biologische grundlagen - informatik.hu-berlin.de · – nature, 1 seite ... org2: aua ucu ugg gac...
55
Biologische Grundlagen ©Silke Trißl, April 2005
Transcript of biologische grundlagen - informatik.hu-berlin.de · – nature, 1 seite ... org2: aua ucu ugg gac...

Biologische Grundlagen
©Silke Trißl, April 2005

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 2
Überblick – Biologie
n Organismenn Aufbau von Zellen
– Prokaryoten und Eukaryotenn Genom und DNAn Transkription
– DNA → RNA → Proteinn Proteinen Regulatorische und metabolische Netzwerken Human Genome Project
n 2. Teil: Überblick – Techniken
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 3
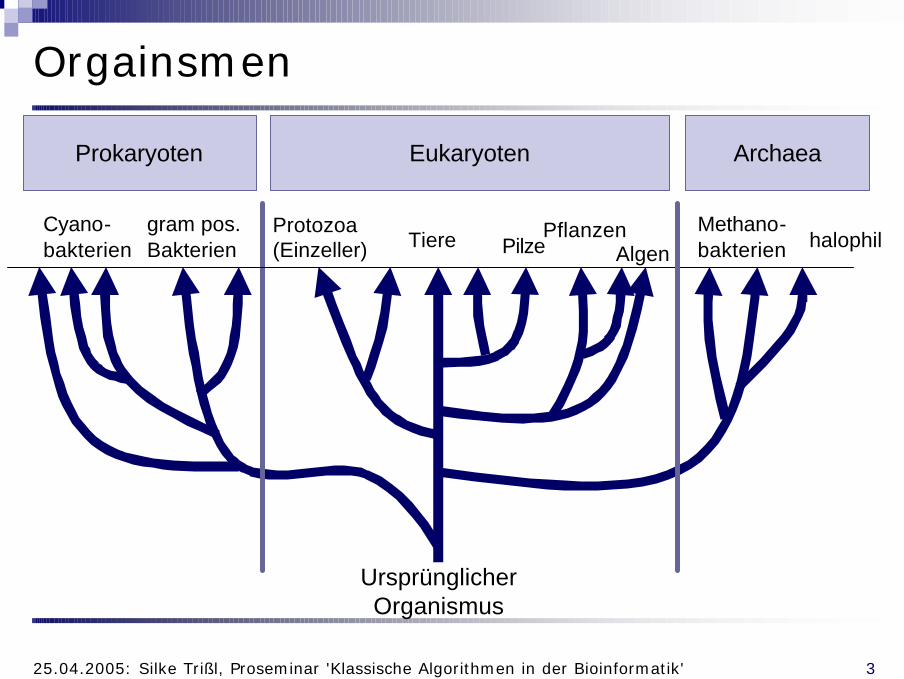
Orgainsmen
Ursprünglicher Organismus
Protozoa(Einzeller) Tiere
Algen
EukaryotenProkaryoten
gram pos.Bakterien
Cyano-bakterien
Archaea
halophilMethano-bakterien
PflanzenPilze

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 4
Beispiele
n Escherichia coli– Bakterium (Prokaryot)– lebt im Darmtrakt von
Tieren und Menschen– Modellorganismus der
Biologie
n Saccharomyces cereviciae– Eukaryot– Bäckerhefe– bildet Sporen
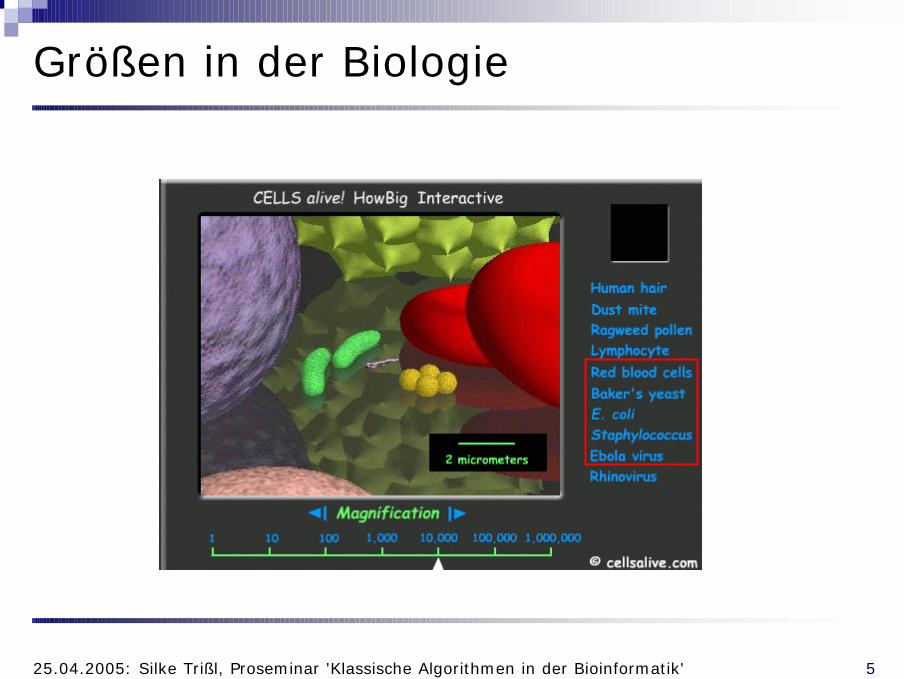
1µm5µm
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 5
Größen in der Biologie
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 6
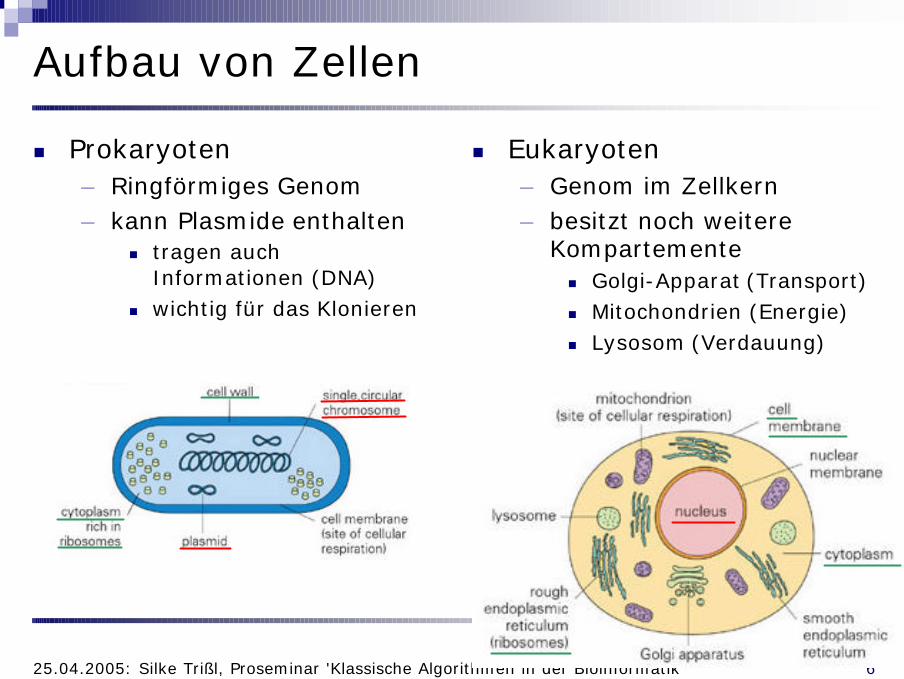
Aufbau von Zellen
n Prokaryoten– Ringförmiges Genom– kann Plasmide enthalten
n tragen auch Informationen (DNA)
n wichtig für das Klonieren
n Eukaryoten– Genom im Zellkern– besitzt noch weitere
Kompartementen Golgi-Apparat (Transport)n Mitochondrien (Energie)n Lysosom (Verdauung)

