
![Redaktionsschluß: 12. 7. 1996 Bibliographia … · BIBLIOGRAPHIA ORIBATOLOGICA [ISSN: 0863-1794] Zusammengestellt von Thomas Schwalbe In den ABHANDLUNGEN UND BERICHTEN DES NATURKUNDEMUSEUMS](https://static.fdokument.com/doc/165x107/5b97492609d3f27a7a8cbe76/redaktionsschluss-12-7-1996-bibliographia-bibliographia-oribatologica-issn.jpg)






Sprachen
Seiten
Rechtliche


1
OC I Vorlesung, zweiter Teil (Skriptversion 1.4)
Verantwortliche:
Prof. Dr. Harald Schwalbe, email: [email protected] Institut für Organische Chemie und Chemische Biologie Johann Wolfgang Goethe-Universität Frankfurt Marie-Curie-Str. 11 N160, 314, 3. Stock D-60439 Frankfurt Telephon: ++49 69 7982 9737 Telefax: ++49 69 7982 9515 http://schwalbe.org.chemie.uni-frankfurt.de/ Dipl. Chem. Jan Ferner, email: [email protected]
Literaturverweis:
Aspekte der Organischen Chemie von Quinkert, Egert und Griesinger [Querverweis
gekennzeichnet mit Kapitel …]
Organic Chemistry von Clayden, Greeves, Warren, and Wothers [Querverweis
gekennzeichnet mit Chapter …]
Wie sollten Sie diese Vorlesungsmitschrift verwenden?
Jede Vorlesung ist in dieser Mitschrift einheitlich strukturiert: Wir stellen zunächst die
wesentlichen Inhalte jeder Vorlesungsstunde zusammen. Dies soll Ihnen helfen, den
roten Faden der Vorlesung besser verstehen zu können. Dann kommt der
Mitschriftteil jeder Vorlesung. Am Ende jeder Vorlesungsstunde sind Fragen
zusammengefasst. Diese halten sich eng an das während der Vorlesung Gesagte.
Sie sollten nach Besuch der Vorlesung und nach der Durchsicht Ihrer eigenen
Mitschrift in der Lage sein, diese Fragen zu beantworten. Die Mitschrift hat vor allem
zwei Bücher genutzt, nämlich das Buch „Aspekte der Organischen Chemie“ von
Quinkert, Egert, Griesinger und das englische Buch „Organic Chemistry“ von
Clayden, Greeves, Warren, and Wothers, auf die mit „Kapitel in Quinkert“ bzw.
„Chapter in Warren“ hingewiesen wird. Eine aktive Bearbeitung dieser beiden Bücher
kann das Vorlesungsskript nicht ersetzen.
Schließlich finden Sie auch die Übungen, die Sie mit dem Vorlesungsassistenten
durchgearbeitet haben. Diese Übungen können manchmal ein wenig schwieriger
sein als die direkten Fragen. Unter einer Vorlesungsstunde meinen wir eher die

2
Summe von Dingen, die inhaltlich zusammengehören, und weniger tatsächlich das,
was wir innerhalb von 90 Minuten besprechen konnten.
Trotz der Mitschrift raten wir Ihnen, während der Vorlesung mitzuschreiben; einmal,
weil die Vorlesung sich verändert, neue Dinge hinzukommen und alte eventuell
weniger intensiv besprochen werden, zum anderen, weil Sie lernen müssen, die
Strukturen von Verbindungen zu zeichnen. Nur durch die Übung werden Sie in der
Lage sein, dieses Werkzeug der Chemie sicher anwenden zu können. Schließlich
raten wir Ihnen, in die Vorlesung zu kommen, denn das gesprochene Wort kann
durch kein geschriebenes ersetzt werden und gerade in der Kombination aus
Vorlesung, Nacharbeiten in der Mitschrift und Auseinandersetzung und Ergänzung
mittels der eigenen Mitschrift ergibt sich ein nachhaltiger Lerneffekt. Die Konzepte,
die Sie in der OCI-Vorlesung erlernt haben, werden Sie in Ihrem Studium der
Chemie, der Biologie, der Biochemie und der Physik brauchen.
Dank:
Die Vorlesung ist Teil des Lehrkonzepts der Organischen Chemie, dass von den
Professoren Quinkert, Egert, Engels, Göbel, Griesinger, Rehm, Schneider, Schwalbe
und Kessler ausgearbeitet wurde.

3
OC I Vorlesung, zweiter Teil ....................................................................................... 1
1 Das qualitative MO-Modell............................................................................... 4
1.1 Die Grenzen des klassischen Strukturmodells der Organischen Chemie. 5
1.2 Das Benzol-Problem................................................................................. 8
1.3 Atom- und Molekül-Orbitale .................................................................... 19
1.4 Deutung der bisher unverständlichen Fälle durch das MO-Modell ......... 42
2 Reaktionen..................................................................................................... 47
2.1 Einführung .............................................................................................. 47
2.2 Verwendung von „gebogenen Pfeilen“.................................................... 57
2.3 Carbonylchemie...................................................................................... 59
2.4 Bildung und Reaktion von Enolen und Enolaten..................................... 94
2.5 Diels – Alder – Reaktion ........................................................................127
3 Übungsblätter im Wintersemster 2003 / 2004...............................................134

4
1 Das qualitative MO-Modell
Erste Vorlesungsstunde
Inhalte:
I. Die Grenzen des klassischen Strukturmodells werden an 4 Beispielen
aufgezeichnet:
a.) Amidbindung,
b.) anomerer Effekt,
c.) Inversionsbarriere der Halogenaziridine,
d.) Benzol.
II. Das Benzolproblem wird eingehend diskutiert, nämlich:
a.) Strukturproblem, mesomere Grenzstrukturen
b.) Stabilitätsproblem, Resonanzstabilisierung
III. Die experimentelle Bestimmung der Stabilität von Verbindungen und die
Berechnung der Stabilität von Verbindungen mittels Inkrementsystemen wird
eingeführt.
Achtung:
In der ersten Vorlesungsstunde wird noch nicht aufgelöst, wie man mit Hilfe des
qualitativen MO-Modells zu einer ‚neuen’ Sicht von Strukturen kommt. Dies passiert
in der zweiten und dritten Vorlesungsstunde.

5
Kapitel 8.1. in Quinkert et al.
1.1 Die Grenzen des klassischen Strukturmodells der Organischen Chemie
Im ersten Teil der Vorlesung haben Sie das klassische Strukturmodell und seine
Leistungsfähigkeit in bezug auf die Vorhersage von Konstitution und Konfiguration
organischer Verbindungen kennen gelernt.
Das klassische Strukturmodell stößt bei einer Reihe von Fragen an seine Grenzen,
die mit Hilfe des qualitativen MO-Modells beantwortet können. Das MO-Modell ist
darüber hinaus ein wichtiges Bindeglied zum Verständnis der Reaktivität von
Verbindungen, die man zur Entwicklung von Reaktionen vorhersagen muß.
1.1.1 Die Amidbindung
a)
H N
O
H
H O N
H
H
H
b)
Abbildung 1: (a) Formamid und Rotation um Carbonyl-Amidstickstoff-Einfachbindung, (b)
Proteinrückgratatome mit Definition der Diederwinkel φ und ψ
Abbildung 1a zeigt die Strukturformel des Formamids, es stellt die einfachste
Verbindung mit einer Amidbindung dar. Die Amidbindung ist das zentrale
Strukturelement in Polypeptiden, die die einzelnen Aminosäuren verknüpft
(Abbildung 1b). Die Amidbindung kann in zwei Formen vorliegen, wie in Abbildung 1a
gezeigt.
Nach dem klassischen Strukturmodell der Organischen Chemie gibt es zwischen
dem Carbonylkohlenstoff- und dem Amidstickstoffatom eine Einfachbindung. Um
solche Einfachbindungen gibt es normalerweise freie Drehbarkeit. Tatsächlich findet

6
man aber folgendes: Röntgenstrukturanalyse und Mikrowellenspektroskopie zeigen:
6 Atome der Amidgruppierung liegen in einer Ebene, d.h. die Amidbinding ist planar
(Abbildung 1). Die Rotation um die C-N Bindung hat eine hohe Barriere. Weder die
Planarität noch die hohe Rotationsbarriere sind nach dem klassischen
Strukturmodell erklärbar.
1.1.2 Der anomere Effekt
O OH
OHOH
OH
OH
O
OHOH
OH
CH2OHOH
O
OHOHOH
OH
OH
β-D-Glucopyranose
cyclo seco
α-D-Glucopyranose
cyclo
1
2
3
4
5
6
2 11
3
4 5
6
Abbildung 2: Gleichgewicht der D-Glucose in Lösung, die β-D-Glucopyranose und die α-D-
Glucopyranose unterscheiden sich durch die Konfiguration am so-genannten anomeren Zentrum C1,
sie sind Epimere und im speziellen hier Anomere.
D-Glucose (C6H12O6) liegt in Lösung in einem Gleichgewicht vor: nämlich zwischen
der zyklischen β-D-Glucopyranose, der offenkettigen seco-Verbindung und der
zyklischen α-D-Glucopyranose (Abbildung 2). α-D-Glucopyranose und β-D-
Glucopyranose unterscheiden sich nur anhand der Konfiguration des anomeren
Zentrums (C1). Das offenkettige seco-Isomere liegt zu weniger als 1% vor.
Die Lage des Gleichgewichts zwischen dem axialen (α-D-Glucopyranose) und dem
äquatorialen Anomeren (β-D-Glucopyranose) an C1 in Abhängigkeit des
Substituenten an C1 ist in Tabelle 1 aufgeführt.
Substituent an C1 % axiales Anomer
D-Glucopyranose OH 36
Methyl-D-glucopyranosid OMe 67
Penta-O-acetyl-D-glucopyranosid OAc 86
Tetra-O-acetyl- D-glucopyranosyl-chlorid Cl 94
Tabelle 1: Anteil des α-Anomeren im Gleichgewicht (siehe Abbildung 2)

7
⇒ Der anomere Effekt ist im Rahmen des klassischen Strukturmodells der
Organischen Chemie nicht erklärbar.
1.1.3 Inversionsbarrieren von N-Halogenaziridinen
NRR´
R´´
Abbildung 3: Räumliche Struktur eines tertiären Amins (R ≠ R’ ≠ R’’) mit freiem Elektronenpaar.
Amine mit drei unterschiedlichen Substituenten NRR´R´´ sind chiral, aber eine
Trennung in die Enantiomeren gelang lange Zeit nicht.
Eschenmoser konnte das unten angegebene N-Halogenaziridin in Enantiomere
trennen (D. Felix, A. Eschenmoser, Angew. Chem. (1968), 80, 197).
⇒ Der Einfluss des Chlorsubstituenten auf die Erhöhung der Inversionsbarriere
ist im Rahmen des klassischen Strukturmodells der Organischen Chemie nicht
erklärbar.
NCl
N
Cl
Abbildung 4: Beispiel eines in die Enantiomeren trennbaren N-Halogenaziridins.
1.1.4 Symmetrie (D6h) und thermodynamische Stabilität des Benzols (C6H6)
Die Symmetrie (D6h) und die thermodynamische Stabilität des Benzols sind
unerwartet. Sie haben zu einem so genannten Benzolproblem geführt, das mit Hilfe
des MO-Modells erklärt werden kann.

8
1.2 Das Benzol-Problem
1.2.1 Strukturvorschlag der Konstitution des Benzols
Kapitel 14.1. in Quinkert et al.
Kekulé (1865): Kekulé postuliert, dass Benzol ringförmig aufgebaut sei und
alternierende C-C-Einfach- und C=C-Doppelbindungen aufweise.
Abbildung 5: Kekulé Strukturmodell für das Benzol: Sechsring mit alternierenden C-C-Einfach- und
C=C-Doppelbindungen.
Falsifizierungsexperiment: Das Postulat der alternierenden, aber lokalisierten C-C-
Einfach- und C=C-Doppelbindungen führt zwingend zu einer Vorhersage von
möglichen Reaktionsprodukten, wenn man einen, zwei oder drei Substituenten im
Benzolring einführt; also ein H-Atom durch ein Atom X oder Y ersetzt. Man kann also
die Maximalzahl an Konstitutionsisomeren für symmetrische Disubstitution (zwei
gleiche Substituenten X werden eingeführt, Abbildung 6) oder für unsymmetrische
Disubstitution (zwei ungleiche Substituenten X und Y werden eingeführt, Abbildung
7) vorhersagen. Das Modell kann mittels chemischer Reaktionen falsifiziert werden.
Was ist die Anzahl der symmetrisch substituierten Benzole?
X
XX
XX
X
XX
X
X
1 2
3
4
5
6
Abbildung 6: Erwartete Anzahl symmetrisch mono- (2) und disubstituierter Benzolderivate (3-6).
9
Experimentell findet man nur 3 statt 4 vorhergesagter Konstitutionsisomere für X,X-
disubstituierte Benzole (Moleküle 3 und 4 sind identisch)
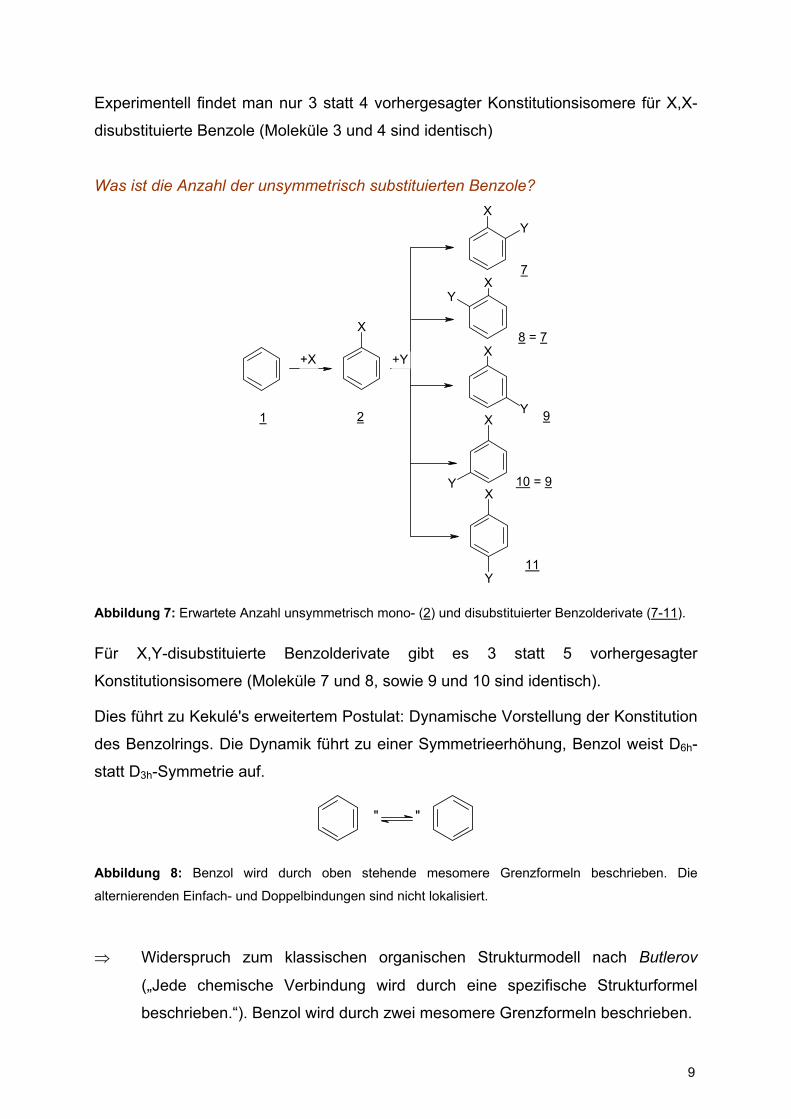
Was ist die Anzahl der unsymmetrisch substituierten Benzole?
X
XY
XY
X
Y
X
Y
X
Y
+X
1 2
7
8 = 7
9
11
+Y
10 = 9
Abbildung 7: Erwartete Anzahl unsymmetrisch mono- (2) und disubstituierter Benzolderivate (7-11).
Für X,Y-disubstituierte Benzolderivate gibt es 3 statt 5 vorhergesagter
Konstitutionsisomere (Moleküle 7 und 8, sowie 9 und 10 sind identisch).
Dies führt zu Kekulé's erweitertem Postulat: Dynamische Vorstellung der Konstitution
des Benzolrings. Die Dynamik führt zu einer Symmetrieerhöhung, Benzol weist D6h-
statt D3h-Symmetrie auf.
" "
Abbildung 8: Benzol wird durch oben stehende mesomere Grenzformeln beschrieben. Die
alternierenden Einfach- und Doppelbindungen sind nicht lokalisiert.
⇒ Widerspruch zum klassischen organischen Strukturmodell nach Butlerov
(„Jede chemische Verbindung wird durch eine spezifische Strukturformel
beschrieben.“). Benzol wird durch zwei mesomere Grenzformeln beschrieben.

10
Robinson hat die Schreibweise des Rings für die delokalisierten Einfach- und
Doppelbindungen eingeführt (Abbildung 9).
Abbildung 9: Robinson: „Bindungen im Benzol werden durch einen Kreis in einem regulären
Sechseck beschrieben.“
Eine interessante Alternative zum Robinson’schen Vorschlag gibt A. von Bayer
(Abbildung 10):
Abbildung 10: A. v. Bayer (anlässlich des Benzolfests 1890): "von den C-Atomen der Koordinations-
zahl 3 verschwindet die 4te Valenz für unsere Wahrnehmung."
1.2.2 Betrachtung der Energie des Benzols
Struktur und Energie charakterisieren eine Verbindung!
⇒ Benzol-Problem hat einen strukturellen Aspekt (D6h statt D3h Symmetrie) und
einen energetischen Aspekt (Stabilität, "Resonanzstabilisierung").
In diesem Zusammenhang:
Was bedeutet stabiler?
- A ist stabiler als B, wenn für die Reaktion A B die freie Reaktionsenthalpie
∆rG0>0 ist.
- P ist relativ zu Y stabiler als Q relativ zu Z, wenn für die freien Enthalpien der
folgenden Reaktionen gilt:
P X + Y ∆rG10
Q X + Z ∆rG20
∆rG10 > ∆rG2
0

11
1.2.3 Reaktionsenthalpie, Bildungsenthalpie, Bindungsenthalpie
Definitionen: Reaktionsenthalpie:
Die Reaktionsenthalpie wird mit dem Symbol ∆H bezeichnet und gibt die als
Reaktionswärme abgegebene Energie einer chemischen Reaktion wieder. Ist
der Wert ∆H negativ, wird Wärme freigesetzt und man spricht von einer
exothermen Reaktion. Endotherme Reaktionen benötigen die Zufuhr von
Wärme, ∆H hat positives Vorzeichen.
Bildungsenthalpie:
Ein bequemer Weg, Reaktionsenthalpien zu berechnen, geht von
tabellarischen Werten aus, die wir Standard-Bildungsenthalpien nennen. Die
Standard-Bildungsenthalpie ist der ∆H-Wert, der zur Bildung von 1 mol reiner
Substanz aus den reinen Elementen unter Standard-Bedingungen gehört.
Bindungsenthalpie:
Die Energie, die zum Aufbrechen der Bindung eines zweiatomigen Moleküls
benötigt wird, ist die Dissoziationsenergie. Die Energie wird in Kilojoule pro
Mol Bindungen angegeben. Geht man zu mehratomigen Molekülen, so lassen
sich mittlere Bindungsenthalpien bestimmen. Diese besagen, wie viel Energie
im Mittel eingesetzt werden muss, um eine X–Y-Bindung homolytisch zu
spalten.
Vergleich von E-But-2-en (12) und Z-But-2-en (13)
12 13
Abbildung 11: Die beiden Stereoisomere des But-2-ens, das E-But-2-en 12 und das Z-But-2-en 13.
12 ist ein Diastereomeres von 13, eines der Diastereomere wird stabiler sein als das
andere. Welches?

12
Folgende Experimente geben darüber Aufschluss:
1.) Bestimmung der Verbrennungsenthalpien ∆cH (bei konstantem Druck p) in
einer Kaloriemeterbombe (oder der Reaktionsenergie ∆cU (bei konstantem
Volumen V))
E-But-2-en + 6 O2 4 CO2 + 4 H2O ∆cH0 = -2706.2 [kJ/mol]
Z-But-2-en + 6 O2 4 CO2 + 4 H2O ∆cH0 = -2710.4 [kJ/mol]
⇒ Das E-Isomere ist um 4.2 kJ/mol stabiler als das Z-Isomere.
2.) Bestimmung der Hydrierungsenthalpien ∆rH0:
E-But-2-en + H2 Butan ∆rH0 = -114.9 [kJ/mol]
Z-But-2-en + H2 Butan ∆rH0 = -119.1 [kJ/mol]
⇒ Das E-Isomere ist um 4.2 kJ/mol stabiler als das Z-Isomere, die experimentell
bestimmte relative Stabilität von E-But-2-en und Z-But-2-en ist also
unabhängig von der experimentellen Methode.
Bei Kenntnis der Verbrennungsenthalpien eines Kohlenwasserstoffs, von Graphit
und von Wasserstoff kann man die Bildungsenthalpie des Kohlenwasserstoffs
berechnen:
Verbrennungsenthalpien:
4 CO2 + 4 H2O (Z)-C4H8 + 6 O2 - ∆cH0 = +2710.4 [kJ/mol]
4 CO2 + 4 H2O (E)-C4H8 + 6 O2 - ∆cH0 = +2706.2 [kJ/mol]
4 C (Graphit) + 4 O2 4 CO2 ∆cH0 = -1574.0 [kJ/mol]
4 H2 + 2 O2 4 H2O ∆cH0 = -1143.3 [kJ/mol]
⇒ Bildungsenthalpien:
4 C (Graphit) + 4 H2 (Z)-C4H8 ∆fH0 = - 6.9 [kJ/mol]
4 C (Graphit) + 4 H2 (E)-C4H8 ∆fH0 = -11.1 [kJ/mol]

13
4 C + 4 H2
(E)-But-2-en
∆fH0 = -11.1 kJ/mol
4 CO2 + 4 H2O
∆cH0 = -2706.2 kJ/mol
(Z)-But-2-en
∆fH0 = -6.9 kJ/mol
∆cH0 = -2710.4 kJ/mol
Abbildung 12: „Heßscher Satz der konstanten Wärmesummen“
Modell: Bildungsenthalpie eines Moleküls setzt sich additiv aus Bildungsenthalpie-
inkrementen zusammen (Prinzip des chemischen Baukastens).
Formel 1: ∑=∆ Bausteinenaren submolekul aus eträgeEnthalpieb konst.(Molekül)0Hf
Tabelle 2: Bindungsenthalpiewerte als Bildungsenthalpieinkremente. Cd = C-Atom mit der
Koordinationszahl 3. Für die Berechnungen werden die (C=C)- und die (C=O)-Gruppe jeweils als
Einheit mit der Koordinationszahl 4 bzw. 2 betrachtet.
14
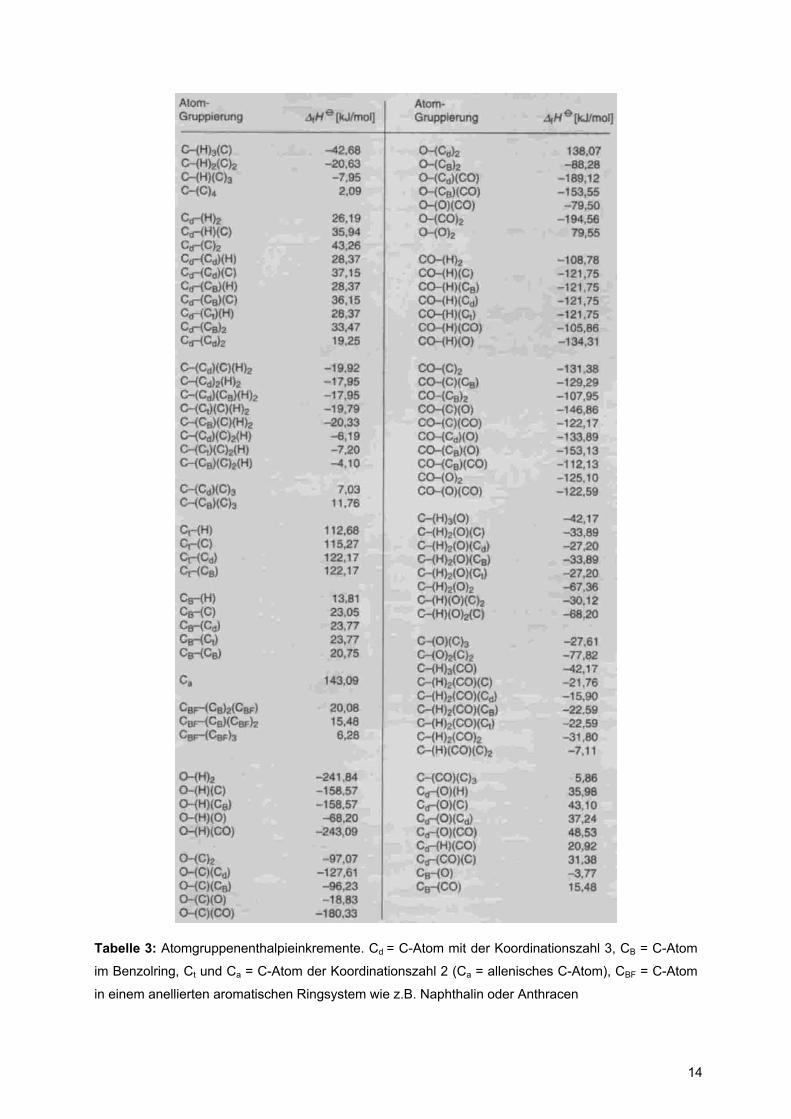
Tabelle 3: Atomgruppenenthalpieinkremente. Cd = C-Atom mit der Koordinationszahl 3, CB = C-Atom
im Benzolring, Ct und Ca = C-Atom der Koordinationszahl 2 (Ca = allenisches C-Atom), CBF = C-Atom
in einem anellierten aromatischen Ringsystem wie z.B. Naphthalin oder Anthracen
15
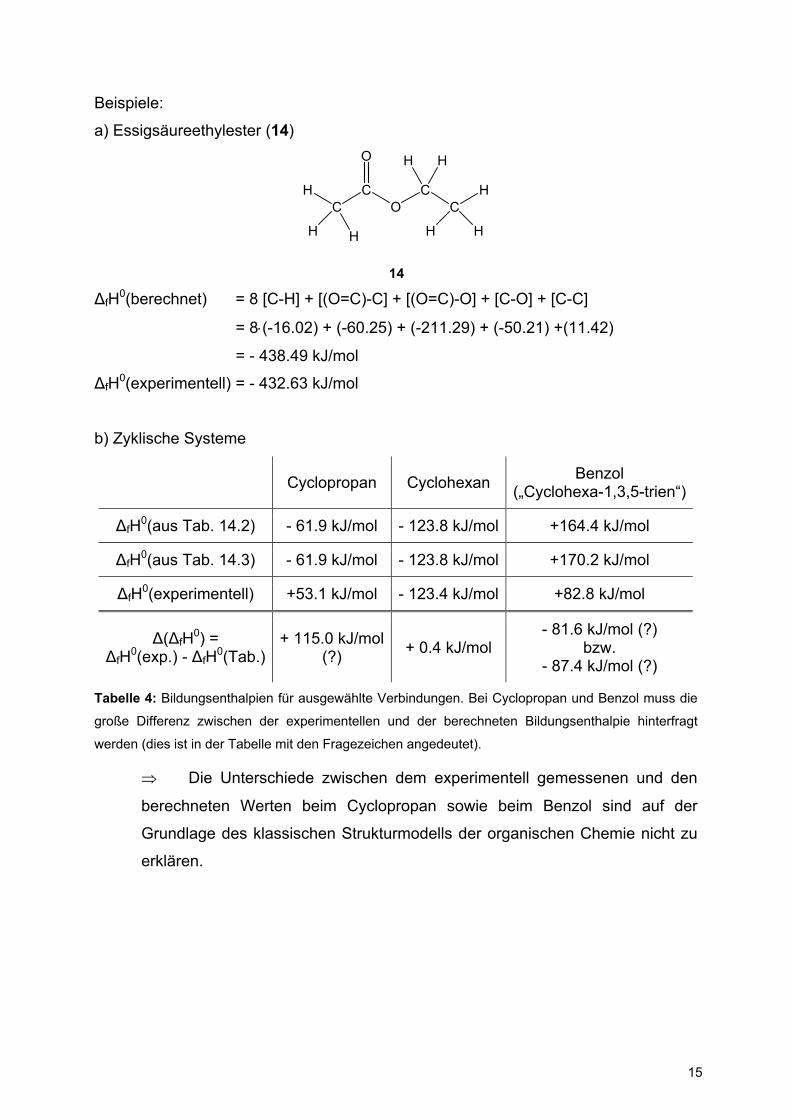
Beispiele:
a) Essigsäureethylester (14)
HC
CO
CC
H
H H
O H H
H H
14 ∆fH0(berechnet) = 8 [C-H] + [(O=C)-C] + [(O=C)-O] + [C-O] + [C-C]
= 8⋅(-16.02) + (-60.25) + (-211.29) + (-50.21) +(11.42)
= - 438.49 kJ/mol
∆fH0(experimentell) = - 432.63 kJ/mol
b) Zyklische Systeme
Cyclopropan Cyclohexan Benzol („Cyclohexa-1,3,5-trien“)
∆fH0(aus Tab. 14.2) - 61.9 kJ/mol - 123.8 kJ/mol +164.4 kJ/mol
∆fH0(aus Tab. 14.3) - 61.9 kJ/mol - 123.8 kJ/mol +170.2 kJ/mol
∆fH0(experimentell) +53.1 kJ/mol - 123.4 kJ/mol +82.8 kJ/mol
∆(∆fH0) = ∆fH0(exp.) - ∆fH0(Tab.)
+ 115.0 kJ/mol(?) + 0.4 kJ/mol
- 81.6 kJ/mol (?) bzw.
- 87.4 kJ/mol (?)
Tabelle 4: Bildungsenthalpien für ausgewählte Verbindungen. Bei Cyclopropan und Benzol muss die
große Differenz zwischen der experimentellen und der berechneten Bildungsenthalpie hinterfragt
werden (dies ist in der Tabelle mit den Fragezeichen angedeutet).
⇒ Die Unterschiede zwischen dem experimentell gemessenen und den
berechneten Werten beim Cyclopropan sowie beim Benzol sind auf der
Grundlage des klassischen Strukturmodells der organischen Chemie nicht zu
erklären.
16
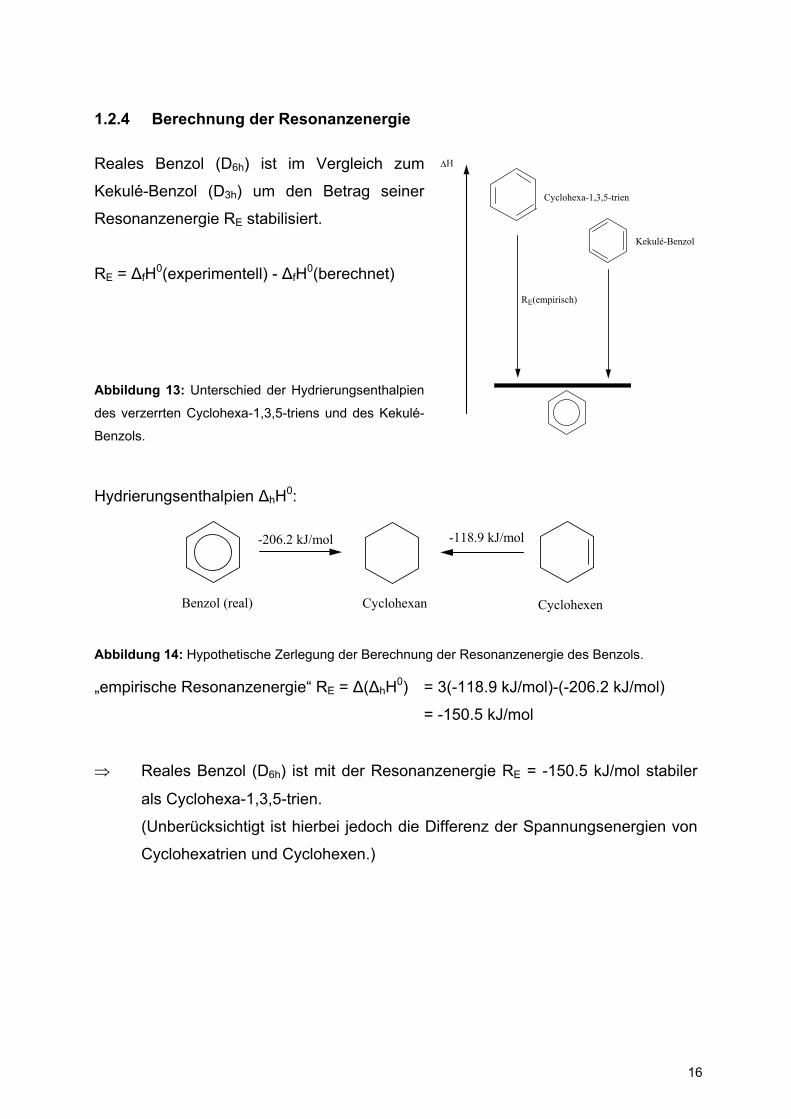
1.2.4 Berechnung der Resonanzenergie
Reales Benzol (D6h) ist im Vergleich zum
Kekulé-Benzol (D3h) um den Betrag seiner
Resonanzenergie RE stabilisiert.
RE = ∆fH0(experimentell) - ∆fH0(berechnet)
Abbildung 13: Unterschied der Hydrierungsenthalpien
des verzerrten Cyclohexa-1,3,5-triens und des Kekulé-
Benzols.
Hydrierungsenthalpien ∆hH0:
Benzol (real) Cyclohexan Cyclohexen
-206.2 kJ/mol -118.9 kJ/mol
Abbildung 14: Hypothetische Zerlegung der Berechnung der Resonanzenergie des Benzols.
„empirische Resonanzenergie“ RE = ∆(∆hH0) = 3(-118.9 kJ/mol)-(-206.2 kJ/mol)
= -150.5 kJ/mol
⇒ Reales Benzol (D6h) ist mit der Resonanzenergie RE = -150.5 kJ/mol stabiler
als Cyclohexa-1,3,5-trien.
(Unberücksichtigt ist hierbei jedoch die Differenz der Spannungsenergien von
Cyclohexatrien und Cyclohexen.)
Kekulé-Benzol
Cyclohexa-1,3,5-trien
∆H
RE(empirisch)

17
Fragen:
I. Die Grenzen des klassischen Strukturmodells wurden an 4 Beispielen auf-
gezeichnet. Fassen Sie zusammen, welche Aspekte für die Punkte a.)-d.) nicht mit
dem klassischen Strukturmodell im Einklang stehen.
a.) Amidbindung (Sie müssen hier 2 Punkte benennen können)
b.) anomerer Effekt (Sie müssen hier 2 Punkte benennen können)
c.) Inversionsbarriere der Halogenaziridine (Sie müssen hier 1 Punkt benennen
können)
d.) Benzol (Sie müssen hier 3 Punkte benennen können)
II. Spezifische Fragen:
a.) Die Amidbindung spielt eine entscheidende Rolle bei der Konformation von
Proteinen. Diskutieren Sie die Konsequenzen für die Einstellung der Rückgrat-
winkel in Proteinen. Welche zwei Möglichkeiten gibt es für den Rückgratwinkel ω
und welche findet man vor allem? Für welche Aminosäure(n) findet man
Ausnahmen (Lesen Sie im Quinkert-Buch nach)?
b.) Können Sie sich vorstellen, dass auch andere Strukturelemente ähnlich wie die
Amideinheit planar sind? Welche?
c.) Der anomere Effekt hängt von der Art des Substituenten ab. Womit wird dieser
Effekt nach Ihrem jetzigen Wissenstand korrelieren?
d.) Lesen Sie in einem Anorganikbuch (z.B. Hollemann-Wiberg) über die Ammoniak-
uhr nach. Sind Phosphorverbindungen mit drei unterschiedlichen Substituenten
einfacher oder schwerer in die Enantiomere zu separieren?
e.) Nehmen Sie an, Sie führen eine Reaktion durch, bei der Sie eine Carbonsäure-
gruppe in das Molekül Methoxybenzol einführen. Welche konstitutionsisomeren
Verbindungen erwarten Sie? Welche Konstitutionsisomere erwarten Sie, wenn
Sie einen zweiten Chlorsubstituenten in Chlorbenzol einführen?
f.) Wie können Sie die relative Stabilität von zwei Verbindungen experimentell
bestimmen?

18
Zweite Vorlesungsstunde
Inhalte:
I. Verbindungen werden durch Bindungen zwischen Atomen zusammengehalten.
Elektronen zwischen Atomkernen stellen diese Bindungen dar. In Reaktionen werden
Bindungen gebildet und gebrochen. In der zweiten Vorlesungsstunde wird in die
Beschreibung der chemischen Bindung und in grundlegende Konzepte eingeführt.
II. Zunächst werden die Atomorbitale (s-, p-, d-, f-Orbitale) eingeführt, diese sind
Ihnen aus der Grundvorlesung bekannt: s-, p-, d-, f- etc. Orbitale sind die exakten
Lösungen des Wasserstoffatoms und werden auch für größere Systeme als
Näherung (!) verwendet.
III. Dann werden Mehrelektronensysteme vorgestellt, das einfachste ist das
Heliumatom, ein Kern und zwei Elektronen. Zur Beschreibung dieses Atoms werden
die unter II. verwendeten Orbitale als Näherung (!) benutzt.
IV. Wir führen die Regeln für die Besetzung von Atomorbitalen mit Elektronen ein:
Pauliprinzip, Hund’sche Regel.
V. Wir beschreiben Moleküle mittels der Orbitale, die wir von Einelektronen- und
Mehrelektronenatomen kennen gelernt haben mittels des LCAO-MO-Ansatzes.
Achtung:
In der zweiten Vorlesungsstunde werden viele theoretische Konzepte eingeführt, die
zu Beginn sehr komplex sind. Verlieren Sie nicht den Atem, sondern behalten Sie im
Auge, dass wir letztlich eine ‚einfache’ Beschreibung erarbeiten wollen, die es uns
ermöglicht, organische Reaktionen und Reaktivitäten von Molekülen vorherzusagen.

19
1.3 Atom- und Molekül-Orbitale
Kapitel 8.3. in Quinkert et al
1.3.1 „Bindungen sind aus Elektronen gemacht“
Grundfragen: Wie beschreiben wir Elektronen? Wie beschreiben wir Bindungen? Wie
können wir aus Atomen aufgebaute Moleküle beschreiben?
Zur Beantwortung dieser Fragen gehen wir von einfachen Systemen, das einfachste
Atom ist das Wasserstoffatom 1H (ein Proton, ein Elektron), das einfachste Molekül
ist das Wasserstoffgas H2, zu komplexeren Molekülen über. Dabei sehen wir, dass
ein chemisches Baukastenmodell sehr aussagekräftig ist.
Die Bewegung von Teilchen kleiner Masse werden nicht durch die Newton´sche
Gesetze der Mechanik beschrieben, sondern durch die Schrödingergleichung. Dies
ist eine Konsequenz der Heisenberg’schen Unschärferelation, die auftritt, wenn Ort
und Impuls eines atomaren Teilchens gleichzeitig genau beschreiben werden sollen
(Lesen Sie dazu im Kapitel 8.3 in Aspekte der Organischen Chemie und in
weiterführenden Lehrbücher der PC, z.B. Hensen). Die Lösung der
Schrödingergleichung kann für das Wasserstoffatom 1H exakt angegeben werden, ist
aber für größere Atome (z.B. He) oder Moleküle häufig nur mittels Näherungs-
verfahren möglich. Wir wollen z.B. die Aufenthaltswahrscheinlichkeit eines Elektrons
berechnen, also wie wahrscheinlich ist es, ein Elektron in einem Abstand r0 vom Kern
entfernt zu finden. Diese Frage wird davon abhängen, welches Atom man anschaut,
d.h. wie viele Protonen sich im Kern befinden, oder mit anderen Worten, wie groß die
Kernladung Z ist und wie viele Elektronen sich in äußeren Schalen befinden. Die
Aufenthaltswahrscheinlichkeit z.B. eines Elektrons wird durch die „psi“-Funktionen
Ψ(r,t) beschrieben.
Die Aufenthaltswahrscheinlichkeit bzw. Wahrscheinlichkeitsdichte, also die
Wahrscheinlichkeit W, ein Teilchen (z.B. ein Elektron) im Volumenelement dV
anzutreffen, ist gegeben durch:
Formel 2: dVdW
=ΨΨ=Ψ *2
(Ψ* ist die komplex konjugierte Funktion zu Ψ).

20
Typische und Ihnen bekannte Ψ-Funktionen sind die s-, p-, d-, f-Orbitale, die die
Lösung des Wasserstoffatoms darstellen (siehe unten ausführlicher erklärt). Jedes
Orbital kann durch jeweils maximal zwei Elektronen besetzt werden (s. unten).
Ψ-Funktionen sind orthonormiert:
Formel 3: ∫⎩⎨⎧
==≠=
=ΨΨjifür 1jifür 0* dVji
⇒ Volumenintegral = 0 für unterschiedliche Funktionen
Volumenintegral = 1 für gleiche Funktionen, weil die Wahrscheinlichkeit,
ein Elektron im gesamten Raum anzutreffen, 100 %
ist.
Das bedeutet:
Ein Elektron kann sich nicht gleichzeitig in einem s- und einem p-Orbital befinden.
a.) es gibt keinen Überlapp zwischen einem Elektron, das sich in einem s-Orbital
befindet, und einem Elektron, das sich in einem p-Orbital befindet
(Volumenintegral = 0 für unterschiedliche Funktionen).
b.) ein s-Elektron befindet sich irgendwo innerhalb des gesamten Volumens des
s-Orbitals (Volumenintegral = 1 für gleiche Funktionen).
Man kann jetzt für verschieden große Schalen um ein Zentralatom angeben, wie dort
die Wahrscheinlichkeit ist, ein Elektron innerhalb dieses Volumenelements
anzutreffen.
1.3.2 Einelektronenatome (z.B. Wasserstoffatom)
e-
r
x
y
z
ϕ
ϑ
Z+
Abbildung 15: Definition der Lage des Elektrons im Potentialfeld eines Z+-fach geladenen
Zentralatoms (H, Z=1; He, Z=2) mittels kartesischer oder polarer Koordinaten.

