
Scriptum - Helmholtz Zentrum München€¦ · Während der Zentrifugation wandern die Blutzellen...
32
In diesem Blockkurs wird ganz auf praktische Tätigkeit gesetzt. Nach einer kurzen Einführung machen wir Sie mit der Durchführung vielfältiger Experi- mente aus der Zell- und Molekularbiologie sowie mit Tiermodellen vertraut. Auf diese Weise gewinnen Sie am Beispiel der aktuellen translationalen Lun- genforschung Einblicke in die modernen Methoden eines Forschungslabors. Damit ist dieser Kurs auch ein idealer Einstieg bzw. Entscheidungshilfe für eine experimentelle Doktorarbeit. Am Ende des Kurses berichten und inter- pretieren Sie die von Ihnen erhobenen Daten. Die Lernziele dieser Veranstaltung sind: Einführung in Zellkultur, Durchfluss- zytometrie, Zellseparation, Life Cell Imaging, RNA-Isolierung, quantitiative PCR, Genotypisierung mittels PCR, Tracheotomie und Lungenfunktion an der Maus, Bronchoalveoläre Lavage und Histologie Wahlfach 2. Studienabschnitt Einführung in wissenschaftliche Methoden und wissenschaftliches Arbeiten im Labor am Beispiel der modernen Lungenforschung 06. August – 10. August 2012 Comprehensive Pneumology Center, Großhadern Max-Lebsche-Platz 31, 1.OG, Seminarraum Weitere Informationen: Dr. Thomas Hofer, [email protected] PD Dr. Susanne Krauss-Etschmann, [email protected] oder im elektronischen Vorlesungsverzeichnis Scriptum
Transcript of Scriptum - Helmholtz Zentrum München€¦ · Während der Zentrifugation wandern die Blutzellen...

In diesem Blockkurs wird ganz auf praktische Tätigkeit gesetzt. Nach einer kurzen Einführung machen wir Sie mit der Durchführung vielfältiger Experi-mente aus der Zell- und Molekularbiologie sowie mit Tiermodellen vertraut. Auf diese Weise gewinnen Sie am Beispiel der aktuellen translationalen Lun-genforschung Einblicke in die modernen Methoden eines Forschungslabors.Damit ist dieser Kurs auch ein idealer Einstieg bzw. Entscheidungshilfe für eine experimentelle Doktorarbeit. Am Ende des Kurses berichten und inter-pretieren Sie die von Ihnen erhobenen Daten.Die Lernziele dieser Veranstaltung sind: Einführung in Zellkultur, Durchfluss-zytometrie, Zellseparation, Life Cell Imaging, RNA-Isolierung, quantitiative PCR, Genotypisierung mittels PCR, Tracheotomie und Lungenfunktion an der Maus, Bronchoalveoläre Lavage und Histologie
Wahlfach 2. Studienabschnitt
Einführung in wissenschaftlicheMethoden und wissenschaftlichesArbeiten im Labor am Beispiel der
modernen Lungenforschung
06. August – 10. August 2012 Comprehensive Pneumology Center, GroßhadernMax-Lebsche-Platz 31, 1.OG, SeminarraumWeitere Informationen: Dr. Thomas Hofer, [email protected] Dr. Susanne Krauss-Etschmann, [email protected] oder im elektronischen Vorlesungsverzeichnis
Scriptum

Wir begrüßen Sie herzlich zu unserem Wahlfach im 2. Studienabschnitt„Experimentelle Pneumologie Live“
Dieses Wahlfach wird veranstaltet vomComprehensive Pneumology Center,gegründet von den Kooperations-Partnern:
Helmholtz Zentrum München,Deutsches Forschungszentrumfür Gesundheit und Umwelt
Ludwig-Maximilians-Universität Münchenmit dem Klinikum der Universität
Asklepios Fachkliniken, München-Gauting
2
Grußwort
Liebe Studentinnen und Studenten,
im Bereich der modernen biomedizinischen Forschung sind heute neue Ansätze zum Verständnis der Pathomechanismen von einzel-nen Erkrankungen wichtig, um innovative Möglichkeiten zur Prä-vention und Therapie von chronischen Erkrankungen zu entwickeln und zu etablieren.
Für chronische Lungenerkrankungen gilt das im speziellen, da diese zwar weit oben in der Rangliste der häufigsten Erkrankungen ste-
hen (die chronisch obstruktive Lungenerkrankung COPD ist die vierthäufigste To-desursache weltweit), kausale Therapien aber nach wie vor rar sind.
Das Translationszentrum für Lungenforschung CPC (Comprehensive Pneumology Center) in München wurde gegründet, damit Experten aus Medizin und Lebenswis-senschaften gemeinsam grundlegende Mechanismen von chronischen Lungener-krankungen, wie z. B. COPD, Lungenfibrose, Lungenkrebs, oder Asthma, erforschen und neue Ansätze für deren Prävention, Diagnostik und Therapie entwickeln.Das Kennenlernen von experimentellen Grundlagen in der Zell- und Molekularbiolo-gie, sowie von Tiermodellen zur Erforschung von chronischen Lungenerkrankungen ist ein guter erster Schritt um die Vielfältigkeit der modernen Lungenforschung ken-nenzulernen. Nutzen Sie die Chance moderne Forschung selbst zu erleben – Viel Freude und Er-folg bei uns am CPC!
Prof. Dr. Oliver EickelbergChairman, Experimentelle Pneumologie, Comprehensive Pneumology Center
Fachkliniken München-Gauting

AblaufZu Beginn des Praktikums werden Sie in drei Gruppen eingeteilt, in der unter-schiedliche methodische Aspekte des Fachs Lungenbiologie behandelt werden: Molekularbiologischer, zellbiologischer und tierexperimenteller Teil. Sie durchlaufen nacheinander die drei Gruppen am Montag, Dienstag und Donnerstag. Am Freitag Vormittag stellen Sie ihre Ergebnisse aus den Experimenten vor und präsentieren ihre Interpretation.Veranstaltungsorte sind das CPC am Max-Lebsche-Platz 31 in Grosshadern für die beiden ersten Gruppen, der tierexperimentelle Teil findet am Helmholtz Zentrum in Neuherberg statt (Ingolstädter Landstraße 1, Neuherberg, Gebäude 35, Raum 1003; Lageplan http://www.helmholtz-muenchen.de/fileadmin/GSF/images/an-fahrt/Wegweiser.pdf).
Ihre Betreuer Zellbiologischer Teil Dr. Gerald Burgstaller, Dr. Marion Frankenberger, Dr. Thomas Hofer, Prof. Dr. Peter Nelson, Prof. Dr. Elfriede Nössner
Molekularbiologischer Teil Dr. Stefan Dehmel, PD Dr. Silke Meiners, PD Dr. Susanne Krauss-Etschmann Dr. Ralf Zarbock
Tierexperimenteller Teil Dr. Gerrit John, Dr. Dr. Melanie Königshoff, Dr. Johannes Schwarz, Dr. Tobias Stöger, Dr. Ali Önder Yildirim
Tages- und Zeitübersicht
3
Zellbiologischer Teil Molekularbiologischer Teil Tierexperimenteller Teil Freitag: Auswertg., Prüfg.08:3008:45
09:00
09:15
09:30
09:45
10:00
10:15
10:30
10:45
11:00
11:15
11:30
11:45
12:00
13:30
13:45
14:00
14:15
14:30
14:45
15:00
15:15
15:30
15:45 Beprechung Gel‐Daten (RZ, SD)
16:00
16:15
16:30
16:45
17:00
17:15
Eröffnung und Grußwort (OE)
Einführung Blutzellen, FACS‐Analyse, Versuchsabläufe (MF, TH)
Abschließende praktische Prüfung
Tierpräparation: BAL, Blut‐ und Organentnahme (JS & MK)
Einführung 2: Mausmodelle in der Lungenforschung (MK)
Einführung besonderheiten von microRNA
Durchführung von Immunfärbungen für die Druchflusszytometrie: Monozyten, CD3/4/8, NK‐Zellen, B‐Zellen, / T cells (MF, TH, EN)
Messung der Blutproben am LSRII Teil1: Monozyten (MF)
RNA‐Isolation, Konz‐Bestimmung, Reverse Transkription (RZ, SD)
Einführung 1: Die Lunge von Maus‐und Mensch ‐ Unterschiede und
Gemeinamkeiten. Prinzipien der Lungenfunktionsuntersuchung ander
MAus (AY, TS)
Aufbereitung Blut & BAL: Zellzählung, Cytospins (TS)T‐Zell/ Endothelinteraktion ‐ Rollen/
Adhäsion/Transmigration (PN)
Analyse qPCR Daten (RZ, SD)
Durchführung qPCR (RZ, SD)
Gel‐Elektrophorese (RZ, SD)Messung Blutproben am LSRII Teil2 (MF,TH, EN)
Life Cell Imaging und Fluoreszenz‐Mikroskopie / Zell Migrations Assay (GB)
Tracheotomie und Lungenfunktion (AY & GJ)
Blutabnahme (MF,SK)
Separation von Bultzellen über Dichtegradienten
Ergebnisvorstellung (Studenten) und ‐besprechung Kurstag: Tierversuche
Ergebnisvorstellung (Studenten) und ‐besprechung Kurstag: Zellbiologie
Ergebnisvorstellung (Studenten) und ‐besprechung Kurstag: Molekularbiologie
Einführung 1 (SM). Warum Methoden funktionieren und was sie aussagen
Standard‐PCR, Agarose‐Gel (RZ, SD)
Aufbereitung Organe: Histologie, MACS, FACS (JS & GJ)

