
Theoretische Chemie/Quantenchemie II · Einleitung MD-Simulation Statistische Physik Monte-Carlo...
62
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen Theoretische Chemie/Quantenchemie II Teil III: Mikroskopische Beschreibung und Simulation molekularer Systeme Stefanie Gr¨ afe & Dirk Bender Friedrich-Schiller-Universit¨ at Jena, Institut f¨ ur Physikalische Chemie Sommersemester 2018 St. Gr¨ afe/D. Bender Theoretische Chemie/Quantenchemie II 22
-
Upload
hoangkhanh -
Category
Documents
-
view
218 -
download
0
Transcript of Theoretische Chemie/Quantenchemie II · Einleitung MD-Simulation Statistische Physik Monte-Carlo...

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Theoretische Chemie/Quantenchemie IITeil III: Mikroskopische Beschreibung und Simulation molekularer
Systeme
Stefanie Grafe & Dirk Bender
Friedrich-Schiller-Universitat Jena, Institut fur Physikalische Chemie
Sommersemester 2018
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 22

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Gliederung
5 Einleitung
6 Molekulardynamische Simulation
7 Grundbegriffe der Statistischen Physik
8 Monte-Carlo-Simulationen
9 Fourier-Transformation, Fourier-Reihen und Fourier-Analyse
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 23

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Einleitung
Modellbildungreales System Modellsystem
ExperimentComputer-
simulation
approximative
Theorie
experimentelle Resultate
Resultate fur das Modell
theoretische Vorhersage
Vergleich Vergleich
Test des Modells Test der Theorie
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 24
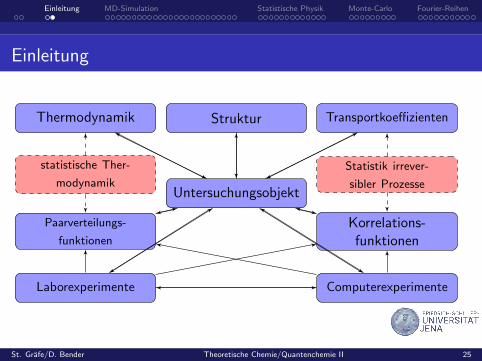
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Einleitung
Untersuchungsobjekt
StrukturThermodynamik Transportkoeffizienten
statistische Ther-
modynamik
Statistik irrever-
sibler Prozesse
Paarverteilungs-
funktionen
Korrelations-funktionen
Laborexperimente Computerexperimente
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 25

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Grundlagen
Grundgedanken
Eine MD-Simulation ist die numerische Losung der mechanischenBewegungsgleichungen eines Systems aus N Teilchen.
Durch Mittelwertsbildung erhalt man die interessierendenphysikalischen Großen (z. B. Verteilungsfunktionen,Diffusionskoeffizienten, Halbwertsbreiten und Linienverschiebungen imSchwingungsspektrum).
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 26
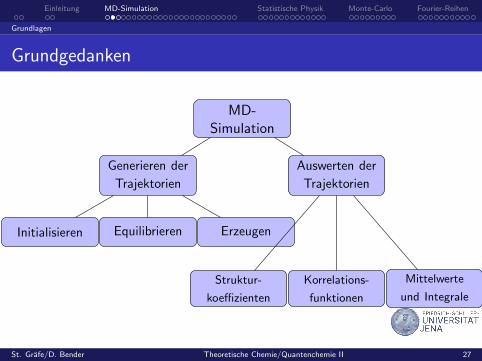
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Grundlagen
Grundgedanken
MD-Simulation
Generieren der
Trajektorien
Auswerten der
Trajektorien
Equilibrieren ErzeugenInitialisieren
Korrelations-
funktionen
Struktur-
koeffizienten
Mittelwerte
und Integrale
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 27

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Grundlagen
Ablauf einer MD-Simulation
START
Berechnung von Orten
und Geschwindigkeiten
Berechnung der Krafte
Auswertung
Laufendeerreicht?
ENDE
ja
nein
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 28

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Intra- und intermolekulare Wechselwirkungspotentiale
Alle makroskopischen Eigenschaften haben ihre Ursache in den Kraften zwischenden elementaren Bausteinen der Materie. Wesentliche Quellen zum Verstandnisdieser Krafte sind:
Streuexperimente (Rontgen-, Neutronenstreuung)
Schwingungsspektroskopie (IR, Raman,. . . )
thermophysikalische Daten (Virialkoeffizienten, spezifische Warme, . . . )
Festkorpereigenschaften (Elastizitatskonstanten, Phononenspektren, . . . )
Hochfrequenzspektroskopie (NMR, EPR)
Es besteht ein komplizierter funktionaler Zusammenhang zwischen dengemessenen Großen und den Wechselwirkungsparametern. SystematischeAnpassung der Modellpotentiale an experimentelle Daten.
U( ~rj ) =∑i
u1(~ri) +∑j>i
u2(~ri, ~rj) +∑k>j>i
u3(~ri, ~rj , ~rk) + . . .
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 29

