
Grundlagen der organischen Chemie Eine Zusammenfassung · Abstract Diese Zusammenfassung - welche...
69
Grundlagen der organischen Chemie Eine Zusammenfassung Christoph Kasper 31. Januar 2005
Transcript of Grundlagen der organischen Chemie Eine Zusammenfassung · Abstract Diese Zusammenfassung - welche...

Grundlagen der organischen Chemie
Eine Zusammenfassung
Christoph Kasper
31. Januar 2005

Abstract
Diese Zusammenfassung - welche sich immer mehr zum Skript entwickelt hat- bezieht sich auf die Vorlesung von Prof. H. Wennemers uber organischeChemie. Der Teil uber Spektroskopie von Prof. U. Sequin wird dabei nichtbehandelt, da dieser bereits ausreichend dokumentiert ist. Oberstes Ziel desTextes ist es, sich auf die Hauptpunkte des Stoffes zu fokusieren und dabeidas Augenmerk v.a. auf eine klare Strukturierung zu richten. Daher werdengewisse zusatzliche Informationen zu gewissen Reaktionen nicht erwahnt.Teilweise wird auf das Buch von P. Bruice, ”Organic Chemistry” verwiesen.Alle die Fehler finden, Fragen oder Anmerkungen haben, konnen mir gerneein Mail an [email protected] schicken.
Das Manuskript wurde mit MiKTeX (LATEXfur Windows) gelayoutet. Eswar mein erster Versuch mit diesem Programm und es durfte daher einigeMangel haben. Wer Anregungen oder Fragen zu LATEXhat, kann mir eben-falls unter der oben erwahnten Adresse schreiben. Allen kann ich die Doku-mentation von Tobias Oetiker (”The not so short introduction to LATEX”)empfehlen.
Ich hoffe die Arbeit ist fur einige von Nutzen!
Christoph Kasper

Contents
1 Die chemische Bindung 11.1 Atomaufbau . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.2 Ionen und ionische Bindung . . . . . . . . . . . . . . . . . . . 11.3 Die kovalente Bindung . . . . . . . . . . . . . . . . . . . . . . 2
2 Sauren und Basen 72.1 Brønsted Sauren und Basen . . . . . . . . . . . . . . . . . . . 72.2 Qualitative Regeln zum Abschatzen des pKs . . . . . . . . . . 82.3 Lewis-Sauren und Lewis-Basen . . . . . . . . . . . . . . . . . 10
3 Substanzklassen und Nomenklatur 11
4 Konformation 134.1 Newman Projektion . . . . . . . . . . . . . . . . . . . . . . . 134.2 Konformation von cyklischen Alkanen . . . . . . . . . . . . . 15
5 Stereochemie 195.1 E/Z- und cis/trans-Isomerie . . . . . . . . . . . . . . . . . . . 195.2 Chiralitat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
6 Substitutionsreaktionen 236.1 SN2-Reaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . 236.2 Abhangigkeit der SN2-Reaktion von Nu, R-L und LM . . . . 256.3 SN1-Reaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . 276.4 Abhangigkeit der SN1-Reaktion von Nu, R-L und LM . . . . 296.5 SN1 oder SN2? . . . . . . . . . . . . . . . . . . . . . . . . . . 29
7 Eliminierungsreaktionen 307.1 E1-Reaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . 307.2 E2-Reaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . 317.3 SN1/SN2 oder E1/E2? . . . . . . . . . . . . . . . . . . . . . . 33
8 Additionsreaktionen an Alkenen 348.1 Allgemeiner Mechanismus und Markovnikov Regel . . . . . . 348.2 Hydroborierung . . . . . . . . . . . . . . . . . . . . . . . . . . 368.3 Wagner-Meerwein Umlagerung . . . . . . . . . . . . . . . . . 388.4 Addition von H2 und Halogenen . . . . . . . . . . . . . . . . 38
9 Aromatizitat 419.1 Resonanz und Huckel-Regel . . . . . . . . . . . . . . . . . . . 419.2 Reaktionen an aromatischen Verbindungen . . . . . . . . . . 42
i

10 Carbonylverbindungen 4510.1 Nukleophile Addition . . . . . . . . . . . . . . . . . . . . . . . 45
10.1.1 Reversible Reaktionen . . . . . . . . . . . . . . . . . . 4610.1.2 Irreversible Reaktionen . . . . . . . . . . . . . . . . . 51
10.2 Oxidation von Alkoholen . . . . . . . . . . . . . . . . . . . . . 5210.3 Reaktionen an der α-Position von Carbonylverbindungen . . 53
10.3.1 Keto-Enol-Tautomerie . . . . . . . . . . . . . . . . . . 5310.3.2 Aldolreaktion . . . . . . . . . . . . . . . . . . . . . . . 5610.3.3 Michael Reaktion, Robinson Annelierung Knoevenagel 59
A Rangordnung wichtiger Gruppen 62
B Verzeichnis der Bilder und Reaktionen 63
ii

1 DIE CHEMISCHE BINDUNG 1
1 Die chemische Bindung
1.1 Atomaufbau
Ein Atom besteht aus einem Kern (aus Protonen und Neutronen) undmehrerer Schalen, welche mit Elektronen besetzt sind. Entscheidend furReaktivitatsbetrachtungen eines Atoms sind die Valenzelektronen, welchesich in der aussersten besetzten Schale befinden. Die Schalen bestehen auseinem oder mehrerern Orbitalen. Orbitale sind Wellenfunktionen, welche dieAufenthaltswahrschinlichkeit der Elektronen beschreibt. Die einzelnen Elek-tronen besetzen zuerst immer die energetisch tiefsten (gunstigsten) Orbitale.Die genaue Abfolge der Besetzung der Orbitale wird durch das Pauliprinzipund die Hundsche Regel festgelegt.
• Aufbauprinzip: 1s 2s 2p 3s etc.
• Pauliprinzip: Ein Orbital besitzt maximal 2 Elektronen.
• Hundsche Regel: Orbitale gleicher Energie (Bsp.: px py pz) werdenzuerst einzeln besetzt.
Atom Atomnr. ElektronenkonfigurationH 1 1s1
C 6 1s2 2s2 2p2 (z.B.: 2px1 2pz
1)F 9 1s2 2s2 2p5 (z.B.: 2px
2 2py2 2pz
1)Na 11 1s2 2s2 2p6 3s1
Table 1: Beispiele von Elektronenkonfigurationen
Figure 1: Elektronenkonfiguration des Kohlenstoffs
1.2 Ionen und ionische Bindung
Eine weitere Regel besagt, dass eine Schale am stabilsten (energetisch gunstig)ist, wenn sie voll besetzt ist. Im Falle der zweiten und dritten Periode be-deutet dies, dass sich in der Valenzschale 8 Elektronen befinden mussen (Ok-tettregel). Die Edelgase besitzen bereits im neutralen Zustand diese Kon-figuration und sind deshalb ausserst reaktionstrage (stabil). Alle anderen

1 DIE CHEMISCHE BINDUNG 2
Atome sind hingegen gezwungen Elektronen abzugeben oder aufzunehmenum eine gefullte Valenzschale zu erreichen. Die neu entstehenden Speziessind entweder positiv oder negativ geladen und werden als Ionen bezeichnet.
Figure 2: Beispiele von Ionen
Da sich entgegengesetzt geladene Ionen anziehen, scheint es logisch, dasssie sich zusammenlagern. Das dadurch entstehende Salz wird durch ionis-che Bindungen zusammengahalten. Konzentriert man sich auf zwei Partnereiner ionischen Bindung, kann diese auch als komplette Abgabe eines Elek-trons des einen Partners an den anderen betrachtet werden.
1.3 Die kovalente Bindung
Anhand des unter 1.2 besprochenen Phanomens musste das Kohlenstoffatomvier Elektronen abgeben oder ebensoviele aufnehmen um eine volle Valen-zschale zu erhalten. Dies scheint energetisch hochst ungunstig. Tatsachlichgelingt es dem Kohlenstoff trotzdem den stabileren Zustand zu erreichen,jedoch nicht durch Abgabe oder Aufnahme von Elektronen, sondern durchausbilden von kovalenten Bindungen. Bei der kovalenten Bindung kann manbildlich von einer gemeinsamen Nutzung von Elektronen sprechen. Dadurcherreichen beide Partner eine gefullte Valenzschale und erfullen die Oktet-tregel.
Figure 3: Molekule mit kovalenten Bindungen
Die Ausbildung einer kovalenten Bindung ist energetisch gunstig. AmPunkt der geringsten potentiellen Energie kann man die Bindungslange(Atomkern zu Atomkern) ablesen.
Sind an einer kovalenten Bindung zwei gleiche Atome beteiligt, so istdie Bindung apolar. Sind jedoch zwei unterschiedliche Atome beteiligt, so

1 DIE CHEMISCHE BINDUNG 3
Figure 4: Potentialkurve von zwei H-Atomen als Funktion des Kernabstands
zieht einer der Partner die Elektronen starker zum Kern. Dadurch entstehteine polare Verbindung, deren Polaritat durch die Differenz der Elektronega-tivitaten der beteiligten Partner charakterisiert werden kann. Polare Molekulebesitzen oft ein Dipolmoment das sich wie folgt definiert:
µ = e · d (1)
µ hat die Einheit Debey (D). In der Formel steht e fur die Partialladungauf einem der beiden Bindungspartner und d fur die Distanz zwischen denbeiden Partnern.
Nicht jedes Molekul mit polaren Bindungen besitzt ein Dipolmoment.Oftmals fuhrt die raumliche Anordnung der Bindungen zu einer Aufhebungder Dipolmomente (Bsp. CO2).
Figure 5: Dipolmomente: H2O = 1.5 D, HCl = 1.1 D, CO2 = 0.0 D
Es stellt sich nun die Frage wie sich das Phanomen der kovalentenBindung mit der Erkenntnis uber den Atomaufbau vereinbaren lasst. Alseinfachstes Beispiel betrachten wir den Wasserstoff. Das einzelne Wasser-stoffatom besteht einzig aus einem halbbesetzten 1s-Orbital. Bei der Aus-bildung einer Bindung uberlagern sich diese Orbitale. Dabei entstehen

1 DIE CHEMISCHE BINDUNG 4
wiederum zwei neue Ornitale (die Anzahl Orbitale bleibt immer erhalten!).Diese Orbitale bezeichnet man nun als Molekulorbitale. Eines der Molekulorbitaleist im Vergleich zu den 1s-Orbitalen energetisch gunstiger (Bindendes Molekulorbital),das andere um denselben Betrag ungunstiger (Antibindendes Molekulorbital).Die Bindung wird als σ-Bindung und die Molekulorbitale deshalb auch alsσ-Orital bzw. als σ*-Orbital bezeichnet. Die beiden Elektronen besetzennun nach denselben Regeln wie zuvor die Molekulorbitale. Dies lasst sicham besten durch ein Molekulorbitaldiagramm darstellen (Bild 6).
Figure 6: Molekulorbitaldiagramm des Wasserstoffs
Geht man beim Molekul CH4 (Methan) analog vor, so musste man an-nehmen, dass zwei H-Atome sich mit dem 2s-Orbital uberlagern und dieanderen beiden mit den zwei halbbesetzten p-Orbitalen. Dies wurde zu un-terschiedlichen Bindungswinkeln fuhren. Man weiss jedoch, dass sich dieH-Atome genau in den Ecken eines Tetraeders anordnen (Bild 7). Folglichkann das Modell fur die Bildung von Methan nicht stimmen.
Figure 7: Tetraederform des Methans
Das Phanomen, welches dieses Problem lost, wird als Hybridisierungbezeichnet. Dabei uberlagern sich die drei p-Orbitale und das s-Orbital zuvier neun, gleichwertigen Hybridorbitalen. In diesem Fall bezeichnet mandie Hybridisierung als sp3-Hybridisierung und die Orbitale als sp3-Orbitale,weil sie aus einem s-Orbital und 3 p-Orbitalen gebildet wurden.
Da die 4 sp3-Orbitale gleichwertig sind, ordnen sie sich gleichmassig im

1 DIE CHEMISCHE BINDUNG 5
Figure 8: sp3-Hybridisierung des Kohlenstoffs
Raum an. Dadurch erklart sich die charakteristische Tetraederform desMethans.
Naturlich sind auch sp2- bzw. sp-Hybridisierungen moglich. Bei der sp2-Hybridisierung bleibt ein p-Orbital im ursprunglichen Zustand. Geometrischliegen die sp2-Orbitale in einer Ebene, auf welcher das p-Orbital senkrechtsteht. Wahrend die sp2-Orbitale σ-Bindungen eingehen, bildet das p-Orbitalzusammen mit einem anderen p-Orbital andere Bindungen aus. Diese beze-ichnet man als π-Bindungen. Ein Beispiel ist das Ethen, bei welchem dieDoppelbindung aus einer σ-Bindung und einer π-Bindung besteht.
Figure 9: sp2-Hybridisierung des Kohlenstoffs am Beispiel von Ethen
Bei der sp-Hybridisierung bleiben zwei p-Orbitale in ihrem ursprunglichenZustand. Beide konnen wie bei der sp2-Hybridisierungen π-Bindungen einge-hen. Ein Beispiel dafur ist das Ethin, bei welchem die Dreifachbindung auseiner σ-Bindung und 2 π-Bindungen besteht.
Dabei ist zu vermerken, dass σ-Bindungen starker sind als π-Bindungen.

1 DIE CHEMISCHE BINDUNG 6
Figure 10: sp-Hybridisierung des Kohlenstoffs am Beispiel von Ethin

2 SAUREN UND BASEN 7
2 Sauren und Basen
2.1 Brønsted Sauren und Basen
Brønsted definierte Sauren und Basen wie folgt:
• Sauren sind Verbindungen, die H+ abgeben.
• Basen sind Verbindungen, die H+ aufnehmen.
Figure 11: Reaktion von HCl in Wasser
Sauren sind unterschiedlich stark, da das Proton, welches abgegebenwird, nicht bei allen Verbindungen gleich stark gebunden ist. Ebenso dieBasen, da sie unterschiedliche Affinitaten fur Protonen besitzen. Folglich giltes einen Weg zu finden, die Saure- bzw. Basenstarke quantitativ zu bestim-men. Eine Moglichkeit ist die Herleitung einer Sauredissoziationskonstantemithilfe der Gleichgewichtskonstanten. Betrachten wir die folgende Reak-tion, wobei HA die Saure darstellt.
Figure 12: Symbolische Saure-Basen-Reaktion
Diese Reaktion stellt ein Gleichgewicht dar. Folglich konnen wir dieGleichgewichtskonstante darstellen als:
K =[H3O
+][A−][H2O][HA]
(2)
In wassrigen Losungen bleibt die Konzentration des Wassers nahezu kon-stant. Daher wird sie in die Konstante einbezogen. Die neue Konstante wirdals Sauredissoziationskonstante (Ks oder Ka) bezeichnet.
Ks = K · [H2O] =[H3O
+][A−][HA]
(3)

2 SAUREN UND BASEN 8
Um handlichere Zahlen zu erhalten, nimmt man den negativen Zehner-logarithmus der Sauredissoziationskonstante (kurz: pKs bzw. pKa).
pKs = − log Ks (4)
Anhand des pKs-Wertes kann man nun auf die Starke einer Saure schliessen.
Sehr starke Sauren pKs < 1Mittelstarke Sauren pKs = 1-5Schwache Sauren pKs = 5-15Sehr schwache Sauren pKs > 15
Table 2: pKs-Werte von Sauren
Neben der Starke einzelner Verbindungen ist oftmals auch die Konzen-tration von H+ oder besser H3O+ gefragt. Um diesen Wert zu bestimmen,verwendet man den pH-Wert.
pH = − log [H3O+] (5)
Auch hier steht das ”p” wieder fur den negativen Zehnerlogarithmus umhandlichere Zahlen zu erhalten.
2.2 Qualitative Regeln zum Abschatzen des pKs
Im Folgenden werden einige Regeln aufgelistet, welche einem qualitativeAnhaltspunkte uber die Saurestarke liefern konnen. Mithilfe dieser Regelnst es moglich Sauren zu vergleichen und nach ihrer Starke zu ordnen.
1. Je starker die Saure, desto schwacher ist die konjugierte Base.
2. Je stabiler die konjugierte Base, desto starker ist die Saure.
Saure CH4 NH3 H2O HFpKa 50 36 15.7 3.2Konj. Base CH−3 NH−2 HO− F−
Stabilitat gering hoch
Table 3: Die Saurestarke korreliert mit der Stabilitat der konj. Base
3. Induktive Effekte haben ebenfalls Einfluss auf die Saurestarke. DaFluor die grosste Elektronegativitat besitzt, wird die negative Ladungauf dem Sauerstoff am besten stabilisiert. Aus Regel 2 ist bereitsbekannt, dass eine stabilere Base eine starkere konjugierte Saure be-sitzt.

