
Synthesi s of Pyridon e Derivatives - Max Planck...
5
This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution 4.0 International License. Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschung in Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht: Creative Commons Namensnennung 4.0 Lizenz. Synthesis of Pyridone Derivatives MICHAEL Condensation with Ethyl Cyanoacetate, Cyanoacetamide and Acetoacetamide H. H. Z O O R O B a n d E. S. ISMAIL Faculty of Science, Mansoura University, National Research Centre, Dokki, Cairo, Egypt (Z. Naturforsch. 31b. 1680-1684 [1976]; received May 31, 1976) 2-Cinnamoylbenzimidazole, GRIGNARD Reagents, Azachromone, Pyrazole MICHAEL condensation of 2-cinnamoylbenzimidazoles (la-h) with ethyl cyanoacetate, cyanoacetamide and acetoacetamide led to the formation of 2(1 H)pyridone derivatives (2a-c), (3a-c), 4a-c), and (5a-c). Compounds (2 a-c) added GRIGNARD reagents at the nitrile group to give 5 a-c. CLAISEN condensation of 5a-c with ethyl acetate gave the corresponding diketo derivatives (6a-c), respectively. Cyclization of 6 b with ethanolic hydrogen chloride gave the 8-azachromone (7). Both 6 and 7 gave the same pyrazole derivative (8) upon the reaction with Phenylhydrazine. In the present work the interest emphasized the synthesis of new compounds of 2-substituted benzimidazole derivatives. Since these types of compounds had demonstrated biological activity in different areas of chemotherapy 1-7 . For these considerations, a new series of hetero- cyclic binary systems including the 2-benzimidazole moiety as well as pyridone moiety were synthesized. The a,/S-unsaturated ketones represent an active intermediate in the synthesis of heterocyclic com- pounds, hence we synthesized some chalcone analogues (la-h) in which one or the two phenyl rings is replaced by heterocyclic rings. m I-A- jtji lc: Ar = C 6 H 4 -2-OH, ld: Ar = C 6 H 4 -4-CH 3 , le: Ar = C 6 H 3 -2-Cl-4-N0 2 , If: 8 Ar = C 6 H 5 , 1 g: 8 Ar = C 6 H 4 -4-OCH 3 , lh: 8 Ar - C 6 H 4 -4-N(CH 3 ) 2 . la-h were prepared by CLAISEN-SCHMIDT con- densation of the appropriate aldehyde namely, 2-thiophenealdehyde, furfural, salicylaldehyde, p- tolualdehyde, 2-chloro-4-nitrobenzaldehyde, anis- aldehyde, and p-N-dimethylaminobenzaldehyde Requests for reprints should be sent to Dr. H. H. ZOOROB , Faculty of Science, Mansoura University, Chemistry Department, Mansoura, Egypt. with 2-acetylbenzimidazole 9 using methanol as a solvent and piperidine as base. 1 a-e were shown to be the arylidenes of 2-acetyl- benzimidazole from their IR spectra. They show the stretching frequency in the range 1685-1665 cm -1 characteristic for conjugated carbonyl group 10 . Here, we deal with the synthesis of 2-pyridones via the base catalyzed condensation of ethyl cyano- acetate and 2-cinnamoylbenzimidazole derivatives (lf-h in the presence of ammonium acetate (molar ratio 1:1:6) at 150-170 °C for 10 hours. Treating the reaction mixture with ethanol yielded two fractions: a) The ethanol insoluble part afforded the 3-cyano- pyridones (2a-c). Ar OOL-C HXT H H 2 2 a: Ar = C 6 H 5 , 2b: Ar = C 6 H 4 -4-OCH 3 , 2 c: Ar = C 6 H 4 -4-N(CH 3 ) 2 . b) The ethanol soluble part: gave on fractional crystallization (i) 3-cyanohexahydropyridones (3 a-c). Ar H H 3 3 a: Ar = CeH 5 , 3 b: Ar = C 6 H 4 -4-OCH 3 , 3c: Ar = C 6 H 4 -4-N(CH 3 ) 2 .