21
Die Position des Elektrons kann entweder durch die kartesischen Koordinaten x,y,z
oder durch die Kugelkoordinaten Radius r sowie die Winkel ϑ und ϕ beschrieben
werden (s. Abbildung 15).
Die Schrödingergleichung beschreibt den Aufenthalt dieses einen Elektrons. Für das
Wasserstoffatomproblem - 1 Elektron im Potential eines geladenen Kerns (vergleich-
bar mit der Bahn eines Monds um sein Zentralgestirn) - gibt es Lösungen, folgendes
halten wir bezüglich dieser Funktionen fest
- Es gibt eine unendlich große Anzahl möglicher Lösungsfunktionen Ψ
(Funktionen 1s, 2s,2p, 3s,3p,3d, etc)
- Die Lösungsfunktionen haben diskrete Energien.
- Lösungsfunktionen, auch Zustandsfunktionen genannt, heißen auch Orbitale.
E [eV]
0
-3.4 Z2
-13.6 Z2
1s
2s 2px 2py 2pz
Abbildung 16: Energetische Lage der Lösungen des Wasserstoffatomproblems. Die Energie der
Orbitale mit n=2 ist gleich und beträgt -3.4Z2 [eV], d.h. die Energie der 2s und 2p Orbitale ist für das
Wasserstoffatomproblem entartet.
1.3.2.1 Grundzustand des H-Atoms
Ψ1s besitzt den niedrigsten Energieeigenwert ⇒ Grundzustand des H-Atoms
Die komplex konjugierte der Ψ1s ist gleich Ψ1s (Ψ1s* = Ψ1s).
Formel 4: dW = Ψ1s* Ψ1s dV = Ψ1s Ψ1s dV
Die Wahrscheinlichkeit, ein Elektron in irgendeinem Volumenelement dV mit Abstand
r vom Kern zu finden, ist gegeben durch:
Formel 5: dW = Ψ1s Ψ1s 4πr2 dr
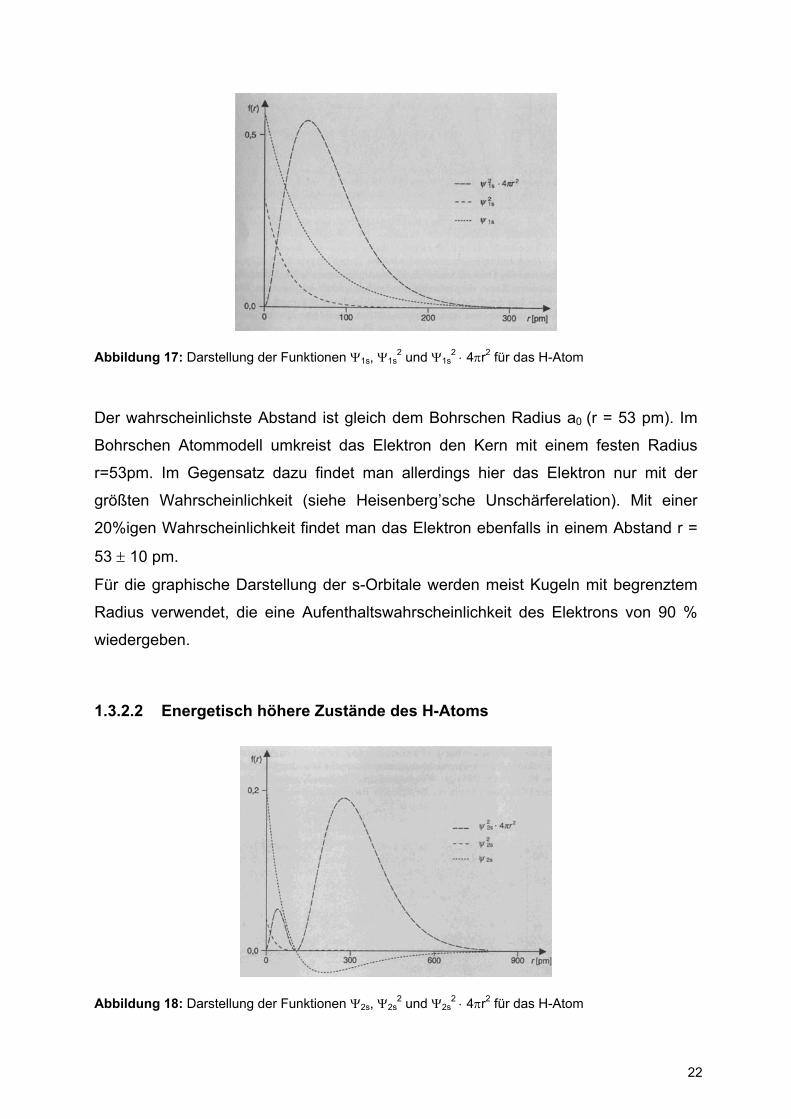
22
Abbildung 17: Darstellung der Funktionen Ψ1s, Ψ1s2 und Ψ1s
2 ⋅ 4πr2 für das H-Atom
Der wahrscheinlichste Abstand ist gleich dem Bohrschen Radius a0 (r = 53 pm). Im
Bohrschen Atommodell umkreist das Elektron den Kern mit einem festen Radius
r=53pm. Im Gegensatz dazu findet man allerdings hier das Elektron nur mit der
größten Wahrscheinlichkeit (siehe Heisenberg’sche Unschärferelation). Mit einer
20%igen Wahrscheinlichkeit findet man das Elektron ebenfalls in einem Abstand r =
53 ± 10 pm.
Für die graphische Darstellung der s-Orbitale werden meist Kugeln mit begrenztem
Radius verwendet, die eine Aufenthaltswahrscheinlichkeit des Elektrons von 90 %
wiedergeben.
1.3.2.2 Energetisch höhere Zustände des H-Atoms
Abbildung 18: Darstellung der Funktionen Ψ2s, Ψ2s2 und Ψ2s
2 ⋅ 4πr2 für das H-Atom
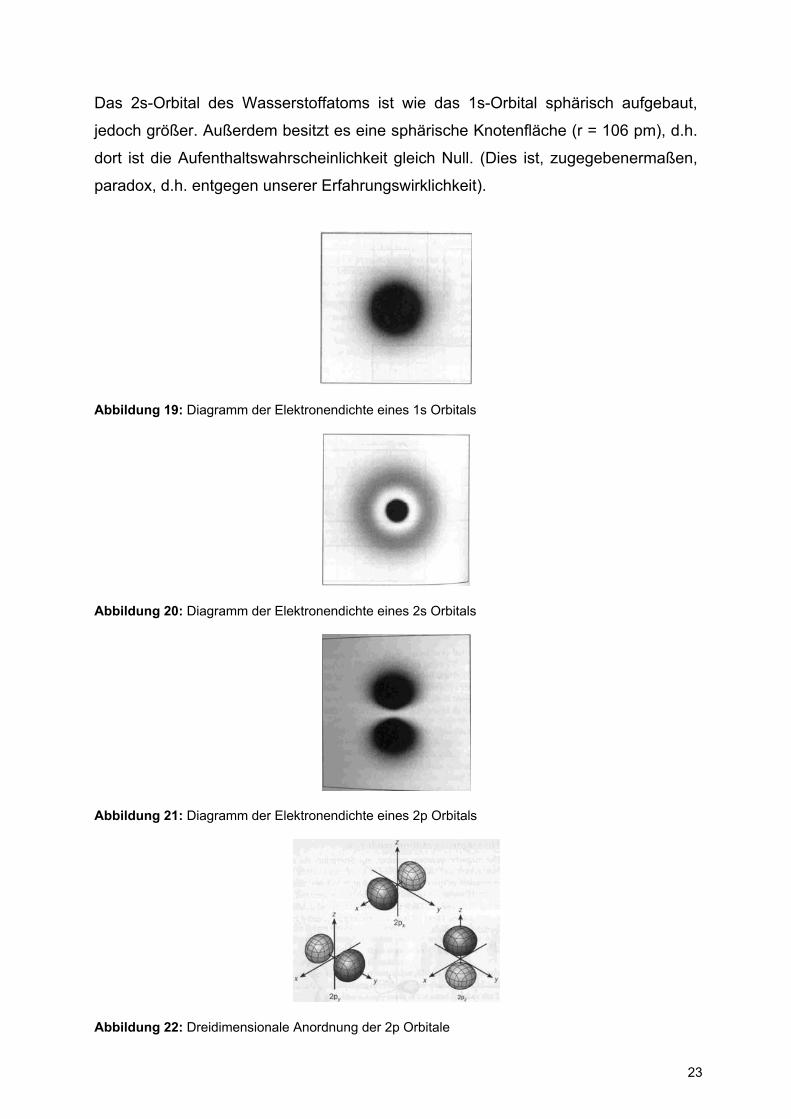
23
Das 2s-Orbital des Wasserstoffatoms ist wie das 1s-Orbital sphärisch aufgebaut,
jedoch größer. Außerdem besitzt es eine sphärische Knotenfläche (r = 106 pm), d.h.
dort ist die Aufenthaltswahrscheinlichkeit gleich Null. (Dies ist, zugegebenermaßen,
paradox, d.h. entgegen unserer Erfahrungswirklichkeit).
Abbildung 19: Diagramm der Elektronendichte eines 1s Orbitals
Abbildung 20: Diagramm der Elektronendichte eines 2s Orbitals
Abbildung 21: Diagramm der Elektronendichte eines 2p Orbitals
Abbildung 22: Dreidimensionale Anordnung der 2p Orbitale

24
1.3.3 Mehrelektronensysteme
Einelektronensystem: In einem Einelektronensystem umkreist das Elektron den
Atomkern und spürt das Coulombpotential des Atomkerns.
Das vom Elektron gespürte Coulombpotential ist nur auf den Kern gerichtet
+r
e_
Abbildung 23: Einelektronensystem
Mehrelektronensystem: In einem Mehrelektronensystem umkreisen z.B. zwei
Elektronen den Atomkern und spüren demzufolge neben dem anziehenden Potential
des Atomkerns auch das abstoßende Potential des zweiten Elektrons.
Das Coulombpotential setzt sich vektoriell aus dem Elektronen- und dem
Kernpotential zusammen.
e1_
r1r2
Z+
e2
_
Abbildung 24: Zweielektronensystem
⇒ Systeme aus 3 Teilchen (Sonne, Zentralgestirn und Mond oder Proton und
zwei Elektronen) sind weder klassisch mechanisch noch quantenmechanisch
exakt lösbar, aber mittels Näherungsverfahren mit jeder gewünschten
Genauigkeit lösbar!
Man kommt also zu Näherungsverfahren, zunächst zur Orbital-Näherung.

25
1.3.3.1 Orbital-Näherung
Die Orbitalnäherung für Mehrelektronensysteme beruht auf zwei Näherungen:
Nullte Näherung:
• Es gibt keine Elektron/Elektron-Wechselwirkungen
Jedes Elektron „spürt“ nur den Atomkern einer Ladung, nicht aber die
Abstoßung durch andere Elektronen.
• Lösung der Schrödingergleichung für jedes der n Elektronen setzt sich
zusammen aus den Lösungen des Einelektronensystems nach dem
Prinzip des chemischen Legokastens. Man nennt dies einen
Produktansatz:
( ) )(...)()(,...,, 2211ln
21 nnäherungOrbita
n rrrrrr ϕϕϕ ⋅⋅⋅⎯⎯⎯⎯⎯ →⎯Ψ
(ϕx(rx) sind wasserstoff-ähnliche Einelektronenfunktionen)
Ein solcher Produktansatz führt mit gewissen Einschränkungen zu brauchbaren
Vorhersagen.
Erste Näherung:
• Die reale Kernladung wird durch eine „effektive“ Kernladung
ersetzt.
Jedes Elektron im Mehrelektronenatom „spürt“ eine reduzierte
Kernladung aufgrund der Anwesenheit weiterer Elektronen. Die
Elektronenkorrelation (des Ortes, der Spins etc.) bleibt unberück-
sichtigt.
1.3.3.2 Besetzungsregeln und Elektronenstruktur
Wie werden unterschiedliche Orbitale (1s, 2s, 2p, etc.) eines Mehrelektronensystems
durch Elektronen besetzt?
- Pauli-Prinzip: 2 Elektronen können nur dann durch gleiche Einelektronen-
funktionen beschrieben werden, wenn sie sich durch den Spin unterscheiden.
- Elektronen besetzen paarweise Orbitale von niedrigster zur höchsten Energie.
- Hund´sche Regel: Bei Besetzung energiegleicher (entarteter) Orbitale ist
Anordnung mit Elektronen gleichen Spins am stabilsten.

26
Beispiel:
Abbildung 25: Beispiel zu Besetzungsregeln (6 Elektronen, z.B. bei Kohlenstoff)
1.3.4 Moleküle
Wir haben die Mehrelektronensystem in einem Baukastenprinzip aus den Orbitalen
für Einelektronensysteme zusammengesetzt. In einem gedanklichen analogen Schritt
setzen wir Moleküle, die aus Atomen aufgebaut sind, aus Orbitalen zusammen, die
die einzelnen Atome beschreiben. Diesen Ansatz nennt man den LCAO-MO–Ansatz.
1.3.4.1 LCAO-MO–Ansatz (Linear Combination of Atomic Orbitals to
Molecular Orbitals)
Wie kommen wir von Atomen zu Molekülen?
H2-MolekülR1 R2
r2r1
Abbildung 26: Modell des Wasserstoffmoleküls
Die Beschreibung eines Moleküls umfasst mehrere Schritte:
Ein Molekül besteht aus m Atomkernen + n Elektronen (2 Atomkerne und 2
Elektronen für das H2-Molekül). Um das System vollständig beschreiben zu können,
brauchen wir im Prinzip die Koordinaten aller Atomkerne und aller Elektronen.
Formel 6: ( )n21m21 r,...,r,r,R,...,R,RΨ
(R1-Rm sind Atomkernkoordinaten, r1-rn sind die Elektronenkoordinaten)

27
Aufgrund der sehr viel kleineren Masse der Elektronen (und damit der Trägheit)
passen sich die Elektronen verzögerungsfrei der Umorientierung des Kerngerüsts an.
D.h.: Die Beschriebung der Kernkoordinaten (R1-Rm) kann von der Beschreibung der
Elektronenkoordinaten (r1-rn) abgetrennt werden. Die Gesamt-Psifunktion (Formel 6)
kann in ein Produkt von Psifunktionen, die nur die Kernbewegung (χ) und die nur die
Elektronenbewegung (ψ’) beschreiben (Formel 7), aufgeteilt werden. Diese
Aufteilung oder Separation der Kernbewegung von der Elektronenbewegung nennt
man die „Born-Oppenheimer-Näherung“.
Formel 7: ( ) ( )n21m21n21m21 r,...,r,r )R,...,R,Rr,...,r,r,R,...,R,R ´( ΨΨ ⋅→ χ
- χ(Rx) beschreibt das fixierte Kerngerüst
- Die Radien rx stehen für die Elektronenkoordinaten im fixierten Kerngerüst
Tabelle 5: Vergleich von Atom-Orbitalen und Molekül-Orbitalen
AO MO
AOs unterscheiden sich nur in
Ausdehnung und Energie von Atom zu
Atom, nicht aber in der Geometrie
Das effektive Feld der Kerngerüste
unterscheidet sich von Molekül zu
Molekül und von Konformation zu
Konformation. MOs werden im
Baukastenprinzip aufgebaut.
1.3.4.2 Orbital-Symmetrie
Orbitale werden klassifiziert nach ihrem Symmetrieverhalten (siehe Gruppentheorie-
vorlesungen).
Ψ´ = c · Ψ Symmetrie-Operator c wandelt Ψ in Ψ´ um.
c = ± 1 c = + 1 → symmetrisch
c = - 1 → antisymmetrisch

28
Tabelle 6: Symmetrie-Verhalten des 2px-Orbitals
MOs gehören zur selben Symmetriepunktgruppe wie das Molekül, das sie
beschreiben.
1.3.4.3 Wechselwirkungsdiagramme
Wie kommt es zur Bindung in einem H2-Molekül?
Dazu müssen wir gedanklich verfolgen, wie zwei H-Radikale langsam aufeinander
sich zu bewegen.
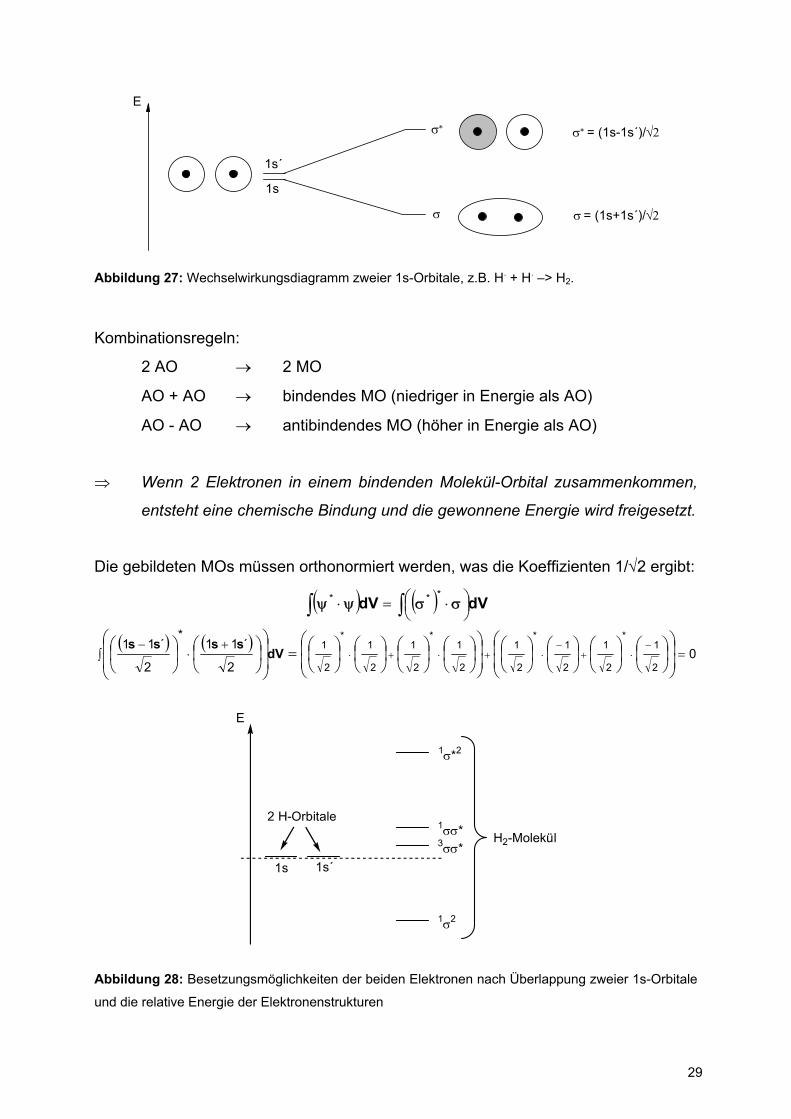
29
1s´
1s
σ
σ∗
E
σ∗ = (1s-1s´)/√2
σ = (1s+1s´)/√2
Abbildung 27: Wechselwirkungsdiagramm zweier 1s-Orbitale, z.B. H. + H. –> H2.
Kombinationsregeln:
2 AO → 2 MO
AO + AO → bindendes MO (niedriger in Energie als AO)
AO - AO → antibindendes MO (höher in Energie als AO)
⇒ Wenn 2 Elektronen in einem bindenden Molekül-Orbital zusammenkommen,
entsteht eine chemische Bindung und die gewonnene Energie wird freigesetzt.
Die gebildeten MOs müssen orthonormiert werden, was die Koeffizienten 1/√2 ergibt:
( ) ( )( ) ( )
02
´11*
2
´11
2
1*
2
1
2
1*
2
1
2
1*
2
1
2
1*
2
1
***
=∫+
⋅−
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛=⎟
⎟
⎠
⎞
⎜⎜
⎝
⎛⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞⎜
⎝⎛ ⋅=⋅
−⋅+
−⋅+⋅+⋅
∫∫
dVssss
dVdV σσψψ
1s 1s´
2 H-Orbitale
1σ*2
1σσ*3σσ*
1σ2
E
H2-Molekül
Abbildung 28: Besetzungsmöglichkeiten der beiden Elektronen nach Überlappung zweier 1s-Orbitale
und die relative Energie der Elektronenstrukturen

30
Jedes Elektron besitzt einen quantenmechanischen Spin s von ±½. Der Gesamtspin
S eines Atoms oder eines Moleküls berechnet sich aus der Summe der einzelnen
Spins (S = Σs).
Aus dem Gesamtspin wird durch die Formel 2S+1 die Spin-Multiplizität bestimmt,
welche durch eine links hochgestellte Ziffer angegeben wird.
Gesamtspin S Spin-Multiplizität Bezeichnung 0 1 Singulett 1 3 Triplett
Abbildung 29: Energieänderung der σ- und σ*-MOs in Abhängigkeit vom (Kern–Kern)-Abstand
Abbildung 29 zeigt die Energieänderung der σ- und σ*-MOs in Abhängigkeit vom
(Kern-Kern)-Abstand. Der Gleichgewichtsabstand erklärt sich durch den Kompromiss
aus möglichst großer Überlappung der beteiligten AOs und der gegenseitigen
Abstoßung der positiven Kerne.
Fragen:
1. Was bedeuten streng genommen s-,p-,d-,f-Orbitale?
2. Welche Vereinfachung führt zur Born-Oppenheimer-Nährung?

31
3. Nennen Sie die drei Regeln, mit deren Hilfe Elektronen die einzelnen Orbitale
eines Mehrelektronensystems besetzen.
4. Nennen Sie Unterschiede zwischen MOs und AOs? Von welchen Eigenschaften
eines Moleküls hängen MOs ab?
5. Wieviele MOs kann man aus 2 AOs konstruiere? Wie unterscheiden sich bindende
und die antibindende Wechselwirkungen?
Dritte Vorlesungsstunde
Inhalte:
I. In der zweiten Vorlesungsstunde haben wir die Beschreibung von Atomen im
Rahmen des MO-Modells diskutiert. Für das H2-Molekül haben wir die Kombination
von Atomorbitalen (s-Orbitalen) der H-Radikale zu Molekülorbitalen (σ-Orbitalen)
eingeführt. In der dritten Vorlesungsstunde werden die Kombinationsmöglichkeiten
weiterer Atomorbitale einführen.
II. Insbesondere wird auch in die Hybridisierung von s- und p-Orbitalen eingeführt.
Dies ist wichtig, um Kohlenstoffverbindungen der Koordinationszahl 2, 3 und 4 zu
verstehen.
III. Es wird am Beispiel des linearen und des gewinkelten Methylen (CH2) eingeführt,
wie die jeweilige Geometrie eines Moleküls die Kombination von Atomorbitalen
beeinflusst.
IV. Für Systeme mit konjugierten π-Systemen, also z.B. Systemen mit konjugierten
Doppelbindungen, kann man von MO-Modell zum HMO-Modell übergehen. In die
Vorhersage der π MO-Orbitale wird eingeführt.
V. In der ersten Vorlesungsstunde haben wir die Grenzen des klassischen
Strukturmodells an vier Beispielen aufgezeichnet. In der dritten Vorlesungsstunde
werden die Phänomen aus der Sicht des MO- und des HMO-Modell diskutiert.
VI. Regeln für aromatische und antiaromatische Verbindungen werden eingeführt.

32
1.3.4.4 Regeln für die Wechselwirkung von AO-Orbitalen
Es können nur solche Atom-Orbitale miteinander in Wechselwirkung treten, die (i) in
bezug auf Drehung oder Rotation gleiches Symmetrieverhalten besitzen und die (ii)
energetisch nah beieinander liegen. Letzteres erklärt, weshalb Bindungen zumeist
aus Orbitalen mit gleicher oder benachbarter Hauptquantenzahl entstehen.
Energetische Begründung:
∆E
AO MO
∆E∆E
AO MO AO MOE
Abbildung 30: Wechselwirkungsdiagramme von Orbitalen mit steigendem Unterschied in ihren
Energieniveaus. Mit zunehmendem energetischem Abstand der AOs wird die stabilisierende
Wechselwirkung ∆E zwischen AOs und MOs immer kleiner.
Für bindende Wechselwirkungen sind gewisse Symmetrievoraussetzungen
vonnöten.
s pz pzpx
Abbildung 31: Symmetrieverbotene Kombinationen von Atom-Orbitalen
Der positive Überlapp zwischen den Volumina mit gleichem Vorzeichen wird durch
den negativen Überlapp des gleich großen Volumenanteils mit entgegen gesetztem
Vorzeichen kompensiert, so dass insgesamt keine Stabilisierung zustande kommt.

33
+
+
2s+2p:
σ∗
σ
2p+2p:
end-on
2pσ-MOs
2sσ-MOs
bindend
antibindend
antibindend
bindend
side-on
2pπ-MOs
bindend
antibindend
+
Abbildung 32: Symmetrie-erlaubte Kombinationen von s- und p-Atom-Orbitalen
Zur Klassifizierung in σ- und π-Bindung muss berücksichtigt werden, welche
Symmetrie die entstandenen MOs relativ zur Bindungsachse aufweisen. Sind die
MOs rotationssymmetrisch zur Bindungsachse, spricht man von σ-Orbitalen, wenn
sie antisymmetrisch sind, von π-Orbitalen. π-Orbitale stehen senkrecht auf der
Bindungsachse, im Falle von zwei π-Orbitalen auch senkrecht zueinander.
1.3.4.5 Hybridisierung von Atom-Orbitalen
Linearkombinationen von Atomorbitalen innerhalb eines Atoms führen zu neuen
Atomorbitalen, so genannten Hybridorbitalen.
Am Beispiel der 2s- und der drei 2p-Orbitale eines Kohlenstoffatoms soll dies gezeigt
werden:

34
+ + +
2s 2px 2py 2pz
4
sp3-Hybridorbital
Abbildung 33: sp3-Hybridisierung
Aus 4 AO entstehen 4 Hybridorbitale. Diese 4 Hybridorbitale eines Atoms nehmen
die strukturell günstigste Geometrie ein, nämlich die, bei der alle Orbitale den
größtmöglichen Abstand zueinander besitzen, einen Tetraeder (vgl. Gillespie-
Nyholm-Modell, z. B. Hollemann-Wiberg).
Weitere Hybridorbitale sind möglich durch Herausnahme von p-Orbitalen aus der
Linearkombination:
+ + +
2s 2px 2py 2pz
3
sp2-Hybridorbital
+
2pz
Abbildung 34: sp2-Hybridisierung, Geometrie: trigonal planar.
Diese Hybridorbitale können nun wiederum mit Orbitalen anderer Atome bzw.
Atomgruppen in Wechselwirkung treten und somit Molekül-Orbitale bilden.
Abbildung 35: Darstellung der σ- und π-Bindungen im Ethen

35
Abbildung 36: sp-Hybridisierung, Geometrie: linear
Abbildung 37: Darstellung der σ- und π-Bindungen im Ethin
1.3.4.5.1 Wechselwirkungsdiagramm für linear angenommenes Methylen
CH H
C2 C2
C∝ Symmetrie D∝h
Abbildung 38: Lineares Methylen und seine Symmetrieoperationen
Zur Vereinfachung wird für das lineare Methylen eine Symmetrie von D2h
angenommen.
Das linear angeordnete Methylen lässt sich symmetrisch in zwei Fragmente
auftrennen, 1. das Fragment C-Atom und 2. das Fragment gedehntes H2-Molekül.
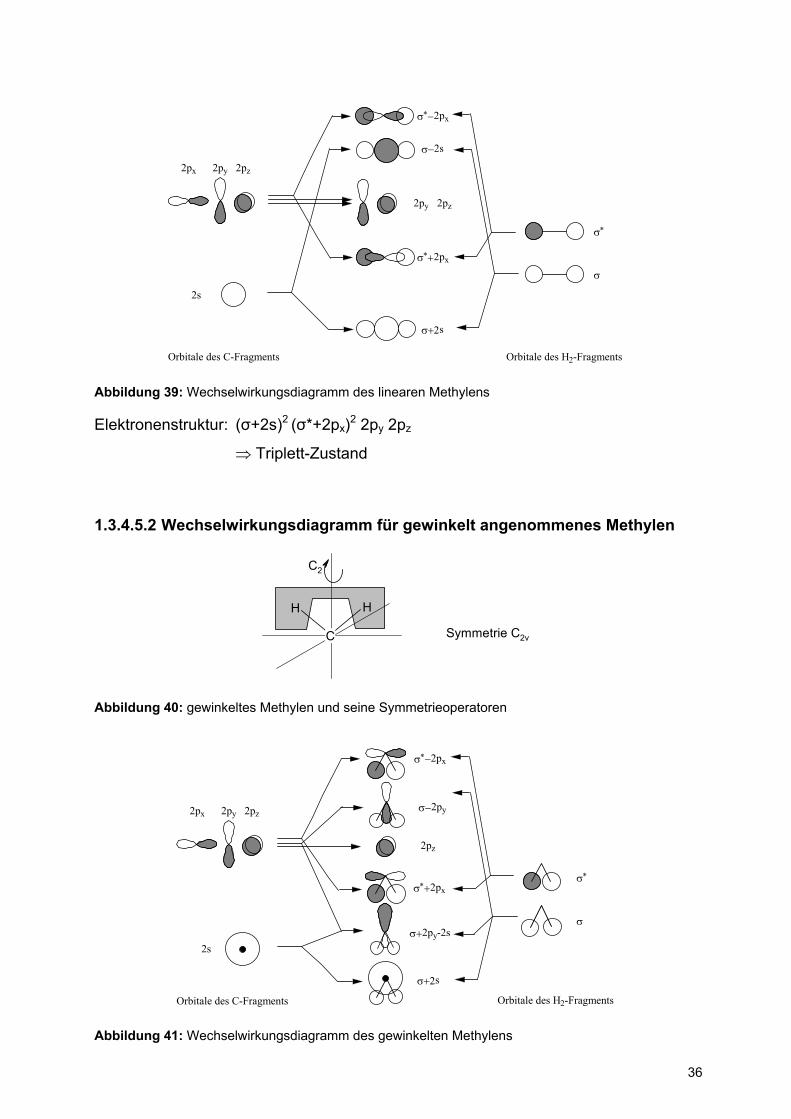
36
2s
2px 2py 2pz
2py 2pz
σ∗−2px
σ∗+2px
σ−2s
σ+2s
σ
σ∗
Orbitale des C-Fragments Orbitale des H2-Fragments
Abbildung 39: Wechselwirkungsdiagramm des linearen Methylens
Elektronenstruktur: (σ+2s)2 (σ*+2px)2 2py 2pz
⇒ Triplett-Zustand
1.3.4.5.2 Wechselwirkungsdiagramm für gewinkelt angenommenes Methylen
C
H H
C2
Symmetrie C2v
Abbildung 40: gewinkeltes Methylen und seine Symmetrieoperatoren
2s
2px 2py 2pz
2pz
σ∗−2px
σ∗+2px
σ−2py
σ+2s
σ
σ∗
Orbitale des C-Fragments Orbitale des H2-Fragments
σ+2py-2s
Abbildung 41: Wechselwirkungsdiagramm des gewinkelten Methylens

37
• 2py hat keinen Beitrag, da unsymmetrisch → nichtbindendes Orbital
• 2px hat gleiches Symmetrieverhalten wie σ* → 2px ± σ*
• σ, 2s, 2py weisen das gleiche Symmetrieverhalten auf
→ Dreierkombination, da man sonst 4 MOs
schaffen würde, was verboten wäre.
Elektronenstruktur: (σ+2s)2 (σ+2py-2s)2 (σ*+2px)2
⇒ Singulett-Zustand
1.3.4.5.3 Walsh-Diagramme
Mit Walsh-Diagrammen sollen die Änderungen der nodalen und energetischen
Eigenschaften der MOs verdeutlicht werden, wenn die Kernanordnung geändert wird.
• Korrelationslinien verbinden Orbitale mit gleichem Symmetrieverhalten
(gemäß den Symmetrie-Operationen beider Kernanordnungen)
Abbildung 42: Walsh-Korrelationsdiagramm für XH2 (X entspricht einem Atom aus der zweiten
Periode)
• Korrelationslinien dürfen sich nur für Orbitalpaare unterschiedlicher Symmetrie
kreuzen
Elektronenstruktur Molekül-Struktur Beispiel φ1
2φ22 linear H Be H
φ12φ2
2φ32 nicht-linear H
CH
Singulett!
φ12φ2
2φ32φ4
2 nicht-linear OH H
Tabelle 7: Strukturvorhersagen von XH2-Molekülen

38
1.3.4.6 Das HMO-Modell (Hückel Molecular Orbitals) und seine Vereinfachungen
1.3.4.6.1 Die (σ/π)-Trennung
σ-Orbitale können von π-Orbitalen getrennt werden.
- Anwendung nur auf π-Elektronensystem
- ebenes Molekül bzw. Molekülteil (xy-Ebene)
- s, px, py sind symmetrisch bezüglich der Molekülebene, während pz
antisymmetrisch angeordnet ist
→ keine Mischung zwischen den s-, px-, py-Atomorbitalen und den pz-
Atomorbitalen.
getrennte Wechselwirkungsdiagamme:
• symmetrische σ-MOs
• antisymmetrische π-MOs
Gesamtenergie Eπ des π-Elektronensystems:
Formel 8: ∑=i
iibE επ
bi: Besetzungszahl (0, 1 oder 2) des i-ten MOs; εi: Energie des i-ten MOs
Die Energie εi der π-MOs ergibt sich aus dem Coulombintegral α, welches die
freiwerdende Energie beschreibt, wenn ein Elektron das 2pz-AO des C-Atoms
besetzt, und dem Resonanzintegral β, welches die freiwerdende Energie darstellt,
wenn sich ein Elektron über weitere benachbarte AOs in bindenden MOs
delokalisiert. Beide Werte sind kleiner als Null.
E
α
Ε2=α−β
Ε1=α+β
2pz
π∗
π
Abbildung 43: HMO-Energieniveau-Diagramm für Ethen

39
Ethen: Gesamtenergie des π-Systems Eπ = 2(α+β)
Energiegewinn ∆Eπ = 2β
1.3.4.6.2 Mnemotechnische Hilfsmittel zur Beschreibung der HMO-Energie-Schemata (Gedächtnisstütze)
A. Monozyklische, konjugiert-ungesättigte Systeme nach Frost-Musulin:
Es werden Kreise mit einem Radius von 2|β| gezeichnet und die zyklischen
Moleküle mit der Spitze nach unten in diese gestellt, so dass die Ecken
(Atome) des Moleküls auf dem Kreis liegen. Eine horizontale Projektion der
Schnittpunkte gibt die Lage der Energieniveaus wider.
Abbildung 44: Mnemotechnisches Hilfsmittel nach Frost-Musulin zur Beschreibung von MO-Energie-
schemata für zyklische, konjugiert-ungesättigte Systeme vom Hückel-Typ (|β| ist die verwendete
Energieeinheit)

40
Besetzung aller bindenden MO mit je zwei Elektronen führt zu Systemen mit
(4n+2) π-Elektronen in abgeschlossenen Elektronenschalen und maximaler
Stabilisierung. Hierbei ist n eine ganze natürliche Zahl.
Abbildung 45: Zyklisch-konjugierte Moleküle bzw. Ionen mit (4n+2) π-Elektronen
In Systemen mit 4n Elektronen kommt man zu Biradikalen im Triplettzustand.
Abbildung 46: Zyklisch-konjugierte Moleküle bzw. Ionen mit 4n π-Elektronen
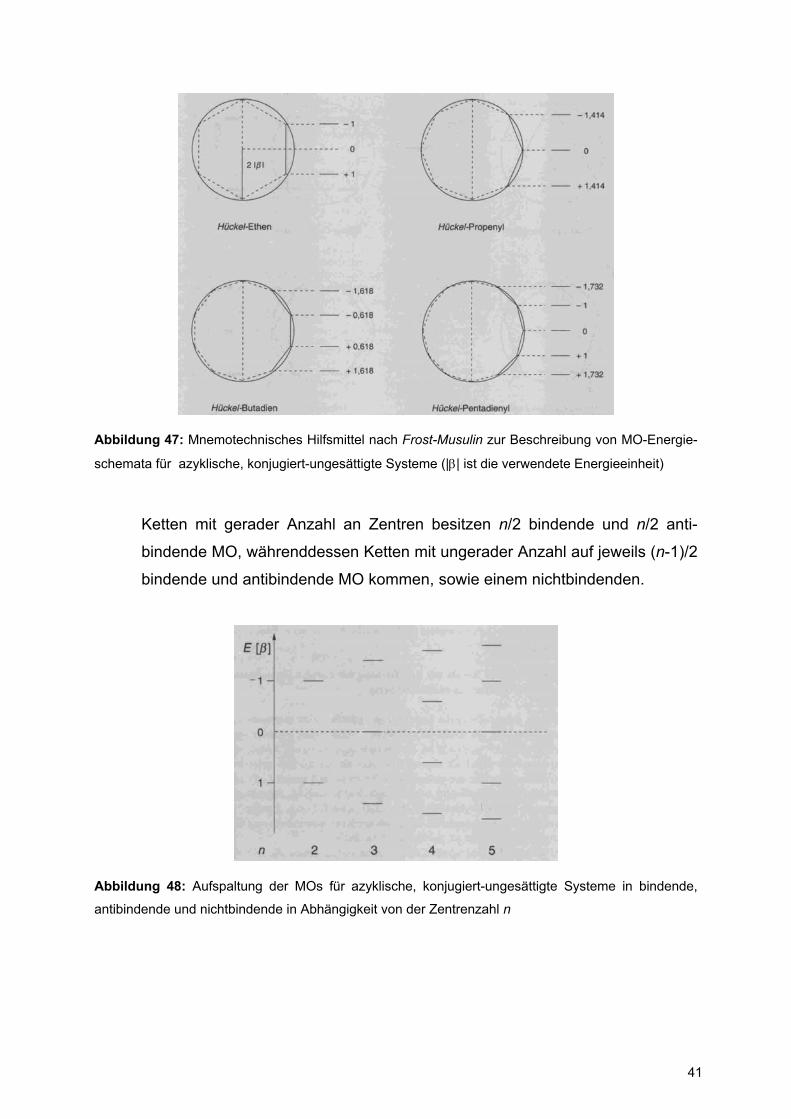
B. azyklische, konjugiert-ungesättigte Systeme nach Frost-Musulin
Für acyclische π-Systeme mit n Zentren werden Polygone mit 2n+2 Zentren in
die Kreise gezeichnet, wobei der azyklische Teil in die rechte Hälfte gesetzt
wird und die unten aufliegende Spitze nicht Bestandteil des Moleküls ist.
41
Abbildung 47: Mnemotechnisches Hilfsmittel nach Frost-Musulin zur Beschreibung von MO-Energie-
schemata für azyklische, konjugiert-ungesättigte Systeme (|β| ist die verwendete Energieeinheit)
Ketten mit gerader Anzahl an Zentren besitzen n/2 bindende und n/2 anti-
bindende MO, währenddessen Ketten mit ungerader Anzahl auf jeweils (n-1)/2
bindende und antibindende MO kommen, sowie einem nichtbindenden.
Abbildung 48: Aufspaltung der MOs für azyklische, konjugiert-ungesättigte Systeme in bindende,
antibindende und nichtbindende in Abhängigkeit von der Zentrenzahl n

42
1.3.4.6.3 Regeln für konjugiert-ungesättigte periplanare Moleküle
- π-Elektronensysteme aus n pz-AOs haben n π-MOs
- im Molekülorbital niedrigster Energie weisen alle Atomorbitale gleiches
Vorzeichen auf
- Zahl der Knotenflächen steigt mit zunehmender Einelektronenenergie
(bei Azyklen ist die Zahl der Knotenflächen um eine Einheit geringer als die
Laufzahl des MOs)
Abbildung 49: HMO-Satz von Benzol mit Angabe der Summe der bindenden(b) und antibindenden(a)
Wechselwirkungen, sowie der Zahl der Knotenflächen
Tabelle 8: HMO-Sätze für C2h- und C2v-Buta-1,3-dien unter Anwendung obiger Regeln (Σ AO-WW:
Summe der bindenden (b) und antibindenden (a) Wechselwirkungen)
1.4 Deutung der bisher unverständlichen Fälle durch das MO-Modell
Knotenflächen
3
2
1
0
43
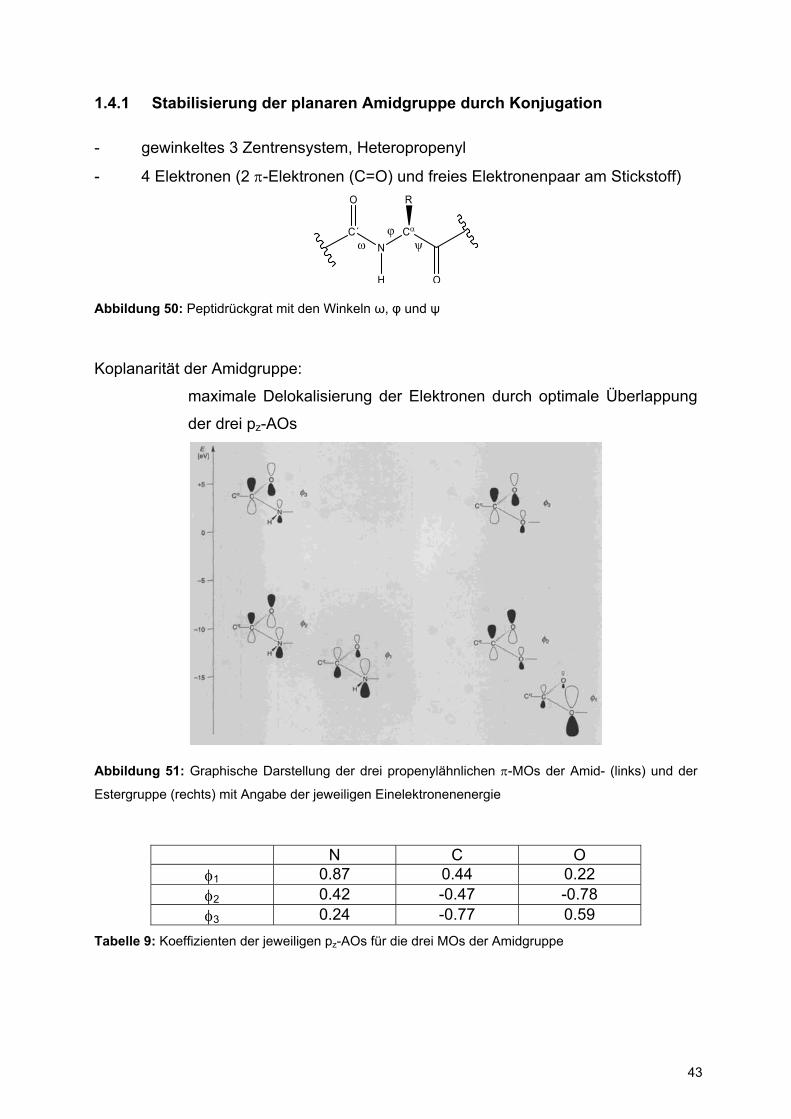
1.4.1 Stabilisierung der planaren Amidgruppe durch Konjugation
- gewinkeltes 3 Zentrensystem, Heteropropenyl
- 4 Elektronen (2 π-Elektronen (C=O) und freies Elektronenpaar am Stickstoff)
C´N
Cα
O
H O
R
ωϕ
ψ
Abbildung 50: Peptidrückgrat mit den Winkeln ω, φ und ψ
Koplanarität der Amidgruppe:
maximale Delokalisierung der Elektronen durch optimale Überlappung
der drei pz-AOs
Abbildung 51: Graphische Darstellung der drei propenylähnlichen π-MOs der Amid- (links) und der
Estergruppe (rechts) mit Angabe der jeweiligen Einelektronenenergie
N C O φ1 0.87 0.44 0.22 φ2 0.42 -0.47 -0.78 φ3 0.24 -0.77 0.59
Tabelle 9: Koeffizienten der jeweiligen pz-AOs für die drei MOs der Amidgruppe
44
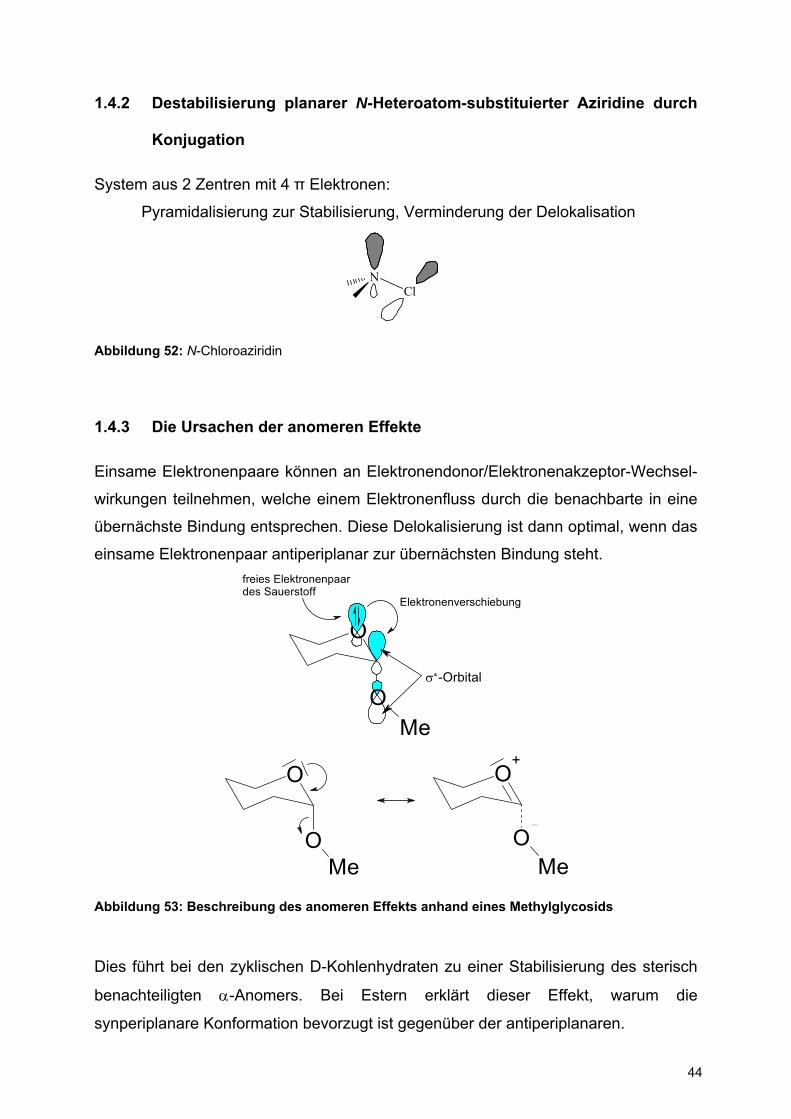
1.4.2 Destabilisierung planarer N-Heteroatom-substituierter Aziridine durch
Konjugation
System aus 2 Zentren mit 4 π Elektronen:
Pyramidalisierung zur Stabilisierung, Verminderung der Delokalisation
NCl
Abbildung 52: N-Chloroaziridin
1.4.3 Die Ursachen der anomeren Effekte
Einsame Elektronenpaare können an Elektronendonor/Elektronenakzeptor-Wechsel-
wirkungen teilnehmen, welche einem Elektronenfluss durch die benachbarte in eine
übernächste Bindung entsprechen. Diese Delokalisierung ist dann optimal, wenn das
einsame Elektronenpaar antiperiplanar zur übernächsten Bindung steht.
O
OMe
O+
OMe
O
OMe
freies Elektronenpaardes Sauerstoff
σ∗-Orbital
Elektronenverschiebung
Abbildung 53: Beschreibung des anomeren Effekts anhand eines Methylglycosids
Dies führt bei den zyklischen D-Kohlenhydraten zu einer Stabilisierung des sterisch
benachteiligten α-Anomers. Bei Estern erklärt dieser Effekt, warum die
synperiplanare Konformation bevorzugt ist gegenüber der antiperiplanaren.

45
O
O
sp
O
O
ap
Abbildung 54: synperiplanare und antiperiplanare Konformationen von Estern
1.4.4 Annulene
- monozyklische, konjugiert-ungesättigte Kohlenwasserstoffe
- Molekularformel (CH)2m
- Elektronenstruktur führt zur Klassifizierung in zwei Untergruppen:
• (4n+2) π-Elektronen → aromatisch
• 4n π-Elektronen → antiaromatisch
1.4.4.1 Annulene mit (4n+2) π-Elektronen: „aromatische Moleküle“
Kapitel 14.4. in Quinkert et al.
- ungerades m in der Molekularformel (CH)2m
- Prototyp: Benzol (CH)6 → m = 3
6 π-Elektronen → n = 1
⇒ HMO-π-Elektronenenergie von 8β
1.4.4.2 Annulene mit 4n π-Elektronen: „antiaromatische Moleküle“
- gerades m in der Molekularformel (CH)2m
- Beispiele: (Z,Z)-Cyclobuta-1,3-dien [4]-Annulen
(Z,Z,Z,Z)-Cycloocta-1,3,5,7-tetraen [8]-Annulen

46
Abbildung 55: Isomerisierung von Cyclobutadien
Cyclobutadien besitzt eine HMO-π-Elektronenenergie von 4β, während dies bei Buta-
1,3-dien 4.5 β beträgt. Daher ist Cyclobutadien antiaromatisch. Im Grundzustand
weist es Singulett-Charakter und eine rechteckige Geometrie auf, während die
Topomerisierung der rechteckigen Isomere eine quadratische Übergangsstruktur
durchläuft.
Fragen:
1. Welche Regeln gelten für die Wechselwirkung von AO’s?
2. Wie kommen Sie zu sp-, sp2-, sp3-Hybridisierung? Wieviele AOs sind an einem sp-
Hybridorbital beteiligt (sp2-, sp3)? Welche weitere Orbitale besitzt der Kohlenstoff?
Welche Bindungsgeometrie finden Sie bei sp-, sp2-, sp3-Hybridisierung? Wie
korreliert dies mit dem Gillespie-Nyholm-Modell?
3. Von welchen Grundannahmen geht das HMO-Modell aus?
4. Zeichnen Sie die HMO-Energieschemata mit dem Satz von HMO für Benzol und
für Butadien!
5. Wann spricht man von aromatischen und wann von antiaromatischen
Verbindungen?