2 SAUREN UND BASEN 9
Figure 13: pKa von Essigsaurederivaten und induktiver Effekt
4. Ethanol besitzt einen pKs von 15.9 wahrend die Essigsaure einen pKs
von 4.76 besitzt. Wie lasst sich dieser markante Unterschied erklaren?
Figure 14: Ethanolat und mesomere Grenzstrukturen des Acetats
Entscheidend fur den markanten Unterschied ist wiederum die Sta-bilitat der Base. Das Acetat ist stabiler als das Ethanolat, da Reso-nanz auftritt. Durch die Resonanz wird die negative Ladung auf zweiZentren verteilt, was zu einer Stabilisierung fuhrt.
5. Auch die Grosse des Ions hat einen Einfluss auf dessen Stabilitat. Dieswird ersichtlich bei den Halogeniden. Da Fluorid den kleinsten Io-nenradius besitzt, ist die negative Ladung schlechter verteilt als beimIodid. Folglich ist Iodwasserstoff die starkste Saure.
Saure HF HCl HBr HIpKa 3.2 -7 -9 -10Konj. Base F− Cl− Br− I−
Stabilitat gering hoch
Table 4: Kleine Ionen haben eine hohere Elektronendichte und sind daherweniger stabil

2 SAUREN UND BASEN 10
2.3 Lewis-Sauren und Lewis-Basen
Lewis definierte Sauren und Basen in einem weiteren Sinn als Brønsted. BeiLewis geht es nicht um Protonenaufnahme oder -abgabe, sondern um freieElektronenpaare und Akzeptoren fur solche:
• Lewis Base = Verbindung mit einem reaktiven, freien Elektronenpaar.
• Lewis Saure = Verbindung mit einem Akzeptor fur freie Elektronen-paare.
Die Brønsted-Sauren und -Basen sind dabei eingeschlossen.
Figure 15: Illustration von Lewis-Sauren und -Basen

3 SUBSTANZKLASSEN UND NOMENKLATUR 11
3 Substanzklassen und Nomenklatur
Fur die korrekte Benennung eines Stoffes bedarf es der Grundekenntnisseder Namen einiger Substanzklassen. Eine solche Auflistung wurde in denubungen verteilt. In der folgenden Tabelle sind zudem die wichtigstenVerbindungsklassen aufgefuhrt. Im weiteren gilt es zu beachten, dass vieleStoffe einen Trivialnamen besitzen. Dieser ist meist kurzer und einfacherals der offizielle Name nach IUPAC (vor allem bei grossen Molekulen) undwird deshalb haufiger verwendet. Zwei Beispiele fur Trivialnamen sind dieEssigsaure und Glycerin.
Figure 16: Essigsaure und Glycerin
Der Nachteil der Trivialnamen ist es, dass man vom Namen keine Ruckschlusseauf die Struktur ziehen kann. Deshalb konnen die Namen nach IUPAChaufig aufschlussreicher sein. Die Namen werden nach einer strikten Sys-tematik gebildet und sind somit eindeutig. Die Grundstruktur der Namenbesteht aus vier Teilen:
Vorsilbe - Stamm - Endung 1 - Endung 2
Der Stamm bezeichnet sozusagen den Ruckgrat des Stoffes. Im Nor-malfall benennt der Stamm die langste C-Kette des Molekuls. Die ersteEndung gibt an, ob die C-Kette gesattigt ist oder allfallige Doppel- undDreifachbindungen enthalt. Die zweite Endung bezeichnet die hochstrangigefunktionelle Gruppe. Rangniedrige Gruppen werden in der Vorsilbe ver-merkt. Anschliessend ist eine Tabelle angefugt, welche die Rangordnung derwichtigsten funktionellen Gruppen auflistet. Zu den jewiligen Teilen des Na-mens mussen vorangehend Zahlen angefugt werden, falls die Verknupfungder einzelnen Gruppen nicht eindeutig ist. Zudem konnen Bezeichner furdie Konfiguration hinzukommen (Z/E, cis/trans und S/R). Im Anhang isteine Tabelle mit der Rangordnung der wichtigsten fubktionellen Gruppenangehngt.
Im folgenden soll anhand einiger Beispiele die korrekte Benennung illus-triert werden. Zudem soll die richtige Nummerierung des Stamms aufgezeigtwerden.
Zur Hervorhebung der drei Alkoholgruppen am Glycerin wird vor dieEndung -ol das Zahlwort tri angefugt. Analog verhalt es sich fur 2 (di) und4 (tetra) gleiche Gruppen.

3 SUBSTANZKLASSEN UND NOMENKLATUR 12
Figure 17: Essigsaure und Glycerin mit Namen nach IUPAC
Figure 18: Zwei weiter Beispiele fur Nomenklatur nach IUPAC
Der Start der Nummerierung wird so gewahlt, dass die hochste funk-tionelle Gruppe moglichst kleine Zahlen erhalt (Im ersten Beispiel beginntdie Nummerierung rechts, damit die -OH Funktionalitat die Nummer 2 statt4 erhalt). Eine Doppelbingung vom C2 zum C3 wird mit der Abkurzung 2-en bezeichnet. Da im zweiten Beispiel eben diese Bindung nicht drehbarist, musste im Namen angegeben werden wie die Konfiguration ist. In un-serem Beispiel stehen die Gruppen hoherer Prioritat E(ntgegen) zueinander.Folglich liegt das E-Isomer vor. Auf die genaue Verwendung von E/Z (undauch cis/trans, S/R) wird im nachsten Kapitel eingegangen.

4 KONFORMATION 13
4 Konformation
4.1 Newman Projektion
Strukturformeln von Molekulen geben genaue Auskunft uber die Verknupfung(Konstitution) der einzelnen Atome im Molekul. Jedoch besitzen die meis-ten Molekule die Moglichkeit der Drehung einzelner Gruppen. Dies fuhrtzu unterschiedlichen Konformeren. uber die genaue Anordnung der Grup-pen (Konformation) geben uns die Strukturformeln jedoch keine Auskunft.Wie sollen wir uns das Molekul nun vorstellen? Welches ist die energetischgunstigste Struktur? Diese Fragen lassen sich mithilfe der sogenannten New-man Projektion - zumindest qualitativ - leicht beantworten. Am Beispiel desEthans lasst sich die Methode leicht erklaren. Haben wir ein Ethanmolekulin der linken Konformation, so lasst es sich durch Drehung um 60 (um dieC-C-Bindung) leicht in eine neue Konformation uberfuhren. Naturlich sindnicht nur diese Positionen Konformere, sondern auch jegliche Positionen,welche wahrend der 60-Drehung durchlaufen werden.
Figure 19: Zwei mogliche Konformationen des Ethans
Es stellt sich nun die Frage, welches der beiden Konformere das ener-getisch gunstigere ist. Dazu Drehen wir das gan ze Molekul nun so, dass wirentlang der C-C-Bindung blicken. Diese Beschreibung der Molekule nenntman die Newman Projektion.
Figure 20: Newman Projektionen der beiden Ethankonfomere
4 KONFORMATION 14
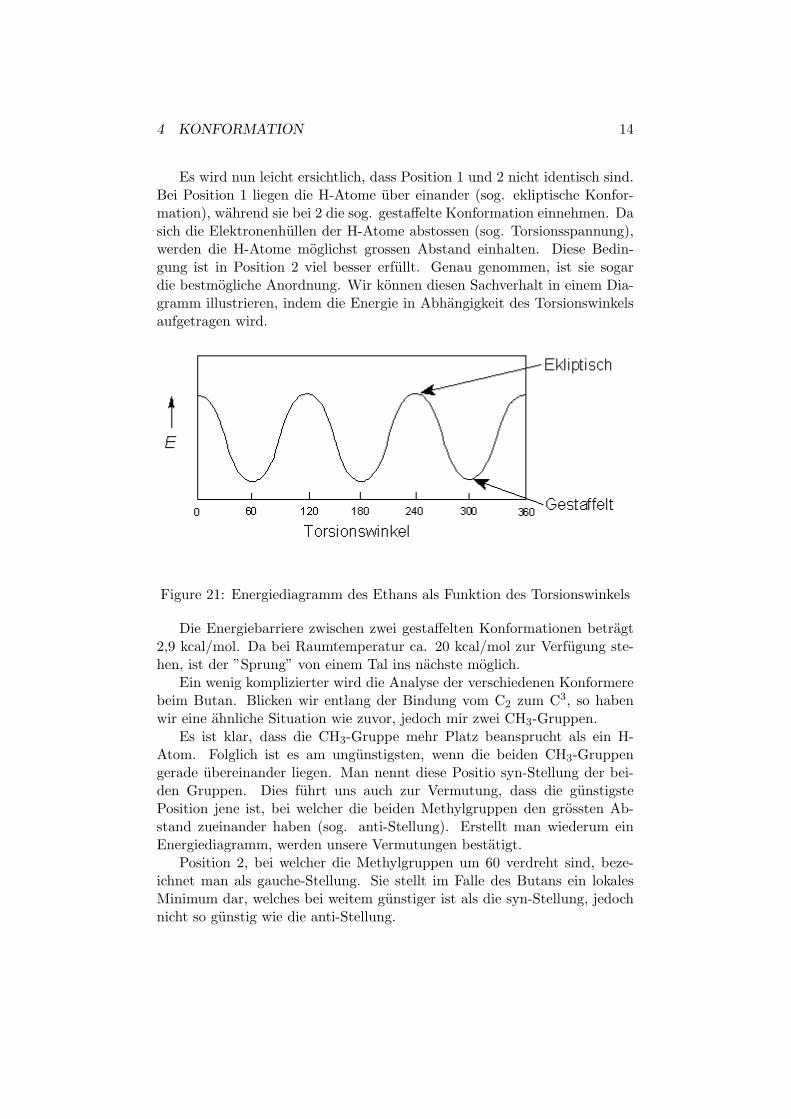
Es wird nun leicht ersichtlich, dass Position 1 und 2 nicht identisch sind.Bei Position 1 liegen die H-Atome uber einander (sog. ekliptische Konfor-mation), wahrend sie bei 2 die sog. gestaffelte Konformation einnehmen. Dasich die Elektronenhullen der H-Atome abstossen (sog. Torsionsspannung),werden die H-Atome moglichst grossen Abstand einhalten. Diese Bedin-gung ist in Position 2 viel besser erfullt. Genau genommen, ist sie sogardie bestmogliche Anordnung. Wir konnen diesen Sachverhalt in einem Dia-gramm illustrieren, indem die Energie in Abhangigkeit des Torsionswinkelsaufgetragen wird.
Figure 21: Energiediagramm des Ethans als Funktion des Torsionswinkels
Die Energiebarriere zwischen zwei gestaffelten Konformationen betragt2,9 kcal/mol. Da bei Raumtemperatur ca. 20 kcal/mol zur Verfugung ste-hen, ist der ”Sprung” von einem Tal ins nachste moglich.
Ein wenig komplizierter wird die Analyse der verschiedenen Konformerebeim Butan. Blicken wir entlang der Bindung vom C2 zum C3, so habenwir eine ahnliche Situation wie zuvor, jedoch mir zwei CH3-Gruppen.
Es ist klar, dass die CH3-Gruppe mehr Platz beansprucht als ein H-Atom. Folglich ist es am ungunstigsten, wenn die beiden CH3-Gruppengerade ubereinander liegen. Man nennt diese Positio syn-Stellung der bei-den Gruppen. Dies fuhrt uns auch zur Vermutung, dass die gunstigstePosition jene ist, bei welcher die beiden Methylgruppen den grossten Ab-stand zueinander haben (sog. anti-Stellung). Erstellt man wiederum einEnergiediagramm, werden unsere Vermutungen bestatigt.
Position 2, bei welcher die Methylgruppen um 60 verdreht sind, beze-ichnet man als gauche-Stellung. Sie stellt im Falle des Butans ein lokalesMinimum dar, welches bei weitem gunstiger ist als die syn-Stellung, jedochnicht so gunstig wie die anti-Stellung.

4 KONFORMATION 15
Figure 22: Konformere des Butans
Figure 23: Energie der moglichen Butankonformere als Funktion des Tor-sionswinkels
4.2 Konformation von cyklischen Alkanen
Im folgenden wollen wir die Konformation in Ringsystemen (cyklischen Alka-nen) betrachten. Wir starten dabei mit dem Cyclopropan und gehen hochbis zum Cyclohexan.
Cyclopropan
Die Skelettformel zeigt ein gleichseitiges Dreieck. Die C-C-Bindungenstehen also in einem 60-Winkel zueinander. Die C-Atome des Rings sindjedoch sp3 hybridisiert und bevorzugen daher einen Tetraederwinkel (109,5).Diese Diskrepanz ist energetisch naturlich hochst ungunstig. Man bezeichnetsie als Winkelspannung. Das Molekul weicht der Spannung insofern aus,indem sich die C-C-Bindungen nach aussen ”wolben”. Dies fuhrt dazu, dass

4 KONFORMATION 16
sich der Winkel von 60 bis auf 104 erhoht. Die sog. Bananenbindungen sinddadurch schwacher als gewohnliche σ-Bindungen.
Figure 24: Strukturformel und Newman Projektion des Propans
Naturlich liegt auch beim Cyclopropan eine Torsionsspannung vor, welchejedoch im Vergleich zur Winkelspannung klein ist uns sich nicht verhindernlasst (keine Drehung moglich).
Cyclobutan
Die quadratische Form des Cyclobutan, postuliert einen 90-Winkel. Diesfuhrt dazu, dass die Winkelspannung weitaus geringer ist, als noch beimCyclopropan. Hingegen stellt sich heraus, dass das Molekul der Torsionss-pannung ausweicht, indem es eine ”Ecke” um 25 aus der Ebene knickt.
Figure 25: Strukturformel und raumliche Struktur des Butans
Cyclopentan
Auch hier fuhrt die Torsionsspannung dazu, dass das Molekul nicht pla-nar ist. Es treten zwei Konformere auf.
Bei der ersten Konformation (sog. Briefumschlag, envelope) ist ein C-Atom aus der Ebene geknickt. In der zweiten Konformation befindet sichein C-Atom uber der Ebene und ein C-Atom darunter. Man bezeichnet dieseAnordnung auch als Twist.