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Quantenmechanik der zwischenmolekuren Wechselwirkung
HAB = HA + HB + VAB
VAB = −∑i∈Aβ∈B
Zβ∣∣∣~Rβ − ~ri∣∣∣ −∑j∈Bα∈A
Zα∣∣∣~Rα − ~rj∣∣∣+∑i∈Aj∈B
1
|~rj − ~ri|+∑α∈Aβ∈B
ZαZβ∣∣∣~Rβ − ~Rα
∣∣∣HABΨAB = EABΨAB
HAΨA = E0AΨA
HBΨB = E0BΨB
U(~RAB) = EAB(~RAB)−(E0A + E0
B
)
R
UAustausch-WW
van-der-WaalsES-, Induktions- und Dispersions-WW
kleine Abstande: elektrostatischeAbstoßung
mittlere Abstande: Abstoßung ∼=Anziehung, alle WW-Typen:
elektrostatischeMultipol-Multipol-WWPolarisations-WWDispersions-WW
große Abstande: im Wesentlichenanziehende Dispersionswechselwirkung
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 30

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Klassische Ansatze
Mechanische Modelle zur Nahordnung von Flussigkeiten
1. Potential harter Kugeln
U(r) =
∞, r ≤ σ0, r > σ
U(r)
rσ
2. Rechteckpotential
U(r) =
∞, r < σ
−ε, σ < r < rmax
0, r > σ
U(r)
rσε
rmax
Bedeutung der Parameter:σ rmax εTeilchendurchmesser Wirkungsbereich der attraktiven Wechselwirkung Starke der attraktiven Wechselwirkung
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 31

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Storungstheorie der langreichweitigen Wechselwirkung
Storungstheorie 1. Ordnung:
u(1)2 (~rAB) =
⟨Ψ0AΨ0
B
∣∣∣ VAB ∣∣∣Ψ0AΨ0
B
⟩Beschreibt die elektrostatische Wechselwirkung zwischen den ungestorten
Ladungsverteilungen A und B. Kann attraktiv oder repulsiv sein.Storungstheorie 2. Ordnung:
u(21)2 = −
∑a6=0
∣∣∣⟨Ψ0AΨ0
B
∣∣∣ VAB(~rAB)∣∣∣Ψa
AΨ0B
⟩∣∣∣2EaA − E0
A
−∑b6=0
∣∣∣⟨Ψ0AΨ0
B
∣∣∣ VAB(~rAB)∣∣∣Ψ0
AΨbB
⟩∣∣∣2EbB − E0
B
u(22)2 = −
∑a6=0b6=0
∣∣∣⟨Ψ0AΨ0
B
∣∣∣ VAB(~rAB)∣∣∣Ψa
AΨbB
⟩∣∣∣2EaA − E0
A + EbB − E0B
Beitrage der dynamischen Polarisation und Dispersionswechselwirkung (Korrelation).Immer attraktiv.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 32

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Potentialfunktionen bei mittleren und großen Abstanden
Fur mittlere und große Abstande sind Quanteneffekte vernachlassigbar.Der Rechenaufwand einer quantenmechanischen Potentialberechnung istunverhaltnismaßig groß.
=⇒Potentialverlauf durch analytische Funktionen annahern
attraktive Wechselwirkung:Multipolentwicklungsinnvoller Ansatz:
uPD2 = −C6
r6
Hohere Potenzen werden vernachlassigt.Sutherland-Potential:
U(r) =
∞, r ≤ σ− crm , r > σ
repulsive Wechselwirkung:Fur kurze Abstande dominiert dieabstoßende Wechselwirkung(Austauscheffekte).Uberlappintegral ∝ exp(−ζr)haufig ∝ r−n angenahert:
U(r) =d
rn, r < σ
d und n (meist 9 < n < 15) bestimmendie Starke der Abstoßung
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 33

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Potential der nichtbindenden Wechselwirkung
r/σ
U(r)/ε
01
Unb(~r) = U el(~r) +C12
r12− C6
r6
(allgemeines) Lennard-Jones-Potential LJ(m,n)
U(r) =1
n−m
(nn
mm
) 1n−m
ε((σ
r
)n−(σr
)m)meist LJ(6,12) U(r) = 4ε
((σr
)12 − (σr
)6)Buckingham-Potential U(r) = b exp(−ar)− c
r6− d
r8
U(r) =
ε
1− 6α
(6α
exp(α(
1− rrmax
))−(
rrmax
)6), r ≥ rmax
∞, sonst
U el Wechselwirkung zwischen eventuell vorhandenen permanenten Multipolen(Ion-Ion-, Ion-Dipol-, Dipol-Dipol-WW,.. . . ).
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 34