47
Vierte Vorlesungsstunde
Inhalte:
I. Wir führen in die Klassifizierung von Reaktionen ein.
II. Am Beispiel der Reaktion von H2 mit D führen wir in das Konzept der
Reaktionskoordinate und Aktivierungsenergie ein.
III. Wir diskutieren den Einfluß der Energiedifferenz von Edukt und Produkt auf die
Lage des Gleichgewichts einer Reaktion.
IV. Wir diskutieren die Triebkraft einer Reaktion. Dies ist ein wichtiges Konzept, das
Sie häufig anwenden werden, um zu verstehen, wieso Reaktionen in einer gewissen
Richtung ablaufen.
V. Wir führen in die Begriffe ‚thermodynamisch kontrollierte’ und ‚kinetisch
kontrollierte’ Reaktion ein. Für den Begriff einer kinetisch kontrollierten Reaktion
müssen wir in die Theorie des Übergangszustands einführen. Hierbei diskutieren wir,
welchen Einfluß die Änderung der Aktivierungsenthalpie auf die Reaktionsrate
besitzt.
2 Reaktionen
2.1 Einführung
Strukturen Bindungen Strukturumwandlung (Stereochemie) (Reaktivität) (Reaktionen)
Reaktion:
Ausgangssubstanz
Reaktand (Katalysator)
Edukt (Enzym)
Substrat
Produkt

48
Ziele beim Studium von Reaktionen:
- Vorhersage der Reaktionsprodukte ?A X⎯→⎯
- Synthesebedingungen BA ?⎯→⎯
- Retrosynthese B? ?⎯→⎯
Reaktionstypen:
⎟⎟⎠
⎞⎜⎜⎝
⎛⎯⎯⎯⎯⎯⎯ →⎯⎪⎭
⎪⎬
⎫
2
1Bindungender Änderung
vv
ät Selektivit und
(v)t Reaktivitä von abhängig
rungIsomerisie -Atomen von Aufnahme -Atomen von Abspaltung -
Bindungsspaltung:
X Y
X Y
X+
Y
+
+
homolytisch (Radikale)
heterolytisch (Ionen)
polarisierte Bindungen, Partialladungen:
C Cl C Oδ−δ+ δ+ δ−
⇒ Angriff durch Elektrophil E+ (z.B. H+, Br+)
Nukleophil Nu- (z.B. OH-)
2.1.1 Einfache Reaktionen: Austauschreaktion (vergl. Lehrbücher der
Physikalischen Chemie)
Austausch eines Protons im Wasserstoffmolekül gegen ein Deuterium:
H H D H D H
. . .+ +
rHH
rHD
Abbildung 56: Gleichung der Reaktion eines Wasserstoffmoleküls mit einem Deuterium-Radikal

49
Der erste Schritt der Reaktion H2 + D -> HD + H ist die Ausbildung eines höher-
energetischen Übergangskomplexes HHD, der in Richtung der Edukte oder Produkte
zerfallen kann. Während sich das Deuteriumatom dem einen H-Atom nähert (rHD wird
kleiner), vergrößert sich der Abstand der Atome im H2-Molekül (rHH wird größer). Der
Reaktionsverlauf wird auf einer Energiehyperfläche G(rHH,rHD) wiedergegeben. Unter
der Annahme, dass der HHD-Komplex linear ist, hängt diese Energiehyperfläche in
guter Näherung nur vom Abstand rHH und rHD ab. Damit Atome und Moleküle
reagieren können, müssen sie eine Aktivierungsenergie besitzen, die zu einem
‚erfolgreichen’ Stoß führt.
Der Übergangskomplex ist zunächst experimentell nicht erfassbar, wird aber als
existent angenommen und als im Gleichgewicht befindlich mit Edukten und
Produkten. Dies erlaubt eine Ableitung der Aktivierungsenergie (siehe weiter unten)
aus thermodynamischen Größen (H, S).
Abbildung 57: Verlauf der potentiellen Energie während der Reaktion H–H + D⋅ → H⋅ + H–D, solche
Energiehyperflächen wurden von H. Eyring, H. Geishiweritz, C.E. Sun berechnet (J.Chem. Phys. 3,
768 (1935)).
Abbildung 58: Zusammenhang zwischen Energieverlauf einer Reaktion und den Potentialen für
Bindungsbruch und Bindungsbildung

50
Abbildung 59: Energiediagramm einer Einstufenreaktion
Es gilt für den Übergangszustand (ÜZ):
- Sattelpunkt des Reaktionsverlaufs
- kein isolierbares Zwischenprodukt
- Lebensdauer τÜZ ~ 10-12 s
- Energiedifferenz zwischen Edukten und ÜZ → Aktivierungsenthalpie ∆GE→ÜZ
- Energiedifferenz zwischen Edukt und Produkt → Reaktionsenthalpie ∆GE→P
Für eine Auftragung der Energie als Funktion der Reaktionskoordinate (RKT) gilt:
Die Reaktionskoordinate ist definiert als der Verlauf einer Reaktion von z.B. zwei
Reaktanden, bei denen sich die Reaktanden entlang einer Trajektorie niedrigster
Energie aufeinander zu bewegen (Pfeilstriche in Abbildung 57).
2.1.2 Gleichgewichtsreaktionen, Ungleichgewichtsreaktionen
Gleichgewichtsreaktion Ungleichgewichtsreaktion • reversibel • Produkte bestimmt durch
thermodynamisches Gleichgewicht K• thermodynamische Produktkontrolle
• irreversibel • Produkte bestimmt durch Selektivität
verschiedener Reaktionen • kinetische Produktkontrolle
Tabelle 10: Eigenschaften von Gleichgewichts-/Ungleichgewichtsreaktionen
Beispiel:
H+
RCOOH + R´OH RCOOR´ + H2O
Abbildung 60: Veresterung als Beispiel einer Gleichgewichtsreaktion
Kleine Mengen an Säure beschleunigen die Gleichgewichtseinstellung, nicht aber die
Gleichgewichtslage: Katalysator!
51
Gleichgewichtskonstante: [ ][ ][ ][ ]R´OHRCOOH
OHRCOOR´K 2=
Die Gleichgewichtskonstante K ist unter anderem abhängig von Lösungsmittel,
Temperatur, Druck, sowie den Konzentrationen von Edukten und Produkten.
Abbildung 61: Energiediagramm einer katalysierten und einer unkatalysierten Reaktion
Beziehung von ∆G zu K:
Formel 9
ST-HGe K
Kln RTG
RTG
∆∗∆=∆=
−=∆∆
−
Beispielwerte:
A B [ ][ ]ABK = T = 298 K (25 °C)
∆G (kcal mol-1) K %-Anteil von A im GG + 5.0 2.14⋅10-4 99.98 + 3.0 6.29⋅10-3 99.38 + 2.0 3.41⋅10-2 96.71 + 1.0 1.85⋅10-1 84.42 + 0.5 4.3⋅10-1 69.95
0 1 50 - 5.0 4.67⋅103 0.0214
Tabelle 11: Zahlenwerte von ∆G und K einer reversiblen Reaktion, GG: Gleichgewicht
Bei großen Beträgen von ∆G wird eine Reaktion praktisch irreversibel.
unkatalysiert
katalysiert
G
RKT
∆∆Gunkat→kat
enthalpischer Beitrag entropischer Beitrag

52
2.1.3 Triebkraft chemischer Reaktionen
Typische Bindungsenergien [kcal mol-1]:
C – C 81 O – O 33 C = C 148 N – N 38 C ≡ C 194 N ≡ N 226 C – O 84 C ≡ O 257 C = O 172 O = CO 128 C – H 100 O – H 110
Als Triebkraft einer chemischen Reaktion dient meist die Bildung sehr stabiler
funktioneller Gruppen oder kleiner Fragmentmoleküle, z.B. CO, N2, CO2,
Carbonylgruppe, R3P=O. So ist C=C weniger als doppelt so stabil wie C–C, während
C=O mehr als doppelt so stabil ist wie C–O.
2.1.4 Irreversible Reaktionen
Für vom Betrag nach große Werte von ∆G oder für Reaktionen, in denen ein
Reaktionspartner aus der Reaktion entweicht (Bsp. Gas) gilt: A → B, da diese keine
Gleichgewichtsreaktion, sind im Prinzip 100 % Ausbeute möglich.
BA k⎯→⎯
Abbildung 62: Energiediagramm einer irreversiblen Reaktion mit der freien Aktivierungsenergie ∆G‡
∆G‡ ist die freie Aktivierungsenthalpie. Ohne Barriere (∆G‡ =0) würde die Reaktion
mit diffusionskontrollierter Geschwindigkeitskonstante ablaufen.
Aber: Meist sind mehrere Produkte möglich!
BAB´ kk´ ⎯→⎯⎯⎯←
∆G
RKT
A
B
∆∆GA→B
A‡
∆∆G‡ freie Aktivierungsenthalpie

53
Produktverhältnis wird durch k und k´ gegeben.
⇒ „Kinetisch kontrollierte Reaktion“
In Abbildung 63 sind zwei mögliche Reaktionsprodukte für A aufgezeigt, B und B’. B’
ist stabiler als B, so dass unter Gleichgewichtsbedingungen das Verhältnis [B]/[B’]
aus der Stabilität von B und B’ berechnet werden kann. Wählt man für die
Durchführung einer Reaktion Bedingungen, bei denen A im Gleichgewicht mit B und
B’ steht, dann nennt man die Reaktion ‚thermodynamisch kontrolliert’.
Die freien Aktivierungsenthalpien ∆G‡B und ∆G‡
B’ unterscheiden sich. Das weniger
stabile B ist mit einer geringeren freien Aktivierungsenergie mit A ‚verknüpft’ als das
stabilere B’ (∆G‡B < ∆G‡
B’). D.h., führt man in einer Reaktion nur soviel Energie zu,
dass die Barriere zu B überwunden werden kann (wieviel, ist eine Funktion der
Temperatur, siehe unten), so kann man selektiv nur B erhalten oder zumindest B
anreichern. Eine solche Reaktion nennt man ‚kinetisch kontrolliert’.
2.1.5 Theorie des Übergangszustands
Wir betrachten die Reaktion von A nach B mit der Geschwindigkeitskonstanten k1
und dem Übergangszustand A‡.
A A‡K‡ k‡
B v ~ [A] k1 v ~ [A‡] k‡
[ ][ ]
≠∆
−≠≠≠≠
≠
=== kkKkk1RTG
eA
A
∆G
RKT
A
B
∆G‡B
B´
∆G‡B´
∆∆G‡B,B´
Abbildung 63: Energiediagramm zweier irreversibler Konkurrenzreaktionen

54
Formel 10: RTG
e≠∆
−=
hTkk B
1 Eyring-Gleichung
cal) (in T 57.4
G-T lg32.10k lg 1
≠∆+=
Beispielwerte (T = 298 K):
∆G‡ [kcal mol-1] t1/2 10 ~10 µs 15 ~10 ms 20 ~1 min 25 ~20 h 30 ~10 Jahre
Tabelle 12: Halbwertszeiten von Reaktionen mit der Aktivierungsenergie ∆G‡
⇒ Reduktion von ∆G‡ um 5 kcal mol-1 liefert eine drastische
Beschleunigung der Reaktion. Sie können nun ∆∆G‡ für zwei
mögliche Reaktionen in Reaktionsraten umrechnen.
Fragen:
1. Was versteht man unter homolytischer und heterolytischer Bindungsspaltung?
2. Was versteht man unter einer Reaktionskoordinate?
3. Stellen Sie zusammen, was Sie über den Übergangszustand einer Reaktion
wissen.
4. Nehmen Sie an, die Produkte einer Reaktion sind 5 kcalmol-1 stabiler als die
Edukte. Wo liegt dann die Gleichgewichtskonstante?
5. Wie unterscheiden sich thermodynamisch kontrollierte und kinetisch kontrollierte
Reaktionen?
55
Fünfte Vorlesungsstunde
Inhalte:
I. Wir führen in die Begriffe Nukleophile (nukleophil) und Elektrophile (elektrophil) ein.
II. Wir diskutieren die Verwendung von gebogenen Pfeilen zur Bilanzierung von
Elektronenwanderungen im Verlauf von Reaktionen. Elektronenquelle und
Elektronenabfluß sind zentrale Begriffe.
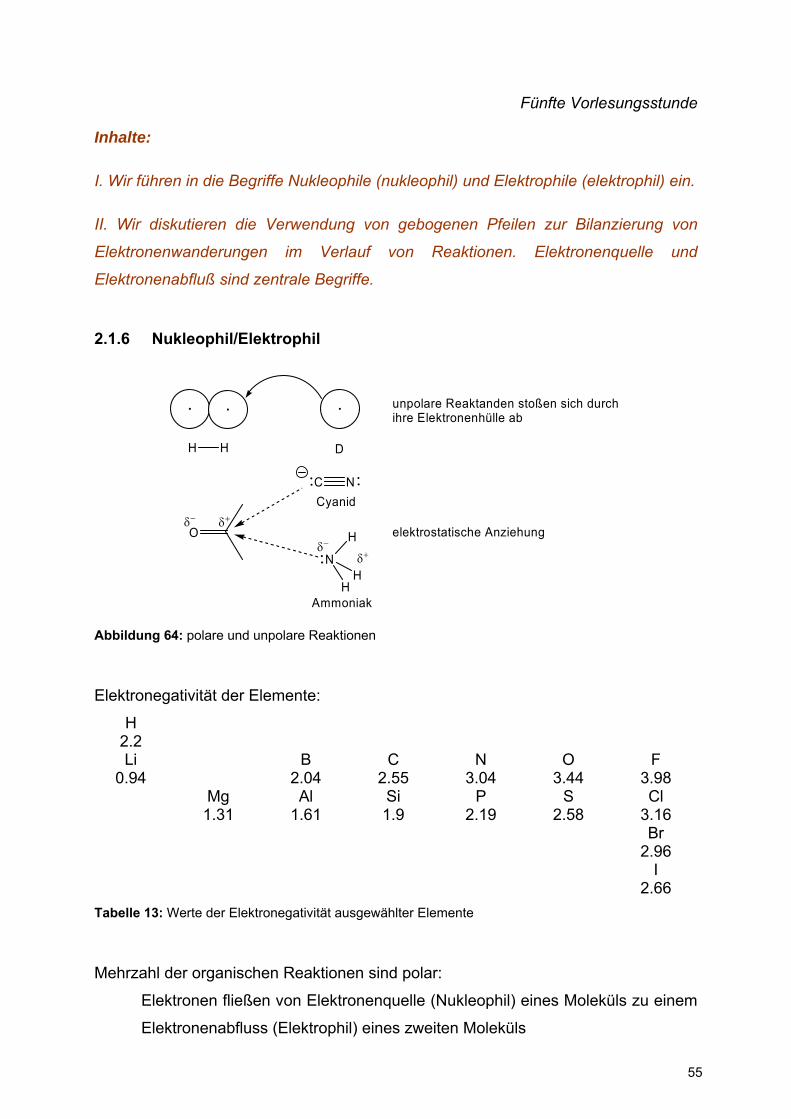
2.1.6 Nukleophil/Elektrophil
O
unpolare Reaktanden stoßen sich durch ihre Elektronenhülle ab
δ− δ+
C NCyanid
N
H
HH
Ammoniak
elektrostatische Anziehungδ−
δ+
H H D
Abbildung 64: polare und unpolare Reaktionen
Elektronegativität der Elemente:
H 2.2
Li 0.94
B 2.04
C 2.55
N 3.04
O 3.44
F 3.98
Mg 1.31
Al 1.61
Si 1.9
P 2.19
S 2.58
Cl 3.16
Br 2.96
I 2.66
Tabelle 13: Werte der Elektronegativität ausgewählter Elemente
Mehrzahl der organischen Reaktionen sind polar:
Elektronen fließen von Elektronenquelle (Nukleophil) eines Moleküls zu einem
Elektronenabfluss (Elektrophil) eines zweiten Moleküls
56
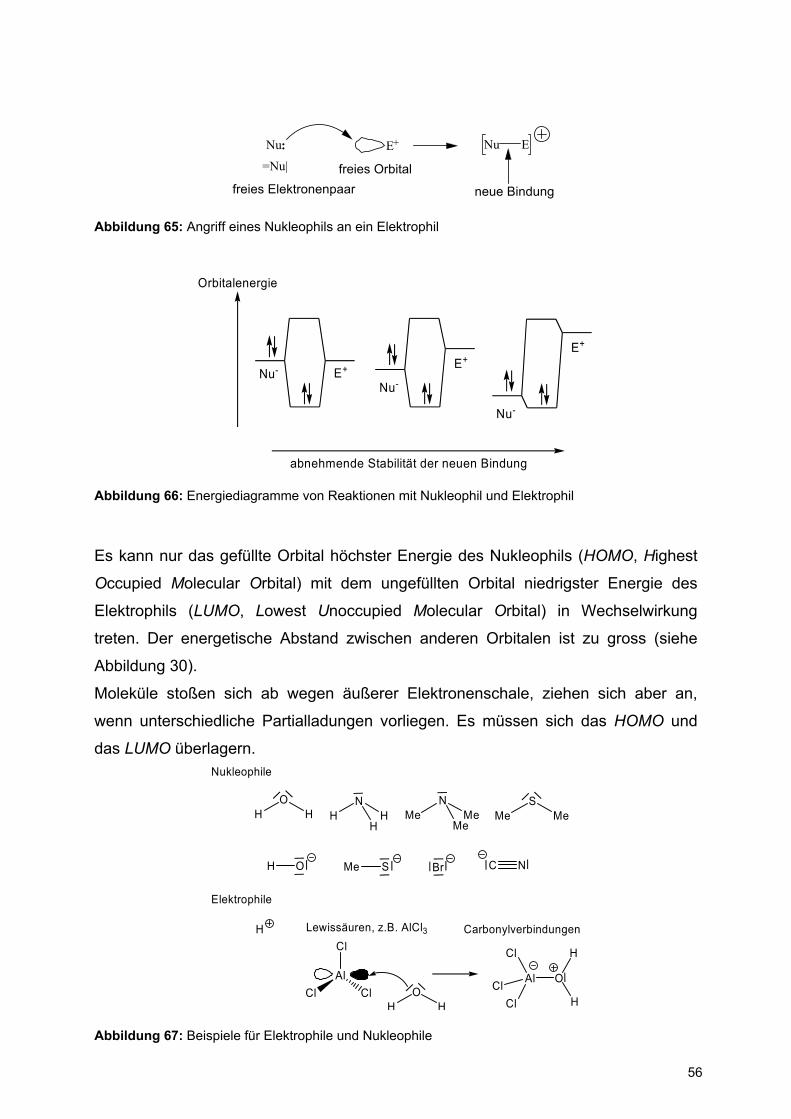
Nu: E+ ENu
=Nu|
freies Elektronenpaarfreies Orbital
neue Bindung
Abbildung 65: Angriff eines Nukleophils an ein Elektrophil
Orbitalenergie
Nu- E+
Nu-
E+
Nu-
E+
abnehmende Stabilität der neuen Bindung
Abbildung 66: Energiediagramme von Reaktionen mit Nukleophil und Elektrophil
Es kann nur das gefüllte Orbital höchster Energie des Nukleophils (HOMO, Highest
Occupied Molecular Orbital) mit dem ungefüllten Orbital niedrigster Energie des
Elektrophils (LUMO, Lowest Unoccupied Molecular Orbital) in Wechselwirkung
treten. Der energetische Abstand zwischen anderen Orbitalen ist zu gross (siehe
Abbildung 30).
Moleküle stoßen sich ab wegen äußerer Elektronenschale, ziehen sich aber an,
wenn unterschiedliche Partialladungen vorliegen. Es müssen sich das HOMO und
das LUMO überlagern.
HO
H HN
HH
MeN
MeMe
MeS
Me
H O Me S Br C N
Nukleophile
Elektrophile
H Lewissäuren, z.B. AlCl3 CarbonylverbindungenCl
AlClCl
HO
H
Al O
Cl
Cl
Cl
H
H
Abbildung 67: Beispiele für Elektrophile und Nukleophile
57
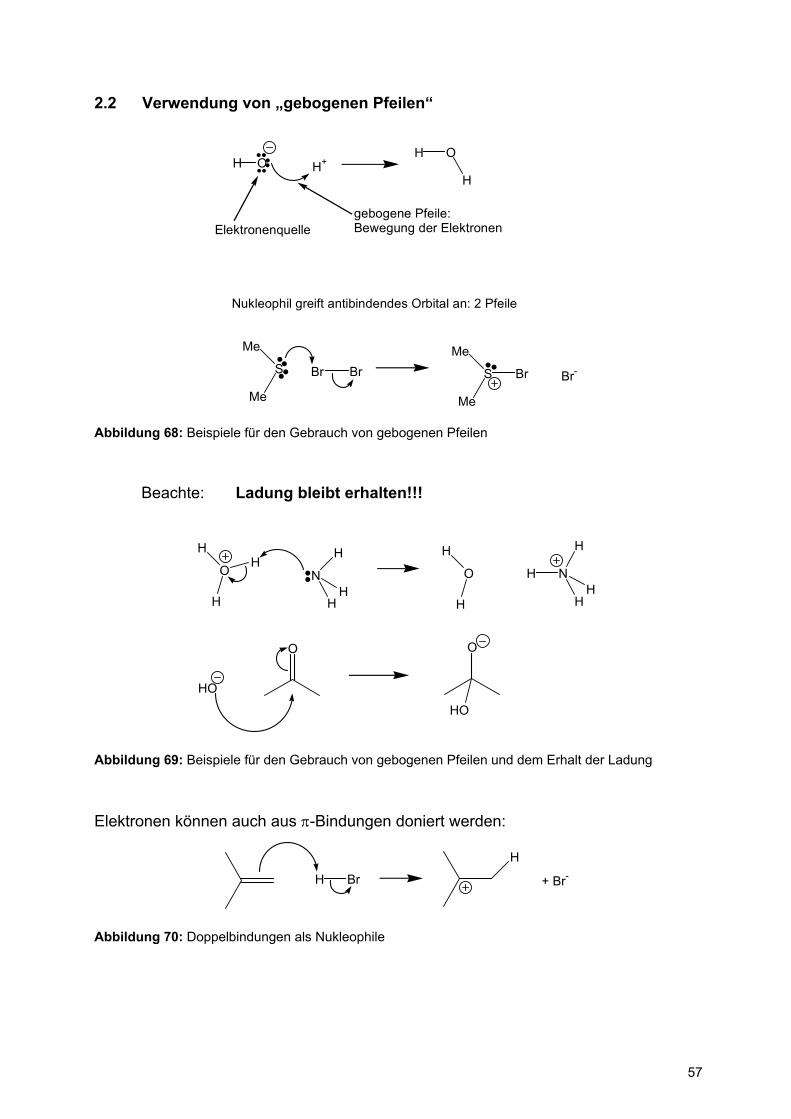
2.2 Verwendung von „gebogenen Pfeilen“
H O H+
Elektronenquelle
H O
H
gebogene Pfeile:Bewegung der Elektronen
Nukleophil greift antibindendes Orbital an: 2 Pfeile
Me
S
Me
Br BrMe
S
Me
Br Br-
Abbildung 68: Beispiele für den Gebrauch von gebogenen Pfeilen
Beachte: Ladung bleibt erhalten!!!
H
O H
H
N
H
HH
H
O
H
H N
H
H
H
HO
O O
HO
Abbildung 69: Beispiele für den Gebrauch von gebogenen Pfeilen und dem Erhalt der Ladung
Elektronen können auch aus π-Bindungen doniert werden:
H Br
H
+ Br-
Abbildung 70: Doppelbindungen als Nukleophile
58
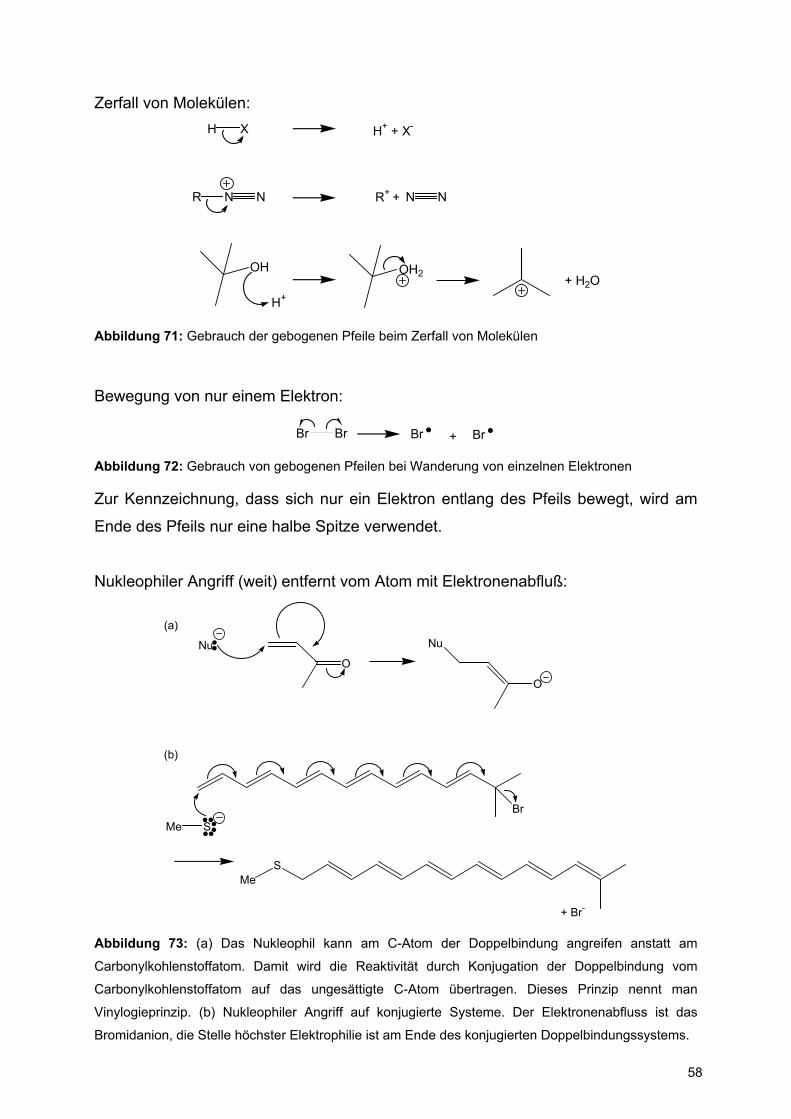
Zerfall von Molekülen:
R N
H X H+ + X-
N R+ + N N
OH
H+
OH2 + H2O
Abbildung 71: Gebrauch der gebogenen Pfeile beim Zerfall von Molekülen
Bewegung von nur einem Elektron:
Br Br Br + Br
Abbildung 72: Gebrauch von gebogenen Pfeilen bei Wanderung von einzelnen Elektronen
Zur Kennzeichnung, dass sich nur ein Elektron entlang des Pfeils bewegt, wird am
Ende des Pfeils nur eine halbe Spitze verwendet.
Nukleophiler Angriff (weit) entfernt vom Atom mit Elektronenabfluß:
ONu Nu
O
BrMe S
SMe
+ Br-
(a)
(b)
Abbildung 73: (a) Das Nukleophil kann am C-Atom der Doppelbindung angreifen anstatt am
Carbonylkohlenstoffatom. Damit wird die Reaktivität durch Konjugation der Doppelbindung vom
Carbonylkohlenstoffatom auf das ungesättigte C-Atom übertragen. Dieses Prinzip nennt man
Vinylogieprinzip. (b) Nukleophiler Angriff auf konjugierte Systeme. Der Elektronenabfluss ist das
Bromidanion, die Stelle höchster Elektrophilie ist am Ende des konjugierten Doppelbindungssystems.
59
Achtung: Bei der Konstruktion von Reaktionsmechanismen muss die Oktettregel bei den Elementen der 2.Periode (B, C, N, O) eingehalten werden.
Intramolekulare Reaktionen:
HSO
A
A
B
B
A
S
OH
BS
OH
Abbildung 74: Verwendung gebogener Pfeile bei intramolekularen Reaktionen
2.3 Carbonylchemie
Chapter 6 in Warren et al.
Die Carbonylgruppe ist durch ihre starke Polarisierung ein gutes Beispiel für
Reaktionen mit Elektrophilen und Nukleophilen. Während die Nukleophilie des
Carbonylsauerstoffs durch seine hohe Elektronegativität gering ist, prägt die starke
Elektrophilie des Carbonylkohlenstoffatoms die Reaktivität dieser funktionellen
Gruppe.
C O
C O
HOMO
LUMO
π
π∗
Abbildung 75: Energiediagramm einer Carbonylgruppe
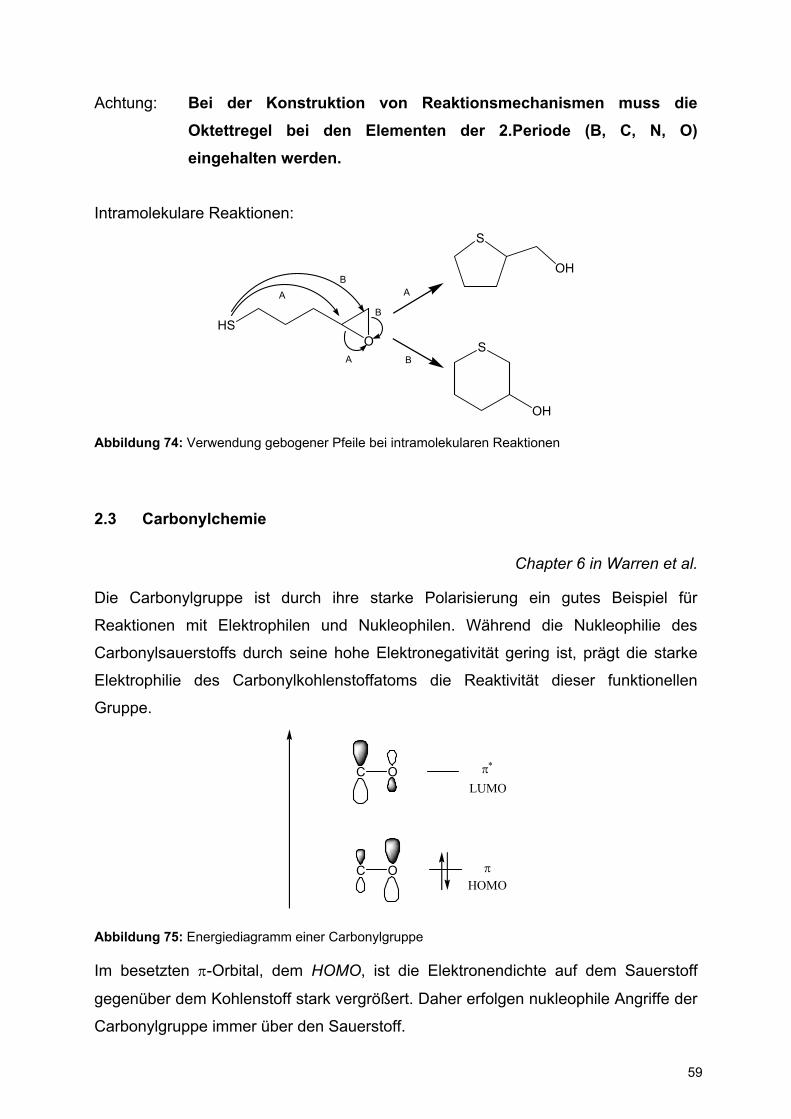
Im besetzten π-Orbital, dem HOMO, ist die Elektronendichte auf dem Sauerstoff
gegenüber dem Kohlenstoff stark vergrößert. Daher erfolgen nukleophile Angriffe der
Carbonylgruppe immer über den Sauerstoff.

60
Dagegen ist der Orbitallappen im unbesetzten π*-Orbital, dem LUMO, am Kohlenstoff
größer, so dass Nukleophile sich hier anlagern.
Nu O ONu
Elektronen-quelle
Elektronen-abfluss
NuO
Elektronen-quelle
Elektronen-abfluss
H+ O
H
ONu H
Abbildung 76: Nukleophile und elektrophile Reaktionen von Carbonylgruppen.
Fragen:
1. Was versteht man unter Elektronenquelle und Elektronenabfluß?
2. Wie interpretieren Sie diese Begriffe im Rahmen des MO-Modells?
3. Machen Sie sich klar, dass immer, IMMER die Elektronen wandern! Machen Sie
sich auch klar, dass wo Elektronenquelle, auch ein Elektronenabfluß da ist.
4. Zeichnen Sie die gebogenen Pfeile und zeichnen Sie KONSEQUENT (nicht
mogeln!!) die resultierenden Produkte.
5. Denken Sie daran: der Kohlenstoff ist nie fünfbindig.

61
Sechste Vorlesungsstunde
I. Die Carbonylgruppe ist ein wichtiges Elektrophil. Reaktionen von Carbonylgruppen
mit verschiedensten Nukleophilen (C-,N-,O-,S-Nukleophile) werden diskutiert.
II. Die Reaktionen werden in der Hinreaktion als auch in ihrer retrosynthetischen
Zerlegung diskutiert.
Bei der Carbonylgruppe handelt es sich um eine starke elektrophile Einheit. Als
Nukleophile können verschiedenste Spezies angreifen, z.B. C-Nukleophile, O-
Nukleophile, S-Nukleophile oder H-Nukleophile.
2.3.1 Cyanid als C-Nukleophil
Das Cyanid ist ein bekanntes Beispiel für ein C-Nukleophil.
R H
O NaCNH2SO4
H2O
OH
R HNC
OH
NC HR
+
Cyanhydrin
R H
OO
R HNC
H+
N C
OH
R HNC
rac
Abbildung 77: Synthese von Cyanhydrinen und der Mechanismus
Addition an Carbonylverbindungen:
1.) nukleophiler Angriff
2.) Protonierung des resultierenden Anions
Resultat: Knüpfung einer neuen C-C-Bindung
62
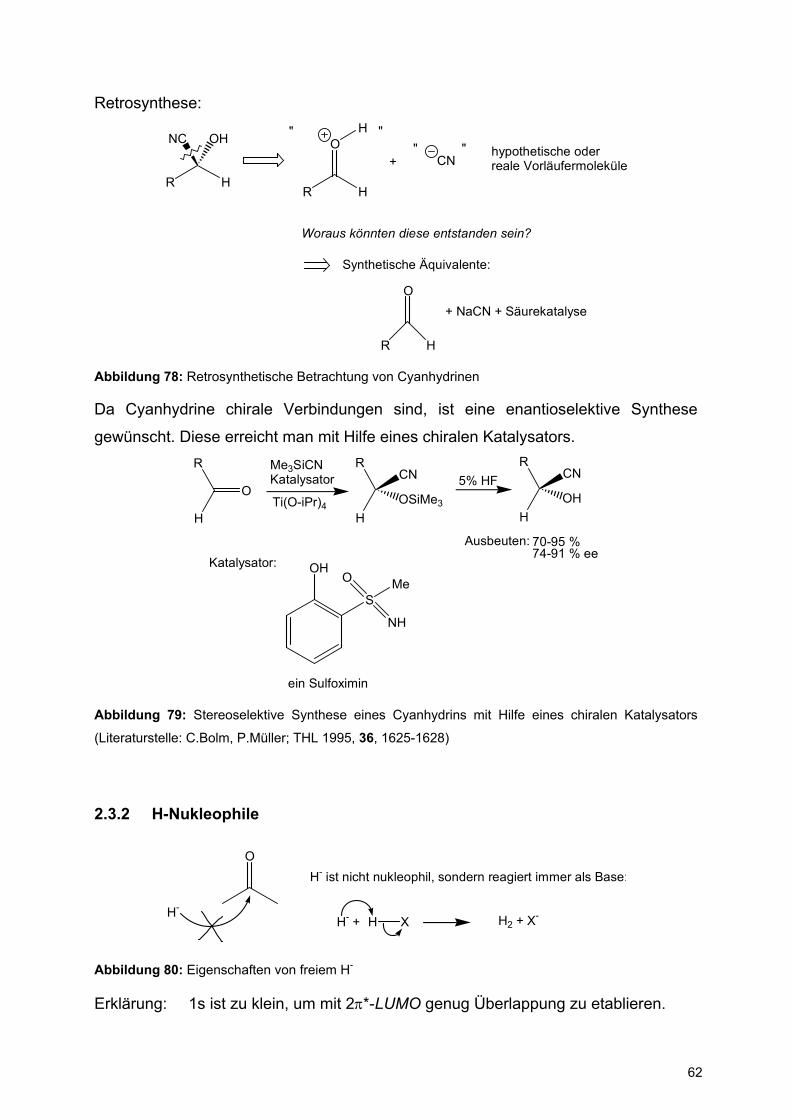
Retrosynthese:
R H
NC OH
R H
OH" "
+ CN" " hypothetische oder
reale Vorläufermoleküle
Woraus könnten diese entstanden sein?
Synthetische Äquivalente:
R H
O+ NaCN + Säurekatalyse
Abbildung 78: Retrosynthetische Betrachtung von Cyanhydrinen
Da Cyanhydrine chirale Verbindungen sind, ist eine enantioselektive Synthese
gewünscht. Diese erreicht man mit Hilfe eines chiralen Katalysators.
O
H
R Me3SiCNKatalysator
Ti(O-iPr)4 OSiMe3
H
RCN 5% HF
OHH
RCN
Katalysator: OH
S
NH
MeO
ein Sulfoximin
Ausbeuten: 70-95 %74-91 % ee
Abbildung 79: Stereoselektive Synthese eines Cyanhydrins mit Hilfe eines chiralen Katalysators
(Literaturstelle: C.Bolm, P.Müller; THL 1995, 36, 1625-1628)
2.3.2 H-Nukleophile
O
H-
H- ist nicht nukleophil, sondern reagiert immer als Base:
H- + H X H2 + X-
Abbildung 80: Eigenschaften von freiem H-
Erklärung: 1s ist zu klein, um mit 2π*-LUMO genug Überlappung zu etablieren.

63
Synthetisches Äquivalent: Natriumborhydrid NaBH4
H
B
H
H H
Na
R
O
HB
H
H H+
R H
H O
rac
R H
H O B
H
H
HR
O
H
R H
H O BH2
+
R H
H O
BH2(OCH2R)2 B(OCH2R)4Boronsäureester
Na
Na
Na
Abbildung 81: Reaktion von Natriumborhydrid als synthetisches Äquivalent eines H--Nukleophils mit
einem Aldehyden
Zur Isolierung des Reduktionsprodukts wird der Boronsäureester z.B. mittels
Methanol im Überschuss hydrolysiert.
R H
H O BH4-n
n R´
OHR H
H On + R´ O BH4-n
H
R H
H OHn +
n
R´ O BH4-nn
Abbildung 82: Hydrolyse des Borsäureester-Zwischenprodukts mittels eines Alkohols im Überschuss
zum reduzierten Produkt.
Resultat: Aldehyd wird zum primären Alkohol reduziert.
Keton wird zum sekundären Alkohol reduziert.
rac
R H
H OH
R
O
H
Reduktion
+I -I
64
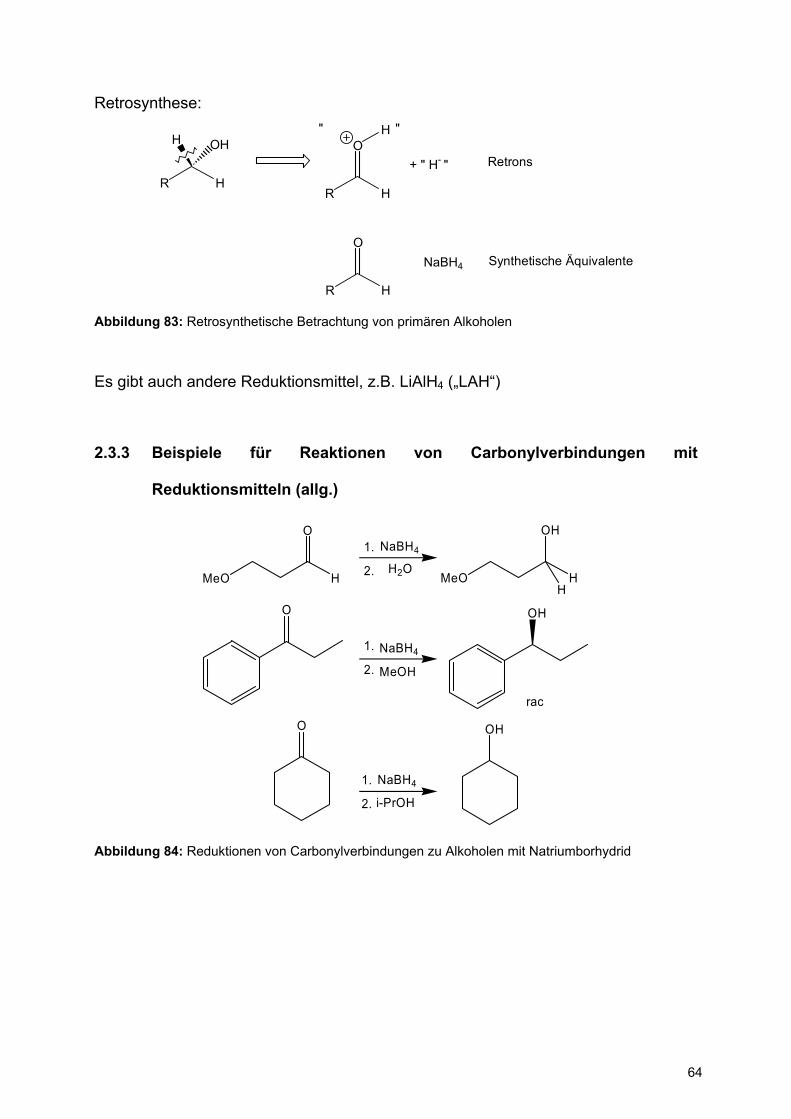
Retrosynthese:
R H
H OH
R
O
H
H" "
+ " H- " Retrons
R
O
H
NaBH4 Synthetische Äquivalente
Abbildung 83: Retrosynthetische Betrachtung von primären Alkoholen
Es gibt auch andere Reduktionsmittel, z.B. LiAlH4 („LAH“)
2.3.3 Beispiele für Reaktionen von Carbonylverbindungen mit
Reduktionsmitteln (allg.)
MeO H
ONaBH4
H2OMeO H
OH
HO
NaBH4
MeOH
OH
rac
O
NaBH4
i-PrOH
OH
1.
2.
1.
2.
1.
2.
Abbildung 84: Reduktionen von Carbonylverbindungen zu Alkoholen mit Natriumborhydrid
65
R Cl
O
>R O
O
R´
O
>R H
O
>
>R R´
O
R O
O
R´
R O
O
R´
>R O
O
H
R O
O
H
Säurechlorid Säureanhydrid Aldehyd Keton
Ester freie Carbonsäure
>R NR2
O
R NR2
O
Säureamid
>R O
O
R O
O
Carboxylat
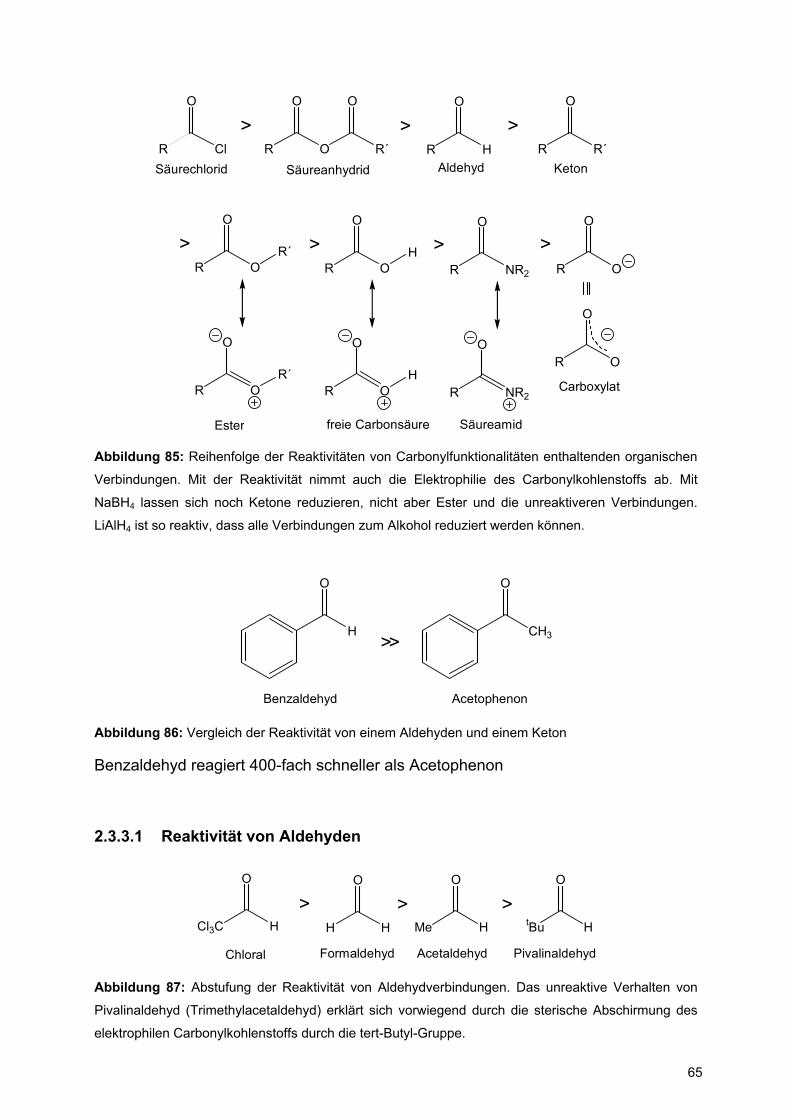
Abbildung 85: Reihenfolge der Reaktivitäten von Carbonylfunktionalitäten enthaltenden organischen
Verbindungen. Mit der Reaktivität nimmt auch die Elektrophilie des Carbonylkohlenstoffs ab. Mit
NaBH4 lassen sich noch Ketone reduzieren, nicht aber Ester und die unreaktiveren Verbindungen.
LiAlH4 ist so reaktiv, dass alle Verbindungen zum Alkohol reduziert werden können.
H
O
CH3
O
Benzaldehyd Acetophenon
>>
Abbildung 86: Vergleich der Reaktivität von einem Aldehyden und einem Keton
Benzaldehyd reagiert 400-fach schneller als Acetophenon
2.3.3.1 Reaktivität von Aldehyden
H H
O
Cl3C H
O
>Me H
O
>tBu H
O
>
Chloral Formaldehyd Acetaldehyd Pivalinaldehyd
Abbildung 87: Abstufung der Reaktivität von Aldehydverbindungen. Das unreaktive Verhalten von
Pivalinaldehyd (Trimethylacetaldehyd) erklärt sich vorwiegend durch die sterische Abschirmung des
elektrophilen Carbonylkohlenstoffs durch die tert-Butyl-Gruppe.
66
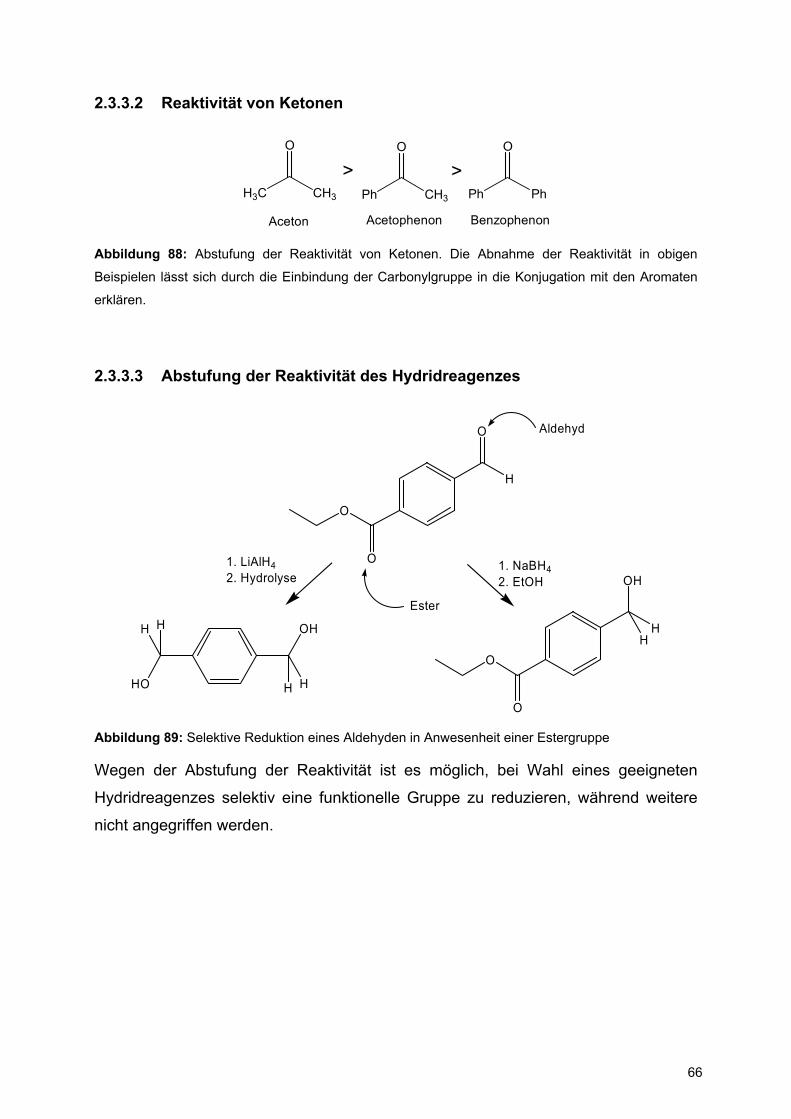
2.3.3.2 Reaktivität von Ketonen
Ph CH3
O
H3C CH3
O
>Ph Ph
O
>
Aceton Acetophenon Benzophenon
Abbildung 88: Abstufung der Reaktivität von Ketonen. Die Abnahme der Reaktivität in obigen
Beispielen lässt sich durch die Einbindung der Carbonylgruppe in die Konjugation mit den Aromaten
erklären.
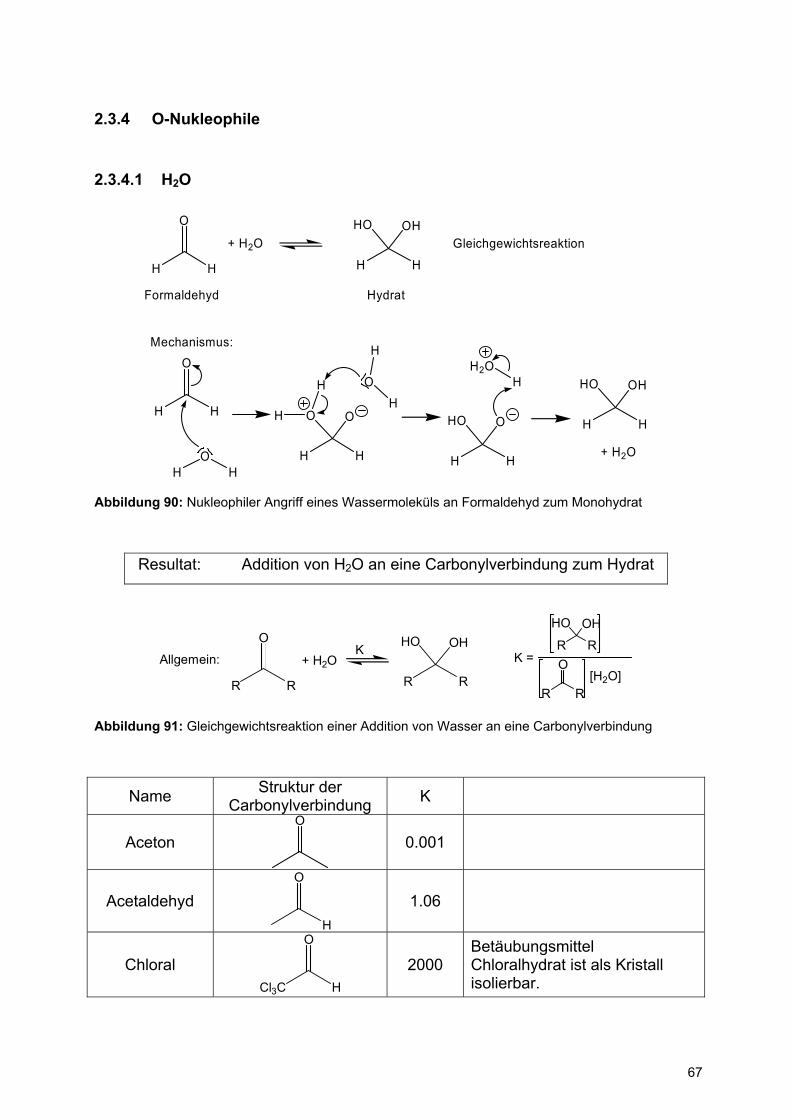
2.3.3.3 Abstufung der Reaktivität des Hydridreagenzes
O
O
H
O
Ester
Aldehyd
O
O
H
OH
H
1. NaBH42. EtOH
1. LiAlH42. Hydrolyse
HO H
OH
H
H H
Abbildung 89: Selektive Reduktion eines Aldehyden in Anwesenheit einer Estergruppe
Wegen der Abstufung der Reaktivität ist es möglich, bei Wahl eines geeigneten
Hydridreagenzes selektiv eine funktionelle Gruppe zu reduzieren, während weitere
nicht angegriffen werden.
67
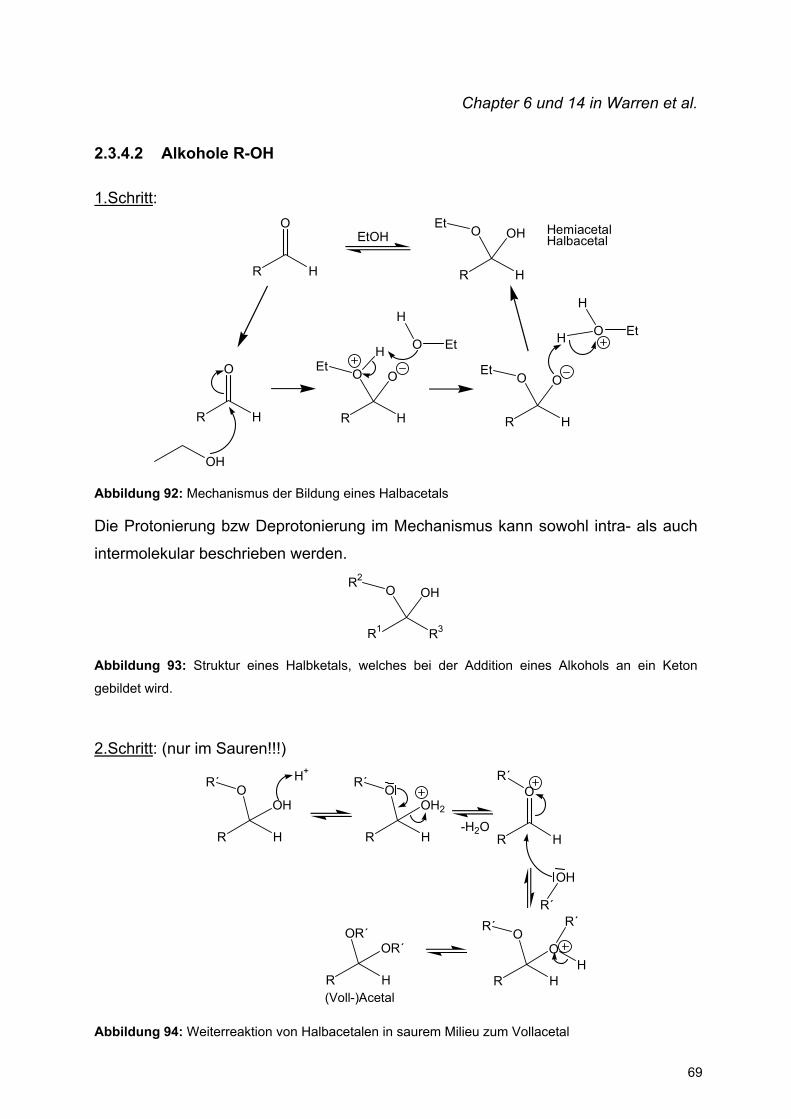
2.3.4 O-Nukleophile
2.3.4.1 H2O
O
H H
+ H2OOH
H H
HOGleichgewichtsreaktion
Mechanismus:O
H H
OH H
O
H H
O
H
H
OH
H
O
H H
HO
H2OH OH
H H
HO
+ H2O
Formaldehyd Hydrat
Abbildung 90: Nukleophiler Angriff eines Wassermoleküls an Formaldehyd zum Monohydrat
Resultat: Addition von H2O an eine Carbonylverbindung zum Hydrat
O
R R
+ H2OOH
R R
HOAllgemein:
KO
R R
OH
R R
HO
K =[H2O]
Abbildung 91: Gleichgewichtsreaktion einer Addition von Wasser an eine Carbonylverbindung
Name Struktur der Carbonylverbindung K
Aceton O
0.001
Acetaldehyd O
H 1.06
Chloral O
Cl3C H 2000
Betäubungsmittel Chloralhydrat ist als Kristall isolierbar.

