







Sprachen
Seiten
Rechtliche


1
Bioinformatik I: Grundlagen der Gentechnik
Prof. Jörg Kudla (Freisemester) Prof. Dr. Antje von SchaewenInstitut für Biologie &Biotechnologie der PflanzenSchlossplatz 7Email: [email protected] [email protected]
Schwerpunkte:
Vorlesung 1: Einführung & Enzyme der GentechnikVorlesung 2: Methoden der GentechnikVorlesung 3: Vektoren & KlonierungVorlesung 4: ReportergeneVorlesung 5: Transgene OrganismenVorlesung 6: Genomik und moderne MethodenVorlesung 7: Proteine
Klonierung: Das Grundlegende Experiment der Gentechnologie

2
Animation Plasmid cloning
Klonierungsvektoren - das Prinzip

3
Klonierungsvektoren
- Vektoren zum Klonieren genomischer DNA
- ermöglichen Vermehrung und Analyse von bestimmten DNA-Fragmenten
- Klonierungskapazität ?- Handhabbarkeit ?- Anwendung ?- Stabilität der klonierten DNA?
Handhabbarkeit versus Klonierungskapazität (DNA-Fragmente)Plasmide max. 8 kb ( 1- 5 kb)Lambda-Phagen max. 25 kb ( 10- 20 kb)Cosmide max. 44 kb ( 30- 44 kb)P1-Phagen max. 100 kb ( 75- 100 kb)BAC max. 300 kb (100- 300 kb)YAC max. 1000 kb (100-1000 kb)
BAC: bacterial artificial chromosomeYAC: yeast artificial chromosome
Plasmide• Klonierungskapazität bis 8 kb
• leichte Handhabbarkeit
• relativ stabil
• einfache Transformation von E. coli
• einfache Vermehrung in E. coli
• einfache Plasmid-Isolierung
• hohe DNA-Ausbeuten (ori-abhängig)
• einfache Insert-Analyse
• für versch. Organismen
Selektionsmarker
Reporter-Gen
Vektor + Insert Trafo in E.coli
Ligase
Klonierung:
Ligation DNA-IsolierungVermehrung in E.coli
ori MCS

4
Shuttle-Vektoren
E. coliS. cerevisiae
Der Phage Lambda ()

5
• Temperenter E. coli -Phage
• Kopf und Schwanz (nicht kontraktil)
• DNA (linear, 48.502 bp)
• Komplexe Regulation
Der (Bakterio-) Phage Lambda ()
Bacterium - altgriechisch βακτήριον „Stäbchen“Phage - altgriechisch φαγεῖν „fressen“Temperent - lateinisch Temperantia „Mässigung“
Der Phage Lambda - Lytischer und lysogener Zyklus

6
Der Phage Lambda - Plaque-Bildung auf Bakterienrasen
Phagenplatte Bakterienplatte
Der Phage Lambda - Regulation von lytischem vs. lysogenem Zyklus
schematisch

7
Das Genom des Phagen Lambda ()
detailliert
17 kb 17 kb 14 kb
halb-schematischcos cohesive site (or end)cosmid half phage half plasmid
Das Genom des Phagen Lambda ()

8
Der Phage Lambda ()
left arm right armstuffer fragmentcos cos
Infektion der Wirtszelle
VermehrungLyse der Wirtszelle
LytischerZyklus
DNA des Phagen Lambda: 48,5 kb
cohesive site cohesive site
Replacement Vektoren
Nach Entfernen des stuffer Fragments können DNA-Fragmente, d.h. 10-23 kb Inserts, eingebracht werden
Restriction digest
9
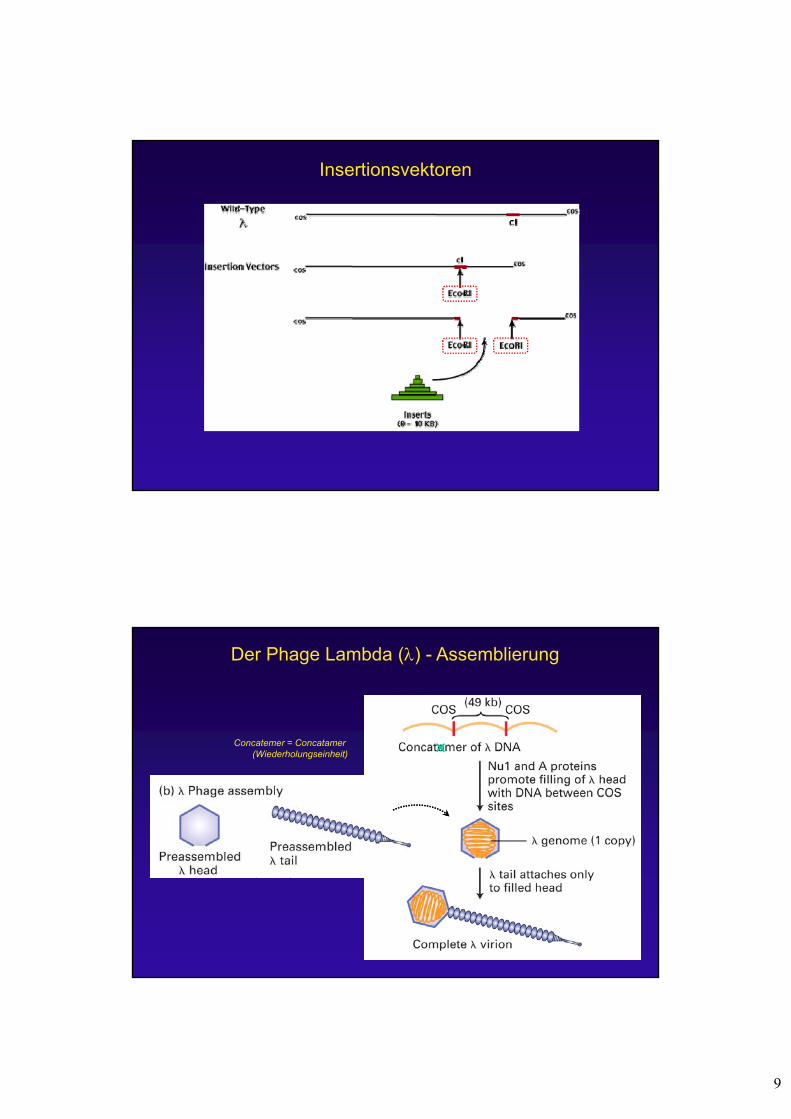
Insertionsvektoren
Concatemer = Concatamer(Wiederholungseinheit)
Der Phage Lambda () - Assemblierung

