







Sprachen
Seiten
Rechtliche


Praktikum
Biochemie II
WS 2010/2011
3-wöchiges Praktikum im Hauptstudium der Biochemie
Paul-Ehrlich-Institut Paul-Ehrlich-Str. 51-59
D-63225 Langen Tel.: 06103-77-0
Wichtig: Alle Teilnehmer bitte am Montag, den 22.11.2010 um 8:30 Uhr an der Pforte des Paul-Ehrlich-Institutes melden

BCII am PEI, 22.11.2010-09.12.2010
2
Praktikumsleitung: Prof. Dr. Christian J. Buchholz Tel.: 06103-774011 Email: [email protected] Prof. Dr. Ralf Tönjes Tel.: 06103-774010 Email: [email protected] Beteiligte Arbeitsgruppen Abt. Medizinische Biotechnologie (Leiter Prof. Dr. Cichutek): Fachgebiet 6/5 „Virale Gentransferarzneimnittel“ (Prof. Dr. Buchholz) Fachgebiet 6/2 „Nicht-virale Gentransferarzneimittel“ (Prof. Dr. Schweizer) Fachgebiet 6/3 „Tissue Engineering, Zelltherapeutika“ (Prof. Dr. Schumann) Fachgebiet 6/4 „Xenogene Zelltherapeutika“ (Prof. Dr. Tönjes) Sachgebiet 6/01 „Rekombinante Masernviren und Impfstoffe“ (Dr. Mühlebach) Abt. Immunologie Fachgebiet 3/01 Dr. Kirberg Fachgebiet 3/3 „Morphologie“ (Dr. Boller) Abt. Virologie Sachgebiet 2/01 „Forschung beim Abteilungsleiter“, (Dr. Schwantes/Dr. Suezer) Abt. Veterinärmedizin Fachgebiet 4/1 „Bakterielle Impfstoffe und Immunsera“ (Dr. Lößner) Fachgebiet 4/3 „Virusimpfstoffe II“ (Dr. Bastian) Nachwuchsgruppen NG1 Experimentelle Allergiemodelle (Dr. Toda) NG2 Neue Vakzinierungsstrategien und frühe Immunantworten (Dr. Waibler)

BCII am PEI, 22.11.2010-09.12.2010
3
Themenblöcke des Praktikums:
Exp. 1: Immunologische Nachweismethoden I Gr. 1-3: Labor Kirberg Gr. 4-6: Labor Toda
Exp. 2: Immunologische Nachweismethoden II
Gr. 1-3: Labor Schweizer Gr. 4-6: Labor Waibler
Exp. 3: Virusquantifizierung/siRNA
Gr. 1-3: Labor Toenjes Gr. 4-6: Labor Schwantes/Suezer
Exp. 4: Zellengineering (Bakterien und Säugerzellen)
Gr. 1-3: Labor Lößner/Bastian Gr. 4-6: Labor Schumann
Exp. 5: Viraler Gentransfer
Gr. 1-3: Labor Buchholz Gr. 4-6: Labor Mühlebach
Exp. 6: Elektronenmikroskopie
Gr. 1-6: Labor Boller
Tierhausbesichtigung Dr. Plesker, Dr. Coulibaly, Fr. Völker
Protokolle: Jede Gruppe (also A1, B1, usw.) fertigt zu jedem Experiment ein Protokoll an. Das Protokoll wird bis spätestens 16.12.10 an den jeweiligen Versuchsbetreuer per eMail geschickt. Das Protokoll wird einmal korrigiert (üblicherweise auf Papier). Das Protokoll wird entsprechend den Korrekturen überarbeitet. Danach muss eine akzeptable Endversion vorliegen, die dann abgezeichnet wird. Achtung, nur wenn Ihre erste Protokollversion bereits halbwegs akzeptabel ist, wird eine Korrekturrunde ausreichend sein!
BCII am PEI, 22.11.2010-09.12.2010
4
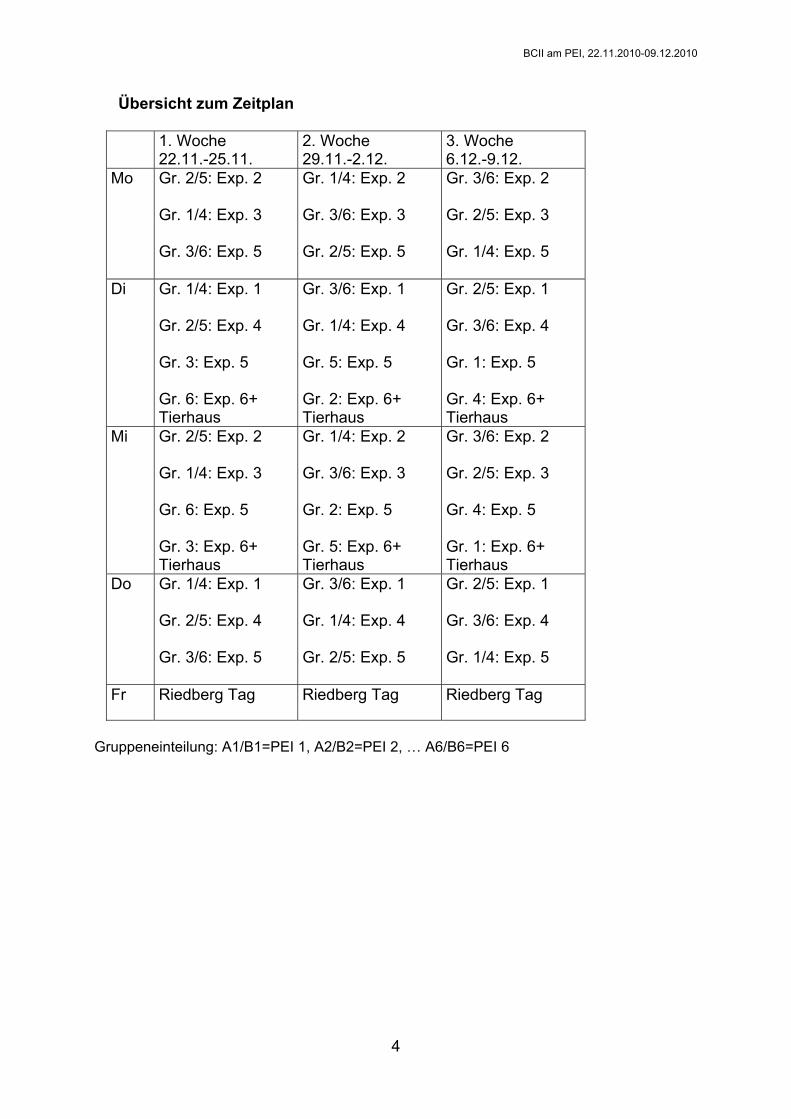
Übersicht zum Zeitplan
1. Woche 22.11.-25.11.
2. Woche 29.11.-2.12.
3. Woche 6.12.-9.12.
Mo Gr. 2/5: Exp. 2 Gr. 1/4: Exp. 3 Gr. 3/6: Exp. 5
Gr. 1/4: Exp. 2 Gr. 3/6: Exp. 3 Gr. 2/5: Exp. 5
Gr. 3/6: Exp. 2 Gr. 2/5: Exp. 3 Gr. 1/4: Exp. 5
Di Gr. 1/4: Exp. 1 Gr. 2/5: Exp. 4 Gr. 3: Exp. 5 Gr. 6: Exp. 6+ Tierhaus
Gr. 3/6: Exp. 1 Gr. 1/4: Exp. 4 Gr. 5: Exp. 5 Gr. 2: Exp. 6+ Tierhaus
Gr. 2/5: Exp. 1 Gr. 3/6: Exp. 4 Gr. 1: Exp. 5 Gr. 4: Exp. 6+ Tierhaus
Mi Gr. 2/5: Exp. 2 Gr. 1/4: Exp. 3 Gr. 6: Exp. 5 Gr. 3: Exp. 6+ Tierhaus
Gr. 1/4: Exp. 2 Gr. 3/6: Exp. 3 Gr. 2: Exp. 5 Gr. 5: Exp. 6+ Tierhaus
Gr. 3/6: Exp. 2 Gr. 2/5: Exp. 3 Gr. 4: Exp. 5 Gr. 1: Exp. 6+ Tierhaus
Do Gr. 1/4: Exp. 1 Gr. 2/5: Exp. 4 Gr. 3/6: Exp. 5
Gr. 3/6: Exp. 1 Gr. 1/4: Exp. 4 Gr. 2/5: Exp. 5
Gr. 2/5: Exp. 1 Gr. 3/6: Exp. 4 Gr. 1/4: Exp. 5
Fr Riedberg Tag Riedberg Tag Riedberg Tag
Gruppeneinteilung: A1/B1=PEI 1, A2/B2=PEI 2, … A6/B6=PEI 6

BCII am PEI, 22.11.2010-09.12.2010
5
Theoretischer Teil BCII Praktikum (jeweils 9.00-10.00, wenn nicht anders angegeben) 22.11.10 Vorbesprechung/Einweisung (8.30 Hörsaal) 23.11.10 Schweizer: Retroviren und retrovirale Vektoren (Hörsaal) 24.11.10 Buchholz/Mühlebach: Oberflächenengineering von viralen Vektoren für Anwendungen in der Molekularen Medizin (Hörsaal) 25.11.10 Toda: Molecular mechanisms of allergic diseases: understanding of signal transduction in inflammatory cells (Hörsaal) 29.11.10 Kirberg: (11.00-12.00 Hörsaal)
30.11.10 Waibler: FACS-Analyse von Immunzellen im Blut (H4 Seminarraum) 01.12.10 Schumann: Transponierbare Elemente im humanen Genom und ihre biomedizinische Anwendung (H4 Seminarraum) 02.12.10 Tönjes: Genome, endogene Retroviren und Suppression der viralen Genexpression durch siRNA (H4 Seminarraum) 06.12.10 Schwantes/Suezer: Virustitration und Pockenvirus-Biologie (Hörsaal) 07.12.10 Boller: Mikroskopie und Elektronenmikroskopie: Ein Überblick über Methoden und Möglichkeiten (Hörsaal) 08.12.10 Lößner/Bastian: Maßgeschneiderte bakterielle Lebendimpfstoffe (Hörsaal) 09.12.10 Abschlußbesprechung (Hörsaal, 15.00-16.00)
BCII am PEI, 22.11.2010-09.12.2010
6
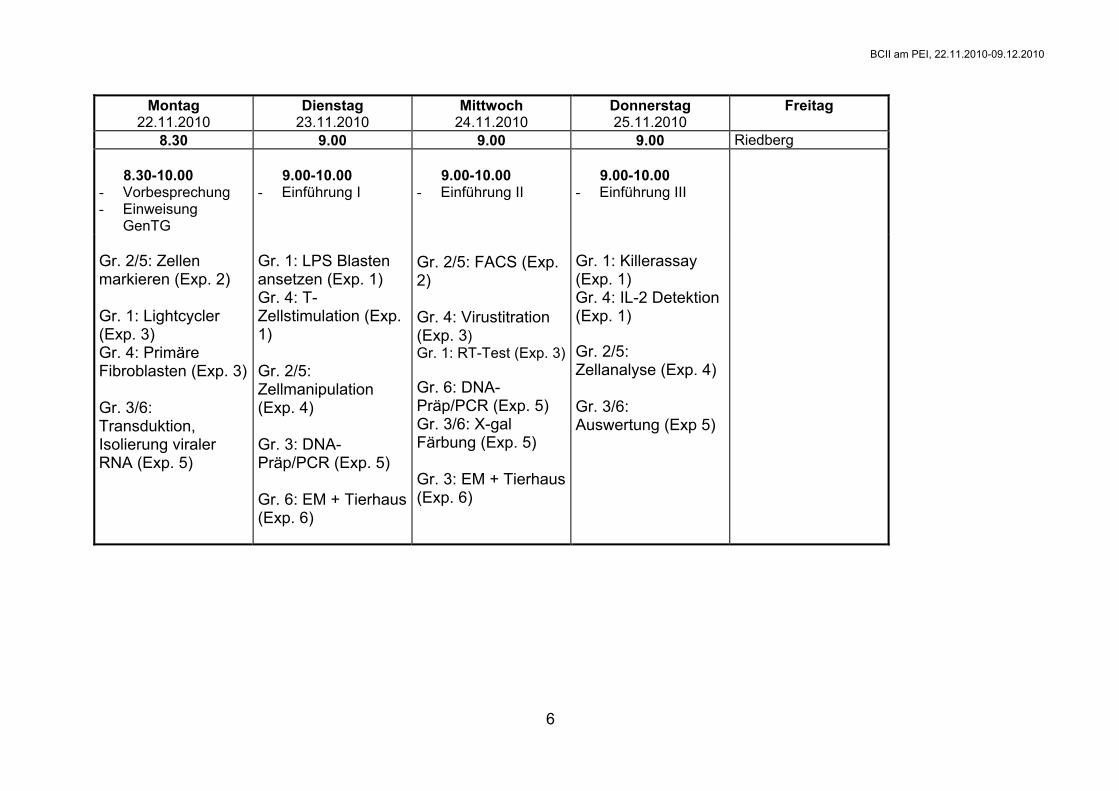
Montag 22.11.2010
Dienstag 23.11.2010
Mittwoch 24.11.2010
Donnerstag 25.11.2010
Freitag
8.30 9.00 9.00 9.00 Riedberg 8.30-10.00 - Vorbesprechung - Einweisung
GenTG
9.00-10.00 - Einführung I
9.00-10.00 - Einführung II
9.00-10.00 - Einführung III
Gr. 2/5: Zellen markieren (Exp. 2) Gr. 1: Lightcycler (Exp. 3) Gr. 4: Primäre Fibroblasten (Exp. 3) Gr. 3/6: Transduktion, Isolierung viraler RNA (Exp. 5)
Gr. 1: LPS Blasten ansetzen (Exp. 1) Gr. 4: T-Zellstimulation (Exp. 1) Gr. 2/5: Zellmanipulation (Exp. 4) Gr. 3: DNA-Präp/PCR (Exp. 5) Gr. 6: EM + Tierhaus (Exp. 6)
Gr. 2/5: FACS (Exp. 2) Gr. 4: Virustitration (Exp. 3) Gr. 1: RT-Test (Exp. 3) Gr. 6: DNA-Präp/PCR (Exp. 5) Gr. 3/6: X-gal Färbung (Exp. 5) Gr. 3: EM + Tierhaus (Exp. 6)
Gr. 1: Killerassay (Exp. 1) Gr. 4: IL-2 Detektion (Exp. 1) Gr. 2/5: Zellanalyse (Exp. 4) Gr. 3/6: Auswertung (Exp 5)
BCII am PEI, 22.11.2010-09.12.2010
7
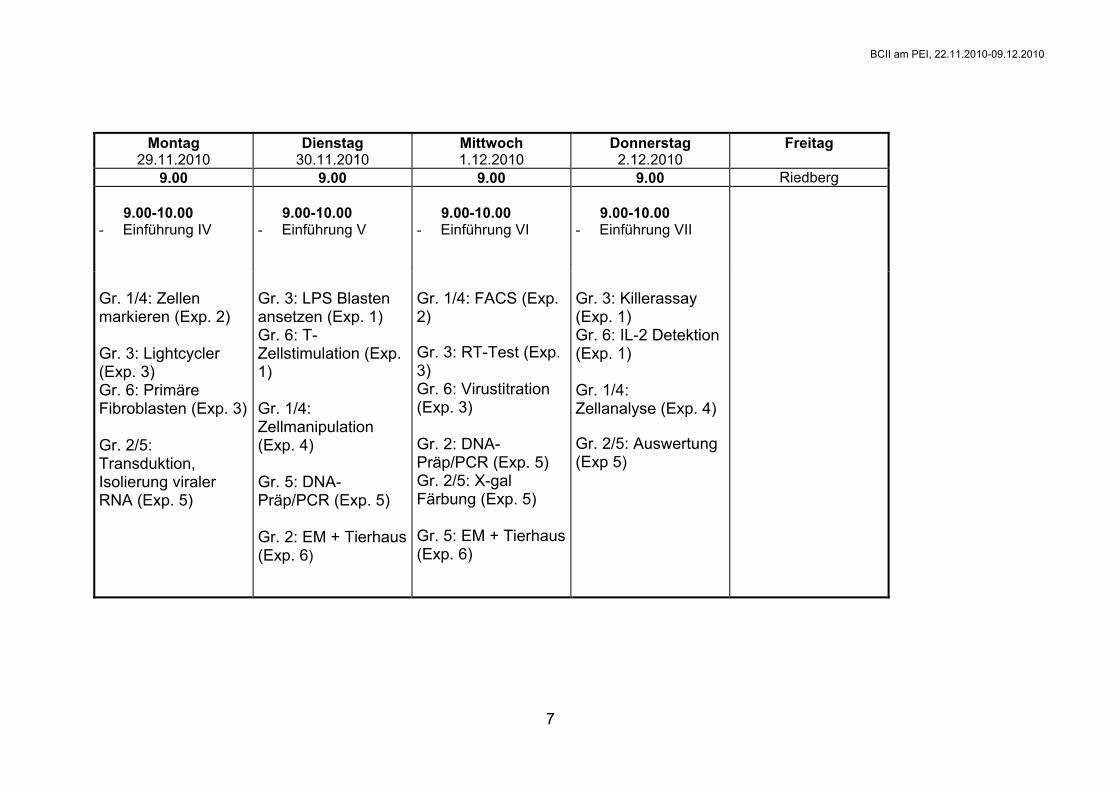
Montag 29.11.2010
Dienstag 30.11.2010
Mittwoch 1.12.2010
Donnerstag 2.12.2010
Freitag
9.00 9.00 9.00 9.00 Riedberg 9.00-10.00 - Einführung IV
9.00-10.00 - Einführung V
9.00-10.00 - Einführung VI
9.00-10.00 - Einführung VII
Gr. 1/4: Zellen markieren (Exp. 2) Gr. 3: Lightcycler (Exp. 3) Gr. 6: Primäre Fibroblasten (Exp. 3) Gr. 2/5: Transduktion, Isolierung viraler RNA (Exp. 5)
Gr. 3: LPS Blasten ansetzen (Exp. 1) Gr. 6: T-Zellstimulation (Exp. 1) Gr. 1/4: Zellmanipulation (Exp. 4) Gr. 5: DNA-Präp/PCR (Exp. 5) Gr. 2: EM + Tierhaus (Exp. 6)
Gr. 1/4: FACS (Exp. 2) Gr. 3: RT-Test (Exp. 3) Gr. 6: Virustitration (Exp. 3) Gr. 2: DNA-Präp/PCR (Exp. 5) Gr. 2/5: X-gal Färbung (Exp. 5) Gr. 5: EM + Tierhaus(Exp. 6)
Gr. 3: Killerassay (Exp. 1) Gr. 6: IL-2 Detektion (Exp. 1) Gr. 1/4: Zellanalyse (Exp. 4) Gr. 2/5: Auswertung (Exp 5)
BCII am PEI, 22.11.2010-09.12.2010
8
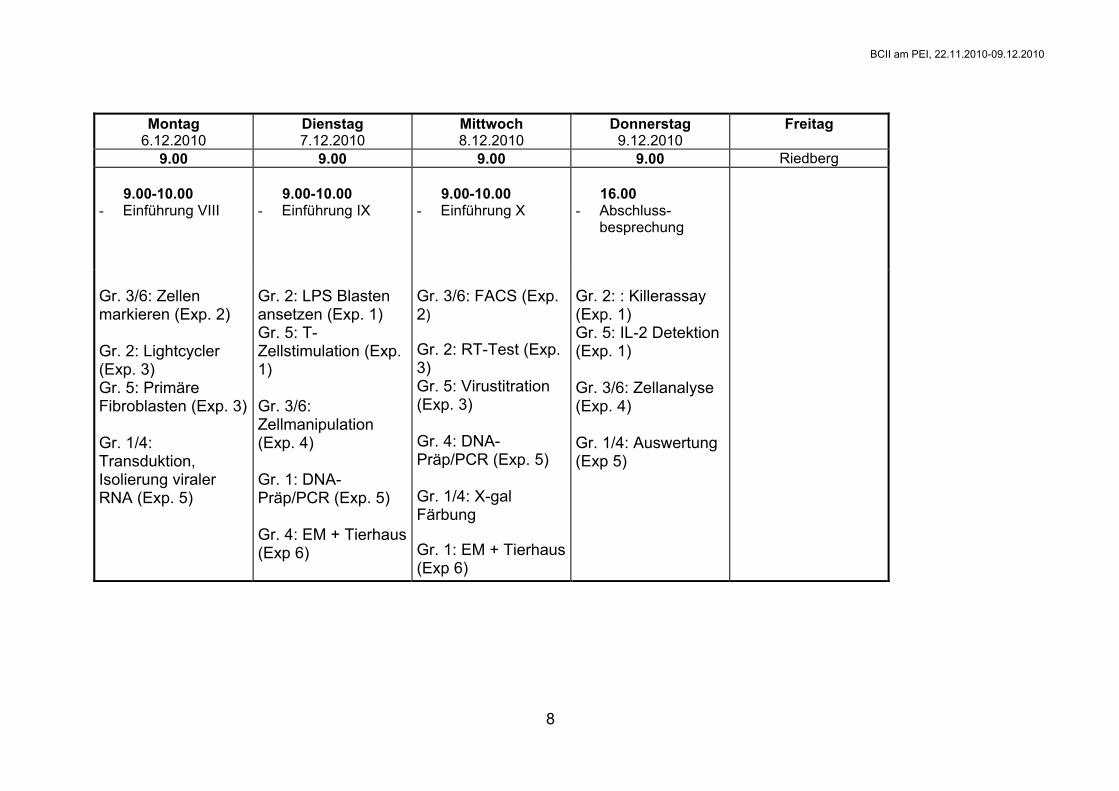
Montag
6.12.2010 Dienstag 7.12.2010
Mittwoch 8.12.2010
Donnerstag 9.12.2010
Freitag
9.00 9.00 9.00 9.00 Riedberg 9.00-10.00 - Einführung VIII
9.00-10.00 - Einführung IX
9.00-10.00 - Einführung X
16.00 - Abschluss-
besprechung
Gr. 3/6: Zellen markieren (Exp. 2) Gr. 2: Lightcycler (Exp. 3) Gr. 5: Primäre Fibroblasten (Exp. 3) Gr. 1/4: Transduktion, Isolierung viraler RNA (Exp. 5)
Gr. 2: LPS Blasten ansetzen (Exp. 1) Gr. 5: T-Zellstimulation (Exp. 1) Gr. 3/6: Zellmanipulation (Exp. 4) Gr. 1: DNA-Präp/PCR (Exp. 5) Gr. 4: EM + Tierhaus (Exp 6)
Gr. 3/6: FACS (Exp. 2) Gr. 2: RT-Test (Exp. 3) Gr. 5: Virustitration (Exp. 3) Gr. 4: DNA-Präp/PCR (Exp. 5) Gr. 1/4: X-gal Färbung Gr. 1: EM + Tierhaus (Exp 6)
Gr. 2: : Killerassay (Exp. 1) Gr. 5: IL-2 Detektion (Exp. 1) Gr. 3/6: Zellanalyse (Exp. 4) Gr. 1/4: Auswertung (Exp 5)

BC II
WS 2010/2011
Experiment 1:
Immunologische Nachweismethoden (I)
Jörg Kirberg Masako Toda Gruppen 1,2,3 Gruppen 4,5,6
[email protected] [email protected]
Paul-Ehrlich-Institut Paul-Ehrlich-Str. 51-59
D - 63225 Langen Tel.: 06103-77-0

BCII am PEI, 22.11.2010-09.12.2010
10
Teil Kirberg : Zytotoxischer T Lymphozyten Test (Killer Assay)
Zytotoxische T Lymphocyten (CTL - cytotoxic T lymphocytes) erkennen Zielzellen
über ihren T Zell Rezeptor (TCR - T cell receptor). Natürliche Liganden sind dabei die
MHC Moleküle (Major Histocompatibility Complex molecules) welche Peptide
präsentieren. Bindet der TCR eines CTL spezifisch an diesen Liganden (MHC +
Peptid), kommt es zur Induktion des Effektor-Mechanismus, bei dem durch Fas-
und/oder Perforin/Granzyme B-vermittelte Apoptose die Zielzelle abstirbt. In vivo sind
zytotoxische T Zellen für die Immunantwort gegenüber intrazellulären Pathogenen,
z.B. Viren, oder auch Transplantaten oder Tumoren bedeutsam.
CTL können gegen viele Antigene erzeugt werden. Dazu gehören z.B.
(I) Alloantigene des Haupthistokompatibilitätskomplexes (MHC),
(II) Antigene welche auf Polymorphismen zwischen verschiedenen Individuen
zurückzuführen sind (Minor Histocompatibility Antigens),
oder
(III) Antigene welche auf syngenen Zellen in Folge von Infektion, Mutation ('fremd’-
Peptide) oder nach Derivatisierung mit Hapten, präsentiert werden.
CTL entstehen aus ihren ruhenden, nicht zytolytisch aktiven Vorläufern (pCTL -
precursorCTL) nach Stimulierung ihres TCRs innerhalb weniger Tage. In vivo z.B. in
Folge einer Immunisierung oder in vitro während der Co-Kultur mit Zellen, die den
spezifischen Peptid/MHC-Komplex präsentieren.
Ohne vorhergehende in vivo Immunisierung lassen sich in der Regel in vitro nur CTL
gegen MHC-Alloantigene oder gegen bestimmte Haptene (z.B. Trinitrophenol, TNP)
die an syngenen Zellen gekoppelt wurden erzeugen. Vermutlich liegt dies an der
hohen pCTL Frequenz welche spezifisch für diese Antigene sind.
Nach in vivo Immunisierung und in vitro Co-Kultur lassen sich CTL auch gegen
andere Antigene nachweisen (z.B. virale Peptide, Minor Histokompatibility Antigens).
Ex vivo können CTL kurz nach dem Höhepunkt einer viralen Infektion nachgewiesen
werden.
Prinzipiell können alle Zelltypen als Zielzellen (in der Folge Targets genannt) für CTL
(Effectors) verwendet werden. Experimentell am einfachsten zu beherrschen sind
Lymphoblasten, Zellen aus Zellkultur oder Tumorzellen.
Zwei Verfahren werden oft für die Messung der Target-Zell-Zytolyse angewendet:
51Cr-release oder JAM (just another method). Bei dem 51Cr-release Test werden die

BCII am PEI, 22.11.2010-09.12.2010
11
Target Zellen mit radioaktivem 51Cr (in Form von Na251CrO4) markiert und die CTL
vermittelte Zytolyse der Targets führt zur Freisetzung von 51Cr in den Überstand:
CrO42- CrO4
2-
PM
Cr O72-
PM
2
Bei dem JAM wird die Target Zell DNA mit radioaktivem 3H markiert indem
3H-Thymidin zu Zellen gegeben wird welche proliferieren. Nach CTL vermittelter
Zytolyse wird gemessen ob die DNA der Target Zellen in Folge von Apoptose
degradiert wurde. Dazu werden die Zellen mit Wasser durch einen Glasfiberfilter
gewaschen. Hochmolekulare genomische DNA bindet an den Filter während DNA
Fragmente nicht zurückgehalten werden.
Anschließend wird die erhaltene radioaktive Menge ausgezählt und eine CTL
vermittelte Zytolyse/Apoptosis in Bezug zu Kontrollen gesetzt, welche die spontane
und maximal mögliche Zytolyse/Apoptosis ermittelt.
Kursprogramm
Im Kurs soll ein Killer Assay gegen allogene MHC Moleküle durchgeführt werden.
Dazu werden allo-MHC spezifische CTL aus pCTL (Responders) während einer Co-
Kultur mit bestrahlten allogenen Zellen (Stimulators) innerhalb von fünf bis sieben
Tagen erzeugt. Diese Kulturen werden auch als primäre gemischte Lymphozyten
Kultur bezeichnet (1°-MLR - mixed lymphozyte reaction).
Als Quelle für die verschiedenen Lymphozyten dienen Milzzellen von Mäusen mit
unterschiedlichem MHC Haplotyp. Dabei sind C57BL/6 (abgekürzt B6) Mäuse vom
H-2b MHC Haplotyp und BALB/c (B/c) Mäuse vom H-2d MHC Haplotyp.
Lymphozyten aus B6 Mäusen exprimieren daher auf ihrer Zelloberfläche MHC
Moleküle des H-2b Allels, während solche aus BALB/c (B/c) Mäusen MHC Moleküle
des H-2d Allels aufweisen.
Als Target Zellen im Killer Assay (s.u.) dienen aus den beiden Mausstämmen
abstammende Tumorzellinien oder B Zell Lymphoblasten. Letztere werden durch
Stimulation mit LPS (Lipopolysaccarid) für 48 Stunden erhalten (LPS Blasten).
Der Killer Assay wird (siehe Protokoll) hier ohne radioaktive Nuklide durchgeführt.
Stattdessen werden die Target Zellen (LPS Blasten oder Tumorzellen) mit einem

BCII am PEI, 22.11.2010-09.12.2010
12
fluoreszierenden Farbstoff [CFDA(SE)] kovalent markiert. Dies ermöglicht die
Unterscheidung zu den Zellen aus der 1°-MLR; vor allem den CTLs.
Die durch CTL erfolgte Zytolyse wird anschließend im Durchflußcytometer (FACS -
fluorescent activated cell sorter) ermittelt. Sie ist zum einen erkennbar an der
Abnahme von grossen Lymphoblasten relativ zu einer Zunahme von kleinen Zellen
(Apoptotic bodies) innerhalb der fluoreszierenden Target Zell Population. Zum
anderen kann die Zytolyse anhand der Zunahme von fluoreszierenden Target Zellen
mit perforierten Membranen bestimmt werden. Letzteres kann mittels einer Färbung
mit PI (propidium iodide) erfolgen: Nach dem die Zellmembranen bei der Apoptose
durchlässig wurden kann PI in den Zellkern eindringen. PI interkaliert dort in die DNA
und weisst in dieser Form eine deutlich erhöhte Fluoreszens auf.

