
4 4 ##/ 9#: : '* : #> )KKKg@2A E 9# ' 9#! I $/ 'E 9 ' 9...
8
This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution 4.0 International License. Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschung in Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht: Creative Commons Namensnennung 4.0 Lizenz. Übergangsmetall-substituierte Acylphosphane und Phosphaalkene, XXXIY [1] Methylendiyl(thioxo)- und MethyIendiyl(selenoxo)-A5,<r3-phosphorane als //-Liganden in [(j/5-C5H5) 2 Fe2(CO)3 ]-Komplexen. Synthese und Struktur von (i/5-C5H5)(CO)Fe0/-CO)[^-C(PHrBu)(SMe)]Fe(i75-C5H5) Transition Metal Substituted Acylphosphanes and Phosphaalkenes, XXXIV [1] Methylenediyl(thioxo)- and Methylenediyl(selenoxo)-P,a3-phosphoranes as //-Ligands in [(775-C5H5)2Fe2(CO)3] Complexes. Synthesis and Structure of (7;5-C5H5)(CO)FeCa-CO)[a-C(PHrBu)(SMe)]Fe(^5-C5H5) Lothar Weber*, Iris Schumann, Matthias H. Scheffer, Hans-Georg Stammler, Beate Neumann Fakultät für Chemie der Universität Bielefeld, Universitätsstr. 25, D-33615 Bielefeld Herrn Prof. Dr. G. Hüttner zum 60. Geburtstag gewidmet Z. Naturforsch. 52b, 655-662 (1997); eingegangen am 14. März 1997 Isophosphaalkyne Complexes, Oxidation, X-Ray The isophosphaalkyne complex (775-C5H5)2(CO)2Fe2CM-CO)(//-C=PMes) (2a) was conver ted into the methylenediyl(thioxo)- and methylenediyl(selenoxo)-A5,cr3-phosphorane comple xes (775-C5H5)2(CO)2Fe2(M-CO)(M-C=P(E)Mes) (5a: E = S; 5b E = Se) by oxidation with sulfur or selenium. Reaction of the ^-carbyne complex [(/ 7 5-C 5H 5) 2(CO) 2Fe 2(w-CO)(w- CSMe)]+S 03CF3 (1) with fBuP(H)SiMe3 in the presence of DBU afforded the ^-phosphino- carbyne complex f? 7 5-C<;FIsWCO):>Fe; 7 |#-CP(HyBu]f#-SMe] (12). //-Carbene complex (/7 5-C5H5)(CO)Fe(a-CO){//-C(PHrBu)(SMe)}Fe(? 7 5-C 5H 5) (11) was isolated as an initial pro duct of this conversion and fully characterized by single crystal X-ray analysis. Einleitung Theoretische Rechnungen [2] wie auch eine Se rie fehlgeschlagener Syntheseversuche [3] unter streichen die große Instabilität von Isophosphaal- kinen RP=C im Vergleich zu den mittlerweile gut untersuchten Phosphaalkinen RC^P [4], Kürzlich berichteten Angelici et al. über Syn these und Strukturbestimmung eines ersten zwei kernigen Platinkomplexes I mit einem halbver- brückenden Isophosphaalkin-Liganden (Gl. (1)) [5], CI2C=P— Mes* L = PEt3 PtL.4 = ,ci Pt -------- Pt c*p Mes* ( 1 ) plexes 1 mit Aryl(silyl)phosphanen ein alternati ver und komplementärer Syntheseweg zu Isopho- sphaalkin-Komplexen vom Typ 2 existiert (Gl. (2)) [6], 4c0 SMe V.^Fe f 'cp II 0 1 T Aryl Ary(P(H)SiMe 3 DBü 3 oc c° r ^ r ' Fe ------- Fe Cp c Cp II 0 2 ( 2) Aryl a 2,4,6 -Me 3 C6H2 b 2,4,6— iPr^Cgh^ c 2,4,6— iBu^Cgh^ d 2,4,6-(CF 3 ) 3 C6H2 Wir konnten später zeigen, daß in der basenin duzierten Kondensation des ^-Carbindieisenkom- Sonderdruckanforderungen an Prof. Dr. L. Weber. In ersten orientierenden Versuchen zur chemi schen Reaktivität von 2 konnten durch Umsetzung mit [(Z)-Cycloocten]Cr(CO)5 bzw. Fe2(CO)9 rjl- Komplexe vom Typ 3 gewonnen werden. Demge genüber entsteht bei der Behandlung von 2a mit (Ph3P)2Pt(?7 2-C 2H 4) Komplex 4 [7]. 0932-0776/97/0500-0655 $06.00 © 1997 Verlag der Zeitschrift für Naturforschung. All rights reserved. D
Transcript of 4 4 ##/ 9#: : '* : #> )KKKg@2A E 9# ' 9#! I $/ 'E 9 ' 9...

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Übergangsmetall-substituierte Acylphosphane und Phosphaalkene, XXXIY [1] Methylendiyl(thioxo)- und MethyIendiyl(selenoxo)-A5,<r3-phosphorane als //-Liganden in [(j/5-C5H5)2Fe2(CO)3]-Komplexen. Synthese und Struktur von (i/5-C5H5)(CO)Fe0/-CO)[^-C(PHrBu)(SMe)]Fe(i75-C5H5)Transition M etal Substituted Acylphosphanes and Phosphaalkenes, XXXIV [1] M ethylenediyl(thioxo)- and M ethylenediyl(selenoxo)-P ,a3-phosphoranes as //-Ligands in [(775-C5H 5)2Fe2(CO)3] C om plexes. Synthesis and Structure of (7;5-C5H5)(CO)FeCa-CO)[a-C(PHrBu)(SM e)]Fe(^5-C5H5)
Lothar W eber*, Iris Schum ann, M atthias H. Scheffer, H ans-G eorg Stammler,Beate N eum annFakultät für Chem ie der U niversität B ielefeld , Universitätsstr. 25, D-33615 B ielefeld
Herrn Prof. Dr. G. H üttner zu m 60. G eburtstag gew idm et
Z. Naturforsch. 52b, 6 5 5 -6 6 2 (1997); eingegangen am 14. März 1997
Isophosphaalkyne Com plexes, O xidation, X -R ay
The isophosphaalkyne com plex (775-C5H5)2(CO)2Fe2CM-CO)(//-C=PMes) (2a) was converted into the m ethylenediyl(th ioxo)- and m ethylenediyl(selenoxo)-A 5,cr3-phosphorane com plexes (775-C5H 5)2(CO)2Fe2(M -CO)(M -C=P(E)M es) (5a: E = S; 5b E = Se) by oxidation with sulfur or selenium . R eaction o f the ^-carbyne com plex [(/75-C5H 5)2(C O )2Fe2(w-CO)(w- C SM e)]+S 0 3CF3 (1) with fB uP (H )SiM e3 in the presence o f D B U afforded the ^-phosphino- carbyne com plex f?75-C<;FIsWCO):>Fe;7|# -C P (H yB u]f# -SM e] (12). //-C arbene com plex (/75-C5H 5)(C O )Fe(a-C O ){//-C (PH rB u)(SM e)}Fe(?75-C5H 5) (11) was isolated as an initial product o f this conversion and fully characterized by single crystal X-ray analysis.
