
medgen2017·29:306–313 ... · Hemihyperplasie reichen. Es können verschiedene Gewebe betroffen...
8
Schwerpunktthema: Tumorsyndrome medgen 2017 · 29:306–313 https://doi.org/10.1007/s11825-017-0153-3 Online publiziert: 23. Oktober 2017 © Der/die Autor(en) 2017. Dieser Artikel ist eine Open-Access-Publikation. Isabel Spier · Stefan Aretz Institut für Humangenetik Universitätsklinikum Bonn, Bonn, Deutschland Überwuchssyndrome durch Mutationsmosaike im PI3K-AKT-Signalweg Einleitung Somatische Mosaike aktivierender Mu- tationen im Phosphatidylinositol-3- Kinase/AKT/mTOR-Signalweg (PI3K- AKT-Signalweg) wurden mittlerweile bei einem breiten Spektrum segmenta- ler Überwuchssyndrome als ursächlich beschrieben. Hierbei sind insbesondere das PIK3CA-assoziierte Überwuchs- spektrum (PIK3CA-Related Overgrowth Spectrum, PROS) sowie das durch eine spezifische Mutation im AKT1-Gen bedingte Proteus-Syndrom hervorzu- heben. Die genetische Abklärung und Diagnosesicherung ist allerdings schwie- riger und komplexer als bei klassischen hereditären Krankheitsbildern. PI3K-AKT-Signalweg und phänotypisches Spektrum Der PI3K-AKT-Signalweg spielt eine wichtige Rolle bei der Regulation von Zellproliferation, -metabolismus und -überleben sowie bei der Angiogene- se (. Abb. 1) und ist eng verbunden mit anderen, onkogenetisch relevanten Signalwegen, insbesondere dem RAS- Signalweg [41]. Heterozygote Keimbahn- mutationen in einigen Genen dieser Si- gnalwege stellen die Ursache bekannter, autosomal-dominant erblicher Tumor- syndrome (Cowden-Syndrom, tuberöse Sklerose) oder syndromaler Erkrankun- gen mit einem erhöhten Tumorrisiko (RASopathien) dar. Somatische Muta- tionen in zahlreichen der beteiligten Gene sind essenziell an der Tumorent- stehung beteiligt. Spezifische somatische Mutationen im PIK3CA-Gen (kodiert für die 110-kD katalytische alpha-Unter- einheit des PI3K-Proteinkomplexes) und AKT1-Gen sind hierbei über eine Akti- vierung des Signalwegs mit vermehrter Proliferation sowie verminderter Apop- tose an der Tumorgenese beteiligt. Als somatische Mosaike können diese Mu- tationen in variabler Ausprägung zur Entwicklung von Überwuchssyndromen führen (. Tab. 1). Somatische Mutationsmosaike Bei einem somatischen Mutationsmosa- ik trägt lediglich ein Teil der Körperzellen die Mutation. Es handelt sich somit in der Regel um Mutationen, die postzygotisch während der Embryonalentwicklung auſtreten (Übersicht zu Mosaiken unter Kurth und Grimm [22]). Daher wird in diesen Fällen auch von postzygotischen Mutationen/Mosaiken gesprochen [36]. Mutationsmosaike werden zunehmend häufiger als Ursache genetisch bedingter Krankheitsbilder erkannt, was neben der größeren klinischen Aufmerksamkeit vor allem an empfindlicheren Nachweisme- thoden liegt. Bei autosomal-dominanten Tumorsyndromen wie z. B. der fami- liären adenomatösen Polyposis (FAP), der Neurofibromatose Typ 1 (NF1) oder der tuberösen Sklerose (TSC) ist eine gewisse Wahrscheinlichkeit für das Auf- treten eines Mosaiks aufgrund der hohen Neumutationsrate gegeben. Bei Krank- heitsbildern, die durch hochpenetrante, aktivierende Mutationen in Onkogenen bedingt sind, wäre eine durchgehende heterozygote Keimbahnmutation ver- mutlich meistens früh letal, weshalb sol- che Mutationen im klinischen Kontext prinzipiell nur in Mosaikform vorliegen [12]. Da jedoch bei dem letztgenannten Pathomechanismus der Mutationsgrad auch in den untersuchten betroffenen Geweben häufig gering ist (meist <20 %, häufig auch <10 %), ist zu vermuten, dass die mutierten Zellen die Fähigkeit ha- ben, die normalen Zellen im Sinne eines parakrinen Signals für den Überwuchs zu „rekrutieren“ [21, 27]. Auf Mosaiken beruhende Krankheits- bilder treten in der Regel sporadisch auf, d. h. es handelt es sich um Einzelfälle in der Familie. Wenn bei einem autosomal- dominanten Tumorsyndrom eine Neu- mutation in Mosaikform vorliegt, kann das Wiederholungsrisiko für zukünſtige Kinder einer erkrankten Person nicht si- cher eingeschätzt werden. In den meis- ten Fällen ist von einem geringen Risiko auszugehen, in Abhängigkeit vom Zeit- punkt des Auſtretens der Mutation und der beteiligten Keimblätter können die Keimzellen aber ebenfalls betroffen sein und ein Risiko von bis zu 50 % bestehen. Bei den durch aktivierende Mutationen in Onkogenen bedingten Krankheitsbil- dern, bei denen das Vorliegen einer he- terozygoten Keimbahnmutation nicht le- bensfähig wäre, ist die Geburt eines be- troffenen Kindes hingegen weitestgehend ausgeschlossen. Molekulargenetische Diagnostik Die Identifizierung von Mutationsmo- saiken ist eine diagnostische Herausfor- derung, da die ursächlichen Mutationen in Leukozyten-DNA häufig entweder gar nicht nachweisbar sind oder weit jenseits der Nachweisgrenze klassischer Methoden wie der Sanger-Sequenzie- rung (~10 % Mutationsanteil) liegen 306 medizinische genetik 3 · 2017
Transcript of medgen2017·29:306–313 ... · Hemihyperplasie reichen. Es können verschiedene Gewebe betroffen...

Schwerpunktthema: Tumorsyndrome
medgen 2017 · 29:306–313https://doi.org/10.1007/s11825-017-0153-3Online publiziert: 23. Oktober 2017© Der/die Autor(en) 2017. Dieser Artikel isteine Open-Access-Publikation.
Isabel Spier · Stefan AretzInstitut für Humangenetik UniversitätsklinikumBonn, Bonn, Deutschland
Überwuchssyndromedurch Mutationsmosaikeim PI3K-AKT-Signalweg
Einleitung
Somatische Mosaike aktivierender Mu-tationen im Phosphatidylinositol-3-Kinase/AKT/mTOR-Signalweg (PI3K-AKT-Signalweg) wurden mittlerweilebei einem breiten Spektrum segmenta-ler Überwuchssyndrome als ursächlichbeschrieben. Hierbei sind insbesonderedas PIK3CA-assoziierte Überwuchs-spektrum (PIK3CA-Related OvergrowthSpectrum, PROS) sowie das durch einespezifische Mutation im AKT1-Genbedingte Proteus-Syndrom hervorzu-heben. Die genetische Abklärung undDiagnosesicherung ist allerdings schwie-riger und komplexer als bei klassischenhereditären Krankheitsbildern.
