
Journal für Reproduktionsmedizin und Endokrinologie - kup.at · (DOC) is accumulated, thus...
8
Offizielles Organ: AGRBM, BRZ, DVR, DGA, DGGEF, DGRM, DIR, EFA, OEGRM, SRBM/DGE Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, A-3003 Gablitz Journal für Reproduktionsmedizin und Endokrinologie – Journal of Reproductive Medicine and Endocrinology – Andrologie • Embryologie & Biologie • Endokrinologie • Ethik & Recht • Genetik Gynäkologie • Kontrazeption • Psychosomatik • Reproduktionsmedizin • Urologie Indexed in EMBASE/Excerpta Medica/Scopus www.kup.at/repromedizin Online-Datenbank mit Autoren- und Stichwortsuche Genetic Causes in Adrenal or Ovarian Hyperandrogenism in the Reproductive Years Bals-Pratsch M, Seifert B, Ortmann O J. Reproduktionsmed. Endokrinol 2009; 6 (1), 19-23
Transcript of Journal für Reproduktionsmedizin und Endokrinologie - kup.at · (DOC) is accumulated, thus...

Offizielles Organ: AGRBM, BRZ, DVR, DGA, DGGEF, DGRM, DIR, EFA, OEGRM, SRBM/DGE
Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, A-3003 Gablitz
Journal für
Reproduktionsmedizin und Endokrinologie– Journal of Reproductive Medicine and Endocrinology –
Andrologie • Embryologie & Biologie • Endokrinologie • Ethik & Recht • Genetik Gynäkologie • Kontrazeption • Psychosomatik • Reproduktionsmedizin • Urologie
Indexed in EMBASE/Excerpta Medica/Scopus
www.kup.at/repromedizinOnline-Datenbank mit Autoren- und Stichwortsuche
Genetic Causes in Adrenal or Ovarian Hyperandrogenism
in the Reproductive Years
Bals-Pratsch M, Seifert B, Ortmann O
J. Reproduktionsmed. Endokrinol 2009; 6 (1), 19-23

T h o m a s S t a u d i n g e r
M a u r i c e K i e n e l
ECMO
für die Kitteltasche
Copyright 2018
Thomas Staudinger - Herausgeber
2. Auflage
Ab sofort in unserem Verlag
Krause & PacherneggGmbH
Bestellen Sie noch heute Ihr Exemplar aufwww.kup.at/cd-buch/75-bestellung.html
Thomas Staudinger Maurice Kienel
ECMOfür die Kitteltasche
2. Auflage Jänner 2019ISBN 978-3-901299-65-078 Seiten, div. Abbildungen19.80 EUR

Genetic Causes in Adrenal or Ovarian Hyperandrogenism in the Reproductive Years
J Reproduktionsmed Endokrinol 2009; 6 (1) 19
Genetic Causes in Adrenal or OvarianHyperandrogenism in the Reproductive Years
M. Bals-Pratsch1, B. Seifert1, O. Ortmann2
Hyperandrogenism and androgenisation frequently occur within families so that a monogenic cause such as congenital adrenal hyperplasia (CAH) mustbe considered. For the most part, classical CAH is first diagnosed and treated on the basis of common sex development disorders in early childhood bypediatric endocrinologists. In contrast, the non-classic “late-onset CAH” is mostly symptomatic by androgenisation from the age of puberty. For bothforms, the final diagnosis relies on the evidence of a CYP21 gene mutation. Even though the incidence of CAH in newborns with a ratio of 1:5000 is rare,the frequency of heterozygotes in central Europe is relatively prevalent with 1:50. In patients with hyperandrogenism of adrenal origin who desirechildren, a CYP21 mutation should therefore be excluded. In case of a known mutation carrier, the CYP21 gene diagnostic also should be applied on thepartner. If both partners are genetic carriers, a dexamethasone (DEX) therapy of the pregnant woman should be considered very early in each caseimmediately after determining a pregnancy to prevent serious virilization of the external genitalia of female CAH fetuses. A common differential diagno-sis is androgenisation through ovarian hyperandrogenism. For many years, candidate genes for the polycystic ovary syndrome (PCOS) have been investi-gated. The search focuses on genes involved in the regulation of insulin, follicle maturation as well as androgen secretion and regulation. Especiallypromising is a gene polymorphism on chromosome 19p13.2 in association with insulin resistance and disordered β-cell functions. Examinations ofpolymorphisms in FSH receptor genes make one expect that the effective gonadotropin dose in ovarian stimulation can be individually determined beforetreatment. Thus, severe hyperstimulation syndromes in PCOS patients possibly can be avoided. One might anticipate that new results in moleculargenetic PCOS research in the upcoming years will lead to significant improvements in treatment strategies. J Reproduktionsmed Endokrinol 2009; 6(1): 19–23.