Transcript of Synthesi s of Pyridon e Derivatives - Max Planck...

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Synthesis of Pyridone Derivatives
MICHAEL Condensation with Ethyl Cyanoacetate,
Cyanoacetamide and Acetoacetamide
H . H . Z O O R O B a n d E . S . I S M A I L
Faculty of Science, Mansoura University, National Research Centre, Dokki, Cairo, Egypt
(Z. Naturforsch. 31b. 1680-1684 [1976]; received May 31, 1976)
2-Cinnamoylbenzimidazole, GRIGNARD Reagents, Azachromone, Pyrazole
MICHAEL condensation of 2-cinnamoylbenzimidazoles (la-h) with ethyl cyanoacetate, cyanoacetamide and acetoacetamide led to the formation of 2(1 H)pyridone derivatives (2a-c), (3a-c), 4a-c), and (5a-c). Compounds (2 a-c) added G R I G N A R D reagents at the nitrile group to give 5 a-c. CLAISEN condensation of 5a-c with ethyl acetate gave the corresponding diketo derivatives (6a-c), respectively. Cyclization of 6 b with ethanolic hydrogen chloride gave the 8-azachromone (7). Both 6 and 7 gave the same pyrazole derivative (8) upon the reaction with Phenylhydrazine.
In the present work the interest emphasized the synthesis of new compounds of 2-substituted benzimidazole derivatives. Since these types of compounds had demonstrated biological activity in different areas of chemotherapy1-7.
For these considerations, a new series of hetero-cyclic binary systems including the 2-benzimidazole moiety as well as pyridone moiety were synthesized.
The a,/S-unsaturated ketones represent an active intermediate in the synthesis of heterocyclic com-pounds, hence we synthesized some chalcone analogues (la-h) in which one or the two phenyl rings is replaced by heterocyclic rings.
m I-A- j t j i
l c : Ar = C6H4-2-OH, ld: Ar = C6H4-4-CH3, l e : Ar = C6H3-2-Cl-4-N02, I f : 8 Ar = C6H5 , 1 g : 8 Ar = C6H4-4-OCH3, lh : 8 Ar - C6H4-4-N(CH3)2.
la -h were prepared by C L A I S E N - S C H M I D T con-densation of the appropriate aldehyde namely, 2-thiophenealdehyde, furfural, salicylaldehyde, p-tolualdehyde, 2-chloro-4-nitrobenzaldehyde, anis-aldehyde, and p-N-dimethylaminobenzaldehyde
Requests for reprints should be sent to Dr. H. H. ZOOROB , Faculty of Science, Mansoura University, Chemistry Department, Mansoura, Egypt .
with 2-acetylbenzimidazole9 using methanol as a solvent and piperidine as base.
1 a-e were shown to be the arylidenes of 2-acetyl-benzimidazole from their IR spectra. They show the stretching frequency in the range 1685-1665 cm-1
characteristic for conjugated carbonyl group10. Here, we deal with the synthesis of 2-pyridones
via the base catalyzed condensation of ethyl cyano-acetate and 2-cinnamoylbenzimidazole derivatives (lf-h in the presence of ammonium acetate (molar ratio 1:1:6) at 150-170 °C for 10 hours. Treating the reaction mixture with ethanol yielded two fractions:
a) The ethanol insoluble part afforded the 3-cyano-pyridones (2a-c). Ar
OOL-CHXT H H
2 2 a: Ar = C6H5 , 2b: Ar = C6H4-4-OCH3, 2 c: Ar = C6H4-4-N(CH3)2.
b) The ethanol soluble part: gave on fractional crystallization (i) 3-cyanohexahydropyridones (3 a-c). Ar
H H 3
3 a: Ar = CeH5, 3 b: Ar = C6H4-4-OCH3, 3c: Ar = C6H4-4-N(CH3)2.

