
Thermal Analysis User Information for Users Com · Thermal Analysis Information for Users User Com...
24
Thermal Analysis Information for Users User Com 25 Einleitung Basislinien werden in der thermischen Analyse vorwiegend zur Integration von Peaks eingesetzt. Die Peakflä- che wird dabei durch die Messkurve und die eingesetzte virtuelle oder reale Basislinie bestimmt. Ebenso wird die Peaktemperatur am Ort des maximalen Abstands zur Basislinie definiert. Extrapolierte Basislinien sind wichtig für die Bestimmung von Glasübergangstemperaturen und von Onset-Temperaturen von Effekten. Da Zur Bestimmung einer Umwandlungs- oder Reaktionsenthalpie muss eine korrekte Basis- linie gewählt werden. Diese sollte die DSC-Kurve wiedergeben, die gemessen würde, wenn keine Umwandlungsenthalpie vorhanden wäre. Wie diese Basislinien gewählt wer- den und welcher Basislinientyp zum Einsatz kommt, wird an einigen charakteristischen Beispielen dargelegt. Wahl der Basislinien Dr. Rudolf Riesen Sehr geehrter Kunde, Es freut uns ausserordentlich, dass Sie uns als Leser des UserComs immer wieder Beiträge zur Publikation zukommen lassen. Die Thermische Analyse kann dank neuen Techniken und besseren Messleistungen in noch mehr Gebieten als bisher eingesetzt werden. Dank gekoppelten Techniken wie Gasanalyse, Mikroskopie oder Chemilumineszenz stehen auch viel mehr Informationen zur Verfügung, die die Interpretation der Messergebnisse in vielen Fällen erleichtert. Wir glauben, dass wir Sie auch dieses Mal wieder auf neue und interessante Gebiete oder Techniken hinwei- sen können. Inhalt 1/2007 TA Tipp - Wahl der Basislinien 1 Applikationen - Bestimmung des Noack- Verdampfungsverlusts von Schmiermitteln mittels TGA 7 - Thermische Analyse zur Charakterisierung von polymorphen Modifikationen 9 - Analyse von Schmelzprozessen mittels TOPEM ® 13 - Charakterisierung von Delivery Systemen mittels Thermo- gravimetrie 18 Tipps und Hinweise - Kleine Probeneffekte im DSC ein- facher aufspüren und auswerten 21 Daten - Exhibitions 23 - Courses and Seminars 23
Transcript of Thermal Analysis User Information for Users Com · Thermal Analysis Information for Users User Com...

Thermal AnalysisInformation for Users
User Com
25
EinleitungBasislinien werden in der thermischen Analyse vorwiegend zur Integration von Peaks eingesetzt. Die Peakflä-che wird dabei durch die Messkurve und die eingesetzte virtuelle oder reale Basislinie bestimmt. Ebenso wird die Peaktemperatur am Ort des maximalen Abstands zur Basislinie definiert. Extrapolierte Basislinien sind wichtig für die Bestimmung von Glasübergangstemperaturen und von Onset-Temperaturen von Effekten. Da
Zur Bestimmung einer Umwandlungs- oder Reaktionsenthalpie muss eine korrekte Basis- linie gewählt werden. Diese sollte die DSC-Kurve wiedergeben, die gemessen würde, wenn keine Umwandlungsenthalpie vorhanden wäre. Wie diese Basislinien gewählt wer-den und welcher Basislinientyp zum Einsatz kommt, wird an einigen charakteristischen Beispielen dargelegt.
Wahl der BasislinienDr. Rudolf Riesen
Sehr geehrter Kunde,Es freut uns ausserordentlich, dass Sie uns als Leser des UserComs immer wieder Beiträge zur Publikation zukommen lassen. Die Thermische Analyse kann dank neuen Techniken und besseren Messleistungen in noch mehr Gebieten als bisher eingesetzt werden. Dank gekoppelten Techniken wie Gasanalyse, Mikroskopie oder Chemilumineszenz stehen auch viel mehr Informationen zur Verfügung, die die Interpretation der Messergebnisse in vielen Fällen erleichtert.Wir glauben, dass wir Sie auch dieses Mal wieder auf neue und interessante Gebiete oder Techniken hinwei-sen können.
Inhalt 1/2007
TA Tipp
- Wahl der Basislinien 1
Applikationen
- Bestimmung des Noack- Verdampfungsverlusts von Schmiermitteln mittels TGA 7
- Thermische Analyse zur Charakterisierung von polymorphen Modifikationen 9
- Analyse von Schmelzprozessen mittels TOPEM® 13
- Charakterisierung von Delivery Systemen mittels Thermo- gravimetrie 18
Tipps und Hinweise
- Kleine Probeneffekte im DSC ein- facher aufspüren und auswerten 21
Daten
- Exhibitions 23
- Courses and Seminars 23

2 METTLER TOLEDO UserCom 1/2007
TA-T
ipp
Abbildung 1: Wahl von interpo-lierten DSC-Basis-linien (endotherme Richtung nach oben).
in der Literatur und in Normen der Be-griff „Basislinie“ zum Teil unterschied-lich definiert wird oder unterschiedliche Begriffe für ein und dasselbe verwendet werden, sind diese hier zusammengestellt und mit Erläuterungen versehen. Welche Regeln zur Wahl der Basislinien gelten und welche Art von Basislinie für eine optimale Auswertung von DSC-Kurven gewählt werden soll, zeigen verschiedene Applikationsbeispiele.
TerminologieBegriffe zur thermischen Analyse sind in verschiedenen Normen zusammenge-stellt und erläutert. Da die Definitionen nicht immer identisch sind, sind die für die nachfolgende Basislinien-Diskussion verwendeten Begriffe hier speziell aufge-führt. Weitere Definitionen sind im Buch von Höhne [1] sowie in den erwähnten Normen (ISO [2], DIN [3], ASTM [4, 5]) zu finden. Die bevorzugten Begriff sind hervorgehoben, andere Begriffe sind ebenfalls aufgeführt.
Blank, Blindkurve, Leerkurve, Nulllinie [3], instrumentelle Basislinie [2]: TA-Kurve, die unter gleichen Bedingungen gemessen wird wie die Probe, aber ohne diese Probe, wobei die Tiegelmassen gleich sein müssen. Solche Kurven sind für Bestimmungen der spezifischen Wär-mekapazität unabdingbar.Bemerkung: Zum Teil wird unter Leer-kurve (zero line [1]) auch eine Kurve verstanden, bei der weder Probe noch Tiegel eingesetzt sind.
Probenblank: TA-Kurve, die von einer „ausreagierten“ Probe erhalten wird, meist durch zweites Aufheizen (2. Run) unter gleichen Bedingungen. Der beim ersten Aufheizen (1. Run) gemessene Ef-fekt zeigt sich dabei nicht mehr.
Basislinie (auch Probenbasislinie [2]): Teil der TA-Kurve, die keine Umwand-lungen oder Reaktionen zeigt. Dies ist eine isotherme Basislinie, wenn die Tem-peratur konstant gehalten wird. Eine dy-namische Basisline wird erhalten, wenn die Temperatur durch Heizen oder Küh-len verändert wird. Die Basislinie hängt von der Wärmekapazität der Probe (bei leerer Referenz) und vom Blank ab.Bemerkung: Umgangssprachlich wird damit auch die für die Integration ver-wendete virtuelle Basislinie gemeint.
Virtuelle Basisline [2]: Eine imaginä-re Linie im Bereich einer Reaktion oder Umwandlung, die die DSC-Kurve zeigen würde, wenn keine Reaktions- oder Um-wandlungsenthalpie vorhanden wäre. Interpolierte Basisline [1]: Sie wird kons-truiert, indem die gemessene Kurve vor und nach dem Peak verbunden wird.Extrapolierte Basisline: Die gemessene Kurve vor oder nach dem thermischen Effekt wird verlängert.Welche Typen von einzelnen virtuellen Basislinien üblicherweise eingesetzt wer-den, wird bei den Anwendungen erklärt.
Wahre Basislinie: Die Basislinie kann im Bereich der Reaktion oder Umwand-
lung den physikalischen Tatsachen ent-sprechend berechnet oder sogar gemes-sen werden.
Einflüsse auf die BasislinieBei den Kurveninterpretationen und nu-merischen Auswertungen sollten immer auch die Einflüsse der Messbedingungen auf die DSC-Kurve und damit auf die Basisline berücksichtigt werden. Zudem sollte der Verlauf des Blanks und dessen Reproduzierbarkeit bekannt sein. Einflussgrössen und deren mögliche Ver-änderungen während einer Umwandlung können sein [1]:
Masse, Form und Struktur der Probe, z.B. Pulver oder Folie;Wärmeleitfähigkeit und Kontakt der Probe zum Tiegelboden, z.B. zerfliesst ein Pulver während des Schmelzens;Wärmeübergang vom Tiegel zum Sensor, z.B. Verformung des Tiegels durch entstehenden Innendruck oder ausfliessende Proben;Heizrate, z.B. bei Übergang zu isother-men Bereichen;Thermische Vorgeschichte von Probe und Messsystem.
Ist die Wahl der Basisline schwierig, dann hilft manchmal eine Untersuchung der gemessenen Probe und des Tiegels im Hinblick auf die oben erwähnten Punkte weiter.
Konstruktionsprinzip von virtu-ellen BasislinienDas Grundprinzip zur Erstellung von vir-tuellen Basislinien ist folgendes: Die interpolierte Basislinie zur Bestim-mung von Umwandlungs- und Reakti-onswärmen soll aus der DSC-Kurve vor dem thermischen Effekt fliessend (tan-gential) abzweigen und nach dem Effekt ebenso in die Messkurve einmünden. Anschaulich erklärt, verhält sich dies wie die Flugbahn eines Flugzeugs beim Ab-heben oder beim Landen. Abweichungen davon gibt es aus besonderen Gründen; diese werden bei den Beispielen beschrie-ben.Abbildung 1 zeigt, wie das Grundprinzip angewendet wird.
1 unsinnig; 2 gut (Linie), 1 ungenügend (horizontale Gerade); 2 gut (Integralbasislinie, ev. Spline),
1.
2.
3.
4.
5.
a)b)
3METTLER TOLEDO UserCom 1/2007
gut (tangentiale Integralbasislinie, ev. Spline-Basislinie), Schmelzen mit exothermer Zersetzung, 1 gut (Gerade bis zum Schnitt mit der DSC-Kurve); 2 eher willkürlich, da die DSC-Kurve die Summe der gleichzeitig ablaufenden Vorgänge abbildet, zwei überlagerte Peaks, z.B. Eutekti-kum und Schmelzpeak der Hauptkom-ponente, 1 gut für das Gesamtintegral, 2 gut für die Integration des ersten Peaks (Peak aufsitzend interpretiert, Spline-Basislinie).
Die Übergangslinie von einer Tangente zur anderen kann verschiedene Formen
c)
d)
e)
annehmen und als Gerade oder als sig-moidale Kurve dargestellt werden. Welche Art von interpolierter Basisline gewählt wird, hängt in erster Linie von den physi-kalischen Bedingungen oder chemischen Veränderungen ab:
Die spezifische Wärmekapazität der Probe cp ändert sich während der Um-wandlung praktisch nicht oder sie ver-ändert sich linear mit der Temperatur.Die Umwandlung ist mit einer signifi-kanten Änderung der Wärmekapazität verbunden.Der Wärmeübergang zur Probe ändert sich während der Umwandlung.
•
•
•
Die Masse der Probe verändert sich während der Umwandlung.
Um diese Veränderungen der DSC-Kurve während einer Umwandlung zu berück-sichtigen, stehen in der STARe-Software mehrere Basislinientypen zur Verfügung. Die Tabelle 1 beschreibt diese Auswahl und die typischen Anwendungen.
Die extrapolierten virtuellen Basislinien sind die Tangenten an die Messkurve bei den Auswertegrenzen, wie sie auch bei den Basislinien für die Interpolation ver-wendet werden. Typische Anwendungen
•
Tabelle 1: Liste der virtuellen Basislinientypen für die Integration.
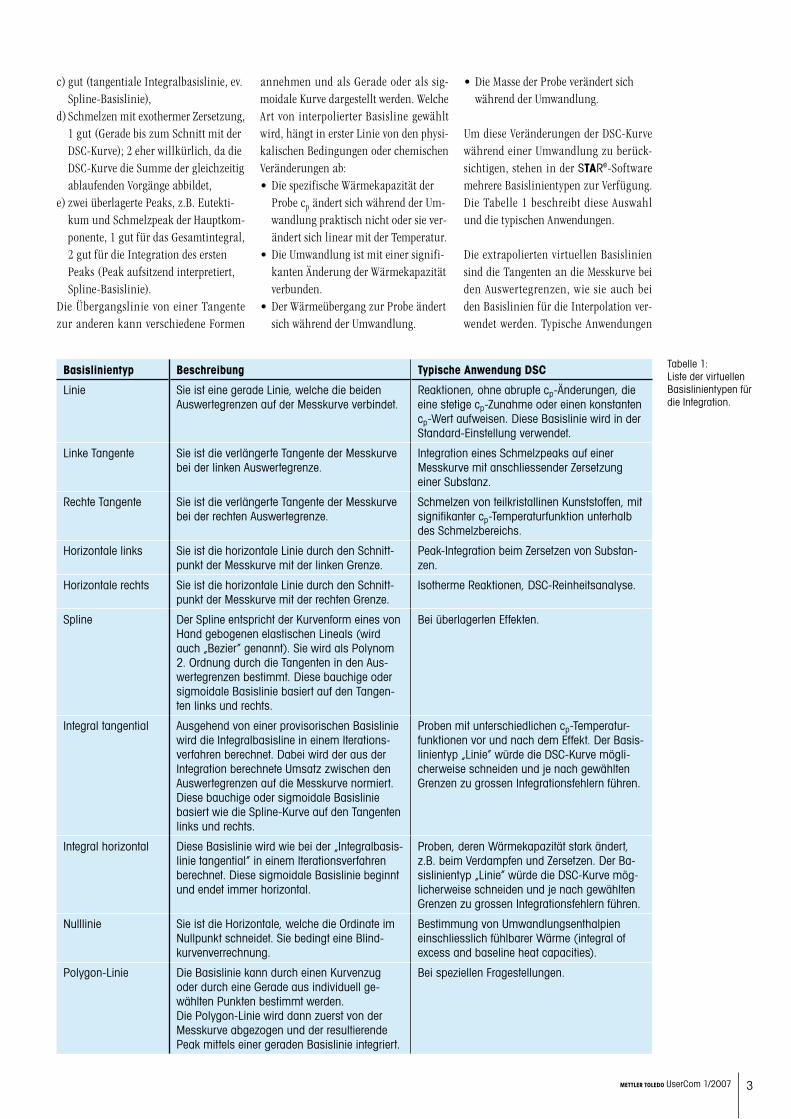
Basislinientyp Beschreibung Typische Anwendung DSC
Linie Sie ist eine gerade Linie, welche die beiden Auswertegrenzen auf der Messkurve verbindet.
Reaktionen, ohne abrupte cp-Änderungen, die eine stetige cp-Zunahme oder einen konstanten cp-Wert aufweisen. Diese Basislinie wird in der Standard-Einstellung verwendet.
Linke Tangente Sie ist die verlängerte Tangente der Messkurve bei der linken Auswertegrenze.
Integration eines Schmelzpeaks auf einer Messkurve mit anschliessender Zersetzung einer Substanz.
Rechte Tangente Sie ist die verlängerte Tangente der Messkurve bei der rechten Auswertegrenze.
Schmelzen von teilkristallinen Kunststoffen, mit signifikanter cp-Temperaturfunktion unterhalb des Schmelzbereichs.
Horizontale links Sie ist die horizontale Linie durch den Schnitt-punkt der Messkurve mit der linken Grenze.
Peak-Integration beim Zersetzen von Substan-zen.
Horizontale rechts Sie ist die horizontale Linie durch den Schnitt-punkt der Messkurve mit der rechten Grenze.
Isotherme Reaktionen, DSC-Reinheitsanalyse.
Spline Der Spline entspricht der Kurvenform eines von Hand gebogenen elastischen Lineals (wird auch „Bezier” genannt). Sie wird als Polynom 2. Ordnung durch die Tangenten in den Aus-wertegrenzen bestimmt. Diese bauchige oder sigmoidale Basislinie basiert auf den Tangen-ten links und rechts.
Bei überlagerten Effekten.
Integral tangential Ausgehend von einer provisorischen Basislinie wird die Integralbasisline in einem Iterations-verfahren berechnet. Dabei wird der aus der Integration berechnete Umsatz zwischen den Auswertegrenzen auf die Messkurve normiert. Diese bauchige oder sigmoidale Basislinie basiert wie die Spline-Kurve auf den Tangenten links und rechts.
Proben mit unterschiedlichen cp-Temperatur-funktionen vor und nach dem Effekt. Der Basis-linientyp „Linie” würde die DSC-Kurve mögli-cherweise schneiden und je nach gewählten Grenzen zu grossen Integrationsfehlern führen.
Integral horizontal Diese Basislinie wird wie bei der „Integralbasis-linie tangential” in einem Iterationsverfahren berechnet. Diese sigmoidale Basislinie beginnt und endet immer horizontal.
Proben, deren Wärmekapazität stark ändert, z.B. beim Verdampfen und Zersetzen. Der Ba-sislinientyp „Linie” würde die DSC-Kurve mög-licherweise schneiden und je nach gewählten Grenzen zu grossen Integrationsfehlern führen.
Nulllinie Sie ist die Horizontale, welche die Ordinate im Nullpunkt schneidet. Sie bedingt eine Blind- kurvenverrechnung.
Bestimmung von Umwandlungsenthalpien einschliesslich fühlbarer Wärme (integral of excess and baseline heat capacities).
Polygon-Linie Die Basislinie kann durch einen Kurvenzug oder durch eine Gerade aus individuell ge-wählten Punkten bestimmt werden. Die Polygon-Linie wird dann zuerst von der Messkurve abgezogen und der resultierende Peak mittels einer geraden Basislinie integriert.
Bei speziellen Fragestellungen.