Key words: PCOS, androgenisation, virilization, CAH, candidate genes, functional genetic polymorphisms, CYP21
Received: January 1, 2009; accepted after revision: January 20, 2009.From the 1Fertility Center Regensburg and the 2Department of Gynaecology and Obstetrics, University Hospital Regensburg, GermanyCorrespondence: PD Dr. med. Monika Bals-Pratsch, Fertility Center Regensburg, D-93047 Regensburg, Hemauer Straße 1; e-mail: [email protected]
Hyperandrogenism
The accumulation of hirsutism withinfamilies and balding in male familymembers suggests a joint genetic cause.Hirsutism, acne and loss of hair are clini-cal signs of hyperandrogenism in the re-productive years. These bodily symp-toms also are described as androgenisa-tion, which is to be distinguished fromvirilization with clitoral hypertrophy upto the point of disorders of sex develop-ment (DSD). A hypertrichosis on theother hand is not androgen-dependentbut rather ethnically conditioned.
Hyperandrogenism affects not only skinand hair, but also leads to abnormal men-strual cycles and infertility as well asdisorders in the metabolism of carbo-hydrates and lipids. Like hirsutism andbalding, polycystic ovaries, insulin re-sistance and obesity also appear morefrequently within affected families.Metabolic disorders are likely the com-mon cause for abnormal menstrual cy-cles and hyperandrogenism. A raisedsensitivity of target cells as well as in-creased or reduced local activity ofenzyme systems in the target cells cancause hyperandrogenism.
Androgens are mainly produced in theadrenal glands and in the ovaries as wellas in fatty tissue. The most important an-drogens are testosterone and DHEAS.The localization of androgen-produc-ing tumors is signaled by DHEAS(Fig. 1) for adrenal tumors and by testo-sterone for ovarian tumors. Suspicion of
a tumor exists if values are > 800 μg/dlfor DHEAS and > 200 ng/dl for testo-sterone. In patients with androgenisationand hyperandrogenism with suspicion ofneoplasms, imaging diagnostics of theadrenal glands and ovaries and, if indi-cated, of the pituitary gland are obliga-tory as well as diagnostics for internal
Figure 1. Clinical work-up of hyperandrogenism
For personal use only. Not to be reproduced without permission of Krause & Pachernegg GmbH.

20 J Reproduktionsmed Endokrinol 2009; 6 (1)
Genetic Causes in Adrenal or Ovarian Hyperandrogenism in the Reproductive Years
medicine and endocrinology, for exam-ple in suspicion of Cushing’s syndrome(adrenal) or Cushing’s disease (central).The diagnostics should be performedinterdisciplinarily with gynecologistsand internists specializing in endocrinol-ogy. If tumor occurrence is excluded,androgenisation either through medi-cation containing androgens such asGynodian® Depot or functional disordercan be considered for the differentialdiagnosis.
Functional HyperandrogenismThe most common cause of androgenisa-tion in the reproductive years is func-tional hyperandrogenism. Throughhyperandrogenism, follicle atresia andanovulation frequently occur with themorphological correlative of polycysticovaries. Mainly congenital hyperplasiaof adrenals (CAH) in its various charac-teristics and polycystic ovary syndrome(PCOS) are to be distinguished from oneanother. However, CAH is seldom andPCOS frequent.