H. H. Zoorob-E. S. Ismail • Synthesis of Pyridone Derivatives 1681
(ii) 3-Carbethoxy-2-aminopyridine (4a-c).
o u u V r 5 H 4
4 a : Ar = C6H5 , 4b: Ar = C6H4-4-0CH3 , 4 c : A r = C 6 H 4 - 4 -N(CH 3 ) 2 .
In support for the structure assignment for 2 a-c are the following: a) Their correct molecular weights (osmometrically). b) Their IR spectra showed the carbonyl stretching
frequency in the range 1706-1649 cm-1, NH frequency in the range 3080-2729 cm-1 (which are characterized for the stretching frequencies in 2(1 H)pyridone11-13 and the C=N frequency at 2227 cm-1.
c) Their correct analytical data. Also, the structure of 4 a-c was supported by
their IR spectra. They showed well defined ab-sorption bands attributable for -NH (3289 cm-1
broad), C=0 (1684 cm-1) and C=NH (1613-1577 cm-1).
A possible explanation for the formation of 2, 3 and 4 via the reaction of ethyl cyanoacetate with 1 is the assumption that the reactants undergo M I C H A E L addition, followed by cyclization to form the pyridone derivative. Moreover, these reactions are accompanied with dehydrogenation.
The structure of 2 a-c was established by indepen-dent synthesis. Thus condensation of lf-h with cyanoacetamide in boiling butanol and few drops of piperidine as catalyst, gave 2a-C.
Furthermore, G R I G N A R D reaction on 2 a-c with subsequent decomposition and hydrolysis was studied. Optimum result have been found with the use of four moles of G R I G N A R D reagents to one mole of 3-cyanopyridone derivatives (2a-c). Thus, treat-ment of 2 a-c with methylmagnesium iodide yielded the corresponding 3-acetylpyridone derivatives (5a-c), respectively.
O u u i n r 3 H H
5
5 a: Ar = C6H5 , 5b : Ar = C6H4-4-OCH3, 5 c : Ar = C6H4-4-N(CH3)2.
In support for the products 5 a-c are the follow-ing: The IR spectra of 5 a-c are characterized by the band (strongest in the spectrum) at 1640-1620 cm-1 attributed to C=0 (amide), a relatively weak band at the range 1525-1505 cm-1
is present and is probably due to the skeletal C=C stretching frequency and at the range 3200-3000 cm-1 due to NH group.
The NMR spectrum of 5 a showed the following-assignments : a) r — 2.35 (for the acetyl-CH3 protons, singlet), b) r = 8.37-7.60 (for the aromatic protons, mul-
tiplet), c) r — 8.55 (for the NH protons, singlet).
The structure of 5 a-c, however, was rigidly established by independent synthesis. Thus conden-sation of 1 f-h with acetoacetamide in boiling alcoholic hydrogen chloride yielded 5 a-c, respec-tively.
Derivatives of 8-azachromone (4-oxo-4H-pyrano-[2,3-b]-pyridine are of interest as aza-analogue of chromone, since many chromones posses pharma-cological activity.
C L A I S E N condensation of 5 a-c with ethyl acetate in presence of sodium hydride or sodium dust gave the corresponding acetoacetyl derivatives (6a-c).
Ar
6
6 a: Ar = C6H5 , 6b: Ar = C6H4-4-OCH3, 6c: Ar = C6H4-4-N(CH3)2.
The IR spectrum of the acetoacetyl derivative (6 a) is characterized by the bands at 1620 cm-1
(C=0 amide) and at 1580 cm"1 (C=C). Cyclization of 6 b with ethanolic hydrogen chloride
gave 2-methyl-5-(25-anisyl)-7-(2-benzimidazolyl)-4H-pyrano-[2,3-b]pyridine-4-one (7).
7
Both 6 b and 7 gave the same pyrazole derivative (8) on reaction with Phenylhydrazine.
C0.CH2 .C0CH3 !=0

1682 H. H. Zoorob-E. S. Ismail • Synthesis of Pyridone Derivatives 1682
£-4 -OCH 3
— i T V c h 3 OH N _ N - C 6 H 5 H 8
The building of the pyrazole structure is expected in such reaction. The product 8 is soluble in alkali indicating a pyridone nucleus.