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 7
Funktion der DNA
n DNA: Träger von Erbinformationen– Codiert für funktionelle Produkte wie Proteine oder RNA
n Genom: Gesamtheit der DNA in einer Zelle– Sprich: alle Gene in einer Zelle
n Millionenfach kopiert– Ohne wesentliche Veränderung– Wird an Tochterzellen weitergegeben

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 8
Jahrestag
n 25.04.1953– James Watson, Francis Crick– „Molecular Structure of Nucleic Acids“– Nature, 1 Seite– „This structure has two helical chains
each coiled around the same axis“n Basiert auf Arbeiten von Wilkins &
Franklin n Nobelpreis 1962
– „for their discoveries concerning themolecular structure of nucleic acids and its significance for information transfer in living material“
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 9
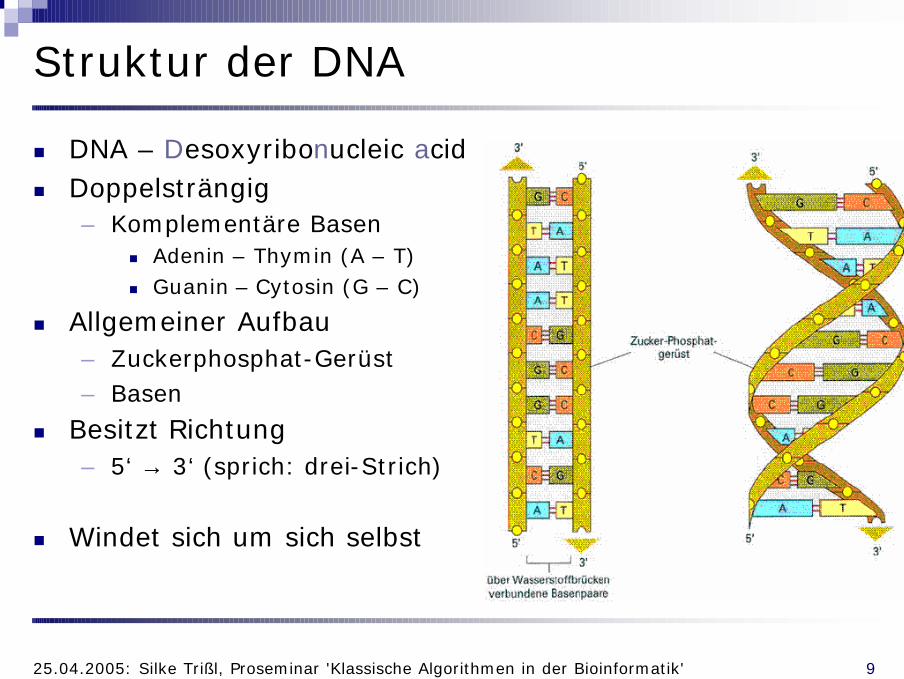
Struktur der DNA
n DNA – Desoxyribonucleic acidn Doppelsträngig
– Komplementäre Basenn Adenin – Thymin (A – T)n Guanin – Cytosin (G – C)
n Allgemeiner Aufbau– Zuckerphosphat-Gerüst– Basen
n Besitzt Richtung– 5‘ → 3‘ (sprich: drei-Strich)
n Windet sich um sich selbst

