Proteolytic Processing of the Receptor-like Protein Tyrosine

Zlotos, Bender & HolzgrabeMuscarinic receptor agonists and antagonists Muscarinic receptor agonists and
antagonists
Darius P Zlotos, Wiebke Bender & Ulrike Holzgrabe
Institut für Pharmazie und Lebensmittelchemie, BayerischeJulius-Maximilians-Universität, Am Hubland, 97074 Würzburg, Germany
Although four different subtypes of the muscarinic acetylcholine (ACh)receptor with functional correlates are known to exist (function for M5 isstill unclear), all muscarinic agonists and antagonists in clinical practiceonly show very weak selectivity. Therefore, intensive investigations are inprogress to develop subtype selective ligands. This review describes thefirst M1 agonists and antagonists of the presynaptic M2 receptor, which canbe used in the treatment of Alzheimer’s disease (AD), and M3 antagonists,which will be useful in the treatment of urinary urge incontinence. Inaddition, muscarinic agonists were found to exhibit analgesic effects andM4 antagonists may be useful in the treatment of movement disorders.However, the latter two pharmacological findings will need more researchwork to become established in clinical trials.
Keywords:Alzheimer’s disease, irritable bowel syndrome, muscarinicanalgesics, urinary urge incontinence
Exp. Opin. Ther. Patents (1999)9(8):1029-1053
1. Introduction
As one part of the peripheral nervous system, the parasympathetic systemcontrols the function of a wide variety of organs, i.e., the accommodation ofthe eyes, the dilatation of the bronchus, the tone of intestine and bladder,the function of the heart, the salivary glands and bronchadentis. Thesedifferent functions are mediated by different muscarinic receptor subtypesM1, M2, M3, and M4, which are specifically located in the organs1. The M1subtype is mainly found in the central nervous system (CNS) and theforebrain, the M2 subtype in the heart, the M3 subtype in the glands and thesmooth muscles, and the M4 subtype in the striatum (for details see [1]).This heterogeneity of the muscarinic receptors opens up the possibility toselectively influence the functions of the organism using specific ligands.Interestingly, with the exception of pirenzepine (applied to the treatment ofulcers), most of antagonists, such as atropine and scopolamine, andagonists, e.g., carbachol, in clinical practice bind more or lessnon-specifically to all receptor subtypes and, thus, induce the entirespectrum of parasympatholytic and parasympathomimetic effects, respec-tively. For over ten years, it is aimed to find ligands which bind selectively toone subtype of the muscarinic receptor. Since the structure of the subtypes
10291999 © Ashley Publications Ltd. ISSN 1354-3776
Review
1. Introduction
2. Muscarinic agonists
3. Muscarinic receptorantagonists
4. New concepts: allostericmodulation of the ligandbinding to the receptorprotein
5. Expert opinion
Bibliography
Patents
http://www.ashley-pub.com
1 In vivo, four receptors M1 - M4 are well-characterised in functionalpharmacological studies. However, five muscarinic receptor subtypes m1 - m5 arecloned and expressed in various cell types. Capital letters stand for studies withhomogenates and functional investigations, small letters for studies with clonedreceptors.
Expert Opinion on Therapeutic Patents

is highly conserved in the binding region for agonistsand antagonists, respectively, it is difficult to develophighly subtype-selective ligands. However, somedrugs are known that exhibit a preference for certainsubtypes (see Table 1). The toxin MT7 has a highaffinity to the M1 subtype, tripitramine to the M2subtype, and the toxin MT3 to the M4 subtype. Amongthe M3 ligands no compound which binds exclusivelyto this receptor subtype had been reported until now[2]. Taken together, these ‘subtype-prevalent’ ligandsare pharmacological tools rather than therapeutics.
This review covers the development of the last fiveyears. In this period of time, M1 agonists attractedinterest in the treatment of AD, which goes along witha shortage of ACh and, thus, an understimulation ofM1 receptors. Central muscarinic agonists were foundto show analgesic effects, M2 antagonists may haveutility as antibradycardiac drugs and cognitionenhancer, M3 antagonists in the treatment of theirritable bowel syndrome, urinary urge incontinenceand asthma, and M4 antagonists in the treatment ofmovement disorders such as Parkinson’s disease (PD)and Huntington’s chorea.
2. Muscarinic agonists
Research work on muscarinic agonists is mainlyfocussing on their action in the CNS with the hope totreat patients suffering from cognitive disorders suchas AD. Most recent work has concentrated on thesearch for M1 (m1) selective muscarinic agonists,because the selective stimulation of M1 receptorsshould compensate the ACh deficit and therefore, bebeneficial in AD. For earlier literature, the reader isreferred to the comprehensive review [3].
An additional novel trend in exploring muscarinicagonists is the development of compounds having an
antinociceptive effect. Here, it is aimed to separate thedose giving the analgesic effect and the dose causingthe parasympathomimetic side-effects. Although nomuscarinic analgesic is undergoing the clinical trial atthe moment, there are some candidates which couldserve as an alternative to opioids in the managementof pain.
Since almost all compounds are developed as M1selective agonists, the discussion here followsstructure characteristics.
2.1 Arecoline related compounds
Compounds derived from the naturally-occurringagonist arecoline (1) are mainly attracting interest forthe treatment of AD. Arecoline shows no subtypeselectivity and is characterised by metabolic instabilitywhich is due to the hydrolysis sensitive ester group.Thus, arecoline has only low clinical efficacy. Themost pursued strategy for improving the pharmacol-ogical profile of arecoline has been the replacementof the ester group with bioisosteric five-memberedheterocyclic rings such as oxadiazole, thiadiazole ortetrazole.
The 1,2,5-thiadiazole derivative xanomeline tartrate(2a) (Memcor®, Lumeron®) is reported to beundergoing Phase II clinical trials in AD patients [4,5].Xanomeline (Lilly, Novo Nordisk) is a selective M1receptor agonist with a high functional selectivity(IC50 = 6 nM at M1 receptors from rabbit vas deferens;EC50 = 3 µM at M2 receptors from guinea-pig atria;weak partial agonist in guinea-pig ileum; no activity inguinea-pig bladder). The functional m1 efficacy of63% was measured as the stimulation of phosphoi-nositide (PI) hydrolysis in BHK cells transfected withthe human m1 receptor relative to carbachol. Incontrast to non-selective muscarinic agonists, whichproduce several undesirable parasympathomimeticeffects in animals and humans, e.g., hypothermia,
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1030 Muscarinic receptor agonists and antagonists
Table 1: Muscarinic receptor subtypes M1 - M5 and their antagonists classified by affinities [2].