PI3K-AKT-Signalweg undphänotypisches Spektrum
Der PI3K-AKT-Signalweg spielt einewichtige Rolle bei der Regulation vonZellproliferation, -metabolismus und-überleben sowie bei der Angiogene-se (. Abb. 1) und ist eng verbundenmit anderen, onkogenetisch relevantenSignalwegen, insbesondere dem RAS-Signalweg[41].HeterozygoteKeimbahn-mutationen in einigen Genen dieser Si-gnalwege stellen die Ursache bekannter,autosomal-dominant erblicher Tumor-syndrome (Cowden-Syndrom, tuberöseSklerose) oder syndromaler Erkrankun-gen mit einem erhöhten Tumorrisiko(RASopathien) dar. Somatische Muta-tionen in zahlreichen der beteiligtenGene sind essenziell an der Tumorent-stehung beteiligt. Spezifische somatischeMutationen im PIK3CA-Gen (kodiertfür die 110-kD katalytische alpha-Unter-
einheit des PI3K-Proteinkomplexes) undAKT1-Gen sind hierbei über eine Akti-vierung des Signalwegs mit vermehrterProliferation sowie verminderter Apop-tose an der Tumorgenese beteiligt. Alssomatische Mosaike können diese Mu-tationen in variabler Ausprägung zurEntwicklung von Überwuchssyndromenführen (. Tab. 1).
SomatischeMutationsmosaike
Bei einem somatischen Mutationsmosa-ik trägt lediglich einTeil derKörperzellendieMutation.Eshandelt sich somit inderRegel um Mutationen, die postzygotischwährend der Embryonalentwicklungauftreten (Übersicht zu Mosaiken unterKurth und Grimm [22]). Daher wird indiesen Fällen auch von postzygotischenMutationen/Mosaiken gesprochen [36].Mutationsmosaike werden zunehmendhäufiger als Ursache genetisch bedingterKrankheitsbilder erkannt, was neben dergrößerenklinischenAufmerksamkeitvorallem an empfindlicheren Nachweisme-thoden liegt. Bei autosomal-dominantenTumorsyndromen wie z. B. der fami-liären adenomatösen Polyposis (FAP),der Neurofibromatose Typ 1 (NF1) oderder tuberösen Sklerose (TSC) ist einegewisse Wahrscheinlichkeit für das Auf-treten einesMosaiks aufgrund der hohenNeumutationsrate gegeben. Bei Krank-heitsbildern, die durch hochpenetrante,aktivierende Mutationen in Onkogenenbedingt sind, wäre eine durchgehendeheterozygote Keimbahnmutation ver-mutlich meistens früh letal, weshalb sol-che Mutationen im klinischen Kontextprinzipiell nur in Mosaikform vorliegen[12]. Da jedoch bei dem letztgenannten
Pathomechanismus der Mutationsgradauch in den untersuchten betroffenenGeweben häufig gering ist (meist <20%,häufig auch <10%), ist zu vermuten, dassdie mutierten Zellen die Fähigkeit ha-ben, die normalen Zellen im Sinne einesparakrinen Signals für den Überwuchszu „rekrutieren“ [21, 27].
AufMosaikenberuhendeKrankheits-bilder treten in der Regel sporadisch auf,d. h. es handelt es sich um Einzelfälle inder Familie. Wenn bei einem autosomal-dominanten Tumorsyndrom eine Neu-mutation in Mosaikform vorliegt, kanndas Wiederholungsrisiko für zukünftigeKinder einer erkrankten Person nicht si-cher eingeschätzt werden. In den meis-ten Fällen ist von einem geringen Risikoauszugehen, in Abhängigkeit vom Zeit-punkt des Auftretens der Mutation undder beteiligten Keimblätter können dieKeimzellen aber ebenfalls betroffen seinund ein Risiko von bis zu 50% bestehen.Bei den durch aktivierende Mutationenin Onkogenen bedingten Krankheitsbil-dern, bei denen das Vorliegen einer he-terozygotenKeimbahnmutationnicht le-bensfähig wäre, ist die Geburt eines be-troffenenKindeshingegenweitestgehendausgeschlossen.
MolekulargenetischeDiagnostik
Die Identifizierung von Mutationsmo-saiken ist eine diagnostische Herausfor-derung, da die ursächlichen Mutationenin Leukozyten-DNA häufig entwedergar nicht nachweisbar sind oder weitjenseits der Nachweisgrenze klassischerMethoden wie der Sanger-Sequenzie-rung (~10% Mutationsanteil) liegen
306 medizinische genetik 3 · 2017

p110p85
PI3K
PIP3PP
PPP
PIP2P
P
PDK1P
AKT
P
P
mTOR
Zellproliferation/-überleben, Angiogenese
PTEN
RTK
GF
Abb. 18 PI3K-AKT-Signalweg:PIK3CAkodiert fürdie110-kDkatalytischealpha-UntereinheitdesPI3K-Proteinkomplexes,diedurch Bindung einesWachstumsfaktors (GF) an eine Rezeptor-Tyrosinkinase (RTK) aktiviertwird und Phosphatidylinositol-(4,5)-bisphosphat (PIP2) zu Phosphatidylinositol(3,4,5)-triphosphat (PIP3) umwandelt. Diese Reaktionwird von PTENantagonisiert. PIP3 führt zur Phosphorylierung von PDK1, daswiederumAKT phosphoryliert unddamit Einfluss auf dieRegulationdesZellzyklus/derApoptose,MetabolismusundAngiogenesenimmt.AKTwirdauchdurcheinePhosphorylierungdes PDK2-Komplexes, der u. a.mTORumfasst, aktiviert
und der Mutationsgrad selbst im be-troffenen Zielgewebe gering sein kann.Durch den Einsatz neuer technischerAnsätze hat sich die Sensitivität inzwi-schen allerdings deutlich verbessert. MitNGS-Verfahren (. Abb. 2a) kann gezielteine besonders hohe Lesetiefe (coverage)angestrebt werden (deep sequencing), umauch Mosaikgrade um 1% nachweisenzu können. Als sehr empfindliche Me-thode hat sich auch die Sequenzierungmit single molecule Molecular Inversi-on Probes (smMIP) herausgestellt [1,13]. Zum gezielten Nachweis bestimm-ter Hotspot-Mutationen können auchandere Methoden eingesetzt werdenwie Restriktions-Fragment-Längenpo-lymorphismus-Analyse (RFLP), digitaldroplet PCR (ddPCR), SNaPshot-Analy-se, Mass-ARRAY [17].
Für den primären Nachweis einesursächlichen somatischen Mosaiks istin der Regel die Untersuchung von be-troffenem Gewebe notwendig, bei derentweder auf eine gezielte Biopsie oderoperativ entferntes Gewebe zurückge-griffen wird. In letzterem Fall ist häufigkein frisches Gewebe verfügbar, son-dern nur formalinfixiertes, in Paraffin
eingebettetes Material (FFPE). Da dieQualität von aus FFPE-Material extra-hierter DNA häufig eingeschränkt ist,wird der Nachweis von (niedriggradi-gen) Mosaiken auch dadurch erschwert.Bei einer kutanen Manifestation ist esmanchmal möglich, mit dem Skalpellvorsichtig Hautschuppen abzukratzenund aus dem Abradat DNA zu isolie-ren. Darüber hinaus kann auch eineHautstanze mit der Untersuchung vonFibroblasten für die Diagnosestellunghilfreich sein [29].
