
Praktikum Biochemische Untersuchungsmethoden für ... · Produktion des Green Fluorescent Protein...
44
1 Praktikum Biochemische Untersuchungsmethoden für Studierende der Pharmazie Dr. John Heiker, Dr. Irene Coin und Ulrike Grafe Leipzig, Februar/März 2019
Transcript of Praktikum Biochemische Untersuchungsmethoden für ... · Produktion des Green Fluorescent Protein...

1
Praktikum Biochemische Untersuchungsmethoden für Studierende der Pharmazie
Dr. John Heiker, Dr. Irene Coin und Ulrike Grafe
Leipzig, Februar/März 2019

2
INHALTSVERZEICHNIS
1. Allgemeine Hinweise Seite
Verhaltensregeln im Labor Hinweise zur Anfertigung der Protokolle Haftungsregelungen
3 4 6
2. Versuche
Versuch 1 Methoden der Proteinbestimmung
8
Versuch 2 Prinzipien der Aktivitätsbestimmung von Enzymen
13
Versuch 3 Kinetik enzymatischer Reaktionen inklusive Enzymhemmungen
19
Versuch 4 Ionenaustausch-Chromatographie als Methode zur Anreicherung von Proteinen
29
Versuch 5 Polyacrylamidgel-Elektrophorese
33
Versuch 6 Produktion des Green Fluorescent Protein (GFP) in E. coli
38
3. Literatur 49

3
Allgemeine Hinweise
Verhaltensregeln im Labor Laborarbeiten bringen durch den Umgang mit Chemikalien und Geräten sowie biologischem Material Gefahren mit sich, die bei nicht sachgemäßer Handhabung zu Gesundheitsschäden führen können. Daher muss bei allen Laborarbeiten die Sicherheit am Arbeitsplatz gewährleistet sein.
Folgende Maßnahmen sind ist besonders zu beachten:
Aufgrund des Umgangs mit gefährdenden Substanzen ist während einer Schwangerschaft bzw. der Stillzeit keine Teilnahme am Praktikum möglich!
In den Praktikumsräumen muss generell ein Labormantel, festes Schuhwerk sowie gegebenenfalls weitere Schutzausrüstung (Handschuhe/ Schutzbrillen) getragen werden. Die Laborkittel müssen nach dem Praktikum im Saal verbleiben.
Im Labor darf nicht gegessen, getrunken oder geraucht werden.
Achten Sie auf Sauberkeit und Ordnung: ein unordentlicher Arbeitsplatz führt leichter zu Unfällen und häufig auch zum Misslingen des Experimentes! Der Arbeitsplatz wird bei Versuchsende aufgeräumt und gereinigt übergeben.
Das Pipettieren von Lösungen geschieht ausschließlich mit Pipettierhilfen - nicht mit dem Mund ansaugen!
Informieren Sie sich über die Toxizität der verwendeten Substanzen. Im Skript und in den Vorbesprechungen wird auf Gefahrstoffe aufmerksam gemacht.
Alle Gefäße für Versuchsansätze, Chemikalien oder Lösungen sind sorgfältig mit wasserfestem Stift zu beschriften. Das betrifft auch Gefäße für mikrobiologische Experimente, wie z.B. Kultivierungsröhrchen, Schüttelkolben oder Petrischalen.
Beachten Sie die Vorschriften für den Umgang mit GVOs. Diese dürfen außerhalb des Praktikumsraumes nur in dafür vorgesehenen Behältern transportiert werden. Kulturüberstände dürfen generell nicht im Ausguss entsorgt werden: sie müssen gesammelt und anschließend autoklaviert werden, ebenso alle nicht mehr benötigten mikrobiologischen Materialien. Dafür stehen im Praktikumssaal gesonderte Behälter zur Verfügung.
Bedienen Sie kein Gerät ohne vorherige Einweisung. Die Schäden von Fehlbedienungen belaufen sich meist auf mehrere hundert Euro!
Das Öffnen von unter Strom stehenden Elektrophoresekammern ist lebensgefährlich!
Bei Kühlzentrifugen wird nach Entnahme der Proben stets der Deckel wieder geschlossen. Wird die Zentrifuge nach Arbeitsende ausgeschaltet, bleibt der Deckel offen, damit sie austrocknen kann.
Informieren Sie sich über die vorhandenen Sicherheitseinrichtungen (z. B. Feuerlöscher, Verbandskästen, Notbrause, Augendusche usw.)
Die Benutzung von Mobiltelefonen sowie das Abspielen von Tonträgern ist im Praktikumsraum, Hörsaal bzw. Seminarraum während der Veranstaltungen nicht gestattet.
Da zu allen Versuchen Vorbesprechungen durchgeführt werden, wird pünktlicher Praktikumsbeginn vorausgesetzt. Für dringende „Erledigungen“ können die Versuchspausen genutzt werden.

4
Hinweise zur Anfertigung der Protokolle
Allgemeines
Im Biochemie-Praktikum wird in Gruppen gearbeitet (in der Regel 3 Studierende).
Beachten Sie, dass bei einer Gruppenarbeit alle Mitglieder der Gruppe die gleiche Verantwortung für das Aufzeichnen, die Auswertung und die Darstellung der Versuchsergebnisse tragen. Dies akzeptieren Sie mit Ihrer Unterschrift auf dem Deckblatt zum jeweiligen Protokoll. Sollte ein Gruppenmitglied ausscheiden, kann das kein Entschuldigungsgrund für fehlende Protokolle sein.
Die pro Versuch während des Praktikums aufgezeichneten Rohmessdaten müssen vom Praktikumsleiter mit Datum und Unterschrift abgezeichnet werden. Diese Aufzeichnungen sind dann als Anhang zum jeweiligen Protokoll abzugeben.
Wenn Daten für Teilversuche von anderen Gruppen übernommen wurden, muss dies unbedingt angegeben werden; alle Berechnungen müssen selbständig pro Gruppe durchgeführt werden!
Zu jedem Versuch ist ein Gruppenprotokoll anzufertigen (max. 10 Seiten). Die Protokolle sind ~1 Woche nach Praktikum gesammelt in einer Email an die Praktikumsleiter zu schicken.
Die Protokolle sind grundsätzlich mit Computer zu erstellen und die Seiten sind zu nummerieren. Beachten Sie unbedingt korrekte Formatierungen (hoch/tiefgestellt, Sonderzeichen, Tabellen ...)!
Gravierende Fehler in den Protokollen müssen korrigiert werden. Die Korrekturen sind zusammen mit den Originalprotokollen nochmals vorzulegen
Nutzen sie die Rechtschreibkorrektur von Word! Schreiben Sie vollständige, grammatikalisch korrekte Sätze!
Geben Sie die KORREKTEN Einheiten für alle Werte an, die Sie im Protokoll erwähnen!!
Bei Abgabe per email muss zwingend im Dateinamen und „Betreff“ die Versuchs-, Gruppen- und Kursnummer sowie der „Klarnamen“ des Absenders angegeben werden. Beispiel: V1_Gr2_KursA_HanSolo
Gliederung und Inhalt der Protokolle
Protokolle als Fließtext (nicht 2-spaltig!) verfassen; Schreibweise: 1,5-zeilig
Bei Material und Methoden ist auch die „Durchführung“ der Experimente präzise und nachvollziehbar zu beschreiben. Bei Materialien darf auf das Skript verwiesen werden, ALLERDINGS sind Abweichungen zum Skript oder gruppenspezifische Bedingungen, welche sich unter Umständen bei der Durchführung der Versuche ergeben, zu dokumentieren.
Prinzipiell wird jedes Protokoll wie eine wissenschaftliche Publikation gegliedert; auch die graphischen Darstellungen und Tabellen sollen in ihrer Form und Beschriftung diesen Anforderungen genügen.
Bedenken Sie, dass das Erstellen von Protokollen eine Übung für das Verfassen wissenschaftlicher Arbeiten ist!

5
1. Deckblatt
Das Deckblatt muss folgende Angaben enthalten:
Nummer und Titel des Versuches
Gruppennummer, Namen und Unterschriften der Gruppenmitglieder
Versuchsdatum, Abgabedatum, Name und Jahr des Praktikums (z.B. Pharmazie WS 14/15; 1. bzw. 2. Kurs)
2. Einleitung
Die Einleitung enthält eine KURZE UND KLARE theoretische Einführung und Aussagen zum Inhalt und Zweck des Versuches. Der theoretische Hintergrund der angewandten Methode(n) soll KURZ erläutert werden. (max. 1 Seite).
3. Material und Methoden
Bei Material darf auf das Skript verwiesen werden. Die „Durchführung“ der Experimente ist präzise und nachvollziehbar zu dokumentieren.
Sie müssen im Punkt „Literatur“ auf das Skript verweisen.
4. Ergebnisse und Diskussion
Die Darstellung der Ergebnisse erfolgt in der Regel in Form von Abbildungen und Tabellen, wobei im dazugehörigen Text wesentliche Sachverhalte beschrieben werden. Folgende Formalien sind dabei einzuhalten:
Abbildungen erhalten stets eine geeignete Unterschrift, die den Inhalt der Abbildung sinnvoll wiedergibt, sowie eine Legende. Die Legende muss eine präzise Zuordnung des Dargestellten zu einem Versuchsteil ermöglichen und eventuell vorhandene Beschriftungen/Kurvensymbole definieren. Graphen sollen möglichst in Excel oder einem anderen geeigneten Programm (z.B. GraphPadPrism) in vernünftiger Größe erstellt werden.

6
Die Abbildungen und Tabellen erhalten innerhalb jedes Versuchs eine fortlaufende Nummerierung, auf die im Text verwiesen werden muss.
Tabellen erhalten eine Überschrift; sollte eine Legende erforderlich sein, steht diese unter der Tabelle. Ergeben sich die Daten in der Tabelle durch Rechnungen, so muss im Text ein nachvollziehbares Rechenbeispiel angegeben werden. Achten Sie auf eine angemessene Zahl von Kommastellen.
Anlagen zum Protokoll wie etwa Spektrenausdrucke oder Elektrophoresebilder werden nicht akzeptiert. Diese zum Protokoll gehörende Abbildungen sind mit Abbildungsunterschrift und -Legende an entsprechender Stelle ins Protokoll einzufügen.
Vergessen Sie nicht, Maßeinheiten anzugeben, wo diese erforderlich sind!
Informieren Sie sich nochmals über Konzentrationsangaben und die Angabe von Enzymaktivitäten:
∘ µM = mikromolar = µmol/l; Mikromol ist keine Konzentrationsangabe!
∘ verwenden Sie mM bzw. µM anstelle von 10-3 M bzw. 10-6 M
∘ % -Angaben (v/v; w/v) beziehen sich auf 100 Einheiten des Lösungsmittels
Beim Arbeiten mit Verdünnungen müssen Sie wissen, dass es eine 1:1-Verdünnung nicht gibt (wohl aber ein Mischen im Verhältnis 1:1); bei der Angabe "Verdünnung 1:2" bezieht sich die 2 auf die Gesamtzahl der gemischten Teile – d.h. 1 + 1.
Wenn ein Versuch aus mehreren Teilexperimenten besteht, ist es sinnvoll, die Teilresultate gleich nach deren Darstellung zu diskutieren und kritisch zu bewerten. In diesem Fall kann im Protokoll ein einheitlicher Gliederungspunkt „Ergebnisse und Diskussion“ gewählt werden. Trennt man zwischen Ergebnisteil und Diskussionsteil, was prinzipiell möglich ist, muss man streng darauf achten, dass man im Textfluss beide Teile nicht vermischt. Oftmals ist das schwer einzuhalten. In jedem Fall hilft auch die Nutzung geeigneter Zwischenüberschriften.

7
Haftungsregelungen
Am Institut für Biochemie gelten für studentische Praktika seit dem SS 07 folgende Haftungsregelungen:
1.) Jede/r Student/in muss für Glasbruch und andere selbst verschuldete Sachschäden in voller Höhe selbst aufkommen. Der Abschluss einer studentischen Haftpflichtversicherung wird empfohlen.
2.) Bei Gruppenpraktika haftet jede/r Student/in der Gruppe, wobei alle Student/innen einer Gruppe für die ausgehändigten Geräte quittieren.
3.) In welcher Form die Ausgabe und Rücknahme der Geräte erfolgt, entscheidet die jeweilige Praktikumsleitung.
4.) Die Student/innen werden vor Beginn eines Praktikums über diese Regelung sowie über Kosten von Geräten und Verbrauchsmaterialien informiert.
5.) Die Bezahlung und Aushändigung entsprechender Quittungen wird im Sekretariat des Instituts für Biochemie über eine Praktikumskasse abgewickelt. Der Abschluss einer privaten Haftpflichtversicherung wird empfohlen.