4 KONFORMATION 17
Figure 26: Strukturformel und zwei mogliche Konformere des Pentans
Cyclohexan
Auch Cyclohexan liegt nicht planar vor, sondern in der Sessel-Konformation(chair). Dies fuhrt dazu, dass die Seitengruppen in der Newmanprojektionperfekt gestaffelt sind. Die grossten Gruppen sind die weiterfuhrenden C-Atome des Rings, welche sich also in gauche-Stellung befinden.
Figure 27: Strukturformel und Sesselkonformation des Hexans
Durch die Konformation konnen die H-Atome in zwei verschiedenen Po-sitionen stehen. Man bezeichnet sie als aequatoriale und axiale Positionen.
Beim Cyclohexan ist Ringinversion moglich. Dabei werden die H-Atomein axialer Position in die aequatoriale Position uberfuhrt und umgekehrt.Das folgende Energiediagramm zeigt den Weg der Ringinversion auf, welcheruber mehrere ubergangszustande fuhrt.

4 KONFORMATION 18
Figure 28: Ringinversion und Energiediagramm das Cyclohexans
Die grosste Energiebarriere (Sessel zu Twist) betraagt beim Cyclohexan12 kcal/mol. Folglich findet Ringinversion bei Raumtemperatur statt.
Figure 29: Die Methylgruppe hat in aequitorialer Position einen grosserenAbstand zu den C-Atomen des Rings
Die beiden Sesselformen sind beim Cyclohexan energetisch gleich gunstig.Wird jedoch ein H-Atom durch eine Methylgruppe ersetzt, ist dies nichtmehr der Fall. Der Grund dafur liegt in der Lage der Methylgruppe imVergleich zu den C-Atomen des Rings. In der Newman Projektion wird er-sichtlich, dass es energetisch gunstiger ist, die Methylgruppe in aequitorialePosition zu stellen.

5 STEREOCHEMIE 19
5 Stereochemie
Wir haben im letzten Kapitel Konformation behandelt. Zwei Konformereunterscheiden sich einzig in der raumlichen Anordnung ihrer Gruppen. DurchDrehen von Bindungen konnen sie ineinander uberfuhrt werden. Ebenfallsbekannt, ist die Konstitutionsisomerie. Dabei haben zwei Molekule iden-tische Summenformeln, wobei die Verknupfung der Atome unterschiedlichist. So kann die Summenformel C2H6O sowohl fur Ethanol (CH3CH2OH)stehen, als auch fur Dimethylether (CH3OCH3). Eine weitere Form derIsomerie ist die Stereoisomerie, welche wir im Folgenden behandeln werden.
5.1 E/Z- und cis/trans-Isomerie
Durch Doppelbindungen und in Ringsystemen ist ein freie Drehung umBindungen im Allgemeinen nicht moglich. Folglich sind zwei Molekule mitunterschiedlicher Anordnung ihrer Gruppen bei einer Doppelbindung keineKonformere, da sie nicht durch Drehung ineinander uberfuhrt werden konnen.Wir mussen folglich die zwei Molekule unterscheiden und auch unterschiedlichbenennen. Wir tun dies, indem wir die Doppelbindung von ”oben” betra-chten und die Stellung der ranghochsten Gruppen betrachten (Bestimmungder ranghochsten Gruppe s. Paula Bruice, Organic Chemistry p. 119ff).Stehen die ranghochsten Gruppen zueinander, so stellt man dem Namen einZ voran. Stehen sie entgegen, so wird ein E voran gestellt.
Figure 30: Die zwei Stereoisomere von Pent-2-en
Die beiden C-Atome der Doppelbindung werden stereogene Zentren genannt,falls die Gruppen an beiden C unterschiedlich sind (ansonsten gabe es keineZ/E-Isomerie).
Ahnlich verhalt es sich mit der Anordnung der Gruppen in Ringsyste-men. Jedes C-atom des Rings geht zwei weitere Bindungen ein, wobei eineGruppe uber, die andere unter der Ringebene zu liegen kommt. Liegenwie bei der Dopplebindung zwei stereogene C-Atome vor, so gibt es zweiMoglichkeiten die Gruppen anzuordnen. Liegen beide Gruppen uber bzw.unter der Ringebene, bezeichnen wir dies als cis-Stellung. Befindet sich eine

5 STEREOCHEMIE 20
Gruppe uber, die andere unter der Ringebene, so wird dies als trans-Stellungbezeichnet.
Figure 31: Beispiel von cis/trans-Isomerie
Auch hier wird die Stellung der ranghochsten Gruppen betrachtet.Sowohl die E/Z-Isomerie, als auch die cis/trans-Isomerie zahlen zur Gruppe
der Stereoisomerie.
5.2 Chiralitat
Eine weitere Form der Stereoisomerie ist die Chiralitat. Dabei handelt essich um Molekule und deren Spiegelbilder. In den meisten Fallen stellt sichheraus, dass die beiden Molekule identisch sind (sprich zur Deckung gebrachtwerden konnen). Liegt jedoch ein C-Atom vor, welches vier verschiedeneSubstituenten besitzt, so ist es nicht mehr moglich die beiden Molekulezur Deckung zu bringen. Wir bezeichnen ein solches C-atom als chiralesZentrum. In Skelettformeln wird ein chirales C-Stom haufig mit einem *gekennzeichnet. Die beiden Spiegelbilder werden als Enantiomere bzw. alsEnantiomerenpaar bezeichnet.
Figure 32: Bei einem chiralen C-Atom kann man durch Spiegeln die Enan-tiomere erhalten
Enantiomere scheinen auf den ersten Blick gleich, konnen jedoch inVerbindung mit Enzymen ganz anders reagieren und haben teilweise un-terschiedliche Geruche. Zudem sind Flussigkeiten, in denen das Verhaltnisder Enantiomere nicht 1:1 ist, optisch aktiv.

5 STEREOCHEMIE 21
Um Enantiomere in der Nomenklatur zu unterscheiden, gibt es eineVorgehensweise, welche die Konfiguration der Chiralitatszentren genau festhalt:Zuerst werden die Substituenten nach denselben Regeln wie bei der Z/E-Isomerie nummeriert (s. Buch p. 119ff). Nun wird das Molekul so gedreht,dass die rangniedrigste Gruppe ”hinter” dem C* zu liegen kommt. Durchdie Nummerierung ist es jetzt moglich eine Drehrichtung einzuzeichnen vonGruppe 1 zu 2 und zu 3.
Figure 33: Korrekte Benennung des Enantiomerenpaars
Ist die Drehung im Uhrzeigersinn, so wird sie als R-Drehung bezeich-net und der Molekulname damit erweitert. Ist die Drehung in der anderenRichtung, so ist es eine S-Drehung und der Molekulname wird entsprechenderweitert.
Wie schon erwahnt sind Enantiomere optisch aktiv. Dabei dreht einesdie Ebene von linear polarisiertem Licht um einen spezifischen Winkel nachrechts (+), wahrend das andere um denselben Winkel nach links (-) dreht.Liegt jedoch ein 1:1-Gemisch der Enantiomere vor, so verschwindet die op-tische Aktivitat. Man bezeichnet ein solches Gemisch als Racemat. Es giltsich zu merken, dass S/R und -/+ nicht korrelieren. Man kann also von derKonfiguration beim C* nicht auf die Drehrichtung schliessen.
Es ist naturlich auch denkbar, dass in einem Molekul zwei oder nochmehr chirale C-Atome vorliegen konnen. Auf kombinatorische Weise lasstsich leicht errechnen, dass ein Molekul mit zwei chiralen Zentren in vierverschiedenen Formen auftreten muss.
Wir erhalten diese, indem wir die Ausgangsform spiegeln und somitzwei Enantiomere erhalten. Zudem konnen wir nun an einem C* zwei Sub-stituenten austauschen und dann erneut spiegeln, wodruch wir ein zweitesEnantiomerenpaar erhalten. Die Molekule 1/3, 1/4, 2/3 und 2/4 werden alsDiastereomere bezeichnet. Diastereomere sind keine Enantiomere (wie 1/2oder 3/4) und konnen durch Vertauschen zweier Substituenten an einem C*

5 STEREOCHEMIE 22
Figure 34: Bei zwei chiralen C-Atomen erhalt man 4 Stereoisomere
ineinander uberfuhrt werden.
Figure 35: Die Molekule der ersten Spieglung sind identisch
Fur n chirale Zentren gilt die Regel, dass 2n Stereoisomere existieren.Die Regel versagt jedoch, wenn es moglich ist eine Symmetrieebene durchdas Molekul zu legen. In diesem Fall sind die beiden Moglichkeiten mit R/Sbzw. S/R identisch. Es treten also nur drei statt vier Stereoisomere auf.Man bezeichnet solche Molekule als Mesoverbindungen.

6 SUBSTITUTIONSREAKTIONEN 23
6 Substitutionsreaktionen
6.1 SN2-Reaktion
Wir haben im Kapitel uber Sauren bereits die folgende Reaktion kennengel-ernt. Es handelt sich hierbei um eine Reaktion zwischen einer Lewis-Saureund einer Lewis-Base.
Figure 36: OH− als Nukleophil greift das partiell positiv geladene C desMethyliodids an
Die Lewis-Base (hier OH−) hat freie Elektronenpaare und in diesemFall sogar eine negative Ladung. Folglich reagiert sie gerne mit (partiell)positiv geladenen Molekulen. Daher bezeichnet man Lewis-Basen auch alsnukleophil. Die Lewis-Saure ist gerade das Gegenteil. In unserem Beispielerhalt das C-Atom des Methyliodids durch die hohe Elektronegativitat desIod-Atoms eine partielle positive Ladung. Damit wird es zum idealen Part-ner fur die Lewis-Base. Man bezeichnet die Lewis-Saure daher auch alselektrophil.
Die Begriffe nukleophil und elektrophil sind sehr nutzlich, da fast im-mer partielle Ladungen vorhanden sind und man somit die Molekule einfachcharakterisieren kann. Auch die folgende Reaktion ist eine Interaktion zwis-chen einer nukleophilen und elektrophilen Spezies.
Figure 37: Beispiel eines nukleophilen Angriffs mit Inversion der Stereo-chemie
Betrachtet man die Reaktion sieht man, dass sich die Anordnung umdas chirale C-Atom andert. Man bezeichnet dies als Inversion der Stereo-chemie. Da wir als Produkt auschliesslich dieses Isomer erhalten, wird die

6 SUBSTITUTIONSREAKTIONEN 24
Reaktion als stereospezifisch bezeichnet. Zudem zeigt sich, dass die Reak-tionsgeschwindigkeit von beiden Reaktionspartnern abhangt.
Figure 38: Allgemeine Reaktionsgleichung einer nukleophilen Substitution
Schreiben wir die Reaktion in der allgemeineren Form (L steht fur leavinggroup bzw. Abgangsgruppe), ergibt sich fur die Reaktionsgeschwindikeitfolgender Zusammenhang.
ν = k[Nu−][R− L] (6)
Es zeigt sich, dass diese Art von Reaktion sehr haufig ist. Anscheinendhandelt es sich um eine bimolekulare, nukleophile Substitution. Daher beze-ichnet man den Reaktionstyp kurz als SN2-Reaktion (N fur nukleophil, 2 furbimolekular).
Wie wir bereits festgestellt haben, tritt bei einer SN2-Reaktion Inversionauf. Es stellt sich nun die Frage wieso dies geschieht und keine anderenStereoisomere auftreten. Betrachten wir den Machanismus der Reaktionwird einiges klar.
Figure 39: Mechanismus der SN2-Reaktion
Das nukleophile Teilchen greift auf der Gegenseite der Abgangsgruppean. Dadurch entsteht ein instabiler Ubergangszustand, in welchem das C-Atom sp2 hybridisiert ist. Die Bindungsbildung und der Bindungsbruchfinden dabei gleichzeitig (sog. konzertiert) ab. Daher ist die Reaktiongeschwindigkeitvon beiden Reaktionspartnern abhangig. Nun tritt die Abgangsgruppe ausund die weiteren Substituenten des C-Atoms knicken wie ein Regenschirmauf die andere Seite um. Durch dieses Umknicken resultiert die Inversionam chiralen C-Atom.
Fur die SN2-Reaktion lasst sich ein Energiediagramm zeichnen, welchesden Ablauf der Reaktion wiederspiegelt. Dabei braucht es einen gewissenBetrag an Aktivierungsenergie um den Ubergangszustand, welcher instabilist, zu erreichen. Anschliessend wird bei der Bindungsbildung wieder En-ergie frei.

6 SUBSTITUTIONSREAKTIONEN 25
Figure 40: Energiediagramm der SN2-Reaktion
6.2 Abhangigkeit der SN2-Reaktion von Nu, R-L und LM
Wie wir bereits festgehalten haben, hangt die Reaktionsgeschwindigkeit derSN2-Reaktion sowohl vom Nuklophil, als auch von R-L ab. Zudem spieltauch das Losungsmittel (LM), in welchem die Reaktion ablauft eine Rolle,da es Edukte oder Produkte stabilisieren kann.
Nukleophile sind naturlich nicht alle gleich stark. So wird eine negativgeladene Spezies viel schneller reagieren, als eine neutrale Spezies, welchenur freie Elektronenpaare besitzt. Die folgende Tabelle fuhrt die relativeNukleophilie einiger Spezies im Verhaltnis zu Wasser (= 1) auf.
NC− 126000 N3− 8000
HS− 126000 NH2− 8000
I− 80000 Cl− 1000OH− 16000 F− 80Br− 10000 CH3OH 1
Table 5: Beispiele relativer Nukleophilie zu Wasser (= 1)
Haufig korreliert die Nukleophilie mit der Basizitat (starke Base = starkesNukleophil). Es ware jedoch falsch Basizitat und Nukleophilie gleichzuset-zen. So reagieren bei einer Base die s-Orbitale, wahrend bei einer nuk-leophilen Reaktion die p-Orbitale reagieren.
Eine weitere Abstufung der Reaktivitat von Nukleophilen entsteht durchdie sterischen Eigenschaften einer Spezies. Beim nukleophilen Angriff sinddie weiteren Substituenten des C-Atoms ”im Weg”. Braucht das Nukleophilnun selbst viel Platz, so trit sterische Hinderung auf und die Reaktions-geschwindigkeit kann massiv reduziert werden. Ein gutes Nukleophil ist

6 SUBSTITUTIONSREAKTIONEN 26
also moglichst klein. Der Effekt wird anhand verschieden grosser Alkoho-late ersichtlich.
Figure 41: Sterische Hinderung fuhrt zu einer Verlangsamung der Reaktion
Ebenso verhalt es sich mit dem Rest von R-L. Ist er gross (z.B.: tertiaresC), so entsteht erhebliche sterische Hinderung beim nukleophilen Angriff unddie RG wird herabgesetzt.
Rest CH3 CH2CH3 CH(CH3)2 C(CH3)3Relative RG 1 0.033 8.3·10−4 ≈0
Table 6: Relative RG von unterschiedlich grossen Resten (CH3 = 1)
Ebenfalls geschwindigkeitsbestimmend sind die Eigenschaften der Ab-gangsgruppe. Da sie nach der Reaktion als L− vorliegt, ist es wichtig, dassL− stabil ist. Ansonsten wird der Austritt von L energetisch ungunstig undfolglich die RG der SN2-Reaktion erheblich herabgesetzt. Die Stabilitat vonL− korreliert mit der Starke der konjugierten Saure. Ist diese stark (tieferpKa), so wird L− eine stabile Spezies sein (so der Fall fur die Halogenide).OH− hingegen wird eine schlecht Abgangsgruppe sein, da Wasser nur eineschwache Saure ist.
Wie schon angedeutet, kann das LM Edukte oder Produkte stabilisieren.Gehen wir von einem polaren LM aus, so wird dieses sich an (partiell)geladene Molekule anlagern und somit stabilisieren. Dies kann auf dieReaktionsgeschwindigkeit unterschiedliche Auswirkungen haben. Startenwir zum Beispiel mit geladenen Edukten, so werden diese stabilisiert (dieSolventhulle schirmt die Ladung nach aussen ab). Der ubergangszustandwird neutral sein und folglich weniger stark stabilisiert. Dies fuhrt dazu,dass die Aktivierungsenergie erhoht und somit die RG langsamer wird. Fureine solche Reaktion verwendet man also besser ein apolares LM. Umgekehrtist es, wenn wir mit ungeladenen Stoffen starten, welche nicht stabilisiertwerden. Die Aktivierungsenergie wird also nicht wesentlich verandert. Dadie Reaktion aber geladene Molekule hervorbringen wird, werden diese sta-bilisiert und folglich die Energie erhoht, welche frei wird. Dies bewirkt ein

