Vorlesung „Einführung in die...
40
Grundlagen der Polymerwissenschaften für Studierende des Studiengangs Nano-Engineering l1 Vorlesung „Einführung in die Polymerwissenschaften“ für Studenten des Studiengangs "Nano-Engineering" Christian Mayer Sommersemester 2016 Vorlesung: Freitag, 13:00 bis 15:00, MD 162 Übungen: Freitag, 15:00 bis 16:00, MD 162 Beginn der Vorlesung: Freitag, den 15. April 2016 Ende der Vorlesung: Freitag, den 22. Juli 2016 Inhalt: 1 Einführung (Polymere, Makromoleküle, Monomereinheiten) 2 Struktur von Makromolekülen 2.1 Konstitution, Konfiguration und Konformation 2.2 Die mittlere Molmasse eines Polymers 3 Herstellung von Polymeren (Polymerisationsreaktionen) 3.1 Radikalische Polymerisation 3.2 Anionische Polymerisation 3.3 Kationische Polymerisation 3.4 Polykondensation und Ringöffnung 4 Makromoleküle in Lösung 4.1 Konformation eines gelösten Makromoleküls 4.2 Charakterisierung gelöster Makromoleküle 5 Makromoleküle in einer Polymerschmelze 5.1 Das Fließverhalten einer Polymerschmelze 5.2 Umformung von flüssigen Polymeren 6 Makromoleküle in festem Polymer 6.1 Amorphe und kristalline Strukturen 6.2 Dynamische Prozesse in festen Polymeren 6.3 Mechanische Eigenschaften von Polymeren 6.4 Thermische Zersetzung von Polymeren 7 Polymere in der Mikro- und Nanotechnologie 7.1 Anwendung in der Lithografie: Resist-Materialien 7.2 Nanostrukturierte Oberflächen 7.3 Spontane Strukturbildungen von amphiphilen Polymeren 7.4 Polymere Nanopartikel 7.5 Technische Anwendungen biologischer Polymere
-
Upload
truongminh -
Category
Documents
-
view
221 -
download
0
Transcript of Vorlesung „Einführung in die...

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l1
Vorlesung „Einführung in die Polymerwissenschaften“
für Studenten des Studiengangs "Nano-Engineering"
Christian Mayer
Sommersemester 2016
Vorlesung: Freitag, 13:00 bis 15:00, MD 162 Übungen: Freitag, 15:00 bis 16:00, MD 162
Beginn der Vorlesung: Freitag, den 15. April 2016 Ende der Vorlesung: Freitag, den 22. Juli 2016
Inhalt:
1 Einführung (Polymere, Makromoleküle, Monomereinheiten)
2 Struktur von Makromolekülen 2.1 Konstitution, Konfiguration und Konformation 2.2 Die mittlere Molmasse eines Polymers
3 Herstellung von Polymeren (Polymerisationsreaktionen) 3.1 Radikalische Polymerisation 3.2 Anionische Polymerisation 3.3 Kationische Polymerisation 3.4 Polykondensation und Ringöffnung
4 Makromoleküle in Lösung 4.1 Konformation eines gelösten Makromoleküls 4.2 Charakterisierung gelöster Makromoleküle
5 Makromoleküle in einer Polymerschmelze 5.1 Das Fließverhalten einer Polymerschmelze 5.2 Umformung von flüssigen Polymeren
6 Makromoleküle in festem Polymer 6.1 Amorphe und kristalline Strukturen 6.2 Dynamische Prozesse in festen Polymeren 6.3 Mechanische Eigenschaften von Polymeren 6.4 Thermische Zersetzung von Polymeren
7 Polymere in der Mikro- und Nanotechnologie 7.1 Anwendung in der Lithografie: Resist-Materialien 7.2 Nanostrukturierte Oberflächen 7.3 Spontane Strukturbildungen von amphiphilen Polymeren 7.4 Polymere Nanopartikel 7.5 Technische Anwendungen biologischer Polymere
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l2
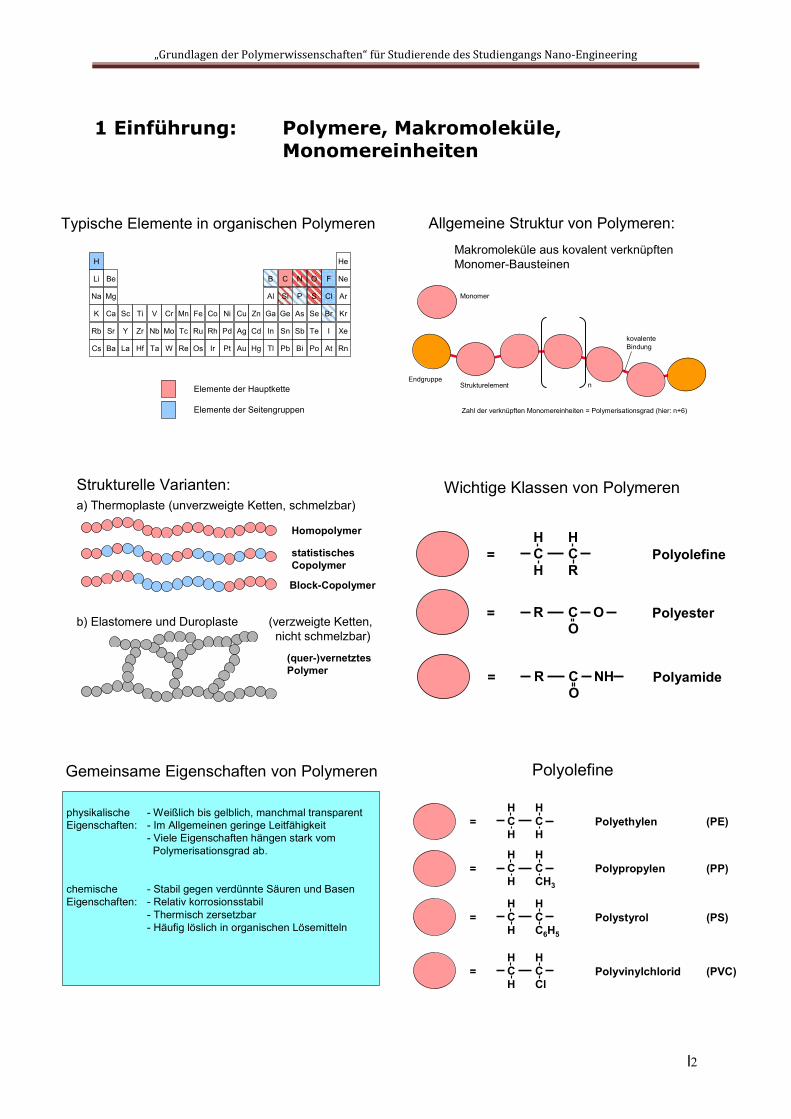
1 Einführung: Polymere, Makromoleküle, Monomereinheiten
Sc TiK Ca Mn FeV Cr Cu ZnCo Ni Br KrGa Ge As Se
Na Mg Cl ArAl Si P S
H
Be F NeB C N OLi
He
Y ZrRb Sr Tc RuNb Mo Ag CdRh Pd I XeIn Sn Sb Te
La HfCs Ba Re OsTa W Au HgIr Pt At RnTl Pb Bi Po
Typische Elemente in organischen Polymeren
Elemente der Hauptkette
Elemente der Seitengruppen
Allgemeine Struktur von Polymeren:Makromoleküle aus kovalent verknüpften Monomer-Bausteinen
Endgruppen
Zahl der verknüpften Monomereinheiten = Polymerisationsgrad (hier: n+6)
kovalenteBindung
Strukturelement
Monomer
Strukturelle Varianten:a) Thermoplaste (unverzweigte Ketten, schmelzbar)
b) Elastomere und Duroplaste (verzweigte Ketten, nicht schmelzbar)
statistischesCopolymer
Block-Copolymer
Homopolymer
(quer-)vernetztesPolymer
Wichtige Klassen von Polymeren
H H C CH R
= Polyolefine
R C OO
= Polyester
R C NHO
= Polyamide
Polyolefine
H H C CH H
= Polyethylen (PE)
H H C CH Cl
= Polyvinylchlorid (PVC)
H H C CH CH3
= Polypropylen (PP)
H H C CH C6H5
= Polystyrol (PS)
- Weißlich bis gelblich, manchmal transparent- Im Allgemeinen geringe Leitfähigkeit- Viele Eigenschaften hängen stark vom Polymerisationsgrad ab.
- Stabil gegen verdünnte Säuren und Basen- Relativ korrosionsstabil - Thermisch zersetzbar- Häufig löslich in organischen Lösemitteln
Gemeinsame Eigenschaften von Polymeren
physikalische Eigenschaften:
chemische Eigenschaften:

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l3
Übungsblatt 1
1) Wenn Sie die in Polymeren vorkommenden chemischen Elemente mit denen anderer Materialien (Metalle, Keramik, Glas) vergleichen, was fällt Ihnen dabei auf? Welche Besonderheit zeichnet die chemischen Elemente der Polymere aus?
2) Welche Funktionen erfüllen Biopolymere in der belebten Natur? Benennen Sie drei Beispiele für Biopolymere mit möglichst unterschiedlichen bio-logischen Aufgabenstellungen.
3) Was unterscheidet in der exakten Definition ein Monomer von einem Strukturelement?
4) Was unterscheidet bei der Betrachtung eines Makromoleküls die als Endgruppen bezeichneten Bauelemente von den Strukturelementen innerhalb der Kette?
5) Warum kann man Gegenstände, die aus einem Elastomer hergestellt sind (z.B. Autoreifen) nicht stofflich wiederverwerten? Erklären Sie das zugrundeliegende Problem anhand der molekularen Struktur des Elasto-mers.
6) Benennen Sie die aus folgenden Strukturelementen gebildeten Polymere und erwähnen Sie dabei gegebenenfalls die Gruppe von Polymeren, zu denen sie gehören:
CH H C CH C6H5
H H C CH Cl
H CH C CH CH3
CH2
C5H10 C NHO
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l4
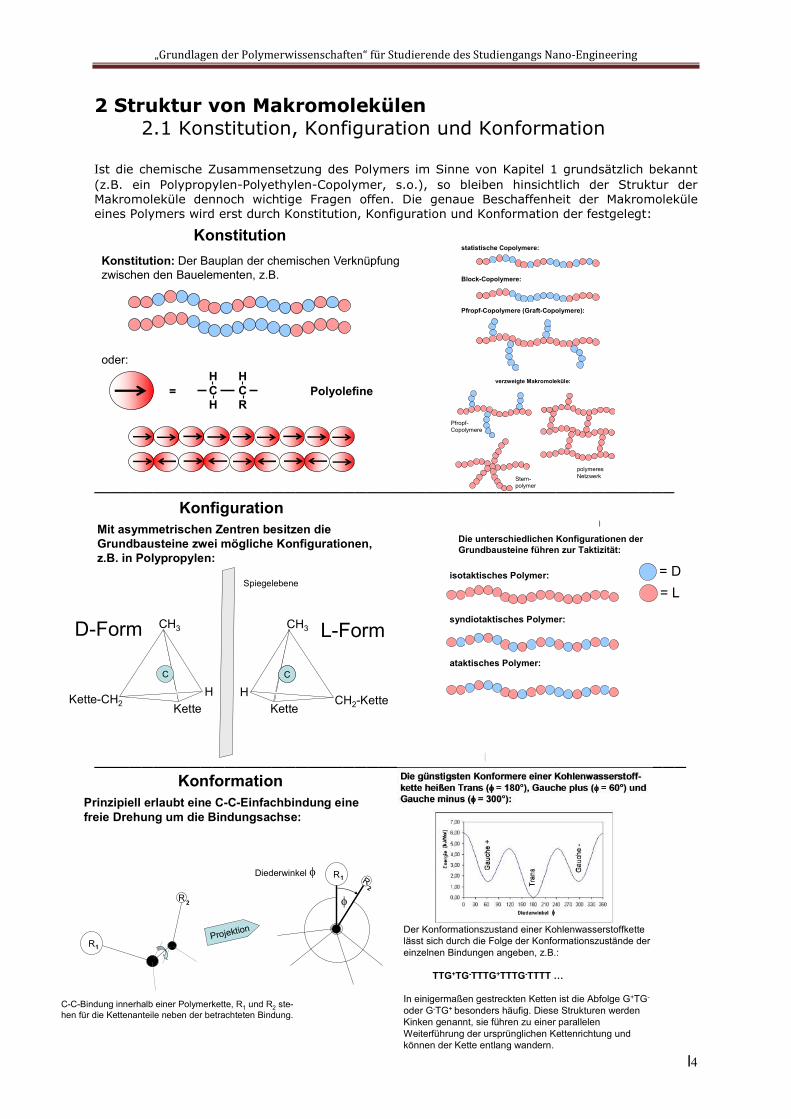
2 Struktur von Makromolekülen 2.1 Konstitution, Konfiguration und Konformation
Ist die chemische Zusammensetzung des Polymers im Sinne von Kapitel 1 grundsätzlich bekannt (z.B. ein Polypropylen-Polyethylen-Copolymer, s.o.), so bleiben hinsichtlich der Struktur der Makromoleküle dennoch wichtige Fragen offen. Die genaue Beschaffenheit der Makromoleküle eines Polymers wird erst durch Konstitution, Konfiguration und Konformation der festgelegt:
_________________________________________________
__________________________________________________
KonstitutionKonstitution: Der Bauplan der chemischen Verknüpfung zwischen den Bauelementen, z.B.
oder:H H C CH R
= PolyolefineKonstitution
verzweigte Makromoleküle:
Pfropf-Copolymere
polymeresNetzwerkStern-
polymer
KonfigurationMit asymmetrischen Zentren besitzen dieGrundbausteine zwei mögliche Konfigurationen, z.B. in Polypropylen:
C
CH3
HKette-CH2 Kette
C
CH3
HKette
CH2-Kette
Spiegelebene
D-Form L-Form
KonfigurationDie unterschiedlichen Konfigurationen der Grundbausteine führen zur Taktizität:
isotaktisches Polymer:
syndiotaktisches Polymer:
ataktisches Polymer:
= D= L
KonformationPrinzipiell erlaubt eine C-C-Einfachbindung eine freie Drehung um die Bindungsachse:
R
R1
2
RR12
Diederwinkel
C-C-Bindung innerhalb einer Polymerkette, R1 und R2 ste-hen für die Kettenanteile neben der betrachteten Bindung.
Projektion Der Konformationszustand einer Kohlenwasserstoffkette lässt sich durch die Folge der Konformationszustände der einzelnen Bindungen angeben, z.B.:
TTG+TG-TTTG+TTTG-TTTT …
In einigermaßen gestreckten Ketten ist die Abfolge G+TG-
oder G-TG+ besonders häufig. Diese Strukturen werden Kinken genannt, sie führen zu einer parallelen Weiterführung der ursprünglichen Kettenrichtung und können der Kette entlang wandern.
Konstitutionstatistische Copolymere:
Block-Copolymere:
Pfropf-Copolymere (Graft-Copolymere):

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l5
Übungsblatt 2
1) Was ist ein Block-Copolymer? Wie unterscheidet sich ein Block-Copolymer von einem statistischen Copolymer? Welche Voraussage könnten Sie für das Kristallisationsverhalten beider Varianten treffen?
2) Ein Monomer mit fünf reaktiven Resten wird zusammen mit einem großen Überschuss eines Monomers mit zwei reaktiven Resten zur Polymerisation gebracht. Welche Art von Polymer könnte dabei entstehen?
3) Welche Voraussetzung muss eine Monomermischung erfüllen, damit ein polymeres Netzwerk entstehen kann? Wie können Sie die Netzwerkdichte steuern?
4) Angenommen, ein Polymerlieferant möchte Ihnen zu einem hohen Preis syndiotaktisches Polyethylen verkaufen. Warum sollten Sie bei diesem Angebot sehr misstrauisch werden?
5) Welchen Vorteil besitzt ataktisches Polypropylen gegenüber isotaktischem Polypropylen? Bei welchen Anwendungen könnten Sie diesen Vorteil nutzen?
6) Ist es möglich, ein syndiotaktisches Polymer in ein isotaktisches Polymer umzuwandeln, ohne die Polymerkette dabei zu spalten?
7) Wie unterscheiden sich wohl die Häufigkeiten von gauche-Konformationen zwischen den Polymeren Polyethylen, Polypropylen und Polystyrol? Postulie-ren Sie jeweils die Verläufe der potentiellen Energie gegen den Diederwinkel in einem gemeinsamen Diagramm.
8) Benennen Sie folgende Polymerstrukturen:
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l6
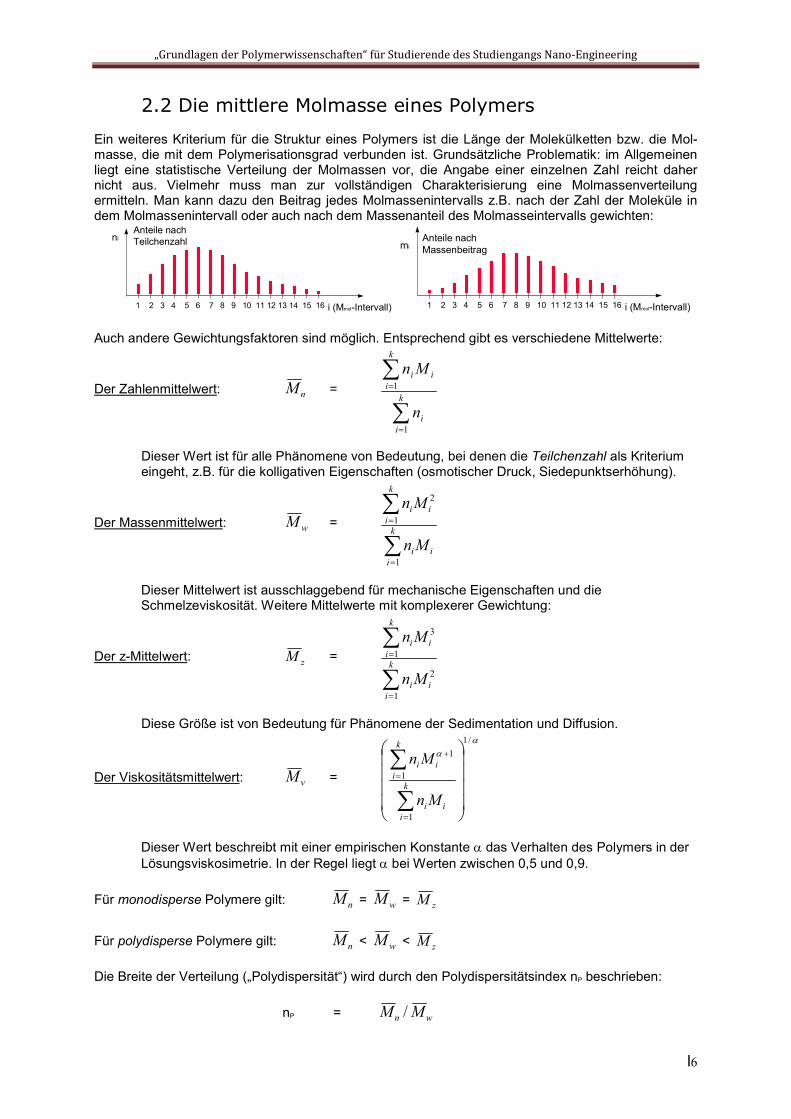
2.2 Die mittlere Molmasse eines Polymers
Ein weiteres Kriterium für die Struktur eines Polymers ist die Länge der Molekülketten bzw. die Mol-masse, die mit dem Polymerisationsgrad verbunden ist. Grundsätzliche Problematik: im Allgemeinen liegt eine statistische Verteilung der Molmassen vor, die Angabe einer einzelnen Zahl reicht daher nicht aus. Vielmehr muss man zur vollständigen Charakterisierung eine Molmassenverteilung ermitteln. Man kann dazu den Beitrag jedes Molmassenintervalls z.B. nach der Zahl der Moleküle in dem Molmassenintervall oder auch nach dem Massenanteil des Molmasseintervalls gewichten:
Auch andere Gewichtungsfaktoren sind möglich. Entsprechend gibt es verschiedene Mittelwerte:
Der Zahlenmittelwert: nM =
k
ii
k
iii
n
Mn
1
1
Dieser Wert ist für alle Phänomene von Bedeutung, bei denen die Teilchenzahl als Kriterium eingeht, z.B. für die kolligativen Eigenschaften (osmotischer Druck, Siedepunktserhöhung).
Der Massenmittelwert: wM =
k
iii
k
iii
Mn
Mn
1
1
2
Dieser Mittelwert ist ausschlaggebend für mechanische Eigenschaften und die Schmelzeviskosität. Weitere Mittelwerte mit komplexerer Gewichtung:
Der z-Mittelwert: zM =
k
iii
k
iii
Mn
Mn
1
2
1
3
Diese Größe ist von Bedeutung für Phänomene der Sedimentation und Diffusion.
Der Viskositätsmittelwert: vM =
/1
1
1
1
k
iii
k
iii
Mn
Mn
Dieser Wert beschreibt mit einer empirischen Konstante das Verhalten des Polymers in der Lösungsviskosimetrie. In der Regel liegt bei Werten zwischen 0,5 und 0,9.
Für monodisperse Polymere gilt: nM = wM = zM
Für polydisperse Polymere gilt: nM < wM < zM
Die Breite der Verteilung („Polydispersität“) wird durch den Polydispersitätsindex nP beschrieben:
nP = wn MM /
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 i (Mmol-Intervall)
niAnteile nach Teilchenzahl
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 i (Mmol-Intervall)
miAnteile nach Massenbeitrag

