
Molekulare Systematik der Gattung Suaeda …nbn:de:hebis:... · Die gegenwärtige Gliederung geht...
155
Molekulare Systematik der Gattung Suaeda (Chenopodiaceae) und Evolution des C 4 -Photosynthesesyndroms Inaugural-Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) im Fachbereich Naturwissenschaften der Universität Kassel vorgelegt von: Peter Wolfram Schütze aus Halle/Saale Kassel, November 2008
Transcript of Molekulare Systematik der Gattung Suaeda …nbn:de:hebis:... · Die gegenwärtige Gliederung geht...

Molekulare Systematik der Gattung Suaeda (Chenopodiaceae) und
Evolution des C4-Photosynthesesyndroms
Inaugural-Dissertation
zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften (Dr. rer. nat.)
im Fachbereich Naturwissenschaften
der Universität Kassel
vorgelegt von:
Peter Wolfram Schütze
aus
Halle/Saale
Kassel, November 2008

2
Betreuer: Prof. Dr. Kurt Weising,
Prüfungskommission: Prof. Dr. Kurt Weising (1. Gutachter)
Prof. Dr. Helmut Freitag (2. Gutachter)
Prof. Dr. Ewald Langer (Beisitzer)
Dr. Frank Blattner (Beisitzer)
Tag der mündlichen Prüfung: 17. Februar 2009

Inhaltsverzeichnis
3
Inhaltsverzeichnis
1. Einleitung ........................................................................................................................................ 5 1.1. Vorbemerkungen.................................................................................................................... 5 1.2. Charakteristik der Suaedoideae............................................................................................. 6
1.2.1. Systematischer Überblick.............................................................................................. 6 1.2.2. Biologie, Klassifikationsmerkmale und Verbreitung der Sippen.................................... 9 1.2.3. Besonderheiten im Photosyntheseweg....................................................................... 12 1.2.4. Evolutionäre Trends innerhalb der Suaedoideae........................................................ 14 1.2.5. Theorien zur Sippenevolution - eine Synthese ........................................................... 16
1.3. Themenentwicklung und Stand der Forschung ................................................................... 18 2. Material und Methoden ................................................................................................................. 22
2.1. Systematische Methoden und Prinzipien............................................................................. 22 2.1.1. Organismenbezogene Methoden................................................................................ 22 2.1.2. Molekülbasierte Methoden .......................................................................................... 22
2.2. Pflanzenmaterial, Kulturversuche ........................................................................................ 25 2.2.1. Auswahl der Arten ....................................................................................................... 25 2.2.2. Aufsammlung, Kulturversuche und Materialkonservierung......................................... 25
2.3. Gewinnung molekularer Merkmale ...................................................................................... 27 2.3.1. DNA-Isolation .............................................................................................................. 27 2.3.2. Gelelektrophorese ....................................................................................................... 28 2.3.3. DNA-Regionen und Primer.......................................................................................... 29 2.3.4. PCR-Methodik ............................................................................................................. 32 2.3.5. DNA-Sequenzierung ................................................................................................... 34
2.4. Auswertung der molekularen Daten..................................................................................... 36 2.4.1. Datenauswahl.............................................................................................................. 36 2.4.2. Sequenz-Alignment ..................................................................................................... 36 2.4.3. Phylogenetische Berechnungsverfahren .................................................................... 38 2.4.4. Molekulare Uhr und Zeitdatierung............................................................................... 45 2.4.5. Test- und Überprüfungsverfahren ............................................................................... 47
2.5. Bewertung morphologischer Merkmale ............................................................................... 49 3. Ergebnisse.................................................................................................................................... 51
3.1. Ausprägung und Variabilität der molekularen Merkmale..................................................... 51 3.1.1. DNA-Isolation, Amplifikation und Sequenzierung ....................................................... 51 3.1.2. Strukturmerkmale der untersuchten DNA-Regionen .................................................. 52
3.2. Phylogenetische Rekonstruktion.......................................................................................... 55 3.2.1 Stellung der Suaedoideae innerhalb der Chenopodiaceae ........................................ 55 3.2.2. ITS-Phylogenie der Suaedoideae ............................................................................... 59 3.2.3. Chloroplasten-Phylogenie der Suaedoideae .............................................................. 68 3.2.4. Phylogenie und Diversität der Sektion Brezia ............................................................. 75
3.3. Altersdatierung und Molekulare Uhr .................................................................................... 81 3.4. Evolution von C4-Photosynthese und Blatttypen ................................................................. 87 3.5. Merkmalsentwicklung anhand molekularer Phylogenien..................................................... 93

Inhaltsverzeichnis
4
4. Diskussion .................................................................................................................................... 97 4.1. Qualität, Aussagekraft und Grenzen der Daten und Methoden........................................... 97
4.1.1. Auswahl und Umfang der Sippen................................................................................ 97 4.1.2. Vergleichende DNA-Sequenzierung ........................................................................... 98 4.1.3. Phylogenetische Rekonstruktionsverfahren und Datierungsmethoden ...................... 99
4.2. Inter- und intraspezifische Variabilität ................................................................................ 100 4.3. Phylogenie und Artbildung innerhalb der Suaedoideae .................................................... 101
4.3.1. Molekulare Evolution ................................................................................................. 101 4.3.2. Radiation, Gruppenbildung und Speziation............................................................... 103 4.3.3. Hybridisierung und Polyploidisierung ........................................................................ 104
4.4. Merkmalsentwicklung innerhalb der Suaedoideae ............................................................ 107 4.4.1. Systematisch relevante Merkmale und ihre Evolution .............................................. 107 4.4.2 Evolution der C4-Photosynthese ............................................................................... 111
4.5. Ableitung systematischer Einheiten ................................................................................... 111 4.5.1. Position und Gliederung der Suaedoideae ............................................................... 111 4.5.2. Arten und systematische Gruppen in der Gattung Suaeda ...................................... 112
4.5.2.1. Brezia-Gruppe ........................................................................................................... 112 4.5.2.2. Schoberia-Gruppe inkl. Alexandra lehmannii............................................................ 113 4.5.2.3. Fruticosa-Gruppe ...................................................................................................... 113 4.5.2.4. Physophora-Gruppe.................................................................................................. 113 4.5.2.5. Suaeda-Gruppe......................................................................................................... 114 4.5.2.6. Schanginia-Gruppe mit Borszczowia aralocaspica................................................... 114 4.5.2.7. Suaeda glauca .......................................................................................................... 114 4.5.2.8. Bienertia .................................................................................................................... 114
4.6. Vorschläge zur Neugliederung der Suaedoideae .............................................................. 115 5. Zusammenfassung ..................................................................................................................... 118 6. Literaturverzeichnis..................................................................................................................... 121 Anhang ................................................................................................................................................ 131
Anhang 1 Abbildungsverzeichnis............................................................................................... 131 Anhang 2 Tabellenverzeichnis................................................................................................... 132 Anhang 3 Abkürzungsverzeichnis: ............................................................................................ 133 Anhang 4 Verzeichnis der Chemikalien, Lösungen und Reaktionskits ..................................... 134 Anhang 5 Materialtabelle ........................................................................................................... 135 Anhang 6 Zusätzliche Stammbäume der phylogenetischen Rekonstruktion ............................ 145 Danksagung................................................................................................................................ 154 Selbständigkeitserklärung........................................................................................................... 155

Einleitung
5
1. Einleitung 1.1. Vorbemerkungen Pflanzen verfügen über ausgeprägte und hoch differenzierte Anpassungen an ihre
Umweltbedingungen. Die Anpassungen der Pflanzen, letztendlich ihre unterschiedliche Morphologie
und Lebensweise, sind das Ergebnis lang andauernder Adaption an Umwelteinflüsse durch die
fortschreitende Auslese optimaler Typen. Aufgabe systematischer Forschung an Organismen ist zum
einen die Beschreibung der historischen und rezenten Vielfalt, zum anderen die Ergründung der
Entwicklungswege zur heutigen Diversität. Systematik ordnet die Vielfalt und stellt Kategorien und
Systeme auf. Dabei ist aber die wichtigste Kategorie - diejenige der Art - heute umstrittener denn je.
Anders ist es kaum zu erklären, dass eine Vielzahl von Vorstellungen über den Begriff der Art existiert,
die den Anspruch des Universellen erheben, aber dennoch oft die Prägung des Bearbeiters erkennen
lassen. Wie die Untersuchungsobjekte unterliegt dabei auch ihre Wissenschaft einer ständigen
Entwicklung und ist abhängig vom Stand der technischen Entwicklung und in nicht geringem Maße
von herrschenden gesellschaftlichen Denkmodellen. Standen in früherer Zeit statische Modelle bereit,
die die Natur als etwas Unveränderliches, Ewiges erklärten, so hat sich gegenwärtig das dynamische,
die Evolution berücksichtigende Modell als Denkrichtung durchgesetzt.
Die systematische Forschung an Organismen hat mittlerweile durch die technischen Entwicklungen
einen Stand erreicht, der wesentlich tiefere Einblicke in die Entwicklung der Sippen erlaubt. Möglich
wurde dies durch Einbeziehung von molekularen Merkmalen der Desoxy-Ribonukleinsäure (DNA), die
als Erbinformationsträger eine mit dem äußeren Erscheinungsbild des Organismus nur bedingt
korrelierte eigene evolutionäre Geschichte besitzt. Da davon ausgegangen wird, dass dieser
evolutionäre Prozess nach modellierbaren Kriterien abläuft und aus der Analyse des rezenten
Diversitätsspektrums rekonstruiert werden kann, steht der evolutionsbiologisch ausgerichteten
systematischen Forschung ein weiteres, für bestimmte Fragestellungen sogar entscheidendes Mittel
zur Verfügung. Mit Einführung der molekularsystematischen Methoden wurden bisher gültige Systeme
grundlegend verändert. Als Beispiel dafür soll die Neugliederung der Angiospermen basierend auf
DNA-Merkmalen angeführt werden (Chase et al. 1993, The Angiosperm Phylogeny Group 2003).
Die vorliegende Arbeit beschäftigt sich mit der Phylogenie, Systematik und Merkmalsentwicklung der
zur Familie der Gänsefußgewächse (Chenopodiaceae) gehörenden Gattung Suaeda sowie ihren
unmittelbaren Verwandten. Wie kaum eine andere Pflanzengruppe haben es die Vertreter dieses
Verwandtschaftskreises geschafft, extreme und für normales Pflanzenwachstum kaum geeignete
Lebensräume zu besiedeln. Dies sind vor allem Habitate mit hohen Ionenkonzentrationen im Boden
(Salzstandorte), extremen täglichen und jährlichen Temperaturschwankungen sowie hoher
Strahlungsintensität. Diese Habitate finden sich vor allem in den primär waldfreien kontinentalen
Trockengebieten sowie entlang von Meeresküsten. In Anpassung an diese extremen
Lebensbedingungen wurde eine Vielzahl von physiologischen und anatomischen Besonderheiten
entwickelt, so z. B. effektive Mechanismen zur Regulation des Ionenhaushalts in der Pflanze,
Minimierung der Blattflächen und Zunahme des Sukkulenzgrades sowie Modifikationen der
Photosynthese (C4-Weg mit mannigfaltigen blattanatomischen Typen).

Einleitung
6
Im Gegensatz zu der starken anatomischen Differenzierung im Blattbereich steht eine auffallende
Merkmalsarmut und Uniformität im äußeren Erscheinungsbild der Gattung Suaeda. Dies ist eine der
hauptsächlichen Ursachen dafür, dass bis heute kein praktikables taxonomisches Konzept für die
gesamte Gruppe vorliegt und die tatsächliche Artenzahl nicht bekannt ist. Unter den genannten
äußeren Einflüssen setzt sich offenbar ein optimierter Morphotyp durch, von dem wenig
Abweichungen toleriert werden. Einige der morphologischen Merkmale weisen aber in Abhängigkeit
von Standortsfaktoren eine hohe intraspezifische Plastizität auf. Suaeda und die verwandten Taxa
gelten daher als taxonomisch sehr schwierige Gruppe, die in den meisten Monographien und
Florenwerken sowie in der geobotanischen Literatur nicht korrekt dargestellt ist.
Das Hauptziel der vorliegenden Arbeit besteht daher in der Aufklärung von Status und Abgrenzung
der Arten einschließlich ihrer systematischen und phylogenetischen Beziehungen, der Nachzeichnung
wichtiger Ereignisse in der Sippenbildung im Kontext zu erdgeschichtlichen Ereignissen sowie der
stammesgeschichtlichen Ableitung wichtiger Merkmale. Sie bildet damit eine der Grundlagen für die
zukünftige Darstellung in Florenwerken sowie für eine Gesamtrevision der Gattung Suaeda. Erstmalig
werden molekularsystematische Methoden in dieser Gruppe etabliert und angewandt.
1.2. Charakteristik der Suaedoideae 1.2.1. Systematischer Überblick Die Gattung Suaeda in der aktuellen taxonomischen Auffassung umfasst weltweit ungefähr 100 Arten
(Iljin 1936b, Kühn et al. 1993, Schenk & Ferren 2001) und wurde in der Vergangenheit in bis zu neun
Sektionen eingeteilt (Moquin-Tandon 1840, Volkens 1894, Ulbrich 1934, Iljin 1936a, Tzvelev 1993,
Schenk & Ferren 2001). Nach Ulbrich (1934) gehört Suaeda innerhalb der Chenopodiaceae zu der
Unterfamilie der Suaedoideae und darin zur Tribus Suaedeae. Zu dieser Tribus wurden noch die
monotypischen Gattungen Hypocylix, Helicilla, Brezia, Alexandra und Calvelia gestellt. Die zweite
Tribus der Unterfamilie enthält die beiden monotypischen Gattungen Borszczowia und Bienertia. Nach
der letzten zusammenfassenden Arbeit von Kühn et al. (1993) wird die gesamte Gruppe mit einer
Vielzahl weiterer Gattungen zur Unterfamilie der Salsoloideae vereinigt und darin als Tribus Suaedeae
geführt, die neben Suaeda nur noch die Arten Alexandra lehmannii, Borszczowia aralocaspica und
Bienertia cycloptera als Vertreter der jeweils monotypischen Gattungen enthält. Brezia heterophylla,
Helicilla altissima und Calvelia pterantha werden heute zu Suaeda, Hypocylix kerneri zu Salsola
gerechnet.
Die enge Zuordnung der Suaedoideae zu den Salsoloideae stützt sich vor allem auf
Embryomerkmale. Auf der Form des Embryos beruhte die lange Zeit gültige Unterteilung der
Chenopodiaceae in Spirolobeae (spiralig aufgerollter Embryo) und Cyclolobeae (ring- oder
hufeisenförmiger Embryo). Damit gehörten die Salicornioideae als Cyclolobeae nicht in die
unmittelbare Verwandtschaft der Suaedoideae. Bereits Ulbrich (1934) zweifelt aber die Gültigkeit
dieser Unterteilung an und auch die aktuellen molekularphylogenetischen Untersuchungen an den
Chenopodiaceae/Amaranthaceae durch Kadereit et al. (2003) relativieren die Bedeutung der
Embryomerkmale für die Großsystematik. Die molekularen Stammbäume auf Basis des rbcL-Gens
sprechen für eine nahe Verwandtschaft der Suaedoideae mit den Salicornioideae.
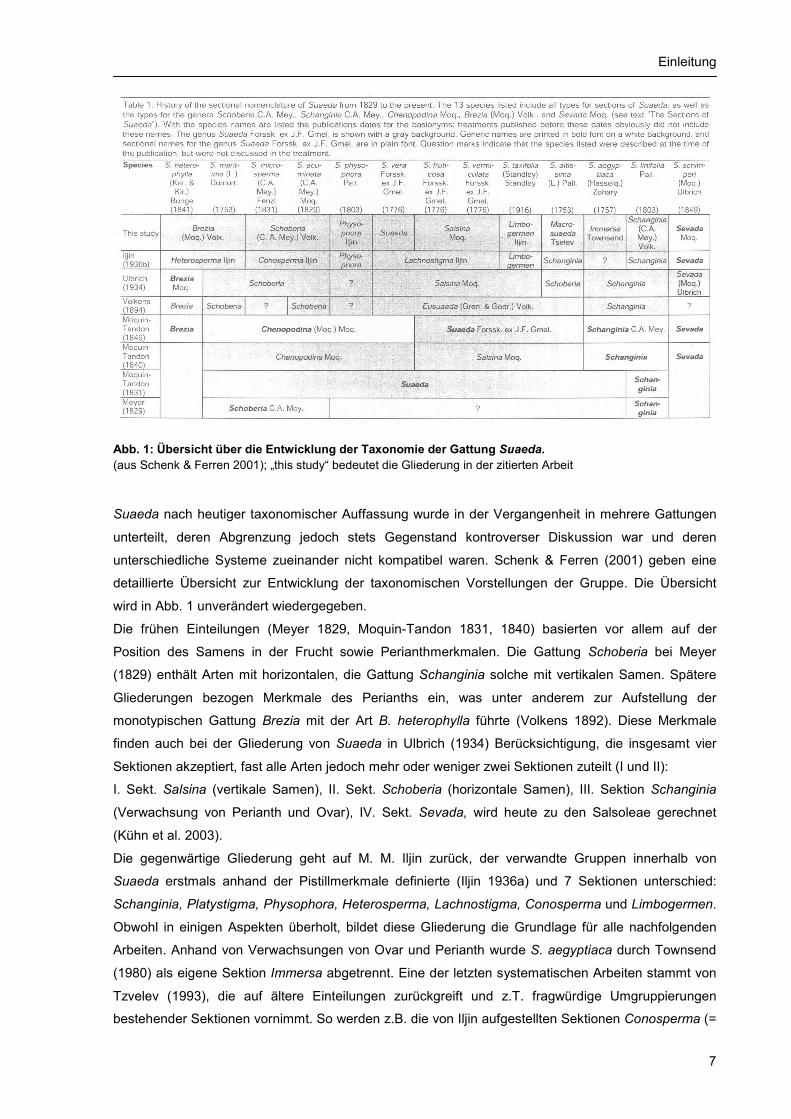
Einleitung
7
Abb. 1: Übersicht über die Entwicklung der Taxonomie der Gattung Suaeda. (aus Schenk & Ferren 2001); „this study“ bedeutet die Gliederung in der zitierten Arbeit
Suaeda nach heutiger taxonomischer Auffassung wurde in der Vergangenheit in mehrere Gattungen
unterteilt, deren Abgrenzung jedoch stets Gegenstand kontroverser Diskussion war und deren
unterschiedliche Systeme zueinander nicht kompatibel waren. Schenk & Ferren (2001) geben eine
detaillierte Übersicht zur Entwicklung der taxonomischen Vorstellungen der Gruppe. Die Übersicht
wird in Abb. 1 unverändert wiedergegeben.
Die frühen Einteilungen (Meyer 1829, Moquin-Tandon 1831, 1840) basierten vor allem auf der
Position des Samens in der Frucht sowie Perianthmerkmalen. Die Gattung Schoberia bei Meyer
(1829) enthält Arten mit horizontalen, die Gattung Schanginia solche mit vertikalen Samen. Spätere
Gliederungen bezogen Merkmale des Perianths ein, was unter anderem zur Aufstellung der
monotypischen Gattung Brezia mit der Art B. heterophylla führte (Volkens 1892). Diese Merkmale
finden auch bei der Gliederung von Suaeda in Ulbrich (1934) Berücksichtigung, die insgesamt vier
Sektionen akzeptiert, fast alle Arten jedoch mehr oder weniger zwei Sektionen zuteilt (I und II):
I. Sekt. Salsina (vertikale Samen), II. Sekt. Schoberia (horizontale Samen), III. Sektion Schanginia
(Verwachsung von Perianth und Ovar), IV. Sekt. Sevada, wird heute zu den Salsoleae gerechnet
(Kühn et al. 2003).
Die gegenwärtige Gliederung geht auf M. M. Iljin zurück, der verwandte Gruppen innerhalb von
Suaeda erstmals anhand der Pistillmerkmale definierte (Iljin 1936a) und 7 Sektionen unterschied:
Schanginia, Platystigma, Physophora, Heterosperma, Lachnostigma, Conosperma und Limbogermen.
Obwohl in einigen Aspekten überholt, bildet diese Gliederung die Grundlage für alle nachfolgenden
Arbeiten. Anhand von Verwachsungen von Ovar und Perianth wurde S. aegyptiaca durch Townsend
(1980) als eigene Sektion Immersa abgetrennt. Eine der letzten systematischen Arbeiten stammt von
Tzvelev (1993), die auf ältere Einteilungen zurückgreift und z.T. fragwürdige Umgruppierungen
bestehender Sektionen vornimmt. So werden z.B. die von Iljin aufgestellten Sektionen Conosperma (=

Einleitung
8
Schoberia) und Heterosperma (= Brezia) zu einer gemeinsamen Sektion (Schoberia) vereinigt und
anhand von Merkmalen des Teilblütenstandes die monotypische Sektion Macrosuaeda mit der Art S.
altissima unterschieden. Schenk & Ferren (2001) diskutieren ausführlich die Benennungsgeschichte
der Gruppe und stellen eine erweiterte Gliederung der Gattung Suaeda vor. Basierend auf
morphologischen Merkmalen werden demnach 9 Sektionen akzeptiert, die früher teilweise als
separate Gattungen geführt wurden. Die aktuelle Gliederung basiert im wesentlichen auf dem System
von Iljin, berücksichtigt aber auch neuere Untersuchungen besonders zu C4-Photosynthese und
Blattanatomie, da sich diese Merkmale als hoch spezifisch erwiesen haben (s. Kap. 1.2.3.). Für einige
Sektionen wird die Typussippe festgelegt. Folgende Sektionen werden aufgestellt:
Suaeda sect. Brezia (Moq.) Volk.; - Typus: Suaeda heterophylla (Kar. & Kir.) Bunge repräsentative Arten: S. australis, S. calceoliformis, S. corniculata, S. esteroa, S. heterophylla, S.
heteroptera, S. japonica, S. linearis, S. maritima, S. mexicana, S. occidentalis, S. olufsenii, S.
patagonica, S. prostrata, S. przevalskyi, S. puertopenascoa, S. rolandii
Suaeda sect. Immersa Townsend; - Typus: Suaeda aegyptiaca (Hasselq.) Zoh.
einzige Art: S. aegyptiaca
als weitere Sippe könnte S. arcuata zu dieser Sektion gehören
Suaeda sect. Limbogermen Iljin; - Typus Suaeda taxifolia Standl. repräsentative Arten: S. argentinensis, S. californica, S. conferta, S. divaricata, S. foliosa, S. nigra, S.
palmeri, S. tampicensis, S. taxifolia
Suaeda sect. Macrosuaeda Tzvelev; - Typus: Suaeda altissima (L.) Pall.
einzige Art: S. altissima
Suaeda sect. Physophora Iljin; - Typus Suaeda physophora Pall. einzige Art: S. physophora
Suaeda sect. Salsina Moq.; - Typus: Suaeda vermiculata Forssk ex J. F. Gmel.
repräsentative Arten: S. vermiculata, S. monoica, S. fruticosa, S. moschata, S. pruinosa, S.
monodiana, S. nudiflora
Suaeda sect. Schanginia (C. A. Mey.) Volk.; - Typus: Suaeda linifolia Pall.
repräsentative Arten: S. linifolia, S. glauca, S. paradoxa
Suaeda sect. Schoberia (C. A. Mey.) Volk.; - Typus Schoberia acuminata C. A. Mey.
repräsentative Arten: S. microsperma, S. acuminata, S. baccifera, S. carnosissima, S. splendens
Suaeda sect. Suaeda; Typus: Suaeda vera Forssk. ex J. F. Gmelin
einzige Art: S. vera

Einleitung
9
Abb. 2: Suaedoideae, Habitate und Wuchsformen. 1 Suaeda heteroptera, Bestand nahe Dauria (Russland); 2 S. spicata: Mittelmeerküste nahe Ebro-Delta (Spanien); 3 Bienertia cycloptera: Sproßabschnitt mit Früchten (Phot. H. Akhani); 4 Suaeda heterophylla: oberer Teil eines Sprosses; 5 Suaeda vera (frischgrün) und S. mollis (graugrün), Phot. H. Freitag, Kanaren
Die artenreichste Sektion, gleichzeitig aber auch diejenige mit den größten taxonomischen
Unklarheiten, ist Brezia. Zu dieser Sektion gehören die taxonomisch kritischen Taxa S. maritima, S.
salsa, S. prostrata und S. corniculata. Aktuelle zusammenfassende Darstellungen der gesamten
Gattung existieren nicht. Von mehreren Sippen ist die Sektionszugehörigkeit bisher ungeklärt (S.
palaestina, S. ifniensis, S. arbusculoides etc.). Die einzige bisher publizierte Revision der Gattung
Suaeda liegt über 150 Jahre zurück (Moquin-Tandon 1840).
1.2.2. Biologie, Klassifikationsmerkmale und Verbreitung der Sippen
.
Die Suaedoideae vereinen einjährige Kräuter, Zwergsträucher oder kleine Bäume (S. monoica).
Ausdauernde Sippen sind stets verholzt und immergrün, einjährige krautig und nur gelegentlich an der
Stängelbasis verholzt. Die Art der Verholzung unterscheidet sich zwischen unterschiedlichen
Gruppen. Hoch entwickelte, ausdifferenzierte Verholzung findet sich bei den strauchigen Sippen der
Sektionen Salsina und Limbogermen, (bei S. monoica Differenzierung in Kern- und Splintholz), viel
1 2
3 4 5

Einleitung
10
weniger entwickelt und meist auf den basalen Teil der Sprossachse beschränkt ist sie bei den
verholzten Sippen der Sektion Brezia. Die Sprosse sind oft in mehreren Ordnungen verzweigt und
tragen im apikalen Teil zahlreiche Blütenstände. Äußerlich sehr einfach gebaut sind die ungeteilten,
linealischen, zylindrischen oder halbzylindrischen Blätter. Sie gehören zum äquifazialen Typ, in dem
das Mesophyll zylinderartig um das Leitbündelsystem angeordnet ist. Die anatomische
Differenzierung ist eng mit den verschiedenen Photosynthesewegen verknüpft (s. Kap. 1.2.3). Ihre
Anordnung am Spross ist im Gegensatz zu vielen Salicornioideae stets wechselständig. Im Bereich
des Blütenstandes sind sie meist verkürzt, morphologisch aber nicht grundsätzlich von den
Laubblättern des Stängels verschieden.
Die Blüten stehen zu je 3-5(-20) in blattachselständigen cymösen Teilblütenständen, die nach Ulbrich
(1934) Schraubel sind. Die Beurteilung ist jedoch schwierig, da die Blütenstandsachsen mit
Ausnahme weniger Arten (S. altissima) so stark verkürzt sind, dass sie praktisch nicht sichtbar sind.
Dennoch wird der Charakter als Blütenstand an den stets vorhandenen Bracteolen deutlich. Suaeda
besitzt trockenhäutige, Bienertia krautige Bracteolen, ein wichtiges Unterscheidungsmerkmal zu den
Salicornioideae, die über keine Bracteolen verfügen. Als Besonderheit sind die Blütenstandsachsen
im Falle von S. altissima und einigen weiteren Sippen teilweise mit dem Stiel des Tragblattes
verwachsen (partielle Recaulescenz). Mit diesem Merkmal wurde unter anderem die Sektion
Schanginia von Iljin (1936a) definiert. Die Blüten sind klein und unscheinbar und besitzen keinerlei
Schau- oder andere Attraktionseinrichtungen für Insekten.
Die Blütenhülle ist ein einfaches, fünfteiliges, grünes Perianth. Die Perianthzipfel sind entweder
gerundet oder gekielt und gelegentlich mit flügelartigen Strukturen, apikalen oder basalen
Auswüchsen versehen, was sich besonders deutlich an der Frucht zeigt. Form und
Verwachsungsgrad der Perianthzipfel untereinander als auch mit dem Ovar sind sippenspezifisch und
von hoher taxonomischer Relevanz. Markante horizontale Flügel haben beispielsweise Suaeda
heterophylla und Bienertia cycloptera, bei Bienertia sind die einzelnen Segmente zu einem mehr oder
weniger geschlossenen Ring verwachsen. Auffallende basale Flügel besitzen S. heteroptera und S.
kulundensis. Durch zugespitzte und manchmal hornartig verlängerte Perianthzipfel sind S. corniculata,
S. pannonica, S. tschujensis und S. patagonica charakterisiert. Aufgrund der Förderung des
Größenwachstums eines Perianthzipfels erscheinen die Blüten oft stark asymmetrisch, dies
charakterisiert vor allem S. corniculata. Vertikale Flügel, allerdings meist nur auf zwei der fünf
Tepalen, kommen bei Alexandra lehmannii und Suaeda pterantha (= Calvelia p.) vor. Die
Perianthzipfel sind meist frei, selten untereinander verwachsen (S. linifolia, S. paradoxa).
Verwachsungen von Perianth und Ovar kennzeichnen auch Suaeda aegyptiaca, S. arcuata, Bienertia
und Borszczowia aralocaspica. Das Ovar ist rundlich bis eiförmig, kegelförmig oder auch
flaschenförmig gestreckt, bei einigen Sippen an der Spitze eingebuchtet und trägt 2-3 Narben. Selten
sind die Narben verbreitert und dadurch undeutlich getrennt (S. vera). Ein echter Griffel ist nicht
ausgebildet. Anzahl und Form der Narben sowie ihre Insertion im Ovar sind wichtige
gruppengliedernde Merkmale und wurden von Iljin (1936a) zur Sektionsgliederung verwendet. Jede
Blüte entwickelt nur einen Samen. Die Testa ist bei normal ausgebildeten Samen verhärtet, schwarz
gefärbt und bei einigen Arten mit sehr charakteristischer Netzzeichnung versehen. Eine Besonderheit,
die bei sehr vielen Arten auftreten kann, ist die Heteromorphie der Samen.

Einleitung
11
Abb. 3: Suaeda, Blüten- und Samenmerkmale. 1 S. altissima, Teilblütenstand mit verlängerten Blütenstandsachsen; 2 S. eltonica: Teilblütenstände mit trockenhäutigen Bracteolen; 3 S. corniculata: Samen mit Oberflächenskulpturierung (Phot M. Lomonosova); 4 S. kulundensis: Perianth mit basalen Flügeln; 5 S. corniculata: asymmetrisches Perianth mit einem vergrößerten Tepalum ; 6 S.maritima: einfaches Perianth
Neben hartschaligen Samen kommen, oft auf der gleichen Pflanze, dünnschalige, wesentlich größere
Samen mit grünem Embryo vor. Samengröße und Zeichnung der Samenschale sind systematisch
bedeutsame Merkmale.
Die Suaedoideae sind weltweit verbreitet mit Häufung in den temperaten Zonen, fehlen jedoch in der
borealen, arktischen und antarktischen Florenzone. Suaeda als artenreichste Gattung der Unterfamilie
besitzt auch das größte Gesamtareal. Das rezente Diversitätszentrum der gesamten Gruppe befindet
sich in einem Gebiet zwischen Mittelmeerraum und Zentralasien.
Als Hygrohalophyten besiedeln alle Arten salzhaltige Standorte entlang der Meeresküsten und im
Binnenland, sind dort jedoch auf zumindest periodische Vernässung angewiesen. Sie können in
Bereiche vordringen die nur von wenigen, hochspezialisierten Pflanzenarten besiedelt werden und
haben bedeutenden Anteil an der halophytischen Vegetation natürlich waldfreier, wechsel- oder
dauernasser Standorte.
Chorologische Daten liegen bisher nur in sehr unzureichendem Maße vor. So wird die Gattung bei
Meusel et al. (1965) nicht behandelt. Einzig Iljin (1936a) bringt eine aufsummierte Verbreitungskarte
der von ihm anerkannten Sektionen, was einen Überblick über grundlegende Verbreitungsmuster
erlaubt. Danach haben nur die Sektionen Heterosperma und Lachnostigma (entspricht etwa den
Sektionen Brezia und Salsina) größere Areale, während die übrigen Gruppen nur sehr eng umgrenzte
Verbreitungsgebiete besitzen. Die Gesamtverbreitung von Suaeda maritima wird von Hultén & Fries
(1986) dargestellt, doch liegt dieser Darstellung eine falsche taxonomische Auffassung zu Grunde. Die
exakte Verbreitung ist gegenwärtig nur von sehr wenigen Sippen mit eng begrenztem Areal bekannt
und lässt sich nur aus einzelnen Florenwerken rekonstruieren.
1 2 3
4 5 6

Einleitung
12
1.2.3. Besonderheiten im Photosyntheseweg Ökologie und Verbreitung vieler Chenopodiaceae bedingen zwangsläufig die Optimierung der
Photosynthese, in der genannten Pflanzenfamilie ausschließlich über den C4-Weg. C4-Photosynthese
kommt innerhalb der Landpflanzen nur bei den Angiospermen vor und kennzeichnet etwa 3% aller
Arten (Sage 2004), die allerdings für 20-30% der Biomasseproduktion der Erde verantwortlich sind
(Gillion & Jakir 2001). Mit etwa 550 Arten (Edwards et al. 2004) stehen die Chenopodiaceae
hinsichtlich der Artenzahlen nach Poaceae (4600 spp.) und Cyperaceae (1350 spp.) an dritter Stelle.
Es wird angenommen, dass sie innerhalb der Chenopodiaceae entwicklungsgeschichtlich älter ist als
bei allen anderen Gruppen. Sage (2004) gibt als Maximalalter 15-21 Mill. Jahre an, während Fisher et
al. (1997) die C4-Entstehung innerhalb Suaeda in der obersten Kreide- bzw. frühen Tertiärzeit
postulieren. Damit würde der Prozess allerdings eng mit der Entstehung der Angiospermen
zusammenfallen.
C4-Pflanzen besitzen bei Wassermangel, hohen Temperaturen (> 25°C), hoher Sonneneinstrahlung
und geringerem CO2-Gehalt der Luft physiologische Vorteile gegenüber C3-Pflanzen. Die genannten
Faktoren begünstigen und selektieren Arten mit C4-Photosynthese, die aufgrund höherer
Assimilationsraten, eines höheren Temperaturoptimums und effizienterer Wassernutzung
konkurrenzstärker sind. Die C4-Photosynthese unterscheidet sich von der C3-Photosynthese vor allem
durch die Trennung von CO2-Fixierung und Calvin-Zyklus und der Bildung von C4-Körpern (Aspartat,
Malat) während der CO2-Fixierung. Dies ist immer verbunden mit funktionalen Differenzierungen in
den beteiligten Zellen und Chloroplasten. Am Beispiel von Suaeda monoica wurde nachgewiesen,
dass die CO2-Fixation und C4-Körperbildung in den Zellen des Palisadenparenchyms (primary carbon
assimilation, PCA) unter Beteiligung der PEP-Carboxylase stattfindet. Die C4-Körper werden
anschließend in die Bündelzellen transportiert (photosynthetic carbon reduction, PCR, Kranzzellen), in
denen der Calvin-Zyklus stattfindet (Shomer-Ilan et al. 1979). Kranzzellen weisen verdickte oder
suberin-verstärkte Zellwände auf, um die CO2-Diffusion zu unterbinden. Dies ist notwendig für die
Effektivität der CO2-Reduktion.
Innerhalb von Suaeda ist die C4-Photosynthese durch den NAD-ME-Typ realisiert (Fisher et al. 1997),
der durch die Aspartatbildung charakterisiert ist. Nach Pyankov et al. (1997) wird der Aspartat-
Metabolismus (NAD-ME) als ursprünglich angesehen und kennzeichnet z.B. Suaedeae und
Atripliceae. Daraus sollte sich als abgeleitete Form der Malat-Metabolisums vieler Salsoleae ableiten.
Der biochemische Weg der C4-Photosynthese ist mit histologisch hoch differenzierten, anatomisch
allerdings sehr unterschiedlichen Blatttypen gekoppelt, von denen allein vier verschiedene bei den
Suaedoideae vorkommen (Freitag & Stichler 2000, Freitag & Stichler 2002, Kadereit et al. 2003,
Schütze et al. 2003). Dabei handelt es sich keineswegs um die innerhalb der Angiospermen weit
verbreitete C4-Anatomie mit den typischen, jeweils ein Leitbündel umschließenden Bündelscheiden-
Zellen sondern um die Ausbildung zylindrischer Chlorenchym-Scheiden, die die Gesamtheit aller
Leitbündel und meist auch noch ein innen liegendes Wasserspeichergewebe umgeben. Die
Zuordnung zu einem photosynthetischen Typus ist für die Suaedoideae weitgehend klar, C3/C4-
Intermediate wie bei einigen Salsoloideae (Pyankov 1997) kommen nicht vor.
Die folgende Übersicht gibt anhand von Blattquerschnitten einen Überblick über die vier innerhalb der
Suaedoideae vorkommenden C4-Blatttypen:
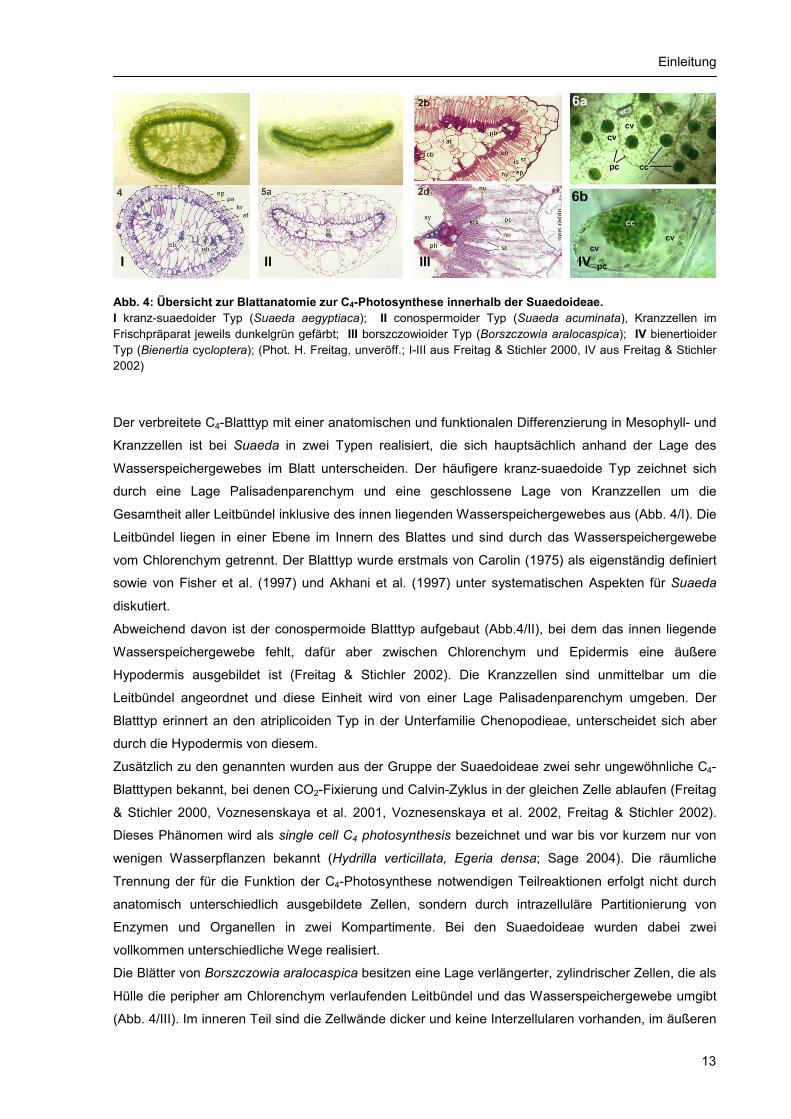
Einleitung
13
Abb. 4: Übersicht zur Blattanatomie zur C4-Photosynthese innerhalb der Suaedoideae. I kranz-suaedoider Typ (Suaeda aegyptiaca); II conospermoider Typ (Suaeda acuminata), Kranzzellen im Frischpräparat jeweils dunkelgrün gefärbt; III borszczowioider Typ (Borszczowia aralocaspica); IV bienertioider Typ (Bienertia cycloptera); (Phot. H. Freitag, unveröff.; I-III aus Freitag & Stichler 2000, IV aus Freitag & Stichler 2002)
I II III IV
Der verbreitete C4-Blatttyp mit einer anatomischen und funktionalen Differenzierung in Mesophyll- und
Kranzzellen ist bei Suaeda in zwei Typen realisiert, die sich hauptsächlich anhand der Lage des
Wasserspeichergewebes im Blatt unterscheiden. Der häufigere kranz-suaedoide Typ zeichnet sich
durch eine Lage Palisadenparenchym und eine geschlossene Lage von Kranzzellen um die
Gesamtheit aller Leitbündel inklusive des innen liegenden Wasserspeichergewebes aus (Abb. 4/I). Die
Leitbündel liegen in einer Ebene im Innern des Blattes und sind durch das Wasserspeichergewebe
vom Chlorenchym getrennt. Der Blatttyp wurde erstmals von Carolin (1975) als eigenständig definiert
sowie von Fisher et al. (1997) und Akhani et al. (1997) unter systematischen Aspekten für Suaeda
diskutiert.
Abweichend davon ist der conospermoide Blatttyp aufgebaut (Abb.4/II), bei dem das innen liegende
Wasserspeichergewebe fehlt, dafür aber zwischen Chlorenchym und Epidermis eine äußere
Hypodermis ausgebildet ist (Freitag & Stichler 2002). Die Kranzzellen sind unmittelbar um die
Leitbündel angeordnet und diese Einheit wird von einer Lage Palisadenparenchym umgeben. Der
Blatttyp erinnert an den atriplicoiden Typ in der Unterfamilie Chenopodieae, unterscheidet sich aber
durch die Hypodermis von diesem.
Zusätzlich zu den genannten wurden aus der Gruppe der Suaedoideae zwei sehr ungewöhnliche C4-
Blatttypen bekannt, bei denen CO2-Fixierung und Calvin-Zyklus in der gleichen Zelle ablaufen (Freitag
& Stichler 2000, Voznesenskaya et al. 2001, Voznesenskaya et al. 2002, Freitag & Stichler 2002).
Dieses Phänomen wird als single cell C4 photosynthesis bezeichnet und war bis vor kurzem nur von
wenigen Wasserpflanzen bekannt (Hydrilla verticillata, Egeria densa; Sage 2004). Die räumliche
Trennung der für die Funktion der C4-Photosynthese notwendigen Teilreaktionen erfolgt nicht durch
anatomisch unterschiedlich ausgebildete Zellen, sondern durch intrazelluläre Partitionierung von
Enzymen und Organellen in zwei Kompartimente. Bei den Suaedoideae wurden dabei zwei
vollkommen unterschiedliche Wege realisiert.
Die Blätter von Borszczowia aralocaspica besitzen eine Lage verlängerter, zylindrischer Zellen, die als
Hülle die peripher am Chlorenchym verlaufenden Leitbündel und das Wasserspeichergewebe umgibt
(Abb. 4/III). Im inneren Teil sind die Zellwände dicker und keine Interzellularen vorhanden, im äußeren

Einleitung
14
dünnwandiger und Interzellularen vorhanden. Zwei morphologisch unterschiedene Typen von
Chloroplasten finden sich in unterschiedlichen Mengen im inneren und äußeren Teil der Zelle. Anhand
von Immunolokalisation der wichtigsten Enzyme wurde die räumliche Trennung der Enzymaktivität
nachgewiesen. Der innere Teil kann damit als PCR-Kompartiment, der äußere Teil analog als PCA-
Kompartiment identifiziert werden (Sage 2002). Der Blatttyp wurde als borszczowioid für Borszczowia
aralocaspica beschrieben (Freitag & Stichler 2000).
Auf anatomisch ganz anderem Weg ist der Prozess der single cell C4 photosynthesis in der Gattung
Bienertia realisiert. Die Blätter besitzen 2-3 Lagen gleichartiger chloroplastenführender Zellen, die ein
zentrales Wasserspeichergewebe und die Leitbündel umgeben. Die Anordnung der Leitbündel
unterscheidet sich deutlich vom borszczowioiden Typ. Die Zellen sind kompartimentiert in einen
zentralen und einen außen liegenden Teil (Abb. 4/IV), die Differenzierung der Enzyme und
Chloroplasten ist derjenigen von Borszczowia vergleichbar. Der Blatttyp wurde als bienertioid für
Bienertia cycloptera beschrieben (Freitag & Stichler 2002). Die beiden genannten Gattungen enthalten
die einzigen bekannten Landpflanzen mit diesem C4-Typ.
Hypothesen über die Evolution der bis dahin bekannten Blatttypen finden sich bei Carolin et al. (1975).
Demnach leitet sich der kranz-suaedoide Blatttyp von dem austrobassioiden bzw. corispermoiden Typ
ab. Allerdings wurde der conospermoide Blatttyp dort noch nicht berücksichtigt. Akhani et al. (1997)
postulieren unabhängige Entstehungen der beiden Kranz-C4-Blatttypen aus C3-Vorfahren. Freitag &
Stichler (2002) halten trotz ähnlichen Photosyntheseweges aufgrund des fundamental
unterschiedlichen Blattbaus unabhängige Entwicklungen von Borszczowia und Bienertia für
wahrscheinlich, obwohl beide der Tribus Bienertieae zugeordnet werden (Ulbrich 1934). Die Ableitung
des bienertioiden C4-Blatttyps von dem in der Gattung Suaeda vorkommenden austrobassioiden C3-
Blatttyp über zunehmende Zelldifferenzierung und Zunahme des Sukkulenzgrades wird als
nachvollziehbarer Weg der Evolution angenommen. Ein Bindeglied zwischen beiden Typen könnten
die hochgradig sukkulenten C3-Blätter von Suaeda crassifolia darstellen (s. Kap. 3.4.).
Der borszczowioide Blatttyp leitet sich wahrscheinlich direkt von C3-Vorfahren mit mehrschichtigem
Mesophyll ab (Freitag & Stichler 2000) und ähnelt durch die Anordnung der Leitbündel dem
salsoloiden Typ. Andere morphologische Merkmale wie die trockenhäutigen Bracteolen von
Borszczowia sprechen jedoch eindeutig für die Zugehörigkeit zu den Suaedoideae.
In Anbetracht der stark differierenden Blatttypen kann von einer viermaligen unabhängigen C4-
Entstehung ausgegangen werden (Freitag & Stichler 2002). Für C4-C3-Reversals gibt es keine
Hinweise (Fisher et al. 1997).
1.2.4. Evolutionäre Trends innerhalb der Suaedoideae Die Suaedoideae verfügen über eine Reihe von Gruppenmerkmalen, die für das Verständnis der
Sippen- und Gattungsevolution von Interesse sind. Die teilweise extremen Lebensbedingungen
erzwingen eine enorme Anpassung der Pflanzen an diese. Ein konstantes Merkmal aller Sippen ist die
Salztoleranz bzw. eine ausgeprägte Bindung an hochsalinare, speziell chloridreiche Standorte.
Experimentell unter salzarmen Bedingungen gezogene Pflanzen sind in allen Teilen schwächer
entwickelt und bilden weit weniger Blüten aus. Nach der Evolution dieser physiologischen Fähigkeit

Einleitung
15
war es den Pflanzen möglich, bisher vegetationsfreie Habitate zu besiedeln, in denen Konkurrenz
weitgehend ausgeschlossen ist. Die ursprüngliche Lebensstrategie der Suaedoideae ist mit hoher
Wahrscheinlichkeit die einjährige, was auch für die gesamten Chenopodiaceae angenommen wird
(Stebbins 1974: 251). Mit der Annuellen-Strategie, die auf der massenhaften Erzeugung von
Individuen innerhalb kurzer Zeit beruht, können die Pflanzen Pionierstandorte sehr effektiv besiedeln
und sind dort auch sehr erfolgreich. Verbunden ist diese Lebensweise mit ausgeprägter Tendenz zu
Autogamie, Vereinfachung der Blütenorgane und der Möglichkeit zu extremer Reduktion von Kormus
und Lebensdauer. Zwergpflanzen haben bereits in den Achseln der Cotyledonen Blütenstände und
können innerhalb weniger Tage funktionsfähige Blüten entwickeln. Mit dieser Lebensstrategie dringen
die Suaedoideae am weitesten in kältere Gebiete vor, da die ungünstigen Perioden als Samen
überdauert werden können. Die verholzten ausdauernden Sippen bleiben auf wärmere Klimazonen
beschränkt, da keine Überdauerungsmechanismen (Laubfall, Frostschutz) ausgebildet wurden. Die
Entwicklung der perennierenden Lebensweise, stets mit Verholzung sowie oft stärker xeromorphen
Blättern gekoppelt, ist mit großer Wahrscheinlichkeit ein abgeleitetes Merkmal, das sicher in
verschiedenen Gruppen unabhängig entstanden ist und die Besiedlung trockenerer Standorte
ermöglichte. Als Übergangsform könnte die basale Verholzung mancher einjähriger Sippen
angesehen werden. Es ist nicht ausgeschlossen, dass sich diese Entwicklungsrichtung auch
umgekehrt hat und aus Mehrjährigen wieder Einjährige entstanden - ein häufig auftretender Prozess
in der Evolution der Angiospermen.
Die Laubblätter weisen äußerlich, im Gegensatz zu der enormen Variabilität im anatomischen Bau (s.
Kap. 1.2.3) kaum Besonderheiten auf. Der aequifaciale Bau und ihre mehr oder weniger vertikale
Stellung ist ein Optimaltyp unter strahlungsreichen, ariden Bedingungen. Blätter sind stets
ausgebildet, Reduktionen wie bei den Salicornioideae kommen nicht vor. Gelegentlich zeigen sie eine
Tendenz zur Verbreiterung der Blattspreite (S. acuminata, Alexandra lehmannii).
Blüten- und Ausbreitungssystem weisen keine besonderen Anpassungen auf. Die ursprünglich wohl
freien Perianthzipfel der Blüten weisen in einigen Fällen Verwachsungen auf (S. ifniensis, S.
aegyptiaca). Gelegentlich ausgebildete Flügel erreichen bei weitem nicht die Größen vieler
Salsoloideae und scheinen damit kaum wesentlich die Effektivität der Samenausbreitung zu erhöhen.
Alle Blütenmerkmale weisen auf Anemo- und Autogamie als hauptsächlich realisierte
Bestäubungsarten hin. Diese Bestäubungsform wird auch von den relativ langen Filamenten
unterstützt, die stets die geöffnete Blüte überragen und die doppelte Länge der Perianthzipfel
erreichen können. Besonders lange Filamente mit ausgeprägten Narben-Papillen besitzen die Arten
der Sektion Salsina. Die bereits erwähnte Tendenz zu Autogamie kommt oft vor, ist aber keineswegs
die Regel. Um die Selbstbestäubung der stets synözischen Blüten zu verhindern, ist bei den
Suaedoideae Dichogamie in Form einer schwach ausgeprägten Proterogynie entwickelt. Ein anderer
Weg ist die Ausbildung funktional getrenntgeschlechtiger Blüten die jeweils Staub- oder Fruchtblätter
nur noch rudimentär ausbilden. Bei vielen Sippen kommen diese Blüten am Beginn und Ende der
Blühphase vor, bei einigen Arten treten sie sogar obligatorisch auf (S. monoica, s. Freitag 2001: 112).
Möglicherweise dient auch die Bildung einer griffelähnlichen Struktur bei manchen Sippen (S. spicata)
der Förderung der Allogamie. Ausbreitungshilfen an den Früchten sind nur gelegentlich entwickelt.
Dazu zählen die schon erwähnten Flügel (S. heterophylla, Bienertia, Alexandra), wahrscheinlich aber

Einleitung
16
auch das an der Frucht schwammartig vergrößerte Perianth bei S. aegyptiaca und S. arcuata. Die
Einrichtungen dienen alle zur Förderung der Windausbreitung. Von großer Bedeutung scheinen auch
passive Formen der Zoochorie zu sein, bei denen Samen durch Phytophage endozoochor oder
ektozoochor (Vögel) ausgebreitet werden. Dies scheint bei der Fernausbreitung eine entscheidende
Rolle zu spielen.
Relativ gut bekannt sind Ploidiestufen innerhalb der Suaedoideae, doch ist aufgrund der umstrittenen
Taxonomie einiger wichtiger Arten die Zuordnung mancher publizierter Chromosomenzahlen wenig
glaubhaft. Ausführlichere Untersuchungen liegen aufgrund der meist geringen Größe der
Chromosomen nicht vor. Als Chromosomen-Grundzahl der Suaedoideae wie auch der gesamten
Chenopodiaceae wird x=9 angegeben (Kühn et al. 1993, Krahulcová & Tomšovic 1997). Davon
ausgehend sind tetra-, hexa-, okto- und dekaploide Stufen bekannt geworden (Akhani et al. 1997,
Krahulcová & Tomšovic 1997, Lomonosova & Krasnikov 1993, Lomonosova et al. 2005, Lomonosova,
unpubl. Daten). Für einige Arten wurden mehrere Ploidiestufen publiziert (S. vera, S. fruticosa, S.
„corniculata“ u.a.), doch handelt es sich bei einigen Angaben offenbar um Verwechslungen. Wenig
bekannt ist über die Herkunft der Polyploidie aus Allo- oder Autopolyploidisierung doch könnte hier ein
Schlüssel zum Verständnis hochpolyploider Arten liegen. Der Trend zu höheren Ploidiestufen
kennzeichnet fast alle Gruppen, insbesondere jedoch die einjährigen, relativ merkmalsarmen Vertreter
der Sektion Brezia und stellt möglicherweise eine Form der Speziation dar, die sich nicht unmittelbar
in morphologischer Differenzierung niederschlägt. Interessant ist die Tatsache, dass die relativ isoliert
stehende Bienertia cycloptera diploid geblieben ist (Al-Turki et al. 2000).
1.2.5. Bisherige Theorien zur Sippenevolution - eine Synthese Trotz zahlreicher systematischer und vergleichend morphologischer Studien ist relativ wenig über die
Evolution der Suaedoideae bekannt. Erst mit der Entdeckung der photosynthetischen Vielfalt
innerhalb der Chenopodiaceae und speziell innerhalb der Suaedoideae entstanden Thesen über
mögliche Evolutionswege der betroffenen Sippen, die mit der Einführung molekularer Methoden der
Verwandtschaftsrekonstruktion überprüft werden konnten. Erst dadurch wurden wichtige
verwandtschaftliche Zusammenhänge in den Chenopodiaceae sichtbar (Kadereit et al. 2003).
Unzweifelhaft ist die Tatsache, dass die Radiation der Chenopodiaceae eng mit der Entwicklung der
ariden Regionen der Erde verknüpft ist. Shmida (1985) verbindet die Entwicklung der
Chenopodiaceae mit dem Zurückweichen des Tethys-Meeres seit dem Miozän (vor ca. 20 Mio.
Jahren). Mit Sicherheit sind in dieser Zeit viele Linien entstanden doch ist das Alter der
Pflanzenfamilie offenbar wesentlich höher, wie einige Fossilfunde belegen. So ist beispielsweise mit
Salicornites massalongoi aus dem Oligozän ein Vorläufer der rezenten Salicornioideae belegt und
Funde fossiler Pollen aus dem Paläozän (ab. ca. 65 Mio. Jahre) weisen auf ein noch viel höheres Alter
der Chenopodiaceae.
Recht umfangreiche Thesen zur Evolution der altweltlichen Sippen stellen Akhani et al. (1997) anhand
von Blatttyp, Lebensformen und Chromosomenzahlen sowie biogeographischen Aspekten auf. Unter
Berücksichtigung der genannten Merkmale werden innerhalb von Suaeda 11 Gruppen unterschieden
und folgende Entwicklungswege postuliert:

Einleitung
17
Abb. 5: Informelle Klassifikation und mögliche phylogenetische Beziehungen altweltlicher Suaeda-Arten. grau schattierter Rahmen: C4-Arten, einfacher Rahmen: C3-Arten (aus Akhani et al. 1997)
1. Suaeda stammt mit hoher Wahrscheinlichkeit aus NW Afrika während es noch eine
Landverbindung zwischen Afrika und Südamerika gab. Die Ähnlichkeit der afrikanischen mit süd- und
zentralamerikanischen Sippen zeigt die hohe Affinität. In NW-Afrika liegt wahrscheinlich auch das
Entfaltungszentrum der Gruppe im Verlauf des Tertiärs.
2. Die einjährigen Sippen sind polyphyletisch. Die S. maritima-Gruppe leitet sich wahrscheinlich von
einer C3-Gruppe ab (S. vera-Gruppe).
3. Von der S. maritima-Gruppe leiten sich eine C3 (S. glauca)- und eine C4 (S. acuminata)-Gruppe ab,
allerdings könnte sich die S. glauca-Gruppe auch von S. physophora ableiten lassen.
4. S. physophora gilt als isolierte C3-Art mit Ähnlichkeit zu S. vera. Beide schließen sich allerdings
biogeographisch aus. S. physophora zeigt zudem enge Bindungen an S. ifniensis und S. palaestina.
S. ifniensis ist ein C3-Endemit in Marokko und könnte als altes Relikt der Gattung interpretiert werden.
5. Die ursprünglichste Art der S. fruticosa-Gruppe ist möglicherweise S. monoica, von der sich weitere
Sippen der Gruppe ableiten lassen. Für diese Position von S. monoica spricht vor allem deren stark
abweichende Morphologie. Zudem ist S. monoica diploid. Innerhalb der Gruppe kommen neben diplo-
und tetraploiden (S. pruinosa, S. vermiculata), westlich verbreiteten auch polyploide Sippen mit mehr
östlicher Verbreitung vor (S. fruticosa, S. baluchestanica). Die polyploiden Sippen könnten während
der Einwanderung der Sippen aus NW-Afrika nach SW-Asien entstanden sein.
6. Photosynthesewege und Karyotyp lassen darauf schließen, dass die annuellen Arten S. altissima,
S. aegyptiaca und S. arcuata Ableitungen aus der ansonsten durch Zwergsträucher gebildeten S.
fruticosa-Gruppe sind.
7. Die weiteren zentralasiatischen annuellen Gattungen der Suaedeae (Bienertia, Alexandra,
Borszczowia) sind wahrscheinlich Ableitungen gemeinsamer Vorfahren mit Suaeda.
Die folgende Übersicht aus Akhani et al. (1997) stellt zusammengehörende Taxa dar und zeichnet
mögliche Entwicklungswege auf:

Einleitung
18
Durch die Postulierung von Zwergsträuchern als ursprüngliche Sippen impliziert die Hypothese
hinsichtlich der Lebensformen die Ableitung annueller von ausdauernden Sippen sowie die
Entwicklung von C3- zu C4-Photosynthese. Eine Ausnahme bilden die sekundär entstandenen
annuellen Sippen S. aegyptiaca, S. arcuata und S. altissima. Den unterschiedlichen Blatttypen kommt
für die Evolution innerhalb der Suaedoideae eine enorme Bedeutung zu. Da die Blattentwicklung ein
sehr komplexer Prozess ist, erscheint es unwahrscheinlich, dass sich ein Blatttyp mehrfach entwickelt
hat. Die Radiation einiger C4-Gruppen müsste daher nach der Blattentwicklung erfolgt sein. Erfolgreich
diversifiziert haben sich demnach die Sippen mit kranz-suaedoidem und conospermoidem Blatttyp.
Die Entwicklung der single cell C4 photosynthesis führte hingegen bis zum gegenwärtigen Zeitpunkt
nicht zu einer anschließenden Radiation.
1.3. Themenentwicklung und Stand der Forschung Die vorangegangene Darstellung umreißt einerseits einige bemerkenswerte morphologische
Erscheinungen innerhalb der Suaedoideae, andererseits die bestehenden Kenntnisdefizite hinsichtlich
Evolution und Systematik der Sippen. Alle bisherigen Systeme beruhen auf vergleichend-
morphologischer Analyse und berücksichtigen nicht die Entstehungsprozesse und
verwandtschaftlichen Zusammenhänge. Zahlreiche Sippen sind bis zum gegenwärtigen Zeitpunkt
nicht eindeutig charakterisiert. Dies betrifft z.B. Suaeda maritima, die aus weiten Teilen der Holarktis
und sogar von der Südhalbkugel angegeben wird, bei der es sich aber mit Sicherheit um mehrere
Sippen handelt. Weitgehend ungeklärt ist der Formenreichtum der Brezia-Sippen Asiens um S.
corniculata, S. salsa und S. prostrata, wozu auch noch falsche Benennungen in der Literatur
beitragen. Kaum bekannt ist die Stellung der südhemisphärischen Arten sowie die Beziehungen
zwischen eurasiatischen und nordamerikanischen Sippen. Ein Beispiel für fehlerhafte Darstellung, die
sich über Jahrzehnte in der entsprechenden Literatur etabliert hat ist Suaeda fruticosa. Der Name
wurde lange Zeit für Suaeda vera verwendet, so z. B. für die Sektionsgliederung bei Iljin (1936a) und
war lange Zeit als Sammelbezeichnung für viele strauchige Arten verwendet. Da Sippendefinition und
-abgrenzung vielfach unklar sind, fehlt eine korrekte systematische Basis für andere Gebiete wie z.B.
Ökologie, Chorologie oder Physiologie.
Aus den genannten Punkten leiten sich direkt die Themen und Fragestellungen für die vorliegende
Arbeit ab. Zentrales Thema ist die Erweiterung der Kenntnis über Phylogenie und Evolution der Arten
der Suaedoideae. Da diese grundlegenden Fragen anhand der Morphologie bisher nur sehr
unzureichend bekannt sind, werden molekulare Methoden eingeführt und davon bessere Einblicke in
diese Zusammenhänge erwartet. Durch die Rekonstruktion der Phylogenie anhand von DNA-
Merkmalen werden gemeinsame Abstammungslinien definiert und, soweit möglich, in einen zeitlichen
Zusammenhang gebracht. Nur dadurch kann die Artbildung sowie die heutige Diversität und
Verbreitung der Sippen richtig interpretiert werden. Basierend auf den molekularen Phylogenien wird
die Merkmalsentwicklung innerhalb der Gruppe untersucht. Ein Schwerpunkt liegt dabei auf der
Entwicklung der mit der C4-Photosynthese gekoppelten Blatttypen. Folgende Fragenkomplexe sollen
bearbeitet werden:

Einleitung
19
1. Monophyletische Gruppen Haben die aufgrund morphologischer Ähnlichkeiten zu Suaeda gezählten Pflanzen einen
gemeinsamen Ursprung (Monophylie-Annahme) und gehören die weiteren Gattungen der Tribus
dazu? Welche monophyletischen Gruppen bestehen innerhalb von Suaeda?
Dem Kriterium der Monophylie wird in der gegenwärtigen systematischen Forschung ein hoher
Stellenwert eingeräumt. Von einer möglichst großen Zahl an Taxa werden molekulare Daten
gewonnen, mit denen statistisch abgesicherte Phylogenien erstellt werden können. Für Suaeda wird
Monophylie postuliert, Alexandra, Bienertia und Borszczowia sollten aufgrund abweichender
Merkmale getrennte Entwicklungswege darstellen (s. Kap. 1.2.2., 1.2.3.). Die Hypothese wird anhand
verschiedener Phylogeniemodelle getestet. Zu überprüfen ist dabei, ob die monophyletischen
Gruppen mit bisherigen Untergruppen von Suaeda (Sektionen, frühere separate Gattungen)
übereinstimmen und wo Abweichungen auftreten. Bei den nicht zu Suaeda gehörenden Sippen der
Unterfamilie soll geklärt werden, ob sie vollkommen getrennte Entwicklungswege oder Ableitungen
aus bereits diversifizierten Suaeda-Vorfahren darstellen. Für die Einordnung in die Chenopodiaceae
werden Vertreter benachbarter Unterfamilien als Außengruppen einbezogen.
2. Artabgrenzung und Artbildung
Folgt die Evolution der Suaedoideae dem kladistischen Modell oder lassen sich von diesem
abweichende Ereignisse wie z. B. retikulate Evolution nachweisen? Lassen sich Arten anhand der
gewählten molekularen Merkmale umgrenzen? Stimmen molekulare und morphologische Diversität
überein? Lässt sich die Artbildung zeitlich eingrenzen?
Das kladistische Modell wird, da es logisch zwingend erscheint, auch wenn es nicht beweisbar ist, als
Grundannahme vorausgesetzt und in den Berechnungen angewandt. Der phylogenetische Baum,
bestehend aus möglichst vielen Dichotomien (und wenig Polytomien), sollte diesen Prozess
widerspiegeln. Anhand des Stammbaumes werden auch Radiationen sichtbar, die mit
biogeographischen Daten verglichen werden. Eine Möglichkeit zum Nachweis weiterer, der
Aufspaltung teils entgegen wirkender Prozesse (retikulate Evolution, Extinktionsereignisse) ist der
Konsistenzvergleich unabhängiger Phylogenien aus Kern- und Chloroplastengenom. Da retikulate
Evolution ein häufig vorkommendes Ereignis ist, aus dem eine große Zahl der rezenten Arten
hervorgingen, wird angenommen, dass es auch in der Evolution der Suaedoideae eine Rolle gespielt
hat. Als zusätzliche Information werden karyologische Merkmale ausgewertet. Hinsichtlich der
Sequenzdaten wird eine höhere molekulare Variabilität erwartet als sich im Phänotyp der rezenten
Arten widerspiegelt, da nichtkodierende DNA-Regionen verwendet werden. Dies sollte besonders für
die Kern-DNA zutreffen, da hier die Variabilität durch Rekombination noch erhöht wird. Auch der
umgekehrte Fall ist wahrscheinlich, d.h. auf molekularem Niveau nicht unterscheidbare Sippen, da
molekulare und morphologische Evolution nicht gleich ablaufen (s. das Bsp. „lebende Fossilien“).
Dennoch wird eine gewisse Korrelation von Arten mit molekular definierten Kern- und Chloroplasten-
„Typen“ postuliert. Die zeitliche Dimension wird durch Kalibrierung der Stammbäume mittels
Fossilfunden untersucht und mit jeweiligen erdgeschichtlichen Ereignissen in Verbindung gebracht.

