
Spektroskopische Untersuchung des H-Clusters von ... · by analysing the structure of their...
94
Spektroskopische Untersuchung des H-Clusters von photosynthetischen [FeFe]-Hydrogenasen am Beispiel von HydA1 aus Chlamydomonas reinhardtii Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Biologie und Biotechnologie an der Internationalen Graduiertenschule Biowissenschaften der Ruhr-Universität Bochum angefertigt im Institut für Biochemie der Pflanzen Abteilung Photobiotechnologie vorgelegt von Christina Kamp aus Viersen Bochum Oktober 2008
-
Upload
duongxuyen -
Category
Documents
-
view
216 -
download
0
Transcript of Spektroskopische Untersuchung des H-Clusters von ... · by analysing the structure of their...

Spektroskopische Untersuchung des H-Clusters von
photosynthetischen [FeFe]-Hydrogenasen am Beispiel von
HydA1 aus Chlamydomonas reinhardtii
Dissertation zur Erlangung des Grades
eines Doktors der Naturwissenschaften
der Fakultät für Biologie und Biotechnologie
an der Internationalen Graduiertenschule Biowissenschaften
der Ruhr-Universität Bochum
angefertigt im
Institut für Biochemie der Pflanzen
Abteilung Photobiotechnologie
vorgelegt von
Christina Kamp
aus
Viersen
Bochum
Oktober 2008

Spectroscopic characterization of the H-Cluster of the
photosynthetic [FeFe]-Hydrogenase HydA1 of
Chlamydomonas reinhardtii
Dissertation to obtain the degree
Doctor Rerum Naturalium (Dr. rer. nat.)
at the Faculty of Biology and Biotechnology
Ruhr-University Bochum
International Graduate School of Bioscience
Ruhr-University Bochum
Department of Biochemistry of Plants
submitted by
Christina Kamp
from
Viersen, Germany
Bochum
October 2008

Teile der Arbeit wurden bereits veröffentlicht:
Kamp, C., Silakov, A., Winkler, M., Reijerse, E., Lubitz, W. und Happe, T. (2008).
“Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae.”
Biochim Biophys Acta 1777: 410-416.

Inhaltsverzeichnis
Inhaltsverzeichnis Zusammenfassung 1
Summary 2
1. EINLEITUNG 3
1.1. Hydrogenasen als Schlüsselenzyme für die H2-Produktion 3
1.2. Die Struktur von [FeFe]-Hydrogenasen 4
1.3. [FeFe]-Hydrogenasen in Grünalgen 6
1.4. in vivo-Wasserstoffproduktion von Grünalgen 9
1.5. Zielsetzung 10
2. MATERIAL und METHODEN 12 2.1. Organismen 12
2.2. Proteine und Molekulargewichtsmarker 12
2.3. Chemikalien 12
2.4. Gase 13
2.5. Lösungen, Puffer und Medien 13
2.5.1. Tris-Acetat-Phosphat (TAP)-Medium 13
2.5.2. TAP-S-Medium 14
2.5.3. Selbstentschwefelndes Medium 14
2.5.4. 57Fe-Medium 14
2.5.5. TAP-Platten 14
2.6. Sterilisation von Flüssigkeiten und Glasgeräten 15
2.7. Kultivierung der Algen 15
2.8. Bestimmung der Chlorophyllkonzentration 15
2.9. Induktion der Hydrogenaseexpression 16
2.9.1. Anaerobe Induktion 16
2.9.2. Schwefelmangel-Induktion 16
2.10. Arbeiten am Anaerobzelt 17
2.11. Isolierung von [FeFe]-Hydrogenasen 17
2.12. Konzentrierung von Proteinlösungen 19
2.13. Dialyse von Proteinlösungen 19
2.14. Gaschromatographische Bestimmung der in vitro-Hydrogenaseaktivität 19
2.15. Konzentrationsbestimmung von Proteinlösungen 20

Inhaltsverzeichnis
2.16. SDS-Polyacrylamid-Gelelektrophorese 20
2.17. Coomassie-Färbung von SDS-Polyacrylamidgelen 21
2.18. Western Blot und Immunodetektion von Proteinen 21
2.19. Sequenzierung von Proteinen 22
2.20. Biochemische Charakterisierung von [FeFe]-Hydrogenasen 23
2.20.1. Bestimmung des Temperaturoptimums 23
2.20.2. Untersuchung der pH-Toleranz 24
2.20.3. Untersuchung der Salzabhängigkeit 24
2.20.4. Untersuchung der Sauerstoffsensitivität 25
2.20.5. Inhibierung von Hydrogenasen durch Kohlenmonoxid 25
2.21. Biophysikalische Untersuchungen von [FeFe]-Hydrogenasen 26
2.21.1. EPR-Spektroskopie 26
2.21.2. FTIR-Spektroskopie 27
3. ERGEBNISSE 29 3.1. Etablierung eines neuen Reinigungsprotokolls für HydA1 aus C. reinhardtii 29
3.1.1. Induktion der Hydrogenase-Expression 29
3.1.1.1. Anaerobe Induktion 29
3.1.1.2. Schwefelmangel-Induktion 29
3.1.1.3. Optimierung der Schwefelmangel-Induktion 31
3.1.2. Reinigung von HydA1 aus C. reinhardtii 32
3.1.3. Nutzung des entwickelten Reinigungsprotokolls für die Isolierung weiterer
[FeFe]-Hydrogenasen aus C. moewusii und C. submarinum 34
3.1.4. Massenspektrometrische Sequenzanalyse zur Identifizierung der isolierten
HydA-Proteine 36
3.2. Biochemische Charakterisierung der isolierten Hydrogenasen 37
3.2.1. Bestimmung des Temperaturoptimums 37
3.2.2. Vergleichende Analyse zur pH-Toleranz der HydA-Proteine 37
3.2.3. Einfluss der Ionenstärke auf die Enzymaktivität 39
3.2.4. Untersuchung der Sauerstoffsensitivität 39
3.2.5. Inhibierung der Hydrogenasen durch Kohlenmonoxid 40
3.3. Spektroskopische Untersuchung des H-Clusters 43
3.3.1. EPR-Spektroskopie 43
3.3.1.1. Vergleichende Untersuchung der HydA-Proteine 43
3.3.1.2. Der Hox-Zustand von HydA1 aus C. reinhardtii 45

Inhaltsverzeichnis
3.3.1.3. ENDOR- und HYSCORE-Analyse des Hox-CO-Zustands von HydA1 aus
C. reinhardtii 46
3.3.2. FTIR-Spektroskopie 51
3.3.2.1. Photodissoziation des Hox-CO-Zustands von HydA1 aus C. reinhardtii bei
kryogenen Temperaturen 51
3.3.2.2. Isotopenmarkierung der CO-Liganden des H-Clusters mit 13C 52
3.3.2.3. Untersuchung der reduzierten Zustände des H-Clusters 56
3.3.2.4. Spektroelektrochemie an HydA1 aus C. reinhardtii 59
4. DISKUSSION 63 4.1. Expression und Reinigung der [FeFe]-Hydrogenasen aus C. reinhardtii,
C. moewusii und C. submarinum 63
4.1.1. Verbesserte Expression der HydA-Proteine unter Schwefelmangel 63
4.1.2. Das neue Reinigungssystem für HydA1 64
4.2. Die hohe Toleranz der HydA-Proteine gegenüber biochemischen Einflüssen 64
4.3. Untersuchung der O2-und CO-Sensitivität: Die hohe O2-Toleranz von HydA
aus C. submarinum 65
4.4. Die Struktur des H-Clusters von Grünalgen-[FeFe]-Hydrogenasen 66
4.4.1. Der Hox-CO-Zustand 67
4.4.2. Die Photodissoziation von Hox-CO und der Zwischenzustand LI2 71
4.4.3. Der Hox-Zustand 71
4.4.4. Die reduzierten Zustände: Hred1 und Hred2 72
4.4.5. Der katalytische Mechanismus im H-Cluster 73
5. LITERATUR 75
Abbildungsverzeichnis 83
Tabellenverzeichnis 85
Erklärung 86
Danksagung 87
Lebenslauf 88

Zusammenfassung 1
Zusammenfassung Grünalgen-[FeFe]-Hydrogenasen sind monomere, kernkodierte Enzyme, die im Chloroplasten
lokalisiert und über ihren natürlichen Elektronendonor Ferredoxin mit der photosynthetischen
Elektronentransportkette verbunden sind. Sie katalysieren die Umsetzung von Protonen und
Elektronen zu molekularem H2 und werden nur unter Anaerobiose exprimiert. Im Gegensatz zu
bakteriellen [FeFe]-Hydrogenasen bestehen sie nur aus dem katalytisch aktiven Zentrum und
besitzen keine zusätzlichen [FeS]-Cluster. Das macht sie für strukturelle Untersuchungen und
mögliche biotechnologische Anwendungen besonders interessant.
Ziel dieser Arbeit war die Reinigung und strukturelle Charakterisierung der [FeFe]-Hydrogenase
HydA1 aus C. reinhardtii. Dazu sollte zunächst ein neues Expressions- und Reinigungssystem
für HydA1 etabliert und anschließend eine erste strukturelle Untersuchung des katalytisch
aktiven Zentrums mittels EPR- und FTIR-Spektroskopie durchgeführt werden.
Durch die Entwicklung eines neuen Expressions- und Reinigungssystems konnte die
Proteinausbeute von HydA1 im Vergleich zu früheren Arbeiten um das 40-fache gesteigert und
durchschnittlich 500 µg HydA1 mit einer spezifischen Aktivität von 741 µmol H2 min-1 mg-1
isoliert werden. Zusätzlich war es erstmals möglich, auch die Hydrogenasen HydA1 aus
C. moewusii mit einer spezifischen Aktivität von 1598 µmol H2 min-1 mg-1 Protein, doppelt so
hoch wie die von HydA1 aus C. reinhardtii, und HydA aus C. submarium in vergleichbarer
Menge zu isolieren. Eine Untersuchung der Proteine in Bezug auf ihre biochemischen
Eigenschaften ergab, dass HydA aus C. submarinum nicht nur eine höhere Toleranz gegenüber
pH-Wert und hoher Ionenstärke aufweist, sondern auch erheblich sauerstofftoleranter ist als die
Hydrogenasen aus C. reinhardtii und C. moewusii. Durch die erstmals möglichen
spektroskopischen Untersuchungen des H-Clusters von HydA1 konnte gezeigt werden, dass das
H-Cluster von Grünalgen-Hydrogenasen ähnlich aufgebaut ist wie das von bakteriellen [FeFe]-
Hydrogenasen. Unterschiede in den detektierten g-Werten und Wellenzahlen, sowie eine
unterschiedliche Reaktion auf Oxidationsmittel und Licht machen allerdings deutlich, dass die
elektronische Struktur und die Symmetrie der Liganden unterschiedlich ist. Während der Hox-
und Hox-CO-Zustand große Ähnlichkeit mit CpI und DdH in dieser Form aufweist, zeigt der
Zwischenzustand LI2 ebenso wie ein zusätzlich detektierter Hred-Zustand eine andere Geometrie.
Dennoch lassen die Ergebnisse auf einen ähnlichen katalytischen Mechanismus für beide
Enzymgruppen schließen.

Summary 2
Summary
Green algal [FeFe]-hydrogenases are nuclear encoded, monomeric enzymes localized in the
chloroplast and linked to the photosynthetic electron transport chain with ferredoxin as their
natural electron donor. They catalyze the reduction of protons and electrons to molecular
hydrogen and are only expressed under anaerobic conditions. In contrast to bacterial
[FeFe]-hydrogenases they only contain the catalytic hydrogen-activating center and no accessory
[FeS]-clusters. Therefore green algal [FeFe]-hydrogenases are especially suited for structure
analyses or biotechnological applications.
In this work the [FeFe]-hydrogenase HydA1 from C. reinhardtii was purified and characterized
by analysing the structure of their catalytic center using EPR- and FTIR-spectroscopic methods.
Newly established and efficient induction and purification protocols yielded 500 µg HydA1 with
a specific activity of 741 µmol H2 min-1 mg-1 corresponding to a 40-fold increase in protein
compared to previous protocols. Similar amounts were also purified from two so far undescribed
hydrogenases, HydA1 from C. moewusii with a specific activity of 1598 µmol H2 min-1 mg-1,
which was surprisingly twofold higher compared to HydA1 from C. reinhardtii, and HydA from
the brakish water green alga C. submarinum.
Biochemical analyses of the isolated HydA proteins indicate that HydA from C. submarinum is
more tolerant to changes in pH or ionic strength as HydA1 from C. reinhardtii and HydA1 from
C. moewusii and surprisingly more stable in the presence of oxygen.
Initial examinations of the active site of HydA1 show that the structure of the H-cluster from
green algal type [FeFe]-hydrogenases seems to be similar to the active sites of bacterial
[FeFe]-hydrogenases. However, differences in the detected g-values, wavenumbers and the
response of the enzyme to the treatment with oxidants and light show that the electronic structure
and the symmetry of the ligands is different. While the Hox and the Hox-CO state is similar to
DdH and CpI, the light-induced intermediate state LI2 and a second detected reduced state, Hred1,
is different. However, the catalytic mechanism seems to be similar in both species.

1. Einleitung 3
1. Einleitung
Molekularer Wasserstoff gilt wegen seines hohen Energiegehalts und seiner umweltfreundlichen
Verbrennung zu Wasser als potentieller Energieträger der Zukunft. Die biologische
Wasserstoffproduktion durch Grünalgen und die genaue Untersuchung der Struktur und
Funktionsweise der in diesen Organismen für die Wasserstoffproduktion zuständigen Enzyme,
der sogenannten Hydrogenasen, ist von zentraler Bedeutung für die Entwicklung zukünftiger
biologisch-basierter Wasserstofftechnologien, da gerade Grünalgen-Hydrogenasen aufgrund
ihres einfachen Aufbaus und ihrer hohen katalytischen Aktivität besonders gut für
biotechnologische Anwendungen geeignet sind (Melis und Happe 2001, Happe et al. 2002).
1.1. Hydrogenasen als Schlüsselenzyme für die H2-Produktion
Hydrogenasen wurden bisher in zahlreichen, sowohl prokaryotischen als auch eukaryotischen
Organismen gefunden. Ihre Funktion ist die Katalyse der reversiblen Redoxreaktion
2 H+ + 2 e- ↔ H2. Dabei können einerseits Protonen und Elektronen beispielsweise zur ATP-
Gewinnung freigesetzt werden, andererseits werden durch die Umsetzung zu molekularem H2
überschüssige Elektronen und Protonen in der Zelle abgebaut. Im lebenden Organismus läuft die
Reaktion meist nur in eine Richtung ab. Je nach ihrer Funktion in der Zelle, H2-Aufnahme oder
H2-Abgabe, können Hydrogenasen im Periplasma oder im Cytoplasma, in gelöster Form oder
membrangebunden vorkommen (Frey 2002, Vignais und Billoud 2007).
Die prinzipielle Einteilung der Hydrogenasen erfolgt aufgrund ihres Metallatoms im aktiven
Zentrum. [NiFe]-Hydrogenasen sind heterodimere Enzyme mit einem dinuklearen [NiFe]-
Cluster innerhalb des aktiven, H2-bindenden Zentrums und hauptsächlich in Prokaryoten zu
finden (Lubitz et al. 2007). Die zweite Gruppe beinhaltet die sogenannten [FeFe]-Hydrogenasen,
welche ein [4Fe-4S]-Cluster verbunden mit einem [2Fe]-Subcluster, das sogenannte H-Cluster
(hydrogen-activating cluster), als aktives Zentrum besitzen. [FeFe]-Hydrogenasen kommen auch
in Eukaryoten und einigen strikt anaeroben Protozoen vor (Adams 1990, Vignais und Colbeau
2004, Ghirardi et al. 2007). Auch Grünalgen-Hydrogenasen gehören zu dieser Klasse. Im
Gegensatz zu [NiFe]-Hydrogenasen sind [FeFe]-Hydrogenasen eher in die H2-Produktion als in
die H2-Oxidation involviert und daher von zentraler Bedeutung für mögliche biotechnologische
Anwendungen. Sie sind im allgemeinen sauerstoffempfindlicher und lassen sich schneller durch
Kohlenmonoxid inhibieren als [NiFe]-Hydrogenasen (Adams 1990, Vignais und Billoud 2007),

1. Einleitung 4
weisen dagegen aber eine etwa hundertfach höhere katalytische Aktivität auf (Happe und
Kaminski 2002). Kürzlich wurde eine dritte Gruppe entdeckt, welche die sogenannten
[Fe]-Hydrogenasen oder [FeS]-Cluster-freien Hydrogenasen umfasst und bisher nur in
methanogenen Archae gefunden wurden (Korbas et al. 2006, Shima et al. 2008). Prominenter
Vertreter dieser Gruppe ist die Hmd (H2-forming methylentetrahydromethanopterin
dehydrogenase), die eine Fe(CO)2-Gruppe verbunden mit einem organischen Cofaktor enthält.
1.2. Die Struktur von [FeFe]-Hydrogenasen
Im Gegensatz zu den [NiFe]-Hydrogenasen ist die Funktionsweise von [FeFe]-Hydrogenasen
bisher weniger gut verstanden. Bislang konnten die Röntgenkristallstrukturen von zwei
bakteriellen [FeFe]-Hydrogenasen aufgelöst werden, CpI aus Clostridium pasteurianum und
DdH aus Desulfovibrio desulfuricans (ATCC 7757) (Abb.1.1.).
Die periplasmatische, heterodimere [FeFe]-Hydrogenase DdH aus D. desulfuricans hat ein
Molekulargewicht von insgesamt 53 kDa und besteht aus dem aktiven Zentrum (H-Cluster) und
zwei weiteren klassischen [4Fe-4S]-Clustern, den sogenannten Ferredoxin-ähnlichen Clustern
oder F-Clustern. Die physiologische Rolle von DdH ist die Oxidation von H2 (Nicolet et al. 1999
und 2000). Die cytoplasmatische, monomere [FeFe]-Hydrogenase CpI aus C. pasteurianum hat
ein Molekulargewicht von 61 kDa und besitzt neben dem H-Cluster vier weitere F-Cluster, zwei
klassische [4Fe-4S]-Cluster nahe am aktiven Zentrum, ein [4Fe-4S]-Cluster mit einem
Histidinliganden und ein [2Fe-2S]-Cluster (Peters et al. 1998). Sie ist ein H2-produzierendes
Enzym.
Spektroskopische Untersuchungen haben gezeigt, dass das katalytische Zentrum beider
Hydrogenasen ähnlich aufgebaut ist (Abb.1.2.). Es besteht aus einem [4Fe-4S]-Cluster, welches
über das Schwefelatom eines Cysteinrestes mit einem [2Fe]-Subcluster verbunden ist. Die
beiden Fe-Atome des [2Fe]-Subclusters sind jeweils mit einem CO- und einem CN-Liganden
assoziiert und im oxidierten Zustand durch einen CO-Brückenliganden verbunden. In der
reduzierten Form ist dieser CO-Ligand terminal an das Fe(2)-Atom gebunden. Das Fe(2)-Atom
besitzt eine freie Koordinationsstelle, die als putative Bindestelle für H2 und Kohlenmonoxid gilt.
Weiterhin sind die beiden Fe-Atome durch einen bisher unbekannten Liganden, vermutlich
Dithiomethylamin, verbunden (Pierik et al. 1998, Peters et al. 1998, Nicolet et al. 1999, Frey
2002, Lubitz et al. 2007).

1. Einleitung 5
Die F-Cluster dienen als Transport-Relais für die bei der Reaktion benötigten Elektronen.
Zusätzlich besitzen beide Enzyme einen Kanal aus hydrophoben Aminosäureresten, der von der
Proteinoberfläche ins aktive Zentrum zum Fe(2)-Atom führt und über den vermutlich
Wasserstoff aufgenommen oder abgegeben wird. Zusätzlich werden Reste im Inneren und an der
Oberfläche des Proteins diskutiert, wie beispielsweise ein Lysinrest nahe am Fe(2)-Atom, die in
den Protonentransfer involviert sind (Peters 1999, Nicolet et al. 2000, Cohen et al. 2005).
H-Cluster
F-Cluster
DdH
Abb. 1.1. Strukturmodell der [FeFe]-Hy
und CpI aus Clostridium pasteu
Die Röntgenkristallstruktur von DdH zeigt
einer kleinen Untereinheit von 11 kDa (
vorhanden. CpI ist ein Monomer von 61
beinhaltet, ist in blau dargestellt, die Dom
dritte Domäne, die ein einzelnes [4Fe-4S
Domäne, die das [2Fe-2S]-Cluster beinhal
CpI
drogenasen DdH aus Desulfovibrio desulfuricans (Nicolet et al. 1998)
rianum (Peters et al. 1998)
ein Dimer, bestehend aus einer großen Untereinheit von 42 kDa (rot) und
türkis). Neben dem H-Cluster sind noch zwei weitere [4Fe-4S]-Cluster
kDa, bestehend aus vier Domänen. Die Domäne, die das aktive Zentrum
äne mit den zwei klassischen [4Fe-4S]-Clustern ist grün eingefärbt. Eine
]-Cluster mit einem Histidinliganden enthält, ist türkis und die vierte
tet, ist violett gekennzeichnet.

1. Einleitung 6
Abb. 1.2. Aufbau des H-Clusters von [FeFe]-Hydrogenasen
Das H-Cluster besteht aus einem [4Fe-4S]-Cluster, das über das S-Atom eines Cysteinrestes mit einem
[2Fe]-Subcluster, koordiniert durch CO- und CN-Liganden, verbunden ist. Im oxidierten Zustand Hox sind die
Fe-Atome Fe(1) und Fe(2) durch eine CO-Brücke verbunden, im reduzierten Zustand Hred bindet der
CO-Brückenligand terminal an das Fe(2)-Atom. Das Fe(2)-Atom besitzt eine freie Koordinationsstelle, an der
vermutlich H2 freigesetzt oder oxidiert wird und das Enzym reversibel durch Kohlenmonoxid inhibiert werden kann.
1.3. [FeFe]-Hydrogenasen in Grünalgen
Grünalgen-Hydrogenasen katalysieren die Umsetzung von Protonen und Elektronen zu
molekularem Wasserstoff und sind mit einem Molekulargewicht von durchschnittlich 48 kDa die
bislang kleinsten entdeckten [FeFe]-Hydrogenasen. Im Gegensatz zu den unter 1.2.
beschriebenen bakteriellen [FeFe]-Hydrogenasen bestehen sie nur aus dem katalytisch aktiven
Zentrum und besitzen keine weiteren [FeS]-Cluster (Florin et al. 2001, Winkler et al. 2002 und
2004, Happe und Kaminski 2002, Forestier et al. 2003). Dies macht sie für strukturelle und
funktionelle Untersuchungen besonders interessant. Sie sind monomere, kernkodierte Enzyme,
die im Chloroplasten lokalisiert und über ihren natürlichen Elektronendonor Ferredoxin (PetF),
ebenfalls ein [FeS]-Protein, mit der photosynthetischen Elektronentransportkette verbunden sind
(Happe et al. 1994, Florin et al. 2001, Happe und Kaminski 2002). Im Gegensatz zu den schon
beschriebenen Hydrogenasen DdH und CpI ist über die Struktur von Grünalgen-Hydrogenasen
noch relativ wenig bekannt. Ein Vergleich der Aminosäuresequenzen verschiedener [FeFe]-
Hydrogenasen zeigt allerdings, dass alle Sequenzmotive, die für das H-Cluster charakteristisch
sind, auch in Grünalgen-Hydrogenasen zu finden sind (Abb.1.3.).
Alle bisher beschriebenen [FeFe]-Hydrogenase-Gene kodieren für Proteine mit einem mehr oder
weniger flexiblen N-Terminus (F-Domäne) und einem homologen, katalytisch relevanten
C-Terminus (H-Domäne). Neben den vier hoch konservierten Cysteinresten, die das [4Fe-4S]-
Cluster koordinieren und es mit dem [2Fe]-Subcluster verbinden, wurden außerdem Reste

1. Einleitung 7
identifiziert, die an der Strukturbildung des H-Clusters beteiligt und in den katalytischen
Protonentransfer involviert sind, sowie die hydrophobe Tasche bilden, in der das katalytische
Zentrum eingebettet ist (Nicolet et al. 2000).
Die Gene von Grünalgen-Hydrogenasen besitzen Exons und Introns sowie Transitpeptid-
Sequenzen unterschiedlicher Länge am N-Terminus, die notwendig sind, um das translatierte
Protein von den Ribosomen zum Chloroplasten zu transportieren (Florin et al. 2001, Forestier
et al. 2003). Ein weiterer Unterschied zu bakteriellen [FeFe]-Hydrogenasen sind zwei
spezifische Peptidinsertionen am N- und C-Terminus, deren Funktion noch unbekannt ist.
Möglicherweise spielen diese zusätzlichen Strukturschleifen eine Rolle bei der
Signaltransduktion oder der Interaktion mit Ferredoxin (Melis und Happe 2001, Ghirardi et al.
2007).
Da die [FeFe]-Hydrogenasen aus Grünalgen keine zusätzlichen F-Cluster besitzen, über die der
Elektronentransport zum aktiven Zentrum stattfinden kann, wird vermutet, dass eine direkte
Wechselwirkung zwischen Ferredoxin und dem H-Cluster stattfindet. Strukturmodelle für die
[FeFe]-Hydrogenase HydA1 aus Chlamydomonas reinhardtii zeigen, dass das Protein eine
negative Oberflächenladung mit einer positiv geladenen Bindenische aufweist (Abb.1.4.). An
dieser Stelle findet vermutlich über elektrostatische Wechselwirkungen die direkte Interaktion
von Ferredoxin mit dem H-Cluster statt (Winkler et al. 2002, Horner et al. 2002). Genaue
Aussagen über den Aufbau und die Funktionsweise von Grünalgen-Hydrogenasen können
allerdings nur durch weitere Untersuchungen erfolgen.