4
Zellbiologischer Teil
Betreuer:Gerald Burgstaller, Marion Frankenberger, Thomas Hofer, Peter Nelson undElfriede Nößner
Versuchsteil A: Leukozyten-Charakterisierung im menschlichen Blut
ÜbersichtDie Zellen des peripheren Bluts haben ihren Ursprung im Knochenmark, wo sie sich aus pluripotenten Stammzellen zu den verschiedenen Subpopulationen weiter ent-wickeln (Abb. 1).
Abb.1
Neben den Erythrozyten und Thrombozyten stellen die Leukozyten die morpholo-gisch und funktionell am stärksten heterogene Zellart dar, die sich weiter in folgen-de Klassen unterteilen läßt.
• Granulozyten o neutrophile Granulozyten o basophile Granulozyten o eosinophile Granulozyten • Lymphozyten o B-Lymphozyten (B-Zellen) o T-Lymphozyten (T-Zellen) PBMC o Natürliche Killerzellen (NK-Zellen) • Monozyten o Classical monocytes CD14++CD16- (ca bis 500/µl Vollblut) o Non-classical monocytes CD14+CD16++ (ca bis 50/µl Vollblut)
5
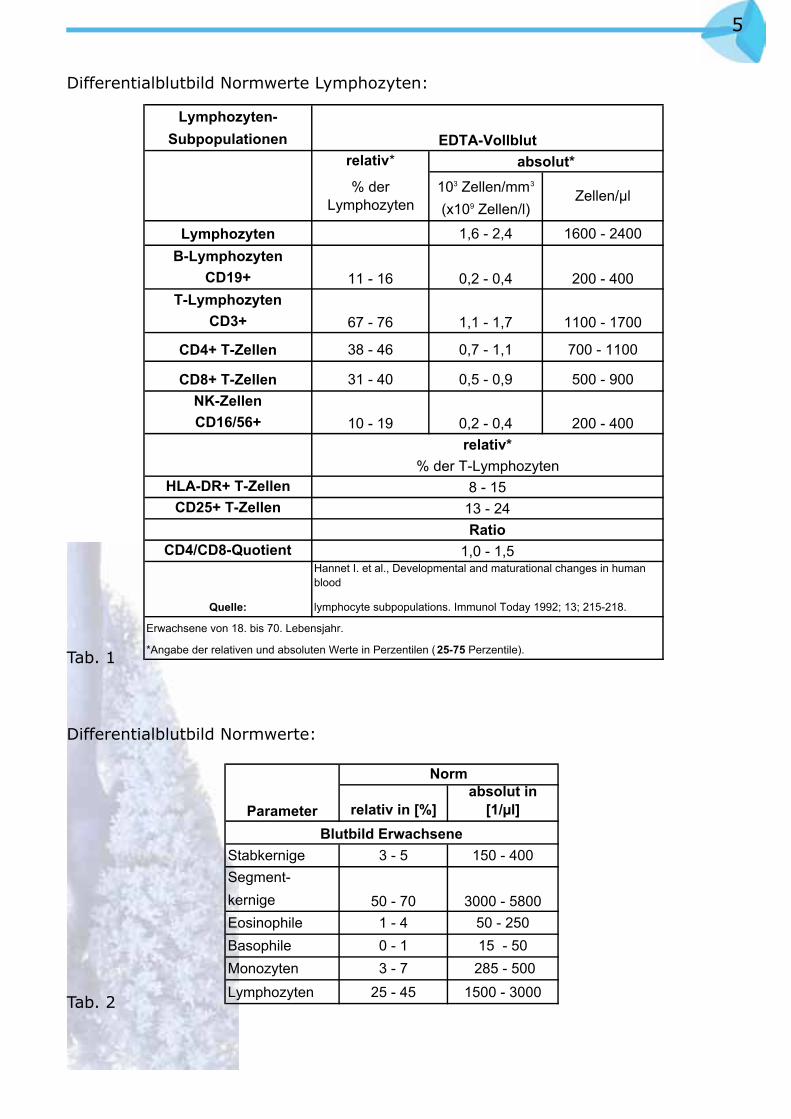
Differentialblutbild Normwerte Lymphozyten:
Tab. 1
Differentialblutbild Normwerte:
Tab. 2
Lymphozyten-Subpopulationen
relativ*
103 Zellen/mm3
(x109 Zellen/l)
Lymphozyten 1,6 - 2,4 1600 - 2400B-Lymphozyten
CD19+T-Lymphozyten
CD3+
CD4+ T-Zellen 38 - 46 0,7 - 1,1 700 - 1100
CD8+ T-Zellen 31 - 40 0,5 - 0,9 500 - 900NK-ZellenCD16/56+
HLA-DR+ T-ZellenCD25+ T-Zellen
CD4/CD8-Quotient 1,0 - 1,5
Quelle:
Hannet I. et al., Developmental and maturational changes in human blood
lymphocyte subpopulations. Immunol Today 1992; 13; 215-218.
Erwachsene von 18. bis 70. Lebensjahr.
*Angabe der relativen und absoluten Werte in Perzentilen (25-75 Perzentile).
relativ*% der T-Lymphozyten
8 - 1513 - 24Ratio
67 - 76 1,1 - 1,7 1100 - 1700
10 - 19 0,2 - 0,4 200 - 400
EDTA-Vollblutabsolut*
Zellen/µl
11 - 16 0,2 - 0,4 200 - 400
% der Lymphozyten
relativ in [%]absolut in
[1/µl]
Stabkernige 3 - 5 150 - 400Segment-kernigeEosinophile 1 - 4 50 - 250Basophile 0 - 1 15 - 50Monozyten 3 - 7 285 - 500Lymphozyten 25 - 45 1500 - 3000
Parameter
Norm
Blutbild Erwachsene
50 - 70 3000 - 5800

6
Ausgangsmaterial für viele immunologische Untersuchungen ist humanes venö-ses Blut. Zahlreiche Nachweise von Oberflächenantigenen können zwar im Vollblut vorgenommen werden, d. h. ohne die Leukozyten in ihre Subpopulationen aufzu-trennen, in vielen Fällen erfordert der Versuchsaufbau aber eine Auftrennung der weißen Blutzellen in ihre Untergruppen.
Humanes Blut enthält neben den roten Erythrozyten, die Zellfraktion der Leukozy-ten, die sich wiederum in Granulozyten, Monozyten plus Lymphozyten aufteilt. Zu den Lymphozyten zählen dann B-Zellen, T- Zellen und NK-Zellen (siehe Abb. 1). Eine morphologische weitere Differenzierung mit dem Mikroskop ist ohne Zuhilfe-nahme von immunologischen Färbemethoden nicht weiter möglich.
Mit der Blutseparation über eine Dichtegradientenzentrifugation erhält man als Endergebnis sog. PBMC = peripheral blood mononuclear cells, also die Gesamtheit aus Lymphozyten plus Monozyten. Die für die meisten immunologischen Fragestel-lungen eher störenden Granulozyten, sowie Erythrozyten und Thrombozyten wer-den mit dieser Methode entfernt. PBMC liefern dann das Ausgangsmaterial für wei-ter analytische Vorgehensweisen wie z.B. das Sortieren von bestimmten Zelltypen (CD4, CD8, B-Zellen) oder die Differenzierung von reiferen Makrophagen aus den monozytären Vorläufern.
Cluster of differentiation (CD) NomenklaturDie verschiedenen Leukozyten tragen unterschiedliche Oberflächenstrukturen (vor allem Rezeptoren) an ihrer Oberfläche, anhand derer sie sich unterscheiden und unterschiedlichen Immunfunktionen zuordnen lassen. Eine Auswahl ist in der unten stehende Tabelle aufgelistet (Quelle: Wikipedia)
CD Zelltyp Funktion des Proteins
CD3 T-Zellen Signaltransduktion, T-Zell-Rezeptor (TCR)CD4 T-Helfer-Zellen Bindung von MHC-IICD8 zytotox. T-Zelle Bindung von MHC-ICD14 Monozyten Bindung von Lipopolysacchariden (LPS)CD16 Phagozyten FcgRIII, bindet Fc-Domäne von IgG, vermittelt PhagozytoseCD19 alle B-Zellen B-Zell-Corezeptor, substanzielle Funktion in B-Zell-Homöostase, -Aktivierung und –DifferenzierungCD45 alle Leukozyten TyrosinphosphataseCD56 NK-Zellen Adhäsion
VorgehensweiseVenöses humanes Blut wird mit einem Gerinnungshemmer (EDTA, Heparin, Citrat) abgenommen und mit gleichem Volumen PBS verdünnt. Diese Mischung schich-tet man über ein Trennmedium (LymphoPrep oder Ficoll) einer definierten Dichte (1.077 g/L) ohne die beiden Phasen zu vermischen.Während der Zentrifugation wandern die Blutzellen durch den Gradienten und sam-meln sich am Ort ihrer spezifischen Dichte, in der sog. Interphase (Abb. 2); Granu-lozyten und Erythrozyten verklumpen durch das Trennmedium und sammeln sich daher im Pellet. Die Interphase mit den enthaltenen PBMC kann nun vorsichtig ge-erntet werden und die Zellen werden durch nachfolgende Waschschritte noch weiter von kontaminierenden Thrombozyten gereinigt und an das nachfolgende Kulturme-dium, das meist FCS (fötales Kälberserum) enthält, gewöhnt.

7
Abb.2
Durchführung
Anleitung Blutseparation- Blut abnehmen (zwei 9 ml EDTA-Röhrchen)- Sterilbank einschalten und mind. 5 Minuten vor Arbeitsbeginn laufen lassen, Arbeitsfläche mit 80% Ethanol reinigen- beim Arbeiten unter der Sterilbank immer hinter den Luftabsaugschlitzen bleiben- zum sterilen Arbeiten Handschuhe tragen, diese mit 80% Ethanol vor Arbeitsbeginn reinigen - Blut in 50 ml Greiner Tube („Bluecap“) geben und mit gleichem Volumen PBS Puffer verdünnen- in zwei Bluecaps je 13 ml Ficoll vorlegen- jeweils mit je 18 ml Blut-Puffer-Gemisch überschichten, dazu Bluecap schräg halten, 20 ml Pipette ganz oben an der unteren Innenwand ansetzen und Blut langsam einlaufen lassen, ohne dass es zu einer Vermischung mit dem Ficoll kommt- Röhrchen austarieren- geschlossenes Bluecap in gegenüber liegenden Plätzen in die Zentrifuge stellen, 30 min bei 800xg, Beschl. 5, Bremse 2 zentrifugieren- Röhrchen vorsichtig aus Zentrifuge entnehmen, ohne Phasen zu vermischen und unter Sterilbank stellen- in den Röhrchen befindet sich eine untere, rot gefärbte und eine durchscheinende, obere Schicht; im unteren Drittel letzterer findet sich eine trübe Phase, die die sog. “Peripheren Blut Mononukleären Zellen“ (PBMC) enthält- mit einer 10 ml Pipette in die obere Phase bis kurz über den PBMC eintauchen und diese durch vorsichtiges und langsames Ansaugen in die Pipette aufnehmen, bis