8
Versuche

9
Versuch 1
Methoden der Proteinbestimmung
Einleitung
Die quantitative Proteinbestimmung ist eine wichtige Technik im biochemischen Labor, die bei verschiedenen Fragestellungen wie z.B. der Beurteilung von Zellaufschlüssen und Proteinreinigungsverfahren, der Bestimmung der spezifischen Aktivität von Enzymen und in kinetischen Experimenten Anwendung findet.
Dazu sind zahlreiche Verfahren entwickelt worden, die sich jeweils unterschiedliche physikochemische Eigenschaften der Proteine zu Nutze machen. Es sind dies im Einzelnen:
der Kolloidcharakter (Trübungsmessung)
das Vorhandensein von Peptidbindungen (Biuret)
der aromatische Charakter einzelner Aminosäureseitenketten (UV-Messung)
die Reaktivität einzelner Aminosäureseitenketten (Lowry)
das Ligandenbindungsvermögen (Bradford)
Im vorliegenden Versuch sollen die Vor- und Nachteile verschiedener Nachweismethoden aufgezeigt werden, um bei entsprechender Fragestellung die optimale Methode auswählen zu können. Folgende Kriterien sollten berücksichtigt werden:
zeitlicher Aufwand
Empfindlichkeit
mögliche Störfaktoren
Vergleichbarkeit bezogen auf die Bestimmung unterschiedlicher Proteine
Das zuverlässigste Verfahren ist die Biuret-Methode, bei der mit einer Farbreaktion die Peptidbindung, die alle Proteine in gleicher Weise besitzen, nachgewiesen wird. In alkalischer Kupfersulfatlösung in Gegenwart von Wein- oder Citronensäure bilden sich rot- bis blauviolett gefärbte Kupferkomplexe, die bei 540 nm photometrisch bestimmt werden können. Obwohl die Methode wenig störanfällig ist (nur Ammoniumionen verfälschen die Werte), wird sie relativ wenig angewandt, da der Substanzbedarf groß ist (1-5 mg). Später eingeführte Mikrobiuret-Verfahren, die weniger Substanz benötigen, haben sich auf breiter Ebene nicht durchgesetzt. Dabei hat dieses Verfahren den großen Vorteil, dass die Bestimmungen nicht durch die Aminosäurezusammensetzung der Proteine beeinflusst werden.
Die Lowry-Bestimmung, deren Substanzbedarf nur bei 5-80 µg liegt, ist eine Kombination aus Biuret- und der Molybdänblau-Reaktion. Dabei bewirken die Seitenketten der Aminosäuren Tyrosin, Tryptophan und Cystein (in geringerem Maß auch Histidin und Phenylalanin) die Reduktion des 6-wertigen Molybdäns einer Heteropolysäure (Folin-Reagenz) zu einem blau gefärbten Mischoxid (MoO2 + MoO3). Ursache für die Erhöhung der Empfindlichkeit ist die Ausbildung der Biuret-Komplexe. Der Vorteil der erhöhten Empfindlichkeit führt allerdings auch zu einer größeren Störanfälligkeit. Zahlreiche häufig eingesetzte Substanzen (z.B. SDS, Triton, EDTA, Mercaptoethanol, verschiedene anorganische Salze) verfälschen die Messwerte. Zudem sind die ermittelten Werte zu einem gewissen Teil auch von der Aminosäure-zusammensetzung des Proteins abhängig. Weicht diese stark von der des Eichproteins ab,

10
kommt es zu fehlerhaften Bestimmungen. Diese Tatsache ist aber nur dann relevant, wenn die zu bestimmende Lösung einen einzigen Proteintyp enthält. Proteinbestimmungen von Roh-extrakten werden davon nicht beeinflusst.
Die Bradford-Methode nutzt die Fähigkeit von Proteinen, Liganden (hier einen Farbstoff) binden zu können. Coomassie-Brilliant-Blau G250 (eigentlich ein Textilfarbstoff!) zeigt bei Proteinbindung eine Verschiebung des Absorptionsmaximums von 465 nm zu 595 nm. Da die entstehenden Farbstoff-Protein-Komplexe sehr stabil sind, wurde diese Substanz (bzw. CBB R250) zunächst nur benutzt, um Proteinbanden in Polyacrylamidgelen anzufärben (siehe Versuch 9). Die Empfindlichkeit dieser Methode liegt bei 5-50 µg Protein. Reduzierend wirkende Substanzen, die besonders bei der Lowry-Bestimmung stören, sind hier ohne Einfluss, allerdings stören Detergenzien sowie Ammoniumsulfat. Der wohl größte Nachteil der Methode besteht darin, dass - deutlicher als bei der Lowry-Methode - Proteine mit unterschied-licher Aminosäurezusammensetzung auch unterschiedliche Mengen an Coomassie binden, was zu Abweichungen der Messwerte vom tatsächlichen Proteingehalt führt. Daher ist die Methode streng genommen nur für die Konzentrationsbestimmung von Proteinmischungen geeignet. Trotz dieser Nachteile hat das Verfahren eine breite Anwendung im Routinelabor gefunden. Ursachen dafür sind die hohe Empfindlichkeit, einfache Durchführung und der geringe Zeitbedarf. Während bei den zuerst aufgeführten Verfahren Inkubationszeiten der Messansätze von mindestens 30 Minuten erforderlich sind, kann man einen Bradford-Ansatz schon nach 5 Minuten photometrisch vermessen.
Photometrische Proteinbestimmungen durch Messungen im UV-Bereich haben den Vorteil, dass sie direkt und ohne Vorbereitung von Inkubationsansätzen vorgenommen werden können. Durch den Gehalt an aromatischen Aminosäuren absorbieren Proteine Licht im Wellenlängenbereich von 260-280 nm, wobei das Tryptophan den größten Beitrag leistet. Eine genaue Konzentrationsbestimmung wäre hier prinzipiell durch Nutzung des Lambert-Beerschen Gesetzes möglich (E= •c•d; Erläuterungen siehe V. 2). Dies kann aber nur dann angewandt werden, wenn es sich um ein reines Protein handelt, dessen Extinktionskoeffizient bei definierter Wellenlänge bekannt ist. Alternativ zum Molaren Extinktionskoeffizienten findet man auch die Angabe E280(0,1%). Dieser sogenannte spezifische Extinktionskoeffizient ist der Extinktionswert, den man für eine 0,1 %ige Lösung (d.h. 1 mg/ml) des gereinigten Proteins ermittelt. Bei vielen Fragestellungen wie z.B. Protein-Aufreinigungen arbeitet man jedoch mit Proteinmischungen, so dass Korrekturverfahren (siehe Teilversuch 3) entwickelt wurden. Diese berücksichtigen zusätzlich noch, dass Proteinrohextrakte häufig mit Nukleinsäuren verunreinigt sind, deren Absorptionsmaximum bei 260 nm liegt. Der Proteinnachweis bei 280 nm ist ein etabliertes Verfahren, das in allen Chromatographie-anlagen Anwendung findet (Durchflussküvetten). Unabhängig von einigen Besonderheiten der UV-Messung ist es natürlich auch bei dieser Technik möglich, eine Eichung des Systems mit einem Standardprotein vorzunehmen, so wie bei den zuvor beschriebenen Farbreaktionen. Als Standardprotein für die Erstellung von Eichkurven wird in der Regel Rinderserumalbumin (RSA bzw. BSA) verwendet.

11
Material Bradford-Reagenz 50 mg Coomassie-Brilliant-Blau G250 werden in 25 ml
Ethanol (95 %) gelöst, danach mit 50 ml Phosphorsäure (85 %) versetzt, gemischt und mit aqua dest. auf 500 ml aufgefüllt. Nach Filtration ist die Lösung gebrauchsfertig. Sie wird in einer dunklen Flasche im Kühlschrank gelagert. Steht fertig zur Verfügung!
Eich- bzw. Probenlösungen Konzentration
Rinderserumalbumin (RSA) 2 mg/ ml
Lysozym (EC 3.2.1.17) 2 mg/ ml
Hefe-Alkoholdehydrogenase (H-ADH; EC 1.1.1.1) 2 mg/ ml
Genomische DNA (E. coli oder H. sapiens) unbekannt
Phosphatpuffer nach Sörensen ( 50 ml) 0,05 M, pH 8,0
Durchführung
1. Erstellen einer Eichkurve nach der Bradford-Methode
Messansatz:
Volumen Substanz
0,04 ml Probenlösung ( je 5-40 µg Protein enthaltend)
2,00 ml Bradford-Reagenz
Es werden 8 Reagenzgläser vorbereitet, in die jeweils 0,04 ml Probenlösung auf den Boden des Glases pipettiert werden. Die Probenlösungen sollten zwischen 5-40 µg RSA enthalten. Danach werden zu jeder Probe 2 ml Bradford-Reagenz pipettiert und gut gemischt. Nach 5-15 min wird die Extinktion der Ansätze bei 595 nm bestimmt, wobei das Photometer gegen einen Leerwert abgeglichen wird. Der Leerwert enthält anstelle der Probenlösung 0,04 ml Puffer.
2. Proteinbestimmung anhand der Bradford-Eichkurve
Bestimmen Sie die Proteingehalte der ausgehändigten Proteinlösungen auf der Basis der unter 1. erstellten Eichkurve. Da Ihnen die Proteingehalte der Lösungen entsprechend der Einwaage gegeben sind, können Sie berechnen, wieviel Lösung einzusetzen ist, um die unter 1. ange-gebenen Rahmenbedingungen einzuhalten. Wählen Sie für jedes Protein 3 Ansätze.
3. UV-Spektren ausgewählter Proteinlösungen
Stellen Sie von den gegebenen Proteinlösungen Verdünnungen in Phosphatpuffer her (Endvolumen 2 ml) und nehmen Sie Spektren im Wellenlängenbereich von 220 nm - 320 nm auf. Vor den Messungen wird das Photometer mit Phosphatpuffer abgeglichen. Von den Einzelproteinen werden Verdünnungen von 1:10 und 1:20 hergestellt und vermessen. Ermitteln Sie aus den Spektren die Extinktionen bei den Wellenlängen 230 nm, 260 nm und 280 nm. Vergleichen Sie beide Verdünnungen.

12
4. UV-Spektrum einer Nukleinsäurelösung
Verdünnen Sie 70 µl der ausgehändigten Nukleinsäurelösung mit 1,93 ml Phosphatpuffer. Nach sorgfältigem Mischen wird ein UV-Spektrum im Wellenlängenbereich von 220-320 nm aufgenommen.
Auswertung
Teilversuch 1
Zeichnen Sie eine Eichkurve für die Bradfordbestimmung, indem Sie die bei 595 nm gemessenen Extinktionen gegen die eingesetzte Proteinmenge (µg) auftragen.
Teilversuch 2
Berechnen Sie die Proteingehalte der Lösungen von ADH, Lysozym und RSA auf der Basis der Bradford-Eichkurve (Angabe in mg/ ml). Diskutieren Sie Ihre Resultate im Zusammenhang mit den Proteingehalten, die über die Einwaage der lyophilisierten bzw. kristallisierten Proteine gegeben war.
Teilversuch 3
Erstellen Sie aus den Spektren eine Tabelle nach folgendem Muster:
Protein E230 E260 E280
Diskutieren Sie die Form der Spektren sowie die Größe der erhaltenen Extinktionen bei den ausgewählten Wellenlängen.
Berechnen Sie die Proteingehalte der Lösungen nach den Korrekturverfahren von Warburg & Christian: P (mg/ml) = 1,55 • E280 – 0,76 • E260
(Biochem.Z. 310 (1941): 384 – 421), sowie nach
Kalb & Bernlohr: P (µg/ ml) = 183 • E230 – 75,8 • E260
(Anal.Biochem. 82 (1977): 362 – 368). Vergessen Sie nicht, den Verdünnungsfaktor zu berücksichtigen!
Vergleichen Sie die erhaltenen Werte mit den Proteingehalten, die über die Einwaage der lyophilisierten Proteine gegeben waren.

13
Teilversuch 4
Vergleichen Sie das Nukleinsäurespektrum mit denen der Proteine.
Berechnen Sie den DNA-Gehalt der Lösung, sowie deren Reinheit:
Konzentrations- und Reinheitsbestimmung von DNA
Die Konzentration und Reinheit einer Nukleinsäure-Lösung wird photometrisch durch Messen der Extinktionen bei 260 und 280 nm ermittelt. Wenn nicht anders angegeben, gilt:
E260 = 1 = 50 µg Doppelstrang- bzw. 40 µg Einzelstrang-DNA/ml
Die Reinheit einer DNA-Lösung wird über den Quotienten E260/E280 bestimmt, der zwischen 1,8 und 2,0 liegen sollte.
Gesamtauswertung
Erstellen Sie eine Tabelle nach folgendem Muster:
Proteintyp
Proteingehalt (mg/ml) nach
Bradford Warburg & Christian
Kalb & Bernlohr
Vergleichen und diskutieren Sie die Größenordnung der erhaltenen Werte auch im Vergleich zu den angegeben Einwaagen. Welche Schlussfolgerungen lassen sich ziehen?
Berechnen Sie, welche Proteingehalte sich ergeben würden, wenn die 3 Proteinlösungen zu gleichen Teilen gemischt würden. Was sagen diese Werte aus?

14
Versuch 2
Prinzipien der Aktivitätsbestimmung von Enzymen
Einleitung
Die Aktivitätsbestimmung von Enzymen ist eine wichtige Technik der Biochemie, die unter anderem benutzt wird, um Stoffwechselvorgänge aufzuklären und Enzyme zu charakterisieren. Ebenso ist sie bedeutsam für die klinisch-chemische Analytik, bei der aus der Höhe der Enzymaktivität relevanter Enzyme einzelne Krankheitsbilder diagnostiziert werden können (z.B. die Proteasen Trypsin und Chymotrypsin Pankreaserkrankungen; Transaminasen Leber- und Gallenwegserkrankungen; α-Hydroxybutyratdehydrogenase Myocardinfarkt).
Prinzipiell wird bei einer Aktivitätsbestimmung die Konzentrationsänderung einer Komponente der Enzymreaktion (umgesetztes Substrat oder entstehendes Produkt) in einem vorgegebenen Zeitintervall untersucht. Die Dimension der Enzymaktivität ist die einer Reaktionsgeschwindigkeit. Sie wird nomenklaturgerecht in mol/s (kat) bzw. µmol/s (µkat) angegeben. Verbreiteter und sinnvoller ist jedoch wegen der Bezugsmöglichkeit auf eine experimentell direkt zugängliche Zeiteinheit die Angabe µmol/min = U (unit). Der Begriff der enzymatischen Aktivität lässt sich klar aus den elementaren Gesetzen der chemischen Kinetik ableiten. Im Gegensatz zur chemischen Katalyse erhält man bei der Enzymkatalyse in Abhängigkeit von der eingesetzten Substratkonzentration verschiedene Geschwindigkeits-konstanten. Dies bedeutet, dass die Analyse solcher Reaktionen auf der Basis einer anderen mathematischen Beziehung erfolgen muss. Ursache hierfür ist, dass jedes Enzym für das umzusetzende Substrat eine definierte Affinität besitzt, wodurch sich vor der chemischen Reaktion (Zusammenstoß der Reaktionspartner) ein Enzym-Substrat-Komplex bildet. Das verwendete Geschwindigkeitsgesetz (Michaelis-Menten-Gleichung) geht von der Annahme aus, dass sich die Konzentration dieses Enzym-Substrat-Komplexes über einen bestimmten Zeitraum nicht ändert (steady state = Fließgleichgewicht).
Die Gültigkeit von „steady state“- Bedingungen muss stets experimentell – also völlig empirisch – ermittelt werden.
Wichtigste Voraussetzung ist dabei die Erzielung linearer Konzentrationsänderungen pro Zeiteinheit.
Zusätzlich muss gesichert sein, dass sich die gemessene Reaktionsgeschwindigkeit unabhängig von der untersuchten Substratkonzentration proportional zur eingesetzten Enzymmenge verhält. Auch dies muss experimentell überprüft werden.
Besonderen Wert besitzen diese Initialgeschwindigkeitsmessungen dann, wenn bei sehr hohen Substratkonzentrationen gearbeitet wird. Unter diesen Bedingungen hängt die Reaktionsgeschwindigkeit (v) allein von der Enzymkonzentration (e0) ab.
v = k+2 • e0 (v wird zur maximalen Geschwindigkeit V)