M1 M2 M3 M4 M5
MT7 (9.8) Tripitramine (9.4 - 9.6) 4-DAMP (8.9 - 9.3) MT3 (8.7) 4-DAMP (8.9 - 9.0)
4-DAMP (8.6 - 9.2) AFDX384 (8.2 - 9.0) Darifenacin (8.4 - 8.9) 4-DAMP (8.4 - 9.4) Darifenacin (8.0 - 8.1)
Tripitramine (8.4 - 8.8) Himbacine (8.0 - 8.3) AFDX384 (7.2 - 7.8) Himbacine (8.0 - 8.8) Tripitramine (7.3 - 7.5)
Pirenzepine (7.8 - 8.5) 4-DAMP (7.8 - 8.4) Tripitramine (7.1 - 7.4) AFDX384 (8.0 - 8.7) Pirenzepine (6.2 - 7.1)
Darifenacin (7.5 - 7.8) Darifenacin (7.0 - 7.4) Himbacine (6.9 - 7.4) Tripitramine (7.8 - 8.2) AFDX384 (6.3)
AFDX384 (7.3 - 7.5) Pirenzepine (6.3 - 6.7) Pirenzepine (6.7 - 7.1) Darifenacin (7.7 - 8.0) Himbacine (6.1 - 6.3)
MT3 (7.1) MT7 (< 6) MT3 (< 6) Pirenzepine (7.1 - 8.1) MT3 (< 6)
Himbacine (7.0 - 7.2) MT3 (< 6) MT7 (< 6) MT7 (< 6) MT7 (< 6)

heavy salivation and tremor, xanomeline did notproduce any of these adverse effects in rats or mice.However, large placebo-controlled trials at tolerabledoses have shown that xanomeline has little effect oncognition while improving psychiatric symptoms [6].Novo Nordisk has described a method for treatingschizophrenia using xanomeline. In addition, theyhave claimed xanomeline as an agent for thetreatment of gastrointestinal disorders.
Exchanging the N-methyltetrahydropyridine ring withvarious azabicyclic ring systems as well as increasingor decreasing the size of the hexyloxy side-chain inxanomeline did not improve the M1 efficacy.
Recently, a structure-activity-relationship (SAR) studyon the side-chain of thiadiazolyltetrahydropyridine(TZTP) has been expanded to more bulky substitu-ents to improve both the m1 selectivity compared toxanomeline and the oral bioavailability [7]. Sincexanomeline is first pass metabolised by the oxidationof the hexyloxy side-chain, it was speculated that thephenyl group would block this metabolism pathway.Benzyloxy-TZTP had no significant effect on PIhydrolysis but increasing the number of methylenegroups between the phenyl group and the oxygenatom resulted in increased M1 efficacy. Similar SARswere observed for bulky alkylthio analogues, where
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1031
N
CH3
N
SN
RN
OCH3
O
CH3
1 Arecoline 2
N
SNN
CH3
S
(CH2)n
F
N
NNN
N
CH3
CH3
NH
NO
OR
3a n = 2
3b n = 3
3c n = 5
4 LU-25-109T 5 O-alkyl-THAO
5a R = methyl
5b R = ethyl
5c R = isopropyl
5d R = propargyl
NH
NO
ORS
NO
OR
N
N
NOCH3
CH3
N
N CH3
O
CH3
HCI
6 O-alkyl-THP7 O-alkyl-DHTO
8
9 R = H
10 R = CH3
11
N
R
N
NOCH3
O CH3
S
O
xanomeline 2a
2b
2c
R
Figure 1: Arecoline related muscarinic agonists.

the phenylpropylthio-TZTP compound (2b) gave51% PI hydrolysis (see Table 2). The efficacy could bedramatically enhanced by the introduction of a triplebond in the side-chain, giving thephenylpropargyloxy-TZTP (2c) (90% PI hydrolysis).
It is hypothesised that the M1 receptor subtype has alarge lipophilic pocket and the occupation of thispocket is essential for M1 activation [7]. The phenyl-propargyloxy and thio side-chain seems to fitperfectly into this cavity. The corresponding1-azabicyclo[3.2.1]octane analogue of (2a)(compound 20) is suggested to be a drug candidate.
With the aim of developing a positron emitting M2selective ligand for use in positron emissiontomography (PET), three fluorinated derivatives ofTZTP (3a-c) have been prepared [8]. PET is able tovisualise the quantities of the M2 receptor subtype inthe living human brain. Since PET studies confirmedthe post-mortem findings in AD patients, thetechnique could be used for an early diagnosis andthe non-invasive monitoring of the drug therapy [8].Only 3a and 3b displayed M2 subtype selectivity invitro. In vivo experiments, [18F] 3b did not showinhibition by the M1 selective antagonist IQNB (19)and displayed low inhibition by the M2 selectiveantagonist (R,S)-fluoromethyl QNB. Thus, [18F] 3bcould be a good PET radiotracer with high affinity andM2 selectivity in vitro. In addition, it shows a highblood-brain barrier transport in vivo.
Yet another useful ester replacement is the tetrazolering, as illustrated by LU-25-109T (4) [9]. With theexception of xanomeline, which was 13 times as
potent at M1, LU-25-109T shows higher affinity for theM1 receptor than most standard agonists. In vitroexperiments provided the following Ki values: 36 nM(vs. [3H]-pirenzepine) on M1 receptors in rat brain¸130nM on cloned human M1 (hm1) receptors expressedon CHO-K1 cells; 340 nM (vs. [3H]-quinuclidinylbenzylate) on M2 receptors in rat brain stem; and 3100nM (vs. [3H]-N-methylscopolamine) on M3 receptorsin rat salivary glands. The selectivity ratio towards M1versus M3 was 44; the M-agonist index (defined as thequotient of Ki values in oxotremorine-M andquinuclidinyl benzilate binding assays) of 4 iscomparable to that of RS-86 (25) and several timeshigher than that of xanomeline. In ex vivo tissuestudies, LU-25-109T was found to be a partial agonistat the rat superior cervical ganglion M1 receptors andat the guinea-pig ileum (M1, M2 and M3), while itacted as an antagonist at the guinea-pig left atriumM2-receptors and on cultured cerebellar granule cellM3 receptors. Unfortunately, Forest Laboratories hasannounced the discontinuation of LU-25-109T for ADfollowing unsatisfactory results of the completedPhase II/III clinical trial. However, new clinical indica-tions, such as urinary incontinence and functionalrecovery from stroke and traumatic brain injury arepossible [10,11].