BCII am PEI, 22.11.2010-09.12.2010
13
1. Arbeits-Tag (Nur Betreuer): Ansetzen der 1°-MLR
Freitag, 20.11.2010 / 27.11.2010 / 4.12.2010
Benötigte Materialien
Sterilbank (Flowhood)
Zentrifuge (bei 5°C)
Responder bzw. Stimulator Zellen (hier Milzzellen aus C57BL/6 bzw. BALB/c
Mäusen; Stimulator Zellen sind zu bestrahlen (137Cs Quelle oder
Röntgenstrahlung; 22 Gy). An diesem Tag evtl. Tumorzellinien auftauen.
CO2 zum Töten der Spendertiere (bzw. per Genickbruch durch Betreuer)
FACS-Puffer (PBS mit 2 % FCS (fetal calf serum)), steril
ACT Puffer (Ammoniumchlorid-Tris Puffer), steril
Trypan Blau Lösung, 0.4 % (w/v)
Medium (SF-IMDM mit % FCS), steril
Plastik Pipetten (10 ml, 5 ml, 2 ml, 1 ml), steril
Pipetten Spitzen, Gelb und Blau, steril
Eppendorf Tubes (1.5 ml) für Verdünnungen bei der Zellzahlbestimmung
14 ml Falcon Röhrchen mit Deckel, steril
6 ml Falcon Röhrchen mit Deckel, steril
2 ml Spritzen, Plastik, steril
Petrischalen (6 cm Ø), steril
T-25 Gewebekulturflaschen mit Normal- oder Filterdeckel, steril
Nylon Netze (100 m, 'grob'), steril
Spritzen mit aufgesetzten 40 m ('fein') Nylon Netzen, steril
Pasteur Pipetten aus Glas, kurz, steril
70 % Ethanol in H20 zum Desinfizieren in Spritzflasche
> 95 % Ethanol zum Abflammen
Instrumente (Schere, Pinzette)
Bechergläser mit 95 % Ethanol für Instrumente
Zählkammer (Neubauer/Bürki)
Gestelle für Röhrchen

BCII am PEI, 22.11.2010-09.12.2010
14
Eisbox
Accuboy oder Pipettierball
Stifte
Pipetten (P20, P200, P1.000)
Pipettierball für Pasteur Pipetten
Versuchsprotokoll
Die Spendertiere werden von den Betreuern mit CO2 getötet. Jede Gruppe erhält
mindestens ein Tier, also eine C57BL/6 (schwarze Fellfarbe) und/oder eine
BALB/c (Albino, d.h. weisse Fellfarbe) Maus.
Alle Arbeitsschritte werden steril ausgeführt !
Entnahme der Milz
Die toten Tiere werden äußerlich mit 70 % Ethanol desinfiziert. Auf der rechten
Flanke liegend erfolgt median ein Einschnitt hinter dem Brustkorb durch die
angehobene Haut. Brustkorb und Peritoneum werden durch das
Auseinanderziehen der Hautlappen exponiert.
Die Instrumente werden kurz (!) abgeflammt. Caudal zu der durch das Peritoneum
sichtbaren Milz wird das Peritoneum geöffnet. Die Milz wird entnommen und in
eine Petrischale mit ca. 10 ml FACS-Puffer gegeben.
Herstellen von Einzelzellsuspension und RBC (red blood cell) - Lyse
Die Milz wird zwischen zwei Nylon Netzen in den Deckel der Petrischale, mit
wenig FACS-Puffer, plaziert. Mit Hilfe des Kolbens einer 2 ml Spritze oder einer
gebogenen Pinzette wird die Milz langsam ausgequetscht. Dazu wird die Milz mit
einer zweiten Pinzette zwischen den beiden Nylon Netzen fixiert.
Die Zellsuspension wird unter mehrmaligem Auswaschen (Pasteur Pipette) der
Nylonnetze in ein 14 ml Falcon Röhrchen übertragen.
Die Zellen werden pelletiert (Zentrifuge 10 min., 1.200 rpm (ca. 300xg))
Die Zellen werden in ca. 2.0 ml ACT-Puffer resuspendiert (Pasteur Pipette) und
sofort für 1 min. 30 sec. bei 37°C inkubiert.

BCII am PEI, 22.11.2010-09.12.2010
15
Sofort danach wird die RBC-Lyse durch Auffüllen des Röhrchens mit FACS-Puffer
gestoppt. Den Deckel des Röhrchens vollständig schliessen. 1-2 x Mischen durch
invertieren.
Die Zellen werden pelletiert (Zentrifuge 10 min., 1.200 rpm (ca. 300xg), 5°C)
Die Zellen werden in 2.0 ml Medium resuspendiert (nicht exzessiv schäumen !).
Die Zellsuspension wird durch filtration (40 m Nylon Netz auf Spritzen) von
Klumpen befreit.
Die Röhrchen auf Eis und geschlossen halten um CO2 Verlust zu minimieren.
Zellzahl Bestimmung
Vorverdünnung ca. 1 : 10 (je nach Zelldichte)
(Dabei besteht 1 : 10 aus 1 Volumen Zellen + 9 Volumen FACS-Puffer !)
Die Vorverdünnung 1 : 2 mit Trypan Blau verdünnen
(d.h. 1 Volumen vor-verdünnte Zellen + 1 Volumen Trypan Blau)
Auf die Zählkammer geben und lebende Zellen bestimmen
Trypan Blau färbt tote Zellen - nicht aber Erythrozyten
Erythrozyten verhalten sich doppelbrechend (charakteristische Ringe)
Für eine Neubauer Kammer gilt:
Zellzahl/ml = (16 große Quadrate) x 2 (Trypan Blau Verd.) x Vor-verd. x 104
C57BL/6: x 106 Zellen/ml (2.0 ml) --> x 106 Zellen total
BALB/c: x 106 Zellen/ml (2.0 ml) --> x 106 Zellen total
Überlegung wieviel Zellen wie verwendet werden
Von jeder Gruppe sollten folgende Kulturen angesetzt werden:
a) 10 x 106 Zellen C57BL/6 ---> 10 x 106 X-bestrahlte BALB/c Zellen
b) 10 x 106 Zellen BALB/c ---> 10 x 106 X-bestrahlte C57BL/6 Zellen
d.h., von jeder Milz werden mindestens 20 x 106 Zellen benötigt, wobei eine Hälfte
davon (10 x 106 Zellen) bestrahlt werden soll, die andere Hälfte nicht.
Falls weniger Zellen vorhanden sind bzw. nur Zellen eines Mausstamms, bitte bei
den KurskollegInnen nachfragen. Bei zu wenigen Zellen nur eine der Kulturen

BCII am PEI, 22.11.2010-09.12.2010
16
nach Wahl ansetzen. Falls noch mehr Zellen benötigt werden bitte bei den
Betreuern nachfragen.
Zum Bestrahlen die Zellen bei den Betreuern abgeben.
Bitte in ca. 3 ml Volumen in 6 ml Falcon Röhrchen. Die Zellen werden mit
2.200 rad (22 Gy) in einer 137Cs Quelle bestrahlt.
Ansetzen der 1°-MLR
10 x 106 Responders werden mit 10 x 106 X-bestrahlten Stimulators in einem
Gesamtvolumen von 8 ml Medium kultiviert.
Zur Kultivation werden die Zellen in senkrecht stehende T-25
Gewebekulturflaschen gegeben und aufrecht bei 37°C, 5 % CO2, für 5 bis 7 Tage
kultiviert. Der Deckel ist im Inkubator nicht ganz zu schliessen, es sei denn es
handelt sich um Kulturflaschen mit Filterdeckel.

BCII am PEI, 22.11.2010-09.12.2010
17
2. Arbeits-Tag (1. Arbeits-Tag für Studenten): LPS Blasten ansetzen
Dienstag, 23.11.2010 / 30.11.2010 / 7.12.2010
Benötigte Materialien (zusätzlich zu denen vom 1. Tag)
LPS Stocklösung (5 mg/ml); 1 : 250 verdünnt zu gebrauchen (= 20 g/ml)
T-75 Gewebekulturflaschen mit Filterdeckel, steril
Versuchsprotokoll
Jede Gruppe erhält mindestens eine C57BL/6 (schwarze Fellfarbe) und/oder eine
BALB/c (Albino, d.h. weisse Fellfarbe) Maus.
Alle Arbeitsschritte werden steril ausgeführt !
Beschreibung der folgenden Arbeitsschritte siehe 1. Arbeits-Tag:
Entnahme der Milz
Herstellen von Einzelzellsuspension und RBC (red blood cell) - Lyse
Zellzahl Bestimmung
C57BL/6: x 106 Zellen/ml (2.0 ml) --> x 106 Zellen total
BALB/c: x 106 Zellen/ml (2.0 ml) --> x 106 Zellen total
Überlegung wieviel Zellen wie verwendet werden
Optimal sollten folgende Kulturen angesetzt werden:
a) 30 x 106 Zellen aus C57BL/6, +LPS
b) 30 x 106 Zellen aus BALB/c, +LPS
Beide Kulturen werden benötigt.
Ansetzen der LPS Blasten
30 x 106 Milz Zellen werden in 30 ml Medium mit 120 l LPS (5 mg/ml) kultiviert.
Zur Kultivierung werden die Zellen in T-75 Gewebekulturflaschen gegeben und bei
37°C, 5 % CO2, für 2 Tage liegend kultiviert. Deckel leicht öffnen bzw. Flasche mit
Filterdeckel verwenden.

BCII am PEI, 22.11.2010-09.12.2010
18
Ansetzen der Tumorzellinien (falls geplant)
Je nach Bedarf an Zellen werden die Zellinien gesplittet und Kulturen in T-75
Gewebekulturflaschen angesetzt.
Zentrifugen auf Raumtemperatur für den folgenden Versuchstag stellen!
3. Arbeits-Tag (2. Arbeits-Tag für Studenten): Killer Assay
[es wird ein längerer Tag !]
Donnerstag, 25.11.2010 / 2.12.2010 / 9.12.2010
Benötigte Materialien (zusätzlich zu denen vom 1. bzw. 2. Arbeits-Tag)
Zentrifuge auf Raumtemperatur am Morgen
Zentrifuge für 96-well Platten
FACS (FACScan bzw. FACSCalibur)
Vortex
Repetitive Pipette
[Eppendorf Mulipette, 1-5 (alt) oder 0.5 - 9 Einheiten (neues Modell)]
Eisbehälter
'Rad' für 6 ml Falcon Röhrchen (oder 8 min. -langsam- per Hand bewegen)
evtl. Tumorzellinien (falls nicht am 2. Tag vorbereitet).
Ficoll Paque (bei Raumtemperatur)
PBS
5(6)-CFDA, SE (Molecular Probes, C-1157) = CFSE , 10 mM in DMSO
Medium + 1 % Saponin (w/v)
[evtl. PI Stocklösung (0.5 mg/ml in H2O)]
14 ml Falcon Röhrchen, Polypropylene
14 ml Konisches (V-Bottom) Polypropylene Röhrchen
Spitzen für Repetitive Pipette (0.5 ml, 2.5 ml)
96-well Platten, U-Boden, mit Deckel
Micron Röhrchen für FACS Analyse
Gestell für Micron Röhrchen

BCII am PEI, 22.11.2010-09.12.2010
19
Pasteur Pipetten aus Glas, lang
Schema für 96-well Platten
Versuchsprotokoll
Isolierung der LPS Blasten über Ficoll Dichtegradienten
Die Kulturen der LPS Blasten von C57BL/6 bzw. BALB/c Mäusen vom 2. Tag
werden geerntet. Jeweils max. 10 ml einer Kultur wird in ein 14 ml Rundboden-
Falcon Röhrchen gegeben – d.h. drei Röhrchen pro Kultur.
In jedes der Röhrchen wird eine lange Pasteur Pipette gegeben. Durch die
Pasteur Pipette werden ca. 3 ml Ficoll unterschichtet.
Zentrifugation: Bei langsamem Anlauf, langsamen Abbremsen !
40 min. 1.600 rpm, 21°C
Die LPS Blasten befinden sich nach dem Zentrifugieren an der Interphase
zwischen Ficoll und Medium. Zum Ernten erst einen Teil des oben befindlichen
Mediums mit einer Pasteur Pipette entfernen. Dann die Blasten mit der Pasteur
Pipette in ein frisches 14 ml Falcon Röhrchen übertragen ohne zu viel von dem
Ficoll mit zu nehmen. Zellen aus gleicher Kultur werden dabei gepoolt. Alle
Schritte nun auf Eis bzw. bei 4°C.
Die LPS Blasten bzw. Tumorzellen 2 x mit PBS waschen (Zentrifugation 10 min.,
1.200 rpm, 4°C).
In 1.2 ml PBS resuspendieren. 1.0 ml in ein 6 ml Falcon Röhrchen geben. Die
übrigen 0.2 ml auf Eis aufbewahren (nicht färben) und etwas Medium (ca. 0.2 ml)
zugeben.
Evtl. die Tumorzellinien ernten. Evtl. teilen sich dazu je zwei Gruppen eine 20 ml
Kultur von P815 oder EL-4 Tumorzellen.
Färbung der LPS Blasten bzw. Tumorzellen mit CFSE
Die verdünnte Lösung des CFSE Farbstoffes ist nur sehr kurz haltbar, daher erst
alles für die folgenden Schritte vorbereiten.
Der CFSE Farbstoff (10 mM Stocklösung) wird 1 : 2.500 in PBS vor-verdünnt (d.h.
0.8 l in 2 ml).

BCII am PEI, 22.11.2010-09.12.2010
20
Zu 1 ml LPS Blasten bzw. Tumorzellen in 6 ml Falcon Röhrchen werden dann
sofort 1 ml frisch vor-verdünnte Farbstofflösung unter ständigem Mischen
(langsam eingestellter Vortex) gegeben.
Färbereaktion: 8 min., bei Raumtemperatur, unter ständigem Mischen (z.B. auf
einem 'Rad' oder -langsam- per Hand).
Nach der Färbung werden die LPS Blasten bzw. Tumorzellen mindestens 2 x mit
FACS-Puffer und einmal mit Medium gewaschen.
[Alternativ: Zentrifugieren durch einen Gradienten. Zellen dazu in PBS oder FACS-
Puffer resuspendieren, mit Medium unterschichten, zentrifugieren (langsamer
Anlauf, langsames Abbremsen), Überstand absaugen, in kleinem Volumen
resuspendieren und in ein frische Röhrchen überführen].
Die gefärbten LPS Blasten bzw. Tumorzellen zuletzt in 1 ml Medium
resuspendieren (ohne Schaum !) und auf Eis aufbewahren.
(Eine Hälfte der Gruppe soll nun beginnen die Effektor-CTLs zu ernten und diese
für den CTL Assay in 96-well Platten auszuplattieren, siehe Pipettierreihenfolge.
Die LPS Blasten bzw. Tumorzellen als Targets werden zuletzt zugegeben. Bitte
sich in das Pipettierschema hineindenken bevor die Betreuer gefragt werden!).
Zelldichte der LPS Blasten bzw. Tumorzellen bestimmen
C57BL/6: x 106 Zellen/ml (1 ml) --> x 106 Zellen total
BALB/c: x 106 Zellen/ml (1 ml) --> x 106 Zellen total
P815: x 106 Zellen/ml (1 ml) --> x 106 Zellen total
EL-4: x 106 Zellen/ml (1 ml) --> x 106 Zellen total
Die LPS Blasten bzw. Tumorzellen auf 2 x 105 Zellen/ml einstellen. Bei 100 l pro
Vertiefung entspricht dies 20.000 LPS Blasten bzw. Tumorzellen pro Vertiefung.
Es werden ca. 2 ml benötigt (also 4 x 105 Zellen - das sollte möglich sein!).

BCII am PEI, 22.11.2010-09.12.2010
21
Ernte der Effektor-CTLs (Killers)
Mit einer Pasteur Pipette werden die Zellen der 1°-MLR Kulturen resuspendiert
und jeweils eine Kultur in ein 14 ml Konisches (V-Bottom) Polypropylen Röhrchen
übertragen.
Die Zellen werden pelletiert (Zentrifugation 10 min., 1.200 rpm, 4°C) und in 1 ml
Medium resuspendiert. Sollten mehrere Kulturen angesetzt worden sein bitte
Zellen gleicher Kultur (B6 -> B/c irrad. oder B/c -> B6 irrad.) zusammengegeben
und dann entsprechend höher verdünnen.
Jede Gruppe benötigt 600 l von jeder der beiden (B6 -> B/c irrad. und
B/c -> B6 irrad.) Kulturen. Evtl. Reste an andere Gruppen verteilen.
Ausplattieren der Effektor-CTLs
In der unten angegebenen Reihenfolge die Zellen entsprechend dem
Pipettierschema ausplattieren. Bitte sich in das Pipettierschema hineindenken
bevor die Betreuer gefragt werden!
Das Pipettieren sollte zügig erfolgen um zu vermeiden das der pH-Wert des
Mediums durch CO2-Verlust zu stark ansteigt ("Pink“-colour).
Prinzipiell wird der CTL Assay in 96-well Platten, mit U-Boden, durchgeführt.
Die einzelnen Bestimmungen erfolgen einfach. Bei Konstanter LPS Target- bzw.
Tumorzellen-Anzahl wird eine Effektor-CTL Verdünnungsreihe in Schritten von
1 : 3 Verdünnungen durchgeführt.
Als positiv bzw. negativ Kontrollen dienen Vertiefungen, in die wohl Target Zellen,
aber keine Effektor-CTL zugegeben werden. Neben den Target Zellen enthalten
diese nur Medium (zur Bestimmung der spontanen Zytolyse) bzw. Medium mit
Saponin (ein Detergens welches zur Lyse führt; zur Bestimmung der maximalen
Zytolyse). Dies erfolgt jeweils für B6 bzw. B/c LPS Targets bzw. Tumorzellen
getrennt.
Pipettierreihenfolge:
1). In die Vertiefungen mit der höchsten Effektor-CTL-Anzahl werden 150 l der
geernteten Effektor-CTLs vorgelegt.
Siehe Pipettierschema: 1 (Einfache Bestimmung) x 4 (B6 bzw. B/c LPS Targets,
zwei Tumorzellinien) x 150 l = 600 l.

BCII am PEI, 22.11.2010-09.12.2010
22
2). Alle anderen Vertiefungen der Effektor-CTL Verdünnungsreihe erhalten 100 l
Medium. Mit einer Pipette wird dann die 1 in 3 Verdünnungsreihe pipettiert (dazu
werden 50 l fortlaufend verdünnt und weitergeführt bzw. am Ende der
Verdünnungsreihe verworfen).
3). negativ Kontrolle: einige Vertiefungen erhalten 100 l Medium (Spontaner
Zelltot, d.h. ohne Effktor-CTLs). Evtl. in zweifacher Anzahl.
4). positiv Kontrolle: einige Vertiefungen erhalten 100 l Medium + 1 % Saponin
(maximaler Zelltot durch Saponin, d.h. ohne Effektor-CTLs).
Sollten die Taget Zellen noch nicht bereit stehen ist die 96-well Platte mit den
CTLs im Inkubator (37°C, 5 % CO2) aufzubewahren.
5). Zuletzt werden zu allen Vertiefungen 100 l der verschiedenen Targets
(B6 bzw. B/c LPS Targets, evtl. von den zwei Tumorzellinien) zugegeben.
6). In die verbleibenden Vertiefungen werden zum einen die noch vorhandenen
LPS Blasten bzw. Tumorzellen gegeben, und in andere Vertiefungen evtl.
Übriggebliebene Reste der Effektor-CTLs. Diese Vertiefungen dienen dazu, nach
der Inkubation genügend Zellen zu erhalten mit deren Hilfe der FACS eingestellt
werden kann.
Schließlich wird die 96-well Platte (mit aufgesetztem Deckel) 5 min. 500 rpm
zentrifugiert. Dann erfolgt die Inkubation bei 37°C, 5 % CO2, für 4 Stunden.
Bestimmung der Zytolyse von fluoreszensmarkierten Blasten
Nach Inkubation werden die Zellen in der 96-well Platte 1 x mit FACS-Puffer
gewaschen (zentrifugation 2 min., 2.000 rpm, Überstand wird durch beherztes
einmaliges Auskippen ('flicken') entfernt, direkt danach weiterhin 'auf dem Kopf'
auf Zellstoff aufsetzen, Zellen vor Zugabe von FACS-Puffer auf dem Vortex
resuspendieren).
Zuletzt die Zellen in 50 l FACS-Puffer resuspendieren und in kalte Micron
Röhrchen überführen. Auf Eis aufbewahren.
Von den Vertiefungen, welche für die FACS Einstellung nur LPS Targets bzw.
Tumorzellen oder nur Effektor-CTLs enthielten, jeweils die Zellen gleicher Art in

BCII am PEI, 22.11.2010-09.12.2010
23
ein Micron Röhrchen zusammen geben. Ebenso von den übrig gelassenen nicht
gefärbten LPS Blasten jeweils ein Micron Röhrchen bereithalten (Kontrolle der
Färbung).
Anschließend mit einem der Betreuer den FACS aufsuchen.
Da normalerweise gut zu sehen ist wie die Zellgrösse nach Zytolyse abnimmt ist
vorläufig eine Zugabe von PI nicht vorgesehen. Falls dies doch gewünscht wird
wie folgt vorgehen: PI erst kurz vor Analyse der Zellen auf dem FACS zugeben.
Dazu wird die PI Stocklösung vor-verdünnt, so das nach Zugabe von z.B. 10 l
eine PI Konzentration von 2.5 g/ml in der Probe resultiert.
Effektor-CTL zu Target (LPS Blasten) Verhältnis (E:T Ratio)
Für die einzelnen Verdünnungen wird dieses Verhältnis errechnet. Dabei wird bei
der Berechnung der Anzahl von Effektor-CTLs pro Vertiefung die Anzahl Zellen,
welche zu Beginn in die 1°-MLR eingesetzt wurden, zugrunde gelegt, unter
Berücksichtigung der verschiedenen Verdünnungs- und Konzentrierungsschritte.
Die Anzahl der Targets pro Vertiefung ergibt sich zu 20.000 (s.o.).
Spezifische Lyse
Im Fall der Messung der Zytolyse per FACS ergibt sich z.B. folgende Möglichkeit:
% specific blast lysis experimental blast % - spontaneous -
x 100
experimental PI %+ -- spontaneous PI %
x 100% specific lysis (PI )
minnimal (saponin) blast %
blast %
spontaneous blast %
+spontaneous PI %
+
maximal (saponin) PI %+ +
Alternativ kann man für jede Messung das Verhältnis aus 'lebende Blasten’ zu
’tote Zellen’ bestimmen und analog in obige Formel setzen (s.u.).
Bei dieser Art der experimentellen Ausführung sollte bei den Ergebnissen der
Prozentsatz an Blasten ohne Behandlung (spontaneous blast % bzw. das
Verhältnis 'lebende Blasten’ zu ’tote Zellen’) bzw. der Prozentsatz von spontan PI+
Zellen als 'Gütewert' mit angegeben werden.

BCII am PEI, 22.11.2010-09.12.2010
24
Die spezifische Lyse wird bei JAM bzw. 51Cr-release wie folgt berechnet:
Um Ergebnisse dieser Assays interpretieren zu können ist es üblich neben der
Standart-Abweichung der experimentellen Ergebnisse folgende Daten mit
anzugeben: 3H-Thymidin Einbau (JAM), spontane 51Cr Freisetzung als
Prozentsatz der total möglichen 51Cr Freisetzung (51Cr-release).
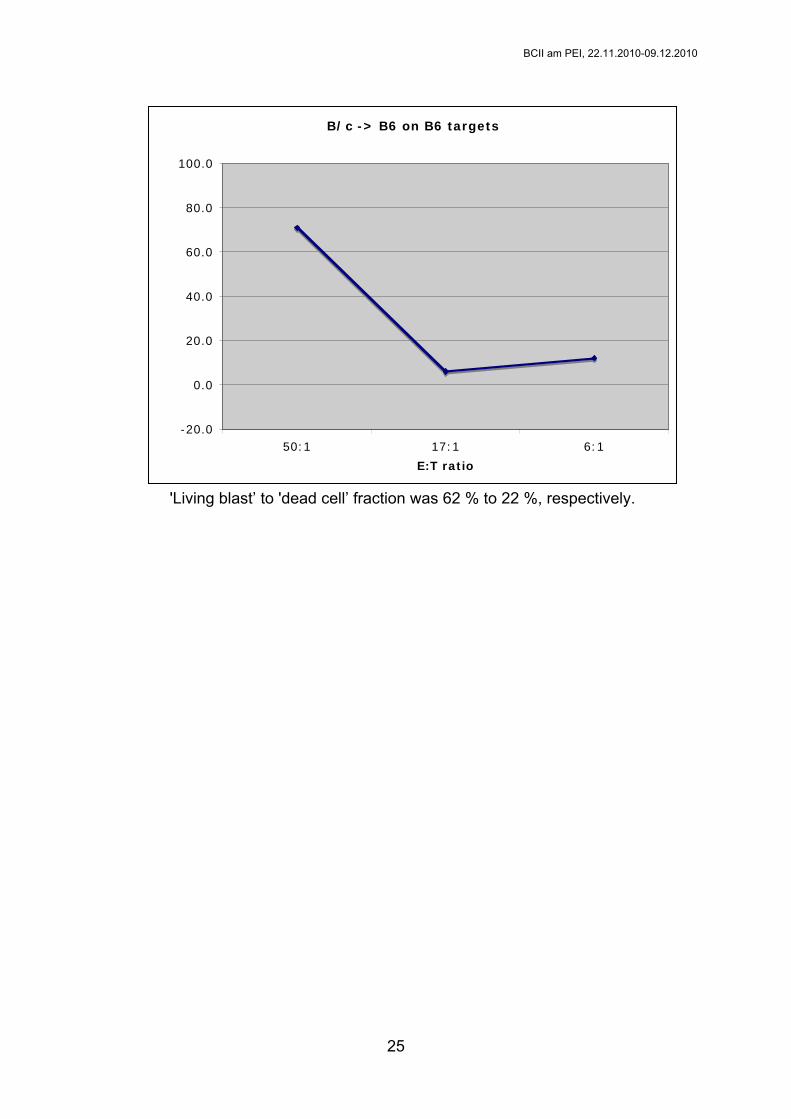
Auswertung
In einem Diagramm wird die spezifische Lyse in Abhängigkeit der E:T Ratio
aufgetragen. Beispiel:
Experimental values
% big cells % small cells % spec. lysis
E/T = 50:1 36 43 70.9 % specific lysis=100*((36/43)-(62/22))/((2/84)-(62/22))
E/T = 17:1 53 20 6.0 % specific lysis=100*((53/20)-(62/22))/((2/84)-(62/22))
E/T = 6:1 62 25 12.1 % specific lysis=100*((62/25)-(62/22))/((2/84)-(62/22))
spontaneous lysis 62 22 0.0 % specific lysis=100*((62/22)-(62/22))/((2/84)-(62/22))
maximal lysis 2 84 100.0 % specific lysis=100*((2/84)-(62/22))/((2/84)-(62/22))
BCII am PEI, 22.11.2010-09.12.2010
25
'Living blast’ to 'dead cell’ fraction was 62 % to 22 %, respectively.
B/c -> B6 on B6 targets
-20.0
0.0
20.0
40.0
60.0
80.0
100.0
50:1 17:1 6:1
E:T ratio

BCII am PEI, 22.11.2010-09.12.2010
26
Teil Toda: Messung der antigen-spezifischen CD4+ T-Zellaktivierung
T-Zellen stellen eine Untergruppe der Lymphozyten dar. Sie spielen eine kritische Rolle in
der adaptiven Immunantwort. T-Zellen werden in zwei große Untergruppen unterteilt,
basierend auf der Expression der Co-Rezeptoren CD4 oder CD8 auf der Zelloberfläche.
CD4+ T-Zellen, auch T-Helfer-Zellen genannt, assistieren anderen Leukozyten bei der
Ausführung ihrer immunologischen Aufgaben: Sie unterstützen die Reifung von B-Zellen in
Plasma-Zellen (Antikörper-produzierende Zellen) und die Aktivierung und Expansion von
CD8+ T-Zellen und Makrophagen. Dagegen differenzieren CD8+ T-Zellen überwiegend in
zytotoxische Zellen welche Virus-infizierte Zellen und Tumor-Zellen eliminieren können.
Für die Aktivierung von naiven CD4+ T-Zellen werden zwei Signale benötigt. Das erste Signal
besteht aus der Interaktion des T-Zell-Rezeptors (TCR) mit Antigen-Peptid/MHC-Klasse II-
Molekül Komplexen, welche auf der Oberfläche von Antigen-präsentierenden Zellen (APCs),
z.B. dendritischen Zellen und Makrophagen, präsentiert werden. Der TCR bindet dabei
neben dem MHC Molekül nur an ein Peptid-Fragment des Antigens, welches im Inneren der
präsentierenden Zelle vorher aus dem Antigen prozessiert wurde. Diese Prozessierung
findet in lysosomalen bzw. endosomalen Kompartimenten der APCs statt. Die Peptide
werden also durch die MHC-Klasse II Moleküle auf der Oberfläche der APCs den CD4+ T-
Zellen präsentiert. Aufgrund der großen Variabilität der möglichen Antigene ist für eine
effektive Immunantwort eine mindestens ebenso große Variabilität der TCRs erforderlich.
Diese wird durch die VDJ-Rekombination der Gen-Elemente der TCR Ketten während der T-
Zell-Reifung im Thymus gewährleistet.
Das zweite Signal zur T-Zell-Aktivierung ist Antigen-unspezifisch und wird als 'Co-Stimulus’
bezeichnet. Die bedeutendsten co-stimulatorischen Signale für die Aktivierung von naiven T-
Zellen werden durch Mitglieder der B7-Familie (B7-1: CD80; B7-2: CD86) vermittelt, welche
ebenfalls auf der Oberfläche von APCs präsentiert werden. Der dazugehörige
Interaktionspartner auf den T-Zellen ist CD28.
Naive T-Zellen exprimieren den IL-2 Rezeptor (IL-2R), welcher aus einer - und einer -
Kette gebildet wird. Dabei hat die -Kette eine niedrige Bindungsaffinität für IL-2. Bei der Co-
Stimulierung von T-Zellen über CD28 mit B7-1 oder B7-2, in Kombination mit einer TCR-
Aktivierung, wird die Synthese von IL-2 induziert. IL-2 ist ein essentieller Wachstumsfaktor
für T-Zellen, der die T-Zellproliferation fördert. Die autokrine und/oder parakrine
Verfügbarkeit von IL-2 ist daher ein entscheidender Schritt für die adaptive Immunantwort.
Es ermöglicht den wenigen für ein bestimmtes Antigen spezifischen T-Zellen sich effizient zu
vermehren. Ohne diese Expansion kann keine effektive Immunantwort ausgeführt werden
(z.B. gegen Pathogene).

