
Bisphosphonatliganden für die zielgerichtete Applikation...
196
Bisphosphonatliganden für die zielgerichtete Applikation liposomaler Trägersysteme an den Knochen: eine Machbarkeitsstudie Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) dem Fachbereich Pharmazie der Philipps-Universität Marburg vorgelegt von Verena Hengst aus Gronau (Westfalen) Marburg/Lahn 2007
Transcript of Bisphosphonatliganden für die zielgerichtete Applikation...

Bisphosphonatliganden für die zielgerichtete Applikation liposomaler Trägersysteme an den Knochen: eine
Machbarkeitsstudie
Dissertation
zur Erlangung des Doktorgrades
der Naturwissenschaften
(Dr. rer. nat.)
dem Fachbereich Pharmazie
der Philipps-Universität Marburg
vorgelegt
von
Verena Hengst
aus Gronau (Westfalen)
Marburg/Lahn 2007

Vom Fachbereich der Pharmazie der Philipps-Universität Marburg als Dissertation am 18.12.2007 angenommen.
Erstgutachter: Professor Dr. T. Kissel
Zweitgutachter: Professor Dr. U. Bakowsky
Tag der mündlichen Prüfung am 19.12.2007

Inhaltsverzeichnis I
Inhaltsverzeichnis
ABKÜRZUNGSVERZEICHNIS........................................................................IV
ABBILDUNGSVERZEICHNIS .........................................................................IX
TABELLENVERZEICHNIS .............................................................................XII
1 EINLEITUNG.............................................................................................. 1
1.1 Generelle Charakteristika des Knochens ....................................................... 4 1.1.1 Aufbau und Funktion des Knochens............................................................... 4 1.1.2 Prozess des Knochenauf- und -abbaus............................................................ 6
1.2 Knochenmetastasen .......................................................................................... 8 1.2.1 Häufig auftretende Knochenerkrankungen..................................................... 9 1.2.2 Entstehung von Knochenmetastasen ............................................................ 10 1.2.3 Prozess der Angiogenese .............................................................................. 12
1.3 Behandlung von Knochenmetastasen ........................................................... 14 1.3.1 Klassische Ansätze zur Therapie von Knochenmetastasen .......................... 15
1.3.1.1 Strahlentherapie .................................................................................... 15 1.3.1.2 Hormontherapie .................................................................................... 16 1.3.1.3 Chemotherapie ...................................................................................... 17
1.3.2 Neue Strategien zur Behandlung von Tumorerkrankungen ......................... 17
1.4 Bisphosphonate ............................................................................................... 22 1.4.1 Struktur ......................................................................................................... 22 1.4.2 Wirkmechanismus ........................................................................................ 24 1.4.3 Indikationen, Nebenwirkungen und Kontraindikationen ............................. 27
1.5 Zielgerichtete Applikation an den Knochen................................................. 28 1.5.1 Trägersysteme basierend auf Bisphosphonaten als knochenaffine Liganden .. ....................................................................................................................... 29 1.5.2 Alternative knochenaffine Liganden ............................................................ 31
1.6 Liposomen als Wirkstoffträgersystem.......................................................... 33 1.6.1 Eigenschaften von Liposomen...................................................................... 33 1.6.2 Ligandmodifizierung von Liposomen .......................................................... 38
2 ZIELE DER ARBEIT ................................................................................ 43
3 MATERIALIEN, GERÄTE UND METHODEN ......................................... 47
3.1 Materialien....................................................................................................... 47 3.1.1 Lipide und andere Membrankomponenten................................................... 47

Inhaltsverzeichnis II
3.1.2 Chemikalien .................................................................................................. 49 3.1.3 Lösungsmittel................................................................................................ 51 3.1.4 Puffer und wäßrige Lösungen....................................................................... 52 3.1.5 Zellkulturmaterialien .................................................................................... 54 3.1.6 Verbrauchsmaterialien .................................................................................. 55
3.2 Geräte............................................................................................................... 57
3.3 Methoden ......................................................................................................... 59 3.3.1 Ligandsynthese ............................................................................................. 59 3.3.2 Zubereitung von ligandmodifizierten Liposomen ........................................ 59 3.3.3 In-vitro-Charakterisierung ............................................................................ 60
3.3.3.1 Bestimmung der Partikelgrösse ............................................................ 60 3.3.3.2 Bestimmung des Zeta-Potentials .......................................................... 61 3.3.3.3 Dynamische Differenz-Kalorimetrie .................................................... 62 3.3.3.4 31Phosphor-Kernresonanz-Spektroskopie............................................. 62 3.3.3.5 Phospholipidbestimmung nach Rouser................................................. 63
3.3.4 Bindung an Hydroxylapatit........................................................................... 64 3.3.4.1 Isothermale Titrationskalorimetrie ....................................................... 65 3.3.4.2 Bestimmung der Bindungsparameter ................................................... 66
3.3.5 Doxorubizin-Beladung („Remote Loading”) ............................................... 67 3.3.5.1 Größenausschlusschromatographie ...................................................... 67 3.3.5.2 Doxorubizin-Gehaltsbestimmung......................................................... 68 3.3.5.3 Doxorubizin-Freisetzung ...................................................................... 68
3.3.6 Zellkultur....................................................................................................... 70 3.3.6.1 Zellkultivierung und Zellzahlbestimmung ........................................... 70 3.3.6.2 Kristallvioletttest................................................................................... 71
3.3.7 In-vivo-Versuche .......................................................................................... 72 3.3.7.1 Art und Haltung der Versuchstiere ....................................................... 72 3.3.7.2 Pharmakokinetik- und Biodistributionsstudien .................................... 73 3.3.7.3 Statistik ................................................................................................. 74 3.3.7.4 Tiermodel für Knochenmetastasen ....................................................... 75 3.3.7.5 „Bioluminescent Reporter Imaging” (BRI) .......................................... 78 3.3.7.6 Lokalisierung von ligandmodifizierten Liposomen.............................. 78
4 ERGEBNISSE UND DISKUSSION.......................................................... 80
4.1 Formulierungsentwicklung von ligandmodifizierten Liposomen .............. 81 4.1.1 Präformulierungen von ligandmodifizierten Liposomen.............................. 84 4.1.2 Zusammensetzung von ligandmodifizierten Liposomen.............................. 85
4.2 Physikochemische Charakterisierung von ligandmodifizierten Liposomen. ........................................................................................................................... 86
4.2.1 Partikelgröße und Zeta-Potential .................................................................. 88 4.2.2 Stabilität im Hinblick auf die Partikelgröße ................................................. 92 4.2.3 Phasenübergangstemperaturbestimmung ..................................................... 93 4.2.4 Ligandquantifizierung................................................................................... 96

Inhaltsverzeichnis III
4.3 Bindung von ligandmodifizierten Liposomen an Hydroxylapatit ............. 98 4.3.1 Einfluss der Ligandkonzentration auf die Bindung an Hydroxylapatit ........ 98 4.3.2 Einfluss von Plasmaproteinen auf die Bindung an HAP............................ 104
4.4 Wirkstoffbeladung von ligandmodifizierten Liposomen .......................... 108 4.4.1 Beladungskapazität ..................................................................................... 110 4.4.2 Physikochemische Charakterisierung......................................................... 111 4.4.3 Stabilität ...................................................................................................... 112 4.4.4 Doxorubizin-Freisetzung ............................................................................ 112
4.5 Zytotoxizität von ligandmodifizierten Liposomen..................................... 115 4.5.1 Bestimmung der Zytotoxizität von ligandmodifizierten, Doxorubizin-beladenen Liposomen versus freiem Doxorubizin ................................................. 116
4.6 Pharmakokinetik und Biodistribution von ligandmodifizierten Liposomen ......................................................................................................................... 120
4.6.1 Einfluss der Ligandkonzentration auf die Zirkulationszeit ........................ 121 4.6.2 Einfluss der DSPE-PEG2000-Dichte auf die Zirkulationszeit ................... 125 4.6.3 Einfluss des DSPE-PEG-Typs auf die Zirkulationszeit.............................. 128
4.7 Zielgerichtete Applikation von ligandmodifizierten Liposomen an den Knochen (Nacktmausmodell)................................................................................... 133
5 ZUSAMMENFASSUNG UND AUSBLICK............................................. 144
6 SUMMARY AND OUTLOOK ................................................................. 152
7 LITERATUR ........................................................................................... 159
8 ANHANG................................................................................................ 176
8.1 Lebenslauf...................................................................................................... 176
8.2 Danksagung/Acknowledgment .................................................................... 178
8.3 Veröffentlichungen ....................................................................................... 180 8.3.1 Publikationen .............................................................................................. 180 8.3.2 Patente......................................................................................................... 180 8.3.3 Vorträge ...................................................................................................... 180 8.3.4 Poster........................................................................................................... 181

Abkürzungsverzeichnis IV
Abkürzungsverzeichnis
ABC-Phänomen „Accelerated Blood Clearance“-Phänomen
AMP Adenosin-monophosphat
APPCCl2p „Adenosine 5´(β, γ-dichlormethylene) triphosphate”
ApppI „Triphosphoric acid 1-adenosin-5´-yl ester 3-(3-methylbut-3-enyl) ester”
Asp6 Hexapeptid der Asparaginsäure
ATCC „American Type Culture Collection“
ATP Adenosin-triphosphat
AUC „Area under the Curve“
BALB-c-nu/nu-Maus Die BALB-c-nu/nu-Maus ist eine immundefiziente Nacktmaus (Stamm BALB-c)
BLP25 liposomales Vakzin (Stimuvax®)
BRI „Bioluminescent Reporter Imaging“
BRU „Bone Remodelling Unit“
BSP „Bone Sialoprotein“
BT-474 Mammakarzinomzellen
bzw. beziehungsweise
ca. circa
CAM „Cell Adhesion Molecule“, Zelladhäsionsmolekül
CDCl3 deuteriertes Chloroform
CHCl3 Chloroform
CHOL Cholesterol
CHOL-BP Cholesteryl-bisphosphonsäure
CHOL-TeOE-BP Cholesteryl-tetraoxyethylen-bisphosphonsäure
CHOL-TOE-BP Cholesteryl-trisoxyethylen-bisphosphonsäure
CMF Zytostatikatherapie bestehend aus der Kombination von Cyclophosphamid, Methotrexat und 5-Fluorouracil

Abkürzungsverzeichnis V
CMV-Luc Vektor, der das Luciferase-Reportergen und den Promotor des Cytomegalie-Virus trägt.
CO2 Kohlenstoffdioxid
d Tag(e)
DC „Dentritic Cells“, dentritische Zellen
DKFZ Deutsches Krebsforschungszentrum
d.h. das heißt
DMEM Dulbecco´s-Modified-Eagle´s Medium
DMPG 1,2-Dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol) (Natriumsalz)
DPPC Dipalmitoyl-phosphatidylcholin
DSC „Differential Scanning Calorimetry“, Dynamische Differenz-Kalorimetrie
DSPE Distearoyl-phosphatidylethanolamin
DSPE-PEG2000 1,2-Distearoyl-glycero-3-phosphoethanolamin-N-[Methoxy(Polyethylen-glykol)2000)]
DSPE-PEG5000 1,2-Distearoyl-glycero-3-phosphoethanolamin-N-[Methoxy(Polyethylen-glykol)5000)]
DU-145 Prostatakarzinomzellen
ECM „Extracellular Matrix“, Extrazellulärmatrix
EDTA Ethylendiamintetraessigsäure
EIA (ELISA) Enzymgekoppelter Immunadsorptionstest („Enzyme-linked ImmunoSorbent Assay“)
EPC Ei-phosphatidylcholin
EPR-Effekt „Enhanced Permeability and Retention”-Effekt
EtOH Ethanol
FAC Zytostatikatherapie bestehend aus der Kombination von 5-Fluorouracil, Doxorubizin und Cyclophosphamid
FCS „Fetal Calf Serum“, Fötales Kälberserum
FDA „Food and Drug Administration”, Amerikanische Aufsichtsbehörde für Lebens- und Arzneimittel
FGF „Fibroblast Growth Factor”, Fibroblast-Wachstumsfaktor

Abkürzungsverzeichnis VI
GM1 Monosialogangliosid GM1
GTP Guanosin-triphosphat
h Stunde(n)
HAP Hydroxylapatit
HBS „HEPES-buffered saline”, HEPES-gepufferte Kochsalzlösung
HCl Salzsäure
HEPES 4-(2-Hydroxyethyl)-1-piperazinethansulfonsäure
HER2 receptor/HER2-Rezeptor
„Human Epidermal Growth Factor Receptor 2”, humaner epidermaler Wachstumsfaktor Rezeptor, Typ 2
5HT3 5-Hydroxytryptamin
IC50 „Half Maximal Inhibitory Concentration”, halbmaximale Hemmkonzentration
IHC Immunhistochemische Analyse
IL-1 Interleukin-1
ITC „Isothermal Titration Calorimetry”, Isothermale Titrationskalorimetrie
i.v. intravenös
appK apparente Bindungskonstante
KG Körpergewicht
LH-RH „Luteinizing Hormone-Releasing Hormone”, Luteinisierendes Hormon-Freisetzendes Hormon
LUV „Large Unilamellar Vesicles”, große unilamellare Liposomen
MCF-7 Mammakarzinomzellen
MCS Micro Carrier Systems GmbH
MDA-MB-435s Mammakarzinomzellen
MEM Minimum-Essential-Medium
MeOH Methanol
Mg2+ Magnesium

Abkürzungsverzeichnis VII
min Minute(n)
MLV „Multilamellar Vesicles”, multilamellare Liposomen
MMM Zytostatikatherapie bestehend aus der Kombination von Mitomycin C, Methotrexat und Mitoxantron
MMP „Matrix Metalloproteinases”, Matrixmetalloproteinasen
MPS Mononukleäres Phagozyten-System
MUC-1 tumorassoziiertes Antigen
MWCO „Molecular Weight Cut Off”
NaCl Natriumchlorid
NBD-PE N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamin Triethylammoniumsalz
NH3 Ammoniak
NH4 Ammonium
OD optische Dichte
PAL-BP Palmitoyl-bisphosphonsäure
PBS „Phosphate-buffered saline”, Phosphat-gepufferte Kochsalzlösung
PCR „Polymerase Chain Reaction”, Polymerasekettenreaktion
PCS „Photon Correlation Spectroscopy”, Photonen-Korrelations-Spektroskopie
PDGF „Platelet-derived Growth Factor”, Plättchen-Wachstumsfaktor
PEG Polyethylenglykol
PGA „Polyglycolid acid”, Polyglykolsäure
PHPMA Poly[N-(2-hydroxypropyl)methacrylamid]
PI Polydispersionsindex
PLA „Polylactid acid”, Polymilchsäure
PLGA „Poly(lactic-co-glycolic) acid”, Poly(milch-co-glykolsäure)
31P-NMR „31P-Nuclear Magnetic Resonance“, 31P-Kernresonanz-Spektroskopie

Abkürzungsverzeichnis VIII
PPES Palmar Plantares Erythrodysästhesie
ppm „parts per million” (Teile pro Millionen)
q.s. quantum satis (so viel, wie notwendig ist)
RES retikuloendotheliales System
RGD Tripeptidsequenz Arginin-Glycin-Asparaginsäure
RLU „Relative Light Unit”
rpm „rotations per minute”, Umdrehungen pro Minute
RPMI 1640 Zellmedium 153SM Samarium-53
sec Sekunde(n)
Ser Serotonin
SD „Standard Deviation”, Standardabweichung
sLex Sialyl-Lewis X 89Sr Strontium-89
SUV „Small Unilamellar Vesicles”, kleine unilamellare Liposomen
TAA tumorassoziiertes Antigen
TGF-α/-β „Transforming Growth Factor-α/-β”, transformierender Wachstumsfaktor-α/-β
Thr Thyrosin
TIMP „Tissue Metalloproteinases”, Metalloproteinasen
TRAP „Tartrate-resistant Acid Phosphatase”, tartratresistente saure Phosphatase
UV ultraviolett
VEGF „Vascular Endothelial Growth Factor”, vaskulärer endothelialer Wachstumsfaktor
z.B. zum Beispiel
Mengen-, Größen- und Gewichtsangaben wurden durch internationale Standards abgekürzt.

Abbildungsverzeichnis IX
Abbildungsverzeichnis
Abbildung 1: Anatomie des Knochens. .......................................................................... 6
Abbildung 2: Entstehung von Knochenmetastasen. ..................................................... 12
Abbildung 3: Ablauf der angiogenen Kaskade............................................................. 14
Abbildung 4: Strukturformel von Pyrophosphat und Bisphosphonat (Quelle: Fleisch, 1998). ............................................................................................................................ 22
Abbildung 5: Räumliche Darstellung von Komplexbildung zwischen einem Bisphosphonat und einem Calciumatom der Knochenoberfläche................................ 24
Abbildung 6: Darstellung von Phospholipidstruktur und Ausbildung von Liposomen in wässriger Umgebung. ................................................................................................... 34
Abbildung 7: Histologische Identifikation eines Ausschnittes vom metastasendurchsetzten, trabekulären Knochen einer Maus. Kollagen-Typ-1-Färbung eines longitudinalen Schnittes von einem langen Knochen, welcher dem linken Hinterbein entnommen wurde. ..................................................................................... 44
Abbildung 8: BALB-c-nu/nu-Maus mit sichtbarer Metastasierung am linken Bein (bei ventraler Ansicht).......................................................................................................... 77
Abbildung 9: Strukturformel der Palmitoyl-bisphosphonsäure.................................... 82
Abbildung 10: Strukturformel der Cholesteryl-bisphosphonsäure............................... 83
Abbildung 11: Strukturformel der Cholesteryl-trisoxyethylen-bisphosphonsäure. ..... 83
Abbildung 12: Strukturformel der Cholesteryl-tetraoxyethylen-bisphosphonsäure. ... 84
Abbildung 13: Langzeitstabilität (9 Monate) im Hinblick auf die durchschnittliche Partikelgröße von ligandmodifizierten Liposomen mit verschiedenen CHOL-TOE-BP-Konzentrationen (1,6 mol%, 6,3 mol%, 14,3 mol%). .................................................. 93
Abbildung 14: DSC-Thermogramme von ligandmodifizierten Liposomen mit folgender Zusammensetzung: A: 28,6 mol% Cholesterol, 14,3 mol% CHOL-TOE-BP; B: kein Cholesterol, 14,3 mol% CHOL-TOE-BP; C: kein Cholesterol, 25,0 mol% CHOL-TOE-BP (abgefahrener Temperaturbereich: von -5 °C bis 80 °C; Scan-Rate: 5 °C/min).......................................................................................................................... 95
Abbildung 15: 31P-NMR-Analyse von ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol%, vor der Extrusion und nach der Extrusion. ...................................................................................................................... 97
Abbildung 16: Titration von ligandmodifizierten Liposomen in eine 0,05-mM-HAP-Suspension: A: 14,3 mol% CHOL-TOE-BP; B: 6,3 mol% CHOL-TOE-BP; C: 1,6 mol% CHOL-TOE-BP. Ligandmodifizierte Liposomen wurden in 150-Sekunden-

Abbildungsverzeichnis X
Intervallen in ein Gefäß titriert (38 Injektionen), welches 1,345 ml von einer 0,05-mM-HAP-Suspension enthielt. ........................................................................................... 102
Abbildung 17: Kumulative Reaktionsenthalpie von ligandmodifzierten Liposomen mit unterschiedlichen CHOL-TOE-BP-Konzentrationen als Funktion von der Zahl der Injektionen. ................................................................................................................. 104
Abbildung 18: Titrationen von ligandmodifizierten Liposomen (20 mM) mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% in Medien bestehend aus 0,05-mM-HAP-Suspension und unterschiedlichen Konzentrationen an Humanplasma (0%, 1%, 5%, 10%)..................................................................................................................... 106
Abbildung 19: Kontrolltitrationen von ligandmodifizierten Liposomen (20 mM) mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% in Medien bestehend aus HBSpH7,4 und unterschiedlichen Konzentrationen an Humanplasma (0%, 1%, 5%, 10%). ........................................................................................................................... 107
Abbildung 20: Doxorubizin-Freisetzung aus ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% bei pH 5,0, pH 6,5 und pH 7,4 (Versuchsbedingungen: 25 °C, HBSpH7,4 50 mmol, 0,7% NaCl). .............................. 115
Abbildung 21: Transport von liposomal verkapseltem Wirkstoff (L-Drug) in die Tumorzelle. ................................................................................................................. 120
Abbildung 22: Pharmakokinetikprofile von ligandmodifizierten Liposomen (4,3 mol% DSPE-PEG2000) mit verschiedenen CHOL-TOE-BP-Konzentrationen (1,6 mol%, 14,3 mol%) und Kontrollliposomen (4,3 mol% DSPE-PEG2000) im Blut nach intravenöser Injektion in die Schwanzvene von Ratten.............................................. 123
Abbildung 23: Organverteilung von ligandmodifizierten Liposomen und Kontrollliposomen. ..................................................................................................... 125
Abbildung 24: Pharmakokinetikprofile von ligandmodifizierten Liposomen, die jeweils 14,3 mol% CHOL-TOE-BP enthielten und sich in ihren DSPE-PEG2000-Konzentrationen unterschieden, sowie von Kontrollliposomen (4,3 mol% DSPE-PEG2000) im Blut nach intravenöser Injektion in die Schwanzvene von Ratten. ..... 127
Abbildung 25: Pharmakokinetikprofile von ligandmodifizierten Liposomen, die jeweils 14,3 mol% CHOL-TOE-BP enthielten und sich in ihrem DSPE-PEG-Typ (DSPE-PEG2000, DSPE-PEG5000) unterschieden, und Kontrollliposomen (4,3 mol% DSPE-PEG2000 bzw. 4,3 mol% DSPE-PEG5000) im Blut nach intravenöser Injektion in die Schwanzvene von Ratten. ................................................................................. 131
Abbildung 26: Organverteilung von ligandmodifizierten Liposomen, die jeweils 14,3 mol% CHOL-TOE-BP enthielten (A: DSPE-PEG2000: 4,3 mol%; B: DSPE-PEG2000: 8,2 mol%; C: DSPE-PEG2000: 13,0 mol%; D: DSPE-PEG5000: 4,3 mol%, G: DSPE-PEG2000: 0 mol%), und Kontrollliposomen (E: DSPE-PEG2000: 4,3 mol%; F: DSPE-PEG5000: 4,3 mol%). ................................................................................. 133
Abbildung 27: Radiografische Aufnahme von untersuchten Mäusen........................ 135

Abbildungsverzeichnis XI
Abbildung 28: Bestimmung des Ausmaßes der Metastasierung in den einzelnen untersuchten Mäusen mittels biolumineszenter Bildgebung. ..................................... 136
Abbildung 29: Visualisierung von Maus und Liposomenverbleib im lebenden Körper (ventrale Ansicht) zu den Zeitpunkten: 5 min, 15 min, 45 min, 2 h, 24 h, 5 d durch eine nicht invasive Detektionsmethode. ............................................................................. 141
Abbildung 30: Visualisierung von Maus und Liposomenverbleib im lebenden Körper (ventrale Ansicht) zu den Zeitpunkten: 10 min, 45 min, 2 h, 24 h, 48 h durch eine nicht invasive Detektionsmethode. ...................................................................................... 142

Tabellenverzeichnis XII
Tabellenverzeichnis
Tabelle 1: Übersicht über verwendete Lipide und Membrankomponenten ................. 47
Tabelle 2: Übersicht über verwendete Chemikalien..................................................... 49
Tabelle 3: Übersicht über verwendete Lösungsmittel .................................................. 51
Tabelle 4: Übersicht über verwendete Puffer und Lösungen ....................................... 52
Tabelle 5: Übersicht über verwendete Zellkulturmaterialien ....................................... 54
Tabelle 6: Übersicht über verwendete Verbrauchsmaterialien..................................... 55
Tabelle 7: Übersicht über eingesetzte Geräte ............................................................... 57
Tabelle 8: Übersicht über die verwendeten Zellen für die Bestimmung der IC50-Werte von Doxorubizin-beladenen, ligandmodifizierten Liposomen. .................................... 70
Tabelle 9: Physikochemische Eigenschaften von ligandmodifizierten Liposomen sowie von Kontrollliposomen, die keinen CHOL-TOE-BP-Liganden aufwiesen.................. 91
Tabelle 10: Thermodynamische Parameter, die die Bindung von ligandmodifizierten Liposomen (20 mM) mit unterschiedlichen CHOL-TOE-BP-Konzentrationen an HAP (0,05 mM) charakterisieren (Versuchsbedingungen: 25 °C, HBSpH7,4 50 mmol, 0,7% NaCl, pH 7,4). ............................................................................................................. 103
Tabelle 11: Übersicht über molare Reaktionsenthalpien bestimmt durch Titrationen von ligandmodifizierten Liposomen (20 mM) mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% in Medien verschiedener Zusammensetzungen (0,05 mM HAP + 0–10% Plasma bzw. HBSpH7,4 + 0–10% Plasma) (Versuchsbedingungen: 25 °C, HBSpH7,4 50 mmol, 0,7% NaCl, pH 7,4).......................................................... 107
Tabelle 12: Ermittlung der Beladungskapazität von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP) nach erfolgter Trennung von nicht verkapselter Wirksubstanz (Säulenchromatographie)..................................................................... 111
Tabelle 13: Physikochemische Eigenschaften von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP), die eine Doxorubizin-Einschlussrate von 99% aufweisen. ................................................................................................................... 111
Tabelle 14: Stabilität von ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% im Hinblick auf die durchschnittliche Partikelgröße und die Verkapselungseffizienz (%) über einen Zeitraum von 1 Woche (die hier dargestellten Werte repräsentieren jeweils einen Mittelwert aus drei Einzelmessungen ± SD) (Lagerungsbedingungen: 2–8 °C, vor Licht geschützt). .................................. 112
Tabelle 15: IC50-Werte für ligandmodifizierte Liposomen, die mit Doxorubizin beladen wurden, für Kontrollliposomen (kein Doxorubizin enthaltend) und für freies Doxorubizin. ............................................................................................................... 117

Tabellenverzeichnis XIII
Tabelle 16: Einfluss der molaren CHOL-TOE-BP-Konzentration auf die „Area under the Curve“ von ligandmodifizierten Liposomen, die 4,3 mol% DSPE-PEG2000 enthielten..................................................................................................................... 124
Tabelle 17: Einfluss der molaren Konzentration von DSPE-PEG2000 auf die „Area under the Curve“ von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP)...................................................................................................................................... 128
Tabelle 18: Einfluss des Molekulargewichts von DSPE-PEG auf die „Area under the Curve“ von ligandmodifizierten Liposomen. ............................................................. 132
Tabelle 19: Vergleich von Fluoreszenzintensitäten, die zu verschiedenen Messzeitpunkten in zwei unterschiedlichen Tieren nach einer intrakardialen Injektion von ligandmodifizierten Liposomen bestimmt wurden. ............................................. 143

Einleitung 1
1 Einleitung
Knochenmetastasen sind eine schwerwiegende, in zahlreichen Fällen auftretende Komplikation von Krebs (vor allem Prostata- und Brustkrebs). Hierbei ist Krebs eine chronische Erkrankung, die von genetischer Prädisposition über verschiedene Formen der malignen Entartung der Zelle bis hin zum Fortschreiten der Erkrankung verlaufen kann. Die meisten Tumore entstehen durch sporadisch auftretende Mutationen im Genom von normalen Körperzellen. Die genetischen Veränderungen haben häufig zur Folge, dass Dysregulationen des Zellstoffwechsels, vermehrte Proliferation und weitere genetische Instabilität dieser Zellen auftreten. Im weiteren Verlauf kommt es zur Ausbildung von sogenannten Zellklonen, die sich in ihren molekularen Profilen unterscheiden. Durch fortlaufende Selektion wachsen diese Klone aus und definieren bzw. verändern dadurch den Charakter des Tumors. 60–80% der Patienten, die unter fortgeschrittenem Brust- oder Prostatakrebs leiden, entwickeln in der Regel Knochenmetastasen. Bei Patienten, die Karzinome in der Lunge oder in der Niere aufweisen, entwickeln 30–50% Knochenmetastasen (Greenlee et al., 2001). Sobald Metastasen im Knochen nachgewiesen werden, besteht in den meisten Fällen nach der aktuellen Kenntnislage nur noch geringe bzw. keine Heilungschance mehr. Knochenmetastasen entwickeln sich bereits sehr früh im Verlauf einer Tumorerkrankung; es kommt jedoch erst spät zur Manifestation der Symptome. Die Metastasierung im Knochen führt zu einer deutlichen Verschlechterung der Lebensqualität, welche mit starken Schmerzen, Kompressionen sowie der Entstehung von pathologischen Frakturen einhergeht.
Bisher erfolgt die Behandlung von Tumorerkrankungen bzw. Knochenmetastasen durch den Einsatz von klassischen Methoden, wie der chirurgische Eingriff, die medikamentöse Chemo- und Hormontherapie sowie die Strahlentherapie. Diese Therapieformen stoßen jedoch schnell an ihre Grenzen, d.h., die Therapieerfolge der klassischen Methoden sind eher gering. Hinzu kommt, dass die Strahlen- und die Chemotherapie mit starken Nebenwirkungen einhergehen. Zu diesen Nebenwirkungen zählen insbesondere der antiproliferative Effekt auf normale Zellen (z.B. Haarfollikel, hämatogene Zellen, Schleimhäute und Zellen der Reproduktionssysteme), starke Übelkeit und substanzspezifische Organtoxizitäten.
In den vergangenen Jahren konnten bahnbrechende Erkenntnisse über die Ursachen von Krebs sowie die molekularen, genetischen und zellulären Grundlagen der

Einleitung 2
Tumorentstehung und der sich häufig anschließenden Metastasierung gesammelt werden. Dadurch wurden bereits vielversprechende innovative Therapieansätze, die auf der Applikation von neuen, hochpotenten Wirksubstanzen basieren, entwickelt und getestet. Es stellte sich jedoch heraus, dass die Anwendung dieser neuen Wirkstoffkandidaten häufig aufgrund von ungeeigneten Eigenschaften, wie z.B. hohe Toxizitäten, nicht ausreichende Pharmakokinetikprofile, Instabilitäten oder schlechte Löslichkeiten sehr eingeschränkt ist. Folglich sind nur geringe Mengen am Zielort applizierbar, so dass ein therapeutischer Erfolg in vivo selten nachgewiesen werden kann. Dies wiederum zeigt die dringende Notwendigkeit auf, die Erforschung von weiteren neuen und effektiveren Therapieoptionen fortzusetzen. Vor diesem Hintergrund erscheint die Verwendung von zielgerichteten Trägersystemen als erfolgversprechende Alternative. Durch die Entwicklung von zielgerichteten Trägersystemen können nachteilige Eigenschaften von Substanzen häufig verbessert werden. Diese Trägersysteme bewegen sich in der Regel in einer Größenordnung des Durchmessers von zehn bis einigen 100 Nanometern. Diese sind in der Lage, Wirkstoff in ihr Inneres einzuschließen sowie diesen gezielt an den Ort der Wirkung zu transportieren und ihn dort freizugeben. Zu diesen sogenannten „Nanocarriern“ gehören auch die Liposomen. Aufgrund ihrer vorteilhaften Eigenschaften, wie Bioabbaubarkeit, gute Veträglichkeit, gehören die Liposomen bereits zu den intensiv und erfolgreich untersuchten Trägersystemen. Die Fähigkeit, sich spontan in wässriger Umgebung zu formen und große Mengen an Wirkstoff zu verkapseln, sowie die Möglichkeit, ihre physikochemischen und biologischen Eigenschaften durch Modifikationen zu verändern, machen sie zu vielseitig einsetzbaren Trägersystemen. So können Liposomen auch mit Ligandmolekülen versehen werden, um die Selektivität für das Zielgewebe zu erhöhen. In der Vergangenheit wurde bereits erfolgreich demonstriert, dass dadurch die Konzentration von verkapselten Substanzen am Wirkort erhöht und die Nebenwirkungen auf nicht betroffene Gewebe reduziert werden können.
Aus den gleichen Motiven heraus befasste sich die vorliegende Arbeit ebenfalls mit der Entwicklung von ligandmodifizierten Liposomen. Ziel hierbei war es, ein liposomales Trägersystem für die zielgerichtete Applikation von hochpotenten Wirkstoffen an den Knochen zu entwickeln. Hierdurch sollte ein neuer Therapieansatz zur Behandlung von Knochenmetastasen geschaffen werden. In diesem Zusammenhang wurde in Form einer Machbarkeitsstudie die Eignung von einem neusynthetisierten, maßgeschneiderten Bisphosphonat als knochenaffines Ligandmolekül untersucht. Schwerpunkte dieser Machbarbeitsstudie waren dabei das

Einleitung 3
Design, die Entwicklung sowie die Überprüfung der Eignung des beschriebenen liposomalen Systems für die angedachte zielgerichtete Applikation in vivo. Die vorliegende Arbeit gliedert sich dabei in folgende Kapitel:
Das Kapitel 1 liefert als einleitendes Kapitel zunächst das Hintergrundwissen zum Thema Knochenphysiologie, um das Dissertationsvorhaben im relevanten Kontext darzustellen. Weiterhin wird in diesem Kapitel neben anderen Knochenkrankheiten das Krankheitsbild der Knochenmetastasen näher erläutert und die bisher etablierten Behandlungsmethoden inklusive neuer Therapiestrategien beschrieben. Zudem wird in der Einleitung auf die Substanzklasse der Bisphosphonate detailliert eingegangen, da diese als essentieller Bestandteil des hier vorgestellten Ligandmoleküls eine wichtige Stellung in der vorliegenden Arbeit einnehmen. Zuletzt wird der Stand der wissenschaftlichen Forschung im Hinblick auf andere Trägersysteme, die Bisphosphonate als knochenaffine Liganden aufweisen, und alternative Ligandmoleküle, die ebenfalls aufgrund ihrer hohen Knochenaffinität, das Targeting von Trägersystemen und Wirkstoffen beispielsweise zum Kochen ermöglichen sollen, recherchiert, um den hier vorgestellten neuen Therapieansatz klar zu positionieren. In diesem Zusammenhang wird außerdem die Ligandmodifizierung von Liposomen behandelt.
Das Kapitel 2 fasst die Ziele der Arbeit zusammen. Kernziel der vorliegenden Arbeit war es, ligandmodifizierte Liposomen zu entwickeln, die sich für die Therapie von Knochenmetastasen eignen. Dabei wurde ein neusynthetisiertes, maßgeschneidertes Bisphosphonsäurederivat als knochenaffines Ligandmolekül in die Membran von Liposomen eingebaut und im Rahmen der Machbarkeitsstudie auf seine Tauglichkeit untersucht. Schwerpunkte dabei waren die physikochemische Charakterisierung des liposomalen Trägersystems inklusive In-vitro-Bindungsstudien an Hydroxylapatit sowie die Untersuchung des Pharmakokinetikprofils und die Überprüfung der Fähigkeit in vivo an die Knochenoberfläche in metastasendurchsetztem Knochengewebe binden zu können.
Das Kapitel 3 listet zunächst die in dieser Arbeit eingesetzten Materialien und Geräte auf und geht zuletzt in detaillierter Form auf die Beschreibung der jeweiligen, teils neuentwickelten, Methoden ein.
Das Kapitel 4 präsentiert die hier generierten Daten, aufgeteilt nach Sinnabschnitten, analysiert und bewertet diese in Form von Teildiskussionen.

Einleitung 4
Zuletzt werden in Kapitel 5 die Ergebnisse, die in der vorliegenden Dissertation erzielt wurden, zusammengefasst. Zudem werden in diesem Kapitel mögliche weitere Untersuchungen in Form eines Ausblickes diskutiert.
1.1 Generelle Charakteristika des Knochens
In diesem Abschnitt werden als Einführung in das vorliegende Dissertationsvorhabens zunächst relevante Aspekte der Knochenanatomie erläutert. Dabei wird insbesondere auf den komplexen Prozess der Knochenbildung durch Osteoblasten und die Knochenresorption durch Osteoklasten eingegangen, um geeignete Voraussetzungen für besseres Verständnis des in dieser Arbeit beschriebenen Vorhabens zu schaffen.
1.1.1 Aufbau und Funktion des Knochens
Der Knochen ist ein lebendes und wachsendes Gewebe, welches zu den Binde- und Stützgeweben gehört. Zu den Funktionen des Knochens zählen die mechanische Unterstützung des Körpers sowie der Schutz von Organen, wie z.B. Gehirn und Lunge. Außerdem ist der Knochen der Hauptspeicherort von körpereigenem Calcium und der Ort der adulten Hämatopoese. Grundsätzlich werden zwei Arten von Knochen unterschieden: Geflechtknochen (Substantia Spongiosa) und Lamellenknochen (Substantia Corticalis bzw. Substantia Compacta). Der Geflechtknochen besteht aus netzförmig angeordneten Knochenbälkchen (Trabekel), zwischen denen Knochenmarkräume liegen. Der Lamellenknochen wiederum besteht aus zahlreichen röhrenformigen Osteonen. Diese Osteone sind aus konzentrisch angeordneten Speziallamellen aufgebaut und enthalten einen axial verlaufenden Havers´schen Kanal, der wiederum die Havers´schen Blutgefäße enthält (Abbildung 1). Zusammen mit der quer zur Längsachse der Osteone verlaufenden Volksmannschen Gefäße, die mit den Gefäßen (Vasa Nutritia) in der inneren und äußeren Knochenhaut (Periost) in Verbindung stehen, gewährleisten sie die Versorgung des Knochens mit Nährstoffen, wie Proteine, Mineralien und Sauerstoff. Fast 80% des Knochens bestehen aus dem Lamellenknochen und die verbleibenden 20% werden aus dem Geflechtknochen gebildet (Marcus, 1994). Der Geflechtknochen enthält im Vergleich zum

Einleitung 5
Lamellenknochen keine Havers´schen Blutgefäße, stattdessen weist dieser neben Knochenzellen blutbildendes Gewebe und Fettgewebe auf, welche sich zwischen den Trabekeln befinden. Außen wird der Knochen vom Periost umgeben, einem speziellen Bindegewebe, welches in der Lage ist, neues Knochengewebe zu bilden. Die Innenräume des Knochens sind mit Endost ausgekleidet, das ebenfalls eine osteogene Gewebeschicht ist. Das Endost kleidet auch die Havers´schen Kanäle aus.
Der Knochen als Binde- und Stützgewebe besteht zum einen aus gewebespezifischen Zellen, den knochenbildenen Zellen (Osteoblasten), den knochenabbauenden Zellen (Osteoklasten) und den Osteozyten, den reifen Osteoblasten, zum anderen aus der gewebespezifischen Interzellularsubstanz. Diese Interzellularsubstanz, auch Matrix genannt, lässt sich in eine organische und eine anorganische Phase unterteilen. Die organische Phase besteht zu 90% aus Kollagen (überwiegend Kollagentyp I) und aus nicht kollagenen Proteinen. Diese Phase verleiht dem Knochen Elastizität, Flexibilität und seine Zugfestigkeit (Robey and Boskey, 1996). Beinahe 75% des Trockengewichts des Knochens werden aus anorganischen Calciumsalzen in Form von Hydroxylapatit gebildet. Apatite haben die allgemeine Formel Ca10(PO4)6X2, worin das X ein Fluoratom (Fluorapatit), eine Hydroxylgruppe (Hydroxylapatit) oder ein Chloratom (Chlorapatit) sein kann. Die anorganische Knochenmatrix besteht hauptsächlich aus Kristallen des Hydroxylapatites Ca10(PO4)6(OH)2, welche Raumgitterstrukturen formen. Das Hydroxylapatit liegt in langen, nadelförmigen Kristallen vor (Durchmesser von ca. 70 Å, Länge variierend von 200–3000 Å), welche durch ihre dichte Anordnung zwischen den Kollagenfibrillen der Matrix in der Länge und Orientierung begrenzt werden (Wang et al., 2005). Dieser Bestandteil des Knochens nimmt in der vorliegenden Arbeit eine wichtige Stellung ein, da das Hydroxylapatit das eigentliche Zielgewebe der hier vorgestellten ligandmodifizierten Liposomen darstellt. Aufgrund der Tatsache, dass die Mineralmatrix eine einzigartige Eigenschaft des Knochens ist, stellt dieses Gewebe ein geeignetes Ziel für zielgerichte Applikationen von therapeutischen Systemen dar. Über die Bisphosphonatliganden sollen die Liposomen der vorliegenden Arbeit idealerweise in der Mineralmatrix verankert werden. Die Ligandmoleküle wurden dazu so konzipiert, dass sie unter anderem Bisphosphonatgruppen aufweisen und somit eine wichtige Voraussetzung für eine effektive Komplexierung der Calciumionen des Hydroxylapatits mitbringen. Weitere Bestandteile der anorganischen Matrix sind Karbonate, Natrium, Magnesium und Eisen. Die anorganische Phase ist aufgrund ihrer Zusammensetzung wiederum für die Härte, Steifheit und Druckfestigkeit des Knochens verantwortlich (Shier et al., 1996).
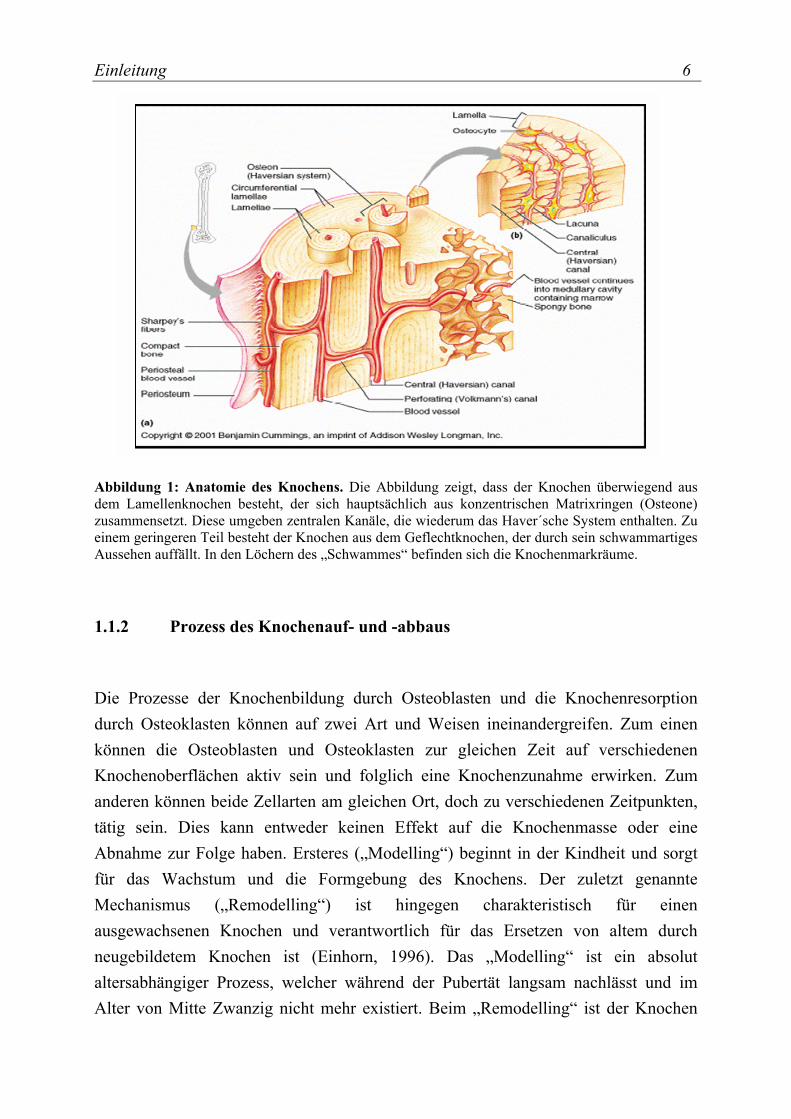
Einleitung 6
Abbildung 1: Anatomie des Knochens. Die Abbildung zeigt, dass der Knochen überwiegend aus dem Lamellenknochen besteht, der sich hauptsächlich aus konzentrischen Matrixringen (Osteone) zusammensetzt. Diese umgeben zentralen Kanäle, die wiederum das Haver´sche System enthalten. Zu einem geringeren Teil besteht der Knochen aus dem Geflechtknochen, der durch sein schwammartiges Aussehen auffällt. In den Löchern des „Schwammes“ befinden sich die Knochenmarkräume.
1.1.2 Prozess des Knochenauf- und -abbaus
Die Prozesse der Knochenbildung durch Osteoblasten und die Knochenresorption durch Osteoklasten können auf zwei Art und Weisen ineinandergreifen. Zum einen können die Osteoblasten und Osteoklasten zur gleichen Zeit auf verschiedenen Knochenoberflächen aktiv sein und folglich eine Knochenzunahme erwirken. Zum anderen können beide Zellarten am gleichen Ort, doch zu verschiedenen Zeitpunkten, tätig sein. Dies kann entweder keinen Effekt auf die Knochenmasse oder eine Abnahme zur Folge haben. Ersteres („Modelling“) beginnt in der Kindheit und sorgt für das Wachstum und die Formgebung des Knochens. Der zuletzt genannte Mechanismus („Remodelling“) ist hingegen charakteristisch für einen ausgewachsenen Knochen und verantwortlich für das Ersetzen von altem durch neugebildetem Knochen ist (Einhorn, 1996). Das „Modelling“ ist ein absolut altersabhängiger Prozess, welcher während der Pubertät langsam nachlässt und im Alter von Mitte Zwanzig nicht mehr existiert. Beim „Remodelling“ ist der Knochen

Einleitung 7
einem ständigen Auf- und Abbau auf der Spongiosa-Oberfläche und in den Haver´schen Kanälen unterworfen, an dem Osteoblasten und Osteoklasten im Zusammenspiel maßgeblich beteiligt sind. Durch die Verformungen des Knochens durch Bewegungen und Beanspruchungen treten Mikrofrakturen auf, die repariert werden müssen (Martin, 2003). Die „Bone Remodelling Unit“ (BRU), zuerst benannt von Frost (Frost, 1964), steuert den Auf- und Abbau-Prozess, welcher insgesamt einen Zeitraum von drei bis vier Monaten in Anspruch nimmt. Es wird angenommen, dass die BRUs durch lokale Stimuli aktiviert werden, da sie örtlich und weitgehend unabhängig voneinander arbeiten. Der neugebildete Knochen wird nach Frost als „Bone Structural Unit“ (BSU) bezeichnet.
Die Knochenresorption beginnt mit der Bindung von aktivierten Osteoklasten an die Knochenoberfläche. Die Bindung an die Oberfläche ist abhängig von spezifischen Integrin-Rezeptoren an der zur Matrix gerichteten Oberfläche der Osteoklasten, die an bestimmte Sequenzen von Matrixproteinen binden (Teitelbaum et al., 1996). An ihrer stoffwechselaktiven Unterseite besitzen Osteoklasten eine spezialisierte Zellmembran, den Bürstensaum, der der Zelle eine große Kontaktfläche zur Knochenoberfläche verschafft. Die Osteoklasten synthetisieren lysosomale Enzyme, wie beispielsweise die tartratresistente saure Phosphatase („Tartrate-resistant Acid Phosphatase“, TRAP) und Kollagenasen, die über den Bürstensaum in den extrazellulären Raum zwischen Knochenmatrix und Bürstensaum ausgeschieden werden. H+-Ionen werden in der Zelle durch das Enzym Carboanhydrase Typ II generiert und mittels einer Ionenpumpe über den Bürstensaum aus der Zelle transportiert (Blair et al., 1989). Die ausgeschiedenen lysosomalen Proteine (Metalloproteinase-1, Kathepsin-K) finden dann das ideale Millieu für ihre proteolytische Aktivität und führen zum Abbau von Kollagenfasern der darunterliegenden Knochenmatrix. Insbesondere das Kathepsin-K ist ein wichtiges Zielenzym für die Hemmung der proteolytischen Wirkung von Osteoklasten (Bossard et al., 1996, Drake et al., 1996). Durch diese resorptive Aktivität produziert der Osteoklast die Howship´schen Resorptionslakunen. Über Aktinfilamente ist der Bürstensaum während des Resorptionsvorganges an der Knochenoberfläche verankert (Baron et al., 1983). Nach Bildung einer Resorptionslakune bewegt sich der Osteoklast über die Knochenoberfläche, um seine Aktivität an anderer Stelle fortzusetzen. Wenn der Osteoklast ruht, dann liegt er in einem Bett von „Lining“-Zellen, den ruhenden Osteoblasten. Sobald der Knochen einen resorptionsstimulierenden Impuls erhält, beginnt die Resorption durch Osteoklasten, indem zunächst die „Lining“-Zellen auseinanderweichen und der Osteoklast den Bürstensaum ausbildet. Osteoklastenaktivierende Substanzen sind

Einleitung 8
beispielsweise Zytokine wie Interleukin-1 und -6, Tumornekrosefaktor-α und -β, Parathormon, transformierender Wachstumsfaktor-α („Transforming Growth Factor-α“, TGF-α) und 1,25-Dihydroxyvitamin D3 (Roodman et al., 1985). Inhibitoren der Osteoklastenaktivität sind unteranderem Calcitonin, Gamma-Interferon und Transformierender Wachstumsfaktor-β („Transforming Growth Factor-β“, TGF-β). Nach Beendigung der Resorptionsphase des „Remodelling“-Prozesses unterliegt der Osteoklast einer morphologischen Apoptose, d.h., dass das Kernchromatin zusammenläuft, der Bürstensaum abgestoßen wird und der Osteoklast sich somit von der Knochenmatrix ablöst. Resorptionshemmende Substanzen, wie Östrogene, Bisphosphonate oder TGF-β, können diese Apoptose initiieren (Hughes et al., 1995). Der Apoptose folgt die Chemotaxis von Osteoblasten sowie deren Weiterentwicklung und Differenzierung, welche dann wiederum die Knochenneubildung nach sich zieht. Dazu werden die Osteoblasten oder ihre Vorläuferzellen durch lokale Faktoren, die während der Resorption freigesetzt wurden, an den defekten Ort gelockt (Yoshikawa, 1998, Raisz, 1999, Troen, 2003). Im Falle von Prekursorzellen schließt sich die Entwicklung und Differenzierung zu reifen Osteoblasten an. Die Resorptionslakunen werden in der Regel vollständig repariert. Der finale Schritt ist das Einstellen der Osteoblastenaktivität, die wahrscheinlich durch Faktoren bewirkt wird, die beim Aufbau freigesetzt werden und diese Aktivität nach und nach herabsenken.
1.2 Knochenmetastasen
Nach einer kurzen Darstellung von verschiedenen und häufig auftretenden Krankheitsbildern, die in der Regel durch den Abbau der Knochenmatrix gekennzeichnet sind, wird im Detail auf die Entstehung von Knochenmetastasen eingegangen. Dabei wird der Prozess der Angiogenese ebenfalls berücksichtigt, da dieser Vorgang für den Fortgang der Metastasierung essentiell ist. Die dadurch gewährleistete Durchblutung ist für die Ansiedlung der ligandmodifizierten Liposomen im metastasierten Knochengewebe möglicherweise auch von Relevanz.

Einleitung 9
1.2.1 Häufig auftretende Knochenerkrankungen
Erkrankungen, die den Knochen betreffen, zeichnen sich durch komplexe Äthiologien aus. Zu den bedeutendsten Knochenerkrankungen zählen Osteoporose, Multiples Myelom, rheumatoide Arthritis und Knochenmetastasen. Alle Krankheitsbilder zeichnen sich neben anderen schwerwiegenden Symptomen durch den Abbau der Knochenmatrix aus. Die Osteoporose wird durch einen Verlust der Knochendichte charakterisiert, welcher zu einer erhöhten Knochenporosität führt. Die Ursache für diese Erkrankung liegt in einem Ungleichgewicht zwischen Knochenauf- und -abbau aufgrund von vermehrtem Abbau durch gesteigerte Osteoklastenaktivität. Dieses Ungleichgewicht ist wiederum in der Regel eine Folge der hormonellen Umstellung des Körpers, die häufig mit einem Östrogenmangel einhergeht, bedingt durch die Wechseljahre. Das Multiple Myelom hingegen gehört zu den B-Zell-Non-Hodgkin-Lymphomen. Es ist gekennzeichnet durch die Proliferation eines neoplastischen Klons von Plasmazellen. Die malignen, differenzierten B-Lymphozyten infiltieren das Knochenmark und hemmen dort die Blutzellbildung. Diese Neoplasie ist unter anderem durch die Gegenwart von osteolytischen Läsionen und Knochenfrakturen als Folge von starker osteolytischer Aktivität charakterisiert. Es wird vermutet, dass Multiple Myelom-Zellen verantwortlich für die Expression von Chemokinen sind, die wiederum den Knochenabbau durch Osteoklasten induzieren. Hyperkalzämie ist zudem häufig eine Folge der durch das Myelom bedingten gesteigerten Knochenresorption und einer eingeschränkten renalen Funktion. Bei der rheumatoiden Arthritis handelt es sich um eine systemische Autoimmunerkrankung, welche hauptsächlich durch die Zerstörung von Knorpel und Knochen gekennzeichnet wird. Eine Vielzahl genetischer und nicht genetischer Faktoren spielt in der Auslösung, aber auch in der Chronifizierung, eine Rolle. Die initiale Immunreaktion, ausgelöst durch ein bisher noch unbekanntes Antigen, ist gefolgt von einer komplexen Kaskade entzündlicher Vorgänge, aus denen lösliche Mediatoren, wie Zytokine und Chemokine, hervorgehen. Diese Mediatoren setzen wiederum Prozesse in Gang, bei denen unter anderem Osteoklasten aktiviert werden, die dann vermehrt den Knochen resorbieren.

Einleitung 10
1.2.2 Entstehung von Knochenmetastasen
Der Prozess der Entstehung von Knochenmetastasen ist sehr komplex. In der Literatur werden dazu zwei Theorien beschrieben. Nach der haemodynamischen Theorie von Ewing (Ewing, 1928) hängt die Entwicklung von Metastasen im jeweiligen Gewebe vom Blutvolumen ab. Diese Theorie fand in manchen Fällen von Darmkrebsmetastasierung Anwendung. Doch in Anbetracht des geringen Blutflusses, der im Knochenmark vorliegt, verglichen mit dem Blutfluß in den Organen, die im geringeren Ausmaß als Zielorgan im Fall von Brust- und Prostatakrebs aufgesucht werden, wie Lunge und Niere, kann die Häufigkeit der Knochenmetastasierung bei diesen Krebstypen nicht einfach erklärt werden (Yoneda, 1998). Der Knochen muss demnach weitere Eigenschaften besitzen, die die Ansiedlung von Metastasen im Knochen begünstigen.
Die „Seed and Soil“-Theorie von Paget (Paget, 1989) scheint dieses Phänomen treffender zu erklären. Die Einzigartigkeit des Knochens im Vergleich zu anderen Zielorganen bei der Streuung von Krebszellen liegt in seiner zellulären und morphologischen Struktur. Der Knochen weist unter anderem eine organische Knochenmatrix auf, die verschiedene Wachstumsfaktoren von osteoblastischer Natur enthält (Paget, 1989, Ribatti et al., 2006, Virk and Lieberman, 2007). Diese Wachstumsfaktoren können als essentielle Nährstoffe von Krebszellen, die sich im Knochen angesiedelt haben, dienen. Nach erfolgter osteoklastischer Knochenresorption im Zuge des physiologischen „Remodelling“-Prozesses werden die Wachstumsfaktoren wahrscheinlich in die Knochenmarkshöhlen freigesetzt. Somit stellt der Knochen eine fruchtbare Umgebung entsprechend der „Seed and Soil“-Theorie dar, welche die Ansiedlung von Mamma- und Prostatakarzinomzellen erklären kann. Diese Theorie zeigt allerdings nicht auf, warum insbesondere diese und nicht andere Krebszelltypen sich dort einnisten. Dies lässt vermuten, dass Prostata- und Brustkrebszellen eine Reihe von intrinsischen Eigenschaften besitzen, die den mehrstufigen Prozess der Entwicklung von Knochenmetastasen, im Zusammenspiel mit der Knochenumgebung ermöglichen. Die spezielle Umgebung in den Sinusoiden des Knochenmarks, charakterisiert durch enge Fenestrae zwischen den Gefäßendothelzellen und der fehlenden Basalmembran, fördert offenbar die Ablagerung von zirkulierenden Tumorzellen, die sich zuvor vom Primärtumor abgelöst haben, in diesen Sinusoiden. Der Prozess wird zudem über

Einleitung 11
Adhäsionsmoleküle und Chemotaxis unterstützt. Es beginnt dadurch der Prozess der Knochenmetastasierung (Abbildung 2) (de Bruyn et al., 1981, Buckwalter et al., 1996, Yoshino et al., 1996, Havens et al., 2006). Durch die Sekretion von proteolytischen Enzymen können die Tumorzellen die sinusoidale Wand durchbrechen und ins Knochenstroma eindringen. In der frühen Phase ihrer Lokalisierung im Knochen binden die neoplastischen Zellen über Adhäsionsmoleküle an das Stroma des Knochenmarks und im weiteren Verlauf an die mineralisierte Matrix des trabekulären Knochens (Bonucci, 2002). Der letzte Schritt in der Kaskade der Knochenmetastasierung ist die Zerstörung der Knochenstruktur (Evans, 1992, Nicolson, 1993, Guise et al., 2005, Raubenheimer and Noffke, 2006). Krebszellen können den Knochen auf zwei Arten beschädigen: durch osteolytische oder osteoblastische (osteosklerotische) Metastasierung. Mammakarzinomzellen entwickeln überwiegend osteolytische Knochenmetastasen, wohingegen Prostatakarzinomzellen zumeist osteoblastische Metastasen entwickeln. Bei der osteolytischen Läsion führen die Metastasen zu einem Ungleichgewicht des „Remodelling“-Vorganges, indem sie eine Erhöhung der Osteoklastenaktivität bewirken. Die Osteoblastenaktivität hingegen bleibt unverändert, so dass ein gesteigerter Knochenabbau auftritt. Bei der blastischen Knochenmetastasierung werden hauptsächlich die Osteoblasten aktiv, ohne dass eine vermehrte Knochenresorption zuvor stattgefunden hat. Folglich kommt es zur gesteigerten Knochenbildung.
Da die metastasierenden Tumorzellen sich nach dem Austritt aus der Blutbahn häufig zunächst im Knochenmark in Gegenden mit hoher Vaskulariät und hoher „Remodelling“-Raten ansiedeln und zumeist nach weiterer Proliferation erst an die Knochenoberfläche gelangen (Bonucci, 2002), fokussiert dieser Ansatz auf die Behandlung von Knochenmetastasen, die sich bereits im fortgeschrittenen Stadium befinden. Mit anderen Worten: dieser Strategie liegen das Ziel der Lebensverlängerung und die Verbesserung der Lebensqualität zugrunde.