10
In vitro Verpacken von DNA in Phagen
Extrakt Extrakt
-Dts -Ets
+ DNA
Herstellung einer Phagen DNA-Bank
Klone finden durchSouthern Blotting, d.h. Hybridisieren mitmarkierten DNA-Sonden

11
Screening einer genomischen Phagen DNA-BankKlone auffinden:
Filter lift von Phagen-Platten & “Southern blot”-Hybridisierungmit markierten DNA-Sonden(nach Lyse & Denaturierung in DNA-Einzelstränge)
Filter aus Nitrocellulose (NC) oder Nylon (stabiler)
Nachweisfrüher radioaktiv, heute auchcolorimetrisch oderchemilumineszent
Re-Infektion von Bakterienzur Vermehrung & Archivierungals Lysat (bei -20/-80 C, stabilisiert mit 10% DMSO, Dimethylsulfoxid)
Phage screening I
Freezer(-20/-80 C)
Maltose transportprotein = Phage receptor
cold room(4 C)

12
Phage screening II
Filter lift assay
Phage screening III
glassPasteur pipette
Primary pick
13
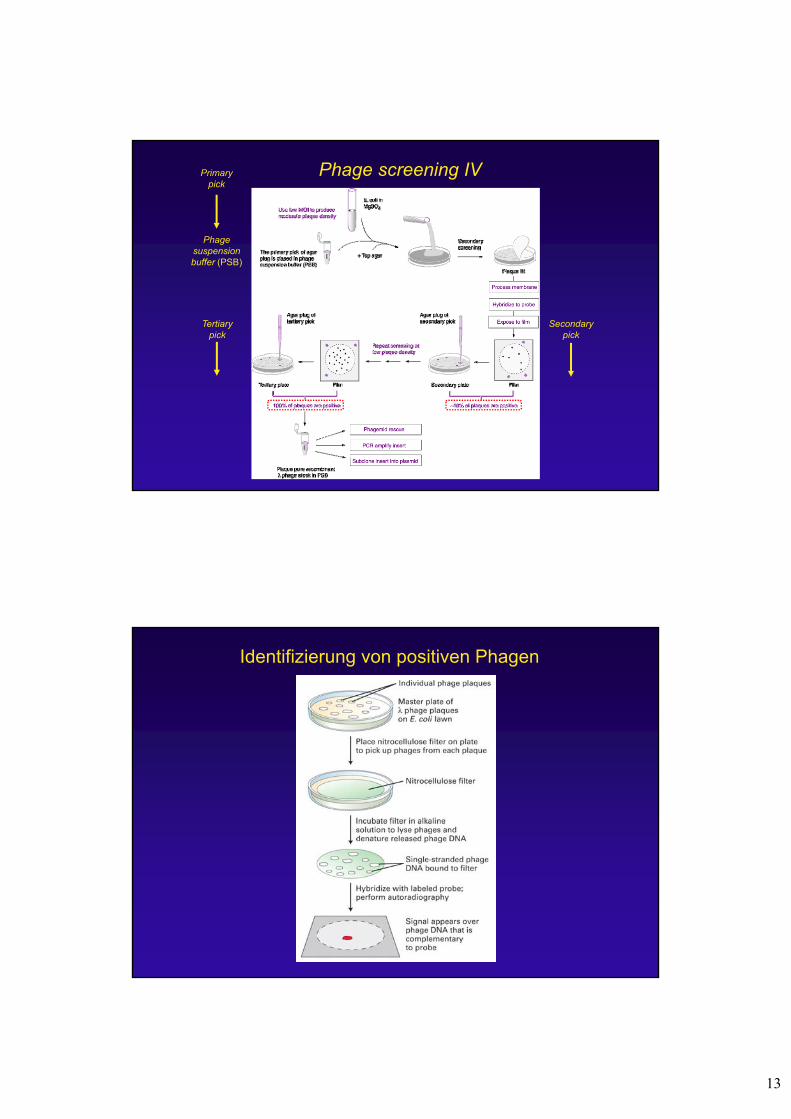
Phage screening IV
Phage suspensionbuffer (PSB)
Primary pick
Secondarypick
Tertiarypick
Identifizierung von positiven Phagen

