Wie funktioniert die Gaschromatographie und was kann man ... · • Gas (GC), Flüssigkeit (LC)...
86
W. Engewald Universität Leipzig Institut für Analytische Chemie Wie funktioniert die Gaschromatographie und was kann man damit machen? Hochschule Zittau (FH) 17.11.2014
-
Upload
duongthien -
Category
Documents
-
view
214 -
download
0
Transcript of Wie funktioniert die Gaschromatographie und was kann man ... · • Gas (GC), Flüssigkeit (LC)...

W. EngewaldUniversität LeipzigInstitut für Analytische Chemie
Wie funktioniert die Gaschromatographie und was kann man damit machen?
Hochschule Zittau (FH)
17.11.2014

Anfänge der modernen ChromatographieMichail (Mikhail) Semenovich Tswett (1872-1919)
Russischer Botaniker und BiochemikerEigenschaften und Zusammensetzung von Chlorophyll
M.S. Tswett, (1906) Ber. Deut. Botan. Ges. 24: 319.
„Wird eine petrolätherische Chlorophylllösung durch eine Säuleeines Adsorptionsmittels durchfiltriert (ich verwendehauptsächlich Calciumcarbonat, welches in engen Glasröhrendicht gestampft wird), so werden die Farbstoffe gemäß derAdsorptionsreihe von oben nach unten in verschieden gefärbtenZonen auseinandergelegt, indem die stärker adsorbiertenFarbstoffe die schwächer zurückgehaltenen weiter nach untenverdrängen. Diese Trennung wird praktisch vollständig, wennman nach dem Durchgange der Farbstofflösung durch dieadsorbierende Säule einen Strom des reinen Lösungsmittelsherstellt. Wie die Lichtstrahlen im Spektrum, so werden in derCalciumcarbonatsäule die verschiedenen Komponenten einesFarbstoffgemisches gesetzmäßig auseinandergelegt, und lassensich darin qualitativ und auch quantitativ bestimmen. Ein solchesPräparat nenne ich ein Chromatogramm und die entsprechendeMethode chromatographische Methode”
Chromatographie-Apparatur von Tswett (1906)

Mikhail Semenovich TSWETT I
1872 – 1919Botaniker, Biochemiker
Studium in Lausanne und Genf : Biologie, Chemie, Physik1896 Promotion an der Universität Genfmit einer Arbeit zur Zell-Physiologie
1896 Rückkehr der Familie nach Russland:→ die in der Schweiz erworbenen
akademischen Grade werden nicht anerkannt
1901 St. Petersburg : 2. Masterarbeit(über Chlorophyll)
1901-1915 Warschau (Universität, Poly-technisches Institut)
Arbeiten über Farbpigmente1910 2. Doktorarbeit (in Kazan eingereicht)
Buch
1915 Evakuierung vor deutschen Truppen

Grundbegriffe der Chromatographie
IUPAC - Definition:Chromatographie ist ein physikalisch-chemisches Trennverfahren, bei dem die
zu trennenden Substanzen zwischen zwei Phasen verteilt werden, von denen eine, die stationäre Phase, festliegt, während die andere, die mobile Phase, sich in einer bestimmten Richtung bewegt.
Stationäre Phase• bewirkt Verzögerung (Retention) der Komponenten• Feststoff oder Flüssigkeit (mit großer Oberfläche)
Mobile Phase• bewirkt Stofftransport• Gas (GC), Flüssigkeit (LC) oder überkritisches Fluid (SFC)• Transport durch Druck, Schwerkraft oder Kapillarkräfte
Detektor-signal
t
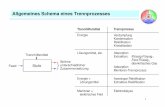
Klassifizierung chromatographischer MethodenAggregatzustand der mobilen Phase (Hauptkriterium)
Trennmechanismus (Separationsfunktion)• Adsorption• Verteilung• Ionenaustausch • Selektive Bindungen (Affinitäts-Chromatographie)• Ausschlusseffekte
Geometrie der Trennanordnung• Säule: Säulenchromatographie (gepackte Säule, Kapillarsäule)• Schicht auf ebener Unterlage: Planarchromatographie
(Dünnschichtchromatographie, Papierchromatographie)
Technik der ChromatogrammentwicklungElutions-, Frontal- oder Verdrängungstechnik
Mobile Phase Stationäre Phase Methode
Gasförmig Flüssig (GLC) – Verteilung
Fest (GSC) -Adsorption
Gaschromatographie
Flüssig Flüssig (LLC) Fest (LSC) Flüssigchromatographie

Gas Versorgung: hochreines Trägergas (He, H2, N2)Injektor: Probenaufgabe (gas, flüssig): mittels Mikroliterspritzen, Autosampler, GasventileTrennsäule: gepackte Säule oder KapillarsäuleSäulenofen: Thermostat ( 30 – 350 °C )Detektor: Universal oder elementspezifischDatenaufnahmesystem
Aufbau eines Gaschromatographen

Trennsäulen
• Innendurchmesser von 2 bis 4 mm • Säule ist vollständig mit Partikel gefüllt
Adsorbens → GSCTrägermaterial mit stationäre Phase (Flüssigkeit) belegt → GLC
•Säulenlänge nur 1 bis 6 m (Strömungs-widerstand)•Säulenmaterial: Kupfer, Stahl, Glas oder Quarzglas
Gepackte Säulen
• Innendurchmesser <1 mm • Stationäre Phase befindet sich auf der Innenwand der Trennsäule•Säulenlänge: 5 bis 100 m•Säulenmaterial: Glas (zerbrechlich), Fused silica (Quarz) aus hochreinem SiO2 mit äußerer Schutzschicht aus Polyimid (flexibel), fused silica beschichteter Edelstahl (hochtemperaturstabil)
Kapillarsäulen
2.50 3.00 3.50 4.00 4.50 5.00 5.50 6.00
50000100000150000200000250000300000350000400000450000500000550000600000650000700000750000800000
Zeit (min) →
Abun
danc
e
C 1
1:0
C 1
2:0 C
13:
0 C 1
4:0
C 1
5:0
C 1
6:0
C 1
7:0
C 1
8:0;
Kp
337°
C
C 2
0:0;
Kp
363°
C
C 2
2:0;
Kp
387°
C
C 1
4:1
C 1
5:1
C 1
6:1
C 1
7:1 C
18:
1
C 1
8:2
C 1
8:3C
18:
3C 1
8:1
C 2
0:1
C 2
0:2
C 2
0:3 C
20:
4
C 2
2:1
C 2
2:2
C 2
4:1
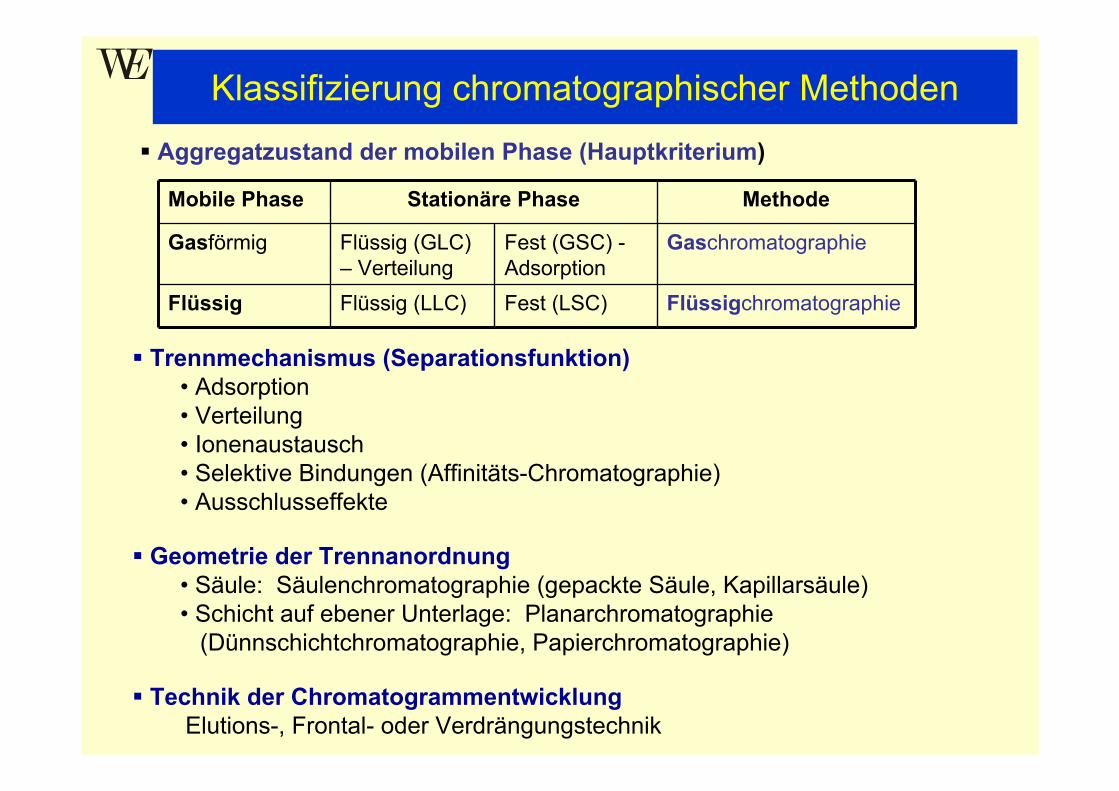
Injektor: KAS 4; 280°CTrägergas: Helium; konst. Fluß; 1,3mL/minSplit: 1:358Säule: high polarity cyanophase (10m x 0,1mm x 0,1µm)Temp.: 40°C; 43°C/min → 185°C; 9,5°C/min → 215°CDetektion: MS (35 - 350amu); TIC
Fettsäuremethylester-Standardgemisch

Liquid – Liquid Verteilung
Verteilung von Substanzen zwischen 2 nicht mischbaren Flüssigkeiten (Wasser / organ.Lösungsmittel) nach ihrer LöslichkeitGrundlage: Nernstscher Verteilungssatz:
W. Nernst, 1891:“Löst sich eine Substanz in zwei verschiedenen, nicht mischbaren Flüssigkeiten,so verteilt sie sich derart, daß das Verhältnis der Konzentrationen konstant ist.”
II
I
Ii
IIi
IIi
IIIIi
Ii
IIiD V
Vmm
VmVm
cc
K ⋅===)(
)(
)(
)(
)(
)(
//
KD Verteilungskoeffizient (Verteilungskonst.) für den Stoff Ici(I), ci(II) Konzentration des Stoffes I in Phase I bzw. IImi(I), mi(II) Menge des Stoffes in Phase I bzw. IIVI, VII Volumen der Phase I oder IIVI / VII Volumenverhältnis der Phasen (Phasenverhältnis β)
Wird die Konzentration in einer Phase verändert, so stellt sich das Gleichgewicht gemäß KD neu ein.
Gültigkeit des Nernstschen Verteilungssatzes:- bei geringen Konzentrationen- wenn die Stoffe in beiden Phasen nur in der gleichen Molekülart vorliegen
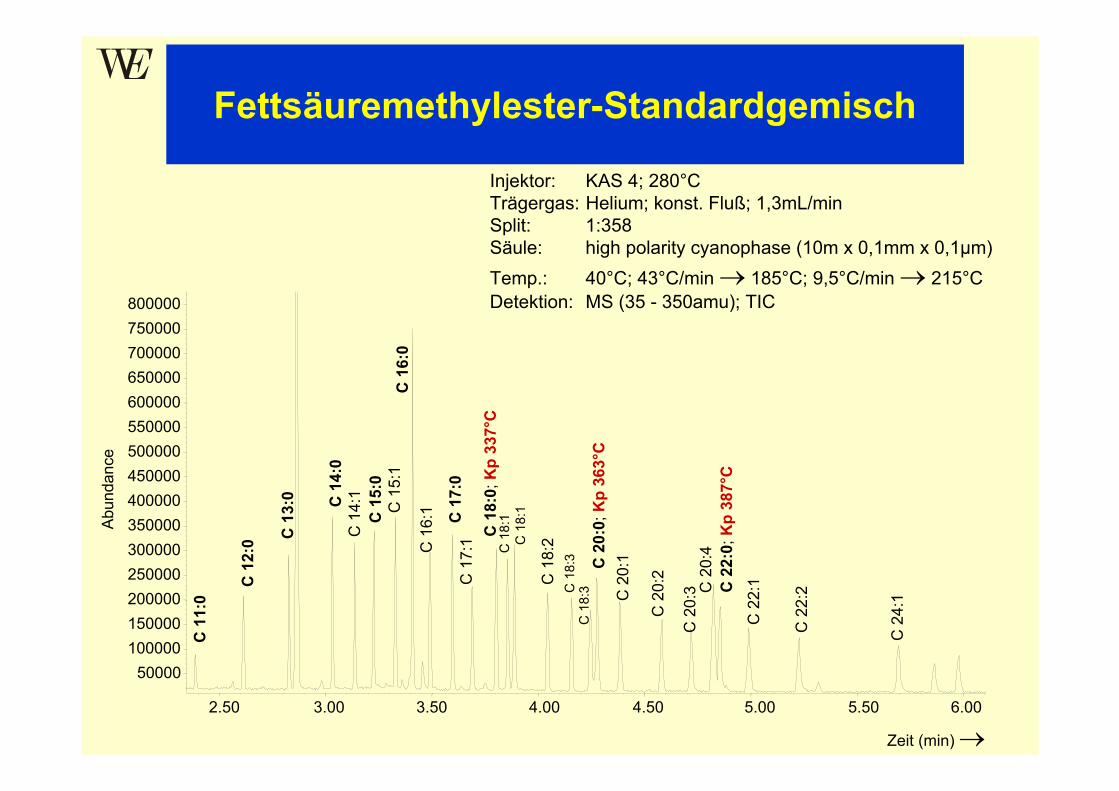
Prinzip der multiplikativen VerteilungTrennung zweier Verbindungen mit Verteilungskoeffizient K1 = 2,0 und K2 = 1,0
Modell:Betrachtung des kontinuierlichen Trennvorgangs als Folge vonEinzelschritten
Zerlegung der Trennsäule in Segmente (Trennstufen, theoretische Böden),in denen jeweils eine Gleichgewichtseinstellung zwischen stationärer undmobiler Phase erfolgen soll:
mobile Phasestationäre Phase
Trennstufe (theoretischer Boden)Volumenverhältnis der Phasen β = VM / VS = 1
Verteilungskoeffizient K = cstat / cmob
K1 = 2,0 = 2:1 α = K1 / K2 = 2K2 = 1,0 = 1:1
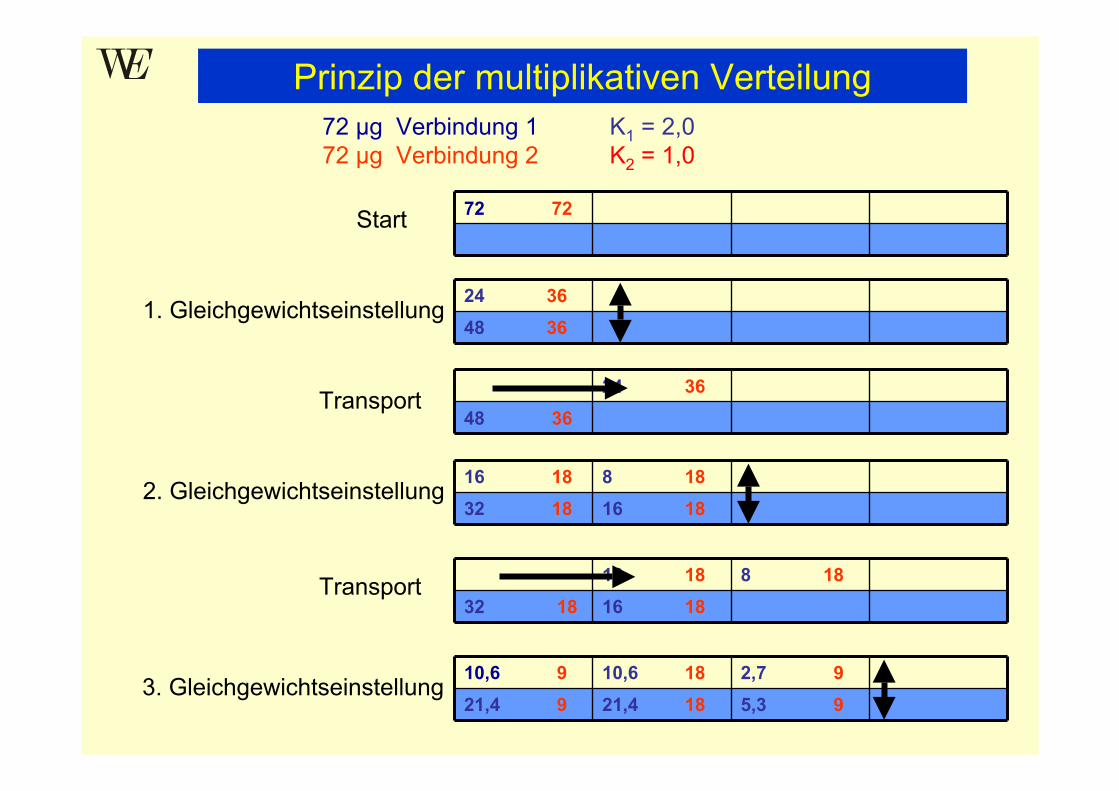
Prinzip der multiplikativen Verteilung72 µg Verbindung 1 K1 = 2,072 µg Verbindung 2 K2 = 1,0
Start 72 72
1. Gleichgewichtseinstellung
Transport
2. Gleichgewichtseinstellung
Transport
3. Gleichgewichtseinstellung10,6 9 10,6 18 2,7 921,4 9 21,4 18 5,3 9
16 18 8 1832 18 16 18
16 18 8 1832 18 16 18
24 3648 36
24 3648 36
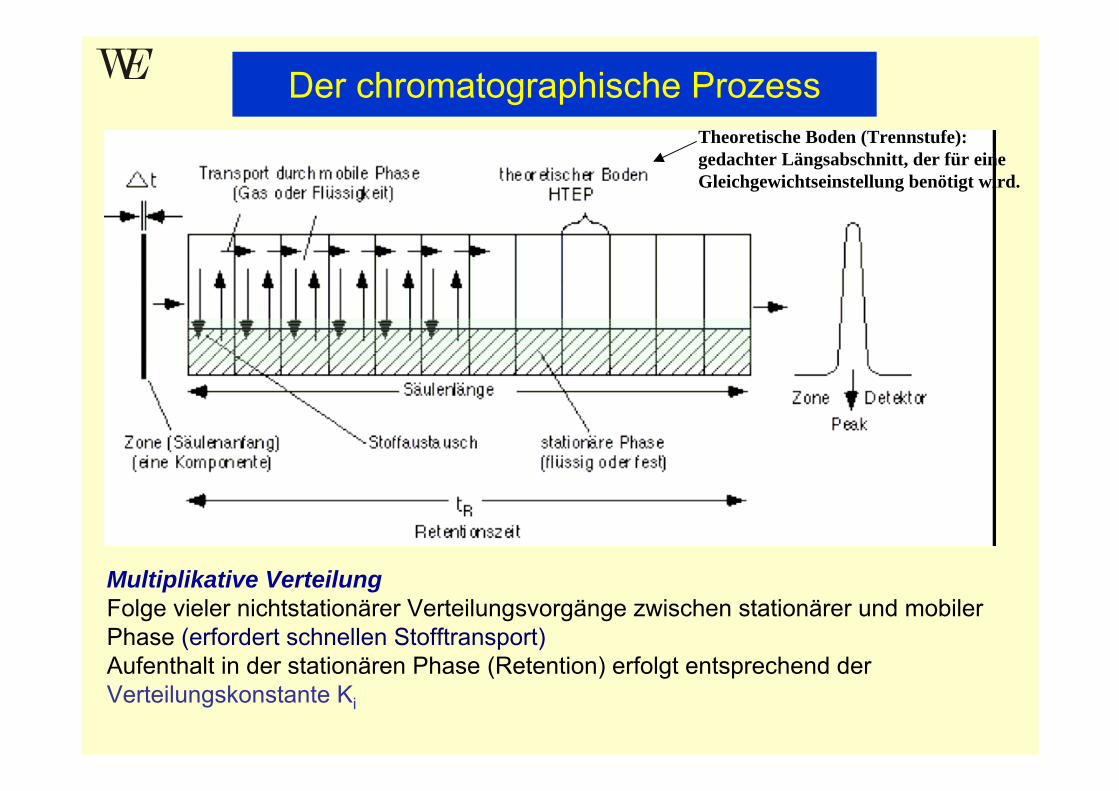
Multiplikative VerteilungFolge vieler nichtstationärer Verteilungsvorgänge zwischen stationärer und mobiler Phase (erfordert schnellen Stofftransport)Aufenthalt in der stationären Phase (Retention) erfolgt entsprechend der Verteilungskonstante Ki
Der chromatographische ProzessTheoretische Boden (Trennstufe): gedachter Längsabschnitt, der für eine Gleichgewichtseinstellung benötigt wird.