68
Formaldehyd O
H H 2280
wasserfreies Formaldehyd? → polymeres Paraformaldehyd
HO O OHn
∆CH2O
Hexafluoro-aceton
O
F3C CF3 1200000
Cyclopropanon O
>> 1
Tabelle 14: Verhältnis einiger Carbonylverbindungen zu ihren Hydraten. Die Gleichgewichtskonstante
K gibt an, wie sehr das Gleichgewicht auf der Seite des Hydrats liegt.
Formaldehyd liegt vorwiegend auf der Seite des Hydrats, da hier keine sterische
Hinderung vorliegt, wenn die Bindungswinkel von 120° zu 109° verkleinert werden.
Bei Chloral erklärt sich die große Zahl von K, da der Substituent CCl3 durch
Elektronenzug (-I – Effekt) die Elektrophilie am Carbonylkohlenstoff verstärkt. Beim
Hexafluoroaceton ist dies noch dramatischer.
Kleine zyklische Ketone, z.B. Cyclopropanon, bevorzugen die hydratisierte Form, da
sich hierbei die Ringspannung vermindert. Bei Cyclopropanon sind die inner-
zyklischen Bindungswinkel auf 60° begrenzt, während das sp2-hybridisierte
Carbonylkohlenstoffatom einen Winkel von 120° anstrebt. In der hydratisierten Form
liegt der bevorzugte Winkel bei 109°, wodurch eine Entspannung des Rings
geschieht.
Retrosynthese:
R R
HO OH
R
O
R
H" "
+ " OH " Retrons
R
O
R
H2O Synthetische Äquivalente
69
Chapter 6 und 14 in Warren et al.
2.3.4.2 Alkohole R-OH
1.Schritt: O
R H
O
R H
OH
O
R H
OEtH
H
O Et
O
R H
OEt
H
H
O Et
EtOH OH
R H
OEt HemiacetalHalbacetal
Abbildung 92: Mechanismus der Bildung eines Halbacetals
Die Protonierung bzw Deprotonierung im Mechanismus kann sowohl intra- als auch
intermolekular beschrieben werden.
OH
R1 R3
OR2
Abbildung 93: Struktur eines Halbketals, welches bei der Addition eines Alkohols an ein Keton
gebildet wird.
2.Schritt: (nur im Sauren!!!)
R H
OOH
R´ H+
R H
OOH2
R´
R H
OR´
-H2O
R´
OH
R H
OO
R´
H
R´
R H
OR´OR´
(Voll-)Acetal
Abbildung 94: Weiterreaktion von Halbacetalen in saurem Milieu zum Vollacetal
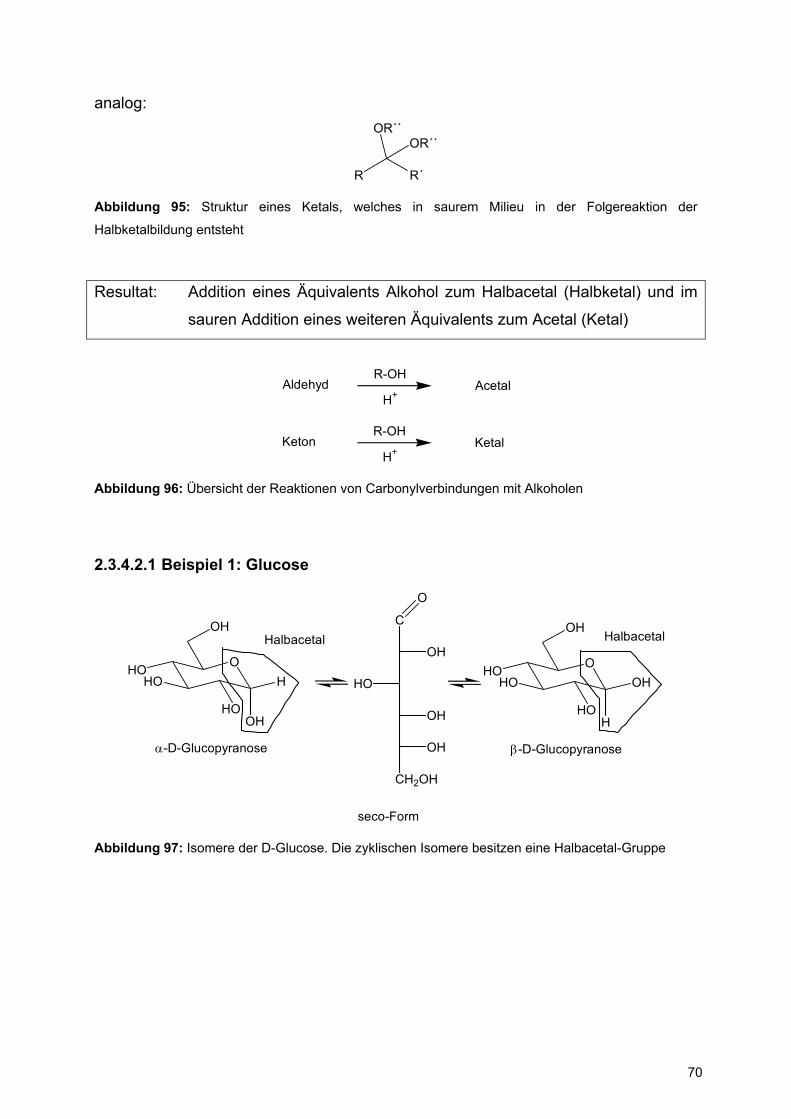
70
analog:
R R´
OR´´OR´´
Abbildung 95: Struktur eines Ketals, welches in saurem Milieu in der Folgereaktion der
Halbketalbildung entsteht
Resultat: Addition eines Äquivalents Alkohol zum Halbacetal (Halbketal) und im
sauren Addition eines weiteren Äquivalents zum Acetal (Ketal)
Aldehyd AcetalR-OH
H+
Keton KetalR-OH
H+
Abbildung 96: Übersicht der Reaktionen von Carbonylverbindungen mit Alkoholen
2.3.4.2.1 Beispiel 1: Glucose
O
OH
HOHO
HOOH
H
α-D-Glucopyranose
HalbacetalC
OH
HO
OH
OH
CH2OH
O
O
OH
HOHO
HOH
OH
β-D-Glucopyranose
Halbacetal
seco-Form
Abbildung 97: Isomere der D-Glucose. Die zyklischen Isomere besitzen eine Halbacetal-Gruppe
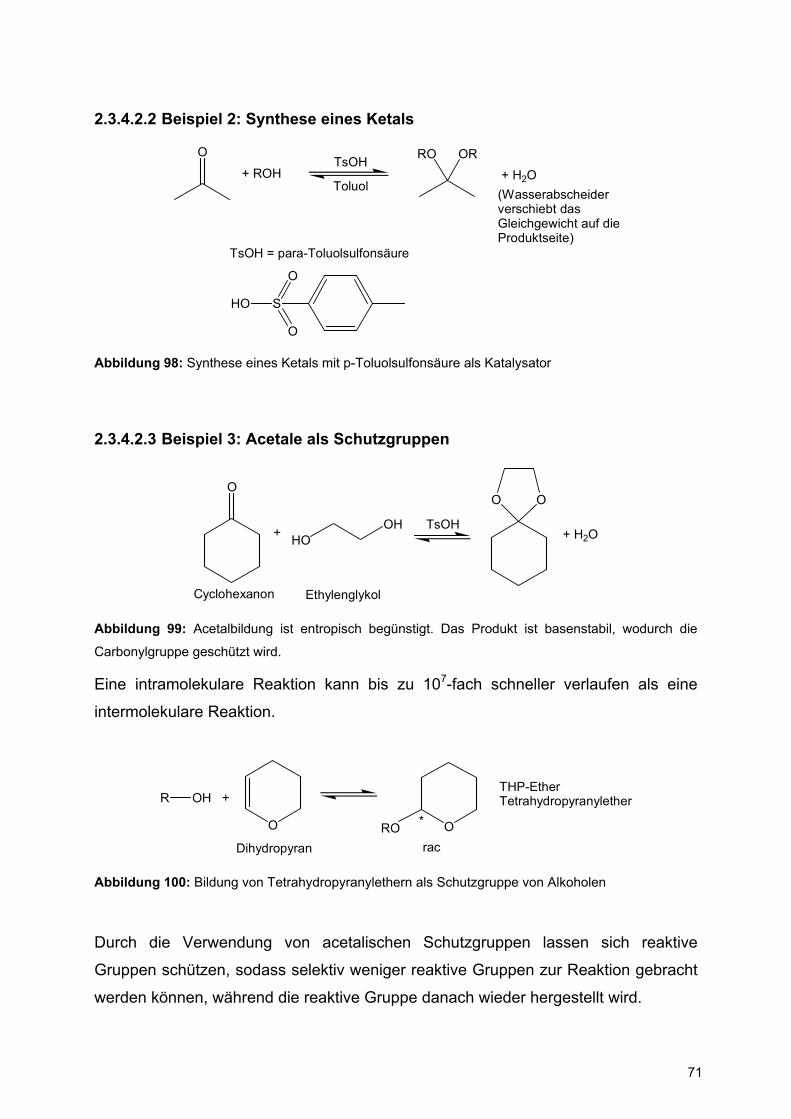
71
2.3.4.2.2 Beispiel 2: Synthese eines Ketals
O
+ ROHTsOH RO OR
+ H2O
TsOH = para-Toluolsulfonsäure
Toluol
S
O
O
HO
(Wasserabscheider verschiebt das Gleichgewicht auf die Produktseite)
Abbildung 98: Synthese eines Ketals mit p-Toluolsulfonsäure als Katalysator
2.3.4.2.3 Beispiel 3: Acetale als Schutzgruppen
O
Cyclohexanon
HOOH+
Ethylenglykol
TsOH
O O
+ H2O
Abbildung 99: Acetalbildung ist entropisch begünstigt. Das Produkt ist basenstabil, wodurch die
Carbonylgruppe geschützt wird.
Eine intramolekulare Reaktion kann bis zu 107-fach schneller verlaufen als eine
intermolekulare Reaktion.
R OH +
O
Dihydropyran rac
THP-EtherTetrahydropyranylether
ORO *
Abbildung 100: Bildung von Tetrahydropyranylethern als Schutzgruppe von Alkoholen
Durch die Verwendung von acetalischen Schutzgruppen lassen sich reaktive
Gruppen schützen, sodass selektiv weniger reaktive Gruppen zur Reaktion gebracht
werden können, während die reaktive Gruppe danach wieder hergestellt wird.
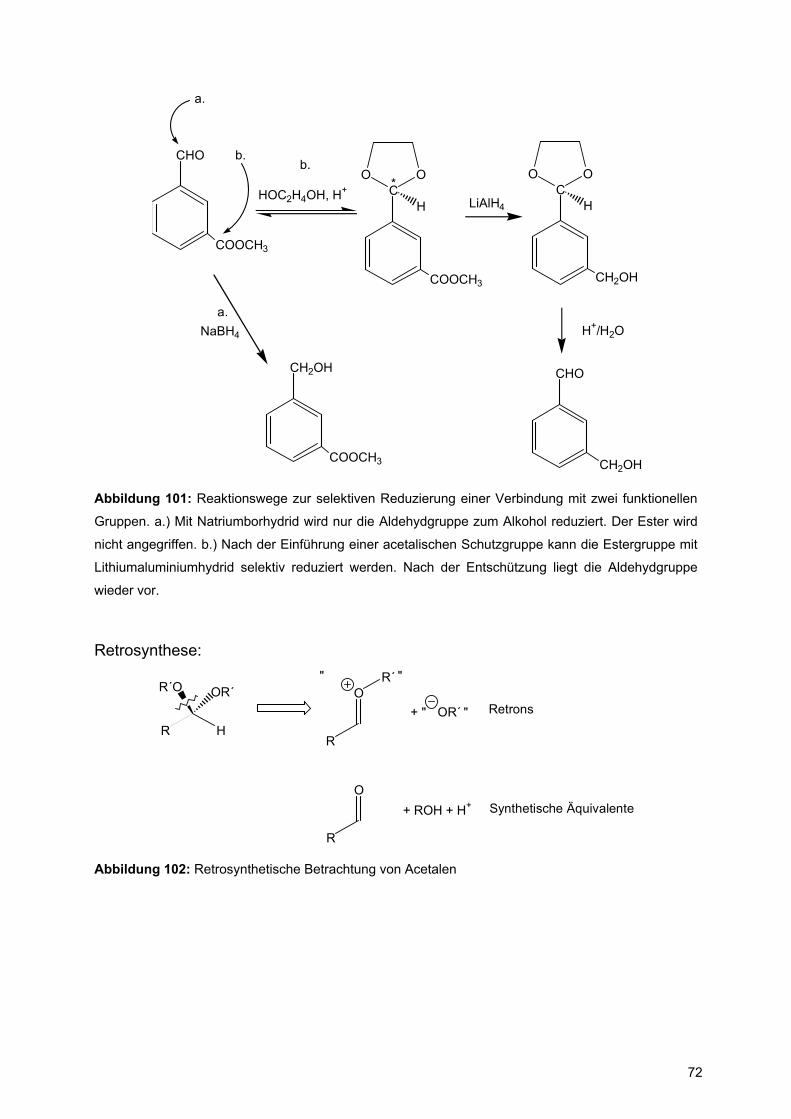
72
CHO
COOCH3
C
COOCH3
O O
H
* C
CH2OH
O O
H
H+/H2O
CHO
CH2OH
a.
HOC2H4OH, H+
b.
LiAlH4
NaBH4
CH2OH
COOCH3
a.
b.
Abbildung 101: Reaktionswege zur selektiven Reduzierung einer Verbindung mit zwei funktionellen
Gruppen. a.) Mit Natriumborhydrid wird nur die Aldehydgruppe zum Alkohol reduziert. Der Ester wird
nicht angegriffen. b.) Nach der Einführung einer acetalischen Schutzgruppe kann die Estergruppe mit
Lithiumaluminiumhydrid selektiv reduziert werden. Nach der Entschützung liegt die Aldehydgruppe
wieder vor.
Retrosynthese:
R H
R´O OR´
R
OR´" "
+ " OR´ " Retrons
R
O+ ROH + H+ Synthetische Äquivalente
Abbildung 102: Retrosynthetische Betrachtung von Acetalen
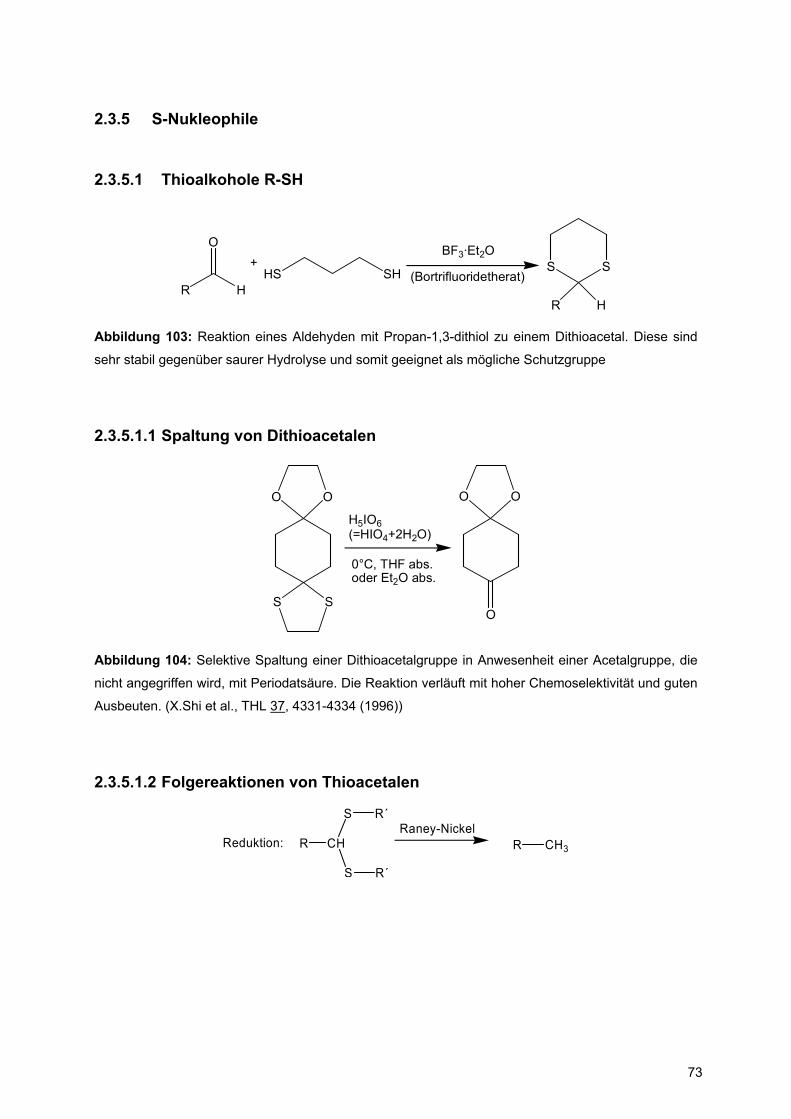
73
2.3.5 S-Nukleophile
2.3.5.1 Thioalkohole R-SH
O
R H
+HS SH S S
R H
BF3·Et2O
(Bortrifluoridetherat)
Abbildung 103: Reaktion eines Aldehyden mit Propan-1,3-dithiol zu einem Dithioacetal. Diese sind
sehr stabil gegenüber saurer Hydrolyse und somit geeignet als mögliche Schutzgruppe
2.3.5.1.1 Spaltung von Dithioacetalen
SS
OO
H5IO6(=HIO4+2H2O)
OO
O
0°C, THF abs.oder Et2O abs.
Abbildung 104: Selektive Spaltung einer Dithioacetalgruppe in Anwesenheit einer Acetalgruppe, die
nicht angegriffen wird, mit Periodatsäure. Die Reaktion verläuft mit hoher Chemoselektivität und guten
Ausbeuten. (X.Shi et al., THL 37, 4331-4334 (1996))
2.3.5.1.2 Folgereaktionen von Thioacetalen
Reduktion: R CH
S
S R´
R´
Raney-NickelR CH3
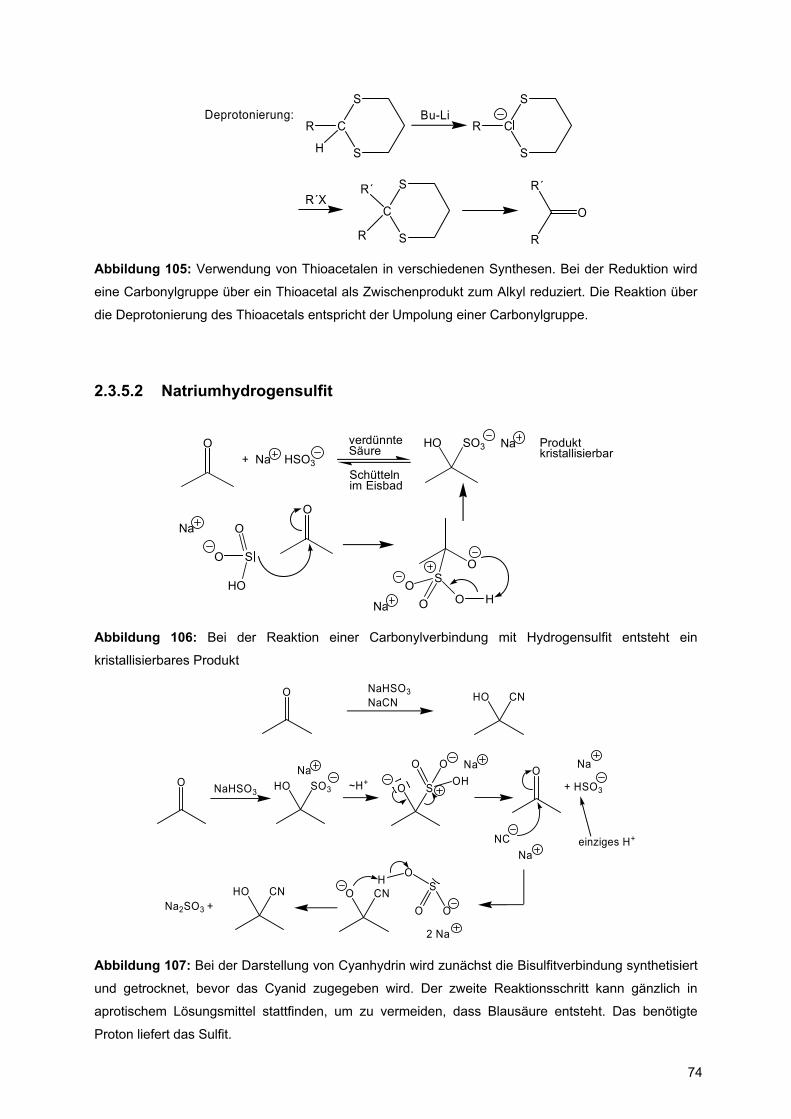
74
Deprotonierung:C
S
S
R
H
Bu-LiC
S
S
R
R´XC
S
S
R
R´
O
R
R´
Abbildung 105: Verwendung von Thioacetalen in verschiedenen Synthesen. Bei der Reduktion wird
eine Carbonylgruppe über ein Thioacetal als Zwischenprodukt zum Alkyl reduziert. Die Reaktion über
die Deprotonierung des Thioacetals entspricht der Umpolung einer Carbonylgruppe.
2.3.5.2 Natriumhydrogensulfit
O+ Na HSO3
O S
O
HO
NaO
OSO
O O HNa
HO SO3 Na
Schüttelnim Eisbad
verdünnteSäure
Produktkristallisierbar
Abbildung 106: Bei der Reaktion einer Carbonylverbindung mit Hydrogensulfit entsteht ein
kristallisierbares Produkt
O NaHSO3NaCN HO CN
O NaHSO3 HO SO3 O S
O OOH
O+ HSO3
NC einziges H+
O CNH O
S
O O
HO CNNa2SO3 +
Na Na
~H+
Na
Na
2 Na
Abbildung 107: Bei der Darstellung von Cyanhydrin wird zunächst die Bisulfitverbindung synthetisiert
und getrocknet, bevor das Cyanid zugegeben wird. Der zweite Reaktionsschritt kann gänzlich in
aprotischem Lösungsmittel stattfinden, um zu vermeiden, dass Blausäure entsteht. Das benötigte
Proton liefert das Sulfit.
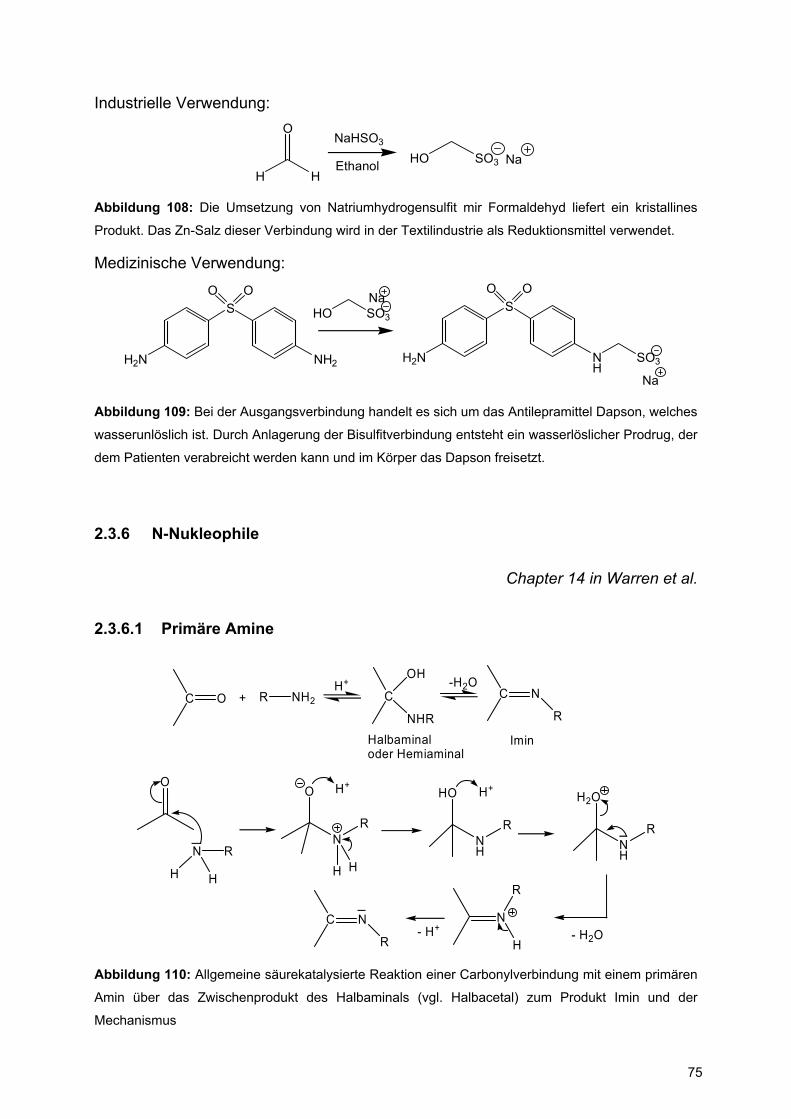
75
Industrielle Verwendung: O
H H
NaHSO3
Ethanol HO SO3 Na
Abbildung 108: Die Umsetzung von Natriumhydrogensulfit mir Formaldehyd liefert ein kristallines
Produkt. Das Zn-Salz dieser Verbindung wird in der Textilindustrie als Reduktionsmittel verwendet.
Medizinische Verwendung:
SOO
H2N NH2
HO SO3
NaS
OO
H2N NH
SO3
Na
Abbildung 109: Bei der Ausgangsverbindung handelt es sich um das Antilepramittel Dapson, welches
wasserunlöslich ist. Durch Anlagerung der Bisulfitverbindung entsteht ein wasserlöslicher Prodrug, der
dem Patienten verabreicht werden kann und im Körper das Dapson freisetzt.
2.3.6 N-Nukleophile
Chapter 14 in Warren et al.
2.3.6.1 Primäre Amine
C O R NH2+H+
C
OH
NHR
C N
R
Halbaminaloder Hemiaminal
Imin
-H2O
O
N R
H H
O
NR
H H
H+HO
NH
R
H+H2O
NH
R
- H2ON
R
H- H+
C N
R
Abbildung 110: Allgemeine säurekatalysierte Reaktion einer Carbonylverbindung mit einem primären
Amin über das Zwischenprodukt des Halbaminals (vgl. Halbacetal) zum Produkt Imin und der
Mechanismus

76
Die Reaktion (Abbildung 110) lässt sich in die folgenden Schritte einteilen:
1. Protonierung von CO
2. Addition des Amins
3. Deprotonierung des Amins
4. säurekatalysierte H2O-Abspaltung
Als Produkt erhält man das Imin. Für R=H kann man die C=NH-Gruppe als Stickstoff-
Analogon zur Carbonylgruppe betrachten. Entspricht R einer organischen Gruppe,
redet man von substituierten Iminen bzw. von einer Schiffschen Base.
Die meisten Imine, vor allem aber unsubstituierte, sind ziemlich unbeständig und
hydrolysieren leicht zurück zum Amin und der Carbonylverbindung. Stabile Imine
erhält man, wenn das Kohlenstoff- oder das Stickstoffatom mit einer Phenylgruppe
substituiert ist, da somit ein konjugiertes System entsteht (Abbildung 111). Außerdem
kann die Reaktion auf die Iminseite verschoben werden, wenn das gebildete H2O von
der Reaktionslösung entfernt wird, z.B. mit einem Wasserabscheider.
CHO + NH2
HC N
+ H2O
Abbildung 111: Durch Konjugation wird das gebildete Imin stabilisiert.
Anstelle von primären Aminen kann man auch folgende Edukte verwenden:
H2N OH N
OHHydroxylamin Oxim
H2N NH2
Hydrazin
N
NH2
+ N
NHydrazon
NO2HN
O2N
H2N 2,4-Dinitrophenylhydrazin(Derivat des Hydrazin, welches früher in der Analytik verwendet wurde, da sich die Hydrazone gut kristallisieren lassen
Abbildung 112: Beispiele weiterer N-Nukleophile und ihrer Produkte aus der Reaktion mit Carbonyl-
verbindungen
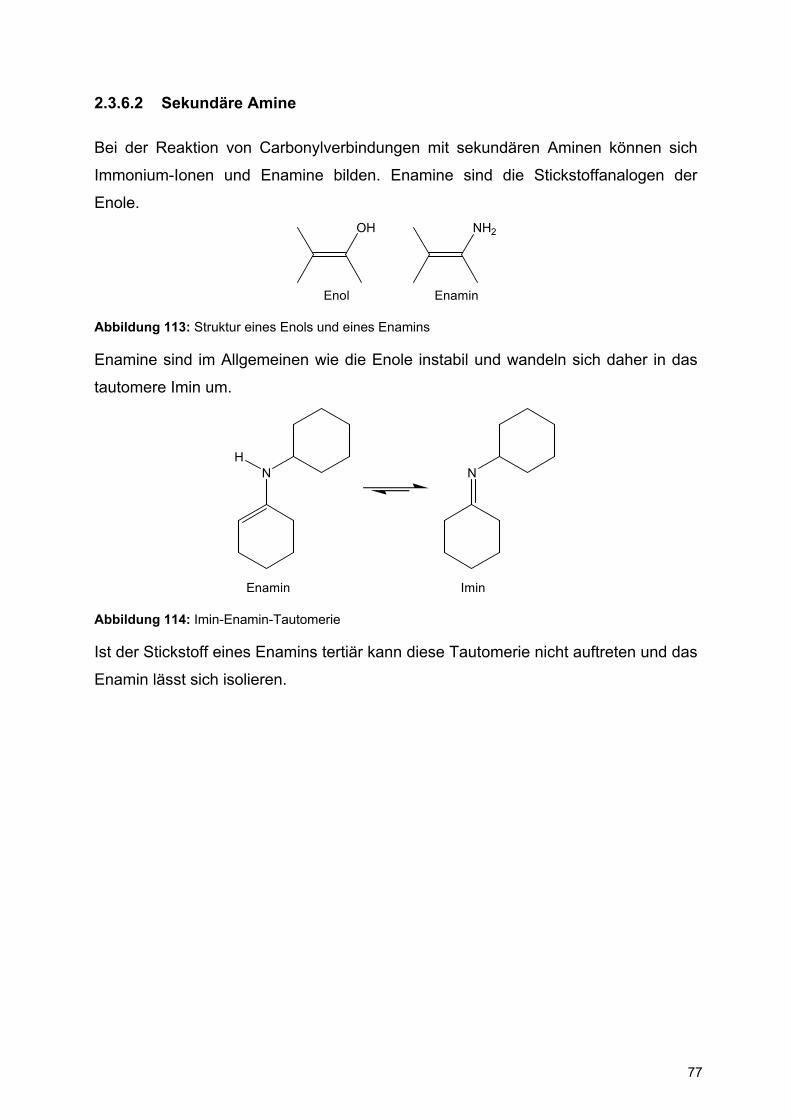
77
2.3.6.2 Sekundäre Amine
Bei der Reaktion von Carbonylverbindungen mit sekundären Aminen können sich
Immonium-Ionen und Enamine bilden. Enamine sind die Stickstoffanalogen der
Enole. OH
Enol
NH2
Enamin
Abbildung 113: Struktur eines Enols und eines Enamins
Enamine sind im Allgemeinen wie die Enole instabil und wandeln sich daher in das
tautomere Imin um.
NH
N
Enamin Imin
Abbildung 114: Imin-Enamin-Tautomerie
Ist der Stickstoff eines Enamins tertiär kann diese Tautomerie nicht auftreten und das
Enamin lässt sich isolieren.

78
O
+ H+
OH
+ R2NH
HO NHR2
~H+
H2O NR2NR2
-H2O
H
- H+
NR2
Immonium-IonEnamin
Abbildung 115: Mechanismus der säurekatalysierten Enaminsynthese. Als Zwischenprodukt entsteht
ein Immonium-Ion, welches bei fehlendem β-H-Atom als Endstufe der Reaktion entsteht.
Beachte: Das β-C-Atom eines Enamins entspricht dem α-C-Atom einer
Carbonylverbindung!
Fragen:
1. Weshalb kann man mit H - nicht direkt nukleophil an einem Kohlenstoff angreifen?
2. Sie kennen die Reaktivität von Carbonylgruppen enthaltenden funktionellen
Gruppen und können deren relative Elektrophilie erklären.
3. Erklären Sie die Lage des Gleichgewichts der Addition von H2O an eine
Carbonylverbindung für Aceton, Acetaldehyd, Formaldehyd, Chloral,
Hexafluoroaceton und Cyclopropanon..
4. Wie kommen Sie von einem Halbacetal zu einem Acetal. Benennen Sie wichtige
Halbacetale.
5. Wie unterscheiden sich die Produkte der Umsetzung von Carbonylverbindungen
mit primären, sekundären und tertiären Aminen.