Einleitung 12
Abbildung 2: Entstehung von Knochenmetastasen. Zunächst lösen sich die Tumorzellen vom Primärtumor ab und gelangen zumeist über neugebildete Blutgefäße in die zentralen Blutgefäße. Nach Erreichen der Sinusoide der Knochengefäße binden die Tumorzellen an die Endothelzellen dieser Gefäße und sind durch die Sekretion proteolytischer Enzyme in der Lage, die Endothelzellwand zu durchbrechen und somit in das Knochengewebe einzutreten. Im Zuge ihrer Vermehrung und ihres Wachstums sind sie in der Lage, die für diese Prozesse notwendige Neoangiogenese zu induzieren. Zudem kann es im weiteren Verlauf zu osteoblastischen oder zu osteolytischen Läsionen kommen.
1.2.3 Prozess der Angiogenese
Um das Wachstum der Krebszellen am Ort der Metastasierung zu ermöglichen, ist eine Reorganisation von Endothelzellen und ihren adhäsiven Eigenschaften für die notwendige Angiogenese erforderlich (Howe et al., 1998, Sanders et al., 1998, Nikitenko and Boshoff, 2006) (Abbildung 3). Die ebenfalls für die Metastasierung notwendige Proliferation der neoplastischen Zellen muss sich proportional zur Neubildung von Blutgefäßen verhalten, um eine ausreichende Versorgung der Zellen mit Nährstoffen zu gewährleisten. Doch in den meisten Fällen kann diese Balance nicht aufrechterhalten werden, da die Zellreplikation oft schneller als die Blutgefäßbildung verläuft. Bereits vor 30 Jahren erkannte Folkman (Folkman, 1971, Folkman, 1975), dass eine Versorgung der Tumorzellen mit einem Blutgefäßsystem unerlässlich für das Wachstum und die Metastasierung von soliden Tumoren ist. Heute ist bekannt, dass die Angiogenese durch einen komplexen, kaskadenartig ablaufenden

Einleitung 13
Vorgang charakterisiert ist, der durch intensive Interaktionen zwischen angiogenen und antiangiogenen Faktoren und der Extrazellulärmatrix („Extracellular Matrix“, ECM) maßgeblich bestimmt wird (Carmeliet, 2000, Jain, 2003). Die Angiogenese wird durch angiogene Faktoren stimuliert. Dabei werden Faktoren, wie der TGF-β, der vaskuläre endotheliale Wachstumsfaktor („Vascular Endothelial Growth Factor“, VEGF), der Plättchen-Wachstumsfaktor („Platelet-derived Growth Factor“, PDGF) und der Fibroblast-Wachstumsfaktor („Fibroblast Growth Factor“, FGF) (Connolly et al., 1989) (Inhibierung dieser Faktoren erfolgt durch Thrombospondin, Protamin, Interferon und Steroiden) von den neoplastischen Zellen freigesetzt, um die Endothelzellen der Kapillargefäße zu aktivieren. Die angeregten Endothelzellen wiederum produzieren dann Proteasen (Gelatinase A und B) (Nguyen et al., 2001) und andere proteolytische Enzyme wie Matrixmetalloproteinasen („Matrix Metalloproteinases“, MMP) und Heparanasen. Diese lösen die Basalmembran auf und bauen die umliegende Extrazellulärmatrix ab. Dadurch ermöglichen sie den Endothelzellen die Migration in das umgebende Gewebe (Kalluri, 2003). Zelladhäsionsproteine, wie Integrine, fördern dabei die Aussprossung eines neuen Gefäßes durch die ECM in Richtung Tumor. Die darauf folgende Proliferation von neuen Endothelzellen führt nach weiterem Endothelzellenwachstum, Kavitation und der Produktion von einer neuen Basalmembran zur der Neubildung eines Kapillargefäßes (Diaz-Flores et al., 1994). Das Resultat einer vollständig ablaufenden angiogenen Kaskade ist ein funktionierendes Blutgefäßsystem, das die Versorgung des Tumors mit Sauerstoff und Nährstoffen garantiert.
Um die notwendige Versorgung auch weiterhin gewährleisten zu können, bleibt der Prozess der Angiogenese während des Tumorwachstums erhalten. Mit anderen Worten: sobald ein Bereich des Tumors hypoxisch oder nekrotisch wird, wird der Vorgang der Gefäßneubildung wieder aktiviert (Carmeliet, 2003). Es wird vermutet, dass die Neoangiogenese die Ansiedlung der ligandmodifizierten Liposomen entscheidend begünstigt. Es ist bereits bekannt, dass kleine Liposomen in der Lage sind, durch das löchrige Endothelium von unvollständig ausgebildeten Blutgefäßen, die Tumore versorgen und im Rahmen von tumorinduzierter Angiogenese entstanden sind, zu diffundieren und aufgrund von mangelnder Lymphdrainage im Tumorgewebe zu verweilen. In diesem Zusammenhang spielen die fenestrierten Blutgefäße im Knochenmark und der hohe „Bone Turnover“, bedingt durch die Metastasierung, sicherlich ebenfalls eine wichtige Rolle.

Einleitung 14
Abbildung 3: Ablauf der angiogenen Kaskade. Die Angiogenese beschreibt den Prozess der Entwicklung von neuen Blutgefäßen zur Versorgung eines Tumors. Generell kann die angiogene Kaskade in drei Schritte unterteilt werden: 1. Proliferation der Endothelzellen; 2. Zusammenbruch der extrazellulären Matrix; 3. Migration der Endothelzellen (Quelle: www.amplab.de/3D-Images/angiogenesis-scheme.gif).
1.3 Behandlung von Knochenmetastasen
Dieser Abschnitt fast zunächst die gängigen, etablierten Therapiemöglichkeiten zur Behandlung von Knochenmetastasen zusammen. Zudem wird der Stand der wissenschaftlichen Forschung im Hinblick auf neue Therapiestrategien zur Behandlung von Tumorerkrankungen, darunter auch Knochenmetastasen, dargestellt, um den hier präsentierten Ansatz klar zu positionieren.

Einleitung 15
1.3.1 Klassische Ansätze zur Therapie von Knochenmetastasen
Neben dem operativen Eingriff, bei dem entweder Tumorzellen entfernt werden, um möglichen Frakturen vorzubeugen oder chirurgisch Metallstäbe, Platten sowie Schrauben eingesetzt werden, gehören die nachfolgenden Optionen heutzutage zu den klassischen Therapieansätzen.
1.3.1.1 Strahlentherapie
Bei der Strahlentherapie werden Krebszellen mit ionisierender Strahlung (X-Strahlen) von hoher Energie beschädigt oder zerstört. Studien zeigen, dass eine Einmaldosis von 8 Gray ausreichend ist, Knochenschmerzen, die als Folge von Metastasen hervorgerufen werden, zu lindern (Janjan, 1997). Bei 70–80% der Patienten stellte sich eine Verbesserung der Symptomatik ein (Hoskin et al., 1992). Ursachen für Schmerzen bei denen die Strahlentherapie indiziert ist, können beispielsweise pathologische Frakturen oder neurologische Komplikationen, wie Rückenmarkskompressionen sein. Eine andere Art der Strahlenbehandlung, auch radiopharmazeutische Therapie genannt, umfasst die i.v.-Injektion von radioaktiven Isotopen, wie Strontium-89, Samarium-153 und Phosphor-32, welche sich im Knochen anreichern. Sie werden ebenfalls zur Linderung der durch Metastasen bedingten Knochenschmerzen eingesetzt. Die zu dieser Art der Therapie eingesetzten Isotope sind β-Strahler, d.h., dass ihre ionisierende Strahlung von niedriger Energie ist, um die Beschädigung von umliegenden, nicht betroffenden Gewebe möglichst zu vermeiden. Der β-Emitter Phosphor-32 reichert sich aufgrund seiner chemischen Struktur im Knochenmark, im trabekulären und kortikalen Knochen an (Lewington, 1993). Das Verhältnis zwischen der Aufnahme in den Tumor und der Aufnahme in den normalen Knochen nach einer intravenösen Applikation beträgt nur 2:1, so dass dieser Strahler nicht so effektiv ist (Lewington, 1993). Strontium-89 (89Sr) wird wegen seiner Ähnlichkeit zu Calcium in den Knochen und zwar hauptsächlich in die Mineralstruktur eingebaut. Von der i.v.-applizierten Dosis werden ca. 50% des 89Sr im Knochen angereichert (Blake et al., 1987). Das therapeutische Verhältnis zwischen der Anreicherung im Tumor und der Anreicherung im roten Knochenmark beträgt 10:1 (Blake et al., 1988). Der β-Strahler Samarium-153 (153SM) wurde bisher an

Einleitung 16
verschiedene knochenaffine Bisphosphonatliganden gekoppelt, um den Transport zum Knochen zu gewährleisten. 153SM gebunden an Ethylen-diamintetramethylenphosphonat (Quadramet®) wurde bereits von der FDA für die Therapie von Metastasen bedingten (osteoblastischer Natur) Schmerzen zugelassen. Ungefähr die Hälfte der verabreichten Dosis bindet aktiv an das Hydroxylapatit und zwar bevorzugt in Gegenden von erhöhter osteoblastischer Aktivität (Bayouth et al., 1994). Das Verhältnis zwischen der Aufnahme in die metastasierten Bereiche und der Aufnahme in den Knochen beträgt 17:1 (Goeckeler et al., 1987).
1.3.1.2 Hormontherapie
Eine Option der systemischen Therapie ist die Hormontherapie. Der Hintergrund dieser Therapieform ist die hormonell bedingte Stimulierung des Tumorwachstums zu unterdrücken. Im Falle von Brustkrebs kann dies durch die operative Entfernung der jeweiligen Quelle der Oestrogen- und der Gonadotropinausschüttung erreicht werden. Dabei erfolgt eine Eierstockentnahme oder Nebennierenentfernung bzw. eine Sektomie der Hypophyse. Eine andere Möglichkeit, das hormonell bedingte Tumorwachstum zu hemmen, ist die Aktivität der Gonadotropine durch die Gabe von Luteinisierendes Hormon-Freisetzendes Hormon (Luteinizing Hormone-Releasing Hormone, LH-RH)-Agonisten, wie Goserelin zu inhibieren. Zudem können die Oestrogenrezeptoren durch Tamoxifen blockiert werden, Androgene die Oestrogene antagonisieren und die Oestrogensynthese durch Aromatasehemmer unterbunden werden. Im Falle von Prostatakrebs basiert die hormonale Therapie auf der Inhibierung der androgenen Stimulation des Tumorwachstums. Am häufigsten wird die bilaterale Orchiedektomie durchgeführt. Außerdem werden LH-RH-Agonisten (Leuprorelide, Buserelin und Goserelin) und Antiandrogene, wie Cyproteronacetat) eingesetzt.

Einleitung 17
1.3.1.3 Chemotherapie
Bei der Therapie von Brustkrebs haben sich zytotoxische Substanzen, wie Anthrazykline (Doxorubizin, Epirubizin), Cyclophosphamide, Methotrexat, 5-Fluorouracil, Mitomycin C und Mitoxantron, bewährt. Häufig werden diese Wirkstoffe auch in Kombination eingesetzt. Gängige Beispiele hierfür sind: CMF (Cyclophosphamid, Methotrexat und 5-Fluorouracil), MMM (Mitomycin C, Methotrexat und Mitoxantron und FAC (5-Fluorouracil, Doxorubizin und Cyclophosphamid). In Fällen von bestehender Anthrazyklin-Resistenz werden heute auch Taxole, wie Paclitaxel und Docetaxel erfolgreich eingesetzt. Doch bisher konnte nicht gezeigt werden, dass die Chemotherapie die Überlebenschance des einzelnen Patienten erhöht. Es stellt lediglich, wie die Mehrheit der anderen aufgeführten Therapieoptionen, eine palliative Alternative dar. Bei der Überlegung, Chemotherapeutika (Zytostatika) in die Therapie mit einzubeziehen, sind die schweren Nebenwirkungen, die diese Substanzen hervorrufen können, zu berücksichtigen. Chemotherapeutika wirken sich besonders negativ auf Gewebe mit hohen Proliferationsraten aus. Somit werden nicht nur die Krebszellen angegriffen sondern es werden auch die Zellen der Haut, Haare und Schleimhäute sowie des Knochenmarks geschädigt. Als Folge kommt es zur Wachstumshemmung dieser Zellen und somit zu Nebenwirkungen, wie z.B. Haarausfall sowie Leuko- und Thrombopenien. Magen-Darm-Störungen, die zumeist mit Übelkeit und Erbrechen einhergehen, gehören ebenfalls zu den unerwünschten Begleiterscheinungen. Eine Möglichkeit diese Nebenwirkungen zu unterdrücken, besteht sicherlich in der Gabe von den hochwirksamen antiemetischen 5-Hydroxytryptamin(5HT3)-Inhibitoren, wie Ondansetron und Granisetron. Bei der Behandlung von Prostatakrebs haben Chemotherapeutika bisher keinen relevanten Erfolg gezeigt.
1.3.2 Neue Strategien zur Behandlung von Tumorerkrankungen
Neue und effektivere Strategien zur Therapie von Tumorerkrankungen und somit in spezifischen Fällen auch zur Therapie von Knochenmetastasen werden stetig weiterentwickelt und auf ihre Wirksamkeit überprüft. Dabei wird unter anderem versucht, bereits bekannte Chemotherapeutika durch die Erprobung von neuen

Einleitung 18
Kombinationen und neuer Verabreichungsschemata effizienter zu nutzen. Außerdem wird angestrebt, die Wirksamkeit durch die Modifikation bekannter Medikamentenstrukturen und durch das Auffinden von völlig neuen Wirkstoffkandidaten zu steigern. Dabei werden mittels „Ultra High Throughput Screening“ biologisch aktive Substanzen aufgespürt und ihre Struktur vor der Synthese durch Molekulares Modellieren so modifiziert, dass sie möglichst genau in die Schlüsselstruktur des biologisch ausgewählten Zielmoleküls passen.
Absolut neue Therapiestrategien setzen auf die Entwicklung von Medikamenten, die auf einzelne molekulare Tumorprofile eingehen. Eine maßgeschneiderte Therapie, basierend auf individuellen molekularen Marker-Expressionsprofilen, zusammen mit ausgereiften Diagnosetechniken, wie Enzymimmunoassays („Enzyme-linked ImmunoSorbent Assay“, ELISA), Immunhistochemische Analyse (IHC) und Polymerasekettenreaktion-Tests („Polymerase Chain Reaction“, PCR) versprechen höhere durchschnittliche Ansprechraten, Überlebensraten sowie auch eine Verbesserung der Lebensqualität infolge verringerter Morbidität. Ein Beispiel für einen Ansatz der individuell angepassten Behandlung von Brustkrebspatientinnen basierend auf therapieentscheidender Diagnostik ist die Tumoranalytik des humanen epidermalen Wachstumsfaktor Rezeptors, Typ 2 („Human Epidermal Growth Factor Receptor 2, HER2 receptor“), welcher sowohl das Tumorwachstum als auch die Metastasierung stimuliert. Um diesen Rezeptor in seiner Aktivität zu hemmen, wurde ein gentechnisch hergestellter monoklonaler Antikörper (Trastuzumab, Herceptin®) entwickelt, der an HER2-Rezeptoren bindet und dadurch die Tumorzellproliferation hemmt. Diese Therapieform ist allerdings nur wirksam, wenn die HER2-Rezeptoren im Übermaß auf der Oberfläche von Brustkrebszellen exprimiert werden. Dieses ist aber nur in etwa 25% der Brusttumoren der Fall. Deshalb ist vor Therapiebeginn der Rezeptorstatus durch Methoden, wie IHC und ELISA-Tests zunächst festzustellen.
Ein weiterer neuer Ansatz zur Bekämpfung von Krebs beinhaltet die Aktivierung des Immunsystems. Dabei sollen Tumorvakzine als neue Klasse aktiver und spezifischer Immuntherapeutika eine gezielte Immunantwort gegen Krebs auslösen. Patienten mit malignen Krankheiten weisen häufig Defizite in ihrer zellulären Immunität auf, und zwar unabhängig von der immunsuppressiven Wirkung von Chemo- oder Radiotherapeutika. Immunsuppression entsteht während der embryonalen Entwicklung (Sotomayor et al., 1996). Dieses Phänomen kann unter anderem eine Folge von fehlenden tumorassoziierten Antigen(TAA)-spezifischen T-Zellen (Sotomayor et al., 1996) sein oder durch die Produktion von immunsuppressiven Faktoren, wie TGF-β

Einleitung 19
und IL-10, die zur Beeinträchtigung der eigenen Immunantwort auf Tumorantigene führt (Wojtowicz-Praga, 1997, Mocellin et al., 2001), ausgelöst werden. Ziel der Tumorimpfstoffe ist, daß sie immunogene Antigene erreichen, die zumeist auf der Oberfläche von Tumorzellen exprimiert werden. Stimuvax® (liposomales Vakzin BLP25) ist beispielsweise entwickelt worden mit der Intention, eine Immunreaktion auf das tumorassoziierte Antigen MUC-1, welches bei epithelialen Tumoren wie z.B. Brust-, Prostata- oder Lungenkrebs überexprimiert wird, zu bewirken. Eine synthetische Lipopeptidsequenz, die einem Teil des Glykoproteins MUC-1 identisch ist, ist für die Immunantwort verantwortlich, wohingegen die Verkapselung in Liposomen den Transport des Impfstoffes zu den antigenpräsentierenden Zellen verbessern soll. Ein Problem der Immuntherapie ist, dass das Immunsystem Krebszellen nicht als fremd erkennt, da diese sich von den normalen Zellen kaum unterscheiden. Somit muss das Tumorvakzin nicht nur eine Immunantwort hervorrufen, sondern auch das Immunsystem in dem Ausmaß stimulieren können, bis es Tumorzellen nicht weiter toleriert sondern attackiert. Deshalb werden Tumorvakzine so beschaffen sein, dass sie erkennbar für das Immunsystem sind. Diese Veränderung umfasst beispielsweise eine Strukturänderung des einzusetzenden Tumorantigens, indem das Gen, welches zu seiner Synthese benötigt wird, zuvor modifiziert wird. Eine andere Strategie sieht auch hier die Applikation eines in einen viralen Vektor verkapselten Genes vor, das zur Synthese eines Tumorantigens vorgesehen ist. Infizierte Krebszellen produzieren dann wesentlich mehr Tumorantigene als nicht infizierte Zellen. Sie werden somit idealerweise für das Immunsytem sichtbarer gemacht. In den letzten Jahren wurde ebenfalls gezeigt, dass die Beladung von antigenpräsentierenden Zellen, den sogenannten dentritischen Zellen ex vivo (DC-Vakzine), eine besonders effiziente Möglichkeit ist, starke Impfreaktionen auszulösen. Derzeit werden zahlreiche Vakzine, die auf verschiedenen Wirkungsmechanismen basieren, in klinischen Studien getestet, um die effektivste Strategie für die einzelnen Tumorarten zu bestimmen (Ueda et al., 2004, von Minckwitz et al., 2005). Trotz der vielversprechenden Ergebnisse gilt es weiterhin, das Tumorvakzin zu finden, das zu langanhaltenden Antworten und zu der vollständigen Remission der Tumorzellen führt.
Ein anderer innovativer Ansatz zur Inhibierung der Entwicklung von Knochenmetastasen beinhaltet die antiangiogene Therapie. Die Hemmung der Bluversorgung eines wachsenden Tumors ist bereits ein gut untersuchter Ansatz in der Tumortherapie. Der umfangreiche Prozess der Angiogenese bietet zahlreiche Angriffspunkte für potentielle Angiogenesehemmer. Ziel der antiangiogenen Therapie

Einleitung 20
ist es, den Tumor indirekt zu zerstören, indem die Nährstoffversorgung und der Abtransport von Stoffwechselmetaboliten durch die Inhibierung der Gefäßneubildung unterbunden werden. Ein Vorteil dieser Art von Behandlung ist, dass die Endothelzellen, die bei der Angiogenese eine sehr wichtige Rolle spielen und somit ebenfalls von hoher Bedeutung bei der Entwicklung antiangiogener Therapeutika sind, selten genetischen Alterationen ausgesetzt sind. Sie stellen deshalb ein homogenes Ziel dar, wohingegen Tumorzellen zumeist genetisch instabile Zellen sind. Es konnten außerdem bereits eine Vielzahl an antiangiogenen Faktoren und Substanzen ausgemacht werden. In diesem Zusammenhang konnten Peyruchaud et al. (Peyruchaud et al., 2003) zum Beispiel in vivo darstellen, dass Angiostatin durch seine antiangiogene Aktivität die Bildung von osteolytischen Knochenmetastasen, die durch Brustkrebs verursacht wurden, verhinderte. Außerdem ist es ihnen gelungen zu zeigen, dass Angiostatin einen direkten, inhibierenden Einfluss auf die Funktion von Osteoklasten hat. Angiostatin ist ein Fragment des Plasminogens und zählt neben Endostatin zu den endogenen Inhibitoren der Angiogenese. Sie hemmen beide selektiv die Endothelzellproliferation, doch der genaue Wirkmechanismus ist bisher noch nicht vollständig geklärt.
Eine völlig andere Strategie, die wiederum spezifisch für die lokale Behandlung von Knochenerkrankungen ist und möglicherweise auch für die Behandlung von Knochenmetastasen eingesetzt werden könnte, beschreibt die Verwendung von modifizierter, implantierbarer Knochensubstanz in Form von Hydroxylapatit. Verschiedene Arbeitsgruppen untersuchten in diesem Zusammenhang Gerüste, Mikrosphären oder Partikel bestehend aus Hydroxylapatit als mögliches implantierbares oder subkutan applizierbares Trägersystem von Wirksubstanzen, wie Antibiotika (Shinto et al., 1992), Chemotherapeutika (Itokazu et al., 1998), Proteine (Mizushima et al., 2006) oder Wachstumsfaktoren (Okubo et al., 2000, Zambonin et al., 2000) zum Knochen. Es hatte sich vorher herausgestellt, dass Hydroxylapatit sich aufgrund seiner hohen Biokompatibilität gut zur Substitution von Knochenmaterial eignet. Dazu wurde häufig die Kombination von biodegradierbaren Polymeren, wie Polylaktidsäure („Polylactid acid“, PLA) (Castro et al., 2003), Polyglykolsäure („Polyglycolic acid”, PGA) (Overbeck et al., 1995) oder Poly(laktid-co-glykolsäure) („Poly(lactid-co-glycolic acid)”, PLGA) (Schnieders et al., 2006) mit HAP untersucht. Die In-vitro- und In-vivo-Versuche dieser Systeme haben bisher vielversprechende Ergebnisse im Hinblick auf die Freisetzungsprofile der jeweils untersuchten Substanzen erzielt. Die Wirksamkeit dieser Therapiestrategie gilt es jedoch in vivo noch aufzuzeigen. Zusammenfassend ist zu sagen, dass bereits zahlreiche potentielle

Einleitung 21
Targets (biochemischer Reaktionsweg, Enzyme, Transkriptionsfaktoren) aus biologischen Prozessen der Angiogenese, Tumorentstehung und Metastasierung untersucht worden sind. Die damit verbundene gezielte Beeinflussung der bei Krebs außer Kontrolle geratenen molekularen Mechanismen führte zu vielversprechenden Ergebnissen. Dennoch ist die Mehrzahl der Therapieansätze auf molekularer Ebene noch in weiter Ferne, so dass die heute klassischen Grundpfeiler der Krebstherapie (Strahlentherapie, chirurgische Entfernung des Tumors, Zytostatikabehandlung) nach wie vor erhalten bleiben.
Mit den in dieser Arbeit vorgestellten ligandmodifizierten Liposomen wird ebenfalls eine neue Form der Therapie von Knochenmetastasen verfolgt. Dabei soll durch die Verkapselung von klassischen, zytostatischen Wirkstoffen in das zielgerichtete, vesikuläre System die hohe Substanztoxizität in sensitivem, gesundem Gewebe vermindert und die niedrige Selektivität für die Tumorregion erhöht werden. Um die Spezifität für das Zielgewebe zu erhöhen, wurden Bisphosphonate als knochenaffine Liganden in die liposomale Membran eingebaut. Der in der vorliegenden Arbeit präsentierten Targeting-Strategie liegt die Theorie zugrunde, eine bewährte und hochwirksame Therapie (Zytostatikabehandlung) durch die Kombination mit einer neuen Technologie, nämlich ligandmodifizierten Liposomen, die auf ihrer Oberfläche maßgeschneiderte Bisphosphonatliganden tragen, verbessern zu können. Dabei hat uns die Tatsache, dass Liposomen sicherlich als bereits umfangreich untersuchtes Trägersystem erachtet werden können, darin bestärkt, diese als Drug-Delivery-System für das hier vorgestellte Konzept auszuwählen. Liposomen gelten in der Regel als untoxisch und bringen durch ihre biodegradierbaren Eigenschaften geeignete Voraussetzungen für ein ideales Trägersystem mit. Im Vergleich zu den zuvor genannten neuen Therapieansätzen sehen wir bei den ligandmodifizierten Liposomen den klaren Vorteil, dass diese möglicherweise, abhängig vom verkapselten Wirkstoff, als breit anwendbares Therapeutikum einzusetzen sind. Die anderen beschriebenen Ansätze sind häufig spezifisch für bestimmte Rezeptoren oder Antigene auf Tumorzellen, wohingegen die ligandmodifizierten Liposomen mit Hilfe der Bisphosphonatliganden zwar spezifisch an die Knochenmatrix binden, doch von dort aus durch den freigesetzten Wirkstoff auf die umliegenden Metastasen wirken sollen. Das Wirkpotential (Sensitivität für die metastasierten Zellen, z.B. Mammakarzinomzellen oder Prostatakarzinomzellen) der verkapselte Substanz bestimmt dann die Wirksamkeit des applizierten Systems. Nach erfolgreicher Demonstration der Fähigkeit, von ligandmodifizierten Liposomen an die Mineralmatrix im metastasierten Knochengewebe binden zu können, könnten diese

Einleitung 22
möglicherweise eine Plattform für die zielgerichtete Applikation von verschiedenen Arzneistoffen an den Knochen darstellen. Die Verkapselung von anderen Substanzen, wie beispielsweise Angiogenesehemmer oder Gentherapeutika, wären alternativ auch denkbar.
1.4 Bisphosphonate
Dieser Abschnitt hebt die Substanzklasse der Bisphosphonate, die in der vorliegenden Arbeit Bestandteil der Targeting-Liganden sind, hervor. Dabei wird zunächst die Struktur dieser Derivate und ihre Bindung an die Mineralmatrix des Knochens näher beschrieben. Außerdem wird ihr Wirkmechanismus im Detail dargestellt. Zuletzt wird auf die derzeitigen Indikationen einschließlich der Nebenwirkungen und Kontraindikationen der Bisphosphonate eingegangen.
1.4.1 Struktur
Bisphosphonate zeichnen sich durch ihre geminale Struktur aus, d.h., dass beide Phosphoratome an das gleiche Kohlenstoffatom gebunden sind. Durch diese Struktur sind sie Analoga des Pyrophosphats, welches anstelle des Kohlenstoffatoms ein Sauerstoffatom aufweist (Abbildung 4).
Abbildung 4: Strukturformel von Pyrophosphat und Bisphosphonat (Quelle: Fleisch, 1998).

Einleitung 23
Die Forschung auf dem Gebiet der Bisphosphonate begann 1968 durch Herbert Fleisch. Seine Arbeitsgruppe entdeckte in den Jahren zuvor, dass anorganisches Pyrophosphat in biologischen Fluiden, wie Plasma und Urin, die Präzipitation von Calciumphosphat verhinderte (Fleisch and Neuman, 1961, Fleisch and Bisaz, 1962). Sie entdeckten, dass Pyrophosphat den Kristallisationsvorgang, die Aggregation und die Auflösung von Calciumphosphatkristallen beeinträchtigte (Fleisch et al., 1966). In In-vivo-Versuchen stellte sich heraus, dass das Pyrophosphat bei parenteraler Gabe ektopische (nicht an typischen Stellen verursachte) Verkalkungen inhibierte und dass es in der Regulation der Knochenresorption involviert war (Schibler et al., 1968). Diese Effekte waren bei oraler Applikation nicht zu beobachten. Ein weiterer Nachteil des Pyrophosphats war die Hydrolyseempfindlichkeit nach parenteraler Gabe, so dass die Suche nach einem analogen, aber stabileren Derivat begann. Zunächst ergab die Untersuchung der Bisphosphonate durch Fleisch et al., dass diese resistent gegenüber enzymatischer Hydrolyse sind und, dass sie dennoch einen ähnlichen Einfluss auf Calciumsalze ausüben. Sie sind dazu geeignet, die ektopischen Verkalkungen und die Knochenresorption zu reduzieren (Fleisch et al., 1970, Shinoda et al., 1983), indem sie an die Knochenoberfläche (Hydroxylapatit) binden. Die P-C-P-Bindung ist in jedem Bisphosphonat-Derivat für die hohe Affinität zum Hydroxylapatit verantwortlich. Diese Art von Bindung stellt die Grundlage der hier vorgestellten Targeting-Strategie dar. Die Substitution der Reste R1 und R2 (Abbildung 4) durch verschiedene Gruppen, verleiht dem einzelnen Derivat unterschiedliche Eigenschaften. Die Fähigkeit, an das Hydroxylapatit zu binden und das Kristallwachstum bzw. die Kristallauflösung von Calciumphosphat zu inhibieren, ist beispielsweise erhöht, wenn R1 durch eine Hydroxylgruppe (OH-Gruppe) ersetzt wird (Etidronat) (Ebetino et al., 1998). Die Substitution von R1 durch die OH-Gruppe führt außerdem dazu, dass ein effektiverer Chelatkomplex zwischen den Calciumionen der Knochensubstanz und den Bisphosphonaten ausgebildet werden kann, da die OH-Gruppe eine Tridentatbindung anstelle einer Bidentatbindung ermöglicht (Abbildung 5) (Ebetino et al., 1998). Um das Bindungspotential von ligandmodifizierten Liposomen an Calciumionen zu erhöhen, wurde diese OH-Gruppe in dem Rest R1 auch bei der Synthese des in dieser Arbeit untersuchten Ligandmoleküls berücksichtigt. Es gibt viele Bisphosphonate, die an der Stelle R2 mit stickstoffhaltigen Gruppen substituiert sind (Pamidronat, Alendronat, Ibandronat). Die Variation der stickstoffhaltigen Reste an dieser Stelle hat einen positiven Einfluss auf die antiresorptive Fähigkeit. Die Substitution von R2
bestimmt die biologische Aktivität des Derivates. Je größer der Rest, desto höher die antiresorptive Potenz (Nancollas et al., 2005). Da bei dem hier vorgestellten

Einleitung 24
Ligandmolekül nicht die pharmakologische Wirkung, sondern das Bindungspotential an den Knochen im Vordergrund steht, wurde die Substitution von R2 durch Stickstoffgruppen nicht in Betracht gezogen. Die Stelle R2 des Liganden wurde stattdessen durch einen Cholesteryl-trisoxyethylen-Rest substituiert, der als Ankermolekül den Einbau des Liganden in die liposomale Membran gewährleisten sollte. Um die Knochenresorption inhibieren zu können, hat sich herausgestellt, dass die P-C-P-Struktur unerlässlich ist. Denn Versuche mit Monophosphaten, mit P-C-C-P-Derivaten oder Substanzen, die eine P-N-P-Gruppe aufwiesen, das Ausmaß der Knochenresorption zurückzufahren, blieben erfolglos (Russell et al., 1970).
Abbildung 5: Räumliche Darstellung von Komplexbildung zwischen einem Bisphosphonat und einem Calciumatom der Knochenoberfläche. Die unterschiedlichen Substitutionen der Reste R1 und R2 sind verantwortlich für die Wirkstärke und die Pharmakokinetik der Bisphosphonate (Bartl, 2001).
1.4.2 Wirkmechanismus
Bisphosphonate inhibieren die Knochenresorption, indem sie in die Knochenmatrix eingebaut werden und durch ihre Bindung an die mineralische Oberfläche von Osteoklasten internalisiert werden. Diese Internalisierung von Bisphosphonaten könnte bei der Freisetzung des hier beschriebenen liposomalen Trägersystems möglicherweise von Relevanz sein. Während der Knochenresorption durch Osteoklasten werden die Bisphosphonatgruppen des Targeting-Liganden mit hoher Wahrscheinlichkeit ebenfalls im Zuge dieses Vorganges internalisiert und die Liposomen als Folge freigesetzt. Es wird vermutet, dass neben dieser Form der Freisetzung der verkapselte

Einleitung 25
Wirkstoff außerdem durch den enzymatischen Abbau der Liposomen freigesetzt wird. Der Einfluss von Bisphosphonaten auf die Osteoklasten umfasst die Inhibierung der Osteoklastenrekrutierung, die Verkürzung der Lebenszeit durch Apoptose und die Inhibierung der Osteoklastenaktivität (Luckman et al., 1998a, Russell, 2007). Bei dem Versuch, den Wirkungsmechanismus der Bisphosphonate bei diesem Vorgang zu erklären, stellte sich heraus, dass diese Substanzen diesbezüglich in zwei Strukturklassen zu unterteilen sind: die pyrophosphatähnlichen und die stickstoffhaltigen Bisphosphonate.
Die Bisphosphonate, die dem Pyrophosphat strukturell ähneln (Bisphosphonate der ersten Generation), werden durch Osteoklasten zu nicht hydrolysierbaren, zytotoxischen Adenosin-triphosphat(ATP)-Analoga abgebaut (Rogers et al., 1992). Clodronat beispielsweise wird zu APPCCl2p („Adenosine 5´(β, γ-dichlormethylene) triphosphate“) metabolisiert, welches in hohen Konzentrationen die mitochondrale ATP/Adenosin-Diphosphat-Translokase hemmt. Dies hat den Verlust des mitochondralen Membranpotentials zur Folge und leitet somit direkt die Apoptose ein (Lehenkari et al., 2002). Der hier vorgestellte Ligand besteht ebenfalls unter anderem aus einem einfachen Bisphosphonat der ersten Generation.
Stickstoffhaltige Bisphosphonate (Bisphosphonate der zweiten Generation) hingegen interferieren mit dem Metabolismus des Osteoklasten, indem sie in den Mevalonsäurezyklus eingreifen. Nach ihrer Internalisierung in die Osteoklasten hemmen sie die Farnesyl-Diphosphat-Synthase, ein Schlüsselenzym des Mevalonsäurezyklus (Luckman et al., 1998b, Roelofs et al., 2006). Die beschriebene Inhibierung führt zur verminderten Synthese von Isoprenoiden, Farnesylpyrophosphat und Geranylgeranylpyrophosphat, welche für die reguläre Prenylierung kleiner Guanosin-triphosphat(GTP)-bindenen Proteine, wie Ras, Rho und Rac, erforderlich sind (Luckman et al., 1998a). Diese signalgebenden Moleküle spielen eine wichtige Rolle in Signaltransduktionswegen von verschiedenen Kinasen und sind verantwortlich für die Regulation der Zellentwicklung sowie für das Überleben der Zelle und der zytoskeletären Organisation. Der Prozess der Prenylierung ist essentiell für die Membranlokalisierung und die Aktivierung der GTP-bindenden Proteine (Rogers, 2003). Wenn diese in ihrer Funktion eingeschränkt werden, führt dies zunächst zur Reduktion ihrer Aktivität bis hin zum Zelltod (Luckman et al., 1998b) (Russell et al., 1999). Insbesondere die Inhibierung der Signalgebung von Ras führt innerhalb der Osteoklasten zu einem defekten intrazellulären Vesikeltransport (Alakangas et al., 2002). Das hat zur Folge, dass die Osteoklasten keinen Bürstensaum

Einleitung 26
mehr ausbilden können, der für die Knochenresorption notwendig ist. Neuere Studien haben ergeben, dass die stickstoffhaltigen Bisphosphonate auch einen anderen Wirkmechanismus aufweisen können. Ähnlich wie die Bisphosphonate der ersten Generation können diese die Entstehung eines intrazellulären ATP-Analogons initiieren („Triphosphoric acid 1-adenosin-5´-yl ester 3-(3-methylbut-3-enyl) ester“, ApppI), welcher ebenfalls direkt die Apoptose bewirken kann (Monkkonen et al., 2006). Zudem können stickstoffhaltige Bisphosphonate die Osteoklastogenese hemmen (Clohisy et al., 2001). Zahlreiche In-vitro-Versuche haben neben der Inhibierung von Osteoklasten durch Bisphosphonate eindeutig eine antitumorale Wirkung unter anderem bei Mammakarzinom-, Pankreaskarzinom- und Osteosarkomzellinien aufgezeigt. (Aparicio et al., 1998, Fromigue et al., 2000, Mackie et al., 2001, Riebeling et al., 2002, Tassone et al., 2003) Gemäß Literatur können Bisphosphonate die Proliferation von Tumorzellen, die Tumorzelladhäsion an der Knochenmatrix reduzieren und den Zelltod von Tumorzellen veranlassen (van der Pluijm et al., 1996, Shipman et al., 1998, Derenne et al., 1999, Senaratne et al., 2000). Den Wirkmechanismus gilt es jedoch noch vollständig aufzudecken. Eine allgemein anerkannte Erklärung lautet aktuell, dass der Effekt auf die Tumorzellen eine Folge von der Inhibierung der Knochenresorption ist. Zum einen wird weniger Knochen abgebaut, folglich steht weniger Raum für die Metastasierung von Tumorzellen zur Verfügung. Zum anderen wird durch die reduzierte Aktivität der Osteoklasten auch die Freisetzung von Wachstumsfaktoren, wie TGF-β und IGFs, heruntergefahren, so dass die Stimulation des Zellwachstums nach dem Prinzip der „Seed and Soil“-Theorie ebenfalls vermindert wird (Mundy and Yoneda, 1995, Guise and Mundy, 1998). Problematisch bei der Anwendung von Bisphosphonaten als antitumorales Agens könnte allerdings sein, dass eine ausreichende Wirkkonzentrationen an Bisphosphonaten aufgrund ihrer pharmakokinetischen Eigenschaften in vivo nur an aktiven Resorptionsflächen, d.h. in Gegenden mit hohem „Bone Turnover“, erzielt werden und ihre Wirkung auf Tumorzellen deshalb limitiert ist. Bisphosphonate haben aufgrund ihrer niedrigen Lipophilie und ihrer negativen Ladung eine geringe Bioverfügbarkeit (Boulenc et al., 1993, Lin, 1996). Sobald sie die Blutbahn erreicht haben, verlassen sie diese auch wieder sehr schnell. Etwa die Hälfte siedelt sich wegen ihrer hohen Affinität zu den Hydroxylapatitkristallen am Knochen an, die andere Hälfte wird in der Regel mit dem Urin ausgeschieden (Jung et al., 1973, Lasseter et al., 2005). Eine relativ neue Studie beschäftigte sich mit der Gewebeverteilung von verschiedenen Bisphosphonaten (Zoledronsäure, Inbandronsäure und Clodronsäure). Die höchsten Konzentrationen wurde erwartungsgemäß zum einen im Knochen

Einleitung 27
detektiert, wobei ein Plateau eine Stunde nach der Applikation erreicht wurde, und zum anderen in der Niere festgestellt. Eine neue Erkenntnis dieser Studie war, dass alle Bisphosphonate auch in der Prostata mit einer Maximalkonzentration nach 30 bis 60 Minuten akkumulierten. In anderen Geweben, wie Lunge, Leber und Milz, wurde hingegen eine vernachlässigbar geringe Akkumulation ermittelt (Fournier et al., 2002).
Sind die Bisphosphonate erst einmal in die Knochenmatrix eingebaut, werden sie im Zuge von Knochenbrüchen oder „Remodelling“-Prozessen freigesetzt. Somit können Verweildauern von bis zu 10 Jahren auftreten (Kasting and Francis, 1992, Rodan et al., 2004). Bisphosphonate reichern sich wegen ihres schnellen Einbaus bevorzugt im kalzifizierten Gewebe und der daraus resultierenden kurzen Zirkulationszeit im Blut an, selten in anderen Geweben an. Vor diesem Hintergrund erklärt sich ihre geringe Toxizität. Eine akute Toxizität besteht nur dann, wenn die Bisphosphonate Komplexe oder Aggregate mit dem Serumcalcium bilden, dadurch den Anteil an ionisiertem Calcium herabsetzen und schließlich eine Hypokalzämie hervorrufen.
In diesem Abschnitt ist abschließend festzuhalten, dass die Bisphosphonate in der vorliegenden Arbeit aufgrund ihrer hohen Bindungsaffinität an den Knochen als Targeting-Liganden und nicht als Therapeutikum vorgesehen werden. Die eingesetzten Bisphosphonatkonzentrationen liegen dabei weit unter den üblich eingesetzten therapeutischen Konzentrationen. Eine inhibierende Wirkung der Bisphosphonate, die möglicherweise dennoch auf die Osteoklasten ausgeübt werden könnte, würde die Wirksamkeit des Trägersystems potenzieren und somit einen zusätzlichen Vorteil erbringen.
1.4.3 Indikationen, Nebenwirkungen und Kontraindikationen
Die Wirksamkeit der Bisphosphonate in der Behandlung von Knochenerkrankungen mit erhöhtem Knochenabbau ist eindeutig belegt. Bei diesen Erkrankungen gehören die Bisphosphonate zur Standardtherapie. In der Onkologie werden sie bisher als adjuvante Therapeutika eingesetzt. Sie sind indiziert bei tumorbedingten Knochenschmerzen, bei tumorbedingter Hyperkalzämie, zur Therapie von Osteolysen und frakturgefährdeten Skelettarealen infolge von Osteoporose. Zudem werden sie zur Therapie von Morbus Paget, zur Prävention tumorbedingter Skelettdestruktion, zur Beeinflussung der Entstehung ossärer und viszeraler Metastasen und zur Hemmung

Einleitung 28
des Tumorwachstums im Knochen eingesetzt (Bartl, 2001). Die Bisphosphonate werden hierzu intravenös appliziert, da die oralen Formen nicht vollständig resorbiert werden oder zur Reizung des gastrointestainalen Traktes führen. In jüngerer Zeit eingeführt wurde beispielsweise das Bondronat® (Ibandronat) zur Verbesserung der Lebensqualität bei Knochenmetastasen. Bei vorschriftsgemäßer Anwendung sind die Bisphosphonate gut verträglich und weisen wenige Nebenwirkungen auf. Als nicht vernachlässigbare Nebenwirkung ist allerdings die Nephrotoxizität zu nennen, d.h., bei einer schnellen Infusion kann es zur Bildung von unlöslichen Komplexen kommen, die potentiell ein akutes Nierenversagen zur Folge haben. Aus diesem Grund werden Bisphosphonate bei intravenösen Gaben sehr langsam und stark verdünnt appliziert. Durch ihre Fähigkeit, Komplexe mit Kationen zu bilden, kann es außerdem zur Komplexierung der Calciumionen im Blut kommen, was möglicherweise die Entstehung von Hypokalzämien zur Folge hat. Durch Maßnahmen, wie Calciumsubstitution oder Rehydrierung, ist die Hypokalzämie jedoch kontrollierbar. Diese Nebenwirkung führt zudem zur Kontraindikation der gleichzeitigen Gabe von Aminoglykosiden, um eine Hypokalzämie zu vermeiden. Außerdem ist eine kombinierte Gabe von Bisphosphonaten und Arzneimitteln, die polyvalente Kationen enthalten, zu unterlassen. Die Neigung zur Komplexbildung ist sicherlich ein entscheidener Nachteil der Bisphosphonate, die bei der Untersuchung der hier vorgestellten ligandmodifizierten Liposomen trotz gering eingesetzter Konzentrationen auch zum Tragen kommen kann.
1.5 Zielgerichtete Applikation an den Knochen
In diesem Abschnitt wird der Stand der wissenschaftlichen Forschung im Hinblick auf bereits untersuchte Wirkstoffträgersysteme zusammengefasst, die ebenfalls Bisphosphonatderivate als Targeting-Liganden aufweisen. Außerdem wird ein Überblick über alternative Liganden dargestellt, die aufgrund ihrer Struktur ebenfalls in der Lage sind, an den Knochen zu binden.

Einleitung 29
1.5.1 Trägersysteme basierend auf Bisphosphonaten als knochenaffine Liganden
Kein therapeutisches Drug-Delivery-System bestehend aus Bisphosphonaten als Targeting-Einheit gekoppelt an ein Trägersystem hat es bisher bis zur Marktreife geschafft. Zugelassen sind derzeit nur die Radiopharmazeutika, und zwar als Diagnostikum oder als palliatives Agens in der Schmerztherapie von Knochenerkrankungen. Aufgrund der bereits bekannten hohen Affinität von Bisphosphonaten zur Knochenmatrix synthetisierten unterschiedliche Arbeitsgruppen Konjugate, die sich aus Bisphosphonaten und nicht knochenspezifischen Therapeutika zusammensetzten, um diesen die Fähigkeit zu verleihen, knochenaffin zu sein. Diese konjugierten Systeme umfassen beispielsweise die Kopplung von Bisphosphonaten an Estradiol (Bauss et al., 1996), Prostaglandin E2 (Gil et al., 1999), Diclofenac (Hirabayashi et al., 2001), Cisplatin (Klenner et al., 1990), Methotrexat (Hosain et al., 1996) und Antibiotika, wie die Fluoroquinolone (Herczegh et al., 2002). Unter ihnen haben einige Konjugate bereits zu vielversprechenden Ergebnissen in präklinischen Studien geführt. In Indikationen, wie Osteoporose, Knochenkrebs und Osteoarthritis, zeigten sie eine verbesserte und verlängerte pharmakologische Wirkung und reduzierten die Nebenwirkungen auf andere, nicht betroffene Gewebe. Weitere Studien müssten genauer präzisieren, welche pharmakologische Aktivität tatsächlich von den einzelnen bisphosphonat-gekoppelten Systemen auf ossäre und extraossäre Gewebe ausgeübt wird, um die Wirksamkeit und Nebenwirkungen dieser Systeme noch besser einschätzen zu können.
Die erfolgreiche Kopplung von Proteinen an Bisphosphonate (Murphy et al., 2007) und ihre Bindung an den Knochen in vivo konnten ebenfalls bereits aufgezeigt werden (Uludag et al., 2000, Uludag and Kousinioris, 2000, Gittens et al., 2005, Murphy et al., 2007). Wang et. al. (Wang et al., 2003) untersuchten nicht nur die Kopplung von Poly[N-(2-hydroxypropyl)methacrylamid] (PHPMA) und Polyethylenglykol (PEG) an Asp6, sondern sie koppelten diese Polymere zudem an Alendronat. Ziel dieser Kopplung war es, ein System zu entwickeln, das Knochentherapeutika über säure- oder enzymempfindliche Spacer an sich bindet. Auch hier ist es ihnen gelungen, erfolgversprechende In-vitro- und In-vivo-Ergebnisse im Hinblick auf eine hohe Bindung am Knochen zu erzielen. Gonzalez et al. (Gonzalez et al., 2002) synthetisierten ein konjugiertes System bestehend aus einem Fulleren und einem

Einleitung 30
Bisphosphonat (C60(OH)16AMBP; 4,4-Bisphosphono-2-(Polyhydroxyl-1,2-dihydro-1,2-methanofulleren[60]-61-carboxamido)buttersäure) mit dem Ziel, es später als Therapeutikum von Knochenerkrankungen einzusetzen, indem sie das Fulleren mit einem geeigneten Wirkstoff beladen. Die Tatsache, dass bereits vielversprechende veröffentlichte Daten zum Thema der zielgerichteten Applikation an den Knochen durch den Einsatz von Bisphosphonatliganden verfügbar sind, hat uns darin bestärkt, die Entwicklung und den Test eines neuen liposomalen Trägersystems in die Tat umzusetzen. Die Zulassung von bisphosphonat-gekoppelten Radiopharmazeutika kann in diesem Zusammenhang sicherlich als „Proof of Concept“ angesehen werden, der beweist, dass Bisphosphonate als bereits etablierte und akzeptierte Targeting-Liganden in der Lage sind, Wirkstoffe zum Wirkort zu transportieren.
In der vorliegenden Arbeit gilt es erstmalig zu zeigen, ob auch ein liposomales System, gebunden an ein neues, maßgeschneidertes Bisphosphonsäurederivat, in der Lage ist, nach einer intravenösen Applikation die Knochenoberfläche zu erreichen. Die Tatsache, dass Bisphosphonate bisher nur als Targeting-Einheit an kleinere Moleküle gebunden wurden, lässt vermuten, dass die physiologischen Gegebenheiten eines Körpers es nicht zulassen. Hirabayashi et. al. sind der Meinung, dass der Zugang zur Knochenoberfläche sehr limitiert ist und dass Wirkstoffträgersysteme wie Liposomen es insbesondere aufgrund ihrer Größe nicht schaffen können, das Zielgewebe zu erreichen (Hirabayashi and Fujisaki, 2003). Wir stellen hingegen die Hypothese auf, dass die ligandmodifizierten Liposomen durch die fenestrierten Blutgefäße, die die Metastasen versorgen, und durch die ebenfalls fenestrierten, sinusoidalen Blutgefäße des Knochenmarks aus der Blutbahn in das betroffene Knochengewebe und danach an die Knochenoberfläche gelangen können. Es wird in diesem Zusammenhang vermutet, dass durch die besonderen physiologischen Gegebenheiten, bedingt durch das Metastasenwachstum im Knochengewebe, ausreichend große Fenestrae vorkommen, die das Entweichen von Liposomen aus der Blutbahn möglicherweise erlauben. Im Vergleich zu den zuvor genannten bisphosphonat-gekoppelten Systemen sehen wir bei dem hier präsentierten System den entscheidenen Vorteil, dass der Wirkstoff in unveränderter Form verkapselt in den Liposomen vorliegt, wohingegen bei den anderen Systemen die Bisphosphonate als Targeting-Liganden direkt an den Wirkstoff (.z.B. Diclofenac, Methotrexat) gebunden wurden. Somit wurden die physikochemischen und pharmakologischen Eigenschaften vermutlich nachteilig verändert. Hinzu kommt, dass in die Liposomen eine Vielzahl von verschiedenen Wirkstoffen (lipophil, hydrophil) verkapselt werden können, wohingegen sicherlich nicht alle Substanzen die direkte Kopplung an Bisphosphonate zulassen. Außerdem

Einleitung 31
sind wir davon überzeugt, den Vorteil der kontrollierten Wirkstofffreisetzung und die Möglichkeit, große Mengen an Wirksubstanz in den Liposomen zu verkapseln, in dem hier vorgestellten System vereinen zu können. Ein weiterer wichtiger Unterschied zu den zuvor genannten bisphosphonat-gekoppelten Substanzen ist, dass auf der Oberfläche der Liposomen eine größere Anzahl an Liganden für die angestrebte Bindung zur Verfügung stehen, so dass eine höhere Wahrscheinlichkeit für die erfolgreiche Bindung gegeben ist. Viele einzelne Ligandmoleküle auf der Liposomenoberfläche könnten durch die Ausbildung von multivalenten Bindungen fester an das Zielgewebe binden.
Die Idee, Trägersysteme bestehend aus Fullerenen einzusetzen, basiert ebenfalls, wie die hier präsentierte Strategie, auf der Kombination von Trägersystem und Bisphosphonatliganden. Doch die Eignung von Fullerenen als Wirkstoffträger gilt es sicherlich noch nachzuweisen. In einer kürzlich erschienenen Veröffentlichung wird ebenfalls die Kombination von einem Trägersystem und Bisphosphonaten als Targeting-Liganden beschrieben. Darin wurde das Design von PLGA-Nanopartikeln, dessen Oberfläche durch Aledronat und PEG modifiziert wurde, vorgestellt (Choi and Kim, 2007). Die Bindungsfähigheit an Hydroxylapatit konnte erfolgreich demonstriert werden, weitere Versuche müssten allerdings noch den Nachweis der In-vivo-Tauglichkeit erbringen.
1.5.2 Alternative knochenaffine Liganden
Die spezielle Zusammensetzung des Knochens, d.h. die Gegenwart von Hydroxylapatit, macht den Knochen zum idealen Ziel für die zielgerichtete Applikation von Wirksubstanzen. Das Ausmaß der Bindung des knochenaffinen Liganden bzw. des mit dem Liganden versehenen Komplexes an die Knochenoberfläche wird durch Faktoren wie die Blutversorgung, die lokale Extravasationsrate in das interstitielle Fluid und die effektiv zur Verfügung stehende Oberfläche des Hydroxylapatites maßgeblich bestimmt. Neben Bisphosphonaten binden die antibiotischen Tetrazykline an den Knochen. Sie formen ebenfalls mit dem Calcium des Hydroxylapatites Chelatkomplexe (Perrin, 1965, Misra, 1991, Kasugai et al., 2000). Aufgrund ihrer hohen Affinität zum Knochen haben diverse Arbeitsgruppen die Tetrazykline als Targeting-Liganden an verschiedene Therapeutika gekoppelt mit dem Ziel, verbesserte Behandlungsmethoden für Krankheiten, die den Knochen

Einleitung 32
betreffen, zu entwickeln. Pierce und Waite (Pierce and Waite, 1987) konjugierten ein Tetrazyklinmolekül beispielsweise an einen Kohlensäure-Anhydrase-Inhibitor, einem Inhibitor der Knochenresorption, und untersuchten die Bindungsfähigkeit des Komplexes an Hydroxylapatit in vitro. Orme and Labro (Orme and Labroo, 1994) bedienten sich einer ähnlichen Strategie und koppelten Tetrazyklin an β-Estradiol. Beide Arbeitsgruppen konnten in vitro eine starke Bindung an Hydroxylapatit nachweisen, doch In-vivo-Versuche wurden jeweils nicht veröffentlicht. Vermutlich konnte zum einem nicht klar aufgezeigt werden, wie der Freisetzungsmechanismus der beiden genannten Konjugate funktioniert, und zum anderen wegen der eher komplizierten chemischen Struktur des Tetrazyklins. Die geringe Stabilität dieses Moleküls während der chemischen Modifizierungen macht es nicht zur geeigneten Substanz als knochenaffiner Targeting-Ligand (Wang et al., 2005).
Die Suche nach strukturell ähnlichen Molekülen mit verbesserten Eigenschaften führte zur Untersuchung von Pyrazolen als knochenaffine Liganden. Willson et al. (Willson et al., 1994) konjugierten 4-Carboxy-3-hydroxy-1,2-pyrazol und Estrogen Hexestrol. Sie zeigten eine hohe Knochenbindungskapazität auf. Ein Nachteil war jedoch, dass der Targeting-Ligand die Bindung von Estrogen an seinen Rezeptor negativ beeinflusste. Da die Knochen-Matrixproteine, wie z.B. Osteocalcin, Osteopontin und Sialoprotein ebenfalls die Fähigkeit haben, das Calcium des Knochenapatits stark zu binden, wurden auch diese als potentielle Liganden untersucht (Steitz et al., 2002, Hoang et al., 2003, Midura et al., 2004). Das Osteopontin beispielsweise bindet über asparaginsäurereiche Domänen an die Hydroxylapatitoberfläche. Der Ser- und der Thr-Teil in Osteopontin sind in der Regel phosphoryliert, und durch diese stark negativgeladenen Gruppen kann das Protein effektiv an das Calcium binden (Wang et al., 2005). Die gewonnenen Erkenntnisse über den Bindungsmechanismus der Knochenproteine veranlassten Kasugai et al., Asparaginsäure als knochenaffinen Liganden einzusetzen (Kasugai et al., 2000). Zunächst konjugierten sie das Hexapeptid der Asparaginsäure (Asp6) an Fluorescein, um den neuen Liganden auf seine Eignung zu testen. Nach erfolgreicher Bindung an den Knochen in vivo, untersuchten sie den therapeutischen Effekt von Asp6 gekoppelt an Estradiol in ovariektomierten Mäuse. Sowohl konjugiertes als auch unkonjugiertes Estradiol zeigte den gleichen Effekt in Bezug auf die Erhaltung der Knochendichte. Das unkonjugierte System erwies sich jedoch als nebenwirkungsreicher (Yokogawa et al., 2001). Wang et al. (Wang et al., 2003) synthetisierten Konjugate bestehend aus Asp6 als Targeting-Ligand gebunden an Polyethylenglykol (PEG) oder Poly[N-(2-hydroxypropyl)methacrylamid (PHPMA) und Fluoresceinisothiocyanat als Modellwirkstoff. Ziel hierbei war es, durch den

Einleitung 33
Einbau der Polymere die Zirkulationszeit der negativgeladenen Asp6 zu verlängern. Ihre Versuche ergaben, dass beide Konjugate prinzipiell an das Knochenapatit in vivo binden. Der Komplex basierend auf PHPMA jedoch zeigte ein höheres Bindungsausmaß aufgrund seiner besseren und verlängerten Zirkulationseigenschaften. Weitere In-vivo- und In-vitro-Versuche müssten zeigen, ob die vielversprechenden, bisher erzielten Ergebnisse zum Asp6-Liganden auch in Zukunft fortgesetzt werden können. Die Tatsache, dass Bisphosphonate im Vergleich zu den hier genannten alternativen Ligandmolekülen zu den umfangreich untersuchten Ligandmolekülen zählen, hat uns darin bestärkt, diese bei der Synthese des hier vorgestellten Liganden als Targeting-Einheit einzusetzen. Die Akzeptanz von Bisphosphonaten als Targeting-Liganden in Form eines Bestandteiles von Radiopharmazeutika erachten wir als weiteres überzeugendes Argument, diese in unser Konzept miteinzubeziehen.
1.6 Liposomen als Wirkstoffträgersystem
Da das in dieser Arbeit vorgestellte Wirkstoffträgersystem auf Liposomen basiert, werden diese im folgenden Abschnitt näher beschrieben. Dabei wird unter anderem ihre Struktur, Zusammensetzung und Herstellung erläutert. Weiterhin wird auf ihre Eigenschaft, als Wirkstoffträger fungieren zu können, detailliert eingegangen. Um die in der vorliegenden Arbeit dargestellte Targeting-Strategie von existierenden Strategien deutlich abzugrenzen, werden zudem bereits untersuchte Ansätze zur Ligandmodifizierung von diversen Liposomen zwecks Tumor-Targeting zusammengefasst.
1.6.1 Eigenschaften von Liposomen
Bangham beobachtete 1964, dass Phospholipide in Wasser spontan konzentrische, vielschichtige Lamellen ausbilden, die durch ein wässriges Kompartiment von einander getrennt waren (Bangham and Horne, 1964) (Abbildung 6). Sessa und Weissmann nannten die von Bangham untersuchten Lipidvesikel im Jahre 1968 zum erstenmal Liposomen (Sessa and Weissmann, 1968). Im Jahre 1970 entdeckten sie

Einleitung 34
dann die potentielle Eignung der Liposomen als Wirkstoffträger, indem sie den Einschluß von Lysosym in Liposomen untersuchten (Sessa and Weissmann, 1970). Liposomen können in ihrer Größe (Durchmesser) von 20 nm bis 10 µm variieren, welche durch die Anzahl der Lamellen (Bilayer) bestimmt wird. Kleine unilamellare Liposomen („Small Unilamellar Vesicles“, SUV) liegen bei einem Durchmesser von 20 bis 100 nm vor, von großen unilamellaren Liposomen („Large Unilamellar Vesicles“, LUV) spricht man bei Durchmessern von 100 nm. Liposomen mit einem Durchmesser von 100 nm bis mehrere µm werden als multilamellare Vesikel („Multilamellar Vesicles“, MLV) bezeichnet. Jede Lamelle besteht aus einer Doppelschicht Phospholipide, bei der die polaren, hydrophilen Kopfgruppen zur Wasserphase hin orientiert sind, während die sich gegenüberstehend, lipophilen Fettsäurereste die lipophile Phase bilden (Rubas and Schreier, 1991). Die hydrophile Kopfgruppe von Phospholipiden besteht aus einem Phosphorsäurediester, der über eine der drei Hydroxylgruppen des Glycerols einerseits und mit Cholin, Ethanolamin, Inositol, Serin oder Glycerol andererseits verknüpft ist. Der lipophile Rest besteht aus zwei Fettsäureketten mit jeweils 14 bis 24 Kohlenstoffatomen, die an die restlichen zwei Hydroxylgruppen des Glycerolmoleküls gebunden sind (Abbildung 6).
Abbildung 6: Darstellung von Phospholipidstruktur und Ausbildung von Liposomen in wässriger Umgebung. Die Phospholipide formen dabei eine Doppelmembran, indem die lipophilen Reste der Lipide sich sich zueinander ausrichten und die hydrophilen Köpfe sich so anordnen, dass sie in die Wasserphase ragen (Quelle: www.cem.msu.edu/~reusch/VirtualText/Images3/bilyrstr.gif).