Molekulare Diffusion
Diffusion (diffundere (lat.): ausbreiten, sich zerstreuen)Durchmischung von verschiedenen miteinander in Berührung befindlichen gasförmigen,flüssigen oder festen Stoffen durch Bewegung der Ionen, Atome, Moleküle oder Kolloid-teilchen hervorgerufen durch Konzentrations-, Temperatur- oder Druckunterschiede bzw.durch äußere Felder.Bei der durch Konzentrationsunterschiede bewirkten Diffusion (Transportdiffusion) bewegensich mehr Moleküle vom Gebiet mit hoher Konzentration, zu einem Gebiet mit niedrigerer Konzentration bis überall die gleiche Konzentration erreicht ist:
Molekulare Diffusion: Zufällige Bewegung von Molekülen in fluiden Medien (Selbstdiffusion)
Diffusionskoeffizient D (m²/s)In binären Systemen ist der Teilchenfluß dem Konzentrationsgradienten direkt proportional
dn/dt = -D(dc/dx) (1. Ficksches Diffusionsgesetz)
Die Proportionalitätskonstante D ist ein Maß für die Wanderungsgeschwindigkeit (pro Einheits-fläche transportierter Stoffmenge) und wird als binärer Diffusionskoeffzient bezeichnet.
hohe Konzentration niedrige Konzentration
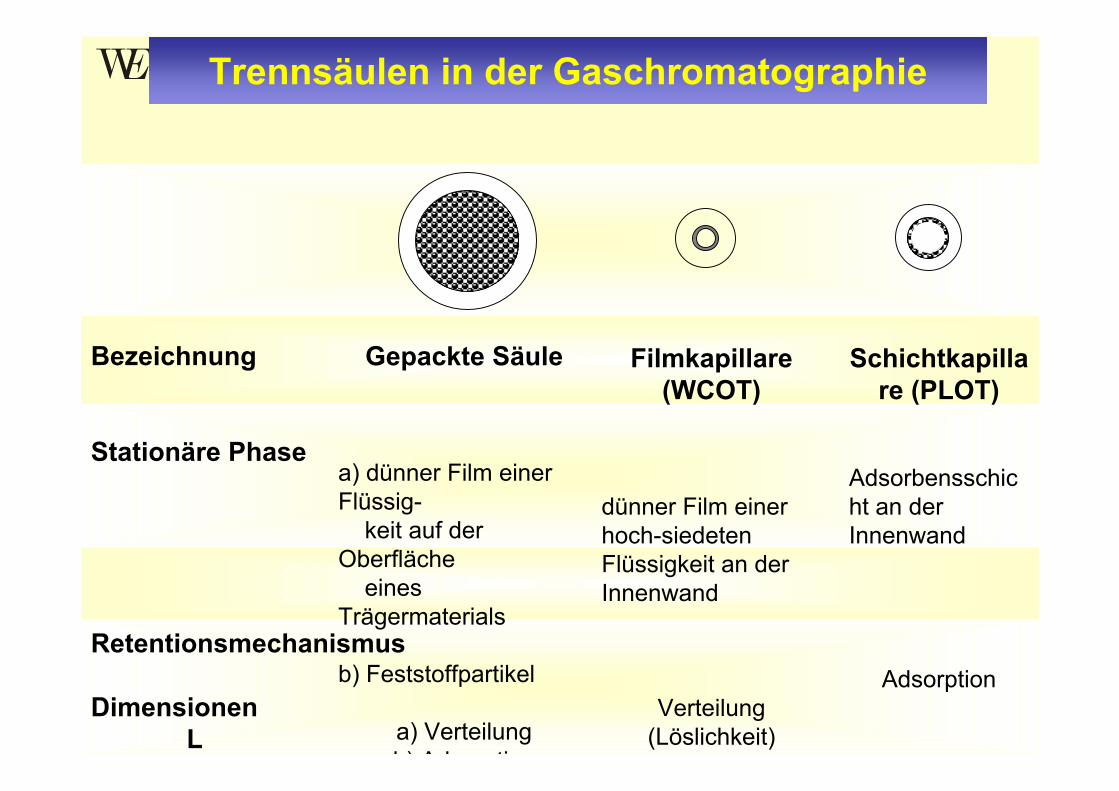
Trennsäulen in der Gaschromatographie
Gepackte Säule
a) dünner Film einer Flüssig-
keit auf der Oberfläche
eines Trägermaterials
b) Feststoffpartikel
a) Verteilungb) Ad ti
Filmkapillare (WCOT)
dünner Film einer hoch-siedeten Flüssigkeit an der Innenwand
Verteilung (Löslichkeit)
Schichtkapillare (PLOT)
Adsorbensschicht an der Innenwand
Adsorption
Bezeichnung
Stationäre Phase
Retentionsmechanismus
DimensionenL
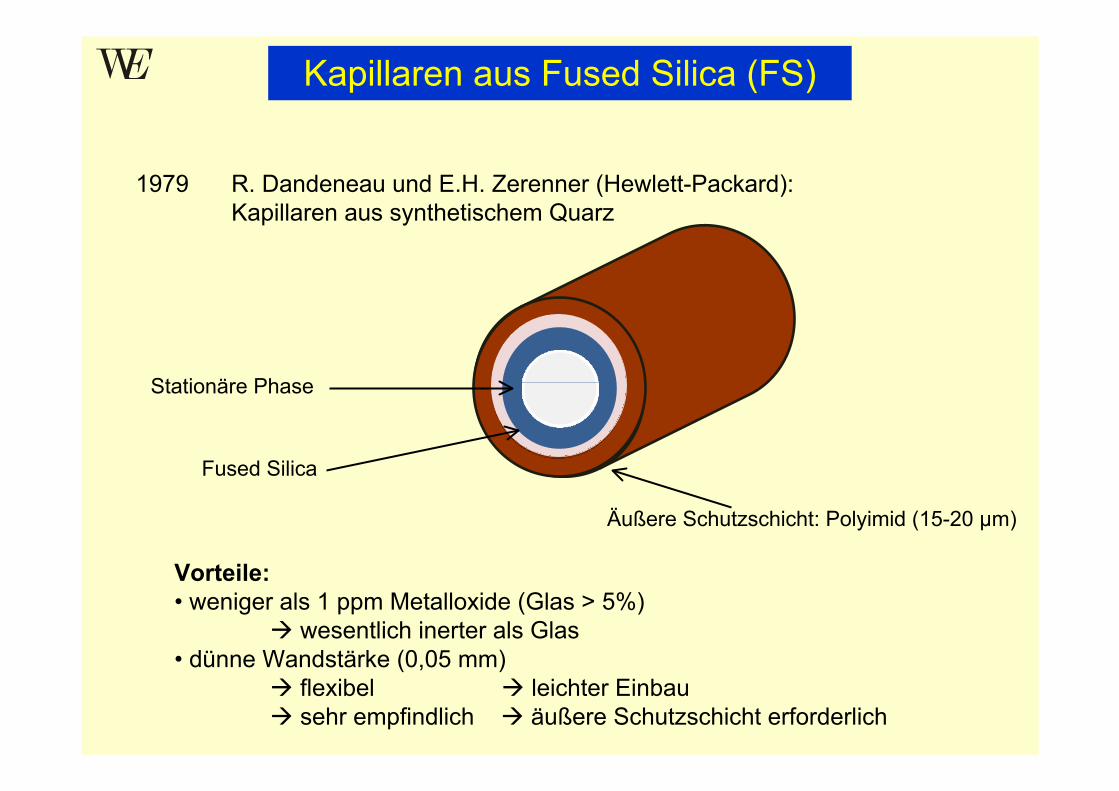
Kapillaren aus Fused Silica (FS)
1979 R. Dandeneau und E.H. Zerenner (Hewlett-Packard):Kapillaren aus synthetischem Quarz
Vorteile:• weniger als 1 ppm Metalloxide (Glas > 5%)
wesentlich inerter als Glas• dünne Wandstärke (0,05 mm)
flexibel leichter Einbausehr empfindlich äußere Schutzschicht erforderlich
Stationäre Phase
Fused Silica
Äußere Schutzschicht: Polyimid (15-20 µm)
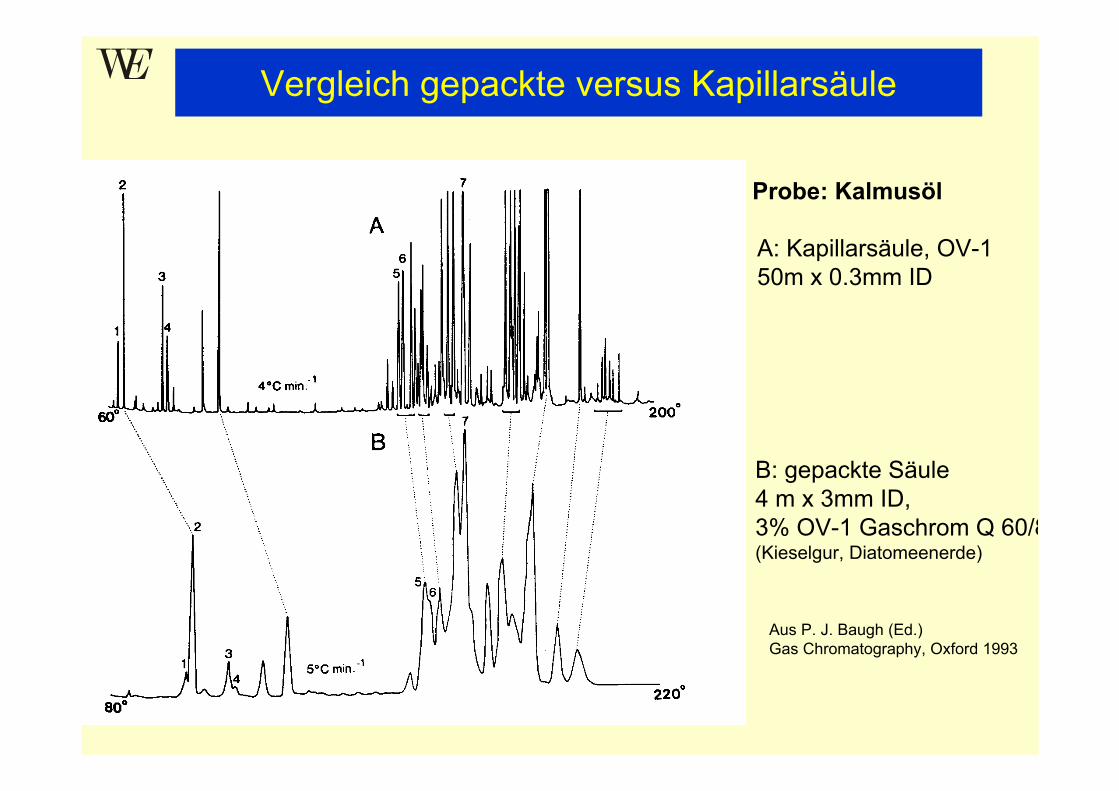
Probe: Kalmusöl
A: Kapillarsäule, OV-150m x 0.3mm ID
B: gepackte Säule4 m x 3mm ID, 3% OV-1 Gaschrom Q 60/8(Kieselgur, Diatomeenerde)
Aus P. J. Baugh (Ed.)Gas Chromatography, Oxford 1993
Vergleich gepackte versus Kapillarsäule

Silikonphasen (Polysiloxane)
Polymere Silikonphasen (Polysiloxane, exakt Poly(organosiloxane))• bestehen aus langen, flexiblen Ketten (Silikonöle, Silikongummi;Molmasse zwischen 5000 und 2 000 000) mit einer helikalen Struktur
• sind die wegen ihrer Vorteile am meisten verwendeten stationären Phasenin der GC
Vorteile• hohe chemische und thermische Stabilität ( hohe Arbeitstemperaturen,geringes Bluten)
• hohe Diffusionskoeffizienten der Analyten (geringer CS-Term)• niedrige Glasübergangstemperatur ( großer Temperaturbereich)• geringe Oberflächenspannung ( gute Filmbildung)• geringe Abnahme der Viskosität mit steigender Temperatur• definierte Struktur und chemische Vielfalt: durch Synthese aus Bausteinenmit verschiedenen funktionellen Gruppen lassen sich Phasen mit unter-schiedlichen Polaritäten / Selektivitäten herstellen(Stabilität bei symmetrisch substituierten Si-Atomen am größten: z.B. Diphenylstatt Methyl-Phenyl)
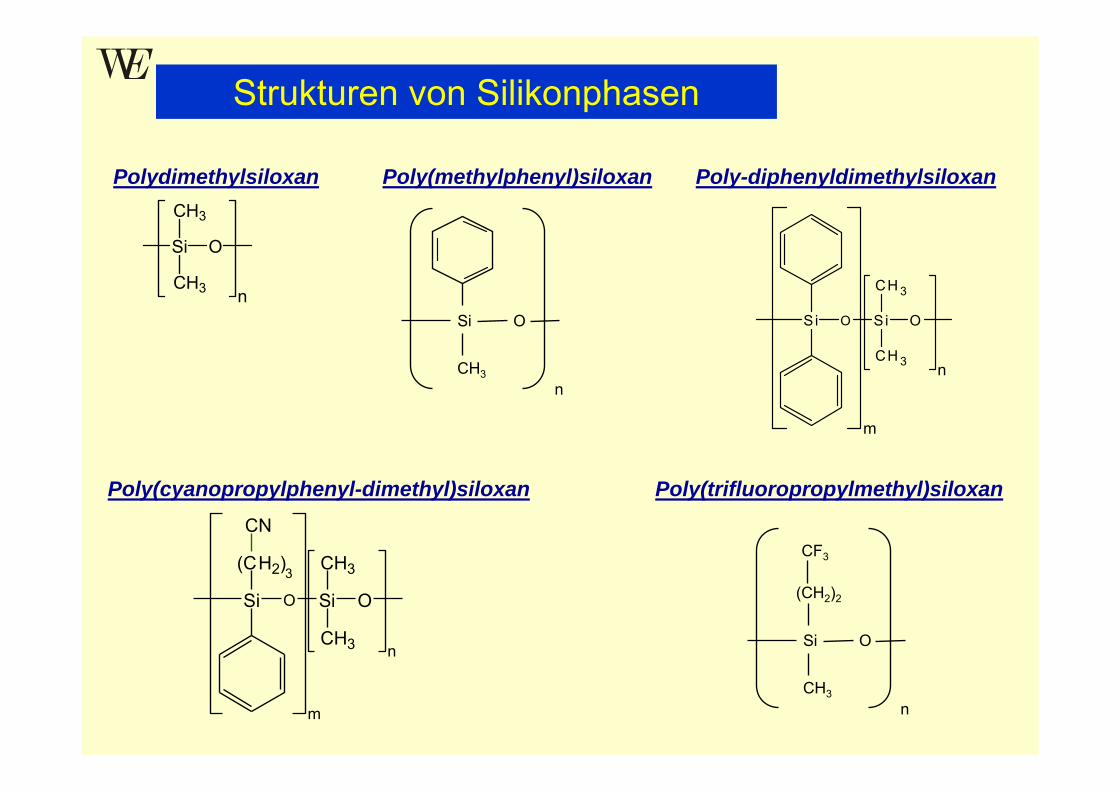
Si
CH3
CH3 n
O
Polydimethylsiloxan Poly-diphenyldimethylsiloxan
O Si
CH 3
CH 3 n
OSi
m
Poly(cyanopropylphenyl-dimethyl)siloxan
O Si
CH3
CH3 n
OSi
(CH2
m
CN
3)
Strukturen von Silikonphasen
Poly(methylphenyl)siloxan
Poly(trifluoropropylmethyl)siloxan
Si O
nCH3
Si O
nCH3
(CH2)2
CF3

