
Kernresonanz-Spektroskopie N uclear M agnetic R esonance-( NMR )-Spektroskopie
12
Kernresonanz-Spektroskopie Nuclear Magnetic Resonance-(NMR)-Spektroskopie Historisches 1938 Messung des Kernspins von I. Rabi 1946 Entdeckung des NMR-Effektes in den Arbeitsgruppen Bloch und Purcell 1952 Nobelpreis für Bloch und Purcell 1965 Beginn der Entwicklung der Fourier-Transformations(FT)-Technik (R. Ernst) 1973 Entwicklung des NMR-Imaging-Verfahrens (MRI) (Einsatz u.a. in der Medizin-Diagnostik als Kernspin-Tomographie) 1991 Nobelpreis für R. Ernst (FT-Methode) 2002 Nobelpreis für K. Wüthrich (Strukturaufklärung von Proteinen mit NMR) 2003 Nobelpreis für Lauterbur und Mansfield (MRI-Verfahren)
description
Kernresonanz-Spektroskopie N uclear M agnetic R esonance-( NMR )-Spektroskopie. Historisches. 1938Messung des Kernspins von I. Rabi 1946Entdeckung des NMR-Effektes in den Arbeitsgruppen Bloch und Purcell 1952 Nobelpreis für Bloch und Purcell 1965 Beginn der Entwicklung der - PowerPoint PPT Presentation
Transcript of Kernresonanz-Spektroskopie N uclear M agnetic R esonance-( NMR )-Spektroskopie

Kernresonanz-SpektroskopieNuclear Magnetic Resonance-(NMR)-Spektroskopie
Historisches
1938 Messung des Kernspins von I. Rabi
1946 Entdeckung des NMR-Effektes in den
Arbeitsgruppen Bloch und Purcell
1952 Nobelpreis für Bloch und Purcell
1965 Beginn der Entwicklung der
Fourier-Transformations(FT)-Technik (R. Ernst)
1973 Entwicklung des NMR-Imaging-Verfahrens (MRI)
(Einsatz u.a. in der Medizin-Diagnostik als Kernspin-Tomographie)
1991 Nobelpreis für R. Ernst (FT-Methode)
2002 Nobelpreis für K. Wüthrich (Strukturaufklärung von Proteinen mit NMR)
2003 Nobelpreis für Lauterbur und Mansfield (MRI-Verfahren)

Der NMR-Effekt basiert auf magnetischen Eigenschaften von Atomkernen. Viele Atomkerne besitzen einen Eigendrehimpuls P, auch Kernspin genannt, der dem Kern besondere magnetische Eigenschaften verleiht. Vereinfacht betrachtet verhalten sich solche Kerne wie kleine Magnete.
Auch die Elemente Wasserstoff und Kohlenstoff, aus denen zum größten Teil die Verbindungen der belebten Natur bestehen, besitzen mit den Isotopen 1H und 13C magnetisch aktive Kerne. Entsprechend können solche Verbindungen mit Hilfe dieser Methode besonders gut analysiert werden.
Der NMR-Effekt
)1( IIP 2
h
I: Drehimpulsquantenzahl(I=0, 1/2, 1, 3/2, 2, 5/2,..., 6)
h: Plancksches Wirkungsquantum
Kern-Drehimpuls P