14
cDNA Banken: 1. Strang (cDNA-Synthese)
Synthese des komplementären Doppelstrangs
15
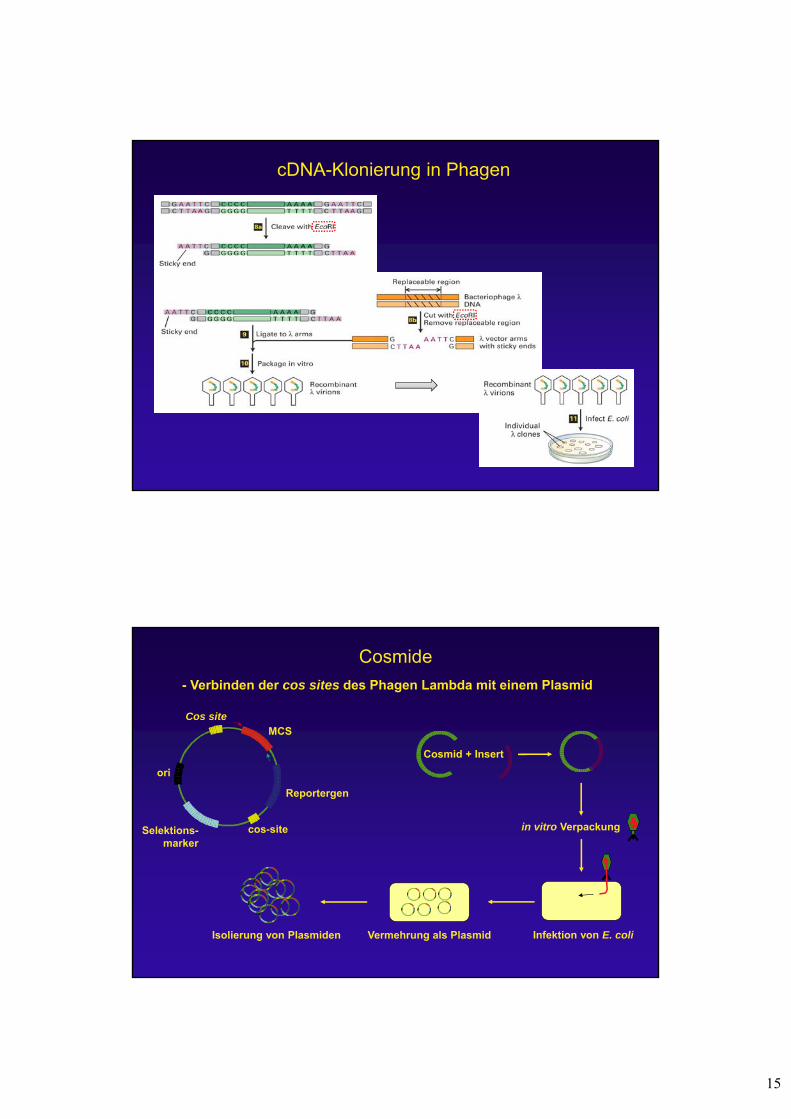
cDNA-Klonierung in Phagen
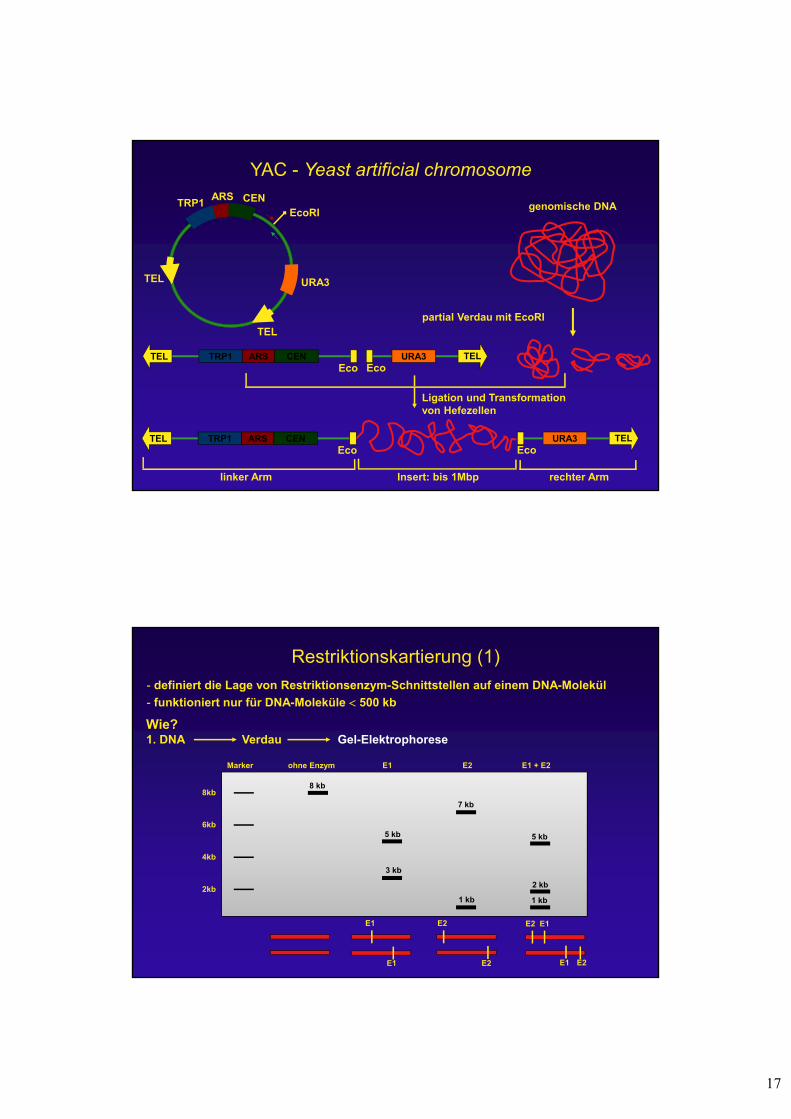
Cosmide
- Verbinden der cos sites des Phagen Lambda mit einem Plasmid
in vitro Verpackung
Infektion von E. coliVermehrung als Plasmid
cos-site
Cos siteMCS
Reportergen
Selektions-marker
ori
Cosmid + Insert
Isolierung von Plasmiden