Zudem kann es bei niedriggradigenMosaiken schwierig sein, den Befundzu validieren, d. h. echte Mutationenvon Sequenzierartefakten oder ggf. auchKontaminationen zu unterscheiden. ZurBestätigung eines Mosaiks sollte diegleiche Mutation in mindestens einemanderen räumlich getrennten Gewebeebenfalls nachgewiesen werden, auchwenn dies im klinischen Alltag nicht im-mer realisierbar ist. Hierzu dient entwe-der ein weiteres betroffenes Gewebe oderNormalgewebe (z. B. DNA aus Leuko-zyten, Mundschleimhaut, Speichel oderUrinsediment). In Normalgewebe solltegezielt nach einem sehr niedriggradigen
Mosaik der im betroffenen ZielgewebeidentifiziertenMutation gesucht werden.Bei den meisten der hier beschriebenenÜberwuchssyndromen ist der Nachweisder postzygotischen Mutation anhandeiner Blutprobe jedoch nicht Erfolgversprechend. Lediglich bei dem schwe-ren Phänotyp des Megalenzephalie-Kapillarfehlbildungen-Polymikrogyrie-Syndroms (MCAP) war die Mutationmeistens in DNA aus Blut oder Spei-chel nachweisbar [36], ebenso bei einemeigenen unpublizierten Fall.
Durch den fehlendenMutationsnach-weis muss eine klinisch gestellte Diagno-se nicht infrage gestellt werden, da inAbhängigkeit des untersuchten Gewebesund der eingesetzten Untersuchungsme-thode nicht jede Mosaikmutation aufge-deckt werden kann, insbesondere wennsie unter der Detektionsschwelle der je-weiligen Methode liegt.
Krankheitsbilder
Das Ausmaß des Überwuchses kannbei den meisten Syndromen sehr varia-bel sein und von einer Makrodaktyliebis zu einer Hemihypertrophie bzw.
medizinische genetik 3 · 2017 307

Hemihyperplasie reichen. Es könnenverschiedene Gewebe betroffen sein,häufig bestehen neben dem Überwuchstypische kutane Veränderungen undvaskuläre Malformationen. Alle hierbeschriebenen Krankheitsbilder tretensehr selten auf. Mögliche Differential-diagnosen sind in . Tab. 2 aufgeführt.
PIK3CA-assoziiertesÜberwuchsspektrum (PROS)
Klinik
Die klinischen Entitäten, die durchPIK3CA-Mutationsmosaike verursachtundmittlerweileunterdemBegriffPROSzusammengefasst werden [17, 18], um-fassen neben den unten ausführlicherbeschriebenen Syndromen CLOVESund MCAP [30] zahlreiche weitere,klinisch definierte Subtypen (. Tab. 1).Durch den Nachweis von PIK3CA-Mu-tationen ist davon auszugehen, dass essich um eine ätiologisch zusammen-gehörende Krankheitsgruppe handelt,die je nach betroffenem Gewebe undAusmaß des Mosaiks (abhängig vomZeitpunkt des Auftretens der Mutati-on in der Embryonalentwicklung) einSpektrum unterschiedlich stark ausge-prägter Manifestationen aufweist [29].Eine Expertengruppe hat diagnostischeKriterien für das PROS vorgeschlagen,die neben demNachweis einer postzygo-tischen PIK3CA-Mutation entweder dasVorliegen von mindestens zwei der fol-genden Merkmale umfasst (segmentalerÜberwuchs, vaskuläre Malformationen,epidermale Nävi) oder das Vorliegenvon passenden isolierten Auffälligkeiten(z. B. Makrodaktylie) [17].
Die klinischen Leitsymptome desPROS sind in . Tab. 3 zusammengefasst.Gemeinsam ist den Krankheitsbilderndarüber hinaus, dass die Symptoma-tik entweder bereits bei Geburt bestehtoder in der frühen Kindheit beginntund einen progredienten Verlauf auf-weist. Eine Progredienz ist jedoch keinzwingendes Kriterium für den Verdachtauf ein Krankheitsbild aus dem „PRO“-Spektrum und nicht so deutlich ausge-prägt wie beim Proteus-Syndrom (sieheunten). Der Überwuchs ist in der Regelasymmetrisch und disproportioniert. In
Zusammenfassung · Abstract
medgen 2017 · 29:306–313 https://doi.org/10.1007/s11825-017-0153-3© Der/die Autor(en) 2017. Dieser Artikel ist eine Open-Access-Publikation.
I. Spier · S. Aretz
Überwuchssyndrome durch Mutationsmosaike im PI3K-AKT-Signalweg
ZusammenfassungEs wurde schon länger vermutet, dasssegmentale Überwuchssyndrome durchsomatische Mutationsmosaike (postzygoti-sche Mutationen) hervorgerufen werden; dieursächlichen genetischen Veränderungenlassen sich aber häufig nur in betroffenemGewebe nachweisen. Durch den Einsatzder Hochdurchsatzsequenzierung (NextGeneration Sequencing, NGS) konnten diegenetischen Ursachen von sich segmentalmanifestierenden Krankheitsbildern in denletzten Jahren zunehmend geklärt werden.Interessanterweise wurden hierdurch beimehreren Entitäten postzygotische aktivie-rende Mutationen im Phosphatidylinositol-3-Kinase/AKT/mTOR-Signalweg (PI3K-AKT-Signalweg) als ursächlich identifiziert. Eshandelt sich insbesondere um das PIK3CA-assoziierte Überwuchsspektrum (PIK3CA-RelatedOvergrowth Spectrum, PROS), zu demneben dem CLOVES-Syndrom (congenitallipomatous overgrowth, vaskuläre Fehlbil-
dungen, epidermale Nävi und Skoliose bzw.Skelettsymptome) und demMCAP-Syndrom(Megalenzephalie-Kapillarfehlbildungen-Polymikrogyrie) mittlerweile vermutlich aucheinige Fälle mit Verdacht auf ein Klippel-Trenaunay-Syndrom gezählt werden können.Beim Proteus-Syndrom dominiert einespezifische kausale Mutation im AKT1-Gen.Auch wenn somatischeMutationen im PI3K-AKT-Signalweg relativ häufig in sporadischenTumoren auftreten, stehen der segmentaleÜberwuchs und weitere Malformationen imVordergrund des phänotypischen Spektrumsder Überwuchssyndrome. Verschiedeneklinisch relevante gut- und bösartigeNeoplasien kommen allerdings gehäuft vor.
SchlüsselwörterSomatische Mutationsmosaike · Postzygoti-sche Mutationen · Segmentaler Überwuchs ·PI3K-AKT-Signalweg
Overgrowth syndromes caused bymosaic mutations in the PI3K-AKT signaling pathway
AbstractIt has long been suspected that segmentalovergrowth syndromes are caused by somaticmosaic mutations (postzygotic mutations);however, the genetic changes can oftenonly be detected in affected tissues. Usinghigh-throughput sequencing methods(next generation sequencing, NGS), thegenetic causes of conditions with segmentalmanifestations have been increasinglyelucidated in recent years. Interestingly,in several entities, postzygotic activatingmutations in the phosphatidylinositol-3-kinase/AKT/mTOR signaling pathway (PI3K-AKT signaling pathway) were identifiedas causative. This is particularly true forthe PIK3CA-related overgrowth spectrum(PROS), including CLOVES syndrome(congenital lipomatous overgrowth, vascularmalformations, epidermal nevi, skeletaland spinal anomalies) and MCAP syndrome
(megalencephaly, capillary malformation,polymicrogyria). In the meantime, somecases of suspected Klippel–Trenaunaysyndrome can probably also be addedto this spectrum. In Proteus syndrome asingle specific mutation in the AKT1 gene iscausative Even though somatic mutationsin the PI3K-AKT pathway arise relativelyfrequently in sporadic tumors, segmentalovergrowth and other malformations areprominent in the phenotypic spectrum ofovergrowth syndromes. However, variousclinically relevant benign and malignantneoplasms often occur.