79
Siebte Vorlesungsstunde
I. Neben dem Cyanidion gibt es auch C-Nukleophile, sogenannte metallorganische
Verbindungen. Sie lernen Grignardverbindungen und Organolithiumverbindungen
kennen..
II. Sie lernen verschiedene Methoden kennen, mit deren Hilfe
Organolithiumverbindungen hergestellt werden können.
III. Als Carbonylelektrophile, die mit metallorganischen Verbindungen reagieren
können, stehen CO2, Ester, Ketone, und Aldehyde zur Verfügung. Sie lernen die
resultierenden Produkte kennen.
IV. Der Mechanismus der Bildung einer Grignardverbindung wird kurz diskutiert.
2.3.7 Metallorganische Verbindungen als C-Nukleophile
Chapter 9 in Warren et al.
Metallorganische Verbindungen haben eine große Bedeutung bei der Bildung von C–
C-Bindungen. Die wesentlichen Reaktionen finden entweder mit Organolithium-
(Abbildung 119) oder mit Grignardverbindungen (Abbildung 117) statt.
Organometallverbindungen besitzen polare C–M-Bindungen (M = Metall):
O
CH H
EN = 3.5
EN = 2.5
Li
CH H
H
EN = 1.0
EN = 2.5
R Li
R MgX
R
R
+ Li
+ MgX
Abbildung 116: Vergleich der Elektronegativitäten bei Organometall- und Carbonylverbindungen und
Aufzeigen der Polarisierung der C–M-Bindung
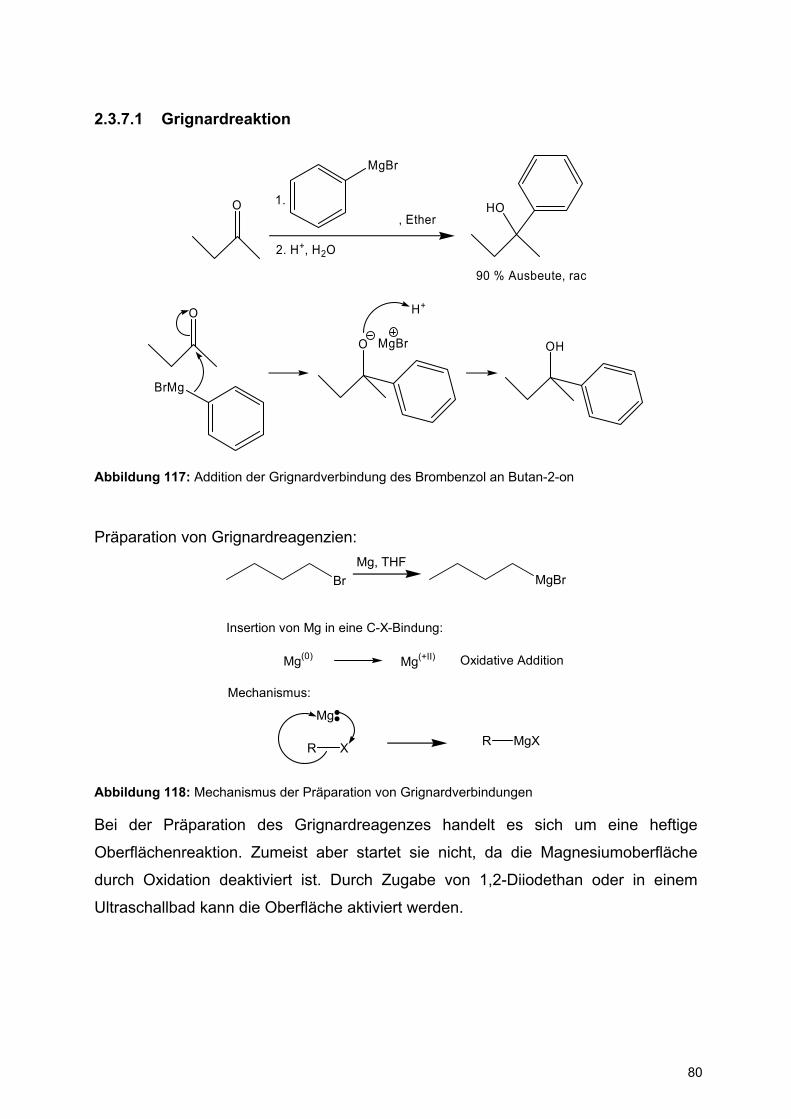
80
2.3.7.1 Grignardreaktion
O
MgBr
2. H+, H2O
1. HO
90 % Ausbeute, rac
, Ether
O
BrMg
O MgBr
H+
OH
Abbildung 117: Addition der Grignardverbindung des Brombenzol an Butan-2-on
Präparation von Grignardreagenzien:
BrMg, THF
MgBr
Insertion von Mg in eine C-X-Bindung:
Mg(0) Mg(+II) Oxidative Addition
R X
Mg
R MgX
Mechanismus:
Abbildung 118: Mechanismus der Präparation von Grignardverbindungen
Bei der Präparation des Grignardreagenzes handelt es sich um eine heftige
Oberflächenreaktion. Zumeist aber startet sie nicht, da die Magnesiumoberfläche
durch Oxidation deaktiviert ist. Durch Zugabe von 1,2-Diiodethan oder in einem
Ultraschallbad kann die Oberfläche aktiviert werden.
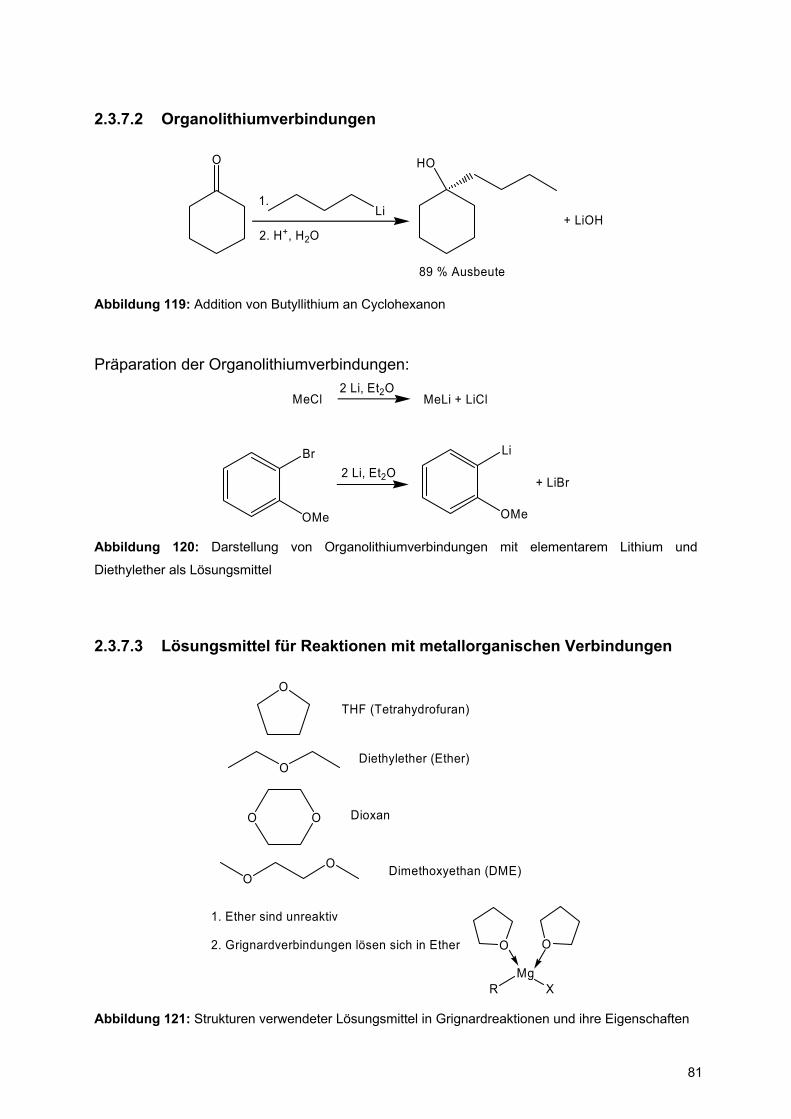
81
2.3.7.2 Organolithiumverbindungen
O
Li1.
2. H+, H2O
HO
89 % Ausbeute
+ LiOH
Abbildung 119: Addition von Butyllithium an Cyclohexanon
Präparation der Organolithiumverbindungen:
MeCl2 Li, Et2O
MeLi + LiCl
Br
OMe
2 Li, Et2O
Li
OMe
+ LiBr
Abbildung 120: Darstellung von Organolithiumverbindungen mit elementarem Lithium und
Diethylether als Lösungsmittel
2.3.7.3 Lösungsmittel für Reaktionen mit metallorganischen Verbindungen
O
THF (Tetrahydrofuran)
ODiethylether (Ether)
OO Dioxan
OO Dimethoxyethan (DME)
1. Ether sind unreaktiv
2. Grignardverbindungen lösen sich in Ether
RMg
X
O O
Abbildung 121: Strukturen verwendeter Lösungsmittel in Grignardreaktionen und ihre Eigenschaften
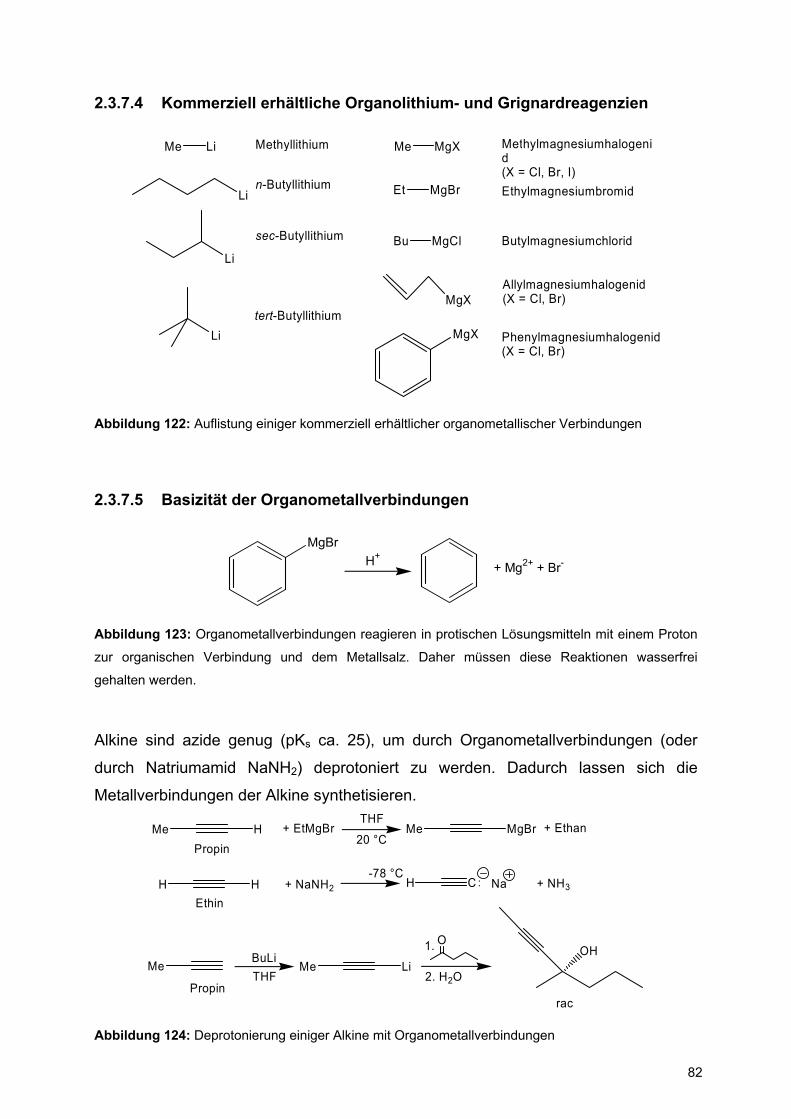
82
2.3.7.4 Kommerziell erhältliche Organolithium- und Grignardreagenzien
Me Li
Li
Li
Li
Methyllithium
n-Butyllithium
sec-Butyllithium
tert-Butyllithium
Me MgX
Et MgBr
Bu MgCl
MgX
MgX
Methylmagnesiumhalogenid(X = Cl, Br, I)Ethylmagnesiumbromid
Butylmagnesiumchlorid
Allylmagnesiumhalogenid(X = Cl, Br)
Phenylmagnesiumhalogenid(X = Cl, Br)
Abbildung 122: Auflistung einiger kommerziell erhältlicher organometallischer Verbindungen
2.3.7.5 Basizität der Organometallverbindungen
MgBrH+
+ Mg2+ + Br-
Abbildung 123: Organometallverbindungen reagieren in protischen Lösungsmitteln mit einem Proton
zur organischen Verbindung und dem Metallsalz. Daher müssen diese Reaktionen wasserfrei
gehalten werden.
Alkine sind azide genug (pKs ca. 25), um durch Organometallverbindungen (oder
durch Natriumamid NaNH2) deprotoniert zu werden. Dadurch lassen sich die
Metallverbindungen der Alkine synthetisieren.
Me H + EtMgBrTHF
20 °CMe MgBr + Ethan
Propin
MeTHF
Me Li
Propin
H H + NaNH2-78 °C
H C Na + NH3
Ethin
BuLiO1.
2. H2O
OH
rac
Abbildung 124: Deprotonierung einiger Alkine mit Organometallverbindungen
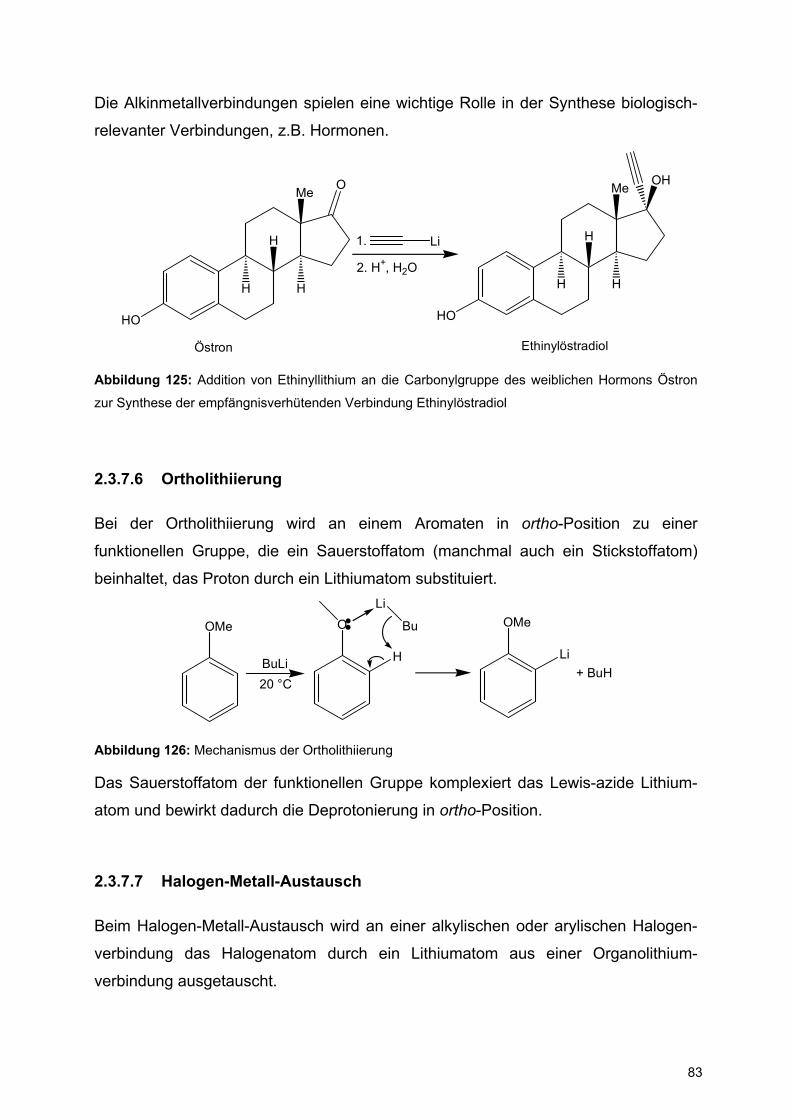
83
Die Alkinmetallverbindungen spielen eine wichtige Rolle in der Synthese biologisch-
relevanter Verbindungen, z.B. Hormonen.
HO
OMe
H
HH
Li1.
2. H+, H2O
HO
OHMe
H
HH
Östron Ethinylöstradiol
Abbildung 125: Addition von Ethinyllithium an die Carbonylgruppe des weiblichen Hormons Östron
zur Synthese der empfängnisverhütenden Verbindung Ethinylöstradiol
2.3.7.6 Ortholithiierung
Bei der Ortholithiierung wird an einem Aromaten in ortho-Position zu einer
funktionellen Gruppe, die ein Sauerstoffatom (manchmal auch ein Stickstoffatom)
beinhaltet, das Proton durch ein Lithiumatom substituiert.
OMe
BuLi20 °C
O
H
Li
Bu OMe
Li+ BuH
Abbildung 126: Mechanismus der Ortholithiierung
Das Sauerstoffatom der funktionellen Gruppe komplexiert das Lewis-azide Lithium-
atom und bewirkt dadurch die Deprotonierung in ortho-Position.
2.3.7.7 Halogen-Metall-Austausch
Beim Halogen-Metall-Austausch wird an einer alkylischen oder arylischen Halogen-
verbindung das Halogenatom durch ein Lithiumatom aus einer Organolithium-
verbindung ausgetauscht.

84
BrBuLi
Li
+ BuBr
Abbildung 127: Reaktion des Halogen-Metall-Austauschs. Das Bromoniumkation wird durch das
Butyllithium abgespalten und durch das Lithiumkation ersetzt. Das gebildete metallorganische Produkt
ist weniger basisch als die Ausgangsverbindung.
Als Halogenide können sowohl Chloride, Bromide als auch Iodide verwendet werden,
während die Reaktionen mit Bromid und Iodid am schnellsten verlaufen.
2.3.7.8 Transmetallierung
Da Organolithiumverbindungen zumeist sehr reaktiv und stark basisch sind, können
sie unerwünschte Nebenreaktionen verursachen. Daher ist es hilfreich, durch Trans-
metallierung weniger reaktive Organometallverbindungen darzustellen.
R MgBr + LiBr R Li R CeCl2 + LiClMgBr2
THF
CeCl3
THF
Abbildung 128: Transmetallierung einer Organolithiumverbindung zu einer Grignard- oder einer
Organocerverbindung
Beispiel der unterschiedlichen Reaktivitäten:
OMe
OMe
O
H H
Li
OMe
OMe
O
H Li
+ Ethin
Organolithiumverbindung
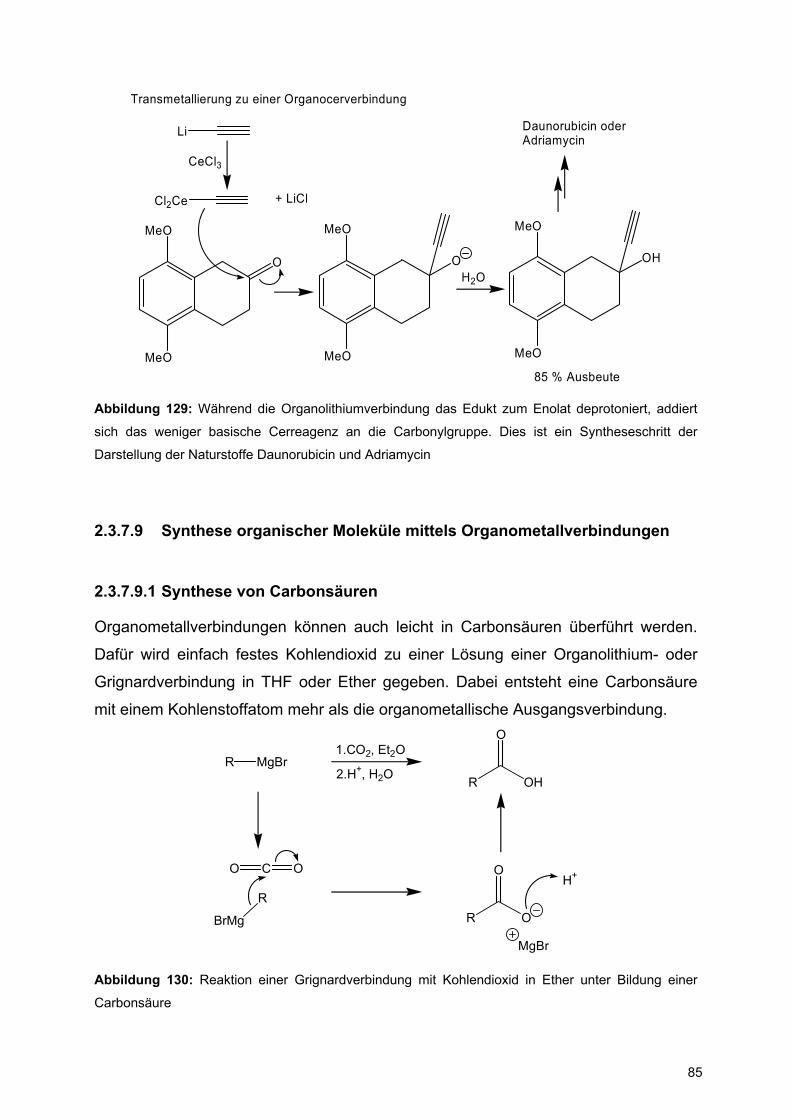
85
MeO
MeO
O
Li
CeCl3
Cl2Ce + LiCl
MeO
MeO
OH2O
MeO
MeO
OH
85 % Ausbeute
Daunorubicin oderAdriamycin
Transmetallierung zu einer Organocerverbindung
Abbildung 129: Während die Organolithiumverbindung das Edukt zum Enolat deprotoniert, addiert
sich das weniger basische Cerreagenz an die Carbonylgruppe. Dies ist ein Syntheseschritt der
Darstellung der Naturstoffe Daunorubicin und Adriamycin
2.3.7.9 Synthese organischer Moleküle mittels Organometallverbindungen
2.3.7.9.1 Synthese von Carbonsäuren
Organometallverbindungen können auch leicht in Carbonsäuren überführt werden.
Dafür wird einfach festes Kohlendioxid zu einer Lösung einer Organolithium- oder
Grignardverbindung in THF oder Ether gegeben. Dabei entsteht eine Carbonsäure
mit einem Kohlenstoffatom mehr als die organometallische Ausgangsverbindung.
R MgBr1.CO2, Et2O
2.H+, H2O
O
R OH
BrMg
R
O C O O
R O
H+
MgBr
Abbildung 130: Reaktion einer Grignardverbindung mit Kohlendioxid in Ether unter Bildung einer
Carbonsäure
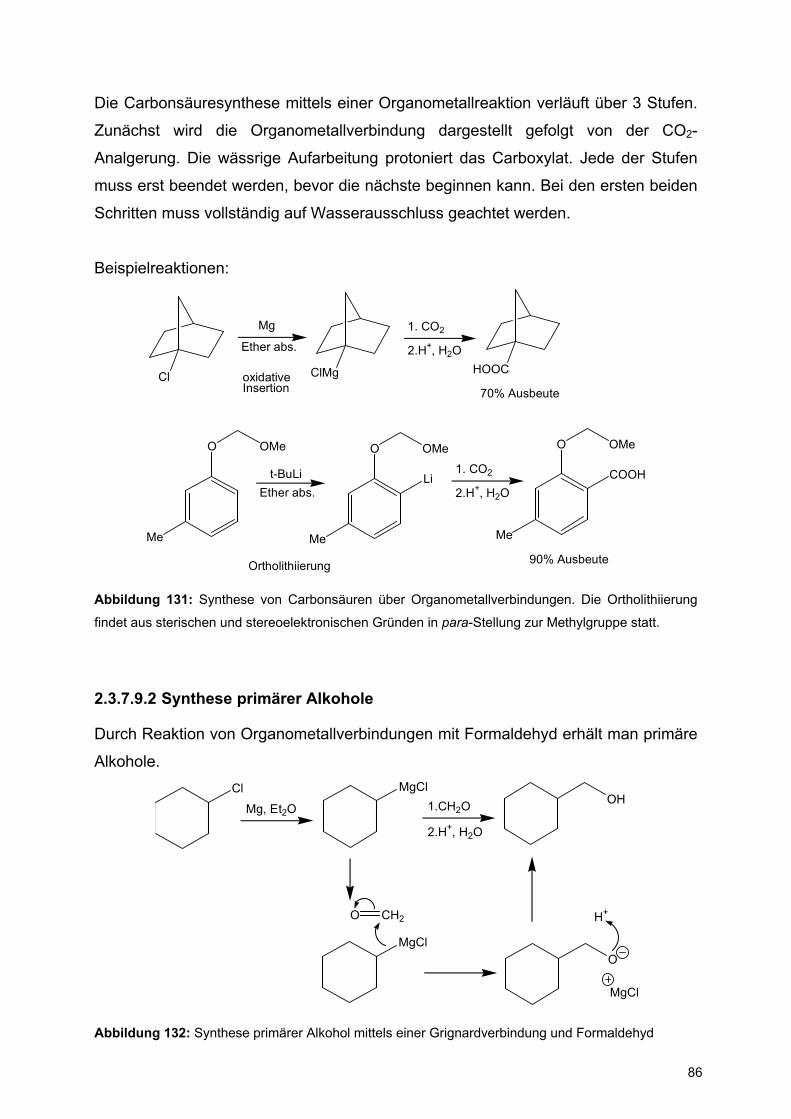
86
Die Carbonsäuresynthese mittels einer Organometallreaktion verläuft über 3 Stufen.
Zunächst wird die Organometallverbindung dargestellt gefolgt von der CO2-
Analgerung. Die wässrige Aufarbeitung protoniert das Carboxylat. Jede der Stufen
muss erst beendet werden, bevor die nächste beginnen kann. Bei den ersten beiden
Schritten muss vollständig auf Wasserausschluss geachtet werden.
Beispielreaktionen:
Cl
Mg
Ether abs.
ClMg
1. CO2
2.H+, H2OHOOC
oxidativeInsertion
Me
O OMe
t-BuLiEther abs.
Ortholithiierung
Me
O OMe
Li1. CO2
2.H+, H2O
Me
O OMe
COOH
70% Ausbeute
90% Ausbeute
Abbildung 131: Synthese von Carbonsäuren über Organometallverbindungen. Die Ortholithiierung
findet aus sterischen und stereoelektronischen Gründen in para-Stellung zur Methylgruppe statt.
2.3.7.9.2 Synthese primärer Alkohole
Durch Reaktion von Organometallverbindungen mit Formaldehyd erhält man primäre
Alkohole. Cl
Mg, Et2O
MgCl1.CH2O
2.H+, H2O
OH
MgCl
O CH2
O
MgCl
H+
Abbildung 132: Synthese primärer Alkohol mittels einer Grignardverbindung und Formaldehyd

87
2.3.7.9.3 Synthese sekundärer (tertiärer) Alkohole
Aldehyde reagieren mit Organometallverbindungen zu sekundären Alkoholen. O
R1 H
1.R2MgBr
2.H+, H2O
OH
R1 R2
BrMg
R2O
R1 H
O
R1 R2
H
H+
MgBr
Abbildung 133: Synthese sekundärer Alkohole bei der Umsetzung einer Grignardverbindung mit
einem Aldehyd
Analog zur Reaktion in Abbildung 133 können mit Ketonen tertiäre Alkohole
dargestellt werden.
2.3.7.9.4 Zusammenfassung
Bei der Addition von Organometallverbindungen an Carbonyle wird die Oxidations-
stufe um eins erniedrigt.
• Addition an CO2 ergibt eine Carbonsäure
• Addition an Formaldehyd ergibt einen primären Alkohol
• Addition an einen Aldehyden ergibt einen sekundären Alkohol
• Addition an ein Keton ergibt einen tertiären Alkohol
2.3.7.10 Mechanismus der Addition von Organometall- an Carbonyl-
verbindungen
Der Mechanismus dieser Reaktionen ist unbekannt. Die Rolle des Metallatoms kann
nicht definiert werden. Nach der heterolytischen Spaltung der C–M-Bindung wird das
Metallkation einfach an den anionischen Sauerstoff koordiniert. Dieses wird dann bei
der Aufarbeitung durch ein Proton ersetzt und als Metallsalz abgetrennt.
Wahrscheinlich trägt das Metallatom in der metallorganischen Verbindung die Rolle
einer Lewis-Säure, die die Carbonylgruppe aktiviert bzw. die Reaktion katalysiert.
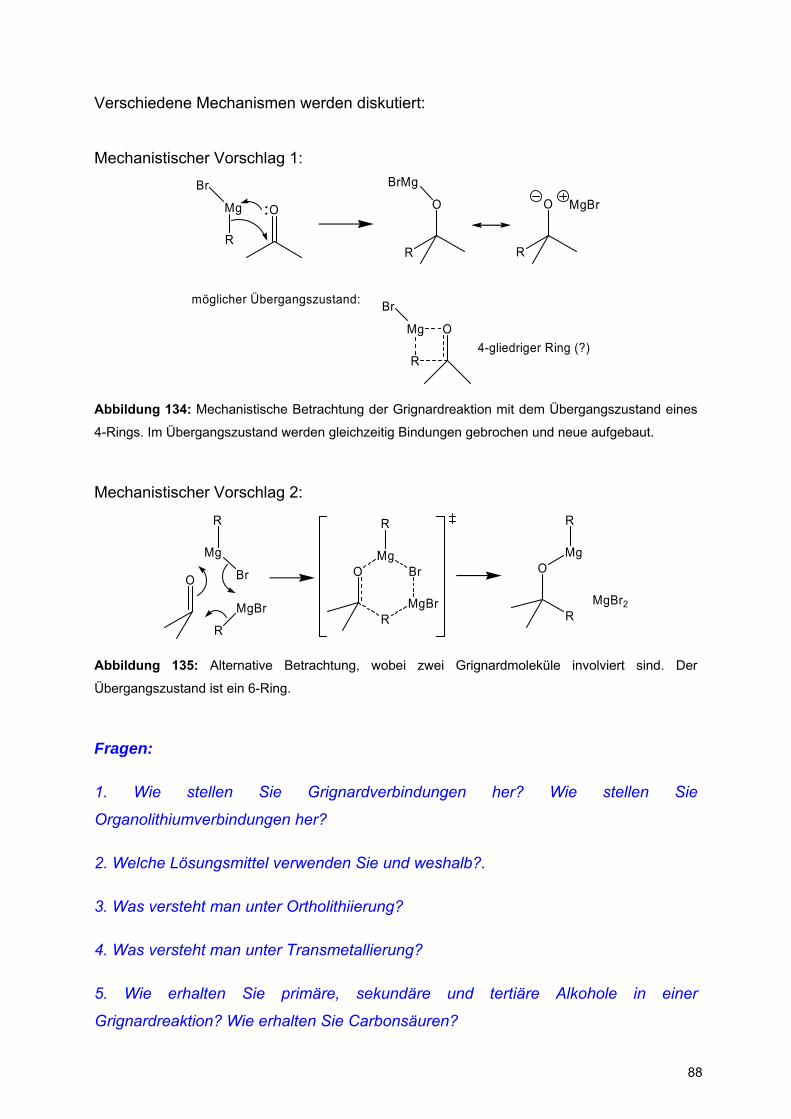
88
Verschiedene Mechanismen werden diskutiert:
Mechanistischer Vorschlag 1: Br
Mg
R
O
R
O
BrMg
R
O MgBr
möglicher Übergangszustand: Br
Mg O
R4-gliedriger Ring (?)
Abbildung 134: Mechanistische Betrachtung der Grignardreaktion mit dem Übergangszustand eines
4-Rings. Im Übergangszustand werden gleichzeitig Bindungen gebrochen und neue aufgebaut.
Mechanistischer Vorschlag 2:
O
R
MgBr
Br
Mg
R
O
RMgBr
BrMg
R
O
RMgBr2
Mg
R
Abbildung 135: Alternative Betrachtung, wobei zwei Grignardmoleküle involviert sind. Der
Übergangszustand ist ein 6-Ring.
Fragen:
1. Wie stellen Sie Grignardverbindungen her? Wie stellen Sie
Organolithiumverbindungen her?
2. Welche Lösungsmittel verwenden Sie und weshalb?.
3. Was versteht man unter Ortholithiierung?
4. Was versteht man unter Transmetallierung?
5. Wie erhalten Sie primäre, sekundäre und tertiäre Alkohole in einer
Grignardreaktion? Wie erhalten Sie Carbonsäuren?

89
Achte Vorlesungsstunde
I. Neben den Grignardverbindungen und Organolithiumverbindungen gibt es auch die
Wittigreagenzien, in denen der Kohlenstoff als Nukleophil reagiert.
II. In dieser Vorlesungsstunde wird die Wittigreaktion im Detail diskutiert, um Ihnen
die Mächtigkeit der Diskussion von Reaktionskoordinaten, kinetischer und
thermodynamischer Kontrolle von Reaktionen vor Augen zu führen.
III. Wir führen Sie in die Bildung und Reaktion von Enolen und Enolaten ein. Enolate
stellen wichtige C-Nukleophile dar, deren Chemie wir in den verbleibenden
Vorlesungsstunden besprechen werden (Stichwort Aldolchemie).
2.3.8 Wittig-Reaktion
Chapter 31 in Warren et al.
Bei der Wittig-Reaktion wird eine Carbonylgruppe in eine C=C-Doppelbindung
überführt. Somit können zwei organische Verbindungen durch eine Doppelbindung
verknüpft werden.
2.3.8.1 Einführung
OPh3P CH2
O
CH2
PPh3
+ Ph3PO
86% AusbeutezyklischesOxaphosphetan
Abbildung 136: Grundschema der Wittig-Reaktion
Entscheidend für die Reaktion ist das Wittig-Reagenz. Dieses wird aus einem
Phosphan gewonnen. Nach Deprotonierung an einem dem Phosphor benachbarten
Kohlenstoffatom erhält man ein C-Nukleophil, genannt Ylid. Dieses lagert sich an den
elektrophilen Carbonylkohlenstoff, während der Phosphor mit dem Carbonyl-
sauerstoff eine Bindung eingeht. In-situ bildet sich dadurch ein zyklisches
Oxaphosphetan. Dieses zerfällt sofort zum thermodynamisch stark begünstigten

90
Phosphinoxid und der neuen organischen Verbindung. Die Bildung des
Phosphinoxids ist die treibende Kraft der Reaktion.
2.3.8.2 Darstellung des Wittig-Reagenzes bzw. des Ylids:
Br
PPh3
H
PPh3
Br
azides Proton
PPh3 PPh3
BuLi
Phosphoniumsalz Ylid Phosphoran
Abbildung 137: Synthese des Wittig-Reagenzes: Triphenylphosphin substituiert das Halogenatom
eines Alkylhalogenids zum Phosphoniumsalz. Dies trägt in direkter Nachbarschaft ein azides Proton,
welches durch eine starke Base abgespalten wird. Somit erhält man das Ylid, das auch als
Phosphoran geschrieben werden kann.
Ylide können isoliert werden, aber zumeist werden sie gleich mit einem Aldehyd oder
Keton umgesetzt.
2.3.8.3 Mechanismus
PPh3
H
Ph
O
Ph3P
CHPh
O
Oxaphosphetan-Intermediat
Ph
O PPh3+
65% Ausbeute
Abbildung 138: Mechanismus der Wittig-Reaktion
2.3.8.4 Stereoselektivität
Bei der Wittig-Reaktion wird eine neue stereogene Einheit erzeugt, eine
Doppelbindung. Die Reaktionen verlaufen auch mit einer ausgeprägten
Stereoselektivität. Diese hängt vom Substituenten des Kohlenstoffatoms am Ylid ab.
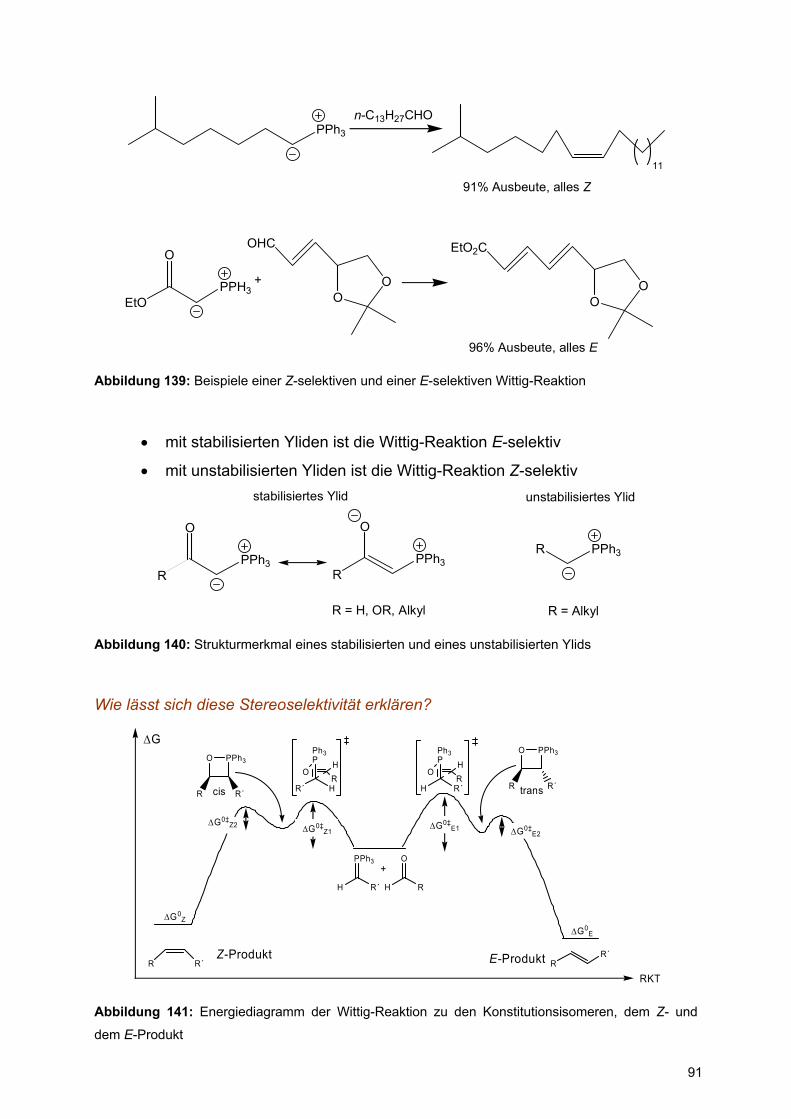
91
PPh3
n-C13H27CHO
11
91% Ausbeute, alles Z
EtO
O
PPH3+
OHC
OO
OO
EtO2C
96% Ausbeute, alles E
Abbildung 139: Beispiele einer Z-selektiven und einer E-selektiven Wittig-Reaktion
• mit stabilisierten Yliden ist die Wittig-Reaktion E-selektiv
• mit unstabilisierten Yliden ist die Wittig-Reaktion Z-selektiv
RPPh3
O
RPPh3
O
R = H, OR, Alkyl
stabilisiertes Ylid unstabilisiertes Ylid
R PPh3
R = Alkyl
Abbildung 140: Strukturmerkmal eines stabilisierten und eines unstabilisierten Ylids
Wie lässt sich diese Stereoselektivität erklären?
PPh3
H R´
O
H R
OHR
Ph3P
H R´
O PPh3
R R´
RR´
OHR
Ph3P
R´ H
O PPh3
R R´
R R´
+
transcis
∆G0‡E1 ∆G0‡
E2
∆G0E
∆G0Z
∆G0‡Z1
∆G0‡Z2
RKT
∆G
Z-Produkt E-Produkt
Abbildung 141: Energiediagramm der Wittig-Reaktion zu den Konstitutionsisomeren, dem Z- und
dem E-Produkt

92
- kinetische Reaktionskontrollebevorzugt Z-Produkt- thermodynamische Reaktionskontrollebevorzugt E-Produkt
∆GE0 < ∆GZ
0 ⇒ E-Produkt ist stabiler
∆GZ10‡ < ∆GE1
0‡
∆GZ20‡ ≈ ∆GE2
0‡
∆GZ20‡ << ∆GZ1
0‡
∆GE20‡ << ∆GE1
0‡
⇒ Thermodynamische Reaktionskontrolle (reversibel)
Was ist das stabilere Produkt?
Kinetische Reaktionskontrolle (irreversibel)
Was benötigt die geringere Aktivierungsenthalpie ∆G0‡?
Beim unstabilisierten Ylid haben wir einen irreversiblen Schritt, so dass die Reaktion
kinetisch-kontrolliert verläuft.
R1 PPh3 R2CHOOPh3P
R1 R2R1 R2
Abbildung 142: Für unstabilisierte Ylide verläuft die Reaktion irreversibel
Die Bildung des Oxaphosphetans verläuft irreversibel, da das Ylid nicht stabilisiert
wird. Der zweite Schritt, der Zerfall des Oxaphosphetans erfolgt stereospezifisch. Aus
dem cis-Oxaphosphetan kann nur das Z-Produkt entstehen.
Warum verläuft die Anlagerung des Ylids an die Carbonylgruppe rechtwinklig?
C O
LUMO derCarbonylverbindung
HOMO desYlids
C
P
Abbildung 143: Darstellung der reagierenden Molekülorbitale während der Oxaphosphetanbildung.
Orbitale gleichen Vorzeichens können in Wechselwirkung miteinander treten, während Orbitale mit
entgegengesetztem Vorzeichen sich abstoßen.
Durch die rechtwinklige Annäherung des HOMOs vom Ylid an das LUMO der
Carbonylgruppe wird eine größtmögliche Überlappung erreicht.
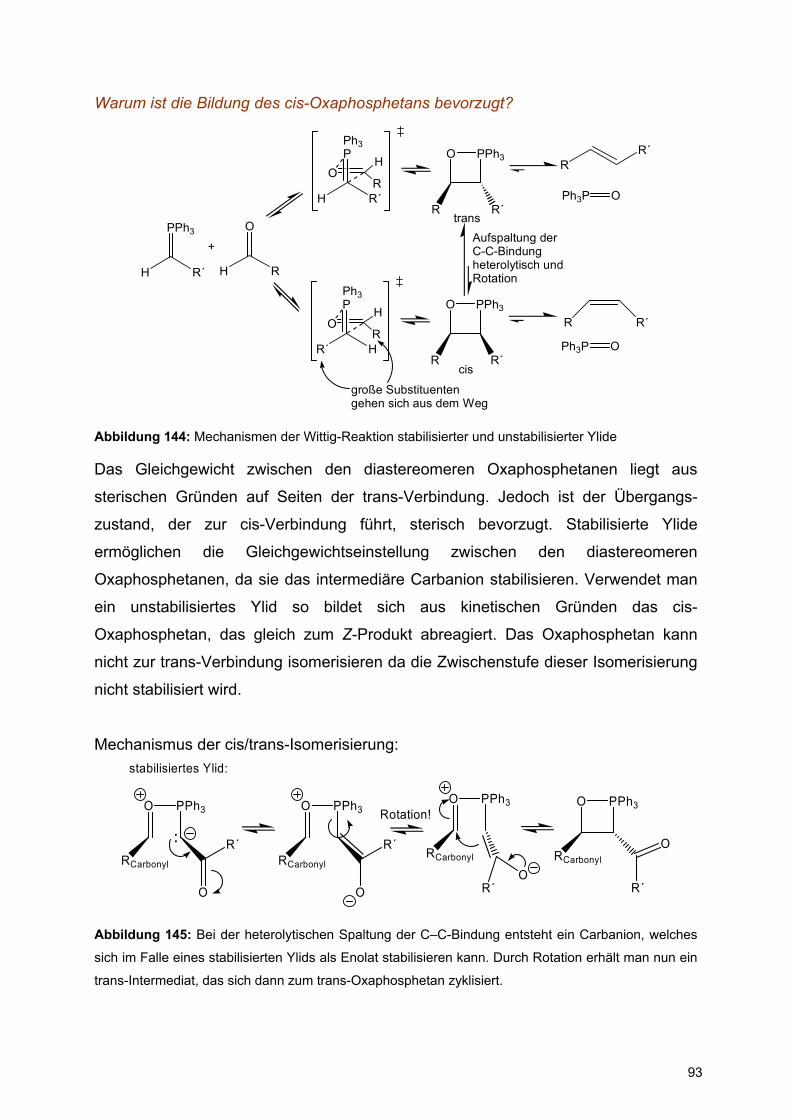
93
Warum ist die Bildung des cis-Oxaphosphetans bevorzugt?
PPh3
H R´
+
O
H R
OH
R
Ph3P
H R´
O PPh3
R R´
RR´
Ph3P O
OH
R
Ph3P
R´ H
O PPh3
R R´Ph3P O
große Substituentengehen sich aus dem Weg
R R´
Aufspaltung derC-C-Bindungheterolytisch undRotation
trans
cis
Abbildung 144: Mechanismen der Wittig-Reaktion stabilisierter und unstabilisierter Ylide
Das Gleichgewicht zwischen den diastereomeren Oxaphosphetanen liegt aus
sterischen Gründen auf Seiten der trans-Verbindung. Jedoch ist der Übergangs-
zustand, der zur cis-Verbindung führt, sterisch bevorzugt. Stabilisierte Ylide
ermöglichen die Gleichgewichtseinstellung zwischen den diastereomeren
Oxaphosphetanen, da sie das intermediäre Carbanion stabilisieren. Verwendet man
ein unstabilisiertes Ylid so bildet sich aus kinetischen Gründen das cis-
Oxaphosphetan, das gleich zum Z-Produkt abreagiert. Das Oxaphosphetan kann
nicht zur trans-Verbindung isomerisieren da die Zwischenstufe dieser Isomerisierung
nicht stabilisiert wird.
Mechanismus der cis/trans-Isomerisierung: stabilisiertes Ylid:
O PPh3
RCarbonyl
O
R´
O PPh3
RCarbonyl
O
R´
O PPh3
RCarbonyl
R´O
O PPh3
RCarbonyl
R´
O
Rotation!
Abbildung 145: Bei der heterolytischen Spaltung der C–C-Bindung entsteht ein Carbanion, welches
sich im Falle eines stabilisierten Ylids als Enolat stabilisieren kann. Durch Rotation erhält man nun ein
trans-Intermediat, das sich dann zum trans-Oxaphosphetan zyklisiert.
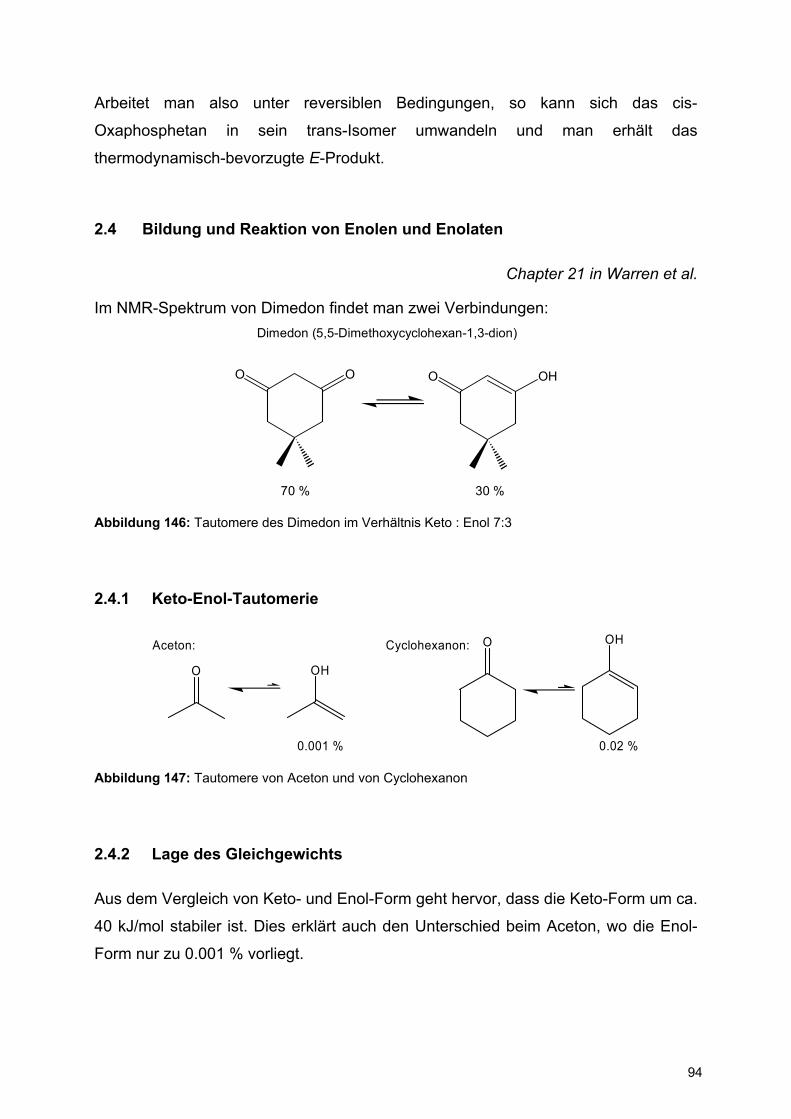
94
Arbeitet man also unter reversiblen Bedingungen, so kann sich das cis-
Oxaphosphetan in sein trans-Isomer umwandeln und man erhält das
thermodynamisch-bevorzugte E-Produkt.
2.4 Bildung und Reaktion von Enolen und Enolaten
Chapter 21 in Warren et al.
Im NMR-Spektrum von Dimedon findet man zwei Verbindungen:
O O O OH
70 % 30 %
Dimedon (5,5-Dimethoxycyclohexan-1,3-dion)
Abbildung 146: Tautomere des Dimedon im Verhältnis Keto : Enol 7:3
2.4.1 Keto-Enol-Tautomerie
O OH
0.001 %
Aceton: Cyclohexanon: O OH
0.02 %
Abbildung 147: Tautomere von Aceton und von Cyclohexanon
2.4.2 Lage des Gleichgewichts
Aus dem Vergleich von Keto- und Enol-Form geht hervor, dass die Keto-Form um ca.
40 kJ/mol stabiler ist. Dies erklärt auch den Unterschied beim Aceton, wo die Enol-
Form nur zu 0.001 % vorliegt.
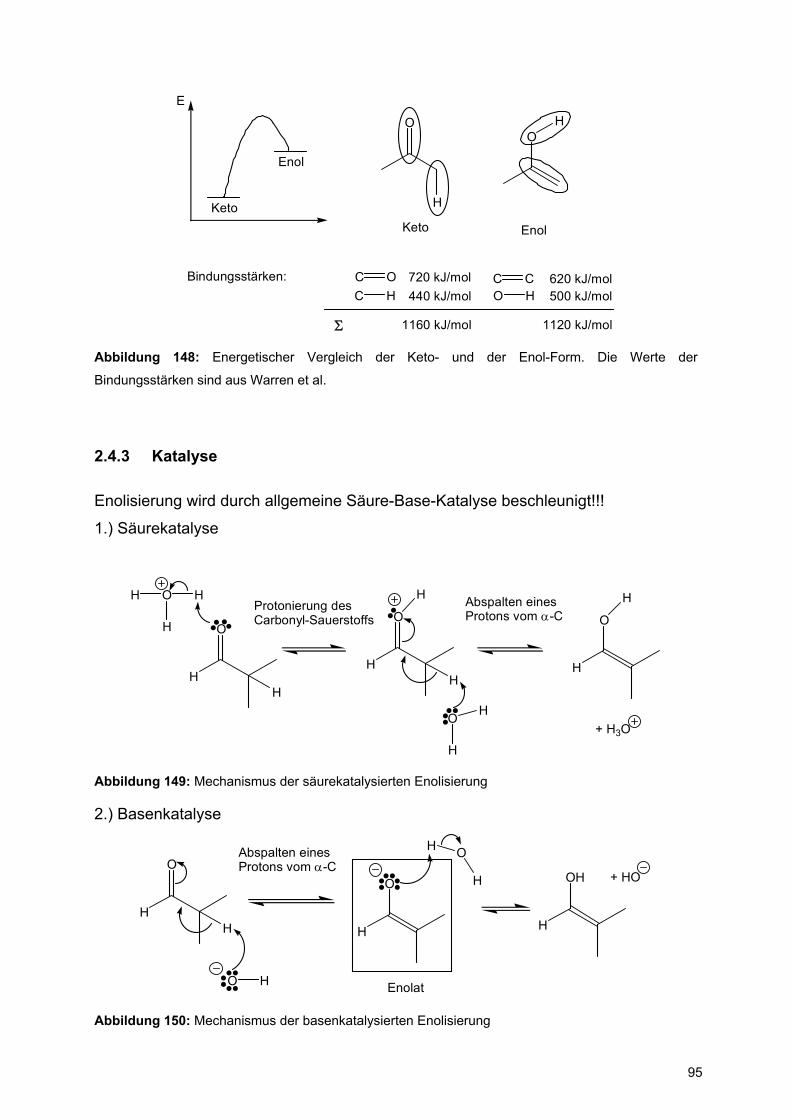
95
E
Keto
Enol
O
H
OH
Keto Enol
C O 720 kJ/molC H 440 kJ/mol
C C 620 kJ/molO H 500 kJ/mol
Σ 1160 kJ/mol 1120 kJ/mol
Bindungsstärken:
Abbildung 148: Energetischer Vergleich der Keto- und der Enol-Form. Die Werte der
Bindungsstärken sind aus Warren et al.
2.4.3 Katalyse
Enolisierung wird durch allgemeine Säure-Base-Katalyse beschleunigt!!!
1.) Säurekatalyse
O
HH
H O
H
HProtonierung desCarbonyl-Sauerstoffs O
HH
H
O
H
H
Abspalten einesProtons vom α-C O
H
H
+ H3O
Abbildung 149: Mechanismus der säurekatalysierten Enolisierung
2.) Basenkatalyse
O
HH
O H
O
H
H O
H
Enolat
OH
H
+ HO
Abspalten einesProtons vom α-C
Abbildung 150: Mechanismus der basenkatalysierten Enolisierung
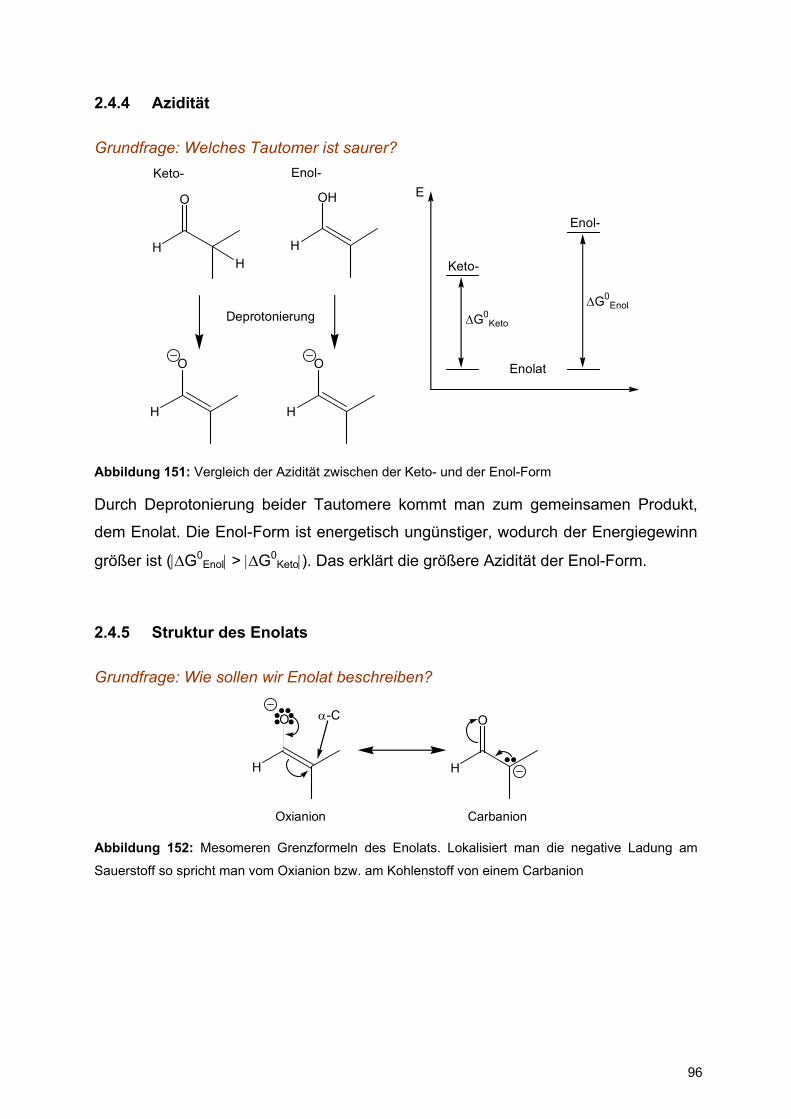
96
2.4.4 Azidität
Grundfrage: Welches Tautomer ist saurer? Keto-
O
HH
OH
H
Enol-
Deprotonierung
O
H
O
H
Enolat
Keto-
Enol-
∆G0Keto
∆G0Enol
E
Abbildung 151: Vergleich der Azidität zwischen der Keto- und der Enol-Form
Durch Deprotonierung beider Tautomere kommt man zum gemeinsamen Produkt,
dem Enolat. Die Enol-Form ist energetisch ungünstiger, wodurch der Energiegewinn
größer ist (|∆G0Enol| > |∆G0
Keto|). Das erklärt die größere Azidität der Enol-Form.
2.4.5 Struktur des Enolats
Grundfrage: Wie sollen wir Enolat beschreiben?
O
H
α-C
Oxianion
O
H
Carbanion
Abbildung 152: Mesomeren Grenzformeln des Enolats. Lokalisiert man die negative Ladung am
Sauerstoff so spricht man vom Oxianion bzw. am Kohlenstoff von einem Carbanion

97
Diese Beschreibung ist analog zum Allylsystem:
HOMO
O O
HOMO
Allylsystem Enolatsystem
Elektronendichteam α-C größer
Abbildung 153: Analogie zwischen einem Allylsystem und einem Enolatsystem. Während im
Allylsystem die Elektronendichte symmetrisch verteilt ist, liegt sie beim Enolat im niedrigen π-MO auf
dem Sauerstoff und im HOMO auf dem α-Kohlenstoff.
Fragen:
1. Wie stellt man ein Wittigreagenz her?
2. Welche Regeln für die Stereoselektivität der Wittigreaktion kennen Sie?
3. Wie können Sie ein Enol erzeugen? Wie können Sie ein Enolat erzeugen?
4. Ist eine Ketoverbindung oder eine Enolverbindung acider? Merken Sie sich das
Prinzip der Erklärung, es lässt sich auf viele Fragestellungen anwenden!
5. Wie können Sie im MO-Modell das Enolat beschreiben?