4 METTLER TOLEDO UserCom 1/2007
TA-T
ipp
der extrapolierten Basislinien sind die Bestimmung der:
Glasübergangstemperaturextrapolierten Onset-Temperatur (zum Teil auch als erstes Abweichen von der Messkurve)Stufenhöhe
Bei allen Auswertungen, bei denen ex-trapolierte Tangenten verwendet werden, muss darauf geachtet werden, dass de-ren Berechnung nicht durch Artefakte auf der Messkurve oder Signalrauschen gestört wird und somit eine falsche Lage aufweist.
AnwendungsbeispieleIn Ergänzung zu Abbildung 1 sind im Folgenden einige charakteristische Bei-spiele zur Wahl des entsprechenden kor-
••
•
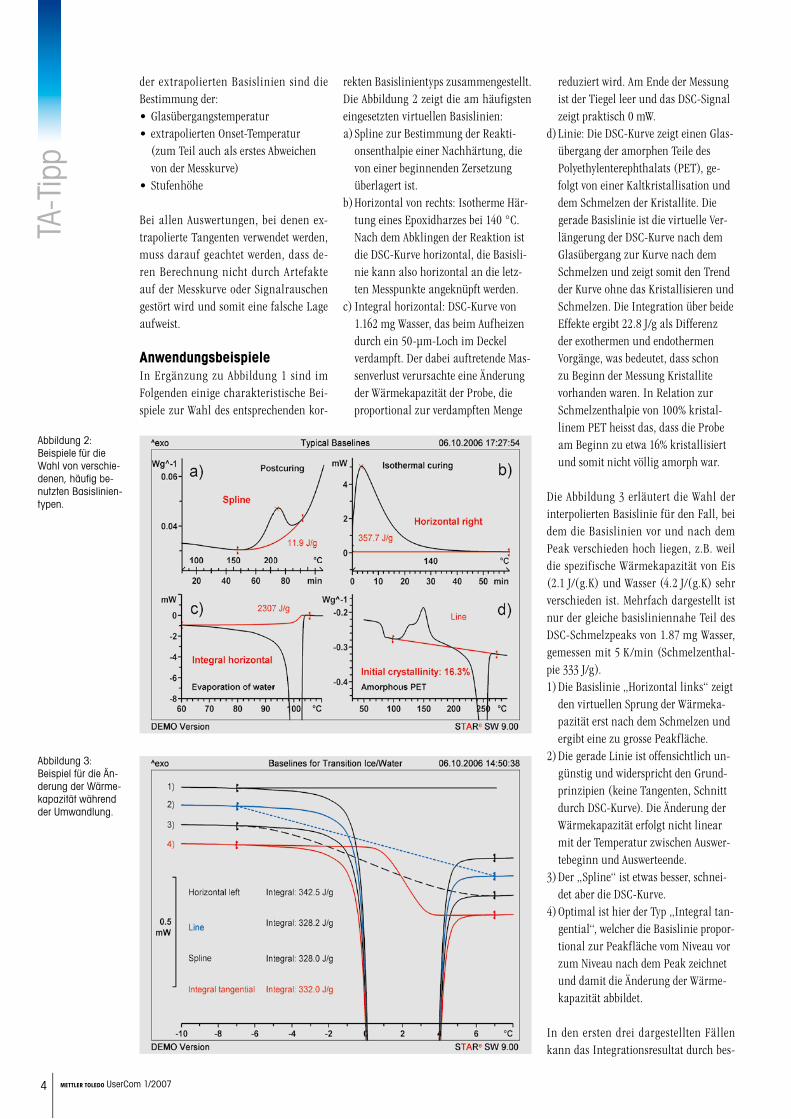
rekten Basislinientyps zusammengestellt. Die Abbildung 2 zeigt die am häufigsten eingesetzten virtuellen Basislinien:
Spline zur Bestimmung der Reakti-onsenthalpie einer Nachhärtung, die von einer beginnenden Zersetzung überlagert ist.Horizontal von rechts: Isotherme Här-tung eines Epoxidharzes bei 140 °C. Nach dem Abklingen der Reaktion ist die DSC-Kurve horizontal, die Basisli-nie kann also horizontal an die letz-ten Messpunkte angeknüpft werden.Integral horizontal: DSC-Kurve von 1.162 mg Wasser, das beim Aufheizen durch ein 50-µm-Loch im Deckel verdampft. Der dabei auftretende Mas-senverlust verursachte eine Änderung der Wärmekapazität der Probe, die proportional zur verdampften Menge
a)
b)
c)
reduziert wird. Am Ende der Messung ist der Tiegel leer und das DSC-Signal zeigt praktisch 0 mW.Linie: Die DSC-Kurve zeigt einen Glas-übergang der amorphen Teile des Polyethylenterephthalats (PET), ge-folgt von einer Kaltkristallisation und dem Schmelzen der Kristallite. Die gerade Basislinie ist die virtuelle Ver-längerung der DSC-Kurve nach dem Glasübergang zur Kurve nach dem Schmelzen und zeigt somit den Trend der Kurve ohne das Kristallisieren und Schmelzen. Die Integration über beide Effekte ergibt 22.8 J/g als Differenz der exothermen und endothermen Vorgänge, was bedeutet, dass schon zu Beginn der Messung Kristallite vorhanden waren. In Relation zur Schmelzenthalpie von 100% kristal-linem PET heisst das, dass die Probe am Beginn zu etwa 16% kristallisiert und somit nicht völlig amorph war.
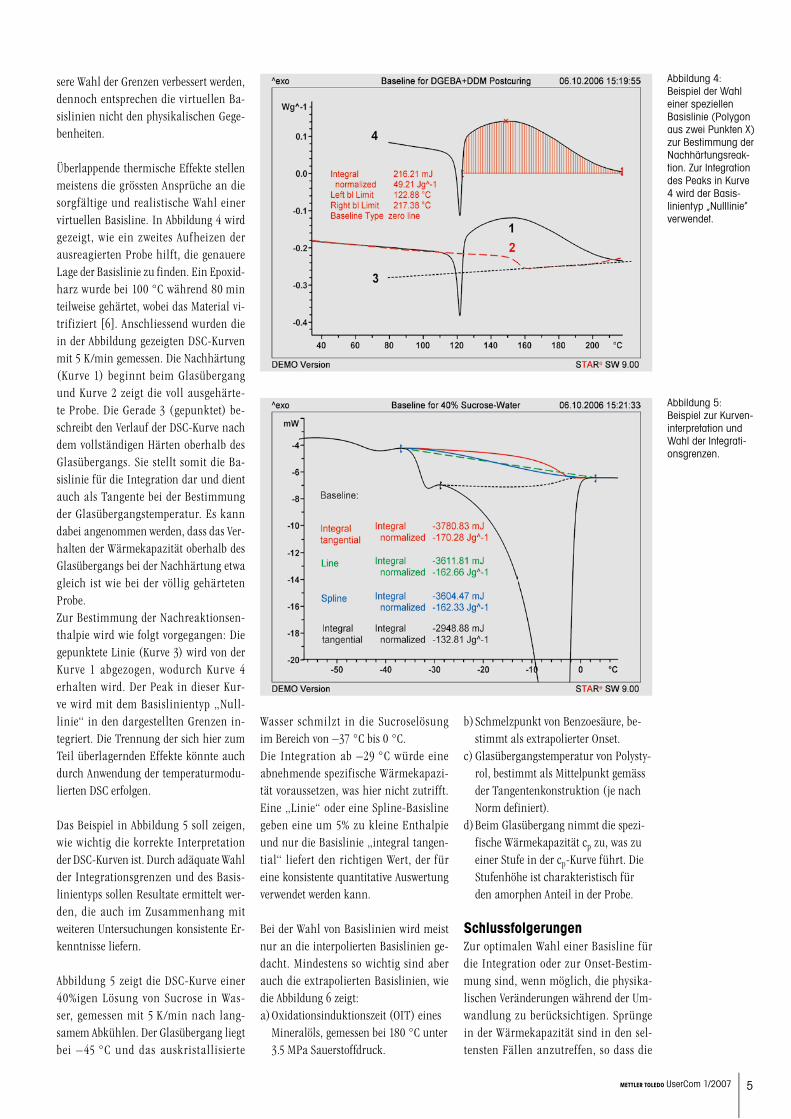
Die Abbildung 3 erläutert die Wahl der interpolierten Basislinie für den Fall, bei dem die Basislinien vor und nach dem Peak verschieden hoch liegen, z.B. weil die spezifische Wärmekapazität von Eis (2.1 J/(g.K) und Wasser (4.2 J/(g.K) sehr verschieden ist. Mehrfach dargestellt ist nur der gleiche basisliniennahe Teil des DSC-Schmelzpeaks von 1.87 mg Wasser, gemessen mit 5 K/min (Schmelzenthal-pie 333 J/g).
Die Basislinie „Horizontal links“ zeigt den virtuellen Sprung der Wärmeka-pazität erst nach dem Schmelzen und ergibt eine zu grosse Peakfläche. Die gerade Linie ist offensichtlich un-günstig und widerspricht den Grund-prinzipien (keine Tangenten, Schnitt durch DSC-Kurve). Die Änderung der Wärmekapazität erfolgt nicht linear mit der Temperatur zwischen Auswer-tebeginn und Auswerteende.Der „Spline“ ist etwas besser, schnei-det aber die DSC-Kurve. Optimal ist hier der Typ „Integral tan-gential“, welcher die Basislinie propor-tional zur Peakfläche vom Niveau vor zum Niveau nach dem Peak zeichnet und damit die Änderung der Wärme-kapazität abbildet.
In den ersten drei dargestellten Fällen kann das Integrationsresultat durch bes-
d)
1)
2)
3)
4)
Abbildung 2: Beispiele für die Wahl von verschie-denen, häufig be-nutzten Basislinien-typen.
Abbildung 3: Beispiel für die Än-derung der Wärme-kapazität während der Umwandlung.
5METTLER TOLEDO UserCom 1/2007
sere Wahl der Grenzen verbessert werden, dennoch entsprechen die virtuellen Ba-sislinien nicht den physikalischen Gege-benheiten.
Überlappende thermische Effekte stellen meistens die grössten Ansprüche an die sorgfältige und realistische Wahl einer virtuellen Basisline. In Abbildung 4 wird gezeigt, wie ein zweites Aufheizen der ausreagierten Probe hilft, die genauere Lage der Basislinie zu finden. Ein Epoxid-harz wurde bei 100 °C während 80 min teilweise gehärtet, wobei das Material vi-trifiziert [6]. Anschliessend wurden die in der Abbildung gezeigten DSC-Kurven mit 5 K/min gemessen. Die Nachhärtung (Kurve 1) beginnt beim Glasübergang und Kurve 2 zeigt die voll ausgehärte-te Probe. Die Gerade 3 (gepunktet) be-schreibt den Verlauf der DSC-Kurve nach dem vollständigen Härten oberhalb des Glasübergangs. Sie stellt somit die Ba-sislinie für die Integration dar und dient auch als Tangente bei der Bestimmung der Glasübergangstemperatur. Es kann dabei angenommen werden, dass das Ver-halten der Wärmekapazität oberhalb des Glasübergangs bei der Nachhärtung etwa gleich ist wie bei der völlig gehärteten Probe. Zur Bestimmung der Nachreaktionsen-thalpie wird wie folgt vorgegangen: Die gepunktete Linie (Kurve 3) wird von der Kurve 1 abgezogen, wodurch Kurve 4 erhalten wird. Der Peak in dieser Kur-ve wird mit dem Basislinientyp „Null-linie“ in den dargestellten Grenzen in-tegriert. Die Trennung der sich hier zum Teil überlagernden Effekte könnte auch durch Anwendung der temperaturmodu-lierten DSC erfolgen.
Das Beispiel in Abbildung 5 soll zeigen, wie wichtig die korrekte Interpretation der DSC-Kurven ist. Durch adäquate Wahl der Integrationsgrenzen und des Basis-linientyps sollen Resultate ermittelt wer-den, die auch im Zusammenhang mit weiteren Untersuchungen konsistente Er-kenntnisse liefern.
Abbildung 5 zeigt die DSC-Kurve einer 40%igen Lösung von Sucrose in Was-ser, gemessen mit 5 K/min nach lang-samem Abkühlen. Der Glasübergang liegt bei –45 °C und das auskristallisierte
Wasser schmilzt in die Sucroselösung im Bereich von –37 °C bis 0 °C. Die Integration ab –29 °C würde eine abnehmende spezifische Wärmekapazi-tät voraussetzen, was hier nicht zutrifft. Eine „Linie“ oder eine Spline-Basisline geben eine um 5% zu kleine Enthalpie und nur die Basislinie „integral tangen-tial“ liefert den richtigen Wert, der für eine konsistente quantitative Auswertung verwendet werden kann.
Bei der Wahl von Basislinien wird meist nur an die interpolierten Basislinien ge-dacht. Mindestens so wichtig sind aber auch die extrapolierten Basislinien, wie die Abbildung 6 zeigt:
Oxidationsinduktionszeit (OIT) eines Mineralöls, gemessen bei 180 °C unter 3.5 MPa Sauerstoffdruck.
a)
Schmelzpunkt von Benzoesäure, be-stimmt als extrapolierter Onset.Glasübergangstemperatur von Polysty-rol, bestimmt als Mittelpunkt gemäss der Tangentenkonstruktion (je nach Norm definiert).Beim Glasübergang nimmt die spezi-fische Wärmekapazität cp zu, was zu einer Stufe in der cp-Kurve führt. Die Stufenhöhe ist charakteristisch für den amorphen Anteil in der Probe.
SchlussfolgerungenZur optimalen Wahl einer Basisline für die Integration oder zur Onset-Bestim-mung sind, wenn möglich, die physika-lischen Veränderungen während der Um-wandlung zu berücksichtigen. Sprünge in der Wärmekapazität sind in den sel-tensten Fällen anzutreffen, so dass die
b)
c)
d)
Abbildung 4: Beispiel der Wahl einer speziellen Basislinie (Polygon aus zwei Punkten X) zur Bestimmung der Nachhärtungsreak-tion. Zur Integration des Peaks in Kurve 4 wird der Basis-linientyp „Nulllinie” verwendet.
Abbildung 5: Beispiel zur Kurven-interpretation und Wahl der Integrati-onsgrenzen.

6 METTLER TOLEDO UserCom 1/2007
virtuelle Basislinie auch ohne Ecken und unstetigen Verlauf konstruiert werden muss. Eine korrekte Wahl der Basislinie bedingt auch immer eine vorausgehende, kon-sistente Kurveninterpretation [7]. Dazu gehört auch, dass die Integrationslimiten der Aufgabenstellung entsprechend sorg-fältig gewählt werden.Die hier an Beispielen von DSC-Mes-sungen besprochenen Regeln und Typen zur Wahl der Basislinien können sinnge-mäss auch auf andere TA-Messtechniken übertragen werden, z.B. zur Integration von Peaks bei der SDTA, der DTG und bei anderen mathematisch abgeleiteten Messkurven.
Literatur[1] G. W. H. Höhne, W. Hemminger and
H.-J. Flammersheim: Differential Scanning Calorimetry, Springer Ver-lag, 1996. Chapter “The DSC Curve”
[2] ISO 11357-1 (1997) Plastics – DSC. General principles
[3] DIN 51005 Thermal analysis; Terms[4] ASTM E473 Standard Terminology
Relating to Thermal Analysis and Rheology
[5] ASTM E 2161 Standard Terminology Relating to Performance Validation in Thermal Analysis
[6] J. Schawe, METTLER TOLEDO UserCom 14, 17
[7] METTLER TOLEDO UserCom mit Beiträgen zur Kurveninterpretation: DSC, UserCom 11 und 12; TGA, UserCom 13
TA-T
ipp
Abbildung 6: Beispiele zu extra-polierten Basis-linien.

AbstractSchmiermittel für Motoren und andere Anwendungen dürfen aus Qualitäts- und Umweltgründen nur eine kleine Ver-dampfungsrate aufweisen. Der Verlust von leicht flüchtigen Komponenten er-höht die Viskosität und somit den Ölver-brauch, die Verkokung und den Abrieb. Für jedes Schmieröl wird deshalb nach genormten Noack-Verfahren der Ver-dampfungsverlust angegeben, der nach den Spezifikationen ILSAC GF-3 und API-SL nicht mehr als 15% betragen darf.
Die ASTM-Norm D6375 zur Bestim-mung des Verdampfungsverlusts von Schmierölen nach Noack [1] beschreibt die entsprechende thermogravimetrische Methode, die die gleichen Resultate wie andere Normen (z.B. ASTM D5800 [2], DIN 51581-1 [3], JPI-5S-41-93 [4]) lie-fert. In diesem Beitrag wird gezeigt, wie der Noack-Verdampfungsverlust mittels TGA im Vergleich zu einem Referenzöl bestimmt wird.
EinleitungDie Zunahme der Lebens- und Ge-brauchsdauer von Schmierstoffen ver-bunden mit höheren Ölumlaufzahlen, längeren Ölwechselintervallen und ge-ringerem Schmierstoffverbrauch führt zu einer Zunahme der Beanspruchung der Schmierstoffe. Höhere Temperaturen verbunden mit kleineren Ölfüllungen und höheren Leistungsdichten führen dazu, dass die Leistungs- und Qualitäts-anforderungen an Schmierstoffe stetig steigen. Damit die Schmiermittel richtig eingesetzt werden, sind sie entsprechend spezifiziert und klassiert.
Die Spezifikationen beschreiben die phy-sikalischen Eigenschaften der Motoröle, wie z.B. Viskosität, Verdampfungsverlust und Scherstabilität. Geprüft wird auch das Leistungsverhalten in Motorentests, wie z.B. Verschleissschutz und Sauberkeit sowie teilweise der Einfluss auf den Kraft-
stoffverbrauch und die Veränderungen des Motoröls während des Betriebs durch Viskositätsänderungen (Eindickung). Die Klassierung erfolgt durch die Organisati-onen ILSAC, API oder SAE (siehe die Ta-belle der Abkürzungen).