Kerne im statischen homogenen Magnetfeld B0
Die Spins der Atomkerne können sich beliebig im Raum ausrichten.
Dies ändert sich jedoch, wenn man die Kerne in ein statisches, homogenes Magnetfeld (B0) bringt. Wie bei zwei Magneten, die man einander annähert, wird die beliebige Ausrichtung im Raum aufgehoben, und es gibt für die Kernspins nur bestimmte Einstellungsmöglichkeiten relativ zum äußeren Magnetfeld.
Beispiel: bei Kernen mit I=1/2 (z.B. 1H) gibt es zwei Ausrichtungsmöglichkeiten: parallel oder antiparallel zum äußeren Magnetfeld.
Zwischen diesen Einstellungen läßt sich durch Energiezufuhr in Form von elektromagnetischer Strahlung geeigneter Wellenlänge hin- und herschalten. Dieser Prozeß wird als Anregung bezeichnet. Die benötigte Strahlungsenergie ist abhängig von
- der Kernsorte (1H, 13C, usw.)- der Stärke des äußeren Magnetfeldes- der chemischen Umgebung der Kerne
Die zur Änderung der Ausrichtung der Spins notwendige Strahlung ist relativ energiearm und liegt im Bereich der Radiowellen (s. elektromagnetisches Spektrum).
Anlegen eines äußerenMagnetfeldes B0
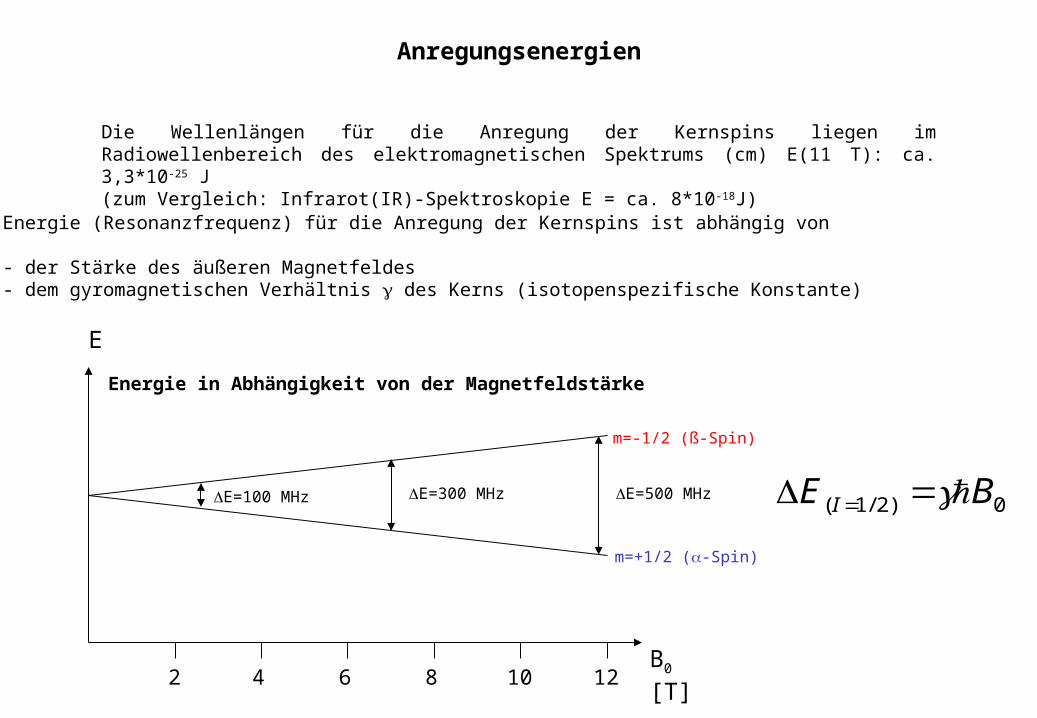
10 128642B0 [T]
E
m=-1/2 (ß-Spin)
m=+1/2 (-Spin)
E=100 MHz E=300 MHz E=500 MHz0)2/1( BE I
Die Wellenlängen für die Anregung der Kernspins liegen im Radiowellenbereich des elektromagnetischen Spektrums (cm) E(11 T): ca. 3,3*10-25 J(zum Vergleich: Infrarot(IR)-Spektroskopie E = ca. 8*10-18J)
Energie (Resonanzfrequenz) für die Anregung der Kernspins ist abhängig von
