
Expression und Isolierung von Domänen der dsRNA abhängigen ... · Die vorliegende Arbeit wurde im...
86
Aus dem Institut für Anatomie I Zentrum für Experimentelle Medizin Universitätsklinikum Hamburg-Eppendorf Direktorin: Prof. Dr. med. G. Rune Projektleiter: Prof. Dr. Süleyman Ergün Expression und Isolierung von Domänen der dsRNA abhängigen Proteinkinase (PKR) für die Aufklärung der Tertiärstruktur mittels NMR- Spektroskopie. DISSERTATION Zur Erlangung des Grades eines Doktors der Medizin dem Fachbereich Medizin der Universität Hamburg vorgelegt von Mirjam A. Debus, geb. in Altenau in Oberbayern 2007
Transcript of Expression und Isolierung von Domänen der dsRNA abhängigen ... · Die vorliegende Arbeit wurde im...

Aus dem Institut für Anatomie I
Zentrum für Experimentelle Medizin
Universitätsklinikum Hamburg-Eppendorf
Direktorin: Prof. Dr. med. G. Rune
Projektleiter: Prof. Dr. Süleyman Ergün
Expression und Isolierung von Domänen der dsRNA abhängigen Proteinkinase (PKR) für die Aufklärung der Tertiärstruktur mittels NMR-
Spektroskopie.
DISSERTATION
Zur Erlangung des Grades eines Doktors der Medizin dem Fachbereich Medizin der Universität Hamburg vorgelegt von
Mirjam A. Debus, geb. in Altenau in Oberbayern
2007

Angenommen vom Fachbereich Medizin der
Universität Hamburg am:
Veröffentlicht mit Genehmigung des Fachbereichs
Medizin der Universität Hamburg
Prüfungsausschuss, der Vorsitzende: Herr Prof. Dr.med. S. Ergün, Institut für Anatomie
Prüfungsausschuss: 2.Gutachter/in:
Prüfungsausschuss: 3.Gutachter/in:

Die vorliegende Arbeit wurde im Rahmen eines wissenschaftlichen
Austauschprogramms mit der Harvard Medical School in Boston, USA angefertigt
und durch das National Institute of Health, NIH, in Bethesda, Maryland gefördert.
Harvard Medical School
Laboratory for Membrane Transport
Department of Cell Biology
240 Longwood Avenue, Boston, MA
Principle Investigator: Prof. MD José Halperin

Mirjam Debus INHALTSVERZEICHNIS
INHALTSVERZEICHNIS 1 ZIELSETZUNG 1 2 EINFÜHRUNG 2 2.1 PKR als Zielmolekül einer antineoplastischen Therapie 2
2.2 Die Proteinstruktur von PKR 3
2.2.1 Die Kinasedomäne von PKR 4
2.2.2 Die dsRNA-bindende Domäne von PKR 5
2.2.3 Die Domäne 3 von PKR 7
2.3 Die Gewebeverteilung von PKR 7
2.4 Die Regulation der Expression von PKR 8
2.5 Mechanismen der Aktivierung von PKR 8
2.5.1 Zellulärer Stress 8
2.5.2 Aktivierende Liganden: dsRNA, PACT, Heparin 9
2.6 Mechanismen der viralen Inhibition von PKR 11
2.7 Substrate von PKR 12
2.8 Biologische Effekte von PKR 12
2.8.1 Inhibition der Translationsinitation 13
2.8.2 Regulierung der Translation bei verschiedenen mRNA Spezies 14
2.8.3 Regulierung der Gentranskription/ Signaltransduktion 15
2.8.4 PKR und Apoptose 15
2.8.5 PKR und Differenzierung 16
2.8.6 PKR und Tumorsuppression 17
3 MATERIAL 19 3.1 Antikörper 19
3.2 Bakterienstämme 19
3.3 Nukleinsäuren 19
3.3.1 Vektoren 19 3.3.2 Oligonukleotide 19
3.4 Enzyme 19
3.5 Puffer und Reagenzien für die DNA-Analytik 20
3.5.1 Reagenzien für die Plasmidisolation 20
3.5.2 Kits für die DNA Isolation 20
3.6 Puffer und Reagenzien für die Proteinaufreinigung 20
3.6.1 Affinitätsmatrizes 20
3.6.2 Puffer für die Isolierung von MBP-Fusionsproteinen 20

Mirjam Debus INHALTSVERZEICHNIS
3.6.3 Puffer für die Isolierung von CBP-Fusionsproteinen 21
3.6.4 Puffer für die Isolierung von PG-Fusionsproteinen 21
3.7 Puffer und Lösungen für die Proteinanalytik 21
3.7.1 BCA-Assay-Reagenzien 21
3.7.2 Coomassie Blue Stain-Reagenzien 21
3.7.3 Silberfärbungsreagenzien 22
3.8 Chemikalien 22
3.9 Geräte 23
3.8. Software 23
3.9. Datenbanken 23
4 METHODEN 24 4.1 Subklonierung von DNA-Fragmenten in bakterielle Vektoren 24
4.1.1 Plasmid Isolation 24
4.1.2 Determinierung der DNA-Konzentration 24
4.1.3 Analyse von Plasmid DNA durch Restriktionsverdau 25
4.1.4 Amplifikation von Genfragmenten durch PCR 25
4.1.5 Klonierung eines PCR-Fragments 26
4.1.5.1 Präparation des PCR-Fragments und der Vektor-DNA 26
4.1.5.2 Ligation der PCR Fragmene mit Vektor-DANN 26
4.1.5.3 Transformation von Escherichia coli (DH-5α) 27
4.1.5.4 Selektion von positiven Klonen 27
4.1.5.5 Anzüchtung der Bakterienkultur 27
4.1.5.6 Überprüfung der Präsenz von Inserts 27
4.1.6 Sequenzierung von DNA-Fragmenten 28
4.1.6.1 Die Dideoxy-Kettenterminationsmethode 28
4.1.6.2 Entwurf der Primer 28
4.2 Expression der Zielproteine 29
4.2.1 Induktion durch IPTG 29
4.2.2 Aufbereitung der bakteriellen Kulturen 29
4.2.3 Proteinanalyse 30
4.2.3.1 SDS-Polyacrylamid-Gelelektrophorese 30
4.2.3.2 Bestimmung der Proteinkonzentration 30
4.2.3.3 Proteinfärbung mit Coomassieblau 30
4.2.3.4 Silberfärbung der Proteine 31
4.2.3.5 Immunblotting 31
4.2.3.6 Massenspektrometrie 31
4.3 Proteinisolierung mittels HPLC 32

Mirjam Debus INHALTSVERZEICHNIS
4.3.1 Präparation der bakteriellen Kulturen 32
4.3.2 Affinitätschromatographie MBP-PKR mittels HPLC 33 4.3.2.1 Spaltung des Fusionsproteins MBP-PKR 33 4.3.2.2 Trennung von PKR-Fragment und MBP nach der Spaltung 34
4.3.3 Affinitätschromatographie CBP-PKR 34
4.3.3.1 Induktion der Spaltung mittels Faktor X 34
4.3.3.2 Die Elution des Zielproteins 35
4.3.4 Isolierung von PKR-PG Fusionsproteinen 35 4.3.4.1 Elution von PKR-PG Fusionsproteinen 35
4.4 Nukleare Magnetresonanzspektroskopie 36
5 ERGEBNISSE 37 5.1 In bakterielle Vektoren subklonierte PKR DNA-Fragmente 37
5.1.1 Konstrukte mit N-terminaler Polyhistidinsequenz 38
5.1.2 Mutagenese von PKR K296P zu PKR K296R 42
5.1.3 Exprimierte His-PKR K296R 44
5.2 Generierte Fusionsproteinen 45
5.2.1 Klonierte Fusionsgene 46
5.2.2 Expremierte Fusionsproteine 50
5.3 Isolierte PKR-Fragmente nach HPLC 51
5.3.1 Isolierte MBP-PKR K296R -Fragmente 51
5.3.2 Isolierte CBP-PKR K296R –Fragmente 52
5.3.3 Isolierte PG-PKR K296R –Fragmente 54
5.4 Die NMR-Struktur der Kinasedomäne 54
6 DISKUSSION 56 6.1 NMR-Strukturanalysen von PKR liefern Informationen betreffs der Auto-
inhibition 56
6.2 Für einzelne Domänen von PKR konnten individuelle Expressions- und
Isolationsbedingungen für eine NMR-Analytik etabliert werden 57
6.3 Die Bildung von Fusionsproteinen ermöglichte eine Isolation für NMR-
Strukturanalysen 58
6.4 Anhand der NMR-spektroskopischen Daten lässt sich die Rolle von PKR
Bei der Inhibition der Translation untersuchen 59
6.5 Die strukturanalytischen Daten von PKR bieten einen potenziellen neuen
Ansatz für die antineoplastische Therapie auf der Ebene der Translation 60

Mirjam Debus INHALTSVERZEICHNIS
6.6 Ein antineoplastischer Effekt über eine Aktivierung von PKR lässt eine
verminderte Chemo- und Apoptoseresistenz vermuten 62
6.7 Durch den synergistischen Effekt von Interferonen und PKR kann eine
optimierte Therapie bei verminderter Toxizität erzielt werden 64
7 ZUSAMMENFASSUNG 65 8 ABKÜRZUNGEN 67 9 LITERATUR 69 10 DANKSAGUNG 77 11 LEBENSLAUF 78 12 ERKLÄRUNG 79

1
Mirjam Debus ZIELSETZUNG 1 ZIELSETZUNG Die Zielsetzung dieser Arbeit basiert auf der Annahme einer generell gesteigerten
translationalen Aktivität in Tumorzellen, insbesondere einer gesteigerten Expression von
Onkogenen. Die Translation, auf deren Initiation die von doppelsträngiger RNA (dsRNA)
abhängige Proteinkinase (PKR) einen entscheidenden inhibitorischen Effekt ausübt, erscheint
ein geeigneter potenzieller Angriffspunkt für eine antineoplastische Therapie zu sein. Die
strukturellen Informationen von PKR können dem Design von aktivierenden Molekülen
dienen, die eine antineoplastische Therapie von hoher Spezifität und Effizienz bei geringer
Toxizität versprechen.
PKR ist über unterschiedliche Mechanismen antiproliferativ wirksam. Bisher ist nur wenig
über die strukturellen Eigenschaften von PKR bekannt. Für das genauere Verständnis von
Signaltransduktion und molekularen Interaktionen ist die Kenntnis der dreidimensionalen
Struktur der beteiligten Moleküle essentiell. Ein zentrales Anliegen dieses Projektes war es
daher, herauszufinden, ob strukturelle Informationen für die einzelnen Domänen von PKR
und ihrer Interaktionspartner mittels nukleärer Magnetresonanzspektroskopie hergeleitet
werden können. Hierfür galt es zunächst entsprechend geeignete Expressionskonstrukte sowie
stabile Isolierungsbedingungen für verschiedene Fragmente von PKR zu entwickeln.
Katalytisch inaktive Mutanten unterschiedlicher Domänen sollten an geeignete
Fusionsproteine gekoppelt, in E.coli exprimiert, und schließlich isoliert werden. Für die
Isolierung mittels High Performance Liquid Chromatographie (HPLC) mussten Bedingungen
etabliert werden, die eine hohe Reinheit, ausreichende Quantität sowie Stabilität des Proteins
gewährleisten.
Neben der Entwicklung einer möglichen antineoplastischen Therapie, wäre über die
Aktivierung von PKR ein weiterer therapeutischer Ansatzpunkt in einer potenziellen
antiviralen Therapie gegeben, da PKR ebenfalls einen supprimierenden Effekt auf die virale
Replikation ausübt. Weiterhin würden die strukturellen Informationen eine Aufklärung
molekularer Mechanismen der Signaltransduktion erlauben.

2
Mirjam Debus EINFÜHRUNG 2 EINFÜHRUNG
2.1 PKR als Zielmolekül einer antineoplastischen Therapie
Maligne Tumoren stehen in der westlichen Welt nach den Herz- Kreislauf-Erkrankungen an
zweiter Stelle der Statistik für Todesursachen. Jährlich erkranken in der Bundesrepublik
Deutschland 400 pro 100.000 Einwohner an einem bösartigen Tumor. Seit den ersten
Therapieversuchen maligner Lymphome mit „Senfgas“ (2,2’-Dichlordiethylsulfid) und an
Leukämie erkrankter Kinder mit Aminopterin, einem Folsäureantagonisten durch Gilman
(Yale University) und Farber (Harvard University) nach dem 2.Weltkrieg (Gilman 1963),
(Farber 1948), sind mannigfaltige Versuche unternommen worden, die Chemotherapie zu
optimieren. In der konventionellen antineoplastischen Therapie ist der Erfolg einer
Behandlung mittels zytotoxischer Medikamente und Bestrahlung immer noch durch eine
Vielzahl an Faktoren limitiert. Unter anderem durch eine hohe Toxizität, eine geringe
Spezifität, Chemoresistenz und Verlust von Apoptose. Neuere Strategien basieren daher eher
darauf, das Wissen über die molekularen Veränderungen bei malignen Transformationen der
Zellen zu vertiefen, um so zukünftig eine mehr spezifische, weniger toxische Therapie
entwickeln zu können.
Studien der letzten Jahre geben zunehmend Aufschluss für eine Verbindung zwischen der
Regulation von Proteinsynthese und dem abnormalen Zellverhalten, welches für Tumorzellen
typisch ist (Clemens and Bommer 1999). Der Ablauf der Translation wird durch viele
unterschiedliche Protein-Protein- und Protein-RNA-Interaktionen kontrolliert. Besonders
strikt erfolgt dabei die Regulation bei der Initiierung der Translation, an der diverse
Initiationsfaktoren beteiligt sind. Es existieren mehrere parallel verlaufende Mechanismen, die
mit unterschiedlicher Effizienz die Translation starten. Dabei wird davon ausgegangen, dass
die Inhibition lediglich eines Initiationsweges die Translation herunterreguliert, aber nicht
vollständig unterbindet.
Proto-Onkogene und wachstumsfördernde Proteine werden in Abwesenheit von
Initiationsfaktoren nur spärlich translatiert (Koromilas 1992). Ihre mRNAs besitzen
typischerweise hochkomplexe Sekundärstrukturen, wie z.B. hochkomplexe 5’Untranslated
Regions (5’UTRs), Open Reading Frames (ORFs) und Stopcodons. Proto-Onkogene sind
daher entscheidend auf Faktoren angewiesen, welche die Rekrutierung an die Ribosomen

3
Mirjam Debus EINFÜHRUNG
unterstützen (Kozak 1991). Die Entwicklung von Medikamenten für die Krebstherapie, die
spezifisch einen Reaktionsweg der Translationsinitiation blockieren, versprechen daher
antiproliferativ, aber wenig toxisch zu wirken.
Als ein geeignetes Zielmolekül erscheint die von doppelsträngiger RNA (dsRNA) abhängigen
Proteinkinase (PKR). Durch Phosphorylierung des eukaryotischen Initiationsfaktor 2 (eIF2)
bewirkt PKR eine Blockierung von Faktoren, welche die Initiation der Proteinsynthese,
insbesondere die Synthese von Onkoproteinen, unterstützen (Proud 1995). Die PKR ist ein
durch Typ I Interferon induzierbares Enzym, welches häufig in eukaryonten Organismen
vorkommt (Roberts 1976). Diese Serin/ Threonin spezifische Proteinkinase ist bereits seit
Mitte der siebziger Jahre durch ihre Eigenschaften in der antiviralen Abwehr bekannt
(Clemens 1975). Insbesondere in den letzten Jahren wurde sie mit großem Interesse erforscht,
und viele Kenntnisse betreffend ihrer Expression, Interaktionen mit dsRNA und
Proteinmolekülen, sowie Modi ihrer Aktivierung und Inhibierung wurden erlangt (Proud
1995), (Clemens and Elia 1997). Es konnte gezeigt werden, dass PKR in eine Vielzahl
wichtiger zellulärer Prozesse involviert ist. Sie spielt eine zentrale Rolle in der Regulation von
Translation, Transkription und Signaltransduktion (Clemens and Bommer 1999), (Clemens
and Elia 1997). Sie besitzt die Fähigkeit den Proteinsynthesefaktor eIF2, IκB, einen Inhibitor
des Transkriptionsfaktors NFκB, und andere PKR Moleküle zu phosphorylieren. (De Haro
1996), (Kumar 1994). Ihre antiproliferativen Eigenschaften und die Induktion von Apoptose
sind inzwischen von zentraler Bedeutung (Der 1997), (Jaramillo 1995). Bisher ist es
allerdings nicht gelungen, die komplette dreidimensionale Struktur sowie die strukturellen
Beziehungen zu den entsprechenden Interaktionspartnern von PKR aufzuklären.
2.2 Die Proteinstruktur von PKR
Die Primärstruktur der humanen PKR, abgeleitet von ihrer cDNA, zeigt ein 68 kD Protein mit
551 Aminosäuren (Meurs 1990). Sie weist eine aminoterminale regulatorische Domäne,
sowie eine carboxyterminale katalytische Domäne auf (Abb. 2.1). Innerhalb der katalytischen
Domäne ist die ATP-bindende Region mit dem ATP-akzeptierenden Lysin an Position 296
lokalisiert, welches essentiell für die Phosphatübertragung ist. Mutationen dieses Lysins zu
Arginin oder Prolin führen zur katalytischen Inaktivität. Die regulatorische Domäne vereint
zwei Kopien eines dsRNA-bindenden Motivs, welches reich an basischen Aminosäuren ist.
Sie sind vergleichbar mit bekannten dsRNA Bindungsmotiven (ST Johnson 1992), (Katze

4
Mirjam Debus EINFÜHRUNG
1991). Außerdem ist die regulatorische Domäne für die Lokalisation an den Ribosomen
verantwortlich. Zusätzlich an der Regulation beteiligt ist eine dritte, bisher weitgehend
unerforschte basische Domäne, die Domäne 3 (Aminosäuren 167-248). Die Region von PKR,
die mit eIF2α interagiert, befindet sich zwischen Aminosäure 367 und 551.
R1 R2 KinaseD3
167 248 267 538296
Regulatorische Domäne Katalytische Domäne
367
eIF2α siteLysATP site
Heparin site
PKR Struktur:
Abb. 2.1: Die Struktur von PKR mit den RNA-Bindungsdomänen (R1 und R2), der regulatorische Domäne 3
und der katalytischen Domäne mit dem ATP akzeptierenden Lysin an Position 296.
2.2.1 Die Kinasedomäne von PKR
Die Kinasedomäne umfasst zentral eine Substrat-Spezifitätsregion und zwei Kinase-
Subdomänen an jeder Seite. Eine Sequenzhomologie besteht zu 30 % mit der Kinasedomäne
des basic Fibroblast Growth Factor Rezeptors 2, FGF-2-Rezeptors, dessen Tertiärstruktur
bekannt ist. Darauf basierend wurden Homologiemodelle generiert, um
Sekundärstrukturelemente zu lokalisieren (Abb. 2.2).

5
Mirjam Debus EINFÜHRUNG
Abb. 2.2: PKR Homologiemodell, Wagner et al. 2000 Es finden sich einige konservierte Regionen von bekannten Kinasedomänen anderer
Proteinkinasen (Meurs 1990). Struktur-Funktionsstudien haben gezeigt, dass die Deletion
einer Kinase-Insertsequenz oder Mutationen von Serin 355 die Kinaseaktivität reduzieren
(Craig, Cosentino et al. 1996). Vor kurzem konnte Dar et al. in Kristallisationsstudien zeigen,
dass der C-Terminus der katalytischen Domäne für die Interaktion eIF2α verantwortlich ist
(Dar A C 2005). Einige Proteinkinasen, so auch PKR, werden durch Phosphorylierung in der
Region zwischen zwei Kinase-Subdomänen, dem sogenannten “Activation Loop“ aktiviert. In
dem PKR Activation Loop sind Thr-446 and Thr-451 für eine hohe Kinaseaktivität essentiell.
Mutationen eines dieser Threoninreste zu Alanin beeinträchtigt die translationelle Kontrolle
durch PKR in Hefe- und COS1 Zellen und führt zu Tumorformation in Mäusen.
Autophosphorylierung von PKR und Phosphorylierung von eIF2α sind dadurch gestört
(Romano, Garcia-Barrio et al. 1998).
2.2.2 Die dsRNA-bindende Domäne von PKR
Die Klonierung der humanen PKR-cDNA Anfang der 90’er Jahre hat erstmalig detailliertere
Struktur-Funktionsstudien der Kinase erlaubt (Meurs 1990), (Feng 1992). Auf diese Weise
konnte die Domäne, welche in die Interaktion mit dsRNA und konsekutiver katalytischer
Aktivierung in virusinfizierten Zellen involviert ist, identifiziert werden [Patel,

6
Mirjam Debus EINFÜHRUNG
1992 #207]. Durch die Bindung von dsRNA wird ebenfalls die autokatalytische Aktivität von
PKR induziert (Galabru 1987). Die dsRNA-bindende Domänen (DRBDs) sind innerhalb der
ersten 171 Aminosäuren am N-Terminus lokalisiert (Katze 1991), (Patel 1994). Diese Region
umfasst zwei Folgen eines Motivs, welches häufig in anderen dsRNA-bindenden Proteinen
gefunden wurde (ST Johnson 1992). Die beiden DRBD-Motive unterscheiden sich in ihrer
Fähigkeit, dsRNA zu binden. Obwohl beide Motive benötigt werden, zeigt die DRBD I eine
höhere Affinität für dsRNA. Sie ist reich an basischen und aromatischen Aminosäureresten.
Diese Reste sind in die elektrostatische Interaktionen mit dem Phosphatrückrad der RNA
sowie der Interkalationen zwischen den Nukleotiden involviert. Es wurde gezeigt, dass die
aminoterminalen 24 Reste unentbehrlich für die Bindung und konsekutive Aktivierung durch
dsRNA sind (Patel 1994). Die DRBD I ist außerdem in die Protein-Protein Interaktionen und
die Dimerisierung von PKR-Molekülen involviert (Abb. 2.3).
P K R
P K R
D ie D im e r is ie ru n g v o n P K R d u rc h d s R N A
d s R N A
NNN
C
N C
PKR
PKR
C C
Abb. 2.3: Dimerisierung von PKR über die Bindung von dsRNA.
Die DRBD II interagiert intramolekular mit der Kinasedomäne, was eine “verschlossene”
inaktive Konformation gewährleistet. Dieses wird wahrscheinlich durch Maskierung der
Dimerisationsdomäne erreicht (Nanduri 2000). PKR bindet unabhängig von mRNA an die
40S ribosomale Untereinheit. Es konnte nachgewiesen werden, dass die DRBDs für die
Bindung an die Ribosomen erforderlich sind (Zhu, Romano et al. 1997). Ribosomen selbst
üben einen inhibitorischen Effekt auf PKR aus, da sie mit der dsRNA um die Bindung
konkurrieren (Raine 1998). Die Kristallstrukturen sowie NMR-Strukturen der DRBDs
mehrerer

7
Mirjam Debus EINFÜHRUNG
Proteine, einschließlich der von PKR, sind bestimmt worden (Ryter 1998), (Nanduri 1998).
2.2.3 Die Domäne 3 von PKR
Nur wenig ist über die Struktur-Funktionsbeziehung einer weiteren Domäne, der sogenannten
Domäne 3 (Aminosäuren 234-272), bekannt. Struktur-Funktionsstudien zeigen, dass diese
Domäne ebenfalls in die Regulation des durch PKR-vermittelten translationellen Blocks
involviert ist (Lee 1994). Deletion der Sequenz 234-272 beeinträchtigt sowohl die
apoptotische Aktivität, als auch die Fähigkeit, die Proteinsynthese zu inhibieren (Lee,
Rodriguez et al. 1997). Zurzeit gibt es keine Informationen über die Tertiärstruktur der
Domäne 3. Sie ist reich an basischen Aminosäureresten und besitzt eine Folge von 12
Argininresten, wie sie in heparinbindenden Proteinen wie beispielsweise Apolipoprotein E zu
finden sind (Margalit H 1993). Auch PKR ist in der Lage an Heparin zu binden. Es ist
anzunehmen, dass Domäne 3 die heparinbindende Domäne von PKR ist. Diese Region besitzt
zudem autoregulatorische Eigenschaften (Taylor 1996).
2.3 Die Gewebeverteilung von PKR
PKR wird nahezu ubiquitär in allen Zelltypen exprimiert, allerdings variiert die
Expressionsrate in Abhängigkeit vom Differenzierungsgrad. Hohe PKR-Konzentrationen
wurden in einer Vielzahl von differenzierten Geweben, insbesondere in epithelialen Zelltypen
gefunden. Hingegen konnten in Blasten und unreifen Mesenchymzellen, also in Zellen, die
eine hohe Proliferationsrate aufweisen, PKR nicht nachgewiesen werden. (Haines 1993). Die
Expression von PKR in malignen Tumoren zeigt sich Gewebespezifisch. Häufig werden
niedrige Konzentrationen gefunden. In einigen malignen Tumoren, wie z.B. dem
kolorektalen Karzinom konnten allerdings hohe Level einer dominant negativen Form von
PKR festgestellt werden (Kim, Gunnery et al. 2002).

