
IgE-Antikörper bei blasenbildenden Autoimmundermatosen ... · Epidermolysis bullosa acquisita Die...
104
Aus der Klinik für Dermatologie und Venerologie der Universität zu Lübeck Direktor: Prof. Dr. med. D. Zillikens IgE-Antikörper bei blasenbildenden Autoimmundermatosen: Epitop-mapping beim bullösen Pemphigoid Inauguraldissertation zur Erlangung der Doktorwürde der Universität zu Lübeck - Aus der Medizinischen Fakultät - vorgelegt von Sina Katinka Dresow aus Braunschweig Lübeck 2007
Transcript of IgE-Antikörper bei blasenbildenden Autoimmundermatosen ... · Epidermolysis bullosa acquisita Die...

Aus der Klinik für Dermatologie und Venerologie
der Universität zu Lübeck
Direktor: Prof. Dr. med. D. Zillikens
IgE-Antikörper bei blasenbildenden Autoimmundermatosen:
Epitop-mapping beim bullösen Pemphigoid
Inauguraldissertation
zur
Erlangung der Doktorwürde
der Universität zu Lübeck
- Aus der Medizinischen Fakultät -
vorgelegt von
Sina Katinka Dresow
aus Braunschweig
Lübeck 2007

1. Berichterstatter: Priv.-Doz. Dr. Bernhard F. Gibbs Ph.D 2. Berichterstatter: Priv.-Doz. Dr. med. Holger Hennig
Tag der mündlichen Prüfung: 25.11.2008
Zum Druck genehmigt. Lübeck, den 25.11.2008
gez. Prof. Dr. med. Werner Solbach
- Dekan der medizinischen Fakultät -

meinen Eltern

INHALTSVERZEICHNIS I
Inhaltsverzeichnis I
Abkürzungen III
1. Einleitung 1
1.1. Struktur der Epidermis und Dermis 1
1.2. Bullöse Autoimmundermatosen 3
1.3. Das bullöse Pemphigoid 5
1.3.1. Epidemiologie und Klinik 5
1.3.2. Diagnose 6
1.3.3. Pathogenese 8
1.4. IgE-Antikörper und -Rezeptoren 13
1.5. Bullöses Pemphigoid und IgE-Antikörper 17
1.6. Ziele dieser Arbeit 19
2. Materialien 21
2.1. Verwendete Laborgeräte 21
2.2. Verbrauchsmaterialien 22
2.3. Chemikalien und Farbstoffe 23
2.4. Puffer und Lösungen 24
2.5. Zellkulturmedien und Zusätze 25
2.6. Verwendete Zelllinien und Bakterienstämme 26
2.7. Patientenseren 26
2.8. Kontrollen 27
2.8.1. Negativkontrollen 27
2.8.2. Positivkontrollen 27
2.9. Antikörper 28
3. Methoden 29
3.1. Zellkultur 29
3.1.1. HaCaT-Zellkultur 29
3.1.1.1. Auftauen von eingefrorenen Zellen 29
3.1.1.2. Subkultivierung der Zellen 30

INHALTSVERZEICHNIS II
3.1.1.3. Bestimmung der Zellzahl 31
3.1.1.4. Einfrieren von Zellen 31
3.2. Expression der rekombinanten GST-Fusionsproteine
hBP180 ICD-A, -B, -C, -D 32
3.2.1. Primärkultur 33
3.2.2. Sekundärkultur 33
3.2.3. Proteinextraktion 34
3.2.4. Affinitätsaufreinigung 34
3.2.5. Dialyse und Gefriertrocknung 37
3.3. Proteinisolierung 37
3.3.1. Proteinisolierung aus dem Mediumüberstand kultivierter Keratinozyten 37
3.3.2. Herstellung der Keratinozytenextrakte 38
3.3.3. Photometrische Messung der Proteinkonzentration 38
3.4. Westernblotanalyse/Epitop-mapping 39
3.4.1. SDS-Polyacrylamid-Gelelektrophorese 39
3.4.2. Commassie-Brilliant-Blue- und Silberfärbung der SDS-Gele 40
3.4.3. Western-Blot 41
3.4.4. Immundetektion der transferierten Proteine/Epitop-mapping 42
3.4.5. Auswertung der Bandenmuster 43
4. Ergebnisse 44
4.1. Expression und Affinitätsaufreinigung von rekombinanten GST-
Fusionsproteinen 44
4.2. Erstellung des Patientenkollektivs und der Negativkontrollen 46
4.3. Erhöhte Gesamt-IgE-Spiegel im Serum von BP-Patienten 47
4.4. IgE- und IgG-Autoantikörper-Reaktivität gegen die intrazelluläre
Domäne von BP180 und NC16A1-5 50
4.5. IgE-Autoantikörper-Reaktivität gegen LAD-1 aus dem Mediumüber-
stand kultivierter Keratinozyten 55
4.6. IgE-Autoantikörper-Reaktivität gegen BP180 und BP230 in
Keratinozytenextrakten 57
4.7. IgE-Autoantikörper-Reaktivität gegen den C-terminalen Abschnitt von
BP180 (BP4575) 59

INHALTSVERZEICHNIS III
4.8. Übersicht der Epitoperkennung für IgE auf dem intrazellulären und
extrazellulären Abschnitt von BP180 61
5. Diskussion 64
5.1. Expression und Aufreinigung der verschiedenen rekombinanten
BP180 Polypeptide 66
5.2. IgE- und IgG-Autoreaktivität gegen die extrazelluläre Domäne von
BP180 67
5.2.1. Nachweis von IgE-Autoreaktivität gegen den C-terminalen Abschnitt
von BP180 (BP4575) und gegen LAD-1 69
5.3. IgG-Autoreaktivität gegen einen immundominanten Abschnitt auf der
intrazellulären Domäne von BP180 70
5.4. IgE-Autoreaktivität gegen die intrazelluläre Domäne von BP180 71
5.5. IgE-Autoreaktivität gegen das gesamte BP180-Molekül und BP230 74
5.6. IgE-Autoantikörper besitzen mehrere Epitope auf dem BP180-
Molekül 75
5.7. Ausblick 77
6. Zusammenfassung 79
7. Literaturverzeichnis 81
8. Anhang 91
8.1. Abbildung I: Aufbau des pGEX-1 Vektors 91
8.2. Tabelle I: Primer Sequenzen für die PCR zur Herstellung von cDNA-
Fragmenten des humanen Typ XVII Kollagens/BP180 91
8.3. Tabelle ll: Status der Kontrollpersonen 92
8.4. Tabelle III: Immunoblot-Ergebnisse der Kontrollpersonen 92
Erklärung 93
Danksagung 94
Lebenslauf 95

ABKÜRZUNGEN IV
Abkürzungen
Abb. Abbildung AS Aminosäure(n) BMZ Basalmembranzone BP bullöses Pemphigoid BPAg1 bullöses Pemphigoid Antigen 1 (BP180, Synonym: Kollagen Typ
XVII) BPAg2 bullöses Pemphigoid Antigen 2 (BP230) BSA Bovines Serumalbumin cpm Impulse pro Minute (Counts per minute) C-Terminus/ Carboxy-Terminus/terminal terminal DAB Diaminobenzidin ddH2O bidestilliertes Wasser DEJ dermo-epidermale Junktionszone DIF Direkte Immunfluoreszenz DMSO Dimethylsulfoxid EBA Epidermolysis bullosa aquisita E. coli Eschericha coli EDTA Ethylen-Diamin-Tetra-Acetat ELISA Enzymatischer Immuntest (Enzyme Linked Immunsorbent Assay) et al. et alii FKS Fötales Kälberserum GST Glutathion-S-Transferase h Stunde(n) HRP Horse Radish Peroxidase IF Immunfluoreszenz Ig Immunglobulin IIF Indirekte Immunfluoreszenz IL Interleukin IPTG Isopropyl-ß-D-Thiagalactopyranosid KCE Keratinozytenextrakt kDa Kilodalton LAD Lineare IgA Dermatose LBM Luria-Bertani (nach Miller)- Medium mA Milliampere mAk monoklonaler Antikörper min Minute(n) ml Milliliter (m)M (milli)molar NaCl Natriumchlorid NC nicht-kollagenös nm Nanometer N-Terminus/ Amino-Terminus/terminal terminal PBS Phosphat-buffered saline PG Pemphigoid gestationes PMSF Phenyl-Methyl-Sulfonyl-Fluorid PV Pemphigus vulgaris rpm Revolutions per minute

ABKÜRZUNGEN V
RT Raumtemperatur RZB relative Zentrifugalbeschleunigung SDS(-PAGE) Sodium-Dodecyl-Sulfat(-Polyacrylamid-Gelelektrophorese) Sek. Sekunden SSS Salt-Split-Skin-Technik Tab. Tabelle TBST tris-buffered saline plus tween TEMED N,N,N’,N’-Tetramethylethylendiamin V Volt VP Vernarbendes Pemphigoid µl Mikroliter °C Grad Celsius

EINLEITUNG 1
1. Einleitung
Autoimmunität ist eine normale Eigenschaft des Immunsystems und kein patholo-
gischer Zustand per se. Auch im gesunden Organismus können Antikörper, B- und
T-Zellen mit Spezifität gegen körpereigene Antigene nachgewiesen werden. In
diesem Fall schützen Mechanismen der peripheren Toleranz, z. B. durch das Feh-
len costimulatorischer Signale, vor einer autoimmunologischen Zerstörung der
Gewebe und Zellen. Die zentrale Toleranz beschreibt die Entwicklung der T-Zellen
im Thymus, wo sie durch positive und negative Selektion die Fähigkeit zur Unter-
scheidung von Selbst- und Nicht-Selbst erlernen. Fehler in der zentralen und peri-
pheren Toleranz führen zu Autoimmunkrankheiten. Diese sind gekennzeichnet
durch nichtinfektiöse Entzündungszustände, die zur lokalen Organzerstörung
(z. B. insulinabhängiger Diabetes) oder zu systemischen entzündlichen Erkran-
kungen (z. B. bullöses Pemphigoid, Vaskulitiden) führen. Eine Vielzahl von Auto-
immunkrankheiten, die unterschiedliche Organe betreffen sind beschrieben. Sero-
logisch sind diese Erkrankungen oft mit dem vermehrten Auftreten von Autoanti-
körpern verbunden. Die Inzidenz dieser Erkrankungen liegt bei etwa 3-4%, wobei
Frauen häufiger als Männer erkranken.
Die Immunpathologie der Autoimmunerkrankungen ist sehr unterschiedlich. Es
gibt primäre Antikörper-vermittelte, Immunkomplex-vermittelte sowie T-Zell-
vermittelte Autoimmunkrankheiten. Laut Definition spricht man von einer Auto-
immunerkrankung wenn folgende drei Kriterien erfüllt sind:
1. Klinische Hinweise, 2. Direkter Beweis: passiver Transfer von Antikörpern, 3.
Indirekter Beweis: Immunisierung mit einem Antigen.
1.1. Struktur der Epidermis und Dermis
Die Haut stellt mit 1,5 bis 2 m2 das flächengrößte und schwerste (bis 10 kg) sowie
funktionell vielseitigste Organ des Körpers dar. Sie dient sowohl als Barriere ge-
gen äußere Einflüsse als auch zur Aufrechterhaltung der physikalischen Unver-
sehrtheit. Die Haut schützt den Körper vor chemischen, mechanischen und ther-
mischen Schäden, ist an der Regulation von Körpertemperatur und Wasserhaus-
halt beteiligt und übernimmt Aufgaben in der Immunabwehr.
Die Epidermis besteht aus mehrschichtigem verhornten Plattenepithel, welches
die unterliegende Dermis bedeckt. Die Epidermis wird hauptsächlich aus Keratino-
zyten gebildet, weiterhin findet man Melanozyten, Merkel-Zellen als Mechanore-

EINLEITUNG 2
zeptoren sowie Lymphozyten. Als antigenpräsentierende Zellen fungieren sog.
Langerhanszellen, die noch inaktive Dendritische Zellen darstellen. Sie befinden
sich in der Epidermis der Haut und in Schleimhäuten und differenzieren sich nach
dem ersten Antigen-Kontakt zu Dendritischen Zellen, die das Antigen in der Der-
mis den T-Lymphozyten präsentieren. Die Dermis besteht vorwiegend aus kom-
paktem, fibroelastischem Bindegewebe, in das ein Netzwerk aus Blutkapillaren
und Nerven, sowie Hautanhangsgebilde integriert sind. Epidermis und Dermis sind
durch die dermo-epidermale Junktionszone (DEJ) fest miteinander verbunden. Die
DEJ ist elektronenmikroskopisch aus fünf unterscheidbaren Zonen aufgebaut (sie-
he Abb.1). Auf der epidermalen Seite liegen die Hemidesmosomen der basalen
Keratinozyten, es folgen die Lamina lucida, die Lamina densa, die Sublamina den-
sa und die Verankerungsfibrillen der papillären Dermis. Die Hemidesmosomen der
basalen Keratinozyten sind scheibchenförmige Plaques, die die Proteine BP230
und Plektin beinhalten. Es bestehen Interaktionen zwischen den intrazellulären
Anteilen der transmembranösen Ankerfilamente BP180 und 64-Integrin und den
Proteinen der hemidesmosomalen Plaques (BP230 und Plektin). Der extrazellulä-
re Anteil von BP180 erstreckt sich über die Lamina lucida in die Lamina densa und
interagiert mit Laminin 5, welches den Kontakt zu Verankerungsfibrillen der Lami-
na/Sublamina densa vermittelt. Die Verankerungsfibrillen bestehen hauptsächlich
aus Typ 7 Kollagen. Sie ragen von der Lamina densa über die Sublamina densa
bis in die papilläre Dermis (Borradori and Sonnenberg, 1999; Georgi et al., 2001).
Die Vernetzung der Keratinozyten in der Epidermis erfolgt über Desmoso-
men. Sie bestehen aus intrazellulär gelegenen Plaques, in die zelleigene Zytoke-
ratinfilamente und transmembranöse Cadherinen wie Desmogleine (Dsg) und
Desmocolline (Dsc) verankert sind. Diese desmosomalen Cadherine besitzen zu-
dem extrazelluläre Anteile über die die einzelnen Keratinozyten miteinander ver-
bunden werden.

EINLEITUNG 3
Lamina
lucida
Lamina
densa
Sublamina
densa
Basale
Keratinozyten
Dsg 1
Dsg 3
Dsc 1
DPZFHD
Kollagen Typ lV
Kollagen Typ Vll
BP180
BP230Plektin
ß4
664-Integrin
Laminin 5 p200
Lamina
lucida
Lamina
densa
Sublamina
densa
Basale
Keratinozyten
Dsg 1
Dsg 3
Dsc 1
DPZFHD
Kollagen Typ lV
Kollagen Typ Vll
BP180
BP230Plektin
ß4
664-Integrin
Laminin 5 p200
Abb. 1: Schematische Darstellung des Desmosoms und der Autoantigene der dermo-epidermalen Junktionszone. Dsg:Desmoglein 1, 3 und Dsc:Desmocollin. DP:desmosomaler Pla-que, HD:Hemidesmosomen, ZF:Zytokeratinfilamente. Plektin und BP230 sind intrazelluläre hemi-
desmosomale Proteine. Plektin interagiert mit der 4-Kette des 64-Integrin, das Kontakt zu Lami-nin 5 hat. Das transmembranöse BP180 interagiert mit BP230 und Laminin 5. Laminin 5 vermittelt den Kontakt zu Verankerungsfibrillen in der Lamina/Sublamina densa. p200 ist ein noch nicht voll-ständig molekularbiologisch charakterisiertes Protein der tiefen Lamina lucida, welches das Anti-gen einer weiteren Sonderform der Pemphigoiderkrankungen darstellt. (Abb. nach Dr. C. Sitaru)
1.2. Bullöse Autoimmundermatosen
Die bullösen Autoimmundermatosen sind organbezogene Autoimmun-
erkrankungen, die durch das Auftreten von Autoantikörpern gegen Strukturpro-
teine der Haut gekennzeichnet sind. Diese Strukturproteine sind in Form von
Desmosomen an die interzelluläre Zellhaftung der Keratinozyten sowie als Hemi-
desmosomen an der Haftung der Epidermis an der Basalmembran und Dermis
beteiligt (Sitaru et al., 2004; Yancey, 1995). Die genaue Ätiologie der Autoantikör-
perbildung bei bullösen Autoimmundermatosen ist immer noch unklar.
Die Einteilung der Autoimmundermatosen erfolgt anhand der betroffenen Ziel-
struktur.
Man unterscheidet dabei 4 Hauptgruppen:
1. Pemphiguskrankheiten:
Pemphigus vulgaris
Pemphigus foliaceus
Paraneoplastischer Pemphigus
IgA-Pemphigus

EINLEITUNG 4
2. Pemphigoidkrankheiten:
Bullöses Pemphigoid
Pemphigoid gestationis
Lineare IgA-Dermatose
Vernarbendes Pemphigoid
Lichen planus pemphigoides
3. Dermatitis herpetiformis Duhring
4. Epidermolysis bullosa acquisita
Die blasenbildenden Autoimmundermatosen unterscheiden sich zum einen in den
Zielstrukturen der Autoantikörper, zum anderen in der pathogenetischen Wirkung
dieser Autoantikörper.
Bei den Pemphiguskrankheiten führt die Bindung der Autoantikörper an Desmo-
gleine zu einem direkten Funktionsverlust der Desmosomen. Dadurch kommt es
zur Auflösung der Zell-Zell-Adhäsionen und zur Ausbildung schlaffer intraepider-
maler Blasen ohne wesentliche Entzündungsreaktion (Amagai et al., 1994; Ama-
gai et al., 1992; Rock et al., 1990).
Im Unterschied dazu führen Autoantikörper beim bullösen Pemphigoid und
der Epidermolysis bullosa acquisita zu einer Entzündungsreaktion, die zu einer
subepidermalen Blasenbildung führt. Die Entzündungsreaktion wird dabei durch
den Fc-Anteil der Autoantikörper vermittelt. So haben F(ab)2-Fragmente im Unter-
schied zu kompletten Antikörpern, sowohl in vitro als auch in vivo keine patho-
genetische Wirkung (Sitaru and Zillikens, 2005). Das Bullöse Pemphigoid ist eine
autoimmune blasenbildende Hauterkrankung, die durch Ablagerungen von IgG
und Komplement C3 an der kutanen Basalmambran, Infiltration der oberen Dermis
durch inflammatorische Zellen und Ablösung der basalen epidermalen Schicht von
der Dermis gekennzeichnet ist.
Die Lineare IgA-Dermatose (LAD) ist eine seltene subepidermale blasenbil-
dende Autoimmunerkrankung mit Ähnlichkeit zum bullösen Pemphigoid. Sie ist
gekennzeichnet durch lineare IgA-Ablagerungen an der DEJ.

EINLEITUNG 5
1.3. Das bullöse Pemphigoid
1.3.1. Epidemiologie und Klinik
Das bullöse Pemphigoid (BP) ist die mit Abstand häufigste Form kutaner blasen-
bildender Autoimmunerkrankungen. Seine Inzidenz wird für Deutschland mit jähr-
lich 6 Neuerkrankungen pro 100.000 Einwohnern angegeben, mit einem Anstieg
ab dem 7. Lebensjahrzehnt (Budinger et al., 1998; Zillikens et al., 1995). In der
Gruppe der über 90-jährigen liegt die Inzidenz bei ca. 40 pro 100.000 Einwohnern
(Jung et al., 1999; Schmidt et al., 2000a). Die Erkrankung ist weltweit verbreitet
und zeigt keine ethnische, geographische oder geschlechtsgebundene Prädisposi-
tion. Beim BP besteht eine Assoziation mit dem HLA-DQB1*0301 Komplex (Bu-
dinger et al., 1998; Delgado et al., 1996). Selten sind Kinder mit der Form des ju-
venilen Pemphigoids betroffen (Chimanovitch et al., 2000). Eine Induktion des bul-
lösen Pemphigoids durch Medikamente ist insgesamt weniger gut belegt als beim
Pemphigus (Smith et al., 1993; Venning and Wojnarowska, 1995). Hinsichtlich der
Assoziation des BP mit malignen Tumoren gibt es widersprüchliche Studienergeb-
nisse (Ahmed et al., 1977; Venning and Wojnarowska, 1990).
Zu Beginn der Erkrankung bestehen häufig erythematöse, z. T. urtikarielle Haut-
veränderungen, verbunden mit starkem Juckreiz (Hadi et al., 1988). Manchmal ist
der Juckreiz einziges Symptom, bevor nach Monaten Blasen auftreten (Alonso-
Llamazares et al., 1998). Später entwickelt sich ein klinischer Befund mit prallen,
teils hämorrhagischen Blasen sowie rupturierten Blasen mit unregelmäßig konfigu-
rierten Erosionen und Verkrustungen auf teils normal aussehender Haut (poikilo-
dermatisches Bild). Im Vergleich zum Pemphigus vulgaris sind die Blasen viel ge-
spannter und widerstandsfähiger, da ihre Blasendecke von der gesamten Epider-
mis gebildet wird. Häufig treten die Bläschen/Blasen in Gruppen an einer Körper-
stelle auf (Abb. 2). Prädilektionsstellen der symmetrischen Eruptionen sind die
seitliche Halspartie, die Axillen, die Beugeseiten der Oberarme und Oberschenkel
(Sison-Fonacier and Bystryn, 1986) sowie die Nabelregion. In einigen Fällen sind
auch die Palmae und Plantae im Sinne eines dyshidrosiformen Hand- und Fußek-
zems betroffen. Die Schleimhäute zeigen seltener Blasen (Kippes et al., 1999).
Das BP kann auch unter dem Bild eines klinischen Ekzems, einer Urtikaria oder
einer Prurigo simplex subacuta ganz ohne Blasenbildung verlaufen (Kippes et al.,
1999; Wever et al., 1995).

EINLEITUNG 6
Zum häufig schlechten Allgemeinbefinden der Patienten tragen Schwäche, Fieber,
Appetitlosigkeit und Gewichtsverlust bei. Die Laborbefunde fallen oft durch eine
sekundäre Anämie, Leukozytose mit leichter Eosinophilie sowie einer erhöhten
Blutsenkungsgeschwindigkeit (BSG) auf.
Das BP verläuft chronisch rezidivierend und meist selbstlimitierend mit einer
Dauer von zwei bis zu fünf Jahren (Hadi et al., 1988; Korman, 1987; Wojnarowska
et al., 2002; Wojnarowska et al., 2001). Exazerbationen der Erkrankung verlaufen
meist milder als die Initialperiode (Ghohestani et al., 2001; Korman, 1998). Nach
Abheilung der Läsionen bleiben häufig inflammatorische Hyperpigmentierungen
bzw. postbullöse Milien zurück.
Abb. 2: Klinischer Befund beim bullösen Pemphigoid: Pralle, teils hämorrhagische Blasen, Erosionen und Krusten auf erythematösem Grund am Unterarm eines Patienten (a). BP unter dem klinischen Bild eines dyshidrosiformen Handekzems (b). Sogenannte prämonitorische Variante, die sich über Jahre unter dem Bild eines Ekzems manifestiert (c). Quelle: mit freundlicher Genehmigung aus dem Archiv der Klinik für Dermatologie und Venerologie der Universität zu Lübeck.
1.3.2. Diagnose
Neben dem klinischen Bild und der dermatohistopathologischen Untersuchung
sind die direkte und indirekte Immunfluoreszenz für die Diagnostik bullöser Auto-
immundermatosen von Bedeutung. Als wichtigster Beweis für bullöse Autoimmun-
dermatosen gilt der Nachweis einer Ablagerung von Autoantikörpern in der Haut.
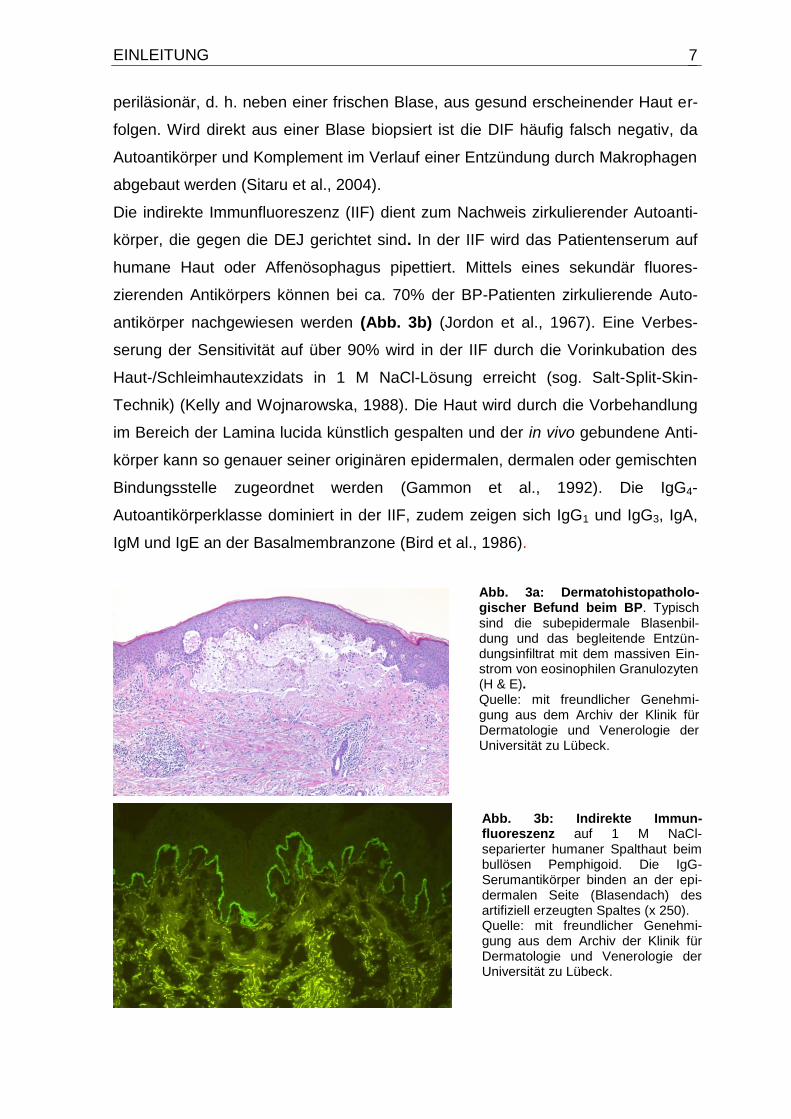
Beim BP findet sich histopathologisch eine subepidermale Spaltbildung mit dich-
tem Entzündungsinfiltrat (Abb. 3a).
Die direkte Immunfluoreszenz (DIF) periläsionaler Haut zeigt bei 90% der Patien-
ten eine homogene lineare Bindung von IgG und in fast allen Fällen eine C3-
Ablagerung (Kippes et al., 1999; Korman, 1998). Für die DIF sollte die Hautbiopsie
EINLEITUNG 7
periläsionär, d. h. neben einer frischen Blase, aus gesund erscheinender Haut er-
folgen. Wird direkt aus einer Blase biopsiert ist die DIF häufig falsch negativ, da
Autoantikörper und Komplement im Verlauf einer Entzündung durch Makrophagen
abgebaut werden (Sitaru et al., 2004).
Die indirekte Immunfluoreszenz (IIF) dient zum Nachweis zirkulierender Autoanti-
körper, die gegen die DEJ gerichtet sind. In der IIF wird das Patientenserum auf
humane Haut oder Affenösophagus pipettiert. Mittels eines sekundär fluores-
zierenden Antikörpers können bei ca. 70% der BP-Patienten zirkulierende Auto-
antikörper nachgewiesen werden (Abb. 3b) (Jordon et al., 1967). Eine Verbes-
serung der Sensitivität auf über 90% wird in der IIF durch die Vorinkubation des
Haut-/Schleimhautexzidats in 1 M NaCl-Lösung erreicht (sog. Salt-Split-Skin-
Technik) (Kelly and Wojnarowska, 1988). Die Haut wird durch die Vorbehandlung
im Bereich der Lamina lucida künstlich gespalten und der in vivo gebundene Anti-
körper kann so genauer seiner originären epidermalen, dermalen oder gemischten
Bindungsstelle zugeordnet werden (Gammon et al., 1992). Die IgG4-
Autoantikörperklasse dominiert in der IIF, zudem zeigen sich IgG1 und IgG3, IgA,
IgM und IgE an der Basalmembranzone (Bird et al., 1986).
Abb. 3b: Indirekte Immun-fluoreszenz auf 1 M NaCl-separierter humaner Spalthaut beim bullösen Pemphigoid. Die IgG-Serumantikörper binden an der epi-dermalen Seite (Blasendach) des artifiziell erzeugten Spaltes (x 250). Quelle: mit freundlicher Genehmi-gung aus dem Archiv der Klinik für Dermatologie und Venerologie der Universität zu Lübeck.
Abb. 3a: Dermatohistopatholo-gischer Befund beim BP. Typisch sind die subepidermale Blasenbil-dung und das begleitende Entzün-dungsinfiltrat mit dem massiven Ein-strom von eosinophilen Granulozyten (H & E). Quelle: mit freundlicher Genehmi-gung aus dem Archiv der Klinik für Dermatologie und Venerologie der Universität zu Lübeck.

EINLEITUNG 8
Für die genauere Charakterisierung der Spezifität zirkulierender Autoantikörper
werden immunserologische Untersuchungen wie Immunoblot, ELISA und Immun-
präzipitation durchgeführt (Hertl and Schuler, 2002).
Für den Immunoblot werden Extrakte aus Epidermis/Dermis, kultivierten Keratino-
zyten bzw. aus dem Überstand kultivierter Keratinozyten oder rekombinanter For-
men dieser Proteine eingesetzt. Darüber hinaus stehen verschiedene sensitive
und spezifische ELISA-Systeme zum Antikörpernachweis zur Verfügung. Unter
Verwendung einer rekombinanten Form der NC16A-Domäne wurde ein sensitiver
und spezifischer ELISA entwickelt, dessen Titer gut mit der Krankheitsaktivität
beim BP korreliert (Zillikens et al., 1997).
1.3.3. Pathogenese
Erste Hinweise auf eine immunologische Pathogenese des BP finden sich 1967 in
den Arbeiten von Jordan et al., die in der Haut und im Serum von BP-Patienten
Autoantikörper nachweisen konnten (Jordon et al., 1967). In den letzten 25 Jahren
wurde eine Vielzahl von Autoantigenen der DEJ beim BP identifiziert, darunter das
Protein BP230, welches erstmals 1981 als Zielantigen beschrieben wurde (Stanley
et al., 1981). Wenige Jahre später konnte das 180 kDa schwere transmembranöse
Ankerprotein BP180, das für die dermal-epidermale Adhäsion verantwortlich ist
(Giudice et al., 1993), als primäres Autoantigen beim BP identifiziert werden (La-
bib et al., 1986). Verschiedene weitere bullöse Erkrankungen wurden mit einer
gegen BP180 gerichteten Autoimmunantwort assoziiert, darunter vernarbendes
Pemphigoid, Pemphigoid gestationis, Schleimhautpemphigoid, Lineare IgA-
Dermatose und Lichen Planus Pemphigoid (Van den Bergh and Giudice, 2003).
Die Klonierung der cDNA für BP180 ermöglichte die weitere Charakterisierung von
Erkennungssequenzen des Autoantigens (Diaz et al., 1990; Giudice et al., 1992;
Stanley et al., 1988).
BP180, auch als Kollagen Typ XVII oder BPAG2 bezeichnet, ist ein 180 kDa gro-
ßes, integrales, transmembranöses Typ ll-Glykoprotein. Es besteht aus 1497 Ami-
nosäuren (AS) und besitzt eine N-terminale intrazelluläre Domäne (466 AS), eine
hydrophobe transmembranöse Domäne (23 AS) und eine C-terminale extrazellulä-
re Domäne (1008 AS) (Giudice et al., 1992; Zillikens and Giudice, 1999). Der ex-
trazelluläre Abschnitt enthält 15 kollagene Untereinheiten, die von 16 nicht-

EINLEITUNG 9
kollagenen (NC-) Domänen durchsetzt sind (Giudice et al., 1992; Zillikens and
Giudice, 1999) (s. Abb. 4).
Kollagen XVll/BP180 wurde 1996 erstmals aus Rinderhaut aufgereinigt und nach
Rotationsbedampfung im Elektronenmikroskop abgebildet. Strukturell besteht das
BP180 Molekül N-terminal aus einem globulären Kopf im Hemidesmosom, einem
zentralen Stab und einem flexiblen Schwanz, der extrazellulär bis in die Lamina
densa reicht (Hirako et al., 1998; Masunaga et al., 1997) und in einer Schlaufe
endet (Nonaka et al., 2000). In der menschlichen Haut liegt BP180 in zwei moleku-
laren Formen vor. Zum einen als komplettes homotrimeres BP180-Molekül, wel-
ches aus drei 180 kDa-1(XVll)-Ketten besteht. Zum anderen als proteolytisches
Spaltprodukt LAD-1 (120 kDa), das weitestgehend der Ektodomäne von BP180
entspricht (Schacke et al., 1998). Die Spaltung erfolgt durch membrangebundene
Metalloproteasen an der Oberfläche von Keratinozyten innerhalb der sechzehnten
nicht-kollagenen Domäne (NC16A), die sich extrazellulär unmittelbar an die Zell-
membran anschließt. Dabei liegt nach Franzke et al. die Spaltungsregion zwi-
schen AS 528 und 547, während Hirako et al. die Position des N-Terminus bei AS
524 oder 528 vermuten (Franzke et al., 2002; Hirako et al., 2003). Dieses Freiset-
zen der Ektodomäne von BP 180 (Shedding) wird von Proteinasen der ADAMs
Familie katalysiert (Sheddasen). Da der Proteinkinase C-Aktivator PMA (Phorbol-
ester) das Shedding von BP180 stimuliert, ist anzunehmen, dass der Prozess über
bestimmte Proteinkinase C-abhängige Signaltransduktionswege gesteuert wird
(Blumer and Johnson, 1994). Dabei handelt es sich entweder um den MAP-
Kinase-Weg (mitogen activated protein kinase), der über Wachstumsfaktoren und
Zytokine wie TNF- und IL-1 aktiviert wird oder um den SAPK-Weg (stress activa-
ted protein kinase), der durch Zellstresseinwirkung wie z. B. UV-Strahlung ausge-
löst werden kann (Blumer and Johnson, 1994). Über die biologische Bedeutung
dieser proteolytischen Abspaltung wird vermutet, dass sie bei der Zellmigration bei
Wachstum und Wundheilung eine Rolle spielt.
Ein weiteres Fragment der BP180 Ektodomäne (AS 531-1209) mit einem Moleku-
largewicht von 97 kDa (LABD97) kann aus der Epidermis extrahiert werden (Zone
et al., 1990). Beide Fragmente sind Hauptantigene der blasenbildenden Hauter-
krankung lineare IgA-Dermatose, daher der Name LAD-1 (linear IgA disease anti-
gen 1) und LABD97 (linear bullouse disease antigen of 97 kDa).