Congenital Adrenal Hyper-
plasia (CAH)
Inherited disorders of adrenal steroido-genesis result from a deficiency in oneout of the five enzymatic steps necessaryfor normal cortisol synthesis (Fig. 2).Precursors of cortisol and aldosteroneaccumulate and DHEAS and testoster-one production increase. The most com-
mon gene mutation CYP21 on chromo-some 6p21.3 causes deficiency of21-hydroxylase and accounts for 95 %of CAH cases. The incidence in new-borns is 1:5,000–15,000. The precursor17-OHP progesterone and androgen pro-ductions increase and cortisol and aldos-terone biosynthesis are limited. 11β-hy-droxylase deficiency is seldom andcaused by CYP11B2 mutation (5 % ofCAH cases). The gene is located onchromosome 8q24.3 and the incidenceof the gene mutation in newborns is1:100,000. The precursor desoxycortisol(DOC) is accumulated, thus affected pa-tients suffer from hypertension. Othergene mutations in the adrenal steroido-genesis such as the 3β-HSD-II-gene mu-tation on chromosome 1p13.1 with defi-ciency of 3β-hydroxy-steroid-dehydro-genase [1] and accumulation of 17-OH-pregnenolone are rare (< 1 % of CAHcases).
The virilizing characteristic of classicCAH (with salt loss or simply viriliza-tion) is the most common cause of disor-ders of sex development. This diagnosisis made by pediatric endocrinologistsbased on symptoms mostly in newbornor infant stages. Nowadays elevated 17-OHP is detected regularly within thenewborn screening in Germany andother Western countries. Prompt treat-ment with glucocorticoids in simple viri-lization CAH (ca 25 %) and alsomineralocorticoids in CAH with salt loss(ca 75 %) has to be initiated to avoid life-
threatening events. The non-classicCAH (late-onset CAH) is mostly symp-tomatic beginning at puberty becausethese patients do not have genital abnor-malities. Instead, these patients aremainly seen first by gynecologists be-cause of menstrual disorders like sec-ondary amenorrhea or oligomenorrhea,hirsutism and acne. In adrenal hyper-androgenism with increased DHEAS,high 17-OHP values in the follicularphase of the cycle indicate CYP21 muta-tions. 17-OHP values do not representany reliable discriminating factors evenin combination with the adrenocortico-tropic hormone (ACTH) test, unless thetest results are generated in a highly spe-cialized endocrine laboratory. Geneticcounseling with molecular genetic diag-nosis is mandatory in confirming thediagnostic of CAH. Patients with CAHshould be regularly seen and treated withhydrocortisone by gynecologists andinternists specialized in endocrinology[2].
CAH and the Desire to HaveChildrenCAH is a monogenic disorder with a fre-quency of 1:50 in heterozygous mutationcarriers in central Europe. The expectedratio of homozygous affected, hetero-zygous or homozygous normal childrenin mutation carrier families according tothe Mendelian rules of inheritance is1:2:1. CAH homozygous females andmales with a heterozygous partner evenface a risk of 50 % for affected children.To improve fertility in CAH femaleswith menstrual disorders the hydrocorti-sone substitution regimen should bechanged to three to four single doses in-cluding a dose at 3 a. m. [3].