KeywordsSomatic mosaic mutations · Postzygoticmutations · Segmental overgrowth · PI3K-AKTsignaling pathway
308 medizinische genetik 3 · 2017

Tab. 1 Phänotypisches Spektrum von Patientenmit PI3K-AKT-Mosaiksyndromen
Gen Krankheitsbilder Referenzen
AKT1 Proteus-Syndrom [26]
AKT2 Lipodystrophie-Syndrommit Hypoglykämie [15]
AKT3 Megalenzephalie-Polymikrogyrie-Polydaktylie-Hydrozephalus-Syndrom (MPPH)a
[36]
Hemimegalenzephalie (HMEG)b [24, 34]
MTOR Hemimegalenzephalie (HMEG)b [24]
PIK3CA Benigne lichenoide Keratose (BLK) [10]
Congenital lipomatous overgrowth, vaskuläre Fehlbildungen, epider-male Nävi und Skoliose bzw. Skelettsymptome (CLOVES)
[21]
Epidermale Nävi [11]
Fazial infiltrierende Lipomatose [28]
Fibroadipöse Hyperplasie oder Überwuchs (overgrowth) (FAO) [25]
Hemihyperplasiemultiple Lipomatose (HHML) [18]
Hemimegalenzephalie (HMEG)b [24, 29]
Klippel-Trenaunay-Syndrom (KTS) [21, 27, 43]
Makrodaktylie [29, 35]
Megalenzephalie-Kapillarfehlbildungen (Capillary Malformation)-Polymikrogyrie-Syndrom (MCAP)
[29, 36]
Seborrhoische Keratose [11]
PIK3R2 Megalenzephalie-Polymikrogyrie-Polydaktylie-Hydrozephalus-Syndrom (MPPH)a
[36]
adurch Mosaikmutationen in AKT3 oder PIK3R2bdurch Mosaikmutationen in AKT3, MTOR oder PIK3CA
einer Studie von 35 Patienten mit soma-tischen Mosaikmutationen in PIK3CAwurde ein bevorzugter linksseitiger unddistaler Überwuchs insbesondere derunteren Extremität sowie eine Dysregu-lation des Fettgewebes bei allen Patien-ten beschrieben (einerseits lipomatöserÜberwuchs sowie andererseits Lipo-atrophie in nicht betroffenen Arealen)[18]. Einem Erklärungsansatz zufolgekönnte der erhöhte Energieverbrauchder betroffenen Gewebe zu einer nega-tiven Energiebilanz der übrigen Gewebeführen [25]. Durch eine Infiltration derMuskulatur und innerer Organe durchdas Fettgewebe scheint es zu einer se-kundären Vergrößerung dieser Gewebezu kommen [18].
Der genaue Mechanismus der Über-wuchsentstehung durch PIK3CA-Muta-tionen ist noch nicht vollständig verstan-den, aber die Rolle, die PI3K in verschie-denen durch Wachstumsfaktoren akti-vierten Signalwegen spielt, ist sicherlichein wesentlicher Faktor [41].
CLOVES-Syndrom
Das CLOVE-Syndrom (kongenitalerlipomatöser Überwuchs [congenitallipomatous overgrowth], vaskuläre Fehl-bildungen, epidermale Nävi) wurde erst-malig 2007 von Sapp et al. beschrieben[39]. Da auch weitereMerkmale wie eineSkoliose oder andere Skelettanomalien(z. B.Makrodaktylie, große Hände/Füße,Sandalenlücke), spinale Auffälligkeiten(z. B. tethered cord, Neuralrohrdefekte)und Epilepsien (seizures) gehäuft vor-kommen, hat Alomari eine Erweiterungdes Akronyms zum CLOVES-Syndromvorgeschlagen [2].
Bei den Patienten steht in der Regeleinedisproportionierte Fettverteilung imVordergrund. Im Gegensatz zu den knö-chernen Verformungen beim Proteus-Syndrom kommt es beim CLOVES-Syn-drominderRegelnurnachausgedehntenchirurgischen Eingriffen zu Verformun-gen. Bei einigen Fällen wurden zusätz-lich eine Hemimegalenzephalie, Chiari-Malformation oder Polymikrogyrie be-obachtet. Die vaskulären Fehlbildungenbeinhalten kapilläre, venöse, lymphati-
sche sowie arteriovenöse Malformatio-nen. Relativ häufig wurden auch renaleFehlbildungen festgestellt. Weiterhin be-steht ein erhöhtes Thromboserisiko.
MCAP
Das Megalenzephalie-Kapillarfehl-bildungen-Polymikrogyrie-Syndrom(MCAP) ist durch eine deutliche Ma-krozephalie/Megalenzephalie mit pro-minenter Stirn und Dolichozephalie ge-kennzeichnet. Außerdem treten kutaneKapillarfehlbildungen auf. Dabei wirdzwar am häufigsten ein Nävus flam-meus im Bereich der Gesichtsmittelliniebeschrieben, es können aber auch gene-ralisierte kutane Kapillarfehlbildungenvorliegen, die an eine Cutis marmorataerinnern. Bei etwa 50% der Patientenbesteht eine Polymikrogyrie; es könnenauch weitere Hirnanomalien auftre-ten (Ventrikulomegalie, Hydrozephalus,Kleinhirntonsillenektopie, Chiari-Mal-formation). Meist besteht eine geistigeBeeinträchtigung, die mild bis schwerausgeprägt sein kann; außerdem findensich gehäuft eine muskuläre Hypotonieund Epilepsien. Poly- und Syndakty-lien wurden ebenfalls beschrieben. BeimanchenPatienten bestehen strukturelleHerzfehler oder Herzrhythmusstörun-gen.
Genetik
Bei den als ursächlich identifiziertensomatischen Mosaiken im PIK3CA-Gen handelt es sich um aktivierendeMissensemutationen in insbesonde-re drei häufigen Mutations-Hotspots:c.1624G>A;p.Glu542Lys und c.1633G>A;p.Glu545Lys in der helikalen Domänesowie c.3140A>G;p.His1047Arg in derKinase-Domäne[17,18,21,36,43].Etwasseltener sind die Missensemutationenc.3140A>T;p.His1047Leu in der Kinase-Domäne sowie c.1258T>C;p.Cys420Argin der C2-Domäne. Es treten jedochauch weitere seltenere Mutationen auf;insbesondere bei Vorliegen einer Me-galenzephalie wurden Mutationen au-ßerhalb der Hotspots nachgewiesen [20,29].
medizinische genetik 3 · 2017 309

Schwerpunktthema: Tumorsyndrome
Abb. 28 aMissensemutation c.1634A>G;p.Glu545Gly in Exon 10des PIK3CA-Gens als niedriggradiges (10%iges)Mosaik;b an derMittellinie des Bauches scharf begrenzter Nävus flammeus auf der rechten Bauchseite bei einemPatientenmitklinischemVerdacht auf ein Klippel-Trenaunay-Syndrom;c zerebriformer Bindegewebsnävus am Fußbei einer 21-jährigenPatientinmitmolekulargenetischgesichertemProteus-Syndrom
Klippel-Trenaunay-Syndrom(KTS)
Klinik
Das Klippel-Trenaunay-Syndrom (KTS)wurde erstmals 1900 beschrieben. Da inder Literatur keine einheitliche Definiti-on Anwendung findet und Überschnei-dungen zu anderen Syndromen bestehen(z. B. Parkes-Weber, Proteus, Hemihy-pertrophie), haben Oduber et al. [32]überarbeitete klinische Kriterien vorge-schlagen: Es soll mindestens eine ange-borene kapilläre oder venöse Malforma-tion vorliegen und ein gestörtes Wachs-tum von Knochen und/oderWeichteilen(meist eine Hypertrophie). Eine Über-sicht der Leitsymptome findet sich in. Tab. 3.