Fettsäuren FS (Fatty acids FA)Natürlich vorkommende Fettsäurenvorwiegend unverzweigte Monocarbonsäuren mit einer geradenAnzahl von C-Atomen (meist zw. C12 und C22)
• Gesättigte Fettsäuren
• Einfach ungesättigte Fettsäuren: cis/trans
• Mehrfach ungesättigte Fettsäuren (polyunsaturated fatty acids PUFA):nicht konjugierte Doppelbindungen; meist in cis-Anordnung und durch eine Methylengruppe voneinander getrennt (methylene-interrupted MI)
Position der Doppelbindungen
• Δ-Nomenklatur: Nummerierung vom Carbonylende ausgehend
• n- oder ω-Nomenklatur: Nummerierung vom Methylende ausgehendbei MI PUFA wird nur Position der ersten Doppelbindung angegeben
• Häufigste PUFA-Familien: n-3 (Linolensäure-Familie)n-6 (Linolsäure-Familie)
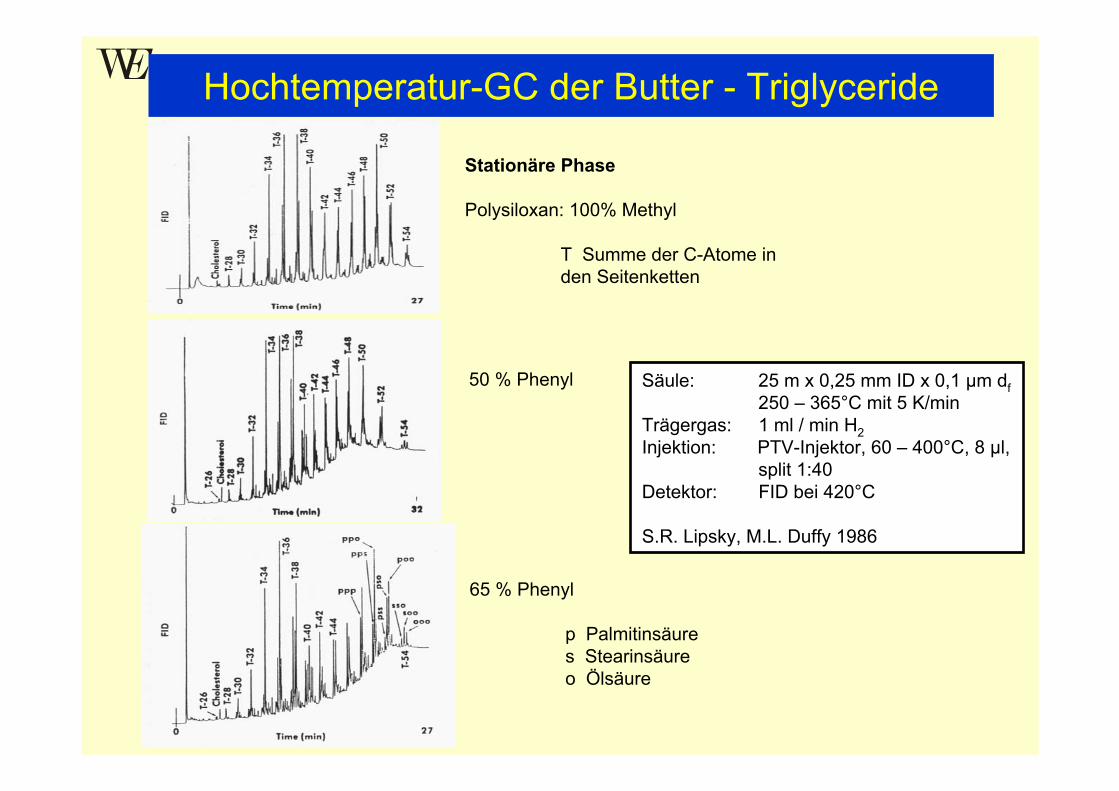
Hochtemperatur-GC der Butter - Triglyceride
Stationäre Phase
Polysiloxan: 100% Methyl
T Summe der C-Atome in den Seitenketten
50 % Phenyl
65 % Phenyl
p Palmitinsäures Stearinsäureo Ölsäure
Säule: 25 m x 0,25 mm ID x 0,1 µm df250 – 365°C mit 5 K/min
Trägergas: 1 ml / min H2Injektion: PTV-Injektor, 60 – 400°C, 8 µl,
split 1:40Detektor: FID bei 420°C
S.R. Lipsky, M.L. Duffy 1986
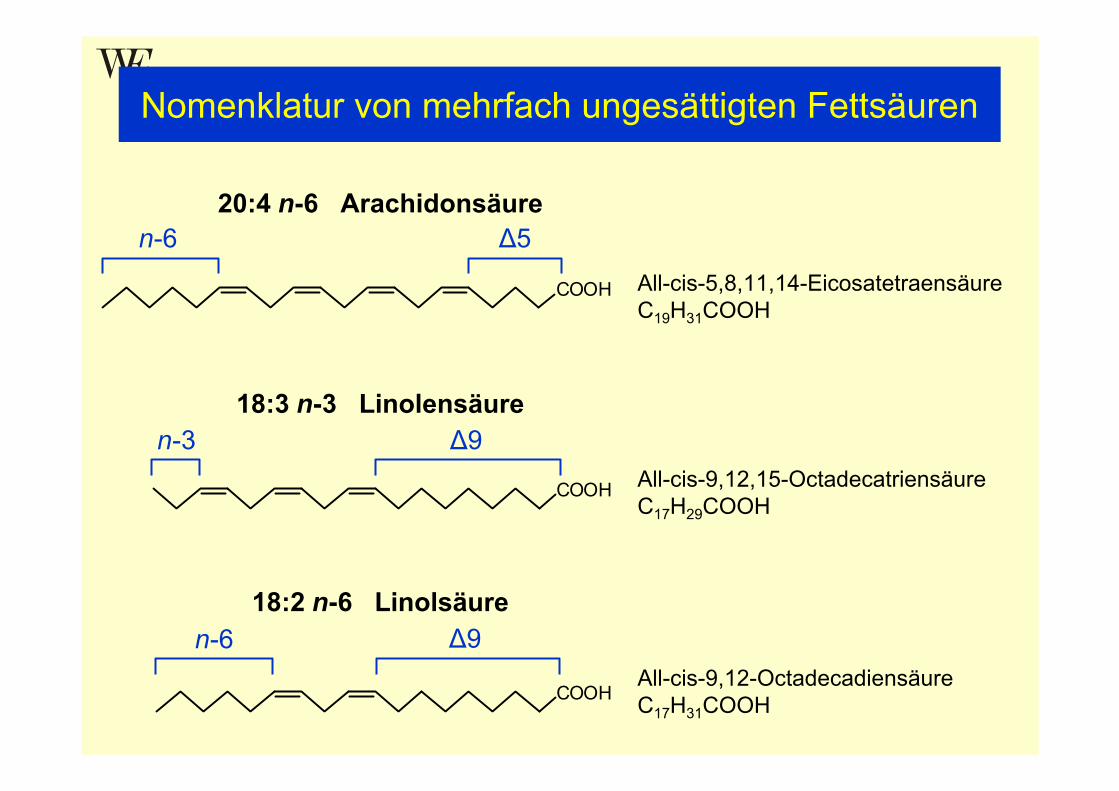
Nomenklatur von mehrfach ungesättigten Fettsäuren
n-6
n-3
n-6
Δ5
Δ9
Δ9
20:4 n-6 Arachidonsäure
18:3 n-3 Linolensäure
18:2 n-6 Linolsäure
All-cis-5,8,11,14-EicosatetraensäureC19H31COOH
All-cis-9,12,15-OctadecatriensäureC17H29COOH
All-cis-9,12-OctadecadiensäureC17H31COOH
COOH
COOH
COOH
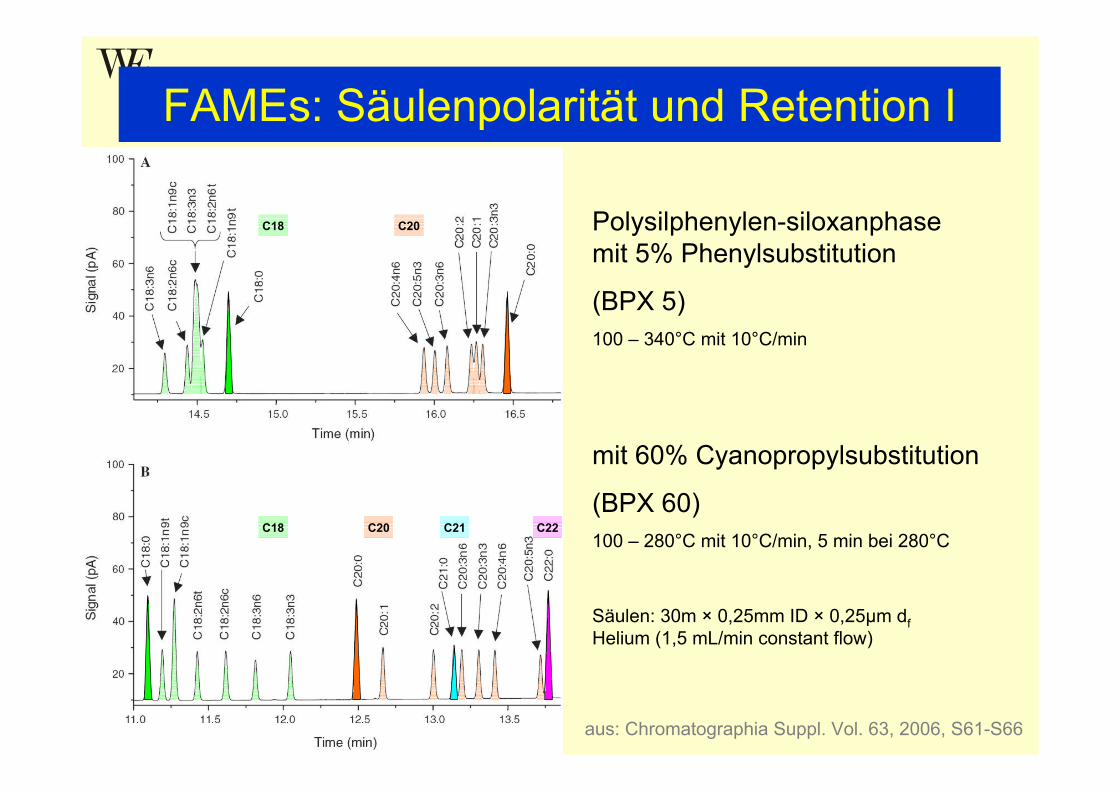
FAMEs: Säulenpolarität und Retention I
Polysilphenylen-siloxanphasemit 5% Phenylsubstitution
(BPX 5)100 – 340°C mit 10°C/min
mit 60% Cyanopropylsubstitution
(BPX 60)100 – 280°C mit 10°C/min, 5 min bei 280°C
Säulen: 30m × 0,25mm ID × 0,25µm dfHelium (1,5 mL/min constant flow)
C18 C20
C18 C20 C22C21
aus: Chromatographia Suppl. Vol. 63, 2006, S61-S66
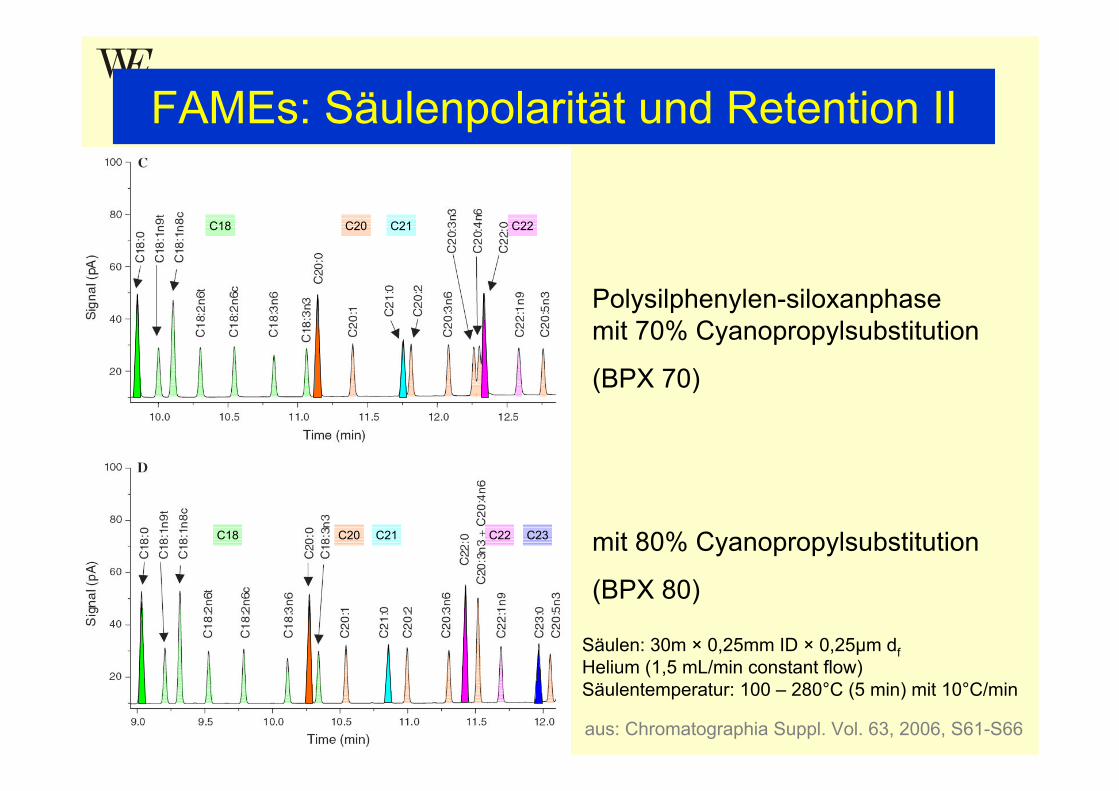
FAMEs: Säulenpolarität und Retention II
mit 80% Cyanopropylsubstitution
(BPX 80)
Polysilphenylen-siloxanphasemit 70% Cyanopropylsubstitution
(BPX 70)
Säulen: 30m × 0,25mm ID × 0,25µm dfHelium (1,5 mL/min constant flow)Säulentemperatur: 100 – 280°C (5 min) mit 10°C/min
aus: Chromatographia Suppl. Vol. 63, 2006, S61-S66
C18 C20 C21 C22
C18 C20 C21 C22 C23

Gas Versorgung: hochreines Trägergas (He, H2, N2)Injektor: Probenaufgabe (gas, flüssig): mittels Mikroliterspritzen, Autosampler, GasventileTrennsäule: gepackte Säule oder KapillarsäuleSäulenofen: Thermostat ( 30 – 350 °C )Detektor: Universal oder elementspezifischDatenaufnahmesystem
Aufbau eines Gaschromatographen
Dosiertechnik