EINLEITUNG 10
Beim bullösen Pemphigoid stellt die sechzehnte nicht-kollagene Domäne
(NC16A), die sich extrazellulär unmittelbar an die Zellmembran des basalen Kera-
tinozyten anschließt, einen immundominanten Abschnitt von BP180 dar (Giudice
et al., 1993; Giudice et al., 1994; Matsumura et al., 1996; Zillikens et al., 1997).
Das Auftreten von Antikörpern gegen NC16A korreliert mit der Schwere der Er-
krankung (Tanaka et al., 1996) und die Höhe des Antikörpertiters korreliert mit der
Krankheitsaktivität (Bernard et al., 1997; Schmidt et al., 2000b).
Auch in Tiermodellen konnte gezeigt werden, dass die IgG-Reaktivität gegen die
NC16A-Domäne eine direkte Rolle in der Entstehung und Progression der Er-
krankung spielt (Liu et al., 1993). Zillikens et al. beschrieben NC16A als ein 73
Aminosäuren langes Protein (AS 490-562) mit einem Molekulargewicht von ca. 35
kDa. In einem Klonierungsversuch unterteilten sie NC16A in 5 Abschnitte zu je ca.
15 Aminosäuren. Auf diesen Abschnitten liegen die Epitope MCW0 bis MCW5 (s.
Abb. 4). BP-Seren reagierten überwiegend mit dem 45 AS langen Bereich nahe
der transmembranen Region der NC16A Domäne (Zillikens et al., 1997) (s. Abb.
4). Das 14 Aminosäuren umfassende MCW1 gilt als Hauptepitop (Matsumura et
al., 1996). Darüber hinaus indiziert der Antikörper gegen MCW1 eine Separation
der DEJ in Gefrierschnitten humaner Spalthaut (Sitaru et al., 2002).
0MCW
1 2 3 4 5NC16A
1 3 4 5
2
0MCW
490 507 521 535 549 562
LAD-1
B C DA
HD490 562
5??
1497
1497
1365 1413
BP180
4575
0MCW
1 2 3 4 5NC16A 1 2 3 4 5NC16A
1 3 4 5
2
0MCW
490 507 521 535 549 562
LAD-1
B C DA
HD490 562
5??
1497
1497
1365 1413
BP180
4575
Abb. 4: Schematische Darstellung des humanen BP180-Proteins. Der N-Terminus liegt intra- und der C-Terminus extrazellulär. Die Intrazellulärdomäne wurde in der vorliegenden Arbeit in 4 Abschnitte unterteilt (s. unten), daran schließt sich die transmembranöse Region. Die Ektodomäne besteht aus 15 kollagenen (in Abb. „dick+grau“ hervorgehoben) und 16 nicht-kollagenen Domänen. Die sechzehnte nicht-kollagene Domäne (NC16A) schließt sich extrazellulär direkt an die Zell-membran an. Zillikens et al. unterteilten die NC16A Domäne in 5 Regionen zu je ca. 15 Aminosäu-ren, die Position der Aminosäuren ist oberhalb der grauen Kästchen angegeben. Unterhalb der fünf Regionen sind als Rauten die verschiedenen Epitope MCW 0 bis 5 angegeben (Zillikens et al., 1997).

EINLEITUNG 11
Eine entscheidende Rolle in der Pathogenese des BPs wird Entzündungszellen
und Entzündungsmediatoren zugeschrieben.
Schon 1982 beschrieben Dvorak et al, dass in der Haut klinischer BP-Läsionen
unterschiedliche histopathologische Veränderungen im Krankheitsverlauf zu er-
kennen sind. Danach sind CD4+-mononukleare Zellen (CD: cluster of differentia-
tion) die ersten infiltrierenden Zellen in der BP-Läsion (Dvorak et al., 1982). Sie
beobachteten zuerst eine Veränderung der Mastzellen mit Verlust der intrazellulä-
ren Granula in der papillären und retikulären Dermis. Nachfolgend ist eine Ein-
wanderung von Lymphozyten und später von eosinophilen und basophilen Granu-
lozyten zu verzeichnen. Die Basophilen befinden sich in einem Fibringerüst und
sind teilweise oder komplett degranuliert (Dvorak et al., 1982). Der Nachweis von
Mastzellen in Gefrierschnitten von BP-Biopsien wurde auch von Dimson et al. er-
bracht (Dimson et al., 2003). Mittels Markierung mit anti-human-Mastzell-Tryptase
und anti-human-IgE konnten in allen Hautbiopsien von BP-Patienten IgE-beladene
Mastzellen in der Dermis nachgewiesen werden. Zusätzlich wurde mittels eines
monoklonalen anti-BP-180-Antikörper, IgE gebundenes BP180 auf der Oberfläche
der Mastzellen nachgewiesen. In experimentellen Versuchen kommt der Mastzell-
degranulation in der Haut eine Schlüsselrolle bei der Infiltration von Neutrophilen
und anschließender subepidermalen Blasenbildung zu (Chen et al., 2002; Chen et
al., 2001). Katayama et al. wiesen erhöhte Histaminspiegel in der Blasenflüssigkeit
der BP-Läsionen nach (Katayama et al., 1984). Dimson et al. konnten außerdem
eine Histaminfreisetzung aus im Blut zirkulierenden Basophilen nach Stimulation
mit rekombinantem BP180 NC16A messen (Dimson et al., 2003). Auch Fairley et
al. wiesen nach Stimulation mit NC16A eine geringe Histaminfreisetzung aus ba-
sophilen Granulozyten nach (Fairley et al., 2005).
Basophile setzen zudem IL-4, IL-13 und CD40L frei. Der CD40Ligand wird
vom CD40-transmembranen Protein auf der Oberfläche von B-Lymphozyten spe-
zifisch gebunden. Ohne Zusammenspiel von CD40L und CD40 können T-Helfer-
Zellen keine B-Zellen aktivieren. Dann ist auch deren Proliferation und Reifung in
Gedächtnis-B-Zellen und Antikörper-sezernierende Zellen unmöglich. T-Helfer
Zellen (Th-1, Th-2) exprimieren den CD4-Corezeptor auf ihrer Oberfläche und er-
kennen Antigene, die ihnen von speziellen antigenpräsentierenden Zellen (dendri-
tische Zellen, Makrophagen, B-Lymphozyten) auf Klasse ll-MHC-Molekülen dar-

EINLEITUNG 12
geboten werden. Durch Ausschüttung von Zytokinen steuern Th-1 und Th-2 Zellen
die Immunantwort.
BP-Läsionen enthalten eine größere Anzahl an T-Lymphozyten, einschließ-
lich T-Helfer-Zellen und zytotoxischen T-Zellen. Darüber hinaus findet man eine
höhere Anzahl an Makrophagen und Langerhanszellen als in normaler humaner
Haut oder Dermatitis herpitiformis Duhring Läsionen (Nestor et al., 1987). Schaller
et al. wiesen im Patientenblut aktivierte T-Zellen nach, deren Menge mit der
Krankheitsaktivität korreliert (Schaller et al., 1990). Es konnte gezeigt werden,
dass während der Initialphase überwiegend Th-1 Zellen an der Zytokinfreisetzung
beteiligt sind (Ghohestani et al., 1998; Laffitte et al., 2001). Th-1 Zellen sezernie-
ren die Zytokine IL-2 und -Interferon, welche Makrophagen und zytotoxische T-
Zelle zur Zerstörung von aufgenommenen Mikroorganismen aktivieren. In der
Phase der Chronifizierung der BP-Läsionen scheinen Th-2 Zellen in der Überzahl
zu sein. Sie stimulieren durch Freisetzung von IL-4, IL-5 und IL-13 die B-Zellen zur
Proliferation und Sekretion von Antikörpern und induzieren den Klassenwechsel
zu IgG4- und IgE-Antikörper (Ghohestani et al., 1998; Laffitte et al., 2005). Diese
Vorgänge weisen auf die Chronifizierung der Erkrankung hin und spiegeln einen
Wechsel in der Th-1/Th-2 Balance wieder (s Abb. 5).
Erst kürzlich konnten Thoma-Uszynski et al. BP180- und BP230-spezifische T-
und B-Zell Proliferationen nachweisen. Sie zeigten, dass BP180 im Gegensatz zu
BP230 immer von T-und B-Zellen erkannt wird, wobei je nach Krankheitsstadien
unterschiedliche Epitope auf dem BP180 Molekül identifiziert werden. Dieser Be-
fund unterstützt die Annahme, dass BP180 aber nicht BP230 als Hauptantigen an
der Krankheitsentwicklung des BP beteiligt ist (Thoma-Uszynski et al., 2006).
Beim BP führt die Bindung der Autoantikörper zu einer Aktivierung des
Komplementsystems an der kutanen Basalmembran. Dadurch entsteht ein che-
motaktischer Gradient, der zur Rekrutierung von Leukozyten führt. Insbesondere
werden die neutrophilen Granulozyten durch die gebundenen Autoantikörper akti-
viert. Sie stoßen reaktive Sauerstoffradikale und proteolytische Enzyme wie Gela-
tinase und Elastase aus, was im Mausmodell zu Blasenbildung führte (Liu et al.,
1995a; Liu et al., 1995b; Liu et al., 1997; Liu et al., 2000; Liu et al., 1998). Auch
Shimanovich et al. zeigten, dass durch eine Inkubation von humanen Gefrier-
schnitten mit IgG aus BP Seren Granulozyten in die DEJ einwandern und eine
subepidermale Spaltung hervorrufen. Sie konnten zeigen, dass Elastase- und Ge-

EINLEITUNG 13
latinase B-Inhibition, die Antikörper-vermittelte dermo-epidermale Separation ver-
hindern (Shimanovich et al., 2004). Dieser Befund impliziert, dass Elastase und
Gelatinase B (auch MPP-9 genannt) essentiell für die Granulozyten gesteuerte
Proteolyse sind, die schließlich zur Trennung der Epidermis und Dermis führt. Die
aus Neutrophilen freigesetzte Elastase induziert durch Spaltung von BP180 direkt
die Blasenbildung (Verraes et al., 2001). Gelatinase B kann von Makrophagen,
Keratinozyten, Throphoblasten und neutrophilen und eosinophilen Granulozyten
sezerniert werden (Lee and Downey, 2001; Westermarck and Kahari, 1999). Es
wird angenommen, dass die Gelatinase B durch eine aus Mastzellen freigesetzte
Chymase C aktiviert wird (Chen et al., 2001). Die aktivierte Gelatinase B fördert
die Blasenbildung entweder durch die Spaltung von Strukturproteinen der DEJ
oder durch die Inaktivierung von Proteaseinhibitoren aus Granulozyten (Liu et al.,
1998; Weiss, 1989).
Eosinophile werden von IL-4 und IL-5 zur Chemotaxis und Differenzierung ange-
regt (Ameglio et al., 1998). IL-4 wird von Th-2 Zellen, aktivierten B-Zellen, Mast-
zellen und basophilen Granulozyten ausgeschüttet.
Da Eosinophile im histologischen Bild einer BP-Blase dominieren (s. Abb. 3a),
muss auf eine entscheidende Beteiligung von IL-4 und damit auch von Basophilen
und IgE-Antikörpern in der Pathogenese des BPs geschlossen werden.
1.4. IgE-Antikörper und -Rezeptoren
Erste Hinweise auf die Existenz eines IgE-Moleküls im Blut einer Allergikers er-
brachten Prausnitz und Küstner (Prausnitz, 1921) wurde das neuartige Antikörper-
Molekül als IgE-Antikörper identifiziert (Ishizaka and Ishizaka, 1967). Es konnte
von den anderen Immunglobulinen IgG, IgM, IgD und IgA getrennt werden und
erhielt seinen Namen IgE aufgrund des Erythems, welches durch ein Allergen auf
sensitiver Haut hervorgerufen wird (Bibel, 1988). Klassischerweise schützen IgE-
Antikörper den Körpers vor Parasiten und sind zudem an der Pathogenese von
Allergieerkrankungen beteiligt. IgE-Antikörper machen mit 50-300 ng/ml im Ver-
gleich zu IgG mit 10 mg/ml nur einen minimalen Anteil der Serum-Antikörper aus
(Sutton and Gould, 1993). Das IgE-Molekül ist ca. 190 kDa schwer und hat eine
mittlere Halbwertszeit von 3 Tagen. IgE-Antikörper sind nicht in der Lage das
Komplementsystem zu aktivieren. Man beobachtet eine stetige Abnahme der IgE-
Antikörper in der zweiten bis achten Lebensdekade.

EINLEITUNG 14
Der IgE-Antikörper bindet an zwei verschiedene Rezeptoren, den high-affinity Re-
zeptor FcRl und den low-affinity Rezeptor FcRll.
FcRl vermittelt die Bindung von IgE-Molekülen auf der Oberfläche von antigen-
präsentierenden Zellen (APC), Gewebe-Mastzellen sowie im Blut zirkulierenden
basophilen Granulozyten. Antigen-Moleküle kreuzvernetzen diejenigen mem-
brangebundenen IgE-Antikörper, die komplementäre Paratope aufweisen. Durch
die Quervernetzung der IgE-Moleküle kommt es u. a. zur Freisetzung von Hista-
min, IL-4, IL-13 und CD40L aus Basophilen (Gibbs, 2005; Sutton and Gould,
1993) sowie von Histamin und einer Vielzahl weiterer Mediatoren (z. B. Protea-
sen, Proteoglykane, TNF, Zytokine wie IL-4, IL-5, IL-8, IL-13) aus Mastzellen
(Gibbs et al., 2001; Gibbs et al., 2000) (s. Abb. 6). Denselben Effekt haben anti-
IgE-Antikörper oder anti-Idiotyp-Antikörper (Gibbs et al., 1996). Die Rezeptoren
können auch direkt mit anti-Rezeptor-Antikörpern kreuzvernetzt werden. Ebenso
führt kovalent verbundenes IgE, wie auch IL-3 konjugiert an C5a zu einer Über-
brückung der Rezeptoren (Dvorak et al., 1981). Eine weitere Möglichkeit der IgE-
abhängigen Stimulation bieten pflanzliche Lectine, parasitäre Antigene und virale
Superantigene (Falcone et al., 1996). Durch Sensibilisierung der Basophilen (Pri-
ming) mit IL-3, GM-CSF und IL-5 sind die Zellen um ein vielfaches empfindlicher
für IgE-vermittelte Stimulation (Baggiolini and Dahinden, 1994; Plaut et al., 1989).
C 1 C 1
V H V H
C 2 C 2
C 3 C 3
C 4
V L V L
C L C L
C 4
Fab - Fragment
Fc - Fragment
Fab - Fragment
Fc - Fragment
Abb. 5: Darstellung des IgE- Antikörpers. Die Antigen-Bindungsstelle ist aus zwei identischen schweren Ketten und zwei identischen leichten Ketten zusammengesetzt. VH: Variable schwere Kette von ungefähr 440 Aminosäuren (h = hea-vy), VL: Variable leichte Kette, die jeweils aus etwa 220 Aminosäuren aufgebaut sind. Die Fc-Region wird ausschließlich durch die schweren
Ketten gebildet (C1- C4). Das Fc-Fragment
des IgE-AK ist abgeknickt und mit einem C4 länger als der IgG-Antikörper. (Abb. nach PD. Dr. B. F. Gibbs)

EINLEITUNG 15
Antigene
Autoantigene
B-Zelle
Basophiler .
IgE
IL-4, IL-13, CD40L
IL-4
Th-2
IL-4
IL-13
CD40L
Th-0
Th-2-
Entwicklung
IL-4
IL-13
Endothel
“AK-class
switching“
IgG IgE
VCAM-1
Einwanderung von
Leukozyten ins Gewebe
Klinische Symptome:
Urtikaria, Rötung, Juckreiz
Histamin
MastzelleAktivierte Gelatinase B
fördert die Blasenbildung
durch Spaltung von
Strukturproteinen der DEJ
bzw. Inaktivierung von
Proteaseinhibitoren (s. Text)
Leukotriene,
Zytokine
Proteasen z.B.
Chymase C
z. B. IL-8 als chemotaktischer Faktor
für neutrophile Granulozyten
Antigene
Autoantigene
B-Zelle
Basophiler .
IgE
IL-4, IL-13, CD40L
IL-4
Th-2
IL-4
IL-13
CD40L
Th-0
Th-2-
Entwicklung
IL-4
IL-13
Endothel
“AK-class
switching“
IgG IgE
VCAM-1
Einwanderung von
Leukozyten ins Gewebe
Klinische Symptome:
Urtikaria, Rötung, Juckreiz
Histamin
MastzelleAktivierte Gelatinase B
fördert die Blasenbildung
durch Spaltung von
Strukturproteinen der DEJ
bzw. Inaktivierung von
Proteaseinhibitoren (s. Text)
Leukotriene,
Zytokine
Proteasen z.B.
Chymase C
Antigene
Autoantigene
B-Zelle
Basophiler .
IgE
IL-4, IL-13, CD40L
IL-4
Th-2
IL-4
IL-13
CD40L
Th-0
Th-2-
Entwicklung
IL-4
IL-13
Endothel
“AK-class
switching“
IgG IgE
VCAM-1
Einwanderung von
Leukozyten ins Gewebe
Klinische Symptome:
Urtikaria, Rötung, Juckreiz
Histamin
MastzelleAktivierte Gelatinase B
fördert die Blasenbildung
durch Spaltung von
Strukturproteinen der DEJ
bzw. Inaktivierung von
Proteaseinhibitoren (s. Text)
Leukotriene,
Zytokine
Proteasen z.B.
Chymase C
z. B. IL-8 als chemotaktischer Faktor
für neutrophile Granulozyten
Abb. 6: Darstellung der IgE-abhängigen Aktivierung der basophilen Granulozyten und Mast-zellen sowie der immunmodulatorischen Wirkung von IL-4 auf die Th-Zellen. FcRI vermittelt die Bindung von IgE auf der Oberfläche von basophilen Granulozyten und wird durch die Querver-netzung zweier IgE-Moleküle aktiviert. Effekte der Zytokin-, Protease- und CD40L-Ausschüttung siehe Text.
Durch die Quervernetzung zweier IgE-Moleküle auf der Oberfläche von basophilen
Granulozyten durch ein Antigen kommt es zur Ausschüttung von IL-4, IL-13, His-
tamin und CD40L aus den Basophilen. Dabei unterstützt IL-4 die Ausreifung der
Th-2-Zelle, die wiederum durch Ausschüttung von IL-4, IL-13 und CD40L die B-
Zelle zur Synthese von Immunglobulinen bzw. Isotypen-switching anregt. IL-4 und
IL-13 sorgen darüber hinaus für die Synthese chemotaktischer Faktoren, wie die
Präsentation von VCAM-1 auf dem Gefäßendothel. Hieran bindet VLA-4, was zur
Einwanderung von T-Zellen, eosinophiler und basophiler Granulozyten selbst in
das betroffene Gewebe führt (Gibbs, 2005) (s. Abb. 6).
FcRll, auch CD23 genannt, ist ein Rezeptor für IgE mit geringer Affinität. CD23 ist
ein 45 kDa Typ ll-Membranprotein. Man unterscheidet zwei Formen des CD23-
Rezeptors, CD23a und CD23b, die sich aufgrund einer 6/7 N-terminalen Amino-
säure unterscheiden. CD23a sitzt auf B-Zellen, Eosinophilen und Langerhanszel-
len. CD23b wird auf Monozyten, Makrophagen und Thrombozyten exprimiert. Die-
ser Rezeptor besitzt je nach Lokalisationsort und Liganden unterschiedliche Funk-
tionen. Der B-Zell FcRlla-IgE-Antigen-Komplex aktiviert zytotoxische Zellen. Da-
rüber hinaus ist CD23 maßgebend an der Feedback-Regulation der IgE-Synthese
beteiligt. IgE und der IgE-Antigen-Komplex inhibieren die Freisetzung von gelös-
tem sCD23 und unterdrücken somit die IgE-Neusynthese (s. Abb. 7a, 7b). Die

EINLEITUNG 16
Produktion von IgE wird gesteigert, wenn sCD23 simultan an CD21 und IgE auf
der Oberfläche von B-Zellen bindet (Aubry et al., 1992; Sutton and Gould, 1993).
Membrangebundenes CD23 ist an der Down-Regulation der IgE-Synthese betei-
ligt. Findet eine Kreuzvernetzung von CD23 auf humanen B-Zellen mit IgE durch
IgE-Antigen-Komplexe oder Anti-CD23-Antikörper statt, wird die B-Zell-
Proliferation und IgE-Synthese gehemmt (Luo et al., 1991; Sherr et al., 1989). Die
Studie von Hibbert et al. zeigte, dass die Bindung von IgE an membrangebun-
denes CD23 eine Abspaltung und Bildung von löslichem CD23 verhindert. Damit
liegt weniger sCD23 vor, welches für die Stimulation der IgE-Synthese benötigt
wird (Hibbert et al., 2005).
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
mIgE
mCD23C1
C1
VH
VH
C2C2
C 3C 3
C 4
VL
VL
CL
CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
Antigen-Komplex
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
mIgE
mCD23C1
C1
VH
VH
C2C2
C 3C 3
C 4
VL
VL
CL
CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
Antigen-Komplex
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
mIgE
mCD23C1
C1
VH
VH
C2C2
C 3C 3
C 4
VL
VL
CL
CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
Antigen-Komplex
C1
C1
VH
VH
C2C2
C 3C 3
C 4
VL
VL
CL
CL
C 4
C1
C1
VH
VH
C2C2
C 3C 3
C 4
VL
VL
CL
CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
Antigen-Komplex
CD21CD21
Abb. 7a: Herunterregulation der IgE-Synthese: „Wettstreit“ zwischen CD23 und CD21 um das membrangebundene IgE auf der Oberfläche der B-Zellen. Die Co-Kreuzvernetzung zwischen membrangebundenem CD23 und membrangebundenem IgE durch einen IgE-Allergen-Komplex führt zur Herunterregulation der IgE-Synthese.
mIgE
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
sCD23
mIgE
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
sCD23
mIgE
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
sCD23
mIgE
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
sCD23
mIgE
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
+
CD21
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
sCD23
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
C1 C1
VH VH
C2 C2
C 3C 3
C 4
VL VL
CL CL
C 4
sCD23
Abb. 7b: Steigerung der IgE-Synthese: Die gelöste Form des CD23 Rezeptors kreuzvernetzt membrangebundenes IgE und CD21 auf der Oberfläche der B-Zelle und führt damit zur Steigerung der IgE-Synthese. Außerdem kommt es durch die Bindung von IgE an membrangebundenes CD23 zur Verhinderung der Protolyse und damit der Bildung von löslichem CD23 (sCD23).

EINLEITUNG 17
Zunehmend werden IgE und Mastzellen wichtige Funktionen bei der Ent-
stehung von Autoimmunerkrankungen und bei der Regulation der Immunantwort
zugewiesen (Frossi et al., 2004). IgE-Autoantikörper wurden bei Patienten mit Au-
toimmunthyreoiditis beschrieben (Guo et al., 1997; Sato et al., 1999). Erhöhte IgE-
Spiegel wurden auch bei Anti-SSA-Antikörper positiven Frauen nach Verlust des
Fetus gemessen (Sekigawa et al., 2004). In beiden Fällen konnte die genaue Rol-
le von IgE nicht aufgeklärt werden. Darüber hinaus weist eine wachsende Zahl
von Studien auf die Bedeutung von IgE-Autoantikörper beim Bullösen Pemphigoid
hin (Chen et al., 2002; Christophoridis et al., 2000; Delaporte et al., 1996; Dimson
et al., 2003; Dopp et al., 2000; Fairley et al., 2005; Provost and Tomasi, 1974).
1.5. Bullöses Pemphigoid und IgE-Antikörper
Das Gesamt-IgE bei BP-Patienten ist in 70% der Fälle erhöht, wobei die BP180
NC16A-Domäne als antigene Determinante für Auto-IgE-Antikörper wirkt (Dimson
et al., 2003). Der IgE-Titer gegen NC16A korreliert sowohl mit der Krankheitsakti-
vität als auch mit der Krankheitsschwere (Delaporte et al., 1996). Fairley et al.
konnten zeigen, dass beim Bullösen Pemphigoid die IgE-Reaktivität gegen den
extrazellulären Teil von BP180 mit der IgG-Reaktivität korreliert. Die IgE-
Antikörper gegen NC16A sind dabei überwiegend gegen die Subdomaine NC16A2
gerichtet (s. Abb. 4). Außerdem beobachteten sie, dass die Histaminfreisetzung
durch basophile Granulozyten mit der IgE-Immunreaktivität gegen NC16A einher-
geht (Fairley et al., 2005). Die Mastzelldegranulation führt zudem zur Ausschüt-
tung von chemotaktischen Faktoren für eosinophile und neutrophile Granulozyten
(Kasahara-Imamura et al., 2001). Die Freisetzung von neutrophilen Elastase und
Matrixmetalloproteinasen, wie Gelatinase B aus Eosinophilen, Neutrophilen und
Makrophagen ist für die Blasenbildung verantwortlich (Gammon et al., 1982;
Stahle-Backdahl et al., 1994; Verraes et al., 2001). In B-Zell Klonen, die Autoanti-
körper gegen BP180 oder BP230 produzieren, kann es zu einem Klassenwechsel
von IgG- zu IgE-Antikörpern kommen (Bernard et al., 1990; Dopp et al., 2000;
Ghohestani et al., 1998). Der Wechsel des Isotypen mit Anlage der IgE-Antikörper
an der Basalmembran mag für die Erhöhung der Eosinophilen im Serum und im
Gewebe und abschließender Aktivierung von Zellen, die den Fc-Rezeptor expri-
mieren verantwortlich sein (Parodi and Rebora, 1992; Schmidt et al., 1995; Soh et
al., 1993), d. h. die IgE-Synthese trägt zur Aktivierung von IgE-tragenden Zellen,

EINLEITUNG 18
wie Eosinophilen und Mastzellen bei (Dubucquoi et al., 1994; Soh et al., 1993).
Gelatinase B wird durch die Bindung von IgE an die FcRl-positiven eosinophilen
Granulozyten freigesetzt und trägt u. a. zur Ablösung der Haut von der Basal-
membran bei, was klinisch als pralle Blase imponiert. Ein weiterer Hinweis für die
Beteiligung von IgE-Antikörpern beim BP wurde von Kraft et al. beschrieben:
Dendritische Zellen können den FcRl unter bestimmten Krankheitsbedingungen
exprimieren und benutzen diesen Rezeptor möglicherweise um IgE-gebundene
Antigene den T-Zellen zu präsentieren (Kraft et al., 1998). Gegen diesen Fc-
Rezeptor-l wurden IgG-Autoantikörper im Serum von BP-Patienten beschrieben
(Fiebiger et al., 1998). Im Gegensatz zu Anti-BP180-IgE-Antikörpern, die eine His-
taminausschüttung bei Mastzellen induzieren (Dimson et al., 2003), verursachen
die IgG-Autoantikörper gegen den FcRl keine Mastzellstimulation (Fiebiger et al.,
1998). Es ist nicht geklärt, wie diese Rezeptor-Antikörper an der Pathogenese des
BP beteiligt sind.
Soh et al. beschreiben die Anreicherung von CD23+-Zellen in dem Infiltrat
der BP-Läsionen (Soh et al., 1993). Schmidt et al. zeigten, dass auch die gelöste
Form des IgE-Rezeptors sCD23 in der Blasenflüssigkeit und im Serum der Patien-
ten erhöht ist. Zudem korreliert der Total IgE-Titer mit freiem sCD23 in den Serum-
Proben von BP-Patienten (Schmidt et al., 1995). CD23 kann als pleiotropes Zyto-
kin betrachtet werden. Delespesse et al. demonstrierten, dass dieser niedrigaffine
Rezeptor CD4+-Zellen stimuliert und als hemmender Faktor für die Einwanderung
von Monozyten/Makrophagen wirkt (Delespesse et al., 1992). Diese beiden Effek-
te erklären, warum in BP-Läsionen CD3+- bzw. CD4+-Zellen 70% der mononuk-
learen Zellen in der Dermis ausmachen, während die Makrophagen an der Basal-
membranzone dominieren (Ambach et al., 1992; Zillikens et al., 1992). Die erhöh-
ten IgE-Spiegel in der Blasenflüssigkeit der Läsionen lassen darauf schließen,
dass IgE aus dem Blut in die Bläschen diffundiert, da B-Zellen in der BP-Läsion
weniger als 1% des Entzündungsfiltrats ausmachen und somit eine lokale IgE-
Sekretion, die mit den erhöhten IgE-Spiegeln im Blut korreliert, unwahrscheinlich
macht (Schmidt et al., 1995).
Kürzlich wurde im Mausmodell gezeigt, dass IgE-produzierende Hybri-
doma-Zellen gegen LABD-97 in implantierter humaner Haut auf SCID-(Severe
Combined Immundeficiency Disorder) Mäusen Blasen hervorrufen. Zudem ließ
sich die Einwanderung von Eosinophilen und eine Mastzelldegranulation in der

EINLEITUNG 19
Läsion nachweisen (Zone et al., 2007). Dieses Phänomen ließ sich jedoch nicht
mit IgG- und IgA-Antikörpern darstellen, was auf die entscheidende Rolle von IgE
beim BP zurückschließen lässt. In früheren Studien galt das in den Keratinozyten
hemidesmosomal gelegene BP230 Protein als hauptsächliche Erkennungsstruktur
der IgE-Autoantikörper (Delaporte et al., 1996; Ghohestani et al., 1998; Soh et al.,
1993).
Neben diesen histologischen und experimentellen Resultaten, weist auch der urti-
karielle Untergrund der Läsionen und die Tatsache, dass die Patienten initial von
starkem Juckreiz geplagt sind daraufhin, dass IgE eine Rolle in der Pathogenese
der Erkrankung spielt.
1.6. Ziele dieser Arbeit
BP180 (Kollagen XVll) ist ein Bestandteil der dermo-epidermalen Junktionszone
und fungiert als Interaktionspartner für hemidesmosomale und extrazelluläre Ma-
trixproteine. BP180 ist das Hauptantigen für Autoantikörper beim bullösen Pem-
phigoid, einer subepidermalen blasenbildenden Autoimmundermatose.
Die klinischen Symptome der BP-Patienten sind gekennzeichnet durch starken
Juckreiz sowie urtikarielle Plaques und weisen damit auf das Mitwirken von Mast-
zellen und basophilen Granulozyten in der Pathogenese dieser Erkrankung hin. In
vorangegangenen Studien wurde von erhöhten Gesamt-IgE-Spiegeln im Blut von
BP-Patienten und von Auto-IgE-Antikörper gegen die NC16A-Domäne berichtet.
Der massive Einstrom von eosinophilen Granulozyten, die u. a. durch IL-4 und
Komplement zur Chemotaxis angeregt werden, weist auf das Mitwirken von IgE-
Antikörpern und Basophilen bei der Blasenentstehung hin.
In mehreren Studien wurde gezeigt, dass BP180 neben NC16A weitere an-
tigene Strukturen sowohl auf der intrazellulären als auch auf der extrazellulären
Domäne enthält. Aus BP-Patientenblut isolierte basophile Granulozyten wurden
mit NC16A stimuliert und führten zu einer kaum messbaren Histaminfreisetzung
und einer nur geringen IL-4-Ausschüttung (mündliche Mitteilung PD Dr. B. F.
Gibbs). In anderen Veröffentlichungen konnte eine Histaminfreisetzung aus Baso-
philen nach Stimulation mit NC16A gemessen werden. Die Befunde lassen vermu-
ten, dass möglicherweise weitere Epitope auf dem BP180 Molekül für IgE-
Autoantikörper existieren. Vor einigen Jahren wurde IgE-Autoreaktivität in BP-
Seren gegen das intrazelluläre Protein BP230 beschrieben. Dies ist ein Hinweis,

EINLEITUNG 20
dass es möglicherweise auch intrazelluläre antigene Bereiche auf BP180 für IgE-
Autoantikörper gibt.
Im Rahmen dieser Arbeit wurden BP-Seren auf ihre IgE-spezifische Reaktivität
gegen rekombinant hergestellte Abschnitte der intra- und extrazellulären Domäne
von BP180 untersucht. Darüber hinaus wurde LAD-1 aus dem Mediumüberstand
von Keratinozyten und Keratinozytenextrakten auf ihre Antigenität für IgE-
Autoantikörper überprüft.