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Potentiale
Weitere Uberlegungen
Potentiale durch außere Felder
molekulare Systeme
starre MolekulmodelleModelle mit inneren Freiheitsgraden (vgl. Kapitel Kraftfeldmethoden)
Valenzkraftfeldinnere KoordinatenMorse-PotentialTorsionsenergie
Wasserstoffbrucken
Bestimmung der Potentialparameter
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 35

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
Verlet-Algorithmus
Zu losen sind die Newtonschen Differentialgleichungen:
miqi = Fi mit Fi =∂U
∂qi.
Addiert man zur Taylorentwicklung von ~ri zum Zeitpunkt t+ δt
~ri(t+ δt) = ~ri + ~viδt+δt2
2~ri(t) + · · ·
die zeitinverse
~ri(t− δt) = ~ri − ~viδt+δt2
2~ri(t)− · · ·
so erhalt man
~ri(t+ δt) = 2~ri(t)− ~ri(t− δt) + δt2~ri(t) .
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 36

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
Verlet-Algorithmus
klassischer Verlet-Algorithmus
die Geschwindigkeit ~vi tritt nicht explizit auf
lasst sich aus der Zentraldifferenz
~vi(t) =~ri(t+ δt)− ~ri(t− δt)
2δt
bestimmen.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 37

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
Geschwindigkeits-Verlet-Algorithmus
Der velocity-Verlet-algorithm (VVA) beseitigt diesen Nachteil.
~ri(t+ δt) = ~ri(t) + δt~vi(t) +δt2
2mi
~Fi(t)
~vi(t+ δt) = ~vi(t) +δt
2mi
(~Fi(t) + ~Fi(t+ δt)
)
vollig aquivalent zum klassischen Verlet-Algorithmus
Vorteil fur beide ist die exakte Reversibilitat.
daraus resultiert eine hohe Stabilitat der Gesamtenergie
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 38

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
mehratomige Molekule
1 verallgemeinerte Koordinaten, numerisches Losen derLagrange-Gleichungen zweiter Art.
2 Ensemble von Atomen, chemische Bindung wird beschrieben durchentsprechende Potentialfunktionen oder verallgemeinertePaarpotentiale
3 Kompromiss ist die Methode der Lagrange-Gleichungen erster Art.Auch hier Ensemble von Atomen, die feste Geometrie wird durchholonome Nebenbedingungen gesichert.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 39
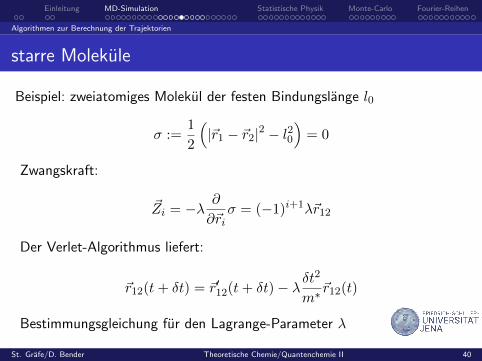
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
starre Molekule
Beispiel: zweiatomiges Molekul der festen Bindungslange l0
σ :=1
2
(|~r1 − ~r2|2 − l20
)= 0
Zwangskraft:
~Zi = −λ ∂
∂~riσ = (−1)i+1λ~r12
Der Verlet-Algorithmus liefert:
~r12(t+ δt) = ~r′12(t+ δt)− λδt2
m∗~r12(t)
Bestimmungsgleichung fur den Lagrange-Parameter λ
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 40
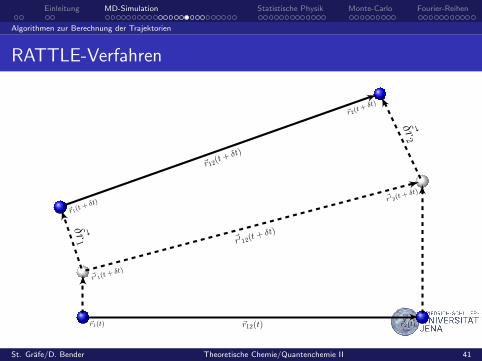
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
RATTLE-Verfahren
~r1(t) ~r12(t) ~r2(t)
~r′ 1(t+ δt)
~r′ 12(t+ δt)
~r′ 2(t+ δt)
~r1(t+ δt)
~r12(t+ δt)
~r2(t+ δt)
~δr1
~δr2
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 41
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
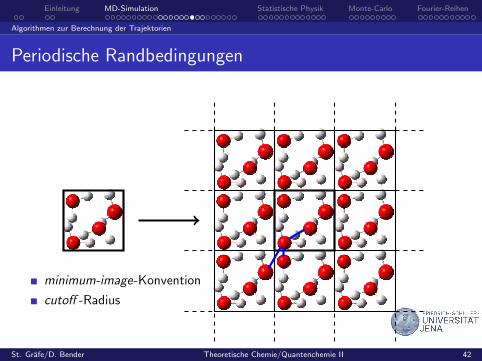
Periodische Randbedingungen
minimum-image-Konvention
cutoff -Radius
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 42