BCII am PEI, 22.11.2010-09.12.2010
27
Um die T-Zell vermittelte Immunität zu untersuchen werden vielfach transgene Mäuse
verwendet, die einen vorgefertigten TCR von bekannter Spezifität in den T Zellen
exprimieren. Dies hat gegenüber normalen [Wildtyp (WT)]-Mäusen den Vorteil, dass in
diesen Tieren T-Zellen einer bestimmten Antigen-spezifität in großer Zahl produziert werden.
Dies vereinfacht die experimentelle Detektion einer Antigen-spezifischen Immunantwort
erheblich. WT-Mäuse haben dagegen eine große Variabilität in ihrer T-Zell-Antigen-Spezifität
(polyklonale TCRs), und haben deswegen nur eine sehr geringe Anzahl von T-Zellen, welche
spezifisch für ein gegebenes Antigen sind.
DO11.10 Mäuse sind ein Beispiel für eine Mauslinie, die einen transgenen TCR trägt. Die
meissten CD4+ T-Zellen aus diesen transgenen Mäusen tragen auf ihrer Oberfläche einen
TCR, der an den Komplex aus den Aminosäuren 323-339 des Ovalbumins (ein
Hauptallergen des Eiweißes) und präsentiert auf I-Ad Klasse II MHC Molekülen bindet.
In diesem Praktikum detektieren wir die Ovalbumin-induzierte T-Zellaktivierung von
Lymphozyten aus DO11.10 Mäusen. Dazu wird den Mäusen die Milz entnommen. Als
sekundäres lymphatisches Organ enthält die Milz sowohl T-Zellen wie APCs. Es wird eine
Einzel-Zellsusupensionen hergestellt und die Zellen werden in vitro mit OVA stimuliert. Falls
OVA-spezifische T-Zellen aktiviert wurden sollte IL-2 sekretiert werden und im Überstand der
Zellen zu detektieren sein. Das produzierte IL-2 wird mittels Enzyme Linked-Immuno-Sorbent
Assay (ELISA) detektiert und quantifiziert.

BCII am PEI, 22.11.2010-09.12.2010
28
1. Arbeits-Tag: Ansetzen der T-Zell Stimulation
Dienstag 23.11.2010 / 30.11.2010 / 7.12.2010
Tag 1
Mäuse
Der verwendete Mausstamm DO11.10 wurde unter spezifiziert pathogen-freien Bedingungen
(SPF) in den S1-Räumen der zentralen Tierhaltung im PEI gezüchtet.
Als Kontrollmäuse werden wildtyp Mäuse (Stamm BALB/c, von Charles River) verwendet.
Die Haltung/Zucht/Tötung der Mäuse erfolgt im Einklang zur relevanten Gesetzgebung. Das
PEI wird durch das Regierungspräsidium Darmstadt dementsprechend überwacht.
Medium
Als Basalmedium wird RPMI 1640, dem 100 U/ml Penicillin, 100 mg/ml Streptomycin und
5x10-5 M 2-Mercaptoethanol zugegeben werden, genutzt. Sollte das Medium kein Glutamin
enthalten, muss diese Aminosäure noch zugegeben werden. Dies ist das "Komplettmedium“:
Materialien für "Komplettmedium“:
RPMI 1640 (Invitrogen) : 500 ml
Penicillin, Streptomycin-Lösung : 5.0 ml
(100x konzentriert Invitrogen)
Glutamine (100x konzentriert PEI) : 5.0 ml
2-Mercaptoethanol (14 M, Sigma Aldrich) : 3.5 µl
Es sind ZWEI verschiedene Medienansätze [(A) mit/ohne Antigen, (B) "Zellkulturmedium“]
daraus zu erstellen:
(A): Dem Komplettmedium wird kein/wird Antigen/Mitogen zugegeben (s.u.)
(B): Dem Komplettmedium wird FCS (fötales Rinderserum) zugegeben:
500 ml Komplettmedium + 50 ml FCS = "Zellkulturmedium“
Herstellung der Antigen-Lösung (A)
- als Antigen wird OVA (Sigma Aldrich; Grade V) verwendet
- Es werden Lösungen mit einer Konzentration von 20, 200 und 2000 g/ml OVA in
Komplettmedium (also ohne FCS) angesetzt
- als T-Zell Mitogen (positive Kontrolle) wird, Concanavalin A (Con A, Sigma Aldrich)
verwendet

BCII am PEI, 22.11.2010-09.12.2010
29
- Es werden Lösungen mit einer Konzentration von 2,0 und 10 g/ml Con A in
Komplettmedium (also ohne FCS) angesetzt
- 100 µl der gewünschten Antigen-Lösung werden in die vorgesehen Wells der 96er
Multiwellplatte einpipettiert
- Während der Präparation der Zellen sollte die Platte bei Raumtemperatur gelagert werden
(sollte die Präperation der Zellen längere Zeit in Anspruch nehmen, ist es besser sie in den
Kühlschrank zu stellen; die Platte sollte aber Raumtemperatur haben, wenn die Zellen
ausgesät werden)
Herstellung der Milz-Zellsuspension
- die Milz wird unter sterilen Bedingungen aus der Maus entnommen, die Arbeit erfolgt unter
der sterilen Bank (Laminar Air Flow)
- das Organ wird in eine Petrischale (Ø 6 cm) mit 5.0 ml Zellkulturmedium gelegt
- um die Zellen der Milz zu erhalten, wird diese mit dem Rücken eines Stempels einer Spritze
zerquetscht. Die Zellen schwimmen nun im Medium.
- Zellsuspension über ein Zellsieb in ein 50 ml Falcon geben
- die Petrischale mit 5.0 ml Zellkulturmedium spülen um restliche Zellen aufzunehmen
- nun wird die Zellsuspension in ein 15 ml Falcon überführt
- mit Zellkulturmedium auf 15 ml auffüllen
- 10 min bei 1300 rpm bei Raumtemperatur (RT) zentrifugieren
- den Überstand verwerfen; etwas Überstand im Falcon belassen, damit das Pellet bedeckt
ist, nun das Falcon schütteln, bis sich das Pellet gelöst hat
- Lysierpuffer zugeben (150 mM NH4Cl; 1-2 ml je Milz) um die Roten Blutzellen zu lysieren, 1
min bei RT inkubieren
- 10 min bei 1300 rpm bei RT zentrifugieren
- der Überstand sollte rot sein, das Pellet weiß
- der Überstand wird verworfen und das Pellet wird in 12 ml RPMI mit 10 % FCS gelöst
- 10 min bei 1300 rpm bei RT zentrifugieren
- der Überstand wird verworfen und der Waschschritt 2 x wiederholt
- die Zellen werden in einer geeigneten Menge Zellkulturmedium gelöst (5 ml je Milz)
- die Zellen werden gezählt und die Zellzahl auf 8 x 106 Zellen eingestellt
- 100 µl der Zellsuspension werden je Well in eine 96er Multiwellplatte einpipettiert
- zusätzlich sollte 100 µl Antigen-Lösungen in den Wells sein, somit ist das Gesamtvolumen
je Well 200 µl

BCII am PEI, 22.11.2010-09.12.2010
30
Platte
jeweils als Dreifachbestimmung/pro gleichen Probenbedingung (z.B.: A1,2,3; A4,5,6;
...)
Milzzell-Kultur
- die Zellen werden in einem CO2-Inkubator bei 37°C , 5% CO2 und 95% relative Luftfeuchte
kultiviert
- Der Zellüberstand wird nach 24 h kultivieren abgenommen
- der Überstand sollte, bis zur ELISA-Messung, bei -20°C gelagert werden

BCII am PEI, 22.11.2010-09.12.2010
31
2. Arbeits-Tag: Detektion von IL-2 mittels ELISA
Donnerstag, 25.11.2010 / 2.12.2010 / 9.12.2010
Tag 3
Beobachtung der Zellen
- an Tag 3 der Zellkultur werden die Zellen unter dem Mikroskop angesehen
- wenn die T-Zellen durch die Antigenstimulation aktiviert wurden, bilden sie „Blasten“,
welche größer sind als nichtaktivierte Zellen
- die Zellen werden unter dem Mikroskop fotografiert
Zytokin ELISA
Die IL-2 Konzentration im Zellkulturüberstand wird mit Hilfe eines ELISA ermittelt.
(Medien/Puffer/Reagenzien untenstehend)
- der anti-Maus-IL-2-Antikörper wird in Coating-Puffer auf eine Konzentration von
0.5 µg/ml verdünnt
- eine ELISA-Platte wird mit 50 µl/well Erstantikörper (=anti-MausIL-2) gecoatet
- die Platte wird über Nacht im Kühlschrank inkubiert
- die Platte wird 3x mit je 150 µl/well Waschpuffer gewaschen
- die freien Bindungsstellen werden durch Zugabe von 100 µl/well Blocking-Puffer und
Inkubation der Platte für 1 h bei RT und 80 rpm auf dem Schüttler abgeblockt
- die Platte wird 3x mit je 150 µl/well Waschpuffer gewaschen
- die Proben und der Zytokin-Standard (von 4000 pg/ml, 2000 pg/ml, 1000 pg/ml …,
serielle Verdünnungsreihe; mindestens 4 verschiedene Konzentrationen werden für
die Standardkurve benötigt, mehr wären besser) werden in RPMI (mit 5 % FCS)
verdünnt. Von den Verdünnungen werden 50 µl/well in die Platte gegeben
- die Platte wird für 2 h bei RT und 80 rpm auf dem Schüttler inkubiert
- die Platte wird 3x mit je 150 µl/well Waschpuffer gewaschen
- der Detektionsantikörper wird in Assay-Puffer auf eine Konzentration von 0.5 µg/ml
verdünnt
- es werden 50 µl/well Detektionsantikörper in die Platte gegeben
- die Platte wird für 1 h bei RT und 80 rpm auf dem Schüttler inkubiert
- die Platte wird 3x mit je 150 µl/well Waschpuffer gewaschen
- Streptavidin konjugiert mit Meerrettichperoxidase (= HRP-Strep) wird 1:4000 in
Assay-Puffer verdünnt
- es werden 50 µl/well verdünnte HRP-Strep Lösung in die Platte gegeben

BCII am PEI, 22.11.2010-09.12.2010
32
- die Platte wird für 30 min bei RT und 80 rpm auf dem Schüttler inkubiert
- die Platte wird mindestens 3x mit je 150 µl/well Waschpuffer gewaschen
- die TMB-Substratlösungen A und B werden im Verhältnis 1:1 gemischt
- es werden 100 µl/well TMB-Substrat in die Platte gegeben
- die Platte wird maximal 30 min bei RT, 80 rpm und im Dunkeln auf dem Schüttler
inkubiert (klar blau)
- zum Abstoppen der Reaktion werden 50 µl/well 1 N H2SO4 in die Platte gegeben
(blau → gelb)
- Messung der Absorption bei 450 nm und 630 nm Referenzfilter im ELISA-Reader
Platten-Layout:
Jede Probe von Tag 1with doppelt im ELISA bestimmt
Puffer und Lösungen für den ELISA
Coating-Puffer:
50 mmol/l Natriumcarbonatpuffer pH 9,6
(1.696 g Na2CO3 + 2.856 g NaHCO3 ad 1 l mit Reinstwasser)
Waschpuffer:
PBS + 0.05 % Tween 20 (250 µl Tween in 500 ml PBS)
Blocking-Puffer:
PBS + 2 % BSA
Medium:
RPMI 1640 + 5 % FCS
Medium wird NUR für die Verdünnung der Proben und des Standards verwendet!!!
Assay-Puffer:
PBS + 0.05 % Tween 20 + 1 % BSA
TMB-Substrat:
Tetramethylbenzidine (TMB) substrate reagent set (BD Bioscience)

BCII am PEI, 22.11.2010-09.12.2010
33
Antikörper
Anti-Maus-IL-2 monklonaler Antikörper (Klon; JES6-1A12, Biolegend )
Biotin markierter anti-murine IL-2 monklonaler Antikörper (Klon; JES6-5H4, Biolegend)
Standard
rekombinantes murines IL-2 (PeproTech)

BCII am PEI, 22.11.2010-09.12.2010
34
Experiment 2: Immunologische Nachweismethoden II
Wichtig
Die Gruppen 1-3 führen das Experiment an humanen Zellen (PBMC) in der AG
Schweizer durch (A), die Gruppen 4-6 an murinen Zellen in der AG Waibler (B).

BCII am PEI, 22.11.2010-09.12.2010
35
Inhalt des Praktikums
In diesem Versuch sollen periphere humane Blutlymphozyten (PBMCs= peripheral blood
mononuclear cells) oder Immunzellen der Maus isoliert und auf charakteristische
Oberflächenmarker mittels FACS-Analyse untersucht werden. Grundsätzlich spielen
Lymphozyten eine bedeutende Rolle für den Ablauf von Immunreaktionen. Normalerweise
sind sie für die erworbene (adaptive) Immunität verantwortlich und dienen der Abwehr z.B.
von Mikroorganismen oder entarteter Zellen im Körper. Die Phänotypisierung erfolgt mittels
Bestimmung populationsspezifischer Zelloberflächenproteine (Clusters of Differentiation
oder CD-Marker). Diese lassen sich durch spezifische Antikörper identifizieren und
durchflußzytometrisch analysieren. Durchflußzytometrische Analysen werden zunehmend
bei zahlreichen immunologischen Fragestellungen auch zu diagnostischen Zwecken
durchgeführt, z.B bei der Immunphänotypisierung und Therapieüberwachung bei HIV-
Patienten.
Im Rahmen des Versuchs mit humanen PBMCs werden die Anteile an B-Lymphozyten
(CD19+), T-Lymphozyten (CD3+) und Monozyten (CD14+) bestimmt. Die beiden
Hauptklassen der T-Lymphozyten (d.h. aller CD3+) lassen sich durch den Nachweis der
Oberflächenmoleküle CD8 (für Zytotoxische T-Zellen) und CD4 (für T-Helferzellen)
voneinander unterscheiden.
Bei den Maus-Experimenten werden Immunzellen aus dem Blut, der Milz und dem
Thymus untersucht. Dafür werden unterschiedliche Mauslinien eingesetzt, bei denen (i) die
vier J-Elemente der variablen Region der schweren Kette von Immunglobulinen deletiert
sind (JHT), (ii) das Recombination Activating Gene 1 inaktiviert ist (RAG1-/-), oder (iii) bei
denen es sich um ganz normale wildtyp Mäuse handelt (WT). Im Rahmen des Versuchs
soll bestimmt werden, in welchen Organen sich B-Lymphozyten (CD19+), T-Lymphozyten
(CD3+) oder antigenpräsentierende Zellen (CD11c+) befinden, und welchen Einfluss die
Deletion der J Elemente oder die von RAG1 auf die Entwicklung der verschiedenen
Immunzelltypen hat.
Prinzip der Durchflusszytometrie
Das FACS (Fluorescence Activated Cell Sorting) stellt eine schnelle Methode zur
Untersuchung von Oberflächenproteinen dar. Hierbei können Oberflächenproteine auf
Zellen detektiert und quantifiziert, und damit Subpopulationen in Zellgemischen untersucht
werden.
Hierfür werden Zellen mit Fluorochrom-markierten monoklonalen Antikörpern gefärbt
und nach Lichtstreuung und Fluoreszenzintensität analysiert. Die Lichtstreuung in 180°
Vorwärtsrichtung (Forward Scatter) gilt als Maß für die Größe der Zellen (kleine dichte
Zellen streuen weniger Licht), die in 90º (Side Scatter) als Maß für die die Granularität.

BCII am PEI, 22.11.2010-09.12.2010
36
Die an die Antikörper gekoppelten Fluoreszenzfarbstoffe wie z.B.
Fluoreszeinisothiozyanat (FITC) und Phycoerythrin (PE) werden bei der gleichen
Wellenlänge (480 nm) von einem Argon-Laser zur Emission von Lichtimpulsen angeregt.
Der Emissionsbereich von FITC (Grünfluoreszenz) liegt zwischen 500 und 570 nm, der von
PE (Rotfluoreszenz) reicht von 570-600 nm.
Abb. 1: Schematische Abbildung des optischen Systems eines FACS-Geräts. Die Zellprobe
(sample) wandert in einer Küvette an einem Argonlaser (488 nm) vorbei. Hierbei wird das Licht zum
einen durch die Zellen gemäß ihrer Größe und Granularität gestreut, zum anderen wird das
Fluorochrom des entsprechend markierten Antikörpers angeregt und emittiert das Licht in einer für
das Fluorochrom typischen Wellenlänge. So genannte Photodetektoren registrieren das emittierte
Licht und setzen dieses in digitale Signale um, die dann mit Hilfe einer entsprechenden Software
dargestellt und analysiert werden können.
Da die Emissionsmaxima von Fluoreszenz 1 (FITC-530 nm) und Fluoreszenz 2 (PE-585
nm) oder Fluoreszenz 3 (z.B. PJ-615 nm) in unterschiedlichen Wellenlängenbereichen
liegen, die von entsprechenden Photodetektoren registriert werden, lassen sich mehrere
Farbstoffe gleichzeitig messen (Mehrfarben-Immunfluoreszenz). Auf Grund der teilweise
überlappenden Emissionsbereiche müssen die Detektoren für die Fluoreszenzen 1 und 2,
so wie 2 und 3 gegeneinander kompensiert werden. Ein elektronisches System, der
Photomultiplier, erhält die Lichtimpulse von der optischen Einheit und übermittelt sie zur
Datenauswertung an den Computer.
BCII am PEI, 22.11.2010-09.12.2010
37
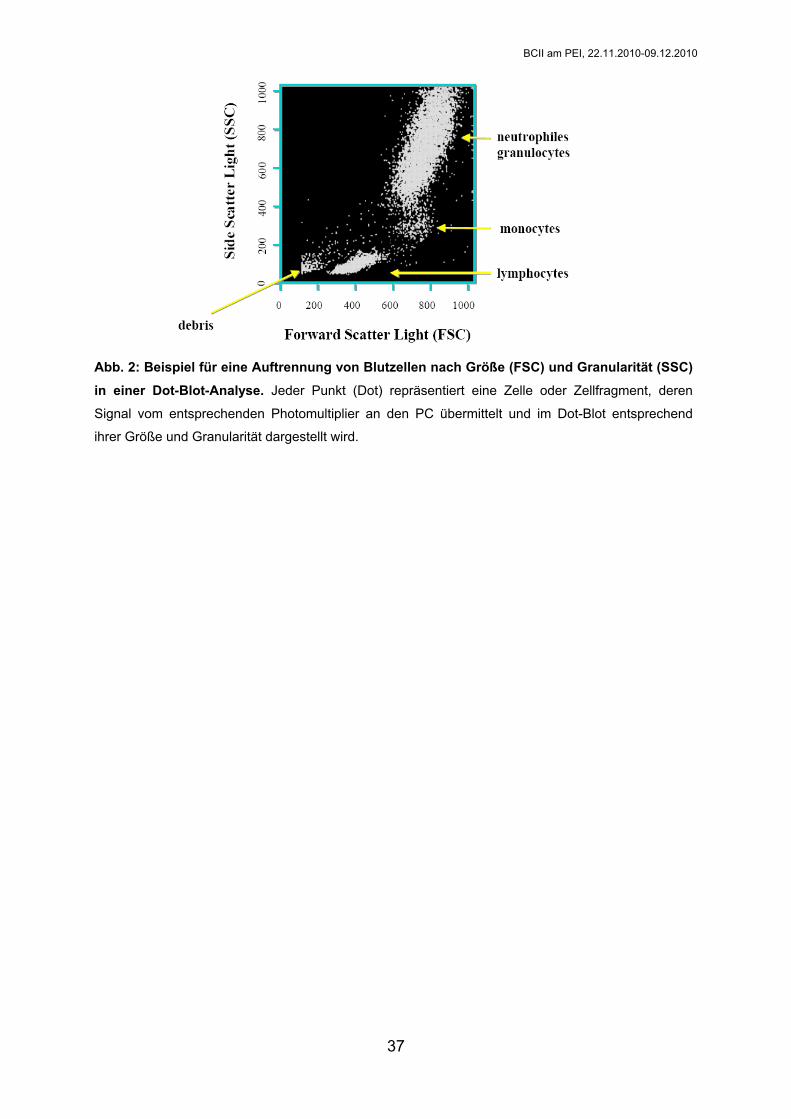
Abb. 2: Beispiel für eine Auftrennung von Blutzellen nach Größe (FSC) und Granularität (SSC)
in einer Dot-Blot-Analyse. Jeder Punkt (Dot) repräsentiert eine Zelle oder Zellfragment, deren
Signal vom entsprechenden Photomultiplier an den PC übermittelt und im Dot-Blot entsprechend
ihrer Größe und Granularität dargestellt wird.

BCII am PEI, 22.11.2010-09.12.2010
38
A) Versuchsablauf humane PBMCs (AG Schweizer)
Teil I PBMC-Präparation und Kultivierung
Teil II FACS-Färbung und -Messung
FACS-Auswertung und Besprechung der Ergebnisse
Teil I
Isolierung von PBMCs aus humanem Vollblut
Die PBMCs werden mittels Dichtegradienten-Zentrifugation (Ficoll-Hypaque) aus frisch
gewonnenem Vollblut eines gesunden Spenders isoliert. Hierbei sammeln sich die
Lymphozyten und Monozyten (PBMC), entsprechend ihrer spezifischen Dichte, in der
Interphase zwischen Überstand (Plasma/Thrombozyten) und Ficoll-Histopaque an. Das
Zellsediment bilden Erythrozyten und Granulozyten, die eine höhere Dichte besitzen (siehe
Abb.).
Durchführung
15 ml Ficoll (Histopaque von Sigma 1077) in einem 50 ml Falcon-Röhrchen vorgelegen
20 ml Vollblut je Falcon vorsichtig über das Ficollkissen schichten.
VORSICHT!: Ist das Vollblut einmal mit Ficoll-Lösung vermischt, ist es unbrauchbar.
die Proben 30 min bei 2.000 rpm und bei Raumtemperatur zentrifugieren, ohne Bremse!
die PBMCs bilden einen milchig-trüben Ring zwischen der oberen Plasmaphase und der
unteren Ficollphase
Interphasenring vorsichtig mit einer 2 ml Pipette abziehen und in ein neues 50 ml
Falcon-Röhrchen pipettieren in dem 10 PBS vorgelegt wurden
um das restliche Ficoll zu entfernen werden die PBMCs mit PBS gewaschen. Falcons
mit 37°C warmem PBS auffüllen!
Zentrifugation bei 1.200 rpm, 10 min, RT (mit Bremse)
Ficoll Ficoll

BCII am PEI, 22.11.2010-09.12.2010
39
um die Erythrozyten vollständig aus der Lymphozytenfraktion zu entfernen, werden
diese mit Ammoniumchloridlösung lysiert.
Überstand verwerfen, PBMCs in 10 ml 0.86%iger Ammoniumchloridlsg.
resuspendieren und 10 min bei 37°C im Wasserbad inkubieren
um das Ammoniumchlorids zu entfernen, wird das Falcon mit warmen PBS aufgefüllt
und die PBMCs erneut bei 1.200 rpm / 10 min abzentrifugiert
Überstand verwerfen und die PBMCs nochmals mit 45 ml PBS resuspendieren und 10
min, bei 1.200 rpm zentrifugieren
Zellpellet in 20 ml RPMI-Medium aufnehmen, Zellzahl bestimmen (s.u.)
Bestimmung der Zellzahl und Zellvitalität
Zur Bestimmung der Zellzahl und der Vitalität der Zellen werden die PBMCs im Verhältnis
1:1 mit Trypanblau gefärbt. Trypanblau wird aufgrund der Membrandurchlässigkeit nur von
den toten Zellen aufgenommen. Die Zellen werden in eine Neubauer Zählkammer überführt
und gezählt. Die Zellkonzentration ergibt sich aus der Multiplikation von Zellzahl,
Verdünnungsfaktor und Kammerfaktor (104).
Beispiel:
Die Neubauerkammer hat in der Mitte ein Kreuz aus engen Linien. Die 4 Eckfelder
(Gruppenquadrate) sind in je 16 Einzelquadrate unterteilt. Zellen in mindestens 2 (bei
besonders wenig Zellen 4) Eckfeldern zählen.
Berechnung (Beispiel):
150 Zellen wurden in 2 Eckfeldern gezählt
15 µl Zellsuspension wurde mit 15 µl Trypanblau verdünnt » Faktor 2
der Kammerfaktor (um vom Einschlussvolumen auf 1 ml umzurechnen) beträgt 10.000
150 : 2 x 2 x 10.000 = 1.5x106 Zellen/ ml
Insgesamt sind von der Zellsuspension 40 ml vorhanden:
1.5 x 106 Zellen x 40 ml = 6x107 Zellen insgesamt

BCII am PEI, 22.11.2010-09.12.2010
40
Kultivierung der PBMCs
Zellen in einer Dichte von 1x106/ml in RPMI Medium (plus 10% FCS, 2 mM Glutamin,
Antibiotikacocktail, 200 U IL-2/ml und 0.22µg PHA/ml) aufnehmen
2 ml der PBMC-Zellsuspension je 6-well aussäen
Teil II
FACS-Färbung und Analyse der PBMCs
Zellen in 15 ml Falcon überführen, wichtig: durch resuspendieren auch die adhärenten
Zellen ablösen
Medium bei 1.200 rpm, 10 min und RT abzentrifugieren
Zellpellet in 5-10 ml PBS aufnehmen
Zellzahl mit Hilfe Neubauer-Zählkammer bestimmen
15 µl Zellsuspension mit 15 µl Trypanblau mischen
5x105 Zellen pro Ansatz in FACS-Röhrchen überführen (insgesamt 10 Ansätze) und die
Röhrchen mit FACS-Waschpuffer auffüllen
bei 1.500 rpm, 5 min und 4°C abzentrifugieren und Überstand verwerfen
das Zellpellet in je 200 µl Waschpuffer aufnehmen und die folgenden
fluoreszenzmarkierten Antikörper zupipettieren (Die Menge der Antikörper wird vom
Kursassistenen vorgegeben)
1. Isotypkontrolle
2. CD19-PE
3. CD3-FITC
4. CD4-Cy5
5. CD8-PE
6. CD3-FITC/CD19-PE
7. CD3-FITC/CD4-Cy5/CD8-PE
die Proben 20 min bei 4°C (im Kühlschrank) inkubieren
zum Waschen der Zellen, die FACS-Röhrchen auffüllen und 5 min, bei 1.500 rpm, 4°C
zentrifugieren
Überstand dekantieren und Zellen erneut in 500µl Fixierlösung (1% PFA)
resuspendieren
FACS-Messung (FACS Galaxy, Software: FloMax):
1. Dot-Plot: FSC/SSC (Zellpopulation festlegen)
2. Geräteeinstellung (Kompensation) mit Hilfe der Einzelfärbungen

BCII am PEI, 22.11.2010-09.12.2010
41
3. Dot-Plot: CD3/CD19
4. Dot-Blot: CD4/CD8 im Gate der CD3+ Zellen
5. Histogramm CD3 und CD19
Aufgaben
Vervollständige die prozentualen Angaben der Lymphozyten-Populationen in folgendem
Schaubild:
1. Wieviel Prozent der Leukozyten sind T-Zellen, wieviel B-Zellen?
2. Wieviel Prozent der T-Zellen sind Zytotoxische T-Zellen bzw. T-Helferzellen?
3a. Beurteile die prozentuale Verteilung der gemessenen Lymphozytenpopulationen des
Spenders.
3b. Wie ist das Verhältnis von CD4+ zu CD8+ und welches Verhältnis ist liegt bei einem
gesunden Donor vor?
3c. Wie verändert sich dieses Verhältnis im Verlauf einer HIV-Infektion?
4. Wo würde man nach Vollblutfärbung in einem FSC/SSC Dot Blot die Granulozten
uund Monozyten finden?
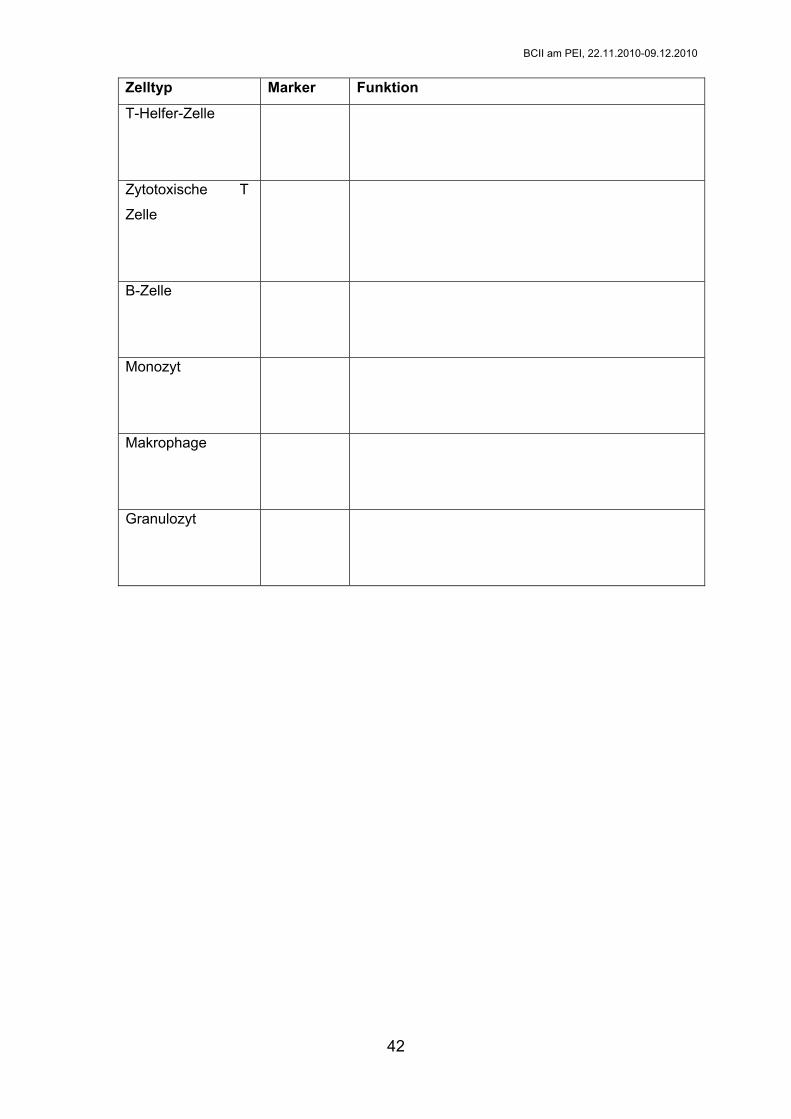
5. Fülle folgende Tabelle aus:
CD-
BCII am PEI, 22.11.2010-09.12.2010
42
Zelltyp Marker Funktion
T-Helfer-Zelle
Zytotoxische T
Zelle
B-Zelle
Monozyt
Makrophage
Granulozyt

BCII am PEI, 22.11.2010-09.12.2010
43
B) Versuchsablauf murine Zellen (AG Waibler)
Teil I Präparation von Blutzellen, Milzzellen und Thymuszellen aus WT, JHT und
RAG1-/- Mäusen
FACS-Färbung
Teil II FACS-Messung
FACS-Auswertung und Besprechung der Ergebnisse
Teil I
Präparation von Blutzellen, Milzzellen und Thymuszellen aus WT, JHT und RAG1-/-
Mäusen
Für dieses Experiment werden je eine WT, JHT und RAG1-/- Maus eingesetzt. Für die
Blutentnahme werden die Tiere kurz betäubt. Nach dem Töten der Tiere werden die
Immunorgane entnommen.
Blut
Die Maus wird durch Inhalations-Narkose mit Isofluran betäubt (erfolgt durch Betreuer)
Das Blut wird retrobulbär abgenommen (erfolgt durch Betreuer)
Das Blut wird in heparinisierte Microcontainer gegeben
15 µl heparinisiertes Blut wird für einen Färbeansatz eingesetzt
Milz
Maus wird durch cervicale Dislokation getötet (erfolgt durch Betreuer)
Maus wird auf die rechte Seite gelegt (Kopf zeigt nach links)
Mit einer stabilen Schere wird eine Öffnung ins Fell geschnitten
Die Öffnung wird weiter aufgerissen
Die Milz wird mit einer chirurgischen Schere und Lidpinzetten entnommen
Die Milz wird in eine Petri-Schale gelegt, ca. 5 ml PBS werden zugegeben und die Milz
wird mit Pipettenspitzen ausgeklopft
Die Milzzellen werden durch Auf- und Abpipettieren mit einer 10 ml Stab-Pipette
resuspendiert, Proben werden mit PBS auf 10 ml aufgefüllt
Zentrifugation (5 min, 1200 rpm), Überstand verwerfen
Zellpellet in 5 ml RBC-Lysis-Puffer resuspendieren und ca. 1 min bei RT inkubieren
Zugabe von 5 ml PBS
Zentrifugation (5 min, 1200 rpm), Überstand verwerfen

BCII am PEI, 22.11.2010-09.12.2010
44
Zellpellet in ca. 10 ml PBS resuspendieren und durch ein 70 µm Filicon filtrieren
Zellzahlbestimmung (siehe unten)
Thymus
Nach Entnahme der Milz wird die Maus auf den Rücken gelegt und der Brustkorb
entlang des Sternums mit einer stabilen Schere geöffnet
Rippenbögen aufziehen
Thymus entnehmen (Vorsicht: nicht ins Herz schneiden)
Der Thymus wird in eine Petri-Schale gelegt, ca. 5 ml PBS werden zugegeben und das
Organ wird wie die Milz ausgeklopft
Thymuszellen werden durch Auf- und Abpipettieren mit einer 10 ml Stab-Pipette
resuspendiert, Proben werden mit PBS auf 10 ml aufgefüllt
Zellsuspension durch ein 70 µm Filicon filtrieren
Zellzahlbestimmung (siehe unten)
Bestimmung der Zellzahl und Zellvitalität
Zur Bestimmung der Zellzahl und der Vitalität der Zellen werden diese seriell mit Trypanblau
verdünnt und gefärbt (1:2, 1:4, 1:8, 1:16). Trypanblau wird aufgrund der veränderten
Membrandurchlässigkeit toter Zellen nur von diesen aufgenommen, lebende Zellen bleiben
farblos. Eine geeignete Verdünnung wird in eine Neubauer Zählkammer überführt und die
lebenden (ungefärbte) Zellen ausgezählt. Die Zellkonzentration ergibt sich aus der
Multiplikation von Zellzahl (Mittelwert aus allen vier Quadranten), Verdünnungsfaktor und
Kammerfaktor (104).
Schematische Darstellung einer Neubauer Zählkammer mit vier
Quadranten à 16 Kleinquadraten.