16
Herstellung einer Cosmid DNA-Bank
BAC - bacterial artificial chromosome
genomische DNA
partiell verdaute DNA
Transformationvon E. coli
DNA-Isolierung Vermehrung in E. coli
MCS
Reportergen
Selektions-marker
(ParA, ParB)ori
Cosmid + Insert
17
YAC - Yeast artificial chromosome
EcoRI
URA3
TEL
TEL
TRP1ARS CEN
TRP1 ARS CENTEL URA3 TELEco Eco
genomische DNA
partial Verdau mit EcoRI
Ligation und Transformation von Hefezellen
TRP1 ARS CENTEL URA3 TELEco Eco
linker Arm rechter ArmInsert: bis 1Mbp
Restriktionskartierung (1)
- definiert die Lage von Restriktionsenzym-Schnittstellen auf einem DNA-Molekül
- funktioniert nur für DNA-Moleküle 500 kb
E1 E2
E1E2
E2
E2
E1
E1
Marker ohne Enzym E1
8kb
E1 + E2
6kb
4kb
2kb
E2
5 kb
8 kb
3 kb
7 kb
1 kb
5 kb
2 kb
1 kb
Wie?1. DNA Verdau Gel-Elektrophorese
18
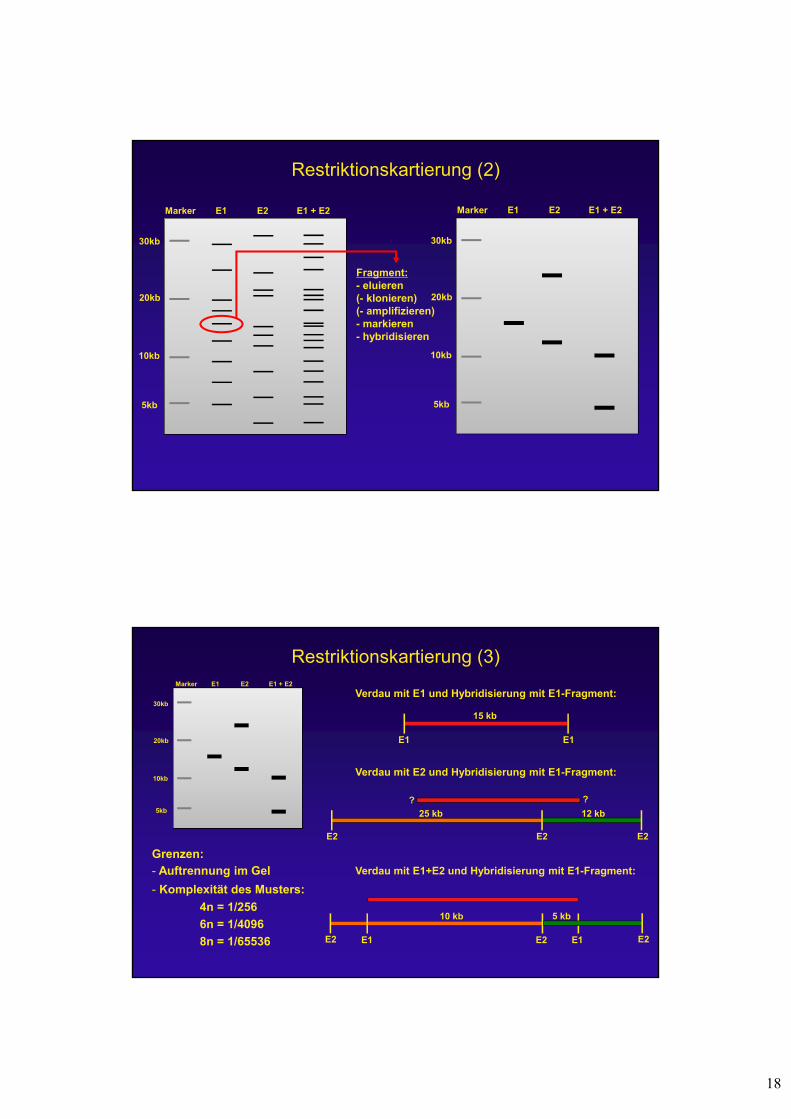
Restriktionskartierung (2)
Marker
30kb
E1 + E2
20kb
5kb
10kb
E1 E2
Fragment:- eluieren(- klonieren) (- amplifizieren)- markieren- hybridisieren
Marker
30kb
E1 + E2
20kb
5kb
10kb
E1 E2
Restriktionskartierung (3)Marker
30kb
E1 + E2
20kb
5kb
10kb
E1 E2
E1 E1
15 kb
E2 E2E2
25 kb 12 kb
Verdau mit E1 und Hybridisierung mit E1-Fragment:
Verdau mit E2 und Hybridisierung mit E1-Fragment:
Verdau mit E1+E2 und Hybridisierung mit E1-Fragment:
Grenzen:
- Auftrennung im Gel
- Komplexität des Musters:
4n = 1/256
6n = 1/4096
8n = 1/65536 E2 E2E1 E1
10 kb 5 kb
E2
??
19
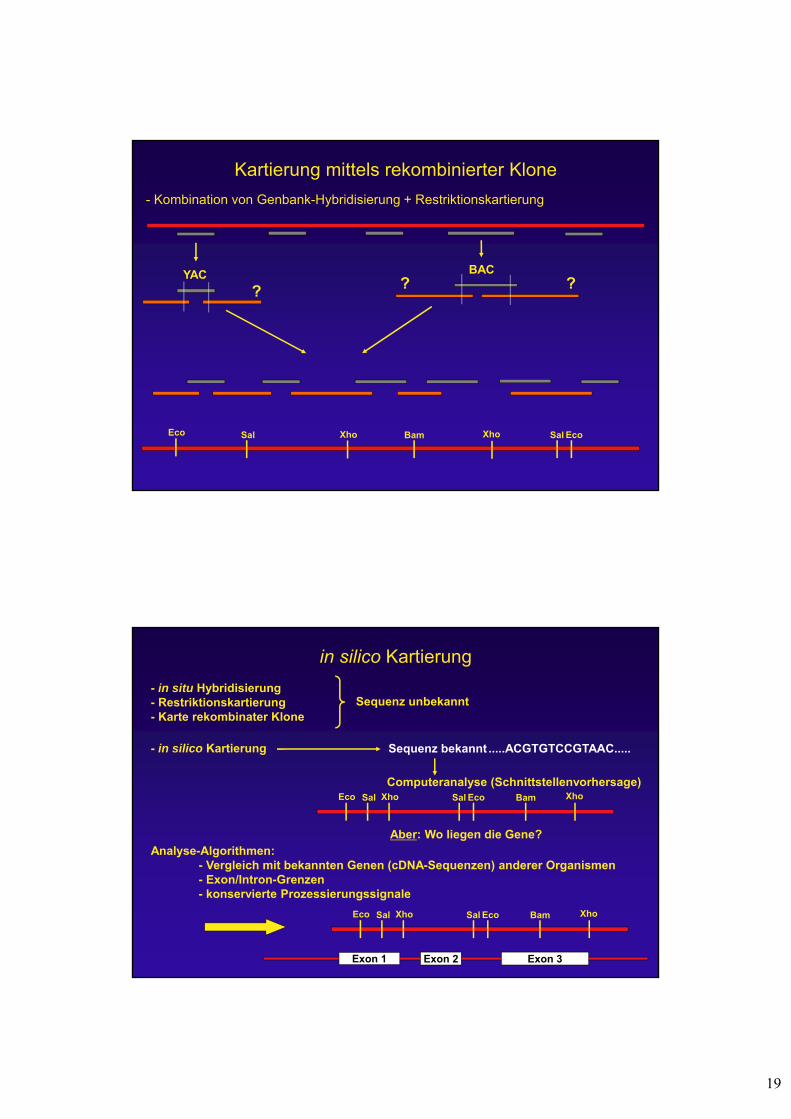
Kartierung mittels rekombinierter Klone
- Kombination von Genbank-Hybridisierung + Restriktionskartierung
Eco EcoSalSal XhoBamXho
YAC BAC
? ? ?
in silico Kartierung
- in situ Hybridisierung- Restriktionskartierung- Karte rekombinater Klone
Sequenz unbekannt
Analyse-Algorithmen:- Vergleich mit bekannten Genen (cDNA-Sequenzen) anderer Organismen- Exon/Intron-Grenzen- konservierte Prozessierungssignale
- in silico Kartierung Sequenz bekannt .....ACGTGTCCGTAAC.....
Aber: Wo liegen die Gene?
Computeranalyse (Schnittstellenvorhersage)EcoEco SalSal XhoBamXho
EcoEco SalSal XhoBamXho
Exon 1 Exon 2 Exon 3

