
Einführung in die Physikalische Chemie Teil 1 ...epc/2012/Kapitel_6.pdf · Erklärungen: • Reale...
17
Einführung in die Physikalische Chemie Teil 1: Mikrostruktur der Materie Einführung in die Physikalische Chemie: Übersicht Kapitel 1: Quantenmechanik Mathematische Grundlagen Schrödingergleichung Einfache Beispiele Kapitel 2: Atome H-Atom Spin Mehrelektronen-Atome und Spektroskopie Kapitel 3: Moleküle Molekülorbitaltheorie Born-Oppenheimer-Potential Kapitel 5: Zwischenmolekulare Kräfte Elektrostatische Eigenschaften von Molekülen Zwischenmolekulare Wechselwirkungen Struktur von Biomolekülen Kapitel 6: Struktur der Materie Reale Gase Kondensierte Phasen Moleküldynamik Mikrokosmos Makrokosmos Kapitel 4: Molekülspektroskopie Bewegungsformen eines Moleküls: Rotationen,Schwingungen, elektron. Bewegung Mikrowellen-, Infrarot- und optische Spektroskopie
Transcript of Einführung in die Physikalische Chemie Teil 1 ...epc/2012/Kapitel_6.pdf · Erklärungen: • Reale...

Einführung in die Physikalische ChemieTeil 1: Mikrostruktur der Materie
Einführung in die Physikalische Chemie: Übersicht
Kapitel 1:Quantenmechanik
Mathematische GrundlagenSchrödingergleichungEinfache Beispiele
Kapitel 2:Atome
H-AtomSpinMehrelektronen-Atome und Spektroskopie
Kapitel 3:Moleküle
MolekülorbitaltheorieBorn-Oppenheimer-Potential
Kapitel 5:ZwischenmolekulareKräfte
Elektrostatische Eigenschaften von MolekülenZwischenmolekulare WechselwirkungenStruktur von Biomolekülen
Kapitel 6:Struktur der Materie
Reale GaseKondensierte PhasenMoleküldynamik
Mikrokosmos
Makrokosmos
Kapitel 4:Molekülspektroskopie
Bewegungsformen eines Moleküls:Rotationen,Schwingungen, elektron. BewegungMikrowellen-, Infrarot- und optische Spektroskopie

Kapitel 6:Struktur der Materie
Übersicht:
Literatur:
Atkins, de Paula, Physikalische Chemie (4. Aufl.), Teile von Kapitel 1,18,19,20Atkins, de Paula, Kurzlehrbuch Physikalische Chemie (4. Aufl.), Teile von Kapitel 1,17,18
6.1 Einführung6.2 Reale Gase6.3 Flüssigkeiten6.4 Feststoffe und Kristalle6.5 Struktur kondensierter Phasen6.6 Moleküldynamik (MD) - Simulationen
Kapitel 6: Struktur der Materie

6.1 Einführung
Fragestellung: Wie bestimmen die Wechselwirkungen zwischen Molekülen die Struktur und den (Aggregat-) Zustand makroskopischer Stoffe ?
Aggregatzustände: fest, flüssig, gasförmig
i i j
j
Prinzip der Paaradditivität: die gesamte potentielle Energie Vtot kann als Summe von Wechselwirkungen zwischen Paaren von Teilchen Vij formuliert werden:
Vtot
=X
alle Paare ij
Vi j (6.1.1)
Vij ist dabei die totale Wechselwir-kungsenergie zwischen zwei Molekülen, also die Summe aller in Kap. 5 diskutierten relevanten Wechselwir-kungspotentialen.
Das Paaradditivitäts-Prinzip gilt auch für zwischenmolekulare Kräfte Fij, da die Kraft definiert ist als
(6.1.2)Fi j = �dVi j/dRintermolekularer Abstand
Mehrteilchen-Wechselwirkungen betragen in vielen Fällen nur wenige Prozent der totalen Wechselwirkungsenergie (Bsp. einer Ausnahme: Wasser H2O. Warum ?).
6.1 Einführung