Genetic counseling should be offered toall couples at risk before getting preg-nant. The possibility of prenatal dexa-methasone (DEX) treatment as an ex-perimental strategy to prevent unwantedvirilization in affected female embryosshould be discussed with pediatriciansand gynecologists experienced in endo-crinology and prenatal medicine. Treat-ment with DEX 20 μg/kg bodyweight inthree divided doses based on prepreg-nancy weight (maximum 1.5 mg daily)is to be started at the time of diagnosinga pregnancy. DEX crosses the placentafrom the mother to the embryo, sup-presses ACTH secretion and preventsexaggerated adrenal androgen produc-
Figure 2. Simplified scheme for adrenal steroidogenesis
Genetic Causes in Adrenal or Ovarian Hyperandrogenism in the Reproductive Years
J Reproduktionsmed Endokrinol 2009; 6 (1) 21
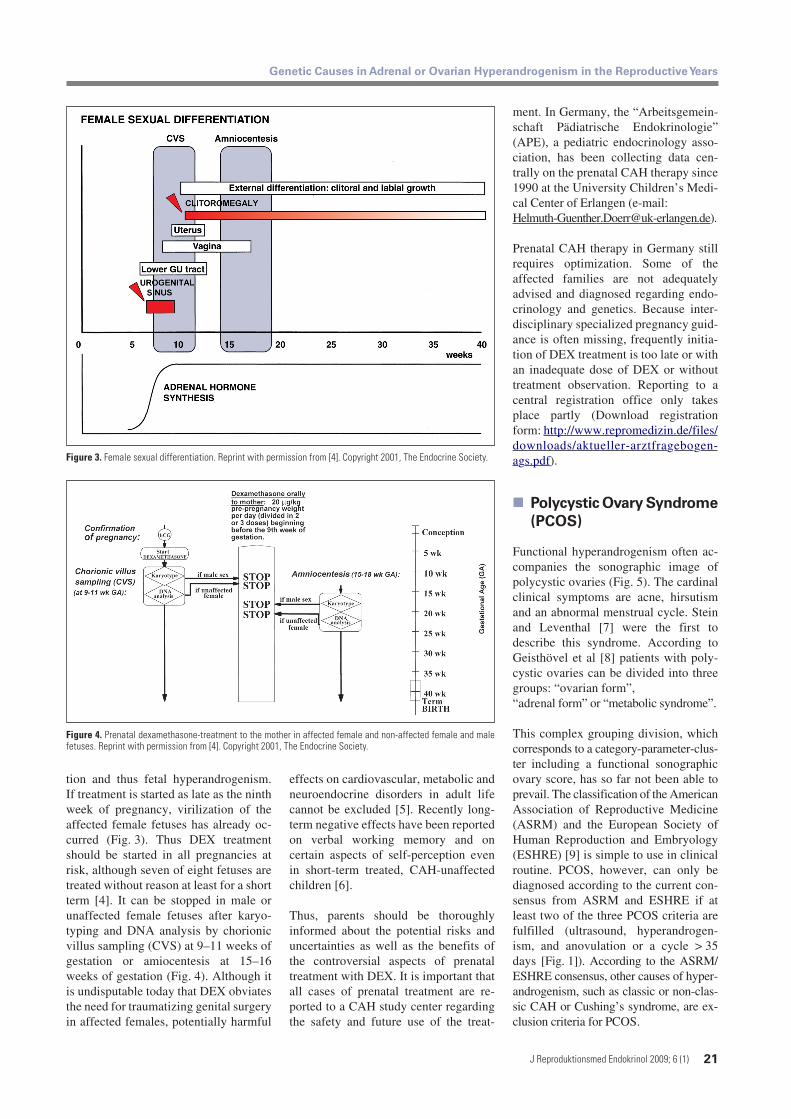
tion and thus fetal hyperandrogenism.If treatment is started as late as the ninthweek of pregnancy, virilization of theaffected female fetuses has already oc-curred (Fig. 3). Thus DEX treatmentshould be started in all pregnancies atrisk, although seven of eight fetuses aretreated without reason at least for a shortterm [4]. It can be stopped in male orunaffected female fetuses after karyo-typing and DNA analysis by chorionicvillus sampling (CVS) at 9–11 weeks ofgestation or amiocentesis at 15–16weeks of gestation (Fig. 4). Although itis undisputable today that DEX obviatesthe need for traumatizing genital surgeryin affected females, potentially harmful
effects on cardiovascular, metabolic andneuroendocrine disorders in adult lifecannot be excluded [5]. Recently long-term negative effects have been reportedon verbal working memory and oncertain aspects of self-perception evenin short-term treated, CAH-unaffectedchildren [6].