Einleitung 35
Faktoren, wie die Größe, die Lamellarität, die Bilayer-Rigidität und die Ladung bestimmen ihre Stabilität und das In-vivo-Verhalten. Die Größe und die Lamellarität können durch das Herstellungsverfahren (Homogenisation, Extrusion, Ultraschallbehandlung) beeinflusst werden. Die Rigidität der Membranen und die Ladung können durch die Wahl der Lipidzusammensetzung verändert werden. Durch den Einbau von Cholesterol beispielsweise kann die Rigidität der Membran erhöht werden. Cholesterol ändert die Permeabiliät und Fluidität der Membran, indem es die Phasenübergangstemperatur (Phasenübergang vom gelartigen zum flüssigen bzw. flüssigkristallinen Zustand) herabsetzt. Die Verringerung der Membranpermeabilität führt sowohl zur erhöhten Lagerstabilität als auch zur verbesserten Stabilität in vivo, da das Cholesterol den Austausch von Phospholipiden mit Lipoproteinen reduziert. Die Vorteile der Liposomen sind, dass sie als Lösungsvermittler von schwerlöslichen, lipophilen Wirkstoffen fungieren können und die verkapselten Wirkstoffe vor dem Abbau durch metabolisierende Enzyme schützen. Durch die Formation einer Schutzhülle um die Wirksubstanz können sie umgekehrt auch den Patienten vor schwerwiegenden Nebenwirkungen schützen, die vom Wirkstoff (beispielsweise Zytostatika) ausgehen. Die Wirkstoffverkapselung in Liposomen im Vergleich zu freier Wirksubstanz erwies sich im Fall von Anthrazyklinen beispielsweise, welche klassische Arzneistoffe in der Tumortherapie darstellen, als klar vorteilhaft im Hinblick auf ihr Nebenwirkungsprofil. Ihre schwerwiegende kardiotoxische Wirkung, bedingt durch schädliche, oxidative Prozesse (ausgehend von den sauerstoffreichen Anthrazyklinen, insbesondere Doxorubizin) in Herzmuskelzellen, ist durch die Verkapselung von Anthrazyklinen in Liposomen nahezu aufgehoben. Dieser Effekt erklärt sich dadurch, dass die Liposomen nicht in der Lage sind, in das Myokardgewebe einzudringen. Außerdem werden weitere Nebenwirkungen wie Alopezie, Neutropenie und Übelkeit durch diese Art von Formulierung aufgrund von veränderten Verteilungsvolumina reduziert (Gill et al., 1995, Maitani et al., 2007). Durch die veränderte Zusammensetzung im Vergleich zur freien Wirksubstanz kommt es zu neuen Nebenwirkungen verursacht durch unterschiedliche Pharmakokinetikprofile. Es konnte gezeigt werden, dass sich kleine Liposomen in der Haut anreichern und als Folge Toxizitätserscheinungen der Haut an Händen und Füssen („Hand Foot Syndrom“, PPES Palmar Plantares Erythrodysästhesie) auftreten (Hwang et al., 1987, Lorusso et al., 2007). Dennoch überwiegen die Vorteile der liposomalen Formulierung von Anthrazyklinen und führen somit zu einer verbesserten Lebensqualität (Gill et al., 1996). Hinzu kommt, dass sie die Wirkung von Wirkstoffen verlängern können, indem sie eine Art Reservoir bilden, das den Wirkstoff nach und

Einleitung 36
nach freigibt. Liposomen können im Hinblick auf ihre Zusammensetzung und ihre In-vivo-Applikation in vier Typen unterschieden werden (Storm and Crommelin, 1998).
Der Typ 1 beschreibt konventionelle Liposomen, die typischerweise aus neutral- und/oder negativgeladenen Lipiden und/oder Cholesterol bestehen. Sie zeichen sich meist durch eine relativ kurze Zirkulationszeit in vivo aus. Nach einer parenteralen Applikation tendieren sie dazu, schnell aus der Blutbahn eliminiert zu werden und in Leber und Milz zu akkumulieren. Verantwortlich für die schnelle Elimination aus der Blutbahn ist die Aufnahme durch das retikuloendotheliale System (RES), auch Mononukleäres Phagozyten-System genannt (MPS), welches in Geweben wie Milz, Knochenmark und vor allem Leber anzutreffen ist (Kaye and Richardson, 1979). Heute ist bekannt, dass das Anheften von Plasmaproteinen, wie beispielsweise Opsoninen, an die liposomale Oberfläche die Phagozytose dieser Liposomen durch Makrophagen begünstigt, die wiederum den Abtransport der Liposomen hauptsächlich in Leber und Milz initiieren. Dieses Phänomen der raschen Clearance und der anschließenden Akkumulation in Geweben des retikuloendothelialen Systems schränkte den Einsatz von Liposomen als Therapeutikum zunächst stark ein.
Den 2. Typ bilden die langzirkulierenden bzw. „Stealth“-Liposomen. Ihr Vorteil ist, dass sie durch den Einbau von hydrophilen Polymeren in ihre Membran vor dem Abbau durch Makrophagen geschützt sind und somit verbesserte Pharmakokinetikprofile aufweisen. Lipide, die für diese sterische Stabilisierung eingesetzt werden, bestehen zumeist aus einer hydrophilen PEG-Kette und einem hydrophoben Molekülteil, welcher als Ankermolekül in die Liposomenmembran eingebaut wird. Das Ankermolekül kann ein Cholesterolrest (Carrion et al., 2001) oder ein Phosphatidylethanolamin (Woodle and Lasic, 1992) sein. In den anfänglichen Versuchen, die Zirkulationszeit von Liposomen zu verlängern, baute man Cholesterol in die Membran von kleinen, unilamellaren Liposomen ein (Kirby et al., 1980). Doch da das Cholesterol die Bluthalbwertszeit nur geringfügig verlängerte, stand die körpereigene Abwehr dem vielseitigen Einsatz von Liposomen als Therapeutikum weiterhin entgegen. Gegen Ende der 80er Jahre entdeckten Allen et al. schließlich, dass der Einbau von Monosialogangliosid (GM1) in die Membran aufgrund seiner voluminösen und hydrophilen Zuckergruppen, die Zirkulationsdauer in Mäusen deutlich verlängerte (Allen and Chonn, 1987). Eine geringe Biokompatibilität stellte sich schnell als limitierender Faktor in der Anwendung von GM1 als Schutz vor dem Abbau von Makrophagen heraus. Weitere Versuchen zeigten nämlich, dass die zuvor

Einleitung 37
beobachtete Verlängerung der Halbwertszeit nicht bei Ratten oder Kaninchen detektiert werden konnte, da diese Tiere wiederum gegen GM1 gerichtete Antikörper entwickelten (Liu et al., 1995). Kurz nach der Entdeckung des richtungsweisenden Effektes, der von dem GM1 ausging, wurde dann die Eignung von PEG-Molekülen als schützendes Agens publiziert (Blume and Cevc, 1990). Neuere Untersuchungen zur Blutverweildauer von pegylierten Liposomen haben jedoch ergeben, dass eine zweite Dosis von pegylierten Liposomen offenbar zu einer dramatisch verkürzten Bluthalbwertszeit und zu einer Akkumulation in der Leber führt. Diese Beobachtung wird auch ABC-Phänomen („Accelerated Blood Clearance“-Phänomen) genannt (Dams et al., 2000, Bendas et al., 2003). Eine weitere wichtige Eigenschaft von langzirkulierenden Liposomen ist, dass sie in der Lage sind, in pathologischen Geweben wie Tumorgewebe oder Entzündungsherde, die sich durch erhöhte Permeabilität ihrer Kapillaren auszeichnen, zu extravasieren und dort zu akkumulieren.
Ein 3. Typ umfasst Liposomen, die spezifische Antibodies auf ihre Oberfläche als Targeting-Einheit tragen (Immunoliposomen), um das Binden der Liposomen am Zielort zu ermöglichen. Konigsberg et al. (Konigsberg et al., 1998) entwickelten beispielsweise Immunoliposomen, indem sie humanes, rekombinates Interleukin-2 (IL-2) als Liganden an die Oberfläche von SUVs für die spezifische Bindung an aktivierte T-Zellen koppelten. Durch die Verkapselung des Immunsuppressivums Methotrexat in diese Liposomen ist es ihnen gelungen aufzuzeigen, dass die Zellproliferation mittels rezeptorvermittelter Endozytose des Wirkstoffes gehemmt werden konnte.
Auf dem Gebiet der Gentherapie wird der 4. Typ von Liposomen untersucht. Die hier eingesetzten Liposomen sind von kationischer Natur und können aufgrund ihrer positiven Ladung an negativgeladene DNA binden (Templeton et al., 1997). Der resultierende Lipid-DNA-Komplex fördert die Internalisierung in die Zelle und die anschließende Exprimierung des kondensierten Plasmids. Aufgrund der strukturellen Vielseitigkeit von Liposomen wurde ihre Anwendung bereits für viele Applikationsarten getestet. Liposomen wurden beispielweise intravenös (Kaiser et al., 2003, Romberg et al., 2006), subkutan (Oussoren et al., 1998), oral (Degim et al., 2006), pulmonal (Krupova et al., 2001, Anabousi et al., 2006, Kleemann et al., 2007) oder auch okular (Fresta et al., 1999) appliziert. Der Vorteil von Liposomen ist, dass sie sowohl mit lipophilen als auch hydrophilen Wirkstoffen beladen werden können. Der lipophile Wirkstoff wird dabei in die Membran eingebaut, wohingegen die

Einleitung 38
hydrophile Wirksubstanz in das wässrige Liposomeninnere eingeschlossen wird. In der Tumortherapie hat sich der Einsatz von Liposomen bereits bewährt. In Europa beispielsweise sind liposomale Präparationen (pegylierte Doxorubizin-Liposomen) wie Doxil® oder Caelyx® zur Anwendung in der Indikation Ovarialkarzinom zugelassen. Außerdem sind einige Liposomenpräparate zur topischen Anwendung auf dem Markt, wie beispielsweise Pevaryl-Lipogel® zur Behandlung von Mykosen oder Diclofenac ratiopharm Gel® für die Behandlung von Entzündungen. Die Tatsache, dass verschiedene liposomale Formulierungen als Therapeutika anerkannt und zugelassen sind, hat uns überzeugt, diese ebenfalls als Trägersystem einzusetzen.
1.6.2 Ligandmodifizierung von Liposomen
Um die Wirksamkeit von liposomalen Formulierungen im Vergleich zu bereits bestehenden Präparationen zu verbessern, werden zumeist die physikochemischen Eigenschaften optimiert. Dazu werden Formulierungsparameter wie Lipidzusammensetzung, Partikelgröße, Fluidität der Lipidmembran, Oberflächenladung, Cholesterolgehalt und sterische Stabilisierung stetig verändert. Eine andere Möglichkeit, den therapeutischen Benefit zu erhöhen, ist der Einbau eines Liganden in die Liposomenmembran, der die Selektivität des liposomalen Systems für den Wirkort verbessert. Die Modifizierung der Liposomenoberfläche mit einem Liganden wird auch als aktives Targeting bezeichnet, wohingegen das passive Targeting auf natürlichen, anatomischen Gegebenheiten oder physiologischen Prozessen basiert, die die Ansammlung von Liposomen vermehrt zulassen bedingt durch ihre physikochemischen Eigenschaften. Beispielsweise neigen Tumore zur Neoangiogese, wenn sie größer werden. Die Vaskulatur dieser Gefäße ist zumeist eher wenig entwickelt, d.h., es entstehen Lücken zwischen den Endothelzellen, und die Basalmembran wird häufig gar nicht ausgebildet. Dieses löchrige Endothelgewebe ermöglicht kleinen partikulären Systemen die Extravasation in das Tumorgewebe. Das Fehlen einer funktionierenden lymphatischen Drainage begünstigt zusätzlich die Akkumulation dieser Systeme im Tumor (Roberts and Palade, 1997). Laut Literatur können die den Tumor versorgenden Blutgefäße für Liposomen mit einem Durchmesser bis zu 400 nm durchlässig sein (Yuan et al., 1995).
Erfolgreiches, aktives Targeting hingegen erfordert beispielsweise ligandspezifische Rezeptoren im Zielgewebe, welche im gesunden Gewebe nicht oder im geringeren

Einleitung 39
Ausmaß vorkommen. Dabei wird das aktive Targeting in Tumorzell-Targeting und in das vaskuläre Targeting unterschieden. Das Targeting zu den Tumorzellen umfasst die Wirkungssteigerung der wirkstoffbeladenen Liposomen, indem die zielgerichtete Applikation durch tumorzellspezifische Liganden erwirkt wird. Bei dem vaskulären Targeting weisen ligandmodifizierte Liposomen eine hohe Affinität zu den Endothelzellen der neugebildeten Blutgefäße auf. Die hier vorgestellte Targeting-Strategie ist ebenfalls eine Form des aktiven Targetings, bei der allerdings nicht die Tumorzellen oder Endothelzellen von Blutgefäßen das primäre Ziel sind, sondern das Hydroxylapatit des Knochens im metastasendurchsetzten Knochengewebe. Sobald die Liposomen an das Hydroxylapatit gebunden haben, sollen sie ihren verkapselten Wirkstoff freigeben, so dass dieser auf die umliegenden Metastasen wirken kann. Um allerdings die Knochenmatrix erreichen zu können, wird das passive Targeting für die ligandmodifizierten Liposomen ebenfalls von essentieller Bedeutung sein. Eine angestrebte Liposomengröße im Bereich 100–200 nm soll die verstärkte Akkumulation der Liposomen in der Region des metastasierenden Knochengewebes aufgrund der erhöhten Kapillarenpermeabilität des befallenen Gewebes gewährleisten. Das passive Targeting wird möglicherweise zudem durch eine eingeschränkte lymphatische Drainage in den betroffenen Gegenden unterstützt und fördert somit die erhöhte Ansammlung der Liposomen in diesen Regionen. Ein klarer Vorteil des Targetings, sowohl des passiven als auch des aktiven, ist, dass der an sich niedrige Therapeutische Index (Therapeutischer Index = Maß für die Sicherheit eines Medikaments, das die Spanne zwischen therapeutischer und toxischer Dosis angibt) von Krebstherapeutika eindeutig verbessert werden kann.
Um ein effizientes Targeting mit ligandmodifizierten Liposomen, wie sie auch in dieser Arbeit eingesetzt werden, zu erzielen, sollte durch den Einbau der Liganden die Zirkulationszeit der Liposomen, d.h., die Wirkung von ebenfalls vorhandenem Polyethylenglykol (PEG) auf der Oberfläche, nicht negativ beeinflusst werden. Umgekehrt ist es nicht wünschenswert, dass das PEG mit dem Zielgewebe interagiert, und somit die Wirkung der Liganden beeinträchtigt. Deshalb ist die Art und Weise der Kopplung von Liganden an die Liposomenoberfläche ein wichtiger Parameter. Es sind gemäß Literatur bereits verschiedene Ansätze untersucht worden. Zum einen können Liganden neben dem PEG auf der Oberfläche angebracht werden (Torchilin et al., 1996). Dazu werden die Liganden häufig kovalent an Phosphatidylethanolamin (z.B. Distearoyl-phosphatidylethanolamin, DSPE) gebunden, einer langkettigen, gesättigten Fettsäure, die dann in der Liposomenmembran verankert wird (Torchilin et al., 2001, Mastrobattista et al., 2002). Auch in dieser Arbeit wurden Liganden neben DSPE-

Einleitung 40
PEG2000 mithilfe eines Cholesterylrestes in die Liposomenmembran verankert. Der Einsatz von Cholesterol als Ankermolekül ist dabei nicht neu (Ishiwata et al., 1995, Zhao et al., 2007). Innovativ jedoch ist die Kombination von Cholesterol als zu verankerndes Derivat und Bisphosphonatmolekülen als Targeting-Einheit (Hengst et al., 2007). Ein Trisoxyethylen-Spacer zwischen der in die Membran verankerte Cholesteryl-Gruppe und der endständigen Bisphosphonsäure-Gruppe soll zudem mögliche Interaktionen zwischen dem DSPE-PEG2000 und den Ligandmolekülen minimieren. Zum anderen können die Ligandfunktionen an die Endgruppen vom PEG-Molekül kovalent gebunden werden. Dabei gibt es drei gängige Möglichkeiten, diese Kopplung auszuführen (Zalipsky et al., 1997, Zalipsky, 1998). Eine Möglichkeit ist, aus endgruppenfunktionalisierten, pegylierten Lipiden (Endgruppenfunktionalisierung durch Amin- (Kirpotin et al., 1997), Thiol- (Allen et al., 1995) oder Hydrazongruppen (Zalipsky, 1993) zunächst Liposomen herzustellen und einen spezifischen Liganden nachträglich anzubringen. Der Nachteil bei dieser Methode ist, dass Gruppen, die nicht reagiert haben, zusätzlich vorhanden sind und weitere Anwendungen stören können. Die zweite Möglichkeit beinhaltet die Synthese eines Ligand-PEG-Lipid-Derivates, welches dann in die Liposomen inkorporiert wird. Hier ist es potentiell nachteilig, dass im Falle von SUVs (Liposomen im Größenbereich von 100 nm) etwas weniger als die Hälfte des Liganden in das Liposomeninnere zeigt, so dass nur ein Ligandanteil von etwa 55–60%, der sich auf der Aussenseite befindet, biologisch aktiv werden kann (Zalipsky et al., 1997). In der vorliegenden Arbeit wurde ebenfalls diese Form des Ligandeinbaus gewählt, da aus unserer Sicht mit dieser Methode das Gelingen des stabilen Einbaus des neusynthesierten Liganden im akzeptablen Ausmaß sehr wahrscheinlich erscheint. Die Tatsache, dass ein großer Teil an Ligandmolekülen bei dieser Methode in das Liposomeninnere gerichtet ist, kann wiederum auch im Hinblick auf die Untersuchungen zur Pharmakokinetik vorteilhaft sein. Eine hohe Liganddichte auf der Liposomenoberfläche erwirkt möglicherweise eine stark negativ geladene Oberfläche, die folglich die Wahrscheinlichkeit drastisch erhöht von Makrophagen erkannt und eliminiert zu werden. Außerdem steigert im gleichen Zusammenhang eine erhöhte Dichte von Liganden auf der Liposomenaussenseite wahrscheinlich die Interaktion zwischen Ligandmolekülen und DSPE-PEG-Ketten, die ebenfalls auf der Oberfläche vorhanden sind. Eine dritte Methode beschreibt die Herstellung von Leerliposomen, die dann nachträglich mit dem Ligand-PEG-Lipid-Konjugat inkubiert werden, und das Derivat sich somit in die Membran einbaut. Vorteile dieser Methode sind, dass bei guter Aufreinigung vor dem Einsatz des Ligandmoleküls keine störenden reaktiven Gruppen vorhanden sind und dass alle Ligandmoleküle auf der

Einleitung 41
Außenseite der Liposomen positioniert sind. Eine Einschränkung dieser Post-Insertions-Methode wiederum ist, dass Fettsäureketten in den Liposomenmembranen den Einbau der Liganden sterisch hindern können, und folglich ein geringes Ausmaß an Insertion erzielt wird (Steenpass et al., 2006). Bei einer unvollständig ablaufenden Kopplungsreaktion könnten zudem unkonjugierte Ligandmoleküle zurückbleiben und die weitere Anwendung der Liposomen maßgeblich beeinträchtigen (Sapra and Allen, 2003).
Liganden können aber auch nicht kovalent an Lipide gekoppelt werden. Dazu wird beispielsweise die sogenannte „Sandwich-Methode“ eingesetzt. Diese Methode beruht auf der Wechselwirkung zwischen Biotin mit Avidin oder Streptavidin (Rivnay et al., 1987, Zavaleta et al., 2007) aufgrund der bekannten hohen Affinität des Glykoproteins Avidin (gewonnen aus Hühnereinweiß) zu dem Vitamin Biotin. Die geeigneten Voraussetzungen des Proteins liegen in der Tetramerstruktur begründet, die aus vier identischen Monomeren aufgebaut ist, welche alle eine starke Bindung zu Biotin eingehen können (Green, 1975, Pugliese et al., 1994). Die Bindung zwischen Avidin und Biotin stellt sich sehr schnell ein und ist über weite pH- und Temperaturbereiche stabil. Streptavidin ist ein Eiweiß, welches aus Streptomyces avidinii isoliert wird, welches trotz seiner strukturellen Unterschiede zu Avidin eine ähnlich hohe Affinität zum Biotin aufweist, und somit ein alternatives Brückenmolekül darstellt. Ein Nachteil dieser „Sandwich-Methode“ ist jedoch, dass die Liganden und die Lipide durch die notwendige Biotinylierung in ihrer angestrebten Funktion beeinträchtigt werden können. Eine mögliche Immunogenität auf die durch dieses Bindungsystem eingebrachten körperfremden Proteine, ausgehend von Mensch und Tier, könnte die Anwendung der „Sandwich-Methode“ limitieren.
Die Vergangenheit hat gezeigt, dass die Ligandkopplung an Liposomen kein trivialer Schritt ist. Beim Design von ligandmodifizierten Liposomen im allgemeinen muss darauf geachtet werden, dass der Einbau des Liganden sowohl die Beladungskapazität als auch wiederum die Freisetzungsrate des Wirkstoffes aus den Liposomen nicht nachteilig beeinflusst. Eine weitere Voraussetzung ist, dass die Bindungsaffinität des Liganden nicht beinträchtigt wird und dass die Liposomen auch mit dem Einbau des Liganden in ihre Membran genügend Stabilität und ausreichende Zirkulationszeiten aufweisen, um die für sie vorgesehene Zielzelle erreichen zu können. Ein anderer wichtiger Aspekt bei der Entwicklung von ligandmodifizierten Liposomen ist, eine Bindung zwischen Liposom und Ligand zu wählen, die weder toxisch noch instabil ist. Ein klarer Vorteil von ligandmodifizierten Liposomen besteht darin, dass relativ

Einleitung 42
wenige Ligandmoleküle pro Liposom für ein effektives Targeting notwendig sind und relativ große Mengen an Wirksubstanz durch die Verkapselung in das Liposom an den Wirkort gebracht werden können. Zudem kann die Bindungsaffinität des Systems zur Zielzelle bzw. zum Zielrezeptor durch die Präsenz von mehreren Liganden auf der Liposomenoberfläche deutlich erhöht werden. Bisher veröffentlichte aktive Targeting-Strategien, bei denen Liposomen involviert waren, zeigen definitiv vielversprechende Ergebnisse (Oku et al., 1996, Schiffelers et al., 2003, Shmeeda et al., 2006, Hatakeyama et al., 2007), dennoch hat das aktive Targeting derzeit sicherlich auch entscheidene Nachteile. Zum einen ist die Kopplung von den oben genannten Liganden häufig ein umfangreiches Verfahren, welches zeit- und kostenaufwendig ist. Oftmals erweist sich auch die Charakterisierung des gewonnenen Systems als sehr schwierig. Eine immunogene Wirkung, die vom Liganden ausgehen kann, ist ebenfalls nicht zu verachten. Hinzu kommt, das Tumorzellen sehr komplexe Zielzellen darstellen, die, jenachdem in welchem Stadium sie sich zum Zeitpunkt der Liposomenapplikation befinden, unterschiedlich in ihrer Sensitivität für das zu bindene System sein können. Ein anderes Problem könnte die Selektivität des entwickelten Systems sein, d.h., das die ligandmodifizierten Liposomen direkt an das erste Epitop binden, das ihnen begegnet, obwohl dieses sich in der Peripherie des Tumors befindet und das Ziel der Liposomen aber beispielsweise der Tumor selbst ist. Bei dem hier vorgestellten Trägersystem wäre es ebenfalls denkbar, dass die Bisphosphonatliganden nicht ausschließlich das Calcium des Hydroxylapatits komplexieren, sondern bereits vor Erreichen des Ziels mit Salzen oder anderen Bestandteilen des Blutes eine Bindung eingehen und durch diese Absättigung möglicherweise in ihrer Bindung eingeschränkt werden.

Ziele der Arbeit 43
2 Ziele der Arbeit
Ziel der vorliegenden Arbeit war es, intravenös applizierbare Liposomen für die zielgerichtete Applikation an den Knochen zu entwickeln. Die Besonderheit dieser Liposomen sollte darin bestehen, dass sie ein neusynthetisiertes Bisphosphonsäurederivat als Liganden auf ihrer Oberfläche tragen, um dem System somit knochenaffine Eigenschaften zu verleihen. Diese ligandmodifizierten Liposomen könnten im nächsten Schritt möglicherweise als Therapieoption von Knochenmetastasen eingesetzt werden. Dabei sollen die wirkstoffbeladenen Liposomen dann aufgrund des hohen „Bone Turnovers“ und der vermehrten Durchblutung der metastasierten Regionen im Knochen mit Hilfe der maßgeschneiderten und knochenaffinen Bisphosphonatliganden an die extrazelluläre Matrix (Hydroxylapatit) binden und den Wirkstoff dort freigeben, so dass dieser idealerweise auf die in der nahen Umgebung liegenden Metastasen wirken kann (die Abbildung 7 gibt die pathologische Situation vom Knochen bei ausgeprägter Metastasierung des umliegenden Gewebes wieder, auf der die hier vorgestellte Targeting-Strategie basiert).

Ziele der Arbeit 44
Abbildung 7: Histologische Identifikation eines Ausschnittes vom metastasendurchsetzten, trabekulären Knochen einer Maus. Kollagen-Typ-1-Färbung eines longitudinalen Schnittes von einem langen Knochen, welcher dem linken Hinterbein entnommen wurde. Eine hohe Knochenmetastasendichte ersetzt hier das interstitielle Gewebe eines trabekulären Knochens; Metastasen und extrazelluläre Knochenmatrix liegen somit dicht beieinander, so dass der aus den verankerten Liposomen freigesetzte Wirkstoff auf die Metastasen wirken könnte. Zudem lässt die Abbildung 7 zahlreiche Blutgefäße innerhalb des betroffenen Gewebes erkennen, die die Zufuhr von ligandmodifizierten Liposomen gewährleisten könnten. Für die hier präsentierte Targeting-Strategie bedeutet es, dass in einem fortgeschrittenen Entwicklungsstadium, so wie es hier dargestellt ist, die pathologischen Gegebenheiten des metastasendurchsetzten Knochengewebes ihre Umsetzbarkeit realistisch erscheinen lassen (Quelle: Dr. Gabri van der Pluijm, Universität Leiden, Niederlande).
Design, Entwicklung und Machbarkeit stellten in diesem Zusammenhang wichtige Pfeiler der Dissertation dar. Die Überprüfung der therapeutischen Wirksamkeit von ligandmodifizierten Liposomen in vivo gehörte dabei nicht zu den Schwerpunkten der Arbeit und wurde folglich nicht getestet. Die Entwicklung des zielgerichteten Systems wird in Abschnitt 4.1 beschrieben. Dabei wurde insbesondere die Machbarkeit des Ligandeinbaus in die liposomale Membran überprüft. Die Vergangenheit hat gezeigt, dass die Inkorporierung von Liganden in Liposomen ein komplexer Vorgang ist, der nicht immer vollständig verläuft. Um das Gelingen des Einbaus in hohem Ausmaß zu garantieren, wurde ein neuer Bisphosphonatligand (CHOL-TOE-BP) synthetisiert, der neben einer Trisoxyethylen-bisphosphonsäure aus Cholesterol, einem wichtigen Bestandteil von Biomembranen, als Ankermolekül bestand. Ein vollständiger Einbau des eingesetzten CHOL-TOE-BP-Liganden in die Liposomenmembran würde im Vergleich zu den meisten bisher untersuchten knochenaffinen Trägersystemen,

Ziele der Arbeit 45
bestehend aus einem Bisphosphonatmolekül gekoppelt an eine Wirksubstanz, den entscheidenen Vorteil bringen, dass eine Vielzahl von Bisphosphonatliganden für die Bindung an Hydroxylapatit zur Verfügung stehen. Somit würde möglicherweise eine Affinitätserhöhung zum Knochen und eine Erhöhung der Bindungsstärke bewirkt werden. Trotz azider und voluminöser Eigenschaften des Ligandmoleküls war ein Ziel der Arbeit, Liposomen mit physiologischem pH-Wert und einer durchschnittlichen Größe von 100–200 nm herzustellen. Der pH-Wert und die Partikelgröße sind wichtige Parameter, die bei der In-vivo-Applikation von Relevanz sein können. Eine ausführliche physikochemische Charakterisierung der neuentwickelten Liposomen, die die Bestimmung der durchschnittlichen Größe, die Messung des Zeta-Potentials, Phasenübergangstemperaturbestimmungen sowie die Ligandquantifizierung mittels 31P-NMR umfasst, ist Bestandteil des Abschnittes 4.2. Im Rahmen der Entwicklung eines zielgerichteten Trägersystems für die Applikation an den Knochen ist die Überprüfung der Bindungsaffinität an die Mineralmatrix unerlässlich. Bisherige Bindungsversuche von liposomalen Systemen, die neben PEG weitere Liganden auf ihrer Oberfläche trugen, haben gezeigt, dass beide Moleküle miteinander interagieren können und einen negativen Einfluss auf die Bindung zum Zielgewebe haben können. Durch die Berücksichtigung eines Spacermoleküls zwischen dem Ankerrest und der Bisphosphonsäure in dem hier vorgestellten Liganden wurde vermutet, dass mögliche Interaktionen mit PEG-Ketten durch die beschriebene Modifikation zu verhindern waren. Deshalb wird dieser Parameter in Abschnitt 4.3 in Form von Hydroxylapatit(HAP)-Bindungsstudien näher untersucht. Basierend auf der bekannten hohen Affinität von Bisphosphonaten zur extrazellulären Matrix des Knochens wurde spekuliert, dass auch der CHOL-TOE-BP-Ligand in der Lage ist, an synthetisches HAP zu binden. Um die In-vivo-Situation im Bindungsexperiment besser nachzustellen, wurden die Bindungsversuche zusätzlich in Gegenwart von humanem Blutplasma durchgeführt. Wir formulierten die Hypothese, dass die Bindung von ligandmodifizierten Liposomen an die Mineralmatrix des Knochens in vivo durch Interaktionen mit verschiedenen Blutplasmabestandteilen wie geladenen Ionen oder Plasmaproteinen nachteilig beeinflusst wurde. Im Hinblick auf eine spätere therapeutische Anwendung von ligandmodifizierten Liposomen sind eine Bestimmung der Verkapselungseffizienz und die Untersuchung der Wirkstofffreisetzung wichtig. Um eine interessante Alternative zu anderen knochenaffinen Trägersystemen darstellen zu können, wurde eine hohe Verkapselungsrate angestrebt. Die meisten bisher untersuchten Systeme für die zielgerichtete Applikation an den Knochen bestanden aus einem Wirkstoff und dem Bisphosphonatderivat als Targeting-Einheit.

Ziele der Arbeit 46
Durch die Verkapselung von großen Wirkstoffmengen könnte die Effektivität maßgeblich gesteigert und die Häufigkeit der Applikation reduziert werden. Für diese Studien (Abschnitt 4.4) wurde Doxorubizin als Modellsubstanz ausgewählt. Gemäß Literatur lässt sich Doxorubizin mittels „Remote Loading“ in hohen Konzentrationen in pegylierte Liposomen verkapseln und auch wieder freisetzen. Es wurde vermutet, dass die CHOL-TOE-BP-Liganden bei gleicher Beladungsmethode die Verkapselungseffizienz aufgrund der nach innen ragenden Ligandmoleküle dahingehend veränderten, dass im Vergleich zu den publizierten Daten eine geringere Einschlussrate erzielt wurde. Bei der Untersuchung der Freisetzung wurde der Einfluss von verschiedenen pH-Werten untersucht, um die In-vivo-Bedingungen, die die Liposomen nach erfolgter Injektion auf dem Weg zum Zielorgan erfahren werden, möglichst realitätsnah nachzustellen. Angestrebt wurde in diesem Zusammenhang eine Freisetzung des Wirkstoffes, die erst am Wirkort, dem metastasendurchsetzten Knochengewebe, erfolgt. In Abschnitt 4.5 wird die Zelltoxizität von wirkstoffbeladenen, ligandmodifizierten Liposomen bestimmt. Aufgrund der bekannten toxischen Wirkung von Doxorubizin auf die ausgewählten Tumorzellen wurde der Wirkstoff auch in dieser Versuchsreihe eingesetzt. Zudem sollte untersucht werden, ob die Verkapselung von Doxorubizin in ligandmodifizierte Liposomen im Vergleich zu freiem Doxorubizin zu einer Potenzierung der antitumoralen Wirkung auf die ausgewählten Zellen führte. Eine erste In-vivo-Evaluierung zur Abschätzung der Umsetzbarkeit der Targeting-Strategie war für die weitere Entwicklung des hier präsentierten zielgerichteten Systems von entscheidender Bedeutung. Die Abschnitte 4.6 und 4.7 richten den Fokus genau auf diese In-vivo-Untersuchungen. Zunächst wurden die Pharmakokinetik und die Biodistribution von ligandmodifizierten Liposomen verschiedener Zusammensetzungen untersucht. Zusammen mit den zuvor generierten Daten wurde anhand dieser Ergebnisse die vielversprechendste Formulierung für die In-vivo-Überprüfung der Fähigkeit, die Knochenoberfläche nach intrakardialer Applikation zu erreichen, ausgewählt. In diesem Zusammenhang wurde angenommen, dass das Nacktmausmodell von Wetterwald et al. (Wetterwald et al., 2002) und eine Fluoreszenzmarkierung der Liposomen dazu geeignet sind, um in einem Pilotversuch die zielgerichtete Applikation im Vergleich zu Kontrollliposomen (ohne CHOL-TOE-BP) sichtbar machen zu können. Es wurde dazu ein lipophiler Farbstoff (NBD-PE) ausgewählt, um sicherzustellen, dass auch das vollständige liposomale Trägersystem verfolgt wird und nicht der gegebenenfalls hinausdiffundierte Farbstoff. Erste Versuche mit dem hydrophilen Fluoreszenzmarker Alexafluor-680 hatten eben dieses unerwünschte Phänomen gezeigt.

Materialien, Geräte und Methoden 47
3 Materialien, Geräte und Methoden
3.1 Materialien
3.1.1 Lipide und andere Membrankomponenten
Tabelle 1: Übersicht über verwendete Lipide und Membrankomponenten
Bezeichnung Abkürzung MW [g/mol] Hersteller
Cholesteryl-bisphosphonsäure CHOL-BP 560,64 Universität Bremen
Cholesteryl-trisoxyethylen-bisphosphonsäure CHOL-TOE-BP 722,84 Universität Bremen
Cholesteryl-tetraoxyethylen-bisphosphonsäure CHOL-TeOE-BP 766,88 Universität Bremen
Palmitoyl-bisphosphonsäure PAL-BP 402,40 Universität Bremen
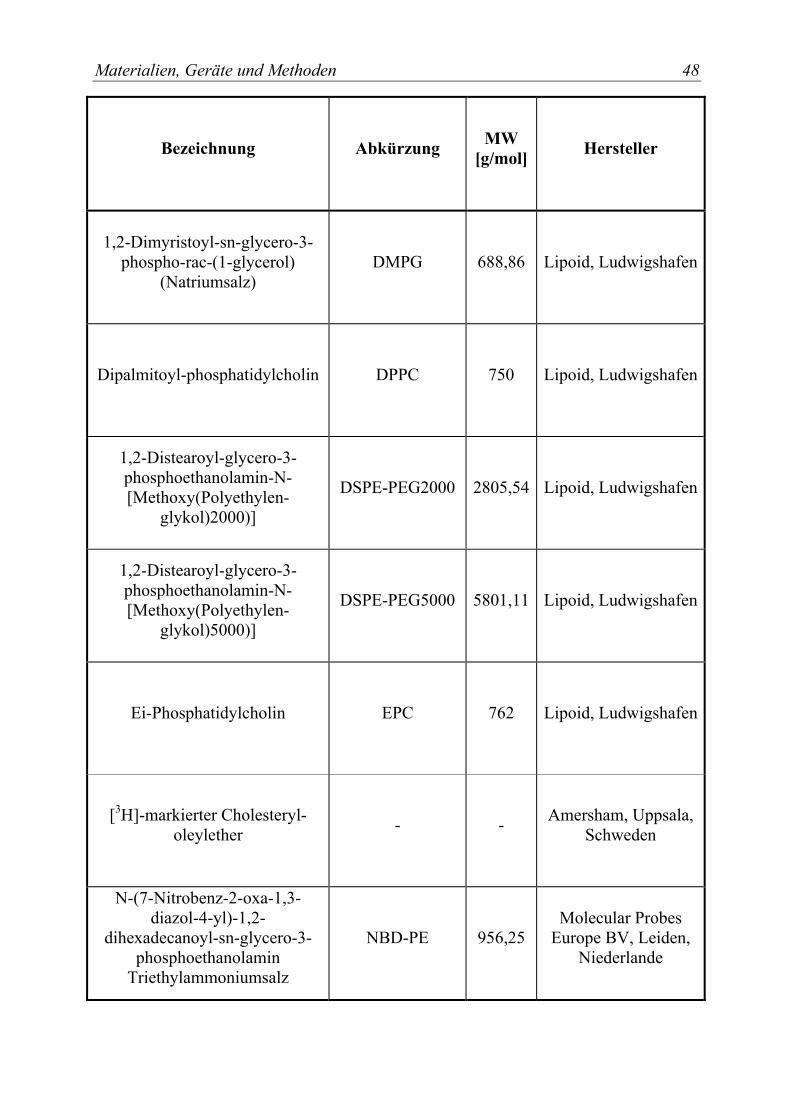
Materialien, Geräte und Methoden 48
Bezeichnung Abkürzung MW [g/mol] Hersteller
1,2-Dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol)
(Natriumsalz) DMPG 688,86 Lipoid, Ludwigshafen
Dipalmitoyl-phosphatidylcholin DPPC 750 Lipoid, Ludwigshafen
1,2-Distearoyl-glycero-3-phosphoethanolamin-N-[Methoxy(Polyethylen-
glykol)2000)]
DSPE-PEG2000 2805,54 Lipoid, Ludwigshafen
1,2-Distearoyl-glycero-3-phosphoethanolamin-N-[Methoxy(Polyethylen-
glykol)5000)]
DSPE-PEG5000 5801,11 Lipoid, Ludwigshafen
Ei-Phosphatidylcholin EPC 762 Lipoid, Ludwigshafen
[3H]-markierter Cholesteryl-oleylether - - Amersham, Uppsala,
Schweden
N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-
dihexadecanoyl-sn-glycero-3-phosphoethanolamin
Triethylammoniumsalz
NBD-PE 956,25 Molecular Probes
Europe BV, Leiden, Niederlande
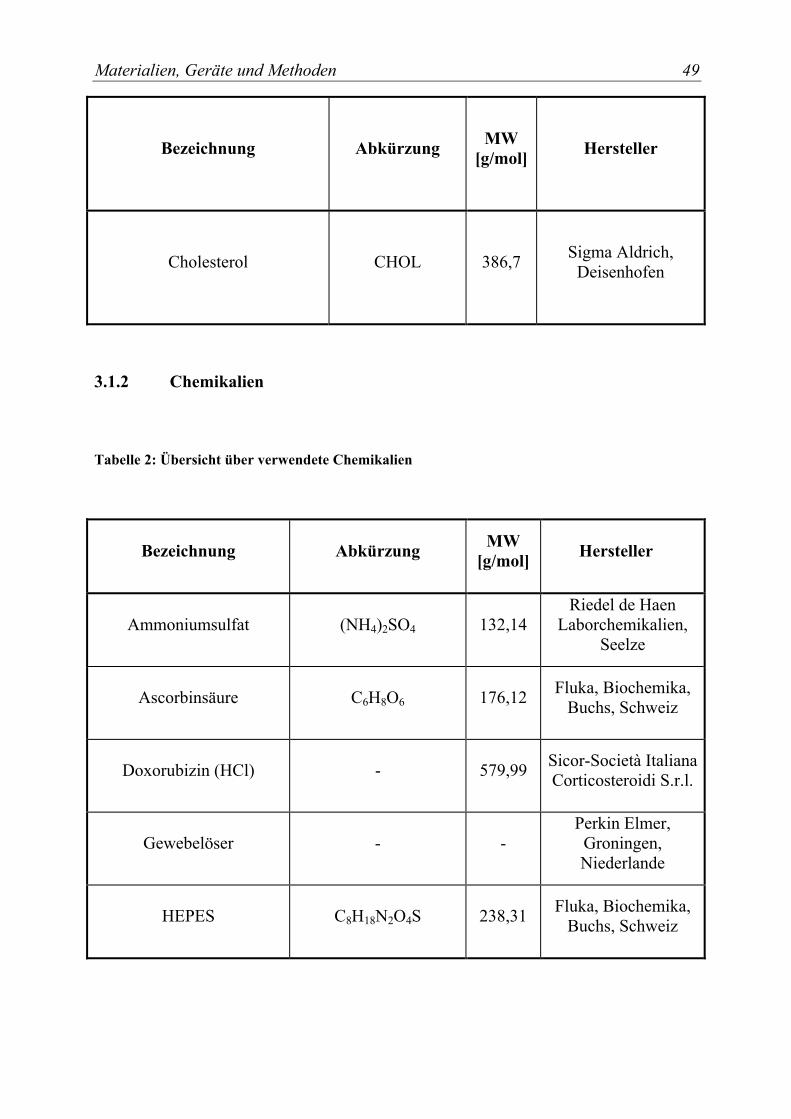
Materialien, Geräte und Methoden 49
Bezeichnung Abkürzung MW [g/mol] Hersteller
Cholesterol CHOL 386,7 Sigma Aldrich, Deisenhofen
3.1.2 Chemikalien
Tabelle 2: Übersicht über verwendete Chemikalien
Bezeichnung Abkürzung MW [g/mol] Hersteller
Ammoniumsulfat (NH4)2SO4 132,14 Riedel de Haen
Laborchemikalien, Seelze
Ascorbinsäure C6H8O6 176,12 Fluka, Biochemika, Buchs, Schweiz
Doxorubizin (HCl) - 579,99 Sicor-Società Italiana Corticosteroidi S.r.l.
Gewebelöser - - Perkin Elmer,
Groningen, Niederlande
HEPES C8H18N2O4S 238,31 Fluka, Biochemika, Buchs, Schweiz

Materialien, Geräte und Methoden 50
Bezeichnung Abkürzung MW [g/mol] Hersteller
Hexa-ammoniummolybdat (NH4)6MO7O24x4H2O 1235,86 Merck, Darmstadt
Hydroxylapatit HAP 1004,60 Callbiochem, CA, USA
Natriumchlorid NaCl 58,44 Fluka, Biochemika, Buchs, Schweiz
Natriumhydroxid NaOH 40,00 Fluka, Biochemika, Buchs, Schweiz
Phosphat-gepufferte Kochsalzlösung PBS - Sigma Aldrich,
Deisenhofen
Phosphatstandard 0,5 mM - - Sigma Aldrich, Deisenhofen
Triton-X-100 (4-1,1,3,3-tetramethylbutyl)phe-nylpolyethylenglykol
C22H38O5 ~ 625 Fluka, Biochemika, Buchs, Schweiz
Ultima-Gold-Scintillation-Cocktail - -
Perkin Elmer, Groningen, Niederlande
Uranylacetat (Dihydrat) UO2(OCOCH3)2x2H2O 425,15 Merck, Darmstadt
Wasserstoffperoxid 35% H2O2 34,01 Sigma Aldrich, Deisenhofen
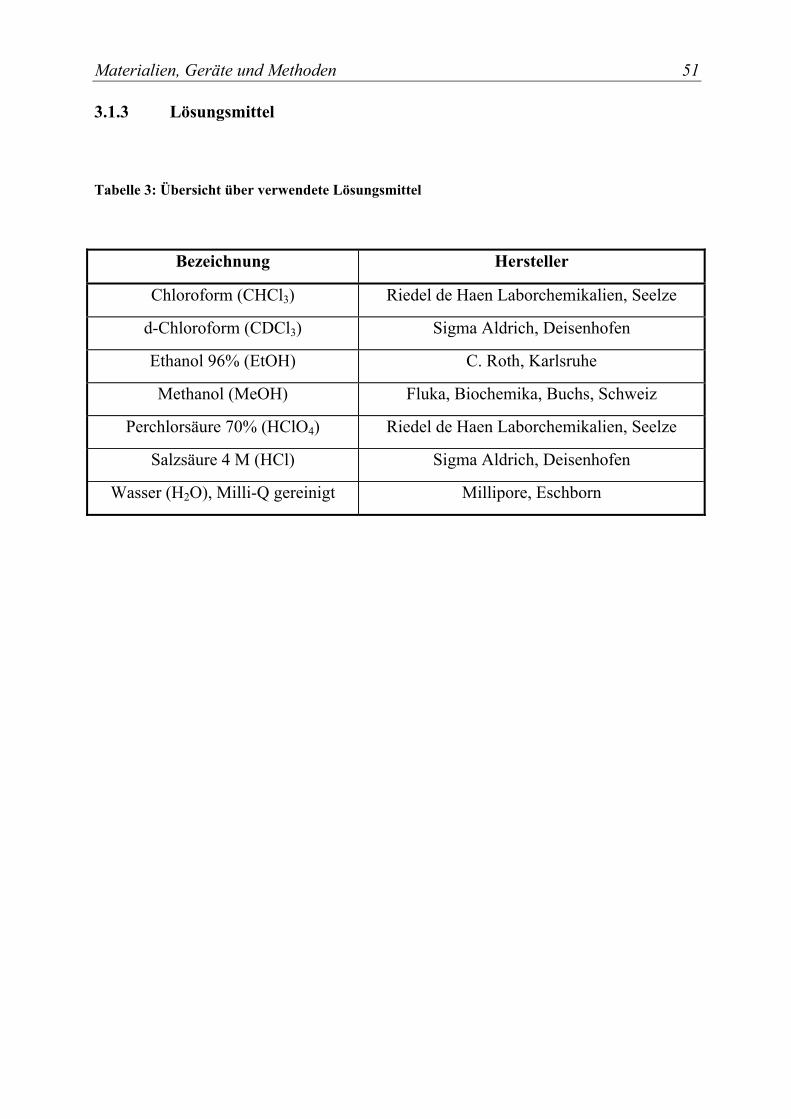
Materialien, Geräte und Methoden 51
3.1.3 Lösungsmittel
Tabelle 3: Übersicht über verwendete Lösungsmittel
Bezeichnung Hersteller
Chloroform (CHCl3) Riedel de Haen Laborchemikalien, Seelze
d-Chloroform (CDCl3) Sigma Aldrich, Deisenhofen
Ethanol 96% (EtOH) C. Roth, Karlsruhe
Methanol (MeOH) Fluka, Biochemika, Buchs, Schweiz
Perchlorsäure 70% (HClO4) Riedel de Haen Laborchemikalien, Seelze
Salzsäure 4 M (HCl) Sigma Aldrich, Deisenhofen
Wasser (H2O), Milli-Q gereinigt Millipore, Eschborn

Materialien, Geräte und Methoden 52
3.1.4 Puffer und wäßrige Lösungen
Tabelle 4: Übersicht über verwendete Puffer und Lösungen
Bezeichnung Komponenten Zusammensetzung/pH
Ammoniumsulfatlösung
240 mM Ammoniumsulfat
1 mM Titriplex
Wasser
3,17 g
0,037 g
ad 100 ml
Ascorbinsäurelösung 5% (w/v) Ascorbinsäure
H2O
1,00 g
ad 20 ml
HCl/Ethanol-Mischung für Doxorubizin Detektion
4 M HCl
96% EtOH
37,8 ml
500 ml
HBS
5 mM HEPES,
1 N NaOH,
H2O
1,19 g
q.s. pH 7,4
ad 1 l
HBSpH7,4
50 mM HEPES,
0,7 % NaCl,
1 M NaOH,
H2O
11, 92 g
7,00 g
q.s. pH 7,4
ad 1 l
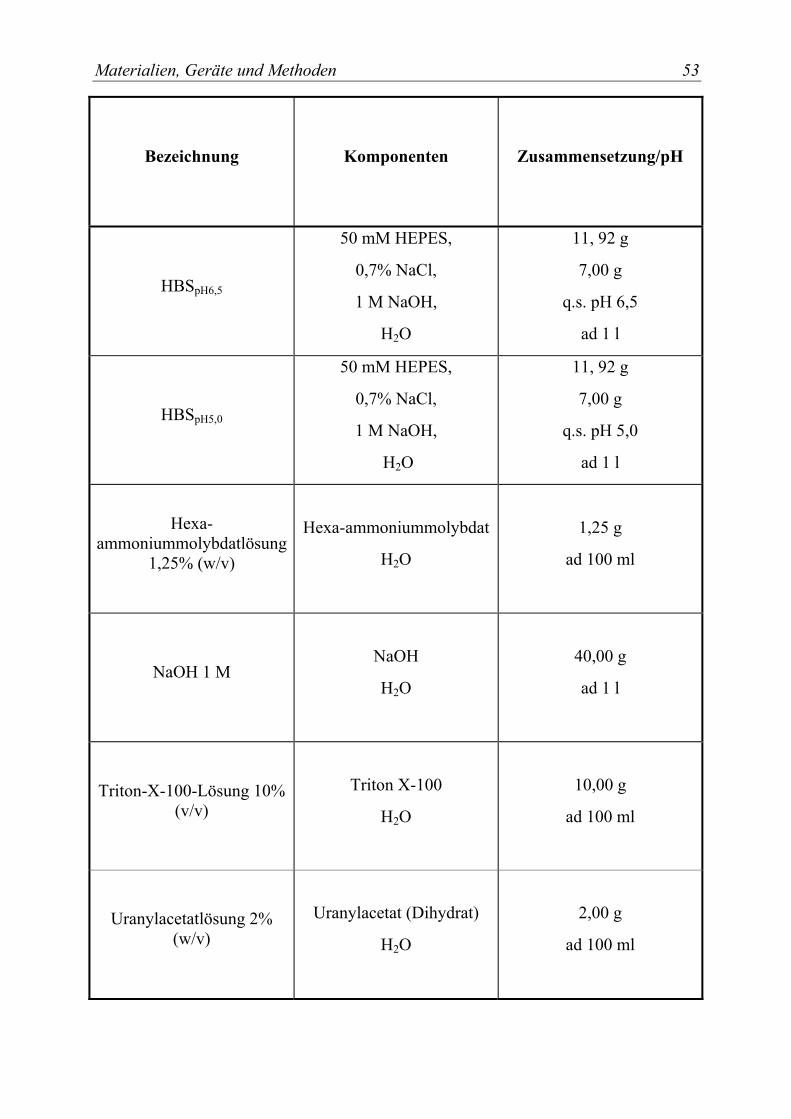
Materialien, Geräte und Methoden 53
Bezeichnung Komponenten Zusammensetzung/pH
HBSpH6,5
50 mM HEPES,
0,7% NaCl,
1 M NaOH,
H2O
11, 92 g
7,00 g
q.s. pH 6,5
ad 1 l
HBSpH5,0
50 mM HEPES,
0,7% NaCl,
1 M NaOH,
H2O
11, 92 g
7,00 g
q.s. pH 5,0
ad 1 l
Hexa-ammoniummolybdatlösung
1,25% (w/v)
Hexa-ammoniummolybdat
H2O
1,25 g
ad 100 ml
NaOH 1 M NaOH
H2O
40,00 g
ad 1 l
Triton-X-100-Lösung 10% (v/v)
Triton X-100
H2O
10,00 g
ad 100 ml
Uranylacetatlösung 2% (w/v)
Uranylacetat (Dihydrat)
H2O
2,00 g
ad 100 ml

Materialien, Geräte und Methoden 54
3.1.5 Zellkulturmaterialien
Tabelle 5: Übersicht über verwendete Zellkulturmaterialien
Bezeichnung Hersteller
Dulbecco´s-Modified-Eagle´s-Medium Biochrom, Basel, Schweiz
Minimum-Essential-Medium Gibco BRL, Life Technologies, Basel Schweiz
RPMI 1640 Gibco BRL, Life Technologies, Basel Schweiz
Fötales Kälberserum Biowhittaker, Verviers, Belgien
cDNA Invitrogen, Breda, Niederlande
pcDNA 3,1-Plasmid Invitrogen, Breda, Niederlande
G418 Gibco BRL Life Technologies, Basel, Schweiz
FuGene6 Roche, Biochemicals, Almere, Niederlande
Tryptanblaulösung 0,4% PAA Laboratories, Cölbe
Trypsin/EDTA-Lösung Gibco BRL, Life Technologies, Basel Schweiz
Formalinlösung 3% Merck, Darmstadt
Kristallviolettlösung 1% Merck, Darmstadt
D-Luciferin Synchem OHG, Felsberg/Altenburg

Materialien, Geräte und Methoden 55
3.1.6 Verbrauchsmaterialien
Tabelle 6: Übersicht über verwendete Verbrauchsmaterialien
Bezeichnung Hersteller
Aluminiumtiegel Perkin Elmer, Groningen, Niederlande
Einmalküvetten (Polystyrol), 4,5 ml C. Roth, Karlsruhe
Einmalpipetten, 1 ml, unsteril C. Roth, Karlsruhe
Eppendorfgefäße (Safe-Lock-Tubes), 2 ml Eppendorf AG, Hamburg
Glaskügelchen, 5 mm Durchmesser Riedel de Haen Laborchemikalien, Seelze
Mikrotiterplatte (96-Well-Platte) Greiner-Bio One, Frickenhausen
NMR-Röhrchen, Welmad, 5 mm, 300 MHz Sigma Aldrich, Deisenhofen
PD-10-Säule GE Healthcare Biosciences AB, Uppsala, Schweden
Pipettenspitzen (epT.I.P.S Standard) Eppendorf AG, Hamburg
Plastikröhrchen Greiner-Bio One, Frickenhausen
Polycarbonatmembranen, 0,1–0,6 µm Porengröße (Nucleopore Tracketch Membrane)
Whatman International Middlesex, UK
Porzellanmurmeln VWR International, Darmstadt
Reagenzgläser, Glas Hecht-Assistant, Sondheim

Materialien, Geräte und Methoden 56
Bezeichnung Hersteller
Slide-A-Lyzer-Dialyse-Kassette, 0,5–3 ml, MWCO: 10000 Pierce, Rockford, USA
Spritzenvorsatzfilter, Minisart, 0,2 µm Sartorius, Göttingen
Zellkulturschalen, Cellstar 658170 Greiner-Bio One, Frickenhausen
Zellkulturflaschen, Cellstar 69016 Greiner-Bio One, Frickenhausen
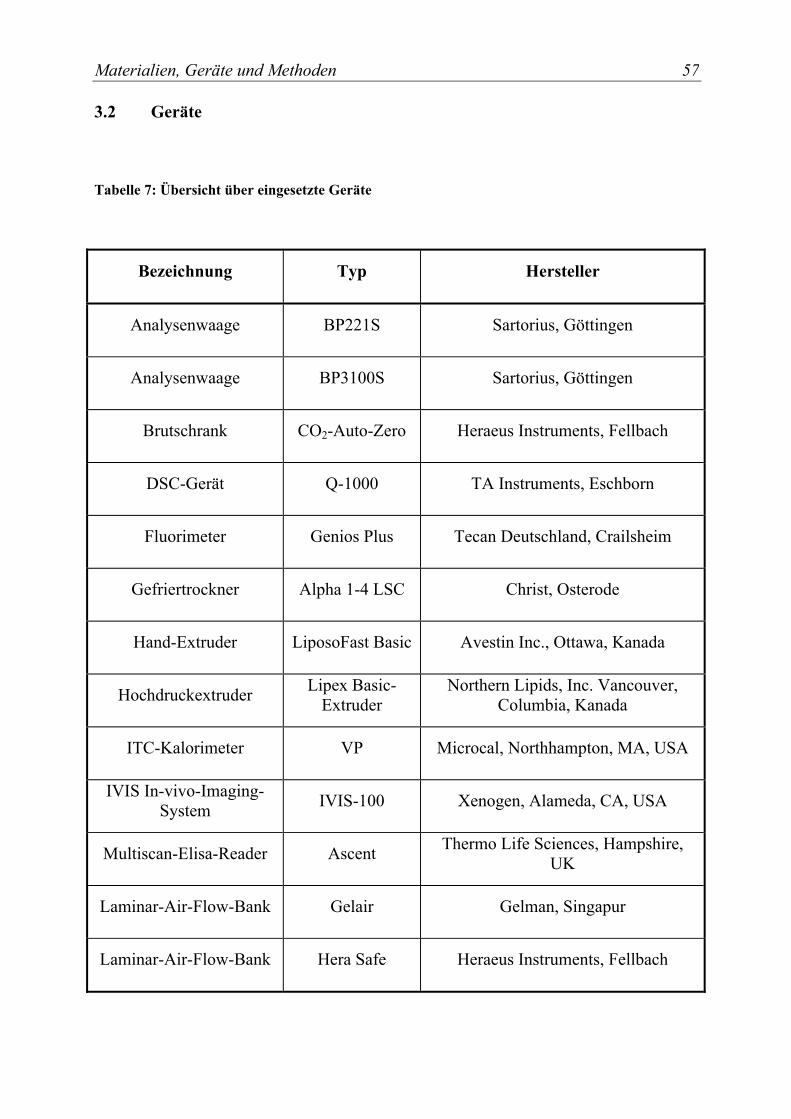
Materialien, Geräte und Methoden 57
3.2 Geräte
Tabelle 7: Übersicht über eingesetzte Geräte
Bezeichnung Typ Hersteller
Analysenwaage BP221S Sartorius, Göttingen
Analysenwaage BP3100S Sartorius, Göttingen
Brutschrank CO2-Auto-Zero Heraeus Instruments, Fellbach
DSC-Gerät Q-1000 TA Instruments, Eschborn
Fluorimeter Genios Plus Tecan Deutschland, Crailsheim
Gefriertrockner Alpha 1-4 LSC Christ, Osterode
Hand-Extruder LiposoFast Basic Avestin Inc., Ottawa, Kanada
Hochdruckextruder Lipex Basic-Extruder
Northern Lipids, Inc. Vancouver, Columbia, Kanada
ITC-Kalorimeter VP Microcal, Northhampton, MA, USA
IVIS In-vivo-Imaging-System IVIS-100 Xenogen, Alameda, CA, USA
Multiscan-Elisa-Reader Ascent Thermo Life Sciences, Hampshire, UK
Laminar-Air-Flow-Bank Gelair Gelman, Singapur
Laminar-Air-Flow-Bank Hera Safe Heraeus Instruments, Fellbach
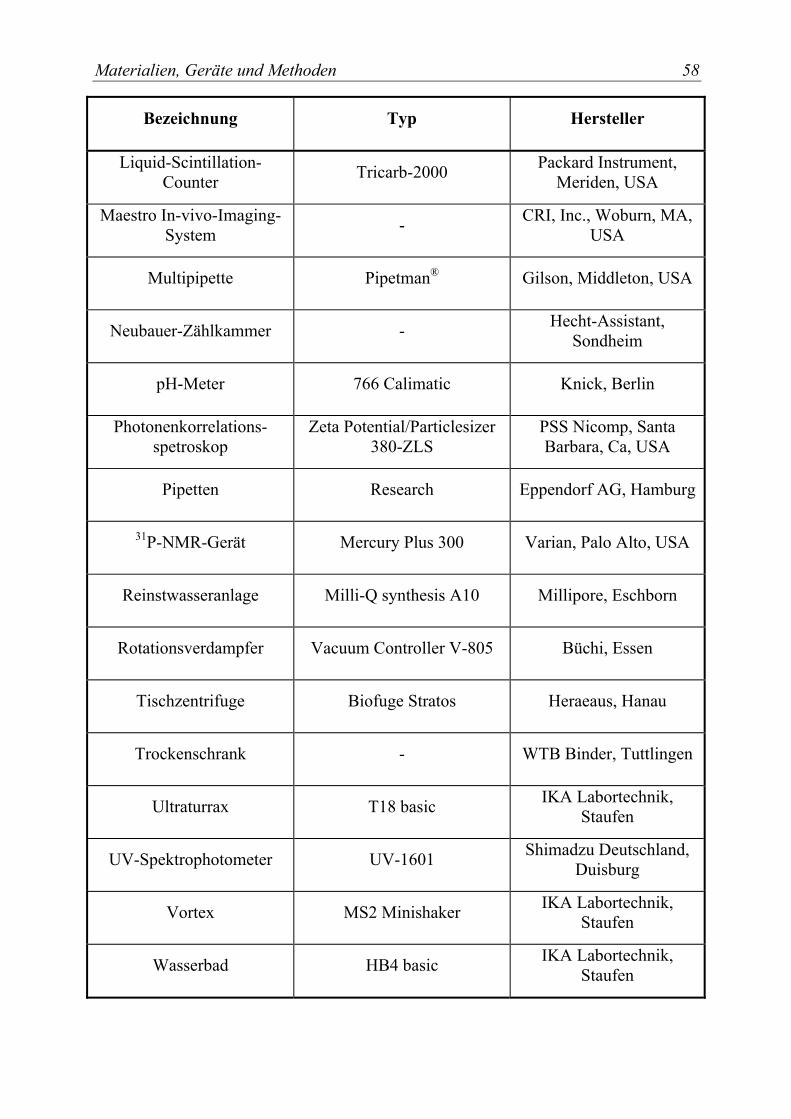
Materialien, Geräte und Methoden 58
Bezeichnung Typ Hersteller
Liquid-Scintillation-Counter Tricarb-2000 Packard Instrument,
Meriden, USA
Maestro In-vivo-Imaging-System - CRI, Inc., Woburn, MA,
USA
Multipipette Pipetman® Gilson, Middleton, USA
Neubauer-Zählkammer - Hecht-Assistant, Sondheim
pH-Meter 766 Calimatic Knick, Berlin
Photonenkorrelations-spetroskop
Zeta Potential/Particlesizer 380-ZLS
PSS Nicomp, Santa Barbara, Ca, USA
Pipetten Research Eppendorf AG, Hamburg
31P-NMR-Gerät Mercury Plus 300 Varian, Palo Alto, USA
Reinstwasseranlage Milli-Q synthesis A10 Millipore, Eschborn
Rotationsverdampfer Vacuum Controller V-805 Büchi, Essen
Tischzentrifuge Biofuge Stratos Heraeaus, Hanau
Trockenschrank - WTB Binder, Tuttlingen
Ultraturrax T18 basic IKA Labortechnik, Staufen
UV-Spektrophotometer UV-1601 Shimadzu Deutschland, Duisburg
Vortex MS2 Minishaker IKA Labortechnik, Staufen
Wasserbad HB4 basic IKA Labortechnik, Staufen

Materialien, Geräte und Methoden 59
3.3 Methoden
3.3.1 Ligandsynthese
Die hier beschriebenen Bisphosphonatliganden, darunter Cholesteryl-bisphosphonsäure, Cholesteryl-trisoxyethylen-bisphosphonsäure, Cholesteryl-tetraoxyethylen-bisphosphonsäure sowie Palmitoyl-bisphosphonsäure wurden von Dr. Oleg Shyshkov (Institut für Anorganische und Physikalische Chemie, Universität Bremen) synthetisiert.
3.3.2 Zubereitung von ligandmodifizierten Liposomen
Ligandmodifizierte Liposomen wurden mit Hilfe der Lipidfilm-Extrusionsmethode hergestellt. Hierzu wurden die berechneten Bestandteile in einen Rundkolben eingewogen und abhängig vom Liganden in unterschiedlichen organischen Lösungsmitteln gelöst. Dabei wurden die Ansätze, die den Liganden CHOL-BP, CHOL-TOE-BP oder CHOL-TeOE-BP enthielten, in Chloroform gelöst. Ansätze, die unter anderem aus dem PAL-BP-Liganden bestanden, wurden hingegen in Methanol gelöst. Das jeweilige Lösungsmittel (Methanol oder Chloroform) wurde am Vakuumrotationsverdampfer bei 40 °C abrotiert. Zur vollständigen Entfernung von Lösungsmittelspuren wurde der Lipidfilm für 30 Minuten unter Stickstoffzufuhr getrocknet. Im Anschluß wurde eine entsprechende Menge an HEPES-Puffer (HBSpH7,4; 50 mmol HEPES, 0,7 % NaCl, pH 7,4) hinzugegeben. Durch Schütteln mit der Hand wurde der Film von der Glaswand des Kolbens abgelöst. Durch das Resuspendieren des Filmes im Puffer entstanden multilamellare Liposomen (MLVs), welche durch Hochdruckhomogenisation verkleinert wurden. Die Liposomendispersion wurde mehrfach mit Hilfe eines Lipex-Extruders durch die folgende Filtersequenz filtriert, so dass unilamellare Liposomen (LUVs) im Größenbereich von 100 bis 160 nm entstanden: 0,6 µm, 0,4 µm, 0,2 µm und 0,1 µm Polycarbonatmembranen. Die Liposomengröße wurde durch PCS-Messungen kontrolliert. Zur Bestimmung des Pharmakokinetikprofils in Ratten wurden ligandmodifizierte Liposomen und Kontrollliposomen mit einer Tracermenge an [3H]-

Materialien, Geräte und Methoden 60
markiertem Cholesteryl-oleylether hergestellt. Für die Lokalisierung dieser Liposomen in BALB-c-nu/nu-Mäusen wurden beide Liposomentypen jeweils mit Fluoreszenz (1 mol% NBD-PE) markiert.
3.3.3 In-vitro-Charakterisierung
3.3.3.1 Bestimmung der Partikelgrösse
Die Größe der Liposomen wurde mittels Photonen-Korrelations-Spektroskopie („Photon Correlation Spectroscopy“, PCS) bestimmt. Die Photonen-Korrelations-Spektroskopie ist ein dynamisches Streulichtverfahren, welches die Bestimmung der Partikelgrößen im Durchmesserbereich von 0,1 bis 5 µm (Herstellerangaben) ermöglicht. Die Probe wird bei dieser Bestimmung mit einem Laser bestrahlt. Die dadurch erwirkte Lichtstreuung wird zeitabhängig erfasst. Schwankungen des Streulichtes sind abhängig von der Brown´schen Molekularbewegung, welche wiederum abhängig von der Größe der zu bestimmenden Partikel ist. Das Streulicht wird in einem Meßwinkel von 90 °C gemessen und mit Hilfe eines Photomultipliers verstärkt. Aus der Fluktuation der Streulichtmuster wird in Abhängigkeit von der Zeit eine Autokorrelationsfunktion erstellt, deren Steilheit vom Diffusionskoeffizienten der Teilchen abhängig ist. Bei bekannter Temperatur und Viskosität des Mediums erhält man als Ergebnis bei der PCS den mittleren hydrodynamischen Teilchendurchmesser sowie den Polydispersionsindex (PI). Der PI ist ein Maß für die Breite der Verteilung. Ein PI-Wert von Null besagt, dass ein absolut monodisperses System vorliegt und ein PI-Wert von 1 beschreibt ein komplett heterodisperses System. Zur Bestimmung der durchschnittlichen Partikelgröße von Liposomenproben wurde ein NicompTM Particlesizer-380-ZLS verwendet. Um die Genauigkeit des Gerätes zu überprüfen, wurden uniforme Standard-Polystyren-Partikel (199 ± 6 nm) (Duke Scientific, Palo Alto, CA, USA) in Wasser vermessen. Liposomenproben, 40 µl gelöst in 2 ml filtriertem HBSpH7,4 (durch einen 0,22 µm Sterilfilter), wurden bei 25 °C vermessen. Jede Probe wurde 15 min lang untersucht, wobei eine Zwischenspeicherung der Ergebnisse jeweils nach 5 min erfolgte. Am Ende lagen drei Einzelmessungen vor, aus denen wiederum ein Mittelwert gebildet wurde. Die Auswertung erfolgte entsprechend einer Gauß-Verteilung (monomodale Verteilung) oder einer Nicomp-Verteilung im

Materialien, Geräte und Methoden 61
Fall von bimodalen Verteilungen mit Hilfe der CW370-Software. Dabei wurden intensitätsgewichtete Verteilungen von Partikelgrößen bestimmt.
3.3.3.2 Bestimmung des Zeta-Potentials
Das Zeta-Potential wird durch die Bestimmung der elektrophoretischen Beweglichkeit der Partikelladung im elektrischen Feld ermittelt. In einer Lösung dispergierte, kolloidale Teilchen sind aufgrund ihrer ionischen und dipolaren Eigenschaften elektrisch geladen. Auf der Oberfläche von Teilchen im flüssigen Medium kommt es zur Adsorption von Ionen in zwei Schichten, bezeichnet als innere und äußere Sternschicht. Diese beiden Schichten sind wiederum von einer weiteren sogenannten diffusen Schicht umgeben, die aus locker gebundenen Gegenionen besteht. Sobald ein elektrisches Feld aufgebaut wird, bewegen sich die geladenen Teilchen auf die entgegengesetzt geladene Elektrode zu. Durch diese Diffusionsbewegung der Teilchen kommt es zur Reibung bzw. zu einer teilweisen Abstreifung der diffusen Schicht. Das elektrische Potential zwischen dem neutralen Umgebungsmilieu und der Scherebene, die nach dem Abstreifen der diffusen Schicht an den Partikeln entsteht, wird als Zeta-Potential bezeichnet. Während der Zeta-Potentialbestimmung wird die Messzelle mit Laserlicht einer bestimmten Frequenz bestrahlt, welches durch die Bewegung der Teilchen gestreut wird. Die Frequenz des gestreuten Laserlichts ist folglich von der Frequenz des ursprünglichen Laserstrahls verschieden (Doppler Shift). Basierend auf dieser Frequenzverschiebung wird die durchschnittliche Partikelgeschwindigkeit berechnet. Die einzelnen Zeta-Potentiale von verschiedenen Liposomenproben wurden mit einem NicompTM Zetaziser-380-ZLS bestimmt. Zur Überprüfung der Geräteeinstellung wurden Standard-Latex-Beads mit einem Zeta-Potential von -50 ± 5 mV (Malvern Ltd, Malvern, UK) vermessen. Das Zeta-Potential einer 0,7 ml-Liposomenprobe (1:100 verdünnt in 5 mM HEPES-Puffer, pH 7,4; HBS) wurde in dreifacher Ausführung bei 25 °C gemessen. Die Ergebnisse wurden mit der CW370-Software ausgewertet.