BCII am PEI, 22.11.2010-09.12.2010
45
FACS-Färbung und Analyse der Proben
In diesem Experiment werden die zuvor präparierten Blut-, Milz- und Thymuszellen aus WT,
JHT und RAG1-/- Mäusen mittel spezifischer, Fluorochrom-gekoppelter Antikörper angefärbt.
Blut
Pro Färbeansatz 15 µl heparinisiertes Blut in FACS-Röhrchen überführen
Zugabe der Antikörper nach dem Schema der Färbeansätze (siehe unten), vortexen
Inkubation der Proben für 15 min bei 4°C (Proben sollen dunkel aufbewahrt werden)
Zugabe von 1 ml Blut-Lyse-Puffer (nicht RBC-Lysis-Puffer)
Inkubation der Proben für 20-30 min bei RT (Proben sollen dunkel aufbewahrt werden)
Zentrifugation (5 min, 1200 rpm), Überstand verwerfen
Zugabe von 1 ml FACS-Puffer
Zentrifugation (5 min, 1200 rpm), Überstand verwerfen und Zellpellet im verbleibenden
Restvolumen resuspendieren (vortexen)
Milz und Thymus
2x105 bis 1x106 Zellen in 50 µl FACS-Puffer pro Färbeansatz in FACS-Röhrchen
überführen (Zellen ggf. vorher auf gewünschte Zellzahl einstellen)
Zugabe der Antikörper nach dem Schema der Färbeansätze (siehe unten), vortexen
Inkubation der Proben für 15 min bei 4°C (Proben sollen dunkel aufbewahrt werden)
Zugabe von ca. 1 ml FACS-Puffer
Zentrifugation (5 min, 1200 rpm), Überstand verwerfen
Zellpellet in 100 µl FACS-Puffer und 100µl 1%iger Paraformaldehyd-Lösung
resuspendieren (vortexen)
Schema der Färbeansätze
1. ungefärbt
2. 1 µl anti-CD3 PE
3. 1 µl anti-CD19 FITC
4. 1 µl anti-CD11c APC
5. 1 µl anti-CD3 PE + 1 µl anti-CD19 FITC + 1 µl anti-CD11c APC
WT Blut: Färbeansätze 1. – 5. durchführen
WT Milz: Färbeansätze 1. – 5. durchführen
WT Thymus: Färbeansätze 1. – 5. durchführen
JHT Blut: Färbeansätze 1. und 5. durchführen
JHT Milz: Färbeansätze 1. und 5. durchführen

BCII am PEI, 22.11.2010-09.12.2010
46
JHT Thymus: Färbeansätze 1. und 5. durchführen
RAG1-/- Blut: Färbeansätze 1. und 5. durchführen
RAG1-/- Milz: Färbeansätze 1. und 5. durchführen
RAG1-/- Thymus: Färbeansätze 1. und 5. durchführen
Die gefärbten Zellen können bis zur Messung am Durchflusszytometer bei 4°C gelagert
werden.
Teil II
FACS-Messung und Auswertung der Daten
Die FACS-Messung der in Teil I isolierten und gefärbten Zellen erfolgt am
Durchflusszytometer LSR II der Firma BD. Die gewonnenen Daten werden mit der BD
FACSDiva ™ Software analysiert. Zur Besprechung der Ergebnisse beantworten Sie bitte
folgende Fragen:
1. Welche Zelltypen können Sie mit Hilfe der verwendeten Antikörper detektieren?
2. Wie sieht die prozentuale Verteilung von T-Zellen, B-Zellen und antigenpräsentierenden
Zellen in Blut, Milz und Thymus aus?
3. Welche absoluten Anzahlen von T- und B-Zellen finden sich in der Milz und im Blut von
Mäusen?
4. Welchen Einfluss hat die Deletion der J Elemente auf die Entwicklung von Immunzellen
in den verschiedenen Organen?
5. Welchen Einfluss hat die Deletion von RAG1 auf die Entwicklung von Immunzellen in
den verschiedenen Organen?

BCII am PEI, 22.11.2010-09.12.2010
47
Experiment 3: Virusquantifizierung/siRNA
Wichtig
Experiment 3 wird an zwei verschiedenen Viren durchgeführt. Die
Gruppen sind wie folgt eingeteilt:
Gruppen 1-3: Reduktion der Virusbelastung in PERV-infizierten humanen
Zellen durch siRNA (AG Toenjes)
Gruppen 4-6: Quantifizierung von Vacciniaviren (AG Schwantes/Suezer)

BCII am PEI, 22.11.2010-09.12.2010
48
Experiment 3: Reduktion der Virusbelastung in PERV-infizierten humanen Zellen durch siRNAs AG Toenjes Ziele des Versuchs: 1. Ermittlung der absoluten Viruslast in infizierten Zell-Linien. 2. Nachweis der spezifischen Reduktion der Virusbelastung durch porzine
endogene Retroviren (PERV) in Zellkultur-Überständen mittels PERV-spezifischer siRNAs.
Theorie: Hemmung der Genexpression durch siRNA 1990 wurde das Posttranscriptional Gene Silencing (PTGS) in Petunien (Solanaceae) gefunden, nachdem Forschergruppen um Mol und Jorgensen versuchten, die Blütenfärbung von Petunien durch das Einbringen eines zusätzlichen Pigmentgens zu verstärken und dabei das genaue Gegenteil erreichten. Es konnte gezeigt werden, dass die Gene nicht nur auf der Ebene der Transkription abgeschaltet wurden, sondern ebenso die zugehörige mRNA schnell abgebaut wurde. Etwas später wurden ähnliche Erkenntnisse bei Pilzen (Neurospora; „Quelling“), Würmern, Fliegen und Mäusen gewonnen (1, 2). Die Technik der RNA Interferenz (RNAi) wurde 1998 durch A. Fire und C. Mello in Caenorhabditis elegans nachgewiesen (1, 2, 3). Doppelsträngige RNA (dsRNA) führte zu einem effizienten und spezifischen Gene-Knockdown. Wird eine doppelsträngige (ds) RNA mit homologer Sequenz zu einer zelleigenen mRNA in eine Zielzelle eingebracht, startet der Vorgang des PTGS, indem Dicer, eine ATP-abhängige Ribonuklease, die dsRNA in viele kleine Stücke schneidet. Es entstehen sogenannte small interfering RNAs (siRNAs). Diese siRNAs wandern zu einem Enzymkomplex, dem sog. RNA-induced-silencing-complex (RISC). Anhand der siRNA wird das komplementäre Stück der zelleigenen mRNA gefunden, zerschnitten und somit die Proteinsynthese des entsprechenden Gens unterbunden (Abb. 1). In höheren Eukaryoten führt lange, dsRNA (mehr als 30 Basenpaare) zu unspezifischen Auswirkungen bzw. zur Apoptose, da diese die enzymatische Zerstörung aller mRNAs und den Stop der Proteinsynthese bewirkt. Erst die Arbeit von S. Elbashir unter der Leitung von T. Tuschl konnte 2001 dieses Problem umgehen, indem kurze dsRNAs von 21 nt Länge verwendet wurden, die am 3’-Ende jeweils 2-3 Nukleotide überstehen. Solche kurzen siRNA-Moleküle können synthetisch hergestellt und in die Zielzellen eingebracht werden. Man steigt dadurch in den Prozess des PGTS zwar erst später ein, erzielt jedoch den gleichen Effekt (1, 2, 4; Abb. 2). Weiterführende Informationen zu siRNA und MicroRNA siehe http://opengenomics.org/CSB2005_RNAiTutorial/3-CSB2005-B&WSlides.pdf
BCII am PEI, 22.11.2010-09.12.2010
49
Die Lage der synthetischen siRNAs innerhalb der Genomstruktur von PERV, die im Versuch verwendet werden, ist in Abb. 3 dargestellt. Die Nukleotidangaben beziehen sich auf eine Referenzsequenz (PERV-B(33)ATG; Acc.No. AJ133816) und bezeichnen die Positionen, die von den siRNAs erkannt werden. Abb. 1:
Modell der molekularen Schritte des Posttranskriptional Gene Silencing (PTGS) http://www.laborjournal.de/rubric/archiv/stichwort/w_02_07.html (2) Abb. 2:
RNA Interferenz (RNAi) mit siRNA http://www.invitrogen.com/content.cfm?pageid=10075 (5)

BCII am PEI, 22.11.2010-09.12.2010
50
Abb. 3
Genomstruktur von PERVPosition der synthetischen siRNA‘s
Nukleotidangaben bezogen auf PERV-B(33)ATG (Czauderna et al. 2000)
3’-LTR
poly A site
env5’-LTR
cap site
PBS (Gly4)
SD
gag pol
SA
1154 2728
gag I1245-1266
si 71414-1435
gag IV2086-2107
gag III1949-1970
gag
Genomstruktur von PERVPosition der synthetischen siRNA‘s
Nukleotidangaben bezogen auf PERV-B(33)ATG (Czauderna et al. 2000)
3’-LTR
poly A site
env5’-LTR
cap site
PBS (Gly4)
SD
gag pol
SA
1154 2728
gag I1245-1266
si 71414-1435
gag IV2086-2107
gag III1949-1970
gag
Genomstruktur von PERVPosition der synthetischen siRNA‘s
Nukleotidangaben bezogen auf PERV-B(33)ATG (Czauderna et al. 2000)
3’-LTR
poly A site
env5’-LTR
cap site
PBS (Gly4)
SD
gag pol
SA
1154 2728
gag I1245-1266
si 71414-1435
gag IV2086-2107
gag III1949-1970
gag
Nukleotidangaben bezogen auf PERV-B(33)ATG (Czauderna et al. 2000)
3’-LTR
poly A site
env5’-LTR
cap site
PBS (Gly4)
SD
gag pol
SA 3’-LTR
poly A site
env
3’-LTR
poly A site
3’-LTR
poly A site
envenv5’-LTR
cap site
PBS (Gly4)
SD
5’-LTR
cap site
PBS (Gly4)
SD
gaggag pol
SA
polpol
SA
1154 2728
gag I1245-1266
si 71414-1435
gag IV2086-2107
gag III1949-1970
gag
11541154 27282728
gag I1245-1266
si 71414-1435
gag IV2086-2107
gag III1949-1970
gag
Zum Versuch: PERV-infizierte humane Zellen (293/PERV-B(33)) werden 48 Std. vor Beginn des Versuches mit PERV-spezifischen siRNAs in verschiedenen Kombinationen und einer nicht PERV-spezifischen siRNA, sowie MOCK (nur Transfektionsreagenz, keine siRNA im Ansatz) transfiziert. Als Negativkontrolle dienen nicht-transfizierte Zellen. Aus den Überständen der transfizierten Zellen wird virale RNA von PERV mittels des High Pure Viral RNA Kit (Roche) isoliert. Daraufhin wird mittels quantitativer One-Step RT-PCR (LightCycler, Roche) die Anzahl der Virionen pro µl Zellkultur-Überstand bestimmt. Aliquots der Überstände werden bei –80°C eingefroren und dienen als Template für einen reversen Transkriptase (RT)-Test (C-type-RT Activity Assay, Cavidi Tech). Es soll gezeigt werden, dass es durch den Einsatz spezifischer siRNA's möglich ist, die Anzahl der Virionen im Zellkultur-Überstand zu verringern und, dass unspezifische siRNAs keinen Einfluss auf die Virusexpression haben.

BCII am PEI, 22.11.2010-09.12.2010
51
Durchführung: ACHTUNG: Die Transfektion wird nicht von den Praktikanten durchgeführt, die folgende Beschreibung dient nur zum besseren Verständnis des Versuches! Pro Reaktionskammer (well) werden am Vortag der Transfektion 2x105 293/PERV-B(33)-Zellen ausgesät. Ansätze (je 1 µg siRNA/well): siRNA 7 (Eurogentec), siRNA I, III u. IV (Dharmacon) bei 2-er-Kombi je 2,5 µl siRNA/Ansatz bei 3-er-Kombi je 1,7 µl siRNA/Ansatz
Transfektion: die zu transfizierenden siRNAs zu 0,1 ml Opti-MEM I (Invitrogen) in
Polystyrolröhrchen geben (A) Mastermix aus 0,9 ml Opti-MEM I und 90 µl Transfektionsreagenz
Lipofectamine ansetzen und vortexen (B) je Probe 0,1 ml B zu A geben, vortexen 15–30 min bei RT inkubieren, damit sich die siRNA-Lipidkomplexe bilden
können in der Zwischenzeit Zellen 1x mit 2 ml Opti-MEM I pro well waschen je 0,8 ml Opti-MEM I zu A/B’s geben (= Transfektionsmix) je 1 ml Transfektionsmix pro well bei 37°C im Brutschrank 4-5 Std. inkubieren Transfektionsmix absaugen je well 2 ml Medium (DMEM + 10% FKS + Streptomycin/Penicillin + L-
Glutamin) zugeben nach 72 Std.: Überstände (Supernatant, SN) zellfrei filtrieren, Isolierung der viralen RNA aus 200 µl SN, SN für RT-Test aliquotieren (je 1x 100 µl)
72 3
4 5
98
106
1
sisi7 + + IV
si I III + IV
si7 + MOCK Neg. control siRNA
untr.Zelle
I III IV
X X
72 3
4 5
98
106
1
sisi7 + + IV
si I III + IV
si7 + MOCK
untr.Zelle
I III IV
X X

BCII am PEI, 22.11.2010-09.12.2010
52
Filtration der Überstände und Aliquotierung: 2x 10 Eppi’s analog den 6-well-plates beschriften (also: si7+I+IV,
siI+III+IV, … ODER von 1-10) [1x 10 Eppi’s für Isolierung viraler RNA, 1x 10 Eppi’s für RT-Test]
10 5-ml-Röhrchen beschriften und in Ständer einsetzen (= Auffanggefäße für filtrierte SN’s)
je ca. 2 ml SN pro well mittels 2-ml-Spritze aufnehmen durch 0.45 µm Sartorius-Filter in vorbereitete 5-ml-Röhrchen filtrieren je 1x 100 µl zellfrei filtrierten SN in vorbereitete Eppi’s geben (für RT-
Test), bei -80°C (Gruppe A) bzw. auf Eis (Gruppe B) lagern
Isolierung viraler RNA (High Pure Viral RNA Kit, Roche): 1) Arbeitslösung herstellen: 5 ml Bindepuffer (grüner Deckel) ergänzt mit
50 µl poly(A) carrier-RNA 2) pro Eppi je 400 µl Arbeitslösung vorlegen, je 200 µl zellfrei filtrierten SN
zugeben und gut mischen (vortexen) 3) Inkubation bei Raumtemperatur (RT) für 10 min 4) High Pure Filter-Tubes in Auffanggefäße setzen und Proben in das
obere Reservoir pipettieren 5) 15 sec bei 10.000 rpm in Eppendorf-Tischzentrifuge zentrifugieren 6) Filter-Tubes in neue Auffanggefäße setzen, alte Auffanggefäße
verwerfen 7) je 500 µl Inhibitor Removal Puffer (Gefäß 3a, schwarzer Deckel) in die
oberen Reservoirs pipettieren (geht am besten mit einer Eppendorf Multipette und 12,5 ml-Combitip)
8) 1 min bei 8.000 rpm zentrifugieren 9) Filter-Tubes in neue Auffanggefäße setzen, alte Auffanggefäße
verwerfen. Es folgen zwei Waschschritte. 10) Für den ersten Waschschritt 500 µl Waschpuffer (blauer Deckel) in die
oberen Reservoirs pipettieren (Eppendorf Multipette, 5 ml-Combitip) 11) 1 min bei 8.000 rpm zentrifugieren 12) Filter-Tubes in neue Auffanggefäße setzen. Für den zweiten
Waschschritt je 400 µl Waschpuffer zugeben. 13) 1 min bei 8.000 rpm zentrifugieren, anschliessend 10 sec bei 13.200
rpm zentrifugieren (um Reste des Waschpuffers zu entfernen) 14) Auffanggefäße verwerfen und Filter-Tubes in sterile, Nuclease-freie 1,5
ml-Eppis einsetzen 15) Je 50 µl Elutionspuffer (Gefäß 4, farbloser Deckel) in die oberen
Reservoirs pipettieren (Eppendorf Multipette, 2,5 ml-Combitip) 16) 1 min bei 10.000 rpm zentrifugieren 17) Filter-Tubes verwerfen, Eppis mit vRNA verschließen und auf Eis stellen
(Gruppe A) bzw. bei –80°C lagern (Gruppe B). Die Proben dienen als Template für die One-Step RT-PCR mittels LightCycler.

BCII am PEI, 22.11.2010-09.12.2010
53
Quantitative One-Step RT-PCR (LightCycler, Roche): Die PCRs werden mittels des QuantiTect SYBR Green RT-PCR Kits (Qiagen, Cat.No.: 204243) durchgeführt. Die PERV RT-PCR dient zur Bestimmung der Virionenzahl im Überstand PERV-infizierter Zellen. 2x QuantiTect SYBR Green RT-PCR Master Mix, RNase-freies Wasser und Primer (F und R, je 10 µM) auftauen (im PCR-Raum), Template-RNAs (vRNA) und externen homologen Standard auf Eis auftauen (im Labor). Der PCR-MasterMix wird im PCR-Raum pipettiert, die Verdünnung des Standards und die Templatezugabe findet im Labor statt. Externen homologen Standard verdünnen (auf Eis; Wasser in Eppis vorlegen): Externer homologer Standard (= 326 bp T7-in-vitro transkribierte RNA): F/R (NdeI) [126 ng/µl] Berechnung der Kopienzahl: 6x1023 [Kopien/µl] x Konz. [g/µl] / MW [g/mol] MW (RNA) = Basepairs x 340 daltons/bp MW = 326 bp x 340 daltons/bp MW = 1,108x105 daltons Für die Konzentration von 126 ng/µl gilt also: 6x1023 x 1,26x10-7 / 1,108x105 = 6,82x1011 copies/µl
Verdünnungsreihe: 1:6,8 (1 µl RNA + 5,8 µl H2O) = 1x1011 copies/µl, dann seriell 1:10 verdünnen von 1010 bis 103
Als Standard einsetzen:103 - 109 copies/µl Proben als Doppelbestimmung einsetzen! MasterMix ansetzen (PCR-Raum!): pro Probe: 5,8 µl RNase-freies Wasser 10 µl 2x QuantiTect SYBR Green RT-PCR Master Mix 1 µl Primer F (0,5 µM Endkonz.) 1 µl Primer R (0,5 µM Endkonz.) 0,2 µl QuantiTect RT Mix 18 µl Gesamtvolumen gesamt (n+2 = 30): 174 µl RNase-freies Wasser 300 µl 2x QuantiTect SYBR Green PCR Master Mix 30 µl Primer F 30 µl Primer R 6 µl QuantiTect RT Mix

BCII am PEI, 22.11.2010-09.12.2010
54
540 µl vortexen 18 µl pro Kapillare + je 2 µl Standard und Template-RNA (erst im Labor zugeben!) Probenreihenfolge: 1: Wasser 15: Probe 4 2: Standard 2 x 103 16: Repl. von Probe 4 3: Standard 2 x 104 17: Probe 5 4: Standard 2 x 105 18: Repl. von Probe 5 5: Standard 2 x 106 19: Probe 6 6: Standard 2 x 107 20: Repl. von Probe 6 7: Standard 2 x 108 21: Probe 7 8: Standard 2 x 109 22: Repl. von Probe 7 9: Probe 1 23: Probe 8 10: Repl. von Probe 1 24: Repl. von Probe 8 11: Probe 2 25: Probe 9 12: Repl. von Probe 2 26: Repl. von Probe 9 13: Probe 3 27: Probe 10 14: Repl. von Probe 3 28: Repl. von Probe 10 Kapillaren mit Deckel verschließen und mit Proben zum LightCycler gehen. Probenkarussell mit Kapillaren beladen und in Zentrifuge kurz zentrifugieren. Probenkarussell in LightCycler platzieren. PCR: RT-Reaktion 50°C 20 min Denaturierung 95°C 15 min Amplifikation (40 cycles) 94°C 15 sec 58°C 15 sec 72°C 16 sec slope 2°C/s, single
measurement Schmelzkurve 95°C 10 sec 65°C 10 sec 97°C 0 slope 0,1°C/s, cont.
measurement Cooling 40°C 30 sec (wenn nicht anders angegeben: slope 20°C/s) Nach Beendigung des PCR-Laufs werden die Proben entsorgt und es erfolgt entweder direkt die Auswertung der Daten oder aber am zweiten Tag (je nach Gruppeneinteilung). RT-Test (C-Type-RT Activity Assay, Protokoll B, Cavidi-Tech)
Bestimmung der Enzymaktivität der reversen Transkriptase im Überstand PERV-infizierter Zellen (d.h. in den Viruspartikeln, die sich im Überstand befinden).

BCII am PEI, 22.11.2010-09.12.2010
55
Sample Dilution Buffer (Flasche B) Reconstitution Buffer (Flasche C2) bei 37°C im Wasserbad auftauen Benötigte Anzahl poly(A)-beschichteter Streifen (siehe Tab.1), Sample Dilution Buffer (2x Flasche B2, weißer Deckel), RT Reaction components (Flasche C1, grüner Deckel, 1 Flasche pro 96-well-plate) und MMuLV-Standard (Flasche D, gelber Deckel, Lot-Nr. 05133) aus –20°C nehmen. Für Gruppe A: Proben aus -80°C nehmen und auf Eis auftauen. Reaktionsmix herstellen: pro Platte 12 ml Reconstitution Buffer (C2) zu einer Flasche C1 geben,
gut mischen und in 50 ml Falcon geben 6 ml Wasser zugeben Sample Dilution Buffer (B) herstellen (je 5 ml aus B1 pro Flasche B2, gut
mischen und Inhalt beider B2‘s wieder in B1 geben, mischen) 6 ml Sample Dilution Buffer (B) in Falcon geben, mischen Poly(A)-Platten vorbereiten: entsprechende Anzahl 8-er-Streifen in 96-well-plate-Rahmen spannen
(Tab. 1) pro well 200 µl Reaktionsmix mit elektronischer Mehrkanalpipette
zugeben Platten mit selbstklebender Folie schließen Präinkubation der Platten für 20-60 min bei 33°C

BCII am PEI, 22.11.2010-09.12.2010
56
Tab. 1: Probenauftragung RT-Test
1
2
3
4
A 1
767 mU/ml
B 2
341 mU/ml
C 3
151 mU/ml
D 4
67,3 mU/ml
E 5
29,9 mU/ml
F 6
13,3 mU/ml
G 7
5,91 mU/ml
H 8
2,63 mU/ml
MMuLV-Standard verdünnen (gilt für Lot-Nr. MUL 05133 !): 420 µl Sample Dilution Buffer (B) zu MMuLV-Lyophilisat geben, gut
mischen 8 Eppi’s vorbereiten, je 250 µl Sample Dilution Buffer (B) vorlegen 200 µl resuspendierten MMuLV Standard ins erste Eppi geben, vortexen Pipettenspitze wechseln, 200 µl aus dem ersten ins zweite Eppi geben,
vortexen Pipettenspitze wechseln, 200 µl aus dem zweiten Eppi ins dritte geben,
vortexen, usw. bis Eppi Nr. 8 RT-Reaktion starten: Poly(A)-Platten aus 33°C-Inkubator nehmen Standard (je 10 µl/well) in die erste Reihe pipettieren (von oben nach
unten abnehmend, entsprechend Tab. 1) Proben zugeben (als Duplikate, Pipettierschema ist selbst zu entwerfen
und in Tab. 1 einzutragen), je 10 µl/well Platten mit selbstklebender Folie verschließen

BCII am PEI, 22.11.2010-09.12.2010
57
Platten für 2-3 Std. bei 33°C inkubieren Platten waschen: Waschpuffer (2.5 ml Waschpufferkonzentrat, 75 ml 10% Triton X-100 ad
1 l H2O) Platte im ELISA-Washer 3x mit 200 µl Waschpuffer pro well waschen
(Programm RT, last strip = 4) Platte umgedreht auf Celltorks aufklopfen Platte kann jetzt entweder eingefroren (-20°C, Gruppe B) oder der
Versuch fortgesetzt werden (Gruppe A) Inkubation mit Product Tracer: 12 ml 1% Triton X-100 zu einer Flasche Product Tracer (O) geben,
vortexen benötigt ca. 10-20 min, bis sich das Lyophilisat gelöst hat, zwischendurch
vortexen je 100 µl Product Tracer pro well zugeben Inkubation bei 33°C für 90 min Inkubation mit AP-Substrat: Flasche mit AP Substrat Puffer bei 37°C im Wasserbad auftauen AP-Substrat Tablette (P1) zu AP Substrat Puffer (Flasche mit Alufolie
umwickeln) geben, vortexen (braucht ca. 10 min zum Lösen) Platte im ELISA-Washer 3x mit 200 µl Waschpuffer pro well waschen
(Programm s.o.) Platte umgedreht auf Celltorks aufklopfen je 125 µl AP-Substrat pro well, Platten mit Kunststoffdeckel schließen, im
Dunkeln 15-30 min inkubieren Platten im ELISA-Reader messen (A405)