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l7
Übungsblatt 3
1) Beschreiben Sie das Energieprofil, das mit der Drehung um eine C-C-Einfach-bindung in einer Polyethylenkette verbunden ist. Erklären Sie die Entstehung der Minima und Maxima der potentiellen Energie und benennen Sie die Konformationen, die den drei Minima zugeordnet werden können.
2) Ist es denkbar, dass die beiden Konformere G+ und G- mit unterschiedlicher Häufigkeit auftreten? Versuchen Sie, solch einen möglichen Fall zu be-schreiben.
3) Warum kann der Massenmittelwert der Molmasse eines Polymers niemals kleiner sein als der Zahlenmittelwert? Begründen Sie diese Tatsache an-schaulich anhand der Darstellungen eines Diagramms für die Molmassen-verteilung.
4) Angenommen, eine Polymerprobe besteht aus einem einzigen Molekül mit der Molmasse 109 g/Mol. Nun fügen Sie ein einzelnes Monomermolekül mit der Molmasse 100 g/Mol hinzu. Wie ändert sich dadurch… a) Das Zahlenmittel der Molmasse b) Das Massenmittel der Molmasse c) Der z-Mittelwert der Molmasse
5) Eine Probe eines Polymers bestehe zu je einem Mol aus Anteilen mit den einheitlichen Molmassen 1000 g/Mol, 2000 g/Mol und 3000 g/Mol. Berechnen Sie das Zahlenmittel, das Massenmittel und den z-Mittelwert der Molmasse. Wie groß wäre in diesem Fall der Polydispersitätsindex?
6) Ist es möglich, dass bei einem gegebenen Polymer der Viskositätsmittelwert und der Massenmittelwert der Molmasse gleich sind, der Zahlenmittelwert dagegen von den beiden erstgenannten abweicht? Begründen Sie an-schaulich (ohne Rechnung).
7) Berechnen Sie den Polydispersitätsindex eines Polymers, das aus einem Mol Monomer der Molmasse 100 g/Mol und einem Mol Polymer der Molmasse 10000 g/Mol besteht.
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l8
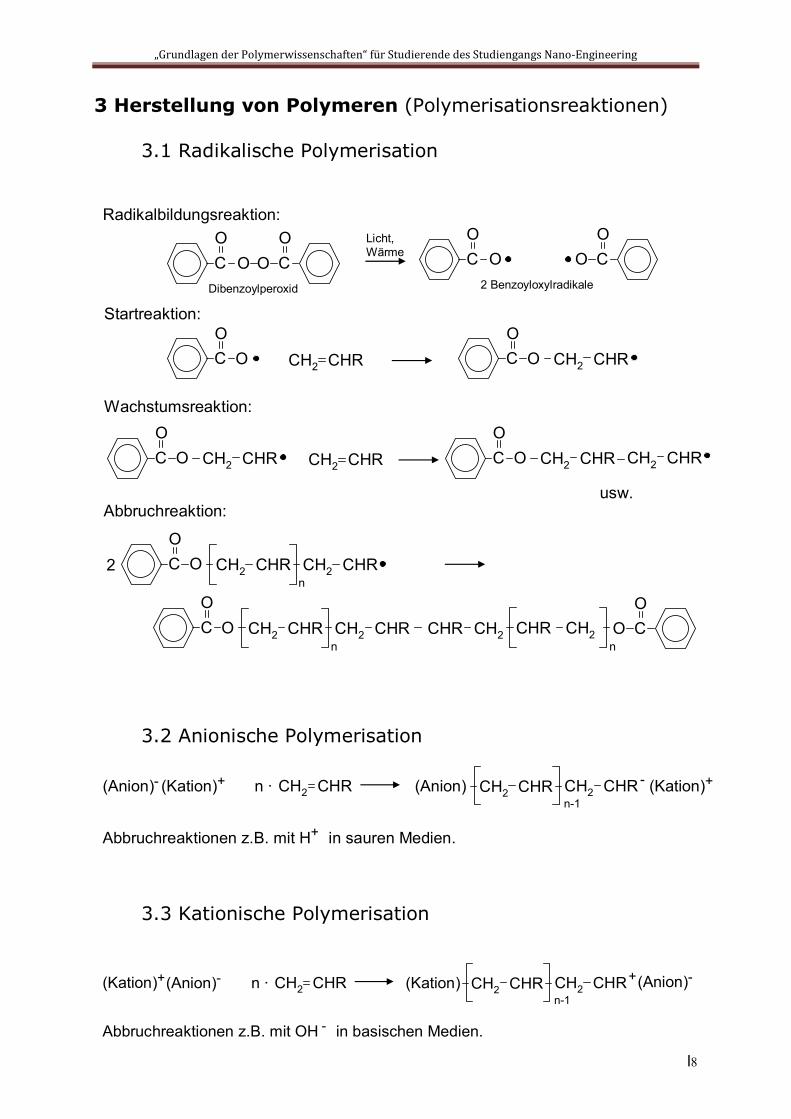
3 Herstellung von Polymeren (Polymerisationsreaktionen)
3.1 Radikalische Polymerisation
3.2 Anionische Polymerisation
3.3 Kationische Polymerisation
Radikalbildungsreaktion:
CO
O CO
ODibenzoylperoxid
Licht, Wärme C
OO C
OO
2 Benzoyloxylradikale
Startreaktion:
CO
O CH2 CHR CO
O CH2 CHR
Wachstumsreaktion:
CH2 CHRCO
O CH2 CHR CO
O CH2 CHR CH2 CHR
usw.Abbruchreaktion:
CO
O CH2 CHR CH2 CHRn
2
CO
O CH2 CHR CH2 CHRn
CHR CH2 CHR CH2n
CO
O
CH2 CHR(Anion)- (Kation)+ n ∙ CH2 CHR CH2 CHR(Anion) - (Kation)+
Abbruchreaktionen z.B. mit H in sauren Medien. +
n-1
CH2 CHR(Anion)-(Kation)+ n ∙ CH2 CHR CH2 CHR(Kation) +(Anion)-
Abbruchreaktionen z.B. mit OH in basischen Medien. -
n-1

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l9
Übungsblatt 4
1) Beschreiben Sie den Verlauf einer radikalischen Polymerisation von Polystyrol unter Einsatz von Wärme und Dibenzoylperoxid als Starterkomponente.
2) Welchen Einfluss nimmt die Konzentration des Starters (z.B. Dibenzoylper-oxid) auf die Molmasse des bei einer radikalischen Polymerisation entste-henden Polymers?
3) Wie können Sie die Zahl der Kettenabbruchreaktionen bei einer gegebenen radikalischen Polymerisation kontrollieren?
4) Die ionische Polymerisation kann schon durch Spuren von Wasser beeinflusst werden, während die radikalische Polymerisation sich dadurch völlig unbeeindruckt zeigt. Geben Sie eine mögliche Erklärung auf der Grundlage der chemischen Eigenschaften eines Wassermoleküls.
5) Welche Bedingung muss das negative Gegenion bei einer kationischen Polymerisation erfüllen, damit tatsächlich ein Polymer mit einer hohen Molmasse entstehen kann?
6) Warum kann man eine anionische Polymerisation durch Ansäuern des Reaktionsmediums kontrolliert stoppen? Wie kann man diesen Einfluss nutzen, um Nanostrukturen (z.B. Nanokapseln) zu erzeugen?
7) Welche Bedeutung besitzt die folgende Reaktion bei einer radikalischen Polymerisation?
8) Welche Eigenschaften des Monomers begünstigen eher eine anionische Polymerisation, welche eher eine kationische Polymerisation? Bei welchem pH-Wert wird eine kationische Polymerisation bevorzugt vorgenommen?
CO
O CH2 CHR CH2 CHRn
2
CO
O CH2 CHR CH2 CHRn
CHR CH2 CHR CH2n
CO
O
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l10
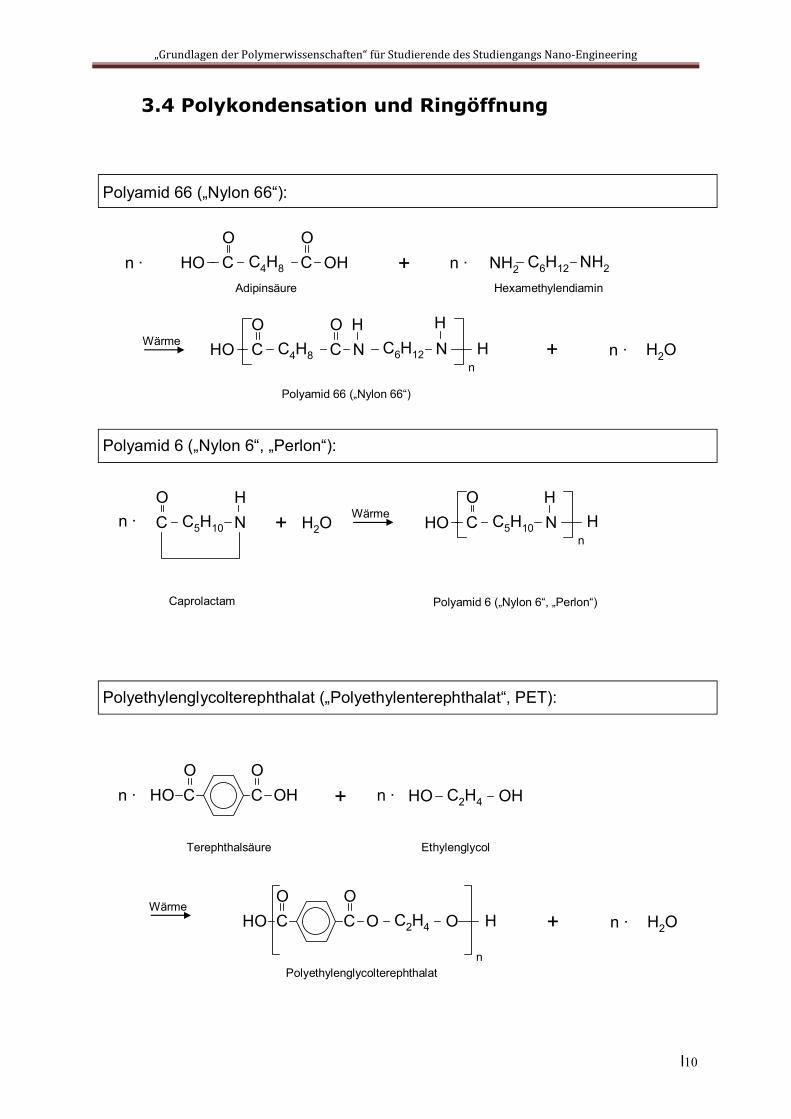
3.4 Polykondensation und Ringöffnung
Polyamid 66 („Nylon 66“):
CO
OHAdipinsäure
Wärme
C4H8CO
HOHexamethylendiamin
C6H12NH2 NH2
CO
N
Polyamid 66 („Nylon 66“)
C4H8CO
HO
n ∙ n ∙
HH
nC6H12 N
H
Polyamid 6 („Nylon 6“, „Perlon“):
NH
Caprolactam
C5H10CO
n ∙ N
Polyamid 6 („Nylon 6“, „Perlon“)
C5H10CO
HOH
Hn
H2OWärme
CO
OHCO
HO
+
+
Polyethylenglycolterephthalat („Polyethylenterephthalat“, PET):
Terephthalsäure
OHHO C2H4
Ethylenglycol
+
WärmeCO
OCO
HO OC2H4 H
n
n ∙ n ∙
Polyethylenglycolterephthalat
H2O+ n ∙
H2O+ n ∙

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l11
4 Makromoleküle in Lösung 4.1 Konformation eines gelösten Makromoleküls
Makromoleküle in Lösung können aufgrund der bei jedem Segment möglichen Konformationen T, G+ und G- (s. Kapitel 2.1) in sehr unterschiedlicher Kettenform auftreten. In guten Lösemitteln tendieren sie dazu, offene, gestreckte Ketten zu bilden. In schlechten Lösemitteln findet man dagegen eher eng geknäuelte, fast kugelförmige Moleküle. Ein weiterer Faktor, der den Grad der Knäuelung prägt, ist die Anwesenheit von Ladungen auf der Polymerkette. Beispielsweise stoßen gleichsinnige Ladungen einander ab und führen zu einem gestreckten Molekül. Der Grad der Knäuelung eines gelösten Makromoleküls kann durch den Kettenendenabstand h ausgedrückt werden. Daneben kann man ausgehend vom Schwerpunkt des Moleküls die räumliche Dichte bestimmen, mit der die Segmente des Makromoleküls pro Volumeneinheit auftreten. Die Verteilungen beider Parameter in einem Polymer sollen im Folgenden genauer beschrieben werden.
4.1.1 Kettenendenabstandsverteilung
Der Wert von h variiert von Molekül zu Molekül, kann aber für eine Polymerlösung in einer Häufigkeits-verteilung W(h) statistisch erfasst werden. Für den Kettenendenabstand h gilt die Verteilungsfunktion
W(h) =
2*
22
2/3
2* 23exp
234
kk lNhh
lN
mit N* als Zahl der Segmente und lk als Länge eines einzelnen Segments. Diese Verteilungsfunktion entspricht in ihrer Form der Geschwindigkeitsverteilung der Teilchen in einem Gas. Die gezeigte Verteilungsfunktion gilt für Moleküle, die innerhalb der Kette keine Wechselwirkungen besitzen. Liegen Abstoßungskräfte z.B. wegen gleichsinniger Ladungen vor, oder besitzt das Lösemittel für das gegebene Polymer eine hohe Qualität, so verbreitert sich die Verteilungsfunktion entsprechend und weicht von oben genannter Gleichung ab.
4.1.2 Segmentdichteverteilung
Bei einem gelösten Makromolekül verteilen sich die Segmente statistisch in der Umgebung des Molekülschwerpunkts. Kennt man den Trägheitsradius der Moleküle, also den Radius einer gedachten Hohlkugel mit demselben Trägheitsmoment, so kann die Dichte P der Segmentverteilung in Form einer Funktion P(r) angegeben werden:
P(r) =
²23exp²
22/3
23*
RrRN
…mit ²R als mittlerem Quadrat des Trägheitsradius, N* als Zahl der Segmente und r als Abstand vom Schwerpunkt. Abstoßende Kräfte innerhalb der Polymerketten oder eine hohe Qualität des Lösemittels haben einen Anstieg des Trägheitsradius und damit – ähnlich wie bei der Kettenendenabstandsverteilung - eine Verbreiterung der Verteilungsfunktion zur Folge.
h
W(h)
h
r
P(r)r