Extending the tetrahydropyridine ring to thecorresponding azepine ring annulated with theisoxazole ring resulted in the so-called THAO(5,6,7,8-tetrahydro-4H-isoxazolo[4,5-c]azepin-3-ol)series. A series of O-alkylated THAO derivatives(5a-d) were found to be very potent muscarinicligands showing EC50 or Ki values in the lownanomolar range and being one order of magnitudemore potent than the respective analogues containingthe THPO (4,5,6,7-tetrahydroisoxazolo[4,5-c]pyridine) (6) or DHTO (6,7-dihydro-4H-thiopyrano[3,4-d]isoxazol) (7) ring systems [12] (see Table 3). Inthis series, 5d proved to be the most potentcompound (IC50 = 8 nM in the [3H]Oxo-M bindingassay). The pharmacological effects of 5a-d werestudied using five cloned human mAChRs m1-m5 andthe functional assay R-SAT (receptor selection andamplification technology). O-Methyl THAO (5a) aswell as O-propargyl-THAO (5d) were partial agonistsat m1 - m5. O-Ethyl-THAO (5b) (partial agonist at m1 -m4, antagonist at m5) was shown to be the mostm1-selective compound.
The compounds 8-10, resulting from the substitutionof the ester group by the 3-(hexyloxy)pyrazinyl groupachieved no improvement in cholinergic agonist
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1032 Muscarinic receptor agonists and antagonists
Table 2: Pharmacological data for thiadiazolyltetrahydro-pyridine derivatives2.
R % max PI, m1§
O-(CH2)3-CH3(xanomeline,2a)
13
O-CH2-Ph 12
O-(CH2)2-Ph 16
O-(CH2)3-Ph 25
O-(CH2)4-Ph 47
S-CH2-Ph 10
S-(CH2)2-Ph 9
S-(CH2)3-Ph (2b) 51
O-CH2-C≡C-Ph (2c) 90
§Stimulation of PI hydrolysis in BHK cells transfected withthe human m1 receptor relative to the carbachol response.

activity over xanomeline [13]. Thus, therapeuticperspectives of the compounds 5-10 were not givenby the authors.
Replacement of the ester group in arecoline with anoxime ether group has lead to milameline HCl (11),which was in Phase III clinical trials for AD but is nolonger in development at the moment (Parke-Davis;Roussel Uclaf) [14]. Milameline is a non-subtype-selective partial muscarinic agonist. After oral admini-stration in rats and mice, milameline was found tohave 2 - 3 times higher potency than arecoline inaddition to a longer duration. A PET study in rhesusmonkeys with [11C]-labelled milameline showed arapid and extensive distribution in regions of the brainwith a high density of muscarinic receptors, whichsupports the findings in rodents.
A novel composition comprising the acetylcholineesterase (AChE) inhibitor tacrine with a muscarinicagonist milameline was tested for the treatment of AD[101]. This new composition was found to reverse thescopolamine-induced decrease in cognitive functionin rhesus monkeys to a higher extent than did equiva-lent doses of milamine or tacrine alone.
In addition, a number of thiadiazole substitutedtetrahydropyridines (50a-d) has been prepared insearch for muscarinic analgesics with strong andselective effects on the gastrointestinal tracts (seeSection 2.5).
2.2 Quinuclidine related compounds
The quinuclidine ring is incorporated in manymuscarinic acting agents. Tritiated 3-quinuclidinylbenzilate ([3H]QNB), a non-selective muscarinicantagonist, is routinely used for receptor bindingstudies. The 3-quinuclidinyl acetate, aceclidine (12) isa moderately potent muscarinic agonist used in thetreatment of glaucoma. Since 3-quinuclidole incorpo-rates the atomic sequence of choline into a semirigidstructure, it has become a popular fragment for thedevelopment of muscarinic agonists.
Talsaclidine fumarate (Boehringer Ingelheim) (13), aquinuclidinyl-propargyl ether has been reported to bein Phase II clinical trials for AD [16-18]. Talsaclidine isa functionally selective M1 agonist with only partialagonist activity at M2 and M3 receptors. Bindingstudies provided Ki values of 6.6, 6.6 and 29.6 µM inrat hippocampus (M1), heart (M2) and lacrimal glands
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1033
Table 3: Pharmacological data forO-alkyl-THAO derivatives5.
Receptor binding, IC50 (mM)
[3H]QNBbrain
[3H]QNBheart
[3H]PZbrain
[3H]Oxo-Mheart
Arecoline 28 1.7 2.1 0.016
Oxotremorine 2.3 0.084 0.093 0.0012
O-Methyl-THAO (5a) 6.4 0.35 0.39 0.010
O-Ethyl-THAO (5b) 0.53 0.14 0.018 0.0034
O-isopropyl-THAO (5c) 0.37 0.36 0.013 0.011
O-Propargyl-THAO (5d) 0.48 0.082 0.023 0.00075
EC50 (mM); % of maximal carbachol response
m1 m2 m3 m4 m5
Arecoline 3.2; 86% 0.025; 105% 0.34; 67% 0.13; 70% 0.60; 77%
Oxotremorine 0.39; 75% 0.019; 105% 0.21; 66% 0.033; 102% 0.055; 74%
O-Methyl-THAO(5a)
0.46; 74% 0.043; 97% 0.62; 72% 0.24; 110% 4.7; 60%
O-Ethyl-THAO (5b) 0.095; 42% 0.041; 85% 0.50; 34% 0.067; 89% 2.1; nd
O-Isopropyl-THAO(5c)
0.17 nd 0.14 nd 0.11
O-Propargyl-THAO(5d)
0.14; 59% 0.0067; 86% 0.13; 70% 0.030; 105% 0.95; 36%
nd: Not determined.

(M3), respectively. In CHO cells expressing high andlow densities of human M1 receptors, talsaclidineeffectively stimulated a PI turnover with an EC50 valueof 4.0 µM and a relative efficacy compared tocarbachol of 85% and 64% in cells expressing high andlow densities, respectively. In contrast, 13 exhibitedvery weak partial agonist activity with an EC50 of 50µM and relative efficacy of 44% in cells expressinghigh densities of M3 receptors, and no activity in cellsexpressing low densities.