Einleitung
Theoretische Rechnungen [2] wie auch eine Serie fehlgeschlagener Syntheseversuche [3] u n te rstreichen die große Instabilität von Isophosphaal- kinen R P = C im Vergleich zu den m ittlerweile gut untersuchten Phosphaalkinen R C ^ P [4],
Kürzlich berichteten Angelici et al. über Synthese und Strukturbestim m ung eines ersten zweikernigen Platinkom plexes I mit einem halbver- brückenden Isophosphaalkin-Liganden (Gl. (1)) [5],
CI2C = P — Mes*
L = PEt3
PtL.4 = ,ciP t --------Pt
c*pM es*
( 1)
plexes 1 mit Aryl(silyl)phosphanen ein a lternativer und kom plem entärer Syntheseweg zu Isopho- sphaalkin-Kom plexen vom Typ 2 existiert (Gl. (2)) [6],
4c0SMe
V . ^ F e f 'c pII0
1
TAryl
A ry (P (H )S iM e 3
D B ü 3
oc c°r ^ r 'F e ------- Fe
Cp c Cp II 02
( 2 )
Aryl
a 2 ,4 ,6 -M e 3C6H2b 2,4,6— iP r^C g h ^c 2,4,6— iB u^C gh^d 2 ,4 ,6 -(C F 3 )3C6H2
Wir konnten später zeigen, daß in der baseninduzierten Kondensation des ^-C arbindieisenkom -
Sonderdruckanforderungen an Prof. Dr. L. W eber.
In ersten orientierenden Versuchen zur chem ischen R eaktivität von 2 konnten durch U m setzung mit [(Z)-Cycloocten]Cr(CO )5 bzw. Fe2(C O )9 rjl- Komplexe vom Typ 3 gewonnen werden. D em gegenüber entsteht bei der Behandlung von 2a mit (Ph3P )2Pt(?72-C2H 4) Komplex 4 [7].
0932-0776/97/0500-0655 $06.00 © 1997 Verlag der Zeitschrift für Naturforschung. All rights reserved. D
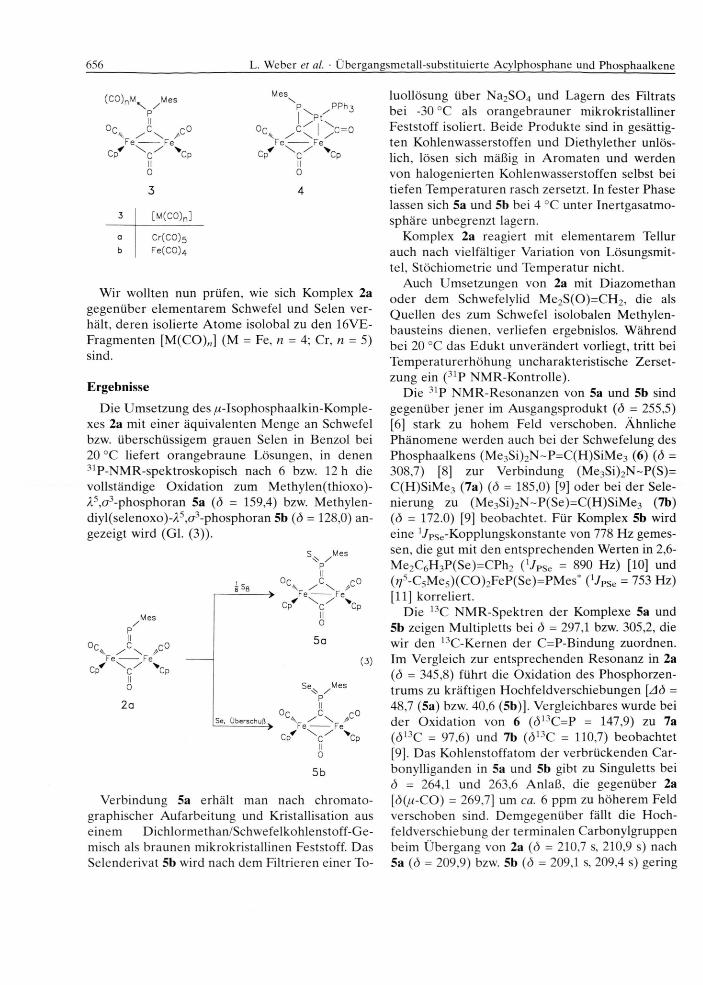
656 L. Weber et al. • Übergangsmetall-substituierte Acylphosphane und Phosphaalkene
(CO)nM^ ^MesPII
Oc C r o / \Fe -------- Ferrf \ / V
Cp c Cp II 03
Mes
PPh-
°C C \ c = 0K. / \ 1 /Fe -------- Fe
Cp c Cp II 04
3 [M (C0)n]
a Cr(CO)5b Fe(C 0)4
Wir wollten nun prüfen, wie sich Komplex 2a gegenüber elem entarem Schwefel und Selen verhält, deren isolierte A tom e isolobal zu den 16VE- Fragm enten [M(CO)„] (M = Fe, n = 4; Cr, n = 5) sind.
