ ∞−∞
∫ ∞−∞
f(vx)f(vy)f(vz)dvxdvydvz
Damit folgt sofort, dass jede Komponente fur sich normiert ist:∫ ∞−∞
f(vx)dvx = 1, etc. (6.7)

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 63
Nun wollen wir die Funktion f(v) explizit ableiten. Dies wird auf eine e-Funktionhinauslaufen, da nur diese so wie in Gl. 6.6 gefordert zerlegbar ist (ea+b+c = eaebec).
Betrachten wir dazu den Logarithmus
ln(F (~v)) = ln(f(vx)) + ln(f(vy)) + ln(f(vz)),
und bilden die partielle Ableitung nach vx:
∂ln(F (~v))∂vx
=dln(f(vx))
dvx
∂ln(F (~v))∂vx
=∂ln(F (~v))
∂v
∂v
∂vx=∂ln(F (~v))
∂v
vxv
da ∂v∂vx
=∂√v2x+v2
y+v2z
∂vx= vx
v (analog fur y,z Komponente). D.h.
1v
∂ln(F (~v))∂v
=1vx
dln(f(vx))dvx
=1vy
dln(f(vy))dvy
=1vz
dln(f(vz))dvz
Nun hangt der 2. Term nur von vx, der 3. nur von vy und der 4. nur von vz ab. Da dies fur allevi gilt, konnen nur gleich sein, wenn sie konstant sind! Aus
1vi
dln(f(vi))dvi
= −2γ
folgt:dln(f(vi)) = −2γvidvi → ln(f(vi)) = −γv2
i + c
oder (mit A = ec)
f(vi) = Ae−γv2i (6.8)
Zur Bestimmung von A verwenden wir die Normierungsbedingung Gl. 6.7.
1 =∫ ∞−∞
f(vi)dvi =∫ ∞−∞
Ae−γv2i dvi = A
√π/γ
D.h.A =
√γ/π

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 64
Wichtige Integrale∫ ∞−∞
e−ax2dx =
√π/a
∫ ∞−∞
x2e−ax2dx =
12
√π/a3∫ ∞
0x3e−ax
2dx =
12a2
∫ ∞0
x4e−ax2dx =
3√π
8a5/2(6.9)
f(vi) =√γ/πe−γv
2i (6.10)
An dieser Stelle sehen wir auch, warum die Konstante negativ (-2γ) gewahlt wurde.Eine positive Konstante wurde keine finite Wahrscheinlichkeit erlauben, sie ware∞.
6.3 Mikrosopischer Druck und Gleichverteilungssatz
Die Geschwindigkeitsverteilung f(vi) ist eine mikroskopische Große die es unserlaubt, alle Eigenschaften eines Systems (s.u), d.h. auch die Makroskopischen,zu berechnen. Es wird oft gesagt, dass sich die Thermodynamik auf die mikro-skopische Theorie reduzieren lasst, d.h. aus ihr ableiten lasst. Dazu mussen wirnoch die Konstante γ bestimmen. Dies geht jedoch nicht ohne Ruckgriff auf dieThermodynamik!Dazu betrachten wir den Druck p, definiert als Kraft/Flache: Mikroskopisch ent-steht der Druck durch Stoße der Teilchen mit der Gefasswand. Bei jedem Stoßwird ein Impuls m∆v ubertragen, d.h. wenn ein Teilchen der Geschwindigkeit vxelastisch an der Wand reflektiert wird, andert sich seine Geschwindigkeit zu -vx,d.h. m∆v = 2mvx. In der Zeit ∆t erreichen die Teilchen mit Geschwindigkeit vxdie Wand, wenn sie sich im Abstand a von ihr befinden (Abb. 26), d.h.
a = vx∆t
Die Kraft auf die Wand ist die Impulsanderung pro Zeit, d.h.:
F =2mvx
∆t=
2mv2x
a
gegeben. Der Druck, der durch ein Teilchen ausgeubt wird, ist:
p =F
A=
2mv2x
aA=
2mv2x
V.
In dem Volumen V= aA befinden sich N Teilchen mit den Geschwindigkeiten vxund -vx, d.h. N/2 Teilchen fliegen in +x Richtung und uben den Druck auf dieFlache A aus:
pges =12
N∑i=1
pi =12
N∑i=1
2mv2xi
V=m
V
N∑i=1
v2xi

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 65
Abbildung 25:
Mit der mittleren quadratischen Geschwindigkeit
< v2x >=
1N
N∑i=1
v2xi
ergibt sichpgesV = mN < v2
x >,
d.h. durch Vergleich mitpV = nRT = NkT
sehen wir sofort:
m < v2x >= kT (6.11)
12m < v2
x >= Exikin ist die kinetische Energie fur einen Freiheitsgrad (xi) EI-NES Teilchens. Jeder Freiheitsgrad eines Teilchens xi,yi,zi hat demnach die gleicheEnergie 1
2 kT, dies wird das Aquipartitiostherorem (Gleichverteilungssatz)genannt. Die gesamte kinetische Energie ist daher:
Ekin =32NkT (6.12)
6.4 Geschwindigkeitsverteilung-2
Im letzten Kapitel hatten wir mit der phanomenologischen Thermodynamikverglichen welche es uns ermoglicht hat, den Mittelwert der Geschwindigkeits-quadrate zu bestimmen:
< v2x >=
kT
m
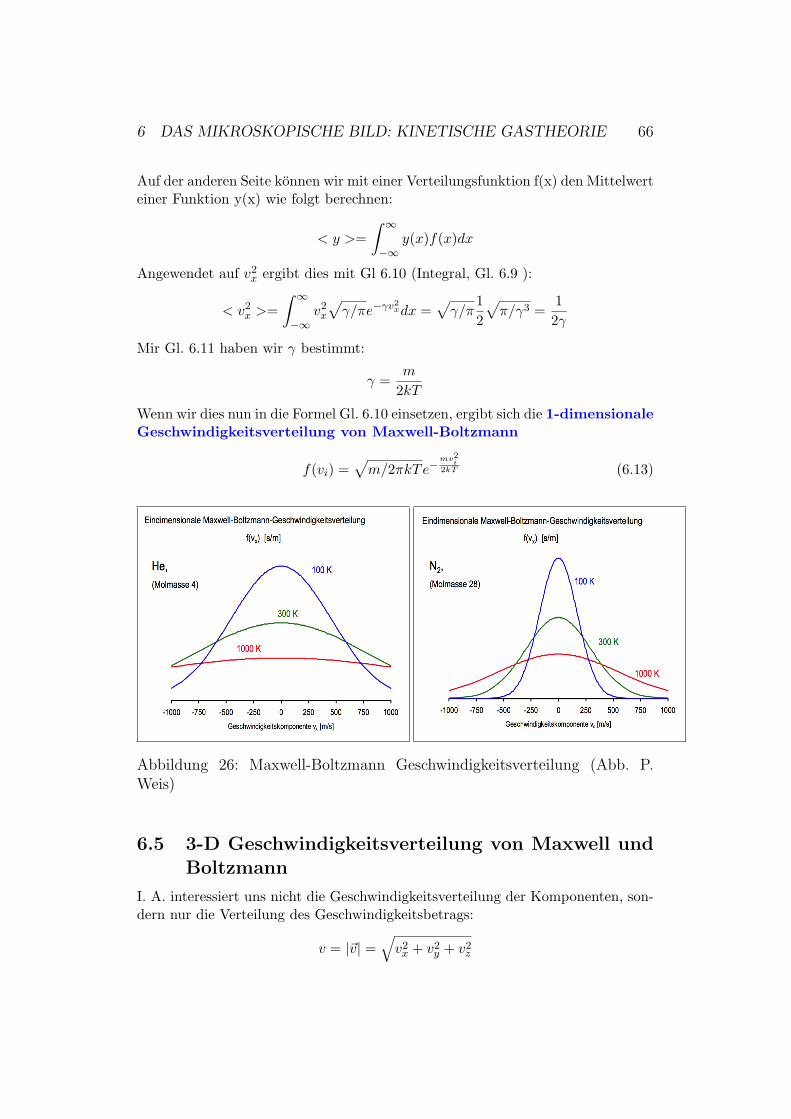
6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 66
Auf der anderen Seite konnen wir mit einer Verteilungsfunktion f(x) den Mittelwerteiner Funktion y(x) wie folgt berechnen:
< y >=∫ ∞−∞
y(x)f(x)dx
Angewendet auf v2x ergibt dies mit Gl 6.10 (Integral, Gl. 6.9 ):
< v2x >=
∫ ∞−∞
v2x
√γ/πe−γv
2xdx =
√γ/π
12
√π/γ3 =
12γ
Mir Gl. 6.11 haben wir γ bestimmt:
γ =m
2kT
Wenn wir dies nun in die Formel Gl. 6.10 einsetzen, ergibt sich die 1-dimensionaleGeschwindigkeitsverteilung von Maxwell-Boltzmann
f(vi) =√m/2πkTe−
mv2i
2kT (6.13)
Abbildung 26: Maxwell-Boltzmann Geschwindigkeitsverteilung (Abb. P.Weis)
6.5 3-D Geschwindigkeitsverteilung von Maxwell undBoltzmann
I. A. interessiert uns nicht die Geschwindigkeitsverteilung der Komponenten, son-dern nur die Verteilung des Geschwindigkeitsbetrags:
v = |~v| =√v2x + v2
y + v2z