Die kapillären Gefäßveränderungenimponieren klinisch als Nävi flammeiund können am ganzen Körper auftre-ten (insbesondere im Bereich der Ex-tremitäten), sehr selten ist das Gesichtbetroffen. Im Bereich des Rumpfes sinddie Nävi flammei häufig scharf begrenztund überschreitenmeist nicht dieMittel-linie (. Abb. 2b). Bei den venösen Mal-formationen treten beispielsweise Phle-bektasien, Varizen, Hypoplasien, Apla-sien, Hypertrophien oder persistierendefetale Venen auf. Ebenso können arterio-
venöseMalformationen oder Malforma-tionen von Lymphgefäßen vorliegen. ImBereichderbetroffenenExtremität findetsich häufig eine oft erhebliche Hypertro-phievonWeichteilgewebeoderKnochen.Diese Hypertrophien können aber auchin Bereichen auftreten, in denen keineNävi flammei vorliegen. Der asymme-trische Überwuchs ist meist schon beiGeburt erkennbar und kann sich als He-mihypertrophie, Hypertrophie einer Ex-tremitätoderaucheinesTeils einerExtre-mität (z. B. Makrodaktylie) präsentieren.Weitere Skelettauffälligkeiten beinhaltenPoly-, Syndaktylien und eine Skoliose.Die intellektuelle Entwicklung ist meis-tens nicht beeinträchtigt.
Genetik
Mittlerweile konnten auch bei einemTeil der Patienten, die phänotypischdem KTS-Spektrum zugeordnet wordenwaren, PIK3CA-Mutationen identifiziertwerden. Kurek et al. [21] fanden bei dreivon 15 Patienten PIK3CA-Mutationen.Da nur zwei Hotspot-Mutationen un-tersucht wurden und keine genauerenklinischen Angaben vorliegen, ist dieAussagekraft allerdings eingeschränkt.Luks et al. wiesen bei 20 von 21 Patientenmit erfüllten klinischen Kriterien für einKTS PIK3CA-Mosaike nach [27]. Da die
Mutationen z. T. nur mit ddPCR nach-gewiesen wurden und mittels NGS nichtnachweisbar waren sowie keine weite-ren Gewebeproben untersucht wurden,müsste diese hohe Mutationsdetektions-rate noch in weiteren Studien bestätigtwerden. In der Studie von Yeung et al.[43]wurde bei einemPatientenmit klini-schen Kriterien eines KTS ebenfalls einePIK3CA-Mutation entdeckt. Aufgrundder inzwischen gehäuft nachgewiese-nen PIK3CA-Mosaike bei Patienten mitden klinischen Kriterien eines KTS undden klinischen Überlappungen ist dasKTS wohl in vielen Fällen dem PROSzuzuordnen.
Proteus-Syndrom
Klinik
Das Proteus-Syndrom ist charakterisiertdurch einen asymmetrischen, dispropor-tionierten und deutlich deformierendenÜberwuchs von einzelnen Körperteilen,zumeist des Skeletts, der Haut, des Bin-degewebes, des Fettgewebes (es kannauch eine regionale Lipoatrophie beste-hen) und des zentralen Nervensystems.Letztlich kann jedoch jedes Gewebe be-troffen sein. In Einzelfällen ist auch eineVergrößerung von inneren Organen be-schrieben. Klinische Diagnosekriterien
310 medizinische genetik 3 · 2017
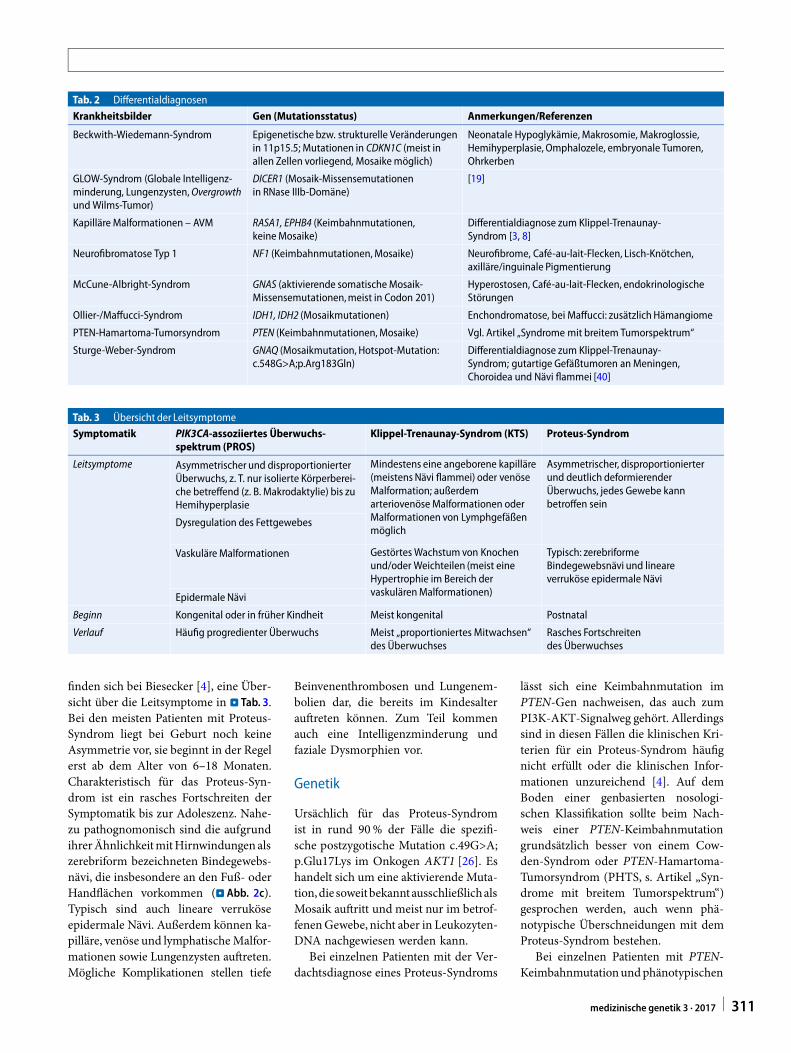
Tab. 2 Differentialdiagnosen
Krankheitsbilder Gen (Mutationsstatus) Anmerkungen/Referenzen
Beckwith-Wiedemann-Syndrom Epigenetische bzw. strukturelle Veränderungenin 11p15.5; Mutationen in CDKN1C (meist inallen Zellen vorliegend, Mosaikemöglich)
Neonatale Hypoglykämie, Makrosomie, Makroglossie,Hemihyperplasie, Omphalozele, embryonale Tumoren,Ohrkerben
GLOW-Syndrom (Globale Intelligenz-minderung, Lungenzysten,OvergrowthundWilms-Tumor)
DICER1 (Mosaik-Missensemutationenin RNase IIIb-Domäne)
[19]
Kapilläre Malformationen – AVM RASA1, EPHB4 (Keimbahnmutationen,keineMosaike)
Differentialdiagnose zum Klippel-Trenaunay-Syndrom [3, 8]
Neurofibromatose Typ 1 NF1 (Keimbahnmutationen,Mosaike) Neurofibrome, Café-au-lait-Flecken, Lisch-Knötchen,axilläre/inguinale Pigmentierung
McCune-Albright-Syndrom GNAS (aktivierende somatischeMosaik-Missensemutationen,meist in Codon 201)
Hyperostosen, Café-au-lait-Flecken, endokrinologischeStörungen
Ollier-/Maffucci-Syndrom IDH1, IDH2 (Mosaikmutationen) Enchondromatose, bei Maffucci: zusätzlich Hämangiome
PTEN-Hamartoma-Tumorsyndrom PTEN (Keimbahnmutationen,Mosaike) Vgl. Artikel „Syndrome mit breitem Tumorspektrum“
Sturge-Weber-Syndrom GNAQ (Mosaikmutation, Hotspot-Mutation:c.548G>A;p.Arg183Gln)
Differentialdiagnose zum Klippel-Trenaunay-Syndrom; gutartige Gefäßtumoren anMeningen,Choroidea und Nävi flammei [40]
Tab. 3 Übersicht der Leitsymptome
Symptomatik PIK3CA-assoziiertes Überwuchs-spektrum (PROS)
Klippel-Trenaunay-Syndrom (KTS) Proteus-Syndrom
Leitsymptome Asymmetrischer und disproportionierterÜberwuchs, z. T. nur isolierte Körperberei-che betreffend (z. B. Makrodaktylie) bis zuHemihyperplasie
Mindestens eine angeborene kapilläre(meistens Nävi flammei) oder venöseMalformation; außerdemarteriovenöseMalformationen oderMalformationen von Lymphgefäßenmöglich
Asymmetrischer, disproportionierterund deutlich deformierenderÜberwuchs, jedes Gewebe kannbetroffen sein
