
Bioinformatik Ringvorlesung SS2005 - UMG · Die Medianzentrierung ist eine globale Methode: Es...
56
Dienstag 21. April 2009 9:15 UMG Georg-August-Universität Göttingen Abt. Medizinische Statistik - Biostatistik Bioinformatik/System Biologie Sommersemester 2009 Prof. Dr. Tim Beißbarth
Transcript of Bioinformatik Ringvorlesung SS2005 - UMG · Die Medianzentrierung ist eine globale Methode: Es...

Dienstag21. April 20099:15
UMG Georg-August-Universität GöttingenAbt. Medizinische Statistik - Biostatistik
Bioinformatik/System Biologie Sommersemester 2009
Prof. Dr. Tim Beißbarth

Tim Beißbarth Bioinformatik
Übersicht
•
Di 14.4.: Vorbesprechung - Microarrays und statistische Datenanalyse
•
Di 21.4.: Normalisierung/Differentielle Genanalyse
•
Di 28.4.: Clustern/Klassifikation
•
Di 8.5.: Gene-Ontologies
•
Di 19.5.: Bayes-Netze

Tim Beißbarth Bioinformatik
Online
•
Vorlesungsslides und R-Skripte: http://www.ams.med.uni-goettingen.de/biostatistics/Sysbio09
•
Lectures Terry Speed, Berkeley: http://www.stat.berkeley.edu/users/terry/Classes/
•
Kurs NGFN „Practical DNA Microarray Analysis”: http://compdiag.molgen.mpg.de/lectures.shtml
•
R/Bioconductor Dokumentation (Vignetten): http://www.bioconductor.org
•
R Tutorial von Günther Sawitzki http://statlab.uni-hd.de/users/gs
•
Google, Pubmed, Wikipedia

Tim Beißbarth Bioinformatik
*****
GeneChip Affymetrix
cDNA microarray
Nylon membrane
Agilent: Long oligo Ink Jet
IlluminaBead Array
CGH
SAGE
VerschiedeneTechnologien
Tim Beißbarth Bioinformatik
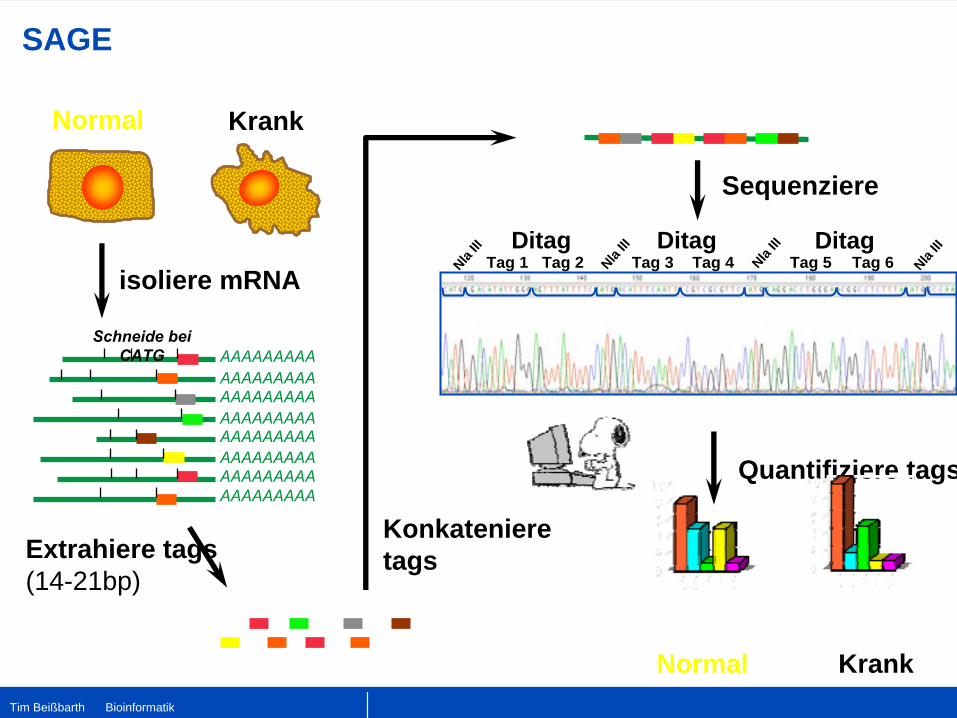
SAGE
Normal Krank
isoliere mRNA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Schneide bei
CATG
Extrahiere tags(14-21bp)
Konkatenieretags
Ditag Ditag
NlaIII
NlaIII
Tag 1 Tag 2 Tag 3 Tag 5 NlaIIIDitag
NlaIII
Tag 4 Tag 6
Sequenziere
Normal Krank
Quantifiziere tags

Tim Beißbarth Bioinformatik
cDNA und Affimetrix (kurze, 25 bp) Oligo Technologien.Lange Oligos (60-75 bp) werden so ähnlich wie cDNA benutzt.
Tim Beißbarth Bioinformatik
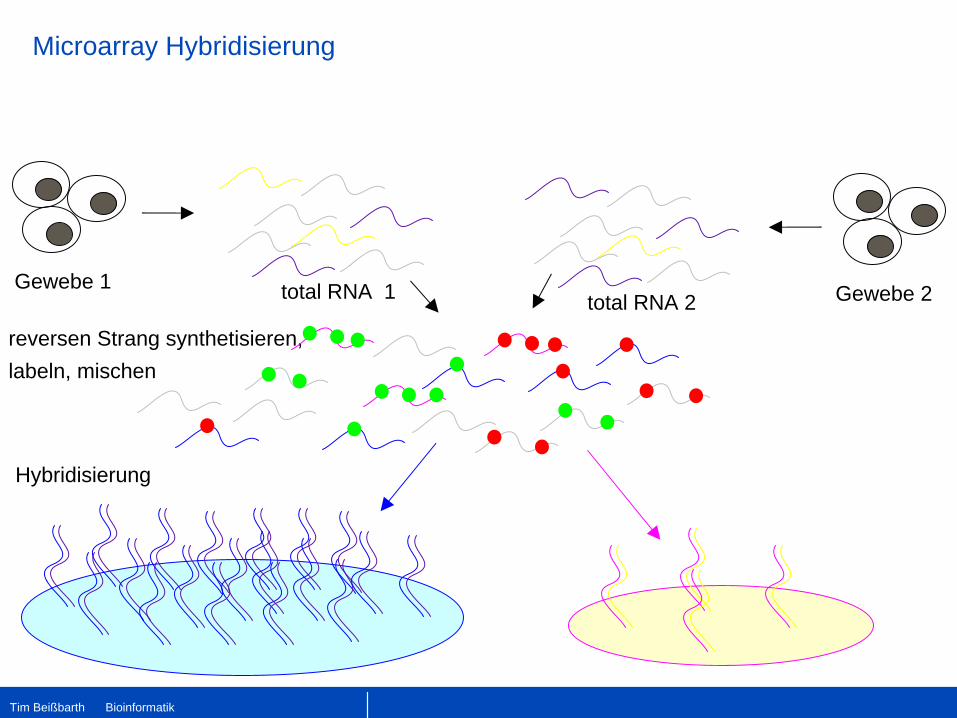
Microarray Hybridisierung
Gewebe 1 total RNA 1 Gewebe 2total RNA 2
reversen Strang synthetisieren,labeln, mischen
Hybridisierung
Tim Beißbarth Bioinformatik
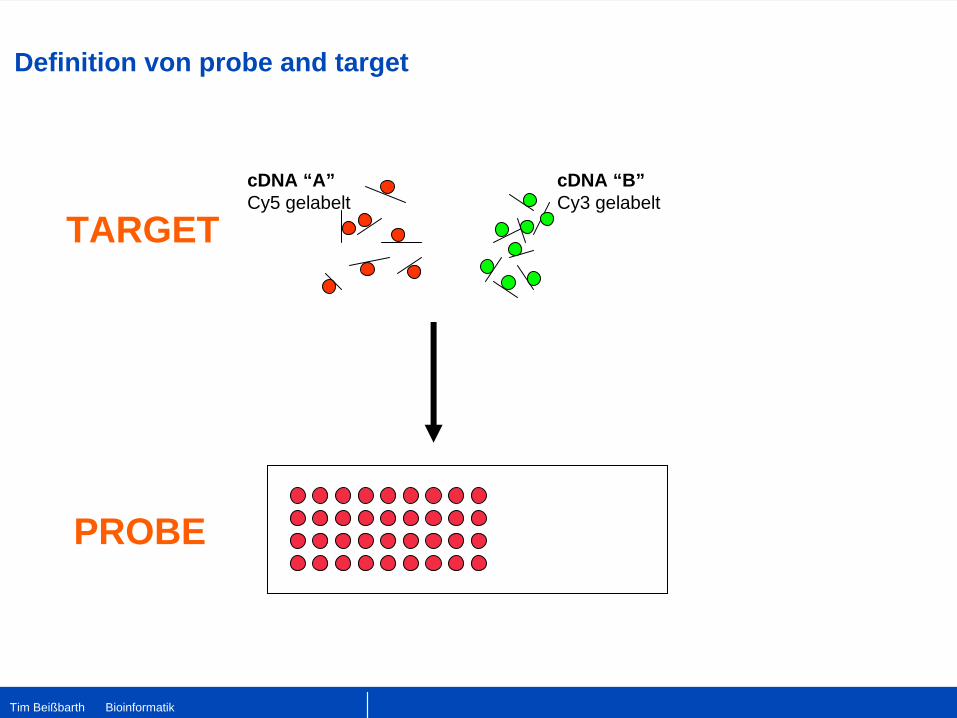
cDNA “A”Cy5 gelabelt
cDNA “B”Cy3 gelabelt
PROBE
TARGET
Definition von probe and target