Probenaufgabe in Kapillar-GC
Probenaufgabe (Injektion, Dosierung)Ziel: Schnelles Einbringen der Probe (oder eines repräsentativen Teils)auf den Säulenanfang
als schmale Substanzzone ohne Veränderung der stofflichen Zusammensetzung (keine Diskriminierung, Adsorption oder Zersetzung)
Probleme bei Kapillarsäulen• kleine Probemengen (0,1 – 100 ng/Komponente)
Manipulation geringer Probemengen• starke Volumenvergrößerung beim Übergang von der Flüssig- in die
Dampfphase (100 – 250 fach)• geringer Volumenfluß der mobilen Phase
lange Verweil- und Überführungszeit
Es gibt keine universelle Probenaufgabetechnik in der KGCIn Abhängigkeit von analytischer Aufgabenstellung sowie Eigenschaften der Probe (Analyt/Matrix) kommen unterschiedliche Dosiervorrichtungen und Dosiertechniken zur AnwendungProbe: gasförmig Gasdosierventil, Gasspritze
flüssig ) Spritzendosierung von Lösungenfest ) Lösungsmittelfreie Dosiertechniken
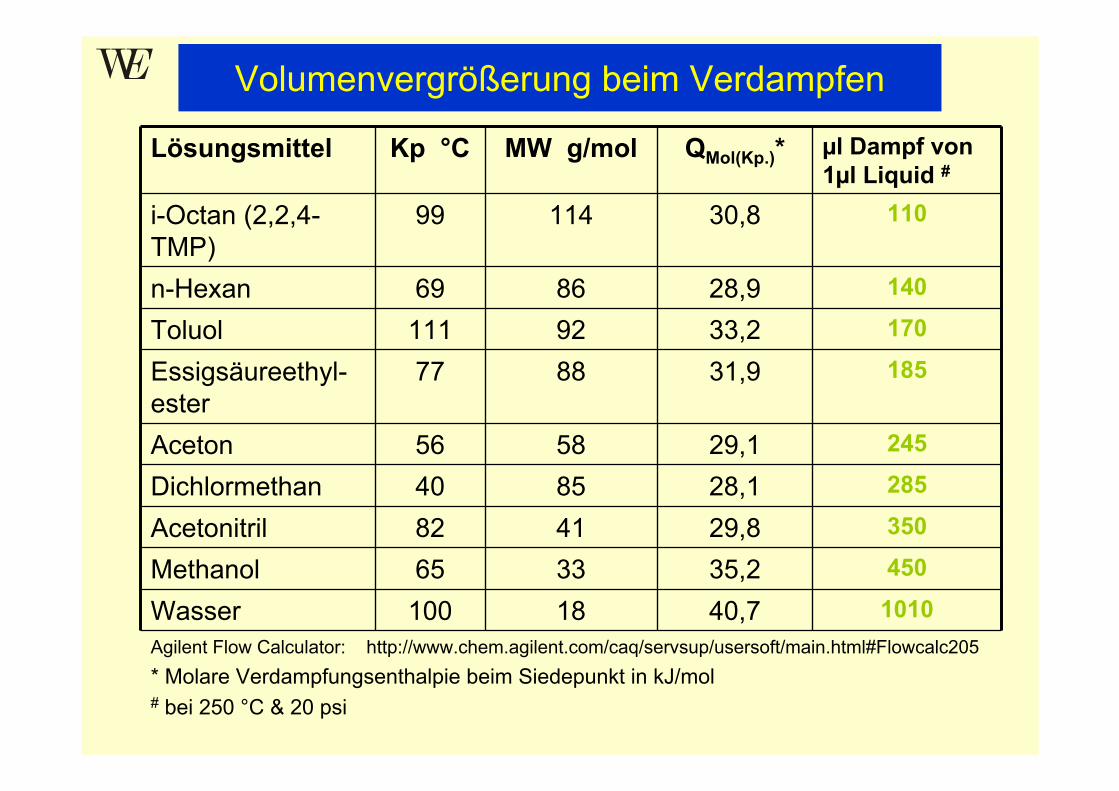
Lösungsmittel Kp °C MW g/mol QMol(Kp.)* µl Dampf von 1µl Liquid #
i-Octan (2,2,4-TMP)
99 114 30,8 110
n-Hexan 69 86 28,9 140
Toluol 111 92 33,2 170
Essigsäureethyl-ester
77 88 31,9 185
Aceton 56 58 29,1 245
Dichlormethan 40 85 28,1 285
Acetonitril 82 41 29,8 350
Methanol 65 33 35,2 450
Wasser 100 18 40,7 1010Agilent Flow Calculator: http://www.chem.agilent.com/caq/servsup/usersoft/main.html#Flowcalc205* Molare Verdampfungsenthalpie beim Siedepunkt in kJ/mol# bei 250 °C & 20 psi
Volumenvergrößerung beim Verdampfen
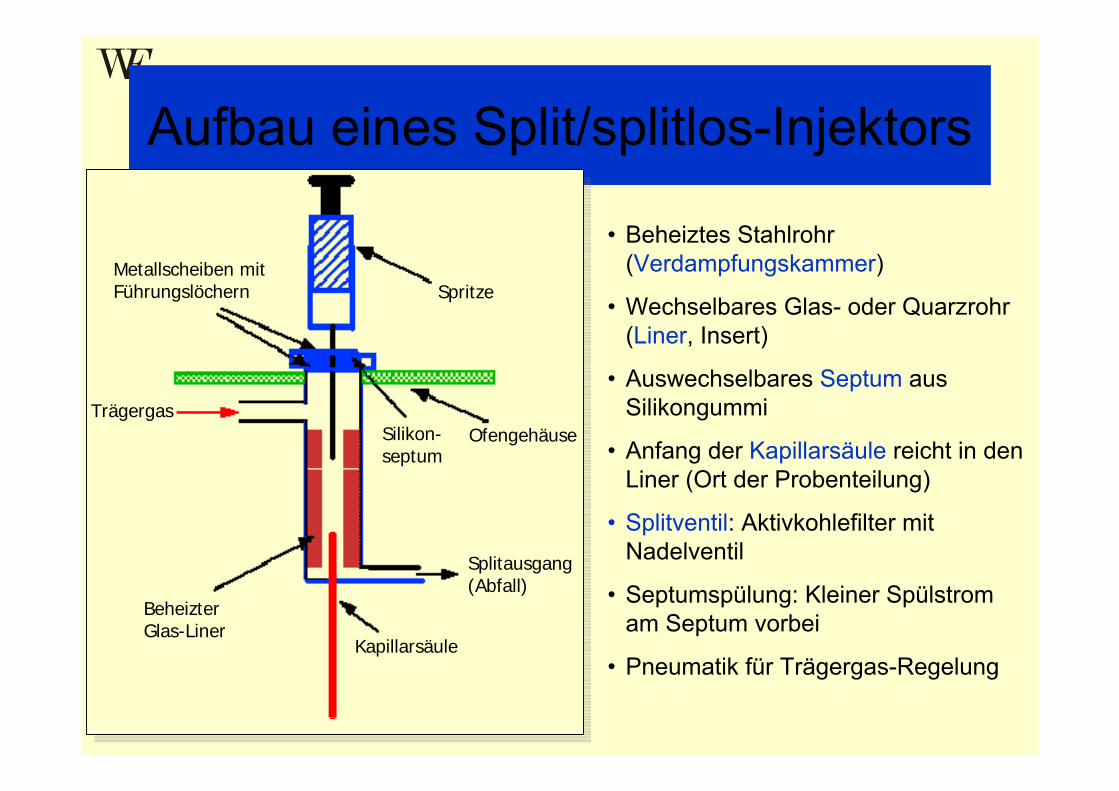
Aufbau eines Split/splitlos-Injektors
Spritze
Silikon-septum
Ofengehäuse
Splitausgang (Abfall)
Kapillarsäule
Beheizter Glas-Liner
Trägergas
Metallscheiben mit Führungslöchern
• Beheiztes Stahlrohr (Verdampfungskammer)
• Wechselbares Glas- oder Quarzrohr (Liner, Insert)
• Auswechselbares Septum aus Silikongummi
• Anfang der Kapillarsäule reicht in den Liner (Ort der Probenteilung)
• Splitventil: Aktivkohlefilter mit Nadelventil
• Septumspülung: Kleiner Spülstrom am Septum vorbei
• Pneumatik für Trägergas-Regelung
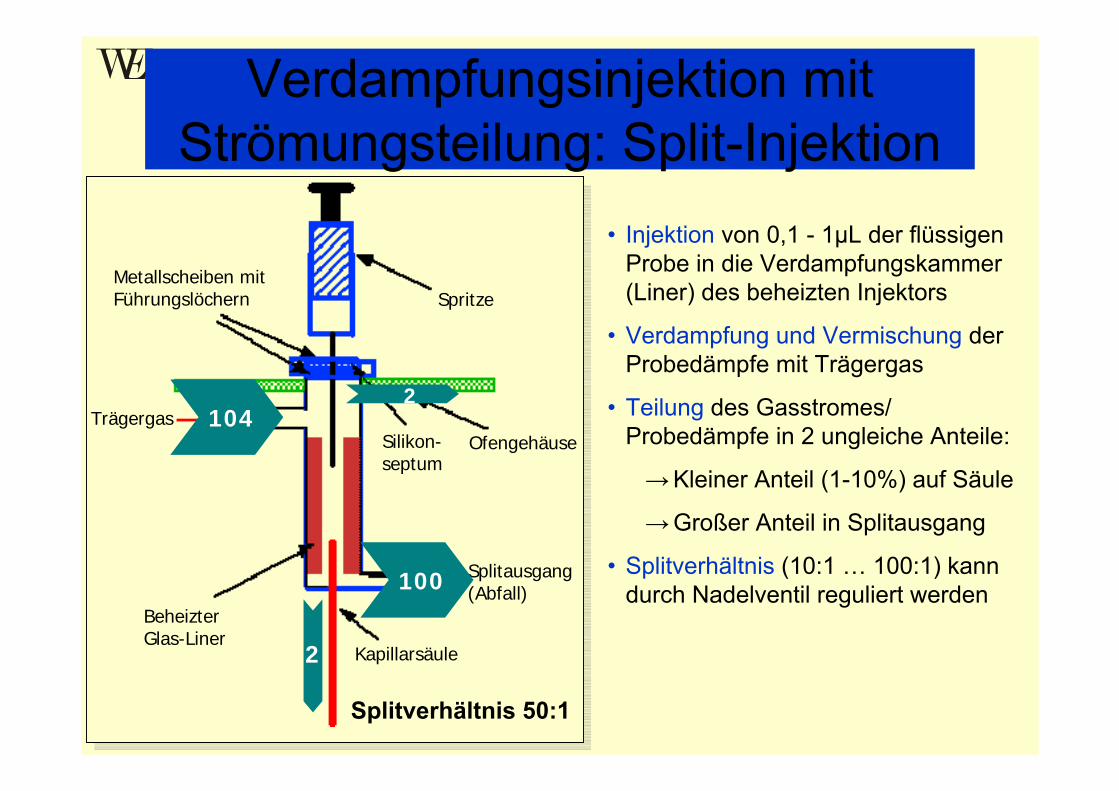
Verdampfungsinjektion mit Strömungsteilung: Split-Injektion
Spritze
Silikon-septum
Ofengehäuse
Splitausgang (Abfall)
Kapillarsäule
Beheizter Glas-Liner
Trägergas
Metallscheiben mit Führungslöchern
• Injektion von 0,1 - 1µL der flüssigen Probe in die Verdampfungskammer (Liner) des beheizten Injektors
• Verdampfung und Vermischung der Probedämpfe mit Trägergas
• Teilung des Gasstromes/ Probedämpfe in 2 ungleiche Anteile:
→Kleiner Anteil (1-10%) auf Säule
→Großer Anteil in Splitausgang
• Splitverhältnis (10:1 … 100:1) kann durch Nadelventil reguliert werden
1042
100
2
Splitverhältnis 50:1
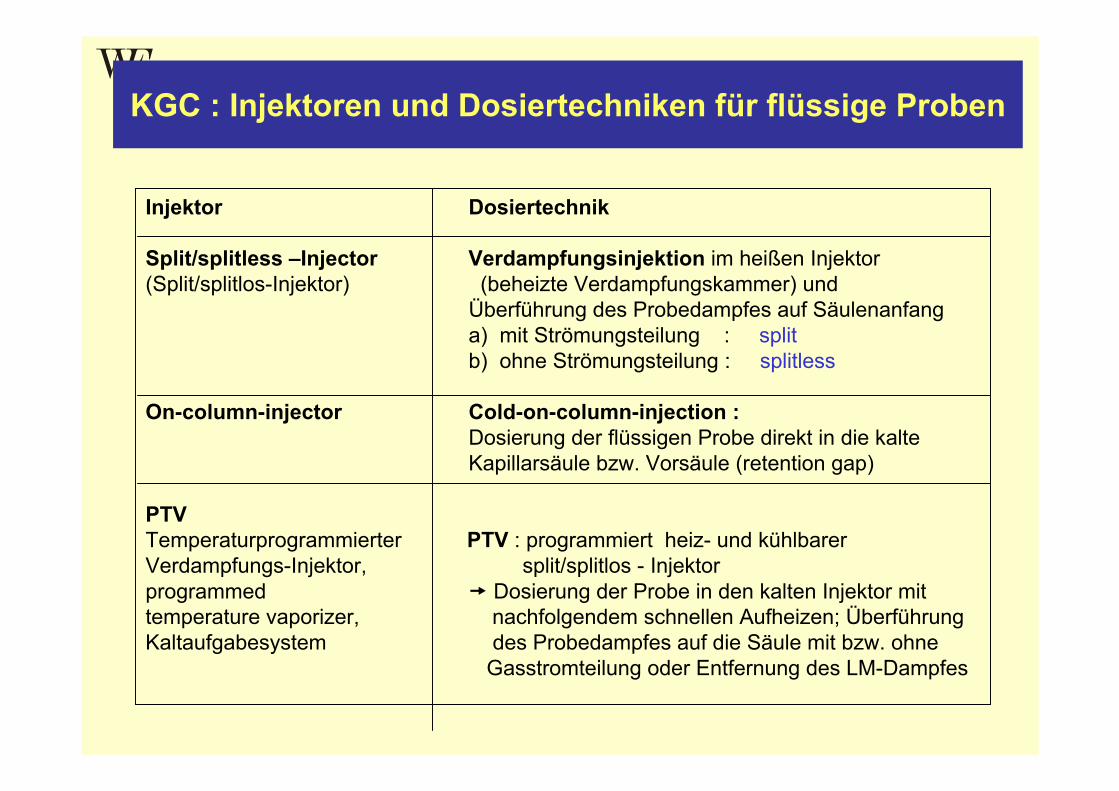
KGC : Injektoren und Dosiertechniken für flüssige Proben
Injektor Dosiertechnik
Split/splitless –Injector Verdampfungsinjektion im heißen Injektor(Split/splitlos-Injektor) (beheizte Verdampfungskammer) und
Überführung des Probedampfes auf Säulenanfanga) mit Strömungsteilung : splitb) ohne Strömungsteilung : splitless
On-column-injector Cold-on-column-injection :Dosierung der flüssigen Probe direkt in die kalteKapillarsäule bzw. Vorsäule (retention gap)
PTVTemperaturprogrammierter PTV : programmiert heiz- und kühlbarer Verdampfungs-Injektor, split/splitlos - Injektorprogrammed Dosierung der Probe in den kalten Injektor mittemperature vaporizer, nachfolgendem schnellen Aufheizen; Überführung Kaltaufgabesystem des Probedampfes auf die Säule mit bzw. ohne
Gasstromteilung oder Entfernung des LM-Dampfes

Ethanolbestimmung in Blut (BAK)
1920 E.M.R. Widmark : „isotherme Destillation“Blutprobe wird in einem Spezialgefäß 1 Stunde auf 60 °C erwärmt,flüchtige Substanzen gelangen in eine Vorlage mit Bichromatlösung,Ethanol wird oxidiertRücktitration des nicht verbrauchten Bichromateswenig spezifisch (oxidierbare Substanzen)
1962 G. Machata (Universität Wien) : GC mit FIDBlutprobe wird auf gepackte Vorsäule dosiert, beim schnellen Aufheizengelangen flüchtige Verbindungen auf Trennsäule, während unverdampfbareRückstände auf Vorsäule verbleibenPackung der Vorsäule muss periodisch ausgewechselt werden
1962 A. C. Curry et al : Headspace – GC
1967 G. Machata und Bodenseewerk Perkin Elmer & Co, Überlingen : F- 40erstes automatisches System für Headspace-GC auf Messe ACHEMAin Frankfurt / M vorgestellt Problem : präzise und reproduzierbare Überführung der Analyten vom Dampfraum der Probengefäße in GC-Säule : balanced–pressure sampling

Prinzip der statischen HS-GC
1. Thermostatisierung der Probe in einem verschlossenen Gefäß bei höherer Temperatur(unterhalb der Siedetemperatur der Analyten)
⇒ Verteilungsgleichgewicht der flüchtigen Verbindungen zwischen Probe und Gasphase
2. Entnahme eines aliquoten Gasvolumens aus dem Dampfraum und Überführung in den GC mittels - gasdichter Spritze- pneumatischer Dosiervorrichtung(Gleichdruck- oder Konstantdruck-Dosierung)
3. Kalibrierung zur quantitativen Analyse interner oder externer Standard, Aufstockung
Headspace-GC HSGC


Headspace-GC-Vial

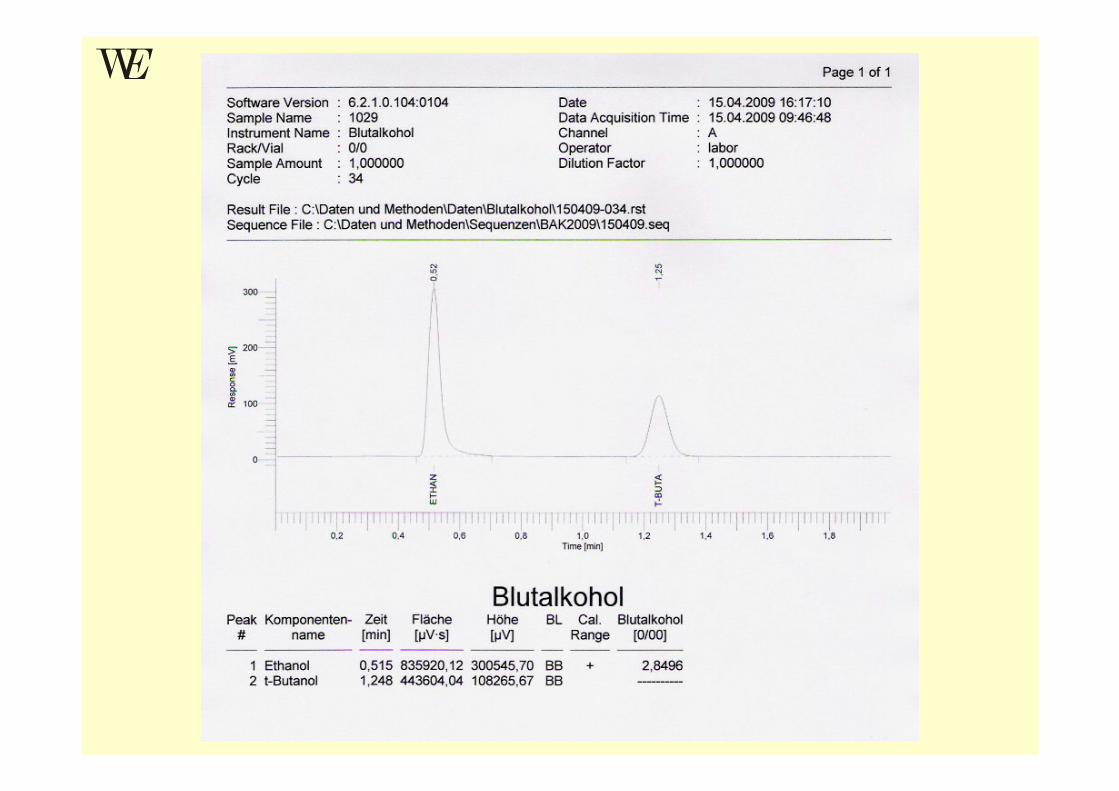
GC-Verfahren zur BAK-Bestimmung
• Headspace-Gaschromatographie nach MACHATA• Statische HSGC mit 22 ml Vials; GC-Detektor: FID• Serumprobe mit internem Standard (tert-Butanol)
versehen, erhitzt und Aliquot des Dampfraumes über Probe wird injiziert
• Ergebnis wird nach Umrechnung Serum ⇒ Blut als Blutalkoholkonzentration in ‰ (=g Ethanol/kg Blut) angegeben

Ethanolnachweis
• 2 unterschiedliche Methoden Vorschrift– ADH-Verfahren und GC-Verfahren– oder 2 x GC-Verfahren auf 2
verschiedenen Geräten mit unterschiedlich polaren Säulen
• jeweils Doppelbestimmungen (4 Einzelwerte)
• von unterschiedlichen Bearbeitern durchgeführt

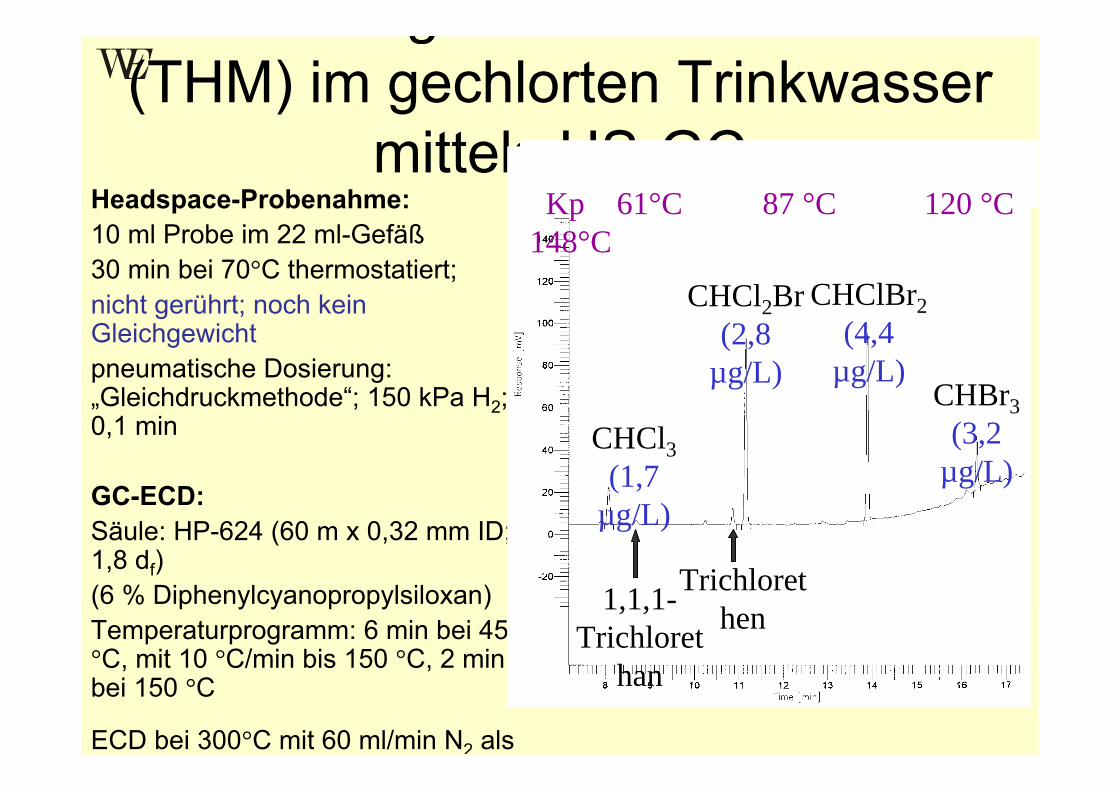
g(THM) im gechlorten Trinkwasser
mittels HS-GCHeadspace-Probenahme:10 ml Probe im 22 ml-Gefäß30 min bei 70°C thermostatiert;nicht gerührt; noch kein Gleichgewichtpneumatische Dosierung: „Gleichdruckmethode“; 150 kPa H2; 0,1 min
GC-ECD:Säule: HP-624 (60 m x 0,32 mm ID; 1,8 df) (6 % Diphenylcyanopropylsiloxan)Temperaturprogramm: 6 min bei 45 °C, mit 10 °C/min bis 150 °C, 2 min bei 150 °C
ECD bei 300°C mit 60 ml/min N2 als
CHCl3(1,7
µg/L)
1,1,1-Trichloret
han
Trichlorethen
CHCl2Br(2,8
µg/L)
CHClBr2(4,4
µg/L)CHBr3
(3,2 µg/L)
Kp 61°C 87 °C 120 °C 148°C