- der Stärke des äußeren Magnetfeldes- dem gyromagnetischen Verhältnis des Kerns (isotopenspezifische Konstante)
Energie in Abhängigkeit von der Magnetfeldstärke
Anregungsenergien
H1H, 2H
H1H, 2H
Li7Li
Li7Li
Na23Na
Na23Na
Be9Be
Be9Be
Mg25Mg
Mg25Mg
K39K
K39K
Ca43Ca
Ca43Ca
Rb87Rb
Rb87Rb
Sr87Sr
Sr87Sr
Cs133Cs
Cs133Cs
Ba137Ba
Ba137Ba
Sc45Sc
Sc45Sc
Ti49Ti
Ti49Ti
V50V
V50V
Cr53Cr
Cr53Cr
Mn55Mn
Mn55Mn
Co59Co
Co59Co
Ni61Ni
Ni61Ni
Cu63Cu
Cu63Cu
Zn67Zn
Zn67Zn
Ga71Ga
Ga71Ga
Ge73Ge
Ge73Ge
As75As
As75As
Y89Y
Y89Y
Zr91Zr
Zr91Zr
Nb93Nb
Nb93Nb
Mo95Mo
Mo95Mo
Ru101Ru
Ru101Ru
Pd105Pd
Pd105Pd
In115In
In115In
Sb121Sb
Sb121Sb
Ln138Ln
Ln138Ln
Hf179Hf
Hf179Hf
Ta181Ta
Ta181Ta
Re187Re
Re187Re
Ir193Ir
Ir193Ir
Au197Au
Au197Au
Bi209Bi
Bi209Bi
Br81Br
Br81Br
Kr83Kr
Kr83Kr
I127I
I127I
B11B
B11B
O 17O
O 17O
Al27Al
Al27Al
S33S
S33S
Ne21Ne
Ne21Ne
Cl35Cl
Cl35Cl
Ar
Ar
He3He
He3He
C13C
C13C
N15N
N15N
F19F
F19F
Si29Si
Si29Si
P31P
P31P
Fe57Fe
Fe57Fe
Se77Se
Se77Se
Te125Te
Te125Te
Xe129Xe
Xe129Xe
Sn119Sn
Sn119Sn
Pb207Pb
Pb207Pb
Tl205Tl
Tl205Tl
Po
Po
At
At
Rn
Rn
Hg199Hg
Hg199Hg
Pt195Pt
Pt195Pt
Rh103Rh
Rh103Rh
Os187Os
Os187Os
W183W
W183W
Tc
Tc
Ag107Ag
Ag107Ag
Cd113Cd
Cd113Cd
Fr
Fr
Ra
Ra
Ac
Ac
Pr141Pr
Pr141Pr
Nd143Nd
Nd143Nd
Sm147Sm
Sm147Sm
Eu153Eu
Eu153Eu
Tb159Tb
Tb159Tb
Er167Er
Er167Er
Tm169Tm
Tm169Tm
Yb171Yb
Yb171Yb
No
No
Pm
Pm
Ce
Ce
Gd157Gd
Gd157Gd
Dy163Dy
Dy163Dy
Ho165Ho
Ho165Ho
Lu175Lu
Lu175Lu
Md
Md
Fm
Fm
Es
Es
Cf
Cf
Bk
Bk
Cm
Cm
Am
Am
Pa
Pa
Th
Th
U235U
U235U
Pu
Pu
Np
Np
Lr
Lr
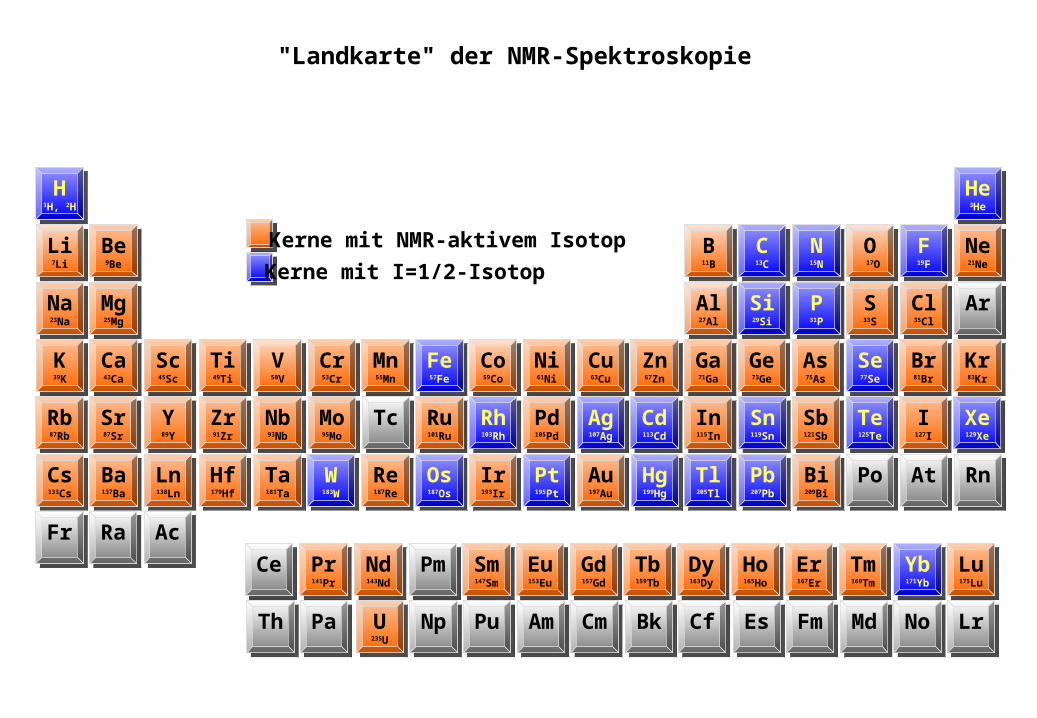
Kerne mit NMR-aktivem Isotop
Kerne mit I=1/2-Isotop
"Landkarte" der NMR-Spektroskopie