Eine der Spezifikationen ist der Verdamp-fungsverlust. Die niedrigmolekularen Be-standteile eines Mineralöls, welche aus Fraktionen unterschiedlicher Kohlen-wasserstoffe verschiedener Kettenlängen und Molekulargewichte besteht, können bei erhöhter thermischer Beanspruchung verdampfen. Dadurch erhöht sich in der Regel die Viskosität des Schmierstoffs. Gleichzeitig wird die Löslichkeit der Addi-tive im Basisöl beeinflusst. Die Verdamp-fung ist für alle Schmierstoffgruppen (z.B. auch bei synthetischen Ölen) von Bedeutung, wenn sie bei höheren Tempe-raturen angewandt werden. So können, z.B. bei Motorölen durch hohe Tempera-turen an den Kolbenringen und am Kol-benunterboden, Verdampfungsverluste auftreten. Sie führen zu unerwünschter Öleindickung und zu erhöhtem Ölver-brauch.
Der Noack-Verdampfungsver-lust nach ASTM D6375Um die Verdampfung quantitativ zu bestimmen, wurde vor Jahrzehnten der Noack-Verdampfungstest unter genorm-ten Bedingungen eingeführt. Zum Bei-spiel wird bei DIN 51581 [3] gemessen, wie gross der Verlust während einer Stun-de bei 250 °C unter Vakuum (2 mbar) ist.
Mit der ASTM Norm D6375 steht ein Standardverfahren zur Verfügung, das entwickelt wurde [5], um die Vorteile der gaschromatischen Methode [6] mit den realistischen Bedingungen der tra-ditionellen Noack-Tests zu verbinden, aber schneller und sicherer ist als beide und zudem mit weniger Probenmaterial durchgeführt werden kann.
Gemäss ASTM D6375 wird eine Probe in einem Tiegel rasch auf 249 °C geheizt und 30 min gehalten und dazu die TGA-Kurve aufgezeichnet. Der Noack-Ver-dampfungsverlust ist dann die Gewichts-abnahme bis zur Noack-Referenzeit. Diese Zeit wird vorher unter den gleichen Versuchsbedingungen mit einem Noack-Referenzöl bestimmt. Wichtig bei diesem Verfahren ist, dass die Probentemperatur rasch auf einen Wert zwischen 247 und 249 °C steigt, aber nicht überschwingt. Um die traditionelle Noack-Methode zu simulieren, wird üblicherweise mit 100 K/min auf 220 °C und dann mit 10 K/min auf 249 °C geheizt. Die Pro-benmenge (ms) wird durch den Tiegel-innendurchmesser (d) gemäss folgender Formel bestimmt:
ms = 350d3
wobei d in cm und ms in mg gemessen wird.
Durchführung einer Noack- BestimmungDie Noack-Verdampfung wurde unter folgenden Bedingungen mit einem METTLER TOLEDO TGA bestimmt:
Tiegel: 100 µl Aluminium ohne Deckel (Innendurchmesser 0.56 cm)Probenmasse: 61 ±3 mgSpül- und Schutzgas: total 80 ml/min LuftNoack-Referenzöl: W4520001 mit 10.93% Gewichtsverlust bis zur Noack- Zeit; Bezugsquelle: Walter Herzog GmbHTemperaturprogramm: 50 °C bis 220 °C mit 100 K/min gefolgt von weiterem Aufheizen auf 249 °C mit 10 K/min und isothermen Halten bei 249 °C. Um die oben erwähnte Bedingung einzuhalten, wurde der Parameter tlag für diesen Tiegel auf null justiert.
Als Probe (Testöl) wurde ein synthetisches Motoröl 5W40 verwendet.
•
••
•
•
Bestimmung des Noack-Verdampfungsverlusts von Schmiermitteln mittels TGADr. Rudolf Riesen
METTLER TOLEDO UserCom 1/2007 7
Appl
ikat
ione
n
8 METTLER TOLEDO UserCom 1/2007
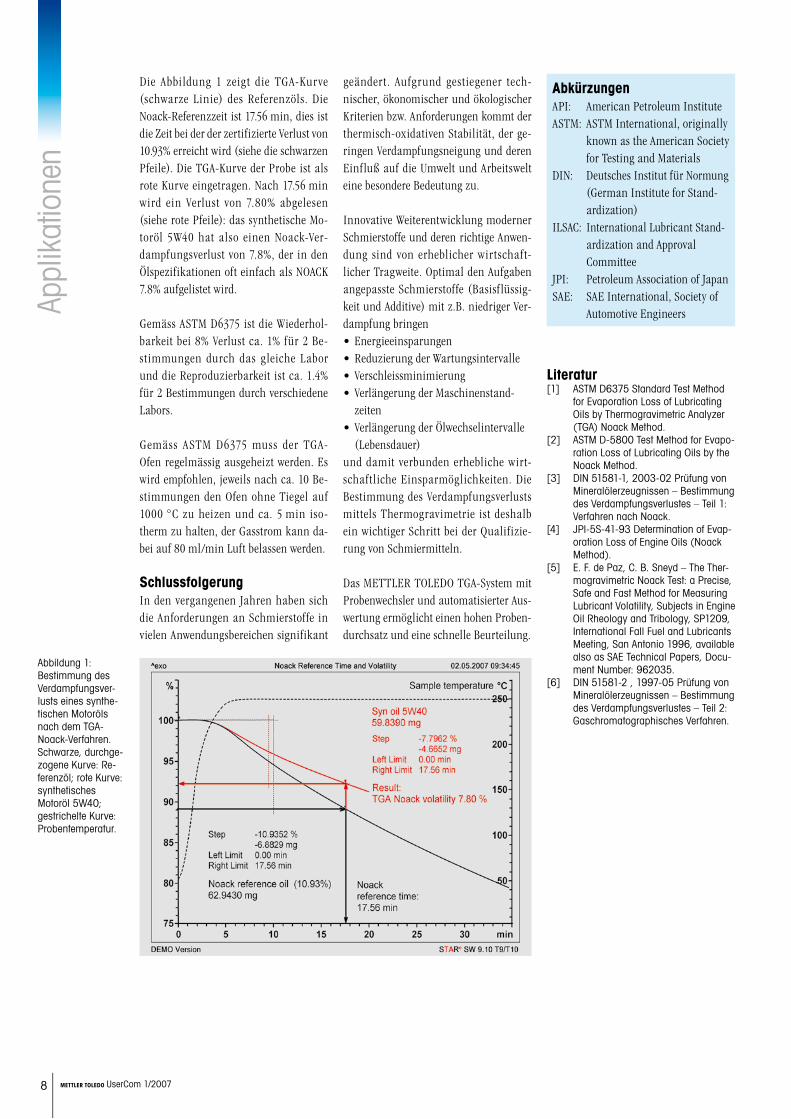
Die Abbildung 1 zeigt die TGA-Kurve (schwarze Linie) des Referenzöls. Die Noack-Referenzzeit ist 17.56 min, dies ist die Zeit bei der der zertifizierte Verlust von 10.93% erreicht wird (siehe die schwarzen Pfeile). Die TGA-Kurve der Probe ist als rote Kurve eingetragen. Nach 17.56 min wird ein Verlust von 7.80% abgelesen (siehe rote Pfeile): das synthetische Mo-toröl 5W40 hat also einen Noack-Ver-dampfungsverlust von 7.8%, der in den Ölspezifikationen oft einfach als NOACK 7.8% aufgelistet wird.
Gemäss ASTM D6375 ist die Wiederhol-barkeit bei 8% Verlust ca. 1% für 2 Be-stimmungen durch das gleiche Labor und die Reproduzierbarkeit ist ca. 1.4% für 2 Bestimmungen durch verschiedene Labors.
Gemäss ASTM D6375 muss der TGA-Ofen regelmässig ausgeheizt werden. Es wird empfohlen, jeweils nach ca. 10 Be-stimmungen den Ofen ohne Tiegel auf 1000 °C zu heizen und ca. 5 min iso-therm zu halten, der Gasstrom kann da-bei auf 80 ml/min Luft belassen werden.
SchlussfolgerungIn den vergangenen Jahren haben sich die Anforderungen an Schmierstoffe in vielen Anwendungsbereichen signifikant
Literatur[1] ASTM D6375 Standard Test Method
for Evaporation Loss of Lubricating Oils by Thermogravimetric Analyzer (TGA) Noack Method.
[2] ASTM D-5800 Test Method for Evapo-ration Loss of Lubricating Oils by the Noack Method.
[3] DIN 51581-1, 2003-02 Prüfung von Mineralölerzeugnissen – Bestimmung des Verdampfungsverlustes – Teil 1: Verfahren nach Noack.
[4] JPI-5S-41-93 Determination of Evap-oration Loss of Engine Oils (Noack Method).
[5] E. F. de Paz, C. B. Sneyd – The Ther-mogravimetric Noack Test: a Precise, Safe and Fast Method for Measuring Lubricant Volatility, Subjects in Engine Oil Rheology and Tribology, SP1209, International Fall Fuel and Lubricants Meeting, San Antonio 1996, available also as SAE Technical Papers, Docu-ment Number: 962035.
[6] DIN 51581-2 , 1997-05 Prüfung von Mineralölerzeugnissen – Bestimmung des Verdampfungsverlustes – Teil 2: Gaschromatographisches Verfahren.
Abbildung 1: Bestimmung des Verdampfungsver-lusts eines synthe-tischen Motoröls nach dem TGA-Noack-Verfahren. Schwarze, durchge-zogene Kurve: Re-ferenzöl; rote Kurve: synthetisches Motoröl 5W40; gestrichelte Kurve: Probentemperatur.
AbkürzungenAPI: American Petroleum InstituteASTM: ASTM International, originally known as the American Society for Testing and MaterialsDIN: Deutsches Institut für Normung (German Institute for Stand- ardization)ILSAC: International Lubricant Stand- ardization and Approval CommitteeJPI: Petroleum Association of JapanSAE: SAE International, Society of Automotive Engineers
geändert. Aufgrund gestiegener tech-nischer, ökonomischer und ökologischer Kriterien bzw. Anforderungen kommt der thermisch-oxidativen Stabilität, der ge-ringen Verdampfungsneigung und deren Einfluß auf die Umwelt und Arbeitswelt eine besondere Bedeutung zu.
Innovative Weiterentwicklung moderner Schmierstoffe und deren richtige Anwen-dung sind von erheblicher wirtschaft-licher Tragweite. Optimal den Aufgaben angepasste Schmierstoffe (Basisflüssig-keit und Additive) mit z.B. niedriger Ver-dampfung bringen
EnergieeinsparungenReduzierung der WartungsintervalleVerschleissminimierungVerlängerung der Maschinenstand-zeitenVerlängerung der Ölwechselintervalle (Lebensdauer)
und damit verbunden erhebliche wirt-schaftliche Einsparmöglichkeiten. Die Bestimmung des Verdampfungsverlusts mittels Thermogravimetrie ist deshalb ein wichtiger Schritt bei der Qualifizie-rung von Schmiermitteln.
Das METTLER TOLEDO TGA-System mit Probenwechsler und automatisierter Aus-wertung ermöglicht einen hohen Proben-durchsatz und eine schnelle Beurteilung.
••••
•
Appl
ikat
ione
n
9METTLER TOLEDO UserCom 1/2007
TGA-FTIR wurde benutzt, um polymor-phe Substanzen zu charakterisieren und zu unterscheiden. Liegen zwei polymor-phe Formen eines Festkörpers vor, deren eine Form schmilzt und die andere bei etwa derselben Temperatur sublimiert oder verdampft, dann kann die gekop-pelte Gasanalyse dazu herangezogen werden, quantitative (Gewichtsverlust) und qualitative (spektrale) Daten zu gewinnen, um diese Festkörper zu ana-lysieren. Zwei Bestandteile, der aktive pharmazeutische Inhaltsstoff Venlafaxin Hydrochlorid (1) und das Trägermaterial 1,1-bis(4-hydroxyphenyl)cyclohexan (2) (Abbildung 1), wurden mit DSC, TGA, Heiztisch-Mikroskopie (HTM) und TGA-FTIR untersucht, um die beim Heizen auftretenden Phasenübergänge zu stu-dieren. Eine Substanz bezeichnet man als polymorph, wenn sie in verschie-denen Kristallstrukturen und damit mit unterschiedlichen physikalischen Eigen-schaften auftreten kann [1].
Venlafaxin Hydrochlorid (Ven-HCl)VenHCl ist das am meisten verkaufte Anti-Depressivum. Venlafaxin, (±)-1-[2(dimethylamino)-1-(4-methoxyphenyl)ethyl]cyclohexanol, angewandt in der Hydrochlorid-Formulierung, liegt in ver-schiedenen polymorphen Modifikationen vor. Sie werden nach ihren Schmelztempe-raturen im DSC als Form 1 (210– 212 °C), Form 2 (208–210 °C), Form 3 (202–204 °C, Phase aus der Schmelze) und
Form 4 (219–220 °C, Hydrat/Alkoholat) klassifiziert. Eine neue Phase (Form 5) wurde während der thermischen Un-tersuchungen dieses Wirkstoffs durch Kristallisation des amorphen, unter Vakuum erhaltenen Sublimations-/Ver-dampfungsprodukts isoliert [2].
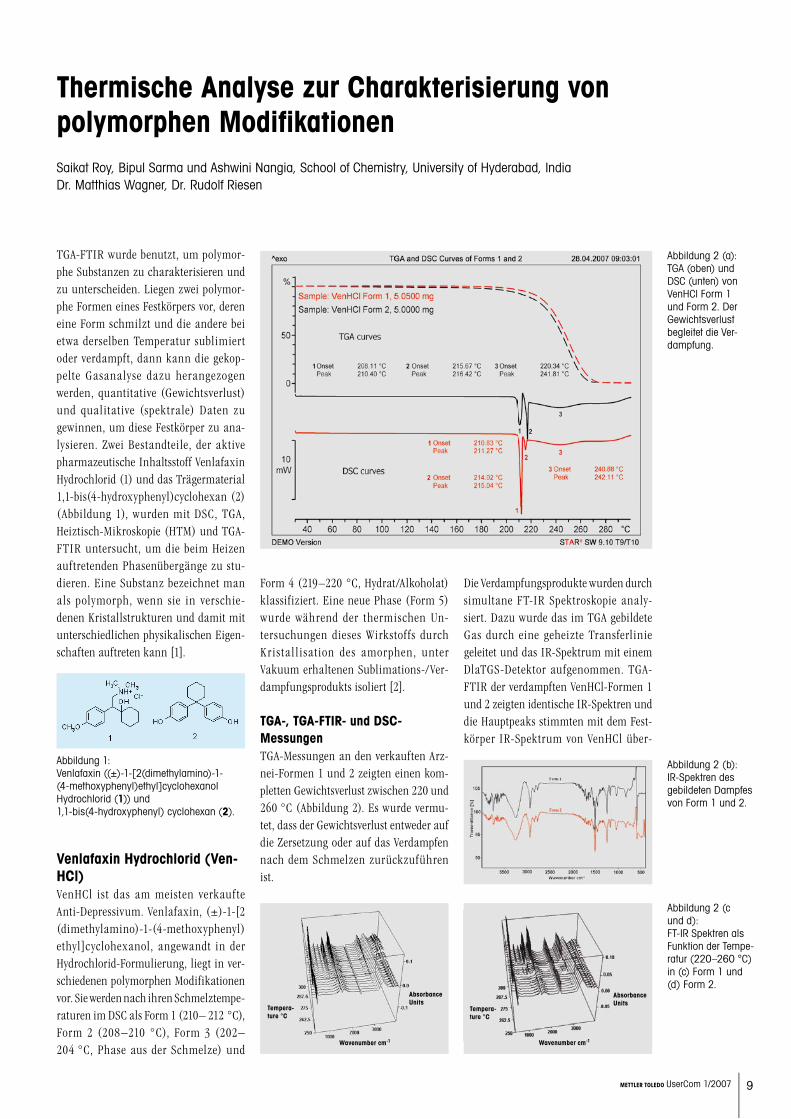
TGA-, TGA-FTIR- und DSC-MessungenTGA-Messungen an den verkauften Arz-nei-Formen 1 und 2 zeigten einen kom-pletten Gewichtsverlust zwischen 220 und 260 °C (Abbildung 2). Es wurde vermu-tet, dass der Gewichtsverlust entweder auf die Zersetzung oder auf das Verdampfen nach dem Schmelzen zurückzuführen ist.
Die Verdampfungsprodukte wurden durch simultane FT-IR Spektroskopie analy-siert. Dazu wurde das im TGA gebildete Gas durch eine geheizte Transferlinie geleitet und das IR-Spektrum mit einem DlaTGS-Detektor aufgenommen. TGA-FTIR der verdampften VenHCl-Formen 1 und 2 zeigten identische IR-Spektren und die Hauptpeaks stimmten mit dem Fest-körper IR-Spektrum von VenHCl über-
Thermische Analyse zur Charakterisierung von polymorphen ModifikationenSaikat Roy, Bipul Sarma und Ashwini Nangia, School of Chemistry, University of Hyderabad, IndiaDr. Matthias Wagner, Dr. Rudolf Riesen
Abbildung 1: Venlafaxin ((±)-1-[2(dimethylamino)-1-(4-methoxyphenyl)ethyl]cyclohexanol Hydrochlorid (1)) und 1,1-bis(4-hydroxyphenyl) cyclohexan (2).
Abbildung 2 (a): TGA (oben) und DSC (unten) von VenHCl Form 1 und Form 2. Der Gewichtsverlust begleitet die Ver-dampfung.
Abbildung 2 (b): IR-Spektren des gebildeten Dampfes von Form 1 und 2.
Abbildung 2 (c und d): FT-IR Spektren als Funktion der Tempe-ratur (220–260 °C) in (c) Form 1 und (d) Form 2.
Tempera-ture °C
AbsorbanceUnits
Wavenumber cm-1
Tempera-ture °C
AbsorbanceUnits
Wavenumber cm-1
10 METTLER TOLEDO UserCom 1/2007
ein. Dies bedeutet, dass VenHCl-Dampf aus beiden Formen nach dem Phasen-übergang bei 214–216 °C freigesetzt wird, ein Phänomen, welches die Ver-dampfung des Festkörpers während des breiten endothermen Effektes bei 220–260 °C begleitet.
Form 2 verflüchtigt sich schneller als Form 1 (TGA).