Trihalomethane (THM) im Trinkwasser• Trinkwasser-Chlorung:
Desinfektion von Rohwasser mit Chlor bzw. chlorhaltigen Desinfektionsmitteln
aber: 1974 wurden in USA und Niederlande Chloroform u.a. Trihalogenmethanverbindungen im Trinkwassergefunden
• Bildung von „Desinfektions-Nebenprodukten“ (DNP)• viele Grund- und Oberflächenwasser enthalten
wasserlösliche organische Verbindungen (Huminstoffe, „gelöster organischer Kohlenstoff“ DOC)
• Chlor als Desinfektionsmittel reagiert auch mit natürlichen organischen Wasserinhaltsstoffen:
- Oxidation, elektrophile Substitution und Additionz. B. „Haloformreaktion“ (Einhorn-Reaktion) ⇒ CHCl3
- Oxidation anderer Halogenionen aus entsprechenden Salzen:
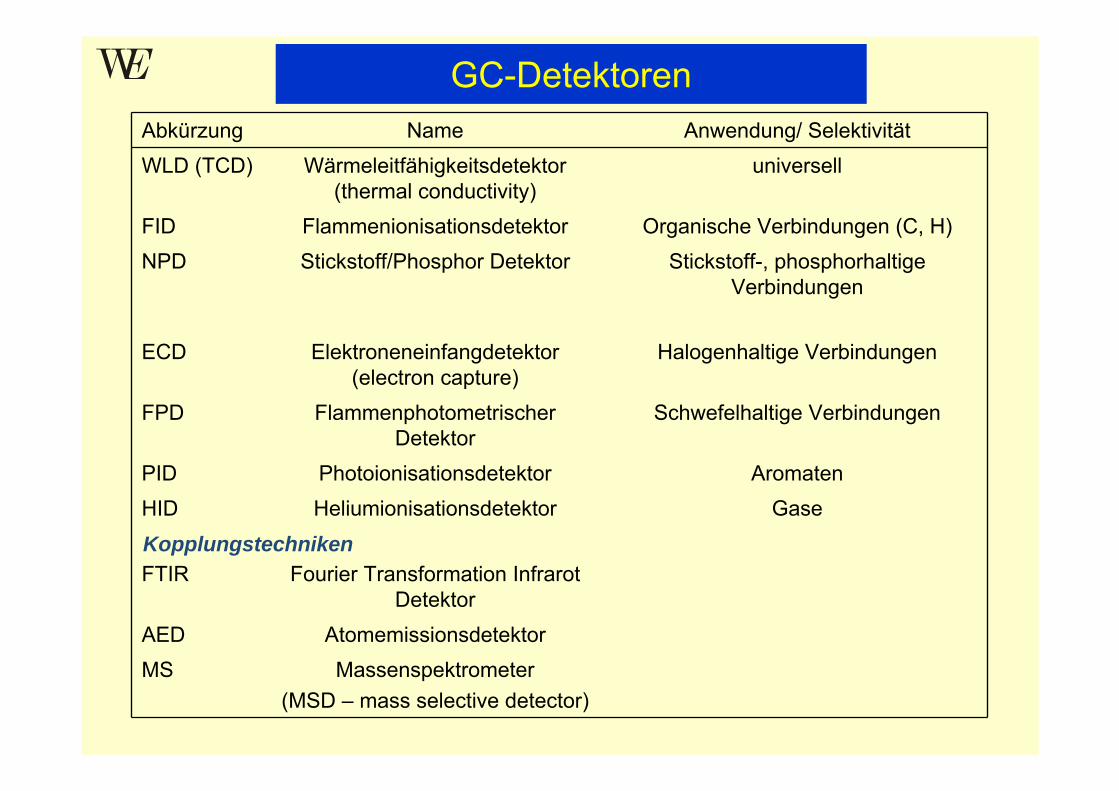
Abkürzung Name Anwendung/ SelektivitätWLD (TCD) Wärmeleitfähigkeitsdetektor
(thermal conductivity)universell
FID Flammenionisationsdetektor Organische Verbindungen (C, H)NPD Stickstoff/Phosphor Detektor Stickstoff-, phosphorhaltige
Verbindungen
ECD Elektroneneinfangdetektor (electron capture)
Halogenhaltige Verbindungen
FPD Flammenphotometrischer Detektor
Schwefelhaltige Verbindungen
PID Photoionisationsdetektor AromatenHID Heliumionisationsdetektor Gase
FTIR Fourier Transformation Infrarot Detektor
AED AtomemissionsdetektorMS Massenspektrometer
(MSD – mass selective detector)
Kopplungstechniken
GC-Detektoren
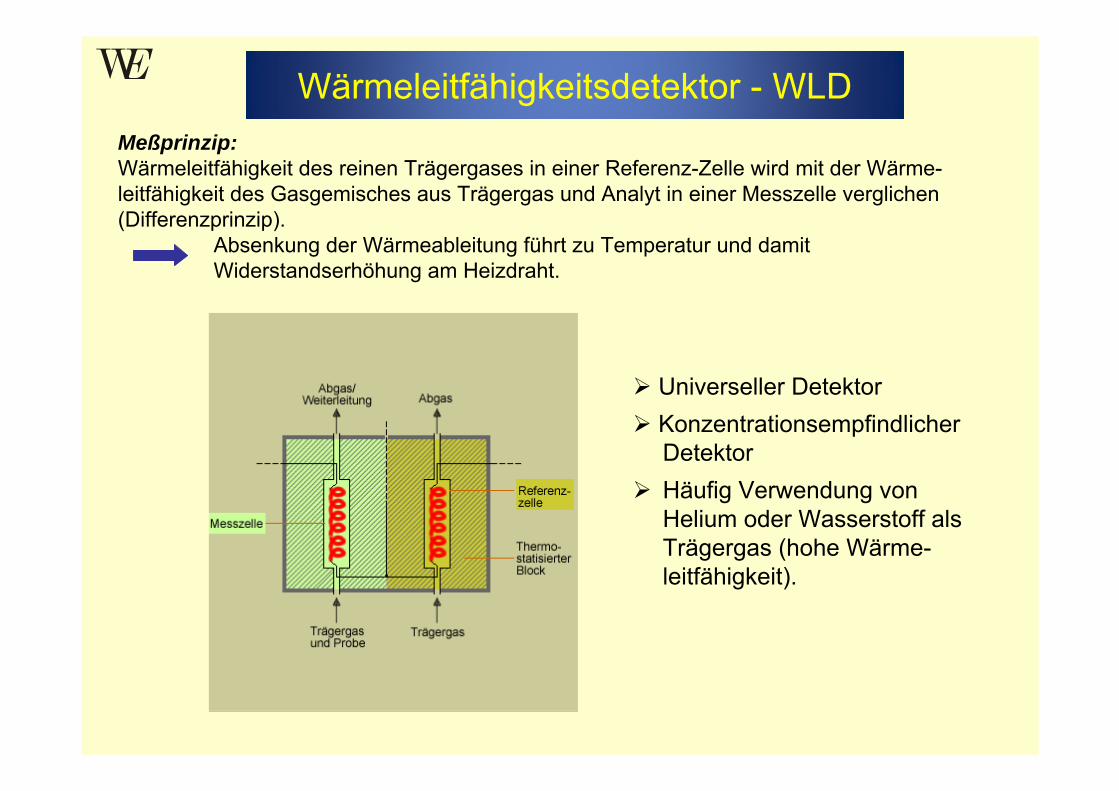
Meßprinzip: Wärmeleitfähigkeit des reinen Trägergases in einer Referenz-Zelle wird mit der Wärme-leitfähigkeit des Gasgemisches aus Trägergas und Analyt in einer Messzelle verglichen (Differenzprinzip).
Absenkung der Wärmeableitung führt zu Temperatur und damit Widerstandserhöhung am Heizdraht.
Universeller DetektorKonzentrationsempfindlicher DetektorHäufig Verwendung von Helium oder Wasserstoff als Trägergas (hohe Wärme-leitfähigkeit).
Wärmeleitfähigkeitsdetektor - WLD
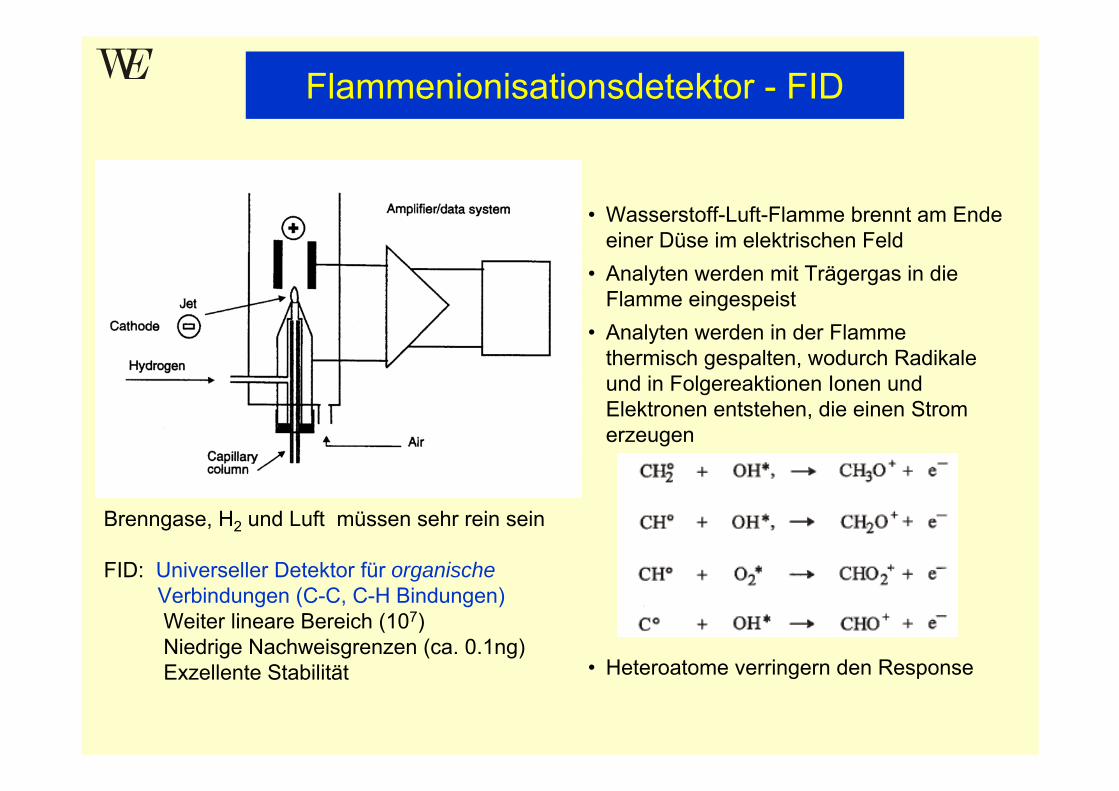
• Wasserstoff-Luft-Flamme brennt am Ende einer Düse im elektrischen Feld
• Analyten werden mit Trägergas in die Flamme eingespeist
• Analyten werden in der Flamme thermisch gespalten, wodurch Radikale und in Folgereaktionen Ionen und Elektronen entstehen, die einen Strom erzeugen
• Heteroatome verringern den Response
Brenngase, H2 und Luft müssen sehr rein sein
FID: Universeller Detektor für organischeVerbindungen (C-C, C-H Bindungen)Weiter lineare Bereich (107)Niedrige Nachweisgrenzen (ca. 0.1ng) Exzellente Stabilität
Flammenionisationsdetektor - FID
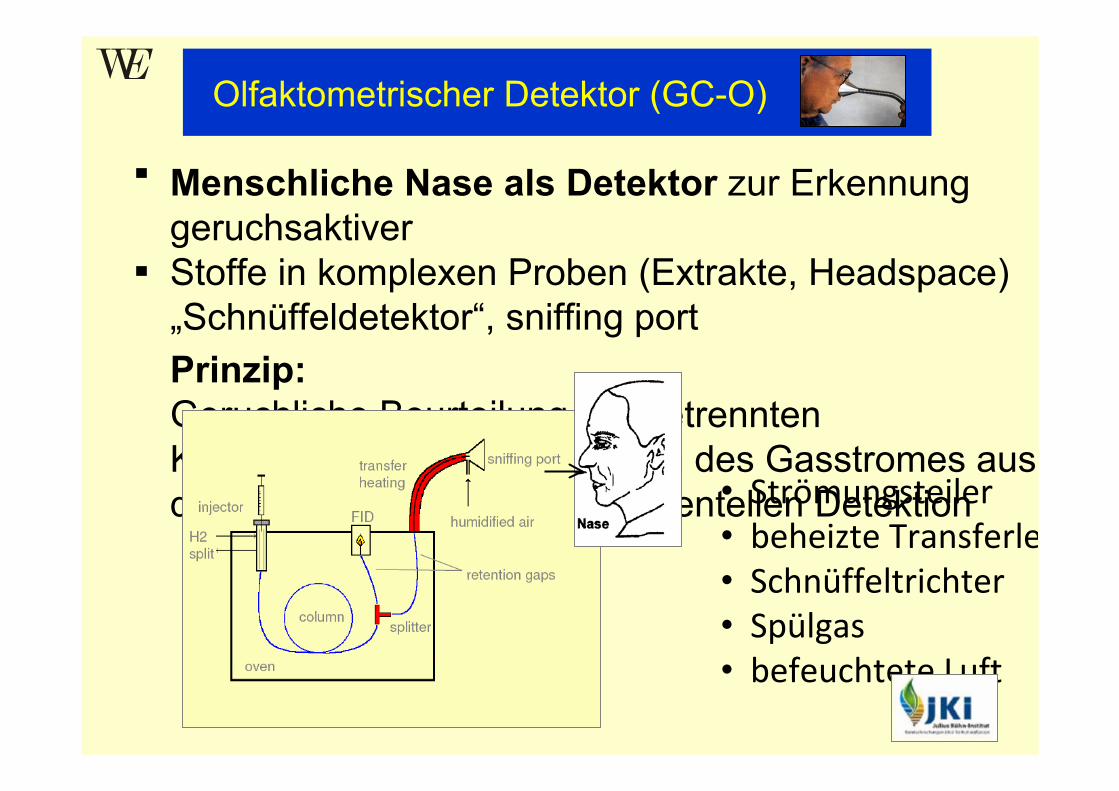
Menschliche Nase als Detektor zur Erkennung geruchsaktiver Stoffe in komplexen Proben (Extrakte, Headspace) „Schnüffeldetektor“, sniffing portPrinzip:Geruchliche Beurteilung der getrennten Komponenten durch Abriechen des Gasstromes aus der Säule simultan zur instrumentellen Detektion
Olfaktometrischer Detektor (GC-O)
• Strömungsteiler• beheizte Transferle• Schnüffeltrichter• Spülgas• befeuchtete Luft

Aufbau• Gasstromteilung am Ende der Trennsäule
a) GC-Detektor : meist FID oder MSb) geheizte Transferleitung zu einem „Schnüffeltrichter“
•Einspeisung von Spülgas (make-up Gas)→ beschleunigter Transfer
•Zumischen von befeuchteter Luft→ verhindert Austrocknung der Nasenschleimhäute
(erhält Sensitivität)
Mitteilung der Geruchswahrnehmung : wann, was, wie stark ?
• Geruchsintensität : schwach … sehr stark→ Tastatur oder Potentiometer :
Signalaufzeichnung, solange Geruch wahrgenommen wird: → Olfaktogramm (Aromagramm)
• Geruchsbeschreibung : Ansage (2. Person oder Head Set)z. B. etherisch, blumig, minzig, faulig, fischig, seifig, stechend …
Olfaktometrischer Detektor II

Gas chromatography – olfactometry (GCO)
Detlef Ulrich, JKI ÖPV
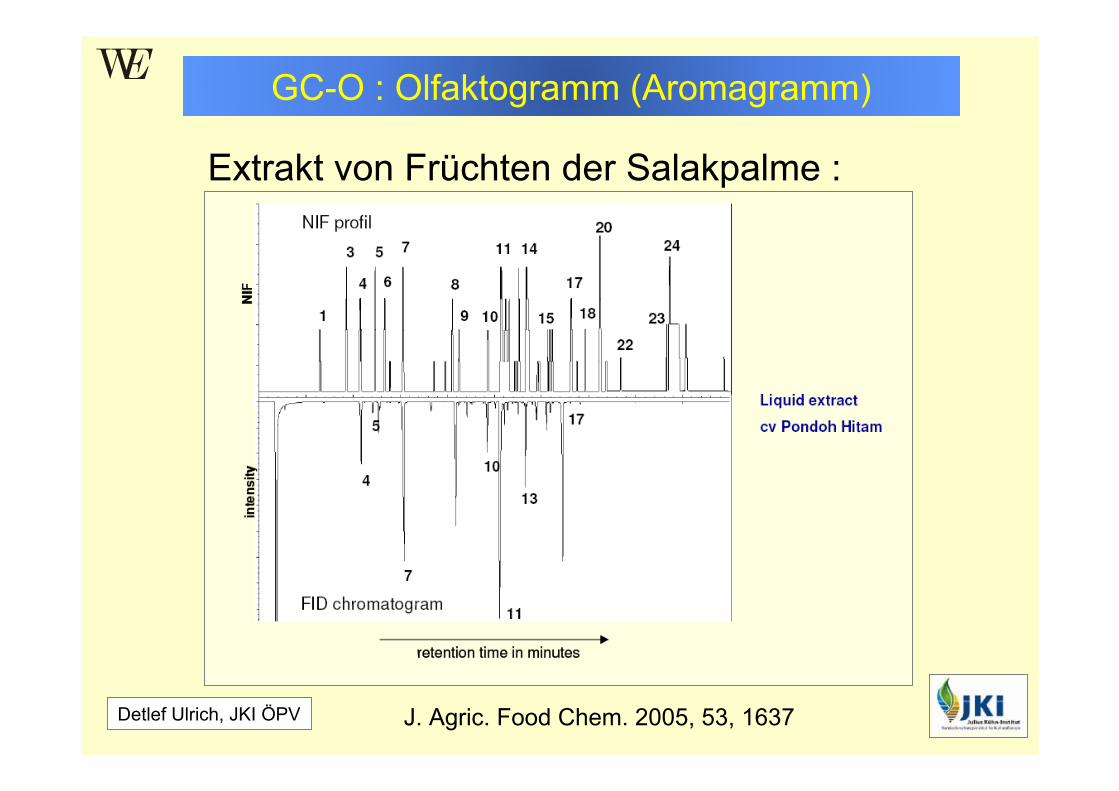
GC-O : Olfaktogramm (Aromagramm)
Detlef Ulrich, JKI ÖPV
Extrakt von Früchten der Salakpalme : Schlangen(haut)frucht
J. Agric. Food Chem. 2005, 53, 1637