20
Genomische Banken 1
Was? - Sammlung von tausenden rekombinanten Klonen, die ein
vollständiges Genom repräsentiert
Wozu? - zur Kartierung von Genomen und Genomabschnitten
- zum Finden und Klonieren eines Gens
- zur Sequenzanalyse (eines Genoms)
Anforderungen: - Repräsentativität (Wahrscheinlichkeit jedes beliebige Gen
zu finden)
- Technisch einfache Handhabbarkeit
- Stabilität der klonierten DNA
Genomische Banken 2
Anzahl der für genomische Banken benötigten Klone:
Spezies Zahl der Klone
Genomgröße (kb) 17 kb-Fragmente 35 kb-FragmenteE. coli 4.639 820 400S. cerevisiae 12.500 2.200 1.075Homo sapiens (Referenz-Genom) 3.500.000 535.000 258.000Zea mais (!) 5.000.000 900.000 450.000A. thaliana (Col Referenz-Genom) 125.000 22.000 10.700
Berechnet nach der Formel: N = ln (1-P)ln (1- a/b)
N = Anzahl der erforderlichen KloneP = Wahrscheinlichkeit, dass jedes Gen vorhanden ist wurde bei dieser Berechnung auf 0,95 gesetzt (d.h. 95%)a = durchschnittliche Größe der DNA-Fragmente in der Genbankb = Gesamtgröße des Genoms

