
Script 19 20 - Ruhr University Bochum · 2019-09-30 · F-Praktikum für Synthesechemie,...
160
F-PRAKTIKUM FÜR SYNTHESECHEMIE WiSe 2019/20 Prof. Dr. Viktoria Däschlein-Gessner und Prof. Dr. Nils Metzler-Nolte Lehrstühle für Anorganische Chemie I und II Ruhr-Universität Bochum
Transcript of Script 19 20 - Ruhr University Bochum · 2019-09-30 · F-Praktikum für Synthesechemie,...

F-PRAKTIKUM
FÜR SYNTHESECHEMIE
WiSe 2019/20
Prof. Dr. Viktoria Däschlein-Gessner
und Prof. Dr. Nils Metzler-Nolte
Lehrstühle für Anorganische Chemie I und II
Ruhr-Universität Bochum

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
1

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
2
Inhaltsverzeichnis Praktikumsordnung
1.1 Konzept 4
1.2 Aufbau des F-Praktikums für Synthesechemie 6
Sicherheit im Labor 10
2.1 Betriebsanweisung 12
2.2 Vor dem Praktikumsbeginn / Sicherheitscheck 18
Präparate
3.1 Magnesium-Anthracen 20
3.2 Bis[bis(trimethylsilyl)-amido]zinn) 24
3.3 Dodecamethylcyclohexasilan 28
3.4 Trimethylammonium-tetradecahydroundecaborat 34
3.5 Natrium Tetrakis[(3,5-Bis(trifluoromethyl)phenyl]borat (NaBArF) 40
3.6 1,1’-Diazidoferrocen 46
3.7 1-Butyl-3-methylimidazolium-cuprat 52
3.8 Triphenylphosphoniumacetonitrilylid 58
3.9 1,3-di-tert-butyl-imidazolin-2-ylidene-[bis-(trimethylsilyl)amido] Sil-
ber(I)
66
3.10 Kupfer benzol-1,3,5-tricarboxylat 72
3.11 1-(4-Iodphenyl)-3-methyl-1H-imidazol-3-ium)-(µ5-pentamethylcyclo-
pentadienyl)-iridiumchlorid
80
3.12 Tris-(2-ammonium-ethyl)-phosphin Trichlorid 88
3.13 Tricyclohexylphosphanverbindungen 94
3.14 Pentlandit 100
3.15 Rutheniumkomplexe 114
3.16 2,2’-Selenobis(ethan-1-tosylamid) &
2,2’ Tellurobis(ethan-1-tosylamid)
120
3.17 Titan-organische Verbindungen 126

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
3
3.18 Holomycin und ROY 134
3.19 Trimethylsilyl(triphenylphosphonium-benzylid) 146
Anlage
Musterprotokoll 152

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
4
Konzept
Vorrangiges Ziel des Synthese-Praktikums ist das Vermitteln von modernen Synthese-
und Analysenmethoden in der Anorganischen und Organischen Molekülchemie; wobei
die traditionellen Grenzen zwischen der AC und OC aufgelöst sind.
Nach Ende dieses Moduls soll der Student/die Studentin in der Lage sein, mehrstufige
Synthesen (anorganische, organische, metallorganische und bioorganische Präpa-
rate) eigenständig durchzuführen und mechanistisch zu interpretieren. Dabei soll vor
allem ein sicherer Umgang mit der Vakuumtechnik, Schutzgastechnik, Trocknung von
Lösemitteln sowie die Anwendung von spektroskopischen Methoden zur Strukturauf-
klärung (IR-, UV-, NMR-Spektroskopie, Massenspektrometrie), Chromatographie und
Diffraktometrie (Pulver und Einkristall) erzielt werden.
Die Techniken und Fertigkeiten werden in ihrer Vielfalt anhand von didaktischen und
forschungsrelevanten Präparaten erworben und vertieft.
Das F-Praktikum für Synthesechemie soll den Übergang von den erworbenen Fertig-
keiten und Kenntnissen in den präparativen Grundpraktika hin zum selbständigen Ar-
beiten in wissenschaftlichen Projekten ermöglichen. Hierbei ist ein wichtiger Aspekt
auch das selbstständige Recherchieren von Synthesevorschriften und sicherheitstech-
nisch relevanter Daten zu erlernen.
Voraussetzung zum Beginn des Praktikums ist der Besuch des vorbereitenden Semi-
nars, das zu Beginn des Wintersemesters eine Einführung in die wesentlichen fortge-
schrittenen Arbeitstechniken und Charakterisierungsmethoden gibt. Zusätzlich muss
vor jedem Versuch ein versuchsspezifisches Testat bestanden werden, bei dem die
theoretischen und sicherheitstechnischen Hintergründe des Versuches nachzuweisen
sind.
Die unten aufgeführten Techniken und Charakterisierungsverfahren werden in ih-
rer Vielfalt anhand von didaktisch und forschungsrelevanten Präparaten aus folgen-
dem Themenverzeichnis erworben und vertieft.
• Organometallchemie der Haupt-und Nebengruppenelemente
• Mehrstufige Synthesen von Liganden
• Komplexchemie
• Clusterverbindungen

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
5
• Katalyseverfahren
• Mehrstufige Synthese von komplexen Molekülen und Biomolekülen
Techniken:
• Vakuumtechnik
• Schutzgastechnik (Schlenktechnik, Substanztransfer in einer Glovebox, Löse-
mitteltransfer unter Schutzgas, Filtration unter Schutzgas, Abfüllen von NMR-
Proben unter Schutzgas)
• Aufreinigungstechniken: Säulenchromatographie, Umkristallisieren, Sublima-
tion, fraktionierte Destillation und fraktionierte Kondensation
• Umgang mit Gefahrstoffen:
selbstentzündliche Reagenzien
Transfer mit Spritze und Septum
Umgang mit toxischen / carcinogenen Substanzen
Umgang mit geruchsbelästigenden Stoffen
Im Mikromaßstab: Umgang mit potentiell explosiven Substanzen
Charakterisierungsmethoden:
• NMR in Lösung und im Festkörper
• Einkristall- und Pulverdiffraktometrie
• IR, UV/VIS
• Dünnschichtchromatographie
• Gaschromatographie
• Massenspektrometrie
• GC/MS-Kopplung

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
6
Aufbau des F-Praktikums für Synthesechemie
Vorbereitungsseminar:
Theoretisches Rüstzeug für Arbeitstechniken und
spektroskopische Methoden in der anorganischen
und organischen Synthesechemie
07.10.2019 – 08.10.2019
Anorganische Synthesechemie :
Kurs I: 10.10.2019 – 12.11.2019
Kurs II: 14.11.2019 – 17.12.2019
Montag 11 - 18 Uhr, Dienstag 10 - 18 Uhr
Donnerstag 13 - 18 Uhr, Freitag 13 - 18 Uhr
Deadline für alle Präparate des AC-Moduls incl. Protokolle
ist zwei Wochen nach dem jeweiligen Kurs
(Kurs I: 26.11.2019, Kurs II: 17.01.2020)
Um 17:30 Uhr sind die Versuche zu beenden, bis 18:00 Uhr müssen die Arbeitsplätze
geräumt sein.
Das Praktikum beginnt am Montag, den 07. Oktober 2019 mit dem Vorbereitungs-
seminar. Die Teilnahme an den Seminaren ist Pflicht. Der erste Versuchstermin wird
der 10. Oktober 2019 sein.
Die Versuche müssen während der angegebenen Praktikumszeit durchgeführt und die
benutzten Geräte und Apparaturen danach in sauberem und unbeschädigtem Zustand
wieder in die zum Versuch gehörenden Schränke eingeräumt werden.
Tische, Abzüge, Ausgüsse, Schränke und Geräte sind stets sauber zu halten. Sau-
berkeit ist eine unerlässliche Voraussetzung für erfolgreiches chemisches Arbeiten.
Zum Reinigen sollte ein saugfähiges Wischtuch bereitgehalten werden.
Vor dem Praktikumsbeginn muss der Inhalt des Schrankes an Hand der beiliegenden
Liste auf Vollständigkeit überprüft werden. Der jeweilige Benutzer haftet für den Inhalt

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
7
des Schrankes. Bei der Benutzung allgemein zugänglicher Geräte wie Öfen, Zentrifu-
gen usw. trägt man sich mit Namen, Datum, Zeit und ggfs. mit den einzustellenden
Temperaturen in die beiliegenden Listen ein.
Das Antestat zu einem Versuch wird nur nach einem Sicherheitskolloquium beim As-
sistenten erteilt. Es werden Kenntnisse über den Versuchsaufbau, den Versuchsab-
lauf, die grundlegende Theorie und Kenntnisse über die beim Versuch eingesetzten
Stoffe mit R+S-Sätzen, über mögliche Gefahrenquellen und Schutzmaßnahmen ver-
langt.
Das Abtestat für den jeweiligen Versuch wird vom betreuenden Assistenten nach Vor-
lage des Laborjournals und Abgabe der angefertigten Präparate erteilt.
Während der Durchführung eines Experiments trägt man alle Rechnungen und Anga-
ben wie Einsatzmengen, Beobachtungen über den Reaktionsablauf, physikalische Pa-
rameter, Skizzierung der Apparatur mit Angabe von Schliffgrößen und -arten in über-
sichtlicher Weise in ein Laborjournal (gebundenes Schreibheft) ein.
Nach Beendigung des Experiments wird aus der Versuchsanleitung und dem La-
borjournal eigenständig in leserlicher Handschrift ein Versuchsprotokoll selbständig
angefertigt. Das Versuchsprotokoll umfasst
1. den Testatbogen als Deckblatt
2. eine kurze theoretische Einführung
3. eine Versuchsbeschreibung mit Zeichnung der verwendeten Apparaturen
4. die Auswertung der Messdaten mit Fehlerbetrachtung
5. die Zusammenfassung
6. die Literaturzusammenstellung
Erfolgreicher Abschluss eines Präparats beinhaltet:
• Sicherheitsgespräch vor dem Versuch
• Durchführung des Versuchs
• Ausbeute: mindestens 50 % der Literaturangabe
• Skizzieren der Versuchsdurchführung im Laborjournal
• Eigenständige Anfertigung eines eigenständigen Versuchsprotokolls in leserlicher
Handschrift

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
8
Das ausgearbeitete Versuchsprotokoll ist eine Woche nach Beendigung des Versuchs
(ggfs. unter Nachreichung der Auswertung von Messergebnissen) vorzulegen. Even-
tuell notwendige Korrekturen der Protokolle sind spätestens eine Woche nach
Beendigung des präparativen anorganischen bzw. organischen Teilmoduls vor-
zulegen.
Ist dies nicht der Fall, gilt der Versuch als nicht durchgeführt und muss zu einem spä-
teren Zeitpunkt vollständig wiederholt werden.
Das Praktikum gilt als erfolgreich abgeschlossen, wenn Präparate, Protokolle und
Schränke ordnungsgemäß abgegeben worden sind.
Alle weiteren Termine (Antestate, Abgabe der Arbeitsplätze etc.) entnehmen Sie bitte
den Aushängen am Schwarzen Brett des Praktikums und den Ankündigungen auf der
Homepage des Praktikums https://www.rub.de/ac1/acf.
Das Praktikum gilt als erfolgreich abgeschlossen, wenn:
• das Vorbereitungsseminar,
• alle Präparate,
• das Abschlussgespräch
erfolgreich abgeschlossen sind.
Ausschluss aus dem Praktikum
Ein Ausschluss aus dem Praktikum findet automatisch statt bei grob fahrlässi-
gem Verhalten.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
9

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
10
Sicherheit im Labor
Verhalten bei Notfällen
Leitwarte Tag und Nacht besetzt: Tel. 23333
Feueralarm:
In den Fluren befinden sich automatische Feuermelder. Das Ansprechen eines
I-Melders wird durch Blinken angezeigt.
Feuermelder befinden sich gegenüber den Aufzügen. Ein Alarm kann auch über die
Leitwarte, Tel. 23333, ausgelöst werden.
Bei Feueralarm muss mit Ausfall der Stromversorgung und mit Ausfall der Lüftung in
den Laboren gerechnet werden!
Die Aufzüge dürfen deshalb nicht benutzt werden! Die Lüftung wird bei Alarm umge-
schaltet, alle Abzüge können abgeschaltet werden. Das Kerntreppenhaus mit den
Aufzügen wird mit Überdruck betrieben und damit rauchfrei gehalten.
Das Gebäude bei Gefahr nur über diese Treppenhäuser verlassen, Aufzüge dürfen
nicht benutzt werden.
Bei Gasaustritt von ätzenden oder stinkenden Verbindungen oder Verschütten von
Chemikalien in Laboratorien ist das Labor zu räumen, bis die Lüftung die Dämpfe ab-
gesaugt hat. Die Fenster dürfen nicht aufgerissen werden, weil der Unterdruck im
Haus dadurch aufgehoben wird und die Dämpfe in die Flure gedrückt werden.
Die Dämpfe oder Gase können nicht nur gefährlich für die Mitarbeiter werden, sondern
auch an den I-Meldern einen Fehlalarm auslösen.
Hausalarm:
Bei Hausalarm (Alarmglocke oder Lautsprecherdurchsage) muss das Gebäude ge-
räumt werden.
Wenn keine unmittelbare Gefahr besteht, alle Apparaturen und Geräte abschalten und
Mäntel und Schlüssel etc. mitnehmen, da das Gebäude für längere Zeit geschlossen
bleiben könnte.
Behinderten beim schnellen Verlassen des Gebäudes behilflich sein!

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
11
Verletzungen:
Über die Leitwarte Tel. 23333 Krankentransport mit oder ohne Arzt anfordern. Ret-
tungsdienst im Norden und im Süden erwarten und einweisen.
Keinen Transport von Schwerverletzten selbst durchführen.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
12
BETRIEBSANWEISUNG
FÜR DAS F-PRAKTIKUM FÜR
SYNTHESECHEMIE
1.) ALLGEMEINE BETRIEBLICHE ANORDNUNGEN
1.1) Die Laborangehörigen haben im Labor die zur Verfügung gestellte Schutzaus-
rüstung zu tragen: stets einen geschlossenen Laborkittel und eine Schutzbrille
bzw. Überbrille, bei Bedarf sonstige Schutzausrüstung. Es ist festes, geschlos-
senes und trittsicheres Schuhwerk zu tragen.
1.2) Laborkittel und Straßenkleidung müssen getrennt aufbewahrt werden.
Für Straßenkleidung sind die abschließbaren Spinde zu benutzen.
1.3) Schreibarbeiten, die nicht den Aufenthalt im Labor erfordern, sind an den dafür
vorgesehenen Schreibarbeitsplätzen außerhalb des Labors durchzuführen.
1.4) Notausgänge, Fluchtwege, Durchgänge, Treppen sowie Zugänge zu Feuerlö-
schern, Notbrausen und Erste-Hilfe-Einrichtungen dürfen nicht zugestellt wer-
den.
1.5) CO2-Löscher, Brandschutzdecken befinden sich im Hauptlabor und auf den Flu-
ren. Augenwaschleitungen an den Spülbecken jeder Laborbox, Feuermelder
gegenüber den Aufzügen.
1.6) Im gesamten Laborbereich und unmittelbar an Laboratorien angrenzenden Flu-
ren besteht Trink-, Ess-, Schmink-, und Rauchverbot. Zum Essen und Trinken
sind die dafür vorgesehenen Räume zu benutzen.
1.7) Der Arbeitsplatz muss ordentlich und sauber gehalten werden. Am Arbeitsplatz
dürfen nur die tatsächlich benötigten Gefahrstoffe und Geräte bereitgehalten
werden.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
13
1.8) Vor Beginn neuer Arbeiten sind unter Verantwortung der Vorgesetzten die Ge-
fährdungen zu ermitteln und die Schutzmaßnahmen festzulegen. Die festgeleg-
ten Schutzmaßnahmen sind einzuhalten. Des Weiteren sind die R- und S-Sätze
aus der Chemikaliendatei und die stoffbezogenen Betriebsanweisungen zu be-
achten.
1.9) Arbeiten mit Gefahrstoffen dürfen nur bei volllaufender Belüftungsanlage durch-
geführt werden. Die Wirksamkeit der Abzüge ist mit den zur Verfügung gestell-
ten Windrädchen zu kontrollieren.
1.10) Bei Ausfall und Störung der Belüftungsanlage darf mit Gefahrstoffen nicht gear-
beitet werden und ist der Aufenthalt in Laboratorien, in denen Gefahrstoffe auf-
bewahrt werden, zu vermeiden.
1.11) Bei Arbeiten in Laboratorien mit Gefahrstoffen muss stets eine weitere, fachlich
qualifizierte Person anwesend sein. Dies ist vom Dienstvorgesetzten zu organi-
sieren. Soweit solche Arbeiten außerhalb der Dienstzeit durchgeführt werden,
bedürfen sie der Genehmigung.
1.12) Glasapparaturen, bei denen die Gefahr des Zerknalls durch Überdruck oder Va-
kuum besteht, sind durch die zur Verfügung gestellten Schutzscheiben, Draht-
käfige oder Folien zu sichern.
1.13) Der Transport von Gasflaschen darf nur mit aufgeschraubter Schutzkappe auf
einem Gasflaschenwagen mit angelegter Sicherheitskette erfolgen. Nach Ar-
beitsschluss müssen von Druckgasflaschen mit aggressiven oder giftigen Ga-
sen Druckminderer und Reduzierventile entfernt und durch Blindstopfen ersetzt
werden. Wenn vorhanden, müssen die Gasflaschen nach Gebrauch unverzüg-
lich in die Sicherheitsschränke zurückgestellt werden.
1.14) Über Nacht laufende Versuche müssen mit der im Abzug vorhandenen Über-
wachungsanlage gesichert werden. Die Anlage ist immer vor Inbetriebnahme
auf ihre Funktionstüchtigkeit zu überprüfen und muss im Bedarfsfall Strom- und
Wasserzufuhr endgültig abschalten.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
14
1.15) Elektrogeräte müssen eine gültige Prüfplakette der RUB-Elektrowerkstatt besit-
zen. Dies ist vor Verwendung der Geräte durch den Mitarbeiter zu prüfen.
1.16) Der Transport von Gefahrstoffen darf nur mit den geeigneten und zur Verfügung
gestellten Griffrollern erfolgen.
1.17) Defekte Geräte dürfen nur gereinigt in die Werkstätten gebracht werden.
1.18) Vor Inspektion durch die Pumpenwerkstatt muss bei allen Pumpen das Öl ab-
gelassen und als verunreinigtes Altöl unter dem Abfallschlüssel 54114 in den
braunen 12l-Behältern entsorgt werden.
1.19) Chemikalien und Lösemittel müssen ordnungsgemäß entsorgt werden. Leere
Chemikaliengefäße, die als Hausmüll entsorgt werden, müssen gereinigt und
die Etiketten sowie Verschlüsse entfernt werden. Glasbruch, sowie Spritzen und
Kanülen dürfen nicht in die Papierkörbe gelangen.
1.20) Auf dem Boden, besonders im Putzbereich, dürfen keine Chemikalien abgestellt
werden.
1.21) Am Arbeitsplatz dürfen Chemikalien nur in Flaschen mit max. 0.5 Liter Inhalt
aufbewahrt und gefährliche Flüssigkeiten nicht über Kopfhöhe abgestellt wer-
den.
1.22) Alle offenen Versuche müssen unter dem Abzug ausgeführt werden.
1.23) In Abzügen, in denen gearbeitet wird, dürfen keine Chemikalien aufbewahrt
werden.
1.24) Chemikalien dürfen nur an den dafür vorgesehenen Plätzen bereitgestellt wer-
den.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
15
2.) SCHUTZ GEGEN GESUNDHEITSSCHÄDLICHE STOFFE
2.1) Explosionsgefahr
Eine Reihe chemischer Substanzen neigt aufgrund ihrer Konstitution zum spontanen
Zerknall (z. B. Perchlorate, Peroxide, bestimmte Nitroverbindungen). Diese Stoffe dür-
fen nur in den dafür vorgesehenen Abzügen, hinter Schutzscheiben oder Drahtkäfigen
mit größter Vorsicht gehandhabt werden. Zusätzlich zur Schutzbrille sind bei diesen
Arbeiten Gesichtsschutz und Handschuhe zu tragen.
2.2) Brennbarkeit
Wegen ihrer explosionsfähigen Dampf-/Luft-Gemische dürfen Lösemittel nur in Kolben
mit aufgesetztem Kühler erhitzt werden. Wird mit selbstentzündlichen oder besonders
feuergefährlichen Stoffen oder mit großen Mengen umgegangen, müssen die dafür
vorgesehenen Feuerlöschmittel griffbereit gehalten werden.
2.3) Giftige, ätzende oder reizende Stoffe
Bei giftigen, ätzenden oder reizenden Stoffen den stoffbezogenen Betriebsanweisun-
gen folgen und wenn nötig, Vollgesichtsschutz, Gummischürze tragen. Müssen zur
Behebung von Stör- und Notfällen Atemschutzmasken mit Filter eingesetzt werden,
sind mindestens 2 weitere eingewiesene Personen erforderlich. Dies ist vom Vorge-
setzten zu organisieren.
3.) VERHALTEN BEI UNFÄLLEN
(siehe auch Merkblatt: Erste Hilfe bei Unfällen sowie Notruf-Übersicht der RUB)
Transport durch Krankenwagen über die Leitwarte anfordern. Bei der Leitwarte genaue
Ortsangabe machen. Ortskundige Personen auffordern, Rettungspersonal einzuwei-
sen.
Rettungswagen sowohl am Gebäudeeingang Nord als auch Süd erwarten.
Bei Unfällen mit Chemikalien auf Begleitzettel Angaben über die Chemikalien sowie
Telefonnummer für Rückfragen mitgeben.
3.1) Nach Einatmen giftiger Gase und Dämpfe - auch bei Verdacht - Betroffene aus
der Gefahrenzone bringen.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
16
3.2) Bei Verätzungen benetzte Kleidung sofort entfernen. Benetzte Hautstellen mit viel
Wasser spülen.
3.3) Chemikalien, die ins Auge gelangt sind, sofort mit viel Wasser aus der Augen-
waschflasche entfernen.
3.4) Bei Verbrennungen Kleidung mit Notdusche oder Feuerlöschdecke ablöschen.
Kleinere Brandwunden mit kaltem Wasser ausgiebig spülen.
3.5) Bei allen Hilfeleistungen auf die eigene Sicherheit achten.
4.) VERHALTEN BEI BRÄNDEN UND GEFAHREN
4.1) Brände sofort melden
- durch Betätigen der Feuermelder, anschließend
- Notruf an die Leitwarte - Tel. 23333 - mit Angabe:
--- wo es brennt
--- was brennt
--- sind Personen akut gefährdet?
4.2) Andere Personen warnen, gefährdete Personen in Sicherheit bringen.
4.3) Gekennzeichnete Fluchtwege benutzen, keinen Aufzug benutzen.
4.4) Möglichst Strom und Gas abschalten (Not-Aus-Schalter für Laborstrom).
4.5) Feuerwehr und Rettungsdienste einweisen.
4.6) Bei Ertönen des Hausalarms: Gebäude räumen und möglichst persönliche Ge-
genstände mitnehmen und wenn möglich, Apparaturen abstellen. Rettung von
Menschen geht vor Sachschutz.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
17
5.) HINWEISE
5.1) Tragen für Verletzte befinden sich auf den Gängen jeder Etage auf der Westseite.
5.2.) Feuermelder befinden sich in den Treppenhäusern gegenüber den Fahrstühlen.
5.3) Strom-Not-Aus-Schalter in den Laboren und auf den Fluren.
5.4) Pulverlöscher sind in den Treppenhäusern angebracht.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
18
Vor dem Praktikumsbeginn
Bitte prüfen Sie!
Können Sie die folgenden Fragen beantworten?
Wo sind
• Notausgang, Nottreppe, allgemeine Fluchtwege?
• Alarmanlagen, Telefon, Notruf, Assistentenzimmer?
• Feuerlöscher, Feuermelder, Atemschutzmasken und –filter?
• Notduschen, Löschdecken?
• Erste-Hilfe-Schränke, Möglichkeit zur Augenspülung?
• Sanitätsraum, Krankentragen?
Wissen Sie zum Beispiel,
• wie und/oder von wem die Medien (Gas, Wasser, Strom) abgeschaltet werden
können?
• dass bei einem Brand kein Aufzug benutzt werden darf?
• dass Feuerlöscher nach jeder Benutzung frisch gefüllt werden müssen?
• dass Druckgasflaschen stets gegen Umfallen gesichert sein müssen?
• was „Selbstschutz“ bedeutet?
• was bei schweren Unfällen zu tun ist?
• welche Chemikalien, mit denen Sie evtl. arbeiten müssen, giftig, explosionsge-
fährlich oder leicht entzündlich sind?
• wo Sicherheitsinformationen zu finden sind?

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
19

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
20
Magnesium-Anthracen
(MgA)
Literatur:
Bogdanović, B.; Liao, S.; Schlichte, K.; Westeppe, U. Synthetic Methods of Organo-
metallic and Inorganic Chemistry, Vol. 1 (Eds.: Herrmann, W. A. Salzer, A.) Thieme,
Stuttgart, 1996. 43.
THF
Mg
+ Mg
O
OO
BrCH2CH2Br
THF

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
21
Chemikalienliste:
Name der Chemikalie Menge
Anthracen 2,5 g (0.014 mol)
Magnesium Pulver – silbrig glänzend - 0.3 g (0.012 mol)
Dibromethan 0.09 g (0.0625 mL, 0.00075 mol)
Tetrahydrofuran 30 mL
Einleitung:
Obwohl die Herstellung des in Tetrahydrofuran löslichem mit Anthracen komplexierten
aktivem Magnesium (MgA) schon seit 1965 bekannt war, entwickelte sich die Magne-
sium-Anthracen-Forschung erst ab 1980. Ausgelöst wurde diese Entwicklung durch
die Verwendung von MgA in einem Katalysatorsystem für die Hydrierung von Magne-
sium unter milden Bedingungen.
Heutzutage werden Magnesium-Anthracen-Verbindungen sowohl aufgrund ihrer viel-
seitigen Synthesepotentiale als auch als wertvolle Katalysatoren für Reaktionen mit
metallischem Magnesium eingesetzt. Elementares Magnesium kann, bei Anwesenheit
von Wasserstoff und katalytischen Mengen von Anthracen, zu hochreaktivem Magne-
siumhydrid überführt werden. Diese Hydride stellen ausgezeichnete Ausgangsverbin-
dungen für die Herstellung von Übergangsmetallverbindungen dar und können auch
für die Bildung von Grignardverbindungen bei extrem milden Bildungen verwendet
werden.
Das aktive MgH2-Mg-System, zugänglich via Phasen-Transfer-Katalyse von Magne-
sium, ist ein hervorragender Wasserstoffspeicher (Metallhydridspeicher) und zeichnet
sich durch hohe massenbezogene Wasserstoffspeicherkapazität und Wärmespeicher-
dichte aus.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit luftempfindlichen Verbindungen
• Lösemitteltransfer unter Inertgasbedingungen

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
22
• Umgang mit reaktiven Organometall-Addukt-Substanzen
• Durchführung heterogener Reaktionen mit Metallen
Charakterisierungsmethoden:
• NMR-Spektroskopie in Lösung
• NMR-Spektroskopie im Festkörper
• IR, UV/Vis
Forschungsrelevante Thematik:
• Magnesium-organische Chemie
• Grignardverbindungen
• Organo-Metallhydride
• Metallhydride als Wasserstoffspeicher
Literatur:
Ramsden, H. E. US. Patent 3354190, 1967; Chem. Abstr. 1968, 68, 114744.
Bogdanovic, B.; Liao, S.; Sikorsky, P.; Spliethoff, B. Angew. Chem. Int. Ed. Engl. 1980,
19, 818.
Bönnemann, H.; Bogdanović, B.; Brinkmann, R.; Spleithoff, B.; He, D.-W. J. Organo-
met. Chem. 1993, 451, 23.
Bartmann, E.; Bogdanović, B.; Janke, N.; Liao, S.; Schlichte, K.; Spliethoff, B.; Treber,
J.; Westeppe, U.; Wilczok, U. Chem. Ber. 1990, 123, 1517.
Bogdanović, B. Acc. Chem. Res. 1988, 21, 261.
Bogdanović, B.; Janke, N.; Kinzelmann, H.-G. Chem. Ber. 1990, 123, 1507.
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon gela-
gert. Der luft- und hydrolyseempfindliche Feststoff Magnesium-Anthracen 3THF wird
unter Inertgas gehandhabt.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
23
Versuchsdurchführung:
Synthese von aktiviertem Magnesium-Anthracen 3THF (MgA)
In einem Schlenkkolben legt man 2,5 g Anthracen (0.014 mol) und 0.3 g Magnesium
(0.012 mol) vor. Dieser Kolben wird offen für 10 min. in einen Trockenschrank (110 °C)
gelegt. Danach wird ein Rückflusskühler aufgesetzt, die ganze Apparatur mehrmals
evakuiert und mit Argon gespült. Anschließend werden trockenes THF (30 mL) und ca.
0.063 mL Dibromethan zugegeben. Die Suspension wird zum Rückfluss gerhitzt, bis
ein Farbumschlag nach dunkelgrün sichtbar ist. Dann wird nach etwa 1 – 1,5 Stunden
refluxiert und für ca. 18 Stunden bei Raumtemperatur weiter gerührt. Nach Beendigung
der Reaktion wird die Suspension über eine Fritte abgesaugt und das verbleibende
orange Pulver mit THF gewaschen und getrocknet. Zurück bleibt ein oranges luftemp-
findliches Pulver.
Literatur-Ausbeute: 4.5 g (94 %)
Kriterium: Ausbeute mindestens 50 %.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
24
(Bis[bis(trimethylsilyl)-amido]zinn)
Sn[N(SiMe3)2]2
Literatur:
Gynane, M. J. S.; Harris, D. H.; Lappert, M. F.; Power, P. P.; Riviere-Baudet, P.
J. Chem. Soc. Dalton Trans. 1977, 2004.
3 [Me3Si]2NH + 3 LiC4H9 → [LiN(SiMe3)2]3 + 3 C4H10
2 [LiN(SiMe3)2]3 + 6 Et2O → 3 [LiN(SiMe3)2 x Et2O]2
[LiN(SiMe3)2 x Et2O]2 + SnCl2 → Sn[N(SiMe3)2]2 + 2 LiCl + 2 Et2O

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
25
Chemikalienliste:
Name der Chemikalie Menge
1,6 M n-Butyllithium in Hexan 8 mL (0.0128 mol)
Hexamethyldisilazan 2.46 g (0.0153 mol)
Zinn(II)chlorid (wasserfrei) 1.2 g (0.0064 mol)
Diethylether 30 - 40 mL
Einleitung:
Ziel dieser Arbeiten ist es, ein spezielles Metallamid des Lithiums und des zweiwerti-
gen Zinns, die aktuell bedeutende Basen und Nucleophilie in der Molekülchemie sind,
herzustellen, zu reinigen und strukturell zu charakterisieren.
Mit zunehmender Elektronegativität des Metalls nimmt der kovalente Bindungsanteil
in der Koordinationsbeziehung zu. Dies wird im Fall der in diesem Versuchsteil be-
schriebenen Amide des Lithiums (überwiegend ionische Li-N Bindungen) und des
Zinns (praktisch kovalente Sn-N Bindungen) deutlich. Zinn(II)amid ist eine Molekülver-
bindung, die durch Destillation gereinigt wird, während Lithiumamid nur einen geringen
Dampfdruck besitzt. Die auffällige schöne Farbe des Zinn(II)amids, im Gegensatz zum
Lithiumamid, hat natürlich mit der besonderen elektronischen Struktur des Zinn(II)-
Zentrums zu tun: das Zinnatom besitzt nur 6 Valenzelektronen wie das Kohlenstoff-
atom im Methandiyl (Trivialname: Methylen), sodass man von einem Stannandiyl-De-
rivat spricht. Das Zinnatom ist zwar elektronisch (nach der Oktettregel) und koordinativ
ungesättigt, doch eine Aggregation wie bei dem entsprechenden Lithiumamid ist nicht
gegeben. Farbe und Assoziationsverhalten solcher Stannandiyle können durch den
elektronischen Einfluss der Liganden am Zinn so eingestellt werden, dass gelbe, rote,
blaue und schwarze Substanzen entstehen, die Lichtfrequenzen stark modulieren kön-
nen. Dies zu verstehen und zu nutzen, ist ein weltweit aktuelles Forschungsthema.
Literatur:
Harris, D. H.; Lappert, M. F. J. Chem. Soc. Chem. Commun. 1974, 895.
Podlech, J. Science of Synthesis 2003, 5, 273.
Driess, M.; Grützmacher, H. J. Angew. Chem. Int. Ed. Engl. 1996, 35, 828.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
26
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit luftempfindlichen Verbindungen
• Lösemitteltransfer unter Inertgasbedingungen
• Umgang mit reaktiven Organometall-Addukt-Substanzen
Charakterisierungsmethoden:
• NMR-Spektroskopie in Lösung
• IR
• UV/Vis
Forschungsrelevante Thematik:
• Zinn-organische Chemie
• Carbenanaloga
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon gela-
gert. Die luft- und hydrolyseempfindlichen Edukte und Produkte werden unter Inertgas
gehandhabt.
Versuchsdurchführung:
Lithium-bis(trimethylsilyl)amid Diethylether:
Ein ausgeheizter (abgewogener!!!) 100 mL Schlenkkolben mit Magnetrührstäbchen
wird mit einer 1.6 M Lösung von Butyllithium in Hexan gefüllt. Unter Argon wird via
Spritze und Septum Hexamethyldisilazan zugegeben, wobei sich die Reaktionsmi-
schung stark erwärmt (Eisbadkühlung). Nach dem Zutropfen wird noch etwa ½ Stunde
gerührt. Das Hexan wird im Unterdruck in eine externe Kühlfalle abkondensiert. Der