8
alle Zellen entnommen sind; darauf achten, dass keine Erythrozyten mit eingesaugt werden- geschlossenes Bluecap bei 800xg, Beschl. und Bremse 9 für 10 min zentrifugiern- Überstand abnehmen (mit der Pipette) und verwerfen- Pellet in 20 ml PBS resuspendieren und erneut zentrifugieren bei 400xg, Beschl. und Bremse 9 für 8 min.- Überstand verwerfen und Pelle je nach Größe in 2-3 ml PBS/2% FCS resuspendieren, dann auf Eis stellen
Zellzahl bestimmen- in zwei Vertiefungen einer 96-well Platte je 75 µl eine Lösung aus 0,4% Trypanblau und 4% Eisessig vorlegen- 25 µl der Zellsuspension in erste Vertiefung geben und gut vermischen, aus dieser Mischung wiederum 25 µl entnehmen und in zweite Vertiefung geben und gut vermischen- Zellsuspension aus zweiter Verdünnung entnehmen und in die Zählkammer auftragen- Zählkammer mit 80% Ethanol reinigen und trocknen und nach anhauchen der Stege Deckglas anbringen; Newtonsche Ringe zeigen an, ob das Deckglas fest sitzt und die Tiefe der Zählkammer richtig eingestellt ist- Zellsuspension nun gut durchmischt in die Zählkammer füllen; nicht unter- oder überfüllen- die vier großen Quadrate auszählen; auf Linien liegende Zellen nicht doppelt zählen (in einem kleinen Quadrat immer nur Zellen auf linker und unteren Linie mitzählen)
- Zellzahl / ml = Zählergebnis / 4 x 16 x 104
- Eppendorf Gefäße (1,5 ml) bereitstellen und beschriften- in FACS-Röhrchen je 1 Mio Zellen pipettieren und auf Eis stellen
Immunfluoreszenz-Färbung im Vollblut(Durchführung während der Zentrifugationszeit des Gradienten)
Reagenzien- Pro Ansatz 100 µl Vollblut- Monoclonale Antikörper in 1:20 Verdünnung- PBS/2% FCS- Lysereagenzien A, B und C für Coulter Q-Prep Workstation

9
Durchführung- Blut im EDTA-tube gut aufsuspendieren (vortexen)- 100 µl Blut entnehmen und in ein 5 ml Falcon-Tube überführen (dabei darauf achten, dass die Röhrchenwand nicht mit Blut benetzt wird)- Zugabe der Antikörper in 1:20 Verdünnung direkt in das Blut- Probe vortexen und für 20 Min auf Eis im Dunkeln inkubieren Lyse der Erys am Coulter Q-Prep- + Zugabe von 800 µl aqua dest. + 1600 µl PBS/2% FCS- + 100 µl Coulter counting beads (1010 beads/µl)- Messung am LSR II Durchflusszytometer
Ansätze:1. CD45-APC/CD14-FITC/CD16-PE/HLA-DR-PC5
Immunfluoreszenz-Färbung aus PBMC
• CD3-PE-Cy7 5 µl• CD56-APC 3,5 µl• CD4-PE 3 µl• CD8-FITC 4 µl• CD14-PB 3 µl• CD19-APC Alexa eFluor 780 3 µl
Durchführung:- 1 x 106 Zellen in 50 µl PBS/2% FCS im FACS Röhrchen- alle Antikörper zugeben- 20 min auf Eis im Dunkeln inkubieren- 1 x waschen mit 500 µl PBS/2% FCS- Zentrifugieren für 6 min bei 4°C und 400 x g- Überstand abnehmen- Pellet in 700 µl PBS/2% FCS resuspendieren und überführen in ein FACS-Messtube- Messung am LSR II
Beispiel einer Messung mit der Multiparameterfärbung
Abb. 3, 1. Teil
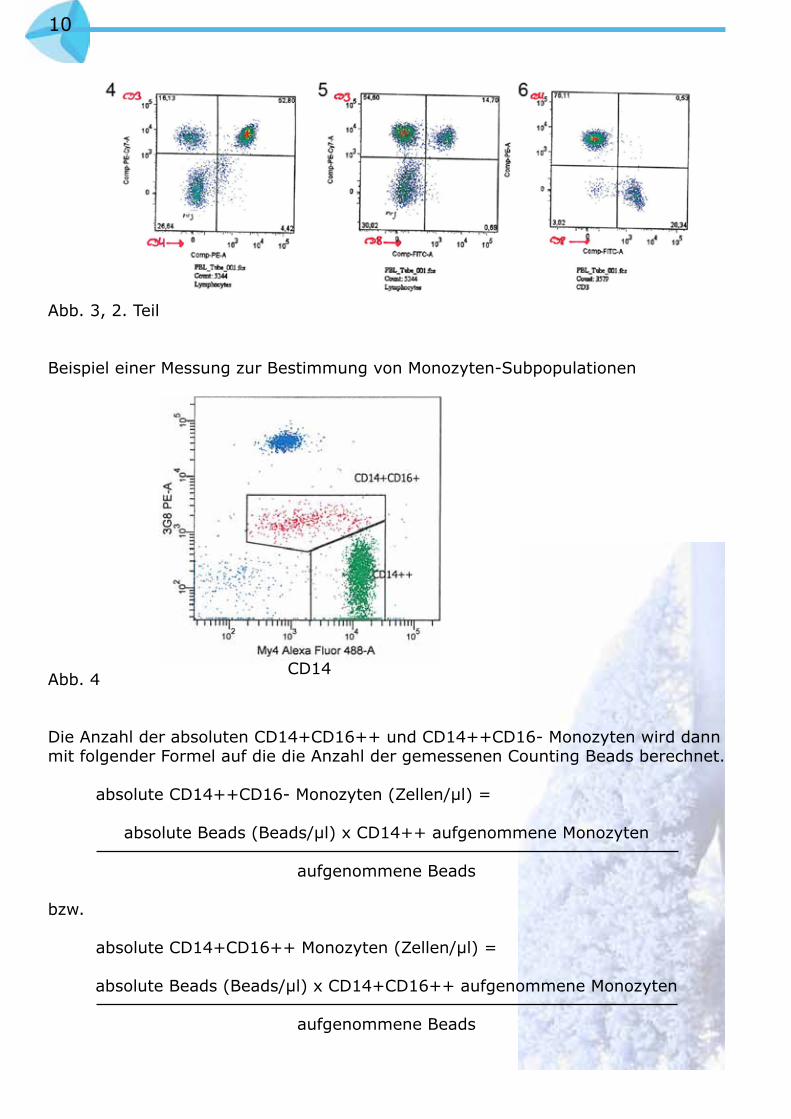
10
Abb. 3, 2. Teil
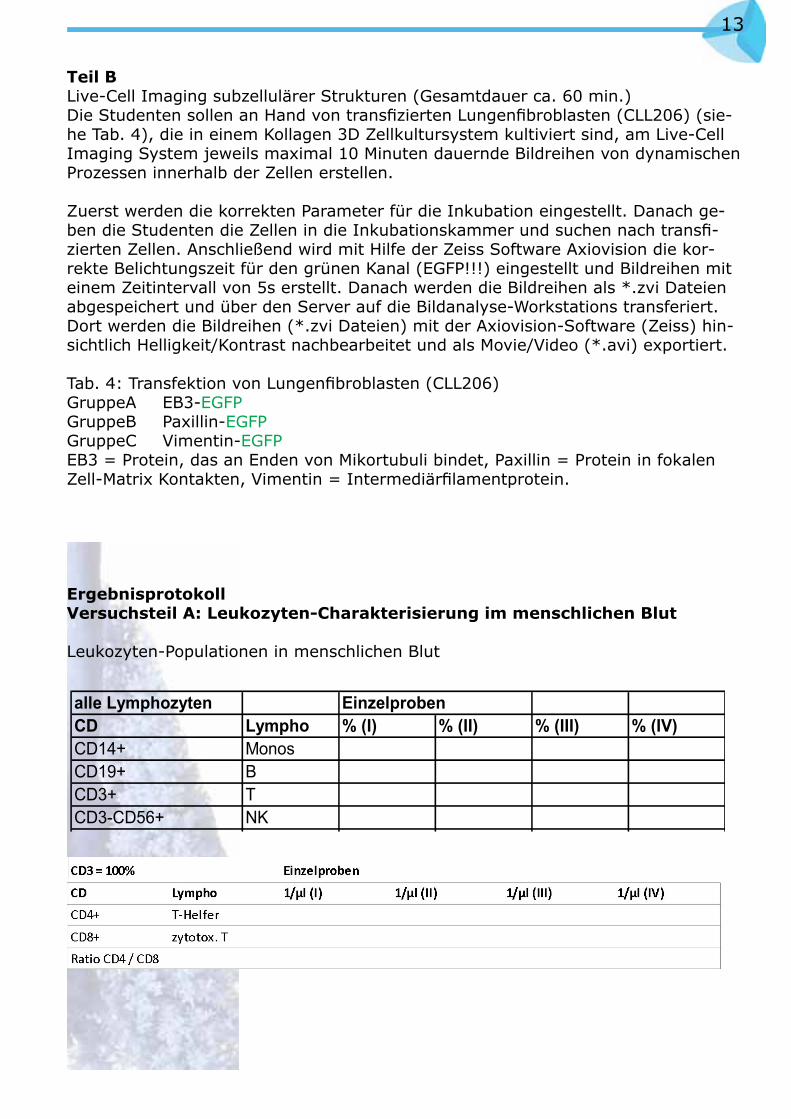
Beispiel einer Messung zur Bestimmung von Monozyten-Subpopulationen
Abb. 4
Die Anzahl der absoluten CD14+CD16++ und CD14++CD16- Monozyten wird dann mit folgender Formel auf die die Anzahl der gemessenen Counting Beads berechnet.
absolute CD14++CD16- Monozyten (Zellen/µl) =
absolute Beads (Beads/µl) x CD14++ aufgenommene Monozyten
aufgenommene Beads
bzw.
absolute CD14+CD16++ Monozyten (Zellen/µl) =
absolute Beads (Beads/µl) x CD14+CD16++ aufgenommene Monozyten
aufgenommene Beads
CD14