Materialien, Geräte und Methoden 62
3.3.3.3 Dynamische Differenz-Kalorimetrie
Die Dynamische Differenz-Kalorimetrie („Differential Scanning Calorimetry“, DSC) ist ein Meßverfahren zur Bestimmung von Wärmestromdifferenzen zwischen einer Probe und einer Referenzprobe, die beide einem vorgegeben Temperatur-Zeit-Programm unterworfen werden. Zur Messung werden ein mit Probe gefüllter und ein leerer Aluminiumtiegel (Referenz) so beheizt, dass beide Tiegel zu jedem Zeitpunkt der Messung dieselbe Temperatur besitzen, d.h., sobald physikalische oder chemische Vorgänge ablaufen, die mit einer Änderung der Reaktionswärme einhergehen, wird die entstehende Temperaturdifferenz zwischen Probe und Referenz durch indivduell benötigte Heizleistungen kompensiert. Somit ist die Wärmestromdifferenz gleich der elektrischen Heizleistungsdifferenz für die Probe und die Referenz. Die Differenz der Wärmeströme wird als Funktion gegen die Temperatur aufgetragen. In der vorliegenden Arbeit wurde mit Hilfe dieser Technik der Einfluss von CHOL-TOE-BP auf den temperaturabhängigen Phasenübergang von DPPC-Liposomen untersucht. Bei niedrigeren Temperaturen weist die Lipidmembran der Liposomen eine feste, gelartige Struktur auf. Bei höheren Temperaturen gehen die membranbildenden Lipide in den flüssigkristallinen Zustand über. Die Messungen wurden mit dem DSC-Gerät Q-1000 von TA Instruments durchgeführt. Dazu wurden 5 µl-Proben in dreifacher Ausführung in Aluminiumtiegel gefüllt und fest verschlossen. Als Referenz dienten leere Aluminiumtiegel. Zur Kühlung des Ofens wurde dieser von Flüssigstickstoff mit einer Flussgeschwindigkeit von 50 ml/min umspült. Es wurde ein Temperaturbereich von -5 °C bis 80 °C abgefahren. Die Scan-Rate betrug 5 °C/min. Die Thermogramme wurden mithilfe der gerätespezifischen Software Universal-V42E, TA Instruments erstellt.
3.3.3.4 31Phosphor-Kernresonanz-Spektroskopie
Das Prinzip der 31Phosphor-Kernresonanz-Spektroskopie („31P-Nuclear Magnetic Resonance“, 31P-NMR) basiert auf der Beobachtung des Verhaltens von Atomkernen im Magnetfeld. Voraussetzung für die Ausführung dieser Art von Messung ist ein permanentes, magnetisches Moment, welches durch den Kernspin der Atome verursacht wird. Durch die Zufuhr von Energie in Form eines Magnetfeldes können

Materialien, Geräte und Methoden 63
Atomkerne in eine energiereichere Stellung übergehen und anschließend unter der Abgabe von Energie in die energieärmere Stellung zurückkehren. Zur Durchführung der Kernspektroskopie wird ein Kernresonanzspektrometer, ein homologes Magnetfeld, ein Hochfrequenzsender zur Bestrahlung der Probe und einen Empfänger, der den Betrag der von den Kernen aufgenommenen Energie misst und im Kernresonanzspektrum registriert, benötigt. Dazu befindet sich die Probe in einem Glasröhrchen, welches um seine Längsachse rotiert, so dass horizontale Feldinhomogenitäten ausgemittelt werden. Die elektromagnetische Strahlung, ausgehend von einer Spule des Hochfreqenzsenders, wirkt senkrecht zur Magnetfeldrichtung auf die Probe ein. Das Proberöhrchen ist von einer Empfängerspule umgeben, welche senkrecht zur Senderspule und zum Magnetfeld steht und die von der Probe ausgehende Strahlung registriert. In der vorliegenden Arbeit wurde die 31P-NMR-Analytik eingesetzt, um die Verkapselungseffizienz des CHOL-TOE-BP-Liganden in die Liposomenmembran im Verhältnis zum Lipid DPPC zu bestimmen. Liposomenproben von 1 ml wurden in dreifacher Ausführung sowohl vor dem Extrusionsschritt als auch nach diesem Prozess entnommen und im Anschluß gefriergetrocknet. Die gefriergetrockneten Rückstände wurden jeweils in 1 ml deuteriertem Chloroform (CDCl3) resupendiert. 300 µl jeder Probe wurde dann mit 300 µl eines zuvor hergestellten internen Standards, bestehend aus 10 mg/ml PAL-BP, gelöst in Methanol, jeweils in einem 31P-NMR-Röhrchen versetzt und für 10 Sekunden gevortext. In Anschluß daran wurden die 31P-NMR-Spektren dieser Mischungen mit einem Varian Mercury-Plus-300-Instrument (300 MHz) aufgenommen. Die Messzeit betrug 8 min 13 sec. Mit Hilfe des Programms VNMR-6.1C wurden die Ergebnisse ausgewertet. Die relative verkapselte Menge an CHOL-TOE-BP und DPPC wurde bestimmt, indem die Integrationswerte der 31P-NMR-Signale vor und nach der Extrusion miteinander verglichen wurden. Das integrierte Signal des PAL-BP-Standards wurde dabei als 100%-Wert gesetzt.
3.3.3.5 Phospholipidbestimmung nach Rouser
Dieser Methode liegt die spektrophotometrische Bestimmung von anorganischem Phosphat zugrunde (Rouser et al., 1970). Dabei werden die Phospholipide der Liposomen zunächst mit Perchlorsäure zu anorganischem Phosphat umgesetzt. Dieses Phosphat wird wiederum zu Phosphomolybdänsäure umgewandelt und diese dann

Materialien, Geräte und Methoden 64
durch die Zugabe von Ascorbinsäure quantitativ in einen blaugefärbten Komplex reduziert, der photometrisch bei 797 nm bestimmt werden kann. Liposomenproben von 3 µl im Falle einer zu erwartenden Phospholipid-Konzentration von 20 µmol/ml wurden in dreifacher Ausführung in phosphatfreie Glasröhrchen pipettiert. Dazu wurde eine Kalibrationsgerade (Doppelbestimmung), hergestellt aus einem 0,5-mM-Phosphatstandard, ebenfalls in phosphatfreie Glasröhrchen pipettiert. Diese Proben wurden dann in einem Trockenschrank oder in einem Heizblock bei 120 °C verdampft. Dann wurden alle Proben mit 300 µl Perchlorsäure versetzt. Die Glasröhrchen wurden im Anschluss mit Porzellanmurmeln verdeckt und erneut in den Trockenschrank bzw. den Heizblock gegeben. Die Röhrchen wurden bei 180 °C solange erhitzt, bis in allen Proben eine klare Lösung zu erkennen war. Nach Erhalt einer klaren Lösung wurden die Proben auf Raumtemperatur abgekühlt. Dann wurden alle Röhrchen mit 1 ml Wasser ergänzt. Außerdem wurden 400 µl einer 1,2%(w/v)-Ammoniummolybdatlösung und dann 400 µl einer frisch zubereiteten 5%(w/v)-Ascorbinsäurelösung hinzupipettiert. Der Inhalt wurde durch kräftiges Schütteln vermischt. Danach wurden die Glasröhrchen erneut mit den Porzellanmurmeln abgedeckt und für 5 min in kochendes Wasser gegeben. Zuletzt wurden die Proben auf Raumtemperatur abgekühlt, in Einmalküvetten überführt und ihre Absorption bei 797 nm mit einem Shimadzu UV-1601-Spektrophotometer spektrophotometrisch bestimmt. Der Phosphatgehalt und somit die Phospholipidkonzentration wurden mit Hilfe der durch lineare Regression ermittelten Geradengleichung der Kalibrationsgerade errechnet.
3.3.4 Bindung an Hydroxylapatit
Bisphosphonate binden an Hydroxylapatit (HAP), dem Hauptbestandteil der mineralischen Phase des Knochens (Fleisch, 2002). Um zu überprüfen, ob ligandmodifizierte Liposomen ebenfalls an HAP binden, wurde eine In-vitro-Methode etabliert, die sich der Isothermalen Titrationskalorimetrie bedient.

Materialien, Geräte und Methoden 65
3.3.4.1 Isothermale Titrationskalorimetrie
Die Isothermale Titrationskalorimetrie („Isothermal Titration Calorimetry“, ITC) bietet die Möglichkeit, sowohl endotherme als auch exotherme Bindungsreaktionen zu messen. Mit der gemessenen Reaktionswärme können dann thermodynamische Eigenschaften wie die Enthalpie H∆ und die apparente Bindungskonstante appK von physikalischen oder chemischen Vorgängen bestimmt werden. Die Hauptbestandteile eines Titrationskalorimeters sind die Injektionsvorrichtung, gefüllt mit einem Reaktionspartner, und die Messzelle, die den anderen Reaktionspartner enthält. Außerdem gibt es noch eine Referenzzelle, die mit dem gleichen Medium befüllt wird wie die Messzelle. Die Injektion des Titranten und die anschließende Mischung mit dem Medium in der Messzelle durch die ständige Rotation der Injektionsvorrichtung verursacht eine Reaktion in der Messzelle, die zur Wärmeentwicklung oder Wärmeabsorption führt. Diese Wärme wird bestimmt, indem die Energie (Js-1) gemessen wird, die erforderlich ist, um die Referenz- und die Messzelle auf der gleichen Temperatur zu halten ( 0=∆T ). Die Bindungsversuche wurden mit einem VP-ITC-Kalorimeter durchgeführt (Microcal, Northhampton, MA). Sowohl die Liposomenproben als auch die HAP-Suspension wurden vor Gebrauch entgast (Thermo-Vac, Microcal, Northhampton, MA). Der ersten Injektion von 0,255 µl einer
20 mM Liposomenprobe ( 0Lipc ) folgten 37 weitere Injektionen dieser Probe von 7,5 µl
in eine Messzelle, gefüllt mit 1,345 ml ( ZelleV ) HAP-Suspension (0,05 mM HAP in
HBSpH7,4) ( 0HAPc ). Die einzelnen Injektionen erfolgten durch eine computergesteuerte
Injektionseinheit des Mikrokalorimeters in einem Intervall von 150 Sekunden. Die Versuchstemperatur betrug 25 °C. Die durch die Verdünnung entstehende Wärmetönung ( dilh ) wurde durch ein Kontrollexperiment bestimmt, indem die Liposomen in den HEPES-Puffer (HBSpH7,4) titriert wurden, der auch für die Herstellung der Liposomen und der HAP-Suspension verwendet worden war. Diese ermittelte Wärmetönung wurde von der gemessenen Wärmeentwicklung, die bei den jeweiligen Titrationen in die HAP-Suspension bestimmt wurde, subtrahiert. Mit Hilfe der Origin-Software (OriginLab) wurden die erhaltenen Titrationskurven bearbeitet.
Um den Einfluss von humanem Blutplasma auf die Bindung von ligandmodifizierten Liposomen an HAP zu untersuchen, wurde die 0,05-mM-HAP-Suspension vor Befüllung der Messzelle jeweils mit 5%, 10% und 30% Plasma versetzt und gut geschüttelt.

Materialien, Geräte und Methoden 66
3.3.4.2 Bestimmung der Bindungsparameter
Durch die Integration der experimentell bestimmten Titrationskurven wurde die Reaktionsenthalpie ( ih∆ ) bestimmt, welche um die Wärmetönung ( dilh∆ ), die durch
Kontrolltitrationen in HBSpH7,4 ermittelt wurde, korrigiert wurde ( dilii hhh −=∆ ). Die
kumulative Reaktionswärme (kumulative Reaktionsenthalpie) wurde nach der
folgenden Formel bestimmt: ∑∆=n
ihn1
, =n Anzahl der Injektionen.
Aus der Gleichung ∑∆=∆n
HAPi nhH1
0 läßt sich die molare Reaktionsenthalpie H∆
berechnen (Seelig, 1997). Nach i Injektionen kann die an die Liposomen gebundene
Menge HAP durch ∑ ∑∆∆==i n
iiHAPi
HAPi
HAP hhnnX1 1
0)()( ausgedrückt werden. Hierbei
bezeichnet )(iHAPn die molare Menge von gebundenem HAP nach i Injektionen. Die
entsprechende freie Menge HAP ( )(ifreec ) in der Messzelle kann mit der folgenden
Gleichung berechnet werden: )( )(0)( 1 iHAPHAP
ifree Xcc −= . 0
HAPC ist dabei die anfängliche
HAP-Konzentration. Der molare Lipidgehalt ( )(iLipn ) an jedem Titrationspunkt kann mit
Hilfe der folgenden Formel berechnet werden: 0)(Lipinj
iLip cVin ××= , worin 0
Lipc die
Startkonzentration des Lipids beschreibt, injV steht für das Injektionsvolumen während
des Titrationsprozesses. Mit den oben bestimmten Parametern kann der Grad der
Bindung ( )(ibX ) nach der Formel ( )ynnX i
Lipi
HAPi
b ×= )()()( ermittelt werden. Y ist in
dieser Gleichung ein Korrekturfaktor, welcher berücksichtigt, dass nur die an der Liposomenoberfläche vorhandenen Liganden die Bindung zum HAP eingehen können.
Gemäß Literatur stehen bei unilamellaren Vesikeln ungefähr 60% des verkapselten Liganden an der Liposomenoberfläche für mögliche Bindungen zur Verfügung (Beschiaschvili and Seelig, 1992, Schote and Seelig, 1998).
Für jeden Punkt der Bindungsisothermen kann eine apparente Bindungskonstante appK
bestimmt werden: )()( ifree
ibapp cXK = .

Materialien, Geräte und Methoden 67
3.3.5 Doxorubizin-Beladung („Remote Loading”)
Die ligandmodifizierten Liposomen wurden über einen transmembranären Ammoniumsulfatgradienten mit Doxorubizin beladen (Haran et al., 1993, Lasic et al., 1995). Der Mechanismus der Wirkstoffaufnahme in das Innere der Liposomen beinhaltet eine pH-unabhängige Wiederverteilung von schwachen Basen. Dazu werden die Liposomen so hergestellt, dass sie im Inneren Ammoniumsulfationen und im externen Medium HEPES-Puffer (HBSpH7,4) aufweisen. Ein kleiner Teil der neutralen Ammoniakionen diffundiert durch die Membran von innen nach außen und schafft somit ein ungepuffertes azides Inneres. Der in das externe Medium gegebene neutrale, membrangängige Wirkstoff wandert nun ins Liposomeninnere, wird dort protoniert und kann durch den Erhalt der Ladung die Membran nicht wieder passieren. Der Austausch zwischen Ammoniumionen und Doxorubizin setzt sich solange fort, bis der Wirkstoff komplett in die Liposomen aufgenommen wurde oder bis die Protonen im Inneren verbraucht sind. Es wurden zunächst ligandmodifizierte Leerliposomen (14,3 mol% CHOL-TOE-BP) mit einer Phospholipidkonzentration von 20 µmol/ml nach der Lipidfilm-Extrusionsmethode hergestellt. Der Lipidfilm wurde hierbei allerdings in 230-mM-Ammoniumsulfatlösung resuspendiert. Um das freie Ammmoniumsulfat außerhalb der Liposomen zu entfernen, wurden die Liposomen in eine Slide-A-Lyzer-Kassette (MWCO: 10000) überführt und in 800 ml HBSpH7,4 bei 4–8 °C für 24 Stunden dialysiert. Der Puffer wurde innerhalb dieser 24 Stunden zweimal durch frischen Puffer ersetzt, um das bereits herausdialysierte Ammoniumsulfat zu entfernen. Nach der Dialyse wurden die Liposomen und die Doxorubizin-Lösung (3,5 mg/ml, w/v) im Verhältnis 1:1 (v/v) in einem Plastikröhrchen gemischt und für 2 Stunden bei 60 °C in einem Wasserbad inkubiert.
3.3.5.1 Größenausschlusschromatographie
Die Größenausschlusschromatographie, auch Gelfiltrationschromatographie genannt, erlaubt die Auftrennung eines Stoffgemisches nach der unterschiedlichen Molekülgröße der Einzelkomponenten. Dabei können kleine Moleküle in die Gelmatrix eindringen, wohingegen große Moleküle an den Poren vorbeiwandern und somit im Eluat als Erstes von der Säule kommen. Die

Materialien, Geräte und Methoden 68
Größenausschlusschromatographie wurde hier durchgeführt, um das nicht verkapselte Doxorubizin vom verkapselten Material abzutrennen. Zunächst wurde dazu die Sephadex-Chromatographie-Säule (PD-10-Säule) mit 20 ml HBSpH7,4 gewaschen. Im Anschluss daran wurden 1,5 ml der wirkstoffbeladenen Liposomen auf die Säule gegeben. Nachdem die Säule trocken gelaufen war, wurden 1 ml HBSpH7,4 auf die Säule gegeben und erneut gewartet, bis dieser in die Säule gelaufen war. Es wurden weitere 3 ml HBSpH7,4 auf die Säule pipettiert und 3 ml eluierte Liposomen in einem neuen Plastikröhrchen aufgefangen.
3.3.5.2 Doxorubizin-Gehaltsbestimmung
Das in den Liposomen verkapselte Doxorubizin wurde spektrophotometrisch bestimmt. Zur Kalibrierung wurde eine Kalibrationsgerade (Doppelbestimmung) im Bereich von 0–30 µg/ml aus einer Doxorubizin-Stammlösung (1 mg/ml HBSpH7,4, w/v) in Einmalküvetten vermessen. Die zu untersuchenden Liposomenproben wurden in dreifacher Ausführung zunächst verdünnt, so dass die Probenkonzentration im linearen Messbereich lag, und dann ebenfalls in Einmalküvetten photometrisch bestimmt. Sowohl die Kalibrationsgerade als auch die Liposomenproben wurden in EtOH/HCl bei 495 nm (Absorptionsmaximum von Doxorubizin) mit einem Shimadzu UV-1601-Gerät vermessen. Durch die Zugabe von EtOH/HCl wurden bei den lipidhaltigen Proben die Liposomenstruktur zerstört, um sicherzustellen, dass diese die Bestimmung nicht stören. Der Doxorubizin-Gehalt wurde mit Hilfe der aus der linearen Regression resultierenden Kalibrationsgeraden und deren Funktion bestimmt.
Der Lipidgehalt wurde nach der in Abschnitt 3.3.3.5 beschriebenen Methode ermittelt.
3.3.5.3 Doxorubizin-Freisetzung
Die Methode zur Bestimmung der In-vitro-Freisetzung von Doxorubizin aus den Liposomen basiert auf den selbstquenchenden Eigenschaften von Doxorubizin bei hohen Konzentrationen. Doxorubizin liegt in hohen Konzentrationen im Liposomeninneren selbstgequencht vor, und weist somit eine geringe Fluoreszenz auf. Außerhalb der Liposomen liegt kaum Doxorubizin vor, so dass dort kein

Materialien, Geräte und Methoden 69
Selbstquenching von Doxorubizin zu erwarten ist, d.h., je mehr Doxorubizin im Zuge des Freisetzungsversuches freigesetzt wird, desto mehr Fluoreszenz wird bestimmbar. Durch die gemessene Fluoreszenzzunahme über die Zeit kann die freigesetzte Menge an Doxorubizin ermittelt werden. Um die Freisetzung von Doxorubizin aus dem Liposomeninnenraum zu untersuchen, wurden ligandmodifizierte Liposomen mit einer Ligandkonzentration von 14,3 mol% hergestellt. Die Freisetzungsversuche wurden bei drei verschiedenen pH-Werten durchgeführt. Dazu wurden die Liposomen mit HBSpH7,4, mit HBSpH6,5 und HBSpH5,0 300-fach verdünnt und in dreifacher Ausführung in 96-Well-Mikrotiterplatten pipettiert. Die Fluoreszenz von Doxorubizin wurde über einen Zeitraum von 2 Stunden mit dem Tecan Genios-Plus-Gerät bestimmt (Anregungswellenlänge: 485 nm, Emissionswellenlänge: 595 nm, Messintervall: 30 sec, Integrationszeit: 40 µs, Schütteldauer der Platte vor jeder Messung: 10 sec, Versuchstemperatur: 25 °C). Zur Bestimmung der 100%igen Freisetzung von Doxorubizin wurden die Liposomen durch die Zugabe von Triton-X 10% (v/v) zerstört und ihre Fluoreszenz gemessen. Die Fluoreszenzbestimmung von Triton-X 10% allein ergab, dass es bei der gewählten Anregungs- und Emissionswellenlänge keinen signifikanten Einfluss auf die Fluoreszenzmessungen hat.
Die freigesetzte Menge an Doxorubizin wurde wie folgt berechnet:
100%0%100
0 ×−−
=II
IIgFreisetzun t
tI Fluoreszenzintensität nach Liposomenzugabe nach der Zeit t
%100I Fluoreszenzintensität nach der Triton-X-Zugabe
0I Fluoreszenzintensität direkt nach der Zugabe der Liposomen

Materialien, Geräte und Methoden 70
3.3.6 Zellkultur
Die Zellkulturversuche wurden unter der Anleitung von Herrn Dr. Amtmann im DKFZ (Deutsches Krebsforschungszentrum) in Heidelberg durchgeführt. Zellen (humane Melanomzellen)
Zellbezeichnung Zelltyp Herkunft Medium Aufteilungsverhältnis bei Passagierung
MCF-7 Mammakarzinom ATCC MEM/10% FCS 1:4
MDA-MB-435s Mammakarzinom ATCC MEM/10% FCS 1:4
BT-474 Mammakarzinom ATCC RPMI
1640/10% FCS
1:4
DU-145 Prostatakarzinom ATCC DMEM 1:4
Tabelle 8: Übersicht über die verwendeten Zellen für die Bestimmung der IC50-Werte von Doxorubizin-beladenen, ligandmodifizierten Liposomen.
3.3.6.1 Zellkultivierung und Zellzahlbestimmung
Die in der Tabelle 8 aufgeführten humanen Melanomzellen wurden jeweils in 10 cm-Zellkulturschalen kultiviert (Versuchsbedingungen: 89% Luftfeuchtigkeit, 37 °C Begasung mit 3% CO2). Die Zellen wurden einmal pro Woche passagiert. Dazu wurde der Zellrasen mit 2 ml einer Trypsin/EDTA-Lösung abgelöst. In dem in der Tabelle 8 aufgeführten Aufteilungsverhältnis wurde die Zellsuspension dann im entsprechenden Medium (Tabelle 8) in 10 cm-Rundschalen ausgesät. Zum Ablösen der Zellen wurden die Zellen bei 37 °C mit 2 ml Trypsin/EDTA-Lösung für 5 Minuten inkubiert, nachdem zuvor das Medium aus der Schale abgesaugt wurde. Durch die Zugabe von 8 ml frischen Mediums wurde die Reaktion unterbrochen. Mit Hilfe von seitlichen Schlägen wurden die abgerundeten Zellen von der Schale abgelöst. Nachdem diese bei 4000 rpm zentrifugiert wurden, wurde das resultierende Zellpellet in 10 ml Medium resuspendiert. Um die Zellzahl zu bestimmen, wurden 20 µl der Zellsuspension mit

Materialien, Geräte und Methoden 71
20 µl einer 0,4%igen Tryptanblaulösung vermischt und in eine Neubauer-Zählkammer gegeben. Unter einem Mikroskop wurden die Zellgesamtzahl und die Anzahl der blaugefärbten Zellen bestimmt. Der Anteil an lebenden Zellen wurde durch den prozentualen Anteil der ungefärbten Zellen repräsentiert.
3.3.6.2 Kristallvioletttest
Der IC50-Wert gibt die Wirkstoffkonzentration an, die nötig ist, um die Tumorzellen absterben zu lassen. Dabei wird die Konzentration des Wirkstoffes gemessen, bei der die Überlebensrate der Zellen auf die Hälfte abgenommen hat. Um die IC50-Werte von Doxorubizin-beladenen, ligandmodifizierten Liposomen im Vergleich zu freiem Doxorubizin zu bestimmen, wurde der Kristallvioletttest durchgeführt. Dieser basiert darauf, dass die Überlebensrate von adhärent wachsenden Zellen in der Gegenwart von den zu testenden Formulierungen ermittelt wird. Insgesamt 2 Millionen der ausgezählten Zellen (siehe 3.3.6.2) wurden in 96-Well-Mikrotiterplatten ausgesät. Einen Tag später wurde zunächst Medium auf die Zellen gegeben, und zwar wie folgt: 180 µl des jeweiligen Mediums (Tabelle 8) wurden in die erste Reihe der Mikrotiterplatten pipettiert, und 150 µl wurden in die verbleibenden Reihen gegeben. Anschließend wurden Verdünnungsreihen der Testformulierungen hergestellt und auf die Zellen der ersten Reihe pipettiert (20 µl, 4-fache Bestimmung jeder Testformulierung). Dann wurden 50 µl entnommen und in die zweite Reihe pipettiert. Nach dreimaligem Mischen wurden 50 µl entnommen und in die Reihe 3 pipettiert. So wurde bis zur Reihe 7 weiterverfahren. Die resultierende Verdünnungsreihe ergab folgende Doxorubizin-Konzentrationen: 10, 3, 1, 0,3, 0,1, 0,03 und 0,01 µg/ml. Die letzte Reihe jeder Platte blieb unbehandelt, d.h., in diese wurde nur Zellkulturmedium pipettiert. Als Kontrollen wurden unbeladene, ligandmodifizierte Liposomen und unbeladene Liposomen, die keinen Liganden aufwiesen, untersucht. Bei 37 °C wurden die Platten inkubiert. Nach zwei Stunden Inkubation wurde das jeweilige Medium entfernt, durch frisches Medium ersetzt und für weitere 48 Stunden bei 37 °C erneut inkubiert. Das Medium wurde entfernt, die Zellen mit 3%iger Formalinlösung fixiert und über Nacht getrocknet. Um die überlebenden Zellen nach der Behandlung mit den Testformulierungen zu bestimmen, wurden 100 µl einer 1%igen Kristallviolettlösung in jede Vertiefung pipettiert. Die noch lebenden Zellen wurden nach einer 10-minütigen Inkubation angefärbt, wohingegen sich die abgetöteten Zellen ablösten und

Materialien, Geräte und Methoden 72
durch die anschließende Behandlung der Platten mit Leitungswasser heruntergewaschen wurden. Die Extinktion wurde bei 595 nm mit einem Ascent Multiscan-ELISA-Reader gemessen. Die IC50-Werte wurden mit Hilfe eines speziellen Programms, das auf Excel basiert, berechnet. Nach Berechnung der Mittelwerte der optischen Dichte (OD) für jede Wirkstoffkonzentration wurde der Nullwert/2 von diesen Mittelwerten abgezogen. Durch den niedrigsten Konzentrationswert, bei dem ein negativer y-Wert erhalten wurde, und dem nächst niedrigsten Wert wurde eine Gerade gelegt, und der Abschnitt auf der x-Achse berechnet, an dem die erhaltene Gerade die x-Achse schneidet. Dieser Wert stellt die IC50 dar.
3.3.7 In-vivo-Versuche
Die Pharmakokinetikversuche wurden in Zusammenarbeit mit Herrn Cor Snel an der Universität Utrecht, Niederlande, durchgeführt.
Die Versuche zur Bestimmung der Lokalisierung von ligandmodifizierten Liposomen im metastasierten Knochengewebe wurden in Zusammenarbeit mit Dr. Gabri van der Pluijm und Herrn Ivo Que an der Universität Leiden, Niederlande fertiggestellt.
3.3.7.1 Art und Haltung der Versuchstiere
3.3.7.1.1 Pharmakokinetik und Biodistribution
Für die Bestimmung der Pharmakokinetik und Biodistribution von ligandmodifizierten Liposomen wurden männliche Wistar-Ratten (Harlan, Horst, Niederlande) mit einem Durchschnittsgewicht von 230 g als Versuchstiere verwendet. Diese wurden bei 25 °C, einer relativen Luftfeuchtigkeit von 60% und einem 12 Stunden Hell-Dunkel-Rhythmus gehalten. Ihre Nahrung bestand aus Wasser und pelletiertem Futter (Altromin, Altromin GmbH, Lage/Lippe) und wurde ihnen ad libitum zugeführt. Vor jeder Injektion von Liposomen sowie vor Blutentnahmen wurden die Tiere mit

Materialien, Geräte und Methoden 73
Isofluran (Isoflo, Abbott AG, Baar, Schweiz) aus einem speziell kalibrierten Verdampfer narkotisiert (inhalative Applikation).
3.3.7.1.2 Tiermodel für Knochenmetastasen
Für das Tiermodel für Knochenmetastasen wurden Nacktmäuse (Charles River Deutschland GmbH, Sulzfeld) mit einem Durchschnittsgewicht von 20 g eingesetzt. Diese wurden in einzelnen ventilierten Käfigen unter sterilen Bedingungen gemäß den „Swiss guidelines for care and use of laboratory animals“ gehalten. Die Nahrung bestehend aus Wasser und pelletiertem Futter (Altromin, Altromin GmbH, Lage/Lippe) wurde den Tieren ad libitum zugeführt. Die Tiere waren 4–6 Wochen alt, als die intraossäre Implantation der Tumorzellen erfolgte. Für diese Implantation wurden die Tiere mit einer Lösung bestehend aus Domitor (1 mg/kg KG, Pfizer, Sandwich, Großbritannien), Climasol (10 mg/kg KG, Gräub, Bern, Schweiz) und Fentanyl (100 µg/kg KG, Krankenhaus-Apotheke des Universitätskrankenhauses von Bern, Schweiz) narkotisiert (intraperitoneale Applikation). Vor weiteren Injektionen bzw. Untersuchungen wurden die Tiere mit einer Lösung bestehend aus Ketamin (10%, Mallinchrodt Veterinary, Burgwedel), Rompun (2%, Bayer Vital, Leverkusen) und PBS im Verhältnis 1:1:1 (Dosis von 50 µl/20 gKG) anästhesiert (intraperitoneale Applikation).
3.3.7.2 Pharmakokinetik- und Biodistributionsstudien
Zur Bestimmung der Pharmakokinetik und der Biodistribution wurde der radioaktive Marker [3H]-Cholesteryl-oleoylether in die Liposomenmembran eingebaut. Einzeldosen von 20 µmol [3H]-markierten, ligandmodifizierten Liposomen (Radioaktivität von ca. 50 kBq) pro kg Körpergewicht (KG) wurden intravenös in die Schwanzvene des Tieres appliziert. Als positive Kontrolle wurden [3H]-markierte Kontrollliposomen ohne Liganden ebenfalls in einer Dosis von 20 µmol/kg KG verabreicht. Jede liposomale Formulierung wurde in einer Gruppe bestehend aus vier Tieren getestet. Zu den Zeitpunkten 2 Minuten sowie 1, 4, 8, 24 und 48 Stunden nach der Injektion wurden Blutproben von 200 µl aus der auf der Gegenseite liegenden

Materialien, Geräte und Methoden 74
Schwanzvene entnommen. Für die Bestimmung der Radioaktivität dieser Blutproben wurden diese wie folgt präpariert: 100 µl der zu analysierenden Probe wurden in 100 µl Gewebslöser gelöst (Perkin Elmer, Groningen, Niederlande) und mit 200–400 µl 35% Wasserstoffperoxid versetzt. Nachdem die Proben über Nacht inkubiert wurden, wurden sie in 10 ml Ultima-Gold-Scintillation-Cocktail (Perkin Elmer, Groningen, Niederlande) mit einem Packard Tricarb-2000-Liquid-Scintillation-Counter vermessen. Nach der Blutentnahme zum Zeitpunkt 48 Stunden wurden die Tiere durch eine Überdosis an Isofluran getötet. Die Organe Leber, Lunge, Nieren und Milz wurden entfernt, mit einem Ultraturrax (IKA Labortechnik, Staufen) in PBS (Lunge, Niere und Milz in jeweils 5 ml PBS, Leber in 25 ml PBS) homogenisiert und ebenfalls, wie oben beschrieben, aufbereitet. Die Radioaktivität der injizierten Dosis wurde mit dem gleichen Gerät bestimmt. Die prozentual injizierte Dosis in den einzelnen Organen/Blutproben wurde berechnet, indem die Radioaktivität der einzelnen Organe/Blutproben durch die Radioaktivität der injizierten Dosis dividiert wurde. Um die prozentual injizierte Dosis im Blut zu berechnen, wurde mit einem Blutvolumen von 20 ml pro Ratte gerechnet.
Berechnung:
%100Pr ××
=Dosisinjizierte
Organ
itätRadioaktivsfaktorVerdünnungitätRadioaktiv
OrganimDosisinjizierteozentual
%100Pr ××
=Dosisinjizierte
Blut
itätRadioaktivsfaktorVerdünnungitätRadioaktivBlutimDosisinjizierteozentual
Für die Ermittlung des zu applizierenden Volumens bei einer gewünschten Dosis von 20 µmol/kg KG Liposomen wurde die unter 3.3.3.5 beschriebene Lipidbestimmung durchgeführt.
3.3.7.3 Statistik
Die Ergebnisse der Pharmakokinetikuntersuchungen wurden durch eine statistische Varianzanalyse (ANOVA) ausgewertet. Die AUC-Daten der Abbildung 22 (Tabelle 16) wurden dabei mit einem ANOVA-Test ausgewertet, bei dem der Einfluss der

Materialien, Geräte und Methoden 75
einzelnen CHOL-TOE-BP-Konzentrationen auf die Zirkulationszeit untersucht wurde. Es wurde dabei ein Vergleich über alle Gruppen (verschiedene CHOL-TOE-BP-Konzentrationen) durchgeführt. Bei der statistischen Auswertung der AUC-Werte aus Tabelle 17 (Abbildung 24) wurden zunächst die Einflüsse der einzelnen DSPE-PEG2000-Konzentrationen in einem Vergleich aller Gruppen (alle getesteten Liposomengruppen) statistisch untersucht. Anschließend wurden die Gruppen paarweise verglichen. Voraussetzung für den paarweisen Vergleich war, dass die Gruppen sich beim ersten Vergleich (Vergleich aller Gruppen) statistisch signifikant unterschieden (p < 0,05). Beim paarweisen Vergleich wurden die Gruppen erst dann als statistisch signifikant bezeichnet, wenn der p-Wert kleiner als 0,05 war und zwar nach der Angleichung für multiples Testen gemäß Bonferroni-Holm. Die gleiche statistische Auswertung wurde mit den AUC-Werten, die in der Tabelle 18 dargestellt sind und anhand der Kinetiken aus Abbildung 25 ermittelt wurden, vorgenommen.
3.3.7.4 Tiermodel für Knochenmetastasen
3.3.7.4.1 Etablierung stabiler Zelllinien
Es wurde zunächst ein Subklon (MDA-231-B) aus einer humanen Mammakarzinomzelllinie MDA-MB-231 (MDA-231) (American Type Culture Collection (Rockville, MD) hergestellt, der nach intrakardialer Inokulation Knochenmetastasen initiierte. Dazu wurden MDA-231-Zellen (1x105/100 µl Phosphatpuffer, PBS) intrakardial in weibliche BALB-c-nu/nu-Mäuse injiziert. Die Entwicklung von osteolytischen Knochenmetastasen wurde mit Hilfe von Radiografie beobachtet. Sobald die Knochenmetastasen eine Größe von ungefähr 2 mm Durchmesser erreicht hatten, wurden diese entnommen, in kleine Stücke geschnitten und in 12-Well-Platten überführt. Nachdem sie drei Tage kultiviert wurden, wurde das Originalgewebe entfernt, und die einzelnen Flächen neuen Zellwachstums wurden mit Hilfe von Klonierungsringen (Fa. Clontech) trypsiniert. Durch die Grenzwertverdünnungsmethode („Limiting Dilution Method“) wurden Einzelklone isoliert. Nach erfolgter Vermehrung wurden diese Zellklone wiederum durch eine speziesspezifische Polymerasekettenreaktion für das humane β2-Mikroglobulin identifiziert (van der Pluijm et al., 2001). Positive Klone wurden für eine weitere

Materialien, Geräte und Methoden 76
intrakardiale Inokulation sowie für die In-vitro-Vermehrung der Zellpopulation ausgewählt. Der MDA-231-B-Subklon ist entstanden, nachdem dieser Zyklus 4-mal durchlaufen wurde. Dieser Klon war in der Lage, nach intrakardialer Inokulation in 100% der Tiere die Entstehung von Knochenmetastasen hervorzurufen. Die Metastasenentstehung wurde durch den Einsatz von Radiografie detektiert. Aufgrund dieser Effizienz wurde der MDA-231-B-Klon für die anschließende stabile Transfektion ausgewählt.
Die Zelllinien zusammen mit den explantierten Fragmenten der Knochenmetastasen wurden in Dulbecco´s-Modified-Eagle´s-Medium kultiviert. Dieses Medium enthielt 4,5 g Glucose/l, 10% fötales Kälberserum und 1% Gentamycin. Die Zellen wurden regelmäßig auf Mykoplasmakontamination getestet.
3.3.7.4.2 Expression von Luciferase-Reportergen
Durch das Einbringen einer Luciferase cDNA (Firefly) in ein pcDNA 3,1-Plasmid wurde ein Vektor, der das Luciferase-Reportergen trägt (CMV-Luc), geschaffen. Die MDA-231-B-Zellen wurden mit 1 µg CMV-Luc (Reportergen-Plasmid) mittels FuGene6 transfiziert. Die Etablierung stabiler Zelllinien erfolgte durch die Zugabe von 800 µg/ml G418 (Life Technologies, Basel, Schweiz). Dabei wurden 25 verschiedene G418-resistente Klone isoliert, subkloniert und auf ihre Luciferase-Aktivität hin überprüft. Sechs Klone zeigten die erfolgreiche Exprimierung von Luciferase-Aktivität. Davon wurde der Klon (MDA-231-B/Luc+) mit der höchsten Aktivität (5–12 RLU/µg Zellprotein) für die folgenden Versuche ausgesucht. Dieser Klon wurde bis zu 40 Passagen kultiviert.
3.3.7.4.3 Induzierung von Metastasenentstehung
Eine einzelne Zellsuspension, bestehend aus MDA-231-B/Luc+-Zellen (1x105/2 µl PBS), wurde in Anlehnung an die Methode von Arguello et al. (Arguello et al., 1988) in die rechte Tibia (dorsale Ansicht) implantiert (Wetterwald et al., 2002). Dazu wurden zwei Löcher mit einem Abstand von 4–5 mm voneinander in das obere Drittel der Tibia mit Hilfe eines dentalen Bohrers (Hedstroem file, 28 mm/30; Maillefer-

Materialien, Geräte und Methoden 77
Instruments, Ballaigues, Schweiz) gebohrt. Der Durchmesser beider Löcher betrug etwa 0,35 mm. Durch das Ausblasen des Knochenmarks wurde Platz innerhalb der Knochenmarkskavitäten geschaffen. Das obere Loch wurde mit Knochenwachs (Ethicon; Johnson & Johnson, Somerville, NJ) geschlossen und die MDA-231-B/Luc+-Zellen (siehe oben) wurden langsam über eine Nadel (30 gauche), welche durch das untere Loch eingeführt wurde, inokuliert. Zuletzt wurde dieses Loch ebenfalls mit Knochenwachs verschlossen. Die Wunde in der Haut, die durch diesen Eingriff verursacht wurde, wurde genäht. Die Entstehung von Knochenmetastasen wurde wöchentlich mittels Radiografie und „Bioluminescent Reporter Imaging“ (siehe Abschnitt 3.3.7.5) überprüft. 5 Wochen nach der Injektion von MDA-231-B/Luc+-Zellen waren ausgeprägte Knochenmetastasen detektierbar.
Abbildung 8: BALB-c-nu/nu-Maus mit sichtbarer Metastasierung am linken Bein (bei ventraler Ansicht). Die Maus war bei dieser Aufnahme zwölf Woche alt.

Materialien, Geräte und Methoden 78
3.3.7.5 „Bioluminescent Reporter Imaging” (BRI)
Das bildgebende System, das in dieser Arbeit eingesetzt wurde, basiert auf dem System, das zuerst von Edinger et al. (Edinger et al., 1999) entwickelt wurde (Wetterwald et al., 2002). Die Emission von Licht in diesem Versuch basiert auf der Oxidationsreaktion von Luciferin unter dem Einfluss von molekularem Sauerstoff und dem Coenzym ATP zu Oxiluciferin, Adenosin-monophosphat (AMP), CO2 und Licht. Diese Reaktion wird von dem Enzym Luciferase katalysiert (Luciferin + ATP + O2 → Oxiluciferin + AMP + CO2 + Licht). Die Reaktion beinhaltet mehrere Reaktionsschritte: zunächst reagiert das Substrat Luciferin mit Mg2+-ATP und bildet Luciferyl-Adenylat. Dieses wird wiederum durch molekularen Sauerstoff oxidiert. Dabei entstehen als Zwischenprodukte zyklisches Peroxid, ein Dioxetanon und ein Molekül ATP. Das Dioxetanon wird durch eine intramolekulare Umstrukturierung decarboxyliert. Als Folge entsteht eine Enol- oder Ketoform des Oxiluciferins, das sich in einem elektronisch angeregten Zustand befindet. Der Übergang vom Oxiluciferin in den energetischen Grundzustand ist mit Lichtemission der Wellenlänge 560–562 nm verbunden, es entsteht grün-gelbes Licht. In Mäusen, die mit MDA-231-B/Luc+-Zellen inokuliert wurden, wurde die Luciferase-Aktivität in Körper mit Hilfe des „In-vivo-Imaging-Systems“ (IVIS 100, Xenogen, Alameda, CA, USA), das mit einer hochempfindlichen Photonendetektionskamera (I-CCD, „Intensified Charged Coupled Device“) ausgestattet war, visualisiert. Dazu wurden die Tiere zunächst anästhesiert (siehe Abschnitt 3.3.7.1.2). Danach wurden ihnen 2 mg D-Luciferin Natriumsalz gelöst in PBS intraperitoneal verabreicht. Die Messungen der biolumineszenten Photonenemission erfolgte 15 min nach der Injektion von D-Luciferin. Die Daten wurden mit Hilfe des Programms „Living Image“ (Xenogen, Alameda, CA, USA) analysiert. Die Werte wurden als „Relative Light Unit“ (RLU) in Photonen/sec ausgedrückt.
3.3.7.6 Lokalisierung von ligandmodifizierten Liposomen
Um den Verbleib von fluoreszenzmarkierten, ligandmodifizierten Liposomen im Vergleich zu fluoreszenzmarkierten, nicht modifizierten Kontrollliposomen nach intrakardialer Applikation in BALB-c-nu/nu-Mäuse zu bestimmen, wurden die Tiere

Materialien, Geräte und Methoden 79
zunächst anästhesiert (siehe Abschnitt 3.3.7.1.2). Jede Formulierung wurde an einer Gruppe von zwei Tieren getestet (weitere Tiere konnten von der Universität Leiden zum Zeitpunkt der Durchführung dieser Experimente nicht zur Verfügung gestellt werden). Nach erfolgter Injektion von 50 µl Liposomenprobe (fluoreszenzmarkierte, ligandmodifizierte Liposomen (14,3 mol% CHOL-TOE-BP), fluoreszenzmarkierte Kontrollliposomen) wurden die Liposomen mit einem Maestro-Gerät (multispektrales, bildgebendes Kamerasystem) (CRI, Inc., Woburn, MA, USA) lokalisiert bzw. visualiert, indem zu verschiedenen Zeitpunkten mit Hilfe der im Gerät integrierten Kamera die Tiere abgelichtet wurden und die Fluoreszenz des signalgebenden Farbstoffes (NBD-PE) in den Liposomen am lebenden Tier nicht invasiv bestimmt wurde. Für die Detektion von NBD-PE wurden Maestro Blue-Filter (M-MSI-FLTR-Blue) eingesetzt (Extinktion: 445–490 nm; Emission: 515 nm longpass). Mit Hilfe der Maestro-Software wurden die Aufnahmen bearbeitet und die Intensität der ermittelten Fluoreszenzsignale bestimmt.
Für die Ermittlung des zu applizierenden Volumens bei einer gewünschten Dosis von 20 µmol/kg KG Liposomen wurde die unter 3.3.3.5 beschriebene Lipidbestimmung durchgeführt.

Ergebnisse und Diskussion 80
4 Ergebnisse und Diskussion
In dieser Arbeit wurden erfolgreich ligandmodifizierte Liposomen für die zielgerichtete Applikation an den Knochen in metastasendurchsetztem Knochengewebe entwickelt und charakterisiert. Der Ligand war dabei so konzipiert, dass er einen Cholesterylrest aufwies, der sich leicht in die Liposomenmembran einbauen ließ und aus einer Bisphosphonsäure bestand, die aufgrund ihrer bekannten Affinität zur Mineralmatrix des Knochens als Targeting-Einheit fungieren sollte. Ein Spacer (Trisoxyethylenkette) zwischen diesen beiden Molekülen wurde eingesetzt, um mögliche unerwünschte sterische Hinderungen durch das Polymer DSPE-PEG2000, welches ebenfalls an der Oberfläche der Liposomen saß, zu vermeiden. Nach zahlreichen Präformulierungsversuchen wurde die vielversprechendste Formulierung für ligandmodifizierte Liposomen, die aus diesen Versuchen hervorging, in weiteren Experimenten eingesetzt. Um dieses liposomale System näher zu charakterisieren, wurden zunächst seine physikochemischen Eigenschaften bestimmt. Fortgefahren wurde mit der Untersuchung der Fähigkeit, an synthetisches Hydroxylapatit in vitro zu binden, um zu überprüfen, ob sich die neuentwickelten, ligandmodifizierten Liposomen für die angestrebte Targeting-Strategie eignen. Ein weiterer Bestandteil der In-vitro-Untersuchungen war die Bestimmung der Beladungskapazität, welche anhand von Doxorubizin ermittelt wurde. Zudem wurde die Freisetzung von Doxorubizin aus den ligandmodifizierten Liposomen evaluiert. Abgeschlossen wurde die In-vitro-Versuchsreihe mit der Bestimmung der In-vitro-Toxizität von Doxorubizin-beladenen, ligandmodifizierten Liposomen gegenüber verschiedenen Zelllinien. In-vivo-Versuche zur Untersuchung der Pharmakokinetik und Biodistribution von unbeladenen, ligandmodifizierten Liposomen sowie zu ihrer Lokalisierung in metastasendurchsetztem Knochengewebe bildeten den Abschluss dieser Arbeit.
Die Autorin fertigte diese Arbeit während ihrer Tätigkeit bei MCS Micro Carrier Systems GmbH (MCS) als externe Doktorandin am Lehrstuhl für Pharmazeutische Technologie und Biopharmazie der Philipps-Universität Marburg an. Im Rahmen ihrer Dissertation verbrachte sie ein Jahr zwecks Forschung an der Universität Utrecht, Niederlande. Dieser Aufenthalt wurde durch das „Galenos“-Stipendium (EST Marie Curie Fellowship) finanziert.

Ergebnisse und Diskussion 81
4.1 Formulierungsentwicklung von ligandmodifizierten Liposomen
Ziel war es, ein liposomales Trägersystem zu entwickeln, dass sich für die zielgerichtete Applikation an Knochen, der von metastasiertem Gewebe umgeben war, eignet. Die Besonderheit der Liposomen sollte darin bestehen, dass sie neusynthetisierte Bisphosphonatderivate auf ihrer Oberfläche als knochenaffine Moleküle aufweisen sollten. Anhand von Vorversuchen wurden zunächst verschiedene neusynthetisierte Bisphosphonatligandmoleküle dahingehend überprüft, ob sie sich stabil in die Liposomenmembran einbauen ließen. Die in den Vorversuchen eingesetzten Liganden bestanden aus folgenden Bisphosphonsäurederivaten, die von der Universität Bremen im Auftrag der Firma MCS Micro Carrier Systems GmbH synthetisiert wurden: Palmitoyl-bisphosphonsäure (PAL-BP) (Abbildung 9), Cholesteryl-bisphosphonsäure (CHOL-BP) (Abbildung 10), Cholesteryl-trisoxyethylen-bisphosphonsäure (CHOL-TOE-BP) (Abbildung 11) und Cholesteryl-tetraoxyethylen-bisphosphonsäure (CHOL-TeOE-BP) (Abbildung 12). Die hier vorliegende Doktorarbeit basiert auf dem von MCS angemeldeten Patent „Bisphosphonat+Cholesterol“ (WO2005070952). Anzunehmen war, dass die speziell angefertigten Bisphosphonatliganden aufgrund ihres Cholesteryl- bzw. Palmitoylrestes über die Lipidfilm-Methode leicht in die Membran verankert werden konnten, da beide Ankermoleküle gewöhnliche Bestandteile von Biomembranen darstellen. Zur Überprüfung wurden diese neusynthetisierten Bisphosphonatliganden zunächst in einer Konzentration von 25,0 mol% in Liposomen, bestehend aus variierenden Lipidzusammensetzungen (EPC oder DPPC) und verschiedenen HEPES-Puffersystemen, inkorporiert und auf ihre Stabilität hin untersucht. Die Stabilitätsuntersuchungen umfassten hierbei die Bestimmung physikochemischer Parameter wie pH und Partikelgröße. Zudem wurde optisch kontrolliert, ob Ausfällungen feststellbar waren. Wichtig bei diesen Entwicklungsversuchen war, die Herstellungsmethode und die liposomale Zusammensetzung so zu wählen, dass es möglich war, Liposomen im Größenbereich von 100–200 nm herzustellen, die zudem einen pH-Wert von etwa 7,4 aufwiesen. Die Entwicklung einer isohydrischen Formulierung wurde angestrebt, da diese Liposomenformulierung für die intravenöse Applikation geeignet sein sollte. Die Grenzwerte für intravenös applizierbare Injektionen liegen zwar zwischen pH 3,0 und pH 10,5, doch um eine schmerzhafte Anwendung zu vermeiden, wurde die Einstellung auf den pH-Wert des Blutes (pH: 7,30–7,45) zum Ziel gesetzt. Die angestrebte Partikelgröße wurde zuvor definiert, da

Ergebnisse und Diskussion 82
die Liposomengröße für die Fähigkeit, die Knochenoberfläche zu erreichen und daran binden zu können, sicherlich eine entscheidende Rolle spielt. Denn um aktiv an die Mineralmatrix binden zu können, muss gewährleistet sein, dass die Liposomen nach intravenöser Applikation durch passives Targeting über fenestrierte Endothelien von Blutgefäßen in metastasendurchsetztem Knochengewebe aus der Blutbahn in das Interstitium gelangen und von dort aus möglicherweise die Mineralmatrix erreichen. Zudem wurde die Größe zuvor als Zielparameter festgelegt, um zu vermeiden, dass die Liposomen in späteren Versuchen auf dem Weg zur Knochenoberfläche bereits aufgrund ihrer Größe von Makrophagen als fremd erkannt und folglich vorzeitig aus der Blutbahn eliminiert werden.
Abbildung 9: Strukturformel der Palmitoyl-bisphosphonsäure.
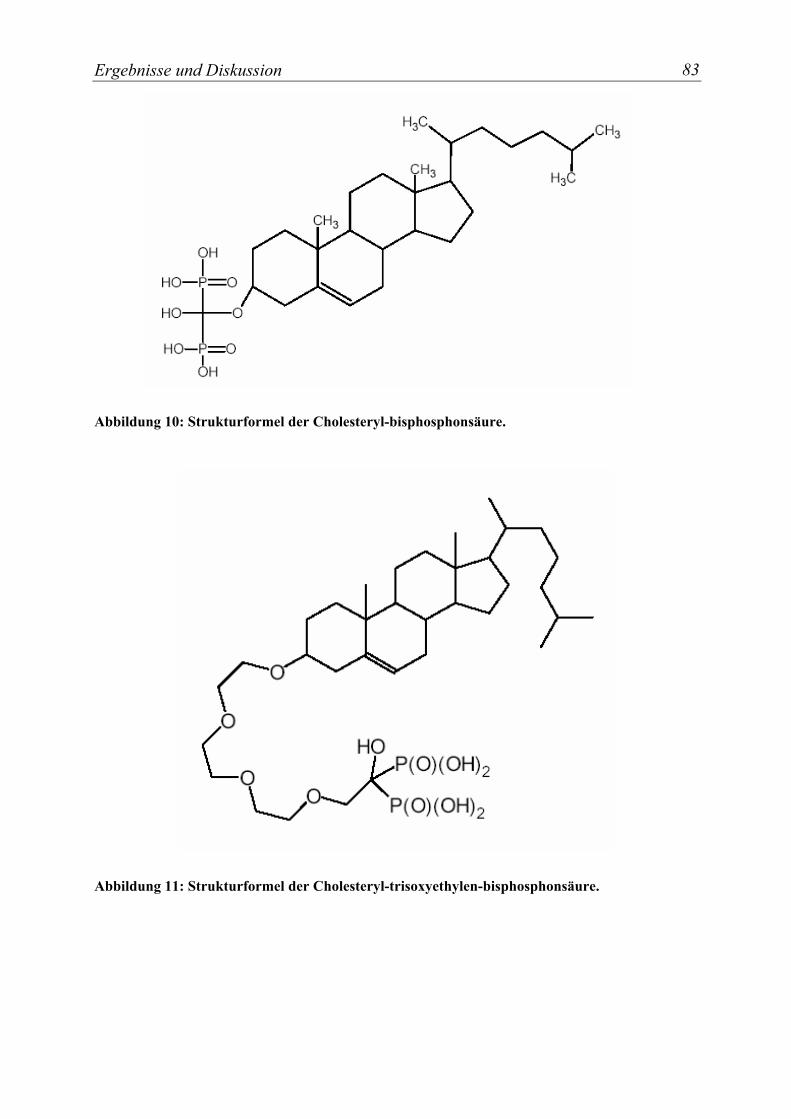
Ergebnisse und Diskussion 83
Abbildung 10: Strukturformel der Cholesteryl-bisphosphonsäure.
Abbildung 11: Strukturformel der Cholesteryl-trisoxyethylen-bisphosphonsäure.