Tim Beißbarth Bioinformatik
Microarrays Geschichte
•
Basiert auf Southern BIot Technologie (Edward Southern, 1975, J. Mol. Biol.)
•
1990: erste high-density Nylonfilter Arrays (Lennon/Lehrach, 1991, Trends Genet., Review)
•
1995: cDNA-Microarrays beschrieben von Schena et al, Science
•
1996: Affymetrix Genechip Technologie beschrieben von Lockhart et al, Nat. Biotechnol.
Tim Beißbarth Bioinformatik
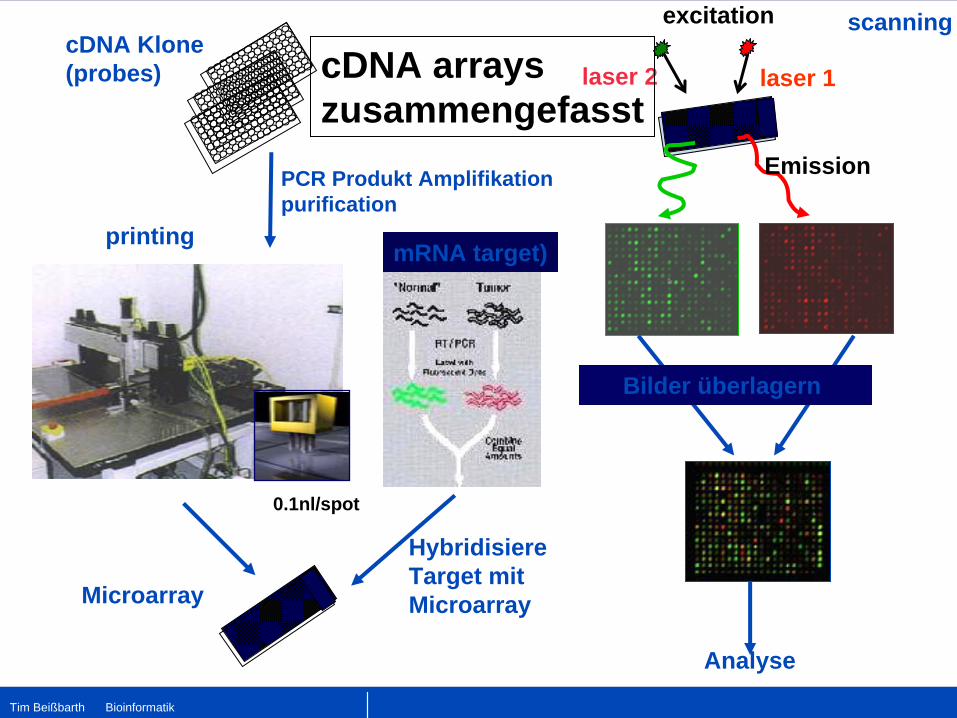
PCR Produkt Amplifikationpurification
cDNA Klone(probes)
printing
Microarray
0.1nl/spot
Hybridisiere Target mit Microarray
mRNA target)
excitation
laser 1laser 2
Emission
scanning
Analyse
Bilder überlagern
cDNA arrayszusammengefasst
Tim Beißbarth Bioinformatik
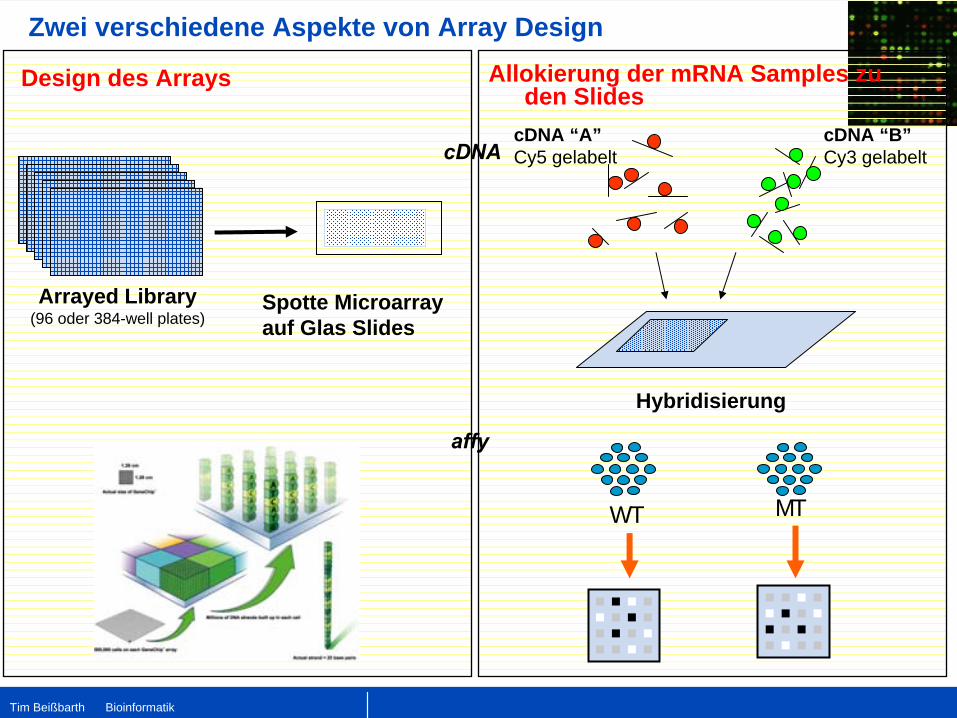
Zwei verschiedene Aspekte von Array Design
Design des Arrays Allokierung der mRNA Samples zu den Slides
Arrayed Library(96 oder 384-well plates)
cDNAcDNA “A”Cy5 gelabelt
cDNA “B”Cy3 gelabelt
Hybridisierung
Spotte Microarrayauf Glas Slides
affy
MTWT
Tim Beißbarth Bioinformatik
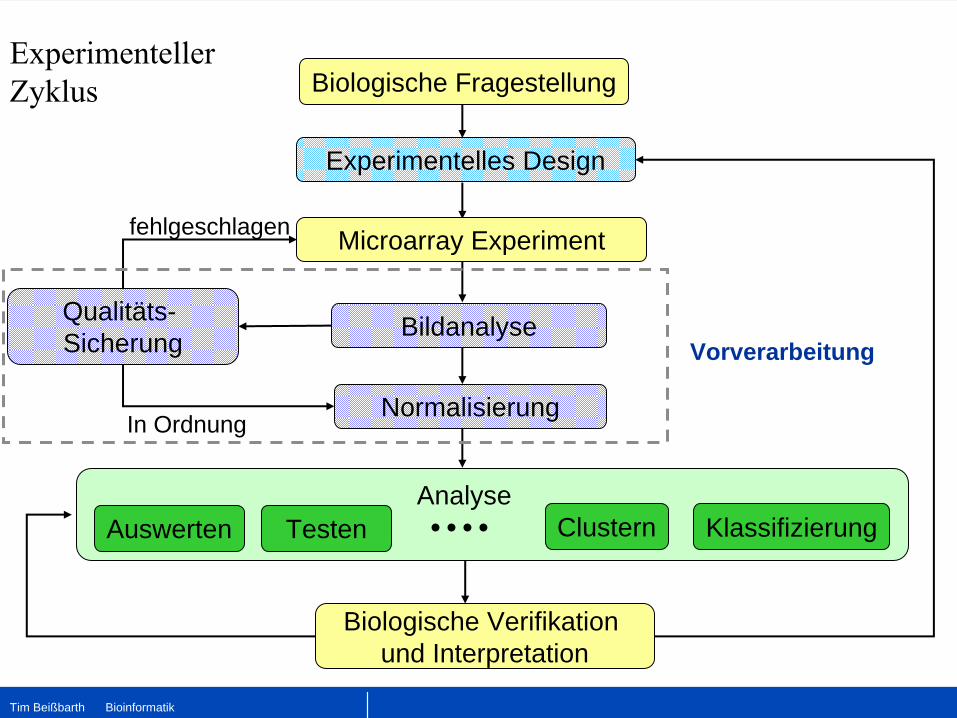
Biologische Verifikation und Interpretation
Microarray Experiment
Experimentelles Design
Bildanalyse
Normalisierung
Biologische Fragestellung
TestenAuswerten KlassifizierungAnalyse
Clustern
Experimenteller Zyklus
Qualitäts-Sicherung
fehlgeschlagen
In Ordnung
Vorverarbeitung