11
Versuchsteil B: Life Cell Imaging, Zell-Migrations-Assay
ÜbersichtLichtmikroskope (v. griechisch μικρός: klein; σκοπεiν: betrachten) sind optische Ge-räte für die visuelle oder bildliche Darstellung von Objekten die unterhalb des Auflö-sungsvermögens des menschlichen Auges liegen.Das Auflösungsvermögen eines klassischen Lichtmikroskops ist unter anderem von der Wellenlänge des verwendeten Lichts abhängig und beträgt im besten Fall 0,2 µm, was bedeutet dass zwei Objekte, die einen Abstand von 0,2 µm haben, noch als getrennt wahrgenommen werden können.
Abhängig von der Beleuchtungstechnik unterscheidet man in der Lichtmikroskopie zwischen Durchlicht- und Auflichtmikroskopie.Die Durchlichtmikroskopie benötigt dünn geschnittene und durchsichtige Präpara-te, da das Licht durch das gesamte Präparat hindurchgeleitet werden muss. Durch verschiedene Färbemethoden oder auch durch verschiedene Kontrastverfahren (Phasenkontrast, Lichtpolarisation, Differentialinterferenzkontrast) können die meist farblosen bzw. grundsätzlich kontrastarmen Strukturen von Zellen und Geweben sichtbar gemacht werden.
Bei der Auflichtmikroskopie wird das Licht durch das Objektiv (= die dem Gegen-stand (Objekt) zugewandte Linse) auf das Präparat geleitet. Am Präparat wird das einfallende Licht reflektiert und wiederum vom Objektiv aufgefangen. Einen Spezi-alfall der Auflichtmikroskopie stellt die Fluoreszenzmikroskopie dar, wobei die Flu-oreszenz ein physikalischer Prozess ist, bei dem ein Farbstoff (Fluorophor) durch einfallendes Licht (= Exzitation) einer bestimmten Wellenlänge angeregt wird (z.B.: blaues Licht) und durch das Aussenden (= Emission) von Licht mit einer nun höhe-ren Wellenlänge reagiert (z.B.: grünes Licht) (Abb.5).
Abb. 5
Einen Spezialfall der Fluoreszenzmikroskopie stellt das Laserscanning Konfokalmi-kroskop (LSM) dar. Das Präparat wird hier mit einem Laserstrahl punktförmig ab-getastet. Durch eine Lochblende (= Pinhole) kann emittiertes Licht aus Ebenen des Präparates, die sich nicht im Fokus befinden, ausgeblendet werden. Dadurch kann eine höhere Bildqualität erreicht beziehungsweise Strukturen in 3D dargestellt wer-den.
Wichtige Werkzeuge in der Fluoreszenzmikroskopie sind mit fluoreszierenden Farb-stoffen gekoppelte Antikörper, mit denen man spezifisch Proteine in der Zelle und somit subzelluläre Strukturen (z.B.: Mitochondrien, Zellmembranen, Mikrotubuli, etc.) zum Leuchten bringen kann.

12
Um die Dynamik von Proteinen in lebenden Zellen zu untersuchen, können Proteine direkt mit fluoreszierenden Farbstoffen markiert oder auch durch molekularbiologi-sche Methoden in Form einer Plasmid-DNA (= ringförmiges DNA Molekül) mit der DNA eines fluoreszierenden Proteins (z.B.: EGFP, mCherry, etc) fusioniert werden. Gefärbte Proteine werden durch Mikroinjektionen in die lebenden Zellen einge-bracht. Plasmid-DNA hingegen muss durch Transfektionsmethoden (z.B.: mit Hilfe von Liposomen) in die lebenden Zellen eingebracht und anschließend zuerst von den Zellen als Protein exprimiert werden.
Für die Beobachtung von lebenden Zellen mit einem Fluoreszenzmikroskop müssen Zellen in kleinen Inkubatoren gehalten werden. Dazu muss die Temperatur konstant auf 37°C beziehungsweise auch der pH Wert des Mediums im physiologischen Be-reich (7.0–7.7) gehalten werden. Durch die Anregung der Fluorophore durch Licht können in den Zellen sogenannte phototoxische Effekte ausgelöst werden, welche die Zellen sterben lassen. Deshalb ist die Anregung durch Licht kurz und auch die Lichtintensität so gering als möglich zu halten. Weiters besteht die Gefahr, dass Flu-orophore durch langen und intensiven Lichteinfluss gebleicht werden.
DurchführungTeil AKonfokale Laser Scanning Fluoreszenz Mikroskopie: Aufnahme einzelner Bildstapel von fixierten und gefärbten Lungenfibroblasten in einem Kollagen 3D Zellkultursys-tem (Gesamtdauer ca. 60 min.)
Den Studenten werden transfizierte und gefärbte CLL206 Lungenfibroblasten (siehe Tab. 3), die in einem Kollagen 3D Zellkultursystem kultiviert wurden, zur Verfügung gestellt. Mit Hilfe eines konfokalen Mikroskopes (ZEISS LSM710) sollen die Studen-ten mit unterschiedlichen Objektivgrössen (10x, 25x, 63x) nach transfizierten Zel-len suchen.
Nach Festlegung eines geeigneten Bereiches sollen die Studenten die Belichtungs-zeit für jeden Fluoreszenzkanal korrekt einstellen, einen z-Bereich festlegen und einen Bildstapel aufnehmen. Anschließend werden die Bildstapel als *.lsm5 Dateien abgespeichert und über den Server auf eine Bildanalyse-Workstation transferiert.
Mit Hilfe der Bildbearbeitungssoftware Imaris (Bitplane) werden die Stapel hin-sichtlich Helligkeit/Kontrast nachbearbeitet, für alle Kanäle eine 3D-Rekonstruktion (Surface, Volume) erstellt und die 3D-Rekonstruktion in Form einer Animation als Movie/Video (avi) exportiert.
Tab. 3: Transfektion/Färbung der Lungenfibroblasten (CLL206)GruppeA EB3-EGFP (grün), Phalloidin (Actin-Filamente, rot), Dapi (Zellkerne, blau)GruppeB Paxillin-EGFP (grün), Phalloidin (Actin-Filamente, rot), Dapi (Zellkerne, blau)GruppeC Vimentin-EGFP (grün), Phalloidin (Actin-Filamente, rot), Dapi (Zellkerne, blau)EB3 = Protein, das an Enden von Mikortubuli bindet, Paxillin = Protein in fokalenZell- Matrix Kontakten, Vimentin = Intermediärfilamentprotein
13
alle Lymphozyten EinzelprobenCD Lympho % (I) % (II) % (III) % (IV)CD14+ MonosCD19+ BCD3+ TCD3-CD56+ NKCD3+CD56+ aktiv.T
Teil BLive-Cell Imaging subzellulärer Strukturen (Gesamtdauer ca. 60 min.)Die Studenten sollen an Hand von transfizierten Lungenfibroblasten (CLL206) (sie-he Tab. 4), die in einem Kollagen 3D Zellkultursystem kultiviert sind, am Live-Cell Imaging System jeweils maximal 10 Minuten dauernde Bildreihen von dynamischen Prozessen innerhalb der Zellen erstellen.
Zuerst werden die korrekten Parameter für die Inkubation eingestellt. Danach ge-ben die Studenten die Zellen in die Inkubationskammer und suchen nach transfi-zierten Zellen. Anschließend wird mit Hilfe der Zeiss Software Axiovision die kor-rekte Belichtungszeit für den grünen Kanal (EGFP!!!) eingestellt und Bildreihen mit einem Zeitintervall von 5s erstellt. Danach werden die Bildreihen als *.zvi Dateien abgespeichert und über den Server auf die Bildanalyse-Workstations transferiert. Dort werden die Bildreihen (*.zvi Dateien) mit der Axiovision-Software (Zeiss) hin-sichtlich Helligkeit/Kontrast nachbearbeitet und als Movie/Video (*.avi) exportiert.
Tab. 4: Transfektion von Lungenfibroblasten (CLL206)GruppeA EB3-EGFP GruppeB Paxillin-EGFP GruppeC Vimentin-EGFP EB3 = Protein, das an Enden von Mikortubuli bindet, Paxillin = Protein in fokalenZell-Matrix Kontakten, Vimentin = Intermediärfilamentprotein.
ErgebnisprotokollVersuchsteil A: Leukozyten-Charakterisierung im menschlichen Blut
Leukozyten-Populationen in menschlichen Blut

14
Anzahl CD14++CD16- und CD14+CD16++ Monozyten im Blut
Zusammenfassung der Ergebnisse
abs. Anzahl Beads (Beads/µl)
gemessene Anzahl Beads Anzahl CD14++CD16‐
Anzahl CD14+CD16++
errechnete Anzahl CD14++CD16‐
errechnete Anzahl CD14+CD16++
Probe 1Probe 2Probe 3Probe 4Probe 5Probe 6Probe 7