Einleitung
20
3. C4-Photosynthesesyndrom
An welchen Punkten in der Stammesgeschichte kam es zur Entwicklung der C4-Photosynthese? Ist
dieser Prozess einmal oder mehrfach unabhängig entstanden? Wie kam es zur Herausbildung der
blattanatomischen Vielfalt? In welche Richtung erfolgte die Photosynthese-Evolution?
Für die Evolution des C4-Photosynthesesyndroms innerhalb der Suaedoideae mit vier histologisch und
funktionell hochgradig spezialisierten C4-Blatttypen sind mehrere Entwicklungsthesen denkbar.
Wahrscheinlich sind zwei Entstehungsereignisse, die jeweils zu den zwei ähnlichen Blatttypen (Kranz-
und nicht-Kranz-Anatomie) führten. Ebenfalls wahrscheinlich sind aufgrund der fundamentalen
anatomischen Unterschiede vier getrennte Entstehungsereignisse. Unwahrscheinlich erscheint
hingegen ein gemeinsamer Ursprung aller C4-Linien, die Entstehung des einen C4-Typs aus einem
anderen oder die Rückentwicklung des hochdifferenzierten C4- zum einfacheren C3-Blatt, da der C3-
Typ in der gesamten Gruppe sehr einheitlich gebaut ist und alle bisherigen Studien keine Hinweise auf
eine Entwicklung in diese Richtung liefern (s. Kap. 1.2.4.). Für mehrfache C4-Evolution mag auch die
Tatsache sprechen, dass dies schon in anderen Gruppen innerhalb nahe verwandter Taxa
nachgewiesen wurde (übrige Chenopodiaceae; Poaceae: Giussani et al. 2001). Aufklärung über diese
Fragestellungen wird aus den Aussagen der molekularen Phylogenien und der Synthese molekularer
und blattanatomischer Daten erwartet. Falls die jeweiligen C4-Typen je einer monophyletischen
Gruppe zugeordnet werden können, sollte auch die Entstehung in dieser Gruppe nur einmal erfolgt
sein. Durch Anwendung neuerer Datierungsmethoden soll der Entstehungsprozess zeitlich
eingegrenzt werden.
4. Merkmalsentwicklung
Welche Merkmale haben gruppierenden Charakter und lassen sich Schlüsselmerkmale definieren, die
die in der molekularen Phylogenie herausgearbeiteten Gruppen definieren? Welche Plastizität weisen
sie auf?
Die teilweise enorme phänotypische Plastizität der Merkmale in Abhängigkeit von verschiedenen
Standortfaktoren wie Wasserhaushalt oder Salinität des Bodens führte in der Vergangenheit zu
konträren taxonomischen Auffassungen. Mittels molekularer Phylogenien und der Synthese
molekularer und morphologischer Merkmale sollen Schlüsselmerkmale für die wichtigen
monophyletischen Gruppen herausgearbeitet werden. Überprüft werden sollen vor allem die bisher in
der Systematik verwendeten Merkmale wie Pistill und Narben (Iljin 1936a), Perianthmerkmale
(besonders die Anhänge), Blattmerkmale und Lebensformen. Die bisher zur Großgliederung
verwendeten Merkmale wie Embryo und Bracteolen sollen für die Gruppe diskutiert werden.
Basierend auf der Baumtopologie aus molekularen Daten sollen mögliche Entstehungswege der
morphologischen Merkmalsentwicklung besprochen werden.

Einleitung
21
Ableitungen
Aufgrund der gewonnenen Erkenntnisse aus der molekularen Analyse wird die aktuelle
Untergliederung von Suaeda nach Schenk & Ferren (2001) überprüft und mit früheren Gliederungen
verglichen. Sollten sich grundsätzliche Änderungen ergeben, werden Vorschläge für eine neue
Gliederung basierend auf monophyletischen Gruppen der molekularen Phylogenien präsentiert. Das
Artkonzept in Suaeda wird aufgrund molekularer Phylogenien überprüft. Besonders für die
taxonomisch sehr schwierige Sektion Brezia mit der „Globalart“ Suaeda maritima werden
entscheidende Verbesserungen für das taxonomische Konzept erwartet.
Die Herausarbeitung wichtiger Merkmale soll die Kenntnis über die Gruppe erweitern und in Zukunft
eine korrekte Darstellung in der Literatur erleichtern. Die Ergebnisse können zudem als Grundlage für
eine Revision der Gattung herangezogen werden.
Mit der vorliegenden Arbeit wird der Versuch unternommen, traditionelle Konzepte aus der
Morphologie, Biologie und Chorologie um molekularbiologisch basierte Daten zu erweitern und daraus
eine Synthese zu entwickeln. Generell geht es um die Einordnung des „Systems Suaeda“ in ein
Raum- und Zeitmuster und dabei um die Nachzeichnung der großen sippenbildenden Ereignisse in
ihrem historischen Kontext, um die rezente Diversität besser verstehen und bewerten zu können.
Die Arbeit ist Bestandteil und wurde gefördert durch das DFG-Projekt „Systematik der
Chenopodiaceae und Evolution des C4-Photosynthesesyndroms“ (Cl 188/1-1 und We 1830/2-1 für G.
Clausing und K. Weising). Durch ein Stipendium des DAAD (Kennz. D/03/26150) wurde ein
Forschungsaufenthalt mit Sammelreise am Zentralen Sibirischen Botanischen Garten Novosibirsk
unterstützt.
Molekularsystematische Untersuchungen an den Suaedoideae wurden bis zum Zeitpunkt der eigenen
Untersuchungen nicht publiziert. Die Ergebnisse der eigenen Arbeiten wurden bereits mehrfach auf
Fachtagungen und in Seminaren präsentiert: Tagung der Deutschen Botanischen Gesellschaft in
Freiburg (2002), Kolloquium des Geobotanischen Institutes der Universität Halle (2002), Tagung der
Russischen Botanischen Gesellschaft in Novosibirsk (2003), 17. Internationaler Botanischer Kongress
in Wien (2005) u.a.
Die wesentlichen Ergebnisse zu Phylogenie und Merkmalsevolution der Suaedoideae wurden bereits
publiziert (Schütze et al. 2003). Eine aktualisierte Fassung und weiterführende Arbeiten sind in
Vorbereitung.

Material und Methoden
22
2. Material und Methoden 2.1. Systematische Methoden und Prinzipien 2.1.1. Organismenbezogene Methoden Für die Bearbeitung systematischer und phylogenetischer Fragestellungen steht gegenwärtig ein
umfangreiches Methodenspektrum zur Verfügung.
Grundlage für Systematik und Taxonomie von Sippen sind nach wie vor die vergleichende
Morphologie und Anatomie sowie die Analyse von Lebenszyklen und räumlichen Verteilungsmustern.
Im Mittelpunkt dieser beschreibenden Methodik steht das Individuum mit seinen spezifischen
Merkmalen, aus der anschließend Gruppierungen und Systeme entwickelt werden. Organismen
werden beschrieben und nachvollziehbar klassifiziert.
Sind damit die rezenten Zustände recht gut zu beschreiben, sind jedoch Prozesse der
Merkmalsevolution oder die Verwandtschaft von Organismen allein aus Morphologie und Anatomie
schwieriger zu rekonstruieren. Morphologische Merkmale von Organismen sind das Ergebnis
komplexer Genwirkungen und zeichnen sich oft durch hohe Plastizität aus. Nicht jede
Merkmalsänderung muss genetische Ursachen haben sondern kann auch eine Modifikation
darstellen. Konvergenzen und umweltbedingte Adaptionen sind zwangsläufig zu erwarten und führen
aufgrund von Ähnlichkeiten zum - manchmal falschen - Rückschluss auf nahe Verwandtschaft. Die
phylogenetische Bewertung von Gestalt und Anatomie der Organismen ist daher oft nicht eindeutig
und wird mit abnehmendem Verwandtschaftsgrad immer unsicherer. Die Entscheidung über die
Ursache von Merkmalsausprägungen kann demnach nicht getroffen werden. Phylogenetisch
relevante Informationen über die Kriterien Homologie, Analogie, ursprüngliche bzw. abgeleitete
Merkmale sind allein aus morphologischen Daten nur beschränkt zu gewinnen.
Ein Morphotaxon ist nicht in jedem Fall objektiv, sondern oft subjektiv, von der Auffassung des
Bearbeiters und Traditionen (Regelwerke) beeinflusst.
2.1.2. Molekülbasierte Methoden Im Gegensatz zu den organismenbezogenen untersuchen molekülbezogene Methoden molekulare
Bestandteile der Organismen, vor allem Nucleinsäuren, Proteine, Enzyme und verschiedene
Stoffwechselprodukte, die zusammenfassend als sekundäre Inhaltsstoffe bezeichnet werden.
Sekundäre Inhaltsstoffe wurden vielfach als Merkmale für Klassifikationen herangezogen, ein Beispiel
dafür ist das System der Angiospermen von Cronquist (1981).
Proteine, Enzyme und sekundäre Inhaltsstoffe sind ähnlich den morphologischen Merkmalen
Erscheinungen des Phänotyps, während es sich bei der Struktur der Nucleinsäuren um den Genotyp,
um „Bauanleitungen“ handelt, aus denen ein Großteil der Informationen für die Ausbildung des
Phänotyps abgeleitet wird. Die Informationen tragen damit primären Charakter und werden auch
vererbt. Diese Eigenschaften machen Nucleinsäuren für die Evolutionsforschung besonders
interessant, sollte doch der Einfluss von Umweltbedingungen nach den Theorien der neutralen
Evolution gering oder ausgeschlossen sein. Weitere Vorteile von Nucleinsäuren liegen in ihrer Struktur
begründet: 1 von 4 möglichen Nukleotiden an definiertem Ort, diskrete Merkmalsausprägung, keine

Material und Methoden
23
Übergangszustände (wie oft bei morphologischen Merkmalen). Die genannten Vorteile sind die
Ursache dafür, dass gegenwärtig auf den Merkmalen von Nukleinsäuren (DNA) basierende Methoden
eine herausragende Bedeutung für die systematische und evolutionsbiologische Forschung erlangt
haben.
Bevor jedoch die DNA als ‚Merkmalslieferant’ für die genannten Fragestellungen herangezogen
werden konnte, waren zahlreiche theoretische und technische Entwicklungen notwendig. Dazu zählen
vor allem die Erkennung der Natur der DNA und ihrer Funktion, die Entwicklung von Theorien über die
Evolution kettenartiger Biomoleküle (Zuckerkandl und Pauling 1965 anhand des Proteins
Hämogolobin, später Anwendung auf DNA) oder die Vorstellungen zur Neutraltheorie der molekularen
Evolution (Kimura 1983). Notwendig war die Etablierung grundlegender Techniken wie die DNA-
Sequenzierung (Sanger 1977), PCR (Mullis et al. 1986) und die Entwicklung von
Berechnungsverfahren bzw. deren Anwendung auf die Auswertung molekularer Merkmale, da
Informationen aus Molekülstrukturen schlecht zur Beschreibung von Organismen verwendet werden
können. Gegenwärtig wird besonders an der Weiterentwicklung von Modellen zur Sequenzevolution
sowie Verfahren zur Altersdatierung gearbeitet und es wird auch im Umkehrschluss versucht, mittels
eines definierten genomischen Profils Sippen zu definieren (barcoding).
Die DNA liegt in pflanzlichen Zellen in drei voneinander weitgehend unabhängigen
Organisationseinheiten vor: Zellkern, Chloroplasten und Mitochondrien. Durch Mutationen
entstehende Sequenzunterschiede zwischen homologen Abschnitten der DNA werden als Merkmale
genutzt. Zu deren Nachweis wurde eine große Anzahl an Verfahren entwickelt, z.B. vergleichende
Sequenzierung, Restriktionsanalysen sowie anonyme Markermethoden wie RAPD und AFLP
(Bachmann 1997, Weising et al. 2005). Studien mit phylogenetischem Schwerpunkt basieren dabei
vor allem auf vergleichender Sequenzierung kürzerer, homologer DNA-Abschnitte aller drei genannter
Genome. Die Wahl der DNA-Region hängt in erster Linie von der Fragestellung ab. Nahe verwandte
Sippen lassen sich nur anhand schnell evolvierender Regionen (intergenische Spacer, Introns,
Mikrosatelliten) differenzieren, während für Großgruppen kodierende Regionen (Gene) verwendet
werden. Am besten geeignet sind DNA-Regionen, auf denen keine oder wenig funktionale Zwänge
liegen, also nichtkodierende Regionen oder in kodierenden Regionen jeweils die 3. Positionen der
Tripletts. Mit DNA-Daten aus nichtkodierenden Regionen steht ein von der Morphologie und Anatomie
unabhängiger Datensatz zur Verfügung, dessen Variabilität als weitgehend unabhängig von adaptiven
Prozessen angesehen werden kann und theoretisch aus dem passenden Evolutionsmodell der
Sequenz erklärt werden müsste.
Die für molekularsystematische Studien am häufigsten verwendete Kernregion ist die ribosomale RNA
kodierende DNA. In Eukaryoten ist sie in funktionellen Einheiten (Cistrons) aus jeweils drei Genen
(18S, 5,8S 26S), zwei internen, die Gene physisch trennenden Spacern (ITS 1 und 2) sowie jeweils
einem externen und nicht-transkribierenden Spacer (ETS, NTS) organisiert. Die einzelnen Einheiten
kommen in sehr zahlreichen Wiederholungen vor und können bis zu 10% Anteil am gesamten Genom
der Pflanze erreichen (Hillis & Dixon 1991, Hamby & Zimmer 1991). Für Fragestellungen auf der
Ebene von Gattungen und Arten hat sich die vergleichende ITS-Sequenzierung etabliert (Baldwin et
al. 1995). ITS gehört nach NTS zu den am schnellsten evolvierenden Regionen der ribosomalen DNA.
Allerdings ist die ITS-Region kein idealer Marker für die Rekonstruktion von Phylogenien (Alvarez &

Material und Methoden
24
Wendel 2003). Die zahlreichen ITS-Kopien stellen paraloge und keine orthologen Sequenzen dar wie
dies bei single-copy-Genen der Fall ist. Zwischen ihnen findet eine intra- und interchromosomale
Homogenisation statt (concerted evolution; Arnheim 1983, Liao 1999), über deren Geschwindigkeit
und Resultate bis jetzt wenig bekannt ist. Es kann zur vollkommenen Durchsetzung eines Typs
kommen, die einzelnen Sequenzen können aber auch mosaikartig neu kombiniert werden. Obwohl
weit verbreitet, kommt concerted evolution nicht in allen Organismen vor, wie am Beispiel der Gattung
Rosa demonstriert werden konnte (Ritz et al 2005). Die ITS-Regionen sind keineswegs funktionslos,
vielmehr werden durch sie in den Transkripten die kodierenden Regionen für die Translation korrekt
positioniert. Dafür sind sie zur Bildung spezifischer Sekundärstrukturen befähigt (Denduangboripant &
Cronk 2001, Hershkovitz & Zimmer 1992, Mayol & Rossello 2001, Ritland et al. 1993, Wesson et al.
1992), die sippenspezifischen Charakter aufweisen. Die Sekundärstrukturen sind entscheidend für
effizientes RNA-processing (van der Sande et al. 1992). Aufgrund dieser Funktionalität kann die
Sequenzevolution nicht als vollkommen unabhängig betrachtet werden und es werden sich auch hier
stärker konservierte und variable Teilregionen nachweisen lassen. ITS wird biparental vererbt, somit
ist unter bedingten Voraussetzungen der Nachweis von Hybridisierung und retikulater Evolution
möglich (Sang et al 1995, Whittall et al. 2000).
Über die Eignung weiterer nuklärer DNA-Regionen für phylogenetische Fragestellungen liegen
gegenwärtig nur wenige Studien vor. Beispiele sind das Alkoholdehydrogenase-Gen (AdhA: Small &
Wendell 2000) sowie die RNA-Polymerase-Gene (RPA2, RPB2, RPD2 intron: Popp et al. 2005). In
beiden Fällen handelt es sich um ‚low copy’-Gene mit ähnlichen evolutionären Phänomenen wie im
Fall der ribosomalen Gene. Da für diese Regionen bis jetzt kaum Vergleichsdaten vorliegen, wurden
sie in der vorliegenden Arbeit nicht untersucht.
Wesentlich mehr DNA-Regionen für phylogenetische Fragestellungen sind vom Chloroplasten
bekannt und erfolgreich angewendet. Aus der großen Zahl sollen als Beispiele genannt werden: rbcL
(Gen), matK (Gen), atpB-rbcL (spacer), trnH-psbA (spacer), rps16 intron, trnT-trnL (spacer), trnL-trnF
(spacer). (Taberlet 1991, Chase et al. 1993, Olmstead & Palmer 1994, Shaw et al. 2005). Der
Unterschied zu Kernmarkern liegt vor allem in der uniparentalen Vererbung der Plastiden sowie kaum
vorhandener Rekombination (Ausnahmen: Ogihara 1988, Palmer et al 1987). Mit den Stammbäumen
auf der Basis von Chloroplasten-Daten können maternale (Angiospermen) oder paternale
(Gymnospermen) Linien rekonstruiert werden. Kombiniert mit Kern-Daten ergeben sich dadurch
Möglichkeiten zur Rekonstruktion retikulater Evolution sowie Hybridisierung. Allerdings scheint es von
den vorherrschenden Vererbungswegen mehrfach Ausnahmen zu geben (McCauley 2007). Nachteile
der Sequenzinformation aus Chloroplasten ist die oftmals hohe Homoplasie der Daten. Bestimmte
Prozesse, die aus dem Vererbungsgang der Plastiden herrühren (selective sweeps, organelle
capture) können die Diversität in Populationen beeinflussen (Ennos et al. 1999). Kaum bekannt ist
außerdem die Interaktion der verschiedenen Genome in der Zelle untereinander wie z.B. horizontaler
Genstransfer (Woloszynska 2004). Die häufigsten Mechanismen der Evolution nichtkodierender DNA-
Regionen und ihre Anwendung in der Pflanzensystematik werden von Kelchner (2000)
zusammenfassend beschrieben.

Material und Methoden
25
2.2. Pflanzenmaterial, Kulturversuche 2.2.1. Auswahl der Arten Phylogenetische Untersuchungen auf Gattungsebene erfordern die Einbeziehung eines möglichst
breiten Spektrums an Taxa, die alle rezenten Entwicklungsrichtungen innerhalb der Gattung
ausreichend repräsentieren und auch die Einordnung der Gattung in größere systematische Einheiten
erlauben sollten. Für die phylogenetischen Berechnungen in der vorliegenden Untersuchung wurden
122 Akzessionen einbezogen, davon 110 Suaeda-Akzessionen, die ca. 68 bekannten Suaeda-Arten
zuzuordnen sind. Damit sind alle bekannten Sektionen nach Schenk & Ferren (2001) in hinreichender
Taxon-Anzahl repräsentiert. Von kritischen Arten wurden dann mehrere Akzessionen verwendet,
wenn morphologische Merkmale eine Differenzierung vermuten ließen. Dies betrifft besonders die
Sektion Brezia, in der noch große Unklarheiten über die Artabgrenzung bestehen.
Neben den drei monotypischen Gattungen der Suaedoideae wurden acht Arten aus den
Salicornioideae sowie eine Art der Salsoloideae ausgewählt, die in den phylogenetischen
Berechnungen als Außengruppen definiert werden und zur Klärung der Stellung der Suaedoideae
innerhalb der Chenopodiaceae herangezogen werden.
Die Bestimmung der Arten erfolgte mangels zusammenfassender Werke anhand der floristischen
Literatur für die jeweiligen geographischen Regionen. Die Nennung der Autorennamen erfolgt der
Einfachheit halber nur in der Tabelle. Alle im Text genannten Namen lauten vollständig wie dort
aufgeführt. Unbekannte Arten werden mit dem Epiteth „sp.“ und der DNA-ID bezeichnet oder mit
vorläufigen Bezeichnungen nach Regionen oder Personennamen (in Anführungszeichen) versehen.
Eine Übersicht über alle verwendeten Proben findet sich in Anhang 5.
Für die Kennzeichnung der Sippen in Stammbäumen und Tabellen wurde der Name und die
vierstellige DNA-Identifikationsnummer kombiniert. Für Taxa, deren Sequenzen aus der Genbank
übernommen wurden, erfolgt die Nennung des Namens sowie einer verkürzten Belegnummer.
2.2.2. Aufsammlung, Kulturversuche und Materialkonservierung In erster Linie wurde auf Material aus Herbarien zurückgegriffen, hier vor allem auf die umfangreiche
Sammlung von H. Freitag aus dem Herbarium der Universität Kassel (KAS). Zahlreiche weitere
Belege aus Zentralasien wurden von M. Lomonosova, Novosibirsk (Central Siberian Botanical
Garden, NS) bereitgestellt. Weitere Belege stammen aus den Herbarien B, BR, GAZI, HUJ, NSW,
LPB, TFC, UTG, W. Einzelne Belege wurden freundlicherweise aus Privatsammlungen zur Verfügung
gestellt: B. Schmalz (Australien), S. Ickert-Bond, T. Borsch (Nordamerika), H. Akhani (Iran), V.
Steinmann (Kalifornien). Die meisten Belege stammen aus jüngeren Aufsammlungen (etwa ab 1994),
nur in Ausnahmefällen wurde auf älteres Herbarmaterial zurückgegriffen.
Für molekularbiologische Analysen ist es von Vorteil Frischmaterial einzusetzen, um negative Effekte
der Degradierung der DNA-Moleküle durch Trocknung auszuschließen. Daher wurden Kulturversuche
durchgeführt sowie Sammelreisen unternommen. Dies erfolgte in den Jahren 2001 und 2003 und
diente neben der Probengewinnung auch dazu, Merkmale, Variabilität und Ökologie einiger Sippen
am Originalstandort zu untersuchen und Samen für Nachzuchten zu gewinnen.

Material und Methoden
26
Die Sippen europäischer Küsten und einiger Binnensalzstellen Deutschlands wurden auf einer
Sammelreise im Herbst des Jahres 2001 intensiv untersucht. Aufgesucht wurden die Küsten
Deutschlands, der Niederlande, Belgiens, Frankreichs, NO-Spaniens und NW-Italiens sowie einige
der Binnensalzstellen Deutschlands in Sachsen-Anhalt. Entlang der Küsten wurden in regelmäßigem
Abstand von ausgewählten Teilpopulationen einige Exemplare entnommen, auffällige Merkmale
notiert, die Pflanzen herbarisiert und Blattproben angefertigt. Ergänzt werden konnte die Sammlung
europäischer Sippen durch Überlassung von Material durch E. Westberg aus der Arbeitsgruppe Prof.
J. W. Kadereit (Universität Mainz). Zahlreiches Material wurde von H. Freitag auf mehreren
Sammelreisen gewonnen: Usbekistan (2000), Kanaren (2001), Italien (1999, 2001), Türkei (1997,
2002). Die wichtigsten zentralasiatischen Sippen konnten auf einer gemeinsamen Expedition im Jahr
2003 nach Südsibirien (Russische Föderation) im Gelände studiert und aufgesammelt werden. Bereist
wurden die Regionen um Novosibirsk, Karasuk, Teile der Republik Tuva sowie die Regionen Burjatien
und Chita.
Frisches Blattmaterial wurde in Silicagel schnellgetrocknet (Chase & Hills 1991). Die Methode erwies
sich als geeignet für sukkulentes Blattmaterial, wenn das Silicagel während der Trocknungsperiode
mehrmals gewechselt wurde. Vollständig getrocknetes Blattmaterial erwies sich über mehrere Jahre
haltbar und für Analysen verwendbar. Alternativ wurden Blattproben im Gelände in hochkonzentrierter
CTAB-Lösung nach einer etwas modifizierten Methode von Rogstad (1992) konserviert (G. Kadereit,
mdl.). In der CTAB-Lösung gelagerte Blattproben lieferten ebenfalls gute Ergebnisse bei der DNA-
Extraktion und waren über längere Zeiträume lagerungsfähig.
Zur Gewinnung lebenden Pflanzenmaterials für DNA-Analysen sowie Beobachtungen zu Morphologie
und Lebenszyklus wurden im Gewächshaus und den Freianlagen der Universität Kassel
Kulturversuche durchgeführt. Das Ziel war eine vergleichende Analyse der Entwicklung von Taxa
verschiedener Herkunft unter identischen Wuchsbedingungen. Die Pflanzen wurden aus Samen
gezogen, die teils während der Sammelreisen vom natürlichen Standort entnommen, teils aus
Kulturen vorangegangener Jahre im Gewächshaus gewonnen wurden. Kulturen wurden in den Jahren
2001 bis 2003 durchgeführt. Jeweils in der ersten Aprilhälfte erfolgte die Aussaat der Samen auf
angefeuchtetem Filterpapier in Petrischalen. Zur Erhöhung der Keimraten wurden einige
Samenproben versuchsweise bei etwa -20°C vor der Au ssaat kältebehandelt, um die jahreszeitlichen
Kältephasen des Originalstandortes zu simulieren. Die Keimlinge wurden bei einer Größe von 2-3 cm
(nach 1-2 Wochen) in Töpfe überführt. Das Wuchsmedium war dabei Gartenerde vom Standort des
Gewächshauses, die mit wenig Humus vermischt und nicht weiter behandelt wurde. Ab dem
Jungpflanzen-Stadium erfolgte eine kontinuierliche Behandlung der Erde mit 0,5 M NaCl-Lösung (1x
wöchentlich) sowie in regelmäßigen Abständen mit einem Universaldünger.
Alle relevanten Pflanzen für die vorliegende Arbeit von Aufsammlungen im Gelände sowie aus
Kulturversuchen wurden herbarisiert und sind als Belege im Herbarium der Universität Kassel (KAS)
hinterlegt.

Material und Methoden
27
2.3. Gewinnung molekularer Merkmale Die molekularen Techniken und Methoden folgen mit wenigen Ausnahmen den Vorgaben in
Sambrook & Russell (2001). Auf Besonderheiten wird an den speziellen Stellen eingegangen.
2.3.1. DNA-Isolation Voraussetzung für Arbeiten an der DNA ist deren Isolation aus der Zelle und Trennung von allen
anderen Zellbestandteilen. Für Pflanzen mit ihren hoch differenzierten Zellwänden ist dabei ein
mehrstufiger Prozess vonnöten, der aus mechanischer Zerkleinerung des Gewebes, Auflösung der
Membranen und Abtrennung aller weiteren Zellbestandteile besteht. Für die Untersuchungen an den
Suaedoideae wurde zwei Verfahren angewendet: Cetyltrimethylammoniumbromid (CTAB)-Isolation
und DNA-Extraktionskits. Das hier verwendete Protokoll der CTAB-Methode ist eine relativ einfache
Variante ohne Phenol-Chloroform-Reinigungsschritt und entspricht in den wesentlichen Schritten dem
in Weising et al. (2005: 100) beschriebenem. Es wurde einer geringeren Menge an Ausgangsmaterial
angepasst. Folgende Schritte waren erforderlich (für die eingesetzten Chemikalien vgl. Anhang):
1. Vorbereiten des Blattmaterials: Maximal 50 mg vollständig trockenes oder 150 mg frisches
Blattmaterial unter Zugabe von Seesand pulverisieren (Mörser und Pistill oder Retsch Kugelmühle MM
300), frisches oder unvollständig getrocknetes Material vorher in flüssigem Stickstoff tieffrieren
2. Zell-Lysis in 400 µl CTAB-Isolationspuffer, 30 min bei 60°C auf Eppend orf Thermomixer (Schütteln)
3. Proteinfällung durch Zugabe von 400 µl Chloroform/Isoamylalkohol, 10 min unter schwachem
Schütteln emulgieren
4. Zentrifugation 15 min bei 9000 rpm zur Phasentrennung (Eppendorf Centrifuge 5804 R)
5. Überführung der oberen, wässrigen Phase in neues Eppendorf-Reaktionsgefäß, Zugabe von 0,6
Vol.% Isopropanol, Ausfällung von CTAB-DNA und Polysacchariden (fädiges Präzipitat)
6. Zentrifugation (15 min bei 12000 rpm)
7. Entfernen des Überstandes, Zugabe von 500 µl 70% Ethanol
8. Erneute Zentrifugation (wie 6.)
9. Entfernen des Überstandes und Trocknung des Pellets (Vakuumkonzentrator Savant Speed Vac®
SPD101B)
10. Lösung des Pellets in 200 µl TE-Puffer (meist über Nacht)
11. RNA-Abbau mit RNAse (Endkonzentration 25 µl/ml) 2 h bei 37°C
12. Zugabe von 100 µl 5M NaCl
13. Polysaccharidfällung durch langsame Zugabe von 73 µl 100% Ethanol (unter Schütteln),
anschließend Inkubation auf Eis (10 min)
14. Abzentrifugation der Polysaccharide 15 min bei 9000 rpm
15. Fällung der DNA im Überstand mit gleichem Volumen 100% Isopropanol
16. Zentrifugation (15 min bei 12000 rpm)
17. Entfernen des Überstandes, Zugabe von 500 µl 70% Ethanol
18. Erneute Zentrifugation (wie 6.)
19. Entfernen des Überstandes und Trocknung des Pellets
20. Lösung des Pellets in 200 µl TE-Puffer

Material und Methoden
28
Modifiziert wurden in Abhängigkeit vom Ausgangsmaterial vor allem die DNA-Fällungsschritte.
Während intakte DNA bereits bei Raumtemperatur und nach sehr kurzer Zeit sichtbar ausfällt, war es
im Falle von erwarteten geringen Konzentrationen oder starker DNA-Degradierung notwendig, die
Fällungszeiten zu erhöhen (teilweise über Nacht) sowie die Temperatur zu verringern (-20°C).
Dadurch konnte die Ausbeute gesteigert werden. Als besonders kritisch erwies sich die
Polysaccharidfällung, da in diesem Schritt größere Mengen DNA ausfallen können.
Alternativ wurden einige Proben mit dem Plant Mini Kit von Qiagen extrahiert. Dies erfolgte vor allem
mit einigen Proben, deren DNA mit CTAB nicht isoliert werden konnte (z.B. Frischmaterial von Suaeda
spicata) sowie für Testzwecke. Die Mengen an Ausgangsmaterial sowie die vorbereitenden Schritte
waren jeweils die gleichen wie bei der CTAB-Methode. Die Isolation erfolgte entsprechend den
Angaben in der Gebrauchsanleitung des Herstellers ohne Modifikationen. Für weitere Analysen wurde
die DNA in TE-Puffer bei 4°C im Kühlschrank gelager t.
Alle DNA-Proben erhielten eine eindeutige, vierstellige Identifikationsnummer. Die Nummerierung für
das Labor Kassel beginnt mit 1001. Bei Wiederholung der Isolation aus der gleichen Pflanze wurde
stets eine neue Nummer vergeben.
2.3.2. Gelelektrophorese Die Gelelektrophorese dient zur Separation von Molekülen unterschiedlicher Größe sowie zum
Nachweis absoluter Mengen anhand von Vergleichsstandards. Für die Arbeit mit DNA werden
Agarose- und Polyacrylamidgele eingesetzt. Benötigt werden dafür die Gelmatrix mit
Aufnahmekammern für die DNA-Lösung, ein Elektrophoresepuffer sowie ein variables elektrisches
Feld.
Agarosegele wurden zum Nachweis hochmolekularer DNA nach der Isolation und zur Kontrolle der
PCR-Amplifikate eingesetzt. Je nach Fragestellung wurden dabei Konzentration und
Pufferbedingungen variiert. Die Aufnahme der DNA erfolgte in einem Bromphenol-Glycerin-
Auftragspuffer, der Nachweis durch Anfärben mit Ethidiumbromid-Lösung und Fluoreszenz im UV-
Licht. Als Elektrophoresepuffer wurde 0,5x TBE-Puffer verwendet.
Die Kontrolle der DNA nach der Extraktion erfolgte auf einem 0,8% Agarose-Gel. Zum Abschätzen der
erzielten Menge an DNA diente ein in fünf Stufen zu je 2, 5, 10, 20 und 40 ng/µl verdünnter Lambda-
DNA-Marker, der auf Agarosegelen ähnliche Laufeigenschaften wie die hochmolekulare DNA hat. Für
die Überprüfung von Menge und Qualität von PCR-Produkten wurde mit Gelkonzentrationen von 1,5%
gearbeitet. Als Standard kam hier der Low DNA Mass Ladder zum Einsatz, mit dem Größen von 100,
200, 400, 800, 1200 und 2000 bp und gleichzeitig Mengen von 10, 20, 40, 80, 120 und 200 ng
nachgewiesen werden können.
Im Falle von unspezifischen PCR-Produkten mussten die kompletten Mengen auf einem speziellen
Agarosegel aufgetrennt werden. Das Gel wurde anschließend mit Ethidiumbromid angefärbt und die
gewünschte Bande unter UV-Licht aus dem Gel ausgeschnitten. Die Herauslösung der PCR-Produkte
aus dem Gel erfolgte mittels des Gel Extraktionskits von Qiagen nach Anleitung des Herstellers. Um
störende Wirkungen des Bors zu vermeiden, wurde anstatt mit TBE-Puffer mit 1x TAE-Puffer
gearbeitet und das Gel aus einer Seakem-LE-Agrose (1%) hergestellt.

Material und Methoden
29
Die Auftrennung der Fragmente nach Sequenzierreaktionen erfolgte auf Polyacrylamidgelen in der
zum Licor-Sequenzierautomaten gehörenden Apparatur. Diese Elektrophorese findet unter für DNA
denaturierenden Bedingungen (hohe Harnstoffkonzentration) statt, da nur einzelsträngige Moleküle
richtig detektiert werden können. In den meisten Fällen war ein Auftrennweg von ca. 35 cm
ausreichend (entspricht 41cm-Gelplatten). Für kritische Proben wurde diese Strecke auf etwa 60 cm
vergrößert (66cm-Gelplatten). Die Gelstärke betrug einheitlich 0,2 mm. Für die 41cm-Gele wurde ein
6%iges Gel aus Fertigmischung (Sequagel XR) verwendet, für die 66cm-Gele ein 3,5%iges, aus den
Grundkomponenten selbst gefertigtes PAA-Gel eingesetzt (s. Anhang).
Die Elektrophorese selbst erfolgte in 1x TBE-Puffer bei 1800 (41 cm) bzw. 3000 V (66 cm) auf dem
Licor-Sequenzierautomat. Unter diesen Konditionen werden etwa 100-120 bp je Stunde detektiert.
Die DNA-Proben wurden für die Elektrophorese mit einem denaturierenden und mit Fuchsin oder
Pararosanilin angefärbten Formamid-Auftragspuffer versetzt.
2.3.3. DNA-Regionen und Primer Von den drei unabhängigen genetischen Einheiten der pflanzlichen Zelle sollten wenigstens zwei für
die phylogenetische Untersuchung ausgewählt werden. Für die vorliegende Arbeit wurden Regionen
aus dem Zellkern und dem Chloroplasten verwendet. Da molekularsystematische Studien an den
Chenopodiaceae am Beginn der eigenen Arbeiten nicht vorlagen, mussten Informationen über
geeignete DNA-Regionen und zugehörige Primer aus thematisch ähnlichen Arbeiten entnommen
werden, die sich vor allem mit Chloroplasten- und Kern-DNA beschäftigen. Die entsprechenden
Primersequenzen stammen aus vergleichenden Studien an bekannten Genomen (Reis, Mais, Tabak
u.a.) und sind in relativ konservativen Genregionen verankert, die die zu untersuchende variable
Region flankieren. Aus diesem Grund sind die Primer meist universell einsetzbar und es ist eine
Abschätzung der zu erwartenden Fragmentlänge möglich.
DNA-Regionen für phylogenetische Studien sollten im Genom als Einzelkopie oder in identischen
Kopien vorliegen. Für nah verwandte Arten wie im Fall der vorliegenden Studie mussten Regionen mit
genügend hohen Substitutionsraten ausgesucht werden, um ausreichend informative Positionen für
die Auswertung zur Verfügung zu haben. Allerdings besteht bei zu hoher Mutationsrate die Gefahr des
„Verrauschens“ der Information aufgrund multipler Mutationen. lm Idealfall sollte die
Basenzusammensetzung annähernd gleich sein da die Gefahr besteht, dass nicht verwandte Taxa
nur aufgrund ähnlicher Basenhäufigkeiten zusammen gruppieren. Aus technischen Gründen sollten
die zu amplifizierenden Fragmente eine maximale Länge von 1000 bp nicht überschreiten, um eine
problemlose PCR und Sequenzierung in einem Schritt zu ermöglichen.
Aus dem Kerngenom wurden die beiden internen transkribierten Spacer (ITS) der ribosomalen DNA
ausgewählt. ITS ist einer der am häufigsten eingesetzten Kernmarker im inter- und intraspezifischen
Bereich, damit ist ein Vergleich der eigenen Ergebnisse mit ähnlichen Arbeiten möglich. Die
Primersequenzen für die Amplifikation von Gesamt-ITS und getrennter ITS1/ITS2-Amplifikation (für
degenerierte DNA) stammen aus der Arbeit von Blattner (1999) und wurden erfolgreich in den
Suaedoideae eingesetzt. Da es sich bei den ribosomalen Genen als Bindungsorte für die ITS-Primer
um hochkonservierte Regionen handelt, waren keine Optimierungsversuche notwendig.

Material und Methoden
30
Hingegen waren für die Auswahl geeigneter Chloroplastenregionen Testreihen erforderlich, da eine
Vielzahl möglicher Regionen für die phylogenetische Analyse zur Verfügung steht und davon zwei mit
passender Variabilität gefunden werden sollten. Aus der Literatur wurden acht mögliche Regionen
ausgewählt. Primersequenzen und PCR-Bedingungen wurden aus den entsprechenden
Originalarbeiten übernommen.
Eine Übersicht über die verwendeten Primer und DNA-Regionen gibt die nachfolgende Tabelle:
Tab. 1: Übersicht der im Auswahlverfahren für die phylogenetische Analyse der Suaedoideae getesteten DNA-Regionen und Primer sowie ihrer Herkunft.
Region Primerpaar 5’ - 3’ Bindungsort sensu
Chloroplast
atpB-rbcL fw:
rv:
GAAGTAGTAGGATTGATTCTC
CAACACTTGCTTTAGTCTCTG
cp atpB
cp rbcL
Xu et al. 2000
atpB-rbcL fw: CAAGTTGATCGGTTAATTAAATAAGAA atpB-rbcL eigene Konstr.
trnH-psbA fw:
rv:
GTTATGCATGAACGTAATGCTC
CGCGCATGGTGGATTCACAAATC
cp trnH
cp psbA
Xu et al. 2000
rps16 intron fw:
rv:
GTGGTAGAAAGCAACGTGCGACTT
TGCGGATCGAACATCAATTGCAAC
cp rps16
cp rps16
Oxelman 1997
psbB-psbH fw:
rv:
AGATGTTTTTGCTGGTATTGA
TTCAACAGTTTGTGTAGCCA
cp psbB
cp psbH
Xu et al. 2000
trnT-trnL fw: (a)
rv: (b)
CATTACAAATGCGATGCTCT
TCTACCGATTTCGCCATATC
cp trnT
cp trnL
Taberlet 1991
trnL intron fw: (c)
rv: (d)
CGAAATCGGTAGACGCTACG
GGGATAGAGGGACTTGAACC
cp trnL
cp trnL
Taberlet 1991
trnL-trnF fw: (e)
rv: (f)
GGTTCAAGTCCCTCTATCCC
ATTTGAACTGGTGACACGAG
cp trnL
cp trnF
Taberlet 1991
trnS-trnG fw
rv:
GATTAGCAATCCGCCGCTTT
TTACCACTAAACTATACCCGC
cp trnS
cp trnG
Xu et al. (2000)
Kern
ITS + 5,8S ITS-A:
ITS-B:
GGAAGGAGAAGTCGTAACAAGG
CTTTCCTCCGCTTATTGAGATG
nc 18S
nc 26S
Blattner 1999
ITS 1 ITS-C: GCAATTCACACCAAGTATCGC nc 5,8S Blattner 1999
ITS 2 ITS-D: CTCTCGGCAACGGATATCTCG nc 5,8S Blattner 1999
Sonstige
M 13 uni: TGTAAAACGACGGCCAGT Fartmann et al 1999
M 13 rev: CAGGAAACAGCTATGACC Fartmann et al 1999

Material und Methoden
31
Die Variabilität der DNA-Regionen zwischen den Arten wurde anhand der sehr begrenzten Auswahl
von 6 Taxa eingeschätzt. Die Auswahl umfasste die Sippen Bienertia cycloptera, Suaeda fruticosa, S.
altissima, S. paradoxa, S. heterophylla, S. crassifolia. Mit dieser Auswahl konnte die Variabilität
sowohl inter- als intragenerisch abgeschätzt werden. Es wurden letztendlich Regionen ausgewählt,
deren Amplifikation ohne Schwierigkeiten möglich war, die keine zu hohe Längenvariabilität aufwiesen
und auch durch eine genügende Anzahl von Punktmutationen eine für den gewählten Rahmen
passende Variabilität erwarten ließen. Um festzustellen, dass es sich bei den eigenen Versuchen
wirklich um die erwarteten DNA-Regionen handelt, wurden die Daten mit den in der Genbank
(National Centre for Biotechnology Information, www.ncbi.nlm.nih.gov) publizierten Sequenzen von
Spinacia oleracea als den Suaedoideae nächst verwandte Sippe überprüft (NCBI acc. no. NC_002202
für das Chloroplastengenom, AF062088 für ITS). Die nachfolgende Tabelle gibt einen Überblick über
die Ergebnisse der vergleichenden Tests aller DNA-Regionen an.
Tab. 2: Tabellarische Übersicht der Ergebnisse des Auswahlverfahrens von DNA-Regionen für die phylogenetische Analyse der Suaedoideae.
Region Ergebnisse Auswahl
atpB-rbcL PCR ++; variable Region mit Punktmutationen und Indels; gut verwendbar +
trnH-psbA PCR +, teilweise unspezifisch (2 Fragmente); extrem längenvariable
Region, zwischen Bienertia und Suaeda in der Mitte nicht alignierbar; nur
bedingt verwendbar
-
rps16 intron PCR +, scheint unspezifisch (2 Fragmente); durchschnittlich variable
Region, Punktmutationen und Indels; gut verwendbar
-
psbB-psbH PCR ++, durchschnittlich variable Region, vorwiegend Punktmutationen;
gut verwendbar
+
trnT-trnL PCR ++, stark variable Region; für Gattungsphylogenie wohl nur bedingt
verwendbar
-
trnL intron PCR ++, variable Region, alignierbar; offenbar gut geeignet -
trnL-trnF PCR o, zu schwache PCR-Produkte, nicht sequenzierbar -
trnS-trnG PCR ++, stark variabler Bereich, lange TA-Microsatelliten; mit Spinacia
nicht alignierbar, möglicherweise falsche Region
-
ITS PCR +, teilweise unspezifisch (mehrere Fragmente), sehr variable Region,
gut geeignet zwischen Arten, Variabilität relativ gering zwischen Arten,
zwischen Gattungen dagegen sehr hoch (innerhalb Chenopodiaceae
kritisch)
+
Basierend auf den Ergebnissen dieser Vorversuche wurden zwei geeignete Chloroplasten-Regionen
(atpB-rbcL, psbB-psbH) und ITS für die Studie ausgewählt. Die atpB-rbcL-Region zeigte bereits in den
Vorversuchen eine höhere Variabilität als die psbB-psbH-Region, so dass zu erwarten war, sowohl auf
Gruppen- als auch auf Artebene verwertbare Ergebnisse zu erhalten. Mit den drei DNA-Regionen

Material und Methoden
32
konnte ein ausreichend großer Datensatz erzeugt werden, um mit gewisser Sicherheit sowohl die
molekulare Phylogenie der Gruppe zu rekonstruieren als auch Artbildungsprozesse zu erklären.
Erst nach Beginn der Arbeiten stellte sich heraus, dass die atpB-rbcL -Region für die Taxa der Sektion
Schoberia mit der üblichen PCR-Technik nicht amplifizierbar ist. Grund dafür ist eine komplexe
Minisatellitten-Struktur am Beginn des spacers, an der die PCR offenbar abbricht. Für diese Sippen
wurden aus dem Alignment bereits vorhandener, ähnlicher Sequenzen ein interner Primer konstruiert,
der die nicht amplifizierbare Region ausschließt. Dadurch gehen etwa 150 bp für die phylogenetische
Analyse verloren.
Sämtliche Primer wurden von den Firmen MWG und metabion synthetisiert. Aufgrund der speziellen
Technik des Licor-Sequenzierautomaten wurde jeder Primer am 5’-Ende mit einer Anhangssequenz
(tail) versehen, die als Bindungsstelle für universelle Sequenzierprimer fungiert. Deren Sequenz ist
von einem M13-Phagen abgeleitet und sollte im Eukaryotengenom keine komplementäre Struktur
besitzen. Die Sequenz wurde dem Licor-Manual entnommen (Fartmann et al. 1999). Beide PCR-
Primer haben jeweils unterschiedliche M13-Anhangssequenzen. An diese Anhänge binden die jeweils
unterschiedlich fluoreszenzmarkierten Sequenzierprimer.
2.3.4. PCR-Methodik Mit den oben beschriebenen Primern wurden alle PCR durchgeführt. Es konnten Standardverfahren
genutzt werden, von denen keine wesentlichen Abweichungen notwendig waren. In der Regel
genügten etwa 25-35 PCR-Zyklen für die Erzeugung von ausreichend konzentriertem PCR-Produkt
für die nachfolgende Sequenzierreaktion (>10ng/µl). Ein typischer PCR-Ansatz betrug insgesamt 50
µl. Für alle PCR wurden verwendet: Taq DNA Polymerase (recombinant), dNTP-Stammlösung, PCR-
Puffer, MgCl2-Stammlösung. Durchgeführt wurden die PCR auf Cyclern der Typen Biometra
TGradient, BioRad ICycler und Perkin Elmer 2400. Zusätzliche Schritte waren für die Amplifikation
stark degenerierter DNA notwendig. Um ausreichend PCR-Produkt zu erhalten, wurde das gering
konzentrierte PCR-Produkt aus der ersten PCR als Templat für eine Reamplifikation unter gleichen
Bedingungen verwendet. Dadurch erhöht sich zwar die Fehlerrate, doch war für einige Proben kein
anderer Weg möglich. Die ITS-Region wurde aus degenerierter DNA in zwei getrennten
Teilreaktionen gewonnen, in denen die gesamte Region von ca. 650 bp in zwei etwa gleich langen
Fragmenten getrennt amplifiziert wird. Dies war in vielen Fällen erfolgreich. Nachfolgend ist das
Standard-Protokoll für die ITS-Amplifikation aufgeführt. Zur Erhöhung der Spezifität wurde das
touchdown-Verfahren angewendet (Don et al. 1991), bei dem die ersten Schritte eine sehr hohe
annealing-Temperatur haben, die schrittweise erniedrigt wird. Weiterhin wurde DMSO in
Konzentrationen bis etwa 10% beigefügt. DMSO unterstützt die Spaltung der
Wasserstoffbrückenbindung und verbessert die PCR GC-reicher Fragmente wie im Fall von ITS. Die
Zusammensetzung eines Reaktionsansatzes ist in folgender Tabelle zusammengestellt:

Material und Methoden
33
Tab. 3: Reaktionsbedingungen der ITS touchdown PCR.
Bezeichnung Konzentr. Stammlösung Endkonzentration
PCR Puffer 10x 1x
MgCl2 50 mM 1,5 mM
dNTP 2,5 mM 200µM
Primer 10 µM 0,25 µM je
taq DNA Polymerase 5 U/µl 1,5 U
DMSO 100% 10%
in 50 µl Ansatz
Die thermischen Bedingungen und Reaktionszeiten für die ITS-PCR sind folgender Übersicht zu
entnehmen:
1x 180“ 94°C
10x 30“ 94°C
30“ 62°C (-0,5 Grad je Zyklus)
90“ 72°C
25x 30“ 94°C
30“ 57°C
90“ 72°C
1x 10’ 72°C
Unter ähnlichen Bedingungen wurde die Chloroplasten-PCR durchgeführt. Hierbei war es nicht
erforderlich, Modifikationen vorzunehmen, da die Primer sehr spezifisch hybridisierten. Allerdings war
die Amplifikation aus degradiertem Material wesentlich schwieriger, da Chloroplasten-DNA in
wesentlich geringerer Menge in den Zellen vorliegt und auch mit Ausnahme der atpb-rbcL-Region für
die Sektion Schoberia keine internen Primer verwendet wurden. Daher liegen nicht für alle Taxa
Chloroplasten-Daten vor.
Tab. 4: Reaktionsbedingungen der Chloroplasten PCR.
Bezeichnung Konzentr. Stammlösung Endkonzentration
PCR Puffer 10x 1x
MgCl2 50 mM 1,5 mM
dNTP 2,5 mM 100µM
Primer 10 µM 0,25 µM je
taq DNA Polymerase 5 U/µl 1,5 U
DMSO 100% 5%
in 50 µl Ansatz

Material und Methoden
34
Der PCR-Zyklus wurde ohne vorangestellte touchdown-Phase durchgeführt:
1x 180“ 94°C
25x 30“ 94°C
30“ 55°C
90“ 72°C
1x 10’ 72°C
Die Reaktionsansätze wurden nach beendigter PCR bei 4°C im Kühlschrank für die weitere
Verwendung gelagert.
2.3.5. DNA-Sequenzierung Die PCR-Fragmente wurden nach der Amplifikation direkt sequenziert, eine Aufreinigung war in der
Regel nicht erforderlich. Sequenziert wurde nach der Kettenabbruchmethode von Sanger (1977). Die
Sequenzierung ist eine etwas modifizierte PCR in der ein Teil der Desoxyribonukleotide (dNTP) durch
2’-3’-Didesoxyribonukleotide (ddNTP) ersetzt wird (meist im Verhältnis 1:200-300). Wird ein ddNTP
anstelle eines dNTP in die Kette eingebaut, bricht die Kettenverlängerung an dieser Stelle ab und
aufgrund der Fragmentlänge erhält man Informationen über das Nukleotid an der Abbruchstelle. Die
Sequenzierreaktion liefert die Fragmente für die Auftrennung auf einem PAA-Gel und der Detektion im
Sequenzierautomaten.
Die Sequenzierung bzw. Fragmentauftrennung erfolgte mit dem Sequenzierautomaten IR 4010 von
Licor. Das Gerät ist mit zwei Detektionslasern ausgestattet, die unterschiedliche Wellenlängen
erfassen (700 und 800 nm). Damit ist es möglich, zwei Auftrennungen gleichzeitig durchzuführen,
sofern diese mit unterschiedlich markierten Sequenzierprimern durchgeführt wurden.
Da mit universalen fluoreszenzmarkierten Primern gearbeitet wurde die keine direkte Detektion der
vier unterschiedlichen Nukleotide zulässt, mussten für die Sequenzierung einer DNA-Probe vier
getrennte Reaktionen durchgeführt werden, die jeweils 1 spezifisches ddNTP enthielten. Verwendet
wurde für die Sequenzierreaktion das komplette Thermo Sequenase™Primer Cycle Sequencing Kit
von amersham Biosciences, das sich als geeignet für alle Reaktionen erwies. Zu dem
Reaktionsansatz wurden nur noch das DNA-Templat, Sequenzier-Primer sowie hochreines Wasser
(für die HPLC) zugesetzt
Die Sequenzierprimer für den Licor-Sequenzierautomaten sind spezielle fluoreszenzmarkierte Primer
(IRDye 700 und IRDye 800), die an den um die tails (s. Kap. 2.3.3) verlängerten PCR-Fragmenten
binden und in der Sequenzierreaktion die Abbruchfragmente in gegensätzlichen Richtungen vom
gleichen Fragment erzeugen. Von jedem PCR-Fragment entstehen damit zwei gegenläufige,
komplementäre Sequenzinformationen. Dies dient zur Kontrolle der erhaltenen Ergebnisse.
Eine einzelne Sequenzierreaktion für den Nachweis eines Nukleotids umfasste insgesamt 6 µl und
enthielt die Komponenten in folgenden Endkonzentrationen:
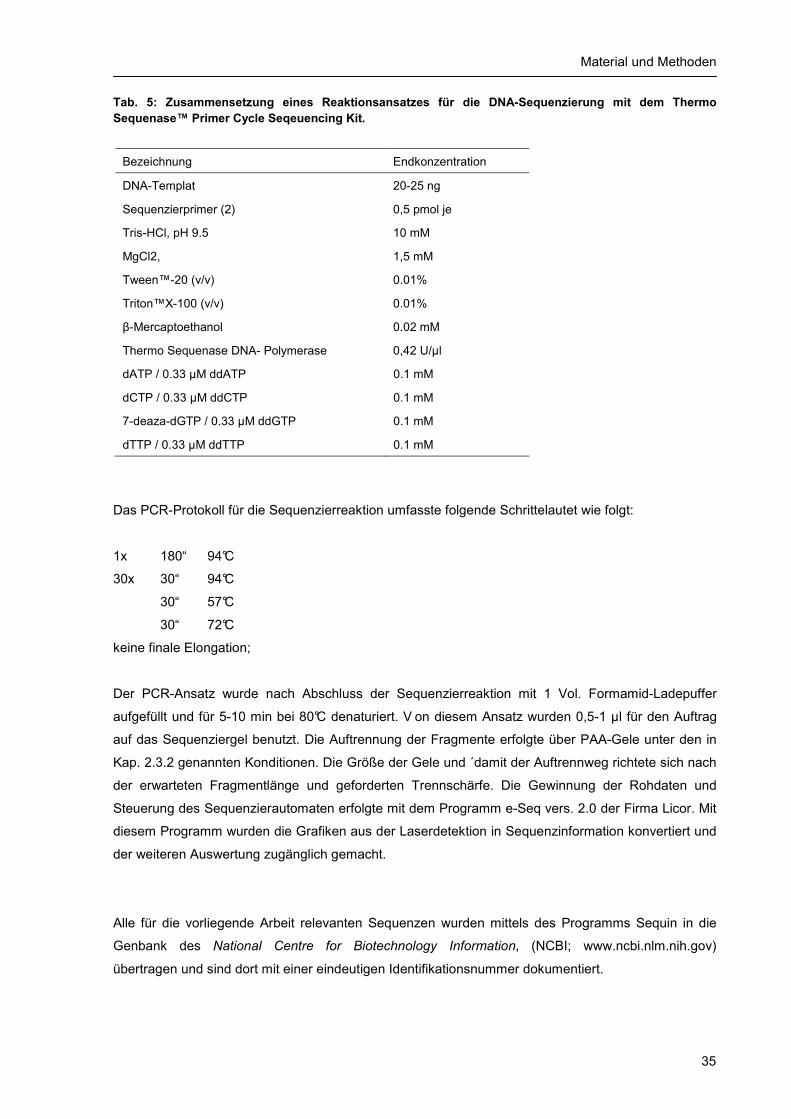
Material und Methoden
35
Tab. 5: Zusammensetzung eines Reaktionsansatzes für die DNA-Sequenzierung mit dem Thermo Sequenase™ Primer Cycle Seqeuencing Kit.
Bezeichnung Endkonzentration
DNA-Templat 20-25 ng
Sequenzierprimer (2) 0,5 pmol je
Tris-HCl, pH 9.5 10 mM
MgCl2, 1,5 mM
Tween™-20 (v/v) 0.01%
Triton™X-100 (v/v) 0.01%
β-Mercaptoethanol 0.02 mM
Thermo Sequenase DNA- Polymerase 0,42 U/µl
dATP / 0.33 µM ddATP 0.1 mM
dCTP / 0.33 µM ddCTP 0.1 mM
7-deaza-dGTP / 0.33 µM ddGTP 0.1 mM
dTTP / 0.33 µM ddTTP 0.1 mM
Das PCR-Protokoll für die Sequenzierreaktion umfasste folgende Schrittelautet wie folgt:
1x 180“ 94°C
30x 30“ 94°C
30“ 57°C
30“ 72°C
keine finale Elongation;
Der PCR-Ansatz wurde nach Abschluss der Sequenzierreaktion mit 1 Vol. Formamid-Ladepuffer
aufgefüllt und für 5-10 min bei 80°C denaturiert. V on diesem Ansatz wurden 0,5-1 µl für den Auftrag
auf das Sequenziergel benutzt. Die Auftrennung der Fragmente erfolgte über PAA-Gele unter den in
Kap. 2.3.2 genannten Konditionen. Die Größe der Gele und ´damit der Auftrennweg richtete sich nach
der erwarteten Fragmentlänge und geforderten Trennschärfe. Die Gewinnung der Rohdaten und
Steuerung des Sequenzierautomaten erfolgte mit dem Programm e-Seq vers. 2.0 der Firma Licor. Mit
diesem Programm wurden die Grafiken aus der Laserdetektion in Sequenzinformation konvertiert und
der weiteren Auswertung zugänglich gemacht.
Alle für die vorliegende Arbeit relevanten Sequenzen wurden mittels des Programms Sequin in die
Genbank des National Centre for Biotechnology Information, (NCBI; www.ncbi.nlm.nih.gov)
übertragen und sind dort mit einer eindeutigen Identifikationsnummer dokumentiert.

Material und Methoden
36
2.4. Auswertung der molekularen Daten Die im folgenden beschriebenen Schritte sind Standardverfahren zur Modellierung molekularer
Phylogenien einschließlich deren statistischer Absicherung. Die Auswahl erfolgte vor allem anhand
zusammenfassender Werke (Felsenstein 2004, Hall 2001, Hillis et al. 1996, Rieppel 1999, Swofford et
al. 1996) sowie durch Vergleich mit thematisch ähnlicher Literatur. Das Ziel aller benutzten Methoden
ist die Rekonstruktion der statistisch am besten abgesicherten Stammbäume für alle untersuchten
Taxa, die als Grundlage für weiterführende Untersuchungen dienen.
2.4.1. Datenauswahl Alignments und Berechnungen wurden für Datensätze unterschiedlicher taxonomischer Ebene sowie
für ITS- und Chloroplastendaten getrennt durchgeführt. Dies erschien aufgrund der hohen Diversität
im gesamten Datensatz sowie unterschiedlicher phylogenetischer Signale notwendig. In den
Berechnungen kombiniert wurden alle Chloroplastendaten, da es sich beim Chloroplastengenom um
eine physische Einheit handelt, in diesem Fall ist dieses Vorgehen legitim. Eine weitere kombinierte
Berechnung erfolgte mit atpB-rbcL/ITS-Daten. Mit dem partition-homogeneity test wurde in jedem Fall
vor einer Kombination die phylogenetischen Signale verglichen und auf Abweichungen geprüft.
Entsprechend dem Ziel der vorliegenden Arbeit wurden Datensätze für drei taxonomische Ebenen
zusammengestellt die sich in etwa an folgenden Einheiten orientieren:
1. Unterfamilie: 22 Taxa Suaedoideae, Salicornioideae, Bassia; enthält nur wichtige,
sektionsspezifische Sippen.
2. Gattung: ein Datensatz für ITS mit 92 Sippen, ein Datensatz für Chloroplasten mit 66 Sippen,
enthält die größte Breite verfügbarer Sequenzen für die Gattung Suaeda.
3. Sektion: als Beispiel wurde hier die Sektion Brezia mit 66 Sequenzen ausgesucht, da in dieser
Sektion die meisten taxonomischen Unklarheiten bestehen und auch das meiste Material zur
Verfügung stand.
Die Unterteilung erschien auch aufgrund der Menge vorhandener Sequenzen und der Darstellbarkeit
der Ergebnisse sinnvoll. Davon abweichend wurde die Bestimmung der Divergenzzeiten und die
Altersdatierung mit einem erweiterten Datensatz vorgenommen, um bereits bekannte
Kalibrierungspunkte zu berücksichtigen.
2.4.2. Sequenz-Alignment Die Sequenzdaten müssen zunächst in eine auswertbare Form gebracht werden, um vorhandene
Unterschiede sichtbar zu machen. Dies geschieht durch das Alignment. Ein Alignment enthält die
Sequenzen in ausgerichteter Form, wodurch das Kriterium der Homologie von Merkmalen hergestellt
wird. Homologe Merkmale sind in diesem Falle die Nukleotide an gleicher Position. Das Alignment ist
ein Modell, welches die Unterschiede zwischen verschiedenen Sequenzen verdeutlicht und

Material und Methoden
37
Längenunterschiede durch Einfügen von Lücken (gap) ausgleicht. Die Konstruktion des Alignments ist
ein Optimierungsprozess, der von mehreren Größen beeinflusst wird: gap open penalty, mismatch
penalty, gap extension penalty. Die genannten Faktoren wirken unmittelbar auf die Definition
homologer Positionen, was für die spätere Analyse von entscheidender Bedeutung ist. Mit diesen
Werten wird ein Optimalwert des Alignments ermittelt, der in diesem Falle zu maximieren ist. Das
Prinzip besteht darin, das Einfügen von Lücken zu erschweren und Fehlpaarungen bis zu einem
gewissen Grad zu ermöglichen, dass ein biologisch sinnvolles Ergebnis entsteht. Ein korrektes
Alignment existiert nicht. Die Konstruktion des Alignments und die Wahl der Parameter hat daher
entscheidenden Einfluss auf die Ergebnisse der phylogenetischen Berechnungen.
Die Sequenzalignments wurden mit dem Programm AlignIR vers. 1.1 der Firma Licor durchgeführt. In
einem ersten Schritt wurden die aus der Sequenzierung mit dem Licor-Sequenzierautomaten
gewonnen komplementären, gegenläufigen Sequenzen für eine DNA-Sequenz durch überlappendes
Alignment vereinigt und daraus eine Konsensus-Sequenz erzeugt. Nicht jede Baseninformation war
exakt zu bestimmen. Dies kann sowohl an Problemen bei der Sequenzierung als auch an wirklich
vorhandenen Doppelungen liegen. Dieser Fall tritt immer dann auf, wenn eine Sequenz in mehreren
Ausprägungen (bzw. Allelen) vorliegt, wie dies in hybridogenen Genomen der Fall sein kann. Unklare
Positionen wurden soweit möglich nach dem IUPAC-Code verschlüsselt. Mit den Konsensus-
Sequenzen aller Taxa wurde das multiple Alignment für die jeweilige DNA-Region konstruiert. In allen
Schritten waren manuelle Editierungen notwendig.
Das endgültige (multiple) Alignment wurde mit dem Programm ClustalX vers. 1.83 (Thompson et al.
1997) konstruiert. Als Alignmentparameter wurden die Einstellungen open gap penalty = 15 und gap
extention penalty = 6,66 gewählt. Durch die Wahl eines Wichtungsfaktors für Transitionen von 0,5 wird
der empirischen Beobachtung Rechnung getragen, dass Transversionen gegenüber Transitionen mit
höheren „Kosten“ verbunden sind. Somit sind bereits im Alignment Parameter der DNA-Evolution
berücksichtigt. Mit den angegebenen Werten werden die Alignments vergleichsweise eng konstruiert,
eine relativ hohe Zahl von Fehlpaarungen toleriert und entstehende Lücken, die als fehlende Daten
gewertet werden, minimiert. Aufgrund der hohen Diversität der Sequenzen war ein eindeutiges
Alignment besonders im Fall von ITS nicht möglich.
Die Längenunterschiede (Insertionen, Deletionen) wurden als zusätzliche Merkmale gewertet, wenn
sie synapomorphen Charakter zeigten. Die Indels können allerdings keinen Wert annehmen sondern
müssen als presence/absence-Charakter (1/0) binär verschlüsselt werden. Technisch wurde das
Problem so gelöst, dass die Sequenzmatrix um die jeweiligen 0/1 Werte erweitert und eine
kombinierte Berechnung durchgeführt wurde. Probleme ergeben sich aus überlappenden Indels in
dem Fall, wenn unterschiedliche Sequenzen in der gleichen Region unterschiedlich lange Indels
haben. Diese Indels wurden nicht gewertet. Besonders bei den Chloroplastendaten lieferten die
kodierten Indels ergänzende Informationen. Allerdings werden in den Berechnungen zwei Datentypen
vereint, was zu Fehlern führen kann und die kombinierte Information ist nur für bestimmte
Berechnungsverfahren (Maximum Parsimony) zu verwerten.
Aus den Alignments wurden mit Hilfe des Programms MEGA version 4 (Tamura et al. 2007) die
Sequenzmerkmale wie z.B. GC-Gehalt, variable Positionen, usw. berechnet.