8
Mirjam Debus EINFÜHRUNG 2.4 Die Regulation der Expression von PKR
Die Transkription von PKR ist nach Behandlung mit Typ I Interferonen um das 5-10 fache
hochreguliert (Roberts 1976). Inwiefern andere Zytokine neben Interferonen in der Lage sein
könnten, PKR zu regulieren, wurde bisher nicht ausreichend untersucht. Es gibt jedoch
Grund zur Annahme für eine Induktion durch TNF-α (Yeung 1996). Andere Beobachtungen
lassen vermuten, dass der transformierende Wachstumsfaktor-β (TGFβ) die
Expression von PKR inhibieren könnte (Salzberg 1995).
2.5 Mechanismen der Aktivierung von PKR
PKR existiert in latenter Form in den meisten Säugerzellen und wird unter anderem während
einer Virusinfektion direkt durch dsRNA aktiviert, welche im Verlauf der viralen Replikation
entsteht. Aber auch diverse Einflüsse, welche zellulären Stress verursachen, haben die
Aktivierung von PKR zur Folge. Die Homodimerisierung ist entscheidend für die PKR
Aktivität in vivo. Eine primäre Funktion der dsRNA, die an die RNA-bindende Domäne
nativer PKR bindet, ist die Förderung der Dimerisierung von PKR-Molekülen mit
konsekutiver Aktivierung der Kinasedomäne (Ung, Cao et al. 2001).
2.5.1 Zellulärer Stress
Die Phosphorylierung von eIF2α durch PKR scheint häufig von einer Signalkaskade
abzuhängen, welche vom endoplasmatischen Retikulum ausgeht. Erhöhte Phosphorylierung
des Initiationsfaktors eIF2α tritt als Reaktion auf unterschiedliche Agenzien auf, welche einen
störenden Einfluss auf das endoplasmatische Retikulum ausüben. Zu ihnen gehören diverse
Ca2+ entleerende Substanzen, wie DTT, Natriumarsenid und Tunicamycin (Prostko 1993),
Clotrimazol (Aktas, Fluckiger et al. 1998), Eicosapentoinsäure (Palakurthi 2000),
Thiazolidinedione (Palakurthi 2001). Die Entleerung von endoplasmatischem Ca2+ führt zur
Induktion von Glukose-regulierten Proteinchaperonen und Hitzeschock-Proteinchaperonen
mit konsekutiver Störung der Proteinfaltung und Modifizierungen innerhalb des Organells,
was die Aktivierung von PKR zur Folge hat (Brostrom, Prostko et al. 1996). Vermutlich
verursacht die Akkumulation von ungefalteten Proteinen einen zellulären Stress, der indirekt
in einer Aktivierung von PKR, und so in einer Herabregulation der Protein- synthese
resultiert. Der genaue Mechanismus ist noch ungeklärt. Es konnte gezeigt werden,

9
Mirjam Debus EINFÜHRUNG
dass durch die Freisetzung von Ca2+ aus intrazellulären Speichern bevorzugt die Synthese und
Expression von G1 Cyclinen inhibiert, was in einem Zellzyklusblock in der G1-Phase
resultiert (Abb. 2.4) (Aktas, Fluckiger et al. 1998).
Protein-synthese
PeIF2α
PeIF2α
eIF2αeIF2α
PKRPKR
G1 Cycline (D,E)
Rb-P
Cycline-cdk
CLTEPATZD
E2
Ca Ca2+2+
G1
E2
S
ER
Abb. 2.4: Die Freisetzung von Ca2+ aus dem endoplasmatischen Retikulum (ER) durch Clotrimazol (CLT), Eicosapentoinsäure (EPA) und Thiazolidinedione (TZD) führt über die Aktivierung von PKR und einer Phosphorylation von eIF2α zur Inhibition der Translationsinitiation. Konsekutiv kommt es zu einer Herabregulation der Proteinsynthese Zellzyklus-relevanter Proteine und so zu einer Blockade in der G1-Phase.
2.5.2 Aktivierende Liganden: dsRNA, PACT, Heparin
Es existiert folgendes Modell für die Reihenfolge von Ereignissen, die zur Aktivierung von
PKR durch dsRNA führt: dsRNA assoziiert mit der dsRNA-bindenden Domäne 1 und 2.
Konsekutiv kommt es zur Freisetzung von aminoterminalen inhibitorischen Sequenzen und
einer aktivierenden Konformationsänderung in PKR. Die Interaktion zwischen PKR
Molekülen ist zudem gesteigert durch Oligomerisation und Assoziation an Ribosomen, was
eine erhöhte lokalisierte Konzentration zur Folge hat (Abb. 2.5) (Vattem, Staschke et al.
2001).

10
Mirjam Debus EINFÜHRUNG
PKRHeparin
PKR
PKR PKR
P
Die Aktivierung von PKR
dsRNA
P
Abb. 2.5: Aktivierungsmodell von PKR: Durch die Bindung von Liganden wie dsRNA oder Heparin erfolgt die
Dimerisierung mit konsekutiver Autophosphorylierung von PKR.
Bis vor kurzem war allerdings unklar, wie die PKR Aktivierung in nicht viral infizierten
Zellen als Reaktion auf die vielfältigen extrazellulären Stimuli vermittelt wird. Vor wenigen
Jahren wurde ein endogener Proteinaktivator für PKR (PACT) identifiziert, und es ist
wahrscheinlich, dass PACT eine entscheidende, obwohl bisher noch undefinierte Rolle in
diesem Prozess spielt. PACT wird als Antwort auf diverse Stresssignale, wie Serumentzug,
Peroxyd- oder Arsenidbehandlung in der Abwesenheit von viralen Infektionen hochreguliert
(Patel and Sen 1998), (Patel 2000), (Williams 1999). PACT wird ubiquitär in niedriger
Konzentration exprimiert, was weder durch Interferone, noch durch dsRNA vermittelt wird.
PACT besitzt typische Domänen, welche für die Vermittlung von Protein-Protein
Interaktionen zwischen den Mitgliedern der PKR-Familie von dsRNA-bindenden Proteinen
bekannt sind und ist in der Lage direkt an PKR zu binden. Ein weiteres PKR aktivierendes
Molekül ist Heparin. Die strukturelle und funktionelle Basis dieser PKR Aktivierung
unterscheidet sich allerdings von derjenigen der dsRNA (Patel 1994), (George 1996). Bis vor
kurzem konnte nur die in vitro Aktivierung durch Heparin nachgewiesen werden, da eine
zelluläre Aufnahme von Heparin in der Regel nicht möglich ist. Jetzt gelang allerdings in
vaskulären glatten Muskelzellen (VSMCs) der Nachweis einer Aktivierung in vivo. Die
Behandlung von VSMCs mit Heparin resultiert in der Aktivierung von PKR durch direkte
Bindung und führt zur Blockade des Übergangs von G1- zu S-Phase. Die Internalisierung und
die Bindung wurde mittels Immunpräzipitationsassays mit einem

11
Mirjam Debus EINFÜHRUNG
PKR Antikörper nach erfolgter Behandlung der VSMCs mit 35S-markiertem Heparin
nachgewiesen (Patel 2002).
2.6 Mechanismen der viralen Inhibition von PKR
Viren haben effektive Mechanismen entwickelt, um die PKR Aktivität während der
Transfektion zu unterdrücken. PKR ist das Zielmolekül für die Regulation durch diverse viral
kodierte inhibitorische Moleküle (Gale and Katze 1998). Adenoviren und Epstein-Bar Viren
generieren ein virales Genprodukt, welches PKR durch direkte Interaktion mit der
aminoterminalen regulatorischen Domäne inaktiviert, und so als kompetitiver Inhibitor von
dsRNA-Bindingsreaktionen funktioniert (Galabru 1989). Daneben existiert auch ein zelluläres
Protein, das sogenannte Tar RNA Binding Protein, welches an die dsRNA-Bindungsdomäne
von PKR bindet (Park, Davies et al. 1994). Virusprodukte von Hepatitis-C Viren und
Baculoviren besitzen die Fähigkeit, die Kinasedomäne von PKR als Pseudosubstrat direkt zu
blockieren (Taylor 2000), (Taylor, Shi et al. 1999). Des Weiteren wurde ein zellulärer PKR-
Inhibitor identifiziert, p58IPK (Barber 1994). p58IPK, ist ein durch Influenzavirus aktiviertes
Co-chaperon, welches wahrscheinlich das Hitzeschockprotein hsp/Hsc70 zur Neufaltung
veranlasst, und so die Kinasefunktion inhibiert (Abb. 2.6) (Melville, Tan et al. 1999).
Inhibition von PKR durch p58 IPK
PKR
PKR
PKR PKR
p58 IPK
P P
Abb. 2.6: Inhibition der Autophosphorylierung von PKR durch das zelluläre Co-Chaperon p58IPK.

12
Mirjam Debus EINFÜHRUNG
Hepatitis C Viren haben verschiedene Mechanismen zur Inhibition von PKR entwickelt. Das
Virusprodukt NS5A, ist in der Lage, Heterodimere mit PKR zu bilden, was die Formation von
PKR-Homodimeren unterbindet (Gale 1997). Der kombinierte Effekt könnte die ausgeprägte
Resistenz bestimmter HCV-Subtypen gegenüber Interferonen, und so die hohe Persistenzrate
dieser Subtypen erklären. Zudem ist ein Sekundäreffekt der PKR-Inhibition eine Stimulation
der Wirtszellenproliferation, was für die Bildung von HCV-assoziierten Hepatozellulären
Karzinomen eine Rolle spielen könnte (Taylor 2000).
HIV-1 hat multiple Mechanismen entwickelt, um die durch PKR und Interferon induzierte
antivirale Reaktion zu umgehen (Barber 1994), (Katze 1990). Das Poliovirus ist in der Lage
PKR zu degradieren (Black 1993).
2.7 Substrate von PKR
PKR katalysiert die intermolekulare Phosphorylierung der α-Untereinheit des
Translationsinitiationsfaktors eIF2, und phosphoryliert außerdem andere PKR Moleküle
sowie den zellulären Inhibitor des Transkriptionsfaktors NF-κB, IκB (Clemens 1994).
(Kumar 1994). Die vielfältigen zellulären Prozesse, in welche PKR involviert ist, lassen die
Existenz weiterer Substrate vermuten. In vitro Experimente haben gezeigt, dass PKR mit dem
Tumorsuppressor p53 assoziiert ist und diesen auch phosphoryliert (Cuddihy, Wong et al.
1999).
2.8 Biologische Effekte von PKR
Studien der letzten Jahre haben die große Vielfalt der durch PKR vermittelten biologischen
Prozesse verdeutlicht. Neben den antiviralen Eigenschaften haben in erster Linie die
verschiedenen wachstumsinhibierenden Prozesse an Interesse gewonnen. Sie werden zum
einen auf der Ebene der Genexpression, zum anderen durch direkte oder indirekte Induktion
der Apoptose vermittelt. Ein weiteres Forschungsfeld bietet die Rolle von PKR bei der
Differenzierung.

13
Mirjam Debus EINFÜHRUNG
2.8.1 Inhibition der Translationsinitation
Die Rekrutierung der tRNA und mRNA zu der kleinen ribosomalen Untereinheit, welcher als
Translationsinitiation bezeichnet wird, unterliegt einer besonders ausgeprägten Regulation.
Dieser Prozess wird vermittelt durch die Translationsinitiationsfaktoren eIF2α, eIF2B und
deren assoziierte Faktoren (Pain 1996). Es bildet sich dabei zunächst ein ternärer Komplex
aus dem Initiationsfaktor 2α (eIF2α), Guanosintriphosphat (GTP) und Methionin-tRNA.
Dieser Komplex bindet an die kleine ribosomale Untereinheit und bildet den
Präinitiationskomplex. Unter Beteiligung weiterer Initiationsfaktoren wird dieser Komplex
am 5’- Ende der mRNA gebunden. Der Prä-Initiationskomplex wandert nun entlang der
mRNA bis zum Startkodon, wo er zusammen mit der ribosomalen 60S-Untereinheit das 80S-
Ribosom bildet und die Translation beginnt (Abb. 2.7).
Die Initiation der Translation
40S
40S
Met-tRNA + eIF2 GTP
46S
40S60S
60S
43S mRNAandere Faktoren
eIF5
eIF2 GDPandere Faktoren
eIF2 GDP
PKR
Abb. 2.7: Die Initiation der Translation.
Während der Initiation wird GTP zu GDP hydrolysiert. Um einen neuen Initiationsvorgang
einleiten zu können, muss GDP durch GTP ausgetauscht werden. Dieser Vorgang wird durch
eIF2B katalysiert und kann durch PKR inhibiert werden. Die Aktivierung von PKR führt zur
Phosphorylierung von eIF2α (Levin 1978). eIF2B bildet dadurch einen festen Komplex

14
Mirjam Debus EINFÜHRUNG
mit eIF2α, wodurch der GDP- GTP Austausch blockiert wird (Abb. 2.8). Dieses resultiert in
einer allgemeinen Verknappung von Translationsinitiationsfaktoren, und führt damit zur
ineffizienten Translation wichtiger zellzyklusregulierender Proteine (wie z.B. Cyclin D1,
Cyclin E). Auf diese Weise kommt es zu einem Wachstumsstillstand in der G1 Phase.
Inh ib ition der T ransla tions in itia tion
eIF2 G TP
eIF2 G D P PK R
eIF2α G D PP
eIF2α G D PP
eIF2B
eIF2B
Abb. 2.8: Die Initiation der Translation wird durch PKR über die Phosphorylierung von eIF2α inhibiert.
2.8.2 Regulierung der Translation bei verschiedenen mRNA Spezies
Die Phosphorylierung von eIF2α dient der Feinregulation der translationellen Effizienz von
verschiedenen mRNA-Subtypen. Proteine haben unterschiedliche mRNA Spezies, die mehr
als hundertfach in ihrer translationellen Effizienz variieren. Substanzen, welche die globale
Rate der Polypetidketteninitiation reduzieren, inhibieren die Translation bevorzugt von
mRNAs mit niedriger konstanter Rate für die Polypeptidketteninitiation. (Lodish 1974). Es
konnte gezeigt werden, dass die primäre Nukleotidsequenz des Initiationscodons und/oder die
Tertiärstruktur der mRNA in der Nähe des Codons die Rate der Ribosomenbindung
bestimmen (Lodish 1970), (Steitz 1969). Viele mRNAs die für wichtige
zellzyklusregulierende Proteine codieren, zeigen eine ineffiziente Translation. Sie weisen zum
Beispiel hochkomplexe 5’ UTRs auf, was die Rekrutierung an die Ribosomen erschwert und
dadurch in einer stärkeren Abhängigkeit von TI-Faktoren resultiert (Kozak 1991).

15
Mirjam Debus EINFÜHRUNG
2.8.3 Regulierung der Gentranskription/ Signaltransduktion
PKR reguliert die Aktivierung verschiedener Transkriptionsfaktoren, wie unter anderen NF-
κB und p53. (Kumar 1997), (Cuddihy, Wong et al. 1999). Auch konnte gezeigt werden, dass
die dsRNA induzierte TNF-α Genexpression in humanen Epithelzellen durch PKR vermittelt
wird. (Meusel 2002).
Dem Transkriptionsfaktor NFκB werden in erster Linie antiapoptotische Wirkmechanismen
zugesprochen. In der letzten Zeit erreicht zunehmend die Induktion proapoptotischer
Signalkaskaden durch NFκB an Relevanz (RyanKM, 2000). NFκB stellt den Knotenpunkt für
die Transkription diverser Gene wie FasL, Fas, IRF-1, IFNβ, Kaspase 1 oder p53 dar. Die
Expression dieser Gene resultiert in der Aktivierung von Kaspasen, und somit in der
Einleitung von Apoptose (Chu 1999), (Gil, Alcami et al. 1999). NF-κB setzt sich aus Dimeren
verschiedener Mitglieder der Rel Proteinfamilie zusammen (Gosh 1998). Die häufigste Form
von NF-κB ist ein Heterodimer von p50 und p65 (RelA), welches im Zytosol durch die IκB
Proteine sequestriert wird. Dieses inhibiert die nukleäre Translokation und die DNA-bindende
Aktivität von NF-κB (Verma 1995). Die Rel/ NF-κB Familie der Transkriptionsfaktoren
steigern die Expression von Genen, welche in immunologische- und inflammatorische
Reaktionen, in Zelldifferenzierung und in die Kontrolle der Apoptose, sowie in andere
wichtige Prozesse involviert sind (Gosh 1998). Es wurde gezeigt, dass die katalytische
Aktivität von PKR für die NF-κB Aktivierung benötigt wird. Die Phosphorylierung von IκBα
durch den über PKR aktivierten IκB Kinase Komplex (IKK) und spätere Degradation des
Proteins ist dabei erforderlich (Gil 2001). 2.8.4 PKR und Apoptose
In zahlreichen Studien wurde PKR eine entscheidende Rolle bei der Apoptose zugeschrieben.
Die Mechanismen sind vielfältig und noch nicht eindeutig geklärt. Als erwiesen gilt aber, dass
verschiene Stimuli für die Induktion von Apoptose durch PKR verantwortlich sind. Bekannte
PKR-Aktivatoren wie dsRNA, Lipopolysaccharide, TNF-α, Serumentzug, IL-3 Entzug, Ca2+-
Entleerung aus dem endoplasmatischen Retikulum, PACT Aktivierung, sowie die
Blockierung von PKR-Inhibitoren waren allesamt in der Lage Apoptose zu induzieren (Gil
and Esteban 2000). Auch wurde eine gesteigerte Phosphorylierung von eIF2α bei der
Apoptoseinduktion beobachtet, was auf eine PKR-Involvierung schließen lässt (Srivastava,

16
Mirjam Debus EINFÜHRUNG
Kumar et al. 1998), (Gil, Alcami et al. 1999). Neuere Studien zeigen, dass PKR zu einem
Zellzyklusblock in der G2/M Phase führt, und die Apoptose in PKR-überexprimierenden
Zellen induziert. Dieser durch PKR induzierte G2/M Block konnte durch ein „Onkogen-like
v-mos“ aufgehoben werden (Dagon 2001). Des Weiteren hat sich gezeigt, dass Fibroblasten
von PKR-/- Mausembryonen gegenüber einer TNFα-induzierten Apoptose resistent sind (Der
1997).
Offenbar spielen viele unterschiedliche Mechanismen bei der Induktion von Apoptose durch
PKR eine Rolle. Zum einen ist PKR durch Regulation der Translation, zum anderen über die
Modulation von Transkription und anderen Signaltranduktionswegen in der Lage Apoptose
einzuleiten. Zum Teil ist ihre Wirkung der Kaspasenkaskade vorgeschaltet beschrieben.
Arbeiten, welche die Signaltransduktionswege in Reaktion auf TNF-α untersuchen, haben zu
dem Ergebnis geführt, dass PKR oder eine PKR-abhängige Signalkomponente auf der
Signalkaskade oberhalb des Death Domain Receptors und der Initiatorkaspase, Kaspase 8,
liegt (Jeffrey, Bushell et al. 2002), (Balachandran 1998). Andererseits wurde auch eine
Kaspase-abhängige PKR Aktivierung gezeigt. Es konnte bewiesen werden, dass PKR in
frühen Stadien der Apoptose proteolysiert und eIF2α phosphoryliert wird. Durch die
Abspaltung einer aminoterminalen inhibitorischen Sequenz ist PKR konstitutiv in einem
aktivem Zustand (Saelens, Kalai et al. 2001).
Der Apoptoseinhibitor Bcl-2 ist in der Lage PKR induzierte Apoptose zu inhibieren. Die
Translation wird dabei jedoch nicht beeinträchtigt. Dieses lässt vermuten, dass Bcl-2 keinen
direkten Einfluss auf PKR hat, allerdings in der nachgeschalteten Signalkaskade noch
oberhalb der CPP32-Proteasekaskade seinen inhibitorischen Effekt ausübt (Lee, Rodriguez et
al. 1997).
2.8.5 PKR und Differenzierung
Zellen, welche die terminale Differenzierung durchlaufen, zeigen deutliche Veränderungen in
ihrem Muster der Genexpression. Es ist wenig über die Mechanismen bekannt, welche die
Translation der meisten Transkripte inhibieren, während die Translation spezifischer mRNAs
in dem Verlauf der Differenzierung aktiviert wird. Es gibt Anhalt dafür, dass mRNAs, welche
Interferone Responsive Elemente (IRES) innerhalb langer, hochstrukturierter und ORF-
beladener 5’UTRs aufweisen, eine Untergruppe umfassen, die während der Differenzierung
spezifisch translational aktiviert wird. Diese spezifische Regulation auf Ebene der Translation

17
Mirjam Debus EINFÜHRUNG
ist nachweislich an die Phosphorylierung von eIF2α und die nachfolgende Inhibition der
Proteinsynthese gekoppelt und somit PKR abhängig (Gerlitz, Jagus et al. 2002).
2.8.6 PKR und Tumorsuppression
PKR scheint ihre antitumorgene Aktivität über die Aktivierung multipler Transduktionswege
zu vermitteln. Sie alle kumulieren in Wachstumsinhibition und Induktion von Apoptose. Der
direkte Zusammenhang zwischen Tumorregression und der Aktivierung von PKR konnte
vielfach nachgewiesen werden.
Anfang der 90er Jahre gelang der Nachweis, dass die Expression einer katalytisch inaktiven
Mutante von PKR in NIH 3T3 Zellen in maligner Transformation resultiert (Koromilas 1992).
Diese Mutanten zeigten einen dominant negativen Effekt auf Wildtyp PKR in vitro,
wahrscheinlich durch Kompetition um die dsRNA Bindung (Sharp, Xiao et al. 1993). Auch
Mutanten mit fehlender dsRNA Bindungsdomäne I imponierten als transdominante
Inhibitoren, die maligne Transformation induzieren (Barber, Jagus et al. 1995). In humanen T
Zell Leukämiezellen fand man eine alternativ gespleißte Form von PKR. Bei ihr resultiert
eine Deletion des Exon 7 (PKRΔ7) in einem trunkiertem Protein mit fehlenden dsRNA-
bindenden Motiven. PKRΔ7 zeigt eine dominant negative Funktion, indem sie die PKR
Autophosphorylierung und eIF2α Phosphorylierung inhibiert (Li 2001).
Erst vor kurzem konnte bewiesen werden, dass der neuentdeckte Tumorsuppressor, das
Melanom Differenzierungs-assoziierte Gen 7 (mda7), die Apoptose in einer Vielzahl von
Tumorzelllinien induziert. Unter anderem führt er zur Regression bei dem kolorektalen
Karzinom und beim Mammakarzinom. Es konnte gezeigt werden, dass die mda7-Expression
in der Induktion und Aktivierung von PKR, mit konsekutiver Phosphorylierung von eIF2α
und Apoptose resultiert (Pataer, Vorburger et al. 2002).
Der tumorsuppressive Effekt von PKR wurde teilweise in Frage gestellt, als bekannt wurde,
dass verschiedene Tumoren, wie Melanome, Kolonkarzinome und Mammakarzinome eine
gesteigerte PKR-Expression und -Aktivität aufweisen (Kim, Gunnery et al. 2002), (Kim,
Forman et al. 2000). Dieses kann zum einen als reaktive Regulation gewertet werden,
andererseits konnte in weiteren Studien der Nachweis erbracht werden, dass bestimmte

18
Mirjam Debus EINFÜHRUNG
Mammakarzinomzellen, MCF7-Zellen, eine gesteigerte PKR inhibierende Aktivität
aufweisen (Savinova 1999).
Die antineoplastischen Eigenschaften von PKR haben die viel diskutierte Frage aufgeworfen,
ob PKR als Tumorsuppressorprotein gewertet werden sollte. Diese Frage konnte bisher nicht
abschließend beantwortet werden. Allerdings muss unterstrichen werden, dass die klassischen
Kriterien eines Tumorsuppressorproteins für PKR nicht zutreffen. Ein Verlust von PKR hat
nicht unweigerlich eine maligne Transformation zur Folge. Beispielsweise wurde in
transgenen Mäusen mit gezielter Mutation der katalytischen Domäne kein Verlust der
tumorsuppressiven Wirkung durch PKR beobachtet (Abraham 1999). In späteren Studien
wurde die Verlässlichkeit dieser Ergebnisse jedoch angezweifelt. Die Resultate mit Zellen
zweier PKR-/- Modelle waren widersprüchlich und verwirrend. Neuere Daten demonstrieren,
dass es sich bei den PKR-/- Modellen um inkomplette Knockoutmäuse handelte (Baltzis, Li et
al. 2002).
In Zusammenschau der genannten Untersuchungen wird die entscheidende Rolle der
Interferon-induzierbaren, dsRNA abhängigen Proteinkinase, PKR, bei diversen zellulären
Prozessen deutlich. Insbesondere die Schlüsselrolle bei der Regulation von Zellwachstum und
Differenzierung sowie ihre potenziellen antineoplastischen Kapazitäten, ferner aber auch ihre
Relevanz in der zellulären Virusabwehr, gaben den Anlass bei dem vorliegenden Projekt, die
Kenntnisse der strukturellen und funktionellen Eigenschaften von PKR zu vertiefen. Da die
dreidimensionale Struktur von PKR und die für die Interaktion mit regulierenden Molekülen
entscheidenden Sequenzen nur in Teilen bekannt war, sollten im Rahmen dieser Arbeit durch
NMR-spektroskopische Untersuchungen wesentliche Informationen diesbezüglich generiert
werden.