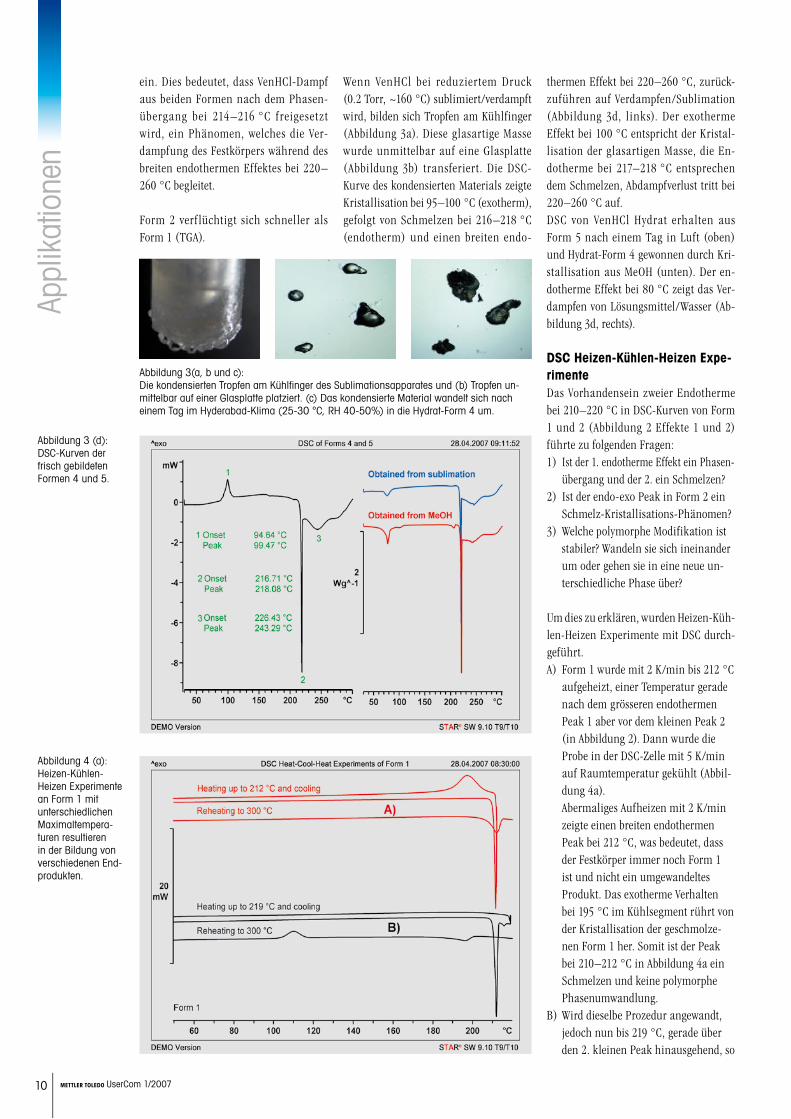
Wenn VenHCl bei reduziertem Druck (0.2 Torr, ~160 °C) sublimiert/verdampft wird, bilden sich Tropfen am Kühlfinger (Abbildung 3a). Diese glasartige Masse wurde unmittelbar auf eine Glasplatte (Abbildung 3b) transferiert. Die DSC-Kurve des kondensierten Materials zeigte Kristallisation bei 95–100 °C (exotherm), gefolgt von Schmelzen bei 216–218 °C (endotherm) und einen breiten endo-
thermen Effekt bei 220–260 °C, zurück-zuführen auf Verdampfen/Sublimation (Abbildung 3d, links). Der exotherme Effekt bei 100 °C entspricht der Kristal-lisation der glasartigen Masse, die En-dotherme bei 217–218 °C entsprechen dem Schmelzen, Abdampfverlust tritt bei 220–260 °C auf. DSC von VenHCl Hydrat erhalten aus Form 5 nach einem Tag in Luft (oben) und Hydrat-Form 4 gewonnen durch Kri-stallisation aus MeOH (unten). Der en-dotherme Effekt bei 80 °C zeigt das Ver-dampfen von Lösungsmittel/Wasser (Ab-bildung 3d, rechts).
DSC Heizen-Kühlen-Heizen Expe-rimenteDas Vorhandensein zweier Endotherme bei 210–220 °C in DSC-Kurven von Form 1 und 2 (Abbildung 2 Effekte 1 und 2) führte zu folgenden Fragen:
Ist der 1. endotherme Effekt ein Phasen-übergang und der 2. ein Schmelzen?Ist der endo-exo Peak in Form 2 ein Schmelz-Kristallisations-Phänomen?Welche polymorphe Modifikation ist stabiler? Wandeln sie sich ineinander um oder gehen sie in eine neue un-terschiedliche Phase über?
Um dies zu erklären, wurden Heizen-Küh-len-Heizen Experimente mit DSC durch-geführt.
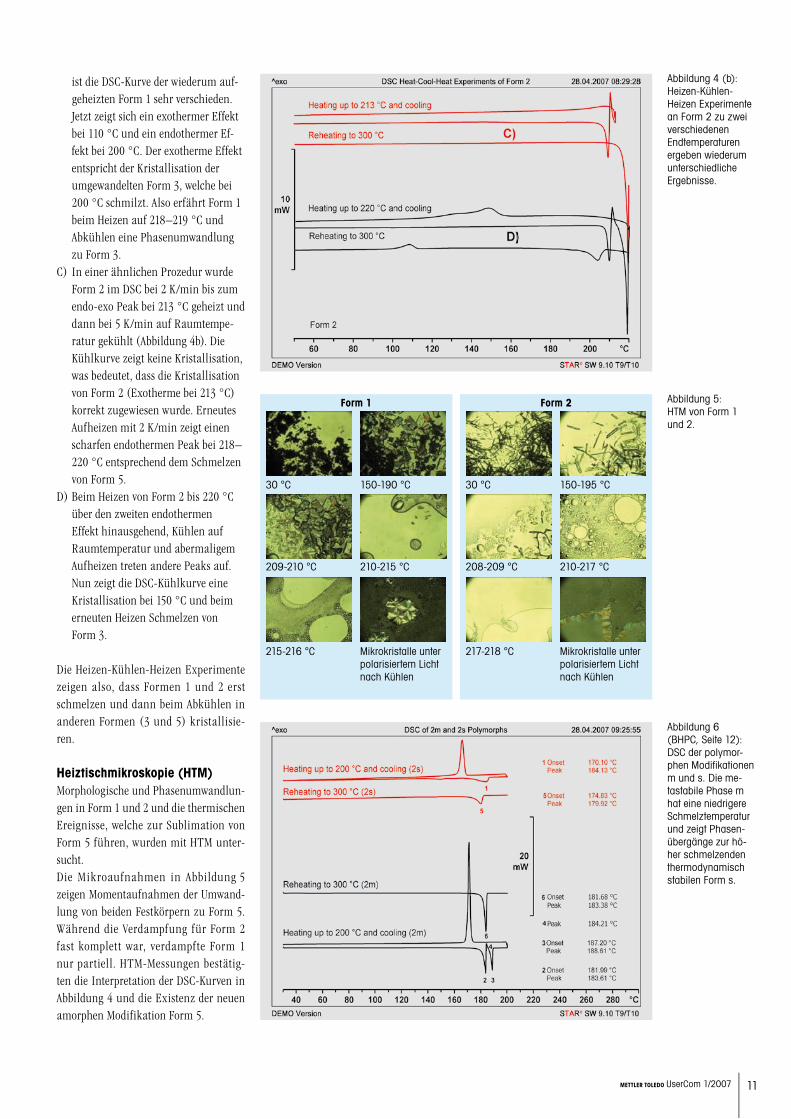
Form 1 wurde mit 2 K/min bis 212 °C aufgeheizt, einer Temperatur gerade nach dem grösseren endothermen Peak 1 aber vor dem kleinen Peak 2 (in Abbildung 2). Dann wurde die Probe in der DSC-Zelle mit 5 K/min auf Raumtemperatur gekühlt (Abbil-dung 4a). Abermaliges Aufheizen mit 2 K/min zeigte einen breiten endothermen Peak bei 212 °C, was bedeutet, dass der Festkörper immer noch Form 1 ist und nicht ein umgewandeltes Produkt. Das exotherme Verhalten bei 195 °C im Kühlsegment rührt von der Kristallisation der geschmolze-nen Form 1 her. Somit ist der Peak bei 210–212 °C in Abbildung 4a ein Schmelzen und keine polymorphe Phasenumwandlung.Wird dieselbe Prozedur angewandt, jedoch nun bis 219 °C, gerade über den 2. kleinen Peak hinausgehend, so
1)
2)
3)
A)
B)
Appl
ikat
ione
n
Abbildung 4 (a): Heizen-Kühlen-Heizen Experimente an Form 1 mit unterschiedlichen Maximaltempera-turen resultieren in der Bildung von verschiedenen End-produkten.
Abbildung 3(a, b und c): Die kondensierten Tropfen am Kühlfinger des Sublimationsapparates und (b) Tropfen un-mittelbar auf einer Glasplatte platziert. (c) Das kondensierte Material wandelt sich nach einem Tag im Hyderabad-Klima (25-30 °C, RH 40-50%) in die Hydrat-Form 4 um.
Abbildung 3 (d): DSC-Kurven der frisch gebildeten Formen 4 und 5.
11METTLER TOLEDO UserCom 1/2007
ist die DSC-Kurve der wiederum auf-geheizten Form 1 sehr verschieden. Jetzt zeigt sich ein exothermer Effekt bei 110 °C und ein endothermer Ef-fekt bei 200 °C. Der exotherme Effekt entspricht der Kristallisation der umgewandelten Form 3, welche bei 200 °C schmilzt. Also erfährt Form 1 beim Heizen auf 218–219 °C und Abkühlen eine Phasenumwandlung zu Form 3.In einer ähnlichen Prozedur wurde Form 2 im DSC bei 2 K/min bis zum endo-exo Peak bei 213 °C geheizt und dann bei 5 K/min auf Raumtempe-ratur gekühlt (Abbildung 4b). Die Kühlkurve zeigt keine Kristallisation, was bedeutet, dass die Kristallisation von Form 2 (Exotherme bei 213 °C) korrekt zugewiesen wurde. Erneutes Aufheizen mit 2 K/min zeigt einen scharfen endothermen Peak bei 218–220 °C entsprechend dem Schmelzen von Form 5.Beim Heizen von Form 2 bis 220 °C über den zweiten endothermen Effekt hinausgehend, Kühlen auf Raumtemperatur und abermaligem Aufheizen treten andere Peaks auf. Nun zeigt die DSC-Kühlkurve eine Kristallisation bei 150 °C und beim erneuten Heizen Schmelzen von Form 3.
Die Heizen-Kühlen-Heizen Experimente zeigen also, dass Formen 1 und 2 erst schmelzen und dann beim Abkühlen in anderen Formen (3 und 5) kristallisie-ren.
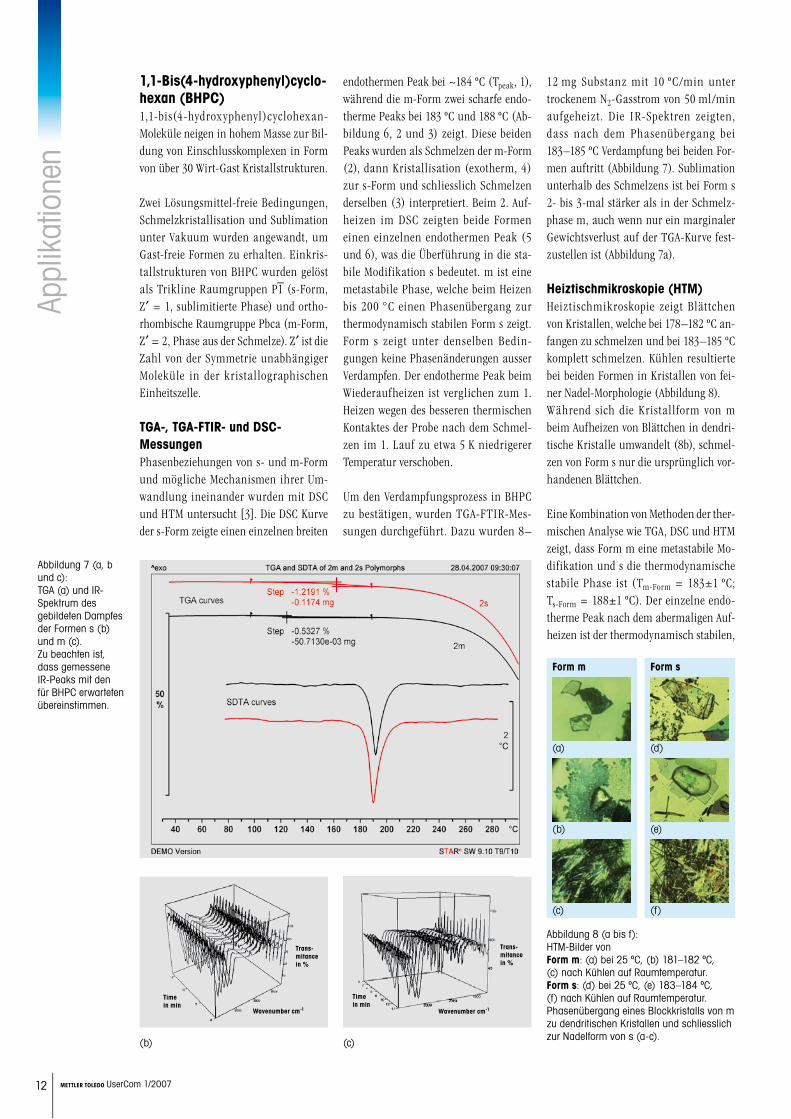
Heiztischmikroskopie (HTM)Morphologische und Phasenumwandlun-gen in Form 1 und 2 und die thermischen Ereignisse, welche zur Sublimation von Form 5 führen, wurden mit HTM unter-sucht. Die Mikroaufnahmen in Abbildung 5 zeigen Momentaufnahmen der Umwand-lung von beiden Festkörpern zu Form 5. Während die Verdampfung für Form 2 fast komplett war, verdampfte Form 1 nur partiell. HTM-Messungen bestätig-ten die Interpretation der DSC-Kurven in Abbildung 4 und die Existenz der neuen amorphen Modifikation Form 5.
C)
D)
Abbildung 5: HTM von Form 1 und 2.
Abbildung 4 (b): Heizen-Kühlen-Heizen Experimente an Form 2 zu zwei verschiedenen Endtemperaturen ergeben wiederum unterschiedliche Ergebnisse.
Abbildung 6 (BHPC, Seite 12): DSC der polymor-phen Modifikationen m und s. Die me-tastabile Phase m hat eine niedrigere Schmelztemperatur und zeigt Phasen-übergänge zur hö-her schmelzenden thermodynamisch stabilen Form s.
Form 1
30 °C 150-190 °C
209-210 °C 210-215 °C
215-216 °C Mikrokristalle unter polarisiertem Licht nach Kühlen
Form 2
30 °C 150-195 °C
208-209 °C 210-217 °C
217-218 °C Mikrokristalle unter polarisiertem Licht nach Kühlen
12 METTLER TOLEDO UserCom 1/2007
12 mg Substanz mit 10 ºC/min unter trockenem N2-Gasstrom von 50 ml/min aufgeheizt. Die IR-Spektren zeigten, dass nach dem Phasenübergang bei 183–185 ºC Verdampfung bei beiden For-men auftritt (Abbildung 7). Sublimation unterhalb des Schmelzens ist bei Form s 2- bis 3-mal stärker als in der Schmelz-phase m, auch wenn nur ein marginaler Gewichtsverlust auf der TGA-Kurve fest-zustellen ist (Abbildung 7a).
Heiztischmikroskopie (HTM)Heiztischmikroskopie zeigt Blättchen von Kristallen, welche bei 178–182 ºC an-fangen zu schmelzen und bei 183–185 ºC komplett schmelzen. Kühlen resultierte bei beiden Formen in Kristallen von fei-ner Nadel-Morphologie (Abbildung 8). Während sich die Kristallform von m beim Aufheizen von Blättchen in dendri-tische Kristalle umwandelt (8b), schmel-zen von Form s nur die ursprünglich vor-handenen Blättchen.
Eine Kombination von Methoden der ther-mischen Analyse wie TGA, DSC und HTM zeigt, dass Form m eine metastabile Mo-difikation und s die thermodynamische stabile Phase ist (Tm-Form = 183±1 ºC; Ts-Form = 188±1 ºC). Der einzelne endo-therme Peak nach dem abermaligen Auf-heizen ist der thermodynamisch stabilen,
Abbildung 8 (a bis f): HTM-Bilder von Form m: (a) bei 25 ºC, (b) 181–182 ºC, (c) nach Kühlen auf Raumtemperatur. Form s: (d) bei 25 ºC, (e) 183–184 ºC, (f) nach Kühlen auf Raumtemperatur. Phasenübergang eines Blockkristalls von m zu dendritischen Kristallen und schliesslich zur Nadelform von s (a-c).
1,1-Bis(4-hydroxyphenyl)cyclo-hexan (BHPC)1,1-bis(4-hydroxyphenyl)cyclohexan-Moleküle neigen in hohem Masse zur Bil-dung von Einschlusskomplexen in Form von über 30 Wirt-Gast Kristallstrukturen.
Zwei Lösungsmittel-freie Bedingungen, Schmelzkristallisation und Sublimation unter Vakuum wurden angewandt, um Gast-freie Formen zu erhalten. Einkris-tallstrukturen von BHPC wurden gelöst als Trikline Raumgruppen P1 (s-Form, Z′ = 1, sublimitierte Phase) und ortho-rhombische Raumgruppe Pbca (m-Form, Z′ = 2, Phase aus der Schmelze). Z′ ist die Zahl von der Symmetrie unabhängiger Moleküle in der kristallographischen Einheitszelle.
