
Kalorimetrische Untersuchungen des Systems ... · Kalorimetrische Untersuchungen des Systems...
146
Kalorimetrische Untersuchungen des Systems Polyethylenglykol/Wasser Von der Fakultät für Naturwissenschaften der Gerhard-Mercator-Universität – Gesamthochschule Duisburg zur Erlangung des akademischen Grades eines Dr. rer. nat. genehmigte Dissertation von Pascal Jablonski aus Mülheim an der Ruhr 27. Mai 2002
Transcript of Kalorimetrische Untersuchungen des Systems ... · Kalorimetrische Untersuchungen des Systems...

Kalorimetrische Untersuchungen des Systems Polyethylenglykol/Wasser
Von der
Fakultät für Naturwissenschaften
der Gerhard-Mercator-Universität – Gesamthochschule Duisburg
zur Erlangung des akademischen Grades eines
Dr. rer. nat.
genehmigte Dissertation
von
Pascal Jablonski
aus
Mülheim an der Ruhr
27. Mai 2002

Die vorliegende Arbeit wurde im Fachgebiet
Angewandte Physikalische Chemie
der Gerhard-Mercator-Universität-GH Duisburg angefertigt.
Berichterstatter: Prof. Dr. W. Borchard
Prof. Dr. A. Golloch
Tag der mündlichen Prüfung: 15. Juli 2002

Mein besonderer Dank gilt Herrn Prof. Dr. W. Borchard für die interessante Auf-
gabenstellung dieser Arbeit, seine fachliche Betreuung und seine stetige Bereit-
schaft zur Diskussion.
Herrn Prof. Dr. A. Golloch möchte ich danken für das Interesse an meiner Arbeit
und die freundliche Übernahme des Korreferats.
Herrn Prof. Dr. D. Eagland danke ich für die Möglichkeit einen Teil der Messun-
gen im Institute of Pharmacy in Bradford durchführen zu können, sowie die hilf-
reiche Organisation meiner Unterbringung in Bradford.
Den Mitarbeitern der Elektronik und Feinmechanik Werkstätten danke ich für
die ständige Hilfsbereitschaft und Unterstützung bei der Konstruktion des
Mischungsaufsatzes.
Den Mitarbeitern des Fachgebietes Angewandte Physikalische Chemie danke
ich für die stete Hilfsbereitschaft und das sehr angenehme Arbeitsklima. Herrn
Dipl. Chem. V. Körstgens danke ich im besonderen für die Durchsicht des
Manuskripts.
Ausserdem gilt mein Dank der Deutschen Forschungsgemeinschaft sowie der
Max Buchner Stiftung, die durch ihre finanzielle Unterstützung die vorliegende
Arbeit ermöglicht haben.

Meiner Frau
Nicole
gewidmet

INHALT
INHALT
1 EINLEITUNG 1
2 THEORETISCHER TEIL 4
2.1 DEFINITIONEN UND KONVENTIONEN...................................................................... 4
2.1.1 Binäre Phasendiagramme............................................................................ 4
2.1.2 Definition des Konzentrationsmaßes............................................................ 9
2.2 QUANTITATIVE BESCHREIBUNG DER KOEXISTENZKURVEN ....................................12
2.2.1 Quantitative Beschreibung der Liquiduskurven ...........................................12
2.2.2 Wechselwirkungsparameter � .....................................................................16
2.2.3 Beschreibung der Liquiduskurven in der Grundmolenbruchskala ................21
3 EXPERIMENTELLER TEIL 24
3.1 DIFFERENTIAL SCANNING CALORIMETRY (DSC) ..................................................24
3.1.1 Aufbau und Messprinzip..............................................................................24
3.1.2 Kalibrierung der DSC ..................................................................................30
3.1.3 Probenvorbereitung.....................................................................................32
3.1.4 Konstruktion von Phasendiagrammen aus DSC-Messkurven......................33
3.2 MISCHUNGSKALORIMETRIE .................................................................................38
3.2.1 Isotherme Messungen mit Hilfe der DSC.....................................................38
3.2.2 Aufbau des Mischungsaufsatzes.................................................................39
3.2.3 Bestimmung von Mischungsenthalpien mit Hilfe der DSC ...........................42
3.3 LKB FLOW CALORIMETER..................................................................................46
3.3.1 Messprinzip und Aufbau..............................................................................46
3.3.2 Versuchsdurchführung ................................................................................48
3.3.3 Integrale Mischungsenthalpie aus Verdünnungsmessungen.......................49

INHALT
4 ERGEBNISSE UND DISKUSSION 53
4.1 PHASENDIAGRAMME DER SYSTEME PEG/WASSER ..............................................53
4.1.1 Phasendiagramm PEG1500/Wasser...........................................................53
4.1.2 Phasendiagramm PEG900/Wasser.............................................................57
4.1.3 Phasendiagramm PEG600/Wasser.............................................................62
4.1.4 Phasendiagramme PEG400/Wasser und PEG300/Wasser.........................64
4.1.5 DSC-Messungen der reinen PEG-Proben...................................................65
4.1.6 Schmelzverhalten von PEG/Wasser Systemen...........................................68
4.1.7 PEG/Wasser Mischkristall ...........................................................................74
4.2 MISCHUNGSENTHALPIE DES SYSTEMS PEG/WASSER...........................................77
4.2.1 Mischungsenthalpien mit dem neuen Mischungskalorimeter .......................77
4.2.2 Mischungsenthalpien mit dem LKB-Flow Kalorimeter..................................89
4.2.3 Mischungsenthalpien der PEG-Wasser Systeme ........................................96
4.3 THERMODYNAMISCHE BESCHREIBUNG DES SYSTEMS AUS PEG UND WASSER ......99
4.3.1 Quantitative Auswertung des Systems PEG1500/Wasser.........................100
4.3.2 Thermodynamische Beschreibung der Liquiduskurve des PEG ................106
4.3.3 Zusammenfassung über die thermodynamische Auswertungen................113
5 ZUSAMMENFASSUNG 118
6 ANHANG 123
6.1 BRECHUNGSINDIZES.........................................................................................123
6.1.1 Brechungsindex des Systems Glykol/Wasser ...........................................123
6.1.2 Brechungsindex des Systems PEG400/Wasser ........................................124
6.2 KALORIMETRISCHE UNTERSUCHUNG DES SYSTEMS AUS PEG900 UND WASSER .125
6.2.1 DSC-Messungen des Systems PEG900/Wasser ......................................125
6.2.2 Auswertung der eutektischen Signale nach Tammann..............................126

INHALT
6.2.3 Phasendiagramm PEG900/Wasser...........................................................127
6.3 KALORIMETRISCHE UNTERSUCHUNG DES SYSTEMS AUS PEG600 UND WASSER .128
6.3.1 DSC-Messungen des Systems PEG600/Wasser ......................................128
6.3.2 Auswertung der eutektischen Signale nach Tammann..............................129
6.3.3 Phasendiagramm PEG600/Wasser...........................................................130
6.4 KALORIMETRISCHE UNTERSUCHUNG DES SYSTEMS AUS PEG400 UND WASSER .131
6.4.1 DSC-Messungen des Systems PEG400/Wasser ......................................131
6.4.2 Phasendiagramm PEG400/Wasser...........................................................132
6.5 KALORIMETRISCHE UNTERSUCHUNG DES SYSTEMS AUS PEG300 UND WASSER .133
6.5.1 DSC-Messungen des Systems PEG300/Wasser ......................................133
6.5.2 Phasendiagramm PEG300/Wasser...........................................................134
7 LITERATUR 135

EINLEITUNG 1
1 EINLEITUNG
Das Mischphasenverhalten binärer Systeme unter Beteiligung von Polymeren
ist von großem Interesse. Bei der Charakterisierung von Polymer-Lösemittel-
Systemen im Hinblick auf ihre physikalisch chemischen Eigenschaften ist das
Vorliegen einer homogenen Lösung bei vielen Untersuchungsmethoden Vor-
aussetzung. Die Kenntnis von heterogenen und homogenen Phasengebieten in
Abhängigkeit vom Druck, von der Temperatur und der Zusammensetzung ist
äußerst relevant für die Genauigkeit bzw. die prinzipielle Anwendbarkeit der
jeweiligen Untersuchungsmethode. Ein weiterer Punkt ist das Ausfrieren ein-
zelner Komponenten in Leitungen chemischer Anlagen, die dadurch blockiert
werden können. Durch die Kenntnis des temperaturabhängigen Phasenver-
haltens der Systeme können solche Kristallisationen, z.B. durch ein gezieltes
Temperieren von Leitungen unterbunden werden. Bei der Temperierung der
Leitungen kann vorhergesagt werden, wie hoch diese erwärmt werden müssen
bzw. ab welchen Temperaturen keine festen Phasen mehr vorliegen. Unnötig
hohe Erwärmungen können so vermieden werden.
Das spezielle Mischungsverhalten von Polyethylenglykol (PEG) verschiedener
molarer Massen mit Wasser ist Gegenstand vieler Untersuchungen gewesen.
Polyethylenglykole zeigen in Wasser ein einzigartiges Verhalten. Innerhalb der
homologen Reihe der Polyether sind sie die einzigen, die in Wasser löslich
sind1,2, wobei diese Eigenschaft auch bei höhermolekularen Spezies
(M > 50.000 g/mol) zu beobachten ist. Polyethylenglykole dienen als Lösemittel
für viele organische und anorganische Verbindungen, was sie – im Zusam-
menhang mit ihrer nicht vorhandenen Toxizität – zu Polymeren von großem
industriellen Interesse macht. Vor allem in der pharmazeutischen Industrie
werden Polyethylenglykole mit ihren geeigneten Löslichkeitseigenschaften als
Trägersubstanzen im Bereich der Medikamentenverabreichung eingesetzt3.
Polyethylenglykole werden hier nicht nur in reiner Form verwendet, oft werden
sie auch als bestimmte Spacer in andere Polymere eingebaut. Dabei werden 1 Davidson R.L.; Handbook of water-soluble gums and resins; McGraw-Hill, New York,
19802 Elias, H.-G.; Makromoleküle; Hütig & Wepf: Basel, Heidelberg, 19713 Kibbe A.H.; Handbook of Pharmaceutical Excipients, Am. pharm association, 1994

EINLEITUNG 2
gezielt die wasserlöslichen Eigenschaften der PEG-Ketten ausgenutzt, wodurch
Medikamente gezielter eingesetzt werden können. Andere wichtige Werkstoffe
sind die auf PEG basierenden Polyurethane, deren Quellverhalten in wässrigen
Medien von besonderem Interesse ist4.
In der Vergangenheit sind mehrere Phasendiagramme bzw. das thermische
Verhalten der PEG/Wasser Systeme mit Hilfe der Differential Scanning Calori-
metry (DSC) untersucht worden5-8. Die molare Masse der verwendeten Poly-
ethylenglykole lag in fast allen Fällen oberhalb von 1000 g/mol. Die Phasen-
diagramme zeigen alle ein eutektisches Schmelzverhalten. Ein Grund für das
Fehlen der Phasendiagramme kann die thermische Analyse der PEG/Systeme
sein, die sich bei molaren Massen unterhalb von 1000 g/mol schwierig gestaltet
im Vergleich zu den Systemen mit Polyethylenglykolen höherer molaren
Masse9. Bei kürzeren PEG-Ketten treten Abweichungen von dem relativ ein-
heitlich in der Literatur beschriebenen Phasendiagramm auf. Im Rahmen dieser
Arbeit werden vor allem die kettenlängenabhängigen Veränderungen der
PEG/Wasser Phasendiagramme dargestellt. Dazu werden mit Hilfe der DSC die
binären Phasendiagramme einiger PEG/Wasser Systeme untersucht. So sollen
die Phasendiagramme schrittweise, in Abhängigkeit von der Kettenlänge des
Polyethylenglykols, ermittelt werden. Es sollten die binären Systeme – vom
Ethylenglykol ausgehend bis zum PEG einer molaren Masse von 1500 g/mol
und Wasser – gemessen werden. So sollte ein vollständiges Bild des Kristal-
lisations- und Schmelzverhaltens des Systems PEG/Wasser in Abhängigkeit
von der Kettenlänge entstehen.
Neben den industriellen Anwendungen sind Polyethylenglykole auch aus
thermodynamischer Sicht sehr interessant, da sie neben ihrer Wasserlöslichkeit
im Bereich der Raumtemperatur, von einer bestimmten molaren Masse an (ca.
1.800 g/mol), bei Temperaturen oberhalb von ca. 100°C, eine geschlossene
4 Frahn S.; Dissertation Duisburg, 19975 Huang L., Nishinari K.; J. Pol. Sci., 39, 2001, 4966 Hager S.L., Macrury T.B.; J. Apl. Pol. Sci., 25, 1980, 15597 Bogdanov B., Mihailov M.; J. Pol. Sci.; 23, 1985, 21498 de Vringer T., Joosten J.G.H., Junginger H.E.; Col. Pol. Sci., 264, 1986, 6239 Dobnik E.; Dissertation Duisburg, 1991

EINLEITUNG 3
Mischungslücke aufweisen. Die thermodynamische Beschreibung dieser
Entmischung ist im allgemeinen nicht einfach10,11.
Für eine konsistente thermodynamische Beschreibung der Gleichgewichts-
kurven ist es notwendig die Exzessgrößen wie die Mischungsenthalpie und die
Mischungsentropie zu berücksichtigen. Diese lassen sich prinzipiell aus den mit
Hilfe der DSC ermittelten Phasendiagrammen berechnen. Es zeigte sich aber,
dass die so ermittelten Werte teilweise nicht einmal vom Vorzeichen her mit den
tatsächlichen Werten übereinstimmten12-14. Bei einer komplizierten Temperatur-
und Konzentrationsabhängigkeit der Exzessgrößen ist es nicht möglich, die in
theoretischen Ansätzen auftretenden relevanten Parameter allein aus dem
Verlauf der Gleichgewichtskurven zu ermitteln. Daher ist es unabdingbar, dass
die Mischungsenthalpien der Systeme getrennt bestimmt werden, um zu einer
quantitativen, konsistenten Beschreibung der Phasengleichgewichtskurven zu
gelangen. Einige Berechnungen an dem binären System n-Hexan/n-Hexadekan
lieferten bereits vielversprechende Ergebnisse12.
Müller hat in seiner Dissertation erste aussichtsreiche Mischungsversuche in
der DSC mit Hilfe von Einspritzvorgängen durchgeführt. Im Rahmen dieser
Arbeit wird eine Methode vorgestellt, mit der es möglich ist, direkt mit Hilfe der
DSC Mischungsenthalpien zu bestimmen. Mit dieser neuen Methode werden
die Mischungsenthalpien der PEG/Wasser Systeme bestimmt.
Die Kombination der kettenlängenabhängigen Mischungsexperimente mit den
differentialkalorimetrischen Messungen wird für ein besseres Verständnis des
Phasenverhaltens von PEG/Wasser und weiteren Mischsystemen, an denen
makromolekulare Substanzen beteiligt sind, sorgen.
10 Fischer V., Borchard W., Karas M.; J. Phys. Chem., 100, 1996, 1599211 Fischer V.; Dissertation Duisburg, 200112 Müller A.; Dissertation Duisburg, 199813 Dobnik E.; Dissertation Duisburg, 199114 Hager S.L., Macrury T.B.; J. Apl. Pol. Sci., 25, 1980, 1559

THEORETISCHER TEIL 4
2 THEORETISCHER TEIL
2.1 Definitionen und Konventionen
2.1.1 Binäre Phasendiagramme
Im folgenden soll ein binäres, isobares, eutektisches Phasendiagramm mit
partieller Mischkristallbildung im festen Bereich erläutert werden. Da lediglich
die Phasengleichgewichte zwischen festen und flüssigen Phasen betrachtet
werden, nennt man diese Phasendiagramme auch Schmelzdiagramme. Der
Druck wird in diesen Diagrammen als konstant vorausgesetzt. Da bei Schmelz-
vorgängen die Umwandlungstemperaturen aufgrund der geringen Kompressibi-
litäten und Ausdehnungskoeffizienten der Festkörper nahezu druckunabhängig
sind, sind geringe Schwankungen des Aussendrucks vernachlässigbar. Auch
bei Messungen von Fest-Flüssig Übergängen in geschlossenen Probengefäßen
können isobare Verhältnisse vorausgesetzt werden, da sich das Volumen beim
Übergang vom Festen zum Flüssigen im allgemeinen nur sehr geringfügig
ändert, vor allem im Vergleich zur Messung von Flüssig-Gasförmig Übergängen
(Siedediagramme).
Das Phasendiagramm in Abbildung 1 zeigt die Koexistenz verschiedener Pha-
sen in Abhängigkeit von der Zusammensetzung und der Temperatur. In dem
hier gezeigten Beispiel wird die Zusammensetzung durch den Molenbruch der
Komponente 2 wiedergegeben (x2). Dieser verläuft auf der Abszisse von 0 bis
1. Alternativ kann auch der Molenbruch der Komponente 1 (x1), angegeben
werden, der auf der Abszisse von 1 bis 0 verläuft (graue Beschriftung). Auf der
Ordinate wird die thermodynamische Temperatur aufgetragen.
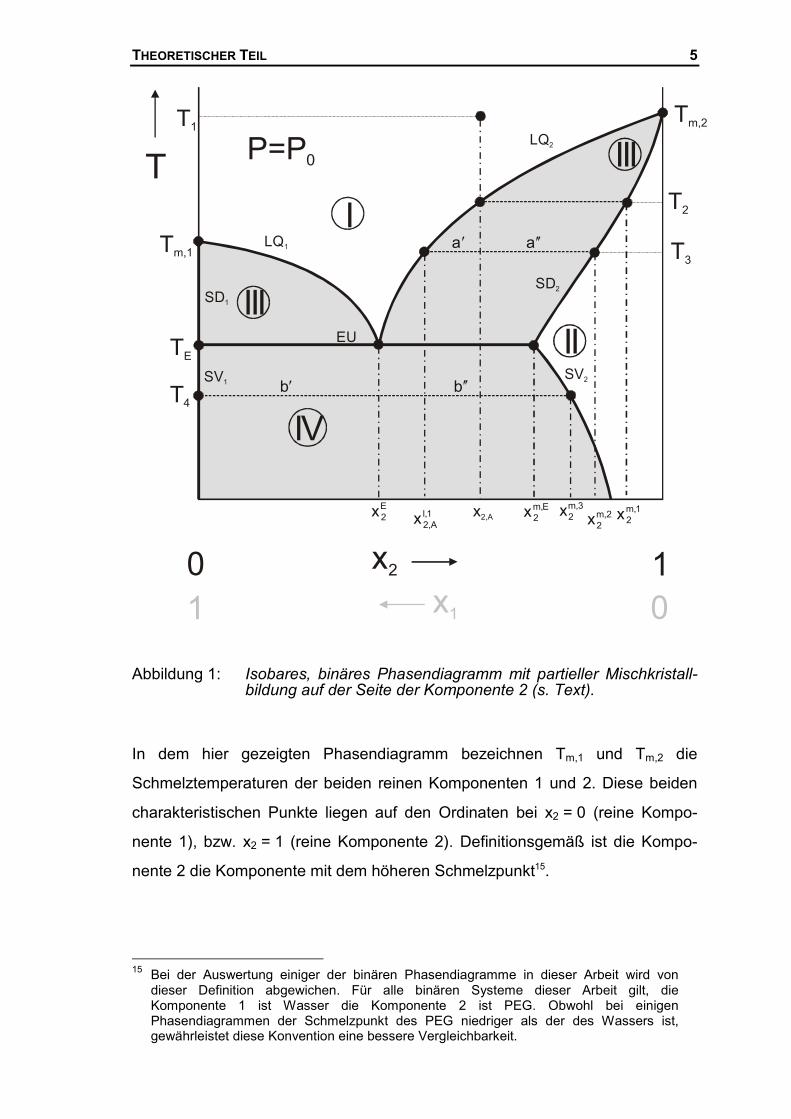
THEORETISCHER TEIL 5
Abbildung 1: Isobares, binäres Phasendiagramm mit partieller Mischkristall-bildung auf der Seite der Komponente 2 (s. Text).
In dem hier gezeigten Phasendiagramm bezeichnen Tm,1 und Tm,2 die
Schmelztemperaturen der beiden reinen Komponenten 1 und 2. Diese beiden
charakteristischen Punkte liegen auf den Ordinaten bei x2 = 0 (reine Kompo-
nente 1), bzw. x2 = 1 (reine Komponente 2). Definitionsgemäß ist die Kompo-
nente 2 die Komponente mit dem höheren Schmelzpunkt15.
15 Bei der Auswertung einiger der binären Phasendiagramme in dieser Arbeit wird von
dieser Definition abgewichen. Für alle binären Systeme dieser Arbeit gilt, dieKomponente 1 ist Wasser die Komponente 2 ist PEG. Obwohl bei einigenPhasendiagrammen der Schmelzpunkt des PEG niedriger als der des Wassers ist,gewährleistet diese Konvention eine bessere Vergleichbarkeit.
x1 01x20 1
Tm,2
Tm,1
TE
T4
T2
T3
T1
T P=P0
I
IIIII
III
IV
LQ1
x2,A
EU
SD1
SV1
LQ2
SD2
SV2
1,m2x2,m
2x1,lA,2x
a� a ��
E2x E,m
2x 3,m2x
b� b ��

THEORETISCHER TEIL 6
Im vorliegenden Schmelzdiagramm unterscheidet man die homogenen Berei-
che (I) und (II) von den heterogenen Bereichen (III) und (IV) (grau unterlegt). Im
Bereich (I) liegt die entsprechende binäre Mischung als homogene Schmelze
vor. Im Bereich (II) liegt eine feste Mischphase vor, diese Mischphase ist reich
an Komponente 2. Die beiden heterogenen Bereiche unterscheiden sich eben-
falls durch ihre Stoffzustände, im Bereich (III) koexistiert eine flüssige mit einer
festen Phase, während im Bereich (IV) zwei feste Phasen vorliegen. Die Linien,
die die homogenen von den heterogenen Phasen trennen sind die Gleich-
gewichtskurven des Schmelzdiagramms. Hier unterscheidet man Liquidus-
kurven (LQ), Soliduskurven (SD) und Solvuskurven (SV). Die jeweilige Gleich-
gewichtskurve auf der Seite der Komponente 1 bekommt den Index 1, die auf
der Seite der zweiten Komponente den Index 2. Die Liquiduskurven geben die
Zusammensetzungen der Lösung wieder, die im heterogenen Gebiet mit einer
festen Phase koexistieren, deren Zusammensetzung von der entsprechenden
Soliduskurve beschrieben wird. Unterhalb der Eutektikalen (EU) liegen nur noch
feste Phasen vor. In diesem Temperaturbereich werden die homogenen Pha-
sen von den heterogenen Phasen durch die Solvuskurven getrennt. Die Sol-
vuskurven geben die Konzentrationen der beiden koexistierenden festen Pha-
sen wieder. Der Unterschied zwischen Solidus- und Solvuskurve ist, dass die
Solvuskurven die Zusammensetzungen der festen Phasen im Fest-Fest Gleich-
gewicht wiedergeben, während die Soliduskurven die Zusammensetzung der
festen Phase im Fest-Flüssig Gleichgewicht wiedergeben.
Auf der Seite der Komponente 1 liegen die Solidus- und die Solvuskurve auf der
Ordinate, d.h. auf der Seite der Komponente 1 liegt keine Mischphasenbildung
vor, in dem analog zum Bereich (II) eine feste Mischphase gebildet wird, die
reich an Komponente 1 ist. Die Lage der Solidus- und der Solvuskurve auf der
Temperaturachse bei x2 = 0 signalisiert, dass sich auf der Seite der Kompo-
nente 1 keine Mischkristalle bilden, sondern die reine Komponente 1 ausfriert.
Die partielle Mischkristallbildung – oder auch Randlöslichkeit im festen Bereich
– wird in diesem Diagrammtyp nur auf der Seite der Komponente 2 gefunden,
was durch den homogenen festen Bereich (II) angedeutet wird. Für das Vor-

THEORETISCHER TEIL 7
liegen einer Mischkristallbildung müssen im wesentlichen drei Bedingungen
erfüllt sein16:
� Gleichheit des chemischen Formeltyps
� Gleichheit des Kristallgittertyps (Isomorphie)
� Gleichheit der Gitterabstände beider Komponenten
Abweichungen von diesen Bedingungen sorgen dafür, dass eine Mischkristall-
bildung lediglich partiell, d.h. in einigen Konzentrationsbereichen, auftritt oder
völlig ausbleibt. Im letzteren Fall lägen in dem Schmelzdiagramm in Abbildung 1
auch die Kurven SD2 und SV2 auf der Ordinate bei x2 = 1 (bzw. x1 = 0).
Im folgenden soll mit Hilfe des Schmelzdiagramms eine Lösung bestimmter
Zusammensetzung, x2,A beschrieben werden, die von einer Temperatur ober-
halb der Liquiduskurven im homogenen Bereich , T1, schrittweise abgekühlt
wird. Wird die Lösung auf die Temperatur T2 abgekühlt, steht die Lösung mit
einer infinitisimal kleinen Mischkristallmenge der Zusammensetzung 1,m2x im
Gleichgewicht.
Wird die Mischung weiter auf Die Temperatur T3 abgekühlt so liegt ein hetero-
genes Gleichgewicht zwischen der Schmelze der Zusammensetzung 1,lA,2x und
einem Mischkristall der Zusammensetzung 2,m2x vor. Mit Hilfe der Abschnitte auf
der Konode (waagerechte Verbindung der Liquiduskurve mit der Soliduskurve)
können gemäß des Hebelgesetzes direkt die entsprechenden Zusammen-
setzungen der flüssigen und der festen Phase angegeben werden:
A,21,l
2
2,m2A,2
Flüssig
Fest
xxxx
aa
nn
�
��
��
�� (1)
Darin bedeuten nFest und nFlüssig die Stoffmengen der festen bzw. flüssigen
Phase, A,2x die Bruttozusammensetzung, 2,m2x und 1,l
2x die Zusammensetzun-
16 Kitaigorodsky A.I.; Mixed Crystals; Springer, Berlin, 1984, Kap. 8-12

THEORETISCHER TEIL 8
gen der Schmelze und des koexistierenden Mischkristalls in der Molenbruch-
skala, sowie a� sowie a �� die in Abbildung 1 gezeigten Hebelarme auf den
Konoden bei der Temperatur T3. Wie aus Gl.(1) zu ersehen ist, verhalten sich
die Stoffmengen in den beiden Phasen umgekehrt proportional zum Verhältnis
der beiden Hebelarme a� sowie a �� .
Bei der Temperatur TE liegt ein invariantes Dreiphasen-Gleichgewicht vor. Die-
ser Punkt ist charakteristisch für ein eutektisches Schmelzdiagramm und wird
als Eutektikum oder eutektischer Punkt bezeichnet. Die Schmelze der Zusam-
mensetzung E2x befindet sich im Gleichgewicht mit einem Mischkristall der
Zusammensetzung E,m2x und einem reinen Kristall der Komponente 1. Nach der
Gibbs’schen Phasenregel kann die Anzahl der Freiheitsgrade mit Hilfe folgen-
der Formel berechnet werden17:
2PKF ��� (2)
F ist die Anzahl der Freiheitsgrade, K ist die Anzahl der Komponenten und P die
Anzahl der koexistierenden Phasen. Nach Gl.(2) kann beim eutektischen Punkt
eines binären Systems ein Freiheitsgrad von 1 berechnet werden, d.h. eine
Zustandsvariable wie die Temperatur, die Konzentration oder der Druck, kann
frei gewählt werden, so dass drei Phasen koexistieren. Unter isobaren Bedin-
gungen fällt der Druck als frei wählbarer Freiheitsgrad weg, was dazu führt,
dass am eutektischen Punkt kein Freiheitsgrad unter isobaren Bedingungen
vorliegt. Somit ist der eutektische Punkt festgelegt (invariant).
Wird die Mischung von der Temperatur T3 weiter unter die eutektische Tempe-
ratur, TE, auf die Temperatur T4 abgekühlt, stellt sich ein Zweiphasengleich-
gewicht ein. In diesem Fall koexistiert ein an Komponente 2 reicher Mischkristall
der Zusammensetzung 3,m2x mit dem reinen Kristall der Komponente 1 (x2 = 0).
Auch in diesem Gleichgewicht kann nach dem Hebelgesetz, Gl.(1), das Men-
17 Gibbs J.W.; Collected Works Vol. I : Thermodynamics, New Haven, 1948

THEORETISCHER TEIL 9
genverhältnis der beiden Mischkristalle mit Hilfe entsprechenden Abschnitte auf
der Konoden, b� und b �� , bestimmt werden.
Neben dem in Abbildung 1 gezeigten binären Phasendiagramm gibt es weitere,
zum Teil wesentlich kompliziertere Typen, auf die hier nicht eingegangen wer-
den soll. Einen Überblick über einen Teil dieser Phasendiagramme gibt die Dis-
sertation von A. Müller18. Desweiteren sind in dem Buch von Tammann grund-
legend einige Phasendiagramme abgehandelt19. Die quantitative Behandlung
von Phasendiagrammen unter besonderer Berücksichtigung der Polymeren
erfolgt in dem Buch von Koningsveld et al.20.
2.1.2 Definition des Konzentrationsmaßes
Im o.g. Kapitel wurde der Molenbruch als Maß für die Zusammensetzung an-
geführt. Dieser ist in einem n-Komponenten-System wie folgt definiert:
��
ii
ii
nn
x (3)
Der Molenbruch wird vor allem in niedrigmolekularen Systemen verwendet, in
denen die Teilchengrößenunterschiede nicht sehr groß bzw. vernachlässigbar
sind. Nehmen die Teilchengrößenunterschiede zu, indem die Differenz der mo-
laren Massen größer wird, kommt es zu einer ungünstigen Gewichtung der
Zusammensetzung in der Molenbruchskala. In der Arbeit von Könneke et al.
werden die Mischungsenthalpien von Wasser mit Glykol, Diglykol und Triglykol
in der Molenbruchskala untersucht21. Vom Glykol zum Triglykol wird der Unter-
18 Müller A.; Dissertation Duisburg, 199819 Tammann G.; Lehrbuch der Heterogenen Gleichgewichte, Friedr. Vieweg & Sohn,
Braunschweig, 192420 Koningsveld R., Stockmayer W.H., Nies E.; Polymer Phase Diagrams, Oxford Univ.
Press, 200121 Könneke H.G., Steinert H., Leibnitz E.;.Z. Phys. Chem., 208, 1957, 147

THEORETISCHER TEIL 10
schied in der molaren Masse zum Wasser hin immer größer. Durch diesen
Unterschied verschiebt sich das Maximum in der Auftragung der Mischungs-
enthalpie gegen die Zusammensetzung in Richtung des Lösemittels. Während
im System Glykol/Wasser das Maximum der Mischungsenthalpie bei einem
Molenbruch des Wassers von 0,6 zu finden ist, verschiebt sich dieses Maximum
beim System Triglykol/Wasser zu einem Wert des Molenbruchs des Wassers
von 0,75.
Nimmt der Unterschied der molaren Massen weiter zu, wird nach Gl.(3) deut-
lich, dass in binären Mischungen stets eine sehr große Stoffmenge der niedrig-
molekularen Komponente zu finden ist, während von der hochmolekularen
Komponente extrem geringe Stoffmengen vorliegen. Aus diesem Grund werden
Systeme mit großen Teilchengrößenunterschieden mit anderen Konzentra-
tionsmaßen als dem Molenbruch beschrieben. Konzentrationsmaße, die die
Teilchengrößenunterschiede mit berücksichtigen sind der Volumenbruch Gl.(4),
und der Grundmolenbruch Gl.(5):
���
ii0
i0i
VV
(4)
Der Volumenbruch gibt den Quotienten aus dem Volumen einer Komponente
V0i und dem Gesamtvolumen der Mischung an. Im Fall des Volumenbruchs
muss bei temperaturabhängigen Untersuchungen darauf geachtet werden, dass
er gegenüber den anderen Konzentrationsmaßen den Nachteil hat, infolge der
thermischen Ausdehnung von der Temperatur abhängig zu sein.
� �
��
iii
ii*i
nrnr
x (5)

THEORETISCHER TEIL 11
Der Volumenbruch wurde im Zuge der molekularstatistischen Theorie von
P.J. Flory und M.L. Huggins für Polymerlösungen eingeführt22-25. Die
höhermolekulare Komponente wird im Volumenbruch in einzelne Segmente
aufgeteilt, deren Größe so bemessen wird, dass jedes Segment der höhermole-
kularen Komponente mit der niedrigmolekularen Komponente (Lösemittel) den
Platz auf einem Gitter tauschen kann. Diese Aufteilung der höhermolekularen
Komponente liefert den Polymerisationsgrad r, der den Teilchengrößenunter-
schied berücksichtigt. Der Polymerisationsgrad gibt an, aus wieviel wiederkeh-
renden Einheiten ein Polymermolekül besteht. Der Polymerisationsgrad in der
Grundmolenbruchskala wird mit dem Index n gekennzeichnet, der Polymeri-
sationsgrad in der Volumenbruchskala wird mit dem Index v gekennzeichnet.
Der Grundmolenbruch gibt den Quotienten aus der Stoffmenge einer Kompo-
nente zur gesamten Stoffmenge der Mischung an, wobei im Unterschied zum
Molenbruch, die höhermolekulare Komponente in einzelne wiederkehrende
Segmente aufgeteilt wird, was auch in diesem Fall den Polymerisationsgrad
eines Polymermoleküls liefert.
Am Beispiel des PEG soll der Polymerisationsgrad kurz erläutert werden:
Polymerisationsgrad (r = 1,2,3.....)
PEG ist aus immer wiederkehrenden Ethylenoxid-Einheiten aufgebaut. Die
Anzahl der Ethylenoxid-Einheiten gibt den Polymerisationsgrad des Moleküls
an. Der Grundmolenbruch ist eine Erweiterung des Molenbruchs. Bei einem 22 Flory P.J.; Principles of Polymer Chemistry, Cornell Univ. Press, New York, 195323 Huggins M.L.; Physical chemistry of high polymers, Wiley & Sons, New York, 195824 Haase R.; Thermodynamik der Mischphasen; Berlin; 1956, Kap. 82 und 8325 Haase R.; Survey of Fundamental Laws; Academic Press, New York; 1971, Kap. 126 Eyring H., Henderson D., Jost W.; Physical Chemistry: An Advanced Treatise, Volume I:
Thermodynamics, Academic Press, New York, 1971
O
H2C
CH2
O
r
HH

THEORETISCHER TEIL 12
Polymerisationsgrad von 1 geht der Grundmolenbruch in den Molenbruch über.
Im o.g. Beispiel liegt bei einem Polymerisationsgrad von 1 reines Ethylenglykol
vor, bei r = 2 liegt Diglykol vor, bei r = 3 Triglykol usw.
Neben dem Grundmolenbruch und dem Volumenbruch wird häufig bei der
Beschreibung polymerer Systeme der Massenbruch als Konzentrationsmaß
verwendet27.
��
ii
ii
mm
w (6)
Der Massenbruch gibt den Quotienten aus der Masse einer Komponente zur
gesamten Masse der Mischung an.
2.2 Quantitative Beschreibung der Koexistenzkurven
2.2.1 Quantitative Beschreibung der Liquiduskurven
Im folgenden soll die quantitative Beschreibung der Liquiduskurven erwähnt
werden. Einschränkend muss bemerkt werden, dass die quantitative Beschrei-
bung nur für den Fall gilt, dass keine Mischkristallbildung vorliegt. Da die alle in
dieser Arbeit verwendeten binären Phasendiagramme PEG/Wasser-Systeme
sind, wird die Ableitung in den folgenden Kapiteln in der Art durchgeführt, dass
die Komponente 1 das Lösemittel Wasser ist und die Komponente 2 das PEG
(s. Fußnote S.5).
Mit Hilfe der Flory-Huggins-Stavermann-van Santen (FHSS) Theorie nicht-ideal-
athermischer Lösungen können die Gleichgewichtskurven in binären Schmelz-
27 Es wird sich im Verlauf dieser Arbeit herausstellen, dass im Fall der binären PEG/Wasser
der Massenbruch den anderen Konzentationsmaßen vorzuziehen ist.

THEORETISCHER TEIL 13
diagrammen quantitativ beschrieben werden. Hierzu wird die Gleichgewichts-
bedingung für ein heterogenes Gleichgewicht zwischen dem Feststoff und der
Lösung formuliert. Dies erfordert die Gleichheit der chemischen Potentiale des
Lösemittels im Feststoff und des chemischen Potentials des Lösemittels in der
Lösung. Im Fall des in Kap.2.1.1 beschriebenen Phasendiagramms wird die
Liquiduskurve des Wassers dadurch beschrieben, dass das spezifische chemi-
sche Potential des Wassers in der Schmelze, L1
~� , gleich dem spezifischen
chemischen Potential des Wassers im reinen Eiskristall, S01
~� , ist (Phasenindex S
für Kristall und L für Lösung):
L1
S01
~~ ��� (7)
Entsprechend kann man für die Liquiduskurve des PEG (Index 2) die Gleichheit
der chemischen Potentiale formulieren, wenn reines PEG mit der Lösung ko-
existiert, dieser Fall entspricht nicht mehr dem vorgestellten Phasendiagramm
in Kap.2.1.1, da in diesem Fall von einer Mischkristallbildung auf der Seite des
PEG ausgegangen wird:
L2
S02
~~ ��� (8)
Das chemische Potential des Wassers in der Lösung, L1
~� , kann durch den fol-
genden Ausdruck aus der statistischen FHSS-Theorie beschrieben werden, der
das chemische Potential in Abhängigkeit von der Zusammensetzung, hier dem
Massenbruch, der Temperatur und dem Molmassenverhältnis des Polymeren
zum Lösemittel in binären Mischphasen beschreibt28-30:
��
���
���
���
�������� 2
2w2w
11
L01
L1 ww
r11wln
MRT~~ (9)
28 Flory P.J.; Principles of Polymer Chemistry, Cornell Univ. Press, New York, 195329 Huggins M.L.; Physical chemistry of high polymers, Wiley & Sons, New York, 195830 Staverman A.J., van Santen J.M.; Rec. Tra. Chim., 60, 1941, 76

THEORETISCHER TEIL 14
Unter Verwendung der Gibbs-Duhem Beziehung, Gl.(10), kann entsprechend
das spezifische chemische Potential des PEG berechnet werden:
0x
xx
xP,T2
2*2
P,T1
1*1 �
��
�
�
��
�
�
�
�
��
�
�
��
�
�
�
�(10)
� �� �21ww1w2
2
L02
L2 wrwr1wln
MRT~~ ��������� (11)
Darin bezeichnen R die universelle Gaskonstante, T die thermodynamische
Temperatur, M1 und M2 die molaren Massen des Lösemittels (Wasser) und des
PEG sowie rw das Verhältnis der Molmasse des PEG M2 zur Molmasse des
Wassers M1. �w ist der FHSS-Wechselwirkungsparameter in der Massen-
bruchskala, in dem alle Abweichungen vom ideal-athermischen Fall berücksich-
tigt werden.
Mit den Gln. (7) bis (11) lassen sich die Differenzen für das heterogene Gleich-
gewicht zwischen Lösung und Kristall berechnen, wenn die durch Gln. (12) und
(13) eingeführten Differenzen bekannt sind:
S01
L01
S/L01
~~~ ������ (12)
S02
L02
S/L02
~~~ ������ (13)
Die beiden chemischen Potentiale des Wassers und des PEG sind nur am
jeweiligen Schmelzpunkt, Tm,1 und Tm,2, gleich. Die Temperaturabhängigkeit vonS/L
i0~�� wird durch Gl.(14) beschrieben31:
� ��
���
����
�
�� T
T P
i0
i,m
i,mi0S/L
i0
i,m
dTT
)T/~(T
)T(~
T)T(~
; i = 1,2 (14)
31 Haase R.; Thermodynamik der Mischphasen, Berlin, 1956

THEORETISCHER TEIL 15
Unter der Voraussetzung, dass die Temperaturabhängigkeit der spezifischen
Schmelzenthalpie, i,mH~� , vernachlässigbar ist, was näherungsweise für kleine
Temperaturbereiche zutrifft, gilt:
��
���
���
��
T1
T1H~
T)T(~
i,mi0
S/Li0 ; ; i = 1,2 (15)
Es wird angenommen, dass das Verhältnis der Wärmekapazitäten im festen
und flüssigen Zustand konstant ist. Durch Zusammenfassung von Gln.(9) bis
(15) erhält man für die Liquiduskurve des Wassers folgenden analytischen Aus-
druck:
��
���
���
���
����
��
�22w2
w1
101
1,m
1,m
wwr11wln
MH~RT
1
TT (16)
Und entsprechend für die Liquiduskurve des PEG:
� �� �21ww1w2
202
2,m
2,m
wrwr1wlnMH~
RT1
TT
������
�
� (17)
Mit den Gln.(16) und (17) stehen quantitative Ausdrücke zur Verfügung, mit
denen der Verlauf der Liquiduskurven in binären Systemen ohne Mischkristall-
bildung berechnet werden kann. Ist der Verlauf der Liquiduskurve bekannt kann
der �w-Parameter berechnet werden, indem alle Abweichungen der realen
(nicht-ideal-athermischen) Mischphase gegenüber dem ideal-athermischen
Bezugszustand erfasst sind.

THEORETISCHER TEIL 16
2.2.2 Wechselwirkungsparameter �
Der �-Wechselwirkungsparameter beschreibt die Abweichungen der realen
hochmolekularen Mischung vom ideal-athermischen Bezugszustand. Der ideal-
athermische Zustand resultiert aus der statistischen FHSS-Theorie hochmole-
kularer Lösungen. Bei der Beschreibung der thermodynamischen Eigenschaf-
ten einer idealen Lösung wird davon ausgegangen, dass die Teilchen gleich
groß sind und die Wechselwirkungsenergien zwischen gleichen Teilchen, e1,1
und e2,2, gleich den Wechselwirkungsenergien zwischen den Teilchen unter-
schiedlicher Spezies ist, e2,1:
e1,1 = e2,2 = e2,1 (18)
Das chemische Potential in Abhängigkeit von der Konzentration nach dem
Ansatz der idealen Lösung lautet in der Molenbruchskala:
ii0i xlnRT���� (19)
Hierin sind R die universelle Gaskonstante, T die thermodynamische Tempe-
ratur, xi ist die Zusammensetzung in der Molenbruchskala und �0i ist das che-
mische Potential des reinen Stoffes. Abweichungen vom idealen Verhalten
können durch einen Faktor vor dem Molenbruch berücksichtigt werden, dem
von Lewis eingeführten Aktivitätskoeffizienten fi32,33. Aufgelöst ergibt dieser
Faktor einen additiven Term in Gl.(18), nämlich iflnRT . Mit Hilfe dieses Terms
wird das chemische Potential einer niedrigmolekularen realen Lösung beschrie-
ben.
Im Gegensatz zum Ansatz der idealen Lösung, in der von gleich großen Teil-
chen ausgegangen wurde, ist es sinnvoller den Bezugszustand einer ideal-
32 Lewis G.N.; J. Amer. Chem. Soc., 30, 1908, 66833 Lewis G.N., Randall M.; Thermodynamics; McGraw-Hill; New York, 1961, Kap. 18

THEORETISCHER TEIL 17
athermischen zu verwenden der von Flory und Huggins mit Hilfe der statisti-
schen Mechanik abgeleitet wurde, und die Teilchengrößenunterschiede zwi-
schen dem Lösemittel und der hochmolekularen Komponente berücksichtigt34-36.
Die Konzentrationsabhängigkeit des chemischen Potentials für ein
hochmolekulares binäres System in der Grundmolenbruchskala ist gegeben
durch:
� �� �*2
*1011 xr11xlnRT ������ (20)
� �� �*1
*2022 xr1xlnRT ������ (21)
r ist der Polymerisationsgrad der hochmolekularen Komponente (s. S. 11).
Die chemischen Potentiale der Komponente 1 und 2 sind nicht symmetrisch wie
im Fall der idealen Lösung, daher wurden in diesem Fall beide chemischen
Potentiale aufgestellt. �2 lässt sich aus �1 mit Hilfe der Gibbs-Duhem Beziehung
Gl.(10) berechnen37.
Abweichungen vom ideal-athermischen Bezugssystem werden wie im Fall der
idealen Lösung durch einen weiteren additiven Term berücksichtigt, dem soge-
nannten FHSS-Wechselwirkungsparameter, �x, in der Grundmolenbruchskala.
Mit Hilfe dieses Parameters der in der ursprünglichen Theorie nur die
Mischungsenthalpie berücksichtigte, wird das chemische Potential des Löse-
mittels folgendermaßen angesetzt:
� �� �2*2x
*2
*1011 xxr11xlnRT �������� (22)
34 Flory P.J.; Principles of Polymer Chemistry; Cornell Univ. Press, New York, 197535 Huggins M.L.; J. Phys. Chem., 46, 1942, 15136 Huggins M.L.; Physical Chemistry of High Polymers; Wiley, New York, 1958, Kap. 5,637 Haase R.; Thermodynamik der Mischphasen, Berlin, 1956

THEORETISCHER TEIL 18
Mit Hilfe von Gl.(10) folgt für �2:
� �� �2*1x
*1
*2022 xrxr1xlnRT �������� (23)
In Gln.(22) und (23) sind alle Abweichungen vom Bezugssystems der ideal-
athermischen Lösung im �x-Wechselwirkungsparameter berücksichtigt. Folglich
gelangt man im Fall von �x = 0 zu einer ideal-athermischen Lösung. Der Ansatz
der eine reale hochmolekulare Mischphase beschreibt, wird auch als nicht-
ideal-athermische Lösung bezeichnet38.
In den Gl.(22) und (23) ist der �x-Wechselwirkungsparameter als konstanter
Parameter angeben. Reale Systeme können allerdings häufig nicht mit Hilfe
von nur einem konstanten Parameter beschrieben werden. In solchen Fällen
kann der �x-Parameter auch als Wechselwirkungsfunktion angesetzt werden,
die abhängig von der Temperatur, dem Druck und der Konzentration ist. Der
einfachste Ansatz für eine Temperaturabhängigkeit der Wechselwirkungsfunk-
tion ist:
Ti
ii�
���� (24)
Da die Wechselwirkungsfunktion abhängig von der Wahl des jeweiligen Kon-
zentrationsmaßes ist, werden die Wechselwirkungsgrößen durch den Index i
gekennzeichnet. i=x kennzeichnet die Wechselwirkungsgrößen in der Grund-
molenbruchskala und i=w kennzeichnet die Wechselwirkungsgrößen in der
Massenbruchskala.
Dieser Ansatz führt zu temperaturunabhängigen Werten für die Mischungs-
enthalpie und -entropie. Die integrale Mischungsenthalpie und -entropie des
Systems können mit Hilfe der Wechselwirkungsfunktion berechnet werden39.
38 Rehage G.; Z. Elektrochem., 59, 1955, 7839 Haase R.; Thermodynamik der Mischphasen, Berlin, 1956

THEORETISCHER TEIL 19
*2
*1x
*M xxRH ������ (25)
*2
*1x
*M xxRS ������� (26)
Die beiden Exzessfunktionen liefern mit dem Ansatz für die �x-Wechsel-
wirkungsfunktion gemäß Gl.(24) zur Konzentration 5,0xx *2
*1 �� symmetrische
Parabeln. An den beiden Gln. (25) und (26) ist zu erkennen, dass die
Mischungsenthalpie nur vom Parameter �x abhängt, und die Mischungsentropie
nur vom Parameter �x abhängt. Da die Mischungsenthalpie und –entropie
jeweils nur von einem Parameter abhängen, spricht man in diesem speziellen
Fall von dem Enthalpieterm �x und dem Entropieterm �x. Bei komplizierteren
Ansätzen für die �x-Wechselwirkungsfunktion sind die einzelnen Wechsel-
wirkungsfunktionen nicht mehr unabhängig voneinander sondern miteinander
gekoppelt. Das wird schon bei dem nächsteinfachen Ansatz für die �x-Wechsel-
wirkungsfunktion deutlich. Der Ansatz
TlnT
xx
xx ���
���� (27)
liefert Exzessfunktionen die beide von �x abhängig sind. Da der Parameter �x
sowohl in der Mischungsenthalpie als auch in der Mischungsentropie auftritt,
kann von einer Aufspaltung der �x-Wechselwirkungsfunktion in einen Enthalpie-
und Entropieterm keine Rede mehr sein.
Werden die Wechselwirkungsfunktionen in der Massenbruchskala angesetzt,
werden die einzelnen Parameter mit dem Index w gekennzeichnet.
Wird der einfache Ansatz für die Temperaturabhängigkeit der �w-Wechsel-
wirkungsfunktion nach Gl.(24) in die Gl.(16) und (17) eingesetzt resultieren die
folgenden quantitative Ausdrücke für den Verlauf der Liquiduskurven:

THEORETISCHER TEIL 20
���
�
���
���
�
��
����
��
���
�
���
����
���
�22w´2
w1
101
1,m
w2
1011,m
wwr1
1wlnMH~
RT1
wMH~
R1T
T (28)
� �� �22ww1w2
202
2,m
21ww
2022,m
wrwr1wlnMH~
RT1
wrMH~
R1T
T�������
��
���
�
�����
���
(29)
mit:
wi Massenbruch der Komponente 1 bzw. 2
R Allgemeine Gaskonstante
rw Molmassenverhältnis von der Komponente 2 zum Lösemittel 1
�w Entropieterm in der Massenbruchskala
�w Enthalpieterm in der Massenbruchskala
i0H~� Spezifische Schmelzenthalpie der reinen Komponente (1 oder 2)
Tm,i Thermodynamische Schmelztemperatur der reinen Komponente
Mi Molare Masse der reinen Komponenten
Die in Gln.(28) und (29) verwendeten Wechselwirkungsgrößen liefern den Ent-
halpie- und Entropieterm in der Massenbruchskala, denen deshalb der Index w
gegeben wird.