19
Mirjam Debus MATERIAL 3 MATERIAL 3.1 Antikörper PKR-Antikörper, C-Terminus, polyclonal . . . . . . . . . . . . . . . . . . . . .Santa-Cruz
PKR-Antikörper, N-Terminus. . . . . . . . . . . . . . . . . . . . . . . . . . . .Santa-Cruz
His-Antikörper. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Chemicon
Sekundärantikörper. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Chemicon
3.2 Bakterienstämme E. coli DH5α . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .I nvitrogen
E. coli BL21DE3gold . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Stratagene
3.3 Nukleinsäuren dATP, dCTP, dGTP, dTTP. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Gibco 3.3.1 Vektoren pBISA PKR K296P GFP I (6000 bp) Aktas et al. pET-15b (5708bp) Novagen pTYB12(7414 bp) New England Biolabs pMal -c2X (6648 bp) New England Biolabs pGEV2 (6382 bp) Wagner et al. Wagner et al. 3.3.2 Oligonukleotide: 5’Δ196-NdeI CGGCAGCCATATGAGCACACTCGCTTCTGAA Gene link, NY5’Δ262-NdeI CGGCAGCCATATGAGG TTTGGCATGGATTT Gene link, NY 5’PKR Δ179- BamHI CGCGGATCCGGTTCTTTTGCTACTACGTGT Gene link, NY5’PKR Δ262- BamHI CGCGGATCCAGGTTTGGCATGGATTTTAAAGAA Gene link, NY3’gev2 XhoI CGGCTCGAGACATGTGTGTCGTTCATT Gene link, NY3’pTYB12 XhoI CGGCTCGAGCTAACATGTGTGTCGTTCATT Gene link, NY5’PKR K296-(Arg)NheI CACCGATCGCCCATGGCTCGAGCTTA Gene link, NY3’PKR Arg1Rev TTAACGCGTCGAATGACGTAAGTCTT Gene link, NY 3.4 Enzyme Restriktionsendonukleasen mit entsprechendem Puffer. . . . . . . . . . New England Biolabs
Elongase mit entsprechendem Puffer. . . . . . . . . . . . . . . . . . . . . . . . . . .Gibco
T4 DNA Ligase, T4 DNA Ligationspuffer. . . . . . . . . . . . . . . . . . . . . . . Gibco

20
Mirjam Debus MATERIAL 3.5 Puffer und Reagenzien für die DNA-Analytik 3.5.1 Reagenzien für die Plasmidisolation Resuspensionspuffer A: Lysepuffer A: 50 mM Tris-Cl, pH 8,0 200 mM NaOH, 10 mM EDTA 1% SDS 100µg/ml RNase A Präzipitationspuffer A: Waschpuffer A: 3,0 M Kaliumacetat, pH 5,5 50 mM MOPS, pH 7,0
1,0 M NaCl Elutionspuffer A: 15% Isopropanol 10 mM Tris-HCl, pH 8,5 Luria-Bertani Medium: 10 g Trypton, 5g Hefeextrakt, 10 g NaCl in 800 ml dH2O; pH 7,0 mit dH2O auf einen Liter aufgefüllt. 3.5.2 Kits für die DNA Isolation DNA-Miniprep . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Qiagen
DNA-Maxiprep . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Qiagen
Qiaquick-DNA-Gelextraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Qiagen
3.6 Puffer und Reagenzien für die Proteinaufreinigung 3.6.1 Affinitätsmatrizes Amylose Resin. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
DEAE-Sepharose . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Chitin Beads (50 –100 µm) . . . . . . . . . . . . . . . . . . . . . . . . New England Biolabs
Nickel Beads. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Ni-Nitrilotriacetat (NTA) Spin Column . . . . . . . . . . . . . . . . . . . . . . . . . Qiagen
3.6.2 Puffer für die Isolierung von MBP-Fusionsproteinen Säulenpuffer M: Elutionspuffer M 1: 20 mM Tris-HCl, pH 7.4 Säulenpuffer M, 200 mM NaCl 10 mM Maltose 1 mM EDTA 1 mM DTT

21
Mirjam Debus MATERIAL Spaltungspuffer M: Dialysepuffer M: Säulenpuffer M, 20 mM Tris-HCl, pH 8.0 1 mg Faktor Xa 25 mM NaCl pro100 mg Fusionsprotein Waschpuffer M: Elutionspuffer M 2: 10 mM Tris -HCl, pH 8.0 20 mM Tris-HCl, pH 8.0 25 mM NaCl Gradient 25mM NaCl - 500mM NaCl 3.6.3 Puffer für die Isolierung von CBP-Fusionsproteinen Zelllysepuffer C: Säulenpuffer C: 20 mM Tris-HCl, pH 8.0 20 mM Tris-HCl, pH 8.0 500 mM NaCl 500 mM NaCl 1 mM EDTA 1 mM EDTA 0.1%Triton X-100 20 µM PMSF. Spaltungspuffer C: 20 mM Tris-HCl, pH 7,5 500 mM NaCl 1 mM EDTA 50 mM DTT 3.6.4 Puffer für die Isolierung von PG-Fusionsproteinen Säulenpuffer G: Elutionspuffer G: 10 mM Imidazol, pH 8,0 20 mM- 250 mM Imidazol, pH 8,0 3.7 Puffer und Lösungen für die Proteinanalytik 3.7.1 BCA-Assay-Reagenzien Reagenz B: Reagenz A: BCA 1% CuSO4- 5 H2O 4% Na2CO3-H2O 2% Aqua dest. Auf 50 ml. Na2C4H4O6-H2O 0,16% NaOH 0,4% Standard Arbeitsreagenz: NaHCO3 0,95% 50 Teile Reagenz A, Aqua dest. auf 1L 1 Teil Reagenz B. 3.7.2 Coomassie Blue Stain-Reagenzien: Coomassie Blue Stain: Coomassie Blue Destain: 1,0 g Coomassie Blue R-250 100 ml Methanol 450 ml Methanol, 450 ml H2O 800 ml H2O 100 ml Eisessigsäure 100ml Eisessigsäure. Aqua dest. auf 1L Aqua dest. auf 1L

22
Mirjam Debus MATERIAL 3.7.3 Silberfärbungsreagenzien Lösung A : Lösung B: 0,4 g Silbernitrat 21 ml 0,36%ige NaOH 4 ml Aqua dest. 1,4 ml Ammoniumhydroxid 14,8 mM Lösung C: Lösung D: Lösung A Tropfenweise zu Lösung B 0,5 ml 1%igeZitronensäure bis das braune Präzipitat aufklart 50 µl 38%iges Formaldehyd Aqua dest. Auf 100 ml Aqua dest. auf 100 ml Haltbarkeit:15 Min. 3.8 Chemikalien Agarose. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Ampicillin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Bicinchoninic Acid (BCA) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Coomassie Brilliantblau R250. . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
DTT. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
D-Glucose. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
EDTA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Ethanol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Essigsäure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Ethidium-Bromid. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Glycerin. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Imidazol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Merck
Isopropyl β-D-1-Thiogalaktopyranosid. . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Isopropanol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Kaliumchlorid. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Kaliumacetat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Maltose. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Methanol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Magnesiumchlorid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Magnesiumsulfat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
MOPS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Natriumacetat. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Natriumchlorid. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Natriumcitrat. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich

23
Mirjam Debus MATERIAL
Natronlauge . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Polyacrylamid. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Salzsäure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sigma-Aldrich
Silbernitrat. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Sodiumdodecylsulfat. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
N,N,NI,NI-Tetramethylethylenediamin (TEMED). . . . . . . . . . . . . . . . Sigma-Aldrich
TRIS-HCl 0,1 M, pH 8,0 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
Triton-X-100. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sigma-Aldrich
3.9 Geräte
Table-top Mikrozentrifuge . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Eppendorf
Maxifuge. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sorvall
Photometer. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Shimadzu
PCR-Maschine. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Eppendorf
High Pressure Cell Disrupter 1,1 KW. . . . . . . . . . . . . . . . . . . . . . . . . Benohtop
Ultrasonicator Tomy UR-150P. . . . . . . . . . . . . . . . . . . . . . . . . . . Seiko, Japan
Elektrophoreseapparat. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .BioRad
BioLogic DuoFlow QuadTec 40 system . . . . . . . . . . . . . . . . . . . . . . . . .BioRad
Kamaras. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Polaroyd, Canon
PH-Meter. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Orion
UV-Lampe. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Fluo Link
NMR-Spektroskop, 600 MHz. . . . . . . . . . . . . . . . Bruker Advance und Varian Inova
3.8. Software Laser GeneTM. . . . . . . . . . . . . . . . . . . . . . . . . . . . . DNASTAR.Inc, Madison
Biologic. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . BioRad
3.9. Datenbanken Preteome Blast Search. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .NCBI

24
Mirjam Debus METHODEN 4 METHODEN 4.1. Subklonierung von DNA-Fragmenten in bakterielle Vektoren
4.1.1. Plasmid Isolation
Plasmid-DNA wurde entsprechend des Qiagen®-Protokolls isoliert. Einzelne bakterielle
Kolonien von E. coli DH5α wurden jeweils in 4 ml Luria-Bertani (LB) Medium (10 g
Trypton, 5g Hefeextrakt, 10 g NaCl in 800 ml dH2O, pH 7,0 auf einen Liter mit ddH2O
aufgefüllt) inokuliert und für 14-16 Stunden unter ständiger Bewegung bei 37 °C inkubiert. 4
ml Kulturen wurden bei 13.000 rpm für 10 Minuten bei 4 °C zentrifugiert. Das Pellet wurde
in 250 µl Resuspensionspuffer A gelöst und in Mikrofugenröhrchen transferiert. Die
Bakterien wurden unter alkalischen Bedingungen wie folgt lysiert: 250 µl Lysepuffer A
wurden hinzugesetzt und die Mikrofugenröhrchen wurden vorsichtig geschwenkt. Die
Lysereaktion dauerte 5 Minuten. Daraufhin wurden 350 µl Präzipitationspuffer A
hinzugesetzt und die Röhrchen wurden sofort 4-6 x vorsichtig geschwenkt. Die Proben
wurden dann für 10 min bei 13.000 rpm zentrifugiert. Der Überstand wurde jeweils auf eine
Affinitätssäule (QIAprep® spin column) gegeben, und für 60 Sekunden bei 4°C und 13.000
rpm zentrifugiert. Der Durchfluss wurde verworfen. Die Affinitätssäule wurde mit 0.5 ml
Waschpuffer A gewaschen, und für 60 Sekunden bei 4°C und 13.000 rpm zentrifugiert. Der
Durchfluss wurde verworfen. Um Reste des Waschpuffers zu entfernen, wurde die Säule für 2
weitere Minuten zentrifugiert. Daraufhin wurde die Säule in ein sauberes 1,5 ml
Mikrofugentube platziert. 50 µl Elutionspuffer A wurden hinzugesetzt. Nach einer
Inkubationszeit von 1 Minute wurde die Säule für 60 Sekunden bei 4°C und 13.000 rpm
zentrifugiert, um die DNA auf diese Weise zu eluieren.
4.1.2 Determinierung der DNA-Konzentration
Die DNA-Konzentration wurde spektrometrisch bestimmt und über die Absorption bei einer
Wellenlänge von 260 nm berechnet. Bei einem 1:50 verdünnten Ansatz entspricht eine OD260
von 1 einer dsDNA-Konzentration von 50 µg/ml. Die Reinheit der Probe wurde durch den
OD260 /OD280 Quotienten bestimmt. Bei hoher Reinheit beträgt er 1,9 bis 2,2.

25
Mirjam Debus METHODEN
4.1.3 Analyse von Plasmid DNA durch Restriktionsverdau
Um das Vorhandensein des richtigen Plasmids zu verifizieren, wurden die Plasmide mit
entsprechenden Restriktionsendonukleasen behandelt. Die Reaktion wurde wie folgt
angesetzt: 2 µg DNA in 10 mM Tris-Cl, pH 8,5 (0,5 µg DNA/µl); je 1 µl der beiden
Endonukleasen ; 2 µl des entsprechenden Puffers; aufgefüllt mit ddH2O auf 20 µl. Die
Inkubationszeit bei 37 °C betrug je nach Aktivität der Enzyme 2-14 Stunden. Die Größe der
resultierenden Fragmente wurde mittels Gelelektrophorese ermittelt. Die Fragmente der durch
die Restriktionsenzymen gespalteten Plasmide wurden in einem 1%igem Agarosegel
separiert, welches 5 mM Ethidiumbromid enthielt. Die Ethidiumbromid-DNA-Komplexe
wurde unter UV Licht sichtbar gemacht, und photographisch dokumentiert.
4.1.4 Amplifikation von Genfragmenten durch PCR
Die Polymerasekettenreaktion (PCR) wurde genutzt, um spezifische DNA-Fragmente direkt
zu amplifizieren. Dabei wurde zunächst die Matrize durch Hitze bei 94°C in einzelsträngige
DNA denaturiert. Zwei synthetische Oligonukleotide, welche zu den 3’ Enden des
entsprechenden DNA Segments komplementär waren, wurden im Überschuss hinzugegeben
und die Temperatur auf 56 °C gesenkt. Die genomische DNA blieb denaturiert, da die
komplementären Stränge eine zu geringe Konzentration aufwiesen, als dass sie sich während
der Inkubationsphase aneinander lagern konnten. Die im Überschuss vorliegenden
spezifischen Oligonukleotide hybridisierten mit den komplementären Sequenzen der
genomischen DNA. Die hybridisierten Oligonukleotide dienten als Primer, welche die DNA-
Synthese mit Hilfe von hinzugesetzten Deoxynukleotiden und einer hitzeresistenten DNA-
Polymerase bei 72 °C initiierten.
Wiederholte Zyklen von Synthese und Denaturierung resultierten in einer exponentiellen
Amplifikation. Die PCR Reaktion wurde wie folgt angesetzt: dNTP 14 µl; 1. Primer 2 µl; 2.
Primer 2 µl; Elongase 6 µl; DNA 1 µl; Puffer 30 µl; ddH2O 95 µl. Der Reaktionsansatz wurde
in 20 µl Portionen aliquotiert und in PCR-Tubes transferiert.

26
Mirjam Debus METHODEN
4.1.5 Klonierung eines PCR-Fragments
4.1.5.1 Präparation des PCR-Fragments und der Vektor-DNA
Die PCR-Fragmente wurden mittels Gelelektrophorese in einem 1%’igen Agarosegel mit 5
mM Ethidiumbromid separiert, und dann entsprechend des Qiagen®-Protokolls aufgereinigt.
Die DNA-Banden wurden unter UV-Licht sichtbar gemacht und mit einem Skalpell aus dem
Agarosegel herausgeschnitten. Drei Volumen des Lysepuffers B wurden zu einem
Gelvolumen (100 mg ~ 100 µl) hinzugesetzt. Die Mixtur wurde dann bei 50 °C für 10
Minuten inkubiert. Zu der vollständig gelösten Probe wurde dann eine äquivalente Menge des
Gelvolumens an Isopropanol hinzugesetzt. Die Mixtur wurde auf eine Affinitätssäule gegeben
und für 1 Minute bei 4 °C und 13.000 rpm zentrifugiert. Der Durchfluss wurde verworfen. Für
den Waschvorgang wurden 0.75 ml des Waschpuffers B auf die Affinitätssäule gegeben und
ebenfalls für 60 Sekunden bei 4 °C und 13.000 rpm zentrifugiert. Der Durchfluss wurde
erneut verworfen, und die Säule wurde für weitere 60 Sekunden bei 4°C und 13.000 rpm
zentrifugiert. Die Säule wurde in ein sauberes 1.5 ml Mikrofugenröhrchen platziert. Für die
Elution der DNA wurden 50 µl Elutionspuffer B zentral auf die Affinitätsmembran gegeben,
und für 60 Sekunden bei 4°C und 13.000 rpm zentrifugiert. Die Konzentration der isolierten
PCR Produkte wurde mittels Gelelektrophorese und Vergleich mit Standards abgeschätzt,
bzw. UV-spektrometrisch bestimmt. Das PCR Produkt sowie 0,5 µg der Vektor-DNA wurden
mit je 10 Einheiten der entsprechenden Restriktionsenzyme bei 37 °C für 12 Stunden verdaut.
Zeigten die Enzyme eine Kreuzreaktion, so wurde ein sequenzieller Verdau durchgeführt.
4.1.5.2 Ligation der PCR Fragmente mit Vektor-DNA
Die Enden der Restriktionsfragmente wurden mittels rekombinant generierter T4-DNA-
Ligase kovalent gebunden. Dieses Enzym katalysiert während der transienten Basenpaarung
von „sticky Ends“ die Formation von 3’-5’-Phosphodiesterbindungen zwischen der 3’-
Hydroxylgruppe des einen, und dem 5’-Phosphat des anderen Restriktionsfragments. Vektor-
DNA und Insert-DNA wurden in einem 1:3 Verhältnis gemischt, 2 µl 10x T4 DNA
Ligationspuffer und 1 µl (~400 units) T4 DNA Ligase wurden hinzugesetzt. Die Mixtur
wurde bei Raumtemperatur für 2 Stunden inkubiert.

27
Mirjam Debus METHODEN
4.1.5.3 Transformation von Escherichia coli (DH 5α)
Für die Transformation wurden kompetente Bakterien verwendet. Da native E. coli-Zellen
nicht in der Lage sind, Plasmide aus dem Medium aufzunehmen, wurden die Bakterien hohen
Konzentrationen von CaCl2 ausgesetzt, was die Zellwand für Fremd-DNA permeabel macht.
E. coli DH5α wurden verwendet, da sie versprechen, qualitativ hochwertige DNA zu liefern.
Die Zellen wurden bei -80 °C in Transformationspuffer A gelagert, und wurden dann für 30
Minuten auf Eis aufgetaut. 10 µl der oben genannten Ligationsmixtur wurden zu je 100 µl der
Zellen hinzugesetzt und für weitere 30 Minuten auf Eis inkubiert. Die Bakterien wurden dann
einer Hitzeschockbehandlung unterzogen, um ihrer Zellwände für Plasmide durchlässiger zu
machen. Es erfolgte dabei eine Erhitzung auf 42 °C für 2 Minuten. Jeweils 1 ml LB-Medium
wurde der Reaktionmixtur zugesetzt, und dann für 60 Minuten bei 37 °C unter ständiger
Bewegung inkubiert. Die Bakterien wurden auf Ampicillin-haltigen LB-Nähragarböden
ausgestrichen und für 14 Stunden bei 37 °C inkubiert.
4.1.5.4 Selektion von positiven Klonen
Die verwendeten Plasmide tragen ein Antibiotikumresistenzgen, welches für β-Laktamase
kodiert, ein Enzym das den β-Laktamring von Ampicillin oder anderen Penicillinderivaten
spaltet. Durch die Exposition gegenüber Ampicillin konnte gewährleistet werden, dass
ausschließlich plasmidtragende Bakterien überleben. Nach 14 Stunden Inkubation bei 36° C
konnten bereits einzelne Kolonien makroskopisch differenziert werden. Die Zellklone wurden
dann wie folgt angezüchtet.
4.1.5.5 Anzüchtung der Bakterienkultur Einzelne Kolonien wurden in 4 ml 100 µg/ml Ampicillin-haltigem LB-Medium inokuliert,
und unter ständiger Bewegung für 12–16 Stunden bei 37° C inkubiert. Es wurden jeweils 20
bis 50 Klone auf diese Weise parallel angezüchtet.
4.1.5.6 Überprüfung der Präsenz von Inserts
Mittels analytischem Restriktionsverdau und PCR wurde das Vorhandensein des gewünschten
Inserts überprüft. Für den analytischen Restriktionsverdau wurde die Plasmid-DNA zunächst

28
Mirjam Debus METHODEN
isoliert und daraufhin mit zwei Endonukleasen behandelt. Die Endonukleasen wurden so
gewählt, dass eine Schnittstelle im Vektor lag und die zweite im Insert. Auf diese Weise
konnte sowohl die Präsenz, als auch die Orientierung überprüft werden. Die resultierenden
Fragmente wurden durch Gelelektrophorese separiert.
Eine Methode, die mit geringerem Aufwand das Screenen einer großen Anzahl von Klonen
erlaubt, ist die PCR. Die Reaktion wurde wie folgt angetzt: 50µl PCR Puffer, 30 µl 1M MgCl,
5 µl 1. Primer, 5 µl 2. Primer, 40 µl 2,5 mMol dNTP, 5 µl Tac Polymerase, 365 µl ddH2O.
Bei 19 µl des Ansatzes wurde je 1 µl der Bakterienkultur als Matrize hinzugegeben. Die
Überprüfung der richtigen Orientierung wurde anschließend bei positiven Klonen durch
Restriktionsverdau durchgeführt.
4.1.6 Sequenzierung von DNA-Fragmenten
4.1.6.1 Die Dideoxy-Kettenterminationsmethode
Für die Sequenzierung der Plasmide wurden vier separate Polymerisationsreaktionen
durchgeführt, jede mit einer niedrigen Konzentration eines der vier ddNTPs in Ergänzung zu
höheren Konzentrationen von normalen Deoxynukleosidtriphosphaten (dNTPs). In jeder
Reaktion wird auf diese Weise das entsprechende ddNTP zufällig in den Positionen des
korrespondierenden dNTPs inkorporiert. Diese Additionen eines ddNTP terminiert den
Polymerisierungsprozess, da das Fehlen einer 3’ Hydroxylgruppe die Addition des nächsten
Nukleotids unterbindet. Die Mixtur der terminierten Fragmente jeder der vier Reaktionen
wurde simultan mittels Gelelektrophorese separiert und autoradiographisch ermittelt. Die
Sequenz der originalen DNA-Matritze wurde direkt aus dem resultierenden Autoradiogramm
abgelesen. Sämtliche DNA Sequenzierungen wurden durch TUFTS-Core Facility, Physiology
Department, TUFTS University, Boston MA durchgeführt.
4.1.6.2 Entwurf der Primer
Die Sequenzierung mittels Dideoxykettenterminationsmethode ist geeignet für DNA-
Moleküle bis zu einer Größenordnung von 500 bp. Entsprechend wurden Primer entworfen,
welche insbesondere die Sequenz der Schnittstellen zwischen Vektor und Insert, aber auch
von neu eingeführten Mutationen überspannten. Auf diese Weise konnte eine Verschiebung

29
Mirjam Debus METHODEN
des Leserahmens ausgeschlossen, bzw. die gewünschte Mutation verifiziert werden. Die
Bestimmung der gesamten Sequenz war meistens nicht notwendig, da spontane Mutationen
nur mit einer sehr geringen Wahrscheinlichkeit auftreten. Die Synthese der Oligonukleotide
wurde durch Gene link, Inc., Hawthorne, NY vorgenommen.
4.2 Expression der Zielproteine
4.2.1 Induktion durch IPTG Einzelne Kolonien wurden in 4 ml Ampicillin-haltigem LB Medium inokuliert. Die Bakterien
wurden für 12–16 Stunden bei 37 °C unter ständiger Bewegung inkubiert. Anschließend
wurden 100 μl der Zellsuspension in 8 ml frischem Ampicillin-haltigem LB-Medium
inokuliert und bis zu dem Erreichen einer optischen Dichte von 0.5 AU bei einer Wellenlänge
von 600 nm bei 37 °C unter ständiger Bewegung inkubiert. Ein Aliquot von 4 ml jeder Kultur
wurde mit IPTG in einer Konzentration von 1 mM versetzt. Über ein auf dem jeweiligen
Vektor kodiertes lac-Operon wurde so spezifisch die Expression des Zielproteins induziert.
Die induzierten Kulturen sowie Negativkontrollen wurden parallel für 4 Stunden bei 37° C
inkubiert.
4.2.2 Aufbereitung der bakteriellen Kulturen
Um die Degradation der Proteine zu reduzieren, wurden die folgenden Schritte bei 4° C
durchgeführt. Nach der Induktion der Expression mit IPTG wurden die Bakterien durch
Zentrifugation bei 12.000 x g für 1 Minute von dem Medium separiert. Der Überstand wurde
durch Aspiration entfernt, während das Pellet in 1 ml eiskaltem 50 mM Tris-HCl pH 7.4
resuspendiert wurde. Die resuspendierten Bakterien wurden erneut zentrifugiert, der
Überstand verworfen und das bakterielle Pellet in 50 µl ddH2O resuspendiert. 30 µl 4 x SDS
Gel loading Puffer wurden hinzugesetzt, welcher eine Lyse der Bakterien bewirkte. Die
unlösliche und die lösliche Fraktion wurden in einem weiteren Zentrifugationsschritt bei
12.000 g für 5 Minuten separiert. Der Überstand wurde in ein frisches Eppendorftube
überführt und bis zur Weiterbearbeitung bei –20° C gelagert. Für die Chromatographie mittels
Elektrophorese, wurden die Proben vor dem Ladevorgang für 10 Minuten bei 95°C
denaturiert.