Ergebnisse und Diskussion 84
Abbildung 12: Strukturformel der Cholesteryl-tetraoxyethylen-bisphosphonsäure.
4.1.1 Präformulierungen von ligandmodifizierten Liposomen
Die Herstellung der Liposomen im Rahmen der Vorversuche erfolgte wie unter 3.3.2 beschrieben. Für den Extrusionschritt wurde dazu entweder der Lipex-Extruder oder der LiposoFast-Handextruder eingesetzt. Die Präformulierungsversuche ergaben (Ergebnisse sind nicht dargestellt), dass der Cholesteryl-bisphosphonsäure-Ligand aufgrund seiner schlechten Löslichkeit ungeeignet war. Mit dem Palmitoyl-bisphosphonsäure-Derivat ließen sich zunächst erfolgreich Liposomen herstellen, die dann aber wegen der kurzen Stabilität nicht für weitere Versuche eingesetzt wurden. Der Einbau von entweder Cholesteryl-trisoxyethylen-bisphosphonsäure (CHOL-TOE-BP) oder von Cholesteryl-tetraoxyethylen-bisphosphonsäure (CHOL-TeOE-BP) erwies sich hingegen als vielversprechend. Die Vorversuche zeigten weiterhin, dass die Bisphosphonsäurederivate wegen ihrer Azidität zu einem starken pH-Wert-Abfall der Liposomenformulierung führten, die das Verwenden eines Puffersystems mit ausreichender Pufferkapazität erforderten. Der anfängliche Gebrauch von 10 mmol HEPES (150 mmol NaCl, pH 7,4) war nicht ausreichend, um den gewünschten pH-Wert von 7,4 in der Liposomendispersion zu erzielen. Der pH-Wert fiel stattdessen aufgrund des Einbaus des aziden Ligandmoleküls auf ca. 3 ab. Durch den Einsatz eines HEPES-Puffers, bestehend aus 50 mmol HEPES, 0,7% NaCl, pH 7,4 (HBSpH7,4), konnte das Problem des pH-Wert-Abfalles gelöst werden. Gemäß der Henderson-Hasselbalch-Gleichung spielen die absoluten Pufferkonzentrationen eine wesentliche Rolle für die Belastbarkeit des Puffers durch Säure- oder Basezusatz. Das Maximum

Ergebnisse und Diskussion 85
der Pufferkapazität liegt bei pkspH = , und die Pufferkapazität ist umso größer, je
konzentrierter die Pufferlösung ist. Aus diesem Grund wurde die Pufferkonzentration erhöht. Weiterhin ergab sich aus den Vorversuchen, dass sich mit dem LiposoFast-Handextruder (Avestin), einer alternativen Homogenisationsmethode zum Lipex-Extruder, ligandmodifizierte Liposomen mit einer Ligandkonzentration von 25,0 mol% im Größenbereich zwischen 100 und 200 nm nur bis zu einer Lipidkonzentration von 13 µmol/ml herstellen ließen. Zum einen kann dies darin begründet liegen, dass bei dem LiposoFast-Handgerät geringere Drücke aufbaubar sind; zum anderen kann auch die kleinere hier zur Verfügung stehende Filteroberfläche eine Ursache für dieses Ergebnis sein. Mit dem Lipex-Extruder hingegen ließen sich Liposomen mit der angestrebten Lipidkonzentration von 20 µmol/ml problemlos herstellen. Ein weiterer Nachteil der Handextrusionsmethode war das kleine Probenvolumen (1 ml), das das Equipment zuließ, so dass die Entscheidung fiel, mit dem Lipex-Extruder fortzufahren. Mit einer Ligandkonzentration (CHOL-TOE-BP, CHOL-TeOE-BP und PAL-BP) von 25,0 mol% und einer Lipidkonzentration von 20 µmol/ml wurde die maximal einbaubare Ligandmenge für jeden Ligandtypen erreicht. Sobald höhere Ligandkonzentrationen eingesetzt wurden, kam es zu Präzipitationen innerhalb des jeweiligen Ansatzes. Sowohl CHOL-TOE-BP als auch CHOL-TeOE-BP erwiesen sich als geeignete Kandidaten für den erfolgreichen Einbau in die Liposomenmembran. Aufgrund ihrer minimalen strukturellen Unterscheidung, und zwar in einer Oxyethylengruppe im Spacermolekül, wurde entschieden, die Versuche zur Überprüfung der Targeting-Strategie zunächst mit einem der beiden Derivate und zwar mit dem CHOL-TOE-BP-Derivat fortzusetzen. Eine erhöhte Oxidationsempfindlichkeit von EPC aufgrund des hohen Grades an nicht gesättigten Fettsäuren gegenüber dem synthetischen Derivat DPPC führte außerdem zu der Entscheidung, mit dem DPPC fortzufahren, obwohl die anfänglichen Stabilitätsuntersuchungen noch keine Instabilität im Fall von ligandmodifizierten EPC-Liposomen feststellen ließen.
4.1.2 Zusammensetzung von ligandmodifizierten Liposomen
Die Vorversuche haben gezeigt, dass das in der vorliegenden Arbeit vorgestellte Konzept ligandmodifizierter, aus maßgeschneiderten Bisphosphonatliganden und Liposomen bestehender Trägersysteme zu funktionieren scheint. Mit Hilfe der

Ergebnisse und Diskussion 86
Lipidfilm-Extrusionsmethode ließen sich insbesondere die Bisphosphonatderivate, die einen Cholesterylrest als Ankermolekül aufwiesen, gut in die Liposomenmembran inkorporieren. Basierend auf den Erkenntnissen, die aus den Vorversuchen im Hinblick auf eine Abschätzung der Eignung von Bisphosphonatderivaten für weitere Versuche gewonnen werden konnten, wurden für die folgenden Untersuchungen ligandmodifizierte DPPC-Liposomen, die das CHOL-TOE-BP-Derivat als Liganden aufwiesen, routinemäßig nach der in Abschnitt 3.3.2 genannten Methode mit einem Phospholipidgehalt von 20 µmol/ml hergestellt. Dabei bewegten sich die CHOL-TOE-BP-Konzentrationen zwischen 1,6 und 25,0 mol% (Tabelle 9). Alle Ansätze enthielten HEPES-Puffer mit der folgenden Zusammensetzung als wäßriges Medium: 50 mmol HEPES, 0,7% NaCl, pH 7,4 (HBSpH7,4).
4.2 Physikochemische Charakterisierung von ligandmodifizierten Liposomen
Das In-vitro- und In-vivo-Verhalten von Liposomen wird maßgeblich durch ihre Größe, Bilayer-Rigidität, Ladung und Morphologie (z.B. unilamellar, multilamellar) bestimmt. Im Hinblick auf das Ziel, eine erfolgversprechende Formulierung zu entwickeln, ist eine physikochemische Charakterisierung im frühen Entwicklungsstadium unerlässlich. In diesem Zusammenhang wurde die erfolgversprechendste Formulierung für ligandmodifizierte Liposomen, die aus den Vorversuchen hervorgegangen war, zunächst in vitro näher untersucht. Dabei wurde der Einfluss von unterschiedlichen CHOL-TOE-BP-Konzentrationen (1,6 mol%–25,0 mol%) auf die durchschnittliche Partikelgröße bestimmt. Auch hier wurde wiederum im Zuge der Herstellung eine Liposomengröße im Bereich von 100–200 nm aus den zuvor genannten Gründen angestrebt. Um vorab zu prüfen, ob sich die CHOL-TOE-BP-Liganden tatsächlich auf der Liposomenoberfläche befanden, wurden Zeta-Potentialmessungen durchgeführt. Aufgrund der negativen Ladung, die von dem CHOL-TOE-BP-Derivat per se ausging, war zu erwarten, dass dieses Molekül abhängig von der eingesetzten Konzentration einen Einfluss auf die Gesamtladung der Liposomen ausüben würde. Außerdem spielt das Zeta-Potential eine wichtige Rolle für die Stabilität des liposomalen Systems und möglicherweise auch für die Verweildauer im Blut. Das Zeta-Potential wurde hierbei jedoch nicht zuvor als konkreter Wert festgelegt. Im Hinblick auf die Stabilität der Liposomen war kein negativer Einfluss

Ergebnisse und Diskussion 87
durch die negative Liposomenoberflächenladung zu erwarten. Stark negativ geladene und stark positiv geladene Partikeloberflächen garantieren zumeist eine ausreichende Stabilität der Dispersion, wohingegen eine schwache Oberflächenladung aufgrund von zu geringer Abstoßung der Teilchen zu Aggregationen der Liposomen führen kann. In Bezug auf die Zirkulationszeit in vivo dagegen kann ein negatives Zeta-Potential zur drastischen Verkürzung der Verweildauer führen. Eine Änderung des pH-Wertes aufgrund der Herstellung von Liposomen mit geringeren CHOL-TOE-BP-Konzentrationen war nach der Auswahl eines stabilen Puffersystems in den Vorversuchen nicht zu erwarten.
Im Rahmen einer Formulierungsentwicklung von Liposomen ist die Überprüfung der Stabilität unverzichtbar. Da die Partikelgröße einen wichtigen Stabilitätsfaktor bei Liposomen darstellt, wurde diese in Form einer Langzeitstabilitätsstudie untersucht. Veränderungen der Bilayer-Fluidität können die Stabilität der Liposomen sowie die vorzeitige Freisetzung des Wirkstoffes zur Konsequenz haben (Sharma and Sharma, 1997). Um sicherzustellen, dass der Einbau der CHOL-TOE-BP-Liganden in die liposomale Membran keinen entscheidenden Einfluss auf die Bilayer-Stabilität hatte, wurde ihr Einfluss auf die Phasenübergangstemperatur des Lipides DPPC, aus dem die Liposomen hauptsächlich bestehen, überprüft. Der Übergang vom flüssigkristallinen Zustand in den Gelzustand stellt einen relevanten stabilisierenden Effekt dar, da die Lipidmembran im Gelzustand praktisch undurchlässig für kleine Moleküle ist (Schreier, 1982). Durch die Zugabe von Cholesterol kann die Rigidität der Liposomenmembran erhöht werden, so dass die Wirkstofffreisetzung von hydrophilen Wirkstoffen gezielt gesteuert und die Stabilität der Liposomen sowohl in vitro als auch in vivo verbessert werden kann. Der Einfluss von Cholesterol wird dabei anhand eines Abfalls der Phasenübergangstemperatur festgemacht. In diesem Zusammenhang wurde spekuliert, dass der CHOL-TOE-BP-Ligand aufgrund seines Cholesterylanteils ebenfalls einen Einfluss auf die Membranrigidität und folglich auf die Phasenübergangstemperatur ausübt. Mit dem quantitativen Nachweis des Liganden in den Liposomen wurde die physikochemische Charakterisierung vervollständigt. Durch die Extrusion hätten nicht gebundene CHOL-TOE-BP-Moleküle von den Liposomen abgetrennt werden können. Ziel der Ligandquantifizierung war es, diesen möglichen Verlust zu bestimmen, doch basierend auf den Beobachtungen der homogenen Filmresuspendierung und der rückstandslosen Extrusion war kaum Verlust zu erwarten. Herkömmliche Phosphatbestimmungen nach Bartlett oder Rouser ließen die präzise Differenzierung zwischen den lipidgebundenen Phosphaten und

Ergebnisse und Diskussion 88
Phosphonatgruppen des Liganden nicht zu, so dass eine 31P-NMR-Methode etabliert wurde, die dies ermöglichte.
4.2.1 Partikelgröße und Zeta-Potential
Die beiden Parameter Partikelgröße und Zeta-Potential wurden mit einem Nicomp Partikelsizer/Zetasizer-380-ZLS gemessen. Die durchschnittliche Partikelgröße aller Liposomentypen, die in Tabelle 9 untersucht wurden, bewegte sich zwischen 120 und 155 nm mit einem Polydispersionsindex variierend von 0,03 bis 0,15. Die Vergangenheit hat bereits gezeigt, dass die Partikelgröße für die Pharmakokinetik eine entscheidende Rolle spielt. Senior et al. konnten aufzeigen, dass Liposomen, die durch einen 400-nm-Filter extrudiert wurden, 7,5-mal schneller aus der Blutbahn eliminiert wurden als Liposomen, die durch einen 200-nm-Filter extrudiert wurden (Senior et al., 1985). Eine zunehmende Partikelgröße führt zu einer vermehrten Aufnahme in das Retikuloendotheliale System (RES) (Abra and Hunt, 1981). Der Einbau von DSPE-PEG in die Liposomenmembran hat zudem in der Vergangenheit gezeigt, dass solche Liposomen, wenn sie sich im Größenbereich von 80–250 nm bewegen, dieser größenspezifischen Elimination durch Makrophagen eher resistent gegenüberstehen (Liu et al., 1992, Woodle and Lasic, 1992). Die ligandmodifizierten Liposomen sollen idealerweise später zur Therapie von Knochenmetastasen eingesetzt werden, und sie sollen in diesem Zusammenhang an die Mineralmatrix des Knochens binden. Bei dieser zielgerichteten Applikation an den Knochen kann die Größe der Liposomen ein limitierender Faktor sein. Um erfolgreiches Knochen-Targeting zu erzielen, müssen die ligandmodifizierten Liposomen mehrere Hindernisse überwinden. Zunächst müssen sie ein ausreichendes Pharmakokinetikprofil aufzuweisen, um an den Wirkort gelangen zu können. Dazu müssten sie das Endothelium der Blutgefäße im Bereich des metastasendurchsetzten Knochens überwinden, das anschließende interstitielle Fluid durchqueren und durch die Lining-Zellen (Lining-Zellen sind ruhende Osteoblasten) penetrieren, um an die Knochenoberfläche binden zu können (Shea and Miller, 2005). Es ist bekannt, dass die Vaskulatur im Knochen fenestriert ist (Howlett et al., 1984, Ghandehari et al., 1997), so dass die Liposomen möglicherweise in der Lage sind, aus der Blutbahn in das Knochengewebe zu gelangen. Die physiologischen Veränderungen im Falle von metastasendurchsetztem Knochengewebe wie gesteigerter „Bone Turnover“ und damit erhöhte Durchblutung des Knochens können

Ergebnisse und Diskussion 89
das Einwandern von ligandmodifizierten Liposomen in das befallene Knochengewebe und das Erreichen der Knochenoberfläche begünstigen. Eine durchlässige Mikrovaskulatur und ein in seiner Funktion beeinträchtigtes lymphatisches System, welche insbesondere in Tumorgewebe und in metastasiertem Gewebe gegeben sind, können das Eintreten der Liposomen in das metastasendurchsetzte Knochengewebe zusätzlich unterstützen (Matsumura and Maeda, 1986, Huang et al., 1992, Seymour, 1992). Dieser Effekt wird auch als „Enhanced Permeability and Retention“-Effekt (EPR-Effekt) bezeichnet (Matsumura and Maeda, 1986, Maeda and Matsumura, 1989). Die Liposomen gelangen dabei durch das diskontinuierliche Endothelium der Tumormikrovaskulatur, das Porengrößen von 100–780 nm aufweist (Yuan et al., 1995, Hobbs et al., 1998), in das Tumorinterstitium, wo sie verweilen und ihren Wirkstoff freigeben. Die Permeabilität des Tumorgewebes ist allerdings stark abhängig von der Tumorart und der Tumorumgebung, wie z.B. der Dichte der Vaskularität (Yuan et al., 1994, Hobbs et al., 1998). Die durchschnittliche Partikelgröße des Trägersystems stellt somit für ein erfolgreiches Targeting an den Knochen einen relevanten Faktor dar. Ligandmodifizierte Liposomen im Größenbereich von 120–155 nm bringen folglich eine wichtige Voraussetzung für das Erhalten eines geeigneten Pharmakokinetikprofils sowie für die zielgerichtete Applikation an den Knochen mit.
Bei den Zeta-Potentialmessungen stellte sich klar heraus, dass der CHOL-TOE-BP-Ligand einen deutlichen Einfluss auf die Oberflächenladung der Liposomen hatte und folglich in die Membran inkorporiert sein musste. Je höher die CHOL-TOE-BP-Konzentration innerhalb der Liposomenformulierung war, desto negativer fiel das Zeta-Potential aus. Nicht pegylierte, ligandmodifizierte Liposomen wiesen beispielsweise ein Zeta-Potential von -58 ± 1 mV auf, wohingegen die Ladung von nicht pegylierten Kontrollliposomen -17 ± 2 mV (Tabelle 9) betrug. Das Zeta-Potential spielt neben anderen Faktoren eine wichtige Rolle bei dem Verbleib der Liposomen in vivo; somit können stark negativ geladene Liposomenoberflächen die Zirkulationszeit in vivo drastisch verkürzen (Gabizon and Papahadjopoulos, 1992). Durch den Einbau von DSPE-PEG in die Liposomenmembran kann wiederum ein abschirmender Effekt auf die negative Ladung bewirkt werden, so dass eine schnelle Elimination von Liposomen aus der Blutbahn durch Opsonisierung häufig verzögert werden kann (Needham et al., 1992, Uster et al., 1996). Aus diesem Grund wurde der Einfluss von DSPE-PEG2000 auf die Oberflächenladung von ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% ebenfalls untersucht. Der Einbau von 4,3 mol% DSPE-PEG2000 in diese Liposomen führte zu einer geringeren

Ergebnisse und Diskussion 90
negativen Ladung (-35 ± 1 mV), wohingegen der Einbau von 13,0 mol% DSPE-PEG2000 eine Ladung von -25 ± 0 mV erzielte (Tabelle 9). Die Zeta-Potentialmessungen zeigten somit den neutralisierenden Einfluss des DSPE-PEGs deutlich auf, so dass es in den In-vivo-Experimenten zu zeigen gilt, ob die Pegylierung der Liposomen vor dem Abbau durch Makrophagen schützt und folglich die Verweildauer im Blut verlängert. Anzumerken ist, dass die hier dargestellten Zeta-Potentialmessungen nur von indikativer Natur sind. Bei einer In-vivo-Applikation würden aufgrund einer anderen Ionenstärke im Blut die Zeta-Potentiale wiederum verändert werden. Die Bestimmung der pH-Werte (Ergebnisse sind nicht dargestellt) hat gezeigt, dass die pH-Werte aller getesteten Formulierungen wegen des stabilen Puffersystems im angestrebten Bereich von etwa 7,4 lagen.
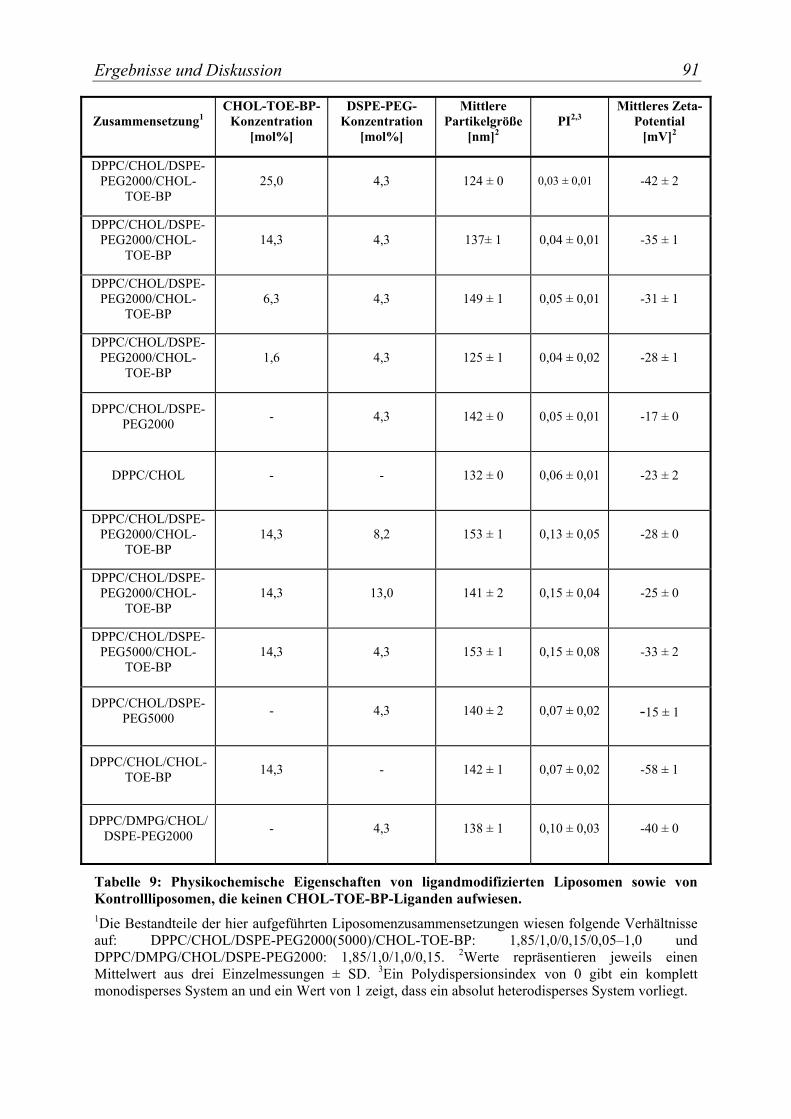
Ergebnisse und Diskussion 91
Zusammensetzung1 CHOL-TOE-BP-
Konzentration [mol%]
DSPE-PEG-Konzentration
[mol%]
Mittlere Partikelgröße
[nm]2 PI2,3
Mittleres Zeta-Potential
[mV]2
DPPC/CHOL/DSPE-PEG2000/CHOL-
TOE-BP 25,0 4,3 124 ± 0 0,03 ± 0,01 -42 ± 2
DPPC/CHOL/DSPE-PEG2000/CHOL-
TOE-BP 14,3 4,3 137± 1 0,04 ± 0,01 -35 ± 1
DPPC/CHOL/DSPE-PEG2000/CHOL-
TOE-BP 6,3 4,3 149 ± 1 0,05 ± 0,01 -31 ± 1
DPPC/CHOL/DSPE-PEG2000/CHOL-
TOE-BP 1,6 4,3 125 ± 1 0,04 ± 0,02 -28 ± 1
DPPC/CHOL/DSPE-PEG2000 - 4,3 142 ± 0 0,05 ± 0,01 -17 ± 0
DPPC/CHOL - - 132 ± 0 0,06 ± 0,01 -23 ± 2
DPPC/CHOL/DSPE-PEG2000/CHOL-
TOE-BP 14,3 8,2 153 ± 1 0,13 ± 0,05 -28 ± 0
DPPC/CHOL/DSPE-PEG2000/CHOL-
TOE-BP 14,3 13,0 141 ± 2 0,15 ± 0,04 -25 ± 0
DPPC/CHOL/DSPE-PEG5000/CHOL-
TOE-BP 14,3 4,3 153 ± 1 0,15 ± 0,08 -33 ± 2
DPPC/CHOL/DSPE-PEG5000 - 4,3 140 ± 2 0,07 ± 0,02 -15 ± 1
DPPC/CHOL/CHOL-TOE-BP 14,3 - 142 ± 1 0,07 ± 0,02 -58 ± 1
DPPC/DMPG/CHOL/DSPE-PEG2000 - 4,3 138 ± 1 0,10 ± 0,03 -40 ± 0
Tabelle 9: Physikochemische Eigenschaften von ligandmodifizierten Liposomen sowie von Kontrollliposomen, die keinen CHOL-TOE-BP-Liganden aufwiesen. 1Die Bestandteile der hier aufgeführten Liposomenzusammensetzungen wiesen folgende Verhältnisse auf: DPPC/CHOL/DSPE-PEG2000(5000)/CHOL-TOE-BP: 1,85/1,0/0,15/0,05–1,0 und DPPC/DMPG/CHOL/DSPE-PEG2000: 1,85/1,0/1,0/0,15. 2Werte repräsentieren jeweils einen Mittelwert aus drei Einzelmessungen ± SD. 3Ein Polydispersionsindex von 0 gibt ein komplett monodisperses System an und ein Wert von 1 zeigt, dass ein absolut heterodisperses System vorliegt.

Ergebnisse und Diskussion 92
4.2.2 Stabilität im Hinblick auf die Partikelgröße
In Abbildung 13 ist die Langzeitstabilität (9 Monate) für ligandmodifizierte Liposomen mit verschiedenen Ligandkonzentrationen (1,6 mol% CHOL-TOE-BP, 6,3 mol% CHOL-TOE-BP, 14,3 mol% CHOL-TOE-BP) dargestellt. Es ist klar erkennbar, dass die durchschnittliche Partikelgröße in allen Fällen über diesen Zeitraum bei einer Lagerungstemperatur von 2–8 °C relativ konstant blieb und auch hier keine Anzeichen für eine baldige Instabilität erkennen ließ. Diese könnte sich beispielsweise durch wachsende Partikelgrößenverteilungen aufgrund von beginnenden Präzipitationen abzeichnen. Zudem lässt die Darstellung dieser Abbildung erkennen, dass die ligandmodifizierten Liposomen mit einer hohen CHOL-TOE-BP-Konzentration (14,3 mol%) im Vergleich zu ligandmodifizierten Liposomen mit einer geringeren CHOL-TOE-BP-Konzentration (6,3 mol% oder 1,6 mol%) im Durchschnitt größere Liposomen aufwiesen. Diese Beobachtung ist in der dargestellten Grafik (Abbildung 13) wahrscheinlich eher ein Zufall. Erfahrungen im Zusammenhang mit der Extrusion von ligandmodifizierten Liposomen haben gezeigt, dass es auch möglich gewesen wäre, die durchschnittliche Größe von Liposomen mit hohen CHOL-TOE-BP-Konzentrationen durch weitere Extrusionsschritte noch ein wenig zu verringern (~ 140 nm) (Ergebnisse sind nicht dargestellt). Dennoch ist anzumerken, dass die Extrusion von Liposomen mit hohen CHOL-TOE-BP-Konzentrationen höhere Drücke und mehr Wiederholungen bei den einzelnen Extrusionsschritten erfordert als bei Liposomen mit geringeren oder gar keinen Ligandkonzentrationen, um die gleiche Partikelgröße zu erhalten. Dies ist möglicherweise darauf zurückzuführen, dass es bei hohen CHOL-TOE-BP-Konzentrationen zu sterischen Abstoßungen unter den einzelnen Liganden kommt. Zudem könnte die negative Ladung (wie bei den folgenden Zeta-Potentialmessungen gezeigt), bedingt durch diesen CHOL-TOE-BP-Liganden, elektrostatische Wechselwirkungen mit den Filtermembranen bewirken, die das Extrudieren nachteilig beeinflussen. Die zusätzlichen Extrusionsschritte hatten zur Folge, dass ein vermehrter Probenverlust zu beobachten war.

Ergebnisse und Diskussion 93
120
130
140
150
160
170
180
0 50 100 150 200 250 300
Tage
Dur
chsc
hnitt
liche
Par
tikel
größ
e [n
m]
14,3 mol% CHOL-TOE-BP 6,3 mol% CHOL-TOE-BP1,6 mol% CHOL-TOE-BP
Abbildung 13: Langzeitstabilität (9 Monate) im Hinblick auf die durchschnittliche Partikelgröße von ligandmodifizierten Liposomen mit verschiedenen CHOL-TOE-BP-Konzentrationen (1,6 mol%, 6,3 mol%, 14,3 mol%). Die Lagerung der Liposomen erfolgte bei 2–8 °C. Die hier dargestellten Werte repräsentieren jeweils einen Mittelwert aus drei Einzelmessungen ± SD.
4.2.3 Phasenübergangstemperaturbestimmung
Um den Einfluss des CHOL-TOE-BP-Liganden auf die Phasenübergangstemperatur von dem Lipid DPPC zu bestimmen, wurden zunächst ligandmodifizierte Liposomen mit hohen Ligandkonzentrationen (14,3 mol% CHOL-TOE-BP und 25,0 mol% CHOL-TOE-BP) untersucht, die kein Cholesterol in ihrer Zusammensetzung aufwiesen. Die Messungen wurden mit einem DSC-Gerät Q-1000 von TA Instruments durchgeführt. Gemäß Literatur weist das DPPC seinen Phasenübergang bei einer Temperatur von etwa 41 °C auf (Fox et al., 2007). Beide Formulierungen wiesen bei eben dieser Temperatur einen Phasenübergang in ihrem Phasendiagramm auf (Abbildung 14: B, C). Bei Liposomen, die in ihrer Zusammensetzung 14,3 mol% CHOL-TOE-BP und 28,6 mol% Cholesterol aufwiesen, ließ sich keine Phasenübergangstemperatur bestimmen (Abbildung 14, A). Die Ergebnisse zeigen somit, dass der Einbau des Liganden in beiden getesteten Konzentrationen keinen entscheidenden Einfluss auf die Phasenübergangstemperatur hatte und deshalb keinen detektierbaren Einfluss auf die Phospholipidkonformation innerhalb der Liposomen

Ergebnisse und Diskussion 94
ausübte. Eine erhöhte Instabilität war aufgrund dieser Beobachtungen nicht zu erwarten. Die Vermutung, dass der Cholesterolanteil im Ligandmolekül möglicherweise die Membranrigidität der Liposomen beeinflusste und die Phasenübergangstemperatur folglich senkte, wurde ebenfalls nicht bestätigt. Die eingesetzte Ligandkonzentration war offenbar zu gering, um einen solchen Effekt deutlich erkennen lassen zu können. Der zusätzliche Einbau von 28,6 mol% Cholesterol in die ligandmodifizierten Liposomen zeigte dann seinen Einfluss auf die Phasenübergangstemperatur (Abbildung 14, A). Diese war durch DSC-Messungen nicht mehr detektierbar, was wiederum den Effekt des Cholesterols auf die Fluidität der Membran widerspiegelte. Es bestätigte sich, dass die Fluidität abnahm und die Liposomenmembranen als Konsequenz starrer in ihrer Struktur wurden. Für zukünftige Versuche ist diese Stabilität von Relevanz, da sie die Liposomen vor dem frühzeitigen Verlust des eingeschlossenen Wirkstoffes schützt (Kirby et al., 1980, Muramatsu et al., 1999).

Ergebnisse und Diskussion 95
Abbildung 14: DSC-Thermogramme von ligandmodifizierten Liposomen mit folgender Zusammensetzung: A: 28,6 mol% Cholesterol, 14,3 mol% CHOL-TOE-BP; B: kein Cholesterol, 14,3 mol% CHOL-TOE-BP; C: kein Cholesterol, 25,0 mol% CHOL-TOE-BP (abgefahrener Temperaturbereich: von -5 °C bis 80 °C; Scan-Rate: 5 °C/min).

Ergebnisse und Diskussion 96
4.2.4 Ligandquantifizierung
Zum quantitativen Nachweis der CHOL-TOE-BP-Liganden in der Formulierung wurden Proben von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP) vor dem Extrusionsschritt und nach vollendeter Extrusion gezogen, gefriergetrocknet, in CDCl3 resuspendiert und vor der Messung mit PAL-BP als internem Standard versetzt. Die Messungen wurden mit einem Varian Mercury-Plus-300-Gerät (300 MHz) durchgeführt. Die Ergebnisse in Abbildung 15 zeigen, dass die neuentwickelte 31P-NMR-Methode eine Auftrennung des Phosphatsignals (Lipid) und des Phosphonatsignals (CHOL-TOE-BP-Ligand) ermöglicht. Die Abbildung 15 zeigt jeweils ein separates Signal für das Lipid DPPC mit einer chemischen Verschiebung zu -0,5 ppm und für den CHOL-TOE-BP-Liganden mit einer chemischen Verschiebung zu 18,5 ppm. Der Zusatz von PAL-BP als internem Standard ermöglichte die relative quantitative Bestimmung des DPPC und des CHOL-TOE-BPs. Dazu wurde die Signalintensität von PAL-BP (chemische Verschiebung zu 22,0 ppm) als 100%-Wert gesetzt, und die Signale der anderen Komponenten wurden zu diesem Wert in Relation gesetzt und ebenfalls in Prozent ausgedrückt. Generell wurde im Zuge der Extrusion in allen Proben ( 3=n ) ein CHOL-TOE-BP-Verlust von 5% bestimmt. Dieses Ergebnis lässt somit eine gute Reproduzierbarkeit der Messung erkennen. In anderen Studien hat sich die 31P-NMR-Methode ebenfalls bereits als reproduzierbare quantitative Analysenmethode bewährt (Sotirhos et al., 1986, Murgia et al., 2003). Ein Nachteil dieser Methode ist sicherlich, dass sie sich nicht ausschließlich auf die Quantifizierung des Ligandenanteils beschränkt, der sich auf der Liposomenoberfläche befindet, sondern auch die in das Liposomeninnere gerichteten Moleküle erfasst. Bei unilamellaren Liposomen, so wie sie in dieser Arbeit bei einer Partikelgröße von etwa 150 nm höchstwahrscheinlich auch vorlagen, befinden sich jedoch laut Literatur etwa 60% des eingebauten Liganden auf der Liposomenoberfläche (Beschiaschvili and Seelig, 1992, Schote and Seelig, 1998).
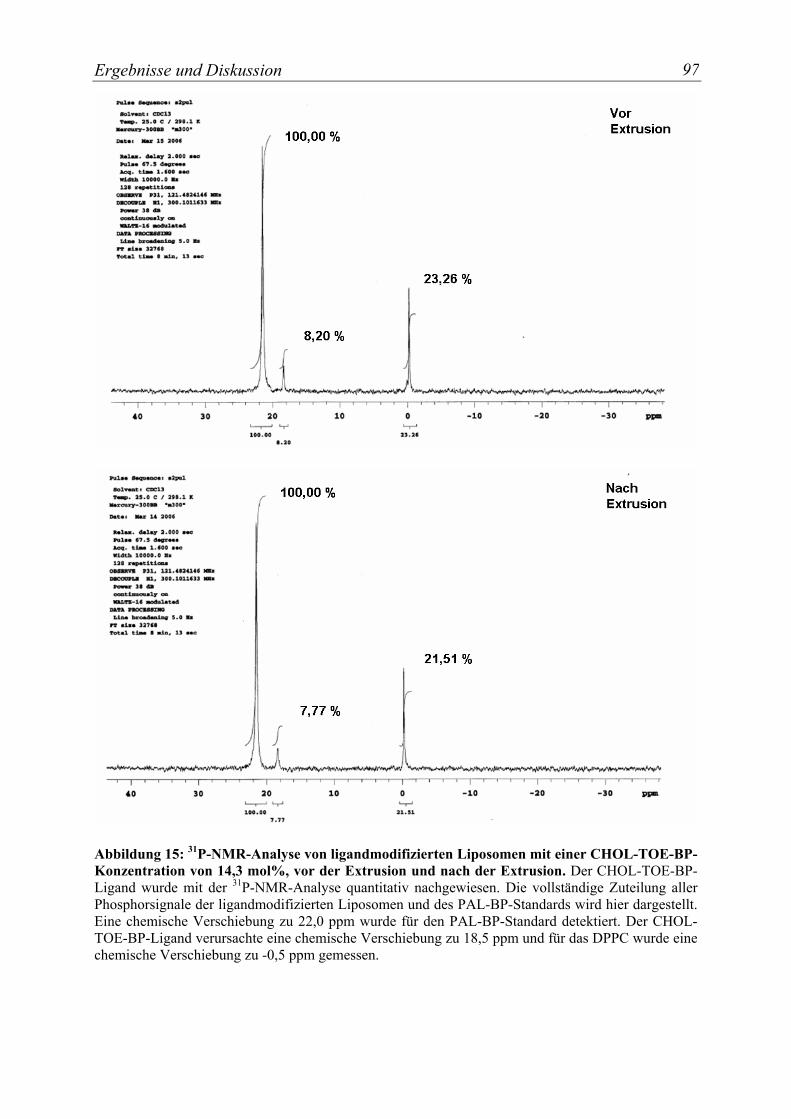
Ergebnisse und Diskussion 97
Abbildung 15: 31P-NMR-Analyse von ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol%, vor der Extrusion und nach der Extrusion. Der CHOL-TOE-BP-Ligand wurde mit der 31P-NMR-Analyse quantitativ nachgewiesen. Die vollständige Zuteilung aller Phosphorsignale der ligandmodifizierten Liposomen und des PAL-BP-Standards wird hier dargestellt. Eine chemische Verschiebung zu 22,0 ppm wurde für den PAL-BP-Standard detektiert. Der CHOL-TOE-BP-Ligand verursachte eine chemische Verschiebung zu 18,5 ppm und für das DPPC wurde eine chemische Verschiebung zu -0,5 ppm gemessen.

Ergebnisse und Diskussion 98
4.3 Bindung von ligandmodifizierten Liposomen an Hydroxylapatit
Mittels Isothermaler Titrationskalorimetrie (ITC) wurde die Bindungsfähigkeit von ligandmodifizierten Liposomen mit CHOL-TOE-BP-Konzentrationen von 1,6 mol% bis 14,3 mol% an Hydroxylapatit (HAP) untersucht, und somit wurde gleichzeitig indirekt bestimmt, ob das CHOL-TOE-BP-Derivat tatsächlich auf der Außenseite der Liposomen saß. Die ITC ist eine thermodynamische Methode zur Bestimmung einer chemischen Reaktion, die initiiert wird, wenn eine zu bindende Komponente zu einem weiteren Reaktionspartner in ein Reaktionsgefäß gegeben wird. Wenn dann eine Bindungsreaktion stattfindet, wird entweder Reaktionswärme absorbiert oder generiert (Heerklotz, 2004). Das Bindungspotential von Bisphosphonaten an Hydroxylapatit wurde bereits von verschiedenen Arbeitsgruppen erfolgreich aufgezeigt (Wingen et al., 1986, Shea and Miller, 2005). Aufgrund dieser Daten wurde die Hypothese aufgestellt, dass auch der hier vorgestellte CHOL-TOE-BP-Ligand in der Lage ist, an das synthetische HAP zu binden. Das CHOL-TOE-BP-Derivat bestand unter anderem aus einer Trisoxyethylenkette, die als Spacermolekül sterische Interaktionen mit dem ebenfalls auf der Liposomenoberfläche vorhandenen DSPE-PEG2000 möglichst vermeiden sollte. Nach erfolgreichem Binden von ligandmodifizierten Liposomen an HAP sollte in einem weiteren Versuch der Einfluss von verschiedenen Plasmakonzentrationen auf die Bindung dieser Liposomen an HAP untersucht werden. Im Hinblick auf die zielgerichtete Applikation dieser Liposomen an den Knochen könnte diese Art von Untersuchung einen Hinweis geben, ob die Bindung der ligandmodifizierten Liposomen an die Mineralmatrix des Knochens möglicherweise durch In-vivo-relevante Faktoren wie Blutplasmabestandteile behindert wird.
4.3.1 Einfluss der Ligandkonzentration auf die Bindung an Hydroxylapatit
Zur Untersuchung des Einflusses der Ligandkonzentration auf die Bindung an HAP mittels ITC wurden ligandmodifizierte Liposomen mit unterschiedlichen CHOL-TOE-BP-Konzentrationen (1,6 mol%, 6,3 mol%, 14,3 mol%) bei konstanter Temperatur schrittweise in eine Messzelle titriert (38 Injektionen), die eine HAP-Suspension enthielt. Nachdem die Liposomen in die Messzelle injiziert wurden, reagierten das

Ergebnisse und Diskussion 99
HAP und die Liposomen miteinander und zwar in Form einer exothermen Reaktion. Durch die Sättigung der HAP-Bindungsstellen mit dem Fortgang der Titration nahm die Wärmeentwicklung ab, so dass am Ende nur noch das Hintergrundsignal der Wärmetönung, die durch die Verdünnung verursacht wurde, zu messen war. Die Titrationskurven in Abbildung 16 zeigen deutlich, dass die ligandmodifizierten Liposomen an HAP gebunden haben. Jede Titration von Liposomen in die HAP-Suspension verursachte eine Wärmeentwicklung, welche im Zuge der 38 Titrationsschritte abnahm. Die Titrationskurven (Abbildung 16) zusammen mit der kumulativen Darstellung der Ergebnisse (Abbildung 17) verdeutlichen die positive Korrelation zwischen der CHOL-TOE-BP-Konzentration und der Wärmeentwicklung. Je mehr CHOL-TOE-BP-Liganden in die Liposomenmembran inkorporiert wurden, desto höher war die entstehende Reaktionswärme und desto steiler fiel die Bindungskurve aus. Die Titration von Kontrollliposomen mit einem Zeta-Potential von -17 mV zeigte keine Reaktionswärmeentwicklung (die mittels der Titrationsergebnisse berechnete Reaktionsenthalpie war 0 kcal/mol), was darauf hinweist, dass keine Bindung zwischen den Liposomen und HAP stattgefunden hat (Ergebnisse sind nicht dargestellt). Die Tatsache, dass ligandmodifizierte Liposomen an HAP gebunden haben, zeigt außerdem zum einen, dass die Liganden tatsächlich auf der Oberfläche sitzen, und zum anderen, dass das ebenfalls vorhandene DSPE-PEG2000 die HAP-Bindung von CHOL-TOE-BP-Liganden offensichtlich nicht durch mögliche Interaktionen untereinander negativ beinflusste. Die Vergangenheit hat in diesem Zusammenhang gezeigt, dass das PEG, welches mit der Intention eingebaut wurde, die Zirkulationszeit im Blut zu verlängern, zur Abschirmung der ebenfalls vorhandenen Liganden führte und somit einen Aktivitätsverlust der Liposomen bewirkte (Mori et al., 1991, Allen, 1994).
Um den Bindungsmechanismus von ligandmodifizierten Liposomen an HAP klarer herauszuarbeiten, d.h. um zu untersuchen, ob ligandspezifisches Binden durch auf der Oberfläche sitzende CHOL-TOE-BP-Liganden vorlag, wurde zusätzlich die Bindung von stark negativ geladenen, nicht modifizierten Liposomen der Zusammensetzung DPPC/DMPG/CHOL/DSPE-PEG2000 in einem molaren Verhältnis von 1,85/1,0/1,0/0,15 (Zeta-Potential: -40 mV) an HAP untersucht. In diesem Fall wurde keine Affinität zum HAP festgestellt (die Bestimmung der Reaktionsenthalpie ergab ebenfalls einen Wert von 0 kcal/mol), was darauf hindeutet, dass tatsächlich ligandspezifisches Binden bei dem vorliegenden liposomalen System eine große Rolle spielt (Ergebnisse sind nicht dargestellt). Es wird vermutet, dass durch die effektive Konfiguration des Ligandmoleküls, welches durch 2 Phosphonate, die sich ein

Ergebnisse und Diskussion 100
Kohlenstoffatom teilen, charakterisiert ist, ein Chelatkomplex mit einem Calciumatom des HAPs gebildet wird (Van Beek ER, 1998, Nancollas et al., 2005). Die relativ hohen negativen Teilladungen der Phosphate im Ligandmolekül und die positiven Ladungen der Calciumatome des HAPs bestärken diese Annahme. Möglicherweise spielen hier auch andere Bindungsmechanismen wie elektrostatische Wechselwirkungen eine Rolle.
Die bei der Isothermalen Titrationskalorimetrie ermittelte Bindungsisotherme erlaubte außerdem die Bestimmung von thermodynamischen Parametern. In der vorliegenden Arbeit wurden die molare Reaktionsenthalpie ( H∆ ) und die apparente Bindungskonstante ( appK ) (Tabelle 10) (die Berechnung ist in Abschnitt 3.3.4.2 zusammengefasst) als Indikatoren für die Bindungsstärke bestimmt, die bei der Bindung von ligandmodifizierten Liposomen an HAP beobachtet wurde. Die Reaktionsenthalpie H∆ , die sich aus der Integration der ermittelten Titrationspeaks ergibt, ist dabei der Term, der in erster Linie die Bindungsstärke definiert (Luque and Freire, 2002). Die erhaltenen Ergebnisse demonstrieren eine positive Korrelation zwischen H∆ und den verschiedenen CHOL-TOE-BP-Konzentrationen und somit stärkere Bindungen im Fall von Liposomen mit höheren CHOL-TOE-BP-Konzentrationen. Folglich wurden für Liposomen mit höheren Ligandkonzentrationen auch höhere Werte für die Bindungskonstante appK bestimmt (siehe Berechnung in Abschnitt 3.3.4.2). Der Erhalt von Titrationskurven unterschiedlicher Steilheit, abhängig von der eingesetzten Ligandkonzentration, unterstreicht zusätzlich die Beobachtungen zur Bindungsstärke an HAP. Sowohl in den Titrationskurven der Abbildung 16 als auch in der kumulativen Darstellung (Abbildung 17) ist klar erkennbar, dass die Steilheit der Kurve mit der Ligandkonzentration korreliert. Je höher die untersuchte CHOL-TOE-BP-Konzentration, desto steiler ist die Titrationskurve und desto eher ist das Plateau erreicht. Eine steile Titrationskurve demonstriert nach Seelig ein stärkeres Bindungsverhalten (Seelig, 1997) im Vergleich zu einer weniger steilen Kurve. Ein stärkeres Bindungsverhalten kann möglicherweise darin begründet sein, dass im Fall von hohen CHOL-TOE-BP-Konzentrationen polyvalentes Binden stattfindet. Mit anderen Worten: Ein Liposom wurde über mehrere CHOL-TOE-BP-Liganden an HAP gebunden.

Ergebnisse und Diskussion 101
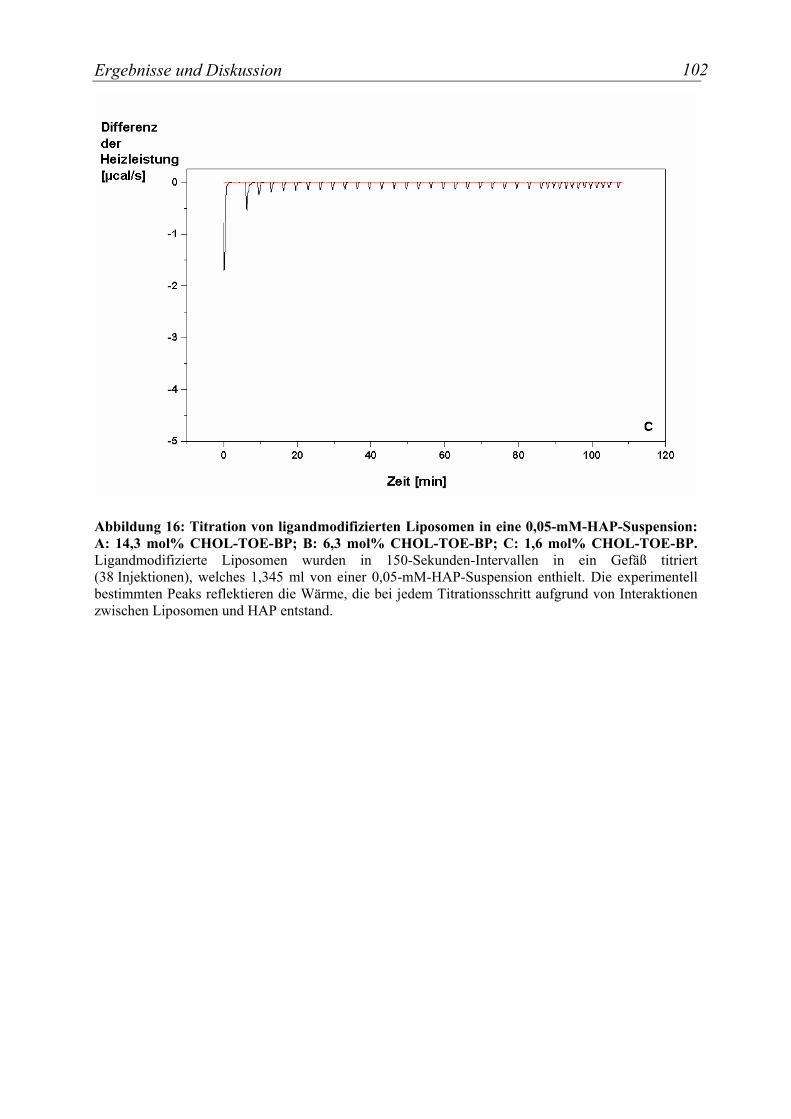
Ergebnisse und Diskussion 102
Abbildung 16: Titration von ligandmodifizierten Liposomen in eine 0,05-mM-HAP-Suspension: A: 14,3 mol% CHOL-TOE-BP; B: 6,3 mol% CHOL-TOE-BP; C: 1,6 mol% CHOL-TOE-BP. Ligandmodifizierte Liposomen wurden in 150-Sekunden-Intervallen in ein Gefäß titriert (38 Injektionen), welches 1,345 ml von einer 0,05-mM-HAP-Suspension enthielt. Die experimentell bestimmten Peaks reflektieren die Wärme, die bei jedem Titrationsschritt aufgrund von Interaktionen zwischen Liposomen und HAP entstand.

Ergebnisse und Diskussion 103
Liposomenzusammensetzung
CHOL-TOE-BP-
Konzentration [mol%]
H∆ [kcal/mol]
appK [M-1] (Mittelwert1)
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 14,3 -17,1 18734
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 6,3 -13,2 5681
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 1,6 -1,3 1874
Tabelle 10: Thermodynamische Parameter, die die Bindung von ligandmodifizierten Liposomen (20 mM) mit unterschiedlichen CHOL-TOE-BP-Konzentrationen an HAP (0,05 mM) charakterisieren (Versuchsbedingungen: 25 °C, HBSpH7,4 50 mmol, 0,7% NaCl, pH 7,4). Die Bestimmung der thermodynamischen Parameter erfolgte nach der Methodenbeschreibung 3.3.4.2. 1Die hier dargestellten Ergebnisse repräsentieren jeweils einen Mittelwert aus einzelnen Bindungskonstanten, die für jeden Punkt der jeweiligen Bindungsisotherme bestimmt werden konnten.

Ergebnisse und Diskussion 104
-1400-1200-1000
-800-600-400-200
0200
0 10 20 30 40
Anzahl der Injektionen
Kum
ulat
ive
Rea
ktio
nsw
ärm
e [µ
cal]
1,6 mol% CHOL-TOE-BP6,3 mol% CHOL-TOE-BP14,3 mol% CHOL-TOE-BP
Abbildung 17: Kumulative Reaktionsenthalpie von ligandmodifzierten Liposomen mit unterschiedlichen CHOL-TOE-BP-Konzentrationen als Funktion von der Zahl der Injektionen. Liposomen wurden in 150-Sekunden-Intervallen in die Reaktionszelle titriert (38 Injektionen), die 1,345 ml einer 0,05-mM-HAP-Suspension enthielt. Durch die Integration der experimentell erhaltenen Titrationspeaks erhält man die Reaktionsenthalpie, die hier kumulativ dargestellt wird.
4.3.2 Einfluss von Plasmaproteinen auf die Bindung an HAP
Die ITC-Technik wurde außerdem zur Untersuchung der Liposomenbindung an HAP unter Einfluss von Blutplasma eingesetzt. Dazu wurde humanes Plasma in verschiedenen Konzentrationen (1%, 5%, 10%) vor Beginn der Titration von ligandmodifizierten Liposomen (14,3 mol CHOL-TOE-BP) zur 0,05-mM-HAP-Suspension in die Messzelle gegeben. Aus den kumulativen Darstellungen der erhaltenen Titrationskurven (Abbildung 18) bzw. der Kontrolltitrationen (Abbildung 19) geht klar hervor, dass die Plasmabestandteile mit den Liposomen reagierten, da die Entstehung der Reaktionswärme mit steigender Plasmakonzentration zunahm. Die in Tabelle 11 dargestellten Werte für die jeweiligen molaren Reaktionsenthalpien bestätigen folglich die folgende Korrelation: Je höher die Plasmakonzentration war, desto negativer fiel der Wert für die molare Reaktionsenthalpie aus. Diese Beobachtung wird zudem durch den Vergleich der Kontrolltitrationen, sprich die Titration von ligandmodifizierten Liposomen in HBSpH7,4 und in das jeweilige HBSpH7,4/Plasma-Gemisch, noch verstärkt. Bei der Titration in HBSpH7,4 war kaum eine Wärmeentwicklung erkennbar (Abbildung 19), wohingegen bei der Titration in

Ergebnisse und Diskussion 105
das HBSpH7,4/Plasma-1%-Gemisch schon eine deutlich stärkere Entstehung von Reaktionswärme zu detektieren war (Abbildung 19). Es ist anzunehmen, dass die im Plasma enthaltenen Komponenten wie Plasmaproteine, Lipide, Cholesterin, Immunglobuline und Ionen (Natrium, Kalium, Calcium, Phosphat und Chlorid) neben dem vorhandenen HAP potentielle Bindungsstellen für ligandmodifizierte Liposomen waren. Da sich bei den Kontrollliposomen (ohne CHOL-TOE-BP-Liganden) kaum eine Wärmeentwicklung detektieren ließ (Ergebnisse sind nicht dargestellt), scheint der CHOL-TOE-BP-Ligand eine wichtige Rolle bei den Interaktionen mit den Plasmabestandteilen zu spielen. Daley-Yates et al. zeigten, dass Bisphosphonate wie Pamidronat beispielsweise Plasmaproteine in vitro binden (Daley-Yates et al., 1989). Die Vermutung liegt nahe, dass das Bisphosphonat auch hier im Plasma vorhandenes Calcium beispielsweise komplexiert oder aufgrund seiner negativen Ladung elektrostatische Wechselwirkungen mit positiv geladenen Plasmakomponenten eingeht. Mit steigender Plasmakonzentration (5% und 10%) nahm auch die Wärmeentstehung bei den Kontrolltitrationen in das entsprechende HBSpH7,4/Plasma-Gemisch stetig zu (Abbildung 19). Die Signalintensitäten dieser Wärmeentwicklungen waren jedoch stets niedriger als die der entprechenden Titrationen in HAP/Plasma-Gemische. Der Vergleich der HAP/Plasma- mit den HBSpH7,4/Plasma-Titrationen zeigt zudem anhand der fehlenden Plateaubildung der Titrationskurve, dass die möglichen Bindungsstellen auf den Plasmakomponenten bzw. auf den HAP-Partikeln noch nicht vollständig besetzt waren und somit weitere Liposomeninjektionen erforderlich waren, um die Bindungsreaktionen zu beenden.
Die angewendete ITC-Methode erreicht in dieser Art von Bindungsassay dennoch sicherlich ihre Grenzen, da nicht eindeutig zwischen den überlagernden Bindungsreaktionen, wie die Bindung der Liposomen an HAP oder an Plasmabestandteile, unterschieden werden kann und somit Schlussfolgerungen eher spekulativer Natur sind. Der Vergleich der Titration von ligandmodifizierten Liposomen in eine 0,05-mM-HAP-Suspension mit der Kontrolltitration in HBSpH7,4
(Tabelle 11) lässt aufgrund der unterschiedlichen molaren Reaktionsenthalpien (molare ReaktionsenthalpieHAP: -15,4 kcal/mol, molare ReaktionsenthalpieHBSpH7,4: 0,6 kcal/mol) klar erkennen, dass die Liposomen dennoch auch an HAP gebunden haben. Doch beim Vorhandensein von Plasma in der Messzelle lässt der geringe Unterschied in den jeweils ermittelten molaren Reaktionsenthalpien von den Referenztitrationen (HBSpH7,4/Plasma) und den entsprechenden Titrationen in HAP/Plasma-Gemische (Tabelle 11) vermuten, dass die Bindung der Liposomen an die Plasmaproteine in diesem Versuch höchstwahrscheinlich die dominierende
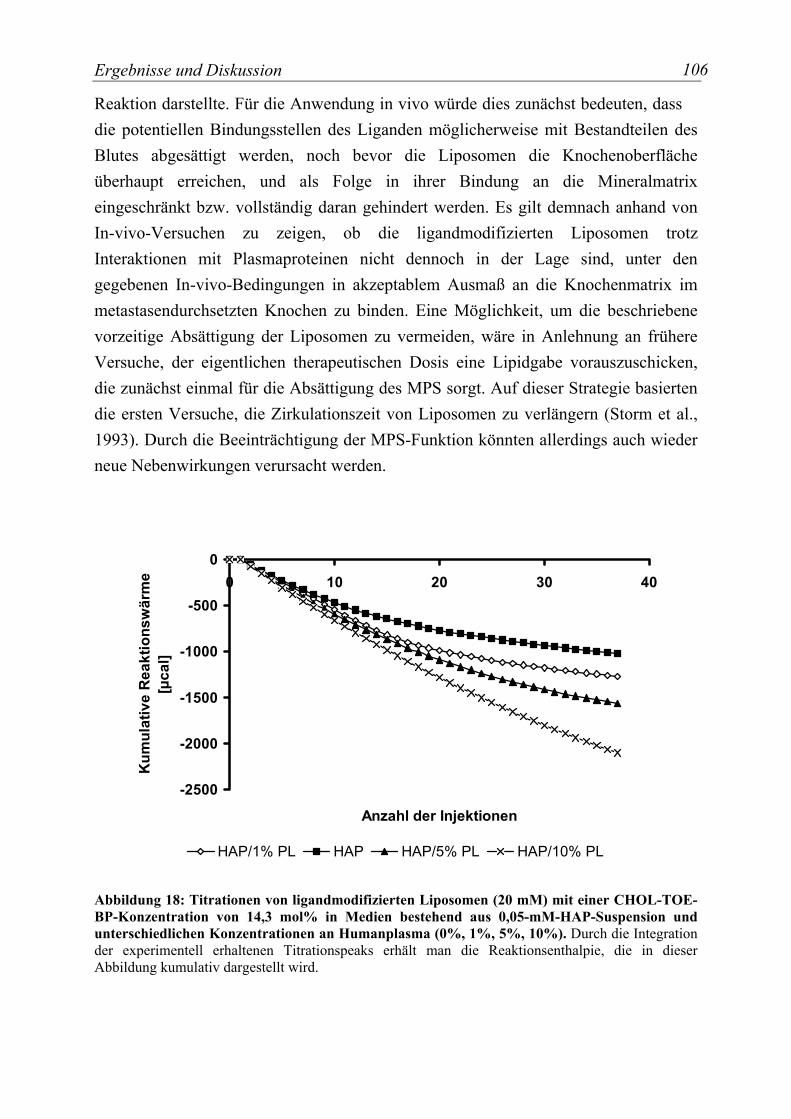
Ergebnisse und Diskussion 106
Reaktion darstellte. Für die Anwendung in vivo würde dies zunächst bedeuten, dass die potentiellen Bindungsstellen des Liganden möglicherweise mit Bestandteilen des Blutes abgesättigt werden, noch bevor die Liposomen die Knochenoberfläche überhaupt erreichen, und als Folge in ihrer Bindung an die Mineralmatrix eingeschränkt bzw. vollständig daran gehindert werden. Es gilt demnach anhand von In-vivo-Versuchen zu zeigen, ob die ligandmodifizierten Liposomen trotz Interaktionen mit Plasmaproteinen nicht dennoch in der Lage sind, unter den gegebenen In-vivo-Bedingungen in akzeptablem Ausmaß an die Knochenmatrix im metastasendurchsetzten Knochen zu binden. Eine Möglichkeit, um die beschriebene vorzeitige Absättigung der Liposomen zu vermeiden, wäre in Anlehnung an frühere Versuche, der eigentlichen therapeutischen Dosis eine Lipidgabe vorauszuschicken, die zunächst einmal für die Absättigung des MPS sorgt. Auf dieser Strategie basierten die ersten Versuche, die Zirkulationszeit von Liposomen zu verlängern (Storm et al., 1993). Durch die Beeinträchtigung der MPS-Funktion könnten allerdings auch wieder neue Nebenwirkungen verursacht werden.
-2500
-2000
-1500
-1000
-500
00 10 20 30 40
Anzahl der Injektionen
Kum
ulat
ive
Rea
ktio
nsw
ärm
e[µ
cal]
HAP/1% PL HAP HAP/5% PL HAP/10% PL
Abbildung 18: Titrationen von ligandmodifizierten Liposomen (20 mM) mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% in Medien bestehend aus 0,05-mM-HAP-Suspension und unterschiedlichen Konzentrationen an Humanplasma (0%, 1%, 5%, 10%). Durch die Integration der experimentell erhaltenen Titrationspeaks erhält man die Reaktionsenthalpie, die in dieser Abbildung kumulativ dargestellt wird.

