
Untersuchung der zeit- und druckabhängigen Expression ... Dr... · Aus dem Veterinär-Anatomischen...
120
Aus dem Veterinär-Anatomischen Institut der Veterinärmedizinischen Fakultät der Universität Leipzig Untersuchung der zeit- und druckabhängigen Expression verschiedener Komponenten der extrazellulären Matrix durch Chondrozyten in vitro Inaugural-Dissertation zur Erlangung des Grades eines Doctor medicinae veterinariae (Dr. med. vet.) durch die Veterinärmedizinische Fakultät der Universität Leipzig eingereicht von Juliane Schneevoigt aus Wernigerode Leipzig, 2015
Transcript of Untersuchung der zeit- und druckabhängigen Expression ... Dr... · Aus dem Veterinär-Anatomischen...

Aus dem Veterinär-Anatomischen Institut
der Veterinärmedizinischen Fakultät der Universität Leipzig
Untersuchung der zeit- und druckabhängigen Expression verschiedener Komponenten
der extrazellulären Matrix durch Chondrozyten in vitro
Inaugural-Dissertation
zur Erlangung des Grades eines
Doctor medicinae veterinariae (Dr. med. vet.)
durch die Veterinärmedizinische Fakultät
der Universität Leipzig
eingereicht von
Juliane Schneevoigt
aus Wernigerode
Leipzig, 2015

Mit Genehmigung der Veterinärmedizinischen Fakultät der Universität Leipzig
Dekan: Prof. Dr. Manfred Coenen
Betreuer: PD Dr. Mahtab Bahramsoltani
Gutachter: PD Dr. Mahtab Bahramsoltani
Veterinär-Anatomisches Institut, Veterinärmedizinische Fakultät der Universität Leipzig
Prof. Dr. Johanna Plendl
Institut für Veterinär-Anatomie, Fachbereich Veterinärmedizin der Freien Universität Berlin
Tag der Verteidigung: 22.09.2015

Für meine Familie


Inhaltsverzeichnis
I
Inhaltsverzeichnis
Liste der Abkürzungen ................................................................................................................................. VI
1 Einleitung ...............................................................................................................................................1
2 Literaturübersicht ..................................................................................................................................2
2.1 Entwicklung des Knorpelgewebes..................................................................................................2
2.1.1 Kondensation mesenchymaler Stammzellen .........................................................................2
2.1.2 Proliferation und Differenzierung von Chondroprogenitorzellen ..........................................3
2.1.3 Terminale Differenzierung zu Chondrozyten .........................................................................3
2.2 Aufbau und Zusammensetzung des Knorpelgewebes ...................................................................4
2.2.1 Die Komponenten des Knorpelgewebes ................................................................................4
2.2.1.1 Die Zellen des Knorpelgewebes .........................................................................................4
2.2.1.2 Die extrazelluläre Matrix des Knorpelgewebes .................................................................5
2.2.1.2.1 Die amorphe Grundsubstanz des Knorpelgewebes ....................................................5
2.2.1.2.2 Die Fasern des Knorpelgewebes..................................................................................6
2.2.1.2.2.1 Die kollagenen Fasern des Knorpelgewebes ........................................................6
2.2.1.2.2.2 Die elastischen Fasern des Knorpelgewebes ........................................................8
2.2.2 Der hyaline Gelenkknorpel .....................................................................................................8
2.2.3 Der Faserknorpel ................................................................................................................. 11
2.2.4 Der elastische Knorpel ......................................................................................................... 12
2.3 Dynamik des hyalinen Gelenkknorpels ....................................................................................... 12
2.3.1 Dynamik des hyalinen Gelenkknorpels unter mechanischen Einflüssen ............................ 12
2.3.2 Dynamik des hyalinen Gelenkknorpels unter chemischen Einflüssen ................................ 14
2.3.3 Seneszenz als Einflussfaktor auf die Dynamik des hyalinen Gelenkknorpels ..................... 14
2.4 Defekte und Regeneration des Knorpelgewebes ....................................................................... 15
2.4.1 Defekte und Regeneration des hyalinen Gelenkknorpels ................................................... 15
2.4.2 Defekte und Regeneration des Faserknorpels .................................................................... 18
2.4.3 Defekte und Regeneration des elastischen Knorpels .......................................................... 18
2.5 Strategien zur Behandlung von Knorpeldefekten ....................................................................... 18
2.5.1 Knochenmarkstimulierende Verfahren ............................................................................... 19
2.5.2 Autologe Knorpelknochentransplantation .......................................................................... 19
2.5.3 Autologe Knorpeltransplantation ........................................................................................ 20
2.6 In-vitro-Kultivierung von Chondrozyten ..................................................................................... 21
2.6.1 Einfluss der In-vitro-Kultivierung auf Chondrozyten ........................................................... 21

Inhaltsverzeichnis
II
2.6.2 Einsatz von Druck bei der In-vitro-Kultivierung von Chondrozyten .................................... 22
3 Zellen, Materialien, Methoden ........................................................................................................... 24
3.1 Zellen und für deren Kultivierung verwendete Lösungen .......................................................... 24
3.1.1 Chemikalien und Lösungen für die Zellkultur ...................................................................... 24
3.1.2 Zusammensetzung des Kulturmediums .............................................................................. 24
3.1.3 Zusammensetzung des Einfriermediums ............................................................................ 25
3.1.4 Zusammensetzung des Spezialgasgemisches für die Kultivierung der Chondrozyten
unter Druck .......................................................................................................................... 25
3.2 Materialien .................................................................................................................................. 25
3.2.1 Chemikalien und Reagenzien .............................................................................................. 25
3.2.2 In der Proteinanalytik verwendete Antikörper, Kontrollseren und Marker ....................... 26
3.2.2.1 Primärantikörper ............................................................................................................ 26
3.2.2.2 Sekundärantikörper ........................................................................................................ 26
3.2.2.3 Kontrollserum ................................................................................................................. 26
3.2.2.4 Marker für SDS-PAGE und Western Blot ........................................................................ 26
3.2.3 In der PCR eingesetzte Nukleotide und Enzyme, sowie für die Klonierung und Plasmid-
Präparation verwendete Materialien .................................................................................. 27
3.2.3.1 Nukleotide ...................................................................................................................... 27
3.2.3.2 Enzyme............................................................................................................................ 27
3.2.3.3 Materialien für die Klonierung und Plasmid-Präparation .............................................. 28
3.2.4 Zusammensetzung von Gebrauchslösungen für die Immunzytochemie ............................ 28
3.2.5 Zusammensetzung von Gebrauchslösungen für die PCR .................................................... 29
3.2.6 Zusammensetzung von Gebrauchslösungen für die Proteinanalytik .................................. 29
3.2.7 Verbrauchsmaterialien ........................................................................................................ 31
3.2.8 Geräte .................................................................................................................................. 32
3.2.8.1 Laborgeräte .................................................................................................................... 32
3.2.8.2 Bioreaktor ....................................................................................................................... 33
3.3 Methoden ................................................................................................................................... 34
3.3.1 Zellkultur .............................................................................................................................. 34
3.3.1.1 Auftauen der Chondrozyten ........................................................................................... 34
3.3.1.2 Kultivierung der Chondrozyten unter Normaldruck ....................................................... 34
3.3.1.3 Kultivierung der Chondrozyten unter erhöhtem Druck ................................................. 34
3.3.1.4 Subkultivierung der Chondrozyten ................................................................................. 35

Inhaltsverzeichnis
III
3.3.1.5 Kryokonservierung der Chondrozyten ............................................................................ 35
3.3.2 Immunzytochemische Untersuchungen ............................................................................. 35
3.3.3 RNA- und Proteinisolierung ................................................................................................. 36
3.3.3.1 Probengewinnung .......................................................................................................... 36
3.3.3.2 Isolierung der RNA .......................................................................................................... 36
3.3.3.2.1 Qualitätskontrolle der RNA mittels Spektralphotometrie........................................ 37
3.3.3.2.2 Qualitätskontrolle der RNA mittels Gelelektrophorese ........................................... 37
3.3.3.3 Isolierung der Proteine ................................................................................................... 38
3.3.4 Transkriptanalyse ................................................................................................................ 39
3.3.4.1 DNase-Verdau ................................................................................................................. 39
3.3.4.2 Reverse Transkription (RT) ............................................................................................. 39
3.3.4.3 Qualitative PCR ............................................................................................................... 40
3.3.4.4 Quantitative PCR............................................................................................................. 41
3.3.4.4.1 Herstellung der Standards ........................................................................................ 41
3.3.4.4.1.1 Herstellung der Medien und Nährböden .......................................................... 41
3.3.4.4.1.2 Hinzufügen eines Adenosin(A)-Überhangs ....................................................... 41
3.3.4.4.1.3 cDNA-Aufreinigung ............................................................................................ 42
3.3.4.4.1.4 Klonierung ......................................................................................................... 42
3.3.4.4.1.5 Plasmid-Präparation .......................................................................................... 43
3.3.4.5 Quantitative Real Time-PCR ........................................................................................... 44
3.3.5 Proteinanalyse ..................................................................................................................... 45
3.3.5.1 SDS-PAGE ........................................................................................................................ 45
3.3.5.2 Western Blot ................................................................................................................... 46
3.3.5.3 Detektion ........................................................................................................................ 48
3.3.5.4 Stripping der Antikörper und erneute Inkubation .......................................................... 48
3.3.5.5 Semiquantitative Analyse des Western Blot .................................................................. 49
3.3.6 Graphische Darstellung und statistische Methoden ........................................................... 49
4 Ergebnisse ........................................................................................................................................... 50
4.1 In-vitro-Kultivierung der Chondrozyten unter Normaldruck ...................................................... 50
4.1.1 Morphologie in-vitro-kultivierter Chondrozyten unter Normaldruck ................................. 50
4.1.2 Immunzytochemischer Nachweis der Expression von Kollagen Typ I und II in-vitro-
kultivierter Chondrozyten unter Normaldruck ................................................................... 51

Inhaltsverzeichnis
IV
4.1.3 Nachweis der Aggrekan-, Kollagen Typ I-, und Kollagen Typ II-mRNA-Expression in-
vitro-kultivierter Chondrozyten unter Normaldruck ........................................................... 53
4.1.4 Nachweis der Aggrekan-, Kollagen Typ I- und Kollagen Typ II-Proteinexpression in-vitro-
kultivierter Chondrozyten unter Normaldruck ................................................................... 57
4.2 In-vitro-Kultivierung von Chondrozyten unter erhöhtem Druck ................................................ 59
4.2.1 Einfluss der Frequenz des Mediumwechsels auf die Vitalität in-vitro-kultivierter
Chondrozyten ...................................................................................................................... 60
4.2.2 Versuchsaufbau für die Untersuchung in-vitro-kultivierter Chondrozyten unter
erhöhtem Druck .................................................................................................................. 61
4.2.3 Morphologie in-vitro-kultivierter Chondrozyten unter erhöhtem Druck ........................... 63
4.2.4 Nachweis der Aggrekan-, Kollagen Typ I- und Kollagen Typ II-mRNA-Expression in-vitro-
kultivierter Chondrozyten unter erhöhtem Druck .............................................................. 65
4.2.4.1 Effekte der Höhe des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten ........................... 66
4.2.4.2 Effekte der Dauer des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten ........................... 68
4.2.4.3 Effekte der Zelldichte auf die Aggrekan-, Kollagen Typ I- und Kollagen Typ II-mRNA-
Expression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck .......................... 71
4.2.5 Nachweis der Aggrekan-, Kollagen Typ I- und Kollagen Typ II-Proteinexpression in-vitro-
kultivierter Chondrozyten unter erhöhtem Druck .............................................................. 73
4.2.5.1 Effekte der Höhe des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-Proteinexpression in-vitro-kultivierter Chondrozyten .......................... 73
4.2.5.2 Effekte der Dauer des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-Proteinexpression in-vitro-kultivierter Chondrozyten .......................... 75
5 Diskussion ........................................................................................................................................... 78
5.1 Diskussion der Untersuchungsmethoden ................................................................................... 78
5.1.1 Varianz der Ergebnisse der PCR........................................................................................... 78
5.1.2 Einsatz spezifischer Antikörper für den Nachweis großer Strukturproteine ...................... 79
5.1.2.1 Einsatz spezifischer Antikörper für den Nachweis großer Strukturproteine in der
Immunzytochemie.......................................................................................................... 79
5.1.2.2 Einsatz spezifischer Antikörper für den Nachweis großer Strukturproteine im
Western Blot .................................................................................................................. 80
5.1.3 Einsatz statistischer Methoden in einer Pilotstudie mit geringem Stichprobenumfang .... 82
5.2 Diskussion der Ergebnisse ........................................................................................................... 83
5.2.1 Einfluss der Kultivierungszeit auf in-vitro-kultivierte Chondrozyten .................................. 83

Inhaltsverzeichnis
V
5.2.1.1 Einfluss der Kultivierungszeit auf die Morphologie in-vitro-kultivierter Chondrozyten 83
5.2.1.2 Einfluss der Kultivierungszeit auf die Aggrekan-, Kollagen Typ I- und Kollagen Typ II-
Expression in-vitro-kultivierter Chondrozyten ............................................................... 83
5.2.2 Einfluss nutritiver Faktoren auf die Vitalität in-vitro-kultivierter Chondrozyten ................ 86
5.2.3 Einfluss hydrostatischen Drucks auf in-vitro-kultivierte Chondrozyten .............................. 87
5.2.3.1 Einfluss hydrostatischen Drucks auf die Morphologie in-vitro-kultivierter
Chondrozyten ................................................................................................................. 87
5.2.3.2 Einfluss hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und Kollagen Typ
II-Expression in-vitro-kultivierter Chondrozyten ............................................................ 88
5.2.4 Kontinuierliche versus diskontinuierliche Druckbedingungen ............................................ 90
5.2.5 Dedifferenzierung in-vitro-kultivierter Chondrozyten - Anwendbarkeit und Limitation
genutzter Strategien ............................................................................................................ 91
5.3 Empfehlungen für die In-vitro-Kultivierung von Chondrozyten unter erhöhtem
hydrostatischen Druck ................................................................................................................ 92
5.4 Möglichkeiten der Analyse druckbehandelter Chondrozyten und deren Nutzung in der
klinischen Anwendung- ein Ausblick ........................................................................................... 94
6 Zusammenfassung .............................................................................................................................. 95
7 Summary ............................................................................................................................................. 97
8 Literaturverzeichnis ............................................................................................................................ 99
9 Danksagung ...................................................................................................................................... 108

Liste der Abkürzungen
VI
Liste der Abkürzungen
A Adenosinrest
ACAN Aggrekan
ACT autologe Knorpeltransplantation
ANOVA One Way Analysis of Variance
APS Ammoniumpersulfat
bar Einheit des Drucks
BMP Bone Morphogenetic Protein
BMP-2 Bone Morphogenetic Protein-2
BMP-7 Bone Morphogenetic Protein-7
bp Basenpaare
BSA Bovines Serum Albumin
bzw. beziehungsweise
ca. circa
CBM Chondrozyten Basal Medium
CBM+ Chondrozytenwachstumsmedium
CD44 Cluster of Differenciation 44
cDNA complementäre DNA
CO2 Kohlenstoffdioxid
COL1A1 Kollagen Typ I, α1-Kette
COL1A2 Kollagen Typ I, α2-Kette
COL2A1 Kollagen Typ II, α1-Kette
CT Anzahl der PCR-Cyclen, die zu dem Zeitpunkt erreicht sind, an dem sich das
Fluoreszenzsignal gerade deutlich vom Hintergrund abhebt
d Tage
DAB Diaminobenzidin
dATP Desoxyadenosintriphosphat
dCTP Desoxycytidintriphosphat
DEPC Diethylpyrocarbonat
dGTP Desoxyguanosintriphosphat
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
dNTPs Nukleotide (dATP, dTTP, dCTP und dGTP)
DPBS Dulbecco's Phosphatgepufferte Kochsalzlösung 1x
DTT Dithiothreitol
dTTP Desoxythymidintriphosphat
ECM Extrazelluläre Matrix
E.coli Escherichia coli, Bakterium
EDTA Ethylendiamintetraacetat
FACIT Fibril-Associated Collagens with Interrupted Triple helices

Liste der Abkürzungen
VII
FBS Fetales Bovines Serum
FGF-2 Fibroblast-Growth-Factor-2
GAG Glykosaminglykan
ggf. Gegebenenfalls
h Stunden
HBSS Hank's Salzlösung
HBV Hepatitis-B-Virus
HCl Salzsäure
HCV Hepatitis-C-Virus
HIF-α Hypoxia-inducible Factor-α
HIV Humanes Immundefizienz-Virus
HRP Meerrettichperoxidase
Hz Hertz, Einheit der Frequenz
IGF-1 Insulin-like Growth Factor-1
IL-1 Interleukin-1
IL-6 Interleukin-6
IL-7 Interleukin-7
IL-8 Interleukin-8
Inkl. Inklusive
kDA Kilo-Dalton
KNS Kaninchen Normalserum
LB Luria Broth
LI Lumineszenz-Intensität
M arithmetisches Mittel MACT Matrix assozierte autologe Knorpeltransplantation
MMP Matrixmetalloproteinasen
MMP-13 Matrixmetalloproteinase 13
mRNA messenger RNA
MSZ Most Superficial Zone
MRT Magnetresonanztomograph
MSC Mesenchymale Stammzelle
n Stichprobenumfang
OA Osteoarthritis
OCT autologe Knorpelknochentransplantation
Oligo(dT)18 Oligonukleotid aus 18 dTTP
P statistische Power (Teststärke)
PBS Phosphatgepufferte Kochsalzlösung
PCR Polymerasekettenreaktion
pDNA Plasmid DNA
Pextern externer auf den Knorpel einwirkender Druck
PHydr hydrostatischer Druck
PKollagen Spannungsdruck der kollagenen Fasern

Liste der Abkürzungen
VIII
POsm osmotischer Druck
PSchwell Schwelldruck
PG Proteoglykan
PTHrP Parathormon-related-Peptide
PTHrP-R Parathormon-related-Peptide-Rezeptor
PVDF Polyvinylidenfluorid
qPCR quantitative PCR
RLI relative Lumineszenz-Intensität
RNA Ribonukleinsäure
RT Reverse Transkription
RT-qPCR Reverse Transkription-quantitative PCR
s Sekunden
SASP Senescense-Associated Secretory Phenotype
SDS Natriumdodecylsulfat
SDS-PAGE Natriumdodecylsulfat-Polyacrylamid Gelelektrophorese
sog. sogenannte
TAE-Puffer Tris, Essigsäure, EDTA-Puffer
TBS Tris-gepufferte Kochsalzlösung
TE-Puffer Tris-EDTA (TE)-Puffer
TEMED N,N,N′,N′-Tetramethylethylendiamin
TGF-β1 Transforming Growth Factor-β1
TNF-α Tumor Necrosis Faktor-α
TNS Trypsin Neutralisationslösung
Tris-Base Tris(hydroxymethyl)-aminomethan
Tris-HCl Tris(hydroxymethyl)aminomethanhydrochlorid
UV Ultraviolett

1 Einleitung
1
1 Einleitung
Der hyaline Knorpel bildet als schmaler Überzug der Gelenkflächen eine wichtige Grundlage für die
Bewegung des Organismus. Die Wahrnehmung seiner Funktion - der primären Druckübertragung auf den
subchondralen Knochen (Stoßdämpfungsfunktion) - erfolgt im Wesentlichen durch seine spezielle
extrazelluläre Matrix, die aus einem zonal organisierten Maschenwerk kollagener Fasern, in welches ein
hoher Anteil von Proteoglykanen mit assoziiertem Wasser eingelagert ist, besteht (BUCKWALTER
et al. 2005, MANSFIELD et al. 2008, SALOMON et al. 2005, POOLE 1997, SCHULZ und BADER 2007). Die
Integrität dieser extrazellulären Matrix erfordert in vivo eine stetige dynamische Anpassung entsprechend
der einwirkenden Druckbelastungen, welche durch die Chondrozyten, die Zellen des hyalinen Knorpels,
organisiert wird (ALFORD 2005, IKENOUE et al. 2003, LUPPA 2000, SCHULZ und BADER 2007).
Da es sich bei den Chondrozyten um postmitotische Zellen handelt und dem hyalinen Knorpel eine
Vaskularisation fehlt, unterbleibt bei einem Verlust der Integrität der extrazellulären Matrix in Folge eines
Traumas eine Defektregeneration. Im weiteren Verlauf kommt es zu einer biomechanischen Überbelastung
des umliegenden Knorpels, zum fortschreitenden Funktionsverlust des Gewebes und als Langzeitfolge zur
Osteoarthrose. Da diese Defekte des hyalinen Knorpels einen Großteil der Erkrankungen des
Bewegungsapparats darstellen und trotz intensiver Bemühungen die heute vorhandenen operativen
Möglichkeiten der Therapie von Knorpeldefekten noch keine befriedigenden funktionellen
Langzeitergebnisse bieten, besteht ein hohes medizinisches Interesse an der Entwicklung und
Weiterentwicklung innovativer Therapieansätze. Einen solchen Therapieansatz stellt dabei die Autologe
Chondrozyten Transplantation (ACT) dar, bei der in-vitro-kultivierte Chondrozyten in einen Knorpeldefekt
eingebracht werden mit dem Ziel, neuen hyalinen Knorpel zu bilden (BUCKWALTER und MANKIN 1998,
FRITSCH et al. 1999, MARCACCI et al., 2013, WONG und CARTER 2003). In den letzten Jahrzehnten zeigte
sich jedoch, dass die Entwicklung solcher Therapieansätze ein detaillierteres Verständnis des hyalinen
Knorpelgewebes und speziell dessen Biodynamik erfordert (BRUNS und STEINHAGEN 2000).
Ziel dieser Arbeit war es, im Rahmen einer Pilotstudie ein Zellkultursystem zu entwickeln, welches die
Untersuchung der Biodynamik von Chondrozyten in vitro ermöglicht. Im Fokus stand dabei der Einfluss der
Kultivierungszeit und des hydrostatischen Drucks auf die Morphologie der Chondrozyten sowie deren
Expression von Aggrekan, Kollagen Typ I und II als knorpelspezifische Komponenten der extrazellulären
Matrix. Die durch diese Untersuchungen gewonnenen Erkenntnisse sollen der Weiterentwicklung
zellbasierter Therapieansätze von Knorpeldefekten, wie der ACT, dienen bzw. dazu genutzt werden, um die
bei der ACT transplantierten Chondrozyten in Richtung der Expression einer chondrozytenspezifischen
extrazellulären Matrix zu modifizieren, welche das Potenzial besitzt, den Druckbedingungen des Gelenks
standzuhalten (MARTINEK und IMHOFF 2003).

2 Literaturübersicht
2
2 Literaturübersicht
2.1 Entwicklung des Knorpelgewebes
Während der Entwicklung des Bewegungsapparates entstehen die Chondrozyten aus mesenchymalen
Progenitorzellen. Die Chondrozyten sind einerseits die für die Synthese des Knorpels zuständigen Zellen.
Andererseits liefern sie die für die spätere Knochenentwicklung essentiellen Knorpelanlagen. Embryonal
kommt dem Knorpelgewebe eine Platzhalterfunktion in der enchondralen Ossifikation des Knochens zu,
sodass dessen erste Entwicklungsabschnitte mit der Entwicklung des Knorpelgewebes identisch sind. Die
Entwicklung des Knorpelgewebes wird schematisch in die Phasen der Kondensation mesenchymaler
Stammzellen, Proliferation von Chondroprogenitorzellen und deren terminale Differenzierung zu
Chondrozyten unterteilt. An diesem Prozess sind unterschiedliche Wachstumsfaktoren beteiligt (Abb. 1)
(GOLDRING et al. 2006).
Abb. 1: Schematische Darstellung der Entwicklung des Knorpelgewebes. Die wichtigsten Wachstums- und Differenzierungsfaktoren sind oberhalb, die Bestandteile der extrazellulären Matrix unterhalb der Pfeile dargestellt; adaptiert nach SINGH und SCHWARZBAUER (2012), GOLDRING et al. (2006).
2.1.1 Kondensation mesenchymaler Stammzellen
Die mesenchymalen Stammzellen (MSCs) initiieren den Entwicklungsprozess des Knorpelgewebes. Im
Bereich des Kopfes entstammen diese der Neuralleiste, wohingegen für die Bildung des Achsenskeletts die
MSCs aus den Sklerotomen des paraxialen Mesoderms zuständig sind. Das Knorpelgewebe der Gliedmaßen
geht aus den MSCs der Somatopleura des lateralen Mesoderms hervor (GOLDRING et al. 2006). Die MSCs
synthetisieren zunächst Hyaluronsäure, Fibronektin und nicht knorpelspezifische Kollagene (Kollagen
Typ I, III, V). In den Bereichen der Knorpel- und Knochenentwicklung kommt es zu einem Anstieg der
Hyaluronidaseaktivität und somit einer Verringerung der Hyaluronsäure-Konzentration in der
extrazellulären Matrix (ECM). In den sich auf diese Weise einander annähernden Zellen steigt die
Expression von Transforming Growth Factor-β1 (TGF-β1), welches die Bildung von Zelladhäsionsmolekülen
induziert, wodurch die Anzahl der Zell-Zellkontakte zunimmt (DELISE et al. 2000, FORTIER et al. 2011). Als
weiterer Effekt des TGF-β1 auf die MSC sinkt deren Synthese und Sekretion von Kollagen Typ I in den
Zellen. Die beschriebenen Veränderungen führen zu einer erhöhten Zelldichte pro Volumeneinheit ohne
Proliferation der Zellen. Dieser als Kondensation bezeichnete Vorgang bedingt die Ausbildung der
skelettalen Blasteme (COLE 2011, DELISE et al. 2000, GOLDRING et al. 2006, GOLDRING 2012).

2 Literaturübersicht
3
2.1.2 Proliferation und Differenzierung von Chondroprogenitorzellen
Aus dem Kondensationsprozess gehen Osteochondroprogenitorzellen hervor. Die zentralen Zellen der
skelettalen Blasteme bilden eine chondrogene Zelllinie, deren Zellen als Chondroprogenitorzellen
bezeichnet werden. Die peripher liegenden Zellen differenzieren zu Osteoprogenitorzellen (SINGH und
SCHWARZBAUER 2012).
Die Chondroprogenitorzellen sind gekennzeichnet durch eine verminderte Expression von TGF-β1 und der
autokrinen Stimulation durch Insulin-like Growth Factor-1 (IGF-1), Bone Morphogenic Proteins (BMPs),
insbesondere BMP-2, und Fibroblast Growth Factor-2 (FGF-2), welche den proliferativen Status dieser
Zellen erhalten und für eine Veränderung der Synthese der Komponenten der ECM verantwortlich sind. So
führen IGF-1 und BMP-2 zur vermehrten Bildung und Abgabe der immaturen Splicing-Variante des Kollagen
Typ II (Kollagen Typ IIA) sowie Kollagen Typ IX und XI, während durch FGF-2 die Produktion von Aggrekan
stimuliert wird (GOLDRING et al. 2006, GOLDRING 2012). Das so entstehende Gewebe wird als Vorknorpel
bezeichnet, dessen Zellen als charakteristisches Merkmal das Parathormon-related-Peptide (PTHrP) bilden
(GOLDRING 2012, MABVUURE et al. 2012, SINGH und SCHWARZBAUER 2012). Diese sich noch im
teilungsfähigen Entwicklungsstadium zwischen Chondroprogenitorzellen und Chondrozyten befindenden
Zellen werden im deutschsprachigen Raum als Chondroblasten bezeichnet (MABVUURE et al. 2012). In
Anlehnung an die internationale Literatur, in der sich diese Einteilung kaum wieder findet und die Begriffe
sogar synonym verwendet werden (COLE 2011, MABVUURE et al. 2012, SINGH und SCHWARZBAUER 2012),
wird in dieser Arbeit für Knorpelzellen, die das Chondroprogenitor-Stadium verlassen haben, einheitlich der
Begriff Chondrozyt genutzt.
2.1.3 Terminale Differenzierung zu Chondrozyten
Die terminalen Differenzierungsprozesse im Knorpel werden insbesondere durch eine Verschiebung der
Expression der BMPs zugunsten von BMP-7 hervorgerufen. Diese beinhalten die Hemmung der
Proliferationsfähigkeit der Chondrozyten, eine verminderte Synthese von Fibronektin und den Isotyp-
Switch auf die mature Splicing-Variante Kollagen Typ IIB. Dieses wird im Allgemeinen als Kollagen Typ II
bezeichnet (DELISE et al. 2000, FORTIER et al. 2011). Insgesamt kommt es zu einer Zunahme der ECM,
welche sich in dem nun differenzierten Knorpel hauptsächlich aus den Komponenten Aggrekan, Kollagen
Typ II, IX und XI zusammensetzt. Hierbei rücken die Chondrozyten immer weiter auseinander, runden sich
ab und verkürzen ihre Fortsätze. Dies führt zu der typischen Morphologie des adulten Knorpels, in dem die
Chondrozyten, deren Zellfortsätze nur wenig in die ECM hineinreichen, einzeln oder in Gruppen vorliegen
(DELISE et al. 2000, KNUDSON und KNUDSON 2001). Während die Anzahl der Zell-Zell-Kontakte in diesem
Prozess sinkt, werden verstärkt Zellkontakte zur ECM ausgebildet. Insbesondere durch die Bindung an
Aggrekan über den Hyaluronsäure-Rezeptor CD44 wird die Organisation der perizellulären Matrix gesteuert
(DELISE et al. 2000).
Durch die Bildung von Kavitäten innerhalb des Vorknorpels entstehen die Gelenkspalten. Die an diese
Gelenkspalten grenzenden Chondrozyten exprimieren den PTHrP-Rezeptor (PTHrP-R), wodurch unter der
Wirkung von PTHrP die Teilungsfähigkeit dieser als artikulär bezeichneten Chondrozyten erhalten bleibt
(GOLDRING 2012, MABVUURE et al. 2012). Durch die weitere Proliferation der artikulären Chondrozyten
und die Sekretion der knorpelspezifischen ECM kommt es zum appositionellen Dickenwachstum des
Knorpels. Mit dem Einsprossen der Blutgefäße und den damit einwandernden Chondroklasten und
Osteoblasten in den Vorknorpel entstehen Ossifikationszentren. Das primäre Ossifikationszentrum befindet

2 Literaturübersicht
4
sich diaphysär, während die sekundären im Bereich der Epiphysen auftreten und den artikulären Knorpel in
einen gelenkspaltnahen Gelenkknorpel und eine epiphysäre Wachstumsfuge teilen (DELISE et al. 2000,
KNUDSON und KNUDSON 2001, MABVUURE et al. 2012).
Während die PTHrP-Synthese und damit die Proliferationsfähigkeit der Chondrozyten im Bereich der
Wachstumsfuge erhalten bleibt, wird die PTHrP-Synthese im Gelenkknorpel durch den im Rahmen des
Wachstums zunehmenden mechanischen Druck gehemmt und damit die Proliferationsfähigkeit der
Chondrozyten allmählich verringert (DONKELAAR und HUISKES 2007, HOLLANDER et al. 2010). So sind im
juvenilen Gelenkknorpel eine verbleibende Population von proliferativen Chondrozyten in der
oberflächlichen Zone des hyalinen Gelenkknorpels nachweisbar, deren Anzahl mit zunehmendem Alter
abnimmt und in geringer Konzentration auch im adulten Organismus erhalten bleibt (DOWTHWAITE et al.
2004, LOTZ und LOESER 2012).
2.2 Aufbau und Zusammensetzung des Knorpelgewebes
2.2.1 Die Komponenten des Knorpelgewebes
Das Knorpelgewebe gehört wie auch das Knochengewebe zu den Binde- und Stützgeweben, welche sich
stets aus Zellen und ECM zusammensetzen.
Embryonales Bindegewebe Bindegewebe Retikuläres Lymphoretikuläres Bindegewebe Bindegewebe Hämoretikuläres Bindegewebe Fettgewebe Faseriges Lockeres Bindegewebe Bindegewebe Straffes Straffes geflechtartiges Bindegewebe Bindegewebe Straffes parallelfaseriges Bindegewebe Binde- und Stützgewebe Knorpelgewebe Stützgewebe Knochengewebe
Der Knorpel gilt als zellarmes Gewebe, da der Anteil der ECM überwiegt (LUPPA 2000). Der Knorpel grenzt
sich von den Bindegeweben durch seine höhere Festigkeit und vom Knochengewebe durch die geringere
Mineralisation ab (SALOMON et al. 2005). Er besitzt durch seinen hohen Gehalt an Wasser und
Glykosaminoglykanen (GAGs) eine gewisse elastische Verformbarkeit, die es ermöglicht Druck- und
Scherkräfte abzufedern (KATTA et al. 2008, KNUDSON und KNUDSON 2001).
2.2.1.1 Die Zellen des Knorpelgewebes
Die spezifischen Zellen des Knorpelgewebes sind die Chondrozyten, die aus den MSCs hervorgegangen sind.
Durch sie werden sowohl die Fasern als auch die amorphe Grundsubstanz synthetisiert und in die
Umgebung abgegeben. Da der Anteil der Chondrozyten im Knorpelgewebe gering ist (2-8% in Abhängigkeit

2 Literaturübersicht
5
vom Alter), bringt jede einzelne Zelle in diesem Gewebe eine enorme Syntheseleistung auf (ALFORD 2005,
LUPPA 2000).
Einmal differenzierte Chondrozyten stellen ortständige Zellen dar, die wenige Zell-Zell- jedoch zahlreiche
Zell-Matrix-Kontakte aufweisen und nicht in der Lage sind, sich in der ECM zu bewegen (MARLOVITS und
VÉCSEI 2000). Zusätzlich besitzen Chondrozyten Mechanorezeptoren, so genannte „connexin
hemichannels“, welche eine zelluläre Transduktion der Druckbelastung ermöglichen. Diese fungieren als
solche auch in der Membran einer primären Stereozilie, welche an der Zelloberfläche der Chondrozyten zu
finden ist. Diese Rezeptoren induzieren intrazelluläre Signalwege, welche eine Modifikation der
metabolischen Aktivität und somit eine Veränderung der Expression der Komponenten der ECM
entsprechend des hyalinen Knorpels (siehe 2.2.2) hervorrufen (BUCKWALTER et al. 2005, GARCIA und
KNIGHT 2010, KNIGHT et al. 2009, WHITFIELD 2008).
2.2.1.2 Die extrazelluläre Matrix des Knorpelgewebes
Die von den Chondrozyten gebildete ECM setzt sich zusammen aus der amorphen Grundsubstanz sowie
den kollagenen und elastischen Fasern.
2.2.1.2.1 Die amorphe Grundsubstanz des Knorpelgewebes
Die amorphe Grundsubstanz des Knorpelgewebes besteht im Wesentlichen aus Proteoglykanen (PGs) und
freier Hyaluronsäure sowie an diese gebundenes Wasser. Die PGs enthalten ein Kernprotein, an das über so
genannte Linker-Proteine aus repetitiven Disaccharideinheiten aufgebaute GAGs kovalent gebunden sind.
Die Namensgebung der PGs erfolgt einerseits auf der Basis des Kernproteins (z.B. Perlekan, Syndekan,
Aggrekan) und andererseits anhand des am meisten enthaltenen GAGs (z.B. Dermatansulfat-,
Heparansulfat-, Keratansulfat-, Chondroitinsulfat-PG). Dabei sind die Kernproteine unspezifisch für die an
sie bindenden GAGs (ASPERG 2012, SCHULZ und BADER 2007). Die Disacharideinheiten binden an eine
zentrale Kohlenstoffkette und bilden so riesige Moleküle. PGs können membrangebunden vorkommen
(z.B. Syndekan, Glypikan) oder aufgrund des Fehlens einer Membrandomäne nach ihrer Synthese sezerniert
werden (z.B. Perlekan, Aggrekan) (KNUDSON und KNUDSON 2001). Hyaluronsäure ist ein GAG, welches
auch ohne Bindung an ein Kernprotein vorliegen kann. Es verlässt die Zelle im Gegensatz zu allen anderen
GAGs ohne Passage des Golgi-Apparates und kann sowohl als membrangebundenes als auch als lösliches
Molekül vorkommen (COUCHMAN und PATAKI 2012).
Die PGs des Knorpelgewebes bestehen überwiegend aus den GAGs Keratansulfat und Chondroitinsulfat,
wobei Aggrekan das häufigste Kernprotein darstellt. Das Aggrekan als Kernprotein besteht aus drei
Globulin-Domänen (G1, G2, G3), einer interglobulinen Domäne, die G1 und G2 verbindet, sowie zwei
Bindungsdomänen für GAGs zwischen G2 und G3. Durch die N-terminale Vernetzung (G1) der Aggrekane
untereinander über Tenascin lagern sich diese zu einer Struktur aus etwa 50 Monomeren zusammen,
sodass sich das im Knorpel reichlich vorhandene Aggrekan zu einer Struktur aus etwa 50 Monomeren
zusammenlagert. Das so gebildete Polymer bindet C-terminal an ein Hyaluronsäurefilament. Die Vorgänge
der Vernetzung untereinander und der Bindung an Hyaluronsäure werden als Aggregation bezeichnet. Die
Chondrozyten sezernieren die Komponenten der PGs und steuern deren Auf- und Abbau. Bei intakter
Knorpeloberfläche werden die beim Abbau anfallenden GAGs für die Resynthese der PGs verwendet. Wird
jedoch die Knorpeloberfläche beschädigt, diffundieren die GAGs in die Synovia und gehen für den erneuten
Aufbau von PGs verloren (ASPBERG 2012, HABENHAUER et al. 2003, LUPPA 2000, KNUDSON und KNUDSON
2001, SCHULZ und BADER 2007).

2 Literaturübersicht
6
Die GAG-Seitenketten besitzen viele negativ geladene Gruppen, die im Gesamtmolekül zu einer starken
Polarität und damit zu einer hohen Wasserbindungsfähigkeit führen. In Abhängigkeit von Länge und Typ
der GAG-Seitenketten wird das Gesamtmolekül somit von einer mehr oder weniger starken Hydrathülle
umgeben (KNUDSON und KNUDSON 2001).
2.2.1.2.2 Die Fasern des Knorpelgewebes
Im Knorpelgewebe unterscheidet man die durch besondere Zugfestigkeit gekennzeichneten kollagenen und
die für Dehnungsbelastungen wichtigen elastischen Fasern. Dabei sind kollagene Fasern sowohl im Knorpel
als auch in anderen Geweben strukturgebend und stabilisierend (GELSE et al. 2003). Elastische Fasern
dagegen kommen relativ selten vor. Sie spielen im elastischen Knorpel und anderen Geweben, die eine
gewisse elastische Verformbarkeit benötigen, wie beispielsweise dem elastischen Nackenband, eine Rolle
(KIELTY et al. 2002).
2.2.1.2.2.1 Die kollagenen Fasern des Knorpelgewebes
Die kollagenen Fasern dienen den PGs einerseits als Gerüst. Auf der anderen Seite schützen die PGs durch
ihre Anlagerung an die kollagenen Fasern diese vor dem Abbau durch Kollagenasen. Die den kollagenen
Fasern zugrunde liegenden Kollagene sind eine heterogene Gruppe von Molekülen, denen ein
tripelhelikaler Molekülbereich gemein ist, wobei sie sich in Funktion und Gewebeverteilung erheblich
unterscheiden (GELSE et al. 2003).
Die Synthese von Kollagen beginnt mit der Bildung einzelner Ketten von jeweils ca. 1000 Aminosäuren, von
denen jede dritte Glycin ist. Entsprechend dieser Gesetzmäßigkeit wird das Kollagen mit der Formel
(Gly-X-Y)n beschrieben, wobei X und Y sehr häufig Prolin und Lysin darstellen (Primärstruktur). Im
endoplasmatischen Retikulum kommt es zur Ascorbinsäure-abhängigen Hydroxylierung von Prolin- und
Lysinresten sowie der Glykosylierung einiger Hydroxylysinreste. Nach der Ausbildung der Sekundärstruktur
wird diese als -Kollagenkette bezeichnet. Anschließend entsteht im Golgi-Apparat aus jeweils drei dieser
-Ketten durch Wasserstoffbrückenbindungen zwischen den Hydroxyprolinresten eine rechtsdrehende
Tripelhelix, wobei Glycin in Richtung des Zentrums der Tripelhelix orientiert ist. Dieses als Prokollagen
(Tertiärstruktur) bezeichnete Molekül wird in den Extrazellularraum abgegeben (GELSE et al. 2003). Als
Kollagen klassifiziert, weist jedes Prokollagen-Molekül zumindest eine tripelhelikale Kollagendomäne sowie
weitere helikale und nicht helikale Bereiche auf. Die dabei vorhandenen N-terminalen 7S-Domänen und
C-terminalen, nicht kollagenen Domänen (NC1) beeinflussen wesentlich die Bildung der Tripelhelix und
werden erst nach deren Formation entfernt. So sind die NC1-Domänen dafür verantwortlich, welche
-Ketten sich miteinander verbinden und wie die fibrillären Anteile des Kollagens organisiert werden
(EYRE 2003, MACDONALD et al. 2006, PARSONS et al. 2006).
Extrazellulär lagern sich fünf tripelhelikale Prokollagene versetzt zu Mikrofibrillen (Kollagenfibrillen) und
letztlich zu kollagenen Fasern (Suprastruktur) zusammen (Abb.2). Die Bildung der Fibrillen erfolgt durch die
Ausbildung von kovalenten Bindungen zwischen Hydroxylysinresten sowie Wasserstoffbrückenbindungen
zwischen Hydroxyprolinresten verschiedener Ketten (Quervernetzungen) (Abb. 2).

2 Literaturübersicht
7
Abb. 2: Die molekulare Modellstruktur einer Kollagenfibrille. Dargestellt sind die Quervernetzungen (cross-links) des Kollagen Typ II die durch Kollagen Typen IX und XI ausgebildet werden; aus: EYRE (2002).
Bei der Zusammenlagerung der Kollagenketten können Homotrimere aus identischen Ketten (z.B. Kollagen
Typ II) und Heterotrimere aus unterschiedlichen Ketten (z.B. Kollagen TypI, IX, XI) entstehen (Tab. 1)
(GELSE et al. 2003, YAMAUCHI und SRICHOLPECH 2012).
Tab. 1: Kollagentypen, ihre Genlokalisation und molekulare Zusammensetzung, adaptiert nach: GELSE et al. (2003) Kollagen-Typ Gene (Lokalisation) Molekulare Zusammensetzung
Fibrillen-bildende Kollagene
I COL1A1 (17q21.31–q22) (α1(I))2 α2(I)
II COL2A1 (12q13.11–q13.2) (α1(II))3
XI COL11A1 (1p21) α1(XI) α2(XI) α3(XI)
Fibril-Associated Collagens with Interrupted Triple helices (FACIT)
IX COL9A1 (6q13) COL9A2 (1p33–p32.2)
α1(IX) α2(IX) α3(IX)
Die bekannten 27 Kollagen Typen werden in verschiedenen Gruppen zusammengefasst. Trotz ihres
ähnlichen Aufbaus haben sie spezifische Aufgaben in verschiedenen Geweben. So unterscheiden sie sich in
ihren immunologischen Eigenschaften und ihrem Verhalten in der Zell-Zell- und Zell-Matrix-Interaktion
(MARK 2006). Für die Struktur und Funktion des Knorpels sind die Fibrillen-bildenden Kollagen Typen I, II
und XI sowie das zu den Fibrillen-assoziierten Kollagenen mit unterbrochenen Tripelhelices (Fibril-
Associated Collagens with Interrupted Triple helices, FACIT) gehörende Kollagen Typ IX bedeutend (GELSE
et al. 2003). Der im Körper am häufigsten vorkommende Kollagen Typ I besteht als Heterotrimer aus zwei
α1(I)- Ketten und einer α2(I)- Kette. Er erreicht durch seine Faserstärke von 25-400 nm eine starke
Zugfestigkeit und tritt daher neben dem Knorpel v.a. in Sehnen, Bändern und Knochen auf (GELSE et al.
2003).
Im Knorpelgewebe stellt Kollagen Typ II einen weiteren Vertreter der Fibrillen-bildenden Kollagene dar.
Hierbei handelt es sich um ein Homotrimer aus drei identischen α1(II)-Ketten, wobei die jeweiligen
α1-Ketten der Kollagen Typen I ((α1(I)) und II (α1(II)) keine Splicingvarianten darstellen, sondern
unterschiedlichen Chromosomen entstammen. Ergänzend finden posttranslationale Modifikationen des

2 Literaturübersicht
8
Kollagen Typ II in Form von Hydroxylierungen des Lysins sowie anschließende Glykosylierungen verglichen
mit Kollagen Typ I in deutlich größerem Umfang statt, sodass dessen Interaktion mit den PGs erleichtert
wird. Diesem Umstand geschuldet ist auch sein Vorkommen im Glaskörper und Corneaepithel außerhalb
des Knorpels (GELSE et al. 2003).
Auch bei Kollagen Typ XI handelt es sich um ein Fibrillen-bildendes Kollagen, das in geringer Menge im
Knorpel vorhanden ist und eine starke Ähnlichkeit zu Kollagen II aufweist, da beide Splicingvarianten
desselben Gens darstellen. Es lagert sich zwischen Kollagen Typ II-Moleküle ein und bindet mit seinem
abknickenden Ende ein weiteres Kollagen Typ II-Molekül, sodass Quervernetzungen entstehen, deren
Gesamtheit der Kollagenfaser ihre Stabilität verleiht (Abb.2) (GELSE et al. 2003, FERNANDES et al. 2007,
MARK 2006, MATYAS et al. 1997).
Daneben ist das zur Gruppe der FACIT gehörende Kollagen Typ IX für die Verbindung des äußeren Bereichs
der Fibrillen verantwortlich. Kollagen Typ IX-Moleküle lagern sich periodisch an Kollagen Typ II an und
bilden untereinander mit Hilfe ihrer nicht-helikalen Anteile kovalente Bindungen aus und tragen dadurch
zur Quervernetzung bei (EYRE 2003, GELSE et al. 2003). Des Weiteren zeigen andere nicht-helikale Bereiche
des Moleküls starke Interaktionen mit den PGs, insbesondere Aggrekan, sodass die weitere Anlagerung von
Fibrillen und damit der Faserdurchmesser auf 15-100 nm begrenzt wird (CHANGOOR et al. 2011, EYRE
1995).
2.2.1.2.2.2 Die elastischen Fasern des Knorpelgewebes
Die elastischen Fasern zeichnen sich durch eine stark reversible Verformbarkeit aus, die dem Gewebe, in
dem sie enthalten sind, eine hohe Widerstandsfähigkeit gegenüber Zugkräften ermöglicht. Die Rückkehr in
den Ausgangszustand erfolgt dabei energieunabhängig durch passive Rückstellkräfte (KIELTY et al. 2002).
Im Verlauf der Entwicklung elastischer Fasern erfolgt eine Einlagerung von Tropoelastin, der löslichen
Vorstufe des Elastins, in ein vorgeformtes Netzwerk aus Fibrillin-haltigen Mikrofibrillen. Die
Tropoelastinmonomere zeigen nach ihrer Sekretion eine Tendenz zur Aggregation. Sie bilden einen Kern
aus stark vernetztem Elastin, der von einem außen liegenden Mantel aus Mikrofibrillen umschlossen wird
(WAGENSEIL und MECHAM 2007). Die zentralen Tropoelastinmonomere werden über Desmosine sowie
Isodesmosine und die peripheren Mikrofibrillen durch Quervernetzungen zusammengehalten. Die intra-
und intermolekularen Faltungen der Mikrofibrillen stellen die Rückstellkräfte für die Elastizität der Fasern
zur Verfügung (KIELTY et al. 2002).
2.2.2 Der hyaline Gelenkknorpel
Frischer hyaliner Knorpel hat eine milchig-bläuliche Farbe und erscheint im Gegenlicht „durchscheinend“,
woher sich der Begriff hyalin ableitet. In Form des Gelenkknorpels kommt dieses vollständig avaskuläre und
nicht sensibel innervierte Gewebe in allen großen Gelenken des Körpers und während des Wachstums in
den Epiphysenfugen der langen Röhrenknochen vor. Daneben tritt hyaliner Knorpel auch im Bereich der
Nase in Form der Cartilagines nasi externi, Cartilagines nasi laterales dorsales et ventrales sowie der
Cartilago septi nasi sowie beim Pferd auch in der Cartilago alaris auf. Die Cartilagines tracheales und
bronchiales, sowie die Kehlkopfknorpel - mit Ausnahme der Cartilago epiglottica - sind einige weitere
Beispiele für das Vorkommen des hyalinen Knorpels im Bereich des Atmungsapparats. Sein Vorkommen im
Bewegungsapparat beschränkt sich nicht nur auf die synovialen Gelenke, sondern schafft auch eine
kontinuierliche Verbindung zwischen Knochen in Form der Synchondrosen, z.B. die Synchondroses
sternales und die Synchondrosis intermandibularis (SALOMON et al. 2005, SCHULZ und BADER 2007).
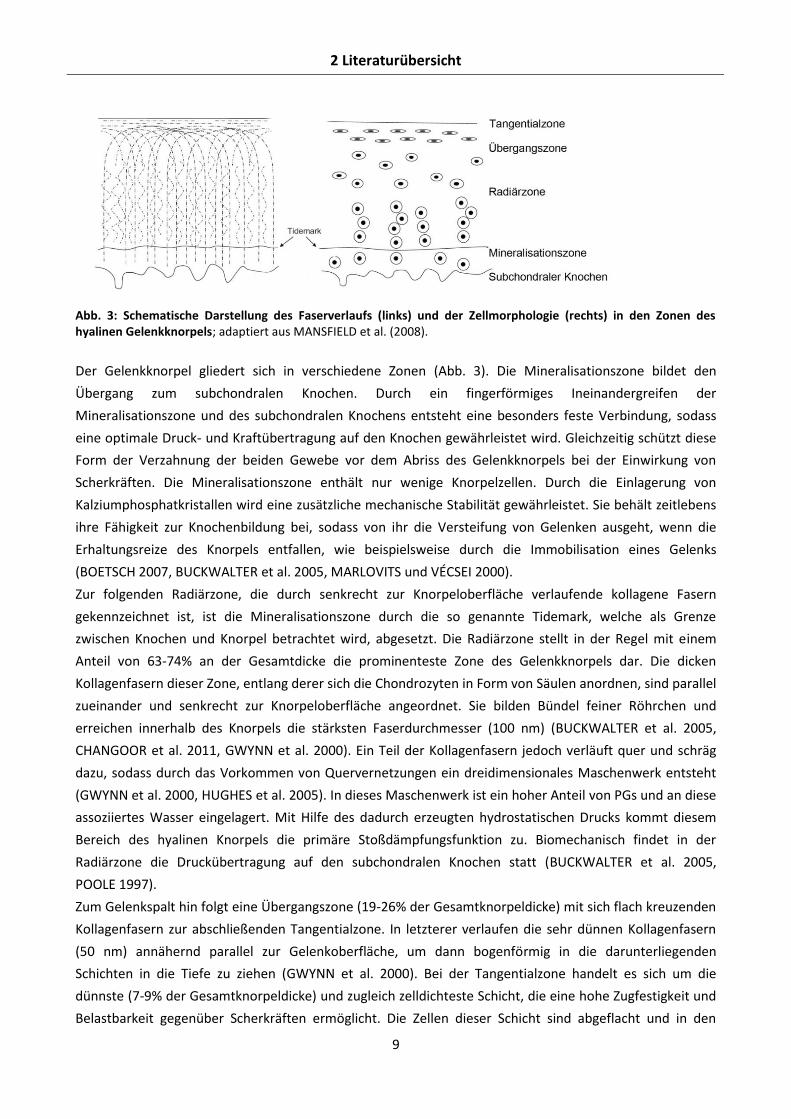
2 Literaturübersicht
9
Abb. 3: Schematische Darstellung des Faserverlaufs (links) und der Zellmorphologie (rechts) in den Zonen des hyalinen Gelenkknorpels; adaptiert aus MANSFIELD et al. (2008).
Der Gelenkknorpel gliedert sich in verschiedene Zonen (Abb. 3). Die Mineralisationszone bildet den
Übergang zum subchondralen Knochen. Durch ein fingerförmiges Ineinandergreifen der
Mineralisationszone und des subchondralen Knochens entsteht eine besonders feste Verbindung, sodass
eine optimale Druck- und Kraftübertragung auf den Knochen gewährleistet wird. Gleichzeitig schützt diese
Form der Verzahnung der beiden Gewebe vor dem Abriss des Gelenkknorpels bei der Einwirkung von
Scherkräften. Die Mineralisationszone enthält nur wenige Knorpelzellen. Durch die Einlagerung von
Kalziumphosphatkristallen wird eine zusätzliche mechanische Stabilität gewährleistet. Sie behält zeitlebens
ihre Fähigkeit zur Knochenbildung bei, sodass von ihr die Versteifung von Gelenken ausgeht, wenn die
Erhaltungsreize des Knorpels entfallen, wie beispielsweise durch die Immobilisation eines Gelenks
(BOETSCH 2007, BUCKWALTER et al. 2005, MARLOVITS und VÉCSEI 2000).
Zur folgenden Radiärzone, die durch senkrecht zur Knorpeloberfläche verlaufende kollagene Fasern
gekennzeichnet ist, ist die Mineralisationszone durch die so genannte Tidemark, welche als Grenze
zwischen Knochen und Knorpel betrachtet wird, abgesetzt. Die Radiärzone stellt in der Regel mit einem
Anteil von 63-74% an der Gesamtdicke die prominenteste Zone des Gelenkknorpels dar. Die dicken
Kollagenfasern dieser Zone, entlang derer sich die Chondrozyten in Form von Säulen anordnen, sind parallel
zueinander und senkrecht zur Knorpeloberfläche angeordnet. Sie bilden Bündel feiner Röhrchen und
erreichen innerhalb des Knorpels die stärksten Faserdurchmesser (100 nm) (BUCKWALTER et al. 2005,
CHANGOOR et al. 2011, GWYNN et al. 2000). Ein Teil der Kollagenfasern jedoch verläuft quer und schräg
dazu, sodass durch das Vorkommen von Quervernetzungen ein dreidimensionales Maschenwerk entsteht
(GWYNN et al. 2000, HUGHES et al. 2005). In dieses Maschenwerk ist ein hoher Anteil von PGs und an diese
assoziiertes Wasser eingelagert. Mit Hilfe des dadurch erzeugten hydrostatischen Drucks kommt diesem
Bereich des hyalinen Knorpels die primäre Stoßdämpfungsfunktion zu. Biomechanisch findet in der
Radiärzone die Druckübertragung auf den subchondralen Knochen statt (BUCKWALTER et al. 2005,
POOLE 1997).
Zum Gelenkspalt hin folgt eine Übergangszone (19-26% der Gesamtknorpeldicke) mit sich flach kreuzenden
Kollagenfasern zur abschließenden Tangentialzone. In letzterer verlaufen die sehr dünnen Kollagenfasern
(50 nm) annähernd parallel zur Gelenkoberfläche, um dann bogenförmig in die darunterliegenden
Schichten in die Tiefe zu ziehen (GWYNN et al. 2000). Bei der Tangentialzone handelt es sich um die
dünnste (7-9% der Gesamtknorpeldicke) und zugleich zelldichteste Schicht, die eine hohe Zugfestigkeit und
Belastbarkeit gegenüber Scherkräften ermöglicht. Die Zellen dieser Schicht sind abgeflacht und in den