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 10
DNA
n Grundbausteine– Phosphatgruppe– Zucker (Desoxyribose)– Base
n 5‘-Ende: Phosphatn 3‘-Ende: Desoxyribose
(Hydroxylgruppe)
5‘
3‘
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 11
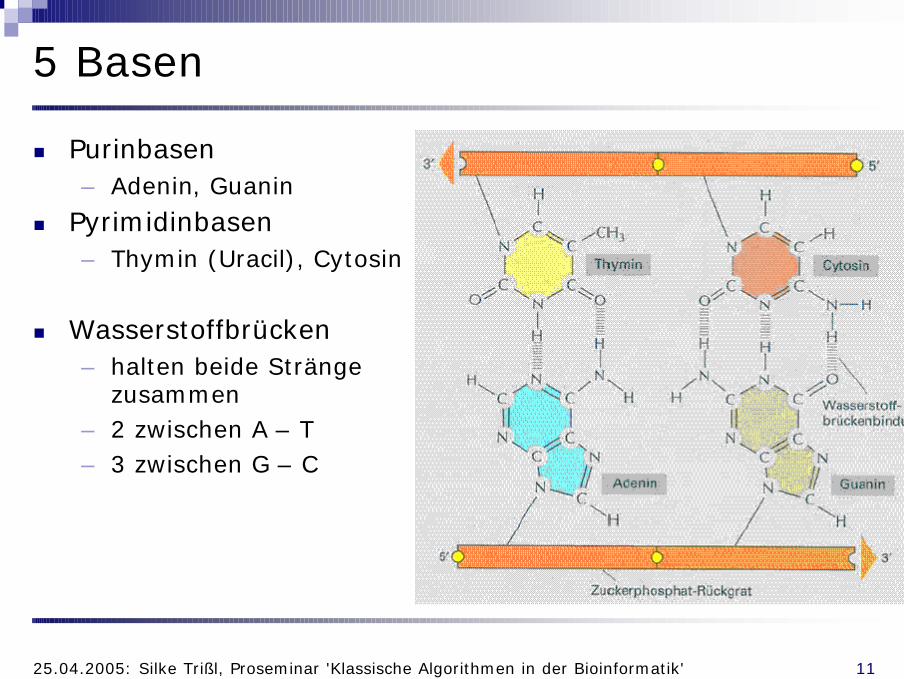
5 Basen
n Purinbasen– Adenin, Guanin
n Pyrimidinbasen– Thymin (Uracil), Cytosin
n Wasserstoffbrücken– halten beide Stränge
zusammen– 2 zwischen A – T – 3 zwischen G – C
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 12
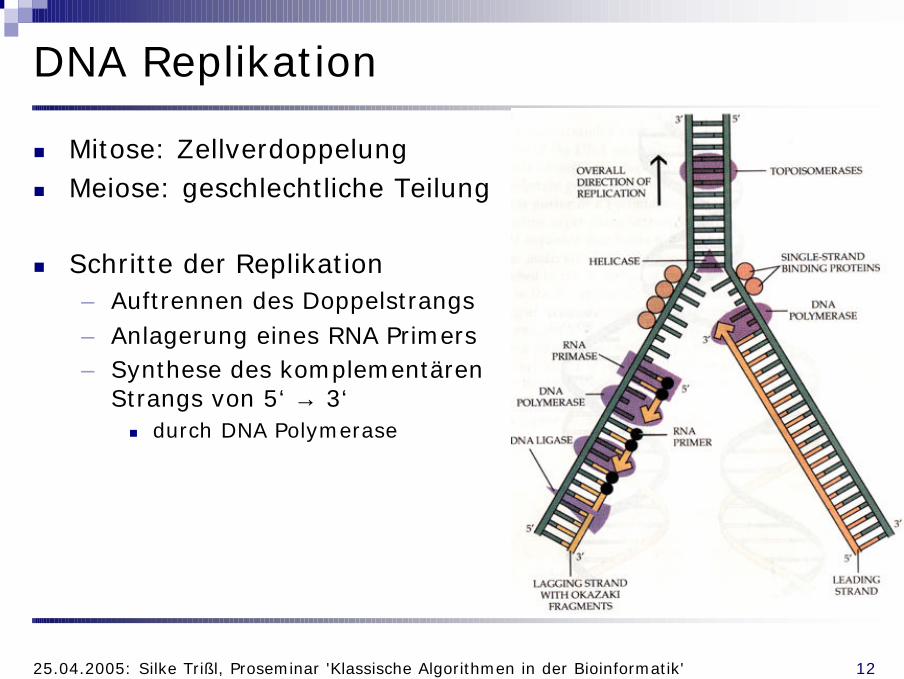
DNA Replikation
n Mitose: Zellverdoppelungn Meiose: geschlechtliche Teilung
n Schritte der Replikation– Auftrennen des Doppelstrangs– Anlagerung eines RNA Primers– Synthese des komplementären
Strangs von 5‘ → 3‘n durch DNA Polymerase
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 13
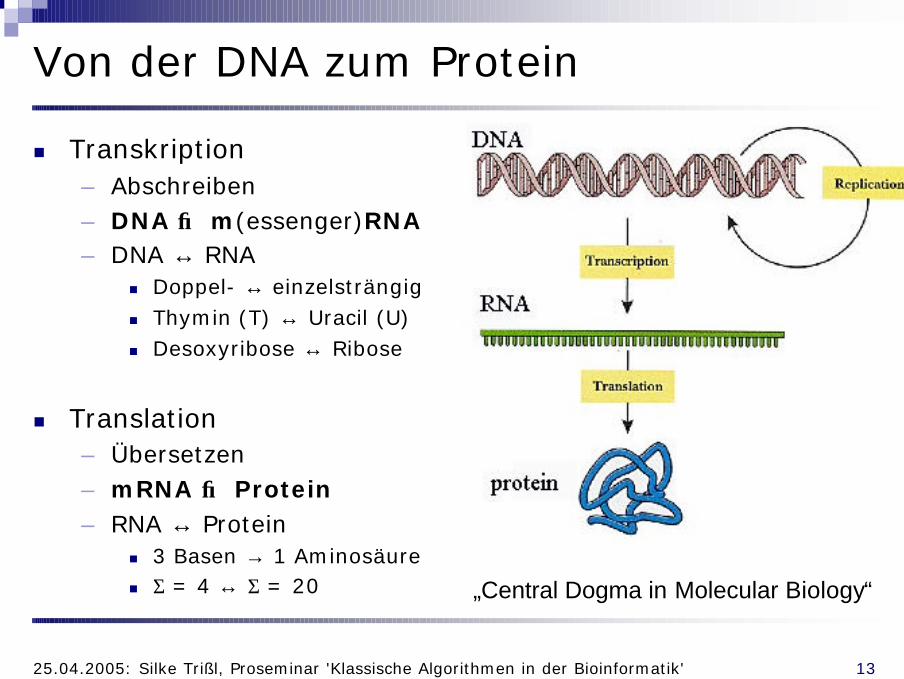
Von der DNA zum Protein
n Transkription– Abschreiben– DNA → m(essenger)RNA– DNA ↔ RNA
n Doppel- ↔ einzelsträngign Thymin (T) ↔ Uracil (U)n Desoxyribose ↔ Ribose
n Translation– Übersetzen– mRNA → Protein– RNA ↔ Protein
n 3 Basen → 1 Aminosäuren Σ = 4 ↔ Σ = 20 „Central Dogma in Molecular Biology“
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 14
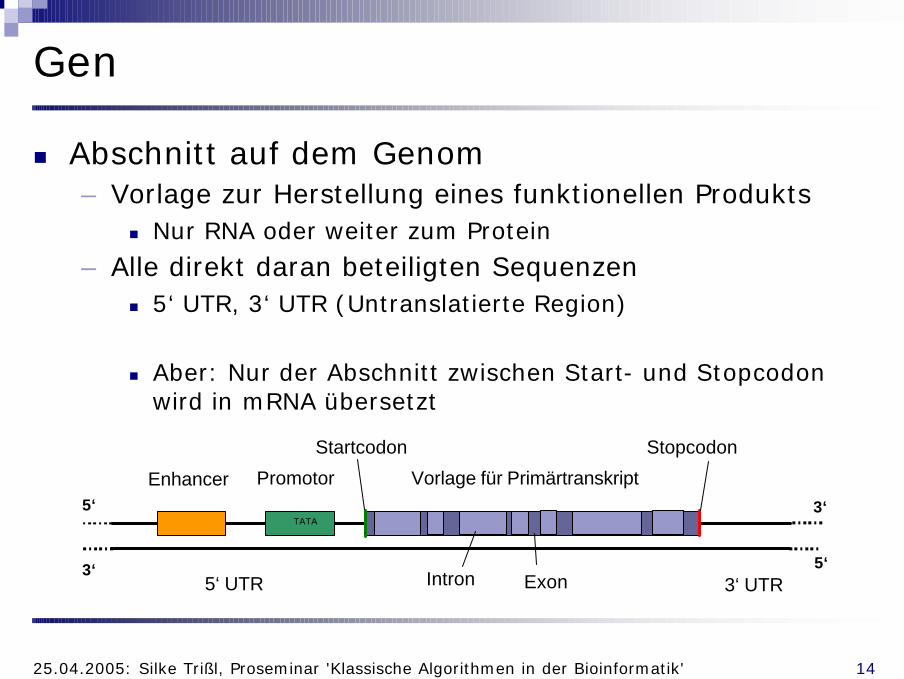
Gen
n Abschnitt auf dem Genom– Vorlage zur Herstellung eines funktionellen Produkts
n Nur RNA oder weiter zum Protein
– Alle direkt daran beteiligten Sequenzenn 5‘ UTR, 3‘ UTR (Untranslatierte Region)
n Aber: Nur der Abschnitt zwischen Start- und Stopcodonwird in mRNA übersetzt
5‘
5‘
3‘
3‘
Enhancer Promotor
Startcodon Stopcodon
Vorlage für Primärtranskript
5‘ UTR 3‘ UTR
TATA
Intron Exon
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 15
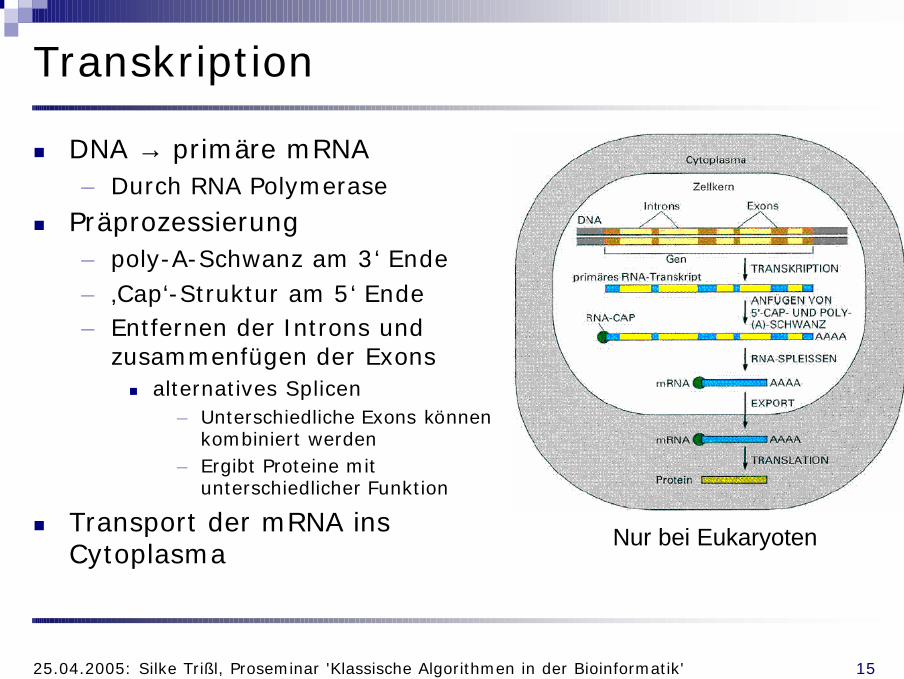
Transkription
n DNA → primäre mRNA– Durch RNA Polymerase
n Präprozessierung– poly-A-Schwanz am 3‘ Ende– ‚Cap‘-Struktur am 5‘ Ende– Entfernen der Introns und
zusammenfügen der Exonsn alternatives Splicen
– Unterschiedliche Exons können kombiniert werden
– Ergibt Proteine mit unterschiedlicher Funktion
n Transport der mRNA ins Cytoplasma
Nur bei Eukaryoten