Thus, parents should be thoroughlyinformed about the potential risks anduncertainties as well as the benefits ofthe controversial aspects of prenataltreatment with DEX. It is important thatall cases of prenatal treatment are re-ported to a CAH study center regardingthe safety and future use of the treat-
ment. In Germany, the “Arbeitsgemein-schaft Pädiatrische Endokrinologie”(APE), a pediatric endocrinology asso-ciation, has been collecting data cen-trally on the prenatal CAH therapy since1990 at the University Children’s Medi-cal Center of Erlangen (e-mail:[email protected]).
Prenatal CAH therapy in Germany stillrequires optimization. Some of theaffected families are not adequatelyadvised and diagnosed regarding endo-crinology and genetics. Because inter-disciplinary specialized pregnancy guid-ance is often missing, frequently initia-tion of DEX treatment is too late or withan inadequate dose of DEX or withouttreatment observation. Reporting to acentral registration office only takesplace partly (Download registrationform: http://www.repromedizin.de/files/downloads/aktueller-arztfragebogen-ags.pdf).
Polycystic Ovary Syndrome
(PCOS)
Functional hyperandrogenism often ac-companies the sonographic image ofpolycystic ovaries (Fig. 5). The cardinalclinical symptoms are acne, hirsutismand an abnormal menstrual cycle. Steinand Leventhal [7] were the first todescribe this syndrome. According toGeisthövel et al [8] patients with poly-cystic ovaries can be divided into threegroups: “ovarian form”,“adrenal form” or “metabolic syndrome”.
This complex grouping division, whichcorresponds to a category-parameter-clus-ter including a functional sonographicovary score, has so far not been able toprevail. The classification of the AmericanAssociation of Reproductive Medicine(ASRM) and the European Society ofHuman Reproduction and Embryology(ESHRE) [9] is simple to use in clinicalroutine. PCOS, however, can only bediagnosed according to the current con-sensus from ASRM and ESHRE if atleast two of the three PCOS criteria arefulfilled (ultrasound, hyperandrogen-ism, and anovulation or a cycle > 35days [Fig. 1]). According to the ASRM/ESHRE consensus, other causes of hyper-androgenism, such as classic or non-clas-sic CAH or Cushing’s syndrome, are ex-clusion criteria for PCOS.
Figure 4. Prenatal dexamethasone-treatment to the mother in affected female and non-affected female and malefetuses. Reprint with permission from [4]. Copyright 2001, The Endocrine Society.
Figure 3. Female sexual differentiation. Reprint with permission from [4]. Copyright 2001, The Endocrine Society.

22 J Reproduktionsmed Endokrinol 2009; 6 (1)
Genetic Causes in Adrenal or Ovarian Hyperandrogenism in the Reproductive Years
Candidate GenesFor many years, there has been a searchfor candidate genes for PCOS. In accord-ance with common clinical symptomsand findings like obesity, resistance toinsulin, disorders in metabolizing lipids,hyperandrogenism, anovulation and ten-dency to miscarry, uncountable genes incorrelation with these symptoms wereexamined (Fig. 6). Research particularlyconcentrates on genes in the regulationof insulin, the maturation of follicles andandrogen secretion and effect [10]. Theresults are up to now inconsistent andcontradictory because the areas of re-search are based on different definitionsof PCOS as well as only on a smallnumber of cases [11].
A potential candidate gene is the insulingene, which influences the secretion andeffect of insulin. The promoter of theinsulin gene determines its expression.A VNTR (variable number of tandemrepeats) polymorphism exists here. Thenumber of repeats influences the expres-
Figure 6. Suggested aetiological factors in PCOS. Reprint with permission from [10].