MATERIALIEN 21
2. Materialien
2.1. Verwendete Laborgeräte
Airsterilisator-US PATS Binder GmbH, Tuttlingen
Begasungsbrutschrank Haereus Instruments GmbH, Hanau
Biophotometer, 8,5 mm Eppendorf AG, Hamburg
Bio Vortex V1 lab4you GmbH, Berlin
Blockthermostat BT 5320 Eppendorf-Netheler-Hinz GmbH,
Hamburg
Dampfsterilisator Webeco GmbH, Bad Schwartau
Elektrische Feinwaage PT 150 Sartorius AG, Göttingen
Elektrophoresekammer Sub-Cell GT Bio-Rad Laboratories GmbH, München
Eppendorf Pipetten Eppendorf Referencen, Hamburg
Gefrierkombination (+4 °C, -20 °C) Liebherr-International AG, Bulle, Schweiz
Gefriertruhe C660 (-80 °C) New Brunswick Scientific, England
Gelkassetten Bio-Rad Laboratories GmbH, München
Inkubator, CERFOMATIS B. Braun Biotech International,
Melsungen
Kühlkammer Viessmann GmbH & Co. KG, Allendorf
Kühlzentrifuge Megafuge 1,0 Haereus Instruments GmbH, Hanau
Laborabzug captairchem Erlab Laboreinrichtungen GmbH &
Co. KG, Wangen
Lyophilisator Leybold-Heraeus, Hanau
Magnetrührer MSH 300 lab4you GmbH, Berlin
Mikroskop Wilovert S Helmut Hund GmbH, Wetzlar
Mini-PROTEAN 3 Gelkammer Bio-Rad Laboratories GmbH, München
Mini Trans-Blot Cell Gelkassette Bio-Rad Laboratories GmbH, München
Netzgerät PowerPac HC Bio-Rad Laboratories GmbH, München
Netzgerät Power Supply EPS 3500 Pharmacia GmbH, Ratingen
Neubauer Zählkammer Brand GmbH & Co. KG, Wertheim
pH-Meter ph526 MultiCal WTW, Weilheim
Schütteltisch Duomax 1030 Heidolph Instruments, Nürnberg
Sonopuls UW 2070 Bandelin electronic, Berlin

MATERIALIEN 22
Sterilbank, Biowizard Kojar W. H. Mahl, Trendelburg
Tischzentrifuge (Rotor: Sorvall 3328) Haereus Instruments GmbH, Hanau
Überkopfschüttler (Kühlraum) Heidolph Instruments, Nürnberg
Vakuumpumpe XF54 23050 Millipore Corporation, Massachusetts,
USA
Wasserbad WTH 500 Hecht-Assistent, Sondheim
Zentrifuge, AvantiJ-25 Beckman Coulter, California, USA
2.2. Verbrauchsmaterialien
Aluminiumfolie Paclan A. Hartenstein Laborbedarf, Würzburg
Blot-Filterpapier Schleicher und Schuell GmbH, Dassel
Blutabnahmesystem SMonovette Sarstedt AG & Co, Nümbrecht
Chromatographie-Säule, Poly-Prep Bio-Rad Laboratories GmbH, München
Dialyseschlauch Roth, GmbH & Co, Karlsruhe
Einfrierbox, Nalgene, IDL Sarstedt AG & Co., Nümbrecht
Einmal-Kanülen Becton Dickinson GmbH, Heidelberg
Einmal-Küvetten UVette Eppendorf AG, Hamburg
Erlenmeyerkolben A. Hartenstein Laborbedarf, Würzburg
Gelloader-Spitzen (1-200 µl) A. Hartenstein Laborbedarf, Würzburg
Gewebekultur-Petrischalen Becton Dickinson GmbH, Heidelberg
Glutathion-Sepharose 4 Fast Flow Amersham Biosciences, Piscatway,
USA
Kryoröhrchen (2 ml) Greiner Bio-One GmbH, Frickenhausen
Nitrozellulose Membran (0,45 µm) Bio-Rad Laboratories GmbH, München
QuixSep, Micro Dialyser Carl Roth GmbH & Co., Karlsruhe
Reaktionsgefäße 0,5 ml, 1,5 ml, 2,0 ml Eppendorf AG, Hamburg
Serologische-Pipetten 5 ml, 10 ml, 25 ml Biochrom AG, Berlin
Skalpellklingen (einmal) Carl Roth GmbH & Co., Karlsruhe
Zellkulturflaschen 25 cm2 Greiner Bio-One GmbH, Frickenhausen
Zellkulturflaschen 75 cm2, 175 cm² Becton Dickinson GmbH, Heidelberg
Zellschaber Sarstedt AG & Co., Nümbrecht
Zentrifugenflaschen 500 ml Biochrom AG, Berlin
Zentrifugenröhrchen 1,5 ml, 2 ml Sarstedt AG & Co., Nümbrecht
Zentrifugenröhrchen 15 ml, 50 ml Biochrom AG, Berlin

MATERIALIEN 23
2.3. Chemikalien und Farbstoffe
Acrylamid 4K-Lösung (30%) AppliChem, Darmstadt
Ammonium Persulfate (APS) Sigma-Aldrich Chemie GmbH,
Deisenhofen
β-Mercaptoethanol Merck KGaA, Darmstadt
Bio-Rad Protein Assay Bio-Rad Laboratories GmbH, München
Borsäure AppliChem, Darmstadt
Bovines Serum Albumin (BSA) Sigma-Aldrich Chemie GmbH,
Deisenhofen
Bromphenolblau 0,2% Merck Eurolab GmbH, Darmstadt
Coomassie Brilliant Blue (R250) Bio-Rad Laboratories GmbH, München
di-Kaliumhydrogenphosphat (K2HPO4) Merck KGaA, Darmstadt
di-Natriumhydrogenphosphat (Na2HPO4) Merck KGaA, Darmstadt
Dodecylsulfat Natriumsalz (SDS) Merck Schuchardt OHG, Hohenbrunn
EDTA Dinatriumsalz Dihydrat Carl Roth GmbH & Co., Karlsruhe
Essigsäure 99-100% J. T. Baker, Deventer-Holland
Ethanol 70% Apotheke UKSH, Campus Lübeck
Fast Green Sigma-Aldrich Chemie GmbH,
Deisenhofen
Glutathion Sigma-Aldrich Chemie GmbH,
Deisenhofen
Glycerol AppliChem, Darmstadt
Glycin Carl Roth GmbH & Co., Karlsruhe
Kaliumchlorid (K+Cl-) Merck KGaA, Darmstadt
Magermilchpulver Merck KGaA, Darmstadt
Methanol 99,8% J. T. Baker, Deventer, Holland
Natriumchlorid (NaCl-) J. T. Baker, Deventer, Holland
Ortophosphorsäure Merck KGaA, Darmstadt
Phenylmethansulfonylfluorid (PMSF) Sigma-Aldrich Chemie GmbH,
Deisenhofen
Protein-Standard (farbmarkiert) Bio-Rad Laboratories, California, USA
Salzsäure (HCl) Merck KGaA, Darmstadt
SDS-PAGE-Standard (SDS-6H) Sigma-Aldrich Chemie GmbH,
Deisenhofen

MATERIALIEN 24
TEMED Carl Roth GmbH & Co., Karlsruhe
Trichloressigsäure (TCA) Carl Roth GmbH & Co., Karlsruhe
Tris Base Merck KGaA, Darmstadt
Tris HCl Carl Roth GmbH & Co., Karlsruhe
Trypan Blue Sigma-Aldrich Chemie GmbH,
Deisenhofen
Tween 20 (M = 1227,72 g/mol) AppliChem, Darmstadt
Wasserstoffperoxid Merck KGaA, Darmstadt
2.4. Puffer und Lösungen
TBST 2 mM: 12,1 g/l Tris Base; 40 g/l NaCl; Tween20 0,5%
v/v in ddH2O; pH 7,5 mit 2 M HCl eingestellt
PBS 10-fach: 80 g/l NaCl; 2 g/l KCl; 14,4 g/l Na2HPO4; K2HPO4
in ddH2O
Puffer zur Affinitätsaufreinigung/Säulenchromatographie:
Borat-Puffer 0,1 M: 6,183 g/l Borsäure; 29,22 g/l NaCl in ddH2O; pH
8,5 mit 2 M NaOH eingestellt
Essig-Puffer 0,1 M: 6 ml Essigsäure; 29,22 g/l NaCl in ddH2O; pH 4
mit 2 M NaOH eingestellt
Elutionspuffer 20 mM: 0,31 g Glutathion ad 50 ml PBS; pH 9 mit 2 M
NaOH eingestellt
Puffer zur SDS-Gelelektrophorese:
SDS-Probenpuffer 5-fach, reduzierend: 60 mM Tris-HCl pH 6,8; 3% w/v SDS;
50 mM Mercaptoethanol; 10% v/v Glycerol;
0,003% w/v Bromphenolblau
Laufpuffer 5-fach: 15 g/l Tris Base; 72 g/l Glycin; 5 g/l SDS in
ddH2O
Tris-Glycin 10-fach: 144,13 g/l Glycin; 30,3 g/l Tris Base in ddH2O
Transferpuffer: 100 ml Tris-Glycin 10-fach; 200 ml Methanol; 700
ml ddH2O.

MATERIALIEN 25
2.5. Zellkulturmedien und Zusätze
Luria- Bertani-Medium nach Miller (LB-Medium):
Minimales Medium zur Anzucht von E. coli
10 g/l Bacto-Trypton; 5 g/l Hefe Extrakt; 10 g/l NaCl wurden in ddH20 gelöst und
mit 2 M NaOH auf pH 7,5 eingestellt. Anschließend wurde das Medium für 20 min
bei 120 °C und 2 bar autoklaviert. Für die selektive Aufzucht von E.coli enthielt das
LB-Medium Carbenicillin in einer Endkonzentration von 50 µg/ml.
Keratinozyten Growth Medium 2 (KGM2): Firma: PromoCell GmbH, Heidelberg
Kultivierung von HaCaT-Zellen
Vor Verwendung wurde dem Medium Supplement zugegeben, das aus 2 ml BPE-
15 (Bovine pituitary extract); 62,5 ng/500µl hEGF (human epidermal growth fac-
tor); 165 µg/500 µl HC (Hydrocortison); 2,5 mg/500 µl Insulin; 195 µg/500 µl Epi-
nephrin; 5 mg/500 µl Transferrin und 150 µl CaCl2-0,5/0,15 besteht. Ggf. wurden
dem Kulturmedium 100 Units Penicillin/ml und 100 µg Streptomycin/ml zugesetzt.
Zelleinfrierlösung:
Zum Einfrieren von Zelllinien bei -80 °C
10% v/v DMSO (Dimethylsulfoxid) in PBS sterilisiert
50% v/v FCS
40% v/v Keratinozytenwachstumsmedium
Ammoniumsulfat-Puder (APS) Sigma-Aldrich Chemie GmbH,
Deisenhofen
Carbenicillin Dinatriumsalz Carl Roth GmbH & Co., Karlsruhe
Dimethylsulfonat (DMSO) Sigma-Aldrich Chemie GmbH,
Deisenhofen
Dulbecco’s Phosphate Buffer (PBS) PAA Laboratories GmbH, Cölbe
Fötales Kälberserum (FCS) PAA Laboratories GmbH, Cölbe
Hefe-Extrakt Difco Mikrobiology, Kansas City, USA
Isopropyl-D-beta-thyogalactopyranoside AppliChem, Darmstadt
Keratinozytenwachstumsmedium 2 PromoCell GmbH, Heidelberg
L-Ascorbinsäure Sigma-Aldrich Chemie GmbH,
Deisenhofen

MATERIALIEN 26
Penicillin/Streptomycin PAA Laboratories GmbH, Cölbe
(10.000 Units/ml/ 10 mg/ml)
Phenylmethylsulfonyl-Fluorid (PMSF) AppliChem, Darmstadt
SupplementPack PromoCell GmbH, Heidelberg
Trypsin/EDTA Lösung 0,05%/ 0,02% (w/v) Biochrom KG,Berlin
Trypton/Pepton Carl Roth GmbH & Co., Karlsruhe
2.6. Verwendete Zelllinien und Bakterienstämme
In der Arbeit wurden Keratinozyten der Linie HaCaT (Human adult low Calcium
high Temperature) verwendet (Boukamp et al., 1988). Herkunft: Dr. med. Fusenig,
DKFZ, Deutsches Krebsforschungszentrum, Heidelberg.
HaCaT-Zellen dienen als Modell für humane Keratinozyten (Boukamp P, 1988;
Ryle, CM 1989). Im Gegensatz zu normalen Keratinozyten wachsen HaCaT-
Zellen in konventioneller Kultur, das bedeutet ohne organotypische Co-Kultur-
bedingungen wie z. B. Fibroblasten.
Für die Expression der rekombinanten Proteine GST-hBP180-ICD-A-D, -NC16A,
und -BP4575 (C-terminaler Abschnitt) wurde der E. coli-Expressionsstamm No-
vaBlue verwendet;
Genotyp: endA1, hsdR17 (rK12-, m K12+), supE44, thi-1, recA1, gyrA96, relA1, lac,
F´ [proAB+,laclqZM15::Tn10(TetR)]. (Eine genauere Erklärung der Angaben und
Kurzbezeichnungen für den Genotyp findet sich in einschlägiger Literatur über
Mikrobiologie). Herkunft: Merck KGaA -Biosciences, Darmstadt
2.7. Patientenseren
Für die vorliegende Arbeit wurden die Seren von 20 nicht vorbehandelten Patien-
ten mit der Diagnose bullöses Pemphigoid untersucht. Alle Patienten wiesen typ-
ische klinische Manifestationen des bullösen Pemphigoids auf. Mindestens eine
der Screening-Untersuchungen (ELISA, DIF, IIF) musste positiv sein (s. Tab. 3).
Von den 20 Patienten waren 5 männlich und 15 weiblich. Das Alter lag zwischen
57 und 94 Jahren, mit einem Durchschnittsalter von 83 Jahren. Die DIF periläsio-
naler Hautbiopsien und die IIF in Salt-Split-Skin-Technik wurden wie beschrieben
durchgeführt (Zillikens et al, 1996).

MATERIALIEN 27
Die Blutproben wurden den Patienten vor Einleitung der Therapie entnommen. Die
Personen wurden vor der Blutentnahme über die Studie aufgeklärt und haben eine
Einwilligungserklärung zur Verwendung ihrer Seren an dieser Untersuchung un-
terschrieben.
Das Blut wurde 8 min bei 15 °C und 3600 rpm (Zentrifuge Heraeus Megafuge 1,0,
Rotor 7570, durchschnittliche RZB= 600 x g) zentrifugiert und das Serum abpipet-
tiert und aliquotiert. Die Seren wurden bis zur weiteren Verwendung bei -20 °C
gelagert. 500 µl jeder Serumprobe wurde zur Gesamt-IgE-Bestimmung mittels
Fluoreszenz-Enzym-Immunoassay und SX1-Analyse ins Labor der Immunolo-
gischen Klinik der Universität zu Lübeck gegeben. SX1 ist eine Abkürzung für
Mischallergene und beinhaltet die acht häufigsten Allergene. Bei positiven SX1, d.
h. spezifischen IgE-Werten >0,35 kU/l erfolgte die weitere Aufschlüsselung nach
diesen Allergenen (s. Tab. 4).
2.8. Kontrollen
2.8.1. Negativkontrollen
Als Negativkontrollen dienten insgesamt 10 Seren. Davon stammte ein Serum von
einer durch IIF, DIF und immunserologischer Untersuchung gesicherten PV-
Patientin. Die anderen 9 Kontrollpatienten wurden nicht selektiert bzw. wiesen kei-
nerlei klinische Hinweise auf ein bullöses Pemphigoid auf, so dass diese keinen
diagnostischen Untersuchungen in der Dermatologischen Universitätsklinik zu Lü-
beck unterzogen wurden. Die Geschlechterverteilung männlich:weiblich war 2:8.
Das Alter der Kontrollpersonen lag zwischen 28 und 93 Jahren (s. Tab. I im An-
hang) und ähnelt somit in der Alters- und Geschlechtszusammensetzung der Pa-
tientengruppe. Die Kontrollpersonen waren mit keinerlei Medikamenten, die die
IgE-Reaktivität beeinflussen könnten, vorbehandelt.
2.8.2. Positivkontrollen
Als Positivkontrolle für die rekombinanten an GST gekoppelten Proteine wurde der
polyklonale Kaninchenantikörper SA8009, der gegen GST-Bestandteile von
BP180 gerichtet ist, verwendet. Außerdem dienten vorcharakterisierte BP-Seren
als Kontrolle. Zur Detektion der BP180-Spaltprodukte, LAD1 und LABD97, diente
das Serum eines bekannten LAD-Patienten mit hoher IgA-Reaktivität. Dieses wur-
MATERIALIEN 28
de auch als Positivkontrolle für die Reaktivität gegen Keratinozytenextrakt ver-
wendet.
Der polyklonale Kaninchen-Anti-GST-BP180 NC16A2-4-Antikörper (SA8009) und
die vorcharakterisierten Patientenseren wurden bis zur Verwendung bei -80 °C
gelagert.
2.9. Antikörper
Tab. 1: Verwendete Antikörper mit Angabe der eingesetzten Konzentrationen. AK: Antikörper; *SA8009, (Sitaru et al., 2002).
Antikörper/ Konjugation Spezifität Verdünnung
Polyklonal Kaninchen IgG-HRP (DakoCytomation, Dänemark)
Human IgG
1:500 in TBST/BSA 1%
Monoklonal Maus IgG (Sigma-Aldrich, Deutschland)
Human IgE 1:1000 in TBST/BSA 1%
Polyklonal Kaninchen IgG-HRP (DakoCytomation, Dänemark)
Human IgA 1:200 in TBST/BSA 1%
Polyklonal Kaninchen IgG-HRP (DakoCytomation, Dänemark)
Anti-Maus AK
1:500 in TBST/Magermilch 5%
Polyklonal Kaninchen IgG* (aus immunisierten Kaninchen- Eurogentec, Belgien)
Human BP180 1:200 in TBST/BSA 1%
Polyklonal Ziege IgG-HRP (DakoCytomation, Dänemark)
Anti-Kaninchen AK
1:1000 in TBST/BSA 1%

METHODEN 29
3. Methoden
3.1. Zellkultur
Alle Zellkulturarbeiten wurden unter einer sterilen Werkbank durchgeführt. Nach
kurzer Lüftung und dem Erreichen der vollen Leistung der Werkbank wurde die
Arbeitsfläche mit Ethanol 70%ig ausgewischt. Alle Arbeiten wurden mit sterilen
Instrumenten durchgeführt. Die verwendeten Materialien wie Eppendorf-
Reaktionsgefäße, Pipettenspitzen, PBS, Aqua bidest. wurden im Dampfsterilisator
(Webeco) für 20 min bei 121 °C autoklaviert. Nicht autoklavierbare/hitzelabile Ge-
räte wurden regelmäßig mit 70% Ethanol gereinigt. Zur Vermeidung von Verunrei-
nigungen wurde immer nur mit einer Zelllinie zurzeit unter der Sterilbank gearbei-
tet.
3.1.1. HaCaT-Zellkultur
3.1.1.1. Auftauen von eingefrorenen Zellen
Zum Anlegen einer neuen Flüssigkultur wurden die in Stickstoff gelagerten tiefge-
frorenen HaCaT-Zellen ca. 15-20 min auf Eis aufgetaut und die Kryoröhrchen für
45 Sekunden im 37 °C warmen Wasserbad geschwenkt. Die Zellen wurden erneut
auf Eis gekühlt und die Zellsuspension in Keratinozyten-Growth-Medium 2
(KGM2) resuspendiert und in ein Falkonröhrchen überführt. Nach Zentrifugation
für 7 Minuten bei 20 °C und 1200 rpm (Zentrifuge Heraeus Megafuge 1,0, Rotor
7570, durchschnittliche RZB= 300 x g) wurde das Pellet in frischem vorgewärmten
KGM2 gelöst und ca. 500.000 Zellen in 25 cm2 Zellkulturflaschen ausgesät. Die
Kultivierung der HaCaT-Zellen erfolgte im Begasungsbrutschrank (Heraeus
GmbH) bei 37 °C, 90% Luftfeuchtigkeit und einem CO2-Gehalt von 5%. Zur An-
zucht wurden HaCaT-Zellen als Adhäsionskultur in KGM2 nach Zugabe von Sup-
plement gehalten. Zur Neutralisation des EDTA und Trypsins nach der Umsetzung
in größere Kulturflaschen wurde 10% fetales Kälberserum (FCS) dazugegeben.
Das FCS wurde zuvor bei 56 °C für 30 min hitzeinaktiviert. Um die Kollagensyn-
these der Keratinozyten zu beschleunigen wurde bei einer Zellkonfluenz von 50%
Ascorbinsäure in einer Endkonzentration von 100 µg/ml dazupipettiert.
Um Kontaminierungen zu Erkennen und die Konfluenz der Zellen zu beurteilen,
wurden die Zellkulturen täglich unter dem Mikroskop betrachtet. Ein Medium-
wechsel erfolgte 14 h nach Aussaat in neue Kulturflaschen und dann alle 48 h,

METHODEN 30
sofern die optimale Zelldichte nicht überschritten wurde. Hierzu wurde die Kultur-
flasche leicht geschwenkt und das Nährmedium mit einer Serologischen-Pipette
(Biochrom) abgesaugt. Nicht adhärente bzw. tote Zellen wurden somit entfernt.
Der Zellrasen sollte nicht durch Berühren mit der Pipettenspitze beschädigt wer-
den. Das entnommene Volumen wurde durch frisches zuvor im Wasserbad ange-
wärmtes Keratinozyten-Wachstumsmedium ersetzt.
3.1.1.2. Subkultivierung der Zellen
Die Zelllinie HaCaT unterliegt einem festen Wachstumszyklus, wobei eine kons-
tante Zellteilung nach 7 Tagen erreicht wird. Nach ca. 24 h treten die HaCaT-
Zellen in die exponentielle Wachstumphase ein, dabei hält das unlimitierte Wach-
stum im Mittel 72 h an (Boukamp et al., 1988; Wanner et al., 1999). Bei einer Zell-
konfluenz von 80-90% wurden die Kulturen passagiert, um die Zellen in der expo-
nentiellen Wachstumsphase zu halten. Dazu wurde das Zellkulturmedium wie
oben beschrieben vollständig entfernt. Die Ablösung der HaCaT-Zellen von der
Oberfläche erfolgte durch Zugabe von Trypsin/EDTA-Lösung (0,05%/0,02% (w/v)).
Die Kulturflaschen wurden mehrmals geschwenkt und für 3-5 min im Brutschrank
inkubiert. Der Grad der Ablösung wurde mikroskopisch kontrolliert, um die Ein-
wirkzeit möglichst gering zu halten und so toxische Effekte des Trypsins zu ver-
meiden. Das Ablösen der Zellen wurde gegebenenfalls mechanisch durch mehr-
faches Klopfen der Kulturflasche unterstützt. Nachdem sich die Zellen zum Groß-
teil gelöst hatten, wurde die enzymatische Proteolyse durch Zugabe von 10% FCS
gestoppt. Die Zellen wurden mittels Serologischer-Pipette in ein steriles 20 ml
Zentrifugenröhrchen überführt und bei 20 °C mit 1200 rpm für 7 min zentrifugiert
(Zentrifuge Heraeus Megafuge 1,0, Rotor 7570, durchschnittliche RZB= 300 x g).
Der Überstand wurde vorsichtig mit Hilfe einer Vakuumpumpe entfernt. Das Zell-
pellet wurde anschließend in frischem Medium resuspendiert und in neue sterile
Zellkulturflaschen ausgesät. Nach ungefähr 6 h sollten die HaCaT-Zellen adhärent
sein, nach weiteren 6 h musste das Medium gewechselt werden, da verbliebene
Reste des Trypsin/EDTA die Zellen schädigen.

METHODEN 31
3.1.1.3. Bestimmung der Zellzahl
Die Zellzählung erfolgte in einer Neubauer-Zählkammer. Der Farbstoff Trypanblau
reichert sich in toten Zellen an und führt zu einer mikroskopisch nachweisbaren
Blaufärbung der Zellen. Gesunde, vitale Zellen erscheinen ungefärbt, da sie in der
Lage sind den Farbstoff über aktive Mechanismen aus dem Zytoplasma auszu-
schleusen. Zur Herstellung der Trypanblaulösung wurden 0,5% (w/v) Trypanblau
und 0,9% (w/v) NaCl in PBS gelöst. Es wurden 20 µl der HaCaT-Zellsuspension
mit 20 µl Trypanblaulösung in einem Reaktionsgefäß gemischt und etwa 5 min bei
RT inkubiert. Dann wurde die Lösung in eine Neubauer-Zählkammer pipettiert und
mindestens 4 Großquadranten ausgezählt. Die Zellzahl wurde nach folgender
Formel berechnet:
mlntenGroßquadraderZahl
Zellzahl
mlZellen 1102 4
3.1.1.4. Einfrieren von Zellen
Zur langfristigen Lagerung der HaCaT-Zellen wurden Gefrierstocks angelegt. Die-
se wurden in flüssigem Stickstoff gelagert und konnten bei Bedarf wieder aufge-
taut werden. Hierzu wurden die HaCaT-Zellen in Kultur in der exponentiellen
Wachstumsphase mit Trypsin/EDTA (0,05%/0,02% (w/v)) gelöst und die Zellzahl
bestimmt. Pro 107 Zellen/ml wurde ein 1,5 ml Kryoröhrchen vorbereitet. Die Zell-
suspension wurde bei 20 °C und 1200 rpm für 7 min (Zentrifuge Heraeus Megafu-
ge 1,0, Rotor 7570, durchschnittliche RZB= 300 x g) zentrifugiert. Das Zellpellet
wurde mit 50% FCS, 40% Keratinozytenwachstumsmedium und 10% Dimethylsul-
foxid (DMSO) suspendiert und je 1 ml gleichmäßig auf die 1,5 ml Kryoröhrchen
verteilt. Das Einfrieren der Zellen erfolgte schrittweise in einer Styroporbox. Zuerst
wurden die Kryoröhrchen 1 h langsam auf -20 °C abgekühlt, dann über Nacht bei
-80 °C eingefroren und daraufhin zur Langzeitkryokonservierung in Behältern mit
flüssigem Stickstoff archiviert.

METHODEN 32
3.2. Expression der rekombinanten GST-Fusionsproteine
hBP180 ICD-A, -B, -C, -D.
Zur Generation der GST-BP180 ICD-A (AS 1-133), -B (AS 103-266), -C (234-398),
-D (367-452) und GST-BP180 4575 (AS 1365-1413) Fusionsproteine wurde das
bakterielle Expressionsystem pGEX-6P-1 (Firma: Amersham Biosciences, Pisca-
taway/USA) verwendet (s. Abb. I, Anhang). Die cDNA Sequenzen, die für die 4
überlappenden Fragmente der intrazellulären Domäne des BP180 (Typ XVII Kol-
lagen) kodieren, wurden wie beschrieben in dem prokaryonten-Expressionsvektor
pGEX-6P-1 eingebaut und in E. coli exprimiert (Sitaru et al., 2005).
1 133
A B C D
490
266
234 398
367 452
B C D
1
103
HD1497
1 133
A B C D
490
266
234 398
367 452
B C D
1
103
HD1497
Abb. 8: Schematische Darstellung der intrazellulären Domäne des humanen BP180-Proteins. Für das Epitop-mapping wurden die überlappenden Fragmente A, B, C, D der intrazellu-lären Domänen hergestellt (siehe Material und Methoden).
Die Primer für die Polymerase-Kettenreaktion (PCR) wurden von MWG Biotech
(Ebersberg, Germany) bezogen (s. Tab. I, Anhang). Die cDNA-Fragmente wurden
durch Reverse Transkription von Gesamt-RNA humaner epidermaler Extrakte ge-
wonnen. Schnittstellen für Bam H l und Sal I wurden durch die Primer eingebaut
(verwendete Primer siehe Anhang). Die Plasmide wurden freundlicherweise von
Dr. Cassian Sitaru zur Verfügung gestellt und enthielten die in Abb. 8 beschrie-
benen Fragmente der BP180 Intrazellulärdomäne zusammen mit der jeweils C-
terminal fusionierten Glutathion-S-Transferase (GST). Der plasmidtragende E.coli-
Stamm für die 4 Abschnitte der ICD und dem C-terminalen Abschnitt BP180 4575

METHODEN 33
war NovaBlue. Als Nährmedium für die E. coli Stämme wurde Luria-Bertani Me-
dium verwendet (LB-Medium).
3.2.1. Primärkultur
Unter der sterilen Werkbank wurden 20 ml LB-Medium in ein 50 ml Zentrifugen-
röhrchen abgefüllt, dazu wurden 20 µl einer 1.000 x Stammlösung Carbenicillin
pipettiert. Die folgenden Arbeiten erfolgten außerhalb der sterilen Werkbank. Von
dem bei -80 °C eingefrorenen Bakterienstock wurde mit einer Filterpipettenspitze
an der obersten Eisschicht leicht gekratzt, um die Spitze dann in das autoklavierte
LB- Medium abzuwerfen. Die angelegte Primärkultur wurde über Nacht im vorge-
wärmten Inkubator (Heraeus) bei 220 rpm und 37°C inkubiert. Dabei war darauf zu
achten, dass der Deckel leicht geöffnet ist, so dass der Sauerstoff den E. coli-
Zellen zum Wachstum zur Verfügung stand.
3.2.2. Sekundärkultur
Nach ca. 12 h wurden jeweils 500 ml des LB-Mediums in zwei 1 Liter-
Erlenmeyerkolben gegeben, Carbenicillin in einer Endkonzentration von 50 µg/ml
zugesetzt und mit je 10 ml der Primärkultur beimpft. Das Antibiotikum diente zur
Stabilisierung der Plasmide. Die Kultur wurde bis zu einer OD600 (optical density
bei 600 nm; Biophotometer, Eppendorf) von 0,5-0,6 im Bakterienschüttler (He-
raeus) bei 220 rpm, 37 °C angezogen. Anschließend wurde die Proteinexpression
durch Zugabe von IPTG in einer Endkonzentration von 0,5 mM induziert und die
Kultur für weitere 3-4 h bei gleichen Konditionen im Schüttler inkubiert.

METHODEN 34
3.2.3. Proteinextraktion
Die rekombinanten Proteine werden von E. coli als lösliches zytoplasmatisches
Protein innerhalb der Zelle abgelagert. Deshalb ist zur Isolierung der rekombinan-
ten Proteine die Lyse der Bakterienzelle notwendig, wobei die Proteine solubilisiert
werden.
Alle Schritte zur Aufreinigung des GST-Fusionsproteins wurden auf Eis oder im
Kühlraum bei 4 °C durchgeführt. 250 ml der Expressionskulturen/Bakterien-
suspensionen wurden in 500 ml Zentrifugenbecher überführt und bei 4 °C für 10
min bei 17.700 x g zentrifugiert. Der Überstand wurde verworfen. Das Zellpellet
wurde in 6 ml PBS resuspendiert und evtl. bis zur weiteren Verarbeitung über
Nacht bei -20 °C eingefroren. Nach dem Auftauen der Zellsuspension wurde unter
dem Abzug 1 mM PMSF (348 mg PMSF in 20 ml Isopropanol) dazupipettiert und
die Bakterien bis zur vollständigen Lyse mit Ultraschall bei mittlerer Intensität und
einer Pulsdauer von 20 sec in 3 Zyklen behandelt (Sonopuls HD-70, Bandelin,
Berlin). Zwischen den Zyklen lagen 30 sek. Pausen, in denen nochmals 1 mM
PMSF hinzugefügt wurde. PMSF ist ein Proteaseinhibitor, der beim Aufschluss
kultivierter Zellen unerwünschten Proteinabbau durch ebenfalls im Lysat befind-
liche Serin-Proteasen verhindert.
Unlösliche Bestandteile wurden durch Zentrifugation für 30 min und 13.000 rpm
(Tischzentrifuge Heraeus, Festwinkelrotor Sorvall ‚3328) bei 4 °C entfernt. Die er-
haltenen Überstände wurden in ein Zentrifugenröhrchen pipettiert und ggf. bis zum
nächsten Verarbeitungsschritt bei -20 °C gelagert.
3.2.4. Affinitätsaufreinigung
Glutathion-S-Transferase (GST) und GST-Fusionsproteine lassen sich mittels Af-
finitätschromatographie aus Bakterienlysat aufreinigen. Das GST bindet dabei an
das an Sepharose 4 Fast Flow gekoppelte Glutathion (Amersham). Es findet je-
doch keine Umsetzung statt, da das zweite Substrat fehlt. Die Elution erfolgt durch
Zugabe hoher Konzentrationen reduzierten Glutathions, das kompetitiv an GST
der Fusionsproteine bindet. Die Säulenchromatographie erfolgt nach dem Proto-
koll des Herstellers Amersham Biosciences bei 4 °C. 5 ml 75% Glutathion-
Sepharose 4 Fast Flow in ddH2O wurden in die Chromatographie-Säule gefüllt.
Nach dem Absetzen der Sepharose wurde die Säule mit 5-10 Säulenvolumina
PBS equilibriert. Das klare Gesamtlysat wurde langsam auf Eis aufgetaut. Danach

METHODEN 35
erfolgte die Inkubation der Glutathion-Sepharose 4 Fast Flow mit dem löslichen
Bakterienlysat für 1 h bei 4 °C auf dem Überkopfschüttler. Anschließend wurde die
Proteinlösung in einem 15 ml Zentrifugenröhrchen gesammelt und die Säule
zweimal mit 15 ml PBS gewaschen. Die Waschlösungen mit den ungebundenen
Proteinen wurden auf Eis gestellt. Zur Eluierung der gebundenen Fusionsproteine
wurde reduzierter Glutathionpuffer verwendet. Das Eluat wurde in je 1000 µl auf-
geteilt und zunächst bei -80 °C gelagert. Von den verschiedenen Aufreini-
gungsstufen wurden sechzehnmal jeweils 1000 µl Fraktionen entnommen. Die
Qualität der Überexpression und Aufreinigung der bakteriellen Fusionsproteine
GST-BP180 ICD-A, -B, -C, -D erfolgte mittels SDS-PAGE mit anschließender Sil-
ber- und Coomassie-Brilliant-Blue-Färbung und Western-Blot. Von der 1-3 und
jeder zweiten Fraktion wurden je 50 µl mit 20 µl 5x SDS-Probenpuffer versetzt und
auf 97 °C erhitzt. Als Molekulargewichtsstandard wurden 8 µl SDS-PAGE Marker
für Molekulargewichte von 30-200 kDa (SDS-6H, Sigma) auf das Gel aufgetragen
und zur Kontrolle wurde 5 µl reines GST eingesetzt. Die Proteinproben, die bei der
Western-Blot-Analyse die stärksten Banden lieferten bzw. die stärkste Antikörper-
reaktion mit Kaninchen-anti-hBP180 NC16A2-4-GST (SA8009) hervorriefen, wur-
den gemischt, dialysiert und gefriergetrocknet.
Zur Regeneration wurde die Glutathion-Agarose-Säule mit je 10 ml folgender Lö-
sungen gewaschen: Glutathion; PBS pH 7,2; Borat-Puffer; PBS pH 7,2; Essig-
säure- Puffer; PBS pH 7,2. Bis zum nächsten Gebrauch wurde die Säule mit PBS
mit Natriumacidzusatz gefüllt bei 4 °C untergebracht.

METHODEN 36
Induktion der
Proteinexpression
Glutathion Sepharose Fast Flow
3-4 h Inkubation,
10 min zentrifugieren
+ IPTG
2 h Inkubation
E.coli Kultur mit
pGEX-BP180
FragmentenPellet in PBS,
sonifizieren
PBS
Glutathionpuffer
Durchlauf; Waschung 1+2
Entfernung der ungebundenen E.coli-Proteine
Bindung der GST-Fusionsproteine an der GST-Agarose Säule
Isolierung der BP180 Fragmente
Analyse mittels SDS-PAGE
Proteinkonzentration (280 nm)
Immunoblot
E.coli- Protein
Glutathion
GST-Fusionsprotein
Induktion der
Proteinexpression
Glutathion Sepharose Fast Flow
3-4 h Inkubation,
10 min zentrifugieren
+ IPTG
2 h Inkubation
E.coli Kultur mit
pGEX-BP180
FragmentenPellet in PBS,
sonifizieren
PBS
Glutathionpuffer
Durchlauf; Waschung 1+2
Entfernung der ungebundenen E.coli-Proteine
Bindung der GST-Fusionsproteine an der GST-Agarose Säule
Isolierung der BP180 Fragmente
Analyse mittels SDS-PAGE
Proteinkonzentration (280 nm)
Immunoblot
E.coli- Protein
Glutathion
GST-Fusionsprotein
Abb. 9: Herstellung der rekombinanten GST-Fusionsproteine.