15
Geschwindigkeitsmessungen (Aktivitätsbestimmungen) sind dann bei Berücksichtigung von standardisierten Rahmenbedingungen letztlich Mengenbestimmungen für das Enzym. Dividiert man den Wert durch die eingesetzte Enzymmenge (in µmol), so erhält man entsprechend obiger Gleichung die reaktionslimitierende Geschwindigkeitskonstante k+2, die ein echtes Maß für die effektive Wirksamkeit des Enzyms ist. Diese Größe wird häufig auch als Wechselzahl (turnover number) des Enzyms bezeichnet. Diese Möglichkeit kann natürlich nur dann genutzt werden, wenn das untersuchte Enzym sauber ist und seine molekularen Eigenschaften bekannt sind (siehe Versuch 3). Im Regelfall besteht jedoch die Aufgabe darin, den Gehalt eines Enzyms in einem Proteingemisch (Geweberohextrakt) zu ermitteln. In diesem Fall erfolgt ein Bezug auf den Proteingehalt der Lösung. Die erhaltene spezifische Aktivität wird zweckmäßigerweise in U/mg angegeben.
Zur quantitativen Proteinbestimmung eignen sich verschiedene Methoden, die in V1 erläutert sind. Die Bestimmung spezifischer Aktivitäten erfolgt in Testansätzen, die dem jeweiligen Enzym in optimaler Weise (hinsichtlich pH-Wert, Ionenart und -konzentration des verwendeten Puffers) angepasst sind. Zu beachten ist außerdem, dass das Enzymsubstrat in Sättigungskonzentration (a >> Km) eingesetzt wird. Am einfachsten gestalten sich die Messungen von Initialgeschwindigkeiten, wenn das Substrat oder das Produkt der Enzymreaktion spektral auffällig ist. Dann kann die Reaktion kontinuierlich im Photometer verfolgt werden. Kommt es bei einer Enzymreaktion zu keiner Veränderung optischer Parameter, gibt es in einigen Fällen die Möglichkeit, gekoppelte Reaktionen durchzuführen. So kann man die Ursprungsreaktion an eine Farbstoffreaktion (dieser Versuch) oder an eine zweite Enzymreaktion koppeln. Voraussetzung dafür ist allerdings, dass die Indikatorreaktion deutlich schneller abläuft als die eigentlich zu bestimmende Reaktion.
Zur Berechnung der Substratumsätze benutzt man das Lambert-Beersche Gesetz: -log (I/I0)λ = ε•c•d (mit I/I0 = Durchlässigkeit), wobei für den negativen dekadischen Logarithmus der Durchlässigkeit der Begriff der Extinktion (E) eingeführt wurde. Die Extinktion ist proportional zur Konzentration c der zu vermessenden Substanz bei entsprechender Wellenlänge λ und Schichtdicke d. Der Extinktionskoeffizient ε ist die jeweilige Stoffkonstante mit der Dimension M-1•cm-1 (molarer Extinktionskoeffizient) beziehungsweise mM-1•cm-1 bzw. cm2/µmol (millimolarer Extinktionskoeffizient).
Die spezifische Aktivität ist außerdem eine hilfreiche Größe bei der Einschätzung der Reinheit eines Enzympräparates und lässt Aussagen zur Substratspezifität von Enzymen zu.

16
Material Teilversuche 1 +2
Phosphatpuffer nach Sörensen ( 100 ml) 0,05 M; pH 8 (= Testpuffer)
Phosphatpuffer nach Sörensen ( 50 ml) 0,05 M; pH 7; zusätzlich 0,05 % RSA (= Enzym-Verdünnungspuffer)
NAD+ 12 mg/ml in aqua dest.
Sorbinalkohol-Lösung ca. 4,5 mM in Phosphatpuffer
Hefe-ADH 1 mg/ml, in 0,05 M Phosphatpuffer pH 7
Ethanol p.A. Teilversuch 3
Tris-HCl Puffer ( 50 ml) 0,05 M; pH 8
Benzoyl-D,L-arginin-p-nitroanilid (BNA) 12,2 mg/ml in DMSO (reinst)
N-Succinyl-Ala-Ala-Pro-Phe-4-nitroanilid (SPNA)
50 mg in 3 ml Methanol lösen und mit Tris-HCl-Puffer auf 25 ml auffüllen
Chymotrypsin 2 mg/ml in Tris-HCl-Puffer
Trypsin 2 mg/ml in Tris-HCl-Puffer
Durchführung
Der gesamte Versuch ist in drei Teilabschnitte gegliedert, die das Umfeld der Aktivitäts-bestimmung von Enzymen von verschiedenen Seiten beleuchten sollen.
1. Beobachtung des zeitlichen Ablaufs einer Enzymreaktion über den Nachweis von Substraten und Produkten
Dehydrogenasen benutzen NAD+ oder NADP+ als Coenzym, das in der Oxidationsreaktion des jeweiligen Enzyms reduziert wird. Da das reduzierte Coenzym im Gegensatz zur oxidierten Form ein Absorptionsmaximum bei 340 nm hat, sind Dehydrogenasereaktionen leicht photometrisch zu erfassen (optischer Test), unabhängig davon, welche spektralen Eigenschaften das jeweilige Substrat des Enzyms hat, dessen Umsatzgeschwindigkeit untersucht werden soll. Voraussetzung für die Nutzung des optischen Testes ist das Vorliegen stöchiometrischer Umsätze von Substrat und Cosubstrat. Dies soll am Beispiel der Hefe-Alkoholdehydrogenase (ADH; EC 1.1.1.1) demonstriert werden.
Reaktionsschema: C2H5OH + NAD+ ⇌ C2H4O + NADH + H+
Das Enzym ist in der Lage, neben seinem natürlichen Substrat Ethanol eine Vielzahl homologer Alkohole zu oxidieren, so auch den Sorbinalkohol, der wie sein Oxidationsprodukt Sorbinaldehyd ein Absorptionsmaximum im UV-Bereich besitzt (λmax = 228 nm bzw. 280 nm).

17
Versuchsaufbau
In eine Quarzküvette werden die unter „Testansatz“ angegebenen Komponenten mit Ausnahme des Alkohols pipettiert, gut gemischt und das Photometer im zu untersuchenden Spektralbereich (380-210 nm) abgeglichen. Der Start der Reaktion erfolgt durch Zugabe der Alkohollösung (20 µl; schnell und gut mischen; sofort (!) das erste Spektrum aufnehmen; Zeit notieren). Weitere Spektren sind im Minutenabstand (bis 5 min) und danach alle 5 min (bis maximal 20 min) aufzunehmen. Notieren Sie die Nummer der Datei, unter der das jeweilige Spektrum im Photometer gespeichert wurde. Nach Abschluss der Messungen werden die gespeicherten Einzelspektren im Photometer aufgerufen und die Extinktionswerte im Maximum der spektral nachweisbaren Substrate und Produkte notiert. Danach werden zur Dokumentation für das Protokoll alle aufgenommenen Spektren in einer Abbildung zusammengefasst und ausgedruckt.
Testbedingungen:
Testansatzvolumen = 2 ml; d = 1 cm; λ = 380-210 nm; εNADH = 6,22 cm2/µmol; εAldehyd = 28 cm2/µmol; εAlkohol = 22 cm2/µmol
Testansatz:
1,955 ml Testpuffer
10 µl NAD+
15 µl ADH
20 µl Sorbinalkohol
Auswertung
Erstellen Sie Extinktions-Zeit-Diagramme für die spektral erfassbaren Komponenten der Enzymreaktion und benennen Sie die Phasen der Reaktion. Welche Phase wäre geeignet, die spezifische Aktivität der ADH für Sorbinalkohol zu ermitteln?
Benutzen Sie die Extinktionswerte der Komponenten aus dem Spektrum nach 20 min und berechnen Sie auf der Basis des Lambert-Beerschen Gesetzes die Menge an gebildetem Sorbinaldehyd und NADH sowie die Menge an umgesetztem Alkohol pro Küvette (in nmol). Um die Menge an umgesetzten Alkohol genau bestimmen zu können, benötigen Sie dessen Ausgangskonzentration, die Sie dem ersten Spektrum entnehmen müssen.
Können Sie die Stöchiometrie der Reaktion nachweisen? Was beeinflusst aus ihrer Sicht die Genauigkeit der Einzelbestimmung?
Was erwarten Sie, wenn die Reaktion im Messansatz noch weitere 60 Minuten verfolgt würde?

18
2. Ermittlung von Rahmenbedingungen zur Bestimmung der spezifischen Aktivität der Hefe-ADH auf der Basis des optischen Testes
Die Bestimmung spezifischer Aktivitäten erfolgt wie in der Einleitung beschrieben auf der Basis von Initialgeschwindigkeitsmessungen, wobei für jedes Enzym und jedes Substrat die Rahmenbedingungen dazu empirisch ermittelt werden müssen.
Finden Sie unter Nutzung des vorgegebenen, schon optimierten Testansatzes den Konzentrationsbereich der ADH, der eine verlässliche Aktivitätsbestimmung zulässt (lineare Extinktions-Zeit-Diagramme bestimmter Größenordnung). Stecken Sie den Rahmen der nutzbaren Enzymmenge für einen Messzeitraum von 2 Minuten zunächst grob ab (Verdopplung, Halbierung usw.; unter Umständen ist auch eine Vorverdünnung der Enzymlösung erforderlich). Wählen Sie danach 5 Enzymkonzentrationen aus und bestimmen Sie jeweils die Reaktionsgeschwindigkeit. Photometer, die über ein Kinetik-Programm verfügen, berechnen nach erfolgter Messung die Extinktionsänderung pro Minute (∆E/min), eine der Enzymaktivität analoge Größe.
Bei den Messungen ist so vorzugehen, dass zunächst Puffer und Substrate in eine Spezialglasküvette pipettiert und gemischt werden. Mit dieser Küvette wird das Photometer abgeglichen und danach die Reaktion durch Zupipettieren der Enzymlösung gestartet (erneut gut mischen!). Nach jeder Messung die Küvette gründlich mit aqua dest. spülen! Testbedingungen Testansatzvolumen = 2 ml; d = 1 cm; λ = 340 nm; εNADH = 6,22 cm2/µmol
Testansatz (TA)
x ml Testpuffer (je nach Enzymlösung 1,79 - 1,835 ml) 100 µl NAD+
60 µl Ethanol x µl Enzymlösung (5 – 50 µl)
Auswertung
Berechnen Sie aus den ∆E/ min-Werten über das Lambert-Beersche Gesetz die Reaktionsgeschwindigkeit in µmol/min/Testansatz und tragen Sie diese gegen die Enzymmenge im Testansatz auf. Aus dem Anstieg der Gerade ergibt sich die spezifische Aktivität (Angabe in µmol/min/mg = U/ mg).
Wenn die Rahmenbedingungen ermittelt sind, kann bei genügend praktischer Erfahrung die spezifische Aktivität auch aus jeder Einzelmessung bestimmt werden. Überprüfen Sie das an 2 Beispielen.
Berechnen Sie die spezifische Aktivität der ADH für Sorbinalkohol aus Teilversuch 1 (Tangentenverfahren). Bewerten Sie die Größenordnung dieser Aktivität im Vergleich zum Substrat Ethanol. Ließe sich der Wert erhöhen?

19
3. Nachweis der Substratspezifität von Enzymen am Beispiel von Proteasen
Kann ein Enzym verschiedene homologe Substrate umsetzen (siehe ADH), so gibt die Höhe der jeweiligen spezifischen Aktivität Auskunft über bevorzugte Substrate und somit auch über die Stoffwechselfunktion des Enzyms.
Am Beispiel der Verdauungsenzyme Trypsin (EC 3.4.21.4) und Chymotrypsin (EC 3.4.21.1) soll demonstriert werden, dass trotz gleicher natürlicher Substrate und Reaktions-mechanismen unterschiedliche Spezifitäten vorliegen können. Eine einfache photometrische Bestimmung der Enzymaktivität dieser proteolytischen Enzyme unter Nutzung ihrer natürlichen Substrate ist nicht möglich. Deshalb wurden synthetische Substrate entwickelt, bei deren hydrolytischer Spaltung ein gefärbtes Produkt freigesetzt wird. In diesem Teilversuch soll mit 2 synthetischen Substraten gearbeitet werden. Wenn Sie im Vorfeld überlegen, was diese beiden Verbindungen unterscheidet und wie sich die Unterschiede auf die Höhe der Enzymaktivität auswirken könnten, lässt sich der Messaufwand minimieren.
Testen Sie beide Substrate mit beiden Enzymen unter Beachtung der nach Teilversuch 2 zu beachtenden Rahmenbedingungen. Für die Messungen wird zunächst ein Substrat ausgewählt und nacheinander mit beiden Enzymen getestet (jeweils mit Trypsin beginnen). Danach erfolgt dieselbe Messung mit dem zweiten Substrat. Achtung: In diesem Teilversuch nutzen sie Plastik(Einweg)küvetten. Testbedingungen Testansatzvolumen 2 ml; d = 1 cm; t = 1 min; λ = 405 nm; εNitroanilin = 9,96 cm2/µmol
Testansatz (TA)
x ml Tris-HCl-Puffer (je nach Enzymlösung 1,85 – 1,895 ml)
100 µl Substratlösung
x µl Enzymlösung (5 – 50 µl)
Auswertung
Berechnen Sie aus den erhaltenen ∆E/min-Werten die spezifischen Aktivitäten von Trypsin und Chymotrypsin gegenüber den vorgegebenen synthetischen Substraten.
Diskutieren Sie die unterschiedlichen Größenordnungen.
Welche Aussagen zur Substratspezifität beider Enzyme lassen sich treffen?
Wie könnte man die Aktivität der Proteasen mit einem natürlichen Substrat bestimmen?