A new muscarinic partial agonist with functionalselectivity for M1 receptors, sabcomeline hydrochlo-ride (14) (Memric™, SmithKline Beecham) contains acyanooximether group which is a novel esterbioisoster among the muscarinic agents [19-21]. 14has been reported to undergo Phase III clinicaltest ing. The compound displaced [3H]-oxotremorine-M from muscarinic receptors in the ratbrain with high affinity (IC50 = 14 nM), a potencysimilar to that of oxotremorine-M (IC50 = 13 nM), butexhibited low affinity for cholinergic nicotinicreceptors and other neuroceptors. In studies, usingcloned human muscarinic receptors, sabcomeline
possessed approximately equal affinity in displacing[3H]QNB from all muscarinic receptor subtypes. Infunctional models in vitro, 14 caused maximaldepolarisation of the rat superior cervical ganglion atlow concentrations (300 nM) (M1-mediated effect),while producing a lower maximal effect than the highefficacy agonists oxotremorine-M and carbachol onM2-mediated release of ACh and M3-mediatedsmooth muscle contraction (guinea-pig ileum),respectively. The pharmacological profile seen invitro was reflected in vivo through potent cognition-related activity (M1-induced increase in hippocampalEEG power) combined with low efficacy, comparedwith arecoline or oxotremorine, on induction ofbradycardia (M2-mediated response), hypotension(via M3-mediated vasorelaxation) and tremor(thought to be mediated by M3 receptors). Interest-ingly, in a patent, a novel transdermal patchformulation comprising sabcomeline for the treatmentand/or prophylaxis of dementia is claimed [102].About 200 thiadiazole derivatives were found to bemuscarinic cholinergic agents which could be usefulas stimulants of the cognitive function of the forebrainand hippocampus. The compounds may not onlyhave utility in the treatment of AD, but also asanalgesic agents and for the treatment of glaucomaand gastrointestinal motility [103]. The affinity for themuscarinic receptors was determined by inhibition ofthe specific binding of [3H]-oxotremorine. Forexample, compound 15 exhibits an IC50 value of 1.00nM. The M1-affinity of 15 (IC50 = 0.6 nM) wasdetermined in rat cerebral cortex membranes.
American Home Products Corp. has synthesised anumber of quinuclidine pyrazinyl ethers (16) asmuscarinic agonists which should be useful fortreating neurological disorders [104]. The compoundswere tested for their affinity to muscarinic receptors inCHO cells in competition with [3H]QNB.
By going from the tetrahydropyridine derivatives (8)to the corresponding 3-quinuclidinyl compounds 17,significant improvement in efficacy and potency wasobtained [9]. Like 8 and xanomeline, 17a was afunctionally selective M1 agonist that showed greaterfunctional selectivity than 17b. The improvedfunctional selectivity of 17a over 17b could be attrib-uted to the additional binding interactions betweenthe hexyloxy side-chain of 17a and the M1 receptor,which is impossible for 17b. Although 17a may showM1 functional selectivity comparable to xanomeline, itis a less efficacious and potent M1 agonist thanxanomeline.
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1034 Muscarinic receptor agonists and antagonists
N
O
N S
N
SS
S
N
OCH
N
CN
NO
CH3
HCI
N
O CH3
O
N
N
NR
N
O
N
N
SR
NO
I
OHO
NO
OHO
I
12 Aceclidine 13 Talsaclidine
14 Sabcomeline 15
16 17a R = O-hexyl
17b R = H
18 IQNP 19 IQNB
Figure 2: Muscarinic agonists incorporating the quinuclidinylring.

In the search for agents for the single-photonemission-computed tomography (SPECT) of cerebraland cardiac mAChR receptor densities, four stereoiso-mers of (R)-(-)-3-quinuclidinyl-α-hydroxy-α-(1-iodo-1-propen-3-yl)-α-phenylacetate IQNP (18) havebeen synthesised [22]. IQNP is an analogue of 4-IQNB(19), a high affinity muscarinic antagonist. Theroutine use of 4-IQNB in SPECT studies is limitedbecause of the low radiolabelling yield and theprolonged time period required between administra-tion and scanning (24 h) due to the slow clearance ofnon-specific binding. In contrast, IQNP is readilyradioiodinated with high specific activity anddemonstrates significant cerebral uptake and rapidvascular clearance. (E)-(-)-(-) IQNP demonstrated thehighest receptor subtype specificity between the m1(KD = 0.38 nM) and the m2 molecular subtype (KD =29.6 nM). In vivo biodistribution studies demonstratedthat iodine-125-labeled (E)-(-)-(+)-(18) clearedrapidly from the brain and heart. In contrast, iodine-125-labelled (E)-(-)-(-)-(17), (Z)-(-)-(-)-(18) and(Z)-(+)-(-)-(18) have a high uptake and retention inmAChR rich areas of the brain. It was also observedthat (E)-(-)-(-)-18 demonstrated a subtype selectivity
in vivo with retention in M1 (m1, m4) mAChR areas ofthe brain. In addition (Z)-(-)-(-)-18 also showed asignificant uptake in tissues containing the M2 (m2)AChR subtype. Taken together, the iodine125-labelled isomers of the (E)-(-)-(-)- and (Z)-(-)-(-)-IQNP are attractive candidates for clinicalstudies of the evaluation of mAChR density by SPECT.
Some thiadiazole substituted quinuclidines are verypotent muscarinic analgesics (see Section 2.5).
2.3 Compounds incorporating1-aza-3-norbornane or1-azabicyclo[3.2.1]octane ring systems
Systematic variation of the side-chain and theazacyclic ring of xanomeline has led to the discoveryof potent muscarinic agonists with robust M1 efficacy[4,5]. The most selective M1/M2 compound in thisser ies was the phenylpropargyl thio-1-azabicyclo[3.2.1] octane endo analogue (20) whichwas suggested to be a drug candidate for AD (m1:EC50 = 1 nM, % max PI BHK = 98%, A9L; m2: EC50 >1000 nM, CHO) [7]. The propylthio analogue (21)exhibited an unexpected antipsychotic-like activity
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1035
Table 4: Pharmacological data for phenylpropargyl-oximeether derivatives22.
R IC50 (nM) PI(% Carbachol)
cAMP(% Inhibition)
CMD QNB m2/m1
Carbachol 6.7 33000 0.05
H 22a 46 5012 4.8 Hm1: 40%Hm3: 8%Hm5: 1%
Hm2: 0%Hm4: 0%
p-OCH3 110 13101 8.0
m-OCH3 22b 28 6927 250 Hm1: 51%Hm3: 6%Hm5: 11%
Hm2: 0%Hm4: 0%
o-OCH3 219 17658 4.2
p-F 30 6092 5.0
m-F 37 4925 5.0
o-F 28 4443 5.2
p-Cl 53 6300 6.2
m-Cl 19 3882 6.2
o-Cl 12 1305 3.6
m-NO2 28 6399 3.1
p-CH3 89 11089 2.7
m-CH3 28 5195 -
m-CF3 32 4237 1.7
3,4-di-OCH3 140 42449 -
3,4-(OCH2O) 23 2547 -

[23]. Thus, the use of potent and highly selectivemuscarinic receptor ligand may be an important newapproach in the medical treatment of schizophrenia.