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l12
Übungsblatt 5
1) Warum entsteht bei der Herstellung von Polyamid 66 im Allgemeinen Wasser? Wie nennt man die damit verbundene Polymerisationsreaktion?
2) Welchen Vorteil bietet die Herstellung von Polyamid 6 aus Caprolactam im Vergleich zur Herstellung von Polyamid 66 aus Adipinsäure und Hexa-methylendiamin?
3) Polymermoleküle mit zwei reaktiven Enden bilden nur äußerst selten Ringmoleküle, obwohl sie dazu prinzipiell in der Lage wären. Der Grund liegt in der Konformation des gelösten Makromoleküls. Warum ist vor diesem Hintergrund eine chemische Reaktion zwischen den beiden Endgruppen eines Makromoleküls relativ unwahrscheinlich? Begründen Sie mit Hilfe der Verteilungsfunktion zum Kettenendenabstand.
4) Angenommen, Polymer A hat genau doppelt so viele Segmente wie Poly-mer B. Trotzdem finden Sie bei den Polymeren A und B völlig identische Kettenendenabstandsverteilungen W(h) vor. Was können Sie unter diesen Umständen über das Verhältnis der Segmentlängen lk beider Polymere aussagen?
5) Welche Möglichkeiten haben Sie, um ein Polymer in eine gestreckte Kon-formation zu bringen, wenn Sie sowohl die chemische Beschaffenheit des Polymers als auch die des Lösemittels verändern können?
6) Für viele Anwendungen in der Nanotechnologie hätte man gerne Polymer-partikel, die sich wie harte, feste Kugeln mit einem genau definierten Ra-dius verhalten. Warum kann diese Idee mit gelösten Polymermolekülen nur unvollkommen verwirklicht werden? In welcher Beziehung weicht das gegebene System vom Idealbild ab?
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l13
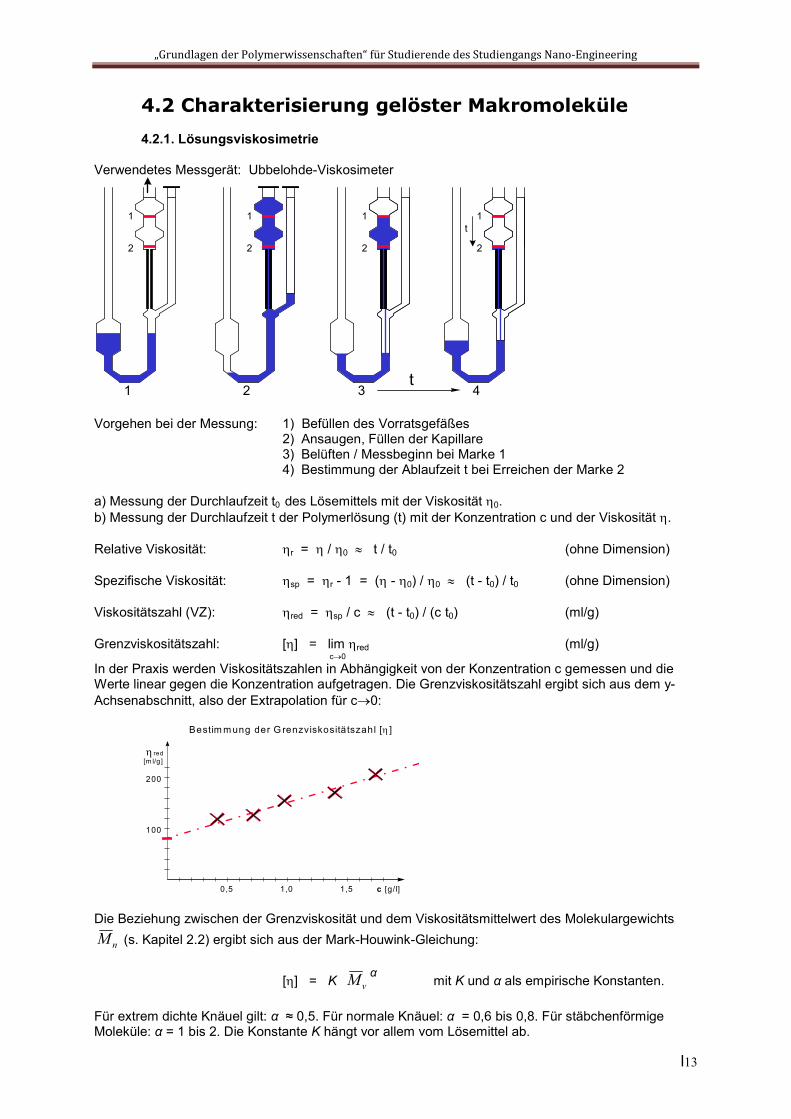
4.2 Charakterisierung gelöster Makromoleküle4.2.1. Lösungsviskosimetrie
Verwendetes Messgerät: Ubbelohde-Viskosimeter
Vorgehen bei der Messung: 1) Befüllen des Vorratsgefäßes 2) Ansaugen, Füllen der Kapillare 3) Belüften / Messbeginn bei Marke 1 4) Bestimmung der Ablaufzeit t bei Erreichen der Marke 2
a) Messung der Durchlaufzeit t0 des Lösemittels mit der Viskosität 0. b) Messung der Durchlaufzeit t der Polymerlösung (t) mit der Konzentration c und der Viskosität .
Relative Viskosität: r = / 0 t / t0 (ohne Dimension)
Spezifische Viskosität: sp = r - 1 = ( - 0) / 0 (t - t0) / t0 (ohne Dimension)
Viskositätszahl (VZ): red = sp / c (t - t0) / (c t0) (ml/g)
Grenzviskositätszahl: [] = lim red (ml/g)c0
In der Praxis werden Viskositätszahlen in Abhängigkeit von der Konzentration c gemessen und die Werte linear gegen die Konzentration aufgetragen. Die Grenzviskositätszahl ergibt sich aus dem y-Achsenabschnitt, also der Extrapolation für c0:
Die Beziehung zwischen der Grenzviskosität und dem Viskositätsmittelwert des Molekulargewichts
nM (s. Kapitel 2.2) ergibt sich aus der Mark-Houwink-Gleichung:
[] = K vM α mit K und α als empirische Konstanten.
Für extrem dichte Knäuel gilt: α ≈ 0,5. Für normale Knäuel: α = 0,6 bis 0,8. Für stäbchenförmige Moleküle: α = 1 bis 2. Die Konstante K hängt vor allem vom Lösemittel ab.
1
2
1
2
1
2
1
2
1 2 3 4t
t
c [g/l]
red
0,5 1,0 1,5
[m l/g]
100
200
Bestim m ung der G renzviskositätszahl [ ]

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l14
4.2.2. Gelpermeationschromatographie (GPC)
Ermöglicht die Analyse der Molekulargewichtsverteilung Basiert auf dem Prinzip der Flüssigkeitschromatographie Als Säulenmaterial dient ein Gel aus mikroporösen Partikeln
Prinzipieller Versuchsaufbau (typisch für alle Methoden der Chromatographie):
Vorrats-gefäß mitLösemittel
Pumpe
Injektor
Säule
Detektor
EDV
Fraktionen-Sammler
Substanz
Das Prinzip des Trennvorgangs ist die Gelpermeation, d.h. das Eindringen der hochmolekularen Kom-ponente in die Poren des Gels. Die Sortierung erfolgt damit nach der Molekülgröße, daher andere Bezeichnung des Verfahrens:
Size Exclusion Chromatography („Größen-Ausschluss-Chromatographie“) oder SEC.
Das zur Trennung verwendete Gel besitzt feine Poren, die eine statistische Größenverteilung auf-weisen. Passiert ein in Lösung befindliches Polymermolekül das Gel, so steht ihm in der Säule ein ganz bestimmtes freies Volumen zur Verfügung, das von seinem Molekulargewicht (bzw. seiner Größe) abhängt. Für große Moleküle ist dieses freie Volumen klein, für kleine Moleküle groß (s. Skizze).
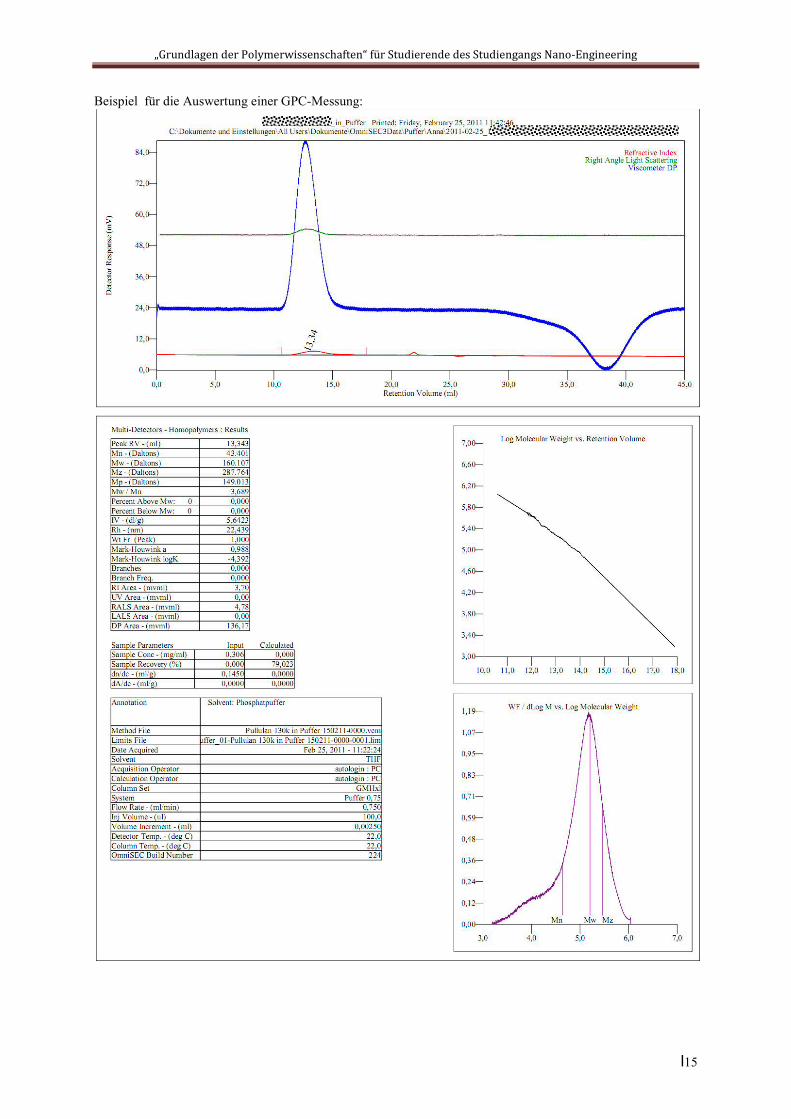
Die in die Säule eingebrachten Teilchen sind bestrebt, sich in diesem Volumen gleichmäßig zu vertei-len. Daraus ergibt sich eine größenabhängige Aufenthaltsdauer in der Säule: große Teilchen passie-ren das Säulenmaterial schneller als kleine. Oder anders gesagt: zur Passage durch die Säule benötigt man für große Moleküle ein kleineres Lösemittelvolumen als für kleine. Dieses zur Passage benötigte Lösemittelvolumen nennt man das Elutionsvolumen (engl. „retention volume“). Die Konzentration der Polymerteilchen im Eluat wird üblicherweise über die Veränderung des Brechungsindex, der Lichtstreuung oder der Viskosität der Lösung detektiert. Man erhält als Ergebnis dann Diagramme der entsprechenden Detektorsignale gegen das Elutionsvolumen (s. unten).
Moderne SEC-Anlagen können sich über die Auswertung des Lichtstreuungssignals selbst kalibrieren. Dazu wird die Lichtstreuung der einzelnen Fraktionen analysiert, was einen Absolutwert für die Partikelgröße ergibt. Diese wiederum lässt sich in die Molmasse der Fraktion umrechnen. Führt man diese Molmassenbestimmung für alle gefundenen Fraktionen durch, so erhält man durch Extrapolation eine Kalibrationskurve, welche das Elutionsvolumen mit der Molmasse verknüpft (Beispiel s. unten). Auf diese Weise können sämtliche Mittelwerte der Molmasse sowie der Polydispersitätsparameter und die Mark-Houwink-Konstanten K und α bestimmt werden:
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l15
Beispiel für die Auswertung einer GPC-Messung:

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l16
4.2.3. Osmometrie
Die Molmasse eines gelösten Polymers kann auch über die Messung des osmotischen Drucks der Lösung bestimmt werden. Bei der Messung des osmotischen Drucks geht man folgendermaßen vor: eine halbdurchlässige Membran trennt eine Kammer mit reinem Lösemittel von einer weiteren Kammer mit der Lösung des Polymers ab. Die dann in der Polymer-haltigen Kammer gemessene Druckerhöhung entspricht dem osmotischen Druck Π:
Das Entstehen des osmotischen Druckes kann in Analogie zum Druck eines Gases verstanden werden: die gelösten Teilchen prallen im Verlauf ihrer thermischen Bewegung immer wieder gegen die Membran, so dass eine kontinuierliche Impulsänderung, d.h. eine Kraft resultiert. Da diese Kraft proportional zur Fläche ist und senkrecht auf diese Fläche wirkt, kann sie flächenbezogen betrachtet und als Druck bezeichnet werden. Es verhält sich so, als wäre das gelöste Polymer ein Gas und als wäre das Lösemittel überhaupt nicht vorhanden.
Wie alle kolligativen Eigenschaften (Siedepunktserhöhung, Gefrierpunktserniedrigung, Dampfdruck-erniedrigung) hängt auch der osmotische Druck im Idealfall lediglich von der Anzahl der gelösten Teilchen ab, nicht von deren Natur oder deren Verhalten. Aus der Anzahl der Teilchen kann man bei bekannter Einwaage des Polymers wiederum die Molmasse ermitteln, wobei man in diesem speziellen Fall (dies gilt für alle kolligativen Eigenschaften) das Zahlenmittel der Molmasse erhält. Für ideale Lösungen gilt (mit n = m/Mn und m = cV):
= V
nRT =
nMcRT
In Wirklichkeit muss man aber auch die Wechselwirkungen zwischen benachbarten Polymermolekülen berücksichtigen. Im Allgemeinen findet man, dass der osmotische Druck etwas kleiner ist, als er eigentlich gemäß der Teilchenzahl sein sollte, da sich benachbarte Moleküle anziehen. Für solche nicht-idealen Lösungen verwendet man häufig den Ansatz einer Virialgleichung:
= cRT (1/Mn + A2c + A3c² + …)
Je größer der so genannte zweite Virialkoeffizient A2 ausfällt (höhere Virialkoeffizienten A3 A4 … werden meistens vernachlässigt), desto mehr weicht die vorliegende Polymerlösung vom idealen Verhalten ab, d.h. umso größer sind die Wechselwirkungen zwischen den gelösten Makromolekülen.
Videos: animation: how osmosis workshttp://highered.mheducation.com/sites/0072495855/student_view0/chapter2/animation__how_osmosis_works.html
Prinzip der Osmometrie
Lösemittel Lösemittel + Polymer
osmotischer Druck