30
Mirjam Debus METHODEN
4.2.3 Proteinanalyse
4.2.3.1 SDS-Polyacrylamid- Gelelektrophorese
Für die elektrophoretische Auftrennung der löslichen Proteinbestandteile von 15-60 kD wurde
ein 12%iges Sodiumdodecylsulfat-Polyacrylamidgel generiert. Durch die Polymerisation von
löslichen Acrylamidmonomeren in Polyacrylamidketten und simultaner Kreuzvernetzung der
Ketten, wurden die zwischen zwei Glasplatten gegossenen Gele in eine semisolide Matrix
überführt. Das vollständig polymerisierte Gel wurde dann in einen BioRad®
Elektrophoreseapparat überführt und die Konduktionskammern mit Tris-Glyzin-Puffer
geflutet.
Die präformierten Taschen wurden mit je 60 µl der aufbereiteten Proteinproben sowie
vorgefärbten Standartmarkern geladen. Eine Separation erfolgte bei 180 mV und 500 mA für
etwa eine Stunde bei Raumtemperatur.
4.2.3.2 Bestimmung der Proteinkonzentration
Der Bicinchoninic Acid (BCA) Assay ist eine quantitative Methode um den Proteingehalt in
einer Lösung zu bestimmen. Dabei werden 100 µl der Probe mit 2 ml eines BCA haltigen
Reagenz für 2 Stunden bei Raumtemperatur inkubiert. Anschließend wird die A562 im
Spektrophotometer bestimmt und mit einer Standartkurve verglichen. Die Sensitivitätsrate
reicht von 10-1200 µg/ml.
4.2.3.3 Proteinfärbung mit Coomassieblau
Die mittels SDS-PAGE separierten Polypeptide wurden simultan für 30 Minuten unter
ständiger Bewegung bei Raumtemperatur mit Methanol-Eisessig fixiert und mit Coomassie
Brilliantblau R250, einem Triphenylmethanfarbstoff gefärbt. Die Sensitivität reicht von 0,1-1
µg pro Proteinbande. Die Auswaschung der überschüssigen Farbe erfolgte durch wiederholte
Behandlungen mit Methanol-Wasserlösungen in absteigender Konzentration über 16-48
Stunden.

31
Mirjam Debus METHODEN
4.2.3.4 Silberfärbung der Proteine
Die Silberfärbung ist im Vergleich zu der Färbung mit der Coomassie Brilliant Blue-Färbung
5-10 x sensitiver. Es werden Proteinmengen von 2-10 ng pro Bande (Giulian, 1983) sichtbar
gemacht. Das SDS-Polyacrylamidgel wurde über eine Stunde in 50 % Methanol/ 10%
Essigsäure fixiert. Nach einer Spülung mit Aqua dest wurde das Gel in einer
Silbernitratlösung für 15 Minuten. unter kontinuierlicher Bewegung gefärbt. Nach einem
weiterem Waschschritt mit Aqua dest erfolgte die Entwicklung in einer formaldehyd-haltigen
Lösung für etwa 10 Minuten.
4.2.3.5 Immunoblotting
Für die Immunoblotanalyse (Westernblot) wurde die lösliche Fraktion der Proteinproben
zunächst mittels SDS-PAGE separiert. Es folgte ein Transfer der Proteinbanden auf eine
Nitrozellulosemembran mittels einer semi-trockenen Elektrophoreseprozedur bei 26 mA.
Die Membran wurde mit 5 % Milch-TBST für 5 Stunden unter ständiger Bewegung bei
Raumtemperatur geblockt. Eine Inkubation mit dem primären Antikörper, gelöst in 5 %
Milch-H2O, pH 7,4 in einem Mischungsverhältnis von 1:1000, erfolgte bei 4° C über Nacht.
Die Membran wurde mit TBST gewaschen, drei Mal für jeweils 20 Minuten und dann mit
dem enzymtragenden Sekundärantikörper für weitere vier Stunden inkubiert. Anschließend
wurde die Membran erneut mit TBST gewaschen. Ein luminiszierendes Substrat einer
Meerrettichperoxidase wurde für 2 Minuten auf die Membran gegeben und dann sofort ein
Kodak X-OMAT Film für 3-60 Sekunden belichtet.
4.2.3.6 Proteinanalyse mittels Massenspektrometrie
Die Methode der Massenspektrometrie eignet sich für die Identifizierung der Proteine über
deren Peptidmassenprofil. Bei der Time-of-Flight Massenspektrometrie werden die gelösten
Proteine und Peptide mit einer organischen Säure versetzt. Anschließend erfolgt die
Auftragung und Trocknung auf einer Metallfläche. Mittels Laser werden die Proteine ionisiert
und können in Abhängigkeit von ihrer Fluggeschwindigkeit detektiert werden. Die
Fluggeschwindigkeit ist umgekehrt proportional zu der Masse und direkt proportional zu der
Ladung der Proteine. Auf diese Weise können Proteine in einer Konzentration > 1 femtomol

32
Mirjam Debus METHODEN
mit einer Fehlerquote von < 0,1 % detektiert werden. Sämtliche massenpektrometrischen
Untersuchungen wurden im Department of Cell Biology, Harvard Medical School
durchgeführt.
4.3 Proteinisolierung mittels High Performance Liquid Chromatography (HPLC)
Affinitätschromatographie
Tag PKR-Fragment
Tag
Tag
Tag
PKR-Fragment
PKR-Fragment
PKR-Fragment
1
2
3
4
Matrix
Matrix
Abb. 3.1: Transiente Bindung des Affinitätstags an die Matrix. 2: Elution mit kompetitivem Substrat. 3:
Enzymatische Spaltung des Fusionsproteins. 4: Separation der Fusionspartner.
4.3.1 Präparation der bakteriellen Kulturen
Von jeder E. coli BL21DE3 Kolonie wurde jeweils 1 Liter Zellkultur angesetzt. Die
spezifische Expression des Zielproteins wurde mit dem Zusatz von IPTG erzielt (siehe oben).
Die Separation von Medium und Bakterien erfolgte mittels Zentrifugation mit 4000 x g für 20
Minuten bei 4° C. Die bakteriellen Pellets wurden zunächst bei -20° C gelagert. Zur weiteren
Verarbeitung wurden die Pellets in dem entsprechenden Lyse-Puffer resuspendiert und mit
den Proteaseinhibitoren PMSF, Aprotenin und Leupeptin in einer Konzentration von jeweils
1mM versetzt.
Für die mechanische Zelllyse wurde ein spezieller Hochdruckapparat verwendet. Dabei
werden die Bakterien mit Hochdruck durch eine Edelstahlkapillare gepresst und durch den
Aufprall auf eine kalte Keramikplatte zum Bersten gebracht. Die lysierten Bakterien wurden

33
Mirjam Debus METHODEN
anschließend in kalten Zentrifugenröhrchen aufgefangen und mit 9000 x g für 30 min bei 4° C
zentrifugiert. Der Überstand wurde direkt bei 4° C weiterverarbeitet, um eine Degradation
weitestgehend zu verhindern.
4.3.2. Affinitätschromatographie MBP-PKR mittels HPLC
Für die Isolierung von PKR-MBP Fusionsproteinen wurde eine amylosehaltige
Affinitätsmatrix verwendet. Um eine Degradation der Amylose durch bakterielle Amylasen
zu verhindern, wurde durch den Zusatz von 2 g Glukose pro Liter LB-Medium eine
Repression der bakteriellen Maltosegene erziehlt. Dem Säulenpuffer M wurde EDTA
zugesetzt, um Proteasen, welche Ca2+ als Cofaktor benötigen zu inaktivieren. Der Zusatz von
DTT sollte ein Formation von Disulfidbrücken unterbinden. Die Bakteriolyse wurde wie oben
beschrieben durchgeführt, nachdem das bakterielle Pellet in 20 ml Säulenpuffer M
resuspensiert wurde. Anschließend wurde die lösliche Proteinfraktion wie oben beschrieben
gewonnen.
Die lösliche Fraktion der E. coli-Extrakte wurde im Verhältnis 1:5 mit Säulenpuffer M
verdünnt. Eine 2,5 x 10 cm Glassäule wurde mit 15 ml der Amylosematrix befüllt und mit
Säulenpuffer getränkt. Mit diesem Volumen kann theoretisch eine Ausbeute von bis zu 45 mg
Fusionsprotein pro Liter bakterieller Kultur zu erzielt werden. Die Säule wurde mit 100 ml
des verdünnten Zellextraktes bei einer Flußrate 0.5 ml/min beladen. Anschließend wurde der
ungebundene Anteil mit Säulenpuffer ausgewaschen. Eluiert wurde dann mit 10 mM Maltose
und in 10-20 Fraktionen á 3 ml aufgefangen. Simultan wurde die Proteinkonzentration bei
einer Wellenlänge von 280 nm ermittelt. Die Elutionsfraktionen wurden gepoolt und mittels
Spinprep Konzentrator (30K) auf etwa 1 mg/ml konzentriert.
4.3.2.1 Spaltung des Fusionsproteins MBP-PKR
40 µl Fusionsprotein (1 µg/µl) wurde mit 2 µl Faktor Xa versetzt. Um die besten Konditionen
zu ermitteln, wurden bei jeweils 5 µl Proben Inkubationszeiten von 2, 4, 6 und 24 Stunden bei
Raumtemperatur und 4° C durchgeführt. 5 µl 2 x SDS-PAGE Sample Puffer wurden zu jeder
5 µl Probe hinzugesetzt. Die Proben wurden für 5 Minuten auf 95 °C erhitzt und mittels SDS-
PAGE aufgetrennt. Die Bande mit dem erwarteten Molekulargewicht wurde mittels
Massenspektrometrie analysiert.

34
Mirjam Debus METHODEN
4.3.2.2 Trennung von PKR-Fragment und MBP nach der Spaltung Für die Trennung der zwei Fusionspartner erfolgte eine weitere Affinitätschromatographie. Es
wurde das Prinzip einer Ionenaustausch-Chromatographie angewandt. Zunächst wurden die
mit Faktor Xa behandelten Fragmente gegen 20 mM Tris-HCl, 25 mM NaCl, pH 8,0
dialysiert, um die Protease und andere Verunreinigungen zu entfernen. Die Dialyse erfolgte
dreimal für jeweils zwei Stunden bei 4 °C. Eine 1 x 10 cm Glassäule wurde mit 6 ml der
DEAE-Sepharose befüllt und 3 Mal mit 20 ml 10 mM Tris -HCl, 25 mM NaCl, pH 8,0
gespült. Das gespaltene Fusionsprotein wurde mit einer Flussrate von 0,5 ml/min geladen.
Schrittweise wurde mit jeweils 25 ml eines Gradienten von 25 - 500 mM NaCl 20 mM Tris-
HCl, pH 8,0 eluiert. Das Eluat wurde in 1 ml Fraktionen aufgefangen während simultan die
Absorbtion (AU) bei einer Wellenlänge von 280 nm gemessen wurde. Bei einer NaCl-
Konzentration von 100-150 mM sowie ca 400 mM wurde jeweils ein scharfer Peak
nachgewiesen. Die entsprechenden Fraktionen wurden nach Präzipitation mit Aceton mittels
SDS- PAGE analysiert. 4.3.3 Affinitätschromatographie CBP-PKR
Die Aufreinigung der CBP-PKR-Fusionsproteine erfolgte mittels IMPACT-System (Intein
Mediated Purification with an Affinity Chitin-binding Tag). IMPACT ist ein
Proteinaufreinigungssystem, welches die induzierbare Selbstspaltungsaktivität eines
Proteinsplicingelements, Intein, für die Trennung des Zielproteins von dem Affinitätspeptid
nutzt. Auf diese Weise kann die Spaltung mittels Protease sowie ein zusätzlicher
Aufreinigungsschritt vermieden werden. Die hohe Affinität der Chitin-bindende Domäne
(CBD) zu Chitinmolekülen erlaubt eine effiziente Isolierung des Fusionsprotein aus dem
Zellextrakt. 20 ml der Chitinbeads wurden bei 4° C mit 200 ml Säulenpuffer C getränkt. Die
Affinitätssäule wurde mit löslichem Zellextrakt mit einer Flussrate von 1 ml/min beladen. Es
folgte ein Waschschritt mit 200 ml 1 M NaCl Säulenpuffer C mit einer Flussrate von 1
ml/min. Die hohe Salzkonzentration kann die unspezifische Bindung von Proteinen
reduzieren.
4.3.3.1 Induktion der Spaltung
Das Zielprotein wird von der Chitinsäule freigegeben, wenn der CBP- Inteinanhang sich einer
Selbstspaltung unterzieht. Induziert wurde dieser mit einer schnellen Flutung der Säule mit 60

35
Mirjam Debus METHODEN
ml Spaltungspuffer C pH 7,5, welcher 50 mM DTT enthält. Die Inkubation mit dem
Spaltungspuffer C erfolgte bei 16 °C über 16-40 Stunden.
4.3.3.2 Die Elution des Zielproteins
Die Elution des Zielproteins erfolgte mittels Säulenpuffers mit einer Flussrate von 1 ml/min.
Während der Selektion der Fraktionen á 2,5 ml wurden die Konzentration simultan UV-
spektrometrisch bei einer Wellenlänge von 280 nm bestimmt. Eine Analyse der Proben
erfolgte zunächst mittels SDS-PAGE. Bei sehr niedrigen Proteinkonzentrationen wurden
jeweils 0,2 ml der Probe mit 0,6 ml Aceton versetzt und für die Präzipitation für 30 min. auf –
20° C gebracht. Um die Spaltungseffizienz zu quantifizieren, wurde die Affinitätsmatrix nach
der Prozedur ebenfalls mittels SDS-PAGE analysiert.
4.3.4 Isolierung von PKR-PG Fusionsproteinen
Das 6-Histidin-Affinitätspeptid des Protein G-Fusionsprotein besitzt eine hohe Affinität
gegenüber Metallchelatoren wie Nickel-Nitrilotriacetat. 20 ml Ni-Nitrilotriacetic acid (NTA)
Superflow Affinitätsmatrix wurden mit jeweils 20 ml mit Säulenpuffer G 10 mM Imidazol,
pH 8,0 getränkt und anschließend 1,5 x 10 cm Glassäulen beschickt. Alle folgenden Schritte
wurden bei 4° C durchgeführt. Die Säule wurde mit 200 ml Säulenpuffer bei einer Flussrate
von 2 ml/min äquilibriert. Die lösliche Fraktion des Zelllysates wurde mit einer Flussrate von
0,25 ml/min appliziert. Anschließend wurde mit 200-400 ml Säulenpuffer G 10 mM Imidazol,
pH 8,0 gewaschen bis UV-spektrometrisch bei einer Wellenlänge von 280 nm eine stabile
Nulllinie erzielt wurde.
4.3.4.1 Elution von PKR-PG Fusionsproteinen
Die Elution der PKR-PG Fusionsproteine erfolgte mit Elutionspuffer G 20 mM- 250 mM
Imidazol, pH 8,0 bei 4° C mittels einer schrittweise gesteigerten Gradientenelution über vier
Stunden. Die Kollektion in 2,5 ml Fraktionen wurde bei simultaner UV-spektrometrischer
Bestimmung der Konzentration bei einer Wellenlänge von 280 nm durchgeführt. Eine weitere
Aufreinigung wurde mittels Dialyse über 30 K Membranen für 24 Stunden bei 4° C
durchgeführt. Auf diese Weise konnte eine Eliminierung des Imidazols und der entstandenen
Degradationsprodukte erzielt werden. Die Analyse der Proben erfolgte zunächst mittels SDS-

36
Mirjam Debus METHODEN
PAGE und Westernblot. Anschließend wurde die Reinheit mittels Massenspektrometrie
verifiziert.
4.4 Nukleare Magnetresonanzspektroskopie
Alle NMR experimente wurden bei 25 °C mittels Bruker Advance und Varian Inova 600
MHz Instrumenten durchgeführt. Für die 15N-markierte Proteinlösung in 93 % H2O, 7% 2H2O
Puffer wurde die 3D 15N-nucleare Overhauser enhancement Spektroskopie- heteronukleare
singuläre Quantum Koherenz (3D 15N-NOESY-HSQC) angewandt. NMR-Titrationen wurden
mit 100-200 µM Proben 15N-markiertem Proteins unter gradueller Addition einer
konzentrierten Stocklösung von unmarkiertem Bindungspartner durchgeführt. Sämtliche
NMR-Studien sowie die Spektrumanalyse wurde von der kollaborierenden Arbeitsgruppe
Wagner et al. prozessiert.

37
Mirjam Debus ERGEBNISSE
5 ERGEBNISSE Die Serin-Threoninkinase PKR ist maßgeblich an zellulären Prozessen wie
Zellzyklusregulation und Apoptose beteiligt. Aufgrund ihrer antiproliferativen Eigenschaften
stellt sie ein potenzielles Zielmolekül für antineoplastische Substanzen dar. Obwohl die
Relevanz der PKR bezüglich ihrer tumorsuppressiven Wirkung vielfach bewiesen wurde, ist
es bisher nicht gelungen die Kinase für strukturanalytische Zwecke zu isolieren. Die stabile
Expression sowie die Isolation von PKR haben in der Vergangenheit mannigfaltige Probleme
aufgezeigt. Bis dato konnte eine für die nukleäre Magnetresonanzspektroskopie (NMR)
geeignete Isolierung des Proteins nicht gelingen, da es zum einen zu groß, zum anderen zu
wenig löslich für die Isolierung und NMR-Analytik ist. Daneben erschwerte eine hohe
Toxizität gegenüber Bakterien die Überexpression in prokaryoten Systemen. Ziel dieses
Projektes war es daher, geeignete Expressionskonstrukte sowie stabile
Isolierungsbedingungen für verschiedene Fragmente von PKR zu generieren. Katalytisch
inaktive Mutanten unterschiedlicher Domänen sollten an geeignete Fusionsproteine gekoppelt
in E.coli BL21DE3 exprimiert und schließlich isoliert werden. Da die Struktur der Amino-
terminalen regulativen Domäne weitgehend identisch mit bekannten RNA-Bindungsdomänen
ist, war bei dem vorliegenden Projekt die katalytische Domäne sowie die regulativen Domäne
3 von zentralem Interesse. Für die Isolierung mittels High Performance Liquid
Chromatographie (HPLC) mussten für jedes der Fusionsproteine individuelle Bedingungen
etabliert werden, welche eine hohe Reinheit sowie Stabilität der Proteine gewährleisten.
Anschließend erfolgte die für die NMR-spektroskopische Analytik erforderliche
Inkorporation mit radioaktiven Isotopen.
5.1. In bakterielle Vektoren subklonierte PKR DNA-Fragmente Es wurden Konstrukte generiert, die sowohl die individuellen Domänen, als auch das gesamte
Carboxy-terminale Segment enthalten (Abb. 5.1). Die Länge der Fragmente wurde variiert,
um die Konditionen der besten Expression zu finden. Anhand von Homologiemodellen
wurden Schnittsequenzen ausgewählt, die sich in nicht-konservierten Regionen finden.

38
Mirjam Debus ERGEBNISSE
1 100 200 300 400 500 551
Δ197
Δ227
Δ262
Δ282
167-248
PKRRD1 RD2 RD3 I VIV VI VII XI
Abb. 5.1: Die Struktur von PKR und ihre C-terminalen Deletionsfragmente mit der regulatorischen Domäne 3
und der Kinasedomäne Δ179, Δ227, Δ262 und Δ282. 5.1.1 Konstrukte mit N-terminaler Polyhistidinsequenz Die cDNA der PKR-Fragmente wurde aus dem Plasmid pBISA PKR K296L mittels
Endonukleasen herausgeschnitten und in geeignete Vektorsysteme subkloniert. Der Vektor
pET15b enthält eine aminoterminale Histidinsequenz, welche dem indirekten
Proteinnachweis und der Isolierung dient. Außerdem besitzt der Vektor einen T7-Promoter
mit induzierbarem lac-Operon sowie ein Ampicillin-Resistenzgen (Abb. 5.2).
T7 Prom oter
pET-15b 5708bp
PKR lacIH is-Tag Am pr
Abb. 5.2: Vektorschema: pET15b mit dem subklonierten PKR-Fragment. T7 Promoter. His-Tag: Histidin-Affinitätsanhang. LacI: Lac-Operon. Ampr: Ampicillin-Resistenzgen.

39
Mirjam Debus ERGEBNISSE
PKR Δ262 u. Δ196
Nde I
BamH I
pET-15b(5708 bp)
lacI (866-1945)
ori(
3882
mcs
Ap (46
43-5
500)
Mlu I (1216)
Bcl I (1230)
BstE II (1379)
Apa I (1427)
BssH II (1627)
Hpa I (1722)
PshA (2061)
Bpu10 I(2926)
BspM I (2399)Msc I (2792)
Nru I (2319)
Eag I (2284)
Bsm I (1216)
Tth111 I (3565)
BsaA (3572)
Acc I (3590)
Bst1107 I (3591)
Bsa I (4774)
Alw I (4236)
BspLU11 I (3820)
Sap I (3704)
Xho I (324)
BamH I (319)
EcoRI I (5706)
Hind III (29)
Cla I (24)
Ssp I (1216)
Aat II (1216)
Sca I (1216) Nde I (331)
Ahd I (4713)
Pvlu I (1216)
Pst I (1216)
Bpu1102 I (276)
EcoN I (/51)Sph I (691)SgrA I (535)
Bgl II (494)Xba I (428)
Nco I (389)
Abb. 5.3: Vektorkarte: pET15b (5708 bp). Die Inserts (750-900 bp) wurden an den Schnittstellen von BamHI und NdeI subkloniert. LacI: Lac-Operon. Mcs: Multiple cloning site. Ap: Ampicillin-Resistenzgen. Die DNA Fragmente Δ196 wt und Δ262 wt wurden an den Schnittstellen BamHI und NdeI
der Multiplen Cloning Site des Vectors pET15b subkloniert (Abb. 5.3). Die Amplifikation der
DNA- Fragmente PKR wt Δ196 und PKR wt Δ262 erfolgte mittels PCR (Abb. 5.4). Vektor
und Insert wurden mit den Restriktionsenzymen BamHI und NdeI behandelt und isoliert.
0 ,9 K b0 ,7 5 K b
1 2 3
5 ,7 5 K b
Abb. 5.4: DNA Gel: Vektor pET15b und Insert: PKR wt PCR-Produkte. Spur 1: PKR wt Δ196 (900 bp). Spur 2: pET15b (5708 bp). Spur 3: PKR wt Δ262 (750 bp).