6 SUBSTITUTIONSREAKTIONEN 27
Beschleunigung der Reaktion.
6.3 SN1-Reaktion
Wie im vorherigen Kapitel gesehen ist eine Substitution an einem tertiarenKohlenstoff nach SN2 nicht moglich. Die folgende Reaktion findet jedochtrotzdem statt. Es sicherlich eine Substitution, jedoch kaum nach SN2.
Figure 42: Reaktion eines tertiaren C-Atoms mit Wasser
Variiert man die eduktkonzentrationen, stellt man fest, dass die Reak-tionsgeschwindigkeit unabhangig von [Nu] ist. Dies fuhrt zur folgendenReaktionsgeschwindigkeit:
ν = k[R− L] (7)
Daraus lassen sich nun Schlusse auf den Mechanismus ziehen. In einemersten Schritt wird das Bromid abgespalten und es entsteht ein Carbokation(als Zwischenprodukt). Diese Spezies ist sehr reaktiv und reagiert deshalbsehr schnell mit dem Nukleophil (hier Wasser). Die Positive Ladung wirduber Deprotonierung abgegeben, wodurch wir zum Produkt gelangen.
Figure 43: Die Abspaltung der Abgangsgruppe bestimmt die Reaktions-geschwindigkeit

6 SUBSTITUTIONSREAKTIONEN 28
Das Abspalten des Bromids benotigt eine relativ hohe Aktivierungsen-ergie, wodurch dieser Schritt sehr langsam verlauft. Der nukleophile Angriffund die Deprotonierung hingegen verlaufen sehr schnell. Daher ist also dererste Schritt der geschwindigkeitsbestimmende, was sich auch in der obigenGleichung wiederspiegelt. Das Energiediagramm der SN1-Reaktion ist etwaskomplizierter. Wir haben mit dem Carbokation nun ein Zwischenproduktund folglich zwei Ubergangszustande.
Figure 44: Energiediagramm der SN1-Reaktion mit einem Zwischenprodukt
Besonders interessant ist, dass wir bei der SN1-Reaktion keine Stere-ospezifitat feststellen. Dies hangt damit zusammen, dass Bindungsbruchund -bildung nicht mehr gleichzeitig verlaufen. Wir erhalten daher ein Car-bokation als Zwischenprodukt, wobei der Kohlenstoff sp2 hybridisiert ist.Folglich ist ein p-Orbital, welches senkrecht zur Molekulebene steht, nichtbesetzt. Die Chance eines nukleophilen Angriffs von ”oben” bzw. ”un-ten” ist nun gleich gross. Wir werden also 50% des einen 50% des anderenStereoisomers erhalten, also ein Racemat.
Figure 45: Beide Angriffe sind gleich wahrscheinlich: Racemisierung
Starten wir mit einer optisch aktiven Substanz, wird das Reaktions-gemisch seine optische Aktivitat zunehmend verlieren.

6 SUBSTITUTIONSREAKTIONEN 29
6.4 Abhangigkeit der SN1-Reaktion von Nu, R-L und LM
Wie wir vorher festgestellt hatten, ist die RG nicht von [Nu] abhangig. DieWahl des Nukleophils hat also keinen Einfluss auf RG. Liegen jedoch mehrereNukleophile vor, so wird das Strarkste das ”Rennen” um das Carbokationmachen.
Der Rest an der Abgangsgruppe sollte - im Gegensatz zur SN2 - moglichstgross sein. Dies ruhrt daher, dass ein Carbokation energetisch sehr ungunstigist. Ist der Kohlenstoff jedoch hoch substituiert (min. sekundar), so kanndie positive Ladung uber mehrere Zentren verteilt werden. Dadurch wirddas Zwischnprodukt stabilisiert und somit die RG erhoht.
Figure 46: Die Verteilung der positiven Ladung uber mehrere Zentren sta-bilisiert das Molekul
Fur die Abgangsgruppe gelten dieseleben Regeln wie bei der SN2-Reaktion.Je stabiler L−, desto schneller die Reaktion.
Polare Losungsmittel stabilisieren das positiv geladene Carbokation. Dadurchwird die RG ebenfalls erhoht.
6.5 SN1 oder SN2?
Es lasst sich nicht immer voraussagen, welcher der beiden Reaktionstypenauftritt. Haufig entstehen auch Gemische, weil beide Reaktionen ablaufen.Es gibt jedoch gewisse Faustregeln, nach welchen man abschatzen kann,welches der bevorzugte Reaktionstyp sein wird.
• Ist der Kohlenstoff primar (oder CH3) so wird SN2 dominieren.
• Ist der Kohlenstoff tertiar so wird SN1 dominieren.
• Ist der Kohlenstoff sekundar und die Substituenten sterisch anspruchsvolldominiert SN1, ansonsten SN2.
• Liegen gute Nukleophile vor, wird SN2 bevorzugt.
• In polaren LM wird eher SN1 bevorzugt.

7 ELIMINIERUNGSREAKTIONEN 30
7 Eliminierungsreaktionen
7.1 E1-Reaktion
Setzt man Brommethan mit Ethanol um, so erwarten wir eine SN1-Reaktion,welche zum tert-Butylethylether fuhrt. Jedoch erhalten wir neben diesemProdukt noch 20% eines Alkens.
Figure 47: Es entsteht nicht ausschliesslich das Substiutionsprodukt
Offensichtlich wurd dabei nicht substituiert, sonder nur eliminiert (DasBr und ein H). Dies ist moglich, weil das Zwischenprodukt der SN1-Reaktionein Carbokation ist. Durch die positive Ladung, erlangen die H-Atome derSubstituenten eine hohere Aciditat. Beim Vorhandensein einer Base kanndaher - anstatt eines nukleophilen Angriffs - eine Deprotonierung stattfinden.Dadurch entsteht ein Alken, welches wieder ungeladen ist. Im Falle unseresBeispiels liegt mit Ethanol eine schwache Base vor, welche das Proton ab-strahiert. Viel besser lauft die Reaktion jedoch mit einer starken Base wieEthanolat. Damit konnen wir die Ausbeute an Eleminierungsprodukt auf93% erhohen.
Figure 48: Durch Zugabe einer starken Base wird die Ausbeute an Alkenerhoht
Die kinetische Betrachtung der Reaktion ergibt, dass die Reaktion uni-molekular verlauft. Sie hangt einzig von der Konzentration an R-L ab:
ν = k[R− L] (8)
Aufgrund dieser Erkenntnis wird die Reaktion als E1-Reaktion bezeich-net. Analog zur SN1-Reaktion tritt als Zwischenprodukt das Carbokation

7 ELIMINIERUNGSREAKTIONEN 31
auf. Abspaltung der Abgangsgruppe und Deprotonierung erfolgen also nichtgleichzeitig.
Betrachten wir nun dieselbe Reaktion mit 2-Brom-2-methylbutan alsEdukt, erhalten wir wiederum ein Produktgemisch. Das Hauptprodukthat eine hoher substituierte Doppelbindung (mehr weiterfuhrende Alkyl-Reste) und wird als Saitzevprodukt bezeichnet. Das Nebenprodukt hat eineweniger substituierte Doppelbindung und wird als Hofmannprodukt beze-ichnet. Die beiden Produkte entstehen, da die Deprotonierung entweder anden Methylgruppen oder an der Ethylgruppe stattfinden kann. Da ein hohersubstitiertes Alken stabiler ist, wird das Seitzev-Produkt bevorzugt.
Figure 49: Die Doppelbindung des Seitzev Produkt ist dreifach substituiertund deshalb stabiler
Man bezeichnet dieses Verhalten der E1-Reaktion als Regioselektivitat.
7.2 E2-Reaktion
Setzt man tert-Butylbromid mit 1 M NaOCH2CH3 erhalt man wie zuvor 2-Mathylpropen. Jedoch erfolgt die Reaktion nun nicht mehr mono-, sondernbimolakular.
ν = k[R− L][Base] (9)
Man bezeichnet die Reaktion daher als E2-Reaktion. Analog zur SN2-Reaktion erfolgt der Austritt der Abgangsgruppe und die Deprotonierunggleichzeitig (konzertiert).
Im weiteren tritt bei E2-Reaktionen (analog zu SN2) auch Stereospezi-fitat auf. So entsteht in der folgenden Reaktion ausschliesslich das E-Isomer.
Auf den ersten Blick scheint es unlogisch, dass kein Z-Isomer entsteht.Damit aus den sp3-Orbitalen bei der Umhybridisierung zwei p-Orbitaleentstehen, welche anschliessend die π-Bindung bilden konnen, sollten sie op-timaler Weise parallel zueinander stehen. Diese Konformation ist moglich,wenn sie einen 0 Winkel (syn-Stellung) oder einen 180 Winkel (anti-Stellung)zueinander einnehmen. Die syn-Eliminierung wurde in unserem Beispiel

7 ELIMINIERUNGSREAKTIONEN 32
Figure 50: Durch Stereospezifitat entsteht ausschliesslich das E-Isomer
zum Z-Isomer fuhren, wahrend anti-Eliminierung zum E-Isomer fuhrt. Be-trachtet man jedoch die Newman Projektion, so wird ersichtlich, dass insyn-Stellung eine ekliptische Konformation vorliegt. In anti-Stellung hinge-gen die energetisch gunstige gestaffelte Konformation.
Figure 51: Stehen das zu abstrahierende Proton und die Abgangsgruppeanti, so liegt eine gestaffelte Konformation vor.
Dies fuhrt dazu, dass syn-Eliminierung nicht auftritt und wir folglichauschliesslich das E-Isomer erhalten. Ist in einem Molekul weder syn-, nochanti-Elimination moglich, so wird keine Eliminierung stattfinden.
Wie wir schon bei E1-Reaktionen gesehen haben, tritt auch bei der E2-Reaktion dieselbe Regioselektivitat auf. Auch hier ist das hoher substituierteProdukt (Seitzev) im Allgemeinen bevorzugt. Jedoch kann das Verwendeneiner sterisch anspruchsvollen Base dazu fuhren, dass das Hofmannproduktbevorzugt auftritt, da dieses Proton ”einfacher zuganglich” ist. Auch imFalle einer schlechten Abgangsgruppe wird Hofmann bevorzugt, da durchden langsameren Bindungsbruch der Austrittsgruppe kurzfristig eine neg-ative Partialladung auftritt. Diese wird (gerade umgekehrt zu positivenLadungen) auf primaren Kohlenstoffen am besten stabilisiert, was das Hof-mannprodukt bevorzugt.

7 ELIMINIERUNGSREAKTIONEN 33
7.3 SN1/SN2 oder E1/E2?
Haufig ist es nicht moglich zu sagen, welche der Reaktionen nun ablauft.In diesen Fallen erhalt man im Versuch haufig Produktgemische (in gewis-sen Verhaltnissen). Im folgenden sind jedoch einige Richtlinien angegeben,anhand derer man die REaktionsweise abklaren kann.
• Wird eine starke Base verwendet, die schwachen oder keinen nuk-leophilen Charakter hat, so wird Elimination erfolgen.
• Liegt hingegen ein starkes Nukleophil vor, welches nicht basisch reagiert,so findet Substitution statt.
• Ist der Rest tertiar und die Reaktion findet unter hoher Temperaturstatt, so wird E2 gegenuber SN2 vorgezogen.
• Ist der Rest jedoch primar, so wird bevorzugt SN2 ablaufen.
• Zu merken ist, dass bei E2-Reaktionen das abstrahierte Proton unddie Abgangsgruppe syn oder anti stehen mussen. Ist dies nicht derFall, so findet kaum E2 statt.
• Verwendet man polare Losungsmittel, so sind die Reaktionen mit einemCarbokation als Zwischenprodukt bevorzugt (also E1 oder SN1).

8 ADDITIONSREAKTIONEN AN ALKENEN 34
8 Additionsreaktionen an Alkenen
8.1 Allgemeiner Mechanismus und Markovnikov Regel
Wie eine Base kann auch ein Alken das Proton von HBr abstrahieren, indemdie π-Bindung der Doppelbindung gebrochen wird. Dadurch entsteht einCarbokation, welches von einem Nukleophil (z.B. Br−) angegriffen werdenkann. Unter dem Strich wurde somit HBr an das Alken addiert.
Figure 52: Mechanismus der Additionsreaktion und allgemeine Reaktions-gleichung
Wie bei einer SN1-Reaktion erhalten wir als Zwischenprodukt wiederumein Carbokation. Die Analogie ist auch im Energiediagramm ersichtlich,welches dem der SN1 sehr ahnlich sieht. Der geschwindigkeitsbestimmendeSchritt ist die Abstrahierung des Protons (1. Reaktionsschritt). Die Ak-tivierungsenergie hangt daher stark von der Starke der Saure ab. Auch imFalle der Additionsreaktion stellt sich die Frage der Stereospezifitat, welchejedoch einfach zu beantworten ist. Da ein Carbokation als Zwischenproduktvorliegt, sind beide moglichen Angriffe - analog zu SN1 - gleich wahrschein-lich. Wir erhalten daher, falls chirale C-Atome vorliegen, razemische Pro-dukte.
Bei der Addition wird in einem ersten Schritt ein Proton abstrahiert undan ein C-Atom der Doppelbindung addiert. Es stellt sich daher die Frage,an welches der beiden C-Atome es binden wird. Um dies abzuklaren, be-trachtet man die unterschiedlichen Zwischenprodukte der beiden moglichenReaktionswege. Es wird sicherlich der Weg vorgezogen, welcher ein stabil-eres Zwischenprodukt hat. Da es sich dabei um ein Carbokation handelt,konnen wir uns in erster Linie auf die Stabilisierung der positiven Ladungkonzentrieren. Wie fruher schon festgestellt, wird eine positive Ladungdurch einen tertiaren Kohlenstoff am besten stabilisiert, wahrend eine pos-itive Ladung auf einem primaren Kohlenstoff energetisch ungunstig ist. Inunserem Beispiel ist die positive Ladung also unterschiedlich gut stabilisiert.Der Reaktionsweg uber das primare Carbokation wird also nicht auftreten

8 ADDITIONSREAKTIONEN AN ALKENEN 35
Figure 53: Energiediagramm einer Additionsreaktion mit Carbokation alsZwischenprodukt
und wir erhalten somit nur ein Produkt.
Figure 54: Nur die Addition uber das stabilere Carbokation erfolgt
Die Additonsreaktion ist also regioselektiv. Markovnikov hat dieses Ver-halten in der Markovnikov Regel festgehalten:
Der Wasserstoff addiert an den Kohlenstoff der Doppelbindung, welcherbereits mehr Wasserstoffatome tragt.
Synthetisch gesehen ist die Additionsreaktion sehr interessant. So konnteman durch Addition von Wasser an ein Alken Alkohole synthetisieren. Je-doch haben wir oben festgehalten, dass die Aktivierungsenergie stark vonder Saurestarke abhangt. Wasser hat aber lediglich einen pKa von 15.5. Fol-glich ware eine enorm hohe Aktivierungsenergie erforderlich um die Reak-