TGA-, TGA-FTIR- und DSC-MessungenPhasenbeziehungen von s- und m-Form und mögliche Mechanismen ihrer Um-wandlung ineinander wurden mit DSC und HTM untersucht [3]. Die DSC Kurve der s-Form zeigte einen einzelnen breiten
endothermen Peak bei ~184 ºC (Tpeak, 1), während die m-Form zwei scharfe endo-therme Peaks bei 183 ºC und 188 ºC (Ab-bildung 6, 2 und 3) zeigt. Diese beiden Peaks wurden als Schmelzen der m-Form (2), dann Kristallisation (exotherm, 4) zur s-Form und schliesslich Schmelzen derselben (3) interpretiert. Beim 2. Auf-heizen im DSC zeigten beide Formen einen einzelnen endothermen Peak (5 und 6), was die Überführung in die sta-bile Modifikation s bedeutet. m ist eine metastabile Phase, welche beim Heizen bis 200 °C einen Phasenübergang zur thermodynamisch stabilen Form s zeigt. Form s zeigt unter denselben Bedin-gungen keine Phasenänderungen ausser Verdampfen. Der endotherme Peak beim Wiederaufheizen ist verglichen zum 1. Heizen wegen des besseren thermischen Kontaktes der Probe nach dem Schmel-zen im 1. Lauf zu etwa 5 K niedrigerer Temperatur verschoben.
Um den Verdampfungsprozess in BHPC zu bestätigen, wurden TGA-FTIR-Mes-sungen durchgeführt. Dazu wurden 8–
Appl
ikat
ione
n
Abbildung 7 (a, b und c): TGA (a) und IR-Spektrum des gebildeten Dampfes der Formen s (b) und m (c). Zu beachten ist, dass gemessene IR-Peaks mit den für BHPC erwarteten übereinstimmen.
Form m Form s
(a) (d)
(b) (e)
(c) (f)
Time in min
Trans-mitancein %
Wavenumber cm-1
Time in min
Trans-mitancein %
Wavenumber cm-1
(b) (c)

13METTLER TOLEDO UserCom 1/2007
höher schmelzenden kristallisierten Pha-se zuzuschreiben. TGA-FTIR bestätigt das Verdampfen/Sublimieren von BHPC.
ZusammenfassungPhasenbeziehungen zwischen VenHCl-Modifikationen sind im Schema (Abbil-dung 9) zusammengefasst. Zusätzlich zu den gemessenen quantitativen und reproduzierbaren Daten an schon vorher bekannten vier Formen 1–4 von VenHCl wurde eine neue durch Verdampfen und Kristallisieren gewonnene Form 5 iden-tifiziert. Diese ist unter inerten Bedin-gungen kurzlebig (einige Stunden bis maximal ein Tag stabil). Sie wandelt sich an der Luft in die Hydrat-Form 4 und unter trockenen Bedingungen in Form 1 um.
TGA, DSC und Heiztischmikroskopie zeigen, dass der 5 K niedriger schmel-zende Festkörper der Form m von BHPC der metastabilen Modifikation und die s-Form der thermodynamisch stabilen Phase entspricht. Der einzelne endother-me Peak nach dem abermaligen Aufhei-zen beider Formen ist der thermodyna-
mischen stabilen, höher schmelzenden Phase zuzuschreiben, welche auch durch Verdampfen/Sublimation erhalten wer-den kann.
Zusätzlich zu den gezeigten Applikati-onen wurde TGA-FTIR benutzt, um zwi-schen Anilin- und Phenoleinschluss in einem Gast-selektiven Wirte-Netzwerk zu unterscheiden [4].
Literatur[1] W. C. McCrone, in Physics and Chem-
istry of the Organic Solid State, Vol. 2, D. Fox, M. M. Labes and A. Weiss-berger (Eds.), Wiley Interscience: New York, 1965, pp. 725–767.
[2] S. Roy, S. Aitipamula and A. Nangia, Cryst. Growth Des. 2005, 5, 2268–2276.
[3] B. Sarma, S. Roy, and A. Nangia, Chem. Commun. 2006, 4918–4920.
[4] S. Aitipamula and A. Nangia, Chem. Eur. J. 2005, 11, 6727–6742.
Abbildung 9: Schema der Pha-senübergänge in VenHCl-Modifikati-onen 1–5.
EinleitungDas Messen und Interpretieren von Schmelzprozessen mittels Methoden der Temperaturmodulierten DSC (TMDSC) gehört zu den eher anspruchsvollen Auf-gaben in der thermischen Analyse. Das mag vielleicht der Grund sein, dass man in der Literatur einige Vorschläge finden kann, die einer kritischen Hinterfragung
nicht standhalten. Trotzdem können mit der TMDSC interessante und wichtige In-formationen zum Schmelzverhalten ge-wonnen werden, die sonst nur schwer zu erhalten sind.
In diesem Artikel wollen wir ausgehend von den Grundlagen des Schmelzverhal-tens aus Referenz [1] an Beispielen auf-
zeigen, wie das Schmelzverhalten mittels TOPEM® untersucht werden kann.
Grundlagen der Temperaturmo-dulierten DSC
Messprinzip und Voraussetzungen an die MessungBei der TMDSC wird einem konventi-onellen Temperaturprogramm (Heizen und Kühlen mit konstanter Rate oder iso-therme Bedingungen) eine kleine Tem-peraturstörung (Modulation) überlagert. Beim Auswertealgorithmus geht man da-von aus, dass sich die Probenreaktionen auf das konventionelle Temperaturpro-gramm und die Modulation gegenseitig
Es werden Voraussetzungen der Analyse von Schmelzprozessen mittels TOPEM® besprochen. Bei Einhaltung dieser Voraussetzungen werden im reversierenden Wärmestrom Prozesse gemessen, die unter Gleich-gewichtsbedingungen ablaufen. Prozesse, mit Unterkühlung oder Über-hitzung werden im nicht-reversierenden Wärmestrom erfasst. Diese Separierung ermöglicht eine Klassifizierung von Schmelzprozessen und die Unterscheidung unterschiedlich stabiler Kristallstrukturen.
Analyse von Schmelzprozessen mittels TOPEM®
Dr. Jürgen Schawe

14 METTLER TOLEDO UserCom 1/2007
nicht beeinflussen. Man kann dann den unterliegenden Anteil (vom konventio-nellen Temperaturprogramm) und den Anteil durch die Modulation separieren. Während der unterliegende Anteil des Wärmestroms (totaler Wärmestrom) wie bei der konventionellen DSC alle Infor-mationen beinhaltet, enthält der Anteil, der durch die Modulation erzeugt wird, nur Informationen über Prozesse, die der Modulation mehr oder weniger folgen können.
Bei allen Modulationstechniken müssen die Messbedingungen so gewählt werden, dass die Messung und Auswertung unter linearen und nahezu stationären Bedin-gungen erfolgen. Das bedeutet, dass das Ergebnis unabhängig von der Intensität (Amplitude) der Modulation ist und sich der totale Wärmestrom während einer des relevanten Auswertefensters (Periode) nicht wesentlich ändert. Die Qualität der Messung verbessert sich mit der Verrin-gerung der unterliegenden Heizrate. Ins-besondere bei der Analyse von Schmelz-prozessen sind kleine Modulationen zu wählen, da sonst Artefakte gemessen wer-den, die zu Fehlinterpretationen führen. TOPEM® ist eine moderne TMDSC-Tech-nik, die sich bezüglich der Art der Modu-lationsfunktion und der Auswertung von der konventionellen TMDSC unterschei-det. Bei TOPEM® wird eine stochastische Funktion zur Modulation verwendet. Die Intensität der Modulationsfunktion ist eine Impulshöhe. Die Auswertung erfolgt durch eine Korrelationsanalyse von ge-messenem Wärmestrom und Heizrate in einem wählbaren Auswertefenster. [2, 3].
Totaler, reversierender- und nicht-reversierender WärmestromBei allen TMDSC-Techniken werden aus dem gemessenen Wärmestrom drei Wär-mestromkomponenten ermittelt. Das sind der Totale Wärmestrom Φtot, der rever-sierende Wärmestrom (engl. reversing heat flow) Φrev und der nicht-reversie-rende Wärmestrom (engl. non-reversing heat flow) Φnon.
Bei der konventionellen TMDSC wird der Totale Wärmestrom aus dem gemessenen Wärmestrom durch Mittlung über min-destens eine Periode erhalten. Aus der modulierten Komponente bestimmt man den reversierenden Wärmestrom. Der nicht-reversierende Wärmestrom ist die Differenz:
Bei TOPEM® erfolgt die Auswertung mit-tels Korrelationsanalyse von Wärmestrom und Heizrate. Man erhält die Komponen-te des gemessenen Wärmestroms, der mit der Heizrate korreliert und eine andere Komponente, die nicht mit der Heizrate korreliert. Die nicht korrelierende Kom-ponente ist der nicht-reversierende Wär-mestrom Φnon. Der reversierende Wär-mestrom wird aus dem korrelierenden Wärmestromanteil ermittelt [3]. Aus bei-den Grössen wird der totale Wärmestrom berechnet:
Dieser Unterschied erscheint gering, er-möglicht aber, wie unten beschrieben, bei TOPEM® eine Konsistenzüberprü-fung der Messung.
Sensibler und latenter Wärme-stromFormal besteht der Wärmestrom aus zwei Komponenten: dem sensiblen (oder fühlbaren) Wärmestrom Φs und dem la-tenten Wärmestrom Φl [3, 4]. Der latente Wärmestrom ist nicht ex-plizit von der Temperatur sondern von der Kinetik des thermischen Ereignisses abhängig. Ein Beispiel ist die Härtungs-reaktion eines Klebstoffes, während der durch Temperaturänderung die Probe nicht mehr zu ihrem Ausgangszustand zurückgebracht werden kann. Es wird nur die Reaktionsgeschwindigkeit verän-dert.Der sensible Wärmestrom hängt explizit von der Heizrate ab. Ein Beispiel ist der Wärmestrom in eine inerte Probe, der proportional zur Heizrate ist. Der Propor-tionalitätsfaktor ist die Wärmekapazität.
GrundlagenAusgangspunkt ist die Beschreibung von Schmelz- und Kristallisationsprozessen mittels der Freien Enthalpie aus Referenz [1]. Ein entsprechendes Diagram ist in Ab-bildung 1 dargestellt. Die rote, schwar-ze und grüne Kurve ist die freie Enthal-pie der Schmelze, des Kristalls und des Glases. Die gestrichelten Kurven kenn-zeichnen Zwischenzustände. Die Kurve mit der jeweils kleinsten Freien Enthalpie charakterisiert den stabilen Zustand. Alle anderen Zustände sind metastabil. Das System ist bestrebt, den stabilen Zustand einzunehmen, wird dabei aber durch ki-netische Prozesse (wie z.B. Keimbildung) behindert.
Prozesse bei einer TOPEM®-Messung sind in Abbildung 1 durch blaue Ellipsen oder Pfeile gekennzeichnet. Es handelt sich dabei um Prozesse, die unter qua-sistabilen, metastabilen und instabilen Bedingungen ablaufen:
Bei den quasi stabilen Prozessen im (lokalen) Gleichgewicht handelt es sich um die Messung der Wärmeka-pazität, ohne das ein weiteres ther-misches Ereignis abläuft.Bei Prozessen unter metastabilen Be-dingungen verlässt das System das ak-tuelle lokale Gleichgewicht nur wenig. Es handelt sich dabei z.B. um Glasü-bergänge oder Schmelz- und Kristal-lisationsprozesse nahe der aktuellen
•
•
Appl
ikat
ione
n
Abbildung 1: Schematisches Dia-gramm der Freien Enthalpie als Funk-tion der Temperatur. Es sind Prozesse eingezeichnet, die unter quasi sta-bilen, metastabilen und instabilen Bedingungen ab-laufen.

15METTLER TOLEDO UserCom 1/2007
lokalen Gleichgewichtsbedingungen, wie sie etwa im Schmelzbereich von verunreinigten Stoffen auftreten (s. [1]). Diese Prozesse können durch geringe Temperaturänderung praktisch rück-gängig gemacht werden.Bei Prozessen mit grosser Änderung der Freien Enthalpie startet das Sys-tem im metastabilen Gleichgewicht und „fällt“ in den neuen stabileren Zustand. Durch kleine Temperatur-änderungen wird der Prozess kaum beeinflusst. Beispiele sind Kristallisati-onsprozesse nach hinreichend grosser Unterkühlung oder Schmelzprozesse von Kristallen mit Überhitzung.
Beschreibung von sensiblem und latentem WärmestromNehmen wir an, dass in einer Probe zwei unterschiedliche Prozesse ablaufen, die jeweils mit einem Ordnungsparameter x beschrieben werden. Beim Schmelzen be-schreibt x den Grad der Unordnung und ändert sich von x = 0 (idealer Kristall) zu x = 1 (equilibrierte Schmelze). Der Pro-zess mit dem Ordnungsparameter xme soll nahe am lokalen Gleichgewicht ab-laufen. Der andere Prozess beginnt fern vom Gleichgewicht und hat den Ord-nungsparameter xi. Für den gemessenen Wärmestrom gilt:
cp ist die spezifische Wärmekapazität, dT/dt ist die Heizrate, ∆hme und ∆hi sind die den entsprechenden Prozessen zuge-ordneten spezifischen Umwandlungsen-thalpien. Da der Prozess (me) nahe am lokalen Gleichgewicht abläuft, kann xme der kleinen Temperaturmodulation fol-gen. Für diesen Fall kann geschrieben werden:
Beim Nicht-Gleichgewichtsprozess (i) folgt der Ordnungsparameter nicht der kleinen, von der Modulationsfunktion bestimmten Temperaturänderung ∆T. Daher gilt:
•
Durch Einsetzen von Gl.(4) in (3) erhält man für den gemessenen Wärmestrom
Der erste Term in Gl. (6) ist eine expli-zite Funktion der Heizrate. Es handelt sich hier um den sensiblen Wärmestrom, der die Prozesse umfasst, die nahe am Gleichgewicht ablaufen. Der latente Wär-mestrom wird durch den letzten Term beschrieben. Er umfasst die Prozesse, die fern vom Gleichgewicht starten.
Wärmestromseparation bei TOPEM®
Die mittels TOPEM® gemessenen re-versierenden und nicht-reversierenden Wärmestrome können bei Beachtung von Linearität und Stationarität im Rahmen der Messgenauigkeit dem sensiblen und latenten Wärmestrom zugeordnet werden:
Messbedingungen und deren Überprüfung
LinearitätDa die Auswertemethodik bei der TMDSC auf der Analyse von linearen Systemen aufbaut, muss das Messprogramm so ge-wählt werden, dass der gemessene Wär-
mestrom linearen Bedingungen genügt. Das ist der Fall, wenn der reversierende Wärmestrom unabhängig von der Inten-sität der Temperaturstörung (Impulshö-he oder Amplitude) ist. Die Grösse der maximalen Intensität ist von den Materi-alien und den zu untersuchenden Ereig-nissen abhängig. Beim Schmelzen liegt die Linearitätsgrenze meistens unterhalb 0.1 K. Am Beispiel des Schmelzens von (PET) ist in Abbildung 2 ein Beispiel für einen Linearitätstest dargestellt. Es wur-den zwei Proben mit ähnlicher Masse mit unterschiedlich grosser Impulshöhe mit einer unterliegenden Heizrate von 0.3 K/min gemessen. Die Impulshöhe war ±5 mK und ±50 mK. Die reversierenden Wärmestromkurven in Abbildung 2 sind unabhängig von der Impulshöhe. Ledig-lich das Rauschen wird bei der kleinen Modulationsintensität grösser. Die blaue Kurve ist die Differenz beider Φrev-Kur-ven. Dieses Material kann mit einer Im-pulshöhe von ±50 mK gemessen werden.
StationaritätIn einem Auswertefensters sollte sich der totale Wärmestrom wenig ändern. Diese Bedingung kann insbesondere bei hö-heren Heizraten während relativ scharfer thermischer Ereignisse nicht immer ein-gehalten werden. Bei TOPEM® gibt es im Gegensatz zur konventionellen TMDSC eine Möglichkeit, Kurvenbereiche zu erkennen, in denen eine Interpretation kritisch ist. Dazu ist ein Vergleich von gemessenem und totalem Wärmestrom nötig. Abbildung 3 zeigte diese Kurven im
Abbildung 2: Test der Lineari-tätsbedingung am Beispiel von PET, das bei 170 °C kri-stallisiert wurde. Oben: Messkurven; unten: Reversie-render Wärmestrom und die Differenz beider Kurven (blau).
16 METTLER TOLEDO UserCom 1/2007
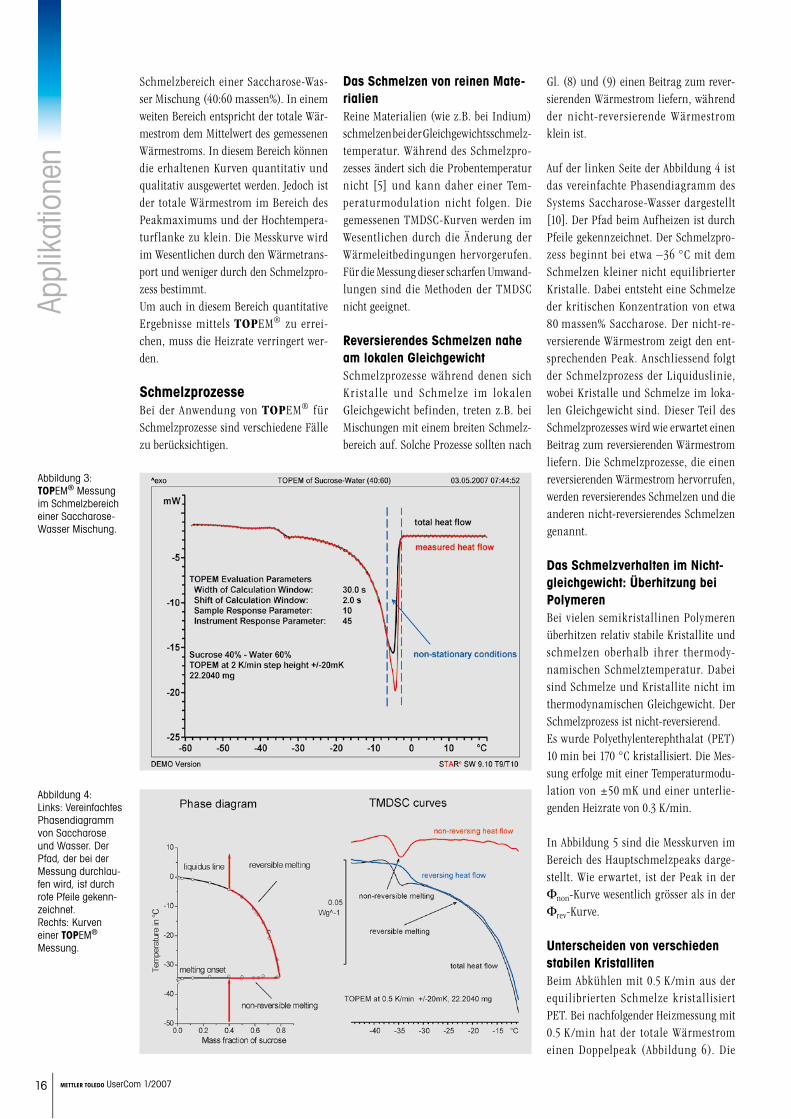
Schmelzbereich einer Saccharose-Was-ser Mischung (40:60 massen%). In einem weiten Bereich entspricht der totale Wär-mestrom dem Mittelwert des gemessenen Wärmestroms. In diesem Bereich können die erhaltenen Kurven quantitativ und qualitativ ausgewertet werden. Jedoch ist der totale Wärmestrom im Bereich des Peakmaximums und der Hochtempera-turflanke zu klein. Die Messkurve wird im Wesentlichen durch den Wärmetrans-port und weniger durch den Schmelzpro-zess bestimmt. Um auch in diesem Bereich quantitative Ergebnisse mittels TOPEM® zu errei-chen, muss die Heizrate verringert wer-den.