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 16
Translation
n Übersetzung – Nukleotidsequenz der mRNA– zu Aminosäuresequenz der Proteine
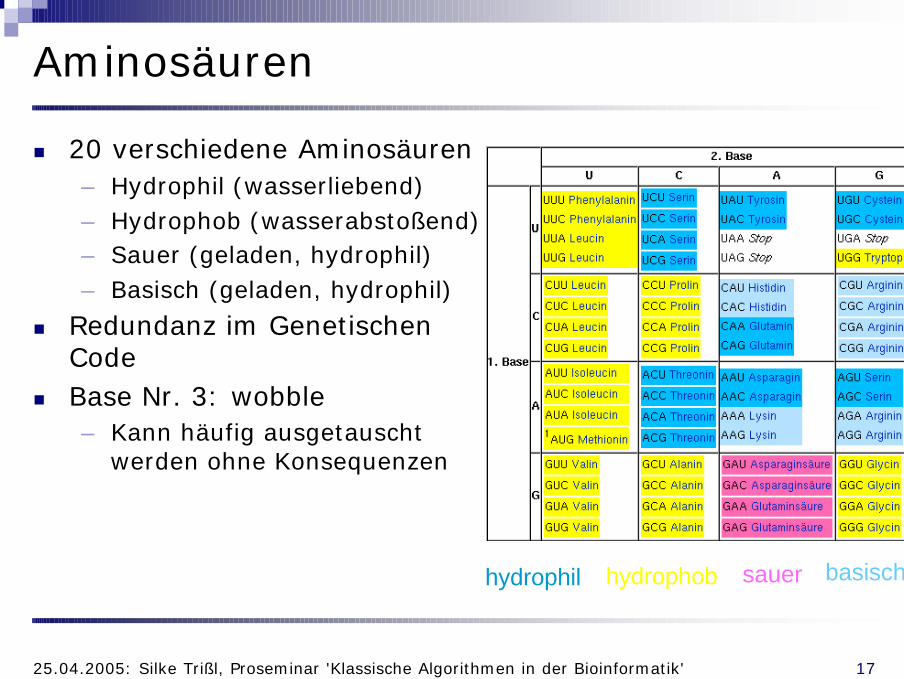
n Je 3 Basen (Codon) codieren für 1 Aminosäure
n Wie viele mögliche Kombinationen?– Triplett → 3 Stellen– 4 mögliche Buchstaben (A, T (U), G, C)– 43 = 64 mögliche Kombinationen– Aber nur 20 Aminosäuren
n Redundanz im genetischen Code
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 17
Aminosäuren
n 20 verschiedene Aminosäuren– Hydrophil (wasserliebend)– Hydrophob (wasserabstoßend)– Sauer (geladen, hydrophil)– Basisch (geladen, hydrophil)
n Redundanz im Genetischen Code
n Base Nr. 3: wobble– Kann häufig ausgetauscht
werden ohne Konsequenzen
hydrophil hydrophob sauer basisch

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 18
Redundanz im Code
Org1: AUU AGU UGG GAU AAA UUA GCUOrg2: AUA UCU UGG GAC AAG CUG GCCOrg3: AUC UCU UGG GAU AAG CUU GCGOrg4: AUU AGC UGG GAC AAA CUC GCAOrg5: AUC UCC UGG GAU AAG UUG GCU
Protein: Ile Ser Trp Asp Lys Leu Ala

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 19
Proteine Grundgerüst
n Synthese erfolgt am Ribosom– tRNA
n Besitzt Anticodonn Hat Aminosäure
n Einzelne Aminosäuren durch Peptidbindung verbunden– kovalente Verbindung
n Abfolge an Aminosäuren= Primärstruktur
mRNA
Ribosom
Anti-codon
Amino-säure
tRNA

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 20
Sekundärstruktur von Proteinen
n a-Helix– Wasserstoffbrücken
n ß-Faltblatt– Wasserstoffbrücken