15
Molekularbiologischer Teil
Betreuer:Stefan Dehmel, Silke Meiners, Susanne Krauss-Etschmann und Ralf Zarbock
Versuchsteil A: Geschlechts- und Genotypbestimmung mittels PCR
ÜbersichtGentechnisch veränderte Mäuse stellen für die biomedizinische Grundlangenfor-schung ein wichtiges Modellsystem dar. Im Gegensatz zu ihren nicht-genetisch veränderten Artgenossen (sog. Wildtyp-Mäuse) besitzen diese Tiere Modifikationen einzelner Gene.
Eine Art der Modifikation ist die Möglichkeit ein Gen gezielt auszuschalten, was im Laborjargon als „knockout“ bezeichnet wird. Dabei wird die Basensequenz eines Gens entweder komplett deletiert oder in Teilen so verändert, dass kein funktionel-les Protein mehr von diesem Gen gebildet werden kann. Diese Tiere gelten dann als defizient für das Protein, das von dem deletierten Gen kodiert wird.
Da alle somatischen Zellen über einen doppelten Chromosomensatz verfügen (ein väterlicher und ein mütterlicher Satz), liegt jedes Gen innerhalb einer Zelle in zwei Kopien vor. Sind beide Kopien identisch (beide wildtyp oder beide deletiert), han-delt es sich um homozygote Mäuse bzgl. des untersuchten Gens. Sind die Kopien unterschiedlich, so spricht man von heterozygoten Mäusen und die Genorte auf dem mütterlichen und väterlichen Chromosom unterscheiden sich somit. In diesem Zusammenhang spricht man auch von unterschiedlichen Allelen eines Gens und das Vorhandensein bestimmter Allele bestimmt den Genotyp (Wildtyp, homozygot-defi-zient oder heterozygot).
Die Kenntnis des Genotyps ist eine grundlegende Voraussetzung vieler Experimente in der biomedizinischen Forschung und die Methodik zur Identifikation des Genoty-ps soll deshalb hier dem interessierten Studenten näher gebracht werden.
Ziel dieses Versuchs ist die molekulare Geschlechtsbestimmung sowie die Bestim-mung des Genotyps für das Gen Tbx21 von Versuchstieren. Dieses Gen, das auch als T-bet (T-box expressed in T cells) bezeichnet wird, kodiert den Transkriptions-faktor TBX21, der eine wichtige Rolle bei der Ausprägung Th1-spezifischer Immu-nantworten spielt.In speziellen Situationen ist neben der Bestimmung des Genotyps auch die Kennt-nis des Geschlechts wichtig. So ist zum Beispiel in neonatalen Mäusen eine Bestim-mung des Geschlechts anhand äußerer Merkmale noch nicht möglich und muss deshalb auf molekularer Ebene durchgeführt werden. Für diese Untersuchungen werden den Mäusen Schwanzbiopsien entnommen und daraus genomische DNA präpariert. Mit spezifischen Primern werden dann mit Hilfe der PCR (Polymerase-Kettenreaktion) geeignete Zielsequenzen amplifiziert und auf einem analytischen Agarosegel nachgewiesen. Das Vorhandensein einzelner Banden mit einer bestimm-ten Größe ermöglicht den Rückschluss auf das Geschlecht (männlich/weiblich) bzw. den Tbx21-Genotyp (wildtyp / heterozygot / homozygot-defizient).
DurchführungJeder Student erhält zu Beginn des Versuchs Tail-Lysate mit der genomischen DNA

16
von drei Mäusen (A, B und C) und soll am Ende des Versuchstages Auskunft über das Geschlecht sowie den Tbx21-Genotyp von Maus A, B und C geben. Um diese Informationen zu ermitteln setzt jeder Student zunächst folgenden PCR-Mastermix in der angegebenen Reihenfolge an:
1x 6x MM10x PCR ThermoPol buffer 2,5 µl 15,0 µl25 mM MgCl2 2,0 µl 12,0 µl1,25 mM dNTP-Mix 4,0 µl 24,0 µlPrimermix 6,0 µl 36,0 µlBraun-H2O 8,3 µl 49,8 µl1 unit Taq-Polymerase 0,2 µl 1,2 µl
Gesamtvolumen 23,0 µl 138,0 µl
Im nächsten Schritt werden je 23,0 µl PCR-Mastermix in 5 beschriftete (A, B, C, - und +) 0,2 ml PCR-Reaktionsröhrchen vorgelegt und je 2,0 µl der Lysate A-C in das jeweilige Röhrchen pipettiert. In die mit „-“ und „+“ beschrifteten Röhrchen wird Wasser (Negativkontrolle) bzw. ein Lysat einer Maus mit bereits bekanntem Genotyp und Geschlecht (Männlich, Tbx21+/-, Positivkontrolle) pipettiert. Alle Pi-pettierschritte sind auf Eis durchzuführen. Sind alle Ansätze pipettiert, werden die Proben in das PCR-Gerät überführt und mit folgendem Programm gestartet: CNTRL TUBE, LID=105°C, WAIT, AUTO
1 T = 95°C 0:03:00 Gründliches Denaturieren der genomischen DNA2 T = 95°C 0:00:30 Denaturieren genomischer DNA und Amplifikate3 T = 62°C 0:00:45 Binden der Primer an die jeweilige Zielsequenz4 T = 68°C 0:00:30 Elongation der Zielsequenz durch die Taq-Polymerase5 GOTO 2 REP 35 Wdh. der Schritte 2 – 4, 35x6 T = 68°C 0:05:00 Final Elongation um alle Reaktionen abzuschließen7 T = 10°C HOLD Temp. wird bis zur Entnahme der Proben gehalten End Ende des Programms
Während das Programm läuft (ca. 2h) stellen die Studenten ein Agarose-Gel zur elektrophoretischen Auftrennung der PCR-Produkte her und bereiten sechs beschrif-tete 1,5 ml Reaktionsröhrchen (A, B, C, -, + und L) mit jeweils 10 µl 2x Ladepuffer vor.
Der Ladepuffer enthält mit DNA interkalierenden SybrGreen-Farbstoff, um die DNA auf dem Agarose-Gel im UV-Licht sichtbar zu machen. SybrGreen steht wie Ethi-diumbromid im Verdacht Krebs erregend zu sein. Deshalb sind beim Umgang mit Ladepuffer spezielle Nitrilhandschuhe zu tragen und auf saubere Arbeitsweise zu achten. Bis zum Abschluss der PCR werden die Studenten über die theoretischen Hintergründe zur PCR informiert (Zeit für Fragen, etc.).
Nach Beendigung der PCR werden die Proben aus dem Gerät entnommen. Nun werden je 10 µl des PCR-Produktes zu den vorbereiteten Tubes mit Ladepuffer gegeben. Das mit „L“ beschriftete Tube wird mit 10 µl DNA-Standard versetzt und dient auf dem Gel als Größenreferenz. Die Mischung (20 µl) wird in die Taschen des Agarose-Gels pipettiert und die PCR-Produkte werden durch anlegen einer Span-nung von 90V für ca. 70 min aufgetrennt. Nach dieser Zeit werden die nach Größe

17
aufgetrennten PCR-Produkte mittels UV-Beleuchtung sichtbar gemacht und foto-grafisch erfasst. Jeder Student erhält ein Gelbild und soll dem Betreuer Geschlecht und Genotyp von Maus A, B und C mit-teilen.
Zur Analyse benötigen die Studenten folgende Daten:
Wildtyp-Bande: 148 bpBande bei Verlust von Tbx21: 189 bpBande für Y-Chromosom: 226 bpBande für X-Chromosom: 255 bp
Folgendes Gelbild soll als Beispiel fürdie Geschlechts- und Genotyp-bestimmung dienen.
Versuchsteil B: Genotypbestimmung mittels quantitativer PCR
ÜbersichtBei der quantitativen PCR, kurz qPCR, ist die Amplifikation und der Nachweis der PCR-Produkte simultan in einem Reaktionsgefäß möglich. Ursprünglich werden für die quantitative PCR spezielle fluorogene Sonden (sog. TaqMan Probes) verwendet, die im mittleren Teil des Amplikons an die Zielsequenz binden.Die 5‘-3‘ Exonukleaseaktivität der DNA Polymerase wird dazu genutzt, einen Quen-cher, der kovalent mit der Sonde verbunden ist und normalerweise das Fluores-zenzsignal des Fluorochromes der Sonde unterdrückt, abzuspalten und so das Fluo-reszenzsignal zu erhöhen.
Bei jedem neuen Zyklus der PCR-Reaktion erhöht sich so die Intensität des Flu-oreszenzsignals. Diese Intensitätszunahme kann innerhalb eines geschlossenen Reaktionsgefäßes gemessen werden. Daher besteht bei der quantitativen PCR, im Gegensatz zur herkömmlichen PCR, bei der es sich um eine reine Endpunktanalyse handelt, die Möglichkeit, die Amplifikation des Produktes in jedem Zyklus zu verfol-gen. Daher wird die qPCR auch oft als „real-time“ PCR bezeichnet.Mit dieser Methode kann also ermittelt werden, ob ein Produkt schon sehr früh (hohe Ausgangsmengen), oder erst zu einem relativ späten Zeitpunkt (niedrige Ausgangsmengen) über die Nachweisgrenze amplifiziert wird.
Statt der TaqMan Sonden kann man zur Quantifizierung von PCR-Produkten auch Farbstoffe verwenden, die den DNA-Doppelstrang spezifisch binden. So wird für die Versuche in diesem Praktikum der Farbstoff SYBR Green 1 benutzt. Dieser Farbstoff verfügt über eine hohe Sensitivität und Spezifität für DNA-Doppelstränge. Durch Bindung von SYBR Green 1 an den DNA-Doppelstrang erhöht sich die Fluoreszenz des Farbstoffs um das bis 100-fache.Dieses Fluoreszenzsignal wird genutzt, um den Amplifikationsprozeß im Laufe der PCR darzustellen. Genauso wie im ursprünglichen 5‘-Exonucleaseassay wird die Flu-oreszenz über den Verlauf der Reaktion aufgezeichnet. Aufgrund der Tatsache, dass im SYBR Green Assay jede doppelsträngige DNA zu