THEORETISCHER TEIL 21
2.2.3 Beschreibung der Liquiduskurven in der Grundmolenbruchskala
Werden die Liquiduskurven in binären Systemen ohne Mischkristallbildung in
der Grundmolenbruchskala ausgewertet, liefert diese Auswertung andere quan-
titative Ausdrücke für den Verlauf der Liquiduskurven als in den Gln.(15) und
(17) bzw. Gln.(28) und (29). Es genügt nicht, lediglich den Massenbruch durch
den umgerechneten Grundmolenbruch in den entsprechenden Auswerte-
gleichungen zu ersetzen. Die anderen Abweichungen resultieren zum einen aus
einem geänderten Ausdruck für den Polymerisationsgrad der in der Grund-
molenbruchskala wie folgt definiert ist:
02
2x
MM
r � (30)
Wobei M2 die molare Masse des Polymeren und M02 die molare Masse des
Grundbausteins darstellt. Weiterhin wird die spezifische Schmelzenthalpie des
Wassers, 01H~� , in der Massenbruchskala durch die molare Schmelzenthalpie,
01H� , in der Grundmolenbruchskala ersetzt. Anstelle der spezifischen
Schmelzenthalpie des Polymeren, 02H~� , aus Gln. (17) und (29) wird nicht die
entsprechende molare Größe verwendet, sondern die auf den Polymerisations-
grad des Polymeren bezogene grundmolare Schmelzenthalpie, G,2H� . Diese
Größe entspricht der molaren Schmelzenthalpie des sich wiederholenden
Grundbausteins des Polymeren unter Vernachlässigung der Endgruppen. Diese
Größe hat den Vorteil, dass sie für hohe Polymerisationsgrade eine konstante
Größe darstellt40.
Mit diesen Bedingungen erhält man folgende Ausdrücke zur quantitativen
Beschreibung der Liquiduskurven in der Grundmolenbruchskala, wenn man in
Gl.(8) S02
~� durch das spezifische chemische Potential des Polymeren im festen
Mischkristall, S2
~� , ersetzt:
40 Dobnik E.; Dissertation Duisburg, 1991

THEORETISCHER TEIL 22
���
�
���
���
�
��
����
��
�2*
2x*2
x
*1
01
1,m
1,m
xxr1
1xlnH
RT1
TT (31)
� �� �2*1xx
*1x
*2
G,2
2,m
2,m
xrxr1xlnH
RT1
TT
������
�
� (32)
Gl.(31) beschreibt analog zu Gl.(16) die Liquiduskurve des Wassers; Gl.(32)
beschreibt analog zu Gl.(17) den Verlauf der Liquiduskurve des Polymeren
unter der Voraussetzung, dass keine Mischkristallbildung im festen Bereich
vorliegt.
Auch bei den beiden Gln.(31) und (32) kann der Wechselwirkungsparameter
gemäß Gl.(24) angesetzt werden, wobei man eine einfache Temperaturab-
hängigkeit des Wechselwirkungsparameters berücksichtigt, der zu temperatur-
unabhängigen Werten der Exzessentropie und –enthalpie führt (vergl. S.16):
Tx
xx�
���� (33)
Die so erhaltenen Wechselwirkungsfunktionen stimmen nur bedingt mit denen
der Auswertung in der Massenbruchskala überein. Die Abhängigkeit der Wech-
selwirkungsgrößen von der Wahl des Konzentrationsmaßes hat Dobnik in
seiner Dissertation behandelt, in der er die Wechselwirkungsfunktion in der
Massenbruch-, Grundmolenbruch- und Volumenbruchskala miteinander ver-
gleicht. Der Unterschied in den einzelnen Skalen führt dazu, dass der Wech-
selwirkungsparameter zum Vergleich mit anderen Autoren immer in der jeweils
gleichen Konzentrationsskala bestimmt werden muss. Andernfalls muss der
Wechselwirkungsparameter in die entsprechenden Konzentrationsmaße umge-
rechnet werden41.
41 Adames W., Breuer W., Michalczyk A., Borchard W.; Eur. Pol. J., 25, 1989, 947

THEORETISCHER TEIL 23
Wird der Ansatz Gl.(33) in Gln.(31) und (32) eingesetzt, erhält man die folgen-
den quantitativen Ausdrücke für den Verlauf der Liquiduskurve des Wassers
(Index 1) bzw. den Verlauf der Liquiduskurve des PEG (Index 2):
���
�
���
���
�
��
����
��
���
�
���
����
���
����
�
22x2
x1
01
1,m
x2
201
1,m
xxr1
1xlnH
RT1
xHR
1TT (34)
���
�
���
���
�
��
����
��
���
�
���
����
���
����
�
21x1
x2
G,2
2,m
x2
1xG,2
2,m
xxr1
1xlnH
RT1
xrHR
1T
T (35)
EXPERIMENTELLER TEIL 24
3 EXPERIMENTELLER TEIL
3.1 Differential Scanning Calorimetry (DSC)
3.1.1 Aufbau und Messprinzip
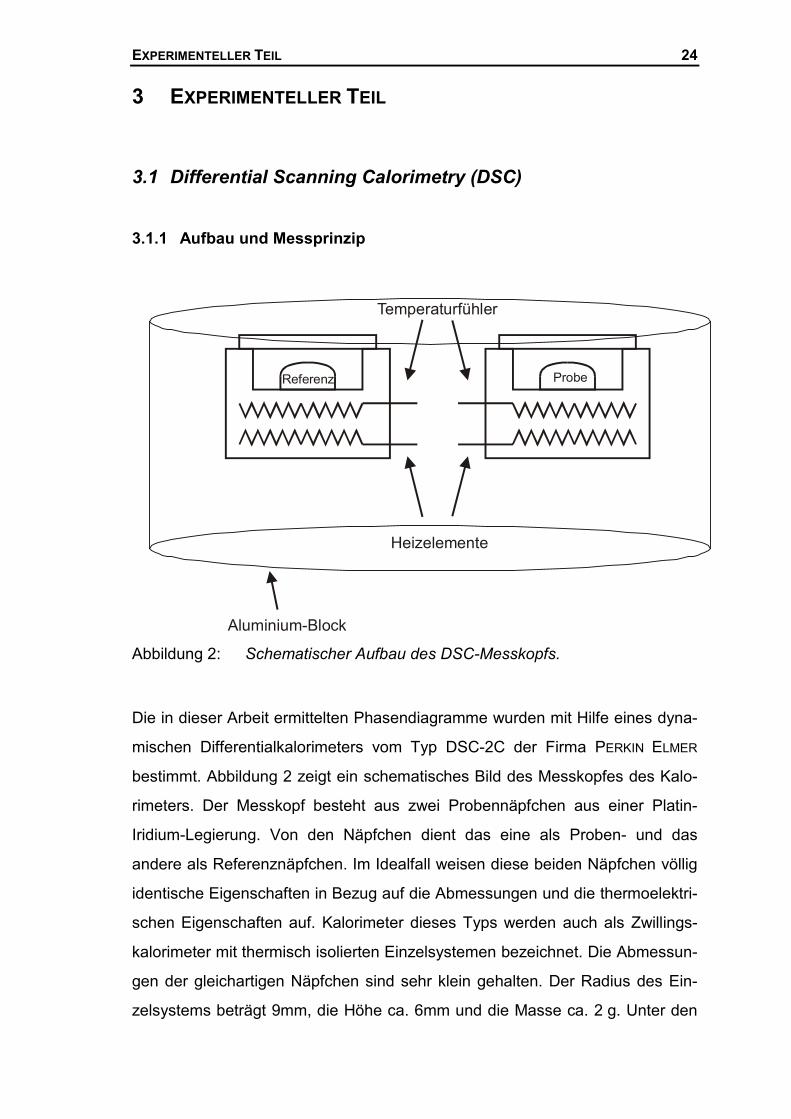
Abbildung 2: Schematischer Aufbau des DSC-Messkopfs.
Die in dieser Arbeit ermittelten Phasendiagramme wurden mit Hilfe eines dyna-
mischen Differentialkalorimeters vom Typ DSC-2C der Firma PERKIN ELMER
bestimmt. Abbildung 2 zeigt ein schematisches Bild des Messkopfes des Kalo-
rimeters. Der Messkopf besteht aus zwei Probennäpfchen aus einer Platin-
Iridium-Legierung. Von den Näpfchen dient das eine als Proben- und das
andere als Referenznäpfchen. Im Idealfall weisen diese beiden Näpfchen völlig
identische Eigenschaften in Bezug auf die Abmessungen und die thermoelektri-
schen Eigenschaften auf. Kalorimeter dieses Typs werden auch als Zwillings-
kalorimeter mit thermisch isolierten Einzelsystemen bezeichnet. Die Abmessun-
gen der gleichartigen Näpfchen sind sehr klein gehalten. Der Radius des Ein-
zelsystems beträgt 9mm, die Höhe ca. 6mm und die Masse ca. 2 g. Unter den
Referenz Probe
Temperaturfühler
Heizelemente
Aluminium-Block

EXPERIMENTELLER TEIL 25
Näpfchen sind untereinander jeweils zwei getrennte durch eine Schicht Alumi-
niumoxid isolierte Platindrähte angeordnet. Der obere Platindraht dient als
Widerstandsthermometer, während der untere als Heizdraht dient.
Die beiden Einzelsysteme sind thermisch voneinander getrennt in einem massi-
ven Aluminiumblock positioniert und mit einem Aluminiumdeckel fest verschlos-
sen. Unter dem Aluminiumblock ist eine Tieftemperatureinrichtung angebracht,
die mit flüssigem Stickstoff gespült werden kann. Der Aluminiumblock wird auf
eine konstante Temperatur von -150°C eingestellt. Beide Näpfchen werden von
einem internen Gasstrom (trockenes Argon) gespült. Der Druck des Spülgases
ist sehr gering und dient einem intensiven konvektiven Wärmeaustausch der
Probennäpfchen mit der Umgebung. Der Spülgasfluss ist nicht geeignet, gas-
förmige Produkte vom Näpfchen abzuführen. In der DSC sollten deshalb keine
chemische Reaktionen im geschlossenen Messkopf untersucht werden, bei
denen im größeren Umfang gasförmige Produkte entstehen. Auch Verdamp-
fungsmessungen sollten im geschlossenen Messkopf nicht durchgeführt wer-
den. Wird anstelle von Argon Helium als Spülgas verwendet, kann die Tem-
peratur des Aluminiumblockes auf eine konstante Temperatur von -194°C ein-
gestellt werden. Die Temperatureinstellung erfolgt mit Hilfe eines Magnetventils,
das gekoppelt an eine Temperiereinheit den Stickstoffstrom regelt. Der nutz-
bare Temperaturbereich der DSC-2C erstreckt sich somit von etwa -170 bis
630°C. Die obere Grenze wird durch die Verwendung von Aluminium als Pro-
bengefäß (Probenpfännchen) vorgegeben, welches einen Schmelzpunkt von
660°C hat und mit der Platin-Iridium-Näpfchen legieren würde. Durch die Ver-
wendung von Stahlpfännchen kann der Messbereich auf höhere Temperaturen
(900°C) ausgedehnt werden.
Der gesamte Messkopf befindet sich innerhalb einer Glovebox, die ständig mit
trockenem Stickstoff gespült wird. Hierdurch werden störende Kondensationen
von Luftfeuchtigkeit am Messkopf verhindert, die die Messungen beeinträchti-
gen würden.

EXPERIMENTELLER TEIL 26
Abbildung 3: Darstellung unterschiedlicher Betriebsarten eines Kalorimeters;(1) Aussenwelt; (2) Umgebung; (3) Messsystem; (4) Wärme-widerstand.
Die Betriebsart des DSC Kalorimeters wird als isoperibol bezeichnet, was
bedeutet, dass die Umgebungstemperatur der Probennäpfchen konstant ist,
wobei die Temperatur des Messsystems variieren kann42. Dies ist durch den
temperierten Aluminiumblock gewährleistet, der auf einer konstanten
Temperatur gehalten wird.
Abbildung 3 zeigt den Unterschied zwischen isothermen, adiabatischen und
isoperibolen Kalorimetern. Bei jedem Kalorimeter lässt sich zwischen der Um-
gebung und den Messsystem unterscheiden. Beim isothermen Kalorimeter ist
der Wärmewiderstand zwischen der Umgebung und dem Messsystem ver-
schwindend klein, was bedeutet, dass Wärme vom Messsystem direkt mit der
Umgebung ausgetauscht wird. Im adiabatischen Fall ist der Wärmewiderstand
idealerweise unendlich groß, d.h. es findet kein Wärmeaustausch zwischen
Messsystem und Umgebung statt. Im isoperibolen Fall hat dieser Wärmewider-
stand einen endlichen, definierten Wert43. Auch das später in dieser Arbeit
beschriebene Mischungskalorimeter von LKB arbeitet im isoperibolen Betrieb.
42 Hemminger W., Höhne G.; Grundlagen der Kalorimetrie, VCh: Weinheim, 197943 Kubaschewski O., Hultgren R.,;Metallurgical and Alloy Thermochemistry in: Experimental
Thermochemistry. Vol.II, Interscience Publishers, New York, 1962
4
31
2
EXPERIMENTELLER TEIL 27
Für genaue, zuverlässige kalorimetrische Messungen ist es nicht unbedingt
notwendig, das Messsystem thermisch zu isolieren und so die Verlustwärme
klein zu halten. Es ist hingegen notwendig diese Verlustwärme in reproduzier-
barer Weise abhängig vom Temperaturunterschied zwischen Messsystem und
Umgebung zu kennen.
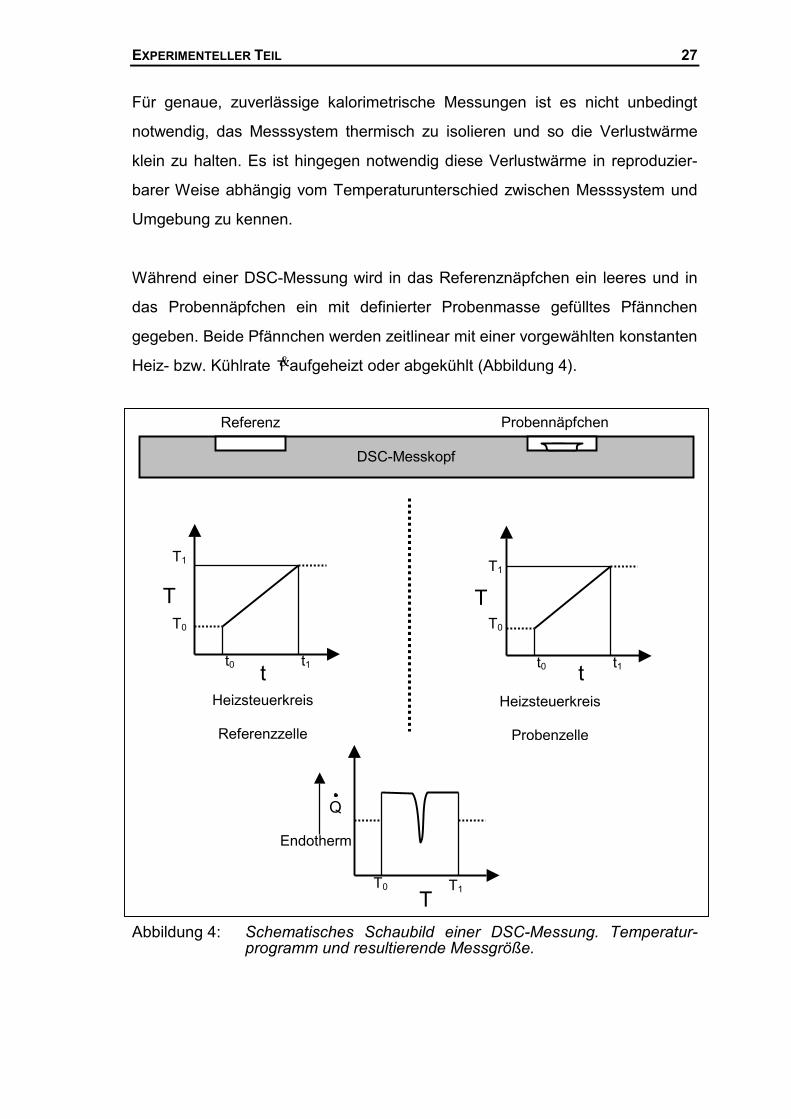
Während einer DSC-Messung wird in das Referenznäpfchen ein leeres und in
das Probennäpfchen ein mit definierter Probenmasse gefülltes Pfännchen
gegeben. Beide Pfännchen werden zeitlinear mit einer vorgewählten konstanten
Heiz- bzw. Kühlrate T�aufgeheizt oder abgekühlt (Abbildung 4).
Abbildung 4: Schematisches Schaubild einer DSC-Messung. Temperatur-programm und resultierende Messgröße.
ProbennäpfchenReferenz
Heizsteuerkreis
Referenzzelle
Heizsteuerkreis
Probenzelle
tt0 t1
T0
T1
T
t0 t1
T0
T1
T
t
DSC-Messkopf
T0 T1T
Q
Endotherm

EXPERIMENTELLER TEIL 28
PtT
T��
�
�
��
�
�
�
�� (36)
Die Heizleistung beider Näpfchen wird dabei so geregelt, dass diese stets die
gleiche Temperatur besitzen. Durch die geringen Abmessungen der Näpfchen
und dem Konvektions-Gasstrom ist ein guter Wärmetransport gewährleistet. So
sind in der DSC Heiz- bzw. Kühlraten von 0,3 bis 320 K/min realisierbar. Ein
zweiter Steuerkreis kompensiert eventuelle Temperaturdifferenzen zwischen
Referenz- und Probennäpfchen infolge einer exo- oder endothermen Reaktion
im Probenpfännchen. Die Differenzleistung dieses zweiten Steuerkreises bzw.
der damit verbundene differentielle Wärmefluss Q� wird als elektrische Span-
nung angezeigt und abhängig von der Zeit registriert. Bei dem Beispiel in
Abbildung 4 ist ein exothermes Signal dargestellt, dass aufgrund einer chemi-
schen Reaktion oder einer Kaltkristallisation im Temperaturbereich T0 < T < T1
während des Aufheizen in der Probe stattfindet.
Bei der Messung eines mit bestimmter Masse gefüllten Probenpfännchens
gegen ein leeres Referenzpfännchen ist das so ermittelte Signal direkt propor-
tional zur spezifischen Wärmekapazität )T(c~P der Probe.
Tm)T(Q)T(c~P �
�
�
� (37)
Da in das Probenpfännchen aufgrund der Wärmekapazität der Probe ständig
mehr Energie zur Temperaturkonstanz eingebracht werden muss als in das
Referenzpfännchen, sollte der differentielle Wärmestrom direkt die Wärme-
kapazität wiedergeben. Tatsächlich zeigt sich aber eine sehr große Abhängig-
keit der Basislinie von der Lage des Pfännchens im Näpfchen. Wiederholende
Messungen ergaben eine starke Streuung der Lage der Basislinie. Auch die
Kontaktfläche zwischen Pfännchen und Probe spielt eine große Rolle beim

EXPERIMENTELLER TEIL 29
Wärmetransport, aus diesen Gründen ist lediglich der relative Verlauf der Wär-
mekapazität mit Hilfe der DSC zu bestimmen.
Die Fläche unter dem Signal im Bereich des Phasenübergangs ist gleich der
Umwandlungsenthalpie. Da sich die Wärmekapazität der Probe beim Phasen-
übergang ändert, ändert sich auch der Verlauf der Basislinie (Sprung). In den
meisten Fällen ist der Sprung jedoch im Vergleich zur Phasenumwandlungs-
wärme so gering, dass man mit guter Näherung eine Gerade als Approximation
zeichnen kann und so die Fläche unter dem Signal bestimmen kann. Bei den
Messungen in dieser Arbeit wurden die Umwandlungsintegrale unter den Mess-
signalen immer mit dieser Näherung bestimmt. Wird dieser Sprung größer,
muss die Basislinie mit anderen Methoden konstruiert werden44-49.
44 E. Dobnik; Dissertation Duisburg, 199145 A. Müller; Dissertation Duisburg, 199846 Hemminger W., Cammenga H.; Methoden der thermischen Analyse, Springer, Berlin,
198947 Hemminger W., Sarge S.M.; J. Therm. Anal., 37, 1991, 145548 Hemminger W., Höhne G.; Calorimetry, VCH, Weinheim, 198449 Hemminger W., Höhne G.; Grundlagen der Kalorimetrie, VCh: Weinheim, 1979
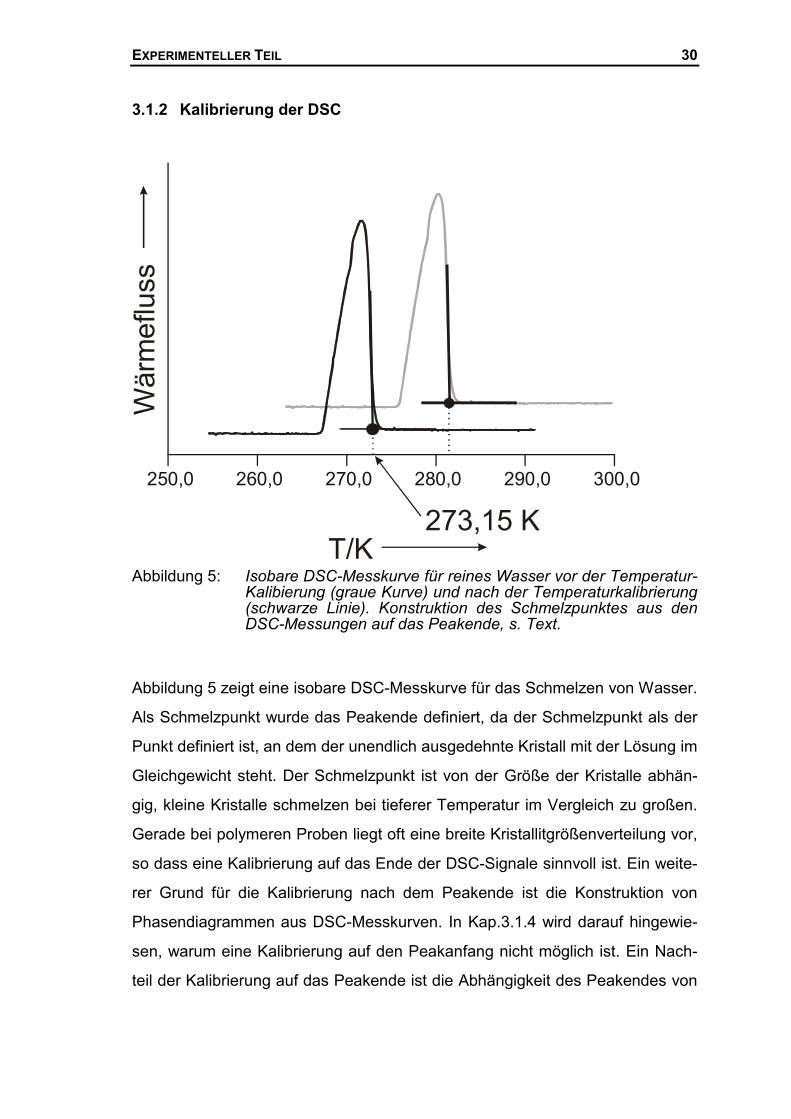
EXPERIMENTELLER TEIL 30
3.1.2 Kalibrierung der DSC
Abbildung 5: Isobare DSC-Messkurve für reines Wasser vor der Temperatur-Kalibierung (graue Kurve) und nach der Temperaturkalibrierung(schwarze Linie). Konstruktion des Schmelzpunktes aus denDSC-Messungen auf das Peakende, s. Text.
Abbildung 5 zeigt eine isobare DSC-Messkurve für das Schmelzen von Wasser.
Als Schmelzpunkt wurde das Peakende definiert, da der Schmelzpunkt als der
Punkt definiert ist, an dem der unendlich ausgedehnte Kristall mit der Lösung im
Gleichgewicht steht. Der Schmelzpunkt ist von der Größe der Kristalle abhän-
gig, kleine Kristalle schmelzen bei tieferer Temperatur im Vergleich zu großen.
Gerade bei polymeren Proben liegt oft eine breite Kristallitgrößenverteilung vor,
so dass eine Kalibrierung auf das Ende der DSC-Signale sinnvoll ist. Ein weite-
rer Grund für die Kalibrierung nach dem Peakende ist die Konstruktion von
Phasendiagrammen aus DSC-Messkurven. In Kap.3.1.4 wird darauf hingewie-
sen, warum eine Kalibrierung auf den Peakanfang nicht möglich ist. Ein Nach-
teil der Kalibrierung auf das Peakende ist die Abhängigkeit des Peakendes von
250,0 260,0 270,0 280,0 290,0 300,0
273,15 KT/K
Wär
mef
luss

EXPERIMENTELLER TEIL 31
der Probenmasse. Aus diesem Grund wurden bei allen Messungen stets die
gleichen Massen eingewogen.
Der Schmelzpunkt des Wassers in Abbildung 5 wurde ermittelt, indem die
rechte Flanke des DSC-Peaks linear extrapoliert wurde und der Schnittpunkt
mit der linearen Verlängerung der Basislinie als Schmelzpunkt angegeben
wurde. Die Temperaturkalibrierung erfolgte mit Wasser (Tm,1 = 273,15 K) und
Benzoesäure (Tm,2 = 395,5 K). Prinzipiell ist es mit dem neuen Steuerprogramm
der DSC möglich bis zu 10 verschiedene Substanzen zur Temperatur und
Enthalpiekalibrierung heranzuziehen. Mit Hilfe der beiden ermittelten Umwand-
lungstemperaturen lässt sich die „wahre“ Temperatur der DSC ermitteln. Durch
die Kalibriertemperaturen wird bei den online-Messungen durch das Steuer-
programm direkt die „wahre“ Temperatur des Kalorimeters errechnet und ange-
zeigt.
Nach der Temperaturkalibrierung wird die Enthalpiekalibrierung durchgeführt.
Anders als bei der Temperaturkalibrierung, bei der ein linearer Zusammenhang
zwischen der „wahren“ und der von der DSC anzeigten Temperatur besteht,
muss lediglich ein Korrekturfaktor bestimmt werden. Dazu wird eine weitere
Messung mit reinem Wasser durchgeführt. Die Fläche unter dem Messsignal
wird bestimmt, indem die Basislinie vom Anfang des Signals bis zum Ende
linear extrapoliert wird. Durch den Quotienten aus der wahren Umwandlungs-
enthalpie und der gemessenen Umwandlungsenthalpie, expm
0m H~/H~ �� , wird der
Korrekturfaktor bestimmt. Nach Berechnung des Korrekturfaktors, wird in den
Messungen der differentielle Wärmefluss online durch den Kalibrierfaktor korri-
giert.
Die Temperatur- und Enthalpiekalibrierung ist abhängig von der Heizrate und
der verwendeten Pfännchensorte. Durch das Steuer- und Auswerteprogramm
kann die Kalibrierung separat für jede Heizrate durchgeführt und abgespeichert
werden, daneben kann auch die Pfännchensorte – Aluminium-Dampfdruck-
oder Stahlhochdruckpfännchen – gewählt werden. Für jede Heiz- bzw. Kühlrate

EXPERIMENTELLER TEIL 32
und Pfännchensorte wird eine Kalibrierdatei erstellt, auf die bei den jeweiligen
Messungen zurückgegriffen werden kann.
3.1.3 Probenvorbereitung
Es wurden Polyethylenglykole mit den molaren Massen 200, 300, 400, 600, 900
und 1500 g/mol verwendet. Die eingesetzten Polyethylenglykole wurden ein-
heitlich von der Firma Aldrich bezogen. Zur Herstellung der wässrigen Lösun-
gen wurde destilliertes Wasser verwendet.
Für die DSC-Messungen der PEG/Wasser-Systeme wurden Mischungen über
dem gesamten Konzentrationsbereich angesetzt. Hierzu wurde zunächst das
PEG und die entsprechende Menge Wasser in ein Hochdruck-Gläschen einge-
wogen und im Trockenschrank für ca. 3 h homogenisiert. Die Temperatur des
Trockenschrankes wurde so gewählt, dass man sich in jedem Fall ca. 15 K über
der entsprechenden Liquidustemperatur befand und so eine homogene Lösung
gewährleistet wurde.
Die bei Raumtemperatur flüssigen Mischungen wurden nach dem Abkühlen mit
Hilfe einer Spritze in Aluminium Dampfdruckpfännchen an einer Mikrowaage
eingewogen. Die verwendete Waage, PERKIN ELMER Autobalance AD-2Z, hat
eine Genauigkeit von � 0,01 mg. Es wurde darauf geachtet, dass die Proben-
masse in den Pfännchen konstant im Bereich von 6 � 0,2 mg lag. Nach dem
Einwiegen der Proben wurden direkt die entsprechenden DSC-Messungen
durchgeführt. Die Proben wurden – wenn nicht anders erwähnt – mit einer Kühl-
rate von 10 K/min auf eine Temperatur von 170 K abgekühlt und aufheizend mit
einer Heizrate von 5 K/min im Temperaturbereich von 180 K bis 300 K gemes-
sen.

EXPERIMENTELLER TEIL 33
3.1.4 Konstruktion von Phasendiagrammen aus DSC-Messkurven
Aus den experimentell ermittelten )T(c~P -Kurven der verschiedenen Lösungen
kann ein binäres Phasendiagramm in Abhängigkeit von den beiden Variablen,
Temperatur (T) und Zusammensetzung (w2), konstruiert werden. Dieses Ver-
fahren soll schematisch an einem binären Phasendiagramm erläutert werden.
Exemplarisch wird wieder ein eutektisches Schmelzdiagramm mit einseitiger
partieller Mischkristallbildung auf der Seite der Komponente 2 (Index 2) darge-
stellt. Bei dem schematischen Diagramm in Abbildung 6 handelt es sich um den
gleichen wie in Kap.1 beschriebenen Typ. Eine Mischkristallbildung auf der
Seite des Lösemittels (Index 1) wird ausgeschlossen. Auf der Abszisse ist der
Massenbruch der Komponente 2, auf der Ordinate ist die Temperatur aufgetra-
gen. LQ1 und LQ2 sind die Liquiduskurven, SD1 und SD2 die Soliduskurven und
SV1 und SV2 die Solvuskurven des binären Phasendiagramms.
Abbildung 6 Schematische Darstellung eines aus experimentell bestimmtenDSC-Kurven ermittelten binären Phasendiagramms. Die DSC-Kurven sind in das Phasendiagramm eingezeichnet, wobei dieTemperaturachse des Phasendiagramms mit der der DSC-Kur-ven identisch ist.
w2
0 1w2,m w2,E
T
E
LQ2LQ1Tm,1
TE
Tm,2
SD2
SD1
SV 2SV1

EXPERIMENTELLER TEIL 34
TE ist die invariante eutektische Umwandlungstemperatur, Tm,1 und Tm,2 sind die
Schmelzpunkte der reinen Komponenten 1 und 2. Die Liquiduskurven schnei-
den sich am eutektischen Punkt, E, bei der eutektischen Temperatur TE und der
eutektischen Zusammensetzung w2,E. Der eutektische Punkt beschreibt das
invariante Dreiphasen-Gleichgewicht, wobei sich bei der Temperatur TE die
Lösung mit der Zusammensetzung w2,E im Gleichgewicht mit dem reinen Kristall
der Komponente 1 und einem an Komponente 2 angereichertem Mischkristall
der Zusammensetzung w2,m befindet.
An den experimentellen Aufheizkurven erkennt man bei den Massenbrüchen
w2 = 0, w2 = 1 und w2 = w2,E lediglich ein scharfes endothermes Messsignal.
Aus dem Ende der Signale können die Schmelzpunkte der beiden reinen Kom-
ponenten und die eutektische Temperatur bestimmt werden. In den Bereichen
0 < w2 < w2,E und w2,E < w2 < w2,m sind zwei endotherme Schmelzsignale zu
erkennen.
Das erste scharfe Signal liegt unabhängig von der Zusammensetzung immer
bei der gleichen Temperatur und wird daher dem invarianten, eutektischen
Schmelzen zugeordnet. Die Fläche unter dem Signal wird in Richtung des eu-
tektischen Punktes im Konzentrationsbereich 0 < w2 < w2,E größer und im Kon-
zentrationsbereich w2,E < w2 < w2,m wieder kleiner. Bei der eutektischen Zu-
sammensetzung w2,E ist die Fläche unter dem Signal am größten. Wie in Kap.1
gezeigt hat man laut Gibbs’scher Phasenregel im isobaren Fall beim eutekti-
schen Schmelzen keinen Freiheitsgrad. Daher wird auch vom invarianten, eu-
tektischen Schmelzen gesprochen.
Das zweite Signal erstreckt sich über einen wesentlich größeren Temperatur-
bereich; in dem Bereich 0 < w2 < w2,E schmilzt der verbleibende Teil an reiner
Komponente 1 auf, dabei ändert sich ständig die Bruttozusammensetzung der
Lösung, was auch eine Schmelzpunktsänderung mit sich bringt. In dem Bereich
w2,E < w2 < w2,m schmilzt nach der eutektischen Temperatur ein an Komponente
2 reicher Mischkristall auf, wodurch sich auch hier ständig die Zusammen-

EXPERIMENTELLER TEIL 35
setzung der Lösung ändert. Bei den Signalen erkennt man, das sie stetig an-
steigen und erst beim Erreichen der Liquidustemperatur stark abfallen. Ober-
halb der Liquidustemperatur sind alle Kristalle aufgeschmolzen und es liegt eine
homogene Lösung vor. Im Bereich w2,m < w2 < 1 erkennt man nur noch ein
Liquidussignal, das sich von der Solidus- bis zur Liquidustemperatur erstreckt.
Entlang der Liquiduskurve liegt im isobaren Fall laut Gibbs’scher Phasenregel
ein Freiheitsgrad von 1 vor, was bedeutet, dass entweder die Temperatur oder
die Zusammensetzung im isobaren Fall frei wählbar ist. Dieser Freiheitsgrad
führt im Gegensatz zum eutektischen Schmelzen zu einem Schmelzbereich und
damit zu wesentlich breiteren Signalen.
Da die Signale im Anfangsbereich für w2,m < w2 < 1 eine sehr geringe Steigung
aufweisen, ist der Punkt, an dem die Basislinie zuerst verlassen wird, nur mit
einer großen Ungenauigkeit zu bestimmen. Aus diesem Grund wurde auf eine
experimentelle Bestimmung der Soliduskurve in dieser Arbeit verzichtet.
Die Form der Signale spricht auch hier dafür, bei der Bestimmung von binären
Phasendiagrammen auf das Peakende zu kalibrieren. Der Peakanfang des
breiten Liquidussignals liegt bei der eutektischen Temperatur, was eine Aus-
wertung nach dem Peakanfang in diesem Fall unsinnig erscheinen lässt, da in
diesem Fall die eutektische Temperatur, TE, gleich der Liquidustemperatur
wäre. Es kann alternativ auch auf das Peakmaximum kalibriert werden. Ein
Überblick ist in der Arbeit von Bogdanov et al. gegeben. Die binären Phasen-
diagramme wurden sowohl aus dem Peakanfang, dem Peakmaximum und dem
Peakende bestimmt50.
Solvuskurven lassen sich mit Hilfe von DSC-Messungen nicht bestimmen. Im
Fall der Solvuskurve handelt sich um ein Fest-Fest-Gleichgewicht. Da die Diffu-
sion in Festkörpern nur langsam ablaufen und sich innerhalb der experimen-
tellen Messzeit dadurch neue Gleichgewichte nicht schnell genug einstellen
können, lassen sich Solvuskurven mit DSC-Messungen nicht bestimmen.
50 Bogdanov B., Mihailov M.; J. Pol. Sci., 23, 1985, 2149

EXPERIMENTELLER TEIL 36
Da es zum einen häufig zu Überlagerungen der Signale kommt, und zum ande-
ren die Liquidussignale sehr breit und dementsprechend sehr gering ausge-
prägt sein können, ist die Bestimmung der Größen w2,m und w2,E in einigen Fäl-
len sehr schwierig oder nicht eindeutig aus den experimentellen Messkurven
abzuleiten. Zur besseren Bestimmung der beiden wichtigen Punkte im binären
Phasendiagramm, trägt man die spezifische, eutektische Umwandlungsenthal-
pie gegen die Zusammensetzung auf. Diese Darstellung wurde 1903 erstmals
von Tammann eingeführt und wird daher auch als Tammannsches Dreieck
bezeichnet51. Abbildung 7 zeigt das Tammannsche Dreieck für das oben ange-
führte Beispiel eines isobaren, binären Phasendiagramms mit einseitiger
Mischkristallbildung. Danach ergeben sich zwei Geraden mit unterschiedlichem
Vorzeichen, die sich bei der Konzentration w2,E schneiden.
Abbildung 7 Schematische Auftragung der spezifischen, eutektischenUmwandlungsenthalpie gegen die Zusammensetzung.
Die Schnittpunkte der Geraden mit der Abszisse ergeben die koexistierenden,
festen Phasen bei der invarianten eutektischen Temperatur TE. Das Beispiel in
51 Tammann G.; Z. Anorg. Chem., 37, 1903; 303
w2,e0 1w2,m
w2
eH�
T=TE

EXPERIMENTELLER TEIL 37
Abbildung 7 zeigt, dass bei der Temperatur TE ein reiner Kristall der Kompo-
nente 1, w2 = 0, mit einem Mischkristall der Zusammensetzung w2,m und einer
eutektischen Schmelze der Zusammensetzung w2,E im Gleichgewicht steht.
Eine quantitative Beschreibung des Tammannschen Dreiecks wurde von Müller
und Borchard gegeben52. Hierzu werden für beide Geraden Formeln aufgestellt,
die für Systeme mit partieller Mischbarkeit im festen Zustand gelten.
� �mix02e,201e,2S,2e,2
S,22e H~H~wH~)w1(
wwww
H~
1
1��������
�
��� (38)
� �mix02e,201e,2e,2S,2
2S,2e H~H~wH~)w1(
wwww
H~
2
2��������
�
��� (39)
In den Gln.(38) und (39) ist w2 der Massenbruch der Komponente 2; 1S,2w ,
2S,2w und w2,e sind die Massenbrüche der Mischkristalle und der Schmelze die
bei der invarianten eutektischen Temperatur im Gleichgewicht stehen. eH~� ist
die spezifische eutektische Umwandlungsenthalpie, 01H~� und 02H~� sind die
spezifischen Umwandlungsenthalpien der beiden reinen Komponenten 1 und 2.
mixH~�� beschreibt einen Term von Mischungsenthalpien, der für das jeweilige
binäre System charakteristisch ist.
Gl.(38) beschreibt die linke Gerade mit positiver Steigung, während Gl.(39) für
die rechte Gerade mit negativer Steigung gilt. Es ist zu erkennen, dass bei die-
ser quantitativen Auswertung der eutektische Punkt nicht nur von den spezi-
fischen Umwandlungsenthalpien der beiden reinen Komponenten abhängt,
sondern auch von der Mischungsenthalpie des Systems.
52 Müller, A., Borchard, W.; J. Phys. Chem., 101, 1997, 4283

EXPERIMENTELLER TEIL 38
3.2 Mischungskalorimetrie
3.2.1 Isotherme Messungen mit Hilfe der DSC
Durch Umbauten der DSC-2C von Perkin Elmer wurde ständig das Spektrum
der thermodynamisch bestimmbaren Daten erweitert. Hierzu zählt der Einbau
der Tieftemperatureinrichtung, wodurch es möglich wurde, DSC-Messungen ab
einer Temperatur von –150°C durchzuführen. Später wurde die DSC mit Hilfe
einer Analog-Wandler Karte an einen PC angeschlossen53. Mit Hilfe des PCs ist
es heute möglich alle Daten, wie Probenmasse, Heizrate und Pfännchensorte
vor der Messung zu berücksichtigen und das Messsignal direkt umzurechnen.
Die DSC-Messung kann zusätzlich online am Bildschirm verfolgt werden. Die
herausragenste Neuerung ist aber die Möglichkeit, direkt online den differen-
tiellen Wärmestrom im isothermen Modus zu messen und in Echtzeit am Bild-
schirm zu verfolgen. Mit Hilfe dieser Neuerung ist es möglich geworden, thermi-
sche Ereignisse in der DSC im isothermen Modus auszuwerten. Diese thermi-
schen Ereignisse können langsam ablaufende chemische Reaktionen sein, wie
das Aushärten von Harzen, oder das isotherme Verdampfen einer Flüssigkeit in
der DSC sein.
Eine andere Möglichkeit bietet sich, falls von außen Substanzen in das DSC-
Näpfchen eingebracht werden. Hierzu ist notwendig, mit einem offenen DSC-
Messkopf zu arbeiten. Durch die Glovebox ist der Messkopf von äußeren Stö-
rungen ausreichend abgeschirmt, so dass Messungen im offenen DSC-Mess-
kopf möglich und reproduzierbar sind. Messungen von Verdampfungs-
enthalpien von reinem Wasser und von Wasser aus Lösungen aus dem offe-
nem Messkopf zeigten eine sehr gute Reproduzierbarkeit54.
53 Herrn Dipl. Chem. Michael Kischel und Herrn Dr. Andreas Müller-Blecking danke ich für
die Arbeiten, die beim Anschluss der DSC mit dem PC auftraten und vor allem für diedamit verbundenen, aufwendigen Programmierarbeiten.
54 Borchard W., Jablonski P.; Dechema Forschungsbericht, 2000, 157

EXPERIMENTELLER TEIL 39
Es kann der differentielle Wärmestrom im isothermen Modus registriert werden,
der zum Temperaturausgleich der eingebrachten Probe nötig ist, so können mit
Hilfe der DSC im Sinne eines von Oelsen vorgestellten Drop-Kalorimeters55
Wärmekapazitäten von Flüssigkeiten bestimmt werden. Es hat sich gezeigt,
dass das Einbringen von Flüssigkeiten in das DSC-Mess-Näpfchen sehr vor-
sichtig geschehen muss. Ein herabfallender Tropfen in das DSC-Näpfchen oder
ein Anstoßen des Näpfchen dissipiert Energie, so dass eine Auswertung der
DSC-Messung unmöglich wird. Für das Einbringen von Flüssigkeiten in das
DSC-Näpfchen wurde ein spezieller Spritzenaufsatz für die DSC konstruiert.
3.2.2 Aufbau des Mischungsaufsatzes
Für den Spritzenaufsatz wurde zunächst die alte, undichte Glovebox gegen
eine neue, größere ausgetauscht. Innerhalb der Glovebox über dem offenen
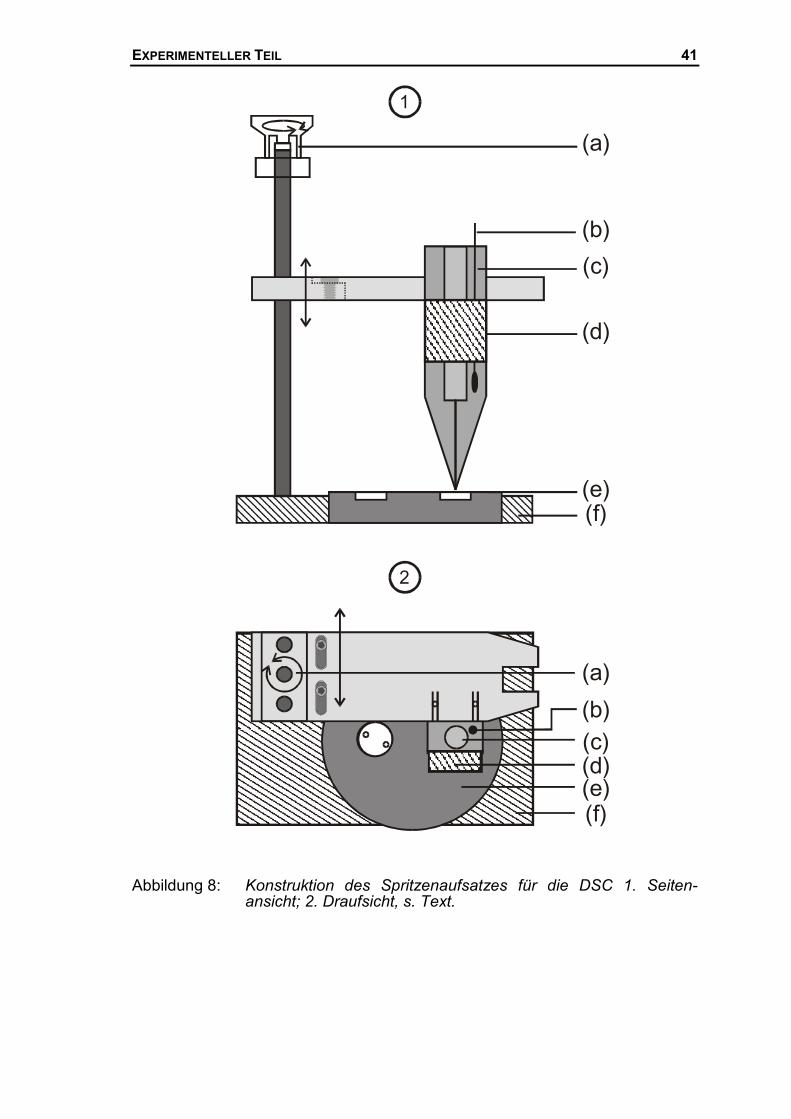
Messkopf wurde ein temperierbarer Spritzenaufsatz installiert. Abbildung 8 zeigt
eine schematische Zeichnung des Mischungsaufsatzes. Dieser Aufsatz besteht
aus einer Grundplatte aus Aluminium (f), die den DSC-Messkopf (e) genau
einfasst, und mit Hilfe von zwei Schrauben an diesem fixiert ist. Auf dieser
Grundplatte sind auf der rechten Seite zwei vertikale Führungsachsen ange-
bracht, in deren Mitte eine Gewindestange positioniert ist. Mit Hilfe eines Elek-
tromotors (a) lässt sich die zweite Aluminiumplatte in vertikaler Richtung bewe-
gen. Die Geschwindigkeit des Motors lässt sich beliebig variieren, wodurch ein
sehr genaues Manövrieren in vertikaler Richtung gewährleistet ist. Die zweite
Aluminiumplatte besteht aus zwei Teilen. Der obere ist über zwei Schrauben
und zwei Langlöcher mit der unteren verbunden und kann so auch in horizon-
taler Richtung hinter den Messkopf bewegt werden. An dieser in zwei Richtun-
gen beweglichen Aluminiumplatte wird der eigentliche Spritzenhalter (c) über
zwei Bohrungen befestigt. Der Spritzenhalter besteht ebenfalls aus einem Alu-
miniumblock, in den eine Bohrung gefräst ist, deren Abmessung genau den
Abmessungen einer �L-Spritze aus der HPLC Technik entspricht. Dieser Alu- 55 Oelsen W., Rieskamp K. H., Oelsen O.; Archiv für das Hüttenwesen, 26, 1955, 253