METHODEN 37
3.2.5. Dialyse und Gefriertrocknung
Die Dialyse der rekombinant hergestellten Proteine erfolgte gegen PBS pH 7, bei
4 °C über Nacht und langsamen Rühren unter Verwendung von permeablen Cellu-
loseschläuchen (Roth, Karlruhe) mit einer Ausschlussgrenze von 12-14 kDa. Der
PBS-Puffer wurde mindestens dreimal in regelmäßigen Abständen durch frischen
Puffer ersetzt. Nach der Dialyse wurde die Proteinlösung aus dem Dialyse-
schlauch in 50 ml Reaktionsgefäße überführt und bei -80 °C eingefroren. An-
schließend wurden die Proben 48 h gefriergetrocknet. Das Lyophilisat wurde in
möglichst geringen Mengen PBS gelöst, der Proteingehalt spektralphotometrisch
bei OD280 gemessen und mit PBS auf eine Proteinkonzentration von 1 mg/ml ver-
dünnt. Die Lösungen wurden bis zur weiteren Verwendung bei -80 °C gelagert.
3.3. Proteinisolierung
3.3.1 Proteinisolierung aus dem Mediumüberstand kultivierter
Keratinozyten
In dem Mediumüberstand kultivierter Keratinozyten befinden sich die proteoly-
tischen Spaltprodukte LAD-1 und LABD97 des hemidesmosomalen Antigens
BP180.
Zum Transfer der Zellen wurde das Medium vollständig abgezogen und auf Eis
gestellt. Die Passage der Zellen erfolgte wie oben beschrieben. Um Zelldebris zu
entfernen wurde der Mediumüberstand 7 min bei 1200 rpm (Zentrifuge Heraeus
Megafugefuge 1,0, Rotor 7570, durchschnittliche RZB= 300 x g) abzentrifugiert.
Der klare Überstand wurde in ein Zentrifugenröhrchen abgefüllt, mit 5 mM EDTA,
1 mM PMSF versetzt und bis zur weiteren Verwendung bei -80 °C gesammelt.
600 ml des Keratinozyten-Growth-Medium-Überstandes wurden langsam auf Eis
aufgetaut. Nach vollständigem Auftauen wurden die enthaltenen Proteine mit
Ammoniumsulfat-Puder präzipitiert. Dazu wurde das Salz unter Rühren bei 4 °C in
mehren Schritten langsam hinzugegeben, um eine grobe Ausflockung zu verhin-
dern. Damit eine vollständige Auflösung der Salzkristalle erfolgte, wurde das Prä-
zipitat über Nacht bei 4 °C gerührt.
Am nächsten Tag wurde die Suspension in 30 ml Zentrifugenröhrchen gefüllt und
für 90 min bei 4 °C und 36.300 x g pelletiert. Das Pellet wurde in 500 µl PBS re-
suspendiert und zur Resuspension aller Pellets in die weiteren Röhrchen trans-

METHODEN 38
feriert. Die Proteinsuspension wurde mittels QuixSep, Micro Dialyzer (Roth, Karl-
sruhe) über Nacht bei 4 °C gegen PBS dialysiert (Dialysemembran Ausschluss-
grenze 12-14 kDa), um das Ammoniumsulfat aus der Suspension zu entfernen.
3.3.2 Herstellung der Keratinozytenextrakte
Nach der dritten Passage der HaCaT-Zellen wurde der gleichmäßige Zellrasen mit
einer Lösung aus 2% SDS, 5% Beta-Mercaptoethanol, 2 mmol/l PMSF, 2 mM
EDTA in 62,5 mM Tris HCl (pH 6,8) lysiert. Anschließend wurden die Zellen mit
einem Zellschaber vom Boden der Kulturflasche gelöst und in ein 50 ml-
Zentrifugenröhrchen überführt. Alle Arbeiten wurden auf Eis durchgeführt, die
Zentrifuge wurde auf 4 °C gekühlt. Das Lysat wurde 3 mal für 20 sec sonikiert, zur
Entfernung der Zelldebris bei 20.000 x g zentrifugiert, aufgeteilt und bei -80 °C
gelagert.
3.3.3. Photometrische Messung der Proteinkonzentration
Die Konzentration von LAD-1 im aufgereinigten Mediumüberstand kultivierter Ke-
ratinozyten, sowie die Proteinkonzentration im Keratinozytenextrakt und in der
Suspensionen der rekombinant hergestellten Proteine wurde photometrisch bei
280 nm im Biophotometer (EpendorfAG, Hamburg) ermittelt. PBS-Puffer diente
dabei als Leerwert. Die Proteinkonzentrationen der Proben ließen sich mit der Be-
stimmung des Wertes nach Lambert Beer durch folgende Formel abschätzen:
OD280 × = Protein mg/ml
Es wurde näherungsweise Albumin=0,65 mg/ml verwendet.
Die erhaltenen Protein-Lösungen wurden mit PBS auf 1 mg Protein/ml eingestellt
und bei -80 °C archiviert. Die Probe wurde anschließend mit SDS-Puffer 5-fach zu
4 Teilen Proteinlösung und 1 Teil SDS-Puffer versetzt. Zur qualitativen Kontrolle
wurde ein Teil der Proteine im SDS-PAGE elektrophoretisch aufgetrennt und im
Immunoblot untersucht.

METHODEN 39
3.4. Westernblot Analyse/Epitop-mapping
3.4.1. SDS-Polyacrylamid-Gelelektrophorese
Die Auftrennung der Proteinen erfolgte nach der Laemmli-Methode (Laemmli,
1970) unter Verwendung eindimensionaler, diskontinuierlicher Zweikomponenten-
SDS-Polyacrylamid-Gele (Mini-Gele, 10 x 7 cm). Die Proben wurden mit 5-fach
SDS-Probenpuffer verdünnt, 2 min auf 98 °C erhitzt, kurz bei 13.000 rpm zentrifu-
giert (Tischzentrifuge Heraeus, Festwinkelrotor #3328) und bis zum Auftragen auf
Eis gekühlt. Um die Proben zu fokussieren, wurde ein Sammelgel dem eigent-
lichen Trenngel vorgeschaltet. Die SDS-Polyacrylamid-Gele wurden je nach Grö-
ße der aufzutrennenden Proteine verschieden dicht präpariert (s. Tab. 3a, b).
Unmittelbar nach dem Gießen des Trenngels in eine vertikale Gel-Kassette (Mini-
Protean 3 Cell, BioRad) mit 0,75 mm Spacern, wurde das Gel mit Aqua dest. über-
schichtet, um das Trenngel zu pressen und eine gleichmäßige Oberfläche für das
Sammelgel zu erhalten. Nach einer Polymerisationszeit von ca. 5 min wurde das
Wasser abgekippt, das Sammelgel darüber gegossen und ein Probenkamm in das
Sammelgel gedrückt. Für die intrazellulären Domänen von BP180 mit einem Mo-
lekulargewicht von ca. 40 kDa wurden 12%-ige Trenngele und 5%-ige Sammelge-
le verwendet. Die BP180 intrazellulären Domänen A bis D wurden der Reihenfolge
nach auf ein Taschengel geladen. Als Kontrolle wurde GST und NC16A aufgetra-
gen. Zur Abschätzung der Molekulargewichte diente ein farbmarkierter Protein-
standard (Bio-Rad, München) mit folgender Zusammensetzung: Myosin (206
kDa), ß-Galactosidase (133 kDa), Rinder-Serumalbumin (84 kDa), Carbo-
anhydrase (41 kDa), Soybean trypsin inhibitor (31 kDa), Lysozyme (17 kDa), Apro-
tinin (7 kDa). Um den Transfer auf die Nitrozellulosemembran zur kontrollieren,
erfolgte die Färbung der Nitrozellulosemembran mit Fast-Green und die Detektion
der Banden mit dem Kaninchen-Serum SA8009, gewonnen durch Immunisierung
mit hBP180-NC16A2-4 (Sitaru et al., 2002). Je nach Intensität der Bande wurde
die aufgetragene Menge erhöht oder erniedrigt.
Für LAD1, KCE und BP180 4575 wurde ein Preparations-Gel (Prep-Gel) herge-
stellt. Pro SDS-Gel wurden 250 µl/500 µg Protein aufgetragen. Als Standard wur-
de der SDS-PAGE-Molekulargewichtsmarker für 30-200 kDa (SDS-6H, Sigma)
geladen. Die Gelelektrophorese erfolgte in der Mini-PROTEAN 3 Elektrophorese-
Kammer mit SDS-PAGE-Laufpuffer. Bis zum Einlauf der Proteine in das Trenngel
erfolgte die Elektrophorese für 15 min bei 20 mA, danach konstant bei 50 mA so-
METHODEN 40
lange, bis die Farbfront des SDS-Puffers den unteren Gelrand erreicht hatte (ca.
75 min.).
Tab. 3a: Gelzusammensetzung% zur Trennung der unterschiedlichen epidermalen Proteine.
Protein
BP180 ICD (40 kDa )
KCE (180 kDa)
LAD-1 (120 kDa)
BP180 4575 (50 kDa)
% Trenngel 12 7,5 7,5 15
% Sammelgel 5 4 4 5
Tab. 3b: Zusammensetzung der verwendeten SDS-Polyacrylamid-Gele
Gelbestandteile Trenngel Sammelgel
7,5% 12% 15% 4% 5%
Aqua bidest 4,15 ml 2,65 ml 2,35 ml 3,1 ml 2,94 ml
1,5M Tris pH 8,8 1,35 ml 1,35 ml 2,5 ml - -
0,5M Tris pH 6,8 - - - 700 µl 700 µl
30% Acrylamid 2,5 ml 4 ml 5 ml 650 µl 812 µl
100% Glycerol 500 µl 500 µl - 250 µl 250 µl
1M Glycin 1 ml 1 ml - - -
0,5M EDTA pH8 - - - 40 µl 40 µl
10% SDS 400 µl 400 µl 100µl 200 µl 200 µl
TEMED 12 µl 12 µl 12 µl 12 µl 12 µl
10% APS 150 µl 150 µl 12 µl 100 µl 100 µl
3.4.2. Coomassie-Brilliant-Blue- und Silberfärbung der SDS-
Gele
Zur Sichtbarmachung der Proteine in den SDS-Gelen wurden die Trenngele für
1 h unter Bewegung in Coomassie-Brillant-Blau (R250) Färbelösung (2g/l Coo-
massie-Brilliant-Blue R250, 450 ml Methanol, 100 ml Essigsäure 100% ad 1000 ml
ddH2O) gebracht und anschließend über 1 h in Entfärbelösung (450 ml Methanol,
100 ml Essigsäure 100% ad 1000 ml ddH2O.) entwickelt. Das Gel wurde über
Nacht zwischen zwei Blättern Zellophanpapier getrocknet und diente somit zur
Dokumentation der Ergebnisse.
Zusätzlich wurde zur Visualisierung der Proteinbanden eine Silberfärbung durch-
geführt. Diese eignet sich besonders, um die Reinheit eines Proteins zu überprü-
fen (s. Abb. 11). Um die Hintergrundfärbung zu minimieren sowie zur Steigerung
der Sensitivität wurde das SDS-Gel vorher mit Coomassie-Brillant-Blue (R250)

METHODEN 41
angefärbt. Darauf erfolgte die Silberfärbung unter Verwendung eines nach Blum et
al. (Blum et al., 1987) modifizierten Protokolls unter Nutzung der angegebenen
Lösungen und Einhaltung der Inkubationszeiten:
Nach der Fixierung der Proteine mittels 10% Acetat, 30% Ethanol, 60% ddH2O
über 60 min erfolgte die Inkubation in einer Sensibilisatorlösung aus 6,8 g NaAc x
3 H2O (0,5 M), 30 ml Ethanol, 2 ml Glutaraldehyd 25 %ig, 0,6 g Na-Thiosulfat x 5
H2O ad 100 ml ddH20 über 30 min. Anschließend wurde das Gel für 3 x 10 min in
ddH2O gewaschen und danach für 30 min in die Silberfärbelösung gelegt. Zur Ver-
ringerung der Hintergrundfärbung wurde nach der Silberbehandlung kurz mit
ddH2O gewaschen bevor die Entwicklung des Gels mit 2,5 g Na2CO3, 30µl 37%ig
Formalaldehyd (ad 0,01) ad 100 ml ddH2O erfolgte. Unter Beobachtung wurde die
Reaktion nach 5-15 min abgestoppt. Hierbei blieb das Gel für 5-10 min in der Ab-
stopplösung (1,6 g Glycin, 0,42 g Dinatrium-EDTA, ad 100 ml ddH2O) und wurde
daraufhin nochmal für 3 x 10 min mit ddH2O gewaschen.
3.4.3. Western-Blot
Für die nachfolgende Immunreaktion wurden die über SDS-PAGE aufgetrennten
Proteine im Tank-Blot-Verfahren auf eine Nitrozellulosemembran (0,45 µm Poren-
durchmesser, Bio-Rad, München) übertragen. Hierzu wurden nach der Elektro-
phorese die Glasplatten voneinander getrennt und das Sammelgel vom Trenngel
abgetrennt. Auf die Kathodenseite der Blotkassette (cell, Bio-Rad, München) wur-
den zwei Faserkissen, 2 Gel-Blotting Filterpapiere (Schleicher & Schüll), das
Trenngel und die Nitrozellulosemembran sandwichartig aufgebaut. Dabei wurde
darauf geachtet, dass sich zwischen den Schichten keine Luftblasen befanden.
Die Faserkissen und Papiere dienten als Ionenreservoir bei der Elektrophorese (s.
Abb. 10). Die Kassetten wurden zwischen die beiden Flächenelektroden der
Kammer in Transferpuffer eingespannt. Das Methanol im Puffer entfernt das SDS
aus dem Protein-Detergenz-Komplex und erhöht die Bindung der SDS-freien Pro-
teine an die Membran. Der Transfer der Proteine vom Trenngel auf die Nitrozellu-
lose erfolgte bei 30 V und 50 mA bei 4 °C für 12 h. (Towbin et al., 1979). Der Er-
folg des Transfers wurde durch anschließende Färbung mit Fast Green (1g Fast
Green, 100 ml Methanol, 50 ml Essigsäure 100% ad 1000 ml ddH2O) kontrolliert.
Die Membran wurde 2 min in der Färbelösung geschwenkt, dann mit ddH2O wie-

METHODEN 42
der entfärbt, auf Filterpapier getrocknet und bis zur weiteren Verwendung bei 4°C
aufbewahrt.
Kathode
Schwamm
Schwamm
Anode
Filterpapier
Filterpapier
SDS- Gel
Nitrozellulose- Membran
Tra
nsfe
r
Kathode
Schwamm
Schwamm
Anode
Filterpapier
Filterpapier
SDS- Gel
Nitrozellulose- Membran
Tra
nsfe
r
Abb. 10: Modell des „Sandwiches“ im Western-Blot.
3.4.4. Immundetektion der transferierten Proteine/
Epitop-mapping
Die Nitrozellulosemembranen mit transferierten LAD-1, KCE, BP180 4575 wurden
in 2-3 mm dicke Streifen geschnitten, beschriftet und in einer Inkubationsschale
gesammelt. Die Membran mit ICD-A, -B, -C, D-, GST, NC16A1-5 wurde als solche
belassen und in eine Gewebe-Petrikulturschale 5 cm gelegt. Die Membranen wur-
den kurz in TBST gewaschen und anschließend für 1 h bei RT in 5% Magermilch-
pulver/TBST-Lösung inkubiert, um unspezifische Bindungen der Antikörper zu
vermeiden. Nach dem Abgießen der Magermilch und Waschen mit TBST erfolgte
die Inkubation mit dem Primärantikörper. Dazu wurde das Serum für die IgG-
Detektion 1:100 mit TBST/BSA 1%, Magermilch 3% verdünnt, für die IgE-
Detektion wurde das Serum 1:2 mit TBST/BSA 1% gemischt. Die Membranen
wurden über Nacht bei 4 °C oder für 3 h bei RT auf einem Schüttler unter leichten
Bewegungen mit dem Primärantikörper inkubiert. Anschließend wurden ungebun-
dene Antikörper durch dreimaliges kurzes Waschen mit TBST und dreimaliges
Waschen für 5 min mit TBST entfernt. Im Anschluss erfolgte die Inkubation mit
den sekundären Antikörpern. Für die IgG-Detektion wurde das Serum 1:500 mit
TBST/BSA 1% verdünnt, für die IgA-Detektion 1:200 sowie für die IgE-Erkennung
1:1000. Diese anti-human-Antikörper verblieben für 1 h bzw. der Maus-anti-human
IgE-Antikörper für 2 h auf der Membran. Anschließend wurde der Waschvorgang
wie oben beschrieben wiederholt. Die Blots zum Nachweis von IgE-Antikörper
wurden noch für 1 h mit einem Kaninchen-anti-Maus-Antikörper-HRP inkubiert.
Dieser Antikörper wurde in einer Verdünnung von 1:500 in TBST/Blockingpuffer

METHODEN 43
5% angesetzt. Alle Inkubationen und Waschschritte erfolgten auf dem Schüttler.
Das an den Zweitantikörper bzw. dritten Antikörper gekoppelte Enzym Meerrettich-
Peroxidase (HRP: „Horse Radish Peroxidase“) erzeugte in Anwesenheit seines
Substrates Chemilumineszenz. Dazu wurde eine Lösung von Diamino-
benzidintetrahydrochlorid (DAB, Merck) angesetzt (1 DAB-Puffertablette wurde in
15 ml Aqua bidest gelöst). Kurz bevor die Lösung über die Membranen gegossen
wurde, musste 20 µl Wasserstoffperoxid (H2O2) zugesetzt werden. Die HRP und
H2O2 katalysieren die Umsetzung von Luminol in seine oxidierte Form, bei der ei-
ne Lumineszenz detektiert werden kann. Die Membranen wurden in der Reak-
tionslösung geschwenkt; beim ersten Auftreten von sichtbaren Banden im Moleku-
largewichtsbereich der entsprechenden Antigene wurde die Enzymreaktion ge-
stoppt, indem die Blots in Aqua bidest. gewaschen wurden. Sämtliche Arbeiten mit
dem DAB-Puffer erfolgten unter dem Laborabzug nach dem Herstellerhinweis.
3.4.5. Auswertung der Bandenmuster
Die Auswertung der Bandenverteilung erfolgte visuell. Die Intensität der Fluores-
zenz der Banden wurde mittels der Anzahl der „+“ beschrieben. Keine Reaktion
der Antikörper gegen das Protein wurden mit „-“ gekennzeichnet. Patientenseren,
die im Immunoblot Banden beim GST-Protein zeigten wurden in der Auswertung
der Ergebnisse nicht miteinbezogen. Um die Beurteilung zu objektivieren, wurden
die Blots verblindet von einer unabhängigen Person ausgewertet. Zur weiteren
statistischen Auswertung wurden der Chi-Quadrat-Test und der Mann-Whitney-U-
Test herangezogen. Der Chi-Quadrat-Vierfeldertest ist ein statistisches Testver-
fahren und dient dazu die Häufigkeit eines Merkmals in zwei statistisch unabhän-
gigen Gruppen zu vergleichen und zugleich die Frage nach der Signifikanz des
Testes zu beantworten. Hiermit lässt sich eine Aussage über die Abhängigkeit der
Reaktion des IgE-Antikörpers mit den ICD A-D, GST und NC16A im Vergleich zu
den Ergebnissen mit dem IgG-Autoantikörper machen.
Der Mann-Whitney-U-Test ist ein parameterfreier statistischer Test. Er dient zur
Überprüfung der Signifikanz der Übereinstimmung zweier Verteilungen, also ob
zwei unabhängige Verteilungen, z. B. der Gesamt-IgE-Spiegel der BP-Seren und
der Negativkontrollen, zu derselben Grundgesamtheit gehören.

ERGEBNISSE 44
4. Ergebnisse
4.1. Expression und Affinitätsaufreinigung von rekombinanten
GST-Fusionsproteinen
Die cDNA Fragmente von 4 Abschnitten der intrazellulären Domäne von BP180
wurden in den Vektor pGEX-6P-1 (Amersham Biosciences) eingebaut (s. An-
hang). Es resultierten vier rekombinante Vektoren, die zur Proteinexpression im
E.coli Stamm NovaBlue kloniert wurden. Die rekombinanten Proteine wurden
durch Bakterienlyse freigesetzt, über eine Glutathion-Säulenchromatographie auf-
gereinigt und im 15% SDS-Gel elektrophoretisch aufgetrennt. Zur Expressions-
kontrolle wurde das Gel anschließend mit Silbernitrat angefärbt (Abb. 11)
LS 1 2 3 4 5
ICD-C
200
11697
66
45
31
[kDa]
LS 1 2 3 4 5
ICD-C
200
11697
66
45
31
[kDa]
Am Beispiel von ICD-C (AS 234-398) lässt sich in Abb. 11 die Instabilität der re-
kombinanten Proteine erkennen. Mehrere stark angefärbte Banden im niederen
Molekulargewichtsbereich lassen auf die Degradierung des Proteins schließen.
Um dieses zu verhindern wurde bei erneuter Expression dieser Proteine der Ein-
fluss von Proteinaseinhibitoren bei der Proteinisolierung untersucht. Dabei zeigte
sich, dass ein kommerzieller Proteinaseinhibitorcocktail im Vergleich zu PMSF +
EDTA einen Abbau weitgehend verhindert (s. Abb. 11).
War die Expressionskontrolle positiv, wurde das Proteineluat gesammelt, gegen
ddH20 dialysiert, gefriergetrocknet und mit PBS auf eine Endkonzentration von 1
mg/ml eingestellt. Die so erhaltenen BP180 Fragmente ICD-A, -B, -C, -D sowie
NC16A1-5 und die GST-Kontrolle wurden elektrophoretisch aufgetrennt, auf eine
Nitrozellulosemembran übertragen und zur Kontrolle mit Fast-Green angefärbt.
Abb. 11: Silberfärbung des SDS-Page Gel. Kontrolle der Affinität-saufreinigung am Beispiel von ICD-C. LS: Längenstandard; 1: Protein-extrakt aus E.coli, 2: Durchlauf, 3: GST, 4: ICD-C, 5: ICD-C mit comp-lete-mini Proteinaseinhibitor, Ro-che Applied Science, Mannheim. Man bemerke, dass in Spur 4 meh-rere Proteinbanden im niederen kDa-Bereich zu sehen sind, die Degradierungsprodukte des Pro-teins darstellen.

ERGEBNISSE 45
Anschließend wurde als weitere Kontrolle ein Immunoblot mit Kaninchen-anti-
GST-BP180 NC16A2-4 (SA8009) und enzymmarkiertem Ziegen-anti-Kaninchen
als Sekundärantikörper sowie anschließender Visualisierung mittels DAB durchge-
führt (s. Abb. 12). Durch die Immunoblotanalyse konnten die Mengen an gelade-
nem Protein überprüft werden und gegebenenfalls ein Angleichen der Konzen-
trationen der vier Fragmente vorgenommen werden.
1 2 3 4 5 6
31
40[kDa]
31
40
(A)
(B)
1 2 3 4 5 6
31
40[kDa]
31
40
(A)
(B)
31
40[kDa]
31
40
(A)
(B)
In der SDS-PAGE migrierten auf 15%-igen Gelen (Abb. 12) GST-hBP180 ICD-A
bei 41 kDa, GST-hBP180 ICD-B bei 38/43 kDa, GST-hBP180 ICD-C bei 44 kDa
und GST-hBP180 ICD-D bis zu 41 kDa. Am linken Rand in Abbildung 12(A) er-
kennt man den farbmarkierten Molekulargewichtsmarker. Die Fusionsproteine
migrierten somit etwas abweichend zu ihrer berechneten Größe von 41; 44,9;
49,17; 39,29 kDa. In der Fast-Green Färbung wiesen die ICD-Fragmente A, B, C,
und D, wie schon in der Silberfärbung dargestellt (Abb. 11), zusätzliche Banden
mit niedrigerem Molekulargewicht auf. Das rekombinant hergestellte NC16A1-5
färbte sich auf der Mem-bran sehr schwach an, auch zeigten sich keine doppelten
Banden im niederen Molekulargewichtsbereich, was auf weniger proteolytischen
Abbau dieser nicht kollagenösen extrazellulären Domäne hinweist.
In der Kontrolle mit dem Kaninchenserum SA8009 wurde eine Vielzahl von Ban-
den detektiert. Dieser Befund lässt sich durch die Reaktion des Kaninchen-anti-
GST-hBP180 NC16A2-4 AK mit GST-gekoppelten Degradierungsprodukten der
Proteine erklären.
Abb. 12: Beurteilungsreferenz für Immunoblots. Anfärbung der Proteine mit Fast Green (A), analog dazu wurde ein Immunoblot (B) mit dem SA8009-Antikörper durchgeführt. Dieser polyklonale Antikörper detektiert GST bzw. GST-Fusionsptoteine. Links Markierung des Längenstan-dards. Für die Beurteilung der Blots wurden nur bestimmte Ban-den herangezogen (Rahmenmar-kierungen). Spur 1: GST-ICD-A, Spur 2: GST-ICD-B, Spur 3: GST-ICD-C, Spur 4: GST-ICD-D, Spur 5: GST, Spur 6: GST-NC16A. Bei ICD-B wurden 2 Banden als positiv gewertet.

ERGEBNISSE 46
4.2. Erstellung des Patientenkollektivs und der
Negativkontrollen
20 Patienten (15 weibliche und 5 männliche) mit einem Durchschnittsalter von 83
Jahren nahmen an dieser Studie teil. Alle 20 Patienten waren nicht systemisch
immunsuppressiv vorbehandelt. Sie wiesen typische klinische und histologische
Merkmale des BP auf. Zusätzlich wurde jedes Patientenserum wie folgt charak-
terisiert:
1. Durch den Nachweis von IgG-Antikörpern- und C3-Ablagerungen an der Ba-
salmembran (DIF)
2. Durch den Nachweis von zirkulierenden IgG-Antikörpern gegen die Basal-
membran auf Normalhaut (IIF) und auf kochsalzbehandelter Haut (Salt-Split-Skin
IIF)
3. Durch den Nachweis einer Reaktivität im BP180-NC16A-ELISA.
4. Durch eine Immunoblotanalyse mit positiver Reaktivität gegen BP180.
Es zeigten sich bei 18 von 19 mittels DIF untersuchter Patienten IgG-Antikörper
bzw. C3-Ablagerungen an der Basalmembranzone. Mittels IIF am Substrat Affen-
ösophagus konnten bei 16 von 18 untersuchten Patienten initial zirkulierende IgG-
und z. T. IgA-Antikörper nachgewiesen werden. Zudem ließen sich bei allen 19
untersuchten Patienten IgG-Antikörper und zusätzlich bei 5 Seren IgA-Antikörper
im Dach der artifiziell erzeugten Blase nachweisen (IIF in Salt-Split-Skin-Technik
(Gammon et al., 1984)). Der NC16A-ELISA zeigte bei 16 von 18 untersuchten Se-
ren positive Ergebnisse (IgG gebunden >9 U/ml), dabei lagen die gebundenen
IgG-Antikörper bei 16,8 bis >1000 U/ml. In allen Patientenseren wurde somit mit
mindestens einem positiven Ausfall in den durchgeführten autoimmundiagnosti-
schen Tests ein BP bestätigt (s. Tab. 3).
Die 9 Negativkontrollen (normale humane Seren) waren in der Alters-, Ge-
schlechtszusammensetzung vergleichbar mit dem Patientenkollektiv und wiesen
keinerlei klinische Hinweise auf ein bullöses Pemphigoid auf, so dass diese keiner
diagnostischen Untersuchungen unterzogen wurden (s. Tab. II im Anhang).
Das PV-Serum (1 Negativkontrolle) zeigte in der IIF mit Affenösophagus
IgG-Antikörper, die SSS war negativ, der Dsg 1 ELISA war negativ, und der Dsg 3
ELISA waren positiv mit 694 U/ml (normal <14 U/ml). Außerdem zeigte die DIF
IgG- und C3-Ablagerungen im Interzellularraum der Epidermis.
ERGEBNISSE 47
Tab. 3: Patientenergebnisse der Routineuntersuchung in der Autoimmundiagnostik der Dermatologie der Universität zu Lübeck und Würzburg. BP: bullöses Pemphigoid mit Patien-tennummerierung, w: weiblich, m: männlich, IIF: indirekte Immunfluoreszenz mit Affenösophagus als Substrat für den Autoantikörper-Nachweis, SSS: Salt-Split-Skin-Technik, DIF: direkte Immun-fluereszenz, n. u.: nicht untersucht, ELISA mit rekombinanten BP180 NC16A (negativ <9 U/ml), +: positiver Nachweis von IgG-, C3 Ablagerungen an der Basalmembranzone periläsionaler Haut-biopsien.
Patientennr. Geschlecht Alter (J.) IIF SSS ELISA U/ml
DIF
BP-1 w 81 n.u. IgG 3699+ IgG+C3
BP-2 w 80 - IgG n.u. IgG+C3
BP-3 w 84 IgG IgG 264+ n.u.
BP-4 w 81 IgG IgG >1000+ IgG+C3
BP-5 w 76 IgG,IgA IgG,IgA 27,6+ IgG<C3
BP-6 w 93 IgG IgG 92,4+ C3
BP-7 m 83 - IgG 0,2 IgG+C3
BP-8 w 87 IgG IgG,IgA 37,5+ IgG+C3
BP-9 m 89 IgG IgG,IgA 51,7+ IgG<C3
BP-10 w 82 IgG IgG 16,8+ IgG+C3
BP-11 w 94 IgG IgG,IgA 366+ IgG+C3
BP-12 m 92 IgG IgG,IgA 8,4 IgG+C3
BP-13 m 80 n.u. n.u. n. u. IgG+C3
BP-14 w 57 - IgG,IgA 38,2+ IgG+C3
BP-15 m 92 IgG IgG 623+ IgG+C3
BP-16 w 83 IgG IgG 26,5+ IgG<C3
BP-17 w 88 IgG IgG 576.5+ IgG<C3
BP-18 w 92 IgG IgG 141+ -
BP-19 w 66 IgG,IgA IgG,IgA 53,3+ IgG+C3
BP-20 w 83 IgG IgG 92+ C3
4.3. Erhöhte Gesamt-IgE-Spiegel im Serum von BP-Patienten
Die Gesamt-IgE-Spiegel im Blut der untersuchten BP-Patienten und der Negativ-
kontrollen wurden mittels Fluoreszenz-Enzym-Immunoassay bestimmt (Labor der
Immunologie der Universität zu Lübeck). Dabei wurden bei 18 von 20 unbehan-
delten Patienten erhöhte IgE-Spiegel gemessen. Der Mittelwert der Gesamt-IgE-
Spiegel bei den BP-Patienten lag bei 2730 kU/l (im Bereich von 14,3–9430 kU/l
mit einem Ausreißer bei 20835 kU/l). Die Kontrollpersonen wiesen mittlere IgE-
Werte von 67 kU/l auf (Bereich 4,82–92,5 kU/l mit einem Ausreißer bei 397 kU/l).
Die Serum-Gesamt-IgE-Spiegel bei den BP-Patienten war damit im Vergleich zu
der Kontrollgruppe signifikant erhöht (p<0,05 im zweiseitigen Mann-Whitney-U-
Test) (s. Abb. 13).

ERGEBNISSE 48
Abb. 13: Serum Gesamt-IgE-Spiegel bei unbehandelten BP-Patienten (n=20) und gesunder in der Alters- und Geschlechtszusammensetzung vergleichbarer Normalseren als Negativ-kontrollen (n=10). Die Daten sind als Boxplot dargestellt; die Box zeigt das 5% Quartil und das 95% Quartil sowie den Median. Die Whisker zeigen Minimum und Maximum. Ausreißer der jeweili-gen Gruppe sind als Kreuz eingezeichnet. Das Kästchen stellt das arythmetrische Mittel dar.
Erhöhte IgE-Spiegel (d. h. Werte >100 kU/l) sind typisch für eine atopische Dia-
these. Um diese als mögliche Ursache der hier gefundenen IgE-Erhöhung abzu-
klären, wurde ein Atopie-Screening durchgeführt (inhalative Mischallergene, Mi-
schung SX1). Diese Mischung testet spezifisches IgE gegen Lieschgras (g6),
Roggen (g12), Birke (t3), Beifuß (w6), Katzenschuppen (e1), Hundeschuppen
(e5), Cladosporium Herbarum (m2) und Dermatophagoides pteronyssinus (d1).
Über Kreuzreaktivitäten werden fast alle Gras- und die häufigsten Baum und Kräu-
terpollen miterfasst. Bei der Gesamtauswertung zeigte sich, dass 7 der 18 Patien-
ten mit erhöhten Gesamt-IgE-Spiegeln, spezifische IgE-Antikörper gegen die
Mischallergene (SX1) aufwiesen, d. h. das spezifische IgE war >0,35 KU/l (RAST-
Klasse 1). Bei 6 Patienten erfolgte daraufhin eine Aufschlüsselung dieser Aller-
gene. Bei den nicht an BP erkrankten Personen waren 2 von 10 in der SX1-
Untersuchung positiv und wurden genauer klassifiziert.