Several differently substituted phenylpropargyloxime-ether incorporating the 1-azanorbornane ring(22) have been described as potent muscarinicagonists [24]. Muscarinic receptor binding assays wereconducted using [3H]-QNB to label antagonist sitesand [3H]-cis-methyldioxolane (CMD) to label agonistsites in membrane preparations from the ratneocortex. Selectivity for m1 over m2 muscarinicsubtypes was determined estimating the affinity ofcompounds for m1 and m2 receptor subtypes labelledby [3H]-QNB in CHO cells selectively expressinghuman m1 and m2 receptors (see Table 4). The abilityof the most selective compound (22b) and the parentoxime (22a) to stimulate PI accumulation or inhibitforskolin-stimulated accumulation of cAMP in CHOcells selectively expressing human m1 and m2receptors was determined.
A number of 1-azanorbornane pyrazinyl ethers hasbeen synthesised as muscarinic agonists useful fortreating of neurological disorders [20]. Thecompounds were tested for their affinity to muscarinicreceptors in CHO cells in competition with [3H]QNBand for their ability to stimulate the hydrolysis of PI inCHO cells. The compound (23), having the highestaffinity and activity, gave values of 2 nM (Ki, M1), 4.5nM (Ki, M2), 0.4 nM (EC50, M1) and 16 nM (EC50, M2).
Several 1-azanorbornane N-oxides, exemplified by24, have been described [105]. Interestingly, the newcompounds are disclosed to bind to central M1receptors and should alleviate the symptoms ofneurological illness associated with ACh deficiency,like memory loss in senility, the neurologicalsymptoms of PD, Down’s syndrome or dementiapugilistica.
A non-opioid analgesic (51) has been reported to be acandidate for clinical evaluation in the treatment ofirritable bowel syndrome (see Section 2.6).
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1036 Muscarinic receptor agonists and antagonists
NO
NOCH
3
N
N
N S
O
CH3
20
21
22
23
24
N
N
SN
S
N
N
SN
SCH
3
N
O NH
CH3
SO
+-
Figure 3: Muscarinic agonists incorporating the 1-aza-3-norbornane and 1-azabicyclo[3.2.1]octane ring system.
N
N
O
O
CH3
CH3
O
N
N
CH2
CH3
CH3
O
N
N
OCH3
CH3
NH
N
N
OCH3
CH3
O
N
OCH3
CH3
O
N
NHCOR
CH3
CH3
25 RS 86 26 YM-796
27 28
29 30
Figure 4: Muscarinic agonists incorporating a spiro structure.

2.4 Spiro compounds
Increasing the rigidity of the muscarinic pharma-cophore is a strategy that has been occasionally usedin order to increase receptor subtype selectivity andwhich often affects the potency and efficacy of thecompounds. The conformationally restricted spirocompound RS-86 (25) was one of the first muscarinicagonists to be tested in AD patients. Since RS-86 is apotent muscarinic agonist, but with preference for theM2 receptor; attempts have been made to change theprofile towards M1 selectivity. A major improvementin profile compared to RS-86 represents the (-)enantiomer of YM-796 (26), which is reported to beundergoing Phase II clinical evaluation for thetreatment of AD [25,26]. 26 is a potent muscarinicpartial agonist with functional selectivity for M1receptors. The affinity for M1 and M2 receptors wasdetermined in terms of ability to displace[3H]pirenzepine, an M1 selective ligand, from a ratcortex membrane preparations (Ki = 1.2 µM) and[3H]QNB from rat cerebellum membrane (Ki = 2.1µM), respectively (see Table 5). YM-796 was alsoactive in a functional model of M1 receptor activation,i.e., the stimulation of the PI hydrolysis in rathippocampal slices. Functional studies in transfectedcells expressing the gene for M1 and M2 receptorsalso indicated high functional selectivity of YM-796for M1 over the M2 subtype. Moreover, 26 exhibitedpotent anti-amnesic activity with higher selectivityover cholinergic side-effects, namely hypothermiaand salivation, as compared with RS-86.
In order to find compounds with high M1 receptorbinding affinity, a high M1 versus M2 receptorselectivity, and an anti-amnesic effect separated frommuscarinic side-effects, SAR were studied in a seriesof spiro compounds related to YM-796 and RS-86 [27].Compound 27a was found to have the best pharma-cological profile. Increasing the size of theN-substituent in the piperidine ring of 27 increasedthe selectivity in binding affinities for M1 over M2receptors but resulted in loss of M1 agonistic activityor anti-amnesic activity. Compound 28 exhibited onlylow affinity for M1 receptors, suggesting that a basicnitrogen atom is not tolerated in M1 receptor bindingas a substitute for an oxygen atom or a carbonyl groupat the corresponding position of 27 or RS-86. None ofthe described agents exhibited high selectivity for theanti-amnesic effect over induction of hypothermiacompared to YM 796.
At the moment, it is difficult to judge whether theanti-amnesic effect can be properly separated fromthe muscarinic adverse effects in order to be utilisedfor therapeutic purposes.
Compound 29, designed by incorporating the tetrahy-drofurane ring moiety of muscarone into anazaspirodecan skeleton exhibited potent muscarinicactivities (Ki = 7 nM vs. [3H]oxotremorin) in vitro andin vivo, but no M1/M2 selectivity [28]. The biologicalactivity of both enantiomers of 29 has been character-ised [29]. The R-isomer is a low affinity agonist whichis selective for M1 and M3 receptors by virtue ofintrinsic activity. In contrast, the S-isomer is a
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1037
Table 5: Pharmacological data for spiro-compounds26-28.
Binding data: K i (mM) Anti-amnesic effecta Hypothermia
[3H]PZ [ 3H]QNB MED mg/kg, sc. EDD2°Cemg/kg, sc.
(±)YM-796b 1.6 3.1 0.03 8
YM-796 26 1.2b 2.1b 0.03b 23c
R = CH3 27ad 1.6 0.74 0.1 3
R = C2H5 27bd 0.28 0.34 > 1 1
R = n-C3H7 27cd 0.33 1.2 > 3 > 30
R = iso-C3H7 27dd 0.43 2.9 0.01 > 30
R = allyl 27ed 0.42 nd nd > 30
28d 25 - - -
a Ameliorating effects on scopolamine-induced impairment of rat passive avoidance testb Fumarate was usedc In mice, tartrate monohydrate was usedd Hydrochloride was usede The dose required to reduce the rectal temperature by 2°C in male ICR mice

relatively high affinity M3 agonist with 10-fold higherbinding selectivity for M3 than M2 or M1 receptorsubtypes. As in case of YM-796, the biological activityof the racemate resides predominantly in theS-enantiomer.