EXPERIMENTELLER TEIL 40
minium-Spritzenhalter ist thermisch isoliert und verfügt an der Vorder- und
Rückseite über je ein Peltierelement (d). Diese Peltierelemente sind an einen
Regler angeschlossen, der mit Hilfe eines Pt-100 Widerstandsthermometers (b)
die Temperatur des Aluminium-Spritzenhalters regelt. Temperaturen von ca.
-10°C bis 40°C können so eingestellt werden. Prinzipiell kann der Regler direkt
durch Umpolung der Peltierelemente von Heizen auf Kühlen umschalten. Da
bei zu großen Temperatursprüngen aber die Keramikoberflächen der Pel-
tierelemente brechen können, wurden die Peltier-Elemente stets entweder als
Heiz- oder Kühlelemente benutzt.
Mit Hilfe des temperierten Spritzenaufsatzes kann eine Flüssigkeit mit Hilfe ei-
ner HPLC-Spritze in das Näpfchen im DSC-Messkopf eingespritzt werden.
Durch den Elektromotor kann die Spitze sehr genau an die Oberfläche des
Mess-Näpfchens herangefahren werden. Dadurch ist es möglich Tropfen ins
Näpfchen „gleiten“ zu lassen und ein Fallen der Tropfen aus großer Höhe zu
verhindern. Die zweite horizontale Achse dient dazu nach der Injektion und dem
Herauffahren des Spritzenaufsatzes, diesen hinter den Messkopf zu schieben
und freien Zugang zum Messsystem zu haben.
EXPERIMENTELLER TEIL 41
Abbildung 8: Konstruktion des Spritzenaufsatzes für die DSC 1. Seiten-ansicht; 2. Draufsicht, s. Text.
(a)
(c)(b)
(d)
(e)(f)
1
2
(a)
(f)
(b)(c)
(e)(d)

EXPERIMENTELLER TEIL 42
3.2.3 Bestimmung von Mischungsenthalpien mit Hilfe der DSC
Die Durchführung und Kalibrierung des Mischungskalorimeters bei der Bestim-
mung von Mischungsenthalpien soll hier am Beispiel des Systems Ethylen-
glykol/Wasser beschrieben werden.
Zunächst muss gewährleistet werden, dass die Temperatur im Spritzenaufsatz,
der des DSC-Mess-Pfännchens entspricht. Hierzu wird Ethylenglykol bei einer
Temperatur von 25°C für 15 Min. im Spritzenaufsatz temperiert. Die Temperatur
des DSC-Mess-Näpfchens wird ebenfalls auf ca. 25°C eingestellt. Ein genauer
Temperaturabgleich zwischen Spritzenaufsatz und DSC-Mess-Näpfchen wird
mit Hilfe von Einspritzmessungen durchgeführt, wobei die Temperatur des
DSC-Mess-Näpfchens variiert wird. Die Temperatur des DSC-Mess-Näpfchens
ist wesentlich leichter zu ändern als die des Spritzenaufsatzes, welcher wieder
für 15 Min. temperiert werden müsste, die neue Temperatur des Mess-
Näpfchens stellt sich hingegen in wenigen Sekunden ein. Nach dem Start des
isothermen Messprogramms wird der Spritzenaufsatz langsam an die Ober-
fläche des Mess-Näpfchens heruntergefahren und vorsichtig ein Tropfen des
Ethylenglykols eingespritzt. Abbildung 9 zeigt ein idealisiertes, isothermes DSC-
Diagramm für das Einspritzen von Ethylenglykol in das leere Mess-Näpfchen.
Aufgetragen ist der differentielle Wärmestrom, �
Q , gegen die Zeit, t. t0signalisiert den Zeitpunkt des Einspritzens. Im Fall der gepunkteten Kurve 1 ist
die Temperatur des DSC-Näpfchens höher als die des Spritzenaufsatzes. Es
resultiert ein endothermer Peak für das Erwärmen des Ethylenglykols.
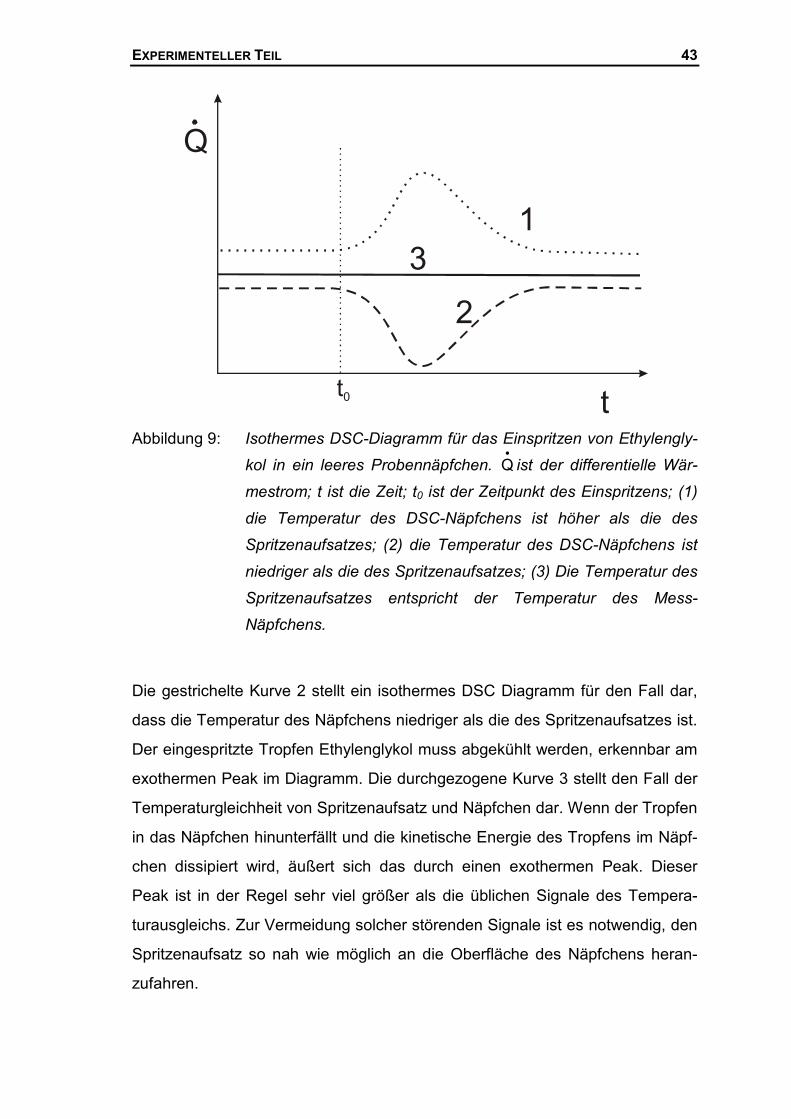
EXPERIMENTELLER TEIL 43
Abbildung 9: Isothermes DSC-Diagramm für das Einspritzen von Ethylengly-kol in ein leeres Probennäpfchen.
�
Q ist der differentielle Wär-mestrom; t ist die Zeit; t0 ist der Zeitpunkt des Einspritzens; (1)die Temperatur des DSC-Näpfchens ist höher als die desSpritzenaufsatzes; (2) die Temperatur des DSC-Näpfchens istniedriger als die des Spritzenaufsatzes; (3) Die Temperatur desSpritzenaufsatzes entspricht der Temperatur des Mess-Näpfchens.
Die gestrichelte Kurve 2 stellt ein isothermes DSC Diagramm für den Fall dar,
dass die Temperatur des Näpfchens niedriger als die des Spritzenaufsatzes ist.
Der eingespritzte Tropfen Ethylenglykol muss abgekühlt werden, erkennbar am
exothermen Peak im Diagramm. Die durchgezogene Kurve 3 stellt den Fall der
Temperaturgleichheit von Spritzenaufsatz und Näpfchen dar. Wenn der Tropfen
in das Näpfchen hinunterfällt und die kinetische Energie des Tropfens im Näpf-
chen dissipiert wird, äußert sich das durch einen exothermen Peak. Dieser
Peak ist in der Regel sehr viel größer als die üblichen Signale des Tempera-
turausgleichs. Zur Vermeidung solcher störenden Signale ist es notwendig, den
Spritzenaufsatz so nah wie möglich an die Oberfläche des Näpfchens heran-
zufahren.
tt0
Q
1
23
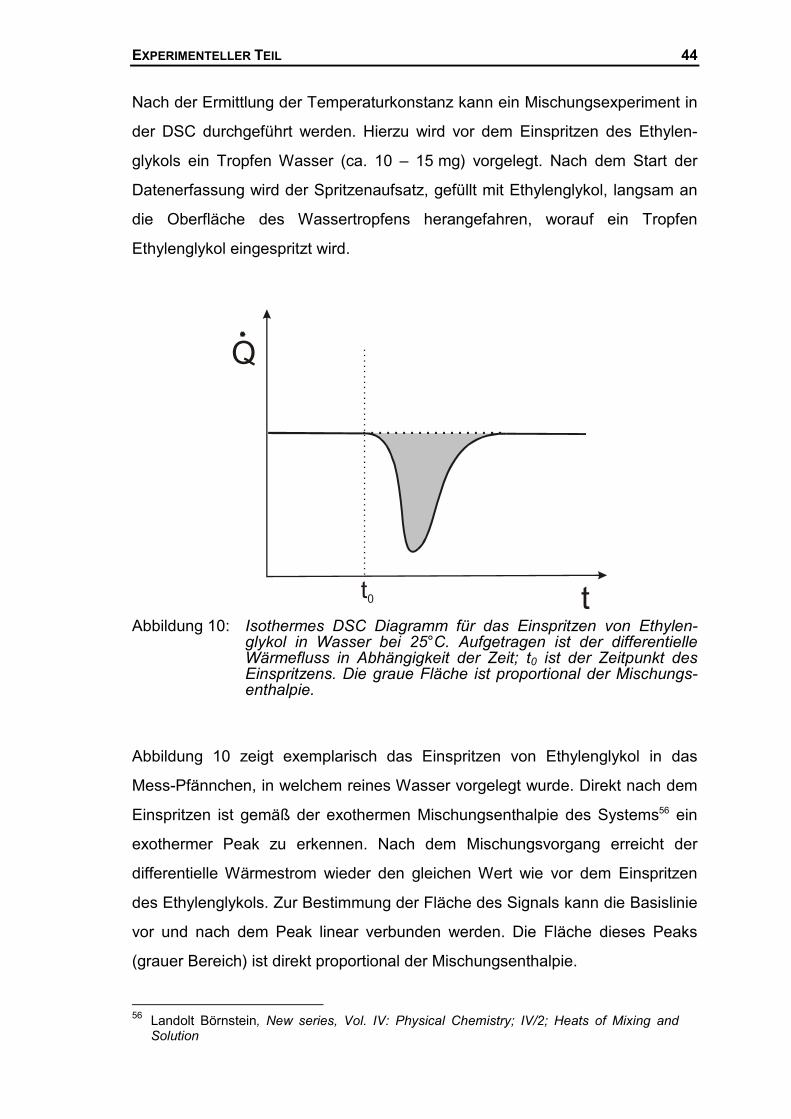
EXPERIMENTELLER TEIL 44
Nach der Ermittlung der Temperaturkonstanz kann ein Mischungsexperiment in
der DSC durchgeführt werden. Hierzu wird vor dem Einspritzen des Ethylen-
glykols ein Tropfen Wasser (ca. 10 – 15 mg) vorgelegt. Nach dem Start der
Datenerfassung wird der Spritzenaufsatz, gefüllt mit Ethylenglykol, langsam an
die Oberfläche des Wassertropfens herangefahren, worauf ein Tropfen
Ethylenglykol eingespritzt wird.
Abbildung 10: Isothermes DSC Diagramm für das Einspritzen von Ethylen-glykol in Wasser bei 25°C. Aufgetragen ist der differentielleWärmefluss in Abhängigkeit der Zeit; t0 ist der Zeitpunkt desEinspritzens. Die graue Fläche ist proportional der Mischungs-enthalpie.
Abbildung 10 zeigt exemplarisch das Einspritzen von Ethylenglykol in das
Mess-Pfännchen, in welchem reines Wasser vorgelegt wurde. Direkt nach dem
Einspritzen ist gemäß der exothermen Mischungsenthalpie des Systems56 ein
exothermer Peak zu erkennen. Nach dem Mischungsvorgang erreicht der
differentielle Wärmestrom wieder den gleichen Wert wie vor dem Einspritzen
des Ethylenglykols. Zur Bestimmung der Fläche des Signals kann die Basislinie
vor und nach dem Peak linear verbunden werden. Die Fläche dieses Peaks
(grauer Bereich) ist direkt proportional der Mischungsenthalpie.
56 Landolt Börnstein, New series, Vol. IV: Physical Chemistry; IV/2; Heats of Mixing and
Solution
tt0
Q

EXPERIMENTELLER TEIL 45
Für die Bestimmung der spezifischen Mischungsenthalpie sind nach der Mes-
sung noch zwei andere Größen zu bestimmen. Das gemessene Signal ist pro-
portional der Masse und abhängig von der Konzentration der Mischung. Nach
dem Mischungsvorgang muss die genaue Masse und die Konzentration der
Mischung bestimmt werden. Durch die online-Messung lässt sich das Ende
eines Mischungsvorganges ermitteln. Nach dem Ende des Mischungsvorgan-
ges wird der Spritzenaufsatz hochgefahren und hinter den Messkopf gescho-
ben. Das Mess-Pfännchen wird von der DSC in ein Schnappdeckelglas gege-
ben und ausserhalb der DSC gewogen.
Bei den in dieser Arbeit verwendeten Systeme bot sich an, die Konzentration
mit Hilfe eines Abbé-Refraktometers zu bestimmen, mit dem es möglich ist,
sehr schnell den Brechungsindex des gemischten Tropfens (ca. 15 mg) zu
bestimmen. Vor den Messungen wurde der Brechungsindex der gemessenen
Systeme in Abhängigkeit von der Konzentration bestimmt (s. Anhang 1). Es ist
zu erkennen, das der Brechungsindex in Abhängigkeit des Massenbruchs sehr
gut durch eine Gerade angenähert werden kann. Die Bestimmung der Masse
und der Konzentration sollte relativ rasch nach dem Mischungsexperiment
erfolgen, da bei längerer Lagerung der Systeme die Konzentration und die
Masse durch das Verdampfen des Wassers aus der Mischung verändert wer-
den. Der Zeitraum zwischen dem Ende des Mischens und der Konzentrations-
bestimmung wurde konstant gehalten und nahm ca. 90 s in Anspruch. Durch
Verdampfungsmessungen auf der Mikrogramm Waage wurde die Verdamp-
fungsgeschwindigkeit des Wassers aus den Mischungen ermittelt. Während der
90 s ergibt sich ein Fehler von max. 2 % in der Konzentration der Mischung.
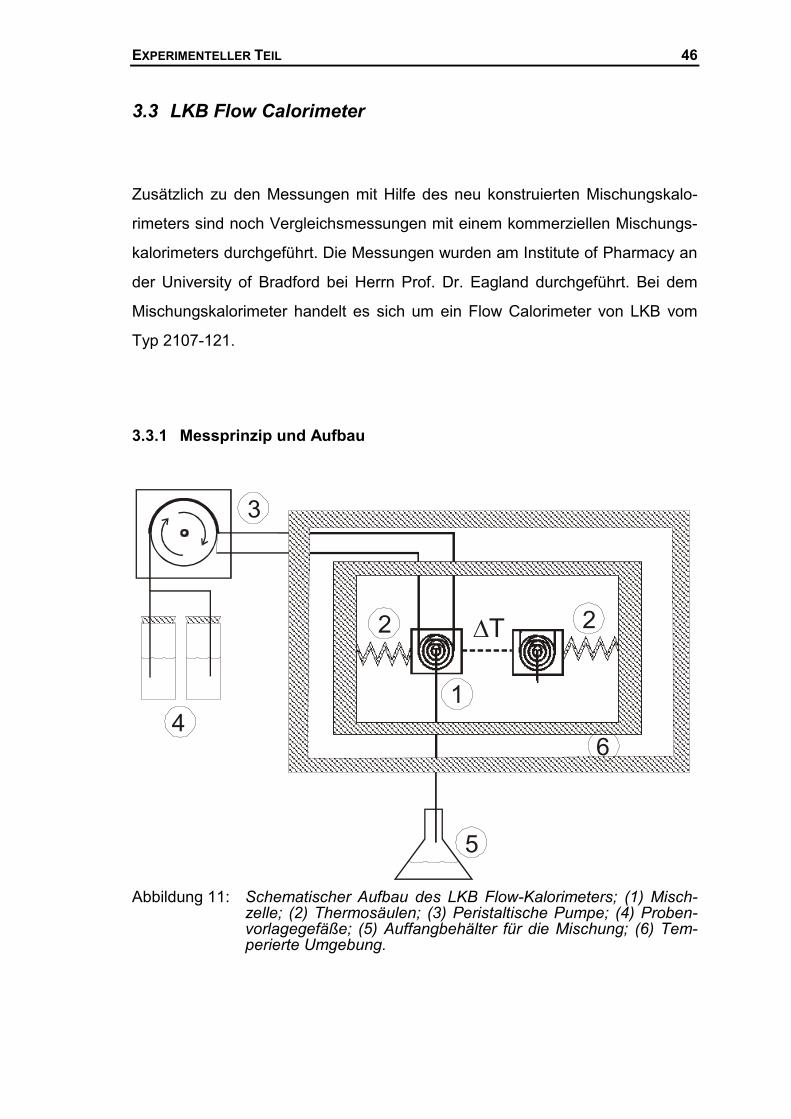
EXPERIMENTELLER TEIL 46
3.3 LKB Flow Calorimeter
Zusätzlich zu den Messungen mit Hilfe des neu konstruierten Mischungskalo-
rimeters sind noch Vergleichsmessungen mit einem kommerziellen Mischungs-
kalorimeters durchgeführt. Die Messungen wurden am Institute of Pharmacy an
der University of Bradford bei Herrn Prof. Dr. Eagland durchgeführt. Bei dem
Mischungskalorimeter handelt es sich um ein Flow Calorimeter von LKB vom
Typ 2107-121.
3.3.1 Messprinzip und Aufbau
Abbildung 11: Schematischer Aufbau des LKB Flow-Kalorimeters; (1) Misch-zelle; (2) Thermosäulen; (3) Peristaltische Pumpe; (4) Proben-vorlagegefäße; (5) Auffangbehälter für die Mischung; (6) Tem-perierte Umgebung.
�T
5
64
3
1
22

EXPERIMENTELLER TEIL 47
Das LKB 2107 – 121 Flow Calorimeter ist mit zwei Zellen ausgestattet, in denen
thermische Reaktionen ablaufen können. Wie bei der DSC handelt es sich auch
hier um ein Zwillingskalorimeter. Die Zelle in der zwei Flüssigkeiten oder Sus-
pensionen gemischt werden, wird Mischzelle genannt (1), während die andere
als Referenzzelle dient. Eine Vielzahl von Temperatur-Kontroll- Systemen in-
nerhalb des Kalorimeters gewährleisten eine Temperaturstabilität von min-
destens � 0,0001°C. Zusätzlich ist das Kalorimeter in einem temperierten Raum
bei 24,7�0,2°C untergebracht. Beide Zellen sind mit Thermoelementen ausge-
stattet und jeweils über eine Thermosäule (2) mit der Umgebung thermisch ver-
bunden. Mit Hilfe einer peristaltischen Pumpe (3) können zwei Flüssigkeiten
aus Probengefäßen (4) durch das temperierte Kalorimeter zur Mischzelle
gepumpt werden Auf dem Weg von der Pumpe zur Mischzelle werden die Flüs-
sigkeiten im Kalorimeter temperiert. Nach dem Mischen in der Mischzelle wird
die Mischung in einem Probengefäß aufgefangen (5).
Werden in der Mischzelle zwei Flüssigkeiten gemischt und es kommt zu einem
exo- oder endothermen Ereignis, resultiert ein Temperaturunterschied, �T, zwi-
schen der Referenz- und der Mischzelle, worauf es zu einem Wärmefluss über
die Thermosäulen von der Mischzelle zur Umgebung (exotherm), oder umge-
kehrt (endotherm) kommt. Dieser Temperaturunterschied impliziert eine elek-
tromotorische Kraft die proportional zur Temperaturdifferenz ist und von einem
Schreiber aufgezeichnet wird.
Das Kalorimeter wird mit Hilfe einer eingebauten regelbaren, elektrischen Hei-
zung und dem resultierenden Wärmeeffekt gemäß der Jouleschen Wärme kali-
briert. Die Heizleistung in J/s, Pkal, der eingebauten Heizung lässt mit Hilfe des
Widerstands der Heizung, R, und dem Heizstrom, I, ausrechnen.
RIP 2kal �� (40)
Aus dieser Heizleistung resultiert ein Ausschlag am Schreiber in mm, die direkt
einer definierten Wärmemenge zugeordnet werden kann.

EXPERIMENTELLER TEIL 48
3.3.2 Versuchsdurchführung
Es werden zwei Probengefäße vorbereitet, das eine wird mit reinem Wasser
das andere wird mit einer PEG-Lösung bzw. reinem PEG befüllt und gewogen.
Die PEG Lösungen wurden vorher in einem Hochdruckgläschen angesetzt und
zur Homogenisierung 1 h auf 60°C erwärmt. Die Zusammensetzung der PEG-
Lösungen mit einer mittleren molaren Masse des PEG von � 400 g/mol deckten
den gesamten Konzentrationsbereich ab. Bei höheren molaren Massen musste
darauf geachtet werden, dass die Lösungen bei Raumtemperatur noch flüssig
sind, daher sind bei den längerkettigen PEG-Wasser Systemen lediglich Ver-
dünnungsmessungen durchgeführt worden.
Durch das Kalorimeter wird permanent reines Wasser durch die beiden Zulei-
tungen gepumpt, bis eine konstante Basislinie erreicht wird. Zur Durchführung
eines Mischungs- oder Verdünnungsexperimentes werden die beiden Zuleitun-
gen jeweils in die Probengefäße gesteckt, worauf das reine Wasser bzw. die
PEG-Lösung durch das Kalorimeter gepumpt wird. Die Pumprate variierte von
ca. 15 bis ca. 0,375 g/h pro Zuleitung. Bei hoher Pumprate (ca. 15 g/h) dauert
es ca. 25 min. bis die Lösung und das reine Wasser die Mischzelle erreicht
haben. In dieser Zeit ist eine Temperierung der Flüssigkeiten im Kalorimeter
gewährleistet. Nachdem eine ausreichende Menge durch die Leitungen
gepumpt wurde, wurden beide Zuleitungen wieder mit reinem Wasser gespült.
Die Zeiten, in der die Proben gepumpt wurden, wurden gestoppt, so kann durch
Rückwägung der Probengefäße für jedes Probengefäß eine genaue Pumprate
in g/h angegeben werden. Mit Hilfe der beiden Pumpraten läßt sich die Kon-
zentration der resultierenden Mischung ermitteln.
Beim Mischen der Flüssigkeiten in der Mischzelle wird aufgrund der exother-
men Mischungsenthalpie Wärme frei. Nachdem das Kalorimeter kalibriert ist,
kann man einem bestimmten Messwert direkt die Wärmeleistung in J/s zuord-
nen. In Kombination mit der Pumprate in g/s kann dieser Wert in die Mischungs-
oder Verdünnungswärme in J/g umgerechnet werden.

EXPERIMENTELLER TEIL 49
Wurde die Konzentration der Lösung erhöht, und damit die Mischungsenthalpie
größer, musste der Empfindlichkeitsbereich des Kalorimeters herabgesetzt
werden. Bei geringster Empfindlichkeit kann zusätzlich die Pumprate herabge-
setzt werden, falls die gemessene Enthalpie zu groß ist.
3.3.3 Integrale Mischungsenthalpie aus Verdünnungsmessungen
Beide Zuleitungen des Kalorimeters liefen über eine peristaltische Pumpe,
dadurch war die Pumprate für beide Leitungen nahezu gleich. Die Konzentra-
tion der Mischung wurde in der Art variiert, dass in dem ersten Probengefäß
reines Wasser vorgelegt wurde und in dem anderen Probengefäß die Konzen-
tration der PEG-Lösung verändert wurde. Es zeigte sich, dass in guter Nähe-
rung von einer gleichen Pumprate in g/h für beide Leitungen ausgegangen wer-
den kann. Das bedeutet, dass der Massenbruch der resultierenden Mischung
genau die Hälfte der eingesetzten PEG-Lösung beträgt. Es wurde der Konzen-
trationsbereich 0 < w2 � 0,5 gemessen. Sobald im zweiten Probengefäß nicht
mehr reines PEG, sondern eine wässrige Lösung vorgelegt wird, kann lediglich
die Verdünnungsenthalpie gemessen werden. Es ist aber möglich, aus den
differentiellen Verdünnungswärmen die integrale Mischungsenthalpie zu
bestimmen. Die Enthalpie und auch die Mischungsenthalpie ist eine Zustands-
funktion. Daher ist sie vom Wege unabhängig. Entscheidend sind die Aus-
gangszustände und der, nur von der Konzentration abhängige, Endzustand.

EXPERIMENTELLER TEIL 50
Abbildung 12: Darstellung der Ermittlung der integralen Mischungsenthalpieaus den Messungen der Verdünnungsenthalpie.
Abbildung 12 zeigt schematisch den Verlauf der Mischungsenthalpie für den
exothermen Fall, wobei die Mischungsenthalpie eine zu w2 = 0,5 symmetrische
Parabel widerspiegelt. w2,0 ist die Ausgangskonzentration der PEG-Lösung, die
mit dem reinen Wasser gemischt wird. Bei gleicher Pumprate des Wassers und
der PEG-Lösung ist der Massenbruch der resultierenden verdünnten Mischung
nach der Messung genau die Hälfte des Massenbruchs der Ausgangskonzen-
tration ( 0,221
m,2 ww � ). Statt der gesamten integralen Mischungsenthalpie für
das System, � �m,2M wH~� , misst man lediglich die Verdünnungsenthalpie, M1H~
� .
Die Differenz zwischen der integralen Mischungsenthalpie und der gemessenen
Verdünnungswärme, M1H~
� , ist genau die Hälfte der integralen Mischungs-
enthalpie der Konzentration der Ausgangslösung, � �0,2M wH~� .
� � M2M1m,2M H~H~wH~ ����� (41)
w2
�1 MH
�H (w )M 2,m
�H (w )M 2,0
�2 MH
�HM
w2,0w2,m

EXPERIMENTELLER TEIL 51
Als empirischer Ansatz für die Mischungsenthalpie wird eine Reihenentwicklung
nach Scatchard verwendet57:
��
���
N
0k
k2k21M )w21(AwwH~ (42)
MH~� ist die integrale Mischungsenthalpie, w1 bzw. w2 sind die Massenbrüche
der resultierenden Mischung, wobei der Zusammenhang w2 = 1-w1 gilt. Mit dem
empirischen Ansatz können für das Beispiel in Abbildung 12 die integralen
Mischungsenthalpien für den Massenbruch der eingesetzten PEG-Lösung,
� �0,2M wH~� , und der resultierenden verdünnten Lösung, � � � �2w
Mm,2M0,2H~wH~ ��� ,
berechnet werden:
� � � � � ���
����
N
0k
k0,2k0,20,20,2M w21Aww1wH~ (43)
� � � � � ���
����
N
0k
k0,2k2
w2
w2
wM w1A1H~ 0,20,20,2 (44)
Die integrale Mischungsenthalpie der resultierenden Mischung setzt sich aus
der gemessenen Verdünnungswärme, M1H~
� , und der Hälfte der integralen
Mischungsenthalpie der eingesetzten Lösung, M2H~
� , zusammen. Die Reihen-
entwicklung für M2H~
� ist gegeben durch:
� � � � � ���
������
N
0k
k0,2k2
w0,20,2M2
1M2 w21Aw1wH~H~ 0,2 (45)
Danach lässt sich die differentielle Verdünnungswärme in Abhängigkeit von der
Ausgangskonzentration darstellen:
57 Scatchard G.; Chem. Rev., 44, 1949, 7

EXPERIMENTELLER TEIL 52
� � � � � � � � ��
���
������� ��
��
N
0k
k0,2k0,2
N
0k
k0,2k2
w2
wM1 w1Aw1w1A1H~ 0,20,2 (46)
Bricht man nach dem zweiten Glied ab, erhält man:
� � � �� � � � � �� �� �0,2100,20,2102w
2w
M1 w21AAw1w1AA1H~ 20,2��������� (47)
ersetzt man 0,221
m,2 ww � erhält man zusammengefasst:
� �� �� �m,2m,21m,20m,2M1 w3w21AwAwH~ ���� (48)
Mit Hilfe von Gl.(47) können aus den Messungen der Verdünnungsenthalpie mit
Hilfe des LKB Mischungskalorimeters die Werte für A0 und A1 bestimmt werden.
Mit diesen empirischen Werten kann gemäß Gl.(41) der komplette Verlauf der
integralen Mischungsenthalpie in Abhängigkeit des Massenbruchs beschrieben
werden. Für das Beispiel in Abbildung 12 wäre A1 = 0, was zu der zur Konzen-
tration w2 = 0,5 symmetrischen Parabel führt.
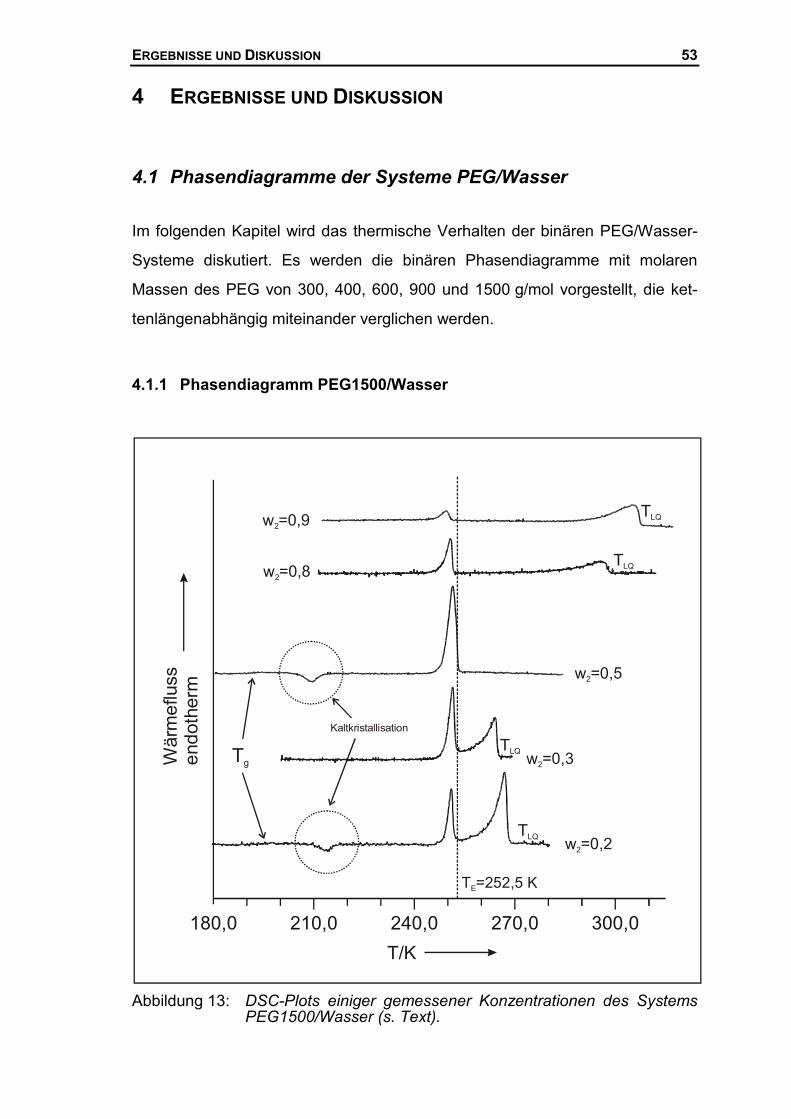
ERGEBNISSE UND DISKUSSION 53
4 ERGEBNISSE UND DISKUSSION
4.1 Phasendiagramme der Systeme PEG/Wasser
Im folgenden Kapitel wird das thermische Verhalten der binären PEG/Wasser-
Systeme diskutiert. Es werden die binären Phasendiagramme mit molaren
Massen des PEG von 300, 400, 600, 900 und 1500 g/mol vorgestellt, die ket-
tenlängenabhängig miteinander verglichen werden.
4.1.1 Phasendiagramm PEG1500/Wasser
Abbildung 13: DSC-Plots einiger gemessener Konzentrationen des SystemsPEG1500/Wasser (s. Text).
180,0 210,0 240,0 270,0 300,0
Wär
mef
luss
endo
ther
m
T/K
Tg
TLQ
TLQ
TLQ
TLQ
Kaltkristallisation
T =252,5 KE
w =0,22
w =0,32
w =0,52
w =0,82
w =0,92

ERGEBNISSE UND DISKUSSION 54
Abbildung 13 zeigt exemplarisch DSC-Messungen verschiedener Konzentratio-
nen des binären Systems PEG1500/Wasser. Alle Mischungen sind mit einer
Kühlrate von 10 K/min abgekühlt worden und danach mit einer Heizrate von
5 K/min gemessen worden. Man detektiert eine in dieser Vergrößerung
schwach ausgeprägte Glasstufe bei 193 K, die je nach System unterschiedlich
deutlich ist. Im Anschluss ist in einigen Fällen ein exothermes Signal zu erken-
nen, was auf eine unvollständige Kristallisation während des Abkühlens hin-
weist. Das exotherme Signal für die Kaltkristallisation liegt deutlich vor dem, für
die Ermittlung des Phasendiagramms relevanten, Temperaturbereich von 240 K
bis 320 K. Es kommt in diesem System zu keiner Überlagerung der Signale für
die Kaltkristallisation und der Schmelzsignale. Auf eine aufwendigere Tempe-
rung der Proben, wie sie bei der Bestimmung des Phasendiagramms
PEG1000/Wasser angewendet werden muss58, kann hier verzichtet werden.
Bei den Messungen im Konzentrationsbereich 0 < w2 < 0,93 sind zwei endo-
therme Signale zu erkennen. Das erste Signal ist sehr scharf und liegt immer
nahezu bei der gleichen Temperatur und wird dem invarianten eutektischen
Schmelzen zugeordnet. Die Temperatur des Peakendes des eutektischen
Signals erhöht sich leicht bis zum eutektischen Punkt und fällt danach wieder
leicht ab. Aus den Messungen lässt sich die eutektische Zusammensetzung
gemäß des Tammannschen Dreiecks mit w2,E = 0,53 bestimmen (Abbildung
14). An der Auswertung gemäß des Tammannschen Dreieck lässt sich auch
erkennen, dass auf der Seite des Wassers, w2 = 0, keine Mischkristallbildung
vorliegt, während auf der Seite des PEG die Bildung eines Mischkristalls beob-
achtet werden kann. Die Zusammensetzung des Mischkristalls ist w2,m = 0,93.
Dieser Mischkristall steht mit dem reinen Eiskristall und der Lösung mit der
Zusammensetzung w2,E = 0,58 bei der eutektischen Temperatur TE = 252,5 K
im Gleichgewicht.
58 Dobnik E.; Dissertation Duisburg, 1991
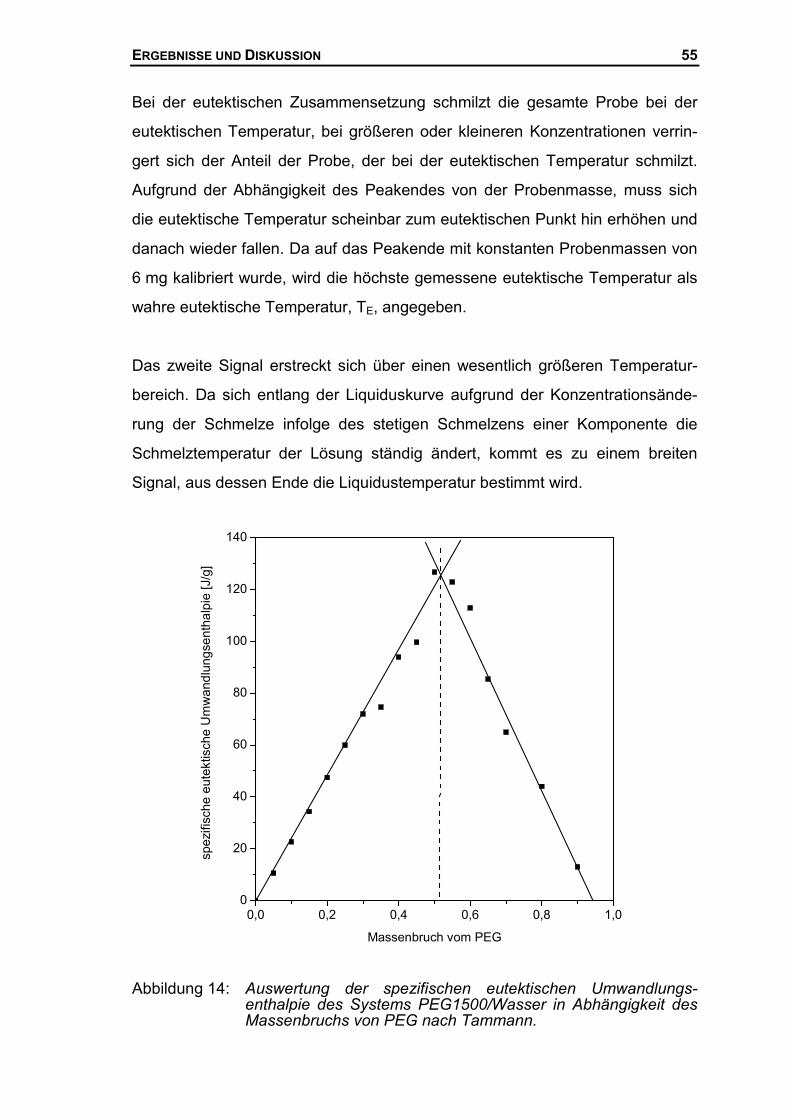
ERGEBNISSE UND DISKUSSION 55
Bei der eutektischen Zusammensetzung schmilzt die gesamte Probe bei der
eutektischen Temperatur, bei größeren oder kleineren Konzentrationen verrin-
gert sich der Anteil der Probe, der bei der eutektischen Temperatur schmilzt.
Aufgrund der Abhängigkeit des Peakendes von der Probenmasse, muss sich
die eutektische Temperatur scheinbar zum eutektischen Punkt hin erhöhen und
danach wieder fallen. Da auf das Peakende mit konstanten Probenmassen von
6 mg kalibriert wurde, wird die höchste gemessene eutektische Temperatur als
wahre eutektische Temperatur, TE, angegeben.
Das zweite Signal erstreckt sich über einen wesentlich größeren Temperatur-
bereich. Da sich entlang der Liquiduskurve aufgrund der Konzentrationsände-
rung der Schmelze infolge des stetigen Schmelzens einer Komponente die
Schmelztemperatur der Lösung ständig ändert, kommt es zu einem breiten
Signal, aus dessen Ende die Liquidustemperatur bestimmt wird.
Abbildung 14: Auswertung der spezifischen eutektischen Umwandlungs-enthalpie des Systems PEG1500/Wasser in Abhängigkeit desMassenbruchs von PEG nach Tammann.
0,0 0,2 0,4 0,6 0,8 1,00
20
40
60
80
100
120
140
spez
ifisc
he e
utek
tisch
e U
mw
andl
ungs
enth
alpi
e [J
/g]
Massenbruch vom PEG
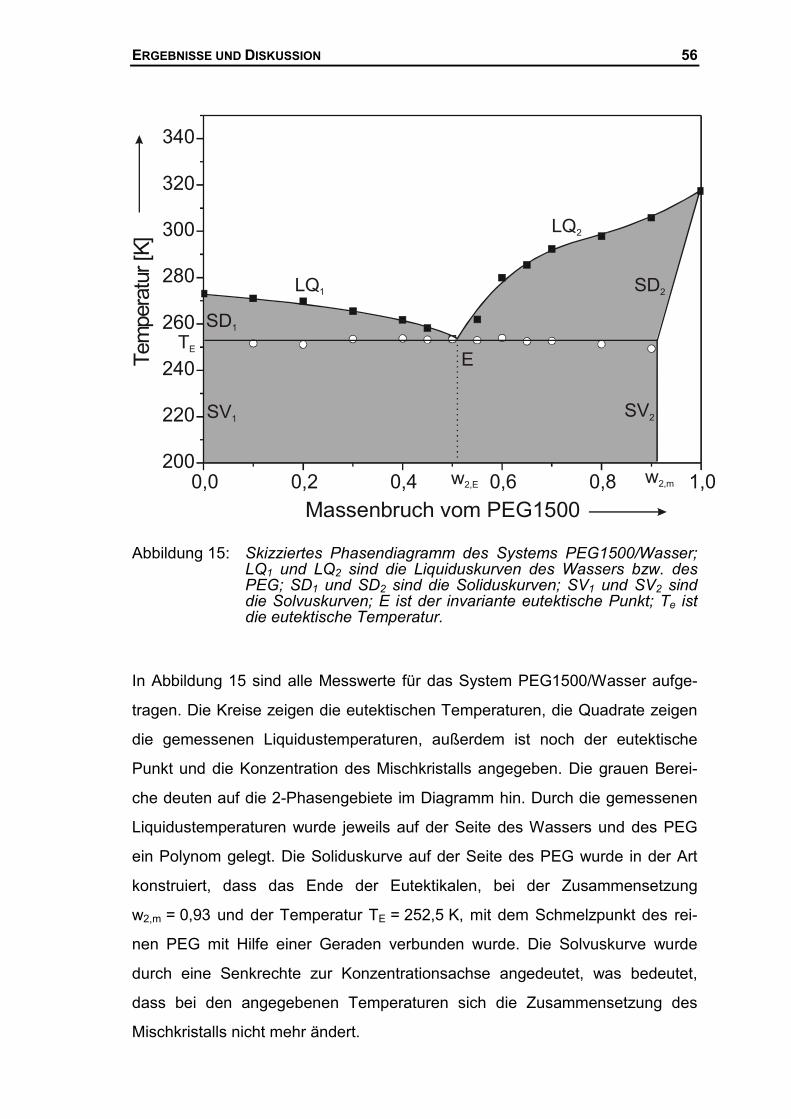
ERGEBNISSE UND DISKUSSION 56
Abbildung 15: Skizziertes Phasendiagramm des Systems PEG1500/Wasser;LQ1 und LQ2 sind die Liquiduskurven des Wassers bzw. desPEG; SD1 und SD2 sind die Soliduskurven; SV1 und SV2 sinddie Solvuskurven; E ist der invariante eutektische Punkt; Te istdie eutektische Temperatur.
In Abbildung 15 sind alle Messwerte für das System PEG1500/Wasser aufge-
tragen. Die Kreise zeigen die eutektischen Temperaturen, die Quadrate zeigen
die gemessenen Liquidustemperaturen, außerdem ist noch der eutektische
Punkt und die Konzentration des Mischkristalls angegeben. Die grauen Berei-
che deuten auf die 2-Phasengebiete im Diagramm hin. Durch die gemessenen
Liquidustemperaturen wurde jeweils auf der Seite des Wassers und des PEG
ein Polynom gelegt. Die Soliduskurve auf der Seite des PEG wurde in der Art
konstruiert, dass das Ende der Eutektikalen, bei der Zusammensetzung
w2,m = 0,93 und der Temperatur TE = 252,5 K, mit dem Schmelzpunkt des rei-
nen PEG mit Hilfe einer Geraden verbunden wurde. Die Solvuskurve wurde
durch eine Senkrechte zur Konzentrationsachse angedeutet, was bedeutet,
dass bei den angegebenen Temperaturen sich die Zusammensetzung des
Mischkristalls nicht mehr ändert.
W2,m1
0,0 0,2 0,4 0,6 0,8 1,0200
220
240
260
280
300
320
340Te
mpe
ratu
r[K]
Massenbruch vom PEG1500w2,mw2,E
E
SD1
SD2
SV2
LQ2
SV1
LQ1
TE

ERGEBNISSE UND DISKUSSION 57
Bei dem gemessenen System PEG1500/Wasser lässt sich bestätigen, dass es
sich um ein eutektisches Schmelzdiagramm mit partieller Mischbarkeit auf der
Seite des PEG handelt.
4.1.2 Phasendiagramm PEG900/Wasser
Anhang 6.2.1 zeigt einige DSC-Messkurven für das System PEG900/Wasser
bei verschiedenen Konzentrationen. Zunächst wurden auch diese Proben mit
einer Kühlrate von 10 K/min auf die Starttemperatur von 170 K abgekühlt und
danach mit einer Heizrate von 5 K/min gemessen. Wie auch bei dem zuvor be-
schriebenen System PEG1500/Wasser erkennt man eine Glasstufe bei 193 K.
Im Anschluss wird auch in diesem System eine Kaltkristallisation gemessen, die
in diesem System sehr nahe bei dem ersten endothermen Schmelzpeak liegt
(s. Abbildung 16). Der erste Schmelzpeak liegt immer bei der gleichen Tem-
peratur und wird dem invarianten eutektischen Schmelzen zugeordnet, aus
dessen Ende die eutektische Temperatur bestimmt wird. Die eutektische
Temperatur von 242,8 K in diesem System ist 9,7 K geringer als im System
PEG1500/Wasser. Im mittleren Konzentrationsbereich – in der Nähe des eutek-
tischen Punktes – kommt es zu einer Überlagerung der exothermen Signale der
Kaltkristallisation und der endothermen, eutektischen Schmelzsignale. Die
Flächenbestimmung in diesen Fällen gestaltet sich als sehr schwierig, da der
Verlauf der Basislinie vom Anfang bis zum Ende des eutektischen Signals nicht
mehr konstruiert werden kann. Eine Auswertung der spezifischen eutektischen
Umwandlungsenthalpie in Abhängigkeit von der Zusammensetzung zur
genauen Bestimmung des eutektischen Punktes kann unter diesen Umständen
nicht mehr durchgeführt werden. In einigen Arbeiten, die ungeachtet dieser
Probleme die Flächen unter diesen Signalen bestimmen und gegen die
Zusammensetzung auftragen, erkennt man, dass die eutektische Umwand-