ERGEBNISSE 49
Die RAST-Klassen-Einteilung richtet sich nach der Höhe des IgE-Antikörpertiters
gegen das untersuchte Antigen (s. Tab. 4). In der RAST-Klassifikation lagen 4 Se-
ren im Bereich der mittleren, nur ein Serum im hohen spezifischen IgE-Bereich.
Tab. 4: Immunologische Diagnostik der BP-Patientenseren und Kontrollseren. Bestimmung der gesamten Immunglobuline der Klasse IgE; Bestimmung spezifischer IgE-Antikörper gegen Mischal-lergene (SX1). Bei positivem Ausfall, d.h. >0,35 KU/l (RAST-Klasse 1), Aufschlüsselung der Mischallergene und wiederum Einteilung in RAST-Klassen. Klassen-Identifikation: <0,35 kU/l RAST-Klasse 0 0,35-0,70 kU/l RAST-Klasse 1 0,70-3,50 kU/l RAST-Klasse 2 3,50-17,50 kU/l RAST-Klasse 3 17,50-50,00 kU/l RAST-Klasse 4 50,00-100,00 kU/l RAST-Klasse 5 >100,00 kU/l RAST-Klasse 6
Patientennr. Geschlecht Alter(J.) Gesamt-IgE (kU/l)
Inhalative Allergene (SX1)
RAST- Klasse
BP-1 w 81 2178.0+ pos. n.u.
BP-2 w 80 14.30 neg. -
BP-3 w 84 9430.00+ neg. -
BP-4 w 81 154.00+ neg. -
BP-5 w 76 4349.0+ pos. 3 für d1
BP-6 w 93 1046.0+ neg. -
BP-7 m 83 3138.0+ pos. 2 für g6
BP-8 w 87 790.00+ neg. -
BP-9 m 89 2785.0+ pos. 2 für d1
BP-10 w 82 842.00+ pos. 5 für t3
BP-11 w 94 658.00+ pos. 0
BP-12 m 92 1902.0+ neg. -
BP-13 m 80 1482.0+ neg. -
BP-14 w 57 123.00+ neg. -
BP-15 m 92 417.00+ neg. -
BP-16 w 83 742.00+ neg. -
BP-17 w 88 20835+ pos. 2 für m2
BP-18 w 92 1734.0+ neg. -
BP-19 w 66 69.90 neg. -
BP-20 w 83 1929.0+ neg.
NHS-1 w 70 397.00+ neg. -
NHS-2 m 28 16.10 neg. -
NHS-3 w 80 92.50 neg. -
NHS-4 m 72 28.60 pos. 1 für d1
NHS-5 w 77 18.60 neg. -
NHS-6 w 93 35.90 pos. 1 für g12
NHS-7 w 85 44.50 neg. -
NHS-8 w 79 19.40 neg. -
NHS-9 w 43 4.82 neg. -
PV-01 K w 45 11.80 neg. -
Mittels zweiseitigem Mann-Whitney-U-Test konnte festgestellt werden, dass es
einen signifikanten Unterschied in der Höhe des Gesamt-IgE-Spiegels bei SX1-
positiven bzw. SX1-negativen Patienten gibt (p<0,05) (s. Abb. 14).
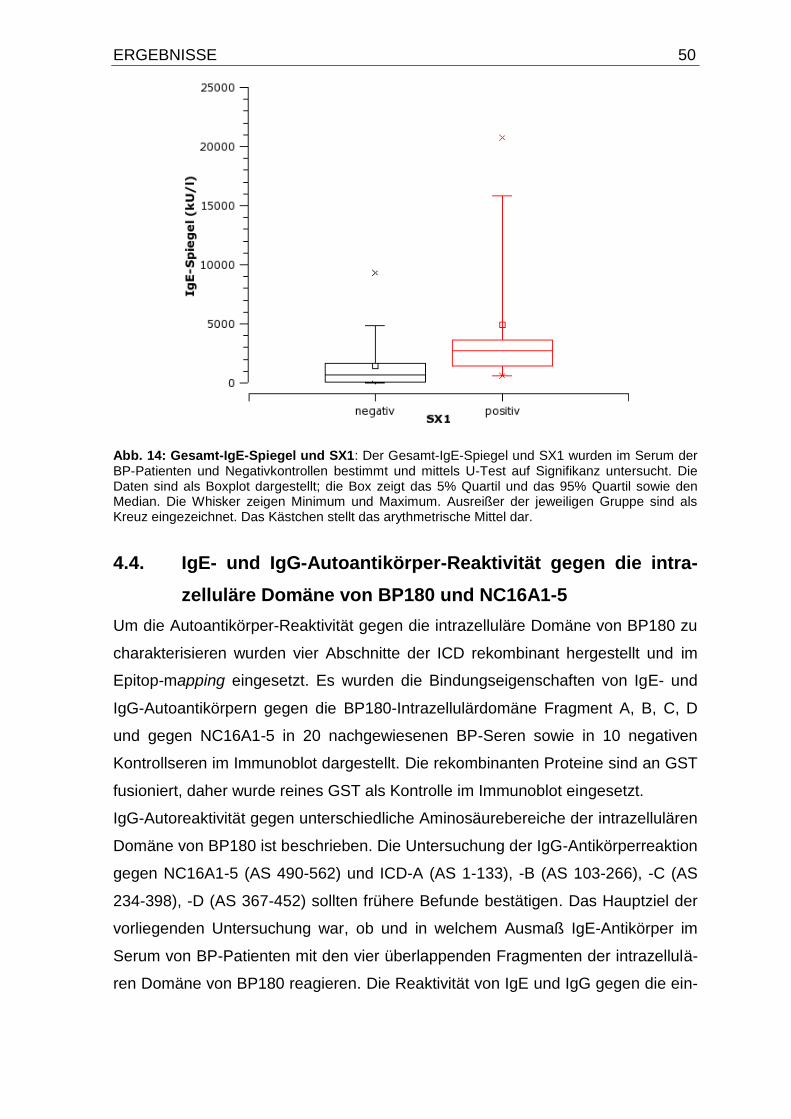
ERGEBNISSE 50
Abb. 14: Gesamt-IgE-Spiegel und SX1: Der Gesamt-IgE-Spiegel und SX1 wurden im Serum der BP-Patienten und Negativkontrollen bestimmt und mittels U-Test auf Signifikanz untersucht. Die Daten sind als Boxplot dargestellt; die Box zeigt das 5% Quartil und das 95% Quartil sowie den Median. Die Whisker zeigen Minimum und Maximum. Ausreißer der jeweiligen Gruppe sind als Kreuz eingezeichnet. Das Kästchen stellt das arythmetrische Mittel dar.
4.4. IgE- und IgG-Autoantikörper-Reaktivität gegen die intra-
zelluläre Domäne von BP180 und NC16A1-5
Um die Autoantikörper-Reaktivität gegen die intrazelluläre Domäne von BP180 zu
charakterisieren wurden vier Abschnitte der ICD rekombinant hergestellt und im
Epitop-mapping eingesetzt. Es wurden die Bindungseigenschaften von IgE- und
IgG-Autoantikörpern gegen die BP180-Intrazellulärdomäne Fragment A, B, C, D
und gegen NC16A1-5 in 20 nachgewiesenen BP-Seren sowie in 10 negativen
Kontrollseren im Immunoblot dargestellt. Die rekombinanten Proteine sind an GST
fusioniert, daher wurde reines GST als Kontrolle im Immunoblot eingesetzt.
IgG-Autoreaktivität gegen unterschiedliche Aminosäurebereiche der intrazellulären
Domäne von BP180 ist beschrieben. Die Untersuchung der IgG-Antikörperreaktion
gegen NC16A1-5 (AS 490-562) und ICD-A (AS 1-133), -B (AS 103-266), -C (AS
234-398), -D (AS 367-452) sollten frühere Befunde bestätigen. Das Hauptziel der
vorliegenden Untersuchung war, ob und in welchem Ausmaß IgE-Antikörper im
Serum von BP-Patienten mit den vier überlappenden Fragmenten der intrazellulä-
ren Domäne von BP180 reagieren. Die Reaktivität von IgE und IgG gegen die ein-

ERGEBNISSE 51
zelnen Epitope der Intrazellulärdomäne bzw. dem NC16A-Abschnitt von BP180
sollten dann auf eine mögliche Korrelation untersucht werden.
Die rekombinanten Proteine GST-hBP180-ICD-A, -B, -C, -D, GST-
hBP180NC16A1-5 und zur Kontrolle GST wurden mittels 15% SDS-PAGE fraktio-
niert, auf eine Nitrozellulosemembran transferiert und mit den zu untersuchenden
Seren inkubiert. Daraufhin erfolgte die Inkubation mit den sekundären Antikörpern:
HRP-konjugierter Maus-anti-Human-IgG- bzw. Maus-anti-human-IgE-Antikörper.
Eine Kreuzreaktion von anti-Maus-IgG mit humanem IgG sowie mit anderen Im-
munglobulinen und den Proteinen auf der Membran wurde ausgeschlossen, indem
der Maus-anti-human-IgE-Antikörper in einem Versuch weggelassen wurde. Für
die IgE- Erkennung musste zusätzlich die Inkubation mit HRP-konjugiertem anti-
Maus-Antikörper durchgeführt werden. Die Sichtbarmachung der Banden erfogte
mit DAB. Die Auswertung der Bandenverteilung erfolgte visuell. Die Intensität der
Fluoreszenz der Banden wurde wie folgt beschrieben: - keine Reaktion; + schwa-
che Reaktion; ++ mittelstarke Reaktion; +++ starke Reaktion der Autoantikörper
gegen das Protein. Die Blots wurden zur weiteren Objekti-vierung verblindet von
einer unabhängigen Person ausgewertet.
Ein BP-Serum zeigte IgE- und IgG-Antikörper-Bindungen an Glutathion-S-
Transferase (GST), ein weiterer Patient wies IgG-Antikörper gegen GST auf. Bei-
de wurden daher nicht in die Auswertung der Ergebnisse mit einbezogen. (s. Abb.
15 und Tab. 5)

ERGEBNISSE 52
35
[kDa]
NHS9
35
35
35
IgG IgE
BP3
BP15
BP4
BP7
NHS2
35
40
40
40
40
40NHS6
40
35
40
35
1 2 3 4 5 6 1 2 3 4 5 6
BP6
40
35
35
[kDa]
NHS9
35
35
35
IgG IgE
BP3
BP15
BP4
BP7
NHS2
35
40
40
40
40
40NHS6
40
35
40
35
1 2 3 4 5 6 1 2 3 4 5 6
BP6
40
35
Abb. 15: IgE- und IgG-Autoantikörper-Reaktivität gegen die 4 Abschnitte der intrazellulären Domäne von BP180 und NC16A1-5. 1: ICD-A, 2: ICD-B, 3: ICD-C, 4: ICD-D, 5: GST-Kontrolle, 6: NC16A1-5. BP: bullöses Pemphigoid; NHS: normales humanes Serum; rechts markieren die Pfeil-spitzen den Molekulargewichtsbereich.
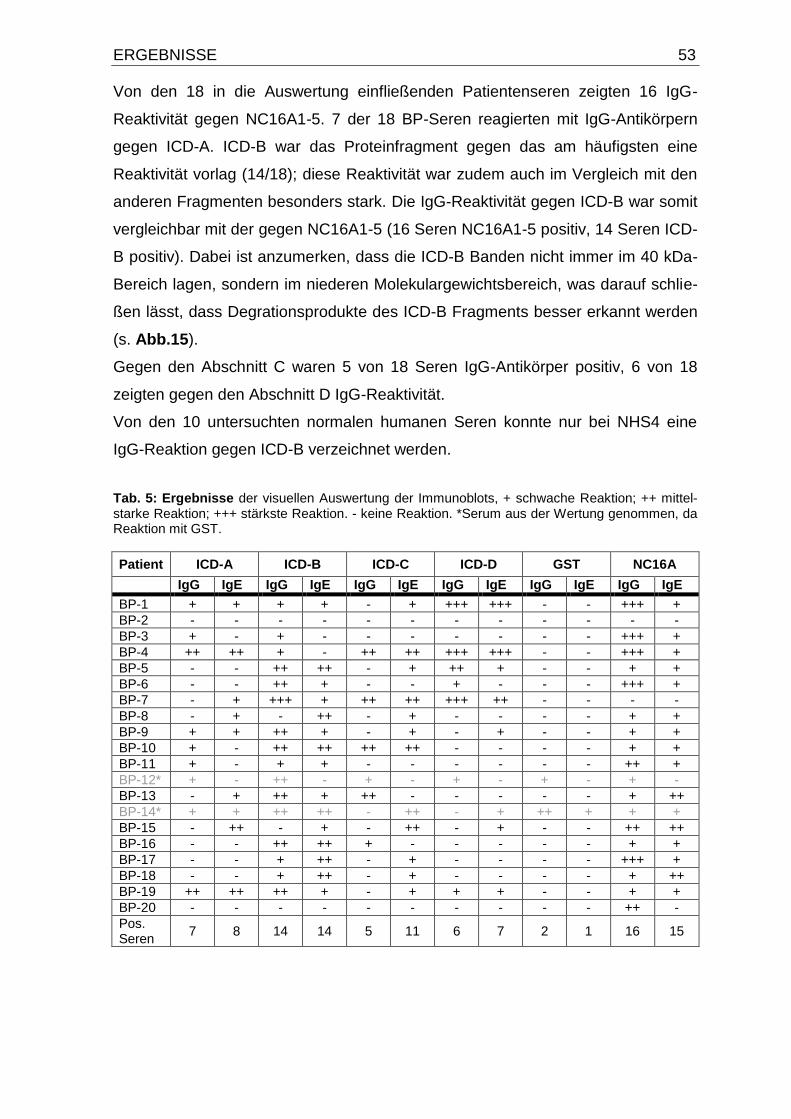
ERGEBNISSE 53
Von den 18 in die Auswertung einfließenden Patientenseren zeigten 16 IgG-
Reaktivität gegen NC16A1-5. 7 der 18 BP-Seren reagierten mit IgG-Antikörpern
gegen ICD-A. ICD-B war das Proteinfragment gegen das am häufigsten eine
Reaktivität vorlag (14/18); diese Reaktivität war zudem auch im Vergleich mit den
anderen Fragmenten besonders stark. Die IgG-Reaktivität gegen ICD-B war somit
vergleichbar mit der gegen NC16A1-5 (16 Seren NC16A1-5 positiv, 14 Seren ICD-
B positiv). Dabei ist anzumerken, dass die ICD-B Banden nicht immer im 40 kDa-
Bereich lagen, sondern im niederen Molekulargewichtsbereich, was darauf schlie-
ßen lässt, dass Degrationsprodukte des ICD-B Fragments besser erkannt werden
(s. Abb.15).
Gegen den Abschnitt C waren 5 von 18 Seren IgG-Antikörper positiv, 6 von 18
zeigten gegen den Abschnitt D IgG-Reaktivität.
Von den 10 untersuchten normalen humanen Seren konnte nur bei NHS4 eine
IgG-Reaktion gegen ICD-B verzeichnet werden.
Tab. 5: Ergebnisse der visuellen Auswertung der Immunoblots, + schwache Reaktion; ++ mittel-starke Reaktion; +++ stärkste Reaktion. - keine Reaktion. *Serum aus der Wertung genommen, da Reaktion mit GST.
Patient ICD-A ICD-B ICD-C ICD-D GST NC16A
IgG IgE IgG IgE IgG IgE IgG IgE IgG IgE IgG IgE
BP-1 + + + + - + +++ +++ - - +++ +
BP-2 - - - - - - - - - - - -
BP-3 + - + - - - - - - - +++ +
BP-4 ++ ++ + - ++ ++ +++ +++ - - +++ +
BP-5 - - ++ ++ - + ++ + - - + +
BP-6 - - ++ + - - + - - - +++ +
BP-7 - + +++ + ++ ++ +++ ++ - - - -
BP-8 - + - ++ - + - - - - + +
BP-9 + + ++ + - + - + - - + +
BP-10 + - ++ ++ ++ ++ - - - - + +
BP-11 + - + + - - - - - - ++ +
BP-12*
+ - ++ - + - + - + - + -
BP-13 - + ++ + ++ - - - - - + ++
BP-14* + + ++ ++ - ++ - + ++ + + +
BP-15 - ++ - + - ++ - + - - ++ ++
BP-16 - - ++ ++ + - - - - - + +
BP-17 - - + ++ - + - - - - +++ +
BP-18 - - + ++ - + - - - - + ++
BP-19 ++ ++ ++ + - + + + - - + +
BP-20 - - - - - - - - - - ++ -
Pos. Seren
7 8 14 14 5 11 6 7 2 1 16 15
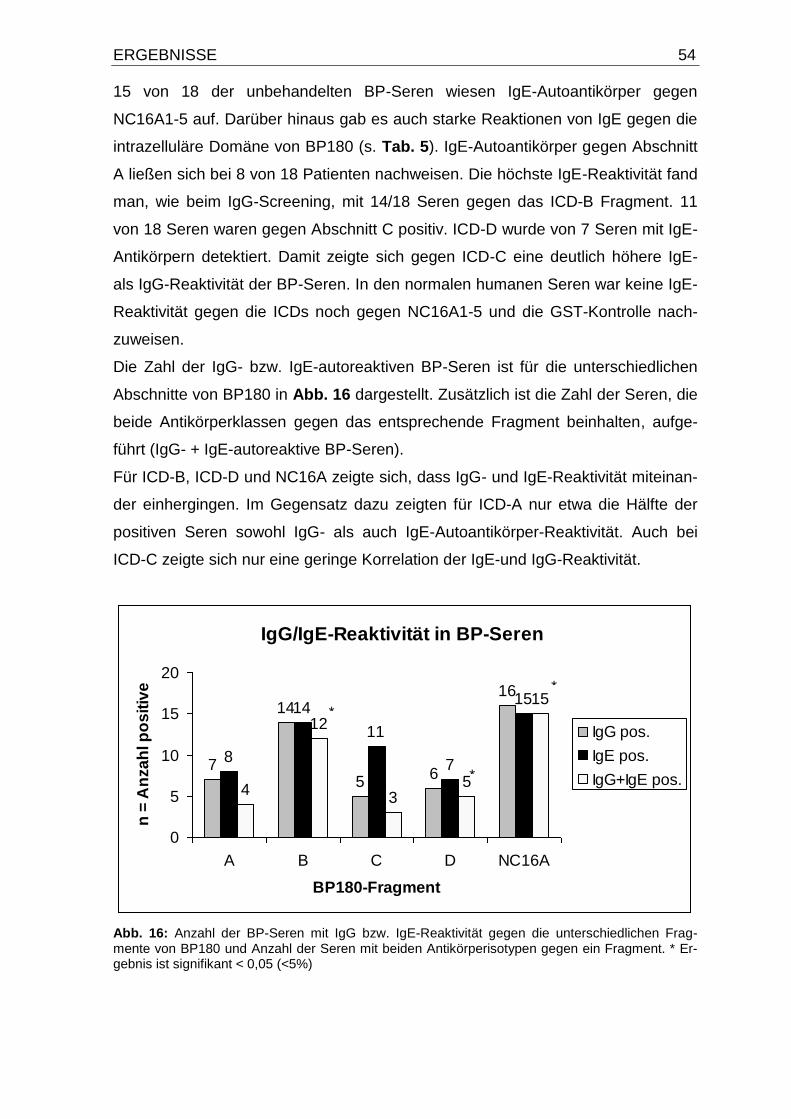
ERGEBNISSE 54
15 von 18 der unbehandelten BP-Seren wiesen IgE-Autoantikörper gegen
NC16A1-5 auf. Darüber hinaus gab es auch starke Reaktionen von IgE gegen die
intrazelluläre Domäne von BP180 (s. Tab. 5). IgE-Autoantikörper gegen Abschnitt
A ließen sich bei 8 von 18 Patienten nachweisen. Die höchste IgE-Reaktivität fand
man, wie beim IgG-Screening, mit 14/18 Seren gegen das ICD-B Fragment. 11
von 18 Seren waren gegen Abschnitt C positiv. ICD-D wurde von 7 Seren mit IgE-
Antikörpern detektiert. Damit zeigte sich gegen ICD-C eine deutlich höhere IgE-
als IgG-Reaktivität der BP-Seren. In den normalen humanen Seren war keine IgE-
Reaktivität gegen die ICDs noch gegen NC16A1-5 und die GST-Kontrolle nach-
zuweisen.
Die Zahl der IgG- bzw. IgE-autoreaktiven BP-Seren ist für die unterschiedlichen
Abschnitte von BP180 in Abb. 16 dargestellt. Zusätzlich ist die Zahl der Seren, die
beide Antikörperklassen gegen das entsprechende Fragment beinhalten, aufge-
führt (IgG- + IgE-autoreaktive BP-Seren).
Für ICD-B, ICD-D und NC16A zeigte sich, dass IgG- und IgE-Reaktivität miteinan-
der einhergingen. Im Gegensatz dazu zeigten für ICD-A nur etwa die Hälfte der
positiven Seren sowohl IgG- als auch IgE-Autoantikörper-Reaktivität. Auch bei
ICD-C zeigte sich nur eine geringe Korrelation der IgE-und IgG-Reaktivität.
IgG/IgE-Reaktivität in BP-Seren
7
14
56
16
8
14
11
7
15
4
12
35
15
0
5
10
15
20
A B C D NC16A
BP180-Fragment
n =
An
zah
l p
os
itiv
e
IgG pos.
IgE pos.
IgG+IgE pos.
*
*
*
Abb. 16: Anzahl der BP-Seren mit IgG bzw. IgE-Reaktivität gegen die unterschiedlichen Frag-mente von BP180 und Anzahl der Seren mit beiden Antikörperisotypen gegen ein Fragment. * Er-gebnis ist signifikant < 0,05 (<5%)
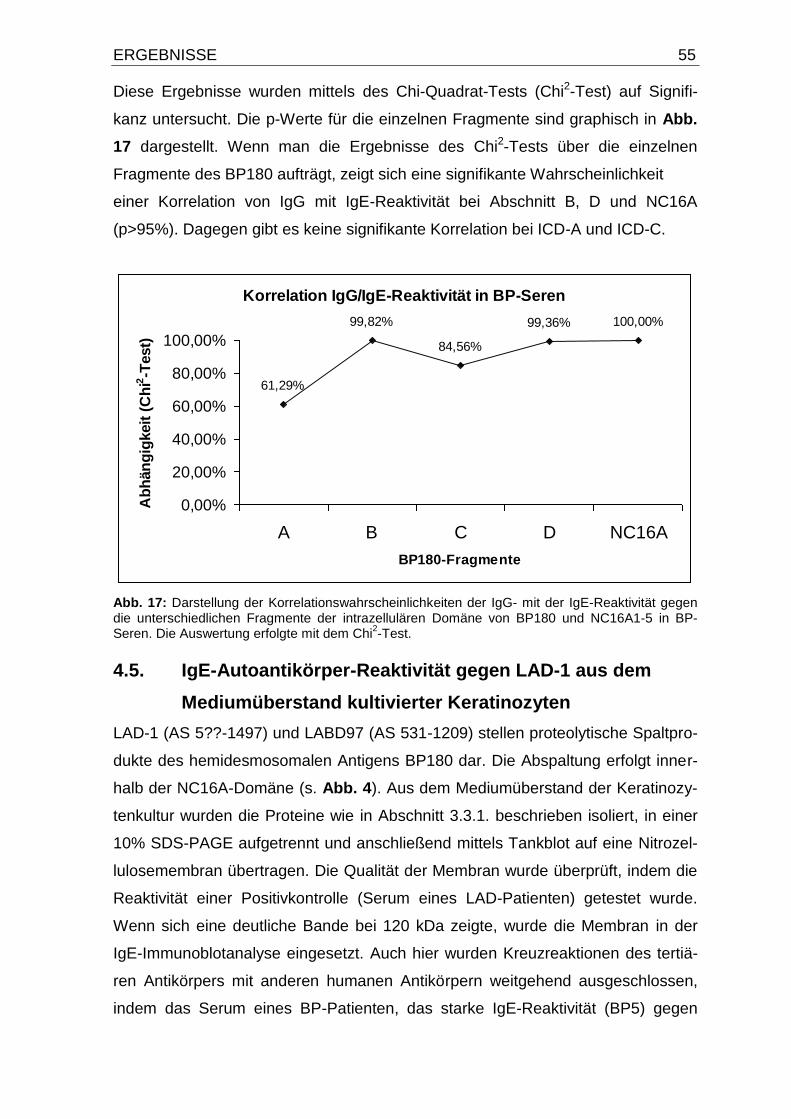
ERGEBNISSE 55
Diese Ergebnisse wurden mittels des Chi-Quadrat-Tests (Chi2-Test) auf Signifi-
kanz untersucht. Die p-Werte für die einzelnen Fragmente sind graphisch in Abb.
17 dargestellt. Wenn man die Ergebnisse des Chi2-Tests über die einzelnen
Fragmente des BP180 aufträgt, zeigt sich eine signifikante Wahrscheinlichkeit
einer Korrelation von IgG mit IgE-Reaktivität bei Abschnitt B, D und NC16A
(p>95%). Dagegen gibt es keine signifikante Korrelation bei ICD-A und ICD-C.
Korrelation IgG/IgE-Reaktivität in BP-Seren
84,56%
99,82% 99,36% 100,00%
61,29%
0,00%
20,00%
40,00%
60,00%
80,00%
100,00%
A B C D NC16A
BP180-Fragmente
Ab
hän
gig
keit
(C
hi2
-Test)
Abb. 17: Darstellung der Korrelationswahrscheinlichkeiten der IgG- mit der IgE-Reaktivität gegen die unterschiedlichen Fragmente der intrazellulären Domäne von BP180 und NC16A1-5 in BP-Seren. Die Auswertung erfolgte mit dem Chi
2-Test.
4.5. IgE-Autoantikörper-Reaktivität gegen LAD-1 aus dem
Mediumüberstand kultivierter Keratinozyten
LAD-1 (AS 5??-1497) und LABD97 (AS 531-1209) stellen proteolytische Spaltpro-
dukte des hemidesmosomalen Antigens BP180 dar. Die Abspaltung erfolgt inner-
halb der NC16A-Domäne (s. Abb. 4). Aus dem Mediumüberstand der Keratinozy-
tenkultur wurden die Proteine wie in Abschnitt 3.3.1. beschrieben isoliert, in einer
10% SDS-PAGE aufgetrennt und anschließend mittels Tankblot auf eine Nitrozel-
lulosemembran übertragen. Die Qualität der Membran wurde überprüft, indem die
Reaktivität einer Positivkontrolle (Serum eines LAD-Patienten) getestet wurde.
Wenn sich eine deutliche Bande bei 120 kDa zeigte, wurde die Membran in der
IgE-Immunoblotanalyse eingesetzt. Auch hier wurden Kreuzreaktionen des tertiä-
ren Antikörpers mit anderen humanen Antikörpern weitgehend ausgeschlossen,
indem das Serum eines BP-Patienten, das starke IgE-Reaktivität (BP5) gegen
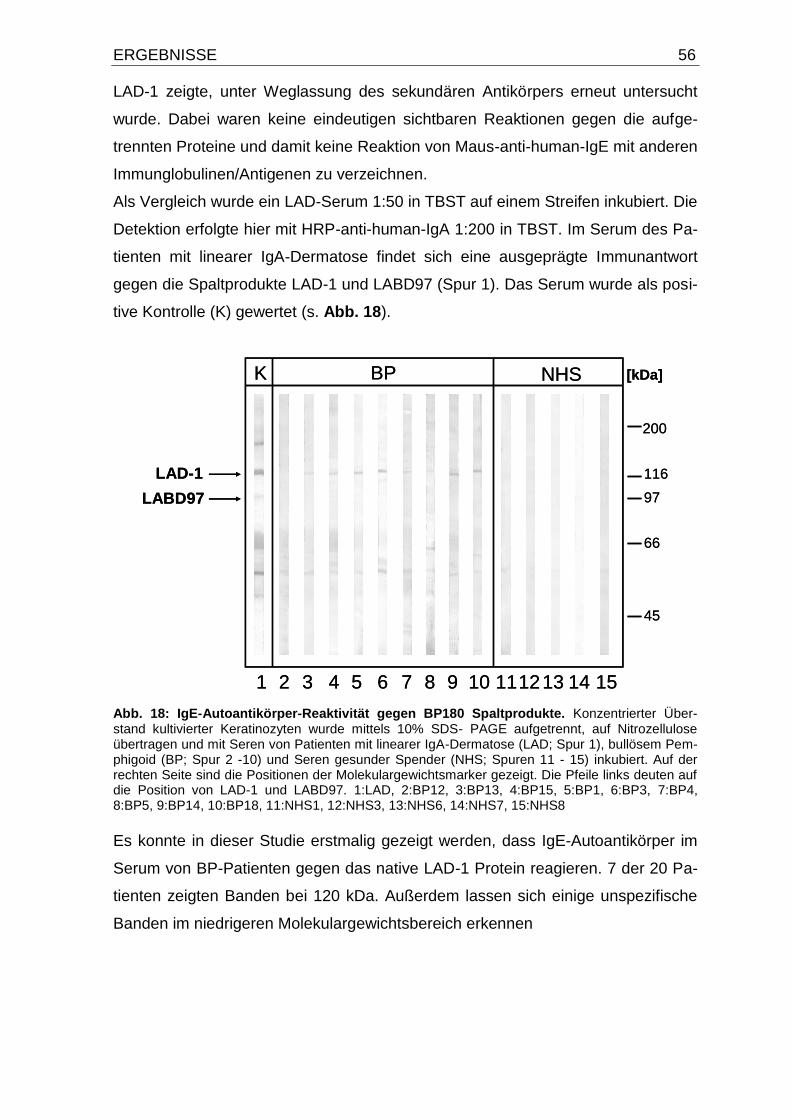
ERGEBNISSE 56
LAD-1 zeigte, unter Weglassung des sekundären Antikörpers erneut untersucht
wurde. Dabei waren keine eindeutigen sichtbaren Reaktionen gegen die aufge-
trennten Proteine und damit keine Reaktion von Maus-anti-human-IgE mit anderen
Immunglobulinen/Antigenen zu verzeichnen.
Als Vergleich wurde ein LAD-Serum 1:50 in TBST auf einem Streifen inkubiert. Die
Detektion erfolgte hier mit HRP-anti-human-IgA 1:200 in TBST. Im Serum des Pa-
tienten mit linearer IgA-Dermatose findet sich eine ausgeprägte Immunantwort
gegen die Spaltprodukte LAD-1 und LABD97 (Spur 1). Das Serum wurde als posi-
tive Kontrolle (K) gewertet (s. Abb. 18).
1 2 3 4 5 6 7 8 9 10 111213 14 15
LAD-1
LABD97
116
200
97
66
45
BP NHSK [kDa]
1 2 3 4 5 6 7 8 9 10 111213 14 15
LAD-1
LABD97
116
200
97
66
45
BP NHSK [kDa]
Abb. 18: IgE-Autoantikörper-Reaktivität gegen BP180 Spaltprodukte. Konzentrierter Über-stand kultivierter Keratinozyten wurde mittels 10% SDS- PAGE aufgetrennt, auf Nitrozellulose übertragen und mit Seren von Patienten mit linearer IgA-Dermatose (LAD; Spur 1), bullösem Pem-phigoid (BP; Spur 2 -10) und Seren gesunder Spender (NHS; Spuren 11 - 15) inkubiert. Auf der rechten Seite sind die Positionen der Molekulargewichtsmarker gezeigt. Die Pfeile links deuten auf die Position von LAD-1 und LABD97. 1:LAD, 2:BP12, 3:BP13, 4:BP15, 5:BP1, 6:BP3, 7:BP4, 8:BP5, 9:BP14, 10:BP18, 11:NHS1, 12:NHS3, 13:NHS6, 14:NHS7, 15:NHS8
Es konnte in dieser Studie erstmalig gezeigt werden, dass IgE-Autoantikörper im
Serum von BP-Patienten gegen das native LAD-1 Protein reagieren. 7 der 20 Pa-
tienten zeigten Banden bei 120 kDa. Außerdem lassen sich einige unspezifische
Banden im niedrigeren Molekulargewichtsbereich erkennen

ERGEBNISSE 57
Patient BP180 LAD-1
IgE- Reaktivität
BP-1 +++
BP-2 -
BP-3 +++
BP-4 +
BP-5 -
BP-6 -
BP-7 -
BP-8 -
BP-9 -
BP-10 -
BP-11 -
BP-12 -
BP-13 ++
BP-14 +++
BP-15 +++
BP-16 -
BP-17 -
BP-18 +++
BP-19 -
BP-20 -
Positive Seren1
6/20
Die Seren von gesunden Kontrollpatienten zeigten keine spezifische IgE-
Antikörper-Reaktivität gegen LAD-1 aus Mediumüberstand kultivierter Keratinozy-
ten. Eine Erkennung des Spaltproduktes LABD97 von Auto-IgE-Antikörper aus
Seren von BP-Patienten konnte nicht nachgewiesen werden. Es fiel jedoch die
Reaktivität des LAD1-Serums mit IgA- und vieler BP-Seren mit IgE-Antikörpern
gegen ein Protein im 50 kDa-Bereich auf. Jedoch zeigt auch ein gesundes Kon-
trollserum an dieser Stelle eine Bande.
Die Ergebnisse des Versuchs sind in Tab. 6 zusammengefasst. Eine repräsentati-
ve Auswahl an Blots ist in Abb. 18 dargestellt.
4.6. IgE-Autoantikörper-Reaktivität gegen BP180 und BP230 in
Keratinozytenextrakten
Neben Keratinozytenkulturüberstanden wurden auch Keratinozytenextrakte getes-
tet. Die Keratinozytenextrakte wurden wie in Abschnitt 3.3.2. beschrieben herge-
stellt, mittels 7,5% SDS-PAGE aufgetrennt und auf Nitrozellulosemembran über-
tragen. Es erfolgte eine Inkubation eines Nitrozellulosestreifens mit Serum eines
Patienten mit linearer IgA-Dermatose (LAD) (s. Abb.19). Alle 20 BP-Seren wurden
auf die IgE-Reaktivität gegen Keratinozytenextrakte getestet. Die Detektion der
Tab. 6: Ergebnisse der visuellen Auswer-tung der Immunoblots, + Reaktion; ++ starke Reaktion; +++ stärkste Reaktion. - keine Reaktion. *Serum aus der Wer-tung genommen, da Reaktion mit GST; 1 Anzahl der positiven Seren/totale Anzahl
der getesteten Seren.
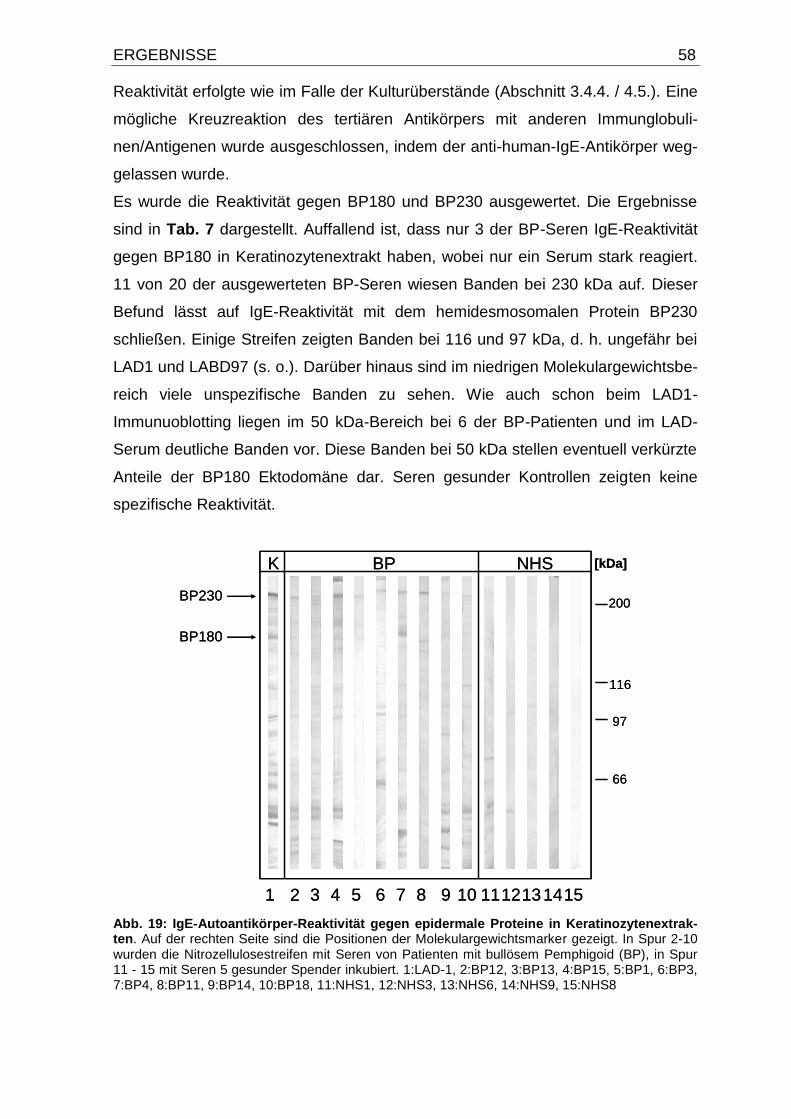
ERGEBNISSE 58
Reaktivität erfolgte wie im Falle der Kulturüberstände (Abschnitt 3.4.4. / 4.5.). Eine
mögliche Kreuzreaktion des tertiären Antikörpers mit anderen Immunglobuli-
nen/Antigenen wurde ausgeschlossen, indem der anti-human-IgE-Antikörper weg-
gelassen wurde.
Es wurde die Reaktivität gegen BP180 und BP230 ausgewertet. Die Ergebnisse
sind in Tab. 7 dargestellt. Auffallend ist, dass nur 3 der BP-Seren IgE-Reaktivität
gegen BP180 in Keratinozytenextrakt haben, wobei nur ein Serum stark reagiert.
11 von 20 der ausgewerteten BP-Seren wiesen Banden bei 230 kDa auf. Dieser
Befund lässt auf IgE-Reaktivität mit dem hemidesmosomalen Protein BP230
schließen. Einige Streifen zeigten Banden bei 116 und 97 kDa, d. h. ungefähr bei
LAD1 und LABD97 (s. o.). Darüber hinaus sind im niedrigen Molekulargewichtsbe-
reich viele unspezifische Banden zu sehen. Wie auch schon beim LAD1-
Immunuoblotting liegen im 50 kDa-Bereich bei 6 der BP-Patienten und im LAD-
Serum deutliche Banden vor. Diese Banden bei 50 kDa stellen eventuell verkürzte
Anteile der BP180 Ektodomäne dar. Seren gesunder Kontrollen zeigten keine
spezifische Reaktivität.
116
200
97
66
1 2 3 4 5 6 7 8 9 10 1112131415
BP NHSK
BP180
[kDa]
BP230
116
200
97
66
1 2 3 4 5 6 7 8 9 10 1112131415
BP NHSK
BP180
[kDa]
BP230
Abb. 19: IgE-Autoantikörper-Reaktivität gegen epidermale Proteine in Keratinozytenextrak-ten. Auf der rechten Seite sind die Positionen der Molekulargewichtsmarker gezeigt. In Spur 2-10 wurden die Nitrozellulosestreifen mit Seren von Patienten mit bullösem Pemphigoid (BP), in Spur 11 - 15 mit Seren 5 gesunder Spender inkubiert. 1:LAD-1, 2:BP12, 3:BP13, 4:BP15, 5:BP1, 6:BP3, 7:BP4, 8:BP11, 9:BP14, 10:BP18, 11:NHS1, 12:NHS3, 13:NHS6, 14:NHS9, 15:NHS8