ErgebnisseDie U m setzung des /i-Isophosphaalkin-Kom ple-
xes 2a mit einer äquivalenten Menge an Schwefel bzw. überschüssigem grauen Selen in Benzol bei 20 °C liefert orangebraune Lösungen, in denen 31P-N M R-spektroskopisch nach 6 bzw. 12 h die vollständige O xidation zum M ethylen(thioxo)- A5,a3-phosphoran 5a ((3 = 159,4) bzw. M ethylen- diyl(selenoxo)-A5,a3-phosphoran 5b (d = 128,0) angezeigt wird (Gl. (3)).
gs8 oC ,c0
Mes
0C *c°\ / V Cp c Cp
II02a
.Mes
pII
C p" X CX 'C p II 0
5a
(3)
Sev Mes
P
Se, Überschuß,,C0Oc Cv / \ ,
Fe -------- Fe
Cp c CpII0
5b
V erbindung 5a erhält man nach chrom atographischer A ufarbeitung und Kristallisation aus einem D ichlorm ethan/Schw efelkohlenstoff-G emisch als braunen m ikrokristallinen Feststoff. Das Selenderivat 5b wird nach dem Filtrieren einer To
luollösung über N a2S 0 4 und Lagern des Filtrats bei -30 °C als orangebrauner m ikrokristalliner Feststoff isoliert. Beide Produkte sind in gesättigten Kohlenwasserstoffen und D iethylether unlöslich, lösen sich mäßig in A rom aten und werden von halogenierten Kohlenwasserstoffen selbst bei tiefen Tem peraturen rasch zersetzt. In fester Phase lassen sich 5a und 5b bei 4 °C unter Inertgasatm o- sphäre unbegrenzt lagern.
Komplex 2a reagiert mit elem entarem Tellur auch nach vielfältiger Variation von Lösungsmittel, Stöchiom etrie und Tem peratur nicht.
Auch U m setzungen von 2a mit D iazom ethan oder dem Schwefelylid M e2S (0 )= C H 2, die als Q uellen des zum Schwefel isolobalen M ethylenbausteins dienen, verliefen ergebnislos. W ährend bei 20 °C das E dukt unverändert vorliegt, tritt bei T em peraturerhöhung uncharakteristische Z ersetzung ein (31P NM R-K ontrolle).
Die 31P N M R -R esonanzen von 5a und 5b sind gegenüber jen er im Ausgangsprodukt (ö = 255,5) [6] stark zu hohem Feld verschoben. Ähnliche Phänom ene werden auch bei der Schwefelung des Phosphaalkens (M e3Si)2N ~P=C(H )SiM e3 (6) (<5 = 308,7) [8] zur Verbindung (M e3Si)2N~P(S)= C (H )SiM e3 (7a) (<5 = 185,0) [9] oder'bei der Sele- nierung zu (M e3Si)2N ~P(Se)=C (H )SiM e3 (7b) (ö = 172.0) [9] beobachtet. Für Komplex 5b wird eine ’/psg-K opplungskonstante von 778 Hz gemessen, die gut mit den entsprechenden W erten in 2,6- M e2C6H 3P(Se)=C Ph2 ( 7 PSe = 890 Hz) [10] und (775-C5M e5)(C O )2FeP(Se)=PM es* (Vpse = 753 Hz) [11] korreliert.
Die 13C N M R -Spektren der Komplexe 5a und 5b zeigen M ultipletts bei d = 297,1 bzw. 305,2, die wir den 13C-K ernen der C=P-Bindung zuordnen. Im Vergleich zur entsprechenden Resonanz in 2a (ö = 345,8) führt die O xidation des Phosphorzentrum s zu kräftigen Hochfeldverschiebungen [Ad = 48,7 (5a) bzw. 40,6 (5b)]. Vergleichbares wurde bei der O xidation von 6 (d 13C=P = 147,9) zu 7a ((513C = 97,6) und 7b (<513C = 110,7) beobachtet [9], Das Kohlenstoffatom der verbrückenden Car- bonylliganden in 5a und 5b gibt zu Singuletts bei ö = 264,1 und 263,6 A nlaß, die gegenüber 2a [d(w-CO) = 269,7] um ca. 6 ppm zu höherem Feld verschoben sind. D em gegenüber fällt die Hoch- feldverschiebung der term inalen Carbonylgruppen beim Ü bergang von 2a (d = 210,7 s, 210,9 s) nach 5a (ö = 209.9) bzw. 5b (d = 209,1 s, 209,4 s) gering

L. Weber et al. • Übergangsmetall-substituierte Acylphosphane und Phosphaalkene 657
aus. Diese D aten lassen den Schluß zu, daß die neuartigen //-Methylendiyl-chalcogeno-A5,a3-phos- phoran-Liganden schwächere D onatoren und bessere A kzeptoren sind als der //-Isophosphaalkin- Ligand in 2a.
Ein vergleichbares Bild wird aus den IR -Spektren von 2a, 5a und 5b im Bereich der Carbonylva- lenzschwingungen gewonnen. Die intensiven r(C O )-B anden für die term inalen Carbonylligan- den in 2a (v = 1978, 1949 c m 1) sind in 5a und 5b um 20 ± 1 bzw. 13 ± 1 cm '1 zu kürzeren W ellen verschoben. Die Valenzschwingung der verbrük- kenden Carbonylgruppe erfährt bei der O xidation von 2a (v(CO) = 1792 c m 1) ebenfalls eine hypsochrome Verschiebung von A v = 22 ± 1 c m 1.
In den M assenspektren von 5a und 5b wird je weils das M olekülion als Peak mit dem höchsten m /z-Wert (520 bzw. 568) beobachtet.
Die durch Schwefelung oder Selenierung gewonnen Methylendiylchalcogeno-A5,o3-Liganden sind ebenso wie das Isophosphaalkin M esP=C im freien Zustand unbekannt und den Y len(phospho- ranyliden)- und Im ido(phosphoranyliden)carbe- noiden 8a [12] und 8b [13] an die Seite zu stellen.
M e s ^ ^ C (S iM e 3)2 M es1 M e s* M e s * N x Me s *
P' %C
C \ ' \ i ( T H F ) 3
8a
cS X
Li CI
8b
+ /P h3P
Das 8b zugrunde liegende C arben konnte als stabiles Triphenylphosphanaddukt 9 abgefangen w erden [13].
Im folgenden sollte geprüft werden, ob auch am Phosphoratom alkylierte Isophosphaalkin-K om - plexe in Anlehnung an Gl. (2) zugänglich sind. Dazu haben wir zunächst Komplex 1 mit äquim o- laren M engen an ?BuP(H)SiM e3 in A cetonitril in Gegenwart von DBU um gesetzt und die R eaktion mittels 31P N M R-Spektroskopie verfolgt. Es zeigte sich, daß 1 und das Phosphan vor der Zugabe der Stickstoffbase unverändert nebeneinander vorliegen. Nach Zugabe von D B U ist das Signal von ?BuP(H)SiMe3 bei d = -85,5 nahezu verschwunden und wird durch ein Singulett bei d = 20,6 ersetzt. Im ^ -g e k o p p e lte n Spektrum spaltet dies zu einem D ublett von M ultipletts mit '/p n = 383,1 Hz auf. Wir ordnen dieses Signal Komplex 10 zu. Nach 14 h R ühren bei 20 °C zeigt das 31P{1H}
N M R -Spektrum neben dem Signal bei ö = 20,6 zusätzlich Singuletts bei <5 = -7,9 und -17,4. Nach zweitägigem R ühren erkennt man nur noch das Signal des Endproduktes 12 bei ö = -7,9. Bei 40 °C ist die Reaktion schon nach 4 h beendet (Gl. (4)).