Material und Methoden
38
2.4.3. Phylogenetische Berechnungsverfahren Die Rekonstruktion verwandtschaftlicher Zusammenhänge erfolgt anhand von gerichteten
Stammbäumen und geschieht im wesentlichen nach zwei methodischen Ansätzen. Phänetische
Verfahren (Sneath & Sokal 1973) gruppieren Taxa nach Ähnlichkeiten bzw. Divergenzen ohne
Berücksichtigung des Entstehungsweges. Kladistische Verfahren rekonstruieren aus den
beobachteten Merkmalsunterschieden gemeinsam abgeleitete Gruppen und stellen diesen Prozess in
Kladogrammen dar. Alle kladistischen Prinzipien gehen auf die Theorien von Hennig (1950) zurück,
wonach sich eine Art bzw. eine Population in zwei Tochterarten bzw. -populationen aufspaltet, die als
Schwestergruppen bezeichnet werden. Die Schwestergruppen teilen gemeinsam abgeleitete
(synapomorphe) Merkmale. Erforderlich ist auch die Annahme, dass sich alle Organismen auf einen
ursprünglichen Organismus zurückführen lassen, der am Beginn bzw. der Wurzel des Stammbaumes
steht.
Das Ziel der auf rezenten Merkmalen basierenden Phylogenie ist die Ermittlung eines Kladogrammes
(=Stammbaumes), das die Verwandtschaft und Evolution seiner Endglieder am besten wiedergibt. Ein
Kladogramm besteht aus einer Vielzahl hierarchisch angeordneter Bifurkationen, die den Weg der
Artbildung und Evolution widerspiegeln und Schwestergruppenbeziehungen aufzeigen sollen. Jeder
Knoten hat darin die Bedeutung eines Aufspaltungsereignisses. Die Äste verdeutlichen die
Merkmalsentwicklung eines untergeordneten Knotens oder eines terminalen Taxons. Mit der Astlänge
wird die jeweilige individuelle Merkmalszahl ausgedrückt. Der Idealfall der phylogenetischen
Rekonstruktion ist genau ein Kladogramm, das die phylogenetischen Verwandtschaftsbeziehungen
aller Taxa eindeutig bestimmt. In den meisten Fällen resultiert Merkmalsinkongruenz aber darin, dass
mehrere Kladogramme gefunden werden, die in einigen Gabelpunkten übereinstimmen, in anderen
sich aber unterscheiden. Die Verwandtschaftsbeziehungen sind damit für einige Taxa nicht eindeutig
bestimmt.
Methodische Ansätze für die Rekonstruktion von Stammbäumen können in Distanz- und
Optimierungsmethoden unterteilt werden. Mit Distanzmethoden wie z.B. neighbor joining (Saitou & Nei
1987) oder UPGMA (Sneath & Sokal 1973) werden Sequenzen oder Binärdaten aus fingerprint-
Analysen paarweise verglichen und Distanzwerte berechnet. Aus den erhaltenen Werten wird eine
Distanzmatrix gebildet und daraus nach speziellen Clusterverfahren ein Stammbaum rekonstruiert.
Der resultierende Stammbaum ist für dieses Verfahren eindeutig. Im Gegensatz zu den
Distanzmethoden benötigen Optimierungs- oder diskrete Methoden wie Maximum Parsimony (Farris
1970), Maximum Likelihood (Felsenstein 1981) oder die komplexeren Verfahren der Bayesianischen
Analyse zwei Schritte. Als erstes wird ein Optimalitätskriterium (z. B. Evolutionsmodell) festgelegt, mit
dem im zweiten Schritt ein gegebener Baum optimiert wird. Die Methoden greifen direkt auf Merkmale
(z. B. Sequenzdaten) zurück und haben damit eine wesentlich breitere Informationsbasis als
Distanzverfahren. Alle in der vorliegenden Arbeit verwendeten Methoden der phylogenetischen
Rekonstruktion haben die Erstellung von optimierten Kladogrammen und ihre Bewertung zum Ziel.
Anhand einer gut gestützten molekularen Phylogenie können Merkmale rekonstruiert und bewertet
sowie bei Vorliegen geeigneter Fossilien Altersdatierungen vorgenommen werden. Dafür ist die
Ermittlung von Nukleotid-Substitutionsraten notwendig. Werden diese als konstant ermittelt

Material und Methoden
39
(molekulare Uhr), kann die Phylogenie kalibriert werden und bekommt damit eine zeitliche
Komponente.
Distanzen und Evolutionsmodelle
Mit Distanzen wird ein Ähnlichkeitsmaß für Sequenzpaare ausgedrückt, aus dem unmittelbar auf die
evolutionäre Geschichte geschlossen werden kann. Verschiedene Möglichkeiten der Distanzbildung
sind beschrieben worden, die mehr oder weniger auf die Spezifik des DNA-Moleküls und seine
Dynamik eingehen.
Die einfachste Möglichkeit der Distanzbildung ist die Ermittlung des Anteils unterschiedlicher
Nukleotidpositionen und die Übertragung in prozentuale Werte. In diesem Fall spricht man von
unkorrigierten oder p-Distanzen. Die besonderen Eigenschaften der DNA-Merkmale werden bei
diesem Verfahren nicht berücksichtigt. Diese Art der Distanzbildung bedeutet eine starke Reduzierung
des in dem Datensatz enthaltenen Informationsgehaltes, da beispielsweise gleiche Distanzwerte aus
unterschiedlichen Sequenzpaaren entstehen können. Da Mehrfach- und Rückmutationen
unberücksichtigt bleiben, ist die Methode fehlerbehaftet. Besonders bei steigender Divergenzzeit wird
die Zahl tatsächlich aufgetretener Mutationen systematisch unterschätzt. Daraus können Fehler bei
der nachfolgenden phylogenetischen Rekonstruktion entstehen.
Evolutionsmodelle sind durch bestimmte empirisch beobachtete und festgelegte Parameter korrigierte
Distanzen. Das Hinzufügen von Korrekturparametern ist nachvollziehbar, da die DNA-Divergenz in
einem stochastischen Prozess mehr oder weniger unabhängiger Einzelereignisse entstanden ist und
die Parameter diesen Prozess nachträglich modellieren sollen. Es wird der Natur des DNA-Moleküls
eher gerecht, die 4 möglichen Merkmalszustände (=Nukleotide) und ihre zeitlichen
Veränderungsprozesse gesondert zu bewerten. Aus der Vielzahl beschriebener Evolutionsmodell
sollen einige wichtige kurz charakterisiert werden.
Das 1-Parameter-Modell von Jukes & Cantor (1969) hat als Grundannahme gleiche Substitutionsraten
und gleiche Nukleotidfrequenzen (je 0,25). Das von Kimura (1980) vorgeschlagene Modell setzt
gleiche Nukleotidfrequenzen voraus, erlaubt aber zwei Substitutionstypen und berücksichtigt
Unterschiede in der Änderungswahrscheinlichkeit von Transitionen und Transversionen. Das F81-
Modell von Felsenstein (1981) erlaubt bei gleichem Substitutionstyp unterschiedliche
Nukleotidfrequenzen. Darüber hinaus existieren zahlreiche weitere, spezielle Modelle, die alle
spezifische Substitutionsraten und Nukleotidfrequenzen zulassen. Das unabhängigste Modell mit den
meisten freien Parametern ist das general time reversible-Modell (Lanave et al. 1984). Bei dem GTR-
Modell weisen Transitionen, Transversionen und alle 6 möglichen Substitutionsraten (AC, AG, AT,
CG, CT, GT) eigene Wahrscheinlichkeiten auf und es werden unterschiedliche Basenhäufigkeiten
berücksichtigt.
DNA-Evolutionsmodelle können anhand einer 4x4-Matrix dargestellt werden, die die
Substitutionsraten für alle vier Merkmalszustände (Nukleotide) enthält. Jede Nukleotidhäufigkeit bzw.
–frequenz wird durch einen prozentualen Wert ausgedrückt. Weiterhin ist zu berücksichtigen, dass
Substitutionsraten meist variabel sind. Die Variabilität wird durch eine Gamma-Verteilung beschrieben.
Diese Verteilung hat einen Formparameter a, der den Umfang der Variation der Substitutionsraten
unter den Nukleotiden einer Sequenz kennzeichnet. Je kleiner der Wert des Formparameters, um so

Material und Methoden
40
höher ist die Variationsbreite der Substitutionsraten. Ein weiterer Parameter ist der Anteil invariabler
Positionen an der Gesamtzahl der Nukleotide (pinv).
Evolutionsmodelle müssen als Grundlage der Maximum Likelihood–Methode für jeden Datensatz
separat ermittelt werden. Sie werden nicht direkt berechnet sondern stammen aus empirischen
Beobachtungen. Die Werte für die variablen Parameter werden aus dem gegebenen Alignment
abgeschätzt. Einfache Modelle erklären die Sequenzevolution meist nur unzureichend, komplexe,
parameterreiche Modelle sollten genauer sein. Da die Parameterschätzung jedoch ebenfalls mit
Ungenauigkeiten behaftet ist, sind zu komplexe Modelle meist nicht optimal. Es muss daher ein
Modell gefunden werden, das so einfach wie möglich und so komplex wie nötig ist, Die Auswahl des
optimalen Evolutionsmodells erfolgt durch den likelihood ratio test (s. Kap. 2.4.5.) und wurde mit der
Software Modeltest 3.7 (Posada & Crandall 1998) für jedes Alignment separat durchgeführt. Zuerst
wurden in PAUP anhand der im Programmumfang von Modeltest enthaltenen Testdatei
(modelblock.txt) die Parameter aller 56 Evolutionsmodelle für das jeweilige Alignment bestimmt,
anschließend mit Modeltest das optimale Modell ermittelt. Es wird getestet, ob sich die gegebenen
Daten durch das jeweils einfachere Modell genauso gut erklären lassen wie durch das kompliziertere.
Das beste Modell wird jeweils ausgewählt, wobei auch mehrere gleichwertige bestimmt werden
können. Alternativ wird das beste Evolutionsmodell durch das Akaike Informations-Kriterium ermittelt,
ein Verfahren, was dem hierarchischen likelihood ratio test aufgrund eines unabhängigeren Ansatzes
unter gewissen Bedingungen überlegen ist (Posada & Buckley 2004).
Für eine vorläufige Einschätzung der Sequenzdivergenz wurden für einen ausgewählten Datensatz
paarweise und durchschnittliche Kimura-2-Parameter-Distanzen berechnet. Dies erfolgte mit dem
Programm MEGA 4.
Neighbor joining Die neighbor joining Methode berechnet aus paarweisen Distanzen unter Minimierung der
Knotenabstände und Astlängen einen phylogenetischen Baum. Aufgrund der geringen Rechenzeiten
gibt neighbor joining-Baum einen ersten Überblick über die phylogenetischen Zusammenhänge und
wird oft als Startbaum für MP- oder ML-Analysen genutzt. NJ arbeitet besonders effektiv bei geringen
Sequenzdivergenzen (Holder & Lewis 2003). Der Berechnung liegt als erster Schritt eine
Distanzmatrix zu Grunde, die durch Berechnung absoluter Ähnlichkeiten oder anhand eines
Evolutionsmodells ermittelt wird. Meist geschieht dies nach dem Jukes-Cantor- oder Kimura-2-
Parameter-Modell, doch können auch korrigierte Distanzen verwendet werden, wodurch die
Ergebnisse unter phylogenetischen Gesichtspunkten verbessert werden. Das Ergebnis des neighbor
joining ist genau ein Dendrogramm, dessen Astlängen die Distanzen zwischen Gruppen und
Endgliedern widerspiegeln. Neighbor joining ist primär ein numerisches Verfahren, erlaubt jedoch
durch das Minimierungsprinzip im Gegensatz zu reinen Clusterverfahren Hinweise auf Verwandtschaft
und Evolutionsraten.
Maximum Parsimony Das Parsimonie-Prinzip bedeutet Sparsamkeits-Prinzip oder die Bevorzugung der einfachsten Lösung
als die beste. Parsimonie als Verfahren in der phylogenetischen Rekonstruktionen greift direkt auf die

Material und Methoden
41
Sequenzdaten zurück und ermittelt denjenigen Baum als den besten, der mit der kürzesten Anzahl
von evolutionären Veränderungen die vorhandene Diversität erklärt. Die Gesamtzahl der evolutiven
Veränderungen entspricht der Länge des Baumes, der zu minimieren ist. Die Länge ist aber auch
abhängig von der Zahl der Taxa, der Divergenz und der Zahl der Merkmale. Parsimonie setzt keine
Annahmen über den Prozess der Sequenzevolution voraus, dennoch werden Annahmen über die
Merkmalstransformation getroffen, anhand derer sich die unterschiedlichen Auffassungen
unterscheiden (z. B. Wagner-Parsimonie, Dollo-Parsiomonie). Als Konsens wird heute ein Konzept der
allgemeine Parsimonie (Swofford et al 1996) verwendet.
Damit Maximum Parsimony angewandt werden kann, müssen aus dem Sequenzalignment in einem
ersten Schritt parsimony-informative Positionen ermittelt werden. Dies sind homologe Positionen, die
bei wenigstens 2 terminalen Taxa oder Gruppen gleiche Merkmalsänderungen aufweisen. Diese
werden als gemeinsam abgeleitet (synapomorph) gewertet. Singuläre Merkmalsänderungen
(Autapomorphien) bleiben unberücksichtigt. Die Synapomorphien werden zur Konstruktion der
Schwestergruppenbeziehungen benutzt. Auftretende Widersprüche, die es sehr oft gibt, werden durch
die Prozesse Reversion, Konvergenz oder Parallelismus erklärt. Diese letztgenannten Merkmale sind
Homoplasien, die als zusätzliche Schritte zur Erklärung der Daten angesehen werden (Hall 2001). Die
Maximum Parsimony-Methode wird bei sehr hohen Distanzen ungenau (long branch attraction), da
Rück- und Doppelmutationen nicht richtig bewertet werden und scheinbare Synapomorphien
entstehen können.
Das optimale Ergebnis ist der sparsamste Baum (most parsimonious tree), meist jedoch gibt es eine
große Zahl gleich langer, sparsamer Bäume. Aus allen kürzesten Bäumen werden durch einen
weiteren Schritt Konsensus-Bäume ermittelt die nur die Verzweigungen wiedergeben, die in allen
(strict consensus) oder in wenigstens 50% aller kürzesten Bäume auftreten (majority rule tree). Die
Verzweigungen der Konsensus-Bäume gelten als relativ gesichert. Allerdings gehen bei der
Konsensusbildung Informationen verloren, da alternative Verzweigungen nicht sichtbar werden und
Astlängen nicht berücksichtigt werden.
Zur Bewertung der gefundenen Bäume werden verschiedene Werte ermittelt, die Aussagen über die
Qualität der errechneten Phylogenien ermöglichen und Vergleiche mit anderen Daten erlauben. Die
Länge des Baumes entspricht der Anzahl von Merkmalstransformationen, ist aber abhängig von der
Merkmalszahl und eignet sich daher nur bedingt für Vergleiche.
Mit den so genannten goodness of fit Indizes wird ausgedrückt, wie gut die errechneten
Baumtopologien den zugrunde liegenden Datensatz repräsentieren. Der Konsistenz-Index
(consistency index, CI, Kluge & Farris 1969) ist ein Durchschnittswert für das Auftreten von
Homoplasien in einem Datensatz. Er wird für jede Merkmalsänderung, die mehrfach unabhängig
auftritt (Homoplasie) als Quotient aus eins und der Anzahl der unabhängigen Merkmalsänderungen
berechnet. Eine hohe Anzahl uninformativer Merkmale (z. B. Autapomorphien) verfälscht das
Ergebnis, daher wird der CI oft auch ohne diese Merkmale berechnet. Die CI-Werte in der
vorliegenden Arbeit entsprechen dieser Berechnungsweise. Der Retentions-Index (retention index, RI,
Farris 1989) beschreibt das Verhältnis der vorhandenen zur maximal möglichen Anzahl von
unabhängigen Merkmalstransformationen.

Material und Methoden
42
Je näher der Wert beider Indices ist, desto besser ist auch die Passung der Merkmale und desto
robuster ist die phylogenetische Hypothese. CI und RI hängen jedoch auch vom Probenumfang ab.
Maximum Likelihood
Die von Felsenstein (1981) erstmals für die Rekonstruktion von Phylogenien eingeführte Methode ist
ein Optimierungsverfahren, das im Gegensatz zu Parsimonie eine Baumtopologie ermittelt, die mit der
höchsten Wahrscheinlichkeit die Entstehung eines gegebenen Datensatz erklärt. In der
phylogenetischen Analyse sind dies die Sequenzunterschiede im Alignment. Der Baum verdeutlicht
die Wahrscheinlichkeit, mit dem gewählten Evolutionsmodell den gegebenen Datensatz „zu erreichen“
und ist im statistischen Sinne ein Schätzer. Im Prinzip wird die Wahrscheinlichkeit ermittelt, mit der
Nukleotide bzw. komplette Sequenzen in andere Zustände evolvieren können. Dafür ist die Definition
eines Evolutionsmodells erforderlich. Die Berechnung erfolgt für jede Position separat, die
Wahrscheinlichkeit eines Baumes errechnet sich aus dem Produkt der Einzelwahrscheinlichkeiten für
jede Position bzw. der Summe ihrer log likelihood-Werte. Der absolute Wert des
Wahrscheinlichkeitsproduktes ist sehr klein. Für eine bessere Darstellung wird daher der natürliche
Logarithmus dieses Wertes verwendet. In der Berechnung wird dieser Wert minimiert. Das Ziel ist es,
den Baum mit dem höchsten Wahrscheinlichkeitswert zu finden, daher auch Maximum Likelihood. Das
Ergebnis der Analyse ist genau ein Baum, in Ausnahmefällen auch mehrere.
Berechnungen mit PAUP Die Methoden Neighbor Joining, Maximum Parsimony und Maximum Likelihood wurden sämtlich mit
dem Programm PAUP 4.10b (Swofford 2002) gerechnet. Alle Einstellungen, Optionen und Befehle
entsprechen den Angaben im PAUP manual (Entwurf Juli 2002; http://paup.csit.fsu.edu). Als
Alternative zu PAUP wurde MEGA 4 für die Evaluation einiger Berechnungsergebnisse
herangezogen.
Für beide Methoden, MP und ML, werden exakte und heuristische Suchverfahren angewendet. Die
kürzesten bzw. wahrscheinlichsten Bäume können nicht direkt ermittelt werden, vielmehr müssen alle
möglichen Baumtopologien berechnet und daraus die optimale gewählt werden. Dies ist mit der
derzeit verfügbaren Hardware nur bei einer Taxonzahl bis maximal 12 vollständig möglich. Die in
diesem Fall angewendeten Methoden sind exhaustive search und branch-and-bound. Beide Verfahren
garantieren das Auffinden der optimalen Baumtopologie. Für mehr als 12 Taxa muss eine heuristische
Suche gewählt werden. Bei dieser Methode werden an einen Startbaum (zufällig erzeugt, neighbor
joining-Baum) durch Knotenbildung an beliebigen Sequenzpositionen neue Taxa angeknüpft und die
Länge des Baumes berechnet. Mit dem kürzesten Baum wird dieser Vorgang mit dem nächsten,
zufällig gewählten Taxon wiederholt usw. Anschließend wird durch Verlagerung von Ästen (branch
swapping) versucht, eine noch sparsamere Baumtopologie zu finden. Dafür stehen die Methoden
nearest neighbor interchange (NNI), subtree pruning and regrafting (SPR) und tree bisection-
reconnection (TBR) zur Verfügung. Die letztgenannte Methode wurde in den Analysen verwendet. Die
heuristische Suche garantiert nicht das Auffinden der kürzesten oder wahrscheinlichsten
Baumtopologie, vielmehr kann ein lokales Optimum erreicht werden, das durch die Methodik nicht
verlassen werden kann.

Material und Methoden
43
Für die ML-Analyse wurden die von Modeltest gefunden Parameter (Zahl der Substitutionstypen,
Substitutionsraten, Nukleotidfrequenzen, Gamma-Formparameter, Anteil invariabler Positionen)
vorgegeben und die heuristische Suche mit einem neighbor joinig tree als Startbaum durchgeführt.
Die Zahl der Replikate betrug 10. Das Vorliegen konstanter Substitutionsraten (molekulare Uhr) wurde
nicht vorausgesetzt. Da ML sehr zeitaufwendig ist wurde die Suche beendet, wenn sich der -ln L -Wert
über einen längeren Zeitraum nicht mehr verringerte. Der erhaltene Baum wurde mit Astlängen
gespeichert.
Die Optionen der heuristischen Suche für die Maximum Parsimony-Analyse waren: Erhalt des
Startbaumes durch stepwise addition, zufälliges Hinzufügen neuer Sequenzen (addseq=random),
TBR branch swapping, acctran. Die Möglichkeit, Annahmen über die Mutationshäufigkeiten (z.B.
Codonpositionen der Tripletts) in der Analyse zu berücksichtigen, wurde im vorliegenden Fall nicht
angewendet, da der größte Teil der Sequenzinformationen aus nichtkodierenden und nur ein
verschwindend geringer Teil aus kodierenden Regionen stammt. Alle Merkmale wurden in der
Berechnung daher gleich gewichtet (equal weight). Lücken im Alignment (gaps) wurden generell als
„nicht vorhanden“ (missing) gewertet. Die alternative Möglichkeit, d. h. die Definition eines
zusätzlichen Merkmals (newstate) führte aufgrund sehr langer Indels besonders in den Chloroplasten-
Alignments zu fehlerhaften bzw. unwahrscheinlichen Ergebnissen. Positionen mit mehr als einem
möglichen Merkmalszustand in einer Sequenz wurden als unbekannt gewertet (multistate=uncertain).
Alle Bäume wurden als nicht gewurzelt (unrooted) berechnet. Die statistischen Parameter consistency
index und retention index sowie die absolute Länge wurden für die MP-Bäume erhoben.
Bayesianische Statistik Der Maximum Likelihood-Methode verwandt ist die Bayesianische Analyse, die mit der Software
MrBayes (Huelsenbeck & Ronquist 2001) als eine weitere Methode für die phylogenetische
Rekonstruktion zugänglich gemacht wurde und auf dem Bayes-Theorem basiert (Yang & Rannala
1997). ML ermittelt den Stammbaum, der mit der höchsten Wahrscheinlichkeit die Entstehung der
beobachteten Merkmale erklärt, die Bayesianische Analyse hingegen berechnet die
Wahrscheinlichkeit eines Baumes auf Grundlage der Daten. Aus den Daten konstruierte
Baumtopologien (bzw. Likelihood-Werte) werden als Annahmen oder a-priori-Wahrscheinlichkeiten
ermittelt. Ziel ist es, unter Einbeziehung der priors die a-posteriori-Wahrscheinlichkeitsverteilung
(posterior probabilities) zu ermitteln. Da die a-posteriori-Wahrscheinlichkeiten nicht direkt berechnet
werden können, werden sie aus Zufallsstichproben geschätzt, in denen die Bäume proportional zu
ihrer Wahrscheinlichkeit vertreten sind. Dies erfolgt mittels der sogen. Metropolis-coupled Markov-
Chain-Monte Carlo-Simulation (Larget & Simon 1999, Altekar et al. 2004). Die Zustände der Markov-
Ketten sind die phylogenetischen Bäume. Jede Kette startet mit einer willkürlichen Zusammenstellung
an Parameterwerten, Astlängen und Baumtopologien und berechnet eine hohe Zahl an Generationen
(meist 1 Mio. oder mehr). In jeder Generation werden Parameter geändert und neue Baumtopologien
errechnet. Sind diese wahrscheinlicher als die vorangegangene, werden diese zwischengespeichert.
Die Übergangswahrscheinlichkeit zu einem bestimmten neuen Baum wird von dem Verhältnis seiner
Wahrscheinlichkeit zu der seines Vorgängers bestimmt. Am Ende gerät die Kette in einen stationären
Zustand, in dem sie um einen Wert fluktuiert (burn-in-Phase). Zur Vermeidung lokaler Optima (ähnlich

Material und Methoden
44
ML und MP) werden meist vier unabhängige Ketten gerechnet (cold chains, hot chains) die Parameter
austauschen und somit lokale Optima überspringen können. Das Ergebnis der Bayesianischen
Analyse ist ein 50% majority rule-Konsensusbaum, in dem für jede Verzweigung die posterior
probabilities angegeben sind. Diese entsprechen dem Anteil mit dem die betreffende Verzweigung in
allen rekonstruierten Bäumen aufgetreten ist.
Für die Bayesianische Analyse wurde das Programm MrBayes 3.1 (Ronquist & Huelsenbeck 2003)
verwendet. Als Optionen wurden zunächst die Standardoptionen gewählt. Zwei vollkommen
unabhängige Läufe wurden ausgehend von Zufallsbäumen gerechnet. Vier taktweise „erhitzte“ Ketten
wurden für jeden Lauf meist bis zu einer Generationenzahl von 1 Mio. gerechnet, die
Samplingfrequenz betrug 100. Die Rechnung wurde beendet, wenn die Standardabweichung der split
frequencies unter 0,01 gefallen war. Dies ist ein Maß für die Ähnlichkeit der gespeicherten Bäume aus
den beiden unabhängigen Läufen. Bei der Aufsummierung der Ergebnisse (sumt, sump) wurden die
ersten 25% der Bäume verworfen.
Außengruppen und Bewurzelung und Darstellung der Bäume
Phylogenetische Berechnungen nach genannten Prinzipien erzeugen primär ungerichtete
Dendrogramme. Am Beginn der Berechnungen ist auch gar nichts anderes möglich, da der
ursprüngliche Zustand meist nicht bekannt ist und falsche Annahmen des ursprünglichen Zustandes
zu fehlerhaften Berechnungen führen können. Um dennoch eine Richtung festzulegen, gibt es
verschiedene Möglichkeiten. Eine davon ist die Annahme einer konstanten Evolutionsrate (molecular
clock assumption). Unter dieser Annahme muss ein Punkt existieren, von dem aus alle Enden des
Baumes in gleicher Zeit erreicht werden können. Dieser Punkt ist die Wurzel oder der ursprüngliche
Zustand. Diese Annahme liegt dem midpoint rooting zu Grunde, bei dem die Wurzel in die Mitte der
am weitesten entfernten Endglieder gesetzt wird. Da aber oftmals keine konstanten Evolutionsraten
vorliegen, führt die Methode zu falschen Ergebnissen was insbesondere bei Datierungsmethoden die
Ergebnisse verfälschen kann.
Eine weitere Möglichkeit ist die Definition einer Außengruppe, die allen anderen Taxa (als
Innengruppe bezeichnet) als Schwestergruppe gegenübergestellt und eine Polarisierung der
Merkmale erreicht wird (outgroup rooting). Damit ist die Richtung der Aufspaltung festgelegt.
Außengruppe bedeutet jedoch nicht den ursprünglichen Zustand, sondern eine (gedachte) parallele
Ableitung mit der Innengruppe von jenem. Daher kann die Außengruppe auch nach den
Berechnungen festgelegt werden. Gemeinsame Merkmale von Außen- und Innengruppe werden als
Symplesiomorphien bewertet, Merkmale der Innengruppe als potentielle Synapomorphien zur
Beurteilung der Verwandtschaft der Innengruppen-Taxa.
Für alle hier präsentierten Bäume wurde daher die Methode des outgroup rooting verwendet. Die
Darstellung kann mit oder ohne Umsetzung der proportionalen Astlängen erfolgen. In der
entsprechenden Software (PAUP, MEGA) werden dafür die Bezeichnungen Phylogramm und
Kladogramm benutzt. In MP-Bäumen entspricht die Astlänge den Merkmalsänderungen, in ML-
Bäumen den Einzelwahrscheinlichkeiten. Für die Präsentation der Phylogenien wurden Phylogramme,
für die Merkmalsableitung aus Übersichtlichkeitsgründen Kladogramme verwendet.

Material und Methoden
45
Darstellungsweisen mit proportionaler definierter Festlegung der Knotenabstände (Chronogramme)
wurden für die Altersdatierung gewählt.
2.4.4. Molekulare Uhr und Zeitdatierung Die Bestimmung evolutionärer Zeiten gehört zu den wichtigsten Ableitungen aus der phylogenetischen
Rekonstruktion und liefert weitere Erklärungen für Speziations- und Extinktionsereignisse sowie die
Erklärung rezenter Verbreitungsmuster der Organismen. Die Fragen zur Zeitdatierung umfassen das
absolute phylogenetische Alter der rezenten Arten und Verwandtschaftsgruppen, d.h. die Festlegung
des Aufspaltungszeitpunktes von Schwestergruppen (Divergenzzeiten) sowie die Geschwindigkeit
dieser Prozesse, ausgedrückt in Substitutionsraten. Mit diesen Informationen kann der Art oder
systematischen Gruppe neben der morphologischen und biogeographischen eine weitere, die zeitliche
Komponente hinzugefügt werden.
Die zeitliche Datierung von Organismen ist mit großen Unsicherheiten behaftet, da sie relativ kurze
Generationszeiten haben und auf ihre Vorfahren nur induktiv, niemals direkt geschlossen werden
kann. Die Vorfahren sind zudem nur durch lückenhaften, zufällig entstandenen und aufgefundenen
Fossilbestand dokumentiert. In der Geologie wird absolute Altersbestimmung z.B. mit radiometrischen
Methoden durchgeführt, da die Zerfallsraten einiger Isotope bekannt sind. Ein Beispiel dafür ist die
Radiocarbon-Methode, mit der absolute Alter bis etwa 70000 Jahre bestimmt werden können (Stanley
1994: 116). Godwin (1969) beschreibt die Eignung pflanzlicher Fossilien für diese Methode.
Für Organismen wurde neben der Analyse des Fossilbestandes eine weitere Methode entwickelt, die
auf den empirischen Beobachtungen beruht, dass langkettige Makromoleküle wie Proteine oder
Nukleinsäuren sich mit einer bestimmten Rate verändern. Erstmals wurde dies anhand der Evolution
von Hämoglobin nachgewiesen (s. Kap. 1.3.2) und die molecular clock hypothese entwickelt, die von
konstanten Evolutionsraten ausgeht (Zuckerkandl & Pauling 1965). Zahlreiche Untersuchungen an der
DNA zeigten jedoch, dass es keine universale molekulare Uhr sondern zahlreiche lokale Uhren gibt
(Wu & Li 1985). Anhand der Substitutionsraten der DNA können demnach keine absoluten, sondern
nur relative Zeiträume ermittelt werden. Für die absolute Festlegung von Substitutionsraten, d.h.
Kalibrierung der molekularen Uhr, müssen nach wie vor Fossilien oder bedeutende erdgeschichtliche
Ereignisse verwendet werden.
Für Zeitdatierungen auf Basis phylogenetischer Bäume müssen diese gewurzelt sein und
Informationen über die Substitutionsraten, ausgedrückt in Astlängen, enthalten. Mittels likelihood ratio
test und relative rate test (s. Kap. 2.4.5) wird ermittelt, ob auf einem definierten Signifikanzniveau
gleiche Substitutionsraten vorliegen. Werden keine gleichen Substitutionsraten gefunden, die
molecular clock-Hypothese also abgelehnt, können die Astlängen des Baumes nicht mit
Divergenzzeiten gleichgesetzt werden. Weitere Schritte sind dann erforderlich, um modellhaft die
unterschiedlichen Substitutionsraten anzugleichen. Dafür wurden mehrere Verfahren entwickelt, unter
anderem Nonparametric Rate Smoothing (NPRS, Sanderson 1997) und Penalized Likelihood (PL,
Sanderson 2002). Mit den angeglichenen Substitutionsraten können ultrametrische Bäume erzeugt
werden, in denen die Knotenabstände das relative Alter der Linien repräsentieren. Zur Bestimmung
der absoluten Zeiten müssen ein oder mehrere Knoten zeitlich festgelegt werden (Kalibrierung). Dies

Material und Methoden
46
erfolgt gewöhnlich mit dem ältesten Fossil, das der Gruppe zugeordnet werden kann. Mit der
Kalibrierung wird das Minimalalter der Knoten festgelegt.
Zur Altersdatierung wurde in Anlehnung an die Arbeiten von Kadereit et al (2003, 2006) jeweils ein
atpB-rbcL- und ein ITS-Baum berechnet, die einige zusätzliche Sequenzen enthalten (s. Kap. 3.3). Für
die phylogenetische Rekonstruktion wurde der Methode Maximum Likelihood der Vorzug gegeben, da
sich die jeweiligen Ergebnisse zwischen ML und MP nicht grundlegend unterscheiden und mit ML im
Gegensatz zu MP nur eine einzige wahrscheinlichste Baumtopologie erzeugt wird, die für die
Datierung der Divergenzereignisse genutzt werden kann. Die erhaltenen Bäume wurden mit einer
vorher bestimmten Außengruppe gewurzelt. Mit den Ergebnissen des relative rate tests und des
likelihood ratio tests wurde entschieden, ob weitere Glättungsmethoden erforderlich sind. Die
korrigierten Distanzen wurden zur Konstruktion ultrametrischer Bäume benutzt, in denen Knoten
kalibriert wurden.
Penalized likelihood Die von Sanderson (2002) entwickelte Methode ist ein Verfahren zur Angleichung stark divergierender
Substitutionsraten. PL wurde in der vorliegenden Arbeit angewandt, weil die beiden Tests auf
Ratenkonstanz signifikante Unterschiede nachwiesen und mit PL Datierungsereignisse in
benachbarten Gruppen vorgenommen wurden (Salicornioideae: Kadereit et al. 2005, 2006). Dadurch
sind direkte Vergleiche möglich. PL belegt jede individuelle Substitutionsrate mit einer Strafkorrektur,
dem Glättungsparameter. Dieser beeinflusst direkt die Substitutionsraten und damit das Alter der
Knoten.
Verwendet wurde das Programm r8s v. 1.7 (Sanderson 2004). Da die Kalibrierungspunkte als
Intervalle vorgegeben wurden („constrained“), wurde als Methode Penalized Likeilhood mit dem
truncated-Newton (TN)-Algorithmus gewählt. In einem ersten Schritt wurde zunächst der optimale
Glättungsparameter mittels Kreuzvalidierung bestimmt. Dazu wurden die Kalibrierungspunkte für die
jeweiligen Knoten vorgegeben. Die Anzahl unterschiedlicher Werte für den Glättungsparameter betrug
dabei 8, die Abstände jeweils 0,5 im Exponent (100 -103). Der optimale Glättungsparameter (als log-
Wert) wurde aus der von r8s generierten Übersicht für den minimalen Vorhersagefehler übernommen.
Mit diesem Glättungsparameter wurde in einem zweiten Schritt das eigentliche PL-Verfahren
durchgeführt und die Alter aller Knoten rekonstruiert.
Kalibrierung Für die Kalibrierung der Divergenzzeiten innerhalb der Suaedoideae wurde auf die umfangreichen
Vorarbeiten innerhalb der Chenopodiaceae zurückgegriffen (Hohmann et al. 2006, Kadereit et al.
2003, 2005, 2006). Einige wichtige Kalibrierungspunkte der Chenopodiaceae und Salicornioideae
wurden übernommen, da für Suaeda keine eigenen Fossilfunde vorliegen. Folgende Fossilien bzw.
Datierungspunkte wurden übernommen und für die eigenen Untersuchungen vorgegeben:
1. Pollen von Chenopodiopollis multiplex aus dem Paläozän (65-56,5 Mio Jahre; USA; Nichols &
Traverse 1971): Wurzel der Chenopodioideae
2. Salicornites massalongoi aus dem Oligozän (35,4-23,3 Mio Jahre, Italien, Collinson et al. 1993):
Salicornioideae mit gegenständiger Beblätterung

Material und Methoden
47
Als zusätzlicher Kalibrierungspunkt für den atpB-rbcL-Stammbaum wurde der aus rbcL-Daten
abgeleitete Zeitpunkt für die Abspaltung der Salsoloideae von den Suaedoideae/Salicorioideae
übernommen (41,3-37,6 Mio. Jahre). Entsprechend der bei Kadereit et al. (2006) beschriebenen
Methode wurde für die absolute Datierung „Krone“ und „Stamm“ der kalibrierten Linie zeitlich bestimmt
und damit ein Intervall definiert. Genauere Aussagen sind nicht möglich. Mit dem in der
Kreuzvalidierung bestimmten Glättungsparameter und den absoluten Zeitangaben wurden die
Chronogramme mit den datierten Knoten erzeugt und Substitutionsraten ermittelt.
2.4.5. Test- und Überprüfungsverfahren
bootstrapping Eine häufig genutzte Methode zur Beurteilung der Robustheit eines Kladogrammes ist das
bootstrapping. Angewendet auf Sequenzdatensätze wird bei dieser Methode aus einem vorhandenen
Datensatz durch zufällige Merkmalsauswahl ein neuer Datensatz gleicher Länge konstruiert und
daraus ein Stammbaum rekonstruiert. Dabei werden Merkmale auch verdoppelt, andere nicht
berücksichtigt. Dieser Vorgang wird mehrfach wiederholt (mindestens 500, meist 1000 oder mehr).
Der prozentuale Anteil des Vorhandenseins einer Verzweigung in allen Bäumen stellt den bootstrap-
Wert dar und ist ein Maß für die Robustheit der bezeichneten Verzweigung. Da bootstrap mit
Zufallsdatensätzen arbeitet für die es kein Evolutionsmodell gibt, wird als Rekonstruktionsmethode
Maximum Parsimony verwendet.
Über die Beurteilung der absoluten Werte existieren unterschiedliche Auffassungen. Dargestellt
werden Werte oberhalb 50%, als gut gestützt gelten aber nur Verzweigungen mit Werten über 70 bzw.
90% (Felsenstein 1985, Efron et al. 1996).
Der Nachteil liegt bei der Beurteilung von Verzweigungen die zwar sicher, aber durch wenige oder nur
ein gemeinsames Merkmal gestützt sind. Diese Verzweigungen werden durch bootstrapping meist
unterbewertet.
Das bootstrapping für alle Datensätze wurde mit PAUP durchgeführt. Die Zahl der Replikate betrug in
der Regel 1000, die Zwischenspeicherung von Bäumen wurde auf 100 begrenzt, um den Zeitaufwand
in Grenzen zu halten. Bootstrap-Werte über 50% wurden in die MP-Bäume übertragen.
partition-homogeneity test Der partition-homogeneity test (Farris et al 1994) vergleicht die phylogenetische Information von
Teildatensätzen und wird zu Beurteilung möglicher Abweichungen verwendet. Es ist eine Teststatistik,
mit der die Nullhypothese (Partitionen ungleich) bestätigt oder verworfen werden soll. Dazu werden
aus den vorhandenen Daten künstliche Partitionen erstellt, daraus Phylogenien errechnet und
verglichen. Der erhaltene Testwert p beschreibt den Anteil kürzerer oder gleich langer Bäume im
Vergleich zu jenen aus den Originalpartitionen. Der prozentuale Wert dient zugleich als
Signifikanzniveau, auf dem die Nullhypothese (Partitionen ungleich) bestätigt oder verworfen wird. Ist
der p-Wert beispielsweise höher als 0,01, wird die Unterschiedlichkeit auf 99%-Signifikanzniveau

Material und Methoden
48
abgelehnt. Partitionen können dann kombiniert werden, wodurch sich das phylogenetische Signal
verbessern sollte.
Der partition-homogeneity test wurde mit PAUP für die Teildatensätze (ITS, atpB-rbcL, psbB-psbH)
getrennt und teilweise kombiniert (Chloroplast/ITS) durchgeführt. Die Zahl der Replikate betrug jeweils
100. Als Rekonstruktionsverfahren wurde wegen der hohen Anzahl an Taxa Maximum Parsimony
gewählt. Die Ergebnisse wurden für die Entscheidung über mögliche Kombination von DNA-Regionen
herangezogen.
Relative rate test Der relative rate test ist eine Methode um zu testen, ob ein Molekül in zwei unabhängigen Linien mit
der gleichen Rate evolviert. Er wurde erstmals für den Ratenvergleich der Proteinevolution entwickelt
(Sarich & Wilson 1973) und später für den Vergleich von DNA-Substitutionsraten genutzt (Wu & Li
1985). Es handelt sich um einen paarweisen Vergleich in dem getestet wird, ob die Substitutionsraten
eines Moleküls a bezogen auf ein Referenzmolekül c gleich sind mit der Substitutionsrate eines
Moleküls b (bezogen auf die gleiche Referenz). Die Unterschiede werden auf Signifikanz geprüft.
Dafür wird ein t-Test für den Vergleich unabhängiger Mittelwerte eingesetzt. Unterscheiden sich die
Werte signifikant (95% Signifikanzniveau), wird die Nullhypothese, dass beide Werte gleich sind,
verworfen. Beide Linien evolvieren dann mit unterschiedlichen Raten. Der relative rate test kann nur
prüfen, ob beide Linien mit der gleichen Rate evolvieren. Es kann keine Aussage über konstante
Evolutionsraten getroffen werden, was für die Evaluierung der molecular clock-Hypothese
entscheidend ist.
Der relativ rate test zur Abschätzung der Ratenunterschiede wurde auf die Datensätze zur
Altersdatierung (ITS, atpB-rbcL, vgl. Kap. 3.3) angewandt. Verwendet wurde dazu das Programm
RRTree (Robinson-Rechavi & Huchon 2000), mit dem der Test anhand eines gegebenen
phylogenetischen Baumes durchgeführt werden kann. Die Distanzberechnung erfolgte mit der Kimura-
2-Parameter-Methode. Als Referenzbaum wurde der für die Alterdatierung rekonstruierte Maximum-
Likelihood-Baum gewählt. Sind die errechneten „exakten Wahrscheinlichkeiten“ größer als das
gegebene Signifikanzniveau (0,05), ist der Ratenunterschied nicht signifikant. Beide Sequenzen
evolvieren dann mit der gleichen Rate.
likelihood ratio test Mit dem likelihood ratio test (Felsenstein 1981) in Phylogenien wird überprüft, ob ein komplexeres
Modell gegenüber einem einfacheren Modell die Wahrscheinlichkeit einer Baumtopologie signifikant
verbessert. Er entspricht einem chi-Quadrat-Anpassungstest zur Bewertung der Übereinstimmung
zwischen zwei alternativen Hypothesen. Verglichen werden die Likelihood-Werte aus der Analyse. Er
ist nur gültig, wenn hierarchisch verknüpfte Modelle verglichen werden. Dies liegt vor, wenn sich das
komplexere Modell von dem einfacheren nur durch einen oder mehrere Parameter unterscheidet. Das
Hinzufügen von weiteren Parametern sollte einen höheren Likelihood-Wert bewirken, die Erhöhung ist
aber ab einem bestimmten Punkt nicht mehr signifikant.

Material und Methoden
49
Die Formel für den likelihood ratio test lautet: LR = 2*(lnL1-lnL2), wobei L1=Likelihood der
Nullhypothese (einfaches Modell) und L2=Likelihood der Alternativhypothese (parameterreicheres
Modell) sind. Berücksichtigt werden muss noch die Anzahl der Freiheitsgrade. Mit der erhaltenen
Differenz kann durch Vergleich mit Tabellenwerten auf einem definierten Signifikanzniveau (95%)
ermittelt werden, ob signifikante Unterschiede bestehen. Ist dies der Fall, ist das komplexere Modell
zu bevorzugen. Der LRT wurde in folgenden Untersuchungen angewendet:
1. Auswahl des optimalen Evolutionsmodells für die ML-Analyse
In diesem Fall wird mittels likelihood ratio test geprüft, ob ein komplexes Evolutionsmodell die
Wahrscheinlichkeit einer Baumtopologie gegenüber einem einfacheren Modell signifikant erhöht. Ist
der Unterschied signifikant, so ist das komplexere Modell zu bevorzugen. Der Test wurde mit dem
Programm Modeltest 3.7 durchgeführt.
2. Test auf Annahme gleicher Substitutionsraten (molekulare Uhr)
Geprüft wird bei dieser Anwendung auf signifikante Unterschiede in den Wahrscheinlichkeitswerten
zweier phylogenetischer Bäume mit identischen Evolutionsmodellen und Topologien unter Annahme
einer molekularen Uhr oder Zulassung variabler Substitutionsraten. Unter der Nullhypothese ist der
phylogenetische Baum gewurzelt und die Substitutionsraten werden als gleich vorausgesetzt. Daher
ist dies das einfachere Modell. Unter der alternativen Hypothese kann die Substitutionsrate jedes
Astes unabhängig variieren. Die Modelle unterschieden sich um 2 Parameter. Die Anzahl der
Freiheitsgrade ist bei dieser Anwendung mit s-2 festgelegt, wobei s die Anzahl der Taxa ist. Ist der
Unterschied zwischen den alternativen Hypothesen signifikant, wird die Nullhypothese konstanter
Substitutionsraten abgelehnt. Eine Gleichsetzung von Substitutionsraten und Divergenzzeiten ist dann
unzulässig und es müssen für die Altersdatierung Glättungsmethoden wie z. B. penalized likelihood
angewendet werden. Die Aussage über Annahme oder Verwerfen der molecular clock-Hypothese
unterscheidet sich gegenüber dem relative rate test insoweit, dass bei dem LRT der gesamte
Datensatz bewertet wird und nicht nur einzelne Sequenzpaare. Die Ergebnisse beider Tests können
somit gegensätzliche Aussagen liefern.
Die likelihood-Werte mit und ohne Annahme einer molekularen Uhr wurden mit der Maximum
Likelihood-Methode in PAUP ermittelt und anschließend die Differenzen beider Werte gebildet. Der
kritische Wert der chi-Quadrat-Verteilung wurde mit der CHIINV-Funktion in Microsoft EXCEL XP für
die jeweilige Anzahl an Freiheitsgraden für das gegebene Signifikanzniveau berechnet und mit dem
erhaltenen Differenzwert verglichen. Wenn der Differenzwert größer war als der kritische Wert chi-
Quadrat-Wert wurde damit der Nachweis inhomogener Substitutionsraten erbracht.
2.5. Bewertung morphologischer Merkmale Morphometrische Untersuchungen und deren statistische Auswertung wurden nicht durchgeführt. Die
Beurteilung der Merkmale erfolgte nach den Angaben in der entsprechenden floristischen Literatur,
anhand von Originalbeschreibungen sowie durch Untersuchungen an Herbar- und Lebendmaterial
visuell und beschreibend.

Material und Methoden
50
Für die Charakterisierung der Arten wurden vor allem folgende Merkmale berücksichtigt: Struktur des
Perianths im fruchtenden Zustand, Samengröße, Ovarform, Narbenanzahl, Verzweigung,
Internodienlänge, Verholzungsgrad, Lebenszyklus, Länge der vegetativen Phase und Blühdauer. Die
Blattmerkmale mit den spezifischen Gewebetypen wurden überwiegend aus der Literatur
übernommen. Aus der Literatur bzw. unveröffentlichten Daten (Lomonosova, Freitag, mdl.) stammen
auch die Angaben über die Chromosomenzahlen (Ebrahimzadeh et al. 1994, Ferren & Schenk 2008,
Freitag & Lomonosova 2006, Freitag et al. 1996, Lomonosova & Krasnikov 1993, Lomonosova et al.
2008, Subramanian 1988, Krahulcová & Tomšovic 1997).
Neben der Beurteilung der einzelnen Merkmale wurde eine Generalisierung angestrebt, um Gruppen
definieren zu können. Anhand der von der Merkmalsentwicklung unabhängigen molekularen
Phylogenien nichtkodierender Regionen wurden als Resultat charakteristische Merkmale dargestellt.
Da es sich nicht um diskrete Merkmale sondern meist um Merkmalskomplexe handelt, wurde auf
statistische Verfahren zu Merkmalsrekonstruktion und Rekonstruktion ancestraler Zustände verzichtet.
Diese Untersuchungen bleiben einer erweiterten morphologischen Studie vorbehalten.

Ergebnisse
51
Abb. 6: Nachweis von genomischer DNA aus der CTAB-Isolation von unterschiedlichen Geweben mittels Agarosegel-Elektrophorese. Auftrag jeweils 5µl DNA-Lösung; Vergleichstandard (rechts): 2, 5, 10, 20, 40 ng/µl (Auftragsmenge 5µl)
3. Ergebnisse 3.1. Ausprägung und Variabilität der molekularen Merkmale 3.1.1. DNA-Isolation, Amplifikation und Sequenzierung Beide verwendeten Methoden, CTAB-Verfahren und Qiagen-Extraktionskit, eigneten sich zur
erfolgreichen Isolation hochmolekularer DNA aus pflanzlichem Gewebe. Da in den untersuchten
Pflanzen offenbar keine störenden Sekundärstoffe vorkommen und auch der hohe Gehalt an NaCl im
pflanzlichen Gewebe den Isolationsprozess nicht beeinträchtigt, war die Qualität der DNA nicht von
der Isolationsmethode, sondern vom Konservierungszustand des Pflanzenmaterials abhängig. Die
qualitativ beste, hochmolekulare DNA wurde aus Frischmaterial, CTAB-konserviertem und in Silicagel
getrocknetem Material erhalten, während die DNA aus luftgetrocknetem Herbarmaterial meist stärker
degradiert, d. h. zu kürzeren Fragmenten abgebaut ist. Ein Grund dafür ist der durch den
Sukkulenzgrad stark verzögerte Trocknungsprozess, während dessen DNAsen die langkettigen
Moleküle effektiv abbauen können. Die folgenden Elektrophorese-Bilder verdeutlichen beispielhaft die
Ergebnisse der DNA-Isolation und erlauben Rückschlüsse auf deren Qualität.
Hochmolekulare DNA verhält sich in der Elektrophorese hinsichtlich der Laufeigenschaften wie der
Vergleichsstandard, während die Fragmente degradierter DNA aufgetrennt werden. Dies zeigt sich
z.B. in Abb. 6 (linkes Bild; 1., 3. und 4. Bahn). Der Hauptteil der DNA besteht aus sehr kurzen
Fragmenten, längere Moleküle sind nur noch in sehr geringen Konzentrationen vorhanden. Aus DNA
dieser Qualität sind mittels Standard-PCR kaum längere Fragmente amplifizierbar. Verglichen mit dem
Mengenstandard ergibt sich damit unter idealen Bedingungen eine DNA-Ausbeute von bis zu 8 µg aus
150 mg eingesetztem Frischmaterial. Oftmals waren die erzielten Mengen jedoch geringer.

Ergebnisse
52
3.1.2. Strukturmerkmale der untersuchten DNA-Regionen Sequenzdaten wurden für ITS, atpB-rbcL und psbB-psbH erzeugt. Nicht für alle Akzessionen wurden
alle drei Sequenzen erhalten, da vor allem die Chloroplasten-Sequenzen nicht für alle Sippen
amplifizierbar war. Für einen direkten Vergleich der Parameter wurde ein weitgehend kongruenter
Datensatz aus 74 Sequenzen zusammengestellt, anhand dessen die Merkmale zwischen den
unterschiedlichen Regionen verglichen wurden. Das Alignment umfasst die 66 Sequenzen der
Suaedoideae aus Kap. 3.2.3 sowie 8 Außengruppen-Sequenzen aus Kap. 3.2.1 und enthält damit 74
Sequenzen. Durch diese Zahl ergibt sich ein repräsentativer Überblick. Die in den molekularen
Phylogenien gefundenen Gruppierungen werden bei der Merkmalsbewertung berücksichtigt. Die
Innengruppe besteht damit aus den Gattungen Suaeda, Alexandra und Borszczowia (s.
nachfolgenden Text).
Die gesamte ITS-Region besitzt eine durchschnittliche Länge von etwa 630 bp. Aus stärker
degradiertem Pflanzenmaterial wurde die Sequenz in zwei getrennten Reaktionen erzeugt und ITS1-
und ITS2-Sequenzen zu einer einzigen kombiniert. Fast alle Sippen haben individuelle ITS-
Sequenzen, identische kommen praktisch nicht vor. Alle Nukleotide waren zweifelsfrei zu
identifizieren, Hinweise auf Sequenzunterschiede zwischen den Paralogen wurden nicht gefunden.
ITS weist innerhalb der untersuchten Gruppe eine vergleichsweise geringe Längenvariabilität auf. Die
längsten ITS1-Sequenzen haben Kalidium foliatum (249bp), Suaeda ifniensis und S. carnosissima
(245bp), die kürzesten S. prostrata (235bp). Die längsten ITS2-Sequenzen haben Bienertia cycloptera
(232bp) und Suaeda fruticosa (229bp), die kürzesten Tecticornia australasica (217bp) und Suaeda
linifolia (223). Die 5,8 S-Region hat für alle Taxa die gleiche Länge von 164bp. Die Längen möglicher
Alignments liegen je nach Wahl der Parameter im Größenbereich zwischen 674 (minimal) und 690
Positionen. Die Variabilität ist ungleich verteilt. Weitgehend einheitlich ist die kodierende 5,8 S-Region.
Hoch variabel hingegen sind ITS1 und ITS2, wobei es auch in den spacer-Regionen relativ
konservierte Regionen gibt, was mit der Sekundärstrukturbildung zusammenhängt. Die hoch variablen
Abschnitte sind demnach die loop-Bereiche, die am wenigsten evolutionären Zwängen unterliegen.
Indels erreichen bei Wahl eines „engen“ Alignments im Durchschnitt 1-4 bp Länge, in dem besonders
längenvariablen ITS2 bis zu 8 bp. Dies verursacht wiederum eine sehr hohe Zahl an Fehlpaarungen
der Nukleotide, die als Merkmalstransformationen für die Berechnungen verwendet werden. Durch
diese Einstellungen des Alignments erreichen die paarweisen Distanzen enorm hohe Werte von 40-
50% Unterschied. Demgegenüber steht eine Variabilität in der 5,8 S-Region von etwa 6%. Daraus
wird ersichtlich, dass die Ergebnisse der molekularen Phylogenie vorwiegend auf der Variabilität der
nichtkodierenden Regionen beruhen und durch geringe Veränderungen der Alignmentparameter
beeinflusst werden können. Um den Fehler aus zu hoher Variabilität etwas zu verringern wurden die
detaillierten phylogenetischen Berechnungen mit ITS (s. Kap. 3.2.2) mit nur einer Sequenz als
Außengruppe durchgeführt.
ITS2 ist in Teilen so variabel, dass an diesen Stellen ein zweifelsfreies Alignment nicht möglich ist. In
hochpolyploiden Genomen (10x) bricht die Sequenzierreaktion an dieser zudem noch GC-reichen
Region ab. Die nachfolgende Grafik verdeutlicht dieses Phänomen.

Ergebnisse
53
TGCA
T
C
T
C
C
C
C
C
C
A
T
C
C
T
G
C
A
Abb. 7: Ausschnitt aus dem Alignment einer GC-reichen, hoch variablen Region des ITS2-Alignments der Suaedoideae. 1 Sequenzalignment mit uneindeutiger Homologisierung von Basenpositionen; 2 Abbruch der Sequenzierreaktion an dieser Region in dekaploiden Genomen (Suaeda kulundensis, 2n=90)
Abb. 8: Ausschnitt aus dem atpB-rbcL-Alignment ausgewählter Taxa der Suaedoideae. Dargestellt ist der mehrfach wiederholte polyA-Microsatellit bei Suaeda eltonica und Alexandra lehmannii,; kennzeichnendes Merkmal der atpB-rbcL-Region in der Sektion Schoberia.
1 2
Als sehr längenvariabel mit Unterschieden von bis zu 300 bp in absoluten Längen erwies sich die
atpB-rbcL-Region Die Amplifikation dieser Region war z.T. mit erheblichen Schwierigkeiten
verbunden, da atpB-rbcL zwei längenvariable Mikrosatelliten enthält, an der die Strangsynthese
während der PCR nicht korrekt ausgeführt wird oder gänzlich abbricht. Die Sequenzinformationen
beruhen daher meist nicht auf der Synthese beider Komplementärsequenzen. Für einen Teil der
Sippen konnten keine Sequenzen erhalten werden, da das Ausgangsmaterial zu stark degradiert war.
Am kürzesten ist der Bereich bei Alexandra lehmannii (494 bp) sowie einigen Arten der Sektion
Salsina (S. asphaltica, S. vermiculata, S. aegyptiaca, S. arcuata), am längsten bei Suaeda linifolia
(793 bp) und Salicornia europaea (799 bp). Das Alignment hat eine Länge von 1022 bp und ist nur
durch Bildung langer Indels konstruierbar. Die Indels, die z.T. synapomorphen Charakter tragen,
gleichen die verkürzten Sequenzen aus und erreichen Längen von mehreren hundert bp. Diese
Bereiche fallen für die Bewertung von Merkmalstransformationen aus. In etwa 75 bp Entfernung vom
Ende des rbcL-Gens haben alle Sequenzen eine Mikrosatellitenstruktur (poly-A), die in
Größenordnungen von 8 - 16 Wiederholungen längenvariabel ist. Bei den Arten der Sektion Schoberia
und Alexandra ist dieses Mikrosatellitenmotiv viermal wiederholt und bildet einen Minisatelliten. PCR
und Sequenzierung brechen an dieser Region meist ab.

Ergebnisse
54
Die aus der psbB-psbH-Region generierten Sequenzen umfassen zwei Genabschnitte (psbT, 102
bp; psbN, 68 bp) und drei intergenische spacer (psbB-psbT, psbT-psbN, psbN-psbH) inklusive 41 bp
des psbB-Gens. Die Längenvariabilität ist wesentlich geringer als bei atpB-rbcL und beschränkt sich
auf die spacer-Regionen. Noch stärker als bei atpB-rbcL zeigen einige Indels synapomorphen
Charakter und kennzeichnen verwandte Gruppen. Die längste Sequenz hat Suaeda altissima (636
bp), die kürzesten S. glauca (607 bp) und Bassia hyssopifolia (600 bp). Längere Mikrosatelliten
kommen nicht vor. Die Variabilität der Genregionen beträgt weniger als 50% der spacer-Regionen und
beruht auf Punktmutationen. Merkmal beider Chloroplasten Regionen ist die im Vergleich zu ITS wesentlich geringere Variabilität.
Die Distanzwerte erreichen durchschnittlich nur ein Drittel derjenigen von ITS. Viele Sippen mit
unterschiedlichen ITS-Sequenzen besitzen identische Chloroplastensequenzen. Die nachfolgende
Tabelle gibt einen Vergleich der Sequenzmerkmale, Alignmentparameter und Merkmalsbewertung für
die Maximum Parsimony-Analyse für alle drei untersuchten DNA-Regionen.
Tab. 6: Sequenzmerkmale und Alignmentcharakteristika für einen vergleichbaren Datensatz aus 74 Sequenzen der Suaedoideae, Salicornioideae und Bassia hyssopifolia. Werte gruppiert nach verwandten Gruppen der molekularen Phylogenien (s. Text); ti/tv: Verhältnis von Transitionen zu Transversionen)
atpB-rbcL psbB-psbH ITS1 5.8S ITS2 ITS
Suaeda, Alexandra, Borszczowia
Länge Alignment 1022 684 266 164 246 276
Längenvariabilität 494-799 600-636 235-249 164 217-232 615-643
variable Pos. 185 8 160 16 116 292
parsimony informativ 103 60 125 9 89 223
GC-Gehalt 27,9 32 60,5 54,8 62,5 59,7
ti/tv 0,55 0,76 1,67 2,59 1,73 1,58
K2P-Divergenz
Mittelwert % 3,7 2,9 22,9 1,8 15,4 14,0
Variationsbereich % 0 - 10,0 0 - 6,2
0 - 47,3 0 - 6,4 0 - 30,0 0 - 25,2
Suaedoideae, Salicornioideae, Bassia
Längenvariabilität
variable Pos. 282 143 191 20 153 364
parsimony informativ 147 84 155 12 118 285
GC-Gehalt (Mittelwert in %) 28,1 32,9 60,3 54,9 62,6 59,7
ti/tv 0,57 0,79 1,68 2,19 1,64 1,53
K2P-Distanzen
Mittelwert % 4,52 3,37 27,4 1,9 18,0 16,0
Variationsbereich % 0-12,4 0-9,36 0-51,5 0 - 6,4 0 - 40,2 30,8

Ergebnisse
55
3.2. Phylogenetische Rekonstruktion 3.2.1 Stellung der Suaedoideae innerhalb der Chenopodiaceae Die insgesamt recht umfangreichen und sehr variablen Sequenz-Datensätze machen zur Klärung der
unterschiedlichen Fragestellungen differenzierte Berechnungsverfahren notwendig. Um die
phylogenetische Positionen der Suaedoideae innerhalb der Chenopodiaceae festzulegen, wurden
zunächst mit einer reduzierten Auswahl von 22 Taxa die Berechnungen durchgeführt. Zu dieser
Auswahl gehören jeweils die Typus-Sippe aller Sektionen von Suaeda nach Schenk & Ferren (2001),
Alexandra, Borszczowia und Bienertia als weitere Gattungen der Suaedoideae, ausgewählte Vertreter
der verwandten Unterfamilie Salicornioideae sowie Bassia hyssopifolia als Außengruppe. Da für
Suaeda taxifolia keine zweifelsfreien Daten vorlagen, wurde stattdessen S. divaricata als Vertreter der
Sektion Limbogermen verwendet. Mit diesen Ergebnissen können die detaillierteren Analysen der
Suaedoideae (Kap. 3.2.2, 3.2.3) mit einer minimalen Zahl an Außengruppen weitergeführt werden, um
die aus den z.T. extremen Divergenzwerten besonders bei ITS resultierenden Fehler zu minimieren.
Die für die ausgewählten Taxa erzeugten Alignments haben folgende Längen: atpB-rbcL 954 bp,
psbB-psbH 670bp, ITS 658 bp. Aufgrund von Unsicherheiten wurden 62 bp des atpB-rbcL-Alignments
und 8 bp des psbB-psbH-Alignments aus der weiteren Berechnung ausgeschlossen.
Ein Test auf signifikante Heterogenität zwischen den Regionen erfolgte mittels partition-homogeneity
test nach der in Kap. 2.3 beschriebenen Methodik. Der Test ergab folgende Werte: atpB-rbcL vs.
psbB-psbH P=0,62; atpB-rbcL/psbB-psbH vs. ITS P=0,01. Signifikante Heterogenität auf 99%-
Signifikanzniveau liegt daher nur zwischen den cp-Regionen und ITS vor. Die cp-Daten konnten daher
kombiniert werden, ohne dass es zu Veränderung der phylogenetischen Aussagen kommen würde.
ITS und psbB-psbH weisen sowohl kodierende als auch nichtkodierende Bereiche auf. Die
unterschiedlichen funktionalen Bereiche wurden bei den Berechnungen nicht in Partitionen unterteilt,
da die kodierenden Regionen nicht oder nur sehr wenig variabel sind und einen geringen Einfluss auf
die resultierenden Phylogenien haben. Die Berechnungen wurden mit den Methoden Maximum
Parsimony und Maximum Likelihood durchgeführt.
Von insgesamt 1624 bp des cp-Alignments waren 328 bp variabel und von diesen 121 bp parsimony-
infomative. Die MP-Analyse aus diesen Daten resultiert in 3 kürzesten Bäumen mit einer Länge von
429 Schritten (CI=0,69, RI=0,86), die sich nur geringfügig unterscheiden. Einer der Bäume ist in Abb.
9 B dargestellt. Als optimales Evolutionsmodell für die ML-Analyse wurde von Modeltest K81uf+G mit
folgenden Parametern ausgewählt: Nukleotidfrequenzen A=0,3271, C=0,1461, G=0,1455, T=0,3812;
Substitutionsraten AC=1 AG=1,3564 AT=0,2969 CG=0,2969 CT=1,3564 GT=1; Anteil invariabler
Positionen: 0, Merkmalsverteilung: Gamma, Formparameter: 0,6825. Die Analyse ergab genau einen
wahrscheinlichsten Baum mit einem -ln L 4670,7418. Die Topologie entspricht einem kürzesten MP-
Baum und wird daher nicht gesondert dargestellt.
Von den 658 bp des ITS-Alignments waren 320 bp variabel und von diesen 224 bp parsimony-
infomative. Die MP-Analyse resultierte in genau einem kürzesten Baum von 857 Schritten Länge
(CI=0,51 RI=0,63). Diese Tatsache ist in Anbetracht der hohen Divergenzwerte bemerkenswert. Als
optimales Evolutionsmodell für die ML-Analyse wurde von Modeltest TrN+G mit folgenden Parametern
ausgewählt: Nukleotidfrequenzen A=0,2523, C=0,2510, G=0,2507, T=0,2460; Substitutionsraten
AC=1 AG=1,7147 AT=1 CG=1 CT=3,9685 GT=1; Anteil invariabler Positionen: 0, Merkmalsverteilung:

Ergebnisse
56
Gamma, Formparameter: 0,3957. Die Analyse ergab genau einen Baum mit einem -ln L von
4812,815. Die Baumtopologie unterscheidet sich an mehreren Stellen, die im Text gesondert
beschrieben werden.
Die erhaltenen Phylogramme lassen folgende grundlegende Aussagen über die Phylogenie der
Suaedoideae zu:
1.) Im Vergleich der beiden Baumtopologien ist festzustellen, dass die untersuchten DNA-Regionen
aus Kern und Chloroplasten sehr ähnliche Aussagen liefern. Dies ist für die Beurteilung der
Verwandtschaftsverhältnisse von entscheidender Bedeutung.
2.) Mit Bassia hyssopifolia (Salsoloideae) als Außengruppe sind Suaedoideae und Salicornioideae in
der Umgrenzung von Kühn et al. (1993) Schwestergruppen. Dieser Befund stimmt mit der
phylogenetischen Analyse der Chenopodiaceae und Amaranthaceae (Kadereit et al. 2003) überein.
Auffällig ist die relativ geringe statistische Unterstützung dieser Schwestergruppenbeziehung und der
Monophylie von Suaeda im ITS-Baum (bootstrap 72%). Dies liegt sicherlich in der z.T. enorm hohen
Variabilität der ITS-Region begründet, die für das gewählte taxonomische Niveau wahrscheinlich
Fehler verursacht. Die weniger variablen cp-Regionen unterstützen die Monophylie mit 98% deutlich.
3.) Bemerkenswert ist die enorme Diversität innerhalb der Suaedoideae bzw. der Gattung Suaeda, die
sogar diejenige der benachbarten Unterfamilie Salicornioideae bei weitem übersteigt. Die relative
Uniformität in der Morphologie ließ dies kaum erwarten.
4.) Suaeda ist die artenreichste Gattung der Suaedoideae und bildet mit den monotypischen
Gattungen Alexandra und Borszczowia eine monophyletische Gruppe. Dies ist ein aus
morphologischer Sicht völlig unerwartetes Resultat, jedoch eindeutig und durch alle
Berechnungsverfahren gestützt. Die Unterschiede in Blatt- und Blütenmorphologie rechtfertigten
bisher die Abgrenzung als eigenständige, wenngleich zu Suaeda nahe verwandte Gattungen. Beide
Arten erscheinen in der molekularen Phylogenie als separate Entwicklungen aus einer in diesem Fall
paraphyletischen Gattung Suaeda.
5.) Die Gattung Bienertia mit nach neuesten Studien von Akhani et al. (2005) zwei Arten nimmt eine
Zwischenstellung zwischen Suaedoideae und Salicornioideae ein. Sie gehört eindeutig nicht zu der
aus Suaeda, Alexandra und Borszczowia gebildeten monophyletischen Einheit. ITS-Daten weisen
Bienertia als Schwestergruppe der Salicornioideae aus, nach den Chloroplasten-Daten hingegen ist
Bienertia Schwestergruppe zu dem von Suaeda, Alexandra und Borszczowia geformten Clade. Beide
Zuordnungen werden jeweils auch durch relativ hohe bootstrap-Werte gut gestützt. Für ITS beträgt die
Unterstützung 72%, für die Chloroplastendaten sogar 100%.
6.) Die wichtigste Unterteilung der Gattung Suaeda erfolgt zwischen S. heterophylla (Sektion Brezia)
und allen übrigen Sektionen einschließlich Alexandra und Borszczowia. Brezia erscheint gegenüber
der restlichen Gattung als stark isolierter Zweig, was durch sehr lange Äste ausgedrückt wird. Die