Figure 5. Polycystic ova-ries in a PCOS patient
sion of the insulin gene. Most womencarry class-I alleles with an average of40 repeats or class-III alleles with amean of 157 repeats. An association be-tween the existence of class-III alleles ofthe insulin gene and PCOS is probable.This would explain the risk for hyper-insulinism and type-II diabetes in PCOSpatients [12].
A further candidate gene is the insulin-like growth factor (IGF). The homo-zygotic mutation of the IGF2 gene withtwo G alleles of the Apa1 variant is asso-ciated with PCOS [13]. The existence ofthe G allele possibly causes a raisedIGF2 expression and secretion in theliver. IGF2 stimulates the production ofandrogens in the ovaries and adrenalglands and thus possibly explains an as-pect of the pathomechanism in PCOS.On the other hand, our data showed nosignificant difference for the genome aswell as for the allele distribution in theIGF2 gene among 219 PCOS patientsand 152 healthy subjects [14].
The CYP11a gene codes the cytochrome450scc as the key enzyme for the andro-gen biosynthesis and metabolism. It islocalized on chromosome 15. WhileGharani et al. [15] found an associationbetween the 4R gene type polymorphismin the CYP11a gene and hyperandrogen-ism in PCOS patients, San Millán et al.[16] could not prove a correlation.
The examination of 37 candidate genesin 150 PCOS families showed an asso-ciation with PCOS only for the follista-tin gene [17]. As opposed to activin, fol-listatin inhibits follicular growth, in-creases ovarian androgen production,suppresses the endogenous FSH leveland inhibits the release of insulin. Theseeffects are characteristic of PCOS. Up tonow, this data could not be confirmed[18].
In the meantime, with the D19S884allele 8 (A8) within the intron 55 offibrillin 3 (FBN3) gene on chromosome19p13.2, a possible PCOS candidategene has been identified by a workgroupfrom Chicago [19]. In this examinationof 412 PCOS families, the D19S884polymorphism was associated with re-sistance to insulin and a defective β-cellfunction of the pancreas.
From a clinical perspective, not onlythe pathogenesis of the PCOS is of sig-nificance but also the treatment options,particularly in infertility. The interac-tion between FSH and its receptor playsa key role in ovarian stimulation withgonadotropins. Polymorphisms in FSHreceptor genes regarding the ovarianresponse were examined by severalworkgroups.
Looking at the genotype of the FSHreceptor gene polymorphism p.N680S,patients with a slight or increased ovar-ian response to FSH stimulation can beidentified before the beginning of thestimulation cycle [20]. Withdrawal fromtreatment and severe ovarian hyper-stimulation syndrome therefore can pos-sibly be avoided in the future.
The years to come will be exciting be-cause genetic diagnostics will have in-creasing clinical significance in theclarification of PCOS, resulting in im-proved treatment strategies for internalmedicine, endocrinology and reproduc-tive medicine.

Genetic Causes in Adrenal or Ovarian Hyperandrogenism in the Reproductive Years
J Reproduktionsmed Endokrinol 2009; 6 (1) 23
References:
1. Pang S, Wang W, Rich B, David R, Chang YT, Carbunaru G,Myers SE, Howie AF, Smillie KJ, Mason JI. A novel nonstopmutation in the stop codon and a novel missense mutation inthe type II 3β-hydroxysteroid dehydrogenase (3β-HSD) genecausing, respectively, nonclassic and classic 3β-HSD deficiencycongenital adrenal hyperplasia. J Clin Endocrinol Metab 2002;87: 2556–63.
2. Bals-Pratsch M, Ortmann O, Felberbaum R. Weibliche Steri-lität durch Erkrankungen der Hypophyse, der Schilddrüse undNebenniere. In: Diedrich K (Hrsg). Endokrinologie und Repro-duktionsmedizin III. Urban & Schwarzenberg, München, Wien,Baltimore, 1998; 226–37.
3. Möller H. Chronopharmacology of hydrocortisone and 9 al-pha-fluorhydrocortisone in the treatment for congenital adre-nal hyperplasia. Eur J Pediatr 1985; 144: 370–3.