The NMR spectrum of 8 reveals the presence of three methyl protons which appeared as singlet at r — 7.61 ppm.
Experimental* All melting points were uncorrected and were
taken in a G A L L E N K A M P electric melting point apparatus and B O E T I U S melting point microscope.
The IR spectra were performed on a C A R L - Z E I S S Jena Infracord Spectrophotometer model "UR 10" using KBr.
The NMR spectra were obtained in deuterotri-fluoroacetic acid solution with the Varian Associates model "A-60".
2-(3-Substituted acryloyl)benzimidazoles (la-h) To a mixture of 2-acetylbenzimidazole (1.60 g,
0.01 mole), methanol (20 ml) and aldehyde (0.01 mole), four drops of piperidine were added. * The nomenclature of the compounds described in
this paper follows the rules of the American Chemi-cal Society and the Chemical Abstracts.
The reaction mixture was refluxed for 6 hours. The precipitated crystalline mass upon cooling was filtered off, recrystallized from the proper solvent to give la-h (cf. Table I). 1,2-Dihydro-2-oxo-4- (substituted )-6-(2-benzimidazolyl)nicotinonitrile (2 a-c) 3-Cyano-4- (substituted )-6-(2-benzim idazolyl) -hexahydro-2-pyridone (3 a-c) Ethyl 2-amino-4- (substituted)-6-(2-benzimidazolyl) nicotinic acid (4 a-c) Method (A): (without solvent):
A mixture of ethyl cyanoacetate (4.5 g, 0.04 mole), chalcone analogues (lf-h) (0.04 mole) and am-monium acetate (17.48 g, 0.24 mole) was heated at 160-170 °C for 8 hours (oil bath) and then allowed to cool. The yellow precipitate that obtained was washed with water, dried and then triturated with ethanol to give two parts:
i) Ethanol insoluble part which was isolated and recrystallized from the proper solvent to give 2 a-c (cf. Table II).
ii) Ethanol soluble part was concentrated and allowed to stand overnight at room temperature. An oily product was separated and triturated with cold ether, the resulting precipitate was crystallized from the proper solvent to give 3 a-c (cf. Table III). The mother liquor was diluted with water and the formed precipitate was recrystallized from the proper solvent to give 4 a-c (cf. Table IV). Method (B): (in butanol):
A mixture of ethyl cyanoacetate (4.5 g, 0.04 mole), chalcone analogues (lf-h) (0.04 mole) and am-
Table I. 2-(3-Substituted acryloyl)benzimidazoles (la-e).
Analysis [ %] Compound m.p. Yield Solvent Formula Carbon Hvdrogen Nitrogen
[°C] [%] of Cryst. Calcd Found Calcd Found Calcd Found
l a 222 71 ethanol C I 4 H I 0 N 2 S O 66.14 66.05 3.99 4.14 1 1 .05 10.89 lb 216 69 methanol C14H10N2O0 70.58 70.57 4.50 4.67 11 .76 12.09 l c 105 73 chloroform/pet. C17H14N2O 71.86 71.50 5.34 5.18 10.68 10.28
ethei ld 205 82 1,2-dichloro CI6HI2N202 71.42 70.96 5.26 5.46 10.52 10.32
ethane le 244 78 methanol CieHioNsOsCl 58.71 58.74 3.05 2.84 12.84 13.20
Table I I . l,2-Dihydro-2-oxo-4-substituted-6-(2-benzimidazolyl)nicotinonitrile (2a-c).
Analysis [%] Compound m.p. Yield Solvent Formula Carbon Hydrogen Nitrogen
[cC] [%] of Cry st. Calcd Found Calcd Found Calcd Found
2a above 350
25 acetic acid CI9HI2N40 73.07 72.98 3.84 4.16 17.94 18.12
2b above 350
22 acetic acid C20H14N4O 70.17 70.47 4.09 4.23 16.37 16.52
2 c above 28 D . M . F . C21H17N5O 70.98 70.61 4.78 4.86 19.71 19.62

H. H. Zoorob—E. S. Ismail • Synthesis of Pyridone Derivatives
Table I I I . 3-Cyano-4-substituted-6-(2-benzimidazolyl)hexahydro-2-pyridone (3a-c).