2 Literaturübersicht
10
oberflächenparallelen Kollagenfaserverlauf eingelagert (ALFORD 2005, CHANGOOR et al. 2011, EYRE 2002,
SICZKOWSK und WATT 1990). Im Gegensatz zu den dicht gepackten Kollagenfibrillen ist der Gehalt an PGs
hier vergleichsweise gering (POOLE 1997). An der Oberfläche der Tangentialzone schließt sich eine dünne
(10 µm), zellfreie Schicht aus oberflächenparallelen Kollagenfasern an, die auch als Lamina splendens
bezeichnet wird. Neben Kollagen Typ II wurden in diesem Bereich Kollagen Typ I und III sowie elastische
Fasern nachgewiesen (FUJIOKA et al. 2013, HE et al. 2013). Die Grenze zum Gelenkspalt besteht aus einer
Schicht von Phospholipiden, Hyaluronsäure und dem Glykoprotein Lubrizin, welche sowohl von den
Synovialozyten als auch von den oberflächlichen Chondrozyten produziert werden (LOTZ und
LOESER 2012). Durch die hohe Konzentration an Phospholipiden, die als Schmierstoffe wirken, wird die
Reibung zwischen den Gelenkoberflächen reduziert (KATTA et al. 2008). Das Vorhandensein dieser Schicht
ist bedingt durch das Fehlen eines vaskularisierten Perichondriums - eine Besonderheit, die den
Gelenkknorpel von den anderen Knorpeltypen unterschiedet (SCHULZ und BADER 2007). Von einigen
Autoren wird diese Schicht mit der Lamina splendens als „Most Superficial Zone“ (MSZ) zusammengefasst.
Die Tangentialzone und die MSZ stellen eine Barriere gegenüber der Synovia dar, die den Austausch kleiner
Moleküle zulässt und den größerer Moleküle verhindert. Somit erfolgt kein Efflux von PGs, gleichzeitig
können beispielsweise Antikörper nicht in den Gelenkknorpel eindringen, wodurch dieser vor dem
Immunsystem abgeschirmt wird (BUCKWALTER et al. 2005).
Der Gehalt an PGs ist in der Tangentialzone am geringsten und nimmt in Richtung Radiärzone zu,
wohingegen in der Mineralisationszone kaum noch PGs vorhanden sind (HUGHES et al. 2005). Die
hydrodynamischen Belastungsmerkmale des Gelenkknorpels werden durch die Balance zwischen der
Hydratation der PGs und dem Widerstand, den das kollagene Netzwerk ihrer Ausdehnung entgegensetzt,
bedingt (POOLE 1997).
Im hyalinen Knorpel liegen die Chondrozyten, deren Anteil am Gesamtvolumen 2% beträgt, einzeln oder in
kleinen Gruppen, welche als Chondrone bezeichnet werden und die primäre strukturelle, funktionelle und
metabolische Einheit des Knorpels darstellen (ALFORD 2005). Diese und die sie umgebende perizelluläre
Matrix bilden ein Territorium. Lediglich in der Tangentialzone bilden die Chondrozyten keine Chondrone
und weisen keine spezifische perizelluläre Matrix auf (BUCKWALTER et al. 2005, BRUCKNER und VAN DER
REST 1994, POOLE 1997).
Der Gelenkknorpel ist nicht vaskularisiert, sodass die Ernährung der Chondrozyten hauptsächlich durch
Diffusion aus der Synovia erfolgen muss und entsprechend langsam vonstatten geht. Dadurch wird ein
hypoxisches Milieu geschaffen, in dem die anaerobe Energiegewinnung dominiert. Nur in den knochennah
gelegenen Bereichen erfolgt eine Ernährung des Gelenkknorpels auch durch die Blutgefäße des
subchondralen Knochens (ALFORD 2005).
Hyaliner Knorpel weist unter allen Knorpelarten den geringsten Anteil an Fasern und einen hohen Anteil
amorpher Grundsubstanz auf. Dabei sind Kollagen Typ II, IX, und XI die wichtigsten Kollagentypen im
hyalinen Knorpel und machen zwei Drittel der Trockensubstanz aus (EYRE 2002). Bei einem Wassergehalt
von 60-85% entfallen somit auf die kollagenen Fasern etwa 10-30% des Nassgewichts. Dabei stellt Kollagen
Typ II, welcher die grundsätzlichen Faserverläufe bestimmt, 90-95% des im hyalinen Knorpel vorhandenen
Kollagens (BUCKWALTER et al. 2005). Daneben liegen in geringeren Anteilen Kollagen Typ IX und XI vor, die
durch Quervernetzungen in Form von kovalenten Bindungen und molekularen Wechselwirkungen die
Vielzahl der Kollagen Typ II-Fibrillen stabilisieren (EYRE 2002). Die PGs, welche hauptsächlich
Chondroitinsulfat- und Keratansulfat-Seitenketten enthalten, und nicht-kollagene Proteine nehmen einen

2 Literaturübersicht
11
Anteil von 3-10% am Nassgewicht ein. Die PGs liegen zu 90% in aggregierter Form vor, wohingegen nur
geringe Anteile auf nicht aggregierte (7%) und kleine PGs (3%) entfallen (BUCKWALTER et al. 2005, SCHULZ
und BADER 2007). Dieses Ausmaß der Aggregation der PGs im Gelenkknorpel ist maßgeblich dafür
verantwortlich, dass dieser zu etwa 70% aus Wasser besteht, wodurch dessen einzigartige
Druckaufnahmefunktion gewährleistet wird (ASPBERG 2012, KIANI et al. 2002, STÄRKE 2008).
Die perizellulären Territorien des Gelenkknorpels enthalten hohe Gehalte an PGs, die auf Aggrekan als
Kernprotein basieren, sowie Hyaluronsäure und kleinere Link-Proteine. Das hier häufiger als in den
Interterritorien vorkommende GAG Chondroitinsulfat ist größer, besitzt mehr Ladungen als Keratansulfat
und bedingt dadurch eine höhere Wasserbindungsfähigkeit der Territorien (LUPPA 2000). In diesem Bereich
entsteht ein Netz feiner Kollagen Typ II-Fasern unter Mitwirkung von Kollagen Typ IX, dessen Präsenz eine
weitere Anlagerung von Kollagen Typ II-Fibrillen inhibiert. Durch diese Begrenzung des Faserdurchmessers
des Kollagen Typ II entstehen dicht gelagerte Einfassungen um die Chondrozyten (BRUCKNER und VAN DER
REST 1994, MARK 2006). Von den Territorien weg laufen dickere Bündel kollagener Fasern in radiärer
Orientierung, um schließlich sehr kompakte Fasern zu bilden, die in den Interterritorien zu finden sind. In
diesen Bereichen liegt eine höhere Konzentration von keratansulfatreichen PGs vor (BONNEMANN et
al. 2000, MARK 2006, OLSEN 1997).
2.2.3 Der Faserknorpel
Der Faserknorpel kommt physiologisch im Bereich des Bewegungsapparats als Menisci articulares des
Kniegelenks zur Kontaktflächenvergrößerung inkongruenter Gelenkoberflächen vor. Zusätzlich dient er in
diesen und den Anuli fibrosi der Zwischenwirbelscheiben sowie den Disci articulares der Kiefergelenke der
Kompensation von Scherkräften und starken mechanischen Zugbelastungen. Das Vorkommen des
Faserknorpels beschränkt sich jedoch nicht auf die Gelenke. Er ist auch als Einlagerung in stark
beanspruchte Sehnen und Bänder nachweisbar und unterstützt neben dem ohnehin hohen Gehalt an
Kollagen Typ I in diesen Geweben deren Zugfestigkeit (BENJAMIN und RALPHS 1998, STÄRKE 2008).
Im Faserknorpel sind die kollagenen Fasern im Gegensatz zur amorphen Grundsubstanz stark
überrepräsentiert und in Richtung der Zugbelastung ausgerichtet. Aufgrund der morphologischen Nähe
zum straffen, kollagenfaserreichen Bindegewebe wird dieser auch als Bindegewebsknorpel bezeichnet
(MAKRIS et al. 2011).
Die Fähigkeit des Gewebes zur Druckaufnahme ist aufgrund des geringeren PG-Gehalts wesentlich geringer
als die des hyalinen Knorpels. Durch die spezielle Anordnung der Kollagenfaserbündel und den
Gesamtquerschnitt des jeweiligen Faserknorpels werden jedoch bei diesen Gelenken punktuell einwirkende
Druckkräfte in Zugspannungen umgewandelt, welche auf die Gesamtfläche des Faserknorpels verteilt
werden können. Diese Umwandlung ermöglicht die Stoßdämpfungsfunktion des Faserknorpels
(MAKRIS et al. 2011, STÄRKE 2008).
Die wenigen Chondrozyten des Faserknorpels sind entsprechend der Zugrichtung der Kollagenfasern
zwischen diese eingebettet. Daher liegen sie hintereinander angeordnet, einzeln oder in Gruppen von zwei
Zellen und bilden keine Chondrone. In den äußeren Bereichen der Disci, Menisci und Anuli fibrosi
erscheinen sie gestreckt und spindelförmig, während sie in den tieferen Regionen eine ovoide Morphologie
aufweisen. Die Chondrozyten des in Sehnen und Bänder eingelagerten Faserknorpels erscheinen groß und
oval. Für die Zellen des Faserknorpels werden entsprechend der Zellmorphologie mit steigender
Konzentration der sie umgebenden ECM die Begriffe Chondrozyten, intermediäre Zellen,

2 Literaturübersicht
12
Fibrochondrozyten oder Fibrozyten verwendet (BENJAMIN und RALPHS 1998, DETAMORE et al. 2006,
MAKRIS et al. 2011).
Faserknorpel weist unter den Knorpelarten den höchsten Anteil an Fasern und die geringste Konzentration
an amorper Grundsubstanz auf. Die kollagenen Fasern stellen mit 60-80% einen mit dem hyalinen Knorpel
vergleichbaren Anteil an der Trockensubstanz dar, wobei sich jedoch die Zusammensetzung unterscheidet.
Innerhalb der Kollagenfraktionen bildet Kollagen Typ I, welches dicke Faserbündel bildet, mit 80% den
Hauptanteil, wohingegen Kollagen II nur in geringer Konzentration gefunden wird. Die als dicke Bündel
vorliegenden Kollagen Typ I-Fasern sind für die Zugfestigkeit dieses Gewebes verantwortlich (CHEUNG
1987, SCHULZ und BADER 2007). Die PGs bilden mit 2-8 % der Trockensubstanz in der ECM nur eine kleine
Fraktion (MAKRIS et al. 2011). Trotz des im Vergleich zum hyalinen Knorpel deutlich geringeren PG-Gehalts
erreicht der Faserknorpel einen Wassergehalt von 70-75%. Dies liegt darin begründet, dass die auf
Aggrekan als Kernprotein basierenden PGs einen hohen Anteil Chondroitinsulfat (50-80%) aufweisen
(BENJAMIN und RALPHS 1998, SCHULZ und BADER 2007). Allerdings kann die Verteilung der kollagenen
Fasern und PGs des in Sehnen und Bändern eingelagerten Faserknorpels hiervon abweichen. In denen, die
neben der Zugbelastung gleichzeitig auch starken Druckkräften ausgesetzt sind, werden höhere Anteile an
Kollagen Typ II und Aggrekan nachgewiesen (BENJAMIN und RALPHS 1998).
Das Vorkommen von Mechanorezeptoren und Nozizeptoren sowie von Blutgefäßen unterscheidet den
Faserknorpel vom hyalinen Knorpel. Allerdings nimmt die zu Beginn der Entwicklung vollständige
Vaskularisierung im Rahmen des Alterungsprozesses kontinuierlich ab (MAKRIS et al. 2011, STÄRKE 2008).
2.2.4 Der elastische Knorpel
Der elastische Knorpel bildet das Stützgewebe der Ohrmuscheln, der Epiglottis und der kleinen Bronchien
sowie von Teilen der Cartilago arytaenoidea der Larynx. Er ist von einem Perichondrium umgeben. Die
Chondrozyten, deren Anteil am Gesamtvolumen 2% beträgt, liegen einzeln oder in Zweiergruppen vor und
bilden mit der sie umgebenden ECM Chondrone. Die PGs liegen in geringer Konzentration vor und sind in
ein feines Netzwerk aus Kollagen Typ II-Fasern eingelagert. Die Hauptkomponente des elastischen Knorpels
bilden die elastischen Fasern, wobei die Menge an elastischen Fasern, die in das kollagene Grundgerüst
eingewoben sind, die Flexibilität des Gewebes bestimmt. Die elastischen Fasern, welche sich bei
Zugbelastungen auf das doppelte ihrer Ausgangslänge dehnen lassen und mit Hilfe der Rückstellkräfte
energieunabhängig in diese zurückgeführt werden können, bedingen die besonderen Eigenschaften dieses
Gewebes. (KIELTY et al. 2002, SCHULZ und BADER 2007).
2.3 Dynamik des hyalinen Gelenkknorpels
Der hyaline Gelenkknorpel darf nicht als statisches Konstrukt verstanden werden. Vielmehr ist dieses
Gewebe in vivo verschiedenen biophysikalischen Einflüssen ausgesetzt, die eine dynamische Anpassung
erfordern. Dabei werden mechanische von chemischen Faktoren unterschieden. (SCHULZ und BADER 2007)
2.3.1 Dynamik des hyalinen Gelenkknorpels unter mechanischen Einflüssen
Auf den Gelenkknorpel wirkt ein hydrostatischer Druck, der je nach Belastung zwischen 1 und 200 bar
variiert. Die bei dessen mechanischer Belastung hervorgerufene Kompression des Gewebes ist essentiell für
seine Struktur und Integrität (IKENOUE et al. 2003).
Die viskoelastischen Eigenschaften des Gelenkknorpels werden mit Hilfe des biphasischen Modells
beschrieben. Eine Phase bilden die kollagenen Fasern der ECM, welche für den Spannungsdruck PKollagen

2 Literaturübersicht
13
verantwortlich sind. Die andere Phase setzt sich zusammen aus den PGs und dem durch sie gebundenen
Wasser, welche den hydrostatischen Druck hervorrufen (PHydr) sowie den nicht-kollagenen Proteinen und
die an diese assoziierten Ionen, welche den osmotischen Druck (POsm) aufbauen. Im Gelenkknorpel bauen
der hydrostatische Druck und der osmotische Druck abzüglich des Spannungsdrucks der kollagenen Fasern
den Schwelldruck des Gelenkknorpels auf. Daraus ergibt sich folgende Gleichung:
PSchwell = PHydr + POsm - PKollagen
Dem Schwelldruck steht der von extern auf den Gelenkknorpel wirkende mechanische Druck (PExtern)
entgegen. Sobald dieser zunimmt, kommt es zu einer vermehrten Umverteilung von Wasser in weniger
belastete Bereiche des Knorpelgewebes. Dadurch wird die externe Druckbelastung initial durch den
hydrostatischen Druck kompensiert bzw. erfolgt eine Flüssigkeitsverschiebung innerhalb des Knorpels.
Dabei kommt es auch zu einer Wasserumverteilung zwischen PG-gebundenem und nicht PG-gebundenem
Wasser innerhalb des Gelenkknorpels, sodass sich zwischen hydrostatischem und osmotischem Druck ein
Gleichgewicht eingestellt. Dauert die Belastung länger, kommt es zum langsamen Flüssigkeitsausstrom aus
der ECM, welcher solange aufrechterhalten bleibt bis der hydrostatische Druck null erreicht und der
gesamte externe Druck von den PGs und kollagenen Fasern übernommen wird (LUPPA 2000, SCHULZ und
BADER 2007). Durch diesen Vorgang kommt es zur Abnahme des Knorpelvolumens innerhalb von 3-12
Minuten (ECKSTEIN et al. 2001). Bei nachlassender Belastung erfährt das Wasser eine erneute
Umverteilung, sobald der externe mechanische Druck den Schwelldruck unterschreitet (LUPPA 2000,
SCHULZ und BADER 2007). Der Ausgangszustand wird aufgrund der langsamen Rückflussrate durch die
Gelenkoberfläche nach ca. 90 Minuten wieder erreicht. Bei hoher kontinuierlicher hydrostatischer
Druckbelastung nimmt die Knorpeldicke nach 1 Minute um 3%, nach 8 Minuten um 11% und nach
4 Stunden um 57% ab (HERBERHOLD et al. 1999). Den Verschiebungen sowohl des freien als auch des
gebundenen Wassers folgen gleichsam die darin gelösten Stoffe (z.B. Glukose, Lactat, Sauerstoff), sodass
die Versorgung mit Nährstoffen und die Entsorgung von Stoffwechselendprodukten an die
biomechanischen Belastungen gekoppelt sind (LUPPA 2000, MARLOVITS und VÉCSEI 2000, POOLE 1997).
Der zonale Aufbau des Knorpels ist für die Bewältigung der im Gelenk wirkenden Kräfte essentiell. Da die
Tangentialzone bei Belastung Flüssigkeit abgibt und sich dadurch ihre parallel zur Oberfläche
ausgerichteten Kollagenfasern verdichten, bildet sie eine effektive Abdichtung der Knorpeloberfläche.
Damit wird der Flüssigkeitsausstrom in der Radiärzone bei zyklischer Belastung vernachlässigbar gering, da
dieser auch subchondral durch das Knochengewebe eingeschränkt wird. Durch die gleichmäßige Verteilung
des hydrostatischen und osmotischen Drucks kommt es unter dem Kontaktbereich zu einer uniformen
Kraftübertragung auf den gesamten Gelenkknorpel und in der Folge auf die vollständige subchondrale
Knochenfläche (ECKSTEIN et al. 2001, LUPPA 2000).
Neben den vertikal wirkenden Kompressionskräften, die hauptsächlich durch den hydrostatischen und
osmotischen Druck kompensiert werden, ist der Gelenkknorpel auch horizontal wirkenden Scherkräften
ausgesetzt. Diesen sich in Form von Zugkräften im Gewebe manifestierenden Kräften wirkt hauptsächlich
der Spannungsdruck der kollagenen Fasern entgegen (WONG und CARTER 2003). Für die
Widerstandsfähigkeit gegenüber den Scherkräften sorgen sowohl die dicht gelagerten und
oberflächenparallel ausgerichteten kollagenen Fasern der Tangentialzone als auch die Verzahnung der
Mineralisationszone mit dem subchondralen Knochen. Zudem tragen die Abgabe von Flüssigkeit aus der
Tangentialzone sowie die Phospholipide, das Lubricin und die Hyaluronsäure der MSZ zur Reduktion der
Reibungskräfte bei und vermindern somit die auf die Gelenkfläche einwirkenden Scherkräfte. Aufgrund des

2 Literaturübersicht
14
wesentlich geringeren Anteils an zugfestem Kollagen Typ I zeigt der hyaline Knorpel eine höhere
Empfindlichkeit gegenüber Scherkräften als der Faserknorpel (BUCKWALTER et al. 2005, LOTZ und LOESER
2012, KATTA et al. 2008).
2.3.2 Dynamik des hyalinen Gelenkknorpels unter chemischen Einflüssen
Die Dynamik des Gelenkknorpels wird auch durch die An- oder Abwesenheit von Sauerstoff sowie dessen
Konzentrationsgradienten beeinflusst. Die hierbei über verschiedene Zytokine vermittelten Effekte führen
zu Veränderungen in der Menge und Zusammensetzung der ECM (FORTIER et al. 2011).
Durch die Limitierung der Ernährung des Gelenkknorpels über die Synovia weist dieser eine langsame
Stoffwechselrate auf. Da hiervon auch der Sauerstofftransport betroffen ist, liegt ein vorwiegend anaerober
Metabolismus vor. Die mit Hilfe von Mikroelektroden messbare Sauerstoffspannung des Gewebes liegt in
der Tangentialzone bei 7,5% und sinkt auf bis zu 1% nahe der Tidemark ab (SCHULZ und BADER 2007). Die
hypoxischen Bedingungen bewirken die Bildung von Hypoxia-inducible Factor 1α (HIF-1α), welcher eine
hemmende Wirkung auf die Chondrozytenproliferation hat und die Expression verschiedener Zytokine
induziert. Unter diesen spielen TGF-β1, BMP-2, BMP-7, IGF-1 und FGF-2 die wichtigste Rolle. Die additiven
Effekte dieser Zytokine steigern die synthetische Aktivität der Chondrozyten und führen somit bei
moderater Belastung zu einer physiologischen Dickenzunahme des Gelenkknorpels (ALFORD 2005,
DANIŠOVIČ et al. 2012, HANSEN et al. 2001, MABVUURE et al. 2012, SCHULZ und BADER 2007).
Die Zytokine TGF-β1, BMP-7 und IGF-1 führen zur verstärkten Synthese aller Komponenten der ECM des
Knorpelgewebes, wobei IGF-1 den stärksten Effekt hat. Gleichzeitig wird die ECM vor dem Abbau geschützt,
indem BMP-7 die Aktivität der Matrixmetalloproteinase 13 (MMP-13) vermindert. BMP-2 hingegen hat eine
selektive Wirkung auf die Syntheseaktivität der Chondrozyten. Es bewirkt eine gesteigerte Expression von
Kollagen Typ II und beschleunigt die Umsatzrate von Aggrekan (COUCHMAN und PATAKI 2012,
EKENSTEDT et al. 2006). FGF-2 entfaltet im Gelenkknorpel eine dosisabhängige Wirkung. So nimmt seine
auf die PG-Synthese stimulierende Wirkung mit steigender Konzentration ab (FORTIER et al. 2011).
2.3.3 Seneszenz als Einflussfaktor auf die Dynamik des hyalinen Gelenkknorpels
Der Gelenkknorpel unterliegt altersabhängigen charakteristischen Veränderungen, die sowohl die Zellen,
die kollagenen Fasern als auch die PGs betreffen (DEGROOT et al. 1999, LOTZ und LOESER 2012, LUPPA
2000, MALLINGER und STOCKINGER 1987).
Da im Knorpel das Verhältnis von Proliferation und Apoptose physiologisch auf Seiten der Apoptose liegt,
nimmt im Zuge des Alterungsprozesses die Anzahl der Chondrozyten ab. So sinkt diese im humanen
Kniegelenk vom 20. bis zum 90. Lebensjahr um etwa 50%, wobei der größte Anteil des Zellverlusts auf die
Zellen der Tangentialzone entfällt. In Abwesenheit phagozytierender Zellen verbleiben die Reste der
Chondrozyten als apoptotische Körperchen in den Territorien. Die in diesen enthaltene alkalische
Phosphatase spaltet extrazelluläres Pyrophosphat, wodurch verstärkt freies Orthophosphat für die Bildung
von Hydroxylapatit zur Verfügung steht. Dies hat die Mineralisierung des Knorpels zur Folge und vermindert
somit nicht nur dessen Vitalität sondern trägt auch zum Fortschreiten des Umbaus und der Degradierung
bei (JOHNSON und TERKELTAUB 2005).
Die herabgesetzte Sensitivität der Rezeptoren alternder Chondrozyten gegenüber den Zytokinen IGF-1 und
FGF-2 sowie die verminderte Expression von TGF-β1 führen zu einer verringerten Synthese der ECM und
somit zur Minderung der Knorpeldicke. Da hierbei die Quervernetzungen zwischen den Fasern des Kollagen
Typ II erhalten bleiben, steigt deren Konzentration, wodurch eine höhere Steifigkeit des Gewebes

2 Literaturübersicht
15
hervorgerufen wird (LOTZ und LOESER 2012). Diese Veränderungen haben zur Folge, dass es bereits bei
geringen Belastungen zum Zerreißen der Kollagenfasern kommen kann (LUPPA 2000). Im Gegensatz dazu
bleibt die Empfindlichkeit der BMP-7-Rezeptoren auch in alternden Chondrozyten erhalten, sodass vor
allem der hemmender Effekt des BMP-7 auf die MMP-13 der Degeneration des Knorpels Grenzen setzt
(FORTIER et al. 2011).
Im Rahmen des Alterungsprozesses kommt es auch zu Veränderungen des Aufbaus und der Konzentration
der PGs. Einerseits nimmt die Gesamtkonzentration der PGs und die Größe der Kernproteine ab.
Andererseits verändert sich das Verhältnis der GAG-Seitenketten von längeren, stärker geladenen
Chondroitinsulfat-Resten zu kürzeren und weniger geladenen Keratansulfat-Resten. Diese Entwicklung setzt
bereits nach der Geburt ein und vollzieht sich insbesondere in der ersten Lebenshälfte
(DEGROOT et al. 1999, LUPPA 2000, MALLINGER und STOCKINGER 1987, ROUGHLEY und WHITE 1980). Der
mit diesem Prozess einhergehende Verlust des hydrostatischen Drucks im Gelenkknorpel wird im mittleren
Lebensabschnitt durch eine Zunahme an permeablen Teilchen, die den osmotischen Druck bestimmen,
kompensiert. Im hohen Alter nimmt jedoch auch deren Bildung ab, während gleichzeitig die Permeabilität
der Grenzflächen, insbesondere der Tangentialzone, ansteigt. Dadurch wird der Anteil des hydrostatischen
und osmotischen Druckes zur Kompensation des externen Drucks verringert (LUPPA 2000). Da es im
Vergleich zu den PGs in stärkerem Maß zur Verminderung der Anzahl kollagener Fasern kommt, sinkt der
Spannungsdruck, welcher dem hydrostatischen Druck des an die PGs gebunden Wassers entgegenwirkt. In
der Folge quellen die PGs weiter auf, wodurch die Härte und Elastizität des Knorpels verloren gehen. Der
stark gequollene Zustand der PGs hat einen negativen Effekt auf die Integrität der ECM, sodass die
Knorpeloberfläche aufraut, wodurch der Reibungswiderstand und der Abrieb des Gelenkknorpels ansteigen
(KATTA et al. 2008, LOTZ und LOESER 2012). Die Gesamtheit dieser Seneszenz bedingten Dynamik führt
dazu, dass bereits geringe mechanische Belastungen Schäden des Gelenkknorpels hervorrufen
(LUPPA 2000).
2.4 Defekte und Regeneration des Knorpelgewebes
Entsprechend der Morphologie und Funktion der verschiedenen Knorpeltypen unterscheiden sich sowohl
die Art und Ausprägung der Defekte als auch die Regeneration dieser (LOTZ und LOESER 2012).
2.4.1 Defekte und Regeneration des hyalinen Gelenkknorpels
Defekte des Knorpels werden nach Outerbridge in verschiedene Grade, welche erstmals für das Knie
entwickelt wurden, eingeteilt. Neben dem Grad 0 (Normalbefund), existieren 4 weitere Grade, deren
arthroskopisches Erscheinungsbild mit der Histologie der Defekte korreliert (Tab. 2) (OUTERBRIDGE 1961,
MADRY 2008).
Bei Gelenkknorpelläsionen wird generell zwischen partiellen Gelenkknorpeldefekten (Grad 1-3) und
subchondralen, vollständigen Defekten (Grad 4) unterschieden, da sie sowohl hinsichtlich der Regeneration
als auch hinsichtlich der Therapieempfehlungen differieren. Seit Hunters Feststellung 1743 (HUNTER 1743),
dass ulzerierter Knorpel „sich nicht wieder reparieren lässt, wenn er einmal zerstört ist“, wurde die
begrenzte Kapazität des Gelenkknorpels zur Regeneration vielfach beschrieben (MARLOVITS und VÉCSEI
2000).

2 Literaturübersicht
16
Tab. 2: Outerbridge Klassifikation von Knorpeldefekten, nach Outerbridge (1961), MADRY (2008). Grad Befund
Grad 0 Normalbefund, intakter Knorpel
Grad 1 Oberflächliche Erweichung des Knorpels, bei glatter Oberfläche
Grad 2 Aufgefaserte Oberfläche mit Einrissen <50% der Knorpeldicke
Grad 3 Tiefe Fissuren, Ulcus mit instabilen Rändern >50% der Knorpeldicke
Grad 4 Knorpelverlust bis zum subchondralen Knochen
Bedingt durch die Abwesenheit von Blutgefäßen fehlt die in allen anderen Geweben des Körpers
stattfindende Wundheilung, die durch Nekrose, Entzündung und Reparatur gekennzeichnet ist, so dass die
Regeneration des hyalinen Knorpels einen anderen Verlauf nimmt (MARLOVITS und VÉCSEI 2000).
Bei Mikroverletzungen (Grad 1) kommt es zur Schädigung der Lamina splendens und den Chondrozyten der
Tangentialzone. Durch den daraus resultierenden Verlust der Integrität der Kollagenultrastruktur und damit
der kollagenen Fasern kommt es zum Quellen der PGs und somit zur verstärkten Wassereinlagerung, die
den Knorpel aufweicht. Zudem sind nun die darunter liegenden Strukturen angreifbar für Kollagenasen und
Aggrekanasen. Da durch diese Prozesse die abdichtende Funktion der Tangentialzone gegenüber der
Radiärzone verloren geht, ist die Biodynamik des Gelenkknorpels einschränkt. In der Folge wird der
punktuell auf den Gelenkknorpel wirkende Druck nicht mehr gleichmäßig in diesem und dem
subchondralen Knochen verteilt, wodurch es zur Verdickung der subchondralen Knochenplatte im Bereich
unterhalb der Läsion kommt. Wird die Ursache nicht behoben, schreitet der Prozess in die Tiefe fort und
führt zur Ausbildung von Fissuren oder Ulzera (Grad 2-4) und somit zum Verlust der biomechanischen
Eigenschaften des Gelenkknorpels (ALFORD 2005, BRUNS und STEINHAGEN 2000, MARLOVITS und VÉCSEI
2000).
Im Rahmen von partiellen Gelenkknorpeldefekten (Grad 2-3) kommt es innerhalb der ersten 48-72 Stunden
zu einer Nekrose der Chondrozyten. Anschließend zeigen überlebende Chondrozyten nur für kurze Zeit eine
vermehrte metabolische Aktivität, welche die Bildung von Kollagen Typ II und anderen Bestandteilen der
ECM bedingt, um anschließend wieder auf Normalniveau zurückzukehren. Einige Autoren berichten vom
Auftreten eines fibrösen Narbengewebes im Sinne der Reparatur. Allerdings findet sowohl im Rahmen
dieses Prozesses als auch in der Folge keine Defektregeneration statt, wodurch eine Wiederherstellung der
Integrität des Gelenkknorpels unterbleibt (MEACHIM und ROBERTS 1971, MARLOVITS und VÉCSEI 2000).
Die subchondralen, vollständigen Defekte (Grad 4) überschreiten die Tidemark bis in den subchondralen
Knochen und schaffen dadurch Voraussetzungen für die physiologische Reparatur. Durch die Eröffnung des
subchondralen Knochens kommt es zu einer Einblutung und Bildung eines Fibringerinnsels, welches sich gut
am Knochen anhaftet, jedoch keine Adhärenz zum benachbarten Knorpelgewebe zeigt. In den folgenden
Tagen wandern MSCs aus dem subchondralen Knochen in den Defekt ein (MARLOVITS und VÉCSEI 2000,
SHAPIRO et al. 1993). Da durch die Eröffnung des subchondralen Knochens die Sauerstoffspannung
innerhalb des Defekts ansteigt, entfällt der hemmende Einfluss von HIF-1α auf die Proliferation
(DAS et al. 2010). Dadurch sind die sich aus den MSCs differenzierenden Chondroprogenitorzellen in der
Lage, den Defekt innerhalb der nächsten Tage vollständig aufzufüllen und das Fibringerinnsel zu
resorbieren. Im weiteren Verlauf differenzieren diese zu Chondrozyten, die sich stellenweise in
Chondrozytenclustern konzentrieren, andere Bereiche jedoch völlig azellulär lassen. Die Chondrozyten
produzieren PGs sowie Kollagen Typ I und II. Der daraus resultierende Knorpel wird als „hyaline-like“
bezeichnet und füllt nach ca. einem halben Jahr den gesamten Defekt aus (MARLOVITS und VÉCSEI 2000,

2 Literaturübersicht
17
SHAPIRO et al. 1993). Dieser aus der Orthopädie stammende Begriff ist nicht einheitlich definiert (MADRY
und PAPE 2008). Einige Autoren beschreiben damit arthroskopisch und funktionell festen Gelenkknorpel,
der jedoch histologisch die Kriterien des Faserknorpels erfüllt; andere nutzen den gleichen Begriff für
Knorpel, der histologisch dem hyalinen Knorpel sehr ähnlich ist. Vom hyalinen Knorpel unterscheidet ihn
der höhere Anteil an Kollagen Typ I-Fasern sowie der zonale histologische Aufbau (GOYMAN 1999,
KRASNICI et al. 2000). An Stelle einer Integration des neu gebildeten Knorpels in den bestehenden Knorpel
wurde das „Cartilage-flow-Phänomen“ beobachtet, bei dem sich die Defektränder abrunden und gleichsam
in den Defekt „fließen“ (BRUNS und STEINHAGEN 2000). Zwischen neu gebildetem und vorhandenem
Knorpel verbleibt immer ein elektronenmikroskopisch nachweisbarer Spalt, sodass keine Druckverteilung
im Rahmen der Biodynamik des Gelenkknorpels mehr stattfinden kann (MARLOVITS und VÉCSEI 2000,
SHAPIRO et al. 1993). Aus diesem Grund entsteht eine Überlastung, sodass der neu gebildete Knorpel
bereits nach einigen Monaten erste Zeichen der Degeneration erfährt. Die Oberfläche sinkt ein und
splittert; es kommt zur Freilegung der Fasern (Fibrillierung). Nachfolgend erfolgt der Umbau des Gewebes,
welches schließlich der Zusammensetzung und Qualität des Faserknorpels entspricht. Allerdings ist dieser
biomechanisch auf die Kompensation von Zugkräften ausgelegt und weist in Bezug auf
Druckkraftübertragung und Haltbarkeit gegenüber Scherkräften erhebliche Defizite gegenüber dem
hyalinen Knorpel auf. Die Belastung dieses Ersatzgewebes hat wiederum die Entstehung neuer Läsionen zur
Folge (BRUNS und STEINHAGEN 2000, MARLOVITS und VÉCSEI 2000, SHAPIRO et al. 1993).
Im juvenilen Organismus ist ein anderer Verlauf der Defektheilung zu beobachten. Oberflächliche Defekte
zeigen hier eine spontane Heilung. Eine Erklärung hierfür liefern die bei juvenilen Tieren vorhandenen
Chondroprogenitorzellen in der Tangentialzone. Im Gegensatz zu Chondrozyten besitzen diese Zellen die
Fähigkeit zur Proliferation und zur Neochondrogenese (FRITSCH et al. 1999, HOLLANDER et al. 2010). WEI
und MESSNER (1998) zeigten in einem Kaninchenmodell mit tiefen Knorpeldefekten, dass die Konzentration
von TGF-β1 in der Synovia juvenil höher ist als adult. Die Wirkung von TGF-β1 auf Chondroprogenitorzellen
der Tangentialzone bewirkt eine schnelle Auffüllung des Defekts mit „hyaline-like“ Knorpel. Zusätzlich
zeigten juvenile Tiere immer eine vollständige Integrität zum umliegenden Knorpel. Der Einfluss des TGF-β1
auf die Chondrozyten des an den Defekt angrenzenden Knorpelgewebes führt jedoch langfristig in diesem
Bereich zu einer starke Fibrillierung und Osteophytenbildung (WEI et al. 1997).
Das Langzeitresultat aus Knorpeltraumata sowie aus Überbelastung und Gelenksentzündungen stellt die
Osteoarthrose (OA) dar. Diese tritt häufig in Kombination mit weiteren prädisponierenden Faktoren wie der
Seneszenz des Gelenkknorpels oder Diabetes auf (LOTZ und LOESER 2012).
Der Prozess der OA im Gelenkknorpel beginnt mit dem Abbau der aggregierten PGs, wodurch der
hydrostatische Druck sinkt und der osmotische Druck steigt. Da im Rahmen der Überbelastung des Knorpels
zusätzlich die Integrität der Tangentialzone verloren geht, fehlt ihre abdichtende Funktion zur Radiärzone.
Dadurch diffundieren bei Belastung des Knorpels die nicht aggregierten PGs in die Synovia und gehen für
eine erneute Aggregation verloren. Somit wird die ursprüngliche Knorpelhöhe nach Entlastung und
Rückdiffusion des Wassers nicht mehr erreicht. Folgen erneute Überbelastungen schnell aufeinander,
nimmt die Knorpeldicke kontinuierlich ab und der Prozess schreitet bis zum subchondralen Knochen fort
(ASPBERG 2012, LUPPA 2000).
Die aus diesem Vorgang resultierende Verringerung der PG-Konzentration im Gelenkknorpel führt zur
Minderung der Hydrathülle der kollagenen Fasern. Auf diese Weise freigelegtes Kollagen wird für
Kollagenasen, hauptsächlich MMP-13, angreifbar. Da hierbei vorrangig Kollagen Typ IX und XI abgebaut

2 Literaturübersicht
18
werden, nimmt der Grad der Quervernetzung ab und es entsteht eine zusätzliche Destabilisierung des
Kollagenfasernetzes. Dadurch sinkt der Spannungsdruck der kollagenen Fasern ab, wodurch die PGs
aufquellen und zur Aufweichung des Knorpels führen. Schreitet der Abbau des Knorpels durch Kollagenasen
und Aggrekanasen fort, verändern sich die Chondrozyten zu so genannten OA-Chondrozyten, welche
gemäß ihres Funktionszustands als „senescense-associated secretory phenotype“ (SASP) bezeichnet
werden (TROEBERG und NAGASE 2012, VERMA und DALAL 2011). Diese Zellen geben nun pro-
inflammatorische Zytokine (SASP-Faktoren) ab, wie beispielsweise IL-1, IL-6, IL-7, IL-8 und TNF-α, welche
einen Entzündungsprozess hervorrufen und aufrechterhalten (COUCHMAN und PATAKI 2012, FORTIER et
al. 2011). Das Ausmaß der daraus resultierenden Synovitis gilt als Maß für die Schwere und die Progression
der OA (COUCHMAN und PATAKI 2012).
Erreicht der Prozess den subchondralen Knochen, reagiert dieser mit einem belastungsassoziierten
Knochenumbau, der zur Sklerose des subchondralen Knochens und zur Bildung von Osteophyten führt. Der
fehlenden Innervation des hyalinen Knorpels ist der Umstand geschuldet, dass die pathologischen
Veränderungen erst zu diesem Zeitpunkt klinisch evident werden, da sie nun Schmerzen verursachen
(GOLDRING 2012, LOTZ und LOESER 2012, MARTIN und BUCKWALTER 2002, TROEBERG und NAGASE 2012).
2.4.2 Defekte und Regeneration des Faserknorpels
Defekte des Faserknorpels, zumeist Risse entlang des Faserverlaufs der Kollagenfasern, entstehen in der
Regel durch hohe Krafteinwirkungen, die nicht in der Hauptbelastungsrichtung auftreten. Da der
Faserknorpel im Gegensatz zum hyalinen Knorpel sensibel innerviert ist, werden Defekte des Faserknorpels,
wie beispielsweise Meniskusrisse, sofort wahrgenommen. Die Vaskularisierung des Faserknorpels ist direkt
korreliert mit seiner Heilungskapazität (MAKRIS et al. 2011). Dabei sind gut vaskularisierte Bereiche in der
Lage eine normale Wundheilung (Nekrose, Entzündung und Reparatur) zu durchlaufen und ein
funktionsfähiges Gewebe in Form von Faserknorpel zu bilden. In avaskulären Bereichen ist die
Regenerationskapazität eingeschränkt. Das hier entstehende Ersatzgewebe weist für den Faserknorpel eine
zu hohe Konzentration der kollagenen Fasern auf (MAKRIS et al. 2011, STÄRKE 2008).
2.4.3 Defekte und Regeneration des elastischen Knorpels
Im Gegensatz zu den Defekten der bisher genannten Knorpelarten, die in der Regel durch Überlastung
entstehen, handelt es sich bei solchen des elastischen Knorpels um neoplastische Prozesse oder
angeborene Anomalien (Anotie, Mikrotie).
Die wenigen, hoch differenzierten Chondrozyten, der langsame Umsatz der ECM und die kaum
vorhandenen Progenitorzellen schränken die intrinsische Regenerationsfähigkeit des elastischen Knorpels
ein. Das Fehlen einer Vaskularisierung ist wie im hyalinen Knorpel für das Ausbleiben der Wundheilung
verantwortlich, so dass die Regeneration des elastischen Knorpels mit der Bildung von Faserknorpel
einhergeht (CIORBA und MARTINI 2006, MARLOVITS und VÉCSEI 2000, STERODIMAS et al. 2009).
2.5 Strategien zur Behandlung von Knorpeldefekten
Bereits 1941 wurde von P.B. Magnuson das chirurgische Debridement („Shaving“) klinisch angewendet, bei
dem die Glättung der Knorpeloberflächen am Rand der Läsion im Vordergrund steht, wodurch die
mechanische Irritation und folglich die Entzündung reduziert, jedoch keine Reparatur erreicht wird (HORAS
et al. 2000). Seither sind viele konservative und chirurgische Therapieansätze mit unterschiedlichen
Resultaten verfolgt worden (HORAS et al. 2000). Allein deren Vielzahl lässt Rückschlüsse auf die

2 Literaturübersicht
19
sozialmedizinische Bedeutung der Knorpelläsionen und der mit ihnen einhergehenden Arthrose zu. Das
Statistische Bundesamtes ermittelte für das Jahr 2002, dass 2% aller Krankenhausaufenthalte des
Menschen durch die Diagnose Arthrose, davon etwa 50% Gonarthrose und 40% Coxarthrose, verursacht
worden (MERX et al. 2007). Auch etwa 20% aller adulten Hunde werden mit dieser Diagnose auffällig. Bei
alten Hunden (>8 Jahren) wird sie sogar zu 90% gestellt, wobei der Anteil klinisch kranker Hunde weit
darunter liegt (DYCUS et al. 2013). Epidemiologische Daten zeigen für das Auftreten der Osteochondrose,
einer Erkrankung, die mit Knorpeldefekten einhergeht, beim Pferd eine Inzidenz von 10-25% (JEFFCOTT
1996). Bislang gibt es kein Verfahren, das sich aufgrund des Therapieerfolgs durchsetzen konnte. Daher
beschränken sich die Therapieverfahren zur Behandlung von Knorpeldefekten, von denen die wichtigsten
im Folgenden vorgestellt werden, derzeit auf die Verbesserung der Schmerzsymptomatik und die Erhaltung
der physiologische Bewegungsfreiheit (FRITSCH et al. 1999).
2.5.1 Knochenmarkstimulierende Verfahren
Ziel dieser Gruppe von Verfahren ist die Eröffnung der subchondralen Knochenplatte, um das Einwandern
von MSCs zu ermöglichen. Man unterscheidet Bohrung, Abrasion und Mikrofrakturierung, wobei letztere
arthroskopisch durchgeführt werden kann. Die durch die gesetzten Defekte eingewanderten MSCs bilden
ein den Defekt füllendes Ersatzgewebe, welches die für den Faserknorpel typischen Eigenschaften der
Zugfestigkeit besitzt, jedoch nicht die für den Gelenkknorpel typische Widerstandsfähigkeit gegenüber
Druck und Scherkräften aufweist. Da sich jedoch durch die mechanischen und funktionellen Defizite des
Faserknorpels nur kurzzeitig eine Linderung der Symptome einstellt, werden diese Verfahren kontrovers
diskutiert. Zur Optimierung und Weiterentwicklung dieser Verfahren erfolgen derzeit Untersuchungen über
den Effekt einer Kollagenmembran, welche, über dem Defekt aufgebracht, das Abschwimmen der MSCs
nach dem Eingriff verhindern soll (MARTINEK und IMHOFF 2003).
2.5.2 Autologe Knorpelknochentransplantation
Bei der autologen Knorpelknochentransplantation (OCT) wird der geschädigte Knorpel bis tief in die
subchondrale Knochenplatte entfernt. Anschließend werden aus einer nicht gewichttragenden Stelle
desselben Gelenks oder eines anderen Gelenks ein oder mehrere zylindrische Transplantate mit ihrer
knöchernen Grundlage entnommen und in die Defektstelle eingesetzt. Aufgrund der Defekte an der
Spenderstelle bleiben solche Transplantate auf kleine Flächen von 2-3 cm2 beschränkt (WINDHAGER et al.
2004). Die Transplantate sollten die gleiche Größe und Oberflächenneigung wie die Defektstelle aufweisen.
In der Regel unterbleibt auch ein Vernähen, um störende Oberflächen zu vermeiden. In der ersten Phase,
in der die Transplantate am subchondralen Knochengewebe anwachsen, weist das in den Transplantaten
enthaltene Knorpelgewebe eine vollständige Funktionalität auf (HORAS et al. 2000). Allerdings werden im
weiteren Verlauf Wachstums- und Differenzierungsfaktoren in zu geringem Maße exprimiert, so dass die
Chondrozyten der Transplantate inaktiv bleiben. Eine Vermehrung der Chondrozyten unterbleibt,
verhindert somit die vollständige Integration der Knorpel-Knochen-Stanze in die umgebende Knorpelmatrix
und erschwert deren Nährstoffversorgung (FRITSCH et al. 1999). Stattdessen werden die Zwischenräume
durch Faserknorpel aufgefüllt, der die Integration in die Biodynamik des Gelenkknorpels verhindert. Den
biomechanischen Anforderungen ist die Stanze langfristig nicht gewachsen, sodass es zur Transformation
des Transplantats zu Faserknorpel kommt. Da dieser in seinen biomechanischen Eigenschaften den
Kompressionskräften nicht gewachsen ist, kommt es zur arthrotischen Veränderung des Gelenks
(BUCKWALTER und MANKIN 1998, FRITSCH et al. 1999).

2 Literaturübersicht
20
Die Probleme dieses Verfahrens entstehen durch die Langzeitfolgen, die durch den Entnahmedefekt
verursacht werden, sowie durch die Schwierigkeit, das Transplantat ohne Kantenbildung in die Umgebung
einzupassen. Zudem verhindert die Inaktivität der transplantierten Chondrozyten die Langzeitstabilität der
Transplantate (MARTINEK und IMHOFF 2003).
2.5.3 Autologe Knorpeltransplantation
Die autologe Knorpeltransplantation (ACT) wurde 1994 in Schweden entwickelt (BRITTBERG et al. 1994).
Die ACT verläuft in 3 Schritten. In einer ersten Operation wird eine Knorpelbiopsie an einer nicht Gewicht
tragenden Stelle des Gelenkknorpels entnommen. Aus der Knorpelbiopsie werden die Chondrozyten isoliert
und 11-21 Tage in Monokultur bis zur zehnfachen Konzentration expandiert. Anschließend werden die
Zellen durch Trypsinierung gelöst und in Suspension überführt. In einer zweiten Operation wird im Bereich
der Knorpelläsion das Defektmaterial entfernt. Es wird ein Stück Periost entnommen und als autologer
Periostlappen über dem Defekt fest vernäht und dicht verklebt. Mit Hilfe einer Nadel wird die
Zellsuspension (2,6x106 - 5x106 Zellen) unter den Periostlappen injiziert. Die Chondrozyten setzen sich
innerhalb von 24-48 Stunden am Grund des Defekts ab, dedifferenzieren und bilden in dieser biologischen
Kammer ein Knorpelregenerat (BRITTBERG et al. 1994, ERGGELET 2003). Nach der Adhärenz der
transplantierten Chondrozyten proliferieren und redifferenzieren diese unter dem Einfluss der aus dem
Periostlappen stammenden Wachstums- und Differenzierungsfaktoren. Diese Faktoren sorgen für die
Synthese, Sekretion und Organisation der ECM des neu gebildeten Knorpels bis zur Auffüllung des Defekts
(FRITSCH et al. 1999). Dieses aufwändige Verfahren ist bislang die Methode, welche die stärkste Linderung
der Symptomatik zur Folge hat. Allerdings entspricht die Qualität des Regenerats nur in 50% der Fälle den
Anforderungen an einen hyalinen Knorpel (MARTINEK und IMHOFF 2003). Das Verfahren wird mittlerweile
kommerziell angeboten und aufgrund seines vielversprechenden Ansatzes stetig weiterentwickelt.
Die Matrix assoziierte autologe Knorpeltransplantation (MACT) stellt eine dieser Modifikationen dar.
Hierbei werden mit Hilfe des Tissue Engineering Biomaterialien als Träger und Matrizen verwendet, die in
den Defekt eingenäht werden können (MARLOVITS et al. 2004). Die verschiedenen Materialien liefern hier
auch unterschiedliche Ergebnisse. Fibrin-Kleber, der gute Bedingungen für die Matrixsynthese der
Chondrozyten in den ersten Wochen schafft, bleibt später nicht lange genug stabil (VANSUSANTE et al.
1998). Hydroxyapatit als fest einnähbares Trägermaterial führte initial zur Bildung von „hyaline-like“
Knorpel, lockerte sich jedoch im weiteren Verlauf vom knöchernen Untergrund (VANSUSANTE et al. 1998).
Erste Versuche der Nutzung von Biomembranen aus Kollagen Typ I, Hyaluronsäure-Vlies oder Kollagen Typ
I-Gel zeigten dagegen gute Transplantatintegration und Defektauffüllung mit hyalinem Regeneratgewebe
(MARLOVITS et al. 2004). Im Gegensatz zur ACT können bei der MACT nicht unbegrenzt viele Zellen
eingebracht werden, da die Zahl der Chondrozyten, die auf dem Trägermaterial angezüchtet werden
können (106Zellen/cm2), limitiert ist (BENTLEY et al. 2013). Trotzdem zeigten BENTLEY et al. (2013) ähnliche
Heilungsverläufe (bis 1 Jahr) bei kürzerer Operationszeit und geringerer Invasivität, da die verwendeten
porcinen Kollagen-I/III-Membranen den Periostlappen entbehrlich machen. Die Nutzung von Biomaterialien
macht die Verwendung von MACT für große Defekte möglich, wenngleich die Autoren selbst das Fehlen von
Langzeitstudien bemängeln (BENTLEY et al. 2013, ZEIFANG et al. 2010). Die Angaben über die Morphologie
und Stabilität der ACT und MACT variieren je nach Autor. FRITSCH et al. (1999) beschrieben 1999 ein
gebildetes Knorpelregenerat gleicher „Struktur, Zusammensetzung, Druckfestigkeit“ wie der
Gelenkknorpel, eine defektfreie Integration in die Umgebung und eine gute biomechanische Belastbarkeit

2 Literaturübersicht
21
über Jahre und verweisen auf Studien mit 70-96% Behandlungserfolg. ZEIFANG et al. (2010) dagegen sehen
2 Jahre nach Behandlung keinen Unterschied zwischen dem Verfahren der Mikrofrakturierung und ACT
bzw. MACT. In der Analyse von Studien, welche die Qualität von Knorpelreparatur untersuchen, ist deren
hohe Variabilität methodischer Kriterien aufgefallen, sodass JAKOBSEN et al. (2005) zu dem Ergebnis
kommen, dass diese der vorsichtigen Interpretation bedürfen und Vergleiche untereinander schwierig sind,
solange nicht einheitliche Methoden zum Einsatz kommen.
2.6 In-vitro-Kultivierung von Chondrozyten
Die Häufigkeit des Einsatzes isolierter und in-vitro kultivierter Chondrozyten zeigt seit nunmehr zwei
Jahrzehnten eine steigende Tendenz. Diese werden einerseits für die Untersuchung der molekularen
Mechanismen und der Dynamik der Chondrozyten bzw. des Knorpelgewebes genutzt, da in vitro-Systeme
den Vorteil einer einfachen Standardisierung und Durchführbarkeit bieten. Ein weiteres Einsatzgebiet
in-vitro kultivierter Chondrozyten liegt in ihrer Verwendung zur Transplantation im Rahmen der
regenerativen Therapie von Knorpeldefekten. Für die In-vitro-Kultivierung von Chondrozyten werden
Systeme mit atmosphärischen und modifizierten Druckbedingungen verwendet, welche die Morphologie
und Funktion der Zellen auf unterschiedliche Weise beeinflussen (GUILAK und MOW 2000, HARDMEIER et
al. 2009, WONG und CARTER 2003).
2.6.1 Einfluss der In-vitro-Kultivierung auf Chondrozyten
Die Isolierung und In-vitro-Kultivierung von Chondrozyten führt bereits in der ersten Passage der
Primärkultur zu stark miteinander korrelierenden morphologischen und funktionellen Veränderungen der
Zellen, welche einer Dedifferenzierung entsprichen. Dieser Effekt tritt sowohl bei artikulären Chondrozyten,
als auch bei solchen aus Faserknorpel und elastischem Knorpel auf (MABVUURE et al. 2012, SCHULZE-
TANZIL 2009).
In-vitro-kultivierte Chondrozyten bilden nach erfolgter Adhärenz Zell-Zell-Kontakte aus. Im Gegensatz zur in
vivo-Situation, wo diese Kontakte durch die Zunahme der ECM wieder zurückgebildet werden, bleiben
diese in vitro auch bei längerer Kultivierung erhalten. Dies liegt darin begründet, dass ein Großteil der von
den Chondrozyten in vitro produzierten ECM im Rahmen des regelmäßigen Mediumwechsels entfernt wird.
Dadurch kommt es zu einer Veränderung im Aufbau und in der Ausrichtung des Zytoskeletts sowie einer
Zunahme so genannter Stressfasern. In Folge dessen weisen Chondrozyten in vitro nicht die in vivo
charakteristische runde sondern eine lang gestreckte Form auf (SCHULZE-TANZIL 2009).
Die funktionellen Veränderungen der Chondrozyten beziehen sich auf den Grad ihrer Differenzierung,
welche dem des Chondroprogenitor-Stadiums entspricht, und das daraus resultierende Syntheseprofil
(SCHULZE-TANZIL 2009). Die Synthese der ECM der Chondrozyten in vitro ist in Richtung einer deutlich
stärkeren Sekretion des Kollagen Typ I, sowie kleinerer (z.B. Biglycan) oder weniger aggregierter (z.B.
Versikan) PGs verschoben (HARDMEIER et al. 2009, ZEIFANG, 2010). Die Gene für Kollagen Typen II und IX,
sowie solche für PGs, die auf Aggrekan als Kernprotein basieren, sind dagegen herunterreguliert (SCHULZE-
TANZIL 2009, ZEIFANG, 2010). So wird als Index für die Dedifferenzierung in vitro das Verhältnis von
Kollagen Typ I zu Typ II, ebenso wie von Aggrekan zu Versikan herangezogen (BARLIČ et al. 2008).
Bei der In-vitro-Kultivierung von Chondrozyten werden verschiedene Strategien eingesetzt, um der
zellulären Dedifferenzierung entgegenzuwirken bzw. eine Redifferenzierung zu erreichen (BRITTBERG 1994,
ERGGELET 2003, FRITSCH et al. 1999). Erstere besteht in der Aussaat der Zellen in sehr hoher Dichte,
welche eine niedrige TGF-1-Expression der Chondrozyten und eine daraus resultierende höhere

2 Literaturübersicht
22
Empfindlichkeit der Zellen gegenüber IGF-1 und FGF-2 zur Folge hat (SCHULZE-TANZIL 2009). Die weitere
Maßnahme besteht in der Zugabe dieser Zytokine zum Kultivierungsmedium. Dadurch werden die
Chondrozyten durch IGF-1 zur Synthese und Sekretion von Kollagen Typ II, IX und XI und durch FGF-2 zur
Expression von Aggrekan stimuliert (GOLDRING et al. 2006, GOLDRING 2012).
2.6.2 Einsatz von Druck bei der In-vitro-Kultivierung von Chondrozyten
Durch die belastungsbedingte Umverteilung von Wasser innerhalb des Gelenkknorpels erfahren auch die in
ihm enthaltenen Chondrozyten diese einwirkenden mechanischen Kräfte, wodurch ihre
Mechanorezeptoren, die „connexin hemichannels“, stimuliert werden und sich ihre metabolische Aktivität
sowie damit die Zusammensetzung der von ihnen gebildeten ECM verändert (BUCKWALTER et al. 2005,
GARCIA und KNIGHT 2009, KNIGHT et al. 2009, WHITFIELD 2008, WONG und CARTER 2003).
Daher erfolgt die In-vitro-Kultivierung von artikulären Chondrozyten häufig in Bioreaktoren, um
Druckbelastungen zu schaffen und die durch diese ausgelösten Effekte nutzbar zu machen. Analog zu den
Voraussetzungen in vivo werden bei der In-vitro-Kultivierung der Chondrozyten in Bioreaktoren
diskontinuierliche (intermittierende), in Zyklen ablaufende (dynamische) von kontinuierlichen, über einen
gewissen Zeitraum konstant einwirkenden (statischen) Druckbedingungen unterschieden. Grundsätzlich
werden Bioreaktoren in vier Typen unterteilt, welche einerseits Scherkraft, Perfusionsdruck oder
hydrostatischen Druck übertragen oder andererseits eine direkte Druck- bzw. Kraftübertragung auf die
Zellen bewirken (SCHULZ und BADER 2007). Bioreaktoren, die Scherkräfte durch die Flüssigkeitsbewegung
des Mediums erzeugen (Spinner-flask, rotierende Bioreaktoren), finden Anwendung im Bereich des Tissue-
Engineering und der Kultivierung von Knorpelexplantaten (NEBELUNG et al. 2011). Gleiches gilt für das in
der Gewebekultur verwendete Verfahren der direkten Druckübertragung, das für die Untersuchung von
Chondrozyten in 3-D-Kultur (z.B. in Hydrogelen) eingesetzt wird. Hier wird mittels eines Stempels ein
direkter Druck erzeugt (NEBELUNG et al. 2011, SCHULZ und BADER 2007).
Für in-vitro-kultivierte Chondrozyten hat sich die Applikation von hydrostatischem Druck, repräsentativ für
die Druckverhältnisse in der Radiärzone herauskristallisiert. Hierbei wird zwischen zwei Bioreaktor-Typen
unterschieden. In den einen wird der Druck durch Kompression einer Gasphase mit entsprechender
Weiterübertragung in das die Zellen umgebende Medium aufgebaut. Zum anderen besteht die Möglichkeit,
das Medium direkt zu komprimieren. In einem Bioreaktorsystem sind Höhe, Zeitdauer und Frequenz des
einwirkenden Drucks die beeinflussbaren Variablen (SCHULZ und BADER 2007).
In den Studien der gegenwärtigen Literatur variieren die Angaben zu diesen Parametern nicht nur, sondern
widersprechen sich auch in einigen Fällen (ELDER und ATHANASIOU 2009). So zeigten IKENOUE et al. (2003)
bei unter diskontinuierlichem Druck kultivierten Chondrozyten über vier Stunden für vier Tage eine
Steigerung des Aggrekan- und Kollagen Typ II-Gehalts auf der Ebene des Transkripts, während sie bei einem
Tag keinen signifikanten Anstieg feststellten. SUH et al. (1999) dagegen wiesen bei annähend ähnlichen
Bedingungen bereits nach 6 Stunden einen Anstieg von Aggrekan um 40% nach. Einige Untersuchungen
führten zu dem Ergebnis, dass kontinuierlicher hydrostatischer Druck keinen bzw. einen negativen Einfluss
auf die mRNA-Synthese von Aggrekan und Kollagen Typ II hat, wohingegen diskontinuierlicher
hydrostatischer Druck günstige Effekte zeigte (ELDER und ATHANASIOU 2009, SMITH et al. 2000). Dagegen
erzielten TAKAHASHI et al. (1997) bei kontinuierlichem hydrostatischem Druck für 2 Stunden einen Anstieg
der PG-Konzentration. Auch in 3-D-Kulturen aus bovinen Chondrozyten in Agarosegel wurde bei
kontinuierlichem Druck ein Anstieg der Aggrekan- und Kollagen II-mRNA nachgewiesen. Dabei fiel auf, dass

2 Literaturübersicht
23
Effekte bezüglich der Konzentration der PGs in der Regel deutlich stärker ausfallen und schneller erreicht
werden als solche auf die Expression von Kollagen Typ II (TOYODA et al. 2003).
Unter dem Einfluss verschiedener diskontinuierlicher Druckbedingungen wiesen SMITH et al. (2000) bei
100 bar und 1 Hz sowie einer Einwirkungszeit von bis zu 24 Stunden einen Anstieg der GAG-Konzentration
nach. Eine Erhöhung der Expression der Kollagen Typ II-mRNA wurde allerdings nur bei einer Einwirkzeit
von 4 und 8 Stunden festgestellt. In vergleichenden Studien juveniler und adulter equiner Chondrozyten
wurden diese über einen Zeitraum von 5 Wochen kultiviert. Dabei wurden sie einem Druck von 34,4 oder
68,7 bar und einer Frequenz von 0,25 Hz für 20 Minuten alle 4 Stunden ausgesetzt. Hierbei zeigte sich in
den adulten Kulturen bei beiden Druckhöhen eine Zunahme der Konzentration der GAGs und Kollagen Typ
II. Unter dem Einsatz gleicher Kultivierungsbedingungen wurden bei den juvenilen Chondrozyten nur bei
höherem Druck (68,7 bar) die gleichen Effekte beobachtet. Der niedrigere Druck (34,4 bar) führte dagegen
zu einer singulären Erhöhung der GAG-Konzentration und zeigte keine Effekte auf die Kollagen Typ II-
Produktion der Chondrozyten (CARVER und HEATH 1999a, CARVER und HEATH 1999b).
ELDER et al. (2008) untersuchten die Effekte des diskontinuierlichen und kontinuierlichen Drucks (10, 50
und 100 bar) von täglich einer Stunde auf ein 3-D-Agarosegel-Konstrukt in verschiedenen Zeiträumen.
Dabei stellten sie fest, dass bei allen Bedingungen eine Zunahme der Synthese der GAGs zu verzeichnen
war. Ein Anstieg der Kollagenproduktion und damit der Zugsteifigkeit des Gel-Konstrukts wurde hingegen
nur bei kontinuierlichem Druck von 50 und 100 bar beobachtet (ELDER et al. 2008).