18
einem Fluoreszenzsignal führt, dient neben der sorgfältigen Primerauswahl die Schmelzkurvenanalyse als weitere Spezifitätskontrolle. Nach jedem PCR-Lauf wer-den die Dissoziationskurven der entstandenen Produkte aufgezeichnet, so kann das Vorliegen von Primerdimeren und unspezifischen Nebenprodukten, die ein ver-fälschtes Fluoreszenzsignal generieren würden, beurteilt werden.
DurchführungJeder Student erhält zu Beginn des Versuchs RNAs von drei Mäusen (1, 2 und 3) und soll mit Hilfe der hier vorgestellten Methodik die mRNA-Expression des Gens Tbx21 (Wildtyp-Allel) bzw. des Neomycinresistenzgens Neo („Knockout“-Allel) be-stimmen. Heterozygote Mäuse zeigen eine Mischexpression von Tbx21 und NeoR.
RNA-Konzentrationsbestimmung mittels SpektrophotometerZunächst werden die Messsockel des Geräts mit je 2 µl Wasser gereinigt. Danach wird das Gerät durch Messung von 1 µl RNase-freiem Wasser (sog. Blank-Wert) auf Null gesetzt. Nun werden die einzelnen RNA-Proben von den Studenten gemes-sen (jeweils 1 µl). Die erhaltenen Konzentrationen und Werte für Kontaminationen (260/280 Ratio) werden mit den Studenten diskutiert. Im letzten Schritt wird das für die reverse Transkription erforderliche Probenvolumen, d.h. die benötigten µl für 1 µg RNA jeder Probe bestimmt.
Reverse TranskriptionFür die Umschreibung der RNA in cDNA wird der Quantitect-Kit von Qiagen einge-setzt. Zunächst werden je 1 µg der RNA-Proben (Volumen laut Berechnung aus der Konzentrationsbestimmung) mit RNase-freiem Wasser auf 12,0 µl in 0,5 ml Reak-tionsgefäßen (beschriften: 1, 2, 3 und RT-) eingestellt. RNA-Probe #1 soll auch als RT- Kontrolle verwendet werden und wird daher zweimal vorbereitet.
Im nächsten Schritt wird ein Mastermix für die RT-Reaktion nach folgendem Sche-ma in einem 1.5 ml Reaktionsgefäß vorbereitet und bis zur Reaktion auf Eis aufbe-wahrt: 1x 3x MMQS Reverse Transkriptase 1,0 µl 3,0 µl QS RT buffer, 5x 4,0 µl 12,0 µlRT Primermix 1,0 µl 3,0 µl
Gesamtvolumen 6,0 µl 18,0 µl
Nun werden kontaminierende Fragmente genomischer DNA in der RNA-Probe durch Inkubation mit einem DNase (gDNA WipeOut buffer) weitgehend entfernt. Dazu werden zu jeder RNA-Probe 2 µl gDNA WipeOut Buffer zugegeben. Die Ansätze werden durch mehrmaliges auf- und abpipettieren gemischt und bei 42°C inkubiert (Verdau der genomischen DNA durch die DNase).Nach 5 min werden die Proben auf Eis transferiert und 6 µl des vorbereiteten RT-Mastermixes zugegeben.Bei der RT- Kontrolle werden 4,0 µl RT buffer, 1.0 µl RT Primermix und 1,0 µl RNa-se-freies Wasser statt der Reversen Transkriptase zugegeben und durch auf- und abpipettieren gemischt. Die Proben werden für 30 min bei 42°C inkubiert (Reverse Transkription) und danach wird das Enzym durch erhitzen auf 95°C für 3 min inakti-viert. Alle Inkubtionsschritte werden in einem MasterCycler Gradient Gerät mit dem

19
Programm „SDRTTOT“ durchgeführt: CNTRL TUBE, LID=105°C, WAIT, AUTO
1 T = 42°C HOLD 2 T = 42°C 0:05:00 DNase-Verdau genomischer DNA3 T = 42°C HOLD 4 T = 42°C 0:30:00 Reverse Transkription5 T = 95°C 0:03:00 Inaktivieren der reversen Transkriptase6 T = 4°C Hold 7 End Ende des Programms
Die Proben enthalten nun die cDNA und können nach 1:5-Verdünnung mit Wasser für die quantitative PCR verwendet werden.
Quantitative PCR am LightCycler 480 SystemNun werden die mRNA-Expressionslevel relativ zum Referenzgen Hprt bestimmt. Dafür werden drei qPCR-Mastermixe mit den Primern für Tbp, Tbx21-WT und Tbx21-KO (NeoR) für jeweils acht Proben (Proben 1+2 (Triplikate!), RT- und NTC) angesetzt:
1x 9x MMWasser 3,8 µl 34,2 µl 2x SybrGreen Mastermix 10,0 µl 90,0 µlFwd-Primer (10 pmol/µl) 0,6 µl 5,4 µlRev-Primer (10 pmol/µl) 0,6 µl 5,4 µl
Gesamtvolumen 15,0 µl 135,0 µl
Von jedem Mastermix werden 8 Wells auf der 96-Well-Platte mit je 15 µl befüllt (siehe Pipettierschema). Danach werden je 5 µl 1:5 verdünnte cDNA-Probe, RT- Kontrolle oder Negativkontrolle zugegeben. Haben alle Studenten ihre Proben pipet-tiert wird die Platte abzentrifugiert und in das LightCycler-Gerät gestellt.
Pipettierschema für LightCycler 96-Well qPCR-Platte:
1 2 3 4 5 6 7 8 9 10 11 12
A #1 #1 #2 #2 #3 #3 RT- NTC #1 #1 #2 #2 Student1
B #3 #3 RT- NTC #1 #1 #2 #2 #3 #3 RT- NTC
C #1 #1 #2 #2 #3 #3 RT- NTC #1 #1 #2 #2 Student2
D #3 #3 RT- NTC #1 #1 #2 #2 #3 #3 RT- NTC
E #1 #1 #2 #2 #3 #3 RT- NTC #1 #1 #2 #2 Student3
F #3 #3 RT- NTC #1 #1 #2 #2 #3 #3 RT- NTC
G #1 #1 #2 #2 #3 #3 RT- NTC #1 #1 #2 #2 Student4
H #3 #3 RT- NTC #1 #1 #2 #2 #3 #3 RT- NTC
Tpb Tbx21-WT (NeoR)

20
Nun wird die Quantifizierung mit folgendem Programm gestartet:
Denat. & Aktivierung 1x 95°C 10:00 Amplifikation 45x 95°C 00:10 60-62°C 00:15 touch down: delay 5, 0,5°C/cycle 72°C 00:10 78°C 00:01 Messung Schmelzkurve 1x 95°C 00:05 60°C 01:00 99°C - kont. Messung Kühlung 1x 40°C 00:10
Nach Ablauf des Programms (ca. 1,5h) stehen die Daten zur Auswertung zur Verfü-gung und werden von den Studenten unter Anleitung der Betreuer analysiert.
ErgebnisprotokollVersuchsteil A: Geschlechts- und Genotypbestimmung mittels PCR
hier das Gelbild einkleben!
Auswertung des Bandenmusters
Maus A Maus B Maus C
Wildtyp-Bande: 148 bpBande bei Verlust von Tbx21
189 bp
Bande für Y-Chromosom
226 bp
Bande für X-Chromosom
255 bp
Ergebnis:GeschlechtGenotyp Tbx21
Bande Größe Bande vorhanden? (x)

21
Versuchsteil B: Genotypbestimmung mittels quantitativer PCR
Zusammenfassung der Ergebnisse
Probe c [ng/µl] A260/A280 V (1 µg RNA)
Maus 1
Maus 2
Maus 3
Probe relative Expression Tbx21
relative Expression Neo
ermittelter Genotyp
Maus 1
Maus 2
RNA-Konzentrationsbestimmung mittels Spektrophotometer
Quantitative PCR am LightCycler 480 System

22
Tierexperimenteller Teil
Betreuer:Gerrit John , Melanie Königshoff, Johannes Schwarz, Tobias Stöger und Ali Önder Yildirim
Versuchsteil A: Tracheotomie für Lungenfunktion und weitere Dissektion der Maus für Organentnahme
ÜbersichtUm die Immunpathogenese von Erkrankungen weitergehend zu erforschen, sind Tierversuche bzw. Tiermodelle oftmals erforderlich. Tiermodelle liefern relevante, aussagekräftige Daten. Insbesondere die Maus wird als Spezies für unterschiedliche Modelle akuter oder chronischer Lungenerkrankungen verwendet.Darüber hinaus ist die Maus auch aus immunologischer Sicht, insbesondere auf-grund des vielfältigen Angebots an analytischen Werkzeugen (z.B. Antikörper), als sehr gut geeignetes Tiermodell akzeptiert. Der Vorteil der Maus als Modelltier liegt, abgesehen von Größe und Haltungseigenschaften, insbesondere in der Verfügbar-keit einer sehr großen Anzahl an Inzucht-Stämmen mit genetisch exakt definiertem Hintergrund.
Lungenfunktionstests werden insbesondere für Fragestellungen zur Entdeckung und Quantifizierung einer Atemwegs- und Lungenerkrankung sowie zur Beurteilung der Wirksamkeit einer Therapie benötigt. Die invasive Lungenfunktionsmessung am narkotisierten und intubierten Tier stellt derzeit den Goldstandard der Lungenfunk-tionsanalyse dar.Der Vorteil dieses Systems besteht darin, dass Aussagen zur Lungenmechanik, wie Lungendehnbarkeit (dynamische Compliance) und Strömungswiderstand (Resis-tance), getroffen werden können. Diese Parameter werden besonders bei struk-turellen Veränderungen der Atemwege und beim Emphysem beeinflusst. Somit erlaubt die Methode, die vor allem am Ende eines Experiments eingesetzt wird, Veränderungen in Bereichen der Atemwege und des Parenchyms differenziert zu analysieren.
Für die Durchführung der Lungenfunktionsuntersuchungen werden von der Firma Buxco u.a. die zwei Systeme FinePointRC System und Forced Maneuvers System hergestellt. Der Vorteil des FinePointRC Systems ist, dass die Untersuchungen so-wohl bei intubierten als auch bei tracheotomierten Mäusen durchgeführt werden können. Hierbei können bei intubierten Mäusen Lungenfunktionsuntersuchungen im zeitlichen Verlauf an denselben Tieren erfolgen. Das Forced Maneuvers System ist das weltweit einzige Gerät, das die Bestimmung der Einsekundenkapazität (FEV) an Mäusen ermöglicht. Diese Messwerte haben eine große Bedeutung bei der Diagnose chronischer Lungenerkrankungen (z.B. COPD).
DurchführungJeder Student führt eine Tracheotomie sowie nach der Lungenfunktionsmessung die weitere Dissektion der Maus für die Organentnahme nach folgendem Protokoll durch.