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
27
verbleibende Feststoff wird in Diethylether gelöst und über Nacht im Kühlschrank auf-
bewahrt. Das über Nacht auskristallisierte Produkt wird vom überstehenden Lösemittel
separiert und in einem Schlenkkolben aufbewahrt. Das Lösemittel wird bis zur Bestim-
mung der Ausbeue aufbewahrt.
Zur Charakterisierung der Substanz wird ein 1H-NMR- Spektrum angefertigt.
Literatur-Ausbeute: 2.61 g (80 %)
Kriterium: Ausbeute 1.24 g (mindestens 40 %).
Charakterisierung:
1H NMR (CD3CN): δ = 3.42 ppm (q, 8H), 1.12 ppm (t, 12H), 0.04 ppm (s, 36H).
Bis[bis(trimethylsilyl)-amido]zinn:
Lithium-bis[(trimethylsilyl)amid]-Etherat, gelöst in Diethylether, wird langsam zu SnCl2
gegeben. Hierbei setzt eine sofortige Reaktion unter Abscheidung von LiCl und Oran-
gefärbung der Lösung ein. Nachdem die Lösung 2 h gerührt worden ist, wird das Lö-
semittel im Vakuum entfernt und der verbleibende Feststoff mehrfach mit n-Hexan ex-
trahiert. Nach Entfernung der flüchtigen Komponenten im Vakuum bleibt das Rohpro-
dukt als orangegefärbter Feststoff zurück, der durch Vakuumdestillation gereinigt wer-
den muss. Zur Charakterisierung des Produkts wird ein 1H-NMR aufgenommen.
Sdp.: 84 °C/5 mbar; Smp.: 37 - 38 °C
Literatur-Ausbeute: 2.12 g (79 %)
Kriterium: Ausbeute mindestens 40 %.
Charakterisierung: 1H NMR (C6D6): δ = 0.32 ppm (s, 36H)
IR (nujol): ν = 400 cm-1

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
28
Dodecamethylcyclohexasilan
(Me2Si)6 Literatur:
Herrmann/Brauer, Synthetic Methods of Organometallic and Inorganic Chemistry,
Thieme Verlag, 1996, Vol. 2, S. 192.
Stolberg, U. G. Angew. Chem. 1963, 75, 206.
6 Me2SiCl2 + 12 Na/K → (Me2Si)6 + 12 NaCl/KCl

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
29
Chemikalienliste:
Name der Chemikalie Menge
Dimethyldichlorsilan 7.2 g (0.055 mol)
Kalium 3.7 g (0.09 mol)
Natrium 1 g (0.05 mol)
Tetrahydrofuran 45 mL
Ethanol 20 mL
Einleitung:
Wie stark sich das Element Silicium von dem leichteren gruppenhomologen Kohlen-
stoff unterscheidet, wird bei der Gegenüberstellung ihrer Dioxide deutlich: CO2 ist bei
Raumtemperatur und Normaldruck ein farbloses Gas, während SiO2 (Quarz, Sand)
bekanntlich ein polymerer Feststoff ist, der ein essentieller Bestandteil der Erdkruste
ist und auch die Quelle für elementares Silicium in der Technik darstellt.
Die drastischen Unterschiede zwischen der Silicium- und Kohlenstoffchemie wird auch
eindrucksvoll belegt durch die hohe Reaktivität der Silane, die erst herabgesetzt wird,
wenn die hydridischen H-Atome (Si+-H-) durch organische Substituenten ausgetauscht
werden. So ist Tetramethylsilan eine luftstabile Flüssigkeit. Großtechnisch erfolgt die
Knüpfung der Si-C-Bindung in der einfachen Direktsynthese aus elementarem Silicium
und Aryl/Alkylhalogeniden in Gegenwart eines Kupferkatalysators (Müller-Rochow-
Synthese). Die dadurch erhältlichen unterschiedlichen Organohalogensilane sind das
Rückgrat der industriellen Si-Chemie und der Schlüssel zu Siliconen (anorganische
Elastomere).
Aber auch in der molekularen Synthesechemie spielen Organohalogensilane als Aus-
gangsverbindungen für anspruchsvolle Substituentengruppen eine wichtige Rolle. Ste-
risch überladene Substituenten erlangen in der Chemie zusehends Bedeutung als
Hilfsmittel zur Synthese von elementorganischen Verbindungen, in welchen die Zent-
ralatome ungewöhnliche Geometrien sowie kleine Koordinationszahlen aufweisen.
Eine andere Entdeckung in der Si-Chemie war die Synthese der ersten wohlcharakte-
risierten und definierten Polysilane mit langen Si-Si Ketten und jeweils zwei organi-
schen Substituenten pro Si-Atom, Anfang der 60iger Jahre3. Man glaubte, dass die
Stabilität von Polysilanen mit zunehmender Kettenlänge drastisch abnimmt. Heute

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
30
sind Polysilane mit bis zu 40.000 Si-Atomen bekannt! Sie sind eine eminente Verbin-
dungsklasse der "hightech" Industrie und der Schlüssel zu Siliciumcarbidfasern mit
extremen Materialeigenschaften und als Photowiderstände in der Mikrochip-Herstel-
lung unentbehrlich.
Literatur:
Ando, W.; Hojo, F.; Sekigawa, S.; Nakayama, N.; Shimizu, T. Organometallics, 1992,
11, 1009.
Ando, W.; Nakayama, N.; Kabe, Y.; Shimizu, T. Tetrahedron Letters, 1990, 31, 3597.
Pang, Y.; Schneider, A.; Barton, T. J. J. Am. Chem. Soc. 1992, 114, 4920.
Hojo, F.; Sekigawa, S.; Nakayama, N.; Shimizu, T.; Ando, W. Organometallics, 1993,
12, 803.
Sekiguchi, A.; Kinjo, R.; Ichinoha, M. Science 2004, 203, 1755.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Lösemitteltransfer unter Inertgasbedingungen
• Fraktionierte Kristallisation unter Inertgasbedingungen
Charakterisierung:
• Hetereokern-NMR-Spektroskopie
Forschungsrelevante Thematik:
• Siliciumorganische Chemie
• Siliciumorganische Synthesebausteine in der präparativen Chemie

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
31
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon gela-
gert.
Bitte informieren Sie sich über die Schutzmaßnahmen und Verhaltensregeln beim Um-
gang mit Natrium/Kalium-Legierung. Filterrückstände und Rückstände des Reaktions-
ansatzes werden vorsichtig mit einem Gemisch aus Isopropanol und Toluol entsorgt.
Versuchsdurchführung:
Herstellung von Dodekamethylcyclohexasilan
In einen 100-mL-Dreihals-Schlenkkolben mit einem großen Magnetrührstäbchen, ei-
nem Rückflusskühler mit Metallkühlspirale sowie einem mit frisch destillierten THF ge-
füllten Tropftrichter, wird Kalium und Natrium vorgelegt und anschließend gerührt.
Hierbei bildet sich die Na/K-Legierung. Nach Erwärmen auf Raumtemperatur wird die
Legierung mit frisch destilliertem THF versetzt. Anschließend werden rasch ca. 2 g
Dimethyldichlorsilan zugetropft. Nach ein paar Minuten setzt eine exotherme Reaktion
ein und das Lösemittel beginnt zu sieden. Die restliche Menge an Dimethyldichlorsilan
wird innerhalb von 10 - 15 Minuten so zugetropft, dass das THF gerade siedet. Die
Reaktion ist nach ca. 15 Stunden Sieden auf Rückfluss abgeschlossen. Abends unter
Argonzufuhr auf Raumtemperatur abkühlen lassen. Am nächsten Praktikumstag wie-
der auf Rückfluss erhitzen. Nach Abkühlen auf Raumtemperatur wird der Rückstand
abgefrittet und mit THF gewaschen. Die flüchtigen Bestandteile des Filtrats und die
der vereinigten Waschlösungen werden im Unterdruck entfernt. Das kristalline, teil-
weise wachsartige Produkt wird in einem heißen Ethanol/THF 7:1-Gemisch (techn.
Qualität) umkristallisiert.
Literatur-Ausbeute: 1.65 g (51 %)
Kriterium: Ausbeute mindestens 35 %
Eigenschaften: farblose kristalline Substanz

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
32
Charakterisierung:
m. p. 254 - 257 oC
1H-NMR (CDCl3): δ = 0.13 ppm
13C-NMR (CDCl3): δ = -6.12 ppm
Pulverdiffraktogramm

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
33

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
34
Trimethylammonium-
tetradecahydroundecaborat
[B11H14][Me3NH]
Literatur:
Dunks, G. B.; Palmer-Ordonez, K. Inorg. Chem. 1978, 17, 1514.
17 NaBH4 + 20 Et2O·BF3
→ 2 NaB11H14 + 15 NaBF4 + 20 Et2O + 20 H2
NaB11H14 + Me3N⋅HCl →[B11H14][Me3NH] + NaClaq.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
35
Chemikalienliste:
Name der Chemikalie Menge
Natriumboranat 7.5 g (0.2 mol)
Bortrifluorid-Etherat 31.25 g (0.25 Mol, δ = 1.13 g/cm3 )
Trimethylammoniumhydrochlorid 2.85 g (0.035 mol)
Diglyme 300 mL
Sand nach Bedarf
Methanol 100 mL
Ausleihe:
Teflonfett
Versuchsaufbau
N 2 Aceton
S icherheits-
flasche
Trockeneis/
i-Propano l
Einleitung:
Borane (BnHm) sind Borwasserstoffverbindungen (Borhydride), über die als erstes Alf-
red Stock 1912 veröffentlichte. Das sich danach entwickelnde Gebiet der Chemie der
Borane und der verwandten Carbaborane ist eines der wichtigsten Entwicklungsfelder
in der anorganischen Chemie.
Ar
500
mL

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
36
Die Bedeutung der Borane liegt nicht nur in der vielseitigen und äußerst umfangreichen
Chemie, sondern auch in dem Auftreten von unerwarteten Strukturprinzipien. Dies be-
dingt die Notwendigkeit die MO-Theorie der kovalenten Bindung zu, um die ungewöhn-
lichen Stöchiometrien zu erklären.
Zahlreiche neutrale Borane BnHm und Borananionen BnHmy- wurden synthetisiert. Man
unterscheidet die Borane hinsichtlich Stöchiometrie und Struktur in Klassen einzutei-
len. Carbaborane sind Verbindungen, bei denen die zugrundeliegende Struktureinheit
aus einer Anzahl von C- und B-Atomen besteht, die an den Eckpunkten dreiwinkliger
Polyeder liegen. Ihre Strukturen sind eng verwandt mit denen der isoelktronischen Bo-
rane. Carbaborane traten in der Chemie um 1962 in Erscheinung, als die geheimen
Arbeiten der späten fünfziger Jahre zur Veröffentlichung freigegeben wurden.
Carbaborane und die verwandten Metallborane nehmen heute in der Chemie der Ele-
mente eine Schlüsselstellung ein, da sie mit einigen anderen großen Bereichen in der
Chemie zusammenhängen, darunter die Chemie der polyedrischen Borane, die Über-
gangsmetallkomplexe, Metall-Cluster-Verbindungen sowie die metallorganische Che-
mie.
Das in diesem Versuch herzustellende [B11H14][Me3NH] wurde erstmals 1962 aus der
Reaktion von Metallboranten mit B10H14 gewonnen. Auch in der Folgezeit erfolgte die
Synthese immer ausgehend von Boranen wie z. B. B2H6 oder B5H9. Erst durch die
Umsetzung von Natriumboranat mit Bortrifluorid-Etherat besteht ein einfacher Zugang
zu diesem interessanten Borananion welches ein geeigneter Baustein für die Herstel-
lung von mehrkernigen Boran-, Carbaboran- und Metallboran-Cluster darstellt.
Literatur:
Organoborane, Elschenbroich, Ch.; Salzer, A. Teubner Studienbücher Chemie, Teu-
bner Stuttgart A (1988)
Dunks, G. B.; Barker, K.; Hedaya, E.; Hefner, C.; Palmer-Ordonez, K.; Remec, P.
Inorg. Chem. 1981, 20, 1692.
Chapman, R. W.; Kester, J. G.; Folting, K.; Streib, W. E.; Todd, L. J. Inorg. Chem.
1992, 31, 979.
Jemmis, E.; Jayasree, E. G. Acc. Chem. Res. 2003, 36 (11), 816.
Review-Band zu Boranen: Pure and Applied Chemistry 2003 75 (9)

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
37
Was Sie lernen:
Synthesemethoden:
• Schutzgas-, und Vakuumtechnik
• Umgang mit luftempfindlichen Verbindungen
• Umgang mit luftempfindlichen Gasen
• Isolierung einkristalliner luftempfindlicher Substanzen
Charakterisierungsmethoden:
• Heterokern-NMR-Spektroskopie
• Einkristallstrukturaufklärung
• Elekrosprayionisations-Massenspektrometrie
Forschungsrelevante Thematik:
• Borane, Clusterverbindungen
Vorbereitung:
Trocknung von Lösemitteln: Diglyme
Allgemeines zur Sicherheit:
Das bei der Reaktion entstehende wenig stabile, hochentzündliches Gas Diboran wird
durch eine Waschflasche mit Aceton geleitet. Im Versuch werden alle Schliffe mit Tef-
lonfett gefettet, da Siliconfett vom Diglyme angegriffen wird. Der Versuch wird unter
nachgereinigtem Argon in ausgeheizten Apparaturen durchgeführt. Lösemittel werden
nach gängigen Methoden getrocknet und unter Argon gelagert. Der luft- und hydroly-
seempfindliche Feststoff [B11H14][Me3NH] wird unter Inertgas gehandhabt.
Man informiere sich besonders über die Gefahren beim Arbeiten mit Diboran.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
38
Versuchsdurchführung:
Herstellung von NaB11H14
Die Reaktionsapparatur wird nach der Versuchsskizze aufgebaut und mit einem gro-
ßen Knochenrührfisch bestückt. Die Apparatur wird evakuiert, ausgeheizt und mit
Inertgas aus der Schutzgasleitung befüllt. Diese Prozedur wird drei Mal wiederholt, bis
das Innere der Apparatur vollkommen inertisiert, d. h. wasser- und sauerstofffrei ist.
Danach wird im Argongegenstrom das NaBH4 eingefüllt, die Apparatur noch einmal
unter leichtem erwärmen evakuiert und 100 mL Diglyme im Argongegenstrom einge-
füllt. Die entstandene Suspension wird innerhalb von 2 Stunden langsam auf eine
Temperatur von 105 °C angenährt und über Nacht exakt bei 105 °C gehalten. Am
nächsten Tag wird BF3·Et2O vorsichtig in den noch geschlossenen Tropftrichter einge-
füllt. Nun wird zur Ableitung der entstehenden Gase eine mit Trockeneis/i-Propanol
gekühlte Falle und eine Waschflasche mit Aceton zugeschaltet. In der Kühlfalle kon-
densiert Diethylether, welcher noch B2H6 enthält. Deswegen wird der Inhalt der Kühl-
falle am Reaktionsende langsam mit Aceton versetzt, damit das B2H6 abreagiert. Der
Argonstrom wird durch die Apparatur auf eine Blase pro Minute eingeregelt.
Das BF3-Et2O wird über 4,5 Stunden zur Reaktionslösung zugetropft. Während der
ganzen Reaktionszeit darf die Temperatur nicht mehr als 2 °C von 105 °C abweichen.
Es wird eine weitere Stunde bei 105 °C und über Nacht bei Raumtemperatur weiter-
gerührt. Es entsteht eine NaBF4 Diglymesuspension. Im Diglyme ist NaB11H14 gelöst.
Eine große Schutzgasfilterfritte wird mit 500 mL-Schlenkkolben versehen, mit Seesand
befüllt und inertisiert. Auch hier werden alle Schliffe mit Teflonfett gefettet. Die Reakti-
onssuspension wird über einen Teflonschlauch unter Schutzgas in die Filterfritte über-
führt. Das im Reaktionskolben verbliebene NaBF4 wird mit kleinen Portionen Dyglyme
aufgeschlämmt und ebenfalls in die Schutzgasfritte gegossen. Nach Ablaufen der flüs-
sigen Phase wird der Filterkuchen mit weiteren 20 mL Diglyme ausgewaschen.
Eine Kondensationsbrücke wird mit einem 500 mL-Schlenkkolben und einer Schliff-
kappe versehen und inertisiert. Die Schliffkappe wird unter Schutzgas durch den 500
mL-Schlenkkolben mit dem Filtrat ersetzt und der leere Schlenkkolben wird mit flüssi-
gem Stickstoff gekühlt und an die Vakuumanlage angeschlossen. Die Lösung wird auf
67 °C im HV gebracht. Dadurch wird das Diglyme vom NaB11H14 in die gekühlte Vor-
lage abgezogen. Zurück bleibt ein zäher, gelber Rückstand.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
39
Herstellung von [B11H14][Me3NH]
200 mL H2Odest. werden in einem Schlenkkolben im Ultraschallbad unter leichtem Va-
kuum entgast und anschließend eisgekühlt. Der Kolben mit dem Reaktionsrückstand
wird ebenfalls mit Eis gekühlt. Eine Schutzgasfilterfritte wird mit einem 500 mL-
Schlenkkolben, der 2.85 g Trimethylammoniumchlorid enthält, versehen und inerti-
siert. Nun wird der Rückstand im Reaktionskolben in 200 mL des entgasten Wassers
suspensiert. Da NaB11H14 wasserempfindlich ist, sollte der Vorgang durch kräftiges
Schütteln beschleunigt werden und die Temperatur des Lösemittels 20 °C nicht über-
schreiten. Die Suspension wird im Argonstrom in die zuvor vorbereitete Schutzgasfritte
gegeben. Im Auffangkolben kommt es zur Reaktion zwischen Trimethylammonium-
chlorid und im Wasser gelösten [B11H14]-, wobei das in Wasser schwerlösliche
[Me3NH][B11H14] als blassgelber Feststoff ausfällt. Der Feststoff wird abfiltriert, in Me-
thanol bei RT aufgelöst und bei –30 °C auskristallisiert. Die Mutterlauge wird zur er-
neuten Kristallisation eingeengt.
Literatur-Ausbeute: 1.63 g (36 %)
Kriterium: Ausbeute mindestens 18 %.
Charakterisierung: 1H NMR (CD3OD/CDCl3): δ = 4.83 ppm (m, 14 H), 3.93 ppm (s, 9 H), 2,4 ppm (s, 1H)
13C NMR (CD3OD/CDCl3): δ = 49.7 ppm
11B NMR (CD3OD/CDCl3): δ = -15 ppm (m)
ESI-MS: (B11H14)-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
40
Natrium Tetrakis[(3,5-Bis(trifluoromethyl)-
phenyl]borat (NaBArF)
Na{B[3,5-(CF3)2C6H3]4}
Literatur:
Reger, D.; Wright, T.; Little, C.; Lamba, J.; Smith, M. Inorg. Chem. 2001, 40, 3810.
Yakelis, N. A.; Bergman, R. G. Organometallics, 2005, 24, 3579.
CF3
CF3
Br
CF3
CF3
BrMgMg NaBF4
CF3
CF3
B4
Na

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
41
Chemikalienliste:
Name der Chemikalie Menge
Magnesiumspäne 5.12 g (0.210 mol)
Natriumtetrafluoroborat 3,3 g (0.030 mol)
3,5-Bis(trifluoromethyl)bromobenzol 50 g (0.17 mol)
Natriumcarbonat (wasserfrei) 74 g
Diethylether (trocken / abdestilliert / p.a.) 1200 mL
Dichlormethan 100 mL
Magnesiumsulfat Trocknungsmittel
Toluol 240 mL
n-Hexan 40 mL
Einleitung:
Die Stabilisierung von hoch reaktiven elektrophilen Metall- und Nichtmetallzentren, als
auch Lewis-Säure-Base-Komplexe von Metallkationen wird oft durch den Austausch
eines Gegenions durch ein schwach koordinierendes Anion erreicht (WCA, „weakly
coordinating anion“). In vielen Fällen verhalten sich deren klassische Vertreter wie
ClO4-, SO3CF3-, BF4-, BPh4- und PF6- dennoch als schwache Nucleophile und binden
zu starken Elektrophilen (wie beispielsweise in obigen Fällen über die Sauerstoff- bzw.
Halogenatome), was z. B. aus Einkristallröntgenstrukturen ersichtlich ist. Das heißt,
wenn man von "nicht koordinierenden Anionen" spricht, ist typischerweise die Rede
von "schwach" koordinierenden Anionen.
Die Nukleophilie kann aber weiter reduziert werden, z. B. durch Delokalisierung über
eine große Fläche nichtnucleophiler, chemisch robuster Gruppen und die Einführung
elektronenziehender Substituenten, wie z. B. Fluorsubstituenten. Die Koordinations-
kraft eines Anions ist durch seine basischste Gruppierung vorgegeben, sodass ein
WCA immer, wenn auch schwach, mit der nucleophilsten, sterisch zugänglichsten
Gruppen an das Kation koordiniert wird. Bei der Entwicklung von WCA’s ist dies auch
der Ausgangspunkt.
Breite Anwendung finden heute Salze wie NaBArF (Na[B(3,5-C6H3(CF3)2]4), entwickelt
von Kobayashi et al. 1984 und B(C6F5)4-, entwickelt von Massey und Park 1964, die
ausgehend von BPh4- elektronenziehende Fluorsubstituenten enthalten. Beide sind re-
lativ günstig und einfach herstellbar. Im Vergleich ist das letztere zwar stabiler, jedoch

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
42
ist NaBArF weniger nucleophil. Einführung der Anionen Gruppe in Komplexe erfolgt z.
B. durch Halogenaustauchreaktionen mit den Alkalisalzen Na[BArF] oder Li[BArF] bzw.
mit dem Thallium- oder Silbersalz Tl[BArF] und Ag[BArF] oder Protolyse mit der Säure
[H(Et2O)2][BArF].
Eine Alternative zu den eben angeführten Boraten sind poly- oder perfluorierte Al-
koxy(ORF)-und Aryloxy(OArF)-Metallate. Als Zentralatome dienen oxophile und stark
Lewis-saure Metallatome wie z.B. BIII, AlIII und NbV. Gegenüber B(C6F5)4- und verwand-
ten Boraten haben diese Metallate den großen Vorteil, dass sie präparativ einfach und
gefahrlos zugänglich, außerdem sind die Aktivitäten in der Anwendung mindestens
genauso groß.
Die Anwendungsbreite schwach koordinierender Anionen ist enorm. Zum Beispiel er-
lauben sie die Untersuchung von sehr reaktiven kationischen Übergangsmetallkom-
plexen. Weitere Beispiele für ihre Anwendung sind Polymerisation (Olefin-Polymerisa-
tion mit Gruppe-4-Metallocenen und mit Nicht-Metallocen-Katalysatoren), C-H-Aktivie-
rung und ionische Flüssigkeiten, um nur einige zu nennen.
Literatur:
Krossing, I.; Raabe, I. Angew. Chem., 2004, 116, 2116.
Krossing, I.; Raabe, I. Angew. Chem. Int. Ed., 2004, 43, 2066.
Krossing, I. Chem. Eur. J., 2001, 7, 490.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Lösemitteltransfer unter Inertgasbedingungen
• Umgang mit reaktiven Lösemittelgemischen
Charakterisierung:
• Hetereokern-NMR-Spektroskopie
Forschungsrelevante Thematik:

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
43
• Nichtkoordinierende Anionen, WCA’s
• Homogene Katalyse
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Beim Ausheizen sollte das Magnesium sich mit im Kolben befinden. Lösemittel
werden nach gängigen Methoden getrocknet und unter Argon gelagert. Wenn nicht
anders im Text beschrieben wird unter Argon gearbeitet. Insbesondere darf die Reak-
tionsmischung nach der Zugabe von 3,5-Bis(trifluoromethyl)bromobenzol nicht über-
hitzt werden, da die Zwischenprodukte sehr reaktiv und potentiell explosiv sind!
Versuchsdurchführung:
Herstellung von Natrium Tetrakis[(3,5-Bis(trifluoromethyl)phenyl]borat
Ein ausgeheizter und mit Argon gefüllter 1000 mL 3-Hals-Kolben (bestückt mit einem
Rückflusskühler, einem Tropftrichter und - wenn nicht vorhanden - einem Gashahn)
wird mit Magnesiumspänen (5.11 g, 0.21 mol) befüllt und in 150 mL trockenem Diet-
hylether suspendiert. In den Tropftrichter werden 29.4 mL 3,5-Bis(trifluoromethyl)bro-
mobenzol (49.9 g, 0.17 mol) sowie 250 mL trockener Diethylether gegeben. Die ent-
standene Lösung wird langsam (max. 200mL / Stunden), unter starken Rühren der
Magnesiumsuspension, zu dieser hinzugetropft. Nachdem ca. 60 mL dieser Lösung
zugetropft wurden, sollte der Grignard „angesprungen“ sein.
Es sollte auf keinen Fall weitere 3,5-Bis(trifluoromethyl)bromobenzol-Ether-Lösung zu-
gegeben werden, da die Reaktion unkontrolliert starten und unter starker Wärmeent-
wicklung durchgehen könnte! Sobald die Grignard-Reaktion gestartet ist (Trübung und
Braunfärbung der Lösung), wird die restliche 3,5-Bis(trifluoromethyl)bromobenzol-
Ether-Lösung (ca. 220 mL) konstant über 1 - 2 Stunden zugetropft (sodass die Lösung
konstant siedet!) und anschließend für 30 Minuten unter Rückfluss erhitzt. Nachdem
die Reaktionsmischung auf Raumtemperatur abgekühlt ist, werden 3.29 g Natriumtet-
rafluorborat (30.0 mmol) zugebenen und die Reaktionsmischung wird für 2 Tage bei
Raumtemperatur gerührt. Danach wird die Reaktionsmischung (im Folgenden wird an
Luft gearbeitet) unter starkem Rühren auf einen Liter 0.7 M wässrige Na2CO3-Lösung
gegeben (75.0 g, 0.71 mol) unter für eine Stunde gerührt. Anschließend wird abfiltriert

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
44
(am besten mit Unterdruck), und der Rückstand mit 4×50 mL abdestilliertem Diethyl-
ether gewaschen. Das flüssige Phase (Wasser-Ether) wird in einen Scheidetrichter
überführt, mit 3 × 200 mL Diethylether extrahiert und die organische Phase über Mag-
nesiumsulfat getrocknet. Das Lösungsmittel wird unter vermindertem Druck destillativ
entfernt. Weitere Verunreinigungen werden im Hochvakuum (Ölbad T = 100 °C) ent-
fernt. Der erhaltene Feststoff wird in einen Faltenfilter überführt und mit −30 °C gekühl-
tem Dichlormethan gewaschen (5 × 20 mL) und anschließend im Hochvakuum über
Nacht getrocknet. Es wird Natrium-tetrakis(3,5-bis(trifluormethyl)phenyl)borat als
grau/brauner Feststoff erhalten. Sollte die Verbindung nicht sauber sein (Vergleich
NMR und Smp.), wird der Feststoff in wenige mL Ether gelöst (gesättigte Lösung Pro-
dukt-Solvens) und Hexan zugegeben, worauf „NaBArF4“ ausfällt. Das überschüssige
Lösungsmittel wird abkannüliert und der Feststoff im Hochvakuum getrocknet. Zuletzt
können mögliche Verunreinigungen mit wenig Toluol und Hexan entfernt werden. Er-
neutes Trocknen im Hochvakuum ergibt Natrium-tetrakis(3,5-bis(trifluormethyl)phe-
nyl)borat als grau/brauner Feststoff.
Literatur-Ausbeute: 22.3 g (25.10 mmol, 84 %)
Kriterium: Ausbeute mindestens 44 %
Eigenschaften: beige mikrokristalline Substanz, hygroskopisch
Charakterisierung1:
m. p. 330 - 335 oC 1H-NMR (400MHz, Aceton-d6): 7.79 ppm (br s, 8 H), 7.67 ppm (br s, 4 H) 13C{1H}-NMR (100MHz, Aceton-d6): 161.5 ppm (q, 1JB-C = 50,0 Hz), 134.6 ppm (s),
129.1 ppm (q, 2JC-F = 31.5 Hz), 124.5 ppm (q, 1JC-F = 270.0 Hz), 117.5 ppm (s), 11B-NMR (160 MHz, Aceton-d6): -6.6 ppm (s) 19F-NMR (19F-CPD) (235 MHz, Aceton-d6): -60.60 ppm (s).
1 Anmerkung: Anstelle von Aceton-d6 kann alternativ CD3CN als Lösungsmittel ver-
wendet werden.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
45
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
46
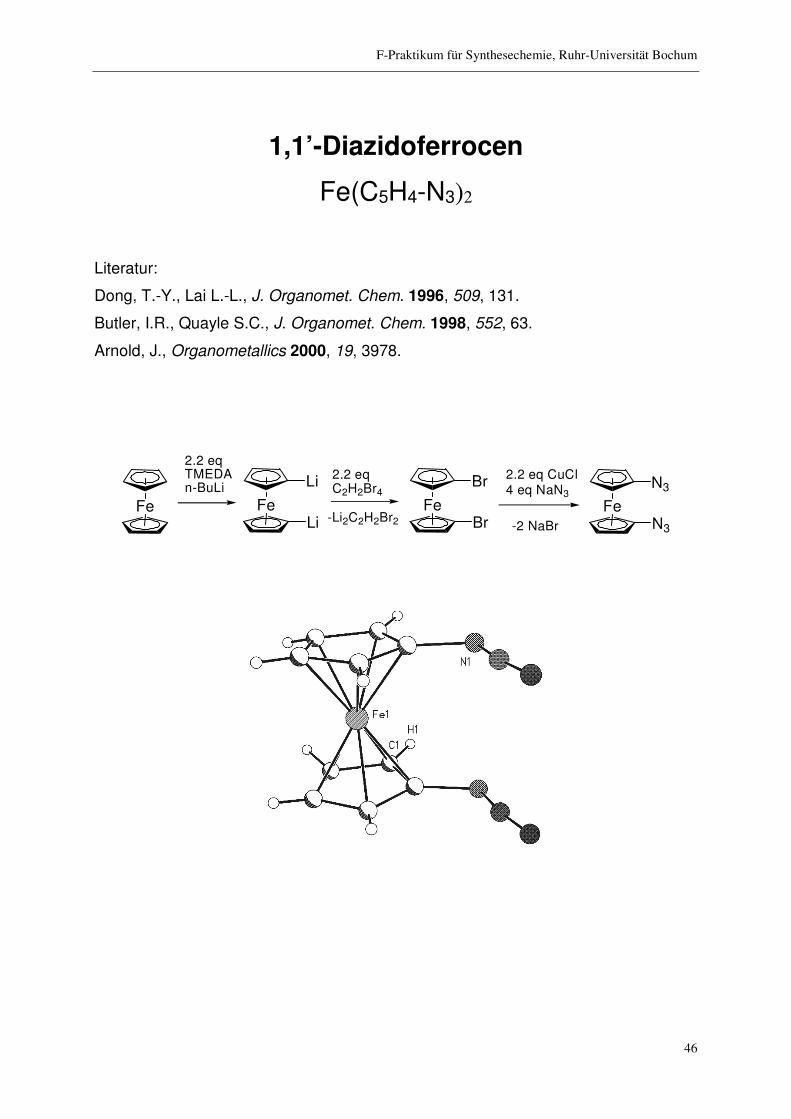
1,1’-Diazidoferrocen
Fe(C5H4-N3)2
Literatur:
Dong, T.-Y., Lai L.-L., J. Organomet. Chem. 1996, 509, 131.
Butler, I.R., Quayle S.C., J. Organomet. Chem. 1998, 552, 63.
Arnold, J., Organometallics 2000, 19, 3978.
Fe Fe Fe
Li
Li
Br
Br
2.2 eqTMEDAn-BuLi
2.2 eqC2H2Br4
Fe
N32.2 eq CuCl4 eq NaN3
N3-Li2C2H2Br2 -2 NaBr