4. New MI, Carlson A, Obeid J, Marshall I, Cabrera MS,Goseco A, Lin-Su K, Putnam AS, Oing Wei J, Wilson RC. Pre-natal diagnosis for congenital adrenal hyperplasia in 532pregnancies. J Clin Endocrinol Metab 2001; 86: 5651–7.
5. Seckl JR. Prenatal glucocorticoids and long-term program-ming. Review. Eur J Endocrinol 2004; 151 (Suppl 3): U49–62.
6. Hirvikoski T, Nordenström A, Lindholm T, Lindblad F, RitzénEM, Wedell A, Lajic S. Cognitive functions in children at riskfor congenital adrenal hyperplasia treated prenatally withdexamethasone. J Clin Endocrinol Metab 2007; 92: 542–8.
7. Stein IF, Leventhal ML. Amenorrhea associated with bilat-eral polycystic ovaries. Am J Obstet Gynecol 1935; 29: 181–91.
8. Geisthövel F, Wacker A, Wetzka B. Funktionelle Androgeni-sierung des peri- und postpuberalen Mädchens sowie derFrau. J Reproduktionsmed Endokrinol 2008; 5: 21–38.
9. The Rotterdam ESHRE/ASRM-Sponsored PCOS ConsensusWorkshop Group. Revised 2003 consensus on diagnostic crite-ria and long-term health risks related to polycystic ovary syn-drome. Fertil Steril 2004; 81: 19–25.
10. Franks S, Gharani N, Waterworth D, Batty S, White D,Williamson R, McCarthy M. The genetic basis of polycysticovary syndrome. Hum Reprod 1997; 12: 2641–8.
11. Franks S. Candidate genes in women with polycystic ovarysyndrome. Fertil Steril 2006; 86 (Suppl 1): S15.
12. Waterworth DM, Bennett ST, Gharani N, McCarthy MI,Hague S, Batty S, Conway GS, White D, Todd JA, Franks S,Williamson R. Linkage and association of insulin gene VNTR
regulatory polymorphism with polycystic ovary syndrome.Lancet 1997; 349: 1771–2.
13. San Millán JL, Cortón M, Villuendas G, Sancho J, Peral B,Escobar-Morreale HF. Association of the polycystic ovary syn-drome with genomic variants related to insulin resistance,type 2 diabetes mellitus, and obesity. J Clin Endocrinol Metab2004; 89: 2640–6.
14. Bals-Pratsch M, Geneidy N, Gruber R, Aslanidis C, SchmitzG, Seifert B. PCO-Syndrom – Genetische Polymorphismen undklinische Befunde. J Reproduktionsmed Endokrinol 2007; 5:253–4.
15. Gharani N, Waterworth DM, Batty S, White D, Gilling-SmithC, Conway GS, McCarthy M, Franks S, Williamson R: Associa-tion of the steroid synthesis gene CYP11a with polycystic ovarysyndrome and hyperandrogenism. Hum Mol Genet 1997; 6:397–402.
16. San Millán JL, Sancho J, Calvo RM, Escobar-Morreale HF.Role of the pentanucleotide (tttta)(n) polymorphism in the pro-moter of the CYP11a gene in the pathogenesis of hirsutism.Fertil Steril 2001; 75: 797–802.
17. Urbanek M, Legro RS, Driscoll DA, Azziz R, Ehrmann DA,Norman RJ, Strauss JF 3rd, Spielman RS, Dunaif A. Thirty-sevencandidate genes for polycystic ovary syndrome: strongest evi-dence for linkage is with follistatin. Proc Natl Acad Sci USA1999; 96: 8573–8.
18. Jones MR, Wilson SG, Mullin BH, Mead R, Watts GF,Stuckey BG. Polymorphism of the follistatin gene in polycysticovary syndrome. Mol Hum Reprod 2007; 13: 237–41.