98
Neunte Vorlesungsstunde
I. Wir diskutieren, wie ein Enolatanion als C-Nukleophil in Reaktionen eingesetzt
wird.
II. Dabei werden zunächst die Alkylierung, also die Umsetzung mit einer R-X
Verbindung besprochen (hierbei steht X für entweder ein Halogen oder auch eine
andere Abgangsgruppe).
III. Dabei führen wir in das Konzept der C- und der O-Alkylierung von Enolaten ein.
IV. Wir diskutieren das Konzept der kinetisch-kontrollierten und der
thermodynamisch-kontrollierten Enolatbildung.
V. Wir führen Enolatanaloga ein, die das Problem der C- bzw. O-Alkylierung bzw. das
Problem der Regioselektivität lösen können.
2.4.6 Chemie des Enolatanions
Chapter 26 in Warren et al.
Enole des Methylethylketons:
OH+
OH OH
+
Z
OH
+
E
Säurekatalysiert:
O OHO O
+
Z
O
+
E
Basenkatalysiert:
Abbildung 154: Produkte der säure- oder basenkatalysierten Enolisierung von Methylethylketon
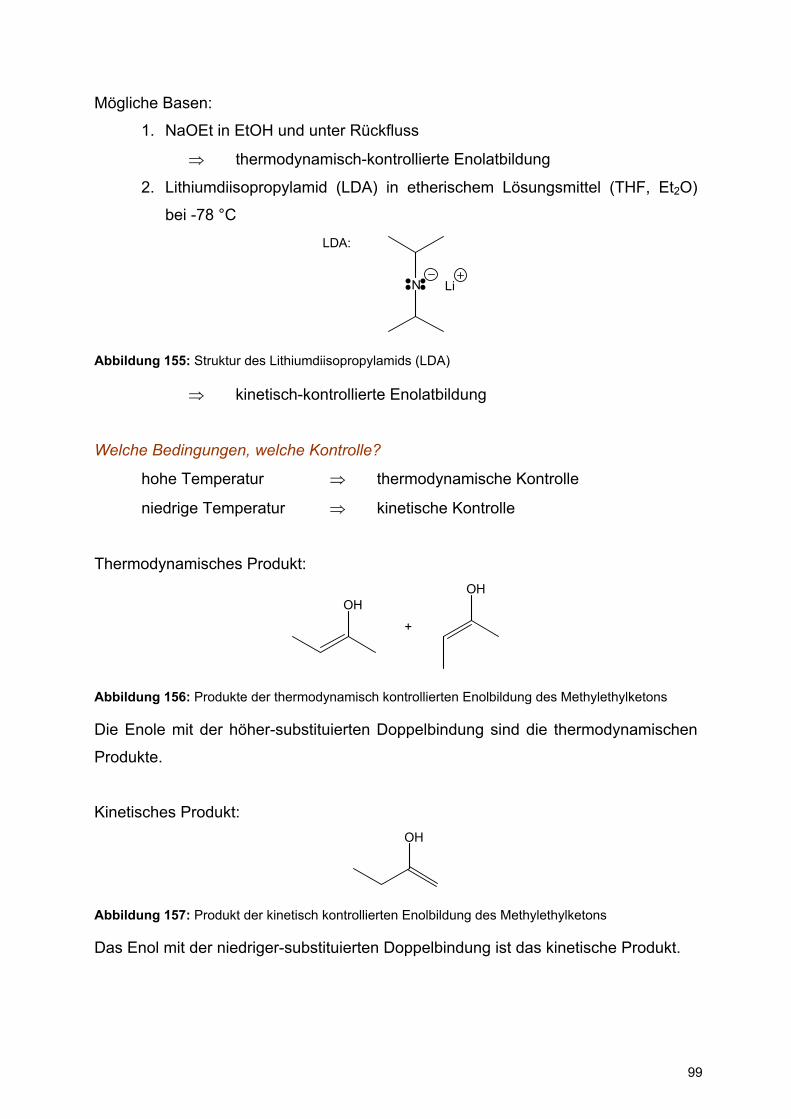
99
Mögliche Basen:
1. NaOEt in EtOH und unter Rückfluss
⇒ thermodynamisch-kontrollierte Enolatbildung
2. Lithiumdiisopropylamid (LDA) in etherischem Lösungsmittel (THF, Et2O)
bei -78 °C
LDA:
N Li
Abbildung 155: Struktur des Lithiumdiisopropylamids (LDA)
⇒ kinetisch-kontrollierte Enolatbildung
Welche Bedingungen, welche Kontrolle?
hohe Temperatur ⇒ thermodynamische Kontrolle
niedrige Temperatur ⇒ kinetische Kontrolle
Thermodynamisches Produkt:
OHOH
+
Abbildung 156: Produkte der thermodynamisch kontrollierten Enolbildung des Methylethylketons
Die Enole mit der höher-substituierten Doppelbindung sind die thermodynamischen
Produkte.
Kinetisches Produkt: OH
Abbildung 157: Produkt der kinetisch kontrollierten Enolbildung des Methylethylketons
Das Enol mit der niedriger-substituierten Doppelbindung ist das kinetische Produkt.
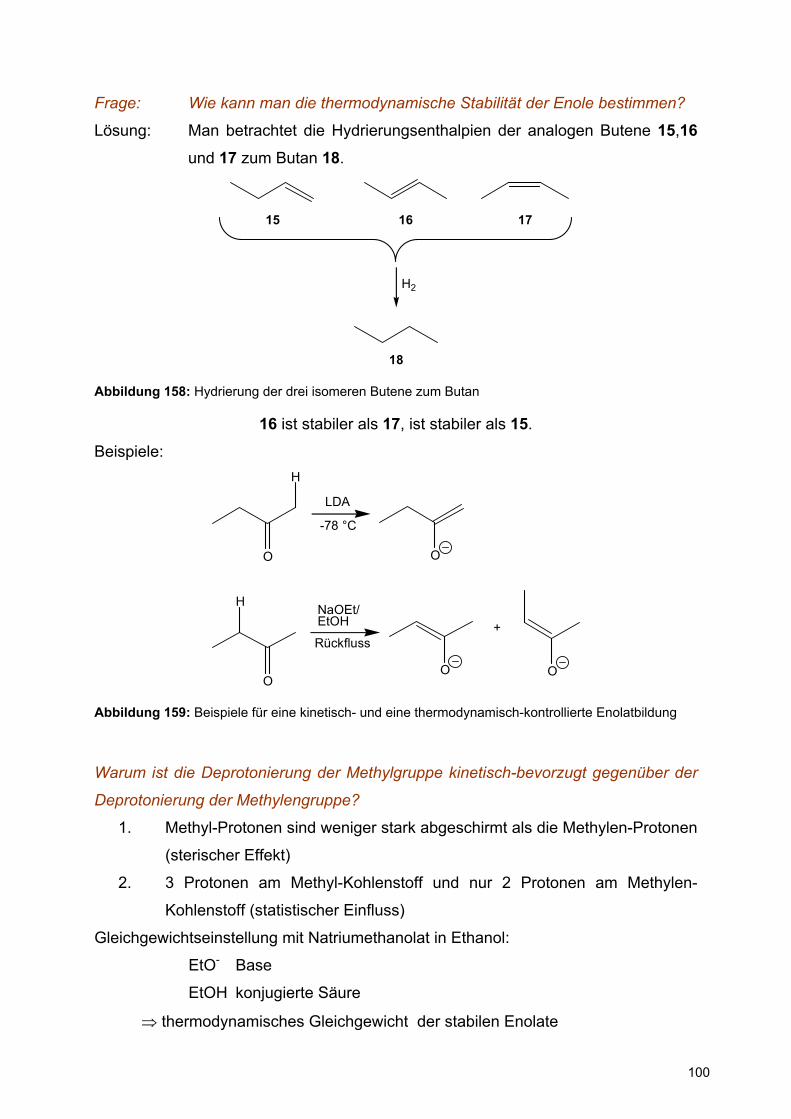
100
Frage: Wie kann man die thermodynamische Stabilität der Enole bestimmen?
Lösung: Man betrachtet die Hydrierungsenthalpien der analogen Butene 15,16
und 17 zum Butan 18.
15 16 17
H2
18
Abbildung 158: Hydrierung der drei isomeren Butene zum Butan
16 ist stabiler als 17, ist stabiler als 15.
Beispiele:
O
H
LDA
-78 °C
O
O
H NaOEt/EtOH
Rückfluss
O O
+
Abbildung 159: Beispiele für eine kinetisch- und eine thermodynamisch-kontrollierte Enolatbildung
Warum ist die Deprotonierung der Methylgruppe kinetisch-bevorzugt gegenüber der
Deprotonierung der Methylengruppe?
1. Methyl-Protonen sind weniger stark abgeschirmt als die Methylen-Protonen
(sterischer Effekt)
2. 3 Protonen am Methyl-Kohlenstoff und nur 2 Protonen am Methylen-
Kohlenstoff (statistischer Einfluss)
Gleichgewichtseinstellung mit Natriumethanolat in Ethanol:
EtO- Base
EtOH konjugierte Säure
⇒ thermodynamisches Gleichgewicht der stabilen Enolate
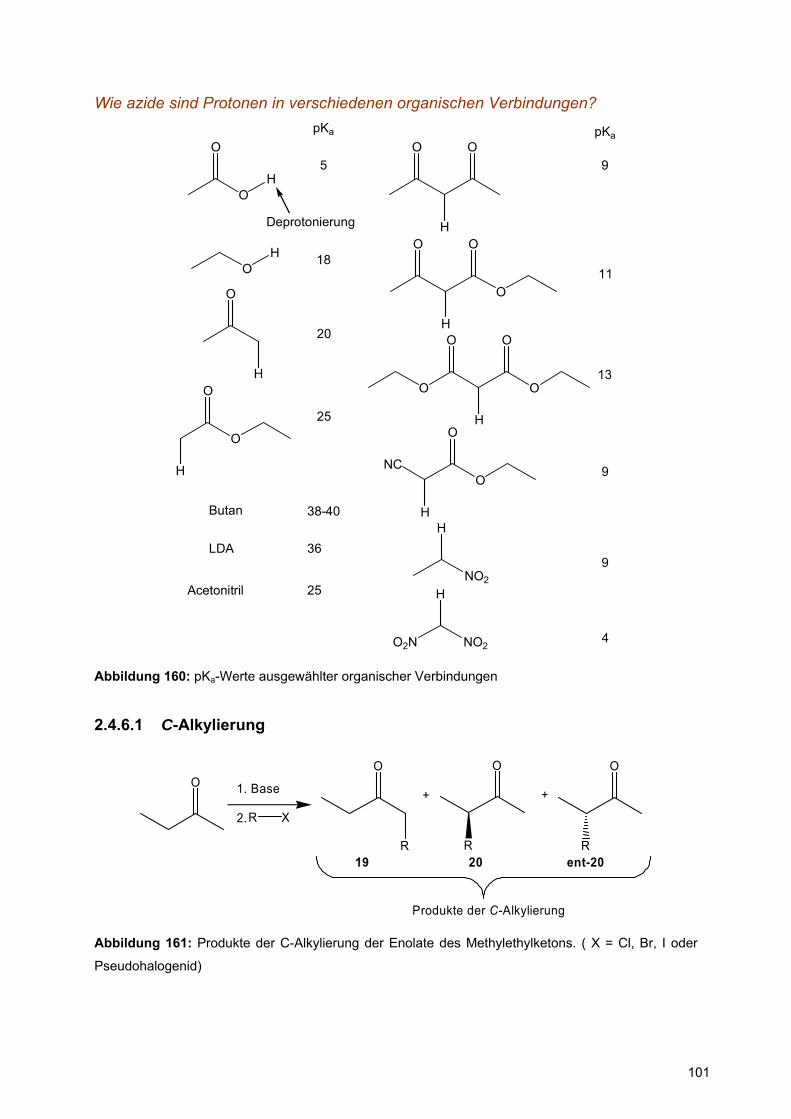
101
Wie azide sind Protonen in verschiedenen organischen Verbindungen?
O
OH
pKa
5O O
HDeprotonierung
9
pKa
OH 18
O
H
20
O
O
H
25
O O
O
HO
O
O
O
H
11
13
Butan 38-40
LDA 36
Acetonitril 25
H
NC
O
O9
H
NO2
9
H
O2N NO24
Abbildung 160: pKa-Werte ausgewählter organischer Verbindungen
2.4.6.1 C-Alkylierung
O 1. Base
R X
O
R
+
O
+
R
O
R19 20 ent-20
Produkte der C-Alkylierung
2.
Abbildung 161: Produkte der C-Alkylierung der Enolate des Methylethylketons. ( X = Cl, Br, I oder
Pseudohalogenid)
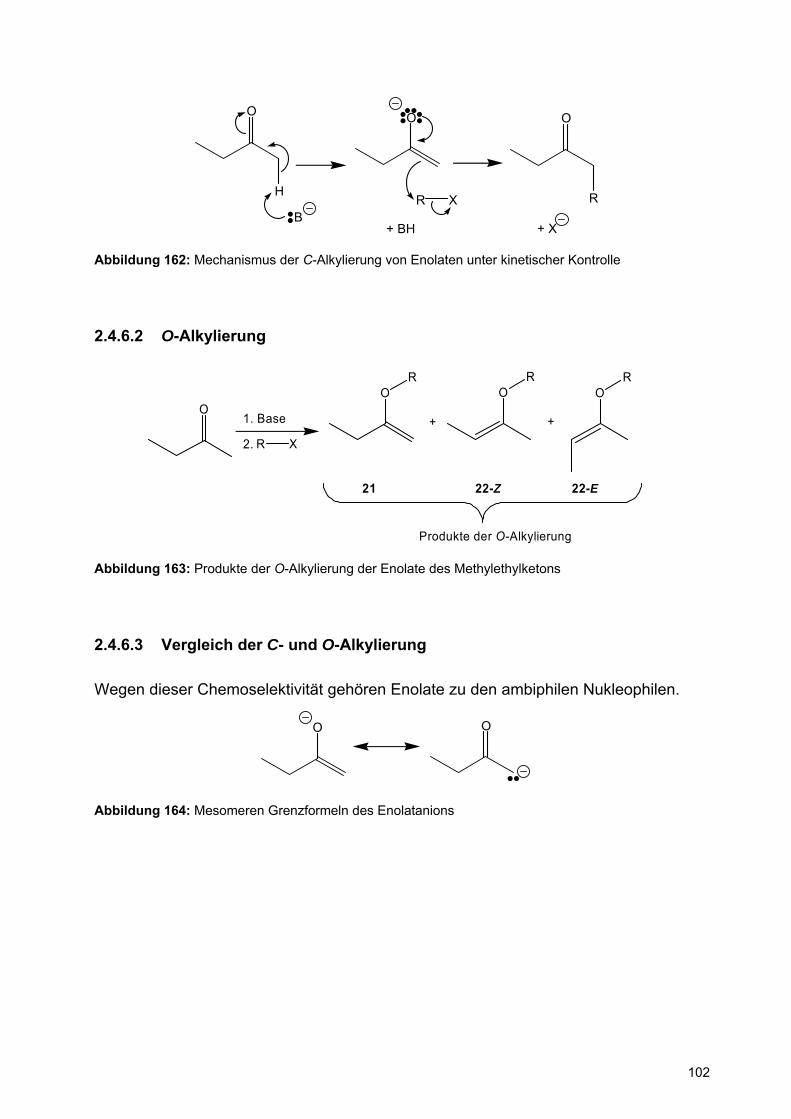
102
O
H
B
O
R X
O
R
+ BH + X
Abbildung 162: Mechanismus der C-Alkylierung von Enolaten unter kinetischer Kontrolle
2.4.6.2 O-Alkylierung
O1. Base
R X
O
+
O
+
O
21 22-Z 22-E
Produkte der O-Alkylierung
R R R
2.
Abbildung 163: Produkte der O-Alkylierung der Enolate des Methylethylketons
2.4.6.3 Vergleich der C- und O-Alkylierung
Wegen dieser Chemoselektivität gehören Enolate zu den ambiphilen Nukleophilen.
O O
Abbildung 164: Mesomeren Grenzformeln des Enolatanions
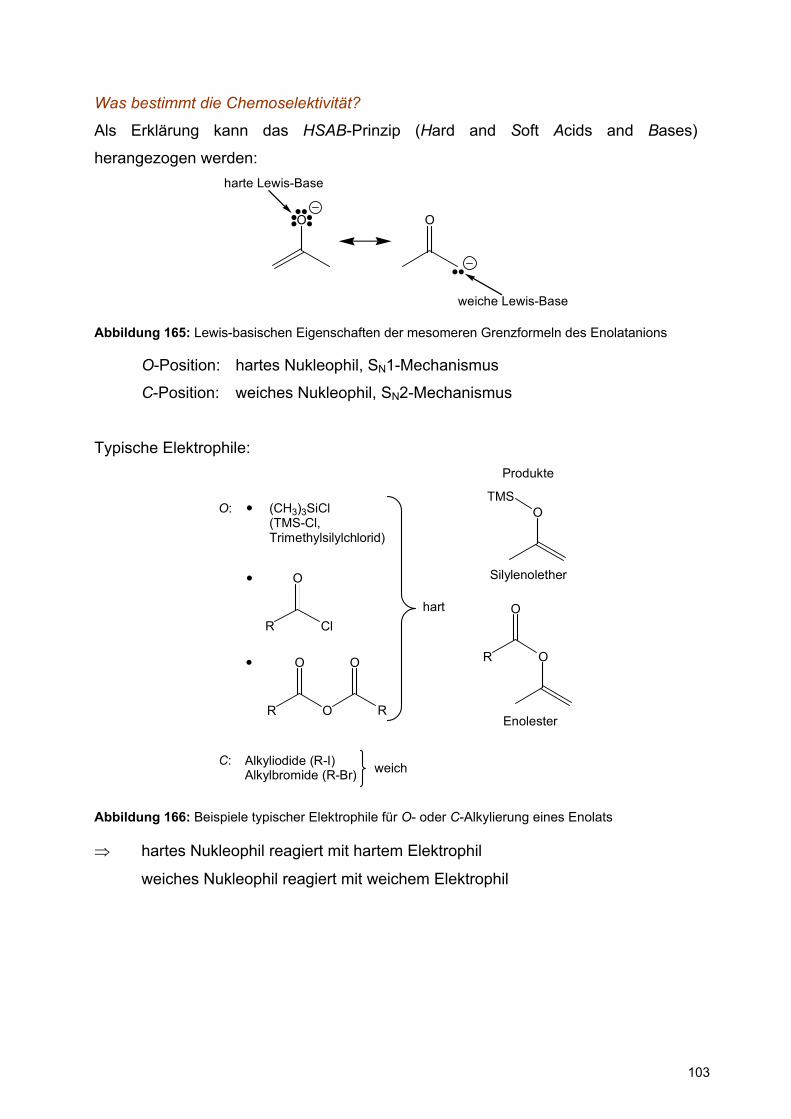
103
Was bestimmt die Chemoselektivität?
Als Erklärung kann das HSAB-Prinzip (Hard and Soft Acids and Bases)
herangezogen werden:
O O
harte Lewis-Base
weiche Lewis-Base
Abbildung 165: Lewis-basischen Eigenschaften der mesomeren Grenzformeln des Enolatanions
O-Position: hartes Nukleophil, SN1-Mechanismus
C-Position: weiches Nukleophil, SN2-Mechanismus
Typische Elektrophile:
O: (CH3)3SiCl(TMS-Cl,Trimethylsilylchlorid)
O
R Cl
O
R O R
O
hart
Produkte
OTMS
Silylenolether
O
O
R
Enolester
C: Alkyliodide (R-I)Alkylbromide (R-Br) weich
Abbildung 166: Beispiele typischer Elektrophile für O- oder C-Alkylierung eines Enolats
⇒ hartes Nukleophil reagiert mit hartem Elektrophil
weiches Nukleophil reagiert mit weichem Elektrophil
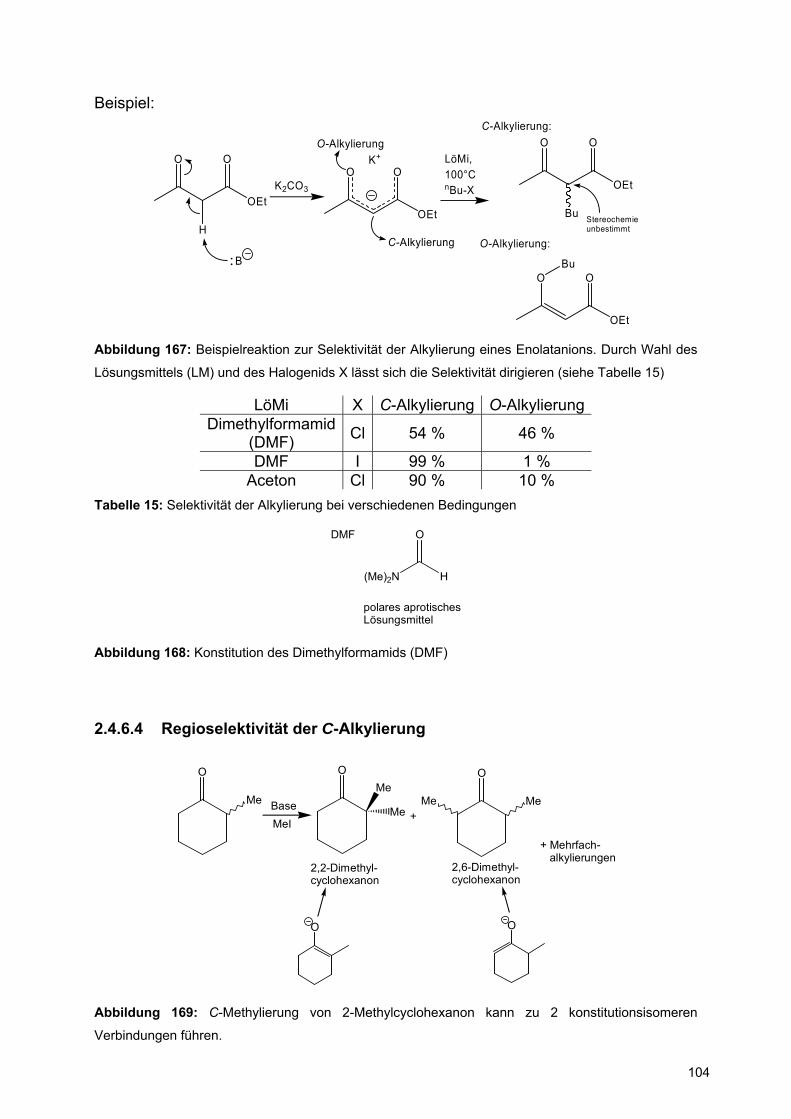
104
Beispiel:
OEt
O O
H
B
K2CO3
LöMi,100°CnBu-X OEt
O O
Bu
C-Alkylierung:
OEt
O O
O-Alkylierung:
Bu
Stereochemieunbestimmt
OEt
O OK+
O-Alkylierung
C-Alkylierung
Abbildung 167: Beispielreaktion zur Selektivität der Alkylierung eines Enolatanions. Durch Wahl des
Lösungsmittels (LM) und des Halogenids X lässt sich die Selektivität dirigieren (siehe Tabelle 15)
LöMi X C-Alkylierung O-Alkylierung Dimethylformamid
(DMF) Cl 54 % 46 %
DMF I 99 % 1 % Aceton Cl 90 % 10 %
Tabelle 15: Selektivität der Alkylierung bei verschiedenen Bedingungen
DMF O
(Me)2N H
polares aprotischesLösungsmittel
Abbildung 168: Konstitution des Dimethylformamids (DMF)
2.4.6.4 Regioselektivität der C-Alkylierung
O
Me BaseMeI
OMe
Me +
O
MeMe
+ Mehrfach- alkylierungen
2,2-Dimethyl-cyclohexanon
2,6-Dimethyl-cyclohexanon
O O
Abbildung 169: C-Methylierung von 2-Methylcyclohexanon kann zu 2 konstitutionsisomeren
Verbindungen führen.
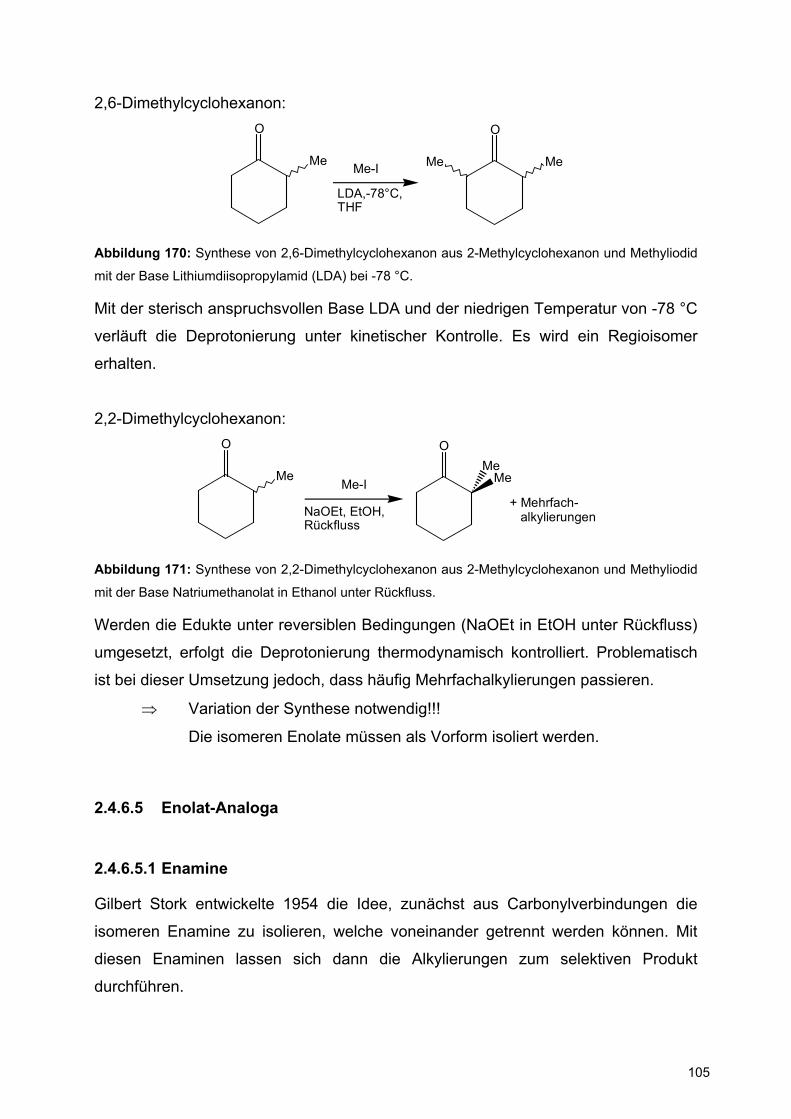
105
2,6-Dimethylcyclohexanon: O
Me
LDA,-78°C,THF
Me-I
O
MeMe
Abbildung 170: Synthese von 2,6-Dimethylcyclohexanon aus 2-Methylcyclohexanon und Methyliodid
mit der Base Lithiumdiisopropylamid (LDA) bei -78 °C.
Mit der sterisch anspruchsvollen Base LDA und der niedrigen Temperatur von -78 °C
verläuft die Deprotonierung unter kinetischer Kontrolle. Es wird ein Regioisomer
erhalten.
2,2-Dimethylcyclohexanon: O
Me
NaOEt, EtOH,Rückfluss
Me-I
O
MeMe
+ Mehrfach- alkylierungen
Abbildung 171: Synthese von 2,2-Dimethylcyclohexanon aus 2-Methylcyclohexanon und Methyliodid
mit der Base Natriumethanolat in Ethanol unter Rückfluss.
Werden die Edukte unter reversiblen Bedingungen (NaOEt in EtOH unter Rückfluss)
umgesetzt, erfolgt die Deprotonierung thermodynamisch kontrolliert. Problematisch
ist bei dieser Umsetzung jedoch, dass häufig Mehrfachalkylierungen passieren.
⇒ Variation der Synthese notwendig!!!
Die isomeren Enolate müssen als Vorform isoliert werden.
2.4.6.5 Enolat-Analoga
2.4.6.5.1 Enamine
Gilbert Stork entwickelte 1954 die Idee, zunächst aus Carbonylverbindungen die
isomeren Enamine zu isolieren, welche voneinander getrennt werden können. Mit
diesen Enaminen lassen sich dann die Alkylierungen zum selektiven Produkt
durchführen.
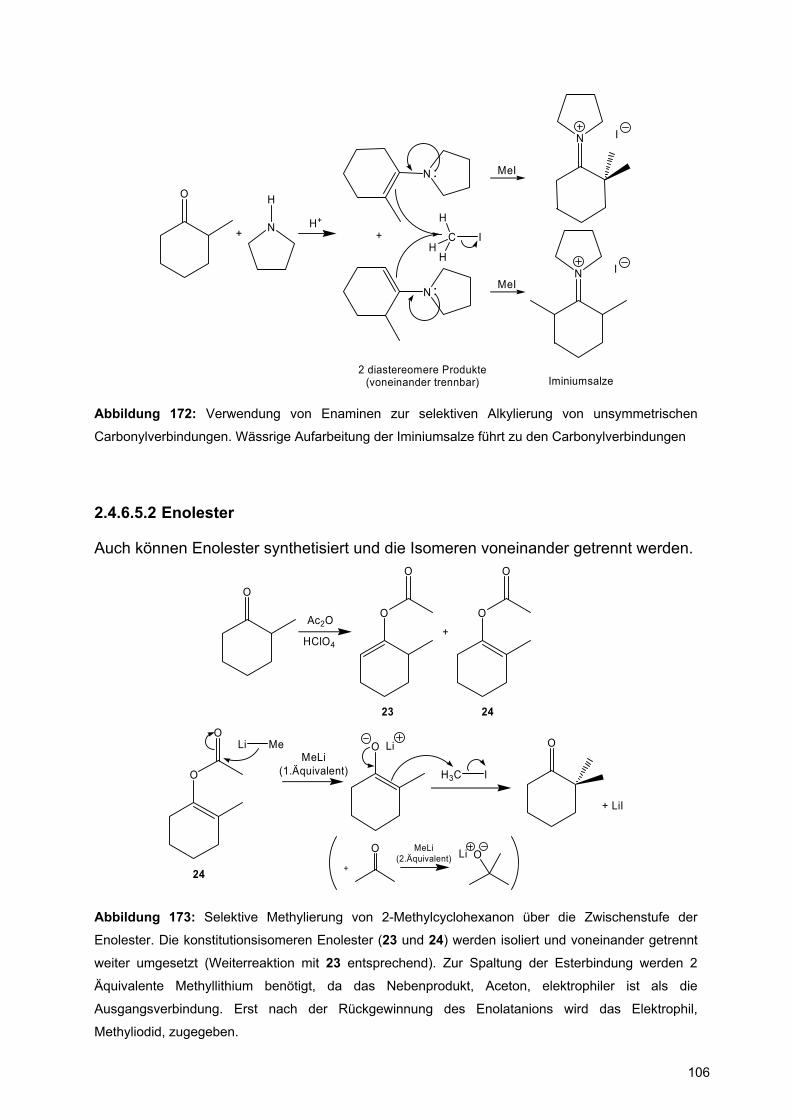
106
H
O
+ N
H
H+
N
N
2 diastereomere Produkte(voneinander trennbar)
C I
N
NMeI
I
I
Iminiumsalze
MeI
+H
H
Abbildung 172: Verwendung von Enaminen zur selektiven Alkylierung von unsymmetrischen
Carbonylverbindungen. Wässrige Aufarbeitung der Iminiumsalze führt zu den Carbonylverbindungen
2.4.6.5.2 Enolester
Auch können Enolester synthetisiert und die Isomeren voneinander getrennt werden.
O
Ac2O O
O
+
O
O
O
O
+
MeLi(1.Äquivalent)
MeLi(2.Äquivalent) O
Li
Li
H3C I
O
+ LiI
HClO4
O
OLi Me
23 24
24
Abbildung 173: Selektive Methylierung von 2-Methylcyclohexanon über die Zwischenstufe der
Enolester. Die konstitutionsisomeren Enolester (23 und 24) werden isoliert und voneinander getrennt
weiter umgesetzt (Weiterreaktion mit 23 entsprechend). Zur Spaltung der Esterbindung werden 2
Äquivalente Methyllithium benötigt, da das Nebenprodukt, Aceton, elektrophiler ist als die
Ausgangsverbindung. Erst nach der Rückgewinnung des Enolatanions wird das Elektrophil,
Methyliodid, zugegeben.

107
2.4.6.5.3 Silylenolether
Zuletzt können die Enolate als Silylenolether abgefangen werden. Silylhalogenide
unterlaufen einer O-Alkylierung durch Enolate. Nach der Trennung der
Konstitutionsisomere erfolgt die Abspaltung des Silylrests mit Methyllithium.
O
TMS-ClOTMS
+
O Li Me
O Li
H3C I
O
+ LiI
Base
Si
Me
Me
Me
MeLi
Abbildung 174: Bei der Reaktion von 2-Methylcyclohexanon mit Trimethylsilylchlorid (TMS-Cl) und
einer Base bilden sich die Silylenolether, die wieder aufgetrennt werden können. Nach der Abspaltung
des Silylrests mit Methyllithium kann das Elektrophil zugegeben werden.
Fragen:
1. Welche Basen nimmt man zur Herstellung von Enolaten? Wie unterscheiden sich
die Basen?
2. Wie erklären Sie die Entstehung des kinetisch bevorzugten Enolats, wie erklären
Sie die Entstehung des thermodynamisch bevorzugten Enolats?
3. Wie erklären Sie die C- bzw. O-Alkylierung des Enolats?
4. Wie können Sie die Regioselektivität der C-Alkylierung kontrollieren?