3 Zellen, Materialien und Methoden
24
3 Zellen, Materialien, Methoden
3.1 Zellen und für deren Kultivierung verwendete Lösungen
Bei den in dieser Arbeit verwendeten Zellen handelt es sich um eine Primärkultur von Chondrozyten des
humanen Gelenkknorpels (Lonza, Basel, Schweiz). Diese Zellen wurden aus dem Kniegelenk eines 50 Jahre
alten männlichen Spenders isoliert (Chargennummer: 5F1452). Sie wurden in der Passage 2 mit einer
Zellzahl von 7,85 x105 Zellen/ml geliefert, wobei der Hersteller die maximal nutzbare Passage auf 15
begrenzte. Entsprechend der Analyse des Herstellers zeigen die Zellen eine Effizienz der Aussaat von 91 %,
eine Verdopplungszeit von 30 h und eine Lebensfähigkeit von 86%. Die Primärkultur wurde seitens des
Herstellers auf Sterilität und das Freisein von Mykoplasmen, HIV, HBV, HCV getestet. Als Kriterium zur
Identifikation der isolierten Zellen als Chondrozyten sollte eine an den Zellen durchgeführte Kollagen
Typ II-Färbung und eine Alzianblau-Färbung zu mehr als 60% positiv ausfallen. Dieses Kriterium wurde
durch die isolierten Chondrozyten dieser Charge erfüllt.
3.1.1 Chemikalien und Lösungen für die Zellkultur
Lösung Hersteller
Chondrozyten Basal Medium (CBM) Lonza, Basel, Schweiz
Dimethylsulfoxid (DMSO) AppliChem GmbH, Darmstadt
Dulbecco's Phosphatgepufferte Kochsalzlösung 1x (DPBS) Gibco life technologies, Darmstadt
Fetales Bovines Serum (FBS) Lonza, Basel, Schweiz
Gentamicinsulfat/Amphotericin-B Lonza, Basel, Schweiz
Hank's Salzlösung (HBSS) Lonza, Basel, Schweiz
Insulin Lonza, Basel, Schweiz
Long-R3 Insulin-like Growth Factor 1 (IGF-1) Lonza, Basel, Schweiz
r-human Fibroblast Growth Factor B (FGF-2) Lonza, Basel, Schweiz
Transferrin Lonza, Basel, Schweiz
Trypsin Neutralisationlösung (TNS) Lonza, Basel, Schweiz
Trypsin-EDTA Lonza, Basel, Schweiz
3.1.2 Zusammensetzung des Kulturmediums
Das Basalmedium CBM bildet zusammen mit den Zusätzen (Chondrocyte Growth Medium Single Quots) das
Chondrozytenwachstumsmedium (CBM+). Die Zusätze wurden hierfür aliquotiert, bei -20 °C gelagert und
dem Medium direkt vor dem Gebrauch zugesetzt.
Komponente Menge
CBM 50 ml
FBS 2,5 ml
FGF-2 250 µl
Gentamicinsulfat/Amphotericin-B 50 µl
Insulin 100 µl
IGF-1 100 µl
Transferrin 50 µl

3 Zellen, Materialien und Methoden
25
3.1.3 Zusammensetzung des Einfriermediums
Komponente Menge
CBM+ 8 ml
DMSO 2 ml
3.1.4 Zusammensetzung des Spezialgasgemisches für die Kultivierung der Chondrozyten unter
Druck
Faktor Menge
Kohlenstoffdioxid (CO2) 5 %
Synthetische Luft ad 100 %
3.2 Materialien
3.2.1 Chemikalien und Reagenzien
Stoff Hersteller
1-Bromo-3-chloropropan Sigma-Aldrich GmbH, Taufkirchen
Acrylamid Carl Roth GmbH & Co. KG, Karlsruhe
Agar-Agar Kobe I Carl Roth GmbH & Co. KG, Karlsruhe
Agarose Carl Roth GmbH & Co. KG, Karlsruhe
Ammoniumpersulfat (APS) Sigma-Aldrich GmbH, Taufkirchen
Aquaclean Karl Hecht GmbH & Co. KG, Sondheim
β-Mercaptoethanol Carl Roth GmbH & Co. KG, Karlsruhe
Bicinchoninsäure-Protein Assay Kit Sigma-Aldrich GmbH, Taufkirchen
Bovines Gamma Globulin Sigma-Aldrich GmbH, Taufkirchen
Bovines Serum Albumin (BSA), Fraktion V Carl Roth GmbH & Co. KG, Karlsruhe
Bromphenolblau Carl Roth GmbH & Co. KG, Karlsruhe
Chloroform Sigma-Aldrich GmbH, Taufkirchen
Coomassie Brilliant Blau G250 Carl Roth GmbH & Co. KG, Karlsruhe
Coomassie Brilliant Blau R250 Carl Roth GmbH & Co. KG, Karlsruhe
Diethylpyrocarbonat (DEPC) AppliChem GmbH, Darmstadt
Essigsäure Carl Roth GmbH & Co. KG, Karlsruhe
Ethanol absolut (96 %) RNase/DNase frei AppliChem GmbH, Darmstadt
Ethidiumbromid 1% AppliChem GmbH, Darmstadt
Ethylendiamintetraacetat (EDTA) Carl Roth GmbH & Co. KG, Karlsruhe
Glycerol Carl Roth GmbH & Co. KG, Karlsruhe
Glycin Carl Roth GmbH & Co. KG, Karlsruhe
Hämatoxylin-Lösung nach Gill II Dr. K. Hollborn & Söhne GmbH & Co. KG, Leipzig
Hefeextrakt Carl Roth GmbH & Co. KG, Karlsruhe
Histokit Carl Roth GmbH & Co. KG, Karlsruhe
Isopropylalkohol Sigma-Aldrich GmbH, Taufkirchen
Kohlenstoffdioxid (CO2) Linde AG, München
Luminol, ECL Crescendo Merck KGaA, Darmstadt
Methanol Carl Roth GmbH & Co. KG, Karlsruhe
Natriumazid Carl Roth GmbH & Co. KG, Karlsruhe
Natriumchlorid Carl Roth GmbH & Co. KG, Karlsruhe
Natriumdodecylsulfat (SDS) Merck KGaA, Darmstadt

3 Zellen, Materialien und Methoden
26
Stoff Hersteller
N,N,N′,N′-Tetramethylethylendiamin (TEMED) Sigma-Aldrich GmbH, Taufkirchen
NP-40 Sigma-Aldrich GmbH, Taufkirchen
Orange G Sigma-Aldrich GmbH, Taufkirchen
Ponceau S Sigma-Aldrich GmbH, Taufkirchen
Proteaseinhibitor Cocktail Set Merck KGaA, Darmstadt
Salzsäure (HCl) VWR International GmbH, Darmstadt
Stickstoff Linde AG, München
Strep-ABC-Lösung, Vectastain ABC Kit Vector Laboratories, Burlingame, USA
SybrGreen I Invitrogen/Life Technologies GmbH, Darmstadt
TriFast peqGOLD PEQLAB Biotechnologie GmbH, Erlangen
Trishydroxymethylaminomethan (Tris) Sigma-Aldrich GmbH, Taufkirchen
Tris-hydrochlorid (Tris-HCl) Sigma-Aldrich GmbH, Taufkirchen
Tris-EDTA (TE)-Puffer (1X) pH 8,0 AppliChem GmbH, Darmstadt
Triton X-100 AppliChem GmbH, Darmstadt
Trypton AppliChem GmbH, Darmstadt
Tween 20 VWR International GmbH, Darmstadt
Wasserstoffperoxid Carl Roth GmbH & Co. KG, Karlsruhe
Xylencyanol Sigma-Aldrich GmbH, Taufkirchen
3.2.2 In der Proteinanalytik verwendete Antikörper, Kontrollseren und Marker
3.2.2.1 Primärantikörper
Antikörper Hersteller
Anti-Human, Aggrekan, ACAN, polyklonal, hergestellt im Kaninchen Acris Antibodies GmbH, Herford
Anti-Human, Kollagen 1α2, COL1A2, monoklonal, hergestellt in der Maus Abnova GmbH, Heidelberg
Anti-Human, Kollagen 2α1, COL2A1, monoklonal, hergestellt in der Maus Abnova GmbH, Heidelberg
Anti-Human, β-Aktin, monoklonal, hergestellt in der Maus Cell Signaling Technology,Boston,USA
3.2.2.2 Sekundärantikörper
Antikörper Hersteller
Kaninchen Anti-Maus IgG (H+L) Meerrettichperoxidase (HRP)-konjugiert Southern Biotech, Birmingham, UK
Kaninchen Anti-Maus IgG (H+L) (HRP)-konjugiert Cell Signaling Technology, Boston, USA
Maus Anti-Kaninchen IgG (H+L) (HRP)-konjugiert Santa Cruz Biotechnology, Inc. Dallas, USA
Kaninchen Anti-Maus IgG (H+L) Biotin-konjugiert Southern Biotech, Birmingham, UK
3.2.2.3 Kontrollserum
Kontrollserum Hersteller
Kaninchen Normalserum (KNS) Dianova GmbH, Hamburg
3.2.2.4 Marker für SDS-PAGE und Western Blot
Marker Hersteller
PageRuler Pre-stained Protein Ladder Thermo-Fisher Scientific GmbH, Schwerte

3 Zellen, Materialien und Methoden
27
3.2.3 In der PCR eingesetzte Nukleotide und Enzyme, sowie für die Klonierung und Plasmid-
Präparation verwendete Materialien
3.2.3.1 Nukleotide
Die folgende Übersicht zeigt die im Rahmen der Transkriptanalyse verwendeten Primer (Herstellung durch
die Fa. Sigma-Aldrich GmbH, Taufkirchen).
Primer Primersequenzen Primerdesign
Aggrekan: ACAN, NCBI Zugriffsnr: NM_001135.3
Primer Sense 5’-GCA CCA TGC CTT CTG CTT CCG-3’ Dr. Bianca M. Bußmann, Sven Reiche, TRM Universität Leipzig, Leipzig Primer
Antisense 5’-CTG GCT CGG TGG TGA ACT CTA GG-3’
Kollagen Typ I: COL1A1, NCBI Zugriffsnr: NM_000088.3
Primer Sense 5’-CAA TGT GGT TCG TGA CCG TGA CC-5’ Dr. Bianca M. Bußmann, Sven Reiche,
TRM Universität Leipzig, Leipzig Primer Antisense
3’-GCT CTC CCT TAG CAC CAG TGT CTC C-3’
Kollagen Typ II: COL2A1, NCBI Zugriffsnr: NM_001844.4
Primer Sense 5’-CAA CAT GGA GAC TGG CGA GAC TTG-3’ Dr. Bianca M. Bußmann, Sven Reiche,
TRM Universität Leipzig, Leipzig Primer Antisense
5’-GAC AGC AGG CGT AGG AAG GTC ATC-3’
36B4: NCBI Zugriffsnr: NM_001002.3
Primer Sense 5’-TCG CTT CCT GGA GGG TGT CCG C-3’ Dr. Claire Fabian, Fraunhofer IZI,
Leipzig Primer Antisense
5’-CTC CAC AGA CAA GGC CAG GAC TCG-3’
Daneben wurden weitere Nukleotide in der PCR verwendet. Hierzu zählen die dNTPs (dATP, dTTP, dCTP und
dGTP) und Oligo(dT)18 sowie der in der Agarosegelelektrophorese eingesetzte DNA-Marker.
Weitere Nukleotide Hersteller
dNTPs Fermentas GmbH, St. Leon-Rot
Oligo(dT)18 Fermentas GmbH, St. Leon-Rot
DNA-Marker Hersteller
GeneRuler DNA Ladder Mix 0,5 µg/µl Thermo-Fisher Scientific GmbH, Schwerte
3.2.3.2 Enzyme
Enzym Hersteller
DNase I Fermentas GmbH, St. Leon-Rot
Platinum taq-Polymerase Invitrogen/Life Technologies GmbH, Darmstadt
Superscript III (Reverse Transkriptase) Invitrogen/Life Technologies GmbH, Darmstadt
SybrGreen Express Master-Mix (taq-Polymerase) Invitrogen/Life Technologies GmbH, Darmstadt
Ribolock RNase-Inhibitor Fermentas GmbH, St. Leon-Rot
3 Zellen, Materialien und Methoden
28
3.2.3.3 Materialien für die Klonierung und Plasmid-Präparation
Material Hersteller
Plasmid Miniprep-Kit MACHEREY-NAGEL GmbH & Co. KG, Düren
TOPO-TA Cloning Kit (pCRII, pCR2.1) Invitrogen/Life Technologies GmbH, Darmstadt
Gene JET PCR Purification Kit Fermentas GmbH, St. Leon-Rot
Bakterien Hersteller
E.coli XL10-Gold
Stratagen, La Jolla, USA Die Herstellung der chemisch kompetenten Bakterien erfolgte durch Claire Fabian, Fraunhofer IZI, Leipzig
3.2.4 Zusammensetzung von Gebrauchslösungen für die Immunzytochemie
Tab. 3: Diaminobenzidin (DAB)-Lösung
Faktor Menge
Diaminobenzidin (DAB) 5 mg
Wasserstoffperoxid 6 µl
Tris-HCl-Puffer (0,1 M, pH 7,6) ad 10 ml
Tab. 4: Phosphatgepufferte Kochsalzlösung (PBS)
Stammlösung (20x)
Faktor Menge
Dinatriumhydrogenphosphat 29,25 g
Kaliumhydrogenphosphat 4,90 g
Natriumchlorid 160,0 g
deionisiertes Wasser ad 1000 ml
Natronlauge ad pH-Wert 7,4
Gebrauchslösung (1x)
Faktor Menge
PBS-Stammlösung 50 ml
deionisiertes Wasser 950 ml
Tab. 5: Proteinblockierungslösung
Faktor Menge
Kaninchen Normalserum (KNS) 5%
Triton X-100 0,3%
PBS ad 100%
Tab. 6: Tris-HCl-Puffer (0,5 M Tris-HCl, pH 7,6)
Faktor Menge
Tris-Base 1,21 g
deionisiertes Wasser ad 100 ml
HCl ad pH 7,6

3 Zellen, Materialien und Methoden
29
3.2.5 Zusammensetzung von Gebrauchslösungen für die PCR
Tab. 7: Orange G-Ladepuffer (6x)
Faktor Menge
Tris-HCl, 1M, pH 7,6 100 µl
0.5 M EDTA, pH 8.0 1200 µl
Orange G (15%) 100 µl
Glycerol 6000 µl
deionisiertes Wasser ad 10 ml
Tab. 8: Tris, Essigsäure, EDTA (TAE)-Puffer
Stammlösung (50x)
Faktor Menge
Tris 242,4 g
Essigsäure 57,2 ml
0,5 M EDTA, pH 8.0 100 ml
deionisiertes Wasser ad 1000 ml
Gebrauchslösung (1x)
Faktor Menge
TAE-Stammlösung 20 ml
Wasser 980 ml
3.2.6 Zusammensetzung von Gebrauchslösungen für die Proteinanalytik
Tab. 9: Ammoniumpersulfat (APS)-Lösung (10%)
Faktor Menge
Ammoniumpersulfat (APS) 1g
deionisiertes Wasser ad 10 ml
Tab. 10: Blockierungslösung 5%
Faktor Menge
Bovines Serum Albumin (BSA) 12,5 g
TBS-T-Puffer ad 250 ml, zentrifugiert und filtriert
Tab. 11: Bovines Serum Albumin (BSA)-Lösung (5%/10%)
Faktor Menge
Bovines Serum Albumin (BSA) 12,5 g/25,0 g
deionisiertes Wasser ad 250 ml
Tab. 12: Coomassie Blau-Färbelösung
Faktor Menge
Coomassie Blau R250 0,125 g
Essigsäure 50 ml
Methanol 200 ml
deionisiertes Wasser ad 500 ml
Tab. 13: Coomassie-Entfärbelösung
Faktor Menge
Methanol 150 ml
Glycerol 25 ml
deionisiertes Wasser ad 500 ml

3 Zellen, Materialien und Methoden
30
Tab. 14: Natriumdodecylsulfat (SDS)-Lösungen (4%/10%/20%)
Faktor Menge
Natriumdodecylsulfat (SDS) 4 g/10 g /20 g
deionisiertes Wasser ad 100 ml
Tab. 15: Laufpuffer
Stammlösung (10x)
Faktor Menge
Tris 30,3 g
Glycin 140 g
deionisiertes Wasser ad 1000 ml
Gebrauchslösung (1x)
Faktor Menge
Stammlösung 100 ml
deionisiertes Wasser 890 ml
SDS-Lösung (10%) 10 ml
Tab. 16: Ponceau S-Färbelösung
Faktor Menge
Ponceau S 1 g
Essigsäure 50 ml
deionisiertes Wasser ad 1000 ml
Tab. 17: Probenpuffer (4x)
Stammlösung
Faktor Menge
Tris 1 M, pH 6,8 2,5 ml
SDS 1 g
Glycerol 4 ml
Bromphenolblau 8 mg
deionisiertes Wasser ad 10 ml
Gebrauchslösung (Probenpuffer (4x))
Faktor Menge
Probenpuffer-Stammlösung 450 µl
β-Mercaptoethanol 50 µl
Tab. 18: Sammelgel-Puffer (0,5 M Tris-HCl, pH 6,8)
Faktor Menge
Tris 6,06 g
deionisiertes Wasser ad 100 ml
HCl ad pH 6,8
Tab. 19: TBS-T-Puffer
Faktor Menge
TBS-Stammlösung 100 ml
Tween 20 1ml
deionisiertes Wasser ad 1000 ml
3 Zellen, Materialien und Methoden
31
Tab. 20: Transferpuffer
Faktor Menge
Laufpuffer-Stammlösung (10x) 80 ml
deionisiertes Wasser 720 ml
Methanol 200 ml
Tab. 21: Trenngel-Puffer (1,5 M Tris-HCl, pH 8,8)
Faktor Menge
Tris 18,17 g
deionisiertes Wasser ad 100 ml
HCl ad pH 8,8
Tab. 22: Tris-gepufferte Kochsalzlösung (TBS)
Stammlösung (10x)
Faktor Menge
Natriumchlorid 292,7 g
Tris 4,24 g
Tris-HCl 26 g
deionisiertes Wasser ad 1000 ml
HCl ad pH 7,6
Tab. 23: Zusammensetzung des Proteaseinhibitorcocktails
Faktor Konzentration Ziel Protease
4-(2-Aminoethyl) benzenesulfonyl fluoridhydrochlorid (AEBSF) 500 µM Serinproteasen
Aprotinin, bovine Lunge, kristallin 150 nM Serinproteasen und Esterasen
E-64 Proteaseinhibitor 1 µM Cysteinproteasen
EDTA, Dinatrium- 0,5 mM Metalloproteasen
Leupeptinhemisulfat 1 µM Cysteinproteasen und Trypsin-like-Proteasen
3.2.7 Verbrauchsmaterialien
Verbrauchsmaterial Hersteller
6, 24-Loch-Zellkulturplatten TPP, Trasadingen, Schweiz
96-Loch-Zellkulturplatten VWR International GmbH, Darmstadt
96-Lochplatte für PCR Cycler Hard Shell, Low profile, white
Bio-Rad Laboratories GmbH, München
Deckgläschen, rund, 8 mm Karl Hecht GmbH & Co. KG, Sondheim
Filterpapier VWR International GmbH, Darmstadt
Glasflaschen Duran Group GmbH, Wertheim/Main
Glaspetrischalen Carl Roth GmbH & Co. KG, Karlsruhe
Kühlakku VWR International GmbH, Darmstadt
Kryoröhrchen 2ml TPP, Trasadingen, Schweiz
Objektträger Karl Hecht GmbH & Co. KG, Sondheim
Parafilm Paul Marienfeld GmbH & Co. KG, Lauda Königshofen
Pasteurpipetten Hirschmann GmbH & Co. KG, Eberstadt
Petrischalen (94x16 mm) Greiner Bio-One GmbH, Frickenhausen
Pinzette Aesculap AG, Tuttlingen
Pipettenspitzen, 10-5000 µl Sarstedt AG &Co., Numbrecht
Pipettenspitzen mit Filter, 10-1000 µl Greiner Bio-One GmbH, Frickenhausen
Pipettenspitzen für Multipette Plus Eppendorf AG, Hamburg

3 Zellen, Materialien und Methoden
32
Verbrauchsmaterial Hersteller
PCR-Gefäße, 0,2 ml Eppendorf AG, Hamburg
Polyvinylidenfluorid (PVDF)-Membran Roche, Basel, Schweiz
Reaktionsgefäße 1,5, 2 ml Eppendorf AG, Hamburg
Serologische Pipetten, 12ml Greiner Bio-One GmbH, Frickenhausen
Serologische Pipetten (RNase/DNase frei), 12 ml Greiner Bio-One GmbH, Frickenhausen
Verschlussfolie Microseal C Optical Series Bio-Rad Laboratories GmbH, München
Zellkulturschalen, rund, 100mm x11mm VWR International GmbH, Darmstadt
Zellkulturschalen, rund, 40mm x11mm TPP, Trasadingen, Schweiz
Zentrifugenröhrchen 15, 50 ml VWR International GmbH, Darmstadt
3.2.8 Geräte
3.2.8.1 Laborgeräte
Gerät Hersteller
Absaugvorrichtung für Zellkulturen schuett biotec GmbH, Göttingen
Analyseprogramm ImageJ Wayne Rasband (gemeinfrei)
Autoklav Systec DX23 Systec Labortechnik GmbH, Wettenberg
Elektrophorese- und Blotting System Mini-Protean Tetra Cell
Bio-Rad Laboratories GmbH, München
Folienschweißgerät vacufix Petra electric GmbH, Geislingen
Gefrierschrank Forma -86 °C ULT Freezer Thermo-Fisher Scientific GmbH, Schwerte
Gefrierschrank Liebherr Premium Liebherr GmbH, Ochsenhausen
Gelgießstation Mini-Protean Tetra Cell Casting Module Bio-Rad Laboratories GmbH, München
Gel-/Membrandetektionssystem Kodak Logic 1500 Imaging
Kodak GmbH, Stuttgart
Gelgießsystem PerfectBlue PEQLAB Biotechnologie GmbH, Erlangen
Gelsystem PerfectBlue PEQLAB Biotechnologie GmbH, Erlangen
Hämozytometer Karl Hecht GmbH & Co. KG, Sondheim
Invertmikroskop Nikon Eclipse TE2000-S Nikon GmbH, Düsseldorf
Kamera SPOT INSIGHT 3 Shot QE für Mikroskop Axioskop VISITRON SYSTEMS GmbH, Puchheim
Kamera Nikon Digital Sight DS-Qi1Mc für Invertmikroskop Nikon
Nikon GmbH, Düsseldorf
Kühlschrank Bosch cooler Robert Bosch GmbH, Stuttgart
Kohlendioxid-Begasungsbrutschrank Hera Cell (Heraeus) Kendro Laboratorys & Products GmbH, Hanau
Manometer Linde AG, München
Mehrkanalpipette Transferpette 10-100µl Brand GmbH & Co KG, Wertheim
Messzylinder Hirschmann Laborgeräte GmbH & Co. KG, Eberstadt
Mikroskop Axioskop Carl Zeiss Microscopy GmbH, Jena
Mikrowelle MS-196VUW LG Electronics, Ratingen
PCR-Blockcycler T-Professional Biometra GmbH, Göttingen
PCR-Cycler Opticon 2 Bio-Rad Laboratories GmbH, München
PCR-Werkbank captair bio Erlab, Köln
Plattform-Schüttler Duo Max 1030 Heidolph Instruments GmbH & Co. KG, Schwabach
Plattenreader Infinite M1000 Tecan, Männedorf, Schweiz
Pipette 1000-5000µl Eppendorf AG, Hamburg

3 Zellen, Materialien und Methoden
33
Gerät Hersteller
Pipette 100-1000µl Eppendorf AG, Hamburg
Pipette 20-200μl Eppendorf AG, Hamburg
Pipette 2-20μl Eppendorf AG, Hamburg
Pipette 0,1-2μl Gilson International, Limburg-Offheim
Pipette 2-10μl Gilson International, Limburg-Offheim
Pipette 100-200μl Gilson International, Limburg-Offheim
Pipette 100-1000µl Gilson International, Limburg-Offheim
Pipette 10-100μl Gilson International, Limburg-Offheim
Pipet-Boy accu-jet pro Brand GmbH &Co KG, Wertheim
Power Supply EV202 PEQLAB Biotechnologie GmbH, Erlangen
Reinraumwerkbank Hera Safe (Thermo Scientific) Kendro Laboratorys & Products GmbH, Hanau
Schüttler Vortex VV3 VWR International GmbH, Darmstadt
SDS-PAGE Mini Protean Tetra Bio-Rad Laboratories GmbH, München
Spektralphotometer Nanodrop 1000 PEQLAB Biotechnologie GmbH, Erlangen
Statistik Programm Sigma Plot Systat Software GmbH, Erkrath
Stepper-Pipette Multipette Plus Eppendorf AG, Hamburg
Steuerungsprogramm SPOT Advanced zur Kamera SPOT INSIGHT 3 Shot QE
SPOT Imaging Solutions, Burroughs, USA
Steuerungsprogramm NIS Elements zur Kamera Nikon Digital Sight DS-Qi1Mc
Nikon GmbH, Düsseldorf
Steuerungsprogramm Molecular Imaging Software zum Gel-/Membrandetektionssystem Kodak Logic Imaging 1500
Kodak GmbH, Stuttgart
Stickstofftank HC 20 tec-lab GmbH, Idstein
Thermomixer Eppendorf AG, Hamburg
Waage ED2025-CN Sartorius AG, Göttingen
Wärmeschrank Binder GmbH, Tuttlingen
Wasserbad P.-D. Industriegesellschaft, Dresden
Zentrifuge Heraeus Biofuge Pico Kendro Laboratory Products, Osterode
Zentrifuge Heraeus Multifuge 3 S-R Kendro Laboratory Products, Osterode
Zentrifuge Mikro 200 R Hettrich Zentrifugen, Tuttlingen
3.2.8.2 Bioreaktor
Für die Kultivierung der Chondrozyten unter Druck wurde ein Bioreaktorsystem der Firma durakult GmbH,
Berlin eingesetzt (Abb. 4). Der Bioreaktor verfügt über eine zylindrische Druckkammer, in die ein
Metallgestell, welches der Halterung der Zellkulturschalen dient, eingesetzt werden kann. Die aus Edelstahl
bestehende Druckkammer mit einer runden Grundfläche von 176 cm2 und einer Höhe von 15 cm schließt
ein Luftvolumen von 0,45 l ein. Sie wird über einen Deckel verschlossen. Den Zustrom von Gas in die
Druckkammer vermittelt ein im Deckel eingelassenes, regelbares Ventil, dem ein Manometer vorgeschaltet
ist. An das Ventil erfolgt der Anschluss eines Druckschlauchs, der den Bioreaktor mit einer Gasflasche mit
dem Spezialgasgemisch verbindet. Ein zweites vorangeschaltetes Manometer an der Gasflasche ermöglicht
eine Regelung des Drucks im Druckschlauch. Der aufgesetzte Deckel wird über zwei den Deckel und den
Zylinder umfassende Metallbacken zusammengehalten, die ihrerseits über einen kontinuierlichen Ring
mittels einer Schraube fixiert werden. Das Ablassen von Gas aus dem Bioreaktor erfolgt über ein zweites

3 Zellen, Materialien und Methoden
34
fein justierbares Ventil (Abstromventil). Der maximal zulässige Druck des Reaktors beträgt 50 bar, wobei
das Schlauchsystem nicht mit mehr als 40 bar belastet werden sollte.
Abb. 4: Darstellung des Druckreaktors und seiner Komponenten
3.3 Methoden
3.3.1 Zellkultur
3.3.1.1 Auftauen der Chondrozyten
Vor dem Auftauen der Zellen wurden Zellkulturschalen (rund, 40 mm x 11 mm) bzw. die Vertiefungen einer
6-Loch-Zellkulturplatte mit 1 ml CBM+ Medium/cm2 überschichtet und 30 min im Brutschrank inkubiert. Die
kryokonservierten Chondrozyten wurden aus dem flüssigen Stickstoff entnommen und zügig im Wasserbad
bei 37 °C aufgetaut. Anschließend wurden die Zellen in einer Konzentration von 104 Zellen/cm2 in den
vorbereiteten Zellkulturschalen ausgesät. Nach 24 h erfolgte ein Mediumwechsel.
3.3.1.2 Kultivierung der Chondrozyten unter Normaldruck
Die Zellen wurden bei 37°C, 5% Kohlenstoffdioxid (CO2), 90% Luftfeuchtigkeit und atmosphärischem
Luftdruck (1,013 bar) in Zellkulturschalen oder in den Vertiefungen einer 6-Loch-Zellkulturplatte kultiviert.
Die Erneuerung des CBM+ Mediums erfolgte alternierend nach 3 und 4 Tagen.
3.3.1.3 Kultivierung der Chondrozyten unter erhöhtem Druck
Die Zellen wurden in einem speziellen Bioreaktorsystem, welches die Applikation eines kontinuierlichen
hydrostatischen Drucks ermöglicht, kultiviert. In dem verwendeten Bioreaktorsystem wurde der Druck
durch die Kompression einer Gasphase mit entsprechender Weiterübertragung in das die Zellen
umgebende Medium aufgebaut (Kapitel 2.6.2). Die Kultivierung erfolgte bei 37 °C, 5% CO2, 90%
Luftfeuchtigkeit in Zellkulturschalen. Im Vergleich zur Kultivierung der Chondrozyten unter Normaldruck
wurden nur der Parameter Druck verändert, wohingegen die Parameter Temperatur, Luftfeuchtigkeit,

3 Zellen, Materialien und Methoden
35
Zusammensetzung der Luft und Erneuerungsfrequenz des Mediums gleich blieben. Für den Druckaufbau
wurde ein Spezialgasgemisch verwendet, welches in seiner Zusammensetzung demjenigen der Luft unter
atmosphärischem Luftdruck (1,013 bar) entspricht (Kap. 3.1.4). Der Luftdruck der Atmosphäre (1,013 bar)
wird im Folgenden gerundet (1 bar) angegeben.
Die in Zellkulturschalen ausgesäten Zellen wurden für die Kultivierung unter erhöhtem Druck in das
Metallgestell des Bioreaktors eingesetzt und mit diesem in die Druckkammer des Bioreaktors verbracht.
Nach Verschluss des Reaktors erfolgte der Zustrom des Spezialgasgemisches über einen Zeitraum von
10 min. Die Zellen verblieben für die jeweils gewünschte Zeitdauer im Bioreaktor. Nach Ende der
gewünschten Druckphase wurde das Abstromventil geöffnet und der Druck über einen Zeitraum von
20 min abgelassen bis das Druckniveau des atmosphärischen Luftdrucks erreicht wurde. Anschließend
wurde der Bioreaktor geöffnet und das Metallgestell mit den Zellkulturschalen entnommen.
3.3.1.4 Subkultivierung der Chondrozyten
Das Medium wurde von der Zellkulturschale abgesaugt und die Zellen mit DPBS gewaschen. Der
anschließenden Zugabe des Trypsin-EDTA (1 ml pro 10 cm2) folgte eine Inkubationszeit von 5 min im
Brutschrank bei 37°C. Nachdem die mikroskopische Kontrolle zeigte, dass sich die Zellen abgelöst hatten,
wurde die Reaktion durch die Zugabe von TNS (1ml pro 10 cm2) gestoppt. Die Zellsuspension wurde in ein
Zentrifugenröhrchen überführt und 5 min bei 1050 g zentrifugiert. Der Überstand wurde abgesaugt und das
Zellpellet mit CBM+ Medium suspendiert. Nach der Zellzählung wurde die Zellsuspension mit einer Dichte
von 104 Zellen/cm2 ausgesät.
3.3.1.5 Kryokonservierung der Chondrozyten
Für die Kryokonservierung wurde die Zellsuspension in einer Konzentration von 106 Zellen/ml verwendet. Je
Kryoröhrchen wurden 500 µl Zellsuspension und 500 µl Einfriermedium pipettiert. Die Zellen wurden in
einer Styroporbox auf -80 °C heruntergekühlt. Nach 24 h wurden die Zellen in flüssigen Stickstoff überführt.
3.3.2 Immunzytochemische Untersuchungen
Für die einzelnen Versuchsbedingungen wurden die Chondrozyten in Zellkulturschalen auf Glasplättchen
ausgesät. Zum jeweiligen Untersuchungszeitpunkt wurde das Medium der Zellkulturschale von den Zellen
entfernt und anschließend zweimal mit 1,5 ml PBS gewaschen. Beim zweiten Waschschritt wurde ein Teil
des PBS belassen, um den folgenden Schritt zu erleichtern. Die Glasplättchen wurden einzeln entnommen,
mit 50 µl eiskaltem Methanol-Aceton (1:1) fixiert und anschließend einzeln in je eine Vertiefung einer 24-
Loch-Zellkulturplatte überführt, in welche zuvor 150 µl PBS vorgelegt wurde. Alle folgenden Waschschritte
wurden mit 150 µl der entsprechenden Lösung durchgeführt. Jedes Glasplättchen in der Vertiefung wurde
zweimal mit PBS gewaschen und anschließend mit 80 µl Proteinblockierungslösung für 20 min bei
Raumtemperatur auf einem Schüttler belassen. Das in der Proteinblockierungslösung enthaltene Triton X-
100 macht die Zellwände permeabel und erlaubt dadurch auch den Nachweis intrazellulären Kollagens. Der
jeweilige Primärantikörper (anti-COL1A2, anti-COL2A1) wurde auf die entsprechende Konzentration
verdünnt.
Tab. 24: Verwendete Primärantikörper (anti-COL1A2, anti-COL2A1) und ihre eingesetzten Konzentrationen bei den immunzytochemischen Untersuchungen in vitro
Primärantikörper Verdünnung
Anti-Human, COL1A2, monoclonal, hergestellt in der Maus 1:100 (10 µg/ml)
Anti-Human, COL2A1, monoclonal, hergestellt in der Maus 1:100 (10 µg/ml)

3 Zellen, Materialien und Methoden
36
Jeweils 50 µl der jeweiligen Antikörperverdünnung wurden nach der Blockierung ohne vorheriges Waschen
auf die Zellen gegeben und diese über Nacht im Kühlschrank bei 4 °C unter Zuhilfenahme einer feuchten
Kammer im Dunkeln inkubiert. Als Negativkontrolle wurde in eine der Vertiefungen PBS und in eine weitere
Kaninchen Normalserum statt des Antikörpers in der gleichen Menge verwendet.
Alle folgenden Schritte wurden bei Raumtemperatur durchgeführt. Der jeweilige Primärantikörper wurde
abgesaugt und jede Vertiefung wurde zweimal mit PBS gewaschen. Anschließend wurde jeweils 50 µl
Biotin-konjugierter Sekundärantikörper pro Vertiefung hinzugefügt.
Tab. 25: Verwendeter Sekundärantikörper und dessen eingesetzten Konzentration bei den immunzytochemischen Untersuchungen in vitro
Sekundärantikörper Verdünnung
Kaninchen Anti-Mouse IgG (H+L) Biotin-konjugiert 1:250 (4 µg/ml)
Nach 30 min Inkubation wurde erneut zweimal mit PBS gewaschen und die Strep-ABC-Lösung für 30 min
inkubiert. Nach Absaugen des Strep-ABC-Komplexes und zweimaligem Waschen mit PBS erfolgte ein
zusätzlicher Waschschritt mit 0,1 M Tris-HCl (pH=7,5). Anschließend wurden die Zellen mit 65 µl DAB-
Lösung überschichtet und 10 min unter mikroskopischer Kontrolle belassen. Nach anschließendem
Waschen mit destilliertem Wasser erfolgte eine Kernfärbung mit Hämatoxylin. Dazu wurde Hämatoxylin-
Lösung nach Gill II für 4 min auf die Zellen gegeben und die Vertiefung anschließend dreimal mit
destilliertem Wasser gewaschen. Eine Teilmenge des destillierten Wassers wurde im letzten Waschschritt
auf den Zellen belassen, um das Entfernen der Glasplättchen aus den Vertiefungen zu erleichtern. Mit Hilfe
einer gebogenen Kanüle und einer Pinzette wurden die Glasplättchen aus den Vertiefungen der 24-Loch-
Zellkulturplatte entnommen, auf Filterpapier kurz getrocknet und anschließend mit Hilfe von Histokit auf
Objektträgern fixiert. Diese wurden vor dem Mikroskopieren zwei Tage zur Aushärtung des Histokit bei
Raumtemperatur belassen.
Die Dokumentation der immunzytochemischen Markierung erfolgte mit dem Steuerungsprogramm SPOT
Advanced für die Kamera SPOT INSIGHT 3 Shot QE. Das System wurde so vorjustiert, dass alle Aufnahmen
einer Vergrößerungsstufe mit der gleichen Lichtintensität und Kontrastierung angefertigt wurden.
3.3.3 RNA- und Proteinisolierung
3.3.3.1 Probengewinnung
Für alle Versuchsbedingungen wurden Chondrozyten in weitere Zellkulturschalen ausgesät, die zu jedem
Zeitpunkt eines Versuchsabbruchs, wie nachfolgend beschrieben behandelt wurden. Das Zellkulturmedium
wurde vollständig abgesaugt. Anschließend wurde 1 ml Trifast Gold in die Zellkulturschale gegeben, 3 min
bei Raumtemperatur belassen und in ein Reaktionsgefäß (1,5 ml) überführt. Die auf diese Weise
entstandenen Proben wurden bis zur Isolierung der RNA und der Proteine bei -80 °C gelagert.
3.3.3.2 Isolierung der RNA
Nach dem Auftauen der Proben wurden diese im Verhältnis 1:5 mit Chloroform versetzt, was bei einem
Probenvolumen der Trifast-Proben von 1 ml einem Volumen von 200 µl Chloroform entspricht. Um eine
ausreichende Durchmischung zu gewährleisten, wurden die Proben 15 s homogenisiert. Darauf folgend
wurden die Proben bei Raumtemperatur 15 min inkubiert und bei 12.000 g bei Raumtemperatur über
15 min zentrifugiert. Anschließend wurde die klare obere Phase, welche die RNA enthielt, in ein neues

3 Zellen, Materialien und Methoden
37
Reaktionsgefäß (2 ml) überführt. In dieses Reaktionsgefäß wurden 500 µl Isopropanol hinzugefügt und nach
kurzer Inversion 90 min bei 4 °C inkubiert. Die Zentrifugation erfolgte bei 12.000 g und 4 °C über einen
Zeitraum von 30 min, woraufhin der Überstand restlos verworfen wurde. Das entstandene RNA-Pellet
wurde anschließend zweimal mit 75%igem Ethanol gewaschen und jeweils bei 12.000 g, 4 °C für 10 min
zentrifugiert. Das gewaschene RNA-Pellet wurde daraufhin für 10 min bei Raumtemperatur getrocknet und
anschließend in 50 µl DEPC-Wasser bei 55 °C auf dem Thermomixer bei einer Frequenz von 300 rpm gelöst.
Der RNA-Gehalt der Proben wurde spektralphotometrisch gemessen und die RNA-Proben bis zur weiteren
Verarbeitung bei -80 °C gelagert.
3.3.3.2.1 Qualitätskontrolle der RNA mittels Spektralphotometrie
Die spektralphotometrische Messung ermöglicht eine ungefähre Messung der isolierten RNA-Menge. Die
Messung der RNA-Absorption erfolgt bei 260 nm (DESJARDINS und CONKLIN 2010). Durch die Messung der
Absorption bei 260 und 280 nm wird eine Aussage über die Reinheit der Probe durch die Absorptions-
Verhältnisse A260/A280 und A260/A230 möglich. Dabei ergibt sich ein Wert von 2 für das Verhältnis
A260/A280, wenn keine Kontamination durch Proteine oder DNA vorliegt. Ein Wert von 2,0 -2,2 bei dem
Verhältnis A260/A280 zeigt, dass keine Kontamination durch Phenol, Polysacharide oder Salze vorhanden
ist (DESJARDINS und CONKLIN 2010). Bei Verhältnissen deutlich außerhalb dieser Werte kann nicht von
einer exakten spektralphotometrischen Messung des RNA Gehalts der Probe ausgegangen werden. Mit
Hilfe dieser Methode wurden extreme Ausreißer von der weiteren Analyse ausgeschlossen.
3.3.3.2.2 Qualitätskontrolle der RNA mittels Gelelektrophorese
Für jede Probe wurde eine RNA-Gelelektrophorese durchgeführt, um zu überprüfen, ob RNA degradiert
vorlag. Für die Herstellung des hierfür benötigten Agarosegels wurde die Agarose mit dem TAE-Puffer in
einem Erlenmeyerkolben gemischt und in der Mikrowelle bis zum Siedepunkt erwärmt. Nach kurzer
Abkühlung des flüssigen Gels im Erlenmeyerkolben unter fließendem Wasser wurde Ethidiumbromid
zugesetzt, wodurch ein Verdampfen desselben verhindert wurde. Das flüssige Gel wurde in die planar
ausgerichtete Gießstation gegossen und 30 min zur Aushärtung belassen. Die Zusammensetzung der Gele
ist in der nachfolgenden Tabelle aufgezeigt.
Tab. 26: Zusammensetzung der Agarosegele für die RNA-Auftrennung
Komponente 1% iges Gel 3% iges Gel
Agarose 3 g 9 g
TAE-Puffer 300 ml 300 ml
Ethidiumbromid 15 µl 15 µl
Die Auftrennung der aus den Proben gewonnenen RNA erfolgte in einem 1 %igem Gel. Das Gel wurde in
das Gelsystem eingelegt und bis zur Markierung mit TAE Puffer aufgefüllt. Auf beiden Seiten wurde ein
Marker aufgetragen, um später die Größe der RNA bestimmen zu können. Es wurden jeweils 5 µl RNA-
Probe mit 5 µl 6-fach konzentriertem Orange G-Ladepuffer gemischt und in die Vertiefungen des
Agarosegels geladen. Im Anschluss wurde das System verschlossen und eine Spannung von 80 V für 1-1,5 h
angelegt bis der Farbstoff die obere Hälfte des Gels durchlaufen hatte.
Die Detektion der RNA-Proben wurde mit Hilfe des Gel/Membrandetektionssystems Kodak Logic 1500
Imaging durchgeführt. Hierfür wurden die Gele nach dem Lauf aus dem Gelsystem entfernt und auf die

3 Zellen, Materialien und Methoden
38
UV-Lichtquelle des Systems gelegt. Die Kameraeinstellungen wurden so gewählt, dass die Bilder bei einer
Belichtungszeit von 0,5 s entstanden.
3.3.3.3 Isolierung der Proteine
Die Isolierung der Proteine erfolgte mit TriFast nach Änderung durch die von Chey et al. (2011)
beschriebenen Ethanol-Bromo-Chloropropan-Wasser-Präzipitation. Nach der Abnahme der wässrigen
Phase für die RNA-Isolierung wurden aus der Phenolphase der Proben die Proteine isoliert. Dafür wurden
der Phenolphase 300 µl Ethanol zur Fällung der DNA hinzugefügt und nach kurzer Inversion und Inkubation
von 5 min bei Raumtemperatur bei 2.000 g und 4 °C für 15 min zentrifugiert. Der Überstand wurde in ein
neues Reaktionsgefäß übertragen. Es wurde mit 300 µl dieses Überstands weitergearbeitet. Der
verbleibende Phenol-Rest wurde für weitere Isolierungen bei -80 °C gelagert. Zu 300 µl der nun
proteinreichen Phenol-Phase wurden nacheinander 700 µl absoluter Ethanol und 200 µl 1-Bromo-3-
chloropan gegeben, wobei nach jeder Zugabe zur einheitlichen Durchmischung homogenisiert wurde. Zur
Phasentrennung führte die folgende Zugabe von 600 µl DEPC-Wasser. Nach erneutem Homogenisieren
wurde bei Raumtemperatur und 12.000 g 5 min lang zentrifugiert, wodurch sich die proteinreiche
Interphase zwischen unterer Phenolphase und oberer wässriger Phase bildete. Die obere wässrige Phase
wurde vollständig abgenommen und eine Fällung des Proteins durch Zugabe von 700 µl absolutem Ethanol
bewirkt. Nach 5-minütiger Zentrifugation bei Raumtemperatur und 12.000 g bildete das Protein am Boden
des Reaktionsgefäßes ein Pellet. Die darüber liegende Phenolphase wurde abgenommen, das entstandene
Pellet 10 min bei Raumtemperatur getrocknet und mit 50-100 µl 4%igem SDS mit Proteaseinhibitorcocktail
(1:100) gelöst. Die Proben wurden zum Lösen der Proteine in einem Thermomixer bei 50 °C und 600 rpm
für 5 min erwärmt. Für die weiteren Schritte wurden diese Proben einmalig bei -80 °C eingefroren und bei
weiterer Bearbeitung wieder aufgetaut.
Zur Quantifizierung des Proteingehalts der Proben wurde eine Proteinbestimmung mit Hilfe des
Bicinchoninsäure-Protein-Assay durchgeführt. Mittels einer Mehrkanalpipette wurden jeweils 200 µl der
BCA-Lösung in die Vertiefungen einer 96-Loch-Zellkulturplatte vorgelegt. Anschließend wurden je
Vertiefung 10 µl der zu messenden Proteinprobe zugegeben. Nach Inkubation bei 37 °C im Wärmeschrank
für 30 min erfolgte die Messung der Absorption bei 562 nm im Plattenreader. Die Bestimmung der
Konzentration der Probe erfolgte mit einer Standardkurve eines BSA-Standards in den Konzentrationen 0,
200, 400, 600, 800, 1000 µg/ml in 4%iger SDS-Lösung in 3-Punktbestimmung (Beispiel siehe Tab. 27). Die
Standardkurve sowie die Koeffizienten der Gleichung wurden bei jeder Messung neu bestimmt (Beispiel
siehe Abb. 5). Die Messungen der Standards und Proben erfolgten in Doppelbestimmung.
Die Proteinmessung wurde zunächst für jede der 3 Proben eines Versuchszeitpunkts einzeln durchgeführt.
Anschließend wurde eine gemischte Sammelprobe aus den Proben eines Versuchsansatzes gebildet,
wodurch die für den Western Blot notwendige Proteinkonzentration von 2 µg/µl erreicht wurde. Proben,
die auch bei der zweiten Isolierung aus dem Phenolrest unter Lösung mit 50 µl 4%iger SDS-Lösung diese
Proteinkonzentration nicht enthielten, wurden in der Sammelprobe nicht berücksichtigt.