BCII am PEI, 22.11.2010-09.12.2010
58
Auswertung:
a) Berechnung der Anzahl der PERV Virionen in den Proben und
Darstellung im Balkendiagramm (Mittelwerte bilden und Standardabweichung [Fehlerbalken] eintragen)
Anhand der PERV-Standardreihe der RT-PCR ermittelt die LightCycler-Software die RNA-Kopien in 2 µl Probe. Viruspartikel enthalten einen doppelten Satz virale Nukleinsäure (2x vRNA), daher muss dieser Wert durch 4 geteilt werden, um die RNA-Kopienzahl pro µl Probe zu erhalten.
RNA-Kopienzahl (in 2 µl) / 4 = Anzahl der Viruspartikel pro µl Probe
b) Berechnung bzw. grafische Auswertung (Balkendiagramm aus
Mittelwerten der Proben, Standardabweichung eintragen) des RT-Tests
c) Vergleich der erhaltenen Daten von RT-PCR und RT-Test Frage: Kann durch spezifische siRNA eine Reduktion der Virusbelastung in PERV-infizierten humanen Zellen qualitativ und quantitativ erreicht werden? Literatur:
1) http://de.wikipedia.org/w/index.php?title=RNA-Interferenz&printable=yes
2) Hien K. RNA-Interferenz (RNAi). Laborjournal-Ausgabe 07/2002 aus http://www.laborjournal.de/rubric/archiv/stichwort/w_02_07.html
3) Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC: “Potent and specific genetic interference by double stranded RNA in Caenorhabditis elegans.” Nature 391, 806-811, (1998).
4) Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T: “Duplexes of 21-nucleotide RNAs mediate RNA interference in mammalian cell culture.“ Nature 411, 494-498 (2001).
5) http://www.invitrogen.com/content.cfm?pageid=10075 6) http://opengenomics.org/CSB2005_RNAiTutorial/3-CSB2005-
B&WSlides.pdf 7) Laborjournal 11/2008, S. 60-63
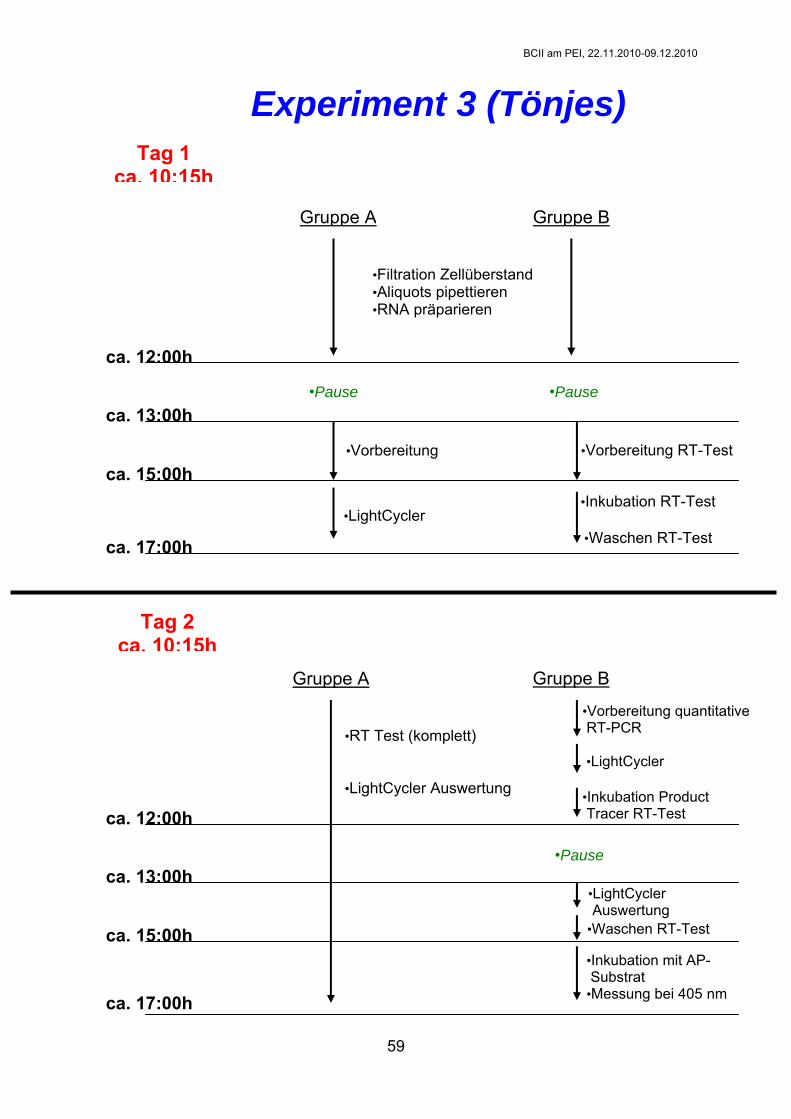
BCII am PEI, 22.11.2010-09.12.2010
59
Experiment 3 (Tönjes) Tag 1
ca. 10:15h
Tag 2 ca. 10:15h
ca. 12:00h
ca. 13:00h
ca. 15:00h
•Filtration Zellüberstand •Aliquots pipettieren •RNA präparieren
ca. 17:00h
ca. 12:00h
ca. 13:00h
ca. 15:00h
ca. 17:00h
Gruppe A
•Pause
•LightCycler
Gruppe A
•RT Test (komplett) •LightCycler Auswertung
•Vorbereitung
Gruppe B
•Pause
Gruppe B
•Vorbereitung RT-Test
•Inkubation RT-Test
•Waschen RT-Test
•LightCycler Auswertung
•LightCycler
•Pause
•Waschen RT-Test
•Inkubation mit AP- Substrat •Messung bei 405 nm
•Vorbereitung quantitative RT-PCR
•Inkubation Product Tracer RT-Test

BCII am PEI, 22.11.2010-09.12.2010
60
Quantifizierung von Vacciniaviren
AG Schwantes/Suezer
Allgemeine Einleitung In diesem Versuch soll die Herstellung einer Virus-Impfpräparation und deren Quantifizierung behandelt werden. Dazu werden einzelne Arbeitsschritte durchgeführt, welche die Herstellung und Anzucht von primären Zellen für die Amplifikation von Viren, die Aufreinigung von Viruspartikeln und die Quantifizierung einer fertigen aufgereinigten Virus-Stammlösung (Virus-Stock) beinhalten. Pockenviren gehören zu den behüllten Viren und besitzen ein doppelsträngiges DNA-Genom. Im Gegensatz zu anderen Viren mit einem DNA-Genom replizieren sie ausschließlich im Zytoplasma der infizierten Zelle. Sie durchlaufen während des Replikationszyklusses verschiedene Reifungsstadien, die in die Bildung verschiedener Formen infektiöser Partikel münden. Diese infektiösen Virionen werden von den unreifen Viren (engl.: immature virion, IV) abgegrenzt und in folgende Klassen eingeteilt: intrazelluläre reife Viren (engl.: intracellular mature virion, IMV), intrazelluläre behüllte Viren (engl.: intracellular enveloped virion, IEV) und extrazelluläre bzw. zellassoziierte behüllte Viren (engl.: extracellular/cell-associated enveloped virion, EEV/CEV). Die extrazellulären behüllten Virionen knospen von infizierten Zellen und sorgen vorrangig für die Verbreitung des Virus innerhalb eines infizierten Wirtsorganismus. Die intrazellulären infektiösen Viren werden zusammen mit infizierten Zellen bzw. Zellresten freigesetzt und dienen vermutlich der Übertragung auf neue Wirte.

BCII am PEI, 22.11.2010-09.12.2010
61
Abbildung 1 Schema des Replikationszyklus von Orthopockenviren. e) frühe, i) intermediäre und l) späte Genexpression, f) virale Fabriken, MV = reifes Viruspartikel, IV = intrazelluläre unreife Viruspartikel (immature virions), IMV = intrazelluläre reife Viruspartikel (intracellular mature virions), IEV = intrazelluläre behüllte Viruspartikel (intracellular enveloped virions), CEV = zellassoziierte behüllte Viruspartikel (cell-associated enveloped virions), EEV = extrazelluläre behüllte Viruspartikel (extracellular enveloped virions). Unter den Pockenviren wurden die Vacciniaviren (VACV) ehemals als Impfstoffe gegen die Pockenerkrankung beim Menschen erfolgreich eingesetzt, was die Ausrottung dieser Erkrankung zur Folge hatte. Heutzutage sind Pocken-Virusimpfstoffe, vor allem als Vektorimpfstoffe, wieder gefragt. Hierbei dient das Pockenvirus lediglich als Träger für Antigene, um damit gegen andere Krankheitserreger oder Erkrankungen eine Immunantwort zu induzieren. Aufgrund der früher gelegentlich beobachteten und teilweise erheblichen Nebenwirkungen der replikationskompetenten Vacciniaviren wurden attenuierte Pockenviren entwickelt, welche ein hohes Sicherheitsprofil aufweisen und die auch bei Menschen mit. Immunsuppression oder bei Schwangeren ohne Risiko verabreicht werden können. Ein solcher wichtiger Impfstoffkandidat ist das in diesem Praktikum verwendete modifizierte Vacciniavirus Ankara (MVA). Im Gegensatz zu VACV ist MVA nicht mehr in der Lage, sich produktiv zu vermehren. Es infiziert noch Zellen und exprimiert alle Gene, besitzt aber einen Block in der Virionen-Morphogenese. Daher bildet es auch in Zellkultur, anders als seine replikationskompetenten Verwandten, keine lytischen Plaques aus, sondern infiziert nur einzelne Zellen. Auch der sonst typische zytopathische Effekt ist nicht vorhanden. Eine Ausnahme bilden dabei primäre Hühnerembryofibroblasten (engl. chicken embryo fibroblast cells, CEF) oder Baby-Hamster-Nierenzellen (engl. baby hamster kidney cells, BHK), auf denen MVA noch replizieren kann und welche auch zur Anzucht von MVA genutzt werden.

BCII am PEI, 22.11.2010-09.12.2010
62
Überblick über den Ablauf der einzelnen Experimente: Tag 1: Experiment 1: Präparation primärer Hühnerembryofibroblasten Experiment 2: Herstellung eines gereinigten Virus-Stocks mittels Sucrose-
Gradienten- Zentrifugation
→ Vorbereitung des Gradienten Experiment 3: Bestimmung des Virus-Titers einer Virus-Präparation mittels
Immunostain und Kristallviolettfärbung
→ Vorbereitung und Titration der Virus-Stammlösungen auf verschiedenen Zellen
Tag 2: Experiment 2: Herstellung eines gereinigten Virus-Stocks mittels Sucrose-Gradienten-
Zentrifugation
→ Beladen und Lauf des Gradienten mit der Virus-Stammlösung; Ernte des gereinigten Virusmaterials
Experiment 3: Bestimmung des Virus-Titers einer Virus-Präparation mittels
Immunostain und Kristallviolettfärbung
→ Fixieren und Färben der Titrationen, Auszählen, Bestimmung des Virustiters

BCII am PEI, 22.11.2010-09.12.2010
63
Versuch 1: Präparation primärer Hühnerembryofibroblasten
Einleitung: Zur Virusanzucht werden sogenannte permissive Zellen benötigt, das sind solche Zellen, die ein produktives Wachstum des jeweiligen Virus erlauben. Dabei erfolgt die Anzucht des MVA auf CEF- bzw. BHK-Zellen und die eines replikationskompetenten Pockenvirus, hier verwenden wir das Mäusepockenvirus (Ectromelievirus, ECTV), auf BS-C-1-Zellen (epitheliale Nierenzellen der grünen Meerkatze). Die Zellen werden mit einer niedrigen MOI (engl. multiplicity of infection, Menge an infektiösen Virionen pro Zelle) infiziert, und 3-4 Tage nach der Vermehrung des Virus können diese geerntet werden. Innerhalb der Zellen befinden sich die meisten infektiösen Virionen, aber auch extrazellulär sind Viren enthalten. Daher werden sowohl der Zellkulturüberstand als auch die Zellen geerntet. Durch wiederholte Einfrieren-Auftauen-Schritte und die Behandlung durch Ultraschall werden die Zellen zerstört und die intrazellulären Viruspartikel freigesetzt und vereinzelt. Durch anschliessende Zentrifugationsschritte werden die Virionen angereichert und gereinigt. CEF-Zellen werden aus den Embryonen von 10 - 11 Tage lang bebrüteten Hühnereiern hergestellt. Da die Embryonen zur Virusanzucht verwendet werden sollten die Eier aus speziell überwachten Haltungen, d.h. aus sogenannten spezifisch-pathogen freien (spf-)Hühnern stammen (z.B. Lohmann Tierzucht GmbH). Zur Präparation der Zellen werden zunächst die Embryonen aus dem Ei entnommen, durch Entfernung des Kopfes getötet und das differenzierte Gewebe (Herz, Leber, Niere) von dem undifferenzierten Fibroblasten-Gewebe (Körper) getrennt. Das Fibroblastengewebe wird anschliessend durch die Methode der fraktionierten Trypsinierung in einzelne Zellen aus dem Zellverband gelöst. Diese noch undifferenzierten Zellen können einige Tage in Kultur gehalten werden, während der sie ein normales Zellwachstum zeigen. Während dieser primären Phase sind sie effektive Produzenten von neuen Viruspartikeln, insbesondere von MVA. Nach einigen Zellteilungen differenzieren die Zellen aus. Dabei verlieren sie die Fähigkeit zur Teilung und sind, vermutlich aufgrund veränderter Genexpressionsmuster innerhalb der Zelle, für eine Anzucht von MVA nicht mehr geeignet. Material:
o PBS, Trypsin o Kulturmedium mit 10 % FCS (fötales Kälberserum) (v/v), 1x NEA (nicht-essentielle
Aminosäuren), 2mM L-Glutamin, Penicillin (60 µg/ml) and Streptomycin (100 µg/ml).
o Kühl-Zentrifuge, Gasbrenner, Feuerzeug o 2-4 Pinzetten, 2 Scheren, steril o Bechergläser 200 ml, 300 ml, 500ml oder 1 l, steril o Bechergläser (400ml), die mit 2 Lagen Gaze bespannt wurden, steril o Erlenmeyerkolben, 300-500 ml, steril o große Petrischalen, steril o Magnetrührer und Magnetrührerstäbchen („Rührfische“), steril o Falcon-Röhrchen (50 ml) o Zellkulturflaschen (T175)

BCII am PEI, 22.11.2010-09.12.2010
64
o 200 ml Spritzen Versuchsablauf:
Vorbereitung: o ca. 20 Eier 10 Tage vor Präparation in einen Brutschrank (37,8°C, 53 % Luftfeuchte)
legen Durchführung (Tag 1): o Eier mittels einer Schier-Lampe auf lebende Embryonen überprüfen o Eier mit Ethanol abspülen o Schale am stumpfen Pol mit Schere aufschlagen und Schalenteile mit steriler
Pinzette entfernen o Eihaut mit neuer steriler Pinzette „wegklappen“ o Embryo mit Pinzette aus dem Ei entnehmen, in einer große, mit PBS gefüllte
Petrischale geben und sofort „köpfen“ (Tierschutz! Kopf wird verworfen) o alle weiteren Eiern so prozessieren o Embryonen in eine frische, mit PBS gefüllte Petrischale überführen und Eingeweide
entfernen o gesäuberte Teile (Rumpf mit Beinen und Flügeln) in eine Petrischale mit frischem
PBS überführen, schwenken, Schritt wiederholen o Embryonenteile zur Zerkleinerung durch eine 20 ml-Spritze direkt in einen 300-
500ml-Erlenmeyerkolben mit sterilen Magnetrührerstäbchen drücken o Zugabe von ca. 2,5 ml Trypsin (37°C) pro Ei o Zugabe von PBS (ad ca. 6,25 ml pro Ei) o Suspension 20-25 min auf dem Magnetrührer bei RT rühren nach der Inkubation erhält man flüssigen Überstand () und Restgewebe ()
zu : o flüssigen Überstand in ein mit steriler Gaze bespanntes Becherglas geben und so
filtrieren (Gazebespannung dazu ein bisschen ins Becherglas zur Vertiefung eindrücken)
o Filtrat in 2 x 50 ml-Falcon-Röhrchen überführen (jeweils gleiche Mengen zum Austarieren der Zentrifuge!)
o Filtrat zentrifugieren, 1.500 rpm, 4°C, 7 min, es entsteht ein lockeres Zellpellet o Überstand vorsichtig in steriles Becherglas abgießen (Pellet schwimmt leicht ab!) und
verwerfen o Waschschritt: Pellets in ca. 15 ml PBS aufnehmen, gut durch Schütteln mit der Hand
mischen (nicht vortexen!!!), beide Zelllösungen vereinen o Zelllösung zentrifugieren, 1.500 rpm, 4°C, 7 min o Waschschritt mit PBS wiederholen o Überstand verwerfen, Pellet in ca. 10 ml CEF-Medium aufnehmen, durch Schütteln
mit der Hand mischen, auf Eis stellen
zu : o Restgewebe im Erlenmeyerkolben (Volumen: ca. 40 ml) o Zugabe von vorgewärmtem Trypsin (37°C, ad ca. 80 ml, ca. 2 ml Trypsin pro Ei) zum
Restgewebe o Zugabe von PBS (ad ca. 125 ml, ca. 6.25ml Gesamtvolumen inkl. Trypsin pro Ei) o Suspension 20 min bei RT rühren nach der Inkubation: flüssiger Überstand (, wieder wie bei verfahren) und Restgewebe ()
zu : o Restgewebe im Erlenmeyerkolben (Volumen: ca. 30 ml)
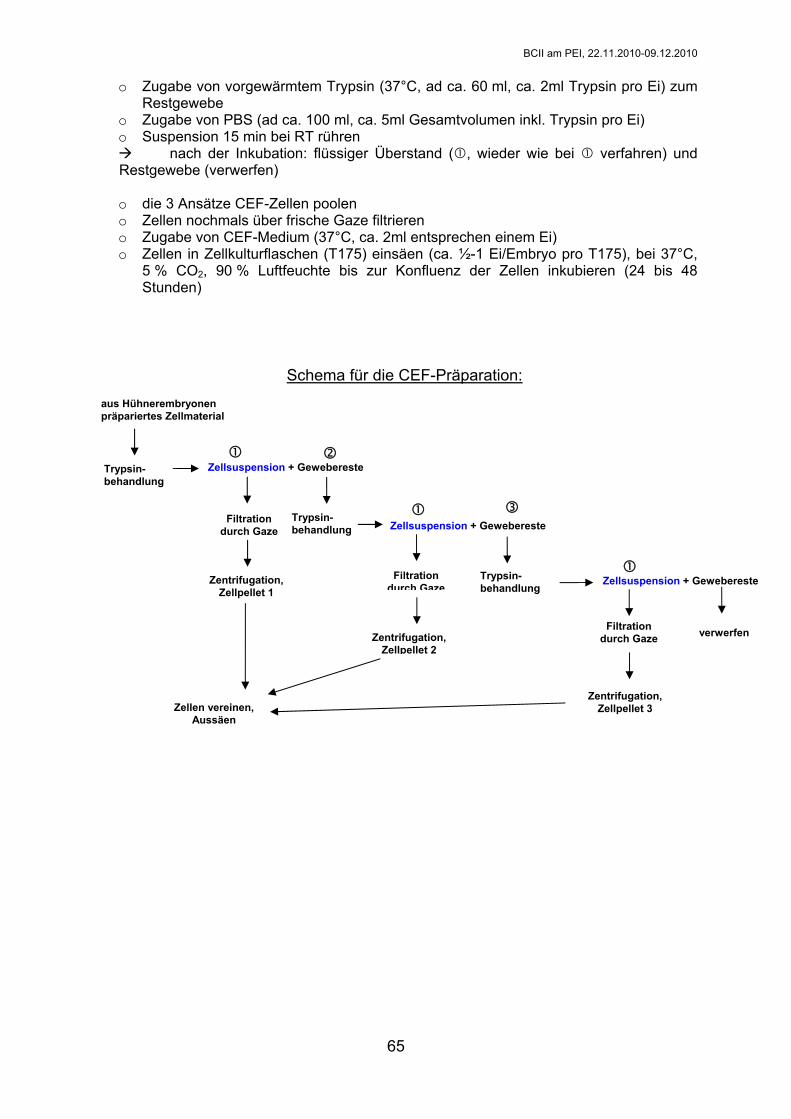
BCII am PEI, 22.11.2010-09.12.2010
65
o Zugabe von vorgewärmtem Trypsin (37°C, ad ca. 60 ml, ca. 2ml Trypsin pro Ei) zum Restgewebe
o Zugabe von PBS (ad ca. 100 ml, ca. 5ml Gesamtvolumen inkl. Trypsin pro Ei) o Suspension 15 min bei RT rühren nach der Inkubation: flüssiger Überstand (, wieder wie bei verfahren) und Restgewebe (verwerfen)
o die 3 Ansätze CEF-Zellen poolen o Zellen nochmals über frische Gaze filtrieren o Zugabe von CEF-Medium (37°C, ca. 2ml entsprechen einem Ei) o Zellen in Zellkulturflaschen (T175) einsäen (ca. ½-1 Ei/Embryo pro T175), bei 37°C,
5 % CO2, 90 % Luftfeuchte bis zur Konfluenz der Zellen inkubieren (24 bis 48 Stunden)
aus Hühnerembryonen präpariertes Zellmaterial
Trypsin- behandlung
Filtration durch Gaze
Zellsuspension + Gewebereste
Zentrifugation, Zellpellet 1
Trypsin- behandlung
Schema für die CEF-Präparation:
Filtration durch Gaze
Zellsuspension + Gewebereste
Zentrifugation, Zellpellet 2
Trypsin- behandlung
Filtration durch Gaze
Zellsuspension + Gewebereste
Zentrifugation, Zellpellet 3
verwerfen
Zellen vereinen, Aussäen

BCII am PEI, 22.11.2010-09.12.2010
66
Versuch 2: Herstellung eines gereinigten Virus-Stocks mittels
Sucrose-Gradienten-Zentrifugation Einleitung: Die Virusaufreinigung erfolgt durch mehrere Zentrifugationsschritte. In einer ersten Zentrifugation werden alle größere Bestandteile (Virionen des Zellkulturüberstandes, Zell-assoziierte Virionen, Zellreste) pelletiert. Dieses Pellet wird nun mit Ultraschall behandelt, um zell-assoziierte Virionen (CEV) freizusetzen. Um die Virionen aufzureinigen, wird das Pellet über ein Sucrosekissen definierter Dichte (36% Sucrose (V/V)) pelletiert. Dabei passieren die Virionen das Kissen, dagegen bleiben größere zelluläre Bestandteile auf oder in dem Kissen liegen/stecken. Das Viruspellet wird weiter aufgereinigt, indem eine Gradientenzentrifugation angeschlossen wird. Der Gradient besteht aus Sucrose zunehmender Dichte (20% bis 50 oder 60% Sucrose). Hier reichern sich die Virionen in einer bestimmten Dichte an und bilden eine Bande aus. Diese Bande wird geerntet und anschliessend werden die Virionen gewaschen, um die Sucrose aus der Viruslösung zu entfernen. Material:
o 50 % (V/V) Sucrose-Lösung in 10mM TrisCl pH 9,0, steril o 20 % (V/V) Sucrose-Lösung in 10mM TrisCl pH 9,0 , steril o UZ-Röhrchen für rote (kleine) Buckets (Beckman Nr. 337986 Polyallomer), Rotor
SW32Ti o 10 mM TrisHCl pH 9,0 o Ultraschallgerät, Pipetten, Ultrazentrifuge Beckman, Kryo-Röhrchen (1-2ml)
Versuchsablauf:
Herstellung des Gradienten (Tag 1): o 50 % und 20 % Sucrose-Lösung zu gleichen Teilen (7 + 7ml) im UZ-Röhrchen
vorsichtig und langsam übereinander schichten, unten die 50%ige, oben die 20%ige Sucrose-lösung
o zu 4°C stellen (12-48h) Gradient bildet sich über Nacht aus (Gradient immer mit äußerster Vorsicht transportieren und behandeln) Beladung und Lauf des Gradienten (Tag 2): o Virus-Suspension (Sucrose-Kissen gereinigt) 3x1 min beschallen (100 %),
zwischendurch gut vortexen o Sucrose-Gradienten mit Virus-Suspension überschichten (max. 1 ml pro Röhrchen,
bzw. Flüssigkeitsspiegel sollte minimal 0,5 cm vom oberen Rand des Röhrchen enfernt sein, sonst kollabieren die Röhrchen ) ACHTUNG: Verwirbelungen vermeiden!!!
o UZ-Röhrchen in Buckets einsetzen und gegeneinander austarieren o zentrifugieren, Rotor SW32Ti, rote Buckets, 13.300 rpm, 4°C, 50 min, kein Bremsen
(„no brake“) nach der Zentrifugation sind eine Bande (, aufgereinigtes Virus) und ein Pellet (, Reste von Virus an Zellwand gebunden & Zelltrümmer) zu erkennen

BCII am PEI, 22.11.2010-09.12.2010
67
zu : o Sucrose über der Bande abpipettieren o Bande vorsichtig mit der Pipette entnehmen und in 50ml Falcon überführen o Zugabe von ca. 20ml 10 mM Tris pH 9,0 zum Waschen o zentrifugieren, Rotor SW32Ti, rote Buckets, 14.000 rpm, 4°C, 60 min, maximales
Bremsen nach der Zentrifugation ist ein Pellet zu erkennen o Überstand vorsichtig abkippen o Pellet in ca. 500µl 10 mM Tris pH 9,0 resuspendieren, bei -80°C lagern
zu : o Nach Entnahme der Bande restliche Sucrose abkippen o Röhrchen umgekehrt auf ein mit Alkohol getränktes Papier stellen o Pellet in ca. 500µl 10 mM Tris pH 9,0 resuspendieren, bei -80°C lagern

BCII am PEI, 22.11.2010-09.12.2010
68
Versuch 3: Bestimmung des Virus-Titers einer Virus-Präparation mittels
Immunostain und Kristallviolettfärbung Einleitung: Die Quantifizierung von Pockenviren wird im Zellkultursystem durchgeführt, wobei mit dieser Methode die infektiösen Partikel (IMV, IEV, EEV und CEV) bestimmt werden. Dabei können normalerweise bei ausreichender Verdünnung der Viruslösung Löcher im Zellrasen beobachtet werden, die jeweils auf die Infektion mit einem einzelnen Virus zurückzuführen sind. Daher bestimmt die Anzahl dieser Löcher (engl. plaques) die Menge an infektiösen Viren und werden entsprechend als sogenannten Plaque-formende Einheiten pro Milliliter (engl. plaque forming units/ml bzw. pfu/ml,) angegeben. Würde man die Quantifizierung mittels des Nachweises der viralen DNA-Genome durchführen, würde man auch Viruspartikel nachweisen, welche zwar ein Genom enthalten, aber nicht-infektiös wären. Diese nicht-infektiösen Viren liegen in höherer Anzahl als die infektiösen Viren vor und bezeichnet den Faktor des Verhältnisses beider Formen als den "Particle-to-Pfu-Ratio". Dieses Verhältnis liegt bei den Pockenviren bei ca. 100:1. Von einer Virussuspension mit unbekannter Menge an (infektiösen) Virionen wird eine Verdünnungsreihe in 10er Schritten angelegt und in einem definierten Volumen (1ml) auf Zellen in einer 6 Loch-Platte gegeben. Zur Verifizierung des Ergebnisses wird stets ein Doppelansatz durchgeführt. Ausgehend von einer -1 Verdünnung, das heißt, einer 10fachen Verdünnung der Virussuspension, bis zu einer -9 Verdünnung, werden die Verdünnungsstufen in jeweils ein Loch einer 6 Loch-Platte im Doppelansatz transferiert. Verdünnungsschema der Viruslösung und Ausbringen in 6-Loch-Platten
Nach 2 Tagen, in denen die Viren sich vermehrt haben, können bereits makroskopisch Löcher im Zellrasen beobachtet werden, die durch die lytische Aktivität und den zytopathischen Effekt der Virusinfektion hervorgerufen wurden. Dabei bilden die Viren in Abhängigkeit vom genutzten Zelltyp unterschiedlich große Plaques aus. MVA bildet dabei auf CEF größere Plaques aus als auf BHK und das ECTV bildet auf BS-C-1 nur kleine, im Mikroskop sichtbare, Plaques aus. Um die Plaques als Herd einer Virusinfektion zu verifizieren, können Färbemethoden die Analyse unterstützen. Insbesondere bei MVA-
10-1 10-2 10-3 10-4 10-5 10-6
-4 -5 -6
-6-5-4
-1 -2 -3
-3 -2 -1
je 1ml
Virus
serielle Verdünnungen

BCII am PEI, 22.11.2010-09.12.2010
69
Infektionen wird auf eine immun-histochemisch vermittelte Färbung mit VACV-spezifischen Antikörpern zurückgegriffen. Der VACV-spezifische Antikörper wird durch einen Zweit- Antikörper detektiert, der gegen die leichte und schwere Kette des ersten Antikörpers gerichtet ist. Der Zweit-Antikörper ist mit der Meerrettichperoxidase (Engl. horse raddish peroxidase, HRP) gekoppelt, welche das Substrat TMB (3,3´,5,5´Tetramethylbenzidine) in Anwesenheit von H2O2 oxidiert, wobei das Substrat in einen blauen Farbstoff umgewandelt wird. Dadurch erscheinen infizierte Zellen dunkelblau. Dagegen werden Plaques von Zellen, die mit replikationskompetenten Viren infiziert wurden, indirekt durch Färbung und gleichzeitiger Fixation der noch intakten Zellen mit Kristallviolett sichtbar gemacht. Hierbei erscheinen die Plaques als weiße Punkte oder Löcher vor violett-blauem Hintergrund. Der Virustiter berechnet sich dabei folgendermaßen: Es wird das Loch, in dem sich mindestens 20-30 Plaques auszählen lassen gewählt und die Anzahl der Plaques gezählt. Dann wird diese Zahl mit dem Verdünnungsfaktor multipliziert und man erhält einen Titer von Plaque-bildenden Einheiten pro ml. Titer = Anzahl Plaques * Verdünnung des ausgezählten Loch´s Die Plaques werden in beiden Doppel-Ansätzen ausgezählt und gemittelt, woraus der endgültige Titer errechnet wird. Vorarbeit:
CEF-Herstellung, Virus-Amplifikation Type 19 Ultrazentrifugation Surcose-Kissen (36% V/V Sucrose in 10mM TrisCl pH 9,0)
Material:
o gereinigter Virus-Stock, dessen Titer bestimmt werden soll o 6-well-Platten mit sekundären CEF Zellen oder BHK-Zellen sowie BS-C-1 Zellen o Kulturmedium mit 2 % FCS (CEF EMEM, BHK RPMI, BS-C-1 DMEM) o Aceton-Methanol Gemisch (1:1) o Kristallviolettlösung (0,1 % Kristallviolett, 20 % Ethanol in H2O) o PBS mit 3 % FCS o rabbit-anti-Vacciniavirus-Antikörper (von Acris) o goat-anti-rabbit-HRP-Antikörper (von JacksonResearch) o PBS o TrueBlue (von Medac) o Ultraschallgerät, Falcon-Röhrchen, Pipetten, Rockerplattform

BCII am PEI, 22.11.2010-09.12.2010
70
Versuchsablauf:
Vorbereitung am Vortag: o Zellen in 6-well-Platten aussäen, so dass sie bis zum nächsten Tag etwa 80 %
Konfluenz erreichen
Durchführung (Tag 1): o Virus-Suspension 3 x 1 min mit Ultraschall (100 %) behandeln, zwischendurch
vortexen o Virus-Verdünnungen ansetzen, zwischendurch gut vortexen:
20 µl Virus + 180 µl Medium 10-1 100 µl Virus-Verdünnung 10-1 + 900 µl Medium 10-2 500 µl Virus-Verdünnung 10-2 + 4.500 µl Medium 10-3 500 µl Virus-Verdünnung 10-3 + 4.500 µl Medium 10-4
usw. bis 10-9
o Medium von Zellen abnehmen o Virus-Verdünnungen auf Zellen ausbringen, je 1 ml/well im Doppelansatz o Zellen bei 37°C, 5 % CO2, 90 % Luftfeuchte 2 h inkubieren o Virus-Verdünnungen abnehmen o 2 ml Kulturmedium/well zufügen o Zellen bei 37°C, 5 % CO2, 90 % Luftfeuchte 48 h inkubieren Durchführung (Tag 2): CEF oder BHK Immunostain: o Medium abnehmen (infektiös! In den Flüssigabfallbehälter und dann autoklavieren) o Zellen mit 1-2 ml/well Aceton-Methanol (1:1) fixieren, 5 min einwirken lassen,
abnehmen (gesonderter Abfall!) und Methanolfilm komplett verdunsten lassen o 2 ml PBS/3 % FCS pro well zufügen, 30 min bei Raumtemperatur (RT) inkubieren o Lösung abnehmen o 1. Antikörper zugeben: 500µl/well, Verdünnung 1:4000 in PBS/3 % FCS o Platte mindestens 1 Std. bei RT auf Rockerplattform inkubieren o Antikörper-Lösung abnehmen o Platte dreimal mit PBS waschen (Volumen ca. 2ml) o 2. Antikörper zugeben: 500µl/well, Verdünnung 1:4000 in PBS/3 % FCS o Platten 45 min bis max. 1 Std bei RT auf Rockerplattform inkubieren o Antikörper-Lösung abnehmen o Platte dreimal mit PBS waschen o Substrat zugeben (500µl/well), auf Rockerplattform färben o nach 10 Minuten Färbe-Lösung abnehmen, mit Aqua dest. 5 Minuten waschen o Aqua dest abnehmen und Platten trocknen lassen
BS-C-1 Kristallviolettfärbung: o Medium abnehmen (infektiös! In den Flüssigabfallbehälter und dann autoklavieren) o 2 ml/well Kristallviolettlösung pro Vertiefung zugeben 5 min inkubieren (= Fixation und
Färbung) o Kristallviolett-Lösung abnehmen (gesonderter Abfall!) o Platte umdrehen auf Zelltork und komplett trocknen lassen
CEF/BHK & BS-C-1: o Titer auszählen (mindestens 30-100 Plaques)
Beispiel: 30 Plaques bei einer Verdünnung von 10-8 Titer 3 x 109

BCII am PEI, 22.11.2010-09.12.2010
71
Experiment 4: Zellengineering (Bakterien und Säugerzellen)
Die Gruppen 1-3 führen das Experiment in der AG Bastian/Lößner,
die Gruppen 4-6 in der AG Schumann durch.