108
Zehnte Vorlesungsstunde
I. Wir diskutieren, wie ein Enolatanion als C-Nukleophil in Reaktionen eingesetzt
wird.
II. 1,3-Dicarbonylverbindungen können decarboxylieren.
III. Wir führen in die Doppelalkylierung von 1,3-Dicarbonylverbindungen ein.
IV. α,β-ungesättigte Carbonylverbindungen heissen auch Enone. Wir diskutieren die
Reaktivität von solchen Enonen, insbesondere die 1,2-Addition und die 1,4-Addition.
V. Wir führen in die Aldolreaktion ein. Wir diskutieren Aldolreaktionen mit
unsymmetrischen Ketonen, die gekreuzte Aldolreaktion, die Aldolreaktion von
Formaldehyd (Stichwort Cannizarro-Reaktion).
2.4.7 1,3-Dicarbonylverbindungen/ β-Dicarbonylverbindungen
R
O O
H H
1 2 3α
β
pKa ~10 pKa ~20
K2CO3
Aceton,Rückfluss
R
O O
R
O O
R
O OK
MeIR
O O
Merac
Abbildung 175: Methylierung von 1,3-Dicarbonylverbindungen.
Die Azidität von Protonen zwischen 2 Carbonylfunktionen ist stark erhöht. Während
Protonen neben einer Carbonylfunktion einen pKa von ca. 20 aufweisen, liegt dieser
bei 1,3-Dicarbonylverbindungen bei ca. 10. Somit ist es bereits möglich, diese mit
schwachen Basen, z.B. Kaliumcarbonat, zu deprotonieren. Die negative Ladung des
Enolats wird über beide Carbonylgruppen delokalisiert und somit das Enolat
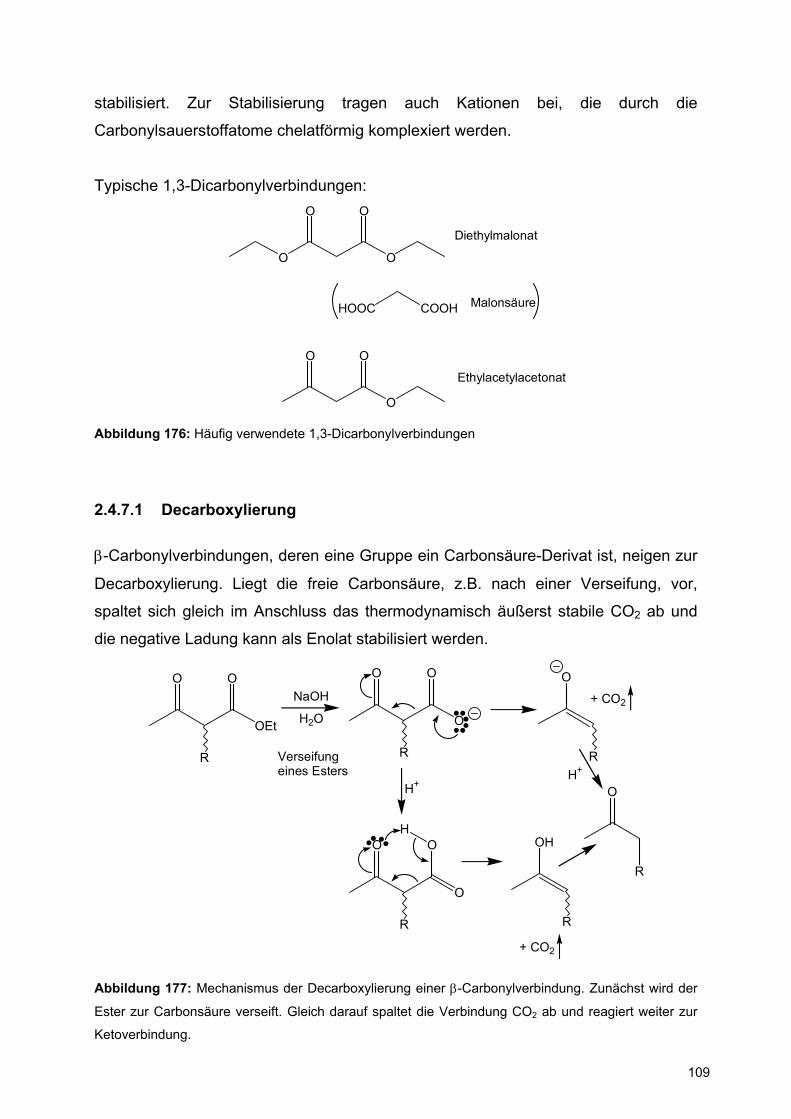
109
stabilisiert. Zur Stabilisierung tragen auch Kationen bei, die durch die
Carbonylsauerstoffatome chelatförmig komplexiert werden.
Typische 1,3-Dicarbonylverbindungen:
O O
O O
Diethylmalonat
HOOC COOH Malonsäure
O
O O
Ethylacetylacetonat
Abbildung 176: Häufig verwendete 1,3-Dicarbonylverbindungen
2.4.7.1 Decarboxylierung
β-Carbonylverbindungen, deren eine Gruppe ein Carbonsäure-Derivat ist, neigen zur
Decarboxylierung. Liegt die freie Carbonsäure, z.B. nach einer Verseifung, vor,
spaltet sich gleich im Anschluss das thermodynamisch äußerst stabile CO2 ab und
die negative Ladung kann als Enolat stabilisiert werden.
OEt
O O
R
NaOH
H2O O
O O
RVerseifungeines Esters
O
R
+ CO2
H+
O
O O
R
HOH
R
+ CO2
OH+
R
Abbildung 177: Mechanismus der Decarboxylierung einer β-Carbonylverbindung. Zunächst wird der
Ester zur Carbonsäure verseift. Gleich darauf spaltet die Verbindung CO2 ab und reagiert weiter zur
Ketoverbindung.
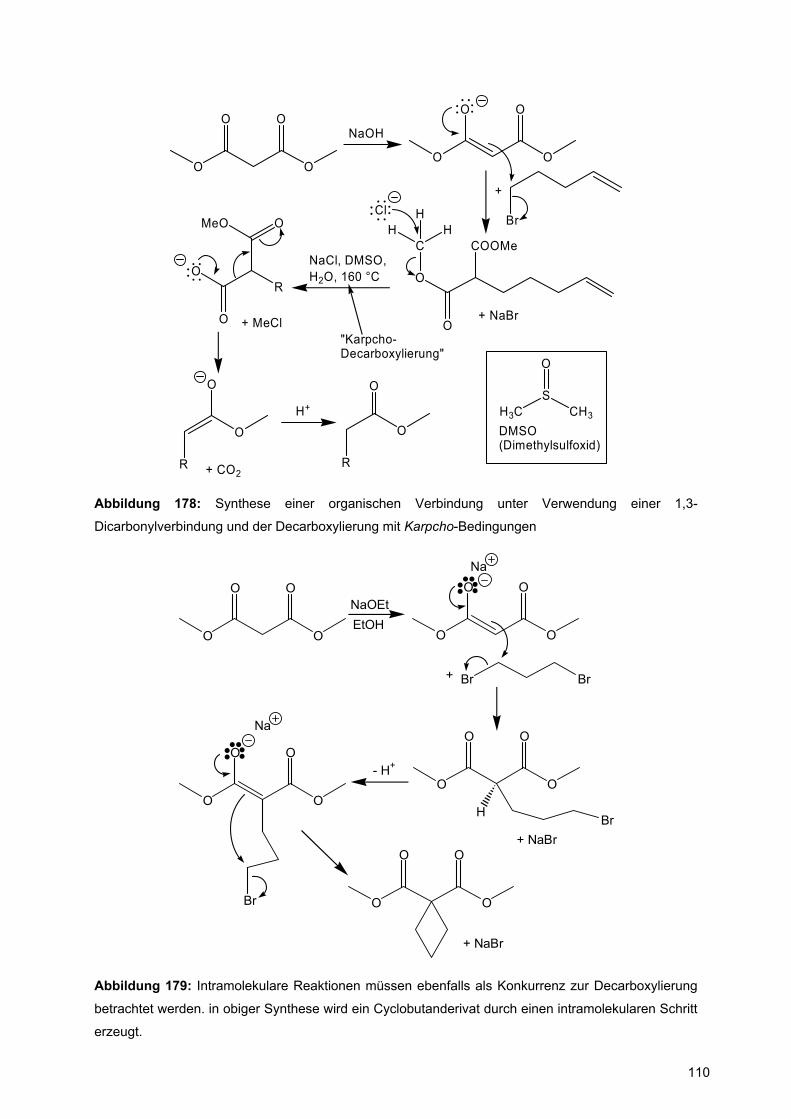
110
O O
O O
H+
+
Br
NaOH
O O
O O
COOMe
+ NaBrO
O
CH H
HCl
R
O
O
OMeO
+ MeCl
NaCl, DMSO,H2O, 160 °C
"Karpcho-Decarboxylierung"
R
O
O
+ CO2R
O
O DMSO(Dimethylsulfoxid)
O
SH3C CH3
Abbildung 178: Synthese einer organischen Verbindung unter Verwendung einer 1,3-
Dicarbonylverbindung und der Decarboxylierung mit Karpcho-Bedingungen
O O
O ONaOEtEtOH
O O
O ONa
+ BrBr
O O
O O
H
+ NaBr
O O
O O
Br
- H+
O O
O O
+ NaBr
Na
Br
Abbildung 179: Intramolekulare Reaktionen müssen ebenfalls als Konkurrenz zur Decarboxylierung
betrachtet werden. in obiger Synthese wird ein Cyclobutanderivat durch einen intramolekularen Schritt
erzeugt.
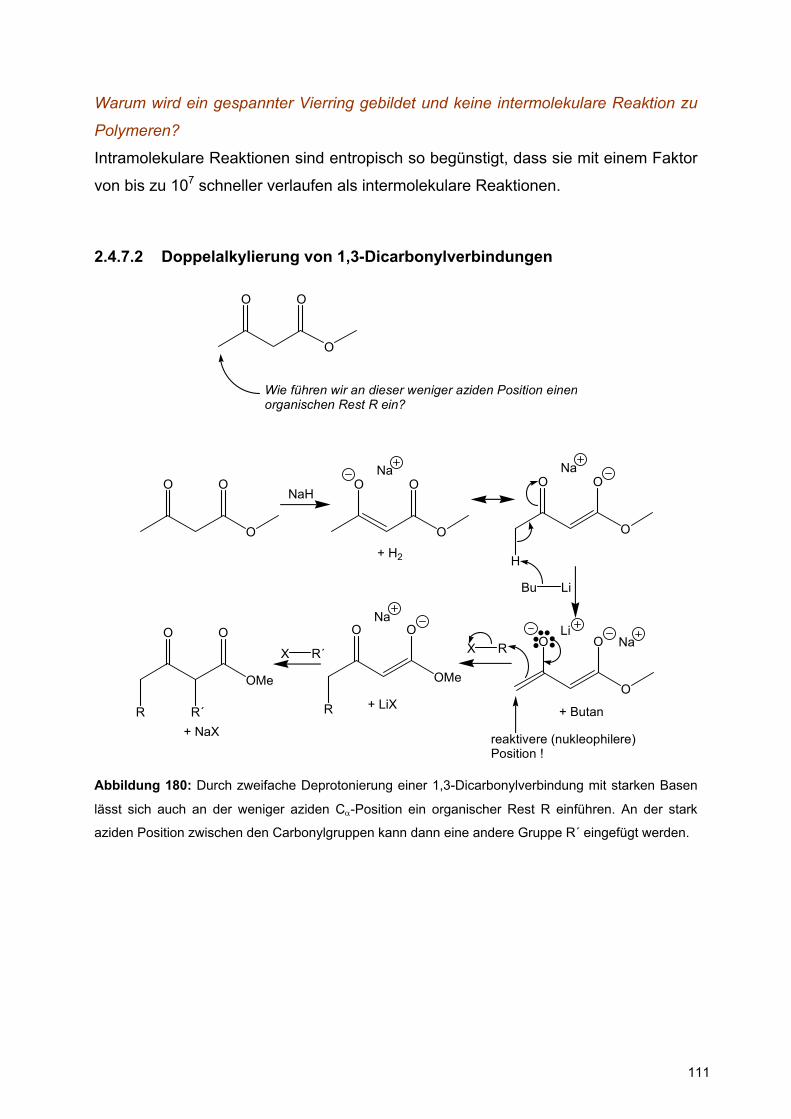
111
Warum wird ein gespannter Vierring gebildet und keine intermolekulare Reaktion zu
Polymeren?
Intramolekulare Reaktionen sind entropisch so begünstigt, dass sie mit einem Faktor
von bis zu 107 schneller verlaufen als intermolekulare Reaktionen.
2.4.7.2 Doppelalkylierung von 1,3-Dicarbonylverbindungen
O
OO
Wie führen wir an dieser weniger aziden Position einenorganischen Rest R ein?
O
OONaH
O
OONa
+ H2
O
OONa
H
Bu Li
O
OO NaLi
+ Butan
X R
OMe
OO
R + LiX
Na
reaktivere (nukleophilere)Position !
X R´
OMe
OO
R R´+ NaX
Abbildung 180: Durch zweifache Deprotonierung einer 1,3-Dicarbonylverbindung mit starken Basen
lässt sich auch an der weniger aziden Cα-Position ein organischer Rest R einführen. An der stark
aziden Position zwischen den Carbonylgruppen kann dann eine andere Gruppe R´ eingefügt werden.
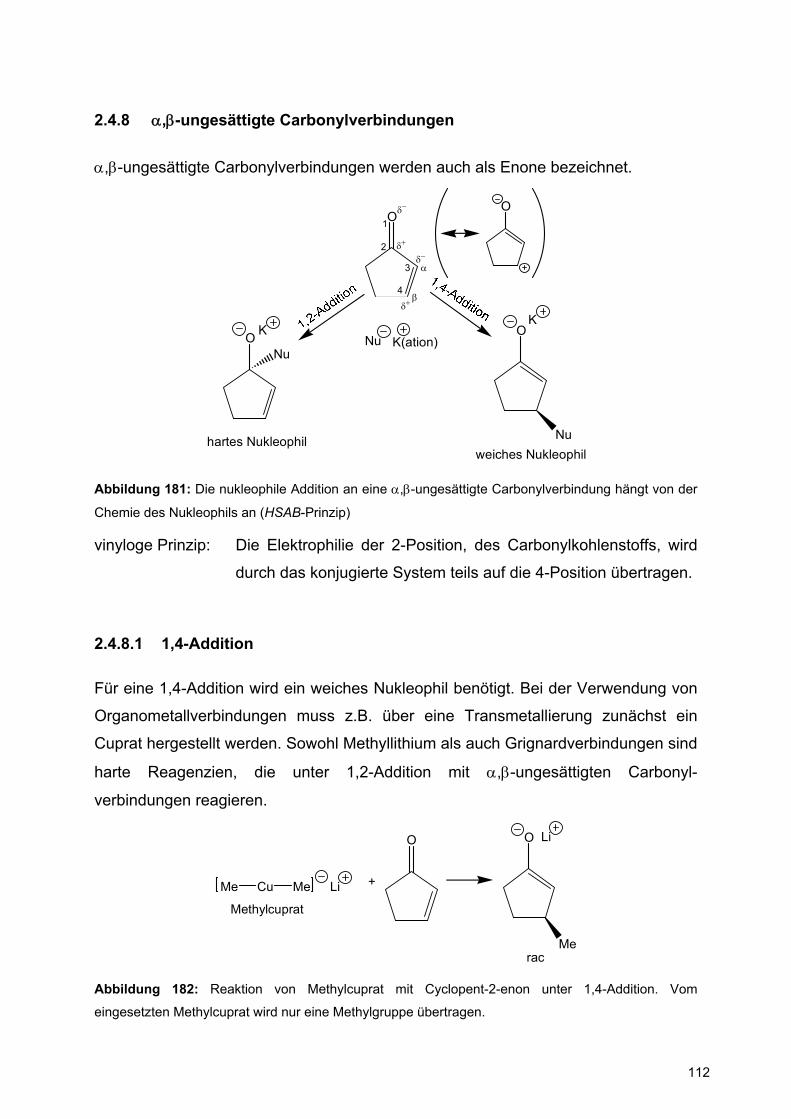
112
2.4.8 α,β-ungesättigte Carbonylverbindungen
α,β-ungesättigte Carbonylverbindungen werden auch als Enone bezeichnet.
OO
1
2
3
4
α
β
δ−
δ−δ+
δ+
Nu K(ation)ONu
K
hartes Nukleophil
OK
Nuweiches Nukleophil
Abbildung 181: Die nukleophile Addition an eine α,β-ungesättigte Carbonylverbindung hängt von der
Chemie des Nukleophils an (HSAB-Prinzip)
vinyloge Prinzip: Die Elektrophilie der 2-Position, des Carbonylkohlenstoffs, wird
durch das konjugierte System teils auf die 4-Position übertragen.
2.4.8.1 1,4-Addition
Für eine 1,4-Addition wird ein weiches Nukleophil benötigt. Bei der Verwendung von
Organometallverbindungen muss z.B. über eine Transmetallierung zunächst ein
Cuprat hergestellt werden. Sowohl Methyllithium als auch Grignardverbindungen sind
harte Reagenzien, die unter 1,2-Addition mit α,β-ungesättigten Carbonyl-
verbindungen reagieren.
Me Cu Me Li
Methylcuprat
+
O O Li
Merac
Abbildung 182: Reaktion von Methylcuprat mit Cyclopent-2-enon unter 1,4-Addition. Vom
eingesetzten Methylcuprat wird nur eine Methylgruppe übertragen.
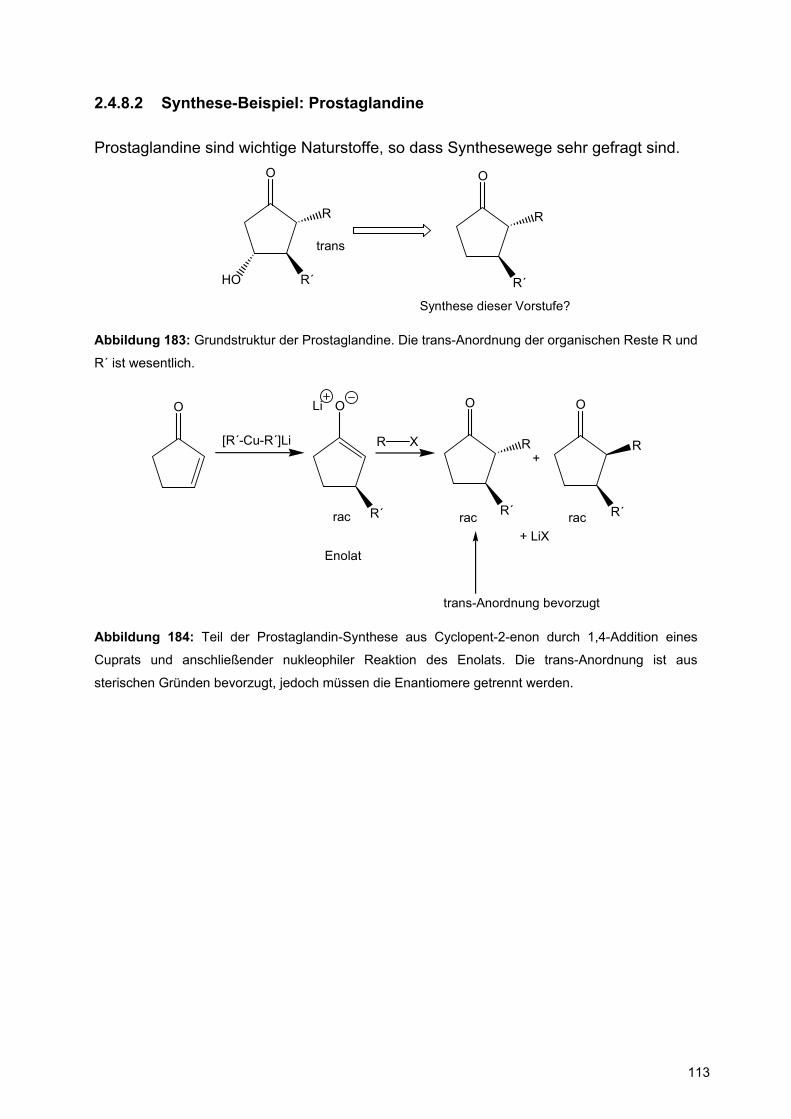
113
2.4.8.2 Synthese-Beispiel: Prostaglandine
Prostaglandine sind wichtige Naturstoffe, so dass Synthesewege sehr gefragt sind. O
HO
R
R´
O
R
R´
Synthese dieser Vorstufe?
trans
Abbildung 183: Grundstruktur der Prostaglandine. Die trans-Anordnung der organischen Reste R und
R´ ist wesentlich.
O
[R´-Cu-R´]Li
O
R´
Li
rac
Enolat
R X
O
R
R´
O
R
R´rac rac+ LiX
+
trans-Anordnung bevorzugt
Abbildung 184: Teil der Prostaglandin-Synthese aus Cyclopent-2-enon durch 1,4-Addition eines
Cuprats und anschließender nukleophiler Reaktion des Enolats. Die trans-Anordnung ist aus
sterischen Gründen bevorzugt, jedoch müssen die Enantiomere getrennt werden.
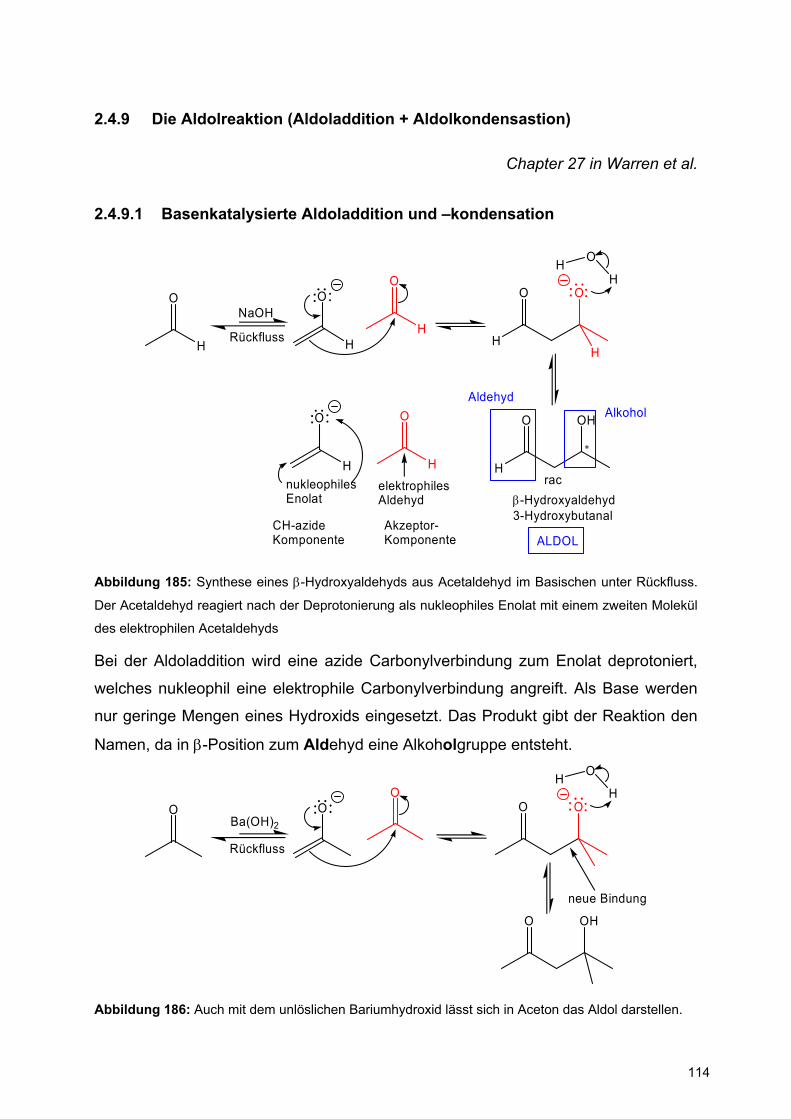
114
2.4.9 Die Aldolreaktion (Aldoladdition + Aldolkondensastion)
Chapter 27 in Warren et al.
2.4.9.1 Basenkatalysierte Aldoladdition und –kondensation
O
H
NaOH
Rückfluss
O
H
O
H
O
H
O
H
H
OH
∗
H
O OH
rac
AlkoholAldehyd
β-Hydroxyaldehyd3-Hydroxybutanal
ALDOL
O
HnukleophilesEnolat
O
H
elektrophilesAldehyd
CH-azideKomponente
Akzeptor-Komponente
Abbildung 185: Synthese eines β-Hydroxyaldehyds aus Acetaldehyd im Basischen unter Rückfluss.
Der Acetaldehyd reagiert nach der Deprotonierung als nukleophiles Enolat mit einem zweiten Molekül
des elektrophilen Acetaldehyds
Bei der Aldoladdition wird eine azide Carbonylverbindung zum Enolat deprotoniert,
welches nukleophil eine elektrophile Carbonylverbindung angreift. Als Base werden
nur geringe Mengen eines Hydroxids eingesetzt. Das Produkt gibt der Reaktion den
Namen, da in β-Position zum Aldehyd eine Alkoholgruppe entsteht.
OBa(OH)2
Rückfluss
OO
O OH
OH
O OH
neue Bindung
Abbildung 186: Auch mit dem unlöslichen Bariumhydroxid lässt sich in Aceton das Aldol darstellen.
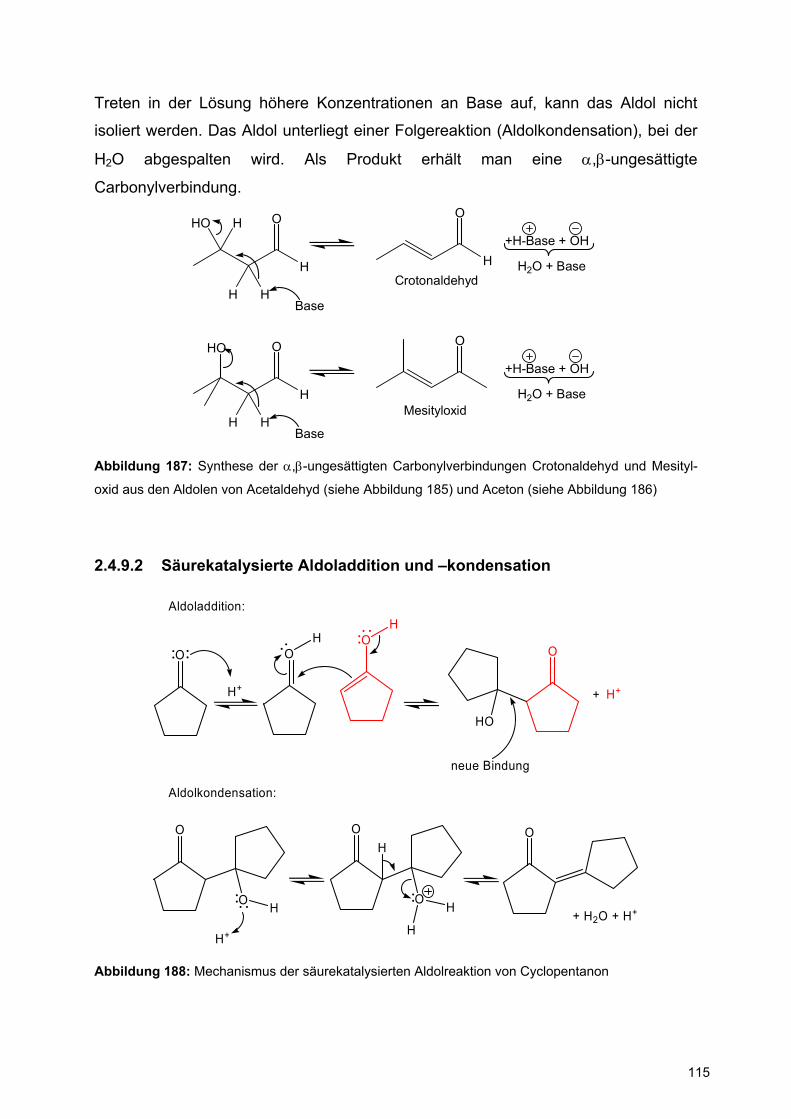
115
Treten in der Lösung höhere Konzentrationen an Base auf, kann das Aldol nicht
isoliert werden. Das Aldol unterliegt einer Folgereaktion (Aldolkondensation), bei der
H2O abgespalten wird. Als Produkt erhält man eine α,β-ungesättigte
Carbonylverbindung.
H
HO H
H H
O
Base
H
O
+H-Base + OH
H2O + BaseCrotonaldehyd
H
HO
H H
O
Base
O
+H-Base + OH
H2O + BaseMesityloxid
Abbildung 187: Synthese der α,β-ungesättigten Carbonylverbindungen Crotonaldehyd und Mesityl-
oxid aus den Aldolen von Acetaldehyd (siehe Abbildung 185) und Aceton (siehe Abbildung 186)
2.4.9.2 Säurekatalysierte Aldoladdition und –kondensation
O
H+
OH O
H
O
HO
+ H+
neue Bindung
Aldoladdition:
Aldolkondensation:
O
O H
H+
O
O H
H
HO
+ H2O + H+
Abbildung 188: Mechanismus der säurekatalysierten Aldolreaktion von Cyclopentanon

116
2.4.9.3 Vergleich der Katalysemechanismen
Beim basenkatalysierten Mechanismus wird die Nukleophilie der einen Komponente
erhöht, während bei der Katalyse mit einer Brønsted-Säure die andere Komponente
elektrophiler gemacht wird.
Außerdem kann man durch geeignete Wahl der Katalyse entweder das Produkt der
Aldoladdition, das Aldol selbst, oder das Produkt der Aldolkondensation, die α,β-
ungesättigte Carbonylverbindung, erhalten.
Produkt Katalyse Addition Kondensation
OH – Aldol (Enon)
H + (Aldol) Enon
Tabelle 16: Bevorzugtes Produkt der säure- oder basenkatalysierten Aldolreaktion
2.4.9.4 Retrosynthese der Aldolreaktion
O
O OH
O
O
+
α,β-ungesättigtesKeton
β-Hydroxy-Keton
Abbildung 189: Retrosynthetische Zerlegung von Mesityloxid
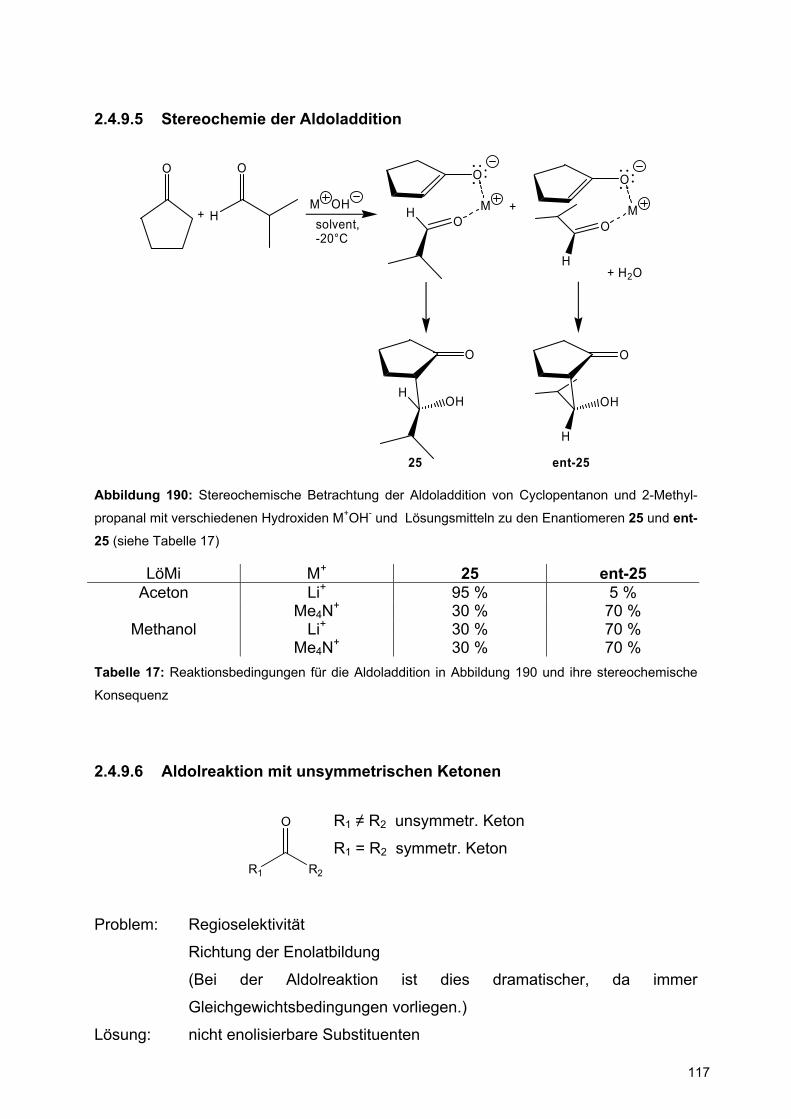
117
2.4.9.5 Stereochemie der Aldoladdition
O O
H+solvent,-20°C
O
MO
H
O
MO
H +
+ H2O
O
OHH
25
O
OH
H
ent-25
M OH
Abbildung 190: Stereochemische Betrachtung der Aldoladdition von Cyclopentanon und 2-Methyl-
propanal mit verschiedenen Hydroxiden M+OH- und Lösungsmitteln zu den Enantiomeren 25 und ent-25 (siehe Tabelle 17)
LöMi M+ 25 ent-25 Aceton Li+ 95 % 5 %
Me4N+ 30 % 70 % Methanol Li+ 30 % 70 %
Me4N+ 30 % 70 % Tabelle 17: Reaktionsbedingungen für die Aldoladdition in Abbildung 190 und ihre stereochemische
Konsequenz
2.4.9.6 Aldolreaktion mit unsymmetrischen Ketonen
O
R1 R2
Problem: Regioselektivität
Richtung der Enolatbildung
(Bei der Aldolreaktion ist dies dramatischer, da immer
Gleichgewichtsbedingungen vorliegen.)
Lösung: nicht enolisierbare Substituenten
R1 ≠ R2 unsymmetr. Keton
R1 = R2 symmetr. Keton
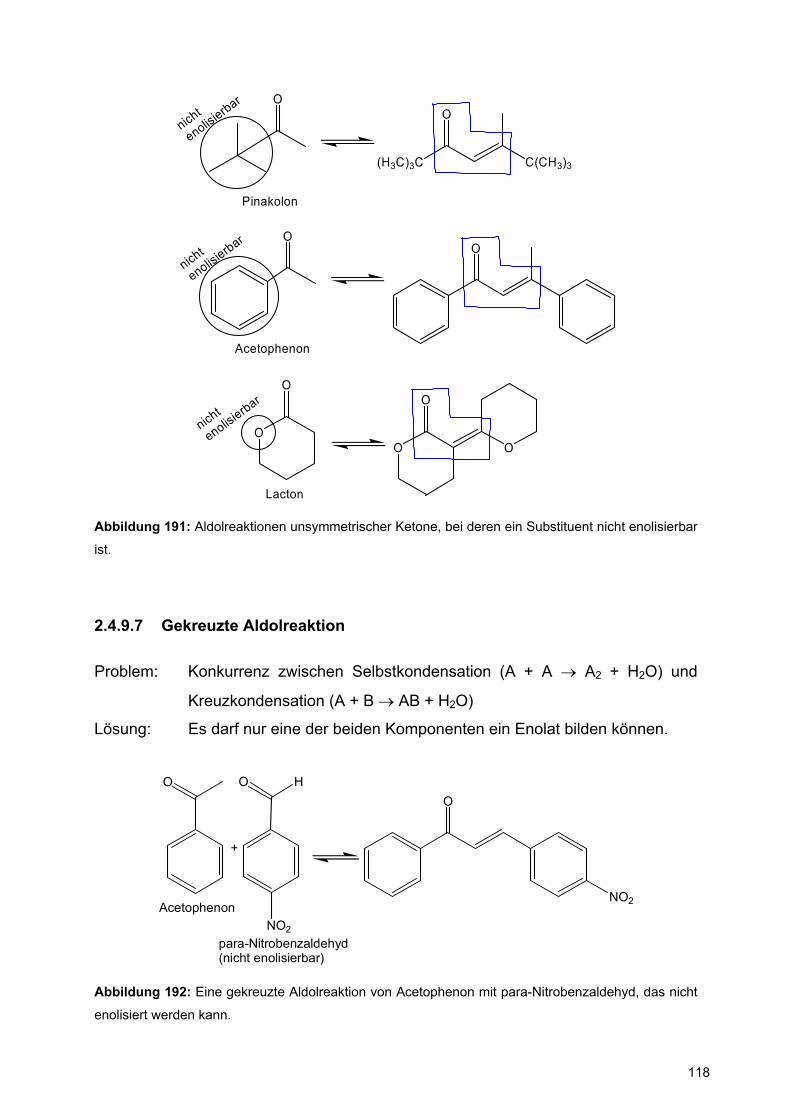
118
O
Pinakolon
nicht
enolisierbarO
(H3C)3C C(CH3)3
O
Acetophenon
nicht
enolisierbarO
O
O
Lacton
O
O O
nicht
enolisierbar
Abbildung 191: Aldolreaktionen unsymmetrischer Ketone, bei deren ein Substituent nicht enolisierbar
ist.
2.4.9.7 Gekreuzte Aldolreaktion
Problem: Konkurrenz zwischen Selbstkondensation (A + A → A2 + H2O) und
Kreuzkondensation (A + B → AB + H2O)
Lösung: Es darf nur eine der beiden Komponenten ein Enolat bilden können.
O
+
Acetophenon
HO
NO2para-Nitrobenzaldehyd(nicht enolisierbar)
O
NO2
Abbildung 192: Eine gekreuzte Aldolreaktion von Acetophenon mit para-Nitrobenzaldehyd, das nicht
enolisiert werden kann.
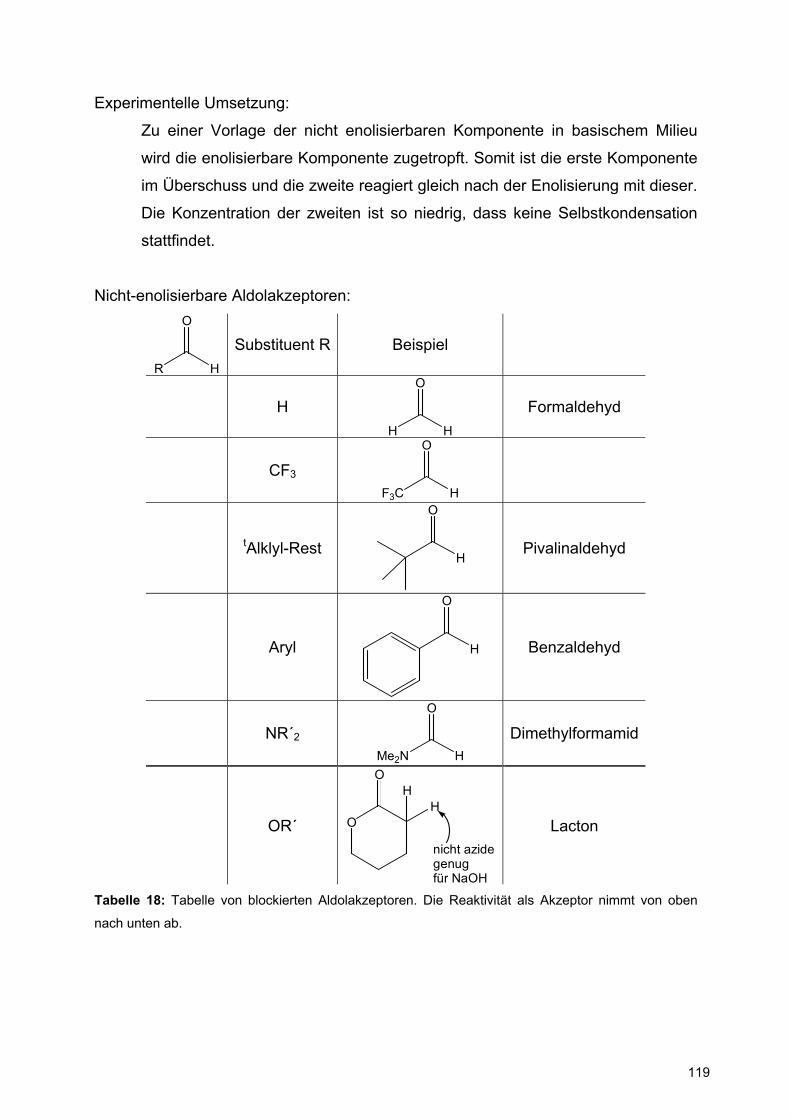
119
Experimentelle Umsetzung:
Zu einer Vorlage der nicht enolisierbaren Komponente in basischem Milieu
wird die enolisierbare Komponente zugetropft. Somit ist die erste Komponente
im Überschuss und die zweite reagiert gleich nach der Enolisierung mit dieser.
Die Konzentration der zweiten ist so niedrig, dass keine Selbstkondensation
stattfindet.
Nicht-enolisierbare Aldolakzeptoren: O
R H Substituent R Beispiel
H O
H H Formaldehyd
CF3 O
F3C H
tAlklyl-Rest
O
H
Pivalinaldehyd
Aryl
O
H
Benzaldehyd
NR´2 O
Me2N H Dimethylformamid
OR´
O
OH
H
nicht azidegenugfür NaOH
Lacton
Tabelle 18: Tabelle von blockierten Aldolakzeptoren. Die Reaktivität als Akzeptor nimmt von oben
nach unten ab.
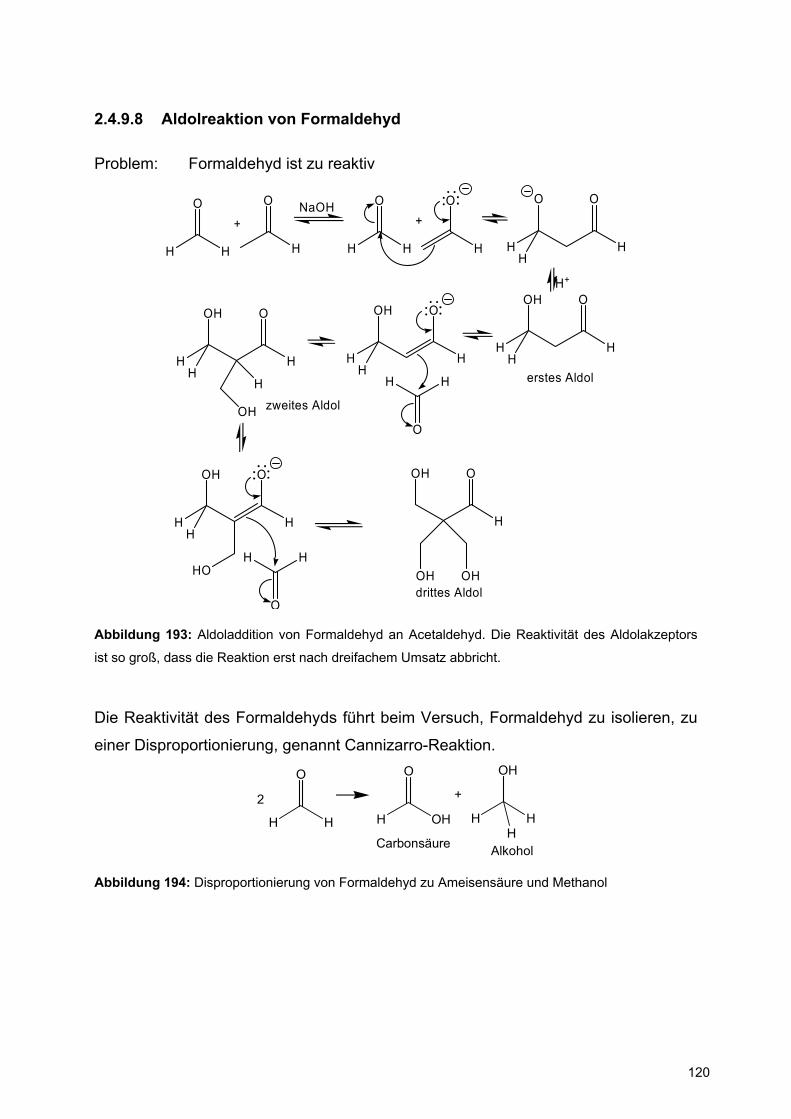
120
2.4.9.8 Aldolreaktion von Formaldehyd
Problem: Formaldehyd ist zu reaktiv
O
H H
O
H
+NaOH O
H H
O
H
+
H H
O
H
O
H+
H H
OH
H
O
erstes AldolH H
OH
H
O
O
HHH H
OH
H
O
H
OH zweites Aldol
H H
OH
H
O
HO
O
HH
H
OOH
OH OHdrittes Aldol
Abbildung 193: Aldoladdition von Formaldehyd an Acetaldehyd. Die Reaktivität des Aldolakzeptors
ist so groß, dass die Reaktion erst nach dreifachem Umsatz abbricht.
Die Reaktivität des Formaldehyds führt beim Versuch, Formaldehyd zu isolieren, zu
einer Disproportionierung, genannt Cannizarro-Reaktion.
O
H H
2
O
H OH
+
OH
H HH
Carbonsäure Alkohol
Abbildung 194: Disproportionierung von Formaldehyd zu Ameisensäure und Methanol
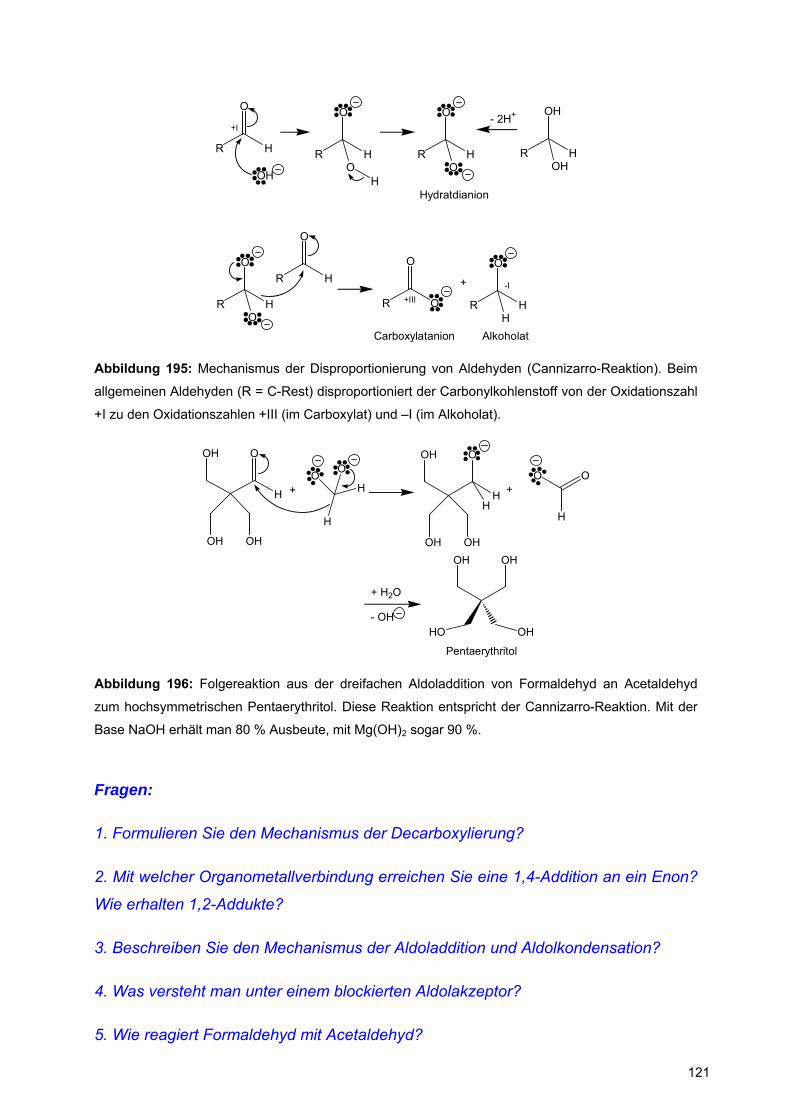
121
O
R H
OH
O
R HO
H
O
R HO
Hydratdianion
OH
R HOH
- 2H+
O
R HO
O
R HO
R O
+
O
R HH
+I
+III-I
Carboxylatanion Alkoholat
Abbildung 195: Mechanismus der Disproportionierung von Aldehyden (Cannizarro-Reaktion). Beim
allgemeinen Aldehyden (R = C-Rest) disproportioniert der Carbonylkohlenstoff von der Oxidationszahl
+I zu den Oxidationszahlen +III (im Carboxylat) und –I (im Alkoholat).
H
OOH
OH OH
OH
H
O
H
OOH
OH OH
H+ +
H
O O
+ H2O
- OH
OH OH
HO OH
Pentaerythritol
Abbildung 196: Folgereaktion aus der dreifachen Aldoladdition von Formaldehyd an Acetaldehyd
zum hochsymmetrischen Pentaerythritol. Diese Reaktion entspricht der Cannizarro-Reaktion. Mit der
Base NaOH erhält man 80 % Ausbeute, mit Mg(OH)2 sogar 90 %.
Fragen:
1. Formulieren Sie den Mechanismus der Decarboxylierung?
2. Mit welcher Organometallverbindung erreichen Sie eine 1,4-Addition an ein Enon?
Wie erhalten 1,2-Addukte?
3. Beschreiben Sie den Mechanismus der Aldoladdition und Aldolkondensation?
4. Was versteht man unter einem blockierten Aldolakzeptor?
5. Wie reagiert Formaldehyd mit Acetaldehyd?
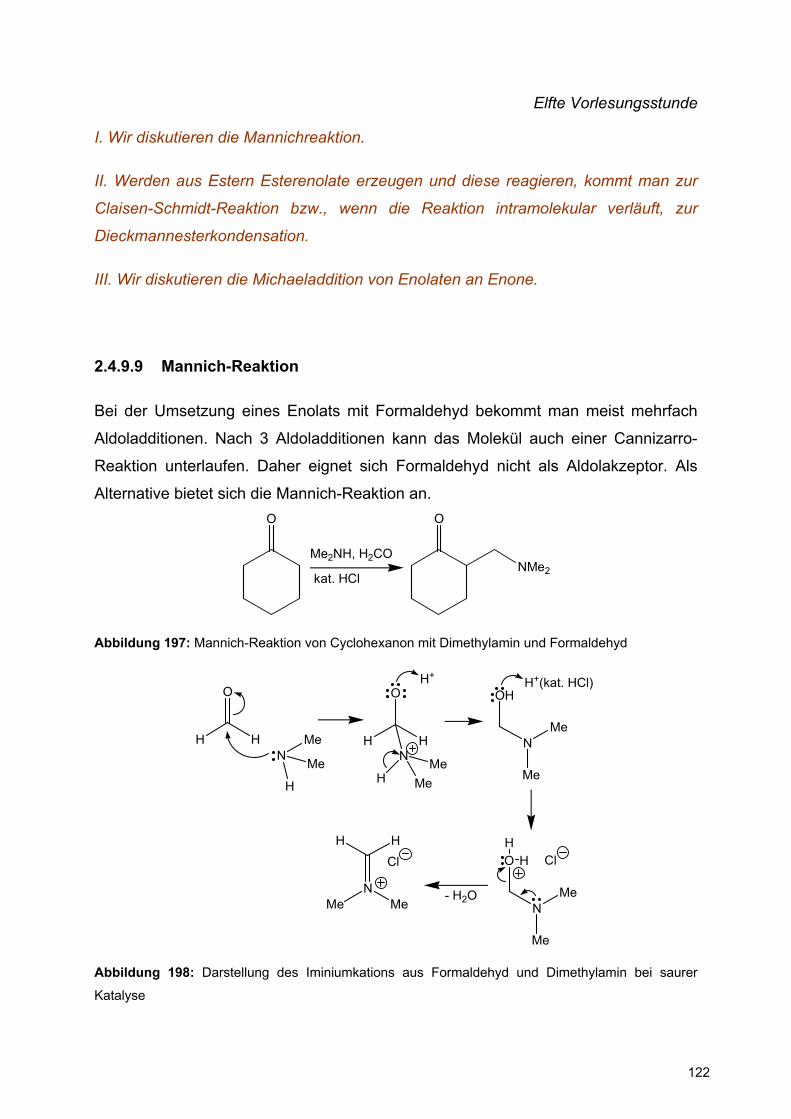
122
Elfte Vorlesungsstunde
I. Wir diskutieren die Mannichreaktion.
II. Werden aus Estern Esterenolate erzeugen und diese reagieren, kommt man zur
Claisen-Schmidt-Reaktion bzw., wenn die Reaktion intramolekular verläuft, zur
Dieckmannesterkondensation.
III. Wir diskutieren die Michaeladdition von Enolaten an Enone.
2.4.9.9 Mannich-Reaktion
Bei der Umsetzung eines Enolats mit Formaldehyd bekommt man meist mehrfach
Aldoladditionen. Nach 3 Aldoladditionen kann das Molekül auch einer Cannizarro-
Reaktion unterlaufen. Daher eignet sich Formaldehyd nicht als Aldolakzeptor. Als
Alternative bietet sich die Mannich-Reaktion an. O
Me2NH, H2CO
kat. HCl
O
NMe2
Abbildung 197: Mannich-Reaktion von Cyclohexanon mit Dimethylamin und Formaldehyd
O
H HN
Me
Me
H
O
H HN
HMe
Me
H+
OH
N
Me
Me
H+(kat. HCl)
O
N
Me
Me
HH
NMe Me
- H2O
ClClH H
Abbildung 198: Darstellung des Iminiumkations aus Formaldehyd und Dimethylamin bei saurer
Katalyse
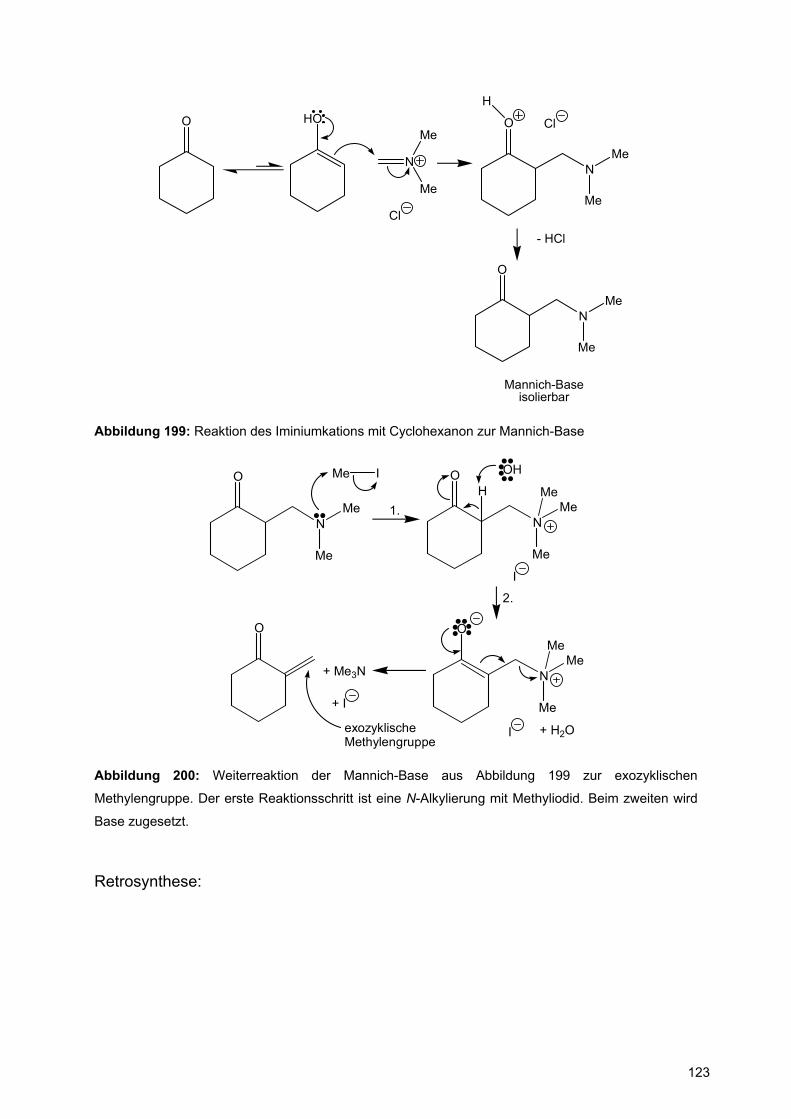
123
O HO
N
Me
Me
Cl
O
H
N
Me
Me
Cl
- HCl
O
N
Me
Me
Mannich-Baseisolierbar
Abbildung 199: Reaktion des Iminiumkations mit Cyclohexanon zur Mannich-Base
O
NMe
Me
Me I
1.
O
NMe
Me
MeH
OH
I
2.
O
NMe
Me
Me
I + H2O
O
+ Me3N
exozyklischeMethylengruppe
+ I
Abbildung 200: Weiterreaktion der Mannich-Base aus Abbildung 199 zur exozyklischen
Methylengruppe. Der erste Reaktionsschritt ist eine N-Alkylierung mit Methyliodid. Beim zweiten wird
Base zugesetzt.
Retrosynthese:
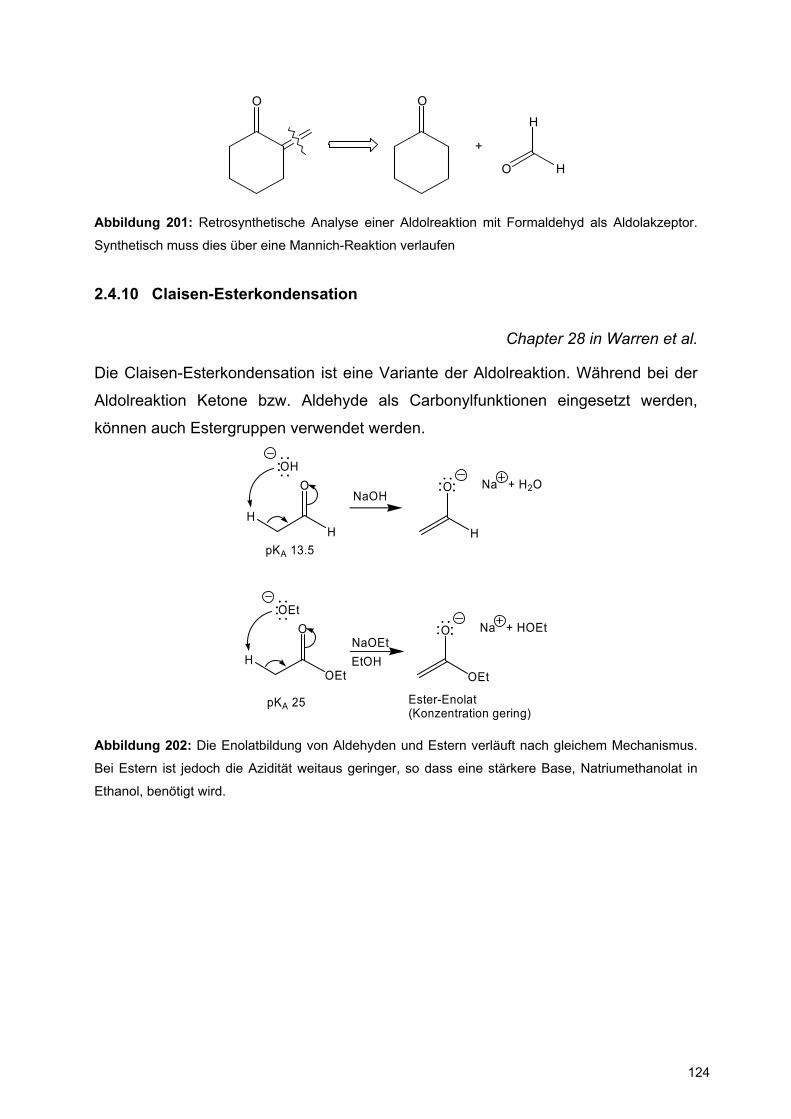
124
O O
H
O H
+
Abbildung 201: Retrosynthetische Analyse einer Aldolreaktion mit Formaldehyd als Aldolakzeptor.
Synthetisch muss dies über eine Mannich-Reaktion verlaufen
2.4.10 Claisen-Esterkondensation
Chapter 28 in Warren et al.
Die Claisen-Esterkondensation ist eine Variante der Aldolreaktion. Während bei der
Aldolreaktion Ketone bzw. Aldehyde als Carbonylfunktionen eingesetzt werden,
können auch Estergruppen verwendet werden.
NaOHO
HH
OHO
H
Na + H2O
NaOEtO
OEtH
OEtO
OEt
Na + HOEt
EtOH
pKA 25 Ester-Enolat(Konzentration gering)
pKA 13.5
Abbildung 202: Die Enolatbildung von Aldehyden und Estern verläuft nach gleichem Mechanismus.
Bei Estern ist jedoch die Azidität weitaus geringer, so dass eine stärkere Base, Natriumethanolat in
Ethanol, benötigt wird.
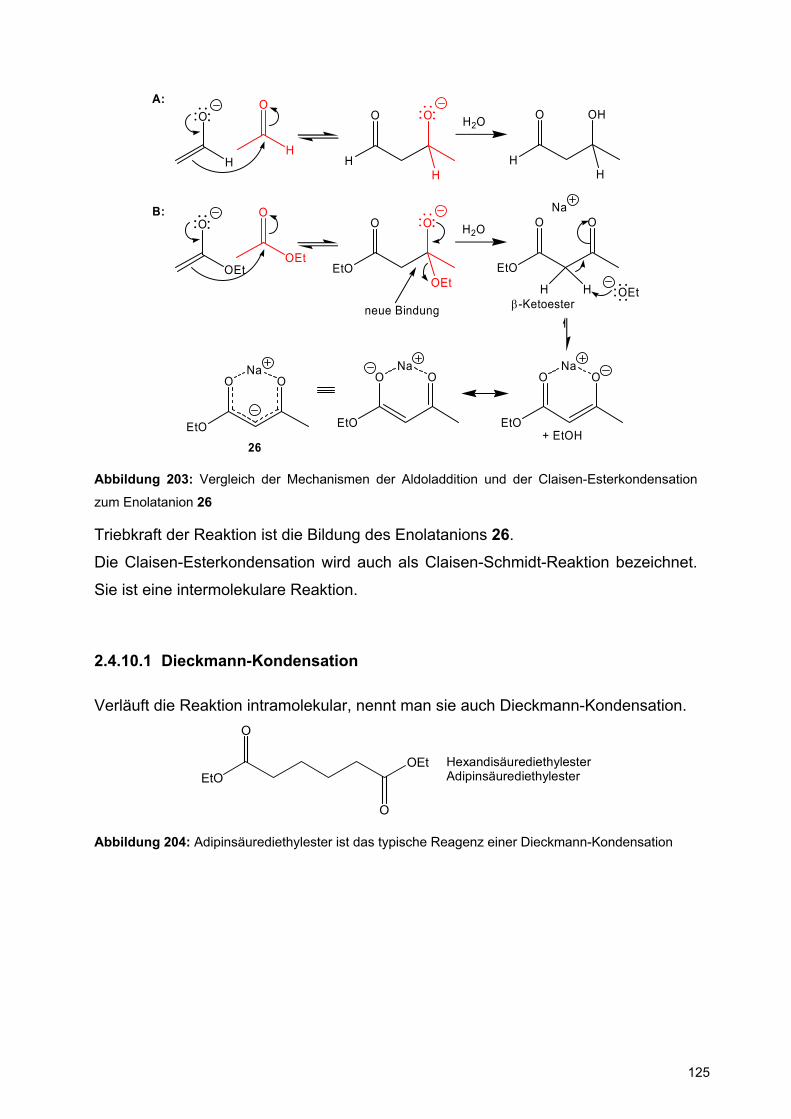
125
O
H
O
HH
O
H
O H2O
H
O
H
OH
O
OEt
O
OEtEtO
O
OEt
O H2O
EtO
O O
neue Bindung
H Hβ-Ketoester
OEt
Na
EtO
O ONa
+ EtOHEtO
O ONa
EtO
O ONa
26
A:
B:
Abbildung 203: Vergleich der Mechanismen der Aldoladdition und der Claisen-Esterkondensation
zum Enolatanion 26
Triebkraft der Reaktion ist die Bildung des Enolatanions 26.
Die Claisen-Esterkondensation wird auch als Claisen-Schmidt-Reaktion bezeichnet.
Sie ist eine intermolekulare Reaktion.
2.4.10.1 Dieckmann-Kondensation
Verläuft die Reaktion intramolekular, nennt man sie auch Dieckmann-Kondensation.
EtOOEt
O
O
HexandisäurediethylesterAdipinsäurediethylester
Abbildung 204: Adipinsäurediethylester ist das typische Reagenz einer Dieckmann-Kondensation
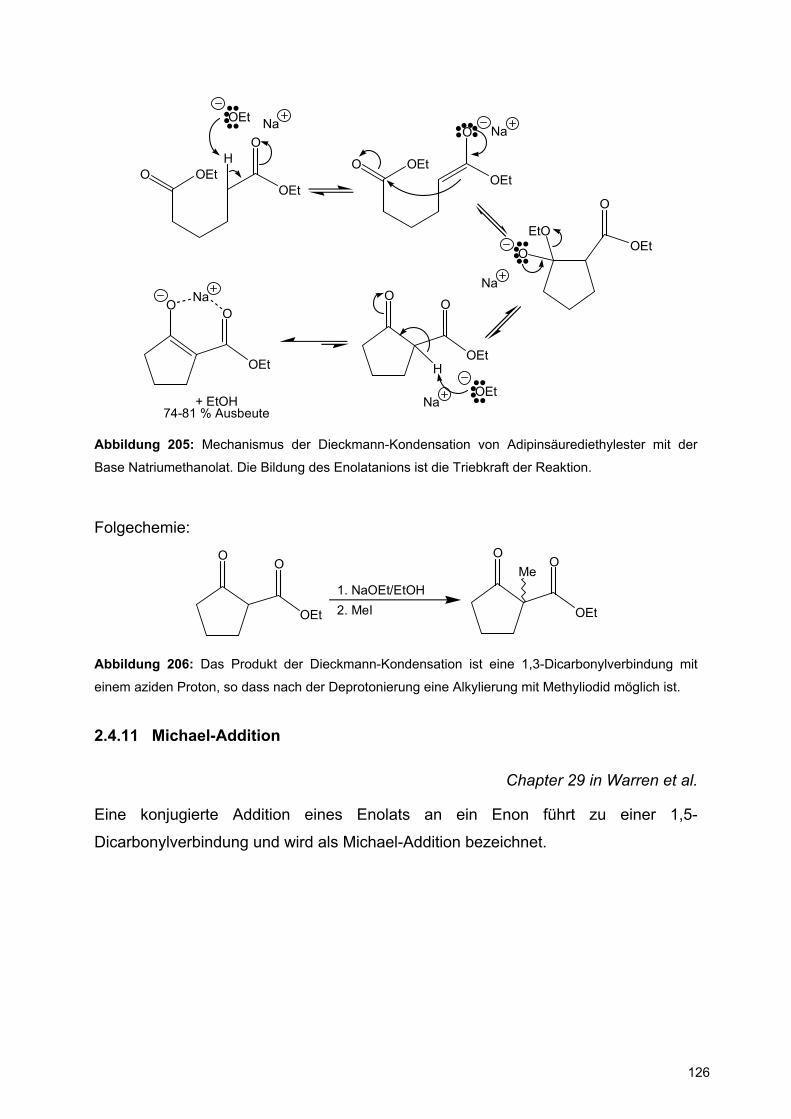
126
O OEtH
O
OEt
OEt Na
O OEt
O
OEt
Na
EtO
O
O
OEt
NaO
O
OEtH
OEtNa
OO
OEt
Na
+ EtOH74-81 % Ausbeute
Abbildung 205: Mechanismus der Dieckmann-Kondensation von Adipinsäurediethylester mit der
Base Natriumethanolat. Die Bildung des Enolatanions ist die Triebkraft der Reaktion.
Folgechemie:
OO
OEt
1. NaOEt/EtOH2. MeI
OO
OEt
Me
Abbildung 206: Das Produkt der Dieckmann-Kondensation ist eine 1,3-Dicarbonylverbindung mit
einem aziden Proton, so dass nach der Deprotonierung eine Alkylierung mit Methyliodid möglich ist.
2.4.11 Michael-Addition
Chapter 29 in Warren et al.
Eine konjugierte Addition eines Enolats an ein Enon führt zu einer 1,5-
Dicarbonylverbindung und wird als Michael-Addition bezeichnet.
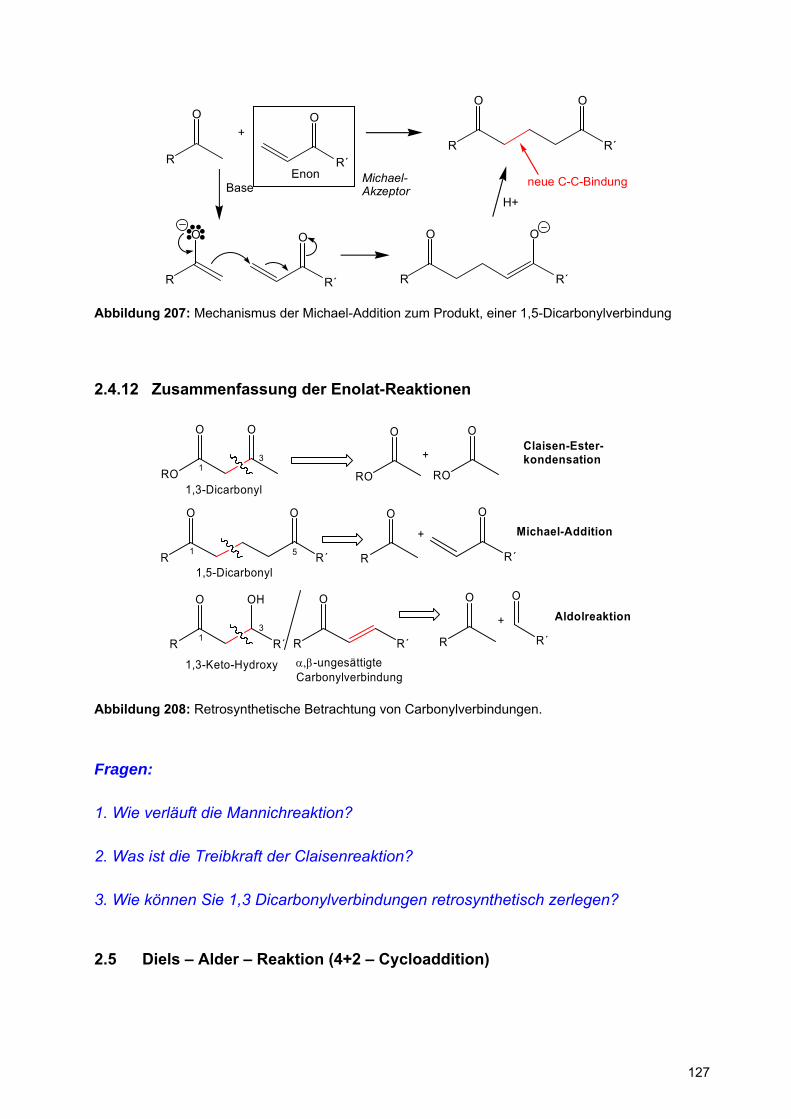
127
O
R
+O
R´R R´
O O
neue C-C-BindungBase
O
R
O
R´ R R´
O O
H+
Enon Michael-Akzeptor
Abbildung 207: Mechanismus der Michael-Addition zum Produkt, einer 1,5-Dicarbonylverbindung
2.4.12 Zusammenfassung der Enolat-Reaktionen
RO
O O
13
1,3-Dicarbonyl
O
RO
O
RO
+Claisen-Ester-kondensation
R
O
1
1,5-Dicarbonyl
O
R
O
R´
Michael-Addition
R´
O
+
R R´
O OH
13
1,3-Keto-Hydroxy
O
R
O
R´
+ Aldolreaktion
5
R R´
O
α,β-ungesättigteCarbonylverbindung
Abbildung 208: Retrosynthetische Betrachtung von Carbonylverbindungen.
Fragen:
1. Wie verläuft die Mannichreaktion?
2. Was ist die Treibkraft der Claisenreaktion?
3. Wie können Sie 1,3 Dicarbonylverbindungen retrosynthetisch zerlegen?
2.5 Diels – Alder – Reaktion (4+2 – Cycloaddition)
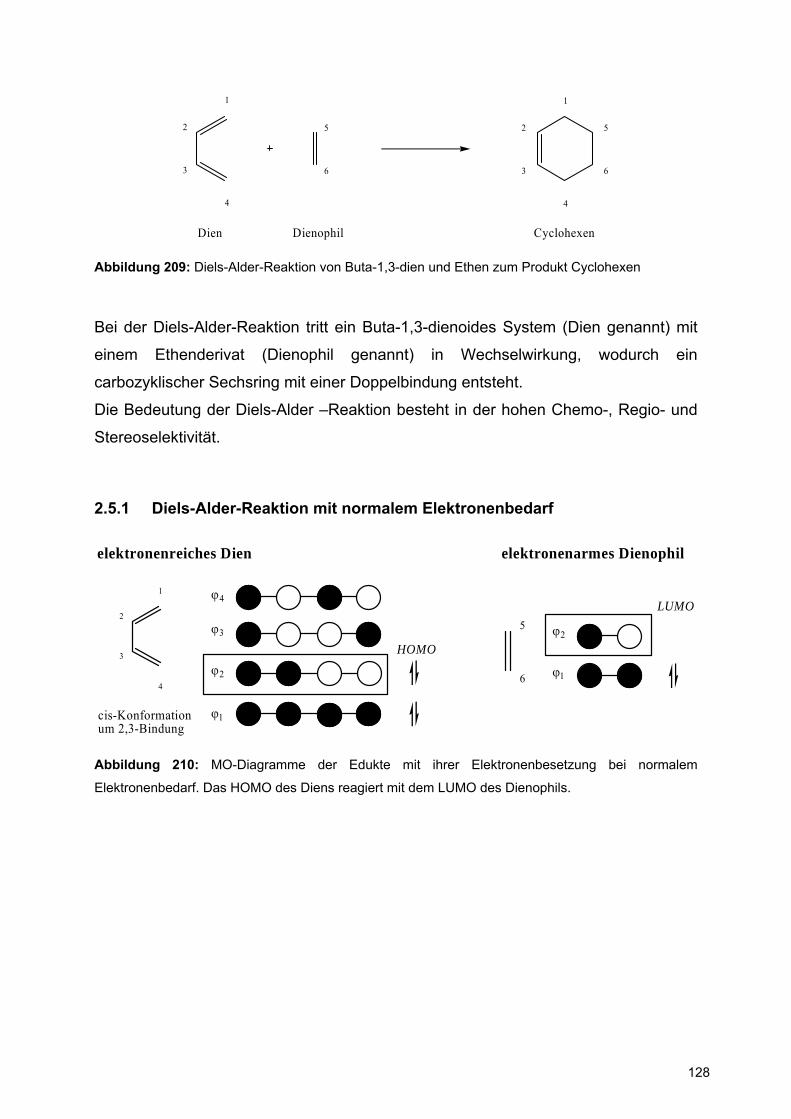
128
4
1
3
2
6
5
4
1
3
2
6
5
Dien Dienophil Cyclohexen
Abbildung 209: Diels-Alder-Reaktion von Buta-1,3-dien und Ethen zum Produkt Cyclohexen
Bei der Diels-Alder-Reaktion tritt ein Buta-1,3-dienoides System (Dien genannt) mit
einem Ethenderivat (Dienophil genannt) in Wechselwirkung, wodurch ein
carbozyklischer Sechsring mit einer Doppelbindung entsteht.
Die Bedeutung der Diels-Alder –Reaktion besteht in der hohen Chemo-, Regio- und
Stereoselektivität.
2.5.1 Diels-Alder-Reaktion mit normalem Elektronenbedarf
4
1
3
2
6
5
cis-Konformationum 2,3-Bindung
elektronenreiches Dien
HOMO
ϕ1
ϕ2
ϕ3
ϕ4
elektronenarmes Dienophil
LUMO
ϕ1
ϕ2
Abbildung 210: MO-Diagramme der Edukte mit ihrer Elektronenbesetzung bei normalem
Elektronenbedarf. Das HOMO des Diens reagiert mit dem LUMO des Dienophils.
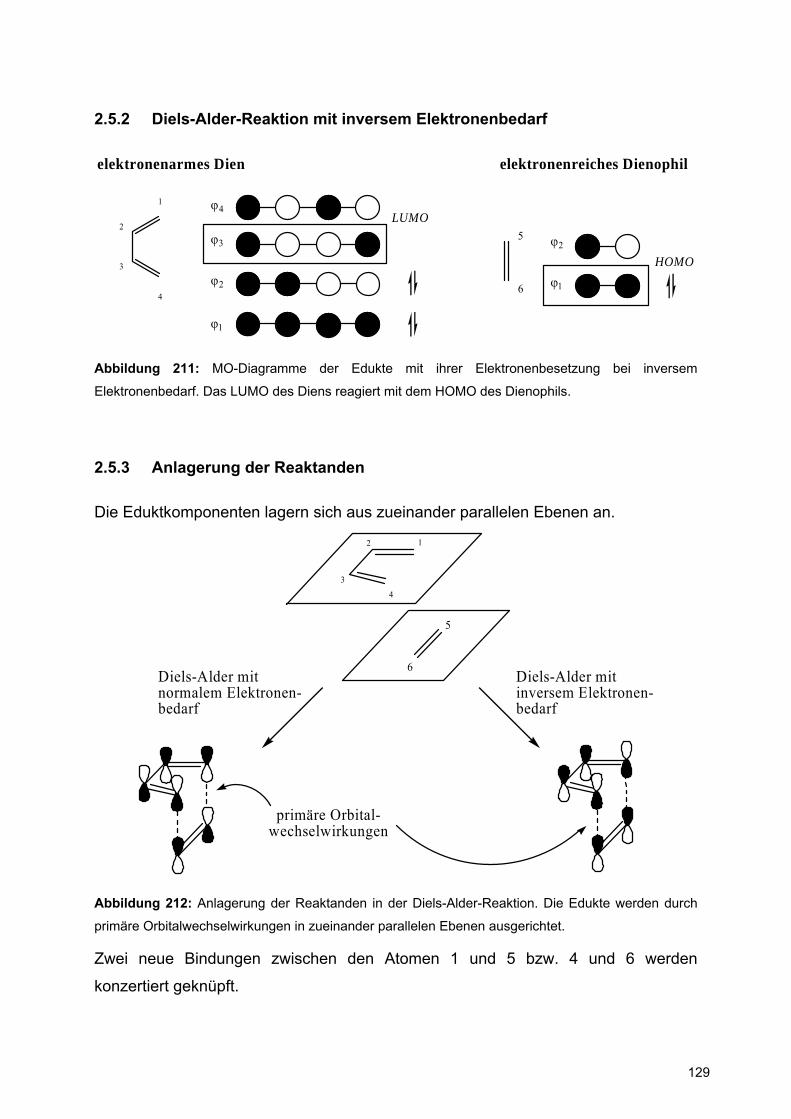
129
2.5.2 Diels-Alder-Reaktion mit inversem Elektronenbedarf
4
1
3
2
6
5
elektronenarmes Dien
LUMO
ϕ1
ϕ2
ϕ3
ϕ4
elektronenreiches Dienophil
HOMOϕ1
ϕ2
Abbildung 211: MO-Diagramme der Edukte mit ihrer Elektronenbesetzung bei inversem
Elektronenbedarf. Das LUMO des Diens reagiert mit dem HOMO des Dienophils.
2.5.3 Anlagerung der Reaktanden
Die Eduktkomponenten lagern sich aus zueinander parallelen Ebenen an.
4
1
3
2
6
5
primäre Orbital-wechselwirkungen
Diels-Alder mitnormalem Elektronen-bedarf
Diels-Alder mitinversem Elektronen-bedarf
Abbildung 212: Anlagerung der Reaktanden in der Diels-Alder-Reaktion. Die Edukte werden durch
primäre Orbitalwechselwirkungen in zueinander parallelen Ebenen ausgerichtet.
Zwei neue Bindungen zwischen den Atomen 1 und 5 bzw. 4 und 6 werden
konzertiert geknüpft.
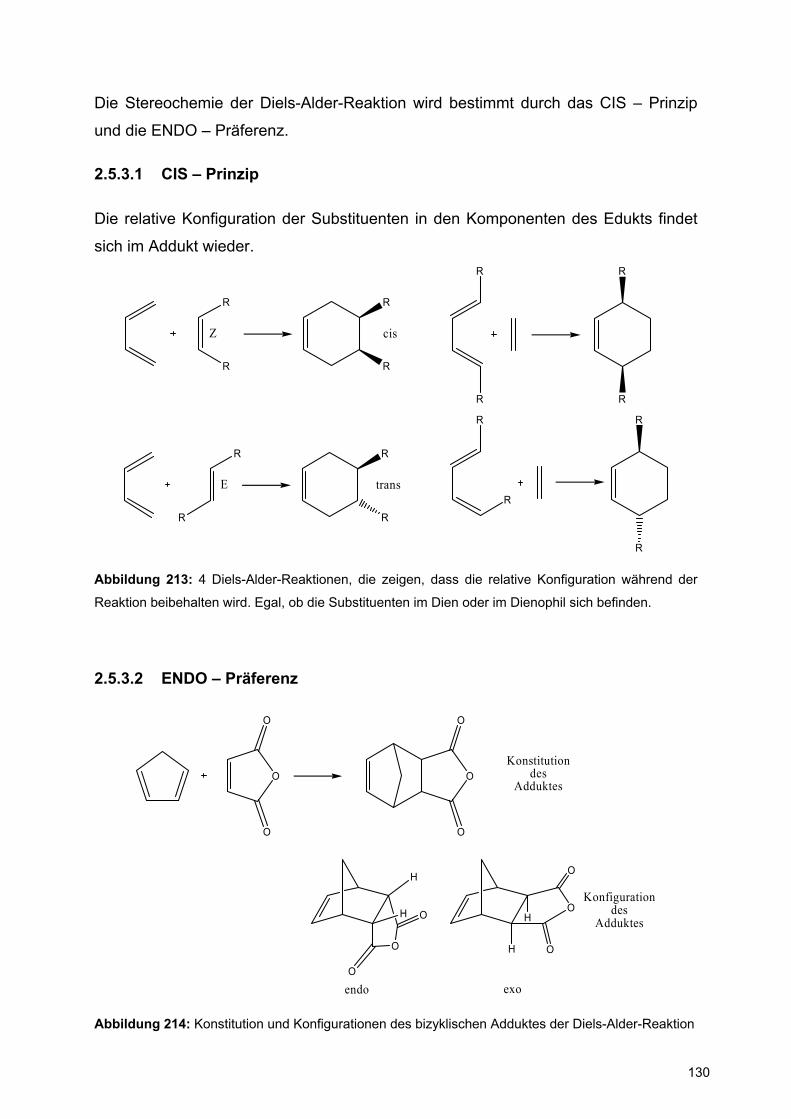
130
Die Stereochemie der Diels-Alder-Reaktion wird bestimmt durch das CIS – Prinzip
und die ENDO – Präferenz.
2.5.3.1 CIS – Prinzip
Die relative Konfiguration der Substituenten in den Komponenten des Edukts findet
sich im Addukt wieder.
R
R
Z
R
R
cis
R
E
R
R
trans
R
R
R
R
R
R R
R
R
Abbildung 213: 4 Diels-Alder-Reaktionen, die zeigen, dass die relative Konfiguration während der
Reaktion beibehalten wird. Egal, ob die Substituenten im Dien oder im Dienophil sich befinden.
2.5.3.2 ENDO – Präferenz
O
O
O
O
O
O
Konstitutiondes
Adduktes
H
H
O
O
OO
O
O
H
H
endo exo
Konfigurationdes
Adduktes
Abbildung 214: Konstitution und Konfigurationen des bizyklischen Adduktes der Diels-Alder-Reaktion