1. Einleitung 8
Abb.1.3. Sequenzalignment von bakteriellen [FeFe]-Hydrogenasen und Grünalgen-[FeFe]-Hydrogenasen
Vergleichend zu DdH und CpI sind die Aminosäuresequenzen der hier untersuchten Hydrogenasen aus
Chlamydomonas reinhardtii, Chlamydomonas moewusii und Chlorococcum submarinum dargestellt. Blau:
homologe Bereiche, rot: Grünalgenspezifische homologe Bereiche, orange: konservierte Cysteinreste, die das
[4Fe-4S]-Cluster koordinieren, violett: Reste, die in den Protonentransfer involviert sind, türkis: Reste, die an der
Strukturbildung des aktiven Zentrums beteiligt sind. Das Sequenzalignment wurde mit dem Programm Jalview Java
alignment editor (Clamp et al. 2004) durchgeführt.

1. Einleitung 9
Abb. 1.4. Strukturmodell und Oberflächenladungsmodell der [FeFe]-Hydrogenase HydA1 aus
Chlamydomonas reinhardtii
Die Modelle wurden mittels der Modellierungssoftware von Swiss Model (http://swissmodel.expasy.org) (Guex und
Peitsch 1997) unter Verwendung der von DdH und CpI bekannten Parameter erstellt. Die α-Helices sind rot, die
β-Faltblätter türkis dargestellt. Die negative Oberflächenladung von HydA1 ist rot, die positiv geladene Nische, in
der vermutlich das Ferredoxin bindet, ist blau eingefärbt.
1.4. in vivo-Wasserstoffproduktion von Grünalgen
Aufgrund ihrer hohen Sauerstoffsensibilität werden Hydrogenasen nur unter anaeroben
Bedingungen exprimiert (Forestier et al. 2003). Die Expression der Hydrogenase und die
Ausbildung des Wasserstoffmetabolismus als Anpassung an Anaerobiose ist für Grünalgen der
einzige Weg, genügend ATP zum Überleben unter diesen Stressbedingungen zu generieren
(Melis et al. 2000, Zhang et al. 2001, Melis und Happe 2001). Vor einigen Jahren wurde
entdeckt, dass die Grünalge C. reinhardtii auch unter Schwefelmangelbedingungen H2
produziert und die dabei gemessene in vitro-Aktivität der [FeFe]-Hydrogenase bedeutend höher
ist, als in einer artifiziell anaerobisierten Kultur (Melis et al. 2000, Zhang et al. 2002).
Eine Kultivierung der Algen in schwefelfreiem Medium führt bei Ausschluss von Sauerstoff und
normaler Belichtung zur Ausbildung eines mehrere Tage anhaltenden photofermentativen
Wasserstoffmetabolismus (Happe et al. 2002). Dabei kommt es neben der Expression der
Hydrogenase und Pyruvat-Format-Lyase als Schlüsselenzyme des Wasserstoffmetabolismus und
der Pyruvat-Gärung (Winkler et al. 2002, Hemschemeier und Happe 2004, Hemschemeier et al.
2008) unter anderem zur Bildung von Sulfat-Transportern mit hoher Sulfat-Affinität (Grossmann
2000), des Enzyms Arylsulfatase, welches Sulfat aus Sulfatestern freisetzen kann, und durch eine
Umverteilung des Schwefels zur Synthese von Zellwandproteinen mit weniger schwefelhaltigen

1. Einleitung 10
Aminosäuren. Weiterhin ist eine Akkumulation von Stärke und die Einstellung des
Zellwachstums zu beobachten (Zhang et al. 2001). Da das Photosystem II abgebaut und die
Lichtenergie auf das Photosystem I konzentriert wird (State I- / State II-Transition), sinkt die
Photosyntheserate rapide. Die Respirationsrate bleibt dagegen konstant. Dadurch wird der in der
Kultur vorhandene Sauerstoff veratmet und es kommt zu anoxischen Bedingungen (Wykoff et al.
1998, Melis und Happe 2001). Durch die Umstellung des Stoffwechsels und die dadurch
verursachte Abwesenheit anderer Elektronensenken wie z. B. des Calvinzyklus entsteht ein
Überschuss an Reduktionsäquivalenten. Die Hydrogenase ist durch Ferredoxin mit der
photosynthetischen Elektronentransportkette verbunden (Florin et al. 2001, Happe und Kaminski
2002). Daher können Elektronen und Protonen aus der Wasserspaltung am Photosystem II über
den Plastochinon-Pool, den Cytochrom-b6 f-Komplex, Plastocyanin, das Photosystem I und
Ferredoxin auf die Hydrogenase übertragen und zu molekularem Wasserstoff umgesetzt werden.
Zusätzlich kann die Hydrogenase überschüssige Elektronen aus dem Abbau der akkumulierten
Stärke, dem Calvin-Zyklus und Fermentationsprodukten abbauen (Happe et al. 2002,
Hemschemeier und Happe 2004). Dies verdeutlicht nicht nur die entscheidende Rolle der
Hydrogenase als H2-produzierendes Enzym sondern auch als Stressprotein in vivo.
1.5. Zielsetzung
Ziel der vorliegenden Arbeit ist die Reinigung und strukturelle Charakterisierung der
[FeFe]-Hydrogenase HydA1 aus C. reinhardtii. Das Enzym konnte bereits 1993 von Happe und
Naber isoliert werden, allerdings war die Proteinmenge zu gering, um die Struktur oder den
katalytischen Mechanismus des Enzyms zu untersuchen. Daher soll zum einen ein neues
Expressionssystem für HydA1, bei dem die Hydrogenase-Expression durch Schwefelmangel
induziert wird, und zum anderen ein neues, effektives Reinigungsprotokoll entwickelt werden,
um das Enzym in ausreichender Menge in nativer Form aus der Alge zu isolieren. Anschließend
sind eine biochemische Charakterisierung des Enzyms, sowie erste Strukturuntersuchungen des
aktiven Zentrums durch Elektronen-Paramagnetische-Resonanz-Spektroskopie (EPR) und
Fourier-Transformations-Infrarot-Spektroskopie (FTIR) vorgesehen. Zusätzlich sollen
hinsichtlich einer späteren biotechnologischen Anwendung zwei weitere, bisher unbeschriebene
Grünalgen-Hydrogenasen isoliert und vergleichend zu HydA1 aus C. reinhardtii untersucht
werden, HydA aus der Brackwasser-Alge Chlorococcum submarinum, die besonders für
biotechnologische Anwendungen interessant sein könnte, da sie in Medien mit hohen

1. Einleitung 11
Salzkonzentrationen kultiviert werden kann, was das Risiko von Kontaminationen vermindert
(Blackwell und Gilmour 1991) und HydA1 aus Chlamydomonas moewusii, die in früheren
Versuchen bei anaerober Adaptation eine im Vergleich zu C. reinhardtii drei mal höhere
in vitro-Hydrogenaseaktivität gezeigt hat (Winkler et al. 2004).

2. Material und Methoden 12
2. Material und Methoden
2.1. Organismen
Algenstamm SAG-Nr.
Chlamydomonas moewusii SAG 24.91
Chlamydomonas reinhardtii 137 SAG 11-32a
Chlorococcum submarinum SAG 2.96
Alle Stämme wurden aus der Sammlung von Algenkulturen der Universität Göttingen (SAG)
bezogen.
2.2. Proteine und Molekulargewichtsmarker
Protein Firma/Herkunft
Goat-Anti-Rabbit HRP-Konjugat Pierce
polyklonales Rabbit-Anti-HydA1-Antiserum Happe et al. 1994
Rinderserumalbumin (BSA) Sigma-Aldrich
Trypsin (Sequencing-Grade) Promega
#SMO441 Prestained Protein Molecular Weight Marker (19-119 kDa) Fermentas
2.3. Chemikalien
Alle verwendeten Chemikalien wurden mit dem höchstmöglichen Reinheitsgrad von den Firmen
Acros Organics, AppliChem, Biomol, Carl Roth, Chemgas, Fluka, J. T. Baker, Merck, Riedel-de
Haen, Serva Electrophoresis, Sigma-Aldrich und Thermo Fisher Scientific bezogen.

2. Material und Methoden 13
2.4. Gase
Gas Firma
Argon (Ar) 5.0 Air Liquide
Formiergas (90% N2, 10% H2) Air Liquide
Kohlenmonoxid (CO) 4.7 Air Liquide 13CO (99% Reinheit) Cambridge Isotope Laboratories
Stickstoff (N2) 5.0 Air Liquide
Wasserstoff (H2) 5.0 Air Liquide
2.5. Lösungen, Puffer und Medien
2.5.1. Tris-Acetat-Phosphat (TAP)-Medium
20 mM Tris
1 ml Spurenelemente
1 ml Kaliumphosphat-Puffer pH 7,2
50 ml Beij´sche Salzlösung
ad 1 l H2O mit Essigsäure auf pH 7,2 einzustellen
Spurenelemente
50 g EDTA Na2-Salz
22 g ZnSO4 x 7 H2O (10,4 g ZnCl für TAP-S-Medium)
11,4 g H3BO3
5,1 g MnCl2 x 4 H2O
1,6 g CoCl2 x 6 H2O
1,6 g CuSO4 x 5 H2O (1,1 g CuCl2 x 6 H2O für TAP-S-Medium)
5 g FeSO4 x 7 H2O (3,6 g FeCl2 x 4 H2O für TAP-S-Medium)
1,1 g (NH4)6Mo7O24 x 4 H2O
ad 1 l H2O Alle Komponenten mit Ausnahme des EDTA Na2-Salzes wurden in H2O gelöst und zusammen aufgekocht.
Nach Abkühlung der Lösung wurde das EDTA Na2-Salz zugegeben und der pH-Wert mit KOH (20% w/v) auf
pH 6,7 eingestellt. Anschließend wurde die Lösung auf 1 l aufgefüllt, autoklaviert und bei 4°C gelagert.

2. Material und Methoden 14
Kaliumphosphat-Puffer pH 7,2
283 mM KH2PO4
717 mM K2HPO4
Beij´sche Salzlösung
8,1 mM MgSO4 (MgCl2 x 6 H2O für TAP-S-Medium)
149,5 mM NH4Cl
6,8 mM CaCl2
2.5.2. TAP-S-Medium
TAP-Medium, in dem alle Sulfatsalze durch die entsprechenden Chloridsalze ersetzt wurden.
2.5.3. Selbstentschwefelndes Medium
TAP-S-Medium mit 75 µM MgSO4 pro l.
2.5.4. 57Fe-Medium
Selbstentschwefelndes Medium, in dem FeCl2 durch 57Fe ersetzt wurde. Bei der Herstellung des Selbstentschwefelden Mediums wurde eine Spurenelementlösung ohne FeCl2 x 4 H2O
verwendet. Nach dem Autoklavieren wurden 0,2 ml 57Fe-Lösung pro l Medium (0,5 µM) zugegeben und der
pH-Wert auf 7,2 eingestellt.
57Fe-Lösung
100 mg 57Fe (at 97% minimum, Chemgas)
25 ml Königswasser
50 ml H2O Das 57Fe wurde in 25 ml Königswasser gelöst und nach Verdünnung mit H2O in einem mit Butylgummi- und
Standard Seal 25-Stopfen (Grace) zukrimpbaren 100 ml Injektionsfläschchen (Thermo Fisher Scientific)
anaerob bei Raumtemperatur gelagert.
2.5.5. TAP-Platten
15 g Agar pro l TAP-Medium

2. Material und Methoden 15
2.6. Sterilisation von Flüssigkeiten und Glasgeräten
Alle temperaturstabilen Lösungen, Puffer, Medien und sonstige Materialien wurden vor
Gebrauch für 20 min bei 121°C und 1 bar autoklaviert (Varioklav® Dampfsterilisator, Thermo
Fisher Scientific). Temperaturinstabile Lösungen wurden mittels einer 0,2 µm Membran
(Filtropur S, Sarstedt) sterilfiltriert. Glasgeräte wurden bei Bedarf für 4 h bei 180°C
hitzesterilisiert (Brutschrank WTC, Binder).
2.7. Kultivierung der Algen
Die Haltung der verschiedenen Algenstämme erfolgte auf TAP-Platten (2.5.) bei einer
Temperatur von 20°C und einer Lichtintensität von 100 µmol Photonen m-2 s-1.
Für die Kultivierung der Algen in Flüssigmedium wurden 200 ml TAP-Medium (2.5.) mit dem
jeweiligen Algenstamm angeimpft und bei 20°C und einer Lichtintensität von 150 µmol
Photonen m-2 s-1 auf einem Schüttler (120 U min-1, Certomat® R, Braun) bis zu einem
Chlorophyllgehalt von 20 µg Chlorophyll ml-1 kultiviert (2.8.). Anschließend wurden 1-2 ml
Algenkultur in frisches TAP-Medium (200 ml oder größere Volumina) überführt und wiederum
bis zu einer Chlorophyllkonzentration von 20 µg Chlorophyll ml-1 inkubiert. Die Kultivierung
von C. moewusii erfolgte in Flaschen unter konstantem Rühren. Das Medium enthielt zusätzlich
10 mM Natriumhydrogencarbonat, welches nach dem Autoklavieren sterilfiltriert zugegeben
wurde (Filtropur S, 0,2 µm, Sarstedt).
2.8. Bestimmung der Chlorophyllkonzentration
Zur Bestimmung der Chlorophyllkonzentration einer Algenkultur wurde 1 ml Kultur pelletiert
(1 min, 13000 rpm, Mini Spin Zentrifuge, Eppendorf) und nach Zugabe von 1 ml Aceton durch
Inkubation für 3 min bei 80°C (Thermo Compact Heizblock, Eppendorf) aufgeschlossen. Nach
Abzentrifugieren der Zelltrümmer (1 min, 13000 rpm, Mini Spin Zentrifuge, Eppendorf) wurde
der Überstand bei 652 nm spektralphotometrisch untersucht (SmartSpec 3000
Spektralphotometer, Bio-Rad). Die Chlorophyllkonzentration wurde wie folgt berechnet:

2. Material und Methoden 16
E652 / ε652 * d = Chlorophyllkonzentration [mg ml-1]
E652: Extinktion bei 652 nm
ε652: Extinktionskoeffizient von Chlorophyll a / b bei 652 nm: 34,5 ml mg-1 cm-1
d: Schichtdicke der Küvette: 1 cm
2.9. Induktion der Hydrogenase-Expression
2.9.1. Anaerobe Induktion
Für die anaerobe Induktion wurden die Algen in TAP-Medium bis zu einer
Chlorophyllkonzentration von 20 µg Chlorophyll ml-1 kultiviert (2.7.), 10 min bei 2000 rpm
abzentrifugiert (Sorvall® RC-5B Refrigerated Superspeed Zentrifuge, DuPont Instruments) und
in 0,1 Volumen frischem TAP-Medium resuspendiert. Anschließend wurden die Zellen
abgedunkelt und mit Argon begast, bis das Maximum der in vitro-Hydrogenaseaktivität erreicht
war (2.14.).
2.9.2. Schwefelmangel-Induktion
Zur Induktion der Hydrogenase-Expression durch Schwefelmangel wurden die Algen in TAP-
Medium bis zu einem Chlorophyllgehalt von 20 µg Chlorophyll ml-1 angezogen (2.7.), 10 min
bei 2000 rpm abzentrifugiert (SorvallR RC-5B Refrigerated Superspeed Zentrifuge, DuPont
Instruments) und anschließend im selben Volumen TAP-S-Medium (2.5.) resuspendiert. Die
weitere Kultivierung erfolgte in Flaschen unter konstantem Rühren. Die Kulturen wurden durch
einen Deckel mit Gummidichtung, durch den mit einer Spritze Proben entnommen werden
konnten, luftdicht verschlossen und bei Raumtemperatur und einer Lichtintensität von < 50 µmol
Photonen m-2 s-1 für 24-48 h auf einem Magnetrührer inkubiert.
Für größere Volumina wurden alternativ 5-10 l selbstentschwefelndes Medium (2.5.) direkt mit
dem jeweiligen Algenstamm angeimpft (5 ml Algenkultur l-1), ebenfalls luftdicht verschlossen
und für ca. 7 Tage bei Raumtemperatur und einer Lichtintensität von < 50 µmol Photonen m-2 s-1
auf einem Magnetrührer kultiviert bis das Maximum der in vitro-Hydrogenaseaktivität erreicht
war (2.14.). Die Kultivierung der Algen in 57Fe-Medium (2.5.) erfolgte analog.

2. Material und Methoden 17
2.10. Arbeiten am Anaerobzelt
Arbeiten, die unter sauerstofffreien Bedingungen stattfinden sollten, wurden an einem
Anaerobzelt (Coy Laboratory Products) mit einer Atmosphäre von 99% N2 und 1% H2
durchgeführt. Restsauerstoff wurde über Palladium-Katalysatoren im Anaerobzelt mit H2 zu H2O
umgesetzt, welches durch Aluminium-Katalysatoren gebunden wurde. Die Regeneration der
Katalysatoren erfolgte nach Bedarf durch Inkubation für 6-12 h bei 125°C (Palladium) bzw.
220°C (Aluminium) (Brutschrank WTC, Binder). Gegenstände und Flüssigkeiten wurden über
eine Schleuse mit angeschlossener Vakuumpumpe (Öl-Drehschieber-Vakuumpumpe DVP Typ
LB25, Toepffer Lab Systems) ins Zelt gebracht. Flüssigkeiten wurden zur Entfernung von
Sauerstoff zunächst 30 min mit Argon begast und anschließend im Vakuum der Schleuse
entgast, bis keine Gasblasen mehr aufstiegen. Anschließend wurden sie ins Zelt geschleust und
vor Benutzung mindestens drei Tage aufbewahrt.
2.11. Isolierung von [FeFe]-Hydrogenasen
Alle Reinigungsschritte wurden unter anaeroben Bedingungen (2.10.) so weit möglich bei 4°C
durchgeführt. Alle Puffer wurden vor Gebrauch, soweit nicht anders angegeben, mit 10 mM
NaDT zum Schutz des Enzyms vor Restsauerstoff versetzt. Zur Zentrifugation größerer
Volumina außerhalb des Anaerobzeltes wurden luftdichte 500 ml Zentrifugenbecher der Firma
Nalgene® Labware verwendet.
Für die Isolierung der Hydrogenase wurden 8 l Schwefelmangelkultur (2.9.2.) mit 25 mM NaDT
(Endkonzentration) versetzt und außerhalb des Zeltes abzentrifugiert (10 min, 2000 rpm,
Sorvall® RC-5B Refrigerated Superspeed Zentrifuge, DuPont Instruments). Anschließend konnte
das Pellet in 30 ml 50 mM Tris-HCl pH 8, 10% Glycerin, 25 mM NaDT gelöst und anaerob bei
- 80°C gelagert werden.
Für den Zellaufschluss wurde das Pellet in 100 ml 50 mM Tris-HCl pH 8,5, 100 mM NaDT, 2%
Triton X-100 (Endvolumen) aufgenommen und 25 min unter Rühren aufgeschlossen. Nach
Abzentrifugieren der Zelltrümmer (20 min, 6000 rpm, 4°C, Sorvall® RC-5B Refrigerated
Superspeed Zentrifuge, DuPont Instruments) wurde der Proteinüberstand mit 40%
Ammoniumsulfat gefällt. Dazu wurde eine 100% gesättigte Ammoniumsulfatlösung nach
Versetzen mit 150 mM NaDT über eine peristaltische Pumpe (Perimax 12, Spetec) gleichmäßig

2. Material und Methoden 18
zugetropft und die Proteinlösung anschließend zur Einstellung des Gleichgewichtes noch 1 h
weitergerührt. Nach Zentrifugation für 1 h bei 6000 rpm und 4°C (Sorvall® RC-5B Refrigerated
Superspeed Zentrifuge, DuPont Instruments) wurde der Überstand mit 75% Ammoniumsulfat
gefällt. Das Präzipitat wurde erneut abzentrifugiert (1 h, 6000 rpm, 4°C, Sorvall® RC-5B
Refrigerated Superspeed Zentrifuge, DuPont Instruments) und das Pellet in 10-15 ml 50 mM
Tris-HCl pH 8,5 gelöst und über Nacht gegen 100 Volumen desselben Puffers dialysiert (2.13.).
Nach Zentrifugation für 10 min (Heraeus-Christ Piccolo, Stufe 4) wurde die Proteinlösung mit
einer peristaltischen Pumpe (Perimax 12, Spetec) auf eine Q-Sepharose Fast Flow-Säule
(30 mm x 60 mm, GE Healthcare), die zuvor mit 5 Volumen 50 mM Tris-HCl pH 8,5 äquilibriert
worden war, aufgetragen (Fließgeschwindigkeit 3 ml min-1). Nach Waschen der Säule mit
100 ml desselben Puffers wurden die Proteine durch einen pH-Gradienten von 50 mM Tris-HCl
pH 8,5 bis 50 mM Kaliumphosphat pH 7 (6-faches Säulenvolumen, Fließgeschwindigkeit
3 ml min-1) und einem nachfolgenden Waschschritt mit 120 ml 50 mM Kaliumphosphat pH 7
eluiert. Für den pH-Gradienten sowie den nachfolgenden Waschschritt wurde eine ÄKTA-
FPLC-Anlage der Firma Amersham Pharmacia Biotech verwendet. Die Hydrogenase
enthaltenden Fraktionen (2.14.) wurden vereint und mit dem 1,5-fachen Volumen 50 mM
Tris-HCl pH 8,5 verdünnt. Anschließend wurde die Proteinlösung auf eine zweite Q-Sepharose
Fast Flow-Säule (20 mm x 35 mm, GE Healthcare), die zuvor mit 5 Volumen 50 mM Tris-HCl
pH 8,5 äquilibriert worden war, aufgetragen (Perimax 12, Spetec, Fließgeschwindigkeit
3 ml min-1). Nach Waschen mit 10 ml 50 mM Tris-HCl pH 8,5 wurde die Hydrogenase durch
einen KCl-Gradienten von 50 mM Tris-HCl pH 8,5 bis 50 mM Tris-HCl pH 8,5 500 mM KCl
(4-faches Säulenvolumen, Fließgeschwindigkeit 3 ml min-1 ÄKTA-FPLC, Amersham Pharmacia
Biotech) eluiert. Die Hydrogenase enthaltenden Fraktionen wurden vereint und mittels vivaspin
6 Zentrifugalkonzentratoren (Sartorius) (2.12.) auf ein Volumen von < 1 ml einkonzentriert
(Heraeus-Christ Piccolo, Stufe 2). Anschließend erfolgte die Abtrennung von noch kleineren in
der Probe vorhandenen Verunreinigungen durch Größenausschlusschromatographie über eine
HiLoadTM 16/60 Superdex 75 prep grade Gelfiltrationssäule (16 mm x 600 mm, GE Healthcare).
Die Säule wurde entsprechend den Herstellerangaben verwendet. Die Elution der Proteine
erfolgte bei 1 ml min-1 (Perimax 12, Spetec) in 50 mM Tris-HCl pH 8,5, 150 mM KCl, 2 mM
NaDT. Die Hydrogenase-Fraktionen wurden vereint und mittels vivaspin 6 Zentrifugal-
konzentratoren (Sartorius) auf 1 ml einkonzentriert (Heraeus-Christ Piccolo, Stufe 2).
Das Protein wurde anschließend in PCR-Gefäße aliquotiert und diese in mit Butylgummistopfen
und Standard Seal 25-Stopfen (Grace) zukrimpbaren 8 ml Rollrandflaschen (ND20, Thermo
Fisher Scientific) anaerob bei - 80°C gelagert.

2. Material und Methoden 19
2.12. Konzentrierung von Proteinlösungen
Zur Konzentrierung von Proteinlösungen wurden vivaspin 6 und vivaspin 500 Zentrifugal-
konzentratoren (10000 MWCO PES) der Firma Sartorius nach Herstellerangaben verwendet.
2.13. Dialyse von Proteinlösungen
Zur Entsalzung von Proteinlösungen in präparativem Maßstab wurden Dialyseschläuche der
Firma Serva Electrophoresis (Membra-CelTM Dialysis Membranes, MWCO 3500, 16 mm
Diameter) nach Herstellerangaben verwendet. Die Dialyse der Proben erfolgte über Nacht gegen
das entsprechende Volumen des gewünschten Puffers.
Für die Dialyse von Proteinlösungen im µl-Maßstab wurde eine Mikrotropfendialyse nach
Marusyk et al. (1980) durchgeführt. Dazu wurde die Proteinlösung auf einen Membranfilter
(Nitrocellulose Membranfilter, Porengröße 0,025 µm, Millipore), der auf der Oberfläche der
entsprechenden Pufferlösung schwamm, pipettiert und nach 30 min wieder abgenommen.
2.14. Gaschromatographische Bestimmung der in vitro-Hydrogenaseaktivität
Die Hydrogenaseaktivität wurde gaschromatographisch über die durch das Enzym produzierte
Wasserstoffmenge bestimmt. Dazu wurde ein 2 ml-Reaktionsansatz, bestehend aus 60 mM
K-Phosphat pH 6,8, 100 mM NaDT, 10 mM Methylviologen und 2-15 µl Proteinlösung, im
Anaerobzelt in ein 8 ml-Testgefäß (8 ml-Rollrandflasche ND20, Thermo Fisher Scientific)
pipettiert und dieses durch einen Suba Seal 25-Rotgummistopfen (Sigma-Aldrich) luftdicht
verschlossen. Für die Bestimmung der in vitro-Hydrogenaseaktivität von Algenkulturen wurden
100 µl Kultur und zusätzlich 1% Triton X-100 in den Ansatz gegeben. Nach 3 minütiger
Entgasung mit Argon wurden die Testgefäße 20 min bei 37°C im Schüttelwasserbad (julabo
SW-20C, Julabo Labortechnik) inkubiert. Danach wurden mit einer 1 ml Kunststoffspritze
(NormJect®, Henke Sass Wolf) 200 µl Gasphase in den Gaschromatographen (GC-2010,
Shimadzu) injiziert und die gebildete Wasserstoffmenge bestimmt. Zur Ermittlung des
Umrechnungsfaktors für die durch die Software des Gerätes ausgegebene Flächeneinheit [area]

2. Material und Methoden 20
in die entsprechende Wasserstoffmenge [nmol] wurde das Gerät mit H2 geeicht. Es ergab sich
ein Umrechnungsfaktor von 135 areas pro nmol H2. Die Hydrogenaseaktivität wurde wie folgt
berechnet:
µmol H2 * 40a / 20b / mg Chlorophyll = Aktivität [µmol H2 min-1 mg-1 Chlorophyll]
µmol H2 * 40a / 20b / mg Protein = spezifische Aktivität [µmol H2 min-1 mg-1 Protein]
a: Injektion von 200 µl Gasphase bei 8 ml Gefäßvolumen
b: 20 min Inkubation
2.15. Konzentrationsbestimmung von Proteinlösungen
Zur Bestimmung der Proteinkonzentration wurde ein Bio-Rad-Mikroassay nach Bradford (1976)
durchgeführt. Die Durchführung erfolgte nach Herstellerangaben unter Verwendung von BSA
als Standard.
2.16. SDS-Polyacrylamid-Gelelektrophorese
Zur Trennung von Proteinen nach ihrem Molekulargewicht wurden SDS-Polyacrylamid-
Gelelektrophoresen nach Laemmli (1970) durchgeführt. Zur Analyse wurden Gele mit einem
15% igen Trenngel (15% Acrylamid-Bisacrylamid 37,5:1, 375 mM Tris-HCl pH 8,8, 0,1% SDS)
und einem 6% igen Sammelgel (6% Acrylamid-Bisacrylamid 37,5:1, 125 mM Tris-HCl pH 6,8,
0,1% SDS) verwendet. Die Proteinproben wurden vor der Elektrophorese mit ¼ Volumen 4x
SDS-Probenpuffer und 3 µl β-Mercaptoethanol versetzt und 3 min bei 95°C denaturiert (Thermo
Compact Heizblock, Eppendorf). Anschließend erfolgte die Elektrophorese bei 60 V in einer
Mini-Protean® 3 Cell Elektrophoresekammer der Firma Bio-Rad. Die Proteine wurden
anschließend durch Färbung mit Coomassie Brilliant Blau R250 sichtbar gemacht (2.17.).