8 ADDITIONSREAKTIONEN AN ALKENEN 36
tion zu starten. Setzt man jedoch neben Wasser noch H3O+ ein, so lauftdie Reaktion sehr gut.
Figure 55: Mithilfe von H3O+ als Katalysator kann aus einem Alken einAlkohol synthetisiert werden
Betrachtet man die Reaktion so stellt man fest, dass H3O+ am Schlussder Reaktion wiedrum auftritt. Folglich wirkt H3O+ als Katalysator indieser Reaktion.
8.2 Hydroborierung
Naturlich kommt auch in der Synthese von Alkoholen die Markovnikov-Regel zum Tragen und wir erhalten nur tert-Butanol. Um iso-Butanol zusynthtisieren, mussen wir also anders vorgehen. Die Reaktion, durch welchewir jeweils das andere Pordukt (das sog. Anti-Markovnikov Produkte er-halten) nennt man Hydroborierung. Dabei setzt man ein Alken mit BH3
(Boran) um. Betrachtet man die Reaktion, so stellt man fest, dass sie stere-ospezifisch ist. Das H und das BH2 stehen nach der Addition immer synzueinander. Zuvor hatten wir aber festgehalten, dass Additionsreaktionenkeine Stereospezifitat aufweisen. Folglich muss hier ein anderer Mechanis-mus vorliegen.
Figure 56: Die Addition von BH3 erfolgt uber einen zyklischenUbergangszustand

8 ADDITIONSREAKTIONEN AN ALKENEN 37
Grund fur die Stereospezifitat ist, dass die Addition des H-Atoms und desBH2 konzertiert (gleichzeitig) verlauft. Dadurch werden die beiden Grup-pen immer auf derselben Seite (also in syn-Stellung) addiert. Zudem stellenwir fest, dass die Markovnikovregel nicht gilt. Das H-Atom addiert an dashoher substituierte C-Atom. Der Grund dafur liegt im ubergangszustand,in welchem die Doppelbindung gebrochen und die beiden Gruppen addiertwerden. Boran besitzt ein freies p-Orital (also eine Elektronenlucke) undreagiert folglich als Lewis-Saure. Es fullt seine Elektronenlucke mit den π-Elektronen der Doppelbindung. Dadurch entsteht zwischenzeitlich eine posi-tive Partialladung auf dem ”ehemaligen” Alken (sozusagen ein Carbokation-ahnlicher Zustand). Diese wird wie wir wissen, auf dem hoher substituiertenKohlenstoff besser stabilisiert, wodurch das Bor an den weniger substitu-ierten Kohlenstoff addieren wird. Anschliessend wird an den Kohlenstoffmit der partiellen positiven Ladung ein Hydrit (H−) addiert.
Nach der ersten Addition bleibt die Reaktion aber nicht stehen. Das BH2
hat (wie auch das BH3) immer noch ein freies p-Orbital. Folglich erfolgt dieAddition auf dieselbe Weise noch zweimal. Ein BH3 reagiert also mit dreiAlkenen.
Figure 57: Zwei weitere Additionen und Oxidation mit Wasserstoffperoxidfuhrt zum Anti-Markovnikov Produkt
Nach den drei Additionen wird nun das Alkylboran mit Wasserstoffper-oxid und Natronlauge umgesetzt. Dabei wird das Bor jeweils durch eineOH-Gruppe ersetzt, wodurch wir zum Alkohol gelangen. Faktisch ist dieseine Oxidation. Der genaue Mechanismus ist kompliziert und kann allenfallsim Buch auf Seite 165f nachgelesen werden.

8 ADDITIONSREAKTIONEN AN ALKENEN 38
8.3 Wagner-Meerwein Umlagerung
Setzt man 3,3-Dimethylbut-1-en mit HBr um, so erhalten wir als Haupt-produkt 1,1,3-Trimethylbromid. Nach den bisherigen Erkenntnissen scheintdies hochst unlogisch. Zwar addiert das H-Atom, der Markovnikov-Regelfolgend, an den tiefer substituierten Kohlenstoff, das Bromid addiert jedochstatt ans nachste C-Atom an den ubernachsten Nachbarn.
Figure 58: Umlagerung fuhrt zu einem stabileren tertiaren Carbokation
Betrachten wir das entstehende Carbokation, so sehen wir dass die posi-tive Ladung auf einem sekundaren Kohlenstoff stabilisiert wird. Energetischware jedoch die Stabilisierung auf einem tertiaren Kohlenstoff zu bevorzu-gen. Um dies zu ermoglichen ”wandert” eine Methylgruppe (faktisch einCH−3 ) an das positiv geladene C-Atom. Somit ist die positive Ladung nunauf einem tertiaren Kohlenstoff stabilisiert. Das Bromid wir nun naturlichdort nukleophil angreifen, wodurch sich die Entstehung des Hauptproduktserklart.
Man bezeichnet diese ”Wanderung” einer CH3-Gruppe als Wagner-MeerweinUmlagerung. Moglich ist nicht nur Umlagerung einer Methylgruppe, son-dern auch eines Hydrids. Jedoch ist die ”Wanderung” immer auf maximalauf den Sprung von einem auf dan nachsten Kohlenstoff beschrankt. Zu-dem muss naturlich durch die Umlagerung eine bessere Stabilisierung derpositiven Ladung resultieren.
8.4 Addition von H2 und Halogenen
Die Additionsreaktionen sind nicht auf H-X oder H2O als Partner beschrankt,sondern konnen auch mit Halogenen und im Speziellen mit Wasserstoff erfol-gen. Die letztere Reaktion bedarf jedoch eines Palladium/Kohle-Kondensators(heterogener Kondensator). Da die Addition nur auf der Oberflache des
8 ADDITIONSREAKTIONEN AN ALKENEN 39
Kondensators ablauft, ist die Reaktion stereospezifisch. Die beiden H-Atomewerden stets in syn-Stellung addiert.
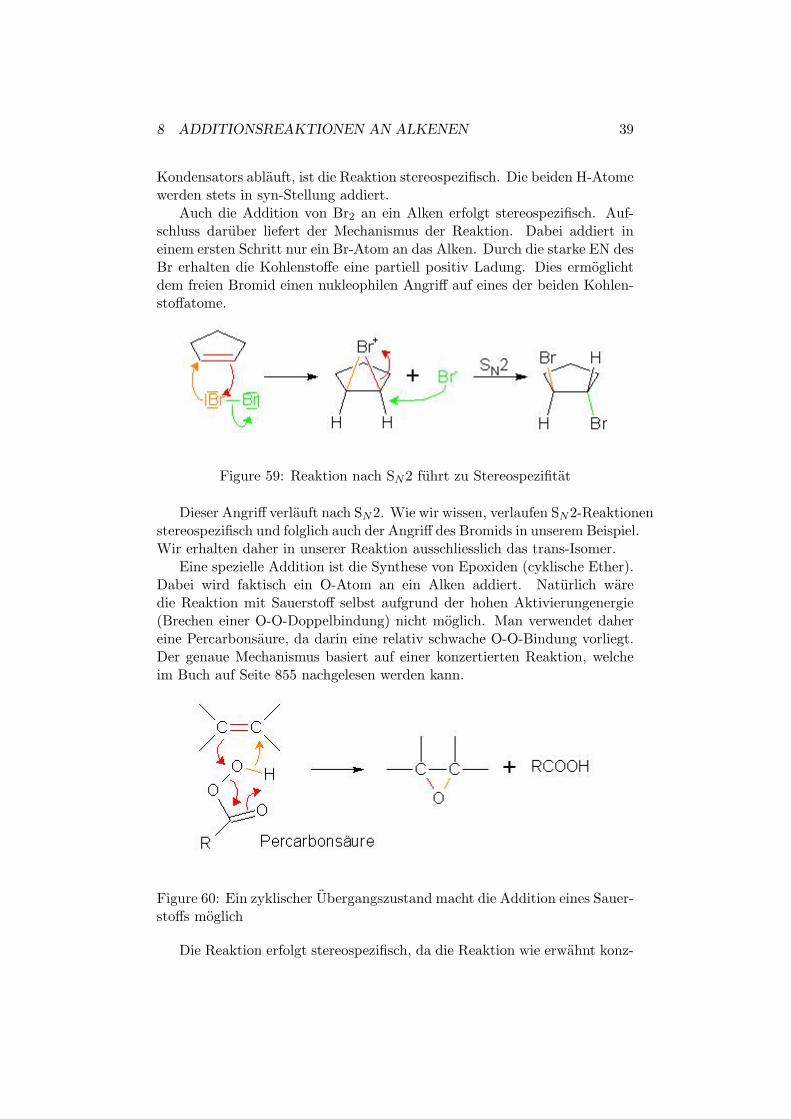
Auch die Addition von Br2 an ein Alken erfolgt stereospezifisch. Auf-schluss daruber liefert der Mechanismus der Reaktion. Dabei addiert ineinem ersten Schritt nur ein Br-Atom an das Alken. Durch die starke EN desBr erhalten die Kohlenstoffe eine partiell positiv Ladung. Dies ermoglichtdem freien Bromid einen nukleophilen Angriff auf eines der beiden Kohlen-stoffatome.
Figure 59: Reaktion nach SN2 fuhrt zu Stereospezifitat
Dieser Angriff verlauft nach SN2. Wie wir wissen, verlaufen SN2-Reaktionenstereospezifisch und folglich auch der Angriff des Bromids in unserem Beispiel.Wir erhalten daher in unserer Reaktion ausschliesslich das trans-Isomer.
Eine spezielle Addition ist die Synthese von Epoxiden (cyklische Ether).Dabei wird faktisch ein O-Atom an ein Alken addiert. Naturlich waredie Reaktion mit Sauerstoff selbst aufgrund der hohen Aktivierungenergie(Brechen einer O-O-Doppelbindung) nicht moglich. Man verwendet dahereine Percarbonsaure, da darin eine relativ schwache O-O-Bindung vorliegt.Der genaue Mechanismus basiert auf einer konzertierten Reaktion, welcheim Buch auf Seite 855 nachgelesen werden kann.
Figure 60: Ein zyklischer Ubergangszustand macht die Addition eines Sauer-stoffs moglich
Die Reaktion erfolgt stereospezifisch, da die Reaktion wie erwahnt konz-

8 ADDITIONSREAKTIONEN AN ALKENEN 40
ertiert verlauft. Geht man also von einer Z- bzw. E-Konformation aus, soerhalt man das cis- bzw. trans-Isomer. Epoxide sind von synthetischemNutzen, weil die C-O-Bindungen aufgrund der realtiv hohen Ringspannungeinfach zu brechen sind. Sie konnen zum Beispiel sowohl in basischem, alsauch in saurem Milieu zu Stoffen mit zwei Alkoholfunktionalitaten (sog.Diolen) umgesetzt werden.

9 AROMATIZITAT 41
9 Aromatizitat
9.1 Resonanz und Huckel-Regel
Setzt man Cyclohexen mit HBr um, so erfolgt an der Doppelbindung Addi-tion wie im vorherigen Kapitel besprochen. Auch mit Br2 erfolgt eine Reak-tion. Setzt man jedoch Benzol statt Cyclohexen ein, so erfolgt mit keinemder beiden Reagenzien eine Reaktion. Offensichtlich mussen die Doppel-bidnungen des Benzols stabiler (energetisch gunstig) sein, als die Doppel-bindung im Cyclohexen.
Eine Messung der Bindungslange zeigt, dass es gar keine ”richtigen”Doppelbindungen sind. Wahrend die C=C-Doppelbindung normalerweiseeine Lange von 133 pm aufweist, so sind die sechs Bindungen des Benzolsbereits 139 pm (alle gleich lang!). Eine C-C-Einfachbindung ist noch einmalgrosser, namlich 147 pm. Die Bindungen des Benzols sind von der Langegesehen, also ein Mittelding zwischen Doppel- und Einfachbindung. Zwis-chen den Bindungen des Benzols gibt es keinerlei Unterschiede, obwohl dieSkelettformel dies andeutet.
Grund fur diese Eigenschaften des Benzols ist das Phanomen der Reso-nanz. Die sechs Kohlenstoffatome des Benzols sind alle sp2 hybridisiert. Dasp-Orbital der C-Atome steht senkrecht zur Ringebene. Die 6 Elektronen,welche sich darin befinden, sind nun nicht genau lokalisiert, sondern ober-und unterhalb des Rings verstrichen. Man kann sich also zwei Elektronen-wolken vorstellen, welche uber bzw. unter der Ringebene liegen. Da unsereFormelsprache diese Delokalisierung nicht wahrheitsgemass darzustellen ver-mag, gibt man sogenannte Grenzformeln bzw. Grenzstrukturen an. Dieseentsprechen nicht wirklich der Realitat, geben aber einen gewissen Rahmenzwischen welchem die tatsachliche Gestalt liegt.
Figure 61: Delokalisierte π-Elektronen uber und unter dem Ring und zweimogliche Grenzstrukturen