GC mit biologischem Detektor
Pheromone : Botenstoffe der InsektenA.Butenandt (Nobelpreis 1939)untersuchte in 50er Jahren die Sexuallockstoffe des Seidenspinners (bombyx mori): Weibchen sendet Substanz aus, die die Männchen ingroßer Entfernung anlockt→ Extrakte aus den Drüsen der Weibchen→ Problem : welche Verbindung(en) wirkt(en) als Lockstoff(e) ?
E. Bayer, F. Anders 1959 : Biologische Objekte als Detektoren zur GCNaturwissenschaften 46, 380 (1959)in einer Kammer am Ende der GC-Säule wurde ein Seidenspinner-männchen plaziert→ keine Reaktion bei Elution der meisten Verbindungen→ Erregung, wirbelnder Tanz bei Elution einer Verbindung Butenandt : Strukturaufklärung(E,Z)-10,12-Hexadecadien-1-ol (Bombykol)
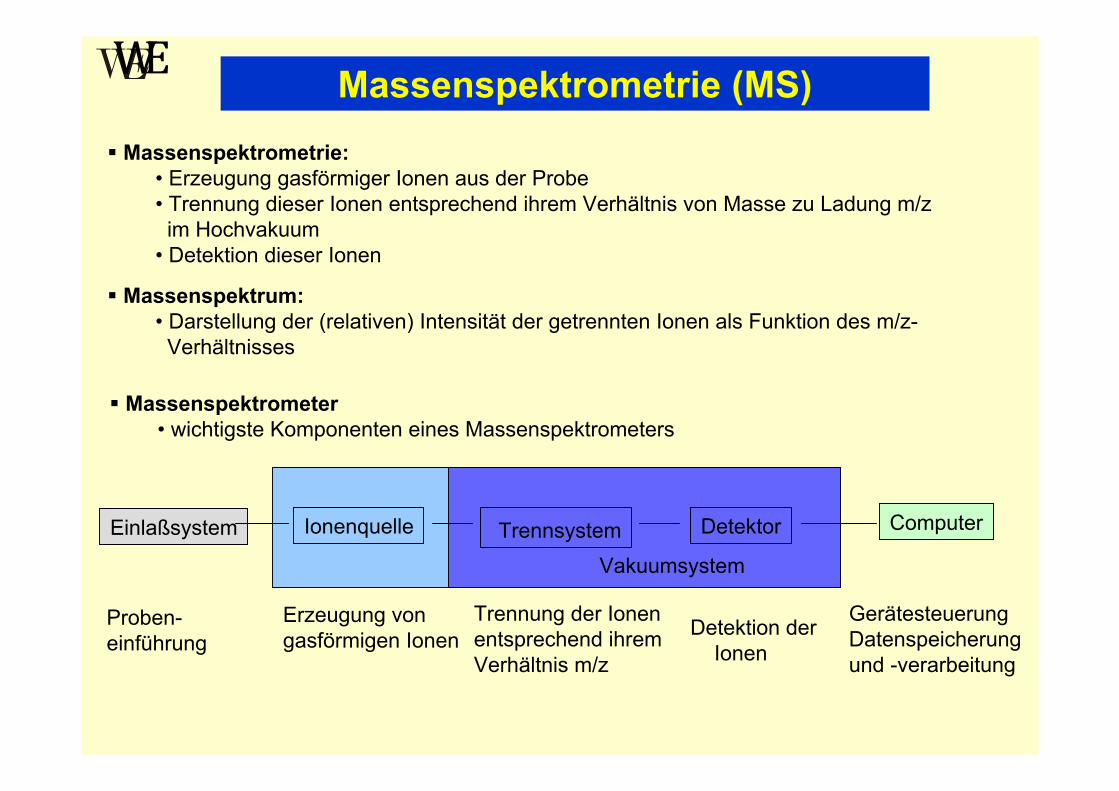
Massenspektrometrie (MS)Massenspektrometrie:
• Erzeugung gasförmiger Ionen aus der Probe• Trennung dieser Ionen entsprechend ihrem Verhältnis von Masse zu Ladung m/z
im Hochvakuum• Detektion dieser Ionen
Massenspektrum:• Darstellung der (relativen) Intensität der getrennten Ionen als Funktion des m/z-
Verhältnisses
Massenspektrometer• wichtigste Komponenten eines Massenspektrometers
Einlaßsystem Ionenquelle Detektor Computer
Proben-einführung
Erzeugung von gasförmigen Ionen
Trennung der Ionen entsprechend ihrem Verhältnis m/z
Detektion der Ionen
Gerätesteuerung Datenspeicherungund -verarbeitung
VakuumsystemTrennsystem
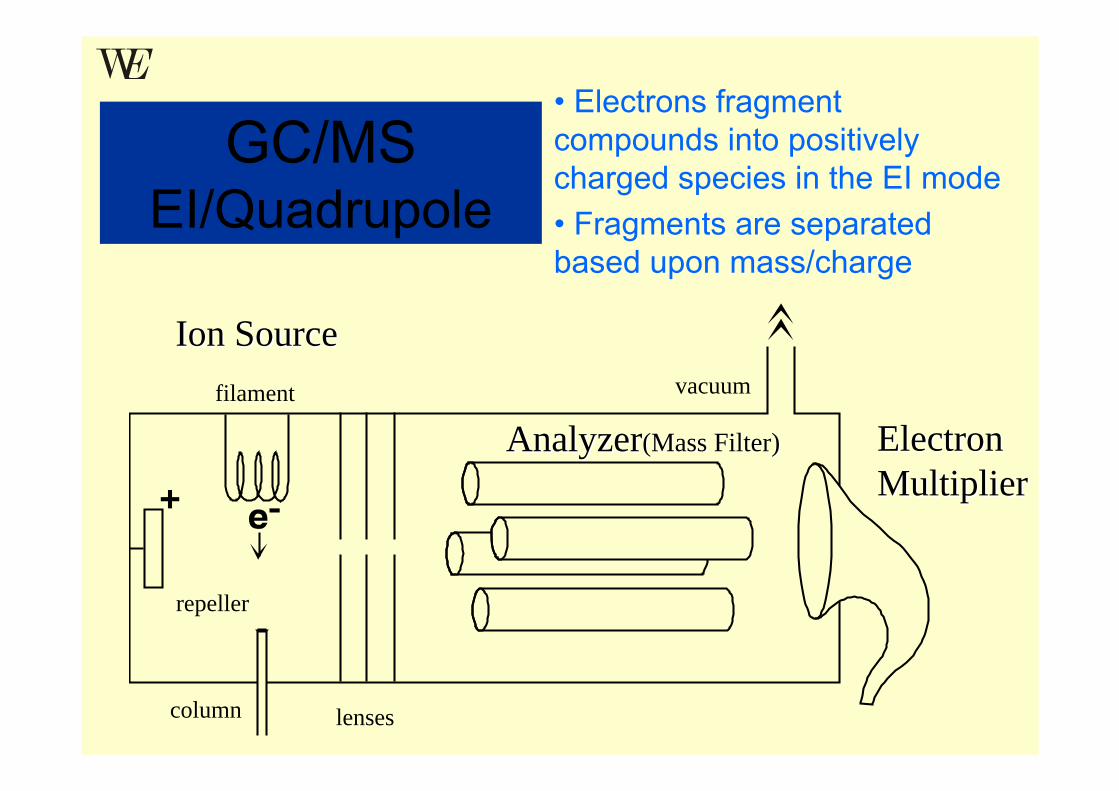
Ion SourceIon Source
AnalyzerAnalyzer(Mass Filter)(Mass Filter) Electron Electron MultiplierMultiplier
GC/MSEI/Quadrupole
column
vacuum
lenses
repeller
filament
• Electrons fragment compounds into positively charged species in the EI mode• Fragments are separated based upon mass/charge
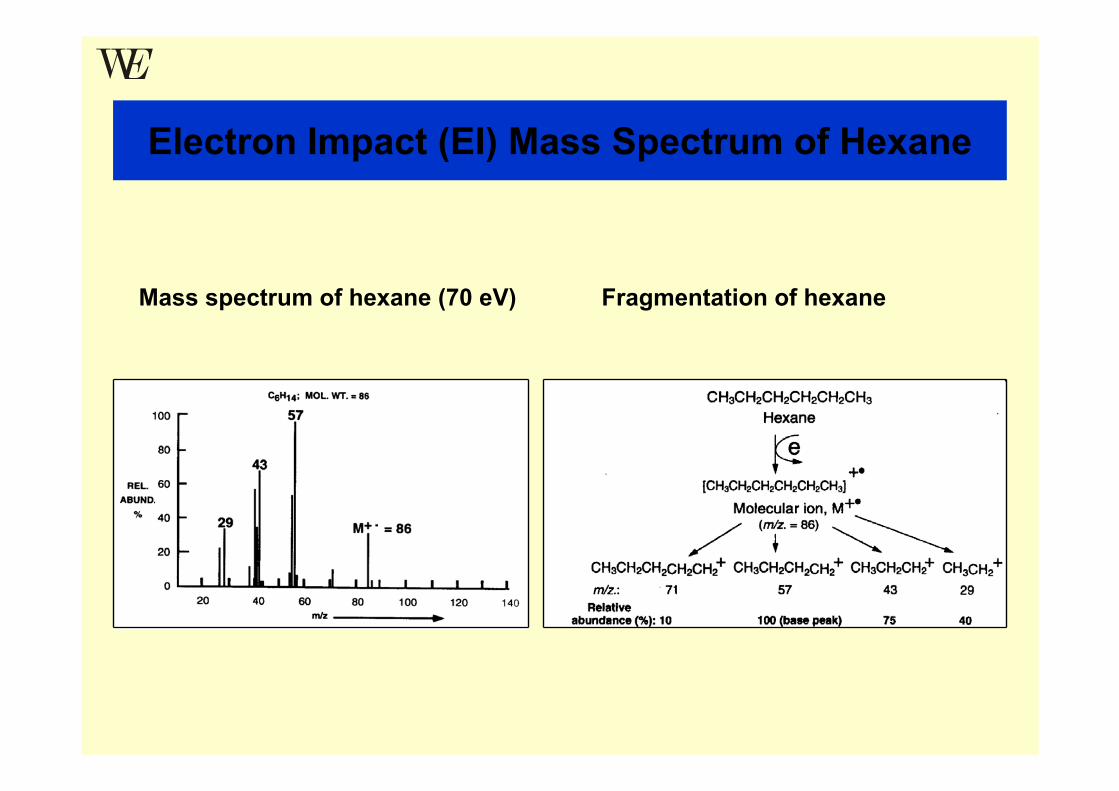
Electron Impact (EI) Mass Spectrum of Hexane
Mass spectrum of hexane (70 eV) Fragmentation of hexane

“Electron Impact” – Mass Spectra
n-Octane C8H18 MW 114
2,2,4-Trimethyl pentane (iso octane) C8H18 MW 114

Größe der Peaks ist Menge der Komponenten in der Probe proportional
Auswertung1. Ermittlung der Peakgröße
Analog-Digital-WandlungMessung der Peakhöhe oder Integration der Peakfläche
2. Kalibrierung und Umrechnung der Peakflächen in Konzentrationen Methoden:• Flächen-Normalisierung (innere Normierung, 100 %-Methode)• Flächen-Normalisierung mit Response-Faktoren• Externe Kalibrierung/Standard• Interner Standard/Kalibrierung (mit Fremdsubstanz)• Standardaddition (Aufstockung, interner Standard mit
analyseneigener Substanz, Spiking)Analyse
Vermeidung bzw. Minimierung der Fehlerquellen in allen Schritten der Analyse
Quantitative Analyse

20 µL Probe
Zugabe interner Standard (Norvaline, 10µL)
Zugabe von Wasser zur Verdünnung der Probe
Zugabe von Propanol and NaOH Lösung, Vortex
Zugabe des Derivatisierungsmittels (Propylchloroformat), Vortex
Zugabe organisches Lösungsmittel zur Extraktion (Isooctan), Vortex
Nach Phasenbildung Injektion eines Aliquot aus der oberen organischen Phase
O
O
Cl+NH2 HR
O OH
NH HR
OO
OO
-CO2
-2 HCl2
cat.
Automatisierte Aminosäureanalyse mittels GC-MS

Automatisierte Aminosäureanalyse mittels GC-MS

Autosampler und Roboter• Autosampler
Ursprünglich zur automatisierten Dosierung flüssiger Probenin GC, HPLC usw.
• Kombination von Autosampler und xyz-RoboterZur Automatisierung komplexer Abläufe (in Steuersoftware des GC eingebunden); vielseitig einsetzbar:– Extraktionstechniken:
Head-space-Extraktion, SPE, SPME, SBSE, in-tube SPME– Probenvorbereitung
Heizen, Kühlen, Schütteln, Filtrieren, Verdünnen, Aufkonzentrieren, Spülen, Derivatisieren (Reagenzzugabe), Zugeben von Standards
– ProbenaufgabeLiquidinjektion LVI Thermodesorption Pyrolyse
4.00 4.50 5.00 5.50 6.00 6.50 7.00 7.50 8.00 8.50 9.00 9.50
50000
100000
150000
200000
250000
300000
350000
400000
450000
500000
Time
Abundance
Tryp
toph
an
Tyro
sine
Hyd
roxy
lysi
ne
His
tidin
eLy
sine
Orn
ithin
e
Glu
tam
ineAm
inop
imel
ic a
cid
Amin
oadi
pidi
c ac
id
Phen
ylal
anin
eG
luta
mat
eH
ydro
xypr
olin
eMet
hion
ine
Asp
arta
teG
luta
min
e-de
com
pTh
iapr
olin
e
Aspa
ragi
neP
rolin
eSe
rine
Thre
onin
eIs
o-Le
ucin
eAl
lo-is
oleu
cine
Leuc
ine
NVa
lβ-
Am
inoi
sobu
tyric
aci
dValin
eα
-Am
inob
utyr
ic a
cid
Gly
cine
Sar
cosi
neAl
anin
e
Cys
tine
Cys
tath
ioni
ne
Prol
ine-
hydr
oxyp
rolin
e
Gly
cine
-pro
line
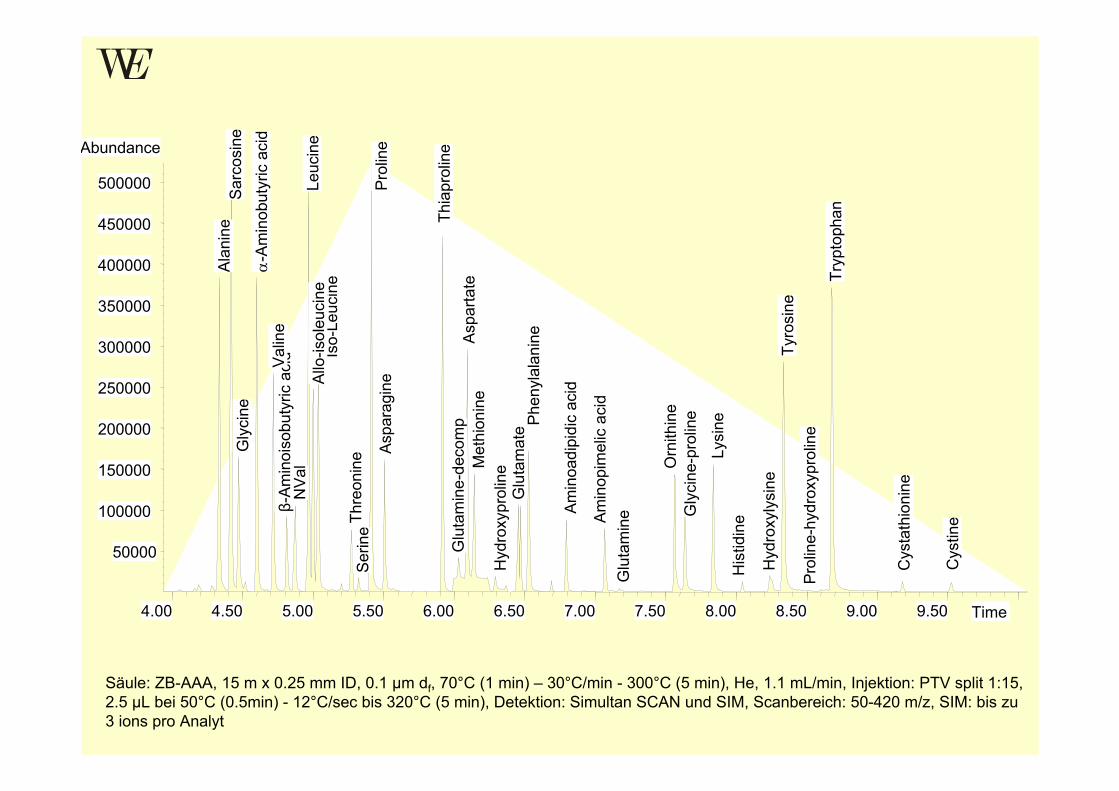
Säule: ZB-AAA, 15 m x 0.25 mm ID, 0.1 µm df, 70°C (1 min) – 30°C/min - 300°C (5 min), He, 1.1 mL/min, Injektion: PTV split 1:15, 2.5 µL bei 50°C (0.5min) - 12°C/sec bis 320°C (5 min), Detektion: Simultan SCAN und SIM, Scanbereich: 50-420 m/z, SIM: bis zu 3 ions pro Analyt