°c*SMeIC.
TFe ------- Fe
rrYCp c Cp II 01
Me
tBuP(H )SiM e3 /DBU
NV <Bu
H tBu MeS P
O r r 0F e --------Fe
Cp C Cp II 0
10
Hvp/ iBu
(4)
,c0s—c> Fe —— Fe'
Cp* V " CpII0
11
°c.
Cp
/ c+xF e ------- Fer v
Me
12
- , 'c °
'Cp
Komplex 12 wird als rotbraunes Pulver in 26% A usbeute isoliert. D ie analog durchgeführten R eaktionen von 1 mit /PrPH 2 oder (M e3Si)3CP(H )- SiM e3 führten zu keinen definierten Produkten. K onstitution und Konfiguration von 12 wurden aus E lem entaranalyse und Spektren abgeleitet.
D as IR -Spektrum der Verbindung zeigt im v(CO )-Bereich nur eine starke breite Bande bei v = 1935 cm '1 für die term inalen Carbonylliganden und eine scharfe schwache A bsorption bei v = 2279 cm '1 für die PH-Valenzschwingung. Auffällig ist das Fehlen von Banden, die von CO -Brücken herrühren. Ein Singulett im ]H N M R-Spektrum von 12 bei ö = 4,12 für die P rotonen beider Cp- Ringe deutet auf eine symmetrische S truktur des M oleküls hin. Die Protonen der S-M ethylgruppe w erden bei <3 = 2,51 als Singulett beobachtet, w ährend die PH -Funktion zu einem D ublett bei (3 = 3,61 (Vph = 265,5 Hz) A nlaß gibt. Interessant ist im 13C{ 1H} N M R-Spektrum ein D ublett bei <3 = 420,0 (V ph = 83 Hz), das wir dem verbrückenden Kohlenstoffatom eines //-Phosphinocarbin-Ligan- den zuordnen. D ie entsprechenden 13C-Kerne w erden in den kationischen Alkylthiocarbin-Kom- plexen 13a und 13b bei ö = 407,9 bzw. 403,1 beobachtet [14].
Im Einklang mit einer symmetrischen M olekülstruk tu r wird ein Singulett bei <5 = 82,9 und ein D ublett bei 220,6 (37pC = 20 Hz) den Cyclopenta- dienyl- bzw. den CO-Liganden zugeordnet. Im protonengekoppelten 31P N M R-Spektrum (C6D 6)

658 L. Weber et al. • Übergangsmetall-substituierte Acylphosphane und Phosphaalkene
SMe
°c\ / PEt3F e ------- Fe
c p ' V CpII0
13a
°cSCH2Ph
C r O\ / + \ AF e ------- Fe
Cp' vcPII0
13b
von 12 wird ein D ublett eines D ezetts bei ö = -6,0 ('/pH = 265 Hz, V PH = 14 Hz) registriert. Aufgrund dieser Befunde schlagen wir für 12 die Struktur eines zweikernigen Eisenkom plexes mit jeweils zwei c/s-ständigen term inalen CO- und Cp-Ligan- den vor. Die M etallatom e werden von einem r-Bu- tylphosphinocarbin- und einem M ethylthiolato-Li- ganden verbrückt. Bricht man das Experim ent bei R aum tem peratur nach 2 h ab, so läßt sich Verbindung 11 nach säulenchrom atographischer A ufarbeitung in Form schwarzer Kristalle isolieren (Gl.(4))-
Das zu 12 isomere Prim ärprodukt 11 zeigt im IR-Spektrum (KBr) intensive CO -Banden bei v = 1941 und 1754 cm '1, die einer term inalen und einer verbrückenden Carbonylgruppe zuzuordnen sind. Die PH-Valenzschwingung wird bei v - 2261 cm '1 als schwache scharfe Bande registriert. Das !H N M R -Spektrum zeigt zwei diskrete Singuletts bei ö = 4,19 und 4,51 für die jeweils 5 Protonen zweier chemisch und magnetisch verschiedener Cp-Li- ganden. Das Singulett für die S-CH3-G ruppe tritt bei erstaunlich hohem Feld (ö - 0,75) auf. Busetto et al. beobachteten bei der photochem ischen De- carbonylierung von 14 zu 15 (Gl. (5)) ebenfalls eine Hochfeldverschiebung der S-M ethylprotonen von <3 = 2,78 nach 1,80 [16].
i(,CNO r C rO
\F e ------- Fe
Cp c Cp II 0
1 4
X °
Me .CN\ **S*-Chi/ \ ----- Fe ------------------- ----- Fe- CO ✓ \ / v
Cp c Cp II 0
15
(5)
Ein D ublett bei d - 5,32 (Vph = 192,3 Hz) rührt von der PH -Funktion her. D ieser W ert fügt sich gut in die entsprechenden N M R -Param eter der /u- Phosphinocarbenkom plexe Cp2(C O )2Fe2(//-CO)- [a-C(CN)PCyH] (CDC13: d = 4,87, 1J PH = 198,0 Hz) und Cp2(C O )2Fe2(J«-CO )[//-C(CN )PPhH ] (CDC13: d = 6,02, 7 p H = 204 Hz) ein [17], Das 13C{'H} N M R-Spektrum von 11 zeigt für die Ring- kohlenstoffatom e der beiden verschiedenen Cp-
Liganden Singuletts bei d = 83,6 und 85,7. Ein Singulett bei d - 218,1 ist dem term inalen CO-Ligan- den zuzuordnen, w ährend die Resonanz für die CO -Brücke bei <3 = 268,8 lokalisiert werden konnte. E in D ublett bei d - 163,2 (Vpc = 108,0 Hz) für das verbrückende C -A tom des /i,?/2-Phos- phino(th iolato)carben-L iganden ist gut mit der entsprechenden Resonanz in [Cp2(C O )2Fe2(/i- C O )[//-C H (PPh2)}] (ö = 159,3 d, 7PC = 60 Hz) vergleichbar [18].