9 AROMATIZITAT 42
Resonanz bringt immer eine Stabilisierung der Struktur mit sich. Vorallem in geladenen Molekulen entscheidet die Verteilung der Ladung ubermehrere Zentren mittels Resonanz stark uber die Stabilitat. Zudem ist Ben-zol planar, da durch den 120 Winkel der sp2 hybridisierten Kohlenstoffe einoptimales Sechseck gebildet wird und folglich keine Ringspannung auftritt.
Die Eigenschaften, welche Benzol auszeichnen, sind nicht einzigartig. Esgibt eine enorme Menge von ahnlichen Verbindungen. Daher werden siezu den sogenannten aromatischen Verbindungen zusammengefasst. Aro-matizitat bringt immer dieselben Charakteristiken mit sich, wodurch mansofort entscheiden kann, ob eine Verbindung aromatisch ist:
• Aromatische Verbindungen sind immer zyklisch.
• Die Doppelbindungen sind vollkommen konjugiert (Die ”Doppelbindun-gen” sind immer durch eine ”Einfachbindung” getrennt).
• Aromatische Verbindungen sind planar.
• Aromatische Veribinungen gehorchen der Huckel-Regel.
Die Huckel-Regel besagt, dass eine aromatische Verbindung 4 ∗ n + 2π-Elektronen besitzen muss (n = 0,1,2,3,...). Anders gesagt muss eine aro-matische Verbindung eine ungerade Anzahl π-Elektronenpaare (”Doppel-bindungen”) besitzen.
Werden Ringe durch andere Atome als C unterbrochen (z.B.: N oder O),so werden die Verbindungen als Heterocyclen oder falls Aromatozitat vor-liegt als Heteroaromaten bezeichnet. Oftmals sind in diesem Fall die freienElektronenpaare ebenfalls Teil des π-Elektronensystems (z.B.: Pyrrol), sodass die Huckel-Regel erfullt wird. Heterocyclen sind in der Natur sehrwichig (Nukleobasen), werden hier jedoch nicht weiter besprochen.
9.2 Reaktionen an aromatischen Verbindungen
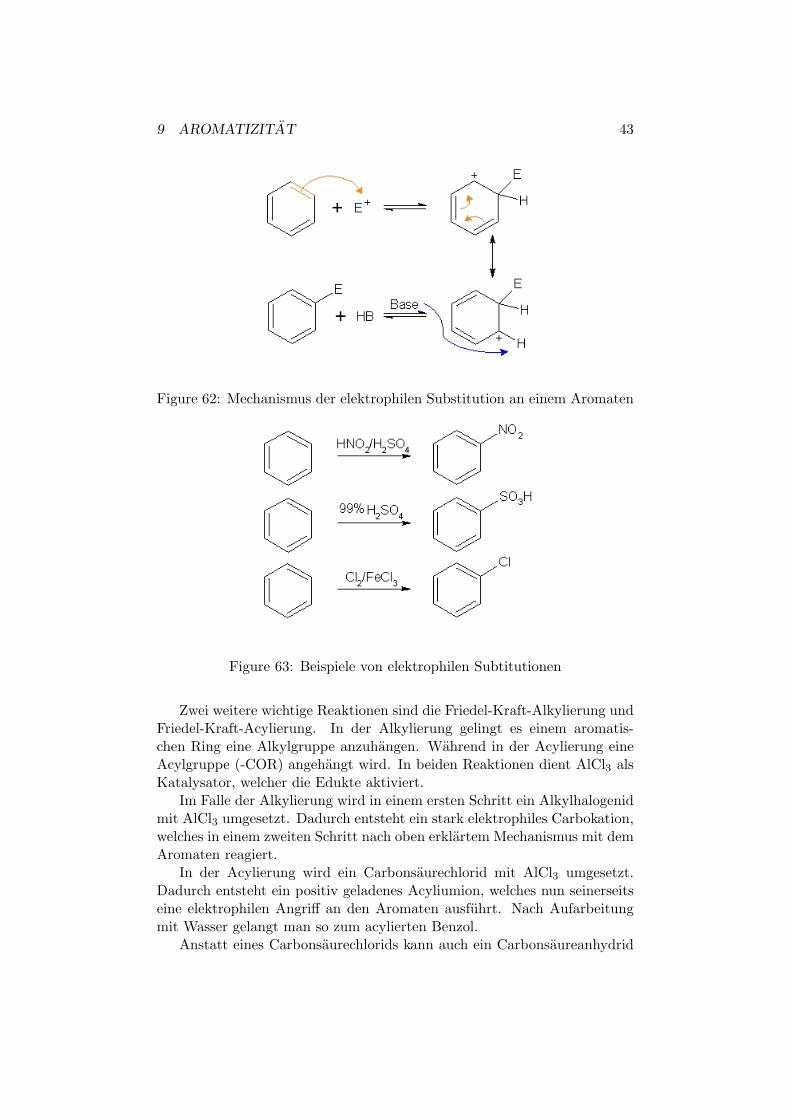
Aromatische Verbindungen sind im Allgemeinen nicht sonderlich reaktiv.Trotzdem sind einige Reaktionen moglich. Eine Moglichkeit ist die elek-trophile Substitution an Aromaten.
Dabei addiert ein Elektrophil in einem ersten Schritt an den Ring. Dadurchentsteht ein Carbokation, welches aufgrund der Resonanzstabilisierung derpositiven Ladung, eine gewisse Stabilitat erreicht. In einem zweiten Schrittwird nun mit Hilfe einer schwachen Base ein Proton abgespalten, woduchman zum Produkt gelangt.
Beispiele fur eine solche Reaktion sind Nitrierung, Sulfonierung undHalogenierung, wobei bei der Letzteren eines der Cl-Atome durch FeCl3(Katalysator) eine postive Ladung erlangt.
9 AROMATIZITAT 43
Figure 62: Mechanismus der elektrophilen Substitution an einem Aromaten
Figure 63: Beispiele von elektrophilen Subtitutionen
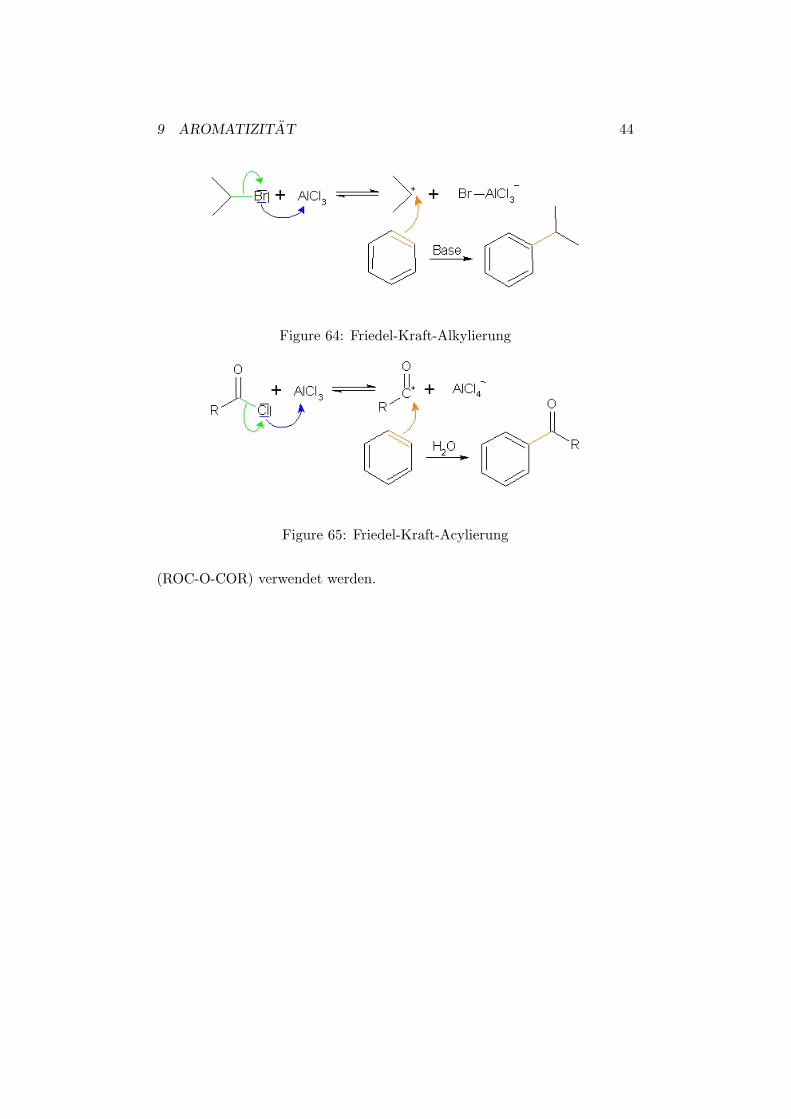
Zwei weitere wichtige Reaktionen sind die Friedel-Kraft-Alkylierung undFriedel-Kraft-Acylierung. In der Alkylierung gelingt es einem aromatis-chen Ring eine Alkylgruppe anzuhangen. Wahrend in der Acylierung eineAcylgruppe (-COR) angehangt wird. In beiden Reaktionen dient AlCl3 alsKatalysator, welcher die Edukte aktiviert.
Im Falle der Alkylierung wird in einem ersten Schritt ein Alkylhalogenidmit AlCl3 umgesetzt. Dadurch entsteht ein stark elektrophiles Carbokation,welches in einem zweiten Schritt nach oben erklartem Mechanismus mit demAromaten reagiert.
In der Acylierung wird ein Carbonsaurechlorid mit AlCl3 umgesetzt.Dadurch entsteht ein positiv geladenes Acyliumion, welches nun seinerseitseine elektrophilen Angriff an den Aromaten ausfuhrt. Nach Aufarbeitungmit Wasser gelangt man so zum acylierten Benzol.
Anstatt eines Carbonsaurechlorids kann auch ein Carbonsaureanhydrid
9 AROMATIZITAT 44
Figure 64: Friedel-Kraft-Alkylierung
Figure 65: Friedel-Kraft-Acylierung
(ROC-O-COR) verwendet werden.

10 CARBONYLVERBINDUNGEN 45
10 Carbonylverbindungen
Die Gruppe der Carbonylverbindungen umfasst eine enorme Menge an wichti-gen Verbindungen. Allen gemeinsam ist die Carbonylgruppe -c=o. Dabeikonzentrieren wir uns auf Aldehyde und Ketone, wahrend Carbonsauren undAmide (und deren Derivate) an diesem Punkt nicht besprochen werden.
Figure 66: Struktur von Aldehyden und Ketonen
Die Nomenklatur verwendet fur Aldehyde die Endung -al, wahrend Ke-tone die Endung -on erhalten. Dabei ist zu beachten, dass Aldehyde nurterminal stehen konnen, wahrend Ketongruppen auch innerhalb der C-Ketteauftreten konnen.
Die Carbonylgruppe besitzt ganz charakteristische Eigenschaften. Auf-grund der Doppelbindung liegen die beiden Reste, das C-Atom und dasO-Atom in einer Ebene. Im weiteren besitzt der Sauerstoff eine grossereElektronegativitat, wodurch die Doppelbindung polarisiert wird. Es liegtsomit eine partiell negative Ladung auf dem Sauerstoff vor, wahrend derKohlenstoff eine partiell positive Ladung tragt. Das damit entstehendeDipolmoment resultiert in zwischenmolekularen Wechselwirkungen. Diesefuhren dazu, dass Aceton erst mit 56C siedet, wahrend das analoge Methyl-propen einen Siedepunkt von -7C besitzt.
10.1 Nukleophile Addition
Die partiell positive Ladung auf dem Kohlenstoff macht einen Angriff einesNukleophils moglich. Da keine Abgangsgruppen das Molekul verlassen konnen,werden die entstehenden Ladungen durch Protonierung (im Falle des Sauer-stoffs) bzw. durch Deprotonierung (im Falle eines neutralen Nukleophils)aufgehoben. Dabei sind die meisten Reaktionen Gleichgeichtsreaktionen undfolglich reversibel. Einige Reaktionen sind hingegen irreversibel, wobei die

10 CARBONYLVERBINDUNGEN 46
Carbonylgruppe reduziert wird (zu Alkoholen oder Alkenen).
10.1.1 Reversible Reaktionen
Das einfachste Nukleophil, welches einen Carbonylkohlenstoff angreifen kann,ist Wasser. Ein freies Elektronenpaar des Sauerstoffs greift dabei den Kohlen-stoff nukleophil an. Die Doppelbindung wird dabei gebrochen und die Elek-tronen auf den Sauerstoff der Carbonylgruppe ubertragen. Durch Umpro-tonierung gelangt man zum Produkt, welches als Hydrat bezeichnet wird.Die Reaktion kann durch Zugabe von Saure beschleunigt werden. In diesemFall wird zuerst der Sauerstoff protoniert. Die entstehende positive Ladungist resonanzstabilisiert und zu einem guten Teil auf dem Kohlenstoff. Diesmacht einen nukleophilen Angriff wahrscheinlicher und damit die Reaktionschneller.
Figure 67: Nukleophiler Angriff von Wasser fuhrt zu Hydraten
Wie vorher gelangt man so zu einem Hydrat und dem Saureaquivalent,welches zu Beginn eingesetzt wurde. Die Saure funktioniert also als Katalysatorder Reaktion.
Die einzelnen Reaktionen sind alle Gleichgewichtsreaktionen. Ob dasGleichgewicht auf Seite der Caronylverbindung oder auf der Seite des Hy-drats liegt, hangt primar von den Resten der Carbonylverbingung ab. Ver-wendet man z.B. Formaldehyd (H2CO), so tritt kaum sterische Hinderung

10 CARBONYLVERBINDUNGEN 47
beim Angriff des Wassers auf und das Gleichgewicht liegt praktisch vollstandigauf der Seite des Hydrats. Aber bereits bei der Reaktion mit Aceton ((CH3)2CO)verhindert die sterische Hinderung durch die beiden Methylgruppen den An-griff des Wassers beinahe vollstandig.
Naturlich ist die nukleophile Addition nicht auf Wasser beschrankt. Auchandere nukleophile konnen mit Carbonylverbindungen auf dieselbe Weisereagieren. Vor allem die Reaktion mit CN− ist interessant, da die entste-henden Cyanhydrine fur weitere Reaktionen dienen.
Figure 68: Nukleophile Addition von CN− und weiterfuhrende Reaktionen
So kann man durch Hydrolyse α-Hydroxycarbonsauren herstellen. Setztman das Cyanhydrin mit LiAlH4 (ein starkes Reduktionsmittel) um, sogelangt man zu β-Hydroxyaminen.
Eine weitere Moglichkeit ist die Addition von Alkoholen an Carbonylverbindun-gen. Der Mechanismus ist derselbe wie bei der Addition von Wasser. Esentstehen dadurch sogenannte Halbacetale. Ein Spezialfall dieser Reaktionist die Ringschliessung in der Glucose. Diese liegt in Losung teilweise offen-kettig vor. Durch einen intramolekularen Angriff der Hydroxygruppe an dieAldehydgruppe wird der Ring geschlossen.
Sind in der Reaktionsgemisch noch Alkoholmolekule vorhanden, so bleibtdie Reaktion nicht auf der Stufe des Halbacetals stehen. Die Alkoholgruppedes Halactetals wird in einem ersten Schritt protoniert (meist wird die Reak-tion unter sauren Bedingungen durchgefuhrt) und anschliessend wird Wasserabgespalten. Dadurch entsteht ein Carbokation, welches wieder nukleophilvon einem Alkoholmolekul angegriffen werden kann. Durch Deprotonierunggelangt man schlussendlich zu sogenannten Acetalen. Die Reaktionen sindweiderum allesamt Gleichgewichte. Jedoch kann man das Gleichgewicht aufdie Seite des Acetals verschieben, indem man Wasser aus dem Reaktions-

10 CARBONYLVERBINDUNGEN 48
Figure 69: Addition von Alkoholen fuhrt zu Acetalen
gemisch eliminiert. Dadurch wird die Abspaltung von Wasser nicht merhumkehrbar und die Reaktion lauft praktisch vollstadig bis zum Acetal.
Acetale konnen in organischen Synthesen als Schutzgruppen dienen. ZumBeispeil bei der Substitution des Bromids von Brompropanon durch CN−.Wird diese Reaktion direkt durchgefuhrt, so erfolgt neben der Substitutionzusatzlich Addition des Cyanids an den Carbonylkohlenstoff. Ist die letztereReaktion unerwunscht, so muss man vor der Umsetzung mit Cyanid die Ke-togruppe in eine weniger reaktive Gruppe umwandeln. Dies ist moglich,indem man das Brompropanon mit Ethan-1,2-diol umsetzt.
Figure 70: Einsetzen eines Diols als Schutzgruppe
Die zwei Alkoholgruppen werden wie zuvor besprochen, unter Abspal-

10 CARBONYLVERBINDUNGEN 49
tung von Wasser, das Acetal bilden. Da sie sich am selben Molekul befinden,entsteht ein 5-Ring, welcher keine weiteren Reaktionen mit Nukleophileneingehen kann. Nun kann die gewunschte Substitution ohne die unerwunschteNebenreaktion erfolgen. Anschliessend kann die Schutzgruppe durch Zugabevon Wasser und Saure hydrolisiert werden (die Acetalreaktion ist ein Gle-ichgewicht, welches durch Wasser verlagert werden kann!) und man gelangtso zum endgultigen Produkt.
Ein weitere mogliche Reaktion ist die Addition eines primaren Amins.Das freie Elektronenpaar des Stickstoffs kann wie zuvor der Alkohol denCarbonylkohlenstoff angreifen. Auch der Rest der REaktion erfolgt genauanalog der Halbacetalbildung. Das Produkt das entsteht kann als Halbami-nal oder Carbinolamin bezeichnet werden. Das Halbaminal kann wiederumweiterreagieren, aber nicht wie zuvor das Halbacetal. Es wird in einem er-sten Schritt zwar wiederum protoniert und anschliessend wird Wasser abges-palten. Nun erfolgt jedoch kein nukleophiler Angriff, sondern es wird de-protoniert und damit eine C=N-Doppelbindung gebildet. Diese Molekulebezeichnet man als Imine.
Figure 71: Addition von primaren Aminen fuhrt zu Iminen
Verwendet man in der vorigen Reaktion ein sekundares Amin, so verlauftsie bis zum Halbaminal identisch. Anschliessend wird ebenfalls die Hydroxy-gruppe protoniert. Da der Stickstoff nun aber kein weiteres Wasserstoffatombesitzt, kann die Deprotonierung nicht wie zuvor am Stickstoff erfolgen. Dadie positive Ladung aufgrund von Resonanz auch teilweise auf dem Kohlen-