An extensive SAR study of a series of straight chained,branched, cyclic alkyl and aromatic acylhydrazones(30) has been conducted [30]. All compounds areef f icacious muscarinic agonists, exhibit inghippocampal PI hydrolysis in the 20 - 50% range ofcarbachol (see Table 6). From this findings, it can be
concluded that an agonist ligand with a longside-chain or bulky group can fit into both thereceptor recognition site (binding site) and the activa-tion site (efficacy site) of the M1 and M3 receptorsubtypes and that they may be excluded from the M2efficacy site by either steric occlusion or detrimentalinteraction. The affinity for the M1 receptor, obtainedfrom the pirenzepine assay, was found to vary widelywith little change in intrinsic activity. This is consistentwith the view that the recognition site for agonists andantagonists differs in muscarinic receptor subtypesand is distinct from the efficacy site. None of the
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1038 Muscarinic receptor agonists and antagonists
Table 6: Pharmacological data for acylhydrazone derivatives30.
R Ki (mM) [ 3H]PZ Intrinsic activity (%, relative to carbachol )
Hip (M1) a Heart (M2)b Tr (M3) c
Parent ketone 3.8 64 100 83
NHCOCH3 100 26 0 91
NCH3COCH3 60 26 nd nd
NHCOCH3d 180 23 100 nd
NHCOC2H5 76 27 nd nd
NHCO-n-Pr 65 20 nd nd
NMeCO-n-Pr 16 39 nd nd
NHCO-i-Pr 28 27 40 nd
NHCO-n-But 41 30 nd nd
NHCO-i-Bu 90 43 nd nd
NHCO-t-But 15.2 40 nd nd
NHCO-n-Pen 25 35 34 nd
NHCO-n-Hex 25 46 nd nd
NHCO-n-Hep 9.5 33 33 nd
NCH3CO-n-Hep 8.8 32 nd nd
NHCO-n-Oct 9.0 4 nd nd
NHCO-n-Nona 14.4 2 nd nd
NHCO-c-Hex 38 32 nd nd
NHCOCH2-c-Hex 30 39 nd nd
NHCO(CH2)3-c-Hex 13.6 23 nd nd
NHCO(CH2)4-c-Hex 8.0 5 nd nd
NHCOPh 78 33 nd nd
NHCOCH2Ph 30 11 nd nd
NHCO(CH2)3Ph 34 32 nd nd
NHCOPh-4-c-Hex 4.8 38 0 78
NHCOCH2-2-Naph 10 20 0 nd
NHCONH2 100 26 51 nd
a Stimulation of PI hydrolysis in rat hippocampal tissueb Inhibition of adenylate cyclase in rat heart membranesc Isometric contractions of the isolated guinea-pig trachead 8-Desmethyl derivativend: Not determined.

described hydrazones exhibited higher affinity andintrinsic activity at M1 receptor subtype compared tothe parent ketone.
2.5 Structurally miscellaneous compounds
A series of quinoline and naphthalene derivativessubstituted with the 1-azabicyclo[3.3.0]octane ring(exemplified by 31-33) were synthesised [31]. Themuscarinic M1 (vs. [3H]pirenzepine) and M2 (vs.[3H]QNB) receptor binding affinity, inhibitory effectson AChE activity and the abilities to improve cognitivefunction (passive avoidance test in scopolamine-induced mice) were evaluated. Most of thecompounds were found to have higher affinities to M1than to M2 receptors. The naphthalene derivativesexhibited much higher M1 affinity than thecorresponding quinolone derivatives. The tetrahy-droacridine derivative (32) had the highest activity inthe ‘quinolone’ series. Compound 33 showing thehighest M1 affinity (Ki = 48 nM) and selectivity(M2/M1 index = 22.9) also exhibited amelioratedscopolamine induced impairment in passiveavoidance tasks and was progressed to further investi-gations. The analogue aniline derivative (34) (M1,IC50 = 0.1 µM; M2, IC50 = 8.3 µM) improved thecognitive function in a passive avoidance tests inpirenzepine-, scopolamine-, cycloheximide- andelectric shock-induced mice and rats and wassuggested as an effective agent for AD [32].
In a patent, the pyrrolotriazine (35) and pyridazinoin-dolizine (36) derivatives have been mentioned asagents for treating cognitive disorders andneurological diseases associated with ACh deficiency(e.g., presenile dementia, senile dementia of the ADtype, PD and Down’s syndrome). The binding affini-ties of the pyrrolotriazine derivatives to M1 receptorsinvestigated using transferred CHO cells ranged fromKi = 0.31 µM for the compound 35 to 21 µM [106].Compound 36 stimulated the hydrolysis of PI in CHOcells expressing M1 receptors by 102.5% relative tocarbachol. Receptor binding studies demonstratedselectivity for M1 versus M2 receptors; Ki values for M1and M2 binding were 1.34 nM and 13.35 nM, respec-tively [107].
Compound 37 consists of a novel heterocyclic ringsystem which may be a novel lead structure fordevelopment of AD drugs [108]. 37 exhibited an IC50value of 2.2 ng/ml and 26 nM in an [3H]oxotremorineand [3H]pirenzepine binding assay in rat cerebralcortex, respectively. Another class of compounds
stimulating the central muscarinic receptors are2-aryl-amidothiazole derivatives exemplified by 38[109]. They can be useful for the treatment ofneurological symptoms associated with PD, Down’ssyndrome, senile pugilistica, dementia and as antipsy-chotic, antidepressant and anxiolytic agents. Receptoraffinities were determined by inhibition of M1[3H]pirenzepine binding in rat cortex and M2 [3H]QNBbinding in rat cerebellum. 38 showed IC50 values of0.054 µM and 3.4 µM at M1 and M2 receptors,respectively.
Several amidine derivatives have been found todisplay high efficacy at M1 receptors and loweractivity at M3 receptors coupled to PI metabolism inA9 L cells [33]. The highest functional M1 versus M3selectivity in this series was observed for thecompounds 39-41. Compound 41 also exhibited lowactivity at M2 receptors. Further evaluation astherapeutic agents for the treatment of AD wasmentioned.