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
langreichweitige Wechselwirkungen
elektrostatische Wechselwirkung
Ewald-Summation
U el =1
2
∑′
~n
N∑i=1
N∑j=1
4πεQiQj|~ri − ~rj + ~n|
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 43

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Algorithmen zur Berechnung der Trajektorien
Simulationen bei konstanter Temperatur
Experimente werden fast immer bei vorgegebener Temperaturdurchgefuhrt
im NVE-Ensemble schwankt die Temperatur aber
Skalierung der Geschwindigkeiten mit dem Faktor√T0/T
Nose-Hoover-Thermostat
~ri =~Fimi− ζ~ri, ζ =
1
Q
( N∑i=1
mi~r2i − 3kBT
)
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 44

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Auswertung der MD-Laufe
Auswertung
Schließen aus Bewegungsablaufen und der raumlichen Anordnungeiniger hundert Teilchen auf makroskopische Eigenschaften
Großen sofort berechnen oder Trajektorien abspeichern
Orts-, Geschwindigkeits- und Orientierungsvektoren der untersuchtenMolekule
ggfs. weitere Großen
Korrelation aufeinanderfolgender Zeitschritte
zeitgeordnete Informationen
statische und dynamische Großen bestimmbar
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 45

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Auswertung der MD-Laufe
Grundlegende Eigenschaften
Kinetische, potentielle und Gesamtenergie
Temperatur uber den Gleichverteilungssatz aus der kinetischenEnergie ⟨
N∑i=1
mi
2~v2i
⟩=
3
2NkBT
Druck aus dem Virial
p =2
3V〈Ekin〉+
1
3V
⟨ N∑i=1
N∑j=i
~Fij~rij
⟩.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 46

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Auswertung der MD-Laufe
Statische Großen (Strukturinformationen)
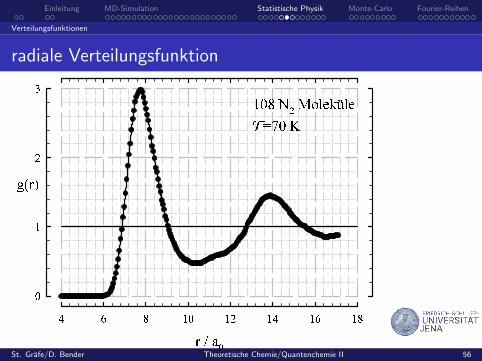
radiale Verteilungsfunktion g(r)
g(r) = %−2
⟨ N∑i=1
N∑′
j=1
δ(~ri)δ(~rj − ~r)
⟩=
V
N 2
⟨ N∑′
i=1
N∑j=1
δ(~r − ~rij)
⟩.
vereinfacht: Zahl der Teilchen in einem Abstand r um ein gegebenesTeilchen – relativ zur entsprechenden Zahl des idealen Gases gleicherDichte und Temperatur
Haufigkeitsverteilung (Histogramm) fur Intervalle der Breite δr
atomare Systeme bzw. Schwerpunktsverteilung von Molekulen
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 47

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Auswertung der MD-Laufe
Dynamische Großen
Korrelationsfunktionen fur Positionen, Geschwindigkeiten,Orientierung, . . .
〈AB(i)〉 =1
jmax
jmax∑j=1
AjBj+i .
in der Praxis: anderes Verfahren
mittlere quadratische Verschiebung (MSD)
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 48
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Auswertung der MD-Laufe
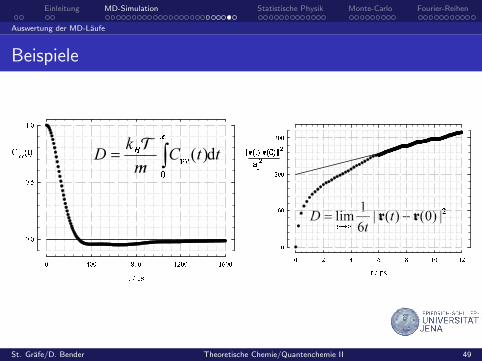
Beispiele
ttCkD d)(
|)0()(|61lim rr tt
D
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 49