10 CARBONYLVERBINDUNGEN 50
stoff liegt, erlangen die Wasserstoffe der Restgruppen eine gewisse Aciditat.Ist eine Base vorhanden, wird also am Kohlenstoffrest deprotoniert und eineC=C-Doppelbindung gebildet. Diese Stoffe werden als Enamine bezeichnet.Enamine sind im Allgemeinen weniger stabil als Imine.
Figure 72: Addition von sekundaren Aminen fuhrt zu Enaminen
Im folgenden werden alle besprochenen reversiblen nukleophilen Addi-tionen an Carbonylverbindungen noch einmal in kurzer Form zusammenge-fasst. Es gilt sich zu merken, dass alle Reaktionsschritte Gleichgewichtsreak-tionen sind. Folglich konnen samtliche Reaktionen auch in die umgekehrteRichtung verlaufen (daher reversible Addition).
Figure 73: Zusammenfassung der reversiblen nukleophilen Additionen

10 CARBONYLVERBINDUNGEN 51
10.1.2 Irreversible Reaktionen
Die folgenden Reaktionen besitzen jeweils mindestens einen Reaktionss-chritt, welcher nicht umkehrbar ist. Daher verlaufen die Reaktionen vollstandigund irreversibel. Im weiteren wird der Carbonylkohlenstoff in samtlichenReaktionen reduziert. Naturlich werden im Laufe der Reaktionen andereVerbindungen oxidiert. Man bezeichnet diese Stoffe als Reduktionsmit-tel. In den folgenden Reaktionen werden LiAlH4, NaBH4 (”H−”-Donoren),Methyllithium bzw. Butyllithium (Organolithiumverbindungen) und R-Mg-X (X = Br, I, Cl - Grigrnardverbindungen). Letztere beiden sind faktischCarbanion-Donoren (R−).
Setzt man eine Carbonylverbindung mit LiAlH4 oder NaBH4 um, sowird ein Hydirid addiert und Anschliessend protoniert.
Figure 74: Oxidation eines Ketons mit Lithium-Aluminium-Hydrid
Startet man mit einem Aldehyd, so gelangt man mit dieser Reaktion zueinem primaren Alkohol. Verwendet man hingegen eine Ketonverbindung,so gelangt man zu einem sekundaren Alkohol. Alkohole sind also reduzierteCarbonylverbindungen (Oxidationszahl des Carbonylkohlenstoffs andert von+I/+II zu -I/0).
Umsetzung einer Carbonylverbindung mit Methyl- oder Butyllithiumfuhrt zur Addition des entsprechenden Carbanions. Nach Protonierunggelangt man so wiederum zu Alkoholen.
Figure 75: Oxidation eines Ketons mit Methyllithium
Wird Formaldehyd verwendet, so gelangt man zu einem primaren Alko-hol, wahrend ein Aldehy bzw. ein Keton zu einem sekundaren bzw. tertiarenAlkohol fuhrt.

10 CARBONYLVERBINDUNGEN 52
Eine etwas kompliziertere Reduktion ist die sogenannte Wittig-Reakktion.Dabei wird zuerst ein Triphenylphosphin mit einem Alkylhalogenid in einerSN2-Reaktion umgesetzt. Anschliessend wird die entstehende Verbindungunter Verwendung einer starken Base (z.B. Butanlithium) deprotoniert, wasuns zu einem sogenannten Ylid fuhrt. Das Ylid setzt man nun zusammen miteiner Carbonylverbindung um, was uber einen zyklischen ubergangszustandzu einem Alken und einem Triphenylphosphinoxid fuhrt.
Figure 76: Mechanismus der Wittig Reaktion
10.2 Oxidation von Alkoholen
Wie wir im zuvor gesehen haben, ist es moglich Carbonylverbindungen zuAlkoholen zu reduzieren. Naturlich ist es unter Verwendung geeigneterOxidationsmittel auch moglich Alkohole zu Carbonylverbindungen zu ox-idieren. Sekundare Alkohole konnen also zu Ketonen oxidiert werden undprimare Alkohole zu Aldehyden. Tertiare Alkohole kann man auf diese Weisenicht oxidieren (Eliminierung aufgrund des fehlenden Wasserstoffsatom nichtmoglich). Als OXidationmittel verwendet man CrO3 in verdunnter Schwe-felsaure (H2SO4), was man als Jones Reagens bezeichnet.
In einem ersten Schritt greift der Alkohol das Chromtrioxid an. DurchUmprotonierung gelangt man zu einem sogenannten Chromsaureester. DurchElimination nach E2 gelangt man zum Aldehyd bzw. Keton und CrO(OH)2.Im Falle des Aldehyds ist die Reaktion jedoch noch nicht beendet. Da wir inverdunnter Schwefelsaure arbeiten, wird ein nukleophiler Angriff von Wasseran den Aldehyd erfolgen und wir erhalten ein Hadrat. Das Hydrat wird nun

10 CARBONYLVERBINDUNGEN 53
Figure 77: Oxidationsprodukte von Alkoholen
nach demselben Mechanismus wie zuvor die Alkohole durch CrO3 oxidiert.Nach der Eliminierung gelangt man so zu einer Carbonsaure.
Will man einen primaren Alkohol zu einem Aldehyd oxidieren, so gelingtdies nur unter Ausschluss von Wasser um die Hydratbildung zu unterbinden.Man verwendet in diesem Fall das Chromtrioxid gelost in Pyrimidin. Dieswird als Collins Reagens bezeichnet.
Da Chrom giftig ist und folglich schwer zu entsorgen, versucht man dieOxidation mit Chromtrioxid zu vermeiden. Eine Alternative bietet die so-genannte Swern Oxidation. Man setzt dabei den Alkohol mit Dimethylsul-foxid, ClCOCOCl und Triethylamin um. Die ersten beiden Stoffe reagierenzu einem Dimethylchlorosulfonium-Ion, welches das eigentliche Oxidation-smittel darstellt. Der genaue Mechanismus kann im Buch auf Seite 852nachgeschaut werden.
10.3 Reaktionen an der α-Position von Carbonylverbindun-gen
10.3.1 Keto-Enol-Tautomerie
Die Kohlenstoffe, welche direkt an die Carbonylgruppe (-c=O) geknupft sind(sog. in α-Position stehen), zeigen eine gewisse Reaktivitat. Dies ruhrt da-her, dass die starke Elektronegativitat des Sauerstoffs bis zu diesem Kohlen-stoff wirkt und ihm bis zu einem gewissen Grad die Elektronen ”entzieht”.Dies fuhrt dazu, dass die H-Atome des α-Kohlenstoffs eine erhohte Aciditatbesitzen. So besitzen die H-Atome des α-Kohlenstoffs des Ethanals einenpKa von 16,7, wahrend die analogen H-Atome in Propen gerade mal einenpKa von 42 aufweisen! Carbonylverbindungen sind also schwache Brønsted-Sauren. Aldehyde und Ketone besitzen einen pKa von 16-20, wahrend Esterbereits wieder einen von 25 besitzen. In Wasser kann somit am α-Kohlenstoff

10 CARBONYLVERBINDUNGEN 54
deprotoniert werden. Die negative Ladung wird resonanzstabilisiert, indemeine C=C-Doppelbindung gebildet wird. Nun kann die Reaktion durch Pro-tonierung am α-Kohlenstoff wieder zuruck zum Keton bzw. Aldehyd laufenoder durch Protonierung des Sauerstoffs zum sogenannten Enol.
Figure 78: Mechanismus der Keto-Enol-Tautomerie
Diese Reaktionen sind Gleichgewichtsreaktionen, wodurch sich jeweilsein Verhaltnis zwischen Carbonylverbindung und Enol einstellt. Man beze-ichnet diese Phanomen daher als Keto-Enol-Tautomerie.
In Wasser liegt die oben beschriebene Reaktion praktisch vollstandig aufder Seite der Carbonylverbindung, da diese stabiler ist. Mit Basen bzw.Sauren kann die Reaktion allerdings auf die Seite des Enols verschoben wer-den.
Figure 79: Keto-Enol-Tautomerie in basischem und saurem Milieu
Beachte, dass in den beiden Fallen die Reaktion genau umgekehrt ablauft.Die Base deprotoniert in einem ersten Schritt und in einem zweiten Schrittwird der Sauerstoff protoniert. Eine Saure protoniert im ersten Schritt unddeprotoniert erst dann den Kohlenstoff.
Interessant ist die Reaktion mit chiralen α-Kohlenstoffen. Wird depro-toniert, so entsteht eine C=C-Doppelbindung, deren Reste in einer Ebene

10 CARBONYLVERBINDUNGEN 55
liegen (planar). Bei der erneuten Protonierung des α-Kohlenstoffs ist nunder ”Angriff” von oben oder unten gleich wahrscheinlich. Versetzt man alsoeine optisch aktive Carbonylverbindung mit einer Base um, so geht die op-tische Aktivitat verloren.
In gewissen Stoffen liegt das Gleichgewicht im Gegensatz zum ”nor-malen” Fall auf der Seite des Enols. Zwei Beispiele sind Phenol und 1,3-Dicarbonylverbindungen. Im ersten Fall liegt in der Enolform ein aromatis-ches System vor (Benzylring). In der Ketoform liegt kein aromatisches Sys-tem vor. Die Enolform ist daher energetisch gunstiger als die Ketoform. DasGleichgewicht liegt daher auf der Seite des Phenols (Enolform). Im Falle der1,3-Dicarbonylverbindungen liegt in der Enolform ein 6-Ring-artige Anord-nung vor, die eine intramolekulare Wasserstoffbrucke ermoglicht. Diese sta-bilisiert die Enolform, so dass sich das Gleichgewicht zumindest teilweise aufdie Seite des Enols verschiebt.
Neben der Tautomerisierung kann es auch zum Austausch der H-Atomedurch Deuterium kommen. Setzt man Ethanal mit D2O und DO− um, sowird am α-Kohlenstoff deprotoniert. Anschliessend tritt gleich wieder dieRuckreaktion ein, indem am Kohlenstoff wieder protoniert wird. Da fastaushliesslich Deuterium zur Verfugung steht, wird ein D+ statt eines H+
ubertragen. Geschieht dies dreimal, so sind alle Wasserstoffatome durchDeuterium-Atome ersetzt worden.
Naturlich kann dies auch mit anderen Stoffen wie Halogeniden erfolgen.Setzt man eine Carbonylverbindung mit I2 um, so ersetzen die Iod-Atomedie Wasserstoffe des α-Kohlenstoffs. Unter basischen Bedingungen werdenalle H-Atome ersetzt, wahrend unter sauren Bedingungen nur ein H-Atomersetzt wird. Der Mechanismus beruht auf einer elektrophilen Addition desHalogenids an die Doppelbindung des Enolats.
Figure 80: Halogenierung von Carbonylverbindungen
Die Einfuhrung des elektronegativen Halogenids hat zur Folge, dass derCarbonylsauerstoff weniger basisch ist und der α-Kohlenstoff noch acider.Im Falle der basischen Reaktion wird damit eine erneute Halogenierung

10 CARBONYLVERBINDUNGEN 56
begunstigt (erster Schritt ist die Deprotonierung am α-Kohlenstoff). Untersauren Bedingungen wird eine weitere Halogenierung durch die kleinere Ba-sizitat des Sauerstoffs verunmoglicht.
Eine weitere Reaktion ist die Alkylierung am α-Kohlenstoff. Durchdie Deprotonierung wird die Carbonylverbindung naturlich zu einem Nuk-leophil. Setzt man das Enolat (bzw. das Carbanion) also mit CH3I um, sowird ein nukleophiler Angriff nach SN2 erfolgen.
Figure 81: Alkylierung von Carbonylverbindungen
Die Reaktion ist jedoch nicht sehr spezifisch, da eine zweite Alkylierungam Produkt moglich ist. Zudem kann die Reaktion naturlich auch am Resterfolgen. Man erhalt daher ein Produktgemisch. Um Monoalkylierung zuerreichen, kann man die Carbonylverbindung in einem ersten Schritt zueinem Enamin reagieren lassen. Die Dopplebindung ist dann ausreichendnukleophil um das Methyliodid anzugreifen. Das entstehende alkylierte Im-moniumion kann nun nicht ein zweites Mal nukleophil reagieren. Hydrolysemit wassriger Schwefelsaure fuhrt dann zur monoalkylierten Carbonylver-bidnung und einem Amin.
Figure 82: Uber ein Enamin erreicht man Monoalkylierung der Car-bonylverbindung
10.3.2 Aldolreaktion
Eine weitere wichtige Reaktion am α-Kohlenstoff ist die Aldolreaktion. Dabeiwird in einem ersten Schritt wiederum am α-Kohlenstoff deprotoniert. Das

10 CARBONYLVERBINDUNGEN 57
Carbanion besitzt einen starken nukleophilen Charakter und kann dahereine Carbonylverbindung angreifen. Wird der Sauerstoff des Produkts pro-toniert, gelangt man zu β-Hydroxyaldehyden oder kurz Aldolen (Al fur dieAldehyd- und ol fur die Alkoholgruppe). Naturlich sind auch β-Hydroxyketonemoglich.
Figure 83: Mechanismus der Aldolreaktion
Auch in saurem Milieu erfolgt die Aldolreaktion. Dabei wird im er-sten Schritt der Sauerstoff protoniert. Anschliessend erfolgt der nukleophileAngriff an die Carbonylverbindung uber die Doppelbindung. Nun wird derSauerstoff wieder deprotoniert, wodurch man zum Aldol gelangt. Im Gegen-satz zur Reaktion im basischen Milieu, kann das Aldol dirket weiterreagieren.Wird die Alkoholfunktion protoniert (im basischen nicht moglich), so entstehteine gute Abgangsgruppe. Es wird also Wasser abgespalten und eine Doppel-bindung unter Deprotonierung des α-Kohlenstoffs gebildet. Man bezeichnetdas entstehende Produkt als α,β-ungesattigtes Aldehyd bzw. Keton.
In der Reaktion dient H3O+ als Katalysator. Zudem wird bei der Reak-tion vom Aldol zum ungesattigten Aldehyd Wasser frei. Durch Zugabe(mehr Aldol) oder Entziehung (mehr ungesattigtes Aldehyd) von Wasserkann die Reaktion also gelenkt werden.
Verwendet man Ketone (mit unterschiedlichen Resten) als Edukt, so sindunter basischen Bedingungen naturlich stets zwei verschiedene Aldolreaktio-nen moglich, da es zwei α-Kohlenstoffe gibt. Indem man die Substituentenan den beiden α-Kohlenstoffen unterschiedlich wahlt, kann man die Depro-tonierung und somit das Hauptprodukt der Aldolreaktionen steuern (Re-giokontrolle). Hangt man an einen der α-Kohlenstoffe einen Phenylrest, sowird dort die Deprotonierung sterisch ungunstiger. Setzt man eine sterischanspruchsvolle Base (z.B. LDA) ein, so wird am anderen Kohlenstoff depro-toniert. Die entstehende Doppelbindung im Enolat ist aber in diesem Fall