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l17
Übungsblatt 6
1) Beschreiben Sie den experimentellen Ablauf einer Viskositätsmessung mit einem Ubbelohde-Viskosimeter. Warum ist die Messung an einer Ver-gleichsprobe (dem reinen Lösemittel) notwendig, um das Polymer zu charakterisieren?
2) Welche Bedeutung hat die Grenzviskositätszahl [η]? Warum ist sie eine geeignetere Kenngröße für das Polymer als die Viskositätszahl selbst?
3) Warum verwendet man in der Gelpermeationschromatographie (GPC) meistens mehrere Detektoren (z.B. einen für Lichtstreuung, einen für die Viskosität und einen für den Brechungsindex)?
4) Warum verbringt ein Makromolekül mit hoher Molmasse mehr Zeit in der GPC-Säule als ein Makromolekül mit geringer Masse?
5) Sie vermessen die Molmassenverteilung eines Polymers mit einer GPC und stellen fest, dass die gemessene Molmasse deutlich kleiner ausfällt als erwartet. Welches Phänomen könnte für diesen Fehler verantwortlich sein?
6) Sie messen zwei verschiedene Polymere A und B mit einem Osmometer und stellen fest, dass der zweite Virialkoeffizient A2 bei Polymer A deutlich größer ausfällt als bei Polymer B. Welche Schlüsse können Sie daraus ziehen?
7) Angenommen, Sie setzen verschiedene Polymerlösungen an, indem Sie jeweils 1 g Polymer in 100 ml Lösemittel auflösen. Wird der osmotische Druck dieser Lösungen mit steigender Molmasse des Polymers größer oder kleiner? Begründen Sie diesen Zusammenhang anschaulich.
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l18
5 Makromoleküle in einer Polymerschmelze 5.1 Viskoelastische Eigenschaften einer
Polymerschmelze
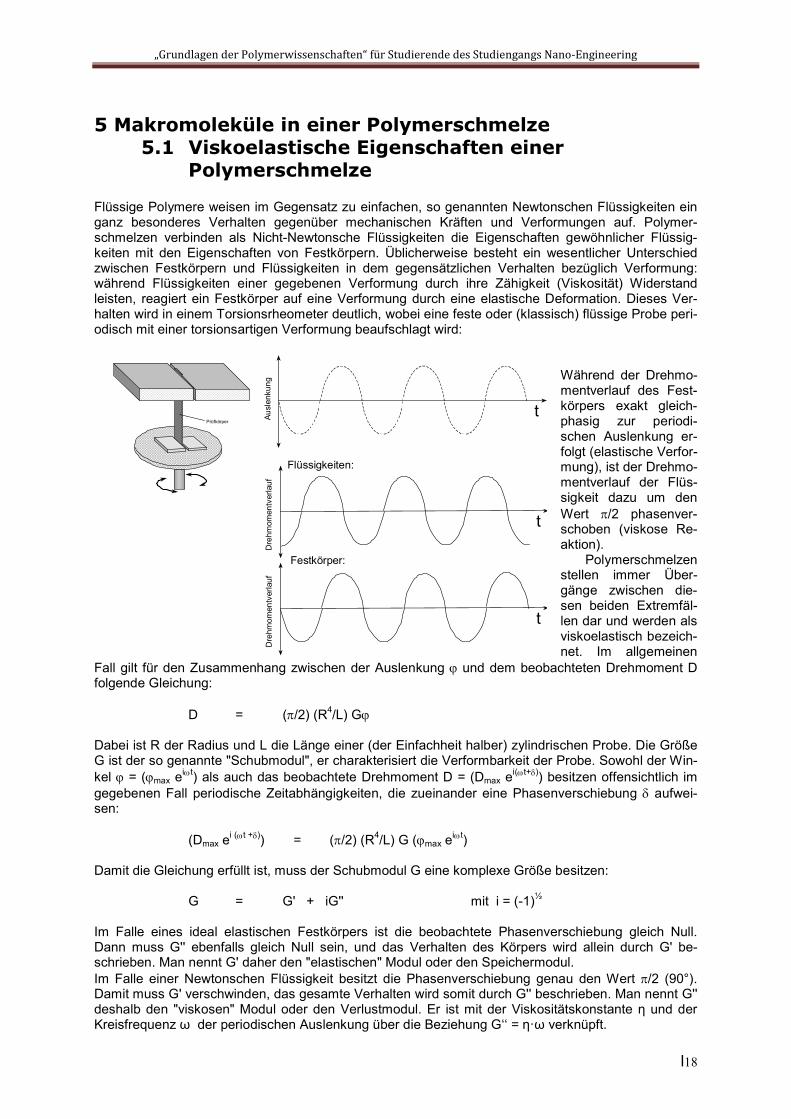
Flüssige Polymere weisen im Gegensatz zu einfachen, so genannten Newtonschen Flüssigkeiten ein ganz besonderes Verhalten gegenüber mechanischen Kräften und Verformungen auf. Polymer-schmelzen verbinden als Nicht-Newtonsche Flüssigkeiten die Eigenschaften gewöhnlicher Flüssig-keiten mit den Eigenschaften von Festkörpern. Üblicherweise besteht ein wesentlicher Unterschied zwischen Festkörpern und Flüssigkeiten in dem gegensätzlichen Verhalten bezüglich Verformung: während Flüssigkeiten einer gegebenen Verformung durch ihre Zähigkeit (Viskosität) Widerstand leisten, reagiert ein Festkörper auf eine Verformung durch eine elastische Deformation. Dieses Ver-halten wird in einem Torsionsrheometer deutlich, wobei eine feste oder (klassisch) flüssige Probe peri-odisch mit einer torsionsartigen Verformung beaufschlagt wird:
Während der Drehmo-mentverlauf des Fest-körpers exakt gleich-phasig zur periodi-schen Auslenkung er-folgt (elastische Verfor-mung), ist der Drehmo-mentverlauf der Flüs-sigkeit dazu um den Wert /2 phasenver-schoben (viskose Re-aktion).
Polymerschmelzen stellen immer Über-gänge zwischen die-sen beiden Extremfäl-len dar und werden als viskoelastisch bezeich-net. Im allgemeinen
Fall gilt für den Zusammenhang zwischen der Auslenkung und dem beobachteten Drehmoment D folgende Gleichung:
D = (/2) (R4/L) G
Dabei ist R der Radius und L die Länge einer (der Einfachheit halber) zylindrischen Probe. Die Größe G ist der so genannte "Schubmodul", er charakterisiert die Verformbarkeit der Probe. Sowohl der Win-kel = (max eit) als auch das beobachtete Drehmoment D = (Dmax ei(t+)) besitzen offensichtlich im gegebenen Fall periodische Zeitabhängigkeiten, die zueinander eine Phasenverschiebung aufwei-sen:
(Dmax ei (t +)) = (/2) (R4/L) G (max eit)
Damit die Gleichung erfüllt ist, muss der Schubmodul G eine komplexe Größe besitzen:
G = G' + iG'' mit i = (-1)½
Im Falle eines ideal elastischen Festkörpers ist die beobachtete Phasenverschiebung gleich Null. Dann muss G'' ebenfalls gleich Null sein, und das Verhalten des Körpers wird allein durch G' be-schrieben. Man nennt G' daher den "elastischen" Modul oder den Speichermodul. Im Falle einer Newtonschen Flüssigkeit besitzt die Phasenverschiebung genau den Wert /2 (90°). Damit muss G' verschwinden, das gesamte Verhalten wird somit durch G'' beschrieben. Man nennt G'' deshalb den "viskosen" Modul oder den Verlustmodul. Er ist mit der Viskositätskonstante η und der Kreisfrequenz ω der periodischen Auslenkung über die Beziehung G‘‘ = η·ω verknüpft.
t
Dre
hmom
entv
erla
uf
tAusl
enku
ng
Festkörper:
t
Dre
hmom
entv
erla
uf
Flüssigkeiten:
Prüfkörper
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l19
Zwischen der Phasenverschiebung δ und dem Verhältnis zwischen G’ und G’’ gilt folgender Zusammenhang:
tan δ = G’’ / G’
Der Tangens der Phasenverschiebung, das „Tangens Delta“, ist ein häufig verwendeter Kennwert für das viskoelastische Verhalten eines Polymers. Polymerschmelzen besitzen meistens ein recht großes „Tangens Delta“, d.h. einen recht hohen Verlustmodul G’’, aber gleichzeitig auch einen gewissen Spei-chermodul G’. Das heißt, dass sie im Gegensatz zu „normalen“ Flüssigkeiten eine gewisse Elastizität aufweisen: versucht man, sie durch eine vorübergehende Krafteinwirkung umzuformen, so „federn“ sie partiell wieder zurück. Man spricht dabei auch von einem „molekularen Gedächtnis“: die ursprüngliche dreidimensionale Form eines Polymerwerkstücks ist in dem molekularen Gefüge in gewisser Weise gespeichert. Gegenstände aus polymeren Materialien können so auch nach langer Zeit in diejenige Form zurückkehren, die der Rohling kurz vor der Umformung besaß.
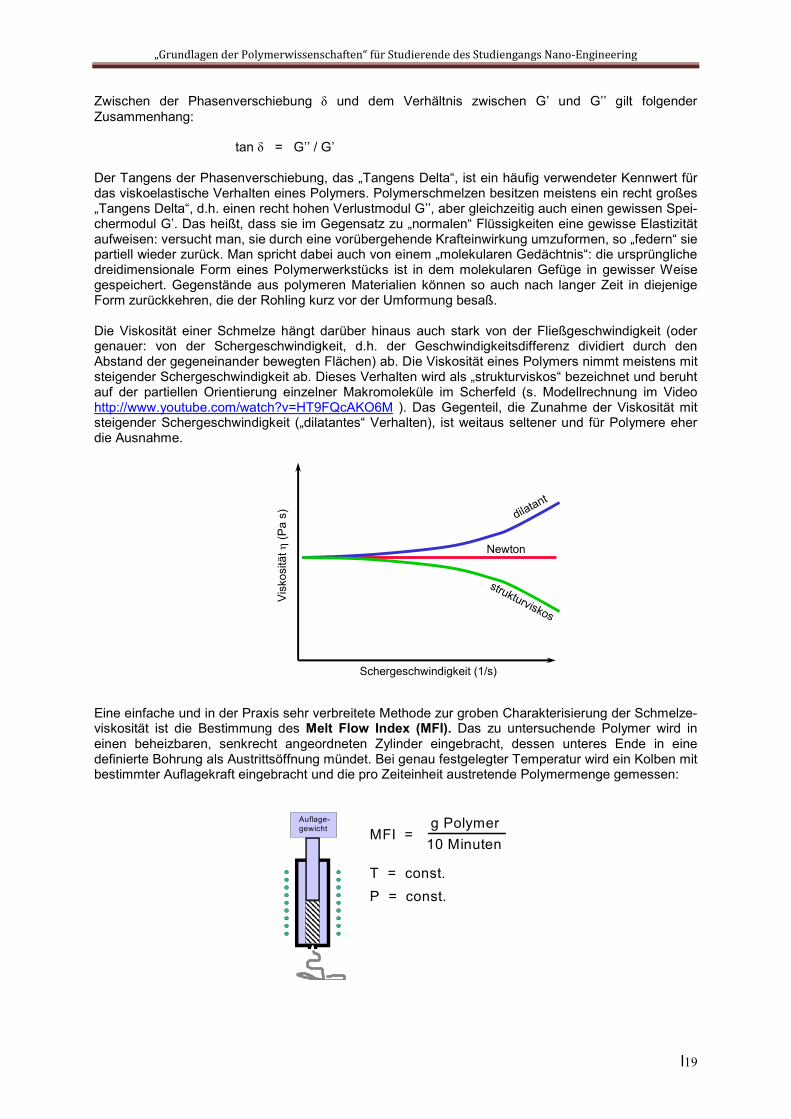
Die Viskosität einer Schmelze hängt darüber hinaus auch stark von der Fließgeschwindigkeit (oder genauer: von der Schergeschwindigkeit, d.h. der Geschwindigkeitsdifferenz dividiert durch den Abstand der gegeneinander bewegten Flächen) ab. Die Viskosität eines Polymers nimmt meistens mit steigender Schergeschwindigkeit ab. Dieses Verhalten wird als „strukturviskos“ bezeichnet und beruht auf der partiellen Orientierung einzelner Makromoleküle im Scherfeld (s. Modellrechnung im Video http://www.youtube.com/watch?v=HT9FQcAKO6M ). Das Gegenteil, die Zunahme der Viskosität mit steigender Schergeschwindigkeit („dilatantes“ Verhalten), ist weitaus seltener und für Polymere eher die Ausnahme.
Eine einfache und in der Praxis sehr verbreitete Methode zur groben Charakterisierung der Schmelze-viskosität ist die Bestimmung des Melt Flow Index (MFI). Das zu untersuchende Polymer wird in einen beheizbaren, senkrecht angeordneten Zylinder eingebracht, dessen unteres Ende in eine definierte Bohrung als Austrittsöffnung mündet. Bei genau festgelegter Temperatur wird ein Kolben mit bestimmter Auflagekraft eingebracht und die pro Zeiteinheit austretende Polymermenge gemessen:
Auflage-gewicht MFI =
g Polymer10 Minuten
T = const.P = const.
Visk
ositä
t (P
a s)
Schergeschwindigkeit (1/s)
Newton
dilatant
strukturviskos

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l20
Übungsblatt 7
1) Warum benötigt man bei der Messung viskoelastischer Eigenschaften einen Deformationsversuch mit periodischer Auslenkung? Welches Ergeb-nis erhält man bei kontinuierlicher Deformation (z.B. konstanter Drehung) a) mit einem idealen Festkörper, b) mit einer idealen Flüssigkeit?
2) Was bedeutet der Term „Tangens Delta“ bei der Charakterisierung eines viskoelastischen Systems? Welches „Tangens Delta“ erwarten Sie bei a) einem idealen Festkörper, b) einer idealen Flüssigkeit, c) einer typischen Polymerschmelze (geschätzt).
3) Warum besitzt eine Polymerschmelze neben den Eigenschaften einer Flüs-sigkeit (Verlustmodul) auch partiell die Eigenschaften eines Festkörpers? Begründen Sie dieses Phänomen unter Berücksichtigung der molekularen Packung und der intermolekularen Wechselwirkungen in einer Poly-merschmelze.
4) Warum ist das Verständnis der viskoelastischen Eigenschaften von beson-derer Bedeutung für die Herstellung von Nanostrukturen aus partiell ge-schmolzenen Polymeren? Betrachten Sie beispielhaft die Herstellung einer nanostrukturierten Polymeroberfläche durch Prägung (Nanoimprinting).
5) Angenommen, Sie möchten eine nanostrukturierte Polymeroberfläche durch Nanoimprinting herstellen. Ihnen stehen dazu zwei Polymere zur Verfügung, die sich nur in dem Wert für Tangens Delta unterscheiden. Welches Polymer würden Sie bevorzugen, das mit größerem oder das mit kleinerem Tangens Delta? Welches Tangens Delta wäre der gewünschte Idealwert?
6) Sie wollen durch Nanoimprinting eine extrem feine Struktur einprägen. Welche Prozessgeschwindigkeit (schnell oder langsam) sollten Sie bevor-zugt wählen, wenn Ihr Polymer sich a) dilatant, b) strukturviskos verhält?
7) Erklären Sie, warum sich die meisten Polymerschmelzen strukturviskos verhalten.

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l21
5.2 Umformung von flüssigen Polymeren
Für die Umformung von flüssigen Polymeren sind zahlreiche Verfahren gebräuchlich. Die wichtigsten großtechnischen Ansätze basieren auf der Anwendung von so genannten Extrudern (s. Abbildung).
Extrusionsanlage für Polymere (nach W. Michaeli, 1992). In der oben eingefügten Skizze ist die nachfolgende Extrusion eines Rohres dargestellt.
Die aus dem Extruder austretende Schmelze kann man beispielsweise portionsweise in eine gegebe-ne Form einspritzen (Spritzguss), zwischen Walzen zu einer flächigen Form pressen (Kalandrieren), in einer Form aufblasen (Blasformung), zu langen Profilen ausformen (Extrusion) oder durch eine enge Düse pressen und den austretenden Polymerfaden verspinnen.
Extruder dienen auch dazu, das flüssige Polymer mit anderen Polymeren oder Additiven zu vermi-schen. Der Mischungsprozess wird auch als Compoundierung bezeichnet, die Mischung als Com-pound. Die entstandene Mischung wird üblicherweise zu einem dünnen Strang extrudiert und anschließend zu kleinen Körnchen („Pellets“) zerhackt („geschreddert“). Die Pellets können dann einem weiteren Umformungsprozess unterworfen werden, dienen also erneut als Rohmaterial bei-spielsweise für eine Extrusion oder einen Spritzgussprozess.
Alternativ kann man Polymere auch portionsweise innerhalb einer Presse aufheizen und unter Druck umformen. Im Bereich der Nanotechnologie fällt beispielsweise das so genannte Nanoimprinting in diese Kategorie (s. Kapitel 7.2).