ERGEBNISSE 59
Tab. 7: Ergebnisse der visuellen Auswertung der Immunoblots, + Reaktion; ++ starke Reaktion; +++ stärkste Reaktion. - keine Reaktion. *Serum aus der Wertung genommen, da Reaktion mit GST;
1 Anzahl der positiven Seren/totale Anzahl der getesteten Seren.
Patient BP230
in Keratinozytenextrakten BP180
in Keratinozytenextrakten
IgE- Reaktivität IgE- Reaktivität
BP-1 + +
BP-2 + -
BP-3 + -
BP-4 + +++
BP-5 ++ -
BP-6 ++ -
BP-7 - -
BP-8 - -
BP-9 - -
BP-10 + -
BP-11 - -
BP-12 + -
BP-13 - -
BP-14 - -
BP-15 - ++
BP-16 - -
BP-17 + -
BP-18 + -
BP-19 - -
BP-20 + -
Positive Seren1 11/20 3/20
4.7. IgE-Autoantikörper-Reaktivität gegen den C-terminalen
Abschnitt von BP180 (BP4575)
Der C-terminale Abschnitt von BP180 (BP4575) umfasst mit einem Molekularge-
wicht von 45 kDa den Aminosäurebereich 1365-1413. Er liegt in der Lamina densa
in der Haut und endet in einer Schlaufenstruktur (s. Abb. 1 und Abb. 2). Dieses
Protein wurde in E.coli rekombinant hergestellt und mir von Dr. med. C. Sitaru zur
Verfügung gestellt. Mittels Immunoblotanalyse wurde wie in Abschnitt 3.4.3. und
3.4.4. beschrieben die IgE- und IgG-Reaktivität von 20 BP-Seren und 10 Negativ-
kontrollen gegen dieses Fragment untersucht. 10 der 18 ausgewerteten BP-Seren
enthielten IgE-Autoantikörper gegen die rekombinant hergestellte Ektodomäne. In
der Regel wurden auf der Membran eine Doppelbande detektiert, welche auf eine
Degradierung des rekombinanten Proteins hinweist. Als Positivkontrolle diente der
aus immunisierten Kaninchen gewonnene anti-GST-Antikörper (SA8009). Die
NHS/PV-Serum wiesen keine IgE-Reaktivität gegen BP4575 auf. Eine Auswahl
repräsentativer Beispiele ist in Abb. 20 dargestellt. Die Tab. 8 zeigt die Ergebnis-
se der Immunoblotanalyse.

ERGEBNISSE 60
BP NHS
1 2 3 4 5 6 7 8 9 10 11 12
BP4575 45
[kDa]K BP NHS
1 2 3 4 5 6 7 8 9 10 11 12
BP4575 45
[kDa]K
66
Abb. 20: IgE-Autoantikörper-Reaktivität gegen den C-terminalen Abschnitt von BP180 (BP4575). Spur 1 zeigt die Positivkontrolle. In Spur 2-9 wurden die Nitrozellulosestreifen mit BP-Seren inkubiert, in Spur 10-12 mit normalen humanen Seren (NHS). 1: SA8009, 2: BP4, 3: BP6, 4: BP5, 5: BP13, 6: BP3, 7: BP15, 8: BP3, 9: BP17
Patient BP180 4575
IgE- Reaktivität
BP-1 -
BP-2 -
BP-3 +
BP-4 ++
BP-5 +
BP-6 -
BP-7 -
BP-8 -
BP-9 +
BP-10 -
BP-11 +
BP-12 (+++)*
BP-13 ++
BP-14 (+++)*
BP-15 +++
BP-16 -
BP-17 +
BP-18 -
BP-19 -
BP-20 -
Positive Seren1
6/18
Tab. 8: Ergebnisse der visuellen Auswer-tung der Immunoblots, + Reaktion; ++ starke Reaktion; +++ stärkste Reaktion; - keine Reaktion. *Serum aus der Wer-tung genommen, da Reaktion mit GST; 1 Anzahl der positiven Seren/totale An-
zahl der getesteten Seren.

ERGEBNISSE 61
4.8. Übersicht der Epitoperkennung für IgE auf dem
intrazellulären und extrazellulären Abschnitt von BP180
Tab
. 9:
Imm
un
ob
lotu
nte
rsu
ch
un
g 2
0 B
P-S
ere
n:
IgE
-Au
toan
tikö
rper-
Reakti
vit
ät
geg
en
BP
180
BP
, b
ullö
ses P
em
ph
igoid
; A
S,
Am
inosä
ure
n;
EC
D,
extr
azellu
läre
Do
mä
ne;
ICD
, in
trazellu
läre
Do
män
e.
1A
S 1
-133 p
räse
ntiere
n d
ie e
rste
n A
S
der
Ep
ito
psequ
enz (
sie
he F
ig.
2);
2S
ere
n im
Test
der
rekom
bin
ante
n P
rote
ine n
icht
gew
ert
et, d
a G
ST
positiv
; 3A
nzah
l der
Sere
n p
ositiv
en S
ere
n/
tota
le A
nzah
l d
er
gete
ste
ten S
ere
n.
*native P
rote
ine in
Kera
tin
iozyte
nextr
akte
n.

ERGEBNISSE 62
Aus den Ergebnissen der Gesamt-IgE-Bestimmung und den mittels Immunoblot-
analysen bestimmten IgE-Autoantikörper-Reaktivitäten in BP-Seren gegen rekom-
binante Abschnitte von BP180 und gegen isolierte native Proteine aus Keratinozy-
tenkulturen wurde versucht eine Gesetzmäßigkeit festzustellen. Die Tab. 9 fasst
die Ergebnisse der Epitopuntersuchung auf dem BP180 Molekül zusammen. Bei
der Betrachtung der Ergebnisse fällt auf:
1. Die BP-Seren 4, 13, 14, 15 zeigten in der Keratinozytenextrakt-, LAD-1- und
BP180 4575-Untersuchung IgE-Reaktionen. Auffällig dabei ist, dass diese Seren
im Gesamtdurchschnitt eher ein niedriges Gesamt-IgE bei negativem SX1 haben.
2. Das Serum BP4 reagiert mit Autoantikörpern gegen alle untersuchten Proteine,
außer gegen ICD-B und wenig gegen NC16A, dabei hat der Patient nur ein gering-
fügig erhöhtes Gesamt-IgE, bei keiner allergischen Disposition.
3. Patient 17 hat extrem hohe Gesamt-IgE-Werte mit Allergieneigung, reagierte
wenig mit IgE-AK gegen die BP180 Proteine.
4. BP3 hat ein hohes Gesamt-IgE, ein negatives SX1 und hat außer mit LAD1,
BP180 4575 keine IgE-Reaktionen.
5. Patienten, die keine IgE-Reaktivität gegen NC16A zeigten (3 von 18), haben
auch keine Reaktivität gegen die anderen untersuchten Abschnitte auf der extra-
zellulären Domäne von BP180.
6. Nur BP7 reagierte als einziges Serum mit dem gesamten intrazellulären Frag-
ment, jedoch ohne Banden bei NC16A und negativem NC16A-ELISA.
7. Zwei Seren (BP2 und BP20) zeigten gegen keinen der untersuchten Abschnitte
von BP180 IgE-Antikörper.
8. Zu erwähnen ist, dass nur 3 Seren IgE-Reaktivität gegen das gesamte BP180-
Molekül zeigten, jedoch die unterschiedlichen Abschnitte von BP180 in viel größ-
erer Anzahl erkannt wurden.
9. Sehr hohes Gesamt-IgE im Blut bei negativem SX1 ließ keine sichtbare Stei-
gerung der IgE-Reaktivitäten gegen BP180 Fragmente erkennen (siehe BP3). 10.
Auf der anderen Seite führten erhöhte Gesamt-IgE-Spiegel (>20.000 kU/l) mit
Nachweis von spezifischem IgE (BP17) ebenfalls zu Reaktionen gegen BP180
Abschnitte.
11. Bei den Negativkontrollen zeigte sich eine vereinzelte Bande in der IgG-
Reaktion gegen rekombinantes ICD-B, jedoch reagierte dieses Serum nicht mit
IgE-Autoantikörpern gegen diesen Abschnitt. Ansonsten waren keine IgG- und

ERGEBNISSE 63
IgE-Antikörper Reaktionen gegen BP180 im Serum der gesunden Kontrollen
nachzuweisen.
Abschließend lässt sich zusammenfassen, dass BP180 NC16A nicht die einzige
Antigendomäne für IgE-Autoantikörper beim bullösen Pemphigoid darstellt. Es
konnte in dieser Arbeit erstmals gezeigt werden, dass zusätzliche Epitope auf der
intrazellulären Domäne sowie auf dem extrazellulären Abschnitt von BP180 exis-
tieren. Es lassen sich aus den vorliegenden Ergebnissen keine Gesetzmäßigkei-
ten für die IgE-Antikörper-Epitoperkennung auf dem BP-180 Molekül erkennen.
Die IgE-Autoreaktivität gegen die untersuchten Abschnitte der BP180-
Intrazellulärdomäne korreliert teilweise mit der IgG-Autoreaktivität in BP-Seren.

DISKUSSION 64
5. Diskussion
Das bullöse Pemphigoid ist eine organspezifische Autoimmunerkrankung, die mit
subepidermaler Blasenbildung der Haut einhergeht. Sie ist eine Erkrankung des
höheren Lebensalters, ihr Verlauf ist chronisch und durch rezidivierende Schübe
gekennzeichnet. Schon lange ist bekannt, dass beim BP die Autoantikörper vor-
rangig gegen das intrazelluläre Protein BP230 (Tanaka et al., 1990) und das
transmembranöse Protein BP180 der Hemidesmosomen gerichtet sind (Giudice et
al., 1992; Labib et al., 1986; Mutasim et al., 1985; Stanley et al., 1988). Beide
Strukturproteine nehmen eine Schlüsselstellung bei der Verankerung basaler Ke-
ratinozyten in der Basalmembran ein (Borradori and Sonnenberg, 1999; Georgi et
al., 2001). Insbesondere den Autoantikörper gegen das BP180 wird eine enorme
pathogene Relevanz beim Verlust der Adhäsion der basalen Keratinozyten zuge-
wiesen.
BP180 ist ein aus 1497 Aminosäuren aufgebautes Glykoprotein und besteht aus
einem intrazellulär gelegenem globulären Kopf, einem zentralen, die Lamina luci-
da durchspannenden Abschnitt und einer extrazellulären flexiblen, schleifenartigen
Struktur in der Lamina densa (Masunaga et al., 1997; Nonaka et al., 2000). Der
extrazelluläre Abschnitt besteht dabei aus 15 unterbrochenen kollagenen Regio-
nen und 16 nicht-kollagenen Domänen (Zillikens and Giudice, 1999). Die sech-
zehnte nicht-kollagene Domäne (NC16A), die sich extrazellulär unmittelbar an die
Zellmembran des basalen Keratinozyten anschließt, stellt den immundominanten
Abschnitt von BP180 dar (Giudice et al., 1993; Giudice et al., 1994; Matsumura et
al., 1996; Zillikens et al., 1997). Neuere Untersuchungen zeigten darüber hinaus,
dass BP180 neben NC16A weitere antigene Strukturen, sowohl auf der extrazel-
lulären, als auch auf der intrazellulären Domäne enthält (Di Zenzo et al., 2004;
Egan et al., 1999; Mariotti et al., 2004; Murakami et al., 1996; Nakatani et al.,
1998; Nie and Hashimoto, 1999; Perriard et al., 1999; Schumann et al., 2000;
Thoma-Uszynski et al., 2006). Dieses erklärt die Beobachtung von Zillikens et al.,
dass die Antikörper-Reaktivität in BP-Seren, die mit der NC16A-Domäne vorinku-
biert waren, nicht vollständig ihre Aktivität gegen die ECD des BP180 verloren (Zil-
likens, Rose et al. 1997).
Die Bindung der Autoantikörper an die Strukturproteine der Haut führt zu einer
Aktivierung von Komplement und dadurch zur Rekrutierung von Leukozyten an
der Basalmembran. Neutrophile Granulozyten stoßen reaktive Sauerstoffradikale

DISKUSSION 65
und proteolytische Enzyme wie Gelatinase B und Elastase aus somit wird die
Funktion der Hemidesmosomen gestört und die Epidermis hebt sich von der Ba-
salmembran ab.
Neben Autoantikörpern der Klasse IgG wurde in der letzten Zeit immer häufiger
auf die pathologische Relevanz der IgE-Autoantikörper beim bullösen Pemphigoid
hingewiesen (Chen et al., 2002; Christophoridis et al., 2000; Delaporte et al.,
1996; Dimson et al., 2003; Dopp et al., 2000; Fairley et al., 2005; Provost and To-
masi, 1974). Gegenüber anderen bullösen Autoimmundermatosen werden beim
BP erhöhte Gesamt-IgE-Werte im Blut gefunden (Arbesman et al., 1974; Bows-
zyc-Dmochowska and Dmochowski, 2002; Dimson et al., 2003) . Durch die IgE-
Antikörper lässt sich der Einstrom von eosinophilen Granulozyten erklären, die im
histologischen Bild einer BP-Blase überwiegen bzw. der Nachweis von basophilen
Granulozyten in Gefrierschnitten der Haut. Erhöhte IgE-Spiegel in der Blasenflüs-
sigkeit von BP-Patienten wurden nachgewiesen. Auch die Bindung zirkulierender
IgE-Antikörper entlang der Basalmembran konnte in einigen BP-Fällen gezeigt
werden (Parodi and Rebora, 1992; Soh et al., 1993).
Die Ergebnisse der vorliegenden Arbeit implizieren die pathogenetische Relevanz
der IgE-Autoantikörper gegen BP180. 18 von 20 der untersuchten Patientenseren
zeigten erhöhte Gesamt-IgE-Werte, eine ähnlich hohe Anzahl, wie auch von an-
deren Autoren beschrieben (Arbesman et al., 1974; Bowszyc-Dmochowska and
Dmochowski, 2002; Dimson et al., 2003). Von den Negativkontrollen wies nur eine
Serumprobe erhöhte IgE-Werte auf. Um falsch-positive IgE-Werte aufgrund einer
atopischen Disposition auszuschließen, wurde mittels ImmunoCap 100 (Firma:
Phadia, Freiburg) ein Atopen-Screening (Mischung SX-1) durchgeführt. Die Be-
funde des Sreening legen nahe, dass die erhöhten IgE-Titer und die Erkrankung
mit der blasenbildenden Autoimmundermatose BP zusammenhängen (siehe Ab-
schnitt 4.3.). Da Juckreiz und urtikarielle Plaques der Blasenbildung beim BP häu-
fig vorausgehen, könnte dies im Zusammenhang mit den erhöhten Serum-
Gesamt-IgE-Spiegeln bei unbehandelten BP-Patienten ein Hinweis dafür sein,
dass IgE möglicherweise eine Rolle in der frühen Phase des bullösen Pemphi-
goids zukommt.
Zu erwähnen ist, dass ein erhöhtes IgE auch andere Ursachen haben kann, z. B.
parasitäre Infektion, Hyper-IgE-Syndrom, Erkrankungen mit T-Zell-Dysfunktion

DISKUSSION 66
(AIDS, M. Hodgkin, Non-Hodgkin-Lymphome der T-Zell-Reihe), maligne Tumore,
IgE-Gammopathie, infektiöse Mononukleose und andere virale Erkrankungen.
Zum großen Teil handelte es sich aber um Patienten von der Station 10c der
Dermatologie des UKSH, Campus Lübeck, klinisch lag kein Anhalt für oben ge-
nannte Krankheiten vor.
5.1. Expression und Aufreinigung der verschiedenen
rekombinanten BP180 Polypeptide
In der vorliegenden Arbeit sollten die antigenen Determinanten auf der extrazellu-
lären sowie insbesondere der intrazellulären Domäne von BP180 für Autoantikör-
per der Isotypen IgG und IgE charakterisiert werden und eventuelle Unterschiede
in der Reaktivität beider Antikörperklassen gegen einzelne Abschnitte des BP180
aufgezeigt werden.
Um eine Kartierung der Epitope der intrazellulären Domäne von BP180 durchfüh-
ren zu können, wurde sie willkürlich in vier sich überlappende Fragmente unter-
teilt. Die Proteine wurden in E.coli exprimiert, über Affinitätschromatographie auf-
gereinigt und im Western-Blot ihrer Größe nach aufgetrennt. ICD-A, -B, -C, -D
migrierten um wenige kDa unterschiedlich zu ihrer berechneten Größe. Die Pro-
teine konnten in der Immunoblotanalyse eingesetzt werden, da deutliche Banden
im 40 kDa-Bereich auf der Membran zu erkennen waren. In einem Kontroll-
Western-Blot mit dem Kaninchen Serum SA8009 (Sitaru et al., 2002), gewonnen
durch Immunisierung mit GST-BP180 NC16A2-4, reagierten die Proteine positiv
(s. Abb. 12, Abschnitt 4.1.). Darüber hinaus wiesen die Fragmente A, B, C, D zu-
sätzliche schmale Banden mit niedrigerem Molekulargewicht auf. Diese Banden
lassen sich am ehesten als Degradierungsprodukte der Proteine deuten (s. Er-
gebnisse).
Die Instabilität der Proteinabschnitte der BP180-Intrazellulärdomäne deutet dar-
aufhin, dass durch die willkürliche Unterteilung einer Proteineinheit in vier Frag-
mente die Ausbildung von stabilen Proteindomänen gestört wird. Weiterere Effekte
dieser Unterteilung sind zudem, dass sich zum einen verschiedene konformatio-
nelle Epitope nicht ausbilden können, zum anderen Neoepitope freigelegt werden.
Dennoch zeigten die durchgeführten Experimente, dass die Fragmente A, B, C, D
der intrazellulären BP180 Domäne zur Analyse der Reaktivitäten geeignet waren.

DISKUSSION 67
Der rekombinant hergestellte C-terminale Abschnitt GST-BP180 4575 (AS 1365 -
1413) zeigte in der Immunoblotanalyse eine Doppelbande, welche ebenfalls auf
ein Degradierungsprodukt hinweist. Im Unterschied dazu zeigte rekombinantes
NC16A1-5 eine deutlich höhere Stabilität. In der Dissertation von J. Korn wird aus
den Ergebnissen einer Konformationsanalyse durch einen trypsin-vermittelten Pro-
teinabbau die Vermutung angestellt, dass die nicht-kollagenen Abschnitte eher
gegen einen proteolytischen Angriff von Trypsin geschützt sind als die kollagenen
Abschnitte des BP180 (Korn, 2006). Auch in der vorliegenden Arbeit war im Ge-
gensatz zu kollagenem BP4575 bei NC16A kaum eine Degradierung zu verzeich-
nen.
5.2. IgE-Autoreaktivität und IgG-Autoreaktivität gegen die
extrazelluläre Domäne von BP180
Um die Stellung der IgE- und IgG-Autoantikörper in der Pathophysiologie des bul-
lösen Pemphigoids weiter zu klären, wurden die Antigenbindungsstellen auf der
extrazellulären des Antigens BP180 untersucht.
15 der 18 unbehandelten BP-Patienten wiesen in der vorliegenden Untersuchung
IgE-Autoantikörper gegen NC16A1-5 auf, ein im Vergleich zu anderen Arbeiten
höherer Anteil. Bei Fairly et al. zeigten 5 von 10 unbehandelten BP-Patienten IgE-
Antikörper gegen NC16A1-5. Dabei wird sowohl von IgG als auch von IgE haupt-
sächlich die NC16A2 Subdomäne (Abb. 4) erkannt (Fairley et al., 2005). Dimson
et al. diagnostizierten bei 16 von 23 unbehandelten Patienten mit BP, IgE-
Autoantikörper gegen BP180, davon reagierten 15 Patienten gegen NC16A. Bei
einem BP-Serum wird vermutet, dass dessen IgE-Autoantikörper mit anderen De-
terminanten auf der Ektodomäne reagierten (Dimson et al., 2003). Dopp et al. wie-
sen bei 55% der BP-Seren IgE-Autoantikörper gegen NC16A nach. Sie beschrie-
ben zudem, dass die Höhe der Serum-Titer dieser Immunglobuline die Krank-
heitsaktivität widerspiegelt (Dimson et al., 2003; Dopp et al., 2000; Fairley et al.,
2005). Christophoridis et al. fanden dagegen bei 100% der untersuchten BP-Seren
IgE-Antikörper gegen die Ektodomäne von BP-180 (Christophoridis et al., 2000).
Möglicherweise beruhen diese zum Teil deutlichen Differenzen auf der unter-
schiedlichen Verdünnung der Patientenseren oder sind auf unterschiedliche
Krankheitsstadien des untersuchten Patientenkollektivs zurückzuführen. Auch

DISKUSSION 68
kann es am Testsystem liegen, d. h. am eingesetzten rekombinant hergestellten
Protein.
Wie von Fairley et al. beschrieben ist das spezifische Profil der IgG- und
IgE-Autoantikörper gegen unterschiedliche Abschnitte des NC16A-Moleküls sehr
ähnlich (Fairley et al., 2005). Auch in der vorliegenden Arbeit zeigten 16 der 18
Patienten in der Immunoblotanalyse IgG-Antikörper gegen NC16A. Keiner der Ne-
gativkontrollen zeigte IgG-Banden im 35 kDa-Bereich. Dieses Ergebnis stimmt mit
den Aussagen vieler Studien überein, die NC16A als Hauptantigene Determinante
BP180 beschreiben. Parallel dazu wurde in der Immundiagnostik der Dermatolo-
gie UKSH, Campus Lübeck ein NC16A ELISA durchgeführt. Hierbei wiesen eben-
falls 16 der 18 untersuchten Patientenseren einen positiven NC16A ELISA auf,
wobei ELISA- und Immunblot-Ergebnisse nicht immer korrelieren. Ein Grund da-
für kann eine mögliche Veränderung des Antigens durch die Hitzeeinwirkung im
Rahmen des Immunoblotten sein. Außerdem werden in der ELISA-Diagnostik Ab-
schnitte von NC16A1-5 eingesetzt und das Protein kann daher von BP-Seren im
ELISA und Immunoblot unterschiedlich erkannt werden.
Das C-terminale Ende des BP180 gilt als weiteres Antigen für BP-Autoantikörper.
Es entspricht ungefähr dem Aminosäure-Bereich ab AS 1320 und besitzt eine
Schlaufenstruktur in der Lamina densa (Nonaka et al., 2000). Di Zenso et al. iden-
tifizierten 2 überlappende Epitope auf diesem C-terminalen Anteil des BP180 Mo-
leküls (AS 1331-1387 und AS 1354-1406). 19% der untersuchten BP-Serum-
Proben hatten an diese Abschnitte gebunden (Di Zenzo et al., 2004). Diese Bin-
dungsregion (AS 1331-1406) ist mit denen aus der Studie von Murakami et al.
(AS1365-1413) vergleichbar (Murakami et al., 1998). Auch in der Veröffentlichung
von Nakatani et al. wird die Reaktivität von IgG-Autoantikörper gegen das Fusi-
onsprotein BP180 4575 (AS 1365-1413) beschrieben. Dabei reagierten 23,5% der
BP-Seren mit diesem Abschnitt (Nakatani et al., 1998).
Auch in der vorliegenden Arbeit wurde das BP180-Molekül auf weitere Au-
toantikörper-Bindungsstellen auf dem C-terminalen Abschnitt untersucht. Dabei
wurde mit dem rekombinant hergestellten GST-Fusionsprotein BP180 4575 gear-
beitet. Das Augenmerk wurde in dieser Arbeit auf die IgE-Reaktivität mit diesem
Abschnitt gelegt (s. unten).

DISKUSSION 69
Des Weiteren wurde in dieser Studie die IgE-Reaktivität gegen LAD-1 un-
tersucht. Dieses 120-kDa-Protein entspricht weitgehend der extrazellulären Do-
mäne von BP180 (AS 5??-1497) und entsteht durch proteolytische Abspaltung
aus dem Gesamtprotein. Die Position des N-Terminus des 120 kDa Fragments
kann bis heute nicht eindeutig festgelegt werden (Franzke et al., 2002), es wird die
Aminosäure 524 oder 528 vermutet (Hirako et al., 2003). Im Westernblot konnte
vor einiger Zeit IgG-Autoreaktivität in BP-Seren mit LAD-1 nachgewiesen werden
(Hirako et al., 1998; Pas et al., 1997; Schmidt et al., 2000a; Schmidt et al., 2000b).
5.2.1. Nachweis von IgE-Autoreaktivität gegen den C-terminalen
Abschnitt von BP180 (BP4575) und gegen LAD-1
8 der 18 in dieser Arbeit gewerteten BP-Seren weisen IgE-Autoantikörper gegen
den rekombinant hergestellten C-terminalen Abschnitt BP180 4575 auf. Interes-
sant ist, dass 1 von 3 IgE-Anti-NC16A negativen Seren, eine IgE-Bande gegen
BP180 4575 lieferte (s. Tab. 9) und somit ein Indiz dafür ist, dass neben NC16A
noch andere Epitope auf dem extrazellulären Abschnitt des BP180 Molekül exis-
tieren.
Neben diesen Untersuchungen zur IgG-/IgE–Reaktivität gegen rekombinant her-
gestellte BP180-Fragmenten ist in der vorliegenden Arbeit die IgE-Autoantikörper
Reaktivität in BP-Seren mit dem natürlichen proteolytischen Spaltfragmenten LAD-
1 und LABD97 des BP180 Moleküls im Immunoblot untersucht worden.
Es konnte erstmals gezeigt werden, dass unbehandelte BP-Patienten (7 von 20)
IgE-Autoantikörper gegen LAD-1 aufweisen. Die Negativkontrollen zeigten keine
Reaktivität. LABD97 (AS 531-1209) wurde von den BP-Seren nicht erkannt. Da
das proteolytische Spaltprodukt LAD-1 ein nativ isoliertes Protein darstellt und
nicht rekombinant hergestellt wurde, ist seine Erkennung durch IgE-Autoantikörper
besonders interessant. Da der BP180 4575 (AS 1365-1413) Abschnitt ein Teil der
LAD-1 Domäne (AS 5??-1497) darstellt, würde man vermuten, dass Patienten mit
Reaktivität gegen 4575 auch mit LAD-1 reagieren. Jedoch nur 4 der 8 Patienten
mit Anti-4575-Antikörpern reagierten mit LAD-1. Die 4 anderen Patienten zeigten
nur Banden bei 4575, 2 Patienten hatten IgE-Antikörper gegen LAD und keine
Reaktion bei BP4575. Diese Beobachtung lässt darauf schließen, dass LAD-1
durch die proteolytische Abspaltung seine Form verändert.