Ergebnisse
57
starke Separation von den übrigen Suaeda-Arten führt zu der vergleichsweise geringen Bootstrap-
Unterstützung im ITS-Baum. Die Distanz zwischen Brezia und den übrigen Gruppen von Suaeda ist
wesentlich höher als zwischen unterschiedlichen Gattungen der Salicornioideae.
7.) Die verbleibende Gruppe innerhalb von Suaeda lässt sich in mehr basal stehende und stärker
abgeleitete Arten gliedern. Basal und am weitesten isoliert steht in jedem Fall S. glauca. Zu den
basalen Arten gehören weiterhin S. vera, S. linifolia und Borszczowia aralocaspica. Die Beziehungen
zwischen diesen Arten werden durch bootstrap-Werte nicht oder mit kaum über 50% nur sehr schlecht
gestützt. Eindeutig ist lediglich die enge Verwandtschaft von Borszczowia mit S. linifolia. Stärker
abgeleitet erscheinen die C4-Arten der bisherigen Sektionen Schoberia, Salsina, Macrosuaeda,
Limbogermen und Immersa. Eindeutig belegt ist die nahe Verwandtschaft von Alexandra lehmanni
und der Sektion Schoberia sowie die nahe Verwandtschaft der vier C4-Arten S. altissima, S.
aegyptiaca, S. vermiculata und S. divaricata. Die Position von S. physophora wechselt hingegen in
Abhängigkeit von der betrachteten DNA-Region.
8.) Die Zugehörigkeit der Taxa zu terminalen Clades ist bei beiden Datensätzen identisch, variabel ist
nur die Beziehung dieser Clades zueinander. Die Beziehungen sind zudem statistisch überwiegend
schlecht oder gar nicht abgesichert.
9.) Die am weitesten isolierten Vertreter der Salicornioideae sind Kalidium und Allenrolfea, die sich
auch morphologisch von anderen Sippen der Unterfamilie unterscheiden (bes. Blattmerkmale).
Innerhalb der Salicornioideae bilden die hier einbezogenen Salicornia-Sippen mit den australischen
Arten Tecticornia australasica und Halosarcia peltata eine gemeinsame Linie. Ähnliche Ergebnisse mit
wesentlich erweitertem Datensatz fanden Kadereit et al (2005). Aufgrund der geringen Zahl an Daten
sind aber weiterführende Aussagen zur Phylogenie der Salicornioideae im Rahmen dieser Arbeit nicht
sinnvoll.
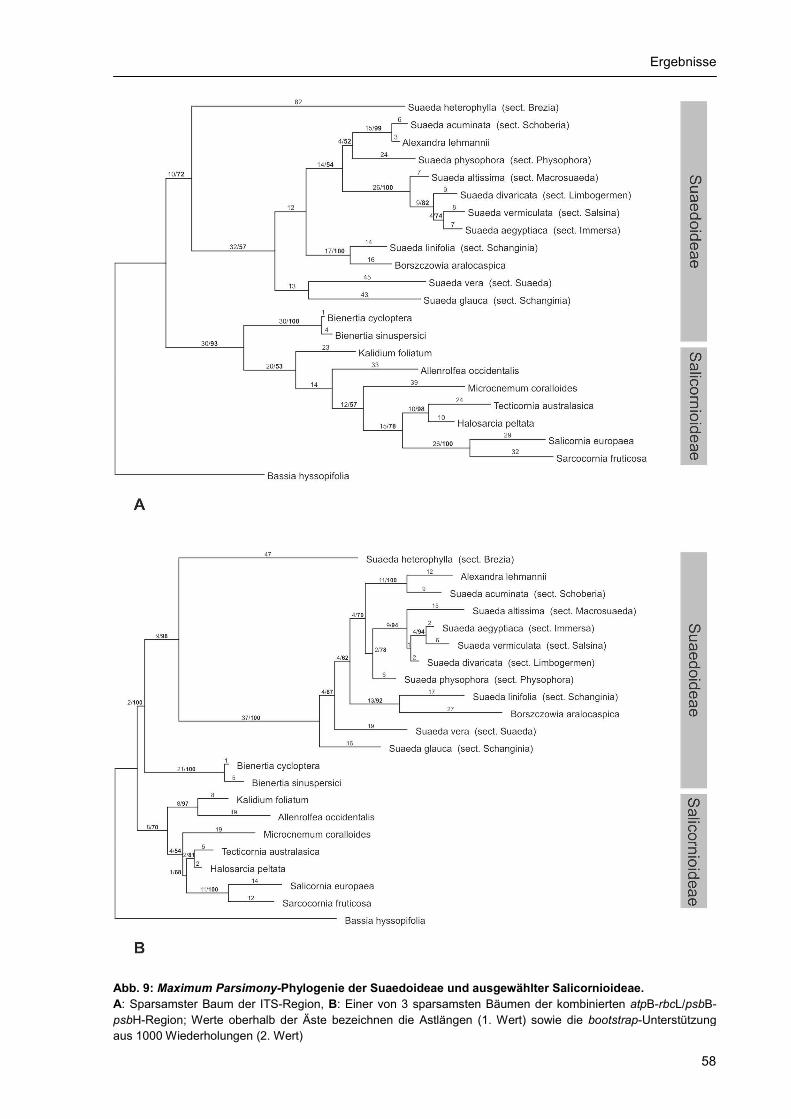
Ergebnisse
58
Abb. 9: Maximum Parsimony-Phylogenie der Suaedoideae und ausgewählter Salicornioideae. A: Sparsamster Baum der ITS-Region, B: Einer von 3 sparsamsten Bäumen der kombinierten atpB-rbcL/psbB-psbH-Region; Werte oberhalb der Äste bezeichnen die Astlängen (1. Wert) sowie die bootstrap-Unterstützung aus 1000 Wiederholungen (2. Wert)

Ergebnisse
59
3.2.2. ITS-Phylogenie der Suaedoideae Basierend auf den Ergebnissen von Kap. 3.2.1 wurde jeweils mit Chloroplasten- und Kerndatensätzen
getrennt die phylogenetische Analyse der Suaedoideae durchgeführt. Aufgrund der hohen Variabilität
besonders im ITS wurde die Zahl der Außengruppen dabei minimiert, da bei der gegebenen Distanz
das Alignment an hochvariablen Positionen teilweise mit Unsicherheiten behaftet und nicht eindeutig
ist. Für die Bewurzelung der Bäume wurde als Außengruppe jeweils die Gattung Kalidium gewählt, die
aufgrund ihrer isolierten und basalen Stellung innerhalb der Salicornioideae den Suaedoideae am
ähnlichsten ist.
Für die Berechnung der ITS-Phylogenie wurde ein erweiterter Datensatz von 92 Taxa der
Suaedoideae zusammengestellt. Darin sind alle Taxa der Cloroplasten-Phylogenie enthalten, erweitert
um eine Reihe von Sippen, für die bisher keine Chloroplasten-Daten vorliegen, die aber für das
Verständnis der Phylogenie durchaus bedeutsam sind.
Das für die Berechnung verwendete ITS-Alignment besitzt eine Länge von 674 bp mit 344 variablen
Positionen. Davon waren 280 parsimony-informative. Verwendet wurden trotz der auftretenden
Unsicherheiten die Merkmale des gesamten Alignments. Eine Einbeziehung synapomorpher Indels
war aufgrund der Diversität nicht notwendig. Die MP-Analyse mit diesen Merkmalen ergab aufgrund
der hohen Taxazahl eine so hohe Anzahl kürzester Bäume, dass mit der verfügbaren
Rechnerkapazität die endgültige Anzahl nicht bestimmt werden konnte. Die Länge der kürzesten
Bäume beträgt 1039 Schritte, die Homoplasieindices haben für alle Bäume die Werte CI=0,49 und
RI=0,91. Die erhaltenen Baumtopologien unterscheiden sich geringfügig in der Anordnung einiger
Clades. Aus allen möglichen kürzesten Bäumen wurden zur Veranschaulichung jeweils die
Konsensus-Bäume konstruiert, werden aber nicht dargestellt. Für die statistische Absicherung wurde
das bootstrapping unter den Bedingungen von MP durchgeführt. Die Zahl der Wiederholungen betrug
1000. Die Zwischenspeicherung von Bäumen wurde auf 100 begrenzt, um die Berechnung in einem
überschaubaren Zeitaufwand zu halten. Bootstrap-Werte von >50% wurden in den ausgewählten MP-
Baum übertragen (Abb. 10). Für die ML-Analyse wurde von Modeltest als optimales DNA-
Evolutionsmodell GTR+I+G mit folgenden Parametern ermittelt: Nukleotidfrequenzen A=0,2051
C=0,2884 G=0,2840 T=0,2225; Substitutionsraten AC=1,1733 AG=2,3794 AT=1,6167 CG=0,3995
CT=4,6540 GT=1,0000; Anteil invariabler Positionen: 0,3714, Merkmalsverteilung: Gamma,
Formparameter: 1,7583. Die Analyse ergab mit dem gewählten Modell genau einen
wahrscheinlichsten Baum mit einem -ln L=6453,341. Der Baum ist in Abbildung 11 dargestellt. Die
Bayesianische Analyse wurde mit 1 Mio. Generationen gerechnet und die posterior probabilities des
50% majority rule-Baumes auf den ML-Baum übertragen. Die Baumtopologien waren mit wenigen
Ausnahmen innerhalb terminaler Gruppen identisch. Die Konsensus-Bäume der MP-Analyse sowie
die Bäume aus der Bayesianischen Analyse finden sich für Vergleichszwecke im Anhang.
Die phylogenetische Analyse der ITS-Region der Suaedoideae einschließlich der gewählten
Außengruppe erbrachte unabhängig von den gewählten Berechnungsverfahren und den
veränderlichen Parametern eine Reihe sehr konsistenter Aussagen. Die in den Berechnungen aus
Kap. 3.2.1 einbezogenen Schlüsseltaxa sind im erweiterten Set Bestandteile mehr oder weniger
diversifizierter Clades. Generell ist dabei festzustellen, dass die Zuordnung von Sippen zu einzelnen
Clades praktisch konstant ist. Variabel sind hingegen wie schon erwähnt die phylogenetischen

Ergebnisse
60
Zusammenhänge dieser Clades untereinander. Auffallend ist weiterhin die im Vergleich zur
Morphologie extreme Variabilität, wodurch eine sehr differenzierte Gliederung möglich ist. Basierend
auf den erhaltenen Baumtopologien sind über die Gliederung der Suaedoideae anhand von ITS
folgende Aussagen zu treffen:
1.) Die 3 Gattungen der Suaedoideae Suaeda, Alexandra und Borszczowia bilden eine
monophyletische Einheit. Die Gattung Bienertia gehört nicht in diesen engeren Verwandtschaftskreis.
2.) Die um die beiden genannten Gattungen erweiterte ursprüngliche Gattung Suaeda teilt sich in zwei
sehr deutlich getrennte Verwandtschaftskreise auf, die hier als Brezia- und Suaeda-Clade benannt
werden. Diese Zweiteilung ist durch sämtliche Berechnungs- und Überprüfungsmethoden eindeutig
belegt. Dem Brezia-Clade gehören alle Arten der bisherigen Sektion Brezia an. Zu dem Suaeda-Clade
gehören weitgehend alle anderen bisher zu Suaeda gestellten Sippen sowie Alexandra lehmannii und
Borszczowia aralocaspica.
3.) Brezia-Clade: Die Zusammengehörigkeit dieser Sippen ist eindeutig und vollständig gestützt
(100% bootstrap). Es sind, soweit bisher zugeordnet, die Sippen der Sektion Brezia sowie einige
bisher nicht klassifizierte Sippen wie S. arbusculoides, deren Zugehörigkeit aber aus morphologischer
Sicht zu der Sektion zu erwarten war. Es handelt sich um die Sektion mit der weitesten Verbreitung
weltweit, auch wenn die Taxonomie der Sippen keineswegs klar ist. Die molekulare Diversität selbst
innerhalb dieses Teils von Suaeda ist sehr hoch, so dass eine Differenzierung in untergeordnete
Verwandtschaftskreise gut möglich ist. Deutlich wird eine sehr tiefe Zweiteilung des Brezia-Clades:
Eine Gruppe um die Art S. corniculata, die hier als corniculata-Subclade bezeichnet wird, bildet die
Schwestergruppe zu den verbleibenden Arten. Die letztere Gruppe lässt sich wiederum in zwei
monophyletische Gruppen aufteilen. Nach wichtigen zugehörigen Arten werden diese als maritima-
und prostrata-Subclade benannt, woraus sich insgesamt eine Dreigliederung des Brezia-Clades
ergibt. Die beschriebene Unterteilung ist eindeutig und wird durch hohe bootstrap-Werte gestützt.
4. Suaeda-Clade: Die Gruppe erscheint als völlig isolierte, für sich gesehen aber monophyletische
Einheit innerhalb der Gattung Suaeda. Im Unterschied zu dem Brezia-Clade ist die Zahl gemeinsamer
Merkmale aber viel geringer, was darauf schließen lässt, dass die Differenzierung innerhalb dieser
Gruppe wesentlich früher eingesetzt haben muss. Die ITS-Sequenzen weisen eine extreme Diversität
auf, wodurch sich Unsicherheiten im Alignment und den damit rekonstruierten Stammbäumen
ergeben. Die Unterteilung erfolgt in 5 Subclades mit einer zweifelhaften Position im Fall von S. glauca.
Nach den wichtigsten Arten bzw. Sektionen werden diese hier als Schoberia-, fruticosa-, physophora-,
vera- und Schanginia-Subclade bezeichnet und verdeutlichen damit schon z.T. die Analogie zu den
bisher bestehenden Sektionen. Es ist charakteristisch, dass die jeweiligen Gruppen durch sehr hohe
bootstrap-Werte, die Verwandtschaft der Subclades untereinander allerdings überhaupt nicht gestützt
wird. Im strict concensus-Baum der MP-Analyse wird lediglich eine nähere Verwandtschaft von
Schoberia-, fruticosa-, und physophora-Subclade belegt, was damit die Ergebnisse aus Kap 3.2.1
bestätigt. Das Verhältnis der übrigen Gruppen wird nicht aufgelöst und verbleibt damit als Polytomie.
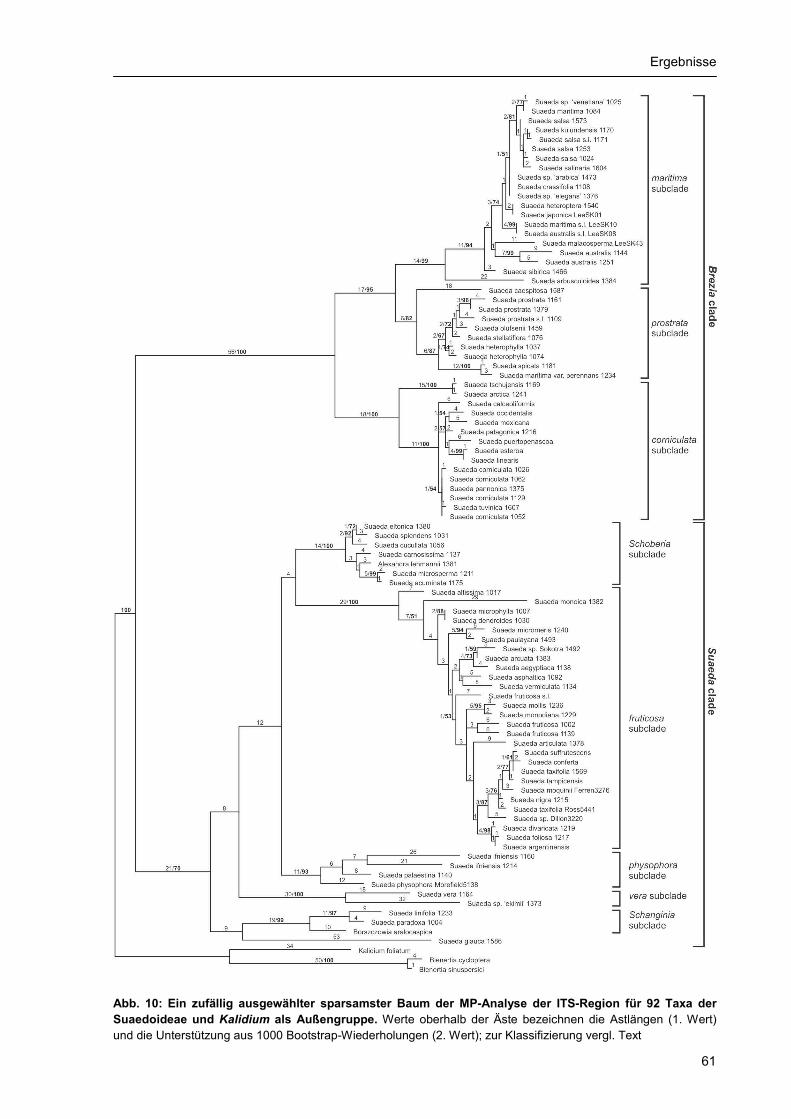
Ergebnisse
61
Abb. 10: Ein zufällig ausgewählter sparsamster Baum der MP-Analyse der ITS-Region für 92 Taxa der Suaedoideae und Kalidium als Außengruppe. Werte oberhalb der Äste bezeichnen die Astlängen (1. Wert) und die Unterstützung aus 1000 Bootstrap-Wiederholungen (2. Wert); zur Klassifizierung vergl. Text
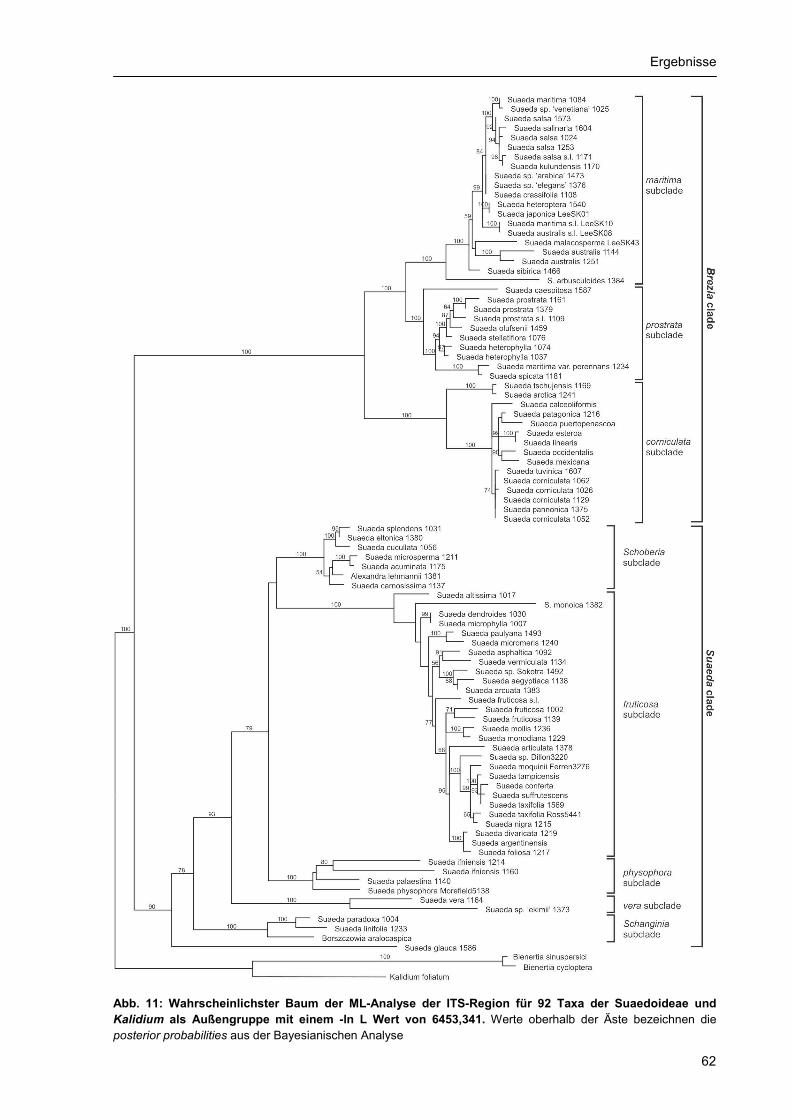
Ergebnisse
62
Abb. 11: Wahrscheinlichster Baum der ML-Analyse der ITS-Region für 92 Taxa der Suaedoideae und Kalidium als Außengruppe mit einem -ln L Wert von 6453,341. Werte oberhalb der Äste bezeichnen die posterior probabilities aus der Bayesianischen Analyse

Ergebnisse
63
Differenzierung der Subclades
3.a.) Maritima-Subclade
Die bekannteste Sippe des Subclades ist S. maritima, für die eine weltweite Verbreitung angenommen
wird, was aber mit großer Wahrscheinlichkeit nicht zutrifft. Obgleich die molekulare Verwandtschaft
des Subclades mit 99% Unterstützung sehr gut belegt ist, lassen sich kaum morphologische
Merkmale finden, die diese Gruppe in besonderer Weise kennzeichnen würden. In diesem Subclade
finden sich neben eurasiatisch verbreiteten auch die beiden einzigen australischen Sippen der
Gattung: S. arbusculoides und S. australis. Am stärksten isoliert von allen anderen steht die
australische S. arbusculoides, eine der wenigen ausdauernden, verholzten Brezia-Sippen. Ähnlich
isoliert, aber nicht näher mit S. arbusculoides verwandt erscheint S. australis, eine tropisch-
südhemisphärisch verbreitete, ebenfalls ausdauernde Sippe. Als Bindeglied zu den beiden
südhemisphärischen Sippen steht eine bisher unbekannte Sippe aus Zentralasien/Südsibirien, die erst
kürzlich als S. sibirica beschrieben wurde (Lomonosova et al. 2008). Damit wird ziemlich sicher belegt,
dass die Besiedlung Australiens zweimal unabhängig voneinander erfolgt sein muss und in beiden
Fällen mit einer Änderung der Lebensform (Verholzung) einhergegangen ist, da S. sibirica annuell ist.
An diese Sippen schließen sich südostasiatisch verbreitete an, deren taxonomischer Status allerdings
nicht geklärt ist. Während S. malacosperma als Sippe eindeutig charakterisiert ist, handelt es sich bei
S. australis s.l. und S. maritima s.l. (nach Lee et al. 2007) mit Sicherheit nicht um die so bezeichneten
Arten, wenngleich um einen eigenständigen Sequenztyp innerhalb des maritima-Subclades. Weitere
Sippen des Clades sind neben S. crassifolia und S. heteroptera (die molekular identisch mit S.
japonica ist) eine Reihe noch nicht beschriebener, morphologisch aber unterscheidbarer Sippen für
die hier vorläufige Namen gebraucht werden (S. ‚arabica’ und S. ‚elegans’). S. crassifolia weist eine
für Brezia ungewöhnliche Blattanatomie auf (s. Kap. 3.4.).
Ein interessantes Ergebnis sind die Positionen von S. maritima im engeren Sinn und S. salsa. Beide
Arten sind molekular deutlich charakterisiert und bilden mehr oder weniger diversifizierte
Schwesterclades. Die Trennung der beiden Sippen war bis jetzt unklar. Im Falle von S. salsa fällt
schon bei der hier einbezogenen Auswahl eine stärkere molekulare Diversität auf. Die Benennung
orientiert sich vor allem an der geographischen Herkunft. Mit S. salsa (1253) wird dabei eine Sippe
aus dem Gebiet der Typuslokalität bezeichnet. Innerhalb von S. salsa steht die erst kürzlich
beschriebene Polyploid-Sippe S. kulundensis, auf deren Entstehungs-Hypothese in der Diskussion
noch eingegangen wird. Mit S. salinaria liegt eine mittel- und südosteuropäisch binnenländisch
verbreitete Sippe vor, die eindeutig nicht zu S. maritima s.str. gehört. Von S. maritima (1084) wird hier
eine weitere, morphologisch unterschiedene Sippe (S. ‚venetiana’) abgetrennt, was auch durch
geographische Herkünfte bedingt ist.
3.b.) prostrata-Subclade
Als Schwestergruppe zu dem maritima-Subclade vereint diese Gruppe drei unterschiedlich stark
diversifizierte Linien: caespitosa, spicata und prostrata/heterophylla. Eine stark isolierte Stellung
nimmt die südafrikanisch verbreitete, ausdauernde S. caespitosa ein, die auch morphologisch stark
von den übrigen Arten abweicht. Damit wird eine erdgeschichtlich sehr alte Auftrennung dieses

Ergebnisse
64
Verwandtschaftskreises deutlich. Die Sippe hat sich offenbar nicht weiter diversifiziert, da nähere
Verwandte fehlen. Verbindendes Merkmal des übrigen prostrata-Subclades ist eine ausgesprochen
reiche Verzweigung der Infloreszenzen bzw. der gesamten Pflanzen. Dies tritt besonders bei der
Typusart der Sektion Brezia - S. heterophylla- sehr deutlich hervor, die zu diesem Subclade gehört.
Auffällig in dieser Gruppe sind weiterhin die im Verhältnis zu anderen Sippen auffallend kleinen
Perianthien mit manchmal auftretenden flügelartigen Auswüchsen. Am stärksten diversifiziert ist S.
prostrata, die im Baum mit drei unterschiedlichen ITS-Typen vertreten ist. Als S. prostrata im engeren
Sinne (von der Typuslokalität) wird Nr. 1379 festgelegt. Davon deutlich unterschieden ist die
kleinasiatisch verbreitete Form (1109). Die Schwesterart zu S. prostrata ist S. olufsenii, eine Sippe
zentralasiatischer Hochgebirge. Nahe verwandt mit S. prostrata gruppieren S. heterophylla und S.
stellatiflora, beide Sippen mit ausgeprägten Flügeln am ausdifferenzierten Perianth, was der
Hauptgrund für die Aufstellung einer eigenen Gattung war (Brezia heterophylla bei Ulbrich 1934). Die
Stellung innerhalb von Suaeda in der S. prostrata-Verwandtschaft ist jedoch eindeutig und rechtfertigt
nicht die Abtrennung von S. heterophylla als eigene Gattung. Alle genannten Sippen weisen mehrere
molekular gut unterscheidbare Typen auf, so dass die Definition von Arten noch weiterer
Untersuchungen bedarf. Den geschilderten Sippen mit fast ausschließlich asiatisch-kontinentaler
Verbreitung steht als Schwestergruppe S. spicata gegenüber, ein vorwiegend westmediterran
verbreiteter Typ, welcher eine der drei Linien des prostrata-Subclades bildet. Suaeda spicata wird in
zahlreichen Floren mit S. maritima vereint, deren phylogenetische Stellung aber zweifelsfrei außerhalb
der S. maritima-Verwandtschaft liegt. Diese Zuordnung war bisher nicht bekannt. S. spicata weist eine
sehr hohe intraspezifische molekulare Diversität auf (s. Kap. 3.2.4). Zu S. spicata im weiteren Sinne
gehört auch die einzige ausdauernde Brezia Europas, S. maritima var. perennans (1234), eine
ausschließlich an den europäischen Atlantikküsten sowie auf den Kanaren verbreitete Sippe.
3.c.) corniculata-Subclade:
Von den übrigen Arten geschieden bilden die Sippen der S. corniculata-Verwandtschaft eine sehr
isolierte Gruppe. Charakteristisches morphologisches Merkmal sind die fast immer auftretenden
hornartigen Auswüchse des Perianths. Als deutlich isolierte Gruppe erscheinen S. arctica und die erst
kürzlich von Lomonosova & Freitag (2003) aus den Hochlagen des Altai neu beschriebene Sippe S.
tschujensis. Die hohe Zahl eigenständiger Merkmale dieser Linie lässt auf eine relativ frühe
Abtrennung dieser Sippen vom Rest der Gruppe schließen. Die Verwandtschaft ist eindeutig und
100% bootstrap belegt. Im verbleibenden Teil des corniculata-Subclades gruppieren zum einen
verschiedene Typen der fast rein asiatischen S. corniculata, zum anderen aber auch
überraschenderweise alle amerikanischen Sippen der Sektion Brezia, von denen immerhin 7 Arten
einbezogen werden konnten. Die Gruppenbildung deutet darauf hin, dass wenigstens zwei
neuweltliche Besiedlungsereignisse stattgefunden haben müssen. S. calceoliformis als isoliert
stehende Sippe scheint sich dabei unabhängig von allen anderen entwickelt zu haben. Im
Stammbaum bildet sie mit den übrigen amerikanischen Sippen und S. corniculata s.l. eine Polytomie.
Es existieren jedoch auch mögliche MP-Baumtopolgien, in denen S. calceoliformis basal zu allen
anderen Sippen steht (inklusive S. corniculata s.l.). Die übrigen amerikanischen Sippen bilden eine gut
gestützte monophyletische Gruppe, eine Differenzierung in nord- und südamerikanische Arten tritt

Ergebnisse
65
dabei allerdings nicht hervor. Lediglich zu der geographisch isolierten S. patagonica existieren keine
ähnlichen, gruppierenden Sequenzen. Auch in dieser Gruppe gibt es mit S. esteroa und S. linearis
wieder ausdauernde Sippen.
Die morphologisch und karyologisch recht diverse S. corniculata weist nur zwei ITS-Typen auf, die
sich zudem nur durch eine Punktmutation unterscheiden. Der häufige Typ, durch 1062 repräsentiert,
bezeichnet die S. corniculata-Sippen von eher niederliegendem bis aufsteigendem Wuchs mit sehr
asymmetrischen, hornartig verlängerten Perianthzipfeln (s. Kap 3.5) und großem Areal von
Zentraleuropa bis Ostsibirien. Eine identische ITS-Sequenz weist auch S. pannonica auf, der einzige
mitteleuropäische Vertreter dieser Verwandtschaftsgruppe. Der abweichende Typ (1026) gehört
möglicherweise zu einer geographisch begrenzten Variante. Bei S. corniculata s.l. steht der
morphologischen Variabilität anders als bei den anderen Sippen keine molekulare Diversität
gegenüber. Durch eine singuläre 6bp-Insertion charakterisiert ist die erst kürzlich beschriebene S.
tuvinica (1607) aus Zentralasien charakterisiert.
4.a.) Schoberia-Subclade
In dieser Gruppe finden sich die annuellen C4-Sippen, die bisher in der Sektion Schoberia vereint
wurden. Die Gruppierung wird zu 100% gestützt. Eine enge Verwandtschaft dieser Arten wird auch
durch die morphologischen Daten belegt. Die molekulare Diversität ist, gemessen an anderen
Gruppen, relativ gering, was für eine relativ junge Radiation sprechen dürfte. Die Unterteilung des
Clades folgt in etwa geographischen Regionen. Jeweils näher verwandt sind S. splendens, S. eltonica
und S. cucullata und S. acuminata mit S. microsperma. Stärker isoliert erscheint hingegen S.
carnosissima. Die gesamte Gruppe besiedelt ein relativ kleines Areal und auch die Arten haben nur
jeweils sehr begrenzte Verbreitungsgebiete. Außer den mittel- und SW-asiatisch verbreiteten Sippen
gibt es mit S. splendens nur eine europäisch-mediterrane. Vollkommen unerwartet in dieser C4-
Gruppe ist die Stellung der morphologisch stark abweichenden C3-Art Alexandra lehmannii, eine
morphologisch von Suaeda anhand der einzigartigen Perianthmerkmale unterschiedene Sippe. Die
Aussagen der molekularen Daten sind hier nicht eindeutig. Es existieren Baumtopologien mit basaler
Position von Alexandra in Bezug auf den Schoberia-Subclade als auch mit genesteter Position
innerhalb dieser Gruppe. Die hohe Ähnlichkeit der Alexandra-Sequenz gegenüber Schoberia-
Sequenzen lässt aber als Konsens durchaus die Position von Alexandra innerhalb von Schoberia zu.
Sie ist den kontinental verbreiteten Sippen S. acuminata und S. microsperma am ähnlichsten.
4.b.) fruticosa-Subclade
Zu dieser sehr diversen und artenreichen monophyletischen Gruppe gehören alle C4-Arten mit kranz-
suaedoidem Blatttyp, die bisher unter den Sektionen Macrosuaeda, Immersa, Limbogermen und
Salsina klassifiziert wurden. Die Annahme einer nahen Verwandtschaft war aufgrund der
morphologischen Daten wahrscheinlich, bisher jedoch nicht in die Klassifikation der Gattung
umgesetzt. Ihre Zusammengehörigkeit wird durch die Stammbäume zu 100% gestützt, allerdings ist
die interne Gliederung nicht sicher zu belegen. Relativ isoliert innerhalb dieses Clades stehen S.
altissima und S. monoica. Die südeuropäisch-westasiatisch verbreitete S. altissima weist
morphologische und karyologische Besonderheiten auf, die isolierte Stellung war daher zu erwarten.

Ergebnisse
66
Sie ist im Gegensatz zu den meisten übrigen Arten des Subclades einjährig. S. monoica weist nach
den molekularen Daten enge Beziehungen zu zwei weiteren strauchigen, ausdauernden Arten auf: S.
dendroides und S. microphylla. In einigen Baumtopologien der MP-Analyse bilden diese 3 Arten einen
gemeinsamen Clade, doch besitzt S. monoica zahlreiche Autapomorphien und dadurch einen sehr
langen singulären Ast. Die nächste Gruppe sind zwei NO-afrikanisch verbreitete Arten, S. paulayana
und S. micromeris, die relativ eng miteinander verwandt sind. Eine weitere Gruppe bilden S.
asphaltica, S. aegyptiaca, S. vermiculata und S. arcuata und eine bis jetzt noch nicht näher definierte
Sippe von Sokotra (1492). Dies ist bemerkenswert, da in dieser Gruppe ausdauernde und einjährige
Sippen vereinigt sind und S. aegyptiaca aufgrund von Blütenmerkmalen bisher als eigene Sektion
Immersa geführt wurde. Die molekulare Analyse rechtfertigt diese Sonderstellung nicht. Die
verbleibenden Arten gehören zu den bisherigen Sektionen Salsina und Limbogermen. An dieser Stelle
findet sich im Stammbaum das Ergebnis einer weiteren neuweltlichen Radiation in Form zahlreicher
Sippen mit unterschiedlichen ITS-Typen. Im Gegensatz zu den amerikanischen Brezia-Sippen
diversifizieren sich die nord- und südamerikanischen Sippen des fruticosa-Clades innerhalb zwei
getrennter Verwandtschaftskreise. An der Basis aller amerikanischen Arten steht die SW-afrikanisch
verbreitete S. articulata. Davon ausgehend entwickelt sich der Südamerika-Clade mit den nahe
verwandten Sippen S. argentinensis, S. divaricata und S. foliosa, womit alle südamerikanischen
Sippen dieser Gruppe repräsentiert sind. Der Nordamerika-Clade ist mit 7 Sippen vertreten, allerdings
ist hier die Systematik noch nicht geklärt, die Namen haben daher nur vorläufigen Charakter. Dies
betrifft vor allem S. taxifolia und S. moquinii/S. nigra. Eine Zwischenstellung nimmt eine Sippe aus
Peru ein (Suaeda sp. Dillon 3220), die an der Basis der nordamerikanischen Sippen steht und noch
nicht näher definiert werden konnte. Im MP-Baum bilden nord- und südamerikanische Clades
Schwestergruppen, im ML-Baum wird diese Beziehung nicht aufgelöst, sie bilden gemeinsam mit S.
articulata eine Polytomie.
Verbindendes Element zwischen den alt- und neuweltlichen Arten ist S. fruticosa, die mit drei
Sequenztypen im Stammbaum vertreten ist und in deren Verwandtschaft auch S. mollis und S.
monodiana gehören. Da es sich bei S. fruticosa um eine polymorphe, weit verbreitete Art handelt, sind
weitere Untersuchungen notwendig.
4.c.) physophora-Subclade
Zu dieser Gruppe gehören neben der namengebenden S. physophora die Zwergsträucher S. ifniensis
und S. palaestina, deren Sektionszugehörigkeit bisher umstritten war. Die Verwandtschaft mit S.
physophora ist jedoch in den molekularen Stammbäumen sehr gut gestützt (93% bootstrap) und auch
durch morphologische Merkmale begründet. S. ifniensis der Kanaren (1160) und S. ifiniensis des
afrikanischen Festlandes (1214) unterscheiden sich deutlich, so dass möglicherweise auch von zwei
Sippen ausgegangen werden kann. Die Stellung dieser Gruppe innerhalb der Suaedoideae-
Phylogenie kann durch ITS-Daten allerdings nicht eindeutig definiert werden. Es existieren
Baumtopologien mit Schwestergruppenstellung sowohl zum fruticosa- als auch zum Schoberia-
Subclade. In Konsensus-Bäumen ist daher die Stellung dieser drei Clades nicht aufgelöst (Polytomie).
Mit Sicherheit scheint es sich aber nicht um eine basale Gruppe innerhalb des Suaeda-Clades zu
handeln.

Ergebnisse
67
4.d.) vera-Subclade
Ebenfalls sehr isoliert und nur mit zwei Arten repräsentiert stehen S. vera und eine bisher unbekannte
Art, die hier vorläufig als S. ‚ekimii’ bezeichnet wird. Beide Arten teilen sehr charakteristische
Narbenmerkmale (s. Kap. 1.2.2), was ihre Eigenständigkeit innerhalb Suaeda schon morphologisch
deutlich unterstreicht. Es sind mit großer Wahrscheinlichkeit basale Sippen, doch ist auch ihre
Stellung durch ITS-Daten nicht eindeutig belegt. Es existieren Baumtopologien mit basaler Stellung
des Suaeda-Subclades gegenüber den anderen Gruppen des Suaeda-Clades.
S. vera beschränkt sich in ihrer Verbreitung auf das Mittelmeergebiet, S. ‚ekimii’ ist bis jetzt nur aus
der östlichen Türkei belegt (H. Freitag, 2002) und bisher nicht beschrieben. Nähere Verwandte von S.
vera waren bisher nicht bekannt, sie bildete bisher die monotypische Sektion Suaeda. Taxonomische
Unklarheiten führten in der Vergangenheit immer wieder zu Verwechslungen zwischen S. vera und S.
fruticosa doch handelt es sich um vollkommen verschiedene Sippen und die molekularen Daten
belegen die unterschiedliche stammesgeschichtliche Ableitung.
4.e.) Schanginia-Subclade
In dieser relativ isoliert stehenden Gruppe sind höchst unterschiedliche Sippen vereint. Zum einen die
einander noch recht ähnlichen S. paradoxa und S. linifolia, mit ihnen jedoch auch die blattanatomisch,
blütenmorphologisch und physiologisch vollkommen abweichende C4-Art Borszczowia aralocaspica.
Diese Art ist der einzige Vertreter mit einer speziellen Form der single cell C4 phyotosynthesis und
dem borszczowioiden Blatttyp (s. Kap. 1.2.3). Die Zusammengehörigkeit dieser drei Arten wird durch
erhebliche morphologische Unterschiede zwischen Borszczowia zu S. linifolia und S. paradoxa nicht
gestützt, ist jedoch anhand der molekularen Daten eindeutig. Es handelt sich insgesamt um eine sehr
isoliert stehende basale Gruppe, die auch molekular stark von anderen Gruppen abweicht,
untereinander jedoch sehr starke Ähnlichkeit aufweist. Die bootstrap-Unterstützung für diesen
Subclade beträgt 100%, so dass kaum alternative Möglichkeiten angenommen werden können.
4.f.) Suaeda glauca
Die Art stellt die am stärksten isolierte dar und hat keine näheren Verwandten. Die bisherige Stellung
innerhalb der Sektion Schanginia wird von den molekularen Ergebnissen nur insofern gestützt, dass in
der MP-Baumtopologie S. glauca mit dem Schangina-Subclade einen durch bootstrap-Werte nicht
unterstützten gemeinsamen Clade bilden. Die Diversität der Einzelsequenzen ist jedoch extrem hoch,
die Anzahl gemeinsamer Merkmale hingegen sehr gering, so dass die Clade-Bildung zweifelhaft
bleibt. ML- und Bayes-Analyse unterstützen diese Gruppierung nicht und weisen S. glauca als
gesonderte Linie und ersten Abzweig des Suaeda-Clades aus. Eine nähere Verwandtschaft mit der
habituell ähnlichen Suaeda altissima, wie in einigen Arbeiten postuliert, besteht nach molekularen
Daten nicht.

Ergebnisse
68
3.2.3. Chloroplasten-Phylogenie der Suaedoideae Mit 66 Sequenzen der Suaedoideae und der Außengruppe Kalidium wurde für atpB-rbcL ein
Alignment von 1006 bp, für psbB-psbH ein Alignment von 666 bp erzeugt. Für die psbB-psbH -Region
liegen von S. caespitosa, S. physophora, S. sp. (Sokotra) und S. nigra keine Sequenzen vor. Ihre
Positionierung innerhalb der Chloroplasten-Bäume beruht damit ausschließlich auf atpB-rbcL-Daten.
Die Abweichungen gegenüber dem ITS-Datensatz kommen zum einen dadurch zustande, dass für
einige Sippen Chloroplasten-Sequenzen aufgrund schlecht konservierten Pflanzenmaterials nicht
generiert werden konnten, zum anderen durch die nachträgliche Einbeziehung nicht selbst erstellter
ITS-Sequenzen vor allem amerikanischer Sippen (V. Steinmann). Die Unterschiede betreffen vor
allem den corniculata- und den fruticosa-Subclade.
Vorangegangene Untersuchungen hatten ergeben, dass die Aussagen der Phylogenien beider
Chloroplasten-Regionen ähnliche Resultate zeigen, die Auflösung bei der psbB-psbH-Region jedoch
schlechter ist als bei atpB-rbcL (Schütze et al. 2003). Daher wurden beide Regionen für die Analyse
kombiniert. Der partition-homogeneity test mit 100 Replikaten zwischen atpB-rbcL und psbB-psbH
ergab einen Wert von P=0,06. Dies bedeutet, dass auf einem Signifikanzniveau von 99% von einem
gleichen phylogenetischen Signal ausgegangen werden kann. Die Kombination beider Regionen sollte
daher nicht zu einer Erhöhung der Homoplasie führen sondern die Aussagen eher verbessern.
Aus der Berechnung ausgeschlossen wurden wegen Unsicherheiten oder teilweise fehlender Daten
134 (atpB-rbcL) und 17 (psbB-psbH) Positionen. Vier synapomorphe Indels wurden für die
Parsimonie-Berechnung als 0/1-Merkmale kodiert. Von insgesamt 292 variablen Positionen des
gesamten Alignments waren 183 parsimony-informative. Mit diesen Merkmalen wurden unter
Maximum Parsimony 17932 kürzeste Bäume mit einer Länge von 404 Schritten erzeugt. Einer der
Bäume ist in Abbildung 12 dargestellt. Die Homoplasieindices für alle Bäume betragen CI=0,71,
RI=0,96. Konsensus-Bäume wurden aus allen Einzelbäumen generiert und befinden sich im Anhang.
Im Anhang sind auch die Bäume der Bayesianischen Analyse sowie der getrennten Berechnungen für
atpB-rbcL und psbB-psbH dargestellt um die Unterschiede in Baumtopolgie und Auflösung
darzustellen. Die folgende Tabelle gibt einen Überblick über die Alignments und die Ergebnisse der
MP-Analyse der getrennten und kombinierten Chloroplasten-Regionen.
Tab. 7: Übersicht über die Alignmentparameter und Ergebnisse der Maximum Parsimony Analyse der ausgewählten Chloroplasten-Regionen.
Region atpB-rbcL psbB-psbH kombiniert
Alignment (bp) 1006 (davon 134 ausgeschlossen)
666 (davon 17 ausgeschlossen)
1672 (davon 151 ausgeschlossen)
variabel 196 100 292
parsimony-informative 122 65 183
Anzahl sparsamster Bäume 792 nicht berechenbar 17932
Länge der sparsamsten Bäume (Schritte)
270 133 404
CI 0,72 0,71 0,71
RI 0,96 0,96 0,96

Ergebnisse
69
Als optimales Evolutionsmodell für die Maximum Likelihood-Analyse wurde von Modeltest K81uf+G
mit folgenden Parametern ausgewählt: Nukleotidfrequenzen A=0,3287 C=0,1439 G=0,1494 T=0,3781;
Substitutionsraten AC=1 AG=1,1831 AT=0,1722 CG=0,1722 CT=1,1831 GT=1; Anteil invariabler
Positionen: 0, Merkmalsverteilung: Gamma, Formparameter: 0,4329. Die Analyse ergab genau einen
wahrscheinlichsten Baum mit einem -ln L=4681,2589. Der Baum ist in Abbildung 13 dargestellt.
Für die Bayesianische Analyse wurden 1000000 Generationen mit einer Speicherfrequenz von 100
gerechnet. Am Ende betrug die Standardabweichung der split frequencies 0,0065 und blieb damit
deutlich unter dem Maximalwert von 0,01. Von den zwischengespeicherten 10000 Bäumen wurden
2500 (25%) eliminiert und aus den verbleibenden 7500 ein 50% majority rule Baum generiert. Da die
Topologie des Baumes der ML-Topologie sehr ähnlich war, wurden die posterior probabilities in den
letzteren übertragen.
Folgende von den bei ITS getroffenen abweichenden Aussagen ergeben sich aus der Chloroplasten-
Analyse:
1. Im Unterschied zu der ITS-Region weisen beide Chloroplasten-Regionen eine wesentlich geringere
molekulare Diversität auf, was unmittelbar in einer schlechteren Auflösung der Bäume resultiert. Ein
Teil der Sippen mit unterschiedlichen ITS-Sequenzen weist identische Chloroplasten-Sequenzen auf.
2. Trotz der geringeren Diversität sind die Baumtopologien der ITS- und Chloroplasten-Datensätze
einander sehr ähnlich, in grundsätzlichen Aussagen unterscheiden sie sich nicht.
3. Die Unterschiede zwischen den Baumtopologien aus allen drei gewählten
Rekonstruktionsverfahren sind gering, so dass konsistente Aussagen zur Phylogenie getroffen werden
können.
4. Die Positionierung von Bienertia als Schwestergruppe zu dem Suaeda-Alexandra-Borszczowia-
Clade wird auch in den veränderten Datensätzen deutlich.
5. Die Haupttrennung in Brezia- und Suaeda-Clade entspricht den bei ITS gefundenen Ergebnissen
und wird auch in der Chloroplasten-Phylogenie deutlich und mit hohen bootstrap-Werten gestützt (94-
100%). Bis auf eine Ausnahme im Brezia-Clade werden alle anhand der ITS-Bäume aufgestellten
Subclades auch im Chloroplasten-Baum reproduziert. Die Zuordnung der terminalen Taxa zu den
Subclades ist mit diesen innerhalb des Suaeda-Clades identisch, innerhalb des Brezia-Clades
insofern verschieden, da hier keine deutliche Dreiergliederung erkennbar ist.
6. Differenzierung des Brezia-Clades
Die außerordentlich hohe Differenzierung des Clades mittels ITS-Daten wird im Chloroplasten-Baum
keineswegs reproduziert. Eine Unterteilung des Brezia-Clades anhand von Chloroplastenmarkern ist
aufgrund der geringen molekularen Differenzierung nicht eindeutig möglich. Für die meisten Taxa sind

Ergebnisse
70
Abb. 12: Ein zufällig ausgewählter sparsamster Baum der MP-Analyse von 66 kombinierten atpB-rbcL/psbB-psbH-Sequenzen der Suaedoideae und Kalidium als Außengruppe. Werte oberhalb der Äste bezeichnen die Astlängen (1. Wert) und die Unterstützung aus 1000 Bootstrap-Wiederholungen (2. Wert)
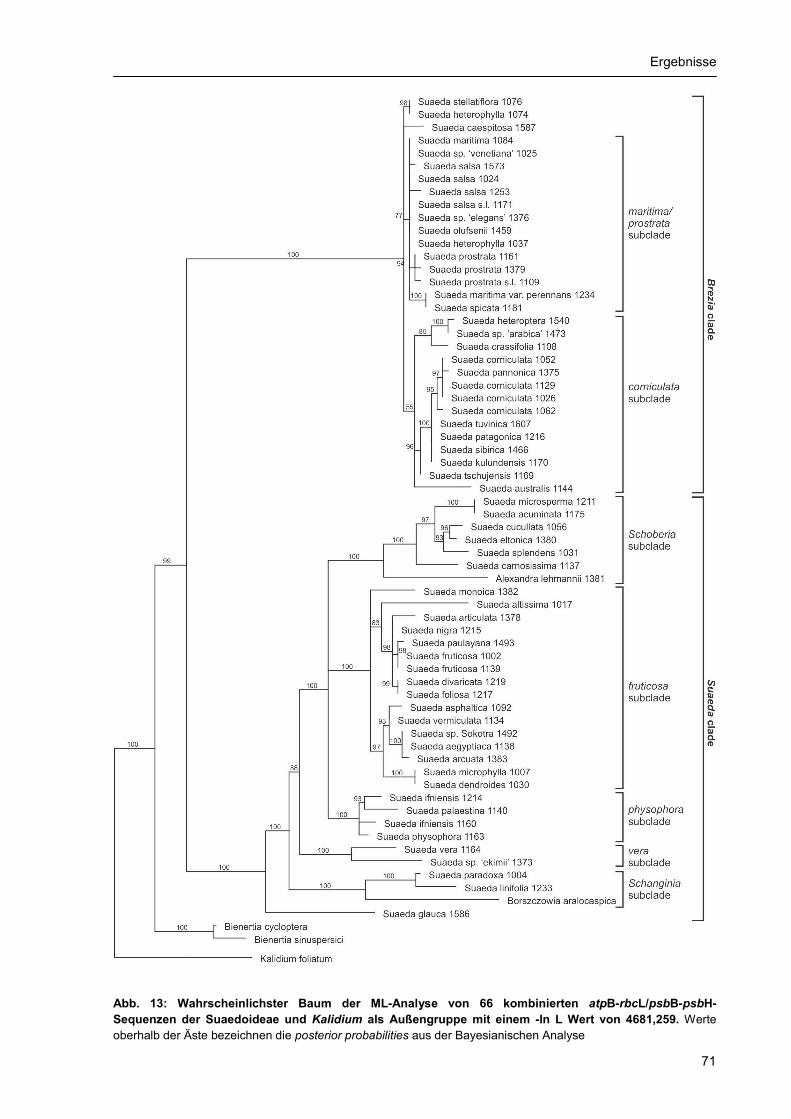
Ergebnisse
71
Abb. 13: Wahrscheinlichster Baum der ML-Analyse von 66 kombinierten atpB-rbcL/psbB-psbH-Sequenzen der Suaedoideae und Kalidium als Außengruppe mit einem -ln L Wert von 4681,259. Werte oberhalb der Äste bezeichnen die posterior probabilities aus der Bayesianischen Analyse

Ergebnisse
72
atpB-rbcL und psbB-psbH-Region identisch. Da die molekularen Merkmale offenbar homoplastisch
sind, ist die untergeordnete Gruppenzugehörigkeit der Sippen variabel, was sich in zahlreichen
möglichen sparsamsten MP-Baumtopologien ausdrückt. Durch bootstrap-Werte wird die Unterteilung
des Brezia-Clades praktisch überhaupt nicht gestützt. Die wenigen variablen Merkmale resultieren in
einer schlecht gestützten Zweiergruppierung, für die auch die bootstrap-Werte kaum höher als 60%
sind. Die aus den ITS-Bäumen ersichtlichen maritima- und prostrata-Subclades werden dabei
statistisch nicht unterstützt. Die gesamte Gruppenbildung beruht jeweils nur auf zwei synapomorphen
Merkmalen. Bei der Beurteilung ist jedoch zu berücksichtigen, dass einige für die Klassifikation
möglicherweise wichtigen Sequenzen (S. arbusculoides, nordamerikanische Sippen) fehlen.
6.a.) maritima/prostrata-Subclade
Die Gruppe enthält die meisten Sippen aus den maritima- und prostrata-Subclades der ITS-Analyse
mit Ausnahme von 3 Sequenzen: S. heterophylla 1074, S. stellatiflora 1076 und S. caespitosa. Aus
der weitgehend undifferenzierten Gruppe sind nur zwei Gruppierungen aus dem ITS-prostrata-
Subclade zu erkennen: S. spicata/S. maritima var. perennans (1234, 1181) und S. prostrata s.l. (1109,
1379, 1161). S. stellatiflora (1076) sowie eine der S. heterophylla-Sequenzen (1074) sind identisch,
aber deutlich von allen anderen unterschieden. Eine auffallende Inkonsistenz tritt bei der Stellung von
S. ‚elegans’ (1376) auf. Während die ITS-Sequenz identisch mit S. crassifolia ist, sind die
Chloroplasten-Sequenzen identisch mit der undifferenzierten maritima-Gruppe.
6.b.) corniculata-Subclade
Vergleichbar mit den Aussagen der ITS-Bäume bilden S. tschujensis, S. patagonica, S. tuvinica und
S. corniculata s.l. einen gemeinsamen und gut gestützten Clade. Die Gruppierung beruht sowohl auf
Nukleotid-Mutationen als auch auf synapomorphen Indels. Ein Widerspruch gegenüber den ITS-Daten
ist die Position von S. kulundensis und S. sibirica in diesem Clade, die nach den ITS-Daten in die
Verwandtschaft von S. salsa (maritima-Subclade) gehören. S. corniculata s.l. (incl. S. pannonica) teilt
sich in drei unterschiedliche Sequenztypen auf, währenddessen es bei ITS nur zwei sind. Die
Zuordnung und Gruppierung der einzelnen Sequenzen ist zudem nicht konsistent. S. patagonica als
einzige amerikanische Sippe weist mit S. tuvinica, S. sibirica und S. kulundensis identische
Sequenzen auf. Die Position der übrigen amerikanischen Sippen bleibt aufgrund des Fehlens von
Daten unklar. Als Bestandteil des Subclades können weitere Sippen interpretiert werden, die aufgrund
von ITS-Daten zu dem maritima-Subclade gehören: S. australis (1144), S. heteroptera (1540), S.
crassifolia (1108) und S. ‚arabica’ (1473). Die genannten Arten bzw. Sequenzen gehören aber nicht
unmittelbar zu der beschriebenen Gruppe um S. corniculata s.l. sondern nehmen gesonderte
Positionen ein. S. australis ist die Sequenz mit der höchsten Zahl individueller Merkmale des
gesamten Clades.
7. Differenzierung des Suaeda-Clades
Die Unterteilung des Suaeda-Clades in 5 Subclades sowie die isoliert stehende S. glauca
unterschiedet sich prinzipiell nicht von den Ergebnissen der ITS-Analyse. Die Ergebnisse aller drei
Rekonstruktionsverfahren MP, ML und Bayes sind weitgehend kongruent. Alle Subclades sind durch

Ergebnisse
73
100% bootstrap und posterior probability gestützt. Die Diversifizierung dieses Clades ist wie bei ITS
wesentlich höher, die Anzahl gemeinsamer Merkmale demgegenüber begrenzt. Die Beziehungen
zwischen den einzelnen Clades ähneln sich ebenfalls, sind aber besser unterstützt. Auch in den
Chloroplasten-Bäumen nimmt S. glauca eine basale Stellung ein. Der Schanginia-Subclade zweigt als
nächste Gruppe ab, während der Suaeda-Subclade eine Schwesterposition zu allen übrigen Gruppen
des Clades einnimmt. Die Stellung dieser basalen Gruppen ist relativ gesichert mit hohen bootstrap-
Werten sowie eindeutigen MP-Bäumen mit jeweils 100% Konsens. Insofern ist dies eine verbesserte
Aussage gegenüber derjenigen der ITS-Bäume. Nicht aufgelöst werden hingegen die Beziehungen
der drei Subclades Schoberia, fruticosa und physophora. Sie können nur als Polytomie bewertet
werden.
7.a.) Schoberia-Subclade
Die Strukturierung entspricht derjenigen im ITS-Subclade. Jeweils S. splendens/S. cucullata/S.
eltonica und S. acuminata/S. microsperma bilden gemeinsame Gruppen, wobei S. acuminata und S.
microsperma identische Sequenzen aufweisen. S. carnosissima nimmt eine isolierte Stellung ein. Ein
gravierender Unterschied gegenüber ITS ist die Position von Alexandra lehmannii im Verhältnis zum
Schoberia-Clade. In den Chloropasten-Bäumen nimmt Alexandra eine deutliche, basale Stellung ein
und ist nicht in die Gruppe eingeschlossen. Diese isolierte Stellung war aufgrund der morphologischen
Daten eher zu erwarten gewesen. Wie alle Schoberia-Sequenzen weist auch Alexandra in atpB-rbcL
eine charakteristische Minisatellitenstruktur auf, die jedoch strukturell deutlich verschieden von
Schoberia ist und schon aus der Sequenzstruktur als unabhängige Entwicklung gedeutet werden
kann. Die Position von Alexandra ist eindeutig und wird zu 100% durch bootstrap gestützt.
7.b.) fruticosa-Subclade
Der Subclade enthält aufgrund des Fehlens von Daten deutlich weniger Arten als im ITS-Baum. S.
monoica und S. altissima werden auch anhand von Chloroplasten-Daten als isolierte Sippen der
gesamten Verwandtschaftsgruppe sichtbar. Die Zahl von autapomorphen Sequenzmerkmalen ist bei
beiden Sequenzen deutlich höher als bei allen anderen Sequenzen des Subclades. Die übrigen Arten
verteilen sich - anders als in den ITS-Bäumen - auf zwei deutlich getrennte Clades, die mit S. monoica
eine Polytomie bilden. Einer der Clades vereint die SW-asiatisch-mediterranen Sippen S. asphaltica,
S. vermiculata, S. aegyptiaca, S. arcuata, S. microphylla und S. dendroides. Die Gruppierung ist stabil
und bleibt auch in den MP-Konsensus-Bäumen erhalten. Die nähere Verwandtschaft dieser Sippen
wurde bereits aus den ITS-Daten deutlich. In dem anderen Clade finden sich die Arten mit größerem
Areal, inklusive der amerikanischen und südafrikanischen. Dazu gehören S. articulata, S. fruticosa, S.
divaricata, S. nigra und S. foliosa. Beide Clades sind durch bootstrap-Werte (50 und 74%) nur
schwach gestützt, dafür sind aber die Werte für die posterior probabilites höher (97 und 98%). Im ML-
Baum ist S. altissima Bestandteil des 2. Clades, was aber durch MP-Daten nicht unterstützt wird. Die
beiden Sequenzen von S. fruticosa stehen auch im Chloroplasten-Baum an unterschiedlichen
Positionen was für eine möglicherweise stärkere Differenzierung der Art spricht. Die amerikanischen
Sippen bilden keinen einheitlichen Clade.

Ergebnisse
74
7.c.) physophora-Subclade
Dass S. ifniensis keineswegs einheitlich ist wird auch im Chloroplasten-Baum deutlich. In diesem
Baum sind sich S. palaestina und S. ifniensis des afrikanischen Festlandes (1214) ähnlicher als
diejenige der Kanarischen Inseln (1160). Die Sequenzähnlichkeit folgt hier eher geographischen
Regionen als traditionellen Artgrenzen. Die Gruppenbildung ist nur schlecht gestützt (58% bootstrap),
bleibt aber auch in den MP-Konsensbäumen durchweg erhalten. S. physophora unterscheidet sich am
stärksten von allen anderen Sequenzen.
7.d.) vera-Subclade
Die Stellung des Subclades an der Basis aller abgeleiteten Subclades wird durch alle
Berechnungsverfahren gestützt. Bei Maximum Parsimony kommt diese Position jedoch nur bei
Einbeziehung der 0/1-kodierten Indels zustande, nur anhand von Basenmutationen wird die
Beziehung zu dem Schanginia-Subclade nicht aufgelöst. Die Sequenzen beider Arten S. vera und der
unbeschriebenen S. ‚ekimii’ unterscheiden sich mit 20 Merkmalen sehr deutlich. Auch morphologisch
sind die Arten deutlich getrennt (ausdauernder Zwergstrauch bzw. annuelle, krautige Art)
7.e.) Schanginia-Subclade
Die Beziehungen der drei enthaltenen Sippen S. linifolia, S. paradoxa und Borszczowia aralocaspica
sind identisch mit den ITS-Ergebnissen. Anhand der Chloroplasten-Sequenzen wird die isolierte
Stellung von Borszczowia noch deutlicher. Bei geringerer Gesamtdiversität des Datensatzes weist
Borszczowia in der MP-Analyse mit 22 Astmerkmalen mehr als doppelt so viele Autapomorhpien auf
wie in der ITS-Phylogenie.
7.f.) Suaeda glauca
Die separate Position innerhalb des Subclades wird mit 82% bootstrap unterstützt. Suaeda glauca ist
der erste Abzweig des vergleichsweise stark diversifizierten Suaeda-Clades und weist wenig
Ähnlichkeiten mit anderen Sequenzen auf. Eine nähere Beziehung zu dem Schanginia-Subclade wie
in manchen ITS-Baumtopologien wird von den Chloroplasten-Phylogenien nicht belegt.

Ergebnisse
75
3.2.4. Phylogenie und Diversität der Sektion Brezia Über Status und Abgrenzung von Sippen innerhalb der Sektion Brezia war bis jetzt relativ wenig
bekannt und zahlreiche fehlerhafte Darstellungen sind publiziert worden. Insbesondere unter dem
Taxon „Suaeda maritima“ wurden in der Vergangenheit undifferenziert zahlreiche Sippen
zusammengefasst und damit die vorhandene Diversität meist nicht annähernd erfasst. Der
Schwerpunkt der hier vorgelegten erweiterten Darstellung für die Sektion liegt auf der Beschreibung
des europäischen Artenspektrums.
Aus der phylogenetischen Analyse der Suaedoideae wurde bereits die Monophylie der Sektion Brezia
und ihre recht isolierte Stellung innerhalb der gesamten Gattung deutlich. Brezia-Sippen formen einen
der zwei Haupt-Clades der gesamten Gattung, der von dem Suaeda-Clade durch eine hohe Zahl
synapomorpher Sequenzmerkmale unterschieden ist. Die ITS-Region als hoch variabler Marker für
inter- und intraspezifische Variabilität wurde dabei bereits ausgewählt. Mit diesen Informationen wurde
ein erweiterter Datensatz von Brezia-Sippen phylogenetisch analysiert. Die Aussage der verfügbaren
Chloroplasten-Daten zur Gliederung von Brezia weicht in einigen Fällen erheblich von derjenigen der
ITS-Daten ab. Da diese Differenzen nur durch Hybridisierung erklärt werden können, erfolgt eine
zusammenfassende Gegenüberstellung in der Diskussion (s. Kap. 4.3).
Mit einem Alignment von 67 Sequenzen und 649 bp wurde eine MP-Analyse durchgeführt. Von 225
variablen Positionen waren 138 parsimony-informative. Mit diesen Merkmalen wurden 120 kürzeste
Bäume mit einer Länge von 378 Schritten ermittelt. Die Werte für die Homoplasieindices betrugen
CI=0,64 RI=0,93. Ein zufällig ausgewählter Baum ist in Abb. 14 dargstellt. Die ML-Analyse wurde mit
folgenden Parametern durchgeführt: ausgewähltes Modell TrN+G, Nukleotidfrequenzen A=0,2312
C=0,2486 G=0,2654 T=0,2548; Substitutionsraten AC=1 AG=2,569 AT=1 CG=1 CT=8,1572 GT=1;
Anteil invariabler Positionen: 0, Merkmalsverteilung: Gamma, Formparameter: 0,4956. Die Analyse
ergab genau einen wahrscheinlichsten Baum mit einem -ln L=2975,6884.
Die erhaltenen Baumtopologien unterscheiden sich innerhalb von MP sowie zwischen MP und ML nur
unwesentlich. Sie bestätigen und erweitern die Ergebnisse der ITS-Gesamtanalyse. Deutlich wird
auch im erweiterten Baum die Dreigliederung des Brezia-Clades mit der starken Separierung des
corniculata-Subclades. Die Erhöhung der Zahl einbezogener Sequenzen brachte in den meisten
Fällen auch eine Erhöhung der Diversität, was als Diskussionsgrundlage für die Artabgrenzung von
Bedeutung ist.
1. maritima-Subclade 1.a.) maritima-Gruppe
Innerhalb dieser Gruppe wurden 6 ITS-Typen gefunden. Als S. maritima s.str. wird der an
europäischen Küsten mit Ausnahme Osteuropas vorherrschende Typ bezeichnet (1084, 1200).
Zahlreiche weitere, hier nicht einbezogene Sequenzen von europäischen Küsten sind mit diesem
identisch. Die zugehörigen Pflanzen haben als Merkmale meist abgerundete Perianthien ohne jegliche
Anhänge, relativ lockere, wenigblütige Teilblütenstände und einen vergleichsweise geringen
Verzweigungsgrad der Infloreszenz. Soweit aus der bis jetzt dokumentierten Variabilität hervorgeht,
folgt die Verteilung geographischen Regionen. Während an nord- und westeuropäischen Küsten nur
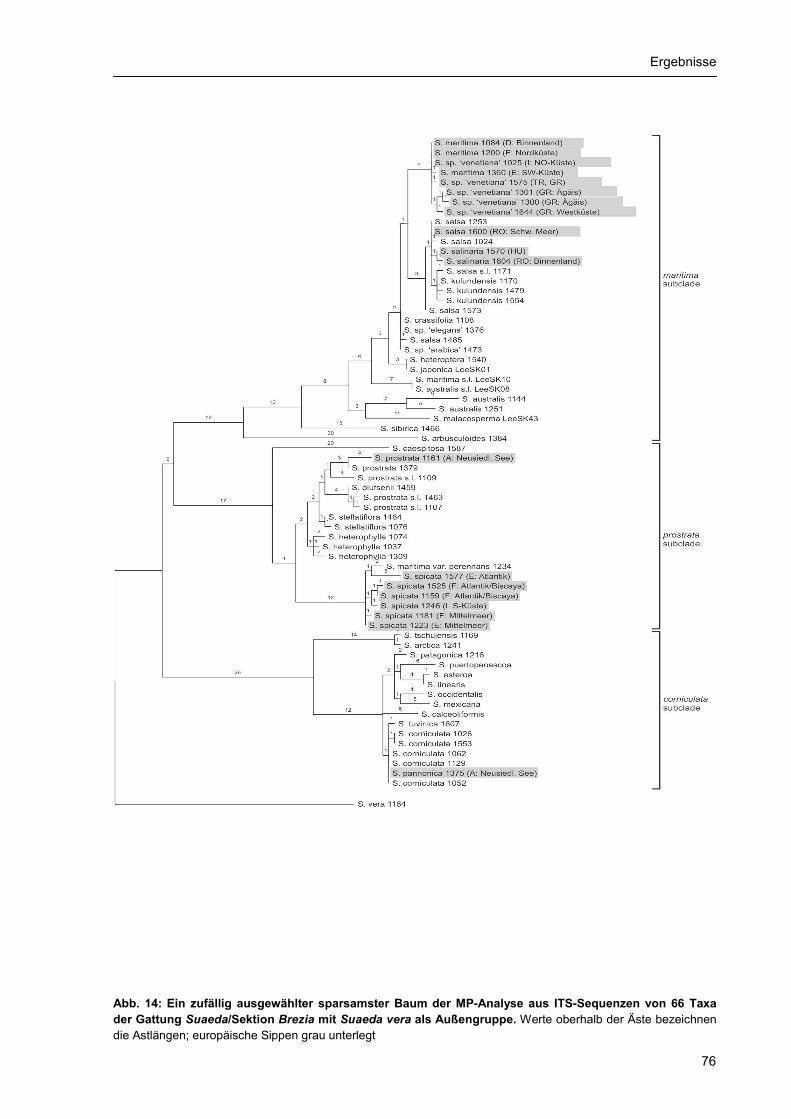
Ergebnisse
76
Abb. 14: Ein zufällig ausgewählter sparsamster Baum der MP-Analyse aus ITS-Sequenzen von 66 Taxa der Gattung Suaeda/Sektion Brezia mit Suaeda vera als Außengruppe. Werte oberhalb der Äste bezeichnen die Astlängen; europäische Sippen grau unterlegt

Ergebnisse
77
ein ITS-Typ vorkommt, ist die Diversität in der Mittelmeerregion höher mit einem Schwerpunkt im
östlichen Teil. Die Unterschiede sind dennoch vergleichsweise gering. Sequenz 1360 weist eine A/T-
Transversion in ITS2 auf, 1575 die einzige ITS1-Mutation (C/A-Transversion). Bei 1301, 1300 und
1644 handelt es sich um Anhäufung von C/T-Transitionen im ITS2, die im Stammbaum bereits zur
Bildung eines unabhängigen Clade führen. Die Art und Weise der Variabilität lässt auf mehrfach
unabhängig entstandene Linien schließen, die sich aus einem weit verbreiteten Typ ableiten lässt. Mit
der Sippenbezeichnung ‚S. venetiana’ soll bereits angedeutet werden, dass S. maritima auch
morphologisch keineswegs uniform ist. ‚S. venetiana’ bezeichnet Pflanzen mit sehr starkem
Verzweigungsgrad und späterer Blühperiode von Küsten des mittleren und östlichen Mittelmeeres.
Eine Zuordnung zu ITS-Typen ist allerdings nicht eindeutig möglich. Der molekular definierte S.
maritima-Typ wurde außerhalb Europas bzw. des Mediterrangebietes nicht nachgewiesen.
1.b.) salsa-Gruppe
S. salsa erscheint ähnlich S. maritima als eine hinsichtlich ITS sehr diverse Gruppe. Die
Schwestergruppenbeziehung zu der maritima-Gruppe ist eindeutig und gut unterstützt. Vergleichbar
mit S. maritima tritt auch in dieser Gruppe ein offenbar weit verbreiteter Grundtyp auf, von dem es
mehrfach unabhängige Ableitungen gibt. Der weit verbreitete Grundtyp ist durch die Sequenzen 1253
und 1600 repräsentiert und steht für S. salsa aus dem west- und zentralasiatischen
Hauptverbreitungsgebiet. Von diesem durch eine G/T-Transversion unterschieden ist 1024, eine
kleinasiatische Form. Die nur aus dem südosteuropäischen Binnenland bekannte S. salinaria fällt
erwartungsgemäß in die Verwandtschaft von S. salsa und ist mit zwei geringfügig durch C/T-
Transitionen verschiedene Sequenzen vertreten. Eine eigenständige Entwicklung aus S. salsa und
von dieser als deutlich abgesetzte Gruppe erkennbar stellt die erst kürzlich erkannte Sippe S.
kulundensis dar. Dieser Sippe werden vier ITS-Typen zugeordnet, die sich jeweils nur durch singuläre
Mutationen unterscheiden. Dabei handelt es sich um G/T-Transversionen an unterschiedlichen
Positionen in ITS1 und ITS2.
1.c.) crassifolia-Gruppe
Die mit vier Proben vertretene Gruppe weist bis auf 1485 vollkommen identische ITS-Sequenzen auf.
Ihre Position ist basal zu den abgeleiteten salsa/maritima-Clades. Bisher beschrieben ist nur die
morphologisch gut charakterisierte S. crassifolia, die übrigen Sequenzen gehören Sippen an, die
morphologisch abweichen und näherer Untersuchung bedürfen. Mit S. salsa 1485 findet sich die
Sequenz einer morphologisch eigentlich in den S. salsa-Clade gehörenden Sippe isoliert von dieser.
1.d.) heteroptera-Gruppe
Zu dieser Gruppe gehören zwei relativ unterschiedliche Sequenztypen, die zur Gliederung des
gesamten Subclades beitragen. Es handelt sich um S. japonica/S. heteroptera sowie um zwei
südostasiatische Küstensippen, die bei Lee et al (2007) aIs S. maritima und S. australis geführt
werden. Aus den Phylogenien wird jedoch deutlich, dass die Benennung für beide Arten nicht korrekt
ist und andere Namen gefunden werden müssen. Je zwei Sippen weisen identische ITS-Sequenzen
auf, der Status aller Sippen muss durch weitere taxonomische Untersuchungen geprüft werden.