2. Material und Methoden 21
4x SDS-Probenpuffer Elektrophoresepuffer
200 mM Tris-HCl pH 6,8 25 mM Tris
8% SDS 250 mM Glycin
40% Glycerin 0,1% SDS
0,04% Bromphenolblau pH 8,8
2.17. Coomassie-Färbung von SDS-Polyacrylamidgelen
Die Färbung von Proteinen in Polyacrylamidgelen erfolgte durch den Farbstoff Coomassie
Brillant Blau R250. Dazu wurden die Gele zunächst 30 min in Färbelösung und anschließend zur
Entfärbung des Hintergrunds in Entfärbelösung unter Schütteln (Rotamax 120, Heidolph)
inkubiert.
Färbelösung Entfärbelösung
50% (v/v) Methanol 10% (v/v) Methanol
10% (v/v) Essigsäure 10% (v/v) Essigsäure
0,1% (w/v) Coomassie Brillant Blau R250
2.18. Western Blot und Immunodetektion von Proteinen
Der spezifische Nachweis von HydA1 aus C. reinhardtii erfolgte durch Western Blot und
nachfolgende Detektion mit einem spezifischen Antikörper. Dazu wurden die Proteine
gelelektrophoretisch getrennt (2.16.) und anschließend in einer Transferapparatur (Fast-Blot
B44-Kammer, Biometra) für 20 min bei 0,24 A auf eine Nitrocellulose-Membran (0,45 µm,
Schleicher & Schuell) übertragen. Nach dem Transfer wurde die Membran 30 min bei
Raumtemperatur in Blockingpuffer und anschließend 2 h bei 4°C mit dem primären Antikörper
(polyklonales Rabbit-Anti-HydA1-Antiserum gegen HydA1 aus C. reinhardtii, Happe et al.
1994, 1:5000 in Blockingpuffer) inkubiert. Danach wurde die Membran dreimal 10 min mit
PBS-Puffer, 0,1% Tween 20 gewaschen und anschließend 1 h bei Raumtemperatur mit dem
sekundären Antikörper (Goat-Anti-Rabbit HRP-Konjugat, Pierce, 1:5000 in Blockingpuffer)
inkubiert. Im Anschluss wurde die Membran zweimal mit PBS-Puffer, 0,1% Tween 20 und

2. Material und Methoden 22
einmal mit PBS-Puffer gewaschen und in Äquilibrierungs-Puffer äquilibriert. Die Detektion
erfolgte mit dem SuperSignal West Dura Extended Substrat-Kit der Firma Pierce gemäß
Herstellerangaben und einem Chemilumineszenz-Detektor (FluorChem 8800) der Firma Alpha
Innotech.
Transferpuffer PBS-Puffer Blockingpuffer Äquilibrierungs-Puffer
20% Methanol 4 mM NaH2PO4 PBS-Puffer 100 mM Tris-HCl pH 9,5
25 mM Tris 16 mM Na2HPO4 2% Milchpulver 100 mM NaCl
192 mM Glycin 115 mM NaCl 0,1% Tween 20 50 mM MgCl2
2.19. Sequenzierung von Proteinen
Die massenspektrometrische Identifizierung von HydA1 aus C. reinhardtii erfolgte mittels
MALDI-TOF-Analyse nach Karas et al. (1987) und Rosenfeld et al. (1992) in Zusammenarbeit
mit Dr. Frank Fischer (Institut für Biochemie der Pflanzen, Ruhr-Universität Bochum). Nach
SDS-Polyacrylamid-Gelelektrophorese (2.16.) und anschließender Coomassie-Blau-Färbung
(2.17.) wurde die entsprechende Proteinbande aus dem Gel ausgeschnitten und zur Entfärbung in
100 µl Waschpuffer 15 min bei 37°C inkubiert (Brutschrank, WTC Binder). Anschließend wurde
das Gelstück im Vakuumkonzentrator (Super Speed Concentrator, Bachofer) getrocknet. Es
folgte die Zugabe von 10 µl Trypsin-Lösung (12,5 ng/µl in 25 mM NH4HCO3) und eine
Inkubation der Probe bei 37°C über Nacht. Nach Zugabe von 10 µl Elutionspuffer und
20 minütiger Inkubation im Ultraschallbad wurde der Überstand massenspektrometrisch
untersucht. Es wurden 0,6 µl Überstand auf einem Probenträger mit 0,6 µl Matrixlösung
vermischt und getrocknet. Danach folgte die Analyse am Voyager DE-Pro Massenspektrometer
(Applied Biosystems) im automatischen Modus der Einstellungen aus der Doktorarbeit von
Frank Fischer (2006). Die erhaltenen Fragmentmassen wurden unter http://expasy.org/sprot/
ausgewertet.
Waschpuffer Elutionspuffer Matrixlösung
25 mM NH4HCO3 50% (v/v) Acetonitril 10 mg/ml Cyano-4-hydroxycinnam-Säure
50% (v/v) Acetonitril 0,5% (v/v) TFA 1% (v/v) TFA
60% (v/v) Acetonitril

2. Material und Methoden 23
Zur Identifizierung von HydA aus C. submarinum und HydA1 aus C. moewusii wurde eine
Sequenzierung der Proteine nach Jensen et al. (1998) in Zusammenarbeit mit Dr. Markus
Piotrowski (Institut für Pflanzenphysiologie, Ruhr-Universität Bochum) durchgeführt. Nach
SDS-Polyacrylamid-Gelelektrophorese (2.16.) und anschließender Coomassie-Blau-Färbung
(2.17.) wurden die entsprechenden Proteinbanden aus dem Gel ausgeschnitten zur Entfärbung
zwei Mal im doppelten Volumen 50% (v/v) Acetonitril für 15 min unter Schütteln inkubiert
(Thermo Compact Heizblock, Eppendorf). Anschließend wurde die Lösung entfernt und
1 Volumen Acetonitril zugegeben. Nach 5 minütiger Inkubation und Entfernung der Lösung
erfolgte die Zugabe von 1 Volumen 100 mM NH4HCO3. Nach 5 min wurde zusätzlich
1 Volumen Acetonitril zugegeben und die Gelstücke weitere 15 min unter Schütteln inkubiert.
Danach wurde die Flüssigkeit vollständig abgenommen und die Gelstücke im
Vakuumkonzentrator (Super Speed Concentrator, Bachofer) getrocknet. Es folgte die Zugabe
von 30 µl Trypsin-Lösung (10 ng/µl in 50 mM NH4HCO3), die nach 10 minütiger Inkubation auf
Eis wieder entfernt wurde. Die Gelstücke wurden mit 25 mM NH4HCO3 bedeckt und bei 37°C
über Nacht inkubiert (Brutschrank WTC, Binder). Zur Extraktion der Peptide wurden die
Ansätze anschließend 2 min im Ultraschallbad inkubiert und der Überstand in ein neues
Reaktionsgefäß überführt. Zur vollständigen Elution der Peptide aus dem Gel wurden die
Restansätze mit 1 Volumen 25 mM NH4HCO3 versetzt, 15 min unter Schütteln inkubiert, mit
1 Volumen Acetonitril versetzt und nach weiteren 15 min erneut im Ultraschallbad inkubiert.
Der Überstand wurde abgenommen und ebenfalls in ein neues Reaktionsgefäß überführt. Die
Gelstücke wurden in 1 Volumen Acetonitril, 5% (v/v) Ameisensäure 15 min unter Schütteln
inkubiert, die Flüssigkeit abgenommen, mit den restlichen Überständen vereint und auf ein
Volumen von 15 µl einkonzentriert. Die Proben wurden anschließend bei -20°C gelagert. Die
massenspektrometrische Untersuchung und die nachfolgende Auswertung erfolgte mit dem
Q-TOF-2 Hybridmassenspektrometer und der zugehörigen Software von Micromass.
2.20. Biochemische Charakterisierung von [FeFe]-Hydrogenasen
2.20.1. Bestimmung des Temperaturoptimums
Zur Bestimmung des Temperaturoptimums der [FeFe]-Hydrogenasen wurden in vitro-
Aktivitätstests wie unter 2.14. beschrieben durchgeführt. Die Reaktionsansätze wurden jeweils
15 min bei Temperaturen von 20-70°C im Schüttelwasserbad inkubiert und die gebildete

2. Material und Methoden 24
Wasserstoffmenge gaschromatographisch bestimmt. Für jeden Temperaturschritt wurden von
drei Proteinchargen jeweils Doppelbestimmungen durchgeführt.
2.20.2. Untersuchung der pH-Toleranz
Zur Bestimmung des pH-Optimums der [FeFe]-Hydrogenasen wurden in vitro-Aktivitätstests
mit unterschiedlichen Reaktionspuffern wie unter 2.14. beschrieben durchgeführt. Alle
Puffersubstanzen wurden in einer Konzentration von 50 mM angesetzt, mit KCl auf eine
Ionenstärke von 150 mM eingestellt und konnten bei 37°C verwendet werden. Die Berechnung
der benötigten Stoffmengen erfolgte mit Hilfe des online verfügbaren Puffer-Kalkulators der
Universität Liverpool (http://www.liv.ac.uk/buffers/buffercalc.html). Die Reaktionsansätze
wurden jeweils 15 min bei 37°C im Schüttelwasserbad inkubiert und die gebildete
Wasserstoffmenge gaschromatographisch bestimmt. Für jeden Puffer wurden
Doppelbestimmungen mit je drei unterschiedlichen Proteinchargen durchgeführt.
pH-Wert Puffersubstanz
MES Bis-Tris K-Phosphat Hepes Mops Tris Tricin
5,5 +
6,0 + + +
6,5 + + +
7,0 + + +
7,5 + + +
8,0 + + +
8,5 + +
9,0 + +
2.20.3. Untersuchung der Salzabhängigkeit
Zur Untersuchung des Einflusses von Salz auf die Aktivität von [FeFe]-Hydrogenasen wurden
in vitro-Aktivitätstests, die statt K-Phosphat pH 6,8 50 mM Tris-HCl pH 7,5 mit
unterschiedlichen Salzkonzentrationen (0-2 M KCl) als Reaktionspuffer enthielten, wie unter
2.14. beschrieben durchgeführt. Die Reaktionsansätze wurden jeweils 15 min bei 37°C im
Schüttelwasserbad inkubiert und die gebildete Wasserstoffmenge gaschromatographisch

2. Material und Methoden 25
bestimmt. Für jeden Puffer wurden Doppelbestimmungen mit je zwei unterschiedlichen
Proteinchargen durchgeführt.
2.20.4. Untersuchung der Sauerstoffsensitivität
Zur Untersuchung der Sauerstoffsensitivität der [FeFe]-Hydrogenasen wurden in vitro-
Aktivitätstests wie unter 2.14. beschrieben durchgeführt. Es wurden je 1 ml 60 mM K-Phosphat
pH 6,8, 2 mM NaDT und 2-15 µl Proteinlösung im Anaerobzelt in 8 ml-Testgefäße
(8 ml-Rollrandflasche ND20, Thermo Fisher Scientific) pipettiert und diese durch Suba Seal 25-
Rotgummistopfen (Sigma-Aldrich) lufticht verschlossen. Anschließend wurden mit einer 1 ml
Kunststoffspritze (NormJect®, Henke Sass Wolf) außerhalb des Zeltes definierte Mengen Luft
(0-3 ml) in die Testgefäße gespritzt und diese anschließend 15 min auf Eis unter Schütteln
inkubiert. Nach 3 minütiger Entgasung mit Argon wurden die restlichen Komponenten des
Reaktionsansatzes zugegeben, die Gefäße nochmals 3 min mit Argon entgast und die Ansätze für
20 min bei 37°C im Schüttelwasserbad inkubiert. Anschließend wurde die gebildete
Wasserstoffmenge wie beschrieben gaschromatographisch bestimmt. Es wurden von zwei
Proteinchargen jeweils Doppelbestimmungen durchgeführt.
2.20.5. Inhibierung von Hydrogenasen durch Kohlenmonoxid
Um die Reaktion von [FeFe]-Hydrogenasen mit Kohlenmonoxid zu untersuchen, wurden
in vitro-Aktivitätstests wie unter 2.14. beschrieben durchgeführt. Dazu wurden luftdicht
verschlossene Testsansätze, die je 1 ml 60 mM K-Phosphat pH 6,8, 2 mM NaDT und 2-15 µl
Proteinlösung enthielten, außerhalb des Anaerobzeltes unterschiedlich lange (0-25 min) mit CO
begast (ca. 125 µl CO sec-1), anschließend 3 min mit Argon entgast und nach Zugabe der
restlichen Komponenten des Reaktionsansatzes und 3 minütiger Argonbegasung für 20 min bei
37°C im Schüttelwasserbad inkubiert. Anschließend wurde die gebildete Wasserstoffmenge wie
beschrieben gaschromatographisch bestimmt.
Für eine genauere Bestimmung der CO-Sensitivität der Hydrogenasen wurden in einer weiteren
Versuchsreihe je 1 ml 60 mM K-Phosphat pH 6,8, 2 mM NaDT und 2-15 µl Proteinlösung wie
unter 2.20.4. beschrieben in 8 ml-Testgefäße pipettiert und diese durch Suba Seal 25-
Rotgummistopfen lufticht verschlossen. Anschließend wurden mit einer 1 ml Kunststoffspritze
aus einer mit CO befüllten Bürette definierte Mengen CO (0-3 ml) in die Testgefäße gespritzt
und diese anschließend 15 min auf Eis unter Schütteln inkubiert. Nach 3 minütiger Entgasung

2. Material und Methoden 26
mit Argon, Zugabe der restlichen Komponenten des Reaktionsansatzes und nochmaliger
Entgasung mit Argon wurden die Ansätze für 20 min bei 37°C im Schüttelwasserbad inkubiert
und die gebildete Wasserstoffmenge gaschromatographisch bestimmt. Es wurden von zwei
Proteinchargen jeweils Doppelbestimmungen durchgeführt.
2.21. Biophysikalische Untersuchungen von [FeFe]-Hydrogenasen
2.21.1. EPR-Spektroskopie
Die EPR-spektroskopische Untersuchung der [FeFe]-Hydrogenasen erfolgte in Zusammenarbeit
mit Dr. Alexey Silakov (Max-Planck-Institut für Bioanorganische Chemie, Mülheim an der
Ruhr). Für die Untersuchung wurden die Proteinproben mittels vivaspin 500
Zentrifugalkonzentratoren (Sartorius) auf ein Volumen von 30 µl und eine Konzentration von
100 µM eingestellt (2.12.) und anschließend falls notwendig dialysiert (2.13.). Eine Begasung
der Proben mit CO, Ar oder H2 erfolgte außerhalb des Anaerobzeltes. Dazu wurde die Probe in
ein PCR-Gefäß überführt und dieses offen in einer 8 ml-Rollrandflasche (ND20, Thermo Fisher
Scientific) positioniert. Nach Verschließen des Gefäßes mit einem Suba Seal 25-
Rotgummistopfen (Sigma-Aldrich) erfolgte die Begasung für 20 min über eine Kanüle.
Anschließend wurde die Probe mit Hilfe einer 0,5 ml-Gastight®-Spritze (Hamilton Medical) in
ein EPR-Röhrchen (Quartz, Innendurchmesser 2 mm) überführt, in flüssigem Stickstoff
schockgefroren und anschließend außerhalb des Zeltes vermessen.
Die Aufnahme der Spektren erfolgte an einem Bruker ELEXSYS E580 Q-Band Spektrometer
mit einer SuperQ-FT Mikrowellenbrücke und der zugehörigen Software von Bruker Optic bei
ca. 34 GHz und einer Temperatur von 20 K nach der Methode des 2 Puls-Elektronenspinechos
(Hahn 1950, Schweiger et al. 2001). Dabei wird ein Elektronenspinecho als Funktion des
externen Magnetfeldes nach zwei Mikrowellenpulsen (π/2 und π) aufgezeichnet. Die
Verzögerung zwischen den Mikrowellenpulsen wurde auf τ = 360 ns, die Länge des
Mikrowellenpulses π/2 auf 36 ns und die von π auf 68 ns eingestellt. Für die Messungen wurde
ein selbstgebauter, leicht überkuppelter zylindrischer TE011 Resonator (Sinnecker et al. 2004),
ähnlich wie bei Sienkiewicz et al. (1996) beschrieben, verwendet. Kryogene Temperaturen
wurden durch Verwendung eines Oxford CF935 Kryostaten auf der Basis von flüssigem Helium
erreicht. Die Belichtung der Proben erfolgte innerhalb des EPR-Setups im Kryostaten bei 40 K

2. Material und Methoden 27
durch Verwendung eines OPO Laser Systems (Quantel, Brilliant Serie) bei 531 nm mit 8 mJ pro
Puls.
Für die zur Untersuchung der 57Fe-Hyperfeinwechselwirkungen durchgeführten Davies
ENDOR- (Davies 1974) und HYSCORE-Messungen wurde ein selbsteingerichtetes
Datenaufnahmesystem verwendet, das auf der SpecMan-Software basiert (Epel et al. 2005). Die
Radiofrequenzimpulse wurden durch einen SMT02 Radiofrequenzgenerator (Rhode &
Schwartz) erzeugt und durch einen ENI 3200 RF solid-state Amplifier verstärkt. Die
Durchführung der ENDOR- und HYSCORE-Messungen und die Auswertung der Spektren
erfolgte in Analogie zu den Angaben aus der Doktorarbeit von Alexey Silakov (2007).
2.21.2. FTIR-Spektroskopie
Die FTIR-spektroskopische Untersuchung der [FeFe]-Hydrogenasen erfolgte in Zusammenarbeit
mit Dr. Alexey Silakov (Max-Planck-Institut für Bioanorganische Chemie, Mülheim an der
Ruhr). Für die Untersuchung wurden die Proteinproben auf ein Volumen von 20 µl und eine
Konzentration von 300-400 µM eingestellt und wie unter 2.21.1. beschrieben vorbereitet.
Die Messungen erfolgten an einem Bruker IFS 66 v/s FTIR-Spektrometer mit MCT-Detektor
und der zugehörigen Software Opus von Bruker Optic. Die Probenkammer wurde während der
Messung mit N2 gespült, um die Bildung von CO2 und Wasserdampf zu vermeiden. Die
Spektren wurden im doppelseitigen Vorwärts-Rückwärts-Modus aufgenommen und die Apertur
auf 1,5 mm eingestellt. Für die Messungen wurde eine luftdicht verschließbare Küvette mit
CaF2-Fenstern verwendet. Kryogene Temperaturen wurden durch Verwendung eines Oxford
Optistat CF Kryostaten, welcher mit CaF2- und Saphir-Fenstern ausgestattet ist, auf der Basis
von flüssigem Helium erreicht. Die Belichtung der Proben erfolgte innerhalb des IR-Setups
durch eine Lampe mit 200 Watt.
Für die Spektroelektrochemischen Messungen wurde eine temperierbare, optisch transparente
Elektrochemie-Dünnschicht-Zelle (OTTLE-Zelle) nach Moss et al. (1990) mit einem sich
zwischen zwei CaF2-Fenstern und der Probenlösung befindlichen Goldnetz als Arbeitselektrode,
einer Platinelektrode als Gegenelektrode und einer Ag/AgCl-Elektrode als Referenzelektrode
verwendet. Das Redoxpotential der Zelle wurde über einen an die Elektroden angeschlossenen
M270 Potentiostaten (EG&G Princeton Applied Research) und der zugehörigen Software M270
kontrolliert. Die Temperatur der Messzelle wurde durch einen RML 6 Thermostaten (Lauda)
konstant gehalten (4°C). Um alle gemessenen Potentiale wie standardmäßig üblich gegen die
Standardwasserstoffelektrode (NHE) anzugeben, wurde das Potential der Ag/AgCl-Elektrode vor

2. Material und Methoden 28
und nach jeder Messung durch Aufnahme eines Voltammogramms von Methylviologen
bestimmt, und die gemessenen Potentiale nachträglich um die Differenz zwischen Ag/AgCl-
Elektrode und Standardwasserstoffelektrode (hier experimentell ermittelt: - 240 mV) korrigiert.
Die Aufnahme der Spektren erfolgte über einen Potentialbereich von - 510 bis 100 mV in
Schritten von 25-50 mV nach einer elektrochemischen Äquilibrierungszeit von 10 min bei jedem
Schritt bei 4°C. Die Auswertung der Spektren erfolgte mit Hilfe des Programms MatLabTM. Die
Bestimmung des Mittelpunktspotentials der einzelnen Redoxübergänge erfolgte unter
Verwendung der Nernst-Gleichung (Atkins 1994).

3. Ergebnisse 29
3. Ergebnisse
3.1. Etablierung eines neuen Reinigungsprotokolls für HydA1 aus
C. reinhardtii
3.1.1. Induktion der Hydrogenase-Expression
3.1.1.1. Anaerobe Induktion
Die Induktion der Hydrogenase-Expression ist durch anaerobe Induktion (Happe et al. 1993)
oder durch Schwefelmangelbedingungen (Melis et al. 2000, Zhang et al. 2002, Winkler et al.
2002) möglich. Bei dem artifiziellen System der anaeroben Adaptation werden anaerobe
Stressbedingungen durch Aufkonzentrierung der Algen und Begasung mit Argon geschaffen, die
Unterbindung der endogenen Sauerstoffproduktion erfolgt durch Abdunklung der Zellen. Das
Maximum der in vitro-Hydrogenaseaktivität ist nach ungefähr 4 h erreicht und liegt für
C. reinhardtii bei ca. 3 µmol H2 min-1 mg-1 Chlorophyll. Ähnliches gilt für C. submarinum. Die
für C. moewusii bestimmte maximale in vitro-Hydrogenaseaktivität ist um das Vierfache höher
(Daten nicht gezeigt).
3.1.1.2. Schwefelmangel-Induktion
Für die Schwefelmangel-Induktion wurden die Algen zunächst in TAP-Medium bis zu einem
Chlorophyllgehalt von 20 µg Chlorophyll ml-1 kultiviert, anschließend in TAP-S-Medium
überführt, luftdicht verschlossen und bei geringer Belichtung für 24-48 h inkubiert. Bei
C. reinhardtii ist das Maximum der in vitro-Hydrogenaseaktivität nach 48 h erreicht und mit
70 µmol H2 min-1 mg-1 Chlorophyll um das 25-fache höher als bei der anaeroben Induktion
(Abb. 3.1. A).
Auch bei C. moewusii und C. submarinum funktioniert die Methode Schwefelmangel besser als
die Methode der anaeroben Adaptation. C. moewusii erreicht ähnliche Aktivitätswerte wie
C. reinhardtii, C. submarinum mit durchschnittlich 35 µmol H2 min-1 mg-1 Chlorophyll etwa die
Hälfte. Das Maximum der in vitro-Hydrogenaseaktivität ist bei beiden im Gegensatz zu
C. reinhardtii schon nach 24 h erreicht (Abb. 3.1. A).

3. Ergebnisse 30
A
B Abb. 3.1. Induktion der Hydrogenase-Expression in C. reinhardtii, C. moewusii und C. submarinum durch Schwefelmangel A Die Algen wurden in TAP-Medium bis zu einem Chlorophyllgehalt von 20 µg ml-1 angezogen und anschließend
in TAP-S-Medium überführt. Danach wurden die Kulturen luftdicht verschlossen und bei geringer Belichtung für
96 h unter Rühren inkubiert. Zu den angegeben Zeitpunkten wurden je 100 µl Kultur steril entnommen und die
Entwicklung der in vitro-Hydrogenaseaktivität gaschromatographisch bestimmt.
B Bei dem optimierten System der Schwefelmangel-Induktion wurde selbstentschwefelndes Medium direkt mit
C. reinhardtii, C. moewusii und C. submarinum angeimpft und die Kulturen unter anaeroben Bedingungen für
10 Tage bei geringer Belichtung unter Rühren kultiviert. Es wurden täglich 100 µl Kultur entnommen und die
Entwicklung der in vitro-Hydrogenaseaktivität gaschromatographisch bestimmt.