Metaboliten-ProfilanalyseUntersuchung sehr komplex zusammengesetzter biologischer und medizinischer Proben→ Große stoffliche Vielfalt (viele Verbindungsklassen),
weiter Polaritätsbereich→ Großer Konzentrationsbereich
Erfassung möglichst vieler Stoffwechselprodukte in einem Analysenlaufbekannte und unbekannte/unerwartete Verbindungenniedermolekulare Verbindungen
Bestimmung der relativen Konzentrationsänderungenvon veränderten Proben (Umwelteinflüsse, gentechnische Manipulation) im Vergleich zu parallel genommenen Referenzproben(keine absolute Konzentrationsbestimmung!)
Anforderungen an Analysenmethode:schnell (hoher Probendurchsatz), empfindlich, automatisiert, reproduzierbar, robust
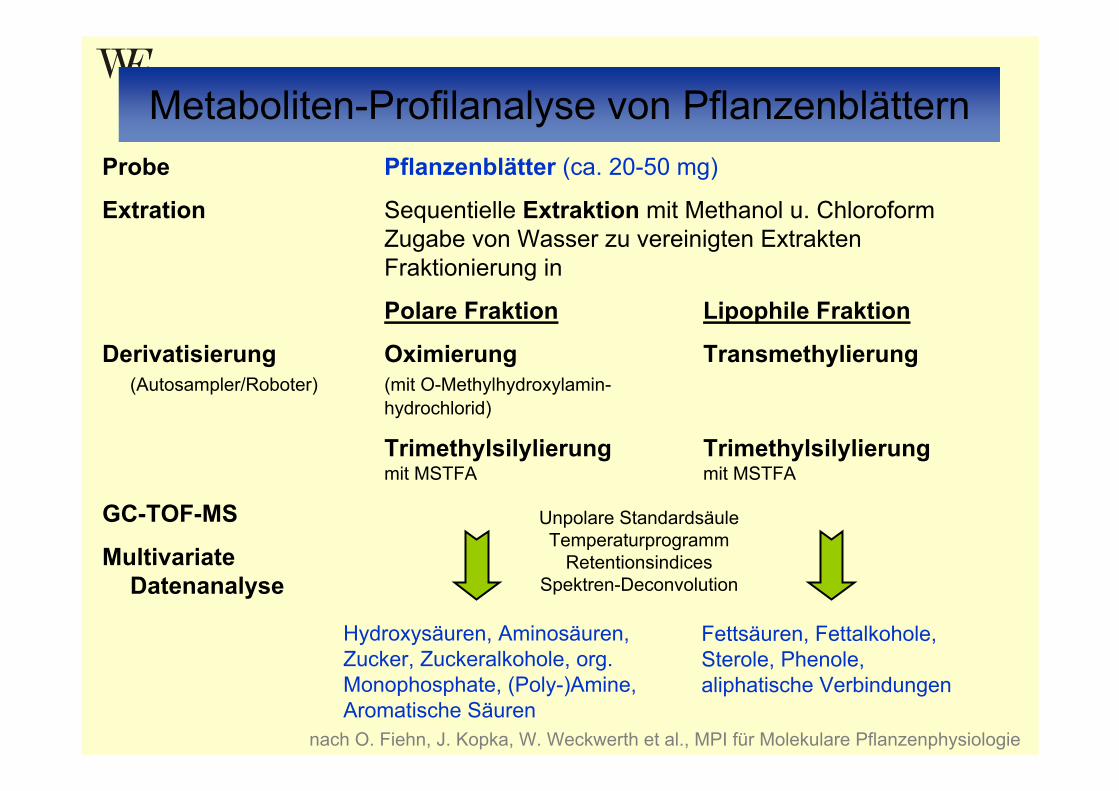
Metaboliten-Profilanalyse von PflanzenblätternProbe Pflanzenblätter (ca. 20-50 mg)
Extration Sequentielle Extraktion mit Methanol u. ChloroformZugabe von Wasser zu vereinigten ExtraktenFraktionierung in
Polare Fraktion Lipophile Fraktion
Derivatisierung Oximierung Transmethylierung(Autosampler/Roboter) (mit O-Methylhydroxylamin-
hydrochlorid)
Trimethylsilylierung Trimethylsilylierungmit MSTFA mit MSTFA
GC-TOF-MS
MultivariateDatenanalyse
Hydroxysäuren, Aminosäuren, Zucker, Zuckeralkohole, org. Monophosphate, (Poly-)Amine, Aromatische Säuren
Fettsäuren, Fettalkohole, Sterole, Phenole, aliphatische Verbindungen
nach O. Fiehn, J. Kopka, W. Weckwerth et al., MPI für Molekulare Pflanzenphysiologie
Unpolare StandardsäuleTemperaturprogramm
RetentionsindicesSpektren-Deconvolution
Weckwerth et al. 2001 Proc. ASMSWeckwerth et al. 2004 PNASWeckwerth et al. 2004 ProteomicsBoldt et al. 2005 Plant CellMorgenthal et al. 2005 MetabolomicsMorgenthal et al. 2005 Biosystems
aliphaticsalcoholsacids
HO -acids NH2 -acids
monosacch..fatty acids
sugar ~phosph..~alcohols
disacch..HO-fatty acids
trisacch..sterols
aliphaticsalcoholsacids
HO -acids NH2 -acids
monosacch..fatty acids
sugar ~phosph..~alcohols
disacch..HO-fatty acids
trisacch..sterols
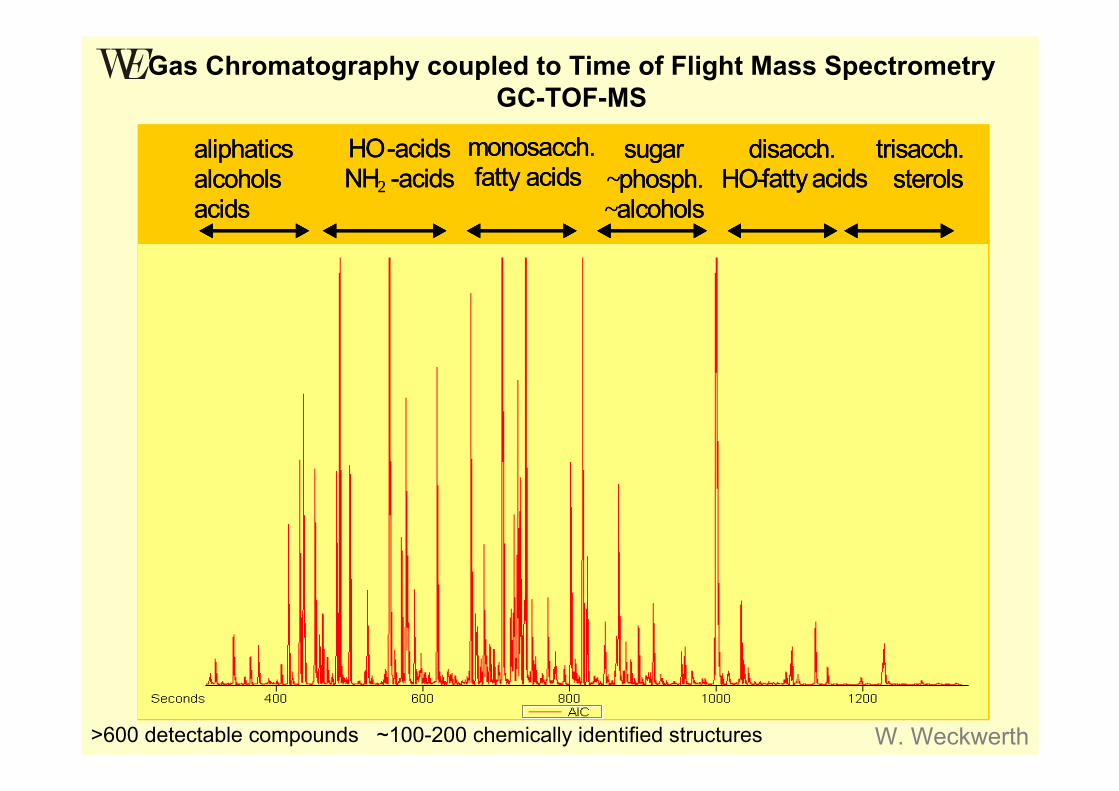
Gas Chromatography coupled to Time of Flight Mass SpectrometryGC-TOF-MS
>600 detectable compounds ~100-200 chemically identified structures W. Weckwerth
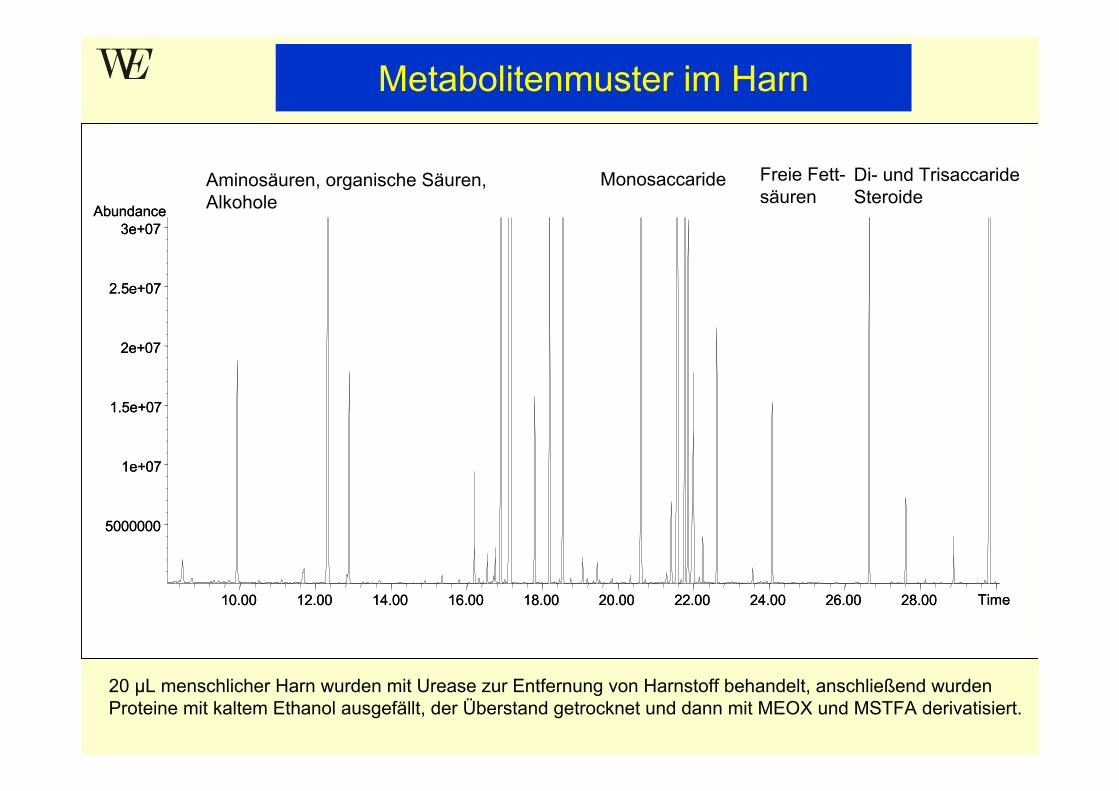
Aminosäuren, organische Säuren, Alkohole
Monosaccaride Freie Fett-säuren
Di- und TrisaccarideSteroide
20 µL menschlicher Harn wurden mit Urease zur Entfernung von Harnstoff behandelt, anschließend wurden Proteine mit kaltem Ethanol ausgefällt, der Überstand getrocknet und dann mit MEOX und MSTFA derivatisiert.
10.00 12.00 14.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00
5000000
1e+07
1.5e+07
2e+07
2.5e+07
3e+07
Time
Abundance
10.00 12.00 14.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00
5000000
1e+07
1.5e+07
2e+07
2.5e+07
3e+07
Time
Abundance
Metabolitenmuster im Harn
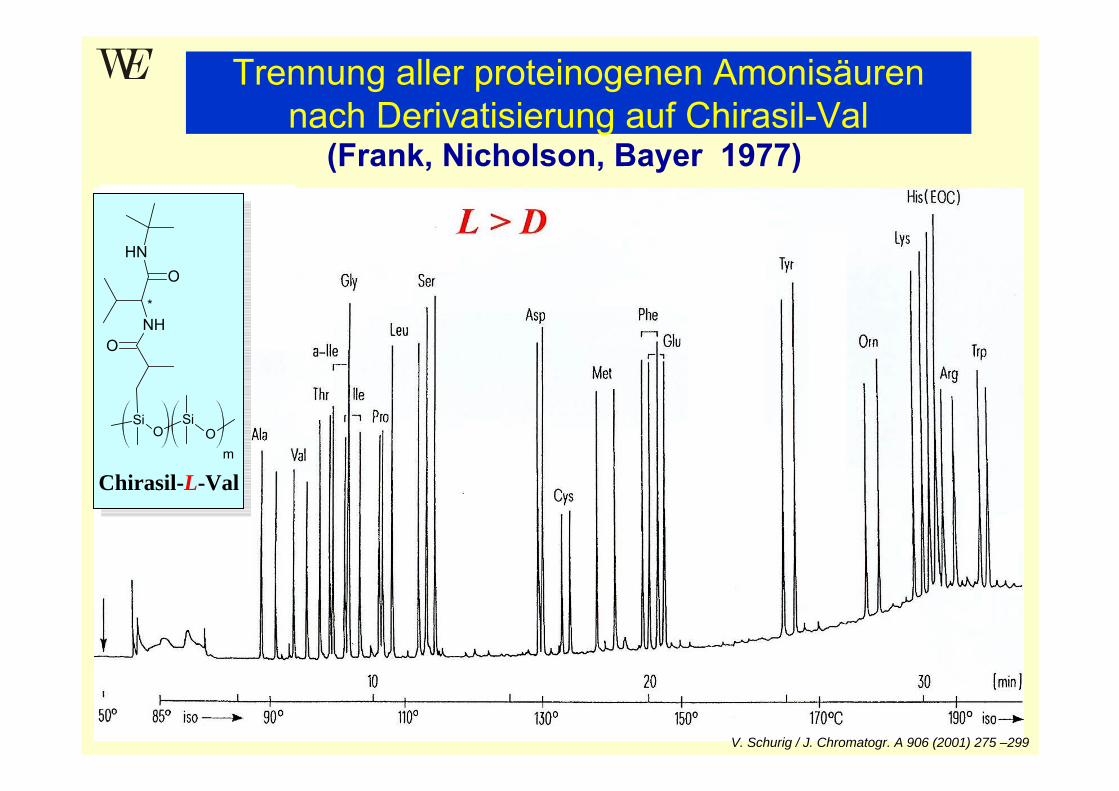
OSi
OSi
NH
ONH
O
*
m
Chirasil-L-Val
(Frank, Nicholson, Bayer 1977)
V. Schurig / J. Chromatogr. A 906 (2001) 275 –299
Trennung aller proteinogenen Amonisäuren nach Derivatisierung auf Chirasil-Val

Landegerät „Philae“ (Rosetta Lander)
http://d1jqu7g1y74ds1.cloudfront.net/wp-content/uploads/2014/09/philae.jpg

Rosetta - Mission
Rosetta - Mission zum Komet 67P• 03/2004: Start der europäischen Raumsonde „Rosetta“ zum Komet
Tschurjumov-Gerassimenko (67P) in 480 km Entfernung• 08/2014: nach 6,4 Mrd. km wurde in eine elliptische Umlaufbahn des
Kometen eingeschwenkt• 12.11.2014: Landung des Landegerätes „Philae“ auf Kometenoberfläche
Benannt nach Stein von Rosetta (Rosetta-Stein):Fragment einer steinernen Stele von 196 v. Chr. mit einer Inschrift in3 versch. Sprachen, 1799 im Nildelta bei ägyptischer Stadt Rosettagefunden ermöglichte gemeinsam mit einem Tempelobelisk von der Nil-Insel Philae die Entzifferung der ägyptischen Hieroglyphen
Kometen – Schweifsterne im Sonnensystem:• bestehen aus Gestein, Staub und Eis: km – große „schmutzige Schneebälle“
leuchtender Schweif (Koma) durch Gas und Staub bei geringerer Entfernung zur Sonne durch Ausgasung und Staubwolken
• entstanden am Anfang unseres Sonnensystems vor ca. 4,5 Mrd. Jahrenwertvolle Erkenntnisse erwartet („Urmaterie“ u.a.)

Landegerät „Philae“ (Rosetta Lander)
12 verschiedene Messgeräte an Bord, darunter
• unterschiedliche Spektrometer
• 2 miniaturisierte GC/MS zur Untersuchung des Bodens und
der Atmosphäre: a) allgemeine Suche nach
organischen Verbindungen
b) Suche nach enantiomeren Aminosäuren:Automatische Extraktion, Derivatisierung und Trennung an 3 verschiedenechiralen Trennsäulen
http://www.cnes.fr/automne_modules_files/standard/public/p11223_35fa30dcb12db70a044488566f7f8096philae_david_ducros.png
http://www.esa.int/var/esa/storage/images/esa_multimedia/images/2013/12/philae_s_instruments_black_background/13466951-4-eng-

Gaschromatographie IMethode der Wahl für die Trennung flüchtiger VerbindungenDampfdruck der Analyten bei Säulentemperatur > 0,1 torr;Säulentemperaturen bis 350 °C
Begrenzte AnwendbarkeitThermisch stabile, verdampfbare Verbindungen (unpolar, schwach polar): MW < 600 (1000) Dalton Kp < 500°C C-Zahl < 50
Erweiterung des Anwendungsbereiches durch• Derivatisierung polarer Verbindungen zu weniger polaren, flüchtige
Derivaten• Definierte thermische Zersetzung der Probe und GC der flüchtigen
Pyrolyseprodukte: Pyrolyse-GC
☺ Übergang in die Gasphase ist elegante Möglichkeit zur Abtrennung der Analyten von unverdampfbarer bzw. schwerflüchtiger Probematrix
DosierungVerschiedene Injektoren und Techniken zur• Dosierung von verdünnten Lösungen: splitless, cold on-column, PTV• Dosierung großer Probenvolumina (LVI)• Lösungsmittelfreie („trockene“) Dosierung

Gaschromatographie II• Hohes Trennvermögen
Kapillarsäulen: L = 25-60mPeakkapazität: 100 – 500(...1000)
N = 100.000 – 300.000 α > 1,02
Temperaturprogrammierte Arbeitsweise → Proben mit weitem Siedebereich
• GC/MSKopplung von 2 „Gasphasentechniken“MS als universal- oder massenselektiver Detektor zur Identifizierung und Quantifizierung (schnell, robust, nachweisstark, großer linearer Bereich)⇒ leistungsfähige und konkurrenzlose Kombination zur A l flü hti

Wichtige Anwendungsgebiete der GC I
• Kohlenwasserstoffanalytik: Erdölraffinerie, PetrolchemieRaffineriegas (LPG; C1 – C5/C6) ⇒
Durchbruch der GCPetrolchemische Grundchemikalien
Prozeß-GCKomplexe KW-Gemische (z.B. Benzin)
• IndividuenanalyseKapillar-GC
• Stoffgruppenanalyse (z.B. PONA, C3 – C12)Mehrsäulentechnik

Wichtige Anwendungsgebiete der GC II
• Geruchs- und Aromastoffe (Flavours), Ätherische Öle„alles, was riecht“
⇒ unerwartet viele Inhaltsstoffe in Ätherischen Ölen
⇒ Erkennung von Verfälschungen
• Umweltanalytik: Wasser, Boden, Luft, Nahrungsmittel• Pestizid-Rückstände in Pflanzen, Tieren,
Nahrungsmitteln⇒Rückstandsanalytik ⇒ Electron Captur
D t t ECD

Derivatisierung in der GC
Chemische Derivatisierunggezielte chemische Veränderung der Probe
Veränderung der Funktionalität der Analyten
Ziele der Derivatisierung
Verbesserung / Ermöglichung der
a) Chromatographierbarkeit:Überführung nichtflüchtiger / polarer / ionischer / labiler / reaktiver Verbindungen in flüchtige / stabile Derivate (Beseitigung aktiver H-Atome)meist Silylierung, Alkylierung, Acylierung
b) Detektierbarkeit (Spurenanalyse):Einführung gut detektierbarer Gruppen oder Elemente zur Anwendung selektiver Detektorenz.B. Einführung von Fluoratomen ECD
MS / NC / GC-AED

Mückenplage, Botenstoffe und GC I
• Problem:
→ Verhalten von Insekten wird durch Botenstoffe gesteuert
• Botenstoffe (Signalstoffe, Semiochemicals):
Warum werden manche Menschen häufig von Mücken gestochen und andere gemieden?