Ergebnisse
78
1.e.) S. sibirica
Die bisher in S. corniculata s.l. eingeschlossene Sippe nimmt eine Zwischenstellung zwischen den
beiden australischen Linien ein. Mehrere untersuchte Sequenzen waren identisch, so dass von einem
einheitlichen ITS-Typ ausgegangen werden kann. Zu diesem existieren keine ähnlichen Sequenzen,
die Position innerhalb des maritima-Subclades ist relativ stabil, die Beziehung zur australis-Gruppe
allerdings durch bootstrap-Werte nicht gestützt.
1.f.) australis-Gruppe
Analog den anderen Sippen des Clades erscheint auch S. australis keineswegs als molekular
einheitlicher Typ. Die zwei ausgewerteten ITS-Sequenzen weichen mit insgesamt 14
unterschiedlichen Merkmalen sehr deutlich voneinander ab, was durch lange individuelle Äste sichtbar
wird. Die Belege stammen aus geographisch getrennten Regionen West- und Nordaustraliens. Die
Zugehörigkeit von S. malacosperma zu dieser Gruppe ist durch bootstrap–Werte nicht unterstützt. Es
handelt sich um eine deutlich von S. australis unterschiedene Sippe, die in einigen MP-
Baumtopologien als vollkommen eigenständige Linie auftritt. Lee et al (2007) fanden drei geringfügig
unterschiedene ITS-Sequenztypen.
1.g.) S. arbusculoides
Von dem australischen Endemiten S. arbuculoides konnte nur eine Sequenz ausgewertet werden, die
am stärksten von allen anderen des maritima-Subclades abweicht und deshalb sehr stabil in basaler
Position steht. Die Sequenz weist gegenüber dem restlichen Clade 30 Autapomorphien auf. Ähnliche
Sequenzen sind nicht bekannt.
2. prostrata-subclade
2.a.) S. caespitosa
Die Sippe steht ähnlich wie S. arbusculoides an der Basis eines gesamten Subclades und ist durch
eine vergleichbare Zahl von Autapomorphien im ITS (20) gekennzeichnet. Die Zuordnung zum
prostrata-Subclade ist jedoch zweifelsfrei und durch einen bootstrap-Wert von 86% gestützt. Ähnliche
Sequenzen wurden nicht gefunden, die nächst verwandte Sippe des Subclades ist die sehr diverse
Suaeda spicata.
2.b.) spicata-Gruppe
Als Bestandteil des prostrata-Subclades stellt die in vielen Florenwerken nicht von S. maritima
getrennte Gruppe stammesgeschichtlich eine vollkommen separate Ableitung dar und hat mit S.
maritima s.str. wenig gemein. Zu dieser Gruppe gehört neben S. spicata auch die einzige europäische
ausdauernde Brezia, S. maritima var. perennans. S. spicata ist ein Beispiel für sehr hohe
intraspezifische Diversität des ITS, für die sich so gut wie keine morphologische Entsprechung findet.
Daher werden alle Sippen auch als S. spicata benannt. Die Variabilität betrifft ITS1 und ITS2
gleichermaßen. Die Gruppenbildung folgt, soweit bekannt, keinem geographischen Muster.

Ergebnisse
79
2.c.) heterophylla-Gruppe
Die morphologisch gut charakterisierte S. heterophylla bildet die Schwestergruppe zu der
Verwandtschaft von S. prostrata und ist von diesen Sippen sehr deutlich getrennt. Die drei Sequenzen
unterscheiden sich ausschließlich durch C/Transitionen vorwiegend in ITS2.
2.d.) prostrata-Gruppe
Zu dieser außerordentlich diversen Gruppe gehören neben bisher drei bekannten Arten (S.
stellatiflora, S. prostrata, S. olufsenii) wahrscheinlich mehrere weitere. Die morphologisch S.
heterophylla ähnelnde S. stellatiflora scheint näher mit S. prostrata s.l. verwandt. Die osteuropäische
S. prostrata s.str. ist mit zwei unterschiedlichen Sequenzen vertreten, 1161 und 1379. S. prostrata
wird damit eindeutig als Bestandteil der europäischen Flora belegt (1161 aus Österreich). Weitere, bis
jetzt als S. prostrata bezeichnete Sippen weichen morphologisch (Wuchsform) und in ihrem
Verbreitungsgebiet von S. prostrata s.str. ab und sind auch durch ITS-Daten von dieser getrennt. Dies
betrifft die kleinasiatische (1109) und eine Zahl zentralasiatisch verbreiteter Formen (1107, 1463), die
nähere Beziehungen zu der zentralasiatischen Gebirgssippe S. olufsenii aufweisen.
3. corniculata-Subclade
3.a.) tschujensis-Gruppe
Zwei nahe verwandte Sippen, S. arctica und die erst kürzlich neu beschriebene S. tschujensis stehen
weit isoliert vom Rest des Subclades in basaler Stellung. Beide Sequenzen sind sich sehr ähnlich und
unterscheiden sich nur um 2 Transversionen (A/G, T/G) in ITS2. Demgegenüber steht eine Divergenz
von 26 Merkmalen gegenüber der Schwestergruppe. Die Merkmale sprechen für eine sehr lange
Abtrennung schon vor der Besiedlung Amerikas, da sich die rezenten amerikanischen und asiatischen
Sippen viel ähnlicher sind.
3.b.) patagonica-Gruppe
Die sehr diverse Gruppe vereint mit S. patagonica, S. puertopenascoa, S. esteroa, S. linearis, S.
occidentalis und S. mexicana die meisten amerikanischen Sippen der Sektion Brezia, soweit sie hier
berücksichtigt werden konnten. Es handelt sich um insgesamt 6 unterschiedliche Sequenzen, deren
Beziehungen untereinander aber nicht eindeutig sind. Durch bootstrap (99%) wird lediglich die enge
Verwandtschaft von S. esteroa und S. linearis unterstützt, was auch morphologisch begründet ist.
Geographische Muster wie z.B. eine Trennung in nord- und südamerikanische Sippen spiegeln sich
hingegen nicht in der Verwandtschaft der Sippen wider.
3.c.) S. calceoliformis
Die einzige mehr oder weniger separate Entwicklung stellt die Sequenz von S. calceoliformis dar. Sie
weist eine vergleichsweise hohe Zahl von Autapomorphien auf und gruppiert mit keinem Clade. In
einigen MP-Baumtopologie steht S. calceoliformis an der Basis der patagonica-Gruppe, diese Stellung
wird aber durch ML und Bayes nicht unterstützt, so dass eine unabhängige Entwicklung angenommen
werden kann.

Ergebnisse
80
4. corniculata-Gruppe
In dieser Gruppe wurden trotz Auswertung einer Vielzahl (hier nicht dargestellter) Taxa nur drei ITS-
Typen nachgewiesen, die zudem nur geringfügig durch Einzelmutationen verschieden sind. Damit
steht diese Gruppe in starkem Kontrast zu allen übrigen Gruppen des Brezia-Clades. Die Unterteilung
folgt wieder geographischen Regionen. Ein (mit S. pannonica) von Mitteleuropa bis Zentralasien
verbreiteter, häufiger Typ (1062, 1052, 1129) ist von einem Typ mit rein zentralasiatischer Verbreitung
(1026, 1553) getrennt. Die Umsetzung in ein taxonomisches Konzept ist hier noch nicht endgültig
geklärt. Bisher unterschieden wird nur die auch morphologisch eigenständige S. tuvinica. Mit S.
pannonica wird ein Vertreter der S. corniculata-Verwandtschaft eindeutig als Bestandteil der
europäischen Flora ausgewiesen, der mit S. maritima in keinem näheren Zusammenhang steht.
Anhand des Stammbaums wird deutlich, dass an den Küsten und Inland-Halobiotopen Europas
sieben unterschiedliche Sippen der Sektion Brezia vorkommen: S. maritima s.str., S. maritima var.
perennans, S. salsa, S. spicata, S. prostrata, S. salinaria, S. pannonica, die z.T. Teil bezüglich des
ITS sehr divers sind. Hinzu kommt die bisher nicht hinreichend untersuchte S. „venetiana“. Die
Diversität der ITS-Sequenzen ist im Vergleich zu den morphologischen Unterschieden
unwahrscheinlich hoch.

Ergebnisse
81
3.3. Altersdatierung und Molekulare Uhr Mit modifizierten Sequenzalignments wurden getrennt für ITS und atpB-rbcL Maximum Likelihood-
Analysen durchgeführt und mit den erhaltenen Bäumen Substitutionsraten und Divergenzzeiten
berechnet. Das atpB-rbcL-Alignment wurde dabei um einige Taxa der Salsoloideae und
Chenopodieae erweitert, um einen basalen Kalibrierungspunkt der gesamten Chenopodiaceae
einzuschließen (Kadereit et al. 2003). Das ITS-Alignment enthält mehr Taxa der Suaedoideae und als
Schwestergruppe nur einige Vertreter der Salicornioideae. Aufgrund der hohen Sequenzdivergenz ist
ein sinnvolles Alignment mit weiter entfernten Gruppen nicht zweifelsfrei zu konstruieren. Die
Kalibrierungspunkte wurden aus den Arbeiten von Kadereit et al. (2005, 2006) übernommen und sind
in Tab. 9 aufgeführt. Die Außengruppen wurden nur zur Definition der Wurzel und des basalen Splits
der jeweiligen Clades verwendet, in den Bäumen aber nicht dargestellt, da ihr Alter nicht zu
bestimmen ist. Einige Sequenzen der Suaedoideae mussten aus dem Alignment entfernt werden, da
sie in ihrem terminalen Ast mit einer Nulllänge definiert sind. Dies führt zu Problemen bei dem Prozess
der Ratenglättung.
In einem ersten Schritt musste die Prüfung auf Ratenkonstanz erfolgen. Der paarweise
Substitutionsratenvergleich mit dem relative rate test weist, bezogen auf Bassia hyssopifolia (ITS) und
Beta vulgaris (atpB-rbcL) für einen Teil der Sequenzen signifikante Ratenheterogenität nach.
Ausgedrückt wird dies durch P-Werte von <0,05. Unterschiedliche Substitutionsraten zeigen ca. 10%
der Sequenzpaare des atpB-rbcL-Alignments, aber nur 5% der Sequenzpaare des ITS-Alignments.
Mit unterschiedlichen Raten evolvieren sowohl weit entfernte Sequenzen aus unterschiedlichen
Unterfamilien als auch stark divergierende Sequenzen innerhalb von Sektionen. Die Unterschiede
begründen sich z.T. auch aus dem unterschiedlichen taxonomischen Niveau der einbezogenen
Sippen. Da keine Ratenkonstanz vorliegt, wird die Hypothese des Vorliegens einer molekularen Uhr
für beide DNA-Regionen verworfen.
Zum gleichen Ergebnis für beide Datensätze kommt der likelihood ratio test. Die doppelten –lnL-
Differenzen liegen mit 200,92 für atpB-rbcL und 249,04 für ITS deutlich über den kritischen Werten
von 91,67 (Freiheitsgrade=71) und 113,15 (Freiheitsgrade=90). Die Werte unterscheiden sich damit
signifikant. Dies bedeutet, dass die Baumrekonstruktion unter Annahme einer molekularen Uhr zu
einer signifikant weniger wahrscheinlichen Lösung führt und die Berechnung mit der Zulassung freier
Substitutionsraten eine wahrscheinlichere Topologie ergibt. Die Annahme einer molekularen Uhr wird
auch mit dem likelihood ratio test verworfen.
Aus diesen beiden Ergebnissen folgt zwingend der Einsatz von Glättungsverfahren, um ultrametrische
Bäume zu erzeugen und universale Substitutionsraten bestimmen zu können.
Mit dem in der ML-Analyse erhaltenen Baum wurde daher zunächst mit r8s die Kreuzvalidierung
durchgeführt, um den optimalen Glättungsparameter zu bestimmen. Mit dem Glättungsparameter
wurde das eigentliche Verfahren der Ratenglättung durchgeführt.
Alle vorab bestimmten Parameter von Maximum Likelihood und Penalized Likelihood sind in der
folgenden Tabelle zusammengefasst.
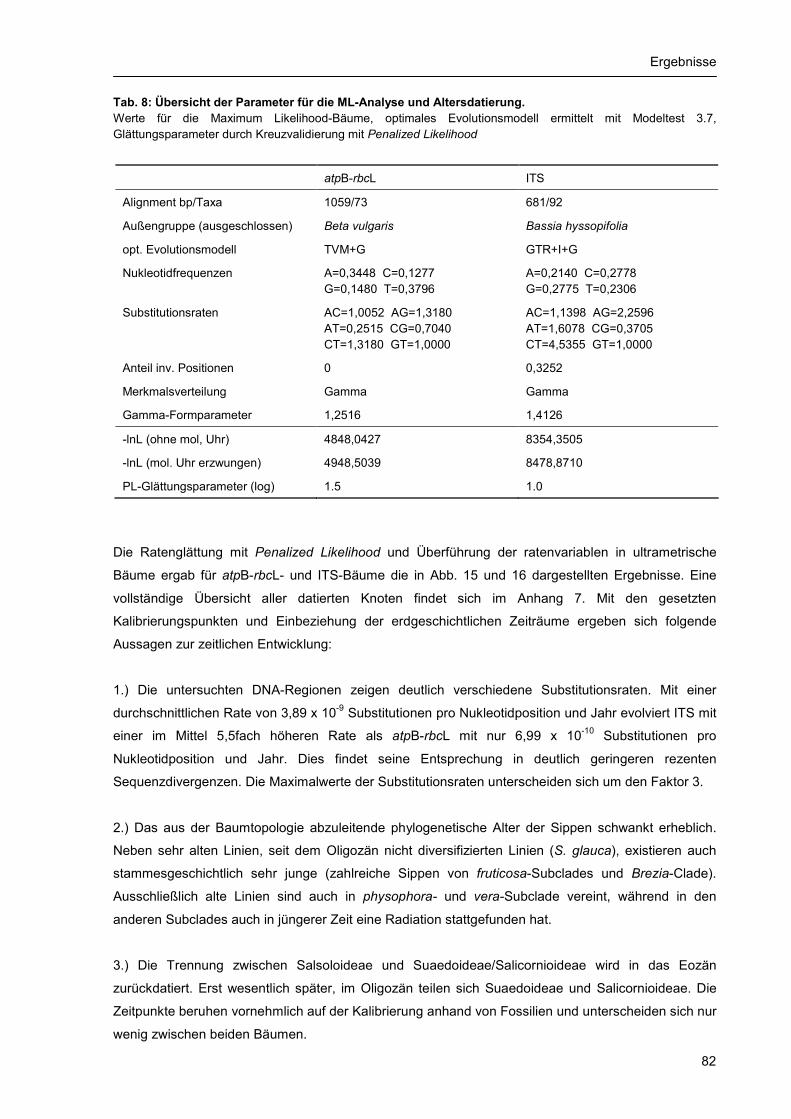
Ergebnisse
82
Tab. 8: Übersicht der Parameter für die ML-Analyse und Altersdatierung. Werte für die Maximum Likelihood-Bäume, optimales Evolutionsmodell ermittelt mit Modeltest 3.7, Glättungsparameter durch Kreuzvalidierung mit Penalized Likelihood
atpB-rbcL ITS
Alignment bp/Taxa 1059/73 681/92
Außengruppe (ausgeschlossen) Beta vulgaris Bassia hyssopifolia
opt. Evolutionsmodell TVM+G GTR+I+G
Nukleotidfrequenzen A=0,3448 C=0,1277 G=0,1480 T=0,3796
A=0,2140 C=0,2778 G=0,2775 T=0,2306
Substitutionsraten AC=1,0052 AG=1,3180 AT=0,2515 CG=0,7040 CT=1,3180 GT=1,0000
AC=1,1398 AG=2,2596 AT=1,6078 CG=0,3705 CT=4,5355 GT=1,0000
Anteil inv. Positionen 0 0,3252
Merkmalsverteilung Gamma Gamma
Gamma-Formparameter 1,2516 1,4126
-lnL (ohne mol, Uhr) 4848,0427 8354,3505
-lnL (mol. Uhr erzwungen) 4948,5039 8478,8710
PL-Glättungsparameter (log) 1.5 1.0
Die Ratenglättung mit Penalized Likelihood und Überführung der ratenvariablen in ultrametrische
Bäume ergab für atpB-rbcL- und ITS-Bäume die in Abb. 15 und 16 dargestellten Ergebnisse. Eine
vollständige Übersicht aller datierten Knoten findet sich im Anhang 7. Mit den gesetzten
Kalibrierungspunkten und Einbeziehung der erdgeschichtlichen Zeiträume ergeben sich folgende
Aussagen zur zeitlichen Entwicklung:
1.) Die untersuchten DNA-Regionen zeigen deutlich verschiedene Substitutionsraten. Mit einer
durchschnittlichen Rate von 3,89 x 10-9 Substitutionen pro Nukleotidposition und Jahr evolviert ITS mit
einer im Mittel 5,5fach höheren Rate als atpB-rbcL mit nur 6,99 x 10-10 Substitutionen pro
Nukleotidposition und Jahr. Dies findet seine Entsprechung in deutlich geringeren rezenten
Sequenzdivergenzen. Die Maximalwerte der Substitutionsraten unterscheiden sich um den Faktor 3.
2.) Das aus der Baumtopologie abzuleitende phylogenetische Alter der Sippen schwankt erheblich.
Neben sehr alten Linien, seit dem Oligozän nicht diversifizierten Linien (S. glauca), existieren auch
stammesgeschichtlich sehr junge (zahlreiche Sippen von fruticosa-Subclades und Brezia-Clade).
Ausschließlich alte Linien sind auch in physophora- und vera-Subclade vereint, während in den
anderen Subclades auch in jüngerer Zeit eine Radiation stattgefunden hat.
3.) Die Trennung zwischen Salsoloideae und Suaedoideae/Salicornioideae wird in das Eozän
zurückdatiert. Erst wesentlich später, im Oligozän teilen sich Suaedoideae und Salicornioideae. Die
Zeitpunkte beruhen vornehmlich auf der Kalibrierung anhand von Fossilien und unterscheiden sich nur
wenig zwischen beiden Bäumen.

Ergebnisse
83
4.) Die Abtrennung von Bienertia als intermediäre Sippe und eine der C4-Linien wird auf ca. auf 31-33
Mio Jahre zurückdatiert. Obwohl ITS- und atpB-rbcL-Bäume unterschiedliche Ableitungen von
Bienertia zeigen, sind die beiden Zeitpunkte durchaus ähnlich. Die Diversifikation von Bienertia in
heute zwei Arten begann hingegen erst im Quartär.
5.) Die Haupt-Trennung von Suaeda in die beiden großen Clades datiert mit ca. 30 Mio. Jahren
ebenfalls auf das Oligozän. Nach ITS-Daten fand dies vor, nach Chloroplasten-Daten erst nach der
beginnenden Diversifikation der Salicornioideae statt. Möglicherweise beeinflusst die unterschiedliche
Position von Bienertia hier die Ergebnisse.
6.) Die Zeitpunkte der Aufspaltung der Hauptclades von Suaeda unterschieden sich deutlich.
Innerhalb des Suaeda-Clades kommt es mit der Abtrennung von S. glauca bereits im Oligozän zu
einer Diversifizierung (24-26 Mio. Jahre), im Brezia-Clade begann dieser Prozess deutlich später. ITS
und atpB-rbcL liefern für Brezia allerdings sehr unterschiedliche Werte. Während der
Aufspaltungsprozess nach ITS-Daten vor 12,4 Mio. Jahren begann, datiert das gleiche Ereignis im
atpB-rbcL-Baum auf 8 Mio. Jahre zurück. Möglicherweise wirken sich hier Unterschiede im sampling
und das Fehlen einiger alter Linien (Suaeda arbusculoides) im Chloroplasten-Baum aus.
7.) Die Diversifizierung des Brezia-Clades kann nur anhand der ITS-Daten sinnvoll rekonstruiert
werden. Die ältesten Abspaltungen sind S. arbusculoides, S. caespitosa, S. spicata und S.
heterophylla, die nach dieser Berechnung mit 8,5-6,8 Mio. Jahren alle noch in das Miozän datieren.
Die wesentliche Differenzierung der drei Subclades erfolgte im Pliozän von 5,3-1,8 Mio. Jahren. Sehr
junge, quartäre Entwicklungen sind hingegen die polymorphen Sippen S. maritima/salsa und S.
corniculata s.l.
8.) Borszczowia aralocaspica als Mitglied einer alten Suaeda-Linie trennt sich von den C3-Sippen des
Schanginia-Subclades im jüngeren Miozän vor 11,21 bzw. 10,37 Mio. Jahren. Auch hier findet sich
wieder eine auffallende Übereinstimmung zwischen beiden Bäumen. Der Zeitpunkt verdeutlicht
zugleich auch das Minimalalter für die Entstehung der C4-Photosynthese in dieser Linie.
9.) Die abgeleiteten Subclades innerhalb von Suaeda (physophora-, fruticosa- und Schoberia-
Subclade) trennen sich im jüngeren Miozän vor etwa 16 Mio Jahren. Welche
Schwestergruppenbeziehung sich daraus entwickelt, bleibt allerdings ungeklärt. Im Chloroplasten-
Baum wird die Beziehung nicht aufgelöst. Für den fruticosa-Subclade kann dieser Zeitpunkt als
Maximalalter für die Entstehung einer C4-Linie angesehen werden, die sich erst viel später
diversifizierte.
9. Die beginnende Diversifizierung der abgeleiteten Subclades wird unterschiedlich rekonstruiert.
Nach ITS-Daten trennten sich die Sippen des physophora-Subclades vor der Aufspaltung von
fruticosa- bzw. Schoberia-Subclade, den Chloroplasten-Daten zufolge erst danach. Die
Diversifizierung des fruticosa-Subclades begann vor derjenigen des Schoberia-Subclades. Die
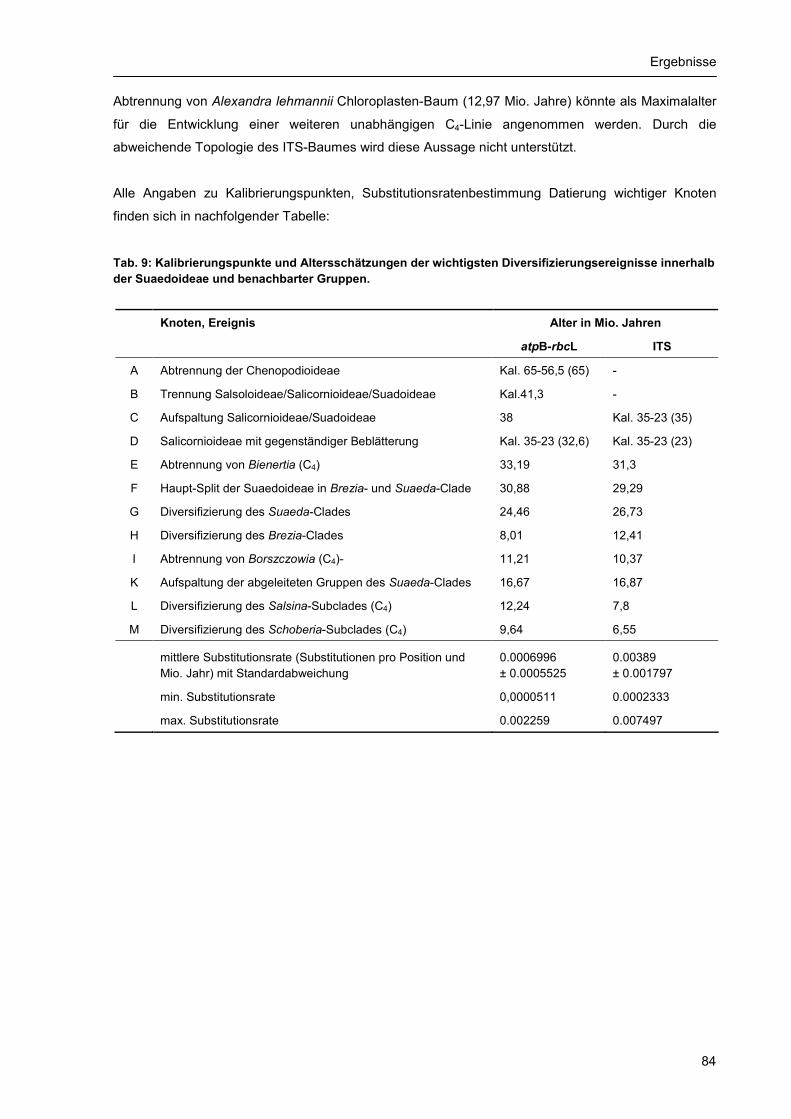
Ergebnisse
84
Abtrennung von Alexandra lehmannii Chloroplasten-Baum (12,97 Mio. Jahre) könnte als Maximalalter
für die Entwicklung einer weiteren unabhängigen C4-Linie angenommen werden. Durch die
abweichende Topologie des ITS-Baumes wird diese Aussage nicht unterstützt.
Alle Angaben zu Kalibrierungspunkten, Substitutionsratenbestimmung Datierung wichtiger Knoten
finden sich in nachfolgender Tabelle:
Tab. 9: Kalibrierungspunkte und Altersschätzungen der wichtigsten Diversifizierungsereignisse innerhalb der Suaedoideae und benachbarter Gruppen.
Knoten, Ereignis Alter in Mio. Jahren
atpB-rbcL ITS
A Abtrennung der Chenopodioideae Kal. 65-56,5 (65) -
B Trennung Salsoloideae/Salicornioideae/Suadoideae Kal.41,3 -
C Aufspaltung Salicornioideae/Suadoideae 38 Kal. 35-23 (35)
D Salicornioideae mit gegenständiger Beblätterung Kal. 35-23 (32,6) Kal. 35-23 (23)
E Abtrennung von Bienertia (C4) 33,19 31,3
F Haupt-Split der Suaedoideae in Brezia- und Suaeda-Clade 30,88 29,29
G Diversifizierung des Suaeda-Clades 24,46 26,73
H Diversifizierung des Brezia-Clades 8,01 12,41
I Abtrennung von Borszczowia (C4)- 11,21 10,37
K Aufspaltung der abgeleiteten Gruppen des Suaeda-Clades 16,67 16,87
L Diversifizierung des Salsina-Subclades (C4) 12,24 7,8
M Diversifizierung des Schoberia-Subclades (C4) 9,64 6,55
mittlere Substitutionsrate (Substitutionen pro Position und Mio. Jahr) mit Standardabweichung
0.0006996 ± 0.0005525
0.00389 ± 0.001797
min. Substitutionsrate 0,0000511 0.0002333
max. Substitutionsrate 0.002259 0.007497

Ergebnisse
85
Abb. 15: Kalibriertes Chronogramm der Suaedoideae sowie ausgewählter Salicornioideae, Salsoloideae und Chenopodieae (als Außengruppen) auf Basis einer ML-Baumtopologie aus 73 atpB-rbcL-Sequenzen. Kalibrierungs- und wichtige Divergenzereignisse s. Tabelle 9; Abk. Q = Quartär, Cp. = Chenopodieae

Ergebnisse
86
Abb. 16: Kalibriertes Chronogramm der Suaedoideae und ausgewählter Salicornioideae auf Basis einer ML-Baumtopologie aus 92 ITS-Sequenzen. Kalibrierungs- und wichtige Divergenzereignisse s. Tabelle 9

Ergebnisse
87
3.4. Evolution von C4-Photosynthese und Blatttypen Die Aufklärung der stammesgeschichtlichen Ableitung der eingangs geschilderten Besonderheiten im
Photosyntheseweg (C4-Photosynthesesyndrom) der Suaedoideae ist wesentlicher Bestandteil der
vorliegenden phylogenetischen Studie. Photosyntheseweg und spezifische Blattmerkmale sind eng
aneinander gekoppelt, die Enstehungswege des C4-Photosynthesesyndroms werden daher anhand
einer Synthese aus molekularer Phylogenie und unterschiedlichen Blatttypen dargestellt.
Im äußerlichen Bau zeigen alle Blatttypen innerhalb der Suaedoideae eine auffallende Ähnlichkeit.
Deutliche Unterschiede zeigen sich hingegen im inneren Bau mit einer spezifischen
Gewebsdifferenzierung. Auf Basis bisheriger blattanatomischer und biochemischer Studien wurden für
die Suaedoideae insgesamt sieben Blatttypen definiert, die sich in anatomischen und physiologischen
Merkmalen sehr deutlich unterscheiden und für bestimmte Gruppen der Unterfamilie typisch sind.
Nach dem Photosyntheseweg werden drei C3-Blattypen und vier C4-Blatttypen unterschieden. Die C4-
Blatttypen unterteilen sich nach Anordnung des C4-Gewebes in jeweils zwei Kranz- und zwei Nicht-
Kranz-Typen (s. Kap. 1.2.3). Die kennzeichnenden Merkmale für jeden Blatttyp sind bei Schütze et al.
(2003) zusammengefasst und werden hier in leicht abgeänderter Form wiedergegeben.
C3-Blattypen 1. Brezia-Typ: ausgesprochen sukkulent, verflacht bis halbzylindrisch, selten zylindrisch; an der
adaxialen Seite gewöhnlich konkav, am Grund blattstielartig verschmälert; Leitbündelsystem (im
Querschnitt) in einer gekrümmten zentralen Ebene angeordnet; 3-4 Lagen Mesophyllzellen zur Mitte
hin stark vergrößert mit abnehmender Chloroplasten-Zahl; die innersten 1(-2) Lagen als
Wasserspeichergewebe ausgebildet, gewöhnlich frei von Chloroplasten und ohne Interzellularen
2. Vera-Typ: vom Grundaufbau wie der Brezia-Typ, von diesem durch schmälere, bikonvexe,
nadelförmige Blätter mit deutlichem Blattstiel unterschieden, deutlich sukkulenter
3. Schanginia-Typ: kaum sukkulent, flach, Leitbündelsystem im Querschnitt in gerader Lage
angeordnet; Zellen der zwei Mesophylllagen in etwa gleich groß und mit gleicher Zahl an
Chloroplasten; Luftzwischenräume durch das gesamte Gewebe
Kranz C4 Blatttypen
4. Salsina-Typ: sehr sukkulent, halbzylindrisch seltener verflacht oder zylindrisch; Leitbündel im
Querschnitt in gerader oder leicht gekrümmter Ebene angeordnet; Hypodermis fehlend; Palisaden-
und Kranzgewebe in randlicher Lage; Kranzzellen von gleicher Größe mit zentripetal angeordneten
Chloroplasten; zwei innere Lagen als Wasserspeichergewebe, ohne Interzellularen und ohne bzw. mit
wenigen Chloroplasten
5. Schoberia-Typ: sehr sukkulent, halbzylindrisch; Hypodermis vorhanden, aus sehr großen
wasserspeichernden Zellen bestehend; Palisaden- und Kranzgewebe die im Querschnitt in
gekrümmter Lage angeordneten Leitbündel umgebend; Kranzzellen sehr verschieden in der Größe,
mit zentrifugal angeordneten Chloroplasten

Ergebnisse
88
Nicht-Kranz C4 Blatttypen 6. Borszczowia-Typ: sukkulent, mehr oder weniger halbzylindrisch, Hypodermis vorhanden; eine
einzige Lage Chlorenchym; einzelne Zellen mit innen liegendem kranzartigen und außen liegendem
palisadenartigen Kompartiment; zahlreiche randlich gelegene Leitbündel, die mit ihrem Phloem dem
Chlorenchym anhaften; zentrales Leitbündel von 1-2 Lagen Wasserspeichergewebe ohne
Chloroplasten und Interzellularen umgeben
7. Bienertia-Typ: sukkulent, halbzylindrisch; Leitbündel im Querschnitt in leicht gekrümmter, zentraler
Ebene angeordnet; Hypodermis fehlend; 1-3 Lagen nicht differenziertes Chlorenchym, jede Zelle mit
einem zentralen, als Kranzzelle fungierenden und einem randlichen, als Palisadenzelle fungierenden
Kompartiment; 2 innere Lagen Wasserspeichergewebe ohne Chloroplasten und Interzellularen
Die beschriebenen Blattypen stellen hochgradig komplexe Merkmale dar, die mit Sicherheit einen
vergleichsweise langen Entwicklungsprozess benötigen und auch längere Zeit erhalten bleiben. Die
Evolution der Blatttypen soll anhand einer abgesicherten Konsensus-Topologie dargestellt werden.
Eine wichtige Frage ist dabei die nach der Zahl der C4-Entstehungsergeignisse. Sie sollten daher
gruppenspezifisch sein. Von fast allen der im Stammbaum dargestellten Taxa konnte die Anatomie
der Blätter untersucht und einem der Blatttypen zugeordnet werden (Freitag & Stichler 2000, 2002,
Freitag, unpubl. Daten), so dass die Gruppenzuordnung gerechtfertigt ist.
Für die Darstellungen zur Evolution der Blatttypen wurde ein kombinierter Datensatz aus allen drei
untersuchten DNA-Regionen ausgewählt, obwohl Chloroplasten- und Kern-Phylogenien z.T.
widersprüchliche Aussagen liefern (Bienertia, Alexandra), was auch durch die Ergebnisse des
partition-homogeneity test unterstrichen wird. Die aus dem kombinierten Datensatz gewonnene
Baumtopologie stellt daher auch eine Kombination der unterschiedlichen phylogenetischen Aussagen
dar. Da für so unterschiedliche Datensätze kein geeignetes Evolutionsmodell gefunden werden kann,
wurde für die phylogenetische Rekonstruktion Maximum Parsimony der Vorzug gegeben und als
Darstellung der strict consensus -Baum gewählt, der nur die wirklich sicheren Verzweigungen enthält.
In dem Datensatz enthalten sind alle Sippen der Chloroplasten-Phylogenie (s. Kap. 3.2.3.) sowie die
Außengruppen aus Kap. 3.1, insgesamt 74 Taxa. Die spezifischen Strukturmerkmale dieses
Datensatzes sind in Tab. 6, Kap. 3.1.2 dargestellt.
Das Alignment für die kombinierte Analyse hat eine Länge von 2382 bp (atpB-rbcL 1022 bp, psbB-
psbH 684 bp, ITS 676 bp). Ausgeschlossen wegen Unsicherheiten oder fehlender Daten wurden 192
bp der cp-Regionen. Von 720 variablen bp waren 424 parsimony-informative. Die MP-Analyse ergab
18 sparsamste Bäume mit einer Länge von 1746 Schritten (CI=0,49, RI=0,88). Die Zahl möglicher
kürzester Bäume ist damit erheblich geringer als in den separaten Analysen. Mit Bassia hyssopifolia
als Außengruppe ergibt sich als Konsensbaum die in Abb. 17 dargestellte Topologie, die als Basis für
die Aufkartierung der verschiedenen Blatttypen verwendet wurde. Für die statistische Absicherung der
Verzweigungen wurde mit dem Gesamtdatensatz eine bootstrap-Analyse mit 1000 Replikaten
durchgeführt. Die schematisierten Blattquerschnitte wurden von H. Freitag entworfen und von D.
Franke (Universität Mainz) erstellt und in der Studie von Schütze et al. (2003) publiziert. Abb. 17 ist
eine erweiterte Variante von Fig. 6 aus der genannten Arbeit.
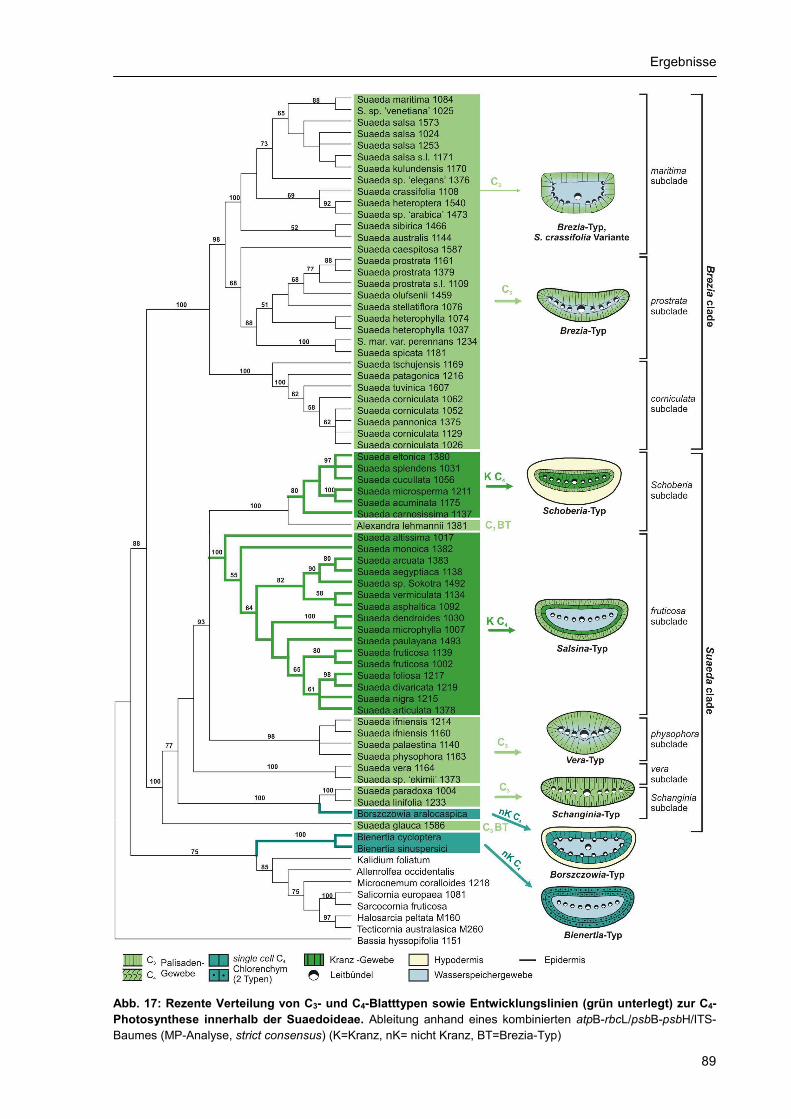
Ergebnisse
89
Abb. 17: Rezente Verteilung von C3- und C4-Blatttypen sowie Entwicklungslinien (grün unterlegt) zur C4-Photosynthese innerhalb der Suaedoideae. Ableitung anhand eines kombinierten atpB-rbcL/psbB-psbH/ITS-Baumes (MP-Analyse, strict consensus) (K=Kranz, nK= nicht Kranz, BT=Brezia-Typ)

Ergebnisse
90
Die Baumtopologie des MP-Konsensusbaumes gibt die Suaedoideen-Gliederung der separaten
Analysen ohne wesentliche Unterschiede wieder. Widersprüchliche Aussagen werden nach
gemeinsamer Merkmalsbewertung unterschiedlich aufgelöst. So wird Bienertia wie in der ITS-Analyse
als Schwestergruppe den Salicornioideae zugeordnet. Alexandra lehmannii nimmt im Gegensatz zur
ITS-Analyse eine isolierte basale Position bezogen auf den Schoberia-Subclade ein, wie dies bereits
in der Chloroplasten-Analyse sichtbar wurde. Die isolierte Stellung von Suaeda glauca gegenüber
dem Schanginia-Subclade wird bestätigt. Die Beziehung der drei abgeleiteten Suaeda-Subclades
(physophora, Schoberia, Salsina) wird im Konsensusbaum nicht aufgelöst. S. altissima ist den ITS-
Daten folgend die am stärksten isolierte Sippe des Salsina-Subclades. Innerhalb des Brezia-Clade
wird die nahe Verwandtschaft von S. crassifolia, S. ’arabica’ und S. heteroptera von
Chloroplastendaten unterlegt. S. kulundensis wird dem maritima-Subclade zugeordnet, was durch
ITS-Daten gestützt wird.
Der dargestellte Baum verdeutlicht die rezente Verteilung unterschiedlicher Blatttypen im Licht der
stammesgeschichtlichen Ableitung und Diversifizierung der beteiligten Sippen. Farblich mit
unterschiedlichen Grüntönen unterlegt wurden im Stammbaum die evolutionären Linien, denen sich
das C4-Photosynthesesyndrom zuordnen lässt. Auf den tatsächlichen Entstehungszeitpunkt kann
daraus nicht geschlossen werden, da historische Merkmalsausprägungen nicht bekannt sind. Die
Darstellung folgt eher einen parsimonischen Ansatz, wonach die Entstehungsereignisse der
komplexen C4-Blätter minimiert sind und sich die Sippen erst nach der Blattentwicklung
diversifizierten. Die Entstehung des C4-Photosynthesesyndroms ist damit lokalisiert zwischen der
Abspaltung einer gedachten ursprünglichen C4- von einer C3-Linie und der ersten Aufspaltung dieser
ursprünglichen C4-Linie.
Die rezente Verteilung der unterschiedlichen Blatttypen ist eng an die in der molekularen Analyse
gefundenen Clades und Subclades gekoppelt. Es handelt sich daher um Merkmale, die eng mit der
Phylogenie und Artbildung in den Suaedoideae verbunden sind und daher auch eine hohe Relevanz
für die Gruppenbildung und Gliederung der Unterfamilie besitzen.
Vorausgesetzt dass die Entwicklungsrichtung stets vom C3- zum C4-Blatt geht, das C4-Blatt somit das
abgeleitete Merkmal darstellt, wird aus der Baumtopologie ersichtlich, dass es innerhalb der
Suaedoideae vier unabhängige Entwicklungen zur C4-Photosynthese mit spezifischen Blatttypen
gegeben haben muss. Drei der vier Entstehungsereignisse kennzeichnen den
Suaeda/Alexandra/Borszczowia-Clade, dort allerdings nur den Suaeda-Subclade. In dem rezent sehr
diversen Brezia-Clade hingegen fand der Prozess der C4-Evolution nicht statt. Ein weiteres C4-
Entstehungsereignis fand in der isoliert zwischen Salicornioideae und Suaedoideae stehenden
Bienertia statt. Es ist weiterhin unzweifelhaft, dass die C4-Linien keinen gemeinsamen Vorfahren
besitzen, sondern jeweils unabhängige Entwicklungslinien darstellen. Damit ist es auch
unwahrscheinlich, dass einerseits lediglich ein ursprünglicher C4-Blatttyp existiert hat, aus dem sich
die rezenten Blatttypen entwickelt haben könnten, andererseits sich ein C4-Blatttyp aus einem
anderen entwickelt haben könnte. Dafür sprechen auch die stets noch vorhanden unmittelbaren C3-
Verwandten der C4-Gruppen.

Ergebnisse
91
Vorkommen der C3-Blatttypen
Brezia-Typ:
Der Blatttyp kennzeichnet hauptsächlich die überwiegend annuellen Sippen des Brezia-Clades (bzw.
der Sektion Brezia). Es handelt sich um einen innerhalb der Chenopodiaceae weit verbreiteten
Blatttyp, der nur wenigen Abweichungen unterworfen ist. Variationen gibt es hinsichtlich des
Sukkulenzgrades. Sukkulenz geht bei der anatomisch etwas abweichenden S. crassifolia mit einer
starken Vergrößerung des inneren Wasserspeichergewebes einher, was insgesamt eine Verdickung
des Blattes bewirkt. Die Leitbündel werden dadurch aus der zentralen Ebene zum adaxialen
Chlorenchym hin verlagert. Da es sich um keinen grundsätzlich abweichenden Typ handelt, wurde
diese spezielle Form als crassifolia-Variante des Brezia-Typs hervorgehoben. Neben S. crassifolia
wurden Tendenzen zu dieser Blattanatomie auch bei den nahe verwandten Sippen S. ‚arabica’ und S.
‚elegans’ beobachtet. Dem Brezia-Typ im weiteren Sinn gehören auch die Blätter von S. glauca und
Alexandra lehmannii an, womit dieser Blatttyp auch innerhalb des Suadea-Clades vorkommt. Das
Blatt von S. glauca zeigt dabei intermediäre Merkmale zum Vera- und Schanginia Typ (s. Kap. 4.4).
Vera-Typ
Der wesentlich sukkulentere Blatttyp ähnelt dem Brezia-Typ und kennzeichnet die meisten der C3-
Sippen des Suaeda-Clades (Suaeda- und physophora-Subclade), die sich damit auch anhand der
Blattanatomie von den C3-Sippen des Brezia-Clades unterscheiden. Es handelt sich fast
ausschließlich um ausdauernde, verholzende Arten. Die Stellung der Sippen im Stammbaum lässt die
Vermutung zu, dass die sukkulenteren C3-Blatttypen eine unmittelbare Weiterentwicklung des Brezia-
Typs sind und dass wiederum Blätter des Vera-Typs den Ausgangspunkt der Kranz- C4-Blatttypen
gebildet haben könnten.
Schanginia-Typ
Innerhalb der C3-Blatttypen stellt der Schanginia-Typ anatomisch eine Sonderentwicklung dar, der
sich durch einen geringeren Sukkulenzgrad und fehlendes Wasserspeichergewebe auszeichnet. Er ist
damit eher untypisch für die Unterfamilie und kommt nur bei den Sippen S. linifolia und S. paradoxa
vor, womit auch anhand der Blattmerkmale die sehr isolierte Stellung dieser Verwandtschaftsgruppe
innerhalb von Suaeda unterstrichen wird. Die Stellung des Schanginia-Clades innerhalb der gesamten
Verwandtschaftsgruppe lässt in Bezug auf die Blattentwicklung den Schluss zu, dass es sich um eine
Vereinfachung der sukkulenten, differenzierten Blattanatomie handelt, die mit dem Verlust des
zentralen Wasserspeichergewebes einher ging.
Vorkommen der C4-Blatttypen
Salsina-Typ
Der Blatttyp kennzeichnet die sehr diverse und weit verbreitete Salsina-Gruppe, zu der in der
traditionellen Systematik vier Sektionen gerechnet werden (Salsina, Macrosuaeda, Immersa und
Limbogermen) und die überwiegend von ausdauernden, verholzenden Arten gebildet wird. Das

Ergebnisse
92
Vorkommen des gleichen, abgeleiteten C4-Blatttyps ist ein Argument gegen eine Unterteilung dieser
Gruppe.
Etwas abweichend vom dargestellten Grundtyp sind die Verhältnisse bei S. monoica. Die Blätter
weisen an den Rändern eine deutliche Lücke im Chlorenchym auf, die auch äußerlich an
durchscheinenden Linien entlang der Blattränder sichtbar wird.
Schoberia-Typ
Der Blatttyp ist kennzeichnend für den Schoberia-Subclade und tritt in keiner anderen Gruppe auf.
Ähnlich dem Salsina-Typ handelt es sich um einen aus Vorfahren des Suaeda-Clade von sukkulenten
C3-Vorfahren abgeleiteten Blatttyp. Die molekularen Daten lassen erkennen, dass es sich bei
Schoberia- und fruticosa-Subclade um näher verwandte Gruppen handelt.
Borszczowia-Typ
Wie bereits aus den vorangegangenen Darstellungen ersichtlich, teilt Borszczowia aralocaspica
gemeinsame Vorfahren mit den Schanginia-Arten S. linifolia und S. paradoxa. Der Borszczowia-
Blatttyp kennzeichnet eine einzige rezente Sippe und stellt eine im gesamten Pflanzereich einmalige
anatomische Erscheinung dar. Die Entwicklung dieses Blatttyps ging nicht mit einer Radiation der
betroffenen Sippe einher. Ein möglicher gemeinsamer Ausgangspunkt für die vollkommen
unterschiedlichen Blatttypen ist jedoch weit schwerer rekonstruierbar als es die relativ nahe
Verwandtschaft in der molekularen Phylogenie implizieren würde.
Bienertia-Typ
Der Blatttyp kennzeichnet die isoliert stehende Gattung Bienertia und stellt wie der Borszczowia-Typ
einen sehr seltenen Sonderweg zur C4-Photosynthese dar. Er kennzeichnet mit Bienertia eine schon
lange vor der Diversifizierung von Suaeda abgetrennte Entwicklungslinie, die intermediär zwischen
Salicornioideae und Suaedoideae steht. Es ist damit faktisch ausgeschlossen und durch die
Ergebnisse der molekularen Phylogenie widerlegt, dass sich die beiden Formen der single cell C4
photosynthesis auf einen gemeinsamen Vorfahren zurückführen lassen.

Ergebnisse
93
3.5. Merkmalsentwicklung anhand molekularer Phylogenien Neben den im vorangegangenen Kapitel aufgezeigten Blattmerkmalen, die in Kombination mit den
biochemischen Eigenheiten der Photosynthese gruppenspezifisch sind, gibt es eine Reihe weiterer,
für die Systematik der Gruppe relevanter Merkmale. Einige dieser in der taxonomischen Literatur oft
kontrovers diskutierten Merkmale sollen anhand der molekularen Phylogenie bewertet werden.
Ähnlich der Darstellung der Blattentwicklung werden auch hier nur die Merkmale der terminalen
Sippen wiedergegeben, da die historischen Zustände der komplexen Merkmale nicht rekonstruierbar
sind. Folgende vier Merkmalskomplexe, die auch in der bisherigen systematischen Literatur z.T. schon
als gruppenspezifisch gelten, wurden ausgewählt:
a.) Pistill-Morphologie (Form des Ovars, Besonderheiten, Narbenzahl und -ausprägung)
b.) Perianth-Morphologie (Formen, Anhänge/Auswüchse, Verwachsungen)
c.) Lebensform, Verholzung, Generationszeiten
d.) Ploidiestufen
Für die Darstellung wurde hier die ML-Topologie des erweiterten ITS-Datensatzes benutzt (s. Kap.
3.2.2.), da sie die meisten Taxa enthält und auch als weitgehender Konsensus unterschiedlicher
Phylogenien interpretiert werden kann. Durch Vergleich mit den Baumtopologien der
vorangegangenen Kapitel lassen sich dort, wo Unterschiede auftreten aber auch alternative Wege der
Merkmalsentwicklung leicht nachvollziehen.
a.) Pistill-Morphologie
Die von Iljin eingeführten Pistill-Merkmale (Iljin 1936a) wurden von H. Freitag an zahlreichen Sippen
untersucht und in fünf unterschiedliche Typen zusammengefasst, die prinzipiell der Iljin’schen
Gliederung entsprechen. Ihre Beschreibung wird hier in unveränderter Form aus Schütze et al. (2003)
wiedergegeben:
1. Brezia-Typ
Narben 2(3), kurz, dick, oft verflacht, mit kurzen Papillen; direkt an der Spitze des verschmälerten
Ovars entspringend, das einen kurzen Griffel darstellt.
2. Schanginia-Typ
Narben (2)3. lang, dünn, mit langen Papillen, aus einer flachen Vertiefung an der Spitze des breit
abgerundeten Ovars entspringend.
3. Vera-Typ
Ovar ähnlich dem Schanginia-Typ, Zahl der Narben schwer zu bestimmen, diese schild- oder
sternförmig verbreitert, Papillen kurz.
4. Schoberia-Typ
Narben (2)3(4), lang und dünn, sehr selten kurz, mit verlängerten Papillen, aus einer tiefen
Einbuchtung mit umgebender kragenartiger Struktur aus einem zusammengezogenen Ovar
entspringend
5. Bienertia-Typ
Ähnlich dem Brezia-Typ, von diesem durch kopfig verdickte Narben unterschieden.

Ergebnisse
94
Die Aufkartierung dieser Merkmale auf die molekularen Stammbäume lässt eindeutig die
systematische Relevanz dieses Merkmalskomplexes erkennen. Die unterschiedlichen Pistill-Typen
kennzeichnen monophyletische Gruppen und stellen für diese synapomorphe Merkmale dar. Der
Brezia-Typ kommt ausschließlich bei dem Brezia-Clade vor. Der morphologisch ähnliche Bienertia-
Typ nur bei der isoliert stehenden Gattung Bienertia. Alle Sippen des Suaeda-Clades hingegen haben
unterschiedliche, vom Brezia-Typ abweichende Pistilltypen entwickelt. Der Schanginia-Typ verbindet
die morphologisch-anatomisch deutlich verschiedenen Sippen des Schanginia-Subclades und
prinzipiell auch S. glauca. S. glauca hat jedoch fast immer 2 Narben. Der Vera-Typ mit der
einzigartigen Narben-Morphologie stellt eine Sonderentwicklung dar und wird nur von den beiden
Arten des Suaeda-Subclades repräsentiert. Verbindendes Merkmal der drei abgeleiteten Subclades
fruticosa, Schoberia und physophora ist der Schoberia-Typ, der die Gruppe unabhängig von der
diversen Blattentwicklung herausstellt.
b.) Perianth-Morphologie
Verwachsungen von Ovar und Perianth sowie morphologische Abweichungen des Perianths wurden
in der Vergangenheit für die Taxonomie hoch bewertet. Auf diesen Merkmalen beruhte in erster Linie
die Beschreibung von Sektionen (Immersa, Schanginia) und Gattungen (Bienertia, Alexandra,
Borszczowia, Brezia heterophylla).
Wie die Synthese mit der molekularen Phylogenie zeigt, können diese Merkmale kaum zur Definition
monophyletischer Gruppen genutzt werden. Relativ vielgestaltig entwickelt sich das Perianth innerhalb
des Brezia-Clades. Mehrere Linien entwickelten hier horizontale Flügel. Bei S. heterophylla
innervieren sie am mittleren Teil des Tepalums und sind besonders stark ausgeprägt. Die nahe
verwandte Sippe S. stellatiflora hat dagegen nur schwach entwickelte dreieckige Auswüchse, die nicht
als echte Flügel gewertet werden. Horizontale Flügel in basaler Position kommen bei S. heteroptera,
S. sibirica und S. kulundensis vor. Beide Arten sind aber nicht näher miteinander verwandt.
Horizontalflügel treten ebenfalls bei der isoliert stehenden Bienertia auf, in diesem Fall sind sie zu
einem geschlossenen Ring verwachsen, was sie auch dadurch von Suaeda unterscheidet. Vertikal
angeordnete Flügel kommen hingegen nur bei der Gattung Alexandra in einem Verwandtschaftskreis,
dessen Perianth ansonsten einheitlich ohne Anhänge oder Umbildungen entwickelt ist, vor.
Möglicherweise gehört S. pterantha, die ebenfalls Vertikalflügel besitzt, in diese Gruppe. Asymmetrien
und hornartige Auswüchse der Tepalen kennzeichnen die Sippen des corniculata-Subclades sowie
die Sippe S. kulundensis. Innerhalb des corniuculata-Subclades zeigen einige Sippen dieses Merkmal
nicht mehr (S. mexicana, S. linearis, S. occidentalis).
Verwachsungen von Blütenhülle und Ovar (S. aegyptiaca, S. arcuata, Bienertia, Borszczowia) ist ein
mehrfach unabhängig entstandenes Merkmal, was ebenfalls nicht zur Kennzeichnung verwandter
Gruppen benutzt werden kann.
c.) Lebensform In der Gattung Suaeda sind einjährige (Therophyten) und ausdauernde, verholzende (Chamaephyten)
Arten bekannt. Die Veränderung der Generationszeit geht immer mit der Entwicklung zu
Zwergsträuchern einher, andererseits zeigen aber auch Annuelle gelegentlich Verholzungstendenzen.

Ergebnisse
95
Da dieser Merkmalskomplex systematische Bedeutung haben sollte, wurde die Lebensformen anhand
der molekularen Phylogenie dargestellt.
Aus dem Stammbaum wird deutlich, dass die beiden Lebensformen sehr unterschiedlich verteilt und
keineswegs gruppenspezifisch sind. An der Basis des Suaeda/Alexandra/Borszczowia-Clades stehen
mit S. glauca, dem Schanginia- und dem weitaus größten Teil des Brezia-Clades annuelle Arten. Auch
die in Chloroplastenbäumen nächst verwandte Bienertia enthält nur annuelle Arten. Dies könnte
bedeuten, dass sich Suaeda von annuellen Vorfahren ableitet. Neben rein annuellen Gruppen
(Schanginia-Clade, Schoberia-Clade) gibt es mit dem physophora-Clade eine ausschließlich
ausdauernde Gruppe. Innerhalb des Brezia-Clades und fruticosa-Clades kommen beide
Lebensformen vor. Innerhalb von Brezia überwiegt die annuelle Lebensform, ausdauernde Sippen
entwickeln sich daraus an 5 unabhängigen Positionen: S. esteroa/linearis, S. caespitosa, S.
arbusculoides, S. australis und S. maritima var. perennans. Der umgekehrte Fall tritt innerhalb des
fruticosa-Subclades auf. Hier überwiegen ausdauernde Formen, von denen sich mit S. aegyptiaca und
S. arcuata annuelle Formen ableiten. Eine Sonderstellung innerhalb dieses Clades nimmt die annuelle
S. altissima ein. Da ursprüngliche Zustände mit den gewählten Markern nicht sinnvoll rekonstruiert
werden können, wird bei der Beurteilung der Lebensformen-Ableitung nach dem parsimonischen
Prinzip argumentiert, wonach die Zahl der Transformationen minimal sein soll. Es können auch
andere Entstehungswege angenommen werden. Das Merkmal Lebensformen ist aber zu variabel, um
daraus Gruppierungen zu bilden, allenfalls auf Ebene der Art kann es zur Sippencharakteristik
verwendet werden.
d.) Ploidiestufe
Vervielfachung des Chromosomensatzes durch Auto- oder Allopolyploidie ist ein im Pflanzenreich weit
verbreitetes Phänomen und nicht unbedingt an Artgrenzen gebunden. Innerhalb der Suaedoideae
sind, ausgehend von der Grundzahl x=9 fünf Ploidiestufen bekannt geworden. Anhand des
Stammbaumes soll dargestellt werden, in welchen Gruppen diploide und in welchen polyploide Sippen
vorkommen. Soweit bis jetzt bekannt, ist ein Großteil der Arten diploid geblieben. Diploide Sippen
finden sich besonders in basaler Position größerer Gruppen (Bienertia, S. glauca, S. linifolia) und auch
untergeordneter Clades (S. monoica, S. altissima, S. tschujensis). Bei der Vervielfachung des
Chromosomensatzes werden Verdopplung und weitere Vervielfachung unterschieden. Die
Verdopplung des diploiden Chromosomensatzes, die zu tetraploiden Genomen führt, lässt sich
mindestens sechs mal unabhängig lokalisieren: S. vera, S. foliosa-Gruppe, S. vermiculata-Gruppe, S.
corniculata-Gruppe, S. spicata und S. salsa/maritima. Damit sind alle Sippen bzw. Gruppen erfasst,
die keine weitere Erhöhung der Ploidiestufe vollzogen haben. Eine weitere Erhöhung der Ploidiestufe
kommt außerhalb von Brezia nur bei S. fruticosa vor, von der zwei Ploidiestufen bekannt sind.
Innerhalb von Brezia zeichnet sich besonders der corniculata-Subclade durch hohe Variabilität der
Ploidiestufen aus. Tetra- bis dekaploide Sippen kommen bei S. coniculata s.l. vor, die ITS-Sequenzen
der Karyorassen sind praktisch identisch. Eine ähnliche offenbar unabhängige Entwicklung fand in
den benachbarten, molekular diversifizierten amerikanischen Sippen des Subclades statt, auch hier
möglicherweise bis zur Dekaploide. Dafür fehlt jedoch noch der molekulare Nachweis (S. rolandii). S.
kulundensis und S. sibirica sind weitere unabhängige polyploide Linien.
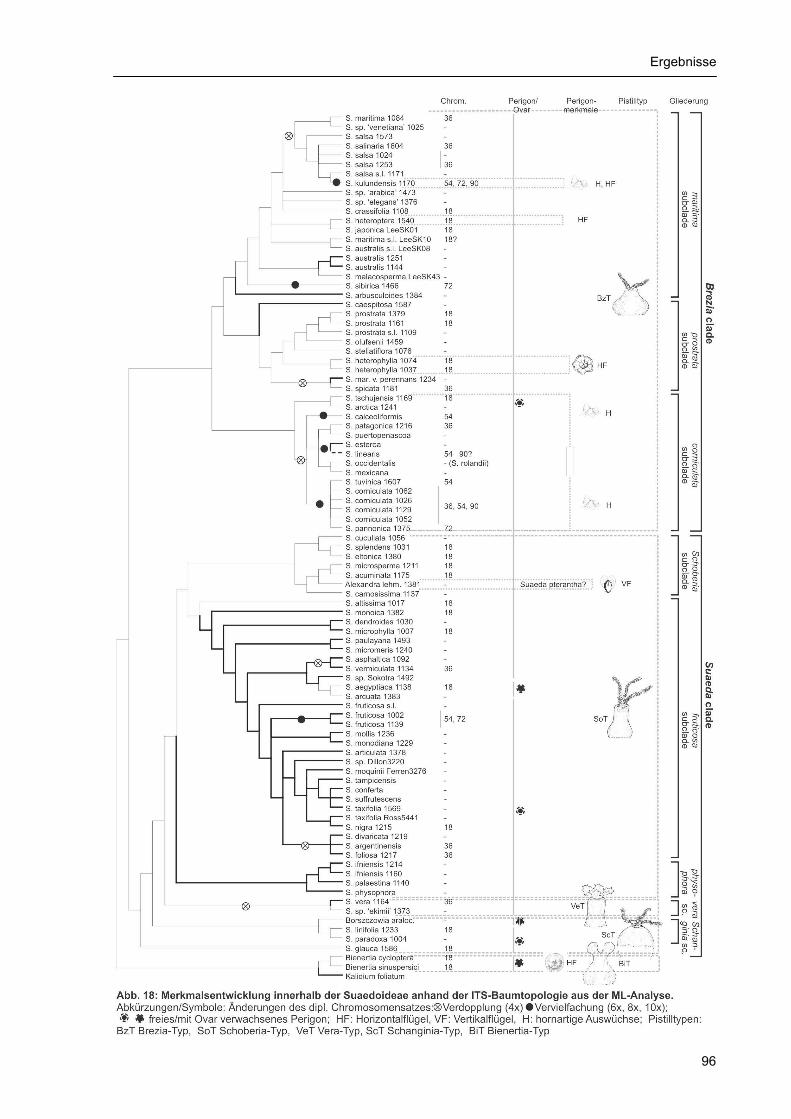
Ergebnisse
96

Diskussion
97
4. Diskussion 4.1. Qualität, Aussagekraft und Grenzen der Daten und Methoden 4.1.1. Auswahl und Umfang der Sippen Die vorgelegte Arbeit basiert auf einer breit angelegten, den gesamten Verwandtschaftskreis der
Suaedoideae weitgehend berücksichtigenden Auswahl. Die Ergebnisse erlauben damit eine
umfassende Sicht auf die molekulare Variabilität im Verhältnis zur morphologischen Konzeption der
Gruppe sowie die Rekonstruktion von statistisch gut abgesicherten verwandtschaftlichen
Beziehungen, die aus der rezenten Diversität ermittelt werden. Es wurde Wert darauf gelegt, alle
aktuell anerkannten Sektionen der Gattung Suaeda (Schenk & Ferren 2001) ausreichend zu
berücksichtigen sowie nahe Verwandte, die mögliche Schwestergruppen darstellen ausreichend
einzubeziehen. Dabei wurden auch aktuelle Ergebnisse der molekularen Phylogenie der
Chenopodiaceae (Kadereit et al. 2003) sowie neuerer taxonomischer Untersuchungen (Akhani et al.
2005, Lomonosova et al. 2008) berücksichtigt. Mit den Gattungen Suaeda, Alexandra, Bienertia und
Borszczowia ist die Unterfamilie der Suaedoideae (Ulbrich 1934) mit Ausnahme der nicht mehr
zugehörigen Gattung Hypocylix komplett repräsentiert. Die monotypischen Sektionen Immersa,
Macrosuaeda, Physophora und Suaeda sind alle einbezogen. Von den artenreicheren Sektionen
wurde die höchst mögliche Probenzahl angestrebt. Besonderer Wert wurde dabei auf die
Repräsentation kritischer taxonomischer Einheiten gelegt. Etwa 40% der einbezogenen Suaeda-
Sippen gehören der Sektion Brezia an, in der die größten taxonomischen Unklarheiten bestehen.
Einige kritische Brezia-Sippen sind durch mehrere Akzessionen belegt, dies betrifft vor allem Suaeda
maritima, S. corniculata, S. prostrata, S. spicata und S. salsa. Fragliche Sektionen, wie die
weitgehend auf geographischer Trennung basierende Sektion Limbogermen, wurden ebenfalls
berücksichtigt.
Als Außengruppen wurden einige Vertreter der Salicornioideae ausgewählt, um die Clades der
phylogenetischen Analyse richtig zu werten und die Variabilität der untersuchten Regionen auch
gruppenübergreifend einschätzen zu können. Als entferntere Außengruppen für die Bewurzelung der
Bäume wurde mit Bassia hyssopifolia ein Vertreter der Salsoloideae gewählt. Basierend auf diesen
Ergebnissen konnte in den erweiterten Phylogenien der Suaedoideae die Außengruppe neu bestimmt
werden (Kalidium), da die Sequenzunterschiede besonders im ITS zwischen Bassia und den
Suaedoideae so hoch sind, dass ein zweifelsfreies Alignment nicht möglich ist. Für die Altersdatierung
anhand von atpB-rbcL wurde zudem das sampling noch um einige Vertreter der Salsoloideae,
Chenopodieae sowie Beta vulgaris erweitert.
Die Ergebnisse basieren auf der Auswertung von insgesamt 294 Sequenzen, davon 99 atpB-rbcL-
Sequenzen, 70 psbB-psbH-Sequenzen und 125 ITS-Sequenzen. Die unterschiedliche Zahl begründet
sich einerseits aus unterschiedlichen Fragestellungen auf unterschiedlicher taxonomischer Ebene,
andererseits aus technischen Schwierigkeiten bei der Generierung von Chloroplasten-Sequenzen. Da
sich ITS als variabelste DNA-Region herausstellte, liegen für diese Region auch die meisten
Sequenzen vor. Die 294 Sequenzen stammen von insgesamt 66 bekannten und 7 weiteren, bisher
nicht beschriebenen Suaeda-Arten. Die Gattung ist daher je nach Gesamtschätzung mit 60-80% aller
Arten repräsentiert.