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 67
Mit dieser hatten wir oben ja begonnen:
F (~v) = f(vx)f(vy)f(vz) =(√
m
2πkT
)3
e−m
2kT(v2x+v2
y+v2z) =
=(√
m
2πkT
)3
e−mv2
2kT (6.14)
Einschub: 2-DBetrachten wir das Problem zunachst in 2-D: Hier ist der Betrag der Geschwindigkeit
v = |~v| =√v2x + v2
y
Die MB-Verteilung aus Gl. 6.14 ist in Abb. 27 (links) gezeigt. Durch Einfuhrung von Polarko-ordinaten kann man vx und vy auch schreiben als:
vx = vcosφ, vy = vsinφ
Damit konnen wir nun eine Koordinatentransformation durchfuhren, und schreiben:
F (vx, vy) = F (v, φ) =(√
m/2πkT)2e−
mv2
2kT
F gibt nun die Wahrscheinlichkeit wieder, dass ein Teilchen eine Geschwindigkeit zwischen v undv + dv hat. Daruberhinaus ist aber auch noch das Verhaltnis von vx und vy uber den Winkelφ bestimmt. Da uns dieses nicht interessiert, konnen wir uber den Winkel integrieren. Dazumussen wir aber das Volumenelement dvx dvy durch die Differentiale dv dφ ausdrucken. Dafurbenotigt man die Funktionaldeterminante,
dvxdvy =∂(vy, vy)∂(v, φ)
dvdφ
∂(vx, vy)∂(v, φ)
=
∣∣∣∣∣∂vx∂v ∂vx∂φ
∂vy∂v
∂vy∂φ
∣∣∣∣∣ =∣∣∣∣cosφ −vsinφsinφ vcosφ
∣∣∣∣ = v
∫ ∞−∞
∫ ∞−∞
F (vx, vy)dvxdvy =∫ ∞
0
∫ π
−πF (v, φ)vdvdφ =
∫ ∞0
2πvF (v)dv
Wir konnen nun die 2D Verteilungsfunktion
G(v) = 2πvF (v)
definieren, die nur die Wahrscheinlichkeit angibt, ein Teilchen mit der Geschwindigkeit zwischenv und v+dv zu finden.
In 3-D fuhren wir Kugelkoordinaten ein, wie in Abb. 27 gezeigt. Das Volu-

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 68
Abbildung 27: 2-D Maxwell-Boltzmann Geschwindigkeitsverteilung (Abb. P.Weis)
menelement wird mit Hilfe der Funktionaldeterminaten zu:
dvxdvydvz = v2sinθdvdφdθ
∫ ∞−∞
F (~v)d3~v =∫ ∞−∞
∫ ∞−∞
∫ ∞−∞
F (~v)dvxdvydvz =∫ ∞
0
∫ π
0
∫ π
−πF (~v)v2sinθdφdθdv
G(v) =∫ π
0
∫ π
−πF (~v)v2sinθdφdθ =
∫ π
0
∫ π
−π
(√m
2πkT
)3
e−mv2
2kT v2sinθdφdθ =
=(√
m
2πkT
)3
e−mv2
2kT v2
∫ π
0sinθdθ
∫ π
−πdφ
G(v) = 4πv2
(√m
2πkT
)3
e−mv2
2kT (6.15)
G(v) ist die 3-d Maxwell-Boltzmann Geschwindigkeitsverteilung und ist in Abb.28 dargestellt.
6.6 Mittelwerte der 3-D Maxwell-Boltzmann Geschwin-digkeitsverteilung
6.6.1 Wahrscheinlichste Geschwindigkeit
Suche das Maximum der Verteilung G(v):
0 =dG(v)dv
=d
dv
(4π(√
m
2πkT
)3
v2e−mv2
2kT
)

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 69
Abbildung 28: 3-D Maxwell-Boltzmann Geschwindigkeitsverteilung (Abb. P.Weis)
0 =d
dv
(v2e−
mv2
2kT
)= 2ve−
mv2
2kT − v22vm
2kTe−
mv2
2kT
vFmax =
√2kTm
6.6.2 Mittlere Geschwindigkeit
< v >=∫ ∞
0vG(v)dv = 4π
(√m
2πkT
)3 ∫ ∞0
v3e−mv2
2kT dv (6.16)
Mit Integral Gl. 6.9[∫∞
0 x3e−ax2dx = 1/(2a2)
]< v >= 4π
( m
2πkT
)3/2 2k2T 2
m2=
√8kTπm
= 1.13vFmax (6.17)
6.6.3 Mittleres Geschwindigkeitsquadrat
< v2 >=∫ ∞
0v2G(v)dv = 4π
(√m
2πkT
)3 ∫ ∞0
v4e−mv2
2kT dv (6.18)
Mit Integral Gl. 6.9[∫∞
0 x4e−ax2dx = 3
√π
8a5/2
]< v2 >=
3kTm

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 70
oder√< v2 > =
√3kTm
= 1.09 < v >= 1.22vFmax
Die Mittelwerte der Geschwindigkeiten sind in Abb. 29 gezeigt.
Abbildung 29: Maxwell-Boltzmann Geschwindigkeitsverteilung (Abb. P.Weis)
6.6.4 Mittlere Relativgeschwindigkeit
Bei der Betrachtung von Kollisionen ist es sinnvoll, die Relativgeschwindigkeit< vr > (s.o.) und die Geschwindigkeit des Massenzentrums zu betrachten.Wir definieren zunachst (Abb. 30):
M = mA +mB µ =mAmB
mA +mB=mAmB
M
−→vA = (vAx, vAy, vAz) vA = |−→vA| =√v2Ax + v2
Ay + v2Az
(analog vB).Betrachten wir nun die Bewegung des Schwerpunkts. Der Gesamtimpuls ist:
M−→vZ = mA−→vA +mB
−→vB.
Umformen gibt die Schwerpunktsgeschwindigkeit:
(a) −→vZ =mA−→vA
M+mB−→vB
M
(a′) −→vZM
mB=mA−→vA
mB+−→vB
(b) −→vr = −→vA −−→vB
(b′) vr = |−→vr | =√
(vAx − vBx)2 + (vAy − vBy)2 + (vAz − vBz)2

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 71
Abbildung 30: Relativgeschwindigkeit vr(rechts) und Geschwindigkeit desMassenzentrums vz
Nun betrachten wir die kinetische Energie in den Laborkoordinaten
Ekin =12mAv
2A +
12mBv
2B (6.19)
In den Schwerpunktskoordinaten sieht die wie folgt aus:
Ekin =12Mv2
z +12µv2
r (6.20)
Beweis:(a’) + b:
−→vZM
mB+−→vr =
mA−→vA
mB+−→vB +−→vA −−→vB | ∗ 1
mA
−→vZ1µ
+−→vrmA
=−→vAµ
−→vA = −→vZ +−→vrmB
M
Analog aus (a”) : −→vZ MmA
= mB−→vB
mA+−→vA folgt mit (a”) - (b):
−→vB = −→vZ −−→vrmA
M
v2A = v2
Z + 2−→vZ−→vrmB
M+ v2
r
m2B
M2
v2B = v2
Z − 2−→vZ−→vrmA
M+ v2
r
m2A
M2
In Gl. 6.19 einsetzen ergibt Gl. 6.20.

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 72
Nach diesen Vorarbeiten konnen wir den Mittelwert der Relativgeschwindigkeitberechnen: Die Wahrscheinlichkeit, dass Teilchen A die Geschwindigkeit vA undTeilchen B die Geschwindigkeit vB hat, ist durch das Produkt der Wahrscheinlich-keiten F(vA)F(vB) gegeben. Den Mittelwert errechnen wir dann:
< vr > =∫ ∞−∞
∫ ∞−∞
vrF (vA)F (vB)d3−→vAd3−→vB =
=∫ ∞−∞
∫ ∞−∞
vrF (vr)F (vZ)d3−→vrd3−→vZ =
=∫ ∞−∞
vrF (vr)d3−→vr(∫ ∞−∞
F (vZ)d3−→vZ)
=
=∫ ∞−∞
vrF (vr)d3−→vr (6.21)
Im dritten Schritt konnten wir die Integrale auseinander ziehen, da die Integrationvon vZ die Koordinate vr nicht betrifft. Im letzten Schritt haben wir ausgenutzt,dass F(vZ) normiert ist.
Beweis:Mit mAmB = µM und Gln. 6.19,6.20 erhalten wir sofort:
F (vA)F (vB) =(√
mA
2πkT
)3
e−mv2
A2kT
(√mB
2πkT
)3
e−mv2
B2kT =
=(√
µ
2πkT
)3(√
M
2πkT
)3
e−mv2
A+mv2A
2kT =
=(√
µ
2πkT
)3(√
M
2πkT
)3
e−µv2r+Mv2
z2kT =
=(√
µ
2πkT
)3(√
M
2πkT
)3
e−µv2r
2kT e−Mv2
z2kT = F (vr)F (vZ)
D.h. die Integrale zerfallen sehr schon in die Relativkoordinaten. Nun mussen wir noch zeigen,dass gilt:
d−→vAd−→vB = d−→vrd−→vZBetrag der Funktionaldeterminante:
| ∂(−→vr ,−→vZ)∂(−→vA,−→vB)
| = 1
∂(−→vr ,−→vZ)∂(−→vA,−→vB)
=
∣∣∣∣∣∂−→vZ∂−→vA
∂−→vZ∂−→vB
∂−→vr∂−→vA
∂−→vr∂−→vB
∣∣∣∣∣ =∣∣∣∣mA/M mB/M
1 −1
∣∣∣∣ = −1