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
47
Chemikalienliste:
Name der Chemikalie Menge
Ferrocen 5 g (0.027 mol)
N,N,N`,N`-Tetramethyl-1,2-ethandiamin 9 mL (0.060 mol)
n-Butyllithium 24 mL (2.5 M Lösung in
Hexan, 0.060 mol)
Diethylether 100 mL
Tetrabromethan 7.0 mL (0.060 mol)
H2O dest. 50 mL
Dichlormethan 100 mL
Methanol 5 mL
Name der Chemikalie Menge
1,1’-Dibromoferrocen 1 g (0.0029 mol)
Natriumazid 754.1 mg (0.0116 mol)
Kupfer(I)chlorid 631.6 mg (0.00638 mol)
Ethanol, 95% 25 mL
H2O dest. 50 mL
Diethylether 100 mL
Einleitung:
Die „Entdeckung“ und nachfolgenden strukturellen Untersuchungen von Ferrocen im
Jahr 1951 können als Geburtsstunde der modernen Organometallchemie angesehen
werden. Durch relativ leicht zugängliche aromatische Substitutionen an den Cyclopen-
tadienyl-Liganden wurde in den letzten Jahrzehnten eine Vielzahl von Ferrocenyl-Ver-
bindungen synthetisiert. Die Stabilität der Ferrocenyleinheit in wässrigem, aerobem
Milieu und die Redoxaktivität mit einem reversiblen Einelektronenübergang machen
diese Verbindungsklasse auch für biologische Anwendungen sehr interessant (van
Staveren, 2004). Eine aktuelle Anwendung sind Biokonjugate von Ferrocen mit DNA
und DNA-Analoga, die für neuartige DNA-Biosensoren mit elektrochemischer Detek-
tion benötigt werden. Hierzu muss die Ferroceneinheit mit dem Biomolekül in geeig-
neter Weise, d. h. selektiv und unter milden, biokompatiblen Bedingungen, verknüpft
werden. Azidoferrocen im Speziellen kann in bioorthogonaler Weise mit Biomolekülen

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
48
verknüpft werden, d. h. die Bindung kommt durch in biologischen Systemen nicht ver-
wendete Reaktionen zustande. Die Reaktion von Aziden mit terminalen Alkinen ist eine
[3+2] Cycloaddition und wird auch als Click-Reaktion bezeichnet. Click-Reaktionen
laufen im Allgemeinen chemoselektiv, unter milden Bedingungen und in hohen Aus-
beuten ab. Diese Reaktion wurde verwendet, um Azidoferrocen an PNA (Peptidnukle-
insäuren, ein künstliches DNA-Analogon mit besonders vielversprechenden Eigen-
schaften) gebunden, was die Entwicklung von elektrochemischen Biosensoren für
DNA und PNA ermöglicht. Die Reaktion von 1,1’-Diazidoferrocen mit alkinsubstituier-
ten Peptiden zu sogenannten Peptidmimetika kann Einblicke in die Struktur von natür-
lichen Peptiden liefern.
Literatur:
van Staveren, D. R.; Metzler-Nolte, N. Chem Rev. 2004, 104, 5931.
Köster, S. D.; Metzler-Nolte, N. Organometallics 2008, 27, 6326.
Hüsken, N.; Metzler-Nolte, N., Bioconjugate Chem. 2009, 20, 1578.
Was Sie lernen:
Synthesemethoden:
• Vakuum- und Schutzgastechnik
• Lösemitteltransfer unter Schutzgas
• Transfer von luftempfindlichen Substanzen mit Spritze und Septum
Charakterisierungsmethoden:
• GC-MS (Gaschromatographie-Massenspektrometrie)
• NMR-Spektroskopie
• Infrarotspektroskopie

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
49
Forschungsrelevante Thematik:
• Organometallchemie der Nebengruppenelemente
• Metallocene
Allgemeines zur Sicherheit:
Der erste Teil des Versuchs (Herstellung von 1,1’-Dibromoferrocen) wird unter nach-
gereinigtem Argon in ausgeheizten Apparaturen durchgeführt. Lösemittel werden nach
gängigen Methoden getrocknet und unter Argon gelagert. Der zweite Teil (Herstellung
von 1,1’-Diazidoferrocen) wird an Luft durchgeführt. Vorsicht: Natriumazid ist sehr to-
xisch und setzt bei der Reaktion mit Säuren HN3 als giftiges Gas frei! Deshalb Azide
unbedingt basisch entsorgen! Bei der Handhabung von getrocknetem 1,1’-Diazidofer-
rocen ist Vorsicht geboten, da Azide potentiell explosiv auf Schlag- oder Hitzeeinwir-
kung reagieren können! Der Ansatz für das 1,1’-Diazidoferrocen darf nicht vergrößert
werden, das Produkt soll nur an Luft und nicht im Vakuum getrocknet werden und darf
nicht über Raumtemperatur erhitzt werden.
Versuchsdurchführung:
Herstellung von 1,1’-Dibromoferrocen
In einem ausgeheizten 250-mL-Zweihalskolben, Thermometer und Abgashahn wird
Ferrocen vorgelegt und im Vakuum getrocknet. Es werden 50 mL trockener Diethyl-
ether hinzugefügt. Zu der gelben Suspension werden 2.2 äquivalente N,N,N`,N`-Tet-
ramethyl-1,2-ethandiamin gegeben und es wird im Eisbad auf etwa 0 C abgekühlt.
Anschließend wird n-Butyllithium so langsam zugegeben, dass die Temperatur nicht
über 10 °C steigt. Nach beendeter Zugabe wird über Nacht bei Raumtemperatur ge-
rührt. Anschließend wird der Reaktionsansatz in einem Aceton/Trockeneisbad auf
etwa –60 C gekühlt und ca. 30 Minuten bei dieser Temperatur gerührt. 2.2 äquivalente
Tetrabromethan werden tropfenweise zugegeben und die Mischung 4 Stunden bei –
60 C gerührt. Die Kühlung wird entfernt und eine weitere Stunde bei Raumtemperatur
gerührt, worauf die zuvor gelbe Lösung eine rote Farbe bekommt. Es werden 50 mL
Wasser (dest.) zugegeben, Diethylether wird abkondensiert und die entstandene Mi-
schung mit 2 x 50 mL Dichlormethan extrahiert. Die organische Phase wird über Nat-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
50
riumsulfat getrocknet, filtriert und im Vakuum vollständig eingeengt. Es bleibt ein dun-
kelrotes Öl, welches in etwa 5 mL Methanol aufgenommen und im Kühlschrank gela-
gert wird. Die entstandenen rotbraunen Kristalle werden filtriert, mit wenig eiskaltem
Methanol gewaschen und im Vakuum getrocknet.
Literatur-Ausbeute: 60 % (5.6 g)
Kriterium: mindestens 30 %
Eigenschaften: rotbraune Kristalle
Charakterisierung: Schmelzpunkt (53 - 55 °C), 1H NMR (CDCl3, 200MHz): δ 4.42 (m, 4 H), 4.17 (m, 4H) 13C NMR (CDCl3, 50MHz): δ 78.3, 72.7, 69.9.
Herstellung von 1,1’-Diazidoferrocen:
In einem Einhalskoben wird 1,1’-Dibromoferrocen in 25 mL Ethanol (95 %) suspen-
diert. Kupferchlorid und eine Lösung von Natriumazid in 5 mL Wasser (dest.) werden
nacheinander hinzugefügt, worauf sich eine dunkelbraune Suspension bildet. Die Re-
aktionsmischung wird bei Raumtemperatur im Dunkeln (Kolben mit Alufolie umwickeln)
48 Stunden gerührt. Es wird in 50 mL Wasser (dest.) gegeben und mit 2 x 50 mL
Diethylether extrahiert. Die organische Phase wird über Natriumsulfat getrocknet, fil-
triert und im Vakuum auf 1 - 2 mL zur Kristallisation eingeengt. Es wird über Nacht im
Kühlschrank aufbewahrt, das kristallisierte Produkt filtriert und im Dunkeln (lichtemp-
findlich!) luftgetrocknet. Wichtig: Produkt nur an der Luft trocknen und keinesfalls über
Raumtemperatur erhitzen!
Literatur-Ausbeute: 59 % (0.46 g)
Kriterium: mindestens 30 %
Eigenschaften: braune, nadelförmige Kristalle
Charakterisierung: 1H NMR (CDCl3, 200 MHz): δ 4.36 (m, 4 H), 4.16 (m, 4H) 13C NMR (CDCl3, 50 MHz): δ 100.5, 66.6, 61.7;
IR (cm-1): 2108 (s).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
51
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
52
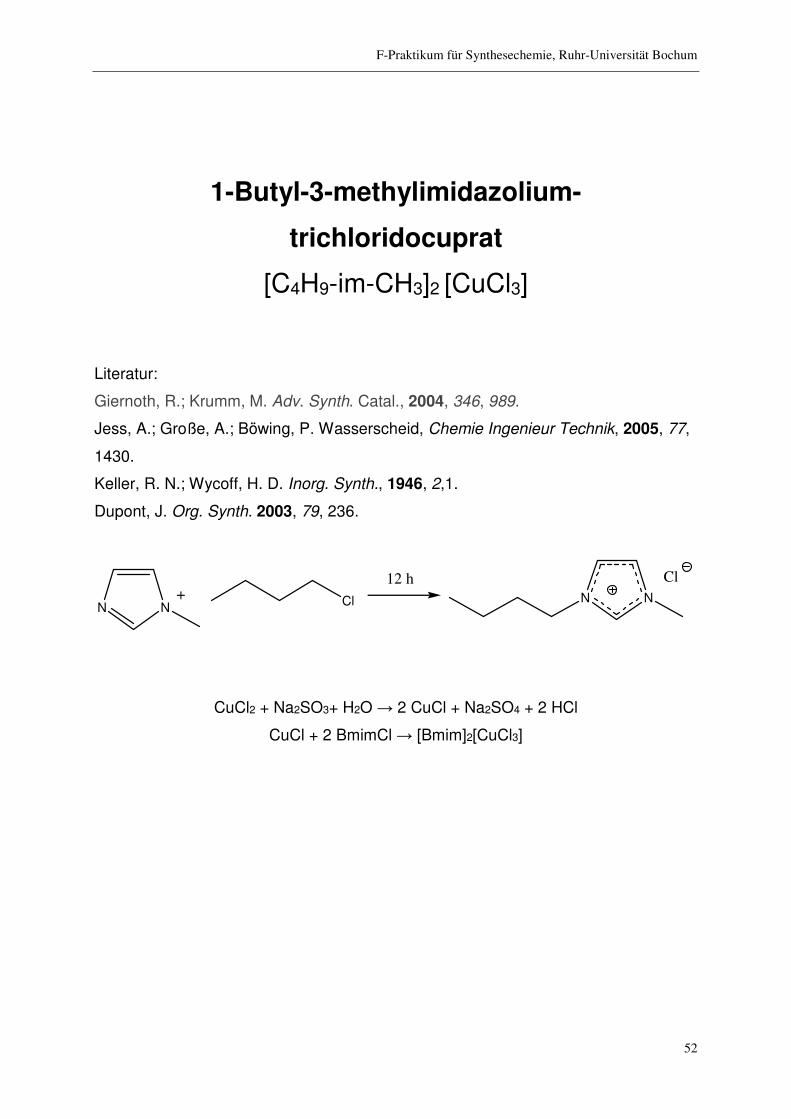
1-Butyl-3-methylimidazolium-
trichloridocuprat
[C4H9-im-CH3]2 [CuCl3]
Literatur:
Giernoth, R.; Krumm, M. Adv. Synth. Catal., 2004, 346, 989.
Jess, A.; Große, A.; Böwing, P. Wasserscheid, Chemie Ingenieur Technik, 2005, 77,
1430.
Keller, R. N.; Wycoff, H. D. Inorg. Synth., 1946, 2,1.
Dupont, J. Org. Synth. 2003, 79, 236.
N N Cl+ N N
Cl12 h
CuCl2 + Na2SO3+ H2O → 2 CuCl + Na2SO4 + 2 HCl
CuCl + 2 BmimCl → [Bmim]2[CuCl3]

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
53
Chemikalienliste:
Name der Chemikalie Menge
1-Chlorbutan 17.5 g, (0.188 mol)
1-Methylimidazol 10.3 g (0.125 mol)
Toluol 50-75 mL
Kupfer(II)-Chlorid 10 g (0,059 mol )
Natriumsulfit
Schweflige Säure
Eisessig
Ethanol
Diethylether
7,6 g (0,060 mol)
10 mL
45 mL
45 mL
45 mL
Einleitung:
Ionische Flüssigkeiten sind Salze, die einen niedrigen Schmelzpunkt, meist unter
100 °C, haben. Sie besitzen einen rein ionischen Charakter und bestehen aus organi-
schen Kationen und organischen oder anorganischen Anionen. Durch die Auswahl der
Anionen und Kationen können die Eigenschaften der Salze reguliert und optimiert wer-
den. Kationen, genauso wie Anionen werden nach ihrer Struktur und Eigenschaften
meistens in vier Gruppen geteilt. Bei Anionen unterscheidet man zwischen den Syste-
men, die sich auf AlCl3, oder solchen Anionen wie [BF4]-, [PF6]- und [SbF6]- basieren.
Zur dritten Gruppe zählen Alkylsulfate und Alkylsulfonate. Die vierte Gruppe bildet sich
aus Anionen des Typs [CF3SO3]-, die sogenannten Triflate, abgekürzt mit [OTf] und
[(CF3SO2)2N]- =� [Tf2N]-(Bis(trifluoromethylsulfonyl)imid). Die wichtigsten Gruppen von
Kationen sind Alkylphosphonium-, Alkylammoniumionen, Alkylimidazolium und Al-
kylpyridinium Kationen. Insbesondere die Anionen haben einen Einfluss auf die Eigen-
schaften der ILs. Sorgfältig ausgewählte Anionen ermöglichen die Einstellung präziser
physikalischen und chemischen Eigenschaften einer IL, die für eine bestimmte Reak-
tion erwünscht ist. Eine der interessantesten Eigenschaften der ionischen Flüssigkei-
ten ist der verschwindend kleine Dampfdruck. So kann das Problem toxischer bzw.
gesundheitsschädlicher Dämpfe sowie Lösemittelverlust minimiert werden. Viele
Übergangsmetallkatalysatoren lösen sich sehr gut, viele organische Substanzen da-
gegen nicht in Ionischen Flüssigkeiten. Das ermöglicht eine zweiphasige Reaktions-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
54
durchführung und somit eine problemlose destillative Abtrennung des Reaktionsgemi-
sches von einem Lösemittel. Dank dieser Eigenschaft können die IL’s wiedergewon-
nen und mehrmals neu verwendet werden. Dies kann im ökologischen Sinne für mo-
difizierte Verfahrenprozesse, z. B. dem BASIL-Prozess bei der BASF, eingesetzt wer-
den. Das Potential von Ils führt dazu, dass diese alternativen Solventien seit den Acht-
zigerjahre des 20ten Jahrhundert als „Green Solvents“ genutzt werden.
Literatur:
Wasserscheid, P.; Kleim, W. Angew. Chem. 2000, 112, 3926.
Chiappe, C.; Pieraccini, D. J. Phys. Org. Chem. 2005, 18, 275.
Qian, W.; Jin, E.; Bao, W.; Zhang, Y. Angew. Chem. Int. Ed. 2005,44, 952.
Maase, M. Multiphase Homogeneous Catalysis 2005, 2, 560.
Mudring, A.-V.; Babai, A.; Arenz, S.; Giernoth, R. Angewandte Chemie (Int. Engl.)
2005, 44(34), 5485.
Dupont J. Et al., Preparation of 1-Butyl-3-Methylimidazolium-bases Romm tempera-
ture ionic liquids, Org. Synth. 2003, 79, 236-243.
Keller, R. N.; Wycoff, H. D. Inorg. Syntheses, Bd 2, 1-4.
Swatloski, R .P.; Spear, S. K.; Holbrey, J. D.; R.D. J. Am. Chem. Soc., 2002, 124,
4974.
Carson, L.; Chau, P. K. W.; Earle, M. J.; Gilea, M. A.; Gilmore, B. F.; Gorman, S. P.;
McCann, M. T.; Seddon, K. R. Green Chemistry, 2009, 11, 492.
Laher, T. M.; Hussey, C.J. Inorg. Chem.,1983, 22, 3247.
Javanshir, S.; Mojtahedi, M. M.; Eslami, J., Current chemistry Letters, 2014, 3, 63.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit metallorganischen Substanzen
• Lösemitteltransfer unter Inertgasbedingungen
Charakterisierungsmethoden:
• NMR-Spektroskopie

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
55
• Elementaranalyse
• UV-Vis-Spektroskopie
• Karl-Fischer-Titration
• Thermoanalyse (DSC)
Forschungsrelevante Thematik:
• Ionische Flüssigkeiten
• Magnetische Flüssigkeiten
Vorbereitungen:
Trocknung von Lösemittel: Acetonitril, Toluol
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon gela-
gert.
Synthese:
1-Butyl-3-methylimidazoliumchlorid
In einem 250 mL-Schlenkkolben werden 1-Chlorbutan und 1-Methylimidazol 12 Stun-
den auf Rückfluss erhitzt. Die Reaktion ist abgeschlossen, wenn sich ein einphasiges
System gebildet hat. In das gelbliche, viskose Öl wird ein 1-Butyl-3-Methylimidazoli-
umchlorid-Impfkristall gegeben. Es werden ca. 25 - 50 mL Toluol hinzugegeben und
das Produkt wird einige Stunden im Kühlschrank bei -50 °C gelagert. Das überste-
hende Lösungsmittel wird im Argongegenstrom entfernt, das weiße kristalline Produkt
wird mit 20 mL Toluol gewaschen und im Unterdruck getrocknet.
Literatur-Ausbeute: 20.2 g (0.116 mol, 92.8 %)
Kriterium: Ausbeute mindestens 45 %

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
56
Charakterisierung:
1H-NMR, CDCl3
δ / ppm Multiplizität Integral δ / ppm Multiplizität Integral
10.61 s 1 4.13 s 3
7.57 s 1 1.91 d 2
7.35 s 1 1.38 tt 2
4.34 t 2 0.96 t 3
2) Kupfer-(I)-chlorid:
In einem Becherglas werden 10 g CuCl2 in 200 mL Wasser vorgelegt. Des Weiteren
wird eine Lösung aus 7.6 g Na2SO3 in 50 mL Wasser angesetzt. Anschließend wird
die Natriumsulfitlösung langsam unter Rühren zu der Kupferchloridlösung hinzugege-
ben. Die Lösung verfärbt sich zunächst braun. Kurze Zeit später fällt das Kupferchlorid
als weißer Feststoff aus. Nach beendeter Zugabe der Lösungen wird das Reaktions-
gemisch für 15 Minuten gerührt. Anschließend wird die Lösung mit 10 mL H2SO3 ver-
setzt. Der weiße Feststoff wird in einer zuvor ausgewogenen, evakuierten Schlankfritte
von der wässrigen Lösung getrennt und mit jeweils 3 Portionen à 15 mL Eisessig, 15
mL Ethanol und 15 mL Diethylether gewaschen.
Dabei ist bei allen Schritten darauf zu achten, dass der Feststoff stets mit Flüssigkeit
noch bedeckt ist. Anschließend wird das nun luftempfindliche Produkt im Vakuum ge-
trocknet.
Kriterium: Ausbeute mindestens 3 g
3) 1-Butyl-3-methylimidazolium-trichloridocuprat 2 eq. von BmimCl werden unter Schutzgasatmosphäre zu dem Kupfer(I)chlorid (1eq.)
hinzugegeben. Das Reaktionsgemisch der beiden Feststoffe wird leicht mit einem

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
57
Heißluftgebläse erwärmt, wobei sich eine farblose Flüssigkeit bildet.
Die Flüssigkeit ist oxidationsempfindlich und färbt sich bei Sauerstoffkontakt braun
(dient als Kriterium).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
58
Triphenylphosphoniumacetonitrilylid
(Y-CN)
Literatur:
Schiemenz, G. P.; Engelhard, H. Chem. Ber. 1961, 94 (3), 578.
Scherpf, T.; Wirth, R.; Molitor, S.; Feichtner, K.-S.; Gessner, V. H., Angew. Chem.
2015, 127, 8662.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
59
Chemikalienliste:
1. Stufe:
Name der Chemikalie Menge
Bromacetonitril 1.45 mL (2.5 g, 20.8 mmol; 1.722 g/
mL (20 °C))
Triphenylphosphan 5.47 g (20.8 mmol)
Toluol 25 mL
Techn. Pentan 45 mL
2. Stufe:
Name der Chemikalie Menge
Phosphoniumsalz 7.65 g (20.0 mmol)
Natriumhydrid 60 % Dispersion in Mineralöl 0.88 g (22.0 mmol)
Pentan 40 mL + x
Tetrahydrofuran 50 mL
Dichlormethan 30 mL + x
Einleitung:
Metallierte Ylide, die sogenannten Yldiide, stellen ein Bindeglied zwischen den dianio-
nischen Methandiiden und den neutralen Bisyliden dar. Diese zeichnen sich formal
durch zwei negative Ladungen am zentralen Kohlenstoffatom und einem positiv gela-
denen Rest aus und können so als X oder L- Ligand mit starker Donorfähigkeit ver-
wendet werden. Während metallierte Ylide heute nahezu unerforscht sind, haben Bi-
sylide in den letzten Jahren aufgrund ihrer besonderen elektronischen Struktur und
Koordinationseigenschaften reges Forschungsinteresse gefunden.
Abbildung 1. Gegenüberstellung von Bisyliden, Yldiiden und Methandiiden.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
60
Paradebeispiel für Bisylide sind die Carbodiphosphorane wie 1, das von der AG Ra-
mirez erstmals 1961 synthetisiert wurde. Schon damals verwendeten die Autoren zwei
Resonanzstrukturen A und B um die Bindungsverhältnisse zu beschreiben. Bei der
heterocumulenen Resonanzstruktur A kommt es zu einer Doppelbindung zwischen
den d-Orbitalen der Phosphoratome und den p-Orbitalen des Kohlenstoffatoms. Des-
wegen muss das Kohlenstoffatom sp hybridisiert und die Struktur linear sein. Im Ge-
gensatz dazu wäre die Struktur der ylidischen Resonanzstruktur B gewinkelt aufge-
baut, weil zwei freie Elektronenpaare am Kohlenstoffatom lokalisiert sind und das Koh-
lenstoffatom formal sp3 hybridisiert wäre. Da zu jener Zeit keine geminalen dimetallier-
ten Derivate isolierten werden konnten, wurde anfänglich zur Beschreibung der Struk-
tur hauptsächlich Resonanzstruktur A verwendet. Neuste Studien und Berechnung
ergaben jedoch, dass die d-Orbitale des Phosphoratoms energetisch zu hoch für eine
Bindung mit den p-Orbitalen des Kohlenstoffatoms liegen, so dass in der Literatur die
ylidische Bindung mit einer P–C-Einfachbindung und elektrostatischen Wechselwir-
kungen bevorzugt wurde. Neuste Arbeiten zeigten jedoch, dass eine Beschreibung
über koordinative Bindung C ebenfalls herangezogen werden kann. Diese Donor-Ak-
zeptor-Wechselwirkung ist bei Metall-Ligand-Interaktionen zu beobachten und kann
mit dem Dewar-Chat-Duncanson-Model erklärt werden.
Abbildung 2. Resonanzstrukturen des Carbodiphosphoran 1.
Während bei Bisyliden die elektronische Struktur ausgiebig diskutiert wurde, ist die
elektronische Struktur sowie die Reaktivität von den Yldiiden nahezu unerforscht. Ei-
nes der wenigen Beispiele ist das von Bestmann und Schmidt 1987 dargestellte Yldiid
2. Sie postulierten auf Grund von IR- und NMR-Daten, dass 2 nicht in der ylidischen
Resonanzstruktur E, sondern in der Resonanzstruktur D vorliegt. Da Cyanid auch als
Ligand für Metallkomplexe verwendet wird, könnte das Yldiid analog zu C in der koor-
dinativen Resonanzstruktur F vorliegen.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
61
Abbildung 3. Resonanzstrukturen des Yldiids 2.
Literatur:
Alcarazo, M. Dalton Trans. 2011, 40 (9), 1839-1845.
Tonner, R.; Öxler, F.; Neumüller, B.; Petz, W.; Frenking, G. Angew. Chem. 2006,
118, 8206.
Himmel, D.; Schnepf, A.; Krossing, I. Angew. Chem. 2014, 126, 6159.
Bestmann, H. J.; Schmidt, M. Angew. Chem. 1987, 99, 64.
Scharf, L. T., Andrada, D. M.; Frenking, G.; Gessner, V. H. Chem. Eur. J. 2017, 23,
4422.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Lösemitteltransfer unter Inertgasbedingungen
Charakterisierungsmethoden:
• Heterokern-NMR-Spektroskopie
• IR-Spektroskopie
Forschungsrelevante Thematik und theoretische Grundlagen:
• Dewar-Chatt-Duncanson Modell
• Ylide und Yldiide

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
62
Allgemeines zur Sicherheit:
Der Versuch wir unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon gela-
gert. Natriumhydrid ist brennbar. Bitte informieren Sie sich über Schutzmaßnahmen
und Verhaltungsregeln beim Umgang mit Natriumhydrid.
Versuchsdurchführung:
Herstellung von (Cyanomethyl)triphenylphosphoniumbromid
In einem 50 mL-Schlenkkolben werden 1.45 mL (2.50 g, 20.8 mmol) Bromacetonitril in
5 mL Toluol gelöst und in einem zweiten 50 mL Schlenkkolben werden 5.47 g (20.8
mmol) PPh3 in 20 mL Toluol gelöst. Die Bromacetonitril-Toluol Lösung wird mit einer
Überführungskanüle zu der PPh3-Toluol Lösung hinzugetropft. Nach beendeter
Zugabe wird ein Rückflusskühler aufgesetzt und die Reaktionslösung wird auf 80 °C
erhitzt. Innerhalb von 15 min kann eine Trübung der anfangs klaren Lösung beobachtet
werden, die sich im weiteren Reaktionsverlauf intensiviert. Nach max. 1.5 Stunden wird
das Reaktionsgemisch auf Raumtemperatur abgekühlt über Nacht rühren gelassen.
Der entstandene farblose Feststoff wird an Luft über einen 50 mL Büchnertrichter bzw.
Glasfritte abfiltriert und dreimal mit je 15 mL technischen Pentan gewaschen und im
Vakuum getrocknet. Das gewünschte Produkt kann als farbloser Feststoff (15.3 g, 40.0
mmol, 96 %) isoliert werden.
Literatur-Ausbeute: 7.65 g (96 %),
Kriterium: Ausbeute mindestens 48 %
Eigenschaften: farbloser luftstabiler lipophiler Feststoff, zersetzt sich langsam an
Luft und in Glasfritten.
Charakterisierung:
IR: 2250 cm-1 31P NMR (162.0 MHz, CDCl3): δ / ppm = 21.9 (s). 1H NMR (400.1 MHz, CDCl3): δ / ppm = 7.92 – 7.86 (m, 6 H, CHPPh), 7.75 – 7.71 (m,
3 H, CHPPh), 7.63 – 7.60 (m, 6 H, CHPPh) 6.33 (d, 2JHP = 15.0 Hz, 2 H, PCH2CN). 13C NMR (75.5 MHz, CDCl3): δ / ppm = 136.0 (d, 4JCP = 3.1 Hz; CPPh,para), 134.4 (d, 2JCP = 10.9 Hz; CPPh,ortho), 130.7 (d, 3JCP = 13.4 Hz; CPPh,meta), 116.3 (d, 1JCP =
88.9 Hz; CPPh,ipso), 112.2 (d, 2JCP = 9.4 Hz; PCH2CN), 17.8 (d, 1JCP = 54.7 Hz;
PCH2CN).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
63
Anmerkung: Der Feststoff sollte sogleich weiter umgesetzt oder unter Argon gela-
gert werden.
Herstellung von Triphenylphosphoniumacetonitrilylid
In einem 100 mL-Schlenkkolben werden 0.88 g (22.0 mmol) Natriumhydrid (60%
Dispersion in Mineralöl) geben und viermal mit 10 mL Hexan oder Pentan gewaschen.
Das heißt: Es wird Hexan oder Pentan auf den Feststoff geben. Dann wird ordentlich
gerührt. Dann lässt man den Feststoff absenken und mit einer Überführungskanüle
wird die überstehende Lösung entfernt. Das restliche Lösungsmittel wird im Vakuum
entfernt. Danach werden erst 50 mL Tetrahydrofuran und dann im Gegenstrom das
Phosphoniumsalz (7.65 g, 20.0 mmol) hinzugeben. Es wird ein Blasenzähler
aufgesetzt. Die Suspension wird so lange gerührt bis keine Wasserstoff Entwicklung
mehr beobachtet werden kann (3 - 4 Tage, länger macht auch nichts). Die überstehen-
de Lösung wird mit Hilfe einer Filterkanüle in einen 250 mL-Schlenkkolben überführt.
Sofern die THF Lösung farblos (nicht braun) und somit sauber ist, wird der
zurückbleibende Feststoff dreimal mit 25 mL Dichlormethan extrahiert und mit
derselben Filterkanüle zu demselben 250 mL-Schlenkkolben geben. Ansonsten
werden zwei Fraktionen gebildet: THF und DCM-Fraktion. Das Lösungsmittel/-
gemisch wird im Vakuum entfernt. Falls das Produkt nicht sauber ist, wird der nicht
weiße Feststoff in Dichlormethan gerade so gelöst und mit Hexan oder Pentan wieder
ausgefällt.
Die überstehende Lösung wird mit einer Überführungskanüle entfernt (1 g lösen sich
in 4 mL Dichlormethan). Das gewünschte Produkt kann als farbloser Feststoff (4.82 g,
16.0 mmol, 80 %) isoliert werden.
Literatur-Ausbeute: 4.82 g (80 %),
Kriterium: Ausbeute mindestens 40 %
Eigenschaften: farbloser luftstabiler lipophiler Feststoff, zersetzt sich schnell an Luft
Charakterisierung: 31P NMR (162.1 MHz, CD2Cl2): δ / ppm = 23.2 (s). 1H NMR (400.3 MHz, CD2Cl2): δ / ppm = 7.68 – 7.60 (m, 9 H, CHPPh,ortho,para), 7.57 –
7.50 (m, 6 H, CHPPh,meta), 1.57 (s, PCHCN).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
64
13C NMR (100.7 MHz, CD2Cl2): δ / ppm = 133.1 (d, 2JCP = 10.1 Hz; CPPh,ortho), 133.0
(d, 4JCP = 2.8 Hz; CPPh,para), 129.4 (d, 3JCP = 12.3; Hz; CPPh,meta), 127.9 (d, 1JCP = 91.8
Hz; CPPh,ipso), 127.3 (d, 2JCP = 7.6 Hz; PCHCN), -2.2 (d, 1JCP = 136.0 Hz; PCHCN).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
65
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
66
1,3-di-tert-butyl-imidazolin-2-ylidene-
[bis-(trimethylsilyl)amido] Silber(I)
[Ag(SItBu)(hmds)]
Literatur:
Boysen, N.; Hasselmann, T.; Karle, S.; Rogalla, D.; Theirich, D.; Winter, M.; Riedl, T.;
Devi, A. Angew. Chem. Int. Ed. 2018, 57, 16224.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
67
Chemikalienliste:
Name der Chemikalie Menge
N,N′-Di-tert-butylethylendiamin 2.76 g, 16.0 mmol
n-Butyllithium (1.6 M in Hexan) 3.52 g, 55.0 mmol
Hexamethyldisilazan 8.88 g, 55.0 mmol
Trimthylorthoformat 40 ml (Überschuss)
HCl (4 M in 1,4-Dioxan) 1.17 g, 32.0 mmol
Silber(I)-chlorid 1.97 g, 13.7 mmol
Ameisensäure ca. 0.2 ml
Diethylether ca. 100 ml
Tetrahydrofuran ca. 100 ml
Hexan ca. 130 ml
Einleitung:
In diesem Versuch soll ein Carben stabilierter Silberkomplex für die Atomlagenab-
scheidung (engl. atomic-layer-deposition, ALD) von Silberdünnschichten hergestellt
werden.
Für die Abscheidung von ultra-dünnen metallischen Schichten bei sehr niedrigen Tem-
peraturen findet eine spezielle Art der chemischen Gasphasenabscheidung (engl. che-
mical vapor deposition, CVD) ihre Anwendung. Bei der ALD werden im Vakuum durch
eine sequentielle Zuführung eines metallorganischen Komplexes (Präkursor) und ei-
nem Co-Reaktanten separiert durch Inertgasspülungen das korrespondierende Metall,
Metalloxid, Nitrid oder Sulfid auf einem geeigneten Substrat, meist Silizium, abgeschie-
den. Im Gegensatz zur klassischen Gasphasenabscheidung kommt es bei der diskon-
tinuierlichen Atomlagenabscheidung zu selbst-limitierenden Reaktionen der Präkurso-
ren mit der Oberfläche. Durch diese selbst-limitierenden Reaktionen auf der Oberflä-
che kann die Dicke der Schicht auf atomarem Maßstab kontrolliert werden.
Als Präkursoren für ALD kommen verschiedene, hoch volatile und reaktive organome-
tallische und metallorganische Komplexe mit Alkyl-, Amid-, Sulfid- oder Alkoxid-basier-
ten Ligandensystemen und reaktiven Gase wie Wasser, Ozon, Sauerstoff oder Was-
serstoff zum Einsatz. Die verwendeten metallorganischen Präkursoren sollten dabei
eine ganze Reihe von verschiedenen Eigenschaften für eine erfolgreiche Anwendung