Diskussion
98
4.1.2. Vergleichende DNA-Sequenzierung Die vergleichende DNA-Sequenzierung hat sich mittlerweile als ein probates und erfolgreiches
Instrument für phylogenetische, populationsbiologische und biogeographische Untersuchungen
etabliert. Für Fragestellungen auf niedrigem taxonomischen Niveau werden nichtkodierende Regionen
bevorzugt, weil auf ihnen weniger funktionelle Zwänge liegen und damit die erwartete Variabilität
höher ist. Mit der Verwendung nichtkodierender Regionen soll gerade ein von den morphologischen
Daten unabhängiger Datensatz gewonnen werden, mit dem das Ergebnis weitgehend zufälliger
Mutationsprozesse rekonstruiert werden kann. Mit der Entwicklung effektiver Mechanismen zur DNA-
Sequenzierung wurde es möglich, immer mehr DNA-Regionen bei phylogenetischen Analysen zu
berücksichtigen und dabei auch unterschiedliche Genome wie Kern und Plastiden zu vergleichen. Als
Beispiele für Multigenanalysen seien genannt: Cuenod et al. (2002), Li et al. (2002), Simmons et al.
(2001), Kadereit et al. (2006), Kapralov et al. (2006). Der Vorteil von Multigenanalysen liegt in der
Vergleichsmöglichkeit der Evolutionswege unterschiedlicher Genome aus dem gleichen Organismus.
Liefern die einzelnen Genome ähnliche phylogenetische Signale, so kann die Kombination zu
verbesserten Aussagen führen. Mit Plastidengenomen können zudem maternale oder paternale Linien
rekonstruiert werden. Für Pflanzen kommt dabei in erster Linie der Chloroplast in Frage, für den
mittlerweile eine Vielzahl an geeigneten Regionen für phylogenetische Studien bekannt ist und auch
viele Sequenzen als Vergleichsmaterial vorliegen (Shaw et al. 2005).
Der Vorteil von Organellen-DNA liegt vor allem darin, dass sie nur in einer Variante in der Zelle
vorhanden sind und Rekombination weitgehend ausgeschlossen ist. Variabilität entsteht vor allem
durch slipped strand-Fehlpaarungen, Insertionen und Deletionen im Zusammenhang mit
Sekundärstrukturbildung, Inversionen verbunden mit hairpin- und stemloop-Strukturen und
Nukleotidsubstitutionen (Kelchner 2000). Rekombination spielt hingegen kaum eine Rolle.
Die vorliegende Studie beruht auf Sequenzunterschieden von drei DNA-Regionen aus Kern und
Chloroplastengenom: ITS, atpB-rbcL und psbB-psbH. Die Regionen haben jeweils eine Länge von
etwa 600-800 bp. Die optimalen Regionen des Chloroplasten-Genoms wurden durch Testreihen mit 6
Taxa aus 8 möglichen Regionen ausgewählt. Es wurden folgende Regionen auf Amplifizierbarkeit und
Variabilität getestet: atpB-rbcL, trnH-psbA, rps16 intron, psbB-psbH, trnT-trnL, trnL intron und trnL-trnF
trnS-trnG. Nur für einen Teil der getesteten Regionen konnten Vergleichsergebnisse erhalten werden.
Um den labortechnischen Aufwand bei der relativ großen Anzahl an Taxa zu begrenzen, wurde die
Auswahl auf zwei DNA-Regionen beschränkt, die einen hohen Anteil an Punktmutationen und
möglichst wenig Längenvariabilität zeigten. Die gewählten Regionen atpB-rbcL und psbB-psbH ließen
sich zumindest aus Frischmaterial problemlos amplifizieren und zeigten im Vortest eine genügende
Zahl an Punktmutationen. Erst im Nachhinein erwies sich die atpB-rbcL-Region als ausgesprochen
längenvariabel, was zu Problemen im Alignment führte. Die Variationsbreite umfasste etwa 300 bp
zwischen den längsten und kürzesten Sequenzen. Die Variabilität der erhaltenen Sequenzen lag mit
Werten von bis zu 12% paarweiser Sequenzdivergenz in vergleichbaren Größenordnungen. Weniger
längenvariabel erwies sich der psbB-psbH-Spacer, der eine noch geringere Variabilität (bis ca. 9%)
zeigte. Die durchschnittliche Variabilität erreichte in den gewählten Regionen nur 20-25% von der bei
ITS. Ähnliche Unterschiede der Sequenzdiversität zwischen atpB-rbcL und ITS fanden Kadereit et al.
(2006) in den Salicornioideae.

Diskussion
99
Die ITS-Region war für die Suaedoideae relativ leicht zu sequenzieren und erwies sich durchweg als
sehr variabel. Die Größenordnungen lagen mit 615-640 bp Länge im durchschnittlichen Bereich der
Angiospermen, die paarweisen Sequenzdivergenzen mit bis zu 50% höher als die üblichen Werte von
10-30% (Baldwin et al. 1995). Ein kennzeichnendes Merkmal sind auch die hohen GC-Gehalte.
Obwohl ITS sehr oft für phylogenetische Studien eingesetzt wird, sind die Aussagen, die daraus
gewonnen werden, nicht uneingeschränkt verwertbar. ITS wird biparental vererbt und wird beeinflusst
durch Homogenisierungsprozesse, die so genannte concerted evolution. Dabei kann sich ein Typ
durchsetzen, es kann aber auch zur partiellen Restrukturierung und Neukombination führen. Wie weit
dieser Prozess schon fortgeschritten ist oder welche Sequenzen wirklich im Genom vorliegen, lässt
sich durch direkte Sequenzierung nur bedingt klären. Nicht klonierte, direkt amplifizierte Sequenzen
sind ein Konsens vieler Zielregionen aus einem oder mehreren Chromosomen (Nieto Feliner &
Roselló 2007).
4.1.3. Phylogenetische Rekonstruktionsverfahren und Datierungsmethoden Die vorgelegten Ergebnisse der Stammbaumrekonstruktion beruhen auf einer Vielzahl von Annahmen
von Voraussetzungen, nach deren Kriterien alle Aussagen zu bewerten sind. Der Prozess der
Evolution und ihre Abbildung in einer Phylogenie der Organismen lässt sich als Prozess nicht
beobachten. Alle Aussagen zur Phylogenie tragen daher hypothetischen Charakter. Die drei zur
Rekonstruktion der Phylogenien benutzten Methoden Maximum Parsimony, Maximum Likelihood und
Bayes’sche Analyse beruhen alle auf Optimalitätskriterien, die sich zwischen den Methoden
grundsätzlich unterscheiden. Maximum Parsimony erklärt den Evolutionsprozess mit der minimal
möglichen Anzahl an Schritten, ohne ein Evolutionsmodell vorauszusetzen und arbeitet
vergleichsweise schlecht bei hohen Distanzen (long branch attraction). Da nur die parsimonie-
informativen Merkmale gewertet werden, geht ein Großteil der Sequenzinformationen verloren. Eine
gewisse Abhilfe schafft hier das Einbeziehen von Längenmutationen durch die Kodierung von Indels.
Es verbleibt natürlich die Frage, ob die Evolution wirklich parsimonisch abläuft. Maximum Likelihood
nutzt alle Merkmale zur Auswertung und optimiert einen gegebenen Baum (z. B. neighbor joining)
anhand eines zuvor ermittelten optimalen Evolutionsmodells. Das Modell kann dabei immer weiter
verfeinert werden, doch führt ein besseres Modell oft nur zu einem besseren Likelihood-Wert, nicht
aber zu einer exakteren Phylogenie (Huelsenbeck & Crandall 1997). Maximum Likelihood sehr ähnlich
ist der Bayes’sche Ansatz, der auf bedingten Wahrscheinlichkeiten basiert (s. Kap. 2.4.3). Alle
Berechnungsverfahren setzen eine Baumstruktur der zu errechnenden Phylogenien voraus. Dies ist
aber oft nicht gegeben. Grund dafür sind Rekombinationsereignisse und retikulate Evolution, die der
Diversifikation, dargestellt durch phylogenetische Bäume, entgegenlaufen. Werden bekannte Hybride
ausgeschlossen und die phylogenetischen Berechnungen nur mit diploiden, nicht hybridogenen
Sippen durchgeführt, sollte dies zu verbesserten Aussagen führen (Blattner 2004). Hybridogene Taxa
können anschließend, sofern ihre Elternarten bekannt sind, als konvergente Linien separat dargestellt
werden.
Die mit den geschilderten Verfahren erzeugten phylogenetischen Bäume waren trotz der Unterschiede
in den Berechnungsverfahren weniger von der Methode, als vielmehr von der Konstruktion des
Alignment abhängig. Besonders drastisch war dies bei dem hoch variablen ITS zu beobachten, bei

Diskussion
100
dem die Erstellung des Alignments für bestimmte Bereiche nicht zweifelsfrei durchgeführt werden
konnte (vgl. Kap. 3.1). Schon geringe Änderungen der Parameter, resultierend in veränderten
Nukleotidgruppierungen, führten zu veränderten Baumtopologien. Im ITS-Stammbaum der
Suaedoideae erwies sich dabei besonders der Suaeda-Clade als instabil hinsichtlich der
Baumtopologien, da die ITS-Sequenzen des Suaeda-Clades deutlich variabler als die des Brezia-
Clades sind. Dies erklärt z.B. auch abweichende Baumtopologien in der Phylogenie der Suaedoideae
bei Lee et al. (2007). Die Baumtopologie des Brezia-Clades war hingegen sehr stabil.
Trotz der geschilderten Unsicherheiten sind die Aussagen der Chloroplasten- und ITS-Stammbäume
grundsätzlich gleich. Die Haupt-Teilung von Suaeda in zwei große Clades, die Untergliederung in
Subclades als monophyletische Gruppen sowie den Einschluss von Borszczowia und Alexandra in
diese Verwandtschaftsgruppe spiegelt sich in allen rekonstruierten Phylogenien wider. Die terminalen
Taxa behalten ihre Gruppenzugehörigkeit bei, nur die Beziehung der Gruppen untereinander variiert.
Widersprüche zwischen Chloroplasten- und ITS-Stammbäumen bestehen nur in der Position von
Bienertia und Alexandra (bei ITS innerhalb von Schoberia).
Der Vergleich des phylogenetischen Signals mit statistischen Tests (partition-homogeneity test) ergab
nur zwischen den Chloroplastenregionen gleiche Ergebnisse, weshalb beide Regionen in den
Analysen auch kombiniert werden konnten. ITS und Chloroplastenregionen liefern hingegen
unterschiedliche phylogenetische Signale, daher sollten die Regionen auch nicht kombiniert werden.
Interessant ist auch die Tatsache, dass ITS1 und ITS2 unterschiedliche Signale liefern. Ein
vergleichsweise durchgeführter Test zwischen beiden Regionen ergab einen signifikanten Unterschied
(P=0,01). Die aus den einzelnen ITS-Regionen rekonstruierten Phylogenien wiesen auch wirklich
abweichende Baumtopologien auf.
Für die Ratenglättung wurde ausschließlich das Verfahren Penalized Likelihood (Sanderson 1997)
eingesetzt, um ultrametrische Bäume zu erhalten. Vergleichbare Untersuchungen haben gezeigt, dass
die Ergebnisse der Raten- Divergenzzeitbestimmung weniger vom Verfahren, als viel mehr von den
Kalibrierungspunkten abhängt (Cabrera 2007).
4.2. Inter- und intraspezifische Variabilität Die Auffassung von Arten als reproduktiv isolierten Gruppen (biologische Art) ist zumindest im
Pflanzenreich nur bedingt gültig und anwendbar. Mit diesem Konzept wäre ein Großteil der
morphologisch unterscheidbaren Arten als eine, dann allerdings sehr variable Sippe aufzufassen. Dies
gilt in besonderem Maße für viele Arten der Gattung Suaeda. Da mit reproduktiv definierten Arten die
vorhandene Diversität kaum ausreichend beschrieben ist, müssen andere Kategorien benutzt werden.
Wird der Entstehungsprozess berücksichtigt dann ist die Artbildung als Prozess zunehmender
evolutionärer Divergenz zu betrachten, der unablässig stattfindet. Grant (1971) schlägt für die
Abkömmlinge einer Ursprungspopulation mit zunehmendem Isolationsgrad folgende Einheiten vor:
Ursprungspopulation - Kolonien A/B - geographische Rassen A/B - Semispecies A/B - sympatrische
Arten A/B - Gattungen A/B. Die in der vorliegenden Arbeit untersuchten Arten sind vor allem anhand
morphologischer Merkmale und eines definierten Verbreitungsgebietes beschrieben worden. In
welchem Stadium der evolutionären Trennung sich diese Sippen befinden, ist natürlich nicht
nachvollziehbar, die Fähigkeit zur Hybridbildung lässt allerdings auf unvollständige reproduktive

Diskussion
101
Isolation schließen. Alle Angaben zur Variabilität beziehen sich auch auf die morphologisch definierten
Sippen im Kontext zu den molekularen Daten.
Die molekulare Variabilität hängt zunächst von der betrachteten DNA-Region ab. Ein einfacher
Vergleich der Distanzmittelwerte zwischen Chloroplasten- und Kernsequenzen der untersuchten
Sippen zeigt im Durchschnitt eine bis zu fünffach höhere Divergenz in ITS im Vergleich zu atpB-rbcL
und psbB-psbH. Dies ist das Ergebnis höherer Substitutionsraten im Kerngenom und wurde schon in
vielen vergleichbaren Studien nachgewiesen (Li et al. 2002, Shepherd et al. 2004). Als Resultat zeigt
sich eine bessere Auflösung der Baumtopologie im ITS (s. Kap. 3.2). Viele der Sippen haben
unterschiedliche ITS- aber identische Chloroplasten-Sequenzen. Besonders kennzeichnend ist dies
für die Sippen der Sektion Brezia. Die Sequenzdiversität in der ITS-Region lässt sich nicht mit den
morphologisch unterscheidbaren Arten in Übereinstimmung bringen. Viele Sippen sind daher
intraspezifisch variabel.
Die Sippe mit der höchsten intraspezifischen Diversität ist zweifelsohne Suaeda spicata. Die in Kap.
3.2.4 aufgezeigte Diversität des S. spicata-Clades mit 7 Sequenztypen ist in Wirklichkeit noch viel
höher. Möller (2005) fand bei einer erweiterten Untersuchung europäischer Sippen insgesamt 14 ITS-
Typen innerhalb dieses Verwandtschaftskreises. Eingeschlossen ist die derzeit noch als Varietät von
S. maritima geführte ausdauernde Sippe (S. maritima var. perennans). Die Unterschiede zwischen
den einzelnen Sequenzen betragen meist nur eine oder wenige Mutationen, trotzdem entsteht im
Stammbaum allein aus den Sequenzen einer Sippe ein eigener Clade. Diese Variabilität findet
allerdings keine Entsprechung in der Morphologie. Der enormen Variabilität im ITS-Bereich steht,
bezogen auf atpB-rbcL und psbB-psbH, nur ein identischer Chloroplasten-Typ für Suaeda spicata
gegenüber, soweit dies aus den vorhandenen Daten geschlossen werden kann. Intraspezifische
Variabilität tritt bei vielen weiteren Sippen auf. So fanden sich bei S. maritima s.l. (inkl. S. ‚venetiana’)
6, bei S. salsa 3, bei S. salinaria 2 und bei S. kulundensis 4 unterschiedliche ITS-Typen.
Der gegenteilige Fall, unterschiedliche Arten mit gleichen Sequenzen, wurde bei der Auswertung der
Sequenzen ebenfalls festgestellt. Ein Beispiel sind drei morphologisch durchaus unterscheidbare
Sippen (S. crassifolia, S. ‚arabica’ und S. ‚elegans’), die identische ITS-Sequenzen aufweisen. Ein
ähnliches Verhalten zeigt ITS in der weit verbreiteten Suaeda corniculata. Eine Vielzahl von mehr oder
weniger verschiedenen Morphospecies (S. corniculata-Formen, 5 Chromosomenrassen, S.
pannonica) besitzt identische ITS-Sequenzen.
4.3. Phylogenie und Artbildung innerhalb der Suaedoideae 4.3.1. Molekulare Evolution Die molekularen Stammbäume rekonstruieren mit den implizierten Verwandtschaftsbeziehungen die
Wege zur rezenten Diversität. Die Geschwindigkeit auf molekularer Ebene lässt sich durch
Substitutionsraten ausdrücken, mit denen der Veränderungsprozess homologer Nukleotidpositionen
beschrieben wird. Dieser Prozess unterscheidet sich zwischen unterschiedlichen Genomen und
Genombereichen, insbesondere zwischen kodierenden und nichtkodierenden Regionen. Bereits Wolfe
et al. (1987) fanden heraus, dass es große Unterschiede hinsichtlich der Substitutionsraten zwischen
Kern- und Plastidengenomen gibt. Bei den untersuchten Angiospermen ermittelten sie
Substitutionsraten von 1,0-3,0 x 10-9 Substitutionen pro Position und Jahr für Chloroplasten und 5-30 x

Diskussion
102
10-9 für das Kerngenom. Li et al. (2002) ermittelten für ITS in den Ericaeae eine durchschnittliche
Substitutionsrate von 1,44 x 10-9. Relativ hohe ITS-Substitutionsraten von 4,8-11,0 x 10-9 wiesen
Kadereit et al. (2005) in den australischen Camphorosmeae nach, viel geringere hingegen im rbcL-
Gen (0,2-0,5 x 10-9).
Eigene Untersuchungen an den Suaedoideae und Salicornioideae ergaben eine mittlere
Substitutionsrate von 3,89 x 10-9 für ITS, die damit etwas geringer ist als diejenige innerhalb der
Camphorosmeae. Die atpB-rbcL-Region evolviert mit einer deutlich geringeren Rate von 6,99 x 10-10 .
Wie dieser kurze Vergleich zeigt, gibt es erhebliche Ratenunterschiede in Abhängigkeit von der
betrachteten Region und vom Umfang der untersuchten Gruppe.
Die Variabilität der untersuchten Chloroplasten-Regionen beruht auf Punktmutationen und
Längenvariationen. Die Längenvariationen entstehen überwiegend durch Verdopplungen kurzer
Abschnitte von durchschnittlich 6-8 bp Länge. Dabei gibt es sowohl singuläre, autapomorphe
Verdopplungen als auch gruppenspezifische, synapomorphe Indels. Diese wurden in der Maximum
Parsimony-Analyse in 0/1-Werte transformiert und in die Berechnung einbezogen.
Weitere strukturelle Merkmale der Chloroplastengenome sind repetitive Elemente in unterschiedlicher
Ausprägung. Mononukleoitidrepeats aus A-(T-)Wiederholungen sind regelmäßige Bestandteile des
Chloroplastengenoms, besonders in Introns und intergenischen Regionen (Weising et al. 2005). Die
atpB-rbcL-Region weist innerhalb der untersuchten Gruppe zwei sehr charakteristische längenvariable
Mononukleotidrepeats (Mikrosatelliten) auf. Einer davon befindet sich in Nähe des rbcL-Gens und
erreicht Größen von (A)8-12 innerhalb Suaeda und bis zu A16 bei Salicornia. Die gefundenen
Längenmuster sind dabei durchaus gruppenspezifisch. Kürzere Varianten kennzeichnen den Suaeda-
Clade, längere den Brezia-Clade, sehr lange Varianten die Salicornioideae. Für eine sinnvolle
Auswertung ist die Variabilität allerdings zu gering. Die repeat-Struktur wirkte sich sehr störend auf
PCR und Sequenzierungsreaktionen aus. Die Sequenzierung wurde ab dieser Struktur meistens
ungenau, so dass ein Großteil der Sequenzinformation nur aus einem Strang abgelesen werden
konnte. In der Sektion Schoberia und Alexandra kam es zur Vervielfachung dieser repeat-Region (s.
Abb. 8, S. 53). Diese Region war mittels PCR meist nicht amplifizierbar, weshalb interne Primer
konstruiert werden mussten. Als gruppenspezifisches Merkmal auf molekularer Ebene erscheint es
jedoch bedeutsam und verdeutlicht einmal mehr die unerwartete Position von Alexandra im Suaeda-
Clade in einem gemeinsamen Subclade mit Schoberia.
Eine weitere repetitive Struktur mit Längevarianten von (T)7-13 befindet sich etwa in der Mitte des
spacers und zeigt innerhalb des Suaeda-Clades längere Varianten als im Brezia-Clade. Sehr lange
synapomorphe Indels weisen die Sippen S. asphaltica, S. vermiculata, S. aegyptiaca und S. arcuata.
auf. Die kürzeste atpB-rbcL-Sequenz hat Alexandra lehmannii, wodurch eine Lücke von etwa 400 bp
im Alignment erzeugt werden muss. Weit weniger längenvariabel ist die psbB-psbH-Region. Allerdings
finden sich auch in dieser Region synapomorphe Indels, die für die Phylogenierekonstruktion genutzt
werden können. Die Variabilität des ITS beruht vorwiegend auf Punktmutationen und kurzen, 1-3 bp
langen Indels. Erstaunlich ist die Tatsache, dass selbst in polyploiden Genomen keine
unterschiedlichen Paralogen nachgewiesen werden konnten, ein Beweis für eine sehr effektive
concerted evolution besonders in dekaploiden Genomen, da dort der Prozess der Homogenisierung
über 10 Chromosomen stattfinden muss.

Diskussion
103
4.3.2. Radiation, Gruppenbildung und Speziation In den molekularen Stammbäumen werden ausgehend von einem gemeinsamen Vorfahren singuläre
Linien und diversifizierte Gruppen sichtbar. Dies ergibt sich aus den für die phylogenetische
Rekonstruktion angewandten kladistischen Modellen. Die Aufspaltung einer gemeinsamen
Ausgangslinie in eine höhere Zahl abgeleiteter Linien kann dabei als Ergebnis von adaptiver Radiation
interpretiert werden, die nach einem Schlüsselereignis wie z.B. Besiedlung eines neuen
Lebensraumes oder der Evolution eines vorteilhaften Merkmals stattfand. Die Radiation ist ein
Prozess, der als Zunahme der Diversität in einem zeitlichen und räumlichen Kontext zu verstehen ist.
Durch Radiation entstehen monophyletische Gruppen, die synapomorphe Merkmale besitzen.
Die wichtigen Aufspaltungen und Radiationen wurden für die Suaedoideae und verwandte Gruppen
rekonstruiert. Der Prozess wird ausschließlich aus den rezenten Sippen und ihren Merkmalen
hergeleitet. Wie viele Sippen wirklich im Verlauf der Evolution entstanden und wieder ausstarben, ist
nicht mehr rekonstruierbar. Bei der Interpretation der Sippenentwicklung ist zu berücksichtigen, dass
zumindest alle rezenten Sippen lichtbedürftige Hygrohalophyten sind, deren Fähigkeit zur aktiven
Fernausbreitung eng limitiert ist. Salzfreie Standorte sowie geschlossene Vegetation (Wälder) stellen
damit erhebliche Ausbreitungsbarrieren dar.
Suaeda teilt sich basal in zwei große Entwicklungslinien bzw. Schwestergruppen, in denen sich alle
rezenten Sippen entwickelten. Diese Haupt-Aufspaltung wird in allen molekularen Phylogenien durch
sehr hohe bootstrap-Werte gestützt und wird zeitlich in das Oligozän datiert (vor ca. 30 Mio Jahren). In
diese Zeit fällt der Rückzug des Tethysmeeres, das zum Ende des Miozäns seinen tiefsten Punkt
erreichte (Stanley 1994). Vorausgesetzt, dass das heutige Diversitätszentrum auch das
Entstehungszentrum der Suaedoideae ist oder diesem zumindest räumlich nahekommt, ist die
Entwicklung daher eng mit dem Rückzug der Tethys und der Vergrößerung terrestrischer saliner
Lebensräume verbunden. Die Kontinentalbewegung war zu diesem Zeitpunkt schon so weit
fortgeschritten, dass die Besiedlung weiter entfernter Kontinente (Nord- und Südamerika, Australien)
nur nachträglich erfolgt sein kann.
Unabhängig von der basalen Aufspaltung zweigt mit Bienertia eine Linie ab, für die sich keine näheren
Verwandten finden ließen und die sich erst im Quartär in zwei Sippen aufspaltet. Warum keine
stärkere Radiation stattgefunden hat, lässt sich nicht erklären, zumindest war aber die Entwicklung der
single cell -C4-Photosynthese in dieser Art kein vorteilhaftes Schlüsselereignis für eine nachfolgende
Arealausweitung oder Sippendifferenzierung. Ihr heutiges Verbreitungsgebiet ist im Vergleich zu
anderen Gruppen relativ beschränkt.
Die Ergebnisse der Zeitdatierung weisen übereinstimmend für die beiden Haupt-Clades von Suaeda
unterschiedliche Zeitpunkte der Diversifizierung aus. Zuerst begann sie innerhalb des Suaeda-Clades,
wesentlich später im Brezia-Clade.
Die Topologie des Brezia-Clades lässt, zumindest nach ITS-Daten, drei weitgehend unabhängige
Radiationen erkennen:
1. Der maritima-Subclade repräsentiert eine vorwiegend ost- und zentralasiatische sowie australische
Gruppe, deren am weitesten nach Westen verbreitete Vertreter das Ergebnis einer sehr jungen
Radiation sind: S. salsa und S. maritima. S. arbusculoides ist die älteste Linie der Gruppe mit dem
längsten singulären Ast.

Diskussion
104
2. Der prostrata-Subclade verbindet mit S. caespitosa und S. spicata Capensis und Mittelmeerraum,
zwei klimatisch ähnliche Regionen. Beides sind sehr alte Linien, die sich möglicherweise bereits im
Miozän abtrennten. S. spicata entfaltet rezent eine außerordentlich hohe Variabilität (Möller 2005). Die
diverse S. prostrata erscheint als Ableitung aus diesen Vorfahren und diversifizierte sich
möglicherweise während der Besiedlung kontinentaler Lebensräume.
3. Der corniculata-Subclade zeigt an der Basis eine alte Aufspaltung in S. tschujensis/S. arctica sowie
den Rest der Gruppe. Die Entwicklung ist als Folge klimatischer Veränderungen vorstellbar, welche
die kälteangepassten Sippen in nördliche Zonen (S. arctica) und Hochgebirgsregionen (Altai: S.
tschujensis) verdrängte. Die räumliche Isolierung zeigt sich in einer beginnenden Differenzierung im
ITS. S. corniculata konnte das Areal bis zur heutigen Größe erweitern, blieb dabei aber genetisch
uniform (einheitlicher ITS-Typ), entwickelte jedoch eine Vielzahl von Ploidiestufen und gelangte durch
long distance dispersal nach Nordamerika, wo eine erneute Radiation stattfand (amerikanische
Brezia-Sippen außer S. maritima).
Eine zweite neuweltliche und ziemlich junge Radiation zeigt die Ableitung der nord- und
südamerikanischen Salsina-Sippen, deren Entstehung in das Pliozän fällt, die sich aber erst im
Quartär diversifizierten. Die nächstverwandte Sippe ist S. articulata (S-Afrika). Das Alter dieser
amerikanischen Sippen ist damit viel jünger als von Akhani (1997) angenommen.
4.3.3. Hybridisierung und Polyploidisierung Hybridisierung und Polyploidsierung sind gerade im Pflanzenreich weit verbreitete Phänomene und
tragen erheblich zur Artbildung und Evolution bei. Polyploidisierung ist wahrscheinlich ein
vorherrschender Faktor bei der sympatrischen Artbildung. Konservative Schätzungen beziffern den
Anteil dieser Form der Artbildung bei Blütenpflanzen auf 2-4% (Otto & Whitton 2000), wahrscheinlich
liegt der Anteil aber höher. Auch in den Chenopodiaceae wurden zahlreiche polyploide Linien
nachgewiesen. So enthält z.B. die Gattung Salsola neben diploiden auch tetra-, hexa- und oktoploide
Taxa (Rilke 1999). Aus der benachbarten Unterfamilie der Salicornioideae sind zahlreiche Polyploide
und mutmaßliche Hybriden bekannt (Shepherd et al. 2004). Besonders die Gattungen Salicornia und
Sarcocornia sind hinsichtlich der Ploidiestufen sehr variabel. Neben tetra- bis oktoploiden Formen
wurden auch tri- und pentaploide nachgewiesen (Kadereit et al. 2006). Eine Vielzahl an Argumenten
spricht dafür, dass Polyploidisierung, Hybridisierung und vor allem die Etablierung hybridogener
Sippen auch innerhalb der Suaedoideae entscheidend zur Diversifikation und Artbildung beigetragen
hat (Lomonosova et al. 2008). Demgegenüber steht die Tatsache, dass für Suaeda nur sehr wenige
Bastarde beschrieben wurden. Ein Beispiel hierfür ist Suaeda x genesiana Pedrol & Castroviejo, ein
Bastard aus S. pruinosa und S. vera (Pedrol & Castroviejo 1988).
Hybridogen entstandene Sippen lassen sich molekular nachweisen, wenn geeignete Marker gefunden
werden. In jungen Hybriden geben polymorphe Loci in Kern-Regionen, wie dem internal transcribed
spacer, Hinweise auf Hybridisierungsereignisse. Mit Hilfe additiver Nukleotide wiesen z. B. Sang et al.
(2005) und Whittal et al. (2000) hybride Sippen nach. Polymorphe ITS-Sequenzen wurden trotz
starker Hinweise auf Hybridisierung bei Suaeda nicht nachgewiesen. Der Homogenisierungsprozess
ist offenbar sehr schnell, vor allem zwischen verschiedenen Chromosomen in hochpolyploiden
Genomen. Da sich bei der Homogenisierung der ITS-Typ der eingekreuzten Sippe ganz oder teilweise

Diskussion
105
durchsetzen kann, geben Inkongruenzen zwischen Stammbäumen von unterschiedlichen Genomen
(z.B. Kern und Chloroplast) Hinweise auf die Elternarten, wenn diese getrennten Linien entstammen.
Dieser Fall wurde bei Suaeda mehrfach beobachtet und wird hier anhand einiger Sippen diskutiert.
Zur Veranschaulichung dient die Gegenüberstellung von einem ITS-Stammbaum (Brezia-Baum aus
Kap. 3.2.4) und einem atpB-rbcL-Baum, soweit die entsprechenden Sequenzen der Akzession
vorhanden waren. Es handelt sich in beiden Fällen um einen zufällig ausgewählten sparsamsten
Baum aus der MP-Analyse. Einige Beispiele werden besprochen.
Suaeda kulundensis Die erst 2008 beschriebene Sippe (Lomonosova et al. 2008) galt lange Zeit als aufrechte Form der
weit verbreiteten S. corniculata und wurde als Unterart geführt (S. corniculata ssp. erecta).
Morphologische Merkmale und in besonderem Maße die molekularen Stammbäume sprechen für eine
hybridogene Entstehung aus S. corniculata und S. salsa. Der aufrechte Wuchs gilt dabei als Merkmal
von S. salsa, das Perianth erinnert in Grundzügen an das von S. corniculata. Die
Chromosomenzahlen weisen die Art als polyploide Sippe aus (2n=72, 90). Die Entstehung der Art
lässt sich daher durch Kreuzung von tetraploiden (S. salsa) mit tetra- bzw. hexaploiden (S.
corniculata) Sippen erklären. Sie ist damit ein allopolyploider Hybrid aus S. corniculata und S. salsa,
wobei S. corniculata die maternale Linie repräsentiert. Sequenzunterschiede und räumliche Trennung
okto- und dekaploider Rassen legen die Vermutung nahe, dass die hier umrissene Art mehrfach
parallel entstanden ist. Die Ausgangssippe S. salsa ist wahrscheinlich durch S. salsa s.l. 1171 noch
rezent vorhanden (maternale salsa-Linie).
Suaeda sibirica Sehr ähnlich, aber durch Einkreuzung einer anderer Paternallinie in S. corniculata, ist die stets
oktoploide S. sibirica entstanden. Ihre Vorfahren sind nicht mehr rekonstruierbar, doch ist
bemerkenswert, dass sie zwischen den beiden australischen Arten steht (S. arbusculoides, S.
australis).
Suaeda spicata Die Beziehungen widerspiegeln die Hybridisierung einer bereits stark diversifizierten S. spicata mit S.
maritima im Mittelmeerraum, wobei 1223 und 1246 die Ausgangssippe S. maritima repräsentieren.
Dieser Hybridisierungsprozess, nachweisbar an einer Vielzahl kombinierter DNA-Typen, wurde von
Möller (2005) ausführlich für den Mittelmeerraum untersucht. Mit weiteren cp-Regionen wurde dabei
auch die hybridogene Natur von S. „venetiana“ erklärt, die durch Hybridisierung von S. maritima und
S. salsa entstanden sein könnte.
Die aufgrund von Inkonsistenzen chloroplastidärer und nukleärer Stammbäume nachgewiesenen
Hybriden belegen damit recht gut, dass die Artbildung und Evolution innerhalb von Suaeda nicht nur
auf dem Hennigschen Modell der Bifurkationen beruht, sondern eher ein retikulater Prozess ist, bei
dem zuvor teilweise sogar lange getrennte Linien durch Hybridisierung wieder vereint werden und sich
sogar neben ihren Ausgangssippen etablieren können.
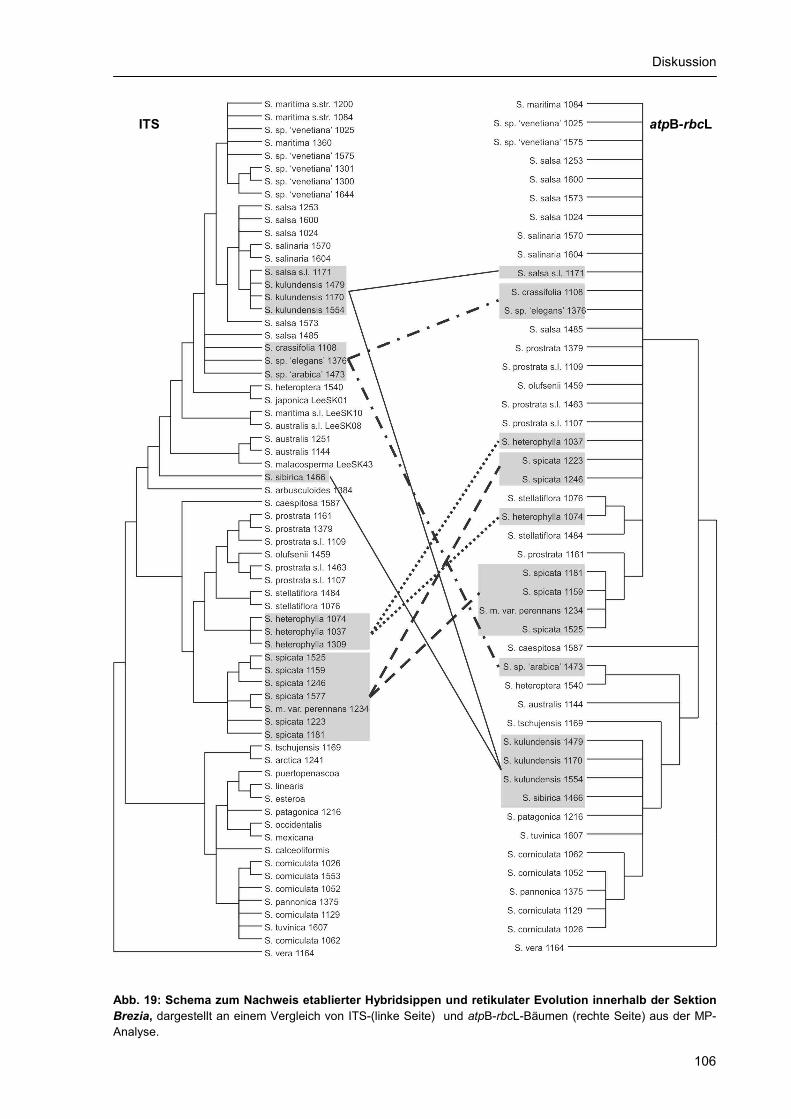
Diskussion
106
Abb. 19: Schema zum Nachweis etablierter Hybridsippen und retikulater Evolution innerhalb der Sektion Brezia, dargestellt an einem Vergleich von ITS-(linke Seite) und atpB-rbcL-Bäumen (rechte Seite) aus der MP-Analyse.
ITS atpB-rbcL

Diskussion
107
4.4. Merkmalsentwicklung innerhalb der Suaedoideae 4.4.1. Systematisch relevante Merkmale und ihre Evolution Morphologische Merkmale sind spezifische Erscheinungen des Phänotyps und werden durch
komplexe, abgestufte Genwirkungen ausgeprägt. Vergleichende Untersuchungen haben gezeigt, dass
die Geschwindigkeiten bzw. Raten molekularer und morphologischer Evolution nicht korreliert sind
(Bromham et al. 2000). Molekulare Diversität und die daraus rekonstruierten Stammbäume finden
daher nicht automatisch eine Entsprechung in morphologisch definierten Arten.
In gewissen Grenzen variieren morphologische Merkmale in Abhängigkeit von umweltbedingten oder
entwicklungsgeschichtlichen Einflüssen, dazu zählen z. B. Nährstoff- und Wasserangebot des
Standorts, Strahlungsintensität, Konkurrenzverhältnisse u.a. Bei den Suaedoideae kommt als
spezifischer Faktor die Salinität hinzu, deren Grad durchaus Einflüsse auf das äußere
Erscheinungsbild der Pflanzen hat. Gut sichtbar wird dies bei direktem Vergleich von in Kultur
gezogenen Pflanzen. Generell lässt sich dabei beobachten, dass Pflanzen identischer Herkunft auf
NaCl-freiem Substrat und unter reduzierter Strahlung (Gewächshaus) einen insgesamt verkümmerten
Wuchs zeigen. Die Blätter sind schlanker und weniger sukkulent, Verzweigungsgrad und Blütenansatz
gering, meist kommt es nicht zur Samenreife. Bestimmte Gewebetypen wie z.B. Wasserspeicher- oder
Abschlussgewebe sind unterentwickelt oder fehlen ganz. Unter Strahlungs- und
Salinitätsbedingungen, die den natürlichen Verhältnissen nahe kommen entwickeln sich die Pflanzen
hingegen normal. Die geschilderte Variabilität tritt auch unter natürlichen Bedingungen auf. Dabei
überlagert die individuelle Plastizität der Merkmale oft die intraspezifische, was die Abgrenzung der
Sippen außerordentlich erschwert. Auf diesen Aspekt wird im Zusammenhang mit der Abgrenzung
von Arten in den meisten systematischen und floristischen Bearbeitungen hingewiesen (Ball &
Akeroyd 1993, Bassett & Crompton 1978, Hopkins & Blackwell 1977, Iljin 1936b, Pedrol & Castroviejo
1988, u.a.). Auf die Plastizität von Merkmalen wird z.B. in den Untersuchungen von Metzing et al.
(1996) und Ungar (1974) eingegangen. Ungar fand starke Variationen in der Wuchsform in
Abhängigkeit von Salinität des Standortes.
Ziel der Arbeit war es, Merkmale zu finden, die trotz aller Plastizität ihrer Anlage nach gleich sind und
zur Charakterisierung von Verwandtschaftsgruppen herangezogen werden können.
Großgruppenmerkmale
Die Verbindung von Salsoloideae und Suaedoideae aufgrund des spiralig aufgerollten Embryos war
seit langem umstritten (Ulbrich 1934). Die molekulare Phylogenie der Chenopodiaceae zeigt eindeutig
verwandtschaftliche Beziehungen auf, die nicht mit der auf Embryomerkmalen beruhenden Gliederung
übereinstimmen.
Von hoher systematischer Relevanz erwiesen sich die Bracteolen, die bei den Suaedoideae
vorkommen, in der Schwestergruppe Salicornioideae jedoch fehlen. Suaeda und Borszczowia
besitzen trockenhäutige, Alexandra lineare, fädige Bracteolen. Die Bracteolen von Bienertia sind
fleischig und grünlich.
Der Besitz von Bracteolen trennt die Suaedoideae von den Salicornioideae, die keine Bracteolen
haben und legt wiederum die Verwandtschaft zu den Salsoloideae nahe. Diese Beziehung wird durch
molekulare Daten nicht unterstützt.

Diskussion
108
Blüten- und Blütenstandsmerkmale
Die Grundstruktur aus 5 Tepalen und 5 Staubblättern ist in den Suaedoideae fast durchweg erhalten,
während es bei den Salicornioideae Reduktionen gibt. Die meisten Vertreter des Suaeda-Clades
besitzen noch das ursprüngliche Perianth mit wenigen Abwandlungen. In einigen Sippen entwickelten
sich flügelartige Anhänge oder hornartige Auswüchse. Die Entwicklung horizontaler Flügel fand bei
den weit entfernten Sippen Bienertia und Suaeda heterophylla statt. Mit diesem Merkmal wurde S.
heterophylla als eigene Gattung Brezia von Suaeda unterschieden (Ulbrich 1934). Basale Flügel
entwickelten S. heteroptera und S. kulundensis, vertikale Flügel Alexandra lehmannii und in
geringerem Umfang Suaeda pterantha. Die Entwicklung von flügelartigen Strukturen an nicht
miteinander verwandten Sippen kennzeichnet das Merkmal als Autapomorphie für die jeweilige Sippe
und kann damit nicht für die Gliederung von Gruppen verwendet werden. Als ursprünglich wohl
synapomorpher Charakter könnte das asymmetrische Perianth mit hornartigen Auswüchsen für den
corniculata-Subclade gewertet werden. Wahrscheinlich hat sich die Perianthform vor der Abtrennung
von S. tschujensis entwickelt. Allerdings fehlt das Merkmal einigen amerikanischen Sippen (S. linearis,
S. mexicana, S. occidentalis), was als sekundärer Verlust gedeutet werden könnte. Die geschilderte
Perianthform kommt ausschließlich im corniculata-Subclade vor. S. kulundensis (maritima-Subclade)
zeigt ähnliche Entwicklungen aufgrund von Hybridisierung. Die Morphologie des Pistill hat sich, wie
bereits von Iljin (1936a) erkannt, als eines der wichtigsten gruppengliedernden Merkmale erwiesen.
Den ursprünglichen Typ repräsentieren dabei Brezia- und Bienertia-Typ, dabei vor allem die
Narbenanzahl (2). Die Darstellung dieses Typs bei Iljin (3 Narben) ist fehlerhaft. Es handelt sich um
eine innerhalb der gesamten Chenopodiaceae weit verbreitete Form die auch bei der
Schwestergruppe der Suaedoideae, den Salicornioideae vorkommt. Der Pistilltyp war damit
wahrscheinlich schon bei den gemeinsamen Vorfahren entwickelt und stellt ein plesiomorphes
Merkmal dar. Die anderen Pistilltypen sind leicht als Weiterentwicklungen dieses Grundtyps
vorstellbar. In diesem Prozess kam es zur Vermehrung der Narbenzahl auf 3 (-4), Vergrößerung der
Papillen und zunehmende Einsenkung der Narbeninsertionsstelle in das an Länge zunehmende Ovar.
Als Zwischenstufe auf diesem Weg könnte das Pistill von S. glauca mit zwei Narben und fehlender
apikaler Vertiefung gedeutet werden. Ähnlich intermediär erscheinen die Pistille des Schanginia- und
Vera-Typs mit kaum ausgeprägter Vertiefung. Die besonders starke Vergrößerung der
Narbenoberfläche bei dem Pistill von S. vera stellt einen evolutionären Sonderweg dar, der
möglicherweise eine Anpassung an Windbestäubung ist. S. physophora besitzt bereits das Schoberia-
Pistill, allerdings sind die Narben stark verkürzt. Das Pistill der mit S. physophora eng verwandten S.
ifniensis entspricht schon weitgehend dem entwickelten Schoberia-Typ (Maire 1962: 115, Fig. 951 C)
und auch S. palaestina besitzt gut entwickelte Narben (Freitag 1989: 155 Fig. 1a). Als Endglied dieser
Entwicklungsreihe könnte das weit entwickelte Pistill von Schoberia- und fruticosa-Subclade
aufgefasst werden. Die Einsenkung der Narben in das Ovar ist am weitesten fortgeschritten,
Narbenlänge und Größe der Papillen haben am meisten zugenommen. Die Zuordnung der
unterschiedlichen Pistilltypen korreliert gut mit den Gruppen der molekularen Phylogenie. Ein
gestrecktes Ovar mit eingesenkten Narben kann damit als synapomorphes Merkmal des Suaeda-
Clades gewertet werden, von dem es mehrere Weiterentwicklungen gibt und das einzigartig innerhalb
der Chenopodiaceae ist.

Diskussion
109
Verwachsungen im Bereich von Blüten und Blütenstand
Mehr oder weniger ausgeprägte Verwachsungen des Perianths mit dem Ovar wurden in der
Vergangenheit zur Abgrenzung von systematischen Gruppen benutzt (Ulbrich 1934, Townsend 1980).
Verwachsungen unterschiedlichen Grades wurden bei der Sektion Schanginia, Immersa und der
Tribus Bienertiae nachgewiesen. Die molekulare Phylogenie zeigt jedoch, dass dieses Merkmal in
vollkommen voneinander getrennten, nicht näher verwandten Gruppen entwickelt wurde. Die
Verwachsung ist daher ein paraphyletisches Merkmal. Bei S. aegyptiaca erscheint durch die
Verwachsung der Fruchtknoten unterständig. Die Art ist aber anhand von Blatt- und Pistillmerkmalen
eindeutig dem fruticosa-Subclade zugehörig.
Eine andere Form der Verwachsung betrifft diejenige der Teilblütenstände mit dem Blattstiel (petiolate
Blütencluster). Dieses Merkmal wurde traditionell der Sektion Schanginia zugeordnet. Iljin (1936b) gibt
5 Sippen mit diesem Merkmal an: S. microphylla, S. altissima, S. glauca, S. paradoxa , S. linifolia.
Nach heutigen Erkenntnissen gehören die mit diesem Merkmal charakterisierten Sippen drei
unabhängigen Verwandtschaftskreisen an (Schanginia- und fruticosa-Subclade, S. glauca). Es ist
deshalb, wie die Verwachsungen von Blütenhülle und Ovar, ein paraphyletisches Merkmal.
Blattentwicklung Der anatomische Bau der Laubblätter stellt neben der Pistillmorphologie eines der wichtigsten
Merkmale für die Klassifizierung sowohl der Suaedoideae als auch der gesamten Chenopodiaceae
dar (Akhani et al. 1997, Fisher et al. 1997, Kadereit et al. 2003, Schütze et al. 2003). Dies wurde
bereits vor der Einführung molekularsystematischer Methoden deutlich (Carolin et al. 1975).
Die gemeinsamen Vorfahren der Suaedoideae und Salicornioideae besaßen mit großer
Wahrscheinlichkeit bereits äquifaziale Laubblätter, die in vielfachen Abwandlungen auch rezent noch
vorkommen. Die Stellung am Spross war sicher wechselständig. Dieses Merkmal hat sich in den
ursprünglichen Salicornioideae und in allen Suaedoideae erhalten. Die abgeleiteten Salicornioideae
entwickelten gegenständige Beblätterung bzw. reduzierten die Blattflächen vollkommen. Die rezenten
Blätter der Salicornioideae (nur Halopeplideae) unterscheiden sich von denjenigen der Suaedoideae.
Die Blätter von Kalidium besitzen peripher am Mesophyll angeordnete Leitbündel (Freitag, unpubl.),
bei fast allen Blatttypen der Suaedoideae sind die Leitbündel hingegen in einer Ebene angeordnet
(Ausnahme: Borszczowia). Diese Leitbündelverteilung weist auch das C4-Blatt von Bienertia auf, das
damit eine nähere Beziehung zu Suaeda erkennen lässt, hinsichtlich der C4-Anatomie aber eine völlig
eigenständige Entwicklung darstellt. Wenige Veränderungen hat das einfache C3-Blatt innerhalb des
Brezia-Clades erfahren, lediglich der Sukkulenzgrad und die Ausbildung des Wasserspeichergewebes
variiert und ist bei einigen Arten stark ausgeprägt (S. crassifolia). Innerhalb des Suaeda-Clades hat
sich demgegenüber eine enorme Vielfalt unterschiedlicher Blatttypen entwickelt, darunter drei C4-
Blatttypen. Durch die Einbeziehung zweier neuer Taxa (Alexandra lehmannii, Suaeda glauca) wurde
erstmals der ursprüngliche Brezia-Blatttyp innerhalb des Suaeda-Clades nachgewiesen. Damit
müssen auch die Aussagen bei Schütze et al. (2003) etwas verändert werden. An der Basis des
Suaeda-Clades steht mit S. glauca eine C3-Art mit einem Blatttyp, der prinzipiell dem Brezia- bzw.
Vera-Typ entspricht, aber wesentlich weniger Leitbündel aufweist und viel zarter gebaut ist (Freitag,
mdl.). Damit ist aber auch noch einmal bestätigt, dass sich die Vielfalt der Blatttypen innerhalb des

Diskussion
110
Suaeda-Clades aus austrobassioiden Vorfahren entstanden ist. In der Schwestergruppe Brezia fand
hingegen keine blattmorphologische Entwicklung statt. Als sehr isolierte Linie zweigt der Schanginia-
Clade ab, an dessen Ende zwei histologisch und funktional sehr verschiedenartige Blatttypen stehen:
das einzigartige C4-Blatt des Borszczowia-Typ und das C3-Blatt des Schanginia-Typs mit reduziertem
Wasserspeichergewebe. Die ursprünglichen C3-Blätter werden mit S. vera und der physophora-
Gruppe zunehmend sukkulenter und entwickeln Blattstiele (Vera-Typ). Aus diesen Vorfahren leiten
sich mit hoher Wahrscheinlichkeit die C4-Blätter mit Kranz-Anatomie ab (Salsina-Typ, Schoberia-Typ).
Lebensform und Verholzung Über die ursprüngliche Lebensform der Suaedoideae können auch anhand der molekularen Daten nur
Hypothesen aufgestellt werden. Die basale Position vieler einjähriger Sippen (Bienertia, Suaeda
glauca, S. linifolia, S. paradoxa, Borszczowia, viele Brezia-Sippen) legt den Schluss nahe, dass sich
die Suaedoideae von annuellen Vorfahren ableiten. Dies widerspricht der Annahme von Akhani
(1997), der als ursprüngliche Sippen Zwergsträucher postuliert. Die von Akhani als ursprünglich
angesehenen Sippen S. ifniensis und S. palaestina gehören zwar einer alten C3-Linie an, die basalen
Abspaltungen begannen jedoch früher. Ein Vergleich mit der Schwestergruppe der Suaedoideae, den
Salicornioideae zeigt dass die ursprünglichsten Vertreter in diesem Verwandtschaftskreis
ausdauernde, verholzte Sippen sind (Kalidium). Die früheste Trennung zwischen Suaedoideae und
Salicornioideae könnte durch Abspaltung annueller Sippen (Suaedoideen-Linie) von ausdauernden
Sippen erfolgt sein. Aus den annuellen Vorfahren der heutigen Suaedoideae entwickelten sich schon
sehr früh ausdauernde, verholzende Sippen, die heute einen Großteil des Suaeda-Clades
ausmachen. Diese Entwicklung setzt mit den Vorfahren der heute noch vorkommenden S. vera ein
(vgl. Abb. 18) und beginnt im mittleren Miozän vor ca. 20 Mio. Jahren. Dieser relativ frühe
Entstehungszeitpunkt verholzter Arten bedingt die hoch differenzierte Holzanatomie bei den Sippen
dieser Verwandtschaft. Vieles spricht dafür, dass sich die ausdauernden, verholzten Sippen weiter
entwickelten und mehrfach wieder Annuelle abspalteten. Dafür kommen der gesamte Schoberia-
Clade, Suaeda altissima und die nahe verwandten S. aegyptiaca und S. arcuata in Frage. Zumindest
für die letztgenannten ist diese Entwicklung nahe liegend und bestätigt damit die Thesen Akhani’s
(1997). Einen umgekehrten Weg nahm die Entwicklung im Brezia-Clade. Die meisten der Sippen sind
einjährig und vieles deutet darauf hin, dass sich davon mehrfach ausdauernde, verholzte Sippen
abgeleitet haben. Die molekulare Phylogenie lässt 5 mögliche Entstehungsereignisse innerhalb von
Brezia zu. Die Entwicklung verholzter Sippen in dieser Gruppe ist höchstens halb so alt wie im
Suaeda-Clade und begann im späten Miozän mit der Entwicklung der südhemisphärischen Arten (S.
arbusculoides, S. caespitosa). S. maritima var. perennans und S. esteroa (z.T. auch S. linearis) sind
sehr junge Ableitungen verholzter Sippen und befinden sich noch in einem Entwicklungsprozess.
Dieses Phänomen wird für die nordamerikanische S. linearis beschrieben: ...“annual, sometimes
persisting in warm regions...“ (Hopkins & Blackwell 1977). Da das Verbreitungsgebiet von den
gemäßigten Zonen der Atlantikküste bis in den subtropischen Bereich reicht, könnte es sich allerdings
auch um zwei verschiedene Sippen handeln. Für S. maritima var. perennans deuten die
Untersuchungen von Möller (2005) an Sippen von den Kanaren und von der portugiesischen
Atlantikküste auf möglicherweise getrennte Entstehungen hin.

Diskussion
111
4.4.2 Evolution der C4-Photosynthese Die C4-Photosynthese als effiziente Anpassung der Assimilationsprozesse an wärmere Klimazonen
stellt ein wichtiges Charakteristikum der Suaedoideae dar und ist mit grundlegenden
Merkmalsentwicklungen gekoppelt. Im Rahmen der vorliegenden Arbeit war zu klären, in welcher
phylogenetischen Beziehung die Arten mit den vier bekannt gewordenen C4-Blatttypen (Carolin et al.
1975, Freitag & Stichler 2000, Freitag & Stichler 2002) stehen und wie oft das gesamte C4-
Photosynthesesyndrom (physiologische und anatomische Merkmale) innerhalb der Suaedoideae
entstanden ist. Die molekularen Phylogenien weisen unzweifelhaft nach, dass die vier Blatttypen aus
getrennten Entwicklungswegen hervorgegangen sind und sich alle von verschiedenen C3-Sippen
ableiten. Bezieht man die Ergebnisse der Altersdatierung ein so wird deutlich, dass die C4-
Entstehungen nicht nur unterschiedliche phylogenetische Herkunft haben sondern auch zu
unterschiedlichen Zeitpunkten erfolgt sein müssen. Diese These bleibt auch dann gültig, wenn weitere
Fossilfunde eine neue Altersschätzung erforderlich machen würden. Die relativen Abstände bleiben
davon unberührt. Die folgende Übersicht stellt noch einmal die aus den kalibrierten Stammbäumen
ermittelten Divergenzzeiten für ITS und atpB-rbcL zusammen:
Blatttyp Abspaltung von C3-Linie Beginn der Diversifizierung Bienertia-Blatttyp (single cell C4 -Typ) 31,3-33,1 Mio J.
Borszczowia-Blatttyp (single cell C4 -Typ) 10,3-11,2 Mio. J.
Salsina-Blatttyp (Kranz-C4-Typ) 16,5 Mio. J. 12,4-7,8 Mio. J.
Schoberia-Blatttyp (Kranz-C4-Typ) 12,9 Mio. J. 9,6 Mio. J.
4.5. Ableitung systematischer Einheiten 4.5.1. Position und Gliederung der Suaedoideae Die Ergebnisse der vorliegenden Arbeit sowie die phylogenetische Studien zu den Chenopodiaceae
(Kadereit et al. 2003) verwerfen die über lange Zeit gültige Unterteilung der Chenopodiaceae anhand
von Embryomerkmalen in Spirolobeae und Cyclolobeae und weisen Suaedoideae und Salicornioideae
als Schwestergruppen aus, zwischen denen die intermediäre Gattung Bienertia steht. Dabei
unterscheiden sich beide Unterfamilien in wichtigen Merkmalen erheblich, so dass als verbindender
Charakter fast nur das ökologische Verhalten herangezogen werden kann (Kadereit et al. 2003). Die
Unterfamilie Suaedoideae in der Fassung von Ulbrich (1934) kann damit mit geringfügigen
Änderungen (s. Kap. 1.2.1) aufrecht erhalten werden und weist im Gegensatz zu der Darstellung bei
Kühn et al. (2003) keine nähere Verwandtschaft zu den Salsoloideae auf. Da die Position von
Bienertia nicht eindeutig ist, kann die Unterfamilie jedoch als paraphyletische Einheit bewertet werden.
Die interne Gliederung der Suaedoideae bedarf jedoch nach den Ergebnissen der molekularen
Phylogenie einer Umgruppierung. Alexandra und Borszczowia sind Teile eines monophyletischen
Clades mit der Gattung Suaeda. Damit ist auch die Gattung Suaeda im traditionellen Verständnis eine
paraphyletische Gruppe. Aufgrund der engen verwandtschaftlichen Beziehungen und der genesteten
Positionen von Borszczowia und Alexandra ist es konsequent, beide Sippen in eine erweiterte

Diskussion
112
Gattung Suaeda einzuschließen, die damit wieder als monophyletische Einheit definiert werden kann.
In den Arbeiten von Schütze et al. (2003, für Borszczowia) und Kapralov et al. (2006, für Alexandra)
wurde diese logische Konsequenz bereits umgesetzt.
4.5.2. Arten und systematische Gruppen in der Gattung Suaeda 4.5.2.1. Brezia-Gruppe Die Gruppe ist molekular und morphologisch gut definiert und entspricht der aktuellen Sektion Brezia (Moq.) Volk. sowie der Sektion Heterosperma bei Iljin (1936a) sowie der Gattung Brezia und einem
Teil der Sektion Schoberia bei Ulbrich (1934). Von 38 bis jetzt bekannten Arten wurden 31 molekular
definiert, einige bisher unbeschriebene konnten anhand der molekularen Daten identifiziert werden.
Nach molekularen Daten umfasst sie alle Arten des in vorliegender Arbeit bezeichneten Brezia-
Clades, der die Schwestergruppe zu allen anderen Suaeda-Arten einschließlich Borszczowia und
Alexandra bildet. Die Zuordnung der Arten zu dieser monophyletischen Einheit ist durch alle Verfahren
der phylogenetischen Rekonstruktion hinweg konsistent und statistisch gut abgesichert. Verbindende
morphologische Merkmale sind die C3-Blätter des Brezia-Typs und der Brezia-Pistilltyp Perianth und
Ovar sind niemals verwachsen. Die Perianthzipfel haben oft charakteristische Flügel ausgebildet oder
sind ungleich in Größe und Gestalt sowie oft gekielt oder hornartig zugespitzt. Es sind überwiegend
annuelle Kräuter, selten auch ausdauernde Zwergsträucher. Die Arten der Sektion besitzen eine
weltweite Verbreitung mit Diversitätszentren in der Holarktis. Der höchste Artenreichtum entfaltet sich
in der submeridionalen und meridionalen Florenzone Eurasiens und Nordamerikas. Wesentlich
artenärmer ist die Sektion auf der Südhalbkugel, wo sie zwar in fast allen Florenreichen vertreten ist,
in jedem allerdings nur mit einer bis wenigen Sippen. Da die tropische Florenzone so gut wie nicht
besiedelt wird, ergibt sich ein bei vielen Verwandtschaftskreisen zu beobachtendes bipolares
Verbreitungsbild (Iljin 1936a: 45).
Die ITS-Daten implizieren eine gut gestützte Dreiteilung dieser Gruppe, die allerdings keine adäquate
morphologische Entsprechung findet. Aus diesem Grund ist es nicht sinnvoll eine ausschließlich auf
ITS-Daten beruhende Unterteilung vorzunehmen zumal diese in den Chloroplasten-Bäumen
weitgehend kollabiert (s. Kap. 3.2.3., 4.3.2.). Die mittels ITS gefundene Dreiergliederung wird zudem
durch hybridogene Sippen überlagert (S. spicata p.p., S. kulundensis, S. sibirica), die aufgrund ihrer
spezifischen Entstehung verschiedene Positionen in Kern- und Chloroplastenbäumen einnehmen.
Das Artkonzept in der Sektion Brezia ist durch die Einbeziehung molekularer Merkmale deutlich
verbessert worden, verbleibt jedoch in der praktischen Umsetzung schwierig. Grund ist die Variabilität
der Merkmale und die Folgen der Hybridbildung. Die molekulare Variabilität kann nicht direkt mit
morphologischer korreliert werden.
Mit den polymorphen Sippen S. corniculata, S. prostrata, S. salsa und S. maritima enthält Brezia die
weit verbreiteten Sippen der altweltlichen Holarktis. S. maritima wird hier wesentlich reduziert auf eine
nord-, mittel- und westeuropäisch verbreitete Sippe, die mit der eher kontinental verbreiteten S. salsa
vikariiert. Ob S. maritima auch in Nordamerika vorkommt, konnte im Rahmen dieser Studie nicht
überprüft werden, da kein Material vorlag. Die Angaben zu Merkmalen und Chromosomenzahl
(2n=36) der nordamerikanischen Pflanzen (Basset & Crompton 1978, Ferren & Schenk 2008) lassen
dies jedoch möglich erscheinen. Die Verbreitung in Europa beschränkt sich auf die ozeanisch

Diskussion
113
geprägten küstennahen Regionen, bereits im österreichisch-ungarischen Tiefland wird sie mit S.
salinaria von einem Vertreter der S. salsa-Verwandtschaft abgelöst und fehlt an der
Schwarzmeerküste. S. salsa repräsentiert die kontinentale Schwestergruppe zu S. maritima und
besitzt ein großes Verbreitungsgebiet von Osteuropa bis Zentralasien. Die nächst Verwandten dieser
beiden Arten sind fast ausschließlich zentral- und ostasiatsche sowie australische Sippen: S
heteroptera, S. japonica, S. malacosperma, S. australis und S. arbusculoides. Überaus diverse
Gruppen sind S. spicata und S. prostrata, in deren Verwandtschaft auch S. heterophylla und S.
stellatiflora gehört.
4.5.2.2. Schoberia-Gruppe inkl. Alexandra lehmannii Der Schoberia-Subclade vereint zwei sehr unterschiedliche Arten bzw. Artengruppen: Die C4-Sippen
der aktuellen Sekt. Schoberia (C. A. Mey.) Volk. und die C3-Art Alexandra lehmannii. Schoberia
enthält ausschließlich einjährige Sippen mit C4-Blättern des Schoberia-Typs und hat als weiteres
Merkmal das Schoberia-Pistill. Die Blätter sind verbreitert und besitzen oft eine Granne. Die weiteste
Verbreitung haben S. acuminata und S. microsperma in Zentral- und Mittelasien, die westlichste Sippe
ist die mediterrane S. splendens. Alexandra lehmannii wurde von Kapralov et al. (2006) als eigene
Sekt. Alexandra (Bunge) Kapralov, Akhani & E. H. Roalson Suaeda zugeordnet und in Suaeda
lehmannii umbenannt. Sie besitzt die weit verbreitete C3-Blattanatomie, charakteristische Vertikalflügel
am Perianth und weicht in Blattstellung und Gestalt deutlich von anderen Suaeda-Arten ab. Suaeda
pterantha, die ebenfalls Vertikalflügel aufweist (Ulbrich 1934, als Calvelia pterantha), gehört
möglicherweise in diese Gruppe, was aber bisher anhand molekularer Daten nicht nachgewiesen
werden konnte.
4.5.2.3. Fruticosa-Gruppe Die monophyletsche Gruppe (fruticosa-Subclade) enthält die früheren Sektionen Immersa,
Macrosuaeda, Salsina und Limbogermen und entspricht der aktuellen Sekt. Salsina Moq.. Die sehr
diverse Sektion vereinigt überwiegend strauchige, selten auch annuelle Sippen mit C4-Blättern des
Salsina-Typs und dem Schoberia-Pistill. Salsina ist neben Brezia die einzige Sektion, die alt- und
neuweltliche Arten enthält.
Altweltliche Arten sind S. altissima, S. monoica, S. vermiculata, S. asphaltica, S. aeqgyptiaca, S.
arcuata, S. microphylla und S. dendroides. Die neuweltlichen Sippen sind durch S. argentinensis, S.
foliosa, S. divaricata (Südamerika) sowie S. taxifolia, S. nigra, S. tampicensis, S. conferta, S.
californica (Nordamerika) und andere vertreten. Die Arten zeichnen in erster Linie geographische
Regionen nach, während die differenzierenden morphologischen Merkmale wie bei vielen Suaeda-
Arten oftmals nicht eindeutig sind. Eine Zwischenstellung nimmt S. fruticosa ein.
4.5.2.4. Physophora-Gruppe Zu dieser Gruppe gehören drei Sippen des physophora-Subclades: S. physophora, S. palaestina und
S. ifniensis. Diese Zusammengehörigkeit war vor den Ergebnissen der molekularen Analyse nicht klar.
Aufgrund der gemeinsamen Ableitung wird damit die bisher als monotypisch bekannte Sekt. Physophora Iljin um zwei Sippen erweitert. Bei den Arten handelt es sich um Zwergsträucher mit