3 Zellen, Materialien und Methoden
39
Tab. 27: Beispiel für die Messung der Absorption der BSA-Standards in 4%iger SDS-Lösung
Standards Absorption der Standards A562nm
Konzentration [µg/ml] Proteinmenge in der Reaktion [µg/Reaktion]
Mittelwert der Absorption(A562nm)
Mittelwert der Absorption korrigiert um A562nm (0 µg/ml)
0 0,0 0,1579 -
200 10,0 0,2588 0,1009
400 20,0 0,3427 0,1847
600 30,0 0,4622 0,3043
800 40,0 0,5222 0,3642
1000 50,0 0,5719 0,4140
Abb. 5: Beispiel einer Standardkurve und deren Gleichung für die BSA-Standards in 4%iger SDS-Lösung unter
Nutzung der in Tab. 27 angegebenen Messwerte
3.3.4 Transkriptanalyse
Die Untersuchung der Expression des Kollagen Typ I und II sowie des Aggrekans auf der Ebene der mRNA
erfolgte quantitativ mit Hilfe der Reverse Transkription-Polymerasekettenreaktion (RT-PCR).
3.3.4.1 DNase-Verdau
Um eine Behinderung der folgenden Schritte durch Reste von genomischer DNA (gDNA) zu vermeiden,
wurde die Probe zunächst einem DNase-Verdau unterzogen. Hierfür wurde ein Mastermix aus DNase-
Puffer und DNase I im Verhältnis 1:1 in einem Volumen von 2 µl in PCR-Gefäße vorgelegt. Anschließend
wurden je Ansatz 8 µl RNA-Probe hinzugegeben und bei 37 °C für 30 min inkubiert. Daraufhin wurde jeweils
1 µl EDTA hinzugefügt, um ein Selbstschneiden der RNA zu verhindern. Abschließend wurde die DNAse I bei
65 °C für 10 min inaktiviert.
3.3.4.2 Reverse Transkription (RT)
Um eine Amplifizierung mittels Polymerasekettenreaktion (PCR) zu ermöglichen wurde nach dem DNase-
Verdau die mRNA zuerst in eine cDNA umgeschrieben. Die Verwendung von Oligo(dT)18 als Primer bewirkte,
dass die gesamte in der Probe enthaltene mRNA umgeschrieben wurde. Für die RT wurde ein linearer
Verlauf im Bereich von 100 ng bis 2 µg nachgewiesen (Arbeitsgruppe Alexandra Stolzing, IZI Fraunhofer,
Leipzig). Die RT geschah in zwei Schritten.
y = 93,662x2 + 76,114x + 0,7595 R² = 0,9913
0,0
10,0
20,0
30,0
40,0
50,0
60,0
0,00 0,05 0,10 0,15 0,20 0,25 0,30 0,35 0,40 0,45
Pro
tein
[µ
g/R
eak
tio
n]
Absorbtion 562 nm
Standardkurve (BSA verdünnt mit 4% SDS)

3 Zellen, Materialien und Methoden
40
1. Preannealing: Als Primer fungierte Oligo(dT)18, der zusammen mit dNTPs (jeweils 10 mmol, insgesamt 3
µl) zugegeben wurde und bei 65 °C für 5 min verblieb, um anschließend auf 4 °C abzukühlen. In diesem
Schritt erfolgte die Aufschmelzung der Sekundärstruktur der mRNA und beim Abkühlen die Anlagerung des
Primers.
2. RT-Reaktion: Nach Zugabe des Mastermix aus Puffer, RNase-Inhibitor und Reverser Transkriptase (Tab.
28) erfolgte die RT-Reaktion für 60 min bei 50 °C. Anschließend wurde das Enzym bei 75 °C für 15 min
inaktiviert und der Ansatz abschließend auf 4 °C heruntergekühlt.
Tab. 28: Zusammensetzung des Mastermix für die RT-Reaktion
Komponente Konzentration Volumen (1x)
Puffer 5 x 4 µl
DTT 0,1 M 1 µl
Ribolock RNase-Inhibitor 1x 0,5 µl
Superscript III (Reverse Transkriptase)
200 U/µl 0,5 µl
Gesamtvolumen 6 µl
3.3.4.3 Qualitative PCR
Für die qualitative PCR wurde die cDNA aus der RT zunächst 1:10 mit TE-Puffer verdünnt, um eine Inhibition
der taq-Polymerase durch das DTT aus der RT-Reaktion zu verhindern. Anschließend wurden die Primer,
sowie dNTPs, PCR-Puffer, Magnesiumchlorid und die taq-Polymerase zur cDNA in ein PCR-Gefäß gegeben
(Tab. 29). Der Reaktionsansatz wurde mit DEPC-Wasser auf 25 µl Reaktionsvolumen aufgefüllt. Hierbei
wurden Primer für Kollagen Typ I, Kollagen Typ II und Aggrekan und 36B4 verwendet.
Tab. 29: Zusammensetzung der PCR Reaktion im Blockcycler
Komponente Konzentration Volumen (1x)
Primer Sense 10 pmol/µl 0,5 µl
Primer Anti-sense 10 pmol/µl 0,5 µl
dNTPs jeweils 10 mM 0,5 µl
Puffer 10 x 2,5 µl
Magnesiumchlorid 50 mM 1,4 µl
Platinum taq-Polymerase 5 U/µl 0,2 µl
cDNA 1:10 5 µl
DEPC-Wasser 14,4 µl
Reaktionsvolumen 25 µl
Anschließend wurden die PCR-Gefäße in den PCR-Blockcycler überführt, in dem das Programm gemäß
Tab. 30 ablief. Die verwendete taq-Polymerase besitzt eine Transferaseaktivität, die einen einzelnen
Adenosinrest (A) an das 3‘ Ende des PCR-Produkts anhängt. Dieser geht jedoch 2 h nach Ende der PCR
verloren.
Tab. 30: Programm der qualitativen PCR
Schritt Temperatur Zeit Zyklen
Denaturierung 95 °C 30 s 1
Denaturierung Annealing Elongation
94 °C 55 °C 72 °C
10 s 30 s 30 s
35
Abkühlung 4 °C 1

3 Zellen, Materialien und Methoden
41
3.3.4.4 Quantitative PCR
3.3.4.4.1 Herstellung der Standards
Für die quantitative PCR wurde zunächst für die Zielgene (Aggrekan, Kollagen Typ I, Kollagen Typ II) und das
Referenzgen (36B4) jeweils ein Standard erstellt. Dieser Plasmidstandard ermöglicht den Einsatz einer
seriellen Verdünnungsreihe an Plasmid-DNA (pDNA) mit bekannter Molekülzahl in der quantitativen PCR
(qPCR) mit deren Hilfe eine Standardkurve zur Berechnung der Molekülzahl der Proben erstellt werden
konnte. Für die Erstellung der Plasmidstandards wurde das PCR-Produkt aus der qualitativen PCR
verwendet.
3.3.4.4.1.1 Herstellung der Medien und Nährböden
Zunächst wurden die in den folgenden Schritten benötigten Medien und Nährböden vorbereitet (Tab. 31,
Tab. 32). Für die Herstellung des Luria Broth (LB)-Ampicillin-Mediums (Tab. 31) wurden Trypton,
Hefeextrakt, Natriumchlorid und Natriumhydroxid-Lösung in destilliertem Wasser gelöst und autoklaviert.
Nach der Abkühlung des so entstandenen LB-Mediums auf 55 °C wurde Ampicillin zugegeben und das LB-
Ampicillin-Medium bis zur Verwendung verschlossen bei 4-7 °C aufbewahrt. Bei der Herstellung der LB-
Ampicillin-Nährböden (Tab. 32) wurde dem LB-Medium hiervon abweichend bereits vor dem Autoklavieren
Agar-Agar zugegeben. Im Anschluss an die Zugabe des Ampicillins wurde der noch flüssige LB-Ampicillin-
Nährboden in die Petrischalen gegossen und dieser nach Aushärtung bis zur weiteren Verwendung bei
4-7 °C gelagert. Das im Rahmen der Transformation genutzte S.O.C. Medium ist Teil des TOPO TA Cloning
Kits und wurde nicht selbst hergestellt.
Tab. 31: Zusammensetzung des LB-Ampicillin-Mediums
Faktor Menge
Trypton 10 g
Hefeextrakt 5 g
Natriumchlorid 8 g
1 M Natriumhydroxid-Lösung 1 ml
Deionisiertes Wasser ad 1000 ml
Ampicillin 10 mg/ml 10 ml
Tab. 32: Zusammensetzung der LB-Ampicillin-Nährböden
Faktor Menge
LB-Medium 500 ml
Agar-Agar 7,5 g,
Ampicillin 10 mg/ml 5 ml
3.3.4.4.1.2 Hinzufügen eines Adenosin(A)-Überhangs
Wenn nicht innerhalb von 2 h mit dem PCR-Produkt weitergearbeitet werden konnte, wurde im ersten
Schritt ein Adenosin(A)-Überhang an die DNA synthetisiert. Hierfür wurden 0,5 µl Platinum taq-Polymerase
und 0,5 µl dNTPs frisch zum Reaktionsansatz aus der PCR hinzugegeben. Der Ansatz wurde erneut in den
Blockcycler überführt, 3 min bei 95 °C und 10 min bei 72 °C belassen sowie abschließend auf 4 °C abgekühlt.
Die verwendete taq-Polymerase hängt durch ihre endogene Transferaseaktivität ein einzelnes A an das
3‘ Ende des PCR-Produkts wieder an.

3 Zellen, Materialien und Methoden
42
3.3.4.4.1.3 cDNA-Aufreinigung
Die cDNA wurde mit dem Gene JET PCR Purification Kit entsprechend der Vorgabe des Herstellers
aufgereinigt. Hierbei erfolgte die Entfernung der Primer, Nukleotide und dNTPs aus der vorangegangenen
PCR über Säulen mit Silicatmembranen. Die negativ geladene DNA bindet durch Zugabe von Ethanol und
chaotropen Salzen an die positiv geladene Silicatmembran, wodurch andere Substanzen ausgewaschen
werden können. Die aufgereinigte cDNA kann im Anschluss mit Wasser wieder von der Säule eluiert
werden.
3.3.4.4.1.4 Klonierung
Ligation 3.3.4.4.1.4.1
Die sich an die cDNA-Aufreinigung anschließende Klonierungsreaktion, in der das PCR-Produkt in einen
Vektor (Plasmid) eingebracht wurde, erfolgte mit dem TOPO TA Cloning Kit für 5 min bei Raumtemperatur
(Tab. 33). Der hierbei verwendete TOPO Vektor liegt mit einem einzelnen Thymidin (T)-Überhang vor, an
den die genutzte Topoisomerase I nach der Spaltung nach 5‘-CCCTT kovalent gebunden bleibt. Die Ligation
des PCR-Produkts mit dem A-Überhang in das Plasmid kann somit am T-Überhang des Vektors von 3‘ nach
5‘ erfolgen.
Tab. 33: Verwendete Komponenten für die Klonierung
Komponente Volumen (1x)
PCR Produkt 2 µl
Salz Lösung 0,5 µl
TOPO Vektor 0,5 µl
Gesamtvolumen 4 µl
Transformation 3.3.4.4.1.4.2
Das durch die Ligation entstandene Plasmid wurde im folgenden Schritt in Bakterien transformiert. Hierfür
wurden zunächst die Nährböden mit LB-Ampicillin-Medium bei 37 °C im Brutschrank vorgewärmt. In einem
Reaktionsgefäß (1,5 ml) wurden 2 µl des Reaktionsansatzes der Ligation zu frisch aufgetauten chemisch
kompetenten E.coli XL10-Gold (50 µl) hinzugegeben und für 30 min auf einem Kühlakku im Kühlschrank
(4 °C) inkubiert. Danach wurde ein Hitzeschock bei 42 °C für 30 s durchgeführt und das Zentrifugenröhrchen
wieder auf den Kühlakku gestellt. Nach der Zugabe von 250 µl S.O.C. Medium wurde das
Zentrifugenröhrchen verschlossen. Auf dem Thermomixer wurden die Bakterien bei 37 °C und 300 rpm für
30 min wachsen gelassen. Im Anschluss wurde der gesamte Ansatz pro Klonierungsreaktion auf einem LB-
Ampicillin-Nährboden ausgestrichen und über Nacht bei 37 °C im Brutschrank gelagert.
Die über Nacht gewachsenen Kolonien wurden über Kolonie-PCR darauf kontrolliert, ob das PCR-Produkt
eingebaut wurde. Dafür wurde mit dem in Tab. 29 beschriebenen PCR-Ansatz gearbeitet, wobei anstelle
der cDNA die Bakterien als Template verwendet wurden. Die gewachsenen Kolonien wurden mit der
Pipettenspitze aufgenommen und auf frische LB-Ampicillin-Nährböden ausgestrichen. Anschließend wurde
der an der Pipettenspitze befindliche Rest einer jeden Kolonie in ein PCR-Röhrchen mit dem PCR-Ansatz
(Tab. 29) gegeben und das Gemisch trituiert. Die neu ausgestrichenen Nährböden wurden erneut über
Nacht bei 37 °C im Brutschrank gelagert. Die Kolonie-PCR wurde wie in Kapitel 3.3.4.3 beschrieben
durchgeführt. Dabei wurden für den pCR 2.1 TOPO-Vektor, der für Aggrekan verwendet wurde,

3 Zellen, Materialien und Methoden
43
M13(-20) forward und M13 reverse als Primer genutzt. Für Kollagen I und II sowie das Referenzgen 36B4
wurde der pCR II TOPO-Vektor und die Primer Sp6 und T7 verwendet.
Tab. 34: Für die Kolonie-PCR verwendete Primer und deren Sequenz in Abhängigkeit von der Auswahl des Vektors
Primer für pCR 2.1-TOPO Vektor (verwendet für Aggrekan)
Primer Sense: M13 (-20) forward 5’-GTA AAA CGA CGG CCA G-3’
Primer Antisense: M13 reverse 5’-CAG GAA ACA GCT ATG AC-3’
Primer für pCR II-TOPO Vektor (verwendet für Kollagen 1/Kollagen 2/36B4)
Primer Sense: Sp6 5’-ATT TAG GTG ACA CTA TAG-3’
Primer Antisense: T7 5’-TAA TAC GAC TCA CTA TAG GG-3’
Das PCR-Produkt der Kolonie-PCR wurde anschließend auf ein Agarose-Gel aufgetragen. Hierfür wurden
5 µl PCR-Produkt und 2,5 µl Orange G-Ladepuffer (6x) gemischt, in die Vertiefungen des Agarosegels
geladen und elektrophoretisch aufgetrennt. Die PCR-Produkte der Bakterienkolonien, in denen das
PCR-Produkt in den Vektor eingebaut wurde, konnten so durch die Anzahl der Basenpaare identifiziert
werden.
Tab. 35: Länge des Kolonie-PCR Produkts ohne Einbau des PCR-Produkts und bei erfolgreichem Einbau des PCR-Produkts in Basenpaaren (bp)
Ziel-/Referenzgen Länge des Kolonie-PCR-Produkts ohne Einbau des PCR-Produkts
Länge des Kolonie-PCR-Produkts bei erfolgtem Einbau des PCR-Produkts
Aggrekan (ACAN) 202 408 bp (206 bp+202 bp)
Kollagen 1 (COL1A1) 187 419 bp (232 bp+187 bp)
Kollagen 2 (COL2A1) 187 388 bp (201 bp+187 bp)
36B4 187 312 bp (125 bp+187 bp)
Die Bakterienkolonien, in denen der Einbau des PCR-Produkts nachgewiesen wurde, wurden selektiert, in
ein Zentrifugenröhrchen mit 5 ml vorgewärmtem LB-Ampicillin-Medium gegeben und bei 37 °C über Nacht
inkubiert.
3.3.4.4.1.5 Plasmid-Präparation
Die Plasmid-Präparation wurde mit dem Plasmid Mini Prep-Kit entsprechend der Vorgabe des Herstellers
durchgeführt. Die geernteten Bakterien aus dem LB-Ampicillin-Medium, bei denen mittels Kolonie-PCR der
Einbau des PCR-Produkts nachgewiesen wurde, wurden einer alkalischen Lyse mit SDS unterzogen. Um eine
optimale Bindung der Plasmid-DNA (pDNA) an die Silicatmembran zu erreichen, wurde vor dem Aufgeben
auf die Säule ein neutralisierender Salzpuffer zugegeben. Die an die Säule gebundene pDNA wurde
mehrmals mit Ethanol gewaschen und abschließend durch Zugabe von Elutionspuffer von der
Silicatmembran gelöst. Die erhaltene pDNA wurde spektralphotometrisch gemessen. Die Reinheit der
Plasmidprobe wurde durch die Absorptions-Verhältnisse A260/A280 und A260/A230 bestimmt. Dabei sollte
A260/A280 bei 1,8-2,0 liegen und A260/A230 sich im Bereich 2-2,22 bewegen. Aus den Messwerten wurde
die Molekülzahl pro 1 µl errechnet, wobei sich die Anzahl der Basenpaare (bp) aus den Basenpaaren für
Plasmid (3931 bp für pCR 2.1-TOPO Vektor und 3973 bp für pCR II-TOPO Vektor) und PCR-Produkt
folgendermaßen zusammensetzt:
pDNA gemessen___
Anzahl der Basenpaare Molekülzahl = x Avogadro‘ sche Konstante

3 Zellen, Materialien und Methoden
44
Tab. 36: Bezeichnungen der Plasmidstandards für Aggrekan, Kollagen Typ I, Kollagen Typ II und 36b4 unter Angabe der für die Berechnung der Molekülzahl notwendigen Anzahl der Basenpaare (bp) des Plasmids und des in dieses Plasmid eingebauten PCR-Produkts
Plasmidstandard Anzahl der Basenpaare (Plasmid+PCR-Produkt)
Aggrekan: pCR 2.1-ACAN 4137 bp (3931 bp+206 bp)
Kollagen Typ I: pCR II-COL1A1 4205 bp (3973 bp+232 bp)
Kollagen Typ II: pCR II-COL2A1 4174 bp (3973 bp+201 bp)
36B4: pCR II-36B4 4098 bp (3973 bp+125 bp)
Die Molekühlzahl wurde auf 1x109 Moleküle/µl mit TE-Puffer eingestellt. Anschließend hergestellte 5 µl
Aliquots wurden bis zur weiteren Verwendung bei -20 °C gelagert.
3.3.4.5 Quantitative Real Time-PCR
In die Vertiefungen der 96-Lochplatte für PCR wurde ein Mastermix (Tab. 37) mit einer Stepper-Pipette
vorgelegt und die cDNA hinzugegeben. Diese wurde zuvor 1:10 mit TE-Puffer (pH 8,0) verdünnt, um eine
Inhibition der taq-Polymerase durch das DTT aus der RT-Reaktion zu verhindern. Je Probe wurden drei
technische Replikate angefertigt. An Stelle der cDNA wurden TE-Puffer (pH 8,0) und DEPC-Wasser als
Negativkontrolle pipettiert. In drei Vertiefungen der 96-Lochplatte wurde eine serielle Verdünnungsreihe
der pDNA aus der Plasmid-Präparation mit 1x107, 1x106, 1x105 Molekülen/µl eingesetzt (Plasmidstandard).
Hierfür wurden die auf 1x109 Molekülen/µl eingestellten 5 µl Aliquots verwendet und die
Verdünnungsreihe für jeden Ansatz frisch mit TE-Puffer angesetzt. Anschließend wurde die 96-Loch-Platte
mit der dazugehörigen Verschlussfolie dicht verschlossen und in den PCR-Cycler Opticon 2 gestellt. Die
qPCR erfolgte entsprechend des in Tab. 38 angegebenen Programms.
Tab. 37: Berechnung der Zusammensetzung der Komponenten der qPCR
Komponente Konzentration Volumen (1x)
SybrGreen Express Master-Mix 2x 10 µl
Primer Sense 10 pmol/µl 0,5 µl
Primer Anti-sense 10 pmol/µl 0,5 µl
SybrGreen 10x 1 µl
DEPC-Wasser 3 µl
cDNA 1:10 5 µl
Gesamtvolumen 20 µl
Tab. 38: Programm für die qPCR
Schritt Temperatur Zeit Zyklen
Aufheizen 50 °C 2 min 1
Denaturierung 95 °C 5 min 1
Denaturierung Annealing Elongation Fluoreszensmessung
95 °C 60 °C 72 °C
3 min 30 s 30 s 2 s
40
Aufheizen 95 °C 1 min 1
Abkühlen 40 °C 1 min 1
Schmelzkurvenanalyse 60-90 °C 1
Abkühlung 30 °C 1
Während der Elongationszyklen bindet der Farbstoff SYBR-Green I unspezifisch an doppelsträngige DNA
(dsDNA). Der gebundene Farbstoff kann durch Licht einer spezifischen Wellenlänge (λmax = 494 nm, blau)

3 Zellen, Materialien und Methoden
45
angeregt werden und emittiert dann Licht der Wellenlänge λmax = 521 nm (grün). Dieses Fluoreszenzsignal
wird am Ende jedes Zyklus gemessen. Es ist proportional zur Menge an der dsDNA im jeweiligen Zyklus.
Zur Quantifizierung wird die Kinetik der PCR-Reaktion genutzt, die durch die Messung der Fluoreszenz
deutlich wird. Bei der qPCR zeigt die PCR-Reaktion und somit das Fluoreszenssignal einen sigmoidalen
Kurvenverlauf. Nach einer flachen Anlaufphase erfolgt die Vermehrung der PCR Produkte exponentiell und
flacht später wieder ab. Der Zeitpunkt während der exponentiellen Phase, an dem sich das
Fluoreszenzsignal gerade deutlich vom Hintergrund abhebt, wurde als Schwellenwert definiert. Die zu
diesem Schwellenwert erreichte Anzahl der Zyklen wird als CT-Wert bezeichnet. Der Schwellenwert wurde
für alle Analysen gleich gesetzt.
Um aus den gemessenen CT-Werten eine absolute Molekülzahl zu erhalten, wurde eine serielle
Verdünnungsreihe mit bekannter Molekülzahl an pDNA mitgeführt. Die zu diesen Verdünnungen 1x107,
1x106, 1x105 Molekülen/µl gemessenen CT-Werte dienten zum einen der Quantifizierung und zum anderen
der Kontrolle der PCR-Reaktion auf Inhibition der Reaktion, da die CT-Werte der Verdünnungsreihe in jedem
qPCR-Ansatz identisch sein mussten.
Um die gemessenen CT-Werte zu normalisieren, wurde das Referenzgen 36B4 für jede Probe mitgeführt.
Das verwendete Referenzgen 36B4 wurde für die chondrogene Differenzierung humaner MSC in der
Arbeitsgruppe Dr. Alexandra Stolzing, IZI Fraunhofer, Leipzig etabliert. Die CT-Werte der Zielgene und des
Referenzgens wurden absolut quantifiziert und anschließend in Relation zueinander gesetzt. Die
Datendarstellung der Zielgene erfolgte im Verhältnis zu 105 Molekülen des Referenzgens.
Nach Abschluss der qPCR wurde die 96-Lochplatte aus dem PCR-Cycler entnommen. Die Verschlussfolie
wurde aufgetrennt, um das Gesamtvolumen des PCR-Produkts (25 µl) vermischt mit 5 µl Orange
G-Ladepuffer (6x) auf ein 3 %iges Agarosegel aufzutragen (Kap. 3.3.3.2.2).
3.3.5 Proteinanalyse
Die Untersuchung der Expression des Kollagen Typ I und II sowie des Aggrekans auf der Ebene des Proteins
erfolgte quantitativ mit Hilfe der Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) und
anschließendem Western Blot. Nach der Quantifizierung des Proteingehalts der Proben mit Hilfe des
Bicinchoninsäure-Protein-Assays wurden die Proben mit 4%iger SDS-Lösung entsprechend so verdünnt,
dass in allen Ansätzen eine Menge von 40 µg Protein in 20 µl Probenvolumen (Konzentration von 2 µg/µl)
vorhanden war.
3.3.5.1 SDS-PAGE
Die SDS-Gele wurden mit Hilfe der Gelgießstation für Mini-Gele Mini-Protean Tetra Cell Casting Module
gefertigt. Hierfür wurden Spacer-Platten mit 1,5 mm Abstandhaltern verwendet, die zusammen mit den
Glasplatten entsprechend der Gebrauchsanweisung des Herstellers in die Station integriert wurden. Das
System wurde vor dem Gießen der Gele mit deionisiertem Wasser auf Dichtigkeit geprüft. Nach dem
Gießen des Trenngels wurde dieses mit 500 µl Isopropanol überschichtet und 30 min auspolymerisiert.
Nach der Polymerisation des Trenngels wurde das Isopropanol entfernt, das Sammelgel gegossen und ein
Kamm (15 Vertiefungen) in dieses eingesetzt. Nach Polymerisation des Sammelgels wurden die fertigen
Gele zwischen den Glasplatten belassen, mit feuchtem Zellstoff und Aluminiumfolie umwickelt und in einer
verschließbaren Plastikfolie bis zur weiteren Verwendung bei 4-7 °C gelagert. Um während der
Gelherstellung eine Kontrolle der Polymerisation mit Hilfe des Gelrests zu ermöglichen, wurden die exakt
benötigten Mengen um eine Zugabe von 5,5 % überschritten.
3 Zellen, Materialien und Methoden
46
Tab. 39: Zusammensetzung des Sammel- und Trenngels.
Faktor 2 Gele (exakt) 2 Gele inkl. Zugabe 4 Gele inkl. Zugabe
Trenngel (10%)
Deionisiertes Wasser 6,1 ml 6,44 ml 12,89 ml
Trenngelpuffer 3,8 ml 4,01 ml 8,03 ml
Bis-Acrylamide (30%) 5,0 ml 5,28 ml 10,56 ml
SDS (10%) 150 µl 158 µl 317 µl
APS (10%) 75 µl 79 µl 158 µl
TEMED 20 µl 21 µl 42 µl
Sammelgel (4%)
Deionisiertes Wasser 3,05 ml 3,30 ml 6,60 ml
Sammelgelpuffer 1,25 ml 1,35 ml 2,71 ml
Bis-Acrylamide (30%) 0,67 ml 0,73 ml 1,45 ml
SDS (10%) 50 µl 54 µl 108 µl
APS (10%) 50 µl 54 µl 108 µl
TEMED 10 µl 11 µl 22 µl
Die auf eine Proteinkonzentration von 2 µg/µl eingestellten Proteinsammelproben wurden im Verhältnis
1:4 mit Probenpuffer (4x), der den Farbstoff Bromphenolblau enthielt, versetzt. Um eine Beeinflussung des
gemessenen Proteingehaltes durch den Gefriervorgang auszuschließen, wurde dieser direkt vor der Zugabe
des Probenpuffers nochmals gemessen. Die Proben wurden bei 90 °C für 5 min auf dem Thermomixer
erwärmt. Dabei lagert sich das im Probenpuffer (4x) enthaltene SDS an die Proteine an und bewirkt eine
negative Ladung aller Proteine, welche somit nach Anlegen einer Spannung in Richtung der Anode wandern
und dadurch allein entsprechend ihrer Größe aufgetrennt werden. Hierfür wurde das Gel in die
Elektrophoresekammer eingesetzt, die Kammer mit Laufpuffer gefüllt und der Kamm aus dem Sammelgel
entfernt. Die Proben wurden in die durch den Kamm entstandenen Taschen pipettiert. In 2 Taschen des
Gels wurde ein Größenmarker mitgeführt. Die Sammelproben eines identischen Versuchszeitpunkts
wurden jeweils in gleicher Reihenfolge auf 3 Gele pipettiert. Anschließend wurde für 30 min eine Spannung
von 80 V angelegt und sichergestellt, dass alle Proben das Trenngel erreicht hatten, bevor die Spannung auf
100 V erhöht wurde. Der Gellauf wurde nach 1,5-2 h, wenn der Farbstoff das untere Ende des Gels erreicht
hatte, beendet.
3.3.5.2 Western Blot
Für den Transfer der Proteine auf die PVDF-Membran wurden die das Gel umgebenden Glasplatten
entfernt und das Sammelgel abgetrennt. Nach Zuschnitt der PVDF Membran entsprechend der Gelgröße
wurde diese aktiviert, indem sie 15 s in Methanol, anschließend 2 min in deionisiertem Wasser und
anschließend 5 min in Transferpuffer gelagert wurde. Für das Blotting wurde die Transferkassette
entsprechend der Darstellung in Abb. 6 zusammengesetzt. Alle Schwämme und Filter wurden vorher 5 min
in Transferpuffer getränkt. Abschließend wurde geprüft, dass keine Luftblasen zwischen den einzelnen
Lagen, insbesondere zwischen den beiden Filtern vorhanden waren.

3 Zellen, Materialien und Methoden
47
Abb. 6: Schematischer Aufbau der Zusammensetzung der Transferkassette; adaptiert nach www.explorer.bio-rad.com)
Die Transferkammer wurde mit Transferpuffer gefüllt und in die Transferkassette eingesetzt. Der Transfer
fand bei Raumtemperatur statt. Um ein Erwärmen zu vermeiden, wurde das System mit Hilfe der
systemeigenen Kühlvorrichtung, die mit Eis befüllt wurde, gekühlt. Somit wurde sichergestellt, dass
während des Transfers eine Temperatur von 30 °C nicht überschritten wurde. Der Transfer der Proteine aus
dem Gel auf die Membran fand bei einer Spannung von 100 Volt für 90 min statt. Der Transfer konnte
anhand der Übertragung des Markers auf die Membran überprüft werden. Um den Erfolg des Transfers
zusätzlich zu prüfen, wurde an dem Gel eine Coomassie-Färbung durchgeführt. Hierfür wurde das Gel für 5
min in der Coomassie Blau-Färbelösung inkubiert. Anschließend wurde das Gel über Nacht in der
Commassie-Entfärbelösung entfärbt und fotografiert.
Im Rahmen der Etablierung dieses Verfahrens wurde die PVDF-Membran direkt nach der Entnahme aus der
Kassette einer Färbung mit Ponceau S unterzogen, um den Proteintransfer zu überprüfen. Hierfür wurde
die Membran 10 min in Ponceau S-Färbelösung inkubiert und anschließend 20 Minuten mit destilliertem
Wasser entfärbt. Die angefärbten Banden auf der PVDF-Membran wurden fotografiert und schließlich mit
PBS entfärbt. Da sich während der Etablierung des Verfahrens ein negativer Einfluss dieser Färbung auf die
anschließende Bindung der Antikörper an die PVDF-Membran zeigte, wurde während der
Versuchsdurchführung von dieser Färbung abgesehen.
Nach Entnahme der PVDF-Membran aus der Kassette wurde diese zur Minderung unspezifischer Bindungen
für 90 min mit 5% BSA-Lösung auf einem Schüttler bei Raumtemperatur blockiert. Anschließend wurde die
Membran mit jeweils 5 ml des auf die jeweilige Konzentration mit 5% BSA-Lösung verdünnten
Primärantikörpers inkubiert. Neben den für den Nachweis der Expression des Kollagen Typ I und II sowie
des Aggrekan verwendeten Antikörpern wurde ein anti-β-Aktin-Antikörper zum Nachweis des
Referenzproteins β-Aktin eingesetzt. Für die Inkubation der Antikörper wurde die PVDF-Membran in eine
Plastikfolie überführt, die nach Zugabe des Antikörpers verschweißt wurde. Anschließend erfolgte die
Inkubation bei Raumtemperatur auf einem Schüttler.
Anschließend wurde die Membran aus der Folie entfernt und auf einem Schüttler drei Mal 10 min mit
TBS-T-Lösung gewaschen. Die Inkubation mit 5 ml des HRP-konjugiertem Sekundärantikörpers erfolgte
ebenfalls unter Zuhilfenahme einer verschweißbaren Plastikfolie bei Raumtemperatur auf einem Schüttler.
Nach der Inkubation wurde die Membran aus der Plastikfolie entfernt und erneut drei Mal für 10 min mit
TBS-T-Lösung und 1 Mal mit TBS gewaschen. Im Anschluss wurden 3 ml Luminol mehrfach über die
Membran pipettiert. Nach 2 Minuten wurde die Membran für die Detektion zwischen zwei Klarsichtfolien
gelegt.

3 Zellen, Materialien und Methoden
48
Tab. 40: Verwendete Primärantikörper (anti-COL1A2, anti-COL2A1, anti-β-Aktin, anti-Aggrekan), deren eingesetzte Konzentrationen und Inkubationszeiten im Western Blot
Primärantikörper Konzentration Inkubationszeit
Anti-Human, Aggrekan, ACAN, polyclonal, hergestellt im Kaninchen 1: 500 3 h
Anti-Human, Kollagen 1α2, COL1A2, monoclonal, hergestellt in der Maus
1: 1000 1 h
Anti-Human, Kollagen 2α1, COL2A1, monoclonal, hergestellt in der Maus
1: 600 3 h
Anti-Human, β-actin, monoclonal, hergestellt in der Maus 1: 1000 1 h
Tab. 41: Verwendeter Sekundärantikörper, deren eingesetzte Konzentrationen und Inkubationszeiten im Western Blot
Sekundärantikörper Konzentration Inkubationszeit
Rabbit Anti-Maus IgG (H+L), HRP-konjugiert, Southern Biotech 1: 5000 1 h
Mouse Anti-Kaninchen IgG (H+L), HRP-konjugiert, Southern Biotech 1: 1600 1 h
Rabbit Anti-Maus IgG (H+L), HRP-konjugiert, Cell Signaling Technology (für β-Actin)
1: 1600 1 h
3.3.5.3 Detektion
Die Detektion des Western Blot erfolgte mit Hilfe des Gel/Membrandetektionssystem Kodak Logic 1500
Imaging und dem dazu gehörigen Steuerungsprogramm Kodak Molecular Imaging Software Tutorial. Für die
Detektion wurde die von den beiden Klarsichtfolien umgebene Membran in das Detektionssystem eingelegt
und zügig mit der Detektion begonnen. Die Kameraeinstellungen wurden so gewählt, dass 7 Bilder bei einer
Kamerabelichtungszeit von 30 s entstanden. Aus diesen Bildern wurde mit Hilfe des Steuerungsprogramms
Kodak Molecular Imaging Software Tutorial ein Summationsbild errechnet, welches dann mit Hilfe des
Analyseprogramms ImageJ ausgewertet wurde.
3.3.5.4 Stripping der Antikörper und erneute Inkubation
Nach der Detektion wurde die Membran gestrippt. Dabei wurden die Antikörper von der Membran gelöst,
sodass diese noch einmal für die Detektion des β-Aktin-Antikörpers verwendet werden konnte. Hierfür
wurde die Membran zwei mal 7 min in einem Plastikgefäß mit Stripping-Puffer bei Raumtemperatur
inkubiert. Daraufhin wurde die Membran zweimal für jeweils 10 min mit PBS-Lösung gewaschen. Die
Waschschritte wurden unter den gleichen Bedingungen mit TBS-T-Lösung für jeweils 5 min wiederholt.
Nach dieser Behandlung konnte mit der Blockierung begonnen werden und der β-Aktin-Antikörper zum
Nachweis des Referenzproteins inkubiert werden.
Das Vorliegen von Interferenzen mit Resten der Primärantikörper, die bei ineffizientem Stripping verbleiben
können, wurde in einem Vorversuch ausgeschlossen. Zusätzlich wurde darauf geachtet, dass das
Molekulargewicht der zu detektierenden Proteinbanden der beiden Detektionen so weit auseinander lag,
dass die mit Hilfe der zweiten primären Antikörperinkubation detektierten Signale nicht durch eventuell
ausgebrannte Signale der ersten primären Antikörperinkubation verfälscht wurden. In den Vorversuchen
konnte ebenfalls gezeigt werden, dass das genutzte Stripping-Protokoll Interferenzen von Resten der
Primärantikörper der Erstinkubation mit der Detektion des Referenzproteins wirksam verhindert. Die zu
detektierenden Proteinbanden des Aggrekan bei 45 kDa konnten daher trotz der Nähe zum
Referenzprotein β-Aktin (42 kDa) eindeutig identifiziert werden. In einigen Fällen kam es verfahrensbedingt

3 Zellen, Materialien und Methoden
49
während der Versuche bei Banden im Randbereich der PVDF-Membran zu schwachen bzw. innerhalb einer
Bande inhomogenen Signalen, welche die semiquantitative Analyse beeinträchtigten.
3.3.5.5 Semiquantitative Analyse des Western Blot
In der Auswertung der optimierten Western Blots zeigten sich mehrere Banden, die durch den jeweiligen
Antikörper spezifisch markiert wurden. Für die Evaluierung der Kollagen Typ I Expression wurden die
Banden des Molekulargewichte 38, 50, 120 kDa, für Kollagen Typ II die Banden bei 65 und 70 kDa und für
Aggrekan bei 45 und 65 kDa ausgewertet. Hingegen wurden die für Kollagen Typ I bei 60 und 170 kDa und
für Kollagen Typ II bei 45 kDa detektierten, unspezifischen Banden nicht berücksichtigt. Die Bande für das
Referenzprotein β-Aktin lag bei einem Molekulargewicht von 42 kDa.
Zur semiquantitativen Analyse der Ergebnisse aus dem Western Blot wurden die Bilder in das
Analyseprogramm ImageJ geladen und der Hintergrund entfernt. Die gemessenen Chemolumineszenz-
Intensitäten der mit Hilfe der Antikörper markierten Zielproteine Kollagen Typ I und II sowie Aggrekan
wurden jeweils im Verhältnis zur Intensität des Referenzproteins β-Aktin gesetzt. Die Ergebnisse, welche
kein Lumineszenz-Signal für β-Aktin lieferten, wurden von der Analyse ausgeschlossen.
3.3.6 Graphische Darstellung und statistische Methoden
Die graphische Darstellung der Ergebnisse der qPCR erfolgt durch Abbildung des arithmetischen Mittels (M)
der biologischen Replikate unter Angabe der Standardabweichung. Aufgrund des geringen
Stichprobenumfangs wird jedoch auch der M der technischen Replikate eines jeden biologischen Replikats
als Punktdarstellung gezeigt.
Ein Ergebnis eines technischen Replikats wurde von der weiteren Analyse ausgeschlossen, wenn es
wesentlich von den beiden anderen technischen Replikaten abwich. Als wesentlich wurde entsprechend
eine Abweichung von >1 CT definiert. Sowohl aufgrund dieser Abweichungen als auch durch den Ausfall
einzelner biologischer Replikate in der qPCR erfolgt die graphische Darstellung unter Angabe der Anzahl (n)
der für deren Erstellung genutzten biologischen Replikate, welche auch in der weiteren Analyse verwendet
wurden.
Für die statistische Analyse der Ergebnisse wurde das Statistik-Programm Sigma Plot verwendet. Es wurden
Berechnungen der M der biologischen Replikate sowie der Standardabweichung durchgeführt. In die
statistische Analyse der Ergebnisse unterschiedlicher Bedingungen wurden die Differenz der Mittelwerte,
die Standardabweichung und der Stichprobenumfang (n) einbezogen, um die statistische Power
(Teststärke, P) der Ergebnisse zu ermitteln (t-Test). Die statistische Analyse kleiner Stichproben (n=2, n=3)
ist fehleranfällig. Daher wurden neben der Analyse der Einzelergebnisse zusätzlich ähnliche
Versuchsbedingungen, bei denen sich kaum Unterschiede zeigten, in Gruppen zusammengefasst und mit
Hilfe der größten aufgetretenen Standardabweichung der Ergebnisse die P berechnet. Zusätzlich wurde mit
Hilfe der Standardabweichung der Ergebnisse der Stichprobenumfang eines Versuchs errechnet, der die P
der Ergebnisse erhöht. Hierbei wurde stets die größtmögliche vorhandene Standardabweichung in die
Analyse einbezogen und eine P von 80 % bei einem Alpha-Fehler von 0,05 angestrebt.
In den Fällen, in denen die P mehr als 80 % bei Gruppierung der Ergebnisse erreichte, erfolgte eine
Untersuchung der Unterschiede auf Signifikanz mit einer „One Way Analysis of Variance“ (ANOVA). Sofern
keine Normalverteilung der Ergebnisse vorlag, wurde die Analyse mit der ANOVA on Ranks (Kruskal-Wallis)
bei einem Signifikanzniveau von <0,05 durchgeführt.

4 Ergebnisse
50
4 Ergebnisse
4.1 In-vitro-Kultivierung der Chondrozyten unter Normaldruck
Im ersten Teil der Studie wurde unter Standard-Zellkulturbedingungen bei Normaldruck (Luftdruck der
Atmosphäre, 1,013 bar) der Einfluss der Zellkulturdauer auf die Morphologie in-vitro-kultivierter
Chondrozyten sowie deren Expression von Aggrekan, Kollagen Typ I und II untersucht. Hierfür wurden diese
über einen Zeitraum von 35 Tagen kultiviert und an den Kulturtagen 1, 2, 3, 4, 7, 14, 21 und 35 nach der
Aussaat phasenkontrastmikroskopisch untersucht. An jedem dieser Untersuchungstage erfolgte zudem der
immunzytochemische Nachweis von Kollagen Typ I und II. Des Weiteren wurden an jedem dieser
Zeitpunkte RNA und Protein zur Analyse der Expression von Kollagen Typ I und II sowie Aggrekan isoliert.
4.1.1 Morphologie in-vitro-kultivierter Chondrozyten unter Normaldruck
Die Chondrozyten stellten sich in vitro am ersten Tag nach der Aussaat als langgestreckte überwiegend
bipolare Zellen dar, die unregelmäßig verteilt adhärent auf dem Boden der Zellkulturschale vorlagen und
nach 6-7 Tagen einen optisch konfluenten Monolayer bildeten (Abb.7 A, Abb. 7 B).
1 T
ag
2 T
age
3 T
age
4 T
age
Abb. 7 A: Humane Chondrozyten in vitro 1, 2, 3 und 4 Tage nach der Aussaat. Phasenkontrastmikroskop, 100 x,
Skalierung: 250 µm.
Mit Zunahme der Zellkonzentration lagerten sich Chondrozyten ab Kulturtag 4 konzentrisch unter Bildung
von Zellwirbeln zusammen oder formten bei paralleler Ausrichtung zwischen diesen Wirbeln verlaufende
Zellstränge. Bis zum Kulturtag 7 zeigten sich die Chondrozyten in den Wirbeln optisch dichter als solche in
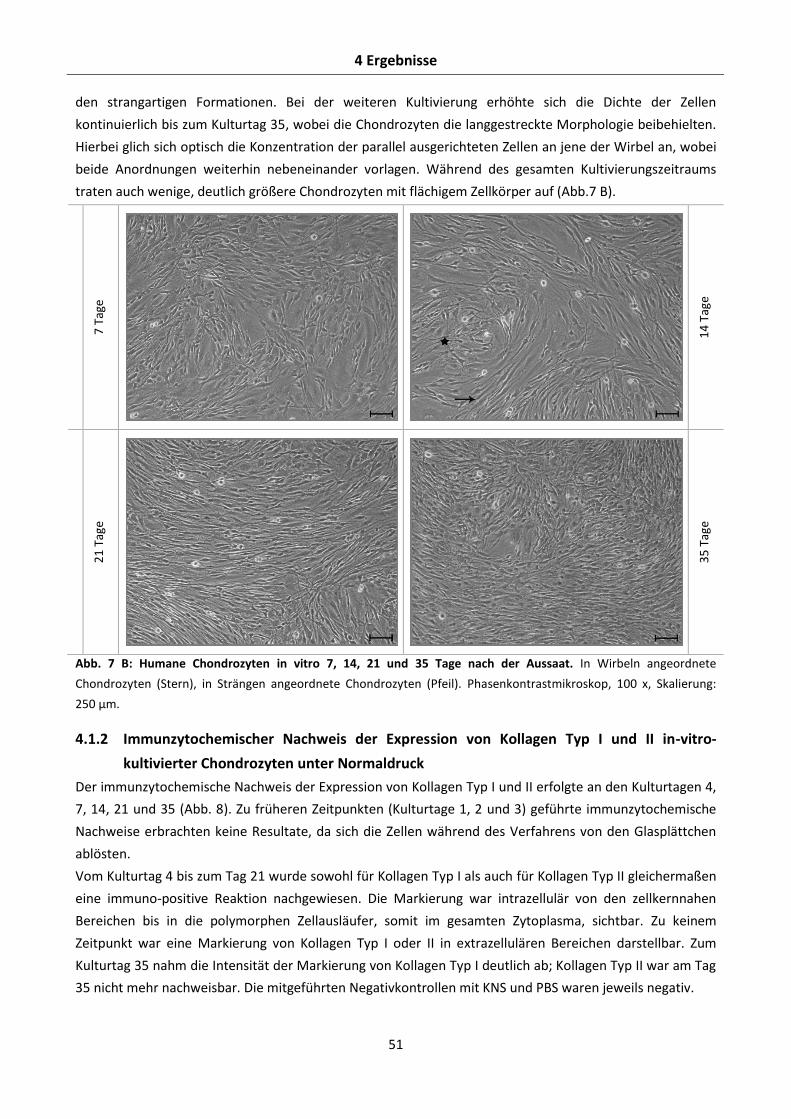
4 Ergebnisse
51
den strangartigen Formationen. Bei der weiteren Kultivierung erhöhte sich die Dichte der Zellen
kontinuierlich bis zum Kulturtag 35, wobei die Chondrozyten die langgestreckte Morphologie beibehielten.
Hierbei glich sich optisch die Konzentration der parallel ausgerichteten Zellen an jene der Wirbel an, wobei
beide Anordnungen weiterhin nebeneinander vorlagen. Während des gesamten Kultivierungszeitraums
traten auch wenige, deutlich größere Chondrozyten mit flächigem Zellkörper auf (Abb.7 B).
7 T
age
14
Tag
e
21
Tag
e
35
Tag
e
Abb. 7 B: Humane Chondrozyten in vitro 7, 14, 21 und 35 Tage nach der Aussaat. In Wirbeln angeordnete
Chondrozyten (Stern), in Strängen angeordnete Chondrozyten (Pfeil). Phasenkontrastmikroskop, 100 x, Skalierung:
250 µm.
4.1.2 Immunzytochemischer Nachweis der Expression von Kollagen Typ I und II in-vitro-
kultivierter Chondrozyten unter Normaldruck
Der immunzytochemische Nachweis der Expression von Kollagen Typ I und II erfolgte an den Kulturtagen 4,
7, 14, 21 und 35 (Abb. 8). Zu früheren Zeitpunkten (Kulturtage 1, 2 und 3) geführte immunzytochemische
Nachweise erbrachten keine Resultate, da sich die Zellen während des Verfahrens von den Glasplättchen
ablösten.
Vom Kulturtag 4 bis zum Tag 21 wurde sowohl für Kollagen Typ I als auch für Kollagen Typ II gleichermaßen
eine immuno-positive Reaktion nachgewiesen. Die Markierung war intrazellulär von den zellkernnahen
Bereichen bis in die polymorphen Zellausläufer, somit im gesamten Zytoplasma, sichtbar. Zu keinem
Zeitpunkt war eine Markierung von Kollagen Typ I oder II in extrazellulären Bereichen darstellbar. Zum
Kulturtag 35 nahm die Intensität der Markierung von Kollagen Typ I deutlich ab; Kollagen Typ II war am Tag
35 nicht mehr nachweisbar. Die mitgeführten Negativkontrollen mit KNS und PBS waren jeweils negativ.
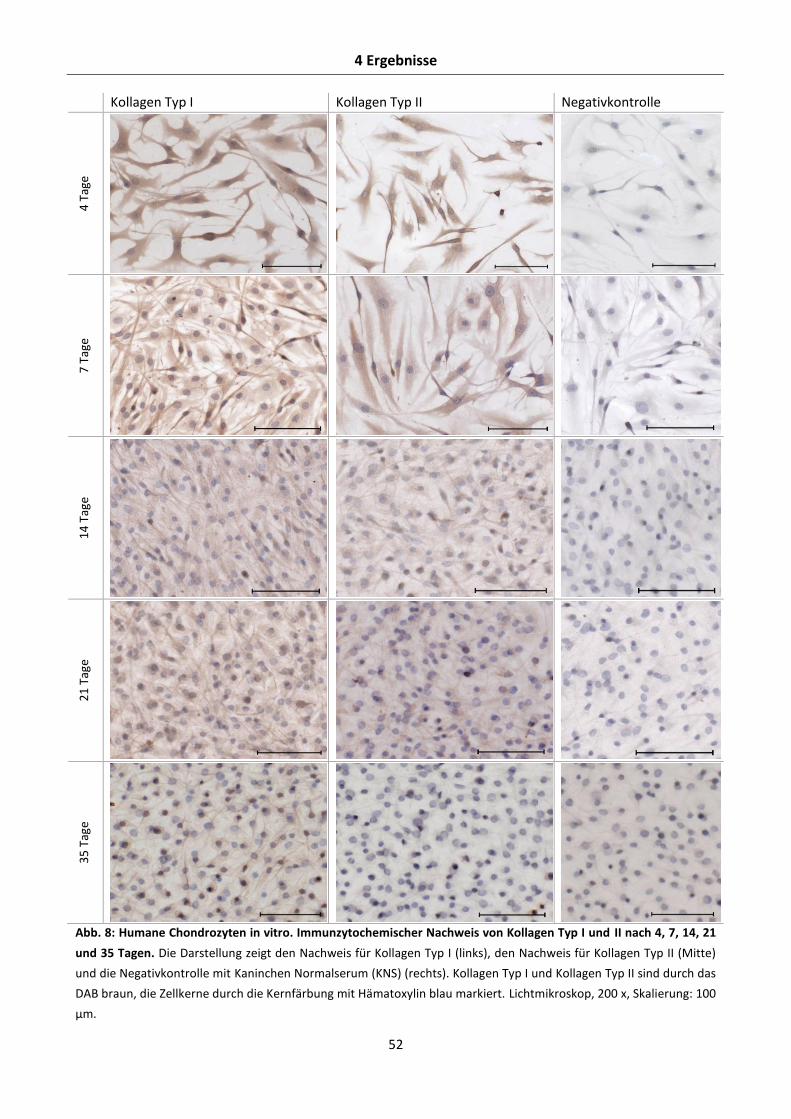
4 Ergebnisse
52
Kollagen Typ I Kollagen Typ II Negativkontrolle
4 T
age
7 T
age
14
Tag
e
21
Tag
e
35
Tag
e
Abb. 8: Humane Chondrozyten in vitro. Immunzytochemischer Nachweis von Kollagen Typ I und II nach 4, 7, 14, 21
und 35 Tagen. Die Darstellung zeigt den Nachweis für Kollagen Typ I (links), den Nachweis für Kollagen Typ II (Mitte)
und die Negativkontrolle mit Kaninchen Normalserum (KNS) (rechts). Kollagen Typ I und Kollagen Typ II sind durch das
DAB braun, die Zellkerne durch die Kernfärbung mit Hämatoxylin blau markiert. Lichtmikroskop, 200 x, Skalierung: 100
µm.