ERGEBNISSE UND DISKUSSION 58
lungsenthalpie im mittleren Konzentrationsbereich abnimmt bzw. verschwin-
det59.
Beim System PEG1000/Wasser konnte Dobnik60 zeigen, dass durch ein relativ
langes Tempern (ca. 2h) unterhalb der eutektischen Temperatur eine Kalt-
kristallisation in den Aufheizkurven unterbunden werden kann. Ein ähnliches
Vorgehen beim System PEG900/Wasser konnte die Kaltkristallisation in den
Proben nicht verhindern. Es wurde versucht die Kaltkristallisation durch sehr
geringe Kühlraten (0,125 K/min) zu unterbinden, was aber auf die exothermen
Signale bei den Messungen keinen Einfluss hatte.
Die Proben wurden jeweils für 2,5 h bei verschiedenen Temperaturen unterhalb
der eutektischen Temperatur von 242,8 K und oberhalb der Glasstufe von
193 K getempert, wobei die jeweiligen Temper-Temperaturen im Abstand von
5 K lagen. Bei keiner Temperung konnte ein Einfluss auf das Kristallisations-
verhalten nachgewiesen werden. Bei allen Versuchen detektierte man im DSC-
Diagramm die Glasstufe gefolgt von einem exothermen Peak vor dem eutek-
tischen Peak.
Abbildung 16 zeigt das Vorgehen, wodurch die Kaltkristallisation vermieden
werden konnte. Zunächst wurden die Proben mit 10 K/min abgekühlt und ledig-
lich bis zum exothermen Peak (213 K) aufheizend mit einer Heizrate von
5 K/min gemessen. Im DSC-Diagramm ist im 1. Lauf die Glasstufe und die Kalt-
kristallisation zu erkennen. Es wurde darauf geachtet, dass die Messung vor
dem eutektischen Signal gestoppt wurde. Nach dieser ersten Messung wurde
die Probe erneut auf die Starttemperatur von 170 K abgekühlt und ein zweites
mal gemessen. Im zweiten Lauf sind keine Glasstufe und keine Kaltkristallisa-
tion mehr zu erkennen. Man detektiert im DSC-Diagramm lediglich die beiden
endothermen Schmelzsignale.
59 Huang L., Nishinari K.; J. Pol. Sci., 39, 2001, 49660 Dobnik E.; Dissertation Duisburg, 1991
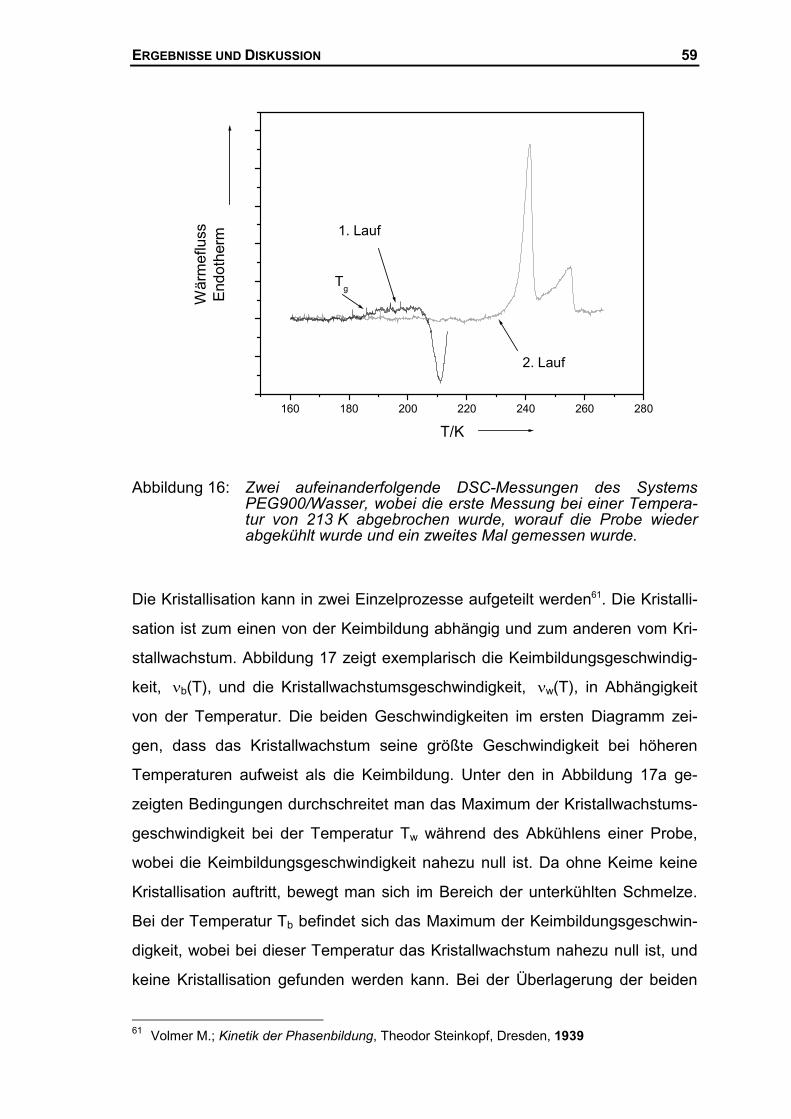
ERGEBNISSE UND DISKUSSION 59
Abbildung 16: Zwei aufeinanderfolgende DSC-Messungen des SystemsPEG900/Wasser, wobei die erste Messung bei einer Tempera-tur von 213 K abgebrochen wurde, worauf die Probe wiederabgekühlt wurde und ein zweites Mal gemessen wurde.
Die Kristallisation kann in zwei Einzelprozesse aufgeteilt werden61. Die Kristalli-
sation ist zum einen von der Keimbildung abhängig und zum anderen vom Kri-
stallwachstum. Abbildung 17 zeigt exemplarisch die Keimbildungsgeschwindig-
keit, ��b(T), und die Kristallwachstumsgeschwindigkeit, ��w(T), in Abhängigkeit
von der Temperatur. Die beiden Geschwindigkeiten im ersten Diagramm zei-
gen, dass das Kristallwachstum seine größte Geschwindigkeit bei höheren
Temperaturen aufweist als die Keimbildung. Unter den in Abbildung 17a ge-
zeigten Bedingungen durchschreitet man das Maximum der Kristallwachstums-
geschwindigkeit bei der Temperatur Tw während des Abkühlens einer Probe,
wobei die Keimbildungsgeschwindigkeit nahezu null ist. Da ohne Keime keine
Kristallisation auftritt, bewegt man sich im Bereich der unterkühlten Schmelze.
Bei der Temperatur Tb befindet sich das Maximum der Keimbildungsgeschwin-
digkeit, wobei bei dieser Temperatur das Kristallwachstum nahezu null ist, und
keine Kristallisation gefunden werden kann. Bei der Überlagerung der beiden
61 Volmer M.; Kinetik der Phasenbildung, Theodor Steinkopf, Dresden, 1939
160 180 200 220 240 260 280
Tg
2. Lauf
1. Lauf
Wär
mef
luss
Endo
ther
m
T/K
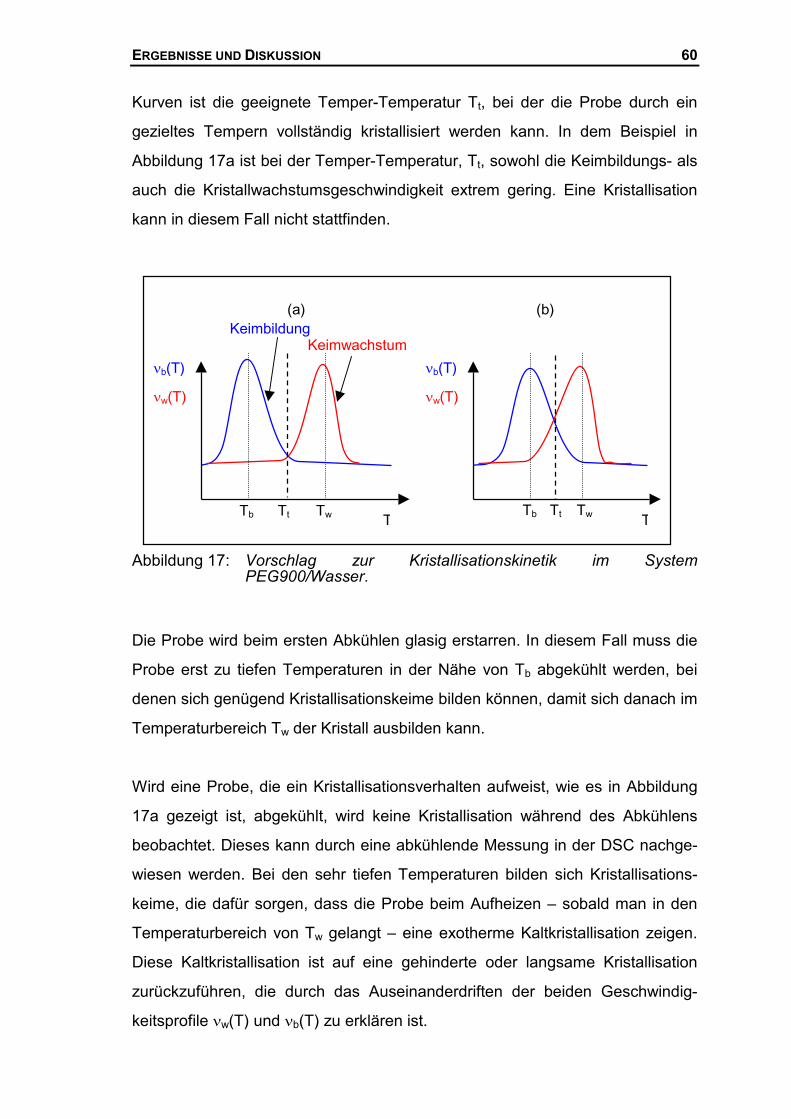
ERGEBNISSE UND DISKUSSION 60
Kurven ist die geeignete Temper-Temperatur Tt, bei der die Probe durch ein
gezieltes Tempern vollständig kristallisiert werden kann. In dem Beispiel in
Abbildung 17a ist bei der Temper-Temperatur, Tt, sowohl die Keimbildungs- als
auch die Kristallwachstumsgeschwindigkeit extrem gering. Eine Kristallisation
kann in diesem Fall nicht stattfinden.
Abbildung 17: Vorschlag zur Kristallisationskinetik im SystemPEG900/Wasser.
Die Probe wird beim ersten Abkühlen glasig erstarren. In diesem Fall muss die
Probe erst zu tiefen Temperaturen in der Nähe von Tb abgekühlt werden, bei
denen sich genügend Kristallisationskeime bilden können, damit sich danach im
Temperaturbereich Tw der Kristall ausbilden kann.
Wird eine Probe, die ein Kristallisationsverhalten aufweist, wie es in Abbildung
17a gezeigt ist, abgekühlt, wird keine Kristallisation während des Abkühlens
beobachtet. Dieses kann durch eine abkühlende Messung in der DSC nachge-
wiesen werden. Bei den sehr tiefen Temperaturen bilden sich Kristallisations-
keime, die dafür sorgen, dass die Probe beim Aufheizen – sobald man in den
Temperaturbereich von Tw gelangt – eine exotherme Kaltkristallisation zeigen.
Diese Kaltkristallisation ist auf eine gehinderte oder langsame Kristallisation
zurückzuführen, die durch das Auseinanderdriften der beiden Geschwindig-
keitsprofile �w(T) und �b(T) zu erklären ist.
�b(T)
�w(T)
KeimbildungKeimwachstum
TTb Tt Tw
�b(T)
�w(T)
TTb Tt Tw
(a) (b)

ERGEBNISSE UND DISKUSSION 61
In Abbildung 17b überlagern sich die beiden Geschwindigkeitsprofile. Im
Bereich Tt Bilden sich nur sehr langsam Kristallisationskeime, welche auch ein
relativ geringes Kristallwachstum zeigen. Durch ein gezieltes Tempern im
Bereich Tt kann die Anzahl der Keime und damit Kristallitgröße beeinflusst wer-
den. Durch dieses Verfahren kann das mechanische Verhalten von Werkstoffen
gezielt beeinflusst werden.
Nach der Kaltkristallisation ist die Probe vollständig kristallisiert. Wird die Probe
ein zweites mal von Temperaturen unterhalb der eutektischen Temperatur
abgekühlt, wird die beim ersten Abkühlen gefundene Glasstufe nicht mehr
detektiert. Bei der zweiten aufheizenden DSC-Messung kann keine Glasstufe
und keine Kaltkristallisation detektiert werden. In diesem Fall können im DSC-
Diagramm nur endotherme Schmelzpeaks detektiert werden.
Erst nach der oben beschriebenen thermischen Vorbehandlung können die
eutektische Signale gemäß des Tammannschen Dreiecks ausgewertet werden
(s. Anhang 6.2.2). Voraussetzung für diese Methode ist, dass die Kaltkristallisa-
tion ausreichend vom Schmelzpeak getrennt ist. Überlagern sich beide Signale,
so dass die Probe simultan kristallisiert und aufschmilzt, ist eine Kristallisation
der Probe durch mehrmaliges Aufheizen und Abkühlen nicht möglich.
Im Phasendiagramm (Anhang 6.2.3) wird deutlich, dass es sich auch in diesem
Fall um ein eutektisches Phasendiagramm mit partieller Mischbarkeit auf der
Seite des PEG handelt. Die Konzentration des Mischkristalls ist w2,m = 0,92. Der
eutektische Punkt liegt bei einer Konzentration von w2,E = 0,583 und einer Tem-
peratur von 242,8 K.

ERGEBNISSE UND DISKUSSION 62
4.1.3 Phasendiagramm PEG600/Wasser
In Anhang 6.3.1 sind DSC-Messungen verschiedener Konzentrationen, die
Auswertung nach Tammann und das Phasendiagramm des Systems
PEG600/Wasser abgebildet.
Bei der Bestimmung des Phasendiagramms des Systems PEG600/Wasser
mussten die Proben in gleicher Weise thermisch vorbehandelt werden, wie es
bei dem System PEG900/Wasser beschrieben wurde.
Im höher konzentrierten Bereich (w2 > 0,62) lässt sich allerdings trotz einer
Temperung und einem zweimaligen Aufheizen kein eutektisches Signal mehr
detektieren, so dass eutektische Signale lediglich bis zu einer Konzentration
von w2 = w2,E = 0,62 gemessen werden konnten. Oberhalb dieser Konzentration
werden nur noch die Liquidussignale gemessen und eine Glasstufe bei 193 K,
die je nach Konzentration unterschiedlich stark ausgeprägt ist.
Die Glasstufen wurden ähnlich des Tammannschen Dreiecks ausgewertet,
indem die Höhe der Glasstufe, pc~� , in Abhängigkeit von der Konzentration auf-
getragen wurde.
Diese Auftragung in Abbildung 18 zeigt, dass die Höhe der Glasstufe ab einer
Konzentration von w2 = 0,6 linear abnimmt. Der Schnittpunkt der linearen
Regression mit der Abszisse bei w2 = 0,92 deutet an, dass ab dieser Konzen-
tration keine Glasstufen mehr gemessen werden und die gesamte Probe als
Mischkristall vorliegt. Dies ist in Übereinstimmung mit dem Schnitt der
Tammannschen Geraden mit der Konzentrationsachse oberhalb der eutekti-
schen Polymerkonzentration.
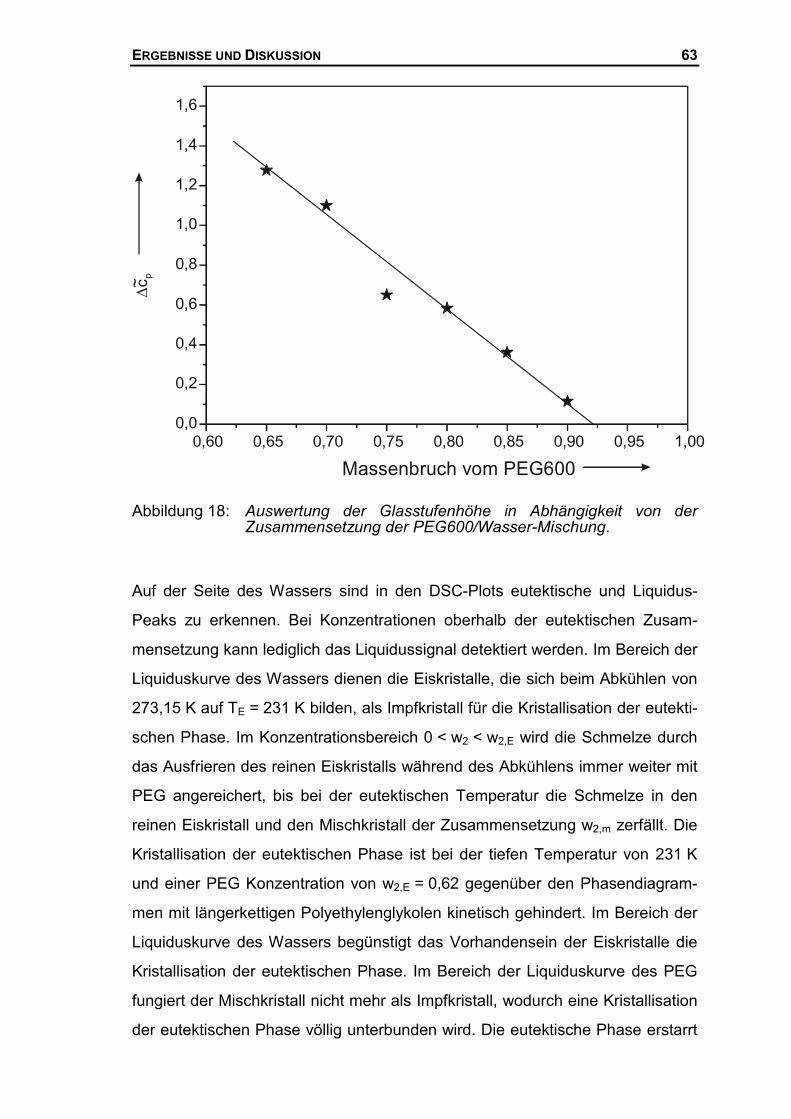
ERGEBNISSE UND DISKUSSION 63
Abbildung 18: Auswertung der Glasstufenhöhe in Abhängigkeit von derZusammensetzung der PEG600/Wasser-Mischung.
Auf der Seite des Wassers sind in den DSC-Plots eutektische und Liquidus-
Peaks zu erkennen. Bei Konzentrationen oberhalb der eutektischen Zusam-
mensetzung kann lediglich das Liquidussignal detektiert werden. Im Bereich der
Liquiduskurve des Wassers dienen die Eiskristalle, die sich beim Abkühlen von
273,15 K auf TE = 231 K bilden, als Impfkristall für die Kristallisation der eutekti-
schen Phase. Im Konzentrationsbereich 0 < w2 < w2,E wird die Schmelze durch
das Ausfrieren des reinen Eiskristalls während des Abkühlens immer weiter mit
PEG angereichert, bis bei der eutektischen Temperatur die Schmelze in den
reinen Eiskristall und den Mischkristall der Zusammensetzung w2,m zerfällt. Die
Kristallisation der eutektischen Phase ist bei der tiefen Temperatur von 231 K
und einer PEG Konzentration von w2,E = 0,62 gegenüber den Phasendiagram-
men mit längerkettigen Polyethylenglykolen kinetisch gehindert. Im Bereich der
Liquiduskurve des Wassers begünstigt das Vorhandensein der Eiskristalle die
Kristallisation der eutektischen Phase. Im Bereich der Liquiduskurve des PEG
fungiert der Mischkristall nicht mehr als Impfkristall, wodurch eine Kristallisation
der eutektischen Phase völlig unterbunden wird. Die eutektische Phase erstarrt
pc�
0,60 0,65 0,70 0,75 0,80 0,85 0,90 0,95 1,000,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Massenbruch vom PEG600

ERGEBNISSE UND DISKUSSION 64
letztendlich glasig bei einer Temperatur von 190 K. In der Nähe des eutek-
tischen Punktes ist der Anteil der glasig erstarrenden Phase am größten, was
dazu führt, dass die Glasstufe in diesem Bereich am deutlichsten ausgeprägt
ist, d.h. pc~� ist in diesem Bereich am größten, und nimmt bis zur Konzentration
w2,m = 0,92 linear ab.
4.1.4 Phasendiagramme PEG400/Wasser und PEG300/Wasser
Bei der Bestimmung des Phasendiagramms PEG600/Wasser wurde der Trend
deutlich, dass mit abnehmender molaren Masse des PEG die Kristallisation der
eutektischen Phase mehr und mehr gehindert ist. Bei den DSC-Messungen des
Systems PEG400/Wasser sind unter keinen Bedingungen mehr eutektische
Signale zu beobachten. Bei den Aufheizkurven zeigt sich im mittleren Konzen-
trationsbereich (0,3 < w2 < 0,9) eine Kaltkristallisation, welche sich mit dem
Liquidussignal überlagert. Durch das Stoppen des DSC-Programms im Bereich
der Kaltkristallisation und ein zweites Abkühlen von Temperaturen unterhalb der
eutektischen Temperatur auf die DSC-Starttemperatur von 180 K, kann die
Kaltkristallisation verhindert werden. Beim zweiten Lauf ist nur noch eine
Glasstufe bei 190 K und ein Liquidussignal zu erkennen. Das bedeutet, dass in
diesem System durch ein 2-maliges Aufheizen die Liquidussignale gemessen
werden können. Eutektische Signale können in diesem System nicht mehr
gemessen werden. Im Konzentrationsbereich 0,62 < w2 < 0,78 können lediglich
noch Glasstufen gemessen werden. In diesem Konzentrationsbereich ist die
Kristallisation derart gehindert, dass sich auch nach umfangreicher thermischer
Vorbehandlung keine Schmelzsignale detektieren lassen.
Im System PEG300/Wasser können wie beim System PEG400/Wasser keine
eutektischen Signale mehr gefunden werden. Auch hier findet man nur noch
Glasstufen und Liquidussignale. Die Glasstufen liegen in diesem System bei
190 K. Es wird auch ein Konzentrationsbereich gefunden, in dem keine Kristal-
lisation stattfindet und die Lösung glasig bei 190 K erstarrt. Dieser Bereich ist im

ERGEBNISSE UND DISKUSSION 65
Vergleich zum System PEG400/Wasser weiter ausgedehnt 0,58 < w2 < 0,98, so
dass auf der Seite des PEG im Phasendiagramm keine Liquiduskurve für das
PEG gefunden wird. Im Phasendiagramm des Systems PEG300/Wasser kann
auf der Seite des PEG der Schmelzpunkt für das reine PEG angegeben wer-
den. Eine Liquiduskurve kann nur auf der Seite des Wassers bestimmt werden.
Einige DSC-Messungen, sowie die Phasendiagramme der Systeme
PEG300/Wasser und PEG400/Wasser sind in Anhang 6.4 und 6.5 abgebildet.
4.1.5 DSC-Messungen der reinen PEG-Proben
In den DSC-Messungen der reinen PEG-Proben ist in vielen Fällen das Auftre-
ten einer „Schulter“ im linken Teil des Schmelzpeaks zu beobachten (Abbildung
19). Wie bei Messungen des eutektischen Signals des Systems Gelatine/Was-
ser62 wurde zunächst davon ausgegangen, dass es sich hier um eine Glasstufe
handelt. Die Glasstufe ist so zu erklären, dass ein Teil des PEG als Kristall
ausfriert, was bedeutet, dass die Beweglichkeit der PEG Ketten in der flüssigen
Phase immer weiter eingeschränkt werden, bis sich die Ketten nicht mehr zu
einem Kristall anordnen können und die verbleibende flüssige Phase schließlich
beim Abkühlen glasig erstarrt. Durch eine Variation der Aufheizgeschwindig-
keiten lässt sich die Form der Glasstufe beeinflussen63,64.
DSC-Diagramme von PEG mit einer molaren Masse von 6000 g/mol zeigen,
dass auch durch ein gezieltes Tempern der Proben die Form des Schmelz-
signals beeinflusst wird. Delahaye et al. konnte mit Hilfe von röntgenografischen
Messungen nachweisen, dass es sich allerdings bei der Schulter im vorderen
Bereich des Schmelzsignals nicht um eine Glasstufe handelt65,66. Bei der 62 Reutner P., Luft B., Borchard W.; Col. Pol. Sci., 263, 1985, 51963 Jablonski P., Golloch A., Borchard W.; Thermochimica Acta, 333, 1999, 8764 Rehage G., Borchard W.; Physics of glassy polymers, Ed. Haward R.N., 1973, 5465 Delahaye N., Duclos R., Saiter J.M.; Int. J. Pharm., 157, 1997, 2766 Saiter J.M., Bayard J., Delahaye N., Varbier S., Vautier C.; J. Therm. Anal., 45, 1995,
1223
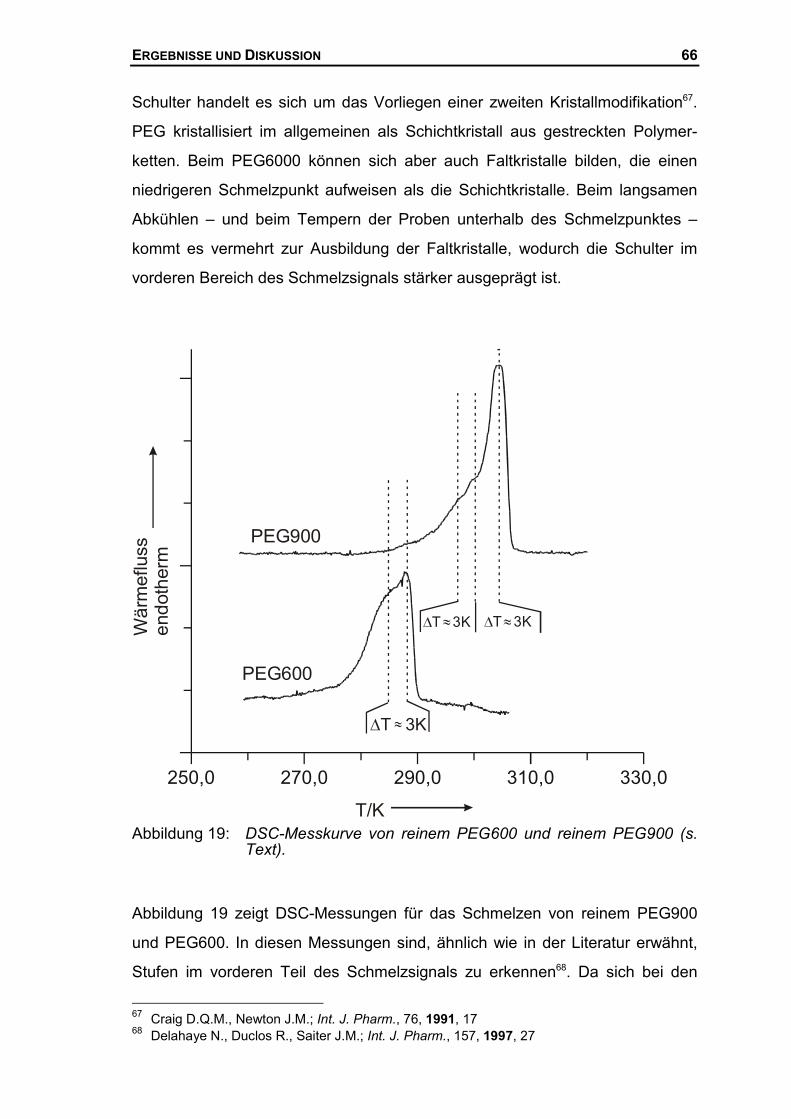
ERGEBNISSE UND DISKUSSION 66
Schulter handelt es sich um das Vorliegen einer zweiten Kristallmodifikation67.
PEG kristallisiert im allgemeinen als Schichtkristall aus gestreckten Polymer-
ketten. Beim PEG6000 können sich aber auch Faltkristalle bilden, die einen
niedrigeren Schmelzpunkt aufweisen als die Schichtkristalle. Beim langsamen
Abkühlen – und beim Tempern der Proben unterhalb des Schmelzpunktes –
kommt es vermehrt zur Ausbildung der Faltkristalle, wodurch die Schulter im
vorderen Bereich des Schmelzsignals stärker ausgeprägt ist.
Abbildung 19: DSC-Messkurve von reinem PEG600 und reinem PEG900 (s.Text).
Abbildung 19 zeigt DSC-Messungen für das Schmelzen von reinem PEG900
und PEG600. In diesen Messungen sind, ähnlich wie in der Literatur erwähnt,
Stufen im vorderen Teil des Schmelzsignals zu erkennen68. Da sich bei den
67 Craig D.Q.M., Newton J.M.; Int. J. Pharm., 76, 1991, 1768 Delahaye N., Duclos R., Saiter J.M.; Int. J. Pharm., 157, 1997, 27
250,0 270,0 290,0 310,0 330,0
T/K
PEG600
PEG900
Wär
mef
luss
endo
ther
m
�T 3K
�T 3K�T 3K
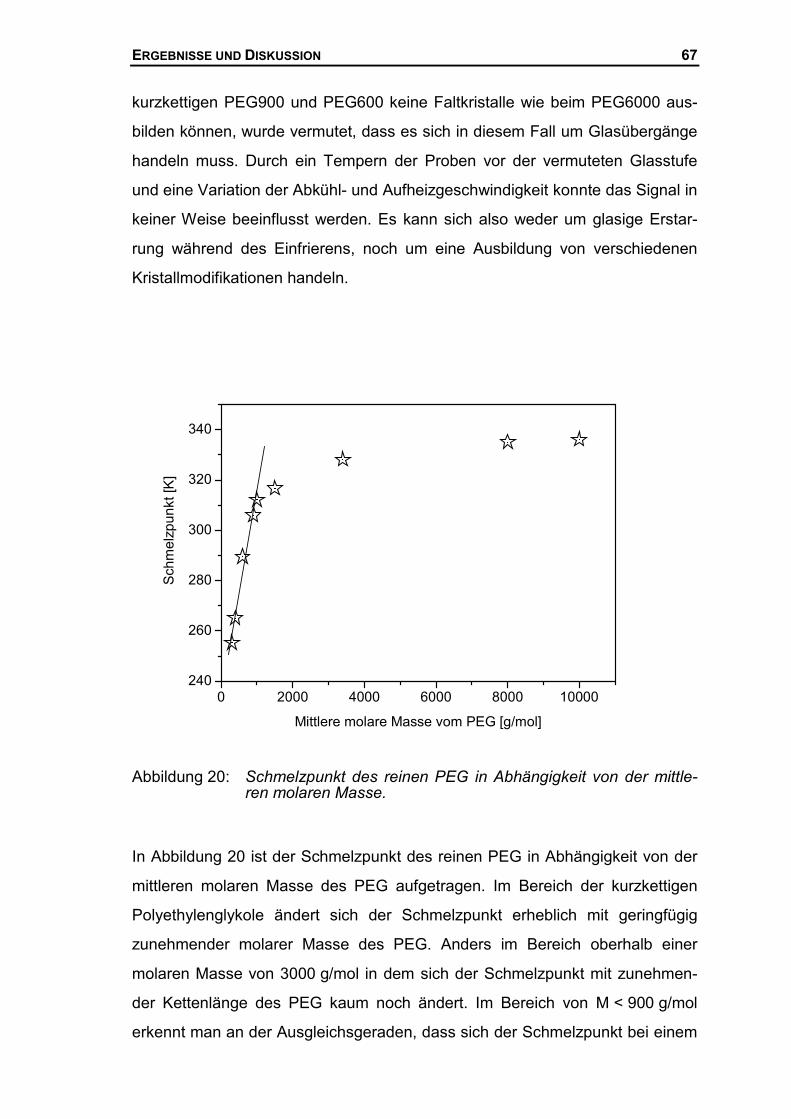
ERGEBNISSE UND DISKUSSION 67
kurzkettigen PEG900 und PEG600 keine Faltkristalle wie beim PEG6000 aus-
bilden können, wurde vermutet, dass es sich in diesem Fall um Glasübergänge
handeln muss. Durch ein Tempern der Proben vor der vermuteten Glasstufe
und eine Variation der Abkühl- und Aufheizgeschwindigkeit konnte das Signal in
keiner Weise beeinflusst werden. Es kann sich also weder um glasige Erstar-
rung während des Einfrierens, noch um eine Ausbildung von verschiedenen
Kristallmodifikationen handeln.
Abbildung 20: Schmelzpunkt des reinen PEG in Abhängigkeit von der mittle-ren molaren Masse.
In Abbildung 20 ist der Schmelzpunkt des reinen PEG in Abhängigkeit von der
mittleren molaren Masse des PEG aufgetragen. Im Bereich der kurzkettigen
Polyethylenglykole ändert sich der Schmelzpunkt erheblich mit geringfügig
zunehmender molarer Masse des PEG. Anders im Bereich oberhalb einer
molaren Masse von 3000 g/mol in dem sich der Schmelzpunkt mit zunehmen-
der Kettenlänge des PEG kaum noch ändert. Im Bereich von M < 900 g/mol
erkennt man an der Ausgleichsgeraden, dass sich der Schmelzpunkt bei einem
0 2000 4000 6000 8000 10000240
260
280
300
320
340
Schm
elzp
unkt
[K]
Mittlere molare Masse vom PEG [g/mol]

ERGEBNISSE UND DISKUSSION 68
Massenunterschied von 44 g/mol – was genau einem Polymerbaustein (CH2-
CH2-O) entspricht – um ca. 3 K ändert. Bei den Polyethylenglykolen mit mola-
ren Massen unterhalb von 1000 g/mol hat das Vorliegen einer polydispersen
Molekulargewichtsverteilung erhebliche Auswirkungen auf das Schmelzver-
halten der Proben. Dementsprechend sind die Schultern in der rechten Flanke
des Schmelzpeaks als Überlagerung mehrerer Schmelzsignale zu interpretie-
ren, die einen Temperaturunterschied von ca. 3 K aufweisen.
Es kann davon ausgegangen werden, dass es sich bei PEG600 und PEG900
nicht um eine einheitliche molare Masse handelt, sondern dass auch um einen
Polymerbaustein kürzere und längere Ketten in nicht vernachlässigbarer Kon-
zentration vorhanden sind. Diese Tatsache wird in den späteren thermodynami-
schen Auswertungen nicht berücksichtigt, es wird weiter von einer binären
Mischung ausgegangen.
4.1.6 Schmelzverhalten von PEG/Wasser Systemen
Das Phasendiagramm des Systems PEG20000/Wasser69 unterscheidet sich
kaum vom Phasendiagramm des Systems PEG35000/Wasser70. In beiden
Phasendiagrammen liegt ein eutektisches Schmelzdiagramm vor, wobei sich
die Lage der charakteristischen Punkte des Phasendiagramms nur unwesent-
lich voneinander unterscheiden. Die eutektische Temperatur beträgt 263 K im
System PEG20000/Wasser und 261 K im System PEG35000/Wasser. Die eu-
tektische Konzentration beträgt w2 = 0,48 bzw. 0,45. Der Schmelzpunkt der
reinen Polyethylenglykole liegt bei 338 K. Der Verlauf der Liquiduskurven ist
ebenfalls annähernd identisch.
Wesentliche Unterschiede werden erst gefunden, wenn die molare Masse der
Polyethylenglykole geringer wird. Mit abnehmender molarer Masse des PEG
69 Bogdanov B., Mihailov M.; J. Pol. Sci., 23, 1985, 214970 Dobnik E.; Dissertation Duisburg, 1991
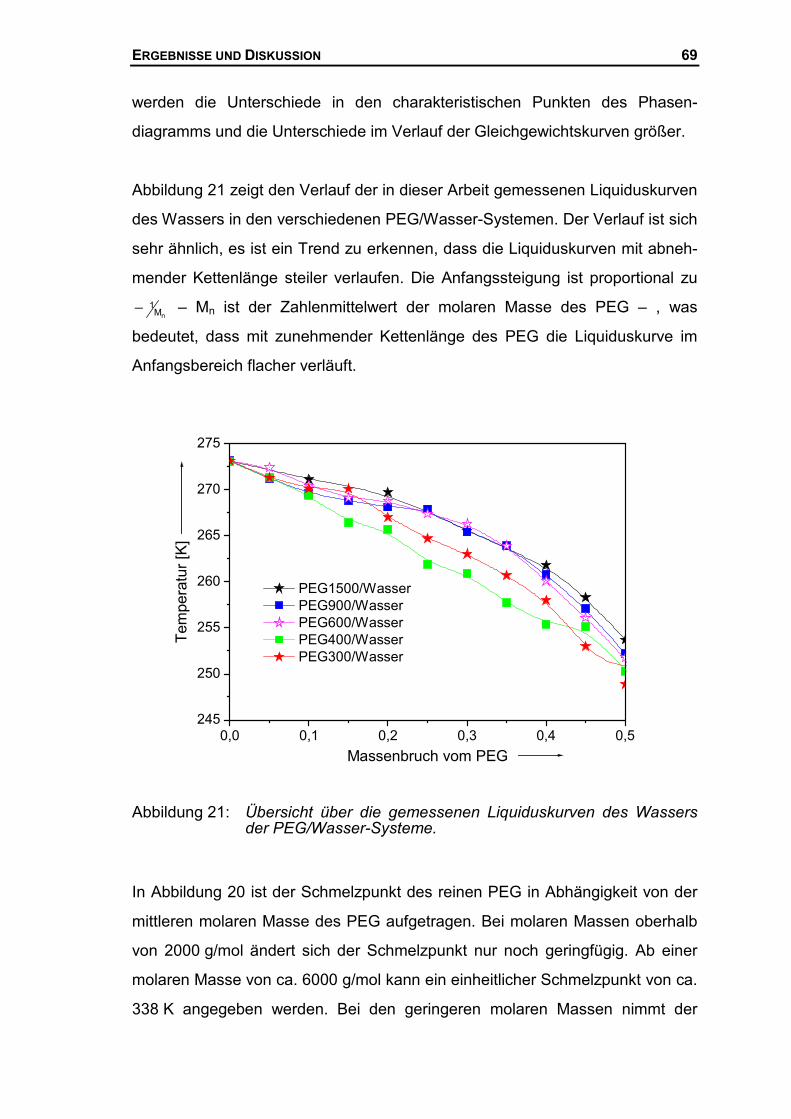
ERGEBNISSE UND DISKUSSION 69
werden die Unterschiede in den charakteristischen Punkten des Phasen-
diagramms und die Unterschiede im Verlauf der Gleichgewichtskurven größer.
Abbildung 21 zeigt den Verlauf der in dieser Arbeit gemessenen Liquiduskurven
des Wassers in den verschiedenen PEG/Wasser-Systemen. Der Verlauf ist sich
sehr ähnlich, es ist ein Trend zu erkennen, dass die Liquiduskurven mit abneh-
mender Kettenlänge steiler verlaufen. Die Anfangssteigung ist proportional zu
nM1
� – Mn ist der Zahlenmittelwert der molaren Masse des PEG – , was
bedeutet, dass mit zunehmender Kettenlänge des PEG die Liquiduskurve im
Anfangsbereich flacher verläuft.
Abbildung 21: Übersicht über die gemessenen Liquiduskurven des Wassersder PEG/Wasser-Systeme.
In Abbildung 20 ist der Schmelzpunkt des reinen PEG in Abhängigkeit von der
mittleren molaren Masse des PEG aufgetragen. Bei molaren Massen oberhalb
von 2000 g/mol ändert sich der Schmelzpunkt nur noch geringfügig. Ab einer
molaren Masse von ca. 6000 g/mol kann ein einheitlicher Schmelzpunkt von ca.
338 K angegeben werden. Bei den geringeren molaren Massen nimmt der
0,0 0,1 0,2 0,3 0,4 0,5245
250
255
260
265
270
275
PEG1500/Wasser PEG900/Wasser PEG600/Wasser PEG400/Wasser PEG300/Wasser
Tem
pera
tur [
K]
Massenbruch vom PEG
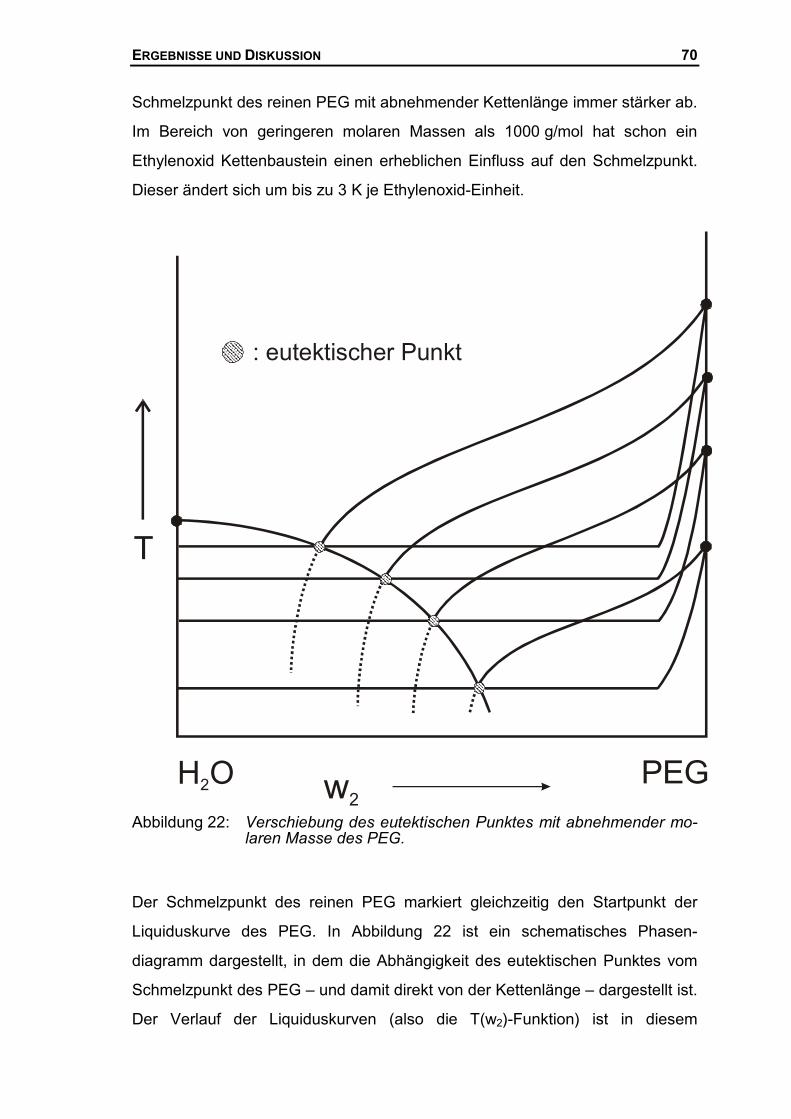
ERGEBNISSE UND DISKUSSION 70
Schmelzpunkt des reinen PEG mit abnehmender Kettenlänge immer stärker ab.
Im Bereich von geringeren molaren Massen als 1000 g/mol hat schon ein
Ethylenoxid Kettenbaustein einen erheblichen Einfluss auf den Schmelzpunkt.
Dieser ändert sich um bis zu 3 K je Ethylenoxid-Einheit.
Abbildung 22: Verschiebung des eutektischen Punktes mit abnehmender mo-laren Masse des PEG.
Der Schmelzpunkt des reinen PEG markiert gleichzeitig den Startpunkt der
Liquiduskurve des PEG. In Abbildung 22 ist ein schematisches Phasen-
diagramm dargestellt, in dem die Abhängigkeit des eutektischen Punktes vom
Schmelzpunkt des PEG – und damit direkt von der Kettenlänge – dargestellt ist.
Der Verlauf der Liquiduskurven (also die T(w2)-Funktion) ist in diesem
H O2 PEG
: eutektischer Punkt
T
w2
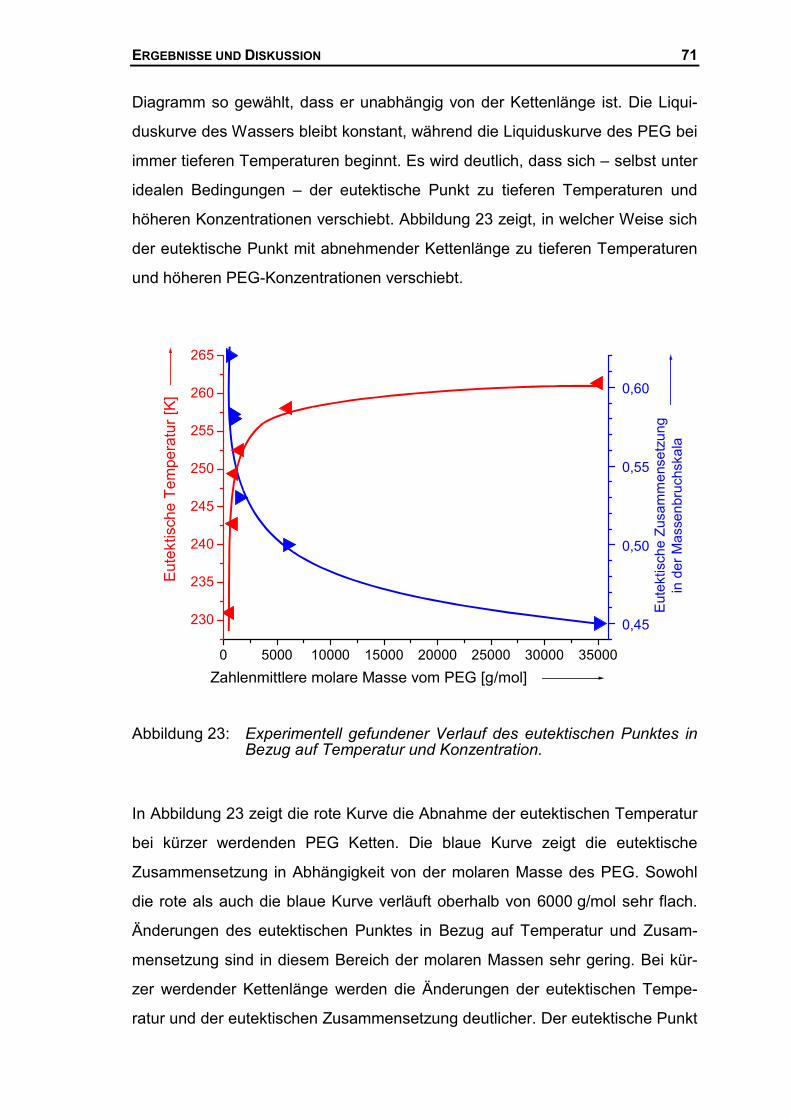
ERGEBNISSE UND DISKUSSION 71
Diagramm so gewählt, dass er unabhängig von der Kettenlänge ist. Die Liqui-
duskurve des Wassers bleibt konstant, während die Liquiduskurve des PEG bei
immer tieferen Temperaturen beginnt. Es wird deutlich, dass sich – selbst unter
idealen Bedingungen – der eutektische Punkt zu tieferen Temperaturen und
höheren Konzentrationen verschiebt. Abbildung 23 zeigt, in welcher Weise sich
der eutektische Punkt mit abnehmender Kettenlänge zu tieferen Temperaturen
und höheren PEG-Konzentrationen verschiebt.
Abbildung 23: Experimentell gefundener Verlauf des eutektischen Punktes inBezug auf Temperatur und Konzentration.
In Abbildung 23 zeigt die rote Kurve die Abnahme der eutektischen Temperatur
bei kürzer werdenden PEG Ketten. Die blaue Kurve zeigt die eutektische
Zusammensetzung in Abhängigkeit von der molaren Masse des PEG. Sowohl
die rote als auch die blaue Kurve verläuft oberhalb von 6000 g/mol sehr flach.
Änderungen des eutektischen Punktes in Bezug auf Temperatur und Zusam-
mensetzung sind in diesem Bereich der molaren Massen sehr gering. Bei kür-
zer werdender Kettenlänge werden die Änderungen der eutektischen Tempe-
ratur und der eutektischen Zusammensetzung deutlicher. Der eutektische Punkt
0 5000 10000 15000 20000 25000 30000 35000
230
235
240
245
250
255
260
265
Eute
ktis
che
Tem
pera
tur [
K]
Zahlenmittlere molare Masse vom PEG [g/mol]
0,45
0,50
0,55
0,60
Eute
ktis
che
Zusa
mm
ense
tzun
gin
der
Mas
senb
ruch
skal
a
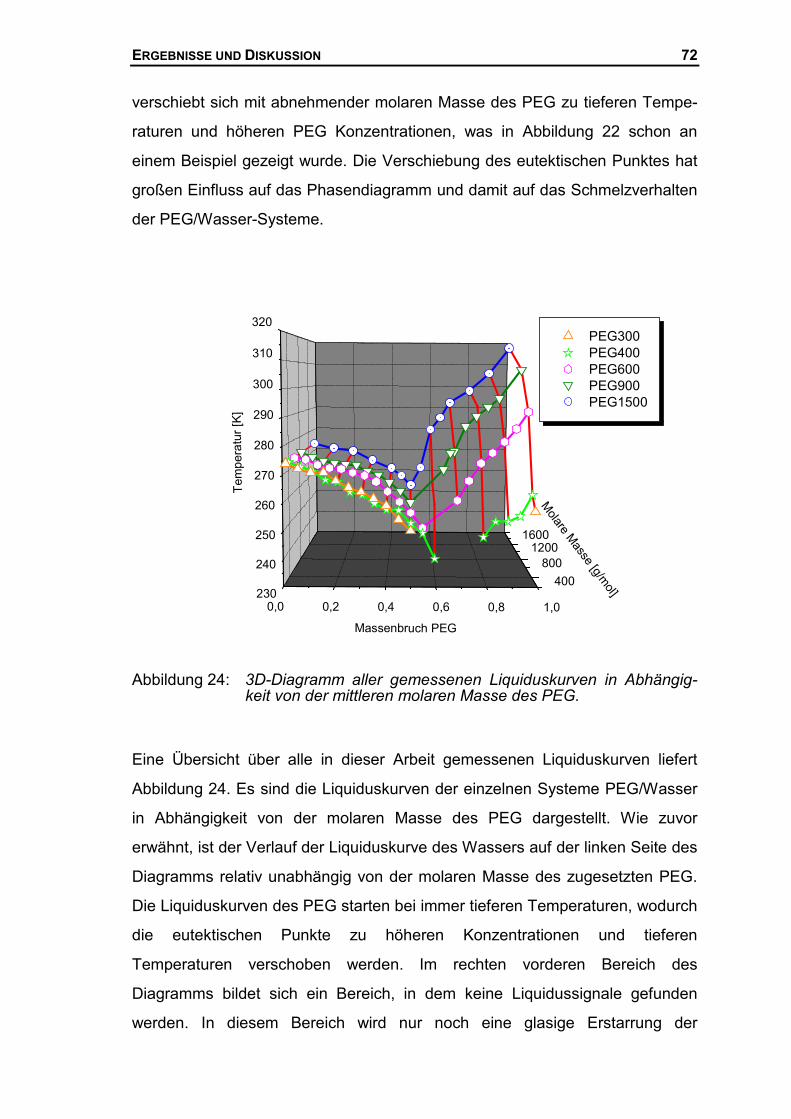
ERGEBNISSE UND DISKUSSION 72
verschiebt sich mit abnehmender molaren Masse des PEG zu tieferen Tempe-
raturen und höheren PEG Konzentrationen, was in Abbildung 22 schon an
einem Beispiel gezeigt wurde. Die Verschiebung des eutektischen Punktes hat
großen Einfluss auf das Phasendiagramm und damit auf das Schmelzverhalten
der PEG/Wasser-Systeme.
Abbildung 24: 3D-Diagramm aller gemessenen Liquiduskurven in Abhängig-keit von der mittleren molaren Masse des PEG.
Eine Übersicht über alle in dieser Arbeit gemessenen Liquiduskurven liefert
Abbildung 24. Es sind die Liquiduskurven der einzelnen Systeme PEG/Wasser
in Abhängigkeit von der molaren Masse des PEG dargestellt. Wie zuvor
erwähnt, ist der Verlauf der Liquiduskurve des Wassers auf der linken Seite des
Diagramms relativ unabhängig von der molaren Masse des zugesetzten PEG.
Die Liquiduskurven des PEG starten bei immer tieferen Temperaturen, wodurch
die eutektischen Punkte zu höheren Konzentrationen und tieferen
Temperaturen verschoben werden. Im rechten vorderen Bereich des
Diagramms bildet sich ein Bereich, in dem keine Liquidussignale gefunden
werden. In diesem Bereich wird nur noch eine glasige Erstarrung der
0,0 0,2 0,4 0,6 0,8 1,0230
240
250
260
270
280
290
300
310
320
400800
12001600
PEG300 PEG400 PEG600 PEG900 PEG1500
Tem
pera
tur [
K]Molare Masse [g/mol]
Massenbruch PEG