6.2 Reale Gase
Ideales Gas: Gasmoleküle werden als nicht-wechselwirkende Teilchen ohne eigenes Volumen betrachtet
pVm = RTZustandsgleichung für das ideale Gas:
mit dem Molvolumen Vm=V/n
(6.2.1)
Probleme:
• Vm→0 wenn T→0 oder p→∞: unphysikalisch, da alle Moleküle ein Eigenvolumen besitzen
• die Wechselwirkungen zwischen den Molekülen werden vernachlässigt → keine Kondensation/Phasenübergänge
Die Van-der-Waals-Gleichung
Die van-der-Waals-Gleichung ist eine (approximative) Zustandsgleichung für reale Gase, die diese beiden Defizite korrigiert:
6.2 Reale Gase

• Das Molvolumen Vm wird um einen Faktor b korrigiert, der dem Eigenvolumen der Gasteilchen entspricht.
• Der Druck wird durch die WW zwischen den Teilchen verringert. Dies wird durch einen Term -a/Vm
2 modelliert.
p =RT
Vm � b�a
V 2m,
✓p +
a
V 2m
◆�Vm � b) = RT (6.2.2)
Tabelle 6.2.1:Van-der-Waals-Koeffizienten für verschiedene Gase (aus Atkins, 4. Aufl.)
6.2 Reale Gase
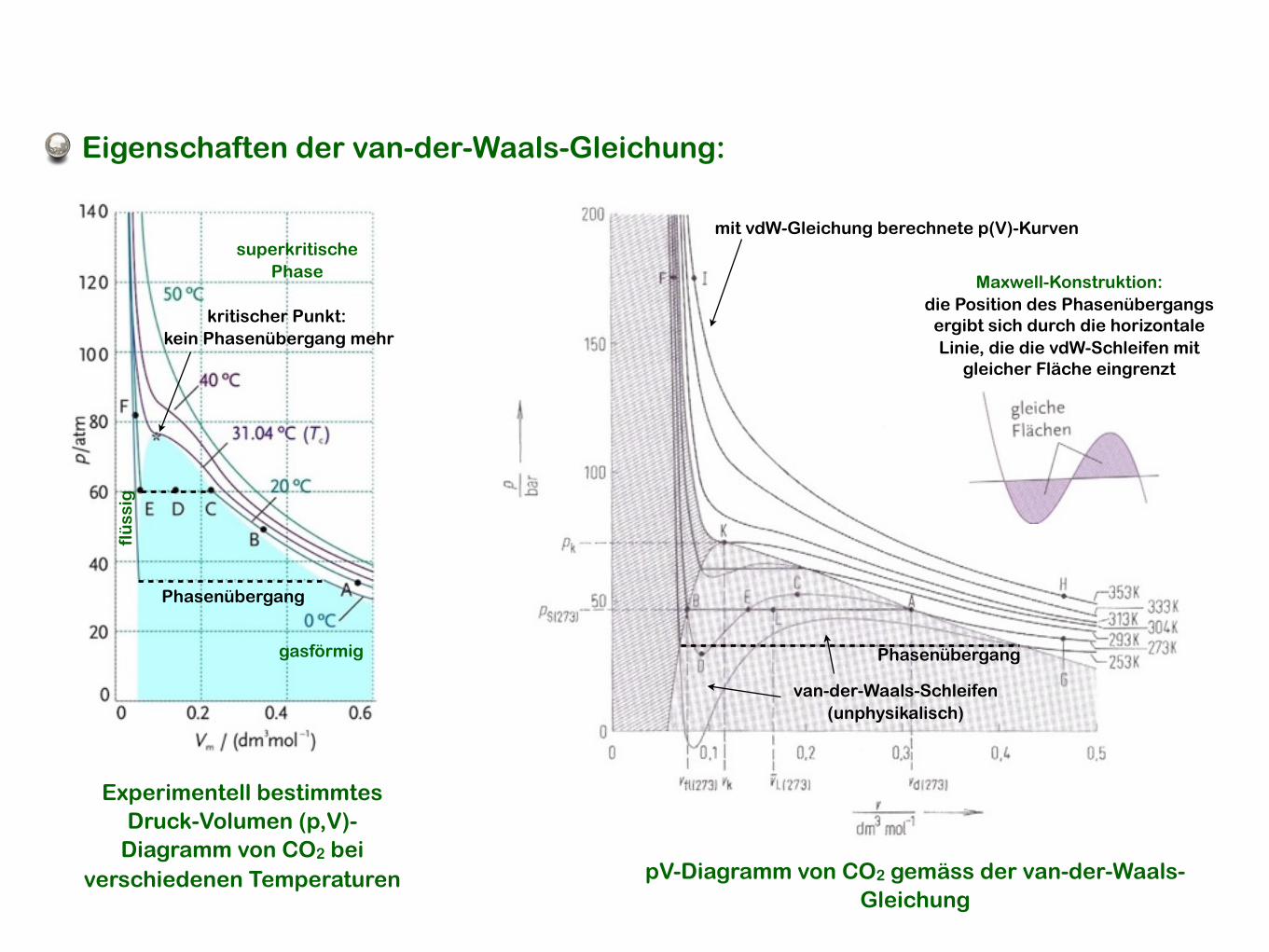
Eigenschaften der van-der-Waals-Gleichung:
Experimentell bestimmtesDruck-Volumen (p,V)-
Diagramm von CO2 bei verschiedenen Temperaturen pV-Diagramm von CO2 gemäss der van-der-Waals-
Gleichung
kritischer Punkt: kein Phasenübergang mehr
flü
ssig
gasförmig
Phasenübergang
superkritischePhase
mit vdW-Gleichung berechnete p(V)-Kurven
van-der-Waals-Schleifen (unphysikalisch)
Phasenübergang
Maxwell-Konstruktion:die Position des Phasenübergangs
ergibt sich durch die horizontale Linie, die die vdW-Schleifen mit
gleicher Fläche eingrenzt
6.2 Reale Gase