3. Ergebnisse 31
3.1.1.3. Optimierung der Schwefelmangel-Induktion
Um die Methode der Schwefelmangel-Induktion im Hinblick auf die geplante Isolierung der
Hydrogenase in großem Maßstab zu optimieren, wurden für die nachfolgenden Versuche keine
zwei Medien mehr, TAP-Medium für die Kultivierung der Algen und TAP-S-Medium zur
Schaffung von Schwefelmangelbedingungen, sondern nur noch selbstentschwefelndes Medium
(2.5.) verwendet. Die direkte Kultivierung in selbstentschwefelndem Medium bietet den Vorteil,
dass bei gleichem Zeitaufwand weniger Medium benötigt wird und der bei großen Volumina
recht aufwändige Zentrifugationsschritt zur Überführung der Algen von TAP-Medium in TAP-
S-Medium wegfällt, wodurch zusätzlich das Risiko für Kontaminationen vermindert wird.
Zusätzlich ist diese Methode durch die allmähliche Anpassung an Schwefelmangelbedingungen
schonender für die Alge (Laurinavichene et al. 2002).
Das selbstentschwefelnde Medium wurde direkt mit dem jeweiligen Algenstamm angeimpft und
die Kulturen luftdicht verschlossen bei geringer Belichtung für 7-8 Tage inkubiert. In dieser Zeit
wachsen die Zellen bis zu einem Chlorophyllgehalt von ca. 20-25 µg Chlorophyll ml-1 und
verbrauchen dabei den im Medium vorhandenen Schwefel und Sauerstoff. Dadurch wird die
Hydrogenase-Bildung induziert, das Maximum der in vitro-Hydrogenaseaktivität ist bei allen
drei Algenstämmen nach 7-8 Tagen erreicht und vergleichbar mit den unter 3.1.1.2.
beschriebenen Werten (Abb. 3.1. B).
Anaerobe Induktion Schwefelmangel-Induktion 5 Stunden 7. Tag 8. Tag
HydA1
Abb. 3.2. Western Blot von HydA1 aus C. reinhardtii, exprimiert unter Schwefelmangel und anaerober
Adaptation
Die anaerobe Induktion und die Schwefelmangel-Induktion wurden wie unter 2.9. beschrieben durchgeführt. Die
Probenentnahme erfolgte jeweils, als das Maximum der in vitro-Hydrogenaseaktivität erreicht war. Es wurde jeweils
eine 10 µg Chlorophyll entsprechende Menge gelelektrophoretisch getrennt und durch Immunodetektion mit einem
spezifisch gegen HydA1 gerichteten Antikörper untersucht.

3. Ergebnisse 32
3.1.2. Reinigung von HydA1 aus C. reinhardtii
Für die Isolierung der Hydrogenase aus C. reinhardtii wurden 8 l Schwefelmangelkultur
verwendet. Die Reinigung erfolgte unter sauerstofffreien Bedingungen im Anaerobzelt. Das
entwickelte Reinigungsprotokoll ist unter 2.11. beschrieben und schematisch in Abb. 3.3.
dargestellt.
Abb. 3.3. Schematische Darstellung des entwickelten Reinigungsprotokolls zur Isolierung von [FeFe]-Hydrogenasen aus Grünalgen
Nach Aufschluss der Zellen durch Triton X-100 wurde der Gesamtproteinextrakt zunächst zur
Entfernung des Chlorophylls und größerer Zellkomponenten mit Ammoniumsulfat gefällt.
Hierbei erfolgte der Ausfall der Hydrogenase im zweiten Fällungsschritt mit 75%
Ammoniumsulfat. Anschließend erfolgte eine Trennung der Proteine mittels Anionenaustausch-
chromatographie über zwei aufeinanderfolgende Q-Sepharose Fast Flow-Säulen. Bei der ersten
Säule wurden die Proteine durch einen pH-Gradienten von pH 8,5-7 eluiert, bei der zweiten
Säule durch einen Salzgradienten von 0-500 mM Kaliumchlorid. Die Hydrogenase konnte in den
Fraktionen ab einem pH-Wert von 7 und einer Salzkonzentration von ca. 300 mM detektiert
werden. Im letzten Schritt erfolgte eine Trennung der Proteine mittels Größenausschluss-
chromatographie über eine HiLoadTM 16/60 Superdex 75 Gelfiltrationssäule. Dabei eluiert die
Hydrogenase bei einer Fließgeschwindigkeit von 1 ml min-1 gemäß ihrem Molekulargewicht von
46 kDa als Monomer nach ca. 58 ml. Das saubere Protein wurde anschließend einkonzentriert
und weiter untersucht. Der typische Verlauf der Reinigung ist in Abb. 3.4. und Tab. 3.1. gezeigt.

3. Ergebnisse 33
kDa M 1 2 3 4 5 6 M 7 8 9 10 11 12 13 14 15
119 79 46 31 24 19
HydA1
Abb. 3.4. SDS-PAGE und Western Blot der verschiedenen Reinigungsstufen von HydA1 aus C. reinhardtii
M: Molekulargewichtsmarker, 1: Proteinextrakt nach Ammoniumsulfatfällung, 2-6: ausgewählte Fraktionen
Q-Sepharose pH-Gradient, M: Molekulargewichtsmarker, 7-9: ausgewählte Fraktionen Q-Sepharose KCl-Gradient,
10: Konzentrierte Probe nach Q-Sepharose KCl-Gradient, 11-13: ausgewählte Fraktionen Gelfiltration Superdex 75,
14: HydA1 1 µg, 15: Western Blot gegen 1 µg gereinigtes HydA1
Die Auftrennung der Proteine erfolgte auf einem 15% igen SDS-Gel mit anschließender Coomassie-Blau-Färbung
bzw. Immunodetektion mit einem spezifisch gegen HydA1 gerichteten Antikörper.
Tab. 3.1. Übersicht über die Reinigung der [FeFe]-Hydrogenase HydA1 aus C. reinhardtii
Gesamtaktivität
[µmol H2 min-1] Gesamtprotein [mg]
Menge [%]
Spezifische Aktivität [µmol H2 min-1 mg-1]
Reinigungsfaktor (-fach)
Gesamtproteinextrakt (8 l) 3780 270 100 14 1 Ammoniumsulfat-Fällung 40-75% 2856 84 48 34 2
1. Anionenaustausch- Chromatographie Q-Sepharose pH-Gradient
1744 8 47 218 16
2. Anionenaustausch- Chromatographie Q-Sepharose KCl-Gradient
533 1,5 25 355 25
Gelfiltration Superdex 75 371 0,5 22 741 53
Aus 8 l Schwefelmangelkultur konnten durchschnittlich 500 µg Hydrogenase mit einer
spezifischen Aktivität von ca. 741 µmol H2 min-1 mg-1 Protein bei Verwendung von reduziertem
Methylviologen als Elektronendonor und einem Reinheitsgrad von 95-100% isoliert werden.
Dies ist eine 40-fach höhere Ausbeute als zuvor bei Happe et al. (1993) beschrieben und nur
etwa 50% weniger als bisher durch die Überexpression in C. acetobutylicum (Girbal et al. 2005)
gewonnen werden konnte. Für die Reinigung konnte ein Reinigungsfaktor von 53 ermittelt
werden, die bestimmte Restaktivität im Vergleich zur Gesamtaktivität des Rohextrakts lag bei

3. Ergebnisse 34
22%. Da der Anteil an Hydrogenase in Schwefelmangelkulturen im Vergleich zur
Gesamtproteinmenge sehr viel höher ist als in einer anaerob induzierten Kultur, ist der
Reinigungsfaktor entsprechend niedrig, was für die Qualität des Expressionssystems spricht.
3.1.3. Nutzung des entwickelten Reinigungsprotokolls für die Isolierung weiterer
[FeFe]-Hydrogenasen aus C. moewusii und C. submarinum
Das für die Isolierung von HydA1 aus C. reinhardtii entwickelte Reinigungsprotokoll konnte
ebenfalls erfolgreich für die Isolierung der bisher noch nicht näher charakterisierten
Hydrogenasen aus C. moewusii und C. submarinum verwendet werden.
Die Hydrogenase aus C. submarium zeigte während des Reinigungsverlaufs ähnliche
Eigenschaften wie HydA1 aus C. reinhardtii. Nach Aufschluss der Zellen und Ammoniumsulfat-
fällung des Gesamtproteinextrakts erfolgte wie bereits beschrieben eine Fraktionierung über
zwei Q-Sepharose Fast Flow-Säulen und eine Größenausschlusschromatographie über
Superdex 75, wobei die Hydrogenase wie HydA1 aus C. reinhardtii bei einem pH-Wert von 7,
einer Salzkonzentration von 300 mM und gemäß ihrem Molekulargewicht als Monomer nach
58 ml eluierte. Da die Zellwand von C. submarinum schwieriger aufzuschließen war als die von
C. reinhardtii, musste die Tritonkonzentration in dem für den Zellaufschluss verwendeten Puffer
auf 2% und die Inkubationszeit auf 25 min erhöht werden. Insgesamt konnten aus 8 l
Schwefelmangelkultur durchschnittlich 500 µg Hydrogenase mit einer spezifischen Aktivität von
ca. 639 µmol H2 min-1 mg-1 Protein und einem Reinheitsgrad von 90-95% isoliert werden (Abb.
3.5. und Tab. 3.2.). Die bestimmte Restaktivität im Vergleich zur Gesamtaktivität des
Rohextrakts lag bei 28%, der ermittelte Reinigungsfaktor hatte einen Wert von 213.
Kleinere Unterschiede konnten bei der Isolierung der Hydrogenase aus C. moewusii festgestellt
werden. Während der Anionenaustauschchromatographie löste sich das Enzym im Gegensatz zu
HydA1 aus C. reinhardtii bereits ab einem pH-Wert von 7,5 und einer Salzkonzentration von
200 mM KCl von den Q-Sepharose-Säulen. Die Trennung mittels Gelfiltration über Superdex 75
verlief dagegen identisch zu den beiden anderen Enzymen. Im Falle von C. moewusii konnten
insgesamt ca. 300 µg Hydrogenase mit einer spezifischen Aktivität von 1598 µmol H2 min-1 mg-1
Protein, doppelt so hoch wie die von HydA1 aus C. reinhardtii, isoliert werden (Tab. 3.3.). Der
Reinheitsgrad des isolierten Enzyms lag ähnlich wie bei C. submarinum bei 90-95% (Abb. 3.5.).

3. Ergebnisse 35
Abb. 3.5. SDS-PAGE der isolierten
M: Molekulargewichtsmarker, 1: Hy
Die Auftrennung der Proteine erfolg
Tab. 3.2. Übersicht über die Reinig
Gesam [µmo
Gesamtproteinextrakt (8 l) Ammoniumsulfat-Fällung 40-75% 1. Anionenaustausch- Chromatographie Q-Sepharose pH-Gradient 2. Anionenaustausch- Chromatographie Q-Sepharose KCl-Gradient Gelfiltration Superdex 75
Tab. 3.3. Übersicht über die Reinig
Gesam
[µmo Gesamtproteinextrakt (8 l) Ammoniumsulfat-Fällung 40-75% 1. Anionenaustausch- Chromatographie Q-Sepharose pH-Gradient 2. Anionenaustausch- Chromatographie Q-Sepharose KCl-Gradient Gelfiltration Superdex 75
kDa M 1 2
119 79 46 31 24 19
[FeFe]-Hydrogenasen aus C. submarinum und C. moewusii
dA C. submarinum 500 ng, 2: HydA1 C. moewusii 500 ng
te auf einem 15% igen SDS-Gel mit anschließender Coomassie-Blau-Färbung.
ung der [FeFe]-Hydrogenase HydA aus C. submarinum
taktivität l H2 min-1]
Gesamtprotein [mg]
Menge [%]
Spezifische Aktivität [µmol H2 min-1 mg-1]
Reinigungsfaktor (-fach)
3081 1027 100 3 1
1248 156 47 8 3
1160 8 46 145 48
536 2 33 268 89
320 0,5 28 639 213
ung der [FeFe]-Hydrogenase HydA1 aus C. moewusii
taktivität l H2 min-1]
Gesamtprotein [mg]
Menge [%]
Spezifische Aktivität [µmol H2 min-1 mg-1]
Reinigungsfaktor (-fach)
2640 240 100 11 1
1728 54 67 32 3
1388 2 58 694 63
574 0,8 28 717 65
479 0,3 23 1598 145

3. Ergebnisse 36
3.1.4. Massenspektrometrische Sequenzanalyse zur Identifizierung der isolierten
HydA-Proteine
Da für C. reinhardtii und C. moewusii in früheren Arbeiten die Existenz von zwei Hydrogenase-
Genen, hydA1 und hydA2, nachgewiesen wurde (Forestier et al. 2003, Winkler et al. 2004),
wurden die isolierten Enzyme zur genauen Identifizierung sequenziert. Dazu wurden die
Proteine gelelektrophoretisch getrennt, mit Trypsin verdaut und die entstandenen Fragmente
anschließend massenspektrometrisch untersucht. Das Ergebnis ist in Abb. 3.6. zu sehen. Im Falle
von C. submarinum konnten drei Peptide identifiziert werden, die wie erwartet eindeutig der
HydA-Sequenz der [FeFe]-Hydrogenase zuzuordnen sind. Bei dem aus C. reinhardtii isolierten
Enzym handelt es sich, wie auch durch Western-Blot gezeigt (Abb. 3.2. und 3.4.), um HydA1. Es
konnten fünf Fragmente bestimmt werden, deren Größe und Sequenz spezifisch für HydA1 ist.
Gleiches gilt für die Hydrogenase aus C. moewusii. Es konnten zwei Peptide nachgewiesen
werden, von denen eins das isolierte Enzym als HydA1 identifiziert. Es wurde somit nur HydA1
aus den Schwefelmangelkulturen isoliert.
Abb. 3.6. Identifizierung der isolierten HydA-Proteine aus C. reinhardtii, C. moewusii und C. submarinum
Die isolierten Proteine wurden mit Trypsin verdaut und die entstandenen Fragmente massenspektrometrisch
untersucht. Die Aminosäuresequenzen der HydA-Proteine wurden mit dem Programm Jalview Java alignment editor
(Clamp et al. 2004) verglichen. Konservierte und homologe Bereiche sind in dunkel- bzw. hellblau markiert. Die
durch die massenspektrometrische Untersuchung identifizierten Sequenzbereiche sind rot gekennzeichnet.

3. Ergebnisse 37
3.2. Biochemische Charakterisierung der isolierten Hydrogenasen
3.2.1. Bestimmung des Temperaturoptimums
Zur genaueren Charakterisierung der isolierten [FeFe]-Hydrogenasen wurde unter anderem die
Temperatursensitivität der Enzyme untersucht. Dabei wurden die Proteine wie unter 2.20.1.
beschrieben jeweils 15 min bei Temperaturen von 20-70°C inkubiert und die Enzymaktivität
anschließend gaschromatographisch über die durch die Enzyme produzierte Wasserstoffmenge
bestimmt. Die Messung zeigte, dass die Aktivität aller Hydrogenasen mit steigender Temperatur
linear zunimmt und bei etwa 60°C ein Maximum erreicht (Daten nicht gezeigt). Ab einer
Temperatur von 70°C sinkt die Aktivität wieder und man misst nur noch ca. 50% der
Maximalaktivität. Dies bestätigt frühere Untersuchungen zu HydA1 aus C. reinhardtii (Roessler
und Lien 1984, Happe et al. 1993) und zeigt, dass die Hydrogenasen aus C. moewusii und
C. submarinum die gleiche hohe Temperaturtoleranz besitzen.
3.2.2. Vergleichende Analyse zur pH-Toleranz der HydA-Proteine
Zur Untersuchung der pH-Toleranz der drei isolierten [FeFe]-Hydrogenasen wurden in vitro-
Aktivitätstests wie unter 2.20.2. beschrieben durchgeführt und die Enzymaktivität wiederum
gaschromatographisch über die durch die Enzyme produzierte Wasserstoffmenge bestimmt. Für
die Aktivitätstests wurden unterschiedliche Reaktionspuffer mit verschiedenen pH-Werten
verwendet. Es wurde ein Bereich von pH 5,5 bis 9 untersucht und für jeden pH-Wert mindestens
zwei unterschiedliche Puffersubstanzen getestet. Das Ergebnis ist in Abb. 3.7. zu sehen.
Alle drei Grünalgen-Hydrogenasen weisen eine hohe pH-Toleranz auf, da im gesamten
untersuchten Bereich Enzymaktivität nachweisbar ist. Das pH-Optimum liegt bei allen drei
Enzymen bei einem pH-Wert von 7. Betrachtet man den gesamten pH-Bereich erkennt man, dass
HydA1 aus C. reinhardtii und HydA1 aus C. moewusii bei allen pH-Werten eine ähnlich hohe
Aktivität aufweisen. HydA aus C. submarinum scheint dagegen etwas aktiver im sauren Bereich
und etwas weniger aktiv im basischen zu sein.

3. Ergebnisse 38
A
B
C
Abb. 3.7. Bestimmung des pH-Optimums von HydA1 aus C. reinhardtii (A), HydA1 aus C. moewusii (B) und
HydA aus C. submarinum (C)
Zur Bestimmung des pH-Optimums wurden in vitro-Aktivitätstests mit unterschiedlichen Reaktionspuffern
durchgeführt. Alle Puffersubstanzen wurden in einer Konzentration von 50 mM angesetzt und mit KCl auf eine
Ionenstärke von 150 mM eingestellt. Die Ansätze wurden jeweils 15 min bei 37°C inkubiert und die gebildete
Wasserstoffmenge gaschromatographisch bestimmt. Für jeden Puffer wurden Doppelbestimmungen mit je drei
unterschiedlichen Proteinchargen durchgeführt. Die Standardabweichung betrug < 15%.

3. Ergebnisse 39
3.2.3. Einfluss der Ionenstärke auf die Enzymaktivität
Der Einfluss der Ionenstärke auf die Aktivität der [FeFe]-Hydrogenasen wurde durch eine
Konzentrationsreihe mit steigenden KCl-Konzentrationen untersucht. Zur Bestimmung der
Enzymaktivität wurden in vitro-Aktivitätstests, die jeweils unterschiedliche Salzkonzentrationen
enthielten, durchgeführt. Alle drei Hydrogenasen weisen eine hohe Toleranz gegenüber Salz bis
zu einer Konzentration von 2 M auf (Daten nicht gezeigt). Die Zugabe von Salz steigert bei allen
drei Enzymen die Aktivität um bis zu 50%. HydA1 aus C. reinhardtii und HydA1 aus
C. moewusii weisen ein Aktivitätsmaximum bei einer Salzkonzentration von 1 M KCl auf, die
maximale Aktivität von HydA aus C. submarinum dagegen ist erst bei einer Konzentration von
1,5 M KCl erreicht.
3.2.4. Untersuchung der Sauerstoffsensitivität
[FeFe]-Hydrogenasen sind extrem sauerstoffempfindlich und werden nur unter anaeroben
Bedingungen exprimiert (Forestier et al. 2003). Um zu untersuchen, ob sich die drei isolierten
Hydrogenasen in ihrer Empfindlichkeit gegenüber Sauerstoff unterscheiden, wurden alle
Enzyme jeweils 15 min in Gegenwart von 2 mM NaDT steigenden Sauerstoffkonzentrationen
ausgesetzt und anschließend die noch vorhandene Enzymaktivität wie bereits beschrieben durch
in vitro-Aktivitätstests gaschromatographisch bestimmt. Das Ergebnis ist in Abb. 3.8. dargestellt.
Wie erwartet sinkt die Aktivität aller drei Hydrogenasen mit steigender Sauerstoffkonzentration
im Testansatz. Dabei zeigen HydA1 aus C. reinhardtii und HydA1 aus C. moewusii eine ähnlich
hohe Sauerstoffempfindlichkeit. Bei einer Konzentration von 2,5% O2 sind bereits nur noch 50%
Enzym aktiv, bei 8% ist keine messbare Aktivität mehr nachweisbar. HydA aus C. submarinum
zeigt dagegen eine deutlich höhere Toleranz gegenüber Sauerstoff. Bei 2,5% O2 sind noch ca.
85% Enzym aktiv, bei 8% sind im Gegensatz zu den Proteinen aus C. reinhardtii und
C. moewusii noch 40% der Ausgangsaktivität vorhanden. Dies stellt zusammen mit der pH-
Toleranz einen weiteren Unterschied dar, der dieses Enzym und den Organismus C. submarinum
besonders interessant für biotechnologische Anwendungen macht.

3. Ergebnisse 40
Abb. 3.8. Sauerstoffsensitiviät von [FeFe]-Hydrogenasen
Die Untersuchung der Sauerstoffsensitivität der [FeFe]-Hydrogenasen wurde wie unter 2.20.4. beschrieben
durchgeführt. Es wurden definierte Mengen O2 in Testansätze mit 60 mM K-Phosphat pH 6,8, 2 mM NaDT und
2-15 µl Proteinlösung gespritzt und diese 15 min inkubiert. Anschließend wurden die restlichen Komponenten des
in vitro-Reaktionsansatzes zugegeben und die gebildete Wasserstoffmenge gaschromatographisch bestimmt. Es
wurden jeweils Vierfachbestimmungen durchgeführt. Die Standardabweichung betrug < 15%.
3.2.5. Inhibierung der Hydrogenasen durch Kohlenmonoxid
Neben der hohen Empfindlichkeit gegenüber Sauerstoff ist die Inhibierung von Hydrogenasen
durch Kohlenmonoxid ein wichtiges Charakteristikum dieser Enzymklasse. Die Inhibierung mit
CO ist im Gegensatz zu der irreversiblen Schädigung des H-Clusters durch O2 reversibel. Bei
Begasung der Proteine mit CO bindet dieses als externer Ligand an das Fe(2)-Atom und
blockiert somit vermutlich den H2-Gaskanal (Pierik et al. 1998). Dieser sogenannte Hox-CO-
Zustand ist relativ stabil und bei anderen Hydrogenasen spektroskopisch bereits gut untersucht
(Lemon und Peters 1999, Albracht et al. 2006). Als Grundlage für die durchzuführenden EPR-
und FTIR-Messungen und zur weiteren Charakterisierung der isolierten Grünalgen-
Hydrogenasen wurde in zwei verschiedenen Versuchsreihen die Reaktion der HydA-Proteine mit
CO untersucht.
Im ersten Versuch wurden Testsansätze mit je 1 ml 60 mM K-Phosphat pH 6,8, 2 mM NaDT
und 2-15 µl Proteinlösung unterschiedlich lange mit CO begast und anschließend die noch
verbliebene Enzymaktivität gaschromatographisch bestimmt (2.20.5.). Das Ergebnis ist in
Abb. 3.9. A zu sehen. Bei allen drei Hydrogenasen sinkt die Enzymaktivität rapide, sobald sie in

3. Ergebnisse 41
Kontakt mit CO kommen. Bereits nach 2 min CO-Begasung ist bei allen drei Hydrogenasen nur
noch ca. 10% der Ausgangsaktivität nachweisbar, eine vollständige Inhibierung ist nach 20 min
erreicht. Eine 40 minütige Begasung der inhibierten HydA-Proteine mit Wasserstoff und Argon
sowie eine 20 minütige Belichtung bei 200 Watt führt zu einer ca. 5% igen Reaktivierung der
Enzyme. Da die Reaktion mit CO insgesamt sehr schnell abläuft und in diesem Versuchsansatz
keine signifikanten Unterschiede zwischen den drei HydA-Proteinen festzustellen war, wurde
eine genauere Untersuchung der CO-Sensitivität der Hydrogenasen analog zu der Untersuchung
der Sauerstoffempfindlichkeit durchgeführt.
In einem zweiten Versuchsansatz wurden alle Enzyme jeweils 15 min steigenden CO-
Konzentrationen ausgesetzt und anschließend die noch vorhandene Enzymaktivität im in vitro-
Aktivitätstest bestimmt (Abb. 3.9. B). Dabei zeigte sich, dass HydA1 aus C. moewusii schneller
durch CO inhibiert wird, als HydA1 aus C. reinhardtii und HydA aus C. submarinum. Im
Gegensatz zu HydA1 und HydA, bei denen bei einer CO-Konzentration von 10% noch 50% der
Ursprungsaktivität und bei 37,5% CO noch ca. 15% Aktivität nachweisbar ist, ist bei
C. moewusii bei 6% CO nur noch 15% der Ausgangsaktivität vorhanden. Bei einer
Konzentration von 25% CO im Testansatz ist das Enzym bereits vollständig inhibiert. Dies lässt
auf einen schnelleren Umsatz des Enzyms schließen, was auch die im Gegensatz zu HydA1 aus
C. reinhardtii und HydA aus C. submarinum höhere spezifische Aktivität belegen würde.

3. Ergebnisse 42
A
B Abb. 3.9. Inhibierung von Hydrogenasen durch Kohlenmonoxid
A Luftdicht verschlossene Testsansätze mit 1 ml 60 mM K-Phosphat pH 6,8, 2 mM NaDT und 2-15 µl
Proteinlösung wurden 0-25 min mit CO begast und nach Zugabe der restlichen Komponenten des Reaktionsansatzes
20 min bei 37°C inkubiert. Die gebildete Wasserstoffmenge wurde anschließend gaschromatographisch bestimmt
(2.20.5.).
B Für eine genauere Bestimmung der CO-Sensitivität wurden definierte Mengen CO in anaerob verschlossene
Testansätze mit 60 mM K-Phosphat pH 6,8, 2 mM NaDT und 2-15 µl Proteinlösung gespritzt und diese 15 min
inkubiert. Anschließend wurden die restlichen Komponenten des in vitro-Reaktionsansatzes zugegeben und die
gebildete Wasserstoffmenge gaschromatographisch bestimmt. Es wurden jeweils Vierfachbestimmungen
durchgeführt. Die Standardabweichung betrug < 15%.