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
68
im korrespondierenden Prozess besitzen. Dazu gehören eine hohe Volatilität bei nied-
rigen Temperaturen, ein definiertes Zerfallsprofil bei gegebenen Prozesstemperatu-
ren, eine unkomplizierte Synthese mit hohen Ausbeuten, eine schwaches toxisches
Potential und speziell für ALD Prozesse eine hohe Reaktivität gegenüber der Oberflä-
che sowie eine hohe Temperaturstabilität. Durch eine geeignete Wahl an Liganden
können nahezu alle Eigenschaften des Präkursors stark beeinflusst werden.
Als Präkursoren für die Abscheidung von Silberdünnschichten existierte bislang nur
eine für die ALD geeignete metallorganische Verbindung [Ag(fod)(PEt3)]. Dieser Sil-
berkomplex besteht aus einem anionischen fluorierten Rückgrat und einem neutralen
Phosphin, welches die Koordinationsumgebung des Silbers sättigt und so eine Bildung
von Dimeren oder Trimeren verhindert. Problematisch ist hierbei jedoch, dass die im
Komplex enthaltenen Elemente wie Sauerstoff, Phosphor und Fluor sich in den Silber-
schichten als Verunreinigungen einlagern können. Dies führt in einer späteren Anwen-
dung (z.B. in Solarzellen als transparente Elektrode) zu Problemen, da diese Verun-
reinigungen die optischen Eigenschaften und die Leitfähigkeit der Silberschicht stark
beeinträchtigen können. Um diese Probleme zu vermeiden und die thermische Stabi-
lität und Reaktivität des Silberkomplexes zu erhöhen wurde ein neuer Silberkomplex
entwickelt, welcher durch die Stabilisierung eines N-heterozyklischen Carbens (NHC)
auf die oben genannten Elemente verzichten kann. Während die Stabilität des Kom-
plexes durch das NHC erreicht wird, kann durch das anionische Hexamethyldisilazanid
eine hohe Reaktivität gegenüber den Oberflächengruppen des Substrats erreicht wer-
den.
Literatur:
Kariniemi, M.; Niinistö, J.; Hatanpää, T.; Kemell, M.; Sajavaara, T.; Ritala, M.; Leskelä,
M. Chem. Mater. 2011, 23, 2901.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
69
Devi, A. Coord. Chem. Rev. 2013, 257, 3332.
Pinna, N.; Knez, M. Atomic Layer Deposition of Nanostructured Materials, Wiley-VCH
Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011.
George, S. M. Chem. Rev. 2010, 110, 111.
Jones, A. C.; Hitchman, M. L. Chemical Vapour Deposition: Precursors, Processes
and Applications, Royal Society of Chemistry, 2008.
Puurunen, R. L. Journal of Applied Physics 2005, 97, 121301.
Leskelä, M.; Ritala, M. Thin Solid Films 2002, 409, 138.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit luft- und lichtempfindlichen Verbindungen
• Vakuumdestillation und Kristallisation unter Schutzgasbedingungen
Charakterisierungsmethoden:
• NMR-Spektroskopie in Lösung (1H, 13C)
Forschungsrelevante Thematik:
• Lithium-organische Chemie
• Silber-organische Chemie
• N-heterozyklische Carbenverbindungen
• Entwicklung von Präkursoren für die Atomlagenabscheidung
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel wie Hexan, Tetrahydrofuran und Diethylether werden nach gängigen
Methoden getrocknet und unter Argon gelagert. Die luft-, und hydrolyseempfindlichen
Edukte und Produkte werden unter Inertgas gehandhabt. Lichtempfindliche Verbin-
dungen werden zusätzlich in Braungläsern gelagert.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
70
Versuchsdurchführung:
Herstellung von 1,3-di-tert-butyl-imidazoliniumchlorid:
In ein sorgfältig ausgeheiztes und mit Magnetrührstäbchen bestücktes Schlenkrohr
werden Trimthylorthoformat (40 mL, Überschuss), N,N′-Di-tert-butylethylenediamin
(2.76 g, 16.0 mmol) gefüllt und die Mischung auf 0 °C heruntergekühlt. Unter starkem
Rühren wird eine Lösung aus HCl in Dioxan (1.17 g, 32.0 mmol, 4 M in 1,4-Dioxan)
langsam mit einer Spritze durch ein auf dem Schlenkrohr aufgestecktes Septum mit
Ausgleichskanüle zugegeben und für 2 Stunden bei Raumtemperatur gerührt. Zur
farblosen Suspension werden ca. 0.5 ml (katalytische Menge) Ameisensäure über eine
Spritze hinzugegeben und unter Zuhilfenahme eines aufgesteckten Rückflusskühlers
wird die Suspension mit einem auf 120 °C geheizten Ölbad so lange refluxiert, bis eine
klare Lösung entstanden ist. Dabei muss je nach Experiment mit mindestens 24 Stun-
den erhitzen gerechnet werden, bis die Reaktion zum Imidazoliumchlorid abgeschlos-
sen ist. Alle volatilen Reaktionsnebenprodukte werden im Unterdruck entfernt, sodass
ein farbloser Feststoff zurückbleibt. Ein weiterer Schritt zur Reinigung des Rohproduk-
tes wird nicht benötigt.
Literatur-Ausbeute: 3.38 g (96 %)
Kriterium: Ausbeute mindestens 90 % von der Literaturausbeute
Eigenschaften: Farbloser bis leicht beiger Feststoff
Charakterisierung: 1H NMR (200 MHz, CDCl3): δ [ppm] = 8.78 (s), 4.02 (s), 1.53 (s).
Herstellung von Lithium-bis(trimethylsilyl)amid Diethylether:
Die Versuchsdurchführung für die Herstellung von Lithium-bis(trimethylsilyl)amid Diet-
hylether ist schon für den Versuch Bis[bis(trimethylsilyl)-amido]zinn beschrieben
worden. Passen Sie die Mengen der Edukte so an, dass Sie unter der Annahme eines
Umsatzes nach angegebener Literaturausbeute ca. 10 g Produkt erhalten. Das Pro-
dukt wird in der Glovebox der AG Devi umgefüllt und aufbewahrt.
Kriterium: Mindestens 6.5 g werden für den folgenden Schritt benötigt.
Eigenschaften: Farbloser und kristalliner Feststoff

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
71
Charakterisierung: 1H NMR (200 MHz, CD3CN): δ [ppm] = 3.42 ppm (q, 8H), 1.12 ppm (t, 12H), 0.04 ppm
(s, 36H).
Herstellung von [Ag(SItBu)(hmds)]:
In ein sorgfältig ausgeheiztes Braunglasschlenkrohr, bestückt mit einem Magnetrühr-
stäbchen, wird Silber(I)-chlorid (1.97 g, 13.7 mmol), 1,3-di-tert-butyl-imidazolinium-
chlorid (3.00 g, 13.7 mmol) und Lithium-bis(trimethylsilyl)amid-Diethylether (6.5 g,
13.7 mmol) zusammen eingewogen. Die Einwaage erfolgt dabei in der Glovebox der
AG Devi. In einem separaten sorgfältig ausgeheiztem Schlenkrohr werden 100 ml Tet-
rahydrofuran auf 0 °C heruntergekühlt. Das gekühlte Tetrahydrofuran wird zu der pulv-
rigen Mischung unter starkem Rühren bei 0 °C mit einer Spritze gegeben. Die klare
Reaktionsmischung wird für mindestens 16 Stunden bei Raumtemperatur gerührt. Das
Lösemittel wird im Vakuum vollständig entfernt. Der feste Rückstand wird mit Hexan
(100 ml) extrahiert. Nach dem vollständigen Absetzen des Feststoffes am Boden des
Schlenkrohrs bei ausgeschalteter Rührung wird die überstehende Lösung unter Be-
nutzung eines Kanülen-Filters in ein sorgfältig ausgeheiztes und mit Magnetrührstab
ausgestattetes Braunglasschlenkrohr kanüliert. Der Rückstand im ersten Kolben wird
nochmals mit 30 ml Hexan gewaschen. Das Lösemittel der vereinten Filtrate wird im
Vakuum solange entfernt, bis eine gesättigte Lösung entsteht. Durch eine Lagerung
des Schlenkrohrs bei –30 °C über Nacht im Kühlschrank bilden sich Kristalle, die an-
schließend von dem überstehenden Hexan über Filtration getrennt werden. Die farb-
losen Kristalle werden im Unterdruck getrocknet und anschließend in einer Glovebox
der AG Devi umgefüllt und gelagert.
Literatur-Ausbeute: 4.5 g (70 %)
Kriterium: Ausbeute mindestens 35 %
Eigenschaften: Farblose Kristalle
Charakterisierung: 1H NMR (400 MHz, Toluol-d8): δ [ppm] = 2.57 (s), 1.24 (s), 1.88 (s), 0.60 (s). 13C NMR (70 MHz, Toluol-d8): δ [ppm] = 205.1 (dd), 55.2 (d), 45.4 (d), 30.9 (d), 7.35 (s).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
72
Kupfer benzol-1,3,5-tricarboxylat
CuBTC / HKUST-1
Literatur:
Klimakow, M.; Klobes, P.; Thünemann, A. F.; Rademann, K.; Emmerling, F. Chem.
Mater. 2010, 22, 5216.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
73
Chemikalienliste:
Name der Chemikalie Menge
1,3,5-Benzoltricarbonsäuren 2,503 g (11,918 mmol)
Kupferacetat Monohydrat 3,564 g (17,875 mmol)
CuBTC ca. 50 mg
Ethanol ca. 500 mL
Einleitung:
Metallorganische Gerüstverbindungen (MOF, metal-organic frameworks) sind poröse
und kristalline Koordinationspolymere. Sie bilden eine recht junge Klasse an Materia-
lien und haben sich in den letzten 20 Jahren zu einer interessanten Stoffklasse anor-
ganischer und organischer Chemiker entwickelt. Dies liegt darin begründet, da sie
nach dem Baukasten-Prinzip zusammengesetzt werden können. Sie bestehen, wie
der Name schon verrät, aus Metallknoten oder Metalloxo-Clustern, die über di- oder
polytopische Linker miteinander verbunden sind. Diese koordinative Bindung führt zur
Ausbildung von mehrdimensionalen Netzwerken. Die Porosität dieser Netzwerke lässt
sich über die Wahl der Linker und Cluster direkt beeinflussen. Wird zum Beispiel eine
verbrückende Phenylgruppe durch eine Biphenylgruppe ersetzt, so vergrößern sich
die Poren des MOFs. Das in diesem Versuch hergestellte CuBTC besteht aus einem
Kupferknoten (aus dimeren Kupferatomen), welcher durch vier Sauerstoffatome der
1,3,5-Benzoltricarbonsäuren koordiniert sind. Dadurch bildet sich ein kubisches Netz-
werk (Fm3�m) mit 9 mal 9 Å großen Poren (grüne Kugeln in der Abbildung). Diese
Poren sind es, die die MOFs zu einer so interessanten Materialklasse machen. So
können diese zum Beispiel mit Gastmolekülen gefüllt werden. Häufige „Gäste“ sind
Gase wie Wasserstoff, Methan und Kohlenstoffdioxid, also Verbindungen die im Zu-
sammenhang mit Energiespeicherung stehen. Aber auch Katalysatoren können in die
Linker oder Metallcluster eingebaut werden oder aber im Netzwerk immobilisiert wer-
den. Durch eine geschickte Wahl der Linker könne MOF weiterhin als Sensoren ein-
gesetzt werden.
In diesem Versuch soll es darum gehen, verschiedenen Synthesetechniken für CuBTC
kennen zu lernen und zu vergleichen. Eine davon ist die sogenannte mechanochemi-
sche Synthese. Mechanochemie beschreibt eine Disziplin der Chemie, in der Reaktion

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
74
durch mechanische Kräfte ausgelöst und aufrechterhalten werden. Besonders interes-
sant wurde dies in den letzten Jahren, da dadurch Reaktionen ohne Lösemittel durch-
geführt werden können. Weiterhin macht ein solche Syntheseführung immer dort be-
sonders Sinn, wo entweder Edukt oder Produkt schwerlöslich sind, da eine Löslichkeit
generell nicht nötig ist. In der Vergangenheit wurde gezeigt das sich dies auch sehr
schön auf MOFs anwenden lässt. So entwickelte die Firma MOF-Technologies eine
der ersten kommerziellen Synthesewege von CuBTC auf der Basis von Mechanoche-
mie. Darüber hinaus werden Sie mit einem theoretischen Modell – einem sog. Kraftfeld
– die Struktur der Verbindung, sowie einer hypothetischen polymorphen Form vorher-
sagen und die berechneten Eigenschaften mit den Ergebnissen Ihrer experimentellen
Charakterisierung vergleichen.
Was Sie lernen:
Synthesemethoden:
• Mechanochemische Synthese in der Kugelmühle
• Umgang mit porösen Materialien
• Insitu Charakterisierung von chemischen Reaktionen
Charakterisierungsmethoden:
• Physisorptionsmessung
• Röntgendiffraktometrie
• Infrarot-Spektroskopie
• Raman-Spektroskopie
Theoretische Methoden:
• Theoretische Modellierung von Strukturen mit einem molekülmechanischen Kraft-
feld
• Topologische Analysen von Netzwerkverbindungen
• Theoretische Vorhersage von Eigenschaften wie Porengrößenverteilungen, Rönt-
gendiffraktogrammen oder Normalmoden

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
75
Forschungsrelevante Thematik:
• Metallorganische Gerüstverbindung
• Gasspeicherung in porösen Materialien
• Simulation von Materialeigenschaften
Literatur:
Furukawa, H.; Cordova, K. E.; O'Keeffe, M.; Yaghi, O. M. Science 2013, 341, 1230444.
Chui; L. C.; Orpen; W. Science 1999, 283, 1148.
Andersen, J.; Mack, J. Green Chem. 2018, 20, 1435.
Amirjalayer, S.; Tafipolsky, M.; Schmid, R. J. Phys. Chem. C 2011, 115, 15133.
Keupp, J.; Schmid, R. Faraday discussions 2018, 211, 79.
Allgemeines zur Sicherheit:
In dem Versuch werden verschiedenen Synthesewege behandelt. Beim Arbeiten mit
mechanischen Geräten sind die gängigen Vorsichtsmaßnahmen einzuhalten. Beim Ar-
beiten mit dem Raman-Laser ist darauf zu achten, dass dieser nur unter den geeigne-
ten Schutzbestimmungen eingesetzt wird.
Vorbereitungen:
Polieren des Plexiglas-Mahlbechers.
Synthese:
(I) CuBTC klassisch:
In einem 250 mL Rundkolben werden 1,3,5-Benzoltricarbonsäuren (2.10 g, 10.0 mmol)
und Kupfer(II) Acetat Monohydrat (2.99 g, 15 mmol) in 100 mL Ethanol gelöst. Die
Lösung wird unter Rückfluss für 2 x 7 Stunden bei 80 °C erhitzt. Das ausgefallene
blaue Pulver wird über eine Fritte abgesaugt und mit 100 mL Wasser und 100 mL
Ethanol gewaschen. Anschließend wird es über Nacht bei 80 °C im Trockenschrank
getrocknet.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
76
Literatur-Ausbeute: 3.74 g (73.4 %)
Kriterium: Ausbeute min. 50 %
(II) CuBTC mechanochemisch:
In einem 14 mL PMMA-Mahlbecher werden 1,3,5-Benzoltricarbonsäuren (0.403 g,
1.918 mmol) und Kupfer(II) Acetat Monohydrat (0.574 g, 2.875 mmol) und eine 10 mm
Mahlkugel aus Zirkonoxid gegeben. Der Mahlbecher wird in die Kugelmühle (MM400)
eingespannt und die Raman-Sonde positioniert. Die Mühle wird 15 Minuten bei 25 Hz
betrieben. Das blaue Pulver wird in eine Fritte überführt und mit 100 mL Wasser und
100 mL Ethanol gewaschen. Anschließend wird es über Nacht bei 80 °C im Trocken-
schrank getrocknet.
Literatur-Ausbeute: 0.824g (84.3 %)
Kriterium: Ausbeute min. 50 %
Charakterisierung:
(I) Stickstoff Physisorption:
Ca. 50 mg der 3 Proben (kommerziell, klassisch und mechanochemisch) werden in
BET-Zellen eingewogen, mit einem Filter versehen und bei 120 °C für 4 Stunden unter
Vakuum aktiviert. Nach der Aktivierung wird die Masse erneut bestimmt, bevor die
Proben bei 77 K im Quadrasorp mit Stickstoff vermessen werden. Neben der BET-
Oberfläche werden auch das Porenvolumen und die Porenradienverteilung bestimmt
und verglichen.
(II) Raman-Spektroskopie:
Während der mechanochemischen Synthese werden konstant Raman-Spektren auf-
gezeichnet. Die anderen beiden Proben werden zusammen mit den Ausgangstoffen
mittels Raman-Spektroskopie analysiert und verglichen. Hierzu wird das getrocknete
Pulver auf den Probenhalter gegeben.
Banden: 1616, 1467, 1009, 830 und 748 cm-1

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
77
(III) PXRD:
Die Proben werden mittels der Scotch-Methode für die PXRD-Messung vorbereitet und
ein Übersichtsdiffraktogramm aufgenommen. Diese werden mit den Referenzdiffrak-
togrammen und den berechneten Werten verglichen.
(IV) Theoretische Rechnungen:
Da MOFs kristalline Strukturen ausbilden, lassen sie sich gut theoretisch modellieren.
Aus diesen Modellen können viele der experimentell zugänglichen Eigenschaften, wie
die, die Sie über die obigen Charakterisierungsmethoden erhalten haben, simuliert
werden. Hierzu muss zunächst die Struktur des zu untersuchenden MOFs aufgebaut
werden. Allerdings können für gegebene Bauelemente verschiedene Netzwerke gebil-
det werden. Man bezeichnet diese auch als Topologien. Für ein 3,4-Netzwerk aus tri-
gonalen, dreifachverknüpften organischen Linkern und quadratisch planaren, vierfach-
verknüpften Kupfer-Dimeren, die wir als Building Blocks (BBs) bezeichnen (siehe Ab-
bildung links!) gibt es zwei hochsymmetrische Topologien, die mit den Kürzeln tbo und
pto bezeichnet werden. Für den Aufbau eines MOFs werden wir diese beiden Topo-
logien benutzen um damit zwei Polymorphe zu erzeugen und beide zu charakterisie-
ren. Diese Methode der Strukturvorhersage, wie dargestellt in der unteren Abbildung,
bezeichnet man als Reverse Topological Approach (RTA). Ihre Aufgabe ist es, die Si-
mulierten Materialeigenschaften mit den experimentellen Daten zu vergleichen und
vorzuschlagen welche der beiden Topologien im Experiment vorliegt.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
78
Aufbau des MOFs:
Um einen MOF in-silico herzustellen benötigen Sie eine dreidimensionale Darstellung
der Topologie sowie die Building Blocks (BBs). Für beides gibt es unter www.mof-
plus.org eine Datenbank, von der Sie benötigten Dateien beziehen können. Die Kon-
struktion selbst führen Sie mit dem weaver Programm durch.
Anschließend optimieren Sie die rein geometrisch konstruierten Strukturen mit einem
speziell für MOFs entwickelten Kraftfeld namens MOF-FF. Hierzu verwenden Sie das
Simulationsprogramm LAMMPS.
Charakterisierung des MOFs:
Die folgenden Schritte führen Sie jeweils für beide Strukturen, die Sie vorher generiert
haben, durch. Zunächst werden sie die Pulverdiffraktogramme der optimierten Struk-
turen (z. B: mit Mercury) simulieren, um diese mit dem experimentell gemessenem zu
vergleichen.
Im nächsten Schritt werden Sie das Porennetzwerk (geometrische Oberfläche, Poren-
größenverteilung und Void Fraction) analysieren. Hierzu werden Sie Zeo++ verwen-
den. Auch diese Daten können z.B. mit experimentell ermittelten Oberflächen und an-
deren Charakteristika verglichen werden.
Im letzten Schritt werden sie das Schwingungsspektrum der generierten MOFs mit
dem Kraftfeld berechnen, um diese mit den gemessenen Raman Spektren zu verglei-
chen und die Schwingungen im Kristall zu verstehen. Dazu werden Sie die Schwin-
gungen mit geeigneten Programmen visualisieren.
Konkrete Arbeitsanweisungen sowie Zugang zu den verwendeten Programmen erhal-
ten sie dann vor Antritt dieses Praktikumsteils von uns.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
79
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
80
1-(4-Iodphenyl)-3-methyl-1H-imidazol-3-ium)-
(µ5-pentamethylcyclopentadienyl)-
iridiumchlorid
[(Cp*)Ir(I-Ph-im-Me)Cl]
Literatur:
Soldevila-Barreda, J. J.; Metzler-Nolte, N. Chem. Rev., 2019, 119, 2, 829.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
81
Chemikalienliste:
Name der Chemikalie Menge
Silbernitrat 175 mg (1,03 mmol) (+ 164 mg)
Natiumhydroxid-Lösung (10M) 10 mL
Iridium(III)chlorid 500 mg (1,67 mmol)
1,2,3,4,5-Pentamethylcyclopenta-1,3-
dien 684,46 mg (5,02 mmol)
Methanol tr. 5 mL
1(4-Iodphenyl)-1H-imidazol 400 mg (1,48 mmol)
Iodmethan 2,1 g (14,8 mmol)
1(4-Iodphenyl)-3-methyl-1H-imidazol-3-
iumiodid (selbst hergestellt) 400 mg (0,986 mmol)
(Ir-Cp*-Dimer) Di-µ-chlorodichloro-
bis[(1,2,3,4,5-n)-1,2,3,4,5-pentamethyl-
2,4-cyclopentadien-1-yl]diiridium (selbst
hergestellt)
386,74 mg (485,42 µmol)
Silberoxid (selbst hergestellt) 60,13 mg (485,42 µmol)
Dichlormethan tr. / p.a. ca. 100 mL
n-Pentan p.a. 15 mL
Diethylether p.a. 15 mL
Tetrahydrofuran p.a. ca. 50 mL
Celite ca. 50 g
Acetonitril p.a. ca. 50 mL
Ethylacetat p.a. ca. 500 mL
Gesättigte NaCl-Lösung (brine) ca. 5 mL
Einleitung:
Nach der Entdeckung von Cisplatin [1] und Anstrengungen, alternative Platinkomplexe
mit ähnlichen antineoplastischen Aktivitäten zu finden, verlagerte sich der Schwer-
punkt der Metallhaltigen Medikamentenforschung vom Platin zu anderen Metallzen-
tren. Hierbei war das Ziel platinbezogene Resistenzmechanismen zu überwinden [2]

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
82
und die potenzielle medizinische Relevanz anderer Metallzentren zu untersuchen. Da
Iridiumkomplexe in ihrem Oxidationszustand +I. quadratische planare Geometrien wie
Cisplatin haben, wurde kurz nach der Entdeckung des Cisplatins, Iridium-Komplexes
mit [Ir(I)(COD)(acac)] eine potentielle antineoplastische Aktivität durch Heilung von
100% der Mäuse mit Ehrlich-Aszites-Sarkom nachgewiesen. Der Wirkmechanismus
von Cisplatin basiert auf der Verdrängung der eher labilen Chloridoliganden durch
Wasser, die dann durch biologische Zielmoleküle wie z. B. DNA, ersetzt werden.
Um die mögliche Verwendung von Iridiumkomplexen als Krebsmedikamente zu unter-
suchen, wurden inzwischen Studien zu verschiedenen Arten von Iridiumkomplexen im
Oxidationszustand +I und +III durchgeführt [3,4]. Iridium hat in seinen beiden biolo-
gisch relevanten Oxidationsstufen +I und +III einige attraktive Eigenschaften. So weist
beispielsweise Ir III mit der elektronischen Konfiguration d6 ausgezeichnete Lumines-
zenzeigenschaften auf, die für die zelluläre Bildgebung genutzt wurden.
Obwohl das erste Beispiel einer zytotoxischen Iridiumverbindung ein Iridiummetall-
zentrum in der Oxidationsstufe +I (d8-Elektronikkonfiguration) besitzt, enthalten die
meisten der aktuellen Beispiele von Iridiumkomplexen, die auf dem Gebiet der medi-
zinischen Chemie untersucht werden, ein Iridium in der Oxidationsstufe +III. Dies ist
insofern nachvollziehbar, da Iridium in der Oxidationsstufe +III als eines der inertesten
Low-Spin d6-Metallionen angesehen wird. Der Ligandenaustausch z.B. durch Biomo-
leküle ist stark vom Ligandsystem abhängig, was bei Wahl des richtigen Ligandsys-
tems zu sehr stabilen Komplexen führt. Im Gegensatz zu Pt II werden Ir I-Verbindun-
gen jedoch leicht zu Ir III-Derivaten oxidiert. Diese Redox-Chemie wird elegant in ka-
talytischen Metalldrogen auf Ir-Basis eingesetzt, wie sie kürzlich von den Gruppen Sa-
dler, Liu und anderen untersucht wurde [5].
Die Änderung der Oxidationsstufe führt zu einer Änderung der Geometrie des Kom-
plexes von quadratisch planar zu oktaedrisch. Dadurch würden zwei weitere Koordi-
nationsstellen für eine mögliche Interaktion mit biologischen Zielmolekülen zu Verfü-
gung stehen. Dieser oxidative Wirkmechanismus wird für andere Metallhaltige Medi-
kamente wie z.B. Ferrocifen, beschrieben. Basierend darauf wurde angenommen,
dass der Wirkmechanismus von zytotoxischen Iridiumkomplexen in ihrer Oxidations-
stufe +I, auf der oxidativen Zugabe von biologischen Zielmolekülen und daraus resul-
tierenden Ausbildung von Ir(III)-Komplexen basieren könnte. Durch diese Änderung
des Oxidationszustandes, würde mit Iridium(III) als eines der inertesten Low-Spin d6-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
83
Metallionen, ein vermeintlich sehr stabiles Bioaddukt entstehen, das die korrekte Funk-
tion des jeweiligen Biomoleküls beeinträchtigen würde.
Literatur:
1. Rosenberg, B.; van Camp, L.; Krigas, T. Nature, 1965, 205, 698.
2. Dilruba, S.; Kalayda, G. V. Chemother Pharmacol, 2016, 77, 1103.
3 Gothe, Y.; Marzo, T.; Messori, L.; Metzler-Nolte, N. Chem. Eur. J., 2016, 22, 12487.
4. Graf, M.; Gothe, Y.; Metzler-Nolte, N.; Czerwieniec, R.; Sünkel, K. J. Organo-
met.Chem., 2014, 765, 46-52
5. Z. Liu, P.J. Sadler, Acc. Chem. Res., 2014, 47, 1174
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit luftempfindlichen Verbindungen
• Mikrowellensynthese, säulenchromatografische Aufreinigung
Charakterisierungsmethoden:
• NMR-Spektroskopie in Lösung
• Dünnschichtchromatografie
Forschungsrelevante Thematik:
• Koordinations-Chemie
• Iridium-organische Chemie
• N-heterocyclische Carbene
• Entwicklung von Metallhaltigen Medikamenten
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel wie Dichlormethan und Methanol werden nach gängigen Methoden