Mückenplage, Botenstoffe und GC II
• Welche flüchtigen Verbindungen werden von Menschen über die Haut freigesetzt?→ Sammlung von „Körperessenz“ von
Versuchspersonen mit unterschiedlicher Affinität zu Mücken
1. Versuchspersonen werden in Al-beschichteten Beuteln eingepackt und gereinigte Luft darüber geleitet, die anschließend über Adsorbentien geführt wird
2. Extraktion der Adsorbentien mit hochreinen Lösungsmittel

Mückenplage, Botenstoffe und GC III
• Welche Verbindungen der „Körperessenz“wirken als Signalstoffe für Mücken?→ Kombination der GC mit
elektrophysikalischen Techniken:GC mit Teilung des Gasstromes am Ausgang der Trennsäule und Paralleldetektion:a) GC-Universaldetektor: FID oder MS →
Substanzerkennungb) „Antennographischer“ Detektor → biologische
Wirkung
Verknüpfung der Antennen von Mücken mit Mikroelektroden→ Wenn Geruchssensor der Mücke durch eine

Mückenplage, Botenstoffe und GC IV
• Resultat• Annahme: Für Mücken attraktive
Personen produzieren Stoffe, die Mücken anlocken
• Ergebnis: Für Mücken unattraktive Personen produzieren Stoffe, die eine abstoßende Wirkungausüben (Interferenzen bei der Suche nach attraktiven Oberflächen)besonders wirksam: 6-Methyl-5-hepten-2-on
Geranylaceton
N
O
N
O O O
N,N-Diethyl-m-toluamid(DEET)
(E)-N-Cyclohexyl-N-ethyl-2-hexenamid
6-Methyl-5-hepten-2-on Geranylaceton
aus: Chemistry World, Sept. 2010, 44-47

Testosteron – gedopt oder körpereigen?Testosteron-DopingDoping mit Testosteron und seinen Prohormonen
fördert Muskelwachstum (Kraft) und Aggressivitätbeschleunigt Erholung nach Training/Wettkämpfenschwer nachweisbar
GC/MS-Nachweis von Testosteron in Urinnach Extraktion und Derivatisierung (Trimethylsilylierung)→ Erlaubt keine Unterscheidung, ob Testosteron vom Körper selbst
synthetisiert oder durch Doping von außen eingeführt wurde, da Testosteron-Gehalt individuell sehr unterschiedlich
T/E-QuotientDonike: Verhältnis Testosteron/Epitestosteron (Stereoisomeren von T;
kein Stoffwechselprodukt von T) ist bei meisten Menschen relativ konst.)
T/E>6 wird als positiv bewertet, wenn dieser Wert deutlich oberhalb desindividuellen T/E-Referenzbereiches des Athleten liegt
Substanzspezifische Isotopenanalytik: KGC-Combustion-IRMS (13C/12C)
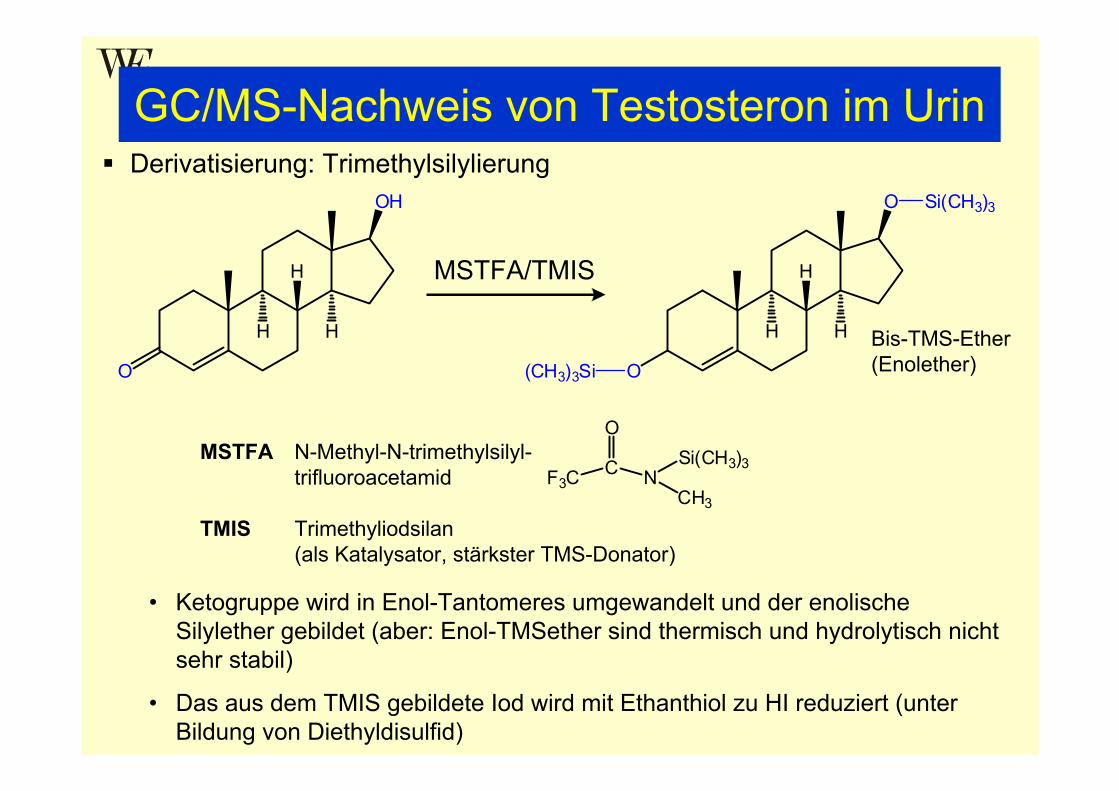
GC/MS-Nachweis von Testosteron im UrinDerivatisierung: Trimethylsilylierung
O
OH
H
H
H
MSTFA/TMIS
O
O
H
H
H
(CH3)3Si
Si(CH3)3
MSTFA N-Methyl-N-trimethylsilyl-trifluoroacetamid
TMIS Trimethyliodsilan(als Katalysator, stärkster TMS-Donator)
• Ketogruppe wird in Enol-Tantomeres umgewandelt und der enolische Silylether gebildet (aber: Enol-TMSether sind thermisch und hydrolytisch nicht sehr stabil)
• Das aus dem TMIS gebildete Iod wird mit Ethanthiol zu HI reduziert (unter Bildung von Diethyldisulfid)
Bis-TMS-Ether(Enolether)
F3CC N
OSi(CH3)3
CH3

Multikomponenten-Analyse
Rückstandsanalytik in Lebens- und Futtermitteln Verschärfte Bestimmungen :→ Nachweis einer steigenden Anzahl von Analyten
in immer geringeren Konzentrationenin immer komplizierteren Matricesin kurzer Zeitbei möglichst geringen Kosten
MultimethodenSpurenanalyse einer möglichst großen Anzahl von Zielverbindungen (targets) in einem Analysenlauf
Anforderungen• empfindliche , einfache, schnelle Methoden
HPLC-MS oder GC-MS im SIM-ModusHPLC-MS/MS oder GC-MS/MS im MRM-Modus
möglichst viele MRM-Übergänge in einem Lauf
• einfache und möglichst universelle Probenvorbereitungz. B. QuEChERS: aber Extrakte enthalten noch Probenmatrix
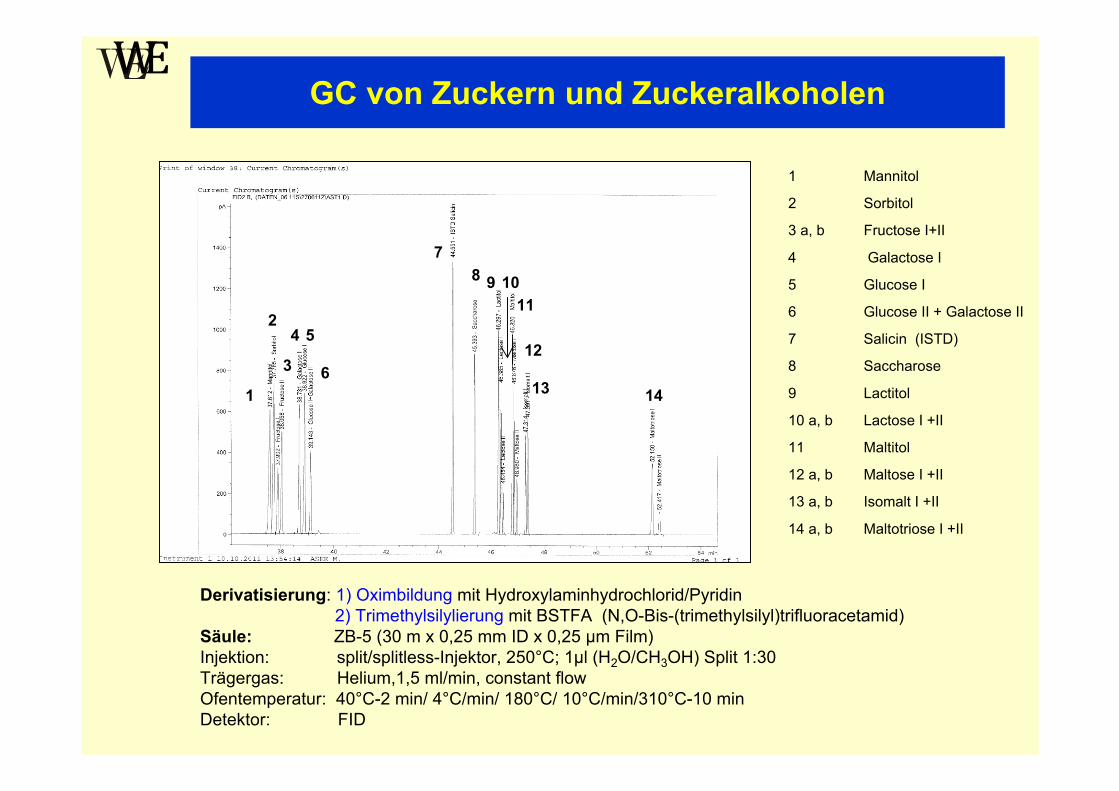
GC von Zuckern und Zuckeralkoholen
2
1 Mannitol
2 Sorbitol
3 a, b Fructose I+II
4 Galactose I
5 Glucose I
6 Glucose II + Galactose II
7 Salicin (ISTD)
8 Saccharose
9 Lactitol
10 a, b Lactose I +II
11 Maltitol
12 a, b Maltose I +II
13 a, b Isomalt I +II
14 a, b Maltotriose I +II
Derivatisierung: 1) Oximbildung mit Hydroxylaminhydrochlorid/Pyridin2) Trimethylsilylierung mit BSTFA (N,O-Bis-(trimethylsilyl)trifluoracetamid)
Säule: ZB-5 (30 m x 0,25 mm ID x 0,25 µm Film)Injektion: split/splitless-Injektor, 250°C; 1µl (H2O/CH3OH) Split 1:30Trägergas: Helium,1,5 ml/min, constant flowOfentemperatur: 40°C-2 min/ 4°C/min/ 180°C/ 10°C/min/310°C-10 minDetektor: FID
1
2
3
4 5
6
78 9 10
11
12
13 14

Bratwürste im Test
Stiftung Warentest (2010) 19 gebrühte Bratwürste verschiedener
Anbieter getestet:• Sensorische Bewertung vor und nach dem Grillen
• Fleischqualität:- Anteil Muskelfleisch- Anteil Bindegewebe- Fettgehalt- Nachweis von ZNS-Gewebe…
• Mikrobiologische Qualität- Hefen- Schimmelpilze- Milchsäurebakterien…

Hintergrund: BSE, ZNS und SRM
• BSE-Krise: 2000/2001 RinderseucheBSE: Bovine Spongiforme Enzephalopathie, Rinderwahn
• schwammförmige Veränderung des Gehirns:Gleichgewichtsstörungen, Krämpfe
• zuerst in GB aufgetreten (Verfütterung von Tiermehl auserkrankten Schafen an Rinder)
• auf Menschen übertragbar (Creutzfeldt-Jacob-Krankheit)• SRM - Spezifiziertes Risikomaterial: Gewebe des zentralenNervensystems (ZNS: Gehirn, Rückenmark) von Rindern, Schafen undZiegen, die älter als 12 Monate sind (potentielle Träger des Erregers)
• EU - Verordnung (EG) Nr. 999/2001: Vorsorge für VerbraucherSRM darf nicht in Fleischerzeugnisse gelangen
Muss während der Schlachtung entfernt und beseitigt werdenKontrolle erforderlich: Bestimmung von Tierart und Tieralter zumZeitpunkt der Schlachtung

Nachweis von SRM in Fleischerzeugnissen
• Immunchemische (ELISA) und molekularbiologische (PCR)VerfahrenZum Nachweis von ZNS-Markersubstanzen (Cholesterin, bestimmteProteine und deren m-RNA)ABER: Ist evtl. nachgewiesenes ZNS-Material tatsächlich Risikomaterial?
Bestimmung von Tierart und Tieralter erforderlich
• GC/MS-Analyse von ZNS-typischen Fettsäuremustern• Nachweis von ZNS-typischen Fettsäuren (Fatty Acids - FA):Cerebronsre. isomere Tetracosensäuren Hydroxytetracosensäure2-OH-C24:0 C24:1ω9, C24:1ω7 C24:1ω9-OH, C24:1ω7-OHAus ihren Verhältnissen können Tierart und –alter bestimmt werden
• ZNS-typische FA liegen als komplex gebundene Lipide vorErfordert mehrstufige Probenvorbereitung und Derivatisierung

GC/MS-Analyse von ZNS-typischen Fettsäuren I
Mehrstufige ProbenvorbereitungZNS-typische FA liegen als komplex gebundene Lipide vor erfordert:
• Extraktion aus Fleischer-zeugnissen
• Abtrennen der Matrix sowie der freien FA, einfachenLipide, Triglyceride …
• Anreicherung• Derivatisierung
• kalte LM-Extraktion mit n-Hexan/MeOH• SPE (supelclean LC-Si) mit n-Hexan/MePropOH gewaschen, Elution mitMeOH
• Zugabe interner Standard• Abdampfen des LM• Methylierung FAME (methanol. HClbei 90°C,1,5h)
• Trimethylsilylierung Hydroxy-FA(TMSIM in DMF bei 100°C, 5min)
• In Hexan aufnehmen
Zeitaufwand: ca. 8 Stunden

GC/MS-Analyse von ZNS-typischen Fettsäuren II
GC/MSSäule: DB5 30m x 0,32mm x 0,25µmTemperaturprogramm:MS-Detektion im SIM-Modus
Auswertung1. Erkennung von ZNS
Cerebronsäure C24:0-OH dient als Markersubstanz für ZNSExterne Kalibrierung, MatrixkalibrierungSchwellwert (cut-off): 2,43 mg/kg = 0,2% ZNS-Gehalt
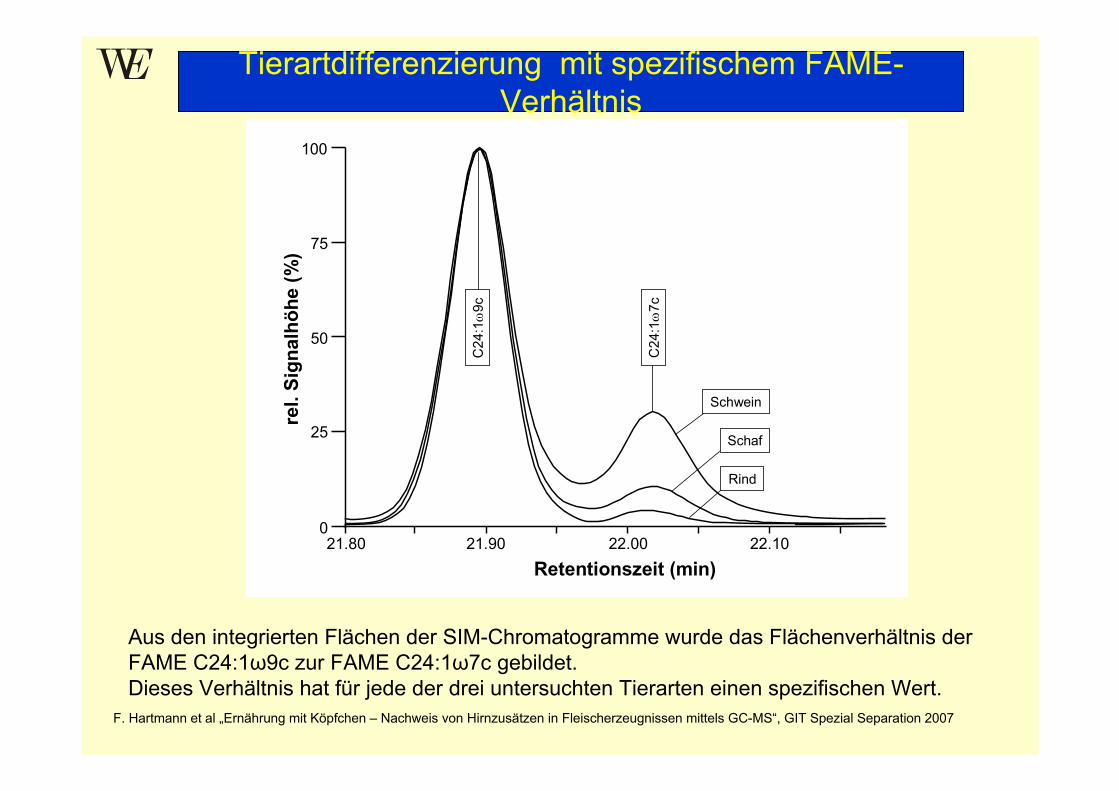
2. Tierartendifferenzierung (bei ZNS-positiven Proben) Charakteristische Peakflächenverhältnisse der ZNS-FA für Rind, Schafund Schwein:A: C24:1ω9/C24:1ω7 B: C24:1ω9-OH/C24:1ω7-OH C: A/B
3. TieralterdifferenzierungMit zunehmendem Alter steigen die Verhältnisse A und B an

18.00 20.00 22.00 24.00 26.00 28.00 30.00 32.0050000
100000150000200000250000300000350000400000450000500000550000600000650000700000750000800000
Retentionszeit [min]
Sign
alhö
he
IS C22:0 d 2,2
C24:1(n-9)
C24:1(n-7)
C24:0
2OH-C24:1(n-9)
2OH-C24:1(n-7)
2OH-C24:0
2OH-C25:0
2OH-C26:1(n-9)
2OH-C26:1(n-7)
2OH-C26:0
SIM-Chromatogramm der Fettsäuremethylester aus einem Schafhirn
Derivatisierung: Veresterung mit MeOH /HCl; Silylierung der Hydroxyfettsäuren mit TMSIM GC: 30 m HP-5MS , 0,32 mm ID, 0,25 µm df
1 µl splitlos (Split 1:15 nach 1 min)Helium (constant flow 1,5 ml/min, v = 32 cm/s)Temperaturprogramm
MS: SIM-ModusFAME RT [min] Ionen [m/z]
Zeitfenster
ab: [min]
C22:0 2,2-D2 19,0 76, 145 18,6
C24:1n9c 22,0 348 21,0
C24:1n7c 22,1 348 21,0
C24:0 22,474, 143, 339,
38222,4
C24:0-OHsil 26,4 73, 411, 455 25,0
M. Grießbach et al „Species and age determination of central nervous system tissue by fatty acid patterns“, J Chromatogr A, 1179 (2008) 69-73
21.90 22.00 22.1021.80
C24
:1ω
9c
Schaf
Schwein
Rind
Retentionszeit [min]
rel.
Sign
alhö
he [%
]
25
0
50
75
100
C24
:1ω
7c
Retentionszeit (min)
rel.
Sign
alhö
he (%
)
Tierartdifferenzierung mit spezifischem FAME-Verhältnis
Aus den integrierten Flächen der SIM-Chromatogramme wurde das Flächenverhältnis der FAME C24:1ω9c zur FAME C24:1ω7c gebildet. Dieses Verhältnis hat für jede der drei untersuchten Tierarten einen spezifischen Wert.
F. Hartmann et al „Ernährung mit Köpfchen – Nachweis von Hirnzusätzen in Fleischerzeugnissen mittels GC-MS“, GIT Spezial Separation 2007

GC und GC/MS in Pharmaanalytik
„Klassische Qualitätskontrolle“Verunreinigungen über 0,05 % in pharmakologisch wirksamen Substanzen(active pharmaceutical ingredients - API`s)
Restlösemittel in pharmazeutischen Produkten : OVI organic volatileimpurities→ Einteilung der LM nach ihrem toxikologischen Risiko in 3 KlassenAllergene Verbindungen
Suspected Allergenes in Essential Oils, Flavours, Fragrances :Substanzliste („Q6-Verbindungen“)Deklarationspflicht , wenn über 0,001 % (10 ppm) für „leave-on“ Produkte
(verbleiben auf der Haut)0,01 % (100 ppm) für „rinse-off“ Produkte
( werden wieder abgespült)
Gentoxische und mutagene Verbindungen in pharmazeutischenProdukten2006 : Grenzwert für gentoxic impurities (GI´s): max. 1,5 µg pro Person und TagListe von Substanzgruppen (z.B. Alkylsulfonate)
PharmaverpackungenExtractables & Leachables (E&L) aus Primärverpackungen von pharma-zeutischen Formulierungen
1ppm = 1mg/kg^