Moderner NMR-Spektrometeraufbaumit supraleitendem Magneten
Sender/Empfänger
Shim/Sweep-Einheit
MagnetpolMagnetpol
Probenröhrchen
Meßkopf
1: He-Dewar2: Probenkopf mit
Sende-/Empfangsspulen3: Probenröhrchen4: Meßkanal5: Shim-Einheit zur Homo-
genisierung des Magnet-feldes
a: Magnetspuleb: Füllöffnung He-Tankc: Füllöffnung N2-Tankd: Dewar-Mäntel
b
Aufbauprinzip eines NMR-Gerätes

Das 1H-NMR-Spektrum
01234 ppm
H C
H
H
C
O
O C
H
H
C
H
H
H
EssigsäureethylesterDas NMR-Spektrum liefertInformationen über:
Anzahl chemisch verschiedenerKerne einer Kernsorte(Beispiel: 3 Sorten von Protonen)
chemische Umgebung bzw.Bindungssituation durch chemischeVerschiebung, der Lage derResonanzen auf derppm-Skala (Beispiel: 1.25, 2.03und 4.12 ppm)
Anzahl der Protonen in derNachbarschaft über die Signal-struktur bzw. FeinaufspaltungSignales durch Kopplungen(Beispiel: grün: 3 Linien,blau: 1 Linie, rot: 4 Linien)
relatives Verhältnis der Protonen zueinander über das Integral(Fläche unter der Kurve) desSignals (Beispiel: grün: 3,blau: 3, rot: 2)
Feinaufspaltung
Integral
0.98 ppm2.03 ppm4.12 ppm

1H-Chemische Verschiebungen von organischen Verbindungen
0123456789101112ppm
C OH
OH
C SH
C NH2
C OH
O
C H
O
N
HN
HH
H
C
H
C
H
H
C O
H
C H
C N
H
C S
H
CH3
CH2CH3/
M CH3
H
H
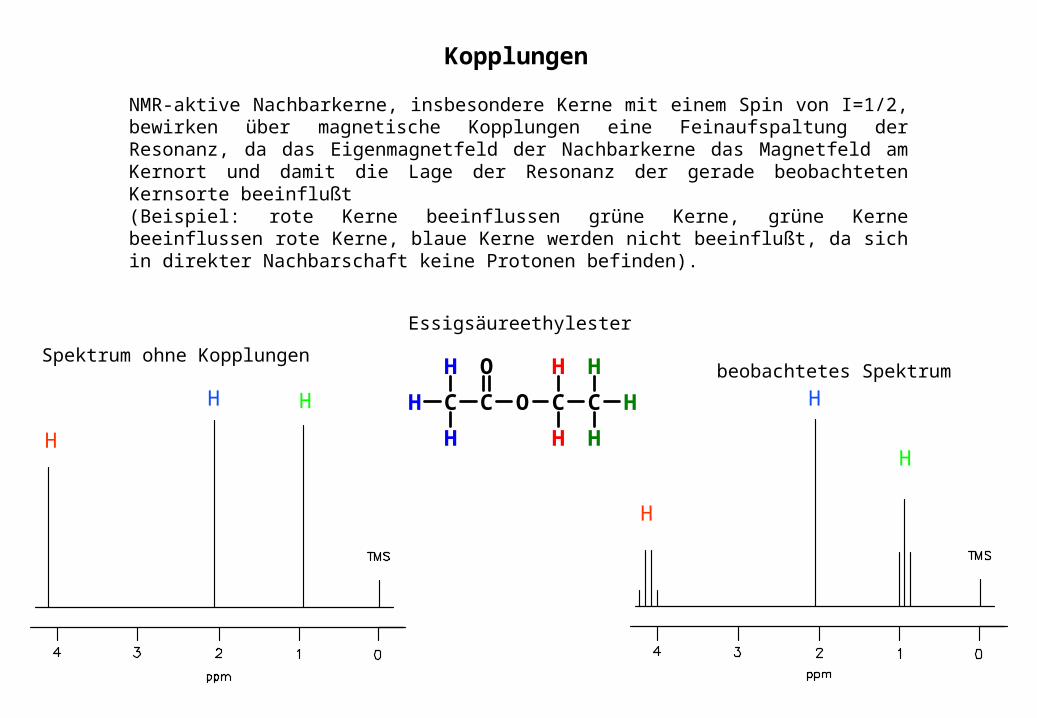
NMR-aktive Nachbarkerne, insbesondere Kerne mit einem Spin von I=1/2, bewirken über magnetische Kopplungen eine Feinaufspaltung der Resonanz, da das Eigenmagnetfeld der Nachbarkerne das Magnetfeld am Kernort und damit die Lage der Resonanz der gerade beobachteten Kernsorte beeinflußt(Beispiel: rote Kerne beeinflussen grüne Kerne, grüne Kerne beeinflussen rote Kerne, blaue Kerne werden nicht beeinflußt, da sich in direkter Nachbarschaft keine Protonen befinden).
Spektrum ohne Kopplungenbeobachtetes Spektrum
H H
H
Kopplungen
H C
H
H
C
O
O C
H
H
C
H
H
H
Essigsäureethylester
H
H
H