40
Mirjam Debus ERGEBNISSE
DH5α kompetente E. coli wurden transformiert und mittels Antibiotikum selektioniert. Die
auf Ampicillin-haltigem Nährboden gewachsenen Kolonien wurden mittels PCR auf das
Vorhandensein der entsprechenden Inserts überprüft. Es fanden sich 9 positive Klone von
pET15b PKR wt Δ196, sowie 3 positive Klone von pET15b PKR wt Δ262 (Abb. 5.5).
pET15B PKRwtΔ194
pET15B PKRwt Δ262
-
0,9 Kb
0,75 Kb
1 5
5
10
1
15 20
10 15 20
Abb. 5.5: PCR-Screening der transformierten E.coli DH5α auf Vorhandensein von Insert PKR wt Δ196 (900 bp) und PKR wt Δ262 (750 bp). PKR wt Δ196 wt positiv: Spur 1, 3, 4, 6, 7, 8, 16, 18, 20. PKR wt Δ262 wt positiv: Spur 2, 3, 8. Nach Auftrennung der Proteine mittels SDS- Polyacrylamidgelelektrophorese zeigte sich
jeweils eine induzierbare Bande bei 58 und 41 kD. Im Westernblot mit einem oligoklonalen
PKR Antikörper sowie einem Histidinantikörper waren die Banden ebenfalls nachweisbar
(Abb. 5.6). Das Expressionssystem erwies sich aber als nicht stabil und inkonstant. Daher
wurde das Wachstums- und Expressionsverhalten der Bakterien unter unterschiedlichen
Konditionen mit variierenden Inkubationszeiten überprüft. Dabei konnte festgestellt werden,
dass die Bakterien in Abhängigkeit von der Inkubationszeit die Fähigkeit, das Zielprotein zu
exprimieren, verlieren. Die Expression der individuellen Domäne 3 war nicht erfolgreich.

41
Mirjam Debus ERGEBNISSE
+
41 kD
58 kD
+-
1 2 3 4 5 6 7 8
A B--- ++
Abb. 5.6: Rekombinant in E.coli exprimierte PKRwt Δ196 und Δ262, IPTG induziert und nicht induziert. SDS-Page, Coomassiefärbung und Westernblot. A Spur 1: nicht induziert PKRwt Δ196 B Spur 5: nicht induziert PKRwt Δ262
Spur 2: induziert PKRwt Δ196 Spur 6: induziert PKRwt Δ262 Spur 3: nicht induziert (PKR-Antikörper) Spur 7: nicht induziert (PKR-Antikörper) Spur 4: induziert (PKR-Antikörper) Spur 8: induziert (PKR-Antikörper)
Die katalytisch aktive PKRwt erwies sich als toxisch gegenüber E.coli. Die transformierten
Bakterien zeigten nach der Induktion mit IPTG ein charakteristisches Zeit- und Temperatur
abhängiges Wachstums- und Expressionsverhalten. Während der ersten acht Stunden fand nur
ein sehr langsames Wachstum statt. Das subklonierte Fragment wurde in dieser Phase
exprimiert. Bei längeren Inkubationszeiten kam es dann zu einer starken Beschleunigung des
bakteriellen Wachstums bei einem gleichzeitigen Verlust der induzierbaren Expression des
subklonierten Fragments. Daraus war zu schließen, dass eine Toxizität von PKRwt gegenüber
E.coli zu einem verminderten Selektionsdruck gegenüber den Bakterien ohne
Ampicillinresistenzgen führt. Nach etwa 8-10 Stunden wurde in ausreichender Konzentration
β-Laktamase in das Medium sezerniert und so den Bakterien ohne Antibiotikumresistenz ein
ungehemmtes Wachstum ermöglicht (Abb. 5.7).
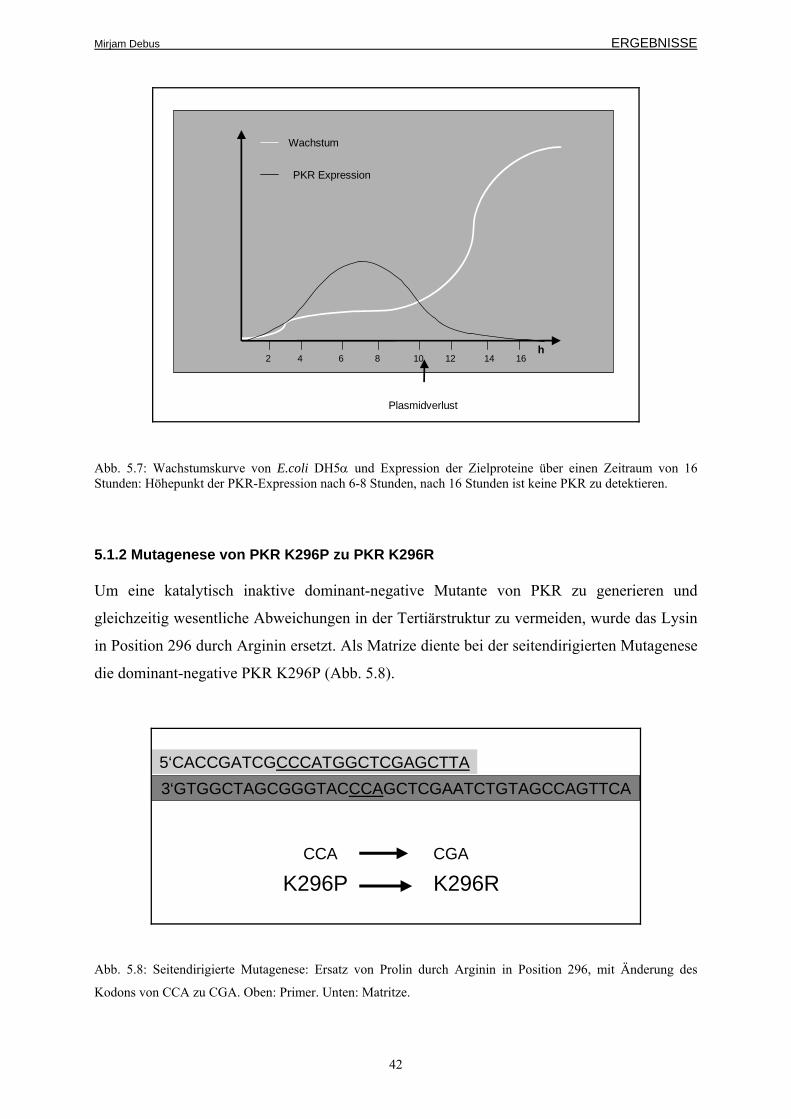
42
Mirjam Debus ERGEBNISSE
h
Plasmidverlust
Wachstum
PKR Expression
2 16141210864
Abb. 5.7: Wachstumskurve von E.coli DH5α und Expression der Zielproteine über einen Zeitraum von 16 Stunden: Höhepunkt der PKR-Expression nach 6-8 Stunden, nach 16 Stunden ist keine PKR zu detektieren. 5.1.2 Mutagenese von PKR K296P zu PKR K296R Um eine katalytisch inaktive dominant-negative Mutante von PKR zu generieren und
gleichzeitig wesentliche Abweichungen in der Tertiärstruktur zu vermeiden, wurde das Lysin
in Position 296 durch Arginin ersetzt. Als Matrize diente bei der seitendirigierten Mutagenese
die dominant-negative PKR K296P (Abb. 5.8).
K296P K296R
3‘GTGGCTAGCGGGTACCCAGCTCGAATCTGTAGCCAGTTCA 5‘CACCGATCGCCCATGGCTCGAGCTTA
CCA CGA
Abb. 5.8: Seitendirigierte Mutagenese: Ersatz von Prolin durch Arginin in Position 296, mit Änderung des
Kodons von CCA zu CGA. Oben: Primer. Unten: Matritze.

43
Mirjam Debus ERGEBNISSE
Mittels geeigneter Primer, welche die zu mutierende Sequenz in Position 296 überspannten
und die Kodierung von CCA zu CAG änderten, wurde ein PCR-Fragment generiert. (Abb. 5.9
A). Das Fragment wurde in den Ursprungsvektor PBISA K296P GFP an den
Restriktionsstellen NheI und MluI subkloniert (Abb. 5.9 B).Verifiziert wurde die erfolgreiche
Ligation durch ein Screening mittels PCR.
2 0 0 b p
3 0 0 b p
6 K b
A B
Abb. 5.9: DNA-Gele: seitendirigierte Mutagenese. A: Spur 1: PCR-Mutationsreaktionsprodukt (200 bp), Spur 2: Negativkontrolle. B: Vektor:pBISA K296 GFP: Restriktionsverdau mit MluI/NheI (6000 bp). Mittels PCR wurden die Kolonien auf das Vorhandensein des entsprechenden DNA
Fragments überprüft. Da die PCR jedoch lediglich über die Präsenz des Inserts, nicht aber
über die Orientierung im Vektor Auskunft gibt, wurde bei den positiven Klonen ein
Restriktionsverdau mit den Endonukleasen XhoI und MluI vorgenommen. Von 14 Klonen
fanden sich drei in korrekter Richtung (Abb. 5.10).
1 2 3
300 bp
Abb. 5.10: DNA-Gel: Restriktionsverdau mit XhoI/ MluI zur Bestimmung der Orientierung. Korrekte Orientierung: 1, 2, 3: Insert 300 bp.

44
Mirjam Debus ERGEBNISSE
Die Sequenzierung mittels dideoxy-Kettenterminationsmethode erlaubt eine exakte
Überprüfung der Sequenz. Hierbei wurden die Primer 5’196 NdeI und 3’ Hind III verwendet.
Die Daten wurden mit Hilfe von Blast Search, einer elektronischen Datensammlung des
National Institute of Health (NIH) interpretiert. Sie ermöglicht den Datenabgleich der
selektierten Nukleotidsequenz mit anderen homologen Sequenzen in der Datensammlung.
Mutationen innerhalb des sequenzierten Fragmentes werden angezeigt. Es sind auf diese
Weise allerdings keine Aussagen über den Leserahmen möglich. Mit Hilfe der Software
Laser Gene, welche die Nukleotidsequenz in drei mögliche Leserahmen übersetzt, konnte die
exakte Rekonstruktion von Leserahmen, Mutation und Aminosäurensequenz erfolgen (Abb.
5.11).
Query: SEGDFSADTSEIXXXXXXXXXXXXXXXXXXXXQRKAKRSLAPRFDLPDMKETKYTVDKRF SEGDFSADTSEI QRKAKRSLAPRFDLPDMKETKYTVDKRF Sbjct: SEGDFSADTSEINSNSDSLNSSSLLMNGLRNNQRKAKRSLAPRFDLPDMKETKYTVDKRF 263 Query: GMDFKEIELIGSGGFGQVFKAKHRIDGKTYVIRRVKYNNEKAEREVKALAKLDHVNIVHY GMDFKEIELIGSGGFGQVFKAKHRIDGKTYVI+RVKYNNEKAEREVKALAKLDHVNIVHY Sbjct: GMDFKEIELIGSGGFGQVFKAKHRIDGKTYVIKRVKYNNEKAEREVKALAKLDHVNIVHY 323 Query: NGCWDGFDYDPETSDDSLESSDYDPENSKNSSRSKTKCLFIQMEFCDKGTLEQWIEKRRG NGCWDGFDYDPETSDDSLESSDYDPENSKNSSRSKTKCLFIQMEFCDKGTLEQWIEKRRG Sbjct: NGCWDGFDYDPETSDDSLESSDYDPENSKNSSRSKTKCLFIQMEFCDKGTLEQWIEKRRG 383 Query: EKLDKVLALELFEQITKGVDYIHSKKLIHRDLKPSNIFLVDTKQVKIGDFGLVTSLK EKLDKVLALELFEQITKGVDYIHSKKLIHRDLKPSNIFLVDTKQVKIGDFGLVTSLK Sbjct: EKLDKVLALELFEQITKGVDYIHSKKLIHRDLKPSNIFLVDTKQVKIGDFGLVTSLK 440 Abb. 5.11: Aminosäurensequenz der Mutationsregion: PKR K296R. 5.1.3. Exprimierte His-PKR K296R Die Fragmente der dominant negativen Mutanten PKR K296R, subkloniert in das
Vektorsystem pET15b ließen sich konstant und induzierbar exprimieren. Das
Löslichkeitsverhalten war allerdings sehr unbefriedigend. Die Fragmente fanden sich so gut
wie ausschließlich in der unlöslichen Fraktion. (Abb. 5.12).
41 kD
4321
Abb. 5.12: His-PKRK296R. SDS-Page, Coomassiefärbung. Spur 1: Zelllysat. Spur 2: Lösliche Fraktion. Spur 3:
Zelllysat nicht induziert. Spur 4: Elutionsfraktion.

45
Mirjam Debus ERGEBNISSE 5.2 Generierte Fusionsproteine 5.2.1 Klonierte Fusionsgene Eine gute Löslichkeit ist für die Isolierung von Proteinen entscheidend. Um diese zu erhöhen,
wurden verschiedene Fusionsproteine generiert. Fusionen wurden gebildet mit einem
Maltosebindungsprotein (MBP), einem Chitinbindungsprotein (CBP) und einem speziell für
diese Zwecke entwickelten Peptid, Protein G (PG) mit N-terminaler Polyhistidinsequenz. Die
Fragmente PKR K296R Δ179, Δ196, Δ227, Δ262 und Δ282 wurden in geeignete bakterielle
Vektorsysteme subkloniert. Als Vektoren wurden verwendet: pTYB12, pMAL –p2X und
pGEV2. Alle Vektoren weisen ein T7-Promoter/ Lac Operator gesteuertes System auf, um
hohe Expressionslevel sowie eine kontrollierte Expression zu erreichen. Außerdem tragen sie
ein Ampicillinresistenzgen, um eine Selektion zu erlauben (Abb.5.13-15).
PKRMBP
Intein
AmprpMal-c2X
6648 bp
PKR
PKR
pTYB12
7414 bpCBD
pGEV2
6382 bpT7 promoter PGlacI Ampr
T7 promoter AmprlacI
lacI
His
T7 promoter
Abb. 5.13: Vektorschema der Fusionsgene mit dem jeweiligen subklonierten PKR-Fragment: pMal-c2X (6648 bp), pTYB12 (7414 bp), pGEV2 (6382 bp). T7 Promoter. His-Tag: Histidin-Affinitätsanhang. MBP: Maltosebindendes Protein. CBD: Chitinbindende Domäne. LacI: Lac-Operon. Ampr: Ampicillin-Resistenzgen.

46
Mirjam Debus ERGEBNISSE
PKR Δ262 u. Δ196Hind III
BamH I
pMAL-c2X (6648 bp )
mcs
Am
pla
cI
lacZ
pBR322 ori
M13 ori
mal
E
Xmn I
EcoR I
BamH IXba I
Sal I
Pst I
Hind III
malE... ATC GAG GGA AGG ATT TCA GAA TTC GGA TCC TCT AGA GTC GAC CTG CAG GCA AGC TTG... lacΖ
pMAL-c2X Polylinker:
Abb. 5.14: Vektorkarten: pMal-c2X (6648 bp). Die Inserts (750-900 bp) wurden an den Schnittstellen von BamHI und HindIII subkloniert. LacI: Lac-Operon. malE: Maltosebindungsgen. Mcs: Multiple cloning site. Amp: Ampicillin-Resistenzgen.

47
Mirjam Debus ERGEBNISSE
PKR Δ262 u. Δ196
Nde I
Xho I
pTY-B12(7414 bp)
mcs
Bsp E I (6703)
Blp I (6625)
Mfe I (5851)
Sac II( 6390)
Bst B I (6327)
Bsr E II ( 3981)
Kpn I (4987)
Sgr AI (4780)
Apa I (3893)
Bss H II (3689)
Bst Z17 I (2480)
Pci I (2310)
Dra III (1319)
Psi I 1194
Swa I 1096
Nru ISpe I
Nde I
Sal I
Nat IEcoR ISma I
PspCM I (3898)
Xho I
PanI I ( 6380)
Stu I ( 6355)
Nco I ( 5958)
Msc I ( 5955)
Nhe I ( 5794)
Acc65 I ( 4987)
Bae I ( 4980)
Xba I ( 4888)
Sph I (4629)
EcoN I (4564)
Mlu I (4100)
Abb. 5.15: Vektorkarte: pTYB12 (7414 bp). Die Inserts (750-900 bp) wurden an den Schnittstellen von XhoI und NdeI subkloniert. Der Vektor enthält Sequenzen für ein Lac-Operon, ein Chitinbindungsprotein, ein Inteintag mit Selbstspaltungsaktivität, eine Multiple Cloning Site sowie ein Ampicillin-Resistenzgen. Die Fragmente mit passendem Linker wurden mittels PCR amplifiziert (Abb. 5.17). Die passenden Primer wurden entsprechend entworfen (Abb .5.16).
5’Δ196-NdeI: CGGCAGCCATATGAGCACACTCGCTTCTGAA
5’Δ262-NdeI: CGGCAGCCATATGAGG TTTGGCATGGATTT
5’PKR Δ179- BamHI: CGCGGATCCGGTTCTTTTGCTACTACGTGT
5’PKR Δ262- BamHI: CGCGGATCCAGGTTTGGCATGGATTTTAAAGAA
3’gev2 XhoI: CGGCTCGAGACATGTGTGTCGTTCATT
3’pTYB12 XhoI: CGGCTCGAGCTAACATGTGTGTCGTTCATT
Abb. 5.16: Primer der PKR-Fragmente.

48
Mirjam Debus ERGEBNISSE
1 2 3 4 5 6
7 5 0 b p
9 0 0 b p
Abb. 5.17: PCR-Produkte der Fragmente K296R Δ 179, Δ 196, Δ 262. DNA-Gel. Spur 1: K296R Δ 179 (Primer: 5’PKR 179 BamHI, 3’gev XhoI) Spur 2: K296R Δ 262 (Primer :5’PKR 262 BamHI, 3’gev XhoI) Spur 3: K296R Δ 179 (Primer: 5’PKR 179 BamHI, 3’HindIII) Spur 4: K296R Δ 262 (Primer: 5’PKR 262 BamHI, 3’HindIII) Spur 5: K296R Δ 196 (Primer: 5'196 NdeI, 3’pTYB12 XhoI) Spur 6: K296R Δ 262 (Primer: 5'262 NdeI, 3’pTYB12 XhoI)
Die Vektoren wurden nach der Amplifikation in E.coli isoliert und mit den entsprechenden
Restriktionsendonukleasen versetzt (Abb. 5.18).
1 2 3
7414 bp6648 bp6382 bp
Abb. 5.18: DNA-Gel: Vektoren nach Restriktionsverdau Spur 1: pGEV2 6382 bp (BamHI/XhoI), Spur 2: pMAL 6648 bp (BamHI/HindIII), Spur 3: pTYB12 7414 bp (NdeI/XhoI).
Einzelne Klone der transformierten Bakterien wurden mittels PCR auf die Existenz der
Vektoren mit den entsprechenden Inserts überprüft (Abb.19).

49
Mirjam Debus ERGEBNISSE
1
2
3
900 bp
900 bp
750 bp
5
6
4
900 bp
750 bp
750 bp
Abb. 5.19: Analytische PCR der bakteriellen Kolonien mit den entsprechenden subklonierten Gensequenzen der unterschiedlichen Fusionsproteine. DNA-Gel. Spur 1 pGEV2 K296R Δ 179 (900 bp). Spur 4 pMal-c2X K296R Δ 262 (750 bp). Spur 2 pGEV2 K296R Δ 262 (750 bp). Spur 5 pTYB12 K296R Δ 196 (900 bp). Spur 3 pMal-c2X K296R Δ 179 (900 bp). Spur 6 pTYB12 K296R Δ 262 (750 bp).
Mit der Dideoxy-Kettenterminationsmethode wurden die Sequenzen überprüft. Mit Hilfe
elektronischer Datenbanken wurden die selektierten Nukleotidsequenzen interpretiert.
Leserahmen, Mutation und Aminosäurensequenz wurden mit der Software Laser Gene
überprüft und waren korrekt.

50
Mirjam Debus ERGEBNISSE
5.2.2 Exprimierte Fusionsproteine Die Fusionsproteine konnten erfolgreich in E.coli BL21DE3 exprimiert werden. Diese
Zelllinie wurde speziell für high-level Proteinexpression entwickelt. Die vorläufige
Proteinanalytik wurde in erster Linie mittels Sodiumdodecylsulfat-Polyacrylamid-
Gelelektrophorese (SDS- Page) vorgenommen. Diese Methode eignet sich für den Vergleich
und die Quantifizierung von Proteinen. Die Proteine werden dabei primär nach ihrem
Molekulargewicht separiert. SDS bindet an die hydrophoben Regionen eines Proteins und
beeinträchtigt die gefaltete Konformation. Als Ergebnis ist die Länge des SDS-
Proteinkomplexes proportional zu seinem Molekulargewicht. Darüber hinaus wurden
Immunoblots mit den entsprechenden Antikörpern angefertigt. Die Fusionsproteine PG-
K296R, MBP-K296R, CBP-K296R konnten mit ihren unterschiedlich langen Fragmenten
erfolgreich exprimiert werden. Alle Fusionsproteine wiesen eine gute Löslichkeit auf (Abb.
5.20).
C B P -P K R Δ 1 96 , Δ 26 2
M B P -P K R Δ 17 9G P -P K R Δ 1 79
8 5 k D5 8 k D
3
7 8 9 1 0 1 1 1 2
4 5 621
9 0 k D
8 3 k D
Abb. 5.20: Exprimierte Fusionsproteine MBP-PKR, CBP-PKR, PG-PKR. SDS-Page, Coomassieblau gefärbt. Die Bestimmung erfolgte jeweils aus Ganzzelllysaten induziert und nicht induziert, sowie der löslichen Fraktion. Spur 1 PG-PKR K296R Δ 179 Zellextrakt induziert. Spur 2 PG-PKR K296R Δ 179 lösliche Fraktion. Spur 3 PG-PKR K296R Δ 179 Zellextrakt nicht induziert. Spur 4 MBP-PKR K296R Δ 179 Zellextrakt induziert. Spur 5 MBP- PKR K296R Δ 179 lösliche Fraktion. Spur 6 MBP- PKR K296R Δ 179 Zellextrakt nicht induziert. Spur 7 CBP- PKR K296R Δ 196 Zellextrakt induziert. Spur 8 CBP- PKR K296R Δ 196 lösliche Fraktion. Spur 9 CBP- PKR K296R Δ 196 Zellextrakt nicht induziert. Spur 10 CBP- PKR K296R Δ 262 Zellextrakt induziert. Spur 11 CBP- PKR K296R Δ 262 lösliche Fraktion. Spur 12 CBP- PKR K296R Δ 262 Zellextrakt nicht induziert.