3. Ergebnisse 43
3.3. Spektroskopische Untersuchung des H-Clusters 3.3.1. EPR-Spektroskopie
3.3.1.1. Vergleichende Untersuchung der HydA-Proteine
Um erste Hinweise auf die Struktur und die vorliegenden Redoxzustände des H-Clusters der drei
isolierten Hydrogenasen zu erhalten, wurden diese EPR-spektroskopisch untersucht. Da nur die
paramagnetischen Zustände des H-Clusters, Hox-CO und Hox, durch EPR detektierbar sind,
wurden die HydA-Proteine sowohl in der isolierten Form als auch in der CO-inhibierten Form
untersucht. Das Ergebnis ist in Abb.3.10. dargestellt.
Der Hox-CO-Zustand aller drei HydA-Proteine zeigt im EPR-Spektrum das für das H-Cluster von
[FeFe]-Hydrogenasen in diesem Zustand charakteristische axiale EPR-Signal (Abb. 3.10. A, D,
F). Für die isolierte Form erhält man dagegen komplexe Spektren, die eine Mischform aus
rhombischem EPR-Signal, das der oxidierten Form des H-Clusters (Hox) zuzuordnen ist, und
axialem Signal zeigen (Abb. 3.10. B, E, F). Da die Proteine nach der Reinigung oftmals nicht in
homogener Form vorliegen, werden solche Mischsignale häufig beobachtet (Bennet et al. 2000).
Dass auch ein axiales Signal vorliegt, obwohl die isolierte Form nicht mit CO inhibiert wurde,
lässt sich dadurch begründen, dass CO-Liganden von zerstörten Clustern dissoziieren und als
freie CO-Moleküle wiederum intakte Enzyme inhibieren können („Kannibalisationseffekt“)
(Albracht et al. 2006). Dadurch liegt ein kleiner Teil der untersuchten Probe zusätzlich im
Hox-CO-Zustand vor, der im EPR-Spektrum das beschriebene axiale Signal zeigt. Eine Begasung
der isolierten Form aus C. reinhardtii mit Wasserstoff scheint wie auch für DdH aus
D. desulfuricans und CpI aus C. pasteurianum gezeigt (Albracht et al. 2006, Chen et al. 2002)
zum reduzierten Zustand des H-Clusters (Hred) zu führen, welcher wie erwartet diamagnetisch
und somit EPR-silent ist. Man erhält im Spektrum nur ein Rauschsignal (Abb. 3. 10. C).
Allerdings bleibt die Probe im Gegensatz zu DdH nach einer Begasung mit Argon zur
Wiederentfernung des H2 und Überführung in den oxidierten Zustand Hox in diesem Zustand, das
EPR-Signal ändert sich nicht.

3. Ergebnisse 44
EP
R A
mpl
itude
Magnetfeld [mT] Abb. 3.10. Q-Band Puls-EPR-Spektren der [FeFe]-Hydrogenase HydA1 aus C. reinhardtii (A, B, C),
HydA aus C. submarinum (D, E) und HydA1 aus C. moewusii (F)
A HydA1 C. reinhardtii Hox-CO-Zustand, 30 µM in 50 mM Tris-HCl pH 8,5, nach Begasung der isolierten Form
mit CO für 20 min
B HydA1 C. reinhardtii isolierte Form, 100 µM in 50 mM Tris-HCl pH 8,5
C HydA1 C. reinhardtii isolierte Form nach Begasung mit H2 für 20 min und Argon für 35 min
D HydA C. submarinum Hox-CO-Zustand, 100 µM in 50 mM Tris-HCl pH 7,5, 150 mM KCl, 2 mM NaDT,
nach Begasung der isolierten Form mit CO für 20 min
E HydA C. submarinum isolierte Form, 100 µM in 50 mM Tris-HCl pH 7,5, 150 mM KCl, 2 mM NaDT
F HydA1 C. moewusii Hox-CO-Zustand, 100 µM in 50 mM Tris-HCl pH 7,5, 150 mM KCl, 2 mM NaDT,
nach Begasung der isolierten Form mit CO für 20 min
Das Spektrum zeigt zusätzlich eine Simulation des Hox-CO-Zustands (blau) und des Hox-Zustands (rot)
Alle Messungen wurden bei einer Temperatur von 20 K durchgeführt. Über jedem Spektrum sind die dazugehörigen
Werte des g-Tensors angegeben (blau: g-Werte für Hox-CO, rot: g-Werte für Hox).

3. Ergebnisse 45
3.3.1.2. Der Hox-Zustand von HydA1 aus C. reinhardtii
Der Hox-Zustand ist im Gegensatz zum Hox-CO-Zustand direkt in den katalytischen Zyklus
eingebunden und spielt daher eine wichtige Rolle für die Bestimmung der Struktur des
H-Clusters. Den oxidierten Zustand von DdH erhält man durch Begasung der inaktiven oder
reduzierten Form mit Argon (Albracht et al. 2006). Dies trifft allerdings wie bereits gezeigt für
HydA1 nicht zu. Weitere Versuche, das Enzym biochemisch zu oxidieren, schlugen ebenfalls
fehl. Die Zugaben von Ferrocyanid (Razavet et al. 2002) und Thionin, welche erfolgreich als
Oxidationsmittel zur Bildung des Hox-Zustandes bei CpI verwendet wurden (Zambrano et al.
1989, Bennet et al. 2000), zerstörten das aktive Zentrum von HydA1. Auch der Verzicht auf
NaDT im letzten Reinigungsschritt führte nur zu einem partiell oxidierten Zustand. Daher wurde
versucht, den Hox-Zustand durch Photodissoziation des Hox-CO-Zustands zu erhalten. Die
Inhibierung von Hydrogenasen mit CO ist reversibel. Durch Einwirkung von Lichtimpulsen bei
kryogenen Temperaturen löst sich das CO wieder ab (Photodissoziation) und der Hox-CO-
Zustand wird in den aktiven Hox-Zustand überführt (Chen et al. 2002, Albracht et al. 2006). Bei
DdH und CpI kann während dieses Prozesses ein Zwischenzustand (LI2) detektiert werden, der
durch eine reversible, partielle Ablösung des CO-Brückenliganden charakterisiert ist, während
der externe, zu entfernende CO-Ligand noch gebunden ist (Chen et al. 2002, Roseboom et al.
2006).
Das Ergebnis der Photodissoziation des Hox-CO-Zustands von HydA1 ist in Abb.3.11. zu sehen.
Das Enzym wurde zunächst mit CO inhibiert und zeigt das für diesen Zustand charakteristische
axiale EPR-Signal (Abb.3.11. A). Durch die anschließende Belichtung löst sich das CO wieder
ab und das EPR-Signal wird rhombisch (Abb.3.11. B). Im Gegensatz zu den bereits bekannten
[FeFe]-Hydrogenasen konnte hierbei kein zusätzlicher Zwischenzustand nachgewiesen werden.
Inkubiert man die Probe nach der Belichtung für 10 min in 200 K kaltem Ethanol erfolgt wieder
eine vollständige Bindung des CO und man erhält wieder Spektrum A.
Tab. 3.4. Überblick über die ermittelten g-Werte
Spezies Hox-CO Hox
C. reinhardtii 2.052 2.007 2.007 2.102 2.040 1.998
C. moewusii 2.052 2.008 2.008 2.103 2.038 1.998
C. submarinum 2.056 2.008 2.008 2.100 2.040 1.998
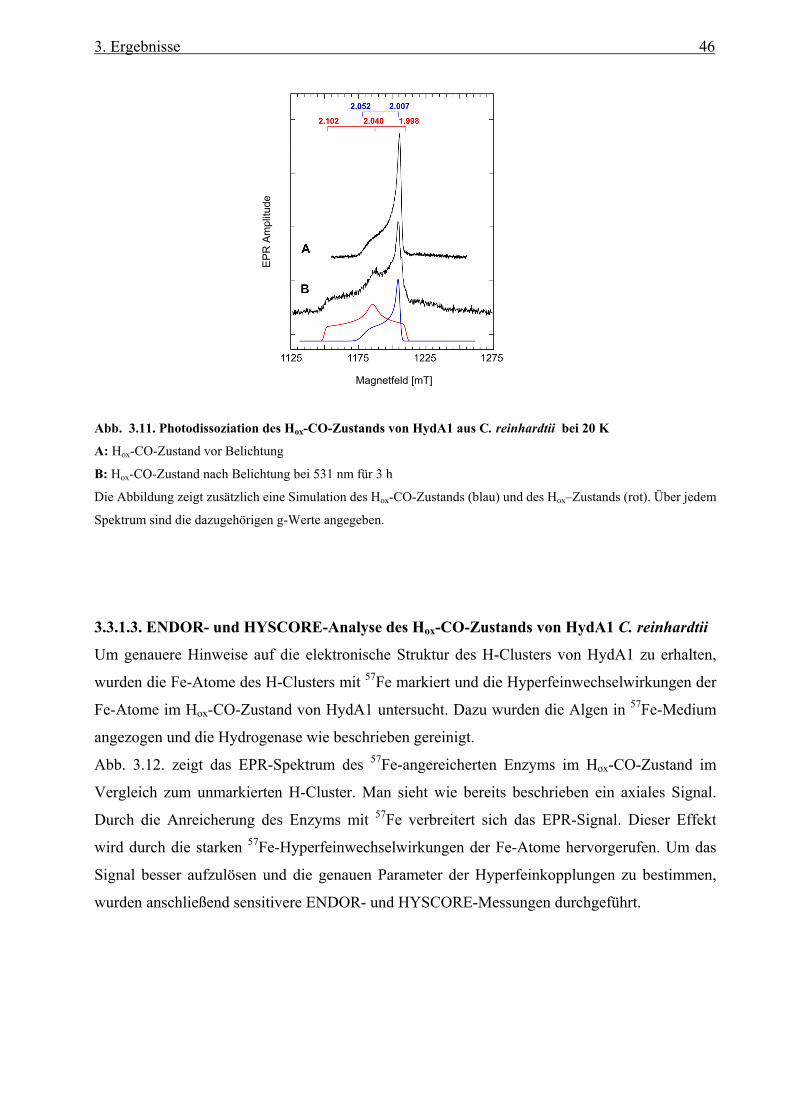
3. Ergebnisse 46
EP
R A
mpl
itude
Magnetfeld [mT] Abb. 3.11. Photodissoziation des Hox-CO-Zustands von HydA1 aus C. reinhardtii bei 20 K
A: Hox-CO-Zustand vor Belichtung
B: Hox-CO-Zustand nach Belichtung bei 531 nm für 3 h
Die Abbildung zeigt zusätzlich eine Simulation des Hox-CO-Zustands (blau) und des Hox–Zustands (rot). Über jedem
Spektrum sind die dazugehörigen g-Werte angegeben.
3.3.1.3. ENDOR- und HYSCORE-Analyse des Hox-CO-Zustands von HydA1 C. reinhardtii
Um genauere Hinweise auf die elektronische Struktur des H-Clusters von HydA1 zu erhalten,
wurden die Fe-Atome des H-Clusters mit 57Fe markiert und die Hyperfeinwechselwirkungen der
Fe-Atome im Hox-CO-Zustand von HydA1 untersucht. Dazu wurden die Algen in 57Fe-Medium
angezogen und die Hydrogenase wie beschrieben gereinigt.
Abb. 3.12. zeigt das EPR-Spektrum des 57Fe-angereicherten Enzyms im Hox-CO-Zustand im
Vergleich zum unmarkierten H-Cluster. Man sieht wie bereits beschrieben ein axiales Signal.
Durch die Anreicherung des Enzyms mit 57Fe verbreitert sich das EPR-Signal. Dieser Effekt
wird durch die starken 57Fe-Hyperfeinwechselwirkungen der Fe-Atome hervorgerufen. Um das
Signal besser aufzulösen und die genauen Parameter der Hyperfeinkopplungen zu bestimmen,
wurden anschließend sensitivere ENDOR- und HYSCORE-Messungen durchgeführt.

3. Ergebnisse 47
57Fe-markierte Probe
unmarkierte Probe
g-Wert Abb. 3.12. Q-Band FID-EPR-Spektrum des 57Fe-angereicherten Hox-CO-Zustands (blau) und des
unmarkierten Hox-CO-Zustands (rot) von HydA1
Die Spektren wurden bei einer Temperatur von 20 K, einer Frequenz von 33,80 GHz und tmw = 1 µs aufgenommen.
Die Zahlen 1-3 zeigen die Feldpositionen an, an denen zusätzlich ENDOR- und HYSCORE-Experimente
durchgeführt wurden.
In Abb. 3.13. ist das Q-Band-ENDOR-Spektrum des 57Fe-angereicherten H-Clusters zu sehen.
Im Falle einer starken Hyperfeinwechselwirkung (|A| > |2υ(57Fe)| ) erscheint das ENDOR-Signal
als Duplett (zentriert um |A/2|) und wird durch Verdopplung der Larmor-Frequenz νFe der 57Fe-Kerne (ca. 1,65 MHz bei 1200 mT) geteilt. Im Falle von HydA1 erkennt man im
niedrigeren Frequenzbereich (< 20 MHz) zwei Sets von Dupletts. Die Peaks in den Dupletts
werden durch die Larmor-Frequenz der 57Fe-Kerne geteilt, was zeigt, dass die detektierten
Signale eindeutig von den 57Fe-Kernen stammen. Basierend auf der Simulation, können vier 57Fe-Hyperfeinkopplungen (A1-A4) aufgelöst werden. Die genauen Parameter der
Hyperfeinkopplungen sind in Tabelle 3.5. aufgelistet. Die Spektren zeigen große Ähnlichkeit zu
den ENDOR-Spektren von DdH (Silakov et al. 2007). Daher sind die beobachteten Signale
vermutlich den 57Fe-Kernen des [4Fe-4S]-Clusters des H-Clusters zuzuordnen.

3. Ergebnisse 48
experimentelle Daten Simulation
Frequenz [MHz] Abb. 3.13. Q-Band Davies-ENDOR-Spektren des 57Fe-angereicherten Hox-CO-Zustands
Die Spektren wurden an verschiedenen Feldpositionen aufgenommen (s. Abb. 3.15.). Experimentelle Bedingungen:
Temperatur: 25 K, trf = 30 µs, Magnetfeldstärke: 1 1205,4 mT, 2 1194,0 mT, 3 1180,0 mT. Die Spektren in rot,
orange, grün und schwarz sind Simulationen der einzelnen 57Fe-Hyperfeinkopplungen A1, A2, A3, A4 des 57Fe-HF-
Tensors (s. Tab. 3.5.). Das violette Spektrum stellt die Summe dieser Simulationen dar. Die Sterne markieren
Messartefakte.
Um zusätzlich die Wechselwirkung schwach gekoppelter Kerne zu untersuchen, wurde die
HYSCORE-Technik angewendet, durch die auch der Frequenzbereich unter 5 MHz aufgelöst
werden kann. In einem HYSCORE-Spektrum erscheinen Signale, die zu schwachen
Hyperfeinkopplungen (|A| < |2υ(57Fe)|) gehören, im (++)- bzw. (--)-Quadranten, wohingegen
starke Hyperfeinwechselwirkungen (|A| > |2υ(57Fe)|) im (+-)- bzw. (-+)-Quadranten detektiert
werden. Abb. 3.14. zeigt die Q-Band HYSCORE-Spektren von HydA1. Um alle Parameter der
Hyperfeinkopplungen aufzulösen, wurden Spektren an verschiedenen Feldpositionen
aufgenommen. Es konnten zwei Sets von Signalen beobachtet werden, die den 57Fe-Kernen des
[2Fe]-Subclusters zugeordnet werden können. Ein Set von Crosspeaks (A5) erscheint im
(+-)-Quadranten des HYSCORE-Spektrums, was zeigt, dass eine starke Kopplung vorliegt
(|A'| > |2υ(57Fe)|). Die Form der Crosspeaks und die Tatsache, dass sie bei Verminderung des
Magnetfeldes breiter werden, lässt eine starke dipolare Verteilung vermuten. Simulationen dieser

3. Ergebnisse 49
Crosspeaks zeigen eine anisotrope 57Fe-Hyperfeinkopplung mit rhombischem Charakter. Ein
anderes Set von Peaks (A6) ist im (++)-Quadranten zu finden. Diese Signale gehören zu einem
anderen, schwach gekoppelten 57Fe-Kern (|A'| < |2υ(57Fe)|). Auch diese Signale sind stark von
der Feldposition abhängig, was eine dipolare Natur der 57Fe-Hyperfeinkopplung vermuten lässt.
Die genauen Parameter des 57Fe-Hyperfein-Tensors sind in Tab. 3.5. dargestellt.
Durch die Hyperfeinkopplungen A1-A6 konnten somit alle sechs Fe-Atome im H-Cluster
aufgelöst werden.
Freq
uenz
[MH
z]
Frequenz [MHz] Abb. 3.14. Q-Band HYSCORE-Spektren des 57Fe-angereicherten Hox-CO-Zustands
Die Spektren wurden an verschiedenen Feldpositionen aufgenommen (s. Abb. 3.15.). Experimentelle Bedingungen:
Temperatur: 25 K, Frequenz: 33,81 GHz, tmw(π/2) = 40 ns, Magnetfeldstärke: 1 1205,4 mT, 2 1194,0 mT, 3
1180,0 mT, Verzögerung zwischen den ersten beiden Pulsen (τ): 1 376 ns, 2 352 ns, 3 300 ns. Die schwarzen und
roten Linien repräsentieren die Frequenzen der Crosspeaks, basierend auf einer Simulation mittels der A5 und A6
Hyperfeinkopplungskonstanten.

3. Ergebnisse 50
ext
Abb. 3.15. Schematische Darstellung der Orientierung des 57Fe-Hyperfein-Tensors des H-Clusters im
Hox-CO-Zustand (Silakov et al. 2007)
Die roten Pfeile repräsentieren die Z-Achse des Hyperfein-Tensors, der blaue Pfeil die Ausrichtung des g-Tensors.
Alle sechs Fe-Atome weisen Spindichte auf, die durch elektronische Austausch-Wechselwirkung zwischen den Fe-
Atomen verursacht wird Die Hyperfeinwechselwirkungen des [4Fe-4S]-Clusters werden durch Austausch-
Wechselwirkung zwischen dem Fe1-Atoms des [4Fe-4S]-Clusters und dem Fep-Atom des [2Fe]-Subclusters
hervorgerufen. Bei Bindung von externem CO ist die Elektronenspindichte am Fep lokalisiert, dadurch ist die
Hyperfeinwechselwirkung am Fep-Atom stärker und am Fed-Atom schwächer.
Tab. 3.5. Überblick über die ermittelten Parameter der Hyperfeinwechselwirkungen der 57Fe-Kerne im
Hox-CO-Zustand
Alle Werte sind in [MHz] angegeben. Die Zuordnung erfolgte in Anlehnung an das für DdH erstellte
Spin-Kopplungs-Modell (Abb. 3.15.)
HFK Ax Ay Az Aiso Zuordnung
A1 28.5 25.0 29.2 27.6 [4Fe-4S]HA2 30.3 23.5 30.5 28.0 [4Fe-4S]H
Fe3-Fe4
A3 34.7 34.7 29.9 33.1 [4Fe-4S]HA4 34.5 37.7 30.9 34.3 [4Fe-4S]H
Fe1-Fe2
A5 5.7 2.9 0.4 3.0 [2Fe]H Fep
A6 3.0 3.0 -2.1 1.3 [2Fe]H Fed

3. Ergebnisse 51
3.3.2. FTIR-Spektroskopie
3.3.2.1. Photodissoziation des Hox-CO-Zustands von HydA1 aus C. reinhardtii bei
kryogenen Temperaturen
Da durch EPR-Spektroskopie nicht alle Redoxzustände des H-Clusters detektiert werden können,
wurde HydA1 aus C. reinhardtii zusätzlich FTIR-spektroskopisch untersucht. Bei einer FTIR-
spektroskopischen Untersuchung von [FeFe]-Hydrogenasen sind im Wellenzahlenbereich von
1800-2100 cm-1 verschiedene Absorptionsbanden zu sehen, die den CN- und CO-Liganden des
H-Clusters zugeordnet werden können.
Hierbei wurde zunächst wieder der für [FeFe]-Hydrogenasen charakteristische Hox-CO-Zustand
untersucht. Vergleicht man das von diesem Zustand aufgenommene FTIR-Spektrum (Abb. 3.16.
A) mit bekannten Spektren von DdH und CpI (Nicolet et al. 2001, Roseboom et al. 2006, Chen
et al. 2002), gehören die Banden bei 2090 cm-1 und 2083 cm-1 zu den CN-Liganden des
proximalen und distalen Fe-Atoms, das Bandenpaar 2013 cm-1 und 1964 cm-1 zu der gekoppelt
auftretenden Streckschwingung des externen und des intrinsischen CO-Liganden des distalen Fe-
Atoms und die Bande bei 1970 cm-1 zu der Schwingung des intrinsischen CO-Liganden des
proximalen Fe-Atoms. Die Bande im niedrigeren Frequenzbereich bei 1807 cm-1 ist dem CO-
Brückenliganden zuzuordnen.
Durch Belichtung bei kryogenen Temperaturen kann der Hox-CO-Zustand im Gegensatz zu den
bisherigen EPR-Experimenten vollständig in den aktiven oxidierten Zustand Hox überführt
werden, der das für diesen Redoxzustand charakteristische Bandenmuster zeigt (Abb. 3.16. D).
Dabei tritt nach 2 min Belichtung ein Zwischenzustand auf. Ein solcher Zwischenzustand
während der Photodissoziation ist wie bereits erwähnt auch für andere [FeFe]-Hydrogenasen
beschrieben und durch eine reversible, partielle Ablösung des CO-Brückenliganden
charakterisiert (Pierik et al. 1998, Roseboom et al. 2006) (Abb. 3.16. B und C). Im
Differenzspektrum (Abb. 3.16. C) erscheinen sechs neue Banden (gelb) zusätzlich zu einer
Bande des Hox-Zustands (blau). Im Gegensatz zum Hox-CO-Zustand sind die CN-Banden stark
zu niedrigeren Wellenzahlen verschoben, was zeigt, dass sich die Geometrie des dinuklearen
Clusters während der Photodissoziation stark ändert. Dies wird wahrscheinlich durch eine
Änderung der Bindung des CO-Brückenliganden verursacht, allerdings zeigt die Bande bei
1817 cm-1, dass dieser nicht dissoziiert ist. Die Bestimmung von vier CO-Streckschwingungen
lässt zudem den Schluss zu, dass im LI2-Zustand dieselbe Anzahl CO-Liganden vorhanden ist
wie im CO-inhibierten Zustand. Daher ist anzunehmen, dass der LI2-Zustand eine Art

3. Ergebnisse 52
Hox-CO-Zustand mit veränderter Geometrie darstellt und aufgrund dessen der Übergang
Hox-CO/LI2 so schnell stattfindet, dass er durch EPR-Spektroskopie nicht nachweisbar war.
Im Hox-Zustand (Abb. 3.16. D) sind drei Banden im Bereich der CN-Liganden, zwei Banden im
Bereich der terminalen CO-Liganden und eine Bande im Bereich der CO-Brücke zu sehen, was
zeigt, dass eine Ablösung des externen, inhibierenden CO-Liganden stattgefunden hat. Da sich
die Koordination des proximalen Fe-Atoms bei der Dissoziation des externen CO-Liganden vom
distalen Fe-Atom nicht ändert, dürfte die Bande bei 2091 cm-1 wie im Hox-CO-Zustand dem
proximalen CN-Liganden und die Bande bei 1966 cm-1 trotz leichter Verschiebung dem
proximalen CO-Liganden zuzuordnen sein. Die Bande bei 1940 cm-1 ist demnach die
Schwingung des distalen CO-Liganden. Für CNd könnten zwei Banden, bei 2079 cm-1 und
2073 cm-1, in Frage kommen. Da die Bande des distalen CO-Liganden eine kleine Schulter in
Richtung 1935 cm-1 aufweist, könnte die Hypothese aufgestellt werden, dass zwei verschiedene
Konformationen des distalen Fe-Atoms vorliegen, was dazu führt, dass sich die CN und CO-
Liganden aufsplitten. Da dies aber bei der EPR-spektroskopischen Untersuchung der
Hydrogenase nicht nachweisbar war, ist zu vermuten, dass diese Konformationsabweichung nur
geringen oder keinen Einfluss auf die Gesamtstruktur des H-Clusters hat.
Im Hox-CO-Zustand scheint die Schwingung des COp wie bei DdH vermutet mit der Schwingung
COd/COext gekoppelt zu sein, da sich die Bande von 1970 cm-1 zu 1966 cm-1 verschoben hat. Ob
dies auch im Hox-Zustand der Fall ist, ist im Spektrum nicht sichtbar. Eine Bestätigung der
richtigen Zuordnung der Absorptionsbanden und Aufschluss über eine solche mögliche
Kopplung kann durch eine isotopische Markierung der CO-Liganden mit 13C gegeben werden,
die im nachfolgenden Versuch gezeigt ist.
3.3.2.2. Isotopenmarkierung der CO-Liganden des H-Clusters mit 13C
Für eine Markierung mit 13C wurde die isolierte Form statt mit CO mit 13CO begast (Abb. 3.17.,
1B). Da durch eine Begasung mit CO der Hox-CO-Zustand entsteht, erwartet man in diesem Fall
die Bildung eines Hox-CO-Zustands, bei dem der externe CO-Ligand markiert ist (Hox-13CO).
Aufgrund der Isotopenmarkierung verschieben sich die CO-Banden im FTIR-Spektrum im
Gegensatz zum normalen Hox-CO-Zustand (Abb. 3.17., 1A) in Abhängigkeit von der reduzierten
Masse des 13CO. Der erwartete Bandenshift lässt sich durch die Teller-Redlich-Produktregel
berechnen:

3. Ergebnisse 53
Abb. 3.16. Photodissoziation des Hox-CO-Zustands von HydA1 aus C. reinhardtii bei 40 K
A Hox-CO-Zustand, 300 µM in 50 mM Tris-HCl pH 7,5, 150 mM KCl, 2 mM NaDT nach Begasung der
isolierten Form mit CO für 20 min
B Hox-CO-Zustand nach Belichtung innerhalb des FTIR-Setups für 2 min (LI2)
C Differenzspektrum A/B: gelb: Banden des Zwischenzustands LI2, blau: Banden des Hox-Zustands
D Hox-CO-Zustand nach Belichtung für 1 h (Hox), nach Inkubation bei 150 K im Dunkeln wird das abgelöste CO
wieder gebunden und man erhält wieder Spektrum A
Π ωi*
= µ n/2 ωi und ωi
*: Schwingungsfrequenz der nicht markierten und n-Mal markierten Moleküle i ωi
µ* µ und µ*: reduzierte Masse der nicht markierten und markierten CO-Gruppen
Für einen einzelnen 12CO/13CO-Austausch (n = 1) kann man den rechten Teil der Gleichung wie
folgt auflösen:
µ 1/2 = 1/16 + 1/13 1/2 = 0,9777
µ* 1/16 + 1/12
Wellenzahl [cm-1]
Da die Bande 2013 cm-1 zu 1992 cm-1 shiftet, kann der Shift der anderen Bande (1964 cm-1)
berechnet werden, da die Schwingung gekoppelt ist:

3. Ergebnisse 54
Π ωi*
= 1992 * x = 0,9777 => x = 1940,6 cm-1
i ωi 2013 * 1964
Im Spektrum 1B ist eine Bande bei 1944 cm-1 zu sehen, was nahezu mit dem berechneten Wert
übereinstimmt. Es handelt sich also eindeutig um die gekoppelte Schwingung COd/COext. Die
Bande bei 1967 cm-1 ist somit wieder dem proximalen CO-Liganden zuzuordnen. Allerdings ist
diese ebenfalls im Gegensatz zum Hox-CO-Zustand um - 3 cm-1 verschoben, was zeigt, dass
diese Schwingung wie schon vermutet nicht unabhängig von der COd/COext-Schwingung ist.
Bezieht man diesen Umstand in die Berechnung mit ein, erhält man für die COd/COext-
Schwingung einen Wert, der exakt mit dem Messwert im Spektrum übereinstimmt:
Π ωi*
= 1992 * 1967 * x = 0,9777 => x = 1943,8 cm-1
i ωi 2013 * 1970 * 1964
Zusätzlich zu den bisher beschriebenen, erwarteten Banden des Hox-13CO-Zustands sind zwei
weitere Banden im Bereich der terminalen CO-Liganden zu sehen (1923 cm-1 und 1985 cm-1),
die nicht eindeutig zugeordnet werden können. Eine mögliche Kontamination mit 13C18O durch
das verwendete 13CO kann ausgeschlossen werden, da eine Markierung der CO-Liganden mit 13C18O zu einer anderen Verschiebung der Banden als im Spektrum zu sehen führen würde.
Der Hox-13CO-Zustand wurde anschließend 1 h bei 200 K belichtet. Der resultierende Zustand ist
der Hox-(13CO)3-Zustand (Abb. 3.17., 1C). Neben den verbliebenen Banden des Hox-13CO-
Zustands erscheinen im Spektrum zwei neue Banden bei 1969 cm-1 und 1928/1920 cm-1, die im
Vergleich zu den entsprechenden Banden des Hox-13CO-Zustands um - 21 cm-1 und zu denen des
Hox-CO-Zustands um - 42 cm-1 verschoben sind. Das bedeutet, dass sowohl COext als auch COd
isotopisch markiert sind. Zusätzlich erkennt man auch Banden des Hox-CO-Zustands (2013 cm-1
und 1964 cm-1). Dies lässt sich dadurch erklären, dass bei einer Belichtung bei 200 K
CO-Liganden ausgetauscht werden können (Roseboom et al. 2006). Wie beschrieben ist die
Schwingung von COd und COext gekoppelt und die von CObr unabhängig. Werden 13COext und
CObr ausgetauscht, würden die Banden der Schwingung COd/COext wieder bei derselben
Wellenzahl detektiert werden wie beim Hox-CO-Zustand, da als externer Ligand wieder
unmarkiertes CO vorliegt. Der CO-Brückenligand dagegen müsste aufgrund der
Isotopenmarkierung in die Region < 1800 cm-1 shiften, was allerdings nicht zu sehen ist, da die
verwendete Küvette eine Nachweisgrenze von 1800 cm-1 hat. Ebenso möglich wäre eine
zusätzliche Markierung des proximalen CO-Liganden mit 13C, was eine daraus resultierende
Bande bei 1926 cm-1 zur Folge hätte. Dies wird allerdings im Spektrum nicht vollständig

3. Ergebnisse 55
aufgelöst. Es ist daher zu vermuten, dass bei einer Belichtung des Hox-13CO-Zustands bei 200 K
ein Austausch der CO-Liganden stattfindet und so alle CO-Liganden mit 13C markiert werden.
Dies wird im weiteren Verlauf des Experiments durch Belichtung des Hox-(13CO)3-Zustands für
1 h bei 40 K bestätigt (Abb. 3.17., 2C).
Eine Belichtung des Hox-13CO-Zustands bei 40 K sollte wie auch in Abb. 3.16. bei Hox-CO
gezeigt zu einer Ablösung des externen 13CO führen. Der resultierende Zustand wäre wiederum
Hox (Abb. 3.17., 2A und B). Dies konnte auch durch die Messung bestätigt werden. Allerdings
sind zusätzlich zu den Banden des Hox-Zustands zwei weitere Banden bei 1955 cm-1 und
1909 cm-1 zu sehen, die sich durch partielles Labelling der CO-Liganden aufgrund des
Ligandenaustauschs während der Belichtung, wie schon bei 200 K gezeigt, erklären lassen. Eine
Markierung von CObr würde die entsprechende Bande von ca. 1800 cm-1 zu 1768 cm-1
verschieben ohne die anderen Schwingungen zu beeinflussen. Da diese aber ebenfalls
verschoben sind, sind vermutlich COd und/oder COp isotopisch markiert. Wären die beiden
Schwingungen unabhängig voneinander, würde die Markierung eines der beiden Liganden die
zugehörige Bande um - 42 cm-1 verschieben. Da aber nur ein Shift von ca. - 21 cm-1 zu sehen ist,
müssen die Schwingungen gekoppelt sein. Durch die Produktregel kann der Shift der einen
Bande berechnet werden, wenn der Shift der anderen bekannt ist. Unter der Annahme, dass sich
die Bande bei 1940 cm-1 durch eine 13C-Markierung des CO-Liganden zu einer Frequenz von
1909 cm-1 verschiebt, verschiebt sich die Bande bei 1966 cm-1 bei einer Kopplung der
Schwingung zu 1953,5 cm-1, was im Spektrum zu sehen ist.
Π ωi*
= 1909 * x = 0,9777 => x = 1953,5 cm-1
i ωi 1940 * 1966
Es liegt somit eine Mischung aus Hox und einem Hox-Zustand vor, bei dem COp oder COd 13C
markiert ist, wobei eine 13COd-Markierung wahrscheinlicher ist, da dies auch schon beim
Hox-(13CO)3-Zustand der Fall war. Nach Erwärmen der Probe auf 200 K und Inkubation für1 h
im Dunkeln bindet das 13CO wieder und man erhält den Ausgangszustand Hox-13CO, was zeigt,
dass alle detektierten Absorptionsbanden zu intaktem Cluster gehören.
Das Spektrum, das man nach Belichtung des Hox-(13CO)3 erhält (Abb.3.17., 2C) ist ebenfalls eine
Art Hox-Zustand, zeigt aber einige Unterschiede. Es sind Banden des nicht markierten Hox sowie
des markierten vorhanden (vgl. Abb.3.17., 2A und B). Zusätzlich erscheinen jedoch zwei weitere
Banden bei 1923 cm-1 und 1896 cm-1. Beide sind ca. - 43 cm-1 vom Bandenpaar 1966/1940 cm-1
entfernt, was bedeutet, dass beide terminalen CO-Liganden 13C markiert sind. Dies wird, unter

3. Ergebnisse 56
der Annahme, dass sich die Bande bei 1940 cm-1 aufgrund des Doppellabellings (13COp/ 13COd,
daher n=2) zu 1896 cm-1 verschiebt, durch die Kalkulation
Π ωi*
= 1896 * x = 0,97772 => x = 1923 cm
-1
i ωi 1940 * 1966
bestätigt. Das Aufwärmen der Probe auf 200 K und eine einstündige Inkubation im Dunkeln
führt anschließend wieder zum Ausgangszustand Hox-(13CO)3.
3.3.2.3. Untersuchung der reduzierten Zustände des H-Clusters
Wie in Abb. 3.18. B zu sehen liegt die Hydrogenase nach der Isolierung in einem Gemisch
oxidiert/reduziert vor. Das Spektrum zeigt Banden des Hox-Zustands (1966 cm-1 und 1940 cm-1)
und Banden, die aufgrund der Ähnlichkeit zu DdH in dieser Form vermutlich dem reduzierten
Zustand Hred zuzuordnen sind, wobei die Bande bei 1888 cm-1 die Schwingung des CObr, die bei
1920 cm-1 die von COd und die bei 1965 cm-1 die von COp darstellen könnte. Die Zuordnung der
CN-Liganden ist schwieriger. Bei DdH ist die Bande im Frequenzbereich von 2041 cm-1 dem
CN-Liganden des distalen Fe-Atoms zuzuordnen, welche sich bei Reduktion der Probe von
2082 cm-1 zu 2041 cm-1 verschiebt, da sich die Oxidationsstufe des distalen Fe-Atoms ändert. In
diesem Spektrum ist eine Bande bei 2028 cm-1 zu sehen, die dieser Schwingung zugeordnet
werden könnte. Eine weitere auffällige Bande ist im Bereich 1931 cm-1 zu sehen, kann aber nicht
zugeordnet werden. Der detektierte reduzierte Zustand ist EPR-silent und hat vermutlich die
Fe-Konfiguration Fe(I)-Fe(I). Im Gegensatz zu den zuvor durchgeführten EPR-Messungen sind
im Spektrum keine auffälligen Banden des Hox-CO-Zustands zu sehen, daher liegt dieser Zustand
vermutlich in so geringem Anteil vor, dass er nicht nachweisbar ist.
Bei DdH führt eine Begasung mit H2 zu einem einzigen klaren reduzierten Zustand (Roseboom
et al. 2006). Die reduzierte, H2-begaste Probe von HydA1 dagegen zeigt ein kompliziertes
Spektrum, in dem insbesondere im Bereich der Schwingungen der CO-Liganden doppelt so viele
Banden vorhanden sind als für einen Einzelzustand zu erwarten wäre (Abb. 3.18. C). Außerdem
sind die Banden im Gegensatz zum in der isolierten Probe detektierten Hred leicht verschoben.
Auch die Bande bei 2028 cm-1 scheint sich in zwei Banden, 2032 cm-1 und 2026 cm-1,
aufzusplitten. Daher sind im reduzierten Zustand höchstwahrscheinlich zwei untereinander und
im Vergleich zu DdH und CpI recht ähnliche Zwischenzustände vorhanden, die durch den

3. Ergebnisse 57
1
Wellenzahl [cm-1]
2
Wellenzahl [cm-1]
Abb. 3.17. Isotopenmarkierung der CO-Liganden mit 13C
1: A Hox-CO-Zustand, 300 µM, nach Begasung der isolierten Form mit CO für 20 min
B Hox-13CO-Zustand, 300 µM, nach Begasung der isolierten Form mit 13CO für 20 min
C Hox-13CO-Zustand nach Belichtung für 1 h bei 200 K (Hox-(13CO)3)
2: A Hox-Zustand, nach Belichtung des Hox-CO-Zustands für 1 h bei 40 K
B Hox-13CO-Zustand nach Belichtung für 1 h bei 40 K
C Belichtung von Hox-(13CO)3 für 1 h bei 40 K
Alle Spektren wurden bei einer Temperatur von 40 K aufgenommen

3. Ergebnisse 58
katalytischen Zyklus im H-Cluster zu begründen sind und wahrscheinlich, da sie mittels EPR
nicht nachweisbar sind, wie DdH und CpI die Fe-Konfiguration Fe(I)-Fe(I) besitzen.
Ein weiterer Versuch, einen klaren Einzelzustand Hred zu erhalten, war die Zugabe von NaDT zu
der isolierten Probe (Abb. 3.18. D). Dies resultierte in einem Spektrum, in dem ungefähr neun
Banden in der Region unter 2000 cm-1 zu sehen sind, die kaum Ähnlichkeit mit den Banden der
vorigen Spektren aufweisen und daher nicht interpretiert werden können. Für einen
Einzelzustand wäre die maximale Anzahl an Banden im Bereich der CO-Schwingungen gleich
der Anzahl an CO-Liganden am dinuklearen Cluster, also für den Hox- und Hred-Zustand drei und
für den Hox-CO-Zustand vier Banden. Daher ist zu vermuten, dass hier drei verschiedene
Zustände vorliegen, unter anderem vielleicht eine Art superreduzierter Zustand wie bei
Roseboom et al. (2006) beschrieben.
Alle Proben lassen sich durch Begasung mit CO vollständig in den Hox-CO-Zustand überführen,
was zeigt, dass alle detektierten Banden und Zwischenformen zu intaktem Cluster gehören.
Wellenzahl [cm-1]
Abb. 3.18. FTIR-Spektren der isolierten und reduzierten Form von HydA1 aus C. reinhardtii A Hox-Zustand, nach Belichtung von Hox-CO für 1 h
B isolierte Form, 300 µM in 50 mM Tris-HCl pH 7,5, 150 mM KCl, 2 mM NaDT
C isolierte Form nach Begasung mit H2 für 20 min
D isolierte Form nach Zugabe von 40 mM NaDT
Alle Spektren wurden bei einer Temperatur von 40 K aufgenommen.

3. Ergebnisse 59
3.3.2.4. Spektroelektrochemie an HydA1 aus C. reinhardtii
Um die einzelnen Redoxzustände des H-Clusters, insbesondere die reduzierten Zustände,
genauer aufzulösen, wurde HydA1 im nachfolgenden Versuch spektroelektrochemisch
untersucht. Dazu wurden in einem Potentialbereich von - 510 mV bis 100 mV FTIR-Spektren
aufgenommen und die spektrale Veränderung des H-Clusters während der Redoxübergänge in
Abhängigkeit von der angelegten Spannung beobachtet. Das Ergebnis ist in den folgenden
Abbildungen (3.19. und 3.20.) dargestellt.
Bei Anlegen eines Potentials von - 260 mV befindet sich das H-Cluster im oxidierten Zustand
Hox, zusätzlich ist noch eine Bande des Hox-CO-Zustands zu sehen (2013 cm-1) (Abb. 3.20. B).
Erniedrigt man das Potential, verschwinden die Hox-Banden und man erhält den reduzierten
Zustand Hred1 mit den Banden 1935 cm-1, 1890 cm-1 und 1793 cm-1. Die Banden, die den CN-
Liganden zuzuordnen sind, werden im Spektrum nicht gut aufgelöst, befinden sich nach
Auswertung des Differenzspektrums aber bei ca. 2083 cm-1 und 2054 cm-1. Der Shift eines der
CN-Liganden zu 2054 cm-1 verdeutlicht, dass beim Übergang Hox/Hred1 eines der beiden
Fe-Atome reduziert wurde. Eine Reduktion der Fe-Atome führt zu einer Elektronenanreicherung
dieser. Dadurch verstärkt sich die Fe-CO-Bindung und die entsprechende
CO-CN-Schwingungsfrequenz wird kleiner. Dieser Effekt kann für alle Banden im Falle des
Hox/Hred1-Übergangs beobachtet werden.
Bei weiterer Erniedrigung des Potentials auf - 510 mV verschwinden die Banden des Hred1 und
neue (2030 cm-1, 2008 cm-1, 1955 cm-1, 1920 cm-1 und 1883 cm-1) erscheinen, die einem zweiten
reduzierten Zustand Hred2 zugeordnet werden. Da im Frequenzbereich 1800 cm-1 keine Bande zu
sehen ist, dafür aber bei 1955 cm-1 eine erscheint, ist zu vermuten, dass der CO-Brückenligand
dissoziiert und im Hred2-Zustand terminal am Fe-Atom gebunden vorliegt. Die Banden bei
2030 cm-1 und 2008 cm-1 gehören vermutlich wieder zu den CN-Liganden. Dass diese im
Vergleich zu Hred1 noch weiter verschoben sind und auch die Banden der beiden anderen
CO-Liganden um ca. - 10 cm-1 verschoben sind, untermauert die Annahme, dass eine
Konformationsänderung des CO-Brückenliganden stattgefunden hat. Die Banden des Hox-CO-
Zustands nehmen während der Reduktion gleichermaßen mit den Banden des Hox-Zustands ab.
Im Anschluss an die Reduktion wurde das Potential erhöht und die Probe wieder oxidiert, wobei
das exakte Erscheinen und Verschwinden von Banden wie bei der Reduktion in umgekehrter
Reihenfolge beobachtet werden konnte.
Bei einem Potential von - 100 mV verschwinden die Banden des Hox-Zustands und andere
erscheinen. Dieser Zustand könnte eine Art überoxidierter Zustand mit der Fe-Konfiguration
Fe(II)-Fe(II) sein, in dem das Enzym inaktiv vorliegt.

3. Ergebnisse 60
A
B
Abb. 3.19. Dreidimensionale Darstellung der Änderung der Absorptionsbanden während der
elektrochemischen Reduktion (A) und Oxidation (B) des H-Clusters von HydA1
A elektrochemische Reduktion in 25-50 mV-Schritten ausgehend von einem Initialpotential von - 260 mV
bei 4°C und pH 7,5 (1000 Scans)
B elektrochemische Oxidation in 25-50 mV-Schritten ausgehend von einem Initialpotential von - 510 mV
bei 4°C und pH 7,5 (2000 Scans)

3. Ergebnisse 61
Abb. 3.20. Ausgewählte FTIR-Spektren bei verschiedenen Potentialen während der elektrochemischen
Reduktion und Oxidation des H-Clusters von HydA1
A überoxidierter Zustand Hsox und Hox-CO-Zustand (die Banden des Hox-CO-Zustands sind hervorgehoben)
B Hox-Zustand
C reduzierter Zustand Hred1
D reduzierter Zustand Hred2
Die Probe wurde zunächst ausgehend von einem Initialpotential von - 260 mV reduziert (blau) und anschließend
wieder oxidiert (rot). Alle Spektren wurden bei einer Temperatur von 4°C aufgenommen. Die Konzentration der
eingesetzten Probe betrug ca. 200 µM in 50 mM Tris HCl pH 7,5, 150 mM KCl, 2mM NaDT.
Die Intensität der Hred2-Banden war während der Oxidation genauso stark wie bei der Reduktion.
Die Signale von Hred1 und Hox dagegen waren etwas kleiner und die Signale des
Hox-CO-Zustands nahmen, gerade auch beim überoxidierten Zustand, weiter zu. Dies zeigt, dass
Teile der Probe während des Experiments durch die Behandlung zerstört werden
(„Kannibalisationseffekt“, Albracht et al. 2006) und könnte zudem ein Zeichen dafür sein, dass
der vollständig reduzierte Zustand Hred2 bei 4°C stabiler ist als Hred1 und Hox.
Im Anschluss an die potentiometrische Titration wurden durch Analyse und Auswertung der
Intensität der für den jeweiligen Zustand charakteristischen CO-Banden die
Mittelpunktspotentiale für den Übergang von einem Redoxzustand in den nächsten ermittelt.

3. Ergebnisse 62
Dabei wurde für den Übergang Hox/Hred1 ein Redoxpotential von - 400 mV und für den
Übergang Hred1/Hred2 ein Potential von - 455 mV bestimmt (Abb. 3.21.).
Abb. 3.21. Intensität charakteristischer CO-Banden bei der elektrochemischen Reduktion und Oxidation
Die Änderungen in der Intensität der Absorptionsbanden während der potentiometrischen Reduktion bzw. Oxidation
bei 4°C und pH 7,5 wurden mit Hilfe der Nernst-Gleichung für einen Elektronenübergang (n=1) ausgewertet. Dabei
wurde für den Übergang Hox/Hred1 ein Mittelpunktspotential von - 400 mV(vs. NHE) und für Hred1/Hred2 ein Potential
von - 455 mV bestimmt. Bei beiden Richtungen der Titration wurden die gleichen Potentiale ermittelt. Für den
Übergang Hox/Hsox konnte kein Redoxpotential bestimmt werden, da die Intensität der Banden für den Hsox-Zustand
nicht der Nernst-Gleichung folgte. Die leeren Kreise stellen die Bandenintensität bei der Oxidation dar, ausfüllte
Kreise die Reduktion.
Tab. 3.6. Überblick über die detektierten FTIR-Banden
Alle Werte sind in [cm-1] angegeben.
Zustand CNp CNd COp COd - COext CObr Hsox 2106 2093 2047 2033 1910 Hox 2091 2076 1966 1940 1799 Hox-CO 2090 2083 1970 2013 1964 1810 Hred1 2083 2054 1935 1890 1793 Hred2 2030 2008 1920 1883 1955 Hox-13CO 2090 2083 1967 1992 1944 1810
Hox-(13CO)3 2090 2083 1964 1967 1970
1969 19281992 1944 2013 1964
1810

4. Diskussion 63
4. Diskussion Primäres Ziel der Arbeit war die Expression, Reinigung und strukturelle Charakterisierung der
[FeFe]-Hydrogenase HydA1 aus C. reinhardtii. Dazu sollte zunächst ein neues Expressions- und
Reinigungssystem für HydA1 etabliert und anschließend eine erste strukturelle Untersuchung
des katalytisch aktiven Zentrums mittels EPR- und FTIR-Spektroskopie durchgeführt werden.
Vergleichend zu HydA1 aus C. reinhardtii wurden dabei zusätzlich die Hydrogenasen HydA1
aus C. moewusii und HydA aus C. submarinum untersucht.
4.1. Expression und Reinigung der [FeFe]-Hydrogenasen aus C. reinhardtii, C. moewusii
und C. submarinum
4.1.1. Verbesserte Expression der HydA-Proteine unter Schwefelmangel
Das in früheren Arbeiten verwendete Verfahren der anaeroben Adaptation zur Induktion der
Hydrogenase-Expression führte zu vergleichsweise niedrigen Expressionsraten und
infolgedessen zu geringen Proteinausbeuten. Daher war eine biochemische Charakterisierung
von Grünalgen-Hydrogenasen nur partiell und eine strukturelle Untersuchung bisher nicht
möglich (Happe et al. 1993, Ueno et al. 1999, Florin et al. 2001, Winkler et al. 2002). Eine
heterologe Expression von Grünalgen-Hydrogenasen in Escherichia coli und Clostridium
acetobutylicum führte zu inaktivem Apoprotein oder zu Protein mit, im Gegensatz zu homolog
exprimierter Hydrogenase, geringerer spezifischer Aktivität (Girbal et al. 2005, King et al.
2006). Daher sollte HydA1 im Hinblick auf die strukturelle Untersuchung des H-Clusters in
nativer Form direkt aus C. reinhardtii isoliert werden.
Zur Verbesserung der Expressionsrate von HydA1 wurde in dieser Arbeit ein neues System, bei
dem Schwefelmangel zur Induktion der Hydrogenase-Expression verwendet wird (Melis et al.
2000, Laurinavichene et al. 2002), angewandt und so modifiziert, dass eine direkte Kultivierung
der Algen in selbstentschwefelndem Medium möglich war, wodurch die Gefahr der
Kontaminationen beim Mediumswechsel, die benötigte Mediumsmenge und der Zeitaufwand
verringert werden konnten (3.1.1.). Die Expressionsrate von HydA1 aus C. reinhardtii konnte
dadurch im Vergleich zu einer anaerob induzierten Kultur erheblich gesteigert (Abb. 3.2.) und
die maximal messbare in vitro-Hydrogenaseaktivität um das 25-fache erhöht werden (Abb. 3.1.).