20
Versuch 3
Kinetik enzymatischer Reaktionen inklusive Enzymhemmungen
Einleitung
1. Allgemeines zur Enzymkinetik
Die Enzymkinetik ist ein biophysikalisches Verfahren, das Geschwindigkeitsmessungen nutzt, um Enzyme zu charakterisieren. Im Prinzip geht es darum, die klassischen kinetischen Enzymparameter Km und Vmax zu bestimmen. Die Kenntnis dieser Größen erlaubt Aussagen zur Funktion der Enzyme in Stoffwechselwegen und zu Regulationsphänomenen. Außerdem sind sie hilfreich, wenn es darum geht, die Nutzungsmöglichkeiten von Enzympräparaten in der Industrie, der Analytik und der Therapie einzuschätzen. Die nachstehende Einleitung kann die Gesamtproblematik nur sehr vereinfacht darstellen; Sie sollten die theoretischen Aspekte der Enzymkinetik daher nochmals in einem Lehrbuch vertiefen.
Allgemein beschreibt die Enzymkinetik den zeitlichen Verlauf einer katalysierten chemischen Reaktion, die davon ausgeht, dass sich aus dem Substrat und dem Katalysator der sogenannte Enzym-Substrat-Komplex (EA) bildet, so dass die Reaktion im einfachsten Fall durch 3 Geschwindigkeitskonstanten charakterisiert wird: k+1 k+2
E + A ⇌ EA → E + P k-1
Während die Bildung des EA-Komplexes durch das Massenwirkungsgesetz mit der Dissoziationskonstante beschrieben werden kann (Kd = k-1/ k+1), ist die Gesamtreaktion charakterisiert durch den Km-Wert (Km= (k-1 + k+2)/ k+1), die Michaelis-Menten-Konstante, benannt nach Leonor Michaelis und Maud Menten (1913), die beide wesentliche Beiträge zur Entwicklung der Grundgleichung der Enzymkinetik, geleistet haben (Michaelis-Menten-Gleichung (MM)). Diese Gleichung beschreibt die Änderung der Reaktionsgeschwindigkeit (v) in Abhängigkeit von der eingesetzten Substratkonzentration (a) bei konstanter Enzymkonzentration (e0).
Michaelis-Menten-Gleichung:
v = V • a / (Km + a)
Diese Gleichung stellt eine Hyperbel dar, die sich bei unendlich hohen Substratkonzentra-tionen dem Grenzwert der Maximalgeschwindigkeit (V) nähert. Der Km-Wert ist dann die Substratkonzentration (a), bei der die Hälfte der maximal möglichen Reaktionsgeschwindigkeit erreicht wird.

21
Während die Größe des Km-Wertes, vereinfacht ausgedrückt, eine Aussage zur Affinität des Enzyms zu seinem Substrat macht, beschreibt die Maximalgeschwindigkeit die katalytische Effizienz der Reaktion. Die Katalyse-Konstante (kkat) einer Reaktion kann man aus der Maximalgeschwindigkeit V berechnen, wenn die Enzymkonzentration bekannt ist, mit der das Experiment durchgeführt wurde (V = kkat•e0). Diese Größe wird auch als Wechselzahl (turnover number) bezeichnet und in der Maßeinheit s-1 angegeben. Ist ein Enzym in der Lage, homologe Substrate umzusetzen, wird häufig der Spezifitätsquotient (kkat/Km) benutzt, um das optimale Substrat zu identifizieren.
Die mathematische Beschreibung einer Enzymreaktion durch die MM-Gleichung ist dann nicht mehr möglich, wenn die Geschwindigkeit der Reaktion nicht oder nicht nur von der Substratkonzentration abhängig ist (wie z.B. bei allosterischen Enzymen), oder wenn es sich um 2-Substrat-Enzyme handelt. Für derartige Fälle wurden andere Gleichungssysteme entwickelt.
Die Bestimmung von Km und V unter Nutzung der MM-Gleichung ist natürlich an die Einhaltung von experimentellen Rahmenbedingungen geknüpft. In jedem Fall muss a >> e0 sein, um die Messungen im Fließgleichgewicht (steady state) durchführen zu können. Bei der Messung von Initialgeschwindigkeiten kann man davon ausgehen, dass die Produktkonzentration (p) im Meßzeitraum nahezu null ist. Das erlaubt die Anwendung der Gleichung auch für reversible Enzymreaktionen, die im Stoffwechsel weitaus häufiger auftreten, als die oben beschriebene irreversible Reaktion. Zusätzlich muss experimentell überprüft werden, ob diese Bedingungen auch bei kleinen Substratkonzentrationen eingehalten werden können (siehe dazu Versuch 2). Wenn es die Löslichkeit der jeweiligen Substrate und die Höhe der Enzymaktivität zulassen, sollte man die Substratkonzentration so abstufen, dass ein Bereich zwischen einer Zehner-potenz unter bzw. über dem Km-Wert abgedeckt ist.
Im vorliegenden Versuch wird ein 2-Substrat-Enzym in vereinfachter Form analysiert. Bei einer 2-Substrat-Reaktion gibt es viel mehr mögliche Geschwindigkeitskonstanten, die sich in den Km-Werten der beiden Substrate A und B widerspiegeln. Das Geschwindigkeitsgesetz für eine 2-Substratreaktion ist die
Cleland-Gleichung :
v = V•a•b / (a•b + KmA•b + KmB•a + KSA•KmB) .
Die Gleichung verdeutlicht, dass die Reaktionsgeschwindigkeit in Abhängigkeit von der Konzentration beider Substrate bestimmt werden muss, um dann wieder auf eine unendlich große Konzentration beider Substrate zu extrapolieren. Man kann nun dieses System vereinfachen, indem man 2 getrennte Experimente durchführt, wobei ein Substrat variiert und das zweite Substrat jeweils in sättigender Konzentration dem Testansatz zugesetzt wird. Die erhaltenen Daten können dann wie für eine 1-Substrat-Reaktion nach der MM-Gleichung ausgewertet werden. Für die Auswertung der Meßdaten gibt es prinzipiell 2 Möglichkeiten: 1. computerbasiert über ein Näherungsverfahren oder 2. die graphische Extrapolation, wobei eine Linearisierung der Hyperbelfunktion vorgenommen wird. Gebräuchlich sind die Transformationen nach Lineweaver-Burk (1/v gegen 1/a), Hanes (a/v gegen a) und Eadie-Hofstee (v gegen v/a).

22
Transformation nach Lineweaver-Burk:
1/v = Km/V • 1/a + 1/V
Transformation nach Hanes
a/v = 1/V • a + Km/V
Transformation nach Eadie-Hofstee
v = -Km • v/a + V
Da es durch die mathematische Behandlung der Messwerte zu einer Verzerrung der Werte kommt, insbesondere bei der doppelt-reziproken Auftragung nach Lineweaver-Burk, erhält man verlässliche Resultate nur dann, wenn man eine Wichtung der Werte vornimmt. Die Linearisierung nach Lineweaver-Burk und Eadie-Hofstee entsprechen den Gleichungen, die zur Beschreibung von Protein-Ligand-Wechselwirkungen genutzt werden (Klotz- bzw. Scatchard-Darstellung).
2. Reversible Enzymhemmungen
Die Charakterisierung der Hemmung von Enzymen durch niedermolekulare Verbindungen ist ein Problemkreis, der in den letzten Jahren an Bedeutung gewonnen hat, da es sich bei zahlreichen Therapeutika und Xenobiotika um Enzyminhibitoren handelt, deren Wirkungsmechanismus man genau kennen möchte. Die Tatsache, dass Enzyme in Gegenwart niedermolekularer Verbindungen ihre Aktivität verlieren können, kann verschiedene Ursachen haben. Zum Beispiel wirken chaotrope Salze oder ionische Detergenzien denaturierend auf die Enzymstruktur oder bestimmte Reagenzien sind in der Lage, die Aminosäureseitenketten zu modifizieren. Die auf diese Weise erhaltenen Aktivitätsverluste sind in der Regel irreversibel und man sollte sie klar von den reversiblen Hemmungen unterscheiden, die in diesem Versuch betrachtet werden. Da es sich bei den reversiblen Hemmungen um Dissoziationsgleichgewichte zwischen dem Enzym und einem Inihibitor handelt, können sie durch Verdünnen oder durch Dialyse der Enzymlösung aufgehoben werden. Der Betrachtung reversibler Hemmungen liegt eine Einteilung zu Grunde, die 1963 von W.W. Cleland vorgenommen wurde. Danach wird differenziert, ob der Inhibitor mit dem freien Enzym, oder mit dem Enzym-Substrat-Komplex in Wechselwirkung tritt. Auf diese Weise kann man zwei Dissoziationsgleichgewichte beschreiben, die jeweils durch eine Dissoziationskonstante charakterisiert sind. Beide Größen werden auch als die Inhibitorkonstanten (Ki1 und Ki2) bezeichnet.
E + I ⇌EI (Ki1)
EA + I ⇌EAI (Ki2)

23
Grundsätzlich werden 3 Hemmungsgrundtypen unterschieden:
Kompetitive Hemmung
Bei diesem Hemmtyp bindet der Inhibitor nur an das freie Enzym, wobei in der Regel eine Konkurrenz um die Substratbindungsstelle (bzw. Cosubstratbindungsstelle) vorliegt. Dies bedeutet, dass der Inhibitor durch das Substrat (bzw. Cosubstrat) vom Enzym verdrängt und die Hemmung damit aufgehoben werden kann. In diesem Fall gibt es nur eine Inhibitorkonstante (Ki1).
Unkompetitive Hemmung
In diesem Fall kann der Inhibitor erst nach der Bindung des Substrates mit dem Enzym in Wechselwirkung treten. Das bedeutet, dass das Enzym durch die Substratbindung seine Konformation ändert und damit die Bindungsstelle für den Inhibitor geschaffen wird. Diese Hemmung kann nicht durch das Substrat bzw. Cosubstrat aufgehoben werden. In diesem Fall ist nur die Inhibitorkonstante Ki2 zu bestimmen.
Nichtkompetitive Hemmung (gemischte Hemmung)
Bei diesem Hemmtyp besitzt der Inhibitor eine Bindungsstelle am Enzym, die er unabhängig von der Substrat- bzw. Cosubstratbindung einnehmen kann. Der nachweisbare Aktivitätsverlust des Enzyms wird somit durch eine Konformationsänderung verursacht, die durch die Inhibitorbindung eintritt. Auch diese Hemmung kann nicht durch das Substrat bzw. Cosubstrat aufgehoben werden. Für diesen Hemmtyp gibt es beide Inhibitorkonstanten Ki1 und Ki2.
Nach der Klassifizierung der Hemmtypen ist es für die Aufklärung eines Wirkmechanismus von Interesse, den Hemmtyp und die Hemmstärke zu bestimmen. Dabei wird mit einer Geschwindigkeitsgleichung gearbeitet, deren Basis die Michaelis-Menten-Gleichung ist: v = V•a / [ Km( 1 + i/ Ki1) + a ( 1 + i/ Ki2) ] Diese „Hemmgleichung“ hat in der Lineweaver-Burk-Transformation dann folgendes Aussehen 1/v = Km/V (1 + i/ Ki1) • 1/a + ( 1 + i/ Ki2) • 1/V Im Analyseverfahren geht man in gleicher Weise vor wie bei der Bestimmung der kinetischen Konstanten: man variiert die Substratkonzentration und misst die jeweilige Reaktionsgeschwindigkeit. Dieses Experiment wird für verschiedene Inhibitorkonzentrationen wiederholt, die im Teilexperiment in jeweils konstanter Konzentration dem Testansatz zugesetzt werden. Als Resultat erhält man in der graphischen Darstellung nach einem Transformationsverfahren (z.B. Lineweaver-Burk) eine Schar von Geraden (Primär-darstellung). Aus der Geschwindigkeitsgleichung ist ersichtlich, dass ein Inhibitor sowohl die Größe des Km-Wertes, als auch den V-Wert (oder beide Konstanten) beeinflussen kann. Welche der Konstanten betroffen ist, zeigt sich im Anstieg und/oder Ordinatenabschnitt der Geraden der Primärdarstellung. Welcher Hemmtyp daraus abgeleitet werden kann, kann der folgenden Tabelle entnommen werden:

24
Hemmtyp Merkmal kompetitiv unkompetitiv nichtkompetitiv Lineweaver-Burk-Darstellung
Schar von Geraden mit einem gemein-samen Schnittpunkt auf der Ordinate
Parallele Schar von Geraden
Schar von Geraden mit einem gemein-samen Schnittpunkt links von der Ordi-nate, im Idealfall auf der Abszisse
durch den Inhibitor beeinflusste Konstante
Km Km und V V
Größe der Ki-Werte 8 • Ki1 ≤ Ki2 8 • KI2 ≤ KI1 KI1 = KI2 (Idealfall)
Nach der allgemeinen Hemmgleichung ist es möglich, durch die mathematische Behandlung der Daten über ein Computerprogramm stets beide Ki-Werte zu berechnen, obwohl es z.B. für die kompetitive Hemmung theoretisch nur einen Ki-Wert (Ki1) gibt. In der letzten Zeile der Tabelle ist eine Groborientierung hinsichtlich der Größenordnung der KI-Werte gegeben, die die Zuordnung des Hemmtyps erleichtert. Diese Groborientierung ist auch dann hilfreich, wenn das Experiment keine völlig eindeutigen Resultate liefert.
Die Größe der Ki-Werte erhält man aus Sekundärdarstellungen. Dabei werden jeweils getrennt die Anstiege und die Ordinatenabschnitte der Geraden der Primärdarstellung gegen die Inhibitorkonzentration aufgetragen. Jede Darstellung liefert wiederum eine Gerade, wobei die Schnittpunkte mit der Abszisse den Ki-Werten entsprechen. Während man für die Primärdarstellung eine Wichtung der Messwerte vornehmen sollte, analysiert man die Sekundärauftragungen über eine lineare Regression.
Im vorliegenden Versuch soll am Beispiel der D-3-Hydroxybutyratdehydrogenase (HBDH) die Hemmwirkung einer substratanalogen Verbindung untersucht werden.
Material Phosphatpuffer nach Sörensen ( 100 ml) 0,05 M; pH 8 D,L-Hydroxybutyrat (Na-Salz) 50 mg/ml aqua dest. (M : 126,1) NAD+ (freie Säure) 12 mg/ml aqua dest. (M : 663,4) D-Laktat (Li-Salz) 2 mg/ml aqua dest. (M : 96) D-3-Hydroxybutyratdehydrogenase aus Pseudomonas putida (HBDH) gentechnisch hergestellt
Suspension in (NH4)2SO4 mit der Konzentration 0,017 mg/ml (MW: 105 840 Da)

25
Durchführung
Teilversuch 1
Im vorliegenden Teilversuch sollen die kinetischen Konstanten der Oxidationsreaktion der D-3-Hydroxybutyratdehydrogenase (HBDH; EC 1.1.1.30) bestimmt werden. Das Enzym katalysiert die stereospezifische Oxidation von D-3-Hydroxybutyrat (D-HB) zu Acetoacetat. Die HBDH ist ein Zweisubstratenzym, es benötigt als Cosubstrat NAD+, das im Reaktionsverlauf zu NADH reduziert wird. Das Enzym ist streng spezifisch für sein Substrat, andere Hydroxysäuren werden nicht umgesetzt.
Messserie A
Zur Bestimmung des Km-Wertes für das Cosubstrat NAD+ wird die Konzentration des Substrates D,L-HB in sättigender Konzentration konstant gehalten (20 mM) und die Konzentration des Cosubstrats variiert. Die jeweiligen Testansatzkombinationen werden mit Ausnahme des Enzyms in eine Küvette pipettiert und gut gemischt. Das Photometer wird abgeglichen, die Reaktion durch Zugabe des Enzyms gestartet (sorgfältig mischen!!) und die Initialgeschwindigkeit (∆E/ min) bestimmt.
Testbedingungen: Testansatzvolumen = 2 ml; d = 1 cm; t = 1 min; λ = 340 nm; ε = 6,22 cm2/µmol
Testansatz 1-A
Volumen Substanz ml Puffer µl NAD+
100 µl D,L-HB µl HBDH

26
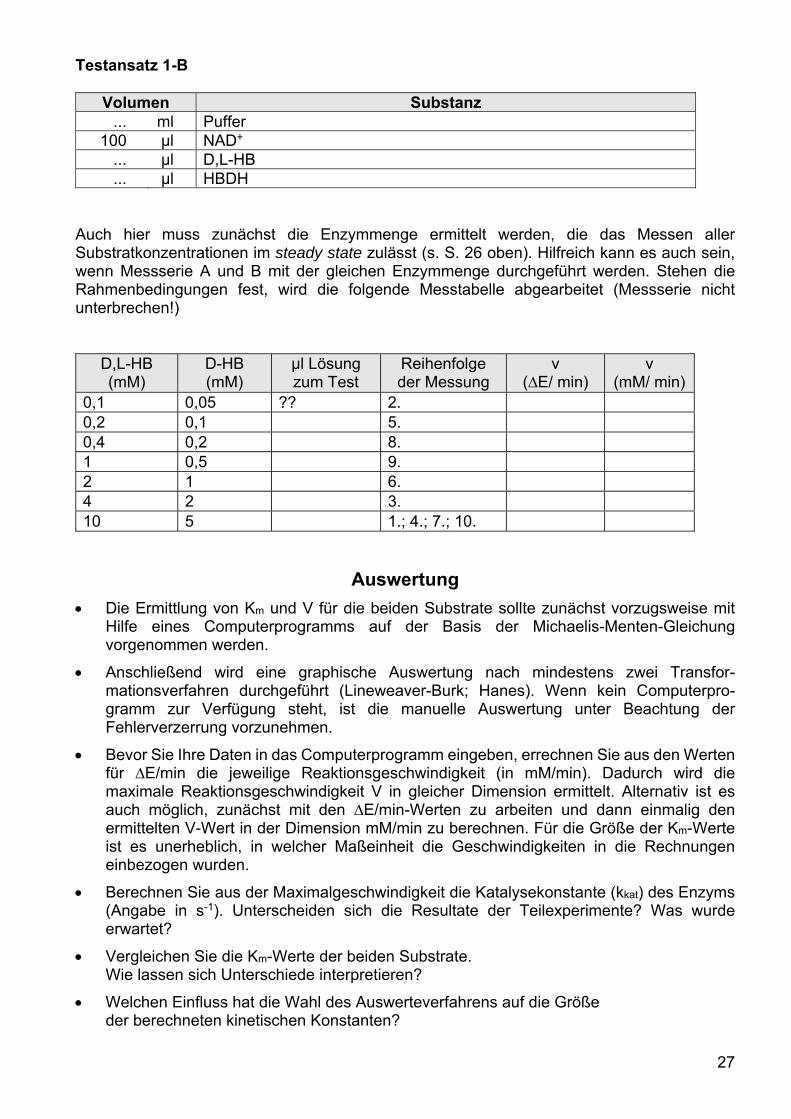
Erstellen Sie sich vor Beginn der Messungen eine Arbeitstabelle nach folgendem Muster:
NAD+ (mM) µl Lösung zum
Test Reihenfolge der
Messung v (∆E/ min) v (mM/ min)
0,009 ?? 2. 0,018 5. 0,045 8. 0,09 9. 0,18 6. 0,27 3. 0,45 1.; 4.; 7.; 10
Um sicherzustellen, dass die Geschwindigkeiten im Zeitraum des steady-state bestimmt werden (siehe Versuch 2), muss zunächst eine geeignete Enzymmenge für die Aktivitätstests festgelegt werden. Dabei ist es von Vorteil, wenn alle gewählten Cosubstratkonzentrationen mit der gleichen Enzymmenge gemessen werden können. Mit folgenden Angaben sollten Sie den geeigneten Rahmen schnell finden: Nutzen Sie Testansatz 1 und variieren Sie das Volumen der Enzymlösung zwischen 10–30 µl. Benutzen Sie das Volumen, mit dem Sie ein ∆E/min in der Größenordnung von 0,13 bis 0,19 bestimmt haben, um den Testansatz 2 zu messen. Wenn auch mit diesem Ansatz ein lineares Extinktions-Zeit-Diagramm mit ausreichendem Anstieg erhalten wird, können Sie die gesamte Wertetabelle abarbeiten. Damit sich bei einer Meßserie kein systematischer Pipettierfehler entwickeln kann, ist die Reihenfolge der einzelnen Messungen bewusst gestreut. Der Testansatz 1 dient zusätzlich als Standardansatz, dessen Aktivität mehrfach bestimmt wird. An der Höhe der jeweils gemessenen Aktivität kann man erkennen, ob das Enzympräparat über den Zeitraum des gesamten Experimentes an Aktivität verliert. Eine solche Überprüfung ist besonders dann von Bedeutung, wenn man mit großen Enzymverdünnungen arbeiten muss. Zusätzlich ist der Testansatz 1, der beide Substrate in Sättigungskonzentration enthält, gegenüber Pipettierfehlern am wenigsten störanfällig (mit Ausnahme der Enzymlösung).
Nach der Festlegung der Rahmenbedingungen sollte die gesamte Meßserie zügig abgearbeitet werden.
Messserie B
Die Bestimmung des Km-Wertes für das Substrat D-HB wird analog zur Messserie A durch-geführt: die Konzentration des Cosubstrats NAD+
wird in sättigender Konzentration konstant gehalten (0,9 mM) und die Konzentration des Substrates variiert. Da für die Messungen nur das Racemat des Substrates zur Verfügung steht, muss bei der Auswertung berücksichtigt werden, dass nur das D-Enantiomer umgesetzt wird.
Testbedingungen: Testansatzvolumen = 2 ml; d = 1 cm; t = 1 min; λ = 340 nm; ε = 6,22 cm2/µmol
27
Testansatz 1-B
Volumen Substanz ... ml Puffer
100 µl NAD+ ... µl D,L-HB ... µl HBDH
Auch hier muss zunächst die Enzymmenge ermittelt werden, die das Messen aller Substratkonzentrationen im steady state zulässt (s. S. 26 oben). Hilfreich kann es auch sein, wenn Messserie A und B mit der gleichen Enzymmenge durchgeführt werden. Stehen die Rahmenbedingungen fest, wird die folgende Messtabelle abgearbeitet (Messserie nicht unterbrechen!)
D,L-HB (mM)
D-HB (mM)
µl Lösung zum Test
Reihenfolge der Messung
v (∆E/ min)
v (mM/ min)
0,1 0,05 ?? 2. 0,2 0,1 5. 0,4 0,2 8. 1 0,5 9. 2 1 6. 4 2 3. 10 5 1.; 4.; 7.; 10.
Auswertung
Die Ermittlung von Km und V für die beiden Substrate sollte zunächst vorzugsweise mit Hilfe eines Computerprogramms auf der Basis der Michaelis-Menten-Gleichung vorgenommen werden.
Anschließend wird eine graphische Auswertung nach mindestens zwei Transfor-mationsverfahren durchgeführt (Lineweaver-Burk; Hanes). Wenn kein Computerpro-gramm zur Verfügung steht, ist die manuelle Auswertung unter Beachtung der Fehlerverzerrung vorzunehmen.
Bevor Sie Ihre Daten in das Computerprogramm eingeben, errechnen Sie aus den Werten für ∆E/min die jeweilige Reaktionsgeschwindigkeit (in mM/min). Dadurch wird die maximale Reaktionsgeschwindigkeit V in gleicher Dimension ermittelt. Alternativ ist es auch möglich, zunächst mit den ∆E/min-Werten zu arbeiten und dann einmalig den ermittelten V-Wert in der Dimension mM/min zu berechnen. Für die Größe der Km-Werte ist es unerheblich, in welcher Maßeinheit die Geschwindigkeiten in die Rechnungen einbezogen wurden.
Berechnen Sie aus der Maximalgeschwindigkeit die Katalysekonstante (kkat) des Enzyms (Angabe in s-1). Unterscheiden sich die Resultate der Teilexperimente? Was wurde erwartet?
Vergleichen Sie die Km-Werte der beiden Substrate. Wie lassen sich Unterschiede interpretieren?
Welchen Einfluss hat die Wahl des Auswerteverfahrens auf die Größe der berechneten kinetischen Konstanten?
28
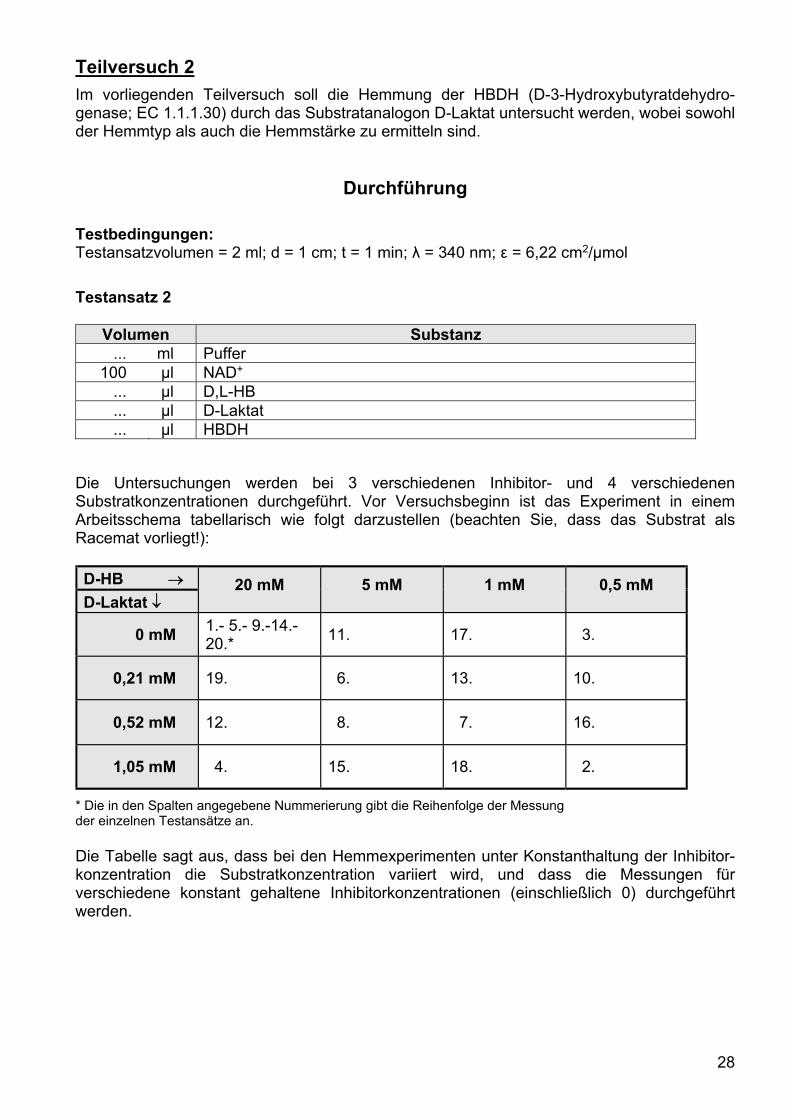
Teilversuch 2
Im vorliegenden Teilversuch soll die Hemmung der HBDH (D-3-Hydroxybutyratdehydro-genase; EC 1.1.1.30) durch das Substratanalogon D-Laktat untersucht werden, wobei sowohl der Hemmtyp als auch die Hemmstärke zu ermitteln sind.
Durchführung
Testbedingungen: Testansatzvolumen = 2 ml; d = 1 cm; t = 1 min; λ = 340 nm; ε = 6,22 cm2/µmol
Testansatz 2
Volumen Substanz ... ml Puffer
100 µl NAD+ ... µl D,L-HB ... µl D-Laktat ... µl HBDH
Die Untersuchungen werden bei 3 verschiedenen Inhibitor- und 4 verschiedenen Substratkonzentrationen durchgeführt. Vor Versuchsbeginn ist das Experiment in einem Arbeitsschema tabellarisch wie folgt darzustellen (beachten Sie, dass das Substrat als Racemat vorliegt!):
D-HB 20 mM 5 mM 1 mM 0,5 mM D-Laktat
0 mM 1.- 5.- 9.-14.- 20.*
11. 17. 3.
0,21 mM 19. 6. 13. 10.
0,52 mM 12. 8. 7. 16.
1,05 mM 4. 15. 18. 2.
* Die in den Spalten angegebene Nummerierung gibt die Reihenfolge der Messung der einzelnen Testansätze an. Die Tabelle sagt aus, dass bei den Hemmexperimenten unter Konstanthaltung der Inhibitor-konzentration die Substratkonzentration variiert wird, und dass die Messungen für verschiedene konstant gehaltene Inhibitorkonzentrationen (einschließlich 0) durchgeführt werden.