Tim Beißbarth Bioinformatik
Gen-expressions Daten
Gene
mRNA Samples
gene-expressions level or ratio für Gen i in mRNA Sample j
M =Log2 (rote Intensität / Grüne Intensität)
Vergleich jeweils zweier Bedingungen.
sample1 sample2 sample3 sample4 sample5 …1 0.46 0.30 0.80 1.51 0.90 ...2 -0.10 0.49 0.24 0.06 0.46 ...3 0.15 0.74 0.04 0.10 0.20 ...4 -0.45 -1.03 -0.79 -0.56 -0.32 ...5 -0.06 1.06 1.35 1.09 -1.09 ...
Gen-expressions Daten für G Gene und n Hybridisierungen. Gene x arrays Daten-matrix:
A =mittel: log2 (rote Intensity), log2 (Grüne Intensität)
Function (PM, MM) von MAS oder RMA
Tim Beißbarth Bioinformatik
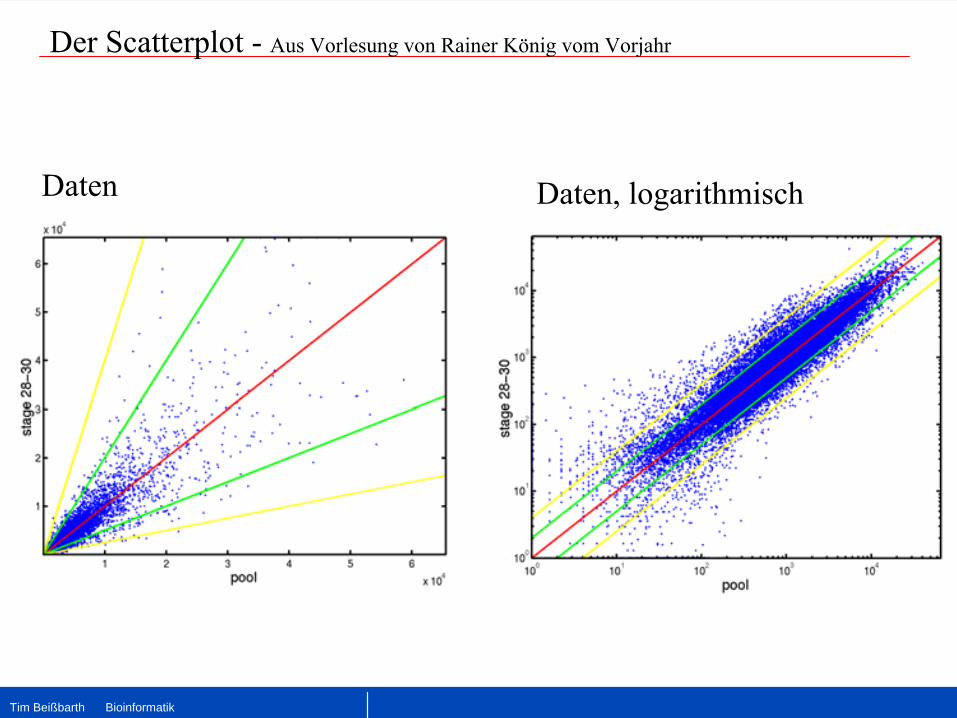
Der Scatterplot
-
Aus Vorlesung von Rainer König vom Vorjahr
Daten Daten, logarithmisch
Tim Beißbarth Bioinformatik
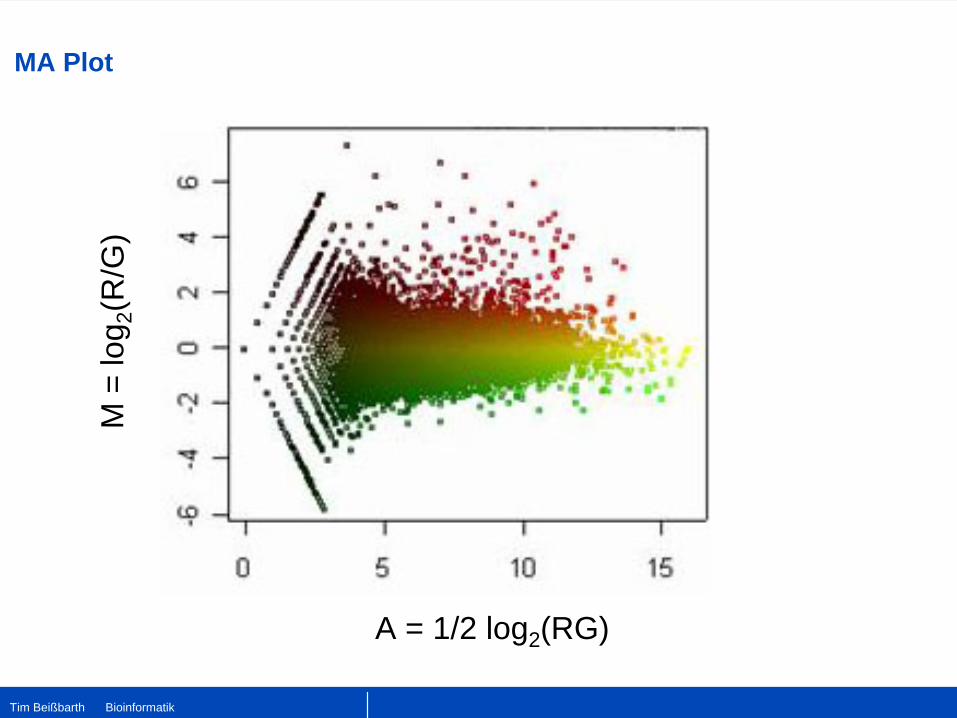
MA Plot
A = 1/2 log2 (RG)
M =
log 2
(R/G
)
Tim Beißbarth Bioinformatik
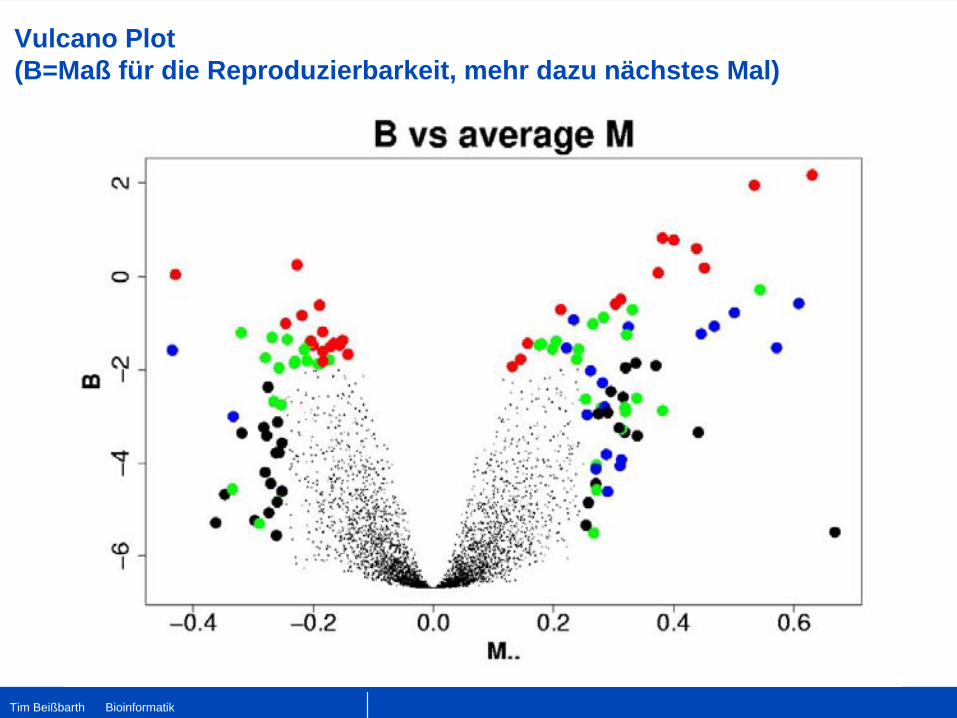
Vulcano Plot (B=Maß für die Reproduzierbarkeit, mehr dazu nächstes Mal)
Tim Beißbarth Bioinformatik
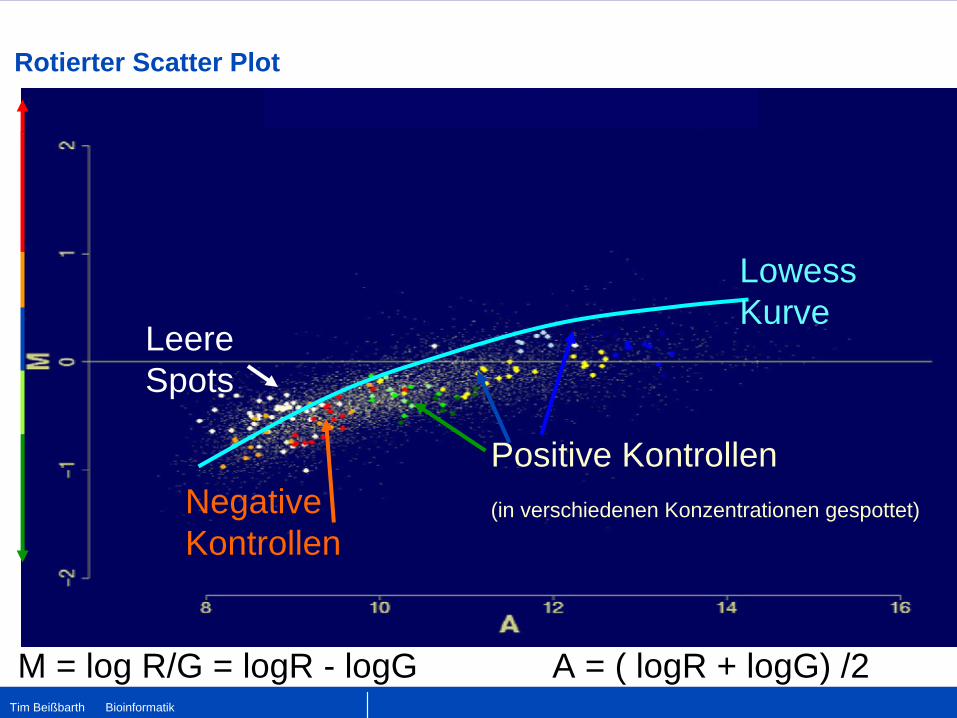
Rotierter Scatter Plot
M = log R/G = logR - logG A = ( logR + logG) /2
Positive Kontrollen(in verschiedenen Konzentrationen gespottet)Negative
Kontrollen
Leere Spots
Lowess Kurve

Tim Beißbarth Bioinformatik
Quellen der Variabilität: Bias und Varianz
“biased” “unbiased”
Schwaches Rauschen
Starkes Rauschen
x