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
84
getrocknet und unter Argon gelagert. Die luft- und hydrolyseempfindlichen Edukte und
Produkte werden unter Inertgas gehandhabt. Besondere Vorsicht gilt bei Iodmethan,
da es in der kanzerogenen Kategorie 2 und zusätzlich in der akuttoxischen
Kategorie 3 (Einatmen) eingestuft ist.
Versuchsdurchführung:
Herstellung von Silberoxid (Ag2O):
In einem 50 mL Rundkolben mit Magnetrührstäbchen werden 10 mL 10 M NaOH-Lö-
sung vorgelegt. Danach wird langsam unter Rühren Silbernitrat (175 mg, 1,03 mmol)
dazugegeben. Bei der Zugabe von AgNO3 fällt ein grau-schwarzer Feststoff (Ag2O)
aus. Die Reaktionsmischung wird für weitere 15 Minuten bei Raumtemperatur gerührt.
Nach Ablauf der Rührzeit wird die Suspension filtriert und mit 15 mL dest. Wasser
gewaschen. Der Feststoff wird anschließend im Vakuum für mind. 3 Stunden getrock-
net. (Achtung: Sowohl das Edukt (AgNO3) als auch das Produkt (Ag2O) sind lichtemp-
findlich. Daher muss der Reaktionskolben und der Aufbewahrungskolben mit Alufolie
umwickelt werden. Silberoxid ist zusätzlich noch luftempfindlich. Deshalb muss es un-
ter Argon gelagert werden. Die Synthese selbst muss nicht unter Schutzgas durchge-
führt werden.)
Literatur-Ausbeute: 100 mg (84 %)
Kriterium: Ausbeute mindestens 50 %
Eigenschaften: grau-schwarzer Feststoff
Charakterisierung: PXRD
Herstellung von Di-µ-chlorodichlorobis[(1,2,3,4,5-n)-1,2,3,4,5-pentamethyl-2,4-cyclo-
pentadien-1-yl]diiridium [(Cp*)IrCl2]2:
Achtung: Mikrowellensynthese!
Ein 10 mL Mikrowellenröhrchen, ausgestattet mit Magnetrührstäbchen, wird mit Iri-
dium(III)chlorid (500 mg, 1,67 mmol) befüllt. Anschließend werden 5 mL trockenes Me-
thanol zugegeben und das Röhrchen mit einem Septum verschlossen. Danach wird
für 5 Minuten Argon durch die Lösung geblubbert. Anschließend wird 1,2,3,4,5-Penta-
methylcyclopenta-1,3-dien (684,46 mg, 5,02 mmol) zu der Reaktionslösung gegeben.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
85
Das Septum wird mit einem Deckel für das Mikrowellenröhrchen ausgetauscht und das
Röhrchen in die Mikrowelle gegeben. Mikrowellen-Programm: „Ir-Dimer“ (140°C, 3
min). Bei der Reaktion entsteht ein orange-roter Feststoff. Das überstehende Lösemit-
tel wird abdekantiert und der Feststoff 3 x mit 5 ml Pentan und anschließend 3 x mit 5
ml Diethylether gewaschen. Der Feststoff wird mit wenig Dichlormethan in einen
Schlenkkolben überführt und das Lösemittel anschließend im Vakuum entfernt und der
Rückstand bei Raumtemperatur im Unterdruck getrocknet.
Literatur-Ausbeute: 500 mg (75 %)
Kriterium: Ausbeute mindestens 50 %
Eigenschaften: Orange-roter Feststoff
Charakterisierung: 1H NMR (200 MHz, DMSO-d6): δ [ppm] = 1,63 (s, 30H).
Herstellung von 1(4-Iodphenyl)-3-methyl-1H-imidazol-3-iumiodid [I-Ph-Im-Me]+ I-:
Ein 10 mL Schlenkrohr/Schlenkkolben mit Magnetrührstäbchen wird im Argon-gegen-
strom mit 1(4-Iodphenyl)-3-methyl-1H-imidazol (400 mg, 1,48 mmol) befüllt und mehr-
mals sekuriert. Anschließend wird Iodmethan (2,1 g, 14,8 mmol) zugegeben und die
Reaktionslösung für 24 h bei Raumtemperatur gerührt. Ein Becherglas wird mit Tetra-
hydrofuran (50 mL) befüllt und die Reaktionslösung wird in das Becherglas überführt.
Dabei fällt ein heller Feststoff aus. Die so entstandene Suspension wird filtriert und mit
Tetrahydrofuran gewaschen. Der Filterkuchen wird in Acetonitril gelöst und in einen
Schlenkkolben überführt. Das Lösemittel im Vakuum entfernt und der Feststoff ge-
trocknet.
Literatur-Ausbeute: 544,8 mg (86 %)
Kriterium: Ausbeute mindestens 50 %
Eigenschaften: farbloser Feststoff
Charakterisierung: 1H NMR (200 MHz, DMSO-d6): δ [ppm] = 9,74 (s, 1H), 8,27 (s, 1H), 8,04 (d, 2H), 7,93
(s, 1H), 7,57 (d, 2H), 3,93 (s, 3H).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
86
Herstellung von 1-(4-Iodphenyl)-3-methyl-1H-imidazol-3-ium)-(µ5-pentamethyl-cyclo-
pentadienyl)-iridiumchlorid [(Cp*)Ir(I-Ph-Im-Me)Cl]:
Achtung: Die angegebenen Mengen beziehen sich auf 400 mg 1(4-Iodphenyl)-3-
methyl-1H-imidazol-3-iumiodid (386,74 mg Ir-Dimer, 60,13 mg Ag2O). Passen Sie
Ihre Mengen bitte entsprechend Ihrer Ausbeute an!
In einem 250 mL Schlenkkolben wird 1(4-Iodphenyl)-3-methyl-1H-imidazol-3-iumiodid
(400 mg, 0,986 mmol) im Argon-Gegenstrom vorgelegt. Nach der Zugabe von Ag2O
(60,13 mg, 485,42 µmol) wird der Schlenkkolben mehrmals sekuriert. Anschließend
werden die beiden Feststoffe in 90 mL trockenem Dichlormethan gelöst und über
Nacht bei Raumtemperatur gerührt. Dabei entsteht eine weiß-trübe Lösung. Anschlie-
ßend wird Di-µ-chlorodichlorobis[(1,2,3,4,5-n)-1,2,3,4,5-pentamethyl-2,4-cyclopenta-
dien-1-yl]diiridium (386,74 mg, 485,42 µmol) zugegeben und wieder über Nacht bei
Raumtemperatur gerührt. Die Reaktionsmischung färbt sich orange. Danach wird die
Reaktionslösung über Celite filtriert und das Lösemittel vom Filtrat im Vakuum entfernt.
Von dem entstandenen Feststoff wird eine Test-DC angefertigt (in 100 % Ethylacetat),
um zu überprüfen ob nur eine oder zwei Spezies entstanden sind. (Da die Edukte un-
terschiedliche Gegenanionen haben kann eine Produktmischung mit verschiedenen
Gegenanionen entstehen) [Rf-Wert vom Chlorid-Komplex: 0,55, Rf-Wert vom Iodid-
Komplex: 0,79] Falls zwei Spezies entstanden sind (sehr wahrscheinlich) wird der
Feststoff in Acetonitril gelöst und mit 10 %, der verwendeten Menge an Acetonitril,
Wasser versetzt. Anschließend wird Silbernitrat (164 mg, 0,965 mmol) zu der Lösung
gegeben. Die Reaktionslösung wird für mind. eine Stunde bei Raumtemperatur ge-
rührt. Dabei wird die Lösung trüb, da AgI ausfällt. Anschließend wird die Suspension
mit ca. 5 mL brine versetzt und für weitere 30 Minuten bei RT gerührt. Danach wird
das Acetonitril im Vakuum entfernt sodass nur die wässrige Phase und der Feststoff
zurückbleibt. Die wässrige Phase wird mehrmals mit Dichlormethan extrahiert. Nach
der Extraktion wird Dichlormethan im Vakuum entfernt und vom Feststoff wird eine
zweite Test-DC angefertigt, um zu überprüfen, ob das gewünschte Produkt phasenrein
vorliegt. Falls nicht, wird das Produkt in 100 % Ethylacetet säulenchromatografisch
aufgereinigt. Die Produktfraktion wird im Vakuum getrocknet.
Ein Teil des Produktes wird in wenig Dichlormethan gelöst und die Lösung zwecks
Kristallisation in den Kühlschrank gestellt.
Literatur-Ausbeute: 360 mg (62 %)
Kriterium: Ausbeute mindestens 40 %

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
87
Eigenschaften: orangefarbener Feststoff
Charakterisierung: 1H NMR (200 MHz, DMSO-d6): δ [ppm] = 7,92 (d, 1H), 7,85 (s, 1H), 7,48 (d, 2H), 7,27
((s), 2H), 3,89 (s, 3H), 1,70 (s, 15H),

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
88
Tris-(2-ammonium-ethyl)-phosphin
Trichlorid
P(CH2CH2NH2)3 · 3 HCl
Literatur:
Sietzen, M.; Batke, S.; Merz, L.; Wadepohl, H.; Ballmann, J. Organometallics 2015, 34,
1118.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
89
Chemikalienliste:
Name der Chemikalie Menge
Natrium 1.60 g (68.6 mmol)
Naphthalin 220 mg (1.71 mmol)
1,2-Dimethoxyethan (DME) 60 mL
Phosphortrichlorid
3-Methyl-3-pentanol
Silyl-geschütztes 2-Chloroethylamin
n-Butyllithium (1.6 M in Hexan)
n-Pentan
1 mL (11.4 mmol)
2.8 mL (22.9 mmol)
7.61 g (34.3 mmol)
15.7 mL (25.2 mmol)
200 mL
Diethylether 190 mL
Ethanol 110 mL
Salzsäure (2 M in Diethylether) 52 mL (104 mmol)
Einleitung:
Tris-(2-aminoethyl)amin (Tren) ist einer der bekanntesten tripodalen Liganden und da-
mit von großem Interesse in der Koordinationschemie. Als tetradentater Ligand bildet
Tren üblicherweise stabile Koordinationsverbindungen mit Übergangsmetallen der
Oxidationsstufen +II und +III, indem die Stickstoff-Donoratome eine sterisch abschir-
mende Tasche um das Metallzentrum bilden (Abbildung 1, A).
Während in der Literatur eine Vielzahl von Koordinationsverbindungen mit Tren be-
schrieben sind, ist kaum etwas über das Phospha-Derivat Tris-(2-aminoethyl)phosphin
(Phospha-Tren) bekannt. Durch den Austausch des Stickstoffatoms im Brückenkopf
durch Phosphor, werden harte und weiche Donor-Eigenschaften vereint. Dieser Hyb-
rid-Charakter erlaubt eine hohe elektronische Flexibilität am Metallzentrum, sodass
eine Vielzahl an Metallen in unterschiedlichen Oxidationsstufen koordiniert werden
können. Ob Phospha-Tren dabei als tetra- oder tridentater Ligand (Abbildung 1, A und
B) agiert, ist noch unklar. Allgemein sind für Trisamido-Phosphine beide Fälle in der
Literatur bekannt und von der Flexibilität des P-C-N-Linkers abhängig.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
90
Abbildung 1. Potentielle Bindungsmodi für die Liganden Tren und Phospha-Tren.
Literatur:
Schrock, R. R. Acc. Chem. Res. 1997, 30, 9.
Kuzu, I.; Krummenacher, I.; Meyer, J.; Armbruster, F.; Breher, F., Dalton Trans. 2008,
5836.
Sietzen, M.; Batke, S.; Merz, L.; Wadepohl, H.; Ballmann, J. Organometallics 2015, 34,
1118.
Sietzen, M.; Wadepohl, H.; Ballmann, J. Organometallics 2014, 33, 612.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Unterdrucktechnik
• Umgang mit Phosphinen
Charakterisierungsmethoden:
• NMR-Spektroskopie
Forschungsrelevante Thematik:
• Phosphine für die Ligandensynthese
• Koordinationschemie
• HSAB-Konzept

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
91
Allgemeines zur Sicherheit:
Der Versuch wird unter Argon und Ausschluss von Sauerstoff durchgeführt. Die luft-
und hydrolyseempfindlichen Intermediate werden unter Argon gehandhabt. Die Ver-
wendung von geschlossenen Apparaturen ist strengstens untersagt.
Da Phosphine einen charakteristischen sehr starken Geruch besitzen, sind Glasgeräte
prinzipiell im Abzug zu säubern und Abfälle nur in geschlossenen Gefäßen zu den
Sammelbehältern zu transportieren. Die anfallenden Reaktionsrückstände sind sach-
gerecht unschädlich zu machen und direkt zu entsorgen.
Versuchsdurchführung:
Ein 250 mL-Dreihalskolben ausgestattet mit Magnetrührstab, Abgashahn und Tropf-
trichter mit Young-Hahn wird sekuriert. Im Argon-Gegenstrom wird der Kolben mit
40 mL DME befüllt und unter Rühren werden Natrium (1.60 g, 68.6 mmol) und Naph-
thalin (220 mg, 1.71 mmol) zugegeben. Zu der dunkelgrünen Lösung wird PCl3 (1 mL,
11.4 mmol) in 5 mL DME zugetropft. Im Anschluss wird das Reaktionsgemisch über
Nacht gerührt, woraufhin es sich schwarz färbt.
In einem nächsten Schritt wird 3-Methyl-3-pentanol (2.8 mL, 22.9 mmol) in 5 mL DME
langsam zu der Lösung hinzugetropft und es wird für 5 Stunden bei Raumtemperatur
gerührt. Die hellbraune Suspension wird auf -78 °C gekühlt und Silyl-geschütztes
2-Chloroethylamin (1 eq., 2.54 g, 11.4 mmol) in 5 mL DME wird zugetropft. Daraufhin
wird das Reaktionsgemisch für 30 Minuten bei -78 °C gerührt, für 2 Stunden bei Raum-
temperatur und für 3 Stunden bei 60 °C.
Das Reaktionsgemisch wird wieder auf -78 °C gekühlt und Silyl-geschütztes 2-Chloro-
ethylamin (1 eq., 2.54 g, 11.4 mmol) in 5 mL DME wird zugetropft, gefolgt von n-Butyl-
lithium (7.9 mL, 12.6 mmol). Nachdem einige Minuten bei -78 °C gerührt wurde, wird
das Reaktionsgemisch für 3 Stunden bei Raumtemperatur gerührt. Das Lösemittel
wird vollständig im Unterdruck entfernt. Der Rückstand wird in 100 mL n-Pentan auf-
genommen und die Suspension unter Inertgas-Bedingungen über Celite filtriert. Das
Filtrat wird im Unterdruck bis zur Trockenen eingeengt. Der gelbe, ölige Rückstandwird
in 150 mL Diethylether aufgenommen und auf -78 °C gekühlt. Wieder werden Silyl-
geschütztes 2-Chloroethylamin (1 eq., 2.54 g, 11.4 mmol) in 5 mL DME, gefolgt von n-
Butyllithium (7.9 mL, 12.6 mmol) zugetropft und das Reaktionsgemisch wird bei Raum-
temperatur über Nacht gerührt.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
92
Das Lösemittel wird im Unterdruck vollständig entfernt. Der Rückstand wird in 100 mL
n-Pentan aufgenommen und die Suspension wird unter Inertgas-Bedingungen über
Celite filtriert. Das Filtrat wird wieder bis zur Trockenen eingeengt. Der Rückstand wird
in 100 mL Ethanol aufgenommen. Die Suspension wird auf 0 °C gekühlt und Salzsäure
(2 M in Diethylether, 52 mL) wird langsam zugetropft. Das Reaktionsgemisch wird für
2 Stunden bei Raumtemperatur gerührt. Der resultierende weiße Feststoff wird bei
0 °C an Luft abfiltriert, mit Ethanol (10 mL) und Diethylether (2 x 20 mL) gewaschen
und anschließend im Unterdruck getrocknet.
Literatur-Ausbeute: 2.55 g (82 %, basierend auf PCl3)
Kriterium: 1.30 g (40 %)
Charakterisierung: 1H-NMR (D2O, 250 MHz): δ = 3.29-3.20 (PCH2CH2NH3Cl, 6 H, m); 2.09-2.02
(PCH2CH2NH3Cl, 6 H, m) ppm. 13C{1H}-NMR (D2O, 63 MHz): δ = 37.0
(PCH2CH2NH3Cl, d); 23.3 (PCH2CH2NH3Cl, d) ppm. 31P{1H}-NMR (D2O, 101 MHz):
δ = -42 ppm.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
93
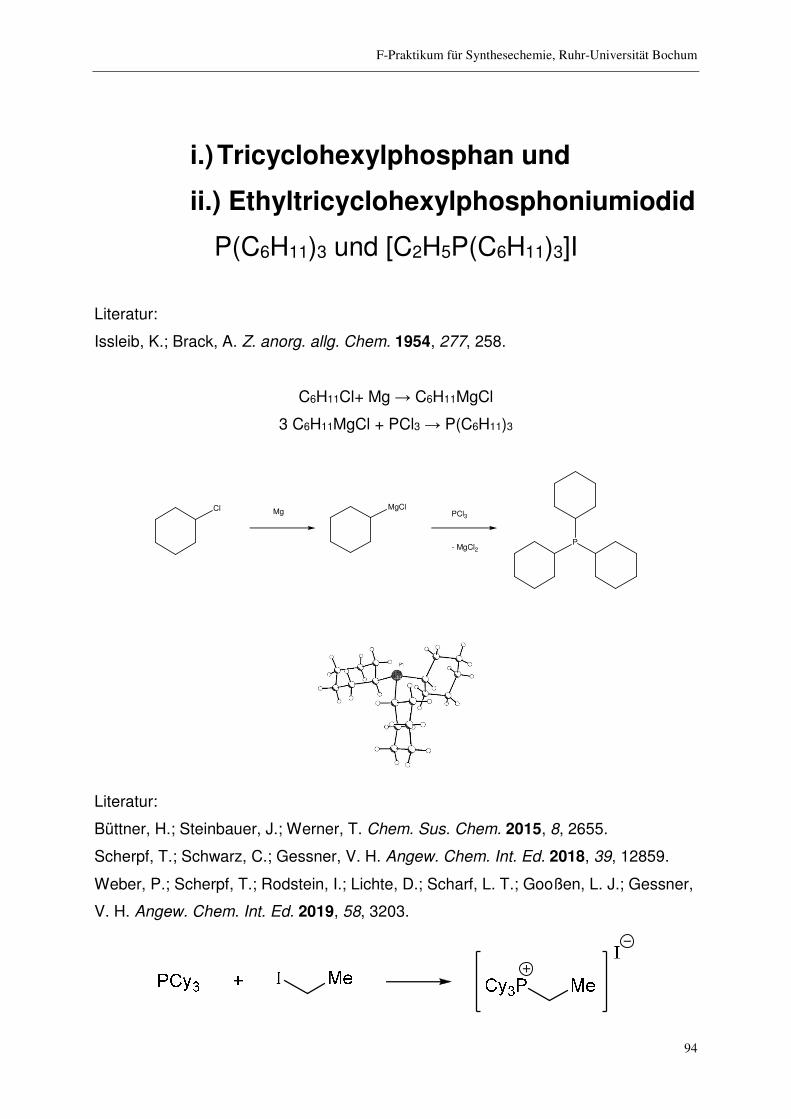
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
94
i.) Tricyclohexylphosphan und
ii.) Ethyltricyclohexylphosphoniumiodid
P(C6H11)3 und [C2H5P(C6H11)3]I
Literatur:
Issleib, K.; Brack, A. Z. anorg. allg. Chem. 1954, 277, 258.
C6H11Cl+ Mg → C6H11MgCl
3 C6H11MgCl + PCl3 → P(C6H11)3
Cl MgMgCl
PCl3
P- MgCl2
Literatur:
Büttner, H.; Steinbauer, J.; Werner, T. Chem. Sus. Chem. 2015, 8, 2655.
Scherpf, T.; Schwarz, C.; Gessner, V. H. Angew. Chem. Int. Ed. 2018, 39, 12859.
Weber, P.; Scherpf, T.; Rodstein, I.; Lichte, D.; Scharf, L. T.; Gooßen, L. J.; Gessner,
V. H. Angew. Chem. Int. Ed. 2019, 58, 3203.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
95
Chemikalienliste:
i.) Tricyclohexylphosphan
Name der Chemikalie M [g/mol] Menge
Chlorcyclohexan 118.6 28.0 mL (236 mmol)
Magnesiumspäne 24.3 6.08 g (250 mmol)
Phosphortrichlorid 137.3 6.24 mL (71.3 mmol)
Iod 126.9 0.1 g
Diethylether 300 mL
Aceton
ii.) Ethyltricyclohexylphosphoniumiodid
Name der Chemikalie M [g/mol] Menge
Tricyclohexylphosphan 280,2 2,0 g (7,2 mmol)
Iodethan 155,9 1,2 g (7,8 mmol)
Toluol ca. 25 ml
Einleitung:
Tricyclohexylphosphan:
Phosphane zählen wegen ihrer vielfältigen Koordinationschemie zu den bedeutends-
ten Liganden der Übergangsmetallchemie und haben entscheidend zu ihrer Entwick-
lung beigetragen. Die Anwendung von Phosphanen als Liganden in der Übergangs-
metallkatalyse hat auch zu einer Vielzahl von neuen Synthesenmöglichkeiten in der
Industrie geführt (z.B. Grubbs-Katalysatoren).
Sie sind im Vergleich zu ihren leichteren Homologen, den Aminen, weiche Basen und
bilden daher stabile Verbindungen mit Übergangsmetallen aus (HSAB-Konzept). Es
können sowohl freie Phosphane (PR3, Oxidationsstufe +III) als auch oxidierte Phos-
phane (z.B. P(=O)R3, P(=S)R3, P(=NR)R3, Oxidationsstufe +V) als Liganden einge-
setzt werden.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
96
Freie Phosphane sind σ-Donoren und π-Akzeptoren, d. h., dass Elektronendichte vom
σ-Orbital in ein unbesetztes d-Orbital des Metalls und von einem besetzten d-Orbital
des Metalls in das σ*-Orbital des Phosphans (π-Symmetrie) gegeben wird. Da die Or-
bitalenergie des σ*-Orbitals bei Phosphanen mit elektronenziehenden Resten niedri-
ger ist, ist deren π-Akzeptorfähigkeit größer.
Vorteilhaft ist, dass die Eigenschaften von Phosphanen je nach Substitutionsmmuster
nach Wunsch eingestellt werden können. Der Tolman Electronic Parameter beschreibt
dabei die elektronischen Eigenschaften bzw. die Donor- bzw. Akzeptorfähigkeit eines
Liganden. Seine Bestimmung erfolgt durch Messung der CO-Schwingung in dem ent-
sprechenden Nickeltricarbonylphosphankomplex mittels Infrarotspektroskopie. Der
Tolman Cone Angle beschreibt den sterischen Anspruch des Phosphans. Durch Ver-
brückung zweier Phosphane können außerdem Chelatliganden erhalten werden, die
zur weiteren Stabilisierung beitragen durch den Bisswinkel charakterisiert werden.
Durch das gezielte Einstellen der Eigenschaften der Phosphanliganden lassen sich so
die Eigenschaften der Metalle in den entsprechenden Komplexen einstellen und somit
ihre Reaktivität/Aktivität kontrollieren. Daher ist die Synthese verschiedener Phos-
phane für die Übergangsmetallkatalyse von größter Wichtigkeit.
Ethyltricyclohexylphosphoniumiodid:
In diesem Praktikum wird die Vorstufe eines elektronenreichen Ylid-funktionalisierten
Phosphans (YPhos) synthetisiert, welches durch die starken Donoreigenschaften, viel-
versprechende Ergebnisse in Übergangsmetall-katalysierten Reaktionen vorweisen
konnte. Erste Einsätze des Liganden in Gold-katalysierten Hydroaminierungen als
auch Palladium-katalysierten Buchwald-Hartwig-Aminierungen waren sehr erfolgreich.
Literatur:
Mason, R.; Meek, D. W. Angew. Chem. 1978, 90, 195.
Tolman, C. A. Chem. Rev. 1977, 77, 313.
Chen, L.; Ren, P.; Carrow, B. P. J. Am. Chem. Soc. 2016, 138, 6392.
Niemeyer, Z. L.; Milo, A.; Hickey, D. P.; Sigman, M. S. Nat. Chem. 2016, 8, 610.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
97
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit reaktiven, organometallischen Substanzen
• Entgasen von Lösemitteln
• Umkristallisation unter Schutzgas
• NMR-Experimente
Charakterisierungsmethoden:
• NMR-Spektroskopie
Forschungsrelevante Thematik:
• Komplexchemie
• Magnesiumorganyle
• Phosphane und Ylid-Substituierte Phosphane
• Komplexchemie und homogene Katalyse
Vorbereitungen:
1. Trocknung von Lösemitteln und Reagenzien
2. Entgasen von Wasser und Aceton
3. Ausheizen der Apparatur
Allgemeines zur Sicherheit:
Der Versuch wird unter Argon in ausgeheizten Apparaturen durchgeführt. Lösemittel
werden nach gängigen Methoden getrocknet und unter Argon gelagert. Die luft- und
hydrolyseempfindlichen Produkte werden unter Inertgas gehandhabt. Phosphortrich-
lorid ist toxisch, daher ist Hautkontakt und Einatmen zu vermeiden. Grignard-Reagen-
zien reagieren stark basisch. Aktiviertes Magnesium muss vorsichtig deaktiviert wer-
den.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
98
Synthese von Tricyclohexylphosphan:
In einen Schlenk-Zweihalskolben mit Rückflusskühler und Tropftrichter werden 6.08 g
Magnesium gegeben und im Unterdruck ausgeheizt. 0.1 g Iod werden im Argongegen-
strom hinzugegeben und über Nacht ohne Lösemittel gerührt. Am nächsten Tag wird
ein Blasenzähler aufgesetzt, 20 mL trockenes Diethylether hinzugegeben und der
Tropftrichter mit 40 mL trockenem Diethylether und 28.0 mL Chlorcyclohexan befüllt.
10 mL werden ohne Rühren hinzugegeben. Sobald die Reaktion startet (Sieden des
Diethylethers, Trübung der Lösung), wird die restliche Lösemittelmenge unter Rühren
langsam hinzugetropft, sodass die Lösung konstant siedet. Nach Beendigung der Zu-
gabe wird die Lösung eine halbe Stunde refluxiert.
In einem zweiten ausgeheizten Dreihalskolben mit Rückflusskühler mit Blasenzähler,
Tropftrichter und Thermometer werden unter Schutzgas 6.24 mL Phosphortrichlorid
und 160 mL trockenem Diethylether vorgelegt. Die Grignard-Lösung wird über eine
Filterkanüle in den Tropftrichter kanüliert. Bei 0° C wird die Grignard-Lösung langsam
zu der Phosphortrichlorid-Lösung hinzugetropft (Temperaturkontrolle!). Nach Beendi-
gung der Zugabe wird die Lösung zwei Stunden refluxiert.
Nach Abkühlen der Reaktionslösung wird diese über eine Schutzgasfritte filtriert. Der
Feststoff wird zweimal mit 100 mL trockenem Diethylether gewaschen. Das Filtrat wird
mit 100 mL entgastem destillierten Wasser gewaschen. Die wässrige Phase wird drei
Mal mit je 60 mL trockenem Diethylether extrahiert. Die vereinigten organischen Pha-
sen werden mit Natriumsulfat getrocknet und abfiltriert. Das Lösemittel wird im Unter-
druck entfernt und der verbleibende Feststoff unter Schutzgas aus entgastem Aceton
(ca. 25 mL) umkristallisiert. Am nächsten Tag wird der Feststoff über eine Umkehrfritte
filtriert, anschließend drei Mal mit je 25 mL kaltem Aceton gewaschen und der Feststoff
im Unterdruck getrocknet. Der Feststoff wird unter Schutzgas (Glovebox) abgewogen
und in einem Schlenkkolben gelagert.
NMR-Experimente:
Zu Tricyclohexylphosphan (20 mg) in deuteriertem Chloroform werden im NMR-Rohr
unter Schutzgas wenige Milligramm Schwefel bzw. ein Tropfen Boran-Tetrahydro-
furan-Addukt gegeben.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
99
Literaturausbeute: 9.4 g (47 %)
Kriterium: Ausbeute mindestens 4.6 g (23 %)
Charakterisierung:
Tricyclohexylphosphan:
Farbloser Feststoff 31P NMR (CDCl3): δ = 9.2 (s) ppm.
Boran-Tricyclohexylphosphan-Addukt: 31P NMR (CDCl3): δ = 28.0 (br) ppm.
Trycyclohexylthiophosphoran: 31P NMR (CDCl3): δ = 62.4 (s) ppm.
Synthese von Ethyltricyclohexylphosphoniumiodid:
Bitte rechnen Sie die Ansätze entsprechend der Menge PCy3, welche Sie im vorheri-
gen Versuch synthetisiert haben.
In einem Schlenk-Kolben wird 2.0 g (7.2 mmol, 1 eq.) Tricyclohexylphosphan in 10 ml
trockenem Toluol gelöst und der Lösung wird über eine Spritze etwa 1.2 g (7.8 mmol,
1.1 eq.) Ethyliodid innerhalb von wenigen Minuten zugetropft. Die Reaktionsmischung
wird für etwa 6 Stunden bei 60 °C gerührt, währenddessen fällt langsam ein weißer
Feststoff aus. Nach 6 Stunden wird die Lösung auf Raumtemperatur aufgewärmt und
über Nacht bei Raumtemperatur gerührt. Anschließend wird der weiße Feststoff über
eine Umkehrfritte filtriert und drei Mal mit jeweils 5 ml Toluol gewaschen. Der Feststoff
wird in der Umkehrfritte über einen ganzen Praktikumstag im Unterdruck getrocknet.
Literatur-Ausbeute: 3.0 g (98 %)
Kriterium: Ausbeute mindestens 50 %
Eigenschaften: Farbloser Feststoff
Charakterisierung: 1H NMR (400 MHz, CDCl3) δ = 1.23 – 1.41 (m, 6H), 1.41 – 1.69 (m, 12H), 1.74 – 1.87
(m, 3H), 1.87 – 1.98 (m), 1.98 – 2.15 (m, 6H), 2.46 – 2.79 (m, 5H). 31P {1H} NMR (162 MHz, CDCl3) δ = 33.3 ppm.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
100
Anorganische Metallsulfide
Pentlandit – Fe4.5Ni4.5S8
Zegkinoglou, I., Zendegani, A., Sinev, I., Kunze, S., Mistry, H., Jeon, H.S., Zhao, J.,
Hu, M.Y., Alp, E.E., Piontek, S., Smialkowski, M., Apfel, U.-P., Körmann, F., Neuge-
bauer, J., Hickel, T., Roldan Cuenya, B. J. Am. Chem. Soc. 2017, 139, 14360.
Abbildung 4: Kristallstruktur des Minerals Pentlandit mit der Stöchiometrie Fe4.5Ni4.5S8. Die Metallatome sind rot, Schwefel dagegen gelb markiert.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
101
Die Hochtemperatursynthese ist eine vielseitige Methode, um zahlreiche anorganische
Feststoffe herzustellen. Die Vorteile sind, dass sich oft mit wenig Aufwand aus den
reinen Elementen und ohne den Einsatz von Lösemitteln die gewünschten Substanzen
herstellen lassen. Auch entfällt meist eine komplizierte Aufreinigung.
Dazu ist aber die genaue Kenntnis des Phasendiagramms der jeweiligen Elementmi-
schung bei unterschiedlichen Temperaturen notwendig. Zusätzlich ist es wichtig, unter
Ausschluss von Feuchtigkeit und Luft zu arbeiten, da es sonst zu unerwünschten Ne-
benreaktionen mit Wasser und Sauerstoff kommen kann, welche eine aufwändige
Nachbehandlung notwendig machen.
Das Präparat, welches mit dieser Methode und in diesem Versuch hergestellt werden
soll, trägt den Namen Pentlandit und gehört zu Gruppe der Metallsulfide. Die genaue
Stöchiometrie des zu synthetisierenden Materials lautet Fe4.5Ni4.5S8.
Das Material findet man auch in der Natur vor - jedoch ist das Verhältnis von Eisen
und Nickel meist variabel und unterscheidet sich stark (je nach Lagerstätte). Die Stö-
chiometrie wird daher oft angegeben mit (Fe,Ni)9S8. Es kommt meist zusammen mit
(Nickel-)Pyrit ((Fe,Ni)0.5S) und andern Eisen-Nickel-Sulfiden vor. Relevant ist diese
Verbindung für die industrielle Gewinnung von Nickel.
Aktuell ist das Mineral Gegenstand der Forschung. Das synthetische Material konnte
als potenter Elektrokatalysator für die Wasserstofferzeugung identifiziert werden. Die
Entdeckung des edelmetallfreien Katalysators ist von großer Relevanz, da Wasserstoff
als alternativer Energieträger zu fossilen Brennstoffen, z.B. in Brennstoffzellenautos,
genutzt werden kann. Aktuell wird der Großteil des Wasserstoffes noch aus fossilen
Brennstoffen hergestellt, was aus ökologischer Sicht sehr problematisch ist. Wasser-
stoff soll jedoch - in nicht allzu ferner Zukunft - ausschließlich durch Elektrolyse von
Wasser bereitgestellt werden. Da dazu noch teure und empfindliche Katalysatoren auf
Platinbasis notwendig sind, ist die Identifikation und Herstellung günstiger und robuster
Alternativen von großer Bedeutung.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
102
Material- und Chemikalienliste:
Name Menge
Synthese
Eisen 976,7 mg (0,01749 mol)
Nickel 1026,3 mg (0,01749 mol)
Schwefel 997,0 mg (0,03109 mol)
Mörser und Pistill Jeweils 1x
Quarzglasampulle 1x
Literatur:
Konkena, B., junge Puring, K., Sinev, I., Piontek, S., Khavryuchenko, O., Duerholt,
J.P., Schmid, R., Tueysuez, H., Muhler, M., Schuhmann, W., Apfel, U.-. Nat. Com-
mun. 2016, 7, 12269.
junge Puring, K., Piontek, S., Smialkowski, M., Burfeind, J., Kaluza, S., Doetsch, C.,
Apfel, U.-P., J. Vis. Exp. 2017,124, E56087.
Was Sie lernen:
Synthesemethoden:
• Vakuumtechnik
• Anorganische Hochtemperatursynthese
• Elektrodenherstellung
Charakterisierungsmethoden:
• Pulver-Röntgendiffraktometrie (PXRD)
• Elektrochemische Analysetechniken in der Anwendung
Cyclovoltammetrie (CV)
• Linearsweep-Voltammetrie (LSV)
• Chronoamperometrie