1683
Compound m.p. Yield Solvent Formula [°C] [%] of Cryst.
3 a 3 5 1 2 0 D . M . F . C I 9 H I 6 N 4 0
3 b 1 9 0 2 3 methanol C 2 0 H I 8 N 4 O 2
3 c 2 1 0 2 1 xylene C 2 1 H I 2 N 5 0
Compound m.p. Yield Solvent Formula [°C] [%] of Cryst.
4 a 205 15 methanol C2 iHi8N402
4 b 342 17 acetone C22H20N4O3
4 c 230 2 1 benzene C 2 3 H 2 3 N 5 0 2
Analysis [ % ] Carbon Hydrogen . Nitrogen
Calcd Found Calcd Found Calcd Found
72.16 72.41 5.06 4.86 17.75 17.47 69.36 68.91 5.20 5.38 16.18 16.32 70.19 70.06 5.84 5.53 19.49 19.42
Analysis [%] Carbon Hydrogen Nitrogen
Calcd Found Calcd Found Calcd Found
70.39 70.62 5.02 4.73 15.64 15.72 68.04 68.34 5 . 15 5.37 14.43 14.58 68.88 69.19 5.73 5.42 17.45 17.28
Table IV. Ethyl 2-amino-4-substituted-6-(2-benzimidazolyl)nicotmic acids (4a-c).
monium acetate (17.48 g, 0.24 mole) in 20 ml butanol was refluxed for 10 hours. The yellow precipitate that obtained on cooling was separated and recrystallized from the proper solvent to give 2 a-c (c/. Table II). Method (C):
A mixture of cyanoacetamide (0.84 g, 0.01 mole), chalcone analogues (1 f-h) (0.01 mole) and two drops of piperidine in 10 ml butanol was refluxed for 10 hours. The most of the alcohol was evaporated, the yellow crystalline mass that separated after cooling was filtered off and dried, recrystallized from the appropriate solvent to give 2a-c (cf. Table II).
No depression occurs in the admixed m.p. be-tween both of each products 2 a-c obtained by methods (B) and (C) and that obtained from method (A). 3-Acetyl-4-substituted-6-(2-benzimidazolyl )-2-[lH[pyridones (5 a-c)
A suspension of 2 a, b and c (0.0072 mole) in dry benzene (50 ml) was added dropwise to an etherial solution of methylmagnesium iodide (prepared from 0.9 g magnesium, 7 g methyl iodide and 40 ml dry ether) the reaction mixture was refluxed on a steam bath for two hours, set aside at room temperature, and then decomposed with a cold solution of hydro-chloric acid (10%). The yellow solid obtained was
crystallized from the proper solvent to give 5 a - c (cf. Table V).
Preparation of an authentic samples of 5 a - c A mixture of acetoacetamide (2.02 g, 0.02 mole),
lf-h (0.02 mole) and (20 ml of 10%) ethanolic hydrogen chloride was heated under reflux for 30 minutes. The reaction mixture was cooled and treated with ammonium hydroxide solution (25%), the solid product was collected and recrystallized from the proper solvent to give 5 a - c (cf. Table V).
No depression occurs in the admixed m.p. be-tween both of each products 5 a - c obtained by the two methods.
l-[ l,2-Dihydro-2-oxo-4-substituted-6-(2-benzimidazolyl)-3-pyridyl]-1,3-butadiones ( 6 a - c ) Method (a):
A mixture of 5a-c (0.0033 mole), ethyl acetate (3 ml; 0.0031 mole), dry dioxane (20 ml) and 50% suspension of sodium hydride in oil (1.2 g) was refluxed for 30 minutes. The mixture was cooled, then stirred into water (50 ml) and 2 N hydrochloric acid (5 ml) was added to give 6a-c (cf. Table VI). Method (b):
A mixture of 5a-c (0.0033 mole), ethyl acetate (excess) (as a solvent and reactant) and granulated
Table V. 3-Acetyl-4-substituted-6-(2-benzimidazolyl)-2-[l HJpyridones (5 a-c).