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l22
6 Makromoleküle in festem Polymer 6.1 Amorphe und kristalline Strukturen
Feste Polymere lassen sich gemäß ihrer molekularen Struktur in drei Gruppen einteilen:
1) Teilkristalline thermoplastische Polymere
2) Elastomere 3) Duroplaste
Während die festen Strukturen bei Elastomeren und Duroplasten bereits bei der Polymerisation ausgebildet werden und sich somit nachträglich nicht beeinflussen lassen, entstehen sie bei den Thermoplasten erst aus der flüssigen Phase heraus (s. dazu ein Video zur Ausbildung von kristallinen Zonen, so genannten Sphäroliten: http://www.youtube.com/watch?v=130sUnjUxmQ). Dabei bilden sich innerhalb des Feststoffes im Allgemeinen regellose, amorphe Bereiche neben geordneten, kristallinen Bereichen (s. ovale Markierungen in der oben gezeigten Darstellung). In bestimmten Fällen erhält man völlig amorphe Polymere, in sehr seltenen Fällen kann man dagegen auch völlig kristalline Polymere (Einkristalle) herstellen.
Der prozentuale Anteil der kristallinen Bereiche (Kristallisationsgrad) in einem Thermoplast kann durch verschiedene Faktoren beeinflusst werden:
a) Abkühlgeschwindigkeit (je größer, desto amorpher) b) Chemische Zusammensetzung (je homogener, desto kristalliner) c) Taktizität (sinkende Kristallinität in der Reihe isotaktisch-syndiotaktisch-ataktisch) d) Verzweigungsgrad (je höher, desto amorpher) e) Bei Nanostrukturen: Größe von Polymerpartikeln (je kleiner, desto amorpher) f) Bei Nanostrukturen: Dicke eines Polymerfilms (je dünner, desto amorpher)
polymere Schmelze teilkristalliner Thermoplast (Sphärolith)umgeben von amorphen Bereichen
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l23
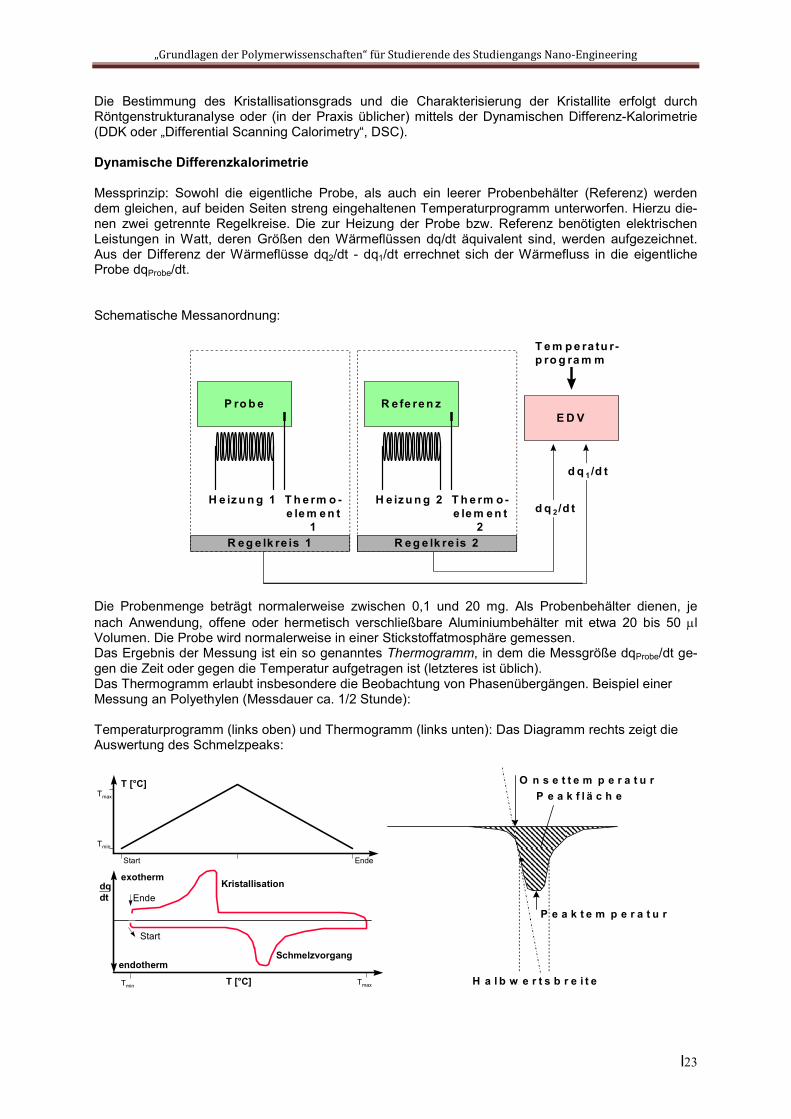
Die Bestimmung des Kristallisationsgrads und die Charakterisierung der Kristallite erfolgt durch Röntgenstrukturanalyse oder (in der Praxis üblicher) mittels der Dynamischen Differenz-Kalorimetrie (DDK oder „Differential Scanning Calorimetry“, DSC).
Dynamische Differenzkalorimetrie
Messprinzip: Sowohl die eigentliche Probe, als auch ein leerer Probenbehälter (Referenz) werden dem gleichen, auf beiden Seiten streng eingehaltenen Temperaturprogramm unterworfen. Hierzu die-nen zwei getrennte Regelkreise. Die zur Heizung der Probe bzw. Referenz benötigten elektrischen Leistungen in Watt, deren Größen den Wärmeflüssen dq/dt äquivalent sind, werden aufgezeichnet. Aus der Differenz der Wärmeflüsse dq2/dt - dq1/dt errechnet sich der Wärmefluss in die eigentliche Probe dqProbe/dt.
Schematische Messanordnung:
Die Probenmenge beträgt normalerweise zwischen 0,1 und 20 mg. Als Probenbehälter dienen, je nach Anwendung, offene oder hermetisch verschließbare Aluminiumbehälter mit etwa 20 bis 50 l Volumen. Die Probe wird normalerweise in einer Stickstoffatmosphäre gemessen. Das Ergebnis der Messung ist ein so genanntes Thermogramm, in dem die Messgröße dqProbe/dt ge-gen die Zeit oder gegen die Temperatur aufgetragen ist (letzteres ist üblich). Das Thermogramm erlaubt insbesondere die Beobachtung von Phasenübergängen. Beispiel einer Messung an Polyethylen (Messdauer ca. 1/2 Stunde):
Temperaturprogramm (links oben) und Thermogramm (links unten): Das Diagramm rechts zeigt die Auswertung des Schmelzpeaks:
R e fe re n z
H e iz u n g 2 T h e rm o -e le m e n t
2
E D V
T e m p e ra tu r-p ro g ra m m
P ro b e
H e iz u n g 1 T h e rm o -e le m e n t
1R e g e lk re is 1 R e g e lk re is 2
d q 1/d t
R e g e lk re is 2
d q 2/d t
T [°C]
dqdt
exotherm
endothermSchmelzvorgang
Kristallisation
Start
Ende
T [°C]
Start Ende
Tmin
Tmax
TmaxTmin
O n s e t t e m p e r a t u rP e a k f l ä c h e
P e a k t e m p e r a t u r
H a l b w e r t s b r e i t e
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l24
Anwendungen der DDK (DSC) bei der Untersuchung von polymeren Materialien:
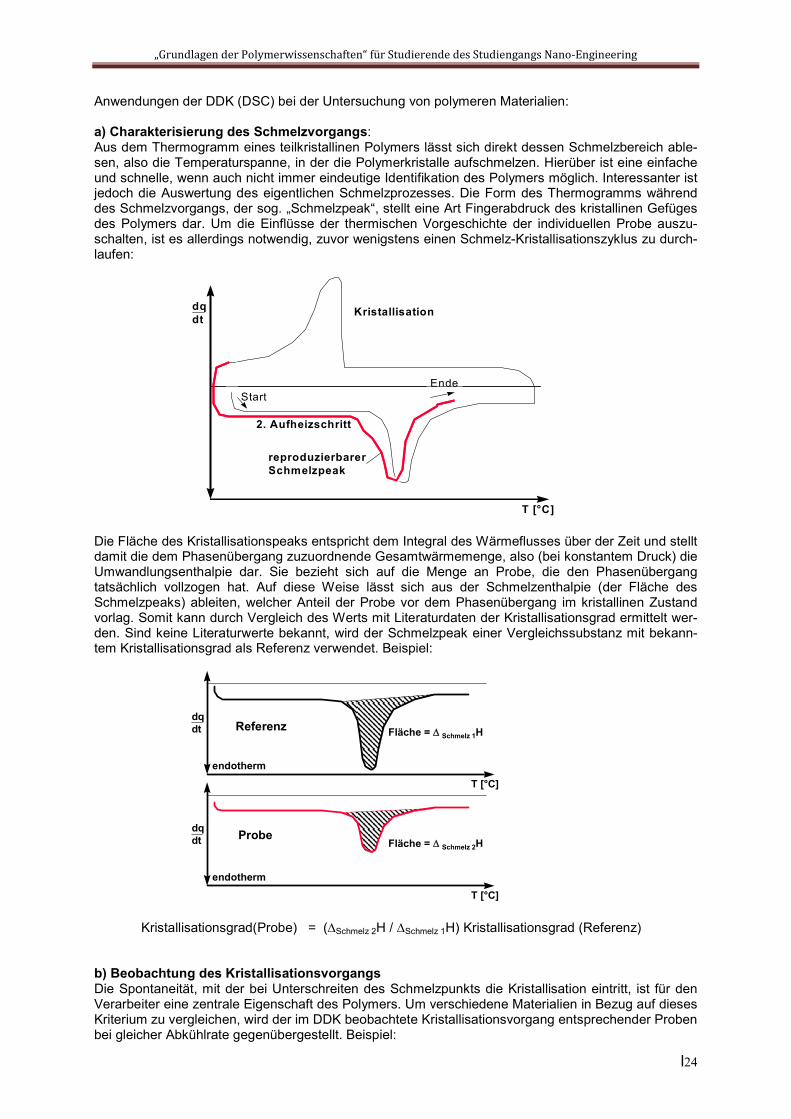
a) Charakterisierung des Schmelzvorgangs: Aus dem Thermogramm eines teilkristallinen Polymers lässt sich direkt dessen Schmelzbereich able-sen, also die Temperaturspanne, in der die Polymerkristalle aufschmelzen. Hierüber ist eine einfache und schnelle, wenn auch nicht immer eindeutige Identifikation des Polymers möglich. Interessanter ist jedoch die Auswertung des eigentlichen Schmelzprozesses. Die Form des Thermogramms während des Schmelzvorgangs, der sog. „Schmelzpeak“, stellt eine Art Fingerabdruck des kristallinen Gefüges des Polymers dar. Um die Einflüsse der thermischen Vorgeschichte der individuellen Probe auszu-schalten, ist es allerdings notwendig, zuvor wenigstens einen Schmelz-Kristallisationszyklus zu durch-laufen:
Die Fläche des Kristallisationspeaks entspricht dem Integral des Wärmeflusses über der Zeit und stellt damit die dem Phasenübergang zuzuordnende Gesamtwärmemenge, also (bei konstantem Druck) die Umwandlungsenthalpie dar. Sie bezieht sich auf die Menge an Probe, die den Phasenübergang tatsächlich vollzogen hat. Auf diese Weise lässt sich aus der Schmelzenthalpie (der Fläche des Schmelzpeaks) ableiten, welcher Anteil der Probe vor dem Phasenübergang im kristallinen Zustand vorlag. Somit kann durch Vergleich des Werts mit Literaturdaten der Kristallisationsgrad ermittelt wer-den. Sind keine Literaturwerte bekannt, wird der Schmelzpeak einer Vergleichssubstanz mit bekann-tem Kristallisationsgrad als Referenz verwendet. Beispiel:
Kristallisationsgrad(Probe) = (Schmelz 2H / Schmelz 1H) Kristallisationsgrad (Referenz)
b) Beobachtung des Kristallisationsvorgangs Die Spontaneität, mit der bei Unterschreiten des Schmelzpunkts die Kristallisation eintritt, ist für den Verarbeiter eine zentrale Eigenschaft des Polymers. Um verschiedene Materialien in Bezug auf dieses Kriterium zu vergleichen, wird der im DDK beobachtete Kristallisationsvorgang entsprechender Proben bei gleicher Abkühlrate gegenübergestellt. Beispiel:
T [°C]
dqdt
Start
reproduzierbarerSchmelzpeak
Kristallisation
Ende
2. Aufheizschritt
T [°C]
dqdt
endotherm
Fläche = Schmelz 2H
T [°C]
dqdt
endotherm
Fläche = Schmelz 1HReferenz
Probe
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l25
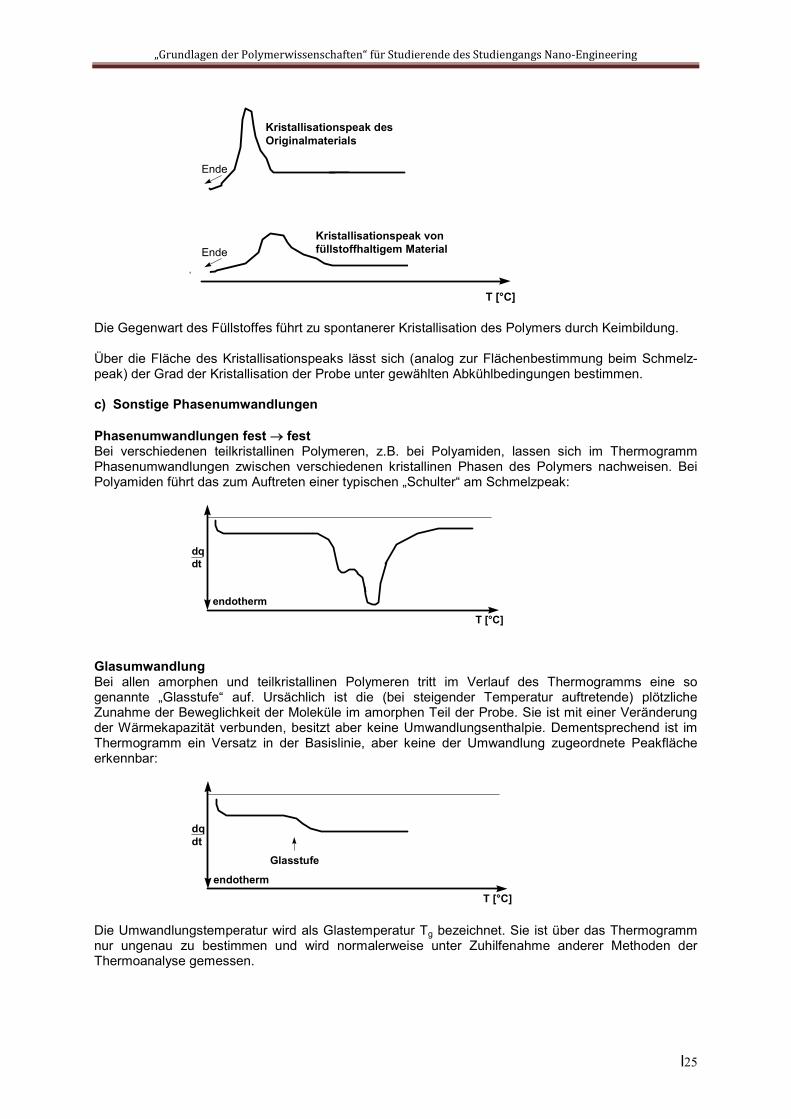
Die Gegenwart des Füllstoffes führt zu spontanerer Kristallisation des Polymers durch Keimbildung.
Über die Fläche des Kristallisationspeaks lässt sich (analog zur Flächenbestimmung beim Schmelz-peak) der Grad der Kristallisation der Probe unter gewählten Abkühlbedingungen bestimmen.
c) Sonstige Phasenumwandlungen
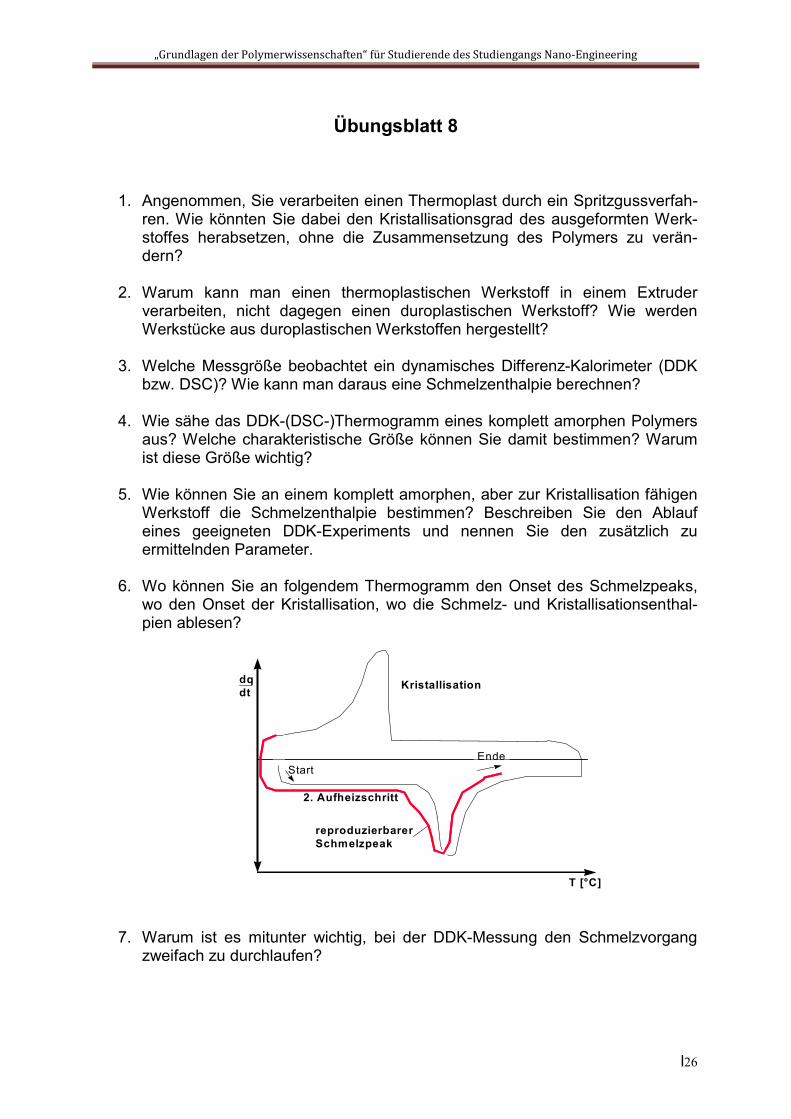
Phasenumwandlungen fest fest Bei verschiedenen teilkristallinen Polymeren, z.B. bei Polyamiden, lassen sich im Thermogramm Phasenumwandlungen zwischen verschiedenen kristallinen Phasen des Polymers nachweisen. Bei Polyamiden führt das zum Auftreten einer typischen „Schulter“ am Schmelzpeak:
Glasumwandlung Bei allen amorphen und teilkristallinen Polymeren tritt im Verlauf des Thermogramms eine so genannte „Glasstufe“ auf. Ursächlich ist die (bei steigender Temperatur auftretende) plötzliche Zunahme der Beweglichkeit der Moleküle im amorphen Teil der Probe. Sie ist mit einer Veränderung der Wärmekapazität verbunden, besitzt aber keine Umwandlungsenthalpie. Dementsprechend ist im Thermogramm ein Versatz in der Basislinie, aber keine der Umwandlung zugeordnete Peakfläche erkennbar:
Die Umwandlungstemperatur wird als Glastemperatur Tg bezeichnet. Sie ist über das Thermogramm nur ungenau zu bestimmen und wird normalerweise unter Zuhilfenahme anderer Methoden der Thermoanalyse gemessen.
Kristallisationspeak desOriginalmaterials
Kristallisationspeak vonfüllstoffhaltigem Material
T [°C]
Ende
Ende
T [°C]
dqdt
endotherm
T [°C]
dqdt
endotherm
Glasstufe
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l26
Übungsblatt 8
1. Angenommen, Sie verarbeiten einen Thermoplast durch ein Spritzgussverfah-ren. Wie könnten Sie dabei den Kristallisationsgrad des ausgeformten Werk-stoffes herabsetzen, ohne die Zusammensetzung des Polymers zu verän-dern?
2. Warum kann man einen thermoplastischen Werkstoff in einem Extruder verarbeiten, nicht dagegen einen duroplastischen Werkstoff? Wie werden Werkstücke aus duroplastischen Werkstoffen hergestellt?
3. Welche Messgröße beobachtet ein dynamisches Differenz-Kalorimeter (DDK bzw. DSC)? Wie kann man daraus eine Schmelzenthalpie berechnen?
4. Wie sähe das DDK-(DSC-)Thermogramm eines komplett amorphen Polymers aus? Welche charakteristische Größe können Sie damit bestimmen? Warum ist diese Größe wichtig?
5. Wie können Sie an einem komplett amorphen, aber zur Kristallisation fähigen Werkstoff die Schmelzenthalpie bestimmen? Beschreiben Sie den Ablauf eines geeigneten DDK-Experiments und nennen Sie den zusätzlich zu ermittelnden Parameter.
6. Wo können Sie an folgendem Thermogramm den Onset des Schmelzpeaks, wo den Onset der Kristallisation, wo die Schmelz- und Kristallisationsenthal-pien ablesen?
7. Warum ist es mitunter wichtig, bei der DDK-Messung den Schmelzvorgang zweifach zu durchlaufen?
T [°C]
dqdt
Start
reproduzierbarerSchmelzpeak
Kristallisation
Ende
2. Aufheizschritt