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
103
Forschungsrelevante Thematik:
• Heterogene Elektrokatalyse
• Wasserelektrolyse
• Synthese anorganischer Katalysatoren
Allgemeines zur Sicherheit:
Das Abwiegen und Mischen der Elemente und des finalen Produkts kann unter atmo-
sphärischen Bedingungen erfolgen. Besondere Sorgfalt sollte beim Umgang mit Nickel
an den Tag gelegt werden. Das Vermahlen der Substanzen sollte stehts unter dem
Abzug erfolgen. Beim Aufbrechen der Ampullen sind schnittfeste Handschuhe zu tra-
gen und die Glasfragmente vom Produkt schnellstmöglich zu entfernen. Auch die Her-
stellung der Elektroden erfolgt mit Schutzhandschuhen, Kittel und Schutzbrille.
Versuchsdurchführung:
Synthese:
Es werden 976,7 mg (0,01749 mol) Eisen-Pulver, 1026,3 mg (0,01749 mol) Nickel-
Pulver und 997,0 mg (0,03109 mol) Schwefel abgewogen und mit Mörser und Pistill
zu einer homogenen Pulvermischung gründlich vermahlen. Das Gemisch wird an-
schließend in eine Quarzglasampulle überführt und für mehrere Stunden evakuiert.
Anschließend wird die Ampulle unter Erhalt des Unterdrucks abgeschmolzen. Mit Hilfe
eines Hochtemperaturofens wird die Ampulle mit 5 °C/min auf 700 °C erhitzt. Diese
Temperatur wird für drei Stunden konstant gehalten, bevor mit einer Heizrate von
10 °C/min auf 1.100 °C geheizt wird. Die Temperatur wird gehalten für 10 Stunden
bevor die Ampulle langsam auf Raumtemperatur abgekühlt wird. Die Ampulle wird auf-
gebrochen und die Glasfragmente vom Produkt getrennt. Das Produkt wird anschlie-
ßend zu einem feinen Pulver gemörsert.
Eigenschaften: äußerlich schwarzer Barren mit metallisch-goldenem Glanz an den
Bruchstellen; nach dem Vermahlen ein dunkel-graues Pulver
Literatur-Ausbeute: >90 %
Kriterium: Ausbeute >60 %
Charakterisierung: PXRD Mo-Kα (2Θ / °): 7.0, 8.1, 11.4, 13.4, 14.0, 16.2, 17.6, 18.1,
19.8, 21.0, 23.0, 24.0, 24.3, 25.7, 26.6, 26.9, 28.2, 29.0.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
104
Elektrodenherstellung und elektrochemische Analyse:
Nach Durchführung der Synthese sowie der Charakterisierung der hergestellten Sub-
stanz wird diese mit Hilfe von einfachen elektrochemischen Methoden untersucht.
Dazu wird zunächst das Material verarbeitet. Es werden 25 - 50 mg des Pulvers mit
Hilfe einer Pressform und einer KBr-Presse zu kleinen Zylindern (3 mm Durchmesser,
min. 2 mm Höhe) gepresst und durch Zuhilfenahme eines leitfähigen 2-Komponenten-
klebers in einem speziellen Teflongehäuse fixiert. Dabei muss darauf geachtet werden,
dass mindestens 0.5 mm des Zylinders aus dem Gehäuse überstehen. Die so herge-
stellten Rohlinge werden im Ofen über Nacht bei 60 °C ausgehärtet. Anschließend
wird der Überstand mit Wasser und Sandpapier (P800) vorsichtig „heruntergeschlif-
fen“, bis dieser fast bündig mit dem Teflongehäuse ist. Der restliche Überstand wird
mit feinem 1 µm-Polierpapier und reinem Wasser vorsichtig abgeschliffen, Zylinder und
Gehäuse bündig sind. Anschließend wird die Oberfläche in einem letzten Schritt mit
0.3 µm-Polierpapier und Wasser poliert, so das eine metallisch glänzende und mög-
lichst glatte Oberfläche entsteht. Die Elektrode wird mit reinem Wasser gründlich ge-
spült und ist danach bereit zu Verwendung.
Die eben hergestellte Arbeitselektrode wird zusammen mit einer Referenzelektrode
(Silber/Silberchlorid in 3 mol/L KCl) und einer Platin-Gegenelektrode in eine gerührte
Elektrolytlösung, bestehend aus 0.5 mol/L Schwefelsäure, eingetaucht. Die Elektroden
werden mit einem Potentiostaten verbunden. Zunächst ist es notwendig, die Arbeits-
elektrode elektrochemisch zu reinigen. Dazu wird die Technik der Cyclovoltammetrie
angewendet. Das Potential wird von 0.2 V bis -0.2 V (versus Ag/AgCl) und zurück an-
gelegt – und zwar mit einer Spannungsvorschubgeschwindigkeit von 100 mV/s. Dies
wird solange wiederholt, bis mindestens die letzten drei bis vier Kurven vollständig
überlappen. In dem Fall kann davon ausgegangen werden, dass die Oberfläche der
Elektrode gereinigt ist (von Rückständen und Oxiden).
Anschließend wird Linearsweep-Voltammetrie durchgeführt. Diese funktioniert ähn-
lich, wie die Cyclovoltammetrie, mit dem Unterschied, dass das Potential nur in eine
Richtung verschoben wird. Der Vorschub zurück zum Startpotential entfällt. Dies er-
folgt im Potentialbereich von -0.2 V bis -0.6 V mit einer Spannungsvorschubgeschwin-
digkeit von 5 mV/s. Insgesamt werden mindestens drei Kurven aufgezeichnet. Das ge-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
105
wählte Potentialfenster deckt den relevanten Bereich für diesen Katalysator ab. Wich-
tig sind die Werte der Überspannung η1 bei -10 mA/cm², bzw. η2 bei -50 mA/cm², da
diese in der Literatur oft zu Vergleichszwecken herangezogen werden.
Zuletzt wird die Stabilität des Katalysators ermittelt. Dazu wird ein chronoamperomet-
risches Experiment durchgeführt. Das bedeutet, dass eine konstante Spannung am
System angelegt und die Stromantwort gemessen wird. Dies erfolgt für eine Dauer von
mindestens 15 Minuten. Das Potential beträgt dabei -0.71 V gegen die Referenzelekt-
rode. Ist der Verlauf des Stroms über langen Zeitraum konstant, ist dies ein Indiz für
einen stabilen Elektrokatalysator/Elektrode.
Zielvorgaben: (CV): Überlappung der kurven spätestens nach 40 CV-Kurven
(LSV): η1 zwischen -0,56 mV bis -0,41 mV bei -10 mA/cm²
η2 zwischen -0,71 mV bis -0,51 mV bei -50 mA/cm²
(Stabilität): Strombetrag: >2 mA; konstant für min. 5 Minuten

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
106
Rutheniumkomplexe
i.) Dichlorobis(tricyclohexylphosphin)
(benzyliden)ruthenium(II) Literatur:
Kadyrov, R.; Rosiak, A. Beilstein J. Org. Chem. 2011, 7, 104.
RuCl3 x H2O [Ru(COD)Cl2]x1,5- COD
[Ru(COD)Cl2]x
1. 2 PCy3, DBU, 2-PrOH2. HCl x Et2O3. TMS
Ru
PCy3
PCy3
Cl
Cl
Ru
PCy3
PCy3
Cl
ClRu
PCy3
PCy3
Cl
Cl
1)
2)
3)
COD = 1,5-Cyclooctadien, PCy3 = Tricyclohexylphosphine, DBU = 1,8-Diazabicyclo(5.4.0)undec-7-en, TMS = Trimethylsilyl
CyclooctanDBU x HCl3 AcetonTMS-Cl
H2O

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
107
Olefin-Metathese (OM) bildet ein starkes, effizientes und vielseitiges Werkzeug in der
organischen Synthese. Im Allgemeinen findet bei der OM ein Austausch von Restgrup-
pen („Umalkylidenierung“, Abb. 1) an Alkenen statt. In Abhängigkeit der Edukte kann
dieser Mechanismus für die verschiedensten Zwecke, z. B. Polymerisation oder Ring-
schluss, eingesetzt werden. Für ihre Fortschritte in der Entwicklung der Metathese
wurden Y. Chauvin, R. Grubbs und R. Schrock 2005 mit dem Nobelpreis für Chemie
ausgezeichnet.
Abbildung 5: Allgemeines Schema der Olefinmetathese – M = Metall-basierter Katalysator
Die Serie der Grubbs-Katalysatoren zählt zu den populärsten OM-Katalysatoren und
wird fortlaufend um weitere Verbindungen ergänzt (aktuell: 3. Generation). Sie umfasst
verschiedene Ruthenium-Carben-Komplexe, die in Relation zu anderen Katalysatoren
(z. B. Schrock-Kat.) stabiler und toleranter gegenüber funktionellen Gruppen sind.
Auch innerhalb der Grubbs-Serie finden sich Aktivitäts- und Stabilitätsunterschiede, so
wurden z. B. in der 2. Generation die Phosphinliganden vollständig substituiert, was
die Stabilität deutlich erhöht hat. Eine zentrale Herausforderung in der Synthese ist die
Bildung eines Ruthenium-Alkyliden-Komplexes. Der Alkyliden-Ligand kann anschlie-
ßend durch komplexere oder mehrzähnige Liganden unter moderaten Bedingungen
ausgetauscht werden. Präparat 2 aus diesem Versuch bildet in diesem Sinne eine
strategische Vorstufe für die Synthese verschiedenster Ruthenium-Carben-Komplexe.
Im Allgemeinen können die Grubbs-Katalysatoren verschiedener Generationen inei-
nander überführt werden (s. Abb.2).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
108
Abbildung 6: Syntheserouten ausgehend von Stufe 2

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
109
Chemikalienliste:
Name der Chemikalie Menge
Stufe 1
RuCl3•H2O 3.00 g (0.0115 mol)
1,5-Cyclooctadien 2.5 mL (0.0229 mol)
Ethanol (abs) 25 mL
Stufe 2
[RuCl2(COD)]x 1.59 g (5.67 mmol)
Tricyclohexylphosphin 3.50 g (12.4 mmol)
1,8-Diazabicyclo[5.4.0]undec-7-en (DBU) 1.85 mL (12.4 mmol)
Trimethylsilyl-Acetylen 2.40 mL (17.0 mmol)
HCl in Et2O (getrocknet) 2.5 mL (13.5 mmol, 5.8 mol/L)
Isopropanol (getrocknet)
THF (getrocknet)
Methanol (getrocknet)
56 mL
85 mL
100 mL
Stufe 3
RuCl2(PCy3)2=Et 1.19 g (1.6 mmol)
Styrol 719 µL (6.3 mmol)
Dichlormethan (getrocknet) 50 mL
Methanol (getrocknet) 60 mL
Literatur:
Schwab, P.; Grubbs, R. H.; Ziller, J. W., J. Am. Chem. Soc. 1996, 118, 100.
Grubbs, R. H.; Wenzel, A. G. Wiley-VCH Verlag. 2015, Vol. 1, 2nd Edition.
Van der Schaaf, P.A.; Kolly, R; Hafner, A. Chem. Commum., 2000, 1045.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Lösemitteltransfer/Filtration unter Inertgasbedingungen

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
110
Charakterisierungsmethoden:
• NMR-Spektroskopie unter Schutzgas
Forschungsrelevante Thematik:
• Organometallchemie der Nebengruppenelemente
• Synthese von Carben-Verbindungen, Grubbs-Katalysatoren
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon aufbe-
wahrt. Getrocknete Lösemittel werden über Molsieb gelagert. NMR-Proben müssen
unter Schutzgas und mit trockenen Lösemitteln hergestellt werden.
Besondere Sorgfalt ist beim Umgang mit Styrol und DBU nötig!
Versuchsdurchführung:
Di-µ-chloro-(η4-Cycloocta-1,5-dien)ruthenium(II) ([RuCl2(COD)]x)
In einem 100 mL-Schlenkkolben mit Rückflusskühler und Abgashahn wird RuCl3•H2O
(3.00 g, 11.47 mmol) in Ethanol (abs., 25 mL) suspendiert. Nach Zugabe von 1,5-Cyc-
loctadien (2.5 mL, 22.95 mmol) wird die Reaktionsmischung 2 mal entgast und an-
schließend 12 Stunden unter Rückfluss erhitzt. Nach Abkühlen auf Raumtemperatur
wird der entstandene Niederschlag an der Luft abfiltriert (Büchnertrichter), mit Diethyl-
ether (2x10 mL) gewaschen und im Ölpumpenvakuum getrocknet.
Eigenschaften: schwer lösliches dunkelbraunes bis schwarzes Pulver
Literatur-Ausbeute: 91 %
Kriterium: Ausbeute mindestens 45 %
Charakterisierung: keine, da das Polymere Produkt in weitestgehend allen Lösemit-
teln unlöslich ist.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
111
Dichlorobis(tricyclohexylphosphin)(ethyliden)ruthenium(II):
Die Hälfte der Ausbeute aus der 1. Stufe wird eingesetzt. Der Ansatz wird dementspre-
chend umgerechnet! Diese Reaktion sollte sorgfältig am Vortag soweit wie möglich
vorbereitet werden. Bei der Zugabe der HCl-Lösung ist zügiges Arbeiten nötig, da die
Lösung leicht aus der Spritze entweicht. Zudem sollte die Lösung weder lange offen
an der Schlenklinie gehandhabt noch mit Argon-Gegenstrom entgast werden, da
HCl(g) schnell ausgast und die Konzentration verringert.
Unter Argon wird in einem ausgeheizten zweihalsigen 250 mL-Schlenkkolben mit
Rückflusskühler [RuCl2(COD)]x (1.59 g, 5.67 mmol) in Isopropanol (56 mL) suspen-
diert. Tricyclohexylphosphin (2.2 eq, 3.5 g, 12.4 mmol) und DBU (2.2 eq, 1.85 mL,
12.4 mmol) werden unter Argon zugegeben. Die schwarze Suspension wird 2 Stunden
auf Rückfluss erhitzt (Farbumschlag → rot). Im Anschluss werden 85 mL THF zuge-
geben und auf exakt (!) 15 °C abgekühlt. HCl in Et2O (13.5 mmol) werden zugegeben
(Farbumschlag → dunkelrot). Die Suspension wird 5 Minuten gerührt. Anschließend
wird Trimethylsilylacetylen (3 eq, 2.4 mL, 17.0 mmol) zugegeben und die Suspension
für 3 Stunden im Eisbad gerührt. Das THF wird im Unterdruck entfernt und das Produkt
wird mittels Kanülenfiltration mindestens 4x mit kaltem Methanol gewaschen.
Eigenschaften: luft-und wasserinstabiles, rosafarbenes Pulver.
Literatur-Ausbeute: 90 %
Kriterium: Ausbeute mindestens 30 %
Charakterisierung: 31P-NMR (CDCl3) – unter Schutzgas - : δ = 35.8 ppm; 1H-NMR
(CDCl3): δ = 19.30 (q, J = 5.6 Hz, 1H), 2.60 (d, J = 5.5 Hz, 3H), 2.60–2.52 (m, 6H),
1.88–1.22 (m, 60H) ppm.
Dichlorobis(tricyclohexylphosphin)(benzyliden)ruthenium(II):
Die gesamte Ausbeute aus der 2. Stufe wird eingesetzt. Der Ansatz wird dementspre-
chend umgerechnet!
In einem ausgeheizten 100 mL-Schlenkkolben wird Styrol (719 µL, 6.28 mmol) in
50 mL Dichlormethan vorgelegt. Stufe 2 (1.19 g, 1.6 mmol) wird zugegeben. In einem
Eisbad wird für 2 Stunden Argon durch das Reaktionsgemisch geleitet (Überdruckven-
til am Septum!). Das Eisbad wird entfernt und für weitere 30 Minuten Argon durchge-
leitet. Im Anschluss wird das Reaktionsgemisch noch 30 Minuten bei RT ohne Argon-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
112
Überdruck gerührt. Zur Aufarbeitung wird erneut mit einem Eisbad gekühlt und Dich-
lormethan im Unterdruck entfernt. Das Produkt wird mit 16 mL vorgekühltem Methanol
aufgeschlämmt und mittels Kanülenfiltration 4 mal gewaschen.
Eigenschaften: braunes bis rosafarbenes Pulver
Literatur-Ausbeute: 89 %
Kriterium: Ausbeute mindestens 35 %
Charakterisierung: 31P-NMR (CD2Cl2): δ 36.63 (s, PCy3). 1H-NMR (CD2Cl2): δ = 20.02 (s, Ru=CH), 8.44 (d,3JHH = 7.6 Hz,o-H of C6H5),7.33 (t,3JHH
= 7.6 Hz,m-H of C6H5), 2.62-2.58, 1.77, 1.67, 1.46-1.39, and 1.25-1.16 (alle m,
P(C6H11)3).

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
113

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
114
ii.) Rac-bis(1,10-phenanthrolin) (dipyrido-
phenazin-11-methylcarboxylat)ruthe-
nium(II)chlorid Sullivan, B. P.; Salmon, D. J.; Meyer, T. J. Inorg. Chem. 1978, 17, 3334.
Norris, M. R.; Concepcion, J. J. ; Glasson, C. R. K. ; Fang, Z ; . Lapides, A. M.;
Ashford, D. L.; Templeton, J. L.; Meyer, T. J. Inorg. Chem. 2013, 52, 12492.
1. Stufe
2. Stufe

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
115
Chemikalienliste:
Name der Chemikalie Menge
Stufe 1
Dichloro(1,5-cyclooctadien)ruthenium(II), polymer 500 mg (1,78 mmol)
1,10-Phenanthrolin 650 mg (3,58 mmol)
1,2-Dichlorbenzol 20 mL
n-Hexan p. a. 160 mL
Stufe 2
cis-Dichlorobis(1,10-phenanthrolin)ruthenium(II) 500 mg (0.88 mmol)
Dipyridophenazin-11-methylcarboxylat 300 mg (0.88 mmol)
Ethanol (getrocknet) 80 mL
Einleitung:
Die Photodynamische Therapie (PDT) ist eine neuartige Technik zur Behandlung be-
stimmter Arten von Krankheiten wie Krebs, Bakterien-, Pilz- und Virusinfektionen. Ei-
ner der wichtigsten Bestandteile der PDT ist der Photosensibilisator (PSs). In der PDT
werden dem Patienten lokal oder systematisch vorzugsweise ungiftige PS verabreicht.
Anschließend wird das PSs bei Bestrahlung mit einer bestimmten Wellenlänge, die
von der Verbindung selbst abhängt, in den Singulett-Zustand S1 angeregt. Der ange-
regte Zustand erfährt entweder einen Energieverlust durch Fluoreszenz oder einen
systemübergreifenden Übergang in den angeregten Triplettzustand T1. Der angeregte
Zustand T1 kann Phosphoreszenz emittieren oder auf unterschiedliche Weise an die
biologische Umgebung interagieren.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
116
Abbildung x: ROS / 1O2 production by Ru(II) complexes.
Das Interesse an Ru-basierten PSs wurde durch die Untersuchung von Multiligand-
Ru(II)-Komplexen, wie Tris(2,2'-bipyridyl)ruthenium(II)-Dichlorid, das hohe Dreifach-
Triplet-Energie- und Elektronenübertragungserträge liefert, angefacht. Ein neuerer An-
satz von Ruthenium PDT verwendet Moleküle wie z. B. [Ru(bpy2)(dppz)]2+ (dppz: Dipy-
rido[3,2-a:2',3'-c]phenazin, bpy: 2,2'-Bipyridin), von denen bekannt ist, dass sie in die
DNA interkalieren. Vertreter dieser Substanzklasse zeigen nur Lumineszenz beim Ein-
lagern in die DNA.
Literatur:
Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100.
Li, W.; Xie, Q.; Lai, L.; Mo, Z.; Peng, X.; Leng, E.; Zhang, D.; Sun, H.; Li, Y.; Mei, W.
et al., Photodiagnosis Photodyn. Ther. 2017, 18, 83.
Lilge, L. Use of Ruthenium Complexes as Photosensitizers in Photodynamic Ther-
apy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017.
Spiller, W.; Kliesch, H.; Wöhrle, D.; Hackbarth, S.; Röder, B.; Schnurpfeil, G. J.
Porphyr. Phthalocyanines 1998, 02, 145.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
117
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
Charakterisierungsmethoden:
• NMR-Spektroskopie
• Fluoreszenzspektroskopie
Forschungsrelevante Thematik:
• Medizinische Anorganik
• Photosensibilisatoren
• Fluoreszenzfarbstoffe für die medizinische Diagnostik
Allgemeines zur Sicherheit:
Die Synthese von Rac-bis(1,10-phenanthrolin)(dipyridophenazin-11-methyl-car-
boxylat)ruthenium(II)chlorid wird unter nachgereinigtem Argon in ausgeheizten Appa-
raturen durchgeführt. Lösemittel werden nach gängigen Methoden getrocknet und un-
ter Argon aufbewahrt. Getrocknete Lösemittel werden über Molsieb gelagert. Nach
Ende der Reaktion kann die Aufarbeitung unter atmosphärischen Bedingungen erfol-
gen.
Versuchsdurchführung:
1.Stufe: cis-Dichlorobis(1,10-phenanthrolin)ruthenium(II) (Ru(phen)2Cl2):
In einem 100 ml Rundkolben werden 500 mg (1,78 mmol) Dichloro(1,5-cycloocta-
dien)ruthenium(II) und 650 mg (3,58 mmol) 1,10-Phenanthrolin vorgelegt und in 20 ml
1,2-Dichlorbenzol suspendiert. Es wird nun ein Rückflusskühler mit Blasenzähler auf-
gesetzt und die Mischung für 24 Stunden bei 160 °C gerührt. Während der Reaktion
verfärbt sich die Lösung violett. Anschließend lässt man die Reaktionslösung auf
Raumtemperatur abkühlen. Die Lösung wird nun filtriert (Faltenfilter) und das Filtrat
wird im Folgenden in 80 mL n-Hexan eingetropft, wobei ein violettes Pulver ausfällt.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
118
Das Produkt wird nun abfiltriert (Büchnertrichter) und mit 2 x 40 mL n-Hexan gewa-
schen. Der verbleibende Feststoff wird anschließend im Vakuum getrocknet.
Eigenschaften: wenig lösliches violettes bis schwarzes Pulver
Literatur-Ausbeute: 95 %
Kriterium: Ausbeute mindestens 45 %
Charakterisierung: 1H NMR (200 MHz, DMSO-d6) δ 10.28 (dd, J = 5.3, 1.3 Hz, 2H),
8.71 (dd, J = 8.1, 1.4 Hz, 2H), 8.43 – 7.99 (m, 8H), 7.75 (dd, J = 5.4, 1.2 Hz, 2H), 7.33
(dd, J = 7.4, 4.7 Hz, 2H).
MS (ESI+): m / z 532.42 [M+H]+.
2.Stufe: Rac-bis(1,10-phenanthrolin)(dipyridophenazin-11-methylcarboxylat)-ruthe-
nium(II)chlorid:
In einem ausgeheizten 250 ml Schlenkkolben werden 500 mg (0,88 mmol) cis-Dichlo-
robis(1,10-phenanthrolin)ruthenium(II) und 300 mg (0,88 mmol) Dipyrido-phenazin-
11-methylcarboxylat vorgelegt und in 80 ml trockenem Ethanol aufgenommen. Es wird
nun ein Rückflusskühler mit Blasenzähler aufgesetzt und die Mischung für 24 Stunden
unter Schutzgasatmosphäre refluxiert. Anschließend lässt man Reaktion auf Raum-
temperatur abkühlen und entfernt das Lösemittel unter vermindertem Druck. Das Pro-
dukt wird unter Vakuum weiter getrocknet. Das entstehende Produkt ist luftstabil und
muss somit nicht unter Schutzgas gehandhabt werden.
Eigenschaften: oranges bis rotes Pulver
Literatur-Ausbeute: 89 %
Kriterium: Ausbeute mindestens 40 %
Charakterisierung: 1H NMR (400 MHz, DMSO-d6) δ 10.15 (ddd, J = 8.3, 4.2, 1.4 Hz, 2H), 9.47 – 9.40 (m,
1H), 9.33 –9.18 (m, 4H), 9.08 (dd, J = 5.4, 1.3 Hz, 2H), 8.99 (q, J = 3.0, 2.3 Hz, 3H),
8.90 – 8.81 (m, 7H), 8.42 (ddd, J = 7.9, 5.4, 2.3 Hz, 2H), 8.28 (ddd, J = 13.6, 6.8,
4.1 Hz, 4H), 4.49 (s, 3H). 13C NMR (101 MHz, DMSO -d6) δ = 166.2, 155.8, 155.7, 154.5, 154, 152.5, 152.3, 149,
148.9, 148.8, 144.9, 142.6, 142.1, 138, 137.8, 134.8, 134.6, 134, 132.6, 132.1, 132,
131.8, 131.5, 131.4, 131.1, 129.2, 129.1, 128.5, 128.4, 127.2, 127.1, 53.3.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
119
Fluoreszenzspektroskopie:
Im Nachgang der Synthese wird der synthetisierte Komplex mittels Fluoreszenzspekt-
roskopie charakterisiert. Die Messungen werden am Fluoreszenzspektrometer im
Praktikum durchgeführt, hierzu werden jeweils 5 mL einer 10 µM Lösung Rac-bis(1,10-
phenanthrolin)(dipyridophenazin-11-methylcarboxylat)-ruthenium(II)chlorid in Wasser
für die Spektroskopie und Acetonitril (HPLC grade) hergestellt (Berechnung). Jeweils
3 mL der Lösungen werden in eine Fluoreszenzküvette gefüllt und diese im Spektro-
meter mit Anregungswellenlänge von 444 nm untersucht. Hierzu wird die Detektion auf
einen Wellenlängenbereich von 454 – 800 nm eingestellt.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
120
2,2’-Selenobis(ethan-1-tosylamid)
&
2,2’ Tellurobis(ethan-1-tosylamid)
Se(CH2CH2NHTs)2 & Te(CH2CH2NHTs)2
Literatur:
Hansheng, X.; Guangya, X.; Jun, W; Xiufang, L. Wuhan Univ. J. Nat. Sci., 1999, 4, 85.
BrNHTs
Se KHSe+ KBH4 TsHNSe
NHTsEtOH EtOH
BrNHTs
Te KHTe+ KBH4 TsHNTe
NHTsEtOH/H2O
EtOH/H2O
Ts = SO2-C6H5-Me

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
121
Chemikalienliste:
Synthese 1: 2,2‘-Selenobis(ethan-1-tosylamid)
Name der Chemikalie Menge
Selen (Pulver) 0.79 g (10 mmol)
Kaliumborhydrid 1.13 g (21 mmol)
N-(2-Bromoethyl)tosylamid 5.30 g (19 mmol)
Ethanol (entgast) 100 mL
Essigsäureethylester nach Bedarf
Diethylether nach Bedarf
Synthese 2: 2,2‘-Tellurobis(ethan-1-tosylamid)
Name der Chemikalie Menge
Tellur (Pulver) 1.28 g (10 mmol)
Kaliumborhydrid 1.63 g (30 mmol)
N-(2-Bromoethyl)tosylamid 5.30 g (19 mmol)
Ethanol (entgast) 100 mL
Wasser (entgast) 6 mL
Essigsäureethylester nach Bedarf
Einleitung:
Selen und Tellur sind zusammen mit Sauerstoff und Schwefel Teil der 16. Gruppe des
Periodensystems. Diese Elemente, insbesondere S, Se und Te, werden im engeren
Sinne „Chalkogene“ genannt, da deren Verbindung oftmals ähnliche chemische und
physikalische Eigenschaften aufweisen. Innerhalb der letzten Dekaden hat die Selen-
und Tellurchemie an enormer Bedeutung gewonnen, was sich an der Anzahl der vielen
neuartigen Organoselen und -tellur Verbindungen messen lässt. Im Vergleich zum
Schwefel sind die entsprechenden Selen- und Tellurverbindung deutlich instabiler, was
über den Halbmetallcharakter der Elemente begründet werden kann. Im direkten Ver-
gleich sind insbesondere die niedervalenten Tellurverbindungen deutlich instabiler ge-
genüber den selenhaltigen Verbindungen.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
122
Organische Chalkogenide und Dichalkogenide gelten als die wichtigsten unter den Or-
ganochalkogenverbindungen. Einer der einfachsten Wege für die Synthese von sym-
metrischen Dialkylchalkogeniden und -dichalkogeniden ist die Alkylierung der entspre-
chenden Alkalimetallsalze von Chalkogenwasserstoff bzw. Dichalkogen-wasserstoff,
die in situ durch die Reduktion der elementaren Chalkogene mit einem geeigneten
Reduktionsmittel erzeugt werden können.
Abbildung 7: Selen-und tellurhaltige Azamakrozyklen.
Die hier synthetisierten Verbindungen 2,2‘-Selenobis(ethan-1-tosylamid) und 2,2‘-Tel-
lurobis(ethan-1-tosylamid) werden in einer anschließenden Ringschlusssynthese zu
den Verbindungen 1 bzw. 2 umgesetzt, welche wiederum nach Abspaltung der To-
sylgruppen als Liganden (3 und 4) für die Komplexierung von Übergangsmetallen ge-
nutzt werden können (Abb. 1).
Literatur:
Hansheng, X.; Guangya, X.; Jun, W.; Xiufang, L. Wuhan Univ. J. Nat. Sci., 1999, 4,
85.
Yamamoto, H.; Oshima, K. Eds., Main Group Metals in Organic Synthesis, Wiley-VCH,
Weinheim, Germany, 2004.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit Halbmetallen
• Umgang mit Chalkogeniden

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
123
Charakterisierungsmethoden:
• NMR-Spektroskopie
• ESI-MS
Forschungsrelevante Thematik:
• Chalkogenhaltiger Baustein für die Ligandensynthese
• Koordinationschemie
• Umwandlung vom metallischen zum nichtmetallischen Charakter
Allgemeines zur Sicherheit:
Der Versuch wird unter Argon/Vakuum und Ausschluss von Sauerstoff durchgeführt.
Die luft- und säureempfindlichen Intermediate werden unter Argon gehandhabt. Die
Verwendung von geschlossenen Apparaturen ist strengstens untersagt. Das bei der
Reaktion entstehende Gas wird über einen Blasenzähler aufgesetzt auf dem Rück-
flusskühler abgeleitet.
Da die intermediär auftretenden Chalkogenide einen charakteristischen sehr starken
Geruch besitzen, sind Glasgeräte prinzipiell im Abzug zu säubern. Die anfallenden
Reaktionsrückstände sind sachgerecht unschädlich zu machen und direkt zu entsor-
gen.
Versuchsdurchführung:
Synthese 1: 2,2‘-Selenobis(ethan-1-tosylamid):
In einem 250-mL Zweihalsschlenkkolben werden 0.79 g Selen (10 mmol) vorgelegt.
Der Kolben wird anschließend mit einem Magnetrührstab, einem Rückflusskühler und
einem Tropftrichter ausgestattet und die Apparatur wird drei Mal sekuriert. (Achtung:
Das Argon-Fluten sollte langsam geschehen, damit das Pulver sich nicht in der ganzen
Apparatur verteilt!) Anschließend werden entgastes EtOH (45 mL) und 1.13 g KBH4
(21 mmol) zugegeben. Das Reaktionsgemisch wird unter Rückfluss erhitzt, wobei eine
Gasentwicklung zu beobachten ist und sich die Farbe von schwarz über rot zu farblos

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
124
verändert (max. 2 Stunden). In der Zwischenzeit werden 5.30 g N-(2-Bromoethyl)to-
sylamid in einem 100 mL Schlenkkolben vorgelegt und der Kolben, ausgestattet mit
einem Magnetrührstab, wird drei Mal sekuriert. Anschließend wird das Amid in entgas-
tem EtOH (55 mL) gelöst und in den Tropftrichter der Apparatur überführt. Sobald die
selenhaltige Lösung farblos ist, kann die Amid-Lösung unter Rückflussbedingungen
zugetropft werden. Das Reaktionsgemisch wird für weitere 12 Stunden unter Rückfluss
erhitzt. Anschließend wird das Gemisch abgekühlt und an Luft geöffnet, einige Minuten
gerührt, über einen Faltenfilter filtriert und vom Lösemittel befreit. (Hinweis: Die Entfer-
nung des Lösemittels kann am Rotationsverdampfer erfolgen.) Der Rückstand wird in
einer minimalen Menge heißem Essigsäureethylester gelöst und auf eine Filtersäule
(Ø 5 cm, 15 cm) aufgetragen und mit Et2O eluiert. Es wird nur die Fraktion, welche das
Produkt enthält, aufgefangen. (Hinweis: Ermittlung der Produktfraktion erfolgt mittels
DC-Tüpfeltest auf UV-aktiven DC-Platten. Es darf mit Hilfe von Druck eluiert werden.)
Die aufgesammelte Produktfraktion wird vom Lösemittel befreit und es bleibt ein farb-
loser Feststoff zurück, welcher mittels 1H-, 13C{1H}-NMR und ESI-MS charakterisiert
wird.
Literatur-Ausbeute: 4.27 g (99 %)
Kriterium: 2.13 g (50 %)
Charakterisierung: 1H-NMR (CDCl3, 250 MHz): δ = 7.74 (HAr, 4 H, d); 7.30 (HAr, 4 H, d); 5.07 (NHTs, 2 H,
t); 3.14 (CH2NHTs, 4 H, q); 2.59 (CH2Se, 4 H, t); 2.43 (CH3, 6 H, s) ppm. 13C{1H}-NMR
(CDCl3, 63 MHz): δ = 143.8, 137.0, 130.0, 127.2 (CAr); 43.1 (CH2NHTs); 24.1 (CH2Se);
21.7 (CH3) ppm.
ESI-MS (Aceton): 498.7 [M + Na]+
Synthese 2: 2,2‘-Tellurobis(ethan-1-tosylamid):
In einem 250-mL Zweihalsschlenkkolben werden 1.28 g Tellur (10 mmol) vorgelegt.
Der Kolben wird anschließend mit einem Magnetrührstab, einem Rückflusskühler und
einem Tropftrichter ausgestattet und die Apparatur wird drei Mal sekuriert. (Achtung:
Das Argon-Fluten sollte langsam geschehen, damit das Pulver sich nicht in der ganzen
Apparatur verteilt!) Anschließend werden entgastes EtOH (45 mL), entgastes H2O
(6 mL) und 1.13 g KBH4 (21 mmol) zugegeben. Das Reaktionsgemisch wird unter
Rückfluss erhitzt, wobei eine Gasentwicklung zu beobachten ist und sich die Farbe