131
Was ist stabiler? Endo- oder exo-Addukt?
endo-Addukt exo-Addukt
1-Brücke
2-Brücke
Abbildung 215: Schematische Darstellung der beiden Addukte mit möglichen sterischen
Behinderungen
⇒ endo-Addukt sterisch anspruchsvoller als exo-Addukt
Allgemein: Es entsteht dasjenige Produkt bevorzugt, das im Übergangszustand mit
einer Sandwich-artigen Vororientierung der Komponenten
korrespondiert, so dass eine maximale Häufung der Doppelbindungen
eintritt.
O
O
O
LUMO
ϕ1
ϕ2
ϕ3
ϕ4
HOMO
ϕ5
ϕ6
Abbildung 216: MO des Dienophils Maleinsäureanhydrid. Maleinsäureanhydrid ist ein
elektronenarmes Dienophil und reagiert mit seinem LUMO bei der Diels-Alder-Reaktion mit normalem
Elektronenbedarf.
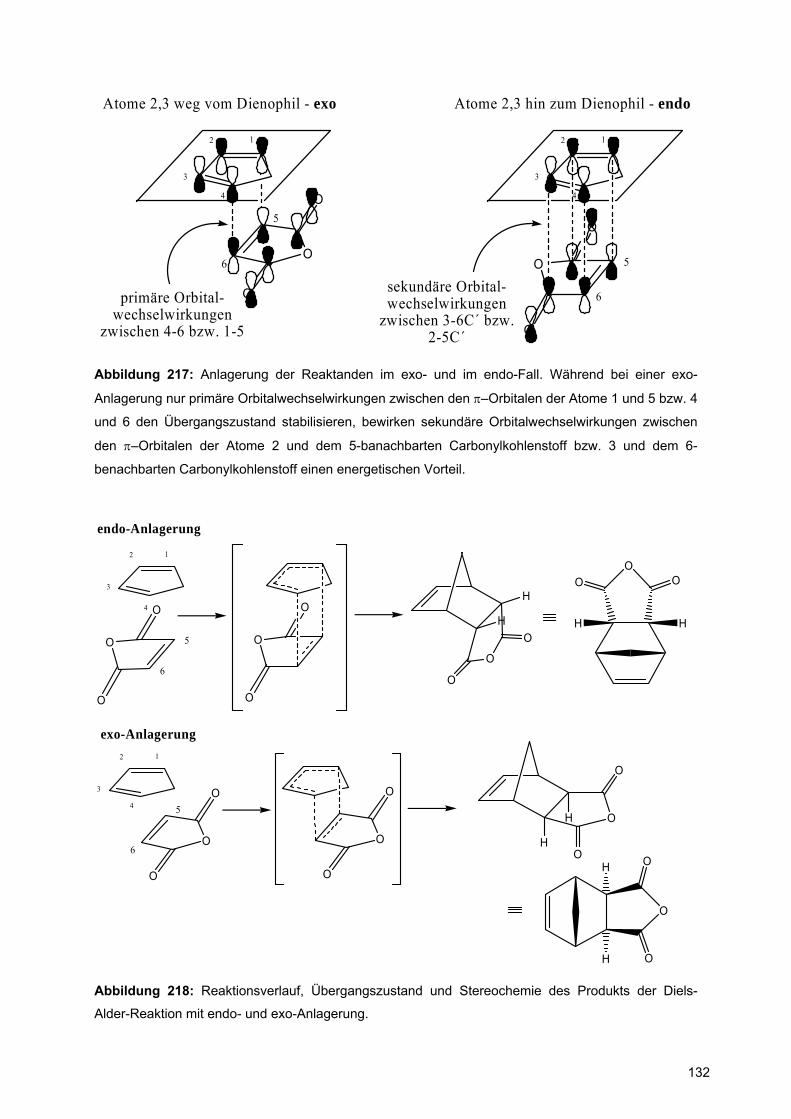
132
4
1
3
2
6
5
primäre Orbital-wechselwirkungen
zwischen 4-6 bzw. 1-5
O
O
O
Atome 2,3 weg vom Dienophil - exo
4
1
3
2
6
5
sekundäre Orbital-wechselwirkungen
zwischen 3-6C´ bzw.2-5C´
Atome 2,3 hin zum Dienophil - endo
O
O
O
Abbildung 217: Anlagerung der Reaktanden im exo- und im endo-Fall. Während bei einer exo-
Anlagerung nur primäre Orbitalwechselwirkungen zwischen den π–Orbitalen der Atome 1 und 5 bzw. 4
und 6 den Übergangszustand stabilisieren, bewirken sekundäre Orbitalwechselwirkungen zwischen
den π–Orbitalen der Atome 2 und dem 5-banachbarten Carbonylkohlenstoff bzw. 3 und dem 6-
benachbarten Carbonylkohlenstoff einen energetischen Vorteil.
4
1
3
2
6
5
endo-Anlagerung
O
O
O
O
O
O
H
H
O
O
O
OO O
H H
4
1
3
2
6
5
O
O
O
O
O
O
H
H
exo-Anlagerung
O
O
O
O
O
O
H
H
Abbildung 218: Reaktionsverlauf, Übergangszustand und Stereochemie des Produkts der Diels-
Alder-Reaktion mit endo- und exo-Anlagerung.

133
+
∆G0endo
∆G0exo
RKT
∆G
exo-Produkt
endo-ProduktO OO
O
O
O
H
H
O
O
O
H
H
Abbildung 219: Energiediagramm der Diels-Alder-Reaktionen von Cyclopenta-1,3-dien mit
Maleinsäureanhydrid zum exo- und endo-Addukt.
Das exo-Addukt der Diels-Alder-Reaktion ist das thermodynamisch stabilere Produkt,
da es weniger sterische Behinderungen aufweist. Jedoch entsteht bei der Diels-
Alder-Reaktion meist das endo-Addukt, da der Übergangszustand durch die
sekundären Orbitalwechselwirkungen eine niedrigere Aktivierungsenergie benötigt.
⇒ endo-Addukt ist das kinetische Produkt

134
3 Übungsblätter im Sommersemester 2007
Übung zur Vorlesung „Organische Chemie I“ (SS 2007) Prof. Harald Schwalbe / Senada Nozinovic
Blatt 1 1) Geben Sie die natürlichen Aminsäuren mit ihrer Struktur und ihre Abkürzungen an.
Fassen Sie Aminosäuren mit ähnlichen Eigenschaften zusammen.
2) Führen Sie eine Monosubstitution (H X) an Pyridin durch.
- Welche möglichen Produkte erhalten Sie nach dem klassischen Strukturmodell? - Wie viele Produkte können Sie tatsächlich isolieren? - Mit welchen Methoden lassen sich die Produkte voneinander unterscheiden?
3) Zeichnen Sie die Glucose in der Ringform (als Sessel) und der offenkettigen Form.
4) Molekül-Orbital-Theorie
Die Dissoziationsenergie des Stickstoffmonoxids NO beträgt 6.5 eV, die des Nitrosylkations
NO+ hingegen 10.6 eV. Erkläre diesen Unterschied mit Hilfe des einfachen MO-Schemas
für homonukleare zweiatomige Moleküle.

135
Übung zur Vorlesung „Organische Chemie I“ (SS 2007) Prof. Harald Schwalbe / Senada Nozinovic
Blatt 2 1) HMO-Modell
a) Wie sind aromatische und antiaromatische Verbindungen definiert? Was spricht
gegen Antiaromaten?
b) Konstruieren Sie mit Hilfe von Frost-Musulin-Diagrammen die HMOs des Cyclohexa-1,3,5-triens und geben Sie die relativen Energien an.
c) Skizzieren Sie für dieses System die Gestalt der π-Molekülorbitale und zeichnen Sie die
Knotenflächen ein.
d) Konstruieren Sie mit Hilfe von Frost-Musulin-Diagrammen die HMOs des Ok-
1,3,5-triens und geben Sie die relativen Energien an.
2) Amid/Ester-Bindung
a) Skizzieren Sie das MO-Schema für die Amidbindung mit den beteiligten
Orbitalen.
b) Skizzieren Sie das MO-Schema für die Esterbindung mit den beteiligten
Orbitalen.
3) Anomerer Effekt beim Zucker
a) Erklären Sie anhand einer Skizze den anomeren Effekt beim Zucker. b) Wie verhält sich der anomere Effekt für die Reihe X = OH, Cl, F?

136
Übung zur Vorlesung „Organische Chemie I“ (SS 2007) Prof. Harald Schwalbe / Senada Nozinovic
Blatt 3 1) Reaktionen
a) Welche Arten von Bindungsspaltung gibt es und von was hängt es ab? b) Was ist ein Katalysator? Nennen Sie Beispiele. c) Wie sind die Eigenschaften von Gleichgewichts- und Ungleichgewichtsreaktionen? d) Berechnen Sie die Lage des Gleichgewichts
bei 37 °C für ∆G = -1, -2, -3 kcal/mol
2) Einschätzung der Azidität/Basizität (sehr wichtig für Vorhersagen von Reaktionen) a) Geben Sie die (ungefähren) pK
a-Werte für die markierten Protonen an und ordnen Sie die
Verbindungen nach steigender Azidität. H
2O (bzw. H
3O
+) HCl NH
4
+
Butan Ethanol
Essigsäure
Ethylacetat Hexan-1,3-dion Aceton
b) Aus welchen Ausgangsverbindungen sind Amide aufgebaut?
Was passiert : + NH3
c) Ordnen Sie folgende Verbindungen nach steigender Nukleophilie ein. R
- RCO
2
- Cl
- R-O
- NH
3
3) Carbonyl-Chemie
a) Nennen Sie mindestens fünf Verbindungsklassen in denen die Carbonylfunktionalität vorkommt. Ordnen Sie diese nach abnehmender Reaktivität. D.h. nach abnehmender Elektrophilie des Carbonylkohlenstoffs.
b) Welche der Carbonylverbindungen kann mit NaBH
4 zu Alkohol reduziert werden?
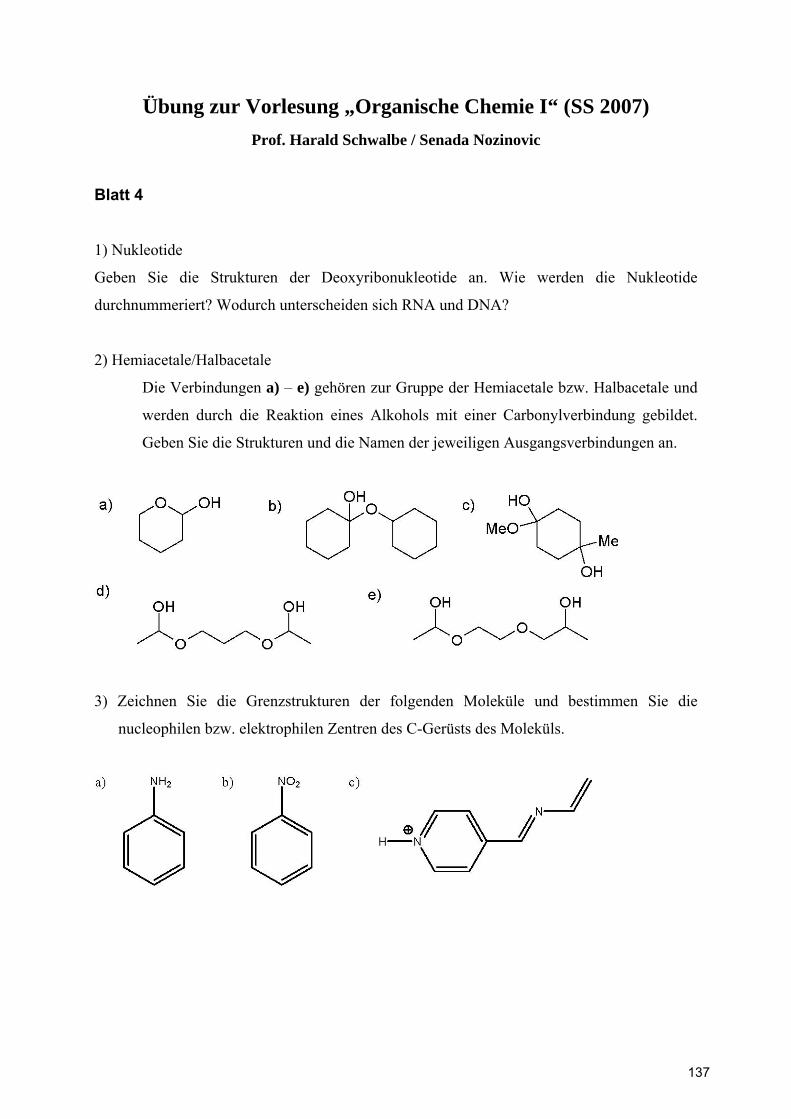
137
Übung zur Vorlesung „Organische Chemie I“ (SS 2007) Prof. Harald Schwalbe / Senada Nozinovic
Blatt 4 1) Nukleotide
Geben Sie die Strukturen der Deoxyribonukleotide an. Wie werden die Nukleotide
durchnummeriert? Wodurch unterscheiden sich RNA und DNA?
2) Hemiacetale/Halbacetale
Die Verbindungen a) – e) gehören zur Gruppe der Hemiacetale bzw. Halbacetale und
werden durch die Reaktion eines Alkohols mit einer Carbonylverbindung gebildet.
Geben Sie die Strukturen und die Namen der jeweiligen Ausgangsverbindungen an.
3) Zeichnen Sie die Grenzstrukturen der folgenden Moleküle und bestimmen Sie die
nucleophilen bzw. elektrophilen Zentren des C-Gerüsts des Moleküls.
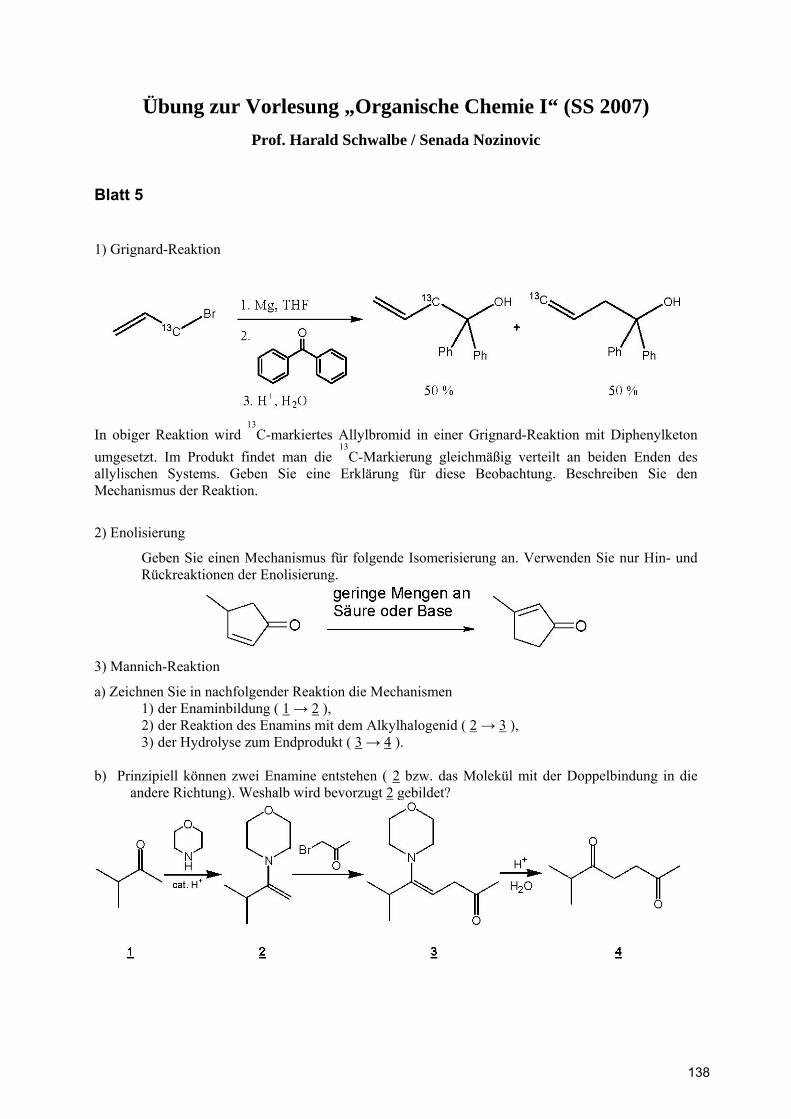
138
Übung zur Vorlesung „Organische Chemie I“ (SS 2007) Prof. Harald Schwalbe / Senada Nozinovic
Blatt 5
1) Grignard-Reaktion
In obiger Reaktion wird 13
C-markiertes Allylbromid in einer Grignard-Reaktion mit Diphenylketon umgesetzt. Im Produkt findet man die
13C-Markierung gleichmäßig verteilt an beiden Enden des
allylischen Systems. Geben Sie eine Erklärung für diese Beobachtung. Beschreiben Sie den Mechanismus der Reaktion.
2) Enolisierung
Geben Sie einen Mechanismus für folgende Isomerisierung an. Verwenden Sie nur Hin- und Rückreaktionen der Enolisierung.
3) Mannich-Reaktion
a) Zeichnen Sie in nachfolgender Reaktion die Mechanismen 1) der Enaminbildung ( 1 → 2 ), 2) der Reaktion des Enamins mit dem Alkylhalogenid ( 2 → 3 ), 3) der Hydrolyse zum Endprodukt ( 3 → 4 ).
b) Prinzipiell können zwei Enamine entstehen ( 2 bzw. das Molekül mit der Doppelbindung in die
andere Richtung). Weshalb wird bevorzugt 2 gebildet?
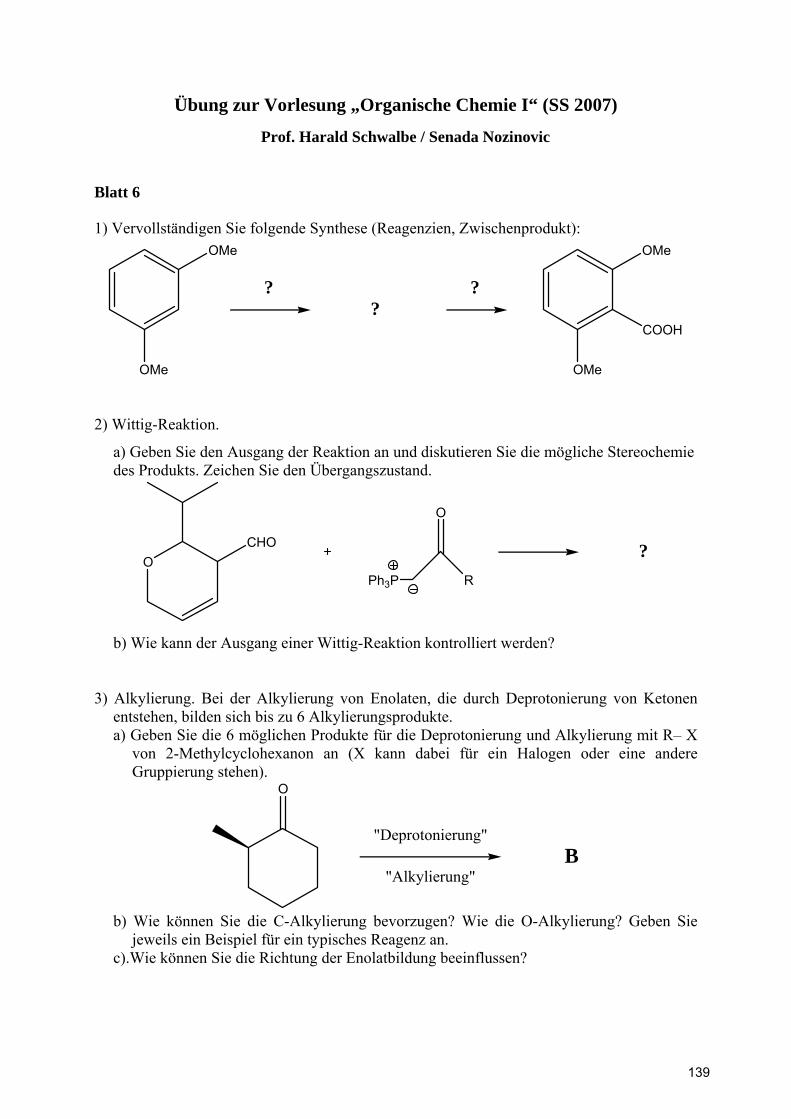
139
Übung zur Vorlesung „Organische Chemie I“ (SS 2007)
Prof. Harald Schwalbe / Senada Nozinovic
Blatt 6
1) Vervollständigen Sie folgende Synthese (Reagenzien, Zwischenprodukt): OMe
OMe OMe
COOH
OMe
??
?
2) Wittig-Reaktion.
a) Geben Sie den Ausgang der Reaktion an und diskutieren Sie die mögliche Stereochemie des Produkts. Zeichen Sie den Übergangszustand.
OCHO
Ph3P R
O
?
b) Wie kann der Ausgang einer Wittig-Reaktion kontrolliert werden?
3) Alkylierung. Bei der Alkylierung von Enolaten, die durch Deprotonierung von Ketonen entstehen, bilden sich bis zu 6 Alkylierungsprodukte. a) Geben Sie die 6 möglichen Produkte für die Deprotonierung und Alkylierung mit R– X
von 2-Methylcyclohexanon an (X kann dabei für ein Halogen oder eine andere Gruppierung stehen).
O
B"Deprotonierung"
"Alkylierung"
b) Wie können Sie die C-Alkylierung bevorzugen? Wie die O-Alkylierung? Geben Sie
jeweils ein Beispiel für ein typisches Reagenz an. c).Wie können Sie die Richtung der Enolatbildung beeinflussen?
Top Related