Dysregulation des Fettgewebes
Vaskuläre Malformationen GestörtesWachstum von Knochenund/oder Weichteilen (meist eineHypertrophie im Bereich dervaskulärenMalformationen)
Typisch: zerebriformeBindegewebsnävi und lineareverruköse epidermale Nävi
Epidermale Nävi
Beginn Kongenital oder in früher Kindheit Meist kongenital Postnatal
Verlauf Häufig progredienter Überwuchs Meist „proportioniertes Mitwachsen“des Überwuchses
Rasches Fortschreitendes Überwuchses
finden sich bei Biesecker [4], eine Über-sicht über die Leitsymptome in . Tab. 3.Bei den meisten Patienten mit Proteus-Syndrom liegt bei Geburt noch keineAsymmetrie vor, sie beginnt in der Regelerst ab dem Alter von 6–18 Monaten.Charakteristisch für das Proteus-Syn-drom ist ein rasches Fortschreiten derSymptomatik bis zur Adoleszenz. Nahe-zu pathognomonisch sind die aufgrundihrerÄhnlichkeitmitHirnwindungenalszerebriform bezeichneten Bindegewebs-nävi, die insbesondere an den Fuß- oderHandflächen vorkommen (. Abb. 2c).Typisch sind auch lineare verruköseepidermale Nävi. Außerdem können ka-pilläre, venöse und lymphatischeMalfor-mationen sowie Lungenzysten auftreten.Mögliche Komplikationen stellen tiefe
Beinvenenthrombosen und Lungenem-bolien dar, die bereits im Kindesalterauftreten können. Zum Teil kommenauch eine Intelligenzminderung undfaziale Dysmorphien vor.
Genetik
Ursächlich für das Proteus-Syndromist in rund 90% der Fälle die spezifi-sche postzygotische Mutation c.49G>A;p.Glu17Lys im Onkogen AKT1 [26]. Eshandelt sich um eine aktivierende Muta-tion,die soweitbekanntausschließlichalsMosaik auftritt und meist nur im betrof-fenenGewebe, nicht aber in Leukozyten-DNA nachgewiesen werden kann.
Bei einzelnen Patienten mit der Ver-dachtsdiagnose eines Proteus-Syndroms
lässt sich eine Keimbahnmutation imPTEN-Gen nachweisen, das auch zumPI3K-AKT-Signalweg gehört. Allerdingssind in diesen Fällen die klinischen Kri-terien für ein Proteus-Syndrom häufignicht erfüllt oder die klinischen Infor-mationen unzureichend [4]. Auf demBoden einer genbasierten nosologi-schen Klassifikation sollte beim Nach-weis einer PTEN-Keimbahnmutationgrundsätzlich besser von einem Cow-den-Syndrom oder PTEN-Hamartoma-Tumorsyndrom (PHTS, s. Artikel „Syn-drome mit breitem Tumorspektrum“)gesprochen werden, auch wenn phä-notypische Überschneidungen mit demProteus-Syndrom bestehen.
Bei einzelnen Patienten mit PTEN-Keimbahnmutationundphänotypischen
medizinische genetik 3 · 2017 311

Schwerpunktthema: Tumorsyndrome
Ähnlichkeiten zum Proteus-Syndrom(z. B. segmentaler Überwuchs, vaskuläreMalformationen und epidermale Nävi)wurde in jeweils mehreren betroffe-nen Geweben zusätzlich entweder einetrunkierende somatische PTEN-Mutati-on gefunden [44] oder ein Verlust desWildtyp-Allels (LOH, loss of heterozygo-sity) nachgewiesen [5]. In diesen Fällenist davon auszugehen, dass der „secondhit“ alsMosaik vorliegt und sichwährendder Embryonalentwicklung ereignet hat.
Tumorerkrankungen
Das Auftreten von PROS-assoziiertenmalignen Tumoren wurde bisher ins-gesamt relativ selten beschrieben. Beifünf Patienten mit PIK3CA-Mutatio-nen im vom Überwuchs betroffenenGewebe wurden Wilms-Tumoren (Ne-phroblastome) oder nephrogene Reste(prämaligne Tumoren) diagnostiziert[9, 14, 18, 21, 27]. In isolierten Wilms-Tumoren wurden bei COSMIC bislangkeine somatischen PIK3CA-Mutationenbeschrieben [7]. Gripp et al. [9] schätz-ten die Häufigkeit von Wilms-Tumorenanhand der vorliegenden Literaturda-ten auf unter 1,6 % (4/258 Patienten);Peterman et al. [33] fanden Wilms-Tu-moren bei vier von 122 Patienten (3,3 %)mit CLOVES-Syndrom (davon zwei mitnachgewiesener PIK3CA-Mutation [21,27]). Somit ist das Erkrankungsrisi-ko zwar gering, aber höher als in derAllgemeinbevölkerung (etwa 1:10.000).Außerdem wurden Wilms-Tumoren beizwei PatientenmitMCAPdokumentiert,bevor die molekulare Ursache bekanntwar [23, 42]. Bei zwei Kindern mit derklinischen Diagnose MCAP traten Me-ningeome auf [6, 31]; bei einer eigenenPatientin mit postzygotischer PIK3CA-MutationundeinerHemihyperplasie derlinken Gesichtshälfte bestanden eben-falls multiple Meningeome. Bisher gibtes keine eindeutigen Hinweise auf eineAssoziation mit anderen Tumoren.
Beim Proteus-Syndrom wurde zwarhäufiger ein breites Spektrum an Tumo-ren wie z. B. Meningeome beschrieben;dies kann jedoch auch durch eine Da-tenverzerrung bei bevorzugter Erfassungschwerer erkrankter Patienten (mit-)be-dingt sein. Als relativ spezifisch werden
die auch in die diagnostischen KriterienaufgenommenenmonomorphenAdeno-me der Parotis und bilateralen Zystade-nome der Ovarien betrachtet [4].