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
125
von schwarz zu violett verändert (1 h). Anschließend werden weitere 0.5 g KBH4 vor-
sichtig zu der siedenden Reaktion hinzugegeben, woraufhin sich die Farbe zu farblos
verändert. In der Zwischenzeit werden 5.30 g N-(2-Bromoethyl)tosylamid in einem
100 mL Schlenkkolben vorgelegt und der Kolben, ausgestattet mit einem Magnetrühr-
stab, wird drei Mal sekuriert. Anschließend wird das Amid in entgastem EtOH (55 mL)
gelöst und in den Tropftrichter der Apparatur überführt. Sobald die tellurhaltige Lösung
farblos ist, kann die Amid-Lösung unter Rückflussbedingungen zugetropft werden. Das
Reaktionsgemisch wird für weitere 12 Stunden unter Rückfluss erhitzt. Anschließend
wird das Gemisch abgekühlt und an Luft geöffnet, einige Minuten gerührt, über einen
Faltenfilter filtriert und vom Lösemittel befreit. (Hinweis: Die Entfernung des Lösemit-
tels kann am Rotationsverdampfer erfolgen.) Der Rückstand wird in einer minimalen
Menge heißem EtOAc gelöst und auf eine Filtersäule (Ø 5 cm, 15 cm) aufgetragen und
mit EtOAc eluiert. Es wird nur die Fraktion, welche das Produkt enthält, aufgefangen.
(Hinweis: Ermittlung der Produktfraktion erfolgt mittels DC-Tüpfeltest auf UV-aktiven
DC-Platten. Es darf mit Hilfe von Druck eluiert werden.) Die aufgesammelte Produkt-
fraktion wird vom Lösemittel befreit und es bleibt ein orangener Feststoff zurück, wel-
cher mittels 1H-, 13C{1H}-NMR und ESI-MS charakterisiert wird.
Literatur-Ausbeute: 4.10 g (82 %)
Kriterium: 2.05 g (41 %)
Charakterisierung: 1H-NMR (CDCl3, 250 MHz): δ = 7.74 (HAr, 4 H, d); 7.30 (HAr, 4 H, d); 5.29 (NHTs, 2 H,
t); 3.21 (CH2NHTs, 4 H, q); 2.65 (CH2Te, 4 H, t); 2.41 (CH3, 6 H, s) ppm. 13C{1H}-NMR
(CDCl3, 63 MHz): δ = 143.7, 137.1, 129.9, 127.2 (CAr); 45.1 (CH2NHTs); 21.7 (CH3);
3.5 (CH2Te) ppm.
ESI-MS (Aceton): 526.0 [M + H]+

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
126
Titan-organische Verbindungen
i.) Dicyclopentadienyltitandichlorid
ii.) Tetrakis-(diethylamido)-titan
i.) Literatur:
Bochmann, M.; „Metallorganische Chemie der Übergangsmetalle“ VCH-Verlag.
Elschenbroich, C.; Salzer, A. „Organometallchemie“ Teubner-Verlag.
Panda, T. K.; Gamer, M. T.; Roesky, P. W. Organometallics 2003, 22, 877.
+ 2Na- H2
Na+2
Na+2 + TiCl4- 2NaCl
Ti
Cl
Cl

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
127
ii) Literatur:
Bradley, D. C.; Gitlitz, M. H. J. Chem. Soc. (A), 1969, 980.
HNEt2 + C4H9LiHexan
-10°C - RTLiNEt2 + C4H10
4 LiNEt2 + TiCl4-10°C - RT
ToluolTi
Et2N
NEt2
NEt2
Et2N+ 4 LiCl

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
128
Chemikalienliste:
i.) Dicyclopentadienyltitandichlorid
1. Stufe:
Name der Chemikalie Menge
Natrium 1.5 g (0.065 mol)
Dicyclopentadien 60 mL
2. Stufe:
Name der Chemikalie Menge
Cyclopentadienylnatrium 4.64 g (0.0526 mol)
Titantetrachlorid 2.9 mL (0.0263 mol)
Toluol 30 mL
Tetrahydrofuran 25 mL
Dichlormethan ca. 300 mL
ii.) Tetrakis-(diethylamido)-titan
Name der Chemikalie Menge
Diethylamin 3.84 g (0.053 mol)
n-Butyllithium in Hexan (1.6 M) 33.1 ml (0.053 mol)
Titan(IV)chlorid 2.33 g (0.0125 mol)
n-Hexan 50 mL
Toluol
Celite
10 mL
Ausleihe:
Soxleth-Apparatur

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
129
Einleitung:
Cyclopentadienyl-Titan-Dichloride:
Polyolefinische Kunststoffe wie Polyethylen und Polypropylen sind wichtige industrielle
Werkstoffe, die noch heute großtechnisch auch über die 1955 von Karl Zieger in Mühl-
heim entdeckte metallkatalytische Polymerisationsreaktion hergestellt werden. Als he-
terogenen Katalysator verwendete Ziegler ein Zwei-Komponenten-Katalysator, beste-
hend aus einem Titanchlorid-Katalysator und einer Aluminium-organischen Verbin-
dung als Co-Katalysator.
Heute werden jedoch in erster Linie homogene „Ziegler-Natta“-Katalysatoren einge-
setzt. Als besonders wirksame Katalysatoren haben sich die Cyclopentadienyl-Metall-
Dichloride der vierten Nebengruppe (Ti, Zr, Hf) erwiesen, welche in gängigen organi-
schen Lösemitteln löslich sind. Der Mechanismus der Polymerisation funktioniert über
eine intramolekulare Insertionsreaktion, bei der sich ein koordiniertes Olefin in die Me-
tall-Alkyl-Bindung einschiebt. Der Kettenabbruch ist letztlich eine β-H-Eliminierung.
Aminosubstituierten Titan-Komplexe:
Derartige Komplexe zeichnen sich durch eine hohe Volatilität und Reaktivität aus, wes-
halb sie sowohl in katalytischen Prozessen als auch als Precursoren für die Abschei-
dung dünner Filme von Metallnitriden benutzt werden. Die auf Halbleiterbauelemente
gestützte Informationstechnik nützt eine Fülle von vergleichsweise einfach strukturier-
ten anorganischen Verbindungen als sogenannte Precursor für den Aufbau der kom-
plexen funktionalen Dünnschichtstrukturen. Die chemische Dampfabscheidung ist da-
für eines der wichtigsten Verfahren. Unter der chemischen Dampfabscheidung ("Che-
mical Vapour Deposition", CVD) versteht man diejenigen Prozesse, bei denen in der
Gasphase transportierte molekulare Komponenten infolge heterogener chemischer
Oberflächenreaktionen zu fest haftenden Filmen umgewandelt werden. Aminosubsti-
tuierte Titankomplexe sind aber nicht nur für CVD-Prozesse interessante Precursoren.
So können z. B. aufgrund der einfachen Substituierbarkeit der Aminogruppen durch
Umsetzung mit Triethanolamin und verschiedenen Organohydroperoxiden supramo-
lekulare peroxohaltige Organotitancluster generiert werden, die hinsichtlich ihrer kata-
lytischen Aktivität in der Alkoholoxidation untersucht werden.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
130
Die einfache Zugänglichkeit dieser Precursoren und die Möglichkeit der Modifikation
der Verbindungen durch Variation der Ligandenklasse ist ein wichtiger Aspekt des vor-
liegenden Experimentes. Des Weiteren wird der Einfluss verschiedenster basischer,
sterisch anspruchsvoller Ligandensysteme auf die Struktur- und Reaktivitätsvielfalt un-
terschiedlichster metallorganischer Komplexsysteme verdeutlicht.
Hitchman, M. L.; Jensen, K. F. Chemical vapor deposition: Principles and applica-
tions, Acad. Press, London, 1993.
Bachmann, K.; Linz, U. Spektrum der Wissenschaft 1992, 9, 30.
Gladfelter, W. L. Chem. Mater. 1993, 5, 1372.
Boche, G.; Möbus, K.; Harms, K.; Marsch, M. J. Am. Chem. Soc. 1996,118, 2770.
Naiini, A. A.; Ringrose, S. L.; Jacobson, R. A.; Verkade, J. G. Inorg. Chem. 1993, 32,
1290.
Rohe, M.; Merz, K., Chem. Commun. 2008, 862.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Umgang mit luftempfindlichen Verbindungen
• Umgang mit reaktiven Organometall-Addukt-Substanzen
Charakterisierungsmethoden:
• NMR-Spektroskopie
Forschungsrelevante Thematik:
• Alkalimetallorganische Chemie
• Titanorganische Chemie
• Komplexchemie
• Precursorsynthese für CVD
Allgemeines zur Sicherheit:
TiCl4 ist sehr feuchtigkeitsempfindlich und hydrolysiert sofort an feuchter Luft. Natrium,
NaCp sind sowohl feuchtigkeits- als auch luftempfindlich. Alle diese Stoffe müssen

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
131
daher unter Schutzgasatmosphäre gehandhabt werden. Dicyclopentadienabfälle wer-
den in einem gesonderten Abfallbehälter entsorgt. Der Versuch wird unter nachgerei-
nigtem Argon in ausgeheizten Apparaturen durchgeführt. Lösemittel werden nach gän-
gigen Methoden getrocknet und unter Argon gelagert.
Butyllithium wird als Reagenz zur Herstellung von lithiertem Diethylamin verwendet.
Es ist eine höchst luftempfindliche und brennbare Chemikalie und muss daher unter
Argon gehandhabt werden. Das Endprodukt Titantetrakisdiethylamid ist ebenfalls luft-
und feuchtigkeitsempfindlich.
Versuchsdurchführung:
i.) Dicyclopentadienyltitandichlorid
1. Stufe: Cyclopentadienylnatrium:
In einem 250 mL-Schlenkkolben werden 1.5 g Natrium (0.065 mol), in - nicht zu kleine
(!) - frisch geschnittenen Stücken eingewogen. Es werden nun 60 mL Dicyclopentadien
hinzugegeben und der Schlenkkolben wird mit einem Rückflusskühler und einem Bla-
senzähler versehen.
Man erhitzt nun auf 170 °C, wobei eine Gasentwicklung einsetzt. Nach Beendigung
der Gasentwicklung (ca. 3 - 4 Stunden) wird das Reaktionsgemisch noch eine halbe
Stunde bei 170 °C gerührt. Anschließend lässt man auf Raumtemperatur abkühlen.
Der entstandene, weiße Feststoff wird nun abgefrittet und dreimal mit 30 mL trocke-
nem n-Hexan gewaschen. Der Feststoff wird im Vakuum getrocknet.
Literatur-Ausbeute: 5.7 g (99 %) (bezogen auf eingesetztes Natrium)
Kriterium: 50 %
2. Stufe: Dicyclopentadienyltitandichlorid:
Es werden 30 mL Toluol in einem ausgeheizten Schlenkkolben vorgelegt und anschlie-
ßend mit 2.9 mL TiCl4 versetzt.
Parallel dazu werden 4.64 g NaCp (0.0527 mol) in ein Schlenkrohr eingewogen und in
25 mL THF gelöst.
Unter Eiskühlung wird nun langsam die NaCp-Lösung zur gerührten TiCl4-Lösung ka-
nüliert. Nach Beendigung des Zutropfens wird das Reaktionsgemisch nun bei Raum-
temperatur über Nacht gerührt. Das Lösemittel wird im Vakuum entfernt und der rote

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
132
Rückstand in der Glove-Box in eine Soxleth-Apparatur gefüllt. Das Rohprodukt wird
nun mit trockenem Dichlormethan extrahiert, wobei der Endpunkt der Extraktion durch
Entfärbung der roten Farbe zu erkennen ist.
Die Lösung wird auf ca. 1/3 des Volumens eingeengt und zur Kristallisation bei -35 °C,
über Nacht gelagert. Der ausgefallene, rote Feststoff wird abgefrittet und mit wenig
n-Hexan gewaschen.
Literatur-Ausbeute: 3.5 g (50 %)
Kriterium: Ausbeute mindestens 25 % 1H-NMR (CDCl3): 6,59 ppm 13C-NMR (CDCl3): 120.5 ppm
Pulverdiffraktogramm
ii.) Tetrakis-(diethylamido)-titan
In einen 100 mL-Schlenkkolben wird eine Mischung aus 25 mL n-Hexan und 33 mL
einer 1.6 molaren n-Butyllithium/Hexan Lösung vorgelegt. Diethylamin wird tropfen-
weise bei 0 °C zu dieser Lösung gegeben und anschließend 1 Stunde bei Raumtem-
peratur gerührt. Das überstehende Lösungsmittel wird im Unterdruck entfernt und der
entstandene Feststoff isoliert und gewogen. Das lithierte Amin wird mit 25 mL n-Hexan
versetzt und zu der auf 0 oC gekühlten Suspension wird die berechnete Menge Titan-
tetrachlorid in 10 mL Toluol gegeben und vier Stunden bei Raumtemperatur gerührt.
Anschließend wird die braune Lösung mittels Schutzgasfritte und Celite filtriert und der
Rückstand zweimal mit 10 mL n-Hexan gewaschen. Das Filtrat und Waschlösung wird
im Unterdruck vollständig eingeengt und das verbleibende Öl einer Vakuumdestillation
unterworfen.
Literatur-Ausbeute: 1.81 g, 0.0055 mol 58 % (bezogen auf TiCl4).
Kriterium: Ausbeute mindestens 30 %
Charakterisierung:
1H-NMR (200 MHz, C6D6): δ = 3.58 (q, NCH2CH3) ; δ = 1.11 (t, NCH2CH3)

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
133
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
134
i) Trifluoro-Holomycin: Metallchelator mit antibakterieller Aktivität
ii) Deuteriertes- 5-Methyl-2-[(2-nitro-phenyl)amino]-3-thiophenecarbonitril (d1-ROY)
i.) Literatur:
Trifluoro-Holomycin – Synthese: Hjelmgaard, T.; Givskovb, M.; Nielsen, J. Org.
Biomol. Chem. 2007, 5, 344.
Holomycin – Metallkoordination: Chana, A. N.; Shiverb, A. L.; Weverc, W. J.; Raz-
vid, S. Z. A.; Traxlere, M. F.; Li, B. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 2717.
ii.) Literatur:
J. Falk, D. W. M. Hofmann, K. Merz, IUCrJ, 2018, 5, 569.
N SCH3
NC
N
OOH
N SCH3
NC
N
OOD
+ CH3OD
- CH3OH

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
135
Chemikalienliste:
Trifluoro-Holomycin, Synthese von 2:
Name der Chemikalie Menge
Hydroxy-Pyrrolon 1 1.0 g
Ammoniumacetat 25 g
Dichlormethan (p.A.) 300 mL (Extraktion)
Na2SO4 (Trocknung nach Extraktion)
Silica-Gel (Chromatographie)
n-Heptan (p.A.) ca. 300 mL (Chromatographie)
Ethylacetat (EtOAc; p.A.) ca. 300 mL (Chromatographie)
Trifluoro-Holomycin, Synthese von 3:
Name der Chemikalie Menge
Amino-Pyrrolon 2 ca. 0.5 g
m-Kresol ca. 1 – 2 mL
Trifluoressigsäure (TFA) ca. 25 – 50 mL
Silica-Gel (Chromatographie)
n-Heptan (p.A.) ca. 500 mL
Essigsäureethylester (p.A.) ca. 700 mL
Trifluoro-Holomycin, Metallkoordinationsstudien mit 3:
Name der Chemikalie/Lösung Konzentration
Trifluoro-Holomycin 3 NMR-Probe in DMSO-d6
Tris-HCl-Puffer, pH 7.4 1 M
Tris(2-carboxyethyl)phosphin (TCEP) 100 mM (Stammlösung in DMSO)
Metallsalz-Lösungen:
Mn(II), Co(II), Ni(II), Cu(II), Zn(II) 20 mM (Stammlösungen in MilliQ-H2O)
Ultrapure MilliQ-H2O (für Verdünnungen)

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
136
d1-ROY
Name der Chemikalie/Lösung Konzentration
ROY 0.1 g
Methanol (techn) 10 mL
d1-Methanol 10 mL
Einleitung:
i) Holomycin als antibakterieller Metallchelator
Die Bioanorganische Chemie ist ein noch relativ junges Gebiet der Anorganischen
Chemie, die sich als interdisziplinärer Forschungszweig mit der Aufklärung der Funk-
tion von Metallen und Hauptgruppenelementen in biologischen Systemen beschäftigt.
Dabei gewinnt auch die Entwicklung neuer Wirkstoffe immer mehr an Bedeutung. Ge-
rade im Anbetracht der rasant steigenden Antibiotikaresistenzen, die jährlich immer
mehr Todesopfer fordern, sind innovative Strategien wichtig.
Um effiziente neue Antibiotika entwickeln zu können, müssen neue Stoffklassen sowie
neue Wirk-Targets erschlossen werden. Das kann durch die Isolierung bereits antimik-
robiell wirkender Naturstoffe und der Aufklärungen zugrundeliegender Wirkmechanis-
men erfolgen. Eine dieser vielversprechenden, aber noch wenig erforschten Natur-
stoffklassen sind die sogenannten Dithiolopyrrolone, wie Holomycin und Thiolutin
(Abb. 1). Diese werden von einigen marinen und Bodenbakterien produziert und wei-
sen eine breite antimikrobielle Aktivität auf, auch gegen multiresistente Pathogene. Der
genaue Wirkmechanismus dieser bicyclischen Naturstoffe ist noch nicht bekannt. Es
konnte aber bereits gezeigt werden, dass das cyclische Disulfid essentiell für die bio-
logische Aktivität ist. Nach Aufnahme in die Zelle wird das Disulfid in der reduktiven
Umgebung des Cytoplasmas wahrscheinlich zum Dithiol reduziert (Abb. 1), welches
dann eine Vielzahl an nukleophilen Reaktionen eingehen kann, die zur Schädigung
essentieller Biomoleküle führen.
Abb. 1. Molekülstrukturen von Dithiolopyrrolonen und Reduktion zum Dithiol.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
137
Neben ihrer nukleophilen Reaktivität fungieren Thiole auch als wichtige Donorgruppen
bei der Koordination von Übergangsmetallionen. In der Zelle übernehmen Übergangs-
metallionen (Mn, Fe, Co, Ni, Cu, Zn) viele wichtige Funktionen, darunter strukturelle,
regulatorische sowie katalytische Funktionen, z. B. in den aktiven Zentren von Metallo-
enzymen. Da Metallionen selbst auch toxisch wirken können, werden sie in der Zelle
sehr strikt reguliert. Störungen des zellulären Metallionen-Gleichgewichtes (Metallho-
möostase) durch z. B. von außen eingebrachte Substanzen kann zu erheblicher Schä-
digung der Zelle führen. Entsprechend stellt die Metallhomöostase in Bakterienzellen
ein vielversprechendes Wirk-Target dar, welches bisher aber kaum adressiert wurde.
Kürzlich konnte gezeigt werden, dass Holomycin, nach Reduktion zum Dithiol, Zn(II)
und andere Übergangsmetallionen chelatisieren und dadurch Metalloenzyme inhibie-
ren kann. Es ist also anzunehmen, dass Holomycin und dessen Analoga nach Auf-
nahme in die Zelle und Reduktion essentielle Metallionen binden und dadurch antibak-
teriell wirken. Die genaue Aufkärung dieser Wirkmechanismen, die wir auch im AK
Neumann verfolgen, wird letztlich eine Optimierung der Molekülstruktur der Dithiolopy-
rrolone ermöglichen und damit eine Steigerung der antibakteriellen Aktivität dieser in-
teressanten Naturstoffklasse.
Im Praktikumsversuch werden Sie Trifluoro-Holomycin 3 synthetisieren, eine syntheti-
sche Vorstufe des eigentlichen Naturstoffes Holomycin. Strukturell unterscheiden sich
die beiden Verbindungen lediglich in der Acetylgruppe, die in Verbindung 3 synthetisch
bedingt noch fluoriert ist. Entsprechend weisen beide Verbindungen ähnliche Eigen-
schaften auf, darunter auch die Koordination von Übergangsmetallionen. Im Versuch
werden Sie sowohl synthetische Aspekte des Naturstoff-basierten Ligandensystems
als auch Methoden zur Untersuchung von Koordinationseigenschaften kennenlernen.
Anhand der Synthese von Trifluoro-Holomycin 3 lernen Sie eine Substitutionsreaktion
zur Synthese eines Amins aus einem Alkohol kennen, die unter Lösungsmittel-freien
Bedingungen erfolgt. Anschließend setzen Sie das Pyrrolon durch Entfernung der
Schutzgruppen zum cyclischen Disulfid um. Dabei lernen Sie die PMB-Gruppe (PMB:
p-Methoxybenzyl) kennen, die als universelle Schutzgruppe sowohl für Amin-, Thiol-
als auch Hydroxy-Funktionen genutzt wird, und damit auch für die Synthese von Lig-
andensystemen von Bedeutung ist. Mit dem synthetisierten Produkt führen Sie
schließlich UV/Vis-basierte Metallkoordinationsstudien durch und untersuchen die Ko-
ordination essentieller Übergangsmetallionen am Dithiolopyrrolon. In diesem Prakti-
kumsversuch erhalten Sie neben der Synthese eines natürlichen, redox-schaltbaren

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
138
Chelators Einblicke in spektroskopische Methoden, die wir für die Aufklärung von phy-
siologisch-relevanten Aktivitätsmechanismen und die Erforschung neuartiger Antibio-
tika-Targets nutzen.
Literatur:
Li, B.; Wever, W. J.; Walsh, C. T.; Bowers, A. A. Nat. Prod. Rep. 2014, 31, 905.
Chana, A. N.; Shiverb, A. L.; Weverc, W. J.; Razvid, S. Z. A.; Traxlere, M. F.; Li, B.
Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 2717.
ii) d1-ROY
Bei der Entwicklung neuer Wirkstoffe ist nicht nur die Aufklärung der Wirkmechanis-
men und potentieller Nebenwirkungen wichtig, sondern auch die Optimierung der Wirk-
stoffstruktur. Eine Möglichkeit die metabolische Stabilität von pharmazeutischen Wirk-
stoffen zu erhöhen besteht in der Verwendung der deuterierten Derivaten. So wurde
2017 Deutetrabenazin als erster deuterierte Wirkstoff durch die FDA (U. S. Food and
Drug Administration) zugelassen.
Olanzapin, ist ein atypisches Antipsychotikum, das in erster Linie zur Behandlung von
Schizophrenie und bipolaren Störungen eingesetzt wird. Dieser pharmazeutische
Wirkstoff wird aus 5-Methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitril, allgemein
bekannt als ROY auf Grund seiner roten, orangefarbenen und gelben polymorphen
Kristalle, synthetisiert. Die kontrollierte Bildung von Polymorphen ist in der chemischen
Industrie von entscheidender Bedeutung da die Gitterenergieunterschiede der erhal-
tenen polymorphen Formen typischerweise sehr gering sind. Über die Hälfte der poly-
morphen Formen zeigen Gitterenergieunterschiede von 2 kJ mol-1 und in nur 5 % der
Fälle mehr als 7,2 kJ mol-1. Diese geringen Energieunterschiede führen bei ROY zu
dem Phänomen gleichzeitig auftretender kristalliner Polymorphe. Eine Möglichkeit die-
ses Phänomen zu umgehen, besteht in der gezielten Synthese und Isolierung von
mono-deuterierten ROY. Der Wasserstoff-Deuteriums-(H/D)-Austausch wird allge-
mein als ein vernachlässigbares Kriterium bei der Bildung von Kristallstrukturen che-
mischer Verbindungen angesehen. Andererseits konnte bereits gezeigt werden, dass
die Aggregation von Molekülen im festen Zustand ausgewählter organischer und an-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
139
organischer Verbindungen sehr empfindlich auf kleine H/D-Veränderung der betrach-
teten Moleküle oder die Verwendung von deuterierten Lösemittel während des Kristal-
lisationsprozesses reagieren kann. Das Phänomen der durch die Isotopensubstitution
induzierten Kristallstrukturveränderungen wird auch als "Isotopenpolymorphismus" be-
zeichnet. Darüber hinaus haben aktuelle Studien gezeigt, dass schweres Wasser Re-
aktionswege und -produkte beeinflussen kann.
Im Falle der gleichzeitig auftretenden Polymorphe von ROY kann durch H/D-Aus-
tausch die intermolekulare Wasserstoffbrücke und damit das Aggregationsverhalten
von ROY so beeinflusst werden, dass ein Polymorph thermodynamisch stabilisiert
wird.
Literatur:
Mullard, A. Nature Reviews Drug Discovery, 2017, 16, 305.
Merz, K.; Kupka, A. Cryst. Growth Des, 2015, 15, 1553.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Lösungsmittel-freie Synthese
• Säulenchromatographie
Charakterisierungsmethoden:
• Dünnschichtchromatographie
• NMR-Spektroskopie in Lösung
Untersuchungsmethoden:
• UV/Vis-Spektroskopie zur Verfolung von Redoxchemie und Metallkoordination
• Arbeiten mit Lösungen im µM-Konzentrationsbereich und mit Micropipetten
Forschungsrelevante Thematik:
• Bioanorganik & Medizinische Anorganische Chemie; essentielle Übergangsme-
talle

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
140
• Naturstoffe mit antimikrobieller Aktivität
• Untersuchung der Aktivitätsmechanismen von Wirkstoffen
• Entwicklung von Antibiotika mit neuartigen, bioanorganischen Wirk-Targets
• William-Irvings-Serie
• Polymorphie
• Wasserstoff/Deuterium Austausch
Allgemeines zur Sicherheit:
Die Synthese wird unter Stickstoff oder Argon durchgeführt. Besondere Vorsicht ist
beim Refluxieren in TFA und der Entsorgung der Säure (Neutralisation der konden-
sierten Säure!) geboten. Ebenso ist zu beachten, dass m-Kresol ein karzinogener Stoff
ist, der leicht über die Haut resorbiert wird; jeglicher Hautkontakt ist zu vermeiden.
Versuchsdurchführung:
Synthese von Amino-Pyrrolon 2:
In einem 250-mL-Schlenkrundkolben mit großem Magnetrührstäbchen wird Ammoni-
umacetat (25 g) vorgelegt und bei 150 °C erhitzt. Nachdem das Salz vollständig ge-
schmolzen ist, wird Hydroxy-Pyrrolon 1 (1.0 g, 1.9 mmol) zugegeben und die Reakti-
onsmischung für 1 Stunde bei 150 °C gerührt. Nach Abkühlen auf Raumtemperatur
wird destilliertes Wasser (100 mL) zugegeben und die wässrige Lösung mit Dichlor-
methan extrahiert (3x 100 mL). Die vereinigten organischen Phasen werden über
Na2SO4 getrocknet und filtriert, und das Lösungsmittel am Rotationsverdampfer ent-
fernt. Die Identität und Reinheit des erhaltenen Produktes (Gemisch aus cis- und trans-
Isomer) wird mithilfe von Dünnschichtchromatographie (DC) überprüft. Anschließend
wird das braune, ölige Produkt mithilfe einer vereinfachten Säulenchromatographie,
d.h. Filtration durch ein Silica-Pad aufgereinigt. Dazu wird eine große Glasfritte mit
Silica, suspendiert in n-Heptan/Essigsäureethylester 3:1, gepackt (Durchmesser:
5 cm, Höhe: 7 cm). Das Produkt, gelöst in möglichst wenig Essigsäureethylester, wird
vorsichtig aufgetragen, und mit dem Eluentsystem n-Heptan/Essigsäureethylester 1:1
eluiert. Die Fraktionen werden in einer Saugflasche aufgefangen und nach DC-Ana-
lyse entsprechend vereinigt, das Lösungsmittel entfernt und das Produkt am Hochva-
kuum getrocknet.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
141
Literatur-Ausbeute: 0.86 g (87 %)
Kriterium: Ausbeute mindestens 45 %
Eigenschaften: Ockerfarbener Feststoff
Charakterisierung: (Gemisch aus cis- und trans-Isomer)
DC: Rf = 0.54 & 0.46, n-Heptan/Essigsäureethylester 1:1 (Detektion mit UV) 1H NMR (250 MHz, CDCl3): δ [ppm] = 3.54 (s, 0.38x 2H, NCH2, isomer A), 3.72 (s,
0.62x 2H, NCH2, isomer B), 3.74–3.82 (m, 11H, OCH3 & SCH2), 4.03 (br s, 0.38x 2H,
NH2, iso A), 4.16 (br s, 0.62x 2H, NH2, iso B), 4.68 (s, 0.62x 2H, SCH2, iso B), 5.09
(s, 0.38x 2H, SCH2, iso A), 5.57 (s, 0.62x 1H, C=CH, iso B), 5.74 (s, 0.38x 1H,
C=CH, iso A), 6.69–7.19 (m, 12H, CHphenyl).
Synthese von Trifluoro-Holomycin 3:
Die angegebenen Mengen beziehen sich auf 0.5 g 2. Passen Sie die Mengen bitte
entsprechend Ihrer Ausbeute an!
In einem 100-mL-Schlenkrundkolben wird Amino-Pyrrolon 2 (0.5 g, 0.9 mmol) vorge-
legt (inkl. Magnetrührstäbchen). TFA (25 mL) und m-Kresol (1 mL, 9 mmol) werden
zugegeben. Nach Aufsetzen eines Rückflusskühlers wird die Lösung für 12 Stunden
unter Argon-Atmosphäre refluxiert (Unterbrechung möglich; unter Argon-Atmosphäre
weiterrühren bis zum nächsten Erhitzen). Anschließend wird die Apparatur geöffnet
und die Lösung wird unter Luftkontakt weitere 30 Minuten gerührt (um eine vollstän-
dige Oxidation zum Disulfid zu bewirken). Nach Abkühlen auf Raumtemperatur wird
die TFA über eine Kühlfalle am Hochvakuum aus der Reaktionslösung entfernt (es
kann währenddessen auf 50 °C erhitzt werden um die Destillation zu beschleunigen).
Die Identität und Reinheit des erhaltenen Produktes wird mithilfe von DC überprüft.
Anschließend wird das braune, ölige Produkt säulenchromatographisch aufgereinigt.
Als kurze Säule dient wiederum eine Glasfritte, die mit Silica, suspendiert in n-Hep-
tan/Essigsäureethylester 3:1, gepackt wird (Durchmesser: 5 cm, Höhe: 7 cm). Das
Produkt, gelöst in möglichst wenig Essigsäureethylester, wird vorsichtig aufgetragen.
Eluiert wird mit einem Gradientensystem: n-Heptan/Essigsäureethylester 3:1 (200 mL)
→2:1 (80 mL) →1:1 (80 mL) →1:2 (80 mL) →1:3 (80 mL) →Essigsäureethylester (ca.
300 mL). Die Fraktionen werden nach DC-Analyse entsprechend vereinigt, das Lö-
sungsmittel entfernt und das Produkt am Hochvakuum getrocknet.
Literatur-Ausbeute: 0.11 g (45 %)

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
142
Kriterium: Ausbeute mindestens 30 %
Eigenschaften: Gelb-orangener Feststoff
Charakterisierung:
DC: Rf = 0.33, n-Heptan/Essigsäureethylester 1:2 (Detektion mit UV) 1H NMR (250 MHz, DMSO-d6): δ [ppm] = 7.32 (s, 1H), 10.92 (br s, 1H), 11.59 (br s,
1H). Bitte heben Sie die Lösung Ihrer NMR-Probe auf! Diese verwenden Sie für die
Metallkoordinationsstudien.
Metallkoordinationsstudien mit 3:
Für die Metallkoordinationsstudien wird eine Stammlösung von Verbindung 3 benötigt.
Dafür dient die NMR-Probe der zweiten Synthesestufe (in DMSO-d6).
Zunächst muss die Konzentration von 3 in dieser Lösung bestimmt werden. Dies er-
folgt mithilfe der UV/Vis-Spektroskopie. Erstellen Sie dazu eine Verdünnungsreihe von
Stammlösung 3 in MilliQ-H2O und bestimmen Sie jeweils den Absorptionswert bei
388 nm. Verwenden Sie mindestens drei unabhängige Verdünnungen (z. B. 1:100,
1:200, 1:250; Totalvolumen: 1 mL); die Absorptionswerte der Verdünnungen sollten
zwischen 0.5–1.0 liegen (linearer Bereich). Berechnen Sie anschließend die Konzent-
ration der Stammlösung mithilfe des Extinktionskoeffizienten von 3, ε(388 nm) =
11 220 M–1 cm–1.
Produkt 3 liegt als Disulfid vor. Für die Metallkoordination muss es zunächst zum
Dithiol reduziert werden. Testen Sie die Reduktion von 3 mit dem Phosphin TCEP
(Tris(2-carboxyethyl)phosphin). Stellen Sie dazu eine Lösung von 50 µM 3 in 75 mM
Tris-HCl, pH 7.4 her (Totalvolumen: 1 mL, Verdünnung mit MilliQ-H2O) und nehmen
Sie das UV/Vis-Spektrum auf. Anschließend geben Sie TCEP zu einer finalen Kon-
zentration von 100 µM zu, mischen die Lösung gut, inkubieren für 3 min, und nehmen
erneut das UV/Vis-Spektrum auf. Sie sollten eine Verschiebung des Absorptionsmaxi-
mums von ca. 380 nm auf ca. 340 nm beobachten.
Um die Chelatierung verschiedener essentieller Metallionen durch Holomycin zu tes-
ten, werden nun Mn(II), Co(II), Ni(II), Cu(II) und Zn(II) zum reduzierten Holomycin-Ana-
logon 3 gegeben. Da reduziertes Holomycin leicht durch Luftsauerstoff oxidiert wird
und entsprechend nur eine kurze Halbwertszeit unter aeroben Bedingungen besitzt,
wird das Dithiol für die Metalltitration jeweils in-situ durch Zugabe von TCEP herge-