Erklärungen:
• Reale Gase durchlaufen bei einem bestimmten Druck, Volumen und Temperatur Phasenübergänge. Diese machen sich im p,V-Diagramm als eine Diskontinuität (horizontale Linie) in der p(V)-Kurve bemerkbar, da sich das Volumen bei einem Phasenübergang in der Regel sehr stark ändert (vgl. Flüssigkeit-Gas).
• Die mit der vdW-Gleichung berechneten p(V)-Kurven weisen im Bereich des Phasenübergangs zwei Schleifen auf, die unphysikalisch sind.
• Im Rahmen der vdW-Gleichung wird die Position des Phasenübergangs durch eine horizontale Linie angenährt, die vdW-Schleifen mit gleicher Fläche einschliesst (Maxwell-Konstruktion).
• Ab einer gewissen Temperatur Tkrit finden keine Phasenübergänge mehr statt. Man beobachtet eine superkritische Phase - weder flüssig noch gasförmig.
• Mehr über Phasenübergänge in Kapitel 14 dieser Vorlesung.
Vor- und Nachteile der vdW-Gleichung:+ einfach und intuitiv- nur eine Näherung, die nicht systematisch verbessert werden kann
6.2 Reale Gase
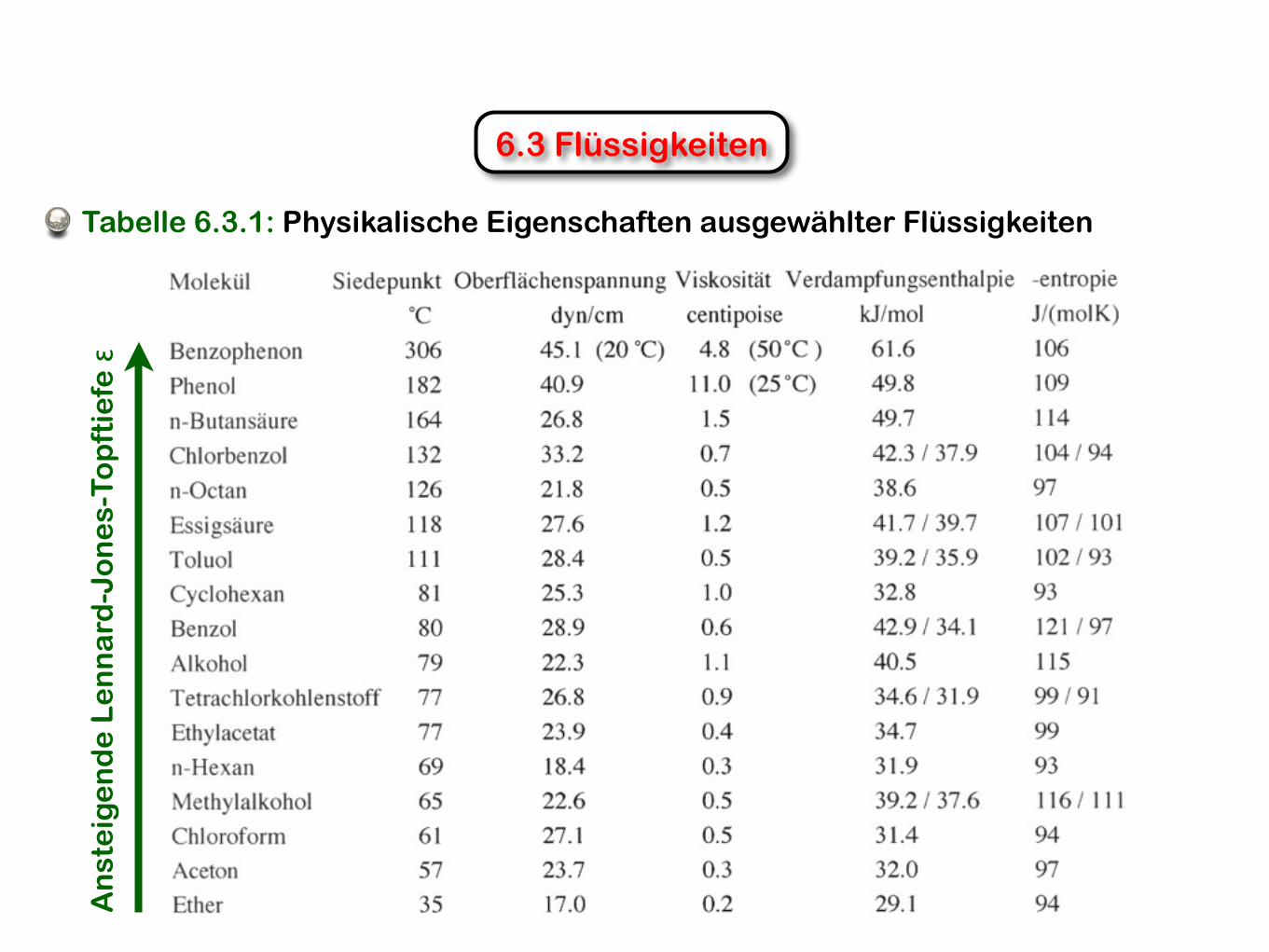
6.3 FlüssigkeitenA
nst
eig
en
de
Le
nn
ard
-Jo
ne
s-To
pft
iefe
ε
Tabelle 6.3.1: Physikalische Eigenschaften ausgewählter Flüssigkeiten
6.3 Flüssigkeiten