Tim Beißbarth Bioinformatik
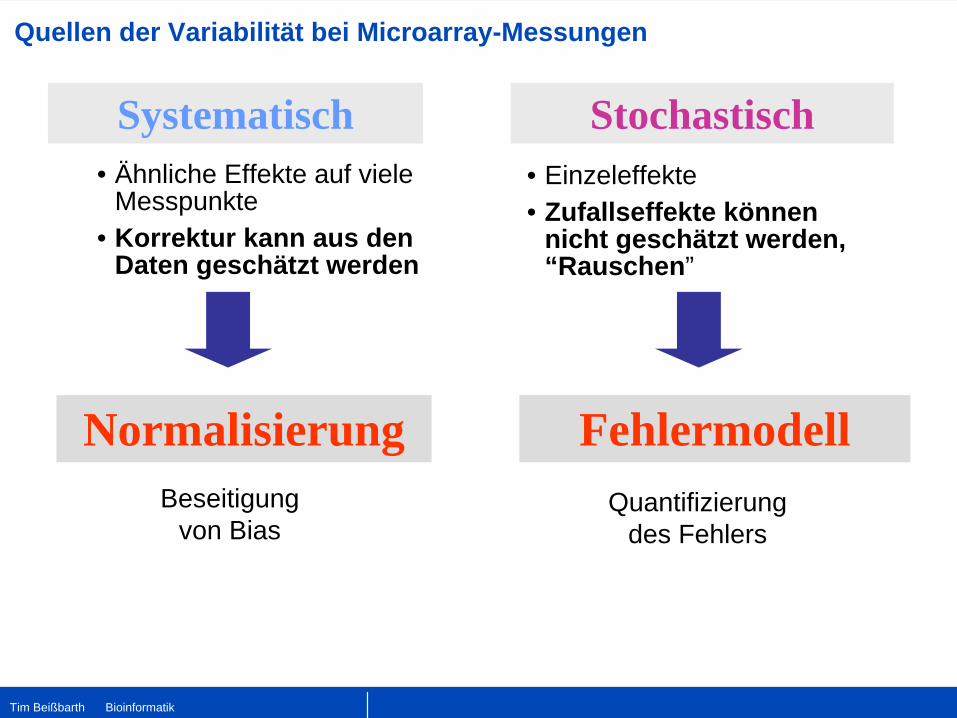
Quellen der Variabilität bei Microarray-Messungen
RNA-Menge in der ProbeRNA-Menge in der Probe
Streu-/HintergrundsignalStreu-/Hintergrundsignal
Spot Größe/-Form
Hybridisierungseffizienz und -Spezifität
Amplifikationseffizienz
Bindung der DNA an die Chipoberfläche
Spotting Effizienz
DNA Qualität RNA Degradation
Bindung der DNA an die ChipoberflächeSpot Größe/-Form
Hybridisierungseffizienz und -Spezifität
Spotting EffizienzAmplifikationseffizienz
RNA DegradationDNA QualitätStreu-/Hintergrundsignal
GewebskontaminationGewebskontaminationEffizienz der RNA Extraktion, Reversen Transkription, LabelingEffizienz der RNA Extraktion, Reversen Transkription, Labeling
• Ähnliche Effekte auf viele Messpunkte
• Korrektur kann aus den Daten geschätzt werden
Systematisch Stochastisch• Einzeleffekte• Zufallseffekte können
nicht geschätzt werden, “Rauschen”
Tim Beißbarth Bioinformatik
Quellen der Variabilität bei Microarray-Messungen
Normalisierung FehlermodellBeseitigung
von BiasQuantifizierung
des Fehlers
• Ähnliche Effekte auf viele Messpunkte
• Korrektur kann aus den Daten geschätzt werden
Systematisch Stochastisch• Einzeleffekte• Zufallseffekte können
nicht geschätzt werden, “Rauschen”
Tim Beißbarth Bioinformatik
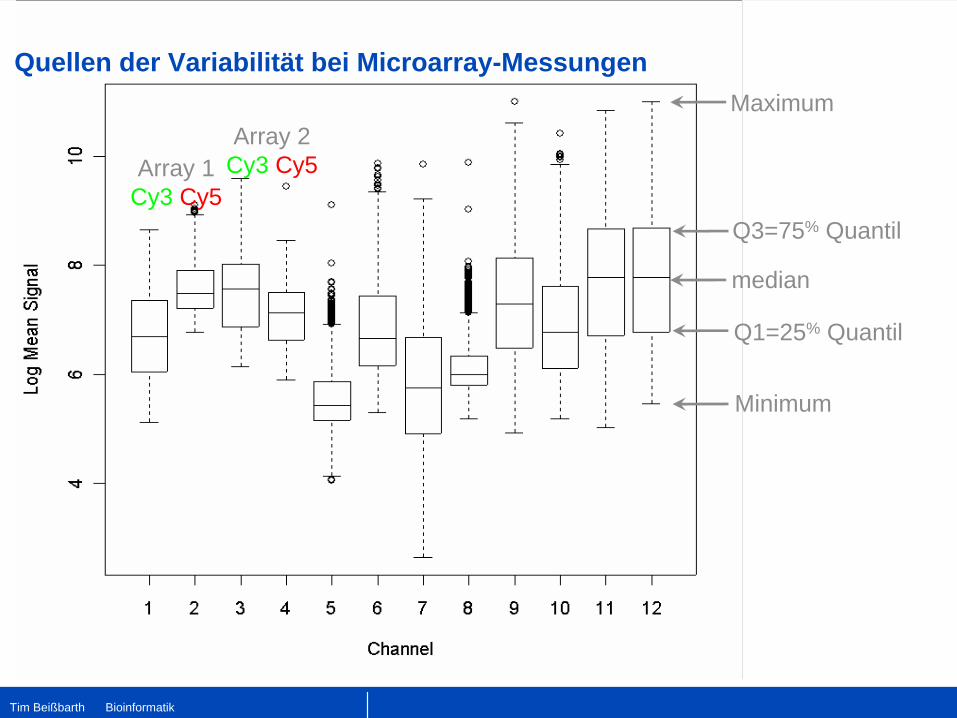
Array 2Cy3 Cy5Array 1
Cy3 Cy5
median
Q3=75% Quantil
Q1=25% Quantil
Minimum
Maximum
Quellen der Variabilität bei Microarray-Messungen
Tim Beißbarth Bioinformatik
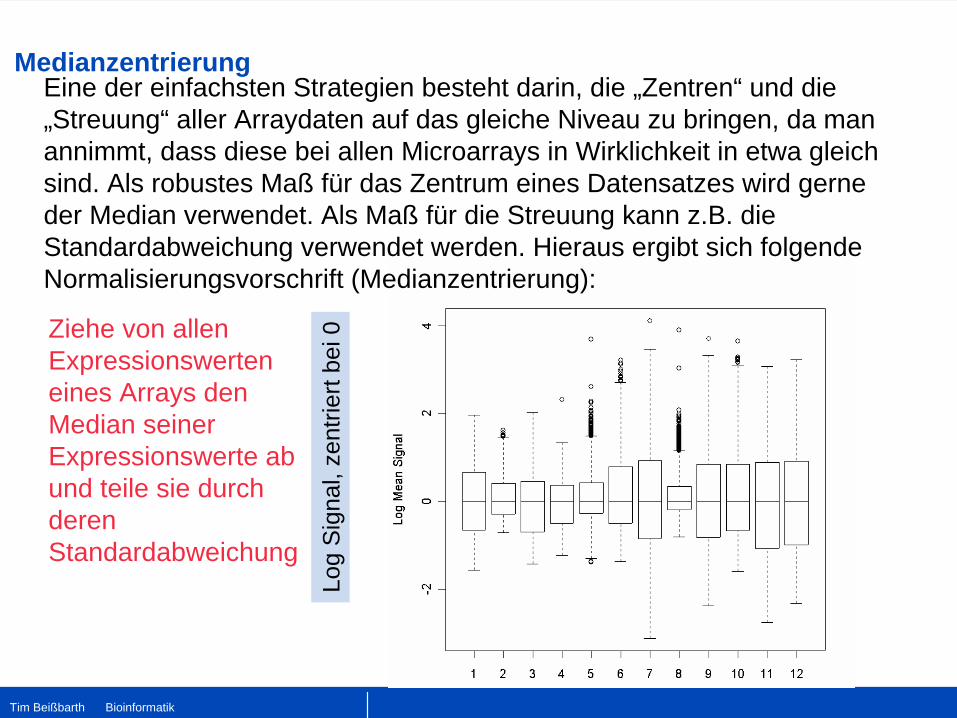
Medianzentrierung
Log
Sig
nal,
zent
riert
bei0
Eine der einfachsten Strategien besteht darin, die „Zentren“ und die „Streuung“ aller Arraydaten auf das gleiche Niveau zu bringen, da man annimmt, dass diese bei allen Microarrays in Wirklichkeit in etwa gleich sind. Als robustes Maß für das Zentrum eines Datensatzes wird gerne der Median verwendet. Als Maß für die Streuung kann z.B. die Standardabweichung verwendet werden. Hieraus ergibt sich folgende Normalisierungsvorschrift (Medianzentrierung):
Ziehe von allen Expressionswerten eines Arrays den Median seiner Expressionswerte ab und teile sie durch deren Standardabweichung
Tim Beißbarth Bioinformatik
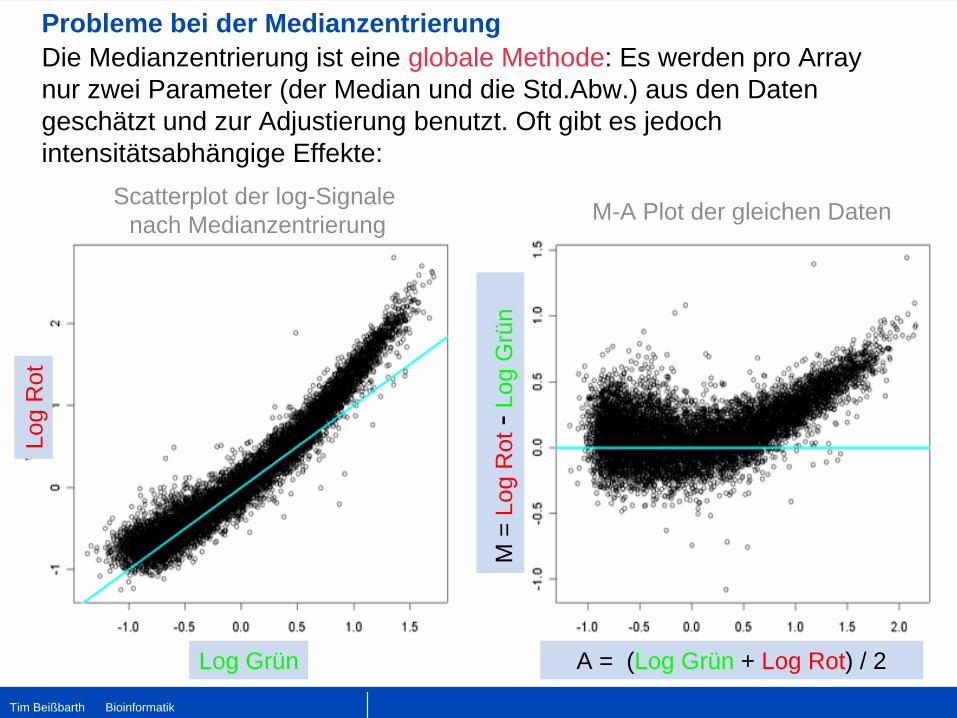
Probleme bei der Medianzentrierung
Log Grün
Log
Rot
Scatterplot der log-Signale nach Medianzentrierung
A = (Log Grün + Log Rot) / 2
M =
Log
Rot
-Lo
g G
rün
M-A Plot der gleichen Daten
Die Medianzentrierung ist eine globale Methode: Es werden pro Array nur zwei Parameter (der Median und die Std.Abw.) aus den Daten geschätzt und zur Adjustierung benutzt. Oft gibt es jedoch intensitätsabhängige Effekte:

Tim Beißbarth Bioinformatik
QuantilnormalisierungDie Grundidee der Quantilnormalisierung ist bestechend einfach:
„Die Histogramme aller Microarrays sehen gleich aus“
Dies ist eine Verschärfung der Hypothese, die der Medianzentrierung zu Grunde lag. Nicht nur das 50%-Quantil=der Median soll in allen Arrays (in etwa) gleich sein, sondern alle
Quantile.
Der Algorithmus lautet:• Ordne die Gene eines jeden Arrays der Größe
nach.
• Sei Mn
der Mittelwert der Gene mit der n-t höchsten Expression. Ersetze den Messwert dieser Gene jeweils durch Mn
.
• Verfahre so für alle Positionen n.
Boxplot nach Quantilnormali-
sierung
•Nachteil der Quantilnormalisierung: In einzelnen Arrays differentiell exprimierte Gene am unteren und am oberen Ende der Messskala werden nivelliert.
Tim Beißbarth Bioinformatik
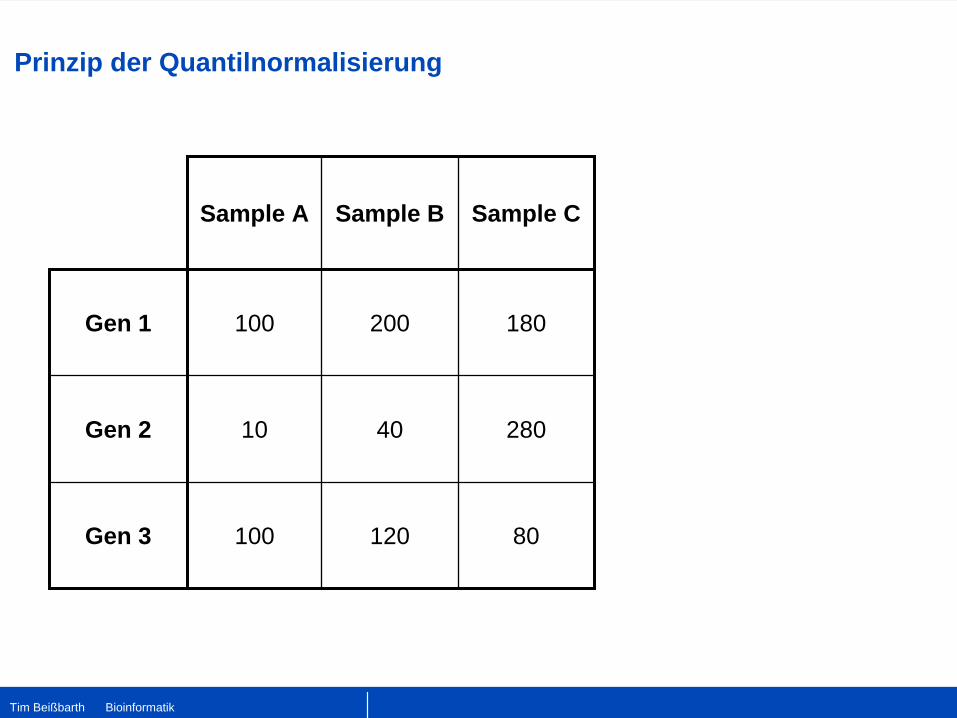
Sample A Sample B Sample C
Gen 1 100 200 180
Gen 2 10 40 280
Gen 3 100 120 80
Prinzip der Quantilnormalisierung