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 73
Das Integral Gl. 6.21 haben wir oben schon berechnet: wir mussen nun wiederPolarkoordinaten fur −→vr einfuhren, uber φ und θ integrieren (fuhrt von F(v) zuG(v))) und erhalten dann wie in den Gln. 6.16,6.17
< vr > =∫ ∞
0vrG(vr)dvr =
√8kTπµ
(6.22)
Das Ergebnis ist sehr interessant: es ist formal identisch zur mittleren Geschwin-digkeit, nur dass v durch vr und m durch die reduzierte Masse µ ersetzt ist.
6.7 Arrhenius 2: Mindestenergie
Die Anzahl der Stoße pro Volumen und Zeit hatten wir oben berechnet (Gl. 6.2):
ZAB = σ< vr >N2A[A][B]
Gerade haben wir < vr > (Gl. 6.22) berechnet, indem wir uber ALLE Relativge-schwindigkeiten, (von ’0’ bis∞) gemittelt haben. Im Gegensatz dazu haben wir beider Diskussion der Arrheniusgleichung festgestellt, dass nur solche Stoße zu einerReaktion fuhren, die eine Mindestenergie Ea haben, d.h. bei denen die Molekuleeine Mindestgeschwindigkeit var besitzen, d.h. uns interessiert eigentlich:
ZaAB = σN2A[A][B]
∫ ∞var
vrG(−→vr)dvr =
= σN2A[A][B]4π
(õ
2πkT
)3 ∫ ∞var
v3re− µv
2r
2kT dvr (6.23)
Die kinetische Energie der Relativbewegung ist:
Er =12µv2
r , → vr =
√2Erµ
→ dvrdEr
=1√
2Erµ
dvr =1√
2ErµdEr → v3
rdvr =2Erµ2
dEr
Dies in obiges Integral einsetzen und mit Hilfe von partieller Integration losen:
ZaAB = σN2A[A][B]4π
(õ
2πkT
)3 2µ2
∫ ∞Ea
Ere−ErkT dEr (6.24)
= σN2A[A][B]
√8kTπµ
(1 +
EakT
)e−
EakT
Qualtiativ entspricht dies schon der Arrhenius Form, jedoch hat der Vorfaktor eine1/T Abhangigkeit, d.h. der Vorfaktor wurde fur kleine Temperaturen sehr grosswerden. Daher mussen wir nochmals nachbessern.

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 74
6.8 Arrhenius 3: Energieabhangiger Stoßparameter
Wie in Abb. 31 gezeigt, finden viele Stoße mit der Versetzung a statt. Fur a=0 istder Impulsubertrag maximal, fur a=d streifen sie sich, es findet kein Impulsubert-rag statt. Fur 0< a > d wird die effektive Relativgeschwindigkeit v′r mit demWinkel θ wie folgt berechnet:
v′r = vrcosθ = vr
√d2 − a2
d
Jetzt quadrieren wir beide Seiten und multiplizieren mit 12µ:
Abbildung 31: Line of centers model (LOC) fur den bimolekularen Stoß
12µv′2r =
12µv2
r
(d2 − a2)d2
→ a2 = d2
(1− E′r
Er
)(E′r = 1
2µv′2r , Er = 1
2µv2r )
Fur E′r ≤ Ea (Aktivierungsenergie) findet keine Reaktion statt, dies definiert einemaximale Versetzung amax:
a2max = d2
(1− Ea
Er
)Nun multiplizieren wir die Gleichung mit π und erinnern uns an die Definition desStreuquerschnitts σ = d2π,
a2maxπ = σ
(1− Ea
Er
):= σ(Er) (6.25)
Fur große Energien kann a großer sein als bei kleinen Energien, der Streuquer-schnitt ist also energieabhangig. Jetzt mussen wir uns aber die Formel fur dieStoßzahl Gl. 6.24 nochmals ansehen. Da σ(Er) energieabhangig ist, mussen wir esnoch ins Integral schreiben:

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 75
ZaAB = N2A[A][B]4π
(õ
2πkT
)3 2µ2
∫ ∞Ea
σ(Er)Ere−ErkT dEr = (6.26)
= σN2A[A][B]4π
(õ
2πkT
)3 2µ2
∫ ∞Ea
(1− Ea
Er
)Ere
−ErkT dEr =
= σN2A[A][B]
√8kTπµ
e−EakT (6.27)
Ergebnis:Wir hatten oben gezeigt, dass bei einer bimolekularen Reaktion
A+B → P
die Abnahme von [A] linear mit ZAB zusammenhangt:
−d[A]dt
=ZaABNA
= k[A][B]
Die mikroskopisch unter den Annahmen der kinetischen Gastheorie hergeleiteteGeschwindigkeitskonstante
k = σNA
√8kTπµ
e−EakT (6.28)
lasst sich gemaß Arrhenius in den praexponentiellen Faktor
A = σNA
√8kTπµ
(6.29)
und den schon oben diskutierten Boltzmannfaktor zerlegen. Die T-Abhangigkeitvon A ist schwach im Vergleich zum Bolzmann Faktor.Dieses k, das eine Temperaturabhangigkeit wie die Arrheniusformel zeigt, konnenwir aus sehr einfachen Prinzipien, allerdings auch mit sehr drastischen Naherun-gen, aus der kinetischen Gastheorie ableiten.
Abb. 32 (links) zeigt den Bereich der MB Geschwindigkeitsverteilung, der zu ei-ner Reaktion fuhrt und Abb. 32 (rechts) illustriert den Effekt der energetischenund LOC Bedingungen auf den Stoßquerschnitt. Abb. 33 vergleicht die berechne-ten und gemessenen Arrhenius Parameter. Die Ubereinstimmung ist quantitativnicht umwerfend, hier scheint die kinetische Gastheorie etwas Essentielles nichtberucksichtigt zu haben. Wir konnen mehrere Probleme unterscheiden:
• Mechanismus: Bei der Reaktion K +Br2 wird der sogenannte Harpunen-mechanismus diskutiert. Hier wird schon vor dem Stoß ein Elektron ubertra-gen, es kommt zu einer elektrostatischen Anziehung zwischen K+ und Br−2 ,was den Stoßquerschitt erhoht.

6 DAS MIKROSKOPISCHE BILD: KINETISCHE GASTHEORIE 76
Abbildung 32:
Abbildung 33: Quelle: Atkins
• Die Naherung von Molekulen als ’harten Kugeln’ ist sicher in zweierlei Hin-sicht ungunstig. Zum Einen konnten wesentlich angessenere VdW Potentialeverwendet werden, zum Anderen ist die Kugelsymmetrie sicher oft eine zudrastische Naherung. Dem wird oft mit Hilfe des sterischen Faktors Rech-nung getragen (s. Atkins).
• In vielen Fallen ist nicht nur die Gesamtenergie beim Stoß entscheidend,sondern auch ihre Verteilung auf die verschiedenen inneren Freiheitsgradeder Molekule. So hangt beispielsweise die Kinetik der Reaktion O + H2 →OH +H von den H2 Schwingungsanregungen ab.

7 THEORIE DES UBERGANGSZUSTANDES 77
7 Theorie des Ubergangszustandes
7.1 Potentialhyperflachen: der Ubergangszustand
Die Berechnung von Ratenkonstanten mit Hilfe der kinetischen Gastheorie hatinteressante qualitative Einsichten in die Kinetik von chemischen Reaktionen ge-bracht, quantitativ scheitert sie fur komplexe Molekule, da sie deren Struktur ver-nachlassigt (Kugelnaherung). Die Reaktionskinetik hangt sowohl von der Form, alsauch von den inneren Freiheitsgraden eines Molekuls ab. Das einfachste Beispielist hier die Reaktion H2 +H → H +H2, wir wollen jedoch allgemeiner
A−B + C → A+B − C
betrachten. Wie diese Reaktion verlauft hangt von der potentiellen Energie derMolekule ab. Die potentielle Energie wird durch die Lage der Atome zueinanderbestimmt, d.h. sie hangt im Prinzip von allen Koordinaten der Molekule ab.In unserem Beispiel wird die potentielle Energie von den Abstanden rAB und rBCabhangen wie auch von dem Winkel φABC , den wir der Einfachheit halber ver-nachlassigen. Abb. 34 zeigt die Energielandschaft fur diese beiden Abstandskordi-
Abbildung 34: Energiehyperflache fur eine triatomare Reaktion (links) 1dSchnitte (rechts) (aus Wedler)
naten. Dieses Energieprofil erinnert an ein Gebirge, man vergleicht daher oft denReaktionsweg von Molekulen mit dem Uberqueren von Gebirgen. Man nennt esauch Energiehyperflache. Molekule werden sich den ’Pass’ mit der geringstenHohe zum Uberqueren ’aussuchen’ (das macht Boltzmann fur sie), d.h. einen Wegder minimalen Energie. Daher wird der dominate Reaktionspfad in Abb. 34 entlang