6.3.1 Siedepunkt
Um ein Teilchen durch Verdampfung aus einer Flüssigkeit zu entfernen, müssen offensichtlich die intermolekularen Anziehungskräfte überwunden werden.
Für viele Flüssigkeiten gilt daher in guter Näherung, dass der Siedepunkt gleich der Lennard-Jones-Topftiefe ε (in Kelvin) ist (s.a. Tabelle 5.3.1).
1/R12
-1/R6
RReq
σ
-ε
V (R) = 4✏
"✓�
R
◆12�
✓�
R
◆6#
6.3 Flüssigkeiten

6.3.2 Oberflächenspannung
Um die intermolekularen Anziehungskräfte zu maximieren, werden Teilchen von der Oberfläche einer Flüssigkeit in ihre Zentrum gezogen:
Folglich versuchen Flüssigkeiten immer, ihre Oberfläche zu minimieren (Tröpfchenbildung). Die Energie ΔE, die benötigt wird, um die Oberfläche A der Flüssigkeit um einen Betrag ΔA zu verändern, ist gegeben durch
(6.3.1)Die Proportionalitätskonstante γ wird als Oberflächenspannung bezeichnet. Einheit [γ]=[J m-2]=[N m-1].
γ steht offensichtlich in Bezug zur Stärke der intermolekularen Wechselwirkungen, die die Oberfläche zu minimieren versuchen: je grösser ε, desto grösser γ.
6.3 Flüssigkeiten

6.3.3 Viskosität
Die Viskosität η bezeichnet den Widerstand einer Flüssigkeit gegenüber einer Deformation ihres Volumens. Einheit: [η]=[Poise]=[10-1 kg m-1 s-1].
Je stärker die intermolekularen WW (d.h., je grösser ε), desto grösser ist die Viskosität.
Die Viskosität wird jedoch auch stark durch andere Parameter wie die Molekülgeometrie beeinflusst.
6.3.4 Verdampfungsenthalpie
Der Wärmeaufwand um eine Flüssigkeit zu verdampfen (Verdampfungsenthalpie) ist offensichtlich ebenfalls proportional zur Lennard-Jones-Topftiefe ε.
6.3 Flüssigkeiten