DISKUSSION 70
5.3. IgG-Autoreaktivität gegen einen immundominanten
Abschnitt auf der intrazellulären Domäne von BP180
Bereits in früheren Arbeiten ist auch die intrazelluläre Domäne von BP180 als Au-
toantigen für BP-Autoantikörper der Klasse IgG beschrieben worden (Di Zenzo et
al., 2004; Perriard et al., 1999). Dabei gibt es nach Perriard et al. mehrere anti-
gene Determinanten auf dieser Domäne, wobei die zentrale Region der ICD den
immundominante Abschnitt für IgG-Autoantikörper darstellt. 59% der BP-Patienten
mit BP180-Reaktivität enthielten IgG-Autoantikörper gegen ein Fusionsprotein aus
der ICD (AS 114-200) und einer Sequenz des K-Ras Proteins. Die Reaktivität von
IgG-Antikörpern in BP-Seren gegen exprimierte ICD des BP180 wurde weiterhin
durch Immunfluoreszenz ermittelt (Perriard et al., 1999). Übereinstimmend mit
diesen Befunden beschrieb Di Zenso et al, dass 21% der getesteten BP-Seren
IgG-Autoantikörpern gegen die ICD-Aminosäureregion 151-203 besitzen, 10,5%
der Patienten erkannten den Aminosäurebereich 299-354. Diese Resultate spre-
chen stark für die Immunogenität der zentralen Region der ICD. Die Idee wurde
zudem durch ihre Beobachtung gestützt, dass Maus-anti-BP180-Antikörper mit
den AS 151-170 reagierten, ein Indiz, dass dieser Bereich auch von Mäusen als
immunogen erkannt wird (Di Zenzo et al., 2004).
In der vorliegenden Arbeit wurde die IgG-Autoreaktivität in 20 nicht medi-
kamentös vorbehandelten BP-Seren gegen die vier rekombinant hergestellten Ab-
schnitte der BP180-Intrazellulärdomäne im Immunoblot untersucht. 18 Seren wur-
den in der Auswertung berücksichtigt. 2 Seren wurden nicht in die Wertung mit
einbezogen, da diese Antikörper gegen das rekombinante Kopplungsprotein Glu-
tathion-S-Transferase aufzeigten. 7/18 der Patienten lassen Banden bei ICD-A
(AS1-133) und 14/18 bei ICD-B (103-266) erkennen. Abschnitt C (234-398) und D
(367-452) werden von IgG-Autoantikörper in 5/18 bzw. in 6/18 der untersuchten
Patientenseren erkannt. Dieses Resultat ist in guter Übereinstimmung mit früheren
Aussage von Perriard et al, dass BP-Seren mehrere Epitope auf dem intrazellulä-
ren Abschnitt des BP180 Proteins erkennen (Perriard et al., 1999) und zeigt, dass
IgG-Autoantikörper zum größten Teil gegen den zentralen Aminosäurebereich
103-266 der ICD von BP180 gerichtet sind.
Die Beobachtung, dass das unmittelbar an der Zellmembran lokalisierte NC16A
die immundominante Region von BP180 darstellt, lässt vermuten, dass Auto-
Antikörper besonders an ICD-D binden, da diese über die transmembranöse

DISKUSSION 71
Komponente des BP180 am nächsten in Kontakt zu NC16A steht. Diese Überle-
gung wurde anhand der vorliegenden Ergebnisse jedoch nicht bestätigten. Sie
unterstützen vielmehr die Aussage von Anderen, dass der zentrale Aminosäurebe-
reich als Hauptantigendeterminante erkannt wird (Di Zenzo et al., 2004; Perriard et
al., 1999). Diese entspräche dem Abschnitt ICD-B in der vorliegenden Arbeit, mit
dem 14 der 18 untersuchten Patientenseren reagierten.
1 der 10 Kontrollseren wies eine starke IgG-Bande gegen ICD-B Bereich auf. Die-
se Reaktivität gesunder Seren gegen die intrazellulären Bestandteile des BP180
lassen sich möglicherweise durch das Erkennen der rekombinant hergestellten, d.
h. nicht in nativer Form vorliegenden Proteine erklären. Darüber hinaus kann mög-
licherweise auch eine hohe Konzentration an IgG im Blut zu Kreuzreaktionen füh-
ren.
5.4. IgE-Autoreaktivität gegen die intrazelluläre Domäne von
BP180
In der vorliegenden Arbeit wurde erstmals gezeigt, dass in BP-Seren IgE-
Autoantikörper gegen die BP180-Intrazellulärdomäne existieren.
8 von 18 Patienten haben IgE-Autoantikörper gegen ICD-A, 14/18 Patienten ge-
gen ICD-B und 7/18 gegen ICD-D. Diese Zahl ähnelt der Anzahl der Seren, die mit
IgG-Autoantikörper gegen Abschnitt A, B und D reagierten. Darüber hinaus zeigte
sich in der vorliegenden Untersuchung, dass gegen Abschnitt C sogar mehr IgE-
(11 positiv von 18 Patienten) als IgG-Reaktivität vorhanden ist (5 positiv von 18).
Überraschend ist die fast gleich hohe Reaktivität der Auto-IgE-Antikörper gegen
ICD-B (14/18) und NC16A1-5 (16/18) des BP180 Moleküls.
3 Seren mit z. T. besonders starker IgE-Reaktion gegen die ICD, wiesen keine
IgE-Reaktion mit NC16A auf. Diese Beobachtung, dass es Patienten mit z. T.
starken Reaktionen gegen die ICD jedoch weder IgG- noch IgE-Autoantikörpern
gegen NC16A1-5 bei gleichzeitig negativen NC16A-ELISA gibt, weist daraufhin,
dass außer NC16A auch andere Abschnitte eine immundominante Rolle sowie
eine pathogenetische Relevanz in der Entstehung des BPs spielen.
Gesetzmäßigkeiten in der Kombination der AK-Reaktionen gegen die intra-
zellulären bzw. gegen die extrazellulären BP180-Fragmente konnten anhand der
Daten dieser Arbeit nicht aufgezeigt werden. Auch die Aussage von Perriard et al.,
dass zwischen der extrazellulären und intrazellulären BP-180-Domäne kein Kreuz-

DISKUSSION 72
reaktivität besteht, ist mit den Ergebnissen der Studie vereinbar (Perriard et al.,
1999). Kein Serum der Kontrollgruppe wies Reaktionen mit IgE-Antikörpern gegen
die rekombinant hergestellten intrazellulären Abschnitte auf.
Wie von Fairley et al. beschrieben, ist das spezifische Profil der IgG- und IgE-
Autoantikörper gegen NC16A sehr ähnlich (Fairley et al., 2005). Dimson et al ver-
öffentlichte 2003, dass im Serum des BP-Patienten IgE-Autoantikörper mit den-
selben Zielantigenen wie IgG-Autoantikörper reagierten (Dimson et al., 2003).
Dieses wurde in der vorliegenden Arbeit bestätigt. Ein Großteil der untersuchten
Seren zeigte ein sehr ähnliches Profil in der Reaktion der IgE- und IgG-
Autoantikörper mit den Abschnitten der BP180-Intrazellulärdomäne und NC16A.
Der Chi-Quadrat-Tests zeigte, dass die Ergebnisse der Bindungseigenschaften
der beiden Antikörperklassen gegen die ICD-B, -D und NC16A1-5 mit sehr hohen
Wahrscheinlichkeiten abhängig voneinander sind (siehe Abschnitt 4.4., Abb. 17).
Zu den Ergebnissen ist anzumerken, dass die IgG- und IgE-Untersuchungen in
einigen Fällen zu breiten Banden im Immunoblot mit den ICDs vor allem bei Ab-
schnitt B und D führten (s. Abschnitt 4.4). Dieses lässt darauf schließen, dass BP-
Seren zum Teil auch mit Abbauprodukten der rekombinant hergestellten Proteine
reagierten.
Die Ähnlichkeit des IgG- und IgE-Bindungsmusters gegen BP180 lässt vermuten,
dass beide Isotypen durch klonale Selektion entstanden sind. Das bedeutet, alle
B-Zellen beginnen in der Initialantwort auf ein Antigen mit der Synthese von IgM.
Durch spezifische Signale wie IL-4, IL-13 und die Bindung von CD40 an seinen
Liganden (CD40L) an der Oberfläche von B-Zellen erfolgt ein Wechsel der Anti-
körperklasse, die die B-Zelle exprimiert (Gibbs et al., 1996; Prussin and Metcalfe,
2003). Durch DNA-Umlagerungen ausgetauschter Gen-Segmente wird die Pro-
duktion eines anderen Antikörper-Isotyps hervorgerufen. Da dieses Isotypen-
switching nicht die vollständig determinierende Region der neuen Antikörperklasse
betrifft, wird die Spezifität dieser Antikörper-Klassen die gleiche sein, wie die Im-
munglobuline, die in der primären Immunantwort dominieren. In einigen Fällen
aber kann die somatische Hypermutation der vollständig determinierenden Region
während des Isotypen-switching zu einer unterschiedlichen Bindungsaffinität zum
Zielantigen führen (Chaudhuri and Alt, 2004).
Da die Konzentration von IgE im Serum eines Patienten ungefähr 10.000-
100.000-fach geringer ist als von IgG, wurde zur Inkubation mit den Proteinen eine

DISKUSSION 73
Verdünnung des Serums von 1:100 für die IgG-Detektion bzw. 1:2 für die Detek-
tion von IgE vorgenommen. Die Konzentration an IgG-Antikörpern im 1:2 verdünn-
ten Serum ist daher sehr hoch und die Immunglobuline konkurrieren eventuell um
die Bindungsstellen auf den rekombinanten Proteinen. Da die IgE-Reaktivität so-
mit nicht detektierbar ist, lässt sich erklären, warum die IgG-Reaktivität in einigen
Fällen nicht mit IgE-Reaktivität korreliert.
Für die Sichtbarmachung der gebundenen IgE-Autoantikörper wurden der
sekundäre Antikörper anti-human-IgE von der Maus und ein tertiärer anti-Maus-
HRP-Antikörper verwendet. Die Ergebnisse der IgE-Autoreaktivität gegen die ICDs
und NC16A lassen sich daher möglicherweise auch erklären, dass einer dieser
Detektionantikörper an gebundene humane Antikörper bindet. Auch wäre ein un-
spezifisches Andocken des sekundären bzw. tertiären Antikörpers an die aufge-
trennten Proteinen möglich. Dieses wurde insoweit ausgeschlossen, indem zu Be-
ginn der Versuche eine Membran mit Serum und folgend mit anti-Maus-Antikörper
unter Auslassung des sekundären Antikörpers inkubiert wurde. Auf dieser Mem-
bran waren keine Banden zu verzeichnen. Auch die starke IgG-Reaktion des Kon-
trollserums gegen ICD-B ohne entsprechende Bande im IgE-Blot schließt eine
Kreuzreaktion weitgehend aus.
In früheren Studien konnte gezeigt werden, dass Autoantikörper in lebende Zellen
eindringen, ihre intrazellulären Ziele erreichen und ihre Funktion erfüllen können
(Alarcon-Segovia 1996). Daher ist es denkbar, dass Autoantikörper gegen die ICD
von BP180 in der Pathogenese des BP von Bedeutung sind. Die ICDs enthalten
Sequenzen, die an der Einbindung des BP180 in den Hemidesmosomalen Plaque
beteiligt sind (Borradori et al., 1997; Hopkinson et al., 1995). Darüber hinaus bein-
haltet die zentrale Region der ICD von BP180 Bindungsstellen, die kritisch für die
Interaktionen des BP180 mit 4-Untereinheit von 64-Integrin sind (Aho and Uitto,
1998; Borradori et al., 1997; Schaapveld et al., 1998). Basierend auf diesen Be-
funden ist es denkbar, dass Autoantikörper gegen die intrazelluläre Domäne von
BP180 in der dermo-epidermalen Spaltbildung beteiligt sind, indem sie die Inter-
aktion von BP180 mit anderen Elementen des Hemidesmosomens stören. An die-
ser Störung könnten nicht nur IgG-und IgE-Antikörper, sondern auch IgE-
assozierte Zellen (sog. allergische Effektorzellen) beteiligt sein. Diese Zellen ver-
ursachen u. a. Symptome wie Juckreiz, Rötung und Urtikaria. Besonders die ba-

DISKUSSION 74
sophilen Granulozyten haben durch Ausschüttung von IL-4, IL-13 und CD40L im-
munmodulatorische Wirkung auf die Th-Zellen (Falcone et al., 2000; Gibbs, 2005;
Gibbs et al., 1996), die in der Pathogenese des BPs mitwirken (Thoma-Uszynski
et al., 2006) (s. Abb. 6).
5.5. IgE-Autoreaktivität gegen das gesamte BP180-Molekül
und BP230
Bei der Betrachtung der Ergebnisse fällt auf, dass die IgE-Reaktivität gegen
BP180 in Keratinozytenextrakten viel geringer ist, als die IgE-Erkennung der un-
terschiedlichen Abschnitte auf dem BP180 Molekül vermuten ließ. Nur 3 von 20
Seren wiesen IgE-Banden gegen BP180 auf, hingegen wird allein schon NC16A
durch 15 von 18 Seren erkannt und auch gegen die ICDs kann eine deutlich höhe-
re IgE-Reaktivität verzeichnet werden. Eine Erklärung dafür könnte sein, dass der
Blot auf Keratinozytenextrakten nicht so sensitiv ist. Vielleicht wurde bei der Her-
stellung der Extrakte der Lysepuffer auch zu lange auf dem Zellrasen belassen,
wodurch evtl. ein beginnender proteolytischer Proteinverdau durch Proteasen
stattfand, so dass BP180 v. a. als Spaltprodukt vorlag. Auch eine Unspezifität der
ICD-Fragmente wäre eine mögliche Erklärung, diese kann jedoch durch die nega-
tiv ausfallenden Normalseren ausgeschlossen werden. Möglicherweise kommt es
durch die willkürliche Abschnittseinteilung der BP180-Intrazellulärdomäne zu einer
Auffaltung der Proteine und somit zu einer neuen bzw. besseren Epitoperkennung.
Auffallend ist auch, dass die Reaktion gegen BP230 in Keratinozytenextrakten fast
viermal so häufig positiv ausfällt wie gegen BP180 (11 positiv von 20). Möglicher-
weise werden intrazellulär gelegene Proteine von IgE-Autoantikörpern verstärkt
erkannt.
Die erhöhte Reaktivität gegen LAD-1 im Vergleich zu BP180 lässt darauf schlie-
ßen, dass LAD-1 durch die proteolytische Abspaltung seine Form verändert und
so vermehrt von Autoantikörpern erkannt wird.

DISKUSSION 75
5.6. IgE-Autoantikörper besitzen mehrere Epitope auf dem
BP180-Molekül
Die Identifikation von mehreren Regionen mit IgE-Antikörper-Reaktivität auf dem
BP180 Molekül konnte in der Arbeit bestätigt werden. Dabei konnten Epitope auf
der IC-Domäne neu beschrieben werden. Eine Vielzahl von Hinweisen zeigen an,
dass im Laufe einer Autoimmunerkrankung die B- und T-Zell-Antworten nicht auf
einzelne immundominante Epitope beschränkt sind, sondern, dass darüber hinaus
zusätzliche Epitope innerhalb eines Proteins erkannt werden (Chan et al., 1998;
Vanderlugt and Miller, 1996). Dieses Phänomen, das als Epitop-spreading be-
zeichnet wird, scheint für die Unterhaltung und Progression der Autoimmunerkran-
kung wichtig zu sein. Die Beobachtung, dass Autoantikörper beim BP zwei Mole-
küle BP180 und BP230 als Ziel haben und darauf eine Vielzahl von Epitopen er-
kennen, lässt vermuten, dass Epitop-spreading ebenfalls beim Krankheitsverlauf
des BP eine Rolle spielt. Zum Epitop-spreading beim BP gibt es unterschiedliche
Hinweise. Es wurde beobachtet, dass Autoantikörper gegen die extrazelluläre
Domäne von BP180 eine Initiatorrolle bei der Entwicklung des BP zukommt, wäh-
rend Antikörper, die gegen zytoplasmatische Antigendeterminanten wie BP230
gerichtet sind, als zweites Ereignis angesehen werden (Giudice et al., 1993; Liu et
al., 1993). Andere Autoren dagegen vermuten aufgrund ihres Epitop-Musters,
dass die Bindung von Autoantikörpern an die ICD in der frühen Phase der Erkran-
kung stattfindet und somit eine bisher unbekannte Bedeutung bei der Auslösung
und Entwicklung der Erkrankung spielen könnten (Di Zenzo et al., 2004). In der
vorliegenden Studie konnte darüber keine Aussage getroffen werden. Um dieses
zu klären, sind weitere Untersuchungen mit BP-Seren unterschiedlicher Erkran-
kungsphasen notwendig.
Die BP180-Intrazellulärdomäne könnte möglicherweise eine pathogenetische Be-
deutung beim BP haben, indem sie „versteckte“ Epitope beinhaltet. Es gibt die
Hypothese, dass bestimmte Epitope eines Proteins nicht vom Immunsystem
wahrgenommen werden, so dass z. B. Autoantikörper hier keine Wirkung zeigen
können, allerdings sich auch keine Toleranz ausbildet. Durch molekulares Mimikry
dieser „versteckten“ Epitope durch z. B. virale oder bakterielle Erreger können An-
tikörper induziert werden. Wenn eine solche „versteckte“ Autoimmunität besteht,
erhöht sich die Wahrscheinlichkeit, dass durch intramolekulares Epitop-spreading
echte pathogene Autoantikörper entstehen. Ein solches Geschehen wird u. a. für

DISKUSSION 76
den endemischen Pemphigus foliaceus (Fogo selvagem) angenommen (Li et al.,
2003).
Es ist möglich, dass es sich bei den ICDs ebenfalls um „versteckte“ Epitope han-
delt, gegen die Autoantikörper gebildet werden, die jedoch nicht pathogen sind.
Durch Epitop-spreading auf die NC16A-Domäne bildet sich dann das volle Krank-
heitsbild, das bullöse Pemphigoid, aus.
Thoma-Uszynski et al. demonstrierten mit ihren Ergebnissen, dass BP180
und zu einem geringeren Anteil BP230 die wichtigsten Antigene für autoreaktive T-
Zellen und IgG-Autoantikörper produzierende B-Zellen beim aktiven BP sind. Da-
bei werden das N-terminale Ende, der mittlere Anteil und das C-terminale Ende
des BP180 weitaus konstanter von T-und B-Zellen erkannt, als das BP230 Pro-
tein. Die BP180-Spezifität der T-und B-Zellen ist sehr ähnlich (Thoma-Uszynski et
al., 2006). Sie berichten weiterhin, dass die unterschiedliche Epitoperkennung auf
BP180 vom Krankheitsstadium abzuhängen scheint; beim ausgedehnten Krank-
heitsbild wird hauptsächlich der N-terminale Teil, beim limitierten Krankheitsbefund
eher der zentrale Teil des Moleküls von T- und B-Zellen erkannt.
In der vorliegenden Arbeit konnte keine Korrelation zwischen dem klinischen Sta-
dium, der Schwere der Erkrankung und der Fein-Spezifität des Anti-BP180-
Antikörpers aufgestellt werden. Hier sind weitere Untersuchungen von BP-
Patienten mit unterschiedlich starker Ausprägung der Erkrankung sowie von Se-
ren, die Patienten im Verlauf ihrer Erkrankung entnommen werden, notwendig.
Gesetzmäßigkeit der IgE-Bindungseigenschaften gegen die Ektodomäne
und den Abschnitten des intrazellulären Anteils des BP180 lassen sich nicht er-
kennen (s. Tab. 9). Als Beispiel sei hier das Patientenserum BP7 angeführt: starke
Reaktion der IgE-Autoantikörper mit ICD, aber keine Banden bei der Unter-
suchung mit NC16A und BP180 4575. Dieses bestätigt die Aussage von Perriard
et al., dass zwischen der extrazellulären und intrazellulären Domäne des BP180
Proteins keine Antigenkreuzreaktivität besteht (Perriard et al., 1999).

DISKUSSION 77
5.7. Ausblick
In der vorliegenden Arbeit konnte gezeigt werden, dass IgE-Autoantikörper multip-
le Autoantigene, mehr als bisher angenommen, erkennen und so in der Pathophy-
siologie des bullösen Pemphigoids beteiligt sind. Es wurde dargestellt, dass außer
NC16A weitere Epitope für IgE-Autoantikörper sowohl auf der extrazellulären als
auch erstmalig beschrieben auf der intrazellulären Domäne existieren. Für die
Reaktion der IgG-Autoantikörper konnte eine Erkennung der intrazellulären Do-
mäne bestätigt werden und eine Übereinstimmung mit den dominanten Aminosäu-
rebereichen auf der BP180-Intrazellulärdomäne aus anderen Arbeiten gefunden
werden. Beide Antikörperisotypen reagieren jedoch nicht unabhängig voneinander
gegen die ICDs, so dass für die alleinige Reaktion der IgE-Autoantikörper und de-
ren genauen Relevanz in der Entstehung des BPs keine Aussage getroffen wer-
den kann. Aus den Ergebnissen vorheriger Veröffentlichungen und anhand der
Ergebnisse der vorliegenden Arbeit lässt sich jedoch stark vermuten, dass auch
Mastzellen und basophile Granulozyten, aktiviert durch spezifische IgE-abhängige
und IgE-unabhängige Trigger, eine Rolle in der Pathogenese des BPs spielen.
Durch die Ausschüttung von IL-4, IL-13 durch Basophile wird die Th2- Immunant-
wort moduliert und es erfolgt ein Antikörperisotypen-switching zu IgE, welches
wiederum basophile und eosinophile Granulozyten aktiviert (Abb. 6).
Aus den Ergebnissen der vorliegenden Arbeit ergeben sich folgende Fragen:
1. Sind die IgE-Autoantikörper gegen Strukturproteine der Haut, die u. a. basophile
und eosinophile Granulozyten aktivieren und die Th2-Immunantwort stimulieren
und somit die weitere Produktion von IgE anregen, die primären Auslöser?
Oder
2. Entstehen die IgE-Autoantikörper erst sekundär, d. h. nach Bindung von IgG an
Strukturproteine der DEJ, Komplementaktivierung und der Einwanderung von
Mastzellen, neutrophiler, eosinophiler und basophiler Granulozyten, die ihrerseits
durch die Ausschüttung von IL-4 ein Antikörperisotypen-switching hervorrufen?
Durch die IgE-Synthese kommt es zu einer weiteren Aktivierung von Fc-Rezeptor
exprimierenden Zellen.
3. Gegen welchen Abschnitt von BP180 werden primär Autoantikörper gebildet,
kommt der BP180-Intrazellulärdomäne eventuell eine sehr viel größere pathoge-
netische Bedeutung zu als bisher angenommen?

DISKUSSION 78
Bei der Beantwortung dieser Fragen könnte ein Nachweis von IgE-
Autoantikörpern gegen BP180 an Gefrierschnitten humaner Haut hilfreich sein.
Auch der histologische Nachweis von basophilen Granulozyten und Mastzellen in
der Haut von BP-Patienten im Bereich von BP-Läsionen könnte weitere Hinweise
geben, in welchem Ausmaß allergische Effektorzellen an der Entstehung des bul-
lösen Pemphigoids beteiligt sind. Zur genaueren Zuordnung der für IgE-Antikörper
pathogenetisch relevanten Krankheitsphase müsste man Patienten im Verlauf be-
obachten. Dabei sollte klinisch auf Juckreiz und Urtikaria geachtet werden. Sero-
logisch sollte man zu unterschiedlichen Zeitpunkten das Gesamt-IgE bestimmen
und wiederholte Immunoblotanalysen durchführen. Weiterhin könnte man vor der
IgE-Diagnostik die IgG-Antikörper aus den BP-Seren entfernen, um Kreuzreaktio-
nen völlig auszuschließen.
Diese Untersuchungen und Beantwortungen der Fragen könnte die Rolle von IgE-
Autoantikörper beim BP und eventuell auch bei anderen Autoimmundermatosen
weiter klären. Damit könnten neue Wege für die pharmakologische Therapie von
blasenbildenden Autoimmundermatosen eröffnet werden.

ZUSAMMENFASSUNG 79
6. Zusammenfassung
Das bullöse Pemphigoid (BP) ist die häufigste subepidermal blasenbildende Auto-
immundermatose beim Menschen. Die Erkrankung ist gekennzeichnet durch IgG-
Autoantikörper gegen das hemidesmosomale Protein BP180 (Typ-XVII-Kollagen),
das für die Verankerung basaler Keratinozyten auf der kutanen Basalmembran
sorgt. Der massiven Einstrom von eosinophilen Granulozyten, der Nachweis von
basophilen Granulozyten und Mastzellen in der Dermis sowie die klinischen Symp-
tome (z. B. Juckreiz) weisen auf die Beteiligung von IgE-Antikörpern in der Patho-
physiologie des BP hin. In der vorliegenden Arbeit sollte mittels Immunoblotdiag-
nostik die Rolle der IgE-Autoantikörper gegen unterschiedliche Epitope auf BP180
aufgeklärt sowie eine mögliche Korrelation der IgE-und IgG-Autoreaktivität über-
prüft werden. Zudem wurde der Serum-Gesamt-IgE-Spiegel der Patienten und
Kontrollen bestimmt und im Zusammenhang mit den Ergebnissen des Epitop-
mappings betrachtet. Dazu wurde die IgE- und IgG-Autoreaktivität gegen ver-
schiedene rekombinant hergestellte intra- (ICD) und extrazelluläre (ECD) Frag-
mente von BP180 sowie gegen aus Keratinozytenkultur isolierte native Abschnitte
von BP180 im Serum von 20 nicht medikamentös vorbehandelten BP-Patienten
und 10 Negativkontrollen untersucht. Die rekombinanten Proteine, hBP180 ICD-A,
-B, -C, -D sowie der C-terminale Abschnitt (BP180 4575), wurden in E.coli expri-
miert. Das proteolytische Spaltprodukt von BP180 (LAD-1) wurde aus dem Me-
diumüberstand kultivierter HaCaT-Zellen isoliert.
Die Gesamt-IgE-Spiegel im Serum der BP-Patienten waren im Vergleich zur Kont-
rollgruppe signifikant erhöht (p<0,05). 15 der 18 ausgewerteten Patienten zeigten
IgE-, 16 IgG-Reaktivität gegen NC16A1-5. Zusätzlich konnten bei 8 der 18 BP-
Seren IgE-Bindungen gegen BP4575 nachgewiesen werden. In der vorliegenden
Arbeit wurde erstmals demonstriert, dass in BP-Seren neben IgG- auch IgE-
Autoantikörper gegen die BP180-ICD existieren. Die Hauptantigendeterminante
liegt dabei auf dem zentralen Abschnitt der ICD (hier ICD-B (AS103-266) 12/18).
Es zeigte sich eine Korrelation der IgE- und IgG-Autoreaktivitäten gegen die Epi-
tope der BP180-ICD Abschnitt B und D. Ebenfalls konnte erstmalig gezeigt wer-
den, dass IgE-Autoantikörper gegen LAD-1 (7 von 20 positiv) gerichtet sind. Die
Kontrollseren zeigten keine IgE-Reaktivität gegen die untersuchten BP180-
Proteine auf. Die Resultate unterstützen die These, dass NC16A nicht das einzige
Epitop für IgE-Autoantikörper beim bullösen Pemphigoid ist. Vielmehr existieren

ZUSAMMENFASSUNG 80
zusätzliche Epitope für IgE-Antikörper auf der ICD und ECD von BP180. Die Hete-
rogenität der Autoimmunantwort gegen BP180, lässt auf ein Epitop-spreading
während des Krankheitsverlaufs schließen. IgE-Autoantikörper spielen vermutlich
eine entscheidende Rolle in der Pathogenese des BPs, indem sie über den Fc-
Rezeptor-I die Aktivität von Leukozyten und Mastzellen beeinflussen.

LITERATURVERZEICHNIS 81
7. Literaturverzeichnis
Ahmed, A.R., Chu, T.M. and Provost, T.T. (1977) Bullous pemphigoid: clinical and serologic evaluation for associated malignant neoplasms. Arch Dermatol, 113, 969.
Aho, S. and Uitto, J. (1998) Direct interaction between the intracellular domains of bullous pemphigoid antigen 2 (BP180) and beta 4 integrin, hemidesmo-somal components of basal keratinocytes. Biochem Biophys Res Commun, 243, 694-699.
Alonso-Llamazares, J., Rogers, R.S., 3rd, Oursler, J.R. and Calobrisi, S.D. (1998) Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol, 37, 508-514.
Amagai, M., Hashimoto, T., Shimizu, N. and Nishikawa, T. (1994) Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vul-garis antigen (Dsg3) produced by baculovirus. J Clin Invest, 94, 59-67.
Amagai, M., Karpati, S., Prussick, R., Klaus-Kovtun, V. and Stanley, J.R. (1992) Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest, 90, 919-926.
Ambach, A., Zillikens, D., Klingert, B., Hartmann, A.A. and Burg, G. (1992) [Im-mune phenotyping of mononuclear infiltrate in bullous pemphigoid]. Hau-tarzt, 43, 81-85.
Ameglio, F., D'Auria, L., Bonifati, C., Ferraro, C., Mastroianni, A. and Giacalone, B. (1998) Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol, 138, 611-614.
Arbesman, C.E., Wypych, J.I., Reisman, R.E. and Beutner, E.H. (1974) IgE levels in sera of patients with pemphigus or bullous pemphigoid. Arch Dermatol, 110, 378-381.
Aubry, J.P., Pochon, S., Graber, P., Jansen, K.U. and Bonnefoy, J.Y. (1992) CD21 is a ligand for CD23 and regulates IgE production. Nature, 358, 505-507.
Baggiolini, M. and Dahinden, C.A. (1994) CC chemokines in allergic inflammation. Immunol Today, 15, 127-133.
Bernard, P., Aucouturier, P., Denis, F. and Bonnetblanc, J.M. (1990) Immunoblot analysis of IgG subclasses of circulating antibodies in bullous pemphigoid. Clin Immunol Immunopathol, 54, 484-494.
Bernard, P., Bedane, C. and Bonnetblanc, J.M. (1997) Anti-BP180 autoantibodies as a marker of poor prognosis in bullous pemphigoid: a cohort analysis of 94 elderly patients. Br J Dermatol, 136, 694-698.
Bibel, D.J. (1988) Milestones in Immunology. Springer Verlag, Berlin. Bird, P., Friedmann, P.S., Ling, N., Bird, A.G. and Thompson, R.A. (1986) Sub-
class distribution of IgG autoantibodies in bullous pemphigoid. J Invest Dermatol, 86, 21-25.
Blum, H., Beier, H. and Gross, J.H. (1987) Improved silver staining of plant pro-teins, RNA and DNA polyacrylamid gels. Electrophoresis, 8, 93-99.
Blumer, K.J. and Johnson, G.L. (1994) Diversity in function and regulation of MAP kinase pathways. Trends Biochem Sci, 19, 236-240.
Borradori, L., Koch, P.J., Niessen, C.M., Erkeland, S., van Leusden, M.R. and Sonnenberg, A. (1997) The localization of bullous pemphigoid antigen 180 (BP180) in hemidesmosomes is mediated by its cytoplasmic domain and seems to be regulated by the beta4 integrin subunit. J Cell Biol, 136, 1333-1347.

LITERATURVERZEICHNIS 82
Borradori, L. and Sonnenberg, A. (1999) Structure and function of hemidesmo-somes: more than simple adhesion complexes. J Invest Dermatol, 112, 411-418.
Boukamp, P., Petrussevska, R.T., Breitkreutz, D., Hornung, J., Markham, A. and Fusenig, N.E. (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol, 106, 761-771.
Bowszyc-Dmochowska, M. and Dmochowski, M. (2002) Immediate hypersensitiv-ity phenomena in bullous pemphigoid: critical concepts. J Med, 33, 189-198.
Budinger, L., Borradori, L., Yee, C., Eming, R., Ferencik, S., Grosse-Wilde, H., Merk, H.F., Yancey, K. and Hertl, M. (1998) Identification and characteriza-tion of autoreactive T cell responses to bullous pemphigoid antigen 2 in pa-tients and healthy controls. J Clin Invest, 102, 2082-2089.
Chan, L.S., Vanderlugt, C.J., Hashimoto, T., Nishikawa, T., Zone, J.J., Black, M.M., Wojnarowska, F., Stevens, S.R., Chen, M., Fairley, J.A., Woodley, D.T., Miller, S.D. and Gordon, K.B. (1998) Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol, 110, 103-109.
Chaudhuri, J. and Alt, F.W. (2004) Class-switch recombination: interplay of tran-scription, DNA deamination and DNA repair. Nat Rev Immunol, 4, 541-552.
Chen, R., Fairley, J.A., Zhao, M.L., Giudice, G.J., Zillikens, D., Diaz, L.A. and Liu, Z. (2002) Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macro-phage-mediated neutrophil infiltration depends on mast cell activation. J Immunol, 169, 3987-3992.
Chen, R., Ning, G., Zhao, M.L., Fleming, M.G., Diaz, L.A., Werb, Z. and Liu, Z. (2001) Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest, 108, 1151-1158.
Chimanovitch, I., Hamm, H., Georgi, M., Kroiss, M., Stolz, W., Apitz, C., Brocker, E.B. and Zillikens, D. (2000) Bullous pemphigoid of childhood: autoantibod-ies target the same epitopes within the NC16A domain of BP180 as autoan-tibodies in bullous pemphigoid of adulthood. Arch Dermatol, 136, 527-532.
Christophoridis, S., Budinger, L., Borradori, L., Hunziker, T., Merk, H.F. and Hertl, M. (2000) IgG, IgA and IgE autoantibodies against the ectodomain of BP180 in patients with bullous and cicatricial pemphigoid and linear IgA bul-lous dermatosis. Br J Dermatol, 143, 349-355.
Delaporte, E., Dubost-Brama, A., Ghohestani, R., Nicolas, J.F., Neyrinck, J.L., Bergoend, H., Janin, A. and Capron, M. (1996) IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol, 157, 3642-3647.
Delespesse, G., Sarfati, M., Wu, C.Y., Fournier, S. and Letellier, M. (1992) The low-affinity receptor for IgE. Immunol Rev, 125, 77-97.
Delgado, J.C., Turbay, D., Yunis, E.J., Yunis, J.J., Morton, E.D., Bhol, K., Norman, R., Alper, C.A., Good, R.A. and Ahmed, R. (1996) A common major histo-compatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A, 93, 8569-8571.
Di Zenzo, G., Grosso, F., Terracina, M., Mariotti, F., De Pita, O., Owaribe, K., Mas-trogiacomo, A., Sera, F., Borradori, L. and Zambruno, G. (2004) Characteri-zation of the anti-BP180 autoantibody reactivity profile and epitope mapping in bullous pemphigoid patients. J Invest Dermatol, 122, 103-110.

LITERATURVERZEICHNIS 83
Diaz, L.A., Ratrie, H., 3rd, Saunders, W.S., Futamura, S., Squiquera, H.L., Anhalt, G.J. and Giudice, G.J. (1990) Isolation of a human epidermal cDNA corre-sponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemides-mosome. J Clin Invest, 86, 1088-1094.
Dimson, O.G., Giudice, G.J., Fu, C.L., Van den Bergh, F., Warren, S.J., Janson, M.M. and Fairley, J.A. (2003) Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol, 120, 784-788.
Dopp, R., Schmidt, E., Chimanovitch, I., Leverkus, M., Brocker, E.B. and Zillikens, D. (2000) IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these im-munoglobulins reflect disease activity. J Am Acad Dermatol, 42, 577-583.
Dubucquoi, S., Desreumaux, P., Janin, A., Klein, O., Goldman, M., Tavernier, J., Capron, A. and Capron, M. (1994) Interleukin 5 synthesis by eosinophils: association with granules and immunoglobulin-dependent secretion. J Exp Med, 179, 703-708.
Dvorak, A.M., Lett-Brown, M., Thueson, D. and Grant, J.A. (1981) Complement-induced degranulation of human basophils. J Immunol, 126, 523-528.
Dvorak, A.M., Mihm, M.C., Jr., Osage, J.E., Kwan, T.H., Austen, K.F. and Win-troub, B.U. (1982) Bullous pemphigoid, an ultrastructural study of the in-flammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J Invest Dermatol, 78, 91-101.
Egan, C.A., Taylor, T.B., Meyer, L.J., Petersen, M.J. and Zone, J.J. (1999) Bullous pemphigoid sera that contain antibodies to BPAg2 also contain antibodies to LABD97 that recognize epitopes distal to the NC16A domain. J Invest Dermatol, 112, 148-152.
Fairley, J.A., Fu, C.L. and Giudice, G.J. (2005) Mapping the binding sites of anti-BP180 immunoglobulin E autoantibodies in bullous pemphigoid. J Invest Dermatol, 125, 467-472.
Falcone, F.H., Dahinden, C.A., Gibbs, B.F., Noll, T., Amon, U., Hebestreit, H., Abrahamsen, O., Klaucke, J., Schlaak, M. and Haas, H. (1996) Human ba-sophils release interleukin-4 after stimulation with Schistosoma mansoni egg antigen. Eur J Immunol, 26, 1147-1155.
Falcone, F.H., Haas, H. and Gibbs, B.F. (2000) The human basophil: a new ap-preciation of its role in immune responses. Blood, 96, 4028-4038.
Fiebiger, E., Hammerschmid, F., Stingl, G. and Maurer, D. (1998) Anti-FcepsilonRIalpha autoantibodies in autoimmune-mediated disorders. Identi-fication of a structure-function relationship. J Clin Invest, 101, 243-251.
Franzke, C.W., Tasanen, K., Schacke, H., Zhou, Z., Tryggvason, K., Mauch, C., Zigrino, P., Sunnarborg, S., Lee, D.C., Fahrenholz, F. and Bruckner-Tuderman, L. (2002) Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. Embo J, 21, 5026-5035.
Frossi, B., De Carli, M. and Pucillo, C. (2004) The mast cell: an antenna of the mi-croenvironment that directs the immune response. J Leukoc Biol, 75, 579-585.
Gammon, W.R., Briggaman, R.A., Inman, A.O., 3rd, Queen, L.L. and Wheeler, C.E. (1984) Differentiating anti-lamina lucida and anti-sublamina densa anti-BMZ antibodies by indirect immunofluorescence on 1.0 M sodium chloride-separated skin. J Invest Dermatol, 82, 139-144.