BCII am PEI, 22.11.2010-09.12.2010
72
Maßgeschneiderte bakterielle Lebendimpfstoffe (Gr. 1-3)
Holger Lößner und Max Bastian (Abteilung Veterinärmedizin)
Bakterielle Lebendimpfstoffe sind seit langem wichtiger Bestandteil des präventiven Gesundheitsschutzes für Mensch und Tier. Darüber hinaus wird die Anwendung lebender rekombinater Bakterien für die Behandlung von Krebs, Autoimmunerkrankungen, Allergien oder Gendefizienzen gegenwärtig intensiv erforscht. Die Immunisierung mit lebenden attenuierten Salmonellen steht exemplarisch für den sicheren und wirksamen Einsatz bakterieller Lebendimpfstoffe insbesondere in der Tiermedizin. Diese Impfstoffe wurden meist durch ungerichtete chemische Mutagenese von Feldisolaten abgeleitet, wobei der vollständige Umfang attenuierender Mutationen bislang unbekannt geblieben ist. Vor dem Hintergrund des zunehmenden Verständnisses der komplexen Wechselwirkungen zwischen Bakterien und dem Wirt sowie der Verfügbarkeit neuer Technologien, z.B. Hochgeschwindigkeitssequenzierung, neue Verfahren der Gensynthese, globale Analysemethoden, Recombineering, wird eine zunehmend rationale Entwicklung von maßgeschneiderten bakteriellen Vektoren für verschiedene medizinische Anwendungen möglich. Eine Seite stellt dabei die Herstellung von geeigneten bakteriellen Trägerstämmen dar, die zuverlässig attenuiert und genetisch stabil sein müssen, sowie phänotypisch markiert und von überflüssiger Stoffwechsellast befreit sein sollten. Dafür sind umfangreiche Modifikationen des bakteriellen Genoms nötig. Eine zweite Seite stellt die jeweilige Programmierung des Trägerstammes für die Expression und Freisetzung von Impfantigenen, therapeutischen Molekülen oder anderweitig unterstützenden Faktoren entsprechend der vorgesehenen Anwendung dar. Hierfür werden oftmals Plasmide in die Bakterien eingebracht oder Expressionskassetten in das bakterielle Genom integriert. Im praktischen Teil wird der Salmonellen-Impfstamm Salmonella enterica Serovar Typhimurium SL7207 gentechnisch auf verschiedene Weise manipuliert und anschließend werden die eingebrachten Modifikationen molekular und funktionell analysiert. Zuerst wird ein Salmonellen-Impfstamm durch ein Minitransposon mit GFP markiert. Die Markierung von Impfstämmen ist für die Entwicklung von Lebendimpfstoffen hilfreich und dient später der leichten Identifizierung solcher Impfstoffe im Feld. Anschließend wird mittels homologer Rekombination das Gen für die Aspartat-Semialdehyd-Dehydrogenase (asd) gezielt deletiert. asd ist essentiell für die Synthese von Diaminopimelischer Säure (DAP), einem wichtigen Bestandteil des Peptidoglykans gramnegativer Bakterien. Eine solche Mangelmutante bietet die Möglichkeit, ein Expressionsplasmid durch Komplementation mit asd zu stabilisieren (balanced lethal system). Mit einem Plasmid werden schließlich zwei Lyseproteine unterschiedlicher Herkunft und Funktion in Salmonellen eingebracht. Das vom Bakteriophagen phiX174 abgeleitete Lyseprotein E ist geeignet, Bakterienzellen durch Eingriff in die Zellwandsynthese zu zerstören. Gekoppelt mit einem speziellen Sensor kann dieses Protein eingesetzt werden, um Impfstämme bei Komplikationen während der Immunisierung zu beseitigen. Andererseits können zytoplasmatisch exprimierte Impfantigene oder therapeutische Faktoren durch die bakterielle Lyse freigesetzt werden. Ein weiteres Lyseprotein ist das porenformende Toxin Listeriolysin O (LLO) aus Listeria monocytogenes. LLO vermittelt die Zerstörung der phagosomalen Membran von Säugerzellen, so dass Listerien kurz nach ihrem Eindringen in die Wirtszelle aus dem Phagosom in das Zytoplasma gelangen. Wird LLO in andere intrazelluläre Bakterien, z.B. Salmonellen, übertragen, so können auch diese Bakterien sich aus einem Phagosom befreien und Antigene direkt in das Zellzytoplasma übertragen.
BCII am PEI, 22.11.2010-09.12.2010
73
Versuchsplan :
Versuche Tag 1 (Dienstag)
Tag 2 (Donnerstag)
A) Transposon-vermittelte chromosomale Markierung eines Salmonellen-Impfstammes
gfp wird Tn7-vermittelt in das Genom des Salmonellen-Impfstammes integriert. Dazu wird eine Konjugations-mischkultur mit dem Salmonellen-Impfstamm, dem Transposasehelferstamm und dem Transposonkarrierstamm hergestellt.
HeLa-Zellen werden mit GFP-markierten Salmonellen infiziert und fluoreszenzmikroskopisch analysiert.
B) Rekombinase-vermittelte Deletion des Gens für die Aspartat-Semialdehyd Dehydrogenase eines Salmonellen-Impfstammes
Ein PCR-Produkt mit kurzen homologen Sequenzen zum chromosomalen Zielgen asd und einem Selektionsmarker wird aufgereinigt und in den -RED-Rekombinase haltigen Salmonellen-Impfstamm transferiert.
Die Gendeletion wird anhand einer Kolonien-PCR sowie biochemisch bestätigt.
C) Transformation eines Expressionsplasmides für zwei lytische Faktoren in einen Salmonellen-Impfstamm
Ein Expressionplasmid für das bakterielle Zellwandlyseprotein E sowie für das Säuger-zellmenbran-lytische Toxin Listeriolysin O werden in einen Salmonellen-Impfstamm eingebracht.
Die induzierbare Protein E-vermittelte Lyse wird anhand der optischen Kulturdichte verfolgt. Die Aktivität von Listeriolysin O wird in einem Hämolyseassay nachgewiesen.
Versuchsdurchführung :
Versuch A
Chromosomale Integration einer konstitutiven GFP-Expressionskassette mittels eines Minitransposons. Im Gegensatz zu den meisten bakteriellen Transposons (Tn) inseriert Tn7 stellenspezifisch in das Genom gramnegativer Bakterien. Die Integrationsstelle attTn7 befindet sich downstream vom essentiellen Gen glmS. Die Transposonenden Tn7-L (150bp) und Tn7-R (90bp) sind die Ansatzpunkte für die Transpositionsproteine und vermitteln die Integration in einer bestimmten Orientierung. Vorbereitung : Die Tn7-Transpositionsproteine befinden sich auf dem mobilisierbaren Helferplasmid pUX-BF13 [1]. Für die Tn7-vermittelte chromosomale Integration wurde ein sogenanntes Minitransposon mit einer GFP-Expressionskassette, einem Selektionsmarker sowie den flankierenden Transposonenden Tn7-L und Tn7-R kloniert (pHL326). Beide Plasmide wurden jeweils in den mobilisierenden E. coli Stamm SMpir transformiert. Der Helferstamm, der Stamm mit dem Minitransposon (Karrierstamm) sowie der Salmonellen-Zielstamm SL7207 wurden auf einer Medienplatte übernacht bei 30°C angezogen.

BCII am PEI, 22.11.2010-09.12.2010
74
Durchführung : Dienstag Von den Medienplatten werden jeweils Bakterien mit einer Impföse gewonnen und in 1ml LB-Medium resuspendiert. Davon werden jeweils 200 µl zur Inokulation von 4 ml LB-Flüssigkulturen verwendet. Die Flüssigkulturen für die beiden E. coli-Stämme werden mit 100 µg/ml Ampicillin supplementiert und für den Salmonellenstamm mit 30 µg/ml Streptomycin. Die Kulturen werden 60 min im Schüttelinkubator bei 150 rpm und 37°C agitiert. Jeweils 1 ml Kultur mit einer OD600 von ca. 1 werden bei 5000 x g für 5 min abzentrifugiert, der Überstand vorsichtig abgesaugt und das Pellet in 8 µl LB-Medium resuspendiert. Die resuspendierten Zellen der drei Stämme werden gemischt und für die Konjugation auf ein Zellulosemischestherfilter, aufgelegt auf einer LB-Agar-Platte, pipettiert. Diese Platte wird für 24 Stunden bei 30°C inkubiert. Mittwoch Bakterien werden vom Filterplättchen abgewaschen und auf LB-Kanamycin-Platten ausgestrichen. Zellen der humanen Zervixkarzinomzellinie HeLa werden in 24-Lochplatten auf runde Deckgläschen eingesäht. Donnerstag Zur Bestätigung der Transposition in das Salmonellengenom wird eine Kolonie-PCR durchgeführt. Dazu wird ein Primer homolog zum Kanamycin-Resistenzgen des Minitransposons und ein Primer homolog zum flankierenden chromosomalen Gen glmS verwendet. Im PCR-Ansatz (50 µl) wird 1 µM je Primer, 200 µM dNTPs, 2,5 U TrueStart Taq-Polymerase und 1 x TrueStart Taq-Puffer (Fermentas) eingesetzt. Mit einer gelben Pipettenspitze wird etwas Bakterienmaterial einer einzelnen Kolonie gewonnen und in dem PCR-Ansatz resuspendiert. Die PCR-Amplifikation erfolgt bei folgenden Temperaturzyklen : 2 min 95°C // 35x 20s 94°C / 20s 55°C / 30s 72°C // 7 min 72°C / 5 min 4°C. Die Größe des amplifizierten Fragmentes wird in einem 1%igen TAE-Agarosegel überprüft. Salmonellen mit der chromosomalen GFP-Kassette werden für die Infektion von HeLa-Zellen eingesetzt. Dazu wird ein positiver Klon in 4 ml LB-Flüssigmedium supplementiert mit 30 µg/ml Streptomycin inokuliert und bei 37°C und 100 rpm agitiert. Im Anschluß werden die HeLa-Zellen mit frischem RPMI-1640-Medium (supplementiert mit 2 mM Glutamin und 5% fötalem Kälberserum) versorgt. Sobald die Bakterien eine OD600 von ca. 0,5 erreicht haben, werden jeweils 1 ml Kultur in Eppis überführt und bei 5000 x g für 5 min abzentrifugiert. Die Bakterien werden dann in RPMI-1640-Medium resuspendiert und in unterschiedlichen Volumina zu den Zellen pipettiert. Die 24-Loch-Platte wird dann 1 min bei 1000 x g zentrifugiert, die Infektionskultur mikroskopisch betrachtet und dann für 30 min im Zellkulturschrank inkubiert. Im Anschluß wird das Medium abgesaugt, die Zellen mit warmen PBS wiederholt gewaschen und frisches Medium supplementiert mit 50 µg/ml Gentamycin zugegeben. Während der folgenden 90 minütigen Inkubation tötet Gentamycin lediglich die extrazellulären Bakterien ab, die intrazellulären Bakterien bleiben unbeeinträchtigt. Schließlich werden die Zellen erneut zweimal mit PBS gewaschen, die Deckgläschen entnommen und auf Objektträgern für die fluoreszenzmikroskopische Analyse eingebettet.
Versuch B
Die gezielte Deletion des asd Gens erfolgt mit Hilfe der RED-Rekombinase des Bakteriophagen Lambda [2]. Vorbereitung : Rekombinase-haltige elektrokompetente Zellen werden vorbereitet. Dazu wird der Salmonellen-Zielstamm SL7207 mit dem RED-Rekombinaseplasmid pKD46 transformiert. In diesem Plasmid befindet sich die RED-Rekombinase unter der Kontrolle des Promotors des Arabinose-Operons aus E.coli. Die Expression der RED-Rekombinase wird in der logarithmischen Wachstumsphase einer Schüttelkultur (200 ml, 30°C, 180 rpm,) durch

BCII am PEI, 22.11.2010-09.12.2010
75
Zugabe von 0,05% (g/v) L-Arabinose induziert. Anschließend werden die Bakterien weitere 90 min kultiviert, dann 10 min auf Eis abgekühlt und 5 min bei 4°C 5000 x g abzentrifugiert. Das Pellet wird in 200 ml eiskaltem Wasser vollständig resuspendiert und nochmals zentrifugiert. Dieser Waschschritt wird ein weiteres Mal unter Verwendung von eiskaltem Glycerinwasser (10% v/v Glycerin) wiederholt. Nach dem Zentrifugieren wird das Pellet in 1 ml Glycerinwasser resuspendiert, auf Eis jeweils 50 µl Zellsuspension (ca. 5x108 Bakterien) in Eppi's aliquotiert und schließlich auf Trockeneis für die Lagerung bei -70°C schockgefroren. Ein lineares DNA-Fragment mit endständigen homologen Sequenzabschnitten und einem Selektionsmarker wird für die Rekombinase-vermittelte Deletion des chromosomalen asd-Gens benötigt. Dieses Fragment wird durch eine PCR erzeugt, in der zwei Primer mit 42bp-langen asd-Homologiesequenzen und ein Plasmid mit dem Selektionsmarker als Template (pKD4) verwendet werden. Im PCR-Ansatz (50µl) werden 1 µM je Primer, 200µM dNTPs, 10 ng Plasmid-Template sowie 2,5 U TrueStart Taq-Polymerase und 1x TrueStart Taq-Puffer (Fermentas) eingesetzt. Die PCR-Amplifikation erfolgt bei folgenden Temperaturzyklen : 2 min 95°C // 35x 20s 94°C / 20s 56°C / 30s 72°C // 7 min 72°C / 5 min 4°C. Primersequenzen : asdFW : ATGGTGAAGGATGCGCCACAGGATACTGGCGCGCATACACAGTG TGTAGGCTGGAGCTGCTTC asdREV : CTACGCCAACTGGCGCAGCATTCGACGCAGCGGCTCGGCGGC CATATGAATATCCTCCTTAG Durchführung : Dienstag Das PCR-Produkt (1562 bp) wird mit dem QIAquick PCR Purification Kit (Qiagen) aufgereinigt, die DNA-Konzentration photometrisch bei einer Wellenlänge von 260 nm bestimmt und die Fragmentgröße in einem 1%igen TAE-Agarosegel bestimmt. Die Konzentration der DNA wird auf 100 ng/µl in Reinstwasser eingestellt. Rekombinase-haltige elektrokompetente Salmonellen werden auf Eis aufgetaut, 200 ng des DNA-Fragmentes mit den Zellen vermischt, in eine vorgekühlte 2 mm Küvette überführt und dann mit dem Bio-rad Gene Pulser elektroporiert. Dafür werden folgende Einstellungen gewählt : Kapazität 25 μF, Widerstand 400 Ω, Spannung 2,5 KV. Direkt nach der Elektroporation wird die Bakteriensuspension in 1 ml SOC-Medium aufgenommen, für eine Stunde bei 37 °C inkubiert und schließlich auf LB-Kanamycin-Platten supplementiert mit 50 µg/ml DAP ausgestrichen. Mittwoch Nach Übernachtinkubation bei 37°C werden einzelne Kolonien auf LB-Kanamycin-Platten mit und ohne DAP überimpft, um den Phänotyp der asd Mutante zu bestätigen. Donnerstag Zur Bestätigung der asd Deletion wird eine Kolonie-PCR durchgeführt. Dazu wird ein Primer homolog zum Kanamycin-Resistenzgen und ein Primer homolog zur asd upstream Region verwendet. Im PCR-Ansatz (50µl) wird 1 µM je Primer, 200 µM dNTPs, 2,5 U TrueStart Taq-Polymerase und 1x TrueStart Taq-Puffer (Fermentas) eingesetzt. Mit einer gelben Pipettenspitze wird etwas Bakterienmaterial einer einzelnen Kolonie gewonnen und in dem PCR-Ansatz resuspendiert. Die PCR-Amplifikation erfolgt bei folgenden Temperaturzyklen : 2 min 95°C // 35x 20s 94°C / 20s 55°C / 30s 72°C // 7 min 72°C / 5 min 4°C. Die Größe des amplifizierten Fragmentes wird in einem 1%igen TAE-Agarosegel überprüft. Der DAP-auxotophe Stamm wird zunächst in 100 ml DAP-supplementiertem LB-Medium angezogen und die optische Dichte verfolgt. Bei einer OD600 von ca. 0,5 werden die Bakterien abzentrifugiert und in DAP-freien LB-Medium resuspendiert. Die bakterielle Zelllyse wird anhand des weiteren Verlaufes der Wachstumskurve verfolgt.

BCII am PEI, 22.11.2010-09.12.2010
76
Versuch C
Plasmidvermittelte Expression des Lyseproteins E vom Bakteriophagen phiX174 sowie des membranlytischen Proteins LLO aus Listeria monocytogenes in einem Salmonellen-Impfstamm. Durchführung : Dienstag Der Salmonellen-Impfstamm SL7207 wird mit einem Expressionsplasmid für Protein E und LLO transformiert. Dieses Plasmid vermittelt die Arabinose-induzierbare Expression von Gen E und eine konstitutive Expression von LLO. Elektrokompetente Bakterien werden mit 200 ng Plasmid-DNA vermischt und analog zu Versuch B elektroporiert. Die transformierten Zellen werden auf LB-Ampicillin-Platten ausgetrichen. Mittwoch Nach Übernachtinkubation bei 37°C werden einzelne Kolonien auf neuen Medienplatten vermehrt. Donnerstag Plasmidhaltige Salmonellen werden von den Medienplatten mit der Impföse gewonnen und in eine 50 ml LB-Schüttelkultur überführt. Die Bei einer OD600 von ca. 0,5 wird die Expression von Lyseprotein E durch Zugabe von 0,05% g/v L-arabinose induziert. Die bakterielle Zelllyse wird anhand des weiteren Verlaufes der Wachstumskurve verfolgt. Zusätzlich werden im Abstand von 30 min jeweils 0,5 ml Proben für den LLO-Hämolyseassay gewonnen. Diesen Proben werden bei 7000 x g für 5 min zentrifugiert, der Überstand abgenommen und auf Eis gelagert. Die Freisetzung von LLO aus Bakterien wird anhand der lytischen Aktivität des Kulturüberstandes gegenüber Schafserythrozyten bestimmt. Zunächst wird zu 0,5 ml Schafsblut 1 ml PBS pH 5,1 zugegeben, leicht gemischt und die Zellen bei 3000 x g für 5 min in der Tischzentrifuge sedimentiert. Dieser Waschschritt wird zweimal wiederholt. Danach werden die Zellen in 1 ml Lyse-PBS (PBS, pH 5,1, 1 mM 2-Mercaptoethanol) resuspendiert. 100 μl dieser Suspension werden mit 3,9 ml Lyse-PBS verdünnt, davon 400 μl mit 400 μl Überstand der Bakterienkultur vermischt und dieses Gemisch 1 h lang bei 37°C inkubiert. Die Proben werden anschließend bei 3000 x g für 5 min zentrifugiert und die Absorption des durch die Erythrozytenlyse freigesetzten Hämoglobins im Überstand photometrisch bei einer Wellenlänge von 541 nm gemessen. Die vollständige Erythrozytenlyse, und damit der maximale Absorptionswert, wird durch die Inkubation mit 1%igen Triton-X-100 in PBS erreicht.
Literatur
1. Bao, Y., Lies, D. P., Fu, H. and Roberts, G. P. (1991) An improved Tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene 109, 167-168.
2. Datsenko, K. A. and Wanner, B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc.Natl.Acad.Sci.U.S.A 97, 6640-6645.

BCII am PEI, 22.11.2010-09.12.2010
77
Transaktivierung der HIV-LTR durch das virale Tat Protein (Gr. 4-6)
Ziele des Versuchs: 1.) Ermittlung der Transfektionseffizienz in der humanen T-Zelllinie A3.01 durch
Transfektion eines GFP-Expressionsplasmids. 2.) Nachweis der LTR-Transaktivierung durch das virale Tat-Protein mittels Reporter-Assay. Theorie: Transaktivierung der HIV-LTR durch Tat: Im HIV-Genom befindet sich vor den viralen Genen der sogenannte LTR-Bereich („long terminal repeats“). Dieser Bereich enthält eine TATA-Box und zahlreiche Bindestellen für verschiedene Transkriptionsfaktoren und stellt damit einen Promotor/Enhancer für die viralen Gene dar. Am LTR-Promotor erfolgt eine effiziente Initiation der Transkription, die Elongation des Transkriptionskomplexes ist allerdings ohne das Tat-Protein („transactivating protein“) stark eingeschränkt. Das Tat-Protein des HI Virus ist ein kleines regulatorisches Protein, dessen Funktion u. a. darin besteht, die Expression der viralen Gene zu kontrollieren. Tat bindet über eine spezifische Interaktion an ein regulatorisches Element der entstehenden mRNA. Dieses Element wird TAR („transactivation-responsive region“) genannt und bildet eine Haarnadelstruktur aus, an die Tat bindet. Über eine assoziierte Protein-Kinase vermittelt Tat die Hyperphosphorylierung der RNA Polymerase II im Elongationskomplex, was zu einer erhöhten Prozessivität des Enzyms führt. Zum Versuch:
Zunächst wird die Transfektionseffizienz bestimmt, indem ein EGFP-Expressions-plasmid („enhanced green fluorescent protein“, pEGFP-N1) transfiziert wird. Die erfolgreich transfizierten Zellen exprimieren das fluoreszierende Protein und können so im Fluoreszenzmikroskop identifiziert werden. Durch Auszählen des Anteils der grün leuchtenden Zellen kann die Transfektionseffizienz ermittelt werden. Zur Untersuchung der LTR-Aktivierung durch das Tat-Protein verwenden wir ein Reportergensystem. Es wird ein Reporterplasmid verwendet, in dem der virale Promotor LTR (inklusive TAR-Region) vor ein Firefly-Luciferasegen geschaltet ist (pGL3-basic-NL4-3-LTR-luc). Tat wird über das Expressionsplasmid pBS- kRSPA-TATNL4-3 in die Zellen eingebracht. Wird am Promotor die Transkription initiiert, kommt es zur Expression des Luciferasegens. Durch Messung der Luciferaseaktivität kann dann eine Aussage über die Stärke der Aktivierung des Promotors getroffen werden. Das Enzym Luciferase bietet sich als Reportergen an, weil seine Aktivität relativ leicht gemessen werden kann. Bei der Umsetzung des Substrats Luciferin wird Fluoreszenzlicht emittiert und kann im Luminometer nachgewiesen werden. In Anwesenheit von ATP, Mg2+ und O2 katalysiert die Luciferase die Oxidation des Luciferins über das Intermediat Luciferyl-AMP zu Oxyluciferin nach folgender Reaktion:
Luciferin + ATP Lucifery-AMP + PP
Lucifery-AMP + O2 Oxyluciferin + Licht
Die Plasmide werden durch transiente Transfektion in die Zellen eingebracht. Hierzu verwenden wir das Transfektionsreagenz DMRIE-C, welches ein Lipid enthält, das mit DNA

BCII am PEI, 22.11.2010-09.12.2010
78
interagieren kann. Es bilden sich Lipid-DNA-Komlexe, die von den Zellen aufgenommen werden können. Zusätzlich wird in alle Ansätze ein Expressionsplasmid für Renilla-Luciferase (phRG-TK) transfiziert. Im Unterschied zur Firefly-Luciferase, die durch den indizierbaren LTR-Promotor reguliert wird, wird die Renilla-Luciferase konstitutiv exprimiert und dient daher als interne Kontrolle. Alle gemessenen Firefly-Luciferaseaktivitäten werden auf den entsprechenden Renilla-Luciferasewert normalisiert. Dadurch können Pipettierfehler und Unterschiede in der Transfektionseffizienz ausgeglichen werden. Da beide Enzyme unterschiedliche Substrate umsetzen, kann zwischen beiden Luciferaseaktivitäten unterschieden werden. Durchführung: 1. Kurstag - Ganztägig Teil 1: Tat- vermittelte Transaktivierung der HIV-LTR: Transfektionen Alle Versuchsansätze werden in drei identischen Ansätzen durchgeführt. - je Ansatz DNA und 0,5 ml Opti-Mem in Polystyrolröhrchen vortexen (dreifache Menge für
Triplikate) - Mastermix (für alle Ansätze): je Ansatz 0,5 ml Opti-Mem und 3,5 µl DMRIE-C - je 0,5 ml Mastermix in ein 6well geben und im gesamten well verteilen - je 0,5 ml DNA-Lösung dazu geben, kurz schütteln - 30 min. bei RT inkubieren, DNA-Lipidkomplexe bilden sich
- A3.01 T-Zellen abzentrifugieren (1200 rpm, 7 min. RT) - in 10 ml RPMI ohne alles resuspendieren - Zellen zählen, mit RPMI auf eine Konzentration von 1x106/ml bringen
- je well 0,5 ml Zellen zugeben (entspricht 5x105 Zellen) - bei 37°C im Brutschrank 4-5h inkubieren - je well 1,5 ml Vollmedium (RPMI+10%FCS+Penicillin+Streptomycin+L-Glutamin)
zugeben Folgende Plasmide werden transfiziert: 1-3) 1 µg pEGFP-N1 + 100 ng phRG-TK
4-6) 0,5 µg pGL3-basic-NL4-3-LTR-luc + 1 µg pBS-kRSPA + 100 ng phRG-TK
7-9) 0,5 µg pGL3-basic-NL4-3-LTR-luc + 0,5 µg pBS-kRSPA + 0,5 µg pBS-
kRSPA-TATNL4-3 + 100 ng phRG-TK
10-12) 0,5 µg pGL3-basic-NL4-3-LTR-luc + 1 µg pBS-kRSPA-TATNL4-3 + 100 ng phRG-TK
Teil 2: Kompartimentierung von zellulären Proteinen Viele virale und zelluläre Proteine sind in der eukaryotischen Wirtszelle nicht gleichmäßig verteilt, sondern in definierten zellulären Kompartimenten lokalisiert. So zum Beispiel findet man retrovirale reverse Transkriptase ausschliesslich im Zytoplasma der infizierten Zelle während retrovirale Integrase auch im Zellkern zu finden ist. Auch eine Vielzahl der von der Wirtszelle kodierten Proteine liegt, je nach ihrer Funktion, entweder im Zellkern oder im Zytoplasma vor. So zum Beispiel findet man den Transkriptionsfaktor OCT-4 (Octamer binding transcription factor 4), der für eine normale Embryonalentwicklung essentiell ist, nur im Zellkern. Oct-4 wird in pluripotenten Stamzellen sowie in Teratokarzinomzelllinien exprimiert, nicht jedoch in ausdifferenzierten Zellen. Im Gegensatz dazu ist das

BCII am PEI, 22.11.2010-09.12.2010
79
Strukturprotein Aktin, das in allen eukaryotischen Zellen vorkommt, nur im Zytoplasma zu finden. Im folgenden Experiment soll die unterschiedliche Lokalisierung von Oct-4 und Aktin mittels Immunofluoreszenz-Analyse anchgewiesen werden: Durchführung: Für die Immunfluoreszenzfärbung werden Teratocarcinomzellen der Linie NCCIT verwendet. Die Zellen wurden bereits am Vortag vom Betreuer auf Deckgläschen ausgesät und über Nacht bei 37°C, 80% Luftfeuchtigkeit und 5% CO2 im Brutschrank weiter kultiviert. Gefärbt werden verschiedene Markerproteine (Oct4, Aktin) und der Zellkern (Dapi-Färbung). - Fixierung der Zellen Das Medium wird zunächst von den Zellen abgesaugt und die Zellen mit 1ml PBS gewaschen. Es folgt die Fixierung mit 0,5 ml 4% Paraformaldehyd/PBS (pH 7.4) auf dem Schüttler bei Raumtemperatur für 15 min. - Permeabilisierung der Zellen Die Paraformaldehyd-Lösung wird abgesaugt und die Zellen für 10 min bei Raumtemperatur auf dem Schüttler in 0,5 ml 1% Triton-X-100/PBS (pH 7.4) inkubiert. - Waschen der Zellen und Blockieren Die Permeabilisierung-Lösung wird vorsichtig abgesaugt und die Zellen 3x2 min in 1 ml PBS auf dem Schüttler bei Raumtemperatur gewaschen. Anschließend werden unspezifische Bindestellen durch Blockieren mit 0,5 ml 5% BSA/ 0,1% Triton-X-100/PBS (pH 7.4) für 30 min bei Raumtemperatur abgesättigt. - Inkubation mit dem Erstantiköper Nach dem Absaugen der Blockierungslösung wird der erste Antikörper zugegeben. Für den Nachweis von Oct 4 wird ein Antikörper aus der Maus verwendet in einer Verdünnung von 1:500 in 5% BSA/PBS (pH 7.4). Es folgt eine Inkubation von 1 Stunde bei Raumtemperatur. Pro Deckgläschen werden 120µl Antikörper-Ansatz benötigt.
- Waschen der Zellen und Inkubation mit dem Zweitantikörper Nach der Inkubation werden die Zellen 3x5 min mit 1 ml PBS gewaschen, um nicht gebundenen Erstantikörper zu entfernen. Anschließend wird mit dem Zweitantikörper im Dunkeln inkubiert. Dabei handelt es sich für Oct4 um einen Fluoreszenz-gekoppelten Anti-Maus-Antikörper (AlexaFluor® 488 goat anti mouse) in einer Verdünnung von 1:1000. Zum Nachweis von Aktin wird ein ebenfalls Fluoreszenz-gekoppelter Phalloidin-Antikörper (AlexaFluor® 633 Phalloidin) in einer Verdünnung von 1:500 eingesetzt. Die Inkubation für 30 min bei Raumtemperatur erfolgt im Dunkeln. Pro Deckgläschen werden 120µl Antikörper-Ansatz benötigt. - Waschen der Zellen und Zellkernfärbung Nach Ende der Inkubationszeit werden die Zellen 3x5 min in 1 ml PBS (pH7.4) gewaschen. Parallel wird die Lösung mit Dapi zum Anfärben der Zellkerne angesetzt. Die Stammlösung hat eine Konzentration von 1mg/ml und wird 1:1000 in PBS (pH7.4) verdünnt. Pro Deckglas werden 500µl verwendet und 2 min bei Raumtemperatur inkubiert. Nicht gebundenes Dapi wird durch Waschen mit PBS (pH 7.4; 3x10 min) bei Raumtemperatur auf dem Schüttler entfernt. - Einbetten der Zellen Zum Abschluss werden die Zellen in Aquapolymount eingebettet. Dazu wird pro Deckgläschen ein Tropfen Aquapolymount auf einen entsprechend beschrifteten Objektträger aufgetropft und die Deckgläschen mit der „Zellseite“ zum Objektträger auf den Tropfen aufgelegt und überschüssiges Einbettmedium vorsichtig abgetupft. Die Proben werden bis zur weiteren Analyse bei 4°C im Kühlschrank gelagert.