M olekülstruktur von 11Das Ergebnis der R öntgenstrukturanalyse von
11 (Einkristalle aus Toluol/Pentan bei -30 °C, Abb. 1 Tab. I) läßt einen „butterfly“-Komplex mit Cp2Fe2-Rückgrat und c/s-ständigen Cp-Ringen erkennen. D er Interplanarw inkel zwischen den E benen aus den A tom en F e (l) , Fe(2), C(6) und F e(l), Fe(2), C(7) beträgt dabei 146,4°. Die Fe-Fe-Bin- dung von 2,502(1) A ist vergleichbar mit jener im K ation [(775-C5H 5)2Fe2(C O )3(C SEt)]+ [2,510(2) A] [19] und liegt zwischen den Fe-Fe-A bständen in cis-[(rj5-C5H 5)(C O )2Fe]2 [2,531(2) Ä] [20] und cis- [(^5-C5H 5)Fe(C O )(C S)]2 [2,482(1) A] [21], D er interessanteste Teil des M oleküls ist der fButylphos- phino(m ethylthiolato)carben-L igand, der die beiden Fe-Z entren über das verzerrt tetraedrische K ohlenstoffatom C(7) unsymmetrisch verbrückt. D er Ligand ist mit F e (l) zusätzlich über eine Fe- S-Bindung von 2,231(1) Ä verknüpft. D ieser Wert
Tab. I. Ausgewählte Bindungslängen (Ä) und Bindungswinkel (°) von 11.
Fe(l)-C(6) 1,849(3) Fe(l)-C(7) 1,910(3)Fe(l)-S(l) 2,2309(8) Fe(l)-Fe(2) 2,5015(7)Fe(2)-C(8) 1,761(3) Fe(2)-C(7) 1,975(3)Fe(2)-C(6) 2,008(3) S(l)-C(7) 1,800(3)S(l)-C(14) 1,809(3) P(l)-C(7) 1,807(3)Pd)-C(15) 1,885(3) P(l)-H(19) 1,19(3)0(1)-C(6) 1,183(4) 0(2)-C(8) 1,139(4)
C(6)-Fe(l)-C(7) 97,46(13) C(6)-Fe(l)-S(l) 91,24(9)C(7)-Fe(l)-S(l) 50,81(8) C(6)-Fe(l)-Fe(2) 52,39(9)C(7)-Fe(l)-Fe(2) 51,06(8) S(l)-Fe(l)-Fe(2) 79,97(2)C(8)-Fe(2)-C(7) 92,62(12) C(8)-Fe(2)-C(6) 89,19(13)C(7)-Fe(2)-C(6) 90.41(12) C(7)-Fe(2)-Fe(l) 48,80(8)C(8)-Fe(2)-Fe(l) 108,89(10) C(6)-Fe(2)-Fe(l) 46,86(9)C(7)-S(l)-C(14) 110.65(13) C(7)-S(l)-Fe(l) 55,34(9)C(14)-S(l)-Fe(l) 108.78(11) C(7)-P(l)-C(15) 108,37(14)C(7)-P(l)-H(19) 99(2) C(15)-P(l)-H(19) 92(2)0(1)-C(6)-Fe(l) 146,0(3) 0(1)-C(6)-Fe(2) 132,9(2)Fe(l)-C(6)-Fe(2) 80,75(11) S(l)-C(7)-P(l) 125,4(2)S(l)-C(7)-Fe(l) 73,85(10) P(l)-C(7)-Fe(l) 126,8(2)S(l)-C(7)-Fe(2) 107,61(13) P(l)-C(7)-Fe(2) 124.34(14)Fe(l)-C(7)-Fe(2) 80,14(10) 0(2)-C(8)-Fe(2) 174.6(3)

L. Weber et al. • Übergangsmetall-substituierte Acylphosphane und Phosphaalkene 659
Abb. 1. M olekülstruktur von 11 im Kristall.
ist gut mit den Fe-S-A bständen, die im Cluster Fe- 3 (C O )9 (/*-Cl)(//3-S-rBu) zu 2,127(4) und 2,226(4) A bestim m t wurden, vergleichbar [22]. Durch d iesen zusätzlichen Kontakt bedingt ist die Bindung F e(l)-C (7 ) [1,910(3) Ä ] deutlich kürzer als der Abstand Fe(2)-C (7) [1,975(3) Ä ], Im K om plex 14, der den //[C (C N )(SM e)]-L igand ohne zusätzliche Fe-S-W echselwirkung besitzt, sind dagegen die Bindungen zwischen dem verbrückenden Carben- kohlenstoffatom und den beiden Fe-Zentren ausgeglichen [1,989(2) und 1,999(2) Ä ] [16].
Gut vergleichbar sind dem gegenüber die C-S- Einfachbindungen, die in 11 1,800(3) [C (7)-S (l)] bzw. 1,809(3) [C (14)-S (l)] Ä und in 14 1,807(2) bzw. 1,805(2) A [16] betragen. D as Schwefelatom ist pyramidal konfiguriert, w ie aus der W inkelsumm e von 274,77° hervorgeht. D ie E benen durch die A tom e C(7), S (l) , F e ( l) und F e ( l) , Fe(2), C(7) schließen dabei einer Interplanarwinkel von111,7° ein, während die erste der beiden E benen nahezu orthogonal auf der E bene durch die A tom e F e ( l) , Fe(2) und C (6 ) steht. D as chirale K ohlenstoffatom C(7) ist zusätzlich über eine C- P-Einfachbindung von 1,807(3) Ä mit der rButyl- phosphino-Gruppe verbunden. D er Standardwert für eine P-C-Einfachbindung wird mit 1,85 A angegeben [23]. Das pyramidal konfigurierte Phosphoratom (W inkelsum m e 299°) trägt neben der rButylgruppe [P (l)-C (15) = 1,885(3) Ä ] noch ein W asserstoffatom [P (l)-H (19 ) = 1,19(3) Ä ].
Es ist interessant, daß sich die Phosphinogruppe auf der konvexen Seite des „butterfly“-G erüstes befindet und zu den Cp-Ringen ds-orientiert ist. D ies ist mit einem stereochem isch einheitlichen exo-A ngriff des Phosphid-Ions auf den ^-Carbinli- ganden von 1 vereinbar. W egen des unterschiedlichen Substitutionsm usters an beiden Fe-A tom en [Fe(2) trägt im G egensatz zu F e ( l) einen term inalen Carbonylliganden] sind die Abstände F e ( l) - C (6 ) [1,849(3) Ä] und F e(2)-C (6) [2,008(3) Ä] zur verbrückenden Carbonylgruppe deutlich verschieden. In 14 werden für die Fe-C-Abstände zur CO Brücke W erte von 1,934(2) und 1,917(2) Ä gem essen. Im Hinblick auf den halbverbrückenden Charakter der Carbonylgruppe C (6 ) -0 ( l) ist es leicht vorstellbar, daß nach dem Bruch der Bindung C (7 )-S (l) der Schwefel das Eisenatom Fe(2) angreift und dabei den Brückenliganden von Fe(2) völlig zum E isenatom F e ( l) verdrängt.