Tim Beißbarth Bioinformatik
Sample A Sample B Sample C
Gen 1 100 200 180
Gen 2 10 40 280
Gen 3 100 120 80
Prinzip der Quantilnormalisierung

Tim Beißbarth Bioinformatik
Sample A Sample B Sample C
100 Gen 1
200 Gen 1
140 Gen 1
10 Gen 2
40 Gen 2
270 Gen 2
100 Gen 3
120 Gen3
70 Gen3
Prinzip der Quantilnormalisierung
Tim Beißbarth Bioinformatik
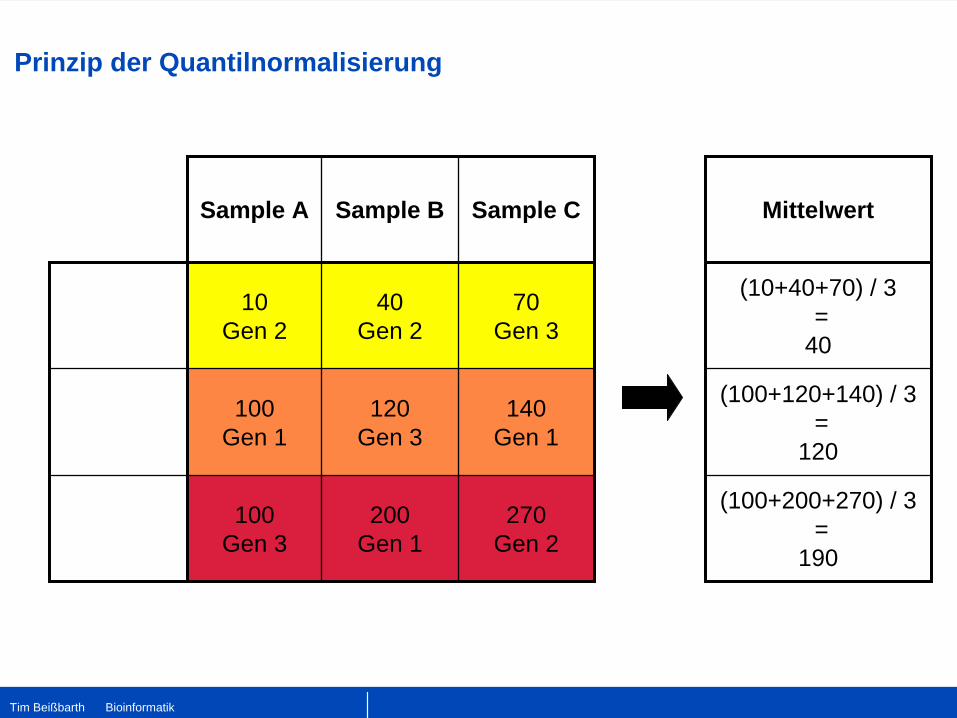
Sample A Sample B Sample C
10 Gen 2
40 Gen 2
70 Gen 3
100 Gen 1
120 Gen 3
140 Gen 1
100 Gen 3
200 Gen 1
270 Gen 2
Mittelwert
(10+40+70) / 3 =
40
(100+120+140) / 3 =
120
(100+200+270) / 3 =
190
Prinzip der Quantilnormalisierung
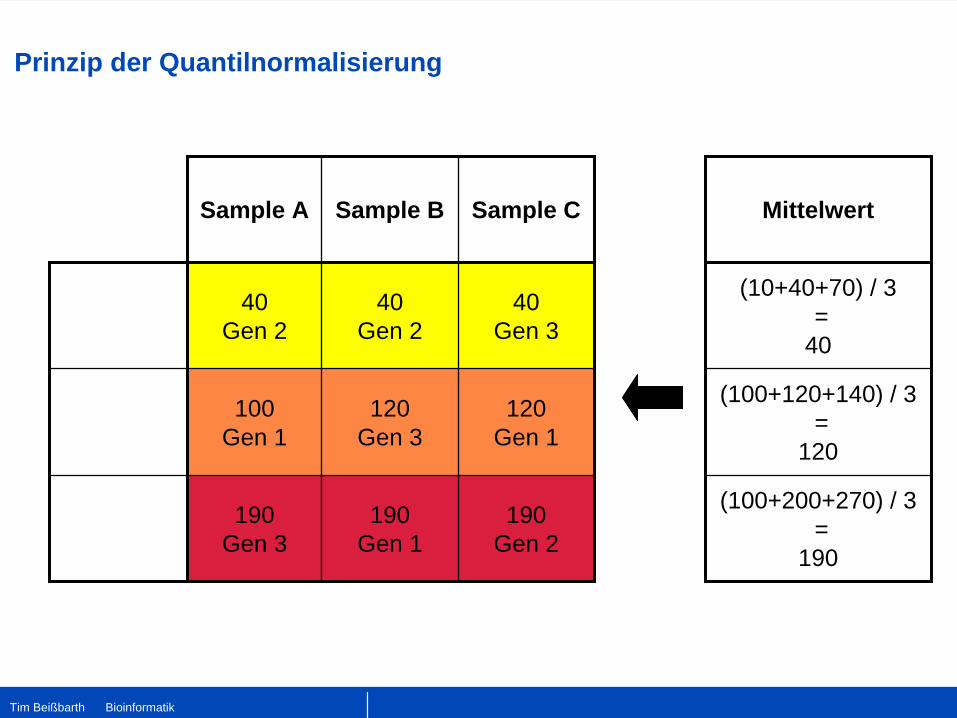
Tim Beißbarth Bioinformatik
Sample A Sample B Sample C
40 Gen 2
40 Gen 2
40 Gen 3
100 Gen 1
120 Gen 3
120 Gen 1
190 Gen 3
190 Gen 1
190 Gen 2
Mittelwert
(10+40+70) / 3 =
40
(100+120+140) / 3 =
120
(100+200+270) / 3 =
190
Prinzip der Quantilnormalisierung

Tim Beißbarth Bioinformatik
Sample A Sample B Sample C
Gen 1 120 (100)
190 (200)
120 (180)
Gen 2 40 (10)
40 (40)
190 (280)
Gen 3 190 (100)
120 (120)
40 (80)
Prinzip der Quantilnormalisierung

Tim Beißbarth Bioinformatik
Varianzstabilisiernde Transformationen•Gegeben sei eine Familie von Zufallsvariablen Xμ
, μ є [a,b],
mit Erwartungswert
•E(Xμ
) = μ. •Die Varianz dieser Zufallsvariablen sei eine Funktion von μ,
•Var(Xμ
) = v(μ).
v(μ)
v(μ)
Xμ
μ μ
v(μ)(Realisierungen von)
Gesucht ist eine Transformation T: IR→IR derart, dass
Var(T(Xμ
)) ≈
const.
Tim Beißbarth Bioinformatik
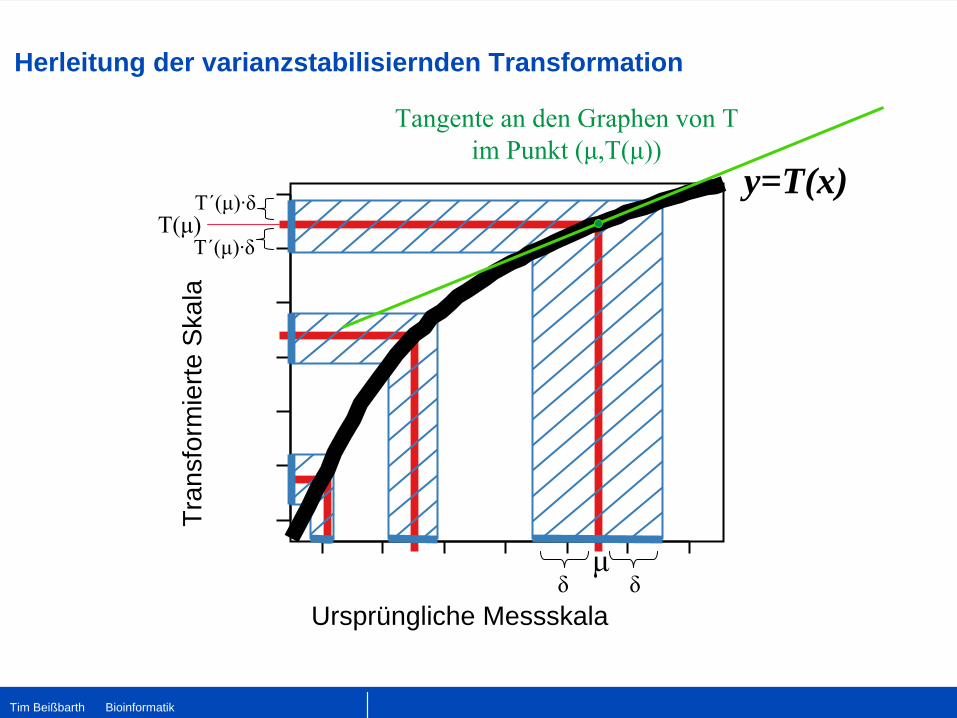
Herleitung der varianzstabilisiernden Transformation
Ursprüngliche Messskala
Tran
sfor
mie
rte S
kala
y=T(x)
μδ δ
Tangente an den Graphen von T im Punkt (μ,T(μ))
T(μ)T´(μ)·δ
T´(μ)·δ
Tim Beißbarth Bioinformatik
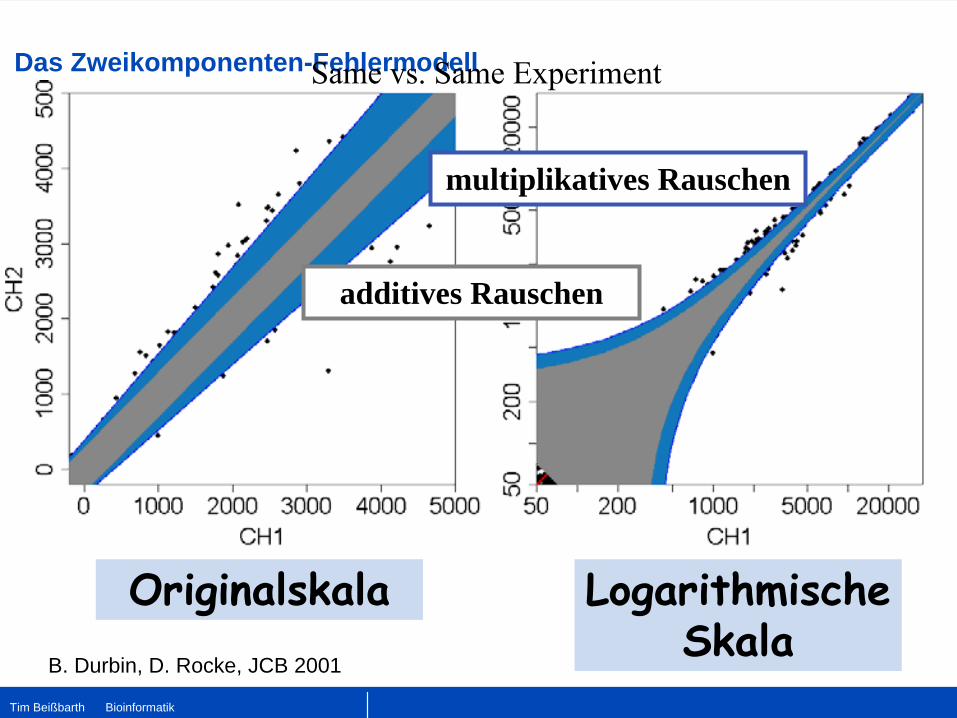
Das Zweikomponenten-Fehlermodell
additives Rauschen
multiplikatives Rauschen
Originalskala Logarithmische Skala
B. Durbin, D. Rocke, JCB 2001
Same vs. Same Experiment