7 THEORIE DES UBERGANGSZUSTANDES 78
Abbildung 35: Konturdiagramm der Energiehyperflache fur eine triatomareReaktion (aus Wedler)
des ’Tals’ uber den Pass sein. Abb. 37 (links unten) zeigt im Konturdiagramm denWeg minimaler Energie. Dieser Weg hat eine effektiv zu uberwindende Barriere(Abb. 37 rechts oben). Ein Molekul mit M Atomen hat 3M Freiheitsgrade (jeweilsx,y,z), jedoch werden nicht alle Koordinaten zur Beschreibung des Reaktionswegesvon Bedeutung sein. Die Koordinaten, die den Reaktionsweg beschreiben werdenReaktionskoordinaten genannt, rAB und rBC sind also im obigen Beispiel dierelevanten Reaktionskoordinaten.
Ein wichtiger Punkt ist der Punkt hochster Energie entlang des Weges minimalerEnergie, der Sattelpunkt, auch Ubergangszustand genannt. Dieser ist durcheine bestimmte Konformation der beiden Molekule charakerisiert, die A-B Bin-dung ist schon ’leicht gelockert’ und die B-C Bindung noch nicht ’ganz geformt’,man kann diese Konformation als (ABC)# bezeichnen,
A...B...C.
Zur genauen Beschreibung einer Reaktion wurde man also diese Energiehyperflachebenotigen, die man mit Hilfe von Quantenchemischen (QC) Rechnungen er-halten kann. Fur einfache Beispiele geht das sehr gut, fur große Molekule wird diesjedoch sehr schnell zu komplex.

7 THEORIE DES UBERGANGSZUSTANDES 79
H2 hat eine Koordinate, die Energiehyperflache ist durch E(r) gegeben, sehr einfachzu rechnen. H2O hat schon 9 Koordinaten, allerdings nur 3 innere Freiheitsgrade(r1, r2, φ, s.u.), d.h. die Energie hangt von drei Koordinaten ab, E(r1, r2, φ), dieseEnergiehyperflache ist in 3-d also schon nicht mehr darstellbar.
I.A. werden wir es mit sehr hochdimensionalen Flachen zu tun haben, die die Ener-gie des Molekuls in Abhangigkeit der Koordinaten angibt E(x1, y1, z1...xM , yM , zM ).I. A nummerieren wir die 3M Koordinaten einfach durch, dies vereinfacht die For-meln:
E = E(x1, x2...x3M )
Wir werden uns dann fur den Ubergangszustand interessieren, dies ist i.A. ein
Abbildung 36: Hyperflachen mit Sattelpunkt 1. Ordnung (f(x, y) = x2− y2)und 2. Ordnung (f(x, y) = −x2 − y2)
Sattelpunkt 1. Ordnung (wie der Alpenpass). Diese Hyperflache hat entlang einerRichtung eine negative Krummung, entlang aller Anderen jedoch eine Positive.(Sattelpunkt 2.Ordnung: Fur 2 Richtungen (Koordinaten) hat die Hyperflache einenegative Krummung. 2D Beispiel: Hugelkuppe) Beispiele fur Sattelpunkte ersterund zweiter Ordnung sind in Abb. 37 gegeben.

7 THEORIE DES UBERGANGSZUSTANDES 80
Einschub: Hessematrix:
H =(
∂E∂xi∂xj
)=
∂E∂x1∂x1
∂E∂x1∂x2
... ∂E∂x1∂x3N
... ... ... ...∂E
∂x3N∂x1
∂E∂x3N∂x2
... ∂E∂x3N∂x3N
Bei einem Sattelpunkt 1. Ordnung hat H 3N-1 positive und 1 negativen Eigenwert.
Beispiele:(a) f(x, y) = x2 − y2
H =(
∂f∂x∂y
)=
(∂f∂x∂x
∂f∂x∂y
∂f∂y∂x
∂f∂y∂y
)=(
2 00 −2
)(b) f(x, y) = −x2 − y2
H =(
∂f∂x∂y
)=
(∂f∂x∂x
∂f∂x∂y
∂f∂y∂x
∂f∂y∂y
)=(−2 00 −2
)Die Werte dieser Matritzen, da sie schon diagonal sind, geben die Krummung der Funktion beif(0,0) in x- und y-Richtung an.
7.2 Aktivierter Komplex
Bei der ReaktionA+B
k1,k−1
C# k2→ P
wird der Ubergangszustand C# als Reaktionsintermediat angenommen, von demwir nun zusatzlich fordern, dass es sich mit den Edukten A und B im (vorge-lagerten) thermodynamischen Gleichgewicht befindet. Wir erhalten die DGLn(k1, k−1 >> k2):
d[C#]dt
= k1[A][B]− k−1[C#] = 0
d[P ]dt
= k2[C#]
Mit der Gleichgewichtskonstanten
K =k1
k−1
erhalten wir:d[P ]dt
= Kk2[A][B]
Aus der Thermodynamik wissen wir, dass sich K aus
∆G = G#C −GA −GB (7.1)

7 THEORIE DES UBERGANGSZUSTANDES 81
Abbildung 37: Freie Aktivierungsenthalpie
berechnet zu:RTlnK = −∆G
Wie kann man nun ∆G fur Molekule berechnen?
7.3 Energie eines Molekuls
Die Gesamtenergie eines Molekuls setzt sich aus folgenden Beitragen zusammen:
• εel: Dies ist die quantenmechanisch berechnete Energie des Molekuls (PCII).Dazu muss man die Schrodingergleichung losen und erhalt die Energie desMolekuls im elektronischen Grundzustand (S0, s.o.). Die Anregungsenergiein die elektronisch angeregten Zustande S1, S2... ist viel großer als die ther-mische Energie (kT), daher liegt das Molekul auch bei 300K nur im S0 vor.
• εtrans: Kinetische Energie der Translationsbewegung des Molekuls das gemaßder Boltzmannverteilung auch von der Temperatur abhangt. Auch diese wirdublicherweise quantenmechnanisch (PCII) berechnet.
• εrot: Kinetische Energie der Rotationsbewegung des Molekuls (QM Berech-nung).
• εvib: Energie der Schwingungsbewegung. Diese wollen wir im Folgenden ge-nauer betrachten.
Die vier Energiebeitrage
ε = εel + εtrans + εrot + εvib (7.2)

7 THEORIE DES UBERGANGSZUSTANDES 82
Abbildung 38: Elektronische, Translations, Rotations und Vibrations Bei-trage zur Energie eines Molekuls bei T6= 0
sind in Abb. 38 grafisch verdeutlicht. Betrachten wir als Beispiel das Molekul H2.Wir hatten im letzten Kapitel die Transformation in die Relativkoordinaten vr undvZ diskutiert und festgestellt, dass sich die kinetische Energie wie folgt schreibenlasst.
Ekin =12mAv
2A +
12mBv
2B =
12Mv2
z +12µv2
r
Die kinetische Energie der Translation ist also
12Mv2
z ,
die Rotation ist ein klein wenig aufwendiger zu berechnen. Dennoch benotigt mandafur nur die atomaren Massen und die Struktur des Molekuls. Wenn das MolekulM Atome hat, so hat es 3M Freiheitsgrade (x,y,z Bewegungen der Atome). AufTranslation und Rotation des Gesamtmolekuls enfallen jeweils 3 Freiheitsgrade,daher gibt es nur noch 3M-6 Freiheitsgrade fur die internen Schwingungsbewegun-gen (lineare Molekule: 3M-5).
Beispiel H2O: Das Wassermolekul hat 3 Atome, also 3x3=9 Freiheitsgrade. NachAbzug der 6 Translations- und Rotationsfreiheitsgrade (ω1, ..., ω6) bleiben noch3 Freiheitsgrade fur interne Vibrationen ubrig. Die Atome schwingen nicht ein-zeln, sondern die Schwingungsmoden sind kollektive Bewegungen der Atome, dieNormalmoden genannt werden, mit den Schwingungsfrequenzen ω7 = 1595cm−1
fur die Winkeldeformationsschwingung und ω8 = 3657cm−1 fur die symmetrischeStreck- und ω9 = 3756cm−1 fur die asymmetrische Streckschwingung.