29
Analog zu Teilversuch 1 muss auch hier die Enzymkonzentration ermittelt werden, die eine Messung im „steady-state“ (Initialgeschwindigkeitsmessungen) zulässt. Dazu wird wie folgt verfahren:
Messung der enzymatischen Reaktion in Anwesenheit der höchsten Konzentration an Hydroxybutyrat sowie in Abwesenheit des Inhibitors (= Messung 1). Die Enzymmenge sollte so gewählt werden, dass sich ein ΔE/min von etwa 0,1 ergibt.
Messung der Reaktion bei Einsatz der gleichen Enzymmenge unter Einsatz der kleinsten Konzentration an Hydroxybutyrat und der größten Inhibitorkonzentration (= Messung 2). Hierbei muss sich ein lineares Extinktions-Zeit-Diagramm ergeben, das eine sicher messbare Extinktionsänderung (∆E/min) zulässt. Notfalls muss die Enzymmenge geändert werden.
Wenn beide Eckpunkte fixiert sind, werden die anderen ∆E/min-Werte möglichst zügig in der im Schema angegebenen Reihenfolge ermittelt.
Auswertung
Da bei den Hemmexperimenten nur die zu ermittelnden Ki-Werte von Interesse sind, ist es nicht erforderlich, die gemessenen ∆E/min-Werte in eine Volumenaktivität umzurechnen.
Zunächst wird von den Messdaten eine Primärdarstellung [Transformationsverfahren nach Lineweaver/Burk (1/v gegen 1/a) oder nach Hanes (a/v gegen a)] erstellt. Dabei erhält man 4 Geraden, die je einer konstant gehaltenen Inhibitorkonzentration (inklusive 0) entsprechen.
Aus der Primärauftragung werden zwei Sekundärauftragungen entwickelt. Dazu werden einmal die Anstiege und zum anderen die Ordinatenabschnitte der Geraden gegen die jeweilige Inhibitorkonzentration aufgetragen.
Die Inhibitorkonstanten Ki1 und Ki2 kann man nun aus den Schnittpunkten der Geraden der Sekundärauftragungen mit der Abszisse ablesen. Welche der Konstanten aus der Darstellung der Anstiege und welche aus der Darstellung der Abschnitte entnommen werden kann, hängt davon ab, welches Transformationsverfahren für die Primärdarstellung gewählt wurde.
Vergleichen Sie die erhaltenen Inhibitorkonstanten. Welcher Hemmtyp liegt vor? Entspricht das Ergebnis den Erwartungen? Begründen Sie das Resultat.

30
Versuch 4
Ionenaustausch-Chromatographie als Methode zur Anreicherung von Proteinen
Einleitung
Bei der Ionenaustausch-Chromatographie nutzt man die positive oder negative Nettoladung der Proteine, um sie über ionische Wechselwirkungen an eine stationäre Phase zu binden. Die dafür eingesetzten Trägermaterialien bestehen aus einem inerten Grundkörper, an den kovalent ladungstragende Gruppen gekoppelt sind. Man unterscheidet dabei zwischen Anionenaustauschern (positiv geladen) und Kationenaustauschern (negativ geladen). Als Trägermaterial werden z.B. vernetzte Kohlehydrate (Cellulose, Dextran, Agarose), Polymethacrylate oder Polystyrol eingesetzt. Je nach den gekoppelten Liganden unterscheidet man zusätzlich zwischen starken und schwachen Kationen- und Anionenaustauschern. Die folgende Tabelle zeigt einige häufig verwendete funktionelle Gruppen:
Ionenaustauschergruppen
Anionenaustauscher Kationenaustauscher
Funktionelle Gruppe Abkürzung Funktionelle Gruppe Abkürzung Diethylaminoethyl (schwach) DEAE Carboxymethyl (schwach) CM Diethylaminopropyl (schwach)
DEAP Sulfonat (stark) S
Quarternäres Aminomethyl (stark)
Q Sulfopropyl (stark) SP
Die Proteinreinigung über Ionenaustauscher kann sowohl im Batch-Verfahren als auch als Säulenchromatographie (wie im vorliegenden Versuch) durchgeführt werden. Generell werden die Proteine bei geringer Ionenkonzentration eines geeigneten Puffers an das Trägermaterial gebunden. Dabei ist zu beachten, dass die Ladung der Proteine vom pH-Wert abhängig ist. Je nach der Zahl der Ladungen eines Proteins binden die Einzelproteine einer Mischung unterschiedlich fest an das Trägermaterial, was letztendlich eine differenzierte Ablösung möglich macht. Proteine mit der gleichen Ladungspolarität wie die Matrix können nach dem Auftragen der Proteinlösung durch Waschen der Säule mit Puffer sofort eluiert und verworfen werden. Das schrittweise Ablösen der Proteine vom Ionenaustauscher erfolgt dann durch Er-höhung der Salzkonzentration im Puffer, was diskontinuierlich oder kontinuierlich in Form verschiedener Gradienten (linear, konvex, konkav) erfolgen kann. Als Salze, die mit den Proteinen um die Bindungsplätze am Träger konkurrieren, verwendet man meist Natrium- oder Kaliumchlorid, da deren Ionen weder strukturbildenden noch chaotropen Charakter besitzen. Alternativ kann auch durch eine Veränderung des pH-Wertes eluiert werden. Für jedes zu reinigende Protein müssen in Vorversuchen die Rahmenbedingungen für Bindung und differenzierte Elution ermittelt werden, um die größte Effizienz (Ausbeute und Reinheit) zu erreichen. Nach jeder Chromatographie muss der Ionenaustauscher für weitere Anwendungen regeneriert werden.

31
Am Beispiel der Reinigung des Green Fluorescent Protein (GFP) soll im nachfolgenden Versuch das Prinzip der Ionenaustausch-Chromatographie demonstriert werden. Durch die Fluoreszenzeigenschaften des GFP kann das Adsorptions- und Elutionsverhalten von Proteinen auf dem Trägermaterial direkt beobachtet werden.
Abb. 4-1: Struktur des GFP. Die 238 aa lange Kette ist als C-Gerüst gezeigt; der Fluorophor besteht aus der Tripeptidsequenz Ser65-Tyr66-Gly67 und ist als Kalottenmodell dargestellt (grün). Das Haupt-Anregungsmaximum liegt bei 395 nm, ein kleineres bei 475 nm. Das Emissionsmaximum liegt bei 509 nm.
Material
GFP-haltiger Proteinrohextrakt siehe Versuch 6
Phosphatpuffer nach Sörensen ( 200 ml) 5 mM, pH 8,0
NaCl-Stammlösung 1 M in Puffer s.o.
Chromatographiesäule (BioRad) d: 1,5 cm; h: 6 cm
DEAE-Sephacel® 6 ml
Schlauchpumpe P1 (Pharmacia) Schlauchdurchmesser = 2,1 mm
UV-Handlampe

32
Durchführung
Die mit dem Trägermaterial DEAE-Sephacel gepackte Säule wird zunächst mit 20 ml Phosphatpuffer äquilibriert (Pumpgeschwindigkeit 1 x 6 = ca. 0,3 ml/min).
Danach werden mit einer Pipette durch Überschichten 2,5 ml des Proteinrohextraktes aufgetragen. Nach dem Einsickern wird die Säule zunächst mit 5 ml Phosphatpuffer gewaschen.
Durchlauf und Waschpuffer werden je als Fraktionen gesammelt.
Der folgende Wasch- und Elutionsprozess wird diskontinuierlich durch Erhöhung der NaCl-Konzentration im Phosphatpuffer unter Nutzung der Schlauchpumpe durchgeführt, wobei jeweils 5 ml-Fraktionen gesammelt werden. Dabei werden die NaCl-Konzentration und die Zahl der Fraktionen wie folgt abgestuft:
Fraktion NaCl (mM) Volumen (ml)
Probe 0 2,5
Durchlauf 0 2,5
Waschpuffer 0 5
Fraktion 1 - 4 80 je 5
Fraktion 5 - 8 100 je 5
Fraktion 9 - x 200 je 5
Vom Rohextrakt und allen Fraktionen wird der Proteingehalt durch UV-Spektroskopie nach der Methode von Kalb & Bernlohr [Anal. Biochem. 82 (1977), 362] bestimmt. Dazu wird ein geeignetes Volumen der Proben auf 2 ml mit Phosphatpuffer aufgefüllt, gut gemischt und die Extinktion bei 230 und 260 nm ermittelt (Schichtdicke der Küvette = 1 cm; Abgleich des Photometers gegen Puffer). Der Proteingehalt der Lösung kann dann nach folgender Beziehung berechnet werden:
Protein (µg/ml) = 183 • E230 – 75,8 • E260
Verfolgen Sie die einzelnen Schritte des Chromatographieprozesses auch durch Beobachtung der Säule unter Bestrahlen mit Licht der Wellenlänge 366 nm (Bindungs-verhalten des GFP am Träger). Notieren Sie die Beobachtungen für das Protokoll.
Eine Probe des Proteinrohextraktes (500 µl) und die Fraktion mit höchster Fluoreszenz bzw. Proteingehalt wird nach Versuchsende eingefroren (Analyse im Versuch 5).
33
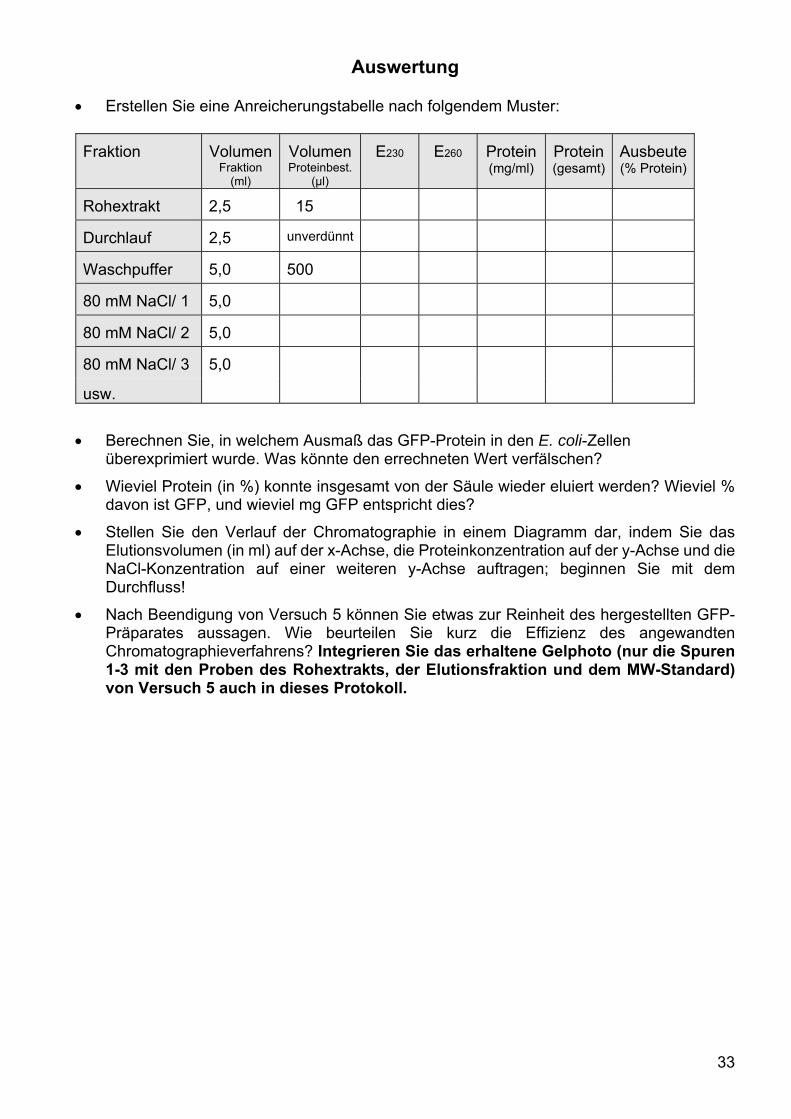
Auswertung Erstellen Sie eine Anreicherungstabelle nach folgendem Muster:
Fraktion Volumen Fraktion
(ml)
Volumen Proteinbest.
(µl)
E230 E260 Protein (mg/ml)
Protein (gesamt)
Ausbeute (% Protein)
Rohextrakt 2,5 15
Durchlauf 2,5 unverdünnt
Waschpuffer 5,0 500
80 mM NaCl/ 1 5,0
80 mM NaCl/ 2 5,0
80 mM NaCl/ 3 5,0
usw.
Berechnen Sie, in welchem Ausmaß das GFP-Protein in den E. coli-Zellen überexprimiert wurde. Was könnte den errechneten Wert verfälschen?
Wieviel Protein (in %) konnte insgesamt von der Säule wieder eluiert werden? Wieviel % davon ist GFP, und wieviel mg GFP entspricht dies?
Stellen Sie den Verlauf der Chromatographie in einem Diagramm dar, indem Sie das Elutionsvolumen (in ml) auf der x-Achse, die Proteinkonzentration auf der y-Achse und die NaCl-Konzentration auf einer weiteren y-Achse auftragen; beginnen Sie mit dem Durchfluss!
Nach Beendigung von Versuch 5 können Sie etwas zur Reinheit des hergestellten GFP-Präparates aussagen. Wie beurteilen Sie kurz die Effizienz des angewandten Chromatographieverfahrens? Integrieren Sie das erhaltene Gelphoto (nur die Spuren 1-3 mit den Proben des Rohextrakts, der Elutionsfraktion und dem MW-Standard) von Versuch 5 auch in dieses Protokoll.