BCII am PEI, 22.11.2010-09.12.2010
80
2. Kurstag – Vormittag: Auswertung der Immunfluoreszenzanalysen zum Nachweis der Kompartimentierung der zellulären Proteine OCT-4 und Aktin 2. Kurstag – Nachmittag:
Tat- vermittelte Transaktivierung der HIV-LTR:
Bestimmung von Luziferaseaktivität und Transfektionseffizienz
Lyse:
- Zellen in 15 ml-Falcons abzentrifugieren (1300 rpm, 7 min.)
- Zellen 1x in PBS waschen
- Pellets in je 100 µl Lysispuffer (PLB, Promega) resuspendieren, in Eppis überführen
- 30 min. auf Eis inkubieren
- mit 13000 rpm abzentrifugieren (10 min., 4°C)
- für Luciferase –Assay je 20 µl in 96well-Platte geben
Messung der Luciferaseaktivität:
Die Substratzugabe erfolgt durch das Luminometer:
- Zugabe von je 50 µl Luciferase Assay Reagenz (Substrat für Firefly-Luciferase)
- nach 2 sec. beginnend wird über 10 sec. die Fluoreszenz gemessen
- Zugabe von 50 µl Stop&Glow (unterdrückt die Firefly-Fluoreszenz und enthält das
Substrat für die Renilla-Luciferase)
- nach 2 sec. beginnend wird über 10 sec. die Fluoreszenz gemessen
Auswertung:
Transfektionseffizienz:
- Bestimmung der Transfektionseffizienz durch Auszählen des Prozentsatzes der
EGFP-exprimierenden Zellen
Einfluss von Tat auf die LTR-Aktivität:
- Normalisierung der Meßwerte auf Renilla-Luciferaseaktivität, Berechnung der Mittelwerte
der Dreifachbestimmungen.
- Erstellen von Säulendiagrammen (Mittelwerte der Dreifachbestimmungen sollen mit
Standardabweichung dargestellt werden), um welchen Faktor erhöht die Tat-Expression
die LTR-Aktivität?
Literatur:
Funktion des Tat Proteins:
http://hiv-web.lanl.gov/content/hiv-db/COMPENDIUM/2000/partI/Karn.pdf

BCII am PEI, 22.11.2010-09.12.2010
81

BCII am PEI, 22.11.2010-09.12.2010
82
Experiment 5:
Gentransfer mit retroviralen Vektoren
Wichtig
Die Gruppen 1-3 führen das Experiment in der AG Buchholz durch, die
Gruppen 4-6 in der AG Mühlebach.
Verlaufsplan: Tag 1: - Transduktion der Zielzellen
- Isolierung viraler RNA
Tag 2: - EGFP
- lacZ-Färbung
- Isolierung zellulärer DNA
Tag 3: - reverse Transkription viraler RNA
- PCR an RNA , cDNA und genomischer DNA

BCII am PEI, 22.11.2010-09.12.2010
83
Versuchshintergrund und -ablauf:
Retrovirale Vektoren, die zur genetischen Veränderung von bestimmten Zielzellen
verwendet werden sollen, können durch verschiedene Methoden hergestellt werden:
zum einen gibt es stabil transfizierte Verpackungszellen, welche alle
Komponenten des Vektors (Transfervektor, Gag/Pol, Env) konstitutiv bilden.
Andererseits können die für diese Vektorbestandteile kodierenden Plasmide transient
(nicht stabil, „vorübergehend“) in geeignete Zelllinien ko-transfiziert (d.h. mit einer
speziellen Technik in die einzelnen Zellen eingebracht) und so „transient
transfizierte Verpackungszellen“ generiert werden (Abb. 1). Hierfür sind 293T-
Zellen (humane embryonale Nierenmarkszellen) aufgrund der hohen erreichbaren
Transfektionseffizienzen besonders geeignet.
Für diesen Versuch wurden in Vorbereitung ausgehend von 293T-Zellen
verschiedene transient transfizierte Vepackungszellen zur Produktion von Murinen
Leukämie Virus (MLV)-abgeleiteten Vektoren hergestellt und die entsprechenden
Vektoren präpariert. Die dabei erzeugten Vektormengen und die Eigenschaften der
Vektorpartikeln werden innerhalb des Praktikums untersucht.
Die zu erzeugenden Vektoren bestehen alle aus MLV-Kapsidpartikeln (Gag/Pol),
unterscheiden sich aber durch die verwendeten Transfervektoren, die
unterschiedliche Reportergene (lacZ/-Gal bzw. gfp/GFP) beinhalten, und die
Hüllproteine, welche das Wirtszellspektrum (Tropismus) der Vektorpartikel durch die
Bindung an spezifische zelluläre Rezeptoren bestimmen (Abb. 2). Folgende
Hüllproteine (Env) sollen hier verwendet werden:
1. Env des ecotropen MLV: Ecotropes MLV (vollständiges Virus, nicht der Vektor!)
repliziert nur in Nagerzellen und nutzt den zellulären Rezeptor MCAT-1 zum
Anbinden und den Eintritt in die Wirtszelle.
2. Env des amphotropen MLV: Amphotropes MLV repliziert in Nagerzellen sowie
auch in humanen Zellen und nutzt den Rezeptor RAM-1 (receptor for amphotropic
MLV 1).

BCII am PEI, 22.11.2010-09.12.2010
84
3. Env des GaLV (Gibbon Ape Leukemia Virus): Das Hüllprotein des GaLV bindet
an den Rezeptor Glvr-1 (GaLV receptor 1), der (wie RAM-1) in fast allen
humanen Zelltypen, jedoch nicht in murinen Zellen gebildet wird. Allerdings
scheint Glvr-1 in einigen für die Gentherapie besonders interessanten
Zellpopulationen (z. B. hämatopoetischen Stammzellen) stärker exprimiert zu
werden als RAM-1. Daher wird GaLV Env zur Pseudotypisierung (Verwendung
eines heterologen Env, d.h. Verwendung eines Hüllproteins eines anderen Virus
auf der Vektoroberfläche) von retroviralen MLV-abgeleiteten Vektorpartikeln
verwendet. Somit verändert man den Vektortropismus und kann in bestimmten
Zielzellen die Gentransfereffizienz erhöhen.
Zunächst wurden durch Lipofektion (= Transfektionsmethode) 293T Zellen mit den
für die Vektorkomponenten kodierenden Plasmiden (Transfervektor,
Verpackungskonstrukt, Hüllproteinkonstrukt) transfiziert. Zwei Tage nach
Transfektion konnten die vektorhaltigen Zellkulturüberstände dieser
Verpackungszellen (viral packaging cells; VPC) „geerntet“ werden. Diese werden
Ihnen im Praktikum zur Verfügung gestellt. Der eigentliche Praktikumsversuch
beginnt dann mit der Transduktion verschiedener Zielzellen, d.h. die Zielzellen
werden mit vektorhaltigem Überstand inkubiert, um die Zielzellen durch Einschleusen
des Transfervektors ins Genom genetisch zu verändern. Zwei Tage nach erfolgter
Transduktion werden die Zielzellen auf die Anwesenheit der transferierten
Reportergene (auf Transfervektor) hin untersucht. Die Menge der generierten
Vektorpartikel und deren Tropismus lassen sich so nachweisen. Im Rahmen dieses
Versuches sollen auch die RNA Genome aus den Vektorpartikeln isoliert, in cDNA
umgeschrieben werden und dann mit PCR detektiert werden.
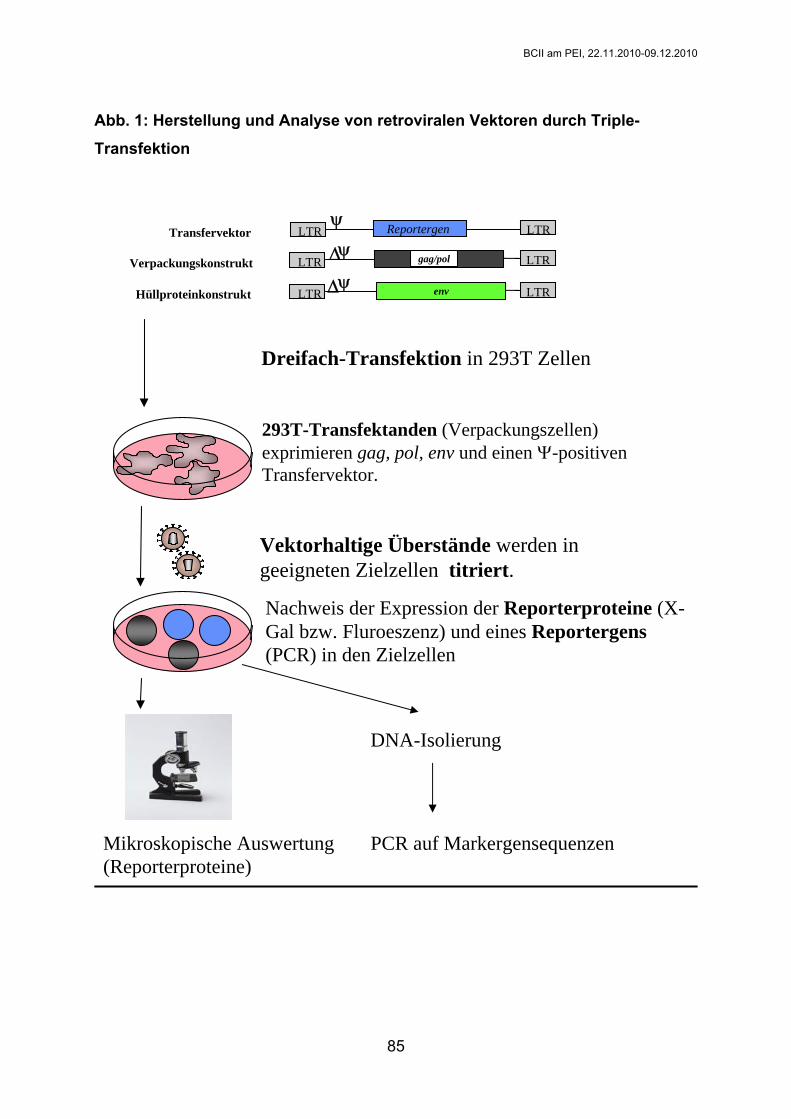
BCII am PEI, 22.11.2010-09.12.2010
85
Abb. 1: Herstellung und Analyse von retroviralen Vektoren durch Triple-
Transfektion
Dreifach-Transfektion in 293T Zellen
Vektorhaltige Überstände werden ingeeigneten Zielzellen titriert.
293T-Transfektanden (Verpackungszellen)exprimieren gag, pol, env und einen -positivenTransfervektor.
Nachweis des Reportergens (Fluoreszenz,bzw. X-Gal-Test)
Mikroskopische Auswertung
Verpackungskonstrukt
Transfervektor LTR LTR
Reportergen
gag/polLTR LTR
Hüllproteinkonstrukt envLTR LTR
gag/pol
Nachweis der Expression der Reporterproteine (X-Gal bzw. Fluroeszenz) und eines Reportergens (PCR) in den Zielzellen
DNA-Isolierung
PCR auf Markergensequenzen Mikroskopische Auswertung (Reporterproteine)

BCII am PEI, 22.11.2010-09.12.2010
86
Vor dem Praktikum vorbereitet und durchgeführt:
Herstellung von MLV abgeleiteten Vektoren
Lipofektion:
24 h vor Versuchsbeginn werden in 35 mm Kulturschalen 6 x 105 Zellen
ausgesät
je 1µg DNA pro Konstrukt (Transfervektor, Verpackungskonstrukt,
Hüllproteinkonstrukt), also insgesamt 3 µg werden in Eppis vorgelegt
(siehe Pipettierschema nächste Seite).
4 µl Lipofektamin PLUS werden in 100 µl DMEM- angesetzt (Eppi) und für
15 min bei RT mit den DNA-Lösungen inkubiert.
je 10 µl Lipofectamin in 100 µl DMEM- werden zugegeben - weitere 15 min
bei RT
nach der Inkubation wird mit DMEM- auf 1 ml aufgefüllt.
die Transfektionslösungen werden auf die Zellen gegeben (Medien zuvor
entfernen).
Inkubation für 2 bis 5 h bei 37° C
Mediumwechsel (komplettes Kulturmedium)
Inkubation über Nacht
Mediumwechsel (je 2 ml komplettes Kulturmedium)
zwei Tage nach Transfektionsbeginn können die Überstände geerntet und
zur Vektorpräparation sowie nachfolgend zur Transduktion der Zielzellen
eingesetzt werden.
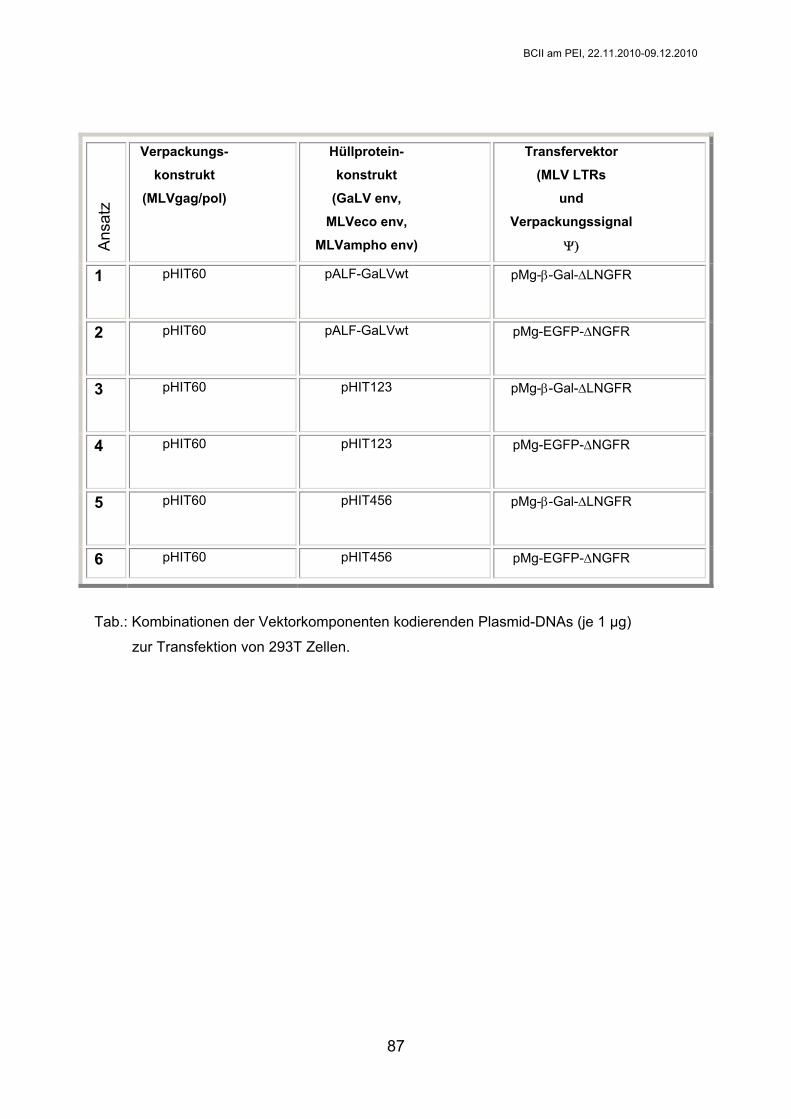
BCII am PEI, 22.11.2010-09.12.2010
87
A
nsat
z
Verpackungs-
konstrukt
(MLVgag/pol)
Hüllprotein-
konstrukt
(GaLV env,
MLVeco env,
MLVampho env)
Transfervektor
(MLV LTRs
und
Verpackungssignal
1 pHIT60
pALF-GaLVwt
pMg--Gal-LNGFR
2 pHIT60
pALF-GaLVwt
pMg-EGFP-NGFR
3 pHIT60
pHIT123
pMg--Gal-LNGFR
4 pHIT60
pHIT123
pMg-EGFP-NGFR
5 pHIT60
pHIT456
pMg--Gal-LNGFR
6 pHIT60 pHIT456 pMg-EGFP-NGFR
Tab.: Kombinationen der Vektorkomponenten kodierenden Plasmid-DNAs (je 1 µg)
zur Transfektion von 293T Zellen.

BCII am PEI, 22.11.2010-09.12.2010
88
Transfervektoren
Verpackungskonstrukt
Hüllprotein(Env)-Konstrukte
pMg -ß- Gal- LNGFR:
LTR LTR
LNGFR
SD SA
lacZ
ATG ATG
SD SA
pMg -EGFP- LNGFR:
LTR LTR
LNGFR egfp
ATG ATG
Fig .2
pHIT60: LTR LTR
ATG
pHIT123: env (ecotrop. MLV)LTR LTR
ATG
pHIT456: env (amphotrop. MLV)LTR LTR
ATG
pALF - GaLVwt :
ATG
env (GaLV )LTR LTR
MLV gag/pol
Die Transfervektoren (pMg--Gal-∆LNGFR, pMg-EGFP--∆LNGFR) besitzen die von MLV abgeleiteten flankierenden LTRs (long terminal repeats), das Verpackungssingnal "", welches die Verpackung der Transkripte in MLV-Kapsidpartikel gewährleistet, sowie einen Splicedonor (SD) bzw. –akzeptor (SA). Als Reportergenprodukte dienen die trunkierte Variante des "low affinity nerve growth factor receptor" (∆LNGFR) sowie -Gal bzw. EGFP. Das Verpackungskonstrukt pHIT60 beinhaltet die Strukturgene gag und pol des MLV, deren Genprodukte in der Lage sind, leere, nicht umhüllte Kapsidpartikel des MLV zu bilden. Die Hüllproteinkonstrukte kodieren für die Hüllproteine des eco- oder amphotrophen MLV bzw. des GaLV. Alle Strukturgene werden von modifizierten LTRs getrieben.

BCII am PEI, 22.11.2010-09.12.2010
89
Versuchsdurchführung:
Alle Arbeiten werden steril und unter der Werkbank durchgeführt
Experiment Transduktion von Zielzellen:
Zielzellen: NIH 3T3, HT1080, und D17 (canine [„Hunde“] Osteosarkomzellen)
3,5x105 NIH 3T3 und 3x105 HT1080 bzw. D17 Zellen wurden in Sechs-Loch-Platten
am Tag zuvor ausgesät und werden zur Verfügung gestellt:
Es werden jeweils 3 Zellkulturplatten benötigt: a) für GFP-Detektion b) für LacZ
Detektion, c) für die Isolierung von zellulärer DNA (s. Schema S. 76).
Vektorhaltige Überstände (SN) wurden vor Beginn des Praktikums generiert und
stehen nun für Transduktionsversuche zur Verfügung. 550 µl einer 1:10
Verdünnung herstellen (nach Absprache mit Versuchsbetreuern).
Zielzellen mit PBS waschen
je 500 µl der unverdünnten und 1:10 verdünnten vektorhaltigen filtrierten
Überstande zu den Zielzellen geben (siehe Schema S. 76; beschriften!).
2h nach Transduktion Zellen mit frischen Medien versorgen.
Weiterhin stehe DNAse-vorbehandelte Vektorüberstände für die Isolierung viraler
RNA zur Verfügung.
Experiment Isolierung von Nukleinsäuren und PCR Detektion:
Isolierung viraler RNAs
Um die viralen RNAs aus den Vektor-Partikeln zu isolieren, wird der vektorhaltige
Überstand direkt eingesetzt und mittels des QIAamp Viral RNA Kit aufgereinigt.
QIAamp viral RNA Mini Kit
The sample is first lysed under highly denaturing conditions to inactivate RNases
and to ensure isolation of intact viral RNA. Buffering conditions are then adjusted to
provide optimum binding of the RNA to the QIAamp membrane, and the sample is
loaded onto the the QIAamp spin column. After contaminants are washed away,
high-quality RNA is eluted in a special RNase-free buffer, ready for direct use or
safe storage.

BCII am PEI, 22.11.2010-09.12.2010
90
Preparation of virus RNA (according to Qiagen)
Use RNase-free material only after the lysis with buffer AVL
Pipet 560µl of prepared Buffer AVL (containing carrier RNA) into a 1.5ml tube
Add 140µl virus-containing supernatant, mix by votexing for 15 sec.
Incubate at RT for 10 min.
Briefly centrifuge tube to remove drops from inside the lid
Add 560µl EtOH (96-100%), mix by vortexing, briefly spin down
apply 630µl of sample to provided column (in a 2-ml collection tube)
centrifuge 1 min (8000rpm) and discard filtrate, place the QIAamp column in a
clean collection tube
apply residual sample to column
centrifuge 1 min (8000rpm) and discard filtrate, place the QIAamp column in a
clean collection tube
add 500µl buffer AW1, centrifuge 1 min (8000rpm) and discard filtrate, place the
QIAamp column in a clean collection tube
add 500µl buffer AW2, centrifuge 3 min (13000rpm) and discard filtrate, place the
QIAamp column in a clean collection tube.
repeat centrifugation to get rid of residual filtrate, place the QIAamp column in a
clean 1.5-ml microcentrifuge tube
add 60µl buffer AVE, incubate 1 min at RT
centrifuge 1 min (13000rpm)
Take off 20 µl for DNase digest (see page 69), store on ice
store rest of eluate at –80°C

BCII am PEI, 22.11.2010-09.12.2010
91
DNAse digestion of RNA
20 µl + 67.5 µl H2O

BCII am PEI, 22.11.2010-09.12.2010
92

BCII am PEI, 22.11.2010-09.12.2010
93

BCII am PEI, 22.11.2010-09.12.2010
94
Experiment Transduktion von Zielzellen:
48h nach Transduktionsbeginn können die Zielzellen mittels
Fluoreszenzmikroskopie (eGFP) bzw. nach Fixierung mittels X-Gal-Test
(-Gal) auf den erfolgten Gentransfer hin untersucht werden.
X-gal-Färbung von Monolayer-Kulturen Fixierlösung: - PBS (ohne Ca und Mg) - 2 % Formaldehyd - 0,2% Glutaraldehyd Stammlösungen: - 5- Bromo-4-Chloro-3-Indolyl ß-D-Galactopyranoside (x-Gal) in DMSO (40mg/ml) - Kaliumhexacyanoferrat III (1M) (Kaliumferricyanid = rotes Blutlaugensalz) - Kaliumhexacyanoferrat II (0,5M) (Kaliumferrocyanid = gelbes Blutlaugensalz) - MgCl2 (1M) Reaktionsmix: - PBS (ohne Ca + Mg) 20ml - X-Gal (40mg/ml) 500µl (=1mg/ml) - R-Ferricyanid (1M) 100µl (=5mM) - R-Ferrocyanid (0,5M) 200µl (=5mM) - MgCl2 (1M) 40µl (=2mM) Ablauf: - Medium von Zellen entfernen - (Zellen 1x PBS waschen) - Fixierlösung auf Zellen geben (6well: +1-2ml), 10Min./RT - 1x (-3x) vorsichtig mit PBS waschen - X-Gal-Lösung (s.o. Reaktionsmix, frisch angesetzt) auf Zellen geben (6well: +1-
2ml) - 37°C stellen (Färbung ist nach einigen Stunden erkennbar – kann aber auch über
Nacht stehen)

BCII am PEI, 22.11.2010-09.12.2010
95
Auswertung des Experiments Transduktion von Zielzellen (eGFP + lacZ):
Die transduzierten Zellen werden unter dem Axiovert (Mikroskop) untersucht und
die Zahl der transduzierten Zellkolonien pro betrachteter Fläche bestimmt.
Hochgerechnet auf die gesamte Fläche der Kulturschale kann die Zahl der
infektiösen Vektorpartikel pro Volumeneinheit Überstand bestimmt werden
(t.u./ml; t.u. = transducing units).
Diese Titer können graphisch dargestellt werden.
Vergleiche die Titer der verschiedenen Vektorpartikel in den
unterschiedlichen Zielzellen;
- Gibt es Unterschiede zwischen den verschiedenen Reportergenen in der
Hinsicht, welche Zellen bei welchen Vektoren positiv werden? Warum?
- Bei den NIH3T3 und HT1080-Zellen handelt es sich um eine humane und eine
murine Zellinie. Welche ist was? (Hinweis: Der Tropismus der verwendeten
Hüllproteine sollte eine Aussage erlauben!)
- Welche Rezeptoren sind auf der caninen Zellinie D17 vorhanden?

BCII am PEI, 22.11.2010-09.12.2010
96
Experiment Isolierung von Nukleinsäuren und PCR Detektion:
Isolierung zellulärer DNAs
Die Gesamt DNA der Vektor-transduzierten Zellen soll isoliert werden und
die Anwesenheit der Vektorsequenzen mit PCR nachgewiesen werden. Wir
nutzen zur Isolierung der DNA das QIAGEN DNeasy Tissue Kit.
DNeasy Tissue procedure:
The kit uses advanced silica-gel-membrane technology for rapid and efficient
purification of total cellular DNA. The buffer system is optimized to allow
direct lysis followed by selective binding of DNA to the to the DNeasy
membrane. DNeasy purified DNA typically has an A260/A280 ratio between
1.7 and 1.9, and is up to 50 kb in size. The DNeasy procedure also efficiently
recovers DNA fragments as small as 100 bp.
Before lysis of cells, these have to be detached from monolayer culture and
washed :
Remove medium
Wash cells 1x with 1 ml PBS/well
Detach cells by adding 500 µl Trypsin-EDTA + 500 µl PBS and
incubation for 5 min at RT
Transfer cells into Eppendorff Tube
Spin down (5 min 2000 rpm)
Wash cells with 1 ml PBS
Spin down (5 min 2000 rpm)
Purification of total DNA from cultured animal cells:
Resupend pellet in 200µl PBS
Add 20µl proteinase K and 200µl buffer AL (do not add proteinase
k directly to buffer AL), mix by votexing
Incubate 56°C 10 min
Add 200 µl EtOH, mix by vortexing
Transfer mixture to DNeasy mini spin column (placed in a 2 ml
collection tube)
spin 1 min at 8000 rpm

BCII am PEI, 22.11.2010-09.12.2010
97
Replace the collection tube, add 500µl buffer AW1
spin 1 min at 8000 rpm
Replace the collection tube, add 500µl buffer AW2
spin 3 min at 14000 rpm
place the column in clean 1.5 ml centrifuge tube
add 200 µl buffer AE on the membrane
incubate 1 min
spin 1 min at 8000 rpm to elute DNA

BCII am PEI, 22.11.2010-09.12.2010
98
Experiment Isolierung von Nukleinsäuren und PCR Detektion:
→ Herstellung von cDNA der viralen RNA
heat one incubator to 80°C and one to 37°C
prepare RT-reaction-Mastermix for all samples: for one sample (=13µl): 5x Buffer 4µl dNTP (10mM) 1µl RNase OUT 0,5µl Reverse Transkriptase 0,5µl DTT 2µl H20 5µl
preparation of the RNA samples: mix 4µl RNA + 1µl (10pmol) Primer + 2µl H2O (control: one sample without template) incubate these 7µl samples 1min at 65°C, then put them directly on ice
add 13µl reaction-mix to each sample, incubate 1h at 42°C
→ Detektion von GFP und lacZ Sequenzen mittels PCR an RNA, cDNA und DNA
prepare PCR-reaction-mix for all samples (additional: one plasmid control to check the PCR and to check for DNA content in the RNA: Instead of cDNA use 0,5µl RNA as Template in the PCR): for one sample=50µl: sample (cDNA or DNA or RNA or H20) in 10µl 10x Buffer 5µl dNTP (10mM) 1µl Primer 1 (10pmol/µl) 1µl Primer 2 (10pmol/µl) 1µl Taq-Polymerase 0,5µl H20 31,5µl
program: 1. 4min 95°C 2. 40sec 95°C 3. 40sec 58°C (Temp depends on the Primer)
4. 1min 72°C (Time depends on expected product length: 1 min / kb) 5. 10min 72°C 6. hold at 4°C
30 repeats from step 2 to 4
Analyse samples on agarose gels.
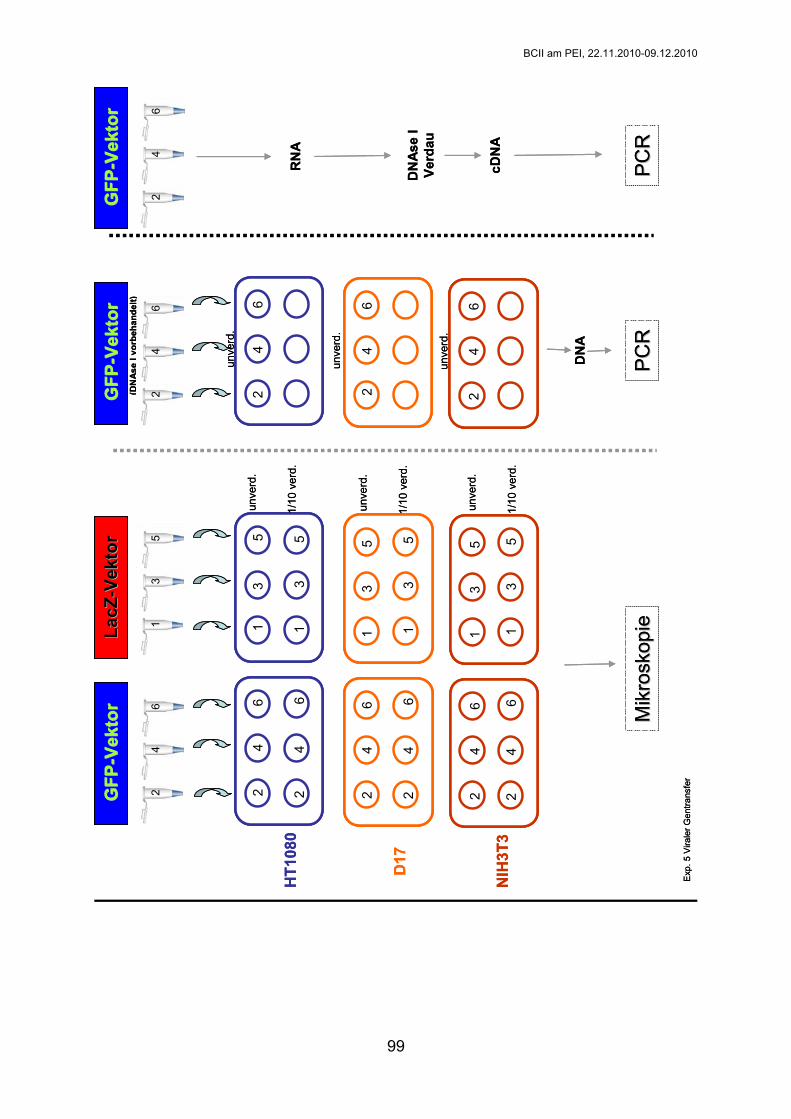
BCII am PEI, 22.11.2010-09.12.2010
99
HT
1080
D17
NIH
3T3
Exp
. 5
Vira
ler
Gen
tra
nsfe
r
2
4
6
1
3
5un
verd
.
1/10
ver
d.
unv
erd
.
1/10
ver
d.
unve
rd.
1/10
ver
d.
Mik
rosk
opie
Mik
rosk
opie
2
4
6
1
3
5
2
4
6
1
3
5
2
4
6
1
3
5
2
4
6
1
3
5
2
4
6
1
3
5
PC
RP
CR
(DN
As
eI
vorb
eha
nd
elt)
2
4
6
2
4
6
2
4
6
unve
rd.
unve
rd.
unve
rd.
RN
A
DN
As
eI
Ver
dau
cD
NA
PC
RP
CR
2
4
6
DN
A
GF
PG
FP
-- Vek
tor
Vek
tor
Lac
ZL
acZ
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
2
4
6
1
3
5
2
4
6
HT
1080
D17
NIH
3T3
Exp
. 5
Vira
ler
Gen
tra
nsfe
r
2
4
6
1
3
5un
verd
.
1/10
ver
d.
unv
erd
.
1/10
ver
d.
unve
rd.
1/10
ver
d.
Mik
rosk
opie
Mik
rosk
opie
2
4
6
1
3
5
2
4
6
1
3
5
2
4
6
1
3
5
2
4
6
1
3
5
2
4
6
1
3
5
PC
RP
CR
(DN
As
eI
vorb
eha
nd
elt)
2
4
6
2
4
6
2
4
6
unve
rd.
unve
rd.
unve
rd.
RN
A
DN
As
eI
Ver
dau
cD
NA
PC
RP
CR
2
4
6
2
4
6
DN
A
GF
PG
FP
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
Lac
ZL
acZ
-- Vek
tor
Vek
tor
Lac
ZL
acZ
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
GF
PG
FP
-- Vek
tor
Vek
tor
2
4
6
2
4
6
1
3
5
1
3
5
2
4
6
2
4
6