Ergebnisse und Diskussion 107
-2000
-1500
-1000
-500
0
500
0 10 20 30 40
Anzahl der Injektionen
Kum
ulat
ive
Rea
ktio
nsw
ärm
e [µ
cal]
HBS/10% PL HBS/5% PL HBS/1% PL HBS
Abbildung 19: Kontrolltitrationen von ligandmodifizierten Liposomen (20 mM) mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% in Medien bestehend aus HBSpH7,4 und unterschiedlichen Konzentrationen an Humanplasma (0%, 1%, 5%, 10%). Durch die Integration der experimentell erhaltenen Titrationspeaks erhält man die Reaktionsenthalpie, die in dieser Abbildung kumulativ dargestellt wird.
Zusammensetzung von Medium in Messzelle H∆ [kcal/mol]
0,05 mM HAP -15,4
0,05 mM HAP + 1% Plasma -19,0
0,05 mM HAP + 5% Plasma -23,5
0,05 mM HAP + 10% Plasma -31,8
HBSpH7,4 0,6
HBSpH7,4 + 1% Plasma -9,1
HBSpH7,4 + 5% Plasma -22,1
HBSpH7,4 + 10% Plasma -27,7
Tabelle 11: Übersicht über molare Reaktionsenthalpien bestimmt durch Titrationen von ligandmodifizierten Liposomen (20 mM) mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% in Medien verschiedener Zusammensetzungen (0,05 mM HAP + 0–10% Plasma bzw. HBSpH7,4 + 0–10% Plasma) (Versuchsbedingungen: 25 °C, HBSpH7,4 50 mmol, 0,7% NaCl, pH 7,4). Molare Reaktionsenthalpien wurden gemäß Methodenbeschreibung 3.3.4.2 errechnet.

Ergebnisse und Diskussion 108
4.4 Wirkstoffbeladung von ligandmodifizierten Liposomen
Ligandmodifizierte Liposomen wurden mit Doxorubizin durch einen transmembranären Protonengradienten beladen. In der Vergangenheit wurde die erfolgreiche Verkapselung von Doxorubizin in Liposomen verschiedener Lipidzusammensetzungen bereits mehrfach demonstriert (Haran et al., 1993, Cheung et al., 1998, Li et al., 1998), so dass die Entscheidung fiel, die Versuche ebenfalls mit Doxorubizin als Modellsubstanz durchzuführen. Im Hinblick auf die Freisetzungsversuche stellte Doxorubizin aus unserer Sicht auch eine gute Substanzwahl dar, da es wegen seiner guten Wasserlöslichkeit die Wirkstofffreisetzung aus den Liposomen nicht von sich aus verzögerte und somit die Ergebnisse nicht verfälschte.
Zur Beladung von Liposomen wurden bereits zahlreiche erfolgreiche „Remote Loading“-Strategien entwickelt. Diese Beladungstechniken basieren alle auf dem Prinzip, dass freies Doxorubizin aufgrund eines Gradienten in das Innere der Liposomen diffundiert, wo dann eine Modifikation der Substanz stattfindet, die die Rückkehr des Doxorubizins nach außen verhindert und somit zur Akkumulation des Wirkstoffes im Liposomeninneren führt. Es wird dabei zwischen folgenden Gradienten unterschieden: pH-, Manganese-, Citrat-, Sulfat- und Phosphatgradienten (Fritze et al., 2006). In dieser Arbeit wurde der Ammoniumsulfatgradient angewendet, in Anlehnung an Versuche, die die Beladung von Liposomen mit Doxorubizin über diesen Gradienten bereits erfolgreich aufgezeigt haben (Haran et al., 1993, Emanuel et al., 1996, Li et al., 1998). Doxil® beispielsweise, eine liposomale Doxorubizin-Formulierung, die sich bereits zur Anwendung am Menschen auf dem Markt befindet, wird ebenfalls durch einen Ammoniumsulfatgradienten beladen. Es wurde die Hypothese aufgestellt, dass der gewählte Gradient ebenfalls eine vielversprechende Option für die Beladung von Doxorubizin in ligandmodifizierte Liposomen darstellt. Der Vorteil eines transmembranären Ammoniumsulfatgradienten gegenüber den anderen Gradienten ist, dass dieser von zwei Mechanismen gesteuert wird, und zwar zum einem von der Doxorubizin-Protonierung und zum anderen von der Doxorubizin-Präzipitation im Liposomeninneren. Die Funktionsweise des Gradienten basiert darauf, dass das Ammoniumion in Wasser zu Ammoniak (NH3) und einem Proton (H+) dissoziiert. Das ungeladene NH3-Molekül ist leicht membrangängig, wohingegen das geladene Proton die Lipidmembran nicht durchqueren kann. Es stellt sich deshalb

Ergebnisse und Diskussion 109
durch die Diffusion des Ammoniaks nach außen schnell ein Ungleichgewicht ein, dass dazu führt, dass im Liposomeninneren ein Protonenüberschuss vorliegt, der den pH-Wert folglich absinken lässt. Die dadurch entstandene saure Umgebung im Inneren der Liposomen führt somit zu einem pH-Gradienten, der die unprotonierte schwache Base membrangängig macht. Das neutrale Molekül wird nach dem Passieren der Membran im Inneren protoniert und akkumuliert dort durch Präzipitation (Mayer et al., 1990). Die geladene Form von Doxorubizin ist nicht in der Lage, durch die Membran zu penetrieren. Der Aufbau des pH-Gradienten beruht auf der Henderson-Hasselbalch-Gleichung. Diese Gleichung nimmt an, dass sowohl innerhalb als auch außerhalb der Membran der gleiche pKs-Wert gegeben ist. Deshalb stellt sich ein Equilibrium der ungeladenen Form innen und außen ein, während sich die geladene Form gemäß dem pH-Gradienten verteilt (Mayer et al., 1986).
Als limitierend bei der Beladung wurde wiederum der Teil der CHOL-TOE-BP-Liganden angesehen, die in das Liposomeninnere ragten. Es wurde angenommen, dass die CHOL-TOE-BP-Liganden möglicherweise aufgrund von voluminösen Gruppen die Beladungskapazität einschränkten. Um einen möglichen Einfluss der CHOL-TOE-BP-Liganden auf diesen zu untersuchenden Parameter feststellen zu können, wurden die in diesem Abschnitt beschriebenen Versuche mit einer hohen Ligandkonzentration (14,3 mol% CHOL-TOE-BP) durchgeführt. Für ein erfolgreich einsetzbares liposomales System ist zum einen eine akzeptable Stabilität im Hinblick auf den verkapselten Wirkstoff wichtig, und zum anderen muss das Trägersystems auch gewährleisten, dass der Wirkstoff am Wirkort ausreichend freigesetzt wird. Aus diesem Grund wurden diese Faktoren in den folgenden Abschnitten näher untersucht. Die Stabilität der beladenen, ligandmodifizierten Liposomen wurde über einen Zeitraum von einer Woche in Bezug auf ihre Partikelgröße und ihren Doxorubizin-Gehalt untersucht. Für die Freisetzungsversuche wurde ebenfalls Doxorubizin als Modellsubstanz eingesetzt. Die Freisetzungsversuche versuchten die Bedingungen, die die Liposomen in vivo auf ihrem Weg zu Wirkort erfahren würden, nachzustellen, d.h., es wurde der Einfluss von verschiedenen pH-Werten auf die Freisetzung des Doxorubizins aus den Liposomen untersucht (pH-Wert: 5,0; pH-Wert: 6,5; pH-Wert: 7,4). Der physiologische pH-Wert des Blutes beträgt 7,4; im Extrazellularraum eines Tumorgewebes sinkt der pH-Wert wegen der erhöhten Zellteilungsrate auf 6,5 ab. Bei Eintritt in das metastasierte Knochengewebe würden die ligandmodifizierten Liposomen möglicherweise mit den pH-Wert-Bedingungen dieses Extrazellularraumes konfrontiert werden. Während der Osteoklastentätigkeit sinkt der pH-Wert in der Umgebung von Resorptionslakunen auf ungefähr 5 ab. Der pH-Wert-Abfall wird

Ergebnisse und Diskussion 110
durch die Aktivierung von Protonenpumpen, die sich auf der Unterseite von Osteoklasten befinden, bewirkt, so dass das darunterliegende Hydroxylapatit im Zuge des Resorptionsvorganges aufgelöst werden kann (Teti et al., 1989, Bozec et al., 2005). Vorausgesetzt die ligandmodifizierten Liposomen sind in der Lage, die Knochenoberfläche zu erreichen, dann ist der pH-Wert, der durch den osteoklastischen Knochenabbau verursacht wird, für die Wirkstofffreisetzung aus diesen Liposomen von Relevanz. Die Literaturrecherche hat ergeben, dass eine pH-Wert-abhängige Freisetzung von Doxorubizin aus gewöhnlichen EPC-Liposomen, die keine speziellen, pH-sensitiven Lipide aufwiesen, möglich ist, so dass die Hypothese aufgestellt wird, dass diese Beobachtung auch auf die in dieser Arbeit vorgestellten Liposomen übertragbar ist (Fritze et al., 2006).
4.4.1 Beladungskapazität
Die ligandmodifizierten Liposomen wurden durch das oben beschriebene effiziente und bereits gut untersuchte „Remote Loading“-Verfahren mit Doxorubizin beladen. Dazu wurden zunächst Leerliposomen wie unter 3.3.2 beschrieben über Extrusion hergestellt. Dabei wurde der Lipidfilm in einer 230-mM-Ammoniumsulfatlösung hydratisiert. Nachdem überschüssiges Ammoniumsulfat vom externen Medium durch Dialyse entfernt worden war, wurde Doxorubizin als amphiphatische schwache Base zu den Liposomen gegeben und für 2 h bei 60 °C vor Licht geschützt inkubiert. Auf diese Art und Weise wurde ein NH4
+-Gradient geschaffen, welcher die schnelle Diffusion von neutralen Ammoniakmolekülen (NH3) durch die Membran hindurch ins äußere Medium ermöglichen sollte. Um die beladenen Liposomen näher zu charakterisieren, wurde zunächst die Verkapselungseffizienz spektrophotometrisch bestimmt. Das molare Verhältnis von Doxorubizin zu Gesamtlipid vor der Beladung betrug 1:3. Nicht verkapselter Wirkstoff wurde durch Säulenchromatographie direkt nach der Beladung entfernt. Der transmembranäre Ammoniumsulfatgradient erwies sich auch hier als schnelle und effiziente Methode. Es wurde eine Einschlusseffizienz des Wirkstoffes in die Liposomen von 99% bestimmt. Der Lipidgehalt wurde mittels der Rouser-Methode ermittelt und betrug nach der Säulenchromatographie (siehe Abschnitt 3.3.5.1) 6,8 µmol/ml (Tabelle 12). Die hohe Verkapselungsrate zeigt, dass der CHOL-TOE-BP-Ligand in den Liposomen bei der Beladung der Liposomen offensichtlich nicht störend interagierte.

Ergebnisse und Diskussion 111
Liposomenzusammensetzung Doxorubizin-Gehalt [µmol/ml]
Gesamtlipidgehalt [µmol/ml]
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP:
1,85/1,0/0,15/0,5 1,5 (± 0,05) 6,8 (± 0,5)
Tabelle 12: Ermittlung der Beladungskapazität von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP) nach erfolgter Trennung von nicht verkapselter Wirksubstanz (Säulenchromatographie). Die hier dargestellten Werte repräsentieren jeweils einen Mittelwert aus drei Einzelmessungen ± SD.
4.4.2 Physikochemische Charakterisierung
Um festzustellen, ob die Wirkstoffverkapselung von Doxorubizin einen Einfluss auf die durchschnittliche Partikelgröße der ligandmodifizierten Liposomen hatte, wurde sowohl vor als auch nach der Beladung die Partikelgröße mittels PCS untersucht. Die in Tabelle 13 dargestellten Ergebnisse zeigen, dass die Partikelgröße bei fast gleichbleibendem Polydispersionsindex aufgrund der Beladung ein wenig zunahm.
Liposomenzusammensetzung
Mittlere Partikelgröße
vor der Beladung [nm]
PI
Mittlere Partikelgröße
nach der Beladung [nm]
PI
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP:
1,85/1,0/0,15/0,5 137 (± 2) 0,04 (± 0,01) 159 (± 1) 0,06 (± 0,03)
Tabelle 13: Physikochemische Eigenschaften von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP), die eine Doxorubizin-Einschlussrate von 99% aufweisen. Die hier dargestellten Werte repräsentieren jeweils einen Mittelwert aus drei Einzelmessungen ± SD.

Ergebnisse und Diskussion 112
4.4.3 Stabilität
Die Stabilität der oben beschriebenen Liposomendispersion, beladen mit Doxorubizin, wurde über einen Zeitraum von 7 Tagen im Hinblick auf ihre Verkapselungseffizienz und ihre Liposomengröße untersucht. Über den Zeitraum dieser Stabilitätsmessungen wurden die Liposomen bei 2–8 °C und vor Licht geschützt gelagert. Die in Tabelle 14 dargestellten Ergebnisse zeigen, dass die Liposomen über den Zeitraum von 7 Tagen kaum Wirkstoff (5%) aus ihrem Inneren freisetzten. Zudem zeigt diese Tabelle, dass die durchschnittliche Partikelgröße über den beschriebenen Zeitraum stabil blieb. Die Frage ist, wie lange die demonstrierte Stabilität anhalten wird. Im Hinblick auf eine spätere Produktentwicklung könnte eine längere Haltbarkeit der wirkstoffbeladenen Liposomen eventuell durch Gefriertrocknung erzielt werden. Bei liposomalen Doxorubizin-Präparaten (Doxil®, Caelyx®), die die Marktreife erlangt haben, hat sich diese Methode bereits bewährt.
Tag Verkapselungseffizienz [%] Mittlere Partikelgröße [nm] PI
0 99 % (± 0,5) 159 (± 1) 0,06 (± 0,03)
1 98 % (± 1,0) 166 (± 2) 0,14 (± 0,03)
3 95 % (± 0,4) 167 (± 2) 0,12 (± 0,05)
5 94 % (± 0,8) 169 (± 1) 0,14 (± 0,02)
7 94 % (± 1,2) 167 (± 2) 0,12 (± 0,04)
Tabelle 14: Stabilität von ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% im Hinblick auf die durchschnittliche Partikelgröße und die Verkapselungseffizienz (%) über einen Zeitraum von 1 Woche (die hier dargestellten Werte repräsentieren jeweils einen Mittelwert aus drei Einzelmessungen ± SD) (Lagerungsbedingungen: 2–8 °C, vor Licht geschützt).
4.4.4 Doxorubizin-Freisetzung
Zur Durchführung des Freisetzungsversuchs wurden die Liposomen 300-fach in Puffermedien verschiedener pH-Werte zunächst verdünnt. Über einen Zeitraum von 2 Stunden wurde direkt im Anschluss die Fluoreszenzintensität des Wirkstoffes

Ergebnisse und Diskussion 113
Doxorubizin vermessen. Neben dem pH-Wert-Einfluss wurde die Freisetzung durch regelmäßige Schütteleinheiten forciert. Die über einen Protonengradienten beladenen, ligandmodifizierten Liposomen zeigten bei einem extraliposomalen pH-Wert von 7,4 eine Wirkstofffreisetzung von bis zu 4%, wohingegen die Freisetzung bei niedrigerem pH-Wert deutlich zunahm. Bei einem pH-Wert von 6,5 wurden bis zu 13% und bei einem pH-Wert von 5,0 bis zu 15% Doxorubizin (Abbildung 20) freigesetzt. Es stellte sich die Frage, welcher Mechanismus zu der Freisetzung geführt hat. Die pH-sensitiven Liposomen sind beispielsweise so konzipiert, dass sie bei einem pH-Wert-Abfall, d.h. nach erfolgter Endozytose in die Zelle, instabil werden, teilweise mit der endosomalen Membran fusionieren und den Wirkstoff dabei in das Zytosol freigeben (Collins et al., 1990, Hong et al., 2002, Karanth and Murthy, 2007). Zumeist bestehen pH-sensitive Liposomen aus Lipiden, die bei neutralem pH-Wert von kegelförmiger Gestalt sind (diese Kegel sind durch kleine polare Kopfgruppen und größere hydrophobe Schwanzteile charakterisiert) und eine lamellare Struktur annehmen. Bei niedrigeren pH-Werten gehen sie in eine hexagonale Phase über, welche mehr Raum für den lipophilen Part des Lipides schafft. Zu diesen Lipiden zählt beispielsweise das Dioleoylphosphatidylethanolamin (Papahadjopoulos, 1968, Cullis and de Kruijff, 1979, de Oliveira et al., 2000, Hong et al., 2002). Da die hier eingesetzten Lipide nicht zu den typischen pH-sensitiven Lipiden gehören, muss der demonstrierten Freisetzung ein anderer Mechanismus zugrundeliegen. Möglicherweise wurden die Lipide in der sauren Umgebung hydrolysiert.
Ein anderer Erklärungsansatz könnte sein, dass das freie, extraliposomale Doxorubizin nun protoniert wurde und es folglich zu einem Ungleichgewicht zwischen der nicht geladenen Form des Doxorubizins innerhalb und außerhalb der Membran kam. Um die Balance des ungeladenen Doxorubizins intraliposomal und extraliposomal wiederherzustellen, müsste die ungeladene Form das Liposomeninnere verlassen. Auf diese Weise könnte es dann als Wirkstoff aktiv werden. Im Vergleich zu der Freisetzung in vitro sind die Mechanismen für die Ruptur bzw. Destabilisierung der Membran von Liposomen in vivo im Allgemeinen ebenfalls bisher nicht vollständig bekannt. Im Fall von pegylierten Doxorubizin-Liposomen (Doxil®) fasst Martin (Martin, 1998) zusammen, dass mehrere Faktoren für die Wirkstofffreisetzung verantwortlich sein können. Dazu zählen beispielsweise die Umgebung des Tumors, Lipasen, die von absterbenden Tumorzellen abgegeben werden, oder andere Enzyme und oxidierende Agenzien. Zudem können phagozytierende Zellen, die sich neben den Tumorzellen aufhalten, die Liposomen metabolisieren und somit das Doxorubizin freigeben (Storm et al., 1988).

Ergebnisse und Diskussion 114
Die Fähigkeit, den verkapselten Wirkstoff aufgrund von pH-Wertänderungen freizugeben, kann sich auf die Effektivität der Liposomen in Bezug auf spätere Anwendungen positiv auswirken, da der pH-Wert im Zielgewebe, dem metastasierten Knochengewebe, im Vergleich zum physiologischen pH deutlich niedriger ist. Im Gegensatz zu gesundem Gewebe sind Tumore durch höhere Teilungsraten gekennzeichnet, verbrauchen somit auch mehr Energie und besitzen deshalb zwangsläufig einen erhöhten Stoffwechsel. Die Azidifizierung des extrazellulären Tumorgewebes kommt dadurch zustande, dass die Tumorzellen versuchen, den erhöhten Energiebedarf zu decken, indem sie vermehrt Glucose zu Milchsäure umsetzen, welche dann über einen H+-Monocarboxylat-Cotransport aus der Zelle transportiert wird (Stubbs et al., 2000). Eine weitere azide Umgebung, mit der die Liposomen konfrontiert werden könnten, ist das Knochenmineral in Resorptionslakunen von Osteoklasten, die diese mineralische Substanz aktiv abbauen. Während des Prozesses der Knochenresorption azidifizieren Osteoklasten mit Hilfe ihres Bürstensaums die unter ihnen liegende Knochenmatrix, um den ebenfalls von ihnen freigesetzten lysosomalen Enzymen eine optimale Umgebung zu schaffen. In diesem Versuch wurde nun bei pH 5,0 eine Freisetzungsrate von 15% nach 2 h detektiert; es bleibt offen, ob der restliche Wirkstoff mit der Zeit ebenfalls noch freigesetzt werden würde. Da bei der Freisetzung in vivo definitv neben dem pH-Wert noch andere Faktoren eine wichtige Rolle spielen (siehe oben), ist davon auszugehen, dass unter In-vivo-Bedingungen mit der Zeit wesentlich mehr Wirkstoff bzw. die gesamte Menge an verkapselter Substanz freigesetzt werden würde. Die Tatsache, dass nicht gleich der gesamte Inhalt der Liposomen freigesetzt wurde, wäre somit für die zukünftige Anwendung insofern von Vorteil, als eine verzögerte Freisetzung und somit eine langanhaltende Wirkung gewährleistet werden würde.
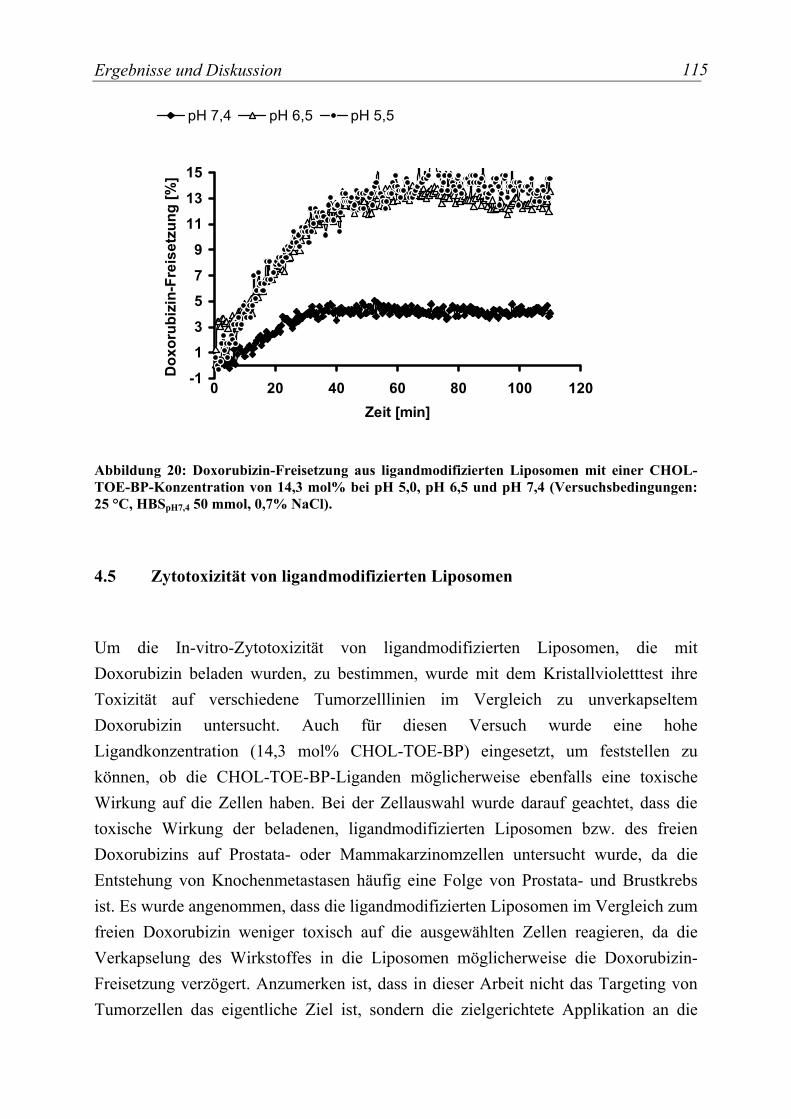
Ergebnisse und Diskussion 115
-1
1
3
5
7
9
11
13
15
0 20 40 60 80 100 120Zeit [min]
Dox
orub
izin
-Fre
iset
zung
[%]
pH 7,4 pH 6,5 pH 5,5
Abbildung 20: Doxorubizin-Freisetzung aus ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% bei pH 5,0, pH 6,5 und pH 7,4 (Versuchsbedingungen: 25 °C, HBSpH7,4 50 mmol, 0,7% NaCl).
4.5 Zytotoxizität von ligandmodifizierten Liposomen
Um die In-vitro-Zytotoxizität von ligandmodifizierten Liposomen, die mit Doxorubizin beladen wurden, zu bestimmen, wurde mit dem Kristallvioletttest ihre Toxizität auf verschiedene Tumorzelllinien im Vergleich zu unverkapseltem Doxorubizin untersucht. Auch für diesen Versuch wurde eine hohe Ligandkonzentration (14,3 mol% CHOL-TOE-BP) eingesetzt, um feststellen zu können, ob die CHOL-TOE-BP-Liganden möglicherweise ebenfalls eine toxische Wirkung auf die Zellen haben. Bei der Zellauswahl wurde darauf geachtet, dass die toxische Wirkung der beladenen, ligandmodifizierten Liposomen bzw. des freien Doxorubizins auf Prostata- oder Mammakarzinomzellen untersucht wurde, da die Entstehung von Knochenmetastasen häufig eine Folge von Prostata- und Brustkrebs ist. Es wurde angenommen, dass die ligandmodifizierten Liposomen im Vergleich zum freien Doxorubizin weniger toxisch auf die ausgewählten Zellen reagieren, da die Verkapselung des Wirkstoffes in die Liposomen möglicherweise die Doxorubizin-Freisetzung verzögert. Anzumerken ist, dass in dieser Arbeit nicht das Targeting von Tumorzellen das eigentliche Ziel ist, sondern die zielgerichtete Applikation an die

Ergebnisse und Diskussion 116
Knochenoberfläche. Von dort aus sollen diese Liposomen idealerweise das Wachstum der benachbarten Metastasen hemmen. Um das neue liposomale System unter diesem Aspekt besser charakterisieren zu können, wurde hier auch der direkte Einfluss von ligandmodifizierten Liposomen auf verschiedene Tumorzellen untersucht.
4.5.1 Bestimmung der Zytotoxizität von ligandmodifizierten, Doxorubizin-beladenen Liposomen versus freiem Doxorubizin
Zur Bestimmung der Zytotoxizität von ligandmodifizierten, Doxorubizin-beladenen Liposomen wurde der Kristallvioletttest, wie unter 3.3.6.3 beschrieben, durchgeführt. Dazu wurden 2 Millionen Zellen pro 96-Well-Platte ausgesät und mit den zu testenden Formulierungen in Form einer Verdünnungsreihe, ausgehend von einer Doxorubizin-Konzentration von 10 µg/ml, versetzt und inkubiert. Nach 48 Stunden wurden die Platten mit Leitungswasser gewaschen, die verbliebenen Zellpellets mit Kristallviolett angefärbt und fixiert und bei 595 nm vermessen. Die IC50-Werte für die Liposomen und das freie Doxorubizin wurden graphisch ermittelt (Abschnitt 3.3.6.3). Die Versuche zeigten, dass das Ausmaß der zytotoxischen Wirkung der freien Wirksubstanz und der ligandmodifizierten Liposomen, die mit Doxorubizin beladen waren, sehr stark von dem getesteten Zelltyp abhing. Im Fall von DU-145-Zellen beispielsweise wurde ein IC50-Wert von 0,07 µg/ml für ligandmodifizierte Liposomen ermittelt, wohingegen bei allen anderen getesteten Zellen ein Acht- bis Elffaches für den IC50-Wert bestimmt wurde (Tabelle 15). Außerdem wirkte sich das freie Doxorubizin toxischer auf drei der vier getesteten Zelltypen aus, und zwar maximal um einen Faktor 4. Im Falle der BT-474-Zellen erwiesen sich die beladenen, ligandmodifizierten Liposomen als potenterer Kandidat. Der IC50-Wert war hier um ein Dreifaches höher als der der freien Wirksubstanz.

Ergebnisse und Diskussion 117
Zelltyp IC50-Werte von ligandmodifizierten Liposomen [µg/ml]
IC50 von freiem Doxorubizin [ug/ml]
MCF-7 0,77 0,19
MDA-MB-435s 0,58 0,45
BT-474 0,56 1,83
DU-145 0,07 0,02
Tabelle 15: IC50-Werte für ligandmodifizierte Liposomen, die mit Doxorubizin beladen wurden, für Kontrollliposomen (kein Doxorubizin enthaltend) und für freies Doxorubizin. Die IC50-Werte wurden durch den Kristallvioletttest anhand von folgenden Zellen ermittelt: MCF-7-Zellen (Mammakarzinom), MDA-MB-435s-Zellen (Mammakarzinom), BT-474-Zellen (Mammakarzinom) und DU-145-Zellen (Prostatakarzinom). Die ligandmodifizierten Liposomen wiesen eine CHOL-TOE-BP-Konzentration von 14,3 mol% auf.
Das freie Doxorubizin wirkte sich prinzipiell zytotoxischer auf die ausgewählten Zellen aus, da der freie Wirkstoff im Vergleich zum liposomal verkapselten Doxorubizin von den Zellen, die beiden Formulierungen über den gleichen Zeitraum ausgesetzt waren, vermutlich schneller aufgenommen wurde. Die Liposomen könnten mit den Zellen auf verschiedene Weisen in Verbindung getreten sein. Eine Möglichkeit ist, dass die Liposomen an die Zellen adsorbiert und ihren Inhalt nach und nach extrazellulär freigegeben haben, der dann von der Zelle aufgenommen wurde und deshalb zu einem Wirkungsverzug führte.
Ein anderer denkbarer Weg wäre, dass die Liposomen endozytotisch internalisiert wurden und den Wirkstoff innerhalb der Zelle freigesetzt haben. (Pagano and Weinstein, 1978, Scherphof, 1991, Higashi and Sunamoto, 1995, Xiong et al., 2005, Yang et al., 2007). Bei dieser Variante würde die Verkapselung der Wirksubstanz in das Liposomeninnere dann offensichtlich zu einer verzögerten Freisetzung innerhalb der Zelle führen, so dass während der Inkubationsdauer von 48 Stunden nicht das gesamte Doxorubizin freigesetzt und folglich eine geringere Toxizität beobachtet wurde.
Die In-vivo-Situation würde jedoch höchstwahrscheinlich ganz anders aussehen, da freies Doxorubizin viel schneller als liposomal verkapseltes Doxorubizin aus der Blutbahn eliminiert werden würde und somit im Vergleich zu verkapseltem

Ergebnisse und Diskussion 118
Doxorubizin nicht in konzentrierten Mengen am Wirkort vorläge. Gabizon et al. zeigten, dass freies Doxorubizin 450-mal schneller eliminiert wurde als in pegylierte Liposomen verkapseltes Doxorubizin (Gabizon et al., 1994). Andere In-vivo-Versuche haben gezeigt, dass Liposomen zumeist nicht direkt von den Tumorzellen aufgenommen werden, sondern ihren Wirkstoff extrazellulär freigeben. Die Wirksubstanz wird somit aus dem interstitiellen Fluid des Tumors als freie Substanz in die Zellen aufgenommen (Abbildung 21) (Sapra and Allen, 2003). Anders ist es bei Liposomen, die so konzipiert sind, dass sie für die zielgerichtete Applikation des liposomalen Systems an die Tumorzelle selbst vorgesehen sind. Diese Liposomen zeichnen sich durch das Vorhandensein von Liganden aus, die wiederum eine hohe Affinität zu internalisierenden Rezeptoren besitzen, die auf dem Tumor sitzen. Es gibt zahlreiche Beispiele, die auf dieser Strategie basieren und zu einer gesteigerten Bioverfügbarkeit und vermehrten Ansammlung des Wirkstoffes innerhalb des Tumors geführt haben (Lundberg et al., 1993, Lee and Low, 1995, Kirpotin et al., 1997).
Bei den BT-474-Zellen erwiesen sich die ligandmodifizierten Liposomen als zelltoxischer. Möglicherweise waren diese Zellen in gewissem Maße resistent gegen die direkte Aufnahme von Doxorubizin. Basierend auf dieser Hypothese wurden im Fall der liposomal verkapselten Substanz zunächst die intakten Liposomen in die Zelle aufgenommen, wo sie dann das Doxorubizin freisetzten und es seine Wirkung entfalten konnte. Die beobachteten Unterschiede bei den IC50-Werten der beiden Formulierungen (freies Doxorubizin und Doxorubizin-beladene, ligandmodifizierte Liposomen) für die verschiedenen Zellen mögen auf die unterschiedliche Sensitivität der einzelnen Zellen zurückzuführen sein. Versuche in der Vergangenheit haben durchaus gezeigt, dass sich Zellen in ihrer Resistenz deutlich unterscheiden können (Fromigue et al., 2000).
Die Kontrollen, bestehend aus unbeladenden Liposomen, zum einen ohne Liganden und zum anderen mit Liganden, zeigten, dass weder die Lipide noch die CHOL-TOE-BP-Liganden eine zytotoxische Wirkung auf die getesteten Zellen ausübten (Ergebnisse sind nicht dargestellt). Gemäß der Literatur haben stickstoffhaltige Bisphosphonate in vitro eine antitumorale Aktivität gegen eine Reihe von Tumorzelllinien bewiesen, und zwar in Konzentrationen von 0,02–100 µmol/l (Clezardin, 2002). Die Inhibierung der Proteinprenylierung in Tumorzellen (kein Vorgang, der die Chelatkomplexbildung voraussetzt) durch stickstoffkonjugierte Bisphosphonate wird als Ursache für die antitumorale Wirkung angesehen (Luckman et al., 1998b, Rogers et al., 1999, Gober et al., 2003). Da der CHOL-TOE-BP-Ligand

Ergebnisse und Diskussion 119
kein Stickstoffmolekül trägt, ist eine antitumorale Wirkung, die vom Liganden ausgeht, unwahrscheinlich. Bisphosphonate der ersten Generation, d.h. Bisphosphonate, die an R2 (Abbildung 4) nicht durch stickstoffhaltige Gruppen substituiert wurden (Etidronat, Clodronat), werden zu nicht hydrolysierbaren, zytotoxischen Adenosin-triphosphat-Analoga metabolisiert. Sie haben in der Regel einen zytotoxischen Einfluss auf Osteoklasten und können die Apoptose von Osteoklasten herbeiführen (Frith and Rogers, 2003). Clezardin zeigte (Clezardin, 2002), dass Clodronat die Reduktion der Überlebensrate von MDA-MB-231-Zellen bewirkte, allerdings nur in relativ hohen Konzentrationen ( lµmolIC /70050 = ).
Ähnliche Ergebnisse erzielte Clezardin ebenfalls mit anderen Mammakarzinom- und Prostatakarzinomzelllinien. Den genauen Wirkmechanismus gilt es hierbei noch zu klären. Die in dieser Arbeit eingesetzte CHOL-TOE-BP-Konzentration betrug 60 µmol/l und lag somit weit unterhalb der zuvor genannten effektiven Konzentration (für Bisphosphonate, die keinen Stickstoff enthalten). Eine zytotoxische Wirkung, die von dem Ligandmolekül ausging, war deshalb nicht zu erwarten.
Um die antitumorale Wirkung von ligandmodifizierten Liposomen zu erhöhen, könnte beispielsweise der Einfluss einer kombinierten Gabe untersucht werden, d.h., die beladenden, ligandmodifizierten Liposomen werden gleichzeitig mit anderen Chemotherapeutika eingesetzt. Für eine spätere Anwendung ist dennoch nicht nur die zytotoxische Wirkung auf die Tumorzellen allein wichtig. Die von dem Präparat ausgehende Toxizität auf das umliegende gesunde Gewebe spielt für die Wirksamkeit ebenso eine entscheidende Rolle.

Ergebnisse und Diskussion 120
Abbildung 21: Transport von liposomal verkapseltem Wirkstoff (L-Drug) in die Tumorzelle. Dieses schematische Diagramm verdeutlicht den möglichen Drug-Delivery-Mechanismus auf zellulärer Ebene: 1. extrazelluläre Freisetzung des Wirkstoffes aus den Liposomen; 2. Diffusion des Wirkstoffes durch das Tumorgewebe; 3. Aufnahme der Substanz in die Tumorzelle; 4. Ein Teil des Wirkstoffes gelangt möglicherweise wieder in den Blutkreislauf.
4.6 Pharmakokinetik und Biodistribution von ligandmodifizierten Liposomen
Zunächst wurde in vivo der Einfluss unterschiedlicher CHOL-TOE-BP-Konzentrationen (1,6 mol% und 14,3 mol% CHOL-TOE-BP) auf die Pharmakokinetik und die Biodistribution von ligandmodifizierten Liposomen, die 4,3 mol% DSPE-PEG2000 enthielten, an Ratten untersucht. Das DSPE-PEG2000 wurde neben den Ligandmolekülen ebenfalls auf der Liposomenoberfläche angebracht, mit der Intention, die Blutverweildauer der Liposomen zu verlängern. Aufgrund der Hydrophilie, die von diesem Polymer ausgeht, können unerwünschte hydrophobe und elektrostatische Wechselwirkungen zwischen Liposomen und zahlreichen Blutbestandteilen sowie die Opsonisierung und die anschließende Aufnahme in Makrophagen möglicherweise reduziert werden (Lasic et al., 1991, Romberg et al., 2007). Es war jedoch zu erwarten, dass die ligandmodifizierten Liposomen trotz des Vorhandenseins von DSPE-PEG2000 in Abhängigkeit von der inkorporierten CHOL-TOE-BP-Konzentration aufgrund der negativen Ladung, die von dem CHOL-TOE-BP-Liganden ausging, aus der Blutbahn eliminiert werden würden. Die HAP-Bindungsversuche, bei denen der Einfluss von Blutplasma auf die Bindung von ligandmodifizierten Liposomen an HAP untersucht wurde, haben in diesem

Ergebnisse und Diskussion 121
Zusammenhang klar gezeigt, dass diese mögliche Interaktion nicht zu vernachlässigen ist.
Das Ziel einer weiteren Untersuchung war, zu testen, ob der vermehrte Einbau von DSPE-PEG2000 in die Liposomenmembran von ligandmodifizierten Liposomen die Verweildauer im Blut verlängerte. Laut Literatur verbessert eine Konzentrationserhöhung bis zu 10 mol% das pharmakokinetische Profil von Liposomen, indem der Polymereinbau in die Membran die Liposomen durch die Bildung einer protektiven Hülle vor Opsonisierung schützt (Arnold et al., 1990). Ein Anteil von mehr als 20 mol% DSPE-PEG2000 führt allerdings zur Destabilisation der Bilayer-Membran, und bei sehr hohen Konzentrationen kann es zur Entstehung von Mischmizellen kommen (Woodle et al., 1992). Basierend auf diesen Daten hofften wir verbesserte Zirkulationszeiten durch den gesteigerten DSPE-PEG2000-Einbau in die liposomale Membran erzielen zu können. Zudem wurde der Einfluss einer Molekulargewichtserhöhung untersucht. Dabei wurde DSPE-PEG2000 durch DSPE-PEG5000 ausgetauscht. Woodle et al. (Woodle et al., 1992, Sandstrom et al., 2007) zeigten zwar anhand von Liposomen, dass kaum ein Unterschied zwischen den beiden Polymeren in Bezug auf die Verweildauer im Blut zu detektieren war, doch wir wollten dieses Ergebnis auch mit unserem System bestätigt wissen.
4.6.1 Einfluss der Ligandkonzentration auf die Zirkulationszeit
Um diese In-vivo-Eigenschaften von ligandmodifizierten Liposomen zu bestimmen, wurde eine Einzeldosis von 20 µmol/kg KG intravenös an Ratten appliziert. Als Kontrolle wurden Liposomen eingesetzt, die keine CHOL-TOE-BP-Liganden enthielten. Durch die Markierung der Liposomen mit [3H]-markiertem Cholesteryl-oleylether konnten die Liposomen in den gezogenen Blutproben quantifiziert werden und somit das Ausmaß der Clearance aus dem Blut zu dem gegebenen Zeitpunkt bestimmt werden. Die Versuche zur Bestimmung der Pharmakokinetik von ligandmodifizierten Liposomen haben den limitierenden Einfluss des Liganden auf die Verweildauer im Blut klar dargestellt. Die Abbildung 22 zeigt dazu deutlich, dass die Liposomen weniger lang in der Blutbahn verweilten, wenn eine hohe CHOL-TOE-BP-Konzentration (14,3 mol%) in die Liposomenmembran inkorporiert wurde, als wenn eine geringere CHOL-TOE-BP-Konzentration (1,6 mol%) in den Liposomen vorlag. Die Kontrollliposomen, die keine CHOL-TOE-BP-Liganden aufwiesen, zirkulierten

Ergebnisse und Diskussion 122
hingegen noch deutlich länger (Abbildung 22). Die errechnete „Area under the Curve“ (AUC0-48h, bestimmt für einen Zeitraum von 48 Stunden) bestätigte diese Beobachtung (Tabelle 16). Es ist somit offensichtlich, dass das Vorhandensein von CHOL-TOE-BP die Clearance-Rate von ligandmodifizierten Liposomen aus dem Blukreislauf erhöht hat. Die Vermutung hat sich somit bestätigt, dass eine stark negativ geladene Oberfläche, die durch den Einschluss des CHOL-TOE-BP-Liganden in hoher Konzentration bewirkt wurde, das Ausmaß der Opsonisierung steigerte. Dadurch wurde die Erkennung durch Makrophagen erhöht, was somit zur Elimierung durch Makrophagen führte. Dieses Phänomen wurde in der Literatur bereits mehrfach beschrieben (Allen et al., 1988, Allen et al., 1989, Levchenko et al., 2002) und wurde so erklärt, dass Interaktionen zwischen bestimmten opsonidierenden Proteinen und den geladenen Kopfgruppen auf der liposomalen Oberfläche stattfinden, welche sowohl Phospholipid-Endgruppen als auch geladene Gruppen von Liganden sein können (Liu et al., 1995, Moghimi and Hunter, 2001). Die durchschnittliche Partikelgröße, die bei der Bestimmung des Pharmakokinetikprofils von Liposomen in der Regel von hoher Bedeutung ist, spielt hier erwartungsgemäß eine untergeordnete Rolle, denn Partikel mit einem Durchmesser von mehr als 200 nm führen typischerweise zu einer erhöhten Clearance, unabhängig davon, ob sie pegyliert sind oder nicht (Moghimi et al., 1993). Da sich die durchschnittliche Größe bei den untersuchten Liposomen zwischen 120 und 155 nm bewegte, ist dieser Aspekt eher zu vernachlässigen.

Ergebnisse und Diskussion 123
0
20
40
60
80
100
120
0 4 8 12 16 20 24 28 32 36 40 44 48
Zeit [h]
% In
jizie
rte
Dos
is
0 mol% CHOL-TOE-BP 1,6 mol% CHOL-TOE-BP14,3 mol% CHOL-TOE-BP
Abbildung 22: Pharmakokinetikprofile von ligandmodifizierten Liposomen (4,3 mol% DSPE-PEG2000) mit verschiedenen CHOL-TOE-BP-Konzentrationen (1,6 mol%, 14,3 mol%) und Kontrollliposomen (4,3 mol% DSPE-PEG2000) im Blut nach intravenöser Injektion in die Schwanzvene von Ratten. Die Ergebnisse werden als prozentualer Anteil der anfänglichen injizierten Dosis an Radioaktivität dargestellt, das geschätzte Blutvolumen betrug 20 ml/Ratte. Die abgebildeten Ergebnisse stellen jeweils einen Mittelwert ± SD dar, gebildet aus vier Ratten pro Zeitpunkt.

Ergebnisse und Diskussion 124
Liposomenzusammensetzung CHOL-TOE-BP-Konzentration [mol%]
AUC0-48h [%ID.h]
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 14,3 111 ± 24
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 1,6 225 ± 45
DPPC/CHOL/DSPE-PEG2000 - 314 ± 53
Tabelle 16: Einfluss der molaren CHOL-TOE-BP-Konzentration auf die „Area under the Curve“ von ligandmodifizierten Liposomen, die 4,3 mol% DSPE-PEG2000 enthielten. Eine einmalige Dosis (20 µmol/kg Körpergewicht) an [3H]-markierten Liposomen wurde i.v. in die Schwanzvene der Ratte injiziert. Die AUC0-48h wurde für jedes einzelne Tier basierend auf der oben dargestellten Pharmakokinetik berechnet und wird als Mittelwert ± SD von vier Tieren dargestellt. p < 0,05 für den Vergleich aller hier genannten Gruppen.

Ergebnisse und Diskussion 125
0
10
20
30
40
50
60
70
80
90
0 1,6 14,3
CHOL-TOE-BP-Konzentration [mol%]
% In
jizie
rte
Dos
is LeberMilz
Abbildung 23: Organverteilung von ligandmodifizierten Liposomen und Kontrollliposomen. Die abgebildeten Ergebnisse stellen jeweils einen Mittelwert ± SD dar, gebildet aus vier Ratten pro Zeitpunkt. % injizierte Dosis bezieht sich auf den prozentualen Anteil der anfänglich injizierten Menge an Radioaktivität (geschätztes Blutvolumen: 20 ml/Ratte).
4.6.2 Einfluss der DSPE-PEG2000-Dichte auf die Zirkulationszeit
Der negative Einfluss der CHOL-TOE-BP-Liganden auf die Zirkulationszeit im Blut führte dazu, dass in einer weiteren Pharmakokinetikstudie der Einfluss eines vermehrten DSPE-PEG2000-Einbaus in Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% auf die Blutverweildauer untersucht wurde. In diesem Zusammenhang wurde zusätzlich eine Gruppe mit ligandmodifizierten Liposomen ohne jegliches DSPE-PEG behandelt, und eine weitere Gruppe erhielt Kontrollliposomen, die keine CHOL-TOE-BP-Liganden, aber wiederum DSPE-PEG2000 aufwiesen. Die Ergebnisse hierzu, dargestellt in Abbildung 24, zeigten, dass eine DSPE-PEG2000-Konzentrationserhöhung von 4,3 mol% über 8,2 mol% auf 13,0 mol% jeweils zu einer Verlängerung der Verweildauer im Blut im Vergleich zu ligandmodifizierten Liposomen ohne DSPE-PEG2000 führte und folglich auch höhere

Ergebnisse und Diskussion 126
AUC0-48h-Werte erzielt wurden (Tabelle 17). Im Falle von Liposomen mit einer hohen DSPE-PEG2000-Konzentration (13,0 mol%) im Vergleich zu Liposomen, die 4,3 mol% DSPE-PEG2000 enthielten, wurde beispielsweise ein beinahe doppelt so hoher AUC0-48h-Wert errechnet. Die statistische Auswertung der hier erzielten Ergebnisse (Tabelle 17) bestätigte diese Beobachtung. Der paarweise Vergleich der Liposomen mit unterschiedlichen DSPE-PEG2000-Konzentrationen untereinander ergab einen statistisch signifikanten Unterschied.
Neben der DSPE-PEG-Dichte ist zudem auch die Konformation der DSPE-PEG-Moleküle auf der Liposomenoberfläche für die „Stealth“-Eigenschaften wichtig. Eine weniger dichte Konformation führt zur „Mushroom“-Konfiguration, bei der die DSPE-PEG-Ketten auf der Oberfläche aufliegen und dennoch unbedeckte Stellen existieren. Bei einer hohen DSPE-PEG-Dichte kommt es zur „Brush“-Konfiguration, bei der aufgrund der hohen Dichte die Mobilität der Ketten beeinträchtigt wird und deshalb der sterische Schutz des DSPE-PEGs nicht mehr vollständig gegeben ist (Storm et al., 1995). Die ideale DSPE-PEG-Konformation auf der Liposomenoberfläche liegt also irgendwo zwischen den beiden genannten Konfigurationen. Problematisch bei einer Erhöhung der DSPE-PEG-Dichte könnte allerdings auch sein, dass die vermehrte Zahl an DSPE-PEG-Molekülen zu sterischen Interaktionen mit den CHOL-TOE-BP-Liganden führt und diese an ihrer Bindung an die Knochenmatrix hindert.

Ergebnisse und Diskussion 127
0
20
40
60
80
100
120
0 8 16 24 32 40 48
Zeit [h]
% In
jizie
rte
Dos
is
4,3 mol% DSPE-PEG2000, kein CHOL-TOE-BP13,0 mol% DSPE-PEG20008,2 mol% DSPE-PEG20004,3 mol% DSPE-PEG20000 mol% DSPE-PEG2000
Abbildung 24: Pharmakokinetikprofile von ligandmodifizierten Liposomen, die jeweils 14,3 mol% CHOL-TOE-BP enthielten und sich in ihren DSPE-PEG2000-Konzentrationen unterschieden, sowie von Kontrollliposomen (4,3 mol% DSPE-PEG2000) im Blut nach intravenöser Injektion in die Schwanzvene von Ratten. Die Ergebnisse werden als prozentualer Anteil der anfänglichen injizierten Dosis an Radioaktivität dargestellt; das geschätzte Blutvolumen beträgt 20 ml/Ratte. Die dargestellten Ergebnisse stellen jeweils einen Mittelwert ± SD dar, gebildet aus vier Ratten pro Zeitpunkt. (Dieser Versuch und die nachfolgende Untersuchung, die den jeweiligen Effekt von DSPE-PEG2000 und DSPE-PEG5000 auf die Zirkulationszeit von ligandmodifizierten Liposomen ermittelten, wurden in einem Tierversuch durchgeführt. Deshalb werden in beiden Graphen (Abbildungen 24, 25) und in beiden AUC0-48h-Tabellen (Tabellen 17, 18) dieselben Kontrollliposomen und dieselben ligandmodifizierten Liposomen, die kein DSPE-PEG aufwiesen, aufgeführt).

Ergebnisse und Diskussion 128
Liposomenzusammensetzung DSPE-PEG-Konzentration [mol%]
AUC0-48h [%ID.h]
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 13,0 149 ± 2
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 8,2 127 ± 3
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 4,3 84 ± 1
DPPC/CHOL/DSPE-PEG2000 4,3 307 ± 2
DPPC/CHOL/CHOL-TOE-BP - 15 ± 2
Tabelle 17: Einfluss der molaren Konzentration von DSPE-PEG2000 auf die „Area under the Curve“ von ligandmodifizierten Liposomen (14,3 mol% CHOL-TOE-BP). Eine einmalige Dosis (20 µmol/kg Körpergewicht) an [3H]-markierten Liposomen wurde i.v. in die Schwanzvene der Ratten injiziert. Die AUC0-48h wurde für jedes einzelne Tier basierend auf der oben dargestellten Pharmakokinetik berechnet und als Mittelwert ± SD von vier Tieren dargestellt. Die statistische Auswertung der hier aufgeführten Gruppen ergab einen signifikanten Einfluss bei allen paarweisen Vergleichen der DSPE-PEG2000-Konzentrationen untereinander, auch nach der Angleichung für multiples Testen gemäß Bonferroni-Holm.
4.6.3 Einfluss des DSPE-PEG-Typs auf die Zirkulationszeit
Um zu prüfen, ob der Austausch des DSPE-PEG2000 durch DSPE-PEG5000, ein Polymer mit einem höheren Molekulargewicht, bei gleichbleibender Konzentration zu einer effektiveren protektiven Hülle um ligandmodifizierte Liposomen (14,3 mol% CHOL-TOE-BP) führt, wurde sowohl das Pharmakokinetikprofil von Liposomen, die 4,3 mol% DSPE-PEG2000 aufwiesen, als auch das Profil von Liposomen, die 4,3 mol% DSPE-PEG5000 enthielten, bestimmt. (Dieser Versuch und die zuvor beschriebene Untersuchung des Einflusses von verschiedenen DSPE-PEG2000-Konzentrationen auf die Zirkulationszeit von ligandmodifizierten Liposomen wurden in einem Tierversuch durchgeführt. Deshalb werden in beiden Graphen (Abbildungen 24, 25) und in beiden AUC0-48h-Tabellen (Tabellen 17, 18) dieselben

Ergebnisse und Diskussion 129
Kontrollliposomen und dieselben ligandmodifizierten Liposomen, die kein DSPE-PEG aufwiesen, als Vergleichsgruppe aufgeführt.) Die Ergebnisse in Abbildung 25 zeigen, dass eine Zirkulationszeitverlängerung von ligandmodifizierten Liposomen, die weder DSPE-PEG2000 noch DSPE-PEG5000 aufwiesen, ebenfalls durch den Einbau von DSPE-PEG5000 in die Liposomenmembran erzielt werden konnte. Der Vergleich der AUC0-48h-Werte (Tabelle 18) beider Liposomenformulierungen, die sich im Molekulargewicht ihres Polymers unterschieden, ergibt, dass diese Werte sich kaum voneinander unterscheiden. Auch der statistische Vergleich der Pharmakokinetikprofile beider Polymere bestätigte einen nicht signifikanten Unterschied. Die in diesem Versuchsabschnitt generierten Daten zeigen zusammengenommen, dass weder eine DSPE-PEG2000-Erhöhung noch eine Molekulargewichtserhöhung dazu führte, dass die CHOL-TOE-BP-Liganden auf der Liposomenoberfläche vollständig abgeschirmt werden konnten. Dies scheint auf den ersten Blick nachteilig für die Zirkulationsfähigkeit der Liposomen in der Blutbahn zu sein, doch im Hinblick auf die Targeting-Strategie, die Liposomen durch Bisphosphonate an den Knochen zu binden, kann dies auch von Vorteil sein bzw. es ist sogar erwünscht, denn freistehende Bisphosphonatgruppen sind für eine effektive Bindung an HAP unerlässlich. In allen Fällen wurden die ligandmodifizierten Liposomen wesentlich schneller aus der Blutbahn eliminiert als die Kontrollliposomen, die keine CHOL-TOE-BP-Liganden an ihrer Oberfläche aufwiesen. Es scheint, dass die negativ geladenen Phosphonat-Endgruppen trotz der ebenfalls vorhandenen PEG-Moleküle für Interaktionen mit Opsoninen leicht zugänglich waren, was eine schnelle Clearance dieser Liposomen aus dem Blut zur Folge hatte. Dennoch bewirkte der Einbau von DSPE-PEG2000 oder DSPE-PEG5000 eine erhöhte Verweildauer der ligandmodifizierten Liposomen in der Blutbahn im Vergleich zu den nicht pegylierten, ligandmodifizierten Liposomen und folglich auch eine Erhöhung der AUC0-48h-Werte. Dies ist sicherlich ein Ergebnis, das dem liposomalen System wertvolle Eigenschaften verleiht. Es gilt nun zu zeigen, ob diese Verweildauer ausreichend ist, um das Zielgewebe – den Knochen im metastasierten Gewebe – erreichen zu können. Das Erreichen des Knochens wird zudem möglicherweise durch seine anatomischen Eigenschaften unterstützt. Knochenmetastasen sind hauptsächlich im hochvaskularisierten roten Knochenmark vorzufinden, welches durch ein umfassendes venöses, sinusförmiges Bett bestimmt wird. Die Sinusoide sind umgeben von fenestrierten Endothelzellen, denen die Basalmembran fehlt und die somit den Zugang von Makromolekülen und „Nanocarriers“ ermöglichen (De Bruyn, 1981, Shea and Miller, 2005).

Ergebnisse und Diskussion 130
Die Ergebnisse der Organverteilung (Abbildungen 23, 26) stimmen mit den erhaltenen Pharmakokinetikprofilen überwiegend überein. Eine höhere CHOL-TOE-BP-Dichte auf der liposomalen Oberfläche und somit eine höhere Oberflächenladung führte zu einer höheren Aufnahme in die Leber und zu einer geringeren Milzaufnahme im Vergleich zu den Kontrollliposomen, die keine Liganden aufwiesen. Unter normalen Bedingungen werden Liposomen nach intravenöser Applikation hauptsächlich von den Zellen des Mononukleären Phagozyten-Systems (MPS) der Leber und der Milz abgebaut. Pegylierte Liposomen, so wie sie in dieser Arbeit auch vorliegen, enden wie konventionelle Liposomen zumeist in den Zellen des MPS, was darin begründet liegt, dass beide Organe ein diskontinuierliches oder fenestriertes Endothelium aufweisen. Zudem enthalten die Leber und die Milz zusammen die größte Population an Makrophagen. Anzumerken ist, dass im Fall der ligandmodifizierten Liposomen, die keinen DSPE-PEG aufwiesen, aufgrund der schnellen Elimination aus der Blutbahn eine höhere Akkumulation in der Leber im Vergleich zu den anderen getesteten Liposomen zu erwarten gewesen wäre (G, Abbildung 26). Möglicherweise wurden hier die Organteile nicht ausreichend homogenisiert, so dass es zu einer falschen Bestimmung der Radioaktivität kam. In der Milz wurde im Vergleich zur Leber stets ein geringerer Anteil an Liposomen ermittelt. Die Unterschiede im Ausmaß bei der Aufnahme zwischen Leber und Milz basieren laut Literatur auf verschiedenen Mechanismen, die bis heute nicht vollständig aufgedeckt werden konnten. Sicher ist, dass die Leber die Liposomen im Gegensatz zur Milz zunächst einmal generell unabhängig von ihrer Größe aufnimmt (Klibanov et al., 1991). Zusätzlich spielen die Hepatozyten abhängig von der Größe und der Zusammensetzung der Liposomen ebenfalls eine große Rolle bei der Elimination der Liposomen aus der Blutbahn (Scherphof and Kamps, 2001). Es scheint, dass die Milz auch hier das selektivere Organ ist, doch es bleibt unklar, was die Ursache für das unterschiedliche Ausmaß der Elimination ist. Zu vermuten ist, dass die bereits genannten Faktoren, wie Größe und Zusammensetzung, auch in diesem Zusammenhang einen relevanten Einfluss haben.