4. Diskussion 64
Zusätzlich war es dadurch möglich, erstmals auch die Hydrogenasen aus den Grünalgenstämmen
C. moewusii und C. submarinum in vergleichbar großer Menge zu exprimieren (Abb. 3.1.).
4.1.2. Das neue Reinigungsprotokoll für HydA1
Aufbauend auf das verbesserte Expressionssystem wurde ein neues Reinigungsverfahren für
HydA1 aus C. reinhardtii etabliert, das weitestgehend auf die Hydrogenasen von C. moewusii
und C. submarinum übertragen werden konnte und somit universell für die Isolierung von
[FeFe]-Hydrogenasen aus Grünalgen ist (3.1.2. und 3.1.3.). Das entwickelte Reinigungsprotokoll
besteht aus einer Ammoniumsulfatfällung gefolgt von drei aufeinanderfolgenden
säulenchromatographischen Trennschritten (Abb. 3.3.) und ist damit im Gegensatz zu bisher
beschriebenen, recht komplizierten Reinigungsvorschriften (Happe et al. 1993, Ueno et al. 1999)
bedeutend kürzer und effektiver. Aus 8 l Schwefelmangelkultur konnten durchschnittlich 500 µg
Hydrogenase mit hoher spezifischer Aktivität, im Falle von HydA1 aus C. moewusii mit
1598 µmol H2 min-1 mg-1 Protein sogar doppelt so hoch wie die von HydA1 aus C. reinhardtii,
isoliert werden (Tab. 3.1., 3.2. und 3.3.). Dies ist eine 40-fach höhere Proteinausbeute im
Vergleich zu früheren Arbeiten (Happe et al. 1993) und nur etwa 50% weniger, als durch die
Überexpression von mit einem StrepTag erweiterten HydA1 in C. acetobutylicum (Girbal et al.
2005) gewonnen werden konnte.
4.2. Die hohe Toleranz der HydA-Proteine gegenüber biochemischen Einflüssen
Durch die in dieser Arbeit isolierten Proteinmengen war es erstmals möglich, die
[FeFe]-Hydrogenasen aus C. reinhardtii, C. moewusii und C. submarinum sowohl biochemisch
als auch strukturell zu charakterisieren. Dabei zeigte sich, dass alle drei HydA-Proteine eine
große Toleranz gegenüber biochemischen Einflüssen wie Temperatur, pH-Wert oder hoher
Ionenstärke zeigen (3.2.).
Alle drei Enzyme haben ein Temperaturoptimum von etwa 60°C und sind in einem pH-Bereich
von pH 5,5-9,0 mit einem Optimum bei pH 7 aktiv. Dabei sind für die Hydrogenasen aus
C. reinhardtii und C. moewusii im gesamten pH-Bereich vergleichbare Enzymaktivitäten
messbar, HydA aus C. submarinum scheint dagegen etwas aktiver im sauren und etwas weniger
aktiv im basischen Bereich zu sein (Abb. 3.7.). Ähnliches konnte auch in früheren Arbeiten für

4. Diskussion 65
die Hydrogenasen aus Chlorococcum littorale (Ueno et al. 1999), Chlorella fusca (Winkler et al.
2002) Scenedesmus obliquus (Florin et al. 2001) und CpI aus C. pasteurianum (Adams und
Mortenson 1984) gezeigt werden.
Zusätzlich wurde, wie auch für bakterielle [FeFe]-Hydrogenasen beschrieben (van Dijk et al.
1980), festgestellt, dass hohe Salzkonzentrationen einen positiven Effekt auf die Enzymaktivität
haben, wenn reduziertes Methylviologen als Elektronendonor verwendet wird (3.2.3.). Bei der
Untersuchung des Einflusses der Ionenstärke auf die Enzymaktivität wurde für die
Hydrogenasen aus C. reinhardtii und C. moewusii ein Aktivitätsmaximum bei einer
Konzentration von 1 M KCl und für C. submarinum von 1,5 M KCl im Reaktionspuffer
bestimmt. Die untersuchten HydA-Proteine sind somit sehr tolerant in Bezug auf hohe
Ionenstärken, allerdings verringert sich die Aktivität in Gegenwart des natürlichen
Elektronendonors Ferredoxin, da die hohe Salzkonzentration vermutlich die elektrostatischen
Wechselwirkungen zwischen Hydrogenase und Ferredoxin stört (Roessler und Lien 1982).
Die allgemein ungewöhnlich hohe Toleranz von Grünalgen-Hydrogenasen gegenüber
biochemischen Einflüssen untermauert ihre entscheidende Rolle als Stressprotein bei der
Umstellung des Stoffwechsels der Alge auf einen photofermentativen Wasserstoffmetabolismus
unter Anaerobiose. C. submarinum kann im Gegensatz zu C. reinhardtii und C. moewusii in
Medien mit hohen Salzkonzentrationen kultiviert werden (Blackwell und Gilmour 1991), was
das Risiko von Kontaminationen vermindert. Die höhere Salztoleranz von HydA könnte ebenso
wie die hohe pH-Toleranz durch den natürlichen Lebensraum der Alge begründet sein.
Brackwasser unterliegt im Vergleich zu reinen Süß- und Salzwassergewässern größeren
Schwankungen sowohl im pH-Wert als auch in der Salzkonzentration. Durch die höhere
Toleranz gerade gegenüber diesen Einflüssen könnte C. submarinum einen Vorteil in vivo
aufweisen und ist deshalb für eine biotechnologische Anwendung besonders interessant.
4.3. Untersuchung der O2-und CO-Sensitivität: Die hohe O2-Toleranz von HydA aus
C. submarinum
Eine weitere charakteristische Eigenschaft von [FeFe]-Hydrogenasen ist ihre hohe
Sauerstoffsensitivität und die Inhibierung durch Kohlenmonoxid. Die Inhibierung mit CO ist im
Gegensatz zu der irreversiblen Schädigung des H-Clusters durch O2 reversibel (Chen et al. 2002,
Albracht et al. 2006). Bei Begasung der Proteine mit CO bindet dieses als externer Ligand an das

4. Diskussion 66
Fe(2)-Atom und blockiert so vermutlich den H2-Gaskanal (Pierik et al. 1998). Eine Bindung von
O2 an das Fe(2)-Atom führt vermutlich zu einer Oxidation des [2Fe]-Subclusters zu Fe (III).
Dadurch gehen CO- und CN-Liganden verloren und das H-Cluster wird irreversibel zerstört
(Lubitz et al. 2007). Zum Teil unterscheiden sich [FeFe]-Hydrogenasen in ihrer Sensitivität
gegenüber O2. So kann beispielsweise DdH aus D. desulfuricans unter aeroben Bedingungen
gereinigt werden und ist nur im aktiven oxidierten Zustand sauerstoffempfindlich, wohingegen
CpI aus C. pasteurianum wie die hier untersuchten Grünalgen-Hydrogenasen strikt anaerob
isoliert werden muss (Adams und Mortenson 1984, Lubitz et al. 2007). Grünalgen-
Hydrogenasen werden im Vergleich zu bakteriellen Hydrogenasen schneller durch O2 und CO
inhibiert, was vermutlich auf das Fehlen zusätzlicher F-Cluster zurückzuführen ist (Adams 1990,
Ghirardi et al. 2007).
Bei der vergleichenden Untersuchung der isolierten HydA-Proteine in Bezug auf ihre CO- und
O2-Sensitivität zeigte sich, dass die HydA1-Proteine aus C. reinhardtii und C. moewusii eine
ähnlich hohe Sauerstoffempfindlichkeit aufweisen, HydA aus C. submarium dagegen wesentlich
sauerstofftoleranter ist (3.2.4. und 3.2.5.). Bei einer Konzentration von 8% O2 sind sowohl
HydA1 aus C. reinhardtii als auch HydA1 aus C. moewusii bereits vollständig inhibiert, während
bei HydA aus C. submarium noch 50% der Ausgangsaktivität vorhanden ist (Abb.3.8.). Dies
macht HydA für weitere Untersuchungen besonders interessant, da im Hinblick auf
biotechnologische Anwendungen insbesondere daran gearbeitet wird, Hydrogenasen durch
Austausch gezielter Aminosäuren sauerstofftoleranter zu machen (Buhrke et al. 2005, Ghirardi
et al. 2007).
In Bezug auf eine Inhibierung mit Kohlenmonoxid zeigen die Hydrogenasen aus C. reinhardtii
und C. submarium ähnliche Eigenschaften, HydA1 aus C. moewusii wird dagegen schneller
inhibiert (Abb. 3.9.). Dies lässt auf einen schnelleren Umsatz des Enzyms schließen, was auch
die im Gegensatz zu HydA1 aus C. reinhardtii und HydA aus C. submarinum höhere spezifische
Aktivität belegen würde.
4.4. Die Struktur des H-Clusters von Grünalgen-[FeFe]-Hydrogenasen
Durch die in dieser Arbeit durchgeführte EPR- und FTIR-spektroskopische Untersuchung des
katalytischen Zentrums von Grünalgen-[FeFe]-Hydrogenasen konnte erstmals gezeigt werden,
dass das H-Cluster aller drei HydA-Proteine prinzipiell ähnlich aufgebaut ist wie das H-Cluster

4. Diskussion 67
der bereits beschriebenen bakteriellen [FeFe]-Hydrogenasen DdH und CpI (3.3.) (Abb. 4.1.).
Unterschiede in den ermittelten g-Werten oder den detektierten Wellenzahlen, sowie eine zum
Teil unterschiedliche Reaktion auf Licht, Oxidationsmittel oder eine Begasung mit Wasserstoff
und Argon zeigen allerdings, dass die elektronische Struktur des H-Clusters und die Symmetrie
der Liganden von Grünalgen- und bakteriellen [FeFe]-Hydrogenasen unterschiedlich ist.
Abb. 4.1. Strukturmodell des H-Clusters von [FeFe]-Hydrogenasen im Hox-Zustand (Silakov et al. 2007)
4.4.1. Der Hox-CO-Zustand
Durch eine Begasung mit CO wird das H-Cluster in den Hox-CO-Zustand überführt, der sowohl
im EPR-Spektrum das für [FeFe]-Hydrogenasen in diesem Zustand charakteristische axiale
Signal als auch das im FTIR-Spektrum für diesen Zustand typische Bandenmuster zeigt
(Abb.3.10. und 3.16.) (Pierik et al. 1998, Lemon et al. 1999, Nicolet et al. 2001, Chen et al.
2002, Albracht et al. 2006, Roseboom et al. 2006). Der Hox-CO-Zustand von Grünalgen-
Hydrogenasen ist somit prinzipiell ähnlich aufgebaut wie der Hox-CO-Zustand von DdH und
CpI. Kleine Unterschiede in den ermittelten g-Werten und detektierten Wellenzahlen zeigen
allerdings, dass Unterschiede in der elektronischen Struktur und der Anordnung der Liganden
vorhanden sind (Tab. 4.1. und 4.2.).
Die genaue Untersuchung der elektronischen Struktur des mit 57Fe markierten Hox-CO-Zustands
durch ENDOR- und HYSCORE-Techniken zeigt, dass die elektronische Struktur des

4. Diskussion 68
Abb. 4.2. Überblick über die verschiedenen Zustände des H-Clusters von HydA1 aus C. reinhardtii
Durch Begasung der isolierten Form mit CO lässt sich das H-Cluster in den CO-inhibierten Zustand Hox-CO
überführen, wobei das externe CO an der freien Koordinationsstelle des Fed-Atoms bindet. Durch Belichtung bei
kryogenen Temperaturen löst sich das CO wieder und man erhält den aktiven oxidierten Zustand Hox, der sich im
Gegensatz zu bakteriellen Hydrogenasen nicht biochemisch herstellen lässt. Der dabei auftretende Zwischenzustand
LI2 ist eine Art Hox-CO-Zustand mit veränderter Geometrie. Unter reduzierenden Bedingungen erhält man zwei
Zustände, Hred1 und Hred2. In Hred2 ist der CO-Brückenligand terminal an Fed gebunden.
Tab. 4.1. Vergleich der ermittelten g-Werte mit CpI (Chen et al. 2002) und DdH (Lubitz et al. 2007)
Spezies Hox-CO Hox
C. reinhardtii 2.052 2.007 2.007 2.102 2.040 1.998
C. moewusii 2.052 2.008 2.008 2.103 2.038 1.998
C. submarinum 2.056 2.008 2.008 2.100 2.040 1.998
D. desulfuricans 2.065 2.007 2.001 2.100 2.040 1.999
C. pasteurianum 2.074 2.011 2.011 2.100 2.040 2.001

4. Diskussion 69
Tab. 4.2. Überblick über die ermittelten FTIR-Banden im Vergleich zu DdH (Roseboom et al. 2006) und
CpI (Chen et al. 2002)
Alle Werte sind in [cm-1] angegeben.
Zustand Spezies CNp CNd COp COd - COext CObrHsox HydA1 2106 2093 2047 2033 1910 Hinact DdH 2106 2087 1983 2007 1848
HydA1 2091 2076 1966 1940 1799 DdH 2093 2079 1965 1940 1802
Hox
CpI 2086 2072 1971 1948 1802 HydA1 2090 2083 1970 2013 1964 1810 DdH 2096 2088 1971 2016 1963 1810
Hox-CO
CpI 2095 2077 1971 2017 1974 1810 Hred DdH 2079 2041 1965 1916 1894 Hred1 HydA1 2083 2054 1935 1890 1793 Hred2 HydA1 2030 2008 1920 1883 1955 Hsred DdH ? ? 1932 1883 1955
HydA1 2090 2083 1967 1992 1944 1810 DdH 2096 2088 1963 1995 1949 1812
Hox-13CO
CpI 2095 2077 1971 2000 1947 1810 HydA1
2090 2083 1964 1967 1970
1969 19281992 1944 2013 1964
1810 Hox-(13CO)3
DdH 2096 2088 1962 1973 1926 1770
Hox-CO-Zustands von HydA1 ähnlich ist wie die elektronische Struktur von DdH, sich aber von
der elektronischen Struktur von CpI unterscheidet (3.3.1.3.) (Tab. 4.3.). Es konnten sechs 57Fe-Hyperfeinkopplungen aufgelöst werden, die in dieser Form auch für DdH detektiert worden
sind (Silakov et al. 2007). Ein Vergleich der für HydA1 und DdH ermittelten Parameter zeigt,
dass die isotropen Teile des Hyperfein-Tensors ähnlich, der dipolare Teil dagegen leicht
unterschiedlich ist (Tab. 4.3.).
Der isotrope Teil des Hyperfein-Tensors spiegelt die ungepaarte Spinpopulation der Fe-Kerne
wieder. Die Ähnlichkeit dieser Werte für beide Spezies zeigt, dass die Verteilung von
ungepaarten Spins gleich ist. Alle sechs Fe-Atome weisen Spindichte auf, die durch
elektronische Austausch-Wechselwirkung zwischen den Fe-Atomen verursacht wird. Bei DdH
wird angenommen, dass das [2Fe]-Subcluster sowohl im oxidierten als auch im CO-inhibierten
Zustand die Konfiguration Fe(I)-Fe(II) aufweist, wobei das paramagnetische Fep(I)-Atom mit
dem formal diamagnetischen [4Fe-4S]2+-Cluster verbunden ist (Silakov et al. 2007). Die
Hyperfeinwechselwirkungen des [4Fe-4S]-Clusters werden durch Austausch-Wechselwirkung
zwischen dem Fe-Atoms, welches das [4Fe-4S]-Cluster mit dem [2Fe]-Subcluster verbindet, und
dem Fep-Atom des [2Fe]-Subclusters hervorgerufen (Popescu und Münck 1999, Silakov et al.

4. Diskussion 70
2007). Im Hox-Zustand ist der ungepaarte Elektronenspin gleichmäßig auf die beiden Fe-Atome
des [2Fe]-Subclusters verteilt. Bei Bindung eines externen CO-Liganden vergrößert sich der
Abstand zwischen den beiden Fe-Atomen des [2Fe]-Subclusters von 2,56 Å auf 2,71 Å (Fiedler
und Brunold 2005), wodurch es zu einer Lokalisierung der Elektronenspindichte am proximalen
Fe-Atom kommt, was anhand einer starken Hyperfeinwechselwirkung am Fep-Atom und einer
schwachen am Fed-Atom detektiert werden kann (Silakov et al. 2007). Dies ist auch bei HydA1
der Fall (Abb. 3.13. und 3.14., Tab. 3.5.).
Der dipolare Teil der Hyperfeinkopplung (Axyz) ist sensitiver in Bezug auf die Umgebung des
Kerns als der isotrope Teil (Aiso). Die hier beobachteten Unterschiede zeigen, dass die
Symmetrie der Liganden der Fe-Atome in DdH und HydA1 unterschiedlich ist, was sich auch in
den unterschiedlichen g-Werten und Wellenzahlen wiederspiegelt (Tab. 4.1. und 4.2.).
Auch für CpI wurde gezeigt, dass die Bindung eines externen CO-Liganden zu einer Änderung
der elektronischen Struktur des H-Clusters führt (Telser et al.1986, Popescu und Münck 1999).
Es wurden vier Hyperfeinkopplungen aufgelöst, die vermutlich zum [4Fe-4S]-Cluster gehören,
allerdings konnte nur eine einzige isotrope Hyperfeinwechselwirkung für das [2Fe]-Subcluster
nachgewiesen werden (Telser et al.1986), was zeigt, dass sich die elektronische Struktur von CpI
von DdH und HydA1 unterscheidet.
Tab. 4.3. Überblick über die ermittelten Werte des 57Fe-Hyperfein-Tensors (Ax, Ay, Az) und des isotropen
Teils (Aiso) im Vergleich zu DdH (Silakov et al. 2007)
Alle Werte sind in [MHz] angegeben.
D. desulfuricans C. reinhardtii HFK Ax Ay Az Aiso Ax Ay Az Aiso Zuordnung
A1 27.8 21.8 30.3 26.7 28.5 25.0 29.2 27.6 [4Fe-4S]HA2 26.7 23.8 30.2 27.0 30.3 23.5 30.5 28.0 [4Fe-4S]H
Fe3-Fe4
A3 35.4 35.0 30.4 33.6 34.7 34.7 29.9 33.1 [4Fe-4S]HA4 34.5 38.4 30.7 34.8 34.5 37.7 30.9 34.3 [4Fe-4S]H
Fe1-Fe2
A5 5.3 4.5 2.2 4.0 5.7 2.9 0.4 3.0 [2Fe]H Fep
A6 2.1 2.1 -1.7 0.8 3.0 3.0 -2.1 1.3 [2Fe]H Fed
Zusätzlich konnte durch FTIR-Spektroskopie gezeigt werden, dass die Schwingung des COp-
Liganden im Hox-CO-Zustand ebenso wie bei DdH und anders als bei CpI mit der Schwingung
COd/COext des distalen und externen CO-Liganden gekoppelt ist, da sich die entsprechende

4. Diskussion 71
Bande bei der Überführung in den Hox-Zustand im Spektrum von 1970 cm-1 zu 1966 cm-1
verschiebt (Abb.3.16. und Tab.4.2). HydA1 hat somit auch in dieser Hinsicht im
Hox-CO-Zustand eine größere Ähnlichkeit mit DdH als mit CpI.
4.4.2. Die Photodissoziation von Hox-CO und der Zwischenzustand LI2
Durch Belichtung des Hox-CO-Zustands bei kryogenen Temperaturen lässt sich das CO wieder
entfernen und das H-Cluster wird in den oxidierten Zustand Hox überführt (Chen et al. 2002,
Albracht et al. 2006, Roseboom et al. 2006). Bei DdH und CpI kann während dieses Prozesses
ein Zwischenzustand (LI2) detektiert werden, der durch eine reversible, partielle Ablösung des
CO-Brückenliganden charakterisiert ist, während der externe, zu entfernende CO-Ligand noch
gebunden ist (Pierik et al. 1998, Chen et al. 2002, Roseboom et al. 2006). Auch bei HydA1 ist
eine Ablösung des externen CO-Liganden durch Photodissoziation des Hox-CO-Zustands
möglich, was im Hox-Zustand resultiert (Abb.3.11. und 3.16.). Bei der Belichtung tritt allerdings
ein Zwischenzustand auf, der sich geometrisch von dem für DdH und CpI beschriebenen
LI2-Zustand unterscheidet (Abb.3.16.). Im FTIR-Spektrum ist zu sehen, dass die CN-Banden des
H-Clusters stark zu niedrigeren Wellenzahlen verschoben sind, was bedeutet, dass sich die
Geometrie des [2Fe]-Subclusters während der Photodissoziation stark ändert. Eine Bande bei
1817 cm-1 zeigt allerdings, dass der CO-Brückenligand im Gegensatz zu DdH und CpI nicht
dissoziiert. Der Zwischenzustand LI2 von HydA1 ist somit eine Art Hox-CO-Zustand mit
veränderter Geometrie.
Durch eine isotopische Markierung der CO-Liganden mit 13C konnte gezeigt werden, dass das
H-Cluster von HydA1 bei Belichtung zum Teil anders reagiert als das H-Cluster von DdH. Bei
DdH ist zu beobachten, dass bei einer Belichtung des Hox-13CO-Zustands bei kryogenen
Temperaturen COd und CObr dissoziieren und durch 13CO ausgetauscht werden (Roseboom et al.
2006). Bei HydA1 werden dagegen alle CO-Liganden des H-Clusters durch 13CO ersetzt (Abb.
3.17.).
4.4.3. Der Hox-Zustand
Der für HydA1 detektierte Hox-Zustand ist nahezu identisch mit dem Hox-Zustand von DdH und
zeigt ebenfalls große Ähnlichkeit zu der oxidierten Form von CpI (Abb.3.10. und 3.16.). Die
ermittelten g-Werte für den Hox-Zustand unterscheiden sich nur geringfügig, bei den detektierten
FTIR-Banden sind kleinere Unterschiede festzustellen (Tab. 4.1. und 4.2.). Durch die

4. Diskussion 72
Untersuchung von 13C markiertem H-Cluster konnte gezeigt werden, dass die Schwingungen von
COp und COd miteinander gekoppelt sind (Abb.3.17.). Im Gegensatz zu DdH und CpI lässt sich
das H-Cluster von HydA1 nur durch Photodissoziation des Hox-CO-Zustands (3.3.1.2.) oder
durch Anlegen eines elektrochemischen Potentials (3.3.2.4.) und nicht biochemisch oder durch
Begasung der reduzierten Form mit Argon in den Hox-Zustand überführen. Die für CpI
verwendeten Oxidationsmittel zerstören das H-Cluster von HydA1, was zeigt, dass der oxidierte
Zustand von HydA1 empfindlicher ist als der von CpI. Dies könnte auf die fehlenden F-Cluster
bei Grünalgen-Hydrogenasen und die direkte Interaktion des H-Clusters mit den verwendeten
Reagenzien zurückzuführen sein.
4.4.4. Die reduzierten Zustände: Hred1 und Hred2
Bei DdH führt die Begasung mit H2 zu einem einzigen reduzierten Zustand, in dem der
CO-Brückenligand terminal an das distale Fe-Atom gebunden ist (Nicolet et al. 2001, Roseboom
et al. 2006). Bei HydA1 erhält man weder durch die Zugabe von NaDT noch durch Begasung
mit H2 einen klaren reduzierten Zustand (Abb. 3.21.), was vermutlich wiederum durch das
Fehlen der zusätzlichen F-Cluster zu begründen ist. Durch das Anlegen eines elektrochemischen
Potentials konnten zwei reduzierte Zustände von HydA1 aufgelöst werden (3.3.2.4.).
Bei einem Potential von - 400 mV erhält man den reduzierten Zustand Hred1 (Abb. 3.20.). Bei
diesem Zwischenzustand ist einer der CN-Liganden verschoben, was zeigt, dass eines der Fe-
Atome reduziert wurde. Dieser Zustand hat aufgrund des im Spektrum detektierten
Bandenmusters und des ähnlichen Mittelpunkspotential von - 400 mV für den Übergang
Hox/Hred1 Ähnlichkeit zum reduzierten Zustand von DdH (Tab. 4.2. und 4.4.). Allerdings ist im
Gegensatz zu DdH die CO-Brücke noch gebunden. Bei einem Potential von - 450 mV erhält man
einen zweiten reduzierten Zustand Hred2, in dem vermutlich wie bei DdH der CO-Brückenligand
dissoziiert ist und nun terminal gebunden an Fed vorliegt. Das für diesen Zustand detektierte
Bandenmuster hat Ähnlichkeit mit dem superreduzierten Zustand Hsred von DdH, allerdings
unterscheiden sich beide in ihrem Mittelpunktspotential (Tab.4.4.). Die Reduktion von HydA1
ist im Gegensatz zu DdH reversibel (Abb.3.19. und Tab.4.4.). Da bei der EPR-spektroskopischen
Untersuchung außer Hox und Hox-CO kein weiterer Zustand nachgewiesen werden konnte, sind
die reduzierten Zustände beide diamagnetisch und haben vermutlich wie DdH und CpI die
Konfiguration Fe(I)-F(I).

4. Diskussion 73
Tab. 4.4. Vergleich der Mittelpunktspotentiale bei der elektrochemischen Titration von HydA1 und DdH
(Roseboom et al. 2006)
D. desulfuricans C. reinhardtii
Übergang Potential (mV) pH 7 pH 8 Übergang Potential (mV)
pH 7,5 Htrans / Hox (2e-) irreversibel - 261 - 301 Hox - 260 Hox / Hred (1e-) - 354 - 395 Hox / Hred1 (1e-) - 400 Hred / Hsred irreversibel - 500 - 540 Hred1 / Hred2 (1e-) - 455
4.4.5. Der katalytische Mechanismus im H-Cluster
Der katalytische Mechanismus von [FeFe]-Hydrogenasen ist im Gegensatz zu
[NiFe]-Hydrogenasen bisher weniger gut verstanden. Für DdH wird angenommen, dass der
Wasserstoff durch einen H2-Gaskanal zum Fed-Atom gelangt, dort an die freie
Koordinationsstelle bindet und heterolytisch gespalten wird (Nicolet 2000, Lubitz et al. 2007).
Die freiwerdenden Elektronen werden auf das [4Fe-4S]-Cluster, die F-Cluster und anschließend
auf einen externen Elektronenakzeptor, die freiwerdenden Protonen auf verschiedene Reste im
Protein, möglicherweise auch den Dithiometylamin-Liganden, übertragen. Verschiedene
Untersuchungen belegen, dass das [2Fe]-Subcluster im Hox-Zustand die Konfiguration
Fep(I)-Fed(II) aufweist, wobei das paramagnetische Fep(I)-Atom mit dem formal
diamagnetischen [4Fe-4S]2+-Cluster verbunden ist. Während des katalytischen Zyklus wechselt
das [2Fe]-Subcluster zwischen der Konfiguration Fep(I)-Fed(II) im Hox- und Fep(I)-Fed(I) im
Hred-Zustand (Silakov et al. 2007, Lubitz et al. 2007). Daraus lässt sich ein Modell ableiten, das
sowohl die H2-Oxidation als auch die H2-Produktion erklärt (Abb. 3.4.) und somit auch auf
HydA1 übertragen werden kann, da die spektroskopische Untersuchung gezeigt hat, dass der
Hox-Zustand von HydA1 wie DdH die Konfiguration Fep(I)-Fed(II) und die reduzierten Zustände
vermutlich die Konfiguration Fep(I)-Fed(I) besitzen (4.4.1. und 4.4.4.). Bei HydA1 läuft der
Mechanismus in umgekehrter Reihenfolge, möglicherweise aber über dieselben Zwischenstufen,
wie bei DdH ab. Die Elektronen werden vermutlich durch Ferredoxin direkt über das [4Fe-4S]-
Cluster auf das Fed-Atom des [2Fe]-Subclusters übertragen. Gleichzeitig werden Protonen über
den Protonentransferpfad zum Fed-Atom transportiert und dort mit den Elektronen zu
molekularem H2 umgesetzt, der dann über den hydrophoben Gaskanal freigesetzt wird (Winkler
et al. 2002).
Da HydA1 nur aus dem katalytisch aktiven H-Cluster besteht und keine zusätzlichen F-Cluster
besitzt, können, wie die Messungen zeigen, in vitro keine Elektronen abgegeben werden.
Dadurch bleibt das Enzym beispielsweise nach einer Begasung mit H2 oder Zugabe von NaDT

4. Diskussion 74
im reduzierten Zustand, lässt sich nicht biochemisch oxidieren und reagiert empfindlicher auf
zugegebene Reagenzien.
aktiv
inaktiv
Abb.4.3. Überblick über die verschiedenen Redoxzustände des H-Clusters und den möglichen katalytischen
Mechanismus von [FeFe]-Hydrogenasen am Beispiel von DdH (Lubitz et al. 2007) Die Zustände, die in den katalytischen Zyklus involviert sind, sind gelb eingerahmt. Für jeden Zustand sind die
Oxidationszahlen der Fe-Atome des [2Fe]-Subclusters und zusätzlich der am Fed-Atom bindende Ligand X in
eckigen Klammern angegeben. Das [4Fe-4S]-Cluster hat jeweils die Oxidationsstufe 2+. Die EPR-aktiven Zustände
Hox und Hox-CO sind rot dargestellt, der diamagnetische Zustand Hred blau.
Eine exakte Aufklärung der Struktur des H-Clusters von Grünalgen-[FeFe]-Hydrogenasen
könnte durch eine für die Zukunft geplante Auflösung und Analyse der Röntgenkristallstruktur
des Proteins erfolgen, genauere Hinweise auf den katalytischen Mechanismus könnte
beispielsweise die Untersuchung von Protonentransfermutanten liefern.