21
Genbanken in verschiedenen Vektoren
bei 95% Wahrscheinlichkeit für das Arabidopsis-Genom notwendig:
Plasmide max. 8 kb ( 1- 5 kb) 5 kb 75.000 KloneLambda-Phagen max. 25 kb ( 10- 20 kb) 17 kb 22.000 KloneCosmide max. 44 kb ( 30- 44 kb) 35 kb 10.700 KloneP1-Phagen max. 100 kb ( 75- 100 kb) 75 kb 5.000 KloneBAC max. 300 kb (100- 300 kb) 200 kb 1870 KloneYAC max. 1000 kb (100-1000 kb) 1 Mb 372 Klone
Genbanken unterschiedlicher Repräsentativität:
Plasmid 95%: 75.000 KlonePlasmid 99%: 115.000 KlonePlasmid 99,9%: 172.700 Klone
Einige Forschungsobjekte aus dem Fachbereich
AG Organismus Seit wann sequenziert?
Bähler Mus musculus 2002
Paul Caenorhabditis elegans 1998
Daphnia pulex Draft genome 2011
Daphnia magna -
Liebau Brugia malayi Draft genome 2007
Onchocerca volvulus -
Klämbt Drosophila melanogaster 2000
Prüfer Taraxacum koksaghyz -
Medicago truncatula 2011
Kudla Arabidopsis thaliana 2000
Nicotiana benthamiana -
Tudzynski Claviceps purpurea -
Moerschbacher Puccinia graminis f. tritici -