51
Mirjam Debus ERGEBNISSE 5.3 Isolierte PKR-Fragmente nach High Performance Liquid Chromatographie (HPLC) Die Isolierung der PKR-Fusionsproteine wurde mittels High Performance Liquid
Chromatographie durchgeführt. Die Prozedur ist computerbasiert. Die Proteinkonzentration
der Fraktionen wird dabei simultan aus der Extinktion bei einer Wellenlänge von 280 nm
ermittelt.
5.3.1 Isolierte MBP-PKR K296R -Fragmente Bei der Elution von MBP-PKR K296R Δ179, Δ227, Δ262 und Δ282 zeigte sich ein deutlicher
Peak in den Elutionsfraktionen. Nach der Spaltung mit der Protease Faktor X wurde eine
Ionenaustauschchromatographie durchgeführt, um eine Trennung der PKR- Fragmente von
MBP, der Protease und anderen Kontaminationen zu erzielen. Ein Nachteil dieser Methode
ist, dass das Zielprotein und MBP gelegentlich gleichzeitig eluiert werden. Dieses war auch
hier der Fall. Photometrisch ließ sich nur ein Peak bestimmen (Abb. 5.21).
DEAE-SepharoseNaCl GradientAmylosematrix
nach Spaltung mit Faktor Xa
1. 2.
ODOD
tt
Abb. 5.21: 1. Elution des Fusionsproteins MBP-PKR. 2. Elution des Fusionsprotein MBP+ PKR nach Spaltung mit Faktor Xa. Bei der Elution mit einem NaCl2-Gradienten kam es zur Elution beider Bindungspartner in der gleichen Fraktion.
In der SDS-Gelelektrophorese war bereits nach der ersten Isolierungsprozedur eine gute
Anreicherung der Fragmente bei zufriedenstellender Reinheit zu sehen (Abb. 5.22). Die
Degradationsprodukte sowie Kontaminationen mit kleineren Proteinen ließen sich durch
wiederholte Dialyseschritte entfernen.
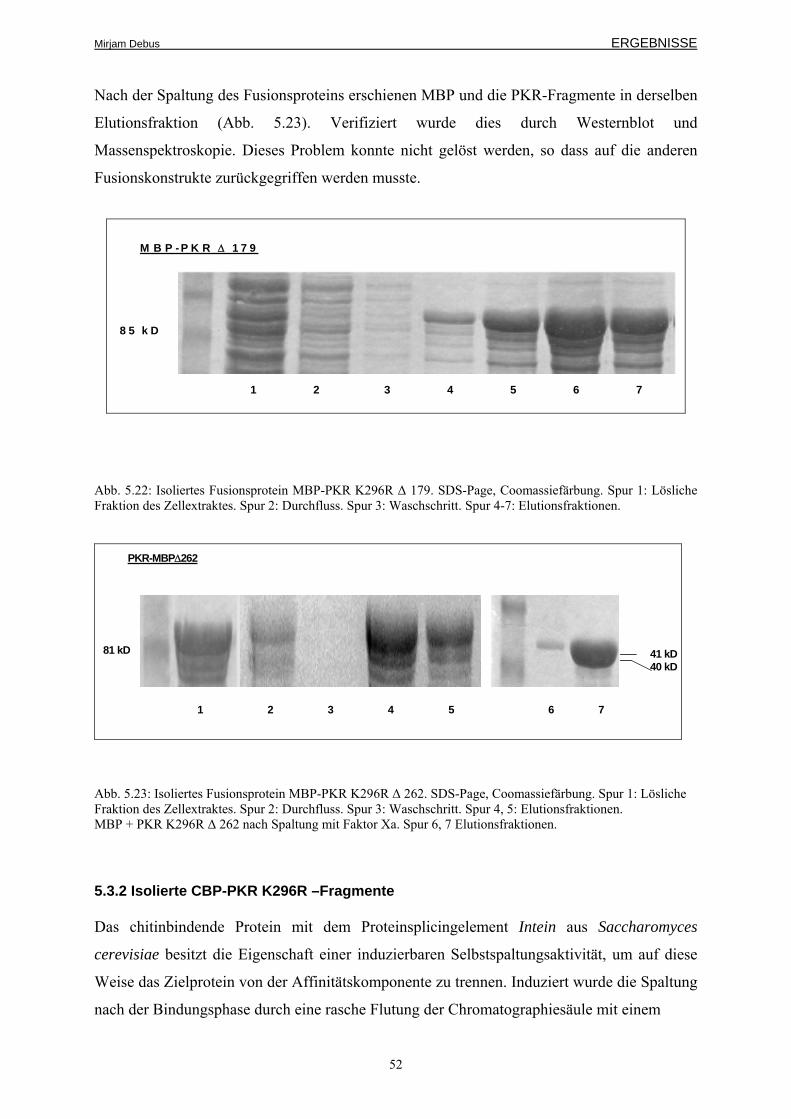
52
Mirjam Debus ERGEBNISSE
Nach der Spaltung des Fusionsproteins erschienen MBP und die PKR-Fragmente in derselben
Elutionsfraktion (Abb. 5.23). Verifiziert wurde dies durch Westernblot und
Massenspektroskopie. Dieses Problem konnte nicht gelöst werden, so dass auf die anderen
Fusionskonstrukte zurückgegriffen werden musste.
M B P - P K R Δ 1 7 9
8 5 k D
654321 7
Abb. 5.22: Isoliertes Fusionsprotein MBP-PKR K296R Δ 179. SDS-Page, Coomassiefärbung. Spur 1: Lösliche Fraktion des Zellextraktes. Spur 2: Durchfluss. Spur 3: Waschschritt. Spur 4-7: Elutionsfraktionen.
PKR-MBPΔ262
81 kD 41 kD40 kD
6 721 3 4 5
Abb. 5.23: Isoliertes Fusionsprotein MBP-PKR K296R Δ 262. SDS-Page, Coomassiefärbung. Spur 1: Lösliche Fraktion des Zellextraktes. Spur 2: Durchfluss. Spur 3: Waschschritt. Spur 4, 5: Elutionsfraktionen. MBP + PKR K296R Δ 262 nach Spaltung mit Faktor Xa. Spur 6, 7 Elutionsfraktionen.
5.3.2 Isolierte CBP-PKR K296R –Fragmente Das chitinbindende Protein mit dem Proteinsplicingelement Intein aus Saccharomyces
cerevisiae besitzt die Eigenschaft einer induzierbaren Selbstspaltungsaktivität, um auf diese
Weise das Zielprotein von der Affinitätskomponente zu trennen. Induziert wurde die Spaltung
nach der Bindungsphase durch eine rasche Flutung der Chromatographiesäule mit einem

53
Mirjam Debus ERGEBNISSE
Thiol, wie in diesem Falle DTT. Die Elution von CBP-K296R Δ196, Δ227, Δ262, zeigte
keinen deutlichen Peak bei der Messung der Extinktion bei einer Wellenlänge von 280 nm. In
der Analyse der Fraktionen mittels SDS-Page zeigte sich eine ineffektive Spaltung der
Fusionsproteine, die auch durch diverse Modifikationen der Induktionsbedingungen nicht
behoben werden konnte. Der Hauptanteil des Zielproteins blieb an der Säule haften (Abb.
5.24).
CBP-PKR Δ179:ce ft el
1 432
res
83 kD
83 kD
A
B
Abb. 5.24: Isoliertes Fusionsprotein CBP-PKR K296R Δ 179. SDS-Page, Coomassiefärbung und Westernblott. Spur 1: Säulenrückstand. Spur 2: Lösliche Fraktion des Zellextraktes. Spur 3: Durchfluss. Spur 4: Elutionsfraktion. 5.3.3 Isolierte PG-PKR K296R -Fragmente Nachdem sich die Fusionsproteine MBP-K296R und CBP-K296R für unsere Zwecke als
ungeeignet erwiesen, zeigte sich das Fusionsprotein PG-K296R als erfolgversprechend. Der
Expressionsvektor wurde speziell für die Expression von Proteinen und Peptiden entwickelt,
die einer NMR-spektroskopischen Analyse zugeführt werden sollen. Vorteilhaft bei diesem
Konstrukt ist, dass eine Spaltung der Fusion nicht unbedingt notwendig ist. Da das Protein G
nur ein sehr geringes Molekulargewicht aufweist, wird die NMR-Analyse nicht beeinträchtigt.
Die Elution der Fragmente PG-K296R Δ179, Δ227, Δ262 und Δ282 mit Imidazolgradienten
zeigte jeweils einen deutlichen Peak bei einer Imidazolkonzentration von ca. 100 mM. Die
Extinktion wurde bei einer Wellenlänge von 280 nm gemessen (Abb.5.25).

54
Mirjam Debus ERGEBNISSE
4 0kD
0 0:00 05 :0004 :0003 :000 2 :000 1 :00
h
-0 ,5
0 ,00
1 ,00
2 ,00
A UIm id a zo l m M25018 01 002 0
Abb. 5.25 Elution des Fusionsproteins PG-PKR K296R Δ 179. Die Elution erfolgte über einen Zeitraum von 6 Stunden mittels Imidazolgradienten. Simultan wurde die AU bei einer Wellenlänge von 280 nm gemessen.
P G -P K R Δ 1 7 9
5 8 k D
8765432 91
Abb. 5.26: Isoliertes Fusionsprotein PG-PKR K296RΔ179. SDS-Page, Coomassiefärbung. Spur 1: Gesamtzelllysat. Spur 2: Lösliche Fraktion des Zellextraktes. Spur 3: Gesamtzelllysat nicht induziert. Spur 4 - 9: Elutionsfraktion. Die SDS-Gelelektrophorese (Abb. 5.26) zeigte eine gute Anreicherung der Fragmente bei
zufriedenstellender Reinheit. Die Degradationsprodukte sowie Kontaminationen mit kleineren
Proteinen ließen sich durch wiederholte Dialyseschritte entfernen, und so die Reinheit
optimieren. Es konnte eine hohe Konzentration von 1,2 µg/µl erreicht werden. Die Intaktheit
und Reinheit des Proteins wurde mittels Massenspektrometrie verifiziert.
5.4 Die NMR-Struktur der Kinasedomäne
Die NMR-Analyse der Kinasedomäne wurde mittles TROSY-15N-heteronukleärer singulärer
Quantum-Koherenz (HSQC) Experimente durchgeführt. 224 (75%) Aminosäuren konnten
lokalisiert und zugeordnet werden. Die restlichen Resonanzen waren stark überlappend und
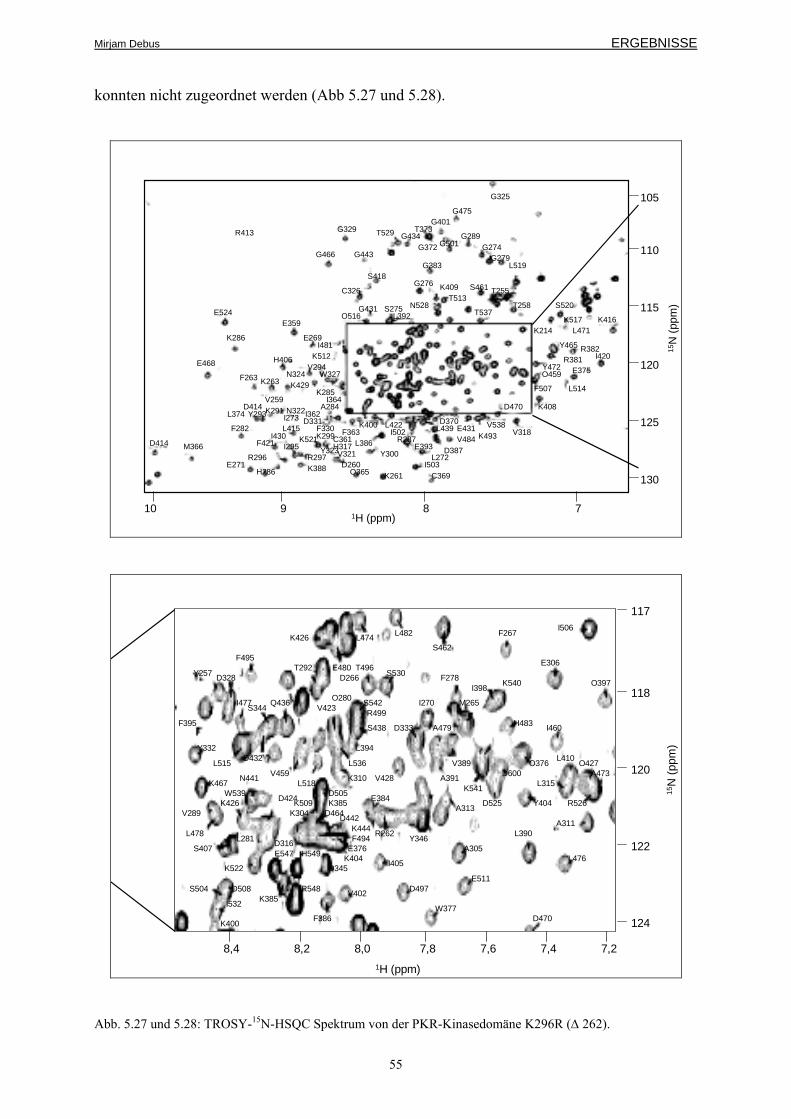
55
Mirjam Debus ERGEBNISSE
konnten nicht zugeordnet werden (Abb 5.27 und 5.28).
D414
F282
E271
M366
E468
K286
E524
R413
F263
H406
I481K512
E269
G329
E359
G466
F363D331I362
I364V259
Y293D414
L374
K263N324
F507
K408
V318
I503O365
R296
H286 K261 C369
O516 L392 T537
R382Y465
Y472
L471K214K517
S520
E375O459
D470
K493
L514
I420
G325
G475
G274G279
L519
S461
G383
G276
S275
S418
C326
G289
G401
K291 A284
K285
W327V294
G431
G501G372
T373G434T529
F421
K388
T513T255
T258
R297
K400
R287I502
E393L386I430
I295 H317K521 C361
L439D370
E431 V538
R381
K429
N528
K409
F330
G443
K416
L422
D260Y300Y323V321 L272
D387V484K299
L415I273N322
15N
( ppm
)
1H (ppm)10
130
125
120
115
110
789
105
K426
Y257 D328
Q436S344
F495T496 S530
L474 L482
S462
D600O376
H483
K540I398
F267 I506
E306
O397
I460
A473O427L410
L315
A311L390
L476
D470
Y404
E480
V423I477
F395
Y332
K426
D432L515V459
L518K467 V428
V289
L478
S407
K522
K400
I532
D508S504
A305
R526
R262
D525
F386
D345
V402
W377
D497
D266T292
D505
K304
D316
R548
H549E547
S438
L394
L536
K310
E384
D333
I270
A479
V389
A391
Y346
K541
I405
E511
R499
A313
S542
F278
M265
K385D464D442
K385
K444
D424K509
F494E376
K404
L281
W539
N441
O280
8,4
124
117
118
120
122
8,2 8,0 7,8 7,6 7,4 7,21H (ppm)
15N
(pp m
)
Abb. 5.27 und 5.28: TROSY-15N-HSQC Spektrum von der PKR-Kinasedomäne K296R (Δ 262).

56
Mirjam Debus DISKUSSION 6 DISKUSSION
Im Rahmen der vorliegenden Arbeit konnten erstmals mittels nuklearer
Magnetresonanzspektroskopie, NMR, dreidimensionale Strukturelemente der Kinasedomäne
von der durch doppelsträngige RNA aktivierten Proteinkinase PKR charakterisiert, und deren
mit den regulativen Domänen interagierenden Regionen lokalisiert werden. Zahlreiche
biochemische Untersuchungen haben gezeigt, dass die Kinasedomäne von PKR in einem
latenten Status durch Interaktionen mit den regulativen Domänen inhibiert wird (Nanduri
2000). Welche Sequenzen jedoch an diesem Prozess beteiligt sind war bisher unbekannt. Die
hier vorgelegten Ergebnisse zeigen, dass die gesamte regulative Domäne an der Inhibition
von PKR beteiligt ist. Die erhobenen NMR-Strukturdaten der Kinasedomäne liefern zudem
die Voraussetzungen für die Charakterisierung der vielfältigen regulativen Interaktionen
dieser Domäne sowie für deren mögliche Modifikation über kleine spezifisch interagierende
Moleküle. Über die damit einhergehende Beeinflussung der Translation
wachstumsregulierender Proteine kann so ein neuer potenzieller Ansatz für eine
antineoplastische Therapie postuliert werden.
6.1 NMR-Strukturanalysen von PKR liefern Informationen betreffs der Autoinhibition
Neben der Charakterisierung der dreidimensionalen Struktur der Kinasedomäne von PKR
stand im Fokus der Untersuchungen, Informationen über deren Dynamik und Interaktionen
mit regulierenden Molekülen zu generieren. Daher wurde unter den gängigen
strukturanalytischen Verfahren die Methode der nuklearen Magnetresonanzspektroskopie
favorisiert. Die NMR-Spektroskopie beschränkt sich nicht nur auf die Bestimmung der
Proteinstruktur, sondern bietet auch die Möglichkeit, über die reine Strukturaufklärung
hinausgehenden Fragestellungen zu untersuchen. Mit dem Wissen der dreidimensionalen
Struktur können funktionelle Fragestellungen wie die Dynamik des Proteins,
Wechselwirkungen mit anderen Molekülen wie kleinen Liganden, Polypeptiden, anderen
Proteinen und DNA-Molekülen sowie Katalysemechanismen, Faltungsverhalten oder
Hydratation des Proteins studiert werden (Riek R 2002). Im Gegensatz zu den durch
Kristallisationsverfahren erhobenen statischen Momentaufnahmen der Struktur hängen viele
NMR-spektroskopische Parameter sowohl von der Bewegung des Moleküls als ganzes, wie
Translations- und Rotationsbewegungen, als auch von internen Bewegungen, wie

57
Mirjam Debus DISKUSSION
Konformationsänderungen und Rotationen von Bindungen ab (Bernado P and 29;125(4):916-
23 2003). Die Messung dieser Parameter lässt daher nicht nur Rückschlüsse auf die
vorliegenden dynamischen Prozesse zu, sie erlaubt auch Aussagen über den zeitlichen Verlauf
dieser Prozesse. Erstmals gelang es anhand von N15-markierten Proben große Teile der PKR-
Kinasedomäne mittels NMR-Spektroskopie zu charakterisieren. In dieser Technik wurde eine
250 μM konzentrierte Proteinlösung in einem magnetischen Feld positioniert, und dabei der
Effekt der unterschiedlichen Radiofrequenzen auf die Resonanz unterschiedlicher Atome
gemessen. Das Verhalten eines jeden Atoms wird dabei durch die benachbarten Atome der
angrenzenden Aminosäuren beeinflusst, wobei die nah beieinander liegenden Aminosäuren
mehr als die entfernteren beeinflusst werden. Aus der Größenordnung des Effektes konnte der
Abstand der Aminosäuren zueinander berechnet, und konsekutiv ein Modell der
dreidimensionalen Struktur des Proteins generiert werden. Die strukturellen Informationen der
Kinasedomäne lieferten die Grundlage für weiterführende Untersuchungen der
Bindungseigenschaften der regulativen dsRNA-bindenden Domänen, DRBD I und II. Durch
Titration der unterschiedlichen Domänen konnte der größte Effekt durch die Addition der
kompletten regulativen Domäne beobachtet werden, was dafür spricht, dass sowohl DRBD I,
als auch DRBD II in entscheidendem Maße an der Regulierung beteiligt sind. Es konnte
nachgewiesen werden, dass die Inhibition der Kinasedomäne durch eine Maskierung der
eIF2α-Substratspezifitätsregion und durch eine allosterische oder direkte Blockierung der
katalytisch aktiven Region erfolgt.
6.2 Für einzelne Domänen von PKR konnten individuelle Expressions- und Isolationsbedingungen für eine NMR-Analytik etabliert werden
In dieser Arbeit gelang es erstmals durch Generierung der katalytisch inaktiven Mutante
K296R das Toxizitätsproblem bei der Überexpression von Wildtyp-PKR in E. coli zu
überwinden, um auf diese Weise eine Expression auf quantitativ hohem Niveau zu erzielen.
Um die Struktur und den Aktionsmechanismus eines Proteins studieren zu können, ist die
Isolation des Moleküls unabdingbar. Da alle der ca. 10.000 unterschiedlichen Proteine,
welche sich in einer Zelle befinden, in Größe, Ladung und Löslichkeitsverhalten variieren,
stellt die Isolation jedes einzelnen Proteins eine große Herausforderung dar [Cristea IM, 2004
#283]. Für biochemische und molekularbiologische Analysen eignen sich am besten
rekombinant generierte Proteine von bakteriellen Expressionssystemen, da diese eine
kostengünstige und zeitsparende Überexpression des entsprechenden subklonierten Proteins

58
Mirjam Debus DISKUSSION
erlauben. Frühere Arbeiten zeigen, dass rekombinante Wildtyp-PKR in E. coli
autophosphoryliert und RNA unabhängig aktiviert wird (McMillan NA 1995), und so bei der
Überexpression einen toxischen Effekt auf E. coli hat (Barber 1991). Die K296-Seitenkette ist
in die Bindung von ATP involviert. Ein Ersatz des Lysins durch Argenin inhibiert den
Phosphattransfer ohne die Bindung von ATP zu beeinflussen. Während die Phosphorylierung
dieser Mutante durch Wiltyp-PKR uneingeschränkt möglich ist, findet eine Dimerisation und
Autophosphorylierung von K296R während der bakteriellen Expression nicht statt. Generelle
Nachteile der bakteriellen Expressionssysteme liegen in der fehlenden posttranslationellen
Modifikation sowie der Bildung von Inclusion Bodies (Singh SM 2005). Da die Faltung eines
Proteins unter anderem von den intramolekularen Wechselwirkungen abhängt, kann es zudem
durch die Einführung von Mutationen zu entscheidenden strukturellen Alterationen kommen.
6.3 Die Bildung von Fusionsproteinen ermöglichte eine Isolation für NMR-Strukturanalysen
Die wahrscheinlich größte Herausforderung bei Proteinstrukturanalysen ist die Gewinnung
adäquater Proteinproben. In dem vorliegenden Projekt gelang es, durch die Etablierung von
individuell angepassten Expressions- und Isolationsbedingungen der einzelnen PKR-
Domänen die Probleme betreffs Löslichkeit- und Stabilität zu überwinden. Neben der
Modifizierung der Pufferlösungen sowie der physikalischen Gegebenheiten führten unter
anderem die Mutagenese der katalytisch aktiven Seite K296R, die Generierung von
trunkierten Domänen sowie die Bildung von Fusionsproteinen zu dem erwünschten Ergebnis.
Es war notwendig, das Protein in großen Mengen zu produzieren, und dabei Konditionen zu
finden, unter welchen das Protein bei Konzentrationen von mindestens 100 µM stabil und
löslich ist. Das häufigste Problem bei diesem Vorhaben ist die robuste Anfertigung der Probe,
da nur ca. 25 % der überproduzierten Proteine biochemisch stabil und somit geeignet für
Strukturanalysen sind (Christendat D 1998). Multiple Herangehensweisen wurden in den
vergangenen Jahren unternommen, um diese Probleme zu umgehen. In einigen Systemen war
die Modifikation der Pufferkonditionen oder die Einführung einer Punktmutation hilfreich
(Bagby S 2001). Allerdings wurden diese Methoden größtenteils durch individuelles
Ausprobieren etabliert, und lassen sich daher nicht ohne weiteres auf andere Systeme
übertragen. Um Löslichkeit, Stabilität und Expressionsrate der Proteine zu erhöhen, hat es
sich als nützlich erwiesen, Fusionsproteine mit hydrophilen Proteinen oder Peptiden zu bilden
(Pryor KD 1997). Die meisten gängigen Fusionsproteine sind allerdings sehr groß, was eine

59
Mirjam Debus DISKUSSION
direkte Strukturanalyse mittels NMR-Spektroskopie unmöglich macht. Für Strukturanalysen
müssen diese Fusionen also wieder gespalten werden, was häufig erneut zu Problemen
betreffs Löslichkeit und Stabilität führt.
Nach aufwendigen Modifikationen der Isolationsbedingungen von PKR und der Entwicklung
unterschiedlicher Expressionssysteme für die einzelnen Domänen, konnten wir von den
Studien der kollaborierenden Arbeitsgruppe G. Wagner et al. profitieren, die inzwischen gute
NMR Ergebnisse mit dem Fusionsprotein Protein G B 1, einer immunglobulinbindenden
Domäne des Streptokokkenproteins G, erzielten (Zhou 2001). Durch die Bildung eines
chimären Proteins mit Protein G B1 gelang es erstmalig, einzelne Domänen von PKR in guter
Stabilität und Löslichkeit sowie ausreichender Quantität für NMR Analysen zu generieren.
Hiermit konnte ein System entwickelt werden, welches die direkte NMR Spektroskopie des
Fusionsproteins durch 1H- 15N Korrelationsspektroskopie erlaubt.
6.4 Anhand der NMR-spektroskopischen Daten lässt sich die Rolle von PKR bei der Inhibition der Translation untersuchen
Mit den im Rahmen dieser Arbeit gewonnenen strukturellen und funktionsanalytischen
Informationen sind die Voraussetzungen für eine exaktere Untersuchung der
Regulierungsprozesse von PKR gegeben. Aufgrund der zentralen Bedeutung von PKR in der
Signaltransduktion wachstumshemmender Prozesse ist sie von zunehmendem Interesse für
die Aufklärung von Mechanismen des Zellwachstums. Einige experimentelle Daten haben
gezeigt, dass die Expression wachstumsregulierender Proteine nicht nur durch die mRNA-
Akkumulation, sondern auch auf der Translationsebene reguliert wird. Extrazelluläre Signale,
welche die Transkription wachstumsregulierender Gene stimulieren, führen ebenfalls zu einer
gesteigerten Expression und Aktivität von Translationsinitiationsfaktoren (Fredrickson 1992).
Auf diese Weise verknüpfen extrazelluläre Signale die Transkription mit der Translation, was
in einer deutlichen Steigerung der Expression wachtumsregulierender Proteine in dem G0-G1
Übergang und in der G1-Phase des Zellzyklus resultiert. Beispielsweise führt eine Stimulation
durch Serum und mitogene Faktoren durch die Aktivierung von
Translationsinitiationsfaktoren zu einer gesteigerten Rate der Translationsinitiation, während
ein Serumentzug die Proteinsynthese auf der Translationsebene deutlich reduziert (Hassell JA
1976). Folglich stellen die Translationsinitiationsfaktoren wesentliche Effektoren der
wachstumskontrollierenden intrazellulären Signalkaskaden dar.