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l27
d) Chemische Reaktionen
Spontane Oxydation bei erhöhter Temperatur: der Oxydations-Onset
Ebenfalls auf einer DSC-Messung beruht ein Standardtest, der die Oxydationsempfindlichkeit eines Polymers misst. Dabei wird die Probe in einer sauerstoffhaltigen Atmosphäre (gewöhnlich Luft) mit konstanter Rate (z.B. 10°C/min) aufgeheizt. Gemessen wird der Onset des bei dem spontanen Oxydationsprozess (bei der Selbstentzündung der Probe) eintretenden exothermen Signals. Der entsprechende Temperaturwert wird „Oxidation Onset Temperature” (OOT) genannt. Daneben wird alternativ auch ein Test bei konstanter Temperatur durchgeführt, bei dem die Zeit bis zur spontanen Oxydation bestimmt wird („Oxidation Induction Time“, OIT).
Vernetzungsreaktionen
In ähnlicher Weise wie bei der Oxydation kann auch der Ablauf einer Vernetzungsreaktion anhand des für gewöhnlich exothermen Signals beobachtet werden. So kann man zum Beispiel bestimmen, bei welcher Temperatur die spontane Vernetzung eintritt oder ob die Vernetzungsreaktion bei einem bereits ausgehärteten Duroplast vollständig abgelaufen ist. Im zweiten Fall wird dann einfach die Reaktionsenthalpie der Restreaktion bestimmt.
6.2 Dynamische Prozesse in festen PolymerenIn ähnlicher Weise, wie flüssige Polymere teilweise Festkörpereigenschaften aufweisen, so findet man bei festen Polymeren viele Phänomene, die sonst für Flüssigkeiten typisch sind. So können oberhalb der Glasstufe eines thermoplastischen, unvernetzten Polymers (s. Kapitel 6.1 c) Wanderungs-bewegungen einzelner Polymermoleküle auftreten. Dabei „kriechen“ Kettenmoleküle in einer sta-tistischen, wurmartig schlängelnden Bewegung durch das Geflecht der Nachbarmoleküle hindurch („Reptation“). Makroskopisch macht sich dieses Phänomen durch mechanische Relaxation bemerk-bar: unter Last zeigt ein polymeres Werkstück ein so genanntes Kriechverhalten („Creeping“), d.h., es verformt sich allmählich und irreversibel. Gleichzeitig besitzt der Werkstoff allerdings auch eine erhöhte Schlagzähigkeit, er reagiert weniger spröde auf plötzliche Verformung und splittert nicht.
Analytisch lässt sich dieses Phänomen am besten mit einem Torsionsrheometer erfassen (s. Kapitel 5.1). Der Tangens der im Verlauf einer periodischen Messung zwischen der Auslenkung und dem Drehmoment auftretenden Phasenverschiebung δ markiert die Relation zwischen Fließvorgängen und elastischer Verformung:
tan δ = G’’ / G’
Ein hoch vernetzter, duroplastischer Werkstoff besitzt ebenso wie ein Thermoplast unterhalb der Glasstufe ein sehr kleines tan δ. Dagegen reagiert ein Thermoplast oberhalb der Glasstufe zuneh-mend „flüssigkeitsähnlich“ auf eine gegebene Last, was sich in einem wachsenden Wert für tan δ bemerkbar macht. Der Sprung im Wert von tan δ gilt als die zuverlässigste Methode für die Bestimmung des Glasübergangs.
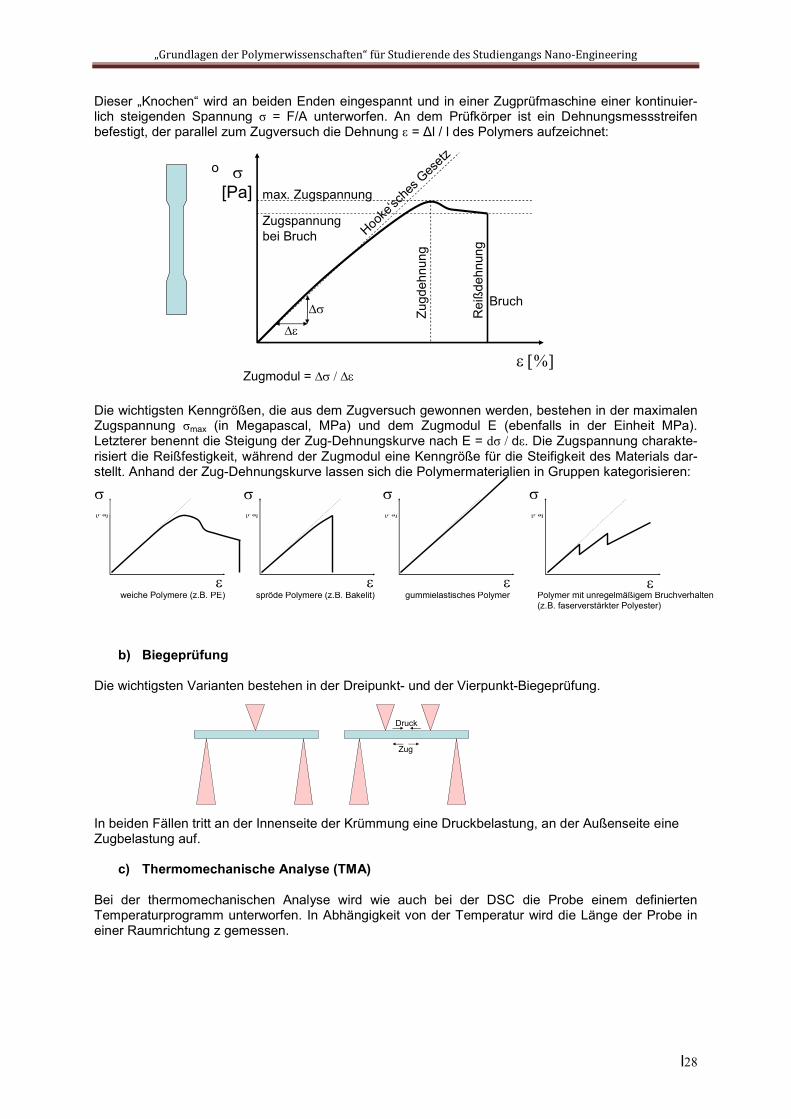
6.3 Mechanische Eigenschaften fester PolymereIm Vordergrund des mechanischen Verhaltens fester Polymere stehen natürlich wieder die viskoelasti-schen Eigenschaften, die mit einem Torsionsrheometer erfasst werden können. Diese sind insbeson-dere bei einer dynamischen Belastung eines Bauteils auch in der Praxis der Werkstoffprüfung von Bedeutung. Allerdings erfährt ein polymerer Werkstoff in vielen Fällen eine rein statische Bean-spruchung, der er in erster Linie gerecht werden muss. Alle in der Technik verwendeten Werkstoffe werden in dieser Hinsicht ausgiebig getestet, wobei je nach Anwendung verschiedene praxisnahe Belastungsformen simuliert werden. Am wichtigsten ist dabei die so genannte Zugprüfung.
a) Zugprüfung Die Prüfkörper weisen zumeist eine „Knochenform“ auf, gleichen also einem Stab mit verbreiterten Enden:
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l28
Dieser „Knochen“ wird an beiden Enden eingespannt und in einer Zugprüfmaschine einer kontinuier-lich steigenden Spannung σ = F/A unterworfen. An dem Prüfkörper ist ein Dehnungsmessstreifen befestigt, der parallel zum Zugversuch die Dehnung ε = Δl / l des Polymers aufzeichnet:
Die wichtigsten Kenngrößen, die aus dem Zugversuch gewonnen werden, bestehen in der maximalen Zugspannung σmax (in Megapascal, MPa) und dem Zugmodul E (ebenfalls in der Einheit MPa). Letzterer benennt die Steigung der Zug-Dehnungskurve nach E = dσ / dε. Die Zugspannung charakte-risiert die Reißfestigkeit, während der Zugmodul eine Kenngröße für die Steifigkeit des Materials dar-stellt. Anhand der Zug-Dehnungskurve lassen sich die Polymermaterialien in Gruppen kategorisieren:
b) Biegeprüfung
Die wichtigsten Varianten bestehen in der Dreipunkt- und der Vierpunkt-Biegeprüfung.
In beiden Fällen tritt an der Innenseite der Krümmung eine Druckbelastung, an der Außenseite eine Zugbelastung auf.
c) Thermomechanische Analyse (TMA)
Bei der thermomechanischen Analyse wird wie auch bei der DSC die Probe einem definierten Temperaturprogramm unterworfen. In Abhängigkeit von der Temperatur wird die Länge der Probe in einer Raumrichtung z gemessen.
[Pa]
weiche Polymere (z.B. PE)
[Pa]
spröde Polymere (z.B. Bakelit)
[Pa]
gummielastisches Polymer
[Pa]
Polymer mit unregelmäßigem Bruchverhalten(z.B. faserverstärkter Polyester)
Zug
Druck
P
o [Pa]
Hooke
‘sche
s Gesetz
max. Zugspannung
Bruch
Zugspannungbei Bruch
Zugd
ehnu
ng
Rei
ßdeh
nung
Zugmodul =
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l29
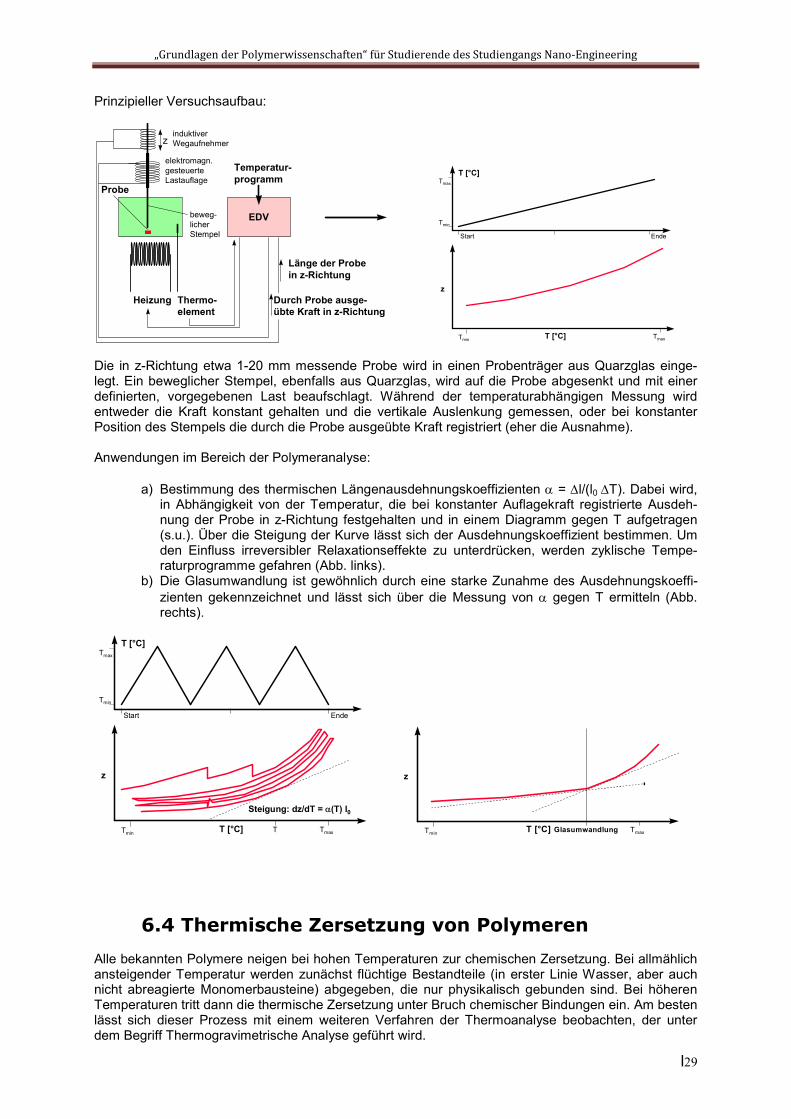
Prinzipieller Versuchsaufbau:
Die in z-Richtung etwa 1-20 mm messende Probe wird in einen Probenträger aus Quarzglas einge-legt. Ein beweglicher Stempel, ebenfalls aus Quarzglas, wird auf die Probe abgesenkt und mit einer definierten, vorgegebenen Last beaufschlagt. Während der temperaturabhängigen Messung wird entweder die Kraft konstant gehalten und die vertikale Auslenkung gemessen, oder bei konstanter Position des Stempels die durch die Probe ausgeübte Kraft registriert (eher die Ausnahme).
Anwendungen im Bereich der Polymeranalyse:
a) Bestimmung des thermischen Längenausdehnungskoeffizienten = l/(l0 T). Dabei wird, in Abhängigkeit von der Temperatur, die bei konstanter Auflagekraft registrierte Ausdeh-nung der Probe in z-Richtung festgehalten und in einem Diagramm gegen T aufgetragen (s.u.). Über die Steigung der Kurve lässt sich der Ausdehnungskoeffizient bestimmen. Um den Einfluss irreversibler Relaxationseffekte zu unterdrücken, werden zyklische Tempe-raturprogramme gefahren (Abb. links).
b) Die Glasumwandlung ist gewöhnlich durch eine starke Zunahme des Ausdehnungskoeffi-zienten gekennzeichnet und lässt sich über die Messung von gegen T ermitteln (Abb. rechts).
6.4 Thermische Zersetzung von PolymerenAlle bekannten Polymere neigen bei hohen Temperaturen zur chemischen Zersetzung. Bei allmählich ansteigender Temperatur werden zunächst flüchtige Bestandteile (in erster Linie Wasser, aber auch nicht abreagierte Monomerbausteine) abgegeben, die nur physikalisch gebunden sind. Bei höheren Temperaturen tritt dann die thermische Zersetzung unter Bruch chemischer Bindungen ein. Am besten lässt sich dieser Prozess mit einem weiteren Verfahren der Thermoanalyse beobachten, der unter dem Begriff Thermogravimetrische Analyse geführt wird.
Heizung Thermo-element
EDV
Temperatur-programm
Probe
elektromagn.gesteuerteLastauflage
induktiverWegaufnehmerz
Länge der Probein z-Richtung
Durch Probe ausge-übte Kraft in z-Richtung
beweg-licherStempel
T [°C]
z
T [°C]
Start Ende
Tmin
Tmax
TmaxTmin
T [°C]
z
T [°C]
Start Ende
Tmin
Tmax
TmaxTmin T
Steigung: dz/dT = (T) l0
T [°C]
z
TmaxTmin Glasumwandlung
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l30
Thermogravimetrische Analyse (TGA)
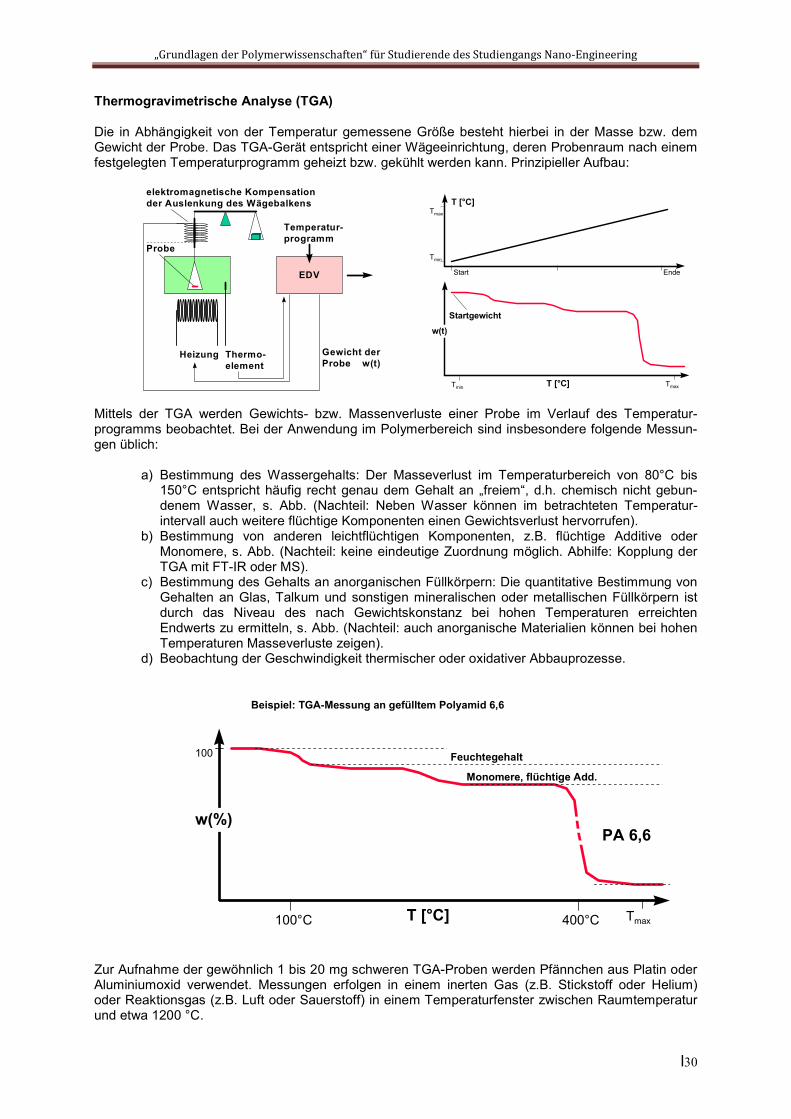
Die in Abhängigkeit von der Temperatur gemessene Größe besteht hierbei in der Masse bzw. dem Gewicht der Probe. Das TGA-Gerät entspricht einer Wägeeinrichtung, deren Probenraum nach einem festgelegten Temperaturprogramm geheizt bzw. gekühlt werden kann. Prinzipieller Aufbau:
Mittels der TGA werden Gewichts- bzw. Massenverluste einer Probe im Verlauf des Temperatur-programms beobachtet. Bei der Anwendung im Polymerbereich sind insbesondere folgende Messun-gen üblich:
a) Bestimmung des Wassergehalts: Der Masseverlust im Temperaturbereich von 80°C bis 150°C entspricht häufig recht genau dem Gehalt an „freiem“, d.h. chemisch nicht gebun-denem Wasser, s. Abb. (Nachteil: Neben Wasser können im betrachteten Temperatur-intervall auch weitere flüchtige Komponenten einen Gewichtsverlust hervorrufen).
b) Bestimmung von anderen leichtflüchtigen Komponenten, z.B. flüchtige Additive oder Monomere, s. Abb. (Nachteil: keine eindeutige Zuordnung möglich. Abhilfe: Kopplung der TGA mit FT-IR oder MS).
c) Bestimmung des Gehalts an anorganischen Füllkörpern: Die quantitative Bestimmung von Gehalten an Glas, Talkum und sonstigen mineralischen oder metallischen Füllkörpern ist durch das Niveau des nach Gewichtskonstanz bei hohen Temperaturen erreichten Endwerts zu ermitteln, s. Abb. (Nachteil: auch anorganische Materialien können bei hohen Temperaturen Masseverluste zeigen).
d) Beobachtung der Geschwindigkeit thermischer oder oxidativer Abbauprozesse.
Zur Aufnahme der gewöhnlich 1 bis 20 mg schweren TGA-Proben werden Pfännchen aus Platin oder Aluminiumoxid verwendet. Messungen erfolgen in einem inerten Gas (z.B. Stickstoff oder Helium) oder Reaktionsgas (z.B. Luft oder Sauerstoff) in einem Temperaturfenster zwischen Raumtemperatur und etwa 1200 °C.
Heizung Thermo-element
EDV
Temperatur-programm
Gewicht der Probe w(t)
Probe
elektromagnetische Kompensation der Auslenkung des Wägebalkens
T [°C]
w(t)
T [°C]
Start Ende
Tmin
Tmax
TmaxTmin
Startgewicht
T [°C]
w(%)
Tmax
100 Feuchtegehalt
Monomere, flüchtige Add.
Beispiel: TGA-Messung an gefülltem Polyamid 6,6
PA 6,6
100°C 400°C