SchmelzprozesseBei der Anwendung von TOPEM® für Schmelzprozesse sind verschiedene Fälle zu berücksichtigen.
Das Schmelzen von reinen Mate-rialienReine Materialien (wie z.B. bei Indium) schmelzen bei der Gleichgewichtsschmelz-temperatur. Während des Schmelzpro-zesses ändert sich die Probentemperatur nicht [5] und kann daher einer Tem-peraturmodulation nicht folgen. Die gemessenen TMDSC-Kurven werden im Wesentlichen durch die Änderung der Wärmeleitbedingungen hervorgerufen. Für die Messung dieser scharfen Umwand-lungen sind die Methoden der TMDSC nicht geeignet.
Reversierendes Schmelzen nahe am lokalen GleichgewichtSchmelzprozesse während denen sich Kristalle und Schmelze im lokalen Gleichgewicht befinden, treten z.B. bei Mischungen mit einem breiten Schmelz-bereich auf. Solche Prozesse sollten nach
Gl. (8) und (9) einen Beitrag zum rever-sierenden Wärmestrom liefern, während der nicht-reversierende Wärmestrom klein ist.
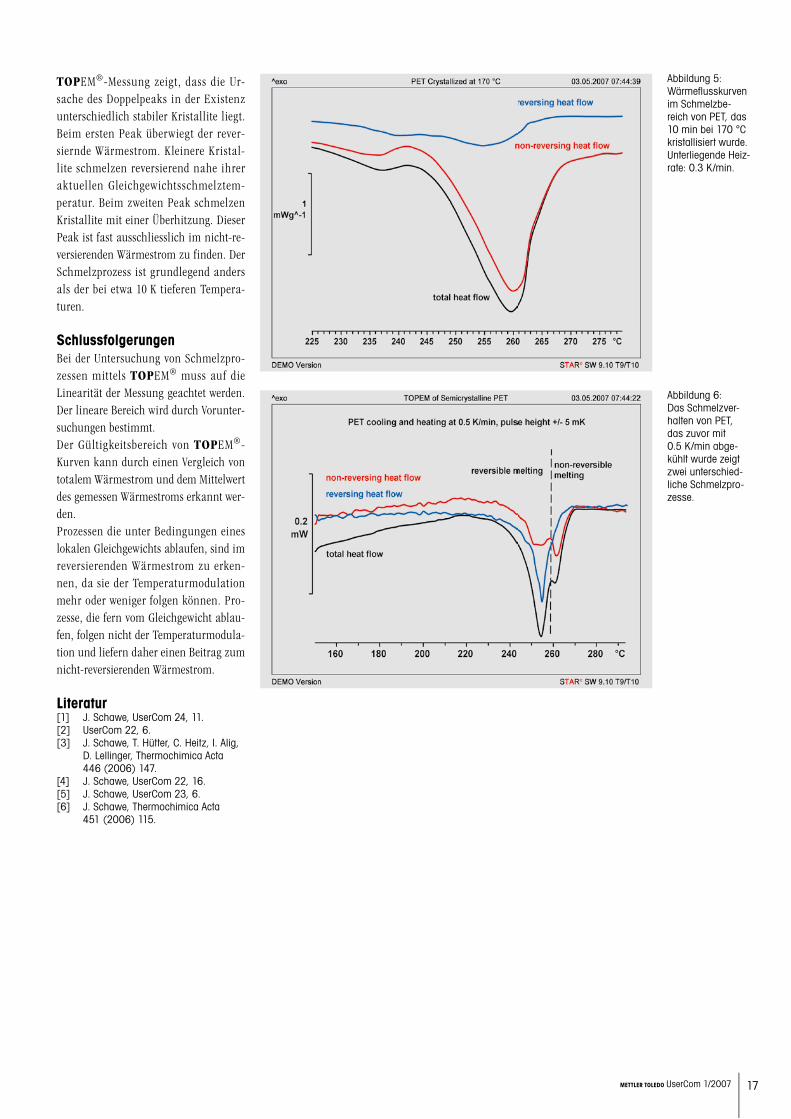
Auf der linken Seite der Abbildung 4 ist das vereinfachte Phasendiagramm des Systems Saccharose-Wasser dargestellt [10]. Der Pfad beim Aufheizen ist durch Pfeile gekennzeichnet. Der Schmelzpro-zess beginnt bei etwa –36 °C mit dem Schmelzen kleiner nicht equilibrierter Kristalle. Dabei entsteht eine Schmelze der kritischen Konzentration von etwa 80 massen% Saccharose. Der nicht-re-versierende Wärmestrom zeigt den ent-sprechenden Peak. Anschliessend folgt der Schmelzprozess der Liquiduslinie, wobei Kristalle und Schmelze im loka-len Gleichgewicht sind. Dieser Teil des Schmelzprozesses wird wie erwartet einen Beitrag zum reversierenden Wärmestrom liefern. Die Schmelzprozesse, die einen reversierenden Wärmestrom hervorrufen, werden reversierendes Schmelzen und die anderen nicht-reversierendes Schmelzen genannt.
Das Schmelzverhalten im Nicht-gleichgewicht: Überhitzung bei PolymerenBei vielen semikristallinen Polymeren überhitzen relativ stabile Kristallite und schmelzen oberhalb ihrer thermody-namischen Schmelztemperatur. Dabei sind Schmelze und Kristallite nicht im thermodynamischen Gleichgewicht. Der Schmelzprozess ist nicht-reversierend.Es wurde Polyethylenterephthalat (PET) 10 min bei 170 °C kristallisiert. Die Mes-sung erfolge mit einer Temperaturmodu-lation von ±50 mK und einer unterlie-genden Heizrate von 0.3 K/min.
In Abbildung 5 sind die Messkurven im Bereich des Hauptschmelzpeaks darge-stellt. Wie erwartet, ist der Peak in der Φnon-Kurve wesentlich grösser als in der Φrev-Kurve.
Unterscheiden von verschieden stabilen Kristalliten Beim Abkühlen mit 0.5 K/min aus der equilibrierten Schmelze kristallisiert PET. Bei nachfolgender Heizmessung mit 0.5 K/min hat der totale Wärmestrom einen Doppelpeak (Abbildung 6). Die
Abbildung 3: TOPEM® Messung im Schmelzbereich einer Saccharose-Wasser Mischung.
Appl
ikat
ione
n
Abbildung 4: Links: Vereinfachtes Phasendiagramm von Saccharose und Wasser. Der Pfad, der bei der Messung durchlau-fen wird, ist durch rote Pfeile gekenn-zeichnet. Rechts: Kurven einer TOPEM® Messung.
17METTLER TOLEDO UserCom 1/2007
TOPEM®-Messung zeigt, dass die Ur-sache des Doppelpeaks in der Existenz unterschiedlich stabiler Kristallite liegt. Beim ersten Peak überwiegt der rever-siernde Wärmestrom. Kleinere Kristal-lite schmelzen reversierend nahe ihrer aktuellen Gleichgewichtsschmelztem-peratur. Beim zweiten Peak schmelzen Kristallite mit einer Überhitzung. Dieser Peak ist fast ausschliesslich im nicht-re-versierenden Wärmestrom zu finden. Der Schmelzprozess ist grundlegend anders als der bei etwa 10 K tieferen Tempera-turen.
SchlussfolgerungenBei der Untersuchung von Schmelzpro-zessen mittels TOPEM® muss auf die Linearität der Messung geachtet werden. Der lineare Bereich wird durch Vorunter-suchungen bestimmt.Der Gültigkeitsbereich von TOPEM®-Kurven kann durch einen Vergleich von totalem Wärmestrom und dem Mittelwert des gemessen Wärmestroms erkannt wer-den.Prozessen die unter Bedingungen eines lokalen Gleichgewichts ablaufen, sind im reversierenden Wärmestrom zu erken-nen, da sie der Temperaturmodulation mehr oder weniger folgen können. Pro-zesse, die fern vom Gleichgewicht ablau-fen, folgen nicht der Temperaturmodula-tion und liefern daher einen Beitrag zum nicht-reversierenden Wärmestrom.
Literatur[1] J. Schawe, UserCom 24, 11.[2] UserCom 22, 6.[3] J. Schawe, T. Hütter, C. Heitz, I. Alig,
D. Lellinger, Thermochimica Acta 446 (2006) 147.
[4] J. Schawe, UserCom 22, 16.[5] J. Schawe, UserCom 23, 6.[6] J. Schawe, Thermochimica Acta
451 (2006) 115.
Abbildung 6: Das Schmelzver-halten von PET, das zuvor mit 0.5 K/min abge-kühlt wurde zeigt zwei unterschied-liche Schmelzpro-zesse.
Abbildung 5: Wärmeflusskurven im Schmelzbe-reich von PET, das 10 min bei 170 °C kristallisiert wurde. Unterliegende Heiz-rate: 0.3 K/min.

18 METTLER TOLEDO UserCom 1/2007
EinleitungVon einem „guten“ Parfüm wird neben einem ansprechenden Geruch erwartet, dass die Duftstoffe möglichst lange mit einer konstanten Intensität wahrnehm-bar bleiben. Aus diesem Grund werden die Duftstoffe in Parfüms neuerdings in sogenannten Delivery Systemen (DS) gekapselt. Damit kann die Abgabe des Duftstoffes kontrolliert werden, was eine Optimierung der Wahrnehmbarkeit des Parfüms bezüglich Intensität und Dauer ermöglicht.
Die Kapselung von Duftstoffen in geeig-neten DS ist demzufolge für Hersteller von Parfüms von grosser Bedeutung.
Um aus der Fülle von möglichen DS die am besten Geeigneten zu identifizieren, braucht man eine rasche und einfache Technik, um das Entweichen des Duft-stoffes aus dem DS und die thermische Stabilität des Gesamtsystems zu beschrei-ben. Die Thermogravimetrie erfüllt diese Anforderungen in hervorragender Weise.
In diesem Beitrag untersuchen wir mit-tels Thermogravimetrie das Entweichen von Romascone® aus 3 unterschiedli-chen DS. Romascone® ist ein Duftstoff, der insbesondere in Parfüms für Frauen Verwendung findet. Als DS wurden Na-nopartikel auf der Basis von vernetztem Vinylacetat verwendet.
ExperimentellesFür die hier beschriebenen Untersuchun-gen wurde ein TGA851/SDTAe mit einem kleinen Ofen von METTLER TOLEDO ver- wendet. Es wurden Probenmassen von typisch et- wa 8 mg (Duftstoff und Nanopartikel zu- sammen) in Tiegeln aus Aluminiumoxid vermessen. Der Gewichtsanteil des DS betrug jeweils 40%. Als Spülgas wurden 20 ml/min Stickstoff verwendet. Die Messungen erfolgten isotherm bei unter-schiedlichen Temperaturen.
Theoretische Grundlagen
Verdunsten von reinen Flüssig-keitenWerden flüchtige Verbindungen (wie beispielsweise Duftstoffe) in der TGA vermessen, so wird ein kontinuierlicher Massenverlust erwartet. Wird davon aus-gegangen, dass in einer TGA die Grenz-schicht zwischen Flüssigkeit und Gaspha-se erreichenden Moleküle einer flüchtigen Verbindung mit dem Spülgas aus dem Ofen der TGA entfernt werden, so ergibt sich unter isothermen Bedingungen eine konstante Massenverlustrate, die durch den Dampfdruck der Verbindung sowie durch den Massentransfer an der Grenz-schicht bestimmt ist; d.h.
Dabei bedeuten m die Masse, k eine Kon- stante, die den Massentransfer an der Grenzschicht zwischen Flüssigkeit und Gasphase beschreibt und Pvap den Dampf-druck.
Verdunsten einer Verbindung aus einer binären MischungIn einer Mischung von 2 Verbindungen sind die chemischen Potenziale der bei-den Verbindungen in der Mischung ge-genüber den chemischen Potenzialen der reinen Verbindungen reduziert. Für ideale Verbindungen (keine Wech-selwirkung, gleiche Grösse der Molekü-le) gilt für den Partialdruck der beiden Verbindungen das Raoult’sche Gesetz, gemäss dem sich die Partialdrücke der beiden Verbindungen entsprechend ihrer molaren Anteile verhalten, d.h.
Hier bedeuten P1 und P2 die Partialdrü-cke der beiden Verbindungen, x1 und x2 deren Molenbruch und P1
0 und P20 die
Dampfdrücke unter Normalbedingun-gen. Für reale Verbindungen und unter der Annahme, dass nur eine Substanz flüchtig ist, gilt für den Partialdruck der flüchtigen Komponente die Näherung von Flory:
Hier steht f1 für den Volumenanteil der flüchtigen Komponente (Lösungsmit-tel) und c für den so genannten Flory-Wechselwirkungsparameter. Damit die Durchmischung der beiden Komponen-ten spontan erfolgt, muss die Mischen-thalpie (hier ausgedrückt durch c) klein sein. c variiert typischerweise zwischen 0 (gutes Lösungsmittel, athermische Mi-schung) und 0.5 (schlechtes Lösungsmit-tel, endothermische Mischung). Liegt der Wechselwirkungsparameter oberhalb von 0.5, wird ein Entmischen des Systems er-wartet.
Falls die Dichte der beiden Verbindungen in der Mischung etwa gleich gross ist, ist der Volumenanteil des Lösungsmittels (f1) identisch mit dessen Massenanteil (w1). Für die Massenverlustrate erhält man so-mit
Wird die erste Ableitung des TGA-Signals als Funktion des Gewichtsanteils des Lö-semittels aufgetragen, so lassen sich aus einem Fit dieser Kurve mit der Funktion gemäss Gl. 4 die Parameter k ·Pvap und c bestimmen. Werden mehrere isotherme Messungen des Verdunstungsverhalten durchgeführt, lässt sich auch die Tempe-raturabhängigkeit der beiden Parameter
Appl
ikat
ione
nCharakterisierung von Delivery Systemen mittels ThermogravimetrieDr. V. Normand, K. Aeberhardt, Firmenich S.A., Genf, Schweiz
19METTLER TOLEDO UserCom 1/2007
untersuchen. Für den Wechselwirkungs-parameter wird dabei der Zusammen-hang
erwartetet. Dabei bedeutet W die Mi-schungsenthalpie des Systems, k ist die Boltzmannkonstante und T die Tempe-ratur.
Verdunsten begrenzt durch DiffusionIn Gleichung 4 wird angenommen, dass die Abdampfrate der flüchtigen Kompo-nente einzig durch deren Partialdruck gegeben ist. In einem DS kann die Ab-dampfrate der flüchtigen Komponente auch durch die Diffusion der flüchtigen Moleküle an die Oberfläche des DS be-grenzt sein. In diesem Fall gilt gemäss dem Fick’schen Gesetz
Dabei steht D für den Diffusionskoeffizi-enten im DS, der hier davon abhängig ge-macht ist, ob das DS flüssig, gummiartig oder glasartig ist. A ist die Austauschflä-che, dc/dr der Konzentrationsgradient im DS und a, a′ und a″ sind Konstanten, die der Abhängigkeit der Diffusion vom Vo-lumen- bzw. Massenanteil der flüchtigen Komponente im DS Rechnung tragen. Besteht das DS wie im hier beschriebenen Fall aus Nanopartikeln, kann angenom-men werden, dass der Konzentrationsgra-dient innerhalb der Partikel nach einer kurzen Zeit konstant ist. In diesem Fall ist die Gewichtsverlustrate proportional zum Diffusionskoeffizienten.
Ergebnisse und DiskussionIm hier untersuchten DS wurde Romas-cone® in Nanopartikeln aus unterschied-lich vernetztem Vinylacetat gekapselt. Bei den Proben A und B wurde der Vernet-zungsgrad so eingestellt, dass das DS im gummielastischen Zustand vorliegt. Bei Probe C wurden sehr stark vernetzte Na-nopartikel verwendet. Dementsprechend lag das DS als Glas vor.