23
VorbereitungPBS 1x;Protease-Inhibitor (Complete Mini, Roche, 11836170001, 1 Tablette in 10ml PBS lösen);Paraformaldehyd (Sigma 441244) – 10% PFA herstellen und mit PBS auf 6%verdünnen (auf Eis!);Blut-Kapillare (Minicap, Hirschmann, 9000205)EDTA Röhrchen (Sarstedt)1ml Spritzen mit 500 µl Protease-Inhibitor (auf Eis!)10ml Spritze und 50ml Falcon mit PBS 1xSchere, Pinzetten, Faden, Tubus, Tupfer und Kanülen zum Fixieren bereit legenZum Fixieren für die Histologie 5ml Spritze mit PFA 6% befüllen2x 1,5ml Eppendorf Tubes für BAL- und RNA-Proben beschriften 15ml Falcon-Tubes für Lungen beschriften und mit ca. 4ml PFA 6% befüllen (auf Eis!)Narkose (Ketamin/Rompun) vorbereiten
Narkotisieren der MausMaus wiegen und je nach Gewicht Narkose aufziehen (1ml Spritze, kurze Kanüle) und intraperitoneal (i.p.) spritzen. (Die Tiefe der Narkose wird durch Kneifen in die Hinterpfote überprüft.)
TracheotomieMaus an den Gliedmaßen fixieren, Kopf überstrecken und an der Schnauze fixieren, Halsbereich mit Ethanol besprühen;Von der Brust Richtung Schnauze das Fell öffnen;Mit zwei Pinzetten Gewebe vor der Trachea vorsichtig (Blutgefäße!) nach links und rechts wegpräparieren;Muskeln entlang der Trachea mit Pinzetten nach oben und unten wegpräparieren; Gebogene Pinzette vorsichtig unter der Trachea hindurchführen und Faden durch-ziehen; Nahe am Kehlkopf mit einer spitzen Schere ein kleines Loch in die Trachea schnei-den und vorsichtig entlang der Trachea vergrößern;Tubus für Lungenfunktion durch leichtes Drehen einführen (bis zum zweiten Ring) und mit Doppelknoten fixieren;è Tier bis zur Lungenfunktionsmessung im Minivent beatmen.
LungenfunktionsmessungUnter Anleitung eines Betreuers werden an den Systemen FinePointRC System und Forced Maneuvers System Lungenfunktionsparameter wie Lungendehnbarkeit (dy-namische Compliance) und Strömungswiderstand (Resistance) sowie die Einsekun-denkapazität (FEV) am tracheotomierten Tier bestimmt. Weitere ausführliche Informationen zur Lungenfunktionsmessung sind in folgender Publikation zu erhalten: Vanoirbeek, J.A., et al., Noninvasive and invasive pulmona-ry function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol, 2010. 42(1): p. 96-104.
Nach Lungenfunktionsmessung erfolgt die weitere Dissektion der Maus für die Organentnahme.
Retrobulbäre BlutentnahmeMaus im Genick festhalten und Fell zurückziehen, bis die Augen hervortreten;

24
Kleine Kapillare in den vorderen Augenwinkel einführen und vorsichtig drehen, bis Blut fließt;Kopf der Maus mit Kapillare nach unten halten und Blut im EDTA-Röhrchen auffan-gen;Beachte: Blutentnahme alternativ an Oberschenkelarterie oder Arterie im Bauch-raum.è Hämatologie (Advia120) nach Blutentnahme
OrganentnahmeMaus fixieren, Halsbereich abdecken und Körper mit Ethanol besprühen;Fell vom Hals an nach unten hin aufschneiden und zu den Seiten hin aufziehen;Bauchhöhle vom Brustkorb nach unten aufschneiden und entlang des Rippenbogens nach links und rechts einschneiden;Nierenarterie durchtrennen und Maus ausbluten lassen;Benötigte Organe (z.B. Milz und Lymphknoten) entnehmen;Zwerchfell anschneiden, damit die Lunge kollabiert, und anschließend vollständig mit Pinzetten entfernen;Brustkorb entlang des Sternums aufschneiden (stumpfe Schere);Rippen links und rechts fixieren;Trachea vollständig frei legen, Thymus entfernen;Beachte: Linke Lunge ist für die Histologie bestimmt, nicht berühren, anheben o.ä.!
Bronchoalveoläre LavageSpritze an Tubus befestigen und PBS/Protease-Inhibitor langsam in die Lunge hin-eindrücken;Spritze wieder langsam aufziehen, eventuell Tubus ein wenig drehen, falls keine Flüssigkeit kommt (insgesamt 3x mit je 500µl spülen);BAL in 1,5ml Tube geben und sofort auf Eis stellen;Beachte: Die Spritze nicht mehrmals aufziehen und wieder reindrücken, da die Lun-ge dadurch beschädigt wird und nicht mehr für die Histologie zu gebrauchen ist!
Lunge für RNA-IsolationFaden unter dem rechten Lungenflügel der Maus durchziehen, linken Flügel nicht berühren!Lungenflügel mit Doppelknoten abbinden;Rechte Lunge in kleinen Stücken abschneiden und sofort in flüssigen Stickstoff ge-ben;Lunge aus dem Stickstoff nehmen und in 1,5ml Tube überführen (ebenfalls in flüssi-gen Stickstoff geben).
Lunge für Histologie fixieren6% PFA mit Spritze über Tubus in die linke Lunge geben;Unterhalb des Tubus einen Faden unter der Trachea durchziehen und abbinden;Spritze und Tubus herausziehen;Mit der Pinzette die Trachea greifen und Lunge entlang der Wirbelsäule heraustren-nen;Lunge in vorbereitetes 15ml-Falcon-Tube mit PFA geben (Fäden nach außen), Tube umdrehen und auf Eis lagern;Beachte: Die Lungen ca. 24h in 6% PFA fixieren, anschließend weiter verarbeiten oder PFA abgießen und durch 4% Formalin ersetzen!

25
Versuchsteil B: Aufbereitung von BAL, Blut und Organen
ÜbersichtDie Bestimmung inflammatorischer und immunologischer Parameter erfolgt auf ver-schiedene Weise (Genexpression, Immunhistochemie, ELISA, Western blot etc.) in den Kompartimenten BAL, Blut und Organen wie Lunge, Milz und Lymphknoten. Die bronchoalveoläre Lavage wird in BAL-Zellen und BAL-Flüssigkeit aufgetrennt. Mittels Cytospin und Färbung erfolgt eine differentielle Entzündungszell-Analyse in der BAL. Die BAL-Flüssigkeit kann mittles ELISA auf Entzündungsmediatoren wie Cytokine und Chemokine hin untersucht werden. Die linke Lunge wird für immunhistochemische sowie quantitativ-morphologische Analysen und die rechte Lunge für Untersuchungen der Genexpression verwen-det. Nach Entwässerung und Einbettung der in PFA fixierten linken Lunge werden immunhistochemische Färbungen (z.B. von Entzündungszellen, Strukturproteinen etc.) an Paraffinschnitten durchgeführt. Präzise und umfassende Aussagen über strukturelle Veränderungen von Zellen, Geweben und Organen können mittels quantitativ-morphologischer Untersuchungen zu Parametern wie Alveolarweite und Gasaustauschoberfläche der Lunge an einem computer-gestützten Forschungsmik-roskop (newCAST-GRID-System, Visiopharm) getroffen werden.
Genexpressionsanalysen werden nach Homogenisierung des Gewebes, RNA-Iso-lation sowie cDNA-Synthese (s. Molekularbiologischer Teil des Praktikums) mittels quantitativer real-time PCR (qPCR) durchgeführt.
Mikroskopische Schnittpräparate besitzen nur einen geringen Eigenkontrast. Um Strukturen besser darzustellen, können eine Vielzahl von Farbstoffen verwendet werden. Häufig verwendete histologische Färbungen sind z.B. HE (Hämatoxylin-Eosin), Azan (Azokarmin/Anilinblau), PAS (Perjodsäure/Schiff’sches Reagenz) oder Toluidinblau.
Viele Standardmethoden verwenden eine Kombination aus einem sauren und einem basischen Farbstoff. Die anionischen Gruppen im Gewebe (z.B. DNA, RNA, sulfa-tierte Glykosaminoglykane) reagieren mit dem basischen Farbstoff (Basophilie), während die kationischen Gruppen (div. Proteine) an den sauren Farbstoff binden (Azidophilie). Die Resultate einiger wichtiger histologischer Färbungen sind in der Tabelle zusammengefasst.
Vor der Färbung muss das Einbettmedium wieder aus den Schnitten entfernt wer-den, da sonst die Farbstoffe nicht binden können. Dies geschieht mit einer abstei-genden Alkoholreihe bis hin zu Wasser. Eine solche Alkoholreihe ist bei in Kunstharz eingebetteten Präparaten nicht notwendig, da der Kunststoff die Färbung nicht beeinflusst. Nach der Färbung werden die Schnitte wiederum entwässert, in dem sie über eine aufsteigende Alkoholreihe in Xylol gebracht werden.
Tab. 1
Färbung FarbstoffZytoplasma Kollagen Elastin
Nukleus(DNA) andere
Hämatoxylin/ HämalaunEosin (HE) Eosin
rot rot rot blau

26
DurchführungCytospin und Färbung von Zellen aus bronchoalveolärer Lavage (BAL)BAL in 1,5 ml Tubes auf Eis.Zentrifugation bei 400 g (ca. 1.200 rpm) und 4 °C für 20 min.BAL wieder auf Eis stellen.Überstände aufteilen auf mind. 2 Tubes mit annähernd gleichen Volumina entspre-chend der Ausbeute, Ausbeute notieren, Tubes erst in Flüssigstickstoff, dann bei -80 °C einfrieren.Zellpellet: Farbe bestimmen und in 500 µl RPMI (ohne Zusätze) resuspendieren und auf Eis stellen.Zellzählung: Zellsuspension (Vortexen nicht vergessen) 1:2 mit Trypanblau in Mik-rotiterplatte verdünnen (z.B. 20 µl + 20 µl), gut mit Pipette mischen und 10 µl der Suspension auf Hemocytometer auftragen.Zellen zählen (4 Großquadrate) im Mikroskop bei 10x Vergrößerung (bei zu hohen Zellzahlen andere Verdünnung wählen); Zellzahl: Mittelwert der 4 Großquadrate x 104/ml, für Gesamtzellzahl Verdünnung und Gesamtvolumen (hier: 500 µl) berück-sichtigen.Für Cytospin: 2 Objektträger (Thermo Scientific Menzel-Gläser, nicht Superfrost) je Probe mit Bleistift beschriften und in Metallklemme mit Filter (Thermo Shandon Filter Cards, in Hälfte geschnitten) und Trichter einspannen und in Cyto-Zentrifuge stellen; max. 30.000 Zellen pro Probe berechnen und entsprechende Volumina auf-tragen (Vortexen nicht vergessen): Mindestvolumen 100 µl, Maximalvolumen 500 µl je Trichter.Zentrifugation: set time 6 min – Enter; set speed 40 x 10 rpm – Enter; Low einstel-len für langsames Abbremsen.Nach Zentrifugation: Objektträger vorsichtig aus der Klemme lösen, ohne die Zellen zu verwischen; Objektträger in Sammelmappe an der Luft trocknen; Cytospin-Trich-ter in Aqua dest. waschen und zum Trocknen aufstellen.
Färbung: Handschuhe anziehen, giftige Farbstoffe! Unter dem Abzug arbeiten und Arbeitsplatz mit Haushaltstüchern mehrlagig abdecken.Färbelösungen in den dafür vorgesehenen Färbeabfall-Kanister entsorgen.Bei H2O immer Leitungswasser nehmen, kein Aqua dest.!Durchführung10 min in May-Grünwald (konz.)2 min wässern in H2O15 min Giemsa (1:20 verdünnen in H2O)2 min wässern in H2OTrocknen lassenEindeckeln (Entellan, Deckgläser 25x25 mm)
Zählung: 200 Zellen pro Objektträger randomisiert in verschiedenen Sichtfeldern auszählen.
Histologie – HE-Färbung
1. Fixierung und Einbettung von GewebeLungengewebe wird in 6% Paraformaldehyd fixiert. Fixierlösungen für Paraffin Einbettungen (Unter dem Abzug arbeiten!):