Tim Beißbarth Bioinformatik
Schätzung der Parameter des Fehlermodells
ε= +iik ika aai
per-sample offset
εik
~ N(0, bi2s1
2)“additive noise”
bi
per-samplenormalization factor
bk
sequence-wiseprobe efficiency
ηik ~ N(0,s22)
“multiplicative noise”
exp( )iik k ikb b b η=
ik ik ik ky a b x= +measured
intensity
= offset
+ gain
×
true
abundance
Tim Beißbarth Bioinformatik
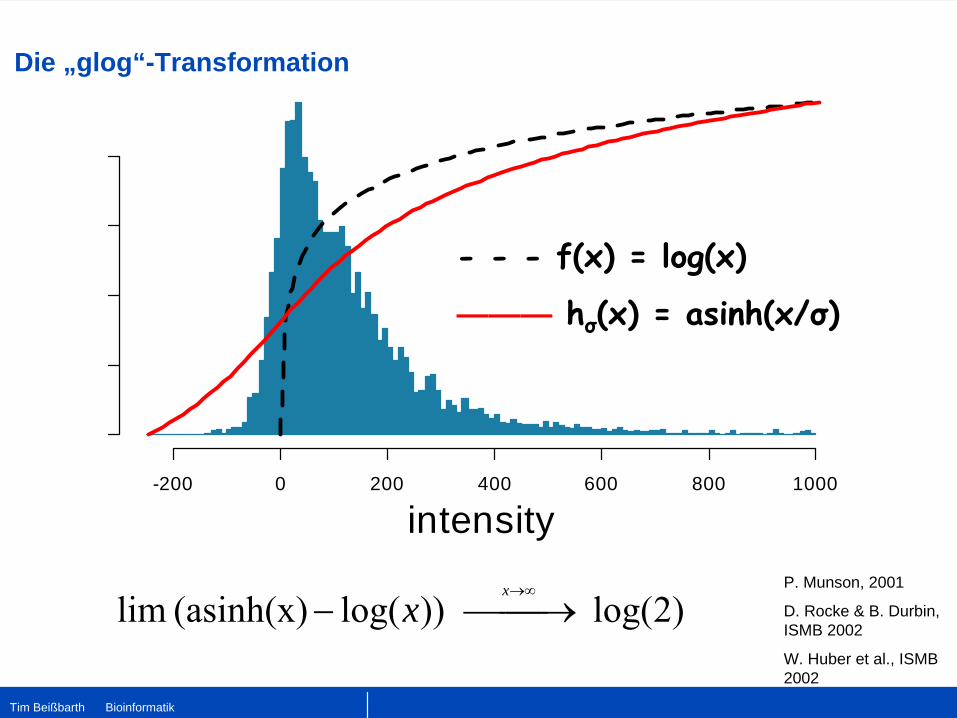
Die „glog“-Transformation
intensity-200 0 200 400 600 800 1000
- - - f(x) = log(x)
——— hσ
(x) = asinh(x/σ)
P. Munson, 2001
D. Rocke & B. Durbin, ISMB 2002
W. Huber et al., ISMB 2002
)2log( ))log((asinh(x) lim ⎯⎯→⎯−→∞x
x
Tim Beißbarth Bioinformatik
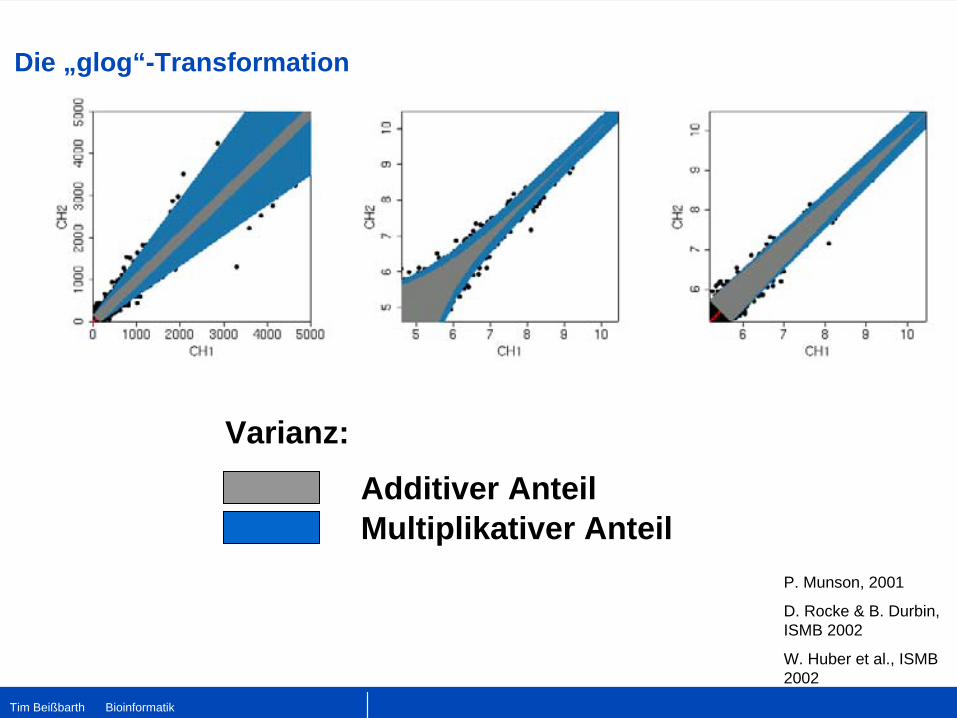
Die „glog“-Transformation
Additiver Anteil
Varianz:
Multiplikativer AnteilP. Munson, 2001
D. Rocke & B. Durbin, ISMB 2002
W. Huber et al., ISMB 2002

Tim Beißbarth Bioinformatik
Literatur, Links
•
Bioconductor vignette for vsn. W.Huber http://www.maths.lth.se/help/R/.R/library/vsn/doc/vsn.pdf•
A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bolstad, B., et al. Bioinformatics
19 2:185-193 (2003)
•
A model for measurement error for gene expression analysis. D. Rocke, B. Durbin. Journal of Computational
Biology, 8:557-569, 2001
•
Variance stabilization applied to microarray data calibration and to the quantification of differential expression. W. Huber, A. von Heydebreck, H. Sültmann, A. Poustka, M. Vingron. Bioinformatics 18 suppl. 1 (2002), S96-S104 (ISMB 2002).
•
Parameter estimation for the calibration and variance stabilization of microarray data. W. Huber, A. von Heydebreck, H. Sültmann, A. Poustka, M. Vingron. Statistical
Applications
in Genetics
and Molecular
Biology
2003 Vol. 2: No. 1, Article
3
•
Error models for microarray intensities. W. Huber, A. von Heydebreck, and M. Vingron. to appear
in: Encyclopedia of Genomics, Proteomics
and Bioinformatics. John Wiley & sons
(2004).
•
Interpretability and Data Transformations for Gene Expression Microarray Data. D. M. Rocke, W. Huber, B. Durbin, A. von Heydebreck, M. Vingron. submitted
(2004).
•
Statistical methods for identifying differentially expressed genes in microarray experiments. S. Dudoit, Y.H. Yang, T.P. Speed, and M.J. Callow. Statistica
Sinica, 12:111-139, 2002.