4 Ergebnisse
53
4.1.3 Nachweis der Aggrekan-, Kollagen Typ I-, und Kollagen Typ II-mRNA-Expression in-vitro-
kultivierter Chondrozyten unter Normaldruck
Der Nachweis der mRNA-Expression für Kollagen Typ I und II sowie für Aggrekan durch Chondrozyten in
vitro erfolgte mittels qPCR.
Im ersten Schritt wurde die Funktion der eingesetzten Primer durch die Darstellung des in der qualitativen
PCR erhaltenen PCR-Produkts im Agarosegel gezeigt (Abb. 9 A). Das entstandene PCR-Produkt wurde
anschließend kloniert. Dabei wurden die Kolonie-PCR-Produkte der Bakterienklone, die für den Einbau des
PCR-Produkts positiv waren, im Agarosegel anhand ihrer Größe von 419 bp (Kollagen Typ I) und 388 bp
(Kollagen Typ II) und 408 bp (Aggrekan) identifiziert (Abb. 9 B). Hierauf basierend wurden zur Erstellung des
Plasmidstandards für die qPCR die positiven Bakterienklone (K) COL1A1-K7 für Kollagen Typ I, COL2A1-K8
für Kollagen Typ II und ACAN-K6 für Aggrekan ausgewählt.
Abb. 9: Aufnahme der Agarosegele der qualitativen PCR und der Kolonie-PCR. A: Aufnahme der Agarosegele der
qualitativen PCR. Auf das Agarosegel wurde das PCR-Produkt der qualitativen PCR für Kollagen Typ I (COL1A1),
Kollagen Typ II (COL2A1) und Aggrekan (ACAN) aufgetragen um die verwendeten Primer zu verifizieren (+ PCR mit
cDNA, - PCR bei Abwesenheit der cDNA (Negativkontrolle)). B: Aufnahme der Agarosegele der Kolonie-PCR. Die
Bakterienklone COL1A1-K7 und COL1A1-K8 konnten in der Kolonie-PCR durch ihre Größe von 419 bp als positiv für
den Einbau des PCR-Produkts für Kollagen Typ I identifiziert werden. Die Bakterienklone COL2A1-K9 und COL2A1-K10
wurden als positiv für den Einbau des PCR-Produkts für Kollagen Typ II mit 388 bp identifiziert. Der Einbau des PCR-
Produkts für Aggrekan mit 408 bp konnte nur für den Bakterienklon ACAN-K6 nachgewiesen werden, wohingegen die
Bakterienklone ACAN-K7, ACAN-K8, ACAN-K9 deutlich kürzere PCR-Produkte bei 202 bp aufwiesen und somit negativ
für dessen Einbau waren. Es wurde eine Negativkontrolle (NK) ohne PCR-Produkt sowie ein Marker zur Bestimmung
der Größe des PCR-Produkts in Basenpaaren (bp) mitgeführt.
Für jeden qPCR-Lauf erfolgte eine Qualitätskontrolle. Hierfür wurde zum einen die Fluoreszenzintensität
der dsDNA in Abhängigkeit von der Zahl der PCR-Zyklen untersucht (Abb. 10 A). Weiterhin wurde eine
Schmelzkurvenanalyse durchgeführt, die das Verhältnis der Änderung der Fluoreszenzintensität über die
Temperaturänderung zeigte (Abb. 10 B). Zusätzlich wurde der Standardgraph für den Plasmidstandard
erstellt (Abb. 10 C) und schließlich das erhaltene PCR-Produkt im Agarosegel aufgetragen (Abb. 11).
B
Mar
ker
Mar
ker
AC
AN
+ -
CO
L2A
1
CO
L1A
1
3000
1000
500
200
Mar
ker
Mar
ker
+ - + -
A
bp
3000 1000
500
200
Mar
ker
Mar
ker
Mar
ker
CO
L1A
1
CO
L2A
1
AC
AN
K7 K8 K9 K10 K6 K7 K8 K9 NK + + + + + - - -
bp
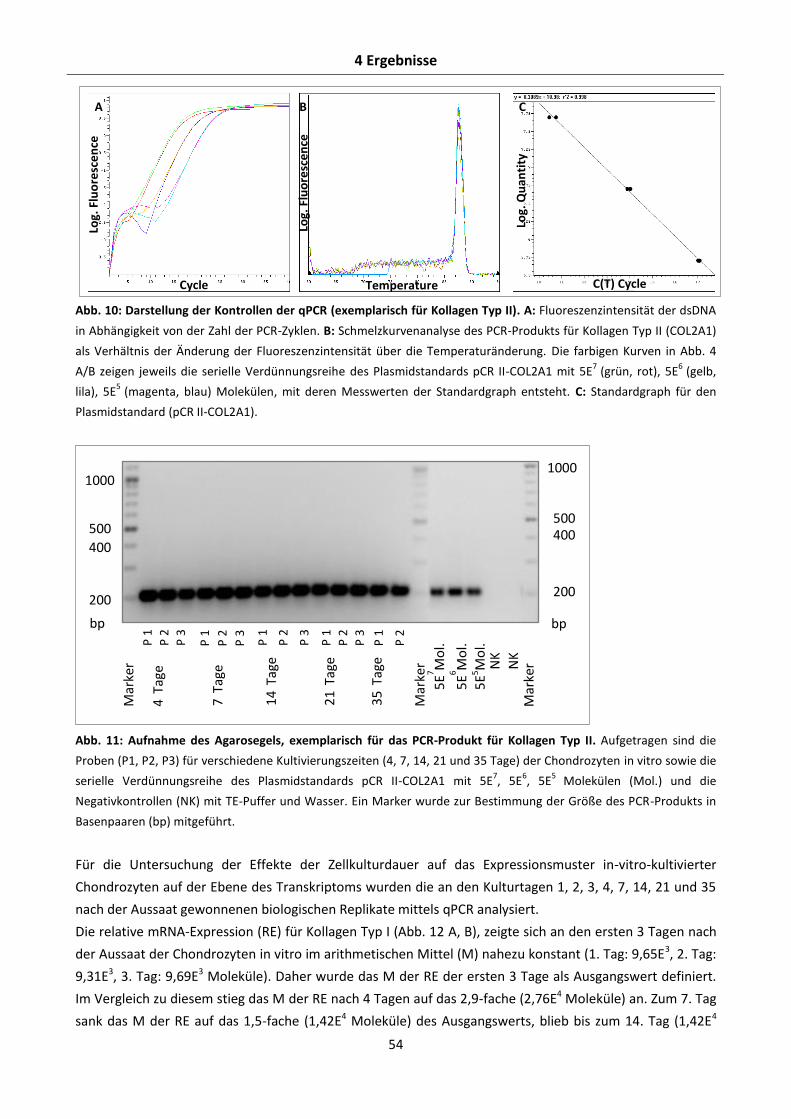
4 Ergebnisse
54
Abb. 10: Darstellung der Kontrollen der qPCR (exemplarisch für Kollagen Typ II). A: Fluoreszenzintensität der dsDNA
in Abhängigkeit von der Zahl der PCR-Zyklen. B: Schmelzkurvenanalyse des PCR-Produkts für Kollagen Typ II (COL2A1)
als Verhältnis der Änderung der Fluoreszenzintensität über die Temperaturänderung. Die farbigen Kurven in Abb. 4
A/B zeigen jeweils die serielle Verdünnungsreihe des Plasmidstandards pCR II-COL2A1 mit 5E7
(grün, rot), 5E6
(gelb,
lila), 5E5
(magenta, blau) Molekülen, mit deren Messwerten der Standardgraph entsteht. C: Standardgraph für den
Plasmidstandard (pCR II-COL2A1).
Abb. 11: Aufnahme des Agarosegels, exemplarisch für das PCR-Produkt für Kollagen Typ II. Aufgetragen sind die
Proben (P1, P2, P3) für verschiedene Kultivierungszeiten (4, 7, 14, 21 und 35 Tage) der Chondrozyten in vitro sowie die
serielle Verdünnungsreihe des Plasmidstandards pCR II-COL2A1 mit 5E7, 5E
6, 5E
5 Molekülen (Mol.) und die
Negativkontrollen (NK) mit TE-Puffer und Wasser. Ein Marker wurde zur Bestimmung der Größe des PCR-Produkts in
Basenpaaren (bp) mitgeführt.
Für die Untersuchung der Effekte der Zellkulturdauer auf das Expressionsmuster in-vitro-kultivierter
Chondrozyten auf der Ebene des Transkriptoms wurden die an den Kulturtagen 1, 2, 3, 4, 7, 14, 21 und 35
nach der Aussaat gewonnenen biologischen Replikate mittels qPCR analysiert.
Die relative mRNA-Expression (RE) für Kollagen Typ I (Abb. 12 A, B), zeigte sich an den ersten 3 Tagen nach
der Aussaat der Chondrozyten in vitro im arithmetischen Mittel (M) nahezu konstant (1. Tag: 9,65E3, 2. Tag:
9,31E3, 3. Tag: 9,69E3 Moleküle). Daher wurde das M der RE der ersten 3 Tage als Ausgangswert definiert.
Im Vergleich zu diesem stieg das M der RE nach 4 Tagen auf das 2,9-fache (2,76E4 Moleküle) an. Zum 7. Tag
sank das M der RE auf das 1,5-fache (1,42E4 Moleküle) des Ausgangswerts, blieb bis zum 14. Tag (1,42E4
Temperature
Log.
Flu
ore
sce
nce
B
Cycle
Log.
Flu
ore
sce
nce
A
Log.
Qu
anti
ty
C(T) Cycle
C
Temperature
Mar
ker
4 T
age
7 T
age
14 T
age
21 T
age
35 T
age
Mar
ker
Mar
ker N
K
NK
5E7 M
ol.
5E6 M
ol.
5E5 M
ol. P
1
P 2
P 3
P 1
P 2
P 3
P 1
P 2
P 3
P 1
P 2
1000 1000
500 500
400 400
200 200
bp bp
P 1
P 2
P 3

4 Ergebnisse
55
Moleküle) nahezu konstant, um bis zum 21. Tag auf das 0,8-fache (8,21E3 Moleküle) des Ausgangswerts
abzusinken. Bei längerer Kultivierungszeit stieg das M der RE zum 35. Tag an und erreichte annährend das
2-fache des Ausgangswerts (2,00E4 Moleküle).
Abb. 12: Relative Expression der mRNA von Kollagen Typ I und II in Abhängigkeit von der Kultivierungszeit durch
Chondrozyten in vitro. A: Darstellung der Ergebnisse der qPCR für Kollagen Typ I und II als Balkendiagramm mit
Mittelwert und Standardabweichung. B, C: Punktdarstellung der arithmetischen Mittel der technischen Replikate
eines jeden biologischen Replikats für Kollagen Typ I (B) und Kollagen Typ II (C). Hierbei ist die Anzahl der biologischen
Replikate (n) angegeben. Die Darstellung der Molekülzahl des Kollagen Typ I und II erfolgt relativ zu 1E5 Molekülen des
Referenzgens 36B4.
0,0E+0
1,0E+4
2,0E+4
3,0E+4
4,0E+4
1 2 3 4 7 14 21 35
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Kultivierungszeit [Tage]
Kollagen Typ I
Kollagen Typ II
0,0E+0
1,0E+4
2,0E+4
3,0E+4
4,0E+4
5,0E+4
1(n=2)
2(n=2)
3(n=2)
4(n=3)
7(n=3)
14(n=3)
21(n=3)
35(n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Kultivierungszeit [Tage]
Kollagen Typ I
0,0E+0
5,0E+3
1,0E+4
1,5E+4
2,0E+4
2,5E+4
3,0E+4
1(n=2)
2(n=2)
3(n=2)
4(n=3)
7(n=3)
14(n=3)
21(n=3)
35(n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Kultivierungszeit [Tage]
Kollagen Typ II
A
B
C

4 Ergebnisse
56
Das M der RE des Kollagen Typ II (Abb. 12 A, C) verhielt sich an den ersten 3 Kulturtagen nach der Aussaat
ebenfalls nahezu konstant mit 1,26E3 am 1. Tag, 1,43E3 am 2. Tag und 1,13E3 Molekülen am 3. Tag, sodass
auch hier das M über die ersten 3 Tage als Ausgangswert diente. Am 4. Tag zeigte sich ein Anstieg auf das
14,2-fache (1,81E3 Moleküle) des M der RE des Ausgangswerts. Am 7. Tag fiel das M der RE auf das 8-fache
(1,07E4 Moleküle) des Ausgangswerts ab. Anschließend blieb das M der RE des Kollagen Typ II bis zum 21.
Tag konstant und stieg bei längerer Kultivierungszeit bis zum 35. Tag auf das 13,8-fache (1,76E4 Moleküle)
des Ausgangswerts an.
Das M der RE des Aggrekan (Abb. 13) erreichte an den ersten 3 Tagen nach der Aussaat Werte von 7,59E0
Molekülen am 1. Tag, 2,62E0 Molekülen am 2. Tag und 6,32E0 Molekülen am 3. Tag. Daher wurde auch hier
das M über die ersten 3 Tage als Ausgangswert definiert. Am 4. Tag stieg das M der RE des Aggrekan auf
das 7,1-fache (4,49E1 Moleküle) des Ausgangswerts, lag am 7. Tag um das 5,3-fache höher (3,34E1
Moleküle) als dieser und blieb bei längerer Kultivierungszeit bis zum Tag 35 annähernd stabil.
Abb. 13: Relative Expression der mRNA von Aggrekan in Abhängigkeit von der Kultivierungszeit durch Chondrozyten
in vitro. A: Darstellung der Ergebnisse der qPCR für Aggrekan als Balkendiagramm mit Mittelwert und
Standardabweichung. B: Punktdarstellung der arithmetischen Mittel der technischen Replikate eines jeden
biologischen Replikats für Aggrekan. Hierbei ist die Anzahl der biologischen Replikate (n) angegeben. Die Darstellung
der Molekülzahl des Aggrekan erfolgt relativ zu 1E5 Molekülen des Referenzgens 36B4.
In der statistischen Analyse der mRNA-Expression der Chondrozyten in Bezug auf Kollagen Typ I, II und
Aggrekan in Abhängigkeit von der Kultivierungszeit wurden die ersten 3 Kulturtage aufgrund der
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
1 2 3 4 7 14 21 35
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Kultivierungszeit [Tage]
Aggrekan
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
7,0E+1
1(n=2)
2(n=2)
3(n=2)
4(n=3)
7(n=3)
14(n=3)
21(n=3)
35(n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Kultivierungszeit [Tage]
Aggrekan
A
B

4 Ergebnisse
57
konstanten Expressionen als Gruppe definiert. Hierdurch wurde statistisch der Stichprobenumfang (n)
erhöht. Die Differenz der Mittelwerte der RE des Kollagen Typ I und Typ II dieser Gruppe zu der RE des
7. Kulturtags wird bei der vorliegenden Standardabweichung der Ergebnisse bei n=2 erkannt, sodass durch
die Gruppierung der Ergebnisse (Tag 1-3 n=6, Tag 7 n=3) der Stichprobenumfang ausreicht, um diesen
Unterschied zu erkennen (Power (P) der Ergebnisse 80 %). Die Differenz der Mittelwerte der RE des
Aggrekan ist niedriger, sodass eine Erhöhung des Stichprobenumfangs auf n=9 notwendig wäre, um eine P
der Ergebnisse von 80 % zu erreichen. In einer weiteren Analyse wurden neben der RE der ersten
3 Kulturtage zusätzlich die RE der Kulturtage 4, 7, 14, 21 und 35 zu einer Gruppe zusammengefasst. Hierbei
lässt sich trotz der auftretenden hohen Standardabweichung die Aussage treffen, dass die Expression von
Kollagen Typ II und von Aggrekan auf Transkriptomebene bei längerer Kultivierung (4-35 Tage) höher liegt
als bei kurzer Kultivierungsdauer (1-3 Tage) (P von 97 %). Für Kollagen Typ I ist die Differenz der
Mittelwerte der Gruppen (1-3 Tage und 4- 35Tage) geringer bei gleichzeitig höherer Variabilität der
biologischen Replikate, sodass sich diese Aussage für Kollagen Typ I nicht treffen lässt.
Unter Berücksichtigung der vorangegangenen statistischen Analysen erfolgte eine Untersuchung der
Unterschiede zwischen diesen zusammengefassten Gruppen auf Signifikanz (ANOVA on ranks), d.h.
zwischen den ersten 3 Kulturtage und dem 7. Kulturtag, sowie zwischen der kurzen Kultivierungszeit (1-3
Tage) und der langen Kultivierungszeit (4-35 Tage). Dabei zeigte sich die Expression von Kollagen Typ I und
II sowie von Aggrekan am 7. Kulturtag signifikant (Signifkanzniveau <0,05) höher als an den Kulturtagen 1-3
und bei langer Kultivierungszeit signifikant höher als bei kurzer Kultivierungszeit (Signifkanzniveau <0,05).
4.1.4 Nachweis der Aggrekan-, Kollagen Typ I- und Kollagen Typ II-Proteinexpression in-vitro-
kultivierter Chondrozyten unter Normaldruck
Der Nachweis der Proteinexpression des Kollagen Typ I und II sowie des Aggrekan durch Chondrozyten in
vitro erfolgte mittels Western Blot.
In Vorversuchen wurde dieses Verfahren für die dabei genutzten spezifischen Antikörper zunächst
optimiert. Hierbei zeigte sich für jedes der Zielproteine nicht nur eine spezifische Bande, sodass für
Kollagen Typ I die Banden bei einem Molekulargewicht von 38, 50, 120 kDa, für Kollagen Typ II bei 65 und
70 kDa sowie für Aggrekan bei 45 und 65 kDa (Abb. 14 A) ausgewertet wurden.
Das Lumineszenz-Signal dieser festgelegten Banden wurde zur Quantifizierung der Lumineszenz-Intensität
(LI) ins Verhältnis zu der des Referenzproteins β-Aktin (Abb. 14 B) gesetzt und somit die Ergebnisse als
relative Lumineszenz-Intensität (RLI) angegeben. Da sich für den ersten Kulturtag nach der Aussaat kein
Lumineszenz-Signal für β-Aktin detektieren ließ, erfolgte die Quantifizierung der Proteinexpression erst ab
dem 2. Tag.
Die Untersuchung der Effekte der Zellkulturdauer auf die Proteinexpression von Kollagen Typ I und II sowie
Aggrekan durch in-vitro-kultivierte Chondrozyten erfolgte mit Hilfe der an den Kulturtagen 1, 2, 3, 4, 7, 14,
21 und 35 nach der Aussaat gewonnenen Proteinproben.
Dabei zeigte sich am 2. Kulturtag für Kollagen Typ I (Abb. 15 A) bei der Betrachtung der 38 kDa-Bande ein
RLI-Wert von 1,00. Dieser stieg zum 3. Tag leicht an (1,31) und fiel am 4. Tag auf 0,39 ab, um anschließend
bis zum 35. Tag, mit Ausnahme des 21. Tags, an dem die RLI niedriger war als am vorherigen
Untersuchungstag 14, wieder bis zu einer RLI von 1,02 anzusteigen. Die 50 kDa-Bande wies am 2. Tag einen
RLI-Wert von 2,37 auf, welcher am 3. Tag auf 2,14 und am 4. Tag auf 1,07 abfiel. Im Anschluss stieg die RLI
bis zum 35. Tag auf 2,03 an, wobei ebenso wie bei der 38 kDa-Bande am 21. Tag die RLI niedriger lag als am
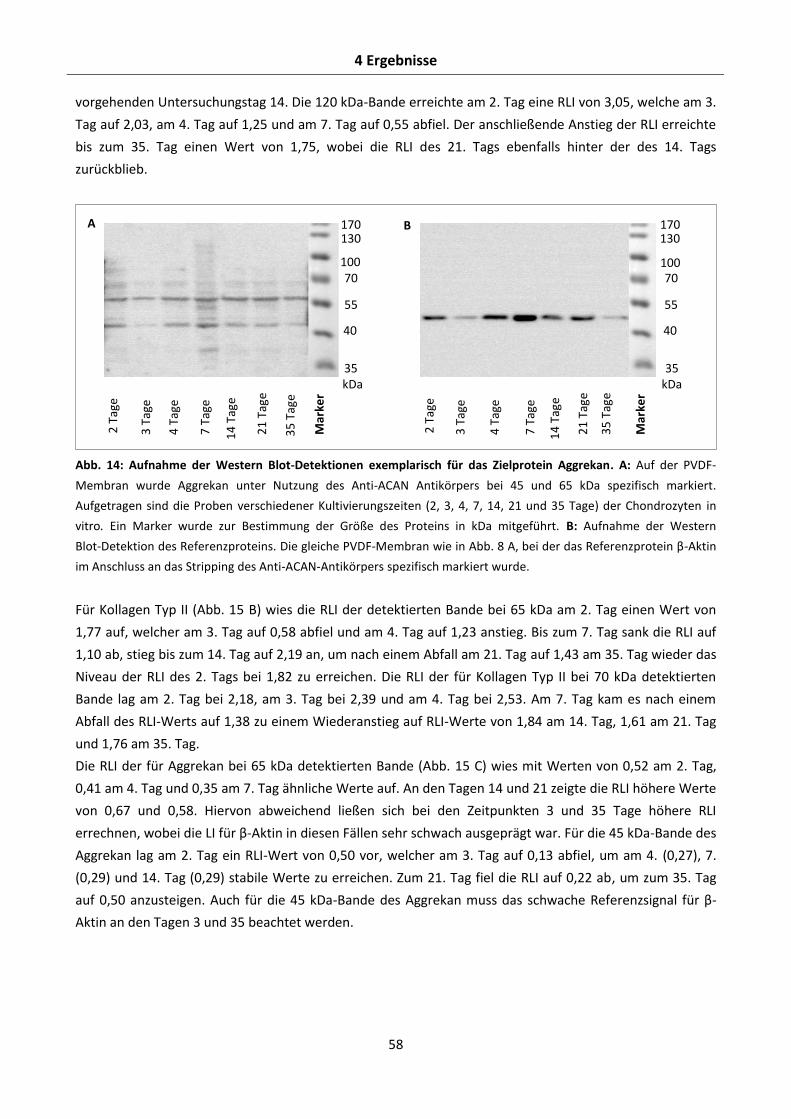
4 Ergebnisse
58
vorgehenden Untersuchungstag 14. Die 120 kDa-Bande erreichte am 2. Tag eine RLI von 3,05, welche am 3.
Tag auf 2,03, am 4. Tag auf 1,25 und am 7. Tag auf 0,55 abfiel. Der anschließende Anstieg der RLI erreichte
bis zum 35. Tag einen Wert von 1,75, wobei die RLI des 21. Tags ebenfalls hinter der des 14. Tags
zurückblieb.
Abb. 14: Aufnahme der Western Blot-Detektionen exemplarisch für das Zielprotein Aggrekan. A: Auf der PVDF-
Membran wurde Aggrekan unter Nutzung des Anti-ACAN Antikörpers bei 45 und 65 kDa spezifisch markiert.
Aufgetragen sind die Proben verschiedener Kultivierungszeiten (2, 3, 4, 7, 14, 21 und 35 Tage) der Chondrozyten in
vitro. Ein Marker wurde zur Bestimmung der Größe des Proteins in kDa mitgeführt. B: Aufnahme der Western
Blot-Detektion des Referenzproteins. Die gleiche PVDF-Membran wie in Abb. 8 A, bei der das Referenzprotein β-Aktin
im Anschluss an das Stripping des Anti-ACAN-Antikörpers spezifisch markiert wurde.
Für Kollagen Typ II (Abb. 15 B) wies die RLI der detektierten Bande bei 65 kDa am 2. Tag einen Wert von
1,77 auf, welcher am 3. Tag auf 0,58 abfiel und am 4. Tag auf 1,23 anstieg. Bis zum 7. Tag sank die RLI auf
1,10 ab, stieg bis zum 14. Tag auf 2,19 an, um nach einem Abfall am 21. Tag auf 1,43 am 35. Tag wieder das
Niveau der RLI des 2. Tags bei 1,82 zu erreichen. Die RLI der für Kollagen Typ II bei 70 kDa detektierten
Bande lag am 2. Tag bei 2,18, am 3. Tag bei 2,39 und am 4. Tag bei 2,53. Am 7. Tag kam es nach einem
Abfall des RLI-Werts auf 1,38 zu einem Wiederanstieg auf RLI-Werte von 1,84 am 14. Tag, 1,61 am 21. Tag
und 1,76 am 35. Tag.
Die RLI der für Aggrekan bei 65 kDa detektierten Bande (Abb. 15 C) wies mit Werten von 0,52 am 2. Tag,
0,41 am 4. Tag und 0,35 am 7. Tag ähnliche Werte auf. An den Tagen 14 und 21 zeigte die RLI höhere Werte
von 0,67 und 0,58. Hiervon abweichend ließen sich bei den Zeitpunkten 3 und 35 Tage höhere RLI
errechnen, wobei die LI für β-Aktin in diesen Fällen sehr schwach ausgeprägt war. Für die 45 kDa-Bande des
Aggrekan lag am 2. Tag ein RLI-Wert von 0,50 vor, welcher am 3. Tag auf 0,13 abfiel, um am 4. (0,27), 7.
(0,29) und 14. Tag (0,29) stabile Werte zu erreichen. Zum 21. Tag fiel die RLI auf 0,22 ab, um zum 35. Tag
auf 0,50 anzusteigen. Auch für die 45 kDa-Bande des Aggrekan muss das schwache Referenzsignal für β-
Aktin an den Tagen 3 und 35 beachtet werden.
3 T
age
Mar
ker
21
Tag
e
35
Tag
e
4 T
age
7 T
age
14
Tag
e
170
70
40
55
35
2 T
age
130
100
kDa
3 T
age
Mar
ker
21
Tag
e
35
Tag
e
4 T
age
7 T
age
14
Tag
e
170
70
40
55
35
2 T
age
130
100
kDa
A B
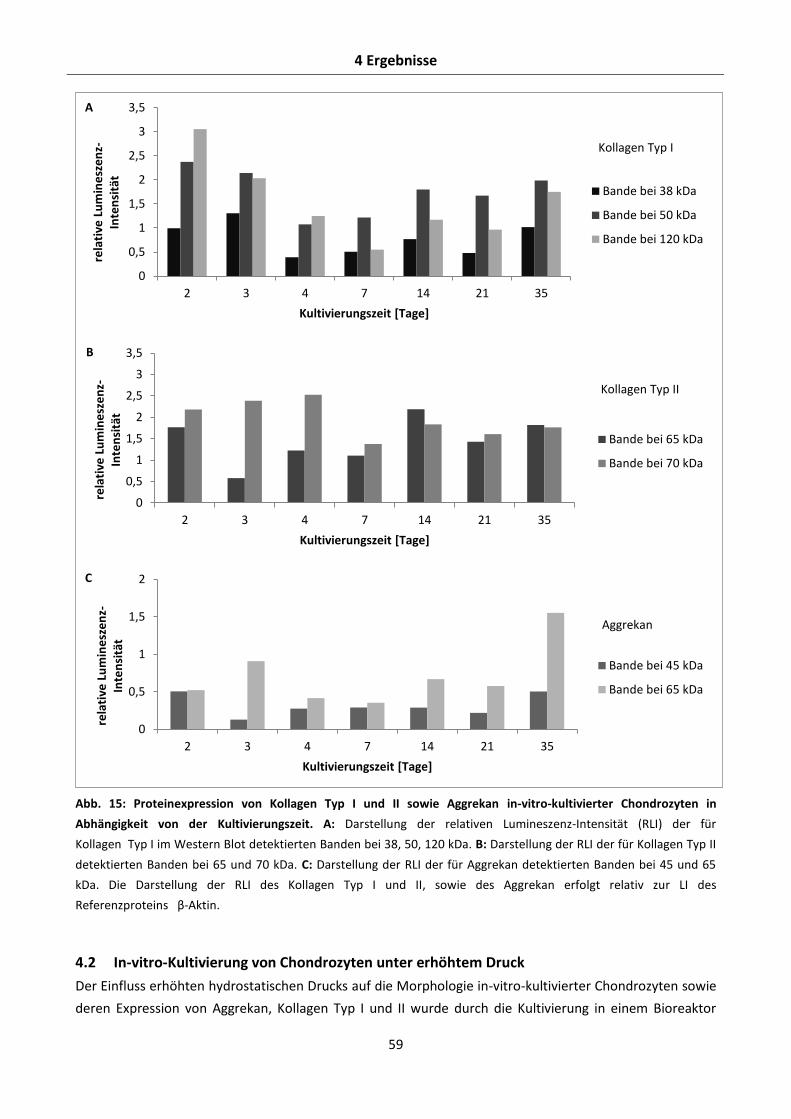
4 Ergebnisse
59
Abb. 15: Proteinexpression von Kollagen Typ I und II sowie Aggrekan in-vitro-kultivierter Chondrozyten in
Abhängigkeit von der Kultivierungszeit. A: Darstellung der relativen Lumineszenz-Intensität (RLI) der für
Kollagen Typ I im Western Blot detektierten Banden bei 38, 50, 120 kDa. B: Darstellung der RLI der für Kollagen Typ II
detektierten Banden bei 65 und 70 kDa. C: Darstellung der RLI der für Aggrekan detektierten Banden bei 45 und 65
kDa. Die Darstellung der RLI des Kollagen Typ I und II, sowie des Aggrekan erfolgt relativ zur LI des
Referenzproteins β-Aktin.
4.2 In-vitro-Kultivierung von Chondrozyten unter erhöhtem Druck
Der Einfluss erhöhten hydrostatischen Drucks auf die Morphologie in-vitro-kultivierter Chondrozyten sowie
deren Expression von Aggrekan, Kollagen Typ I und II wurde durch die Kultivierung in einem Bioreaktor
0
0,5
1
1,5
2
2,5
3
3,5
2 3 4 7 14 21 35
rela
tive
Lu
min
esz
en
z-
Inte
nsi
tät
Kultivierungszeit [Tage]
Kollagen Typ I
Bande bei 38 kDa
Bande bei 50 kDa
Bande bei 120 kDa
A
0
0,5
1
1,5
2
2,5
3
3,5
2 3 4 7 14 21 35
rela
tive
Lu
min
esz
en
z-
Inte
nsi
tät
Kultivierungszeit [Tage]
Kollagen Typ II
Bande bei 65 kDa
Bande bei 70 kDa
B
0
0,5
1
1,5
2
2 3 4 7 14 21 35
rela
tive
Lu
min
esz
en
z-
Inte
nsi
tät
Kultivierungszeit [Tage]
Aggrekan
Bande bei 45 kDa
Bande bei 65 kDa
C
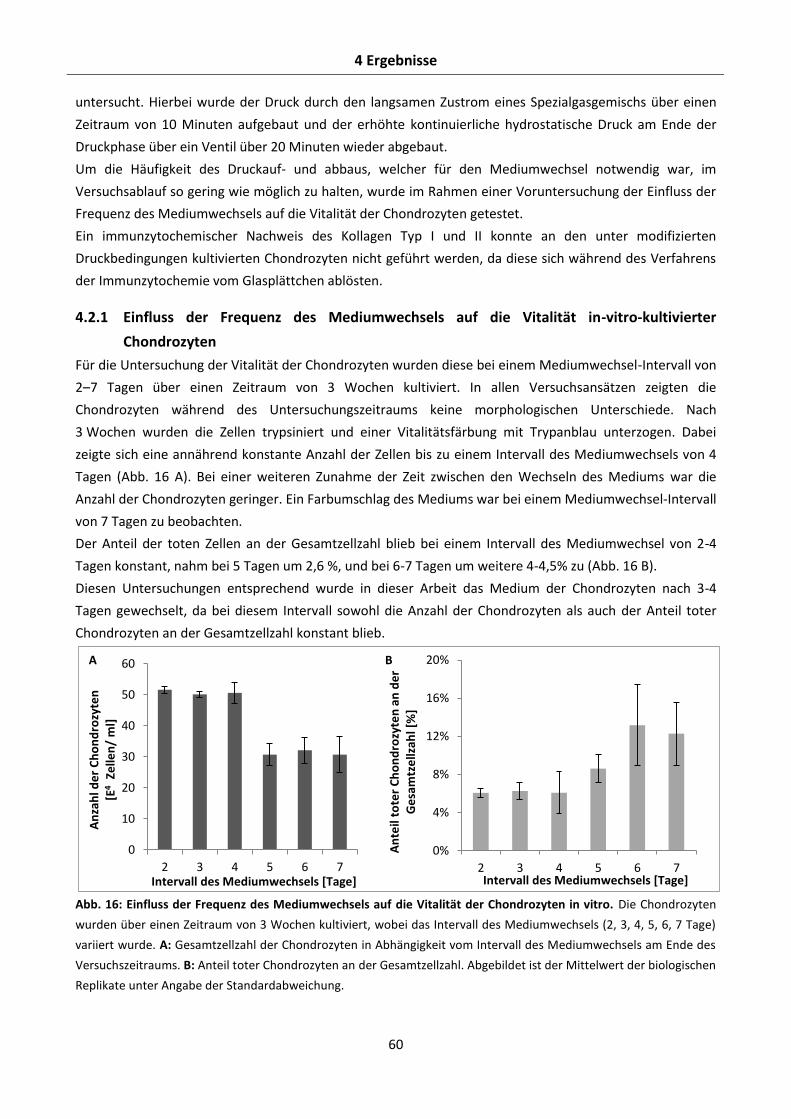
4 Ergebnisse
60
untersucht. Hierbei wurde der Druck durch den langsamen Zustrom eines Spezialgasgemischs über einen
Zeitraum von 10 Minuten aufgebaut und der erhöhte kontinuierliche hydrostatische Druck am Ende der
Druckphase über ein Ventil über 20 Minuten wieder abgebaut.
Um die Häufigkeit des Druckauf- und abbaus, welcher für den Mediumwechsel notwendig war, im
Versuchsablauf so gering wie möglich zu halten, wurde im Rahmen einer Voruntersuchung der Einfluss der
Frequenz des Mediumwechsels auf die Vitalität der Chondrozyten getestet.
Ein immunzytochemischer Nachweis des Kollagen Typ I und II konnte an den unter modifizierten
Druckbedingungen kultivierten Chondrozyten nicht geführt werden, da diese sich während des Verfahrens
der Immunzytochemie vom Glasplättchen ablösten.
4.2.1 Einfluss der Frequenz des Mediumwechsels auf die Vitalität in-vitro-kultivierter
Chondrozyten
Für die Untersuchung der Vitalität der Chondrozyten wurden diese bei einem Mediumwechsel-Intervall von
2–7 Tagen über einen Zeitraum von 3 Wochen kultiviert. In allen Versuchsansätzen zeigten die
Chondrozyten während des Untersuchungszeitraums keine morphologischen Unterschiede. Nach
3 Wochen wurden die Zellen trypsiniert und einer Vitalitätsfärbung mit Trypanblau unterzogen. Dabei
zeigte sich eine annährend konstante Anzahl der Zellen bis zu einem Intervall des Mediumwechsels von 4
Tagen (Abb. 16 A). Bei einer weiteren Zunahme der Zeit zwischen den Wechseln des Mediums war die
Anzahl der Chondrozyten geringer. Ein Farbumschlag des Mediums war bei einem Mediumwechsel-Intervall
von 7 Tagen zu beobachten.
Der Anteil der toten Zellen an der Gesamtzellzahl blieb bei einem Intervall des Mediumwechsel von 2-4
Tagen konstant, nahm bei 5 Tagen um 2,6 %, und bei 6-7 Tagen um weitere 4-4,5% zu (Abb. 16 B).
Diesen Untersuchungen entsprechend wurde in dieser Arbeit das Medium der Chondrozyten nach 3-4
Tagen gewechselt, da bei diesem Intervall sowohl die Anzahl der Chondrozyten als auch der Anteil toter
Chondrozyten an der Gesamtzellzahl konstant blieb.
Abb. 16: Einfluss der Frequenz des Mediumwechsels auf die Vitalität der Chondrozyten in vitro. Die Chondrozyten
wurden über einen Zeitraum von 3 Wochen kultiviert, wobei das Intervall des Mediumwechsels (2, 3, 4, 5, 6, 7 Tage)
variiert wurde. A: Gesamtzellzahl der Chondrozyten in Abhängigkeit vom Intervall des Mediumwechsels am Ende des
Versuchszeitraums. B: Anteil toter Chondrozyten an der Gesamtzellzahl. Abgebildet ist der Mittelwert der biologischen
Replikate unter Angabe der Standardabweichung.
0
10
20
30
40
50
60
2 3 4 5 6 7
An
zah
l de
r C
ho
nd
rozy
ten
[E
4 Z
elle
n/
ml]
Intervall des Mediumwechsels [Tage]
0%
4%
8%
12%
16%
20%
2 3 4 5 6 7
An
teil
tote
r C
ho
nd
rozy
ten
an
de
r G
esa
mtz
ellz
ahl [
%]
Intervall des Mediumwechsels [Tage]
A B

4 Ergebnisse
61
4.2.2 Versuchsaufbau für die Untersuchung in-vitro-kultivierter Chondrozyten unter erhöhtem
Druck
Für die Untersuchung des erhöhten Drucks auf die Morphologie in-vitro-kultivierter Chondrozyten sowie
deren Expression von Aggrekan, Kollagen Typ I und II wurden der Zeitpunkt der Einwirkung des Drucks,
dessen Höhe sowie die Dauer der Druckeinwirkung als Parameter variiert. Zusätzlich wurde der Einfluss der
Zelldichte der Chondrozyten auf diese Parameter unter erhöhtem Druck untersucht. Die sich daraus
ergebenden Versuchsansätze sind in Abb. 17, Abb. 18 und Abb. 19 dargestellt.
Für die Untersuchung der Vorkultivierungszeit unter Normaldruck wurden die Chondrozyten direkt nach
der Aussaat (Abb. 17 A und D), nach 1 Tag bei erfolgter Adhärenz (Abb. 17 B und E) und bei Erreichen der
optischen Konfluenz nach 6 Tagen (Abb. 17 C und F) für die Dauer von 24 h in den Bioreaktor überführt. In
diesem wurden die Chondrozyten für 24 h bei einem Druck von 5 bar (Abb. 17 A-D) und 10 bar (Abb. 17 E-F)
kultiviert, um die Effekte der Höhe des hydrostatischen Drucks zu untersuchen.
Abb. 17: Schema des Versuchsaufbaus zur Untersuchung der Vorkultivierungszeit unter Normaldruck und der
Effekte der Höhe des hydrostatischen Drucks auf in-vitro-kultivierte Chondrozyten. Die Darstellung zeigt die hierbei
untersuchten Druckbedingungen A-F. Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter
Normaldruck (1 bar) 0, 1 oder 6 Tage (d) vorkultiviert. Anschließend wurden die Chondrozyten für 24 h im Bioreaktor
kultiviert, in welchem ein hydrostatischer Druck von 5 bar (schmaler Balken) (A-D) bzw. 10 bar (breiter Balken)(E-F) für
24 Stunden (h) wirkte.
Bei der Betrachtung der Morphologie in-vitro-kultivierter Chondrozyten unter erhöhtem Druck (Kap. 4.2.3)
zeigte sich bei einer Druckeinwirkung von 5 bar für 24 h bereits in einigen Zellkulturen eine Abrundung und
ein Ablösen der Zellen vom Boden der Zellkulturschale im Verlauf der Druckbehandlung. Solche Kulturen
lieferten keine auswertbaren Resultate in der qPCR. Vor dem Hintergrund, ein stabiles Zellkultursystem zu
entwickeln, mit welchem auswertbare Ergebnisse generiert werden können, wurden die Druckbedingungen
bei 10 bar im folgenden Versuch variiert und der Einfluss der Dauer der Druckphase an bei Normaldruck bis
zur optischen Konfluenz vorkultivierten Chondrozyten bei einem Druck von 10 bar für 1,5 h, 3 h, 6 h, 12 h,
24 h, 48 h und mehr als 72 h untersucht (Abb. 18 G-M).

4 Ergebnisse
62
Abb. 18: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Dauer der hydrostatischen Druckphase auf
die Morphologie in-vitro-kultivierter Chondrozyten. Die Darstellung zeigt die hierbei untersuchten Druckbedingungen
G-M. Die bis zur Konfluenz unter Normaldruck vorkultivierten Chondrozyten wurden für 1,5 h (G), 3 h (H), 6 h (I), 12 h
(J), 24 h (K), 48 h (L) und mehr als 72 h (M) bei einem hydrostatischen Druck von 10 bar im Bioreaktor kultiviert.
Schließlich wurde der Einfluss der Zelldichte der Chondrozyten auf deren Morphologie und Expression
unter hydrostatischem Druck untersucht. Hierfür wurden analog zu den Versuchsansätzen Abb. 18 H und I
neben den in normaler Zelldichte (104 Zellen/cm2) ausgesäten Chondrozyten zusätzlich in doppelter
Zelldichte ausgesäte Chondrozyten bei einem Druck von 10 bar und einer Druckphase von 3 h und 6 h im
Bioreaktor kultiviert (Abb. 19 H, I, N, O).
Abb. 19: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Zelldichte der Chondrozyten auf deren
mRNA-Expression bei der Einwirkung eines erhöhten hydrostatischen Drucks. Die Bezeichnung der jeweiligen
Kultivierungsbedingungen entspricht der in Abb. 18. Die in den Versuchen eingesetzten Chondrozyten wurden in
normaler (H, I) und doppelter Zelldichte (N, O) ausgesät und zunächst unter Normaldruck (1 bar) 6 Tage (d)
vorkultiviert. Anschließend wurden die Chondrozyten für 3 h (H, N) und 6 h (I, O) im Bioreaktor kultiviert, in welchem
ein hydrostatischer Druck von 10 bar wirkte.
Für jeden Versuchsansatz (Abb. 17 A-F, Abb. 18 G-M, Abb. 19 H, I, N, O) wurde jeweils eine Kontrollgruppe
von Chondrozyten mitgeführt, die abgesehen von der Druckeinwirkung identisch behandelt wurde.

4 Ergebnisse
63
4.2.3 Morphologie in-vitro-kultivierter Chondrozyten unter erhöhtem Druck
Die Chondrozyten wiesen bei hydrostatischer Druckeinwirkung von 5 bar für die Dauer von 24 h je nach
Dauer der Vorkultivierungszeit morphologische Unterschiede auf. Unter dem Einfluss des direkt nach der
Aussaat wirkenden Drucks (Abb. 17 A) zeigten die Chondrozyten keine Adhärenz zum Boden der
Kulturschale. Es fielen sowohl unregelmäßig begrenzte Chondrozyten, denen die bipolare Zellmorphologie
fehlte, auf, als auch solche Zellen mit größerem, flächigen Zellkörper. Bei diesen wurden zum einen eine
Verdichtung des Chromatins (Karyopyknosis) und zum anderen dessen Zerfall (Karyorrhexis) beobachtet
(Abb. 20).
0 Tage vorkultiviert 1 Tag vorkultiviert 6 Tage vorkultiviert
Ko
ntr
olle
(N
orm
ald
ruck
,1b
ar)
Dru
ck: 5
bar
Dru
ck: 1
0 b
ar
Abb. 20: Untersuchung der Vorkultivierungszeit unter Normaldruck und der Effekte der Höhe des hydrostatischen
Drucks auf die Morphologie in-vitro-kultivierter Chondrozyten. Abgerundete Morphologie der Chondrozyten (Pfeile),
Chondrozyten mit wenigen Kontakten zu Nachbarzellen (Pfeilköpfe). Phasenkontrastmikroskop, 200 x, Skalierung:
250 µm. Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter Normaldruck (1 bar) 0, 1 oder 6
Tage vorkultiviert. Anschließend wurden die Chondrozyten für 24 h im Bioreaktor kultiviert, in welchem ein
hydrostatischer Druck von 5 bar bzw. 10 bar für 24 Stunden (h) wirkte. Zum Vergleich sind zusätzlich die Kontrollen
(Normaldruck, 1 bar) dargestellt.
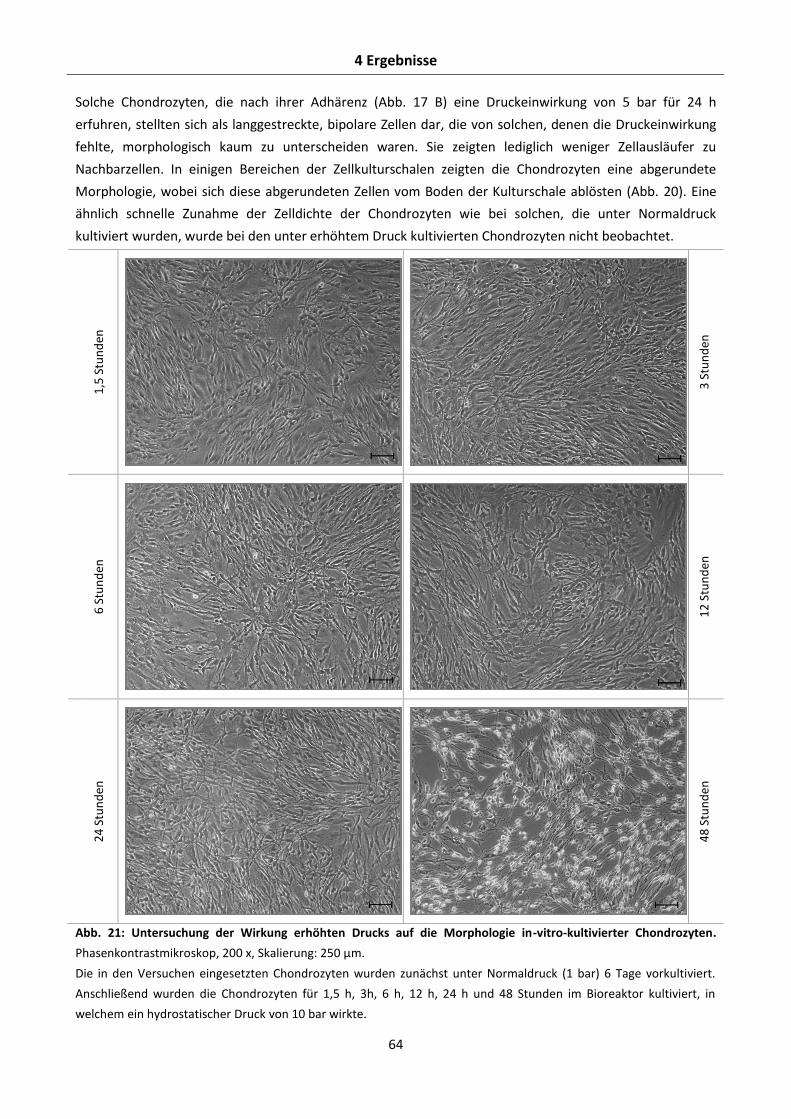
4 Ergebnisse
64
Solche Chondrozyten, die nach ihrer Adhärenz (Abb. 17 B) eine Druckeinwirkung von 5 bar für 24 h
erfuhren, stellten sich als langgestreckte, bipolare Zellen dar, die von solchen, denen die Druckeinwirkung
fehlte, morphologisch kaum zu unterscheiden waren. Sie zeigten lediglich weniger Zellausläufer zu
Nachbarzellen. In einigen Bereichen der Zellkulturschalen zeigten die Chondrozyten eine abgerundete
Morphologie, wobei sich diese abgerundeten Zellen vom Boden der Kulturschale ablösten (Abb. 20). Eine
ähnlich schnelle Zunahme der Zelldichte der Chondrozyten wie bei solchen, die unter Normaldruck
kultiviert wurden, wurde bei den unter erhöhtem Druck kultivierten Chondrozyten nicht beobachtet.
1,5
Stu
nd
en
3 S
tun
den
6 S
tun
den
12
Stu
nd
en
24
Stu
nd
en
48
Stu
nd
en
Abb. 21: Untersuchung der Wirkung erhöhten Drucks auf die Morphologie in-vitro-kultivierter Chondrozyten.
Phasenkontrastmikroskop, 200 x, Skalierung: 250 µm.
Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter Normaldruck (1 bar) 6 Tage vorkultiviert.
Anschließend wurden die Chondrozyten für 1,5 h, 3h, 6 h, 12 h, 24 h und 48 Stunden im Bioreaktor kultiviert, in
welchem ein hydrostatischer Druck von 10 bar wirkte.

4 Ergebnisse
65
Für 6 Tage bei Normaldruck vorkultivierte Chondrozyten (Abb. 17 C) waren nach 24 h Kultivierung bei 5 bar
Druck von solchen, denen die Druckeinwirkung fehlte, optisch kaum zu unterscheiden. In einigen Bereichen
der Zellkulturschalen kam es zu einer Abrundung der Chondrozyten, wobei sich diese abgerundeten
Chondrozyten vom Boden der Zellkulturschale ablösten (Abb. 20).
Die Chondrozyten, auf welche direkt nach der Aussaat ein Druck von 10 bar für 24 h (Abb. 17 D) wirkte,
zeigten ebenfalls keine Adhärenz zum Boden der Zellkulturschale und keine Zunahme der Zelldichte. In
diesen Zellkulturen fehlte die bipolare Morphologie der Chondrozyten. Stattdessen wurde, ebenso wie bei
einer Druckeinwirkung von 5 bar, das Auftreten größerer, flächiger Zellen mit Karyopyknosis und
Karyorrhexis beobachtet.
Nach einer Druckeinwirkung von 10 bar für 24 h auf adhärente Zellkulturen der Chondrozyten (Abb. 17. E)
waren die langgestreckten, bipolaren Zellen von solchen, denen die Druckeinwirkung fehlte, morphologisch
kaum zu unterscheiden. Sie zeigten jedoch weniger Zellausläufer zu Nachbarzellen. Den unter diesen
Druckbedingungen kultivierten Chondrozyten fehlte die schnelle Zunahme der Zelldichte, die bei unter
Normaldruck kultivierten Chondrozyten beobachtet wurde. Bei den unter diesen Bedingungen (10 bar,
24 h) kultivierten subkonfluenten, adhärenten Chondrozyten wurde nur in einzelnen Bereichen der
Zellkulturschalen eine abgerundete Morphologie der Zellen sichtbar, wobei ein Ablösen dieser Zellen vom
Boden der Zellkulturschale im Vergleich zu den bei 5 bar kultivierten Zellen nur selten beobachtet wurde.
Konfluente Zellkulturen der Chondrozyten waren nach 24 h, 10 bar (Abb. 17 F) von solchen, denen die
Druckeinwirkung fehlte, morphologisch kaum zu unterscheiden. In einzelnen Bereichen der Kulturschalen
kam es zu einer Abrundung der Chondrozyten (Abb. 20).
Die Chondrozyten der untersuchten konfluenten Zellkulturen (Abb. 21) waren bei Einwirkung kürzerer
Druckphasen von 1,5 h (Abb. 18 G), 3 h (Abb. 18 H), 6 h (Abb. 18 I) und 12 h (Abb. 18 J) von unter
Normaldruck kultivierten Chondrozyten morphologisch kaum zu unterscheiden. Sie waren durch
langgestreckte, bipolare Zellen charakterisiert (Abb. 21). Zellen mit abgerundeter Morphologie waren bei
Einwirkung kürzerer Druckphasen von unter 24 h kaum zu beobachten (Abb. 21).
Bei einer Wirkung des Drucks für 24 h (10 bar) (Abb. 18 K) wiesen Zellen in einzelnen Bereichen der
Zellkulturschalen die o.g. abgerundete Morphologie der Chondrozyten auf (Abb. 21). Bei einer
Verlängerung der Druckphase auf 48 h (Abb. 18 L) zeigten deutlich größere Anteile der Zellen die
abgerundete Zellmorphologie und Zeichen von Karyopyknosis und Karyorrhexis. Wurde die Dauer der
Druckphase weiter verlängert (>72 h), lösten sich die abgerundeten Chondrozyten vom Boden der
Kulturschale und voneinander. Unter den verbliebenen Chondrozyten nahmen gleichzeitig die Zeichen von
Karyopyknosis und Karyorrhexis sowie der Anteil der Zellen mit vermehrtem Zytoplasma zu. Die
beschriebene Ablösung der Chondrozyten betraf bei 10 bar und einer Dauer der Druckeinwirkung von 24-
72 h nur einzelne Zellkulturschalen (Abb. 18 K, L, M) oder nur umschriebene Bereiche in einer solchen. Bei
einer weiteren Verlängerung der Druckphase auf 98 h war die Ablösung der Chondrozyten in allen
Zellkulturschalen vollständig, wobei die Anzahl karyopyknotischer und karyorrhektischer Zellen deutlich
zunahm und die Dichte der Zellen abnahm.
4.2.4 Nachweis der Aggrekan-, Kollagen Typ I- und Kollagen Typ II-mRNA-Expression in-vitro-
kultivierter Chondrozyten unter erhöhtem Druck
Der Nachweis der Expression der mRNA für Aggrekan, Kollagen Typ I und II durch Chondrozyten in vitro
unter erhöhten Druckbedingungen erfolgte ebenso wie unter Normaldruck (1 bar) (Kap. 4.1.3) mittels

4 Ergebnisse
66
qPCR. Dabei dienten gleich behandelte Chondrozyten unter Normaldruck (1 bar) als Kontrollen.
Chondrozyten, die keine Adhärenz zum Boden der Kulturschale zeigten, lieferten ebenso keine
auswertbaren Ergebnisse in der qPCR wie Zellen, die sich im Laufe der Druckbehandlung vom Boden der
Zellkulturschale ablösten. Sie fanden in dieser Auswertung daher keine Berücksichtigung.
4.2.4.1 Effekte der Höhe des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten
Für die Untersuchung der Effekte der Höhe des hydrostatischen Drucks auf die mRNA-Expression von
Aggrekan, Kollagen Typ I und II durch Chondrozyten in vitro wurden die Zellen 6 d bis zum Erreichen der
Konfluenz vorkultiviert und anschließend im Bioreaktor bei einem Druck von 5 bar und 10 bar für jeweils
24 h weiterkultiviert (Abb. 22).
Abb. 22: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Höhe des hydrostatischen Drucks auf die
mRNA-Expression in-vitro-kultivierter Chondrozyten. Die Bezeichnung der jeweiligen Kultivierungsbedingungen
entspricht der in Abb. 17. Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter Normaldruck
(1 bar) 6 Tage (d) vorkultiviert. Anschließend wurden die Chondrozyten für 24 h im Bioreaktor kultiviert, in welchem
ein hydrostatischer Druck von 5 bar (schmaler Balken) (C) bzw. 10 bar (breiter Balken) (F) für 24 Stunden (h) wirkte.
Bei einer Druckhöhe von 5 bar fiel das M der RE des Kollagen Typ I auf das 0,2-fache (3,40E3 Moleküle) und
bei einer Druckhöhe von 10 bar auf das 0,8-fache (1,09E4 Moleküle) gegenüber der unbehandelten
Kontrolle (1,42E4 Moleküle) ab (Abb. 23 A, B).
Einen ähnlichen Verlauf nahm die mRNA-Expression des Kollagen Typ II (Abb. 23 C, D). Dessen M der RE
sank bei einer Druckhöhe von 5 bar auf das 0,2-fache (2,38E3 Moleküle) und bei einer Druckhöhe von
10 bar auf das 0,8-fache (8,64E3 Moleküle) gegenüber der unbehandelten Kontrolle (1,07E4 Moleküle).
Das M der RE für Aggrekan (Abb. 24 A, B) stieg bei einer Druckhöhe von 5 bar auf das 2,2-fache (7,44E1
Moleküle) und bei einer Druckhöhe von 10 bar auf das 1,7 fache (5,82E1 Moleküle) gegenüber der
unbehandelten Kontrolle an (3,34E1 Moleküle).
In der statistischen Analyse wurden zunächst die Ergebnisse der mRNA-Expression für Kollagen Typ I und II
und Aggrekan der bei 5 und 10 bar kultivierten Chondrozyten im Vergleich zur Kontrollgruppe (1 bar)
untersucht. Dabei wiesen die Ergebnisse der RE des Kollagen Typ I und Kollagen Typ II eine hohe Differenz
der Mittelwerte bei 1 und 5 bar auf. Dadurch erreichte die Aussage, dass es durch die Einwirkung von 5 bar
Druck zu einem Absinken der Kollagen Typ I- und II-Expression kommt bei dem Stichprobenumfang (n=3)
eine P von 99 %. Für einen Druck von 10 bar erzielte diese Aussage nur eine P von 27-31 %, sodass n=9
notwendig wäre, um diese mit einer P von 80 % auch für den höheren Druck treffen zu können. Eine hohe
Differenz der Mittelwerte zeigten auch die Ergebnisse der RE für Aggrekan, wobei diese eine hohe
Standardabweichung aufwiesen. Der Anstieg der mRNA-Expression von Aggrekan durch die Einwirkung von
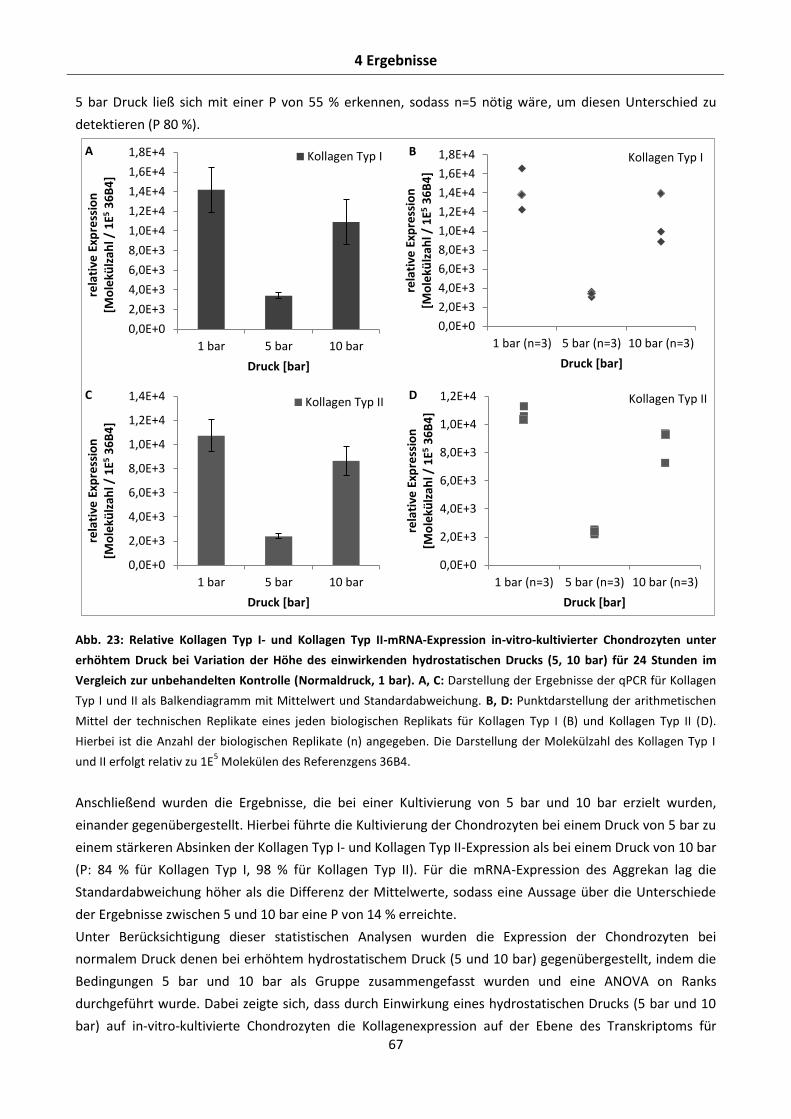
4 Ergebnisse
67
5 bar Druck ließ sich mit einer P von 55 % erkennen, sodass n=5 nötig wäre, um diesen Unterschied zu
detektieren (P 80 %).
Abb. 23: Relative Kollagen Typ I- und Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten unter
erhöhtem Druck bei Variation der Höhe des einwirkenden hydrostatischen Drucks (5, 10 bar) für 24 Stunden im
Vergleich zur unbehandelten Kontrolle (Normaldruck, 1 bar). A, C: Darstellung der Ergebnisse der qPCR für Kollagen
Typ I und II als Balkendiagramm mit Mittelwert und Standardabweichung. B, D: Punktdarstellung der arithmetischen
Mittel der technischen Replikate eines jeden biologischen Replikats für Kollagen Typ I (B) und Kollagen Typ II (D).
Hierbei ist die Anzahl der biologischen Replikate (n) angegeben. Die Darstellung der Molekülzahl des Kollagen Typ I
und II erfolgt relativ zu 1E5 Molekülen des Referenzgens 36B4.
Anschließend wurden die Ergebnisse, die bei einer Kultivierung von 5 bar und 10 bar erzielt wurden,
einander gegenübergestellt. Hierbei führte die Kultivierung der Chondrozyten bei einem Druck von 5 bar zu
einem stärkeren Absinken der Kollagen Typ I- und Kollagen Typ II-Expression als bei einem Druck von 10 bar
(P: 84 % für Kollagen Typ I, 98 % für Kollagen Typ II). Für die mRNA-Expression des Aggrekan lag die
Standardabweichung höher als die Differenz der Mittelwerte, sodass eine Aussage über die Unterschiede
der Ergebnisse zwischen 5 und 10 bar eine P von 14 % erreichte.
Unter Berücksichtigung dieser statistischen Analysen wurden die Expression der Chondrozyten bei
normalem Druck denen bei erhöhtem hydrostatischem Druck (5 und 10 bar) gegenübergestellt, indem die
Bedingungen 5 bar und 10 bar als Gruppe zusammengefasst wurden und eine ANOVA on Ranks
durchgeführt wurde. Dabei zeigte sich, dass durch Einwirkung eines hydrostatischen Drucks (5 bar und 10
bar) auf in-vitro-kultivierte Chondrozyten die Kollagenexpression auf der Ebene des Transkriptoms für
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
1 bar (n=3) 5 bar (n=3) 10 bar (n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Druck [bar]
Kollagen Typ II
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
1,4E+4
1,6E+4
1,8E+4
1 bar (n=3) 5 bar (n=3) 10 bar (n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Druck [bar]
Kollagen Typ I
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
1,4E+4
1 bar 5 bar 10 bar
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Druck [bar]
Kollagen Typ IIC
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
1,4E+4
1,6E+4
1,8E+4
1 bar 5 bar 10 bar
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Druck [bar]
Kollagen Typ IA B
D
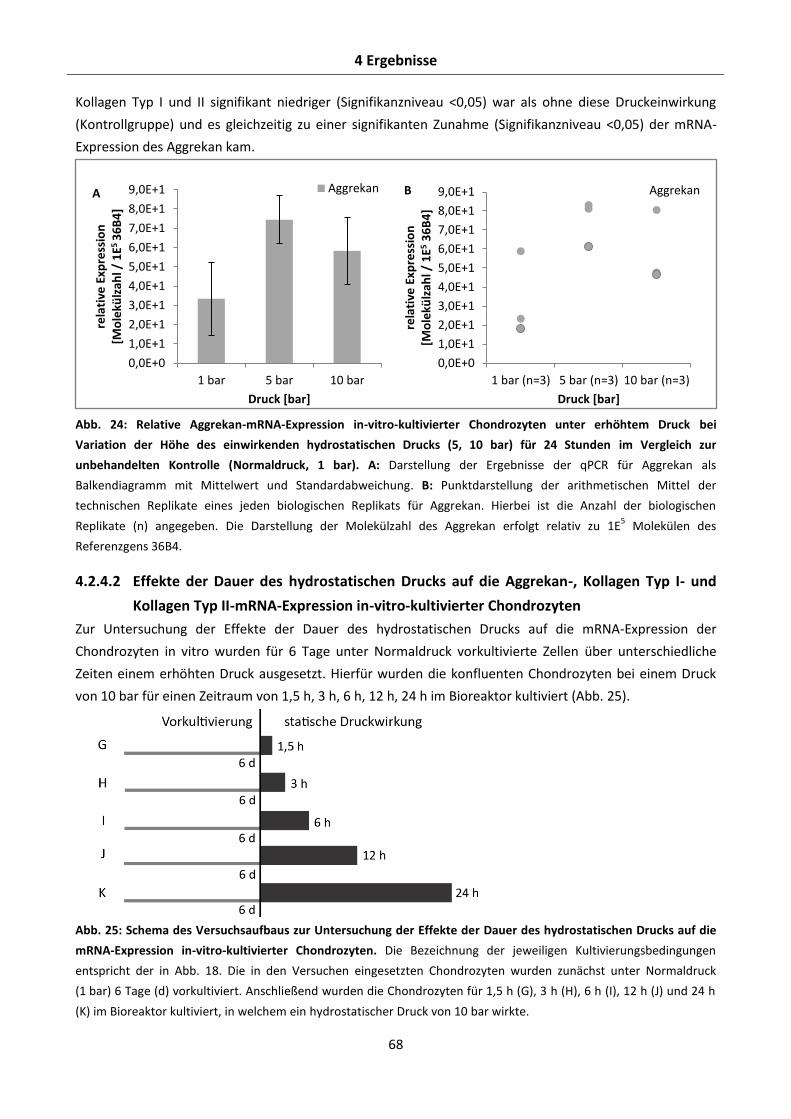
4 Ergebnisse
68
Kollagen Typ I und II signifikant niedriger (Signifikanzniveau <0,05) war als ohne diese Druckeinwirkung
(Kontrollgruppe) und es gleichzeitig zu einer signifikanten Zunahme (Signifikanzniveau <0,05) der mRNA-
Expression des Aggrekan kam.
Abb. 24: Relative Aggrekan-mRNA-Expression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck bei
Variation der Höhe des einwirkenden hydrostatischen Drucks (5, 10 bar) für 24 Stunden im Vergleich zur
unbehandelten Kontrolle (Normaldruck, 1 bar). A: Darstellung der Ergebnisse der qPCR für Aggrekan als
Balkendiagramm mit Mittelwert und Standardabweichung. B: Punktdarstellung der arithmetischen Mittel der
technischen Replikate eines jeden biologischen Replikats für Aggrekan. Hierbei ist die Anzahl der biologischen
Replikate (n) angegeben. Die Darstellung der Molekülzahl des Aggrekan erfolgt relativ zu 1E5 Molekülen des
Referenzgens 36B4.
4.2.4.2 Effekte der Dauer des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten
Zur Untersuchung der Effekte der Dauer des hydrostatischen Drucks auf die mRNA-Expression der
Chondrozyten in vitro wurden für 6 Tage unter Normaldruck vorkultivierte Zellen über unterschiedliche
Zeiten einem erhöhten Druck ausgesetzt. Hierfür wurden die konfluenten Chondrozyten bei einem Druck
von 10 bar für einen Zeitraum von 1,5 h, 3 h, 6 h, 12 h, 24 h im Bioreaktor kultiviert (Abb. 25).
Abb. 25: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Dauer des hydrostatischen Drucks auf die
mRNA-Expression in-vitro-kultivierter Chondrozyten. Die Bezeichnung der jeweiligen Kultivierungsbedingungen
entspricht der in Abb. 18. Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter Normaldruck
(1 bar) 6 Tage (d) vorkultiviert. Anschließend wurden die Chondrozyten für 1,5 h (G), 3 h (H), 6 h (I), 12 h (J) und 24 h
(K) im Bioreaktor kultiviert, in welchem ein hydrostatischer Druck von 10 bar wirkte.
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
7,0E+1
8,0E+1
9,0E+1
1 bar 5 bar 10 bar
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Druck [bar]
AggrekanA
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
7,0E+1
8,0E+1
9,0E+1
1 bar (n=3) 5 bar (n=3) 10 bar (n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Druck [bar]
Aggrekan B