60
Mirjam Debus DISKUSSION
Es existiert eine differentielle Regulation der Genexpression durch die Initiation der
Translation. Viele Daten zeigen, dass nicht alle mRNAs mit derselben Effizienz translatiert
werden. Die Länge des Poly-A-Tails, Interaktionen von Cis-regulatorischen Elementen der
mRNA mit RNA-bindenden Proteinen, Sekundärstrukturen in der Nähe des Initiationskodons
sowie die Gesamtrate der Proteinsynthese beeinflussen die Effizienz der Translation. Ein
weiterer Faktor, der die Translationseffizienz beeinflusst, ist die Struktur des 5’UTRs der
mRNA. Umso komplexer die Struktur des 5’UTRs, umso geringer ist ihre spontane
Expressionsrate (Kozak 1991). Die Translation dieser mRNAs ist in erheblich höherem Maße
von unterstützenden Translationsinitiationsfaktoren abhängig, als mRNAs mit einfachen
5’UTRs (Koromilas 1992). Eine Modulation auf Ebene der Translationsinitiation hätte somit
einen bevorzugten Effekt auf mRNAs mit komplexen 5’UTRs. Es ist bekannt, das mRNAs
von Proto-Onkogenen im besonderen Maße hochkomplexe 5’-Enden aufweisen. Vorherige
Untersuchungen unserer Arbeitsgruppe haben gezeigt, dass Inhibitoren der
Translationsinitiation wie PKR preferentiell die Synthese von wachstumsfördernden
Proteinen wie beispielsweise Cyclin D1 und c-myc inhibieren, während die Translation
anderer Proteine, z.B. β-Aktin und Ubiquitin unbeeinflusst bleibt (Palakurthi 2000).
Zusammenfassend zeigen diese Daten, dass die Translationsinitiation ein streng kontrollierter
Prozess ist, der im Falle einer Aktivierung durch die preferentielle Expression
wachstumsfördernder Proteine substantiell zu einer Stimulation des Zellwachstums beiträgt.
Durch eine gezielte Inhibition über eine Aktivierung von PKR würde hingegen im besonderen
Maße die Expression wachstumsfördernder Proteine unterbunden werden.
6.5 Die strukturanalytischen Daten von PKR bieten einen potenziellen neuen Ansatz für die antineoplastische Therapie auf der Ebene der Translation
Aufgrund ihrer Schlüsselfunktion in der Expression von wachstumsfördernden Proteinen
stellt PKR ein attraktives Zielmolekül für die Entwicklung einer spezifischen
antineoplastischen Therapie dar. Anhand der strukturanalytischen Daten kann postuliert
werden, dass sich theoretisch kleine Moleküle generieren lassen, welche PKR durch direkte
oder indirekte Interaktion aktivieren können. Zahlreiche biochemische Untersuchungen haben
gezeigt, das eine eIF2α-Phosphorylierung durch PKR, und damit die Inhibition der
Translation, häufig über eine Ca2+-Entleerung aus dem endoplasmatischen Retikulum (ER)
getriggert wird. So ist bekannt, dass Agenzien wie DTT, Natriumarsenid und Tunicamycin,
welche die Ca2+-Freigabe aus dem ER induzieren, eine gesteigerte eIF2α-Phosphorylierung

61
Mirjam Debus DISKUSSION
durch PKR erzielen (Prostko 1993). Der genaue Mechanismus der PKR-Aktivierung ist
diesbezüglich noch nicht aufgeklärt. Spekuliert wird, dass es über die Induktion von
Proteinchaperonen zur Störung der Proteinfaltung und zur Akkumulation von ungefalteten
Proteinen kommt, welche in zellulärem Stress und der Aktivierung von PKR resultiert
(Brostrom, Prostko et al. 1996).
In vorangegangenen Untersuchungen konnte unsere Arbeitsgruppe zeigen, dass das
Antimykotikum Clotrimazol (CLT) ebenfalls eine signifikante Entleerung intrazellulärer
Ca2+-Speicher verursacht, welche in der Aktivierung von PKR, in Phosphorylierung von
eIF2α, und somit im Zellzyklusarrest resultiert (Aktas, Fluckiger et al. 1998). Die konsekutive
Blockierung des Zellzyklus in der G0-Phase bewirkt einen Wachstumsstillstand in
Tumorzellen sowohl in vitro als auch in vivo. (Aktas 2004). Aus früheren Studien ist bekannt,
dass Clotrimazol die Rate der Zellproliferation von normalen und Krebszelllinien in vitro
inhibiert und zur Reduktion von Metastasen in SCID Mäusen mit Melanomen führt
(Benzaquen 1995). Ebenso verursacht Eicosapentoinsäure (EPA), einer in Fischöl enthaltenen
vielfach ungesättigten Fettsäure, eine PKR vermittelte Inhibition der Translationsinitiation
(Palakurthi 2000). Ihr werden tumorpräventive Eigenschaften zugesprochen, seitdem bekannt
ist, dass Eskimovölker, welche große Mengen an Fisch konsumieren, eine sehr niedrige
Inzidenz maligner Tumoren aufweisen (Caygill 1996). Es konnte nachgewiesen werden, dass
EPA bevorzugt die Synthese und Expression von G1-Cyclinen inhibiert, was in einem
Zellzyklusblock in der G1-Phase resultiert. Dieser Effekt wird erreicht durch Freisetzung von
Ca2+ aus intrazellulären Speichern, während die Wiederauffüllung durch kapazitativen Ca2+-
Influx verhindert wird (Aktas, Fluckiger et al. 1998). In einem KLN-205-
Plattenepithelkarzinom-Maus-Modell reduzierte die orale EPA Gabe signifikant das
Tumorwachstum. Eine reduzierte Expression von Cyclin D1 führte zu einem G1
Zellzyklusblock (Palakurthi 2000). Kürzlich konnte zudem gezeigt werden, dass auch
Thiazolidinedione (TZDs) zur Aktivierung von PKR führen. TZDs gehören zur Gruppe der
PPARγ Liganden. Sie besitzen bekannte antikarzinogene Eigenschaften (Elstner 1998). Zur
Zeit werden TZDs in klinischen Studien in der Krebstherapie bei Tumoren mit hoher PPARγ
Expression getestet. Bislang wurde angenommen, dass die antiproliferativen Eigenschaften
durch Aktivierung von PPARγ vermittelt werden (Morrison 1999). Neuere Studien zeigen
allerdings, dass TZDs die Proliferation von PPARγ-/- and PPARγ+/+ ES Zellen in gleichem
Umfang reduzieren. TZDs induzieren also einen Zellzyklusblock in der G1 Phase durch einen
PPARγ unabhängigen Mechanismus. Dieser involviert die partielle Entleerung von
intrazellulären Ca2+-Speichern, die Aktivierung von PKR und die Phosphorylierung von

62
Mirjam Debus DISKUSSION
eIF2α und resultiert in der Inhibition der Translationsinitiation (Palakurthi 2001).
Aufgrund ihrer Schlüsselrolle von PKR bei der Translation unterstreichen alle genannten
Ergebnisse die Attraktivität von PKR als pharmakologische Zielstruktur. Neben dem
spezifischen Wirkmechanismus verspricht eine Interaktion auf Ebene der Translation
gegenüber einer konventionellen antineoplastischen Therapie vor allem eine geringere
Toxizität, da hier keine generelle Schädigung der zellulären DNA und damit eine schwere
Genotoxizität induziert wird. Auch können Zellen, welche sich nicht in Teilung befinden
durch eine derartige Therapie erreicht werden. Gleichzeitig wird aber auch die Relevanz einer
grundlegenden Untersuchung der aktivierenden und inhibierenden Prozesse offensichtlich, da
nur auf diese Weise die aus einer Modifizierung resultierende Effekte bewertet werden
können. Wesentliche Informationen zu den Mechanismen einer Autoinhibition von PKR in
ihrer inaktiven latenten Form, konnten im Rahmen dieser Arbeit generiert werden.
Strukturanalytische Daten zeigen, dass beide dsRNA bindende Domänen, DRBD I und II, mit
der Kinasedomäne interagieren. Sie umwickeln sozusagen die Kinasedomäne und blockieren
auf diese Weise sowohl die Dimerisation, als auch die cis- und trans- Autophosphorylierung,
welche für eine Aktivierung von PKR essentiell ist. In Deletionsstudien konnte außerdem
gezeigt werden, dass die DRBD II die eIF2α-Bindungsstelle am C-Terminus der
Kinasedomäne, jedoch nicht die N-terminale Autophosphorylierungseite blockiert. Dieses
impliziert, dass die DRBD II allein zwar die katalytische Aktivität von PKR stört, nicht aber
die Autophosphorylierung. Eine direkte Blockierung durch die DRBD-Kinase-Interaktion
konnte nicht nachgewiesen werden. Der gezeigte Effekt kann daher über zwei möglich
Mechanismen erzielt werden: Entweder über eine Restriktion der katalytisch aktiven Seite
durch Konformationsänderungen, oder alternativ durch ein „Pseudosubstrat“ in der
Verbindungsregion zwischen den dsRNA bindenden Domänen und der Kinasedomäne. Eine
allosterische Regulation ist jedoch wahrscheinlicher.
6.6 Ein antineoplastischer Effekt über eine Aktivierung von PKR lässt eine verminderte Chemo- und Apoptoseresistenz vermuten
Ein nicht unerhebliches Problem in der Therapie von Tumoren begründet sich in einer
generellen Resistenz gegenüber Toxinen. Zum Teil wird der chemoresistente Phänotyp erst
durch die Applikation zytotoxischer Agenzien induziert. Die meisten genotoxischen Agenzien
benötigen für die Induktion des Zelltodes einen intakten Apoptoseapparat (Coultas L, 2001).
Inhibitoren der Translationsinitiation wie PKR haben gegenüber einer konventionellen

63
Mirjam Debus DISKUSSION
Chemotherapie dem Vorteil, ihren antiproliferativen Effekt auch ohne intakte
Apoptosekaskade auszuüben. Zum Teil ist ihr Effekt unabhängig von der durch p53
vermittelten Apoptosekaskade (Vorburger SA 2002).
Maligne Tumore weisen häufig Defekte in Apoptose-regulierenden Proteinen wie p53 oder
dem nukleärem Faktor kappa κB (NFκB) auf (Kaufmann SH, 2004). Aus einer defekten
Apoptosekaskade resultiert oft eine generelle Chemoresistenz. Derzeitig sind noch nicht alle
Signalwege, die in der Apoptose eine Rolle spielen, aufgeklärt. Es ist bekannt, dass der
Prozess des programmierten Zelltodes durch eine Vielzahl an Faktoren reguliert wird. Eine
Dysregulierung dieser Modulatoren kann zu einer malignen Transformation führen. Man kann
zwei Hauptsignalwege unterscheiden. Eine extrinsische oder zytoplasmatische Signalkaskade
wird durch den Fas Todesrezeptor, einem Mitglied der TNF-Rezeptor Superfamilie getriggert
(Zapata JM 2001). Eine zweite Signalkaskade, der intrinsische oder mitochondriale
Signalweg, führt über eine Freisetzung von Cytochrom C aus den Mitochondrien, und
konsekutiv zur Aktivierung des Todessignals (Hockenbery D 1990). Beide Signalkaskaden
konfluieren in einer gemeinsamen Endstrecke, welche in einer Aktivierung von Caspasen und
über die proteolytische Spaltung regulatorischer und struktureller Proteine im Zelltod
resultiert (Scaffidi C 1989).
Einen wesentlichen Induktor der Apoptosekaskade stellt das Produkt des p53
Tumorsupressorgens dar (Benchimol 2001). Nach der Detektion eines DNA-Schadens führt
die p53-Aktivierung zu einem Zellzyklusarrest in der G1-Phase, um eine Reparatur vor dem
Eintritt in die S-Phase zu ermöglichen. Kann der Schaden nicht repariert werden, so triggert
p53 den Eintritt in die Apoptose. Sowohl in Zellkultur als auch in Tiermodellen konnte
gezeigt werden, dass ein funktionelles p53-Genprodukt für den mittels genotoxischer
Agenzien getriggerten programmierten Zelltod essentiell ist (Johnstone RW 2002). Da viele
Tumoren eine p53-Mutation aufzeigen, ist hierin eine entscheidende Ursache der
Chemoresistenz zu sehen.
Ein weiterer wichtiger Regulator der Apoptose ist der Transkriptionsfaktor NFκB, welcher
unter anderem das Schlüsselmolekül FasL traskriptionell reguliert (Wajant 2002). NFκB gilt
in erster Linie als anti-apoptotischer Faktor. Situationsabhängig werden über NFκB aber
sowohl pro-, als auch antiapoptotische Signalwege induziert. Unter anderem konnte gezeigt
werden, dass die NFκB Untereinheit p65/RELA für die durch p53 getriggerte
Apoptosekaskade erforderlich ist (Ryan KM 2000). In Hinblick auf die biologischen Effekte

64
Mirjam Debus DISKUSSION
von PKR sind insbesondere die proapoptotischen Eigenschaften von Interesse. Es wurde
gezeigt, dass die Aktivierung von NFκB durch PKR über den IκB Kinasekomplex (IKK) für
die aptotische Wirkung von PKR entscheidend ist (Gil 2001). Eine wesentliche Rolle bei der
Signalübermittlung spielen dabei Proteine der TNF-Rezeptor assoziierten Faktoren (TRAF)
(Gil 2004).
6.7 Durch den synergistischen Effekt von Interferonen und PKR kann eine optimierte Therapie bei verminderter Toxizität erzielt werden
Interferone wurden ursprünglich als Regulatoren der zellulären Abwehr gegen Virus-
infektionen identifiziert. Interferone spielen aber auch eine entscheidende Rolle in der
Regulation von Zellwachstum und maligner Entartung. (Kailash 2004). Typ I Interferon wird
ubiquitär exprimiert. Die Aktivierung von Interferonrezeptoren durch Ligandenbindung
verursacht die Phosphorylierung der Rezeptoren durch assoziierte Kinasen sowie die
Induktion von Interferon stimulierten Genen (ISG) [Doly , 1998 #280]. Die Aktivität vieler
dieser Gene, wie beispielsweise PKR und 2-5 Adenylatsynthase, ist abhängig von
doppelsträngiger RNA. Die Bindung von dsRNA führt zu Aktivierung von PKR und somit
zur Inhibition der Proteinsynthese.
Typ I Interferone sind für ihre antineoplastischen Eigenschaften, insbesondere bei malignen
Erkrankungen des hämatopoetischen Systems bekannt. Die Induktion von ISG in
Tumorzellen, wie unter anderem PKR, wird zumindest partiell für den antineoplastischen
Effekt verantwortlich gemacht (Kirkwood 2002). In diesem Fall würde die gesteigerte PKR
Aktivität aus einer spontanen Dimerisierung von PKR Molekülen resultieren, welche auftritt,
wenn PKR supraphysiologische Konzentrationen erreicht. Dieses könnte die sehr hohen,
häufig toxischen, erforderlichen Dosen an Interferonen in der Krebstherapie erklären. Die
simultane Therapie von Patienten mit Interferonen und PKR-aktivierenden Agenzien wie
Clotrimazol, Eicosapentanoinsäure, Thiazolidinidionen bzw. direkt aktivierenden kleinen
Moleküle versprechen, die Effizienz bei geringerer Toxizität zu erhöhen. In vitro konnte der
synergistische Effekt von Translationsinitiations-Inhibitoren und Interferonen bereits bestätigt
werden (Caraglia M 2003). Aufgrund dieser Daten gibt es Anhalt dafür, dass eine
Kombinationstherapie mit Interferonen und Translationsinitiations-Inhibitoren nicht nur einen
effektiveren antineoplastischen Effekt als eine Monotherapie erzielt, sondern sich dadurch
auch die Dosis der kostenintensiven sowie nebenwirkungsreichen Interferone reduzieren lässt.

65
Mirjam Debus ZUSAMMENFASSUNG 7 ZUSAMMENFASSUNG Bei dem vorliegenden Projekt geht es um die Modulation der Translation als möglicher
Ansatz in der antineoplastischen Therapie. Im Fokus steht hier die von doppelsträngiger
Virus-RNA (dsRNA) aktivierte Proteinkinase (PKR), welche einen inhibitorischen Effekt auf
die Translation ausübt.
Bei der Proteinbiosynthese ist die Initiation der Translation besonders strikt reguliert. Durch
Inhibition der Translationsinitiation werden bevorzugt G1-Cycline supprimiert, was zu einem
Zellzyklusstillstand in der G1-Phase führt. Substanzen, die modulierend auf dieser Ebene
eingreifen, versprechen antiproliferativ, aber nur gering toxisch zu wirken. Die PKR stellt
somit ein potenzielles Zielmolekül in der antiproliferativen Therapie dar. Zudem spielt PKR
eine Schüsselrolle in der antiviralen Abwehr. Induziert durch Interferon-α und aktiviert durch
dsRNA vermittelt PKR die Herabregulierung viraler Replikation in infizierten Zellen.
Maligne Tumore weisen in der Regel eine gesteigerte translationelle Aktivität, insbesondere
eine gesteigerte Expression von Proto-Onkogenen auf. Die Möglichkeit einer präferentiellen
Herabregulierung von wachstumsfördernden Proteinen auf Ebene der Translationsinitiation
beruht auf der Tatsache, dass Proteine verschiedene messenger Ribonukleinsäuren (mRNA)
Spezies besitzen, die in ihrer translationalen Effizienz hundertfach variieren. Die meisten der
bekannten Proto-Onkogene und Wachstumsfaktoren haben komplexe mRNA Strukturen, die
eine starke translationelle Regulation vermuten lassen. Sie zeigen beispielsweise
hochkomplexe 5’Untranslated Regions (5’UTRs), welche die Rekrutierung der ribosomalen
Untereinheiten erschweren, und dadurch eine stärkere Abhängigkeit von unterstützenden
Faktoren erforderlich machen. Es ist also davon auszugehen, dass Proto-Onkogen-mRNAs im
besonderen Maße von Initiationsfaktoren der Translation abhängig sind.
Ziel dieses Projektes war es, einen Beitrag zu Strukturanalysen einzelner Domänen mittels
nukleärer Magnetresonanzspektroskopie (NMR) zu leisten. Bisher konnte eine für die NMR
geeignete Isolierung des Proteins aus unterschiedlichen Expressionssystemen nicht gelingen,
da es zum einen zu groß für die NMR-Analytik ist, zum anderen erschwerte eine geringe
Löslichkeit die Isolierung. Bei dem vorliegenden Projekt wurden Konstrukte generiert, die
sowohl die individuellen Domänen, als auch das gesamte C-terminale Segment enthalten. Um
die Löslichkeit zu erhöhen, wurden unterschiedliche Fusionsproteine gebildet. Die Isolierung
der Fusionsproteine gelang mittels High Performance Liquid Chromatography (HPLC). Die

66
Mirjam Debus ZUSAMMENFASSUNG
NMR-Analyse der Kinasedomäne wurde mittels TROSY-15N-heteronukleärer singulärer
Quantum-Koherenz (HSQC) Experimente durchgeführt. Auf diese Weise konnten erstmals
mittels NMR dreidimensionale Strukturelemente der Kinasedomäne von PKR charakterisiert,
und deren mit den regulativen Domänen interagierenden Regionen lokalisiert werden. Die
generierten Informationen erlauben nun eine exaktere Untersuchung der Aktionsmechanismen
von PKR und bieten zudem die Basis für die Entwicklung von kleinen aktivierenden
Molekülen zum Zweck einer mehr spezifischen weniger toxischen antineoplastischen sowie
antiviralen Therapie.

67
Mirjam Debus ABKÜRZUNGEN
8 ABKÜRZUNGEN A Ampère Abb Abbildung ApoE Apolipoprotein E Ak Antikörper ATP Adenosintriphosphat AU Absorbance Units BCA Bicinchoninic Acid bp Basenpaar Bcl-2 B-cell lymphoma protein 2 °C Grad Celsius CBP Chitin binding protein CLT Clotrimazol CPP32 Cellular Protease 32 DEAE Diethylaminoethyl dest destillatum DNA Desoxyribonukleinsäure dNTP Desoxyribonukleosid-5’triphosphat dsDNA doppelsträngige DNA DTT Dithiolthreitol E. coli Escherichia coli EDTA Ethylendiamintetraacetat eIF2α eukaryotischer Initiationsfaktor-2α EPA Eicosapentoinsäre FasL FasLigand g Gramm G1 Gap 1 GTP Guanidin-Triphosphat h Stunde HPLC High Performance Liquid Chromatographie HSQC Heteronuclear Single Quantum Coherence IFN Interferon IPTG Isopropyl β-D-1-Thiogalaktopyranosid IRF-1 Interferon regulierender Faktor ISG Interferon stimulierte Gene kD kilo Dalton OD Optical Density l Liter LB Luria Bertanii m milli M Molar M Mitose MBP Maltose binding protein Mda7 Melanom Differenzierungs-assoziiertes Gen 7 mi Minute ml Milliliter mm Millimeter mM Millimolar mAk monoklonale Antikörper mRNA meesenger Ribonuleinsäure mV Millivolt

68
Mirjam Debus ABKÜRZUNGEN µ mikro NFκB Nukleärer Faktor-κB ng Nanogramm nm Nanometer NOESY Nuclear Overhauser Enhancement Spectroscopy OD optische Dichte PAGE Polyacrylamid-Gelelektrophorese PCR Polymerase chain reaction PG Protein G PH -log [H+] PKR Proteinkinase R PMSF Phenylmethylsulfonylfluorid PPARγ Peroxysome Proliferator-Activated Receptor Rel Untereinheit von NFκB RNA Ribonukleinsäure rpm Rotationen pro Minute RT Raumtemperatur SDS Natriumdodecylsulfat TAE Tris-Acetat-EDTA TBS Tris-Base Sodium TEMED N,N,N’,N’-Tetramethylendiamin TNFα Tumor-Nekrosefaktor α Tris Tris(hydroxymethyl) amino-Methan TROSY Transverse Relaxation Optimized Spectroscopy TZD Thiazolidinedione UV Ultraviolet V Volt v-mos Gen einer viralen Serin-Threoninkinase W Watt Abkürzungen für Aminosäuren A Ala Alanin C Cys Cystein D Asp Aspartat E Glu Glutamat F Phe Phenylalanin G Gly Glycin H His Histidin I Ile Isoleucin K Lys Lysin L Leu Leucin N Met Methionin M Asn Asparagin P Pro Prolin Q Gln Glutamin R Arg Argenin S Ser Serin T Thr Threonin V Val Valin W Trp Tryptophan Y Tyr Tyrosin
Abkürzungen für Nukleotide A Adenin C Cytosin G Guanin T Thymin

69
Mirjam Debus LITERATUR 9 LITERATUR Abraham, N. (1999). “Characterization of Transgenic Mice with Targeted Disruption of the
Catalytic Domain oft the Double-stranded RNA-dependent Protein Kinase, PKR.” J Biol Chem 274: 5953-5962.
Aktas, H. (2004). “Translational regulation of gene expression by omega-3 fatty acids.” J.
Nutr. 134: 2487S-2491S. Aktas, H., R. Fluckiger, et al. (1998). “Depletion of intracellular Ca2+ stores,
phosphorylation of eIF2alpha, and sustained inhibition of translation initiation mediate the anticancer effects of clotrimazole.” Proc Natl Acad Sci U S A 95(14): 8280-5.
Bagby S, T. K., Ikura M (2001). “Optimization of protein solubility and stability for protein
nuclear magnetic resonance.” Methods Enzymol 339: 20-41. Balachandran, S. (1998). “Activation of the dsRNA-dependent protein kinase, PKR, induces
apoptosis through FADD-mediated death signaling.” Embo J 17: 6888-6902. Baldwin, E. (2005). “Etoposide, topoisomerase II and cancer.” Curr Med Chem Anticancer Agents 5: 363-72. Baltzis, D., S. Li, et al. (2002). “Functional characterization of pkr gene products expressed in
cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR.” J Biol Chem 277(41): 38364-72.
Barber, G. N., R. Jagus, et al. (1995). “Molecular mechanisms responsible for malignant
transformation by regulatory and catalytic domain variants of the interferon-induced enzyme RNA-dependent protein kinase.” J Biol Chem 270(29): 17423-8.
Barber, G. N., Tomita, J., Hovanessian, A. G., Meurs, E, Katze, M. G. (1991). “Functional
expression and characterization of the interferon-induced double-stranded RNA activated P68 protein kinase from Escherichia coli.” Biochemistry 30: 10356-10361.
Barber, G. (1994). “Regulation of viral and cellular gene expression in cells infected by
animal viruses, including influenza virus and human immunodeficiency virus type 1.” Methods in Virology: 141-168.
Barber, G. N. (1994). Proc Natl Acad Sci U S A 91: 4278-4282. Benchimol, S. (2001). “p53-dependent pathways of apoptosis.” Cell Death Differ 8: 1049-51. Benzaquen, L. (1995). “Clotrimazole inhibits cell proliferation in vitro and in vivo.” Nat
Med 1: 534-540. Bernado P, A. T., Garcia de la Torre J, Akke M, Pons M. and J. A. C. S. J. 29;125(4):916-23
(2003). “Combined use of NMR relaxation measurements and hydrodynamic calculations to study protein association. Evidence for tetramers of low molecular weight protein tyrosine phosphatase in solution.” J Am Chem Soc 125: 916-23.
Bignold, L. (2006). “Alkylating agents and DNA polymerases.” Anticancer Res 26: 1327-36.