Ergebnisse und Diskussion 131
0
20
40
60
80
100
120
0 8 16 24 32 40 48Zeit [h]
% In
jizie
rte
Dos
is
4,3 mol% DSPE-PEG2000, kein CHOL-TOE-BP4,3 mol% DSPE-PEG5000, kein CHOL-TOE-BP4,3 mol% DSPE-PEG50004,3 mol% DSPE-PEG20000 mol% DSPE-PEG
Abbildung 25: Pharmakokinetikprofile von ligandmodifizierten Liposomen, die jeweils 14,3 mol% CHOL-TOE-BP enthielten und sich in ihrem DSPE-PEG-Typ (DSPE-PEG2000, DSPE-PEG5000) unterschieden, und Kontrollliposomen (4,3 mol% DSPE-PEG2000 bzw. 4,3 mol% DSPE-PEG5000) im Blut nach intravenöser Injektion in die Schwanzvene von Ratten. Die Ergebnisse werden als prozentualer Anteil der anfänglichen injizierten Dosis an Radioaktivität dargestellt; das geschätzte Blutvolumen beträgt 20 ml/Ratte. Die dargestellten Ergebnisse stellen jeweils einen Mittelwert ± SD dar, gebildet aus vier Ratten pro Zeitpunkt. (Dieser Versuch und die zuvor beschriebene Untersuchung des Einflusses von verschiedenen DSPE-PEG2000-Konzentrationen auf die Zirkulationszeit von ligandmodifizierten Liposomen wurden in einem Tierversuch durchgeführt. Deshalb werden in beiden Graphen (Abbildungen 24, 25) und in beiden AUC0-48h-Tabellen (Tabellen 17, 18) dieselben Kontrollliposomen und dieselben ligandmodifizierten Liposomen, die kein DSPE-PEG aufwiesen, aufgeführt).
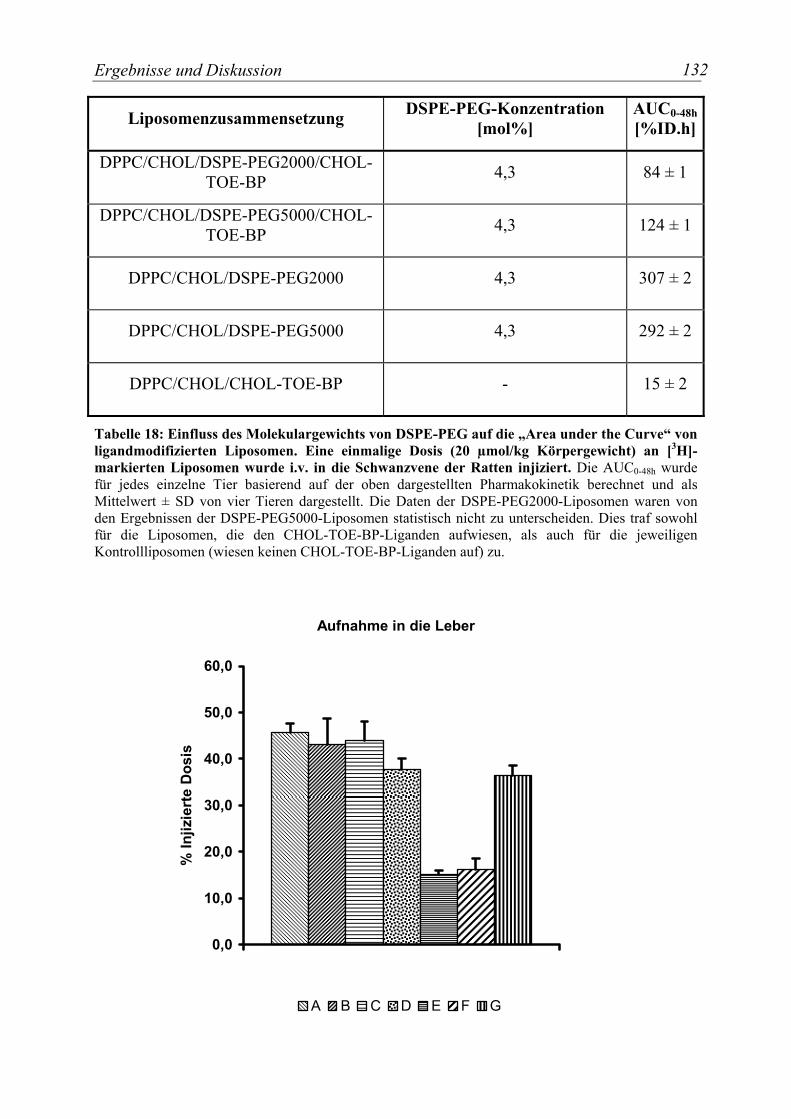
Ergebnisse und Diskussion 132
Liposomenzusammensetzung DSPE-PEG-Konzentration [mol%]
AUC0-48h [%ID.h]
DPPC/CHOL/DSPE-PEG2000/CHOL-TOE-BP 4,3 84 ± 1
DPPC/CHOL/DSPE-PEG5000/CHOL-TOE-BP 4,3 124 ± 1
DPPC/CHOL/DSPE-PEG2000 4,3 307 ± 2
DPPC/CHOL/DSPE-PEG5000 4,3 292 ± 2
DPPC/CHOL/CHOL-TOE-BP - 15 ± 2
Tabelle 18: Einfluss des Molekulargewichts von DSPE-PEG auf die „Area under the Curve“ von ligandmodifizierten Liposomen. Eine einmalige Dosis (20 µmol/kg Körpergewicht) an [3H]-markierten Liposomen wurde i.v. in die Schwanzvene der Ratten injiziert. Die AUC0-48h wurde für jedes einzelne Tier basierend auf der oben dargestellten Pharmakokinetik berechnet und als Mittelwert ± SD von vier Tieren dargestellt. Die Daten der DSPE-PEG2000-Liposomen waren von den Ergebnissen der DSPE-PEG5000-Liposomen statistisch nicht zu unterscheiden. Dies traf sowohl für die Liposomen, die den CHOL-TOE-BP-Liganden aufwiesen, als auch für die jeweiligen Kontrollliposomen (wiesen keinen CHOL-TOE-BP-Liganden auf) zu.
Aufnahme in die Leber
0,0
10,0
20,0
30,0
40,0
50,0
60,0
1
% In
jizie
rte
Dos
is
A B C D E F G

Ergebnisse und Diskussion 133
Aufnahme in die Milz
0,0
10,0
20,0
30,0
40,0
50,0
60,0
1
% In
jizie
rte
Dos
is
A B C D E F G
Abbildung 26: Organverteilung von ligandmodifizierten Liposomen, die jeweils 14,3 mol% CHOL-TOE-BP enthielten (A: DSPE-PEG2000: 4,3 mol%; B: DSPE-PEG2000: 8,2 mol%; C: DSPE-PEG2000: 13,0 mol%; D: DSPE-PEG5000: 4,3 mol%, G: DSPE-PEG2000: 0 mol%), und Kontrollliposomen (E: DSPE-PEG2000: 4,3 mol%; F: DSPE-PEG5000: 4,3 mol%). Die abgebildeten Ergebnisse stellen jeweils einen Mittelwert ± SD dar, gebildet aus vier Ratten pro Zeitpunkt. % injizierte Dosis bezieht sich auf den prozentualen Anteil der anfänglich injizierten Menge an Radioaktivität (geschätztes Blutvolumen: 20 ml/Ratte).
4.7 Zielgerichtete Applikation von ligandmodifizierten Liposomen an den Knochen (Nacktmausmodell)
In einem Pilotversuch wurde in vivo überprüft, ob ligandmodifizierte Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% im Vergleich zu Kontrollliposomen (ohne CHOL-TOE-BP-Ligand) an eine Knochenoberfläche, die von metastasiertem Knochengewebe umgeben ist, binden bzw. im metastasendurchsetzten Knochengewebe akkumulieren können. Es wurde die Hypothese aufgestellt, dass dieses Vorhaben anhand des von Wetterwald et al. (Wetterwald et al., 2002) etablierten Nacktmausmodells realisiert werden könnte, indem die Liposomen zuvor mit dem Fluorenzfarbstoff NBD-PE markiert werden. Um

Ergebnisse und Diskussion 134
sicherzustellen, dass der Fluoreszenzmarker fest in der Liposomenmembran verankert ist und sich nicht während der Versuche ablöst, wurde ein lipidgekoppelter Farbstoff eingesetzt.
Das beschriebene Mausmodell basiert auf der „Bioluminescent Reporter Imaging(BRI)“-Methode, bei der die Expression von Reportergenen, die die biolumineszente Luciferase in vivo kodieren, von außen detektiert wird. Krebszelllinien wurden dazu zuvor mit Luciferase transfiziert, um das lokale Tumorwachstum am Knochen und in anderen Organen am lebenden Tier detektieren zu können. Die Mäuse waren 12 Wochen alt, als mit den Targeting-Versuchen begonnen wurde. Zu diesem Zeitpunkt war die Metastasierung bereits im fortgeschrittenen Stadium, was durch die Entstehung von Osteolysen gekennzeichnet war (Abbildung 27). Das Ausmaß der Metastasierung wurde ebenfalls mittels BRI bestimmt (Abbildung 28).
Die In-vivo-Versuche zur Bestimmung der Pharmakokinetikprofile von ligandmodifizierten Liposomen haben gezeigt, dass Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% relativ schnell aus der Blutbahn von Ratten eliminiert werden. Die In-vitro-Bindungsversuche an HAP haben wiederum verdeutlicht, dass eine hohe Konzentration an CHOL-TOE-BP (14,3 mol%) ein effektives Binden an HAP garantiert. Basierend auf diesen Ergebnissen fiel die Entscheidung, diesen Pilotversuch mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% durchzuführen, in der Annahme, dass zumindest ein kleiner Teil ausreichend lange zirkuliert, um am Zielorgan anzukommen. Es wurde vermutet, dass möglicherweise sowohl die ligandmodifizierten Liposomen als auch die nicht modifizierten Kontrollliposomen in der Lage sind, sich in dem metastasendurchsetzten Knochengewebe durch passives Targeting anzusiedeln. Das passive Targeting wird durch eine verstärkte Akkumulation von langzirkulierenden Liposomen in der Region des Tumor- bzw. des Metastasenwachstums aufgrund von erhöhter Kapillarenpermeabilität des befallenen Gewebes gekennzeichnet. Eine eingeschränkte lymphatische Drainage in den betroffenen Gegenden unterstützt zumeist die erhöhte Ansammlung der Liposomen in diesen Regionen. Diese Vermutung basierte auf der Tatsache, dass die beiden Liposomentypen die gleiche durchschnittliche Partikelgröße von 150 nm aufwiesen, die in der Regel eine gute Voraussetzung für die Akkumulation im metastasierten Gewebe bedingt durch den „Enhanced Permeability and Retention“-Effekt (EPR-Effekt) darstellt. Zudem wurde spekuliert, dass die ligandmodifizierten Liposomen aufgrund ihres knochenspezifischen Liganden eine

Ergebnisse und Diskussion 135
zusätzliche Selektivität für den Knochen beweisen und folglich durch die Bindung an den Knochen zusätzlich aktives Targeting aufzeigen würden. Dieses ligandspezifische Targeting sollte sich durch die vergleichende Untersuchung von nicht modifizierten Kontrollliposomen anhand des ausgewählten Tiermodells demonstrieren lassen, indem die zielgerichtete Applikation durch längere Verweildauern in der betroffenen Knochenregion demonstriert wird, vorausgesetzt, die CHOL-TOE-BP-Liganden bewirken feste Bindungen an Hydroxylapatit.
Abbildung 27: Radiografische Aufnahme von untersuchten Mäusen. Bei allen Tieren ist das fortgeschrittene Stadium der Metastasenentwicklung anhand der Osteolyse im rechten Bein bei dorsaler Ansicht zu erkennen.

Ergebnisse und Diskussion 136
Abbildung 28: Bestimmung des Ausmaßes der Metastasierung in den einzelnen untersuchten Mäusen mittels biolumineszenter Bildgebung. Durch die intraperitoneale Gabe des Substrates Luciferin (2 mg gelöst in PBS) wurde das Luciferase-Gen, mit dem die Tumorzellen zuvor versehen wurden, in vivo aktiviert. Das dadurch emittierte Licht (500 nm Wellenlänge) drang durch das Gewebe des Tieres und konnte somit außerhalb des Körpers mit Hilfe einer hochempfindlichen Photondetektionskamera gemessen werden (ventrale Ansicht). Die Daten wurden mit Hilfe des Programms „Living Image“ (Xenogen, Alameda, CA, USA) analysiert. Die Werte wurden als „Relative Light Unit“ (RLU) in Photonen/sec ausgedrückt.
Um das Targeting von ligandmodifizierten Liposomen in BALB-c-nu/nu-Mäusen zu untersuchen, die Knochenmetastasen an der linken Tibia (bei ventraler Ansicht des Tieres) aufwiesen, wurden diese in den linken Herzventrikel injiziert. Als Kontrolle wurde Kontrollliposomen, die keine CHOL-TOE-BP-Liganden aufwiesen, untersucht. Beide Liposomentypen wurden in einer Dosis von 50 µmol/kg KG an jeweils zwei Tieren appliziert. Aufgrund der Fluoreszenzmarkierung (1 mol% NBD-PE) der Liposomen konnte die Zirkulation der Liposomen am lebenden Tier von außen mit dem Maestro In-vivo-Imaging-System verfolgt werden. Die Ergebnisse in den Abbildungen 29 und 30 zeigen klar, dass die ligandmodifizierten Liposomen im metastasendurchsetzten Knochengewebe akkumulieren. Beim ersten Versuch mit ligandmodifizierten Liposomen stellte sich heraus, dass die Akkumulation erst nach ca. 45 min zu detektieren war (Fluoreszenzintensität von 119112, Tabelle 19 (1. Versuch)). Zu den Messzeitpunkten 5 min und 15 min waren lediglich Fluoreszenzsignale an der Einstichstelle zu messen. Mit der Zeit nahm die Signalintensität in dem betroffenen Knochengewebe zu, da sich dort offenbar weitere

Ergebnisse und Diskussion 137
Liposomen ansammelten. Nach 2 h beispielsweise wurde eine Signalintensität von 266705 (Abbildung 29, Tabelle 19 (1. Versuch)) gemessen. Zum Zeitpunkt 24 h wurden bereits schwächere Fluoreszenzintensitäten gemessen. Nach 5 Tagen konnte immer noch ein Fluoreszenzsignal gemessen werden, welches jedoch noch geringer in der Intensität ausfiel als das vom 24-h-Messpunkt.
Das zweite untersuchte Tier, welches ebenfalls mit der gleichen Dosis an ligandmodifizierten Liposomen behandelt wurde, zeigte ähnliche Ergebnisse (Abbildung 30, Tabelle 19 (2. Versuch)). Die zielgerichtete Applikation an den Knochen bzw. zum metastasendurchsetzten Knochengewebe war auch hier deutlich zu erkennen. Das Intensitätsmaximum des Fluoreszenzsignals fiel in diesem Fall jedoch geringer aus. Zudem wurde dieses Maximum anders als im ersten Versuch (nach 2 h) am 24-h-Messzeitpunkt bestimmt (Fluoreszenzintensität von 168321). Die beschriebenen Unterschiede bei den Ergebnissen könnten in dem unterschiedlichen Ausmaß der Metastasierung (Abbildungen 27, 28) der untersuchten Tiere begründet sein. Die Untersuchung von fluoreszenzmarkierten Kontrollliposomen, die keine CHOL-TOE-BP-Liganden aufwiesen, ergab, dass diese nicht im metastasendurchsetzten Knochengewebe akkumulierten (Abbildung 30). Zum Messzeitpunkt 10 min war bei dieser Kontrolle ein Fluoreszenzsignal im Zielgewebe sichtbar, welches zu den nachfolgenden Messzeitpunkten jedoch nicht mehr feststellbar war. Das Kontrolltier des ersten Targeting-Versuches starb zum Messzeitpunkt 2 h nach der Gabe des Anästhetikums wahrscheinlich aufgrund von Stresserscheinungen. Die Aufnahmen, die bis zu diesem Zeitpunkt gemacht wurden, ließen kein Targeting erkennen (Ergebnisse sind nicht dargestellt). In Vorversuchen konnten bereits ähnliche Beobachtungen gemacht werden – Kontrollliposomen waren im Zielgewebe nicht anzutreffen (Ergebnisse sind nicht dargestellt). Es konnte allerdings auch nicht der Verbleib der Kontrollliposomen in anderen Geweben detektiert werden. Dies trifft für die ligandmodifizierten Liposomen ebenfalls zu, d.h. es war zwar möglich, die Liposomen im metastasierten Gewebe zu lokalisieren, doch in anderen Geweben waren sie anscheinend nicht anzutreffen.
Dennoch hat der Pilotversuch klar demonstriert, dass die zielgerichtete Applikation von ligandmodifizierten Liposomen im Vergleich zu Kontrollliposomen, die kein Targeting aufzeigten, mit Hilfe des ausgewählten Tiermodells dargestellt werden konnte. Zudem ist eine weitere essentielle Fragestellung für die in dieser Arbeit dargestellte Targeting-Strategie zu klären, und zwar, ob die Liposomen tatsächlich an den Knochen gebunden haben. Das ausgesuchte Tiermodell ließ in diesem

Ergebnisse und Diskussion 138
Zusammenhang die Differenzierung zwischen einer Bindung an die Knochenoberfläche und der Ansammlung im metastasendurchsetzten Knochengewebe leider nicht zu. Aufgrund der detektierten Fluoreszenzabnahme ist zu vermuten, dass keine richtige Bindung stattfand. Dies würde bedeuten, dass die Liposomen zunächst im metastasierten Gewebe akkumulierten, indem sie zuvor aus der Blutbahn durch fenestriertes (Sinusoide des Knochenmarks) oder löchriges Endothelium (Blutgefäße von Metastasen) in das metastasierte Gewebe diffundierten und dort dann mit der Zeit abgebaut wurden. Mit anderen Worten: Es hat passives Targeting basierend auf dem EPR-Effekt stattgefunden. Dieser Theorie steht allerdings die Tatsache entgegen, dass die Kontrollliposomen, die keinen Liganden aufwiesen, sich nicht dort ansammelten. Zum Messzeitpunkt 10 min wurde im zweiten Versuch zwar ein Fluoreszenzsignal im Zielgewebe bei der Untersuchung des Kontrolltieres sichtbar, doch als Targeting konnte dies nicht bezeichnet werden, da in den darauffolgenden Messungen dort kein Signal mehr detektierbar war. Deshalb wäre ein weiterer Erklärungsansatz möglich, der besagt, dass hier ein nicht spezifisches Binden von ligandmodifizierten Liposomen an die Knochenoberfläche vorlag, dessen Bindungsausmaß aufgrund von geringer Affinität mit der Zeit nachließ. Anzumerken ist hierbei, dass die In-vitro-Bindungsversuche zuvor gezeigt haben, dass unter anderem eine spezifische Bindung der ligandmodifizierten Liposomen an HAP bestimmt wurde. Bestätigt wird diese Beobachtung durch den bereits intensiv untersuchten Bindungsmechanismus einer Vielzahl von Bisphosphonaten. Diese sind wiederum bekannt als effektive Chelatbildner, die eine hohe Affinität zum Hydroxylapatit des Knochens haben (Fleisch, 1998, Cukrowski et al., 2007). Denkbar wäre aufgrund dieser Beobachtungen auch, dass die Liposomen spezifisch an den Knochen gebunden haben, so dass die CHOL-TOE-BP-Liganden in der mineralischen Knochenmatrix fest verankert wurden und die Liposomen bzw. die Liganden mit der Zeit enzymatisch abgebaut wurden. Der Fluoreszenzfarbstoff wurde folglich freigesetzt, was dazu führte, dass die gemessene Intensität der Fluoreszenz abnahm. Das Ankermolekül des Liganden wäre somit möglicherweise immer noch in der Matrix eingebaut, was nicht ungewöhnlich wäre. Sind Bisphosphonate erst einmal in die Knochenmatrix eingebaut, so werden sie nur dann freigesetzt, wenn der Knochen im Zuge des regulären Knochenauf- und -abbaus bzw. des pathologisch bedingten „Bone Turnovers“ zerstört wird. Die Halbwertszeit verschiedener Bisphosphonate im Skelett bewegt sich bei Mäusen und Ratten zwischen 3 Monaten und 1 Jahr (Wingen and Schmahl, 1985, Monkkonen, 1988, Monkkonen et al., 1990). Beim Menschen können die Bisphosphonate häufig für mehr als 10 Jahre in der Knochenmatrix eingeschlossen sein.

Ergebnisse und Diskussion 139
Die Fluoreszenzabnahme könnte jedoch auch noch eine andere Ursache haben, und zwar könnte die Abnahme der Signalintensität auch durch das sogenannte „Photobleaching“ (Nachlassen der Fluoreszenz nach Bestrahlung) verursacht worden sein. Wenn also tatsächlich eine Bindung der Liposomen an die Knochenmatrix stattgefunden hat, dann würde dieser Versuch zudem aufzeigen, dass das DSPE-PEG2000, welches ebenfalls auf der Liposomenoberfläche saß, auch in vivo die Bindung nicht nachteilig beeinflusste. Das hier beschriebene Tiermodell ermöglichte in diesem Zusammenhang keine quantitative Bestimmung der HAP-gebundenen, ligandmodifizierten Liposomen. Aufgrund der Tatsache, dass keine Akkumulation in der Leber oder Milz detektiert wurde, wäre zu vermuten, dass die ermittelte Ansammlung im metastasierten Gewebe von hoher Konzentration war. Diese Hypothese wird jedoch von der Beobachtung widerlegt, dass der Verbleib der Kontrollliposomen ebenfalls nicht in der Leber oder Milz auszumachen war. Zudem haben die zuvor beschriebenen Pharmakokinetikversuche an Ratten gezeigt, dass von der injizierten Dosis von ligandmodifizierten Liposomen mit einer Ligandkonzentration von 14,3 mol% bereits nach einer Stunde nur noch 35% im Blut feststellbar waren, weil sie höchstwahrscheinlich wegen ihrer stark negativen Ladung vom RES leicht erkannt und als Folge eliminiert wurden. Zu beachten ist hierbei, dass die Pharmakokinetikdaten der Rattenversuche nicht vollständig auf das vorliegende Mausmodell übertragbar sind, da bereits gezeigt wurde, dass nicht nur die Clearance-Rate zwischen diesen beiden Spezies unterschiedlich ist, sondern auch der Mechanismus, der für die Clearance verantwortlich ist (Liu et al., 1995).
Die Ursache für die offenbar unmögliche Detektierbarkeit der Liposomen außerhalb des Tumorgewebes, insbesondere im Fall der Kontrollliposomen, könnte aber auch in einem technischen oder methodischen Problem begründet sein. Möglicherweise absorbierten die Organe das emittierte Fluoreszenzlicht, das von den Liposomen ausging. Es bleibt somit offen, warum der sonstige Verbleib sowohl von ligandmodifizierten Liposomen als auch von Kontrollliposomen in diesem Versuchsaufbau nicht bestimmbar war.

Ergebnisse und Diskussion 140

Ergebnisse und Diskussion 141
Abbildung 29: Visualisierung von Maus und Liposomenverbleib im lebenden Körper (ventrale Ansicht) zu den Zeitpunkten: 5 min, 15 min, 45 min, 2 h, 24 h, 5 d durch eine nicht invasive Detektionsmethode. Die fluoreszenzmarkierten Liposomen wurden dazu intrakardial injiziert. Die injizierte Dosis betrug 50 µmol/kg Körpergewicht. (Die unterschiedlichen Farben der Tiere in den einzelnen Aufnahmen sind zu vernachlässigen, sie werden von dem eingesetzten Gerät verursacht).

Ergebnisse und Diskussion 142
Abbildung 30: Visualisierung von Maus und Liposomenverbleib im lebenden Körper (ventrale Ansicht) zu den Zeitpunkten: 10 min, 45 min, 2 h, 24 h, 48 h durch eine nicht invasive Detektionsmethode. Die fluoreszenzmarkierten Liposomen wurden dazu intrakardial injiziert. Die injizierte Dosis betrug 50 µmol/kg Körpergewicht. (Die unterschiedlichen Farben der Tiere in den einzelnen Aufnahmen sind zu vernachlässigen, sie werden von dem eingesetzten Gerät verursacht).

Ergebnisse und Diskussion 143
Messzeitpunkt Absorption (1. Versuch) Absorption (2. Versuch)
5 min - nicht gemessen
10 min nicht gemessen -
15 min - nicht gemessen
45 min 119112 -
2 h 266705 152751
24 h 103426 168321
48 h nicht gemessen 85719
5 d 71088 nicht gemessen
Tabelle 19: Vergleich von Fluoreszenzintensitäten, die zu verschiedenen Messzeitpunkten in zwei unterschiedlichen Tieren nach einer intrakardialen Injektion von ligandmodifizierten Liposomen bestimmt wurden. Die injizierte Dosis betrug in beiden Fällen 50 µmol/kg Körpergewicht.

Zusammenfassung und Ausblick 144
5 Zusammenfassung und Ausblick
Zahlreiche verschiedene Krebstypen metastasieren zum Knochen, darunter insbesondere Prostata- und Brustkrebs. Derzeitige verfügbare Standardtherapien von Knochenmetastasen umfassen die Chemotherapie, die Hormontherapie und die Strahlentherapie. Ein entscheidender Nachteil dieser Behandlungen ist das Verursachen von schweren Nebenwirkungen in Geweben, die nicht vom Krebs befallen sind. Chemotherapeutika werden beispielsweise oral oder intravenös verabreicht. Als systemisch wirkende Therapeutika gelangen sie in die Blutbahn, um die gestreuten Krebszellen erreichen zu können. Antitumorale Substanzen töten auf diese Weise Krebszellen, aber auch normale Zellen ab und rufen folglich starke Nebenwirkungen hervor. Deshalb muss die Aufmerksamkeit gezielt auf das Bestreben gerichtet werden, die Nebenwirkungen zu vermeiden oder zumindest zu reduzieren. Vor dem Hintergrund, die derzeitige Behandlung von Knochenmetastasen verbessern zu können, wurden im Zuge eines besseren Verständnisses der Knochenbiologie sowie der molekularen Prozesse, die bei der Entwicklung von Knochenmetastasen involviert sind, neue therapeutische Zielzellen bzw. Zielgewebe im Knochen identifiziert, darunter die Knochenoberfläche. Basierend auf diesen Erkenntnissen wurden auch neue potentielle Wirkstoffe entwickelt, die jedoch zumeist keine Affinität zum Knochen aufweisen. Hinzu kommt, dass diese Substanzen häufig für die Anwendung in vivo ungeeignet sind, da sie Stabilitätsprobleme oder nicht ausreichende Pharmakokinetikprofile aufweisen. Um diese Nachteile zu beheben, scheint die Entwicklung von zielgerichteten Trägersystemen ein vielversprechender Ansatz zu sein. Die Vergangenheit hat gezeigt, dass Wirkstoffe mit einer geringen Affinität zum Knochen durch die Kopplung an Bisphosphonate knochenspezifische Eigenschaften erlangen und somit erfolgreich an den Knochen transportiert werden können. In diesem Zusammenhang wird angenommen, dass Bisphosphonate, die als Chelatkomplexbildner gelten, an die Knochenoberfläche binden, indem sie mit den Calciumionen des Hydroxylapatits, dem Hauptbestandteil der Knochenmatrix, stabile Komplexe formen. Die erfolgreiche Darstellung einer verbesserten therapeutischen Wirksamkeit, die durch die Kopplung von Bisphosphonaten als knochenaffine Liganden an einzelne Wirkstoffmoleküle erreicht wurde, die keine Knochenaffinität aufwiesen, hat uns ermutigt, ebenfalls ein knochenaffines Trägersystem zu entwickeln. Die Besonderheit dieses Systems sollte darin bestehen, dass hier neusynthetisierte Bisphosphonatliganden in die Membran von Liposomen inkorporiert werden. Die

Zusammenfassung und Ausblick 145
Entscheidung, Liposomen als Trägersystem zu wählen, basierte auf der Tatsache, dass diese in vielerlei Hinsicht als vorteilhaft erachtet werden. Liposomen gelten beispielsweise als bioabbaubar, nicht immunogen und untoxisch. Aufgrund der Möglichkeit, Wirksubstanzen darin verkapseln zu können, können unerwünschte Nebenwirkungen möglicherweise reduziert werden, und die eingeschlossene Substanz wird gleichzeitig vor vorzeitigem Abbau während der Zirkulation in der Blutbahn geschützt. Wir stellten die Hypothese auf, den therapeutischen Benefit noch weiter steigern zu können, indem mit der Entwicklung von ligandmodifizierten Liposomen, die sich durch das Vorhandensein von Bisphosphonatliganden auf ihrer Oberfläche auszeichnen, ein Trägersystem geschaffen wird, welches die Verkapselung von hochwirksamen Substanzen erlaubt. Somit würde ebenfalls die Spezifität dieser Stoffe für den Knochen erhöht werden können. Vorteile gegenüber den anderen bisher untersuchten Systemen sahen wir darin, durch die Einbeziehung von Liposomen in das Targeting-Konzept zum einen eine große Menge an Wirksubstanz verkapseln zu können und zum anderen durch das Anbringen von zahlreichen neusynthetisierten Bisphosphonatliganden auf der Liposomenoberfläche die Effizienz der zielgerichteten Applikation an den Knochen zu erhöhen.
In der vorliegenden Dissertation wurde die erfolgreiche Entwicklung von ligandmodifizierten Liposomen beschrieben. Dabei wurde der Fokus insbesondere auf die Machbarkeit gerichtet, ein neusynthetisiertes, maßgeschneidertes Bisphosphonatmolekül (CHOL-TOE-BP) in eine liposomale Membran zu inkorporieren, sowie auf die Überprüfung der Eignung des neuentwickelten Trägersystems, als knochenaffines System eingesetzt werden zu können. Die Versuche zur Bestimmung der Einsetzbarkeit der Liposomen umfassten In-vitro-Bindungsstudien an Hydroxylapatit, die Untersuchung des Zirkulationsverhaltens in vivo und die Überprüfung der Fähigkeit, in vivo an den Knochen zu binden bzw. in metastasendurchsetztem Knochengewebe zu akkumulieren. Der eingesetzte Ligand setzte sich aus einem Cholesterylrest, einer Trisoxyethylenkette und einem Bisphosphonsäuremolekül zusammen. Mit Hilfe des Cholesterylrestes wurde der Ligand in der Liposomenmembran verankert, die Trisoxyethyleneinheit diente als Spacermolekül und die endständige Bisphosphonsäuregruppe bildete den eigentlichen Targeting-Liganden zur Bindung an den Knochen.
Formulierungsentwicklung von ligandmodifizierten Liposomen (Abschnitt 4.1). In diesem Abschnitt wurde die Machbarkeit der Herstellung von Liposomen demonstriert, die auf ihrer Oberfläche neusynthetisierte Bisphosphonatmoleküle

Zusammenfassung und Ausblick 146
tragen. Dabei wurden verschiedene Bisphosphonatderivate (Palmitoyl-bisphosphonsäure (PAL-BP), Cholesteryl-bisphosphonsäure (CHOL-BP), Cholesteryl-trisoxyethylen-bisphosphonsäure (CHOL-TOE-BP) und Cholesteryl-tetraoxyethylen-bisphosphonsäure (CHOL-TeOE-BP)) in Liposomen bestehend aus unterschiedlichen Lipiden (Ei-Phosphatidylcholin (EPC), Dipalmitoyl-phosphatidylcholin (DPPC)) direkt über die Lipidfilmherstellung in die Membran eingebaut. Ein nicht gelungener Einbau wurde im Rahmen der Formulierungsfindung anhand von sichtbaren Ausfällungen festgemacht. Neben einem erfolgreichen Ligandeinbau wurde bei der Formulierungsentwicklung zudem ein physiologischer pH-Wert angestrebt, da die Formulierung für die intravenöse Anwendung vorgesehen ist. Um den Liposomen die Möglichkeit einzuräumen, die Blutbahn durch lückenhafte Endothelien von Blutgefäßen (passives Targeting) insbesondere im Zielgewebe verlassen zu können, wurde zudem eine durchschnittliche Liposomengröße von 100–200 nm gefordert. Außerdem ist eine kleine Partikelgröße auch im Hinblick auf den Erhalt längerer Zirkulationszeiten im Blut vorteilhaft. Anhand dieser Zielparameter wurde die vielversprechendste Formulierung für die nachfolgenden Versuche ausgewählt. In diesem Zusammenhang wurde entschieden, mit DPPC-Liposomen fortzufahren, die den Cholesteryl-trisoxyethylen-bisphosphonsäure-Liganden (CHOL-TOE-BP-Liganden) aufwiesen.
Physikochemische Charakterisierung von ligandmodifizierten Liposomen (Abschnitt 4.2). Um ligandmodifizierte Liposomen, die den CHOL-TOE-BP-Liganden in verschiedenen Konzentrationen enthielten, näher zu charakterisieren, wurden zunächst verschiedene physikochemische Eigenschaften untersucht. In diesem Zusammenhang wurden homogene Partikelgrößenverteilungen mit durchschnittlichen Partikelgrößen zwischen 120 und 155 nm (PI: 0,03–0,15) bei unterschiedlichen Ligandkonzentrationen ermittelt. Die Langzeitstabilitätsuntersuchung der Liposomen von 9 Monaten im Hinblick auf die Partikelgröße ergab, dass die Stabilität durch den Ligandeinbau nicht beeinträchtigt wurde. Durch den Einbau eines neuen Liganden bedingte Reaktionen wie liposomale Aggregation oder Verschmelzung von Doppelmembranen (Bilayer-Fusion) hätten die Stabilität von Liposomen beispielsweise drastisch beeinflussen können und waren deshalb für die Entwicklung eines erfolgversprechenden Wirkstoffträgersystems unerwünscht. Die Fähigkeit von ligandmodifizierten Liposomen, an Hydroxylapatit (HAP) zu binden, wurde sicherlich entscheidend von der Liganddichte auf der Liposomenoberfläche bestimmt. Es war jedoch nicht garantiert, dass alle eingesetzten Substanzen, Lipide und CHOL-TOE-BP-Liganden, vollständig in die Liposomen eingebaut würden. Deshalb war es

Zusammenfassung und Ausblick 147
wichtig, die tatsächliche CHOL-TOE-BP-Konzentration innerhalb der liposomalen Formulierung zu ermitteln. Anhand von Zeta-Potential-Messungen konnte vorab eine Inkorporierung des Liganden abhängig von der eingesetzten Konzentration festgemacht werden. Höhere CHOL-TOE-BP-Konzentrationen führten zu negativeren Zeta-Potentialen. Um eine genaue Aussage über das Ausmaß des Ligandeinbaus machen zu können, wurde eine 31P-NMR-Methode etabliert. Es stellte sich heraus, dass fast 100% des CHOL-TOE-BP-Derivates in die liposomale Membran inkorporiert wurden. Die Strategie, den CHOL-TOE-BP-Liganden über Cholesterol in die Membran zu verankern, erwies sich somit als erfolgreich.
Bindung von ligandmodifizierten Liposomen an Hydroxylapatit (Abschnitt 4.3). Da die ligandmodifizierten Liposomen für die zielgerichtete Applikation an den Knochen entwickelt wurden, war die Überprüfung der Fähigkeit, an den Knochen binden zu können, ein wichtiger Bestandteil der vorliegenden Arbeit. Diese Eigenschaft wurde anhand von In-vitro-Bindungsstudien an Hydroxylapatit (HAP), dem Hauptbestandteil der Knochenmatrix, bestimmt. Zu diesem Zweck wurde die Isothermale Titrationskalorimetrie (ITC) herangezogen. ITC ist eine Technik, die für die Darstellung von chemischen Reaktionen geeignet ist, die durch die Zugabe von ligandmodifizierten Liposomen in eine HAP-Suspension initiiert werden. Sobald die Liposomen an HAP binden, wird Wärme generiert. Auch hier stellte sich heraus, dass die CHOL-TOE-BP-Liganden eine wichtige Rolle spielten. Es ließ sich eine Abhängigkeit der untersuchten CHOL-TOE-BP-Konzentration und der detektierten Bindungswärme feststellen. Eine höhere Ligandkonzentration führte zu einer gesteigerten Wärmeentwicklung. Um festzustellen, ob die Oberflächenladung der Liposomen einen relevanten Einfluss auf die Bindung hat, wurde zusätzlich die Bindung von stark negativ geladenen Liposomen und gering negativ geladenen Kontrollliposomen, die jeweils keine CHOL-TOE-BP-Liganden aufwiesen, an Hydroxylapatit untersucht. In beiden Fällen ließ sich keine Bindung an HAP feststellen. Aufgrund dieser Ergebnisse ist zu vermuten, dass der Bindungsmechanismus nicht hauptsächlich auf elektrostatischen Interaktionen basiert, sondern dass der CHOL-TOE-BP-Ligand entscheidend für die Bindung der Liposomen an Hydroxylapatit ist. Die Auswertung der erhaltenen Ergebnisse im Sinne der Thermodynamik (Bestimmung der molaren Reaktionsenthalpie und einer Bindungskonstanten) zeigte eine Zunahme der Bindungsstärke abhängig von der Ligandkonzentration. Diese Beobachtung deutet daraufhin, dass multivalentes Binden stattgefunden hat.

Zusammenfassung und Ausblick 148
Wirkstoffbeladung von ligandmodifizierten Liposomen (Abschnitt 4.4). Im Hinblick auf eine mögliche spätere Anwendung der Liposomen als erfolgversprechende alternative Therapieoption bei der Behandlung von Knochenmetastasen wurden diese über einen Ammoniumsulfatgradienten mit dem Modellwirkstoff Doxorubizin beladen, um zunächst ihre Beladungskapazität und ihre Stabilität nach erfolgter Beladung zu untersuchen. Dabei wurde eine Beladungskapazität von beinahe 100% erzielt, und bei Stabilitätsprüfungen über einen Zeitraum von einer Woche wurde detektiert, dass nur 5% des verkapselten Wirkstoffes aus dem Liposomeninneren freigesetzt wurden. Für ein erfolgreich einsetzbares Trägersystem ist neben der Beladungseffizienz auch die Freisetzung des verkapselten Wirkstoffes im Zielgewebe wichtig. Folglich wurde diese bestimmt. Um bei der Durchführung der Freisetzungsversuche die In-vivo-Bedingungen zu berücksichtigen, die die ligandmodifizierten Liposomen auf dem Weg zur Knochenoberfläche erfahren würden, wurde die Freisetzung bei unterschiedlichen pH-Werten untersucht. Die Versuche hierzu haben klar gezeigt, dass ein Abfall des extraliposomalen pH-Wertes die vermehrte Doxorubizin-Freisetzung aus ligandmodifizierten Liposomen bewirkt.
Zytotoxizität von ligandmodifizierten Liposomen (Abschnitt 4.5). Um eine Vorstellung über die Wirkung zu erhalten, die von wirkstoffbeladenen, ligandmodifizierten Liposomen ausgeht, sind Versuche zur Bestimmung der Zelltoxizität sehr wichtig. In diesem Zusammenhang wurden die IC50-Werte von Doxorubizin-beladenen, ligandmodifizierten Liposomen im Vergleich zur freien Wirksubstanz mittels Kristallvioletttest anhand von verschiedenen Prostatakarzinom- und Mammakarzinomzelllinien in vitro bestimmt. Dabei stellte sich heraus, dass sich der freie Wirkstoff prinzipiell toxischer auf die Zellen auswirkte als das verkapselte Doxorubizin, was vermuten lässt, dass die toxische Wirkung allein vom freien Doxorubizin ausging. Wirkstoff, der in verkapselter Form von den Zellen aufgenommen oder extrazellulär freigesetzt wurde und dann erst in die Zellen gelangte („Bystander-Effekt“), konnte seine zytotoxische Wirkung in der gegebenen Zeit offenbar nicht so schnell entfalten.
Pharmakokinetik und Biodistribution von ligandmodifizierten Liposomen (Abschnitt 4.6). Um den Einfluss der negativ geladenen CHOL-TOE-BP-Liganden auf die Verweildauer im Blut zu untersuchen, wurden die Pharmakokinetik- und Biodistributionsprofile von ligandmodifizierten Liposomen in Abhängigkeit von verschiedenen CHOL-TOE-BP-Konzentrationen bestimmt. Es konnte gezeigt werden, dass der CHOL-TOE-BP-Ligand wiederum eine entscheidende Rolle spielt. Höhere

Zusammenfassung und Ausblick 149
Ligandkonzentrationen führten trotz des Vorhandenseins von 4,3 mol% DSPE-PEG2000 zu verkürzten Verweildauern im Blut. Das Polymer DSPE-PEG2000 ist dafür bekannt, dass es die Opsonisierung von Liposomen verhindern kann, indem es eine dichte schützende „Wolke“ auf der Liposomenoberfläche bildet und somit zu verlängerten Verweildauern im Blut führt. Aus diesem Grund untersuchten wir den Einfluss von DSPE-PEG2000-Konzentrationserhöhungen (8,2 mol% und 13,0 mol% DSPE-PEG2000) auf die Pharmakokinetik von ligandmodifizierten Liposomen mit einer hohen CHOL-TOE-BP-Konzentration (14,3 mol% CHOL-TOE-BP). Die Ergebnisse haben gezeigt, dass durch jede untersuchte Erhöhung des DSPE-PEG2000-Anteils in der Formulierung tatsächlich eine Verlängerung der Verweildauer erzielt werden konnte. Die bewirkten Verlängerungen der Zirkulationszeiten waren dennoch lange nicht vergleichbar mit den Pharmakokinetikprofilen der Kontrollliposomen (ohne CHOL-TOE-BP-Liganden). Mit dem Austausch von 4,3 mol% DSPE-PEG2000 durch ein Polymer mit höherem Molekulargewicht (4,3 mol% DSPE-PEG5000) wurden die In-vivo-Pharmakokinetikversuche abgeschlossen. Es stellte sich heraus, dass das DSPE-PEG5000 im Vergleich zum DSPE-PEG2000 zum Erhalt eines ähnlichen Pharmakokinetikprofils im Fall von ligandmodifizierten Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% führte und somit trotz seiner wahrscheinlich dichter liegenden Ketten keinen Vorteil erbrachte. Festzuhalten ist jedoch, dass trotz des beobachteten negativen Einflusses der CHOL-TOE-BP-Liganden auf die Blutverweildauer der Liposomen dennoch vielversprechende Ergebnisse im Sinne von akzeptablen Zirkulationszeiten erreicht wurden, die das Fortsetzen weiterer In-vivo-Untersuchungen unterstützten.
Zielgerichtete Applikation von ligandmodifizierten Liposomen an den Knochen (Nacktmausmodell) (Abschnitt 4.7). In diesem Abschnitt wurde anhand eines gut etablierten Nacktmausmodells für Knochenmetastasen die Fähigkeit von ligandmodifizierten Liposomen untersucht, an den Knochen im metastasendurchsetzten Knochengewebe binden zu können. In der Hoffnung, wertvolle Ergebnisse aus diesem In-vivo-Versuch gewinnen zu können, wurde versucht, anhand der generierten Pharmakokinetikdaten zusammen mit den ermittelten Ergebnissen der HAP-Bindungsstudien eine sinnvolle Zusammensetzung für diese Zwecke auszuwählen. Die Versuche hierzu haben klar gezeigt, dass ligandmodifizierte Liposomen mit einer CHOL-TOE-BP-Konzentration von 14,3 mol% und einem DSPE-PEG2000-Anteil von 4,3 mol% zu einem hohen Anteil im metastasendurchsetzten Knochengewebe akkumulierten bzw. an den Knochen dieser Gegend gebunden haben. Die Kontrollliposomen hingegen, die aus 4,3 mol% DSPE-

Zusammenfassung und Ausblick 150
PEG2000 bestanden, aber keine CHOL-TOE-BP-Liganden aufwiesen, sammelten sich nicht bzw. nicht in detektierbaren Konzentrationen im betroffenen Knochengewebe an. Diese Beobachtung deutet darauf hin, dass der „Enhanced Permeability and Retention“- Effekt (EPR-Effekt) bei der Ansammlung im betroffenen Knochengewebe keine maßgebliche Rolle spielt. Durch die Fluoreszenzmarkierung der Liposomen konnte ihr Verbleib am lebenden Tier mit einem multispektralen bildgebenden Kamerasystem (Maestro In-vivo-Imaging-System) festgehalten werden. Die für diesen Pilotversuch gewählte Methode ließ jedoch die genaue Lokalisierung der Liposomen nicht zu. Es konnte folglich nicht festgestellt werden, ob die ligandmodifizierten Liposomen tatsächlich an den Knochen gebunden haben oder nur im metastasendurchsetzten Knochengewebe akkumulierten. Doch aufgrund der Tatsache, dass die Kontrollliposomen im metastasierten Knochengewebe nicht detektiert wurden, scheinen die CHOL-TOE-BP-Liganden für die Ansammlung von großer Bedeutung zu sein. Es ist folglich nicht auszuschließen, dass eine CHOL-TOE-BP-vermittelte Bindung der Liposomen an die Knochenmatrix stattgefunden hat.
Fazit. Zusammenfassend ist zu sagen, dass der „Proof of Concept“ mit dieser Arbeit erbracht worden ist. Maßgeschneiderte Bisphosphonatliganden wurden zunächst erfolgreich in liposomale Membranen eingebaut. Neben einer umfangreichen Charakterisierung dieser ligandmodifizierten Liposomen ist es gelungen, ihr Targeting-Potential zu metastasendurchsetztem Knochengewebe in vivo klar zu demonstrieren. Weitere Bestrebungen im Sinne von Untersuchungen der ligandmodifizierten Liposomen als neue Therapieoption für die Therapie von Knochenmetastasen werden somit stark befürwortet.
Ausblick. Durch die erfolgreiche Demonstration der Machbarkeit, Bisphosphonatliganden und Liposomen als Trägersystem zu kombinieren, wurde ein richtungsweisender Ansatz für die Behandlung von Knochenmetastasen entwickelt. Anhand der durchgeführten Studien konnten in diesem Zusammenhang wertvolle Erkenntnisse gewonnen werden. In der Zukunft müssten jedoch weitere Versuche folgen, die den Nachweis erbringen, dass die zielgerichtete Applikation an die Knochenoberfläche tatsächlich gelungen ist. Die genaue Lokalisierung könnte präzisiert werden, indem die Versuchstiere nach der Applikation der Liposomen und erfolgter Ansammlung an der Knochenoberfläche bzw. Akkumulation im Knochengewebe aufgeschnitten und Gewebeproben aus dem metastasendurchsetzten Bereich und der extrazellulären Knochenmatrix aus dieser Gegend auf ihre Fluoreszenz hin untersucht werden. Alternativ könnte hier anstelle des

Zusammenfassung und Ausblick 151
Fluoreszenzfarbstoffes auch ein radioaktiver Marker wie beispielsweise [3H]-Cholesteryl-oleylether eingesetzt werden. Im Rahmen dieser Untersuchungen wäre es zudem interessant, das Ausmaß der knochengebundenen Menge zu bestimmen, um Aussagen über die mögliche Effizienz dieser Liposomen machen zu können. Dazu könnte die Signalintensität des eingesetzten Markers vor der Applikation und bei der Entnahme der Gewebeproben bestimmt werden. Im gleichen Versuch könnte zudem der Einfluss einer DSPE-PEG2000-Konzentrationserhöhung oder einer Verkleinerung der durchschnittlichen Liposomengröße in Betracht gezogen werden. Möglicherweise führt eine Änderung dieser Parameter zu einer vermehrten Bindung von ligandmodifizierten Liposomen an die Knochenoberfläche. Versuche, die den therapeutischen Benefit von ligandmodifizierten Liposomen im Vergleich zu bestehenden Therapieoptionen klar herausarbeiten, sind ebenfalls essentiell. In diesem Zusammenhang könnte der Einfluss von verschiedenen liposomal verkapselten Wirkstoffen auf bereits bestehende Knochenmetastasen und auf die Entstehung neuer Zellen untersucht werden. Hierbei könnte die Technik der biolumineszenten Bildgebung erneut Anwendung finden. Nach erfolgreicher Aktivierung des Luciferase-Gens in Luciferase-transfizierten Tumorzellen durch die Gabe des enstprechenden Substrates Luciferin könnte anhand der dadurch abgegebenen Photonen das Ausmaß des Metastasenwachstums festgestellt werden. Zudem könnte die Wirkung des therapeutischen Agens auf die Metastasen bestimmt werden, indem spezifische Tumormarker wie Zytokine oder angiogene Faktoren, die im neoplastischen Gewebe überexprimiert werden, mittels Enzymgekoppeltem Immunadsorptionstest (EIA) identifiziert und quantifiziert werden. Ein weiterer interessanter Versuch für die Zukunft wäre die Überprüfung, ob eine geringere Liganddichte auf der Liposomenoberfläche ebenfalls ein gezieltes Knochen-Targeting bewirken kann und somit ein mögliches Binden an Hydroxylapatit, welches in gesunden Geweben mit hohem „Bone Turnover“ (Knochen im Wachstum) anzutreffen ist, reduzieren bzw. eliminieren kann.

Summary and outlook 152
6 Summary and outlook
The bone is a common site of metastases for a number of different types of cancer, especially prostate and breast cancer. Today, standard options for the treatment of bone metastases include chemotherapy, hormone therapy and radiation therapy. However, the major drawbacks of these treatments are severe side effects in non-affected tissues. Chemotherapeutics, for instance, are usually applied orally or intravenously. As systemic drugs, they enter the bloodstream in order to reach cancer cells that have spread. Anticancer drugs kill not only cancer cells, but also damage healthy cells, thus leading to serious side effects. Therefore, special attention should be directed to avoiding or reducing side effects. As a result of a better understanding of bone biology and molecular processes involved in the development of bone metastases, several novel therapeutic targets have recently been identified to improve the treatment of bone metastases. New drugs have been developed based on these findings. However, the majority of the newly developed therapeutic agents do not possess sufficient bone specificity. In addition, their application in vivo is often limited due to suboptimal instability or insufficient pharmacokinetics. To overcome such drawbacks, the development of targeted drug delivery systems seems to be a promising approach. Past experience has shown that drugs with a low affinity to bone can be successfully delivered by coupling them to bisphosphonates to increase specificity to bone. Consisting of calcium chelating groups, bisphosphonates are supposed to bind to the bone surface by forming stable complexes with calcium derived from hydroxyapatite, the main component of the bone matrix. The successful demonstration of improving therapeutic efficacy by coupling bisphosphonates as bone-seeking ligand to drug molecules lacking bone specificity encouraged us to develop a bone-targeted drug delivery system composed of bisphosphonates bound to liposomes. The decision to use liposomes as a drug delivery system was driven by the fact that liposomes are considered advantageous in many respects. For example, liposomes are considered biodegradable, non-immunogenic, as well as non-toxic. Due to the encapsulation of drug molecules inside liposomes, harmful side effects may be reduced and, at the same time, the drug may be protected from degradation while circulating in the blood stream. We hypothesized that the therapeutic benefit may be further increased by encapsulating highly potent drugs in bisphosphonate-targeted liposomes, thus delivering a higher amount of drug to the desired site. In addition, a higher amount of ligand present on the liposomal surface would lead to increased

Summary and outlook 153
targeting efficiency compared to other existing bone-targeted systems whereby one drug molecule is coupled to one bisphosphonate ligand.
In this thesis, the successful development of bisphosphonate-targeted liposomes is described. The research project particularly focussed on the feasibility of incorporating a newly-synthesized, tailor-made bisphosphonate ligand into liposomal membranes including its evaluation as bone targeting device. The characterization of bisphosphonate-targeted liposomes comprised the determination of their physicochemical properties, in vitro binding studies to hydroxyapatite, pharmacokinetic studies, and the verification of the bone binding capability in vivo. The ligand was composed of a cholesteryl residue to be incorporated as an anchor into the liposomal membrane, a trisoxyethylene chain as spacer molecule and a bisphosphonic acid as targeting device.
Formulation development of bisphosphonate-targeted liposomes (section 4.1). In this section, the feasibility of preparing bisphosphonate-targeted liposomes is demonstrated. Different bisphosphonate ligands (Palmitoyl-bisphosphonic acid (PAL-BP), Cholesteryl-bisphosphonic acid (CHOL-BP), Cholesteryl-trisoxyethylene-bisphosphonic acid (CHOL-TOE-BP) and Cholesteryl-tetraoxyethylene-bisphosphonic acid (CHOL-TeOE-BP)) were incorporated into the membranes of liposomes varying in lipids (Egg-Phosphatidylcholine (EPC), Dipalmitoyl-phosphatidylcholine (DPPC)) by introducing them directly into the lipid film during liposome preparation. Visible precipitations indicated the failure of incorporation. Since we aimed for an intravenously injectable formulation, we tried to develop bisphosphonate-targeted liposomes with a physiological pH. To enable an uptake of bisphosphonate-targeted liposomes in metastasized bone tissue by selective extravasation through leaky endothelium (passive targeting), we aimed for an average particle size of 100–200 nm. In addition, small particles were considered essential in terms of achieving acceptable pharmacokinetic profiles. Based on these parameters, the most promising formulation was selected for further experiments. We decided to continue with DPPC liposomes containing CHOL-TOE-BP as targeting ligand.
Physicochemical characterization of bisphosphonate-targeted liposomes (section 4.2). Physicochemical properties of bisphosphonate-targeted liposomes with different CHOL-TOE-BP concentrations have been examined to study the influence of the ligand amount on these characteristics. In this respect, homogenous particle size distributions with an average particle size of 120–155 nm (PI: 0.03–0.15) were obtained. Furthermore, we were able to demonstrate that the stability (9 months) of

Summary and outlook 154
liposomes with regards to the average particle size was not negatively influenced by the incorporation of a CHOL-TOE-BP ligand into the membrane. The incorporation of a new ligand could lead to non-desired effects such as liposomal aggregation or bilayer fusion and thereby cause instabilities. The capability of binding to hydroxyapatite (HAP) was undoubtedly dependent on the CHOL-TOE-BP ligand density present on the liposomal surface. However, complete incorporation of the added substances, lipids and CHOL-TOE-BP ligand, into the liposomes was not guaranteed. A loss of material due to extrusion might have occurred for example. It was therefore important to determine the total concentration of CHOL-TOE-BP ligand in the liposomal formulation. Based on zeta-potential measurements, a ligand concentration-dependent influence on the surface charge could be determined. Higher concentrations of CHOL-TOE-BP led to more negative zeta-potential values. A 31P-NMR method was established to identify the incorporated amount of CHOL-TOE-BP. With this method, an incorporation rate of almost 100% could be determined, demonstrating successful ligand incorporation through using cholesterol as anchor molecule.
Binding of bisphosphonate-targeted liposomes to hydroxyapatite (section 4.3). The examination of the binding capability to hydroxyapatite (HAP) was an important study in the thesis, since the bisphosphonate-targeted liposomes were developed for drug delivery to bone. The binding studies were performed in vitro by using Isothermal Titration Calorimetry (ITC). ITC is a technique for monitoring chemical reactions initiated by the addition of bisphosphonate-targeted liposomes to a defined HAP suspension. As soon as the liposomes bound to HAP, heat was generated. The results obtained clearly showed the impact of the CHOL-TOE-BP ligand on liposome binding to HAP: in cases of higher CHOL-TOE-BP concentrations, a higher heat development was detected. To determine whether the surface charge of bisphosphonate-targeted liposomes had a relevant influence on binding, the HAP binding of highly negative-charged liposomes and weakly negative-charged control liposomes, which both lacked the CHOL-TOE-BP ligand, was examined. In both cases, no HAP binding could be observed, indicating that the CHOL-TOE-BP ligand plays an essential role in the binding of bisphosphonate-targeted liposomes to HAP. The evaluation of data from a thermodynamic point of view (determination of the reaction enthalpy and a binding constant) showed an increase in binding strength dependent on ligand concentration, thus leading to the conclusion that multivalent binding had taken place.

Summary and outlook 155
Drug loading of bisphosphonate-targeted liposomes (section 4.4). The drug-loading capacity of bisphosphonate-targeted liposomes was determined with regards to the aim of using them as a promising, alternative therapy option for the treatment of bone metastases. For this purpose, they were loaded with Doxorubicin as a model compound via an ammonium sulphate gradient. A loading capacity of almost 100% was obtained and during stability monitoring (7 days), a Doxorubicin release of only 5% was observed. In addition to loading efficiency, the release of the encapsulated drug at the target site is also a relevant parameter for the development of a promising drug delivery system. Release studies were therefore performed at different pH-values to consider the in vivo conditions that the bisphosphonate-targeted liposomes would encounter on their way to the bone surface. The results have clearly shown that a decrease of the extra-liposomal pH value led to increased Doxorubicin release from the tested liposomes.
Cytotoxicity of bisphosphonate-targeted liposomes (section 4.5). The determination of cytotoxicity in vitro is a good option for gauging the therapeutic efficacy of drug-loaded, bisphosphonate-targeted liposomes. In this respect, IC50-values of Doxorubicin-loaded, bisphosphonate-targeted liposomes in comparison to free Doxorubicin have been determined by crystal violet staining. The experiments have been performed with different prostate and breast cancer cell lines. The free drug generally showed a more toxic effect on the cells compared to the tested liposomes. This indicated the toxic effect caused by the free doxorubicin alone. Doxorubicin molecules that entered the cells in an encapsulated manner, or were released extracellularly, were obviously unable to achieve the same level of activity in the given time.
Pharmacokinetics and biodistributions of bisphosphonate-targeted liposomes (section 4.6). Pharmacokinetic studies including biodistributions considering the examination of bisphosphonate-targeted liposomes with different CHOL-TOE-BP concentrations have been performed to determine the influence of the negatively charged CHOL-TOE-BP ligand on the circulation time in blood. The CHOL-TOE-BP ligand again proved to have a relevant impact on circulation time kinetics. In cases of higher ligand concentrations, faster blood clearance was obtained although 4.3 mol% DSPE-PEG2000 was also present on the liposomal surface. The polymer DSPE-PEG2000 is known to prevent liposome opzonisation by forming a dense and protecting polymeric “cloud” over the liposome surface, thus leading to longer residence time in blood. We therefore examined the influence of a DSPE-PEG2000

Summary and outlook 156
increase (8.2 mol%, 13.0 mol% DSPE-PEG2000) on the pharmacokinetics of bisphosphonate-targeted liposomes with a higher ligand concentration (14.3 mol% CHOL-TOE-BP). The data demonstrated that better pharmacokinetics could be obtained by increasing DSPE-PEG2000 concentrations in bisphosphonate-targeted liposomes, although the observed kinetics were not as good as those of the non-targeted control liposomes containing 4.3 mol% DSPE-PEG2000. The attempt to prolong circulation-time kinetics by increasing the molecular weight of DSPE-PEG from 2000 to 5000 improved the circulation time to the same extend in comparison to the kinetics obtained by testing liposomes containing the same amount of DSPE-PEG2000 and the same amount of CHOL-TOE-BP ligand (14.3 mol%). However, promising results in terms of acceptable circulation-time profiles have been obtained that suggest further in vivo evaluation of the presented drug delivery system, although the negative influence of the CHOL-TOE-BP ligand could be demonstrated.
Bone-targeting of bisphosphonate-targeted liposomes (nude mouse model) (section 4.7). In this section, the bone-targeting capability of bisphosphonate-targeted liposomes was examined in a pilot experiment by using a well-established nude mouse model for bone metastases. Based on the results of the pharmacokinetic studies and the HAP-binding studies, we tried to select a promising formulation for bisphosphonate-targeted liposomes to obtain valuable results with this targeting experiment. The experiments have clearly shown that bisphosphonate-targeted liposomes composed of 14.3 mol% CHOL-TOE-BP and 4.3 mol% DSPE-PEG2000 accumulated to a high extend in metastasized bone tissue or bound to bone in this area. Control liposomes composed of 4.3 mol% DSPE-PEG2000 (without CHOL-TOE-BP ligand), on the other hand, did not accumulate in the affected bone tissue, or at least not in detectable concentrations, indicating that the “Enhanced Permeability Retention Effect” (EPR-Effect) did not play a relevant role in the observed accumulation or binding phenomenon. Fluorescent labelling of the tested liposomes allowed the detection of the liposomes in vivo in the living animal with the help of a multispectral camera system (Maestro In vivo Imaging System). The major drawback of the method was that the precise localisation in bone tissue could not be identified. Real binding to the bone surface could therefore not be determined. The fact that control liposomes did not show detectable accumulation in metastasized bone tissue indicates that the CHOL-TOE-BP ligand is apparently of great significance for accumulation or binding. It cannot therefore be ruled out that ligand-specific binding to the bone matrix took place.

Summary and outlook 157
Conclusion. In summary, this thesis can be considered to have provided the proof of concept. Tailor-made bisphosphonate-ligands have initially been successfully incorporated into the membrane of liposomes. An extensive characterization of these bisphosphonate-targeted liposomes and encouraging in vivo experiments, demonstrating their targeting potential to metastatic bone tissue, strongly recommend further investigation as a new therapy option for the treatment of bone metastases.
Outlook. The successful demonstration of the feasibility of combining bisphosphonate ligands and liposomes as a drug-carrier system may be considered a valuable contribution to the development of new therapy approaches for the treatment of bone metastases. The studies conducted provide an insight into new findings; but additional experiments should supply evidence that bisphosphonate-targeted liposomes effectively bind to the bone surface. The site of accumulation or binding could be determined by measuring the fluorescence of dissected tissue samples taken from bone metastases and bone matrix after sacrification of the animals. Alternatively, a radioactive label such as [3H]-Cholesteryl-oleylether could replace the fluorescent marker. Within the scope of these experiments, it would also be interesting to quantify the bone-bound liposome fraction to gauge the efficiency of the system by measuring the signal intensity of the used label before application and in the removed tissue samples. In the same experiment, the investigation of the influence of an increase in DSPE-PEG2000 concentration, or the impact of a particle size reduction, could be taken into consideration. Eventually, these modifications might lead to an increased binding of bisphosphonate-targeted liposomes to the bone surface. Experiments that clearly elucidate the therapeutic benefit of those liposomes in comparison to existing therapy options are also of particular importance. In this context, the effects of different encapsulated drugs on existing bone metastases and on the formation of metastases could be investigated. Bioluminescent Imaging could again be a suitable technique for this purpose. After successful activation of the luciferase gene in luciferase-transfected cells by the injection of the substrate luciferin, the emission of photons could indicate the extent of metastases growth. The detection and quantification of specific tumour markers, such cytokines, or angiogenic factors that are over expressed in the neoplastic tissue using Enzyme-linked ImmunoSorbent Assay (ELISA), would also be an option for identifying the stage of metastases development. Another interesting experiment would be the examination of liposomes characterized by lower CHOL-TOE-BP ligand densities on their surface to verify whether bone targeting also takes place although lower ligand quantities are present in the formulation. Possible binding to HAP in healthy bone tissue characterized by a

Summary and outlook 158
high bone turn over, due for example to normal growth, could thereby be reduced or even eliminated.

Literatur 159
7 Literatur
Abra, R. M. and Hunt, C. A., 1981. Liposome disposition in vivo. III. Dose and vesicle-size effects. Biochim Biophys Acta, 666, 493-503. Alakangas, A., Selander, K., Mulari, M., Halleen, J., Lehenkari, P., Monkkonen, J., Salo, J. and Vaananen, K., 2002. Alendronate disturbs vesicular trafficking in osteoclasts. Calcif Tissue Int, 70, 40-7. Allen, T. M., 1994. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol Sci, 15, 215-20. Allen, T. M. and Chonn, A., 1987. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett, 223, 42-6. Allen, T. M., Williamson, P. and Schlegel, R. A., 1988. Phosphatidylserine as a determinant of reticuloendothelial recognition of liposome models of the erythrocyte surface. Proc Natl Acad Sci U S A, 85, 8067-71. Allen, T. M., Hansen, C. and Rutledge, J., 1989. Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues. Biochim Biophys Acta, 981, 27-35. Allen, T. M., Brandeis, E., Hansen, C. B., Kao, G. Y. and Zalipsky, S., 1995. A new strategy for attachment of antibodies to sterically stabilized liposomes resulting in efficient targeting to cancer cells. Biochim Biophys Acta, 1237, 99-108. Anabousi, S., Kleemann, E., Bakowsky, U., Kissel, T., Schmehl, T., Gessler, T., Seeger, W., Lehr, C. M. and Ehrhardt, C., 2006. Effect of PEGylation on the stability of liposomes during nebulisation and in lung surfactant. J Nanosci Nanotechnol, 6, 3010-6. Aparicio, A., Gardner, A., Tu, Y., Savage, A., Berenson, J. and Lichtenstein, A., 1998. In vitro cytoreductive effects on multiple myeloma cells induced by bisphosphonates. Leukemia, 12, 220-9. Arguello, F., Baggs, R. B. and Frantz, C. N., 1988. A murine model of experimental metastasis to bone and bone marrow. Cancer Res, 48, 6876-81. Arnold, K., Zschoernig, O., Barthel, D. and Herold, W., 1990. Exclusion of poly(ethylene glycol) from liposome surfaces. Biochim Biophys Acta, 1022, 303-10. Bangham, A. D. and Horne, R. W., 1964. Negative Staining of Phospholipids and Their Structural Modification by Surface-Active Agents as Observed in the Electron Microscope. J Mol Biol, 12, 660-8. Baron, R., Vignery, A. and Horowitz, M. (1983) In Bone and Mineral Research, Vol. Annual 2 (Ed, Peck, W. A.) Elsevier, Amsterdam, pp. 175-242. Bartl, R., 2001. Bisphosphonate. Mannual Supportive Maßnahmen und symptomorientierte Therapie, 184-90. Bauss, F., Esswein, A., Reiff, K., Sponer, G. and Muller-Beckmann, B., 1996. Effect of 17beta-estradiol-bisphosphonate conjugates, potential bone-seeking estrogen pro-drugs, on 17beta-estradiol serum kinetics and bone mass in rats. Calcif Tissue Int, 59, 168-73.