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
143
stellt. Stellen Sie dafür 5 Lösungen von 50 µM 3 in 75 mM Tris-HCl, pH 7.4 her (Total-
volumen je 1 mL; 1 Lösung pro Metallion). Geben Sie jeweils 100 µM TCEP zu und
inkubieren für mindestens 3 Minuten. Nehmen Sie das UV/Vis-Spektrum von reduzier-
tem 3 auf. Geben Sie anschließend das jeweilige Metallion zu einer finalen Konzent-
ration von 50 µM zu, mischen gut, inkubieren für 3 Minuten und nehmen erneut das
UV/Vis-Spektrum auf. Nehmen Sie so die Spektren für alle 5 Metallionen (separat) auf.
Vergleichen Sie nun die erhaltenen Absorptionsspektren. Sie sollten für jedes Metal-
lion ein charakteristisches Absorptionsspektrum erhalten haben und eine Aussagen
treffen können, mit welchen Metallionen reduziertes Holomycin interagiert.
Anschließend soll die Komplexstabilität mit zwei Metallionen, Ni(II) und Zn(II), getestet
werden. Dazu wird jeweils eines der beiden Metallionen mit reduziertem 3 chelatisiert.
Anschließend wird das andere Metallion im Überschuss (10 eq.) zugegeben und ge-
prüft, ob sich die Chelatisierung ändert. Stellen Sie dazu nochmals 2 Lösungen von
50 µM 3 in 75 mM Tris-HCl, pH 7.4 her (1 mL). Inkubieren Sie beide Lösungen mit
100 µM TCEP. Geben Sie anschließend 50 µM Ni(II) bzw. Zn(II), inkubieren und neh-
men die beiden UV/Vis-Spektren auf. Geben Sie nun 500 µM des jeweils anderen Me-
tallions zu (Zn(II) zum Ni(II)-Komplex bzw. Ni(II) zum Zn(II)-Komplex), inkubieren für
3 min und nehmen nochmals die UV/Vis-Spektren auf. Vergleichen Sie die Spektren
vor und nach der Zugabe des zweiten Metallions und diskutieren Sie die relativen Kom-
plexstabilitäten im Protokoll und vergleichen diese mit der William-Irvings-Serie.
d1-ROY
In einem 50 mL Rundkolben mit Magnetrührstäbchen und Rückflußkühler werden O.1
g ROY mit 5mL d4-Methanol versetzt und unter Rühren 20 Minuten auf Rückfluß er-
hitzt. Anschließend wird die Lösung auf Raumtemperatur abgekühlt und der Reakti-
onsansatz über Nacht bei -15°C in der Kühltruhe gelagert. Die auskristallisierte poly-
morphe gelbe d1-ROY-Form wird durch Filtration isoliert und mittels IR-Spektroskopie
charakterisiert.
ROY: ν(N-H): 3303 cm-1, d1-ROY: ν(N-D): 2402 cm-1

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
144
Polymorphie-Screening von ROY und d1-ROY
In den Screening-Experimenten werden in einem 50 mL Rundkolben mit Magnetrühr-
stäbchen und Rückflußkühler 25mg der Substanzen mit 5 mL Methanol (ROY) bzw. 5
mL d4-Methanol (d1-ROY) versetzt, und unter Rühren 10 Minuten auf 50°C erwärmt.
Anschlirßend werden die Lösungen auf Raumtemperatur abgekühlt und über Nacht
bei -15°C in der Kühltruhe gelagert. Die erhaltenen kristallinen Produkte werden filtriert
und mittels Pulverdiffraktometrie charakterisiert.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
145

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
146
Trimethylsilyl(triphenylphosphonium-benzylid)
(PPh3)CPh(SiMe3)
Literatur:
Bestmann, H. J., Bomhard, A. Angew. Chem. 1982, 94, 562.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
147
Chemikalienliste:
Name der Chemikalie Menge
Triphenylphosphan 10.0 g (38.1 mmol)
Toluol tr. 600 ml
Benzylbromid 4.50 ml (38.1 mmol)
Tetrahydrofuran tr. 400 ml
n-BuLi (1.54 M in Hexan) 24.8 ml (38.2 mmol)
Iodotrimethylsilan 4.00 ml (31.4 mmol)
Kalium-tert-butanolat 3.40 g (30.3 mmol)
Einleitung:
Um ylidische Liganden in Hauptgruppenelementverbindungen einzuführen, werden oft
Yldiide verwendet. Yldiide haben in der Regel elektronenziehende Gruppen (z. B. Sul-
fonyl- oder Nitrilgruppen), um die hohe negative Ladung am ylidischen Kohlenstoff-
atom zu stabilisieren. Diese Gruppen können jedoch oftmals - bevorzugt in elektronen-
armen, niedervalenten Hauptgruppenelementverbindungen - zum Hauptgruppenele-
ment koordinieren. Dies ist nicht immer gewünscht, z. B. bei der Synthese von Borini-
umkationen, welche durch die Koordinierung eines weiteren Liganden zu Boreniumka-
tionen werden.
Um eine ungewünschte Koordination zu vermeiden, können ylidische Liganden auch
ausgehend von Yliden ohne koordinierende Gruppen eingeführt werden. Da diese
dann gleichzeitig keine ausreichende Stabilisierung mehr erfahren, um Yldiide zu bil-
den, ist der präparative Weg über diese nicht mehr möglich. Alternativ können Haupt-
gruppenelementhalogenide wie z. B. PCl3 in sogenannten Halosilan-Eliminierungen
mit Silyl-substituierten Yliden umgesetzt werden. Aufgrund der hohen Affinität von Si-
lizium zu den elektrophilen Halogeniden entsteht ein Halosilan und eine Ylid-substitu-
ierte Hauptgruppenelementverbindung. Die Affinität des Siliziums zu Halogeniden
steigt dabei von Iodid über Bromid und Chlorid zu Fluorid.
Ein erster Syntheseweg zur Herstellung des hier verwendeten Silyl-substituierten Ylids
wurde 1982 von Bestmann et al. vorgestellt. Durch Umsetzung des Triphenylphospho-
niumbenzylid mit einem halben Äquivalent ClSiMe3 wurde zunächst das Silyl-substitu-
ierte Phosphoniumsalz erhalten, welches vom Überschuss an Ylid zum Silyl-substitu-
ierten Ylid deprotoniert wurde. Da sich dieser Syntheseweg jedoch als unzuverlässig

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
148
erwies und überdies auch die Hälfte des eingesetzten Ylids als Base verloren geht,
wird bei der hier beschriebenen Synthese eine stöchiometrische Menge ISiMe3 ver-
wendet, das Zwischenprodukt isoliert und die Deprotonierung erfolgt mit einer zusätz-
lichen Base.
Literatur:
Scherpf,, T.; Feichtner, K.-S.; Gessner, V. H. Angew. Chem. Int. Ed. Engl. 2017, 56,
3275.
Mohapatra,C.; Scharf, L. T.; Scherpf, T.; Mallick, B.; Feichtner, K.-S.; Schwarz, C.;
Gessner, V. H. Angew. Chem. Int. Ed. Engl. 2019, 58, 7459.
Schmidpeter, A.; Nöth, H.; Jochem, G.; Schrödel, H.-P.; Karaghiosoff, K. Chem. Ber.
1995, 128, 379.
Schmidpeter, A.; Jochem, G.; Klinger, C.; Robl, C.; Nöth, H. J. Organomet. Chem.
1997, 529, 87.
Bestmann H. J.; Bomhard, A. Angew. Chem. 1982, 94, 562.
Was Sie lernen:
Synthesemethoden:
• Schutzgas- und Vakuumtechnik
• Lösemitteltransfer/Filtration unter Inertgasbedingungen
• Umgang mit reaktiven Lösemittelgemischen
• Transfer von luftempfindlichen Substanzen mit Spritze und Septum
• Temperaturvariation unter Schutzgas
Charakterisierung:
• Heterokern-NMR-Spektroskopie
Forschungsrelevante Thematik:
• Halosilan-Eliminierungsreaktionen
• Niedervalente Hauptgruppenelementverbindungen

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
149
Allgemeines zur Sicherheit:
Der Versuch wird unter nachgereinigtem Argon in ausgeheizten Apparaturen durchge-
führt. Lösemittel werden nach gängigen Methoden getrocknet und unter Argon gela-
gert. Wenn nicht anders im Text beschrieben, wird unter Argon gearbeitet. Bei der
Verwendung von n-BuLi muss besonders sorgsam gearbeitet werden, da es pyrophor
ist. Bei der Zugabe von n-BuLi zur Reaktionslösung muss darauf geachtet werden,
dass sich diese nicht überhitzt.
Geschlossene Gefäße dürfen keinesfalls erhitzt werden.
Versuchsdurchführung:
Triphenylphosphoniumbenzylid
In einem ausgeheizten und mit Argon gefüllten 250-mL-Schlenkkolben wurden 10,0 g
Triphenylphosphan in 100 mL trockenem Toluol gelöst und durch ein Septum mit
4,50 mL Benzylbromid versetzt. Das Gemisch wurde für 5 Stunden bei aufgesetztem
Rückflusskühler mit Blasenzähler auf 80 °C erwärmt. Nachdem die Reaktionslösung
abgekühlt war, wurde das überstehende Toluol abkanüliert und der verbliebene farb-
lose Feststoff einmal mit 100 mL trockenem Toluol gewaschen.
Das restliche Lösungsmittel wird im Unterdruck entfernt und der verbliebene Feststoff
in 100 mL trockenem THF aufgenommen. Zu der Suspension wurden unter Eiskühlung
langsam 24,8 mL 1,54 M n-BuLi in Hexan durch ein Septum zugetropft. Die Lösung
färbte sich dabei dunkelrot. Das Reaktionsgemisch wird 1 Stunde gerührt und nach
Erwärmen auf Raumtemperatur das Lösemittel im Unterdruck entfernt. Der Kolben
wird mit einer Filterkanüle mit einem zweiten Kolben verbunden und der verbliebene
dunkelrote Feststoff in 80 mL Toluol suspendiert und das Toluol auf 110 °C mit einem
Ölbad erhitzt, bis sich nahezu der gesamte Feststoff gelöst hat. Die löslichen Bestand-
teile werden in einer Heißfiltration (110 °C) über die Filterkanüle in den zweiten Kolben
überführt. Das Lösungsmittel wird im Unterdruck entfernt und es verbleibt Triphenylp-
hosphoniumbenzylid, welches in einer Glovebox gewogen und charakterisiert wird.
Literatur-Ausbeute: 12.5 g (93 %)
Kriterium: Ausbeute min. 40 %

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
150
Charakterisierung: 1H NMR (400 MHz, C6D6): δ = 7.75-7.66 (m, 6 H), 7.56-7.48 (m, 3 H), 7.48-7.40 (m, 6
H), 6.74-6.58 (m, 2 H), 6.59-6.45 (m, 2 H), 6.31-6.16 (m, 1 H), 2.44 (d, 2JPH = 18.7 Hz,
1 H) ppm. 31P NMR (162.1 MHz, C6D6): δ = 7.59 ppm.
Trimethylsilyl(triphenylphosphonium-benzylid)
Die nachfolgende Versuchsvorschrift bezieht sich auf eine Ausbeute von 10,0 g
Triphenylphosphoniumbenzylid. Bitte rechnen Sie die Vorschrift entsprechend Ihrer
Ausbeute um.
In einem 500-mL-Schlenkkolben werden in der Glovebox 10.0 g Triphenylphosphoni-
umbenzylid vorgelegt und der Feststoff anschließend in 250 mL trockenem Toluol ge-
löst. Folglich werden 4.00 ml ISiMe3 zur Reaktionslösung mit einer Spritze zugetropft.
Anschließend wird die Suspension über Nacht bei RT gerührt. Dabei fällt ein gelblicher
Feststoff aus. Die überstehende Lösung wird über eine Filterkanüle abfiltriert. Der zu-
rückgebliebene Feststoff wird dreimal mit 15 mLl trockenem Toluol gewaschen. An-
schließend wird der Feststoff im Vakuum bei etwa 45 °C getrocknet, wobei ein gelber
Feststoff erhalten wird.
In einen weiteren Kolben werden 3.40 g KOtBu zugegeben und die Mischung in
250 mL trockenem THF suspendiert. Über eine Filterkanüle wird die basische Lösung
zum gelben Feststoff zugegeben und es wird anschließend über Nacht gerührt. Die
Lösung wird folglich vom entstandenen Feststoff abfiltriert und der Rückstand dreimal
mit 15 mL trockenem THF gewaschen. Die kombinierten THF Lösungen werden im
Unterdruck vom Lösemittel entfernt. Der Feststoff wird in der Glovebox gewogen und
charakterisiert.
Literatur-Ausbeute: 8.10 g (67 %)
Kriterium: Ausbeute min. 30 %
Eigenschaften: rotbrauner Feststoff
Charakterisierung: 1H NMR (400.3 MHz, C6D6): δ = 7.76-7.68 (m, 6 H), 7.34-7.28 (m, 2 H), 7.07-6.95 (m,
11 H), 6.88-6.81 (m, 1 H), 0.22 (s, 9 H) ppm. 31P NMR (162.1 MHz, C6D6) δ = 13.36 ppm.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
151
29Si NMR (79.5 MHz, C6D6): δ = -7.49 ppm.
Die 29Si NMR Daten werden von einem 1H-29Si Korrelationsspektrum erhalten.
Sollten in den NMR-Spektren erhebliche Mengen (über 5 %) an Triphenylphos-
phanoxid oder sonstigen Nebenprodukten (wie z. B. Triphenylphosphan) oder Edu-
ktreste erkennbar sein, so können diese durch Zugabe von einem Äquivalent getrock-
netem ZnCl2 - bezogen auf das Triphenylphosphanoxid - zu einer Lösung in Toluol
entfernt werden (die Menge von Triphenylphosphanoxid wird durch Integration der
NMR-Signale erhalten). Das Gemisch wird über Nacht gerührt, anschließend filtriert
und das Lösungsmittel im Vakuum entfernt.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
152
Musterprotokoll
Jana Musterfrau Datum
Tricyclohexyl(2,4,6-Trimethylbenzyl)-Phosphoniumi-
odid
Literatur
[1] Schmidtpeter, A.; Lochschmidt, S.; Sheldrick, W. S. Angew. Chem. 1985, 97, 214.
[2] Schmidtpeter, A.; Lochschmidt, S. Angew. Chem. 1986, 98, 271.
[3] Schmidtpeter, A.; Lochschmidt, S.; Karaghiosoff, K.; Sheldrick, W. S. J. Chem.
Soc. Chem. Commun. 1985, 1447.
[Anmerkung: Formatierung der Literatur: Angewandte Chemie oder JACS Style]
[Anmerkung: Formatierung Blocksatz, 1.5 Zeilenabstand, Arial, Times New Roman, Calibri;
11 pt – 13 pt]
[Anmerkung: Tempus: Das getan oder beobachtet wurde im Präteritum und Fakten (Bsp: Ver-
schiebungen von Substanzen im NMR, Kristallstrukturen) im Präsens]
Theoretischer Hintergrund:
Basierend auf den Vorbereitungen zum Versuchsantestat soll die Einleitung in das
Thema einführen. Die Länge der Einleitung soll in ihrer Länge (max. eine Seite) dem
übrigen Text angemessen sein und darf nicht zu weitschweifig ausgeführt werden.
Übernehmen Sie keine Textpassagen (keine copy/paste-Aktionen), sondern formulie-
ren Sie mit eigenen Worten den theoretischen Hintergrund zum Präparat.
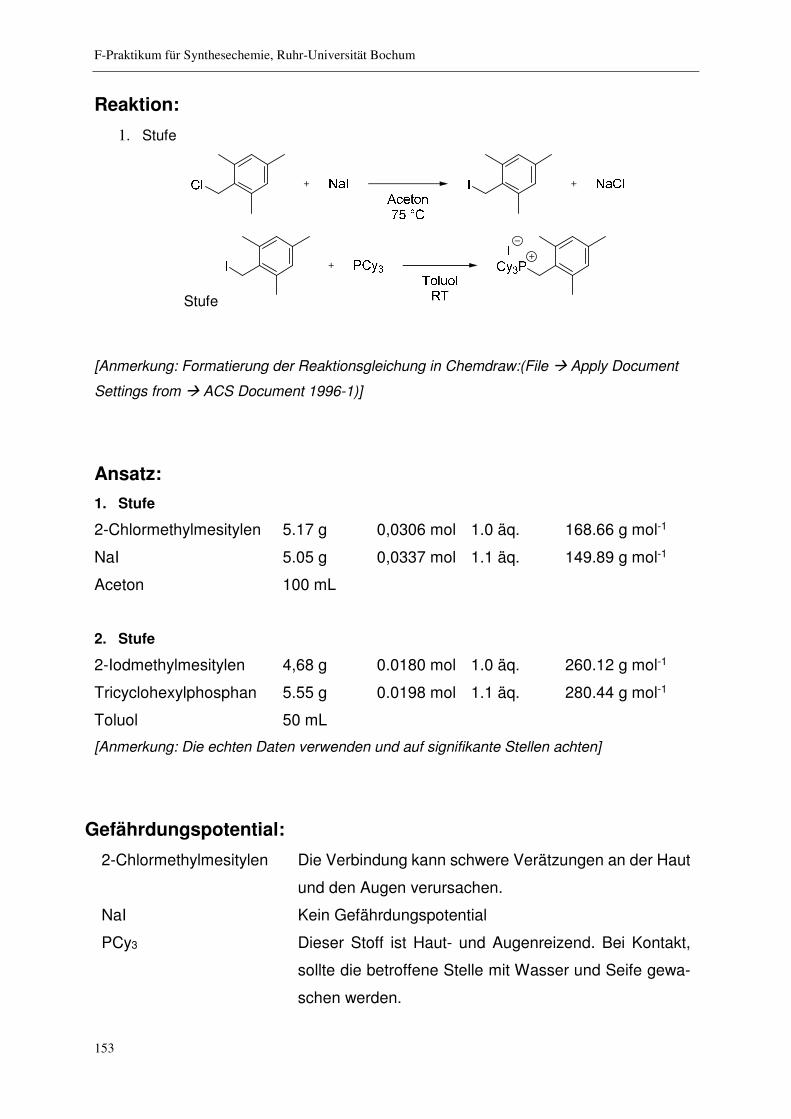
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
153
Reaktion:
1. Stufe
Stufe
[Anmerkung: Formatierung der Reaktionsgleichung in Chemdraw:(File � Apply Document
Settings from � ACS Document 1996-1)]
Ansatz:
1. Stufe
2-Chlormethylmesitylen 5.17 g 0,0306 mol 1.0 äq. 168.66 g mol-1
NaI 5.05 g 0,0337 mol 1.1 äq. 149.89 g mol-1
Aceton 100 mL
2. Stufe
2-Iodmethylmesitylen 4,68 g 0.0180 mol 1.0 äq. 260.12 g mol-1
Tricyclohexylphosphan 5.55 g 0.0198 mol 1.1 äq. 280.44 g mol-1
Toluol 50 mL
[Anmerkung: Die echten Daten verwenden und auf signifikante Stellen achten]
Gefährdungspotential:
2-Chlormethylmesitylen Die Verbindung kann schwere Verätzungen an der Haut
und den Augen verursachen.
NaI Kein Gefährdungspotential
PCy3 Dieser Stoff ist Haut- und Augenreizend. Bei Kontakt,
sollte die betroffene Stelle mit Wasser und Seife gewa-
schen werden.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
154
2-Iodmethylmesitylen Die Verbindung kann schwere Verätzungen an der Haut
und den Augen verursachen.
Aceton Aceton ist brennbar und sollte von Zündquellen fernge-
halten werden. Auch auf Einatmen großer Mengen sollte
verzichtet werden, da dies Schläfrigkeit und Benom-
menheit verursacht.
Toluol Die Flüssigkeit kann über die Haut und den Atemtrakt
aufgenommen werden, wovon akute und chronische
Gesundheitsgefahren ausgehen können. Es sollte des-
halb stets unter einem Abzug gearbeitet werden, Haut-
kontakt sollte mit der Lösung vermieden werden. Toluol
ist brennbar und sollte von Zündquellen ferngehalten
werden.
Tricyclohexyl(2,4,6-Tri-
methylbenzyl)-Phospho-
niumiodid
Ungeprüfter Forschungsstoff. Es muss behandelt wer-
den, als wäre er giftig und krebseregend.
[Anmerkung: Keine bloße Aufzählung von H und P-Sätzen, Wichtige Gefahren zuerst
(Bsp Explosiv vor Gesundheitsgefähredend]
Gefährdungsbeurteilung:
Beim Arbeiten mit Toluol sollte die Abzugsscheibe gut geschlossen sein. 2-Chlorme-
thylmesitylen sowie das Zwischenprodukt 2-Iodmethylmesitylen ist ein reizender Stoff
und deshalb sollte Hautkontakt vermieden werden. Bei Hautkontakt muss die betroffene
Stelle mit Wasser und Seife gewaschen werden.
Entsorgung
2-Chlormethylmesitylen Lösen in Ethanol und dann in Sammelbehälter halogenhal-
tige Lösungsmittel
NaI Sammelbehälter für Feststoffe

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
155
PCy3 vorsichtig mit iso-Propanol neutralisieren und in den Behäl-
ter für organische halogenfreie Lösungsmittel entsorgen
2-Iodmethylmesitylen Sammelbehälter für Feststoffe
Aceton Sammelbehälter für organische halogenfreie Lösungsmittel
Toluol Sammelbehälter für organische halogenfreie Lösungsmittel
Tricyclohexyl(2,4,6-Tri-
methylbenzyl)-Phospho-
niumiodid
Lösen in Ethanol und dann in Sammelbehälter halogenhal-
tige Lösungsmittel
Versuchsdurchführung:
1. Stufe
Ein 250 mL-Schlenkkolben wird mit einem großen Magnetrührstab versehen und eva-
kuiert. Hierzu wird mehrfach Unterdruck an den Kolben angelegt und anschließend mit
Argon gespült. 2-Chlormethylmesitylen (5.17 g, 30.6 mmol, 1,0 äq.) und Natriumiodid
(5,05 g, 33.7 mmol, 1.1 äq.) werden vorgelegt und in 100 mL Aceton suspendiert. Die
Suspension wird drei Stunden bei 75 °C Ölbadtemperatur refluxiert, eine gelbliche
Suspension entstand. Die Suspension wurde abgekühlt und das Aceton wurde im Va-
kuum entfernt. Der Feststoff wurde mit Filterkanüle entfernt. Nach Entfernen des Lö-
sungsmittels wurde das Produkt als gelb-brauner Feststoff isoliert (4.68 g, 18.0 mmol,
59 %).
Ausbeute:
Berechnung der molaren Ausbeute:
n = m
M=
4.68g
260gmol��= 0.0180mol
Berechnung der prozentualen Ausbeute
a = n(Praxis)
n(Theorie)=
0.0180mol
0.0306mol= 0.59 = 59%

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
156
2. Stufe
4.68 g (18.0 mmol, 1.0 äq.) 2-Iodmethylmesitylen sowie 5.55 g (19.8 mmol, 1.1 äq.)
Tricyclohexylphosphan wurden in einem 100 ml Schlenkkolben vorgelegt und in 80
mL trockenem Toluol gelöst. Das Gemisch wurde bei Raumtemperatur über Nacht
gerührt und der resultierende Feststoff über einer Schutzgasfritte filtriert und zwei
Mal mit jeweils 10 mL Toluol gewaschen. Der weiße Feststoff wurde im Vakuum ge-
trocknet und 9.45 g (0.0175 mol, 97 %) vom Produkt konnten erhalten werden.
Berechnung der molaren Ausbeute:
n = m
M=
9.45g
540.2gmol��= 0.0175mol
Berechnung der prozentualen Ausbeute
a = n(Praxis)
n(Theorie)=
0.0175mol
0.0180mol= 0.97 = 97%
Charakterisierung:
[Anmerkung: Phosphor, Fluor, Kohlenstoff und Bor-NMR mit einer Nachkommastelle,
1H-NMR mit zwei Nachkommastellen in der Verschiebung. Kopplungskonstanten
sind mit einer Nachkommastelle anzugeben.]
1) Vergleichsdaten[1]
1. Stufe
1H NMR (500 MHz, CDCl3) δ = 2,25 (s, 3H), 2,33 (s, 6H), 4,48 (s, 2H), 6,85 (s, 2H)
ppm.
2. Stufe 31P {1H} NMR (202 MHz, CD2Cl2) δ =14.6 ppm.
1H NMR (400 MHz, CDCl3) δ = 1.21 – 1.50 (m, 9H), 1.48 – 1.68 (m, 6H), 1.69 – 1.82
(m, 6H), 1.82 – 1.93 (m, 10H), 2.13 (s, 3H), 2.22 – 2.35 (m, 2H), 2.63 (s, 6H), 6.88 (s,
2H) ppm.

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
157
2) erhaltene Daten
1. Stufe
1H NMR (400 MHz, CDCl3) δ = 2.26 (s, 1H), 2.33 (s, 2H), 4.46 (s, 1H), 6.84 (s, 1H)
ppm.
2. Stufe
31P {1H} NMR (162 MHz, CD2Cl2) δ = 14.6, 24.2, 34.0, 46.0 ppm.
1H NMR (400 MHz, CD2Cl2) δ = 0.75 – 1.10 (m, 9H), 1.10 – 1.27 (m, 4H), 1.29 – 1.58
(m, 9H), 1.55 – 1.72 (m, 6H), 1.69 – 1.88 (m, 6H), 1.90 – 2.03 (m, 10H), 2.10 (d, J=1.7,
3H), 2.16 – 2.44 (m, 2H), 2.65 (s, 6H), 6.88 (s, 2H). [Anmerkung: bei NMR mit Verunreinigungen, werden die Produktsignale unterstri-
chen]
3) Auswertung
In der ersten Stufe wurde ein sauberes Produkt erhalten, denn alle NMR-Signale ent-
sprechen den zu erwarteten Signalen im 1H NMR Spektrum.
In der zweiten Stufe erscheinen vier Signale in im 31P {1H} NMR Spektrum. Diese vier
Signalen werden vier Spezies zugeordnet. Das bei 14.6 ppm ist das Produkt. Das bei
24.2 ppm ist das oxidierte Produkt und 34.0 ppm und 46.0 ppm sind unbekannte Pro-
dukte. Die Verhältnisse der Signale sind 58:35:4:3 (14.6:24.2:34.0:46.0).
Im 1H NMR Spektrum sind weitere unbekannte aliphatischen Signale zusehen.
Sonst sind keine Lösungsmittelsignale zu erkennen.
[Anmerkung: Falls Lösungsmittelsignale zu erkennen sind, begründen Sie woher diese
stammen. 1. Tipp: Wenn man nicht mit Pentan gearbeitet hat, werden die Signale nicht
zu Pentan gehören. 2. Tipp: Mit was machen Sie ihre Gefäße sauber]
Fehlerbetrachtung:
Bei Substanz aus dem ersten Schritt handelt es sich um das entsprechende saubere
Produkt. Die Ausbeute ist jedoch mit 59 % relativ niedrig, dass womöglich daran liegt,

F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
158
dass das Produkt nicht vollständig reagiert ist, weil die Reflux-Zeit nicht eingehalten
werden konnte. und längere Reaktionszeit benötigt wurde.
In der zweiten Stufe wurde zwar das Produkt synthetisiert. Aber, es weist starke Ver-
unreinigungen auf. Möglicherweise fand durch Sauerstoffeintritt eine Nebenreaktion
statt, weshalb mehrere zusätzliche Spezies im 31P{1H} NMR Spektrum beobachtet
wurden. Das 1H NMR Spektrum zeigt zusätzliche Signale im aliphatischen Bereich,
welche auf zusätzliche Produkte hindeuten, welche ein Cyclohexylgruppe aufweisen.
Diese Verunreinigungen konnten nicht einfach über eine Filtration nicht entfernt wer-
den, weshalb man weitere Maßnahmen ergreifen müsste, um diese zu entfernen. Ein
Beispiel wäre eine Umkristallisation. Dies wurde aus Zeitmangel nicht durchgeführt.
Anhang:
Spektren [Kompletten Spektralbereich zeigen und nicht irgendwelche Ausschnitte! Alle Signale sollten über MestReNova zugeordnet werden. Die Tabelle rechts auf dem Bild kann über dem Menüpunkt neues MestreNova-Design: View � Parameters � Report |(al-tes MestreNova-Design View � Tables � Parameters aufgerufen werden.]
Abb 1. 1H NMR Spektrum von ICxHx (Stufe 1).
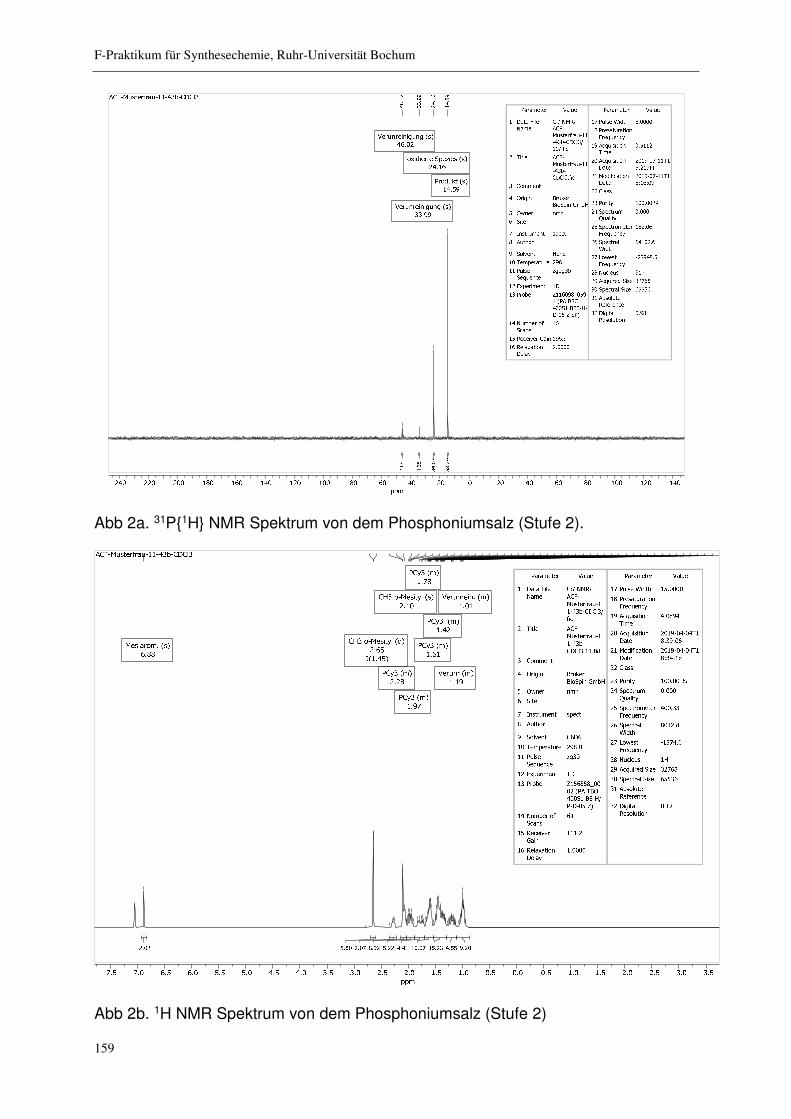
F-Praktikum für Synthesechemie, Ruhr-Universität Bochum
159
Abb 2a. 31P{1H} NMR Spektrum von dem Phosphoniumsalz (Stufe 2).
Abb 2b. 1H NMR Spektrum von dem Phosphoniumsalz (Stufe 2)


