Verdunsten von reinem Romascone®
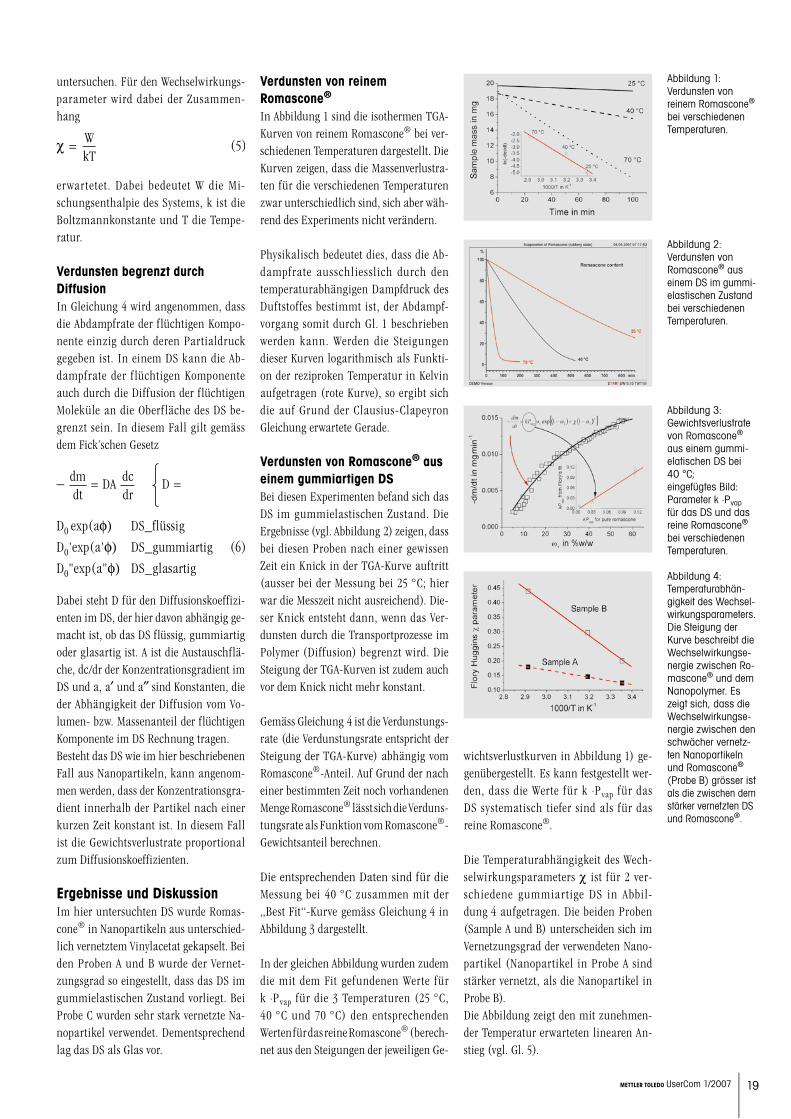
In Abbildung 1 sind die isothermen TGA-Kurven von reinem Romascone® bei ver-schiedenen Temperaturen dargestellt. Die Kurven zeigen, dass die Massenverlustra-ten für die verschiedenen Temperaturen zwar unterschiedlich sind, sich aber wäh-rend des Experiments nicht verändern.
Physikalisch bedeutet dies, dass die Ab-dampfrate ausschliesslich durch den temperaturabhängigen Dampfdruck des Duftstoffes bestimmt ist, der Abdampf-vorgang somit durch Gl. 1 beschrieben werden kann. Werden die Steigungen dieser Kurven logarithmisch als Funkti-on der reziproken Temperatur in Kelvin aufgetragen (rote Kurve), so ergibt sich die auf Grund der Clausius-Clapeyron Gleichung erwartete Gerade.
Verdunsten von Romascone® aus einem gummiartigen DSBei diesen Experimenten befand sich das DS im gummielastischen Zustand. Die Ergebnisse (vgl. Abbildung 2) zeigen, dass bei diesen Proben nach einer gewissen Zeit ein Knick in der TGA-Kurve auftritt (ausser bei der Messung bei 25 °C; hier war die Messzeit nicht ausreichend). Die-ser Knick entsteht dann, wenn das Ver-dunsten durch die Transportprozesse im Polymer (Diffusion) begrenzt wird. Die Steigung der TGA-Kurven ist zudem auch vor dem Knick nicht mehr konstant.
Gemäss Gleichung 4 ist die Verdunstungs-rate (die Verdunstungsrate entspricht der Steigung der TGA-Kurve) abhängig vom Romascone®-Anteil. Auf Grund der nach einer bestimmten Zeit noch vorhandenen Menge Romascone® lässt sich die Verduns-tungsrate als Funktion vom Romascone®-Gewichtsanteil berechnen.
Die entsprechenden Daten sind für die Messung bei 40 °C zusammen mit der „Best Fit“-Kurve gemäss Gleichung 4 in Abbildung 3 dargestellt.
In der gleichen Abbildung wurden zudem die mit dem Fit gefundenen Werte für k ·Pvap für die 3 Temperaturen (25 °C, 40 °C und 70 °C) den entsprechenden Werten für das reine Romascone® (berech-net aus den Steigungen der jeweiligen Ge-
wichtsverlustkurven in Abbildung 1) ge-genübergestellt. Es kann festgestellt wer-den, dass die Werte für k ·Pvap für das DS systematisch tiefer sind als für das reine Romascone®.
Die Temperaturabhängigkeit des Wech-selwirkungsparameters c ist für 2 ver-schiedene gummiartige DS in Abbil-dung 4 aufgetragen. Die beiden Proben (Sample A und B) unterscheiden sich im Vernetzungsgrad der verwendeten Nano-partikel (Nanopartikel in Probe A sind stärker vernetzt, als die Nanopartikel in Probe B). Die Abbildung zeigt den mit zunehmen-der Temperatur erwarteten linearen An-stieg (vgl. Gl. 5).
Abbildung 2: Verdunsten von Romascone® aus einem DS im gummi-elastischen Zustand bei verschiedenen Temperaturen.
Abbildung 1: Verdunsten von reinem Romascone® bei verschiedenen Temperaturen.
Abbildung 3: Gewichtsverlustrate von Romascone® aus einem gummi-elatischen DS bei 40 °C; eingefügtes Bild: Parameter k ·Pvap für das DS und das reine Romascone® bei verschiedenen Temperaturen.
Abbildung 4: Temperaturabhän-gigkeit des Wechsel-wirkungsparameters. Die Steigung der Kurve beschreibt die Wechselwirkungse-nergie zwischen Ro-mascone® und dem Nanopolymer. Es zeigt sich, dass die Wechselwirkungse-nergie zwischen den schwächer vernetz-ten Nanopartikeln und Romascone® (Probe B) grösser ist als die zwischen dem stärker vernetzten DS und Romascone®.
20 METTLER TOLEDO UserCom 1/2007
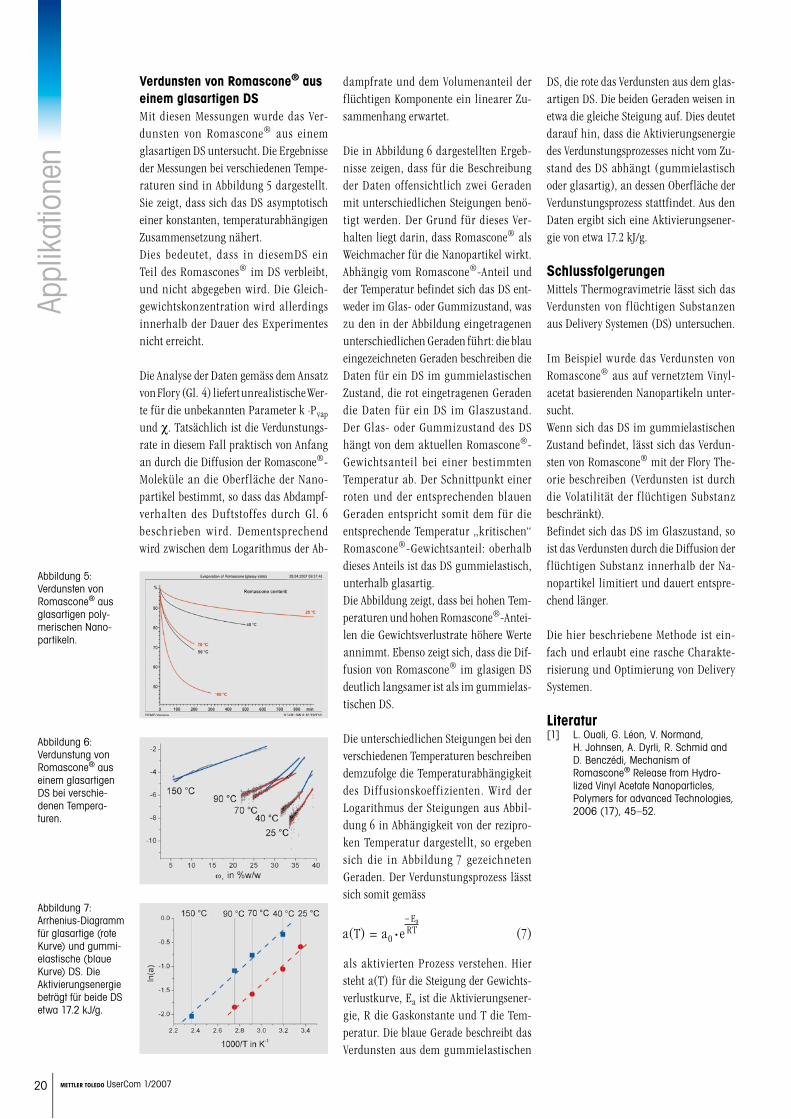
Verdunsten von Romascone® aus einem glasartigen DSMit diesen Messungen wurde das Ver-dunsten von Romascone® aus einem glasartigen DS untersucht. Die Ergebnisse der Messungen bei verschiedenen Tempe-raturen sind in Abbildung 5 dargestellt. Sie zeigt, dass sich das DS asymptotisch einer konstanten, temperaturabhängigen Zusammensetzung nähert. Dies bedeutet, dass in diesemDS ein Teil des Romascones® im DS verbleibt, und nicht abgegeben wird. Die Gleich-gewichtskonzentration wird allerdings innerhalb der Dauer des Experimentes nicht erreicht.
Die Analyse der Daten gemäss dem Ansatz von Flory (Gl. 4) liefert unrealistische Wer-te für die unbekannten Parameter k ·Pvap und c. Tatsächlich ist die Verdunstungs-rate in diesem Fall praktisch von Anfang an durch die Diffusion der Romascone®-Moleküle an die Oberfläche der Nano-partikel bestimmt, so dass das Abdampf-verhalten des Duftstoffes durch Gl. 6 beschrieben wird. Dementsprechend wird zwischen dem Logarithmus der Ab-
dampfrate und dem Volumenanteil der flüchtigen Komponente ein linearer Zu-sammenhang erwartet.
Die in Abbildung 6 dargestellten Ergeb-nisse zeigen, dass für die Beschreibung der Daten offensichtlich zwei Geraden mit unterschiedlichen Steigungen benö-tigt werden. Der Grund für dieses Ver-halten liegt darin, dass Romascone® als Weichmacher für die Nanopartikel wirkt. Abhängig vom Romascone®-Anteil und der Temperatur befindet sich das DS ent-weder im Glas- oder Gummizustand, was zu den in der Abbildung eingetragenen unterschiedlichen Geraden führt: die blau eingezeichneten Geraden beschreiben die Daten für ein DS im gummielastischen Zustand, die rot eingetragenen Geraden die Daten für ein DS im Glaszustand. Der Glas- oder Gummizustand des DS hängt von dem aktuellen Romascone®-Gewichtsanteil bei einer bestimmten Temperatur ab. Der Schnittpunkt einer roten und der entsprechenden blauen Geraden entspricht somit dem für die entsprechende Temperatur „kritischen“ Romascone®-Gewichtsanteil: oberhalb dieses Anteils ist das DS gummielastisch, unterhalb glasartig. Die Abbildung zeigt, dass bei hohen Tem-peraturen und hohen Romascone®-Antei-len die Gewichtsverlustrate höhere Werte annimmt. Ebenso zeigt sich, dass die Dif-fusion von Romascone® im glasigen DS deutlich langsamer ist als im gummielas-tischen DS.
Die unterschiedlichen Steigungen bei den verschiedenen Temperaturen beschreiben demzufolge die Temperaturabhängigkeit des Diffusionskoeffizienten. Wird der Logarithmus der Steigungen aus Abbil-dung 6 in Abhängigkeit von der rezipro-ken Temperatur dargestellt, so ergeben sich die in Abbildung 7 gezeichneten Geraden. Der Verdunstungsprozess lässt sich somit gemäss
als aktivierten Prozess verstehen. Hier steht a(T) für die Steigung der Gewichts-verlustkurve, Ea ist die Aktivierungsener-gie, R die Gaskonstante und T die Tem-peratur. Die blaue Gerade beschreibt das Verdunsten aus dem gummielastischen
DS, die rote das Verdunsten aus dem glas-artigen DS. Die beiden Geraden weisen in etwa die gleiche Steigung auf. Dies deutet darauf hin, dass die Aktivierungsenergie des Verdunstungsprozesses nicht vom Zu-stand des DS abhängt (gummielastisch oder glasartig), an dessen Oberfläche der Verdunstungsprozess stattfindet. Aus den Daten ergibt sich eine Aktivierungsener-gie von etwa 17.2 kJ/g.
SchlussfolgerungenMittels Thermogravimetrie lässt sich das Verdunsten von flüchtigen Substanzen aus Delivery Systemen (DS) untersuchen.
Im Beispiel wurde das Verdunsten von Romascone® aus auf vernetztem Vinyl-acetat basierenden Nanopartikeln unter-sucht. Wenn sich das DS im gummielastischen Zustand befindet, lässt sich das Verdun-sten von Romascone® mit der Flory The-orie beschreiben (Verdunsten ist durch die Volatilität der flüchtigen Substanz beschränkt). Befindet sich das DS im Glaszustand, so ist das Verdunsten durch die Diffusion der flüchtigen Substanz innerhalb der Na-nopartikel limitiert und dauert entspre-chend länger.
Die hier beschriebene Methode ist ein-fach und erlaubt eine rasche Charakte-risierung und Optimierung von Delivery Systemen.
Literatur[1] L. Ouali, G. Léon, V. Normand,
H. Johnsen, A. Dyrli, R. Schmid and D. Benczédi, Mechanism of Romascone® Release from Hydro-lized Vinyl Acetate Nanoparticles, Polymers for advanced Technologies, 2006 (17), 45–52.
Abbildung 5: Verdunsten von Romascone® aus glasartigen poly-merischen Nano-partikeln.
Appl
ikat
ione
n
Abbildung 6: Verdunstung von Romascone® aus einem glasartigen DS bei verschie-denen Tempera-turen.
Abbildung 7: Arrhenius-Diagramm für glasartige (rote Kurve) und gummi-elastische (blaue Kurve) DS. Die Aktivierungsenergie beträgt für beide DS etwa 17.2 kJ/g.
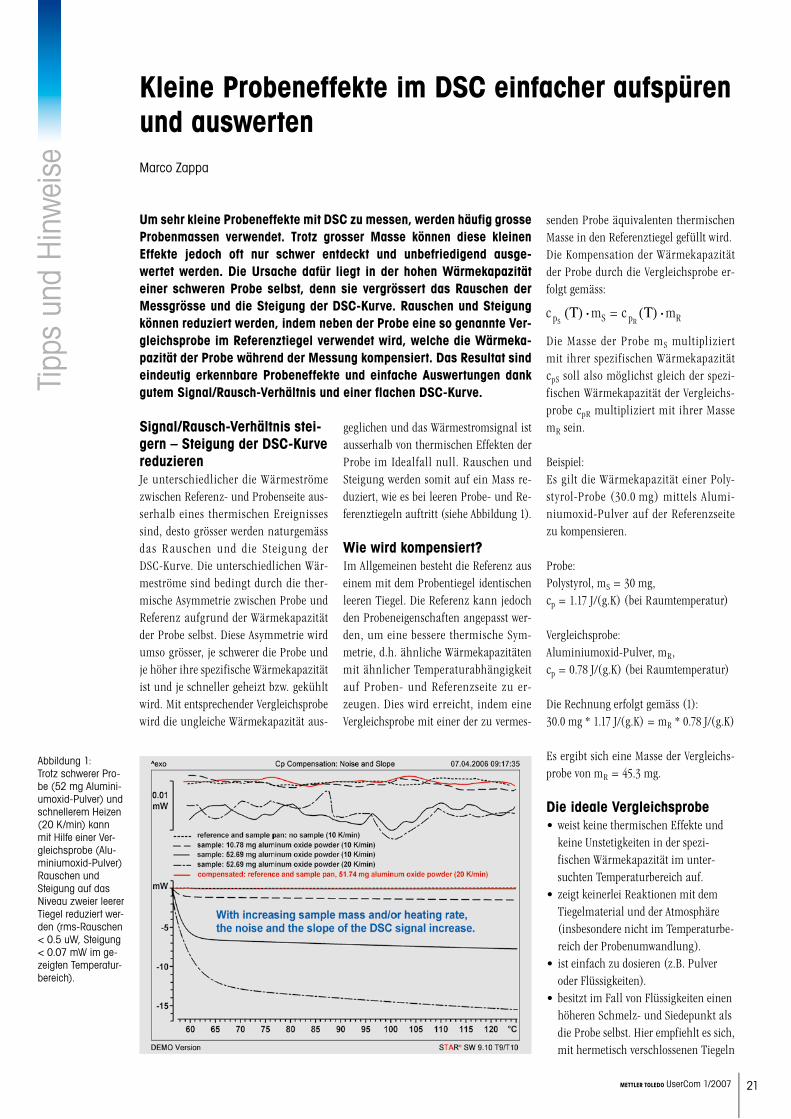
Signal/Rausch-Verhältnis stei-gern – Steigung der DSC-Kurve reduzierenJe unterschiedlicher die Wärmeströme zwischen Referenz- und Probenseite aus-serhalb eines thermischen Ereignisses sind, desto grösser werden naturgemäss das Rauschen und die Steigung der DSC-Kurve. Die unterschiedlichen Wär-meströme sind bedingt durch die ther-mische Asymmetrie zwischen Probe und Referenz aufgrund der Wärmekapazität der Probe selbst. Diese Asymmetrie wird umso grösser, je schwerer die Probe und je höher ihre spezifische Wärmekapazität ist und je schneller geheizt bzw. gekühlt wird. Mit entsprechender Vergleichsprobe wird die ungleiche Wärmekapazität aus-
geglichen und das Wärmestromsignal ist ausserhalb von thermischen Effekten der Probe im Idealfall null. Rauschen und Steigung werden somit auf ein Mass re-duziert, wie es bei leeren Probe- und Re-ferenztiegeln auftritt (siehe Abbildung 1).