Da somatische Mutationen im PI3K-AKT-Signalwegrelativhäufiginverschie-denen Tumoren (PIK3CA z. B. in Ko-lon,Gehirn,Magen,Brust)nachgewiesenwerden und es sich dabei meist eben-falls um die oben genannten Hotspotshandelt, mag es überraschen, dass Mali-gnome bei den hier beschriebenenÜber-wuchssyndromen eher selten beobachtetwerden. Typischerweise treten PIK3CA-Mutationen allerdings erst in späterenStadien der Tumorgenese auf [38], d. h.es handelt sich um Mutationen, die eherdas Tumorwachstum unterstützen. So-mit ist davon auszugehen, dass sich beiPatienten mit einem PI3K-AKT-Mosa-ik-assoziierten Überwuchssyndrom wiebei einem sporadischen Tumor zunächstandere tumorinitiierendeMutationener-eignen müssen.
(Früherkennungs-)Unter-suchungen und Therapie-ansätze
Beim PROS werden zur Früherkennungvon Wilms-Tumoren in Anlehnung andie Empfehlungen beim Beckwith-Wie-demann-Syndrombzw.der isoliertenHe-mihyperplasie abdominelle Ultraschall-untersuchungenalle dreiMonate bis zumAlter von 7–8 Jahren empfohlen [9, 17,33]. Darüber hinaus existieren keine spe-zifischen Früherkennungsempfehlungenbezüglich Tumorerkrankungen. Die Pa-tienten und ihre Eltern sowie die behan-delnden Ärzte sollten bei Auftreten vonunklaren oder unspezifischen Sympto-menentsprechend frühzeitig eineweitereAbklärung anstreben.
Generell sollten bei allen Patientenmit segmentalen Überwuchssyndromenzur Abklärung zusätzlicher Manifesta-tionen und Komplikationen in Abhän-gigkeit von den betroffenen Körperre-gionen entsprechende bildgebende Ver-fahren eingesetzt werden (z. B. MRT desSchädels, der Wirbelsäule usw.) und Un-tersuchungen zur Identifizierung vasku-lärerMalformationen erfolgen [17]. ZumTeil wurde auch von einem erhöhtenThromboserisiko berichtet, sodass ins-
besondere eine adäquate perioperativeAntikoagulation beachtet werden soll-te. Umfang und Zeitpunkt orthopädi-scherundggf.chirurgischerMaßnahmenorientieren sich individuell am AusmaßdesÜberwuchses undmöglicherBegleit-symptome.
Da zur Behandlung von sporadischenTumoren mit somatischen Mutationenim PI3K-AKT-Signalweg bereits ziel-gerichtete medikamentöse Therapiean-sätze zum Einsatz kommen (mTOR-Inhibitoren, z. B. Rapamycin), bestehtdie Hoffnung, dass sich diese in klini-schen Studien auch zur Behandlung derÜberwuchssyndrome mit PI3K-AKT-Mosaiken bewähren [16]. Eine klini-sche Studie in Deutschland ist unterder Leitung von Herrn Prof. Rößler(Universitätsklinikum Freiburg, Pädi-atrische Hämatologie und Onkologie,[email protected])in Vorbereitung [37].
Fazit für die Praxis
4 Bei Vorliegen eines segmentalenÜberwuchses können der Beginn undder Verlauf der Symptomatik eineerste Zuordnung ermöglichen.
4 Häufig bestehen neben dem Über-wuchs kutane Veränderungen, vas-kuläre Malformationen und damitverbunden auch ein erhöhtes Throm-boserisiko.
4 Die Sicherung der Verdachtsdiagnosedurch Nachweis eines somatischenMutationsmosaiks ist häufig nurdurch die Untersuchung von be-troffenem Gewebe möglich. ZumAbgleich sollte aber immer auch eineEDTA-Blutprobe eingesandt werden.
4 Die Untersuchung sollte möglichstan einem Zentrummit entsprechen-der Expertise und den technischenVoraussetzungen (z. B. NGS) nachRücksprache erfolgen, um das bes-te Vorgehen und die Details vonProbenentnahme und -versand zubesprechen.
4 Tumoren treten bei den genanntenÜberwuchssyndromen relativ seltenauf. Früherkennungsuntersuchungenhinsichtlich Wilms-Tumoren werdenempfohlen.
312 medizinische genetik 3 · 2017

4 Ein Wiederholungsrisiko bei Kin-dern der Patienten ist weitgehendausgeschlossen.
Korrespondenzadresse
Dr. med. I. SpierInstitut für HumangenetikUniversitätsklinikumBonnSigmund-Freud-Str. 25, 53127 Bonn,[email protected]
Einhaltung ethischer Richtlinien
Interessenkonflikt. I. Spier undS. Aretz sind andergenetischenDiagnostik und klinischenBetreuungder genannten Patientengruppenbeteiligt. Darüberhinaus bestehen keine Interessenkonflikte.
Dieser Beitragbeinhaltet keine vondenAutorendurchgeführten Studien anMenschenoder Tieren.
Open Access.Dieser Artikelwird unter der CreativeCommonsNamensnennung4.0 International Lizenz(http://creativecommons.org/licenses/by/4.0/deed.de) veröffentlicht, welche dieNutzung, Vervielfäl-tigung, Bearbeitung, VerbreitungundWiedergabein jeglichemMediumundFormat erlaubt, sofernSie den/die ursprünglichenAutor(en) unddieQuelleordnungsgemäßnennen,einenLinkzurCreativeCom-mons Lizenz beifügenundangeben, obÄnderungenvorgenommenwurden.
Literatur
1. Acuna-Hidalgo R, Bo T, KwintMP et al (2015) Post-zygotic Point Mutations Are an UnderrecognizedSource of De Novo Genomic Variation. Am J HumGenet97:67–74
2. Alomari AI (2009) Characterization of a dis-tinct syndrome that associates complex truncalovergrowth, vascular, and acral anomalies: a des-criptivestudyof18casesofCLOVESsyndrome.ClinDysmorphol18:1–7
3. AmyereM,RevencuN,Helaers Retal (2017)Germ-line Loss-of-Function Mutations in EPHB4 Causea Second Form of Capillary Malformation-Arte-riovenousMalformation (CM-AVM2)DeregulatingRAS-MAPKSignaling.Circulation136:1037–1048
4. Biesecker L (2006) The challenges of Proteussyndrome: diagnosis andmanagement. Eur JHumGenet14:1151–1157
5. Caux F, Plauchu H, Chibon F et al (2007) Seg-mental overgrowth, lipomatosis, arteriovenousmalformation and epidermal nevus (SOLAMEN)syndrome is related tomosaic PTEN nullizygosity.Eur JHumGenet15:767–773
6. Conway RL, Pressman BD, DobynsWB et al (2007)Neuroimagingfindings inmacrocephaly-capillarymalformation: a longitudinal study of 17 patients.AmJMedGenetA143A:2981–3008