A series of tropane derivatives, related in structure tobaogongteng A (42a), an alkaloid from a Chineseherb, have been synthesised [34]. 6-β-Acetoxynortr-opane (42b) had weak affinity (Ki = 22 µM) for centralM1 muscarinic receptors in a [3H]QNB binding assaybut extremely high affinity and selectivity for M2muscarinic receptors expressed in CHO cells (Ki = 2.6nM) (see Table 7). 42b had 13-fold lower affinity forM4 receptors, 260-fold lower affinity for M3 receptorsand 8200-fold lower affinity for M1 receptors. Intransfected CHO cells, 42b was an M2 agonist, basedon GTP-elicited decrease in affinity, and a full M4agonist (IC50 = 11 nM), based on inhibition of cyclicAMP accumulation while being a full M1 agonist (EC50= 23 nM) and a partial M3 agonist (EC50 = 3.6 nM),based in both cases on stimulation of PI breakdown.Substitution of the acetoxy group with thecarbomethoxy group (-COOCH3) led to the loss of M2affinity as in the case of 42c and 42d.
Oxotremorine (43) has long been known to act as anunselective potent muscarinic agonist. By means ofclassical isosteric replacement of a methylene groupwith oxygen, compound 44a as well as the relateddimethylamino (44b) and trimethylammonium (44c)derivatives have been prepared [35]. The newcompounds displayed binding affinities comparableto those of the parent molecule with the exception ofthe trimethylammonium salt (44c), which behaved asa full muscarinic agonist and showed a pronouncedselectivity for M2 versus M1 receptor subtypes.
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1039
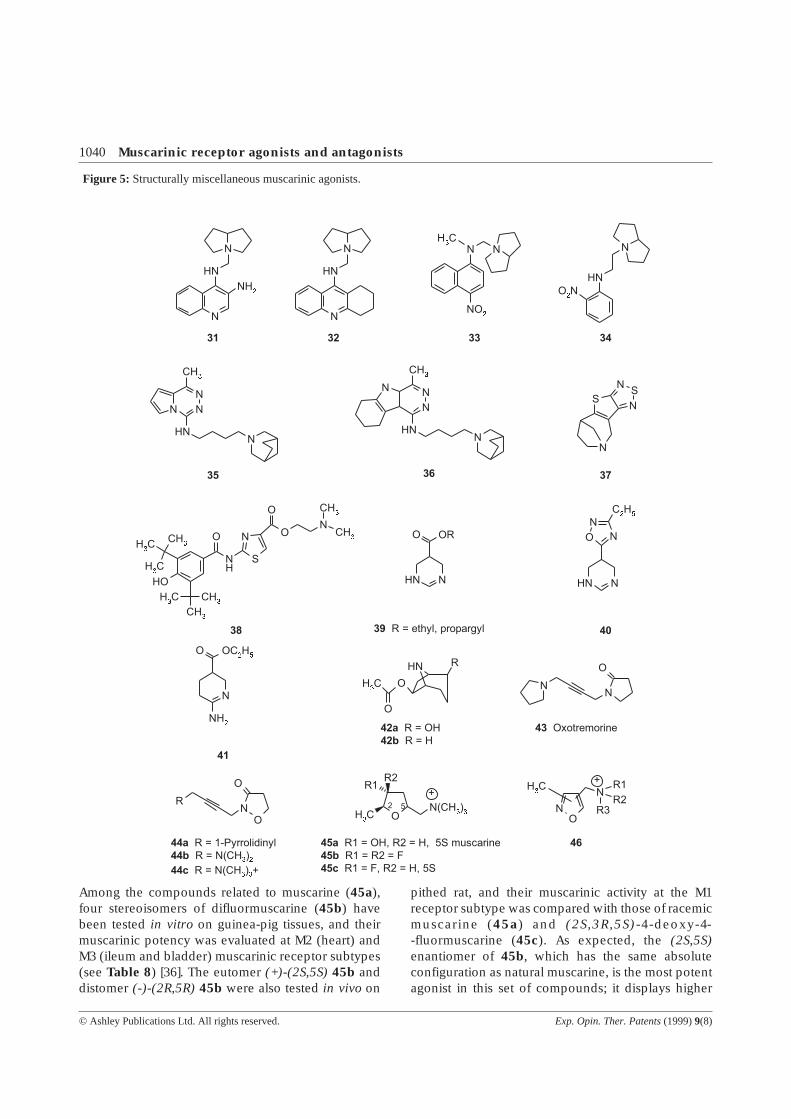
Among the compounds related to muscarine (45a),four stereoisomers of difluormuscarine (45b) havebeen tested in vitro on guinea-pig tissues, and theirmuscarinic potency was evaluated at M2 (heart) andM3 (ileum and bladder) muscarinic receptor subtypes(see Table 8) [36]. The eutomer (+)-(2S,5S) 45b anddistomer (-)-(2R,5R) 45b were also tested in vivo on
pithed rat, and their muscarinic activity at the M1receptor subtype was compared with those of racemicmuscarine (45a) and (2S,3R,5S)-4-deoxy-4--fluormuscarine (45c). As expected, the (2S,5S)enantiomer of 45b, which has the same absoluteconfiguration as natural muscarine, is the most potentagonist in this set of compounds; it displays higher
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1040 Muscarinic receptor agonists and antagonists
N
NH2
NH
N
N
NH
N N NCH3
NO2
NH
N
O2N
N
N
NHN
N
CH3
N
N
NHN
N
CH3
N
SN
SN
31 32 33 34
35 36 37
NH
S
N OO
O
NCH
3
CH3
OH
CH3
CH3
CH3
CH3
CH3
CH3
NH N
ORO O N
N
NH N
C2H
5
N
OC2H
5O
NH2
38 39 R = ethyl, propargyl 40
41
CH3
O
O
NH R
NN
O
NO
R
O
42a R = OH
42b R = H
43 Oxotremorine
44a R = 1-Pyrrolidinyl
44b R = N(CH3)2
44c R = N(CH3)3+
45a R1 = OH, R2 = H, 5S muscarine
45b R1 = R2 = F
45c R1 = F, R2 = H, 5S
46
ON(CH
3)3
R2
CH3
R1+
ON
CH3 N
R1
R2R3
+
2 5
Figure 5: Structurally miscellaneous muscarinic agonists.
potency at the heart (M2 subtype) at variance with(±)-muscarine and 45c, which are more active at theileum (M3 subtype). Difluoromuscarine (+)(2S,5S)45b has 6 and 21 times higher affinity at M2 incomparison to M3 receptor subtypes, respectively. Invivo, (+)-(2S,5S) 45b is 20 and 11 times less activethan (±)-muscarine and 45c at M1 receptor subtype,while at M2 subtype activity reductions amount to 6and 38, respectively. Difluoromuscarines represent avaluable tool to study the heterogenity among M2 andM3 subtypes and to put in evidence for the differencesbetween the muscarinic receptors mediating the heartrate and the heart force, and those of the ileum andbladder.