7 THEORIE DES UBERGANGSZUSTANDES 83
Abbildung 39: Schwingungsmoden des Wassermolekuls
7.3.1 Molekulschwingungen
Betrachen wir nun das H2 Molekul. Die wichtige Koordinate ist der Abstand,als Relativkoordinate xr geschrieben, und die Relativgeschwindigkeit vr. Wichtigist noch die reduzierte Masse µ, die die kinetische Energie der Relativbewegungbestimmt. Um das Problem einfach losen zu konnen, nahern wir das H-H Potentialals harmonisches Potential, d.h. wir modellieren die chemische Bindung zwischenden H’s als Feder (Hooksches Gesetz), siehe Abb. 40 (links):
Epot = 0.5k(xr − x0)2
Die Federkonstante k ist die 2te Ableitung von Epot, d.h.:
k =∂2Epot∂x2
r
Damit konnen wir die Kraftgleichung aufstellen:
F = µa = µxr = −k(xr − x0)
Als Losung erhalten wir die Schwingungsfrequenz:
ω =
√k
µ
(Die Kreisfrequenz ω ist mit der Frequenz uber ω = 2πν verbunden). Die Relativ-bewegung ist dann durch
xr(t)− x0 = Asin(ωt)
(schwingt um x0) beschrieben. Ein klassischer harmonischer Oszillator hatein kontinuierliches Energiespektrum, abhangig von A2:
E = Ekin + Epot =12kA2

7 THEORIE DES UBERGANGSZUSTANDES 84
Abbildung 40: Quantenmechanischer harmonischer Oszillator
Dagegen ist nach der Quantenmechanik die Energie gequantelt, dies gilt auch furden harmonischen Oszillator, wie in Abb. 40 (rechts) skizziert. Die Lage der Ener-gieniveaus wird in der QM (PCII) zu
εi = (i+12
)hν
berechnet. Man findet zum Einen eine endliche Energie selbst fur i=0 (Nullpunkts-energie), als auch ein Energiespektrum mit konstanten Energiedifferenzen hν (Be-sonderheit des harmonischen Oszilators).
Das H2 Molekul hat eine Schwingungsmode, d.h. eine Frequenz, i.A. finden wirfur Molekule mit M Atomen 3M-6 Schwingungsfrequenzen
ν1, ν2...ν3M−6
Ein Molekul besteht dann aus 3M-6 solchen Oszillatoren und hat die Schwingungs-energie:
E(ν1, ν2...) = ε1i + ε2j + ...ε3M−6k
Jede Schwingung kann in einem anderen Grund- oder angeregten Zustand i,j,k...sein.
7.4 Statistische Mechanik
In der Thermodynamik wurde gezeigt, dass die thermische Besetzung der Ener-gieniveaus durch die Boltzmann-Verteilung beschrieben wird:
niN
= pi =e−βεi
q(7.3)

7 THEORIE DES UBERGANGSZUSTANDES 85
mit der Abkurzung β = 1/kT und der Normierungskonstante
q =∑i
e−βεi . (7.4)
pi gibt die Wahrscheinlichkeit, ein Molekul im Vibrationszustand i zu finden, niist dann die Anzahl der Molekule in diesem Zustand, wenn man ein Ensemble vonN Molekulen betrachtet. q wird auch molekulare Zustandssumme genannt.
7.4.1 Molekulschwingungen: Harmonischer Oszilator
Fur H2, ein Molekul mit einer Schwingungsfrequenz ν hatten wir εi = (i+ 12)hν:
qvib =∑i
e−β(i+ 12
)hν = e−12βhν∑i
e−iβhν = e−12βhν∑i
(e−βhν
)i= (7.5)
= e−12βhν 1
1− e−βhν=: qvib0 qvibT
(eab = (ea)b,∑
n xn = 1
1−x). Der erste Term
qvib0 = e−12βhν
betrifft die niedrigste Schwingungsenergie fur i=0, die auch bei T=0 vorhanden ist(im Gegensatz zur klassischen Mechanik). Dieser Term wird daher weiter untender inneren Energie U0 bei 0K zugerechnet. Der zweite Term
qvibT =1
1− e−βhν
betrifft dann alle thermisch angeregten Zustande. Er geht in die freie Enthalpie G(s.u.) ein.
7.4.2 Elektronische Energie
Molekule befinden sich bei 300K i.A. im S0 Zustand, da die Anregungsenergie inden S1 wesentlich grosser als kT ist.
Da nur der elektronische Grundzustand εel0 besetzt ist, erhalt man fur die Summe:
qel = exp(−βε0)
7.4.3 Mittlere (Innere) Energie
(N Molekule):
U =< E >=∑i
niεi = −Nq
dq
dβ(7.6)

7 THEORIE DES UBERGANGSZUSTANDES 86
Dies ist ein wichtiges Ergebnis: wir konnen die innere Energie durch dieZustandssumme ausdrucken! Dies wollen wir im Folgenden fur S und G auchnoch ableiten.
Beweis:
dq
dβ=∑i
d(e−βεi)
dβ= −
∑i
εie−βεi = −q
∑i
εipi = − q
N
∑i
εini
Beispiel: Harmonischer Oszillator
U = −Nq
dqvib
dβ= (−N
q)∑i
(−(i+12
)hν)e−βεi =
= N
(∑i
pi12hν +
∑i
(ihν)pi
)=
= N(12hν) +N < ihν >
Die Innere Energie ist also die Summe aus N mal der Nullpunktsenergie der Mo-lekule (T=0) und N mal dem Mittelwert der Schwingungsenergie.
Beispiel: Elektronische Zustande eines Molekuls
U = −Nqel
dqel
dβ=N
qelε0q
el = Nε0
Die Innere Energie setzt sich demnach aus den N Grundzustandsenergien der NMolekule zusammen.
Beide Beitrage, der Elektronische und der Vibrationsbeitrag, lassen sich mit Quan-tenchemieprogrammen ausrechnen.
7.4.4 Entropie
S = klnW = −Nk∑i
pilnpi (7.7)

7 THEORIE DES UBERGANGSZUSTANDES 87
Beweis:
lnW = n
(N !
n0!n1!....
)= ln(N !)− ln(n0!n1!....) =
= ln(N !)−∑i
ln(ni!) = NlnN −N −∑i
(niln(ni)− ni) =
= NlnN −∑i
niln(ni) =∑i
ni(lnN − ln(ni)) =
= −∑i
niln(niN
)= −
∑i
niln(pi) = −N∑i
piln(pi)
(2. Zeile: Stirling ln(x!) = xlnx - x)
Damit folgt:
S = U/T +Nklnq (7.8)
Beweis:
ln(pi) = ln(e−βεi/q) = −βεi − lnq
−Nk∑i
pilnpi =∑i
Nkpiβεi +Nk∑i
pilnq = βkU +Nklnq
7.4.5 Freie Enthalpie
G = H − TS = U − TS + pV = U − U −NkTlnq + pV (7.9)
Fur ein ideales Gas (pV = NkT= nRT) und mit alnb = lnba:
G = nRT − kT ln(q)N = nRT − kT lnQ (7.10)

7 THEORIE DES UBERGANGSZUSTANDES 88
Erlauterung: was heisst (q)N = Q?Wenn wir N (gleiche) Molekule haben, so kann Molekul 1 im Schwingungszustandi sein, Molekul 2 im Schwingungszustand j, usw., und da die Wahrscheinlichkei-ten statistisch unabhangig sein sollten erhalten wir die Wahrscheinlichkeit fur dasGesamtsystem:
P (i, j, ...k) = p1i p
2j ...p
Nk =
e−βεie−βεj ...e−βεk
q1q2...qN=e−β(εi+εj ...+εk)
q1q2...qN(7.11)
Wir konnen die Zustandsumme fur das Gesamtsystem damit definieren als:
Q = q1q2...qN = (q)N (7.12)
(da q = q1 = q2 = ... = qN fur gleiche Molekule bei gleicher T). Wir sehen also, dieZustandssumme fur ein Gesamtsystem mit Energie
E = εi + εj ...+ εk
ist das Produkt der Zustandssummen der Einzelsysteme (Molekule). Dies gilt auch(a) fur die verschiedenen Energiebeitrage:
ε = εel + εtrans + εrot + εvib
fuhrt also zur Zustandssumme:
q = qelqtransqrotqvib (7.13)
(b) fur Molekule mit M Freiheitsgraden und damit mehreren Schwinungsfrequenzenν1, ν2...:
qvib = qvibν1qvibν1
...
Ist das Produkt der Zustandssummen der Einzelschwinungen.

7 THEORIE DES UBERGANGSZUSTANDES 89
Erlauterung 2: UnunterscheidbarkeitEs gibt aber eine kleine Einschrankung fur Gl. 7.12: sie gilt nur fur klassische Sy-steme, bei denen die Teilchen unterscheidbar sind. Die Formel unterscheidet dieWahrscheinlichkeit fur Teilchen 1 in Zustand i und 2 in j
P (i, j, ...k)
von der WahrscheinlichkeitP (j, i, ...k).
Quantenmechanisch kann man diese Unterscheidung nicht mehr aufrecht erhalten,die beiden Falle sind in der QM identisch. Das fuhrt dazu, dass man die Wahrschein-lichkeit durch N! teilen muss, man findet:
Q =(q)N
N !(7.14)
Gl. 7.13 ist davon aber nicht betroffen, da die unterschiedlichen Energien in der QMweiterhin unterscheidbar sind.
Quantenmechanisch erhalten wir damit:
G = −kTNln( qN
)(7.15)
Beweis:
G = nRT − kT lnqN
N != nRT − kT lnqN + kT lnN ! =
= nRT − kT lnqN + kTNlnN − kTN = −kTNlnq + kTNlnN =
= −kTNln( qN
)(Stirlings Formel, s.o.)
7.5 Freie Enthalpie und Gleichgewichtskonstante
Als wichtiges Ergebnis aus der Statistischen Thermodynamik haben wir:
G = −kTNln( qN
). (7.16)
Man braucht also nur die Zustandssumme fur EIN Molekul ausrechnen, und erhaltdaraus die freie Enthalpie (und damit K).
Die Zustandssumme selbst ist allerdings komplex: Wir hatten zunachst die Auf-spaltung
q = qelqtransqrotqvib,