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Auswertung der MD-Laufe
Fehlerbetrachtung
Auftreten zufalliger Fehlern
die momentanen Werte aller untersuchten Großen (Energie, Druck,etc.) schwanken um ihre Mittelwerte.
nichttrivale Fehlerabschatzung, da aufeinanderfolgende Werte Gieiner Meßgroße G nicht statistisch unabhangig voneinander sind
systematische Fehler
z. B. bedingt durch das Abschneiden des Potentials
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 50

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Phasenraum
Phasenraum
geometrische Lage aller Teilchen zu einem Zeitpunkt
zusatzlich Geschwindigkeiten oder Impulse
Phasenraum, Ortsraum, Impulsraum
Molekul-Phasenraum (µ-Raum)
2f -dimensionaljedes Teilchen ist Punkt im µ-Raumideale Systeme
Gas-Phasenraum (Γ-Raum)
ganzes System als UbermolekulSystem entspricht einem Punkt im Γ-Raumvirtuelle Gesamtheiten
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 51

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Phasenraum
virtuelle Gesamtheiten
1 abgeschlossene Systeme,Systeme ohne Energie- oderMaterieaustausch
2 geschlossene Systeme,Systeme mit Energie-, aberohne Materieaustausch
3 offene Systeme, Systeme mitEnergie- undMaterieaustausch
1 mikrokanonischeGesamtheit, EVN ,% ∝ δ(H − E)
2 kanonische Gesamtheit,NVT , % ∝ exp(− H
kBT )
3 großkanonische Gesamtheit,µVT , % ∝ exp(−H−µN
kBT )
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 52

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Phasenraum
Mittelwerte
Messmittelwert
〈A〉 =1
l∆t
l∑j=0
A(j∆t)
Zeitmittelwert
〈A〉 = limτ→∞
1
τ
∫ τ
0A(q(t), p(t)) dt
Ensemblemittelwert
〈A〉 =
∫∫A(q(t), p(t))%(q(t), p(t)) dq dp
nach der Ergodenhypothese sind Zeit- und Ensemblemittel aquivalent
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 53

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Phasenraum
Phasenraumdichten
Wahrscheinlichkeitsdichte %(~r1, . . . , ~rN , ~p1, . . . , ~pN )
reduzierte n-Teilchendichten
%(n)(~r1, . . . , ~rn, ~p1, . . . , ~pn)
=N !
(N − n)!
∫· · ·∫%(~r1, . . . , ~rN , ~p1, . . . , ~pN ) d3~rn+1 · · · d3~pN
Spezialfall n = 1:∫V
%(1)(~r) d3~r = N .
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 54

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Verteilungsfunktionen
Verteilungsfunktionen
aus den %(n) lassen sich neue Funktionen definieren
g(n)(~r1, . . . , ~rn) :=%(n)(~r1, . . . , ~rn)
n∏i=1
%(~ri)
g(n) heißen n-Teilchenverteilungsfunktionen
Paarverteilungsfunktion g(2) ≡ g(~r1, ~r2)
g reprasentiert die Nahordnunggeht fur große Abstande gegen EinsHangt g nur vom Abstand der beteiligten Teilchen ab, so nennt mansie radiale Verteilungsfunktion.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 55
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Verteilungsfunktionen
radiale Verteilungsfunktion
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 56

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Verteilungsfunktionen
molekulare Paarverteilungsfunktionen
Im Falle molekularer Flussigkeiten tritt an Stelle der radialenVerteilungsfunktion g(r) eine Funktion g(~R, ~Ω1, ~Ω2, ~Ω12), die von denPositionen und Orientierungen der beteiligten Molekule abhangt. Diesemolekularen Paarverteilungsfunktionen (MPDF) sind sehr kompliziertstrukturiert. Zu ihrer Handhabung bieten sich zwei Wege:
1 Entwicklung nach Wignermatrizen (oder fur lineare MolekuleKugelflachenfunktionen)
2 Atom-Atom-Paarverteilung
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 57

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Molekulare Dynamik
Korrelationsfunktionen
Die Molekuldynamik wird beschrieben durch Korrelationsfunktionen(CF).Sie geben die Wechselbeziehung einer Systemeigenschaft zu einemReferenzzeitpunkt t0 mit einer Eigenschaft zu einem anderenZeitpunkt t an. Hierbei kann es sich um gleiche oder verschiedeneEigenschaften handeln.Setzt man die Stationaritat des Systems voraus, so wahlt mangewohnlich t0 = 0 und erhalt die CF in allgemeiner Gestalt:
GAB(t) :=⟨Ai(0)|B∗j (t)
⟩≡⟨AiB
∗j (t)
⟩.
A und B sind hierbei beliebige dynamische Variablen (z. B. ~ri, ~vi, ~Ωi,~ωi, usw.).∗ bedeutet die komplexe Konjugation.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 58