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l31
Übungsblatt 9 1) Angenommen, Sie suchen ein möglichst schlagzähes Material, das im Alltag
auch bei tiefen Temperaturen keine Sprödigkeit aufweisen soll. Welches Poly-mer würden Sie bevorzugt einsetzen?
a. Ein Polymer mit einer hohen oder einer niedrigen Glasumwandlungs-temperatur?
b. Ein stark oder ein schwach vernetztes Polymer? c. Ein Polymer mit einer hohen oder einer niedrigen Reißdehnung?
2) Warum ist die kurz vor dem Bruch erreichte maximale Zugspannung meistens deutlich größer als die „Spannung bei Bruch“?
3) Worin besteht der Vorteil der Vierpunkt-Biegeprüfung gegenüber der Dreipunkt-Biegeprüfung?
4) Warum besitzen sowohl die maximale Zugspannung als auch der Zugmodul die physikalische Einheit des Drucks (Pascal)?
5) Für welche Anwendungen könnte der Unterschied zwischen Zugdehnung und Reißdehnung von größerer Bedeutung sein?
6) Warum besitzt der Zugprüfkörper im Allgemeinen die Form eines Quaders mit verdickten Enden („Knochen“)?
7) Warum erhält man aus dem Spannungs-Dehnungsdiagramm bei einem Biegeversuch stets einen Mittelwert aus Zug- und Druckmodul?
8) Warum kann man selbst bei ideal gummielastischen Polymeren kaum erwarten, dass der Prüfkörper im Zugversuch das Hooke’sche Gesetz fehlerfrei erfüllt?
9) Warum verwendet man im Zugversuch zur Messung der Dehnung ε einen Dehnungsmessstreifen, anstatt sich auf die Relativbewegung der Prüfbacken zu verlassen?
10) Welche Phänomene werden bei der TMA-Messung an einem Polymer im ersten Aufheizvorgang am Diagramm z gegen T erkennbar? Welches Phänomen beeinflusst die Messkurve bei den Wiederholungsmessungen? Wie bestimmen Sie den Längenausdehnungskoeffizienten α des Polymers?
11) Wie bestimmen Sie an einer Polymerprobe mit Hilfe einer TGA den Gehalt an anorganischen Füllstoffen? Welche Probleme können dabei auftreten?
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l32
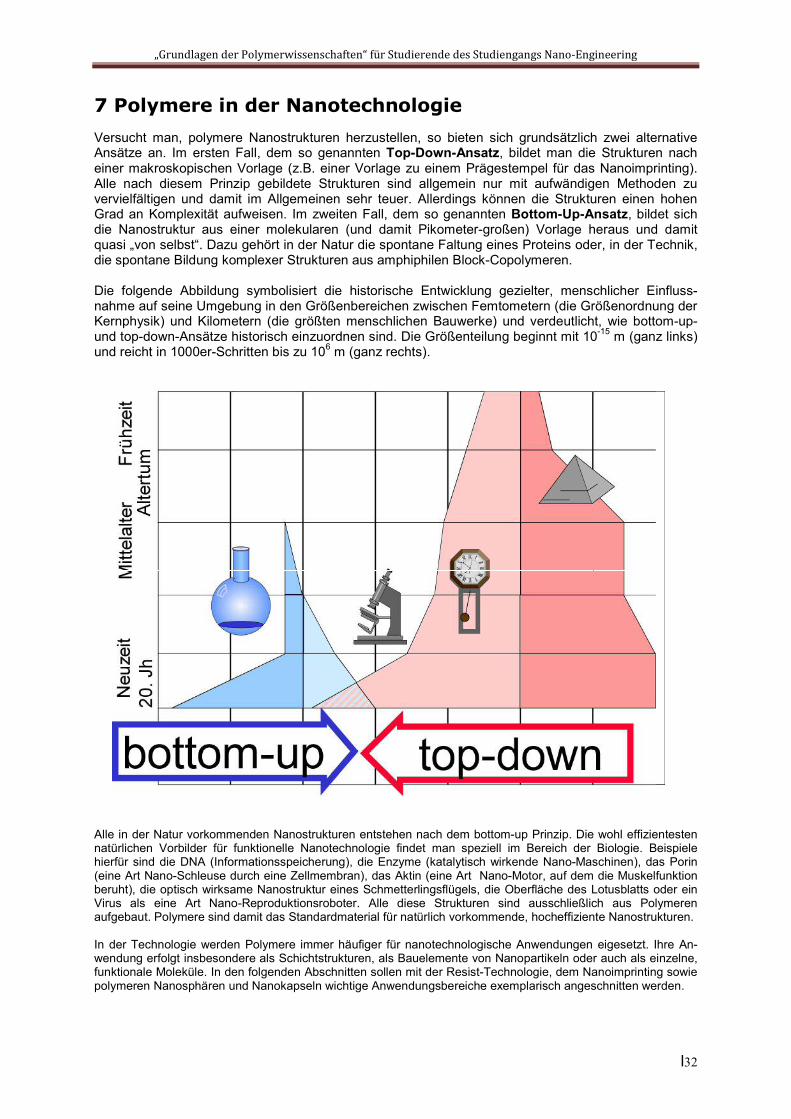
7 Polymere in der Nanotechnologie Versucht man, polymere Nanostrukturen herzustellen, so bieten sich grundsätzlich zwei alternative Ansätze an. Im ersten Fall, dem so genannten Top-Down-Ansatz, bildet man die Strukturen nach einer makroskopischen Vorlage (z.B. einer Vorlage zu einem Prägestempel für das Nanoimprinting). Alle nach diesem Prinzip gebildete Strukturen sind allgemein nur mit aufwändigen Methoden zu vervielfältigen und damit im Allgemeinen sehr teuer. Allerdings können die Strukturen einen hohen Grad an Komplexität aufweisen. Im zweiten Fall, dem so genannten Bottom-Up-Ansatz, bildet sich die Nanostruktur aus einer molekularen (und damit Pikometer-großen) Vorlage heraus und damit quasi „von selbst“. Dazu gehört in der Natur die spontane Faltung eines Proteins oder, in der Technik, die spontane Bildung komplexer Strukturen aus amphiphilen Block-Copolymeren.
Die folgende Abbildung symbolisiert die historische Entwicklung gezielter, menschlicher Einfluss-nahme auf seine Umgebung in den Größenbereichen zwischen Femtometern (die Größenordnung der Kernphysik) und Kilometern (die größten menschlichen Bauwerke) und verdeutlicht, wie bottom-up- und top-down-Ansätze historisch einzuordnen sind. Die Größenteilung beginnt mit 10-15 m (ganz links) und reicht in 1000er-Schritten bis zu 106 m (ganz rechts).
Alle in der Natur vorkommenden Nanostrukturen entstehen nach dem bottom-up Prinzip. Die wohl effizientesten natürlichen Vorbilder für funktionelle Nanotechnologie findet man speziell im Bereich der Biologie. Beispiele hierfür sind die DNA (Informationsspeicherung), die Enzyme (katalytisch wirkende Nano-Maschinen), das Porin (eine Art Nano-Schleuse durch eine Zellmembran), das Aktin (eine Art Nano-Motor, auf dem die Muskelfunktion beruht), die optisch wirksame Nanostruktur eines Schmetterlingsflügels, die Oberfläche des Lotusblatts oder ein Virus als eine Art Nano-Reproduktionsroboter. Alle diese Strukturen sind ausschließlich aus Polymeren aufgebaut. Polymere sind damit das Standardmaterial für natürlich vorkommende, hocheffiziente Nanostrukturen.
In der Technologie werden Polymere immer häufiger für nanotechnologische Anwendungen eigesetzt. Ihre An-wendung erfolgt insbesondere als Schichtstrukturen, als Bauelemente von Nanopartikeln oder auch als einzelne, funktionale Moleküle. In den folgenden Abschnitten sollen mit der Resist-Technologie, dem Nanoimprinting sowie polymeren Nanosphären und Nanokapseln wichtige Anwendungsbereiche exemplarisch angeschnitten werden.
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l33
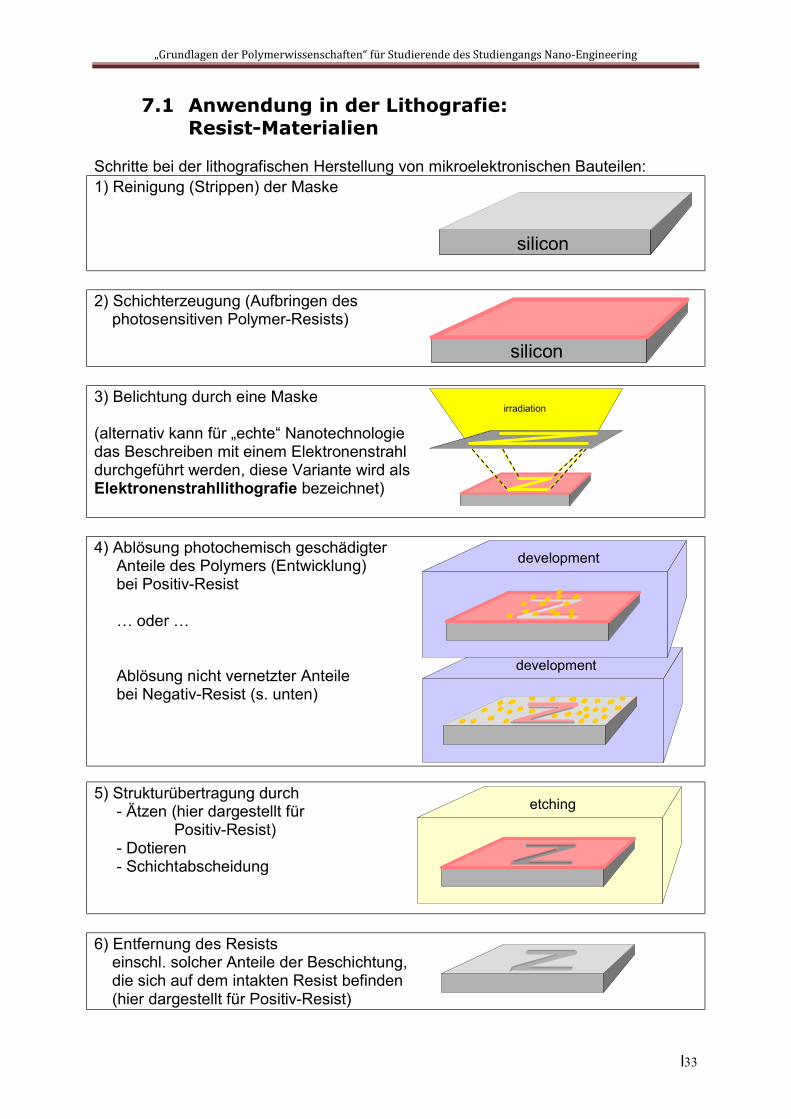
7.1 Anwendung in der Lithografie: Resist-Materialien
Schritte bei der lithografischen Herstellung von mikroelektronischen Bauteilen: 1) Reinigung (Strippen) der Maske
2) Schichterzeugung (Aufbringen des photosensitiven Polymer-Resists)
3) Belichtung durch eine Maske
(alternativ kann für „echte“ Nanotechnologie das Beschreiben mit einem Elektronenstrahl durchgeführt werden, diese Variante wird als Elektronenstrahllithografie bezeichnet)
4) Ablösung photochemisch geschädigter Anteile des Polymers (Entwicklung) bei Positiv-Resist
… oder …
Ablösung nicht vernetzter Anteile bei Negativ-Resist (s. unten)
5) Strukturübertragung durch - Ätzen (hier dargestellt für Positiv-Resist) - Dotieren - Schichtabscheidung
6) Entfernung des Resists einschl. solcher Anteile der Beschichtung, die sich auf dem intakten Resist befinden (hier dargestellt für Positiv-Resist)
silicon
silicon
irradiation
etching
development
development
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l34
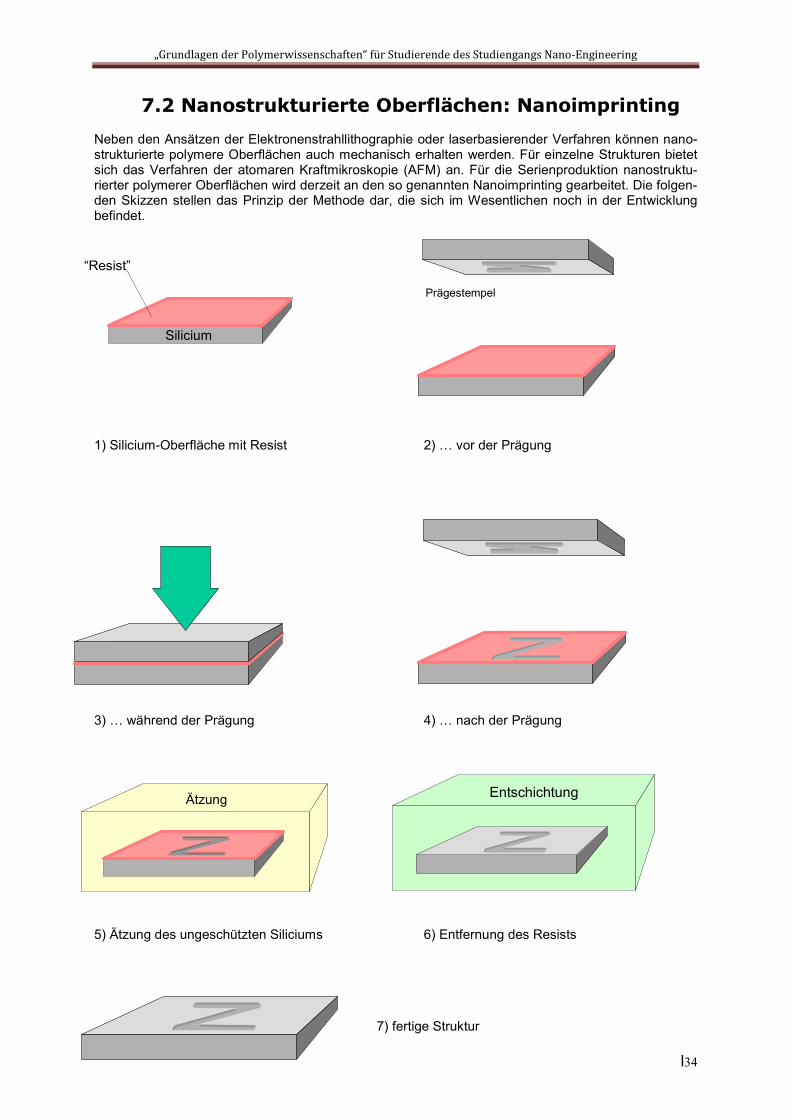
7.2 Nanostrukturierte Oberflächen: NanoimprintingNeben den Ansätzen der Elektronenstrahllithographie oder laserbasierender Verfahren können nano-strukturierte polymere Oberflächen auch mechanisch erhalten werden. Für einzelne Strukturen bietet sich das Verfahren der atomaren Kraftmikroskopie (AFM) an. Für die Serienproduktion nanostruktu-rierter polymerer Oberflächen wird derzeit an den so genannten Nanoimprinting gearbeitet. Die folgen-den Skizzen stellen das Prinzip der Methode dar, die sich im Wesentlichen noch in der Entwicklung befindet.
1) Silicium-Oberfläche mit Resist 2) … vor der Prägung
3) … während der Prägung 4) … nach der Prägung
5) Ätzung des ungeschützten Siliciums 6) Entfernung des Resists
7) fertige Struktur
Silicium
“Resist”
Prägestempel
Ätzung Entschichtung