LITERATURVERZEICHNIS 84
Gammon, W.R., Fine, J.D., Forbes, M. and Briggaman, R.A. (1992) Immunofluo-rescence on split skin for the detection and differentiation of basement membrane zone autoantibodies. J Am Acad Dermatol, 27, 79-87.
Gammon, W.R., Merritt, C.C., Lewis, D.M., Sams, W.M., Jr., Carlo, J.R. and Wheeler, C.E., Jr. (1982) An in vitro model of immune complex-mediated basement membrane zone separation caused by pemphigoid antibodies, leukocytes, and complement. J Invest Dermatol, 78, 285-290.
Georgi, M., Jainta, S., Brocker, E.B. and Zillikens, D. (2001) [Autoantigens of subepidermal bullous autoimmune dermatoses]. Hautarzt, 52, 1079-1089.
Ghohestani, R.F., Cozzani, E., Delaporte, E., Nicolas, J.F., Parodi, A. and Claudy, A. (1998) IgE antibodies in sera from patients with bullous pemphigoid are autoantibodies preferentially directed against the 230-kDa epidermal anti-gen (BP230). J Clin Immunol, 18, 202-209.
Ghohestani, R.F., Novotney, J., Chaudhary, M. and Agah, R.S. (2001) Bullous pemphigoid: from the bedside to the research laboratory. Clin Dermatol, 19, 690-696.
Gibbs, B.F. (2005) Human basophils as effectors and immunomodulators of aller-gic inflammation and innate immunity. Clin Exp Med, 5, 43-49.
Gibbs, B.F., Haas, H., Falcone, F.H., Albrecht, C., Vollrath, I.B., Noll, T., Wolff, H.H. and Amon, U. (1996) Purified human peripheral blood basophils re-lease interleukin-13 and preformed interleukin-4 following immunological activation. Eur J Immunol, 26, 2493-2498.
Gibbs, B.F., Wierecky, J., Welker, P., Henz, B.M., Wolff, H.H. and Grabbe, J. (2001) Human skin mast cells rapidly release preformed and newly gener-ated TNF-alpha and IL-8 following stimulation with anti-IgE and other secre-tagogues. Exp Dermatol, 10, 312-320.
Gibbs, B.F., Wolff, H.H. and Grabbe, J. (2000) Ambroxol inhibits IgE-dependent mediator secretion from human skin mast cells. Inflamm Res, 49 Suppl 1, S17-18.
Giudice, G.J., Emery, D.J. and Diaz, L.A. (1992) Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol, 99, 243-250.
Giudice, G.J., Emery, D.J., Zelickson, B.D., Anhalt, G.J., Liu, Z. and Diaz, L.A. (1993) Bullous pemphigoid and herpes gestationis autoantibodies recog-nize a common non-collagenous site on the BP180 ectodomain. J Immunol, 151, 5742-5750.
Giudice, G.J., Wilske, K.C., Anhalt, G.J., Fairley, J.A., Taylor, A.F., Emery, D.J., Hoffman, R.G. and Diaz, L.A. (1994) Development of an ELISA to detect anti-BP180 autoantibodies in bullous pemphigoid and herpes gestationis. J Invest Dermatol, 102, 878-881.
Guo, J., Rapoport, B. and McLachlan, S.M. (1997) Thyroid peroxidase autoanti-bodies of IgE class in thyroid autoimmunity. Clin Immunol Immunopathol, 82, 157-162.
Hadi, S.M., Barnetson, R.S., Gawkrodger, D.J., Saxena, U., Bird, P. and Merrett, T.G. (1988) Clinical, histological and immunological studies in 50 patients with bullous pemphigoid. Dermatologica, 176, 6-17.
Hertl, M. and Schuler, G. (2002) [Bullous autoimmune dermatoses. 3: Diagnosis and therapy]. Hautarzt, 53, 352-365; quiz 366-357.

LITERATURVERZEICHNIS 85
Hibbert, R.G., Teriete, P., Grundy, G.J., Beavil, R.L., Reljic, R., Holers, V.M., Han-nan, J.P., Sutton, B.J., Gould, H.J. and McDonnell, J.M. (2005) The struc-ture of human CD23 and its interactions with IgE and CD21. J Exp Med, 202, 751-760.
Hirako, Y., Usukura, J., Uematsu, J., Hashimoto, T., Kitajima, Y. and Owaribe, K. (1998) Cleavage of BP180, a 180-kDa bullous pemphigoid antigen, yields a 120-kDa collagenous extracellular polypeptide. J Biol Chem, 273, 9711-9717.
Hirako, Y., Yoshino, K., Zillikens, D. and Owaribe, K. (2003) Extracellular cleavage of bullous pemphigoid antigen 180/type XVII collagen and its involvement in hemidesmosomal disassembly. J Biochem (Tokyo), 133, 197-206.
Hopkinson, S.B., Baker, S.E. and Jones, J.C. (1995) Molecular genetic studies of a human epidermal autoantigen (the 180-kD bullous pemphigoid anti-gen/BP180): identification of functionally important sequences within the BP180 molecule and evidence for an interaction between BP180 and alpha 6 integrin. J Cell Biol, 130, 117-125.
Ishizaka, K. and Ishizaka, T. (1967) Identification of gamma-E-antibodies as a car-rier of reaginic activity. J Immunol, 99, 1187-1198.
Jordon, R.E., Beutner, E.H., Witebsky, E., Blumental, G., Hale, W.L. and Lever, W.F. (1967) Basement zone antibodies in bullous pemphigoid. Jama, 200, 751-756.
Jung, M., Kippes, W., Messer, G., Zillikens, D. and Rzany, B. (1999) Increased risk of bullous pemphigoid in male and very old patients: A population-based study on incidence. J Am Acad Dermatol, 41, 266-268.
Kasahara-Imamura, M., Hosokawa, H., Maekawa, N. and Horio, T. (2001) Activa-tion of Fc epsilon RI-positive eosinophils in bullous pemphigoid. Int J Mol Med, 7, 249-253.
Katayama, I., Doi, T. and Nishioka, K. (1984) High histamine level in the blister fluid of bullous pemphigoid. Arch Dermatol Res, 276, 126-127.
Kelly, S.E. and Wojnarowska, F. (1988) The use of chemically split tissue in the detection of circulating anti-basement membrane zone antibodies in bullous pemphigoid and cicatricial pemphigoid. Br J Dermatol, 118, 31-40.
Kippes, W., Schmidt, E., Roth, A., Rzany, B., Brocker, E.B. and Zillikens, D. (1999) [Immunopathologic changes in 115 patients with bullous pemphigoid]. Hau-tarzt, 50, 866-872.
Korman, N. (1987) Bullous pemphigoid. J Am Acad Dermatol, 16, 907-924. Korman, N.J. (1998) Bullous pemphigoid. The latest in diagnosis, prognosis, and
therapy. Arch Dermatol, 134, 1137-1141. Korn, J. (2006) Kollagen XVII - Expression und Charakterisierung Rekombinanter
Fragmente. Technisch-Naturwissenschaftliche Fakultät. Medizinische Uni-versität zu Lübeck, Lübeck.
Kraft, S., Wessendorf, J.H., Hanau, D. and Bieber, T. (1998) Regulation of the high affinity receptor for IgE on human epidermal Langerhans cells. J Im-munol, 161, 1000-1006.
Labib, R.S., Anhalt, G.J., Patel, H.P., Mutasim, D.F. and Diaz, L.A. (1986) Molecu-lar heterogeneity of the bullous pemphigoid antigens as detected by im-munoblotting. J Immunol, 136, 1231-1235.
Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680-685.

LITERATURVERZEICHNIS 86
Laffitte, E., Burkhard, P.R., Fontao, L., Jaunin, F., Saurat, J.H., Chofflon, M. and Borradori, L. (2005) Bullous pemphigoid antigen 1 isoforms: potential new target autoantigens in multiple sclerosis? Br J Dermatol, 152, 537-540.
Laffitte, E., Skaria, M., Jaunin, F., Tamm, K., Saurat, J.H., Favre, B. and Borradori, L. (2001) Autoantibodies to the extracellular and intracellular domain of bul-lous pemphigoid 180, the putative key autoantigen in bullous pemphigoid, belong predominantly to the IgG1 and IgG4 subclasses. Br J Dermatol, 144, 760-768.
Lee, W.L. and Downey, G.P. (2001) Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med, 164, 896-904.
Li, N., Aoki, V., Hans-Filho, G., Rivitti, E.A. and Diaz, L.A. (2003) The role of in-tramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med, 197, 1501-1510.
Liu, Z., Diaz, L.A., Swartz, S.J., Troy, J.L., Fairley, J.A. and Giudice, G.J. (1995a) Molecular mapping of a pathogenically relevant BP180 epitope associated with experimentally induced murine bullous pemphigoid. J Immunol, 155, 5449-5454.
Liu, Z., Diaz, L.A., Troy, J.L., Taylor, A.F., Emery, D.J., Fairley, J.A. and Giudice, G.J. (1993) A passive transfer model of the organ-specific autoimmune dis-ease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest, 92, 2480-2488.
Liu, Z., Giudice, G.J., Swartz, S.J., Fairley, J.A., Till, G.O., Troy, J.L. and Diaz, L.A. (1995b) The role of complement in experimental bullous pemphigoid. J Clin Invest, 95, 1539-1544.
Liu, Z., Giudice, G.J., Zhou, X., Swartz, S.J., Troy, J.L., Fairley, J.A., Till, G.O. and Diaz, L.A. (1997) A major role for neutrophils in experimental bullous pem-phigoid. J Clin Invest, 100, 1256-1263.
Liu, Z., Shapiro, S.D., Zhou, X., Twining, S.S., Senior, R.M., Giudice, G.J., Fairley, J.A. and Diaz, L.A. (2000) A critical role for neutrophil elastase in experi-mental bullous pemphigoid. J Clin Invest, 105, 113-123.
Liu, Z., Shipley, J.M., Vu, T.H., Zhou, X., Diaz, L.A., Werb, Z. and Senior, R.M. (1998) Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med, 188, 475-482.
Luo, H.Y., Hofstetter, H., Banchereau, J. and Delespesse, G. (1991) Cross-linking of CD23 antigen by its natural ligand (IgE) or by anti-CD23 antibody pre-vents B lymphocyte proliferation and differentiation. J Immunol, 146, 2122-2129.
Mariotti, F., Grosso, F., Terracina, M., Ruffelli, M., Cordiali-Fei, P., Sera, F., Zam-bruno, G., Mastrogiacomo, A. and Di Zenzo, G. (2004) Development of a novel ELISA system for detection of anti-BP180 IgG and characterization of autoantibody profile in bullous pemphigoid patients. Br J Dermatol, 151, 1004-1010.
Masunaga, T., Shimizu, H., Yee, C., Borradori, L., Lazarova, Z., Nishikawa, T. and Yancey, K.B. (1997) The extracellular domain of BPAG2 localizes to an-choring filaments and its carboxyl terminus extends to the lamina densa of normal human epidermal basement membrane. J Invest Dermatol, 109, 200-206.
Matsumura, K., Amagai, M., Nishikawa, T. and Hashimoto, T. (1996) The majority of bullous pemphigoid and herpes gestationis serum samples react with the NC16a domain of the 180-kDa bullous pemphigoid antigen. Arch Dermatol Res, 288, 507-509.

LITERATURVERZEICHNIS 87
Murakami, H., Amagai, M., Higashiyama, M., Hashimoto, K., Chorzelski, T.P., Bhogal, B.S., Jenkins, R.E., Black, M.M., Zillikens, D., Nishikawa, T. and Hashimoto, T. (1996) Analysis of antigens recognized by autoantibodies in herpes gestationis. Usefulness of immunoblotting using a fusion protein representing an extracellular domain of the 180 kD bullous pemphigoid an-tigen. J Dermatol Sci, 13, 112-117.
Murakami, H., Nishioka, S., Setterfield, J., Bhogal, B.S., Black, M.M., Zillikens, D., Yancey, K.B., Balding, S.D., Giudice, G.J., Diaz, L.A., Nishikawa, T., Kiyo-kawa, C. and Hashimoto, T. (1998) Analysis of antigens targeted by circu-lating IgG and IgA autoantibodies in 50 patients with cicatricial pemphigoid. J Dermatol Sci, 17, 39-44.
Mutasim, D.F., Takahashi, Y., Labib, R.S., Anhalt, G.J., Patel, H.P. and Diaz, L.A. (1985) A pool of bullous pemphigoid antigen(s) is intracellular and associ-ated with the basal cell cytoskeleton-hemidesmosome complex. J Invest Dermatol, 84, 47-53.
Nakatani, C., Muramatsu, T. and Shirai, T. (1998) Immunoreactivity of bullous pemphigoid (BP) autoantibodies against the NC16A and C-terminal do-mains of the 180 kDa BP antigen (BP180): immunoblot analysis and en-zyme-linked immunosorbent assay using BP180 recombinant proteins. Br J Dermatol, 139, 365-370.
Nestor, M.S., Cochran, A.J. and Ahmed, A.R. (1987) Mononuclear cell infiltrates in bullous disease. J Invest Dermatol, 88, 172-175.
Nie, Z. and Hashimoto, T. (1999) IgA antibodies of cicatricial pemphigoid sera specifically react with C-terminus of BP180. J Invest Dermatol, 112, 254-255.
Nonaka, S., Ishiko, A., Masunaga, T., Akiyama, M., Owaribe, K., Shimizu, H. and Nishikawa, T. (2000) The extracellular domain of BPAG2 has a loop struc-ture in the carboxy terminal flexible tail in vivo. J Invest Dermatol, 115, 889-892.
Parodi, A. and Rebora, A. (1992) Serum IgE antibodies bind to the epidermal side of the basement membrane zone splits in bullous pemphigoid. Br J Derma-tol, 126, 526-527.
Pas, H.H., Kloosterhuis, G.J., Heeres, K., van der Meer, J.B. and Jonkman, M.F. (1997) Bullous pemphigoid and linear IgA dermatosis sera recognize a simi-lar 120-kDa keratinocyte collagenous glycoprotein with antigenic cross-reactivity to BP180. J Invest Dermatol, 108, 423-429.
Perriard, J., Jaunin, F., Favre, B., Budinger, L., Hertl, M., Saurat, J.H. and Borra-dori, L. (1999) IgG autoantibodies from bullous pemphigoid (BP) patients bind antigenic sites on both the extracellular and the intracellular domains of the BP antigen 180. J Invest Dermatol, 112, 141-147.
Plaut, M., Pierce, J.H., Watson, C.J., Hanley-Hyde, J., Nordan, R.P. and Paul, W.E. (1989) Mast cell lines produce lymphokines in response to cross-linkage of Fc epsilon RI or to calcium ionophores. Nature, 339, 64-67.
Prausnitz, C., Küstner, H. (1921) Sudien über die Überempfindlichkeit. Zentralblatt für Bacteriologie, Infectionskrankheiten und Hygiene Abt.1, 86, 160-169.
Provost, T.T. and Tomasi, T.B., Jr. (1974) Immunopathology of bullous pemphi-goid. Basement membrane deposition of IgE, alternate pathway compo-nents and fibrin. Clin Exp Immunol, 18, 193-200.
Prussin, C. and Metcalfe, D.D. (2003) 4. IgE, mast cells, basophils, and eosino-phils. J Allergy Clin Immunol, 111, S486-494.

LITERATURVERZEICHNIS 88
Rock, B., Labib, R.S. and Diaz, L.A. (1990) Monovalent Fab' immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest, 85, 296-299.
Sato, A., Takemura, Y., Yamada, T., Ohtsuka, H., Sakai, H., Miyahara, Y., Aizawa, T., Terao, A., Onuma, S., Junen, K., Kanamori, A., Nakamura, Y., Tejima, E., Ito, Y. and Kamijo, K. (1999) A possible role of immunoglobulin E in pa-tients with hyperthyroid Graves' disease. J Clin Endocrinol Metab, 84, 3602-3605.
Schaapveld, R.Q., Borradori, L., Geerts, D., van Leusden, M.R., Kuikman, I., Nievers, M.G., Niessen, C.M., Steenbergen, R.D., Snijders, P.J. and Son-nenberg, A. (1998) Hemidesmosome formation is initiated by the beta4 in-tegrin subunit, requires complex formation of beta4 and HD1/plectin, and involves a direct interaction between beta4 and the bullous pemphigoid an-tigen 180. J Cell Biol, 142, 271-284.
Schacke, H., Schumann, H., Hammami-Hauasli, N., Raghunath, M. and Bruckner-Tuderman, L. (1998) Two forms of collagen XVII in keratinocytes. A full-length transmembrane protein and a soluble ectodomain. J Biol Chem, 273, 25937-25943.
Schaller, J., Giese, T., Ladusch, M. and Haustein, U.F. (1990) Interleukin-2 recep-tor expression and interleukin-2 production in bullous pemphigoid. Arch Dermatol Res, 282, 223-226.
Schmidt, E., Brocker, E.B. and Zillikens, D. (1995) High levels of soluble CD23 in blister fluid of patients with bullous pemphigoid. Arch Dermatol, 131, 966-967.
Schmidt, E., Brocker, E.B. and Zillikens, D. (2000a) [New aspects on the patho-genesis of bullous pemphigoid]. Hautarzt, 51, 637-645.
Schmidt, E., Obe, K., Brocker, E.B. and Zillikens, D. (2000b) Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bul-lous pemphigoid. Arch Dermatol, 136, 174-178.
Schumann, H., Baetge, J., Tasanen, K., Wojnarowska, F., Schacke, H., Zillikens, D. and Bruckner-Tuderman, L. (2000) The shed ectodomain of collagen XVII/BP180 is targeted by autoantibodies in different blistering skin dis-eases. Am J Pathol, 156, 685-695.
Sekigawa, I., Seta, N., Yamada, M., Iida, N., Hashimoto, H. and Ogawa, H. (2004) Possible importance of immunoglobulin E in foetal loss by mothers with anti-SSA antibody. Scand J Rheumatol, 33, 44-46.
Sherr, E., Macy, E., Kimata, H., Gilly, M. and Saxon, A. (1989) Binding the low affinity Fc epsilon R on B cells suppresses ongoing human IgE synthesis. J Immunol, 142, 481-489.
Shimanovich, I., Mihai, S., Oostingh, G.J., Ilenchuk, T.T., Brocker, E.B., Opdenak-ker, G., Zillikens, D. and Sitaru, C. (2004) Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bul-lous pemphigoid. J Pathol, 204, 519-527.
Sison-Fonacier, L. and Bystryn, J.C. (1986) Regional variations in antigenic prop-erties of skin. A possible cause for disease-specific distribution of skin le-sions. J Exp Med, 164, 2125-2130.
Sitaru, C., Goebeler, M. and Zillikens, D. (2004) [Bullous autoimmune dermatoses (I): Pathogenesis and diagnosis]. J Dtsch Dermatol Ges, 2, 123-128; quiz 139-140.

LITERATURVERZEICHNIS 89
Sitaru, C., Mihai, S., Otto, C., Chiriac, M.T., Hausser, I., Dotterweich, B., Saito, H., Rose, C., Ishiko, A. and Zillikens, D. (2005) Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII col-lagen. J Clin Invest, 115, 870-878.
Sitaru, C., Schmidt, E., Petermann, S., Munteanu, L.S., Brocker, E.B. and Zillik-ens, D. (2002) Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Der-matol, 118, 664-671.
Sitaru, C. and Zillikens, D. (2005) Mechanisms of blister induction by autoantibod-ies. Exp Dermatol, 14, 861-875.
Smith, E.P., Taylor, T.B., Meyer, L.J. and Zone, J.J. (1993) Antigen identification in drug-induced bullous pemphigoid. J Am Acad Dermatol, 29, 879-882.
Soh, H., Hosokawa, H. and Asada, Y. (1993) IgE and its related phenomena in bullous pemphigoid. Br J Dermatol, 128, 371-377.
Stahle-Backdahl, M., Inoue, M., Guidice, G.J. and Parks, W.C. (1994) 92-kD ge-latinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest, 93, 2022-2030.
Stanley, J.R., Hawley-Nelson, P., Yuspa, S.H., Shevach, E.M. and Katz, S.I. (1981) Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell, 24, 897-903.
Stanley, J.R., Tanaka, T., Mueller, S., Klaus-Kovtun, V. and Roop, D. (1988) Isola-tion of complementary DNA for bullous pemphigoid antigen by use of pa-tients' autoantibodies. J Clin Invest, 82, 1864-1870.
Sutton, B.J. and Gould, H.J. (1993) The human IgE network. Nature, 366, 421-428.
Tanaka, M., Hashimoto, T., Dykes, P.J. and Nishikawa, T. (1996) Clinical manifes-tations in 100 Japanese bullous pemphigoid cases in relation to autoantigen profiles. Clin Exp Dermatol, 21, 23-27.
Tanaka, T., Korman, N.J., Shimizu, H., Eady, R.A., Klaus-Kovtun, V., Cehrs, K. and Stanley, J.R. (1990) Production of rabbit antibodies against carboxy-terminal epitopes encoded by bullous pemphigoid cDNA. J Invest Dermatol, 94, 617-623.
Thoma-Uszynski, S., Uter, W., Schwietzke, S., Schuler, G., Borradori, L. and Hertl, M. (2006) Autoreactive T and B Cells from Bullous Pemphigoid (BP) Pa-tients Recognize Epitopes Clustered in Distinct Regions of BP180 and BP230. J Immunol, 176, 2015-2023.
Towbin, H., Staehelin, T. and Gordon, J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some ap-plications. Proc Natl Acad Sci U S A, 76, 4350-4354.
Van den Bergh, F. and Giudice, G.J. (2003) BP180 (type XVII collagen) and its role in cutaneous biology and disease. Adv Dermatol, 19, 37-71.
Vanderlugt, C.J. and Miller, S.D. (1996) Epitope spreading. Curr Opin Immunol, 8, 831-836.
Venning, V.A. and Wojnarowska, F. (1990) The association of bullous pemphigoid and malignant disease: a case control study. Br J Dermatol, 123, 439-445.
Venning, V.A. and Wojnarowska, F. (1995) Induced bullous pemphigoid. Br J Dermatol, 132, 831-832.

LITERATURVERZEICHNIS 90
Verraes, S., Hornebeck, W., Polette, M., Borradori, L. and Bernard, P. (2001) Re-spective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphi-goid. J Invest Dermatol, 117, 1091-1096.
Wanner, R., Wolff, B., Glowacki, F., Kolde, G. and Wittig, B. (1999) The loss of desmosomes after retinoic acid treatment results in an apparent inhibition of HaCaT keratinocyte differentiation. Arch Dermatol Res, 291, 346-353.
Weiss, S.J. (1989) Tissue destruction by neutrophils. N Engl J Med, 320, 365-376. Westermarck, J. and Kahari, V.M. (1999) Regulation of matrix metalloproteinase
expression in tumor invasion. Faseb J, 13, 781-792. Wever, S., Rank, C., Hornschuh, B., Hashimoto, T., Nishikawa, T., Brocker, E.B.
and Zillikens, D. (1995) [Bullous pemphigoid simulation subacute simple prurigo]. Hautarzt, 46, 789-795.
Wojnarowska, F., Kirtschig, G., Highet, A.S., Venning, V.A. and Khumalo, N.P. (2002) Guidelines for the management of bullous pemphigoid. Br J Derma-tol, 147, 214-221.
Wojnarowska, F., Kirtschig, G. and Khumalo, N. (2001) Treatment of subepidermal immunobullous diseases. Clin Dermatol, 19, 768-777.
Yancey, K.B. (1995) Adhesion molecules. II: Interactions of keratinocytes with epi-dermal basement membrane. J Invest Dermatol, 104, 1008-1014.
Zillikens, D., Ambach, A., Schuessler, M., Dummer, R., Hartmann, A.A. and Burg, G. (1992) The interleukin-2 receptor in lesions and serum of bullous pem-phigoid. Arch Dermatol Res, 284, 141-145.
Zillikens, D. and Giudice, G.J. (1999) BP180/type XVII collagen: its role in ac-quired and inherited disorders or the dermal-epidermal junction. Arch Der-matol Res, 291, 187-194.
Zillikens, D., Rose, P.A., Balding, S.D., Liu, Z., Olague-Marchan, M., Diaz, L.A. and Giudice, G.J. (1997) Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Invest Dermatol, 109, 573-579.
Zillikens, D., Wever, S., Roth, A., Weidenthaler-Barth, B., Hashimoto, T. and Brocker, E.B. (1995) Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol, 131, 957-958.
Zone, J.J., Taylor, T., Hull, C., Schmidt, L. and Meyer, L. (2007) IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Invest Dermatol, 127, 1167-1174.
Zone, J.J., Taylor, T.B., Kadunce, D.P. and Meyer, L.J. (1990) Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J Clin Invest, 85, 812-820.
ANHANG 91
8. Anhang
8.1. Abbildung I Aufbau des pGEX-6P-1 Vektors (website GE Healthcare-formerly Amersham Biosciences)
8.2. Tabelle l
Die Primer Sequenzen für die PCR zur Herstellung von cDNA-Fragmenten des humanen Typ XVII Kollagen/BP180.
Fragment Bp Primer Sequenz
hCOL17A 400 F:ATCGGATCCATAGAAGGAAGAATGGATGTAACCAAGAAAAAC
R:GATCGTCGACTCAGTCCTCTGGTTGAAGAAGATG
hCOL17B 489 F:ATCGGATCCATAGAAGGAAGAGTTACCCGCCATGCGTATGAA
R:GATCGTCGACTCAGCAGGACGCCATGTTGTTT
hCOL17C 492 F:GATCGGATCCATAGAAGGAAGACACAACAACATGACAACCCA
R:GATCGTCGACTCAAGCATTGTAGGCAGCTTGCTT
hCOL17D 369 F:ATCGGATCCATAGAAGGAAGAGGGAAGGTCTTTACAGCCTCC
R:GATCGTCGACTCACTCCGCCAGAGCAATGAG
Bp= Basenpaare, F= forward primer, R= reverse primer
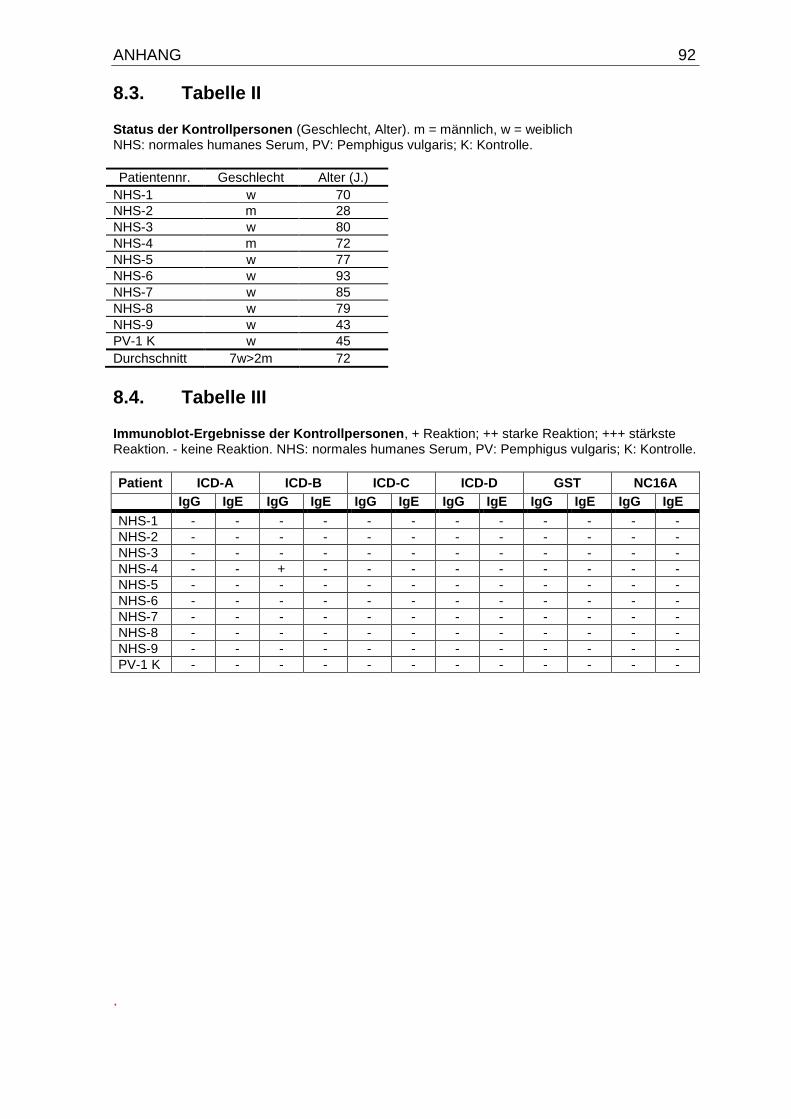
ANHANG 92
8.3. Tabelle II Status der Kontrollpersonen (Geschlecht, Alter). m = männlich, w = weiblich NHS: normales humanes Serum, PV: Pemphigus vulgaris; K: Kontrolle.
Patientennr. Geschlecht Alter (J.)
NHS-1 w 70
NHS-2 m 28
NHS-3 w 80
NHS-4 m 72
NHS-5 w 77
NHS-6 w 93
NHS-7 w 85
NHS-8 w 79
NHS-9 w 43
PV-1 K w 45
Durchschnitt 7w>2m 72
8.4. Tabelle III Immunoblot-Ergebnisse der Kontrollpersonen, + Reaktion; ++ starke Reaktion; +++ stärkste Reaktion. - keine Reaktion. NHS: normales humanes Serum, PV: Pemphigus vulgaris; K: Kontrolle.
Patient ICD-A ICD-B ICD-C ICD-D GST NC16A
IgG IgE IgG IgE IgG IgE IgG IgE IgG IgE IgG IgE
NHS-1 - - - - - - - - - - - -
NHS-2 - - - - - - - - - - - -
NHS-3 - - - - - - - - - - - -
NHS-4 - - + - - - - - - - - -
NHS-5 - - - - - - - - - - - -
NHS-6 - - - - - - - - - - - -
NHS-7 - - - - - - - - - - - -
NHS-8 - - - - - - - - - - - -
NHS-9 - - - - - - - - - - - -
PV-1 K - - - - - - - - - - - -
.

DANKSAGUNG 93
Erklärung
Die vorliegende Arbeit wurde in der Klinik für Dermatologie und Venerologie der
Universität zu Lübeck unter Aufsicht von Herrn PD Dr. Bernhard F. Gibbs durchge-
führt.
Das Forschungsvorhaben wurde von der Ethikkommission der Universität zu
Lübeck mit dem Schreiben vom 10.09.2004 genehmigt (Aktenzeichen: 04-087).
Ich versichere ausdrücklich, dass ich die vorgelegte Dissertation selbstständig,
ohne fremde Hilfe und nur mit den Quellen und Hilfsmitteln angefertigt habe, wie in
der Arbeit angegeben. Die aus der verwendeten Literatur wörtlich oder inhaltlich
entnommenen Textstellen und alle Angaben, die auf mündlichen Auskünften be-
ruhen, sind als solche kenntlich gemacht. Ich versichere außerdem, diese Disser-
tation bisher nicht einem Fachvertreter an einer anderen Hochschule zur Überprü-
fung vorgelegt zu haben und mich nicht anderweitig um Zulassung zur Promotion
beworben zu haben.
Lübeck, im Oktober 2007 Sina Katinka Dresow

DANKSAGUNG 94
Danksagung
Ich danke Herrn Direktor Prof. Dr. med. D. Zillikens für die Möglichkeit meine
Arbeit an der Klinik für Dermatologie und Venerologie durchführen zu dürfen sowie
für die Bereitstellung des dermatologischen Forschungslabors und für die Förde-
rung der Teilnahme am ESDR-Kongress, Paris, September 2006.
Mein großer Dank gilt Herrn PD. Dr. B. F. Gibbs (jetzt Universität Kent, England)
für die freundliche Überlassung des Themas, für die wissenschaftliche Leitung der
vorliegenden Arbeit, für seine stetige Hilfs- und Diskussionsbereitschaft, sowie für
die Übernahme der Begutachtung.
Herzlich bedanke ich mich bei Dr. Cassian Sitaru für die hilfreichen Ratschläge
während der praktischen Arbeit im Labor.
Ganz besonders möchte ich mich auch bei Dr. Andreas Recke für seine unermüd-
liche Unterstützung und aufmunternden Worte bedanken.
Ein herzliches Dankeschön an alle weiteren Mitarbeiter der Abteilung dermatolo-
gische Forschung, besonders an Bianca Hinz, Marina Kongsbak-Reim, Mircea
Chiriak, Josep Herrero, Ute Hoppe, Sidonia Mihai und Florina Olaru. Sie waren
stets bereit mir mit Rat und Tat zur Seite zu stehen. Die Arbeit in der Abteilung hat
mir viel Freude gemacht und ich habe mich dort sehr wohlgefühlt. Frau Schröder
danke ich für ihr immer offenes Ohr und ihre Hilfsbereitschaft.
Dem Labor des Instituts für Immunologie der Universität zu Lübeck danke ich für
die Durchführung der Gesamt-IgE-Bestimmung im Patienten- und Kontrollserum.
Von ganzem Herzen danke ich meinen Eltern, die mich stets unterstützt und moti-
viert haben. Sie haben mir dieses Studium und diese Arbeit ermöglicht und ich
konnte mich auf ihre Hilfe stets verlassen.

LEBENSLAUF 95
Lebenslauf Persönliche Angaben Name: Sina Katinka Dresow Geburtsdatum: 23.06.1980 Geburtsort: Braunschweig
Schulische Ausbildung 1987-1991: Grundschule in Nusse 1991-1998: Lauenburgische Gelehrtenschule in Ratzeburg 1998-2000: Heilwig-Gymnasium in Hamburg 2000: Reifeprüfung
Universitäre Ausbildung 10/2000-10/2002: Studium der Humanmedizin an der Phillips-Universität
zu Marburg 08/2002: Ärztliche Vorprüfung 10/2002-03/2007: Studium der Humanmedizin an der medizinischen Uni-
versität zu Lübeck 08/2003: Erster Abschnitt der ärztlichen Prüfung 09/2005: Zweiter Abschnitt der ärztlichen Prüfung 04/2007: Dritter Abschnitt der ärztlichen Prüfung - Approbation
Klinische Ausbildung Praktisches Jahr: Chirurgie 03/2006-07/2006: Kantonsspital Aarberg/Schweiz, Chefarzt Dr. Ch. de Montmollin Mund-, Kiefer-, Gesichtschirurgie 08/2006-10/2006 Kantonsspital Luzern/Schweiz , Chefarzt Dr. Dr. J. Kuttenberger 10/2006-11/2006 Universitätsklinikum Schleswig-Holstein, Campus Lübeck, Chefarzt Prof. Dr. Dr. P. Sieg Innere Medizin 11/2006-01/2007 Pneumologie und Infektiologie, UK-SH, Campus Lübeck, Leitung Prof. Dr. K. Dalhoff 01/2007-02/2007 Internistische Intensivstation 11a, UK-SH, Campus Lübeck, Leitung Prof. Dr. C. Dodt

LEBENSLAUF 96
Dissertation 10/2004-03/2006 Bearbeitung des experimentellen Themas „IgE-
Antikörper bei blasenbildenden Autoimmundermatosen: Epitop-mapping beim bullösen Pemphigoid“ in der Klinik für Dermatologie und Venerologie der Universität zu Lübeck, Prof. Dr. D. Zillikens. Arbeitsgruppe PD Dr. B. Gibbs.
Publikation/Abstract: Dresow S, Sitaru C, Oostingh GJ, Zillikens D and Gibbs BF (2006) IgE Autoantibodies from Bullous Pemphigoid Patients Target Multiple Antigenic Regions of Typ XVll Collagen. J Invest Dermatol, 126, 22 (Kongress der Eu-ropean Society for Dermatological Research (ESDR), September 2006, Paris, Frankreich).
Mannhagen, im Oktober 2007