ERGEBNISSE UND DISKUSSION 73
Mischungen gefunden. Dieser Konzentrations- und Kettenlängenbereich ist für
verschiedene Anwendungen der Kryo-Konservierung interessant, da Systeme
heruntergekühlt werden können, ohne dass Eiskristalle diese zerstören. Solche
Verfahren sind bei der Konservierung von biologischen Zellen denkbar, wobei
PEG mit geeigneter molarer Masse direkt in die Zellen gespritzt wird. Sobald
man im Konzentrationsbereich ist, in dem keine Kristallisation gefunden wird,
kann die Zelle ohne zerstörende Kristallisation des Zellwassers abgekühlt und
damit konserviert werden71.
Es wird deutlich, dass in allen binären PEG/Wasser-Systemen ein eutektisches
Schmelzverhalten vorliegt, wobei eine Mischkristallbildung nur auf der Seite des
PEG gefunden wird. Auf der Seite des Lösemittels Wasser kann aufgrund der
Auswertungen nach Tammann eine Randlöslichkeit ausgeschlossen werden.
Die Form der Gleichgewichtskurven zeigen erwartungsgemäß einen kontinuier-
lichen Gang mit abnehmender molaren Masse des PEG. Die Steigungen der
Liquiduskurven des Wassers werden mit steigender molarer Masse des PEG
erwartungsgemäß flacher. Am eutektischen Punkt und dem Schmelzpunkt des
reinen PEG sind größere Veränderungen bei Variation der molaren Masse des
PEG zu erkennen. Die Änderungen sind im kurzkettigen Bereich am größten
und nehmen mit zunehmender Kettenlänge ab, so dass ab einer molaren
Masse von ca. 10000 g/mol von einem einheitlichem Phasendiagramm gespro-
chen werden kann. Eine Aufteilung in mehrere Gruppen (kurz-, mittel- und lang-
kettige Polyethylenglykole), wie sie von anderen Autoren vorgeschlagen wird72,
erscheint aufgrund der Systematik, die die Phasendiagramme zeigen, nicht
geeignet, da die entsprechenden Grenzen nicht oder nur sehr vage auszu-
machen sind. Das liegt vor allem am einheitlichen Gang der charakteristischen
Größen des Phasendiagramms mit der molaren Masse des PEG.
71 Franks F. ; Biophysics and Biochemistry at Low Temperatures, Univ. Press., New York,
Cambridge, 198572 Huang L., Nishinari K.; J. Pol. Sci., 39, 2001, 496

ERGEBNISSE UND DISKUSSION 74
4.1.7 PEG/Wasser Mischkristall
Bei den in dieser Arbeit gemessenen vollständigen Phasendiagrammen liegt in
allen Fällen ein Mischkristall auf der Seite des PEG vor, dessen Konzentration
in der Grundmolenbruchskala mit 8,0x*2 � angegeben werden kann, was einem
Massenbruch im System PEG/Wasser von w2 = 0,92 entspricht. Dieser Wert
wird auch in allen Phasendiagrammen von Dobnik gefunden73. Andere in dieser
Arbeit zitierte Arbeiten finden auch die Mischkristallbildung des PEG, verzichten
aber auf eine Auswertung gemäß des Tammannschen Dreiecks. In diesen
Arbeiten wird auch die spezifische eutektische Umwandlungsenthalpie gegen
die Zusammensetzung aufgetragen, woran abgelesen werden kann, dass auch
in diesen Systemen die Mischkristallbildung vorliegt. Im System PEG/Wasser
bildet sich unabhängig von der Kettenlänge des PEG ein Mischkristall der
Zusammensetzung 8,0x*2 � . Auch die Auswertung der Glasstufenhöhe ähnlich
des Tammannschen Dreiecks im System PEG600/Wasser deutet auf die Bil-
dung der festen Mischphase bei der Konzentration w2 = 0,92, was einem
Grundmolenbruch von 8,0x*2 � entspricht.
In diesem Kapitel wurde der Grundmolenbruch verwendet, da an diesem direkt
das Verhältnis PEG- zu Wassermolekülen abgelesen werden kann. 2,0x*1 �
und 8,0x1x *1
*2 ��� führt zu einem Verhältnis 4xx *
1*2 � , was bedeutet, dass
im Mischkristall ein Wasser Molekül von vier Ethylenoxid-Einheiten umgeben
ist. Dieses Verhältnis wird bei allen Kettenlängen gefunden.
Über die genaue Struktur der festen Phase ist in der Literatur nicht viel bekannt.
Es wird häufig das Kristallisationsverhalten der reinen PEG-Kristalle untersucht,
die in verschiedenen Modifikationen auftreten können, die sowohl von der Ket-
tenlänge des PEG als auch von der thermischen Vorgeschichte (Abkühl-
geschwindigkeiten, Temperungen) abhängen. PEG kann in gestreckter Form
oder in Form von Faltkristallen kristallisieren74-79.
73 Dobnik E.; Dissertation Duisburg, 199174 Arlie J.P., Spegt P.A., Skoulios A.E.; Makromol. Chem., 99, 1966, 16075 Strobl G.; The Physics of Polymers, Springer, Berlin, 1996

ERGEBNISSE UND DISKUSSION 75
An den Enden des PEG Kristalls können Kettenenden aus dem Kristall heraus-
ragen oder Schlaufen bilden. Die Position des Wassers wurde in der Ver-
gangenheit in den amorphen Bereich des PEG Kristalls vermutet. Hierzu unter-
suchte Dobnik in seiner Dissertation die Kristallinität des PEG in Zusammen-
hang mit dem Gehalt an Wasser in der Mischphase. Es stellte sich allerdings
heraus, dass der Wassergehalt nicht mit dem Grad der Kristallinität beschrieben
werden kann. Die Tatsache, dass bei verschiedenen Arbeitsgruppen, die unter
verschiedenen Bedingungen messen (Probenmasse, Kalorimeter, Heiz- Kühl-
rate, etc.), stets kettenlängenunabhängig das gleiche Verhältnis von Wasser zu
Ethylenoxid-Einheiten finden, spricht dagegen, dass das Wasser in amorphen
Bereichen zu finden ist.
Die PEG Kristalle besitzen eine helicale Form80,81, daher wurde ebenfalls die
Möglichkeit diskutiert, ob die Wassermoleküle im PEG-Kristall systematisch in
die Helix eingebaut werden. Der systematische Einbau der Wassermoleküle
konnte nicht gefunden werden. Bei den kleineren molaren Massen des PEG
(M < 3000 g/mol) ist die Ausbildung einer Helix eher zweifelhaft, da nicht genü-
gend Einheiten zur Ausbildung einer kompletten Helix zur Verfügung stehen.
Allerdings wird auch bei diesen geringeren molaren Massen das o.g. Verhältnis
gefunden. Die Wassermoleküle könnten die relativ lockere Helix stabilisieren,
was auch bei helicalen Teilstücken denkbar wäre. Bei den in dieser Arbeit
gemessenen PEG/Wasser-Systemen von ca. 5 bis 10 Ethylenoxid-Einheiten
müsste allerdings eine deutliche Abweichung von dem Verhältnis 4:1 vorliegen.
Die wahrscheinlichste Annahme ist, dass das Wasser in die Schichtkristalle des
PEG eindringen kann, wobei ein strenges Molverhältnis gegeben ist. Das Was-
76 Arlie J.P., Spegt P.A., Skoulios A.E.; Makromol. Chem., 104, 1967, 21277 Spegt P.A.; Makromol. Chem., 140, 1970, 16778 Buckley C.P., Kovacs A.J.; Col. Pol. Sci., 254, 1976, 69579 Delahay N., Duclos R., Saiter J.M.; Int. J. Pharm., 157, 1997, 2780 Takahashi Y., Tadokoro H.; Macromolecules, 6, 1973, 67281 Tadokoro H.; Structure of crystalline polymers; Kap. 4.6.4, 99, Wiley & Sons, New York,
1979

ERGEBNISSE UND DISKUSSION 76
sermolekül befindet sich in Hohlräumen, die sich durch die Stapelung der ein-
zelnen PEG-Schichten bilden. In diesen Hohlräumen ist das Wasser insgesamt
von vier Ethylenoxid-Einheiten umgeben. Selbst bei den niedrigen molaren
Massen kann von einer Schichtstruktur der PEG-Kristalle ausgegangen werden,
so dass auch bei den Kürzeren PEG-Ketten das gleiche stöchiometrische Ver-
hältnis gefunden wird, wie bei den längeren PEG-Ketten.
Auch von anderen niedrigmolekularen Verbindungen ist bekannt, dass sie mit
PEG kristalline Komplexe bzw. Mischphasen bilden, die jeweils kettenlängen-
unabhängig die gleiche Zusammensetzung aufweisen. Beispiele hierfür sind
Harnstoff und Thioharnstoff82,83, oder para-dihalogenierte Benzolderivate84-86.
Die genaue Position der entsprechenden Zusätze im Mischkristall kann aller-
dings auch in diesen Fällen nicht angegeben werden.
Das Verhältnis Ethylenoxid-Einheiten zu Wassermolekülen von 4:1 ist auf kei-
nen Fall von der Bildung von Faltkristallen oder der Kristallisation gestreckter
PEG-Ketten abhängig, da in diesem Fall die Mischkristallbildung von der ther-
mischen Vorgeschichte abhinge, mit der man das Verhältnis von Faltkristallen
zu Kristallen gestreckter PEG-Ketten beeinflussen kann87. Ein direkter Einbau in
die PEG-Helix gilt ebenfalls als unwahrscheinlich, da in diesem Fall die Abwei-
chungen im kurzkettigen Bereich vorliegen müssten, da der Endgruppeneinfluss
zunimmt. Die wahrscheinlichste – wenn auch in dieser Arbeit nicht verifizierte –
Position des Wasser, ist systematisch in Zwischenräumen der PEG-Kristall-
schichten, in denen das Wassermolekül von vier Ethylenoxid-Einheiten umge-
ben ist.
82 Bailey F.E.Jr., France H.G.; J. Pol. Sci. A, 49, 1961, 39883 Tadokoro H., Yoshihara T., Chatani Y., Murahashi S.; J. Pol. Sci. A, 2, 1964, 36384 Point J.J., Coutelier C.; J. Pol. Sci., Pol Phys. Ed., 23, 1985, 23185 Point J.J.; J. Inclusion Phen., 6, 1988, 25386 Point J.J., Demaret J.P.; J. Phys. Chem., 91, 1987, 79787 Delahay N., Duclos R., Saiter J.M.; Int. J. Pharm., 157, 1997, 27
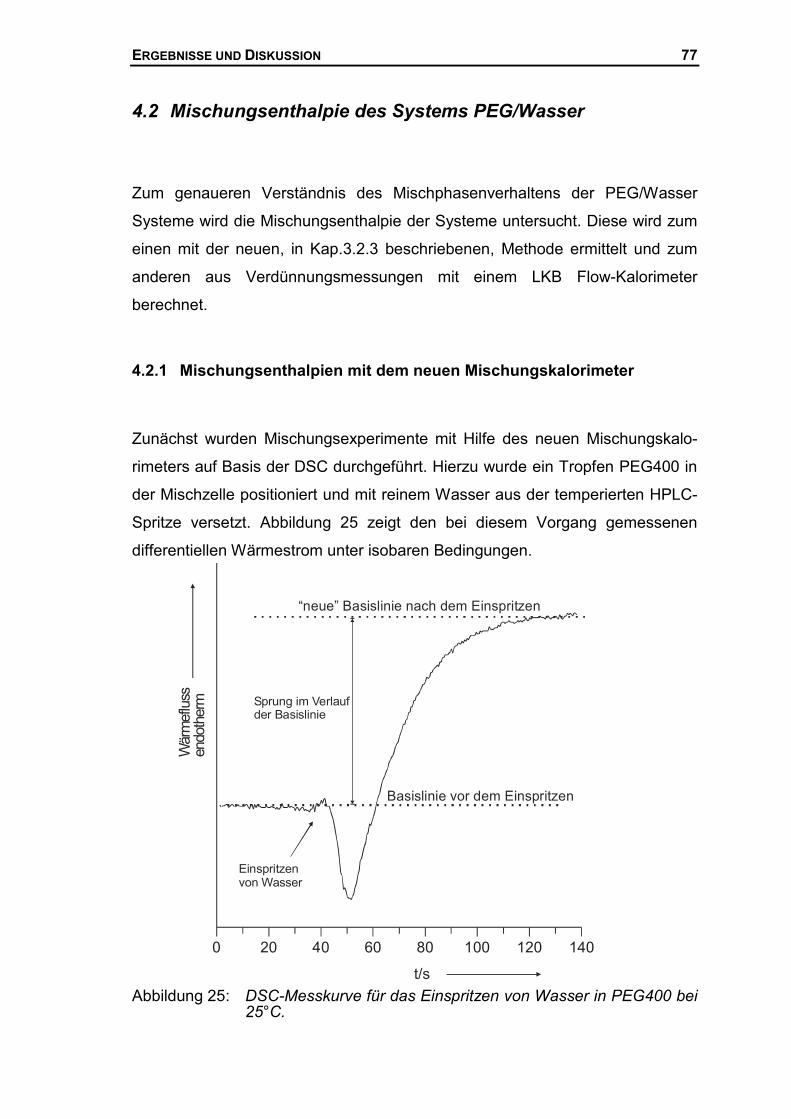
ERGEBNISSE UND DISKUSSION 77
4.2 Mischungsenthalpie des Systems PEG/Wasser
Zum genaueren Verständnis des Mischphasenverhaltens der PEG/Wasser
Systeme wird die Mischungsenthalpie der Systeme untersucht. Diese wird zum
einen mit der neuen, in Kap.3.2.3 beschriebenen, Methode ermittelt und zum
anderen aus Verdünnungsmessungen mit einem LKB Flow-Kalorimeter
berechnet.
4.2.1 Mischungsenthalpien mit dem neuen Mischungskalorimeter
Zunächst wurden Mischungsexperimente mit Hilfe des neuen Mischungskalo-
rimeters auf Basis der DSC durchgeführt. Hierzu wurde ein Tropfen PEG400 in
der Mischzelle positioniert und mit reinem Wasser aus der temperierten HPLC-
Spritze versetzt. Abbildung 25 zeigt den bei diesem Vorgang gemessenen
differentiellen Wärmestrom unter isobaren Bedingungen.
Abbildung 25: DSC-Messkurve für das Einspritzen von Wasser in PEG400 bei25°C.
0 20 40 60 80 100 120 140
t/s
Wär
mef
luss
endo
ther
m
Einspritzen von Wasser
Sprung im Verlauf der Basislinie
Basislinie vor dem Einspritzen
“neue” Basislinie nach dem Einspritzen
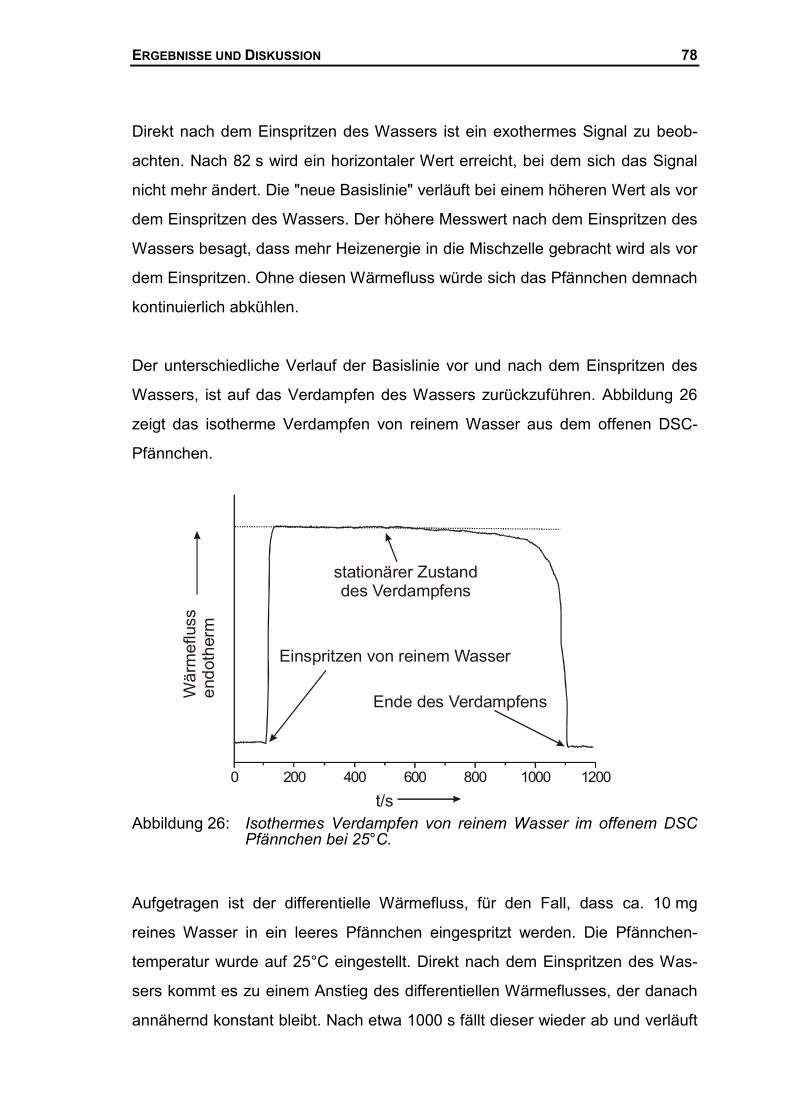
ERGEBNISSE UND DISKUSSION 78
Direkt nach dem Einspritzen des Wassers ist ein exothermes Signal zu beob-
achten. Nach 82 s wird ein horizontaler Wert erreicht, bei dem sich das Signal
nicht mehr ändert. Die "neue Basislinie" verläuft bei einem höheren Wert als vor
dem Einspritzen des Wassers. Der höhere Messwert nach dem Einspritzen des
Wassers besagt, dass mehr Heizenergie in die Mischzelle gebracht wird als vor
dem Einspritzen. Ohne diesen Wärmefluss würde sich das Pfännchen demnach
kontinuierlich abkühlen.
Der unterschiedliche Verlauf der Basislinie vor und nach dem Einspritzen des
Wassers, ist auf das Verdampfen des Wassers zurückzuführen. Abbildung 26
zeigt das isotherme Verdampfen von reinem Wasser aus dem offenen DSC-
Pfännchen.
Abbildung 26: Isothermes Verdampfen von reinem Wasser im offenem DSCPfännchen bei 25°C.
Aufgetragen ist der differentielle Wärmefluss, für den Fall, dass ca. 10 mg
reines Wasser in ein leeres Pfännchen eingespritzt werden. Die Pfännchen-
temperatur wurde auf 25°C eingestellt. Direkt nach dem Einspritzen des Was-
sers kommt es zu einem Anstieg des differentiellen Wärmeflusses, der danach
annähernd konstant bleibt. Nach etwa 1000 s fällt dieser wieder ab und verläuft
0 200 400 600 800 1000 1200
Einspritzen von reinem Wasser
stationärer Zustand des Verdampfens
Ende des Verdampfens
t/s
Wär
mef
luss
endo
ther
m

ERGEBNISSE UND DISKUSSION 79
auf gleichem Niveau wie vor dem Einspritzen des Wassers. Der Anstieg ist auf
das kontinuierliche Verdampfen des Wassers aus dem Pfännchen zu erklären.
Durch die Phasenumwandlung flüssig-gasförmig des Wassers aus dem Pfänn-
chen in die trockene Glovebox wird dem Mess-Näpfchen die Verdampfungs-
enthalpie entzogen. Würde der zweite Heiz- Steuerkreis dieses nicht durch
einen höheren Wärmefluss erwidern, würde sich das Mess-Näpfchen während
des Verdampfens des Wassers abkühlen.
Während des Verdampfens ändert sich ständig die Oberfläche des Wasser-
tropfens im Mess-Näpfchen. Da diese Fläche immer kleiner wird, nimmt die
Verdampfungsrate immer weiter ab. Hierdurch nimmt auch der differentielle
Wärmefluss immer stärker ab. Am Anfang des Verdampfens ändert sich die
Oberfläche und damit der differentielle Wärmefluss kaum, so dass im Anfangs-
bereich von einem stationären Zustand der Verdampfung gesprochen werden
kann. Dieser stationäre Zustand ist nach dem Einspritzen nach 20 s erreicht. Es
ist möglich diesen Anstieg aufgrund der Verdampfung des Wassers beim
Mischungsexperiment zu berücksichtigen. Durch die Bestimmung der Fläche
zwischen der Basislinie und dem endothermen Verdampfungssignal lässt sich
bei Kenntnis der Masse Wasser die spezifische Verdampfungsenthalpie
bestimmen. Die isotherme Verdampfungsenthalpie kann bei verschiedenen
Temperaturen bestimmt werden, wobei die Reproduzierbarkeit bei höheren
Temperaturen (T > 50°C) immer geringer wird, was auf ein zu rasches Ver-
dampfen des Wassers zurückzuführen ist. Neben der Verdampfungsenthalpie
reiner Flüssigkeiten kann auch die Überführungsenthalpie von Wasser (oder
anderen Lösemitteln) aus Mischungen bestimmt werden. Da Wasser in diesem
Fall aus der Mischung in den reinen Dampf überführt wird, spricht man in die-
sem Fall nicht von der Verdampfungsenthalpie, sondern von einer Überfüh-
rungsenthalpie88. Diese beinhaltet auch noch die Mischungsenthalpie des ent-
sprechenden Systems. Mischungsenthalpien sind gegenüber Verdampfungs-
wärmen generell sehr klein, daher kann diese in erster Näherung vernachlässigt
werden.
88 Borchard W., Jablonski P.; Dechema Forschungsbericht, 2000, 157
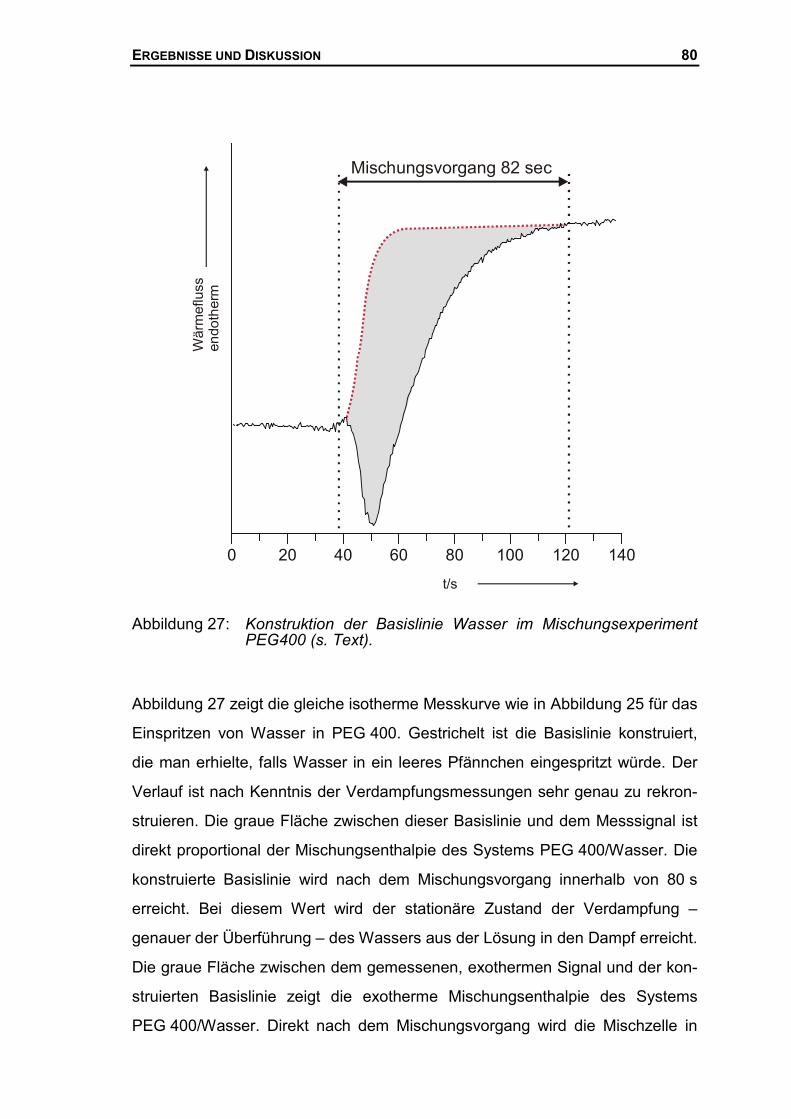
ERGEBNISSE UND DISKUSSION 80
Abbildung 27: Konstruktion der Basislinie Wasser im MischungsexperimentPEG400 (s. Text).
Abbildung 27 zeigt die gleiche isotherme Messkurve wie in Abbildung 25 für das
Einspritzen von Wasser in PEG 400. Gestrichelt ist die Basislinie konstruiert,
die man erhielte, falls Wasser in ein leeres Pfännchen eingespritzt würde. Der
Verlauf ist nach Kenntnis der Verdampfungsmessungen sehr genau zu rekron-
struieren. Die graue Fläche zwischen dieser Basislinie und dem Messsignal ist
direkt proportional der Mischungsenthalpie des Systems PEG 400/Wasser. Die
konstruierte Basislinie wird nach dem Mischungsvorgang innerhalb von 80 s
erreicht. Bei diesem Wert wird der stationäre Zustand der Verdampfung –
genauer der Überführung – des Wassers aus der Lösung in den Dampf erreicht.
Die graue Fläche zwischen dem gemessenen, exothermen Signal und der kon-
struierten Basislinie zeigt die exotherme Mischungsenthalpie des Systems
PEG 400/Wasser. Direkt nach dem Mischungsvorgang wird die Mischzelle in
0 20 40 60 80 100 120 140
t/s
Wär
mef
luss
endo
ther
m
Mischungsvorgang 82 sec

ERGEBNISSE UND DISKUSSION 81
ein verschlossenes Glas gegeben. In Abbildung 26 wird gezeigt, dass ein
Tropfen von ca. 10 mg innerhalb von 1000 s komplett verdampft. Innerhalb der
Mischzeit ändert sich somit die Konzentration der Lösung nur sehr geringfügig.
Nach dem Mischen im Kalorimeter wird außerhalb der DSC mit Hilfe einer
Mikrowaage die Masse der Lösung bestimmt. Die Konzentration wird mit Hilfe
eines Abbé-Refraktometers bestimmt. Hierzu wurde zuvor eine Kalibrierkurve
erstellt, in der der Brechungsindex in Abhängigkeit von der Zusammensetzung
aufgetragen ist. Es zeigte sich bei den verschiedenen Systemen, Glykol/Wasser
(Kalibrierung) und PEG400/Wasser ein linearer Zusammenhang zwischen Bre-
chungsindex und Konzentration der Lösung, der mit folgender Formel beschrei-
ben werden kann89:
)2Komponente(nw)Wasser(nw)Lösung(n 20D2
20D1
20D ���� (49)
mit:
wi Massenbruch vom Wasser (i = 1) bzw. von der zweiten Kom-
ponente (i = 2).20Dn (x) Brechungsindex der Lösung, des Wassers bzw. der Komponente
2 (PEG400) bei 20°C.
Nach der Massen- und Konzentrationsbestimmung können folgende Werte für
die spezifische integrale Mischungsenthalpie des Systems PEG400/Wasser
angegeben werden, die mit Hilfe des neuen Mischungskalorimeters bestimmt
worden sind:
89 Die Brechungsindex-Kurven sind in Anhang 6.1 abgebildet.

ERGEBNISSE UND DISKUSSION 82
Tabelle 1: Ermittelte Integrale Mischungsenthalpien des SystemsPEG400/Wasser bei verschiedenen Konzentrationen.
Konzentration der Lösung (w2) Integrale Mischungsenthalpie [J/g]
0,52 -54,0
0,55 -55,0
0,57 -54,7
Der absolute Fehler der Messmethode liegt bei � 3 J/g. Es zeigt sich, dass im
gemessenen Konzentrationsbereich die Mischungsenthalpien vom System
Ethylenglykol/Wasser zum System PEG400/Wasser deutlich exothermer wer-
den. Dieser Trend lässt sich auch in der Arbeit von Könneke et al. ablesen, in
der Mischungsenthalpien der Systeme Ethylenglykol/Wasser, Diethylen-
glykol/Wasser und Triethylenglykol/Wasser gemessen wurden90. Mit zuneh-
mender Kettenlänge wird die Mischungsenthalpie stärker exotherm (s.
Abbildung 29 undAbbildung 33).
Eine mögliche Fehlerquelle bei der Bestimmung der Mischungsenthalpie ist die
ungenaue Bestimmung der Basislinie in den Messkurven. Dieses Problem wird
umgangen, indem nicht mehr Wasser in PEG400 gespritzt wird, sondern Was-
ser vorgelegt wird und PEG400 eingespritzt wird.
90 Könneke H.G., Steinert H., Leibnitz E., Z. Phys. Chem., 208, 1957, 147
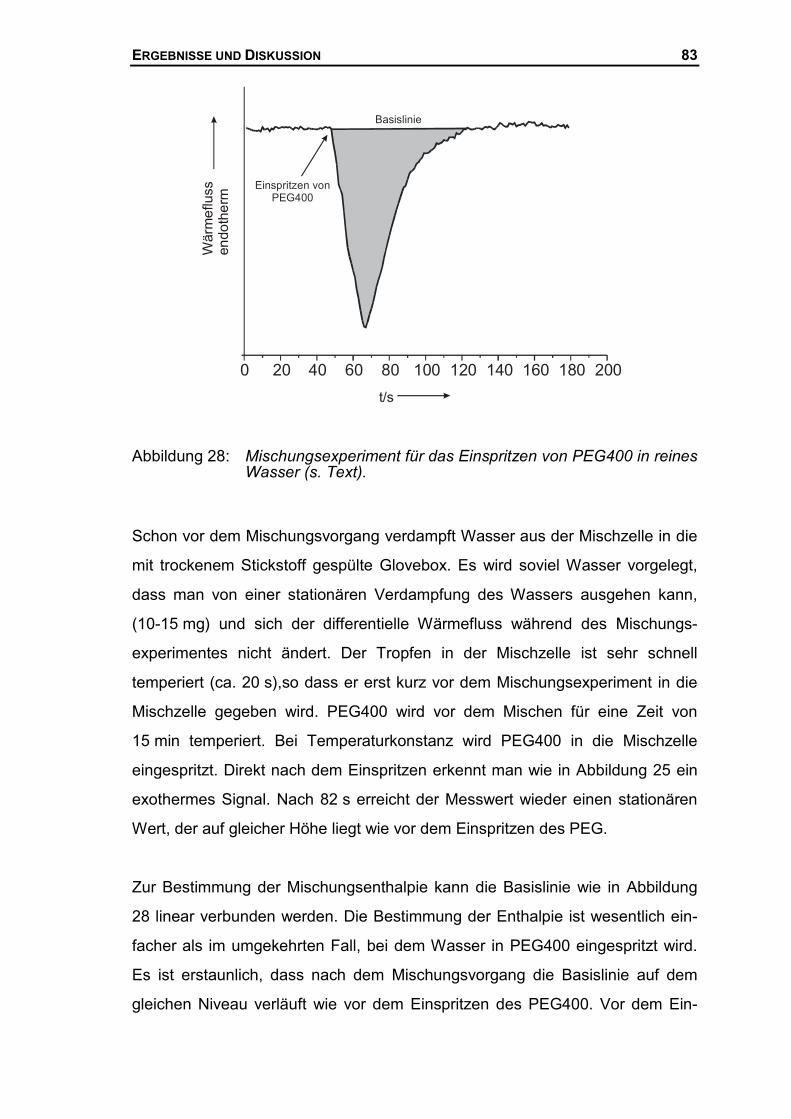
ERGEBNISSE UND DISKUSSION 83
Abbildung 28: Mischungsexperiment für das Einspritzen von PEG400 in reinesWasser (s. Text).
Schon vor dem Mischungsvorgang verdampft Wasser aus der Mischzelle in die
mit trockenem Stickstoff gespülte Glovebox. Es wird soviel Wasser vorgelegt,
dass man von einer stationären Verdampfung des Wassers ausgehen kann,
(10-15 mg) und sich der differentielle Wärmefluss während des Mischungs-
experimentes nicht ändert. Der Tropfen in der Mischzelle ist sehr schnell
temperiert (ca. 20 s),so dass er erst kurz vor dem Mischungsexperiment in die
Mischzelle gegeben wird. PEG400 wird vor dem Mischen für eine Zeit von
15 min temperiert. Bei Temperaturkonstanz wird PEG400 in die Mischzelle
eingespritzt. Direkt nach dem Einspritzen erkennt man wie in Abbildung 25 ein
exothermes Signal. Nach 82 s erreicht der Messwert wieder einen stationären
Wert, der auf gleicher Höhe liegt wie vor dem Einspritzen des PEG.
Zur Bestimmung der Mischungsenthalpie kann die Basislinie wie in Abbildung
28 linear verbunden werden. Die Bestimmung der Enthalpie ist wesentlich ein-
facher als im umgekehrten Fall, bei dem Wasser in PEG400 eingespritzt wird.
Es ist erstaunlich, dass nach dem Mischungsvorgang die Basislinie auf dem
gleichen Niveau verläuft wie vor dem Einspritzen des PEG400. Vor dem Ein-
0 20 40 60 80 100 120 140 160 180 200
Einspritzen von PEG400
Basislinie
t/s
Wär
mef
luss
endo
ther
m

ERGEBNISSE UND DISKUSSION 84
spritzen verdampft reines Wasser aus der Mischzelle, nach dem Einspritzen ist
das Messsignal auf das Verdampfen des Wassers aus der PEG400/Wasser-
Lösung zurückzuführen. Das Niveau der Basislinie ist vor und nach dem
Mischen das gleiche, d.h. dass durch das Verdampfen des Wassers vor und
nach dem Mischen der gleiche differentielle Wärmefluss gemessen wird. Da der
Dampfdruck abhängig von der Konzentration ist, und nach dem Mischen die
Konzentration der PEG400/Wasser-Lösung bei ca. 50 Gew.-% liegt, sollte der
Dampfdruck des Wassers entsprechend herabgesetzt werden und sich damit
auch der differentielle Wärmefluss ändern. Die größere Masse im Pfännchen
kann für diese Beobachtung ausschlaggebend sein. Hauptgrund für den glei-
chen dirfferentiellen Wärmefluss ist der Verlauf der Dampfdruckkurve in Abhän-
gigkeit der Zusammensetzung in Polymer-Systemen. Die Dampfdruckkurve
verläuft über einen großen konzentrationsbereich annähernd horizontal, was
bedeutet, dass der Dampfdruck nicht von der Konzentration abhängig ist. Durch
die größere Masse liegt auch ein größerer Tropfen Lösung vor als vor dem
Mischen. Bei einem reinen Wassertropfen ist zu beobachten, dass die
Verdampfungsrate von der Oberfläche des Wassertropfens abhängig ist. Durch
die Vergrößerung der Masse im Pfännchen und der damit verbundenen größe-
ren Oberfläche des Flüssigkeitstropfens sollte mehr Wasser verdampfen kön-
nen. Durch die Herabsetzung des Dampfdruckes des Wassers in der Lösung
verdampft hingegen weniger Wasser aus der Lösung. Diese beiden Effekte
scheinen sich gegenseitig zu kompensieren, so dass der differentielle
Wärmestrom für das Verdampfen des reinen Wassers der gleiche ist, wie der
für das Überführen des Wassers aus der Lösung in den Dampf. Der Dampf-
druck ist in komplizierter Weise noch von weiteren Faktoren abhängig, auf die
an dieser Stelle nicht genauer eingegangen werden soll91. Das Phänomen, dass
der differentielle Wärmefluss vor und nach dem Mischen der gleiche ist, wurde
auch bei dem Kalibriersystem Ethylenglykol/Wasser gefunden (s. Kap.3.2.3).
91 Tomlinson A., Chylewski Ch., Simon W., Tetrahedron, 19, 1963, 949

ERGEBNISSE UND DISKUSSION 85
Nach der Bestimmung der Masse und der Konzentration nach dem Mischungs-
experiment kann auch für diesen Fall die spezifische integrale Mischungs-
enthalpie angegeben werden.
Tabelle 2: Mischungsenthalpien des Systems PEG400/Wasser. Bestimmt durchEinspritzen von PEG400 in Wasser bei 25°C.
Konzentration der Lösung (w2) Integrale Mischungsenthalpie [J/g]
0,53 -53,7
0,54 -54,8
0,58 -54,0
Ein Vergleich der beiden Methoden zeigt, dass es für die Bestimmung der
Mischungsenthalpie einfacher ist, die flüchtigere Komponente Wasser vorzu-
legen und die weniger flüchtige Komponente – Ethylenglykol oder PEG400 –
mit Hilfe des Mischungsaufsatzes einzuspritzen. Die Basislinie kann in diesem
Fall einfach horizontal verbunden werden, was die Flächenbestimmung extrem
vereinfacht. Im ersten Fall muss zunächst die Basislinie konstruiert werden. Bei
der Messung anderer Systeme muss der Verlauf der Basislinie erneut kalibriert
werden. Ein Vergleich der beiden Verfahren zeigt, dass die bestimmten
Mischungsenthalpien sehr genau übereinstimmen, so dass beide Methoden
angewendet werden können. Die Methode, bei der Wasser eingespritzt wird,
hat den Nachteil, dass die Basislinienkonstruktion aufwendiger ist. Ein Vorteil
dieser Methode ist, dass die zu mischenden Massen exakter bestimmt werden
können, da man sowohl die vorgelegte Masse als auch die Masse Wasser, die
eingespritzt wird, relativ genau abschätzen kann. Auf diese Weise kann schon
im Vorfeld bestimmt werden, in welchem Konzentrationsbereich die Mischungs-
enthalpie des Systems bestimmt wird. Prinzipiell könnte auf diese Weise auch
die spätere Bestimmung der Konzentration mit Hilfe des Brechungsindex ver-
zichtet werden.
Bei der zweiten Methode, wird die Basislinie lediglich linear verbunden. Ein
Nachteil dieser Methode ist das Verdampfen des Wassers aus der Mischzelle

ERGEBNISSE UND DISKUSSION 86
vor dem Mischungsexperiment. Es wird ca. 10 – 15 mg Wasser vorgelegt und
mit einer entsprechenden Menge PEG400 gemischt. Vor dem Mischen der bei-
den Komponenten verdampft das Wasser, was dazu führt, dass die Masse
Wasser, die mit dem PEG400 gemischt wird, nur noch sehr ungenau abge-
schätzt werden kann. Durch die spätere Konzentrations- und Massenbestim-
mung der Mischung ist die genaue Kenntnis der Massen vor dem Mischungs-
experiment nicht relevant.
Bei den Messungen in dieser Arbeit hat sich erstaunlicher Weise gezeigt, dass
das Verdampfen des reinen Wassers aus der Mischzelle annähernd den selben
differentiellen Wärmefluss aufweist wie das Verdampfen des Wassers aus der
Lösung nach dem Mischungsexperiment und somit die Basislinie linear verbun-
den werden kann. Bei anderen Systemen ist es fraglich, ob diese Linearität
gegeben ist, oder die Verdampfung nach dem Mischen einen anderen differen-
tiellen Wärmefluss in der DSC verursacht. Wenn nach dem Mischen ein ande-
rer Wert für das stationäre Verdampfen einer Komponente gefunden wird, muss
die Basislinie in komplizierterer Form konstruiert werden. In diesem Fall muss
zumindest die Konzentrationsabhängigkeit des Dampfdrucks berücksichtigt
werden. Weiterhin müsste die Verdampfungsgeschwindigkeit in Abhängigkeit
von der Oberfläche (der Masse) der Mischung berücksichtigt werden. Dadurch
würde die Basislinienkonstruktion aufwendiger. Nach einer Kalibrierung wie bei
der beschriebenen Methode des Einspritzens von Wasser in PEG400 können
aber auch solche Abhängigkeiten berücksichtigt werden.
Bei der Bestimmung von Mischungsenthalpien mit Hilfe der hier neu beschrie-
benen Methode ist die Verwendung von nicht-flüchtigen Substanzen eine
grundlegende Voraussetzung. Bei den Messungen in dieser Arbeit konnte
gezeigt werden, dass die Mischungsenthalpien wässriger PEG-Systeme mit
hinreichender Genauigkeit bestimmt werden können. Die Flüchtigkeit des Was-
sers kann bei der Kalibrierung und damit bei der Konstruktion der Basislinie
berücksichtigt werden. Bei der Messung von flüchtigen Komponenten werden
die Fehler in der Konzentrationsbestimmung allerdings immer größer, wodurch
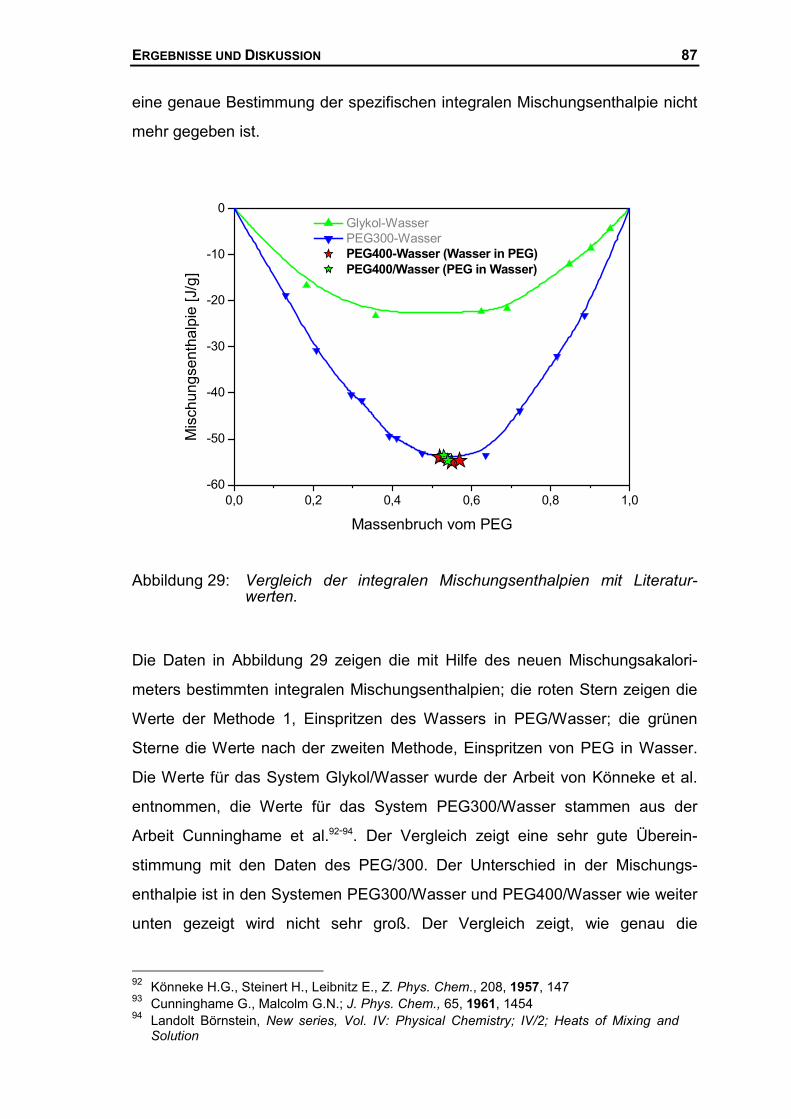
ERGEBNISSE UND DISKUSSION 87
eine genaue Bestimmung der spezifischen integralen Mischungsenthalpie nicht
mehr gegeben ist.
Abbildung 29: Vergleich der integralen Mischungsenthalpien mit Literatur-werten.
Die Daten in Abbildung 29 zeigen die mit Hilfe des neuen Mischungsakalori-
meters bestimmten integralen Mischungsenthalpien; die roten Stern zeigen die
Werte der Methode 1, Einspritzen des Wassers in PEG/Wasser; die grünen
Sterne die Werte nach der zweiten Methode, Einspritzen von PEG in Wasser.
Die Werte für das System Glykol/Wasser wurde der Arbeit von Könneke et al.
entnommen, die Werte für das System PEG300/Wasser stammen aus der
Arbeit Cunninghame et al.92-94. Der Vergleich zeigt eine sehr gute Überein-
stimmung mit den Daten des PEG/300. Der Unterschied in der Mischungs-
enthalpie ist in den Systemen PEG300/Wasser und PEG400/Wasser wie weiter
unten gezeigt wird nicht sehr groß. Der Vergleich zeigt, wie genau die
92 Könneke H.G., Steinert H., Leibnitz E., Z. Phys. Chem., 208, 1957, 14793 Cunninghame G., Malcolm G.N.; J. Phys. Chem., 65, 1961, 145494 Landolt Börnstein, New series, Vol. IV: Physical Chemistry; IV/2; Heats of Mixing and
Solution
0,0 0,2 0,4 0,6 0,8 1,0-60
-50
-40
-30
-20
-10
0 Glykol-Wasser PEG300-Wasser PEG400-Wasser (Wasser in PEG) PEG400/Wasser (PEG in Wasser)
Mis
chun
gsen
thal
pie
[J/g
]
Massenbruch vom PEG

ERGEBNISSE UND DISKUSSION 88
Mischungsenthalpien mit der neuen kalorimetrischen Methode auf Basis der
DSC bestimmt werden können95.
Der größte Vorteil der neuen Methode auf Basis der DSC ist die Verwendung
geringster Massen, welche im Bereich von 10 mg liegen. Soll die Wechsel-
wirkung von beispielsweise pharmazeutischen Wirkstoffen untersucht werden,
stehen oft nur sehr geringe Substanzmengen zur Verfügung. Mit Hilfe der
neuen Methode kann eine exo- oder endotherme Wechselwirkung mit hinrei-
chender Genauigkeit angegeben werden. Durch die eingesetzten geringen
Massen sind die Diffusionswege sehr gering, wodurch eine rasche Mischung
der Komponenten auch ohne zusätzliches Rühren gewährleistet ist. Häufig
treten vor allem bei Verwendung größerer Substanzmengen Probleme beim
Mischvorgang auf. Diese Probleme sind häufig auf die polymerspezifische,
hohe Viskosität zurückzuführen96,97. Hier ist ein weiterer Vorteil des neuen
Mikrokalorimeters hervorzuheben. Durch die Verwendung kleinster Substanz-
mengen und der damit verbundenen kurzen Diffusionswege resultiert eine sehr
kurze Mischzeit. Bei kommerziellen Geräten sind die Massen um den Faktor
100 größer. Zweifelsfrei ist die Genauigkeit dieser Geräte wesentlich größer,
was aber vor allem auf die Verwendung der größeren Massen zurückgeführt
werden kann.
Neben Mischungsenthalpien lassen sich mit Hilfe der neuen Methode auch
Reaktionsenthalpien bestimmen. So kann eine reaktionsfähige Mischung oder
Substanz im Mikrogramm-Bereich (z.B. Acrylsäure) vorgelegt werden, wonach
ein Katalysator (z.B. Radikalstarter) mit dem Spritzenaufsatz hinzugefügt wird.
Neben der Bestimmung von Mischungs- und Reaktionsenthalpien ist es – da
mit einem offenen System gearbeitet wird – möglich, die spezifischen Wärme-
kapazitäten von reinen Substanzen oder Mischungen zu bestimmen. Diese Art
der Bestimmung der spezifischen Wärmekapazität, bei der eine bekannte Pro-
95 Jablonski P., Müller-Blecking A., Borchard W.; J. Therm. Anal. Cal., eingereicht96 Siemens W.; Dissertation Clausthal-Zellerfeld, 197697 Schönert H., Monshausen F.; Col. Pol. Sci., 258, 1980, 578