Experimenteller Teil
A lle U ntersuchungen und Spektrenaufnahm en wurden unter N 2-Schutzgas durchgeführt. D ie verw endeten Lösungsm ittel waren wasserfrei, frisch destilliert und N 2-gesättigt. D ie Verbindungen [(775 -C 5 H 5 )2 (C 0 )2Fe2 (M-C0 )CM-CSMe)]+S 0 3 CF3-[14], [(775 -C 5H 5 )2 (C O )2Fe2 (w-CO)(//-C=PM es)] 2a[6 ] und rBu(M e3Si)PH [15] wurden nach Literaturangaben hergestellt. 1,8-Diazabicyclo[5.4.0]un- dec-7-en (D B U ) wurde gekauft. Sämtliche NM R-

660 L. Weber et al. • Übergangsmetall-substituierte Acylphosphane und Phosphaalkene
Spektren wurden bei 300 K mit Bruker Geräten (A M 300 und AC 100) [Standards: intern M e4Si C H, 1 3C -N M R ), extern 85% H 3P 0 4 (3 1 P-N M R)] registriert. IR: m odifiziertes G itterspektrom eter PE 580 der Fa. Perkin Eimer. M assenspektrom eter: Finnigan M AT 311A (EI, 70 eV ).
(rj5-C5H5)2(CO)2Fe2(fi-CO) (ji-C=P(S)Mes) (5a)
Zu einer Lösung von 0,12 g (0,24 m m ol) 2a in 5 ml B enzol gibt man bei 20 °C eine Lösung von 0,007 g (0,24 m mol berechnet auf S) Schwefel in 1 ml B enzol und rührt 6 h. A nschließend wird das Lösungsm ittel im Vakuum entfernt, der R ückstand in 10 ml CS2 aufgenom m en und die Lösung über eine 10 cm dicke Schicht von silanisiertem K ieselgel filtriert. Es wird zur Trockene eingeengt, der braune Rückstand in 5 ml eines CH 2C12 /CS2- G em isches gelöst und die Lösung im N 2-Strom vorsichtig bis zur ersten Trübung konzentriert. Kristallisieren über Nacht bei -30 °C liefert 0,08 g mikrokristallines braunes 5a ■ CS2 (56% A usbeute). - IR (KBr): 2000 vs [v(C O )j, 1961 s [v(C O )j, 1813 s [v(CO)] cm '1. - IR (C H 2C12): v(C O ) = 2007 vs, 1973 s, 1809 s c m 1. - 1 H -N M R (C D 2C12): (3 = 2,37 (s, 3H, p - C H 3), 2,60 (s, 3H , o- C H 3), 2,94 (s, 3H , o-C H 3), 4,53 (s, 5H, C5H 5), 5,20 (s, 5H, C 5H 5), 7,07 (s, 1H, m -A ryl-H ), 7,10 (s, 1H, m -A ryl-H ). - ^C pH J-N M R (C D 2 C12): (3 = 21,5 (d, 5/p C = 5,0 Hz, p - C H 3), 23,0 (s, br, o-C H 3), 88,3 (s, C5 H 5 ), 89,8 (s, C5 H 5 ), 129,1 (s, m -Aryl-C), 129,3 (s, m -Aryl-C), 131,4 (s, /?-Aryl-C), 140,4 (s, br, o- A ryl-C ), 140,6 (s, br, o-Aryl-C), 141,3 (s, 1-Aryl- C), 209,9 (m, C O ), 264,1 (s, /^-CO), 297,1 (m, C= P). - 3 1 P{'H }-N M R (C 6H 6):<3 = 159,4(s). M S/FD: m /z = 520 (M +).
Aufgrund der Thermolabilität der Verbindung konnte kein solvatfreies Produkt erhalten werden. D ie A nalyse wurde für 5a • CS2 berechnet.
Analyse: C 24H 2 1Fe20 3PS3 (596,29)Ber. C 48,34 H 3,55% ,Gef. C 48,79 H 4,02% .
(rj5-C5H5)2(CO)2Fe2(pi-CO)(fx-C=P(Se)Mes) (5b)
D ie Lösung von 0,12 g (0,24 m m ol) 2a in 4 ml B enzol wird bei 20 °C mit 0,05 g (0,63 m m ol) grauem Selenpulver versetzt und 12 h gerührt. A lle flüchtigen Bestandteile werden im Vakuum entfernt und der Rückstand in 10 ml Toluol aufgenom m en. Es wird über N a 2S 0 4 filtriert und das Filtrat auf 4 ml konzentriert. Kristallisation über Nacht bei -30 °C führt zu 0,07 g (51% ) m ikrokristallinem , orangebraunem 5b. - IR (KBr): v = 2002 vs [v(C O )], 1963 s [v(CO )], 1815 s [r(C O )]
c m 1. - ’H -N M R (C D 2C12): (3 = 2,37 (s, 3H, p- C H 3), 2,58 (d, 4/ PH = 2,1 Hz, 3H , o-C H 3), 2,94 (d, V ph = 1,3 Hz, 3H, o-CH^), 4,55 (s, 5H, C5 H 5), 5,22 (s, 5H , CSH S), 7,05 (d. 4/ PH = 5,0 Hz, 1H, m-Aryl- H ), 7,09 (s, 1H, m -A ryl-H ). - 1 3C{'H}-NM R (C D 2C12): ( 5 - 2 1 ,5 (d, V PC = 7 Hz, p -C H 3), 22,8 (d, 3/p c = 9 Hz, o -C H ,), 23,5 (d, 37PC = 7 Hz, o- C H ,), 88,3 (s, CSH ,), 90,1 (s, CSH 5), 129,1 (s, m- A ryl-C ), 129,2 (s, m -A ryl-C ), 138,3 (d, 4JPC = 7 Hz, p-A ryl-C ), 140,4 (s, o-A ryl-C ), 140,5 (s, o-Aryl-C),141,3 (d, 7 PC = 3 Hz, i-Aryl-C ), 209,1 (d, 3/ PC =11 Hz, C O ), 209,4 (d, 3/ PC = 9 Hz, CO), 263,6 (s, fi-C O ), 305,2 (m, C=P). - 3 ,P{'H )-N M R (C 6H 6): ö = 128,0 s. - 3 1 P{‘H )-N M R (C D 2 C12): ö = 124,0 (s, VpSe = 778 H z), - M S/EI (70 eV): m /z = 568 (M +), 553 (M +-C H 3), 488 (M +-Se), 460 (M +-Se,- C O ), 432 (M +-Se,-2C O ), 404 (M +-Se,3CO ), 338 (M +-Se,-C O ,-C ,H 6), 282 (M +-Se,-3CO,-C 5 H 6 ,-Fe),186 (F e(C 5 H 5)2+), 121 (Fe(C 5 H 5)+).