4 Ergebnisse
69
Die so behandelten Chondrozyten wiesen bei einer Dauer der Druckphase von 1,5 h (8,19E3 Moleküle), 3 h
(6,03E3 Moleküle), 6 h (6,65E3 Moleküle) und 12 h (6,95E3 Moleküle) ähnlich hohe M der RE des Kollagen
Typ I auf, die um das 0,8-fache niedriger lagen als die der jeweiligen unbehandelten Kontrolle. Bei einer
Druckphase von 24 h fiel das M der RE des Kollagen Typ I auf das 0,4-fache (3,18E3 Moleküle) ab
(Abb. 26 A, B).
Ebenso wiesen die M der RE des Kollagen Typ II nahezu konstante Ergebnisse auf. Diese blieben mit 3,33E3
Molekülen bei 1,5 h, 3,08E3 Molekülen bei 3 h, 3,59 E3 Molekülen bei 6 h und 3,11 E3 Molekülen bei 12 h
ebenfalls um das 0,8-fache hinter der jeweilige unbehandelten Kontrolle zurück. Bei einer Druckphase von
24 h fiel das M der RE des Kollagen Typ II auf das 0,7-fache (1,29E3 Moleküle) der Kontrolle ab
(Abb. 26 C, D).
Abb. 26: Relative Kollagen Typ I- und Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten unter
erhöhtem Druck bei Variation der Dauer (1,5 h, 3 h, 6 h, 12 h, 24 h) des einwirkenden hydrostatischen Drucks (10
bar) im Vergleich zur unbehandelten Kontrolle (Normaldruck, 1 bar). A, C: Darstellung der Ergebnisse der qPCR für
Kollagen Typ I und II als Balkendiagramm mit Mittelwert und Standardabweichung. B, D: Punktdarstellung der
arithmetischen Mittel der technischen Replikate eines jeden biologischen Replikats für Kollagen Typ I (B) und Kollagen
Typ II (D). Hierbei ist die Anzahl der biologischen Replikate (n) angegeben. Die Darstellung der Molekülzahl des
Kollagen Typ I und II erfolgt relativ zu 1E5 Molekülen des Referenzgens 36B4
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
1,5 h(n=3)
1,5 h(n=3)
3 h(n=3)
3 h(n=3)
6 h(n=3)
6 h(n=3)
12 h(n=2)
12 h(n=3)
24 h(n=3)
24 h(n=3)
Dauer der Druckeinwirkung [h]
Kollagen Typ I
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
1,4E+4
1,5 h 3 h 6 h 12 h 24 h
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Dauer der Druckeinwirkung [h]
10 bar 1 bar
0,0E+0
1,0E+3
2,0E+3
3,0E+3
4,0E+3
5,0E+3
6,0E+3
1,5 h 3 h 6 h 12 h 24 h
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Dauer der Druckeinwirkung [h]
10 bar 1 barKollagen Typ II
0,0E+0
1,0E+3
2,0E+3
3,0E+3
4,0E+3
5,0E+3
6,0E+3
1,5 h(n=3)
1,5 h(n=3)
3 h(n=3)
3 h(n=3)
6 h(n=3)
6 h(n=3)
12 h(n=2)
12 h(n=3)
24 h(n=3)
24 h(n=3)
Dauer der Druckeinwirkung [h]
Kollagen Typ II
B A
Kollagen Typ I
D C

4 Ergebnisse
70
Das M der RE des Aggrekan wies bei einer Dauer der Druckeinwirkung von 1,5 h (4,93E3 Moleküle) und 3 h
(3,89 E3 Moleküle) um das 1,5- bis 2,5-fache höhere Werte auf als bei der Dauer von 6 h (2,76E3 Moleküle),
12 h (2,07E3 Moleküle) und 24 h (1,76E3 Moleküle) (Abb. 27 A, B). Auch gegenüber der unbehandelten
Kontrolle stieg die RE des Aggrekan bei 1,5 h Druck um das 1,6-fache und bei 3 h Druck um das 2,4-fache
an, fiel bei 6 h auf das 0,8-fache der Kontrolle ab und zeigte bei 12 h sowie 24 h annährend gleich hohe
Werte wie die Kontrolle.
Abb. 27: Relative Aggrekan-mRNA-Expression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck bei
Variation der Dauer (1,5 h, 3 h, 6 h, 12 h, 24 h) des einwirkenden hydrostatischen Drucks (10 bar) im Vergleich zur
unbehandelten Kontrolle (Normaldruck, 1 bar). A: Darstellung der Ergebnisse der qPCR für Aggrekan als
Balkendiagramm mit Mittelwert und Standardabweichung. B: Punktdarstellung der arithmetischen Mittel der
technischen Replikate eines jeden biologischen Replikats für Aggrekan. Hierbei ist die Anzahl der biologischen
Replikate (n) angegeben. Die Darstellung der Molekülzahl des Aggrekan erfolgt relativ zu 1E5 Molekülen des
Referenzgens 36B4.
Da das Expressionsmuster der Chondrozyten in Bezug auf Kollagen Typ I und II bei den Bedingungen 1,5 h,
3 h, 6 h, 12 h nur geringe Differenzen der Mittelwerte aufwies, wurde auf die statistische Auswertung
dieser Daten verzichtet. Um die Unterschiede in Bezug auf die Expression des Aggrekan zu analysieren,
wurden den kurzen Druckzeiten (1,5 und 3 h) lange Druckzeiten (6 h, 12 h und 24 h) gegenübergestellt,
wofür die zugehörigen Ergebnisse als Gruppen zusammengefasst wurden. Die vorhandene Differenz der
Mittelwerte in der Expressionen des Aggrekan bei kurzen und langen Druckzeiten wurde bei der
vorliegenden Standardabweichung der Ergebnisse bei n=8 erkannt. Daher war der Stichprobenumfang trotz
der Gruppierung der Ergebnisse (n=6) zu klein, sodass die höhere Expression des Aggrekan bei kurzen
Druckzeiten nur mit einer P von 78 % erkannt wurde. Mit n=8 würde auch der Anstieg der Expression des
Aggrekan bei kurzzeitigen Druckeinwirkung verglichen mit der Kontrolle erkannt. Der notwendige hohe
Stichprobenumfang lag in der besonders hohen Standardabweichung der M der RE des Aggrekan dieses
Versuchs begründet. Da für die hohe Standardabweichung der für Aggrekan sowohl unter Druck als auch
ohne Druck (Kontrollen) erzielten Ergebnisse bei 3 h und 12 h gegenüber 1,5 h und 6 h keine Ursache im
Versuchsaufbau gefunden werden konnte, wurde auf einen direkten Vergleich der unter erhöhtem
hydrostatischem Druck erzielten Ergebnisse mit denen der Kontrollgruppe verzichtet, da dessen
Aussagekraft fraglich bliebe.
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
7,0E+1
1,5 h 3 h 6 h 12 h 24 h
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
Dauer der Druckeinwirkung [h]
10 bar 1 barAggrekan
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
7,0E+1
1,5 h(n=3)
1,5 h(n=3)
3 h(n=3)
3 h(n=3)
6 h(n=3)
6 h(n=3)
12 h(n=2)
12 h(n=3)
24 h(n=3)
24 h(n=3)
Dauer der Druckeinwirkung [h]
Aggrekan A B

4 Ergebnisse
71
4.2.4.3 Effekte der Zelldichte auf die Aggrekan-, Kollagen Typ I- und Kollagen Typ II-mRNA-
Expression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck
Zur Untersuchung des Einflusses der Zelldichte der Chondrozyten auf deren Expression bei der Einwirkung
eines erhöhten hydrostatischen Drucks wurden Chondrozyten in normaler Zelldichte (104 Zellen/ cm2) und
doppelter Zelldichte ausgesät. Diese wurden nach 6 Tagen Vorkultivierung einem Druck von 10 bar für
entweder 3 h oder 6 h ausgesetzt (Abb. 28 H, I, N, O).
Abb. 28: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Zelldichte der Chondrozyten auf deren
mRNA-Expression bei der Einwirkung eines erhöhten hydrostatischen Drucks. Die Bezeichnung der jeweiligen
Kultivierungsbedingungen entspricht der in Abb. 19. Die in den Versuchen eingesetzten Chondrozyten wurden in
normaler (H, I) und doppelter Zelldichte (N, O) ausgesät und zunächst unter Normaldruck (1 bar) 6 Tage (d)
vorkultiviert. Anschließend wurden die Chondrozyten für 3 h (H, N) und 6 h (I, O) im Bioreaktor kultiviert, in welchem
ein hydrostatischer Druck von 10 bar wirkte.
Dabei zeigten die mit doppelter Zelldichte ausgesäten Chondrozyten sowohl bei 3 h (9,26E3 Moleküle) als
auch bei 6 h (9,71E3 Moleküle) andauerndem Druck einen Anstieg des M der RE des Kollagen Typ I auf das
1,5-fache gegenüber solchen mit normaler Zelldichte (6,03E3 Moleküle bei 3h, 6,65E3 Moleküle bei 6h)
(Abb. 29 A, B). Gleichzeitig fiel das M der RE des Kollagen Typ II der Chondrozyten mit doppelter Zelldichte
(1,10E3 Moleküle bei 3h, 1,28E3 Moleküle bei 6h) auf das 0,4-fache des Wertes ab, den die Chondrozyten
mit normaler Zelldichte erreichten (3,08 E3 Moleküle bei 3h, 3,59E3 Moleküle bei 6h) (Abb. 29 C, D).
Bei einer Druckeinwirkung von 3 h fiel das M der RE des Aggrekan bei doppelter Zelldichte (1,76E1
Moleküle) auf das 0,5-fache gegenüber dem M der RE des Aggrekan bei normaler Zelldichte (3,89E1
Molekülen) ab (Abb. 30 A, B). Bei einer Druckeinwirkung von 6 h zeigte sich ein Anstieg um das 1,3-fache
(3,81E1 Moleküle) verglichen mit Chondrozyten in normaler Zelldichte (2,76E1 Moleküle).
Statistisch gesehen zeigt diese Untersuchung nur Tendenzen auf, da es den Ergebnissen mit n=2 und n=3 an
P (P von 22,1% bzw. 29,8% für Kollagen Typ I, P von 41,9% bzw. 72,3% für Kollagen Typ II und P von 0 bzw.
10 % für Aggrekan) fehlt. Ergebnisse mit einer P von 80% würden Untersuchungen ab n=5 liefern.

4 Ergebnisse
72
Abb. 29: Relative Kollagen Typ I- und Kollagen Typ II-mRNA-Expression in-vitro-kultivierter Chondrozyten unter
erhöhtem Druck (3 h und 6 h, 10 bar) bei Variation der Zelldichte. Zur Evaluierung des Einflusses der Zelldichte
wurden die Chondrozyten in normaler Zelldichte (104 Zellen/ cm
2) und doppelter Zelldichte ausgesät und die Wirkung
des Drucks bei einer Einwirkzeit von 3 h und 6 h an den konfluenten Chondrozyten (6 Tage Vorkultivierung unter
Normaldruck) untersucht. A, C: Darstellung der Ergebnisse der qPCR für Kollagen Typ I und II als Balkendiagramm mit
Mittelwert und Standardabweichung. B, D: Punktdarstellung der arithmetischen Mittel der technischen Replikate
eines jeden biologischen Replikats für Kollagen Typ I (B) und Kollagen Typ II (D). Hierbei ist die Anzahl der biologischen
Replikate (n) angegeben. Die Darstellung der Molekülzahl des Kollagen Typ I und II erfolgt relativ zu 1E5 Molekülen des
Referenzgens 36B4.
0,0E+0
1,0E+3
2,0E+3
3,0E+3
4,0E+3
5,0E+3
doppelteZelldichte
(n=2)
normaleZelldichte
(n=3)
doppelteZelldichte
(n=2)
normaleZelldichte
(n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Dauer der Druckeinwirkung [h]
Kollagen Typ II
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
doppelteZelldichte
(n=2)
normaleZelldichte
(n=3)
doppelteZelldichte
(n=2)
normaleZelldichte
(n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Dauer der Druckeinwirkung [h]
Kollagen Typ I
3 h 6 h
3 h 6 h
D
B
0,0E+0
2,0E+3
4,0E+3
6,0E+3
8,0E+3
1,0E+4
1,2E+4
3 h 6 h
rela
tive
Exp
ress
ion
[
Mo
lekü
lzah
l / 1
E5 3
6B
4]
Dauer der Druckeinwirkung [h]
Kollagen Typ I
doppelte Zelldichte
normale Zelldichte
A
0,0E+0
1,0E+3
2,0E+3
3,0E+3
4,0E+3
5,0E+3
3 h 6 h
rela
tive
Exp
ress
ion
[
Mo
lekü
lzah
l / 1
E5 3
6B
4]
Dauer der Druckeinwirkung [h]
Kollagen Typ II doppelte Zelldichtenormale Zelldichte
C
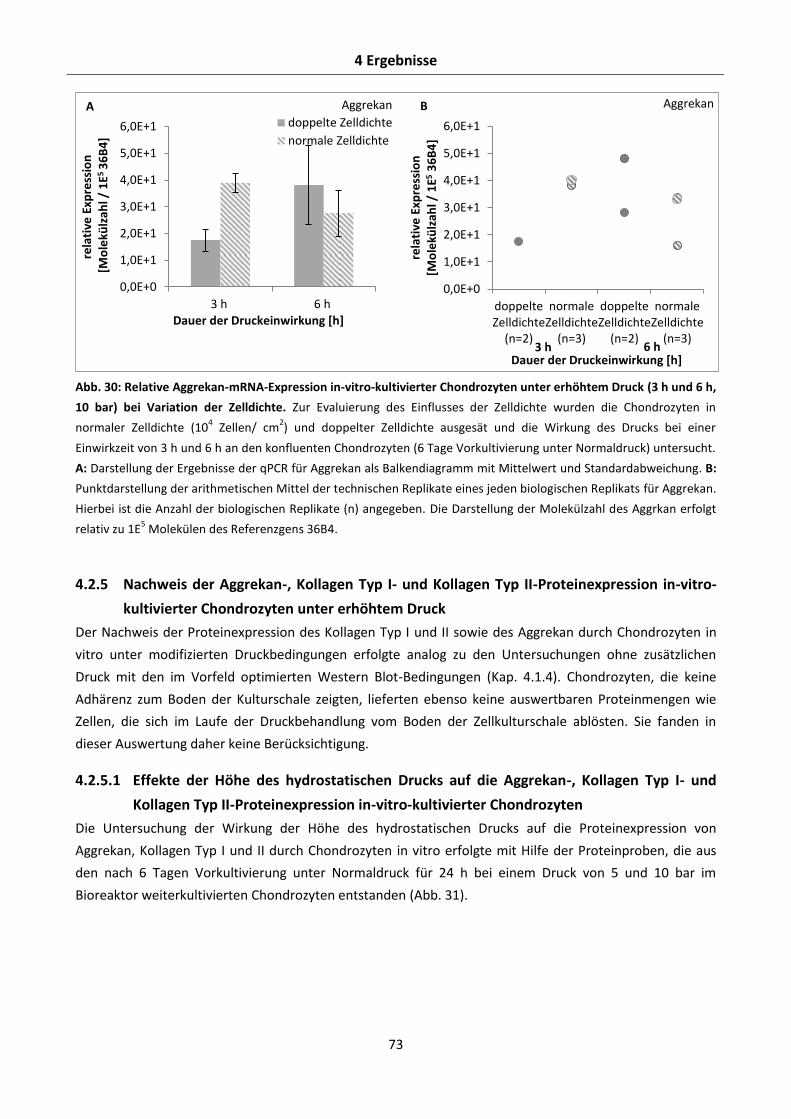
4 Ergebnisse
73
Abb. 30: Relative Aggrekan-mRNA-Expression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck (3 h und 6 h,
10 bar) bei Variation der Zelldichte. Zur Evaluierung des Einflusses der Zelldichte wurden die Chondrozyten in
normaler Zelldichte (104 Zellen/ cm
2) und doppelter Zelldichte ausgesät und die Wirkung des Drucks bei einer
Einwirkzeit von 3 h und 6 h an den konfluenten Chondrozyten (6 Tage Vorkultivierung unter Normaldruck) untersucht.
A: Darstellung der Ergebnisse der qPCR für Aggrekan als Balkendiagramm mit Mittelwert und Standardabweichung. B:
Punktdarstellung der arithmetischen Mittel der technischen Replikate eines jeden biologischen Replikats für Aggrekan.
Hierbei ist die Anzahl der biologischen Replikate (n) angegeben. Die Darstellung der Molekülzahl des Aggrkan erfolgt
relativ zu 1E5 Molekülen des Referenzgens 36B4.
4.2.5 Nachweis der Aggrekan-, Kollagen Typ I- und Kollagen Typ II-Proteinexpression in-vitro-
kultivierter Chondrozyten unter erhöhtem Druck
Der Nachweis der Proteinexpression des Kollagen Typ I und II sowie des Aggrekan durch Chondrozyten in
vitro unter modifizierten Druckbedingungen erfolgte analog zu den Untersuchungen ohne zusätzlichen
Druck mit den im Vorfeld optimierten Western Blot-Bedingungen (Kap. 4.1.4). Chondrozyten, die keine
Adhärenz zum Boden der Kulturschale zeigten, lieferten ebenso keine auswertbaren Proteinmengen wie
Zellen, die sich im Laufe der Druckbehandlung vom Boden der Zellkulturschale ablösten. Sie fanden in
dieser Auswertung daher keine Berücksichtigung.
4.2.5.1 Effekte der Höhe des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-Proteinexpression in-vitro-kultivierter Chondrozyten
Die Untersuchung der Wirkung der Höhe des hydrostatischen Drucks auf die Proteinexpression von
Aggrekan, Kollagen Typ I und II durch Chondrozyten in vitro erfolgte mit Hilfe der Proteinproben, die aus
den nach 6 Tagen Vorkultivierung unter Normaldruck für 24 h bei einem Druck von 5 und 10 bar im
Bioreaktor weiterkultivierten Chondrozyten entstanden (Abb. 31).
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
doppelteZelldichte
(n=2)
normaleZelldichte
(n=3)
doppelteZelldichte
(n=2)
normaleZelldichte
(n=3)
rela
tive
Exp
ress
ion
[M
ole
külz
ahl /
1E5
36
B4
]
Dauer der Druckeinwirkung [h]
Aggrekan
0,0E+0
1,0E+1
2,0E+1
3,0E+1
4,0E+1
5,0E+1
6,0E+1
3 h 6 h
rela
tive
Exp
ress
ion
[
Mo
lekü
lzah
l / 1
E5 3
6B
4]
Dauer der Druckeinwirkung [h]
Aggrekan
doppelte Zelldichte
normale Zelldichte
A B
3 h 6 h
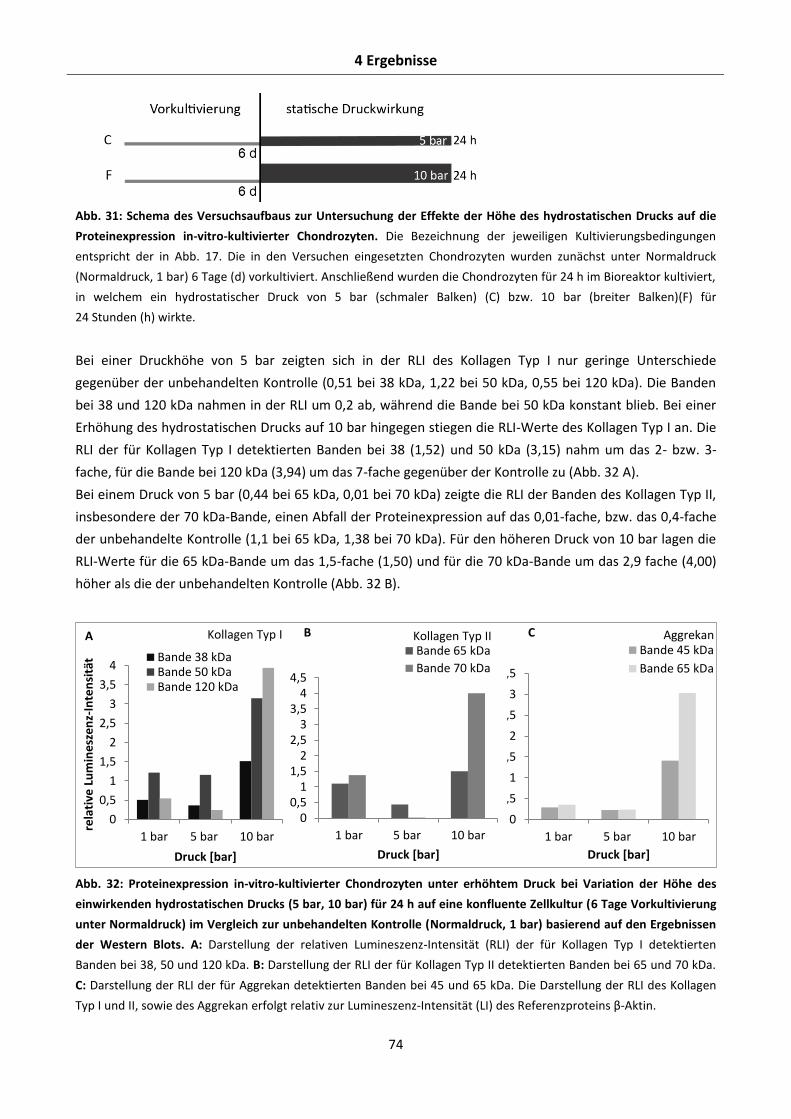
4 Ergebnisse
74
Abb. 31: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Höhe des hydrostatischen Drucks auf die
Proteinexpression in-vitro-kultivierter Chondrozyten. Die Bezeichnung der jeweiligen Kultivierungsbedingungen
entspricht der in Abb. 17. Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter Normaldruck
(Normaldruck, 1 bar) 6 Tage (d) vorkultiviert. Anschließend wurden die Chondrozyten für 24 h im Bioreaktor kultiviert,
in welchem ein hydrostatischer Druck von 5 bar (schmaler Balken) (C) bzw. 10 bar (breiter Balken)(F) für
24 Stunden (h) wirkte.
Bei einer Druckhöhe von 5 bar zeigten sich in der RLI des Kollagen Typ I nur geringe Unterschiede
gegenüber der unbehandelten Kontrolle (0,51 bei 38 kDa, 1,22 bei 50 kDa, 0,55 bei 120 kDa). Die Banden
bei 38 und 120 kDa nahmen in der RLI um 0,2 ab, während die Bande bei 50 kDa konstant blieb. Bei einer
Erhöhung des hydrostatischen Drucks auf 10 bar hingegen stiegen die RLI-Werte des Kollagen Typ I an. Die
RLI der für Kollagen Typ I detektierten Banden bei 38 (1,52) und 50 kDa (3,15) nahm um das 2- bzw. 3-
fache, für die Bande bei 120 kDa (3,94) um das 7-fache gegenüber der Kontrolle zu (Abb. 32 A).
Bei einem Druck von 5 bar (0,44 bei 65 kDa, 0,01 bei 70 kDa) zeigte die RLI der Banden des Kollagen Typ II,
insbesondere der 70 kDa-Bande, einen Abfall der Proteinexpression auf das 0,01-fache, bzw. das 0,4-fache
der unbehandelte Kontrolle (1,1 bei 65 kDa, 1,38 bei 70 kDa). Für den höheren Druck von 10 bar lagen die
RLI-Werte für die 65 kDa-Bande um das 1,5-fache (1,50) und für die 70 kDa-Bande um das 2,9 fache (4,00)
höher als die der unbehandelten Kontrolle (Abb. 32 B).
Abb. 32: Proteinexpression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck bei Variation der Höhe des
einwirkenden hydrostatischen Drucks (5 bar, 10 bar) für 24 h auf eine konfluente Zellkultur (6 Tage Vorkultivierung
unter Normaldruck) im Vergleich zur unbehandelten Kontrolle (Normaldruck, 1 bar) basierend auf den Ergebnissen
der Western Blots. A: Darstellung der relativen Lumineszenz-Intensität (RLI) der für Kollagen Typ I detektierten
Banden bei 38, 50 und 120 kDa. B: Darstellung der RLI der für Kollagen Typ II detektierten Banden bei 65 und 70 kDa.
C: Darstellung der RLI der für Aggrekan detektierten Banden bei 45 und 65 kDa. Die Darstellung der RLI des Kollagen
Typ I und II, sowie des Aggrekan erfolgt relativ zur Lumineszenz-Intensität (LI) des Referenzproteins β-Aktin.
0
0,5
1
1,5
2
2,5
3
3,5
1 bar 5 bar 10 bar
Druck [bar]
Aggrekan Bande 45 kDa
Bande 65 kDa
C
00,5
11,5
22,5
33,5
44,5
1 bar 5 bar 10 bar
Druck [bar]
Kollagen Typ II Bande 65 kDa
Bande 70 kDa
B
0
0,5
1
1,5
2
2,5
3
3,5
4
1 bar 5 bar 10 bar
rela
tive
Lu
min
esz
en
z-In
ten
sitä
t
Druck [bar]
Kollagen Typ I
Bande 38 kDaBande 50 kDaBande 120 kDa
A

4 Ergebnisse
75
Für Aggrekan lag die RLI bei 5 bar Druckeinwirkung für die 45 (0,23) und 65 kDa-Bande (0,24) auf ähnlich
niedrigem Niveau wie die unbehandelte Kontrolle (0,29 bei 45 kDa, 0,35 bei 65 kDa). Bei 10 bar
Druckeinwirkung stieg die Proteinexpression des Aggrekan gegenüber der unbehandelten Kontrolle an. Die
RLI nahm bei 10 bar Druckeinwirkung auf das 4,8-fache für die 45 kDa-Bande (1,41) bzw. 8,6-fache für die
65 kDa-Bande (3,03) zu (Abb. 32 C).
4.2.5.2 Effekte der Dauer des hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und
Kollagen Typ II-Proteinexpression in-vitro-kultivierter Chondrozyten
Für die Untersuchung des Effekts der Dauer der Druckeinwirkung auf die Proteinexpression des Kollagen
Typ I und II sowie des Aggrekan durch die Chondrozyten in vitro wurden die Proteinproben verwendet,
welche aus den nach 6 Tagen Vorkultivierung für 1,5 h, 3 h, 6 h, 12 h und 24 h bei einem Druck von 10 bar
im Bioreaktor weiterkultivierten Chondrozyten gewonnen wurden (Abb. 33).
Abb. 33: Schema des Versuchsaufbaus zur Untersuchung der Effekte der Dauer des hydrostatischen Drucks auf die
Proteinexpression in-vitro-kultivierter Chondrozyten. Die Bezeichnung der jeweiligen Kultivierungsbedingungen
entspricht der in Abb. 18. Die in den Versuchen eingesetzten Chondrozyten wurden zunächst unter Normaldruck (1
bar) 6 Tage (d) vorkultiviert. Anschließend wurden die Chondrozyten für 1,5 h (G), 3 h (H), 6 h (I) und 12 h (J) im
Bioreaktor kultiviert, in welchem ein hydrostatischer Druck von 10 bar wirkte.
Die so behandelten Zellen wiesen bei einer Druckeinwirkung von 1,5 h eine 1,7-fach höhere RLI der für
Kollagen Typ I detektierten Bande bei 37 kDa (0,72) auf als die jeweilige unbehandelte Kontrolle (0,42)
(Abb. 34 A). Die 50 kDa-Bande erreichte mit 0,6 die gleiche RLI wie die Kontrolle, wohingegen für die 120
kDa-Bande (1,53) ähnlich der 37 kDa-Bande eine verglichen mit der Kontrolle (0,95) 1,6-fach höhere RLI
detektiert wurde. Die RLI-Werte der für Kollagen Typ I bei 120 kDa detektierten Bande lag auch verglichen
mit den bei 3 h, 6 h und 12 h erreichten Werten 1,6-fach höher.
Bei längerer Dauer der Druckphase (3 h, 6 h) zeigten sich ähnlich hohe RLI der detektierten Banden des
Kollagen Typ I. Die RLI-Werte lagen bei 3 h Druckphase bei 0,70 für die 37 kDa-, bei 0,96 für die 50 kDa-, bei
1,22 für die 120 kDa-Bande; und bei einer Druckphase von 6 h bei 0,64 für die 37 kDa-, bei 0,79 für die 50
kDa- und bei 1,16 für die 120 kDa-Bande. Somit wurden bei 3 und 6 h für die 37 kDa-Bande verglichen mit
der Kontrolle (0,56 für 3 h, 0,55 für 6 h) 1,2-1,3-fach höhere Werte detektiert. Diese Erhöhung der
RLI-Werte auf das 1,3-fache verglichen mit der Kontrolle (0,76) zeigte auch die 50 kDa-Bande für 3 h (0,96),
wohingegen das Signal bei 6 h Druck mit 0,79 im Vergleich zur Kontrolle (0,82) nahezu konstant blieb. Die
RLI der 120 kDa-Bande bei einer Druckzeit von 3 h und 6 h Kultivierung blieb mit Werten von 1,22 für 3 h
und 1,16 für 6 h ebenfalls nahezu konstant zur unbehandelten Kontrolle (1,27 für 3 h, 1,14 für 6 h).
Während bei 3 h und 6 h Druck verglichen mit der Kontrolle annähernd konstante oder höhere RLI-Werte
der für Kollagen Typ I detektierten Banden vorlagen, wurden bei 12 h für alle Banden der Kontrolle höhere
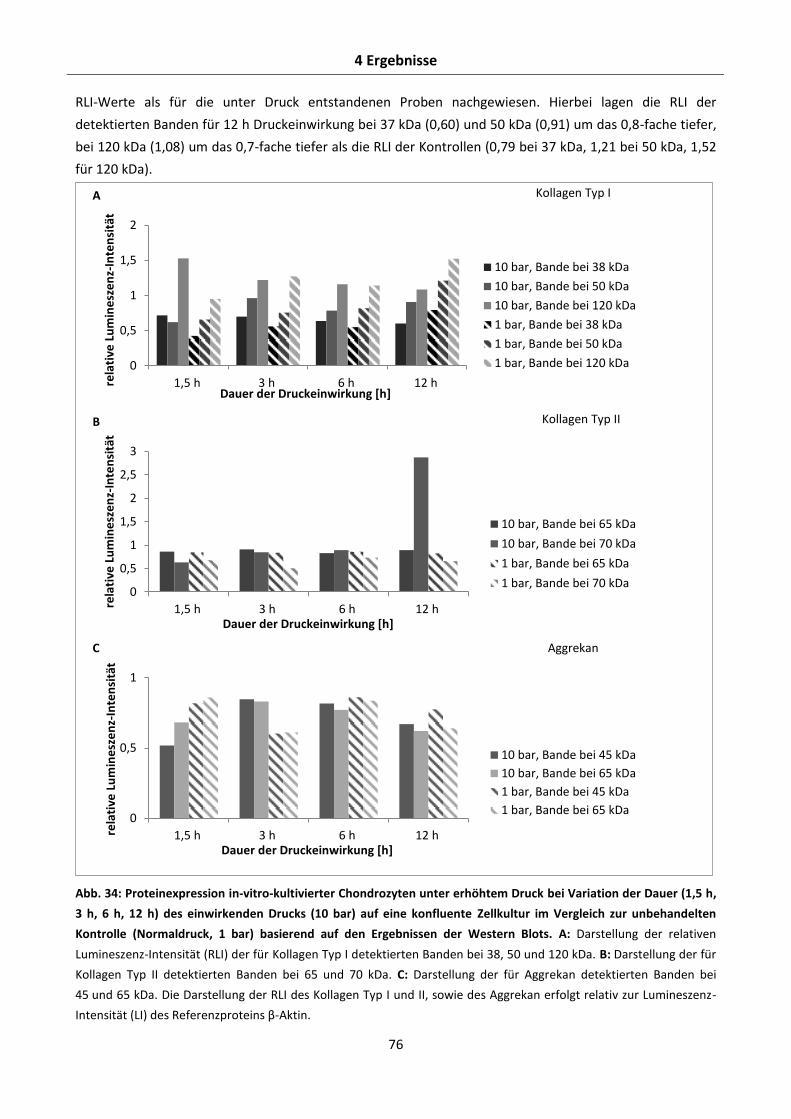
4 Ergebnisse
76
RLI-Werte als für die unter Druck entstandenen Proben nachgewiesen. Hierbei lagen die RLI der
detektierten Banden für 12 h Druckeinwirkung bei 37 kDa (0,60) und 50 kDa (0,91) um das 0,8-fache tiefer,
bei 120 kDa (1,08) um das 0,7-fache tiefer als die RLI der Kontrollen (0,79 bei 37 kDa, 1,21 bei 50 kDa, 1,52
für 120 kDa).
Abb. 34: Proteinexpression in-vitro-kultivierter Chondrozyten unter erhöhtem Druck bei Variation der Dauer (1,5 h,
3 h, 6 h, 12 h) des einwirkenden Drucks (10 bar) auf eine konfluente Zellkultur im Vergleich zur unbehandelten
Kontrolle (Normaldruck, 1 bar) basierend auf den Ergebnissen der Western Blots. A: Darstellung der relativen
Lumineszenz-Intensität (RLI) der für Kollagen Typ I detektierten Banden bei 38, 50 und 120 kDa. B: Darstellung der für
Kollagen Typ II detektierten Banden bei 65 und 70 kDa. C: Darstellung der für Aggrekan detektierten Banden bei
45 und 65 kDa. Die Darstellung der RLI des Kollagen Typ I und II, sowie des Aggrekan erfolgt relativ zur Lumineszenz-
Intensität (LI) des Referenzproteins β-Aktin.
0
0,5
1
1,5
2
1,5 h 3 h 6 h 12 hrela
tive
Lu
min
esz
en
z-In
ten
sitä
t
Dauer der Druckeinwirkung [h]
Kollagen Typ I
10 bar, Bande bei 38 kDa
10 bar, Bande bei 50 kDa
10 bar, Bande bei 120 kDa
1 bar, Bande bei 38 kDa
1 bar, Bande bei 50 kDa
1 bar, Bande bei 120 kDa
A
0
0,5
1
1,5
2
2,5
3
1,5 h 3 h 6 h 12 hrela
tive
Lu
min
esz
en
z-In
ten
sitä
t
Dauer der Druckeinwirkung [h]
Kollagen Typ II
10 bar, Bande bei 65 kDa
10 bar, Bande bei 70 kDa
1 bar, Bande bei 65 kDa
1 bar, Bande bei 70 kDa
B
0
0,5
1
1,5 h 3 h 6 h 12 hrela
tive
Lu
min
esz
en
z-In
ten
sitä
t
Dauer der Druckeinwirkung [h]
Aggrekan
10 bar, Bande bei 45 kDa
10 bar, Bande bei 65 kDa
1 bar, Bande bei 45 kDa
1 bar, Bande bei 65 kDa
C

4 Ergebnisse
77
Bezüglich der Expression des Kollagen Typ II ließen sich nur geringe Unterschiede in den detektierten
Banden bei Variation der Dauer der Druckeinwirkung erkennen (Abb. 34 B). Für die beiden detektierten
Banden bei 65 und 70 kDa wurden bei 1,5 h Druck RLI-Werte von 0,86 bzw. 0,63 und für die Kontrolle 0,84
bzw. 0,51 gemessen. Bei 3 h Druck lag die 65 kDa-Bande (0,91) ebenfalls auf dem Niveau der Kontrolle
(0,84), wohingegen für die 70 kDa-Bande ein verglichen mit der Kontrolle (0,51) 1,7-fach stärkeres Signal
(0,85) detektiert wurde. Während die RLI der 65 kDa-Bande (0,83) bei 6 h Druck ebenfalls konstant zur
Kontrolle (0,86) blieb, zeigte sich auch hier bei 70 kDa (0,85) eine 1,2-fach höhere RLI verglichen mit der
Kontrolle (0,73). Bei einer Druckeinwirkung von 12 h blieb die RLI der bei 65 kDa für Kollagen Typ II
detektierten Bande konstant auf Höhe der für 6 h Druck detektierten Bande (0,89 für 12 h, 0,83 für die
Kontrolle). Hingegen zeigte die 70kDa-Bande für 12 h Druck eine RLI von 2,87, welche 4,4-fach höher lag als
die RLI der Kontrolle (0,66). Hierbei ist zu beachten, dass sich das Signal der Bande des Referenzproteins β-
Aktin sehr schwach und inhomogen innerhalb der Bande darstellte.
Bei der Auswertung der Expression des Aggrekan auf Proteinebene unter Berücksichtigung der Banden bei
45 und 65 kDa wurden nur geringe Unterschiede deutlich (Abb. 34 C). Die RLI der 45 kDa-Bande lag bei
1,5 h Druckeinwirkung für die 45 kDa-Bande (0,52) 0,8-fach und für die 65 kDa-Bande (0,68) 0,6-fach
niedriger als die der unbehandelten Kontrolle (0,82 für 45 kDa, 0,86 für 65 kDa). Bei einer Druckeinwirkung
von 3 h wurde jedoch eine verglichen mit der unbehandelten Kontrolle (0,6 bei 45 kDa, 0,61 bei 65 kDa)
1,4-fach höhere RLI für die beiden detektierten Banden gemessen (0,85 für 45 kDa, 0,77 für 65 kDa). Bei
einem für 6 h wirkenden Druck waren die RLI-Werte der 45 kDa Bande (0,82) und der 65 kDa Bande (0,77)
verglichen mit der Kontrolle nahezu konstant (0,86 für 45 kDa, 0,84 für 65 kDa). Ebenso wiesen die bei 12 h
Druck detektierten Signale (0,67 für 45 kDa, 0,62 für 65 kDa) im Vergleich zur Kontrolle (0,77 für 45 kDa,
0,64 für 65 kDa) kaum Unterschiede auf.

5 Diskussion
78
5 Diskussion
Die heute vorhandenen operativen Möglichkeiten der Therapie von Knorpeldefekten führen überwiegend
zu funktionell unbefriedigenden Langzeitergebnissen (ALFORD 2005). Da die Defekte des Knorpels einen
Großteil der Erkrankungen des Bewegungsapparats darstellen und zudem die Arthrose als Langzeitfolge
nach sich ziehen, besteht ein hohes medizinisches Interesse an der Entwicklung und Weiterentwicklung
innovativer Therapieansätze (BENTLEY et al. 2013, FRITSCH et al. 1999, ZEIFANG et al. 2010). Einen
vielversprechenden Therapieansatz stellt dabei die ACT bzw. MACT dar, bei der in-vitro-kultivierte
Chondrozyten in einen Knorpeldefekt eingebracht werden, mit dem Ziel neuen hyalinen Knorpel zu bilden
(BUCKWALTER und MANKIN 1998, MARCACCI et al. 2013, WONG und CARTER 2003). Bisher existieren
keine geeigneten Bedingungen, um die dabei transplantierten Chondrozyten so zu modifizieren, dass sie
hyalinen Knorpel bilden, welcher langfristig den Druckbedingungen des Gelenks standhält (MARTINEK und
IMHOFF 2003).
Inzwischen zeigte sich jedoch, dass die Entwicklung solcher Therapieansätze ein detaillierteres Verständnis
des Knorpelgewebes und speziell dessen Biodynamik erfordert (ALFORD 2005, BRUNS und STEINHAGEN
2000). Da hierbei ein Fokus auf dem Effekt des auf die Zellen und die ECM des Knorpelgewebes
einwirkenden Drucks liegt, wurde im Rahmen dieser Arbeit die Wirkung verschiedener hydrostatischer
Druckbedingungen auf das Expressionsmuster in-vitro-kultivierter Chondrozyten auf Transkriptom- und
Proteomebene untersucht, um ein Zellkultursystem und Rahmenbedingungen zu etablieren, die gezielt
weitere Untersuchungen ermöglichen. Im Folgenden werden zunächst die dabei genutzten Methoden und
Ergebnisse diskutiert und schließlich daraus resultierende Empfehlungen für die Kultivierung von
Chondrozyten unter erhöhtem hydrostatischem Druck abgeleitet.
5.1 Diskussion der Untersuchungsmethoden
Für die in dieser Arbeit durchgeführten Untersuchungen der Expression in-vitro-kultivierter Chondrozyten
wurden die Methoden PCR, SDS-PAGE/Western Blot und Immunzytochemie etabliert. Die Interpretation
der mit Hilfe dieser Methoden erzielten Ergebnisse sollte unter Berücksichtigung ihrer
methodenspezifischen Besonderheiten erfolgen. Hierzu zählen zum einen die Varianz der Ergebnisse der
PCR und andererseits die aus der Detektion großer Strukturproteine mittels spezifischer Antikörper in
Western Blot und Immunzytochemie resultierenden Abweichungen. Daneben sollten die Möglichkeiten
und Limitationen der statistischen Analysen bei geringem Stichprobenumfang Beachtung finden.
5.1.1 Varianz der Ergebnisse der PCR
Die Ergebnisse der PCR unterliegen einer biologischen und einer technischen Varianz. Die biologische
Varianz liegt zum einen in der Nutzung einer Primärkultur der Chondrozyten in den durchgeführten
Versuchen und zum anderen in individuellen Fehlern begründet. Der Vorteil in der Nutzung einer
Primärkultur im Vergleich zu den ebenfalls nutzbaren Zelllinien liegt vor allem in den geringen
Veränderungen, welche die Primärkultur erfahren hat. Dadurch kommt die Primärkultur den Bedingungen
im Organismus am nächsten, wobei in vitro niemals die Komplexität der in vivo auf die Chondrozyten
einwirkenden Umgebungsfaktoren erreicht werden kann, sodass grundsätzlich Unterschiede zu den unter
physiologischen Bedingungen wachsenden Zellen verbleiben (SCHMITZ 2011). Einen wesentlichen Nachteil
der genutzten Primärkultur stellen die deutlichen individuellen Unterschiede sowohl zwischen den Zellen

5 Diskussion
79
gleicher als auch verschiedener Passagen dar (SCHMITZ 2011). Diese tragen erheblich zur Varianz der
Ergebnisse der biologischen Replikate bei (BUSTIN 2009). Daneben wird die Varianz biologischer Replikate
durch individuelle Fehler erhöht, die auch Unterschiede im Versuchsablauf miteinbeziehen (BRYANT et al.
2011, HUMMERT 2014). Hierzu zählen beispielsweise geringfügige Abweichungen in der Anzahl ausgesäter
Zellen, sowie der Zeitpunkt des letzten Mediumwechsels vor Beginn des Versuchs (GSTRAUNTHALER und
LINDL 2013, SCHMITZ 2011).
Auf die technische Varianz wirkt sich dagegen vor allem die Methodik von der Probenisolation bis zum
PCR-Ergebnis aus. Hierzu zählen neben der Probenisolation und Lagerung der RNA-Proben auch die Qualität
und Quantität der isolierten RNA (FLEIGE et al. 2006). Daneben haben sowohl geringfügige
Konzentrationsunterschiede, beispielsweise durch Pipettierfehler, als auch Temperaturabweichungen
während der RT-Reaktion oder der qPCR im Opticon II-Cycler Einfluss auf die Effizienz dieser Verfahren. Um
die technischen Fehler zu minimieren, wurde zum einen die RNA-Qualität mit Hilfe des
Absorptionsverhältnisses A 260/A 280 spektralphotometrisch beurteilt. Zum anderen wurde durch den
Einsatz von 3 technischen Replikaten jedes biologischen Replikats und durch die für jede qPCR
durchgeführten Kontrollen die Effizienz der PCR (Darstellung der Fluoreszenzintensität der dsDNA in
Abhängigkeit von der Zahl der PCR-Zyklen, Schmelzkurvenanalyse des PCR-Produkts, Standardgraph für den
Plasmidstandard, siehe Kap. 3.1.2.2 ) überprüft (BUSTIN 2009, HUTH 2012).
5.1.2 Einsatz spezifischer Antikörper für den Nachweis großer Strukturproteine
Für den Nachweis von Proteinen mittels Immunzytochemie oder Western Blot stehen heute viele
spezifische Antikörper zur Verfügung. Die Einführung des Phage Display Verfahrens und die Etablierung von
Antikörper-Bibliotheken machen es möglich, Antikörper für fast jedes Antigen zu kreieren. Dabei sind auch
früher als schwierig betrachtete Zielproteine, wie Proteine an der Oberfläche der Zellen und große
Strukturproteine in den Fokus geraten (WILDT 2000). Die speziell in der Analytik großer Strukturproteine
mit Hilfe der Antikörper erreichte Sensitivität und Qualität unterliegt Schwankungen (WILDT 2000). Dies
trifft auch auf die gegen Kollagen Typ I und II sowie Aggrekan gerichteten Antikörper zu, die im Rahmen
dieser Arbeit verwendet wurden.
5.1.2.1 Einsatz spezifischer Antikörper für den Nachweis großer Strukturproteine in der
Immunzytochemie
Im Bereich der Immunzytochemie zeigten sich unterschiedlich starke immuno-positive Reaktionen bei
Einsatz der anti-Kollagen Typ I- und anti-Kollagen Typ II-Antikörper. Hierfür kommen neben der Detektion
von tatsächlichen Unterschieden in der Konzentration der beiden Kollagentypen weitere Ursachen in
Betracht. Dem Versuchsaufbau entsprechend wurde der immunzytochemische Nachweis der Expression
der Chondrozyten in Abhängigkeit von der Länge der Kultivierungszeit zu verschiedenen Zeitpunkten
durchgeführt. Somit können eine unterschiedliche Kinetik der Antikörperbindung durch unterschiedliche
Umgebungstemperaturen oder Unterschiede in der Qualität der Strep-ABC-Lösung bzw. DAB-Lösung als
Einflussfaktoren auf die Stärke der immuno-positiven Reaktionen nicht ausgeschlossen werden. Mit Hilfe
der für jeden Zeitpunkt mitgeführten Negativkontrollen lässt sich sowohl eine unspezifische Bindung des
Sekundärantikörpers als auch eine Kontamination während des Verfahrens ausschließen (MATZ 2003).