Tim Beißbarth Bioinformatik
Differentielle Gene finden
•
Wir haben in n
Experimenten jeweils die differentielle Genexpression zwischen zwei Bedingungen gemessen, z.B. WT/KO.
•
Messwerte M-values für Gen g: x1
, … , xn
•
Hypothese H0 - die Gene sind nicht differentiell:
•
Annahme – die Meßwerte sind Normalverteilt N(μ0
, σ0
).
( ) 0E x =
( )2
221( )2
x
f x eμ
σ
σ π
− −
=
- 3 - 2 - 1 0 1 2 3

Tim Beißbarth Bioinformatik
Differentielle Gene finden
•
Bester Schätzer für μ: (Mittelwert)
•
Bester Schätzer für σ: (Standardabweichung)
•
Erwartete Abweichung von von μ:
Standard error of the mean
1
1 n
ii
x xn =
= ∑
2
1
( )
1
n
ii
x xs
n=
−=
−
∑
x
SEMn
σ=
- 3 - 2 - 1 0 1 2 3
Tim Beißbarth Bioinformatik
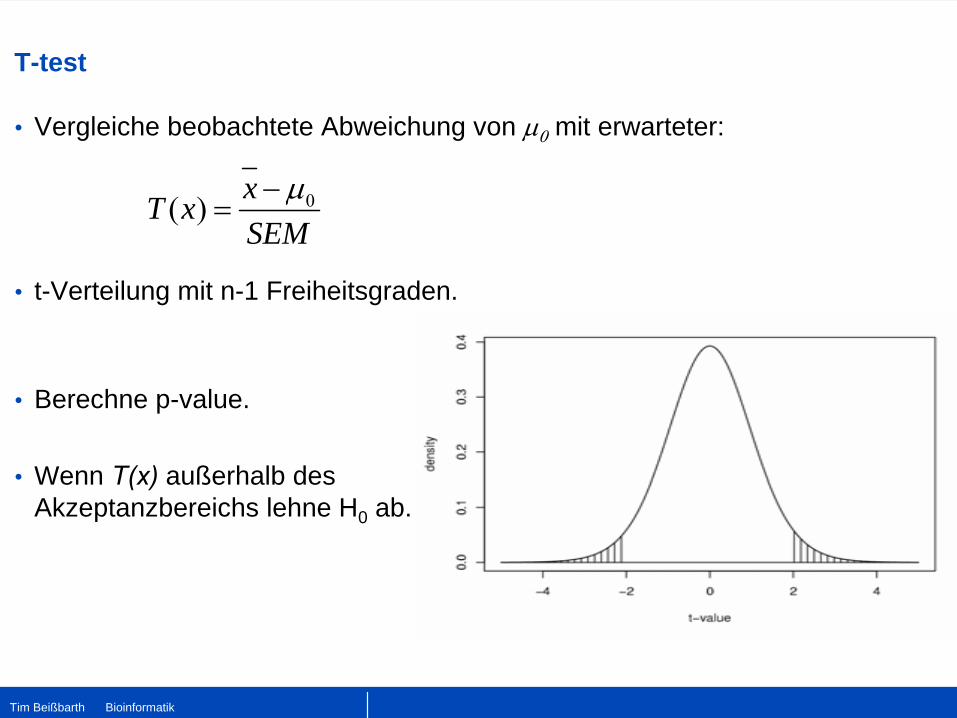
T-test
•
Vergleiche beobachtete Abweichung von μ0
mit erwarteter:
•
t-Verteilung mit n-1 Freiheitsgraden.
•
Berechne p-value.
•
Wenn T(x)
außerhalb des Akzeptanzbereichs lehne H0 ab.
0( ) xT xSEM
μ−=

Tim Beißbarth Bioinformatik
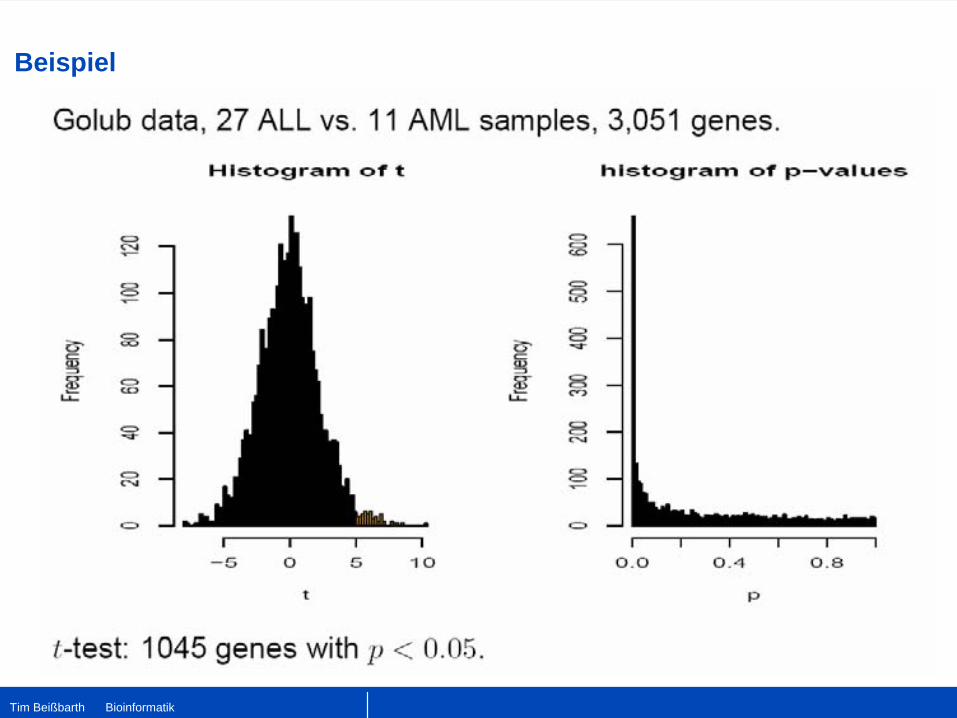
Beispiel 2 – T-test 2
•
Affymetrix Daten von Golub et al, 1998•
38 Tumor Gewebe:
•
27 acute lymphoblastic leukemia (ALL)•
11 acute myeloid leukemia (AML)
•
6817 Gene, 3051 nach filtern•
Expressionswerte von Gen g in ALL x1
, … , xn
und in AML y1
, … , yn
.•
Berechne gemeinsame Varianz:
•
2 Sample T-test:
t-Verteilung mit nx
+ny
-2 Freiheitsgraden.
1 1( , )
x yn n
x yT x ys
−=
+
2 2 2
1 1
1 ( ) ( )2
yx nn
i ii ix y
s x x y yn n = =
⎛ ⎞= − + −⎜ ⎟⎜ ⎟+ − ⎝ ⎠
∑ ∑
Tim Beißbarth Bioinformatik
Beispiel

Tim Beißbarth Bioinformatik
Bedeutung des p-Wertes: Typ I Fehler

Tim Beißbarth Bioinformatik
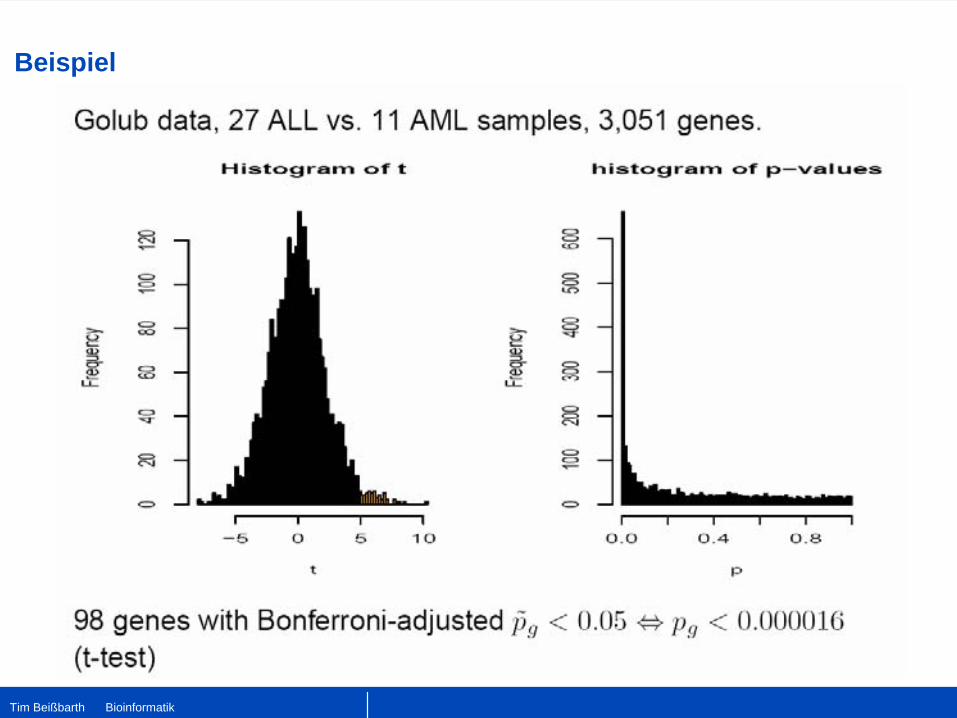
Multiples Testen
•
Problem: Tausende von Hypothesen werden gleichzeitig getestet.
•
Beispiel: Bei 10000 Genen auf dem Chip und einem Cutoff des p-Wertes von 0.01 erwarte ich, daß 10000×0.01=100 Gene einen signifikanten p-Wert p<0.01 haben.
•
Resultat: Ein einzelner p-Wert von 0.01 indiziert nicht mehr unbedingt ein signifikantes Gen. Es gibt eine erhöhte Chance falsch-positive Gene zu finden.
•
Lösung: Man muß die p-Werte für multiples Testen korrigieren.
•
Methode: Einfachstes Verfahren von Bonferroni Multipliziere alle p-Werte mit der Anzahl der Tests.
•
Mehr dazu am Montag 18.4.
Tim Beißbarth Bioinformatik
Beispiel

Tim Beißbarth Bioinformatik
T-test: Variante von Welch
•
Erlaube verschiedene Varianzen in den beiden verschiedenen Stichproben.
•
Wir nehmen an x ~ N(μ1
, σ1
)
und y ~ N(μ2
, σ2
).
•
Testen Hypothese H0 : μ1
= μ2
.
22( , )
yx ssn m
x yT x y −=
+