Analyse: C 23H 2 1 Fe2 0 3PSe (567,04)Ber. C 48,72 H 3,73% ,Gef. C 48,59 H 3,88% .
(V5-C5H 5)2(C O )2Fe[fi-C-P(H)tBu](fi-SCH3) (12)Zur Lösung von 0,22 g (0,41 m m ol) 2a in 20 ml
A cetonitril wird bei 0 °C eine Lösung von 0,06 g (0,41 m m ol) /B uP (H )S iM e3 in 5 ml A cetonitril getropft. Nach beendeter Zugabe läßt man 10 min rühren und versetzt dann mit 0,09 g (0,59 m m ol) D B U , w obei sich die Lösung augenblicklich von tiefrot nach braun verfärbt. Es wird auf 40 °C erwärmt und 4 h gerührt. Lösungsm ittel und alle flüchtigen K om ponenten werden im Vakuum entfernt. Sodann wird der Rückstand in wenig Benzol aufgenom m en und auf silanisiertes K ieselgel aufgezogen . B ei der nachfolgenden Säulenchrom atographie an silanisiertem K ieselgel wird eine rote Z one mit einem H exan/Ether-G em isch (1:4) eluiert. D as Eluat wird vom Lösungsm ittel befreit und der feste Rückstand in Cyclopentan aufgenom m en. B ei -30 °C fällt über Nacht Verbindung12 als rotbraunes Pulver aus (Ausb. 0,05 g, 26% ). - IR (KBr): 2279 w [v(PH )], 2013 sh, 1935 s br [v(CO )] c m 1. - 'H -N M R (C 6D 6): (3 = 1,22 (d, V PH = 14,5 Hz, 9H, fBu), 2,51 (s, 3H, SCH3), 3,61 (d, V PH = 265,5 H z, 1H, PH ), 4,12 (s, 10H, C5H 5).13C -N M R (C 6D 6): (3 = 30,8 (s, C (C H 3)3), 32,3 (d, Vpc = 9 Hz, C (C H ,)0 , 36,2 (s, SC H 3), 82,9 (s, C 5H 5 ), 220,6 (d, V pc = 20 Hz, CO ), 420,0 (d, lJPC = 83 Hz, Fe2 CP). - 3 1 P-NM R (C 6D 6): (5 = -6.0 (d dec, 1/ PH = 265 Hz. 3/ PH =14 Hz). - M S/EI (70 eV ): m /z = 446 (M +), 418 (M +-CO ), 390 (M +- 2C O ), 333 (M +-2CO,-C 4 H 9), 186 (FeCp2+), 121 (FeC p+).

L. Weber et al. ■ Übergangsmetall-substituierte Acylphosphane und Phosphaalkene 661
A nalyse: C 18H 2 3Fe2 0 2 PS (446,11)Ber. C 48,46 H 5,20%,Gef. C 48,31 H 5,07% .
(r f - C , H ,)(C O )F e(u -C O )[(u -C (P (H )tB u ) (SMe)]Fe(r/5-C5H ,) (11)
D ie Lösung von 0,56 g (1,05 m m ol) 2a in 25 ml A cetonitril wird bei 0 °C mit 0,31 g (1,05 m m ol) rB uP(H )SiM e3 versetzt. Nach 15 min Rühren bei0 °C tropft m an eine Lösung von 0,22 g (1,50 m m ol) D B U in 5 ml Acetonitril hinzu. D ie Lösung färbt sich schwarzbraun. Man entfernt die Kühlung und rührt 2h bei Raumtemperatur. D ann werden Lösungm ittel und flüchtige Verbindungen im Vakuum abgezogen. D er Rückstand wird in w enig B enzol aufgenom m en und auf silanisiertes K ieselgel aufgezogen. D ie nachfolgende Säulenchrom atographie an silanisiertem Kieselgel mit H exan/E ther-G em ischen liefert eine schwarze Z one, deren Eluat zur Trockene eingeengt wird. D er Rückstand wird in 5 ml Toluol aufgenom m en, mit 2 ml «Pentan überschichtet und bei -30 °C kristallisiert. Ausb. 0,170 g (36% ) 11 in Form schwarzer Kristalle. - IR (KBr): v = 2261 w [v(PH)], 1941 s [v(C O )], 1754 s [v(m -C O )] cm '1. - !H -NM R (C 6D 6): d = 0,75 (s, 3H , SCH3), 1,39 (d, 3/ PH = 11,4 Hz, 9H , tBu), 4,19 (s, 5H, C5 H 5), 4,51 (s, 5H, C5H 5), 5,32 (d, VpH = 192,3 H z, 1H, PH). - ^ C ^ H j-N M R (C 6 D 6): (3 = 24,7 (s, SCH 3), 30,8 (d, V pc = 11,8 Hz, C (C H 3)3), 33,0 (d, lJ PC = 15,8 Hz, C (C H 3)3), 83,6 (s, C5H5), 85,2 (s, C 5 H 5), 163,2 (d, VpC = 108,0 H z, //-C (PH rB u)(SM e)), 218,1 (s, CO ), 268,8 (s, f i-C O ). - 3 1P{'H )-N M R (C6D 6): ö = 17,5 s.
Analyse: C 18H 23Fe2 0 2PS (446,11)Ber. C 48,46 H 5 ,20% ,Gef. C 48,82 H 5,15% .