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Molekulare Dynamik
Korrelationsfunktionen
Die Indizes i und j beziehen sich auf die Teilchen. Damit kann man dieKorrelationsfunktionen klassifizieren als:
1a) Autokorrelationsfunktionen (ACF) fur A = B,
1b) Kreuzkorrelationsfunktionen (CCF) fur A 6= B,
2a) Einteilchen-CF fur i = j und
2b) Mehrteilchen-CF fur i 6= j
Haufig erweist es sich als sinnvoll, normierte Korrelationsfunktionen unddaraus abgeleitete Korrelationszeiten τAB zu verwenden:
GAB(t) :=〈A(0)B(t)〉〈A(0)B(0)〉
τAB :=
∞∫0
GAB(t) dt .
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 59

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Molekulare Dynamik
Korrelationsfunktionen und Transportprozesse
mikroskopischen Beschreibung von Transportprozessen
CF ist mit makroskopischen Transportkoeffizienten verknupft
Beispiel: Diffusion
verknupft werden die (normierte)Geschwindigkeitsautokorrelationsfunktion (VACF)
G ~vv(t) :=〈~v~v(t)〉〈~v2〉
und der Selbstdiffusionskoeffizient D
Letzteren kann man aus der mittleren quadratischen Verschiebung(MSD) bestimmen:
D = limt→∞
1
6t
⟨|~r(t)− ~r(0)|2
⟩.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 60

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Molekulare Dynamik
Korrelationsfunktionen und Transportprozesse
Diese Einsteinbeziehung folgt unmittelbar aus dem FickschenDiffusionsgesetz.
Die Diffusion ist auch ein Beispiel fur die “random walk” genanntestochastische Bewegung. Fur diese ist bekannt, dass die MSD des
”Spaziergangers“ nach hinreichend langer Zeit eine lineare Funktion
von t wird.
Der Diffusionskoeffizient laßt sich nun auch aus der CF berechnen:
D =kBTm
∞∫0
Gvv(t) dt
Green-Kubo-Relationen
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 61

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Molekulare Dynamik
Korrelationsfunktionen und Spektroskopie
Korrelationsfunktion G(t) ←→ spektrale Leistungsdichte J(ω)
Beide enthalten identische Informationen uber den stochastischenProzess (Wiener-Chintchine-Theorem).
Frequenz- und Zeitdomane sind uber eine Fouriertransformationmiteinander verknupft:
G(t) =
∞∫−∞
J(ω) exp(− iωt) dω
J(ω) =1
π
∞∫0
exp( iωt)G(t) dt .
Ubergang zwischen Dynamik und Spektroskopie
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 62

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Molekulare Dynamik
Spektralmomente
normierte Spektralmomente (n-ter Ordnung)
Mn :=
∞∫−∞
ωnJ(ω)
J(0)dω
Konvergenz der Integrale wird vorausgesetzt. (Beispiel:Lorentzbanden!)
Korrelationsfunktion in eine Taylorreihe entwickeln
G(t) =
∞∑n=0
ann!tn
Spektralmomente stimmen betragsmaßig mit denEntwicklungskoeffizienten uberein an = (− i)nMn.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 63

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Mathematische Statistik
Mathematische Statistik
Mittelwerte
〈A〉 =∑x∈Ω
P (x)A(x)
〈A〉 =
∫∫ΓA(q, p, t)%(q, p, t) dq dp
Gesetz der großen Zahlen
limn→∞
P (∣∣Xn
∣∣ > ε) = 0 (schwach)
P
(lim sup
n→∞
∣∣Xn
∣∣ = 0
)= 1 (stark)
zentraler Grenzwertsatz
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 64

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Mathematische Statistik
Einleitung
1 Aufbau eines adaquaten stochastischen Modells
2 Durchfuhrung einer großen Zahl von Zufallsexperimenten
3 Auswertung dieser Experimente mit statistischen Methoden
4 Interpretation der Schatzwerte als Problemlosung
einfache oder naive Verfahren
importance-sampling -Verfahren
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 65
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Mathematische Statistik
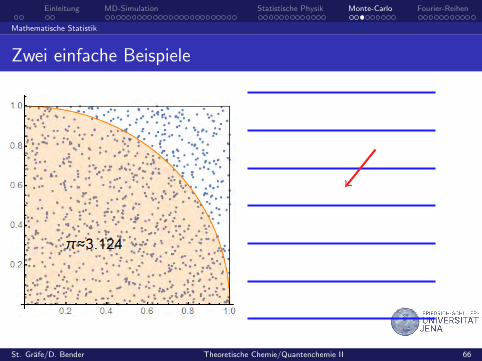
Zwei einfache Beispiele
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 66