Wie wird kompensiert?Im Allgemeinen besteht die Referenz aus einem mit dem Probentiegel identischen leeren Tiegel. Die Referenz kann jedoch den Probeneigenschaften angepasst wer-den, um eine bessere thermische Sym-metrie, d.h. ähnliche Wärmekapazitäten mit ähnlicher Temperaturabhängigkeit auf Proben- und Referenzseite zu er-zeugen. Dies wird erreicht, indem eine Vergleichsprobe mit einer der zu vermes-
senden Probe äquivalenten thermischen Masse in den Referenztiegel gefüllt wird.Die Kompensation der Wärmekapazität der Probe durch die Vergleichsprobe er-folgt gemäss:
Die Masse der Probe mS multipliziert mit ihrer spezifischen Wärmekapazität cpS soll also möglichst gleich der spezi-fischen Wärmekapazität der Vergleichs-probe cpR multipliziert mit ihrer Masse mR sein.
Beispiel:Es gilt die Wärmekapazität einer Poly-styrol-Probe (30.0 mg) mittels Alumi-niumoxid-Pulver auf der Referenzseite zu kompensieren.
Probe: Polystyrol, mS = 30 mg, cp = 1.17 J/(g.K) (bei Raumtemperatur)
Vergleichsprobe: Aluminiumoxid-Pulver, mR, cp = 0.78 J/(g.K) (bei Raumtemperatur)
Die Rechnung erfolgt gemäss (1):30.0 mg * 1.17 J/(g.K) = mR * 0.78 J/(g.K)
Es ergibt sich eine Masse der Vergleichs-probe von mR = 45.3 mg.
Die ideale Vergleichsprobeweist keine thermischen Effekte und keine Unstetigkeiten in der spezi-fischen Wärmekapazität im unter-suchten Temperaturbereich auf.zeigt keinerlei Reaktionen mit dem Tiegelmaterial und der Atmosphäre (insbesondere nicht im Temperaturbe-reich der Probenumwandlung).ist einfach zu dosieren (z.B. Pulver oder Flüssigkeiten).besitzt im Fall von Flüssigkeiten einen höheren Schmelz- und Siedepunkt als die Probe selbst. Hier empfiehlt es sich, mit hermetisch verschlossenen Tiegeln
•
•
•
•
Um sehr kleine Probeneffekte mit DSC zu messen, werden häufig grosse Probenmassen verwendet. Trotz grosser Masse können diese kleinen Effekte jedoch oft nur schwer entdeckt und unbefriedigend ausge-wertet werden. Die Ursache dafür liegt in der hohen Wärmekapazität einer schweren Probe selbst, denn sie vergrössert das Rauschen der Messgrösse und die Steigung der DSC-Kurve. Rauschen und Steigung können reduziert werden, indem neben der Probe eine so genannte Ver-gleichsprobe im Referenztiegel verwendet wird, welche die Wärmeka-pazität der Probe während der Messung kompensiert. Das Resultat sind eindeutig erkennbare Probeneffekte und einfache Auswertungen dank gutem Signal/Rausch-Verhältnis und einer flachen DSC-Kurve.
Kleine Probeneffekte im DSC einfacher aufspüren und auswertenMarco Zappa
Abbildung 1: Trotz schwerer Pro-be (52 mg Alumini-umoxid-Pulver) und schnellerem Heizen (20 K/min) kann mit Hilfe einer Ver-gleichsprobe (Alu-miniumoxid-Pulver) Rauschen und Steigung auf das Niveau zweier leerer Tiegel reduziert wer-den (rms-Rauschen < 0.5 uW, Steigung < 0.07 mW im ge-zeigten Temperatur-bereich).
METTLER TOLEDO UserCom 1/2007 21
Tipp
s un
d H
inw
eise
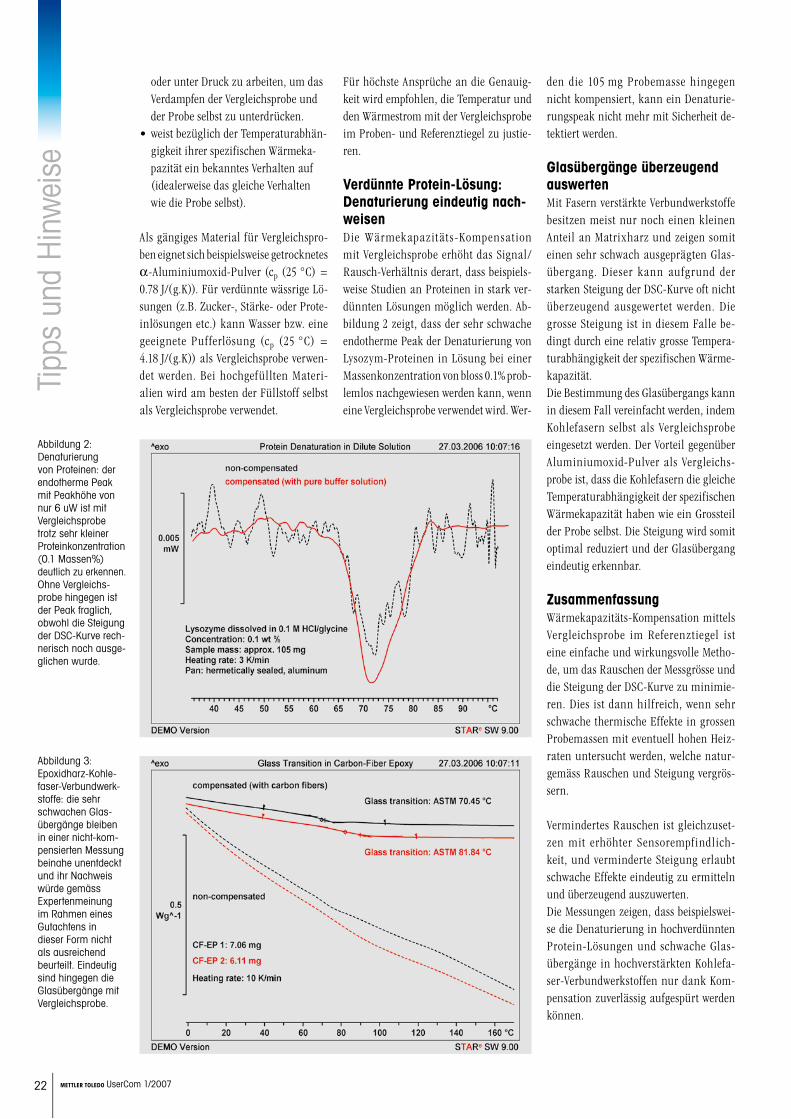
22 METTLER TOLEDO UserCom 1/2007
oder unter Druck zu arbeiten, um das Verdampfen der Vergleichsprobe und der Probe selbst zu unterdrücken.weist bezüglich der Temperaturabhän-gigkeit ihrer spezifischen Wärmeka-pazität ein bekanntes Verhalten auf (idealerweise das gleiche Verhalten wie die Probe selbst).
Als gängiges Material für Vergleichspro-ben eignet sich beispielsweise getrocknetes α-Aluminiumoxid-Pulver (cp (25 °C) = 0.78 J/(g.K)). Für verdünnte wässrige Lö-sungen (z.B. Zucker-, Stärke- oder Prote-inlösungen etc.) kann Wasser bzw. eine geeignete Pufferlösung (cp (25 °C) = 4.18 J/(g.K)) als Vergleichsprobe verwen-det werden. Bei hochgefüllten Materi-alien wird am besten der Füllstoff selbst als Vergleichsprobe verwendet.
•
Für höchste Ansprüche an die Genauig-keit wird empfohlen, die Temperatur und den Wärmestrom mit der Vergleichsprobe im Proben- und Referenztiegel zu justie-ren.
Verdünnte Protein-Lösung: Denaturierung eindeutig nach-weisenDie Wärmekapazitäts-Kompensation mit Vergleichsprobe erhöht das Signal/Rausch-Verhältnis derart, dass beispiels-weise Studien an Proteinen in stark ver-dünnten Lösungen möglich werden. Ab-bildung 2 zeigt, dass der sehr schwache endotherme Peak der Denaturierung von Lysozym-Proteinen in Lösung bei einer Massenkonzentration von bloss 0.1% prob-lemlos nachgewiesen werden kann, wenn eine Vergleichsprobe verwendet wird. Wer-
den die 105 mg Probemasse hingegen nicht kompensiert, kann ein Denaturie-rungspeak nicht mehr mit Sicherheit de-tektiert werden.
Glasübergänge überzeugend auswertenMit Fasern verstärkte Verbundwerkstoffe besitzen meist nur noch einen kleinen Anteil an Matrixharz und zeigen somit einen sehr schwach ausgeprägten Glas-übergang. Dieser kann aufgrund der starken Steigung der DSC-Kurve oft nicht überzeugend ausgewertet werden. Die grosse Steigung ist in diesem Falle be-dingt durch eine relativ grosse Tempera-turabhängigkeit der spezifischen Wärme-kapazität. Die Bestimmung des Glasübergangs kann in diesem Fall vereinfacht werden, indem Kohlefasern selbst als Vergleichsprobe eingesetzt werden. Der Vorteil gegenüber Aluminiumoxid-Pulver als Vergleichs-probe ist, dass die Kohlefasern die gleiche Temperaturabhängigkeit der spezifischen Wärmekapazität haben wie ein Grossteil der Probe selbst. Die Steigung wird somit optimal reduziert und der Glasübergang eindeutig erkennbar.
ZusammenfassungWärmekapazitäts-Kompensation mittels Vergleichsprobe im Referenztiegel ist eine einfache und wirkungsvolle Metho-de, um das Rauschen der Messgrösse und die Steigung der DSC-Kurve zu minimie-ren. Dies ist dann hilfreich, wenn sehr schwache thermische Effekte in grossen Probemassen mit eventuell hohen Heiz-raten untersucht werden, welche natur-gemäss Rauschen und Steigung vergrös-sern.
Vermindertes Rauschen ist gleichzuset-zen mit erhöhter Sensorempfindlich-keit, und verminderte Steigung erlaubt schwache Effekte eindeutig zu ermitteln und überzeugend auszuwerten.Die Messungen zeigen, dass beispielswei-se die Denaturierung in hochverdünnten Protein-Lösungen und schwache Glas-übergänge in hochverstärkten Kohlefa-ser-Verbundwerkstoffen nur dank Kom-pensation zuverlässig aufgespürt werden können.
Tipp
s un
d H
inw
eise
Abbildung 2: Denaturierung von Proteinen: der endotherme Peak mit Peakhöhe von nur 6 uW ist mit Vergleichsprobe trotz sehr kleiner Proteinkonzentration (0.1 Massen%) deutlich zu erkennen. Ohne Vergleichs-probe hingegen ist der Peak fraglich, obwohl die Steigung der DSC-Kurve rech-nerisch noch ausge-glichen wurde.
Abbildung 3: Epoxidharz-Kohle-faser-Verbundwerk-stoffe: die sehr schwachen Glas-übergänge bleiben in einer nicht-kom-pensierten Messung beinahe unentdeckt und ihr Nachweis würde gemäss Expertenmeinung im Rahmen eines Gutachtens in dieser Form nicht als ausreichend beurteilt. Eindeutig sind hingegen die Glasübergänge mit Vergleichsprobe.
METTLER TOLEDO UserCom 1/2006 23
Dat
enExhibitions, Conferences and Seminars – Veranstaltungen, Konferenzen und SeminareEuropean Polymer Congress 2007 July 2-6, 2007 Portoroz, Slovenia12th International Congress on the Chemistry of Cement July 8-13, 2007 Montreal, CanadaFederated Society for Coatings, Coating Woods and Wood Composites for Durability Symposium July 23-25, 2007 Seattle, WA, USANATAS 2007 August 26-29, 2007 State University, Lansing, MI, USAAIM XVIII Convegno Italiano di Scienza e Tecnologia delle Macromolecole September 16-20, 2007 Catania, ItalyBAKELAND Thermosets 2007 (www.baekeland2007.be) September 23-26, 2007 Ghent, BelgiumILMAC 2007 September 25-28, 2007 Basel, SwitzerlandMEDICTA 2007. The 8th Mediterranean Con- ference on Calorimetry and Thermal Analysis September 25-29, 2007 Palermo, ItalyPhandTA10 October 22-24, 2007 Monte Verità, Ascona, SwitzerlandK 2007 October 24-31, 2007 Düsseldorf, Germany
International and Swiss TA Customer Courses:
TA-Kundenkurse
und Seminarein Deutschland
TA Customer Courses and Seminars in Switzerland - Information and Course Registration:TA-Kundenkurse und Seminare in der Schweiz - Auskunft und Anmeldung:Ms Esther Andreato, Mettler-Toledo AG, Analytical, Schwerzenbach,Tel: ++41 44 806 73 57, Fax: ++41 44 806 72 60, e-mail: [email protected]
Courses / KurseSW Basic (Deutsch) 17. September, 2007 SW Basic (English) September 24, 2007TMA (Deutsch) 17. September, 2007 TMA (English) September 24, 2007DMA Basic (Deutsch) 17. September, 2007 DMA Basic (English) September 24, 2007DMA Advanced (Deutsch) 18. September, 2007 DMA Advanced (English) September 25, 2007TGA (Deutsch) 18. September, 2007 TGA (English) September 25, 2007TGA-MS (Deutsch) 19. September, 2007 TGA-MS (English) September 26, 2007DSC Basic (Deutsch) 19. September, 2007 DSC Basic (English) September 26, 2007DSC Advanced (Deutsch) 20. September, 2007 DSC Advanced (English) September 27, 2007TGA-FTIR (Deutsch) 20. September, 2007 TGA-FTIR (English) September 27, 2007SW Advanced (Deutsch) 21. September, 2007 SW Advanced (English) September 28, 2007
Cours et séminaires
d’Analyse Thermique en
France
Cursos y Seminarios
de TAen España
Local TA Customer Courses:
Für nähere Informationen wenden Sie sich bitte an: Frau Petra Fehl, Mettler-Toledo GmbH, Giessen, Tel: ++49 641 507 404, e-mail: [email protected]
Anwenderworkshop DSC 04./05.09.2007 Giessen
Renseignements et inscriptions par: Christine Fauvarque, Mettler-Toledo S.A., 18-20 Av. de la pépinière, 78222 Viroflay Cedex, Tél: ++33 1 3097 1439, Fax: ++33 1 3097 1660
Séminaires Analyses Thermique (gratuit)Paris (ESPCI) 6 juin 2007 Laboratoire Physique ThermiqueToulouse (Université Paul Sabatier) 18 septembre 2007 IMRCPLille 24 septembre 2007
Cours clientsPrincipe de la TMA/DMA 1 octobre 2007 Viroflay (France) Principe de la TGA 4 octobre 2007 Viroflay (France)Principe de la DSC : les bases 2 octobre 2007 Viroflay (France) Logiciel STARe : perfectionnem. 5 octobre 2007 Viroflay (France)Principe de la DSC : perfectionnem. 3 octobre 2007 Viroflay (France)
Para detalles acerca de los cursos y seminarios, por favor, contacte con: Francesc Catala, Mettler-Toledo S.A.E., Tel: ++34 93 223 76 00, e-mail: [email protected]
Aplicaciones del Análisis Térmico Octubre, 3 de 2007 Barcelona Octubre, 9 de 2007 MadridUso del sistema STARe Octubre, 4 de 2007 Barcelona Octubre, 10 de 2007 Madrid

Editorial team
Dr. J. Schawe Dr. R. Riesen J. Widmann Dr. M. Schubnell Dr. M. Wagner Dr. D. P. May Ni Jing Marco Zappa Urs JörimannPhysicist Chem. Engineer Chem. Engineer Physicist Chemist Chemist Chemist Material Scientist Electr. Engineer
www.mt.com/ta
Mettler-Toledo AG, AnalyticalPostfach, CH-8603 SchwerzenbachTel. ++41 44 806 73 87Fax ++41 44 806 72 60 Kontakt: [email protected]
© 05/2007 Mettler-Toledo AGME-51724545, Gedruckt in der SchweizMarCom Analytical
Für mehr Informationen
Per ulteriori informazioni Vi preghiamo di contattare: Simona Ferrari, Mettler-Toledo S.p.A., Novate Milanese, Tel: ++39 02 333 321, Fax: ++39 02 356 2973, e-mail: [email protected]
DSC base 12 Giugno 2007 25 Settembre 2007 Novate MilaneseDSC avanzato 13 Giugno 2007 26 Settembre 2007 Novate MilaneseTGA 14 Giugno 2007 27 Settembre 2007 Novate MilaneseTMA 15 Giugno 2007 28 Settembre 2007 Novate Milanese
For details of training courses and seminars, please contact: Rod Bottom, Mettler-Toledo Ltd, Leicester, Tel: ++44 116 234 5025, Fax: ++44 116 236 5500, e-mail: [email protected]
DSC BASIC September 25, 2007 Leicester
For details of training courses and seminars, please contact: Bryan Yiew, Mettler-Toledo (S) Pte Ltd, 28 Ayer Rajah Crescent, #05 - 01, Singapore 139959Tel: +65-68900011, Fax: +65-68900012, e-mail: [email protected]
DSC BASIC August 2007 November 2007 Singapore
For details of training courses and seminars, please contact: KiHun Lee at Mettler-Toledo Korea, Tel: ++82 2 3498 3500, e-mail: [email protected], Homepage: www.kr.mt.com TA Public Seminar in 2007 October 2007 Seoul October 2007 Ulsan
For details of seminars, please contact: Tomoko Wakabayashi, Mettler-Toledo KK, 3-8 Sanbancho, Chiyoda-ku, Tokyo 102-0075, JapanTel: +81-3-3222-7164, Fax: +82-3-3222-7124, e-mail: [email protected] TA Seminar July 2007 Sendai, Tsukuba, Sapporo, Shizuoka, Tokushima, Ube, FukuokaMT Forum July 2007 TokyoJAIMA Show August 2007 MakuhariTA Annual Meeting October 2007 Sapporo
For further information regarding meetings, products or applications, please contact your local METTLER TOLEDO representative and visit our home page at http://www.mt.com
TA Customer Courses
and Seminarsin the UK
TA Seminars and Trade
Shows in Japan
TA Customer Courses
and Seminars in Singapore
TA School and Seminars
in Korea
Corsi e Seminari di Analisi
Termica in Italia


