Röntgenstrukturanalyse von 11Einkristalle von 11 wurden aus Toluol/«Pentan
bei -30 °C gezüchtet. C 18H 23Fe20 2PS, Kristalldim ensionen 0,80 x 0,40 x 0,30 m m 3, orthorhom bisch, Raum gruppe P 2 12 12 1, a = 11,023(2), b = 12,145(2), c = 13,795(3) Ä , V = 1846,8(6) Ä 3, Z = 4, dber = 1,604 g cm'3, S iem ens-P2r Vierkreisdif- fraktom eter, M oK a-Strahlung, G raphitm onochromator, X = 0,71073 Ä , /z = 1,778 mm"1, T = 173 K. Datensam m lung: («-Scan, 20 max = 55°, 4206 unabhängige R eflexe, (R int = 0,0223). Strukturlösung nach D irekten M ethoden und Verfeinerung nach der M ethode der kleinsten Fehlerquadrate mit Siem ens SH E L X T L -PL U S/SH E L X L -93, 225 Parameter, alle N ichtw asserstoffatom e anisotrop, H (19) isotrop verfeinert, alle w eiteren W asserstoffatom e an berechneten Positionen, /?1 = 0,037, wR2 = 0,076 für 4196 unabhängige R eflexe, RI = I \ \ F 0 \ - IFc ll/2;iF 0 l, w R 2 = [£[w (F 0 2 - Fc2)2] / £[w (F 02)2]]1/2, w 1 = 1/[ct2 (F02) + (0,0315P)2], P = (F0 2 + 2FC2) /3, m axim ale R estelektronendichte 0,3 e Ä ' 3 [24].
D ank
D iese A rbeit wurde in dankenswerter W eise von der D eutschen Forschungsgem einschaft (B onn), dem Fonds der Chem ischen Industrie (Frankfurt/M ain) und der B A S F -A G (Ludwigshafen) unterstützt.
[1] X X X III. M itteilung: L. Weber, S. Uthm ann, B. Torw iehe, R. Kirchhoff, R. Boese, D. Bläser, Organo- m etallics, im Druck.
[2] a) K. K. L ehm an, S. C. Ross, L. L. Lohr, J. Chem. Phys. 82, 4460 (1985);b) M. T. N guyen , T.-K. Ha, J. M ol. Struct. (Theo- chem .) 139, 145 (1986).
[3] a) S. J. G oed e, F. B ickelhaupt, Chem . Ber. 124, 2677 (1991);b) M. Yoshifuji, T. Niitsu, N. Inam oto, Chem. Lett. 1722 (1988);c) L. N. M arkovskii, G. N. Koidan, A . P. M archenko, V. D. R om anenko, M. I. Povolotskii, A . M. Pinchuk, Zh. O bshch. Khim . 59, 2133 (1988).
[4] a) G. B ecker, G. G resser, W. U h l, Z. Naturforsch. 36b, 16 (1981);
b) M. R egitz, Chem . Rev. 90, 191 (1990);c) M. R egitz, P. Binger, A ngew . Chem . 100, 1541 (1988); A ngew . Chem ., Int. Ed. Engl. 27, 1484(1988);d) M. R egitz, M ultiple B onds and Low C oordination in Phosphorus Chemistry, M. R egitz, O. J. Scherer (H rsg.), S. 58, Thiem e, Stuttgart, (1990).
[5] a) H. Jun, V. G. Young, R. J. A ngelici, J. A m . Chem. Soc. 113, 9379 (1991);b) H. Jun, R. J. A ngelici, O rganom etallics 13, 2454(1994).
[6] L. W eber, I. Schum ann, T. Schm idt, H.-G. Stam m ler, B. N eum ann, Z. A norg. Allg. Chem . 619, 1759 (1993).

662 L. Weber et al. • Übergangsmetall-substituierte Acylphosphane und Phosphaalkene
[7] L. W eber, I. Schum ann, H.-G. Stam m ler, B. N eu mann, Chem . Ber. 127, 1349 (1994).
[8] E. N iecke, W. W. Schoeller, D .-A . W ildbredt, A n gew. Chem . 93, 119 (1981); A ngew . Chem ., Int. Ed. Engl. 20, 131 (1981).
[9] E. N iecke, D .-A . W ildbredt, J. Chem . Soc., Chem . Com m un. 72 (1981).
[10] T. A . van der Knaap, M. Vos, F. B ickelhaupt, J. O rganom et. Chem . 244, 363 (1983).
[11] L. W eber, G. M eine, R. B oese, N. Niederprüm , Z. Naturforsch. 43b, 715 (1988).
[12] E. N iecke, P. B ecker, M. N ieger, D. Stalke, W. W. Schoeller, Angew. Chem . 107, 2012 (1995); Angew . Chem ., Int. Ed. Engl. 33, 1849 (1995).
[13] W. Schilbach, V. von der G önna, D. G udat, M. N ie ger, E. N iecke, Angew . Chem . 106, 1037 (1994); A n gew. Chem ., Int. Ed. Engl. 33, 982 (1994).
[14] M. H. Q uick, R. J. A ngelici, Inorg. Chem. 20, 1123(1981).
[15] G. Becker, O. M undt, M. R össler, E. Schneider, Z. Anorg. Allg. Chem . 443, 42 (1978).
[16] L. B usetto, S. Bordoni, V. Zanotti, V. G. A lbano, D. Braga, Gazz. Chim. Ital. 118, 667 (1988).
[17] L. Busetto, L. Carlucci, V. Zanotti, V. G. A lbano, M. M onari, Chem. Ber. 125, 1125 (1992).
[18] S. Bordoni, F. Mazza, V. Zanotti, Inorg. Chim. Acta. 223, 31 (1994).
[19] R. E. Wagner, R. A . Jacobson. R. J. A n gelici, M. H. Q uick, J. O rganom et. Chem. 148, C35 (1978).
[20] R. F. Bryan, P. T. G reene, M. J. N ew lands, D. S. Field, J. Chem. Soc. A 3068 (1970).
[21] J. W. Dunker, J. S. Finer, J. Clardy, R. J. A n gelici, J. O rganom et. Chem. 114, C49 (1976).
[22] A . W inter, L. Zsolnai. G. Hüttner, J. O rganom et. Chem . 232, 47 (1982)
[23] D. E. C. Corbridge, The Structural C hem istry o f Phosphorus, S. 393, E lsevier Scient. Publ. Comp., Am sterdam (1974).
[24] W eitere Kristallstrukturdaten können beim Fachin- form ationszentrum Karlsruhe G m bH , D -76344 Eg- genstein-L eopoldshafen , unter A ngabe der H interlegungsnum m ern C SD 406920, angefordert werden.



![01 - LU · ˘7 (f78(dfgdf((37g 9˘7dfg(fg( 37 ˘7g(97[(ml7g< 38˘ ˘8(7 9< 2l ˘9 7fg8h9 7fg38(gˆ ˘9 9 -g8-˘g82g 9˘7dh(7ˆ83d ˘7 (9˘f78(d9˘2-8(79dg 9 5qn9 5gk9,mg2g ]9 9y8](https://static.fdokument.com/doc/165x107/602f22e92cb00345207c776e/01-lu-7-f78dfgdf37g-97dfgfg-37-7g97ml7g-38-87-9-2l.jpg)