ERGEBNISSE UND DISKUSSION 89
benmasse auf eine messbare Temperatur erwärmt und anschließend in ein
„Kalorimeterbad“ eingebracht wird, geht auf Oelsen zurück98,99. In dem Diffe-
rentialkalorimeter wird die Energie direkt messbar, die erforderlich ist, um wie-
der isotherme Bedingungen herzustellen.
4.2.2 Mischungsenthalpien mit dem LKB-Flow Kalorimeter
Zusätzlich sind noch Mischungsenthalpien des Systems PEG/Wasser mit einem
kommerziellen Flow-Kalorimeter gemessen worden. Wie in Kap. 3.3 gezeigt
wurde, werden mit Hilfe einer peristaltischen Pumpe zwei Flüssigkeiten durch
eine Mischzelle gepumpt. Es können gleiche Volumina von zwei Flüssigkeiten
in der Mischzelle gemischt werden, wodurch die Mischungsenthalpie bestimmt
werden kann. Es wurden direkt die integralen Mischungsenthalpien der
PEG/Wasser Systeme mit den molaren Massen des PEG von 200, 300 und
400 g/mol gemessen. Durch die gleiche Pumprate für das Wasser und das PEG
erhielt man stets eine Mischung der Zusammensetzung w2 = 0,5. Polyethylen-
glykole mit höheren Massen konnten nicht mehr gemessen werden, da PEG600
bereits einen Schmelzpunkt über Raumtemperatur besitzt. Von den höheren
molaren Massen wurden Verdünnungsenthalpien im Konzentrationsbereich
0 < w2 < 0,5 durchgeführt, wobei verschiedene PEG/Wasser-Lösungen mit
reinem Wasser gemischt wurden (s. Kap. 3.3).
98 Oelsen, W., Rieskamp, K.H., Oelsen, O.; Archiv f. Eisenhüttenwesen, 26, 1955, 25399 Oelsen, W., Tebbe, W., Oelsen, O.; Archiv f. Eisenhüttenwesen, 27, 1956, 689
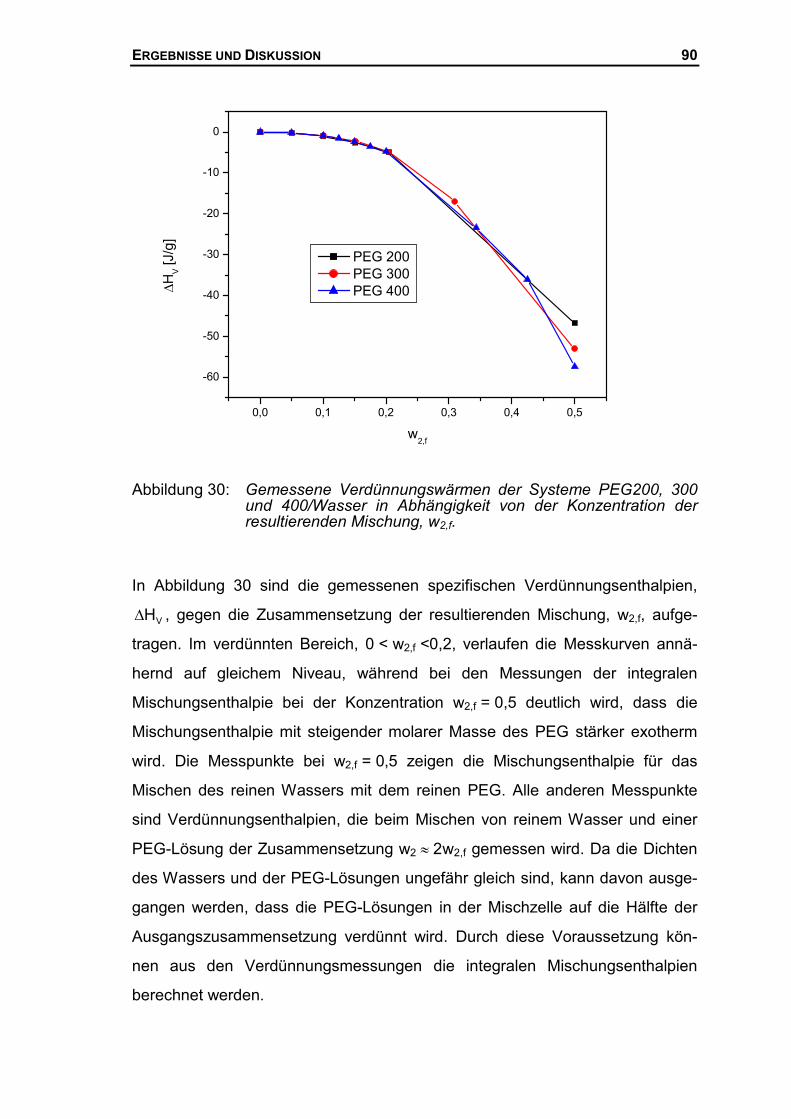
ERGEBNISSE UND DISKUSSION 90
Abbildung 30: Gemessene Verdünnungswärmen der Systeme PEG200, 300und 400/Wasser in Abhängigkeit von der Konzentration derresultierenden Mischung, w2,f.
In Abbildung 30 sind die gemessenen spezifischen Verdünnungsenthalpien,
VH� , gegen die Zusammensetzung der resultierenden Mischung, w2,f, aufge-
tragen. Im verdünnten Bereich, 0 < w2,f <0,2, verlaufen die Messkurven annä-
hernd auf gleichem Niveau, während bei den Messungen der integralen
Mischungsenthalpie bei der Konzentration w2,f = 0,5 deutlich wird, dass die
Mischungsenthalpie mit steigender molarer Masse des PEG stärker exotherm
wird. Die Messpunkte bei w2,f = 0,5 zeigen die Mischungsenthalpie für das
Mischen des reinen Wassers mit dem reinen PEG. Alle anderen Messpunkte
sind Verdünnungsenthalpien, die beim Mischen von reinem Wasser und einer
PEG-Lösung der Zusammensetzung w2 � 2w2,f gemessen wird. Da die Dichten
des Wassers und der PEG-Lösungen ungefähr gleich sind, kann davon ausge-
gangen werden, dass die PEG-Lösungen in der Mischzelle auf die Hälfte der
Ausgangszusammensetzung verdünnt wird. Durch diese Voraussetzung kön-
nen aus den Verdünnungsmessungen die integralen Mischungsenthalpien
berechnet werden.
0,0 0,1 0,2 0,3 0,4 0,5
-60
-50
-40
-30
-20
-10
0
PEG 200 PEG 300 PEG 400�
HV [
J/g]
w2,f
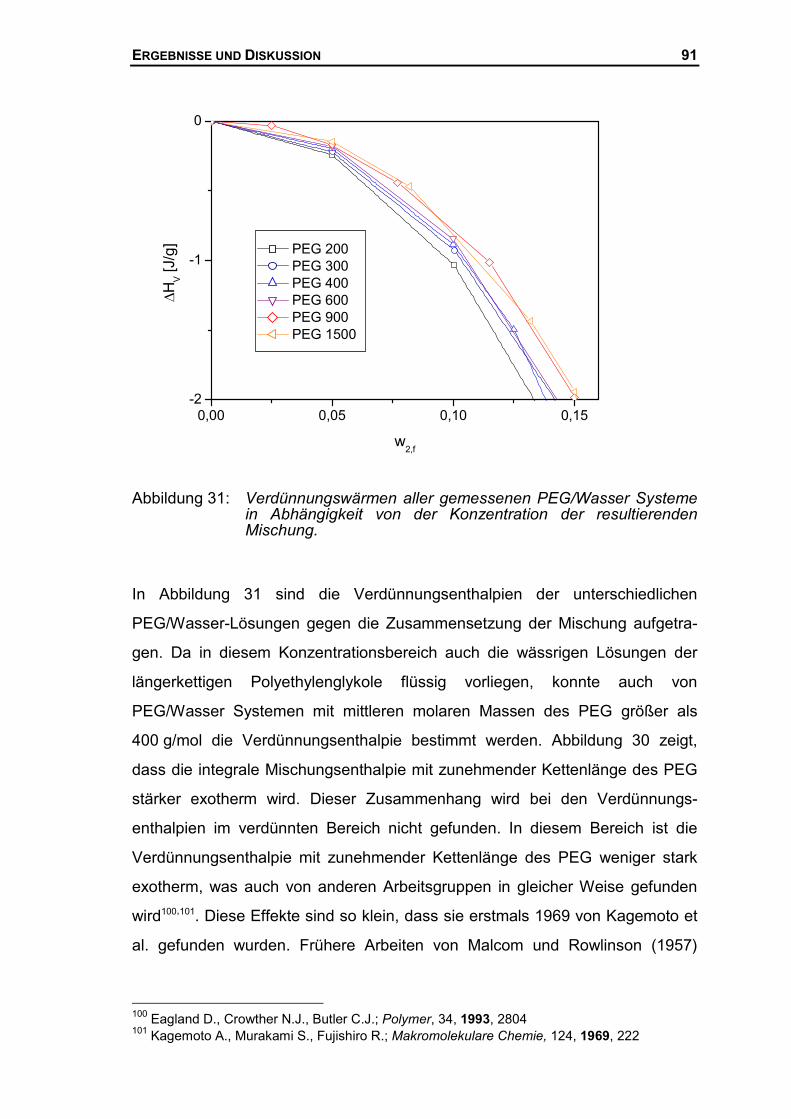
ERGEBNISSE UND DISKUSSION 91
Abbildung 31: Verdünnungswärmen aller gemessenen PEG/Wasser Systemein Abhängigkeit von der Konzentration der resultierendenMischung.
In Abbildung 31 sind die Verdünnungsenthalpien der unterschiedlichen
PEG/Wasser-Lösungen gegen die Zusammensetzung der Mischung aufgetra-
gen. Da in diesem Konzentrationsbereich auch die wässrigen Lösungen der
längerkettigen Polyethylenglykole flüssig vorliegen, konnte auch von
PEG/Wasser Systemen mit mittleren molaren Massen des PEG größer als
400 g/mol die Verdünnungsenthalpie bestimmt werden. Abbildung 30 zeigt,
dass die integrale Mischungsenthalpie mit zunehmender Kettenlänge des PEG
stärker exotherm wird. Dieser Zusammenhang wird bei den Verdünnungs-
enthalpien im verdünnten Bereich nicht gefunden. In diesem Bereich ist die
Verdünnungsenthalpie mit zunehmender Kettenlänge des PEG weniger stark
exotherm, was auch von anderen Arbeitsgruppen in gleicher Weise gefunden
wird100,101. Diese Effekte sind so klein, dass sie erstmals 1969 von Kagemoto et
al. gefunden wurden. Frühere Arbeiten von Malcom und Rowlinson (1957)
100 Eagland D., Crowther N.J., Butler C.J.; Polymer, 34, 1993, 2804101 Kagemoto A., Murakami S., Fujishiro R.; Makromolekulare Chemie, 124, 1969, 222
0,00 0,05 0,10 0,15-2
-1
0
PEG 200 PEG 300 PEG 400 PEG 600 PEG 900 PEG 1500
�H
V [J/
g]
w2,f
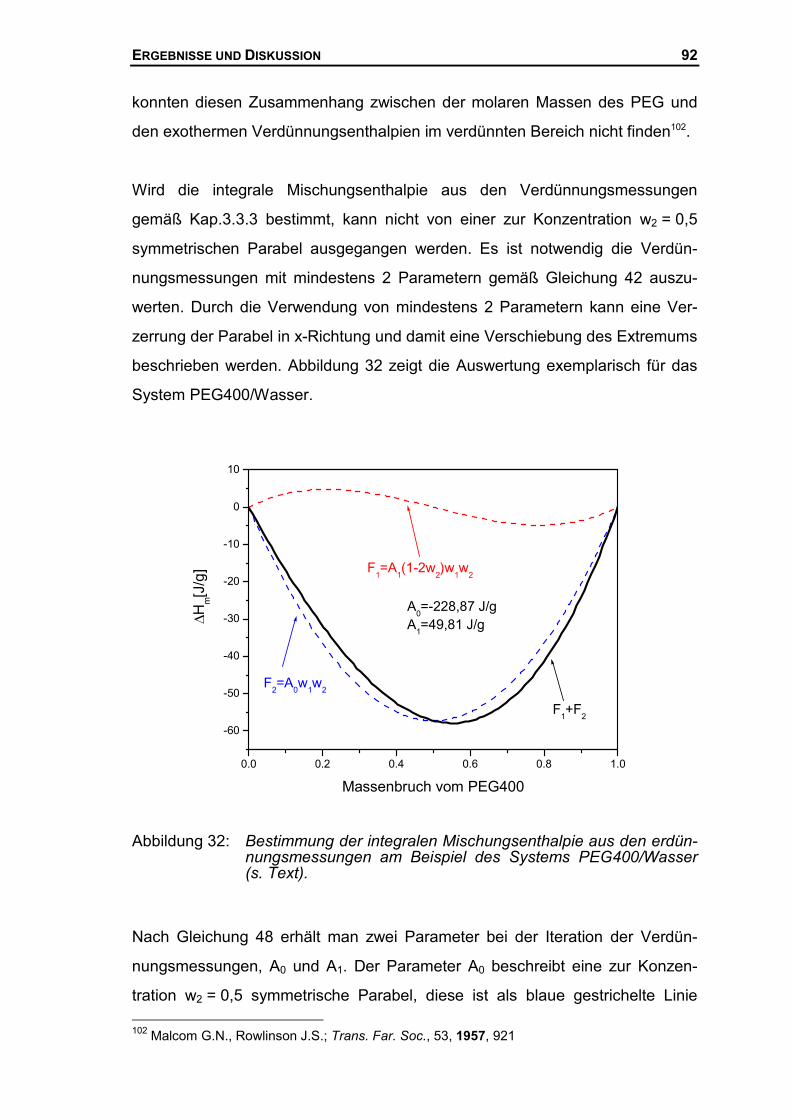
ERGEBNISSE UND DISKUSSION 92
konnten diesen Zusammenhang zwischen der molaren Massen des PEG und
den exothermen Verdünnungsenthalpien im verdünnten Bereich nicht finden102.
Wird die integrale Mischungsenthalpie aus den Verdünnungsmessungen
gemäß Kap.3.3.3 bestimmt, kann nicht von einer zur Konzentration w2 = 0,5
symmetrischen Parabel ausgegangen werden. Es ist notwendig die Verdün-
nungsmessungen mit mindestens 2 Parametern gemäß Gleichung 42 auszu-
werten. Durch die Verwendung von mindestens 2 Parametern kann eine Ver-
zerrung der Parabel in x-Richtung und damit eine Verschiebung des Extremums
beschrieben werden. Abbildung 32 zeigt die Auswertung exemplarisch für das
System PEG400/Wasser.
Abbildung 32: Bestimmung der integralen Mischungsenthalpie aus den erdün-nungsmessungen am Beispiel des Systems PEG400/Wasser(s. Text).
Nach Gleichung 48 erhält man zwei Parameter bei der Iteration der Verdün-
nungsmessungen, A0 und A1. Der Parameter A0 beschreibt eine zur Konzen-
tration w2 = 0,5 symmetrische Parabel, diese ist als blaue gestrichelte Linie 102 Malcom G.N., Rowlinson J.S.; Trans. Far. Soc., 53, 1957, 921
0.0 0.2 0.4 0.6 0.8 1.0
-60
-50
-40
-30
-20
-10
0
10
A0=-228,87 J/gA1=49,81 J/g
F1+F2
F2=A0w1w2
F1=A1(1-2w2)w1w2
�H
m[J
/g]
Massenbruch vom PEG400

ERGEBNISSE UND DISKUSSION 93
abgebildet und stellt die Funktion F1 dar. Der Parameter A1 beschreibt die
Abweichungen von dieser Parabel (rote gestrichelte Linie F2) und sorgt dafür,
dass die resultierende Kurve (schwarze durchgezogene Linie) für die integrale
Mischungsenthalpie nicht mehr symmetrisch ist. Die schwarze Kurve ist die
Summe aus den beiden Funktionen F1 und F2. Diese Auswertung wurde für alle
Verdünnungsmessungen durchgeführt. Die Parameter A0 und A1 sind in
Abhängigkeit von der Kettenlänge des PEG in Tab.2 aufgeführt. Die Kurve für
die resultierende integrale Mischungsenthalpie ist im wesentlichen durch den
Parameter A0 bestimmt. Der Einfluss von A1 ist wesentlich geringer.
Tabelle 3: Parameter A0 und A1 zur Beschreibung der integralen Mischungs-enthalpie aus Verdünnungsmessungen.
M(PEG) [g/mol] 200 300 400 600 900 1500
A0 -186 -210 -228 -222 -196 -168
A1 37 41 49 58 51 37
An den Werten in Tabelle 2 wird deutlich, dass bis zum System PEG400 Was-
ser die Werte für A0 abnehmen, was bedeutet, dass die maximale Mischungs-
enthalpie in diesen Systemen im mittleren Konzentrationsbereich immer stärker
exotherm wird. Der Wert für A1 wird geringfügig größer, wodurch berücksichtigt
wird, dass im verdünnten Bereich die Verdünnungsenthalpie weniger stark
exotherm mit steigender Kettenlänge wird. Die Werte für A0 und A1 bei den
Systemen mit höheren molaren Massen (M > 400 g/mol) weichen von diesem
Trend ab. Die Werte für A0 werden wieder größer und die Werte für A1 werden
kleiner. Die Werte in diesen Systemen sind allerdings wenig aussagekräftig, da
bei diesen Systemen lediglich bis zu einer Konzentration der Mischung von
w2 = 0,25 gemessen wurde. Bei der Auswertung nach Gleichung 48 wird so der
verdünnte Bereich wesentlich stärker berücksichtigt. Der mittlere Bereich wird
allerdings nicht mehr berücksichtigt. Die Kurven lassen sich nur noch im extrem
verdünnten Bereich auswerten. Eine Extrapolation über den gesamten Konzen-
trationsbereich ist bei diesen Systemen nicht möglich.

ERGEBNISSE UND DISKUSSION 94
Selbst mit anderen Drop-Kalorimetern, ist eine direkte Messung der
Mischungsenthalpie bei längerkettigen PEG/Wasser Systemen mit Schwierig-
keiten verbunden. Bei Raumtemperatur liegt PEG als Feststoff vor. Bei einem
Mischungsexperiment gleicher Massen PEG und Wasser überlagert sich die
Schmelzenthalpie des PEG mit der Mischungsenthalpie, so dass die Schmelz-
enthalpie des PEG zuvor separat mit Hilfe einer DSC-Messung bestimmt wer-
den muss. Beim PEG1500 liegt eine Schmelzenthalpie von 170 J/g vor, gleich-
zeitig kann die Mischungsenthalpie mit ca. –60 J/g angegeben werden. Bei der
Überführung des reinen PEG aus dem Kristall in die flüssige PEG/Wasser-
Lösung wird demnach eine endotherme Überführungsenthalpie gefunden. Bei
der genauen Bestimmung der Mischungsenthalpie in diesem System muss die
Temperaturabhängigkeit der Schmelzenthalpie des reinen PEG mit berücksich-
tigt werden.
Aus diesen Gründen wurden aus den Verdünnungsmessungen die integralen
Mischungsenthalpien nur von den PEG/Wasser-Systemen mit einer molaren
Masse des PEG < 600 g/mol ausgewertet.
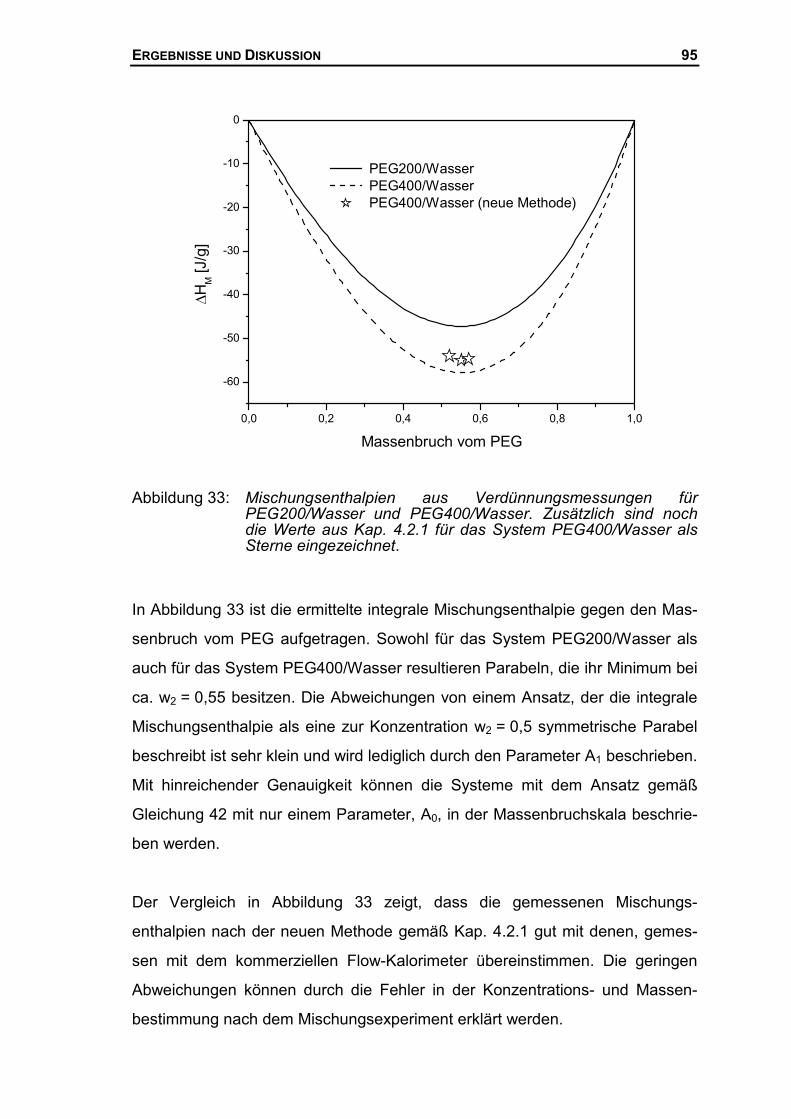
ERGEBNISSE UND DISKUSSION 95
Abbildung 33: Mischungsenthalpien aus Verdünnungsmessungen fürPEG200/Wasser und PEG400/Wasser. Zusätzlich sind nochdie Werte aus Kap. 4.2.1 für das System PEG400/Wasser alsSterne eingezeichnet.
In Abbildung 33 ist die ermittelte integrale Mischungsenthalpie gegen den Mas-
senbruch vom PEG aufgetragen. Sowohl für das System PEG200/Wasser als
auch für das System PEG400/Wasser resultieren Parabeln, die ihr Minimum bei
ca. w2 = 0,55 besitzen. Die Abweichungen von einem Ansatz, der die integrale
Mischungsenthalpie als eine zur Konzentration w2 = 0,5 symmetrische Parabel
beschreibt ist sehr klein und wird lediglich durch den Parameter A1 beschrieben.
Mit hinreichender Genauigkeit können die Systeme mit dem Ansatz gemäß
Gleichung 42 mit nur einem Parameter, A0, in der Massenbruchskala beschrie-
ben werden.
Der Vergleich in Abbildung 33 zeigt, dass die gemessenen Mischungs-
enthalpien nach der neuen Methode gemäß Kap. 4.2.1 gut mit denen, gemes-
sen mit dem kommerziellen Flow-Kalorimeter übereinstimmen. Die geringen
Abweichungen können durch die Fehler in der Konzentrations- und Massen-
bestimmung nach dem Mischungsexperiment erklärt werden.
0,0 0,2 0,4 0,6 0,8 1,0
-60
-50
-40
-30
-20
-10
0
PEG200/Wasser PEG400/Wasser PEG400/Wasser (neue Methode)
�H
M [J
/g]
Massenbruch vom PEG
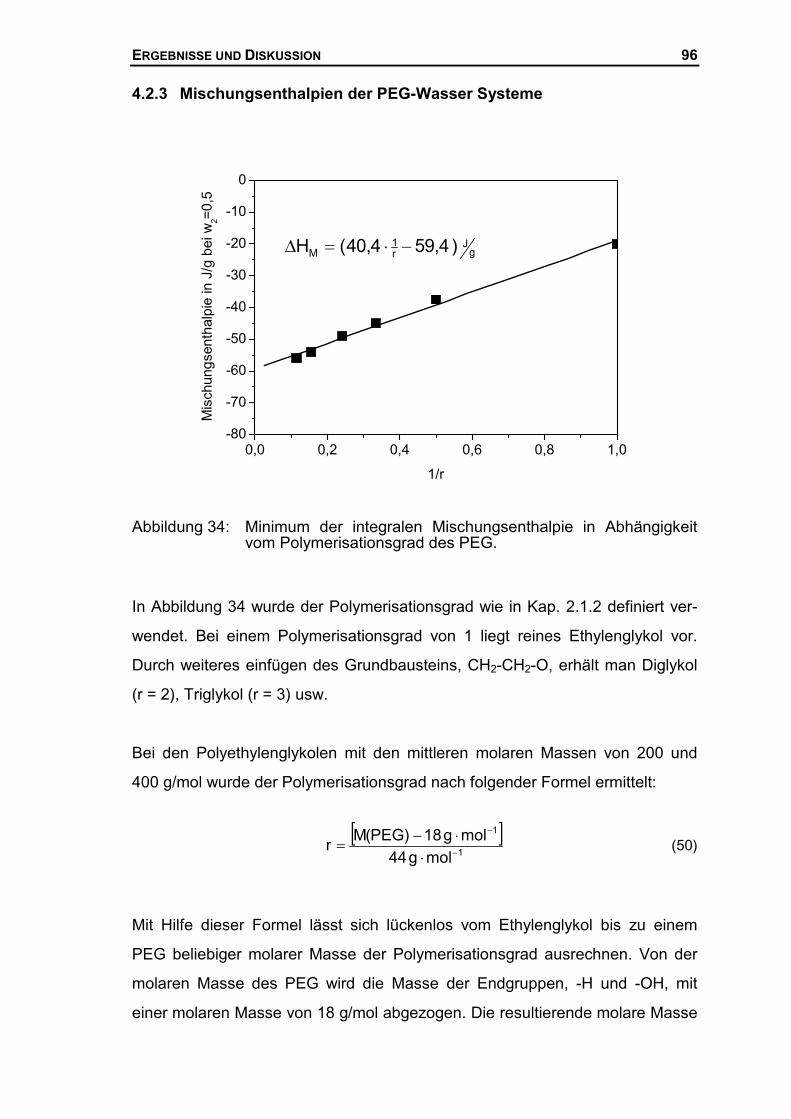
ERGEBNISSE UND DISKUSSION 96
4.2.3 Mischungsenthalpien der PEG-Wasser Systeme
Abbildung 34: Minimum der integralen Mischungsenthalpie in Abhängigkeitvom Polymerisationsgrad des PEG.
In Abbildung 34 wurde der Polymerisationsgrad wie in Kap. 2.1.2 definiert ver-
wendet. Bei einem Polymerisationsgrad von 1 liegt reines Ethylenglykol vor.
Durch weiteres einfügen des Grundbausteins, CH2-CH2-O, erhält man Diglykol
(r = 2), Triglykol (r = 3) usw.
Bei den Polyethylenglykolen mit den mittleren molaren Massen von 200 und
400 g/mol wurde der Polymerisationsgrad nach folgender Formel ermittelt:
� �1
1
molg44molg18)PEG(Mr
�
�
�
��
� (50)
Mit Hilfe dieser Formel lässt sich lückenlos vom Ethylenglykol bis zu einem
PEG beliebiger molarer Masse der Polymerisationsgrad ausrechnen. Von der
molaren Masse des PEG wird die Masse der Endgruppen, -H und -OH, mit
einer molaren Masse von 18 g/mol abgezogen. Die resultierende molare Masse
0,0 0,2 0,4 0,6 0,8 1,0-80
-70
-60
-50
-40
-30
-20
-10
0
gJ
r1
M )4,594,40(H ����
Mis
chun
gsen
thal
pie
in J
/g b
ei w
2=0,
5
1/r

ERGEBNISSE UND DISKUSSION 97
wird durch die des Grundbausteins, 44 g/mol, dividiert. Im Falle des Ethylen-
glykols ergibt sich der Polymerisationsgrad 1. Bei einer molaren Masse des
PEG von 400 g/mol erhält man danach einen Polymerisationsgrad von 8,68.
Beim PEG400 muss von einer Molmassenverteilung ausgegangen werden,
wodurch sich der nicht ganzzahlige Polymerisationsgrad erklären lässt.
Üblich ist es, den Polymerisationsgrad durch den Quotienten aus molarer
Masse des Polymeren und molarer Masse des Grundbausteins zu definieren.
Es wird hier auf die Subtraktion der Endgruppen verzichtet. Bei hohen molaren
Massen des Polymeren sind die Massen der Endgruppen vernachlässigbar.
Behandelt man wie in dieser Arbeit Kettenlängenabhängigkeiten bei sehr
kleinen Polymerisationsgraden müssen die Endgruppen berücksichtigt werden.
Durch die Berücksichtigung der Endgruppen gemäß Gl.(50) kann in Abbildung
34 die Mischungsenthalpie gegen den reziproken Polymerisationsgrad bis zum
Ethylenglykol aufgetragen werden. Für r = 1, Ethylenglykol/Wasser, liegt eine
maximale Mischungsenthalpie von -20 J/g vor, längerkettige Polyethylenglykole
zeigen ein stärker exothermes Mischverhalten. Für unendlich lange PEG-Ket-
ten, 1/r = 0, wird durch Extrapolation eine Mischungsenthalpie von -59,4 J/g
gefunden.
Die Literaturwerte für die Mischungsenthalpie bei einer Temperatur von 25°C
der Systeme Glykol/Wasser, Diethylenglykol/Wasser und Triethylen-
glykol/Wasser103 und die Werte für das System PEG300/Wasser104 zeigen deut-
lich, dass die Mischungsenthalpie mit steigender Kettenlänge stärker exotherm
wird. Könneke et al. tragen die gemessenen Mischungsenthalpien gegen den
Molenbruch des Ethylenglykols, bzw. des Di- und Triethylenglykols, auf. Durch
die Wahl des Molenbruchs als Konzentrationsmaß kommt es zu einer Verzer-
rung der Parabel. Rechnet man die Molenbrüche in die Massenbrüche um,
zeigt sich, dass auch bei diesen Systemen die integrale Mischungsenthalpie
sehr gut durch eine zur Konzentration w2 = 0,5 symmetrische Parabel beschrie- 103 Könnecke H. G., Steinert H., Leibnitz E.; Z. Phys. Chem., 208, 1957, 147104 Cunninghame G., Malcolm G.N.; J. Phys. Chem., 65, 1961, 1454
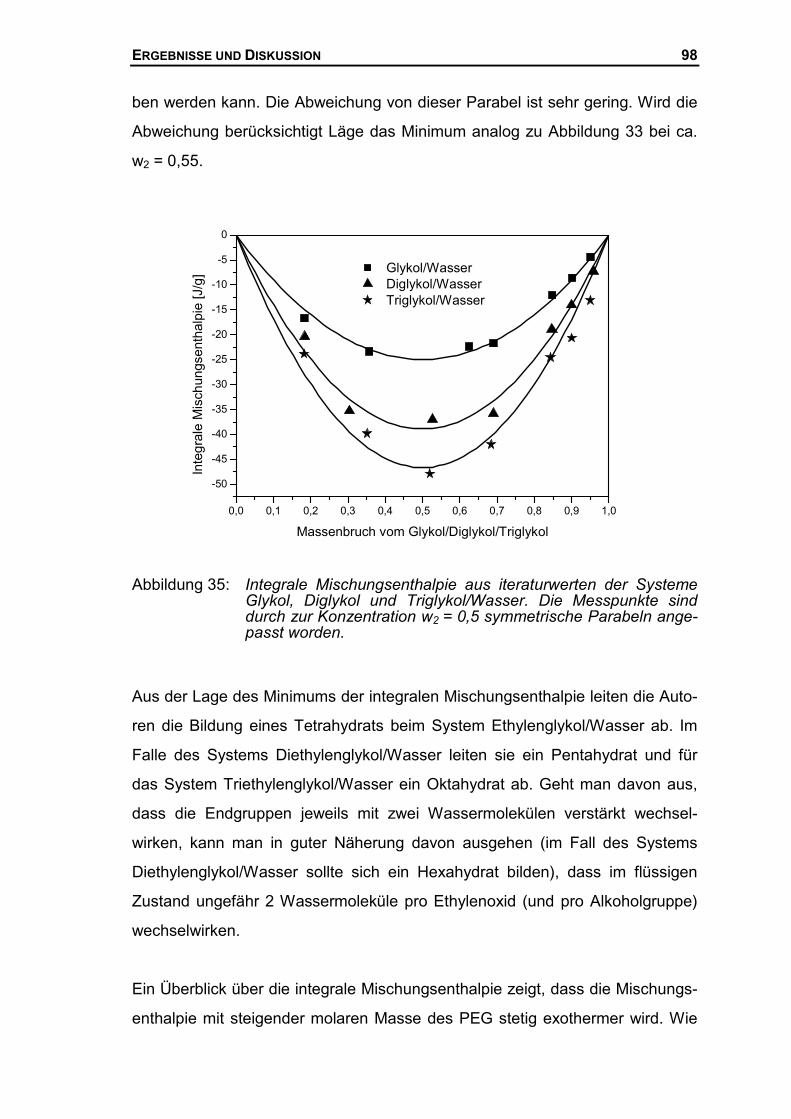
ERGEBNISSE UND DISKUSSION 98
ben werden kann. Die Abweichung von dieser Parabel ist sehr gering. Wird die
Abweichung berücksichtigt Läge das Minimum analog zu Abbildung 33 bei ca.
w2 = 0,55.
Abbildung 35: Integrale Mischungsenthalpie aus iteraturwerten der SystemeGlykol, Diglykol und Triglykol/Wasser. Die Messpunkte sinddurch zur Konzentration w2 = 0,5 symmetrische Parabeln ange-passt worden.
Aus der Lage des Minimums der integralen Mischungsenthalpie leiten die Auto-
ren die Bildung eines Tetrahydrats beim System Ethylenglykol/Wasser ab. Im
Falle des Systems Diethylenglykol/Wasser leiten sie ein Pentahydrat und für
das System Triethylenglykol/Wasser ein Oktahydrat ab. Geht man davon aus,
dass die Endgruppen jeweils mit zwei Wassermolekülen verstärkt wechsel-
wirken, kann man in guter Näherung davon ausgehen (im Fall des Systems
Diethylenglykol/Wasser sollte sich ein Hexahydrat bilden), dass im flüssigen
Zustand ungefähr 2 Wassermoleküle pro Ethylenoxid (und pro Alkoholgruppe)
wechselwirken.
Ein Überblick über die integrale Mischungsenthalpie zeigt, dass die Mischungs-
enthalpie mit steigender molaren Masse des PEG stetig exothermer wird. Wie
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
-50
-45
-40
-35
-30
-25
-20
-15
-10
-5
0
Glykol/Wasser Diglykol/Wasser Triglykol/Wasser
Inte
gral
e M
isch
ungs
enth
alpi
e [J
/g]
Massenbruch vom Glykol/Diglykol/Triglykol

ERGEBNISSE UND DISKUSSION 99
auch bei den Phasendiagrammen, zeigen sich die größten Änderungen im
kurzkettigen Bereich. Schon beim Übergang vom System Glykol/Wasser zu den
Systemen Diglykol/Wasser und Triglykol/Wasser in Abbildung 35 zeigen sich
starke Änderungen bei den integralen Mischungsenthalpien. Diese Änderungen
werden mit zunehmender Kettenlänge immer geringer, was an der Auftragung
in Abbildung 34 zu erkennen ist. Die Daten bezüglich der integralen
Mischungsenthalpie können bei der Wahl des Massenbruchs als Konzentra-
tionsmaß sehr gut durch zur Konzentration w2 = 0,5 symmetrische Parabeln
angepasst werden. Die Übersicht über die Kettenlängenabhängigkeit der inte-
gralen Mischungsenthalpie zeigt deutlich, welche Schwächen der Molenbruch
als Konzentrationsmaß bei Polymer-Systemen aufweist. Wird der Volumen-
oder Grundmolenbruch als Konzentrationsmaß verwendet, sind die Parabeln
nicht mehr symmetrisch, das Minimum liegt aber auch in diesen Diagrammen
immer bei der gleichen Konzentration. Die Mischungsenthalpie des Minimums
kann nach Abbildung 34 nach folgender Formel berechnet werden.
gJ
r1
M )4,594,40(H ���� (51)
4.3 Thermodynamische Beschreibung des Systems aus PEG
und Wasser
Im folgenden Kapitel wird die thermodynamische Beschreibung der
PEG/Wasser-Phasendiagramme behandelt. Nach der Beschreibung der Pha-
sendiagramme in der Massenbruchskala wird ein Vergleich der Daten dieser
Arbeit mit denen anderen Autoren in der Grundmolenbruchskala erfolgen.
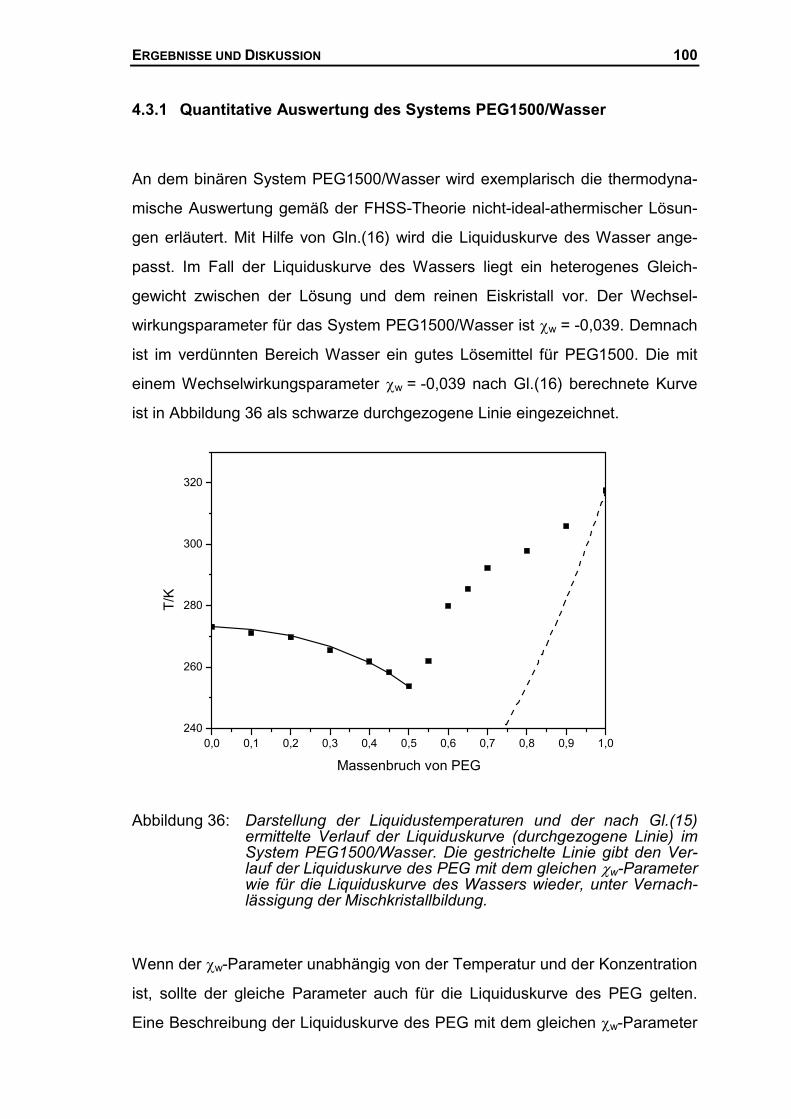
ERGEBNISSE UND DISKUSSION 100
4.3.1 Quantitative Auswertung des Systems PEG1500/Wasser
An dem binären System PEG1500/Wasser wird exemplarisch die thermodyna-
mische Auswertung gemäß der FHSS-Theorie nicht-ideal-athermischer Lösun-
gen erläutert. Mit Hilfe von Gln.(16) wird die Liquiduskurve des Wasser ange-
passt. Im Fall der Liquiduskurve des Wassers liegt ein heterogenes Gleich-
gewicht zwischen der Lösung und dem reinen Eiskristall vor. Der Wechsel-
wirkungsparameter für das System PEG1500/Wasser ist �w = -0,039. Demnach
ist im verdünnten Bereich Wasser ein gutes Lösemittel für PEG1500. Die mit
einem Wechselwirkungsparameter �w = -0,039 nach Gl.(16) berechnete Kurve
ist in Abbildung 36 als schwarze durchgezogene Linie eingezeichnet.
Abbildung 36: Darstellung der Liquidustemperaturen und der nach Gl.(15)ermittelte Verlauf der Liquiduskurve (durchgezogene Linie) imSystem PEG1500/Wasser. Die gestrichelte Linie gibt den Ver-lauf der Liquiduskurve des PEG mit dem gleichen �w-Parameterwie für die Liquiduskurve des Wassers wieder, unter Vernach-lässigung der Mischkristallbildung.
Wenn der �w-Parameter unabhängig von der Temperatur und der Konzentration
ist, sollte der gleiche Parameter auch für die Liquiduskurve des PEG gelten.
Eine Beschreibung der Liquiduskurve des PEG mit dem gleichen �w-Parameter
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0240
260
280
300
320
T/K
Massenbruch von PEG

ERGEBNISSE UND DISKUSSION 101
unter Verwendung der Gl.(17) ergibt die in Abbildung 36 dargestellte gestri-
chelte Kurve, die die Liquiduskurve wegen der Vernachlässigung der Misch-
kristallbildung nicht wiedergeben kann. Weiter unten in diesem Kapitel wird die
Liquiduskurve des PEG unter Berücksichtigung der Mischkristallbildung
thermodynamisch beschrieben.
Die Liquiduskurve des Wassers wird in diesem System sehr genau wiederge-
geben, so dass dazu übergegangen wurde den Wechselwirkungsparameter
gemäß Gl.(24) als temperaturabhängige Größe anzunehmen. Aus der Gleich-
gewichtskurve des Wassers kann die Mischungsenthalpie und –entropie des
Systems bestimmt werden. Eine Auswertung der Liquiduskurve des Wassers im
Systems PEG1500/Wasser liefert für die Größe �w den Wert -2,52 und für �w
den Wert +2,36. Betrachtet man zunächst den Parameter �w, aus dem die
Exzessentropie des Systems nach Gl.(26) berechnet werden kann, fällt auf,
dass sich eine positive Mischungsentropie für das System ergibt. Als nächstes
kann nach Gl.(25) die Mischungsenthalpie des Systems durch den Parameter
�w berechnet werden. Die Mischungsenthalpie des Systems wäre demnach
stark positiv, was auf ein endothermes Mischverhalten schließen ließe. Die
Bestimmung der Mischungsenthalpien in Kap.4.2 zeigten allerdings deutlich,
dass die Mischungsenthalpie eine stark exotherme Größe ist und auf keinen
Fall eine endothermes Mischverhalten vorliegt.
Da die Wechselwirkungsgrößen bei der Wahl eines anderen Konzentrations-
maßes teilweise andere Größen aufweisen, wurde – auch zum besseren Ver-
gleich mit Wechselwirkungsgrößen anderer Autoren – die Auswertung der
Liquiduskurve des Wassers nach Gl.(34) in der Grundmolenbruchskala durch-
geführt. In diesem Fall erhält man für �x den Wert –17,6, und für �x den Wert
+14,8. Auch in diesem Fall liefert die Auswertung Wechselwirkungsgrößen
zweifelhafte Größen für die Mischungsenthalpie und –entropie, auf deren grafi-
sche Auftragung daher verzichtet wird.

ERGEBNISSE UND DISKUSSION 102
Die Auswertung aller in dieser Arbeit gemessenen PEG/Wasser-Systeme nach
Gl.(28) und (34) liefern Wechselwirkungsgrößen, die nicht einmal vom Vorzei-
chen her mit den realen Werten übereinstimmen. E. Dobnik untersuchte in sei-
ner Arbeit die PEG/Wasser-Systeme mit molaren Massen des PEG von 1000,
6000 und 35000 g/mol. Auch in diesen Systemen wurde die Liquiduskurve des
Wassers nach Gl.(34) ausgewertet. Bei den von ihm gemessenen Systemen
stimmten zwar die Vorzeichen der Wechselwirkungsgrößen, die Größen-
ordnung der errechneten Mischungsenthalpie für die Systeme lagen allerdings
in physikalisch nicht sinnvollen Bereichen.
A. Müller hat Phasendiagramme von n-Paraffinen untersucht und auch in die-
sen Systemen die Liquiduskurven nach Gl.(28) und (29) angepasst. Auch in
diesen Systemen zeigte sich, dass aus den Gleichgewichtskurven nicht auf
Wechselwirkungsgrößen wie die Mischungsenthalpie und -entropie geschlos-
sen werden kann. Auch bei den n-Paraffin-Systemen stimmten die Wechsel-
wirkungsgrößen nicht einmal vom Vorzeichen mit den realen messbaren Grö-
ßen überein.
Die ermittelten Exzessfunktionen stimmten somit weder in der Größenordnung
noch vom Vorzeichen her mit den tatsächlichen Messwerten dieser Größen
überein. Es hat sich gezeigt, dass diese Diskrepanz vor allem damit zusam-
menhängt, dass die durch die FHSS-Theorie eingeführten Wechselwirkungs-
funktionen sowohl temperatur-, druck- als auch konzentrationsunabhängig
sind105. Es liegt eine Kopplung der "wahren" Entropie- und Enthalpieanteile
�(T,w2) und �(T,w2) der Wechselwirkungsfunktion �w vor106. Es kann als bewie-
sen gelten, dass man von den Exzessfunktionen auf die einzelnen, durch die
FHSS-Theorie definierten Wechselwirkungsgrößen (wie den Enthalpie-, den
Entropie- und den Wechselwirkungsparameter) schließen kann. Ein Schritt in
die umgekehrte Richtung – also die Berechnung der Exzessfunktionen aus die-
sen Wechselwirkungsgrößen – ist nicht statthaft.
105 Vanhee S., Kiepen F.-H., Brinkmann D., Borchard W., Koningsveld. R., Berghmans, H.;
Macromol. Chem. Phys., 195, 1994, 759106 Müller A.; Dissertation Duisburg, 1998

ERGEBNISSE UND DISKUSSION 103
Aus den o.g. Gründen wurden die Liquiduskurven des Wassers in den in dieser
Arbeit gemessenen Phasendiagrammen in der Art durchgeführt, dass zunächst
mit Hilfe der Mischungsenthalpie-Messungen die einzelnen Werte für � in der
Massenbruchskala nach Gl.(25) errechnet wurden und danach die Liquidus-
kurve des Wassers gemäß Gl.(28) iteriert wurde. Der Parameter �w kann ket-
tenlängenabhängig mit Hilfe des Mischungsenthalpieminimums Gl.(25) berech-
net werden, unter der Voraussetzung, dass das Minimum der Mischungs-
enthalpie bei der Konzentration w2 = 0,5 liegt. Die maximale Mischungs-
enthalpie kann nach Gl.(52) berechnet werden. Der Enthalpieterm �w ist dem-
nach gegeben durch:
25,0RH~Max
Mw
�
��� (52)
Durch die Vorgabe des �w-Parameters muss aus den Liquiduskurven nur noch
ein Parameter – nämlich �w – ermittelt werden.
Tabelle 4: Ermittelte Werte für �w nach Vorgabe von �w durch die Mischungs-experimente.
M(PEG) �w �w
300 -25,55 0,002
400 -26,34 0,015
600 -27,11 0,030
900 -27,61 0,035
1500 -28,00 0,091
Mit zunehmender Kettenlänge des PEG wird die Mischungsenthalpie stärker
exotherm und der Betrag des Parameter �w zufolge größer. Aus dem Parameter
�w lässt sich gemäß Gl.(26) die Mischungsentropie berechnen, die mit zuneh-
mender Kettenlänge immer größer wird.