5 Diskussion
80
5.1.2.2 Einsatz spezifischer Antikörper für den Nachweis großer Strukturproteine im Western
Blot
Die Schwankungen der Umgebungstemperaturen oder die Qualität der verwendeten Lösungen spielen bei
der Verwendung der Antikörper im Western Blot keine Rolle, da das Verfahren zum einen mit den beiden
PVDF-Membranen simultan durchgeführt wurde und zum anderen grundsätzlich nur die auf einer
Membran detektierten Signale miteinander verglichen wurden. Dennoch zeigten sich Unterschiede in der
Stärke der einzelnen detektierten Banden, die nicht unbedingt auf die Versuchsbedingungen zurückgeführt
werden können, sondern auch in der Spezifität und Sensitivität der Antikörper begründet liegen (WILDT
2000). Hierbei sind Unterschiede in der Stärke des detektierten Signals insbesondere auf Abweichungen der
eingesetzten Proteinmenge zurückzuführen, die sich auch durch die erneute Messung der
Proteinkonzentration direkt vor der SDS-PAGE nicht vermeiden ließen (TAYLOR et al. 2013, TAYLOR und
POSCH 2014). Aufgrund dessen erfolgte die Betrachtung der jeweiligen Proteinexpression relativ zu der des
Referenzproteins β-Aktin (MAHMOOD und YANG 2012, FERGUSON et al. 2005). Dennoch birgt die
Ungenauigkeit der Messung der im Western Blot eingesetzten Proteinmenge einen Unsicherheitsfaktor,
welcher bei der Evaluierung der erhaltenen Ergebnisse berücksichtigt werden sollte (TAYLOR und POSCH
2014).
Bei der Durchführung des Western Blots für Kollagen Typ I und II sowie Aggrekan wurde β-Aktin als
Referenzprotein verwendet und angenommen, dass dessen Expression durch die Versuchsbedingungen
nicht beeinträchtigt wird. Für dieses kleine Protein war mit dem hierfür genutzten Antikörper eine einzelne
spezifische Bande detektierbar, die abgesehen von den Randbereichen der PVDF-Membran und dem
Ausfall des Signals in einem Fall eine gleichmäßige Lumineszenz-Intensität (LI) innerhalb der Bande aufwies.
In den Anweisungen des Herstellers des anti-β-Aktin Antikörpers wird das Signal bei 42 kDa erwartet und
wurde in den Versuchen dieser Arbeit bei dieser Molekülgröße auch detektiert. Grundsätzlich
unterschiedlich stellt sich das für die großen Strukturproteine Kollagen Typ I und II sowie Aggrekan
detektierte Signal dar. Mit Hilfe der für diese Proteine verwendeten Antikörper waren jeweils mehrere
Banden nachweisbar.
Im Gegensatz zum anti-β-Aktin-Antikörper wurden die Daten über die Spezifität der Banden des anti-
Kollagen Typ I-Antikörpers seitens des Herstellers nicht mit Hilfe eines Zell- oder Gewebelysats sondern mit
Hilfe des aufgereinigten Immunogens erhoben. Somit wird laut Herstellerinformation keine Aussage zu
weiteren Banden und deren Spezifität getroffen. Neben der laut Herstellerinformation erwarteten Bande
bei 38 kDa waren in den Versuchen dieser Arbeit weitere Banden bei 50, 60, 120, 170 kDa detektierbar. Die
erwartete Bande bei 38 kDa stellt das C-terminale Propeptid (NC1) (Immunogen) dar, welches nach der
Formation der Tripelhelix vom Prokollagen I entfernt wird (EYRE 1980, KOIVULA et al. 2012). Seine
Konzentration trifft somit eine Aussage über die momentane Kollagen Typ I Synthese der Chondrozyten.
Daneben wird auch das N-terminale Propeptid (etwa 15-20 kDa) von der prematuren α2(I)-Kette (130 kDa)
abgespalten, sodass für die mature α2(I)-Kette ein Molekulargewicht von 70-90 kDa verbleibt, wobei die
Angaben über die Molekulargewichte der Varianten der α2(I)-Kette in der Literatur schwanken (BORNSTEIN
und SAGE 1980, FESSLER und FESSLER 1978, GOLDBERG et al. 1972, KOIVULA et al. 2012). Daneben weisen
große Proteine, welche wie die α-Kollagenketten posttranslational Hydroxylierungen und Glykosilierungen
erfahren haben, in der SDS-PAGE und im Western Blot eine Diskrepanz zwischen erwartetem und
detektiertem Molekulargewicht auf (GELSE 2003, COSMA 1998). Unter Berücksichtigung dieser
Abweichungen kann die 120 kDa Bande als premature α2(I)-Kette angesehen werden, die auch

5 Diskussion
81
KNEIßEL (2003) als solche identifizieren konnte. Die Bande, welche bei 50 kDa detektiert wurde, stellt am
ehesten ein Abbauprodukt der α2(I) Kette dar, wie von WU et al. (1991) für die α2(IX) Kette beschrieben.
AIMES und QUIGLEY (1995) erhielten wie in dieser Arbeit ein Abbauprodukt, welches nach gezieltem Abbau
des Kollagen Typ I durch MMPs bei etwa 60 kDa nachweisbar war. Eine Begründung für das Auftreten der
Bande bei 170 kDa konnte nicht gefunden werden, sodass diese als unspezifisch betrachtet wurde. Somit
konnte die zum jeweiligen Versuchszeitpunkt vorhandene Kollagen Typ I-Synthese der Chondrozyten durch
das Lumineszenzsignal der bei 38, 50 und 120 kDa detektierten Banden wiedergegeben werden.
Der anti-Kollagen Typ II-Antikörper wurde wie der anti-Kollagen Typ I-Antikörper an der aufgereinigten
α1(II)-Kette getestet, wobei seitens des Herstellers keine Angaben zum Molekulargewicht der zu
erwartenden Bande vorliegen. Die im Rahmen dieser Arbeit für Kollagen Typ II detektierten Banden liegen
bei 65 und 70 kDa. In der Literatur variieren die Angaben bezüglich der Molekulargewichte der von der
prematuren α1(II)-Kette (140 kDa) abzuspaltenden Propeptide von 23-36 kDa (C-terminal) und 13-25 kDa
(N-terminal) (BURGESON und HOLISTER 1979, EYRE et al. 2003, FESSLER und FESSLER 1978, RUSSEAU et al.
2004). Dagegen wiesen FUKUI et al. (2001) für das N-terminale Propeptid der immaturen α1(IIA)-Kette
sogar ein Molekulargewicht von ca. 45 kDa nach. In ihren Untersuchungen wurde auch deutlich, dass die
N-terminalen Propeptide der α1(II)-Kette nach ihrer Abtrennung von dem tripelhelikalen Teil in der Lage
sind, sich zu Polymeren zusammen zu lagern, die als solche im Western Blot nachgewiesen wurden. Auf
diese Weise ist die in den Versuchen detektierte Bande bei 70 kDa durch ein Dimer N-terminaler
Propeptide erklärbar. Das Phänomen der Polymerbildung existiert auch für die abgespaltenen C-terminalen
Propeptide, sodass MIOSGE et al. (2004) die von ihnen detektierten Doppelbanden zwischen 69 und 79 kDa
mit Dimeren und Trimeren C-terminaler Propeptide erklärten, wobei die kleinere Bande bereits bei 65 kDa
detektiert wurde (DODGE und POOLE 1989). Daher kann das Lumineszenzsignal der detektierten Banden
bei 65 und 70 kDa als geeignet angesehen werden, die zum jeweiligen Versuchszeitpunkt vorliegende
Kollagen Typ II-Synthese der Chondrozyten widerzuspiegeln.
Der anti-Aggrekan-Antikörper wurde wie der gegen Kollagen Typ I gerichtete ebenfalls am aufgereinigten
Immunogen getestet. Der Antikörper erkennt eine Aminosäuresequenz der G3-Domäne des Aggrekan und
zeigt laut Herstellerinformation im Western Blot mehrere Banden, wobei Angaben über deren
Molekulargewicht fehlen. Das Molekulargewicht des Gesamtproteins Aggrekan beträgt 350 kDa. Im
Western Blot werden neben der Detektion des Gesamtproteins häufig sowohl die einzelnen Domänen als
auch deren kleinere Fragmente nachgewiesen (HANDLEY et al. 2001, OSHITA et al. 2004, SANDY und
VERSCHAREN 2003). Die im Rahmen dieser Arbeit bei 45 kDa und 65 kDa detektierten Fragmente der
G3-Domäne entstehen durch Proteolyse des Aggrekan (HANDLEY et al. 2001). Hierfür kommen ADAMTS
und MMPs in Frage, jedoch auch Proteasen, die durch die Proteaseinhibitoren während der
Probenbearbeitung unzureichend inhibiert wurden. Weiterhin spielen auch die während der
Probenisolation oder der SDS-PAGE verwendeten Lösungen bei der Entstehung dieser kleineren
detektierten Fragmente eine Rolle (KIANI et al. 2002, OSHITA et al. 2004, SANDY und VERSCHAREN 2003,
STRUGLICS et al. 2006). Somit wird in Anlehnung an die von LEE et al. (2002) isoliert nachgewiesene
G3-Domäne des Aggrekan bei 48 und 72 kDa und unter Berücksichtigung der starken Glykosilierung des
Proteins angenommen, dass die in dieser Arbeit nachgewiesenen Banden bei 45 und 65 kDa die Expression
von Aggrekan wiedergeben.
Die Bedeutung der Methoden SDS-PAGE und Western Blot in der Proteinanalytik und der dabei
verwendeten spezifischen Antikörper hat in den letzten Jahren deutlich zugenommen. Sie haben sich zu

5 Diskussion
82
einem wichtigen molekularbiologischen Werkzeug weiterentwickelt, welches die Expression der Zellen
nicht nur auf Grundlage des Transkriptoms sondern auch auf Proteinebene zugänglich macht. Auch wenn
diese Methoden empfindlich von der Spezifität und Sensitivität der Antikörper abhängen, die bei der
Auswertung der mit ihnen gewonnenen Ergebnisse beachtet werden sollten, so bieten sich momentan
kaum vergleichbare Alternativen für die Proteinanalytik (TAYLOR et al. 2013, TAYLOR und POSCH 2014,
WILDT 2000).
5.1.3 Einsatz statistischer Methoden in einer Pilotstudie mit geringem Stichprobenumfang
Die statistische Analyse von Untersuchungsergebnissen mit geringem Stichprobenumfang ist kritisch zu
hinterfragen. Eine Überinterpretation der Ergebnisse einer Pilotstudie sollte verhindert werden, wobei
gleichzeitig eine Aussage über den notwendigen Umfang späterer Untersuchungen anzustreben ist.
Daher wurde in dieser Pilotstudie auf die Untersuchung der Unterschiede auf Signifikanz verzichtet.
Stattdessen wurden der Unterschied der Mittelwerte, die Standardabweichung der biologischen Replikate
und der Stichprobenumfang in die statistische Analyse der Ergebnisse unterschiedlicher Bedingungen
einbezogen, um die statistische Power (P) der Ergebnisse zu ermitteln. Neben der Analyse der
Einzelergebnisse wurden diese anschließend in Gruppen mit ähnlichen Versuchsbedingungen, bei denen
auch im Vorfeld kaum Unterschiede zu erwarten waren, zusammengefasst und mit Hilfe der höchsten
aufgetretenen Standardabweichung der Ergebnisse die P berechnet. Anschließend wurde der
Stichprobenumfang eines Versuchs errechnet, welcher in Folgeversuchen zur Erhöhung der statistischen P
bei einem Alpha-Fehler von 0,05 auf 80% führen würde. Nur in den Fällen, in denen die P mehr als 80% bei
Gruppierung der Ergebnisse erreichte, wurde in dieser Arbeit eine Untersuchung der Unterschiede auf
Signifikanz (ANOVA, Signifikanzniveau <0,05) durchgeführt.
Um statistisch auswertbare Daten zu erhalten, wären abhängig von den Versuchsbedingungen 2-9
biologische Replikate ausreichend, um eine P von 80 % zu erreichen und sinnvolle Analysen in Hinblick auf
die Signifikanz der erreichten Unterschiede durchführen zu können. Für die Untersuchungen bei
atmosphärischem Druck, welche die Kollagenexpression betrachten, wären n=2 ausreichend; für solche, die
Aggrekan betrachten, dagegen n=9, da diese einer höheren Standardabweichung unterlagen. Diese
Bedingungen wurden unter Berücksichtigung der Gruppierung der Ergebnisse in dieser Arbeit erfüllt. In
Bezug auf Versuche mit erhöhter Druckbelastung sollte für Untersuchungen der Kollagenexpression n=9
gewählt werden. Hierbei wäre die Betrachtung der Aggrekanexpression ebenfalls ausreichend
berücksichtigt, da für eine P von 80 % n=5-8 ausreichend wären. Diese Kriterien wurden für einige
Ergebnisse dieser Arbeit, auch bei einer Gruppierung der Ergebnisse, nicht erfüllt.
Spätere Versuche sollten zusätzlich berücksichtigen, dass sich Unterschiede in der Höhe der Genexpression
bereits zwischen verschiedenen Proben der gleichen Passage ergeben können. Daher wären
Versuchswiederholungen mit verschiedenen Proben der gleichen Passage empfehlenswert, um die
Vergleichbarkeit der Versuchsergebnisse, die mit verschiedenen Proben der gleichen Passage stattfinden,
zu gewährleisten. Für eine standardisierte Aussage sollten auf diese Arbeit aufbauende Versuche eine
Wiederholung unter gleichen Versuchsbedingungen mit weiteren Primärkulturen beinhalten.

5 Diskussion
83
5.2 Diskussion der Ergebnisse
5.2.1 Einfluss der Kultivierungszeit auf in-vitro-kultivierte Chondrozyten
Im Rahmen der In-vitro-Kultivierung von Chondrozyten werden neben morphologischen Veränderungen
auch Unterschiede in der Gen- und Proteinexpression dieser Zellen beschrieben. Diese Unterschiede
betreffen zum einen die Expression des COL1A1-Gens, welche bedingt durch die Ausbildung von
Stressfasern in vitro bereits ab der ersten Passage wesentlich höher liegen soll als in vivo (HARDMEIER et al.
2009, TEW et al. 2008, ZEIFANG, 2010). Im Gegenzug wurde ein Absinken der Expression spezifischer Gene
des hyalinen Knorpels, wie beispielsweise jener für Kollagen Typ II und Aggrekan, beobachtet (SCHULZE-
TANZIL 2009, ZEIFANG 2010).
Ein Ziel dieser Arbeit war, zu überprüfen, ob sich der in der Literatur beschriebene progressive Verlust
typischer chondrogener Eigenschaften im Rahmen eines längeren Kultivierungszeitraums relativiert und ob
sich innerhalb dieses Kultivierungszeitraums ein geeigneter Zeitpunkt für die Untersuchungen in-vitro-
kultivierter Chondrozyten unter erhöhten Druckbedingungen finden lässt.
5.2.1.1 Einfluss der Kultivierungszeit auf die Morphologie in-vitro-kultivierter Chondrozyten
Ab dem Zeitpunkt, da frisch isolierte Chondrozyten einer Primärkultur erstmals in Zellkulturgefäßen
anhaften und sich zu teilen beginnen, wird ein Prozess in Gang gesetzt, der die Morphologie und das
Expressionsmuster der Zellen verändert. Der im Rahmen dieser Dedifferenzierung stattfindende
progressive Verlust des chondrogenen Phänotyps verläuft bezüglich des Expressionsmusters über mehrere
Passagen, wohingegen die runde Zellmorphologie nicht über die erste Passage hinaus erhalten bleibt
(TEW 2008). Stattdessen lassen sich in-vitro-kultivierte Chondrozyten morphologisch als bipolare,
spindelförmige Zellen beschreiben, welche nach erfolgter Adhärenz Zell-Zell-Kontakte ausbilden. Bei
längerer Kultivierungszeit (ab 4. Kulturtag) zeigt sich eine optische Zunahme der Zell-Zell-Kontakte zu
benachbarten Zellen (SCHULZE-TANZIL 2009). Da sich in den Versuchen dieser Arbeit bei einer Kultivierung
über den 4. Kulturtag hinaus die von SCHULZE-TANZIL (2009) beschriebene bipolare Zellmorphologie nicht
änderte und abgesehen von einer Verdichtung der Chondrozyten im Monolayer keine phasenkontrast-
mikroskopisch sichtbaren Veränderungen der in-vitro-kultivierten Chondrozyten auftraten, erscheint die
alleinige Betrachtung morphologischer Kriterien ungeeignet, um den Einfluss der Kultivierungszeit auf in-
vitro-kultivierte Chondrozyten zu untersuchen.
5.2.1.2 Einfluss der Kultivierungszeit auf die Aggrekan-, Kollagen Typ I- und Kollagen Typ II-
Expression in-vitro-kultivierter Chondrozyten
Im Rahmen der qualitativen Untersuchungen in-vitro-kultivierter Chondrozyten in Abhängigkeit von deren
Kultivierungszeit konnte deren Expression von Kollagen Typ I- und Kollagen Typ II mittels Immunzytochemie
nachgewiesen werden. Dabei war die durch die Ausbildung von Stressfasern bedingte hohe Expression des
Kollagen Typ I (Hardmeier et al. 2009, TEW et al. 2008) qualitativ analog zu den Ergebnissen von
DOMM et al. (2002) über den gesamten untersuchten Kultivierungszeitraum (4.-35. Kulturtag) nachweisbar.
Im Gegensatz zur vorgenannten Studie (DOMM et al. 2002) zeigten in-vitro-kultivierte Chondrozyten dieser
Arbeit die von SCHULZE-TANZIL (2009) und ZEIFANG (2010) beschriebene Expression knorpelspezifischer
Proteine, wie beispielsweise Kollagen Typ II, vom 4.-21. Kulturtag.
Die quantitativen Untersuchungen dieser Arbeit liefern Aussagen über die Änderung der Aggrekan-,
Kollagen Typ I- und Kollagen Typ II-Expression in-vitro-kultivierter Chondrozyten in Abhängigkeit von deren

5 Diskussion
84
Kultivierungszeit. Analog zu den Ergebnissen von TEW et al. (2008) zeigten auch die in dieser Arbeit
kultivierten Chondrozyten zunächst (1.-3. Kulturtag) eine hohe Expression von COL1A1-Genen, wohingegen
spezifische Gene des hyalinen Knorpels, wie COL2A1 und Aggrekan, herunterreguliert vorlagen. Bei längerer
Kultivierung (ab 4. Kulturtag) lag die Expression knorpelspezifischer Gene signifikant höher als an den
ersten Kulturtagen und blieb auch bei längerer Kultivierung (bis 35. Kulturtag) auf diesem Niveau. Dabei fiel
auf, dass die Expression des Kollagen Typ II nach dem initial niedrigen Ausgangswert der ersten 3 Tage am
Tag 4 einen deutlich stärkeren Anstieg aufwies als Kollagen Typ I und die Reduktion der Expression des
Kollagen Typ II am 7. Tag im Vergleich zu Kollagen Typ I geringer ausfiel. Dieses führte dazu, dass sich die
Expression des Kollagen Typ II an die des Kollagen Typ I anglich, um am Tag 21 sogar höhere Werte als
dieser zu erreichen. Das knorpelspezifische Kernprotein Aggrekan, welches etwa im Verhältnis 1:3 zu den
Kollagen Typ II-Fasern im hyalinen Knorpel vorliegt (BUCKWALTER et al. 2005, SCHULZ und BADER 2007),
blieb in seiner Expression in vitro insgesamt deutlich hinter jener der beiden Kollagentypen zurück. Es
erreichte für keinen der Kultivierungszeitpunkte eine ähnlich hohe Expression wie die beiden untersuchten
Kollagentypen, sodass sich in vitro ein Verhältnis Aggrekan zu Kollagen Typ II von 1:320 (berechnet für
7. Kulturtag, 1 bar) ergab.
Insgesamt kann davon ausgegangen werden, dass die Synthese knorpelspezifischer Bestandteile der ECM
zunächst zeitweilig zu Gunsten der Zellteilung in den Hintergrund tritt (GSTRAUNTHALER und LINDL 2013,
SCHULZE-TANZIL 2009, TEW et al. 2008). Anfänglich stand für die frisch ausgesäten, sich schnell teilenden
Chondrozyten die Anpassung an die Bedingungen der Zellkultur im Vordergrund und somit vornehmlich die
Synthese des Kollagen Typ I, welches am Aufbau und an der Ausrichtung des Zytoskeletts, sowie an der
Bildung von Stressfasern beteiligt ist (HARDMEIER et al. 2009, ZEIFANG, 2010). Durch die Zunahme der
Kontakte der Zellen untereinander herrschen Bedingungen, die dem Stadium der Chondroprogenitorzellen
im Anschluss an den Kondensationsprozess entsprechen (GOLDRING et al. 2006, GOLDRING 2012, SINGH
und SCHWARZBAUER (2012). Dieses liefert eine Erklärung dafür, dass zu diesem Zeitpunkt eine
Hochregulierung der Gene für Kollagen Typ II und Aggrekan stattfand und das dabei erreichte
Expressionslevel auch bei langer Kulturzeit aufrechterhalten wurde. Gleichzeitig unterblieb jedoch die für
dieses Stadium typische Herunterregulierung der Gene des Kollagen Typ I bei der Langzeitkultivierung
(4.-35. Kulturtag). Als Ursache hierfür kommen zum einen die weiterhin stattfindenden Zellteilungen in
Betracht, zum anderen erfordert die stetige Entfernung des extrazellulär abgegebenen Kollagen Typ I mit
jedem Mediumwechsel dessen Neusynthese. Hinzu kommt, dass die Mediumwechsel-bedingte Entfernung
der in den Extrazellularraum abgegebenen ECM ein Auseinanderrücken der Chondrozyten verhindert.
Hierdurch werden die Zell-Zell-Kontakte durch die Synthese einer chondrozytenspezifischen ECM nicht, wie
in vivo im Stadium der Chondroprogenitorzellen wieder zurückgebildet, sondern bleiben in vitro auch bei
längerer Kultivierung erhalten (GOLDRING et al. 2006, GOLDRING 2012, SCHULZE-TANZIL 2009). Durch den
in vivo typischen Abfall der TGF-1-Konzentration der Chondroprogenitorzellen im Kondensationsstadium
entfällt dessen hemmender Einfluss auf die Expression der COL1A1-Gene, sodass die Synthese des Kollagen
Typ I aufrechterhalten wird. Gleichzeitig führt dieses Absinken der TGF-1-Konzentration zu einer höheren
Empfindlichkeit der Zellen gegenüber den Zytokinen IGF-1 und FGF-2, die für den Anstieg der
Genexpression des chondrozytenspezifischen Kollagen Typ II und Aggrekan verantwortlich sind
(SCHULZE-TANZIL 2009).
Die stetige Entfernung neu gebildeter ECM liefert auch eine Erklärung für die Detektion annähernd
einheitlicher Proteinexpressionen des Kollagen Typ I und II sowie des Aggrekan zu den verschiedenen

5 Diskussion
85
Zeitpunkten im Western Blot. Da beide Kollagentypen sowie auch Aggrekan hauptsächlich in den
Extrazellularraum sezerniert vorkommen, wurden die α-Ketten der Kollagene und die G3-Domäne des
Aggrekan in hohem Maße auch dort nachgewiesen (MARLOVITS und VÉCSEI 2000). Der Mediumwechsel
verhindert offenbar die Anreicherung der extrazellulären Proteine, sodass eine deutliche Zunahme
chondrozytenspezifischer ECM über den Zeitverlauf der Kultur nicht nachgewiesen wurde. Stattdessen sind
für die Unterschiede in der Proteinexpression des Kollagen Typ I am ehesten Unterschiede der einzelnen
Proben zwischen letztem Mediumwechsel und Probengewinnung entscheidend (GSTRAUNTHALER und
LINDL 2013) und daneben die in Kap. 5.1.2.2 genannten Abweichungen durch die Sensitivität und Qualität
der Antikörper. Aufgrund der Höhe der Unterschiede in der mRNA-Expression der einzelnen untersuchten
Kulturtage, ein Erreichen eines detektierbaren Unterschieds, für welchen ein Unterschied der
Proteinexpression von 30-40 % erforderlich ist, nicht zu erwarten.
Die Ergebnisse des Western Blots lieferten zusätzliche Hinweise darauf, dass die in-vitro-kultivierten
Chondrozyten nicht in ein Entwicklungsstadium übergehen, welches die Knorpelentwicklung mit der
Knochenbildung verbindet und somit entdifferenzieren. Vielmehr ist anzunehmen, dass es sich bei der im
Rahmen dieser Arbeit für Kollagen Typ II bei 65 kDa detektierten Proteinbande um das von FUKUI et al.
(2001) beschriebene Dimer des N-terminale Propeptids der immaturen α1(IIA)-Kette des Kollagen Typ II bei
60-65 kDa handelt, die von Chondroprogenitorzellen exprimiert wird. Die Chondrozyten vollziehen somit
durch die In-vitro-Kultur in ihrer Entwicklung eher „einen Schritt zurück“ ausgehend von den Chondrozyten
hin zu Zellen mit Merkmalen des Stadiums der Chondroprogenitorzellen. Eine angestrebte
Redifferenzierung der Zellen sollte somit nicht darauf abzielen, ein bereits durchlaufenes
Entwicklungsstadium wieder zu erreichen, sondern Methoden zu entwickeln, die eine gerichtete
Weiterdifferenzierung der Zellen unterstützen, um somit deren Expression in Richtung einer
chondrozytenspezifischen ECM zu lenken, die in der Lage ist, in vivo die Stabilität und Integrität des
hyalinen Knorpels zu garantieren.
Entsprechend der Expressionslevel der in dieser Arbeit kultivierten Chondrozyten sollte das von BARLIČ et
al. (2008) als Index für die Dedifferenzierung in-vitro-kultivierter Chondrozyten etablierte Verhältnis von
Kollagen Typ I zu Typ II sowie von Aggrekan zu Versikan kritisch betrachtet werden, insbesondere da es als
ein für die jeweilige Passage der Zellen fest stehender Parameter angesehen wird. Während der
Dedifferenzierungs-Index für Kollagen Typ I/II der im Rahmen dieser Arbeit kultivierten Chondrozyten
insbesondere an den ersten 3 Kulturtagen deutlich zu Gunsten des Kollagen Typ I ausfiel, glich sich die
Expression der beiden Kollagentypen bei längerer Kultivierung einander an. Da die Zunahme der Expression
des Aggrekan mit der des Kollagen Typ II zusammenfällt, ist ein ähnliches Verhalten des Dedifferenzierungs-
Index für Aggrekan/Versikan zu erwarten. Die von BARLIČ et al. (2008) beschriebene Zunahme der
Dedifferenzierung durch Herunterregulierung der Gene für Kollagen Typ II und Aggrekan bei längerer
Kultivierungszeit steht im Kontrast zu den in dieser Arbeit erzielten Ergebnissen. Hierfür kommen
verschiedene Ursachen in Betracht. Zum einen wurden im Rahmen dieser Arbeit Zytokine wie IGF-1 und
FGF-2, die in vivo für die Synthese und Sekretion von Kollagen Typ II und Aggrekan verantwortlich sind, dem
Kulturmedium zugesetzt, zum anderen ein langes Mediumwechsel-Intervall (3-4 Tage) genutzt
(GOLDRING et al. 2006, GOLDRING 2012). Während ersteres die Synthese der Chondrozyten in Richtung
einer chondrozytenspezifischen ECM lenkt, vermag letzteres diese im Extrazellularraum länger
aufrechtzuerhalten, wodurch Kulturbedingungen geschaffen werden, welche der Situation in vivo näher
kommen.

5 Diskussion
86
Die Abhängigkeit des Expressionsmusters und somit des Ausmaßes der Dedifferenzierung
in-vitro-kultivierter Chondrozyten von der Kultivierungszeit ist bei der Planung von Versuchen zu
berücksichtigen. Folglich ist im Sinne der Optimierung des Zellkultursystems die Nutzung
in-vitro-kultivierter konfluenter Chondrozyten für die Untersuchung der Effekte modifizierter
Druckbedingungen sinnvoll, sodass die in dieser Arbeit geplanten Untersuchungen unter erhöhten
Druckbedingungen an optisch konfluenten Chondrozyten der Passage 6 bei einer Vorkultivierungszeit von
6 Tagen ohne Druck durchgeführt wurden. Hierbei wurde auch die geringere Standardabweichung der
PCR-Ergebnisse bei 7 Tagen verglichen mit der bei 4 Tagen berücksichtigt. Da die Chondrozyten nach
Erreichen der optischen Konfluenz bei weiterer Kultivierung (14, 21, 35 Tage) keine Regelmäßigkeit in der
Expression des Kollagen Typ I, II sowie des Aggrekan bei gleichzeitig vorhandener hoher Varianz erkennen
ließen, kann die Nutzung einer Langzeitkultur der Chondrozyten für weitere Versuche nicht empfohlen
werden.
5.2.2 Einfluss nutritiver Faktoren auf die Vitalität in-vitro-kultivierter Chondrozyten
Der Einfluss des Intervalls des Mediumwechsels auf die Kultur der Chondrozyten wurde untersucht, bevor
diese bei modifizierten Druckbedingungen kultiviert wurden. Dies geschah vor dem Hintergrund, die
Beeinflussung der unter erhöhtem Druck stattfindenden Versuche durch nutritive Faktoren möglichst
gering zu halten.
Durch den Stoffwechsel der Chondrozyten ändern sich die Kulturbedingungen fortwährend. Die
Konzentration der Nährstoffe (Glucose, Aminosäuren) im Medium sinkt stetig ab, wohingegen es zu einer
Anreicherung der Stoffwechselendprodukte (Lactat, Ammoniak) kommt. Dadurch verschieben sich der pH-
Wert des Mediums und der Redoxstatus der Zellen. Diese Veränderungen der Zusammensetzung des
Mediums verlaufen zwischen zwei Mediumwechseln kontinuierlich, wohingegen die Chondrozyten zum
Zeitpunkt des Mediumwechsels eine plötzliche Änderung der Kulturbedingungen erfahren
(GSTRAUNTHALER und LINDL 2013). Da die Abnahme der Nährstoffkonzentration und die Zunahme der
Stoffwechselendprodukte im Medium mit der Proliferation der kultivierten Zellen korreliert, sollte der
Mediumwechsel in schnell wachsenden Zellkulturen, zu denen die Chondrozyten zählen, alle 2 Tage
erfolgen (GSTRAUNTHALER und LINDL 2013, TEW et al. 2008). Um eine längere Kultivierung der
Chondrozyten im Bioreaktor bei konstanten Druckbedingungen zu ermöglichen, wurde das Medium der
Chondrozyten zunächst unter atmosphärischem Druck in längeren Abständen erneuert. Dabei zeigte sich,
dass bei einem Mediumwechsel nach bis zu 4 Tagen die Vitalität der Chondrozyten unverändert war. Erst
bei weiterer Verlängerung des Intervalls zwischen zwei Mediumwechseln wurde die Zellausbeute geringer
und die Mortalität der Zellen nahm zu. Der von GSTRAUNTHALER und LINDL (2013) als Indikator für den
Zeitpunkt des Mediumwechsels angeführte Farbumschlag des Mediums durch dessen pH-Wert-Änderung
lässt sich für das verwendete CBM+ Medium nicht nutzen. Der Farbumschlag dieses Mediums fand erst am
7. Kulturtag und somit zu einem Zeitpunkt statt, an dem bereits deutliche negative Effekte auf die Vitalität
der Chondrozyten nachweisbar waren.
Diesen Untersuchungen entsprechend wurde in dieser Arbeit das Medium der Chondrozyten nach 3-4
Tagen gewechselt, da bei diesem Intervall keine negativen Effekte auf die Vitalität und somit die
Syntheseleistung der Chondrozyten zu erwarten waren.

5 Diskussion
87
5.2.3 Einfluss hydrostatischen Drucks auf in-vitro-kultivierte Chondrozyten
Durch die belastungsbedingte Umverteilung von Wasser innerhalb des Gelenkknorpels erfahren auch die
Chondrozyten den auf den hyalinen Gelenkknorpel einwirkenden Druck (WONG und CARTER 2003). Die
zelluläre Transduktion dieser einwirkenden Druckbelastung wird durch Mechanorezeptoren ermöglicht, die
so genannten „connexin hemichannels“ (KNIGHT et al. 2009). Diese versetzen die Chondrozyten in die Lage,
ihre metabolische Aktivität und somit die Expression der Komponenten der ECM zu modifizieren
(BUCKWALTER et al. 2005, GARCIA und KNIGHT 2010, WHITFIELD 2008). Daher zielen In-vitro-Studien
darauf ab, Druckbelastungen zu simulieren, welche die metabolische Aktivität der Chondrozyten und somit
die Zusammensetzung der durch sie synthetisierten ECM optimieren. Hierbei hat sich die Applikation von
hydrostatischem Druck als repräsentativ für die Druckverhältnisse in der Radiärzone des Gelenkknorpels
herauskristallisiert, wobei Scherkräfte, wie sie in vivo auf die Tangentialzone einwirken, unberücksichtigt
bleiben (ELDER 2009). Entsprechend der einfacheren Standardisierung und Durchführbarkeit entstammt
heute ein Großteil der Informationen über die Auswirkungen hydrostatischer Bedingungen auf
Chondrozyten den Untersuchungen in Zell- und Gewebekulturen in Bioreaktoren (GUILAK und MOW 2000,
HARDMEIER et al. 2009, WONG und CARTER 2003). In dem in dieser Arbeit genutzten Bioreaktor wurde der
Druck durch die Kompression einer Gasphase mit entsprechender Weiterübertragung in das die Zellen
umgebende Medium aufgebaut. In dieser Art der Bioreaktorsysteme sind Zeitpunkt, Höhe, Zeitdauer und
Frequenz des einwirkenden Drucks die beeinflussbaren Parameter (SCHULZ und BADER 2007). Mit Hilfe des
für diese Arbeit verwendeten Bioreaktors wurden die Parameter Zeitpunkt, Höhe und Zeitdauer des
einwirkenden Drucks variiert, mit dem Ziel, die metabolische Aktivität der Chondrozyten und somit die
Zusammensetzung der durch sie synthetisierten ECM zu untersuchen.
5.2.3.1 Einfluss hydrostatischen Drucks auf die Morphologie in-vitro-kultivierter Chondrozyten
Analog zu den Ergebnissen von FIORAVANTI et al. (2003) und SMITH et al. (2000) traten auch in dieser
Untersuchung bei kurzen Druckphasen (bis 24 h) keine Veränderungen der bipolaren Morphologie der
Chondrozyten im Vergleich mit den ohne Einwirkung von Druck kultivierten Kontrollen auf. Bei längerer
Dauer der Druckeinwirkung (> 24 h) rundeten sich die Zellen ab und lösten sich schließlich vom Boden der
Zellkulturschale. Dies deutet darauf hin, dass die Synthese der für die Adhärenz der Chondrozyten am
Boden der Kulturschale verantwortlichen Zelladhäsionsmoleküle, wie beispielsweise N-CAMs und
Fibronectin, offenbar nicht in adäquater Menge stattfand (GSTRAUNTHALER und LINDL 2013, SCHMITZ
2011). Im gleichen Zusammenhang traten Zellen auf, die Zeichen von Karyopyknosis und Karyorrhexis sowie
ein Anschwellen des Zytoplasmas aufwiesen. Diese Veränderungen des Kerns und des Zytoplasmas der
Zellen wurden als zytologische Kriterien bezüglich ihres Auftretens im Rahmen des Zelltods herangezogen,
wobei sie den unterschiedlichen Formen des Zelltods im Rahmen der Apoptose, Onkose und Nekrose
zugeordnet wurden (MAJNO und JORIS 1995).
Bei der Apoptose handelt es sich um einen genetisch kontrollierten Prozess, dessen Eintreten durch die
extrazellulären Bedingungen der Zelle modifiziert wird. Hierzu gehören auch physikalische Bedingungen,
wie beispielsweise der einwirkende Druck. Im Rahmen der Apoptose stehen die Kondensation des
Chromatins (Karyopyknosis) und die Abschnürung pyknotischer Bestandteile der Zelle sowie die
Zellschrumpfung im Vordergrund. Im Gegensatz dazu sind die Veränderungen extrazellulärer Bedingungen
der Auslöser der als Onkose bezeichneten Initiation des Zelltods, dem die genetische Kontrolle fehlt.
Pathogenetisch kommt es hierbei zu einem Funktionsverlust membranständiger Adenosintriphosphat

5 Diskussion
88
(ATP)-abhängiger Ionenpumpen und in der Folge zu einem osmotischen Anschwellen der Zellen. Beide
Formen der Initiation des Zelltods führen zur Nekrose, welche morphologisch neben der Karyopyknosis
durch eine Fragmentierung des Zellkerns (Karyorhexis) und dessen Zerfall (Karyolysis) gekennzeichnet ist,
wodurch es letztlich zum Strukturverlust des Zytoplasmas und dessen Zerfall kommt (MAJNO und JORIS
1995).
Als Ursache des auslösenden Funktionsverlusts der Ionenpumpen im Rahmen der Onkose wurden bisher
zumeist die Ischämie-bedingte Verarmung der Zellen an Adenosintriphosphat (ATP), die Hemmung der
Bildung von ATP durch Toxine sowie die Denaturierung der Ionenpumpe (Protein) durch Hitze betrachtet
(MAJNO und JORIS 1995). Daneben ist jedoch auch das druckabhängige Verhalten der Ionenpumpen
in-vitro-kultivierter Chondrozyten untersucht worden. Hierbei konnte gezeigt werden, dass eine
Druckeinwirkung von 10-50 bar bereits im Bereich von Sekunden bis Minuten in der Lage ist, eine reversible
Inhibition der Ionenpumpen auszulösen (HALL 1999).
Die im Rahmen dieser Arbeit bei langer Kultivierung der Chondrozyten unter hydrostatischem Druck (>24 h)
festgestellten morphologischen Veränderungen können somit auf eine umgebungsbedingte Zellschädigung
im Sinne einer Onkose mit anschließender Nekrose zurückgeführt werden. Als Ursache für die
Zellschädigung kann die von HALL (1999) beschriebene Inhibition der Ionenpumpen durch den
einwirkenden hydrostatischen Druck angesehen werden, welche initial ein osmotisch bedingtes
Anschwellen des Zytoplasmas der untersuchten Chondrozyten auslöst (MAJNO und JORIS 1995). Da die
Anzahl der auf diese Weise veränderten Zellen mit zunehmender Dauer der Kultivierung unter Druck
anstieg, ist denkbar, dass die Hemmung der Ionenpumpen zu lange aufrechterhalten wurde und somit bei
mehr als 24-stündiger Druckeinwirkung zytotoxisch auf die Chondrozyten wirkte. Die Hemmung der
Ionenpumpen scheint jedoch, analog zu den Ergebnissen von Hall (1999), in den überlebenden
Chondrozyten reversibel zu bleiben, da diese sich bei anschließender Kultivierung unter atmosphärischem
Druck zumindest morphologisch normal weiterentwickelten.
Während HALL (1999) bei niedrigerem Druck im Bereich von 1-10 bar eine geringere Hemmung der
Ionenpumpen nachwies, setzte der zytotoxische Effekt des Drucks in den Untersuchungen dieser Arbeit bei
5 bar zeitlich eher ein als bei 10 bar. Eine mögliche Erklärung hierfür könnte sein, dass der Druck von 5 bar
deutlich unter dem in vivo intraartikulär wirkenden Druck liegt (BUCKWALTER et al. 2005), sodass eine
adäquate Reaktion der Zellen auf diesen niedrigen Druck nicht zu erwarten war. Da das morphologisch
instabile Verhalten der bei 5 bar kultivierten Chondrozyten in der Zellkultur auch in einer Wiederholung
erhalten blieb, wurde für die weitere Untersuchung des Expressionsmusters bei Variation der
Druckbedingungen eine Druckhöhe von 10 bar favorisiert.
5.2.3.2 Einfluss hydrostatischen Drucks auf die Aggrekan-, Kollagen Typ I- und Kollagen Typ II-
Expression in-vitro-kultivierter Chondrozyten
Die Untersuchung des Einflusses eines erhöhten Drucks auf die Aggrekan-, Kollagen Typ I- und Kollagen
Typ II-Expression erfolgte mittels qPCR und Western Blot. Sie wurde an Chondrozyten bei einer
Vorkultivierungszeit von 6 Tagen (unter Normaldruck) und einer anschließenden maximalen
Kultivierungszeit von 24 h unter erhöhtem Druck im Bioreaktor durchgeführt. Mit dem Ziel, Bedingungen zu
finden, welche die Komposition einer chondrozytenspezifischen ECM positiv beeinflussen, wurden für die
Untersuchung des Einflusses der Druckhöhe des auf konfluente Chondrozyten einwirkenden
hydrostatischen Drucks Druckbelastungen von 5 und 10 bar gewählt, welche physiologischer Weise

5 Diskussion
89
während der normalen Gelenkbewegung erreicht werden (10 bar) oder diese unterschreiten (5 bar)
(IKENOUE et al. 2003).
Während IKENOUE et al. (2003) durch einen hydrostatischen Druck keine Veränderung der Genexpression
chondrozytenspezifischer ECM nachwiesen, zeigen die Ergebnisse dieser Untersuchung analog zu WONG
und CARTER (2003) eine signifikante Zunahme der Expression des Aggrekan durch den einwirkenden Druck,
wohingegen es zu einer signifikanten Abnahme der Genexpression von Kollagen Typ I und II verglichen mit
den Verhältnissen ohne Druck kam. Entsprechend ist bei der Einwirkung hydrostatischen Drucks von 24 h
auf konfluente Chondrozyten eine Steigerung solcher PGs zu vermuten, die auf Aggrekan als Kernprotein
basieren, da dessen Genexpression unter Druck anstieg. Gleichzeitig sank die Expression der kollagenen
Fasern. Eine solche rückläufige Entwicklung der Expression des in vitro überrepräsentierten Kollagen Typ I
ist vorteilhaft, da Kollagen Typ II 90-95% der Kollagenfasern im hyalinen Knorpels stellt, wohingegen
Kollagen Typ I nur in geringem Anteil vorliegt (BUCKWALTER et al. 2005, GELSE et al. 2003). Die
Verminderung der Expression des Kollagen Typ I deutet somit darauf hin, dass die Konzentration der durch
die Chondrozyten in vitro produzierten Stressfasern durch den in diesem Versuch einwirkenden Druck
abnahm (TEW et al. 2008, ZEIFANG et al. 2010). Überträgt man die Situation während der Entwicklung des
Knorpels auf diese Ergebnisse, so ist denkbar, dass die geringe TGF-1-Konzentration der
Chondroprogenitorzellen in vivo, welche die Expression von Kollagen Typ I vermindert, in deren weiterer
Entwicklung, insbesondere nach der Bildung des Gelenkspalts, durch den Gelenkdruck aufrechterhalten
wird (GOLDRING et al. 2006, GOLDRING 2012, FORTIER 2011). Dieser physiologische Druck (10 bar) scheint
bei Einwirkung auf in-vitro-kultivierte Chondrozyten ebenfalls in der Lage zu sein, die TGF-1-Konzentration
zu senken. Hierdurch wäre die niedrigere Kollagen Typ I-Expression der unter Druck kultivierten
Chondrozyten verglichen mit solchen, denen die Druckeinwirkung fehlte, zu erklären (SCHULZE-TANZIL
2009).
Da in vivo auf die das Grundgerüst des hyalinen Knorpels bildenden Kollagen Typ II-Fasern 10-30% und auf
die PGs, welche hauptsächlich auf Aggrekan als Kernprotein beruhen, 3-10 % seines Nassgewichts entfallen,
liegt das Verhältnis Aggrekan zu Kollagen Typ II im hyalinen Gelenkknorpel bei 1:3 (BUCKWALTER et al.
2005, SCHULZ und BADER 2007). Diesem steht in den hier erfolgten Untersuchungen in vitro ein Verhältnis
Aggrekan zu Kollagen Typ II von 1:320 (berechnet für 7. Kulturtag, 1 bar) und somit eine verglichen mit der
Situation in vivo etwa 100-fach höhere Kollagen Typ II-Expression gegenüber, sodass dessen verminderte
Synthese in vitro nicht zwingend als negativer Effekt des Drucks auf die Komposition der ECM in vitro zu
interpretieren ist.
Diese Veränderungen der Chondrozyten auf der Ebene des Transkriptoms ließen sich im Gegensatz zu den
Ergebnissen von IKENOUE et al. (2003) in den Versuchen dieser Arbeit bei 10 bar nachweisen. Die weitere
Reduktion der Höhe des hydrostatischen Drucks auf 5 bar bewirkte ein weiteres Absinken der mRNA für die
kollagenen Fasern, jedoch auch einen höheren Anstieg der mRNA für Aggrekan. Die gegenüber den bei 10
bar kultivierten Chondrozyten erhöhte Aggrekansynthese bei 5 bar deutet eine Zunahme
chondrozytenspezifischer ECM bei geringerem Druck an. Diese Annahme ist in Frage zu stellen, da sie sich
im Wesentlichen auf den Anstieg der Genexpression des Aggrekan bei 5 bar verglichen mit 10 bar stützt,
wobei die hierfür zugrunde liegenden Daten einer hohen Variabilität unterlagen. Daneben ist insbesondere
der Anstieg der mRNA-Expression des knorpelspezifischen Aggrekan in der Analyse des Proteoms bei 5 bar
nicht nachvollziehbar. Vielmehr zeigt sich auf Proteinebene eine deutlich höhere Expression des Aggrekan
bei 10 bar verglichen mit jener bei 5 bar, welche kaum Unterschiede zur Kontrolle zeigte. Neben dieser

5 Diskussion
90
höheren Proteinexpression des Aggrekan lag auch jene der beiden untersuchten Kollagentypen bei 10 bar
verglichen mit 5 bar höher. Die erhöhte Expression des Aggrekan in dieser Untersuchung könnte ebenfalls
mit dem Entwicklungsstadium der Chondroprogenitorzellen in Verbindung gebracht werden, da deren
geringere TGF-1-Konzentration zu einer höheren Empfindlichkeit der Zellen gegenüber den Zytokinen
IGF-1 und FGF-2 führt, welche für den Anstieg der Genexpression der chondrozytenspezifischen
Bestandteile der ECM, wie beispielsweise Aggrekan, verantwortlich sein könnte (FORTIER 2011,
SCHULZE-TANZIL 2009).
Betrachtet man die bei 5 bar in der qPCR und im Western Blot erzielten Ergebnisse im Zusammenhang mit
den bei 5 bar auftretenden morphologischen Veränderungen (Ablösung der Zellen von der Kulturschale),
wird deutlich, dass bei dem unphysiologisch niedrigen Druck von 5 bar das Einstellen der Syntheseleistung
der Chondrozyten im Zuge der Onkose und Nekrose als wahrscheinliche Ursache der geringeren
Kollagenexpression angesehen werden kann. Da sich die Abrundung der Chondrozyten bei 24 h und 5 bar
auch bei der Wiederholung nicht vermeiden ließ, konnte somit die im Vergleich zu 10 bar verringerte
Expression kollagener Fasern auf eine früher einsetzende Nekrose zurückgeführt werden. Daher wurden
die weiteren Untersuchungen bei einer Druckhöhe von 10 bar durchgeführt. Dies geschah vor dem
Hintergrund, Bedingungen zu finden, welche ähnlich hemmende Effekte auf die Kollagenexpression und
fördernde Einflüsse auf die Expression des Aggrekan erreichen wie bei 5 bar und gleichzeitig die Vitalität
der Chondrozyten erhalten.
5.2.4 Kontinuierliche versus diskontinuierliche Druckbedingungen
Die morphologischen Erkenntnisse über die Zytotoxizität der Langzeitkultivierung der Chondrozyten unter
Druck führten zu der Frage, ob die dabei in den Zellen induzierten Signalwege schon zu einem früheren
Zeitpunkt einen negativen Einfluss auf das Expressionsmuster der Chondrozyten im Hinblick auf die
Zusammensetzung der ECM im hyalinen Knorpel zeigen, oder ob dieses erst bei mehr als 24 h Kultivierung
unter Druck der Fall ist. Daher wurde der Zeitraum, innerhalb dessen die Chondrozyten eine
Druckeinwirkung von 10 bar erfuhren, ausgehend von den bisher genutzten 24 h auf 12 h, 6 h, 3 h und 1,5 h
verkürzt.
Hierbei zeigten die bei 10 bar kultivierten Chondrozyten eine etwas geringere Expression der beiden
Kollagentypen verglichen mit den bei atmosphärischem Druck kultivierten Zellen. Trotz der deutlichen
Unterschiede in der Zeitdauer der Druckeinwirkung, der die Chondrozyten ausgesetzt waren, wichen ihre
Kollagenexpressionen sowohl auf der Ebene des Transkriptoms als auch bei Betrachtung des Proteoms
analog zu den Untersuchungen von IKENOUE et al. (2003) kaum voneinander ab. IKENOUE et al. (2003)
wiesen einen Anstieg der Kollagen Typ II-Expression erst bei einer diskontinuierlichen Druckbelastung von
jeweils 4 h Druck an 4 aufeinanderfolgenden Tagen nach. Daneben schlussfolgerten WONG und CARTER
(2003) aus der Untersuchung von Gewebeexplantaten, dass analog zur Situation in vivo die Abhängigkeit
der Kollagenexpression von der Anwesenheit von Scherkräften stärker ist als vom einwirkenden Druck.
Einzig für die Expression des Kernproteins Aggrekan ließ sich in diesem Versuch eine Abhängigkeit der Höhe
seiner Expression von der Dauer der Druckeinwirkung darstellen. So war die Genexpression des Aggrekan
bei kurzer Kultivierung der Chondrozyten unter erhöhtem Druck (<3 h) höher als bei langer Dauer der
Druckeinwirkung, wobei diese Unterschiede auf Proteinebene im Western Blot nicht bestätigt wurden.
Diesen Zusammenhang stellten auch IKENOUE et al. (2003) bei Nutzung eines höheren Drucks für kurz
einwirkende Druckbelastungen heraus. Die erhöhte Expression des Aggrekan ist auch in Zusammenhang

5 Diskussion
91
mit dem von TAKAHASHI et al. (1997) bei kontinuierlichem Druck für 2 h nachgewiesenen Anstieg der
Gesamt-PG-Konzentration zu sehen.
Zusammenfassend stützen die Ergebnisse dieser Arbeit die Hypothese, dass Effekte bezüglich der
Konzentration der PGs in der Regel deutlich stärker ausfallen und schneller erreicht werden als solche auf
die Expression von Kollagen Typ II (TOYODA et al. 2003). Diese Hypothese wird in der Literatur kontrovers
diskutiert. Zum einen, da es im Rahmen der Biodynamik des Gelenkknorpels bei einwirkendem Druck zu
einem Flüssigkeitsausstrom kommt und frühe In-vivo-Untersuchungen Hinweise lieferten, dass ein
Flüssigkeitsverlust ursächlich für eine Verminderung der PG-Synthese ist (BUSCHMANN et al. 1995, GUILAK
und MOW 2000, LUPPA 2000, TAKAHASHI et al. 1997). Andererseits zeigen neuere Studien, dass die bei
moderater Belastung stattfindende physiologische Dickenzunahme des Gelenkknorpels zum Großteil auf
die verstärkte Synthese der PGs zurückgeht (ALFORD 2005, LUPPA 2000). Diese gesteigerte Synthese der
PGs wird maßgeblich durch das Zytokin FGF-2 gesteuert, dessen stimulierende Wirkung mit steigender
Konzentration abnimmt (FORTIER et al. 2011). Es ist denkbar, dass dessen Syntheserate druckabhängig
gesteuert wird und für die bei 1,5 und 3 h Druck vorliegende hohe Expression des Aggrekan verantwortlich
ist. Mit zunehmender Dauer des einwirkenden Drucks kommt es demnach zu einem weiteren Anstieg der
Konzentration des FGF-2, sodass der positive Effekt des kurzzeitig einwirkenden Drucks auf die Expression
des Aggrekan mit zunehmender Dauer seiner Einwirkung wieder rückläufig ist.
Ein Vergleich mit diskontinuierlichen Druckbedingungen ist nur eingeschränkt möglich, da sich die
Ergebnisse solcher Untersuchungen in den genutzten Zellen, den angewendeten Druckbedingungen sowie
in Bezug auf die gewählte Methodik unterscheiden (JORTIKKA et al. 2000, SMITH et al. 1996, TAKAHASHI et
al. 1997). Daneben ist die Abgrenzung diskontinuierlicher hydrostatischer Druckbedingungen, welche durch
die zusätzliche Variable der Frequenz gekennzeichnet sind, von kontinuierlichen hydrostatischen
Druckbedingungen nicht immer klar definiert, sodass die Übergänge mitunter fließend sein können (IKENOE
et al. 2003, SMITH et al. 2000). Auf diese Art betrachtet, weisen die kurzzeitigen Druckbelastungen dieser
Arbeit (1,5 und 3 h) Ähnlichkeiten mit den von IKENOE et al. (2003) und SMITH et al. (2000) genutzten
Bedingungen (4 h pro Tag, bei 1 Tag Kultivierung unter Druck) auf. Die Vergleichbarkeit der Ergebnisse lässt
schlussfolgern, dass auch unter kontinuierlichen Druckbedingungen eine adäquate Expression von Kollagen
Typ II und Aggrekan erzielt werden kann. Dies ist insofern von Vorteil, als dass Bioreaktoren zur
Generierung eines diskontinuierlichen Drucks wesentlich komplexer in Bezug auf den apparativen Aufwand
und deutlich schwieriger in der Vermeidung eines Eintrags von Verunreinigungen sind als Bioreaktoren,
welche einen kontinuierlichen Druck erzeugen (SCHULZ und BADER 2007). Selbstverständlich kommt eine
zyklische Belastung der Chondrozyten, wie sie in diskontinuierlichen Bioreaktoren erreicht wird, der
Situation in vivo näher. Wenn mit kontinuierlichen Bioreaktoren jedoch vergleichbare Ergebnisse in Hinblick
auf die Synthese des Aggrekan erreichbar sind, bleibt zu untersuchen, ob die zyklische Belastung für die
Untersuchung der Druckwirkung auf in-vitro-kultivierte Chondrozyten wesentlich ist und den enormen
apparativen Aufwand rechtfertigt.
5.2.5 Dedifferenzierung in-vitro-kultivierter Chondrozyten - Anwendbarkeit und Limitation
genutzter Strategien
Die bei der In-vitro-Kultivierung von Chondrozyten eingesetzten Strategien, um der zellulären
Dedifferenzierung entgegenzuwirken, fanden auch in dieser Arbeit Anwendung (BRITTBERG 1994,
ERGGELET 2003, FRITSCH et al. 1999). Zum einen wurden dem CBM-Medium Zytokine zugesetzt, um mit

5 Diskussion
92
Hilfe von IGF-1 die Synthese und Sekretion von Kollagen Typ II, und durch FGF-2 die Expression von
Aggrekan zu stimulieren (GOLDRING et al. 2006, GOLDRING 2012). Zum anderen wurden die Chondrozyten
in relativ hoher Zelldichte ausgesät, um die Konzentration des Zytokins TGF-1 gering zu halten und die
daraus resultierende höhere Empfindlichkeit der Zellen gegenüber IGF-1 und FGF-2 auszunutzen (SCHULZE-
TANZIL 2009). Mit einer abschließenden Untersuchung sollte der Einfluss einer weiteren Erhöhung der in
die Kulturschalen ausgesäten Chondrozyten beurteilt werden.
In dieser Untersuchung zeigte sich bei doppelter Zelldichte der Chondrozyten verglichen mit in normaler
Zelldichte ausgesäten Chondrozyten eine 1,5-fach höhere Expression des Kollagen Typ I, bei gleichzeitigem
Rückgang der Expression des Kollagen Typ II auf das 0,4-fache. Somit war durch die Erhöhung der Zelldichte
eine Verschiebung des von BARLIČ et al. (2008) als Index für die Dedifferenzierung in-vitro-kultivierter
Chondrozyten etablierte Verhältnis von Kollagen Typ I zu Typ II in Richtung des Kollagen Typ I nachweisbar.
Der von SCHULZE-TANZIL (2009) beschriebene positive Effekt einer erhöhten Zelldichte auf die
Konzentration des Zytokins TGF-1 und damit auf den Dedifferenzierungs-Index der Chondrozyten konnte
in dieser Arbeit nicht nachgewiesen werden. Daher kann davon ausgegangen werden, dass die These einer
geringeren Dedifferenzierung der Chondrozyten bei erhöhter Aussaatdichte keine Allgemeingültigkeit für
Zellkultursysteme der Chondrozyten, insbesondere für solche unter erhöhten Druckbedingungen, besitzt
oder der beschriebene Effekt einer höheren Aussaatdichte nach oben limitiert ist (GOLDRING 2012,
SCHULZE-TANZIL 2009). Vielmehr ist die Anpassung der Chondrozyten an die In-vitro-Kultivierung und die
anschließende Proliferation der Zellen wesentlich für das Erreichen des Chondroprogenitorstadiums der
Zellen (GSTRAUNTHALER und LINDL 2013, TEW et al. 2008). Die Entwicklung bis zum Erreichen dieses
Stadiums scheint, entsprechend den Untersuchungen dieser Arbeit, somit hauptsächlich zeitabhängig und
konnte weder durch das Einwirken eines Drucks noch durch die mit einer höheren Dichte ausgesäten Zellen
verbundene geringere Notwendigkeit der Proliferation bis zum Erreichen eines konfluenten Monolayers
wesentlich beeinflusst werden. Die durchgeführten Untersuchungen deuten darauf hin, dass das Erreichen
des Chondroprogenitorstadiums und damit die Synthese einer für den hyalinen Knorpel spezifischen ECM in
vitro nicht allein von dem Kontakt der Zellen untereinander, sondern wie auch im Rahmen der Entwicklung
des Knorpelgewebes von einem bestimmten Maß an Proliferation der Chondrozyten abhängig ist.
5.3 Empfehlungen für die In-vitro-Kultivierung von Chondrozyten unter erhöhtem
hydrostatischen Druck
Die Untersuchungen zeigten, dass für die In-vitro-Kultivierung von Chondrozyten unter erhöhtem
hydrostatischem Druck Primärkulturen von Chondrozyten aus dem humanen Gelenkknorpel mit dem
dazugehörigen Chondrozytenwachstumsmedium geeignet sind, wobei die Aussaat der Chondrozyten in
einer Zelldichte von 104 Zellen/ cm2 erfolgen sollte.
Bei der Nutzung des in dieser Arbeit etablierten Zellkultursystems zeigte sich ein Mediumwechsel alle 3-4
Tage als ausreichend, um die Mortalität der Chondrozyten durch Nährstoffmangel oder die Anreicherung
von Stoffwechselendprodukten zu minimieren und gleichzeitig die Entfernung der in den Extrazellulärraum
abgegebenen ECM möglichst gering zu halten.
Die Verwendung von Chondrozyten bis zur Passage 6 kann für die Analyse der Expression des Kollagen Typ I
und II sowie des Aggrekan empfohlen werden. Die Chondrozyten sollten nach dem 1. Auftauen zunächst
einmalig passagiert werden, da hierdurch eine Beeinflussung durch das zytotoxisch wirkende

5 Diskussion
93
Kryoprotektivum DMSO minimiert wird und somit eine auf DMSO begründete Ergebnisheterogenität
ausgeschlossen werden kann (ALMANSOORI et al. 2012).
Die Kultivierung von Chondrozyten unter erhöhtem hydrostatischem Druck, repräsentativ für die
Druckverhältnisse der Radiärzone, sollte mittels eines Bioreaktors erfolgen, der sich aufgrund seiner
Materialeigenschaften für die Zellkultur eignet. Hierbei ist insbesondere zu beachten, dass alle Teile aus
hitzesterilisierbarem Edelstahl bestehen, welche mit der Zellkultur in Berührung kommen (SCHULZ und
BADER 2007). In dieser Arbeit erwies sich ein feinregulierbares Ventil für den Aufbau und den Abbau des
hydrostatischen Drucks als vorteilhaft, welches den Druckabbau über einen Zeitraum von 20 Minuten
ermöglicht und somit den Monolayer schont.
Die Untersuchung der Effekte eines kontinuierlichen hydroststischen Drucks sollten an konfluenten
Chondrozyten (6.-7. Kulturtag) erfolgen. Zu einem früheren Zeitpunkt sind nutzbare Effekte des Drucks
unwahrscheinlich, da die Expression einer für den hyalinen Knorpel spezifischen ECM zugunsten der
Proliferation der Chondrozyten in den Hintergrund tritt. Es zeigte sich, dass vor Erreichen der Konfluenz der
Zeitpunkt des Einsetzens der Synthese nicht durch einen einwirkenden Druck modifizierbar ist.
Die zytotoxischen Effekte des Drucks sind bereits früh durch die gezielte morphologische Beurteilung der
Chondrozyten im Phasenkontrastmikroskop erkennbar. Die abgerundete Morphologie der Chondrozyten
weist bereits vor der Ablösung der Zellen von der Kulturschale auf die initiierte Nekrose hin.
Es bedarf einer gezielten Einstellung der mit Hilfe des Bioreaktors variablen Parameter (Höhe und Dauer
des auf die Chondrozyten einwirkenden Drucks), um Effekte auf das Expressionsmuster zu erzielen, welche
sich der Synthese einer für den hyalinen Knorpel spezifischen ECM annähern. Hierbei ist zu berücksichtigen,
dass die Anwendung eines hohen Drucks, wie er physiologischer Weise im Gelenk während der normalen
Bewegung erreicht wird (10 bar), gegenüber einem unphysiologisch niedrigem Druck (5 bar) zu bevorzugen
ist. Die durchgeführten Untersuchungen zeigten, dass eine Druckeinwirkung von >24 h zu morphologischen
Veränderungen führt, die auf eine Zytotoxizität hindeuten. Diese kann auf eine druckabhängige Inhibition
der Protonenpumpen zurückgeführt werden (HALL 1999). Diese Inhibition der Ionenpumpen wird als
reversibel beschrieben (HALL 1999), jedoch scheinen sich die Chondrozyten nach einer bestimmten Zeit von
dem zurückgebliebenen Zellschaden nicht mehr zu erholen. Daher sollten für die Untersuchung der in
vitro-kultivierten Chondroyzten unter erhöhtem Druck die Vorteile des kurzzeitig einwirkenden erhöhten
Drucks bei 1,5-3 h insbesondere in Bezug auf die Expression des Aggrekan genutzt werden.
Die quantitative Analyse des Transkriptoms unter Nutzung o.g. Primer und die semiquantitative Analyse des
Proteoms unter Verwendung der o.g. Antikörper ist zur Auswertung der Proben hinsichtlich der Expression
von Kollagen Typ I und Kollagen Typ II sowie Aggrekan geeignet.
Um statistisch auswertbare Daten zu erhalten sind, abhängig von den Versuchsbedingungen, 2-9
biologische Replikate ausreichend, um eine P von 80 % zu erreichen und valide Analysen in Hinblick auf die
Signifikanz der erreichten Unterschiede durchführen zu können. Für Untersuchungen bei atmosphärischem
Druck, welche die Kollagenexpression betrachten, ist n=2 ausreichend, für solche die Aggrekan betrachten
dagegen n=9. In Bezug auf Versuche mit erhöhter Druckbelastung sollte für Untersuchungen der Kollagen-
bzw. Aggrekanexpression n=9 gewählt werden. Die Aussagekraft der Ergebnisse profitiert zusätzlich von der
Wiederholung der Untersuchungen an einer weiteren Primärkultur.