22
Genomsequenzierung 1A (shot gun-Sequenzierung)
genomische DNA
gescherte DNA
Klonierung inLambda-Phagen
20 kb
2 kbKlonierung in
Plasmide
z.B. Haemophilus influenzae1.830.137 bp (1,83 Mb); 1743 Gene
(Wikipedia)Bei den Bakterien der Gattung Haemophilus handelt es sich um eine Gruppe stäbchenförmiger gramnegativer Bakterien der Familie der Pasteurellaceae. Die 16 Arten dieser Bakterien sind unbewegliche Stäbchen, die mitunter in den Schleimhäuten von Menschen und Tieren leben und Erkrankungen auslösen können.
genomic DNA digest
„smear“
Klonierung in Lambda-Phagen (20 kb)Klonierung in Plasmide (2 kb)
19687 Klone sequenziert
28643 Sequenzreaktionen
24304 gute Sequenzen (>400 bp)
11.631.485bp (6,3x)
140 Contigs
L1F
x 100 Phagen
L2FL1R L2R L3RL3F
c1
c2 c3c1
Lambda-Phagec2
Endsonden
Genomsequenzierung 1B (shot gun-Sequenzierung)
L = LambdaF = forwardR = reverse
bridge fragments
23
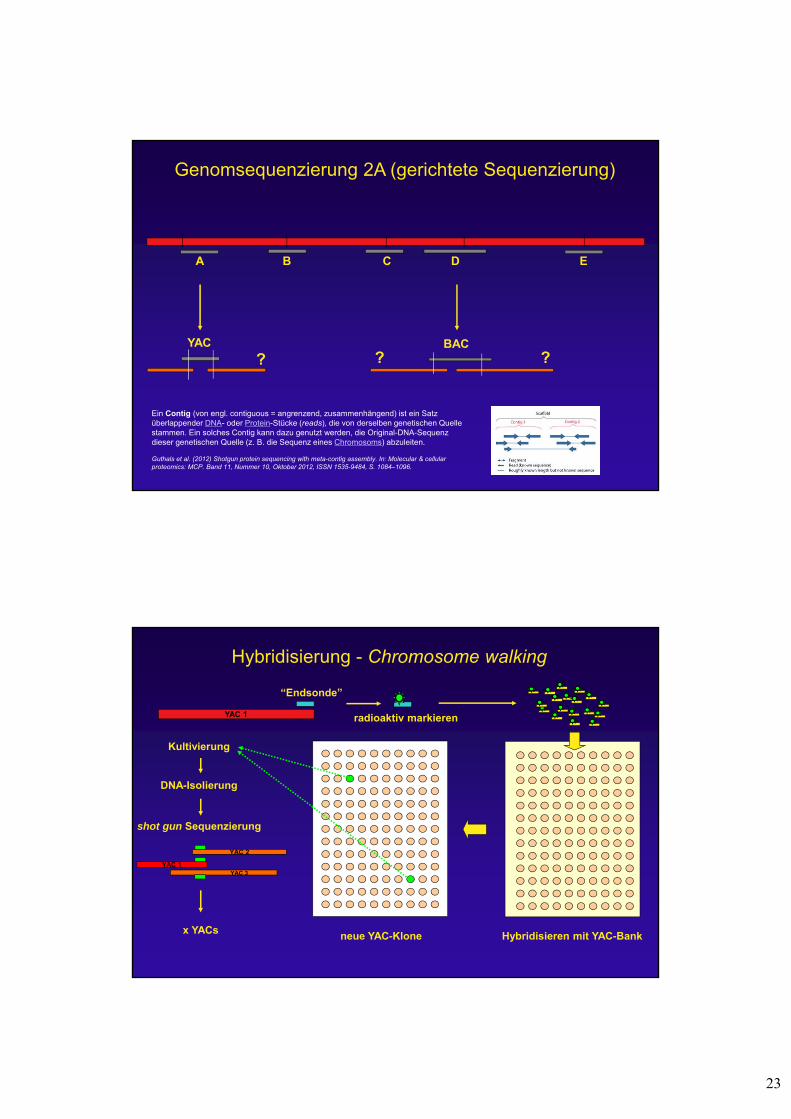
Genomsequenzierung 2A (gerichtete Sequenzierung)
A B C D E
YAC BAC
? ? ?
Ein Contig (von engl. contiguous = angrenzend, zusammenhängend) ist ein Satz überlappender DNA- oder Protein-Stücke (reads), die von derselben genetischen Quelle stammen. Ein solches Contig kann dazu genutzt werden, die Original-DNA-Sequenz dieser genetischen Quelle (z. B. die Sequenz eines Chromosoms) abzuleiten.
Guthals et al. (2012) Shotgun protein sequencing with meta-contig assembly. In: Molecular & cellularproteomics: MCP. Band 11, Nummer 10, Oktober 2012, ISSN 1535-9484, S. 1084–1096.
Hybridisierung - Chromosome walking
YAC 1
Kultivierung
DNA-Isolierung
shot gun Sequenzierung
“Endsonde”
radioaktiv markieren
Hybridisieren mit YAC-Bankx YACs
YAC 2
YAC 1YAC 3
neue YAC-Klone

24
A B C D E
YAC BAC
PCR aufLambda Phagen
(Lysat)
shot gun-Sequenzierung einzelner YACs/BACs(scheren, klonieren in Plasmide)
Assemblierung
Sequenzauswertung
Genomsequenzierung 2B (gerichtete Sequenzierung)
gap
Next generation sequencinghttp://www.medizinische-genetik.de/index.php?id=next-generation-sequencingDie Einführung der Next Generation Sequencing (NGS)-Technologien hat die Etablierung bedeutender, neuer diagnostischer Anwendungen in der täglichen Routine ermöglicht. Obwohl der Einsatz der NGS-Technologie in der klinischen Diagnostik mit Herausforderungen verbunden ist, wird die Umstellung aufgrund der sich bietenden Vorteile dennoch unumgänglich sein. Neben der extrem hohen Sequenzierkapazität ermöglicht NGS die klonale Sequenzierung einzelner Moleküle, eine höhere diagnostische Sensitivität durch parallele Sequenzierung von ganzen Genpanels, vereinfachte Handhabung sowie die parallele und dadurch kostengünstigere Bearbeitung der Proben. Neueste technische Fortschritte haben NGS zu einer verlässlichen Alternative für die klassische Sanger-Sequenzierung entwickelt.
Roche 454Roche Life Sciences war 2005 mit der Vorstellung des GS 20 Sequenziergeräts das erste Unternehmen, das eine Next Generation Sequencing Plattform kommerziell angeboten hat. Die Roche 454 Sequenziersysteme benutzen emulsions-PCR (emPCR) für die klonale Amplifikation der Proben, gefolgt von hochparallelem Pyrosequencing. Während der emPCR wird die zu sequenzierende DNA an spezifische Beads gebunden und klonal amplifiziert. DNA-tragende Beadswerden dann angereichert und in spezielle Reaktionskammern auf so genannten Pico Titre Plates (PTP) befördert. Diese Reaktionskammern enthalten alle für die Sequenzierung nötigen Reagenzien. Anschließend wird sequentiell je eine Art von fluoreszenzmarkierten Desoxyribonucleotiden (dATP, dTTP, dCTP, dGTP) in definierter Reihenfolge zugegeben. Ist ein Nukleotid komplementär zu der Base im DNA-Template, erfolgt ein Einbau und ein Lichtsignal wird detektiert. Sind in der zu sequenzierenden DNA mehrere aufeinanderfolgende, identische Nukleotide vorhanden (Homopolymere) werden mehrere gleiche Nukleotide hintereinander eingebaut und das korrespondierende Lichtsignal ist proportional stärker. Auf dieser Technologie basieren der Roche GS FLX Titanium Sequencer, der GS FLX+, eine verbesserte Version, die längere Leseweiten ermöglicht und die Benchtop Version, der Roche 454 Junior. Mit dem GS FLX und Junior Geräten können durchschnittliche Leseweiten von 450-550 bp erreicht werden, während mit dem FLX+ Upgrade die Leseweiten auf 750-850 bp gesteigert werden können. Mit diesen Geräten kann ein Durchsatz von 350-700 Mb pro 10 Stunden Lauf ( Homo sapiens: 3.500 Mb, ca. 1 Woche)auf den GS FLX Systemen und 30-50 Mb pro 8 Stunden Lauf auf dem GS Junior Gerät erreicht werden.