Analysis [ %] Compound m.p. Yield Solvent Formula Carbon Hydrogen Nitrogen Compound
[°C] [%] of Cryst. Calcd Found Calcd Found Calcd Found
5a 320 92 methanol C2oHi5N302 72.92 72.90 4.55 4.32 12.76 12.58 5b 325 95 ethanol C 2 iHi7N 30 3 70.19 69.96 4.73 5.16 11.69 1 1 . 53 5c 327 89 acetone C22H2qN402 70.43 70.18 5.37 5.40 15.05 15.21
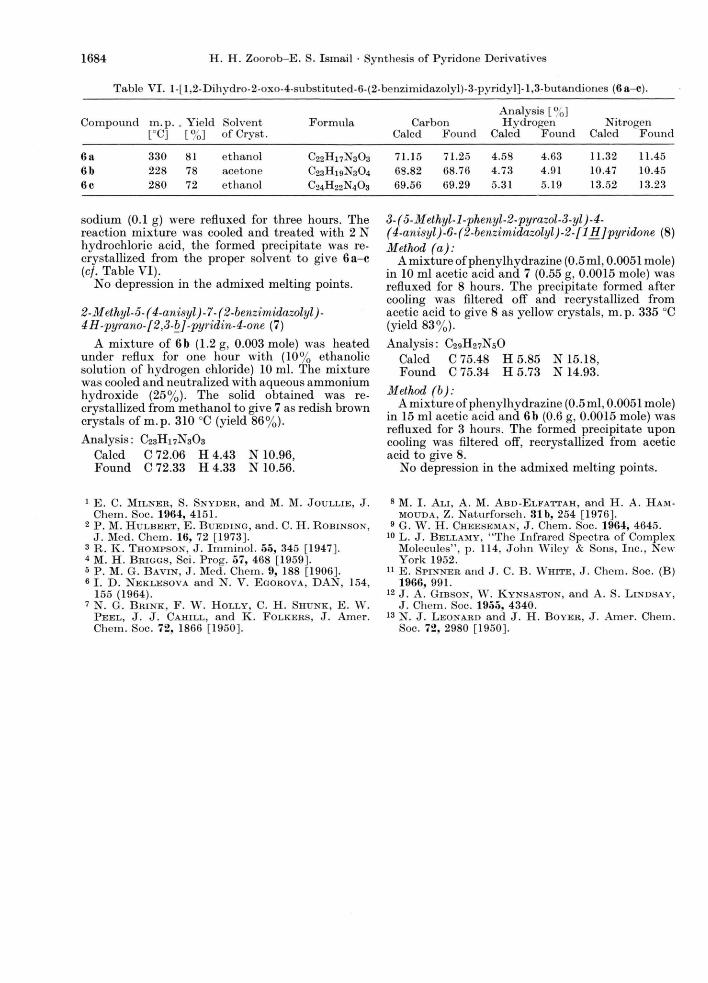
H. H. Zoorob-E. S. Ismail • Synthesis of Pyridone Derivatives 1684
Table VI . l-[1.2-Dihydro-2-oxo-4-substituted-6-(2-benzimidazolyl)-3-pyridyl]-l,3-butandiones (6a-c).
Analysis [%] Compound m.p. . Yield Solvent Formula Carbon Hydrogen Nitrogen
[°C] [%] of Cry st. Calcd Found Calcd Found Calcd Found
6 a 330 81 ethanol C22H 1 7N 303 7 1 . 1 5 71.25 4.58 4.63 1 1 . 32 1 1 .45 6b 228 78 acetone C23H19N304 68.82 68.76 4.73 4.91 10.47 10.45 6c 280 72 ethanol C24H22N403 69.56 69.29 5.31 5.19 13.52 13.23
sodium (0.1 g) were refluxed for three hours. The reaction mixture was cooled and treated with 2 N hydrochloric acid, the formed precipitate was re-crystallized from the proper solvent to give 6 a - c {cf. Table VI).