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 21
Tertiärstruktur
n Tertiärstruktur: Räumliche Anordnung der Sekundärstrukturelemente
1b71 aus Protein Data Bank

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 22
Tertiärstruktur - Kräfte
n Wechselwirkungen zwischen Teilen der Proteinkette
n Disulfidbrücken– kovalente Bindung– sehr stabil E=380 kJ/mol
n Wasserstoffbrücken– sehr häufig E=4 kJ/mol
n Ionenbindung– E=12.5 kJ/mol
n Van-der-Waals– allgemeine Interaktion
zwischen Atomen– E=0,4 kJ/mol
n Hydrophobe Interaktion– Abstoßung vom Wasser

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 23
Quartärstruktur
n Anordnung von mehreren Proteinkettenn Beispiel – Hämoglobin
– Besteht aus 4 Ketten– Wichtig für den Sauerstofftransport im Blut

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 24
Mutationen in DNA – Auswirkungen
n Stille Mutation – keine Auswirkung auf Protein
n Echte Mutation– Ersetzen von Aminosäuren
n durch Ähnliche– keine Auswirkung auf Struktur– keine Auswirkung auf Funktion
n durch ‚Unähnliche‘– Auswirkung auf Struktur– Verlust / Verbesserung der Funktion

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 25
Proteinfunktionen
n Struktur– Zellwand, Membrane, Zellkern, Organellen, …
n Signaltransduktion– Signalerkennung (von Außen), Transduktion, intrazelluläre
Reaktion, …
n Metabolismus– Atmung, Energieproduktion, Nährstoffumwandlung, Abbau von
chem. Substanzen, …
n Housekeeping– Proteinsynthese und -abbau, DNA Verdopplung, Zellzyklus, ...
n Transport– mRNA vom Zellkern zum Ribosom, Proteine vom Ribosome zur
Zellwand, …

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 26
Regulatorische Netzwerke
n Einflüsse regulieren die Transkription von Genen– Äußere (Chemische Stoffe, Temperaturen, Strahlung)– Innere (Stoffwechselprodukte)
n Gene haben einen unterschiedlich hohenExpressionslevel– Menge an vorhandener
mRNA– Ändert sich über Zeit
und Zellart

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 27
Metabolische Netzwerke
n Glykolyse– Umwandlung von Glukose
zu Pyruvat unter Engergiegewinnung
n Start-, Zwischen- und Endprodukte regulieren auch die Transkription

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 28
Human Genome Project
n Begonnen 1986– Geplante Fertigstellung 2005 – Ziel: Finden aller Gene des Menschen
n Weltweite Kollaboration– 20 große Institutionen– In Deutschland: seit 1996 etwas 60 Mbp
n Fertiggestellt– Draft im Jahr 2000: (90% draft, 30% finished, 99.99%
accuracy)– Beinahe fertig 2001 (analyzed draft)– Wirklich fertig 2003

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 29
Menschliche Genom
n Menschen – Homo sapiens– ~ 3.100.000.000 bp (Basenpaare)
n Entspricht ~ 2 m DNA
– Verteilt auf n 22 Chromosomenn + 2 Geschlechtschromosomenn Länge: 50–250 Mbp
– ~ 25.000 Genen (war mal bei ~100.000)
– ~ 150.000 Proteine– ~ 500.000 Proteinformen

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 30
Menschliche Gene
n ~ 25.000 Gene– Niemand weis wirklich wie viele
n Länge zwischen 100bp und 2Mbp (Introns+Exons)n Durschschnittliche Länge der codierenden Region: 1400 bps
– Duchschnittl. Proteinlänge 447 Aminosäurenn Durschnittl. Gen hat 9 Exonsn Nur 3 % des menschlichen Genoms ist kodierend
– Rest: „junk“?– Viele Repeats, Transposons– Regulatorische Elemente– Pseudogene– Chromosomale Struktur: Zentromere und Telomere

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 31
Chromosome bei Eukaryoten
n Stark strukturiert während der Metaphase
– Doppelsträngige DNA– ‚Perlschnurform‘ des Chromatin– Chromatinfibrille– Fibrille wird weiter strukturiert– Chromosom
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 32
gatcaattatagttgacttcagtcctgcctgattcatctccaaaaatgtagtctgcctgattcatctcccaaaaatgtagctccgcttaaaggagctttcaagttgggggtggtgggccattcagtgttgtcactaacagatgcatcttgtgggggtaaaatgtcccaaagtatcttttcttgcttatgttcataagggcgctggtctggaatgtgccacatctgttctcactctgccatggactcctggaccctctgtgtgtccctttgtatcctggtagcgagtgagtcctcatgatttatcatcctcatgctgggcctctgtatagatga
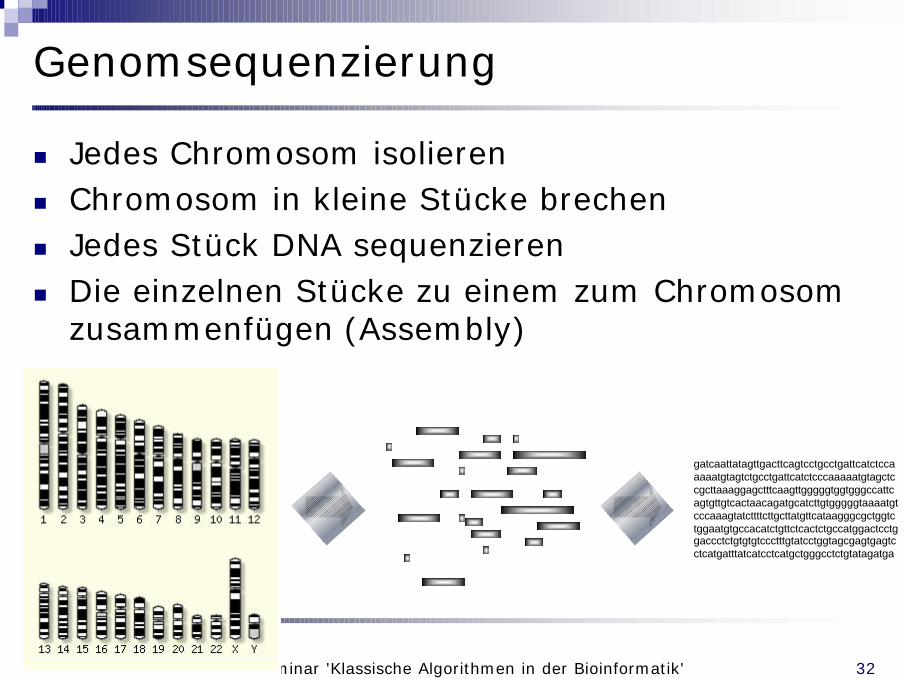
Genomsequenzierung
n Jedes Chromosom isolierenn Chromosom in kleine Stücke brechenn Jedes Stück DNA sequenzierenn Die einzelnen Stücke zu einem zum Chromosom
zusammenfügen (Assembly)