5 Diskussion
94
5.4 Möglichkeiten der Analyse druckbehandelter Chondrozyten und deren Nutzung in der
klinischen Anwendung- ein Ausblick
Die vorliegenden epidemiologischen Daten für die mit Knorpeldefekten einhergehenden Erkrankungen des
Bewegungssystems bei Menschen und Tieren unterstreichen die Notwendigkeit einer adäquaten
Therapieoption (DYCUS et al. 2013, JEFFCOTT 1996, MERX et al. 2007). Bislang konnte sich kein Verfahren
aufgrund seines Therapieerfolgs durchsetzen, sodass die Therapie von Knorpeldefekten allein auf die
Verbesserung der Schmerzsymptomatik und die Erhaltung der physiologischen Bewegungsfreiheit
beschränkt bleibt (FRITSCH et al. 1999, MARTINEK und IMHOFF 2003, SCHULZ und BADER, 2007). Das
vielversprechendste Verfahren stellt das Zellkultur-gestützte Verfahren der ACT/MACT dar (BRITTBERG et
al. 1994, ERGGELET 2003). Dieses kommerziell angebotene Verfahren wird stetig weiterentwickelt.
Gegenwärtig konzentrieren sich diese Weiterentwicklungen hauptsächlich auf die Trägermaterialien
transplantierter Chondrozyten (BENTLEY et al. 2013, ZEIFANG et al. 2010).
Daneben ist die Nutzung der Erkenntnisse der Untersuchung in-vitro-kultivierter Chondrozyten unter
erhöhten Druckbedingungen denkbar. Da die im Rahmen des Verfahrens transplantierten Chondrozyten in
der Lage sind, den Defekt anfänglich aufzufüllen, das neu gebildete Knorpelgewebe jedoch keine Integrität
zur Umgebung und somit keine gute Langzeitstabilität erreicht, sind grundlegende Untersuchungen des
Verhaltens der Chondrozyten unter Druck erforderlich (BRITTBERG 2010, ZEIFANG et al. 2010). Das im
Rahmen dieser Arbeit entwickelte Zellkultursystem ermöglicht die Durchführung solcher Untersuchungen
in vitro unter hydrostatischem Druck. Darauf aufbauenden Studien bietet es ein System, um die
Zusammensetzung der durch in-vitro-kultivierte Chondrozyten synthetisierten ECM zu optimieren und die
für diese verantwortlichen zellulären Mechanismen zu untersuchen. Hierbei sollte ein Fokus auf der
Untersuchung zellulärer Signal-Transduktionsmechanismen liegen, um zu prüfen, ob sich die Konzentration
der Zytokine in vivo wie in vitro tatsächlich so verhalten, wie häufig aus anderen Geweben abgeleitet
wird (FORTIER et al. 2011, SCHULZE-TANZIL 2009).
Daneben wäre basierend auf den Ergebnissen von CARON et al. (2012) die Nutzung einer 3D-Kultur der
Chondrozyten denkbar, in welcher die stetige Entfernung neusynthetisierter ECM unterbleibt. Hierdurch
wird ein Auseinanderrücken der Chondrozyten möglich und die Anzahl der Zell-Zell-Kontakte in vitro
reduziert, sodass es unter dem Einfluss der Zytokine IGF-1 und FGF-2 zu einer stärkeren Expression von
Kollagen Typ II und Aggrekan kommen kann (FORTIER et al. 2011). Die Kombination der 3D-Kultur mit den
in dieser Arbeit etablierten Druckbedingungen birgt somit die Möglichkeit die Synthese in-vitro-kultivierter
Chondrozyten in Richtung einer chondrozytenspezifischen ECM zu lenken und somit Kulturbedingungen zu
schaffen, welche der Situation in vivo näher kommen.
Hieraus resultierend müssen zukünftig die entsprechenden Zytokine nicht dem Zellkulturmedium zu
transplantierender Chondrozyten zugesetzt werden, sondern könnten in spezifischer Konzentration in das
Trägermaterial eingebracht werden. So könnten zelluläre Mechanismen auch nach der Transplantation
hinsichtlich einer biodynamischen Stabilität des Transplantats beeinflusst werden. Daneben ist auch die
Kultivierung der zu transplantierenden Zellen selbst unter optimierten Druckbedingungen mittels eines
Bioreaktors denkbar. Einschränkend bleibt jedoch der Umstand, dass ein In-vitro-System die Komplexität
der Situation der Chondrozyten in vivo nur unvollständig widerzuspiegeln vermag und z.B. die Wirkung von
Scherkräften in diesem Zellkultursystem unberücksichtigt bleibt.

6 Zusammenfassung
95
6 Zusammenfassung
Juliane Schneevoigt
„Untersuchung der zeit- und druckabhängigen Expression verschiedener Komponenten der extrazellulären
Matrix durch Chondrozyten in vitro“
Veterinär-Anatomisches Institut der Veterinärmedizinischen Fakultät der Universität Leipzig
Eingereicht im Juni 2015
98 Seiten, 34 Abbildungen, 41 Tabellen, 153 Literaturangaben
Schlüsselwörter: Knorpel, Chondrozyten, hydrostatischer Druck, Bioreaktor, qPCR
Einleitung
Der hyaline Knorpel gewährleistet die grundlegende Funktion der Druckübertragung innerhalb der
synovialen Gelenke und stellt somit die Grundlage für die Bewegung des Organismus dar. Als schmaler
Überzug der Gelenkflächen ist er in seinem Aufbau an diese Funktion spezifisch angepasst. Dabei bedingt
der hohe Gehalt an Proteoglykanen und das an diese assoziierte Wasser die elastische Verformbarkeit des
hyalinen Knorpels, die es ermöglicht, Druck- und Scherkräfte abzufedern. Die Proteoglykane, die
hauptsächlich auf Aggrekan als Kernprotein basieren, sind in ein Maschenwerk kollagener Fasern
eingelagert, welches im Wesentlichen durch Kollagen Typ II gebildet wird. Diese Zusammensetzung darf
nicht als statisches Konstrukt verstanden werden. Vielmehr ist der hyaline Knorpel in vivo verschiedenen
biophysikalischen Einflüssen ausgesetzt, die eine dynamische Anpassung erfordern. Solche
Anpassungsvorgänge in Form einer Änderung der Zusammensetzung der aus kollagenen Fasern und
Proteoglykanen bestehenden extrazellulären Matrix werden durch die wenigen eingelagerten
Chondrozyten, die Zellen des hyalinen Knorpels, organisiert. Da die reifen Chondrozyten jedoch keine
Zellteilungen aufweisen und dem hyalinen Knorpel eine Vaskularisierung fehlt, ist eine Defektregeneration
kaum möglich, sodass eine Wiederherstellung der Integrität des Gewebes unterbleibt und stattdessen ein
Ersatzknorpel, der Faserknorpel, gebildet wird, welcher den einwirkenden biodynamischen Belastungen
jedoch nicht standhalten kann. Da die Defekte des hyalinen Knorpels einen Großteil der Erkrankungen des
Bewegungsapparats darstellen und zudem die Arthrose als Langzeitfolge nach sich ziehen, besteht ein
hohes medizinisches Interesse an der Entwicklung zellbasierter Therapieansätze, wie der Autologen
Chondrozytentransplantation (ACT). Hierbei werden - bislang mit unterschiedlichen Erfolgen –
in-vitro-kultivierte Chondrozyten mit dem Ziel, neuen hyalinen Knorpel zu bilden, in einen Knorpeldefekt
eingebracht.
Ziele der Untersuchungen
In den letzten Jahrzehnten zeigte sich, dass die Entwicklung von Therapieansätzen zur Behandlung von
Knorpeldefekten ein detaillierteres Verständnis des Knorpelgewebes und speziell dessen Biodynamik
erfordert. Ziel dieser Arbeit war es daher, im Rahmen einer Pilotstudie, ein In-vitro-System zu etablieren,
welches die Untersuchung der Biodynamik der Chondrozyten ermöglicht. Neben der Untersuchung der
Morphologie der Chondrozyten und der durch sie synthetisierten extrazellulären Matrix in Abhängigkeit
von der Kultivierungszeit der Zellen, wurde die Fragestellung bearbeitet, ob durch die Wirkung eines
hydrostatischen Drucks günstige Effekte in Hinblick auf die Expression einer extrazellulären Matrix, wie sie
im hyalinen Knorpel vorliegt, erzielt werden kann.

6 Zusammenfassung
96
Materialien und Methoden
Eine Primärkultur humaner artikulärer Chondrozyten wurde zunächst unter Standardzellkulturbedingungen
und atmosphärischem Druck kultiviert. Die Zellen wurden phasenkontrastmikroskopisch und hinsichtlich
der Verteilung von Kollagen Typ I und II immunzytochemisch untersucht. In den weiteren Versuchen
wurden optisch konfluente Chondrozyten in einen Bioreaktor überführt und weiter unter einem
hydrostatischen Druck von 5 oder 10 bar kultiviert. Dabei wurde die Dauer der Druckeinwirkung auf die
Chondrozyten variiert. Das Expressionsmuster der so kultivierten Chondrozyten wurde quantitativ in
Hinblick auf Kollagen Typ I und II sowie Aggrekan mittels qPCR und Western Blot untersucht. Dabei dienten
jeweils Chondrozyten, die ohne erhöhte Druckbedingungen kultiviert wurden, als Kontrollen. In dieser
Pilotstudie wurden die Proben unter Berechnung der Mittelwerte und Standardabweichung hinsichtlich
ihrer statistischen Power ausgewertet. Neben dieser Analyse der Einzelergebnisse wurden die
Versuchsbedingungen, die kaum Unterschiede in den Ergebnissen aufwiesen, in Gruppen zusammengefasst
und mit Hilfe der größtmöglichen vorhandenen Standardabweichung der Stichprobenumfang eines
Versuchs errechnet, welcher die statistische Power der Ergebnisse bei einem Alpha-Fehler von 0,05 auf 80%
erhöht. In den Fällen, in denen diese Power erreicht wurde, erfolgte eine Untersuchung der Unterschiede
auf Signifikanz („One Way Analysis of Variance“) bei einem Signifikanzniveau < 0,05.
Ergebnisse
Während der In-vitro-Kultivierung der Chondrozyten unter atmosphärischem Druck zeigte die Länge der
Kultivierungszeit weder einen Einfluss auf die phasenkontrastmikroskopisch untersuchte Morphologie der
Zellen noch auf die immunzytochemisch detektierte Verteilung des Kollagen Typ I und II. Die Wirkung eines
erhöhten hydrostatischen Drucks (5 bar, 10 bar) für 24 Stunden führte zu einer Abnahme der Expression
von Kollagen Typ I und Typ II auf das 0,2-0,8-fache bei gleichzeitiger Zunahme der Expression des Aggrekan
auf das 1,7-2,2-fache, verglichen mit der unbehandelten Kontrolle. Dieser Effekt war bei 5 bar ausgeprägter
als bei 10 bar, führte jedoch gleichzeitig zu einer starken Instabilität des Zellkultursystems. Vor diesem
Hintergrund wurde für den höheren Druck (10 bar) die Zeitdauer der Druckeinwirkung verkürzt. Hierbei
konnten bei kurzzeitiger Druckeinwirkung von 10 bar (1,5 und 3 Stunden) bei Erhalt der Zellen ähnliche
Effekte erzielt werden wie für die Bedingung 5 bar, 24 Stunden. Die Expression von Kollagen Typ I und Typ II
sank auf das 0,8-fache, wohingegen ein Anstieg der Aggrekanexpression auf das 1,6-2,4-fache erreicht
wurde. Diese Ergebnisse der qPCR konnten durch die im Western Blot für Kollagen Typ I, II und Aggrekan
detektierte Proteinexpression gestützt werden.
Schlussfolgerungen
Im Rahmen dieser Arbeit wurde ein In-vitro-System etabliert, welches einerseits der Untersuchung des
Einflusses von hydrostatischem Druck auf die Expression von Chondrozyten dienen und andererseits für die
Herstellung von Zelltransplantaten weiter modifiziert werden kann. Die Ergebnisse der Untersuchungen
führten zur Definition von Bedingungen für das In-vitro-System, unter denen die Expression der
extrazellulären Matrix durch die Chondrozyten in Richtung der Zusammensetzung im hyalinen Knorpel
stimuliert werden kann. Dies zeigte sich bei einer Aussaat der humanen Chondrozyten in einer
Konzentration von 104 Zellen/cm2 und einer Vorkultivierungszeit von 6 Tagen unter Normaldruck, gefolgt
von der Kultivierung unter hydrostatischem Druck von 10 bar für 1,5 bis 3 Stunden. Mit Hilfe dieser
Pilotstudie wurde somit ein In-vitro-System etabliert, auf dessen Basis Untersuchungen durchgeführt
werden können, die weiterführende Erkenntnisse zur Biodynamik des hyalinen Knorpels liefern und der
zukünftigen Entwicklung zellbasierter Therapieansätze der Knorpeldefekte, wie der ACT, zu Gute kommen.

7 Summary
97
7 Summary
Juliane Schneevoigt
„Investigation of time- and pressure-dependent expression of different components of the extracellular
matrix by chondrocytes in vitro”
Institute of Anatomy, Histology and Embryology of the University of Leipzig
Submitted in June 2015
98 pages, 34 figures, 41 tables, 153 references
keywords: cartilage, chondrocytes, hydrostatic pressure, bioreactor, qPCR
Introduction
Hyaline cartilage maintains the basic function of transmitting articular pressure load within synovial joints
and therefore provides the basis for the movements of an organism. Being a small coverage of the joint
surface, it shows a composition designed to match this function specifically. A high amount of
proteoglycans and its associated water determines the elastic formability of the hyaline cartilage which
allows the absorbance of pressure and shear forces. These proteoglycans, mainly based on aggrecan as
core-protein, are embedded into a meshwork of collagen fibres, primarily formed of collagen type II. This
composition is not to be understood as a static construct; moreover it is exposed to biophysical forces that
permanently require a dynamic adaptation. This adaptation of the extracellular matrix formed by
proteoglycans and collagen type II is organised by a small number of embedded chondrocytes, the cells of
the hyaline cartilage. As chondrocytes are post-mitotic cells and due to the lack of vascularisation within
hyaline cartilage, there is hardly any chance for regeneration of defects in order to maintain the integrity of
the tissue. The resulting replacement is formed as fibrocartilage, which has not the capability to withstand
the biodynamical forces within the joint. As these defects in hyaline cartilage represent a gross of the
diseases of the musculoskeletal system, there is a high medical interest in the development of innovative
cell-based therapies, as autologous chondrocyte transplantation (ACT) is one. With this type of therapy in
vitro cultivated chondrocytes are seeded into a cartilage defect with the aim of producing hyaline cartilage.
Aims of the study
In the last decades the need for a detailed understanding of the biodynamics of cartilage became obvious
for further development of therapies. The aim of this study was therefore to establish a cell culture system
to provide an insight into the biodynamics of chondrocytes. Aside from the examination of the
differentiation of in vitro cultivated chondrocytes and their synthesis of extracellular matrix as a function of
the cultivation time, another aim of this study was to determine whether the application of hydrostatic
pressure might have beneficial influence on the expression of extracellular matrix components by
chondrocytes in vitro, in accordance with the hyaline cartilage.
Material and methods
Human articular chondrocytes were cultivated in vitro without the application of hydrostatic pressure in
the first place. The cells were observed phase contrast microscopically and the distribution of collagen type
I and II was detected immuncytochemically. In further experiments optical confluent chondrocytes were
transferred to a bioreactor system applying a hydrostatic pressure of 5 or 10 bar with variable time periods
of the pressure applied. Subsequently, the expression of collagen type I, collagen type II and aggrecan was

7 Summary
98
investigated and quantified using qPCR and Western Blot. Chondrocytes cultivated exclusively without the
application of hydrostatic pressure served as controls. In this pilot-study the samples were analysed using
arithmetic mean and standard deviation to evaluate the power statistically. In addition, similar test
conditions with marginal differences were pooled and the necessary sample size to meet a power of 80 %
with an alpha error of 0.05 was calculated using the maximum potential standard deviation. In cases where
this statistic power was obtained, an analysis of significance ("One Way Analysis of Variance”) was carried
out meeting a significance level under 0.05.
Results
During the cultivation of chondrocytes in vitro without hydrostatic pressure the length of the cultivation
time did neither show an effect on the phase contrast microscopical morphology nor on the
immuncytochemically detected distribution of collagen typ I and II. The application of increased hydrostatic
pressure for 24 hours results in a 0.2-0.8-fold decrease of the expression of collagen type I and II and a 1.7-
2.2-fold increase of aggrecan expression compared to the unloaded controls. This effect was more distinct
with 5 bar but was accompanied by instabilities in the cell culture. This is why further investigations
concentrated on the use of 10 bar pressure with subsequently shortened time period of the applied
pressure. With short times of loading (1.5 and 3 hours) a pressure load of 10 bar led to a 0.8 fold decrease
of the expression of collagen type I and II and showed a 1.6-2.4 fold increase of aggrecan expression. These
qPCR results were supported by the protein expression of collagen type I, II and aggrecan detected in
Western Blot.
Conclusions
A cell culture system was established to examine the effect of hydrostatic pressure on the expression of
chondrocytes on the one hand, which can further be modified for the assembly of cell transplants on the
other hand. Subsequently the results of this study led to a definition of cell culture conditions, stimulating
the extracellular matrix production of chondrocytes towards the composition of hyaline cartilage. This was
the case using a seeding density of 104 cells/cm2 and a pre-cultivation time of 6 days of normal pressure,
followed by the application of 10 bar hydrostatic pressure for 1.5-3 h. With the help of this pilot-study a cell
culture system was established to gain more information on biodynamics of hyaline cartilage. Moreover it
is possible that this information will provide a basis for further development of cell based therapies of
cartilage defects, such as ACT.

8 Literaturverzeichnis
99
8 Literaturverzeichnis
Alford JW. Cartilage Restoration, Part 1: Basic Science, Historical Perspective, Patient Evaluation, and
Treatment Options. Am J Sport Med. 2005;33:295–306.
Almansoori K, Prasad V, Forbes J, Law G, McGann L, Elliott J, Jomha, NM. Cryoprotective agent toxicity
interactions in human articular chondrocytes. Cryobiology 2012;64:185–91.
Aimes R, Quigley J. Matrix Metalloproteinase-2 Is an Interstitial Collagenase. J Biol Chem. 1995;270:5872-6.
Aspberg A. The Different Roles of Aggrecan Interaction Domains. J Histochem Cytochem 2012; 60:987-96.
Barlič A, Drobnič M, Maličev E, Kregar-Velikonja N. Quantitative analysis of gene expression in human
articular chondrocytes assigned for autologous implantation. J Orthop Res. 2008;26:847–53.
Benjamin M, Ralphs JR. Fibrocartilage in tendons and ligaments - an adaptation to compressive load. J Anat.
1998;193:481–94.
Bentley G, Bhamra JS, Gikas PD, Skinner JA, Carrington R, Briggs TW. Repair of osteochondral defects in
joints-How to achieve success. Injury. 2013;44:3-10.
Biorad, Schematischer Aufbau der Zusammensetzung der Transferkassette; http://www.bio-rad.com/de-
de/applications-technologies/western-blotting, 01.08.2015, 17:30 Uhr
Boetsch K. Funktionelle Anatomie des Gelenkknorpels [Dissertation med. vet]. München: Ludwig-
Maximilians-Universität München; 2007.
Bonnemann CG, Cox GF, Shapiro F, Wu J, Feener CA, Thompson TG et al. A mutation in the alpha 3 chain of
type IX collagen causes autosomal dominant multiple epiphyseal dysplasia with mild myopathy.
Proc Natl Acad Sci. 2000;97:1212–7.
Bornstein P, Sage H. Structurally distinct collagen types. Ann Res Biochem. 1980;49:957–1003.
Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L. Treatment of Deep Cartilage Defects in
the Knee with Autologous Chondrocyte Transplantation. N Engl J Med. 1994;331:889–95.
Brittberg M. Cell Carriers as the Next Generation of Cell Therapy for Cartilage Repair: A Review of the
Matrix-Induced Autologous Chondrocyte Implantation Procedure. Am J Sport Med.
2010;38:1259–71.
Bruckner P, van der Rest M. Structure and function of cartilage collagens. Microsc Res Tech.
1994;28:378-84.
Bruns J, Steinhagen J. Der Knorpelschaden als präarthrotische Deformität – Biologische Grundlagen.
Z Sportmed. 2000;51:42–7.
Buckwalter J, Mankin H.J, Grodzinsky AJ. Articular Cartilage and Osteoarthritis. Instr Course Lect.
2005;54:465–80.
Buckwalter J, Mankin H. Articular cartilage repair and transplantation. Arthritis Rheum. 1998;41:1331–42.
Burgeson RE, Hollister DW. Collagen heterogeneity in human cartilage: Identification of several new
collagen chains. Biochem. Biophys Res Commun. 1979;87:1124–31.

8 Literaturverzeichnis
100
Buschmann MD, Gluzband YA, Grodzinsky AJ, Hunziker E. Mechanical compression modulates matrix
biosynthesis in chondrocyte/agarose culture. J Cell Sci 1995;108:1497–508.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M et al. The MIQE Guidelines: Minimum
Information for Publication of Quantitative Real-Time PCR Experiments. Clin Chem.
2009;55:611-22.
Bryant PA, Smyth GK, Robins-Browne R, Curtis N. Technical variability is greater than biological variability in
a microarray experiment but both are outweighed by changes induced by stimulation. PLoS ONE
2011;6:e19556.
Caron MMJ, Emans PJ, Coolsen MME, Voss L, Surtel DAM, Cremers A et al. Redifferentiation of
dedifferentiated human articular chondrocytes: comparison of 2D and 3D cultures. Osteoarthr
Cartil. 2012;20:1170–8.
Carver SE, Heath CA. Increasing extracellular matrix production in regenerating cartilage with intermittent
physiological pressure. Biotechnol Bioeng. 1999a;62:166–74.
Carver SE, Heath CA. Semi-Continuous Perfusion System for Delivering Intermittent Physiological Pressure
to Regenerating Cartilage. Tissue Eng. 1999b;5:1–11.
Changoor A, Nelea M, Méthot S, Tran-Khanh N, Chevrier A, Restrepo A. Structural characteristics of the
collagen network in human normal, degraded and repair articular cartilages observed in polarized
light and scanning electron microscopies. Osteoarth Cartilage. 2011;19:1458–68.
Cheung HS. Distribution of Type I, II, III and V in the Pepsin Solubilized Collagens in Bovine Menisci. Connect
Tissue Res. 1987;16:343–56.
Chey S, Claus C, Liebert UG. Improved method for simultaneous isolation of proteins and nucleic acids. Anal
Biochem. 2011;411:164–6.
Ciorba A, Martini A. Tissue engineering and cartilage regeneration for auricular reconstruction. Int J Pediatr
Otorhinolaryngol. 2006;70:1507–15.
Cole A. A review of diversity in the evolution and development of cartilage: The search for the origin of the
chondrocyte. Eur Cells Mat. 2011;21:122–9.
Cosma M, Cardone M, Carlomagno F, Colantuoni V. Mutations in the Extracellular Domain Cause RET Loss
of Function by a Dominant Negative Mechanism. Mol Cell Biol. 1998;18:3321–9.
Couchman JR, Pataki CA. An Introduction to Proteoglycans and Their Localization. J Histochem Cytochem.
2012;60:885–97.
Danišovič Ľ, Varga I, Polák Š. Growth factors and chondrogenic differentiation of mesenchymal stem cells.
Tissue Cell. 2012;44:69–73.
Das R, van Osch G, Kreukniet M, Oostra J, Weinans H, Jahr H. Effects of individual control of pH and hypoxia
in chondrocyte culture. J Orthop Res. 2010;28:537-45.
DeGroot J, Verzijl N, Bank RA, Lafeber FPJG, Bijlsma JWJ, TeKoppele JM. Age-related decrease in
proteoglycan synthesis of human articular chondrocytes: The role of nonenzymatic glycation.
Arthritis Rheum. 1999;42:1003–9.

8 Literaturverzeichnis
101
DeLise AM, Fischer L, Tuan RS. Cellular interactions and signaling in cartilage development. Osteoarth
Cartilage. 2000;8:309–34.
Desjardins P, Conklin D. NanoDrop microvolume quantitation of nucleic acids. J Vis Exp. 2010;45:1–5.
Detamore MS, Hegde JN, Wagle RR, Almarza AJ, Montufar-Solis D, Duke PJ et al. Cell Type and Distribution
in the Porcine Temporomandibular Joint Disc. J Oral Maxillofac Surg. 2006;64:243–8.
Dodge G, Poole A. Immunohistochemical Detection and Immunochemical Analysis of Type II Collagen
Degradation in Human Normal, Rheumatoid, and Osteoarthritic Articular Cartilages and in
Explants of Bovine Articular Cartilage Cultured with Interleukin 1. J Clin Invest. 1989;83:647–61.
Domm C, Schünke M, Christesen K, Kurz B. Redifferentiation of dedifferentiated bovine articular
chondrocytes in alginate culture under low oxygen tension. Osteoarthr. Cartil. 2002;10:13–22.
Donkelaar CC, Huiskes R. The PTHrP–Ihh Feedback Loop in the Embryonic Growth Plate Allows PTHrP to
Control Hypertrophy and Ihh to Regulate Proliferation. Biomech Model Mechanobiol. 2007;6:55–
62.
Dowthwaite GPBJC, Redman SN, Khan IM, Rooney P, Evans DJR, Haughton L et al. The surface of articular
cartilage contains a progenitor cell population. J Cell Sci. 2004;117:889–97.
Dycus DL, Au AY, Grzanna MW, Wardlaw JL, Frondoza CG. Modulation of inflammation and oxidative stress
in canine chondrocytes. Am J Vet Res. 2013;74:983–9.
Eckstein F, Reiser M, Englmeier K, Putz R. In vivo morphometry and functional analysis of human articular
cartilage with quantitative magnetic resonance imaging - from image to data, from data to theory.
Anat Embryol. 2001;203:147–73.
Ekenstedt KJ, Sonntag WE, Loeser RF, Lindgren BR, Carlson CS. Effects of chronic growth hormone and
insulin-like growth factor 1 deficiency on osteoarthritis severity in rat knee joints. Arthritis Rheum.
2006;54:3850-8.
Elder BD, Athanasiou KA. Hydrostatic Pressure in Articular Cartilage Tissue Engineering: From Chondrocytes
to Tissue Regeneration. Tissue Eng Pt B-Rev. 2009;15:43–53.
Elder BD, Athanasiou KA, Egles C. Synergistic and Additive Effects of Hydrostatic Pressure and Growth
Factors on Tissue Formation. PLOS ONE 2008;3:2341.
Erggelet C. Die autologe Chondrozytentransplantation zur Behandlung von Knorpeldefekten des
Kniegelenkes. In: Jerosch J, Heisel J, Imhoff A. Fortbildung Orthopädie, Traumatologie: Die ASG-
Kurse der DGOOC. Darmstadt: Steinkopff; 2003. p. 42–7.
Eyre DR. Structural Analysis of Cross-linking Domains in Cartilage Type XI Collagen. J Biol Chem.
1995;270:18865–70.
Eyre DR. Collagen: Molecular Diversity in the Body's Protein Scaffold. Science 1980; 207:1315–22.
Eyre DR. Collagen of articular cartilage. Arthritis Res. 2002;4:30–5.
Eyre DR. Covalent Cross-linking of the NC1 Domain of Collagen Type IX to Collagen Type II in Cartilage. J Biol
Chem. 2003;279:2568–74.

8 Literaturverzeichnis
102
Ferguson RE, Carroll HP, Harris A, Maher ER, Selby PJ, Banks RE. Housekeeping proteins: a preliminary study
illustrating some limitations as useful references in protein expression studies. Proteomics
2005;5:566–71.
Fernandes RJ, Weis M, Scott MA, Seegmiller RE, Eyre DR. Collagen XI chain misassembly in cartilage of the
chondrodysplasia (cho) mouse. Matrix Biol. 2007;26:597-603.
Fessler J, Fessler L. Biosynthesis of procollagen. Ann Res Biochem. 1978;47:129–52.
Fleige S, Walf V, Huch S, Prgomet C, Sehm J, Pfaffl MW. Comparison of relative mRNA quantification models
and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol Lett 2006;28:1601–
13.
Fortier LA, Barker JU, Strauss EJ, McCarrel TM, Cole BJ. The Role of Growth Factors in Cartilage Repair. Clin
Orthop Relat Res. 2011;469:2706-15.
Fritsch K, Josimovi, Alasevi, O. Chondroneogenese durch autologe Chondrozytentransplantation (ACT).
Arthroskopie 1999;12:43–9.
Fujioka R, Aoyama T, Takakuwa T. The layered structure of the articular surface. Osteoarth Cartilage.
2013;21:1092–8.
Fukui N. Processing of Type II Procollagen Amino Propeptide by Matrix Metalloproteinases. J Biol Chem.
2001;277:2193–201.
Garcia M, Knight MM. Cyclic loading opens hemichannels to release ATP as part of a chondrocyte
mechanotransduction pathway. J Orthop Res. 2010;28:510-5.
Gelse K, Pöschl E, Aigner T. Collagens—structure, function, and biosynthesis. Adv. Drug Delivery Rev.
2003;55:1531-46.
Gstraunthaler G, Lindl T. Zell- und Gewebekultur: Allgemeine Grundlagen und spezielle Anwendungen. 7th
ed. Berlin: Springer Spektrum; 2013.
Goldberg B, Epstein EH, Sherr CJ. Precursors of Collagen Secreted by Cultured Human Fibroblasts. Proc Nat
Acad Sci. 1972;69:3655–9.
Goldring MB. Chondrogenesis, chondrocyte differentiation and articular cartilage metabolism in health and
osteoarthritis. Ther Adv Musculoskelet Dis. 2012;4:269-85
Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem. 2006;97:33–44.
Goyman V. Abrasionsarthroplastik. Orthopäde. 1999;28:11–8.
Guilak F, Mow VC. The mechanical environment of the chondrocyte: a biphasic finite element model of
cell–matrix interactions in articular cartilage. J Biomech. 2000;33:1663–73.
Gwynn A, Wade S, Kaab M, Owen GRH, Richards R. Freeze-substitution of rabbit tibial articular cartilage
reveals that radial zone collagen fibres are tubules. J Microsc. 2000;197:159–72.
Haberhauer G, Dunky A, Feyertag J. Knorpel-Stoffwechsel: Cartilage Oligomeric Matrix Protein (COMP) als
"Prognostikum" der Gelenksknorpel-Destruktion. Journal für Mineralstoffwechsel 2003; 10:17–21.
Hall A. Differential Effects of Hydrostatic Pressure on Cation Transport Pathways of Isolated Articular
Chondrocytes. J Cell Physiol. 1999;178:197–204.

8 Literaturverzeichnis
103
Handley CJ, Tuck Mok M, Ilic MZ, Adcocks C, Buttle DJ, Robinson H. Cathepsin D cleaves aggrecan at unique
sites within the interglobular domain and chondroitin sulfate attachment regions that are also
cleaved when cartilage is maintained at acid pH. Matrix Biol. 2001;20:543–53.
Hansen U, Schünke M, Domm C, Ioannidis N, Hassenpflug J, Gehrke T. Combination of reduced oxygen
tension and intermittent hydrostatic pressure: a useful tool in articular cartilage tissue
engineering. J Biomech. 2001;34:941–9.
Hardmeier R, Redl H, Marlovits S. Effects of mechanical loading on collagen propeptides processing in
cartilage repair. J Tissue Eng Regen Med. 2009:1-11.
He B, Wu J, Chim S, Xu J, Kirk T. Microstructural analysis of collagen and elastin fibres in the kangaroo
articular cartilage reveals a structural divergence depending on its local mechanical environment.
Osteoarthr Cartil. 2013;21:237–45.
Herberhold C, Faber S, Stammberger T, Steinlechner M, Putz R, Englmeier K et al. In situ measurement of
articular cartilage deformation in intact femoropatellar joints under static loading. J Biomech.
1999;32:1287–95.
Hollander AP, Dickinson SC, Kafienah W. Stem Cells and Cartilage Development: Complexities of a Simple
Tissue. Stem Cells. 2010;28:1992–6.
Horas U, Schnettler R, Pelinkovic D, Herr G, Aigner T. Knorpelknochentransplantation versus autogene
Chondrozytentransplantation. Chirurg. 2000;71:1090–7.
Hughes LC, Archer CW, Gwynn IA. The ultrastructure of mouse articular cartilage: Collagen Orientation and
Implications for Tissue functionality. A polarized light and scanning electron microscope study and
review. Eur Cells Mater. 2005;9:68–84.
Hummert C. Analyse und Interpretation der Varianz von Genexpressionsdaten [Dissertation rer. nat.]. Jena:
Friedrich-Schiller-Universität Jena; 2014.
Hunter W. Of the structure and disease of articular cartilages. 1743 Philos Trans R Soc London Biol.
1743;42:514–52.
Huth A. Studien zum mRNA profiling an humanem postmortalem Gewebe [Dissertation rer.nat.]. Würzburg:
Julius-Maximilians-Universität Würzburg; 2012.
Ikenoue T, Trindade MCD, Lee MS, Lin EY, Schurman DJ, Goodman SB et al. Mechanoregulation of human
articular chondrocyte aggrecan and type II collagen expression by intermittent hydrostatic
pressure in vitro. J Orthop Res. 2003;21:110–6.
Jakobsen RB, Engebretsen L, Stauterbeck J. An Analysis of the Quality of Cartilage Repair Studies. J Bone
Joint Surg Am 2005;87:2232-9.
Jeffcott LB. Osteochondrosis - An international problem for the horse industry. J Equine Vet Sci.
1996;16:32–7.
Johnson K, Terkeltaub R. Inorganic Pyrophosphat (PPi) in pathologic Calcification of Articular Cartilage.
Front Biosci. 2005;10:988–97.

8 Literaturverzeichnis
104
Jortikka MO, Parkkinen JJ, Inkinen RI, Kärner J, Järveläinen HT, Nelimarkka LO et al. The Role of
Microtubules in the Regulation of Proteoglycan Synthesis in Chondrocytes under Hydrostatic
Pressure. Arch Biochem Biophys. 2000;374:172–80.
Katta J, Jin Z, Ingham E, Fisher J. Biotribology of articular cartilage-A review of the recent advances. Med
Eng Phys. 2008;30:1349–63.
Kiani C, Chen L, Wu YJ, Yee AJ, Yang BB. Structure and function of aggrecan. Cell Res 2002;12:19–32.
Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci. 2002;115:2817–28.
Kneißel, N. Untersuchung der Chondrogenese humaner mesenchymaler Stammzellen des Knochenmarks in
vitro durch Einbettung in Agarose [Dissertation med.]. Münster: Universität Münster; 2003.
Knight MM, McGlashan SR, Garcia M, Jensen CG, Poole CA. Articular chondrocytes express connexin 43
hemichannels and P2 receptors - a putative mechanoreceptor complex involving the primary
cilium? J Anat. 2009;214:275–83.
Knudson C, Knudson W. Cartilage proteoglycans. Semin Cell Dev Biol. 2001;12:69–78.
Koivula M, Risteli L, Risteli J. Measurement of aminoterminal propeptide of type I procollagen (PINP) in
serum. Clin Biochem. 2012;45:920–7.
Krasnici S, Labza S, Ertel W. Synthetischer Knochen- und Knorpelersatz. In: Jerosch J, Heisel J, Imhoff A.
Fortbildung Orthopädie: Die ASG-Kurse der DGOT. Band 3. Knie. Darmstadt: Steinkopff; 2000. p.
278–83.
Lee V, Chen L, Paiwand F, Cao L, Wu YJ, Imman R Adams, ME; Yang, BB. Cleavage of the Carboxyl Tail from
the G3 Domain of Aggrecan but Not Versican and Identification of the Amino Acids Involved in the
Degradation. Journal of Biological Chemistry 2002;277:22279–88.
Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241-8.
Luppa D. Biochemie und Pathobiochemie des hyalinen Gelenkknorpels. Klin Sportmedizin. 2000; 12:29–39.
Mabvuure N, Hindocha S, Jordan D, S. Khan W. Chondrogenesis and Developments in Our Understanding.
Curr Stem Cell Res Ther. 2012;7:243–59.
MacDonald BA, Sund M, Grant MA, Pfaff KL, Holthaus K, Zon LI et al. Zebrafish to humans: evolution of the
3-chain of type IV collagen and emergence of the autoimmune epitopes associated with
Goodpasture syndrome. Blood. 2006;107:1908–15.
Madry H, Pape D. Autologe Chondrozytentransplantation. Orthopäde.2008; 37:756–63.
Mahmood T, Yang P. Western blot: technique, theory, and trouble shooting. N Am J Med Sci 2012;4:429-34.
Majno G, Joris I. Apoptosis, Oncosis, and Necrosis An Overview of Cell Death. Am J Path 1995;146:3–15.
Makris EA, Hadidi P, Athanasiou KA. The knee meniscus: Structure–function, pathophysiology, current
repair techniques, and prospects for regeneration. Biomat. 2011;32:7411–31.
Mallinger R, Stockinger L. Histochemistry of the extracellular matrix of aging hyaline cartilage. Folia
Histochem Cytobiol.1987;25:129–132.

8 Literaturverzeichnis
105
Mansfield JC, Winlove CP, Moger J, Matcher SJ. Collagen fiber arrangement in normal and diseased
cartilage studied by polarization sensitive nonlinear microscopy. J Biomed Opt. 2008;13:44–57.
Marcacci M, Filardo G, Kon E. Treatment of cartilage lesions: What works and why? Injury 2013;44:115.
Mark K von der. Structure, Biosynthesis and Gene Regulation of Collagens in Cartilage and Bone. In: Seibel
MJ, Robins SP, Bilezikian JP. Dynamics of bone and cartilage metabolism. 2nd ed. San Diego:
Academic Press; 2006. p. 3–40.
Marlovits S, Nürnberger S, Kolonja A, Singer P, Zeller P, Mandl I. Matrixgekoppelte autologe
Knorpelzelltransplantation. Trauma Berufskrankh. 2004;6:314–23.
Marlovits S, Vécsei V. Möglichkeiten zur chirurgischen Therapie von Knorpeldefekten – Teil 1: Grundlagen
der Korpelbiologie und der Heilung von Knorpeldefekten. Acta Chir Austriaca 2000;32:124–9.
Martinek V, Imhoff A. Therapie von Knorpelschäden. Deutsche Zeit Sportmed. 2003;54:70–6.
Martin JA, Buckwalter JA. Aging, articular cartilage chondrocyte senescence and osteoarthritis.
Biogerontology. 2002;3:257–64.
Matyas J, Sandel L, Adams M. Gene expression of type II collagens in chondro-osteophytes in experimental
osteoarthritis. Osteoarthr Cartil. 1997;5:99–105.
Matz S. Methodische Evaluierung der immunhistochemischen Detektion aktivierter Caspase-3 im
Hippokampus als Apoptoseparameter nach inkompletter zerebraler Ischämie bei der Ratte
[Dissertation med. vet.]. München: Ludwig-Maximilians-Universität München; 2003.
Meachim G, Roberts C. Repair of the joint surface from subarticular tissue in the rabbit knee the joint
surface tissue in the rabbit knee sub. J Anat. 1971;109:317–27.
Merx H, Dreinhöfer K, Günther K. Sozialmedizinische Bedeutung der Arthrose in Deutschland. Z Orthop
Unfall. 2007;145:421–9.
Miosge N, Hartmann M, Maelicke C, Herken R. Expression of collagen type I and type II in consecutive
stages of human osteoarthritis. Histochem Cell Biol 2004;122:229–36.
Nebelung S, Ladenburger A, Gavenis K, Stoffel M, Andereya S, Muller-Rath R. Tissue engineering of cartilage
replacement material - mechanical stimulation in the in-vitro cultivation of human chondrocytes.
Z Orthop Unfall 2011;149:52–60.
Olsen BR. Collagen IX. Int J Biochem Cell B. 1997;29:555–8.
Oshita H, Sandy J, Suzuki K, Akaike A, Bai Y, Sasaki T et al. Mature bovine articular cartilage contains
abundant aggrecan that is C-terminally truncated at Ala719-Ala720, a site which is readily cleaved
by m-calpain. Biochem J. 2004;382:253–9.
Outerbridge RE. The etiology of chondromalacia patellae. J Bone Joint Surg Am. 1961;43:752–7.
Parsons KK, Maeda N, Yamauchi M, Banes AJ, Koller BH. Ascorbic acid-independent synthesis of collagen in
mice. J Endocrinol Metab. 2006;290:e1131.
Poole CA. Review. Articular cartilage chondrons: form, function and failure. J Anat. 1997;191:1–13.
Roughley PJ, White RJ. Age-related Changes in the Structure of the Proteoglycan Subunits from Human
Articular Cartilage of the Proteoglycan Subunits. J Biol Chem. 1980;255:217–25.

8 Literaturverzeichnis
106
Rousseau J, Vignon E, Sandel L, Gamero P, Delmas P. Serum levels of type IIA procollagen amino terminal
propeptide (PIIANP) are decreased in patients with knee osteoarthritis and rheumatoid arthritis.
Osteoarthr Cartil. 2004;12:440–7.
Salomon F, Geyer H, Gille U. Anatomie für die Tiermedizin. 2nd ed. Stuttgart: Enke; 2008. p. 27.
Sandy J, Verscharen C. Analysis of aggrecan in human knee cartilage and synovial fluid indicates that
aggrecanase (ADAMTS) activity is responsible for the catabolic turnover and loss of whole
aggrecan whereas other protease activity is required for C-terminal processing in vivo. Biochem J.
2001;358:615–26.
Schmitz S. Der Experimentator: Zellkultur. 3rd ed. Heidelberg: Spektrum Akademischer Verlag; 2011.
p. 71-84.
Schulze-Tanzil G. Activation and dedifferentiation of chondrocytes: Implications in cartilage injury and
repair. Anat Anzeiger. 2009;191:325–38.
Schulz RM, Bader A. Cartilage tissue engineering and bioreactor systems for the cultivation and stimulation
of chondrocytes. Eur Biophys J. 2007;36:539–68.
Shapiro F, Koide S., Glimcher MJ. Cell origin and differentiation in the repair of full-thickness defects of
articular cartilage. J Bone Joint Surg Am. 1993;75:532–53.
Siczkowsk M, Watt FM. Subpopulations of chondrocytes from different zones of pig articular cartilage
Isolation, growth and proteoglycan synthesis in culture. J Cell Sci. 1990;97:349–60.
Singh P, Schwarzbauer JE. Fibronectin and stem cell differentiation - lessons from chondrogenesis. J Cell Sci.
2012;125:3703-12.
Smith RL, Rusk SF, Ellison BE, Wessells P, Tsuchiya K, Carter DR et al. In vitro stimulation of articular
chondrocyte mRNA and extracellular matrix synthesis by hydrostatic pressure. J Orthop Res.
1996;14:53-60.
Smith RL, Lin J, Trindade M, Shida J, Kajiyama G, Vu T et al. Time-dependent effects of intermittent
hydrostatic pressure on articular chondrocyte type II collagen and aggrecan mRNA expression. J
Rehabil R D. 2000;37:153–61.
Stärke C. Form und Funktion von Menisken. Arthroskopie 2008;21:223–8.
Sterodimas A, Faria J de, Correa WE, Pitanguy I. Tissue engineering and auricular reconstruction: a review. J
Plast Reconstr Aesthet Surg. 2009; 62:447–52.
Struglics A, Larsson S, Lohmander L. Estimation of the identity of proteolytic aggrecan fragments using
PAGE migration and Western immunoblot. Osteoarthr Cartil. 2006;14:898–905.
Suh J, Baek GH, Arøen A, Malin CM, Niyibizi C, Evans et al. Intermittent sub-ambient interstitial hydrostatic
pressure as a potential mechanical stimulator for chondrocyte metabolism. Osteoarthr Cartil.
1999;7:71–80.
Takahashi K, Kubo T, Kobayashi K, Imanishi J, Tkigawa M, Arai Y et al. Hydrostatic pressur influences mRNA
exprssion of trnsforming growth factor-β1 and heat shock protein 70 in chondrocyte-like cell line.
J Orthop Res. 1997;15:150–8.

8 Literaturverzeichnis
107
Taylor SC, Berkelman T, Yadav G, Hammond M. A defined methodology for reliable quantification of
Western blot data. Mol. Biotechnol. 2013;55:217–26.
Taylor SC, Posch A. The design of a quantitative western blot experiment. Biomed Res Int 2014;2014:1-9.
Tew SR, Murdoch AD, Rauchenberg RP, Hardingham TE. Cellular methods in cartilage research: Primary
human chondrocytes in culture and chondrogenesis in human bone marrow stem cells. Methods
2008;45:2–9.
Toyoda T, Seedhom BB, Kirkham J, Bonass WA. Upregulation of aggrecan and type II collagen mRNA
expression in bovine chondrocytes by the application of hydrostatic pressure. Biorheolgy.
2003;40:79–85.
Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochem Biophys
Acta. 2012;1824:133–45.
Vansuante J, Buma P, Homminga G, Vandenberg W, Veth R. Chondrocyte-seeded hydroxyapatite for repair
of large articular cartilage defects. A pilot study in the goat. Biomaterials. 1998;19:2367–74.
Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: Key enzymes in osteoarthritis. J Cell Biochem.
2011;112:3507-14.
Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defect Res C. 2007;81:229–40.
Wei X, Gao J, Messner K. Maturation-dependent repair of untreated osteochondral defects in the rabbit
knee joint. J Biomed Mater Res. 1997;34:63–72.
Wei X, Messner K. Age- and injury-dependent concentrations of transforming growth factor-β1 and
proteoglycan fragments in rabbit knee joint fluid. Osteoarthr Cartil. 1998;6:10-18
Whitfield J. The solitary (primary) cilium– A mechanosensory toggle switch in bone and cartilage cells. Cell
Signal. 2008;20:1019–24.
Wildt RMT de, Tomlinson IM, Mundy CR, Gorick BD. Nat. Biotechnol. 2000;18:989–94.
Windhager R, Lüring C, Anders S, Bäthis H, Perlick L, Tingart M et al. Gegenwärtige Praxis der Behandlung
des Knorpelschadens am Kniegelenk - Ergebnisse einer deutschlandweiten Umfrage an
unfallchirurgischen und orthopädischen Kliniken. Z Orthop Ihre Grenzgeb. 2004;142:546–52.
Wong M, Carter D. Articular cartilage functional histomorphology and mechanobiology: a research
perspective. Bone. 2003;33:1–13.
Wu J, Larkll M, Chun L, Eyre DR. Sites of Stromelysin Cleavage in Collagen Types and II, XI, X and XIof
Cartilage. J Biol Chem. 1991;266:5625–8.
Yamauchi M, Sricholpech M. Lysine post-translational modifications of collagen. Essays Biochem.
2012;52:113–33.
Zeifang F, Oberle D, Nierhoff C, Richter W, Moradi B, Schmitt H. Autologous Chondrocyte Implantation
Using the Original Periosteum-Cover Technique Versus Matrix-Associated Autologous
Chondrocyte Implantation: A Randomized Clinical Trial. Am J Sports Med. 2010;38:924–33.

11 Danksagung
108
9 Danksagung
Im Rahmen meiner Promotion habe ich von sehr vielen Menschen Unterstützung erfahren. Nun, da mein
Projekt in dieser Dissertation seinen Abschluss findet, ist es an der Zeit Danke zu sagen.
Danke, Mondy Bahramsoltani für die freundliche Überlassung des Themas. Angefangen bei der
Einarbeitung in die Zellkultur, insbesondere in die Arbeit mit dem Bioreaktor, über ihre Einführung in das
wissenschaftliche Arbeiten bis hin zu ihrer konstruktiven Kritik bei der Erstellung des Manuskripts. Ihre
Ideen bezüglich des Studiendesigns legten den Grundstein für diese Arbeit, gaben mir jedoch auch die
nötige Freiheit diese Ideen während des gesamten Projekts weiterzuentwickeln. Vor allem aber ihre
intensive und professionelle Unterstützung bei der Erstellung des Manuskripts haben maßgeblich zum
Gelingen dieser Arbeit beigetragen.
Danke, Johannes Seeger für die finanzielle Unterstützung des Projekts und die Bereitstellung des
Arbeitsplatzes am Veterinär-Anatomischen Institut.
Danke an die Mitarbeiter des Veterinär-Anatomischen Instituts- Gabriele Lindner für ihre kompetente Hilfe
bei der Etablierung der Immunzytochemie. Johannes Kacza danke ich für die umfangreiche Einarbeitung in
die mikroskopische Bilddokumentation. Bei meinen Mit-Doktorandinnen Katharina Lübbe, Maria Schober
und Jule Kristin Michler möchte ich mich für die außerordentlich gute Zusammenarbeit, die freundliche
Aufnahme im molekularbiologischen Labor und die vielen hilfreichen Tipps bedanken.
Danke an die Mitarbeiter des Fraunhofer Instituts für Zelltherapie- Alexandra Stolzing für die freundliche
Aufnahme in ihre Arbeitsgruppe. Mein ganz besonderer Dank gilt Claire Fabian- neben ihrer fachlichen
Unterstützung in allen Bereichen der Etablierung der PCR, vor allem auch für ihre warmherzige Art, die
mich immer wieder motivierte. Christiane Leovsky für die unzähligen konstruktiven gemeinsamen Stunden,
ob im Labor oder bei Kaffee und Schokolade, die mir immer in guter Erinnerung bleiben werden. Vielen
Dank auch für ihre hilfreichen Anmerkungen bei der Gestaltung des Manuskripts.
Danke, Christian Barucker und Josephine Böhme für die umfassende Einarbeitung in die Methoden
SDS-PAGE und Western Blot.
Danke, Franziska Richter für die wertvolle Unterstützung in der statistischen Analyse der Ergebnisse dieser
Arbeit.
Danke für die technische Unterstützung an Jens Baumgardt und Claudia Keil-Dieckmann von der
Fa. Durakult GmbH. Sie haben mit ihrer Konzeption eines für die Zellkultur optimierten Bioreaktors eine
wesentliche Voraussetzung für diese Arbeit geschaffen. Dies gilt auch für Wolfgang Schilbach, der mit der
technischen Konstruktion eines feinjustierbaren Abstromventils als Erweiterung für den Bioreaktor die
Fortsetzung der Zellkulturarbeiten mit dem Bioreaktor ermöglichte.
Danke, Hans-Dieter Gerstner. Die häufig umfangreiche Freistellung in der Startphase des Projekts, sowie die
spontane Übernahme meiner Bereitschaftsdienste haben diese Arbeit ohnehin erst ermöglicht.
Danke meiner Familie- meinen Eltern Birgit und Gerd Schneevoigt, die mir zusammen mit meinen
Großeltern Anita und Heinz Riehmer das Studium der Veterinärmedizin ermöglichten und mir auch
während der Anfertigung der Doktorarbeit immer unterstützend und liebevoll zur Seite standen.
Meinem Mann Lutz Schneevoigt danke ich von ganzem Herzen für seine unermüdliche Unterstützung, seine
Liebe und Motivation.


