ppm
H=Spineinstellung parallelH=Spineinstellung antiparallel
01234
=Kopplungskonstante J [Hz]
HCHCH2
HHCH
CH2
H
HCHCH2
HHCH
CH2
H
HCHCH2
HHCH
CH2
HHCH
CH2
HHCH
CH2
H
HCH
CH3
HCH
CH3
HCH
CH3
HCH
CH3C CH
H
H
O
O C
H
H
C
H
H
H
Essigsäureethylester
KopplungenDie Spins der Protonen in der Nachbarschaft einer beobachteten Protonensorte können sich entweder parallel oder antiparallel relativ zum äußeren Magnetfeld einstellen. Daraus resultierenden Einstellungsmöglichkeiten ergeben aus der Statistik.Beispiel: CH2-Gruppe (Signal bei 4.12 ppm); für die Einstellung der Protonen in der Nachbar-CH3-Gruppe ergeben sich folgende Einstellungszustände
- alle drei Protonen sind antiparallel ausgerichtet: 1 Möglichkeit- zwei Protonen sind antiparallel und ein Proton ist parallel ausgerichtet: 3 Möglichkeiten- ein Proton ist antiparallel und zwei Protonen sind parallel ausgerichtet: 3 Möglichkeiten- alle drei Protonen sind parallel ausgerichtet: 1 Möglichkeit
Die Größe der Feinaufspaltung (Abstand zwischen den einzelnen Linien in Hz) wird als Kopplungskonstante J bezeichnet.

Multiplizitätsregeln für die Signalstruktur
n = 0 1 Singulettn = 1 1 1 Dublettn = 2 1 2 1 Triplettn = 3 1 3 3 1 Quartettn = 4 1 4 6 4 1 Quintettn = 5 1 5 10 10 5 1 Sextettn = 6 1 6 15 20 15 6 1 Septett
M = 2nI+1M: Anzahl der Linien des Signalsn: Anzahl der koppelnden Kerne
mit gleicher KopplungskonstanteI: Spinquantenzahl
bei I=1/2 vereinfacht sich das zu M=n+1
Anzahl derKopplungspartner
Muster und Intensitätder Linien des Signals
Bezeichnung derLinienstruktur

Heterokerne
Neben dem Proton lassen sich viele andere Kerne NMR-spektroskopisch beobachtenDiese Kerne werden als Heterokerne bezeichnet.Weitere wichtige Kerne mit I=1/2 sind:
Kohlenstoff-Isotop 13 (13C: natürliche Häufigkeit 1.1 %)Fluor-Isotop 19 (19F: natürliche Häufigkeit 100%)Phosphor-Isotop 31 (31P: natürliche Häufigkeit 100%)
Wesentliche Unterschiede zur 1H-NMR-Spektroskopie sind:
- geringere Empfindlichkeit z.B. durch eine geringe natürliche Häufigkeit (erfordert zum Teil erheblich längere Meßzeiten)
- größerer Bereich für die chemische Verschiebung


