BCII am PEI, 22.11.2010-09.12.2010
100
Experiment 6:
GRUNDLAGEN DER ELEKTRONENMIKROSKOPIE
Das Auflösungsvermögen eines optischen Gerätes ist definiert als der Abstand
zweier Punkte, die man gerade noch getrennt wahrnehmen kann. Das theoretische
Auflösungsvermögen des Lichtmikroskopes ist im Wesentlichen bestimmt durch die
Wellenlänge des sichtbaren Lichtes und eine Eigenschaft des Objektives, die
numerische Apertur (NA). Die numerische Apertur ist ein Maß für den Öffnungswinkel
des Objektives. Ernst Abbe fand 1873 die Formel für das Auflösungsvermögen d als
d= 0.61 /NA. Nach der Theorie von de Broglie (Dualismus Welle-Teilchen) kann
man Elektronen als elektromagnetische Wellen betrachten. Die Wellenlänge ist
dann umgekehrt proportional zur Geschwindigkeit des Elektrons. Das theoretische
Auflösungsvermögen des Elektronenmikroskopes berechnet sich danach zu 0,23 nm,
also ca. 1000 mal besser als das des Lichtmikroskopes. In der Praxis der
biologischen Elektronenmikroskopie ist allerdings meistens die Präparation
auflösungsbegrenzend.
Der Aufbau des Elektronenmikroskops entspricht grundsätzlich dem des
Lichtmikroskopes. Beide besitzen zunächst eine Strahlquelle: Der Glühlampe beim
Lichtmikroskop emtspricht beim Elektronenmikroskop eine Kathode, aus der bei
Stromdurchfluß durch Erhitzen Elektronen austreten. Diese werden auf eine auf
Hochspannung gebrachte Anode hin beschleunigt. Die Elektronen treten gebündelt
durch ein Loch in der Kathode und bilden den eigentlichen Strahl. Die Licht- bzw.
Elektronenstrahlen werden durch Kondensorlinsen auf das Präparat gebündelt; die
eigentliche Vergrößerungslinse ist das Objektiv, es entwirft ein Zwischenbild. Dieses
wird entweder - beim Lichtmikroskop - mit dem Okular betrachtet, oder - beim
Elektronenmikroskop - von einem "Projektiv" auf einen fluoreszierenden
Leuchtschirm projiziert. Der Fluoreszenzschirm ist nötig, da das Auge keine
Elektronen wahrnehmen kann, er wandelt die Elektronen in sichtbares Licht um. Da
Elektronen durch Zusammenstoß mit den Molekülen der Luft abgelenkt, gebremst
und adsorbiert würden, herrscht im Elektronenmikroskop Vakuum.
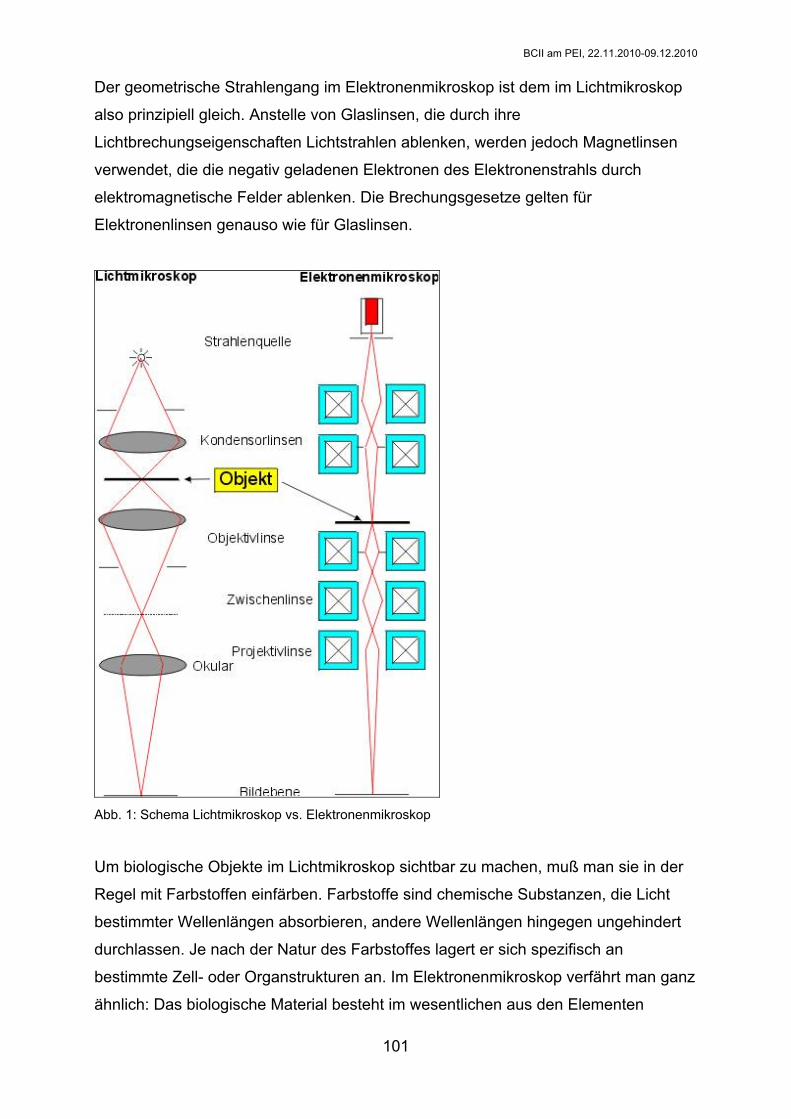
BCII am PEI, 22.11.2010-09.12.2010
101
Der geometrische Strahlengang im Elektronenmikroskop ist dem im Lichtmikroskop
also prinzipiell gleich. Anstelle von Glaslinsen, die durch ihre
Lichtbrechungseigenschaften Lichtstrahlen ablenken, werden jedoch Magnetlinsen
verwendet, die die negativ geladenen Elektronen des Elektronenstrahls durch
elektromagnetische Felder ablenken. Die Brechungsgesetze gelten für
Elektronenlinsen genauso wie für Glaslinsen.
Abb. 1: Schema Lichtmikroskop vs. Elektronenmikroskop
Um biologische Objekte im Lichtmikroskop sichtbar zu machen, muß man sie in der
Regel mit Farbstoffen einfärben. Farbstoffe sind chemische Substanzen, die Licht
bestimmter Wellenlängen absorbieren, andere Wellenlängen hingegen ungehindert
durchlassen. Je nach der Natur des Farbstoffes lagert er sich spezifisch an
bestimmte Zell- oder Organstrukturen an. Im Elektronenmikroskop verfährt man ganz
ähnlich: Das biologische Material besteht im wesentlichen aus den Elementen

BCII am PEI, 22.11.2010-09.12.2010
102
Wasserstoff, Sauerstoff und Kohlenstoff sowie Stickstoff und Phosphor. Das sind
alles Elemente mit niedriger Ordungszahl im Periodensystem der Elemente (niedriger
Kernladungszahl). Da Elektronen vorwiegend mit den Atomkernen wechselwirken,
werden sie von Elementen mit hoher Kernladungszahl (z.B. Schwermetallen)
wesentlich stärker gestreut bzw. absorbiert, als von Elementen mit niedriger
Ordnungszahl. Aus diesem Grunde imprägniert man die Präparate mit
Schwermetallsalzen (z. B. Bleicitrat und Uranylacetat) um genügend Kontrast für die
Beobachtung im Elektronenmikroskop zu erzeugen (man spricht folgerichtig nicht von
"färben", sondern von "kontrastieren").
Während die Farbstoffe für die Lichtmikroskopie bestimmte Wellenlängen
absorbieren, entsteht im Elektronenmikroskop das Bild also durch Streuung der
Elektronen an den Schwermetallatomen. In großem Winkel abgelenkte Elektronen
Abb. 2: Ablenkung der Strahlelektronen in Abhängigkeit von der Kernladungszahl im Präparat und der Hochspannung des Elektronenmikroskopes.
werden durch eine Blende, die „Kontrastblende“, von der Bildentstehung
ausgeschlossen. Präparatstellen, die Schwermetallsalze eingelagert haben
erscheinen im Bild also dunkel. Die Energie, die die Elektronen bei Absorption und
Ablenkung auf das Präparat übertragen wird als Wärme frei, sie kann das Präparat
schnell beschädigen und verändern.
Die für die Praxis wichtigsten Unterschiede zwischen dem Licht- und dem
Elektronenmikroskop bestehen also darin, dass 1. im Elektronenmikroskop Vakuum

BCII am PEI, 22.11.2010-09.12.2010
103
herrscht, 2. anstelle von Lichtstrahlen, welche durch Glaslinsen abgelenkt werden,
Elektronenstrahlen verwendet werden, die durch Magnetlinsen abgelenkt werden..
DDaarraauuss eerrggiibbtt ssiicchh,, wwiiee eeiinn ggeeeeiiggnneetteess PPrrääppaarraatt ffüürr ddaass EElleekkttrroonneennmmiikkrroosskkoopp
bbeesscchhaaffffeenn sseeiinn mmuußß:: EEss mmuußß iimm VVaakkuuuumm uunndd bbeeii BBeessttrraahhlluunngg mmiitt EElleekkttrroonneenn ssttaabbiill
sseeiinn,, uunndd eess mmuußß ggeennüüggeenndd „„kkoonnttrraassttrreeiicchh““ sseeiinn –– aallssoo EElleemmeennttee hhoohheerr
KKeerrnnllaadduunnggsszzaahhll eenntthhaalltteenn –– uumm EElleekkttrroonneenn aabbzzuulleennkkeenn.. AAuuss eerrsstteerreemm rreessuullttiieerrtt
eeiinneess ddeerr HHaauuppttpprroobblleemmee bbeeii ddeerr PPrrääppaarraattiioonn bbiioollooggiisscchheenn MMaatteerriiaallss,, wweellcchheess jjaa
zzuummeeiisstt wweeiitt üübbeerr 9900%% WWaasssseerr eenntthhäälltt:: EEss mmuußß ggeettrroocckknneett wweerrddeenn,, ddaa ddaass WWaasssseerr
ssiicchh iimm VVaakkuuuumm ssooffoorrtt vveerrffllüücchhttiiggeenn wwüürrddee.. EEiinnee TTrroocckknnuunngg iisstt jjeeddoocchh zzuummeeiisstt mmiitt
eeiinneerr DDeeffoorrmmaattiioonn ooddeerr ggaarr ZZeerrssttöörruunngg bbiioollooggiisscchheerr SSttrruukkttuurreenn vveerrbbuunnddeenn.. EEss
wwuurrddeenn ddaahheerr vveerrsscchhiieeddeennee VVeerrffaahhrreenn eennttwwiicckkeelltt,, MMaakkrroommoolleekküüllee,, ZZeelllleenn uunndd
GGeewweebbee sscchhoonneenndd uunntteerr EErrhhaallttuunngg iihhrreerr MMoorrpphhoollooggiiee vvoomm WWaasssseerr zzuu bbeeffrreeiieenn::
LLuuffttttrroocckknnuunngg
GGeeffrriieerrttrroocckkuunngg
KKrriittiisscchh--PPuunnkktt--TTrroocckknnuunngg
EEnnttwwäässsseerruunngg dduurrcchh AAllkkoohhooll uunndd EEiinnbbeettttuunngg iinn eeiinn KKuunnsstthhaarrzz,, mmiitt aannsscchhlliieeßßeennddeerr
AAnnffeerrttiigguunngg vvoonn UUllttrraaddüünnnnsscchhnniitttteenn
EEiinnee AAlltteerrnnaattiivvee bbeesstteehhtt ddaarriinn,, ddiiee OObbjjeekkttee eeiinnzzuuffrriieerreenn uunndd ddaannnn –– iinn ggeeffrroorreenneemm
ZZuussttaanndd –– zzuu bbeettrraacchhtteenn,, bbeeii kklleeiinneenn OObbjjeekktteenn wwiiee MMaakkrroommoolleekküülleenn ooddeerr VViirreenn ddiirreekktt,,
bbeeii ZZeelllleenn ooddeerr GGeewweebbeenn nnaacchh AAnnffeerrttiigguunngg vvoonn GGeeffrriieerrsscchhnniitttteenn.. DDaass PPrroobblleemm ddaabbeeii
iisstt –– nneebbeenn ddeerr TTaattssaacchhee,, ddaassss EEiiss iimm VVaakkuuuumm rreecchhtt sscchhnneellll ssuubblliimmiieerrtt –– ddaassss
WWaasssseerr bbeeiimm GGeeffrriieerreenn EEiisskkrriissttaallllee bbiillddeett,, ddiiee ddiiee SSttrruukkttuurreenn zzeerrssttöörreenn kköönnnneenn.. NNuurr
dduurrcchh uullttrraasscchhnneelllleess EEiinnffrriieerreenn ddeerr PPrroobbee ((VViittrriiffiizziieerreenn)) ooddeerr dduurrcchh ZZuuggeebbeenn eeiinneess
GGeeffrriieerrsscchhuuttzzmmiitttteellss kkaannnn ssttöörreennddee KKrriissttaallllbbiilldduunngg vveerrhhiinnddeerrtt wweerrddeenn..
SScchhnneelllleeiinnffrriieerrvveerrffaahhrreenn ssiinndd rreecchhtt aauuffwwäännddiigg,, eebbeennssoo ddeerr KKrryyoottrraannssffeerr iinn ddaass
ggeekküühhllttee MMiikkrroosskkoopp..
DDiiee ggäännggiiggeenn VVeerrffaahhrreenn zzuumm KKoonnttrraassttiieerreenn bbiioollooggiisscchheerr PPrrääppaarraattee ssiinndd::
NNeeggaattiivvkkoonnttrraassttiieerruunngg
PPoossiittiivvkkoonnttrraassttiieerruunngg
SScchhrräägg-- uunndd KKeeggeellbbeeddaammppffuunngg
BBeeii mmooddeerrnneenn EElleekkttrroonneennmmiikkrroosskkooppeenn kköönnnneenn dduurrcchh eelleekkttrroonneennooppttiisscchhee VVeerrffaahhrreenn,,
MMaatteerriiaalleeiiggeennsscchhaafftteenn aauussggeennuuttzztt wweerrddeenn uumm eeiinneenn aauussrreeiicchheennddeenn KKoonnttrraasstt zzuu

BCII am PEI, 22.11.2010-09.12.2010
104
eerrzziieelleenn.. EEiinnee KKoonnttrraassttiieerruunngg dduurrcchh cchheemmiisscchhee KKoonnttrraassttiieerruunnggssmmiitttteell iisstt ddaannnn nniicchhtt
mmeehhrr zzwwiinnggeenndd nnoottwweennddiigg..

BCII am PEI, 22.11.2010-09.12.2010
105
PRAKTISCHER TEIL
IImm PPrraakkttiikkuumm ssoolllleenn vveerrsscchhiieeddeenn VViirreenn ((VVaacccciinniiaa--VViirruuss,, IInnfflluueennzzaa--VViirruuss uu..aa..)) mmiitt uunntteerrsscchhiieeddlliicchheenn PPrrääppaarraattiioonnssmmeetthhooddeenn pprrääppaarriieerrtt uunndd aannsscchhlliieeßßeenndd iimm EElleekkttrroonneennmmiikkrroosskkoopp vveerrgglliicchheenn wweerrddeenn.. DDaa VViirreenn aauucchh ffüürr ddaass EEMM sseehhrr kklleeiinnee OObbjjeekkttee ddaarrsstteelllleenn,, iisstt eeiinnee aauuffwwäännddiiggee TTrroocckknnuunngg nniicchhtt nnööttiigg.. NNeeggaattiivvkkoonnttrraassttiieerruunngg Beim Negativkontrast werden die Objekte in einen Tropfen eines elektronendichten Kontrastierungsmittels eingebracht, das man eintrocknen läßt. Da biologisches Material in der Regel elektronendurchlässig ist, erscheinen die Strukturen hell vor dem dunklen Untergrund des Kontrastierungsmittels. DDuurrcchhffüühhrruunngg::
eeiinn ffrriisscchh bbeegglliimmmmtteess GGrriidd iinn PPiinnzzeettttee eeiinnssppaannnneenn eeiinneenn TTrrooppffeenn VViirruussssuussppeennssiioonn aauuff ddaass GGrriidd aauuffbbrriinnggeenn VViirruussppaarrttiikkeell 22 MMiinnuutteenn aaddhhäärriieerreenn llaasssseenn nnaacchh ddeerr AAddhhäässiioonnsszzeeiitt 22 mmaall nnaacchheeiinnaannddeerr kkuurrzz mmiitt AAqquuaa--ddeesstt.. wwaasscchheenn üübbeerrsscchhüüssssiiggee FFllüüssssiiggkkeeiitt aann eeiinneemm FFiilltteerrppaappiieerr aabbfflliieeßßeenn llaasssseenn ((VVoorrssiicchhtt!!
nniicchhtt ttrroocckkeenn ssaauuggeenn)) eeiinneenn TTrrooppffeenn 22%% UUrraannyyllaacceettaatt mmiitt PPaasstteeuurrppiippeettttee aauuffppiippeettttiieerreenn nnaacchh 1100 SSeekkuunnddeenn aann eeiinneemm FFiilltteerrppaappiieerrssttrreeiiffeenn ddaass KKoonnttrraassttmmiitttteell vvoollllssttäännddiigg
aabbfflliieeßßeenn llaasssseenn ((FFllüüssssiiggkkeeiittssffiillmm ddaarrff nniicchhtt aabbrreeiißßeenn,, dd..hh.. ddeerr KKoonnttaakktt zzuumm FFiilltteerrppaappiieerr mmuussss ssoollaannggee ggeehhaalltteenn wweerrddeenn,, bbiiss ddiiee FFllüüssssiiggkkeeiitt vvoollllssttäännddiigg aabbggeezzooggeenn iisstt !!))
SScchhrrääggbbeeddaammppffuunngg FFüürr eeiinnee SScchhrrääggbbeeddaammppffuunngg ddeerr OObbjjeekkttee wwiirrdd iinn eeiinneerr BBeeddaammppffuunnggssaannllaaggee iimm VVaakkuuuumm eeiinnee MMiisscchhuunngg aauuss KKoohhllee uunndd PPllaattiinn aallss sseehhrr ffeeiinneess KKoorrnn uunntteerr eeiinneemm sscchhrrääggeenn WWiinnkkeell aauuff ddaass PPrrääppaarraatt ggeeddaammppfftt ((„„EElleekkttrroonneennssttrraahhllvveerrddaammppffuunngg““)).. DDiiee OObbjjeekkttee eerrsscchheeiinneenn ddaannnn ppllaassttiisscchh,, ddaa ssiicchh MMeettaallll vvoorr ddeenn SSttrruukkttuurreenn aannllaaggeerrtt,, ddaahhiinntteerr jjeeddoocchh eeiinn mmeettaallllffrreeiieerr BBeeddaammppffuunnggss--„„SScchhaatttteenn““,, lliieeggtt.. DDuurrcchhffüühhrruunngg::
BBeeddaammppffuunnggssaannllaaggee vvoorrbbeerreeiitteenn eeiinn ffrriisscchh bbeegglliimmmmtteess GGrriidd iinn PPiinnzzeettttee eeiinnssppaannnneenn eeiinneenn TTrrooppffeenn VViirruussssuussppeennssiioonn aauuff ddaass GGrriidd aauuffbbrriinnggeenn VViirruussppaarrttiikkeell 22 MMiinnuutteenn aaddhhäärriieerreenn llaasssseenn nnaacchh ddeerr AAddhhäässiioonnsszzeeiitt 22 mmaall nnaacchheeiinnaannddeerr kkuurrzz mmiitt AAqquuaa--ddeesstt.. wwaasscchheenn ttrroocckknneenn llaasssseenn NNeettzzcchheenn aauuff TTiisscchh ddeerr BBeeddaammppffuunnggssaannllaaggee eeiinnssppaannnneenn,, ddiieesseenn iinn ddeenn
RReezziippiieenntteenn eeiinnbbrriinnggeenn,, EEvvaakkuuiieerreenn,, VVaakkuuuumm << 55xx1100--66,, VVeerrddaammppffeerrkkooppff aauussrriicchhtteenn,, BBeeddaammppffuunnggsswwiinnkkeell eeiinnsstteelllleenn,,
SScchhiicchhttddiicckkeennmmeessssggeerräätt eeiinnsscchhaalltteenn uunndd NNuullllppuunnkktt eeiinnsstteelllleenn BBeeddaammppffeenn,, SScchhiicchhttddiicckkee 22nnmm,, ZZeeiitt ccaa.. 1100 ss,, AAnnllaaggee bbeellüüfftteenn,, PPrrääppaarraattee eennttnneehhmmeenn,, AAnnllaaggee sscchhlliieeßßeenn

BCII am PEI, 22.11.2010-09.12.2010
106
UUllttrraaddüünnnnsscchhnniitttttteecchhnniikk UUmm vvoonn ZZeelllleenn ooddeerr GGeewweebbeenn uullttrraaddüünnnnee SScchhnniittttee aannzzuuffeerrttiiggeenn,, mmuussss mmaann zzuunnääcchhsstt –– mmöögglliicchhsstt sscchhoonneenndd -- ddaass WWaasssseerr dduurrcchh eeiinn aauusshhäärrttbbaarreess KKuunnsstthhaarrzz eerrsseettzzeenn,, wweellcchheess aannsscchhlliieeßßeenndd dduurrcchh WWäärrmmee ooddeerr BBeessttrraahhlluunngg mmiitt uullttrraavviioolleetttteemm LLiicchhtt ppoollyymmeerriissiieerrtt wwiirrdd.. DDiieesseess VVeerrffaahhrreenn ddaauueerrtt iinn ddeerr RReeggeell mmeehhrreerree TTaaggee,, ddaahheerr kkaannnn eess iimm PPrraakkttiikkuumm nnuurr ddeemmoonnssttrriieerrtt wweerrddeenn.. DDuurrcchhffüühhrruunngg::
11.. FFiixxiieerreenn ddeess OObbjjeekktteess 22,,55 %% GGlluuttaarraallddeehhyydd ffüürr 4455’’ bbeeii RRaauummtteemmppeerraattuurr ZZeelllleenn 22 xx mmiitt PPBBSS wwaasscchheenn 22.. NNaacchhffiixxiieerruunngg uunndd KKoonnttrraassttiieerruunngg aamm BBlloocckk PPBBSS aabbzziieehheenn,, uunntteerr ddeemm AAbbzzuugg ccaa.. 11 mmll OOssmmiiuummtteettrrooxxiidd zzuuggeebbeenn uunndd 11 hh
aauuff EEiiss iinnkkuubbiieerreenn 33 xx 55’’ mmiitt PPBBSS wwaasscchheenn 22 %% UUrraannyyllaacceettaattllöössuunngg ggeeffiilltteerrtt zzuuggeebbeenn.. 11 SSttuunnddee bbeeii RRaauummtteemmppeerraattuurr
lliicchhttggeesscchhüüttzztt iinnkkuubbiieerreenn UUrraannyyllaacceettaatt aabbzziieehheenn,, 22 xx 55’’ mmiitt AAqquuaa DDeesstt wwaasscchheenn 33.. EEnnttwwäässsseerruunngg AAqquuaa DDeesstt aabbzziieehheenn,, 3300%% EEtthhaannooll zzuuggeebbeenn uunndd 3300’’ iinnkkuubbiieerreenn.. MMiitt 5500%%,, 7700%%,,
8800%%,, 9900%% uunndd 9966%% EEttOOHH wwiieeddeerrhhoolleenn 22 xx 1155’’ EEtthhaannooll,, wwaasssseerrffrreeii 44.. AAuussttaauusscchh vvoonn EEtthhaannooll ggeeggeenn EEppoonn AAllkkoohhooll//EEppoonn 11::11 uunndd 11::33 jjeewweeiillss 3300’’ iinnkkuubbiieerreenn rreeiinneess EEppoonn 11 SSttuunnddee rreeiinneess EEppoonn üübbeerr NNaacchhtt aamm nnääcchhsstteenn MMoorrggeenn EEppoonn aauussttaauusscchheenn,, 33--44 SSttuunnddeenn iinnkkuubbiieerreenn 55.. EEiinnbbeettttuunngg uunndd PPoollyymmeerriissaattiioonn ZZeellll-- bbzzww.. GGeewweebbsspprroobbee iinn EEppoonn iinn eeiinneerr GGeellaattiinneekkaappsseell eeiinnbbeetttteenn 4488hh bbeeii 6600°°CC ppoollyymmeerriissiieerreenn 66.. HHeerrsstteelllluunngg vvoonn UUllttrraaddüünnnnsscchhnniitttteenn

BCII am PEI, 22.11.2010-09.12.2010
107
DDiiee aauussppoollyymmeerriissiieerrtteenn BBllööcckkcchheenn mmiitt ddeerr TTrriimmmmffrräässee aannssppiittzzeenn uunndd ZZeellllpprroobbee ffrreeiilleeggeenn,, aannsscchhlliieeßßeenndd mmiitt ddeemm UUllttrraammiikkrroottoomm ccaa.. 8800 nnmm ddüünnnnee SScchhnniittttee hheerrsstteelllleenn uunndd aauuff KKuuppffeerrggrriiddss aauuffzziieehheenn..
77.. NNaacchhkkoonnttrraassttiieerruunngg 22%% UUrraannyyllaacceettaattllöössuunngg iinn 7700%% EEtthhaannooll aallss TTrrooppffeenn aauuff eeiinn SSttüücckk PPaarraaffiillmm
ggeebbeenn GGrriidd ddaarraauuff ppllaattzziieerreenn,, SScchhnniittttee nnaacchh uunntteenn,, IInnkkuubbaattiioonn 77 MMiinnuutteenn iimm DDuunnkkeellnn 55 xx iinn aaqq.. BBiiddeesstt.. wwaasscchheenn GGrriiddss aauuff 00,,22%% BBlleeiicciittrraatt iinn aaqq.. bbiiddeesstt.. lleeggeenn,, 22 MMiinnuutteenn IInnkkuubbaattiioonn 55 xx iinn aaqq.. BBiiddeesstt.. WWaasscchheenn,, aannsscchhlliieeßßeenndd üübbeerrffllüüssssiiggeess WWaasssseerr mmiitt
FFiilltteerrppaappiieerr aabbssaauuggeenn..

BCII am PEI, 22.11.2010-09.12.2010
108
HINWEISE ZUM PRAKTIKUM
Zeitplan: Vormittags:
Kurze theoretische Einführung Vorbereitungen (Herstellung der Präparateträger, Beglimmung) Negativkontrastierung Einweisung am Mikroskop, Mikroskopieren und Fotografieren Vorbereiten der Schrägbedampfung (Vakuum)
Nachmittags:
Schrägbedampfung Demonstration einer Einbettung und der Herstellung von Ultradünnschnitten Mikroskopieren und Fotografieren verschiedener Präparate
Mittagspause je nach Bedarf zwischen 12 und 14 Uhr Aufgabenstellung für die praktische Arbeit:
Wie groß sind die verschiedenen Viruspartikel, welche Details erkennt man im Elektronenmikroskop?
Welchen Einfluss hat das Kontrastierungsmittel auf die Darstellung der Präparate?
Wie äußern sich Strahlenschäden an den Präparaten? Welches Kontrastierungsmittel ist stabil, welches weniger?
Welche Strukturdetails sind besonders gut darstellbar mit Negativkontrastierung, welche mit Schrägdedampfung, welche in Ultradünnschnitten?
Welche Zellorganelle erkennt man in Ultradünnschnitten? Welchen Einfluss hat die Größe der Aperturblende auf das Bild? Welchen Einfluss hat die Hochspannung auf das Bild? Was ist Astigmatismus? Wieso kommt es zur Bilddrehung beim Vergrößerungswechsel? Wie verändert sich das Bild, wenn sehr helle oder sehr dunkle Strukturen
vorhanden sind (automat. Kontrastausgleich)? Was ist „förderliche Vergrößerung“?
Top Related