Diskussion
114
sukkulenten C3-Blättern des Vera-Typs. Der Pistill-Typ gehört zum Schoberia-Typ der abgeleiteten
Gruppen. Die drei Sippen besiedeln heute jeweils eng umgrenzte Areale, die sich nicht mehr
überlappen. Der westlichste Vertreter ist S. ifniensis mit wenigen Vorkommen in Marokko und auf den
Kanaren, beide Sippen sind jedoch durch molekulare Merkmale sehr deutlich voneinander
unterschieden. Die Chloroplasten-Bäume zeigen sogar eine nähere Verwandtschaft der afrikanischen
S. ifniensis mit S. palaestina. In dieser Sippe sind damit weitere systematische Untersuchungen
notwendig. S. physophora ist die östlichste Sippe mit einem Verbreitungsgebiet, das ungefähr der
aralokaspischen Florenprovinz entspricht (Iljin 1936a, Meusel & Jäger 1992). Geographisch zwischen
beiden Sippen kommt S. palaestina vor (NO-Afrika, vord. Orient). Freitag (1989) weist auf den
xerohalophytischen Charakter von S. palaestina hin, was sicher auch für die anderen Arten der
Sektion gilt und typisch für die gesamte Sektion ist.
4.5.2.5. Suaeda-Gruppe Die Gruppe umfasst den vera-Subclade und entspricht der aktuellen Sekt. Suaeda mit S. vera und
einer bis jetzt noch nicht beschriebenen Sippe (S. „ekimii“). Die Sektion vereint damit eine
ausdauernde, verholzte Sippe und eine annuelle. Verbindende morphologische Merkmale sind die
Blätter des Vera-Typs und der singuläre Pistilltyp mit verbreiterten bzw. verzweigten Narben.
4.5.2.6. Schanginia-Gruppe mit Borszczowia aralocaspica Die C3-Arten des Schanginia-Subclades bilden die neu gefasste Sekt. Schanginia (C. A. Mey.) Volk.
mit Schanginia-Blatttyp und Schanginia-Pistilltyp. Es handelt sich um annuelle Sippen mit teilweise mit
dem Ovar verwachsenen Tepalen. Aktuell werden nur S. linifolia und S. paradoxa in diese Sektion
gestellt.
Mit ihnen verwandt, morphologisch aber stark abweichend ist Borszczowia aralocaspica, die aufgrund
der Stellung innerhalb Suadea in diese Gattung als eigene Sektion eingeschlossen wurde: Sekt. Borszczowia (Bunge) Freitag & Schütze und daher Suaeda aralocaspica heißt. Mit Schanginia teilt
sie den Pistill-Typ, die Blätter stellen aber eine völlig eigenständige Entwicklung dar.
4.5.2.7. Suaeda glauca Als isolierte Sippe steht S. glauca an der Basis des Suaeda-Clades. Es handelt sich um eine in vielen
Charakteren intermediäre Art, dies betrifft auch wichtige Schlüsselmerkmale wie Blattmorphologie und
Pistilltyp. Aufgrund dieser Sonderstellung und vor allem der Unterschiede gegenüber der Sektion
Schanginia (Blatttyp) sollte sie als eigene Sektion geführt werden. S. glauca ist ausschließlich
ostasiatisch verbreitet.
4.5.2.8. Bienertia Die vollkommen isolierte Gruppe besitzt als ursprüngliches Merkmal den einfachen Pistill-Typ mit 2
Narben, als Sonderentwicklung den Bienertia-Blatttyp mit single cell C4-Photosynthese. Sie gehört
nicht in die engere Verwandtschaft von Suaeda und verbleibt mit zwei Arten (B. cycloptera, B.
sinuspersici) in der Tribus Bienertieae Ulbr.
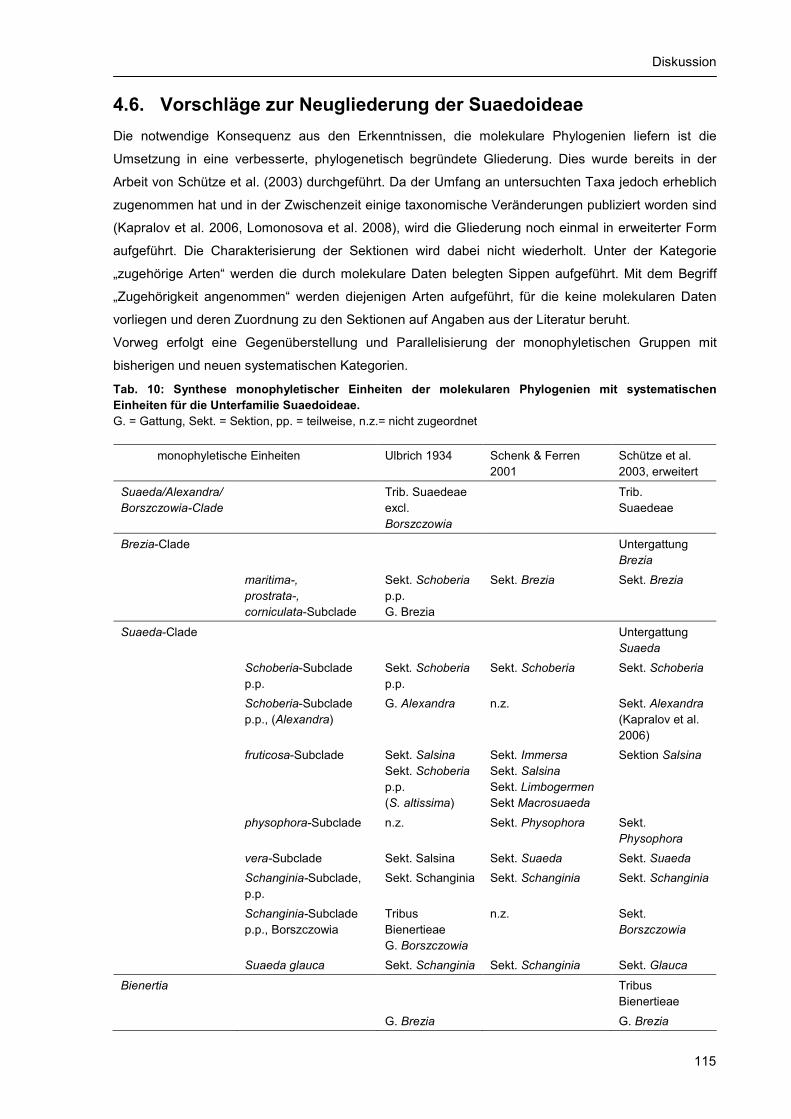
Diskussion
115
4.6. Vorschläge zur Neugliederung der Suaedoideae Die notwendige Konsequenz aus den Erkenntnissen, die molekulare Phylogenien liefern ist die
Umsetzung in eine verbesserte, phylogenetisch begründete Gliederung. Dies wurde bereits in der
Arbeit von Schütze et al. (2003) durchgeführt. Da der Umfang an untersuchten Taxa jedoch erheblich
zugenommen hat und in der Zwischenzeit einige taxonomische Veränderungen publiziert worden sind
(Kapralov et al. 2006, Lomonosova et al. 2008), wird die Gliederung noch einmal in erweiterter Form
aufgeführt. Die Charakterisierung der Sektionen wird dabei nicht wiederholt. Unter der Kategorie
„zugehörige Arten“ werden die durch molekulare Daten belegten Sippen aufgeführt. Mit dem Begriff
„Zugehörigkeit angenommen“ werden diejenigen Arten aufgeführt, für die keine molekularen Daten
vorliegen und deren Zuordnung zu den Sektionen auf Angaben aus der Literatur beruht.
Vorweg erfolgt eine Gegenüberstellung und Parallelisierung der monophyletischen Gruppen mit
bisherigen und neuen systematischen Kategorien.
Tab. 10: Synthese monophyletischer Einheiten der molekularen Phylogenien mit systematischen Einheiten für die Unterfamilie Suaedoideae. G. = Gattung, Sekt. = Sektion, pp. = teilweise, n.z.= nicht zugeordnet
monophyletische Einheiten Ulbrich 1934 Schenk & Ferren 2001
Schütze et al. 2003, erweitert
Suaeda/Alexandra/Borszczowia-Clade
Trib. Suaedeae excl. Borszczowia
Trib. Suaedeae
Brezia-Clade
Untergattung Brezia
maritima-, prostrata-, corniculata-Subclade
Sekt. Schoberia p.p. G. Brezia
Sekt. Brezia Sekt. Brezia
Suaeda-Clade
Untergattung Suaeda
Schoberia-Subclade p.p.
Sekt. Schoberia p.p.
Sekt. Schoberia Sekt. Schoberia
Schoberia-Subclade p.p., (Alexandra)
G. Alexandra n.z. Sekt. Alexandra (Kapralov et al. 2006)
fruticosa-Subclade Sekt. Salsina Sekt. Schoberia p.p. (S. altissima)
Sekt. Immersa Sekt. Salsina Sekt. Limbogermen Sekt Macrosuaeda
Sektion Salsina
physophora-Subclade n.z. Sekt. Physophora Sekt. Physophora
vera-Subclade Sekt. Salsina Sekt. Suaeda Sekt. Suaeda Schanginia-Subclade,
p.p. Sekt. Schanginia Sekt. Schanginia Sekt. Schanginia
Schanginia-Subclade p.p., Borszczowia
Tribus Bienertieae G. Borszczowia
n.z. Sekt. Borszczowia
Suaeda glauca Sekt. Schanginia Sekt. Schanginia Sekt. Glauca
Bienertia Tribus Bienertieae
G. Brezia G. Brezia

Diskussion
116
Unterfamilie Suaedoideae Ulbr. 1934 Tribus 1 - Bienertieae Ulbr 1934:560 emend. Freitag & Schütze
1 Gattung, 2 Arten:
Bienertia cycloptera Bunge ex Boiss., Bienertia sinuspersici Akhani
Tribus 2 - Suaedeae Dumort.
1 Gattung,
Suaeda Forssk ex Scop. (inklusive Alexandra Bunge, Borszczowia Bunge)
Suaeda Untergattung 1 - Brezia (Moq.) Freitag & Schütze
1 Sektion
Suaeda Sektion 1 - Brezia (Moq.) Volk.
Typus: Suaeda heterophylla (Kar. & Kir.) Bunge; mindestens 37 Arten
zugehörige Arten: S. arbusuculoides L. S. Sm., S. arctica Jurtzev & Petrovsky, S. australis Moq., S.
caespitosa Wolley-Dod, S. calceoliformis (Hook.) Moq., S. corniculata (C. A. Mey.) Bunge, S.
crassifolia Pall., S. esteroa Ferren & Whitmore, S. heterophylla (Kar. & Kir.) Bunge, S. heteroptera
Kitag., S. inflata Aellen, S. japonica Makino, S. kulundensis Lomon. & Freitag, S. linearis (S. Elliott)
Moq., S. malacosperma Hara, S. maritima (L.) Dumort., S. maritima var. perennans Maire, S.
mexicana Standl., S. occidentalis S. Watson, S. olufsenii Pauls., S. pannonica (Beck) Graebner, S.
patagonica Speg., S. prostrata Pall., S. puertopenascoa M. C. Watson & Ferren, S. salsa (L.) Pall., S.
salinaria (Schur) Simk., S. sibirica Lomon. & Freitag, S. spicata (Willd.) Moq., S. stellatiflora G. L. Chu,
S. tschujensis Lomon. & Freitag, S. tuvinica Lomon. & Freitag
Zugehörigkeit angenommen: S. albescens Lázaro Ibiza, S. densiflora A. Soriano, S. kossinskyi Iljin, S.
liaotungensis Kitag, S. novae-zelandiae Allan, S. przewalskii Bunge, S. rolandii Basset & Crompton
weitere: mehrere bisher nicht beschriebene (S. ‚arabica’, S. ‚elegans’, S. ‚venetiana’, S.
‚maritima/australis’ aus Korea)
Suaeda Untergattung 2 - Suaeda
9 Sektionen
Suaeda Sektion 2 - Schanginia (C. A. Mey.) Volk.
Typus: Suaeda linifolia Pall.; 2(3) Arten
zugehörige Arten: S. linifolia Pall., S. paradoxa Bunge
Zugehörigkeit angenommen: S. asparagoides (Miq.) Makino
Suaeda Sektion 3 - Borszczowia (Bunge) Freitag & Schütze
Typus: Borszczowia aralocaspica Bunge; 1 Art
zugehörige Arten: Suaeda aralocaspica (Bunge) Freitag & Schütze (Borszczowia aralocaspica Bunge)

Diskussion
117
Suaeda Sektion 4 - Suaeda
Typus: Suaeda vera Forssk.; 2 Arten
zugehörige Arten: Suaeda vera Forssk.
weitere: S. ‚ekimii’ (bisher unbeschrieben)
Suaeda Sektion 5 - Physophora Iljin
Typus: Suaeda physophora Pall.; 3(4) Arten
zugehörige Arten: S. ifniensis Caball., S. palaestina Eig. & Zoh., Suaeda physophora Pall.
weitere: möglicherweise eine weitere Sippe innerhalb von S. ifniensis
Suaeda Sektion 6 - Schoberia (C. A. Mey) Volk.
Typus: Suaeda acuminata (C. A. Mey.) Moq.; 9 (12) Arten
zugehörige Arten: S. acuminata (C. A. Mey.) Moq., S. carnosissima Post., S. cucullata Aellen, S.
eltonica Iljin, S. microsperma (C. A. Mey.) Fenzl, S. splendens (Pourr.) Gren. & Godr.
Zugehörigkeit angenommen: S.confusa Iljin, S. pterantha (Kar. & Kir.) Bunge, S. kareliniana Fenzl, S.
laevissima Kitag., S. turkestanica Litv., S. rigida Kung & G. L. Chu
Suaeda Sektion 7 - Salsina Moq.
Typus: Suaeda vermiculata Fossk. ex J. F. Gmelin.; ca. 30 Arten
zugehörige Arten: S. aegyptiaca Hasselq., S. altissima (L.) Pall. ex J. F. Gmel., S. arcuata Bunge, S.
argentinensis A. Soriano, S. articulata Aellen, S. asphaltica Moq., S. baluchestanica Akhani & Podl.,
S. californica S. Watson, S. conferta (Small) I. M. Johnston, S. dendroides (C. A. Mey.) Moq., S.
divaricata Moq., S. foliosa Moq., S. fruticosa Forssk. ex J. F. Gmel., S. micromeris Brenan, S.
microphylla Pall., S. mollis Desf., S. monodiana Maire, S. monoica Forssk. ex J.F. Gmel., S. moquinii
(Torr.) Greene, S. paulayana Vierh., S. tampicensis Standl., S. taxifolia Standl., S. vermiculata Forssk.
ex J.F. Gmel.(=S. pruinosa Willk. & Lange).
Zugehörigkeit angenommen: S. arguinensis Maire, S. merxmuelleri Aellen, S. moschata A.J. Scott, S.
palmeri Standl., S. plumosa Aellen, S. salina
Suaeda Sektion 8 - Alexandra (Bunge) Kapralov, Akhani & E. H. Roalson
Typus: Alexandra lehmanii Bunge; 1 Art
zugehörige Arten:. Suaeda lehmannii (Bunge) Kapralov, Akhani (Alexandra lehmanii Bunge)
Suaeda Sektion 9 - Glauca sect. nov. zugehörige Art: Suaeda glauca Bunge
Die Sektion muss noch beschrieben werden.

Zusammenfassung
118
5. Zusammenfassung Mit der vorliegenden Arbeit wird eine Synthese aus molekularer Phylogenie und Merkmalsentwicklung
für die Unterfamilie Suaedoideae der Chenopodiaceae präsentiert. Anhand von rekonstruierten
Stammbäumen aus den Sequenzunterschieden von DNA-Abschnitten zweier unabhängiger Genome
(Kern, Chloroplast) werden monophyletische Gruppen herausgearbeitet und die Variabilität der
molekularen Merkmale in Bezug auf die bekannten Arten diskutiert. Insgesamt wurden für alle
molekularen Analysen 294 Sequenzen ausgewertet. Mit 254 Sequenzen von Arten der Unterfamilie
Suaedoideae und 18 Sequenzen von Arten der Unterfamilie Salicornioideae wurde eine vergleichende
molekulare Analyse mit den DNA-Regionen ITS, atpB-rbcL und psbB-psbH durchgeführt. Mit der
Einbeziehung von ca. 65 bekannten Suaeda-Arten sind damit je nach Artauffassung bis zu 80% aller
Arten der Gattung berücksichtigt. Mittels Fossildaten werden die wichtigsten Divergenzereignisse
zeitlich fixiert. Die molekularen Stammbäume dienen weiterhin als Grundlage für die Bewertung der
Arten sowie ihrer morphologischen und anatomischen Merkmale. Ein wichtiger Aspekt bildet dabei die
Entwicklung der C4-Photosynthese mit den zugehörigen Blatttypen.
Die folgenden vier Themenkomplexe bzw. Fragestellungen sollten bearbeitet werden (für ausführliche
Darstellung vgl. Kap. 1.3):
1. Monophyletische Gruppen und ihre Beziehungen
2. Prinzipien der Evolution und Artbildung in der untersuchten Gruppe, Abgrenzung der Arten.
3. Entwicklung des C4-Photosynthesesyndroms
4. Entwicklung und Variabilität systematisch relevanter Merkmale
Die Ergebnisse der auf vergleichender DNA-Sequenzierung beruhenden molekularen Analyse und die
Synthese mit weiteren Daten führen als Resultat der vorliegenden Arbeit zusammenfassend zu
folgenden Ergebnissen:
1.) Die drei sequenzierten DNA-Regionen ITS, atpB-rbcL und psbB-psbH zeigen im Vergleich eine
sehr unterschiedliche Variabilität, ITS ist die variabelste aller Regionen. Die in den Alignments
gefundenen Merkmale in Form von Punkt- und Längenmutationen zwischen den Einzelsequenzen
waren zahlenmäßig ausreichend und qualitativ geeignet, um mit den drei Verfahren Maximum
Parsimony, Maximum Likelihood und Bayes’scher Analyse aussagekräftige und in wesentlichen
Aussagen kongruente molekulare Phylogenien zu rekonstruieren. Die Chloroplasten-Daten wurden für
die Berechnungen kombiniert.
2.) Die beiden DNA-Regionen ITS und atpB-rbcL evolvieren mit sehr unterschiedlichen
Geschwindigkeiten. Die mit der Glättungsmethode PL berechneten durchschnittlichen
Substitutionsraten weisen für ITS eine 5,5fach höhere Substitutionsrate gegenüber atpB-rbcL nach.
Die ITS-Sequenzen sind daher wesentlich diverser und für einige Sippen, bei identischen atpB-rbcL-

Zusammenfassung
119
Sequenzen, unterschiedlich. Eine direkte Homologisierung von molekularer und morphologischer
Variabilität oder die molekulare Limitierung von Arten ist daher nur in einigen Fällen möglich.
3.) Die Gattungen Suaeda, Alexandra und Borszczowia bilden eine monophyletische Gruppe, die den
Salicornioideae als Schwestergruppe gegenübersteht. Dies bestätigt die Ergebnisse der Phylogenie
der Chenopodiaceae (Kadereit et al. 2003). Im traditionellen Verständnis ist damit die Gattung Suaeda
paraphyletisch. Durch taxonomische Umkombination (Schütze et al. 2003. Kapralov et al. 2006)
werden die Gattungen Alexandra und Borszczowia in Suaeda eingegliedert, womit das Monophylie-
Kriterium für die Gattung wieder erfüllt ist. Die molekularen Daten unterstützen diese
nomenklatorische Neubewertung. Der nachfolgend verwendete Gattungsname Suaeda bezieht sich
auf die neue Fassung.
4.) Bienertia gehört nicht in die unter 2.) beschriebene monophyletische Gruppe. Die Stellung dieser
Gattung ist intermediär. Im ITS-Baum bildet sie die Schwestergruppe zu den Salicornioideae, im
Chloroplasten-Baum diejenige zu Suaeda. Da Bienertia aufgrund morphologischer Merkmale Suaeda
ähnlicher ist als den Salicornioideae wird sie in die intern neu gegliederte Unterfamilie der
Suaedoideae einbezogen, die weitgehend der in Ulbrich (1934) vertretenen Auffassung entspricht.
5.) Suaeda teilt sich in zwei sehr deutlich getrennte Gruppen, die in einer neuen Gliederung als
Untergattungen Brezia und Suaeda definiert werden. Die Trennung datiert mit etwa 30 Mio. Jahren in
das Oligozän. Zur Untergattung Brezia gehören alle Arten der Sektion Brezia nach bisheriger
taxonomischer Auffassung, die Untergattung Suaeda vereint alle übrigen Arten und wird in weitere
Sektionen untergliedert.
6.) Die Untergattung bzw. Sektion Brezia zeigt im ITS-Baum eine deutliche Dreigliederung in 3
Subclades, die allerdings von den Chloroplasten-Bäumen nicht verifiziert wird und auch durch
morphologische Merkmale nicht zu rechtfertigen ist. Die Subclades der Untergattung Suaeda
entsprechen in Grundzügen den bisherigen Sektionen und sind durch synapomorphe Merkmale
gekennzeichnet.
Für die Gattung Suaeda leiten sich nach monophyletischen Gruppen oder singulären Linien folgende
Sektionen ab: Brezia, Alexandra, Borszczowia, Schanginia, Schoberia Salsina (inkl. der früheren
Sektionen Limbogermen, Immersa und Macrosuaeda), Suaeda, Physophora und Glauca (neu).
7.) Hybridisierung und Polyploidisierung sind wichtige Prozesse der sympatrischen Artbildung
innerhalb der Suaedoideae und waren wahrscheinlich immer mit Arealerweiterungen gekoppelt.
Mehrere Linien sind durch Vervielfachung des Chromosomensatzes charakterisiert, wobei auch die
recht seltene Form der Dekaploidie erreicht wird. Vieles spricht dafür, dass sowohl Auto- als auch
Allopolyploidie eine Rolle spielt. Auf Autopolyplodie beruhen höchstwahrscheinlich die
unterschiedlichen Chromosomenrassen von S. corniculata. Durch Inkongruenzen zwischen
Chloroplasten- und ITS (Kern)-Stammbäumen konnten einige Arten mit hoher Wahrscheinlichkeit als

Zusammenfassung
120
etablierte, allopolyploide Hybridsippen identifiziert werden (Suaeda kulundensis, S. sibirica). Es ist
damit erwiesen, dass die Diversifizierung durch retikulate Evolution beeinflusst wird.
8.) Die Ergebnisse der molekularen Phylogenien belegen sehr deutlich, dass sich die C4-
Photosynthese innerhalb der Suaedoideae viermal unabhängig mit vier vollkommen unterschiedlichen
Blatttypen entwickelt hat. Dazu gehören zwei Blatttypen mit single cell C4-photosynthesis, ein bis vor
kurzem bei Landpflanzen unbekanntes Phänomen. Innerhalb der Sektionen Schoberia, Salsina und
Borszczowia datiert die Entstehung in das späte Miozän, bei Bienertia entstand die C4-Photosynthese
möglicherweise noch früher.
9.) Als systematisch äußerst bedeutsame Merkmale haben sich die schon von Iljin (1936a) benutzten
Pistill-Formen sowie spezifische Blattmerkmale herausgestellt. Mit diesen Merkmalen können
Sektionen, die auf monophyletischen Gruppen beruhen, gut definiert werden. Synapomorphe
Merkmale des Pistills sind die Zahl und Ausbildung der Narben sowie die Form ihrer Insertion im Ovar.
Bei den Blatttypen sind es vor allem die vier histologisch hoch differenzierten C4-Blatttypen, die als
gemeinsam abgeleitetes Merkmal rezenter, teilweise aufgespaltener Linien gewertet werden.
10.) Die früher z.T. überbewerteten Merkmale des Perianths (Verwachsungen, Flügel, Anhänge und
Umbildungen) können nur zur Beschreibung einzelner Sippen oder lokaler Gruppen herangezogen
werden. Ebenso ist das Merkmal der Lebensformen (Therophyten, Chamaephyten) kaum zur
Charakterisierung von Gruppen geeignet. Wie das Beispiel Brezia sehr deutlich zeigt, kam es allein in
dieser Gruppe mehrfach zur Entwicklung ausdauernder, verholzender Sippen. Der umgekehrte
Prozess fand bei der Entstehung der annuellen S. aegyptiaca und S. arcuata statt.
Mit Hilfe von DNA-basierten Stammbäumen ist es in der vorliegenden Arbeit möglich geworden, die
evolutionäre Geschichte der Gattung nachzuzeichnen und eine auf monophyletischen Gruppen
basierende Gliederung abzuleiten. Damit wird eine Grundlage für ein verbessertes Artkonzept der
Gattung Suaeda geschaffen. Für die praktische Taxonomie ist dies aber nur teilweise bedeutend. Die
morphologisch nachvollziehbare Abgrenzung von Arten bleibt, gerade in den diversen Sektionen
Brezia und Salsina, umstritten und kaum nachvollziehbar. Ein Großteil der Arten hat offenbar keine
interspezifischen Kompatibilitätsschranken, es handelt sich daher um Morpho- oder Semispecies,
möglicherweise sogar nur geographische Rassen, die allerdings mit schnell evolvierenden DNA-
Regionen wie ITS differenzierbar sind.
Ausgehend von den Ergebnissen der vorliegenden Arbeit verbleibt genügend Raum für
weiterführende, vertiefende systematische und populationsbiologische Studien.

Literaturverzeichnis
121
6. Literaturverzeichnis Akhani, H., Barroca, J., Koteyeva, N., Voznesenskaya, E., Franceschi, V., Edwards, G., Ghaffari, S. M. & Ziegler, H. (2005): Bienertia sinuspersici (Chenopodiaceae): a new species from SW Asia
and discovery of a third terrestrial C4 plant without Kranz anatomy. Syst. Bot. 30: 290-301.
Akhani, H., Trimborn, P. & Ziegler, H. (1997): Photosynthetic pathways in Chenopodiaceae from
Africa, Asia and Europe with their ecological, phytogeographical and taxonomical importance. Plant
Syst. Evol. 206: 187-221.
Altekar, G., S. Dwarkadas, S., J. P. Huelsenbeck, J. P. & Ronquist, F. (2004): Parallel Metropolis-
coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 20: 407-415.
Al-Turki, T. A., Filfilan, S. A. & Mehmood, S. F. (2000): A cytological study of flowering plants from
Saudi Arabia. Willdenowia 30: 339-358.
Alvarez, I. & Wendel, J. F. (2003): Ribosomal ITS sequences and plant phylogenetic inference. Mol.
Phyl. Evol. 29 (3): 417-434.
Arnheim, N. (1983): Concerted evolution in multigene families. In: Nei & Koehn (eds.): Evolution of
Genes and Proteins. Sinauer Associates, Sunderland, MA.
Bachmann, K. (1997): Nuclear markers in plant biosystematic research. Opera Bot. 132: 137-148.
Baldwin, B. G., Sanderson, M. J., Porter, J. M., Woiciechowski, M. F., Campbell, C. S. & Donoghue, M. J. (1995): The ITS region of nuclear ribosomal DNA: a valuable source of evidence on
angiosperm phylogeny. Ann. Missouri Bot. Gard. 82: 247-277.
Ball, P.W. & Akeroyd, J.R. (1993): Suaeda Forssk. ex Scop. In: Tutin, T. G. et al. (eds.) Flora
Europaea 1, ed. 2, pp. 123-125. Cambridge University Press, Cambridge, UK.
Bassett, I. J. & Crompton, C. W. (1978): The genus Suaeda (Chenopodiaceae) in Canada. Canad. J.
of Botany 56: 581-591.
Blattner, F. R. (1999): Direct amplification of the entire ITS region from poorly preserved plant
material using recombinant PCR. BioTechniques 27: 1180-1186.
Blattner, F. (2004): Phylogenetic analysis of Hordeum (Poaceae) as inferred by nuclear rDNA ITS
sequences. Mol. Phyl. Evol. 33: 289-299.
Bromham, L., Woolfit, M., Lee, M. S. Y. & Rambaut, A. (2002): Testing the relationships between
morphological and molecular rates of change along phylogenies. Evolution 56 (10): 1921-1930.
Cabrera, J. F. (2007): Phylogeny and historical biogeography of the Australian Camphorosmeae
(Chenopodiaceae). Dissertation. Johannes-Gutenberg-Universität Mainz.
Carolin, R. C., Jacobs, S. W. L. & Vesk, M. (1975): Leaf structure in Chenopodiaceae. Bot. Jahrb.
Syst. 95 (2): 226-255.
Chase, M. W. & Hills, H. H. (1991): Silica gel: An ideal material for field preservation of leaf samples
for DNA studies. Taxon 40: 215-220.
Chase, M. W. et al. (1993): Phylogenetics of seed plants: an analysis of nucloetide sequences from
the plastid gene rbcL. Ann. Missouri Bot. Gard. 80: 528-580.

Literaturverzeichnis
122
Collinson, M. E., Boulter, M. C. & Homes, P. L. (1993): Magnoliophyta ('Angiospermae'). In: Beton,
M. J. (ed.) The Fossil Record, vol. 2 : 809-841. Chapman & Hall, London.
Cronquist, A. (1981): An Integrated System of Classification of Flowering Plants. Columbia Press,
New York
Cuenoud, P., Savolainen, V., Chatrou, L. W., Powell, M., Grayer, R. J. & Chase, M. W. (2002):
Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, apB and
matK DNA sequences. Am. J. Bot. 89 (1): 132-144.
Denduangboripant, J. & Cronk, Q. C. B. (2001): Evolution and alignment of the hypervariable arm 1
of Aeschynanthus (Gesneriaceae) ITS2 nuclear ribosomal DNA. Mol. Phyl. Evol. 20 (2): 163-172.
Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K. & Mattick, J. S. (1991): 'Touchdown' PCR to
circumvent spurious priming during gene amplification. Nucleic Acids Res. 19: 4008.
Ebrahimzadeh, H., Ataei-Azimi, A., Akhani, H. & Noori-Daloii, M. R. (1994): Studies on the
caryology of some species of the genus Suaeda (Chenopodiaceae) in Iran. Journal of Sciences,
Islamic Rep. of Iran 5 (3): 135-138.
Edwards, G. E., Franceschi, V. R. & Voznesenskaya, E. V. (2004): Single cell C4 photosynthesis
versus the dual cell (Kranz) paradigm. Annu. Rev. Plant Biol. 55: 173-196.
Efron, B., Halloran, E. & Holmes, S. (1996): Bootstrap confidence levels for phylogenetic trees. Proc.
Natl. Acad. Sci. USA 93: 7085-7090.
Ennos, R. A., Sinclair, W. T., Hu, X.-S. & Langdon, A. (1999): Using organelle markers to elucidate
the history, ecology and evolution of plant populations. In Hollingsworth, P. M., Bateman, R. M. &
Gornall, R. J. (eds): Molecular systematics and plant evolution. pp. 1-19. Taylor & Francis, London.
Farris, J. S. (1970): Methods for computing Wagner trees. Syst. Zool. 19: 83-92.
Farris, J. S. (1989): The retentions index and the rescaled consistency index. Cladistics 5: 417-419.
Farris, J. S., Källersjö, M, Kluge, A. G. & Bult, C. (1994): Testing Significance of Incongruence.
Cladistics 10 (3): 315-319.
Fartmann, B., Nikoleit. K. & Rehfeldt, K. (1999): Sequencing Brochure by MWG-Biotech AG v. 4.
Felsenstein, J. (1981): Evolutionary trees from DNA sequences: a maximum likelihood approach. J.
Mol. Evol. 17: 368-376.
Felsenstein, J. (1985): Confidence limits on phylogenies: an approach using the bootstrap. Evolution
39: 783-791.
Felsenstein, J. (2004): Inferring Phylogenies. Sinauer Associates, Sunderland, Massachusetts
Ferren, W. R., Schenk, H. J (2008): Suaeda. In: Flora of North America, webressouce;
http://www.efloras.org.
Fisher, D. D., Schenk, H. J, Thorsch, J. A. & Ferren, W. R. (1997): Leaf anatomy and subgeneric
affiliation of C3 and C4 species of Suaeda (Chenopodiaceae) in North America. Am. J. Bot. 84 (9):
1198-1210.
Freitag, H. (1989): Contributions to the chenopod flora of Egypt. Flora 183: 149-173.

Literaturverzeichnis
123
Freitag, H. (2001): Suaeda. In: Ali, S. I. & Qaiser, M. (eds) Flora of Pakistan 204. pp. 104-126. MBG
Press, Karachi and St. Louis.
Freitag, H. & Lomonosova, M. (2006): Typification and identity of Suaeda crassifolia, S. prostrata
and S. salsa, three often confused species of Suaeda sect. Brezia (Chenopodiaceae, Suaedoideae).
Willdenowia 36: 21-36.
Freitag, H. & Stichler, W. (2000): A remarkable new leaf type with unusual photosynthetic tissue in a
Central Asiatic genus of Chenopodiaceae. Plant Biol. 2: 154-160.
Freitag, H. & Stichler, W. (2002): Bienertia cycloptera Bunge ex Boiss., Chenopodiaceae, another C4
plant without Kranz tissues. Plant Biol. 4: 121-132.
Freitag, H., Walter, J. & Wucherer, W. (1996): Die Gattung Suaeda (Chenopodiaceae) in Österreich,
mit einem Ausblick auf die pannonischen Nachbarländer. Ann. Naturhist. Mus. Wien, Suppl. 98B: 343-
367.
Gillion, J. & Jakir, D., (2001): Influence of carbonic anhydrase activity in terrestrial vegetation on the
18O content of atmosphaeric CO2. Science 291: 2584-2587.
Giussani, K. M., Cota-Sanchez, J. H., Zuloaga, F. O. & Kellogg, E. A. (2001): A molecular
phylogeny of the grass subfamily Panicoideae (Poaceae) shows multiple origins of C4 photosynthesis.
Am. J. Bot. 88 (11): 1993-2012.
Godwin, H. (1969): The value of plant materials for radiocarbon dating. Am. J. Bot. 56 (7): 723-731.
Grant, V. (1971): Plant Speciation. Columbia University Press, New York and London.
Hall, B. G. (2001): Phylogenetic Trees Made Easy. A how-to manual for olecular biologists. Sinauer
Associates, Sunderland, Massachusetts, USA.
Hamby, R. K. & Zimmer, E. A. (1991): Ribosomal RNA as a phylogenetic tool in plant systematics. In:
Soltis, P., Soltis, D. & Doyle, J. (eds.): Molecular Systematics of Plants. pp. 50-91. Chapman & Hall,
New York.
Hennig, W. (1950): Grundzüge einer Theorie der phylogenetischen Systematik. Deutscher
Zentralverlag, Berlin.
Hershkovitz, M. A. & Zimmer, E. A. (1996): Conservation patterns in angiosperm rDNA ITS2
sequences. Nucleic Acids Res. 24 (15): 2857-2867.
Hillis, D. M. & Dixon, M. T. (1991): Ribosomal DNA: Molecular evolution and phylogenetic inference.
Quart. Rev. Biol. 66: 411-453.
Hillis, D. M., Mable, K. B. & Moritz, C. (1996): Applications of Molecular Systematics. In: Hillis, D. M.,
Moritz, C. & Mable, B. K. eds.: Molecular Systematics. second ed. Sinauer Associates, Sunderland,
Massachusetts.
Hohmann, S., Kadereit, J. W. & Kadereit, G. (2006): Understanding Mediterranean-Californian
disjunctions: molecular evidence from Chenopodiaceae-Betoideae. Taxon 55 (1): 67-78.
Holder, M. & Lewis, P. O. (2003): Phylogeny estimation: traditional and Bayesian approaches. Nature
Reviews Genetics 4: 275-284.

Literaturverzeichnis
124
Hopkins, C. O. & Blackwell, W. H. (1977): Synopsis of Suaeda (Chenopodiaceae) in North America.
Sida 7: 147-173.
Huelsenbeck, J. P. & Crandall, K. A. (1997): Phylogeny estimation and hypothesis testing using
maximum likelihood. Annu. Rev. Ecol. Syst. 28: 437-466.
Huelsenbeck, J. P. & Ronquist, F. (2001): MRBAYES: Bayesian inference of phylogenetic trees.
Bioinformatics Applications Note 17 (8): 754-755.
Hultén, E. & Fries, M. (1986): Atlas of North European vascular plants: north of the Tropic of Cancer
I-III. Koeltz Scientific Books, Königstein.
Iljin, M. M. (1936a): K sistematike roda Suaeda Forssk. i triby Suaedeae Rchb. (in Russian). [On the
systematics of genus Suaeda Forssk. and tribe Suaedeae Rchb.]. Sov. Bot. 5: 39-49.
Iljin, M. M. (1936b): Chenopodiaceae Less. In: Komarov V. L., Shishkin, B. K. (eds.) Flora SSSR 6.
Akademia Nauk, Moskva/Leningrad, pp. 2-354 (engl. transl. 1985. Singh/Koeltz, Dehra Dun).
Jukes, T. H. & Cantor, C. R. (1969): Evolution of protein molecules. In: Munro, H. M. (ed.):
Mammalian Protein Metabolism.: 21-132. Academic Press, New York.
Kadereit, G., Borsch, T., Weising, K.& Freitag, H. (2003): Phylogeny of Amaranthaceae and
Chenopodiaceae and the evolution of C4-photosynthesis. International Journal of Plant Sciences 164
(6): 959-986.
Kadereit, G., Gotzek, D., Jacobs, S. & Freitag, H. (2005): Origin and age of Australian
Chenopodiaceae. Organisms, Diversity & Evolution 5: 59-80.
Kadereit, G., Mucina, l. & Freitag, H. (2006): Phylogeny of Salicornioideae (Chenopodiaceae):
diversification, biogeography and evolutionary trends in leaf and flower morphology. Taxon 55 (3):
617-642.
Kapralov, M. V., Akhani, H., Voznesenskaya, E. V., Edwards, G., Franceschi, V. & Roalson, E. H. (2006): Phylogenetic relationships in the Salicornioideae / Suaedoideae / Salsoloideae s.l.
(Chenopodiaceae) clade and a clarification of the phylogenetic position of Bienertia and Alexandra
using multiple DNA sequence datasets. Syst. Bot. 31 (3): 571-585.
Kelchner, S. A. (2000): The evolution of non-coding chloroplast DNA and its application in plant
systematics. Ann. Missouri Bot. Gard. 87 (4): 482-498.
Kimura, M. (1980): A simple method for measuring evolutionary rates of base substitutions through
comparative studies of nucleotide sequences. J. Mol. Evol. 16: 111-120.
Kimura, M. (1983): The Neutral Theory of Molecular Evolution. Cambridge University Press,
Cambridge, UK.
Kluge, A. G. & Farris, J. S. (1969): Quantitative phyletics and the evolution of anurans. Syst. Zool.
18: 1-32.
Krahulcová, A. & Tomsovic, P. (1997): Ploidy levels in some European representatives of the
Suaeda maritima group. Preslia 69: 327-332.

Literaturverzeichnis
125
Kühn, U., Bittrich, V., Carolin, R., Freitag, H., Hedge, I. C., Uotila, P. & Wilson, P. G. (1993):
Chenopodiaceae. In: Kubitzki K. (ed.) The families and genera of vascular plants 2. pp. 253-281.
Springer-Verlag, Berlin, New York, London.
Lanave, C., Preparata, G. Saccone, C. & Serio, G. (1984): A new method for calculating
evolutionary substitution rates. J. Mol. Evol. 20: 86-93.
Larget, B. & Simon, D. (1999): Markov chain Monte Carlo algorithms for the Bayesian analysis of
phylogenetic trees. Mol. Biol. Evol. 16: 750-759.
Lee, J.-S., Park, D. S., Ihm, B.-S. & Lee, W. J. (2007): Taxonomic reappraisal on Suaeda australis
(Chenopodiaceae) in Korea based on the mophological and molecular characteristics. Journal of Plant
Biology 50 (6): 605-614.
Li, J., Alexander, J., Ward, T., Del Tredici, P. & Nicholson, R. (2002): Phylogenetic relationships of
Empetraceae inferred from sequences of chloroplast matK and nuclear ribosomal DNA ITS region.
Mol. Phyl. Evol. 25: 306-315.
Liao, D. (1999): Concerted evolution: molecular mechanism and biological implications. Am. J. Hum.
Genet. 64: 24-30.
Lomonosova, M. & Krasnikov, A. A. (1993): Chromosomnye cisla nekotorych predstavitelej
semejstva Chenopodiaceae (Chromosome numbers in some members of the Chenopodiaceae.). Bot.
Zhurn. St. Petersburg 78: 158-159.
Lomonosova, M. N., Krasnikova, S. A., Krasnikov, A. A., Sukhorukov, A. P., Bananova, V. A. & Pavlova, N . S. (2005): Chromosome numbers of Chenopodiaceae species from Russia and
Kazakhstan. Botan. Journ. 7: 34-39.
Lomonosova, M., Brandt, R. & Freitag, H. (2008): Suaeda corniculata (Chenopodiaceae) and
related new taxa from Eurasia. Willdenowia 38: 81-109.
Maire, R. (1962): Flore de l'Afrique du Nord (Maroc, Algerie, Tunisie, Tripoltanie, Cyrenaique et
Sahara). Paul Lechevalier, Paris.
Mayol, M. & Rossello, J. A. (2001): Why nuclear ribosomal DNA spacers (ITS) tell different stories in
Quercus. Mol. Phyl. Evol. 19 (2): 167-176.
Metzing, D., Cordes, H. & Kuhbier, H. (1996): Biosystematische Untersuchungen an Suaeda
maritima (Chenopodiaceae). Drosera 96 (1): 1-26.
Meusel, H. & Jäger, E. J. (1992): Vergleichende Chorologie der zentraleuropäischen Flora. Bd. III.
Gustav Fischer, Jena, Stuttgart, New York.
Meusel, H., Jäger., E. & Weinert, E. (1965): Vergleichende Chorologie der zentraleuropäischen
Flora. Gustav Fischer Verlag, Jena.
Meyer, C. A. (1829): Chenopodeae. In Ledebour, C. F. (ed.) Flora Altaica 1. pp. 370-417. Reimer,
Berlin.
Möller, S. (2005): Speziation und Biogeographie von Suaeda-Arten an europäischen Küsten.
Diplomarbeit, Univeristät Kassel.

Literaturverzeichnis
126
Moquin-Tandon, A. (1831): Essai monographique sur le genre Suaeda et sur les Chénopodées les
plus voisines. Ann. Sci. Nat. (Paris) 23: 278-325.
Moquin-Tandon, A. (1840): Chenopodearum monographica enumeratio. P.-J. Loss, Paris.
Mullis, K., Faloona, S., Scharf, R., Saiki, G., Horn, G. & Ehrlich, H. (1986): Specific enzymatic
amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp. Quant. Biol.
51.
Nichols, D. J. & Traverse, A. (1971): Palynology, petrology, and depositional environments of some
early Tertiary lignites in Texas. Geoscie. & Man. 3: 37-48.
Nieto Feliner, F. & Rosello, J. A. (2007): Better the devil you know? Guidelines for insightful
utilization of nrDNA ITS in species-level evolutionary studies in plants. Mol. Phyl. Evol. 44: 911-919.
Ogihara, Y., Teerachi, T. & Sasakuma, T. (1988): Intramolecular recombination of chloroplast
genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. USA 85:
8573-8577.
Olmstead, R. G. & Palmer, J. D. (1994): Chloroplast DNA systematics: A review of methods and data
analysis. Am. J. Bot. 81 (9): 1205-1224.
Otto, S. P. & Whitton, J. (2000): Polyploid incidence and evolution. Annu. Reviews Genetics 34: 401-
437.
Oxelman, B., Liden, M. & Berglund, D. (1997): Chloroplast rps16 intron phylogeny of the tribe
Sileneae (Caryophyllaceae). Plant Syst. Evol. 206: 393-410.
Palmer, J. D., Nugent, J. M. & Herbon, L. A. (1987): Unusual structure of geranium chloroplast DNA:
A triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat
families. Proc. Natl. Acad. Sci. USA 84: 769-773.
Pedrol, J. & Castroviejo, S. (1988): A proposito del tratamiento taxonomico y nomenclatural del
genero Suaeda Forsskal ex Scop. (Chenopodiaceae) en ""Flora Iberica"". Ann. Jard. Bot. Madrid 45:
93-102.
Pedrol, J. & Castroviejo, S. (1990): Suaeda Forssk. ex Scop. - In Castroviejo & al. (eds.): Flora
Iberica 2, pp. 536-541. Real Jardin Botanico, Madrid.
Popp, M., Erixon, P., Eggens, F. & Oxelman, B. (2005): Origin and Evolution of a circumpolar
polyploid species complex in Silene (Caryophyllaceae) inferred from low copy nuclear RNA
polymerase introns, rDNA, and chloroplast DNA. Syst. Bot. 30 (2): 302-313.
Posada, D. & Buckley, T. R. (2004): Model selection and model averaging in phylogenetics:
Advantages of Akaike Information Criterion and Bayesian approaches over likelihood ratio tests. Syst.
Biol. 53 (5): 793-808.
Posada, D. & Crandall, K. A. (1998): Modeltest: testing the model of DNA substitution. Bioinformatics
14: 817-818.
Pyankov, V. I., Voznesenskaya, E. V., Kondratschuk, A. V & Black, C. C. (1997): A comparative
anatomical and biochemical analysis in Salsola (Chenopodiaceae) species with and without Kranz
type leaf anatomy: A possible reversion of C4 to C3 photosynthesis. Am. J. Bot. 84 (5): 597-606.

Literaturverzeichnis
127
Rieppel, O. (1999): Einführung in die computergestützte Kladistik. Verlag Dr. Friedrich Pfeil,
München.
Rilke, S. (1999): Revision der Sektion Salsola s.l. der Gattung Salsola (Chenopodiaceae).
Schweizerbarth, Stuttgart.
Ritland, C. E., Ritland, K. & Straus, N.A. (1993): Variation in the ribosomal internal transcribed
spacers (ITS1 and ITS2) among 8 taxa of the Mimulus-guttatus species complex. Mol. Biol. Evol. 10:
1273-1288.
Ritz, C. M., Schmuths, H. & Wissemann, V. (2005): Evolution by reticulation: European dogroses
originated by multiple hybridization across the genus Rosa. Journal of Heredity 96 (1): 4-14.
Robinson-Rechavi, M. & Huchon, D. (2000): RRTree: Relative-rate tests between groups of
sequences on a phylogenetic tree. Bioinformatics 16: 296-297.
Rogstad, S. H. (1992): Saturated NaCl-CTAB solution as a means of field prservation of leaves for
DNA analyses. Taxon 41: 701-708.
Ronquist, F. & Huelsenbeck, J. P. (2003): MRBAYES 3: Baysian phylogenetic inference under
mixed models. Bioinformatics 19: 1572-1574.
Sage, R. F. (2002): C4 photosynthesis in terrestrial plants does not require Kranz anatomy. Trends in
Plant Science 7 (7): 283-285.
Sage, R. F. (2004): Transley review. The evolution of C4 photosynthesis. New Phytologist 161: 341-
370.
Saitou, N. & Nei, M. (1987): The neighbor joining method: a new method for reconstructing
phylogenetic trees. Mol. Biol. Evol. 4 (4): 406-425.
Sambrook, J., Russell, D.W. (2001): Molecular Cloning: A Laboratory Manual, 3. Auflage. Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Sande, A. v. d., Kwa, M., Nues, R. v., Heerikhuizen, H. v., Raue, H. A. & Planta, R. J. (1992): Functional analysis of internal transcribed spacer 2 of Saccharomyces cerevisiae ribosomal DNA. J.
Mol. Biol. 223 (4): 899-910.
Sanderson, M. J. (1997): A nonparametric approach to estimating divergence times in the absence of
rate constancy. Mol. Biol. Evol. 14 (12): 1218-1231.
Sanderson, M. J. (2002): Estimating absolute rates of molecular evolution and divergence times: A
penalized likelihood approach. Mol. Biol. Evol. 19 (1): 101-109.
Sanderson, M. J. (2004): r8s, version 1.70 user’s manual available from http://ginger.ucdavis.edu/r8s.
Sang, T., Crawford, D. J. & Stuessy, T. F. (1995): Documentation of reticulate evolution in peonies
(Paeonia) using internal transcribed spacer sequences of nuclear ribosomal DNA: Implications for
biogeography and concerted evolution. Proc. Natl. Acad. Sci. USA 92: 6813-6817.
Sanger, F., Nicklen, S. & Coulson, A. R. (1977): DNA sequencing with chain-terminating inhibitors.
Proc. Natl. Acad. Sci. USA 74: 5463-5467.

Literaturverzeichnis
128
Sarich, V .M. & Wilson A. C. (1973): Generation time and genomic evolution in primates. Science
179: 1144-1147.
Schenk, H. J. & Ferren, W. R. (2001): On the sectional nomenclature of Suaeda (Chenopodiaceae).
Taxon 50: 857-873.
Schmitz-Linneweber, C., Maier, R. M., Alcaraz, J. P., Cottet, A., Herrmann, R. G. & Mache, R. (2001): The plastid chromosome of spinach (Spinacia oleracea): complete nucleotide sequence and
gene organization. Plant Mol. Biol. 45 (3): 307-315.
Schütze, P., Freitag, H. & Weising, K. (2003): An integrated molecular and morphological study of
the subfamily Suaedoideae Ulbr. (Chenopodiaceae). Plant Syst. Evol. 239: 257-286.
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W. S., Miller, J., Siripun, K. C., Winder, C. T., Schilling, E. E. und Small, R. L (2005): The tortoise and the hare II: Relative utility of 21
noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92 (1): 142-166.
Shepherd, K. A., Waycott, M. & Calladine, A. (2004): Radiation of the australian Salicornioideae
(Chenopodfiaceae) - based on evidence from nuclear and chloroplast DNA sequences. Am. J. Bot. 91
(9): 1387-1397.
Shmida, A. (1985): Biogeography of the desert flora. In: Evenaris, M., Noy-Meir, I. & Goodall, D. W.
eds. Hot deserts and arid shrubland. Elsevier, Amsterdam.
Shomer-Ilan, A., Neumann-Ganmorem, R. & Waisel, Y. (1979): Biochemical specialization of
photosynthetic cell layers and carbon flow paths in Suaeda monoica. Plant Physiol. 64: 963-965.
Simmons, M.P., Savolainen, V, Clevinger, C. C. , Archer, R. H., & Davis, J. I. (2001): Phylogeny of
the Celastrales inferred from 26S nuclear ribosomal DNA, phytochrome B, rbcL, atpB, and
morphology. Mol. Phyl. Evol. 19 (3): 353-366.
Small, R. L. & Wendell, J. F. (2000): Phylogeny, Duplication, and Intraspecific Variation of Adh
Sequences in New World Diploid Cottons (Gossypium L., Malvaceae). Mol. Phyl. Evol. 16 (1): 73-84.
Sneath, P. H. A. & Sokal, R. R. (1973): Numerical Taxonomy. Freeman, San Francisco.
Stanley, H. (1994): Historische Geologie. Spektrum Akademischer Verlag, Heidelberg.
Stebbins, G. L. (1974): Flowering Plants. Evolution above the Species Level. The Belknap Press of
Harvard University, Cambridge, Massachusetts.
Subramanian, D. (1988): Cytological studies of some mangrove flora of Tamiland. Cytologia 53: 87-
92.
Swofford, D. L. (2002): PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods).
Version 4. Sinauer Associates, Sunderland, Massachusetts, USA.
Swofford, D. L., Olsen, G. J., Waddell, P. J. & Hillis, D. M. (1996): Phylogenetic inference. In: Hillis,
D. M., Moritz, C. & Mable, B. K. (eds): Molecular systematics. pp. 407-514. Sinauer Associates,
Sunderland, Massachusetts, USA.
Taberlet, P., Gielly, G, Pautou, G. & Bouvet, J. (1991): Universal primers for amplification of three
noncoding regions of chloroplast DNA. Plant Mol. Biol. 17: 1105-1109.

Literaturverzeichnis
129
Tamura, K., Dudley, J., Nei, M. & Kumar, S. (2007): MEGA4: Molecular Evolutionary Genetics
Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24: 1596-1599.
The Angiosperm Phylogeny Group (2003): An update of the Angiosperm Phylogeny Group
classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141: 399-436.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F.& Higgins, D. G. (1997): The
ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality
analysis tools. Nucleic Acids Res. 25: 4876-4882.
Townsend, C. C. (1980): Contributions to the flora of Iraq: XIII. Notes on some genera of
Chenopodiaceae. Kew. Bull. 35: 291-296.
Tzvelev, N. N. (1993): Samjetki o marjewych (Chenopodiaceae) vostotschnoi Ewropy. (Notes on
Chenopodiaceae of Eastern Europe). Ukrainsk. Botan. Zurn. 50: 78-85.
Ulbrich, E. (1934): Chenopodiaceae. In: Engler A. & Prantl K.(eds.) Die natürlichen Pflanzenfamilien,
ed. 2, 16c. pp. 379-584. Duncker & Humblot, Leipzig
Ungar, I. A. (1974): Inland halophytes of the United States. In: Reimold, R. J. & Queen, W. H. (eds.)
Ecology of Halophytes. Academic Press, New York.
Volkens, G. (1892): Chenopodiaceae. In: Engler A. & Prantl, K. (eds.) Die natürlichen
Pflanzenfamilien, ed. 1, 3. pp. 36-91.. Engelmann, Leipzig.
Voznesenskaya, E. V., Franceschi, V. R., Kiirats, O., Artyusheva, E.G., Freitag, H.& Edwards, G. (2002): Proof of C4 photosynthesis without Kranz anatomy in Bienertia cycloptera (Chenopodiaceae).
The Plant Journal 31 (5): 649-662.
Voznesenskaya, E. V., Franceschi, V. R., Kiirats, O., Freitag, H. & Edwards, G. E. (2001): Kranz
anatomy is not essential for terrestrial C4 plant photosynthesis. Nature 414: 543-546.
Weising, K., Nybom, H., Wolff, K. & Kahl, G. (2005): DNA fingerprinting in plants. Principles,
methods and applications., 2. Edition. Taylor and Francis, Boca Raton, London, New York, Singapore.
Wesson, D. M., Porter, C. H. & Collins, E. H. (1992): Sequence and secondary structure
comparisons of the ITS rDNA in mosquitos (Diptera: Culicidae). Mol. Phyl. Evol. 1: 253-269.
Whittall, J., Liston, A., Gisler, S. & Meinke, R. J. (2000): Detecting Nucleotide Additivity from Direkt
Sequences is a SNAP: An Example from Sidalcea (Malvaceae). Plant Biol. 2: 211-217.
Wolfe, K. H., Li, W.-H. & Sharp, P. M. (1987): Rates of nucleotide substitution vary greatly among
plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 84: 9054-9058.
Woloszynska, M., Bocer, T., Mackiewicz, P. & Janska, H. (2004): A fragment of chloroplast DNA
was transferred horizontally, probably from non-eudicots, to mitochondrial genome of Phaseolus. Plant
Mol. Biol. 56: 811-820.
Wu, C.-I & Li, W.-H. (1985): Evidence for higher rates of nucleotide substitution in rodents than in
man. Proc. Natl. Acad. Sci. USA 82: 1741-1745.
Xu, D. H., Abe, J., Sakai, M., Kanazawa, A. & Shimamoto, Y. (2000): Sequence variation of non-
coding regions of chloroplast DNA of soybean and related wild species and its implications for the
evolution of different chloroplast haplotypes. Theor. Appl. Genet. 101: 724-732.

Literaturverzeichnis
130
Yang, Z., &. Rannala, B. (1997): Bayesian phylogenetic inference using DNA sequences: a Markov
chain Monte carlo method. Mol. Biol. Evol. 14: 717-724.
Zuckerkandl, E. & Pauling, L. (1965): Evolutionary divergence and convergence in proteins. In:
Bryson, V. & Vogel, H. J. (eds.) Evolving genes and proteins. 97-166. Academic Press, New York,
USA.

Abbildungsverzeichnis
131
Anhang Anhang 1 Abbildungsverzeichnis Abb. 1: Übersicht über die Entwicklung der Taxonomie der Gattung Suaeda. 7
Abb. 2: Suaedoideae, Habitate und Wuchsformen. 9
Abb. 3: Suaeda, Blüten- und Samenmerkmale. 11
Abb. 4: Übersicht zur Blattanatomie zur C4-Photosynthese innerhalb der Suaedoideae. 13
Abb. 5: Informelle Klassifikation und mögliche phylogenetische Beziehungen altweltlicher Suaeda-Arten. 17
Abb. 6: Nachweis von genomischer DNA aus der CTAB-Isolation von unterschiedlichen Geweben mittels Agarosegel-Elektrophorese. 51
Abb. 7: Ausschnitt aus dem Alignment einer GC-reichen, hoch variablen Region des ITS2-Alignments der Suaedoideae. 53
Abb. 8: Ausschnitt aus dem atpB-rbcL-Alignment ausgewählter Taxa der Suaedoideae. 53
Abb. 9: Maximum Parsimony-Phylogenie der Suaedoideae und ausgewählter Salicornioideae. 58
Abb. 10: Ein zufällig ausgewählter sparsamster Baum der MP-Analyse der ITS-Region für 92 Taxa der Suaedoideae und Kalidium als Außengruppe. 61
Abb. 11: Wahrscheinlichster Baum der ML-Analyse der ITS-Region für 92 Taxa der Suaedoideae und Kalidium als Außengruppe mit einem -ln L Wert von 6453,341. 62
Abb. 12: Ein zufällig ausgewählter sparsamster Baum der MP-Analyse von 66 kombinierten atpB-rbcL/psbB-psbH-Sequenzen der Suaedoideae und Kalidium als Außengruppe. 70
Abb. 13: Wahrscheinlichster Baum der ML-Analyse von 66 kombinierten atpB-rbcL/psbB-psbH-Sequenzen der Suaedoideae und Kalidium als Außengruppe mit einem -ln L Wert von 4681,259. 71
Abb. 14: Ein zufällig ausgewählter sparsamster Baum der MP-Analyse aus ITS-Sequenzen von 66 Taxa der Gattung Suaeda/Sektion Brezia mit Suaeda vera als Außengruppe. 76
Abb. 15: Kalibriertes Chronogramm der Suaedoideae sowie ausgewählter Salicornioideae, Salsoloideae und Chenopodieae (als Außengruppen) auf Basis einer ML-Baumtopologie aus 73 atpB-rbcL-Sequenzen. 85
Abb. 16: Kalibriertes Chronogramm der Suaedoideae und ausgewählter Salicornioideae auf Basis einer ML-Baumtopologie aus 92 ITS-Sequenzen. 86
Abb. 17: Rezente Verteilung von C3- und C4-Blatttypen sowie Entwicklungslinien (grün unterlegt) zur C4-Photosynthese innerhalb der Suaedoideae. 89
Abb. 18: Merkmalsentwicklung innerhalb der Suaedoideae anhand der ITS-Baumtopolgie aus der ML-Analyse
96
Abb. 19: Schema zum Nachweis etablierter Hybridsippen und retikulater Evolution innerhalb der Sektion Brezia.
106
Abb. 20: Konsensus-Baum (50% majority rule) der MP-Analyse von 67 atpB-rbcL-Sequenzen. 145
Abb. 21: Konsensus-Baum (50% majority rule) der MP-Analyse von 63 psbB-psbH-Sequenzen 146
Abb. 22: Konsensus-Baum (50% majority rule) der MP-Analyse von 67 kombinierten atpB-rbcL/psbB-psbH-Sequenzen. 147
Abb. 23: Baumtopologie (50% majority rule-Konsensus) der Bayesianischen Analyse von 67 kombinierten atpB-rbcL/psbB-psbH-Sequenzen. 148
Abb. 24: Konsensus-Baum (50% majority rule) der MP-Analyse von 93 ITS-Sequenzen. 149
Abb. 25: Baumtopologie (50% majority rule-Konsensus) der Bayesianischen Analyse von 93 ITS-Sequenzen. 150
Abb. 26: Kalibriertes Chronogramm von 73 atpB-rbcL-Sequenzen auf Basis einer ML-Baumtopologie mit vollständiger Benennung der Knoten. 151
Abb. 27: Kalibriertes Chronogramm von 92 ITS-Sequenzen auf Basis einer ML-Baumtopologie mit vollständiger Benennung der Knoten. 152

Tabellenverzeichnis
132
Anhang 2 Tabellenverzeichnis Tab. 1: Übersicht der im Auswahlverfahren für die phylogenetische Analyse der Suaedoideae getesteten DNA-Regionen und Primer sowie ihrer Herkunft. 30
Tab. 2: Tabellarische Übersicht der Ergebnisse des Auswahlverfahrens von DNA-Regionen für die phylogenetische Analyse der Suaedoideae. 31
Tab. 3: Reaktionsbedingungen der ITS touchdown PCR. 33
Tab. 4: Reaktionsbedingungen der Chloroplasten PCR. 33
Tab. 5: Zusammensetzung eines Reaktionsansatzes für die DNA-Sequenzierung mit dem Thermo Sequenase™ Primer Cycle Seqeuencing Kit. 34
Tab. 6: Sequenzmerkmale und Alignmentcharakteristika für einen vergleichbaren Datensatz aus 74 Sequenzen der Suaedoideae, Salicornioideae und Bassia hyssopifolia. 54
Tab. 7: Übersicht über die Alignmentparameter und Ergebnisse der Maximum Parsimony Analyse der ausgewählten Chloroplasten-Regionen. 68
Tab. 8: Übersicht der Parameter für die ML-Analyse und Altersdatierung. 82
Tab. 9: Kalibrierungspunkte und Altersschätzungen der wichtigsten Diversifizierungsereignisse innerhalb der Suaedoideae und benachbarter Gruppen. 84
Tab. 10: Synthese monophyletischer Einheiten der molekularen Phylogenien mit systematischen Einheiten für die Unterfamilie Suaedoideae. 115
Tab. 11: Materialtabelle. 135
Tab. 12: Übersicht der Altersschätzungen mittels PL für atpB-rbcL-Baum und ITS-Baum 154

Abkürzungsverzeichnis
133
Anhang 3 Abkürzungsverzeichnis:
A Adenin
bp Basenpaare
C Cytosin
CI Consistency Index (Konsistenzindex)
CTP Cytosin-5’-triphosphat
cp Chloroplast
CTAB Cetyltrimethylammoniumbromid
ddNTP 2’,3’-Didesoxynukleosidtriphosphat
DNA Desoxyribonukleinsäure
dNTP 2’-Didesoxynukleosidtriphosphat
G Guanin
GTP Guanin-5’-triphosphat
ITS: Internal transcribed spacer
ML Maximum Likelihood
MP Maximum Parsimony
NCBI National Center for Biotechnology Information
PL Penalized Likelihood
RI Retentionsindex
rpm Umdrehungen pro Minute
s.l. sensu lato
s.str. sensu stricto
T Thymin
U Unit (Einheit)
UV Ultraviolett
µl Mikroliter
µMol Mikromolar

Chemikalien, Lösungen, Reaktionskits
134
Anhang 4 Verzeichnis der Chemikalien, Lösungen und Reaktionskits
2-Propanol (Isopropanol): 100% Stammkonzentration
Agarose NEEO Ultra-Qualität: (Carl Roth)
Bromphenol-Glycerinpuffer f. d. Elektrophorese: 40% Glycerin, 0.1% Bromphenolblau, 0.1%
Xylencyanol in 0.5x TBE- bzw. 1x TAE-Puffer
Borsäure: pulverförmig (Carl Roth)
Cetyltrimethylammoniumbromid-Puffer (CTAB): 2 % (w/v) CTAB; 1,4 M NaCl; 0,1 M Tris-HCl, pH 8,0;
20 mM EDTA; 1 % (w/v) Polyvinylpyrrolidon (PVP 40); 0,2 % (v/v) β-Mercaptoethanol (MET)
Chloroform/Isoamylalkohol: Mischung im Verhältnis 24:1
dATP, dCTP, dGTP, dTTP (als Na+-Salz): 100mM Stammlösung (Carl Roth)
DMSO: 100% Stammlösung (sigma)
DNA-Auftragspuffer: 40 % Glycerin (v/v); 0,1 % Bromphenolblau (w/v); 0,1 % Xylencyanol (w/v)
DNA Größenstandard: 100 bp-DNA-Marker (invitrogen)
Ethanol 96% f.d. Analyse; 70%
Ethidiumbromid: Arbeitslösung 1µg/ml in 0,5 x TBE-Puffer; (Carl Roth)
Formamid Loading Dye
Harnstoff: kristallin (Carl Roth)
Low DNA Mass Ladder: Größen- und Mengenmarker (Invitrogen)
Magnesiumchlorid (MgCl2): Arbeitslösung 50mM
N2: flüssig, (Linde AG)
NaCl: Arbeitslösung 5M
Polyacrylamid-Gel (PAA) 6% Fertiglösung (Sequagel XR):
PCR-Puffer: 10x (Qiagen)
Qiagen DNeasy® Plant Mini Kit (Qiagen)
Seakem LE Agarose: (Biozym)
TBE-Puffer 0,5x: 45 mM Tris-Borat; 1 mM EDTA
TE-Puffer: 10 mM Tris-HCl, pH 8,0; 1 mM EDTA
Taq DNA Polymerase, Recombinant: 5 units/µl (invitrogen)
Thermo Sequenase™ Primer Cycle Sequencing Kit (Amersham Pharmacia)
Tris HCl
135
Anhang 5 Materialtabelle Tab 11: Materialtabelle. Sequenzen mit * (Genbank-Accessionsnummer) wurden im Rahmen dieser Arbeit nicht selbst generiert SK=Sektionszugehörigkeit entsprechend Kap. 4.6: Ax: Alexandra, Bw: Borszczowia, Bz: Brezia, Gl: Glauca, Py: Physophora, Sc: Schanginia, So: Schoberia, Sl: Salsina, Su: Suaeda
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Suaedoideae
Ax Alexandra lehmannii Bunge
Diomina 5171a (KAS) E Kasachstan: Dzhambul, Czu-Fluss 1381 FJ449756 DQ499432* FJ449821
Bienertia cycloptera Bunge
Akhani 16/11/2000 (KAS) Iran: Kavir Nationalpark 1027 AY181808 AY181935 AY181873
Bienertia sinuspersici Akhani
Akhani 17433 (WS) Iran: Khuzestan, Bandare Mahshahr DQ499373* DQ499434* DQ499349*
Bw Borszczowia aralocaspica Bunge
Ogar 10.2000 (KAS) E Kasachstan, Uigur distr. 1028 AY181807 AY181934 AY181872
So Suaeda acuminata (C. A. Mey.) Moq.
Lomonosova 053a_ (NS, KAS)
Kasachstan: Provinz Almaty, Ushtobe 1175 FJ449757 AY181912 AY181848
Sl Suaeda aegyptiaca (Hasselq.) Zohary
Freitag 30.120 (KAS) Jordanien: Azraq Oase 1138 AY181788 AY181917 AY181853
Sl Suaeda altissima Pall.
Freitag 28.150 (KAS) Kasachstan: Prov. Atyrau, Guryev 1017 AY181785 AY181914 AY181850
Bz Suaeda arbusculoides L. S. Sm.
Jacobs 5873 (NSW) Australien: Queensland, Gladstone 1384 no data - AY181827
Bz Suaeda arctica Jurtzev & V. V. Petrovskii Korobkov & Petrovskyi 04/08/1968 (KAS)
Russland: N-Sibirien, Tschuktschan 1241 - - FJ449785
Sl Suaeda arcuata Bunge
Löffler 1/2001 (W) Iran: Prov. Fars, Maharlu-See 1383 AY181789 AY181918 AY181854
Sl Suaeda argentinensis Soriano
Fortunato 4303 (NY) Argentinien - - FJ449786
Sl Suaeda articulata Aellen
Okaukungo 23/04/1968 (W) Namibia: Etoscha 1378 AY181795 AY181924 AY181860
Sl Suaeda asphaltica Boiss.
Danin 2000 (HUJ) Jordanien: Totes Meer 1092 AY181786 AY181915 AY181851