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Einfache (naive) MC-Verfahren
Einfache (naive) MC-Verfahren
Zufallsexperimente mit gleichverteilten Zufallszahlen
Volumen- und Oberflachenberechnung
zufallig besetzte Gitter, random walk
porose Medien
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 67

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Einfache (naive) MC-Verfahren
Volumenberechnung
Das Volumen und die Oberflache verschmolzener Hartkugelmodelle furMolekule sind prinzipiell elementargeometrisch berechenbar. Mitsteigender Atomanzahl werden diese Berechnungen jedoch schnellunubersichtlich und ineffizient.
1 Molekul in Grundgebiet bekannten Volumens
2 Folge von Zufallskoordinaten im Grundgebiet auswurfeln
3 relative Haufigkeit von Koordinaten im Innern des Molekulskonvergiert gegen das Molekulvolumen
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 68

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Einfache (naive) MC-Verfahren
random walk
unbeschrankter Irrweg
nichtumkehrbarer Irrweg
sich selbst nicht schneidenderIrrweg
1 Einen Gitterplatz mit einemMolekul besetzen
2 Auswahl eines zufalligenNachbarplatzes
3 Ist dieser unbesetzt, springt dasMolekul.
4 Ist er besetzt, verbleibt dasMolekul an seinem Platz.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 69

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
importance sampling Verfahren
Grundlagen
N -Teilchensystem im Volumen V bei der Temperatur Tjedes Teilchen charakterisiert durch einen Satz dynamischer VariablenαiKonfiguration (oder Phasenraumpunkt)X = α1, α2, . . . , αN
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 70

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
importance sampling Verfahren
Grundlagen
Konfigurationsmittel der Große A(X)
〈A〉 =
∫A(X) exp (−βH(X)) dX∫
exp (−βH(X)) dX
im diskreten Falle gilt naherungsweise
〈A〉 ≈ A =
∑Mj=1A(Xj)P
−1(Xj) exp (−βH(Xj))∑Mj=1 P
−1(Xj) exp (−βH(Xj))
Punkte Xj aus dem Phasenraum werden zufallig gemaß derWahrscheinlichkeitsverteilung P (X) ausgewahlt
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 71

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
importance sampling Verfahren
Metropolis-AlgorithmusSTART
Anlegen der Start-
konfiguration X0 =
α1 , α2 , . . . αk0
zufallige Auswahl
eines Teilchens j
zufallige Anderung
αj → αj′
Berechnung von
δH = H(X ′) −H(X)δH ≤ 0?
generiere Zufalls-
zahl z aus J0, 1K
e− δHkBT >
z?
Konfiguration X ′
wird akzeptiert
Konfiguration X ′
wird verworfen
nachster
Schritt
ENDE
nein
ja
ja
neinja
nein
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 72

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Transformation
Definition und grundlegende Eigenschaften
Definition (Fouriertransformation)
Es sei φ(~x) ∈ S(Rn). Dann nennen wir φ mit
φ(~ξ) :=1
(2π)1/2
∫Rn
e− i〈~ξ | ~x〉φ(~x) d~x
die Fouriertransformierte der Funktion φ(~x).
~x = (x1, x2, . . . , xn) ∈ Rn ~ξ = (ξ1, ξ2, . . . , ξn) ∈ Rn⟨~ξ∣∣∣ ~x⟩ =
n∑j=1
ξjxj
Funktionalanalysis
sinnvoll (Integral existiert)
Distributionen (δ)
eineindeutige Abbildung von S(Rn)auf S(Rn)
Umkehrung
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 73

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Transformation
Eigenschaften
”Einfachere“ Definition
Definition (Fouriertransformation)
f(ω) :=
∫ ∞−∞
e− iωtf(t) dt f(t) :=1
2π
∫ ∞−∞
e iωtf(ω) dω
Theorem (Eigenschaften (ohne Beweis))
linearer Operator
stetige Abbildung
Ableitungsregeln
Gaußfunktion ist Fixpunkt
Spiegelsymmetrie
Rucktransformation
Faltungstheorem
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 74