Tim Beißbarth Bioinformatik
Mehr Tests: Wilcoxon-Test (auch Man-Whitney Test)
•
Nicht-parametrischer Test (keine Normalverteilungsannahme) zum Vergleichen von zwei empirischen Verteilungen.
•
Berechne die Ränge der Werte aus beiden Messreihen:
•
Die Teststatistik wird aus der Summe der Ränge berechnet: R1
=6•
Für kleine Stichprobengrößen kann die Verteilung der Teststatistik exakt berechnet werden (i.e. alle Möglichkeiten), für große Stichproben kann eine Approximation durch Normalverteilung benutzt werden.
•
Vorteile: Nicht-parametrisch, robust gegen Ausreißer.•
Nachteile: weniger Mächtig da keine Verteilungsannahmen.
Tim Beißbarth Bioinformatik
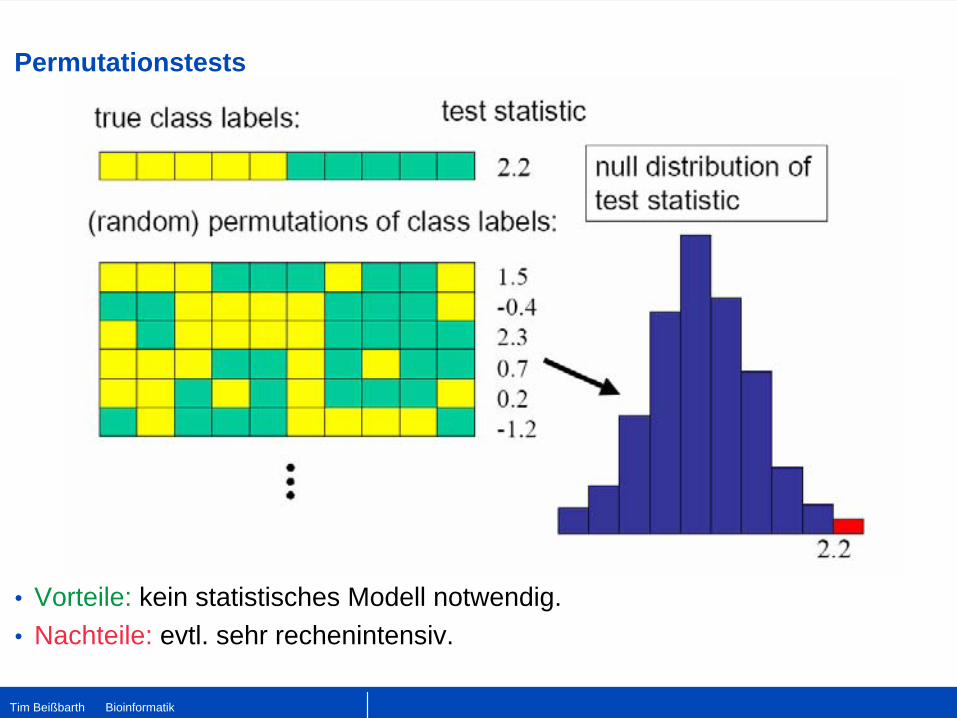
Permutationstests
•
Vorteile: kein statistisches Modell notwendig.•
Nachteile: evtl. sehr rechenintensiv.

Tim Beißbarth Bioinformatik
Problem beim T-test
•
Problem:
•
Es gibt sehr viele Gene (Tests) meistens aber nur sehr wenige Wiederholungen.
•
Als Schätzer für σ2
habe ich s2
benutzt → evtl. zufällige Fehler bei s2.
•
Beispiel: Gen g
wird mit den M-Werten 0.0011, 0.0012 und –0.0009 gemessen → T~12, p~0.007.
•
Merke: Bei sehr kleiner gemessener Varianz wird der Wert für T sehr groß.
•
Fazit: Bei Microarray Experimenten nach Möglichkeit keinen Standard-T- test verwenden.
•
Folgerung: Modifizierte T-Statistik nötig.

Tim Beißbarth Bioinformatik
moderated T-statistics
•
Beim T-test schätzen wir die Varianz für jedes Gen sg2
einzeln. Dies ist evtl. unstabil.
•
Stattdessen versuchen wir nun die Varianz über alle Gene s02
oder Subgruppen von Genen zu schätzen und damit die Varianzschätzung zu korrigieren.
•
Dieser „Fudge Factor“ kann noch durch Faktoren (α, β) unterschiedlich gewichtet werden.
•
Referenz: Efron/Tibshirani, Genet. Epidemiol., 2000•
Software: R/Bioconductor Pakete – limma, siggenes
2 20
( , ) x yT x ys sα β
−=
+

Tim Beißbarth Bioinformatik
Empirical Bayes
•
Methode um über viele Gene moderierte Statistiken zu Berechnen unter Annahme von Prior-Distributions.
•
Verschiedene Varianten existieren. Siehe Efron et al 2001, Lönsted/Speed 2002, ...
•
Beispiel:
Für große n:≈ t
= M.
/ s
B = const + log
2an
+ s 2 + M•2
2an
+ s2 + M•2
1 + nc
⎛
⎝
⎜ ⎜
⎞
⎠
⎟ ⎟
Tim Beißbarth Bioinformatik
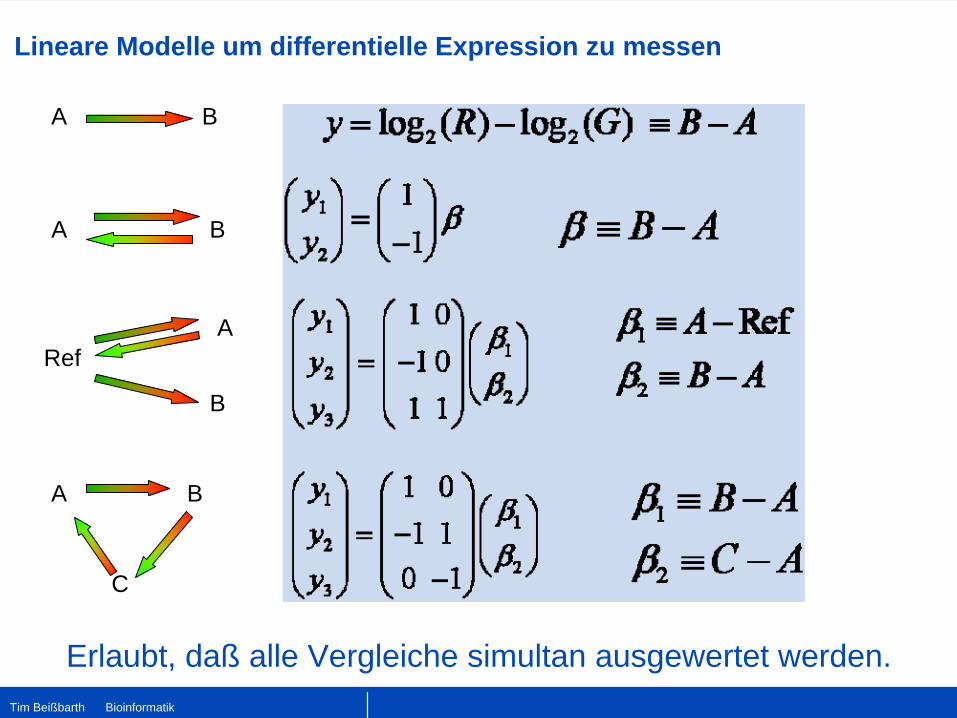
Lineare Modelle um differentielle Expression zu messen
A B
RefA
B
A B
A B
C
Erlaubt, daß alle Vergleiche simultan ausgewertet werden.
Tim Beißbarth Bioinformatik
⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜
⎝
⎛
⎥⎥⎥⎥⎥⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢⎢⎢⎢⎢⎢
⎣
⎡
=
⎟⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
baba
baa
yyyyyyy
2
1
2
1
7
6
5
4
3
2
1
11100100100001001100010010000100100
WT.P11μ
+ a1
MT.P21μ
+ (a1 + a2) + b + (a1 + a2)b
MT.P11μ
+a1+b+a1.b
WT.P21μ
+ a1 + a2
WT.P1μ
MT.P1μ
+ b
1
2
3
4
5
6
7
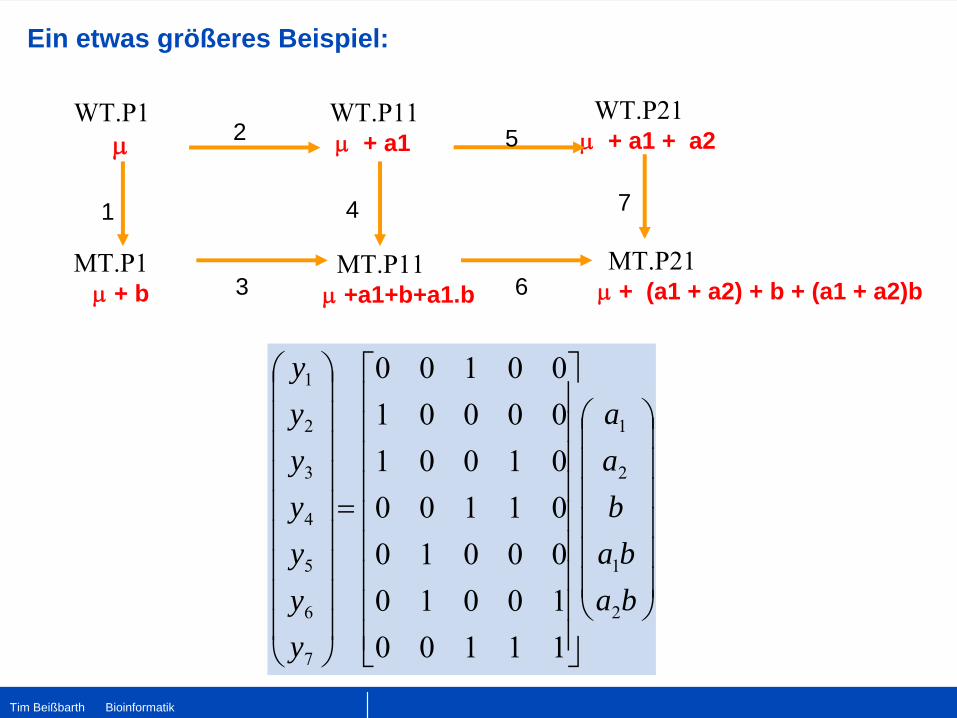
Ein etwas größeres Beispiel:

Tim Beißbarth Bioinformatik
Lineare Modelle berechnen
Entwerfe lineares Modell für jedes Gen g
Schätze Modell durch robuste Regression,least squares oder generalized least squares (in R Funktionen lm, rlm, glm) und erhalte
coefficients
standard deviations
standard errors

Tim Beißbarth Bioinformatik
References
•
T. P. Speed and Y. H Yang (2002). Direct versus indirect designs for cDNA microarray experiments. Sankhya
: The Indian Journal of Statistics, Vol. 64, Series A, Pt. 3, pp 706- 720
•
Y.H. Yang and T. P. Speed (2003). Design and analysis of comparative microarray Experiments In T. P Speed (ed) Statistical analysis of gene expression microarray data, Chapman & Hall.
•
R. Simon, M. D. Radmacher and K. Dobbin (2002). Design of studies using DNA microarrays. Genetic Epidemiology 23:21-36.
•
F. Bretz, J. Landgrebe and E. Brunner (2003). Efficient design and analysis of two color factorial microarray experiments. Biostaistics.
•
G. Churchill (2003). Fundamentals of experimental design for cDNA microarrays. Nature genetics review 32:490-495.
•
G. Smyth, J. Michaud and H. Scott (2003) Use of within-array replicate spots for assessing differential experssion in microarray experiments. Technical Report In WEHI.
•
Glonek, G. F. V., and Solomon, P. J. (2002). Factorial and time course designs for cDNA microarray experiments. Technical Report, Department of Applied Mathematics, University of Adelaide. 10/2002

Tim Beißbarth Bioinformatik
Acknowledgements – Slides geborgt von
•
Achim Tresch
•
Benedikt Brors
•
Wolfgang Huber
•
Anja von Heydebreck
•
Terry Speed
•
Jean Yang


