Literatur 160
Bayouth, J. E., Macey, D. J., Kasi, L. P. and Fossella, F. V., 1994. Dosimetry and toxicity of samarium-153-EDTMP administered for bone pain due to skeletal metastases. J Nucl Med, 35, 63-9. Bendas, G., Rothe, U., Scherphof, G. L. and Kamps, J. A., 2003. The influence of repeated injections on pharmacokinetics and biodistribution of different types of sterically stabilized immunoliposomes. Biochim Biophys Acta, 1609, 63-70. Beschiaschvili, G. and Seelig, J., 1992. Peptide binding to lipid bilayers. Nonclassical hydrophobic effect and membrane-induced pK shifts. Biochemistry, 31, 10044-53. Blair, H. C., Teitelbaum, S. L., Ghiselli, R. and Gluck, S., 1989. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science, 245, 855-7. Blake, G. M., Gray, J. M., Zivanovic, M. A., McEwan, A. J., Fleming, J. S. and Ackery, D. M., 1987. Strontium-89 radionuclide therapy: a dosimetric study using impulse response function analysis. Br J Radiol, 60, 685-92. Blake, G. M., Zivanovic, M. A., Blaquiere, R. M., Fine, D. R., McEwan, A. J. and Ackery, D. M., 1988. Strontium-89 therapy: measurement of absorbed dose to skeletal metastases. J Nucl Med, 29, 549-57. Blume, G. and Cevc, G., 1990. Liposomes for the sustained drug release in vivo. Biochim Biophys Acta, 1029, 91-7. Bonucci, E., 2002. Physiopathology of cancer metastases in bone and of the changes they induce in bone remodeling. Rend. Fiss. Acc. Lincei, 13, 181-246. Bossard, M. J., Tomaszek, T. A., Thompson, S. K., Amegadzie, B. Y., Hanning, C. R., Jones, C., Kurdyla, J. T., McNulty, D. E., Drake, F. H., Gowen, M. and Levy, M. A., 1996. Proteolytic activity of human osteoclast cathepsin K. Expression, purification, activation, and substrate identification. J Biol Chem, 271, 12517-24. Boulenc, X., Marti, E., Joyeux, H., Roques, C., Berger, Y. and Fabre, G., 1993. Importance of the paracellular pathway for the transport of a new bisphosphonate using the human CACO-2 monolayers model. Biochem Pharmacol, 46, 1591-600. Bozec, L., de Groot, J., Odlyha, M., Nicholls, B. and Horton, M. A., 2005. Mineralised tissues as nanomaterials: analysis by atomic force microscopy. IEE Proc Nanobiotechnol, 152, 183-6. Buckwalter, J. A., Glimcher, M. J., Cooper, R. R. and Recker, R., 1996. Bone biology. II: Formation, form, modeling, remodeling, and regulation of cell function. Instr Course Lect, 45, 387-99. Carmeliet, P., 2000. Mechanisms of angiogenesis and arteriogenesis. Nat Med, 6, 389-95. Carmeliet, P., 2003. Angiogenesis in health and disease. Nat Med, 9, 653-60. Carrion, C., Domingo, J. C. and de Madariaga, M. A., 2001. Preparation of long-circulating immunoliposomes using PEG-cholesterol conjugates: effect of the spacer arm between PEG and cholesterol on liposomal characteristics. Chem Phys Lipids, 113, 97-110. Castro, C., Sanchez, E., Delgado, A., Soriano, I., Nunez, P., Baro, M., Perera, A. and Evora, C., 2003. Ciprofloxacin implants for bone infection. In vitro-in vivo characterization. J Control Release, 93, 341-54.

Literatur 161
Cheung, B. C., Sun, T. H., Leenhouts, J. M. and Cullis, P. R., 1998. Loading of doxorubicin into liposomes by forming Mn2+-drug complexes. Biochim Biophys Acta, 1414, 205-16. Choi, S. W. and Kim, J. H., 2007. Design of surface-modified poly (d,l-lactide-co-glycolide) nanoparticles for targeted drug delivery to bone. J Control Release, 122, 24-30. Clezardin, P., 2002. The antitumor potential of bisphosphonates. Semin Oncol, 29, 33-42. Clohisy, D. R., PF, O. K. and Ramnaraine, M. L., 2001. Pamidronate decreases tumor-induced osteoclastogenesis in osteopetrotic mice. J Orthop Res, 19, 554-8. Collins, D., Litzinger, D. C. and Huang, L., 1990. Structural and functional comparisons of pH-sensitive liposomes composed of phosphatidylethanolamine and three different diacylsuccinylglycerols. Biochim Biophys Acta, 1025, 234-42. Connolly, D. T., Heuvelman, D. M., Nelson, R., Olander, J. V., Eppley, B. L., Delfino, J. J., Siegel, N. R., Leimgruber, R. M. and Feder, J., 1989. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest, 84, 1470-8. Cukrowski, I., Popovic, L., Barnard, W., Paul, S. O., van Rooyen, P. H. and Liles, D. C., 2007. Modeling and spectroscopic studies of bisphosphonate-bone interactions. The Raman, NMR and crystallographic investigations of Ca-HEDP complexes. Bone, 41668-78. Cullis, P. R. and de Kruijff, B., 1979. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim Biophys Acta, 559, 399-420. Daley-Yates, P. T., Gifford, L. A. and Hoggarth, C. R., 1989. Assay of 1-hydroxy-3-aminopropylidene-1,1-bisphosphonate and related bisphosphonates in human urine and plasma by high-performance ion chromatography. J Chromatogr, 490, 329-38. Dams, E. T., Laverman, P., Oyen, W. J., Storm, G., Scherphof, G. L., van Der Meer, J. W., Corstens, F. H. and Boerman, O. C., 2000. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther, 292, 1071-9. de Bruyn, J. H., Man in't Veld, A. J., Wenting, G. J., Derkx, F. H. and Schalekamp, M. A., 1981. Haemodynamic profile of captopril treatment in various forms of hypertension. Eur J Clin Pharmacol, 20, 163-8. De Bruyn, P. P., 1981. Structural substrates of bone marrow function. Semin Hematol, 18, 179-93. de Oliveira, M. C., Rosilio, V., Lesieur, P., Bourgaux, C., Couvreur, P., Ollivon, M. and Dubernet, C., 2000. pH-sensitive liposomes as a carrier for oligonucleotides: a physico-chemical study of the interaction between DOPE and a 15-mer oligonucleotide in excess water. Biophys Chem, 87, 127-37. Degim, I. T., Gumusel, B., Degim, Z., Ozcelikay, T., Tay, A. and Guner, S., 2006. Oral administration of liposomal insulin. J Nanosci Nanotechnol, 6, 2945-9. Derenne, S., Amiot, M., Barille, S., Collette, M., Robillard, N., Berthaud, P., Harousseau, J. L. and Bataille, R., 1999. Zoledronate is a potent inhibitor of myeloma cell growth and secretion of IL-6 and MMP-1 by the tumoral environment. J Bone Miner Res, 14, 2048-56. Diaz-Flores, L., Gutierrez, R. and Varela, H., 1994. Angiogenesis: an update. Histol Histopathol, 9, 807-43.

Literatur 162
Drake, F. H., Dodds, R. A., James, I. E., Connor, J. R., Debouck, C., Richardson, S., Lee-Rykaczewski, E., Coleman, L., Rieman, D., Barthlow, R., Hastings, G. and Gowen, M., 1996. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem, 271, 12511-6. Ebetino, F. H., Francis, M. D., Rogers, M. J. and Russell, R. G. G., 1998. Etidronate. Mechanisms of action of etidronate and other bisphosphonates. Rev Contemp Pharma, 9, 233-43. Edinger, M., Sweeney, T. J., Tucker, A. A., Olomu, A. B., Negrin, R. S. and Contag, C. H., 1999. Noninvasive assessment of tumor cell proliferation in animal models. Neoplasia, 1, 303-10. Einhorn, T. A. (1996) In Osteoporosis(Ed, Kelsey, J.) Academic Press, San Diego, California. Emanuel, N., Kedar, E., Bolotin, E. M., Smorodinsky, N. I. and Barenholz, Y., 1996. Preparation and characterization of doxorubicin-loaded sterically stabilized immunoliposomes. Pharm Res, 13, 352-9. Evans, C. W., 1992. Cell adhesion and metastasis. Cell Biol Int Rep, 16, 1-10. Ewing, J. A. (1928) Treatise on tumors, WB Saunders, Philadelphia. Fleisch, H., 1998. Bisphosphonates: mechanisms of action. Endocr Rev, 19, 80-100. Fleisch, H., 2002. Development of bisphosphonates. Breast Cancer Res, 4, 30-4. Fleisch, H. and Neuman, W., 1961. [The role of phosphatase and polyphosphates in calcification of collagen.]. Helv Physiol Pharmacol Acta, 19, C17-8. Fleisch, H. and Bisaz, S., 1962. Isolation from urine of pyrophosphate, a calcification inhibitor. Am J Physiol, 203, 671-5. Fleisch, H., Maerki, J. and Russell, R. G., 1966. Effect of pyrophosphate on dissolution of hydroxyapatite and its possible importance in calcium homeostasis. Proc Soc Exp Biol Med, 122, 317-20. Fleisch, H. A., Russell, R. G., Bisaz, S., Muhlbauer, R. C. and Williams, D. A., 1970. The inhibitory effect of phosphonates on the formation of calcium phosphate crystals in vitro and on aortic and kidney calcification in vivo. Eur J Clin Invest, 1, 12-8. Folkman, J., 1971. Tumor angiogenesis: therapeutic implications. N Engl J Med, 285, 1182-6. Folkman, J., 1975. Tumor angiogenesis: a possible control point in tumor growth. Ann Intern Med, 82, 96-100. Fournier, P., Boissier, S., Filleur, S., Guglielmi, J., Cabon, F., Colombel, M. and Clezardin, P., 2002. Bisphosphonates inhibit angiogenesis in vitro and testosterone-stimulated vascular regrowth in the ventral prostate in castrated rats. Cancer Res, 62, 6538-44. Fox, C. B., Uibel, R. H. and Harris, J. M., 2007. Detecting phase transitions in phosphatidylcholine vesicles by Raman microscopy and self-modeling curve resolution. J Phys Chem B, 111, 11428-36. Fresta, M., Panico, A. M., Bucolo, C., Giannavola, C. and Puglisi, G., 1999. Characterization and in-vivo ocular absorption of liposome-encapsulated acyclovir. J Pharm Pharmacol, 51, 565-76. Frith, J. C. and Rogers, M. J., 2003. Antagonistic effects of different classes of bisphosphonates in osteoclasts and macrophages in vitro. J Bone Miner Res, 18, 204-12.

Literatur 163
Fritze, A., Hens, F., Kimpfler, A., Schubert, R. and Peschka-Suss, R., 2006. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim Biophys Acta, 1758, 1633-40. Fromigue, O., Lagneaux, L. and Body, J. J., 2000. Bisphosphonates induce breast cancer cell death in vitro. J Bone Miner Res, 15, 2211-21. Frost, H. M. (1964) In Bone BiodynamicsLittle, Brown, Boston, MA. Gabizon, A. and Papahadjopoulos, D., 1992. The role of surface charge and hydrophilic groups on liposome clearance in vivo. Biochim Biophys Acta, 1103, 94-100. Gabizon, A., Catane, R., Uziely, B., Kaufman, B., Safra, T., Cohen, R., Martin, F., Huang, A. and Barenholz, Y., 1994. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res, 54, 987-92. Ghandehari, H., Smith, P. L., Ellens, H., Yeh, P. Y. and Kopecek, J., 1997. Size-dependent permeability of hydrophilic probes across rabbit colonic epithelium. J Pharmacol Exp Ther, 280, 747-53. Gil, L., Han, Y., Opas, E. E., Rodan, G. A., Ruel, R., Seedor, J. G., Tyler, P. C. and Young, R. N., 1999. Prostaglandin E2-bisphosphonate conjugates: potential agents for treatment of osteoporosis. Bioorg Med Chem, 7, 901-19. Gill, P. S., Espina, B. M., Muggia, F., Cabriales, S., Tulpule, A., Esplin, J. A., Liebman, H. A., Forssen, E., Ross, M. E. and Levine, A. M., 1995. Phase I/II clinical and pharmacokinetic evaluation of liposomal daunorubicin. J Clin Oncol, 13, 996-1003. Gill, P. S., Wernz, J., Scadden, D. T., Cohen, P., Mukwaya, G. M., von Roenn, J. H., Jacobs, M., Kempin, S., Silverberg, I., Gonzales, G., Rarick, M. U., Myers, A. M., Shepherd, F., Sawka, C., Pike, M. C. and Ross, M. E., 1996. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol, 14, 2353-64. Gittens, S. A., Bansal, G., Zernicke, R. F. and Uludag, H., 2005. Designing proteins for bone targeting. Adv Drug Deliv Rev, 57, 1011-36. Gober, H. J., Kistowska, M., Angman, L., Jeno, P., Mori, L. and De Libero, G., 2003. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med, 197, 163-8. Goeckeler, W. F., Edwards, B., Volkert, W. A., Holmes, R. A., Simon, J. and Wilson, D., 1987. Skeletal localization of samarium-153 chelates: potential therapeutic bone agents. J Nucl Med, 28, 495-504. Gonzalez, K. A., Wilson, L. J., Wu, W. and Nancollas, G. H., 2002. Synthesis and in vitro characterization of a tissue-selective fullerene: vectoring C(60)(OH)(16)AMBP to mineralized bone. Bioorg Med Chem, 10, 1991-7. Green, N. M., 1975. Avidin. Adv Protein Chem, 29, 85-133. Greenlee, R. T., Hill-Harmon, M. B., Murray, T. and Thun, M., 2001. Cancer statistics, 2001. CA Cancer J Clin, 51, 15-36. Guise, T. A. and Mundy, G. R., 1998. Cancer and bone. Endocr Rev, 19, 18-54.

Literatur 164
Guise, T. A., Kozlow, W. M., Heras-Herzig, A., Padalecki, S. S., Yin, J. J. and Chirgwin, J. M., 2005. Molecular mechanisms of breast cancer metastases to bone. Clin Breast Cancer, 5 Suppl, S46-53. Haran, G., Cohen, R., Bar, L. K. and Barenholz, Y., 1993. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta, 1151, 201-15. Hatakeyama, H., Akita, H., Ishida, E., Hashimoto, K., Kobayashi, H., Aoki, T., Yasuda, J., Obata, K., Kikuchi, H., Ishida, T., Kiwada, H. and Harashima, H., 2007. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int J Pharm, 342, 194-200. Havens, A. M., Jung, Y., Sun, Y. X., Wang, J., Shah, R. B., Buhring, H. J., Pienta, K. J. and Taichman, R. S., 2006. The role of sialomucin CD164 (MGC-24v or endolyn) in prostate cancer metastasis. BMC Cancer, 6, 195. Heerklotz, H., 2004. Microcalorimetry of lipid membranes. Journal of Physics: Condensed Matter 16, R441-R467. Hengst, V., Oussoren, C., Kissel, T. and Storm, G., 2007. Bone targeting potential of bisphosphonate-targeted liposomes Preparation, characterization and hydroxyapatite binding in vitro. Int J Pharm, 331, 224-227. Herczegh, P., Buxton, T. B., McPherson, J. C., 3rd, Kovacs-Kulyassa, A., Brewer, P. D., Sztaricskai, F., Stroebel, G. G., Plowman, K. M., Farcasiu, D. and Hartmann, J. F., 2002. Osteoadsorptive bisphosphonate derivatives of fluoroquinolone antibacterials. J Med Chem, 45, 2338-41. Higashi, N. and Sunamoto, J., 1995. Endocytosis of poly(ethylene oxide)-modified liposome by human lymphoblastoid cells. Biochim Biophys Acta, 1243, 386-92. Hirabayashi, H. and Fujisaki, J., 2003. Bone-specific drug delivery systems: approaches via chemical modification of bone-seeking agents. Clin Pharmacokinet, 42, 1319-30. Hirabayashi, H., Takahashi, T., Fujisaki, J., Masunaga, T., Sato, S., Hiroi, J., Tokunaga, Y., Kimura, S. and Hata, T., 2001. Bone-specific delivery and sustained release of diclofenac, a non-steroidal anti-inflammatory drug, via bisphosphonic prodrug based on the Osteotropic Drug Delivery System (ODDS). J Control Release, 70, 183-91. Hoang, Q. Q., Sicheri, F., Howard, A. J. and Yang, D. S., 2003. Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature, 425, 977-80. Hobbs, S. K., Monsky, W. L., Yuan, F., Roberts, W. G., Griffith, L., Torchilin, V. P. and Jain, R. K., 1998. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A, 95, 4607-12. Hong, M. S., Lim, S. J., Oh, Y. K. and Kim, C. K., 2002. pH-sensitive, serum-stable and long-circulating liposomes as a new drug delivery system. J Pharm Pharmacol, 54, 51-8. Hosain, F., Spencer, R. P., Couthon, H. M. and Sturtz, G. L., 1996. Targeted delivery of antineoplastic agent to bone: biodistribution studies of technetium-99m-labeled gem-bisphosphonate conjugate of methotrexate. J Nucl Med, 37, 105-7. Hoskin, P. J., Price, P., Easton, D., Regan, J., Austin, D., Palmer, S. and Yarnold, J. R., 1992. A prospective randomised trial of 4 Gy or 8 Gy single doses in the treatment of metastatic bone pain. Radiother Oncol, 23, 74-8.

Literatur 165
Howe, A., Aplin, A. E., Alahari, S. K. and Juliano, R. L., 1998. Integrin signaling and cell growth control. Curr Opin Cell Biol, 10, 220-31. Howlett, C. R., Dickson, M. and Sheridan, A. K., 1984. The fine structure of the proximal growth plate of the avian tibia: vascular supply. J Anat, 139 ( Pt 1), 115-32. Huang, S. K., Mayhew, E., Gilani, S., Lasic, D. D., Martin, F. J. and Papahadjopoulos, D., 1992. Pharmacokinetics and therapeutics of sterically stabilized liposomes in mice bearing C-26 colon carcinoma. Cancer Res, 52, 6774-81. Hughes, D. E., Wright, K. R., Uy, H. L., Sasaki, A., Yoneda, T., Roodman, G. D., Mundy, G. R. and Boyce, B. F., 1995. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J Bone Miner Res, 10, 1478-87. Hwang, K. J., Padki, M. M., Chow, D. D., Essien, H. E., Lai, J. Y. and Beaumier, P. L., 1987. Uptake of small liposomes by non-reticuloendothelial tissues. Biochim Biophys Acta, 901, 88-96. Ishiwata, H., Vertut-Doi, A., Hirose, T. and Miyajima, K., 1995. Physical-chemistry characteristics and biodistribution of poly(ethylene glycol)-coated liposomes using poly(oxyethylene) cholesteryl ether. Chem Pharm Bull (Tokyo), 43, 1005-11. Itokazu, M., Sugiyama, T., Ohno, T., Wada, E. and Katagiri, Y., 1998. Development of porous apatite ceramic for local delivery of chemotherapeutic agents. J Biomed Mater Res, 39, 536-8. Jain, R. K., 2003. Molecular regulation of vessel maturation. Nat Med, 9, 685-93. Janjan, N. A., 1997. Radiation for bone metastases: conventional techniques and the role of systemic radiopharmaceuticals. Cancer, 80, 1628-45. Jung, A., Bisaz, S., Bartholdi, P. and Fleisch, H., 1973. Influence of pyrophosphate on the exchange of calcium and phosphate ions on hydroxyapatite. Calcif Tissue Res, 13, 27-40. Kaiser, N., Kimpfler, A., Massing, U., Burger, A. M., Fiebig, H. H., Brandl, M. and Schubert, R., 2003. 5-Fluorouracil in vesicular phospholipid gels for anticancer treatment: entrapment and release properties. Int J Pharm, 256, 123-31. Kalluri, R., 2003. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer, 3, 422-33. Karanth, H. and Murthy, R. S., 2007. pH-sensitive liposomes--principle and application in cancer therapy. J Pharm Pharmacol, 59, 469-83. Kasting, G. B. and Francis, M. D., 1992. Retention of etidronate in human, dog, and rat. J Bone Miner Res, 7, 513-22. Kasugai, S., Fujisawa, R. and Waki, Y., 2000. Selective drug delivery systems to bone: small peptide (Asp)6 conjugation. J Bone Miner Res., 15, 936-43. Kaye, S. B. and Richardson, V. J., 1979. Potential of liposomes as drug-carriers in cancer chemotherapy: a review. Cancer Chemother Pharmacol, 3, 81-5. Kirby, C., Clarke, J. and Gregoriadis, G., 1980. Cholesterol content of small unilamellar liposomes controls phospholipid loss to high density lipoproteins in the presence of serum. FEBS Lett, 111, 324-8.

Literatur 166
Kirpotin, D., Park, J. W., Hong, K., Zalipsky, S., Li, W. L., Carter, P., Benz, C. C. and Papahadjopoulos, D., 1997. Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry, 36, 66-75. Kleemann, E., Schmehl, T., Gessler, T., Bakowsky, U., Kissel, T. and Seeger, W., 2007. Iloprost-containing liposomes for aerosol application in pulmonary arterial hypertension: formulation aspects and stability. Pharm Res, 24, 277-87. Klenner, T., Valenzuela-Paz, P., Keppler, B. K., Angres, G., Scherf, H. R., Wingen, F., Amelung, F. and Schmahl, D., 1990. Cisplatin-linked phosphonates in the treatment of the transplantable osteosarcoma in vitro and in vivo. Cancer Treat Rev, 17, 253-9. Klibanov, A. L., Maruyama, K., Beckerleg, A. M., Torchilin, V. P. and Huang, L., 1991. Activity of amphipathic poly(ethylene glycol) 5000 to prolong the circulation time of liposomes depends on the liposome size and is unfavorable for immunoliposome binding to target. Biochim Biophys Acta, 1062, 142-8. Konigsberg, P. J., Godtel, R., Kissel, T. and Richer, L. L., 1998. The development of IL-2 conjugated liposomes for therapeutic purposes. Biochim Biophys Acta, 1370, 243-51. Krupova, Y., Mistrik, M., Bojtarova, E., Sejnova, D., Ilavska, I. and Krcmery, V., Jr., 2001. Liposomal nystatin (L-NYS) in therapy of pulmonary aspergillosis refractory to conventional amphotericin B in cancer patients. Support Care Cancer, 9, 209-10. Lasic, D. D., Martin, F. J., Gabizon, A., Huang, S. K. and Papahadjopoulos, D., 1991. Sterically stabilized liposomes: a hypothesis on the molecular origin of the extended circulation times. Biochim Biophys Acta, 1070, 187-92. Lasic, D. D., Ceh, B., Stuart, M. C., Guo, L., Frederik, P. M. and Barenholz, Y., 1995. Transmembrane gradient driven phase transitions within vesicles: lessons for drug delivery. Biochim Biophys Acta, 1239, 145-56. Lasseter, K. C., Porras, A. G., Denker, A., Santhanagopal, A. and Daifotis, A., 2005. Pharmacokinetic considerations in determining the terminal elimination half-lives of bisphosphonates. Clin Drug Investig, 25, 107-14. Lee, R. J. and Low, P. S., 1995. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta, 1233, 134-44. Lehenkari, P. P., Kellinsalmi, M., Napankangas, J. P., Ylitalo, K. V., Monkkonen, J., Rogers, M. J., Azhayev, A., Vaananen, H. K. and Hassinen, I. E., 2002. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol Pharmacol, 61, 1255-62. Levchenko, T. S., Rammohan, R., Lukyanov, A. N., Whiteman, K. R. and Torchilin, V. P., 2002. Liposome clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. Int J Pharm, 240, 95-102. Lewington, V. J., 1993. Targeted radionuclide therapy for bone metastases. Eur J Nucl Med, 20, 66-74. Li, X., Hirsh, D. J., Cabral-Lilly, D., Zirkel, A., Gruner, S. M., Janoff, A. S. and Perkins, W. R., 1998. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim Biophys Acta, 1415, 23-40.

Literatur 167
Lin, J. H., 1996. Bisphosphonates: a review of their pharmacokinetic properties. Bone, 18, 75-85. Liu, D., Mori, A. and Huang, L., 1992. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim Biophys Acta, 1104, 95-101. Liu, D., Hu, Q. and Song, Y. K., 1995. Liposome clearance from blood: different animal species have different mechanisms. Biochim Biophys Acta, 1240, 277-84. Lorusso, D., Di Stefano, A., Carone, V., Fagotti, A., Pisconti, S. and Scambia, G., 2007. Pegylated liposomal doxorubicin-related palmar-plantar erythrodysesthesia ('hand-foot' syndrome). Ann Oncol, 18, 1159-64. Luckman, S. P., Coxon, F. P., Ebetino, F. H., Russell, R. G. and Rogers, M. J., 1998a. Heterocycle-containing bisphosphonates cause apoptosis and inhibit bone resorption by preventing protein prenylation: evidence from structure-activity relationships in J774 macrophages. J Bone Miner Res, 13, 1668-78. Luckman, S. P., Hughes, D. E., Coxon, F. P., Graham, R., Russell, G. and Rogers, M. J., 1998b. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res, 13, 581-9. Lundberg, B., Hong, K. and Papahadjopoulos, D., 1993. Conjugation of apolipoprotein B with liposomes and targeting to cells in culture. Biochim Biophys Acta, 1149, 305-12. Luque, I. and Freire, E., 2002. Structural parameterization of the binding enthalpy of small ligands. Proteins, 49, 181-90. Mackie, P. S., Fisher, J. L., Zhou, H. and Choong, P. F., 2001. Bisphosphonates regulate cell growth and gene expression in the UMR 106-01 clonal rat osteosarcoma cell line. Br J Cancer, 84, 951-8. Maeda, H. and Matsumura, Y., 1989. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst, 6, 193-210. Maitani, Y., Igarashi, S., Sato, M. and Hattori, Y., 2007. Cationic liposome (DC-Chol/DOPE=1:2) and a modified ethanol injection method to prepare liposomes, increased gene expression. Int J Pharm, 342, 33-9. Marcus, R. (1994) In Osteoporosis(Ed, Kanis, J. A.) Blackwell Science Ltd., Oxford, England. Martin, F. J. (1998) In Medical Applications of Liposomes(Ed, Papahadjopoulos, D.) Elsevier Science BV, New York, pp. 635-688. Martin, R. B., 2003. Fatigue microdamage as an essential element of bone mechanics and biology. Calcif Tissue Int, 73, 101-7. Mastrobattista, E., Crommelin, D. J., Wilschut, J. and Storm, G., 2002. Targeted liposomes for delivery of protein-based drugs into the cytoplasm of tumor cells. J Liposome Res, 12, 57-65. Matsumura, Y. and Maeda, H., 1986. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-92. Mayer, L. D., Bally, M. B. and Cullis, P. R., 1986. Uptake of adriamycin into large unilamellar vesicles in response to a pH gradient. Biochim Biophys Acta, 857, 123-6.

Literatur 168
Mayer, L. D., Tai, L. C., Bally, M. B., Mitilenes, G. N., Ginsberg, R. S. and Cullis, P. R., 1990. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta, 1025, 143-51. Midura, R. J., Wang, A., Lovitch, D., Law, D., Powell, K. and Gorski, J. P., 2004. Bone acidic glycoprotein-75 delineates the extracellular sites of future bone sialoprotein accumulation and apatite nucleation in osteoblastic cultures. J Biol Chem, 279, 25464-73. Misra, D. N., 1991. Adsorption and orientation of tetracycline on hydroxyapatite. Calcif Tissue Int, 48, 362-7. Mizushima, Y., Ikoma, T., Tanaka, J., Hoshi, K., Ishihara, T., Ogawa, Y. and Ueno, A., 2006. Injectable porous hydroxyapatite microparticles as a new carrier for protein and lipophilic drugs. J Control Release, 110, 260-5. Mocellin, S., Wang, E. and Marincola, F. M., 2001. Cytokines and immune response in the tumor microenvironment. J Immunother, 24, 392-407. Moghimi, S. M. and Hunter, A. C., 2001. Recognition by macrophages and liver cells of opsonized phospholipid vesicles and phospholipid headgroups. Pharm Res, 18, 1-8. Moghimi, S. M., Hedeman, H., Muir, I. S., Illum, L. and Davis, S. S., 1993. An investigation of the filtration capacity and the fate of large filtered sterically-stabilized microspheres in rat spleen. Biochim Biophys Acta, 1157, 233-40. Monkkonen, H., Auriola, S., Lehenkari, P., Kellinsalmi, M., Hassinen, I. E., Vepsalainen, J. and Monkkonen, J., 2006. A new endogenous ATP analog (ApppI) inhibits the mitochondrial adenine nucleotide translocase (ANT) and is responsible for the apoptosis induced by nitrogen-containing bisphosphonates. Br J Pharmacol, 147, 437-45. Monkkonen, J., 1988. A one year follow-up study of the distribution of 14C-clodronate in mice and rats. Pharmacol Toxicol, 62, 51-3. Monkkonen, J., Koponen, H. M. and Ylitalo, P., 1990. Comparison of the distribution of three bisphosphonates in mice. Pharmacol Toxicol, 66, 294-8. Mori, A., Klibanov, A. L., Torchilin, V. P. and Huang, L., 1991. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett, 284, 263-6. Mundy, G. R. and Yoneda, T., 1995. Facilitation and suppression of bone metastasis. Clin Orthop Relat Res, 34-44. Muramatsu, K., Maitani, Y., Takayama, K. and Nagai, T., 1999. The relationship between the rigidity of the liposomal membrane and the absorption of insulin after nasal administration of liposomes modified with an enhancer containing insulin in rabbits. Drug Dev Ind Pharm, 25, 1099-105. Murgia, S., Mele, S. and Monduzzi, M., 2003. Quantitative characterization of phospholipids in milk fat via 31P NMR using a monophasic solvent mixture. Lipids, 38, 585-91. Murphy, M. B., Hartgerink, J. D., Goepferich, A. and Mikos, A. G., 2007. Synthesis and in vitro hydroxyapatite binding of peptides conjugated to calcium-binding moieties. Biomacromolecules, 8, 2237-43.

Literatur 169
Nancollas, G. H., Tang, R., Phipps, R. J., Henneman, Z., Gulde, S., Wu, W., Mangood, A., Russell, R. G. and Ebetino, F. H., 2005. Novel insights into actions of bisphosphonates on bone: Differences in interactions with hydroxyapatite. Bone, 38, 617-27. Needham, D., McIntosh, T. J. and Lasic, D. D., 1992. Repulsive interactions and mechanical stability of polymer-grafted lipid membranes. Biochim Biophys Acta, 1108, 40-8. Nguyen, M., Arkell, J. and Jackson, C. J., 2001. Human endothelial gelatinases and angiogenesis. Int J Biochem Cell Biol, 33, 960-70. Nicolson, G. L., 1993. Growth mechanisms and cancer progression. Hosp Pract (Off Ed), 28, 43-53. Nikitenko, L. and Boshoff, C., 2006. Endothelial cells and cancer. Handb Exp Pharmacol, 307-34. Oku, N., Tokudome, Y., Koike, C., Nishikawa, N., Mori, H., Saiki, I. and Okada, S., 1996. Liposomal Arg-Gly-Asp analogs effectively inhibit metastatic B16 melanoma colonization in murine lungs. Life Sci, 58, 2263-70. Okubo, Y., Bessho, K., Fujimura, K., Kusumoto, K., Ogawa, Y. and Iizuka, T., 2000. Osteogenesis by recombinant human bone morphogenetic protein-2 at skeletal sites. Clin Orthop Relat Res, 295-301. Orme, M. W. and Labroo, V. M., 1994. Synthesis of â-estradiol-3-benzoate-17-(succinyl-12á-tetracycline): a potential bone-seeking estrogen. Bioorg Med Chem Lett, 4, 1375-80. Oussoren, C., Eling, W. M., Crommelin, D. J., Storm, G. and Zuidema, J., 1998. The influence of the route of administration and liposome composition on the potential of liposomes to protect tissue against local toxicity of two antitumor drugs. Biochim Biophys Acta, 1369, 159-72. Overbeck, J. P., Winckler, S. T., Meffert, R., Tormala, P., Spiegel, H. U. and Brug, E., 1995. Penetration of ciprofloxacin into bone: a new bioabsorbable implant. J Invest Surg, 8, 155-62. Pagano, R. E. and Weinstein, J. N., 1978. Interactions of liposomes with mammalian cells. Annu Rev Biophys Bioeng, 7, 435-68. Paget, S., 1989. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev, 8, 98-101. Papahadjopoulos, D., 1968. Surface properties of acidic phospholipids: interaction of monolayers and hydrated liquid crystals with uni- and bi-valent metal ions. Biochim Biophys Acta, 163, 240-54. Perrin, D. D., 1965. Binding of tetracyclines to bone. Nature, 208, 787-8. Peyruchaud, O., Serre, C. M., NicAmhlaoibh, R., Fournier, P. and Clezardin, P., 2003. Angiostatin inhibits bone metastasis formation in nude mice through a direct anti-osteoclastic activity. J Biol Chem, 278, 45826-32. Pierce, W. M., Jr. and Waite, L. C., 1987. Bone-targeted carbonic anhydrase inhibitors: effect of a proinhibitor on bone resorption in vitro. Proc Soc Exp Biol Med, 186, 96-102. Pugliese, L., Malcovati, M., Coda, A. and Bolognesi, M., 1994. Crystal structure of apo-avidin from hen egg-white. J Mol Biol, 235, 42-6. Raisz, L. G., 1999. Physiology and pathophysiology of bone remodeling. Clin Chem, 45, 1353-8.

Literatur 170
Raubenheimer, E. J. and Noffke, C. E., 2006. Pathogenesis of bone metastasis: a review. J Oral Pathol Med, 35, 129-35. Ribatti, D., Mangialardi, G. and Vacca, A., 2006. Stephen Paget and the 'seed and soil' theory of metastatic dissemination. Clin Exp Med, 6, 145-9. Riebeling, C., Forsea, A. M., Raisova, M., Orfanos, C. E. and Geilen, C. C., 2002. The bisphosphonate pamidronate induces apoptosis in human melanoma cells in vitro. Br J Cancer, 87, 366-71. Rivnay, B., Bayer, E. A. and Wilchek, M., 1987. Use of avidin-biotin technology for liposome targeting. Methods Enzymol, 149, 119-23. Roberts, W. G. and Palade, G. E., 1997. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res, 57, 765-72. Robey, P. G. and Boskey, A. L. (1996) In Osteoporosis(Ed, Kelsey, J.) Academic Press, San Diego, California. Rodan, G., Reszka, A., Golub, E. and Rizzoli, R., 2004. Bone safety of long-term bisphosphonate treatment. Curr Med Res Opin, 20, 1291-300. Roelofs, A. J., Thompson, K., Gordon, S. and Rogers, M. J., 2006. Molecular mechanisms of action of bisphosphonates: current status. Clin Cancer Res, 12, 6222s-6230s. Rogers, M. J., 2003. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des, 9, 2643-58. Rogers, M. J., Russell, R. G., Blackburn, G. M., Williamson, M. P. and Watts, D. J., 1992. Metabolism of halogenated bisphosphonates by the cellular slime mould Dictyostelium discoideum. Biochem Biophys Res Commun, 189, 414-23. Rogers, M. J., Frith, J. C., Luckman, S. P., Coxon, F. P., Benford, H. L., Monkkonen, J., Auriola, S., Chilton, K. M. and Russell, R. G., 1999. Molecular mechanisms of action of bisphosphonates. Bone, 24, 73S-79S. Romberg, B., Hennink, W. E. and Storm, G., 2007. Sheddable Coatings for Long-Circulating Nanoparticles. Pharm Res. Romberg, B., Oussoren, C., Snel, C. J., Carstens, M. G., Hennink, W. E. and Storm, G., 2006. Pharmacokinetics of poly(hydroxyethyl-l-asparagine)-coated liposomes is superior over that of PEG-coated liposomes at low lipid dose and upon repeated administration. Biochim Biophys Acta, 1768, 737-43. Roodman, G. D., Ibbotson, K. J., MacDonald, B. R., Kuehl, T. J. and Mundy, G. R., 1985. 1,25-Dihydroxyvitamin D3 causes formation of multinucleated cells with several osteoclast characteristics in cultures of primate marrow. Proc Natl Acad Sci U S A, 82, 8213-7. Rouser, G., Fkeischer, S. and Yamamoto, A., 1970. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids, 5, 494-6. Rubas, W. and Schreier, H., 1991. [Liposomes: advances in production-technology and therapy]. Pharm Unserer Zeit, 20, 255-70.

Literatur 171
Russell, R. G., 2007. Bisphosphonates: mode of action and pharmacology. Pediatrics, 119 Suppl 2, S150-62. Russell, R. G., Muhlbauer, R. C., Bisaz, S., Williams, D. A. and Fleisch, H., 1970. The influence of pyrophosphate, condensed phosphates, phosphonates and other phosphate compounds on the dissolution of hydroxyapatite in vitro and on bone resorption induced by parathyroid hormone in tissue culture and in thyroparathyroidectomised rats. Calcif Tissue Res, 6, 183-96. Russell, R. G., Rogers, M. J., Frith, J. C., Luckman, S. P., Coxon, F. P., Benford, H. L., Croucher, P. I., Shipman, C. and Fleisch, H. A., 1999. The pharmacology of bisphosphonates and new insights into their mechanisms of action. J Bone Miner Res, 14 Suppl 2, 53-65. Sanders, R. J., Mainiero, F. and Giancotti, F. G., 1998. The role of integrins in tumorigenesis and metastasis. Cancer Invest, 16, 329-44. Sandstrom, M. C., Johansson, E. and Edwards, K., 2007. Structure of mixed micelles formed in PEG-lipid/lipid dispersions. Langmuir, 23, 4192-8. Sapra, P. and Allen, T. M., 2003. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res, 42, 439-62. Scherphof, G. L. (1991) Handbook of experimental pharmacology, Springer-Verlag, Berlin Heidelberg. Scherphof, G. L. and Kamps, J. A., 2001. The role of hepatocytes in the clearance of liposomes from the blood circulation. Prog Lipid Res, 40, 149-66. Schibler, D., Russell, R. G. and Fleisch, H., 1968. Inhibition by pyrophosphate and polyphosphate of aortic calcification induced by vitamin D3 in rats. Clin Sci, 35, 363-72. Schiffelers, R. M., Koning, G. A., ten Hagen, T. L., Fens, M. H., Schraa, A. J., Janssen, A. P., Kok, R. J., Molema, G. and Storm, G., 2003. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release, 91, 115-22. Schnieders, J., Gbureck, U., Thull, R. and Kissel, T., 2006. Controlled release of gentamicin from calcium phosphate-poly(lactic acid-co-glycolic acid) composite bone cement. Biomaterials, 27, 4239-49. Schote, U. and Seelig, J., 1998. Interaction of the neuronal marker dye FM1-43 with lipid membranes. Thermodynamics and lipid ordering. Biochim Biophys Acta, 1415, 135-46. Schreier, H., 1982. [Liposomes--a new slow-release dosage form. I. Phospholipids; production and characterization of liposomes]. Pharm Unserer Zeit, 11, 97-101. Seelig, J., 1997. Titration calorimetry of lipid-peptide interactions. Biochim Biophys Acta, 1331, 103-16. Senaratne, S. G., Pirianov, G., Mansi, J. L., Arnett, T. R. and Colston, K. W., 2000. Bisphosphonates induce apoptosis in human breast cancer cell lines. Br J Cancer, 82, 1459-68. Senior, J., Crawley, J. C. and Gregoriadis, G., 1985. Tissue distribution of liposomes exhibiting long half-lives in the circulation after intravenous injection. Biochim Biophys Acta, 839, 1-8. Sessa, G. and Weissmann, G., 1968. Phospholipid spherules (liposomes) as a model for biological membranes. J Lipid Res, 9, 310-8.

Literatur 172
Sessa, G. and Weissmann, G., 1970. Incorporation of lysozyme into liposomes. A model for structure-linked latency. J Biol Chem, 245, 3295-301. Seymour, L. W., 1992. Passive tumor targeting of soluble macromolecules and drug conjugates. Crit Rev Ther Drug Carrier Syst, 9, 135-87. Sharma, A. and Sharma, U. S., 1997. Liposomes in drug delivery: progress and limitations. Int J Pharm, 154, 123-40. Shea, J. E. and Miller, S. C., 2005. Skeletal function and structure: implications for tissue-targeted therapeutics. Adv Drug Deliv Rev, 57, 945-57. Shier, D., Butler, D. and Lewis, R. (1996) Hole´s Human Anatomy & Physiology, Wm C Brown Publishers, Dubuque, Iowa. Shinoda, H., Adamek, G., Felix, R., Fleisch, H., Schenk, R. and Hagan, P., 1983. Structure-activity relationships of various bisphosphonates. Calcif Tissue Int, 35, 87-99. Shinto, Y., Uchida, A., Korkusuz, F., Araki, N. and Ono, K., 1992. Calcium hydroxyapatite ceramic used as a delivery system for antibiotics. J Bone Joint Surg Br, 74, 600-4. Shipman, C. M., Croucher, P. I., Russell, R. G., Helfrich, M. H. and Rogers, M. J., 1998. The bisphosphonate incadronate (YM175) causes apoptosis of human myeloma cells in vitro by inhibiting the mevalonate pathway. Cancer Res, 58, 5294-7. Shmeeda, H., Mak, L., Tzemach, D., Astrahan, P., Tarshish, M. and Gabizon, A., 2006. Intracellular uptake and intracavitary targeting of folate-conjugated liposomes in a mouse lymphoma model with up-regulated folate receptors. Mol Cancer Ther, 5, 818-24. Sotirhos, N., Herslof, B. and Kenne, L., 1986. Quantitative analysis of phospholipids by 31P-NMR. J Lipid Res, 27, 386-92. Sotomayor, E. M., Borrello, I. and Levitsky, H. I., 1996. Tolerance and cancer: a critical issue in tumor immunology. Crit Rev Oncog, 7, 433-56. Steenpass, T., Lung, A. and Schubert, R., 2006. Tresylated PEG-sterols for coupling of proteins to preformed plain or PEGylated liposomes. Biochim Biophys Acta, 1758, 20-8. Steitz, S. A., Speer, M. Y., McKee, M. D., Liaw, L., Almeida, M., Yang, H. and Giachelli, C. M., 2002. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol, 161, 2035-46. Storm, G. and Crommelin, D. J., 1998. Liposomes: quo vadis? PSTT, 1, 19-31. Storm, G., Oussoren, C., Peeters, P. A. M. and Barenholz, Y. (1993) In Liposome Technology: Interaction of liposomes with the biological milieu, vol III(Ed, Gregoriadis, G.) CRC, Ann Arbor, pp. 345. Storm, G., Belliot, S. O., Daemen, T. and Lasic, D. D., 1995. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv Drug Deliv Rev, 17, 31-48. Storm, G., Steerenberg, P. A., Emmen, F., van Borssum Waalkes, M. and Crommelin, D. J., 1988. Release of doxorubicin from peritoneal macrophages exposed in vivo to doxorubicin-containing liposomes. Biochim Biophys Acta, 965, 136-45.

Literatur 173
Stubbs, M., McSheehy, P. M., Griffiths, J. R. and Bashford, C. L., 2000. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today, 6, 15-9. Tassone, P., Tagliaferri, P., Viscomi, C., Palmieri, C., Caraglia, M., D'Alessandro, A., Galea, E., Goel, A., Abbruzzese, A., Boland, C. R. and Venuta, S., 2003. Zoledronic acid induces antiproliferative and apoptotic effects in human pancreatic cancer cells in vitro. Br J Cancer, 88, 1971-8. Teitelbaum, S. L., Tondravi, M. M. and Ross, F. P. (1996) In Osteoporosis(Ed, Kelsey, J.) Academic Press, San Diego, California. Templeton, N. S., Lasic, D. D., Frederik, P. M., Strey, H. H., Roberts, D. D. and Pavlakis, G. N., 1997. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol, 15, 647-52. Teti, A., Blair, H. C., Teitelbaum, S., Kahn, A. J., Koziol, C., Konsek, J., Zambonin-Zallone, A. and Schlesinger, P. H., 1989. Cytoplasmic pH regulation and chloride/bicarbonate exchange in avian osteoclasts. J Clin Invest, 83, 227-233. Torchilin, V. P., Narula, J., Halpern, E. and Khaw, B. A., 1996. Poly(ethylene glycol)-coated anti-cardiac myosin immunoliposomes: factors influencing targeted accumulation in the infarcted myocardium. Biochim Biophys Acta, 1279, 75-83. Torchilin, V. P., Levchenko, T. S., Lukyanov, A. N., Khaw, B. A., Klibanov, A. L., Rammohan, R., Samokhin, G. P. and Whiteman, K. R., 2001. p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim Biophys Acta, 1511, 397-411. Troen, B. R., 2003. Molecular mechanisms underlying osteoclast formation and activation. Exp Gerontol, 38, 605-14. Ueda, Y., Itoh, T., Nukaya, I., Kawashima, I., Okugawa, K., Yano, Y., Yamamoto, Y., Naitoh, K., Shimizu, K., Imura, K., Fuji, N., Fujiwara, H., Ochiai, T., Itoi, H., Sonoyama, T., Hagiwara, A., Takesako, K. and Yamagishi, H., 2004. Dendritic cell-based immunotherapy of cancer with carcinoembryonic antigen-derived, HLA-A24-restricted CTL epitope: Clinical outcomes of 18 patients with metastatic gastrointestinal or lung adenocarcinomas. Int J Oncol, 24, 909-17. Uludag, H. and Kousinioris, N., 2000. Bisphosphonate conjugation to proteins as a mean to impart bone affinity. Biotechnol Prog., 16, 258-67. Uludag, H., Gao, T., Wohl, G. R., Kantoci, D. and Zernicke, R. F., 2000. Bone affinity of a bisphosphonate-conjugated protein in vivo. Biotechnol Prog, 16, 1115-8. Uster, P. S., Allen, T. M., Daniel, B. E., Mendez, C. J., Newman, M. S. and Zhu, G. Z., 1996. Insertion of poly(ethylene glycol) derivatized phospholipid into pre-formed liposomes results in prolonged in vivo circulation time. FEBS Lett, 386, 243-6. Van Beek ER, L. C., Ebetino FH, 1998. Binding and antiresorptive properties of heterocycle-containing bispphosphonate analogs: structure-activity relationship. Bone, 23, 437-42. van der Pluijm, G., Vloedgraven, H., van Beek, E., van der Wee-Pals, L., Lowik, C. and Papapoulos, S., 1996. Bisphosphonates inhibit the adhesion of breast cancer cells to bone matrices in vitro. J Clin Invest, 98, 698-705.

Literatur 174
van der Pluijm, G., Sijmons, B., Vloedgraven, H., Deckers, M., Papapoulos, S. and Lowik, C., 2001. Monitoring metastatic behavior of human tumor cells in mice with species-specific polymerase chain reaction: elevated expression of angiogenesis and bone resorption stimulators by breast cancer in bone metastases. J Bone Miner Res, 16, 1077-91. Virk, M. S. and Lieberman, J. R., 2007. Tumor metastasis to bone. Arthritis Res Ther, 9 Suppl 1, S5. von Minckwitz, G., Harder, S., Hovelmann, S., Jager, E., Al-Batran, S. E., Loibl, S., Atmaca, A., Cimpoiasu, C., Neumann, A., Abera, A., Knuth, A., Kaufmann, M., Jager, D., Maurer, A. B. and Wels, W. S., 2005. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res, 7, R617-26. Wang, D., Miller, S. C., Kopeckova, P. and Kopecek, J., 2005. Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev, 57, 1049-76. Wang, D., Miller, S., Sima, M., Kopeckova, P. and Kopecek, J., 2003. Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconjug Chem, 14, 853-9. Wetterwald, A., van der Pluijm, G., Que, I., Sijmons, B., Buijs, J., Karperien, M., Lowik, C. W., Gautschi, E., Thalmann, G. N. and Cecchini, M. G., 2002. Optical imaging of cancer metastasis to bone marrow: a mouse model of minimal residual disease. Am J Pathol, 160, 1143-53. Willson, T. M., Henke, B. R., Momtahen, T. M., Charifson, P. S., Batchelor, K. W., Lubahn, D. B., Moore, L. B., Oliver, B. B., Sauls, H. R., Triantafillou, J. A. and et al., 1994. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J Med Chem, 37, 1550-2. Wingen, F. and Schmahl, D., 1985. Distribution of 3-amino-1-hydroxypropane-1,1-diphosphonic acid in rats and effects on rat osteosarcoma. Arzneimittelforschung, 35, 1565-71. Wingen, F., Sterz, H., Blum, H., Moller, H., Pittermann, W., Pool, B. L., Sinn, H. J., Spring, H. and Schmahl, D., 1986. Synthesis, antitumor activity, distribution and toxicity of 4-[4-[bis(2-chloroethyl)amino]phenyl]-1-hydroxybutane-1 1-bisphosphonic acid (BAD), a new lost derivative with increased accumulation in rat osteosarcoma. J Cancer Res Clin Oncol, 111, 209-19. Wojtowicz-Praga, S., 1997. Reversal of tumor-induced immunosuppression: a new approach to cancer therapy. J Immunother, 20, 165-77. Woodle, M. C. and Lasic, D. D., 1992. Sterically stabilized liposomes. Biochim Biophys Acta, 1113, 171-99. Woodle, M. C., Matthay, K. K., Newman, M. S., Hidayat, J. E., Collins, L. R., Redemann, C., Martin, F. J. and Papahadjopoulos, D., 1992. Versatility in lipid compositions showing prolonged circulation with sterically stabilized liposomes. Biochim Biophys Acta, 1105, 193-200. Xiong, X. B., Huang, Y., Lu, W. L., Zhang, X., Zhang, H., Nagai, T. and Zhang, Q., 2005. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release, 107, 262-75. Yang, T., Choi, M. K., Cui, F. D., Kim, J. S., Chung, S. J., Shim, C. K. and Kim, D. D., 2007. Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release, 120, 169-77.

Literatur 175
Yokogawa, K., Miya, K. and Sekido, T., 2001. Selective delivery of estradiol to bone by aspartatic acid oligopeptide and its effects on ovariectomized mice. Endocrinology, 142, 1228-33. Yoneda, T., 1998. Cellular and molecular mechanisms of breast and prostate cancer metastasis to bone. Eur J Cancer, 34, 240-5. Yoshikawa, H., 1998. [Regulatory mechanisms of bone remodeling]. Nippon Rinsho, 56, 1400-5. Yoshino, K., Tanabe, M., Ohnuma, N. and Takahashi, H., 1996. Histopathologic analysis of bone marrow and bone metastasis in murine neuroblastoma. Clin Exp Metastasis, 14, 459-65. Yuan, F., Leunig, M., Huang, S. K., Berk, D. A., Papahadjopoulos, D. and Jain, R. K., 1994. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res, 54, 3352-6. Yuan, F., Dellian, M., Fukumura, D., Leunig, M., Berk, D. A., Torchilin, V. P. and Jain, R. K., 1995. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res, 55, 3752-6. Zalipsky, S., 1993. Synthesis of an end-group functionalized polyethylene glycol-lipid conjugate for preparation of polymer-grafted liposomes. Bioconjug Chem, 4, 296-9. Zalipsky, S., Mullah, N., Harding, J. A., Gittelman, J., Guo, L. and DeFrees, S. A., 1997. Poly(ethylene glycol)-grafted liposomes with oligopeptide or oligosaccharide ligands appended to the termini of the polymer chains. Bioconjug Chem, 8, 111-8. Zalipsky, S., Gittelman, J., Mullah, N., Qazen, M. M., Harding, J. (1998) (Ed, Gregoriadis, G.) Plenum, New York, pp. 131-138. Zambonin, G., Camerino, C., Greco, G., Patella, V., Moretti, B. and Grano, M., 2000. Hydroxyapatite coated with heaptocyte growth factor (HGF) stimulates human osteoblasts in vitro. J Bone Joint Surg Br, 82, 457-60. Zavaleta, C. L., Phillips, W. T., Soundararajan, A. and Goins, B. A., 2007. Use of avidin/biotin-liposome system for enhanced peritoneal drug delivery in an ovarian cancer model. Int J Pharm, 337, 316-28. Zhao, X. B., Muthusamy, N., Byrd, J. C. and Lee, R. J., 2007. Cholesterol as a bilayer anchor for PEGylation and targeting ligand in folate-receptor-targeted liposomes. J Pharm Sci.

Anhang 176
8 Anhang
8.1 Lebenslauf
Verena Hengst geb. 17. August 1976 in Gronau (Westfalen) Familienstand: ledig Staatsangehörigkeit: deutsch
Berufliche Erfahrung seit 2007 A. Nattermann & Cie. GmbH, ein Unternehmen der Gruppe sanofi-aventis, Köln,
Tätigkeit als Leiterin der Galenik (Industrial Development & MP OTC, Liquids & Semisolids)
2003–2007 MCS Micro Carrier Systems GmbH, Neuss, Tätigkeit als Projektmanagerin (seit 2004) und berufsbegleitende Doktorandin
FOCUS Clinical Drug Development GmbH, Neuss, Ausbildung zur Sachkundigen Person gemäß §15 AMG
2003/2005 Liebig-Apotheke, Düsseldorf, Teilzeittätigkeit als Apothekerin (Chefvertretung) 2002/2003 Fortuna-Apotheke, Köln, Tätigkeit als Apothekerin (Chefvertretung), 4 Monate Flora-Apotheke, Bonn, Tätigkeit als Apothekerin (Chefvertretung), 4 Monate Ausbildung 2005/2006 Universität Utrecht, Niederlande, Auslandsjahr während der Promotion im Rahmen
des EST Marie Curie Stipendiums, Professor Gert Storm, Institut für Pharmazeutische Wissenschaften
2004–2007 Universität Marburg, Promotion, Professor Thomas Kissel, Institut für
Pharmazeutische Technologie und Biopharmazie
• Promotionsvorhaben: “Bisphosphonatliganden für die zielgerichtete Applikation liposomaler Trägersysteme an den Knochen: eine Machbarkeitsstudie“
• Teilnahme in EU-Programm zur Erlangung des Titels “Euro PhD in Advanced Drug Delivery“ (Universität Saarbrücken)

Anhang 177
1996–2000 Rheinische Friedrich-Wilhelm-Universität Bonn, Studium der Pharmazie
(1999: 1. Staatsexamen, 2001: 2. Staatsexamen, 2002: 3. Staatsexamen)
1987–1996 Werner von Siemens Gymnasium, Gronau (Westfalen)
Berufliche Weiterbildung 2004 European Continuing Education Course ”Formulation and Development of poorly
water-soluble drugs”, London, Großbritanien, 22-24 November
2004 GCP-Schulungen bei FOCUS Drug Development GmbH, 17. Juni, 04. November
Praktika 2002 MCS Micro Carrier Systems GmbH, Neuss, Praktikum, 1 Monat
2001/2002 Fortuna-Apotheke, Köln, Praktikum im Rahmen des Praktischen Jahres, 6 Monate
2001 Lilly Forschung GmbH, Hamburg, Praktikum im Rahmen des Praktischen Jahres im
Bereich der Formulierung (Pharmazeutische Technologie), 6 Monate
1999 Bayer AG, Leverkusen, Tätigkeit als studentische Hilfskraft im Bereich der
Qualitätskontrolle, 2 Monate
1998 Flora–Apotheke, Gronau–Epe, Famulatur, 1 Monat
1997 Flora–Apotheke, Gronau–Epe, Famulatur, 1 Monat

Danksagung/Acknowledgment 178
8.2 Danksagung/Acknowledgment
The completion of this dissertational thesis would not have been possible without the generous help of many people.
In particular, I owe a great debt to my supervisor Professor Dr Thomas Kissel from the University of Marburg for supervising my research project. His invaluable scientific advice as well as his inspiring personal support significantly helped me in designing and compiling my work. Very strong and important support was also forthcoming from Professor Dr Gert Storm from the University of Utrecht, the Netherlands. I very much enjoyed the fruitful discussion with him and very much appreciated his contribution. Furthermore, I am also very grateful that Professor Bakowsky from the University of Marburg accepted to take on the role as co-assessor.
I also benefited greatly from constructive ideas and personal support from Dr Christien Oussoren from the University of Utrecht, the Netherlands. Additionally, I cannot sufficiently acknowledge the generous support I have received from my employer, MCS Micro Carrier Systems GmbH. Special thanks in this respect go to Dr Hans Schreier who introduced me to the field of liposomes research and provided significant scientific and personal support.
Fortunately, I had the chance to spend a year at the University of Utrecht to continue my experimental work. Many thanks go to the Galenos Network for offering the required stipend. I also would like to say thank you to the Biopharmacy group, especially to Manuela Banciu, Maryam Amidi, Marjan Fretz, Christianne Rijken, Sabrina Oliveira, Marcel Fens, Holger de Wolf, Frits Flesch and Louis van Bloois. Thank you to all of them for their support and for the pleasant stay. Special thanks go to Cor Snel and Birgit Romberg for their enormous contribution during conduction of animal studies. Birgit deserves additional thanks for her warm welcome to the group and her assistance during my stay in Utrecht.
Strong support was also given by Dr Eefjan Breukink and Dr Inge Visscher of the Department of Biochemistry of Lipids, University of Utrecht, the Netherlands, who introduced me to the technique of Isothermal Titration Calorimetry.

Danksagung/Acknowledgment 179
Many thanks also go to Dr Johan Kemmink of the department of Medicinal Chemistry, University of Utrecht, the Netherlands, who taught performing 31P-NMR-measurements.
I would also like to say thank you to Professor Dr Volker Röschenthaler and Dr Oleg Shyshokov of the Department of Anorganic and Physical Chemistry at the University of Bremen for preparing the synthesis of the bisphosphonate-ligands. Without their help, the realisation of the present research project would not have been possible.
In addition, I would to like to express my gratitude to Dr Gabri van der Plujim and Ivo Que from the University of Leiden, the Netherlands, who provided valuable insights in the fields of bone metastases and Bioluminescent Imaging. They also took the time to perform in vivo studies using their well-established nude mouse model for bone metastases.
Special thanks also go to Dr Eberhard Amtmann and Waltraud Baader of the German Cancer Research Center, Heidelberg, who made it possible for me to perform cell culture experiments under their supervision.
Finally, I would like to express my gratitude to my parents for their enduring care, strong encouragement, and generous support during the course of my education.
Verena Hengst

Veröffentlichungen 180
8.3 Veröffentlichungen
8.3.1 Publikationen
Hengst V, Oussoren C, Kissel T, Storm G, 2007, Bone targeting potential of bisphosphonate-targeted liposomes. Preparation, characterization and hydroxyapatite- binding in vitro, Int J Pharm 331, 224-227
Hengst V, van der Pluijm G, Kissel T, Storm G, Oussoren C, 2008, Bone targeting capability of bisphosphonate-targeted liposomes, in Bearbeitung
8.3.2 Patente
Miterfinder von Patent der Firma MCS Micro Carrier Systems GmbH. Internationale Patentanmeldung PCT/DE2005/000095: Titel “Bisphosphonate + Cholesterol“
8.3.3 Vorträge
6th European Workshop on Particulate Systems, Genf, Schweiz, 23-24 März, 2006, eigener Vortrag zum Thema “Bone-seeking liposomes as drug delivery system for the treatment of bone metastases”
Socrates Intensive Programme “Innvovative therapeutics: from molecules to medicines”, Lovain-La Neuve, Belgien, 3-14 Juli, 2005, eigener Vortrag zum Thema “Treatment of bone metastases with bisphosphonate-targeted liposomes”
2nd Thematic Workshop and Training Course on “Galenos-Towards a European PhD in advanced drug delivery, Cardiff, Großbritannien, 20-22 Oktober, 2005, eigener Vortrag zum Thema “Bisphosphonate-targeted liposomes”
Liposome Workshop, Ameland, Niederlande, 8-12 September, 2003, eigener Vortrag zum Thema “Long-circulating bisphosphonate-targeted liposomes”

Veröffentlichungen 181
8.3.4 Poster
International Workshop on “Cell culture and in vitro models for drug absorption and delivery”, Saarbrücken, 1-10 März, 2006, Posterpräsentation “Bone-seeking liposomes as drug delivery system for the treatment of bone metastases”
European IP-Galenos Course “Skin barrier function: pharmaceutic and cosmetic applications”, Lyon, Frankreich, 12-22 September, 2005, Posterpräsentation “Treatment of bone metastases with long-circulating bisphosphonate-targeted liposomes”


