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 33
DNA schneiden
n Restriktionsenzyme– Erkennen Sequenzabschnitte
auf der DNA– Schneiden an der Stelle
n Blunt (gerader Schnitt)n Sticky (überhängende Enden)
n Länge der Erkennungssequenz steuert die DNA Fragmentgröße– 4 Basen → 256 bp Fragmente– 6 Basen → 4,000 bp Fragmente– 8 Basen → 65,000 bp Fragmente
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 34
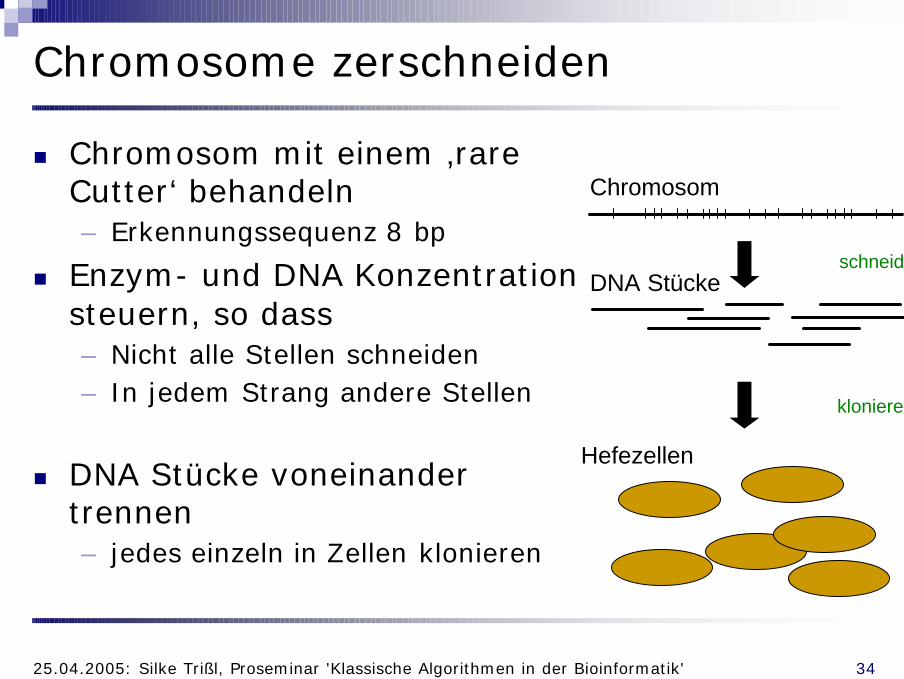
Chromosome zerschneiden
n Chromosom mit einem ‚rare Cutter‘ behandeln– Erkennungssequenz 8 bp
n Enzym- und DNA Konzentration steuern, so dass– Nicht alle Stellen schneiden– In jedem Strang andere Stellen
n DNA Stücke voneinander trennen– jedes einzeln in Zellen klonieren
schneiden
klonieren
Chromosom
DNA Stücke
Hefezellen

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 35
Chromosomen zerkleinern
n YAC – Yeast ArtificialChromosome– Enthält < 1.000.000 bp
chromosomale DNA– Je YAC nur einen Abschnitt
n BAC – Bacterial ArtificialChromosome– < 250.000 bp erneut
geschnittene DNA
n Plasmid– < 4.000 bp– Wird in Bakterien eingebracht
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 36
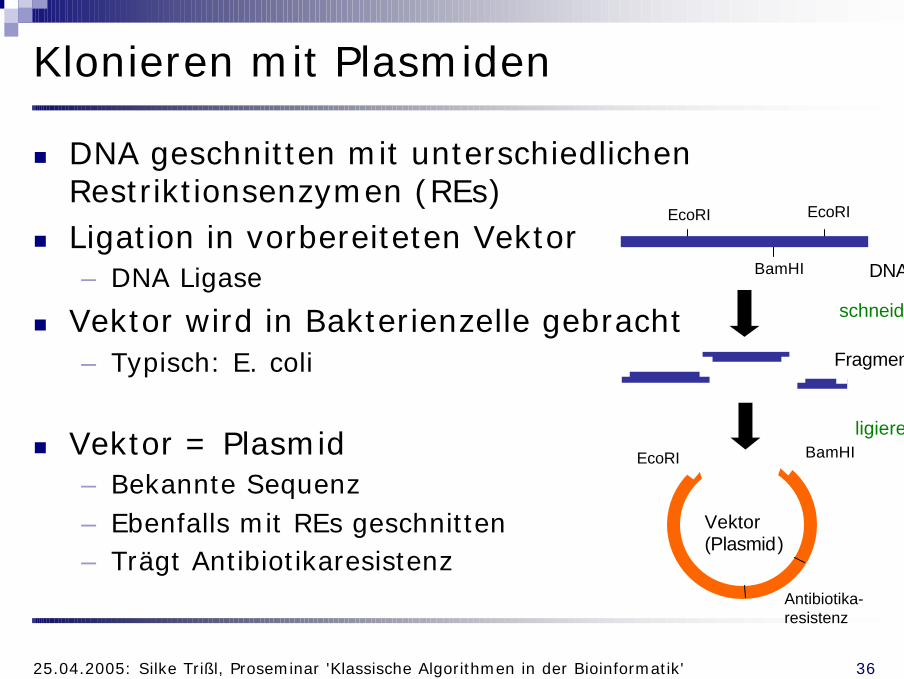
Klonieren mit Plasmiden
n DNA geschnitten mit unterschiedlichen Restriktionsenzymen (REs)
n Ligation in vorbereiteten Vektor– DNA Ligase
n Vektor wird in Bakterienzelle gebracht– Typisch: E. coli
n Vektor = Plasmid– Bekannte Sequenz– Ebenfalls mit REs geschnitten– Trägt Antibiotikaresistenz
BamHI
EcoRI EcoRI
BamHIEcoRI
Antibiotika-resistenz
Vektor(Plasmid)
DNA
schneiden
Fragmente
ligieren

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 37
Klonieren – cont. –
n Jedes Bakterium nimmt nur ein Plasmid aufn Ausplattieren der Bakterien
– Auf Nährbodenn Agarn Nährstoffen Antibiotikum
n Inkubieren (wachsen lassen)– E. coli: 37 °C, über Nacht
n Jede einzelne Bakterie wächstzu einer sichtbaren Kolonie (Klon)– Nur Bakterien mit Plasmid können wachsen