6.4 Ionische Kristalle
Ziel: Wir wollen die Gitterenergie eines Kristall aus den intermolekularen Wechselwirkungsenergien berechnen.
Die Gitterenergie bezeichnet die “Bindungsenergie” eines Kristalls, also die Energiedifferenz zwischen den freien Ionen in der Gasphase und den Ionen im Kristallgitter.
Modellpotential für die WW zwischen einem Anion und einem Kation in einem Kristallgitter:
V =C
Rm�Z2e2
4⇡✏0R
attraktiver Term (Coulomb-WW)
repulsiver Term(empirisch, C>0, m≈9-12)
(6.4.1)
Ladungszahl
Die Topftiefe ε für dieses Modellpotential kann berechnet werden zu (→ Tafel):
✏def=
��V (Req)�� =
Z2e2
4⇡✏0Req
✓1�
1
m
◆(6.4.2)
6.4 Ionische Kristalle

Der Gleichgewichtsabstand Req kann aus der Summe von tabellierten Ionenradien abgeschätzt werden: Req ⇡ R+ + R� (6.4.3)
Lennard - Jones - Parameter
Topftiefe und Kollisionsradius von Dimeren und Siedepunkt der Flüssigkeit
(/k (K) '(pm) Sdp.(K) (/k (K) '(pm) Sdp.(K)
He 10.22 258 4 Cl2 357 412 239
Ne 35.7 279 27 Br2 520 427 332
Ar 124 342 87 CO2 190 400 195
Xe 229 406 166 CH4 137 382 109
H2 33.3 297 20 CCl4 327 588 350
N2 91.5 368 77 C2H4 205 423 169
O2 113 343 90 C6H6 440 527 353
Lennard-Jones-12,6-Potential: V(R) = 4(.{('/R)12 - ('/R)6 }
( = Topftiefe (hier in Kelvin, d.h. k.( hat die Dimension [Energie]), ' = Kollisionsabstand.
Aus J.O.Hirschfelder, C.F.Curtiss, R.Bird, Molecular theory of gases and liquids, Wiley (1954)
Ionen - Radien (pm) (Kristallradien)
Li+ 60 Be2+ 31 B3+ 20 H- 208
Na+ 95 Mg2+ 65 Al3+ 50 F- 136 O2- 140 N3- 171
K+ 133 Ca2+ 99 Sc3+ 81 Cl- 181 S2- 184 P3- 212
Cu+ 96 Zn2+ 74 Ga3+ 62
Rb+ 148 Sr2+ 113 Y3+ 93 Br- 195 Se2- 198 As3- 222
Ag+ 126 Cd2+ 97 In3+ 81
Cs+ 169 Ba2+ 135 La3+ 115 I- 216 Te2- 221 Sb3- 245
Nach Linus Pauling: The Nature of the Chemical Bond, Cornell University Press 1960, p. 514.
Madelung - Konstanten
Gittertyp M____________________________Natriumchlorid 1.747565
Caesiumchlorid 1.76268
Zinkblende 1.63806
Wurtzit 1.641
Fluorit 5.03878
Cuprit 4.11552
Rutil 4.816
E i n f ü h r u n g i n d i e P h y s i k a l i s c h e C h e m i e
K2-6 Bau der Materie
Tabelle 6.4.1: Ionenradien in Kristallen (in pm)
Die molare Gitterenergie des Kristalls EG ergibt sich dann aus der Topftiefe ε zu:
EG = NA✏M (6.4.4)
Die Madelung-Konstante ist ein Proportionalitätsfaktor, der die Abweichung der totalen Gitterenergie von der Summe der Paar-WW-Energien (für ein Mol gleich NA
.ε) korrigiert. Das Paaradditivitätsprinzip ist bei ionischen Kristallen nicht mehr gültig (Warum ?). M hängt von der Symmetrie des Kristalls ab und ist immer grösser als 1 (M>1).
Bsp.: Berechnung der Madelung-Konstante für einen 1D-Kristall → Tafel
6.4 Ionische Kristalle