5. Literatur 75
5. Literatur
Adams, M. W. W. (1990). “The structure and mechanism of iron-hydrogenases.”
Biochim Biophys Acta 1020: 115-145.
Albracht, S. P. J., Roseboom, W. und Hatchikian, E. J. (2006). “The active site of the [Fe-Fe]-
Hydrogenase from Desulfovibrio desulfuricans. Light sensitivity and magnetic hyperfine
interactions as observed by electron paramagnetic resonance.” J Biol Inorg Chem 11: 88-101.
Atkins, P. W. (1994). “Physical Chemistry.” 5th edition, Oxford University Press.
Bennet, B., Lemon, B. J. und Peters, J. W. (2000). “Reversible carbon monoxide binding and
inhibition at the active site of the Fe-Only Hydrogenase.” Biochemistry 39: 7455-7460.
Blackwell, J. R. und Gilmour, D. J. (1991). “Physiological reponse of the unicellular green alga
Chlorococcum submarinum to rapid changes in salinity.” Arch of Microbiol 157: 86-91.
Bradford, M. M. (1976). “A rapid and sensitive method for the quantitation of microgram
quantities of protein using the principle of protein-dye binding.” Anal Biochem 72: 248-254.
Buhrke, T., Lenz, O., Krauss, N. und Friedrich, B. (2005). “Oxygen tolerance of the H2-sensing
[NiFe]-hydrogenase from Ralstonia eutropha H16 is based on limited access of oxygen to the
active site.“ J Biol Chem 280: 23791-23796.
Chen, Z., Lemon, B. J., Huang, S., Swartz, D. J., Peters, J. W. und Bagley, K. A. (2002).
“Infrared Studies of the CO-Inhibited Form of the Fe-Only Hydrogenase from Clostridium
pasteurianum I: Examination of Its Light Sensitivity at Cryogenic Temperatures.“
Biochemistry 41: 2036-2043.
Clamp, M., Cuff, J., Searle, S. und Barton, G. J. (2004). “Jalview Java alignment editor.”
Bioinformatics 20: 426-427.

5. Literatur 76
Cohen, J., Kim, K., King, P., Seibert, M. und Schulten, K. (2005). “Finding Gas Diffusion
Pathways in Proteins: Application to O2 and H2 Transport in CpI [FeFe]-Hydrogenase and the
Role of Packing Defects.” Structure 13: 1321–1329.
Cohen, J., Kim, K., Posewitz, M., Ghirardi, M. L., Schulten, K., Seibert, M. und King, P. (2005).
“Molecular dynamics and experimental investigation of H2 and O2 diffusion in [Fe]-
hydrogenase.” Biochem Soc Trans 33: 80-82.
Davies, E. R. (1974). “New Pulse Endor Technique.“ Phys Lett A A47 (1): 1-2.
Epel, B., Gromov, I., Stoll, S., Schweiger, A. und Goldfarb, D. (2005). “Spectrometer manager:
A versatile control software for pulse EPR spectrometers.”
Concepts Magn Reson B-Magn Reons Eng 26B (1): 36-45.
Fiedler, A. T. und Brunold, T. C. (2005). “Computational studies of the H-Cluster of Fe-Only
Hydrogenases: Geometric, electronic and magnetic properties and their dependence of the
[Fe4S4] Cubane.” Inorg Chem 44: 9322-9334.
Fischer, F. (2006). “Umfassende Charakterisierung des Membranproteoms eines L-Lysin
produzierenden Corynebacterium glutamicum Stammes.“
Lehrstuhl für Biochemie der Pflanzen, Ruhr Universität Bochum.
Florin, L., Tsokoglou, A. und Happe, T. (2001). “A novel type of iron hydrogenase in the green
alga Scenedesmus obliquus is linked to the photosynthetic electron transport chain.”
J Biol Chem 276: 6125-6132.
Forestier, M., King, P., Zhang, L., Posewitz, M., Schwarzer, S., Happe, T., Ghirardi, M. L. und
Seibert, M. (2003). “Expression of two [Fe]-hydrogenases in Chlamydomonas reinhardtii under
anaerobic conditions.” Eur J Biochem 270: 2750-2758.
Frey, M. (2002). “Hydrogenases: Hydrogen-Activating Enzymes.“ ChemBioChem 3: 153-160.

5. Literatur 77
Ghirardi, M. L., Posewitz, M. C., Maness, P., Dubini, A., Yu, J. und Seibert, M. (2007).
“Hydrogenases and Hydrogen Photoproduktion in Oxygenic Photosynthetic Organisms.“
Annu Rev Plant Biol 58: 71-91.
Girbal, L., von Abendroth, G., Winkler, M., Benton, P. M. C., Meynial-Salles, I., Croux, C.,
Peters, J. W., Happe, T. und Soucaille, P. (2005). “Homologous and Heterologous
Overexpression in Clostridium acetobutylicum and Characterization of Purified Clostridial and
Algal Fe-Only Hydrogenases with High Specific Activities.”
Appl Environ Microbiol 71: 2777-2781.
Grossmann, A. (2000). “Acclimation of Chlamydomonas reinhardtii to its nutrient
environment.” Protist 151: 201-224.
Guex, N. und Peitsch, M. C. (1997). “SWISS-MODEL and the SWISS-PDBViewer: an
environment for comparative protein modeling.“ Electrophoresis 18: 2714-2723.
Hahn, E. L. (1950). “Spin Echoes.” Phys Rev 80: 580-594.
Happe, T. und Naber, J. D. (1993). “Isolation, characterisation and N-terminal amino acid
sequence of hydrogenase from the green alga Chlamydomonas reinhardtii.”
Eur J Biochem 214: 475-481.
Happe, T., Mosler, B. und Naber, J. D. (1994). “Induction, localization and metal content of
hydrogenase in the green alga Chlamydomonas reinhardtii.” Eur J Biochem 222: 769-774.
Happe, T. und Kaminski, A. (2002). “Differential regulation of the Fe-hydrogenase during
anaerobic adaptation in the green alga Chlamydomonas reinhardtii.”
Eur J Biochem 269: 1022-1032.
Happe, T., Hemschemeier, A., Winkler, M. und Kaminski, A. (2002). “Hydrogenases in green
algae: do they save the algae´s life and solve our energy problems?” Trends Plant Sci 7: 246-250.
Hemschemeier, A. und Happe, T. (2004). “The exeptional photofermentative hydrogen
metabolism of the green alga Chlamydomonas reinhardtii.” Biochem Soc Trans 33: 3 9-41.

5. Literatur 78
Hemschemeier, A., Jacobs, J. und Happe, T. (2008). “Biochemical and Physiological
Characterization of the Pyruvate-Formate-Lyase Pfl1 of Chlamydomonas reinhardtii, a Typically
Bacterial Enzyme in an Eukaryotic Alga.“ Eukaryotic Cell 7: 518-526.
Horner, D. S., Heil, B., Happe, T. und Embley, T. M. (2002). “Iron hydrogenases-ancient
enzymes in modern eukaryotes.“ Trends Biochem Sci 27: 148-153.
Jensen, O. N., Wilm, A., Schevchenko, A. und Mann, M. (1998). “Sample preparation methods
for mass spectrometric peptide mapping directly from 2-D gels.”
Methods Mol Biol 112: 513-539.
Karas, M., Bachmann, D., Bahr, U. und Hillenkamp, F. (1987). “Matrix-assisted ultraviolet laser
desorption of non-volatile compounds.” Int J Mass Spectrom Ion Process 78: 53.
King, P. W., Posewitz, M. C., Ghirardi, M. L. und Seibert, M. (2006). “Functional Studies of
[FeFe] Hydrogenase Maturation in an Escherichia coli Biosynthetic System.“
J Bacteriol 188: 2163-2172.
Korbas, M., Vogt, S., Meyer-Klaucke, W., Bill, E., Lyon, E. J., Thauer, R. K. und Shima, S.
(2006). “The iron-sulfur cluster-free hydrogenase (Hmd) is a metalloenzyme with a novel iron
binding motif.” J Biol Chem 281: 30804-30813.
Laemmli, U. K. (1970). ”Cleavage of structural proteins during the assembly of the head of
bacteriophage T4.” Nature 227: 680-685.
Laurinavichene, T. V., Tolstygina, I. V., Galiulina, R. R., Ghirardi, M. L., Seibert, M. und
Tsygankov, A. A. (2002). “Dilution methods to deprive Chlamydomonas reinhardtii cultures of
sulfur for subsequent hydrogen photoproduction.” Int J Hydrogen Energy 27: 1245-1249.
Lemon, B. J. und Peters, J. W. (1999). “Binding of Exogenously Added Carbon Monoxide at the
Active Site of the Iron-Only Hydrogenase (CpI) from Clostridium pasteurianum.”
Biochemistry 38: 12969-12973.

5. Literatur 79
Lubitz, W., Reijerse, E. J. und van Gastel, M. (2007). ”[NiFe] and [FeFe] Hydrogenase Studied
by Advanced Magnetic Resonance Techniques.” Chem Rev 107: 4331-4365.
Lubitz, W., van Gastel, M. und Gärtner,W. (2007). ”Nickel Iron Hydrogenases.“
Met Ions Life Sci 2: 279-322.
Marusyk, R. und Sergeant, A. (1980). “A simple method for dialysis of small-volume samples.”
Anal Biochem 105: 403-404.
Melis, A., Zhang, L., Forestier, M., Ghirardi, M. L. und Seibert, M. (2000). “Sustained
photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the
green alga Chlamydomonas reinhardtii.” Plant Physiol 122: 27-135.
Melis, A. und Happe, T. (2001). “Hydrogen Production. Green Algae as a Source of Energy.”
Plant Physiol 127: 740-748.
Moss, D., Nabedryk, E., Breton, J. L. J. und Mäntele, W. (1990). “Redox-linked conformational
changes in proteins detected by a combination of infrared spectroscopy and protein
electrochemistry.” Eur J Biochem 187: 565-572.
Nicolet,Y., Piras, C., Legrand, P., Hatchikian, C. E. und Fontecilla-Camps, J. C. (1999).
“Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an
active site Fe binuclear center.” Structure 7:13-23.
Nicolet,Y., Lemon, B. J., Fontecilla-Camps, J. C. und Peters, J. W. (2000). “A novel FeS cluster
in Fe-only hydrogenases.” Trends Biochem Sci 25: 138-142.
Nicolet,Y., de Lacey, A. L., Vernede, X., Fernandez, V. M., Hatchikian, C. E. und Fontecilla-
Camps, J. C. (2001). “Crystallographic and FTIR Spectroscopic Evidence of changes in Fe
coordination upon reduction of the active site of the Fe-only-hydrogenase from Desulfovibrio
desulfuricans.” J Am Chem Soc 123: 596-1601.

5. Literatur 80
Peters, J. W., Lanzilotta, W. N., Lemon, B. J. und Seefeldt, L. C. (1998). “X-ray Crystal
Structure of the Fe-only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom
resolution.” Science 282: 1853-1858.
Peters, J. W. (1999). “Structure and mechanism of iron-only hydrogenases.”
Curr Opin Struct Biol 9: 670-676.
Pierik, A. J., Hulstein, M., Hagen,W. R. und Albracht, S. P. J. (1998). “A low-spin iron with CN
and CO as intrinsic ligands forms the core of the active site in [Fe] hydrogenases.”
Eur J Biochem 258: 572-578.
Popescu, C. V. und Münck, E. (1999). “Electronic Structure of the H Cluster in [Fe]-
Hydrogenases.” J Am Chem Soc 121: 7877-7884.
Razavet, M., Borg, S. J., George, S. J., Best, S. P., Fairhurst, S. A. und Pickett, C. J. (2002).
“Transient FTIR spectroelectrochemical and stopped-flow detection of a mixed valence {Fe(I)-
Fe(II)}bridging carbonyl intermediate with structural elements and spectroscopic characteristics
of the di-iron sub-site of all-iron hydrogenase.” Chem Commun: 700-701.
Roessler, P. G. und Lien, S. (1984). “Purification of hydrogenase from Chlamydomonas
reinhardtii.” Plant Physiol 75: 705-709.
Roseboom, W., de Lacey, A. L., Fernandez, V. M., Hatchikian, C. E. und Albracht, S. P. J.
(2006). “The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox
properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy.”
J Biol Inorg Chem 11: 102-118.
Rosenfeld J., Capdevielle, J., Guillemont, J. C. und Ferrara, P. (1992). “In-gel digestion of
proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis.”
Anal Biochem 203: 173-179.
Schweiger, A. und Jeschke, G. (2001). “Principles of Pulse Electron Paramagnetic Resonance.”
Oxford University Press.

5. Literatur 81
Shima, S., Pilak, O., Vogt, S., Schick, M., Stagni, M. S., Meyer-Klaucke, W., Warkentin, E.,
Thauer, R. K. und Ermler, U. (2008). “The Crystal Structure of [Fe]-Hydrogenase Reveals the
Geometry of the Active Site.” Science 321: 572-575.
Sienkiewicz, A., Smith, B. G., Veselov, A. und Scholes, P. (1996). “Tunable Q-band resonator
for low temperature electron paramagnetic resonance electron nuclear double resonance
measurements.” Rev Sci Instrum 67: 2134-2138.
Silakov, A. (2007). “Investigation of the active site of the [FeFe] hydrogenase from
Desulfovibrio desulfuricans.” Max-Planck-Institut für Bioanorganische Chemie, Mülheim an der
Ruhr (Heinrich-Heine-Universität Düsseldorf)
Silakov, A., Reijerse, E. J., Albracht, S. P. J., Hatchikian, E. C. und Lubitz, W. (2007). “The
electronic structure of the H-cluster in the [FeFe]-hydrogenase from Desulfovibrio desulfuricans.
A Q-band 57Fe-ENDOR and HYSCORE study.” J Am Chem Soc 129: 11447-11458.
Sinnecker, S., Reijerse, E., Neese, F. und Lubitz, W. (2004). “Hydrogen bond geometries from
electron paramagnetic resonance and electron-nuclear double resonance parameters: Density
functional study of quinone radical anion-solvent interactions.” J Am ChemSoc 126: 3280- 3290.
Telser, J., Benecky, M. J., Adams, M. W. W., Mortenson, L. E. und Hoffmann, B. M. (1986).
“EPR and Electron Nuclear Double Resonance Investigation of Oxidized Hydrogenase II
(Uptake) from Clostridium pasteurianum W5.” J Biol Chem 262: 6589-6594.
Ueno, Y., Kurano, N. und Miyachi, S. (1999). ”Purification and characterization of hydrogenase
from the marine green alga Chlorococcum littorale.” FEBS Letters 443: 144-148.
van Dijk, C., Grande, H. J., Mayhew, S. G. und Veeger, C. (1980). “Properties of the
Hydrogenase of Megasphaera elsdenii.“ Eur J Biochem 107: 251-261.
Vignais, P. M. und Colbeau, A. (2004). ”Molecular Biology of Microbial Hydrogenases.”
Curr Issues Mol Biol 6: 159-188.
Vignais, P. M. und Billoud, B. (2007). ”Occurrence, Classification and Biological Function of
Hydrogenases: An Overview.” Chem Rev 107: 4206-4272.

5. Literatur 82
Winkler, M., Heil, B. und Happe, T. (2002). “Isolation and molecular characterization of the
[Fe]-hydrogenase from the unicellular green alga Chlorella fusca.”
Biochim Biophys Acta 1576: 330-334.
Winkler, M., Hemschemeier, A., Gotor, C., Melis, A. und Happe, T. (2002). “[Fe]-hydrogenases
in green algae: photo-fermentation and hydrogen evolution under sulfur deprivation.”
Int J Hydrogen Energy 2:1431-1439.
Winkler, M., Maeurer, C., Hemschemeier, A. und Happe,T. (2004). “The isolation of green algal
strains with outstanding H2-productivity.” Biohydrogen III 103-115.
Wykoff, D. D., Davies, J. P., Melis, A. und Grossmann, A. R. (1998). “The regulation of
photosynthetic electron transport during nutrient deprivation in Chlamydomonas reinhardtii.”
Plant Physiol 117: 129-139.
Zambrano, I. C., Kowal, A. T., Mortenson, L. E., Adams, M. W. W. und Johnson, M. K. (1989).
“Magnetic circular dichroism and electron paramagnetic resonance studies of hydrogenase I and
II from Clostridium pasteurianum.” J Biol Chem 264: 20974-20983.
Zhang, L., Happe, T. und Melis, A. (2002). “Biochemical and morphological characterization of
sulfur-deprived and H2-producing Chlamydomonas reinhardtii (green alga).”
Planta 214: 552-561.

Abbildungsverzeichnis 83
Abbildungsverzeichnis Abb. 1.1. Strukturmodell der [FeFe]-Hydrogenasen DdH aus Desulfovibrio desulfuricans (Nicolet et al. 1998) und CpI aus Clostridium pasteurianum (Peters et al. 1998) 5 Abb. 1.2. Aufbau des H-Clusters von [FeFe]-Hydrogenasen 6 Abb. 1.3. Sequenzalignment von bakteriellen [FeFe]-Hydrogenasen und Grünalgen-
[FeFe]-Hydrogenasen 8 Abb. 1.4. Strukturmodell und Oberflächenladungsmodell der [FeFe]-Hydrogenase
HydA1 aus Chlamydomonas reinhardtii 9 Abb. 3.1. Induktion der Hydrogenase-Expression in C. reinhardtii, C. moewusii und C. submarinum durch Schwefelmangel 30 Abb. 3.2. Western Blot von HydA1 aus C. reinhardtii, exprimiert unter Schwefelmangel und anaerober Adaptation 31 Abb. 3.3. Schematische Darstellung des entwickelten Reinigungsprotokolls zur
Isolierung von[FeFe]-Hydrogenasen aus Grünalgen 32 Abb. 3.4. SDS-PAGE und Western Blot der verschiedenen Reinigungsstufen von
HydA1 aus C. reinhardtii 33 Abb. 3.5. SDS-PAGE der isolierten [FeFe]-Hydrogenasen aus C. submarinum und
C. moewusii 35 Abb. 3.6. Identifizierung der isolierten HydA-Proteine aus C. reinhardtii,
C. moewusii und C. submarinum 36 Abb. 3.7. Bestimmung des pH-Optimums von HydA1 aus C. reinhardtii (A), HydA1
aus C. moewusii (B) und HydA aus C. submarinum (C) 38 Abb. 3.8. Sauerstoffsensitiviät von [FeFe]-Hydrogenasen 40 Abb. 3.9. Inhibierung von Hydrogenasen durch Kohlenmonoxid 42 Abb. 3.10. Q-Band Puls-EPR-Spektren der [FeFe]-Hydrogenase HydA1 aus C. reinhardtii (A, B, C), HydA aus C. submarinum (D, E) und HydA1 aus C. moewusii (F) 44 Abb. 3.11. Photodissoziation des Hox-CO-Zustands von HydA1 aus C. reinhardtii bei 20 K 46 Abb. 3.12. Q-Band FID-EPR-Spektrum des 57Fe-angereicherten Hox-CO-Zustands (blau) und des unmarkierten Hox-CO-Zustands (rot) von HydA1 47

Abbildungsverzeichnis 84
Abb. 3.13. Q-Band Davies-ENDOR-Spektren des 57Fe-angereicherten Hox-CO- Zustands 48 Abb. 3.14. Q-Band HYSCORE-Spektren des 57Fe-angereicherten Hox-CO-Zustands 49 Abb. 3.15. Schematische Darstellung der Orientierung des 57Fe-Hyperfein-Tensors
des H-Clusters im Hox-CO-Zustand (Silakov et al. 2007) 50 Abb. 3.16. Photodissoziation des Hox-CO-Zustands von HydA1 aus C. reinhardtii bei
40 K 53 Abb. 3.17. Isotopenmarkierung der CO-Liganden mit 13C 57 Abb. 3.18. FTIR-Spektren der isolierten und reduzierten Form von HydA1 aus
C. reinhardtii 58 Abb. 3.19. Dreidimensionale Darstellung der Änderung der Absorptionsbanden
während der elektrochemischen Reduktion (A) und Oxidation (B) des H-Clusters von HydA1 60
Abb. 3.20. Ausgewählte FTIR-Spektren bei verschiedenen Potentialen während der
elektrochemischen Reduktion und Oxidation des H-Clusters von HydA1 61 Abb. 3.21. Intensität charakteristischer CO-Banden bei der elektrochemischen
Reduktion und Oxidation 62 Abb. 4.1. Strukturmodell des H-Clusters von [FeFe]-Hydrogenasen im Hox-
Zustand (Silakov et al. 2007) 67 Abb. 4.2. Überblick über die verschiedenen Zustände des H-Clusters von HydA1
aus C. reinhardtii 68 Abb. 4.3. Überblick über die verschiedenen Redoxzustände des H-Clusters und den
möglichen katalytischen Mechanismus von [FeFe]-Hydrogenasen am Beispiel von DdH (Lubitz et al. 2007) 74

Tabellenverzeichnis 85
Tabellenverzeichnis Tab. 3.1. Übersicht über die Reinigung der [FeFe]-Hydrogenase HydA1 aus C. reinhardtii 33 Tab. 3.2. Übersicht über die Reinigung der [FeFe]-Hydrogenase HydA aus
C. submarinum 35 Tab. 3.3. Übersicht über die Reinigung der [FeFe]-Hydrogenase HydA1 aus
C. moewusii 35 Tab. 3.4. Überblick über die ermittelten g-Werte 45 Tab. 3.5. Überblick über die ermittelten Parameter der Hyperfeinwechselwirkungen
der 57Fe-Kerne im Hox-CO-Zustand 50 Tab. 3.6. Überblick über die detektierten FTIR-Banden 62 Tab. 4.1. Vergleich der ermittelten g-Werte mit CpI (Chen et al. 2002) und DdH
(Lubitz et al. 2007) 68 Tab. 4.2. Überblick über die ermittelten FTIR-Banden im Vergleich zu DdH
(Roseboom et al. 2006) und CpI (Chen et al. 2002) 69 Tab. 4.3. Überblick über die ermittelten Werte des 57Fe-Hyperfein-Tensors
(Ax, Ay, Az) und des isotropen Teils (Aiso) im Vergleich zu DdH (Silakov et al. 2007) 70
Tab. 4.4. Vergleich der Mittelpunktspotentiale bei der elektrochemischen Titration von HydA1 und DdH (Roseboom et al. 2006) 73

ERKLÄRUNG Hiermit erkläre ich, dass ich die Arbeit selbstständig verfasst und bei keiner anderen Fakultät
eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel verwendet habe. Es
handelt sich bei der von mir heute eingereichten Dissertation um fünf in Wort und Bild völlig
übereinstimmende Exemplare.
Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in
keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde.
Bochum, den

DANKSAGUNG Mein besonderer Dank gilt Prof. Dr. Thomas Happe für die Betreuung dieser Arbeit und die
Möglichkeit zur Promotion in der Arbeitsgruppe Photobiotechnologie.
Gleiches gilt für Prof. Dr. Eckhard Hofmann, der freundlicherweise das Zweitgutachten
übernommen hat.
Des weiteren möchte ich mich ganz herzlich bei unseren Kooperationspartnern am Max-Planck-
Institut für Bioanorganische Chemie, Prof. Dr. Wolfgang Lubitz, Dr. Edward Reijerse und im
Besonderen Dr. Alexey Silakov für die Zusammenarbeit und die Möglichkeit der Durchführung
der EPR- und FTIR-spektroskopischen Untersuchungen bedanken.
Weiterhin danke ich Dr. Frank Fischer und Dr. Markus Piotrowski für die Sequenzierung der
isolierten Proteine sowie allen Mitarbeitern und Mitarbeiterinnen der Arbeitsgruppe
Photobiotechnologie.

LEBENSLAUF
Persönliche Daten
Christina Kamp
geboren am 12.1.1980
in Viersen, Nordrhein-Westfalen
Schulbildung
1986-1990
1990-1999
Besuch der Kreuzherren-Grundschule Dülken
Besuch des Albertus-Magnus-Gymnasiums Dülken
Studium und Promotion
1999-2005
2005-2008
Studium der Biologie an der Heinrich-Heine-Universität Düsseldorf
Thema der Diplomarbeit:
Untersuchungen zur Expression und Funktion des E. coli Proteins Rsd, ein
möglicher Antisigmafaktor für σ70
angefertigt im Institut für Physikalische Biologie in der Abteilung
Molekularbiologie der Prokaryoten
Promotion an der Ruhr-Universität Bochum
Thema der Doktorarbeit:
Spektroskopische Untersuchung des H-Clusters von photosynthetischen
[FeFe]-Hydrogenasen am Beispiel von HydA1 aus Chlamydomonas reinhardtii
angefertigt im Institut für Biochemie der Pflanzen in der Abteilung
Photobiotechnologie


