136
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Bz Suaeda australis Moq.
21368 (KAS) W Australien 1251 - - FJ449787
Bz Suaeda australis Moq.
Schmalz 55 (MJG) Australien: New South Wales, Karuah 1144 AY181766 AY181891 AY181826
Bz Suaeda australis Moq. s.l. `Korea`
Lee SK08 (KNU) Südkorea; ex Lee et al. (2007) - - DQ786335
Bz Suaeda caespitosa Wolley-Dod Mucina 28/12/2003 (KAS) Südafrika: W Cape Provinz, Agulhas Plain
1587 FJ449758 - FJ449817
Bz Suaeda calceoliformis (Hook.) Moq.
Schenk 01/10/1997 (RSA) USA - - FJ449788
So Suaeda carnosissima Post
Freitag 30.159 (KAS) Syrien: Palmyra-Oase 1137 AY181783 AY181910 AY181846
Sl Suaeda conferta (Small) I. M. Johnst.
Schenk 06/10/1994 (RSA) USA - - FJ449789
Bz Suaeda corniculata (C. A. Mey.) Bunge
Artemov1999 (NS) Russland: Altaiski Krai 1553 - - FJ449790
Bz Suaeda corniculata (C. A. Mey.) Bunge Lomonosova 051a (NS, KAS) Kasachstan: Provinz Almaty, Distr. Taldy-Kurgan
1026 FJ449759 FJ449833 FJ449820
Bz Suaeda corniculata (C. A. Mey.) Bunge
Lomonosova 71a (NS, KAS) Kasachstan: Provinz Almaty, Ushtobe 1062 AY181779 AY181904 AY181840
Bz Suaeda corniculata (C. A. Mey.) Bunge
Lomonosova 79 (NS, KAS) Russland: Oblast Novosibirsk, Karasuk 1052 AY181780 AY181905 AY181841
Bz Suaeda corniculata (C. A. Mey.) Bunge
Lomonosova 80 (NS, KAS) Russland: Distr. Karasuk (S-Sibirien) 1129 AY181781 AY181906 AY181842
Bz Suaeda crassifolia Pall.
Freitag 30.134 (KAS) Usbekistan: Aidar-Kul-See NW Jizzakh 1108 AY181760 AY181885 AY181820
So Suaeda cucullata Aellen
Freitag 28.729 (KAS) Türkei: Provinz Eskişehir, Polatle 1056 FJ449760 AY181909 AY181845
Sl Suaeda dendroides (C. A. Mey.) Moq.
Freitag 30.127 (KAS) Usbekistan: Provinz Sirdaryo 1030 AY181791 AY181920 AY181856
137
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Sl Suaeda divaricata Moq. Torrico-Peca 101 (LPB, KAS) Bolivien: Depto. Potosi, Prov. Nor Chichas
1219 AY181797 AY181926 AY181863
So Suaeda eltonica Iljin
Freitag 28.242 (KAS) Russland: Oblast Wolgograd, Elton-See 1380 AY181784 AY181911 AY181847
Bz Suaeda esteroa Ferren & S. A. Whitmore
Ferren 2836 (RSA) Mexico - - FJ449791
Sl Suaeda foliosa Moq. R. de Michel 2982 (LPB, KAS) Bolivien: Depto. Oruro, Prov. Eduardo Avaroa
1217 AY181796 AY181925 AY181862
Sl Suaeda fruticosa Forssk. ex J. F. Gmel.
Freitag 21.500 (KAS) Pakistan: Punjab 1002 AY181792 AY181921 AY181857
Sl Suaeda fruticosa Forssk. ex J. F. Gmel.
Freitag 31.138 (KAS) Jordanien: Totes Meer 1139 AY181793 AY181922 AY181858
Sl Suaeda fruticosa s.l. Forssk. ex J. F. Gmel.
Fosberg & Jayasuria 52806 (RSA)
Sri Lanka - - FJ449792
Gl Suaeda glauca Bunge Nechayev & Pavlova 04/10/2003 (KAS)
Russland: Primorski Krai, Küste bei Shkotova
1586 FJ449761 FJ449835 FJ449825
Bz Suaeda heterophylla Bunge
Freitag 30.132 (KAS) Usbekistan: Aidar-Kul 1074 AY181774 AY181899 AY181835
Bz Suaeda heterophylla Bunge Lomonosova 138 (KAS) Kasachstan: Prov. Ost-Kasachstan, Karaberik Gebirge
1309 - - FJ449793
Bz Suaeda heterophylla Bunge Lomonosova 51 (NS, KAS) Kasachstan: Provinz Almaty, Distr. Taldy-Kurgan
1037 AY181776 AY181901 AY181837
Bz Suaeda heteroptera Kitag. Schütze 2003/243 (KAS) Russland: Zabaykalsky Krai, Krasnokamensk
1540 FJ449762 FJ449829 FJ449815
Py Suaeda ifniensis Caball. ex Maire
Podlech 48.924 (KAS) Marokko 1214 AY181801 AY181929 AY181867
Py Suaeda ifniensis Caball. ex Maire
Reys-Betancourt 41074 (TFC) Spanien: Kanarische Inseln, Lanzarote 1160 AY181800 AY181928 AY181866
Bz Suaeda japonica Makino
Lee SK01 (KNU) Südkorea; ex Lee et al. (2007) - - DQ786340*
Bz Suaeda kulundensis Lomon. & Freitag Lomonosova 135 (NS, KAS) Russland: Primorski Krai, Küste bei Shkotova
1554 FJ449763 - FJ449794

138
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Bz Suaeda kulundensis Lomon. & Freitag Lomonosova 195 (KAS) Kasachstan: Prov. Ost-Kasachstan, Distr. Tarbatagay
1479 FJ449764 - FJ449795
Bz Suaeda kulundensis Lomon. & Freitag
Lomonosova 51a (NS, KAS) Kasachstan: Provinz Almaty, Distr. Taldy-Kurgan
1170 AY181764 AY181889 AY181824
Bz Suaeda linearis (Elliott) Moq.
Schenk 06/08/1994 (RSA) USA - - FJ449796
Sc Suaeda linifolia Pall. Freitag 28.092 (KAS) Kasachstan: Prov. West-Kasachstan, Chelkar-See bei Saryumir
1233 AY181805 AY181932 AY181870
Bz Suaeda malacosperma Hara
Lee SK43 (KNU) Südkorea; ex Lee et al. (2007) - - DQ786339*
Bz Suaeda maritima (L.) Dumort. Schütze 10/09/2001 (KAS) Deutschland: Sachsen-Anhalt, Hecklingen
1084 AY181758 AY181883 AY181818
Bz Suaeda maritima (L.) Dumort. Schütze ER167 (KAS) Frankreich: Region Nord-Pas-de-Calais, Etaples
1200 - - DQ899059
Bz Suaeda maritima s.l. (L.) Dumort
Lee SK10 (KNU) Südkorea; ex Lee et al. (2007) - - DQ786336*
Bz Suaeda maritima s.l. (L.) Dumort
Westberg 25/01/SP (KAS) NW Spanien: Illa de Arousa 1360 - - DQ899064
Bz Suaeda maritima var. perennans Maire Reys-Betancourt (TFC 41071) (KAS)
Spanien: Kanarische Inseln, Lanzarote 1234 AY181768 AY181893 AY181829
Bz Suaeda mexicana Standl.
Henrickson 20407 (RSA) Mexico - - FJ449797
Sl Suaeda micromeris Brenan 4007 (Herb. Mogadishu) (MOG)
Somalia 1240 - - FJ449798
Sl Suaeda microphylla Pall.
Freitag 28.686 (KAS) Türkei: Prov. Kars, Tuzluca 1007 AY181790 AY181919 AY181855
So Suaeda microsperma Fenzl Lomonosova 045a (NS, KAS) Kasachstan: Prov. Ost-Kasachstan, Aktogay am Balchaschsee
1211 FJ449765 AY181913 AY181849
Sl Suaeda mollis Delile
TFC 41066 (TFC, KAS) Spanien: Kanarische Inseln, Lanzarote 1236 - - FJ449799
Sl Suaeda monodiana Maire Bornkamm 28/09/1986 (B, KAS)
Ägypten: Qattara depression 1229 - - AY181861

139
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Sl Suaeda monoica Forssk. ex J. F. Gmel.
Léonard 7476 (KAS) Ägypten: Sinai, Taba 1382 AY181794 AY181923 AY181859
Sl Suaeda moquinii Greene
Ferren 3276 (RSA) Mexico - - FJ449800
Sl Suaeda nigra J. F. Macbr.
Ickert-Bond 1122 (ASU, KAS) USA: Arizona, Marcopa Co. 1215 AY181798 - AY181864
Bz Suaeda occidentalis S. Watson
Schenk 03/10/1997 (RSA) USA - - FJ449801
Bz Suaeda olufsenii Paulsen
Lomonosova 236 (NS, KAS) Russland: Republik Altai, Tebeler 1459 FJ449766 FJ449831 FJ449818
Py Suaeda palaestina Eig. & Zohary
Freitag 30.165 (KAS) Jordanien: Totes Meer 1140 AY181799 AY181927 AY181865
Bz Suaeda pannonica Beck
Freitag 27.156 (KAS) Österreich: Burgenland, Neusiedler See 1375 AY181778 AY181903 AY181839
Sc Suaeda paradoxa Bunge
Freitag 30.028 (KAS) Usbekistan: Provinz Sirdaryo 1004 AY181806 AY181933 AY181871
Bz Suaeda patagonica Speg. R. de Michel 2862 (LPB, KAS) Bolivien: Depto. Oruro, Prov. Eduardo Avaroa
1216 AY181782 AY181907 AY181843
Sl Suaeda paulayana Vierh.
Kilian YP3782 (KAS) Yemen Sokotra: Shuab 1493 FJ449767 FJ449834 FJ449823
Py Suaeda physophora Pall. Freitag 28.041 (KAS) Kasachstan: Prov. West-Kasachstan, Chelkar-See bei Saryumir
1163 AY181802 - -
Py Suaeda physophora Pall.
Morefield 5136 (RSA) China - - FJ449824
Bz Suaeda prostrata Pall.
Freitag 27.155 (KAS) Österreich: Burgenland, Neusiedler See 1161 AY181773 AY181898 AY181834
Bz Suaeda prostrata Pall.
Freitag 28.305 (KAS) Russland: Oblast Wolgograd, Pallasovka 1379 AY181772 AY181897 AY181833
Bz Suaeda prostrata s. l. Pall.
Freitag 28.793 (KAS) Türkei: Prov. Aksaray, Sultanhani 1109 AY181769 AY181894 AY181830
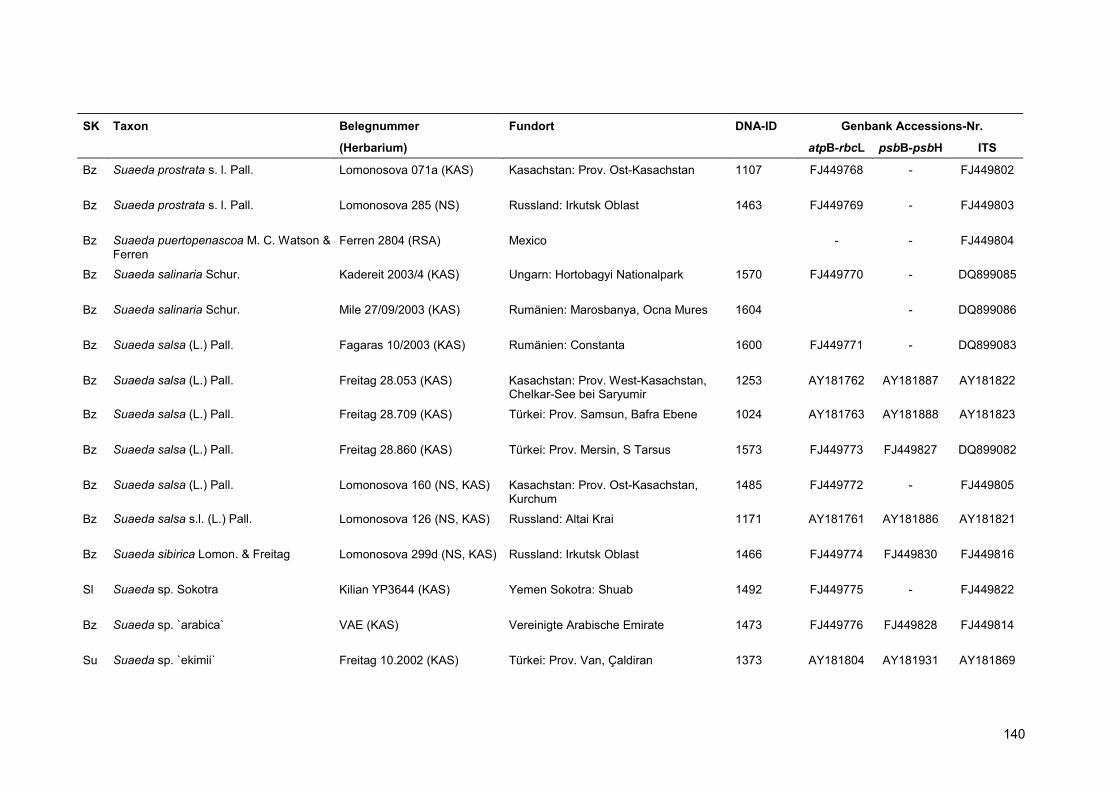
140
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Bz Suaeda prostrata s. l. Pall.
Lomonosova 071a (KAS) Kasachstan: Prov. Ost-Kasachstan 1107 FJ449768 - FJ449802
Bz Suaeda prostrata s. l. Pall.
Lomonosova 285 (NS) Russland: Irkutsk Oblast 1463 FJ449769 - FJ449803
Bz Suaeda puertopenascoa M. C. Watson & Ferren
Ferren 2804 (RSA) Mexico - - FJ449804
Bz Suaeda salinaria Schur.
Kadereit 2003/4 (KAS) Ungarn: Hortobagyi Nationalpark 1570 FJ449770 - DQ899085
Bz Suaeda salinaria Schur.
Mile 27/09/2003 (KAS) Rumänien: Marosbanya, Ocna Mures 1604 - DQ899086
Bz Suaeda salsa (L.) Pall.
Fagaras 10/2003 (KAS) Rumänien: Constanta 1600 FJ449771 - DQ899083
Bz Suaeda salsa (L.) Pall. Freitag 28.053 (KAS) Kasachstan: Prov. West-Kasachstan, Chelkar-See bei Saryumir
1253 AY181762 AY181887 AY181822
Bz Suaeda salsa (L.) Pall.
Freitag 28.709 (KAS) Türkei: Prov. Samsun, Bafra Ebene 1024 AY181763 AY181888 AY181823
Bz Suaeda salsa (L.) Pall.
Freitag 28.860 (KAS) Türkei: Prov. Mersin, S Tarsus 1573 FJ449773 FJ449827 DQ899082
Bz Suaeda salsa (L.) Pall. Lomonosova 160 (NS, KAS) Kasachstan: Prov. Ost-Kasachstan, Kurchum
1485 FJ449772 - FJ449805
Bz Suaeda salsa s.l. (L.) Pall.
Lomonosova 126 (NS, KAS) Russland: Altai Krai 1171 AY181761 AY181886 AY181821
Bz Suaeda sibirica Lomon. & Freitag
Lomonosova 299d (NS, KAS) Russland: Irkutsk Oblast 1466 FJ449774 FJ449830 FJ449816
Sl Suaeda sp. Sokotra
Kilian YP3644 (KAS) Yemen Sokotra: Shuab 1492 FJ449775 - FJ449822
Bz Suaeda sp. `arabica`
VAE (KAS) Vereinigte Arabische Emirate 1473 FJ449776 FJ449828 FJ449814
Su Suaeda sp. `ekimii`
Freitag 10.2002 (KAS) Türkei: Prov. Van, Çaldiran 1373 AY181804 AY181931 AY181869
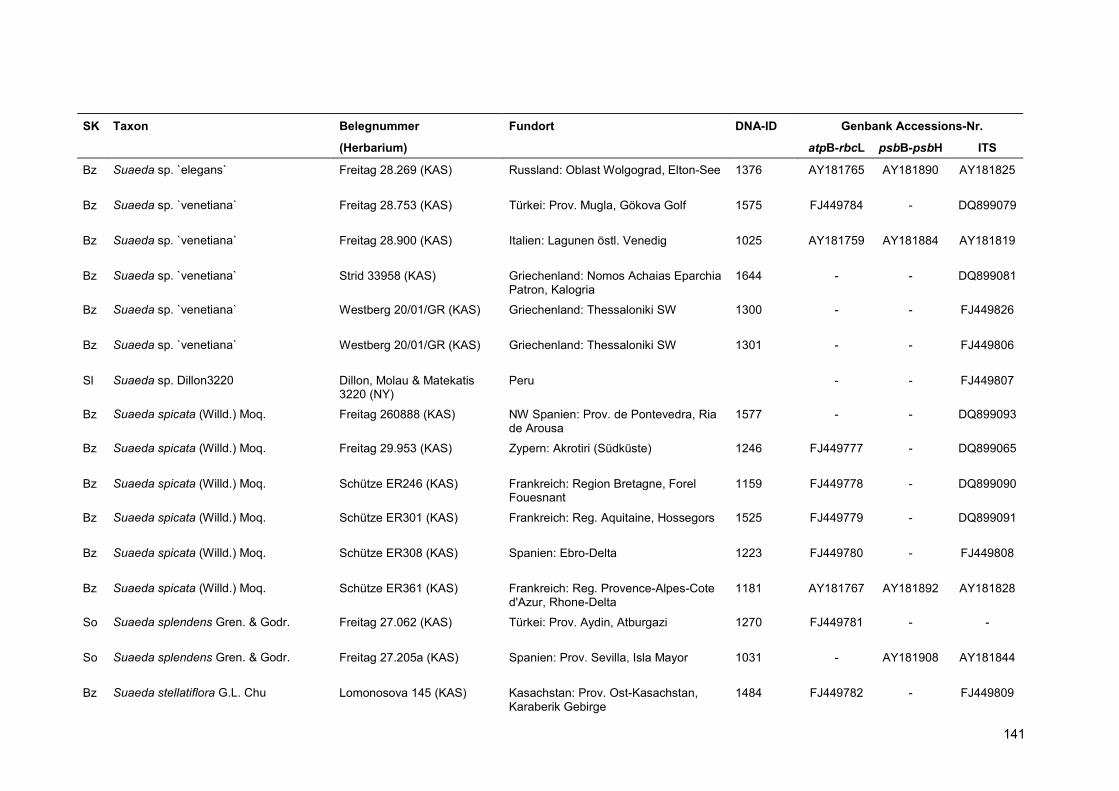
141
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Bz Suaeda sp. `elegans`
Freitag 28.269 (KAS) Russland: Oblast Wolgograd, Elton-See 1376 AY181765 AY181890 AY181825
Bz Suaeda sp. `venetiana`
Freitag 28.753 (KAS) Türkei: Prov. Mugla, Gökova Golf 1575 FJ449784 - DQ899079
Bz Suaeda sp. `venetiana`
Freitag 28.900 (KAS) Italien: Lagunen östl. Venedig 1025 AY181759 AY181884 AY181819
Bz Suaeda sp. `venetiana` Strid 33958 (KAS) Griechenland: Nomos Achaias Eparchia Patron, Kalogria
1644 - - DQ899081
Bz Suaeda sp. `venetiana`
Westberg 20/01/GR (KAS) Griechenland: Thessaloniki SW 1300 - - FJ449826
Bz Suaeda sp. `venetiana`
Westberg 20/01/GR (KAS) Griechenland: Thessaloniki SW 1301 - - FJ449806
Sl Suaeda sp. Dillon3220 Dillon, Molau & Matekatis 3220 (NY)
Peru - - FJ449807
Bz Suaeda spicata (Willd.) Moq. Freitag 260888 (KAS) NW Spanien: Prov. de Pontevedra, Ria de Arousa
1577 - - DQ899093
Bz Suaeda spicata (Willd.) Moq.
Freitag 29.953 (KAS) Zypern: Akrotiri (Südküste) 1246 FJ449777 - DQ899065
Bz Suaeda spicata (Willd.) Moq. Schütze ER246 (KAS) Frankreich: Region Bretagne, Forel Fouesnant
1159 FJ449778 - DQ899090
Bz Suaeda spicata (Willd.) Moq.
Schütze ER301 (KAS) Frankreich: Reg. Aquitaine, Hossegors 1525 FJ449779 - DQ899091
Bz Suaeda spicata (Willd.) Moq.
Schütze ER308 (KAS) Spanien: Ebro-Delta 1223 FJ449780 - FJ449808
Bz Suaeda spicata (Willd.) Moq. Schütze ER361 (KAS) Frankreich: Reg. Provence-Alpes-Cote d'Azur, Rhone-Delta
1181 AY181767 AY181892 AY181828
So Suaeda splendens Gren. & Godr.
Freitag 27.062 (KAS) Türkei: Prov. Aydin, Atburgazi 1270 FJ449781 - -
So Suaeda splendens Gren. & Godr.
Freitag 27.205a (KAS) Spanien: Prov. Sevilla, Isla Mayor 1031 - AY181908 AY181844
Bz Suaeda stellatiflora G.L. Chu Lomonosova 145 (KAS) Kasachstan: Prov. Ost-Kasachstan, Karaberik Gebirge
1484 FJ449782 - FJ449809
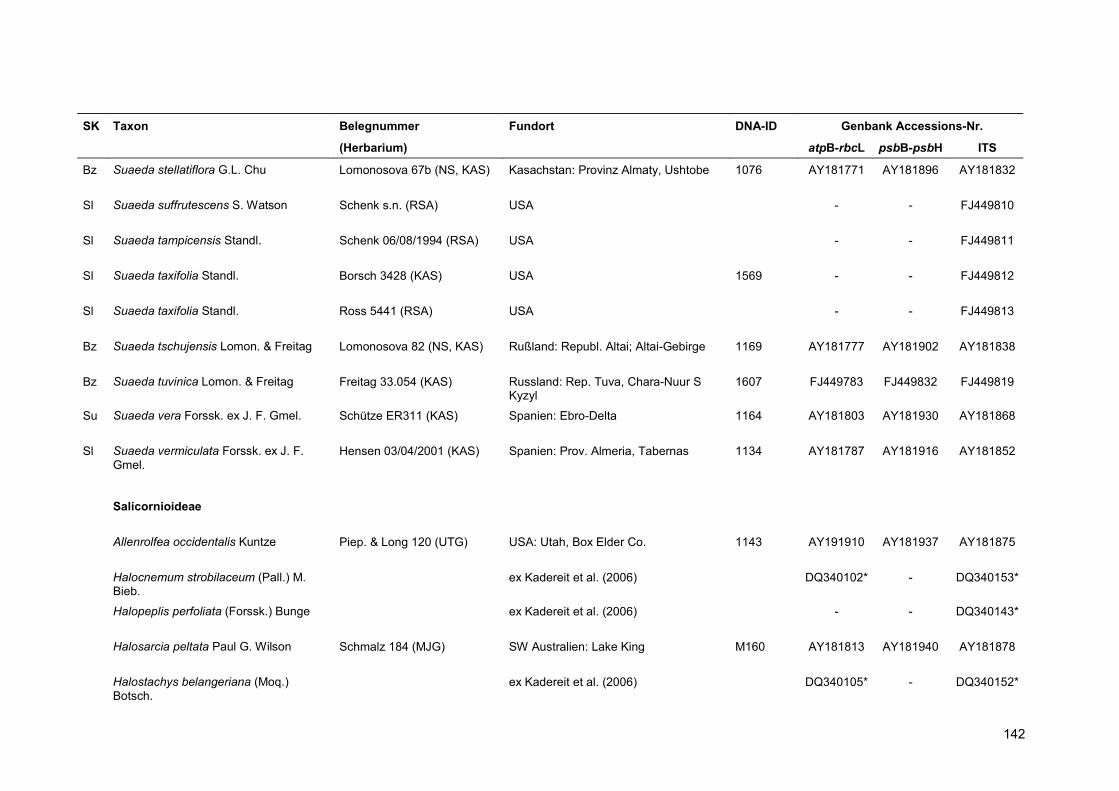
142
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Bz Suaeda stellatiflora G.L. Chu
Lomonosova 67b (NS, KAS) Kasachstan: Provinz Almaty, Ushtobe 1076 AY181771 AY181896 AY181832
Sl Suaeda suffrutescens S. Watson
Schenk s.n. (RSA) USA - - FJ449810
Sl Suaeda tampicensis Standl.
Schenk 06/08/1994 (RSA) USA - - FJ449811
Sl Suaeda taxifolia Standl.
Borsch 3428 (KAS) USA 1569 - - FJ449812
Sl Suaeda taxifolia Standl.
Ross 5441 (RSA) USA - - FJ449813
Bz Suaeda tschujensis Lomon. & Freitag
Lomonosova 82 (NS, KAS) Rußland: Republ. Altai; Altai-Gebirge 1169 AY181777 AY181902 AY181838
Bz Suaeda tuvinica Lomon. & Freitag Freitag 33.054 (KAS) Russland: Rep. Tuva, Chara-Nuur S Kyzyl
1607 FJ449783 FJ449832 FJ449819
Su Suaeda vera Forssk. ex J. F. Gmel.
Schütze ER311 (KAS) Spanien: Ebro-Delta 1164 AY181803 AY181930 AY181868
Sl Suaeda vermiculata Forssk. ex J. F. Gmel.
Hensen 03/04/2001 (KAS) Spanien: Prov. Almeria, Tabernas 1134 AY181787 AY181916 AY181852
Salicornioideae
Allenrolfea occidentalis Kuntze
Piep. & Long 120 (UTG) USA: Utah, Box Elder Co. 1143 AY191910 AY181937 AY181875
Halocnemum strobilaceum (Pall.) M. Bieb.
ex Kadereit et al. (2006) DQ340102* - DQ340153*
Halopeplis perfoliata (Forssk.) Bunge
ex Kadereit et al. (2006) - - DQ340143*
Halosarcia peltata Paul G. Wilson
Schmalz 184 (MJG) SW Australien: Lake King M160 AY181813 AY181940 AY181878
Halostachys belangeriana (Moq.) Botsch.
ex Kadereit et al. (2006) DQ340105* - DQ340152*
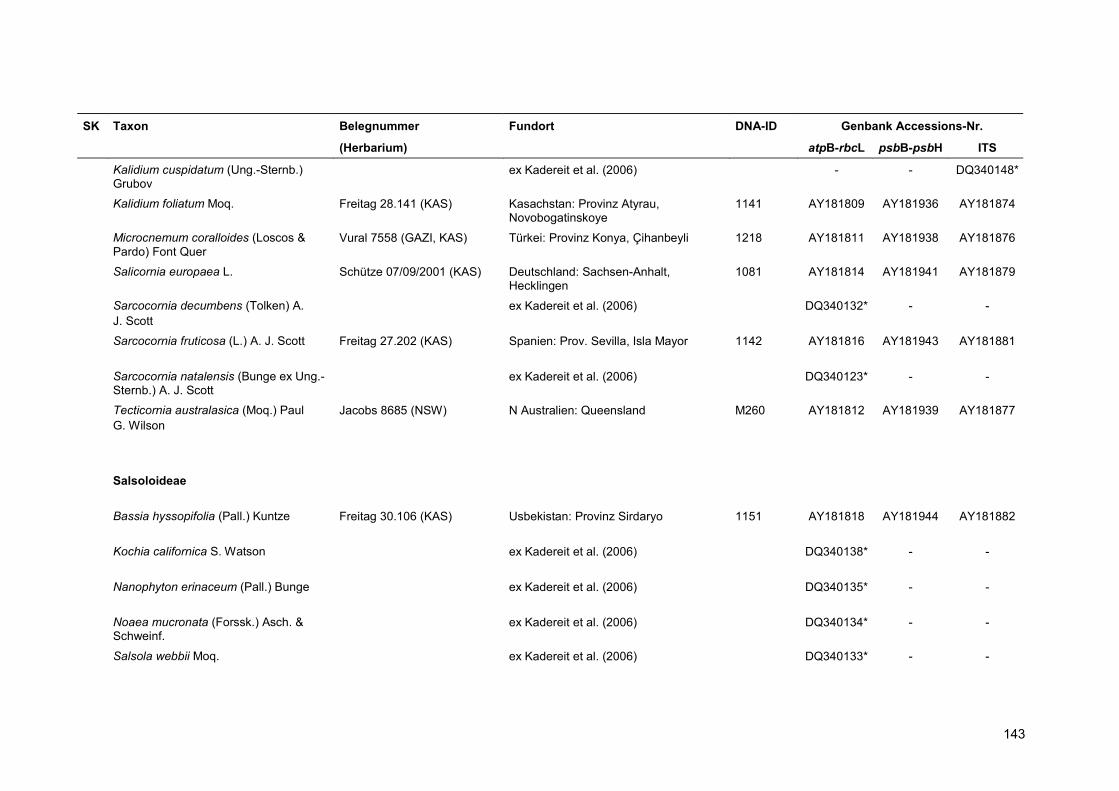
143
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Kalidium cuspidatum (Ung.-Sternb.) Grubov
ex Kadereit et al. (2006) - - DQ340148*
Kalidium foliatum Moq. Freitag 28.141 (KAS) Kasachstan: Provinz Atyrau, Novobogatinskoye
1141 AY181809 AY181936 AY181874
Microcnemum coralloides (Loscos & Pardo) Font Quer
Vural 7558 (GAZI, KAS) Türkei: Provinz Konya, Çihanbeyli 1218 AY181811 AY181938 AY181876
Salicornia europaea L. Schütze 07/09/2001 (KAS) Deutschland: Sachsen-Anhalt, Hecklingen
1081 AY181814 AY181941 AY181879
Sarcocornia decumbens (Tolken) A. J. Scott
ex Kadereit et al. (2006) DQ340132* - -
Sarcocornia fruticosa (L.) A. J. Scott
Freitag 27.202 (KAS) Spanien: Prov. Sevilla, Isla Mayor 1142 AY181816 AY181943 AY181881
Sarcocornia natalensis (Bunge ex Ung.-Sternb.) A. J. Scott
ex Kadereit et al. (2006) DQ340123* - -
Tecticornia australasica (Moq.) Paul G. Wilson
Jacobs 8685 (NSW) N Australien: Queensland M260 AY181812 AY181939 AY181877
Salsoloideae
Bassia hyssopifolia (Pall.) Kuntze
Freitag 30.106 (KAS) Usbekistan: Provinz Sirdaryo 1151 AY181818 AY181944 AY181882
Kochia californica S. Watson
ex Kadereit et al. (2006) DQ340138* - -
Nanophyton erinaceum (Pall.) Bunge
ex Kadereit et al. (2006) DQ340135* - -
Noaea mucronata (Forssk.) Asch. & Schweinf.
ex Kadereit et al. (2006) DQ340134* - -
Salsola webbii Moq.
ex Kadereit et al. (2006) DQ340133* - -
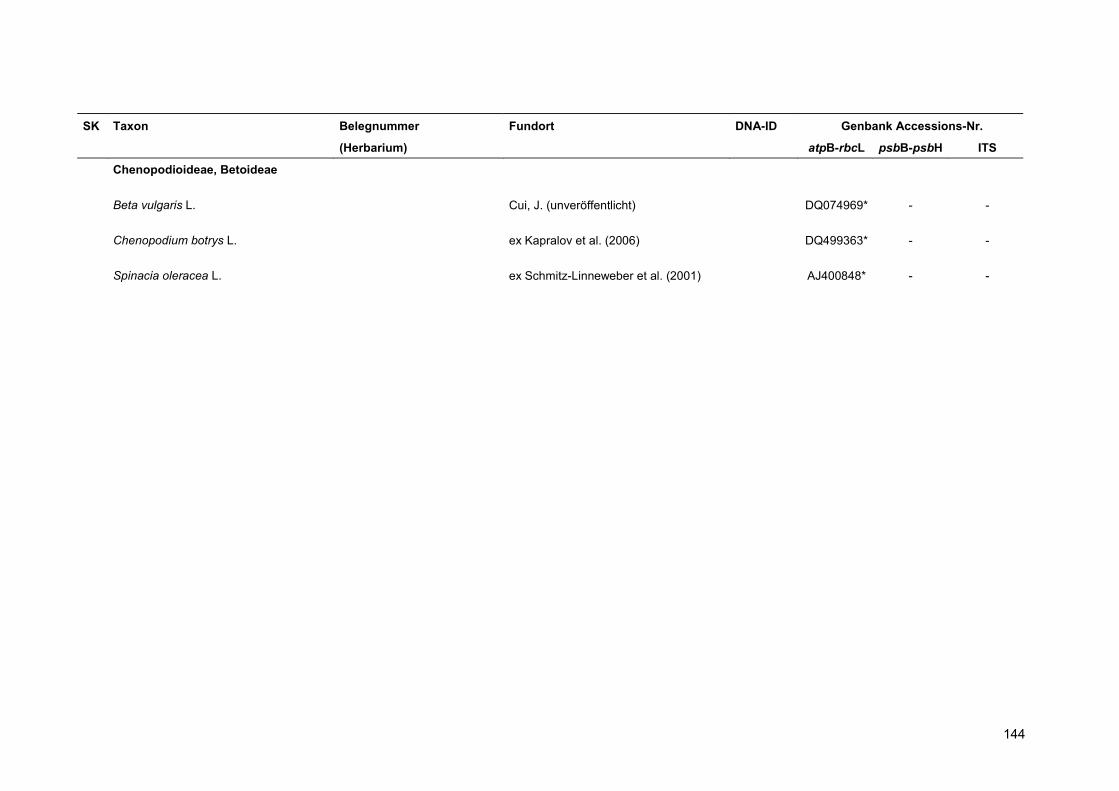
144
SK Taxon Belegnummer Fundort DNA-ID Genbank Accessions-Nr.
(Herbarium) atpB-rbcL psbB-psbH ITS
Chenopodioideae, Betoideae
Beta vulgaris L.
Cui, J. (unveröffentlicht) DQ074969* - -
Chenopodium botrys L.
ex Kapralov et al. (2006) DQ499363* - -
Spinacia oleracea L.
ex Schmitz-Linneweber et al. (2001) AJ400848* - -
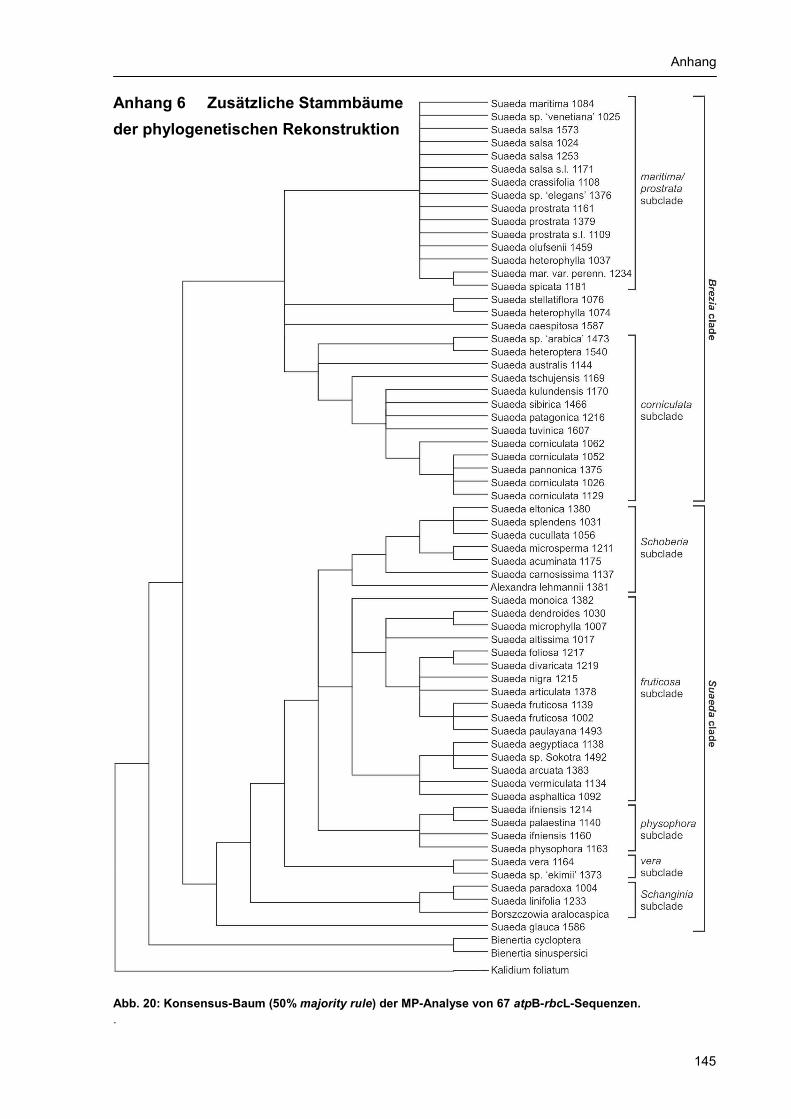
Anhang
145
Abb. 20: Konsensus-Baum (50% majority rule) der MP-Analyse von 67 atpB-rbcL-Sequenzen. .
Anhang 6 Zusätzliche Stammbäume der phylogenetischen Rekonstruktion
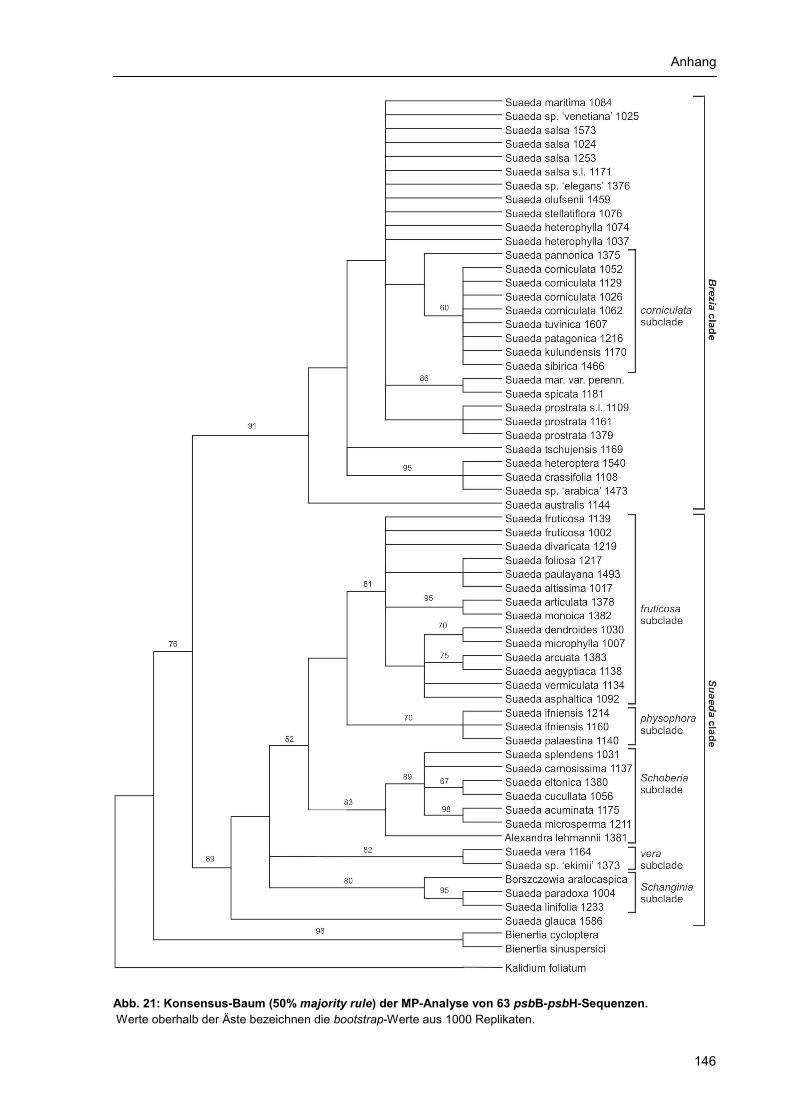
Anhang
146
Abb. 21: Konsensus-Baum (50% majority rule) der MP-Analyse von 63 psbB-psbH-Sequenzen. Werte oberhalb der Äste bezeichnen die bootstrap-Werte aus 1000 Replikaten.
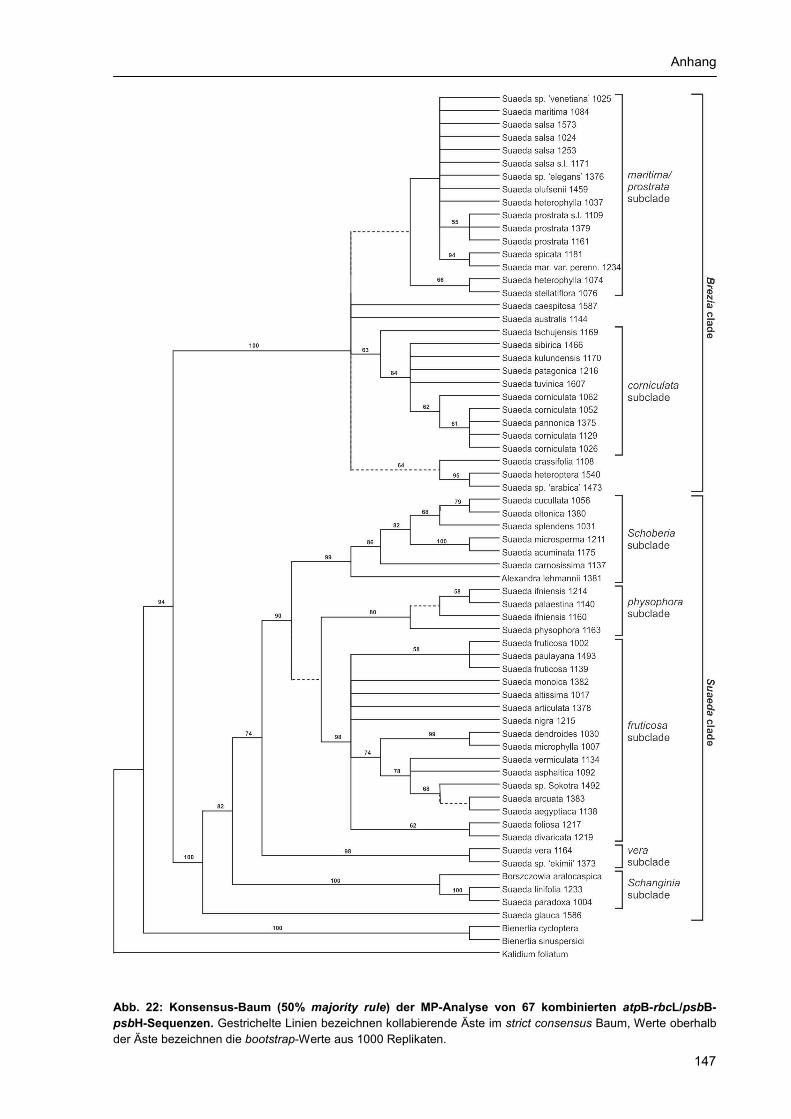
Anhang
147
Abb. 22: Konsensus-Baum (50% majority rule) der MP-Analyse von 67 kombinierten atpB-rbcL/psbB-psbH-Sequenzen. Gestrichelte Linien bezeichnen kollabierende Äste im strict consensus Baum, Werte oberhalb der Äste bezeichnen die bootstrap-Werte aus 1000 Replikaten.
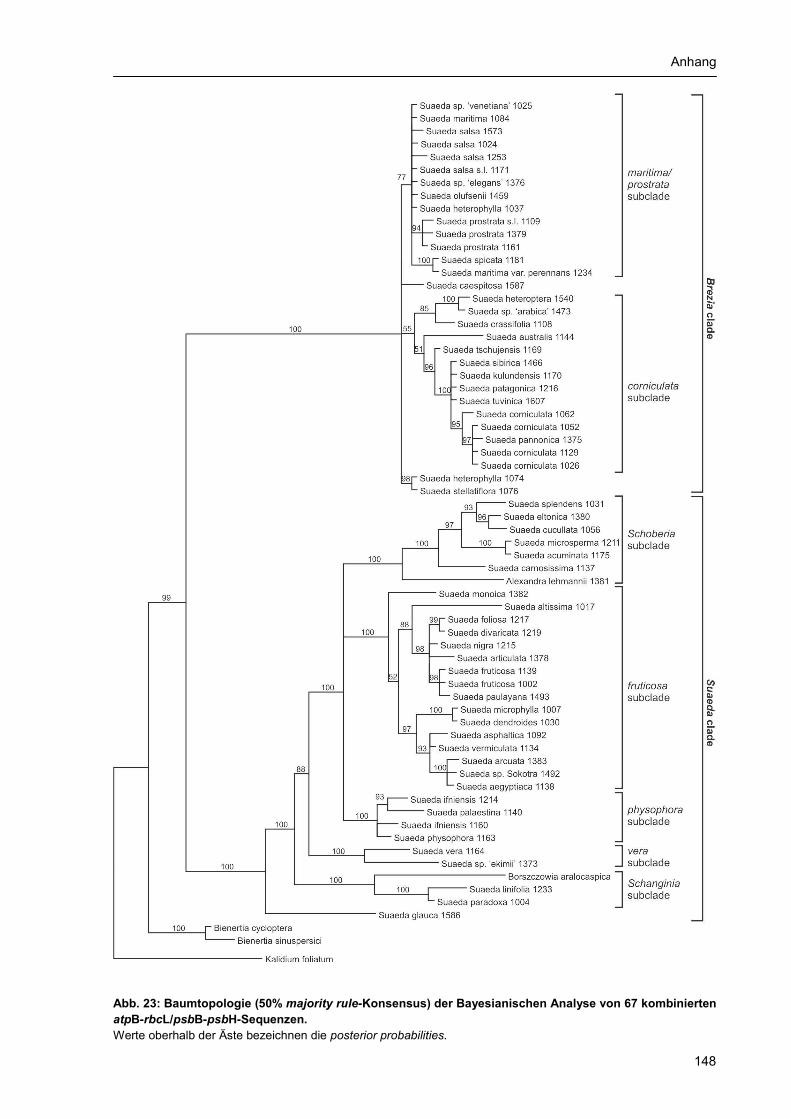
Anhang
148
Abb. 23: Baumtopologie (50% majority rule-Konsensus) der Bayesianischen Analyse von 67 kombinierten atpB-rbcL/psbB-psbH-Sequenzen. Werte oberhalb der Äste bezeichnen die posterior probabilities.

Anhang
149
Abb. 24: Konsensus-Baum (50% majority rule) der MP-Analyse von 93 ITS-Sequenzen. Gestrichelte Linien bezeichnen kollabierende Äste im strict consensus-Baum, Werte oberhalb der Äste bezeichnen die bootstrap-Werte aus 1000 Replikaten.
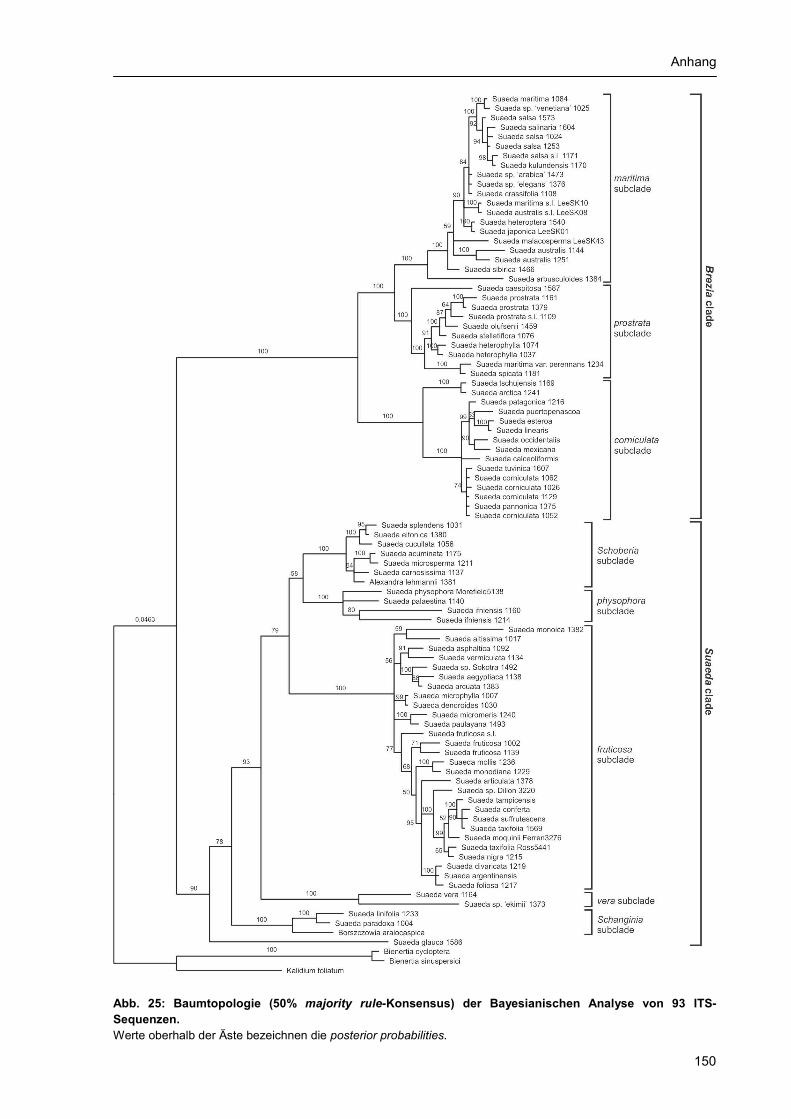
Anhang
150
Abb. 25: Baumtopologie (50% majority rule-Konsensus) der Bayesianischen Analyse von 93 ITS-Sequenzen. Werte oberhalb der Äste bezeichnen die posterior probabilities.
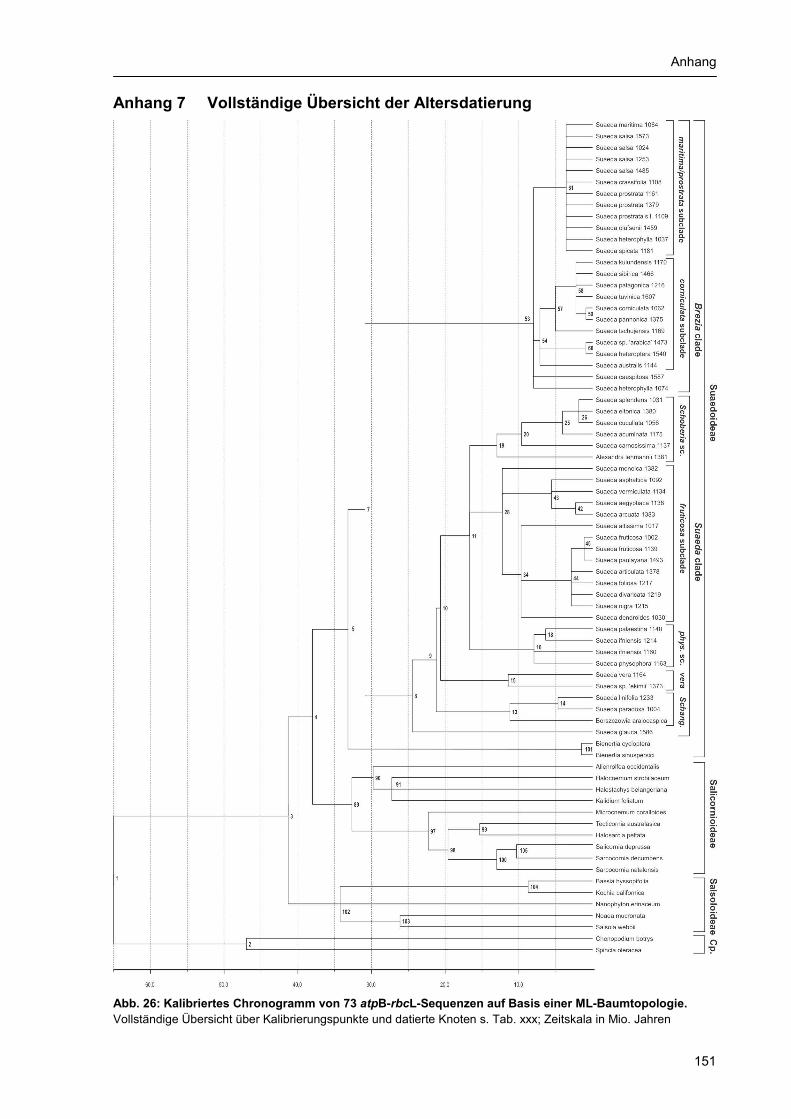
Anhang
151
Abb. 26: Kalibriertes Chronogramm von 73 atpB-rbcL-Sequenzen auf Basis einer ML-Baumtopologie. Vollständige Übersicht über Kalibrierungspunkte und datierte Knoten s. Tab. xxx; Zeitskala in Mio. Jahren
Abb. xx: Vollständige Übersicht über Kalibrierungspunkte und datierte Knoten s. Tab. xxx; Zeitskala in Mio. Jahren ITS Chronogramm
Anhang 7 Vollständige Übersicht der Altersdatierung
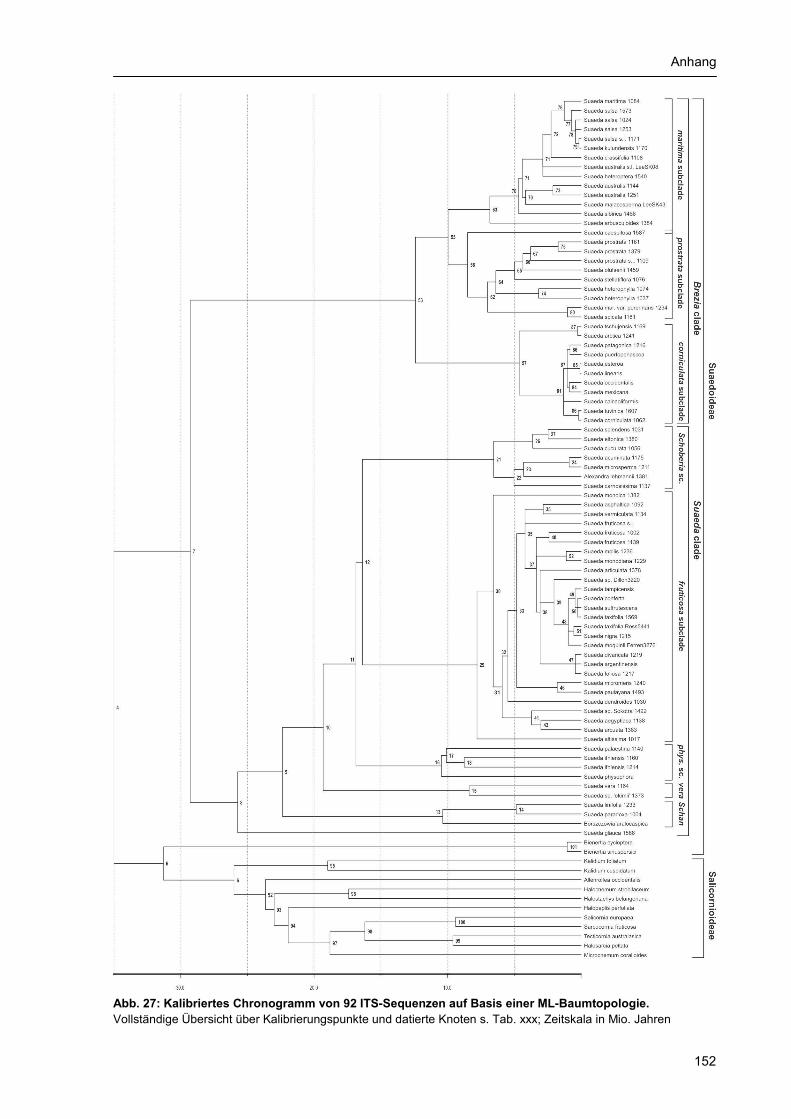
Anhang
152
Abb. 27: Kalibriertes Chronogramm von 92 ITS-Sequenzen auf Basis einer ML-Baumtopologie. Vollständige Übersicht über Kalibrierungspunkte und datierte Knoten s. Tab. xxx; Zeitskala in Mio. Jahren
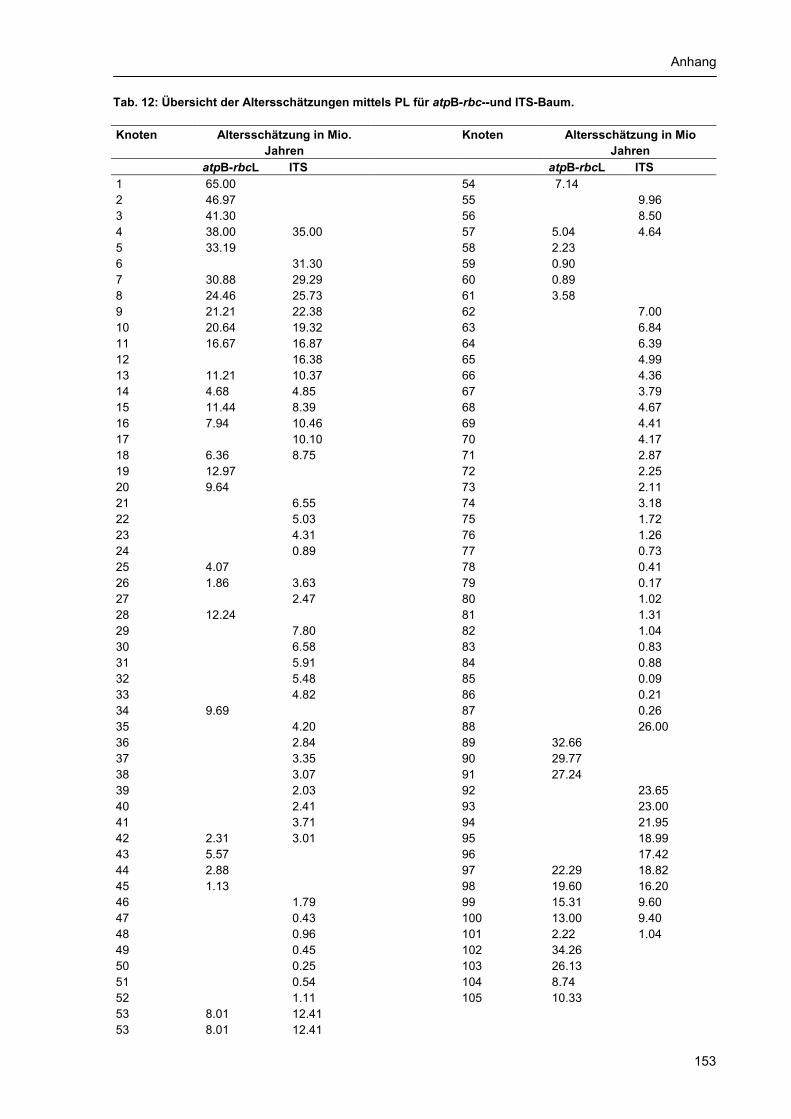
Anhang
153
Tab. 12: Übersicht der Altersschätzungen mittels PL für atpB-rbc--und ITS-Baum. Knoten Altersschätzung in Mio.
Jahren Knoten Altersschätzung in Mio
Jahren atpB-rbcL ITS atpB-rbcL ITS 1 65.00 54 7.14 2 46.97 55 9.96 3 41.30 56 8.50 4 38.00 35.00 57 5.04 4.64 5 33.19 58 2.23 6 31.30 59 0.90 7 30.88 29.29 60 0.89 8 24.46 25.73 61 3.58 9 21.21 22.38 62 7.00 10 20.64 19.32 63 6.84 11 16.67 16.87 64 6.39 12 16.38 65 4.99 13 11.21 10.37 66 4.36 14 4.68 4.85 67 3.79 15 11.44 8.39 68 4.67 16 7.94 10.46 69 4.41 17 10.10 70 4.17 18 6.36 8.75 71 2.87 19 12.97 72 2.25 20 9.64 73 2.11 21 6.55 74 3.18 22 5.03 75 1.72 23 4.31 76 1.26 24 0.89 77 0.73 25 4.07 78 0.41 26 1.86 3.63 79 0.17 27 2.47 80 1.02 28 12.24 81 1.31 29 7.80 82 1.04 30 6.58 83 0.83 31 5.91 84 0.88 32 5.48 85 0.09 33 4.82 86 0.21 34 9.69 87 0.26 35 4.20 88 26.00 36 2.84 89 32.66 37 3.35 90 29.77 38 3.07 91 27.24 39 2.03 92 23.65 40 2.41 93 23.00 41 3.71 94 21.95 42 2.31 3.01 95 18.99 43 5.57 96 17.42 44 2.88 97 22.29 18.82 45 1.13 98 19.60 16.20 46 1.79 99 15.31 9.60 47 0.43 100 13.00 9.40 48 0.96 101 2.22 1.04 49 0.45 102 34.26 50 0.25 103 26.13 51 0.54 104 8.74 52 1.11 105 10.33 53 8.01 12.41 53 8.01 12.41

154
Danksagung
An dieser Stelle möchte ich mich bei allen Menschen bedanken, die direkt oder indirekt zum Gelingen
dieser Arbeit beigetragen haben.
Bei Herrn Prof. Dr. Kurt Weising bedanke ich mich in erster Linie für die Themenstellung und die
Möglichkeit, die gute Infrastruktur des Institutes für alle notwendigen Arbeiten zu nutzen sowie den
vielfältigen Gedankenaustausch und Hilfestellungen bei der Arbeit.
Mein ganz besonderer Dank gilt Herrn Prof. Dr. Helmut Freitag für viele Jahre einer außerordentlich
fruchtbaren und wertvollen Zusammenarbeit. Helmut Freitags grenzenlose Begeisterung für die
Botanik, seine außerordentliche Fachkenntnis sowie den steten Wunsch und vor allem auch die
Fähigkeit dies mitzuteilen und zu diskutieren haben mich ganz entscheidend vorangebracht und vor
allem auch bis zum Schluss durchhalten lassen. Die hier vorgelegte Arbeit hätte ohne Helmut Freitags
morphologische, floristische und systematische Arbeiten, die umfangreiche Materialsammlung sowie
die einzigartige Kenntnis der untersuchten Pflanzengruppe nicht in dieser Form entstehen können.
Allen Mitgliedern der Arbeitgruppe Systematik und Morphologie der Pflanzen möchte ich für das gute
Arbeitsklima und die ständige Hilfs- und Diskussionsbereitschaft bedanken. Ich bedanke mich
besonders bei Irene Diebel, Maria Maier-Stolte und Christine Frohmut.
Gudrun und Martina danke ich für schöne Jahre der Zusammenarbeit am Beginn unserer
Unternehmungen. Ich danke auch allen, die mit mir in Kassel als Praktikanten und Diplomanden
zusammengearbeitet haben, insbesondere Susanne Möller für die schöne Diplomarbeit und eine stets
nette Zusammenarbeit.
Maria Lomonosova aus Novosibirsk danke ich für die unermüdliche Unterstützung mit der Zähigkeit
einer echten Sibirierin, ein ganz besonderer Dank für die wunderschöne Exkursion zu den Steppen
Südsibiriens und an den unvergleichlichen Baikal!
Gudrun Kadereit, Erik Westberg und Doris Franke von der Universität Mainz danke ich für fachliche
Hilfestellungen, für die Überlassung von Pflanzen und für die Anfertigung wichtiger Zeichnungen.
Frank Blattner aus Gatersleben und Prof. Dr. E. Jäger aus Halle danke ich für prägende Erlebnisse
und Erfahrungen während meiner Studentenzeit in Halle.
Eine Arbeit wie die vorliegende ist das Ergebnis eines sehr langen Prozesses, der schon in der
Kindheit mit der Freude und Begeisterung für die umgebende Natur begann. Ich danke hier noch
einmal meinem Großvater Theodor Schütze, der in dem etwa 13jährigen das Interesse für die Botanik
entdeckte und mir Wagner’s „Deutsche Flora“ vererbte, mit welcher er selbst gut 70 Jahre zuvor den
Zugang zur Botanik fand. Damit wurden die ersten Pflanzen bestimmt!
Ich danke weiterhin meinen Eltern, meiner Schwester Annette, Julia und meinen beiden Kindern
Johannes und Josephine für so viele Dinge, dass der Platz zum Aufzählen gar nicht ausreichen
würde.

155
Selbständigkeitserklärung
Hiermit versichere ich, dass ich die vorliegende Dissertation selbständig und ohne unerlaubte Hilfe
angefertigt und andere als die in der Dissertation angegebenen Hilfsmittel nicht benutzt habe. Alle
Stellen, die wörtlich oder sinngemäß aus veröffentlichten oder unveröffentlichten Schriften entnommen
sind, habe ich als solche kenntlich gemacht. Kein Teil dieser Arbeit ist in einem anderen Promotions-
oder Habilitationsverfahren verwendet worden.
Halle, im November 2008 . . . . . . . . . . . . . . . . .
Peter Schütze













![Robert Weltsch Verwandte Wege 1919 [zu Hans Blueher]](https://static.fdokument.com/doc/165x107/577d23921a28ab4e1e9a2d28/robert-weltsch-verwandte-wege-1919-zu-hans-blueher.jpg)