No depression in the admixed melting points.
2-Methyl-5- ( 4-anisyl)-7- ( 2-benzimidazolyl )-4H-pyrano-[2,3-b]-pyridin-4-one (7)
A mixture of 6 b (1.2 g, 0.003 mole) was heated under reflux for one hour with (10% ethanolic solution of hydrogen chloride) 10 ml. The mixture was cooled and neutralized with aqueous ammonium hydroxide (25%). The solid obtained was re-crystallized from methanol to give 7 as redish brown crystals of m.p. 310 °C (yield 86%). Analysis: C23H17N3O3
Calcd C 72.06 H 4.43 N 10.96, Found C 72.33 H 4.33 N 10.56.
3-(5-Methyl-l-phenyl-2-pyrazol-3-yl )-4-(4-anisyl)-6- (2-benzimidazolyl)-2-[ 1H]pyridone (8)
Method (a): A mixture of Phenylhydrazine (0.5ml, 0.0051 mole)
in 10 ml acetic acid and 7 (0.55 g, 0.0015 mole) was refluxed for 8 hours. The precipitate formed after cooling was filtered off and recrystallized from acetic acid to give 8 as yellow crystals, m.p. 335 °C (yield 83%). Analysis: C29H27N5O
Calcd C 75.48 H 5.85 N 15.18, Found C 75.34 H 5.73 N 14.93.
Method (b): A mixture of Phenylhydrazine (0.5ml, 0.0051 mole)
in 15 ml acetic acid and 6b (0.6 g, 0.0015 mole) was refluxed for 3 hours. The formed precipitate upon cooling was filtered off, recrystallized from acetic acid to give 8.
No depression in the admixed melting points.
1 E . C . M I L N E R , S . S N Y D E R , a n d M . M . JOIILLIE , J . Chem. Soc. 1964, 4 1 5 1 .
2 P . M . H U L B E R T , E . B U E D I N G , a n d . C . H . R O B I N S O N , J . Med. Chem. 16, 72 [1973].
3 R . K . T H O M P S O N , J . Imminol. 5 5 , 3 4 5 [ 1 9 4 7 ] . 4 M . H . B R I G G S , Sei. Prog. 5 7 , 4 6 8 [ 1 9 5 9 ] . 5 P. M. G. B A V I N , J . Med. Chem. 9, 188 [1906]. 6 I . D . N E K L E S O V A a n d N . V . E G O R O V A , D A N , 1 5 4 ,
1 5 5 ( 1 9 6 4 ) . 7 N . G . B R I N K , F . W . H O L L Y , C . H . S H U N K , E . W .
P E E L , J . J . C A H I L L , and K . F O L K E R S , J . Amer. Chem. Soc. 72, 1866 [1950].
8 M . I . A L I , A . M . A B D - E L F A T T A H , a n d H . A . H A M -M O U D A , Z. Naturforsch. 3 1 b , 254 [1976].
9 G . W. H. C H E E S E M A N , J . Chem. Soc. 1964, 4645. 1 0 L . J . B E L L A M Y , "The Infrared Spectra of Complex
Molecules", p. 1 14 , John Wiley & Sons, Inc., New York 1952.
1 1 E . S P I N N E R and J . C. B . W H I T E , J . Chem. Soc. ( B ) 1966, 991.
1 2 J . A . G I B S O N , W . K Y N S A S T O N , a n d A . S . L I N D S A Y , J. Chem. Soc. 1955, 4340.
1 3 N. J . L E O N A R D and J . H . B O Y E R , J . Amer. Chem. Soc. 72, 2980 [1950].


