27
6 %ige Paraformaldehyd Stammlösung: 6 g Paraformaldehyd in 100 ml 1x PBS, erhitzen (ca. 70 °C), mit NaOH klären, abkühlen, filtrieren
Zur Vorbereitung der Einbettung in Paraffin wird die Lunge in Agar-Agar eingelegt. Hierzu werden zunächst die beiden Lungenflügel mit der lateralen Seite nach unten gerichtet in jeweils ein Becherglas mit 2%-igem Agar-Agar gelegt und vollständig damit bedeckt. Nach einer Härtungszeit von ca. 60-90 min bei 4°C im Kühlschrank wird der Agar-Agar Block mit den eingelegten Lungen komplett aus dem Becherglas entnommen und in einer Schneidekammer in ca. 2mm dicke Scheiben geschnitten werden. Im Anschluss werden die Lungenscheiben von den Agar-Agar Resten be-freit und die Scheiben in Einbettungskassetten gelegt, welche bis zur Einbettung in 10% Formalin aufbewahrt.
Mithilfe dieser Technik ist gewährleistet, dass die Atemwege völlig zufällig auf die Objektträger verteilt sind („systematic uniform random sampling“ (SURS)). Prinzip des SURS ist es, die Probenauswahl aus einer Menge oder einem Gewebe an einem willkürlichen Punkt zu beginnen und von dort entweder in einem festgelegten Mus-ter oder zufällig weitere Proben auszuwählen. Von den in Paraffin eingelegten Prä-paraten wurden dann mittels Mikrotom Dünnschnitte von 3-5 µm Dicke hergestellt.
2. Entwässerung und Einbetten in Paraffin
Da Paraffin nicht wasserlöslich ist, muss das Wasser aus dem Gewebe zunächst durch Alkohol und schließlich der Alkohol durch Xylol ersetzt werden. Dies geschieht schrittweise durch eine aufsteigende Alkoholreihe (verdünnt aus 100 %igem unver-gällten Ethanol). In der Zwischenzeit das Paraffin schmelzen bei ca. 60 °C.
1. 10 min in A.dest spülen 2. 15-30 min 40 % Ethanol 3. 15-30 min 70 % Ethanol 4. 15-30 min 96 % Ethanol 5. 15-30 min Isopropanol 6. 2 x 15 min Xylol 7. Infiltration des Gewebes in Schnappdeckelgläschen mit flüssigen Paraffin über Nacht bei ca. 60 °C im Bakterienbrutschrank. Paraffin + Gewebe in Formen (z.B. Eiswürfelbehälter) gießen und bei Raumtemperatur aushärten lassen. Achten Sie auf die Lage des Gewebes. Block eventuell mit heißem Spatel auf einen Träger bringen.
3. SchneidenParaffinblöcke bei -20 °C über Nacht lagern (verbessert das Schneiden).Paraffin wird am Rotationsmikrotom geschnitten (eventuell noch mal zwischendurch auf Kühlakku oder auf Eis legen). Der Paraffinblock wird so getrimmt, dass das Gewebestück etwa mittig liegt und die gegenüberliegenden Kanten parallel verlaufen. Dadurch wird gewährleistet, dass Schnittbänder entstehen. Markieren Sie sich den rechten oberen Rand. Folgende Aufsicht sollte entstehen:

28
Ein Trimmen der Kunstoffblöcke ist nicht erforderlich, es entstehen dadurch aber keine Schnittbänder. Die Schnitte werden in ein mind. 40 °C warmes Streckbad überführt und dann auf die Objektträger gezogen. Achten Sie auf die Reihenfolge! Die Schnitte werden vor der Färbung ü.N. bei 37 °C oder für 20 min bei 45-60 °C getrocknet.
Schnittdicke Paraffin: 2-5 µm (im Praktikum: 5-8µm)
4. FärbenDa Paraffin nicht wasserlöslich ist, muss es zunächst durch eine absteigende Alko-holreihe entfernt werden (2x 10 min Xylol, 1x 5 min Isopropanol, 96 %, 90 %, 70 %, 40 % unvergällter Ethanol, dest. Wasser).
HE- Färbung:Zellkerne werden blau gefärbt, Zytoplasma rosa, kollagene Bindegewebsfasern rot, Muskelgewebe rot, Knorpel blauSaures Hämalaun nach Mayer: 1 g Hämatoxylin + 0,2 g Natriumjodat + 50 g Kali-alaun in 1 l dest. Wasser lösen; 50 g Chloralhydrat + 1 g kristalline Zitronensäure. è Gebrauchsfertige Lösung liegt vor (vor Gebrauch filtrieren!)
1. Absteigende Alkoholreihe, dest. Wasser (entfällt bei Kunstharz) 2. 2-5 min Hämalaun nach Mayer (je nach Ergebnis auch länger) 3. 10 min Spülen in fließenden Leitungswasser 4. kurz in dest. Wasser 5. 5 min 1 % Eosin 6. kurz in dest. Wasser ausspülen 7. Aufsteigende Alkoholreihe 70 %, 90 %, 96 % Ethanol, Isopropanol, Xylol (je 3 5 min) Eindeckeln in Eukitt

29
Ergebnisprotokoll
Tierversuchsprotokoll Stand:Beginn:
Versuchsnr: Ende:Verantwortlich:
Versuchsbezeichnung: TV-Nummer:
Untersuchungsdatum:
Maus-Stamm von: Charles River/ ILBD-ZucAlter: Wochen
Maus ID Nr.:________________ Ohr-Nr Käfig-Nr Geburtsdatum:
Gewicht:____________ [g] Narkose:_________________µl Ketamin Rompun
Applikation von Substanz: Dosis: Dauer:
Kontrolle: Cage Control
Intubation / Tracheotomie: (Name)
LUFU- Flexi-Vent: Unters.-Nr.:
LUFU- Buxco1 (Name): Unters.-Nr.:
LUFU- Buxco2 (Name): Unters.-Nr.:
Blutentnahme: (Name)
Methode: a.) - Punktion des Retrobulären Venenplexus -S-Monovette für Serumgewinnungb.) - Schnitt Oberschenkelvene -Eppi für Serumgewinnung
Serumgewinnung: abpipettieren in Anzahl Aliquotsà µl einfrieren: -20^C / -80°C(für ELISA IgE, IgG1, IgG2c)
BAL : (Name)
Spülung: µl X (Anzahl) Recovery: µl
Zentrifugation 20 Min 400g Überstand für ELISA( IL's): Anzahl Aliquotsà µl einfrieren: -20^C / -80°C
Zellpellet in [ml] RPMI 1640 + 5% FKS-Medium / PBS aufgenommen
Pellet-Farbe Verdünnung 1:5 mit Trypanblau
Zählkammer(A) BAL vitale Zellen tote (blaue) Zellen 1. Quadrat2. Quadrat3. Quadrat4. QuadratMittelwert
Zellzahl / 1 ml Medium « (Zellzahl x Verdünnungsfaktor x 104 ;nur vitale Zellen !)
Gesamt-Zellzahl (absolut) « (Zellzahl pro ml PBS x tatsächliches Flkt.-Volumen)Zellzahl / ml BAL « (Gesamt-Zellzahl / Gesamtvolumen BAL-Flkt.)Zellzahl / ml BAL / 100g KGW « (Gesamt-Zellzahl / Gesamtvolumen BAL-Flkt./ KGW [g] x 100)Tote Zellen: [%]
Zellen für Cytospins 50 000 / 6 Min bei 400 U / Min zentrifugieren
50000 x 1000 Mio. = ___________µl x______=______________µl Zellen + _____________µl RPMI /PBS
Cytospins: gefärbt Superfrost -80°C
Sektion: (Name)
Organe: 1/2 Lunge in Stickstoff / -80 °C 1/2 Lunge in PFA 6%ig für Histo
Milz für: Lymphknoten für:
sonstiges:

30
Broncho-Alveoläre lavage (BAL) - Zytologie Untersucher______________________
Färbung: 10 Min .May-Grünwald (Merck 1424) 2 Min. wässern (kein A.dest. !)15 Min. Giemsa 1:20 verdünnt mit Wasser (Merck 9204) 2 Min wässern (s.o.)trocknen lassen, eindeckeln
Allgemeiner Eindruck Qualität des Zytospins:________[%]zerstörte ZellenZelldichte: (zu gering / einzelne / ausreichend / viele / sehr viele / überladen)Qualität der Färbung: (gut / ausreichend / schlecht / unbrauchbar)Auswertbar: (gut / ausreichend / schlecht / nicht )
Auswertung der Zytospins: pro Zytospins im oberen & unteren Drittel je 200 Zellen bestimmen
Makrophagen - normal - mehrkernigLymphozytenNeutrophileEosinophileandere(benennen)
Erythrozyten
Anmerkungen (Kontamination, Einschlüsse,...)
Makrophagen mit / ohne / einzelnen / viele / sehr viele / massenhaft / Partikel Sonstiges
1. Zytospin 2. Zytospin[%] [n/200] [%] [n/200]Zellart [n/200] [%] [n/200]
keine / einzelne / viele/ massenhaft keine /einzelne / viele / massenhaft
[%]

Zusammenfassung der Ergebnisse
31



