10 CARBONYLVERBINDUNGEN 58
Figure 84: Aldolreaktion in saurem Milieu
tiefer substituiert, also energetisch ungunstiger, als die Doppelbindung imanderen Fall. Man benennt daher die beiden Enolate als kinetisches Enolat(energetisch ungunstiger aber schneller ”herstellbar”) und thermodynamis-ches Enolat (energetisch gunstiger, jedoch durch sterische Hinderung ver-langsamte Herstellung).
Will man das thermodynamische Enolat erhalten, so sollte man ster-isch wenig anspruchsvolle Basen (z.B. NaOCH3) verwenden. Zudem kannman die Reaktion langer laufen lassen, so dass sich das thermodynamischeGleichgewicht einstellen kann.
Bei der Aldolreaktion entstehen haufig Produktgemische, welche nichterwunscht sind. Das Phanomen ist auf sogenannte Kreuzkupplungen zuruckzufuhren.Diese treten auf, wenn man zwei Aldehyde miteinander reagieren lasst. Beze-ichnen wir die Edukte als A und B, so werden beide Enolate A− und B−
auftreten. Nun sind vier unterschiedliche nukleophile Angriffe moglich: A−
mit A, A− mit B, B− mit A und B− mit B. Wir erhalten nach der Pro-tonierung als 4 Produkte: AA, AB, BA und BB (AB und BA sind unter-schiedliche Stoffe!). Fur eine ausfuhrliche Reaktion s. Buch p. 809.
Will man nur ein Produkt, so muss man speziell vorgehen. Entwedertragt ein Partner am α-Kohlenstoff keine H-Atome oder einer der Reak-tionspartner ist sterisch anspruchsvoll. Im ersten Fall kann nur einer derReaktionspartner deprotoniert werden. Setzt man den Reakktionspartner,

10 CARBONYLVERBINDUNGEN 59
Figure 85: Regiokontrolle bei der Aldolreaktion
welcher nicht deprotoniert werden kann, zudem im uberschuss ein, so wirddieser Stoff immer als Akzeptor wirken. Man erhalt somit fast auschliesslichdas gewunschte Produkt. Im zweiten Fall ist der nukleophile Angriff beieinem Reaktionspartner aufgrund der sterischen Hinderung (z.B. -C(CH3)3)unmoglich. Gibt man den zweiten Stoff langsam zum Reaktionsgemisch,wird verhindert, dass dieser mit sich selbst reagiert. Auch in diesem Fallwird man beinahe vollstandig zum gewunschten Produkt gelangen.
10.3.3 Michael Reaktion, Robinson Annelierung Knoevenagel
Vorher haben wir α,β-ungesattigte Carbonylverbindungen kennengelernt.Sie haben die spezielle Eigenschaft, dass ein Nukleophil nicht nur am Car-bonylkohlenstoff angreiffen kann, sondern auch am β-Kohlenstoff. Verwen-det man als Nukleophil das Enolat von β-Dicarbonylverbindungen, so wirddie Addition am β-Kohlenstoff bevorzugt. Man bezeichnet diese Addition-sreaktion als Michael Reaktion.
Zu beachten ist, dass beim α,β-ungesattigten Keton die Ladung vomβ-Kohlenstoff zum Sauerstoff ”weitergeleitet” wird, was die Addition erstmoglich macht (Resonanzstabilisierung). Man bezeichnet deshalb die α,β-ungesattigte Carbonylgruppe auch als Michaelsystem. Wurde das Enolatdirekt am Carbonylkohlenstoff angreifen, ware die Ladung ausschliesslichauf dem Sauerstoff und nicht resonanzstabilisiert. Allgemein gilt, dass beider Michael Reaktion stets 1,5-Dicarbonylverbidngungen entstehen.
Genutzt wird die Michael Reaktion unter anderem in der Robinson An-nelierung. In der organischen Chemie ist es von zentraler Bedeutung neueC-C-Bindungen zu bilden. Sowohl die Michael Reaktion, als auch die Al-dolreaktion konnen dies. In der Robinson Annelierung werden sie daher

10 CARBONYLVERBINDUNGEN 60
Figure 86: Mechanismus der Michael Reaktion
hintereinander ausgefuhrt, was uns bereits zu relativ komplexen organis-chen Strukturen fuhren kann. Als Edukte werden ein Keton und eine α,β-ungesattigte Carbonylverbidnung verwendet. Sie reagieren in einer MichaelReaktion zu einer 1,5-Dicarbonylverbindung. Die Aldolreaktion erfolgt nunintramolekular (also innerhalb des Molekuls), was zum Ringschluss fuhrt.
Figure 87: Schritte der Robinson Annelierung
In der Robinson Annelierung entsteht immer ein Ring. Im letzten Schrittder Reaktion wird lediglich noch Wasser abgespalten, wodurch man zumneuen ringformigen α,β-ungesattigte Keton gelangt.
Eine weitere Reaktion ist die sogenannte Knoevenagel Reaktion. Dabeireagiert ein Aldehyd oder Keton ohne α-Wasserstoffe mit einer β-Dicarbonylverbindung.
Die Reaktion verlauft analog zur Aldolreaktion. Interessant ist hinge-

10 CARBONYLVERBINDUNGEN 61
Figure 88: Mechanismus der Knoevenagel Reaktion
gen der letzte Schritt, bei dem Wasser abgespalten wird. Dies scheintim ersten Moment energetisch nicht sinnvoll. Betrachtet man jedoch dieAnordnung der Doppelbindungen, so sieht man, dass nach der Wasserab-spaltung alle Doppelbindungen konjugiert sind. Die π-Elektronen sind fol-glich vom Phenylrest bis zu den beiden Sauerstoffen verstrichen. Dies hateine betrachtliche Stabilisierung des Molekuls zur Folge, was die Wasserab-spaltung erklart.

A RANGORDNUNG WICHTIGER GRUPPEN 62
A Rangordnung wichtiger Gruppen
Verbindungsklasse Formel Vorsilbe EndungKationen -NR3(+), -onium
-OR2(+),-SR2(+)
Carbonsauren -COOH carboxy -carbonsaureSulfonsauren -SO3H sulfo -sulfonsaureCarbonsauresalze -COOMe Me-carboxylato -Me-carboxylatSaureanhydrid -CO-O-CO- - -carbonsaureanhydridCarbonsaureester -COOR R-oxycarbonyl ..carbonsaure..R-esterCarbonsaurehalogenid -CO-X halogenformyl -carbonylhalogenidCarbonsaureamid -CO-NH2 carbamoyl -carboxyamid
-amidSulfonsaureamid -SO2NH2 sulfamoyl -sulfonamidNitrile -CN cyano -carbonitril
-nitrilAldehyde -CHO formyl -carbaldehyd
-alThioaldehyde -CHS thioformyl -carbothialdehydKetone -C=O oxo -onThioketone -S=O thioxo -thionAlkohole, Phenole -OH hydroxy -olAlkoholate Me− Me-oxido Me-olatHydroperoxide -OOH hydroperoxy -hydroperoxidAmine -NH2 amino -aminImine =NH imino -iminAlkin ≡ C ..in -inAlken =C ..en -enAlkan -C ..yl -an
Table 7: Rangordnung wichtiger funktioneller Gruppen
Die vollstandige Tabelle ist unter http://www.kfunigraz.ac.at/ipcwww/Elearning/nomenklatur/grundlagen/seite3text.htmzu finden.

B VERZEICHNIS DER BILDER UND REAKTIONEN 63
B Verzeichnis der Bilder und Reaktionen
List of Figures
1 Elektronenkonfiguration des Kohlenstoffs . . . . . . . . . . . 12 Beispiele von Ionen . . . . . . . . . . . . . . . . . . . . . . . . 23 Molekule mit kovalenten Bindungen . . . . . . . . . . . . . . 24 Potentialkurve von zwei H-Atomen als Funktion des Kernab-
stands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 Dipolmomente: H2O = 1.5 D, HCl = 1.1 D, CO2 = 0.0 D . . 36 Molekulorbitaldiagramm des Wasserstoffs . . . . . . . . . . . 47 Tetraederform des Methans . . . . . . . . . . . . . . . . . . . 48 sp3-Hybridisierung des Kohlenstoffs . . . . . . . . . . . . . . . 59 sp2-Hybridisierung des Kohlenstoffs am Beispiel von Ethen . 510 sp-Hybridisierung des Kohlenstoffs am Beispiel von Ethin . . 611 Reaktion von HCl in Wasser . . . . . . . . . . . . . . . . . . . 712 Symbolische Saure-Basen-Reaktion . . . . . . . . . . . . . . . 713 pKa von Essigsaurederivaten und induktiver Effekt . . . . . . 914 Ethanolat und mesomere Grenzstrukturen des Acetats . . . . 915 Illustration von Lewis-Sauren und -Basen . . . . . . . . . . . 1016 Essigsaure und Glycerin . . . . . . . . . . . . . . . . . . . . . 1117 Essigsaure und Glycerin mit Namen nach IUPAC . . . . . . . 1218 Zwei weiter Beispiele fur Nomenklatur nach IUPAC . . . . . 1219 Zwei mogliche Konformationen des Ethans . . . . . . . . . . . 1320 Newman Projektionen der beiden Ethankonfomere . . . . . . 1321 Energiediagramm des Ethans als Funktion des Torsionswinkels 1422 Konformere des Butans . . . . . . . . . . . . . . . . . . . . . 1523 Energie der moglichen Butankonformere als Funktion des Tor-
sionswinkels . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1524 Strukturformel und Newman Projektion des Propans . . . . . 1625 Strukturformel und raumliche Struktur des Butans . . . . . . 1626 Strukturformel und zwei mogliche Konformere des Pentans . 1727 Strukturformel und Sesselkonformation des Hexans . . . . . . 1728 Ringinversion und Energiediagramm das Cyclohexans . . . . 1829 Die Methylgruppe hat in aequitorialer Position einen grosseren
Abstand zu den C-Atomen des Rings . . . . . . . . . . . . . . 1830 Die zwei Stereoisomere von Pent-2-en . . . . . . . . . . . . . 1931 Beispiel von cis/trans-Isomerie . . . . . . . . . . . . . . . . . 2032 Bei einem chiralen C-Atom kann man durch Spiegeln die
Enantiomere erhalten . . . . . . . . . . . . . . . . . . . . . . 2033 Korrekte Benennung des Enantiomerenpaars . . . . . . . . . 2134 Bei zwei chiralen C-Atomen erhalt man 4 Stereoisomere . . . 2235 Die Molekule der ersten Spieglung sind identisch . . . . . . . 22

LIST OF FIGURES 64
36 OH− als Nukleophil greift das partiell positiv geladene C desMethyliodids an . . . . . . . . . . . . . . . . . . . . . . . . . . 23
37 Beispiel eines nukleophilen Angriffs mit Inversion der Stereo-chemie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
38 Allgemeine Reaktionsgleichung einer nukleophilen Substitution 2439 Mechanismus der SN2-Reaktion . . . . . . . . . . . . . . . . . 2440 Energiediagramm der SN2-Reaktion . . . . . . . . . . . . . . 2541 Sterische Hinderung fuhrt zu einer Verlangsamung der Reaktion 2642 Reaktion eines tertiaren C-Atoms mit Wasser . . . . . . . . . 2743 Die Abspaltung der Abgangsgruppe bestimmt die Reaktion-
sgeschwindigkeit . . . . . . . . . . . . . . . . . . . . . . . . . 2744 Energiediagramm der SN1-Reaktion mit einem Zwischenpro-
dukt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2845 Beide Angriffe sind gleich wahrscheinlich: Racemisierung . . . 2846 Die Verteilung der positiven Ladung uber mehrere Zentren
stabilisiert das Molekul . . . . . . . . . . . . . . . . . . . . . . 2947 Es entsteht nicht ausschliesslich das Substiutionsprodukt . . . 3048 Durch Zugabe einer starken Base wird die Ausbeute an Alken
erhoht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3049 Die Doppelbindung des Seitzev Produkt ist dreifach substi-
tuiert und deshalb stabiler . . . . . . . . . . . . . . . . . . . . 3150 Durch Stereospezifitat entsteht ausschliesslich das E-Isomer . 3251 Stehen das zu abstrahierende Proton und die Abgangsgruppe
anti, so liegt eine gestaffelte Konformation vor. . . . . . . . . 3252 Mechanismus der Additionsreaktion und allgemeine Reak-
tionsgleichung . . . . . . . . . . . . . . . . . . . . . . . . . . . 3453 Energiediagramm einer Additionsreaktion mit Carbokation
als Zwischenprodukt . . . . . . . . . . . . . . . . . . . . . . . 3554 Nur die Addition uber das stabilere Carbokation erfolgt . . . 3555 Mithilfe von H3O+ als Katalysator kann aus einem Alken ein
Alkohol synthetisiert werden . . . . . . . . . . . . . . . . . . . 3656 Die Addition von BH3 erfolgt uber einen zyklischen Ubergangszustand 3657 Zwei weitere Additionen und Oxidation mit Wasserstoffper-
oxid fuhrt zum Anti-Markovnikov Produkt . . . . . . . . . . . 3758 Umlagerung fuhrt zu einem stabileren tertiaren Carbokation . 3859 Reaktion nach SN2 fuhrt zu Stereospezifitat . . . . . . . . . . 3960 Ein zyklischer Ubergangszustand macht die Addition eines
Sauerstoffs moglich . . . . . . . . . . . . . . . . . . . . . . . . 3961 Delokalisierte π-Elektronen uber und unter dem Ring und
zwei mogliche Grenzstrukturen . . . . . . . . . . . . . . . . . 4162 Mechanismus der elektrophilen Substitution an einem Aro-
maten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4363 Beispiele von elektrophilen Subtitutionen . . . . . . . . . . . 4364 Friedel-Kraft-Alkylierung . . . . . . . . . . . . . . . . . . . . 44

LIST OF FIGURES 65
65 Friedel-Kraft-Acylierung . . . . . . . . . . . . . . . . . . . . . 4466 Struktur von Aldehyden und Ketonen . . . . . . . . . . . . . 4567 Nukleophiler Angriff von Wasser fuhrt zu Hydraten . . . . . . 4668 Nukleophile Addition von CN− und weiterfuhrende Reaktionen 4769 Addition von Alkoholen fuhrt zu Acetalen . . . . . . . . . . . 4870 Einsetzen eines Diols als Schutzgruppe . . . . . . . . . . . . . 4871 Addition von primaren Aminen fuhrt zu Iminen . . . . . . . . 4972 scale = 0.85 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5073 Zusammenfassung der reversiblen nukleophilen Additionen . . 5074 Oxidation eines Ketons mit Lithium-Aluminium-Hydrid . . . 5175 Oxidation eines Ketons mit Methyllithium . . . . . . . . . . . 5176 Mechanismus der Wittig Reaktion . . . . . . . . . . . . . . . 5277 Oxidationsprodukte von Alkoholen . . . . . . . . . . . . . . . 5378 Mechanismus der Keto-Enol-Tautomerie . . . . . . . . . . . . 5479 Keto-Enol-Tautomerie in basischem und saurem Milieu . . . . 5480 Halogenierung von Carbonylverbindungen . . . . . . . . . . . 5581 Alkylierung von Carbonylverbindungen . . . . . . . . . . . . 5682 Uber ein Enamin erreicht man Monoalkylierung der Carbonylverbindung 5683 Mechanismus der Aldolreaktion . . . . . . . . . . . . . . . . . 5784 Aldolreaktion in saurem Milieu . . . . . . . . . . . . . . . . . 5885 Regiokontrolle bei der Aldolreaktion . . . . . . . . . . . . . . 5986 Mechanismus der Michael Reaktion . . . . . . . . . . . . . . . 6087 Schritte der Robinson Annelierung . . . . . . . . . . . . . . . 6088 Mechanismus der Knoevenagel Reaktion . . . . . . . . . . . . 61


