34
Versuch 5
Polyacrylamidgel-Elektrophorese
Einleitung
Die Elektrophorese nutzt die Ladungsunterschiede von Proteinen bzw. Nukleinsäuren zu deren Trennung aus. Elektrophoresen können mit präparativer oder analytischer Zielsetzung durchgeführt werden. Als Trägermaterial für die Protein-Elektrophorese haben sich Polyacrylamidgele durchgesetzt, da deren Porengröße durch Variation der Acrylamidkonzentration (3-30 %ige Gele sind herstellbar) leicht steuerbar ist, wodurch zusätzlich zur Trennung nach Ladung ein Molekularsiebeffekt die Trennschärfe erhöht. Der Vernetzer (Crosslinker) N,N’-Methylen-bis-Acrylsäureamid, der zu einem dreidimensionalen Netzwerk führt, wird in einem Mengenverhältnis von 3-5 % der Monomereinwaage beigemischt. Meist werden bei der Polyacrylamid-Gel-Elektrophorese (PAGE) von Proteinen diskontinuierliche Puffer- und Gelsysteme verwendet, die auf Grund des zu Beginn der Elektrophorese auftretenden Konzentrierungseffektes eine hohe Trennschärfe besitzen. Poly-acrylamidgel-Elektrophoresen können unter nativen oder unter denaturierenden Bedingungen durchgeführt werden, wobei letztere Technik weit häufiger angewandt wird, so auch im vorliegenden Versuch. Als Denaturierungsmittel wird das ionische Detergens Natriumdodecylsulfat (engl. sodium dodecyl sulfate = SDS SDS-PAGE) verwendet. Inkubiert man Proteine mit SDS, wird deren Sekundär- und Quartärstruktur weitgehend aufgebrochen. SDS lagert sich über hydrophobe Wechselwirkungen in einem nahezu konstanten Verhältnis (1,4 mg SDS pro mg Protein) an Proteine an (Ausnahmen: Glycoproteine sowie extrem hydrophobe Proteine). Protein-SDS-Komplexe haben eine gestreckte Gestalt. Auf Grund des konstanten Ladungs/Masse-Verhältnisses hängt deren elektrophoretische Beweglichkeit nur noch von der Molekülgröße ab. Disulfidbrücken müssen vor der Elektrophorese reduziert werden. Außerdem müssen zur vollständigen und schnellen Auffaltung der Proteine die Proben kurzzeitig erhitzt werden. Durch Eichung mit kommerziell verfügbaren Molmassestandards kann man mit dieser Technik die Molmasse von Proteinen relativ genau bestimmen. Handelt es sich um oligomere Proteine, so wird mit diesem Verfahren lediglich die Molmasse der Untereinheiten ermittelt. Vorsicht: Acrylamid und der Vernetzer (Bisacrylamid) sind toxisch!! (Nervengift und
Hautreizmittel, im Tierversuch krebserregend und erbgutverändernd): bei der Vorbereitung der Gele Gummihandschuhe tragen !!
35
Material
Elektrophorese-Apparatur Minigel G 41 (Biometra); 1 mm Gelstärke Elektrodenpuffer (pH 8,3) 3 g Tris + 14,4 g Glycin + 1 g SDS in ca. 500 ml aqua
dest. lösen, auf 1 l auffüllen; pH-Wert kontrollieren ( steht fertig zur Verfügung)
Trenngelpuffer (pH 8,8) 18,2 g Tris in ca. 50 ml aqua dest. lösen; nacheinander 0,4 g SDS und 26,4 ml 1 N HCl zugeben und auf 100 ml auffüllen; pH-Wert kontrollieren ( steht fertig zur Verfügung)
Sammelgelpuffer (pH 6,8)
6 g Tris in ca. 80 ml aqua dest. lösen; 0,4 g SDS zugeben, mit 1 N HCl auf geforderten pH-Wert einstellen und mit aqua dest. auf 100 ml auffüllen ( steht fertig zur Verfügung)
Probenpuffer (pH 8,0) 48 mg Tris + 800 mg SDS + 15 mg EDTA in aqua dest. lösen und auf 10 ml auffüllen ( steht fertig zur Verfügung)
-Mercaptoethanol Fertigsubstanz; nur unter dem Abzug verwenden! Glycerin/ Farbstofflösung 0,05 %ige Bromphenolblaulösung in aqua dest.
im Verhältnis 1:1 mit Glycerin (87 %) gemischt ( steht fertig zur Verfügung)
Acrylamid-Stammlösung 40%ige Stammlösung
TEMED: N,N,N`,N`Tetramethylethylendiamin
Katalysatorlösung 0,1 g Ammoniumperoxodisulfat/ml aqua dest. ( frisch herstellen!)
Färbelösung 200 mg Coomassie-Brilliant-Blau R 250 werden in 91 ml Methanol (50%ig) gelöst, danach 9 ml Eisessig zugegeben und die Mischung durch ein Faltenfilter filtriert ( steht fertig zur Verfügung)
Entfärber 50 ml Methanol und 75 ml Eisessig werden mit aqua dest. auf 1 l aufgefüllt ( steht fertig zur Verfügung)
Molmassestandard siehe Abb. 5-1
Proben Stammlösungen kommerzieller Proteine sowie Proben aus Versuch 4 und 6
Rinderserumalbumin und Lysozym (Roth),1 mg/ml; Hefe-ADH (Roche), 1 mg/ml;
Trypsin und Chymotrypsin aus Rinderpankreas (Sigma), 5 mg/ml;
Hydoxybutyratdehydrogenase (HBDH; eigene Herstellung), 1 mg/ml
Mikrokonzentratoren Vivaspin 500 (Sartorius Stedim Biotech GmbH)

36
Durchführung
Herstellung des Polyacrylamidgels
Acrylamid- und Probenpufferlösungen werden Ihnen fertig zur Verfügung gestellt!
Zusammenbau der Apparatur: Die fettfreien Glasplatten (mit Spülmittel und anschließend Ethanol sorgfältig gereinigt) werden nach Einlegen des Dichtungsgummis mit 2 Klemmen aufeinandergepresst und senkrecht gestellt; die Höhe des Trenngels wird bei 6,5 cm mit einem wasserfesten Stift markiert. Der Taschenformer (Kamm) wird ebenso gereinigt.
Trenngel (12 % Acrylamid/Bisacrylamid): 10 ml Trenngellösung werden aus 3 ml Acrylamidstammlösung, 2,5 ml Trenngelpuffer, 10 µl TEMED und 4,5 ml aqua dest. hergestellt (durch Schwenken im Glas gründlich mischen, dabei Luftblasen vermeiden!).
Sammelgel (4 % Acrylamid/Bisacrylamid): 5 ml Gellösung werden aus 0,5 ml Acrylamidstammlösung, 0,625 ml Sammelgelpuffer, 5 µl TEMED und 3,87 ml aqua dest. hergestellt (mischen s.o.!).
Beide Lösungen kühl (aber nicht im Eis!) zwischenlagern.
Zur Polymerisation werden direkt vor dem Gießen jeweils 5 µl Katalysatorlösung pro 1 ml Gellösung eingesetzt (mischen s.o.!).
Gießen des Gels: Mit einer Injektionsspritze wird zunächst das Trenngel zwischen die Glas-platten bis zur Markierung eingefüllt und vorsichtig mit aqua dest. überschichtet. Nach erfolgter Polymerisation (ca. 30 min) wird das Wasser mit einem Fließpapier abgezogen, das Sammel-gel bis zum Plattenende eingefüllt und der Kamm zur Ausformung der Geltaschen eingesetzt (Polymerisation in ~30 min). Die unteren Kanten der Probentaschen werden mit einem Filzstift markiert. Ist das Sammelgel polymerisiert, werden Kamm und Dichtungsgummi vorsichtig entfernt und die Gelplatte kann in der Apparatur befestigt werden. Die Gelplatte wird mit zwei Klammern in die Elektrophoreseapparatur eingepasst und der Elektrodenpuffer in beide Elektrodenkammern eingefüllt. Probenvorbereitung und Elektrophorese Die zu analysierenden Proteine werden Ihnen mit Ausnahme der eigenen Proben aus Versuch 4 in Form von Stammlösungen zur Verfügung gestellt. Die pro Geltasche aufzutragende Proteinmenge richtet sich nach der Zahl der zu erwartenden Banden. Bei reinen Proteinen sind 5 µg pro Spur ausreichend, bei einem Proteinrohextrakt können bis zu 80 µg pro Spur aufgetragen werden. Eine Probentasche fasst maximal 20 µl. Berechnen Sie zunächst, welches Volumen der jeweiligen Proteinlösung sie einsetzen müssen, um die im Auftragsschema angegebene Proteinmenge zu erhalten. Jede Probe muss weiterhin 5 µl Probenpuffer (SDS Denaturieren und negative Nettoladung!), 1 µl β-Mercaptoethanol ( Reduzieren von Disulfidbrücken) und 5 µl Glycerin/Färbelösung ( Viskosität / Verfolgung der Lauffront) enthalten. Stellen Sie dafür zunächst einen Mastermix her Zusammensetzung Mastermix: 50 µl SDS-Probenpuffer werden mit 10 µl β-Mercaptoethanol und 50 µl Glycerin/Farbstofflösung versetzt und kurz gevortext (Achtung: Schaumbildung durch das SDS!). Endkonzentration an SDS in der Probe ist dann 2 %)
Das jeweils berechnete Volumen der Proteinlösung ein Eppendorf-Gefäß pipettiert, dann mit 11µl Mastermix versetzt und mit x µl H2O auf 25 µl aufgefüllt. Nach vorsichtigem Mischen (Schaum vermeiden!) müssen die Proben blau gefärbt sein.
Anschließend werden die Proben kurz anzentrifugiert und danach für 5 Minuten bei 95 °C
inkubiert ( Denaturieren).
37
Achtung: Sind die Proteinlösungen so gering konzentriert, dass die oben genannten Volumenverhältnisse nicht eingehalten werden können, müssen die Proben zunächst eingeengt werden. Für das Konzentrieren werden Ihnen Mikrokonzentratoren zur Verfügung gestellt. Das jeweils benötigte Probenvolumen wird mit aqua dest. auf 100 µl aufgefüllt und in die Mikrokonzentratoren pipettiert. Danach wird bei 13000 rpm ca. 5 Minuten zentrifugiert und das verbleibende Volumen bestimmt. Sollte dieses Volumen für die oben angegebenen Rahmenbedingungen immer noch zu groß sein, muss länger zentrifugiert werden; das Volumen soll 14 µl betragen (evtl. mit ddH2O auffüllen).
Die Proben (je 20 µl) werden danach in der angegebenen Reihenfolge auf das Gel aufgetragen, die Randspur wird freigelassen.
Der Molmassestandard der Firma Thermo Fischer Scientific ist nach Firmenvorschrift gebrauchsfertig vorbereitet (10 µl/ Spur). Auftragsschema SDS-Gel
Spur Probe Proteinmenge (µg) 1 GFP-Rohextrakt (Probe aus V. 6) 60 2 GFP-Probe nach DEAE-Chromatographie (V. 4) 10 3 Standard (10 µl), wird fertig zur Verfügung gestellt x 4 Lysozym 5 5 HBDH 5 6 RSA 5 7 Hefe-ADH 5 8 Trypsin 30 9 Chymotrypsin 20
10 - -
Die Elektrophorese wird bei einer konstanten Spannung von 60 V gestartet, bis die Lauffront das Trenngel erreicht hat (~20 min). Anschließend wird die Spannung auf 160 V erhöht und die Elektrophorese für ~1 h bei Raumtemperatur durchgeführt. Sie wird beendet, wenn die Bromphenolblaufront das untere Gelende erreicht hat.
Proteinnachweis im Gel
Nach dem Lauf werden die Glasplatten aus der Apparatur genommen und vorsichtig getrennt. Das an einer Glasplatte haftende Gel wird vorsichtig in die Färbelösung gelegt und 30 Minuten leicht geschwenkt. Da bei dieser Färbetechnik auch die Gelmatrix angefärbt wird, muss nach der Färbung der nicht gebundene Farbstoff entfernt werden. Dazu wird das Gel nach kurzem Abspülen mit aqua dest. in das Entfärbebad gelegt und ebenfalls vorsichtig geschwenkt. Zugabe eines Schwämmchens zur Aufnahme des gelösten Farbstoffes beschleunigt das Entfärben. Im Anschluss wird das Gel zur Dokumentation fotografiert.