7 THEORIE DES UBERGANGSZUSTANDES 90
um dann noch weiter aufzuspalten:
qvib = qvib0 qvibT
qel und qvib0 hangen beide nicht von T ab, wie wir oben gesehen haben, es liegt alsonahe diese in einen Term fur T=0, U0 zusammenzufassen. Wir bilden
G = −NkT ln(q/N) = −NkT (lnqel + lnqvib0 )−NkT ln(qtransqrotqvibT
N)
= Nε0 +N12hν −NkTln(
qtransqrotqvibTN
)
= U0 −NkTln(qtransqrotqvibT
N) (7.17)
U0 enthalt also die QM Energie und die Energie der Nullpunktsschwingungen.Wie wir oben gesehen haben, gibt es ein unterstes Vibrationsniveau, das auch beiT=0K eine Schwingungsenergie beitragt, wahrend diese beim klassichen Oszillatorverschwindet. Alle thermischen Beitrage sind nun in qtransqrotqvibT /N .Jetzt mussen wir diese Beitrage fur die Molekule A und B und den Ubergangszu-stand C# berechnen, wir definieren:
qA = qtransA qrotA qvibAT
GA = UA0 −NkT ln(qAN
)
(qB, q#C analog).
Damit erhalten wir fur die Differenz der freien Enthalpie in Gl. 7.1:
∆G = GC# −GA −GB = (7.18)
= UC#0 − UA0 − UB0 −
− kTNln
(q#C
N
)+ kTNln
(qAN
)+ kTNln
(qBN
)Mit der Aktivierungsenergie ∆E = UC#
0 − UA0 − UB0 erhalten wir:
∆G = ∆E −NkT ln
(Nq#
C
qAqB
)(7.19)
Umrechnung auf molare Großen q bezog sich bisher auf beliebige Systeme mitN Teilchen. Wir definieren jetzt die molare Zustandssumme (n: Molzahl):
qm =q
n

7 THEORIE DES UBERGANGSZUSTANDES 91
und teilen die Gleichung durch n (NA = N/n,Gm = G/n,Em = E/n):
∆Gm = ∆Em −RTln
(NAq
#mC
qmAqmB
)(7.20)
MitRTlnK = −∆G
erhalten wir fur K:
K =
(NAq
#mC
qmAqmB
)e−
∆EmRT (7.21)
K hat nun noch ein ’kleines’ Problem’, es ist nicht dimensionslos. Um K dimensi-onslos zu machen, multipliziert man K ublicherweise mit der Standardkonzentration[I] = 1mol/l. Dies verandert den numerischen Wert von K nicht, nur seine Dimen-sion.
K ′ =[I][C#][A][B]
daher: K = K’/[I] Mit p0 = [I]RT erhalten wir:
K = K ′/[I] = K ′RT
p0
Unser berechnetes K muss also noch mit RTp0 multipliziert werden, um thermody-
namisch korrekt zu sein. Wir wollen uns dies merken, jedoch im Folgenden mit Kweiterrechnen.
Die Ratenkonstante fur die Produktbildung war:
k = k2K.
K haben wir nun berechnet, jetzt untersuchen wir k2.
7.6 ’Imaginare’ Schwingungsfrequenzen am Ubergangs-zustand
Betrachten wir nun H-H-H am Ubergangszustand: Wir hatten gesehen, dass ent-lang der Reaktionskoordinate das Potential eine negative Krummung hat. Wenndie potentielle Energie jedoch eine negative Krummung 0 > k aufweist (Abb. 41),resultiert eine imaginare Schwingungsfrequenz νI : 1 Fur den in der Abbildung
1Ein Ubergangszustand wird sich also durch eine Hessematrix mit einem negativenEigenwert ausweisen.

7 THEORIE DES UBERGANGSZUSTANDES 92
skizzierten einfachen (1-dimensionalen) Fall erhalten wir:
ωI = 2πνI =
√−|k|µ
= i
√|k|µ.
Abbildung 41: Sattelpunkt: Schwingung mit ’imaginarer Frequenz
Diese ’Schwingung’ fuhrt vom Sattelpunkt entweder zum Produkt oder zu denEdukten zuruck. Die Ratenkonstante k2 wird demnach direkt von dieser Schwin-gungsfrequenz abhangen. Die Einheit dieser Schwingungsfrequenz ist [s−1], dies istauch die Einheit der Ratenkonstante k2. Wir konnen also schreiben:
k2 = νI (7.22)
Der Ubergangskomplex zerfallt also in einer unimolekularen Reaktion in die Pro-dukte. Die Ratenkonstante ist durch die Frequenz νI der Molekulschwingung ge-geben, die entlang der Reaktionskoordinate verlauft.
I. A. interessieren uns grossere Molekule mit einer Vielzahl von Schwingungen.Wir hatten oben gesehen, dass die Zustandssumme fur ein Molekul mit mehrerenSchwingungsmoden mit den Frequenzen ν1, ν2... folgendermassen aussieht:
qvibT = qν1T q
ν2T ...q
νIT ...
Die Zustandssumme des Ubergangszustandes q#mC enthalt also im Vibrationsteil
eine Schwingung mit ’imaginarer’ Frequenz νI , die wir nun abspalten wollen:
q#vibmC = qνIT q
∗vibmC (7.23)
qνIT =∑n
e−nβhνI =1
1− e−βhνI≈ 1
1− (1 + (−βhνI))=kT
hνI

7 THEORIE DES UBERGANGSZUSTANDES 93
(Reihenentwicklung: ex = 1+x+ ...). q∗vibmC ist die (Vibrations-) Zustandsumme furden aktivierten Komplex, bei dem die imaginare Frequenz fehlt.
q#vibmC =
kT
hνIq∗vibmC (7.24)
Da alle anderen Beitrage (rot,trans) zu der Zustandssumme gleich bleiben, konnenwir auch schreiben:
q#mC =
kT
hνIq∗mC (7.25)
7.7 Eyring Gleichung
Wir erhalten damit die Eyring Gleichung:
k = k2K = νIkT
hνI
(NAq
∗mC
qmAqmB
)e−
DeltaEmRT
= NAkT
h
q∗mCqmAqmB
e−∆EmRT (7.26)
kTh = 6.25 ∗ 1012s−1 (T=300K) ist der Frequenzfaktor, e−
∆EmRT ist der Boltzmann-
faktor fur die ’klassische’ Barrierenhohe Em, der Quotient aus den Zustandssum-men kann als ein Entropieterm interpretiert werden. Dabei wurde vernachlassigt,dass in qmC q#vib
mC durch q∗vibmC ersetzt wurde.Wir haben damit das kinetische Problem auf ein Thermodynamisches zuruck-gefuhrt. Die Kunst besteht nun darin, den Ubergangszustand zu lokalisieren, wasinzwischen eine Routineaufgabe fur die Quantenchemie ist.Dies ist ein interessantes Ergebnis, das eine hinreichend genaue Berechnung zulasst:Mit heutigen Quantenchemieverfahren kann man mit guter Genauigkeit die Schwin-gungsfrequenzen der Molekule A, B und C#, und damit die Vibrationszustands-sumen berechnen. Fur q∗vibmC wird bei Bildung der Summe einfach die Schwingungmit imaginarer Frequenz weggelassen. Auch die Berechnung der Rotations- undTranslationsbeitrage zu q stellt keine Schwierigkeit dar. Etwas herausfordernderist die Berechnung von Em, da diese Energiedifferenz im Exponenten steht: Manbenotigt daher sehr genaue QM Methoden, die einen Fehler von ∆E < 1kcal/molin der Energiedifferenz aufweisen:
e−Em+∆ERT = e−
EmRT e−
∆ERT = e−
EmRT ∗ 0.18
Ein Fehler von ∆E = +1 kcal/mol fuhrt also zu einer Unterschatzung der Raten-konstante um etwa 80 %, ein Fehler von ∆E = - 1 kcal/mol uberschatzt sie umden Faktor 5 (bei RT = 0.59 kcal/mol). D.h., mit heutigen Methoden kann mandie Großenordnung der Ratenkonstanten bestimmen. Viel wichtiger sind jedochoft relative Ratenkonstanten, z.B. beim Vergleich von verschiedenen Reaktionen.Hier sind QM Verfahren eine wertvolle Hilfe geworden.