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 38
Zur Erinnerung
n YACs und BACs so geschnitten, dass überlappende Stücke entstehen
schneiden
YAC1 YAC2 YAC3
YAC4YAC5
YAC6YAC7
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 39
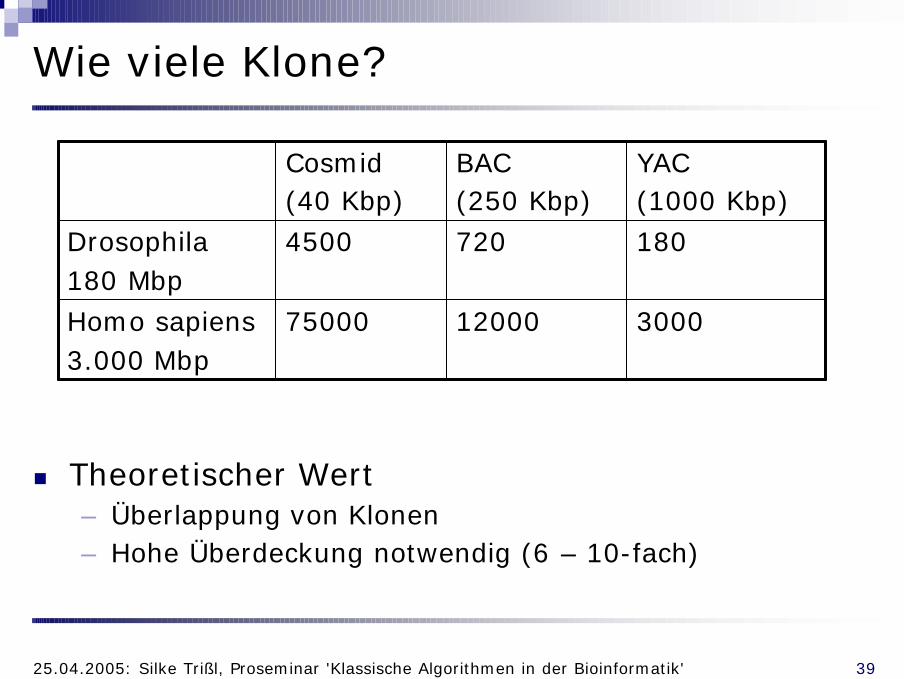
Wie viele Klone?
n Theoretischer Wert– Überlappung von Klonen– Hohe Überdeckung notwendig (6 – 10-fach)
30001200075000Homo sapiens3.000 Mbp
1807204500Drosophila180 Mbp
YAC(1000 Kbp)
BAC(250 Kbp)
Cosmid(40 Kbp)
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 40
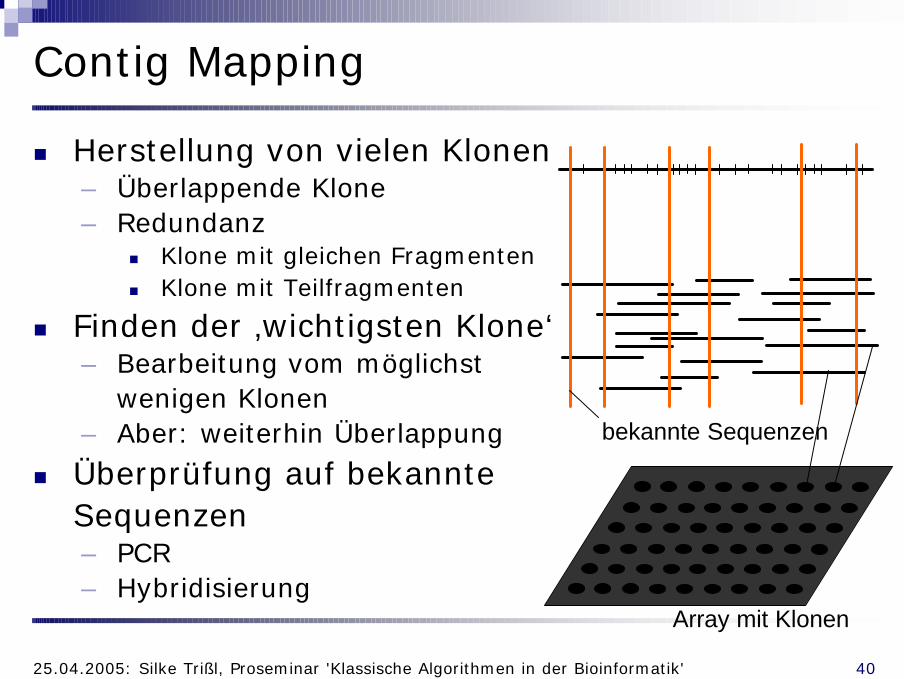
Contig Mapping
n Herstellung von vielen Klonen– Überlappende Klone– Redundanz
n Klone mit gleichen Fragmentenn Klone mit Teilfragmenten
n Finden der ‚wichtigsten Klone‘– Bearbeitung vom möglichst
wenigen Klonen– Aber: weiterhin Überlappung
n Überprüfung auf bekannte Sequenzen– PCR– Hybridisierung
bekannte Sequenzen
Array mit Klonen

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 41
PCR: Polymerase chain reaction
n Vervielfältigen von DNA– in vitro (im Reagenzglas)– Ausgehend von einer Vorlage
n PCR benötigt– DNA Template (doppelsträngig)– 2 DNA Primer
n Einzelsträngign Komplementär zu einer bekannten Sequenzn Zwischen 15 und 25 Nukleotide lang
– DNA Polymerase– dNTP‘s (Desoyxnukleotide)
n dATP, dCTP, dGTP, dTTP

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 42
PCR – Ablauf
Denaturieren– Auftrennen des Doppelstrangs
Annealen– Anlagern der Primer an komplementäre DNA (~ 55 °C)
Elongation– Verlängern des Strangs durch DNA Polymerase
n Vervielfältigung– 1. Zyklus: 2 Doppelstr.– 2. Zyklus: 4 Doppelstr.– …– 20. Zyklus: 1.000.000
1 2 3
95
72
55
T(°C)
t
1
2
3

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 43
Länge von DNA bestimmen
n DNA Negativ geladen– Wandert im elektrischen Feld zum
Pluspol
n Agarose Gel– Hoch vernetzt– Hindert DNA am wandern
n Kurze kommen eher durch als lange DNA Stücke
n DNA wird auf Gel aufgetragen und sichtbar gemacht
n Länge: Vergleich gegen Standard
-
+

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 44
Länge von DNA bestimmen
50
200
400
800
1500
n DNA Negativ geladen– Wandert im elektrischen Feld zum
Pluspol
n Agarose Gel– Hoch vernetzt– Hindert DNA am wandern
n Kurze kommen eher durch als lange DNA Stücke
n DNA wird auf Gel aufgetragen und sichtbar gemacht
n Länge: Vergleich gegen Standard

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 45
DNA Sequenzierung (nach Sanger)
n Abfolge von Basen in DNA Sequenz lesenn Sequenzierreaktion (basiert auf PCR) mit
– DNA Template (doppelsträngig)– 1 Primer– DNA Polymerase– dNTP‘s– ddNTP‘s (Didesoxynukleotide)
n Brechen Kettenverlängerung abn Sind markiert
– Früher: radioaktiv– Heute: fluoreszent (1 Farbstoff je Base)