7. Cosmic http://cancer.sanger.ac.uk/cosmic/browse/tissue.Zugegriffen7.Aug.2017.
8. Eerola I, BoonLM,Mulliken JBetal (2003)Capillarymalformation-arteriovenousmalformation, anewclinical and genetic disorder caused by RASA1mutations.AmJHumGenet73:1240–1249
9. Gripp KW, Baker L, Kandula V et al (2016)Nephroblastomatosis or Wilms tumor in a fourthpatientwitha somaticPIK3CAmutation.AmJMedGenetA170:2559–2569
10. Groesser L, Herschberger E, Landthaler M et al(2012)FGFR3,PIK3CAandRASmutations inbenignlichenoidkeratosis. Br JDermatol166:784–788
11. Hafner C, Lopez-Knowles E, Luis NM et al (2007)Oncogenic PIK3CAmutations occur in epidermalnevi andseborrheic keratoseswitha characteristicmutation pattern. Proc Natl Acad Sci USA104:13450–13454
12. Happle R (1987) Lethal genes surviving bymosaicism: a possible explanation for sporadicbirth defects involving the skin. J Am AcadDermatol16:899–906
13. Hiatt JB, Pritchard CC, Salipante SJ et al (2013)Single molecule molecular inversion probesfor targeted, high-accuracy detection of low-frequencyvariation.GenomeRes23:843–854
14. Hucthagowder V, Shenoy A, Corliss M et al (2017)Utility of clinical high-depth next generationsequencing for somatic variant detection in thePIK3CA-related overgrowth spectrum. Clin Genet91:79–85
15. Hussain K, Challis B, Rocha N et al (2011) Anactivatingmutation of AKT2. HumHypoglycemiaSci334:474
16. Keppler-Noreuil KM, Parker VE, Darling TN etal (2016) Somatic overgrowth disorders of thePI3K/AKT/mTORpathway&therapeuticstrategies.American journal of medical genetics. Part CSeminarsMedGenet172:402–421
17. Keppler-Noreuil KM, Rios JJ, Parker VE et al(2015) PIK3CA-related overgrowth spectrum(PROS): diagnostic and testing eligibility criteria,differential diagnosis, and evaluation. Am J MedGenetA167A:287–295
18. Keppler-Noreuil KM, Sapp JC, Lindhurst MJ et al(2014) Clinical delineation and natural history ofthe PIK3CA-related overgrowth spectrum. Am JMedGenetA164A:1713–1733
19. KleinS, LeeH,GhahremaniSetal (2014)Expandingthe phenotype of mutations in DICER1: mosaicmissense mutations in the RNase IIIb domainof DICER1 cause GLOW syndrome. J Med Genet51:294–302
20. Kuentz P, St-Onge J, Duffourd Y et al (2017)MoleculardiagnosisofPIK3CA-relatedovergrowthspectrum (PROS) in 162 patients and recommen-dations for genetic testing. Genetics in medicine19:989–997
21. Kurek KC, Luks VL, Ayturk UM et al (2012) Somaticmosaic activating mutations in PIK3CA causeCLOVESsyndrome.AmJHumGenet90:1108–1115
22. Kurth I, Grimm T (2014) Mosaike bei monogenenErkrankungen.MedizinischeGenetik26:336–341
23. Lapunzina P, Gairi A, Delicado A et al (2004)Macrocephaly-cutis marmorata telangiectaticacongenita: report of six newpatients and a review.AmJMedGenetA130a:45–51
24. Lee JH, Huynh M, Silhavy JL et al (2012) De novosomatic mutations in components of the PI3K-AKT3-mTORpathwaycausehemimegalencephaly.NatGenet44:941–945
25. Lindhurst MJ, Parker VE, Payne F et al (2012)Mosaic overgrowthwith fibroadipose hyperplasiais caused by somatic activating mutations inPIK3CA.NatGenet44:928–933
26. LindhurstMJ, SappJC, Teer JKetal (2011)Amosaicactivating mutation in AKT1 associated with theProteussyndrome.NEngl JMed365:611–619
27. Luks VL, Kamitaki N, Vivero MP et al (2015)Lymphatic and other vascular malformative/overgrowth disorders are caused by somaticmutations inPIK3CA. JPediatr166:1048–1054
28. Maclellan RA, Luks VL, Vivero MP et al (2014)PIK3CA activating mutations in facial infiltratinglipomatosis. PlastReconstrSurg133:12e–19e
29. Mirzaa G, Timms AE, Conti V et al (2016) PIK3CA-associated developmental disorders exhibitdistinct classes of mutations with variableexpression and tissue distribution. JCI Insight1:e87623
30. Mirzaa G, Conway R, Graham JM Jr, Dobyns WB(2013) PIK3CA-Related Segmental Overgrowth.In: Pagon RA, Adam MP, Ardinger HH, et al (Hrsg)Available from: https://www.ncbi.nlm.nih.gov/books/NBK153722/ Zugegriffen 08. September2017 GeneReviews® [Internet]. Seattle (WA):UniversityofWashington,Seattle, 1993–2017
31. Moore CA, Toriello HV, Abuelo DN et al (1997)Macrocephaly-cutis marmorata telangiectaticacongenita: a distinct disorderwith developmentaldelay and connective tissue abnormalities. Am JMedGenet70:67–73
32. Oduber CE, Van Der HCM, Hennekam RC (2008)Klippel-Trenaunay syndrome: diagnostic criteriaand hypothesis on etiology. Ann Plast Surg60:217–223
33. Peterman CM, Fevurly RD, Alomari AI et al (2017)SonographicscreeningforWilmstumorinchildrenwith CLOVES syndrome.[Epub. Ahead Print).https://doi.org/10.1002/pbc.26684
34. Poduri A, EvronyGD,Cai Xetal (2012) Somatic acti-vationofAKT3causeshemisphericdevelopmentalbrainmalformations.Neuron74:41–48
35. Rios JJ, PariaN, BurnsDKet al (2013) Somatic gain-of-function mutations in PIK3CA in patients withmacrodactyly.HumMolGenet22:444–451
36. Riviere JB, Mirzaa GM, O’Roak BJ et al (2012)De novo germline and postzygotic mutations inAKT3, PIK3R2 and PIK3CA cause a spectrum ofrelated megalencephaly syndromes. Nat Genet44:934–940
37. RösslerJ (2016)SegmentaleÜberwuchssyndrome.Kinder- Jugendmed6:411–416
38. Samuels Y, Velculescu VE (2004) Oncogenicmutations of PIK3CA in human cancers. Cell Cycle3:1221–1224
39. Sapp JC, Turner JT, Van De Kamp JM et al(2007) Newly delineated syndrome of congenitallipomatous overgrowth, vascular malformations,and epidermal nevi (CLOVE syndrome) in sevenpatients. American journal of medical genetics.PartA143A:2944–2958
40. ShirleyMD, TangH, Gallione CJ et al (2013) Sturge-Weber syndrome and port-wine stains causedby somatic mutation in GNAQ. N Engl J Med368:1971–1979
41. Vanhaesebroeck B, Stephens L, Hawkins P (2012)PI3K signalling: the path to discovery andunderstanding.NatRevMolCellBiol13:195–203
42. Wright DR, Frieden IJ, Orlow SJ et al (2009)The misnomer “macrocephaly-cutis marmoratatelangiectatica congenita syndrome”: report of12 new cases and support for revising the nameto macrocephaly-capillary malformations. ArchDermatol145:287–293
43. Yeung KS, Ip JJ, Chow CP et al (2017) SomaticPIK3CAmutations in seven patients with PIK3CA-related overgrowth spectrum. Am J Med Genet A173:978–984
44. ZhouXP,MarshDJ,HampelHetal (2000)Germlineand germlinemosaic PTEN mutations associatedwith a Proteus-like syndrome of hemihyper-trophy, lower limb asymmetry, arteriovenousmalformations and lipomatosis. Hum Mol Genet9:765–768
medizinische genetik 3 · 2017 313


