70
Mirjam Debus LITERATUR Brostrom, C. O., C. R. Prostko, et al. (1996). “Inhibition of translational initiation by
activators of the glucose- regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase.” J Biol Chem 271(40): 24995-5002.
Black, T. L. (1993). “Degradation of the interferon induced 68.000 M protein kinase by
poliovirus requires RNA.” J Virology 67: 791-800. Caraglia M, M. M., Giuberti G, D'Alessandro AM, Baldi A, Tassone P, Venuta S,
Tagliaferri P, Abbruzzese A. (2003). “The eukaryotic initiation factor 5A is involved in the regulation of proliferation and apoptosis induced by interferon-alpha and EGF in human cancer cells.” J Biochem (Tokyo) 133: 757-65.
Caygill, C. P. (1996). “Fat, fish, fish oil and cancer.” Br J Cancer 74: 159-164. Christendat D, S. V., Turnbull JL (1998). “Use of site-directed mutagenesis to identify
residues specific for each reaction catalyzed by chorismate mutase-prephenate dehydrogenase from Escherichia coli.” Biochemistry 10: 15703-12.
Chu, W. M. (1999). Immunity 11: 721-731. Clemens, M. J. (1975). “Inhibition of protein synthesis in rabbit reticolocyte lysates by
double-stranded RNA and oxidized glutathione: indirect mode of action on polypeptide chain initiation.” Proc Natl Acad Sci U S A 72: 1286-1290.
Clemens, M. J. (1994). “Regulation of eukariotic protein synthesis by protein kinases that
phosphorylate initiation factor eIF2.” Mol Biol Reports 19: 201-210. Clemens, M. J. and U. A. Bommer (1999). “Translational control: the cancer connection.” Int
J Biochem Cell Biol 31(1): 1-23. Clemens, M. J. and A. Elia (1997). “The double-stranded RNA-dependent protein kinase
PKR: structure and function.” J Interferon Cytokine Res 17(9): 503-24. Craig, A. W., G. P. Cosentino, et al. (1996). “The kinase insert domain of interferon-induced
protein kinase PKR is required for activity but not for interaction with the pseudosubstrate K3L.” J Biol Chem 271(40): 24526-33.
Cuddihy, A. R., A. H. Wong, et al. (1999). “The double-stranded RNA activated protein
kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro.” Oncogene 18(17): 2690-702.
Dagon, Y. (2001). “Double-stranded RNA-dependent protein kinase, PKR, down-regulates
CDC2/cyclin B1 and induces apoptosis in non-transformed but not in v-mos transformed cells.” Oncogene 20: 8045-8056.
Dar A C, D. T. E., Sicheri F (2005). “Higher-Order Substrate Recognition of eIF2 alpha by
the RNA-Dependent Protein Kinase PKR.” Cell 122: 887-900 De Haro, C. (1996). “The eIF-2alpha kinases and the kontroll of protein sythesis.” Faseb J
10: 1378-1387.

71
Mirjam Debus LITERATUR Der, S. D. (1997). “A double-stranded RNA-activated protein kinase-dependet pathway
mediating stress-induced apoptosis.” Proc Natl Acad Sci U S A 94: 3279-3283. Elstner, E. (1998). “Ligands for peroxysome proliferator-activated receptor gamma and
retenoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice.” Proc Natl Acad Sci U S A 95: 8806-8811.
Farber, S., Diamond, L.K., Mercer, R.D., Sylvester, R.F., Wolff, J.A. (1948). “Temporary
remission in acute leukemia in children produced bei folic antagonist, 4-aminopterylglutamic acid.” New England Yournal of Medicine 238: 787-793.
Feng, G.-S. (1992). “Identification of double-stranded RNA-binding domains in the
interfernon-induced double-stranded RNA-activated p68 kinase.” Proc Natl Acad Sci U S A 89: 5447-5451.
Fredrickson, R. (1992). “Signal transduction and regulation of translation initiation.” Semin
Cell Biol 3: 107-115. Galabru, J. (1987). “Autophosphorylation of the protein kinase dependent on double-
stranded RNA.” J Biol Chem 262: 15538-15544. Galabru, J. (1989). “The binding of double-stranded RNA and adenovirus VAI RNA to the
inetrferon-induced protein kinase.” Eur J Biochem 178: 581-589. Gale, M., Jr. and M. G. Katze (1998). “Molecular mechanisms of interferon resistance
mediated by viral- directed inhibition of PKR, the interferon-induced protein kinase.” Pharmacol Ther 78(1): 29-46.
Gale, M. J., Jr. (1997). “Evidence that hepatitis C virus resistance to interferonis mediated
through repression of the PKR protein kinase by nonstructural 5A protein.” Virology 230: 217-227.
George (1996). “Characterization of the Heparin-Mediated Activation of PKR, the Interferon-
Inducible RNA-Dependent Protein Kinase.”: 180-188. Gerlitz, G., R. Jagus, et al. (2002). “Phosphorylation of initiation factor-2 alpha is required
for activation of internal translation initiation during cell differentiation.” Eur J Biochem 269(11): 2810-9.
Gil, J., J. Alcami, et al. (1999). “Induction of apoptosis by double-stranded-RNA-dependent
protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB.” Mol Cell Biol 19(7): 4653-63.
Gil, J. and M. Esteban (2000). “Induction of apoptosis by the dsRNA-dependent protein
kinase (PKR): mechanism of action.” Apoptosis 5(2): 107-14. Gil, J., J. Rullas, M. A. Garcia, J. Alcami, and M. Esteban (2001). “The catalytic activity of
dsRNA-dependent protein kinase, PKR, is required for NF-kappaB activation.” Oncogene 20: 385-394.
Gil, J., J. Rullas, M. A. Garcia, P. Gomez-Puertas, S. Guerra, J. Rullas, H. Nakano,J. Alcami, and M. Esteban (2004). “TRAF family proteins link PKR with NF-kappa B activation.” Mol Cell Biol 24: 4502-12.

72
Mirjam Debus LITERATUR Gilman, A. (1963). “The initial clinical trial of nitrogen mustard.” Am J Surg 105: 574-8. Gmeiner, W. (2005). “Novel chemical strategies for thymidylate synthase inhibition.” Curr
Med Chem 12: 191-202. Gosh, S. (1998). Annu Rev Immunol 16: 225-260. Haines, G. K. (1993). “Expression of p68 protein kinase as recognized by the monoconal
antibody TJ4C4 during human fetal development.” Tumour Biol 14: 95-104. Hassell JA, E. D. (1976). “The regulation of protein synthesis in animal cells by serum
factors.” Biochemistry 15: 1375-81. Hockenbery D, N. G., Milliman C (1990). “Bcl-2 is an inner mitochondrial membrane
protein that blocks programmed cell death.” Nature 348: 334-336. Jaramillo, M. L. (1995). “The interferon system: a review with emphasis on the role of PKR
in growth control.” Cancer Invest 13: 327-338. Jeffrey, I. W., M. Bushell, et al. (2002). “Inhibition of protein synthesis in apoptosis:
differential requirements by the tumor necrosis factor alpha family and a DNA-damaging agent for caspases and the double-stranded RNA-dependent protein kinase.” Cancer Res 62(8): 2272-80.
Johnstone RW, R. A., Lowe SW (2002). “Apoptosis: a link between cancer genetics and
chemotherapy.” Cell 108: 153-64. Jordan, M. (2002). “Mechanism of action of antitumor drugs that interact with microtubules
and tubulin.” Curr Med Chem Anticancer Agents 2: 1-17. Kailash, C. (2004). “Interferons and interferon inhibitory activity in disease and therapy.”
Exp Biol and Med. 229: 285-290. Katze, M. G. (1990). “The regulation of viral and cellular RNA turnover in cells infected by
eukaryotic viruses including human immunodeficiency virus, HIV 1.” Enzyme 44: 332-346.
Katze, M. G. (1991). “Functional expression and RNA binding analysis of the interferone-
induced,RNA-activated, 68,000-Mr protein kinase in a cell free system.” Mol Cell Biol 11: 5497-5505.
Kim, S. H., A. P. Forman, et al. (2000). “Human breast cancer cells contain elevated levels
and activity of the protein kinase, PKR.” Oncogene 19(27): 3086-94. Kim, S. H., S. Gunnery, et al. (2002). “Neoplastic progression in melanoma and colon cancer
is associated with increased expression and activity of the interferon-inducible protein kinase, PKR.” Oncogene 21(57): 8741-8.
Kirkwood, J. (2002). “Cancer immunotherapy: the interferon-alpha experience.” Semin
Oncol. 29: 18-26. Koromilas, A. E. (1992). “Malignant transformation by a mutant of the IFN-inducible
dsRNA dependent Proteinkinase.” Science 257: 1685-1689.

73
Mirjam Debus LITERATUR Koromilas, A. E. (1992). “mRNAs containing extensive secondary structure in their 5' non-
coding region translate efficiently in cells overexpressing initiation factor eIF-4E.” Embo J 11: 4153-4158.
Kozak, M. (1991). “An analysis of vertebrate mRNA sequences: intimations of translational
controll.” Journal of Cell Biology 115: 887-903. Kumar, K. U. (1994). “Double-stranded RNA-dependent protein kinase activates
transcription factor NF-kappaB by phosphorylating IkappaB.” Proc Natl Acad Sci U S A 91: 6288-6298.
Kumar, K. U. (1997). Embo J 16: 406-416. Lee, S. B. (1994). “The interferon-induced double stranded RNA-activated protein kinase
induces apoptosis.” Virology 199: 491-496. Lee, S. B. D. Rodriguez, et al. (1997). “The apoptosis pathway triggered by the interferon-
induced protein kinase PKR requires the third basic domain, initiates upstream of Bcl- 2, and involves ICE-like proteases.” Virology 231(1): 81-8.
Levin, D. (1978). Proc Natl Acad Sci U S A 75: 1121-1125. Li, S. (2001). “Dominant Negative Function by an Alternatively Spliced Form of the
Interferon-inducible Protein Kinase PKR.” J Biol Chem 276: 13881-13890. Lodish, H. F. (1970). J Mol Biol 50: 689. Lodish, H. F. (1974). “Model for the regulation of mRNA applied to haemoglobin synthesis.”
Nature 251: 385-388. Margalit H (1993). “Comparative analysis of structurally defined heparin binding sequences
reveals a distinct spatial distribution of basic residues.” J Biol Chem 15: 19228-31. McMillan NA, C. B., Hollis B, Toone WM, Zamanian-Daryoush M, Williams BR. (1995).
“Mutational analysis of the double-stranded RNA (dsRNA) binding domain of the dsRNA-activated protein kinase, PKR.” J Biol Chem 270: 2601-6.
Melville, M. W., S. L. Tan, et al. (1999). “The cellular inhibitor of the PKR protein kinase,
P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity.” J Biol Chem 274(6): 3797-803.
Meurs, E. F. (1990). “Molecular cloning and characterization of the human double-stranded
RNA-activated protein kinase induced by interferone.” Cell 62: 379-390. Meusel, T. R. (2002). “Protein kinase R regulates double-stranded RNA induction of TNF-
alpha but not IL-1ß mRNA in human epithelial cells.” J Immunol 186: 6429-6435. Morrison, R. F. (1999). “Role of PPAR gamma in regulating a cascade expression of cyclin
dependent kinase inhibitors, p18(INK 4c) and p21(Waf1/Cip1), during adipogenesis.” J Biol Chem 274: 17088-17097.
Nanduri, S. (1998). Embo J 17: 5458-5465.

74
Mirjam Debus LITERATUR Nanduri, S. (2000). Embo J 19: 5567-5574. Pain, V. M. (1996). “Initiation of protein synthesis in eukaryotic cells.” Eur J Biochem 236:
747-771. Palakurthi, S. S. (2000). “Inhibition of Translation Initiation Mediates the Anticancer Effect
o n-3 Polyunsaturated Fatty Acid Eicosapentaenoic Acid.” Cancer Res 60: 2919-2925. Palakurthi, S. S. (2001). “Anticancer effects of thiazolidinediones are independent of
peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation.” Cancer Res 61: 6213-6218.
Park, H., M. V. Davies, et al. (1994). “TAR RNA-binding protein is an inhibitor of the
interferon-induced protein kinase PKR.” Proc Natl Acad Sci U S A 91(11): 4713-7. Pataer, A., S. A. Vorburger, et al. (2002). “Adenoviral transfer of the melanoma
differentiation-associated gene 7 (mda7) induces apoptosis of lung cancer cells via up-regulation of the double-stranded RNA-dependent protein kinase (PKR).” Cancer Res 62(8): 2239-43.
Patel, R. C. (1992). “Identification of the double stranded RNA-binding domain of the
human interferon-inducible protein kinase.” J Biol Chem 267: 7671-7676. Patel, R. C. (1994). “Role of the amino-terminal residues of the interferon-induced protein
kinase in its activation by double-stranded RNA and Heparin.”: 18593-18598. Patel, R. C. (2000). “PACT, a stress-modulated cellular activator of inerferon-induced
double-stranded RNA-activated protein kinas, PKR.” J Biol Chem 275: 37993-37998. Patel, R. C. (2002). “Contribution of double-stranded RNA-activated Protein Kinase Toward
Antiproliferative Actions of Heparin on Vascular Smooth Muscle Cells.” Arterioscl Thromb Vasc Biol 22: 1439-1444.
Patel, R. C. and G. C. Sen (1998). “PACT, a protein activator of the interferon-induced
protein kinase, PKR.” Embo J 17(15): 4379-90. Prostko, C. R. (1993). “Reversible phosphorylation of eukaryotic initiation factor 2-alpha in
response to endoplasmatic reticular signaling.” Mol Cell Biochem 127/128: 255-265. Pryor KD, L. B. (1997). “High-level expression of soluble protein in Escherichia coli using a
His6-tag and maltose-binding-protein double-affinity fusion system.” Protein Expr Purif. 10: 309-19.
Prostko, C. R. (1993). “Reversible phosphorylation of eukaryotic initiation factor 2-alpha in
response to endoplasmatic reticular signaling.” Mol Cell Biochem 127/128: 255-265. Proud, C. G. (1995). “PKR: a new name and new roles.” Trends Biochem Sci 20(6): 241-6. Raine, D. (1998). “Inhibition of double-stranded RNA-dependent protein kinase PKR by
mammalian ribosomes.” FEBS Lett 436: 343-8.

75
Mirjam Debus LITERATUR Roberts, W. K. (1976). “Interferone-induced inhibition of protein synthesis in L-cell extracts:
an ATP dependent step in the activation of an inhibitor by double-stranded RNA.” Proc Natl Acad Sci U S A 73: 3136-3140.
Romano, P. R., M. T. Garcia-Barrio, et al. (1998). “Autophosphorylation in the activation
loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2.” Mol Cell Biol 18(4): 2282-97.
Riek R, F. J., Bertelsen EB, Horwich AL, Wuthrich K (2002). “Solution NMR techniques for
large molecular and supramolecular structures.” J Am Chem Soc 124: 12144-53. Ryan KM, E. M., Rice NR, Vousden KH (2000). “Role of NF-kappaB in p53-mediated
programmed cell death.” Nature 404: 892-7. Ryter, M. (1998). “Molecular basis of double-stranded RNA-protein interactions: structure of
a dsRNA-binding domain complexed with dsRNA.” Embo J 17(24): 7505-7513. Saelens, X., M. Kalai, et al. (2001). “Translation inhibition in apoptosis: caspase-dependent
PKR activation and eIF2-alpha phosphorylation.” J Biol Chem 276(45): 41620-8. Salzberg, S. (1995). “Interruption of myogenesis by transforming growth factor beta 1 or
EGTA inhibits expression an activity of the myogenic-associated (2'-5') oligoadenylate synthetase and PKR.” Exp Cell Res. 219: 223-32.
Savinova, O. (1999). “Abnormal levels and minimal activity of the dsRNA-activated protein
kinase, PKR, in breast carcinoma cells.” Int J Biochem Cell Biol 31: 175-189. Scaffidi C, F. S., Srinivasan A (1989). “Two CD95 (APO-1/Fas) signaling pathways.” Embo
J 17: 1675-1687. Sharp, T. V., Q. Xiao, et al. (1993). “Reversal of the double-stranded-RNA-induced
inhibition of protein synthesis by a catalytically inactive mutant of the protein kinase PKR.” Eur J Biochem 214(3): 945-8.
Singh SM, P. A. (2005). “Solubilization and refolding of bacterial inclusion body proteins.”
proteins J Biosci Bioeng. 99: 303-10. Srivastava, S. P., K. U. Kumar, et al. (1998). “Phosphorylation of eukaryotic translation
initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA- dependent protein kinase.” J Biol Chem 273(4): 2416-23.
ST Johnson, D. (1992). “A conserved double stranded RNA-binding domain.” Proc Natl
Acad Sci U S A 89: 10979-10983. Steitz, J. A. (1969). Nature 224: 957. Taylor, D. R. (1996). “Autophophorylation sites participate in the activation of the double-
stranded-RNA-activated protein kinase PKR.” Moll. Cell. Biol. 16: 6295-6302. Taylor, D. R. (2000). “Hepatitis C and interferon resistance.” Microbes and Infection 2:
1743-1756.

76
Mirjam Debus LITERATUR
Taylor, D. R., S. T. Shi, et al. (1999). “Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein.” Science 285(5424): 107-10.
Trieb, M. (2004). “Cooperative effects on the formation of intercalation sites.” Nucleic Acids
Res 32: 4696-703. Ung, T. L., C. Cao, et al. (2001). “Heterologous dimerization domains functionally substitute
for the double-stranded RNA binding domains of the kinase PKR.” Embo J 20(14): 3728-37.
Vattem, K. M., K. A. Staschke, et al. (2001). “Mechanism of activation of the double-
stranded-RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2).” Eur J Biochem 268(13): 3674-84.
Verma, I. M. (1995). Genes Dev 9: 2723-2735. Vorburger SA, P. A., Yoshida K, Barber GN, Xia W, Chiao P, Ellis LM, Hung MC, Swisher
SG, Hunt K (2002). “Role for the double-stranded RNA activated protein kinase PKR in E2F-1-induced apoptosis.” Oncogene 21: 6278-88.
Wajant, H. (2002). “The Fas signaling pathway: more than a paradigm.” Science 296: 1635-
1636. Williams, B. R. (1999). “PKR; a sentinel kinase for cellular stress.” Oncogene 18: 6112-
6120. Yeung, M. C. (1996). “An essential role for the interferon-inducible, double-stranded RNA-
activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells.” Proc Natl Acad Sci U S A 93: 12451-12455.
Zapata JM, P. K., Haas E (2001). “A diverse family of proteins containing tumor necrosis
factor receptor-associated factor domains.” J Biol Chem 276: 24242-24252. Zhou, P. (2001). “A solubility-enhancement tag (SET) for NMR studies of poorly behaving
proteins.” J Biomol NMR 20: 11-4. Zhu, S., P. R. Romano, et al. (1997). “Ribosome targeting of PKR is mediated by two double-
stranded RNA- binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2.” J Biol Chem 272(22): 14434-41.

77
Mirjam Debus DANKSAGUNG 7 DANKSAGUNG
Ich möchte mich bei allen bedanken, die mit ihrer Unterstützung, Freundschaft und Liebe
zum Gelingen dieser Arbeit beigetragen haben. Insbesondere danke ich
Herrn Prof. Dr. Süleyman Ergün für seinen grenzenlosen Enthusiasmus und für die
Vermittlung des Kontaktes nach Boston.
Herrn Prof. Dr. José Halperin für seine hohen Ansprüche, für die Konferenz in Bethesda und
das Skilaufen in den White Mountains von New Hampshire.
Herrn Dr. Huseyin Aktas für all die zahlreichen Wortgefechte und Gedankenkriege, für seine
wunderbare Art, mir die Knoten im Kopf zu lösen, und nicht zuletzt dafür, dass ich lernen
durfte, kurdisches Englisch zu verstehen.
Herrn Prof. Dr. Daniel Tosteson dafür, dass er unsere wöchentlichen Labormeetings stets mit
kontroversen Gedankengängen und wohlwollenden Fragen bereichert hat.
Frau Dr. med. Nerbil Kilic, für ihre hilfreiche Unterstützung und ihre Bemühungen die
Promotionsprozedur voran zubringen . Frau Dr. med. Anna Solth, für die Hilfe bei der
Korrektur. Herrn Dr. med. Rainer Günther für alle reflektierenden Gespräche. Frau Dr. med.
Julia Bachmann für die erfrischende Arbeit mit ihr und die korrigierende Durchsicht. Frau Dr.
rer. nat. Dr. med. Sonja Loges und Frau Dr. rer. nat. Susanne Sebens für alle guten
Ratschläge.
Cornelia, Maria, Theresa, Jakob, Charlotte, Britta, Johanna, Christina und Katharina für die
Gewissheit, dass mein Kind all die Jahre unter Eurer Obhut stets gut versorgt war.
meinen Eltern und Geschwistern, die immer für mich da sind, und vor allen Dingen meiner
wundervollen Tochter Milena Sophie, die immer noch so geduldig mit ihrer Mama ist,
obwohl der Besuch im Schwimmbad an unzählbaren Samstagen ausfallen musste.

78
Mirjam Debus LEBENSLAUF 10 LEBENSLAUF
Persönliche Geburtstag: 10.04.1974 Daten Geburtsort: Altenau bei Garmisch Partenkirchen
Familienstand: ledig, eine Tochter, geb. 08.11.2001
Derzeitige Wissenschaftliche Mitarbeiterin. 2. Weiterbildungsjahr Innere Medizin seit 03-2005 Tätigkeit 1. Medizinische Klinik, Universitätsklinikum Kiel Ausbildung Medizinstudium 10-1997 bis 12-2004. Universitätsklinikum Hamburg & Training Physikum: September 1999
Erstes Staatsexamen: März 2002 Zweites Staatsexamen: März 2003 Drittes Staatsexamen: Dezember 2004 Famulaturen: Innere Medizin. 09-02. Klinikum Neustadt, Ostholstein Innere Medizin. 07 u. 08-02. Brigham Women’s Hospital, Harvard Medical School, Boston Pulmonologische Praxis. 03-02. Dres. Koppermann u. Hißnauer , Hamburg Pathologie. 03-00 Institut für Pathologie, Universitätsklinik Hamburg-Eppendorf Molekulare Virologie. 02-00Heinrich-Pette-Institut, Hamburg-Eppendorf Ausbildung in Naturheilkunde. Sept. 1993-Sept. 1996 Homöopathie, Akupunktur und Phytotherapie. Schule für Naturheilkunde, Hamburg
Wissenschaftliche Tätigkeiten Wissenschaftliche Angestellte. Seit 9-05. Labor für Molekulare Gastroenterologie.
Forschungsschwerpunkt: L1CAM bei der Vermittlung von Chemoresistenz und Ausprägung eines malignen Phänotypes in duktalen Pakreaskarzinomen. 1. Medizinische Klinik, Universitätsklinikum Kiel
Doktorarbeit in der Onkologie. September 2000-September 2001 Modulation der Translation als Weg in der antineoplastischen Therapie. Cell Biology Institute, Laboratory for Translational Research. NIH gefördert. Harvard Medical School, Boston, USA
Stipendien Forschungsstipendium 09-00 bis 09-01 der Harvard Medical School, Boston, USA
Publikationen Gelev V, Aktas H, Marintchev A, Ito T, Frueh D, Hemond M, Rovniak D, Debus M, Hyberts S, Usheva A, Halperin J, Wagner G, Mapping of the Auto-inhibitory Interactions of Protein Kinase R by Nuclear Magnetic Resonance. 2006, J Mol Biol.
Sebens-Muerkoester S, Werbing V, Sipos B, Debus M, Witt M, Großmann M,
Leisner D, Koetteritzsch, Kappes H, Kloeppel G, Altevogt P, Foelsch U, Schaefer H, Drug induced expression of the cellular adhesion molecule L1CAM confers anti-apoptotic protection and chemoresistance in pancreatic ductal adenocarcinoma cells. 2006, Oncogene.

79
Mirjam Debus ERKLÄRUNG 11 ERKLÄRUNG
EIDESSTATTLICHE VERSICHERUNG: Ich versichere ausdrücklich, dass ich die Arbeit selbständig und ohne fremde Hilfe verfasst,
andere als die von mir angegebenen Quellen und Hilfsmittel nicht benutzt und die aus den
benutzten Werken wörtlich oder inhaltlich entnommenen Stellen einzeln nach Ausgabe, Band
und Seite des benutzten Werkes kenntlich gemacht habe.
Ferner versichere ich, dass ich die Dissertation bisher nicht einem Fachvertreter an einer
anderen Hochschule zur Überprüfung vorgelegt oder mich anderweitig um Zulassung zur
Promotion beworben habe.
Mirjam A. Debus


