38
Auswertung
1. Beurteilen Sie, ob es im Versuch 6 gelungen ist, das Green Fluorescent Protein in E. coli-Zellen überzuexprimieren.
2. Beurteilen Sie den Grad der Anreicherung des GFP, der durch die Ionenaustausch-Chromatographie (V. 4) erreicht wurde. Ergänzen Sie dieses Resultat im Protokoll zu Versuch 4 (inklusive Gelfoto).
3. Bewerten Sie die Reinheit der zur Verfügung gestellten kommerziellen Proteine.
4. Bestimmen Sie die Molmassen der analysierten Proteine anhand des Molmasse-standards. Dazu erstellen Sie eine Eichkurve, bei der auf der x-Achse die rf-Werte und auf der y-Achse die Molmassen der Standardproteine im logarithmischen Maßstab aufgetragen werden (siehe Anhang). Über eine lineare Regression werden dann die unbekannten Molmassen ermittelt. Alternativ kann das Computerprogramm Kodak Digital Science genutzt werden.
5. Machen Sie Aussagen zu einer möglichen Quartärstruktur der analysierten Proteine.
Abb. 5-1: Protein molecular weight standard
Protein Source
-Galactosidase E. coli
RSA Bovine plasma
Ovalbumin
Chicken egg white
Lactate dehydrogenase Porcine muscle
REase Bsp98I E. coli
-Lactoglobulin Bovine milk
Lysozyme Chicken egg white

39
Versuch 6
Produktion des GREEN FLUORESCENT PROTEIN (GFP) in Escherichia coli
Einleitung Transformation
Gene sind definiert als DNA-Regionen, deren Expression eine bestimmte Eigenschaft in einem Organismus hervorbringt. Die Ausprägung dieser Eigenschaft wird vermittelt durch das Protein, für welches dieses Gen codiert. Genetische Manipulation (genetic engineering) bedeutet, die Eigenschaften eines Organismus durch die Einführung von genetischem Material zu verändern; der eigentliche Vorgang wird Transformation genannt. In diesem Versuch soll ein Fremdgen in E. coli eingeführt werden, das zu einer sichtbaren Veränderung des Phänotyps der Bakterien führt, und zwar auf dem "normalen" Weg der Gen-Expression. Als Vektoren für die Übertragung von Fremdgenen in Organismen nutzt man meist Plasmide. Diese sind relativ kleine zirkuläre, selbstreplizierende DNA-Moleküle mit speziellen Eigenschaften und können als Gen-Transporter (Vektoren) für die Transformation genutzt werden, wenn ein Fremdgen integriert wurde. Um zu gewährleisten, dass eine genügend große Zahl von Zellen ein Plasmid aufnimmt, müssen diese zunächst für den gewählten Übertragungsmechanismus kompetent gemacht werden. Im vorliegenden Versuch erfolgt die Übertragung der Plasmide nach vorheriger Behandlung der Zellen mit CaCl2 und anschließendem Hitzeschock (chemisch kompetente Zellen). Das Fremdgen codiert für ein grün fluoreszierendes Protein (GFP), das ursprünglich aus der biolumineszenten Qualle Aequorea victoria stammt. Im Versuch wird mit dem Vektor pGLO (BioRad) gearbeitet, der das Gen für das GFP bereits enthält. Der Vektor (Abb. 6-1) trägt neben dem Replikations-ursprung (ori) zusätzlich ein Ampicillin-Resistenzgen (bla), welches die Selektion rekombinanter Zellen auf Ampicillin-haltigem Agar ermöglicht.
Außerdem enthält das Plasmid ein Genregulationssystem, über das die Expression des Fremdgens in den transformierten Zellen an- und abgeschaltet werden kann. Das Regulationssystem stammt in diesem Fall aus dem Arabinose-Operon (Abb. 6-2), das die Zellen des Wildtyps befähigt, auf das Nährstoffangebot der Umwelt zu reagieren. Befindet sich Arabinose im Nährmedium, werden die zum Abbau dieser Kohlenstoffquelle erforderlichen Enzyme über die zugehörigen Strukturgene (araB, araA und araD) induziert, wobei die Transkription über den gemeinsamen Promotor (pBAD) kontrolliert wird. In entgegegesetzter Richtung zu den Strukturgenen wird die Sequenz des Regulatorproteins araC abgelesen. In Abwesenheit von Arabinose bindet dieses Protein an den Arabinose-Operator und verhindert damit die Transkription der Strukturgene. Bei Anwesenheit von Arabinose bildet sich ein Komplex aus araC und Arabinose, der die Bindung der RNA-Polymerase an den Promotor (pBAD) begünstigt und die Transkription der Strukturgene initiiert.
Auf dem verwendeten Vektor sind die Strukturgene des Arabinose-Operons durch das Gen des GFP ersetzt, so dass dessen Produktion durch Zusatz oder Weglassen von Arabinose im Medium gesteuert werden kann (Abb. 6-3).

40
Abb. 6-1: Karte des Vektors pGLO (5371 Bp) mit den wichtigsten funktionellen Elementen: ori, bakterieller Replikationsursprung; bla, Ampicillinresistenz; araC, Arabinose-induzierbarer Promotor; GFP, für GFP codierende Region.
Abb. 6-2: Aufbau des Arabinose-Operons
Abb. 6-3: GFP-Expression, gesteuert vom ara-Promotor
Nachweis und Isolation von GFP
Der Nachweis von GFP geschieht über seine Eigenschaft, bei Anregung mit Licht der Wellenlänge 396 bzw. 475 nm grün zu leuchten (Emissionswellenlänge 509 nm). Aus diesem Grund hat das Protein vielfältige Anwendungen in der Molekularbiologie gefunden. Der Fluorophor wird bei der Faltung des Proteins autokatalytisch aus der Aminosäuresequenz Ser65-Tyr66-Gly67 durch Zyklisierung und Oxidation gebildet und ist somit kovalent in der Proteinstruktur verankert. Das GFP ist ein monomeres Protein mit fassförmiger Struktur und besteht aus 238 aa (MW: 26,9 kDa).
Nach Überexpression des Proteins in E. coli werden die Zellen durch Ultraschall aufgeschlossen und das GFP über eine Ionenaustausch-Chromatographie gereinigt (siehe V4).

41
Material
Ampicillin-Stammlösung 50 mg/ml H2O (kalt sterilisiert)
Arabinose Stammlösung 20 % in H2O (kalt sterilisiert)
Transformationslösung 50 mM CaCl2 in 10 mM Tris/HCl-Puffer pH 7,4
Plasmidlösung 1 5,2 ng/µl pGLO in TE-Puffer
Plasmidlösung 2 15,6 ng/µl pGLO in TE-Puffer
Phosphatpuffer ( 50 ml) 50 mM pH 8,0 (nach Sörensen)
LB- Medium 10 g Trypton, 10 g NaCl, 5 g Hefeextrakt in aqua dest. lösen, auf 1 l auffüllen und den pH-Wert mit NaOH auf 7,5 einstellen
LBA-Medium Zusatz von Ampicillin zum sterilen LB-Medium (Endkonzentration: 100 mg/ l)
LBA-Ara-Medium Zusatz von Arabinose zum LBA-Medium (Endkonzentration: 1 %)
Festmedien (Agarplatten) 1,5 % (w/v) Agar im entsprechenden Medium
Durchführung
Herstellung chemisch kompetenter E. coli DH5α-Zellen
Am Vortag 3 ml steriles LB-Medium mit einer vollen Impföse von E .coli DH5α animpfen und über Nacht bei 37 °C und 200 rpm schütteln
300 µl der Kultur in 5 ml frisches LB-Medium überführen und ca. 2 h unter gleichen Bedingungen weiter kultivieren (OD600 ca.0,6 -1,0)
Nacheinander dreimal 1,4 ml Bakterienkultur im selben 2 ml-Eppendorf-Gefäß abzentrifugieren (5 min, 6000 rpm, 4 °C) und Überstände verwerfen
Pellet zweimal in 1,5 ml eiskalter, steriler Transformationslösung resuspendieren, erneut zentrifugieren (siehe oben) und den Überstand verwerfen
1 ml eiskalte Transformationslösung zusetzen und Zellen resuspendieren.
Je 250 µl Zellsuspension in 3 Probengefäße pipettieren und gemäß folgender Tabelle beschriften:
Probe Plasmidmenge pGLO Bezeichnung
1 5,2 ng/µl + pGLO 1 2 15,6 ng/µl + pGLO 2 3 -- - pGLO

42
Transformation der chemisch kompetenten E. coli DH5α
Probe 1: 10 µl Plasmidlösung 1 zusetzen und gut mischen Probe 2: 10 µl Plasmidlösung 2 zusetzen und gut mischen Probe 3: 10 µl Transformationslösung zusetzen (= Kontrolle ohne Plasmid) alle Gefäße auf Eis inkubieren (10 min) 50 Sekunden Hitzeschock bei 42 °C (Thermomixer ohne Schütteln) 2 min auf Eis inkubieren je 500 µl LB-Medium zugeben und die Ansätze 30 min
bei 37 °C schüttelnd inkubieren (Thermomixer; 800 rpm) Nachweis der Transformation und Bestimmung der Transformationsrate
Während der Inkubation der Transformationsansätze werden die benötigten Agarplatten (s. Tabelle) auf dem Boden entsprechend beschriftet
Nach der Kultivierung werden die transformierten Zellen durch Fingerschnipsen bzw. vorsichtiges (!) vortexen homogen suspendiert und je 100 µl der Zellsuspension auf entsprechenden Agarplatten mit Hilfe von kleinen Glaskügelchen gleichmäßig auf der Platte verteilt (siehe Tabelle) und über Nacht bei 37 °C inkubiert
Agarplatte Transformationsansatz LB -pGLO (3) LBA -pGLO (3) LBA +pGLO 1 LBA +pGLO 2 LBA-Ara +pGLO 1 LBA-Ara +pGLO 2
LBA = LB + Ampicillin Datum und Gruppennummer nicht vergessen !
Auszählen der Kolonien bei Tageslicht und unter Licht der Wellenlänge 366 nm Berechnung der Transformationsrate:
Anzahl der Transformanden (µg Plasmid x ml kompetente Zellen)
Anzucht GFP-produzierender rekombinanter Zellen Herstellung von Vorkulturen
Jeweils eine „grüne“ Einzelkolonie auf einer der LBA+Ara-Agarplatten und eine „weiße“ Kolonie auf einer der LBA-Agarplatten wird auf dem Plattenboden mit einem Faserschreiber ringförmig markiert
Zwei 15 ml Kultivierungsröhrchen werden in geeigneter Weise (z.B. „grüne Kolonie“ und „weiße Kolonie“) beschriftet und mit jeweils 2 ml LBA-Ara-Flüssigmedium (aus den Stammlösungen hergestellt) gefüllt
Das Flüssigmedium wird mit den ausgewählten Kolonien angeimpft und bei 37 °C und 200 rpm geschüttelt (ca. 6 Stunden)
Die erhaltenen Zellsuspensionen werden hinsichtlich ihrer Färbung unter UV-Licht verglichen und danach die optische Dichte bei 600 nm bestimmt

43
Herstellung der Hauptkultur
30 ml LBA-Ara-Flüssigmedium (vorgelegt in einem 100 ml Schüttelkolben mit Schikanen) werden mit einem solchem Volumen der Vorkultur versetzt, dass sich eine Animpfdichte der Hauptkultur bei 600 nm von ca. 0,06 ergibt.
Der Kolben wird über Nacht bei 30 °C und 180 rpm geschüttelt Zellaufschluss:
Die optische Dichte der Hauptkultur wird am Folgetag bestimmt und die Zellen durch Zentrifugation geerntet (4 °C, 10 min, 4 000 rpm).
Das Pellet wird in 30 ml Phosphatpuffer resuspendiert (automatische Pipette verwenden) und unter gleichen Bedingungen erneut zentrifugiert (= Waschen der Zellen).
Das Zellpellet wird in 3,5 ml Phosphatpuffer aufgenommen.
Die Zellsuspension wird in ein 5 ml Eppendorf-Gefäß überführt, auf Eis abgekühlt und durch Ultraschall unter ständiger Eiskühlung aufgeschlossen (kleine Sonde, 40 W, Intervalle: 4 x 2 min).
Die Suspension wird auf zwei Eppendorf-Gefäße (2 ml) verteilt und 10 min bei 4 °C und 13 000 rpm zentrifugiert.
Die Überstände werden vom Pellet abpipettiert und vereinigt = Proteinrohextrakt
Pellet und Überstand werden hinsichtlich ihrer Färbung unter UV-Licht verglichen (leuchten Überstand, Zelltrümmer und/oder Inclusion Bodies? War der Aufschluss quantitativ?).
Der erhaltene Rohextrakt wird eingefroren. Er ist das Ausgangsmaterial für die Ionenaustausch-Chromatographie (Versuch 4).
Auswertung
Bewerten Sie, ob die Transformation der Zellen (Transformationsrate in pfu/µg DNA), die Synthese des rekombinanten Proteins sowie der Verlauf des Zellaufschlusses aufgrund der visuellen Analyse den Erwartungen entsprachen.

44
Literatur
C. Mülhardt Der Experimentator: Molekularbiologie/ Genomics Spektrum Akademischer Verlag Heidelberg/ Berlin, 6. Auflage 2009 ISBN 978-3-8274-2036-7 M.R. Green und J. Sambrook Molecular cloning : a laboratory manual Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press 4 th ed.; Vol. 1 - 3 ISBN 978-1-936113-42-2 (Paperback) bzw. -5 (gebunden) T.A. Brown Gentechnologie für Einsteiger Spektrum Akademischer Verlag Heidelberg/ Berlin 5. Auflage 2007 ISBN 3-8274-1830-5 H. Bisswanger Enzymkinetik-Theorie und Methoden Wiley-VCH, 3. Auflage 2005 ISBN 978-3527300969 H.-P. Kleber, D. Schlee und W. Schöpp Biochemisches Praktikum Gustav Fischer Jena 5. überarbeitete Auflage 1997 ISBN 3-437-35020-X H. Rehm Der Experimentator: Proteinchemie/Proteomics Gustav Fischer Stuttgart, 6. Auflage 2009 ISBN 3-437-25750-1 A. Pingoud und C. Urbanke Arbeitsmethoden der Biochemie Walter de Gruyter Berlin/ New York 1997 ISBN 3 11 014696 7 (oder neuere englische Ausgaben) E. Keil und H. Fiedler Klinische Chemie systematisch UNI-MED Bremen 2000 ISBN 3- 89599-128-7 G. Pindur und U. Pindur Klinische Chemie und serologische Laboratoriumsdiagnostik Wissenschaftliche Verlagsgesellschaft mbH Stuttgart 1991 ISBN 3-8047-1153-7 D. Voet, J.G. Voet und C.W. Pratt Lehrbuch der Biochemie Wiley-VCH Weinheim 2002


