25
Roche 454 Sequencing:
(a) Genomische DNA, (b) Fragmentierung und Adapter-Ligation, (c) emPCR I, DNA-Fragment, gebunden an Bead, (d) emPCR II, klonal amplifizierte Fragmente (an Bead), (e) Beads in den Reaktionskammern der PTP-Platte, (f) emittierte Lichtsignale (Pyrogramm).
(Mit freundlicher Genehmigung von Roche Diagnostics GmbH, Mannheim, Deutschland)
(a)
(b)
(d)
(e)
(c) (f)
Prinzip der Pyrosequenzierung
Licht
Schritt 1: Die zu untersuchende DNA wird extrahiert und mit Bisulfit behandelt, um unmethylierte Cytosine in Uracile zu verwandeln. Nach Amplifikation der DNA mittels PCR, durch die alle Uracile durch Thymidine ersetzt werden, wird die Probe denaturiert um einzelsträngige DNA (ssDNA) zu erhalten. Sobald ein Sequenzierprimer an die einzelsträngige DNA gebunden wurde, wird der Komplementärstrang in Gegenwart von Adenosin-5’-Phosphosulfat (APS), Luziferin und den Enzymen DNA-Polymerase, ATP-Sulfurylase, Luziferase und Apyrase gebildet.
Schritt 2: Die Pyrosequenzierung wird durch Zugabe eines der vier Desoxynukleotid-Triphosphate (dNTPs) gestartet. Wenn dieses komplementär zur ersten ungepaarten Base des Hauptstranges ist, katalysiert die DNA-Polymerase seinen Einbau in den entstehenden Strang, wobei Pyrophosphat (PPi) freigesetzt wird. Dessen Menge entspricht der Anzahl eingebauter Nukleotide.
Schritt 3: Freigesetztes PPi wird durch die ATP-Sulfurylase, die APS als Substrat verwendet, zu ATP umgesetzt, welches wiederum die Luziferase-Reaktion antreibt. Dadurch wird Luziferin in Oxyluziferin verwandelt. Dies resultiert wiederum in einem sichtbaren Lichtsignal – dessen Stärke proportional zum verbrauchten ATP ist und mit einer Kamera erfasst werden kann. Mehrere Peaks, die durch den Einbau sukzessiven Einbau mehrerer Nukleotide entstehen, ergeben ein Pyrogramm (siehe Abb.).
Schritt 4: Apyrase, ein Enyzm das Nukleotide degradiert, baut nicht eingebautes ATP und dNTP kontinuierlich ab. So wird das Licht wieder „ausgeschaltet“ und die Reaktionslösung regeneriert. Die Schritte 2 und 3 können nun mit dem nächsten dNTPbegonnen werden.
Schritt 5: Die Zugabe der dNTPs erfolgt einzeln. Desoxyadenosin-alpha-thio-triphosphat (dATPas) wird hierbei als Ersatz für das Desoxyadenosin-triphosphat verwendet, da es von der DNA-Polymerase sehr effizient, von der Luziferase jedoch nicht als Substrat erkannt wird. Der Komplementärstrang wird durch Voranschreiten des Pyrosequenzierungs-Prozesses gebildet. Die Nukleotid-Sequenz kann an den Signal-Peaks aus dem Pyrogramm abgelesen werden.
Prinzip der Pyrosequenzierung

26
Illumina SBS
Schematischer Ablauf der SBS-Methode (2006): Einbau eines komplementären, fluoreszenzmarkierten Nukleotids (Detektion des Lichtsignals), Abspaltung der Terminatorgruppe und zyklusbasierte Sequenzierung (Mit freundlicher Genehmigung von Ilumina Inc., San Diego, CA, U.S.A.)
Nächste Woche
Reportergene
O. Batistic
Top Related