19. Urbanek M, Sam S, Legro RS, Dunaif A. Identification of apolycystic ovary syndrome susceptibility variant in fibrillin-3and association with a metabolic phenotype. J Clin EndocrinolMetab 2007; 92: 4191–8.
20. Simoni M, Tempfer CB, Destenaves B, Fauser BC. Func-tional genetic polymorphisms and female reproductive disor-ders: Part I: Polycystic ovary syndrome and ovarian response.Hum Reprod Update 2008; 14: 459–84.
Practical Aspects
The most common cause of hyper-androgenism in the reproductiveyears is PCOS. For several years re-search has concentrated on identify-ing potential candidate genes forPCOS without a breakthrough yet.Although PCOS is frequent, rarecases of endocrine neoplasms and lateonset CAH have to be distinguishedfrom PCOS. Patients at risk of givingbirth to CAH homozygous femalefetuses should be offered prenataldexamethasone treatment to preventvirilization in affected female em-bryos.

Haftungsausschluss
Die in unseren Webseiten publizierten Informationen richten sich ausschließlich an geprüfte und autorisierte medizinische Berufsgruppen und entbinden nicht von der ärztlichen Sorg-faltspflicht sowie von einer ausführlichen Patientenaufklärung über therapeutische Optionen und deren Wirkungen bzw. Nebenwirkungen. Die entsprechenden Angaben werden von den Autoren mit der größten Sorgfalt recherchiert und zusammengestellt. Die angegebenen Do-sierungen sind im Einzelfall anhand der Fachinformationen zu überprüfen. Weder die Autoren, noch die tragenden Gesellschaften noch der Verlag übernehmen irgendwelche Haftungsan-sprüche.
Bitte beachten Sie auch diese Seiten:
Impressum Disclaimers & Copyright Datenschutzerklärung
Mitteilungen aus der Redaktion
e-Journal-AboBeziehen Sie die elektronischen Ausgaben dieser Zeitschrift hier.
Die Lieferung umfasst 4–5 Ausgaben pro Jahr zzgl. allfälliger Sonderhefte.
Unsere e-Journale stehen als PDF-Datei zur Verfügung und sind auf den meisten der markt-üblichen e-Book-Readern, Tablets sowie auf iPad funktionsfähig.
Bestellung e-Journal-Abo
Haftungsausschluss
Die in unseren Webseiten publizierten Informationen richten sich ausschließlich an geprüfte und autorisierte medizinische Berufsgruppen und entbinden nicht von der ärztlichen Sorg-faltspflicht sowie von einer ausführlichen Patientenaufklärung über therapeutische Optionen und deren Wirkungen bzw. Nebenwirkungen. Die entsprechenden Angaben werden von den Autoren mit der größten Sorgfalt recherchiert und zusammengestellt. Die angegebenen Do-sierungen sind im Einzelfall anhand der Fachinformationen zu überprüfen. Weder die Autoren, noch die tragenden Gesellschaften noch der Verlag übernehmen irgendwelche Haftungs-ansprüche.
Bitte beachten Sie auch diese Seiten:
Impressum Disclaimers & Copyright Datenschutzerklärung
Mitteilungen aus der Redaktion
e-Journal-AboBeziehen Sie die elektronischen Ausgaben dieser Zeitschrift hier.
Die Lieferung umfasst 4–5 Ausgaben pro Jahr zzgl. allfälliger Sonderhefte.
Unsere e-Journale stehen als PDF-Datei zur Verfügung und sind auf den meisten der markt-üblichen e-Book-Readern, Tablets sowie auf iPad funktionsfähig.
Bestellung e-Journal-Abo
Besuchen Sie unsere Rubrik
Medizintechnik-Produkte
InControl 1050 Labotect GmbH
Aspirator 3 Labotect GmbH
Philips Azurion: Innovative Bildgebungslösung
Neues CRT-D Implantat Intica 7 HF-T QP von Biotronik
Artis pheno Siemens Healthcare Diagnostics GmbH


