7 THEORIE DES UBERGANGSZUSTANDES 94
7.8 Verbindung zu Arrhenius
Arrhenius Gleichung:
k = Ae−εa/kT = Ae−Ea/RT (7.27)
Fur die Eyring Gleichung kann man mit
∆G = ∆H − T∆S
zeigen dass gilt (siehe z.B. Atkins) :
Ea = ∆H + 2RT
undA = e2kT
he∆S/R

8 EXPERIMENTELLE TECHNIKEN 95
8 Experimentelle Techniken
Um die Kinetik einer Reaktion untersuchen zu konnen, muss man die Konzentra-tion(en) in Abhangigkeit von der Zeit messen.
8.1 Langsame Reaktionen
Manche Reaktionen benotigen Minuten oder Stunden, um das Gleichgewicht zuerreichen. Es gibt dann mehrere Moglichkeiten, die Konzentrationen zu messen.
Wenn ein Stoff der Reaktion gasformig ist, kann bei konstant gehaltenem Vo-lumen der Druckanstieg als Indikator fur den Fortgang der Reaktion dienen.
Daruber hinaus gibt es viele spektroskopische Techniken (UV/vis, IR, Raman ...)die es erlauben, den Konzentrationsverlauf zeitabhangig zu messen.
Diese Messungen kann man an Fließsystemen vornehmen. Dabei werden die Eduk-te in einer Reaktionskammer gemischt, das Gemisch durchstromt anschliessendein Rohr mit konstanter Geschwindigkeit v. Man kann entlang des Rohres in ver-schiedenen Abstanden x von der Mischkammer die Konzentration messen, diesentspricht verschiedenen Zeitpunkten t= x/v nach der Vermischung.Das Problem bei dieser Technik sind die relativ großen Substanzmengen und langenZeitskalen, die benotigt werden.
8.2 ’stopped flow’ Technik
Schnelle Reaktionen bis hin zu Halbwertszeiten im Millisekunden Bereich konnenmit abgewandelten Fließapparaturen gemessen werden, mit der sogenannten ’stop-ped flow’ Technik (Abb. 42). Diese erlaubt eine sehr effiziente und schnelle Vermi-schung der Reaktanden, zeichnet sich durch ein kleines Probenvolumen aus undkann bei Reaktionen mit Halbwertszeiten bis zu 1 ms angewendet werden. Hierbeiist das Spektrometer ortsfest, es gibt nur ein kleines Probenvolumen und es wirddie Zeitentwicklung der Konzentration am Ort des Spektrometers gemessen. Furschnellere Reaktionen stellt das Mischen den zeitkritischen Schritt dar, man mussalso andere Techniken verwenden.
8.3 Relaxationsmethoden
Das Konzentrationsverhaltnis bei der Reaktion
A B
ist durch die Gleichgewichtskonstante K bestimmt, die u.a. Druck- und Tempe-raturabhangig ist. Schnelle Anderungen von p oder T (Druck-Temperatursprung)

8 EXPERIMENTELLE TECHNIKEN 96
Abbildung 42: ’stopped flow’ Methode (Abb. Atkins)
Abbildung 43: (Abb. P. Weis)
fuhren zu einer abrupten Anderung von K, d.h. das Sytem wird in ein neues GGrelaxieren. Wir hatten oben fur die Reaktion 1. Ordnung mit Ruckreaktion (Abb45):
[A]− [A]∞ = ([A]0 − [A]∞)e−(k1+k−1)t
Man bestimmt damit durch die Kinetik nach dem ’Sprung’ die Summe der Raten-konstanten (k1+k−1), aus Gleichgewichtsmessungen dann K = k1/k−1. Dies gibt 2Gleichungen fur 2 Unbekannte und man erhalt die Ratenkonstanten fur Hin- undRuckreaktion. Ein Temperatursprung (bis 10 K) kann z.B. durch das Entladeneines Kondensators erreicht werden. Dazu benotigt man ein elektrisch leitendesMedium (Elektrolytlosung).

8 EXPERIMENTELLE TECHNIKEN 97
8.4 Blitzlichtphotolyse
Reaktionen mit Halbwertszeiten bis zu 10−5 lassen sich mit Hilfe der Blitzlichtpho-tolyse erzeugen.Hierbei wird eine photochemische Reaktion durch eine erste Blitz-lampe initiiert und durch einen zweiten Blitz werden die Konzentrationsverlaufeverfolgt (Abb. 44). Wesentlich schnellere Reaktionen (bis fs) lassen sich mit Hilfe
Abbildung 44: Blitzlichtphotolyse (Abb: Weis)
von Lasern in sogenannten ’pump-probe’ Methoden verfolgen. Hier wird die Reak-tion durch einen ’pump’ Puls initiiert. Diese Methode ist sehr erfolgreich gewordenda es gelungen ist, immer kurzere Pulse (fs-Pulse) zu erzeugen. Der ’probe’ Pulswird dann zur Analyse der Reaktion zeitverzogert in die Probe gefuhrt, die Zeit-verzogerung erreicht man durch eine Laufzeitverlangerung. Beide Pulse werdengleichzeitig erzeugt, aber der probe Puls lauft uber einen ’Umweg’ zur Probe.
8.5 Geschwindigkeitsfilter
Eine schematische Darstellung des Geschwindigkeitsfilters ist in Abb. 46 gezeigt.Hier verlassen Atome/Molekule einen Ofen der Temperatur T und durchlaufendie Apparatur. Die Geschwindigkeitsverteilung gehorcht der MB- Verteilung, aberje nach Rotationsgeschwindigkeit der Schlitze werden nur Teilchen mit einer Ge-schwindigkeit durchgelassen.
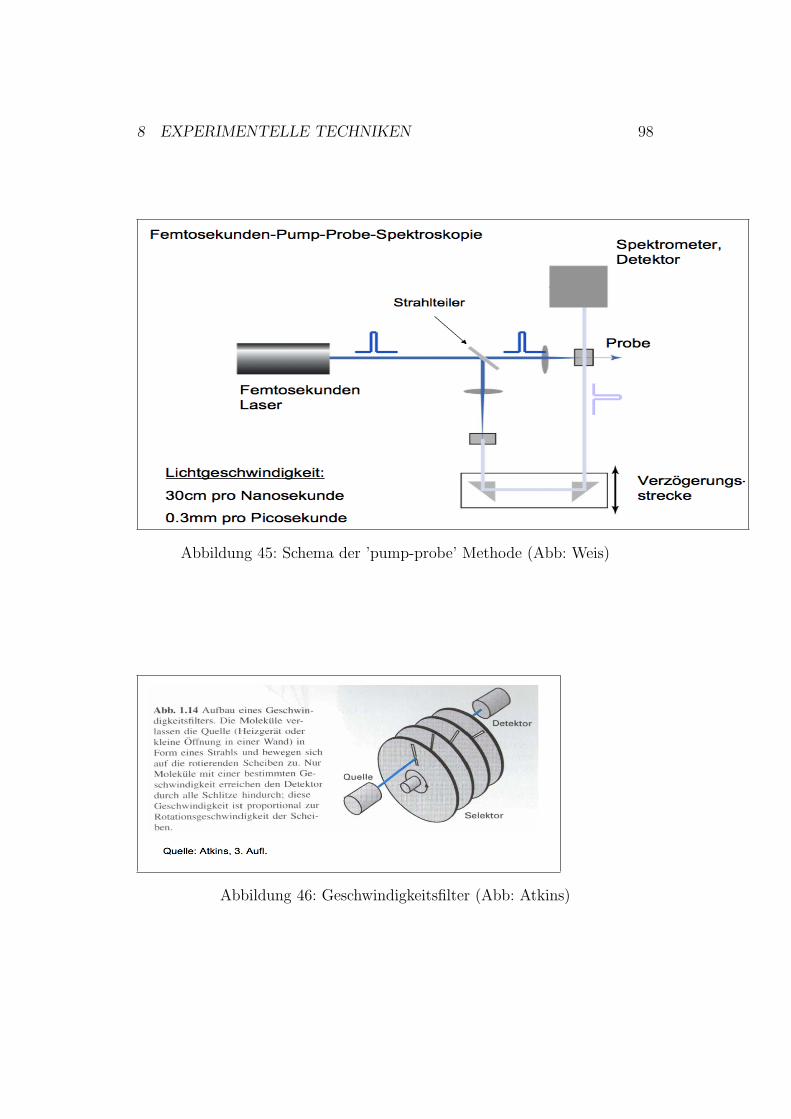
8 EXPERIMENTELLE TECHNIKEN 98
Abbildung 45: Schema der ’pump-probe’ Methode (Abb: Weis)
Abbildung 46: Geschwindigkeitsfilter (Abb: Atkins)

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 99
9 Reaktionen in Flussigkeiten: Diffusion
In einer Flussigkeit sind die Reaktanden A und B jeweils von einer Solvathulleumgeben. In einem ersten Schritt vor der eigentlichen Reaktion findet durch Dif-fusion der Molekule ein Einschluss in eine gemeinsame Solvathulle,
A+Bk1,k−1
AB
und erst in einem zweiten Schritt findet die eigentliche Reaktion statt:
ABk2−→ P
Wir verwenden wieder das QS Prinzip und erhalten (siehe vorgelagertes GG, Mi-chaelis Menten):
d[P ]dt
=k1k2
k−1 + k2[A][B] (9.1)
Grenzfalle:
• k2 >> k−1,d[P ]dt
= k1[A][B]
Die Reaktion ist schneller als die Diffusion, d.h. die Gesamtgeschwindigkeitwird durch die Diffusion kontrolliert.
• k2 << k−1, K = k1k−1
d[P ]dt
= k2K[A][B]
Die Reaktion ist langsamer als die Diffusion, d.h. die Gesamtgeschwindigkeitwird durch die Aktivierungsenergie kontrolliert.
Den zweiten Fall haben wir bisher ausgiebig besprochen, nun wollen wir uns derDiffusion zuwenden und k1 zu bestimmen versuchen.Wir werden zunachst die phanomenologischen Gesetze ableiten, weiter unten wer-den wir eine thermodynamische Betrachtung anschliessen.
9.1 Diffusion (stationar)
Wir betrachten nun den stationaren Fall der Diffusion bei dem ein Konzentrations-gradient zwischen zwei Reservoires zur Diffusion von Teilchen fuhrt. Wir nehmenan, dass die Reservoires sehr groß sind, so dass die Diffusion die Konzentrationennicht andert (Abb. 47), d.h.
c2 − c1
x2 − x1=dc
dx