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 46
DNA Sequenzierung – cont. –
n PCR ergibt DNA Stücke mit unterschiedlicher Länge– Markiert
n Werden über ein Acrylamid-Gel aufgetrennt-
+ Laser

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 47
DNA Sequenzierung – cont –
n Markierten Basen werden durch Laser detektiert– Ergibt 4 Tracefiles (Für jede Base eines)
n Tracefiles werden zum Chromatogrammzusammengesetzt– Leseweite ~ 500 bp

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 48
Großer Fortschritt
n Früher:– Radioaktiv– Handarbeit
n Heute:– 4 Floureszensfarbstoffe– vollautomatisch

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 49
Assembly
n Wir haben jetzt sequenzierte Stücke DNA– ~ 500 bp groß– Überlappend
n Wir wissen, sie kommen aus einem BAC, bzw. einem YAC
n Algorithmisches Problem: Sequence assembly– setze die 500 bp großen Stücke wieder zu einem BAC
bzw. YAC zusammen
n Aber: nicht überall funktioniert diese Strategie– Chromosome walking

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 50
Auswirkungen vom HGP
n Datenflut– Datenbanken & Bioinformatik
n Datenbasis für Finden von Genen– Alle Gene finden– Zusammenwirken von Genen erkennen– Erkennen von Genen, die mit Erbkrankheiten in
Verbindung stehen
n Erkenntnis über ähnliche/gleiche Gene in anderen Organismen– Experimente mit Modellorganismen
n Maus, Hefe, E. coli

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 51
Finden von Genen
n Aus einem Gen entsteht ein funktionelles Produkt (Protein)– mRNA muß existieren
n mRNA durch reverse Transkription in cDNAumschreiben– poly-T Primer– cDNA klonieren und sequenzieren
AAAAACAP5‘ 3‘
5‘TTTTT3‘

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 52
Expressionslevel von Genen
n Microarrays– enthält kurze Abschnitte von bekannten Genen– 30.000 – 100.000 Spots (Proben) pro Array– jeder Spot enthält mehrere Kopien
Array mit DNA Proben
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 53
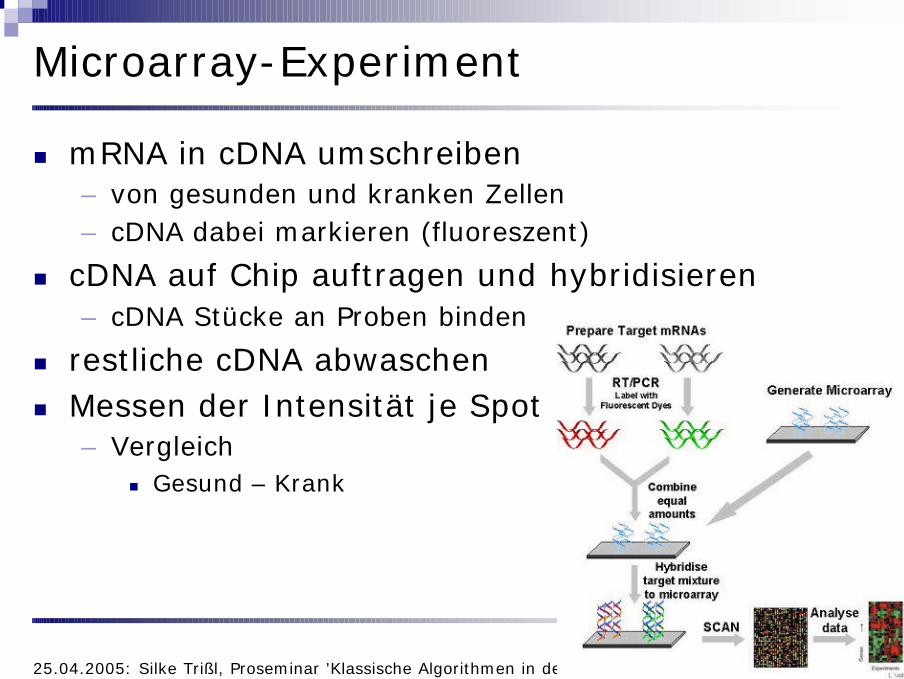
Microarray-Experiment
n mRNA in cDNA umschreiben– von gesunden und kranken Zellen– cDNA dabei markieren (fluoreszent)
n cDNA auf Chip auftragen und hybridisieren– cDNA Stücke an Proben binden
n restliche cDNA abwaschenn Messen der Intensität je Spot
– Vergleichn Gesund – Krank
25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 54
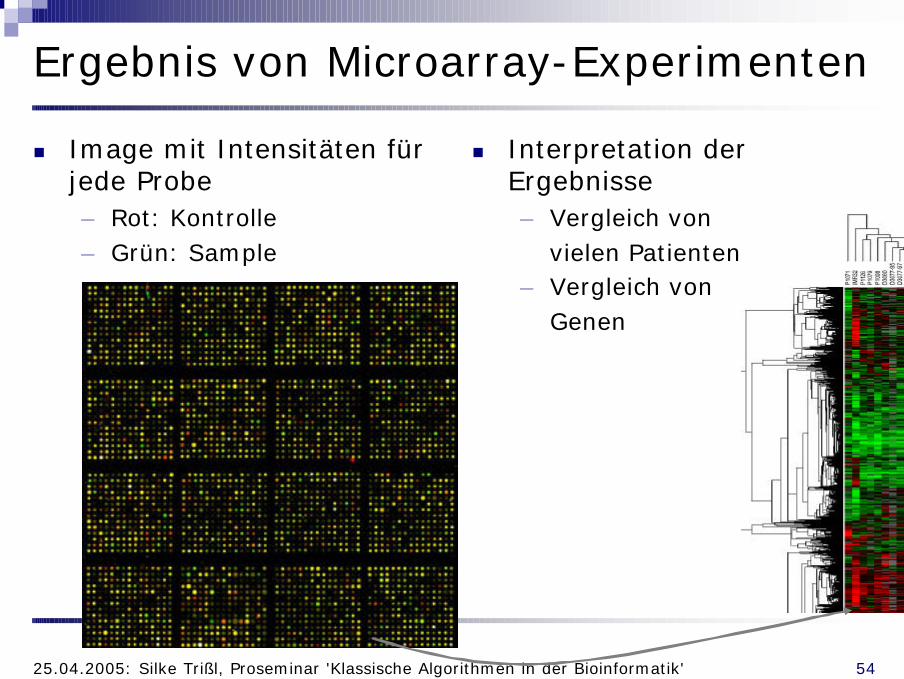
Ergebnis von Microarray-Experimenten
n Image mit Intensitäten für jede Probe– Rot: Kontrolle– Grün: Sample
n Interpretation der Ergebnisse– Vergleich von
vielen Patienten– Vergleich von
Genen

25.04.2005: Silke Trißl, Proseminar 'Klassische Algorithmen in der Bioinformatik' 55
Fragen?