Für einen 3D-Kristall ergibt sich die Madelung-Konstante aus dreifachen Summen über die x,y,z-Achsen, die mathematisch oft schwierig zu berechnen sind. Madelung-Konstanten sind für die verschiedenen Kristalltypen tabelliert.Bsp.: Madelung-Konstante für einen NaCl-Kristall (kubisch dichteste Kugelpackung):
M =1X
i ,j,k=�1(i ,j,k 6=0)
(�1)i+j+kpi2 + j2 + k2
= 1.748 (6.4.5)
Bsp.: Berechnung der Gitterenergie für NaCl → Tafel
6.5 Struktur kondensierter Phasen
Problemstellung: wir suchen ein mathematisches Kriterium, um die räumliche Ordnung von Molekülen in kondensierten Phasen beschreiben zu können.
Die radiale Verteilungsfunktion g(r): die Funktion
gibt die Wahrscheinlichkeit an, ein Molekül auf einer Kugelschale mit unendlich kleiner Dicke dR im Abstand R von einem anderen Molekül zu finden. g(R) wird als radiale Verteilungsfunktion bezeichnet.
Bem.: g(R) ist etwas anders definiert als die radiale Verteilungsfunktion P(r) für das Elektron im H-Atom, s. Abschn 2.1.2
W (R)dR = R2g(R)dR (6.5.1)
6.5 Struktur kondensierter Phasen

g(R) für verschiedene Aggregatzustände:
• Gas: zufällige Bewegung der Gasteilchen⇒ keine Ordnung
• Flüssigkeit: Bildung von Solvatationshüllen⇒ kurzreichweitige Ordnung
• Kristall: Gitterstruktur mit wohldefinierter Symmetrie⇒ langreichweitige Ordnung
• Wie sieht g(R) für ein ideales Gas aus ?
6.5 Struktur kondensierter Phasen

6.6 Moleküldynamik (MD)-Simulationen
Moleküldynamik (MD)- Simulationen sind ein Ansatz, die Bewegungen (Dynamik) einer grossen Anzahl von Molekülen oder auch von sehr grossen Molekülen theoretisch zu modellieren.
Während in Molekülmechanik (MM)- Rechnungen die Potentialfläche eines Moleküls “erforscht” wird, löst man in MD-Simulationen die klassischen (Newtonschen) Bewegungsgleichungen für das betrachtete Ensemble von Molekülen.
6.6 MD-Simulationen
Gemäss dem 2. Newtonschen Gesetz kann die Kraft Fi, die auf das Teilchen i wirkt, wie folgt formuliert werden:
mit ... Nabla-Operator (=Vektor der 1. Ableitungen)~ri =d
dxi,
d
dyi,
d
dzi
�
Das WW-Potential Vij ist dabei in der Regel eine geeignete Kombination aus intra- und intermolekularen (Modell-) Potentialen, s. Abschn. 3.3 und 5.2, 5.3.
Masse vonTeilchen i
Koordinatenvektor von Teilchen i
Wechselwirkungspotentialzwischen Teilchen i und j
Summe über alle Teilchenin der Simulation
Beschleunigungsvektor von Teilchen i
(6.6.1)~Fi = mi~ai = mid2~ri(t)
dt2= �~ri
✓X
i 6=jVi j(~ri ,~rj)
◆

Die Newtonschen Bewegungsgleichungen für die einzelnen Teilchen Fi=miai sind i.A. gekoppelt (da das WW-Potential die einzelnen Teilchen koppelt, s. Gl. (6.6.1)) und werden numerisch mit dem Computer integriert. Als Ergebnis erhält man die Trajektorien (=Bewegungsbahnen=Ort als Funktion der Zeit) ri(t) jedes Teilchens i.
Beispiel (Molecular and Computational Biophysics Group, University of Illinois at Urbana-Champaign):
MD-Simulation des Wasserflusses durch einen Wasserkanal in einer Zellmembran
(Aquaporin)106’000 Atome, einige ns Simulationsdauer
(E. Tajkhorshid et al., Science 296 (2002), 525
Siehe auch Web-Tutorial “Molecular Dynamics Simulation” auf der Vorlesungs-Webseite (Bestimmung von radialen Verteilungsfunktionen g(R) mit MD-Simulationen) sowie PC-Vertiefungsvorlesungen.
6.6 MD-Simulationen


