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 100
Abbildung 47: Konzentrationsdifferenz zwischen zwei Reservoires
Die Teilchenstromdichte J ist definiert als
AnzahlmolTeilchen
Zeit ∗Rohrquerschnittsflache,
J =∆n(x, t)A∆t
d.h. die Anzahl der Mole, die pro Zeiteinheit die Querschnittsflache A passieren.In erster Naherung kann man annehmen, das die Teilchenstromdichte proportionalzum Konzentrationsgradienten (Konzentrationsdifferenz) ist,
J = −D dc
dx(9.2)
Dies ist das 1. Fick’sche Gesetz. D ist der Diffusionskoeffizient (Einheit [m2/s]),den wir weiter unten bestimmen werden (Beispiel Saccarose: D= 5∗10−6cm2/s).
9.2 Diffusion als zeitabhangiger Transport
Nun betrachten wir die Diffusion unter nicht-stationaren Bedingungen, d.h. dieKonzentrationen (an verschiedenen Orten x) werden sich mit der Zeit andern. Umdies zu beschreiben betrachten wir das Volumen ∆V = A∆x in Abb. 48.
∆c∆t
=∆n(x, t)A∆x∆t
− ∆n(x+ ∆x, t)A∆x∆t
Mit (1. Fick’sches Gesetz)
∆n(x, t)A∆t
= J = −D∂c(x, t)∂x

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 101
Abbildung 48: Diffusion durch ein Volumen V (links) und Diffusion einerpunktformigen Anfangsverteilung (Abb. Weis)
gibt dies (∆c∆t →
∂c∂t ):
∂c
∂t=J(x, t)
∆x− J(x+ ∆x, t)
∆x= − D
∆x
(∂c(x, t)∂x
− ∂c(x+ ∆x, t)∂x
)(9.3)
Taylor Entwicklung:
c(x+ ∆x, t) = c(x, t) + ∆x∂c(x, t)∂x
+ ...
∂c
∂t≈ − D
∆x
∂c(x, t)∂x
−∂[c(x, t) + ∆x∂c(x,t)∂x
]∂x
= − D
∆x
(∂c(x, t)∂x
− ∂c(x, t)∂x
+ ∆x∂ ∂c(x,t)∂x
∂x
)
∂c
∂t= −D∂
2c(x, t)∂x2
(9.4)
Dies ist das 2. Ficksche Gesetz. Die Losung der Differentialgleichung hangt vonden Anfangs- und Randbedingungen ab. Fur eine punktformige Anfangsverteilung(bei t=0 sind n Teilchen auf eine kleine Flache A konzentriert) erhalt man (in 1-D)die Konzentration zur Zeit t (Abb. 48 rechts):
c(x, t) =n
A√πDt
e−x2
4Dt

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 102
9.3 Berechnung des Diffusionskoeffizienten D aus derGastheorie
Im Folgenden wollen wir den Diffusionskoeffizienten aus der kinetischen Gastheo-rie berechnen. Wir betrachten der Einfachheit halber nur eine Bewegung in x-Richtung.Dazu benotigen wir folgende Großen:
• Die mittlere Geschwindigkeit in (positive) x-Richtung < vx >:
< vx >=∫ ∞
0vxf(vx)dvx =
√m
2πkT
∫ ∞0
vxe−mv
2x
2kT dvx =
√kT
2πm
• Die mittlere freie Weglange λ. Dies ist der Weg x, den ein Teilchen zwi-schen zwei Kollisionen zurucklegt. Wir hatten oben die Stoßzahl berechnetals z = σ < vr > NA[B]. Wenn wir nur eine Sorte Teilchen betrachtenerhalten wir ([A] = nA/V ):
z = σ < vr > NA[A] = σ < vr >N
V= σ < vr >
p
kT
z ist die Anzahl der Kollisionen pro Zeit, also ist 1/z die Zeit zwischen zweiKollisionen. Damit ist die Strecke, die ein Teichen zwischen zwei Kollisionenzurucklegt
λ =< vx >
z=
< vx >
σ < vr >pkT
=< vx >
< vr >
kT
σp
Da < vr >=√
kT2πµ (µ = 1
2m: reduzierte Masse), erhalten wir:
λ =kT√2σp
(9.5)
• Die mittlere Zeit zwischen 2 Stoßen:
∆t =λ
< vx >.
In der Zeit ∆t durchfliegt die Halfte der Teilchen n (in mol) im markierten Volumen(Abb. 49)
V = λA = A < vx > ∆t
durch die Flache A in positiver x-Richtung,
n−λ =12c−λV =
12c−λA < vx > ∆t.
Dabei sei c die Konzentration (in mol/l). Der Teilchenstrom J ist
J =dieAnzahlMol(Teilchen)
Flache ∗ Zeit,

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 103
Abbildung 49: Diffusion durch ein Volumen zwischen x0−λ und x0 +λ (Abb.Weis)
d.h.J(pos.x−Richtung) = Jp =
n−λA∆t
=12c−λ < vx >
J(neg.x−Richtung) = Jn =nλA∆t
=12cλ < vx >
undJp − Jn =
12
(c−λ − cλ) < vx >
Taylorentwicklung:
c(±λ) ≈ c(0)± λ dcdx
ergibt:
J = Jp − Jn = −12< vx > (2λ
dc
dx) = − < vx > λ
dc
dx
Das 1. Fick’sche Gesetz ist:J = −D dc
dx,
d.h. wir konnen die Konstante D fur die 1-D Bewegung bestimmen:
D =< vx > λ =
√kT
2πmλ.
Fur 3-D ist die Rechnung etwas aufwandiger und man erhalt:
D =34
√kT
2πmλ. (9.6)

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 104
9.4 Thermodynamische Behandlung der Diffusion
Oben haben wir das 1. Fick’sche Gesetz durch Annahme einer Proportionalitatzwischen Teilchenstrom J und Konzentrationsdifferenz aufgestellt. Was ist aberdie Kraft, die in der Diffusion einen Ausgleich der Teilchenkonzentrationen an-strebt?
In diesem Fall ist es kein Enthalphie-, sondern ein Entropiebeitrag, der zur Mi-schung bzw. zum Konzentrationsausgleich fuhrt, die Mischungsentropie. Diese ist(p und T konstant und es wird keine Volumenarbeit geleistet) mit dem chemischenPotential aquivalent.Aus der Thermodynamik wissen wir fur diesen Fall, dass die bei diesem Prozesspro Mol geleistete Arbeit dW gleich dG ist,
dW = dG = dµ.
Wenn µ sich entlang der Koordinate x andert, haben wir:
dµ =(∂µ
∂x
)p,T
dx
Arbeit kann man immer als (generalisierte) Kraft x Weg ausdrucken, d.h.
dW = −FDdx
mit
FD = −(∂µ
∂x
)p,T
Die treibende Kraft, die das System ins Gleichgewicht bringt, ist also der Gradientdes chemischen Potentials und mit
µ = µ0 +RTlnc
−(∂µ
∂x
)= −RT
c
(∂c
∂x
)proportional zum Gradienten der Konzentration.
FD = −RTc
(∂c
∂x
)(9.7)
Diese Kraft (sie ist bezogen auf ein Mol) fuhrt zu der Diffusion der Teilchen,bewirkt also einen Teilchenstrom. Die Viskositat der Flussigkeit bedingt eine Rei-bungskraft, die proportional zur Diffusionsgeschwindigkeit vD und der Diffusions-kraft entgegengerichtet ist (f: Reibungskoeffizient):
FR = −fvD

9 REAKTIONEN IN FLUSSIGKEITEN: DIFFUSION 105
d.h.−FR = FD
fvD = −RTc
(∂c
∂x
)oder
cvD = −RTf
(∂c
∂x
)cvD hat die Dimension (Teilchenzahl/Flache*Zeit) und kann daher mit dem Teil-chenstrom identifiziert werden,
J = cvD = −RTf
(∂c
∂x
)= −D
(∂c
∂x
)Wir haben damit:
D =RT
f(9.8)
und konnen mit Gl. 9.7 schreiben:
vD = −Dc
dc
dx=DFDRT
(9.9)
Die Kraft FD ist auf ein Mol bezogen, pro Molekul hat man:
FmD = −kTc
(∂c
∂x
)=vDkT
D(9.10)
vD =DFmDkT
(9.11)
Die Reibungskraft an einem ungeladenen spharischen Teilchen mit hydrodynami-schen Radius r und dem Viskositatskoeffizienten des Losungsmittels ist:
FmR = −6πηrvD (9.12)
Mit −FmR = FmD bekommen wir:
D =kT
6πηr(9.13)
die Stokes-Einstein Beziehung zwischen dem Viskositatskoeffizienten η unddem Diffusionskoeffizienten D. Fur die diffusionskontrollierte Reaktion fanden wir(k2 >> k−1):
d[P ]dt
= k1[A][B]
k1 ist damit berechenbar und man erhalt (siehe z.B. Atkins):
k1 = 4πNA(DA +DB)rABf (9.14)
DA,B sind die Diffusionskoeffizienten von A und B, rAB = rA + rB und f ist derelektrostatische Faktor. f=1 fur ungeladene Teilchen.


