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l35
7.3 Spontane Strukturbildungen aus amphiphilen Polymeren
Die für die Nanotechnologie interessanteste Eigenschaft von Makromolekülen ist ihre Fähigkeit, sich unter bestimmten Voraussetzungen selbsttätig zu einer definierten Nanometer-großen Überstruktur zu falten. Dies eröffnet die Möglichkeit, Nanostrukturen im bottom-up-Ansatz herzustellen. Das Vorbild sind in dieser Hinsicht Biomoleküle wir die DNA und die Polysaccharide, vor allem aber die Proteine. Diese Polymere, die als lange, Polyamid-ähnliche Ketten eine komplexe Folge von 22 verschiedenen Aminosäuren aufweisen, bilden in wässriger Umgebung spontan nanostrukturierte Knäuel aus. Diese Strukturen sind in der Lage, als Katalysatoren (Enzyme), Membranporen, Aktuatoren, Informations-speicher (molekulares Gedächtnis) oder als dreidimensionale Bauelemente zu fungieren. Der „Bauplan“ für alle diese funktionalen Strukturen liegt einzig und allein in der Folge (Sequenz) der Aminosäuren begründet. Die treibende Kraft hinter der Strukturbildung ist ein Wechsel von hydrophilen und hydrophoben Eigenschaften innerhalb der Polymerkette und die wässrige Umgebung. Das Makromolekül „versucht“ dabei, soviel hydrophile Kettenanteile wir möglich nach außen (zum wässrigen Medium hin) zu kehren und gleichzeitig so viel hydrophobe Kettenanteile wie möglich im Inneren des Knäuels zu verstecken. (s. http://www.youtube.com/watch?v=fvBO3TqJ6FE&feature=related ). Die energetisch günstigste Form des Knäuels ist dann die angestrebte funktionelle Nanostruktur. Daneben ist die Ausbildung von Wasserstoffbrücken und Disulfidbrücken für die Stabilität der entstandenen Struktur entscheidend.
Die ersten technischen Ansätze, dieses Prinzip nachzuahmen, sind bisher ungleich primitiver. Sie bestehen in der Synthese amphiphiler (also gleichzeitig hydrophiler und hydrophober) Block-Copolymere mit einer definierten Folge von hydrophoben und hydrophilen Blöcken. Ähnlich einem Protein faltet sich ein solches Molekül in wässriger Phase derart, dass die hydrophilen Teile A nach außen und die hydrophoben Teile B nach innen weisen. Ein BAB-Block-Copolymer bildet somit in geringer Konzentration Haarnadel-artige Strukturen aus. Bei einfachen AB-Block-Copolymeren kommt es unter bestimmten Bedingungen zur Bildung von kugelförmigen Strukturen (s. folgende Abbildung), bei denen alle hydrophoben B-Enden (blau) zusammen einen kugelförmigen Kern aufbauen, während die hydrophilen A-Segmente (rot) nach außen weisen (Mizellen). Alternativ können unter anderen Randbedingungen auch Doppelschichten entstehen, bei denen zwei innere Lagen aus B-Segmenten mit zwei äußeren Lagen aus A-Segmenten eine Art Sandwich bilden. Alle diese Nanostrukturen entstehen nach einem bottom-up-Ansatz und können deshalb sehr preiswert in großen Mengen hergestellt werden.
Mizellen Doppelschichten
Neben der Strukturbildung aus amphiphilen Molekülen sind auch spontan gebildete Strukturen aus Makromolekülen mit selektiven Bindungspositionen. So könnte beispielsweise eine Kette an einer Position drei Wasserstoffbrücken-Donoren in einem Abstand von jeweils 0,5 nm aufweisen, eine andere dagegen drei Wasserstoffbrücken-Akzeptoren in einem Abstand von ebenfalls jeweils 0,5 nm. Begegnen sich diese beiden Moleküle dann an der entscheidenden Stelle, so kommt es zur Ausbildung von drei Wasserstoffbrücken und damit zur Entstehung eines insgesamt recht stabilen Haftpunkts. Plant man bei der Synthese der Makromoleküle an definierten Stellen solche Haftpunkte ein, so können im bottom-up-Ansatz sehr komplexe Nanostrukturen ausgebildet werden. Vorbild für diese Art der spontanen Strukturbildung ist besonders das Molekül der genetischen Informations-speicherung, die Desoxyribonucleinsäure (DNA).
„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l36
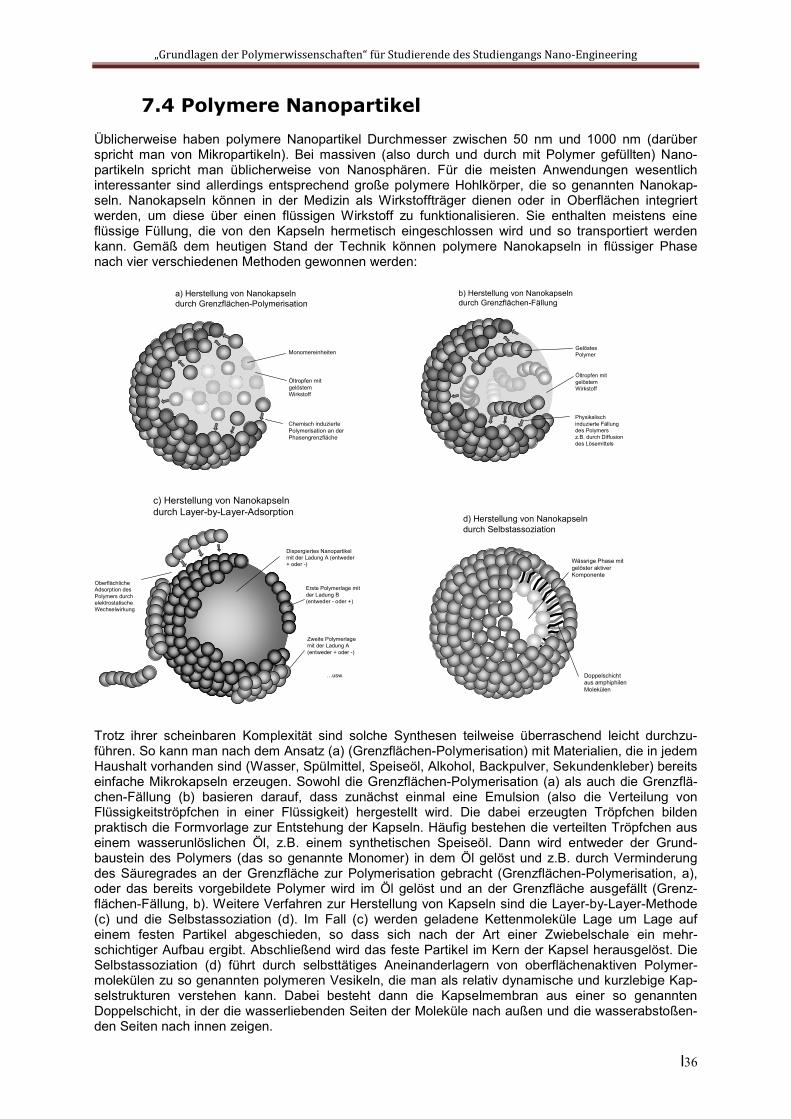
7.4 Polymere NanopartikelÜblicherweise haben polymere Nanopartikel Durchmesser zwischen 50 nm und 1000 nm (darüber spricht man von Mikropartikeln). Bei massiven (also durch und durch mit Polymer gefüllten) Nano-partikeln spricht man üblicherweise von Nanosphären. Für die meisten Anwendungen wesentlich interessanter sind allerdings entsprechend große polymere Hohlkörper, die so genannten Nanokap-seln. Nanokapseln können in der Medizin als Wirkstoffträger dienen oder in Oberflächen integriert werden, um diese über einen flüssigen Wirkstoff zu funktionalisieren. Sie enthalten meistens eine flüssige Füllung, die von den Kapseln hermetisch eingeschlossen wird und so transportiert werden kann. Gemäß dem heutigen Stand der Technik können polymere Nanokapseln in flüssiger Phase nach vier verschiedenen Methoden gewonnen werden:
Trotz ihrer scheinbaren Komplexität sind solche Synthesen teilweise überraschend leicht durchzu-führen. So kann man nach dem Ansatz (a) (Grenzflächen-Polymerisation) mit Materialien, die in jedem Haushalt vorhanden sind (Wasser, Spülmittel, Speiseöl, Alkohol, Backpulver, Sekundenkleber) bereits einfache Mikrokapseln erzeugen. Sowohl die Grenzflächen-Polymerisation (a) als auch die Grenzflä-chen-Fällung (b) basieren darauf, dass zunächst einmal eine Emulsion (also die Verteilung von Flüssigkeitströpfchen in einer Flüssigkeit) hergestellt wird. Die dabei erzeugten Tröpfchen bilden praktisch die Formvorlage zur Entstehung der Kapseln. Häufig bestehen die verteilten Tröpfchen aus einem wasserunlöslichen Öl, z.B. einem synthetischen Speiseöl. Dann wird entweder der Grund-baustein des Polymers (das so genannte Monomer) in dem Öl gelöst und z.B. durch Verminderung des Säuregrades an der Grenzfläche zur Polymerisation gebracht (Grenzflächen-Polymerisation, a), oder das bereits vorgebildete Polymer wird im Öl gelöst und an der Grenzfläche ausgefällt (Grenz-flächen-Fällung, b). Weitere Verfahren zur Herstellung von Kapseln sind die Layer-by-Layer-Methode (c) und die Selbstassoziation (d). Im Fall (c) werden geladene Kettenmoleküle Lage um Lage auf einem festen Partikel abgeschieden, so dass sich nach der Art einer Zwiebelschale ein mehr-schichtiger Aufbau ergibt. Abschließend wird das feste Partikel im Kern der Kapsel herausgelöst. Die Selbstassoziation (d) führt durch selbsttätiges Aneinanderlagern von oberflächenaktiven Polymer-molekülen zu so genannten polymeren Vesikeln, die man als relativ dynamische und kurzlebige Kap-selstrukturen verstehen kann. Dabei besteht dann die Kapselmembran aus einer so genannten Doppelschicht, in der die wasserliebenden Seiten der Moleküle nach außen und die wasserabstoßen-den Seiten nach innen zeigen.
10 Nanometer
Monomereinheiten
Chemisch induziertePolymerisation an derPhasengrenzfläche
Öltropfen mit gelöstem Wirkstoff
a) Herstellung von Nanokapseln durch Grenzflächen-Polymerisation
b) Herstellung von Nanokapseln durch Grenzflächen-Fällung
10 Nanometer
Gelöstes Polymer
Physikalischinduzierte Fällungdes Polymersz.B. durch Diffusion des Lösemittels
Öltropfen mit gelöstem Wirkstoff
c) Herstellung von Nanokapseln durch Layer-by-Layer-Adsorption
10 Nanometer
Dispergiertes Nanopartikel mit der Ladung A (entweder + oder -)
Oberflächliche Adsorption des Polymers durch elektrostatische Wechselwirkung
Erste Polymerlage mit der Ladung B (entweder - oder +)
Zweite Polymerlage mit der Ladung A (entweder + oder -)
…usw.
d) Herstellung von Nanokapseln durch Selbstassoziation
10 Nanometer Doppelschicht aus amphiphilen Molekülen
Wässrige Phase mit gelöster aktiver Komponente

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l37
Ein nicht unerhebliches Problem ist die Strukturaufklärung an solchen Nanokapseln. Die Rasterelek-tronenmikroskopie führt in vielen Fällen nur zu sehr unbefriedigenden Ergebnissen, da sich die oberflächenaktive Substanz wie ein Schleier über die Kapseln legt. Etwas besser gelingt dies mit der so genannten Kryo-Transmissions-Elektronenmikroskopie (kryo-TEM), mit deren Hilfe zumindest der Kern-Schale-Aufbau der Kapseln sichtbar gemacht werden kann. Auch diese Methode lässt jedoch klare Aussagen zum Beispiel zur Beschaffenheit von Hülle und Kern der Kapseln vermissen. Der Nachweis der Kapselstruktur gelingt am besten mit einer relativ jungen Methode, die als Atomare Kraftmikroskopie (oder Atomic Force Microscopy, kurz: AFM) Einzug in das Repertoire der Nano-technologie genommen hat. Hierbei wird die Form der Kapsel mit einer Art nadelfeiner Spitze in einem feinen Raster systematisch abgetastet, so dass mit Hilfe eines Computers ein mechanisch erzeugtes Abbild der Struktur entsteht (folgende Abb. links).
Abbilder einer einzelnen Nanokapsel, die mit Hilfe eines Atomaren Kraftmikroskops (AFM) erzeugt wurden. Das linke Bild zeigt die ursprüngliche Kapsel, das mittlere und rechte Bild dieselbe Kapsel nach einem bzw. zwei Eindrückversuchen.
Mittels der Messspitze gelingt es auch, die Kapsel an einer gewünschten Stelle gezielt einzudrücken und deren Festigkeit zu bestimmen. Man erhält dabei eine Kraft-Weg-Kurve nach Art einer Spannungs-Dehnungs-Kurve (s. Abschnitt 6.3), deren Verlauf Rückschlüsse auf die Flexibilität der Kapselhülle zulässt. So verhalten sich viele Kapseln ähnlich wie beispielsweise ein Tischtennisball: sie leisten viel Widerstand, solange die Hülle an der Eindrückstelle noch nach außen gewölbt ist. Sobald die Kapselmembran die erste Vertiefung aufweist, ist der Widerstand gegen weiteres Eindrücken deutlich geringer. Die letzte Abbildung (Mitte und rechts) zeigt die Form einer Kapsel nach einem bzw. nach zwei solchen Eindrückversuchen. Die im Eindrückversuch bewirkte Verformung ist permanent und lässt Rückschlüsse auf die Flexibilität und die Elastizität der Kapselwand zu. Insbesondere kann sie auch als Beleg dafür herhalten, dass es sich tatsächlich um einen Hohlkörper mit einer flexiblen Wand und einem flüssigen Inhalt handelt. Tritt bei einem solchen Versuch ein Teil des flüssigen Inhalts aus, so führt der Volumenverlust bei gleichzeitigem Erhalt der Oberfläche zu einer typischen Faltung der Kapselhülle, die analytisch ausgewertet werden kann (Abbildung rechts).
7.5 Technische Anwendung biologischer PolymereDie wesentlichen biologisch relevanten Vorbilder für die Nanotechnologie wurden bereits zu Anfang des Kapitels kurz erwähnt. Viele Ansätze in der Nanotechnologie zielen darauf ab, die Funktionen dieser hocheffizienten natürlichen Systeme unter Verwendung technisch verfügbarer Materialien nachzubilden. Ein alternatives Konzept, über das gegenwärtig an verschiedenen Stellen nachgedacht wird, sieht allerdings auch vor, die biologischen Systeme direkt zu nutzen und lediglich zu modifizieren um sie für technische Zwecke einzusetzen. Deshalb sollen Beispiele für funktionelle biologische Nanostrukturen, die bereits angewandt werden oder ein gewisses Potential für zukünftige technische Anwendungen bieten könnten, im Folgenden kurz vorgestellt werden.
Desoxyribonucleinsäure (DNA): Die DNA ist das wichtigste Molekül zur Informationsspeicherung in biologischen Systemen. Sie kommt in allen bekannten lebenden Wesen vor und besitzt die Struktur einer schraubenförmig gewundenen Leiter. Die durchgehenden „Holme“ werden dabei durch abwechselnd auftretende Ribose- und Phosphateinheiten gebildet. Die „Sprossen“ der Leiter setzen an je zwei gegenüberliegenden Ribose-einheiten der an und bestehen aus den so genannten Basenpaaren. Die Reihenfolge der vier ver-schiedenen vorkommenden Basen Adenin, Guanin, Thymin und Cytosin (A, G, T und C) bilden den Code für die Erbinformation jeder Zelle. In jeder „Leitersprosse“ paaren sich die Basen A und T bzw. G und C, so dass bei einer mittigen Trennung aller Sprossen zwei Teilstränge mit komplementärer

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l38
Information entstehen. Durch neue Basenpaarbildung kann so die gespeicherte Information kopiert oder abgelesen werden.
Theoretisch ließe sich die Selektivität der Anbindung an Basenpaargruppen nutzen, um nanoskopi-sche Bauteile zu adressieren und spontan komplexe Schaltungen aufzubauen. Allerdings hat dieser Ansatz die ursprünglichen Erwartungen bisher nicht erfüllt.
Eine Blütezeit erlebt dagegen in jüngerer Zeit das so genannte DNA Origami, das von Paul Rothemund an der CalTech entwickelt wurde. Hierzu wird eine biologisch aus Viren oder Bakterien gewonnene DNA-Kette mit bekannter Sequenz eingesetzt. Diese wird mit kurzen synthetischen Gegenstücken versetzt, die an vorbestimmten Stellen anbinden und innerhalb der Kette Verknüpfun-gen erzeugen. Die ursprüngliche DNA wird dadurch in genau vorbestimmter Weise gefaltet (daher der Name Origami = japanische Faltungskunst), so dass dreidimensionale Faltungsfiguren mit fast belie-biger Form entstehen. Diese können durch weitere synthetische Gegenstücke auch noch miteinander verknüpft werden, so dass größere Nanostrukturen gebildet werden. Auch Kombinationen mit Nanopartikeln sind möglich. Beispiele für DANN-Origami-Strukturen:
Katalytisch wirkende Proteine (Enzyme): Der allergrößte Teil aller biochemischen Reaktionen wird durch katalytisch wirkende Proteine (Enzyme) unterstützt. Viele dieser Enzyme besitzen sehr komplexe Funktionsmechanismen, so dass man durchaus von kleinen Molekül-Manipulatoren sprechen könnte. Die Funktion beruht wesentlich auf der dreidimensionalen Struktur des Proteins, die wiederum wesentlich von der amphiphilen Struktur der Polymerkette gebildet und durch Wasserstoffbrücken und Schwefelbrücken fixiert wird. Natürliche Enzyme werden bereits vielfach technologisch genutzt, wobei man diese sowohl in freier Form (Waschmittel) als auch oberflächlich gebunden einsetzt.
Aktin-Myosin-Komplex: ein nanoskopischer Bewegungsapparat: In einem komplexen Wechselspiel zwischen den Proteinen Aktin und Myosin wird ein Bewegungs-vorgang induziert, der dem Wechselspiel zwischen einem Hebel und einer Zahnstange ähnelt. Auf dieser Grundlage beruht die Funktion des Muskelgewebes (für eine Animation des Bewegungs-vorgangs siehe http://www.youtube.com/watch?v=gJ309LfHQ3M ). Prinzipiell könnte man solche und ähnliche Prozesse einsetzen, um „Nanoroboter“ anzutreiben und ihnen funktionelle Bewegungsprozesse zu ermöglichen.

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l39
Übungsblatt 10
1) Beschreiben Sie die grundsätzlichen Prinzipien des Top-Down-Ansatzes sowie des Bottom-Up-Ansatzes. Erläutern Sie die mit diesen Vorgehens-weisen grundsätzlich verbundenen Vor- und Nachteile. Nennen Sie jeweils ein Beispiel.
2) Warum bereitet die optische Lithografie im Größenbereich der Nanotechno-logie grundsätzliche Probleme? Wie können diese Probleme durch einen geänderten physikalischen Ansatz gelöst werden?
3) Warum ist bei der Strukturbildung im Nanometerbereich die Methode des Nanoimprintings möglicherweise der Lithografie überlegen?
4) Ordnen Sie alle Ihnen bekannten Verfahren der Nanotechnologie den beiden Sparten Top-Down-Ansatz und Bottom-Up-Ansatz zu.
5) Warum besitzt die Technik der Atomaren Kraftmikroskopie neben ihrem Potential in der Analytik nanoskopischer Strukturen auch Anwendung bei der Präparation in der Nanotechnologie? Beschreiben Sie eine mögliche Vorgehensweise.
6) Erläutern Sie das Verfahren der Grenzflächen-Polymerisation, das zur Bildung von Nanokapseln führt. Welche Voraussetzungen muss das verwendete Monomer erfüllen?
7) Welche potentiellen Anwendungen besitzen polymere Nanokapseln?
8) Nennen Sie ein Beispiel für eine biologische Nanostruktur und erläutern Sie, inwiefern diese als Vorbild für technische Anwendungen dienen kann.
10 Nanometer
Monomereinheiten
Chemisch induziertePolymerisation an derPhasengrenzfläche
Öltropfen mit gelöstem Wirkstoff
a) Herstellung von Nanokapseln durch Grenzflächen-Polymerisation

„GrundlagenderPolymerwissenschaften“fürStudierendedesStudiengangsNano-Engineering
l40
Übungsblatt 10, Lösungen
1) Im Fall des Top-Down-Ansatzes bildet man die Strukturen nach einer makros-kopischen Vorlage (z.B. einer Vorlage zu einem Prägestempel für das Nano-imprinting). Alle nach diesem Prinzip gebildete Strukturen sind allgemein nur mit aufwändigen Methoden zu vervielfältigen und damit gewöhnlich sehr teuer. Allerdings können die Strukturen einen hohen Grad an Komplexität aufweisen. Im Fall des Bottom-Up-Ansatzes bildet sich die Nanostruktur aus einer molekularen (und damit Pikometer-großen) Vorlage heraus und damit quasi „von selbst“. Dazu gehört in der Natur die spontane Faltung eines Proteins oder, in der Technik, die spontane Bildung komplexer Strukturen aus amphiphilen Block-Copolymeren oder das DNA-Origami.
2) a) Die Wellenlänge des zur Lithografie verwendeten Lichts beträgt um 250 nm. Feinere Strukturen (z.B. im 10 nm-Bereich) sind daher kaum zu realisieren. b) Eine denkbare Lösung besteht in der mechanischen Prägung der Strukturen in das Resist (Nanoimprinting)
3) Bei der mechanischen Prägung spielt die Wellenlänge des Lichts keine Rolle. Die Grenzen bestehen lediglich in der mechanischen Strukturübertragung und liegen im Bereich der molekularen Faltung der Polymere, also bei etwa 10 nm.
4) Top-Down-Ansätze: Lithografie, Nanoimprinting, AFM Bottom-Up-Ansätze: Nanokapselbildung, spontane Strukturbildung mit Amphiphilen, DNA-Origami.
5) Mittels AFM lassen sich durch gezielte Manipulation von Oberflächen einzelne Strukturen im top-down-Ansatz herstellen. Zum Beispiel können einzelne Nanopartikel zu einem Muster arrangiert oder weiche Oberflächen geritzt werden.
6) a) Zunächst wird eine Emulsion (also die Verteilung von Flüssigkeitströpfchen in einer Flüssigkeit) hergestellt. Die dabei erzeugten Tröpfchen bilden nun die Formvorlage zur Entstehung der Kapseln. Die verteilten Tröpfchen bestehen häufig aus einem wasserunlöslichen Öl, z.B. einem synthetischen Speiseöl. Dann wird der Grundbaustein des Polymers (das betreffende Monomer) in dem Öl gelöst und z.B. durch Verminderung des Säuregrades an der Grenz-fläche zur Polymerisation gebracht. Das Öl wird dabei eingeschlossen. b) Das Monomer muss sich bezüglich seines Polymerisationsverhaltens gut steuern lassen, z.B. durch Veränderung des pH-Werts. Am besten eignen sich die Vorgänge der anionischen oder der kationischen Polymerisation.
7) Nanokapseln können in der Medizin als Wirkstoffträger dienen oder in Oberflächen integriert werden, um diese über einen flüssigen Wirkstoff zu funktionalisieren.
8) Beispiel: Der Aktin-Myosin-Komplex als ein nanoskopischer Bewegungs-apparat. Prinzipiell könnte man ihn einsetzen, um „Nanoroboter“ anzutreiben und ihnen funktionelle Bewegungsprozesse zu ermöglichen.