ERGEBNISSE UND DISKUSSION 104
Es zeigt sich, dass unter Berücksichtigung der Mischungsenthalpie die Liqui-
duskurve des Wassers in den PEG/Wasser-Systemen sehr genau angepasst
werden kann, und dass der Verlauf der Mischungsenthalpie mit zunehmender
Kettenlänge des PEG berechnet werden kann. Da die Wechselwirkungsgrößen
mit dem Quadrat der Konzentration des PEG eingehen – welche auf der Seite
des Wassers klein ist – , ist deren Einfluss auf den Verlauf der Liquiduskurve
entsprechend gering.
Abbildung 37 zeigt die Messwerte für die Liquiduskurve des Wassers beim
System PEG1500/Wasser und zusätzlich verschiedene iterierte Liquiduskurven.
Die grüne Kurve (von der roten und blauen Kurve teilweise verdeckt) zeigt die
Anpassung gemäß einer ideal-athermischen Lösung; die blaue Kurve zeigt die
Anpassung nach Gl.(28) bei gleichzeitiger Bestimmung von �w und �w; die rote
Kurve stellt die Bestimmung von �w dar, wobei �w durch Kenntnis der
Mischungsenthalpie vorgegeben wurde. Die schwarze gestrichelte Linie
entspricht dem Ansatz der idealen-Lösung, r = 1 sowie �w und �w = 0.
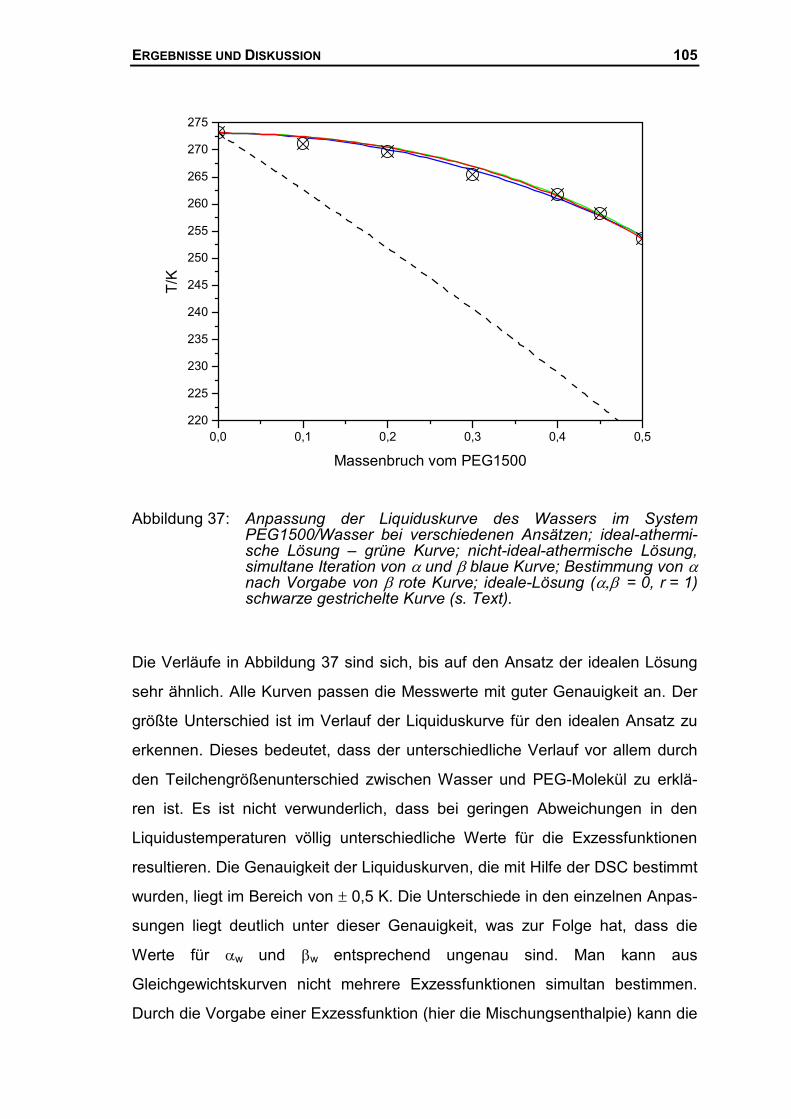
ERGEBNISSE UND DISKUSSION 105
Abbildung 37: Anpassung der Liquiduskurve des Wassers im SystemPEG1500/Wasser bei verschiedenen Ansätzen; ideal-athermi-sche Lösung – grüne Kurve; nicht-ideal-athermische Lösung,simultane Iteration von � und � blaue Kurve; Bestimmung von �nach Vorgabe von � rote Kurve; ideale-Lösung (���� = 0, r = 1)schwarze gestrichelte Kurve (s. Text).
Die Verläufe in Abbildung 37 sind sich, bis auf den Ansatz der idealen Lösung
sehr ähnlich. Alle Kurven passen die Messwerte mit guter Genauigkeit an. Der
größte Unterschied ist im Verlauf der Liquiduskurve für den idealen Ansatz zu
erkennen. Dieses bedeutet, dass der unterschiedliche Verlauf vor allem durch
den Teilchengrößenunterschied zwischen Wasser und PEG-Molekül zu erklä-
ren ist. Es ist nicht verwunderlich, dass bei geringen Abweichungen in den
Liquidustemperaturen völlig unterschiedliche Werte für die Exzessfunktionen
resultieren. Die Genauigkeit der Liquiduskurven, die mit Hilfe der DSC bestimmt
wurden, liegt im Bereich von � 0,5 K. Die Unterschiede in den einzelnen Anpas-
sungen liegt deutlich unter dieser Genauigkeit, was zur Folge hat, dass die
Werte für �w und �w entsprechend ungenau sind. Man kann aus
Gleichgewichtskurven nicht mehrere Exzessfunktionen simultan bestimmen.
Durch die Vorgabe einer Exzessfunktion (hier die Mischungsenthalpie) kann die
0,0 0,1 0,2 0,3 0,4 0,5220
225
230
235
240
245
250
255
260
265
270
275
T/K
Massenbruch vom PEG1500

ERGEBNISSE UND DISKUSSION 106
zweite (Mischungsentropie) sehr genau bestimmt werden. Es bleibt zu
berücksichtigen, dass die zuvor gemachten Auswertungen unter der Annahme
einer vor allem konzentrationsunabhängigen �w-Wechselwirkungsfunktion
gemacht wurden. Die daraus resultierenden Wechselwirkungsparameter sind
unabhängig von der Konzentration und der Temperatur. Messungen von
Mischungsenthalpien bei dem System PEG6000/Wasser zeigen, dass die
Mischungsenthalpien mit steigender Temperatur immer kleiner werden107. Diese
Temperaturabhängigkeit wurde in der Auswertung vernachlässigt, da die
Abweichungen bei tiefen Temperaturen, und der Temperaturbereich über den
iteriert wurde, sehr klein ist.
4.3.2 Thermodynamische Beschreibung der Liquiduskurve des PEG
Bei der Beschreibung der Liquiduskurve des PEG gemäß Gl.(28) wurde ein
heterogenes Gleichgewicht zwischen der Lösung und dem reinen PEG-Kristall
vorausgesetzt. Die Auswertung der DSC-Messungen ergaben, dass unabhän-
gig von der Kettenlänge ein Mischkristall mit der Konzentration von w2 = 0,92
auf der Seite des PEG vorliegt. Das Vorliegen des Mischkristalls muss in der
thermodynamischen Beschreibung berücksichtigt werden. Mit dem einfachen
Ansatz eines heterogenen Gleichgewichts zwischen Lösung und PEG Kristall
kann die Liquiduskurve mit keinem Wert für �w beschrieben werden. Eine
eigene Iteration der Liquiduskurve des PEG nach Gl.(28) ergibt ebenfalls keine
sinnvolle Anpassung der Messwerte. Das die Liquiduskurve des PEG mit die-
sen Ansätzen nicht beschrieben werden kann, wird auch in anderen Arbeiten
deutlich108,109. Der Grund für die mangelnde Beschreibung ist in der Vernachläs-
sigung der Mischkristallbildung auf der Seite des PEG zu finden.
107 Cunninghame G., Malcolm G.N.; J. Phys. Chem., 65, 1961, 1454108 Dobnik E.; Dissertation Duisburg, 1991109 Hager S.L., Macrury T.B.; J. Apl. Pol. Sci., 25, 1980, 1559

ERGEBNISSE UND DISKUSSION 107
Es ist möglich die Liquiduskurve des PEG gekoppelt mit der entsprechenden
Soliduskurve zu beschreiben. Die anfängliche heterogene Gleichgewichts-
bedingung lautet in diesem Fall:
L1
S1
~~ ��� (53)
und L2
S2
~~ ��� (54)
Wobei Si
~� und Li
~� das spezifische chemische Potential der Komponente i (i = 1
für Wasser und i = 2 für PEG) in der kristallinen (Index S) und in der flüssigen
Mischphase (Index L) bedeuten. Bei der Berücksichtigung der Mischkristall-
bildung müssen, anders als bei Systemen ohne Mischkristallbildung, beide
Bedingungsgleichungen erfüllt sein110. Alle Größen in Gln.(53) und (54) sind
abhängig von der Temperatur und der Zusammensetzung.
Die chemischen Potentiale werden in der Grundmolenbruchskala folgender-
maßen angesetzt:
L*1
L*1
L01
L1 fxlnRT)P,T( ���� (55)
L*2
L*2
L02
L2 fxlnRT)P,T( ���� (56)
cr*1
cr*1
cr01
cr1 fxlnRT)P,T( ���� (57)
cr*2
cr*2
cr02
cr2 fxlnRT)P,T( ���� (58)
aus Gln.(53), (55) und (56) folgt:
���
����
���� cr*
1cr*
1
l*1
l*1l
01cr01 fx
fxlnRT (59)
entsprechend aus Gln.(54), (57) und (58):
���
����
���� l*
2l*
2
cr*2
cr*2cr
02l02 fx
fxlnRT (60)

ERGEBNISSE UND DISKUSSION 108
Mit Hilfe von Gl.(59) unter der Voraussetzung (14) folgt:
���
����
��
���
�
����
�
1,m
1,ml*
2l*
2
cr*2
cr*2
T1
T1
RH
fxfxlnRT (61)
mit ���
����
��
��
1,m
1,m1 T
1T1
RH
(62)
folgt aus Gl.(61) für Komponente 1:
)(expffxx 1cr*1
l*1l*
1cr*
1 ����
����
�� (63)
Für die zweite Komponente kann leicht die entsprechende Formel abgeleitet
werden:
)(expffxx 2cr*2
l*2l*
2cr*
2 ����
����
�� (64)
mit ���
����
��
�
1,m
1,m2 T
1T1
RH
(65)
Die beiden Gln.(64) und (65) sind bis auf die Aktivitätskoeffizienten und Grund-
molenbrüche identisch mit Ansätzen von Haase111 und Prigogine112.
Mit Hilfe der Gln.(64) und (65) können die Größen �1 und �2 für jede Tempera-
tur entlang der Liquiduskurve berechnet werden. Mit Hilfe dieser temperatur-
abhängigen Werte für �1 und �2 können danach die Verhältnisse der Aktivitäts-
koeffizienten berechnet werden, wenn für jede Liquidustemperatur die Konzen-
trationen der flüssigen und der festen Phase bekannt ist. Die Konzentration der
flüssigen Phase ist gegeben durch die mit Hilfe der DSC bestimmten Liquidus-
110 Müller A.; Dissertation Duisburg; 1998111 Haase R.; Thermodynamik der Mischphasen; Springer, Berlin, 1956112 Prigogine I., Defay R., Chemical Thermodynamics, Longmans Green & Co, London, 1952
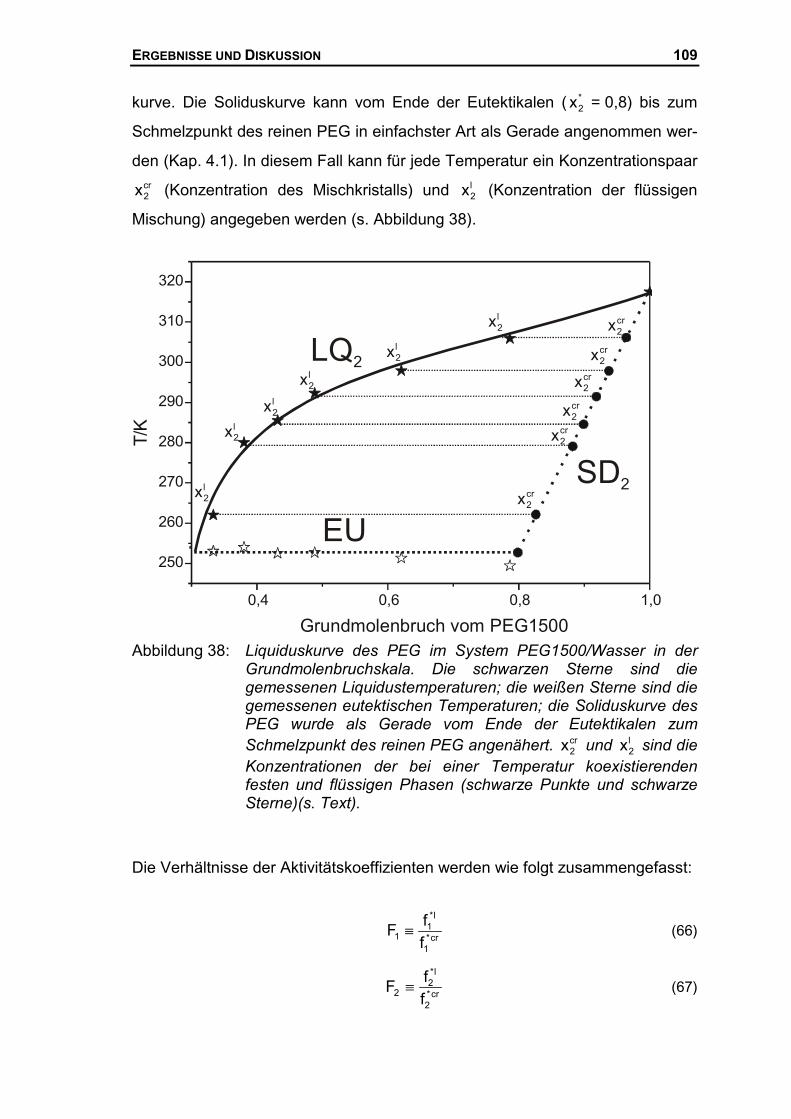
ERGEBNISSE UND DISKUSSION 109
kurve. Die Soliduskurve kann vom Ende der Eutektikalen ( *2x = 0,8) bis zum
Schmelzpunkt des reinen PEG in einfachster Art als Gerade angenommen wer-
den (Kap. 4.1). In diesem Fall kann für jede Temperatur ein Konzentrationspaarcr2x (Konzentration des Mischkristalls) und l
2x (Konzentration der flüssigen
Mischung) angegeben werden (s. Abbildung 38).
Abbildung 38: Liquiduskurve des PEG im System PEG1500/Wasser in derGrundmolenbruchskala. Die schwarzen Sterne sind diegemessenen Liquidustemperaturen; die weißen Sterne sind diegemessenen eutektischen Temperaturen; die Soliduskurve desPEG wurde als Gerade vom Ende der Eutektikalen zumSchmelzpunkt des reinen PEG angenähert. cr
2x und l2x sind die
Konzentrationen der bei einer Temperatur koexistierendenfesten und flüssigen Phasen (schwarze Punkte und schwarzeSterne)(s. Text).
Die Verhältnisse der Aktivitätskoeffizienten werden wie folgt zusammengefasst:
cr*1
l*1
1 ffF � (66)
cr*2
l*2
2 ffF � (67)
0,4 0,6 0,8 1,0
250
260
270
280
290
300
310
320
T/K
Grundmolenbruch vom PEG1500
SD2
LQ2
EU
l2x
l2x
l2x
l2x
l2x
l2x
cr2x
cr2x
cr2x
cr2x
cr2x
cr2x

ERGEBNISSE UND DISKUSSION 110
Bei Kenntnis der Verhältnisse der Aktivitätskoeffizienten in der flüssigen und der
festen Phase können aus den Funktionen F1 und F2 die Wechselwirkungs-
parameter crx� und l
x� ermittelt werden. crx� und l
x� sind temperatur- und kon-
zentrationsabhängige Wechselwirkungsgößen des Systems im Kristall (cr) und
in der Lösung (l). Hierzu werden die Aktivitätskoeffizienten nach dem Ansatz
der nicht-ideal-athermischen Lösung angesetzt:
2l*2
lx
l*2
l*1 )x(x
r11fln ����
���
� (68)
2l*2
lx
l*2
l*2 )x(rx)r1(fln ���� (69)
2cr*2
crx
cr*2
cr*1 )x(x
r11fln ����
���
� (70)
2cr*2
crx
cr*2
cr*2 )x(rx)r1(fln ���� (71)
Sind die Funktionen F1 und F2 bekannt, können crx� und l
x� bestimmt werden.
Hierzu werden Gln.(68) bis (71) unter Verwendung von (66) und (67) umge-
formt, wodurch folgender Ausdruck für crx� und l
x� resultiert:
� �� �2l*
12cr*
22l*
22cr*
1
2l*2
2l*1
cr*2
l*2
2l*22
2l*11cr
x)x()x()x()x(r
)x()x()xx)(1r()x(Fln)x(Flnr�
������� (72)
� �� �2cr*
22l*
12cr*
12l*
2
2cr*2
2cr*1
cr*2
l*1
2cr*22
2cr*11l
x)x()x()x()x(r
)x()x()xx)(1r()x(Fln)x(Flnr�
������� (73)
Nach der Auswertung kann ein konzentrations- und temperaturabhängiger
Wechselwirkungsparameter für die flüssige Mischung und den Mischkristall
angegeben werden. Die Berechnungen ergeben, dass die Wechselwirkungs-
gößen in diesem Fall vor allem für höhere Temperaturen sehr groß werden
5 < )T,x( 2l/cr
� < 150 für Temperaturen zwischen 260 und 305 K113.
113 Jablonski P., Borchard W.; In Vorbereitung
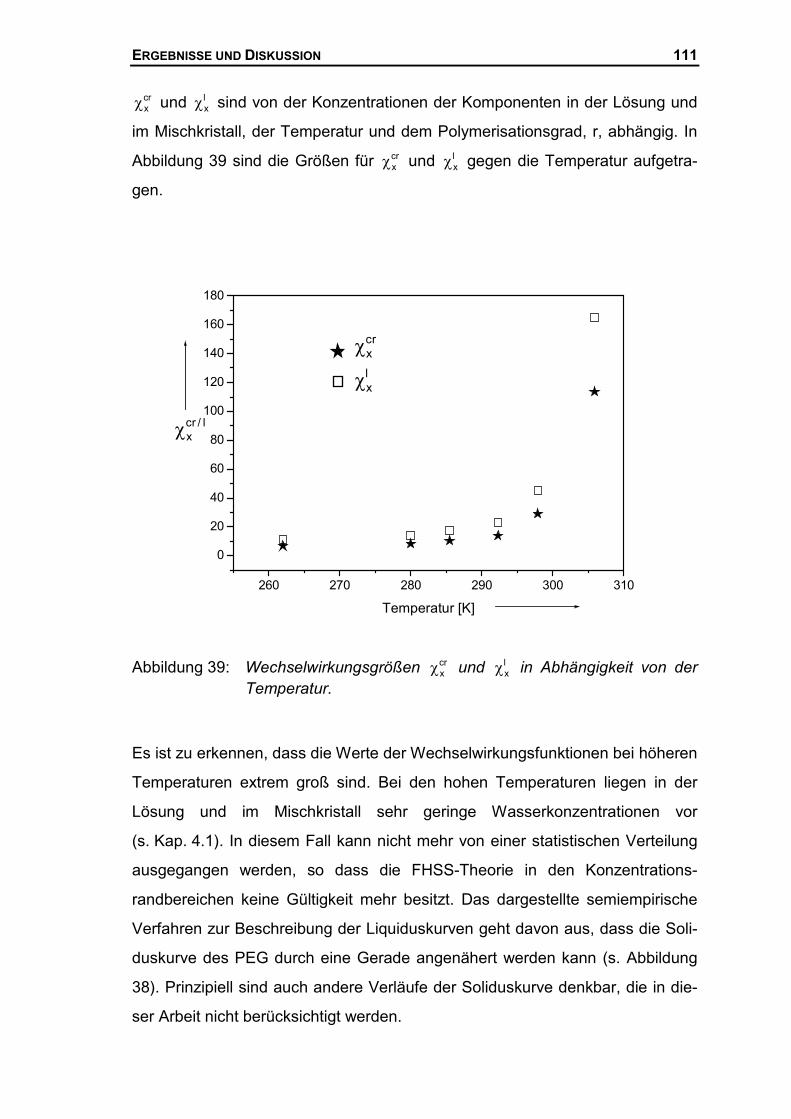
ERGEBNISSE UND DISKUSSION 111
crx� und l
x� sind von der Konzentrationen der Komponenten in der Lösung und
im Mischkristall, der Temperatur und dem Polymerisationsgrad, r, abhängig. In
Abbildung 39 sind die Größen für crx� und l
x� gegen die Temperatur aufgetra-
gen.
Abbildung 39: Wechselwirkungsgrößen crx� und l
x� in Abhängigkeit von derTemperatur.
Es ist zu erkennen, dass die Werte der Wechselwirkungsfunktionen bei höheren
Temperaturen extrem groß sind. Bei den hohen Temperaturen liegen in der
Lösung und im Mischkristall sehr geringe Wasserkonzentrationen vor
(s. Kap. 4.1). In diesem Fall kann nicht mehr von einer statistischen Verteilung
ausgegangen werden, so dass die FHSS-Theorie in den Konzentrations-
randbereichen keine Gültigkeit mehr besitzt. Das dargestellte semiempirische
Verfahren zur Beschreibung der Liquiduskurven geht davon aus, dass die Soli-
duskurve des PEG durch eine Gerade angenähert werden kann (s. Abbildung
38). Prinzipiell sind auch andere Verläufe der Soliduskurve denkbar, die in die-
ser Arbeit nicht berücksichtigt werden.
260 270 280 290 300 310
0
20
40
60
80
100
120
140
160
180
lx
crx
�
�
l/crx�
Temperatur [K]

ERGEBNISSE UND DISKUSSION 112
In umgekehrter Weise kann auch der Verlauf der Soliduskurve nach Gln.(72)
und (73) berechnet werden, unter der Voraussetzung, dass die Liquiduskurve
und der Verlauf der Wechselwirkungsfunktionen crx� und l
x� bekannt sind. Die
Liquiduskurve kann mit Hilfe der Messwerte in Abbildung 38 interpoliert werden,
so dass für jede Temperatur eine Liquiduskonzentration angegeben werden
kann. Wird der Verlauf der Wechselwirkungsfunktionen vorgegeben, kann für
jede Temperatur eine entsprechende Soliduskonzentration berechnet werden.
Werden die Verläufe der Wechselwirkungsfunktionen gemäß Abbildung 39
interpoliert, resultiert für den Verlauf der Soliduskurve des PEG der beschrie-
bene lineare Verlauf analog zur Abbildung 38. Bei anderen Verläufen der
Wechselwirkungsfunktionen resultieren zwangsläufig auch andere Verläufe der
Soliduskurve. Da die FHSS-Theorie in den Konzentrationsrandbereichen keine
Gültigkeit mehr besitzt und der Verlauf der Soliduskurve in Rahmen dieser
Arbeit nicht gemessen werden kann, wurde auf weitere Berechnungen der Ver-
läufe der Soliduskurven verzichtet.
Müller konnte zeigen, dass der �w-Parameter für verschwindend geringe Kon-
zentrationen w2 im binären System Hexan/Hexadecan in den Randbereichen
extrem groß wird. Auch in dem System PEG1500/Wasser ist dieser Trend zu
erkennen. Die wahre �w-Wechselwirkungsfunktion ist in komplizierter Weise von
der Konzentration, der Temperatur und dem Druck abhängig. Die �(T,P,w2)-
Funktion kann aus den wahren Wechselwirkungsgrößen beliebig genau
abgleitet werden. Diese Berechnung ist sehr ausführlich für das System
Hexan/Hexadecan beschrieben worden114. Mit Hilfe dieser �(T,P,w2)-Funktion
können die Gleichgewichtskurven beschrieben werden. Der Schritt in umge-
kehrte Richtung führt allerdings zwangsläufig zu einem Informationsverlust, da
– im Fall der Schmelzdiagramme – aus der zweidimensionalen T(w2) Ebene der
Liquiduskurve auf den höherdimensionalen �(T,P,w2)-Raum projeziert wird.
114 Müller A, Borchard W.; in Vorbereitung

ERGEBNISSE UND DISKUSSION 113
4.3.3 Zusammenfassung über die thermodynamische Auswertungen
Ein Vergleich mit anderen Autoren im Fall der Liquiduskurve des PEG ist wegen
des Mangel an Daten nicht möglich ist. In den zitierten Arbeiten, die sich mit
dem Phasenverhalten des Systems PEG/Wasser (und auch anderen organi-
schen Lösemitteln) beschäftigen ist die Konzentrationsabhängigkeit des �w-
Parameters nicht berücksichtigt worden115,116. Die Mischkristallbildung auf der
Seite des PEG wird ebenfalls in keiner kalorimetrischen Arbeit erwähnt. Durch
die Berücksichtigung der Mischkristallbildung resultiert die in Kap.4.3.2 abge-
leitete Beschreibung mit Wechselwirkungsparametern, die sowohl von der
Temperatur als auch von der Konzentration abhängig sind. Die Autoren tragen
zwar die spezifische eutektische Umwandlungsenthalpie in Abhängigkeit von
der Zusammensetzung gemäß des Tammannschen Dreiecks auf, verzichten
aber darauf, die Messwerte durch die entsprechenden Geraden anzupassen,
wobei direkt die Mischkristallbildung auf der Seite des PEG bewiesen wird.
Vielmehr zwingen sie den rechten Teil des Tammannschen Dreiecks durch die
Konzentration w2 = 1, was bedeuten würde, dass reine PEG-Kristalle gebildet
werden und keine Randlöslichkeit vorliegt117,118,119. Eine derartige Kurven-
konstruktion ist auch gemäß der quantitativen Auswertung des Tammannschen
Dreiecks von Müller und Borchard nicht zu erklären (Gln.(38) und (39))120.
Dementsprechend kann ein Vergleich der thermodynamischen Daten lediglich
mit den in Kap.4.3.1 gefundenen Daten in der Grundmolenbruchskala stattfin-
den. Dobnik hat in seiner Arbeit bereits seine Daten für die �x-Wechsel-
wirkungsfunktion mit denen anderer Arbeiten verglichen121. Hierzu verglich er
seine Ergebnisse mit den Wechselwirkungsfunktionen nicht nur mit solchen, die
mit Hilfe von kalorimetrischen Messungen gewonnen wurden122-124, sondern
115 Hager S.L., Macrury T.B.; J. Apl. Pol. Sci., 25, 1980, 1559116 Smith P., Pennings A.J.; Polymer, 15, 1974, 413117 Hey M.J., Ilett S.M.; J. chem. Soc. Far. Trans., 87, 1991, 3671118 Bogdanov B., Mihailov M.; J. Pol. Sci., 23, 1985, 2149119 Huang L., Nishinari K.; J. Pol. Sci.: Part B, 39, 2001, 496120 Müller, A., Borchard, W.; J. Phys. Chem., 101, 1997, 4307121 Dobnik E.; Dissertation Duisburg, 1991122 Schönert H., Monshausen F.; Col. Pol. Sci., 369, 1980, 578

ERGEBNISSE UND DISKUSSION 114
auch mit denen, die mit anderen Methoden und teilweise in anderen Tempera-
turbereichen ermittelt wurden (Quellungsmessungen125) (Entmischungsversu-
che126).
Die �x-Parameter bei Temperaturen von 300 K streuen recht stark, was auf die
unterschiedlichen Methoden und die verschiedenen Polymere zurückzuführen
ist. Bei den Untersuchungen der Netzwerke, wurden Polyethylenglykole mit den
mittleren molaren Massen von 400, 600 und 1000 g/mol untersucht. An den
Mischungsexperimenten in dieser Arbeit erkennt man deutlich, dass in diesem
Kettenlängenbereich der Endgruppeneinfluss sehr groß ist. Wird die Alkohol-
endgruppe durch einen Netzwerkknotenpunkt ersetzt, kann nicht mehr davon
ausgegangen werden, dass die selben Werte für die Mischungsenthalpie und –
entropie gefunden werden.
In Abbildung 40 sind die temperaturabhängigen Werte für �x für die in dieser
Arbeit gemessenen PEG/Wasser-Systeme in der Grundmolenbruchskala gegen
die reziproke Temperatur aufgetragen. Aus den Mischungsexperimenten kann
eine grundmolare maximale Mischungsenthalpie (pro Grundbaustein) mit
-2613 J/mol* angegeben werden, was einem Enthalpieterm von �x = -1257 ent-
spricht. In guter Näherung liefert dieser Wert den gemittelten Wert für �x von
3,11, welcher auch mit den Werten von Hager et al. und Dobnik übereinstim-
men. Zusätzlich sind die Werte für �x eingezeichnet die Saeki aus Ent-
mischungsversuchen ermittelt hat.
In Abbildung 40 ist die �x-Wechselwirkungsfunktion gegen die reziproke thermo-
dynamische Temperatur aufgetragen und kann im Temperaturbereich von ca.
270 K sehr gut durch eine Gerade angenähert werden was auch in verschiede-
nen anderen Arbeiten deutlich geworden ist. Bei höheren Temperaturen verläuft
die �x-Wechselwirkungsfunktion durch ein Maximum, ohne dass die
123 Michalczyk A., Borchard W.; Eur. Pol. J., 25, 1989, 947124 Hager S.L., Macrury T.B.; J. Apl. Pol. Sci., 25, 1980, 1559125 Steinbrecht U.; Dissertation Duisburg, 1991126 Saeki F., Kuwahara N., Kaneko M.; Polymer, 17, 1976, 685
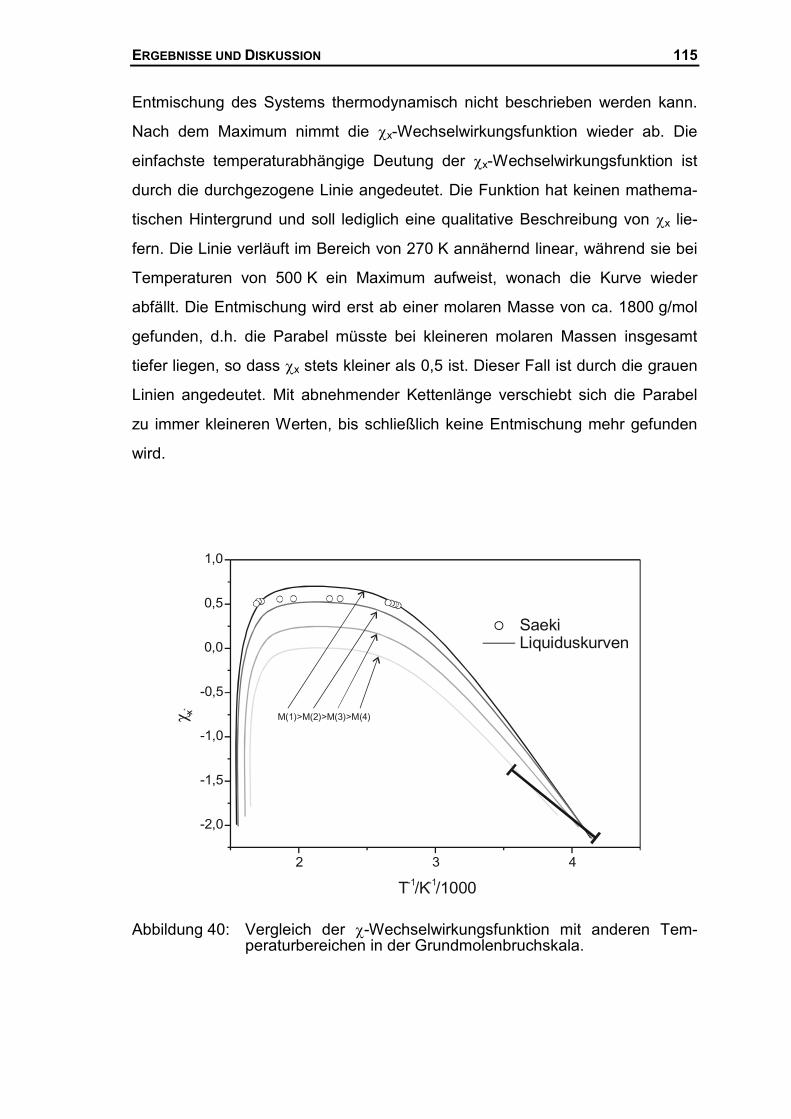
ERGEBNISSE UND DISKUSSION 115
Entmischung des Systems thermodynamisch nicht beschrieben werden kann.
Nach dem Maximum nimmt die �x-Wechselwirkungsfunktion wieder ab. Die
einfachste temperaturabhängige Deutung der �x-Wechselwirkungsfunktion ist
durch die durchgezogene Linie angedeutet. Die Funktion hat keinen mathema-
tischen Hintergrund und soll lediglich eine qualitative Beschreibung von �x lie-
fern. Die Linie verläuft im Bereich von 270 K annähernd linear, während sie bei
Temperaturen von 500 K ein Maximum aufweist, wonach die Kurve wieder
abfällt. Die Entmischung wird erst ab einer molaren Masse von ca. 1800 g/mol
gefunden, d.h. die Parabel müsste bei kleineren molaren Massen insgesamt
tiefer liegen, so dass �x stets kleiner als 0,5 ist. Dieser Fall ist durch die grauen
Linien angedeutet. Mit abnehmender Kettenlänge verschiebt sich die Parabel
zu immer kleineren Werten, bis schließlich keine Entmischung mehr gefunden
wird.
Abbildung 40: Vergleich der �-Wechselwirkungsfunktion mit anderen Tem-peraturbereichen in der Grundmolenbruchskala.
M(1)>M(2)>M(3)>M(4)
2 3 4
-2,0
-1,5
-1,0
-0,5
0,0
0,5
1,0
Saeki Liquiduskurven
�x*
T-1/K-1/1000

ERGEBNISSE UND DISKUSSION 116
Eine quantitative Beschreibung für die Parabel wird in dieser Arbeit nicht gege-
ben werden, da auch diese Beschreibung die Werte für �x unter Berücksichti-
gung der Mischkristallbildung, die extrem großen Werte für �x in den Konzen-
trationsrandbereichen nicht erklären würde.
Anhand mehrerer unterschiedlicher thermodynamischer Auswertungen weiß
man, wie eine konsistente Beschreibung des Systems PEG/Wasser aussehen
müsste. Jede thermodynamische Beschreibung beschränkt sich auf einen klei-
nen Temperaturausschnitt und ermittelt in diesem die Temperaturabhängigkeit
der �-Wechselwirkungsfunktion. In Abbildung 40 wird deutlich, dass die �x-
Wechselwirkungsfunktion nicht einfach durch eine Gerade angepasst werden
kann, da sie die Werte bei hohen Temperaturen nicht beschreiben kann. Im
Temperaturbereich der Liquiduskurven erhält man eine sehr genaue Beschrei-
bung des Systems, welche sogar die einzelnen Exzessfunktionen berücksich-
tigt. Eine Extrapolation zu hohen Temperaturen ist allerdings nicht statthaft und
führt zu unsinnigen Werten der �x-Wechselwirkungsfunktion. Eine konsistente
Beschreibung muss die Temperaturabhängigkeit der einzelnen Exzessgrößen
(Mischungsenthalpie und -entropie) berücksichtigen, die mit zunehmender
Temperatur kleiner werden. Eine derartige Beschreibung, die die Konzentra-
tions- und Temperaturabhängigkeit kettenlängenabhängig berücksichtigt, muss
sehr kompliziert werden. Die Druckabhängigkeit der �x-Wechselwirkungsfunk-
tion wird immer noch vernachlässigt, was gerade bei hohen Temperaturen zu
fragwürdigen Ergebnissen führen kann. Im System PEG/Wasser gibt es ohne
Zweifel einen systematischen Gang der Exzessgrößen in Abhängigkeit von der
Kettenlänge, der Temperatur und der Konzentration. Die Kettenlängenab-
hängigkeit der Mischungsenthalpie wurde in dieser Arbeit untersucht, wobei
diese temperaturunabhängig angenommen wurde, was sie jedoch nicht ist. Die
Abweichungen sind allerdings so minimal, dass sie bei der Beschreibung der
Liquiduskurven keine Rolle spielt. Soll die Mischungsenthalpie bei sehr hohen
Temperaturen berücksichtigt werden, wird die Temperaturabhängigkeit größer,
so dass das System ein endothermes Mischverhalten zeigt (Entmischung).

ERGEBNISSE UND DISKUSSION 117
Für viele Beschreibungen ist es eher hinderlich, eine Theorie anzuwenden, die
alle Abhängigkeiten berücksichtigt, da diese zu kompliziert wird. Oft, wie im Fall
der Liquiduskurven in dieser Arbeit, genügt es eine Teiltheorie anzuwenden, die
den untersuchten Temperaturbereich sehr gut beschreibt. Es ist aber klar, dass
die Beschreibung nur für den entsprechenden Temperaturbereich gilt. Die Ver-
einfachungen lassen sich auf keinen Fall auf andere Temperaturbereiche über-
tragen, so dass dort wieder eine andere Teiltheorie Anwendung finden muss.
Im Fall des Systems PEG/Wasser wären das die entsprechenden Theorien zur
Entmischung127,128.
127 Fischer V., Borchard W.; J. Phys. Chem. B, 104, 2000, 4463128 Fischer V.; Dissertation Duisburg, 2001

ZUSAMMENFASSUNG 118
5 ZUSAMMENFASSUNG
Das Thema der Arbeit ist das thermische Verhalten von PEG/Wasser-Syste-
men, wobei der Hauptschwerpunkt auf die binären Phasendiagramme gerichtet
ist, in denen die molare Masse des PEG unter 1500 g/mol liegt. Hierzu wurden
die binären Schmelzdiagramme einiger PEG/Wasser-Systeme mit Hilfe der
DSC bestimmt. Die molaren Massen der Polyethylenglykole lagen bei 300, 400,
600, 900 und 1500 g/mol.
Bei den DSC-Messungen der einzelnen Proben der PEG/Wasser-Systeme
zeigte sich, dass nur bis zu einer molaren Masse von 1500 g/mol ohne Pro-
bleme das Schmelzdiagramm ermittelt werden kann. Bei kleineren molaren
Massen kommt es in allen Fällen bei den aufheizenden DSC-Messungen zu
einer Überlagerung der exothermen Kaltkristallisation und des ersten eutek-
tischen Schmelzsignals. Zur genauen Bestimmung des eutektischen Punktes
und dem Nachweis der Mischkristallbildung wurde ein spezielles thermisches
Verfahren zur vollständigen Kristallisation der Mischungen vorgestellt. Dieses
zeigte, dass ein Tempern unterhalb des eutektischen Schmelzsignals keine
Auswirkungen auf das Kristallisationsverhalten der Proben hat. Alle Mischungen
müssen zweimal abgekühlt werden und gemessen werden, wobei erst bei der
zweiten Messung die eutektischen Signale ausgewertet werden können.
Die Schmelzdiagramme der Systeme zeigen einige Gemeinsamkeiten. Wie in
Phasendiagrammen unter Beteiligung höhermolekularer Polyethylenglykole
liegt ein eutektisches Schmelzverhalten vor. In allen Fällen wird eine Mischkri-
stallbildung auf der Seite des PEG gefunden. Eine Mischkristallbildung auf der
Seite des Wasser kann für alle Systeme ausgeschlossen werden, was bedeu-
tet, dass beim Abkühlen der Mischungen reines Wasser ausfriert.
Die Liquiduskurven des Wassers ähneln sich sehr stark, was nicht nur auf den
in dieser Arbeit untersuchten Bereich der molaren Masse des PEG zutrifft. Es
wird deutlich, dass die Liquiduskurven mit zunehmender Kettenlänge des PEG

ZUSAMMENFASSUNG 119
flacher verlaufen. Da die Anfangssteigung der Liquiduskurven im Anfangs-
bereich proportional dem negativen reziproken Wert des Zahlenmittelwertes der
molaren Masse ist, sind diese Unterschiede beim Vergleich der kurzkettigen
PEG/Wasser-Systeme größer als im Bereich der höhermolekularen PEG/Was-
ser-Systeme. Der Unterschied im Verlauf der Liquiduskurve des Wasser mit
Hilfe der DSC kann hier kaum noch gemessen werden.
Die Verläufe der Liquiduskurven des PEG ähneln sich ebenfalls, allerdings lie-
gen die Kurven in unterschiedlichen Temperaturbereichen. Der Beginn der
Liquiduskurve wird durch den Schmelzpunkt des reinen PEG definiert. Dieser
ändert sich ab einer molaren Masse von 10.000 g/mol nicht mehr und kann
konstant mit 338 K angegeben werden. Bei kürzer werdendem PEG verschiebt
sich der Schmelzpunkt zu immer tieferen Temperaturen. Im Bereich unter
900 g/mol ist die Abhängigkeit von der molaren Masse so groß, dass der
Schmelzpunkt bei der Verlängerung oder Verkürzung um eine Ethylenoxid-
Einheit sich um 3 K erhöht bzw. erniedrigt. Die Schmelzdiagramme der reinen
Proben zeigen eine Überlagerung verschiedener Polyethylenglykole deren
Schmelzpunkte ca. 3 K auseinander liegen.
Durch den immer tiefer liegenden Schmelzpunkt des reinen PEG verschiebt
sich der eutektische Punkt zu tieferen Temperaturen und höheren PEG-Kon-
zentrationen. Diese Verschiebung hat erhebliche Auswirkungen auf das Kristal-
lisationsverhalten der Proben. Im Bereich von 1500 g/mol ist die Kristallisation
gehindert, so dass für eine vollständige Kristallisation eine spezielle thermische
Vorbehandlung nötig wird. Im Bereich von 400 g/mol wird keine Kristallisation
der eutektischen Phase gefunden, der eutektische Punkt ist zu so hohen Kon-
zentrationen und gleichzeitig tiefen Temperaturen verschoben, dass die Kristal-
lisation der eutektischen Phase völlig ausbleibt. In diesen Schmelzdiagrammen
wird nur eine glasige Erstarrung bei 193 K detektiert. Diese Glastemperatur ist
unabhängig von der Kettenlänge des PEG. Im Schmelzdiagramm des Systems
PEG300/Wasser wird ein Konzentrationsbereich gefunden, in dem weder eu-
tektische noch Liquidussignale detektiert werden. In diesem Konzentrations-

ZUSAMMENFASSUNG 120
bereich erstarrt die gesamte Probe glasig bei der systemcharakteristischen
Glastemperatur von 193 K. Dieser Bereich ist interessant für Anwendungen im
Bereich der Kryo-Konservierung. Biologische Zellen können mit PEG geeigne-
ter molarer Masse bis zu einer genau definierten Konzentration versetzt wer-
den, wonach sie beim späteren Einfrieren (Konservieren) nicht durch Eis-
kristalle zerstört werden.
In den PEG/Wasser-Systemen ab einer molaren Masse von 600 g/mol können
die spezifischen eutektischen Signale gemäß des Tammannschen Dreiecks
ausgewertet werden. Auf der Seite des PEG bildet sich ein PEG/Wasser-Misch-
kristall der kettenlängenunabhängig immer bei der gleichen Konzentration von
92 Gew.-% liegt. Im Fall des Systems PEG600/Wasser wurde diese Konzentra-
tion nach Auswertung der Glasstufenhöhe in Abhängigkeit von der Zusammen-
setzung bestätigt. Der Massenanteil entspricht einem molaren Verhältnis der
Ethylenoxid-Einheiten zu Wassermolekülen von 4:1. Es wird vermutet, dass
sich die Wassermoleküle zwischen den einzelnen PEG-Kristallschichten befin-
den, wodurch diese Stöchiometrie festgelegt ist.
Zur genauen thermodynamischen Beschreibung der PEG/Wasser Systeme ist
die Kenntnis der Mischungsenthalpie Voraussetzung. Im Rahmen dieser Arbeit
wurde eine neue Methode vorgestellt, mit der es möglich ist, Mischungs-
enthalpien von zwei Flüssigkeiten zu bestimmen. Dazu wurde ein temperierba-
rer Spritzenaufsatz konstruiert, mit dem definierte Mengen einer Flüssigkeit in
das DSC-Pfännchen eingespritzt werden können. Aus dem differentiellen Wär-
mestrom im isothermen Modus kann die Wärmetönung beim Mischen gemes-
sen werden. Die Ergebnisse wurden mit Messungen mit einem kommerziellen
Flow-Kalorimeter und mit Literaturdaten verglichen, wobei eine Genauigkeit für
das neue Kalorimeter von � 3 J/g angegeben werden konnte. Mit dem Kalori-
meter können Mischungsenthalpien geringster Massen (ca. 10 mg) bestimmt
werden. Einschränkungen sind die Verwendung von nicht zu flüchtigen Flüssig-
keiten. Aufgrund des absoluten Fehlers von � 3 J/g können Mischungsenthal-
pien vor allem in den Konzentrationsbereichen bestimmt werden, in denen sie

ZUSAMMENFASSUNG 121
am größten sind. Nicht nur Mischungsenthalpien lassen sich mit der neuen
Methode bestimmen, prinzipiell ist es auch möglich, Reaktionsenthalpien,
Wärmekapazitäten oder Verdampfungsenthalpien zu bestimmen, was das
Spektrum der mit der DSC bestimmbaren Größen erheblich vergrößert.
Die Messungen der Mischungsenthalpien und die Auswertung verschiedener
Literaturdaten zeigt, dass die Mischungsenthalpie des Systems PEG/Wasser
sehr gut mit symmetrischen Parabeln in der Massenbruchskala beschrieben
werden können. Die Mischungsenthalpie wird mit zunehmender Kettenlänge
des PEG stärker exotherm.
Unter Berücksichtigung der Mischungsenthalpien wurden die Liquiduskurven
des Wassers angepasst. Während bei gleichzeitiger Ermittlung des Enthalpie-
und Entropieterms aus den Gleichgewichtskurven physikalisch unsinnige Werte
für diese resultieren, kann die Mischungsentropie nach Vorgabe der
Mischungsenthalpie sehr gut bestimmt werden. Eine Übersicht über die Anpas-
sung der Liquiduskurven zeigt den geringen Einfluss des Enthalpie- und Entro-
pieterms. Die Liquiduskurven werden in allen Fällen sehr genau angepasst. Die
Abweichungen in den Verläufen der Liquiduskurven nach den verschiedenen
Ansätzen der Wechselwirkungsfunktion sind wesentlich geringer als die Abwei-
chungen in den DSC-Messungen. Der Verlauf der Liquiduskurve des Wasser
kann bereits sehr gut durch den Ansatz der ideal-athermischen Lösung
beschrieben werden.
Für den Verlauf der Liquiduskurve des PEG unter Berücksichtigung der Misch-
kristallbildung wird eine Ableitung vorgeschlagen, die konzentrations- und tem-
peraturabhängige Wechselwirkungsgößen in der Lösung und im Mischkristall
berücksichtigt. Die semiempirische Auswertung zeigt, dass die Wechsel-
wirkungsgrößen im Konzentrationsrandbereich sehr große Werte annehmen.
Gründe hierfür sind zum einen das Versagen der FHSS-Theorie in den Kon-
zentrationsrandbereichen da in diesen nicht mehr von einer statistischen Ver-
teilung ausgegangen werden kann, und zum anderen der ungewisse Verlauf