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Transformation
Fourier-Paare
Durch Fourier-Transformationen gekoppelte Großen, wie z.B.
Zeit und (Kreis-)Frequenz
Ort und Impuls
Gitter und reziprokes Gitter
. . .
Beispiel
FT
f(t− a) e− iaω f(ω)
e iatf(t) f(ω − a)
f(at) 1|a| f(ω
a)
f (n)(t) ( iω)nf(ω)
FT
e−a2t2 1√
ae−
12aω2
e−a|t|√
2π
aω2+a2
1t2+a2
√π2
1a
e−a|ω|
u sinc
FT
e iat√
2πδ(ω − a)
tn in√
2πδ(n)(ω)Θ(t)
√π2
(1
iπω+ δ(ω)
)
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 75

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Reihe und Fourier-Analyse
Definition der Fourier-Reihe
f(t) =
∞∑k=0
(ak cosωkt+ bk sinωkt)
Hierin sind b0 = 0 und ωk = 2πkT .
Einige Basisfunktionen: links sin, rechts cos, k = 1, 2, 3
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 76
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Reihe und Fourier-Analyse
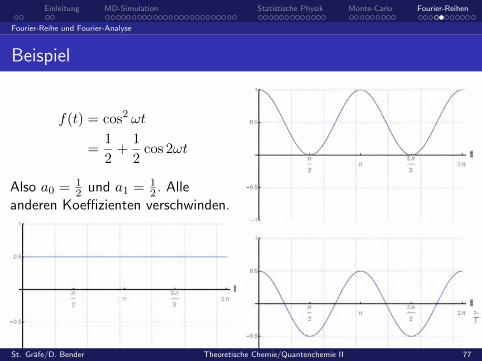
Beispiel
f(t) = cos2 ωt
=1
2+
1
2cos 2ωt
Also a0 = 12 und a1 = 1
2 . Alleanderen Koeffizienten verschwinden.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 77

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Koeffizienten
Symmetrieuberlegungen
2 cosα cosβ = cos(α− β) + cos(α+ β)
2 sinα sinβ = cos(α− β)− cos(α+ β)
2 sinα cosβ = sin(α− β) + sin(α+ β)
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 78

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Koeffizienten
Fourier-Koeffizienten
ak =
1T
T/2∫−T/2
f(t) dt falls k = 0,”Mittelwert“
2T
T/2∫−T/2
f(t) cosωkt dt falls k = 1, 2, 3, . . .
bk =2
T
T/2∫−T/2
f(t) sinωkt dt falls k = 1, 2, 3, . . .
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 79

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Koeffizienten
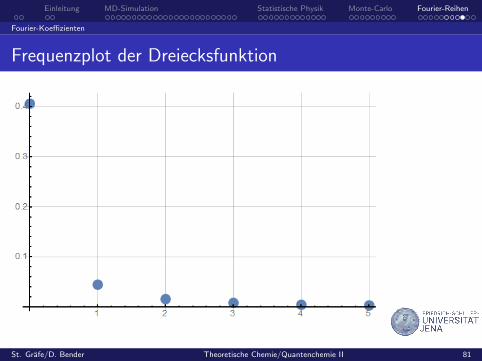
Beispiel: Dreiecksfunktion
f(t) =
1 + 2
T t fur − T2 ≤ t ≤ 0
1− 2T t fur 0 ≤ t ≤ T
2
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 80
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Fourier-Koeffizienten
Frequenzplot der Dreiecksfunktion
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 81

Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Probleme und Grenzen der Fourier-Entwicklung
Dirichletscher Integralkern und Einheitsstufe
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 82
Einleitung MD-Simulation Statistische Physik Monte-Carlo Fourier-Reihen
Probleme und Grenzen der Fourier-Entwicklung
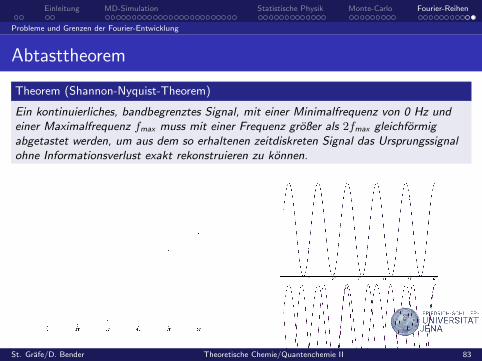
Abtasttheorem
Theorem (Shannon-Nyquist-Theorem)
Ein kontinuierliches, bandbegrenztes Signal, mit einer Minimalfrequenz von 0 Hz undeiner Maximalfrequenz fmax muss mit einer Frequenz großer als 2fmax gleichformigabgetastet werden, um aus dem so erhaltenen zeitdiskreten Signal das Ursprungssignalohne Informationsverlust exakt rekonstruieren zu konnen.
St. Grafe/D. Bender Theoretische Chemie/Quantenchemie II 83


