ZUSAMMENFASSUNG 122
der Soliduskurve des PEG. Dieser wurde in dieser Arbeit als Gerade angege-
ben, wobei ein Beweis für einen derartigen Verlauf nicht gegeben werden kann.
Andere kompliziertere Verläufe sind denkbar, die die Wechselwirkungsgrößen
in der Lösung und im Mischkristall beeinflussen.
Ein Vergleich der Wechselwirkungsgrößen mit denen anderer Autoren zeigt,
dass im Bereich der Raumtemperatur das System PEG/Wasser sehr gut durch
einen einfachen Ansatz der Wechselwirkungsfunktion beschrieben werden
kann, und in anderen Temperaturbereichen ein anderer Ansatz gewählt werden
muss.
Ein allgemeiner Ansatz für die Wechselwirkungsfunktion des Systems
PEG/Wasser der alle Phänomene wie die Mischkristallbildung oder die
Mischungslücke bei höheren Temperaturen berücksichtigt, kann nicht gegeben
werden. Es wurde gezeigt, welchen Verlauf diese Wechselwirkungsfunktion
haben muss. Für eine allgemeine konsistente Beschreibung des Phasen-
verhaltens des Systems PEG/Wasser müssen kettenlängenabhängige
Exzessfunktionen abhängig von der Temperatur und der Zusammensetzung
unter Berücksichtigung der Polydispersität erstellt werden.
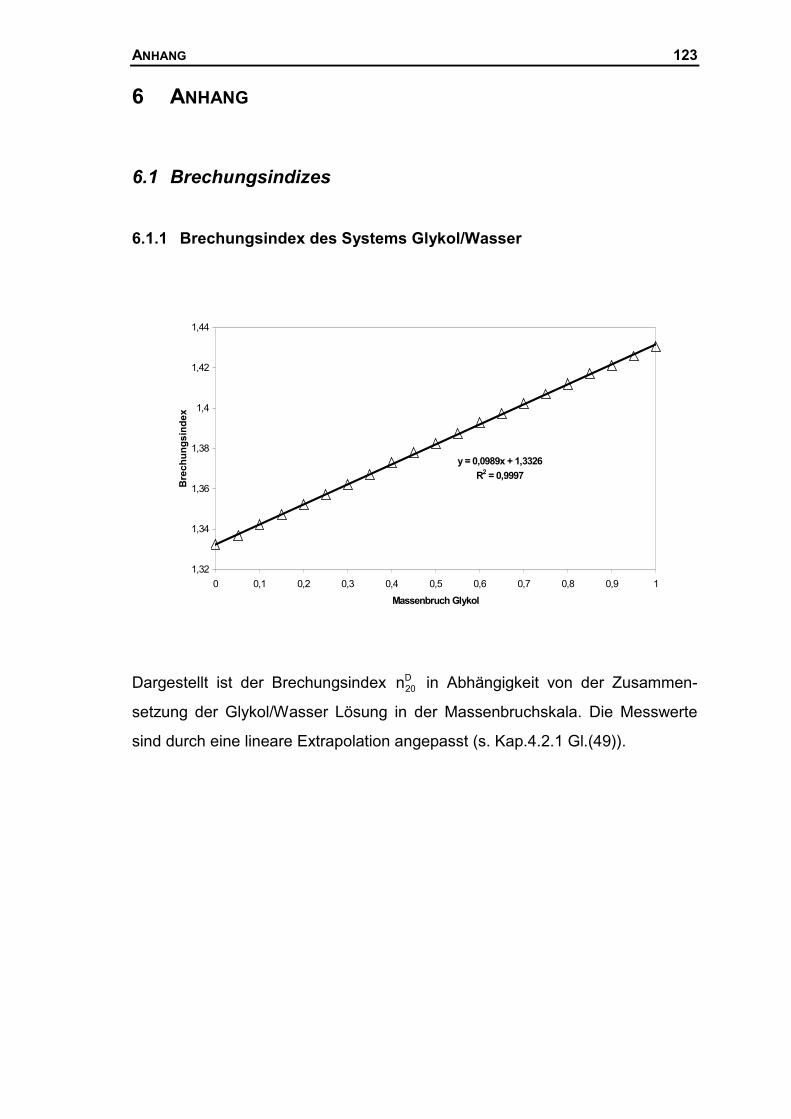
ANHANG 123
6 ANHANG
6.1 Brechungsindizes
6.1.1 Brechungsindex des Systems Glykol/Wasser
Dargestellt ist der Brechungsindex D20n in Abhängigkeit von der Zusammen-
setzung der Glykol/Wasser Lösung in der Massenbruchskala. Die Messwerte
sind durch eine lineare Extrapolation angepasst (s. Kap.4.2.1 Gl.(49)).
Brechungsindex in Abhängigkeit der Zusammensetzungfür das System Glykol - Wasser
y = 0,0989x + 1,3326R2 = 0,9997
1,32
1,34
1,36
1,38
1,4
1,42
1,44
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1Massenbruch Glykol
Bre
chun
gsin
dex
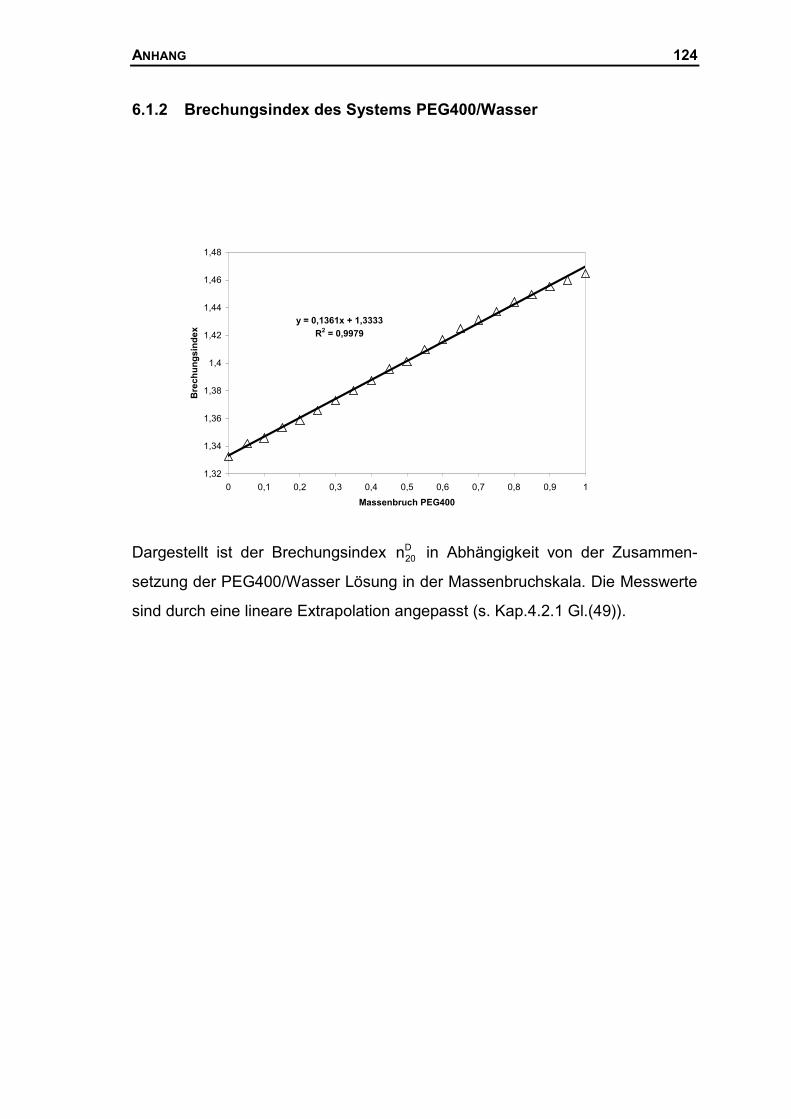
ANHANG 124
6.1.2 Brechungsindex des Systems PEG400/Wasser
Dargestellt ist der Brechungsindex D20n in Abhängigkeit von der Zusammen-
setzung der PEG400/Wasser Lösung in der Massenbruchskala. Die Messwerte
sind durch eine lineare Extrapolation angepasst (s. Kap.4.2.1 Gl.(49)).
Brechungsindex in Abhängigkeit der Zusammensetzungfür das System PEG400 - Wasser
y = 0,1361x + 1,3333R2 = 0,9979
1,32
1,34
1,36
1,38
1,4
1,42
1,44
1,46
1,48
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1Massenbruch PEG400
Bre
chun
gsin
dex
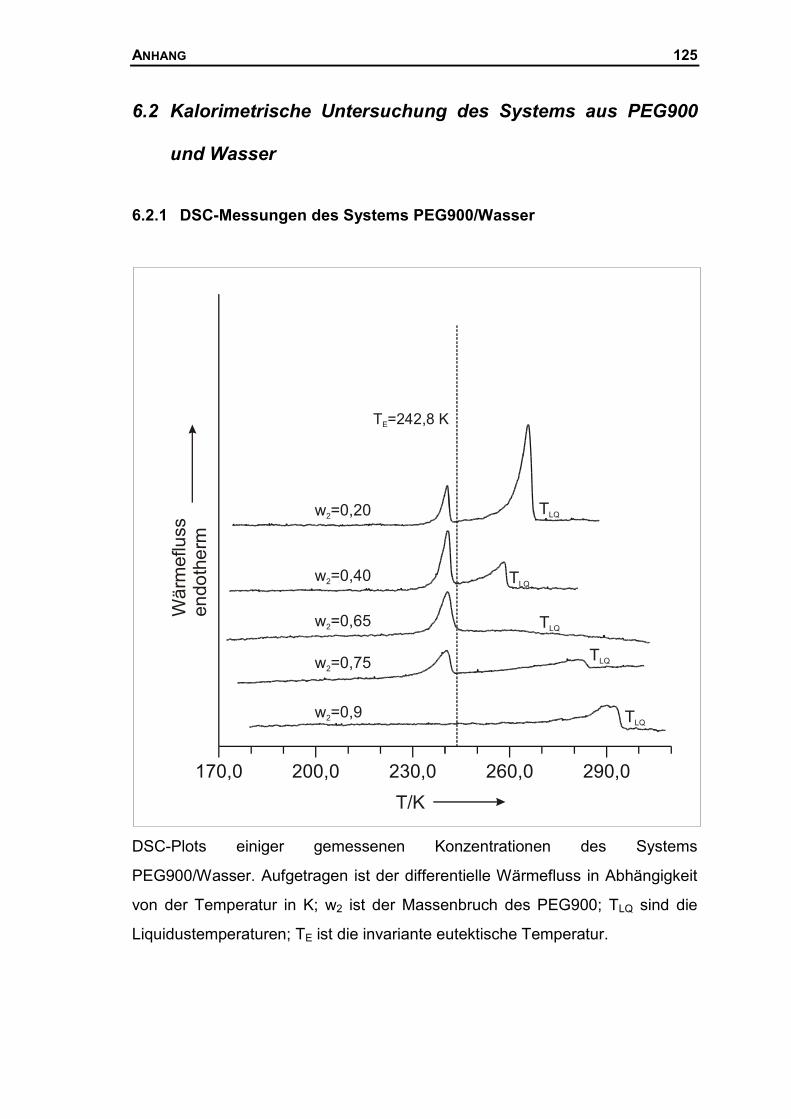
ANHANG 125
6.2 Kalorimetrische Untersuchung des Systems aus PEG900
und Wasser
6.2.1 DSC-Messungen des Systems PEG900/Wasser
DSC-Plots einiger gemessenen Konzentrationen des Systems
PEG900/Wasser. Aufgetragen ist der differentielle Wärmefluss in Abhängigkeit
von der Temperatur in K; w2 ist der Massenbruch des PEG900; TLQ sind die
Liquidustemperaturen; TE ist die invariante eutektische Temperatur.
170,0 200,0 230,0 260,0 290,0
Wär
mef
luss
endo
ther
m
T/K
T =242,8 KE
w =0,202
w =0,652
w =0,752
w =0,402
w =0,92 TLQ
TLQ
TLQ
TLQ
TLQ
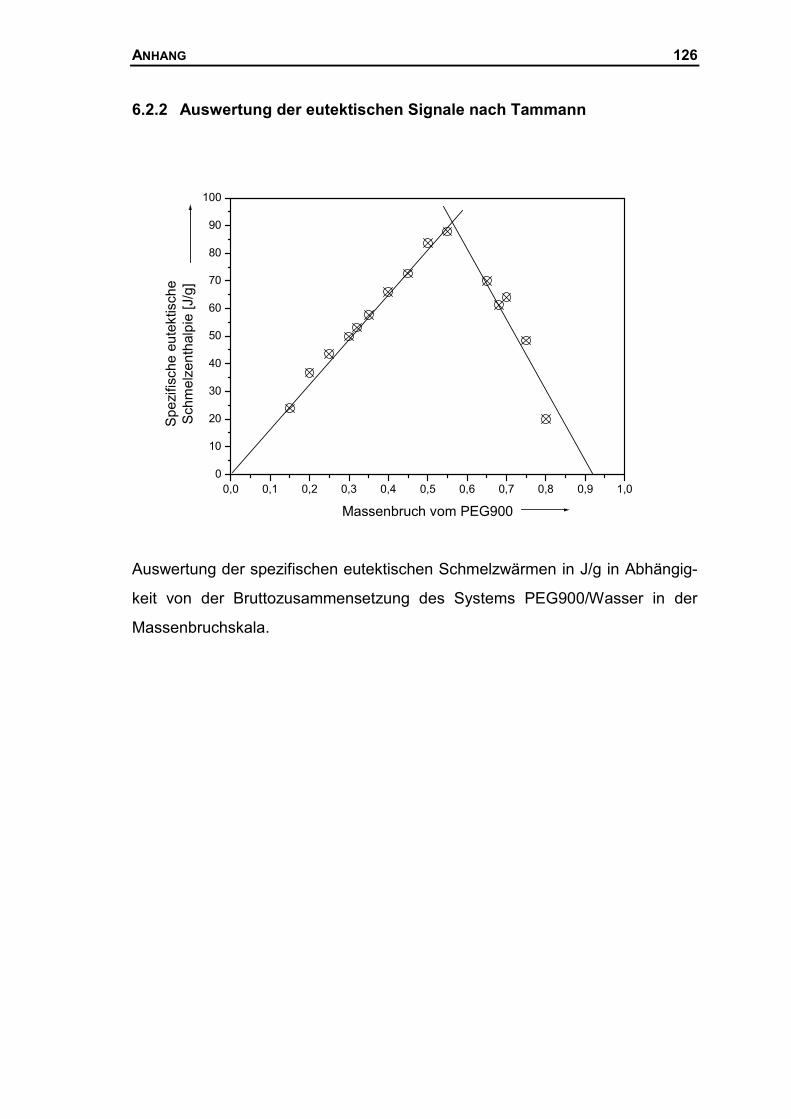
ANHANG 126
6.2.2 Auswertung der eutektischen Signale nach Tammann
Auswertung der spezifischen eutektischen Schmelzwärmen in J/g in Abhängig-
keit von der Bruttozusammensetzung des Systems PEG900/Wasser in der
Massenbruchskala.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
10
20
30
40
50
60
70
80
90
100
Spez
ifisc
he e
utek
tisch
eSc
hmel
zent
halp
ie [J
/g]
Massenbruch vom PEG900
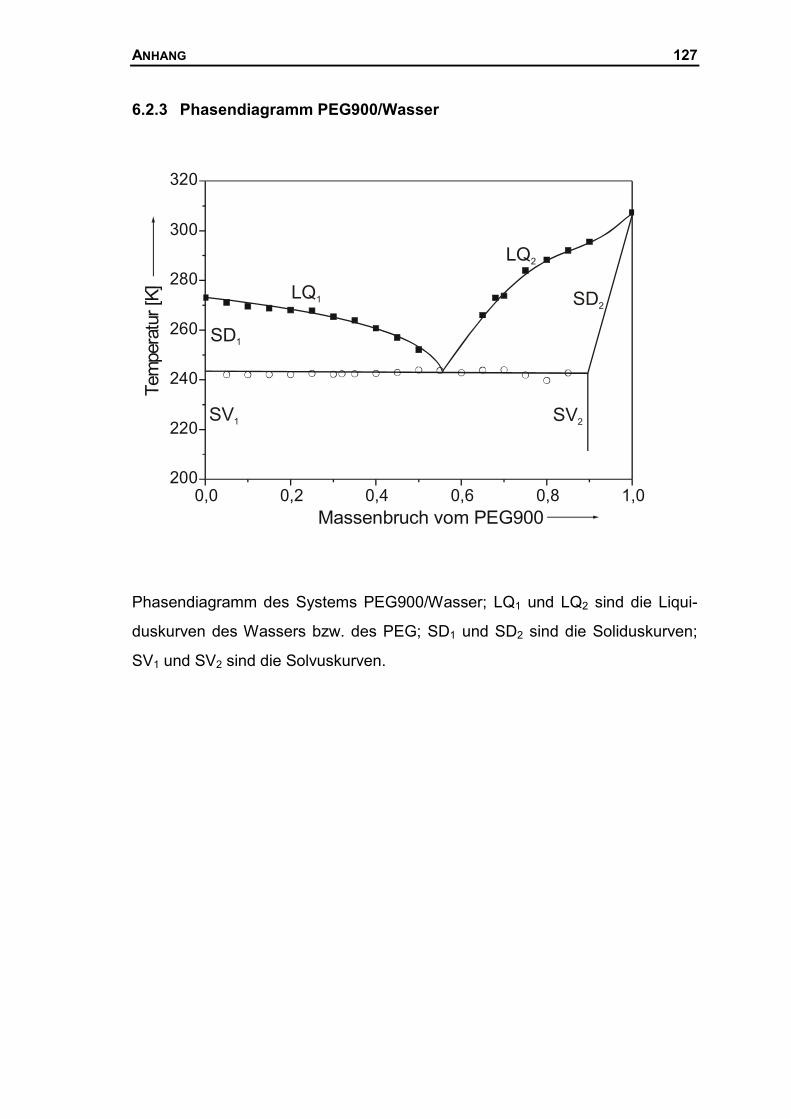
ANHANG 127
6.2.3 Phasendiagramm PEG900/Wasser
Phasendiagramm des Systems PEG900/Wasser; LQ1 und LQ2 sind die Liqui-
duskurven des Wassers bzw. des PEG; SD1 und SD2 sind die Soliduskurven;
SV1 und SV2 sind die Solvuskurven.
0,0 0,2 0,4 0,6 0,8 1,0200
220
240
260
280
300
320Te
mpe
ratu
r[K]
Massenbruch vom PEG900
SD1
SV1
LQ1 SD2
LQ2
SV2
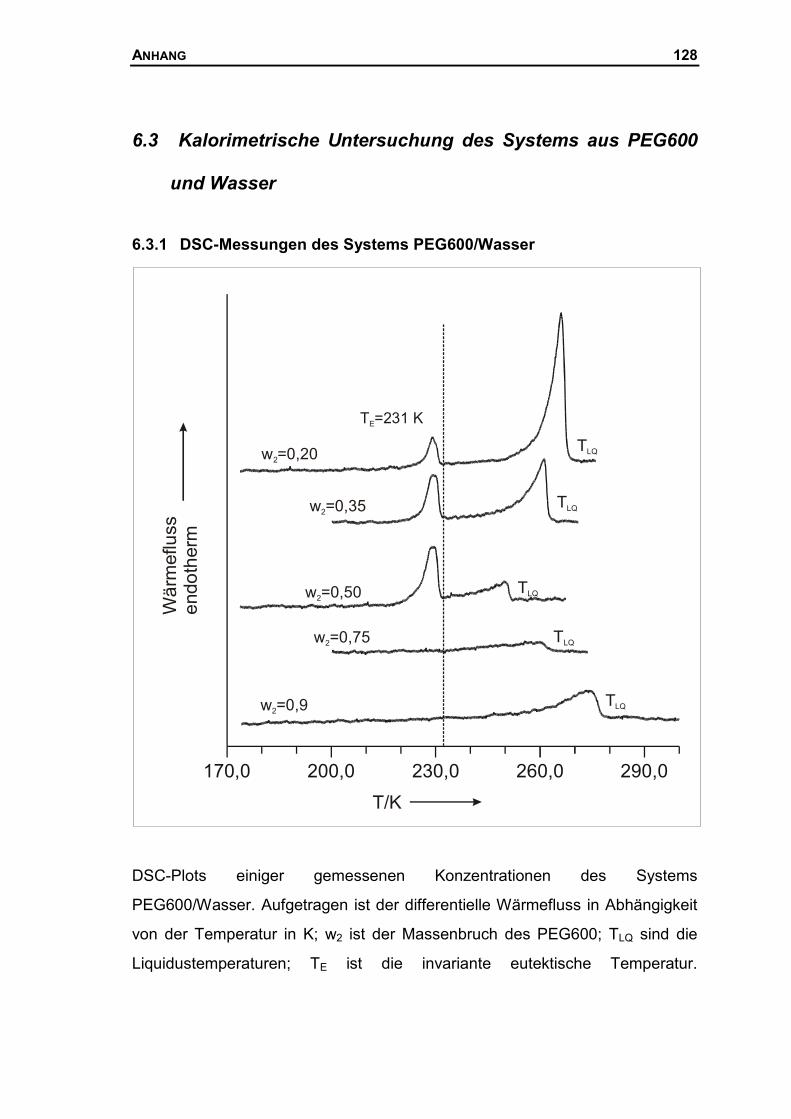
ANHANG 128
6.3 Kalorimetrische Untersuchung des Systems aus PEG600
und Wasser
6.3.1 DSC-Messungen des Systems PEG600/Wasser
DSC-Plots einiger gemessenen Konzentrationen des Systems
PEG600/Wasser. Aufgetragen ist der differentielle Wärmefluss in Abhängigkeit
von der Temperatur in K; w2 ist der Massenbruch des PEG600; TLQ sind die
Liquidustemperaturen; TE ist die invariante eutektische Temperatur.
Wär
mef
luss
endo
ther
m
T/K
T =231 KE
w =0,202
w =0,502
w =0,752
w =0,352
w =0,92TLQ
TLQ
TLQ
TLQ
TLQ
170,0 200,0 230,0 260,0 290,0
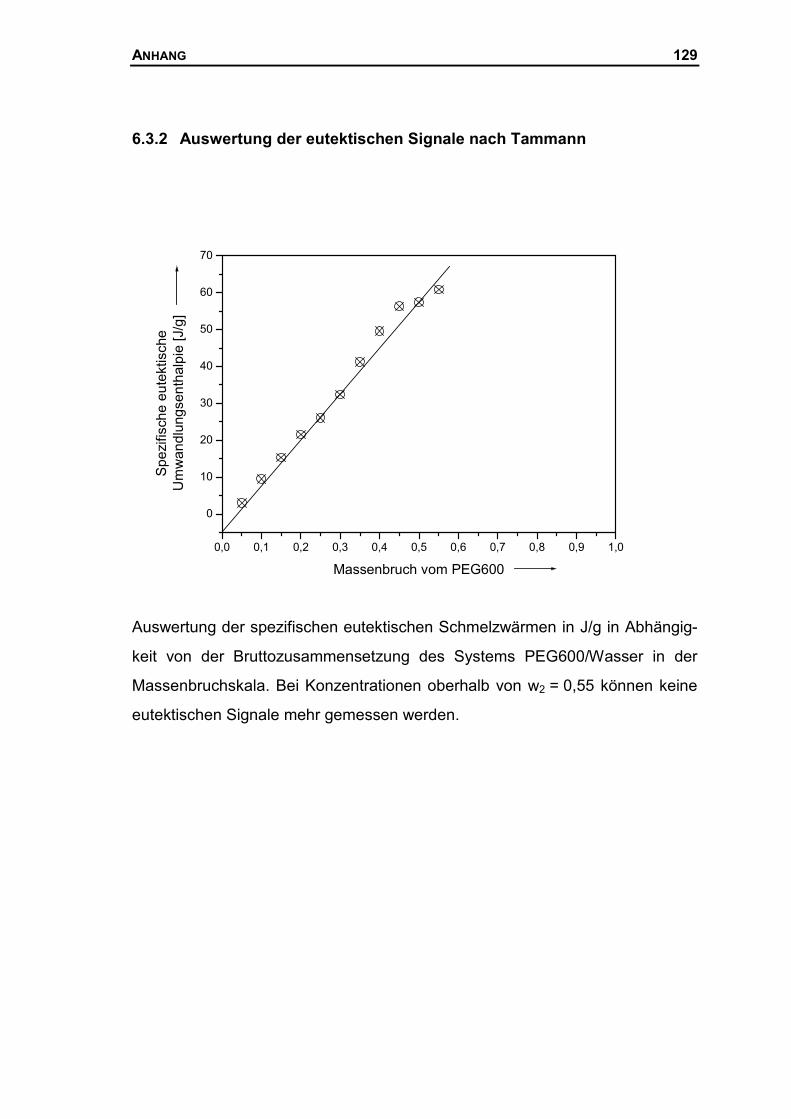
ANHANG 129
6.3.2 Auswertung der eutektischen Signale nach Tammann
Auswertung der spezifischen eutektischen Schmelzwärmen in J/g in Abhängig-
keit von der Bruttozusammensetzung des Systems PEG600/Wasser in der
Massenbruchskala. Bei Konzentrationen oberhalb von w2 = 0,55 können keine
eutektischen Signale mehr gemessen werden.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
0
10
20
30
40
50
60
70
Spez
ifisc
he e
utek
tisch
eU
mw
andl
ungs
enth
alpi
e [J
/g]
Massenbruch vom PEG600
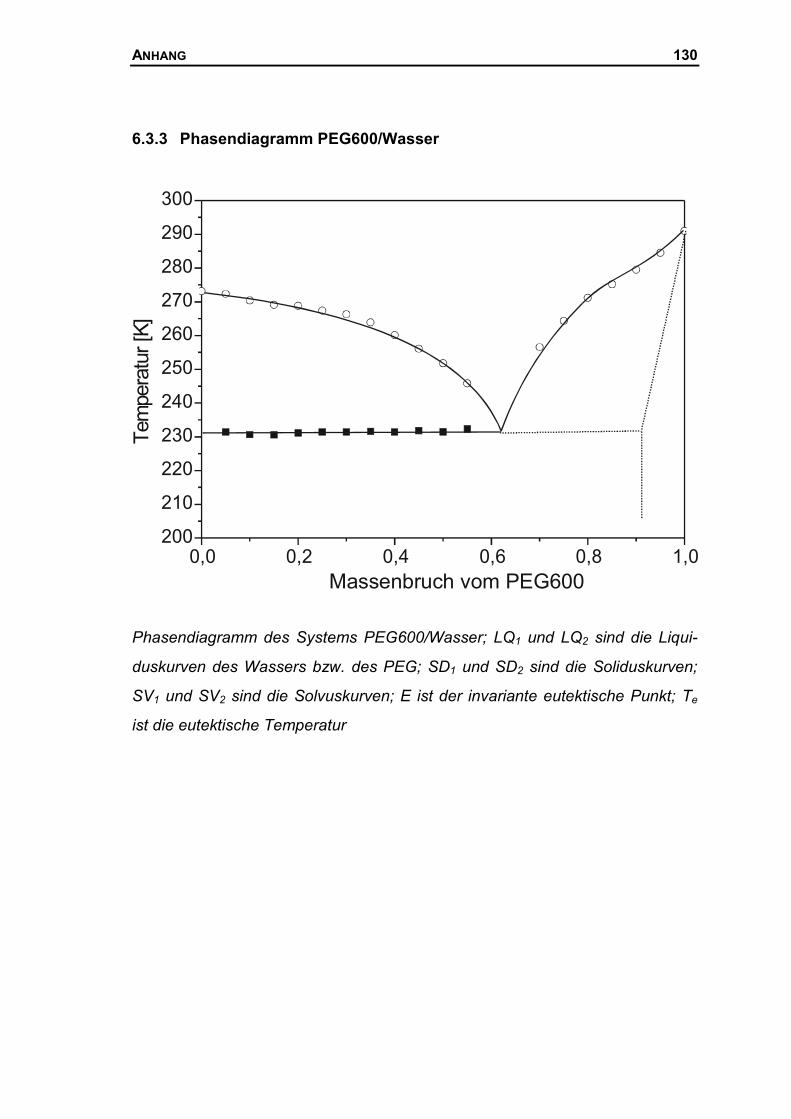
ANHANG 130
6.3.3 Phasendiagramm PEG600/Wasser
Phasendiagramm des Systems PEG600/Wasser; LQ1 und LQ2 sind die Liqui-
duskurven des Wassers bzw. des PEG; SD1 und SD2 sind die Soliduskurven;
SV1 und SV2 sind die Solvuskurven; E ist der invariante eutektische Punkt; Te
ist die eutektische Temperatur
0,0 0,2 0,4 0,6 0,8 1,0200
210
220
230
240
250
260
270
280
290
300
Tem
pera
tur[
K]
Massenbruch vom PEG600
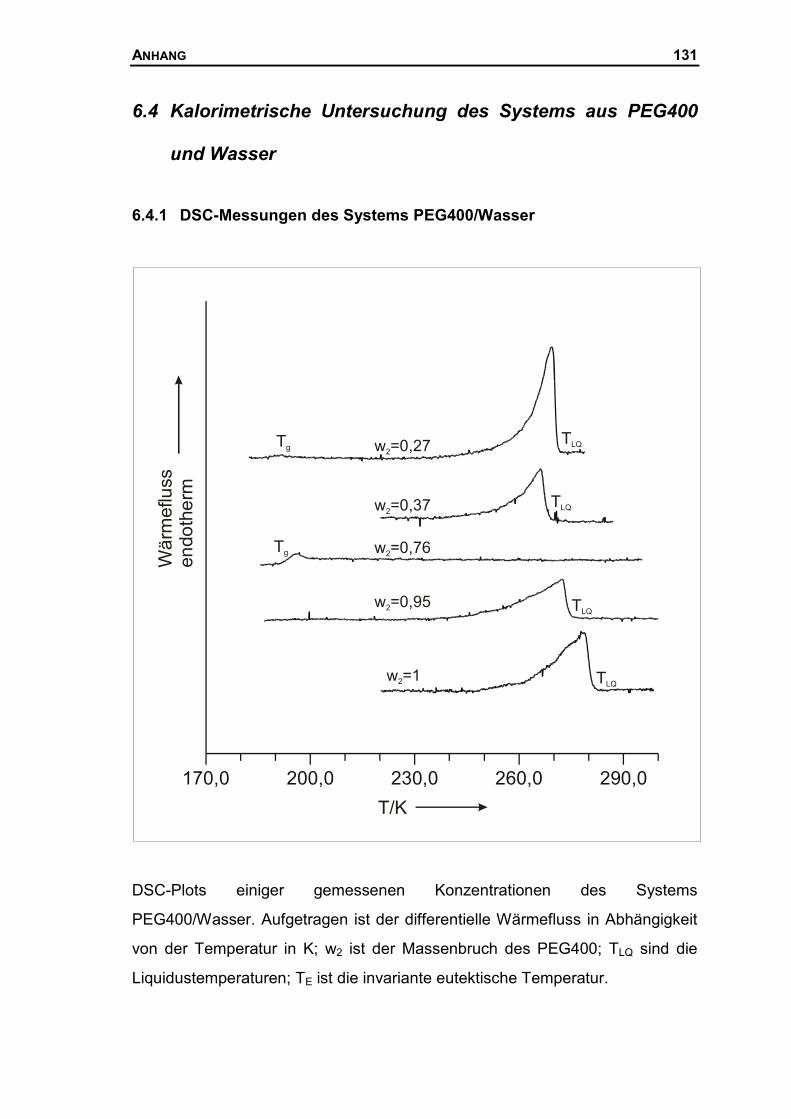
ANHANG 131
6.4 Kalorimetrische Untersuchung des Systems aus PEG400
und Wasser
6.4.1 DSC-Messungen des Systems PEG400/Wasser
DSC-Plots einiger gemessenen Konzentrationen des Systems
PEG400/Wasser. Aufgetragen ist der differentielle Wärmefluss in Abhängigkeit
von der Temperatur in K; w2 ist der Massenbruch des PEG400; TLQ sind die
Liquidustemperaturen; TE ist die invariante eutektische Temperatur.
170,0 200,0 230,0 260,0 290,0
Wär
mef
luss
endo
ther
m
T/K
TLQ
TLQ
TLQ
TLQ
Tg
Tg
w =0,372
w =0,272
w =0,952
w =12
w =0,762
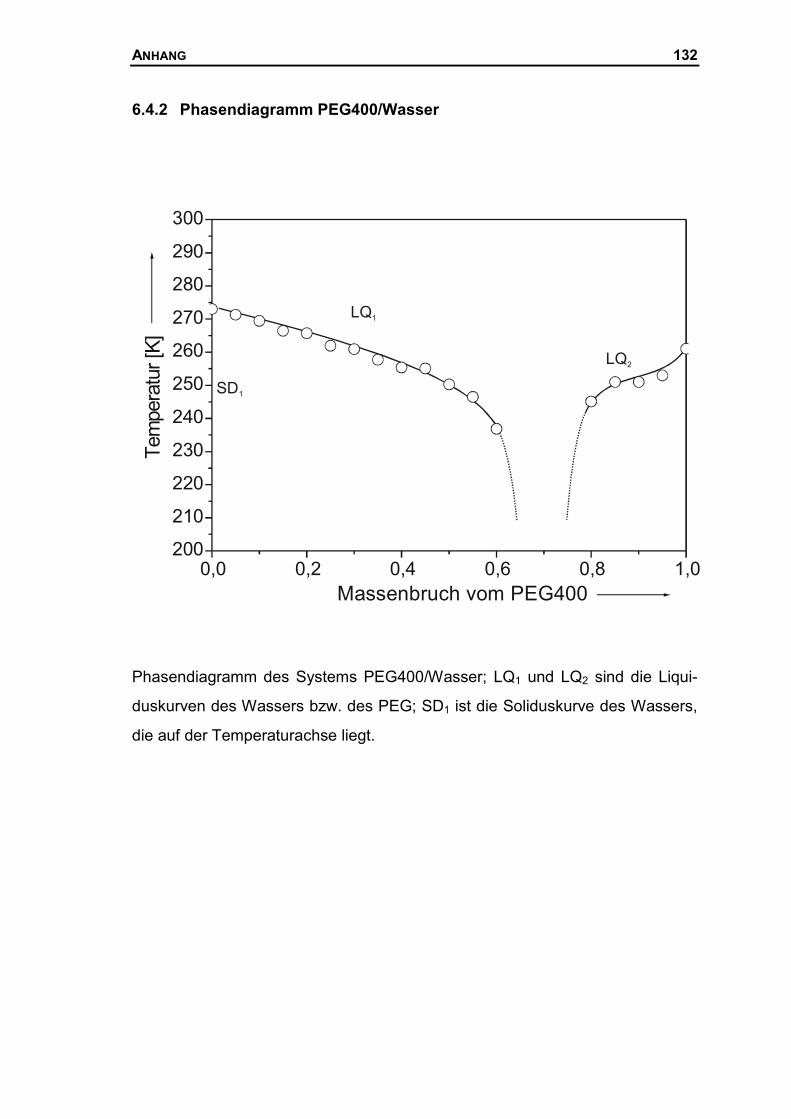
ANHANG 132
6.4.2 Phasendiagramm PEG400/Wasser
Phasendiagramm des Systems PEG400/Wasser; LQ1 und LQ2 sind die Liqui-
duskurven des Wassers bzw. des PEG; SD1 ist die Soliduskurve des Wassers,
die auf der Temperaturachse liegt.
LQ1
LQ2
SD1
0,0 0,2 0,4 0,6 0,8 1,0200
210
220
230
240
250
260
270
280
290
300
Tem
pera
tur[
K]
Massenbruch vom PEG400
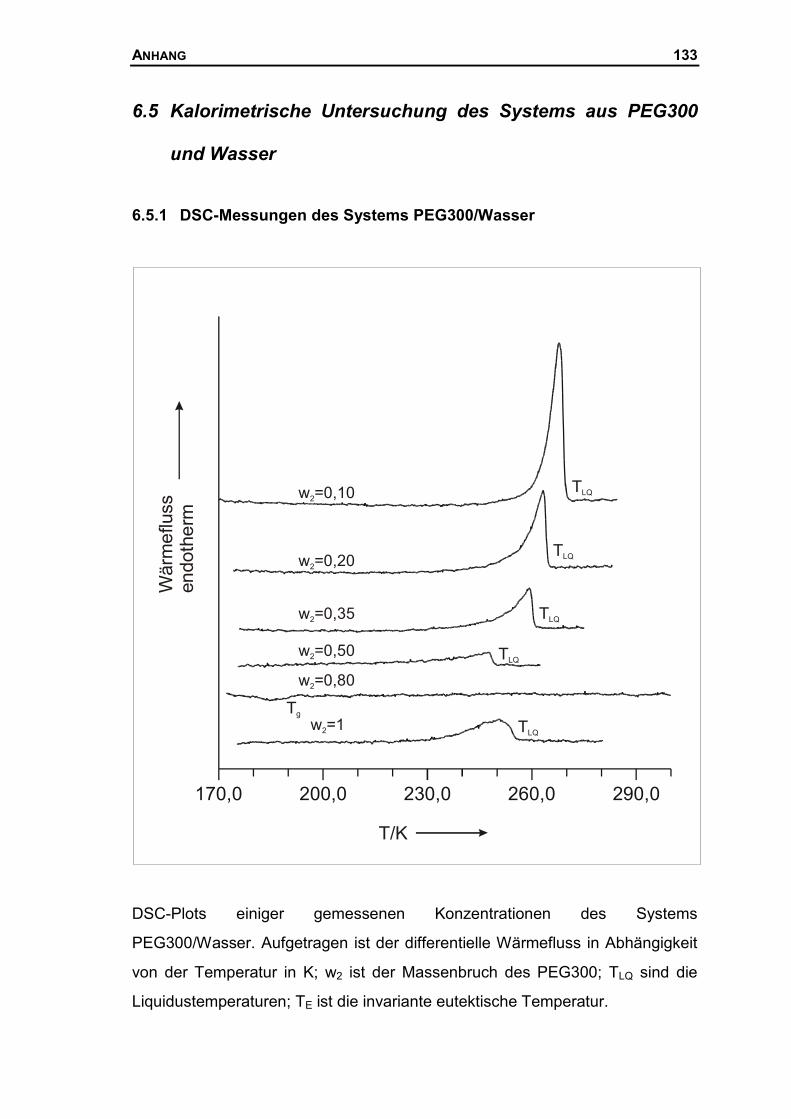
ANHANG 133
6.5 Kalorimetrische Untersuchung des Systems aus PEG300
und Wasser
6.5.1 DSC-Messungen des Systems PEG300/Wasser
DSC-Plots einiger gemessenen Konzentrationen des Systems
PEG300/Wasser. Aufgetragen ist der differentielle Wärmefluss in Abhängigkeit
von der Temperatur in K; w2 ist der Massenbruch des PEG300; TLQ sind die
Liquidustemperaturen; TE ist die invariante eutektische Temperatur.
Wär
mef
luss
endo
ther
m
T/K
w =0,202
w =0,102
w =0,502
w =0,802
w =0,352
w =12
TLQ
TLQ
Tg
TLQ
TLQ
TLQ
170,0 200,0 230,0 260,0 290,0
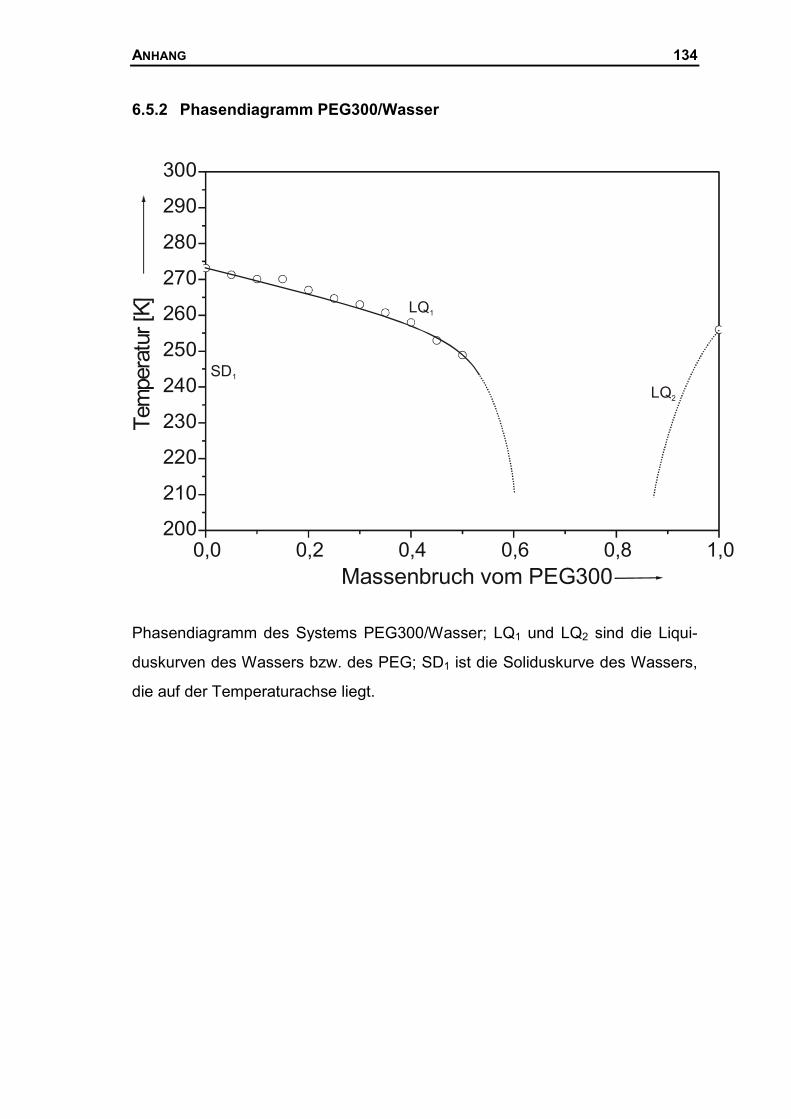
ANHANG 134
6.5.2 Phasendiagramm PEG300/Wasser
Phasendiagramm des Systems PEG300/Wasser; LQ1 und LQ2 sind die Liqui-
duskurven des Wassers bzw. des PEG; SD1 ist die Soliduskurve des Wassers,
die auf der Temperaturachse liegt.
0,0 0,2 0,4 0,6 0,8 1,0200
210
220
230
240
250
260
270
280
290
300Te
mpe
ratu
r[K]
Massenbruch vom PEG300
LQ1
LQ2
SD1

LITERATURVERZEICHNIS 135
7 LITERATUR
Adames W., Breuer W., Michalczyk A., Borchard W.; Eur. Pol. J., 25, 1989, 947
Arlie J.P., Spegt P.A. Skoulios A.E.; Makromol. Chem., 104, 1967, 212
Arlie J.P., Spegt P.A. Skoulios A.E.; Makromol. Chem., 99, 1966, 160
Bailey F.E.Jr., France H.G.; J. Pol. Sci. A, 49, 1961, 398
Bogdanov B., Mihailov M.; J. Pol. Sci., 23, 1985, 2149
Borchard W., Jablonski P.; Dechema Forschungsbericht, 2000
Buckley C.P., Kovacs A.J.; Col. Pol. Sci., 254, 1976, 695
Craig D.Q.M., Newton J.M.; Int. J. Pharm., 76, 1991, 17
Cunninghame G., Malcolm G.N.; J. Phys. Chem., 65, 1961, 1454
Davidson R.L.; Handbook of water-soluble gums and resins; McGraw-Hill, New
York, 1980
de Vringer T., Joosten J.G.H., Junginger H.E.; Col. Pol. Sci., 264, 1986, 623
Delahaye N., Duclos R., Saiter J.M.; Int. J. Pharm., 157, 1997, 27
Dobnik E.; Dissertation Duisburg, 1991
Eagland D., Crowther N.J., Butler C.J.; Polymer, 34, 1993, 2804
Elias, H.-G.; Makromoleküle; Hütig & Wepf: Basel, Heidelberg, 1971
Eyring H., Henderson D., Jost W.; Physical Chemistry: An Advanced Treatise,
Volume I: Thermodynamics, Academic Press, New York, 1971
Fischer V., Borchard W.; J. Phys. Chem. B, 104, 2000, 4463
Fischer V., Borchard W., Karas M.; J. Phys. Chem., 100, 1996, 15992
Fischer V.; Dissertation Duisburg, 2001
Flory P.J.; Principles of Polymer Chemistry, Cornell University Press, Ithaca –
N.Y., 1953
Frahn S.; Dissertation Duisburg, 1997

LITERATURVERZEICHNIS 136
Franks F. ; Biophysics and Biochemistry at Low Temperatures, Univ. Press.,
New York, Cambridge, 1985
Gibbs J.W.; Collected Works Vol. I : Thermodynamics, New Haven, 1948
H. G. Könnecke, H. Steinert and E. Leibnitz; Z. Phys. Chem., 208, 1957, 147
Haase R.; Survey of Fundamental Laws; Academic Press, New York; 1971,
Kap. 1
Haase R.; Thermodynamik der Mischphasen; Springer, Berlin, 1956
Hager S.L., Macrury T.B.; J. Apl. Pol. Sci., 25, 1980, 1559
Hemminger W., Cammenga H.; Methoden der thermischen Analyse, Springer,
Berlin, 1989
Hemminger W., Höhne G.; Calorimetry, VCH, Weinheim, 1984
Hemminger W., Höhne G.; Grundlagen der Kalorimetrie, VCh: Weinheim, 1979
Hemminger W., Sarge S.M.; J. Therm. Anal., 37, 1991, 1455
Hey M.J., Ilett S.M.; J. chem. Soc. Far. Trans., 87, 1991, 3671
Huang L., Nishinari K.; J. Pol. Sci., 39, 2001, 496
Huggins M.L.; J. Phys. Chem., 46, 1942, 151
Huggins M.L.; Physical chemistry of high polymers, Wiley & Sons, New York,
1958
Jablonski P., Borchard W.; In Vorbereitung
Jablonski P., Golloch A., Borchard W.; Thermochimica Acta, 333, 1999, 87
Jablonski P., Müller-Blecking A., Borchard W.; J. Therm. Anal. Cal., eingereicht
Kagemoto A., Murakami S., Fujishiro R.; Makromolekulare Chemie, 124, 1969,
222
Kibbe A.H.; Handbook of Pharmaceutical Excipients, Am. pharm association,
1994
Kitaigorodsky A.I.; Mixed Crystals; Springer, Berlin, 1984, Kap. 8-12

LITERATURVERZEICHNIS 137
Koningsveld R., Stockmayer W.H., Nies E.; Polymer Phase Diagrams, Oxford
Univ. Press, 2001
Könneke H.G., Steinert H., Leibnitz E.; Z. Phys. Chem., 208, 1957, 147
Kubaschewski O., Hultgren R.,;Metallurgical and Alloy Thermochemistry in:
Experimental Thermochemistry. Vol.II, Interscience Publishers, New York, 1962
Landolt Börnstein, New series, Vol. IV: Physical Chemistry; IV/2; Heats of
Mixing and Solution
Lewis G.N., Randall M.; Thermodynamics; McGraw-Hill; New York, 1961
Lewis G.N.; J. Amer. Chem. Soc., 30, 1908, 668
Malcom G.N.; Rowlinson J.S.; Trans. Far. Soc., 53, 1957, 921
Michalczyk A.; Borchard W.; Eur. Pol. J., 25, 1989, 947
Müller A.; Dissertation Duisburg, 1998
Müller, A., Borchard, W.; J. Phys. Chem., 101, 1997, 4283
Müller, A.; Borchard, W.; J. Phys. Chem., 101, 1997, 4307
Müller-Blecking A, Borchard W.; in Vorbereitung
Oelsen W., Rieskamp K. H., Oelsen O.; Archiv für das Hüttenwesen, 26, 1955,
253
Oelsen, W.; Tebbe, W.; Oelsen, O.; Archiv f. Eisenhüttenwesen, 27, 1956, 689
Point J.J., Coutelier C.; J. Pol. Sci., Pol Phys. Ed., 23, 1985, 231
Point J.J., Demaret J.P.; J. Phys. Chem., 91, 1987, 797
Point J.J.; J. Inclusion Phen., 6, 1988, 253
Prigogine I., Defay R.; Chemical Thermodynamics, Longmans Green & Co,
London, 1952
Rehage G., Borchard W.; Physics of glassy polymers, Ed. Haward R.N., 1973,
54
Rehage G.; Z. Elektrochem., 59, 1955, 78
Reutner P., Luft B., Borchard W.; Col. Pol. Sci., 263, 1985, 519

LITERATURVERZEICHNIS 138
Saeki F., Kuwahara N., Kaneko M.; Polymer, 17, 1976, 685
Saiter J.M., Bayard J., Delahaye N., Varbier S., Vautier C.; J. Therm. Anal., 45,
1995, 1223
Scatchard G.; Chem. Rev., 44, 1949, 7
Schönert H., Monshausen F.; Col. Pol. Sci., 258, 1980, 578
Siemens W.; Dissertation Clausthal-Zellerfeld, 1976
Smith P., Pennings A.J.; Polymer, 15, 1974, 413
Spegt P.A.; Makromol. Chem., 140, 1970, 167
Staverman A.J., van Santen J.M.; Rec. Tra. Chim., 60, 1941, 76
Steinbrecht U.; Dissertation Duisburg, 1991
Strobl G.; The Physics of Polymers, Springer, Berlin, 1996
Tadokoro H., Yoshihara T., Chatani Y., Murahashi S.; J. Pol. Sci. A, 2, 1964,
363
Tadokoro H.; Structure of crystalline polymers; Kap. 4.6.4, 99, Wiley & Sons,
New York, 1979
Takahashi Y., Tadokoro H.; Macromolecules, 6, 1973, 672
Tammann G.; Lehrbuch der Heterogenen Gleichgewichte, Friedr. Vieweg &
Sohn, Braunschweig, 1924
Tammann G.; Z. Anorg. Chem., 37, 1903; 303
Tomlinson A., Chylewski Ch., Simon W., Tetrahedron, 19, 1963, 949
Vanhee S., Kiepen F.-H., Brinkmann D., Borchard W., Koningsveld. R.,
Berghmans, H.; Macromol. Chem. Phys., 195, 1994, 759
Volmer M.; Kinetik der Phasenbildung, Theodor Steinkopf, Dresden, 1939

Lebenslauf
Name: Pascal JablonskiGeboren am: 26.08.1972 in Mülheim an der RuhrFamilienstand: verheiratet
SCHULBILDUNG
1979 – 1983 Grundschule in Duisburg Meiderich1983 – 1992 Max Planck Gymnasium in Duisburg Meiderich
Abschluss: Allgemeine Hochschulreife
STUDIUM
10/1993 – 02/1996 Grundstudium Chemie an der Gerhard-Mercator-Universität DuisburgAbschluss: Diplom-Vorprüfung Chemie
03/1996 – 10/1998 Hauptstudium Chemie an der Gerhard-Mercator-Universität DuisburgAbschluss: Diplomprüfung Chemie
seit 11/1998 Promotion an der Gerhard-Mercator-Universität Duisburg


![Untersuchungen an Gläsern des Systems CaF2-Al2O3 2 und ... · 46 und 50 % [2]. Jahrelang herrschte die Meinung vor, dass Amalgam ohne Bedenken verwendet werden kann. Aber schon 1927](https://static.fdokument.com/doc/165x107/5e8fd39b7d2706096a1ba763/untersuchungen-an-glsern-des-systems-caf2-al2o3-2-und-46-und-50-2-jahrelang.jpg)