A series of 3-, 4- and 5-aminomethyl isoxazoles substi-tuted with one or two additional methyl groups (46)have been synthesised in order to investigate thestructural requirements, i.e., heterocyclic moiety,regiochemistry and length of an aminoalkyl unit, formuscarinic activity [37]. Assays on isolated rabbit vasdeferens (M1 receptor subtype), isolated guinea-pigatrium (M2 receptor subtype) and ileum (M3 receptorsubtype) showed 10 - 1000 times lower activities thanfor the corresponding furane or oxadiazole deriva-tives. No therapeutic perspectives have been given bythe authors.
2.6 Muscarinic analgesics
The antinociceptive activity of cholinergic agonists inanalgesic tests is well described in the literature [38].The fact that analgesic effects of classical muscarinicagonists like arecoline or oxotremorine were antago-nised by muscarinic antagonists but not by opioidantagonists, demonstrates that muscarinic analgesia ismediated directly through muscarinic receptors andnot indirectly through opioid systems. Despite muchhigher antinociceptive efficacy than morphine, theparasympathomimetic cholinergic side-effects such asbradycardia, hypotension, diarrhoea, urination,salivation and lacrimation, precluded the clinicalutility of muscarinic agonists. However, the recentdevelopment of subtype selective muscarinic agonistshas provoked a great interest in the discovery ofmuscarinic analgesics which are devoid of theunwanted effects.
Recently, it was hypothesised that muscarinicanalgesia is mediated by central muscarinic receptors.The analgesic effects of oxotremorine, pilocarpine,arecoline, aceclidine, RS-86, xanomeline (hexyloxy-TZTP) 2a, hexylthio-TZTP, propoxy-TZTP and3-Cl-propylthio-TZTP were investigated by use of themouse acetic writhing, grid shock, hot plate and tailflick test [39]. These agonists were also evaluated fortheir ability to displace [3H]oxotremorine and[3H]pirenzepine binding and for their functional
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1041
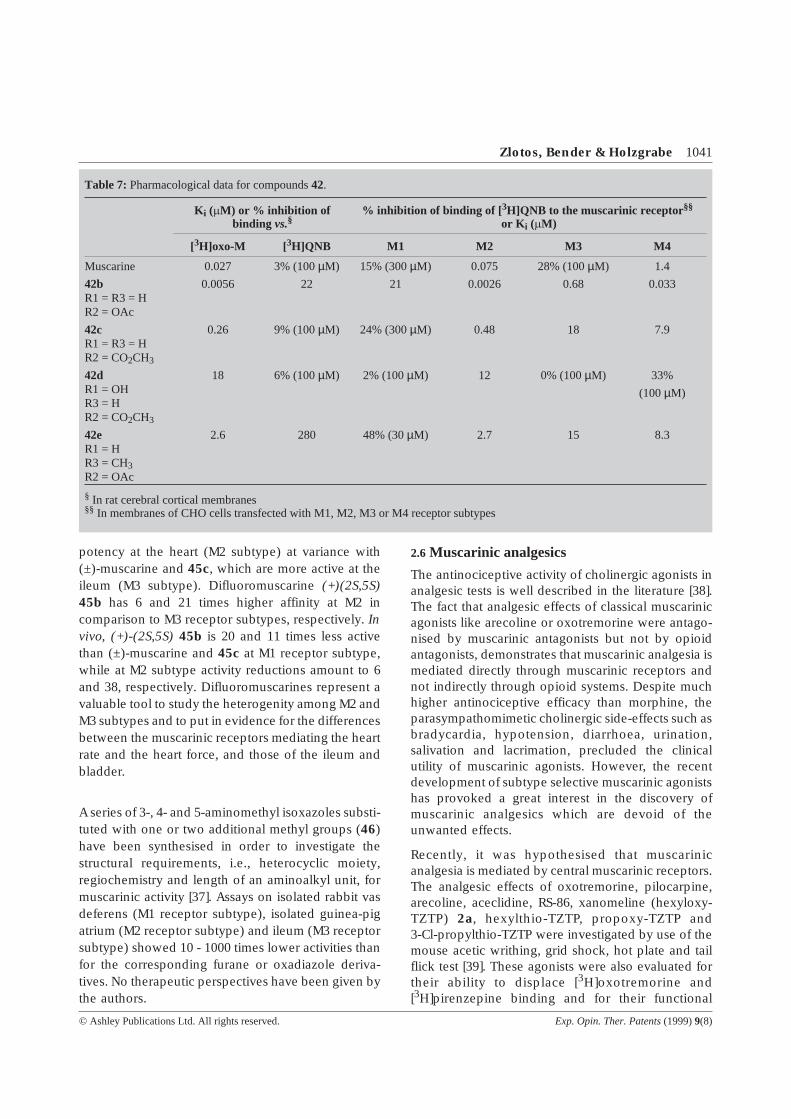
Table 7: Pharmacological data for compounds42.
Ki (mM) or % inhibition ofbinding vs.§
% inhibition of binding of [ 3H]QNB to the muscarinic receptor§§
or K i (mM)
[3H]oxo-M [ 3H]QNB M1 M2 M3 M4
Muscarine 0.027 3% (100µM) 15% (300µM) 0.075 28% (100µM) 1.4
42bR1 = R3 = HR2 = OAc
0.0056 22 21 0.0026 0.68 0.033
42cR1 = R3 = HR2 = CO2CH3
0.26 9% (100µM) 24% (300µM) 0.48 18 7.9
42dR1 = OHR3 = HR2 = CO2CH3
18 6% (100µM) 2% (100µM) 12 0% (100µM) 33%
(100µM)
42eR1 = HR3 = CH3R2 = OAc
2.6 280 48% (30µM) 2.7 15 8.3
§ In rat cerebral cortical membranes§§ In membranes of CHO cells transfected with M1, M2, M3 or M4 receptor subtypes
selectivity at M1, M2 and M3 receptors. Allcompounds produced dose-dependent antinocicep-tive effects in all of the mouse analgesia tests. Theeffects of oxotremorine in the writhing test were fullyantagonised by the muscarinic antagonist scopola-mine, but only part ia l ly antagonised bymethylscopolamine and unaffected by the opioidantagonist naltrexone. 3-Cl-Propylthio-TZTP andpropoxy-TZTP had virtually no effect at the M1receptor subtype as measured by human m1 clonedexpressed in baby hamster kidney cells or the rabbitvas deferens assay. These compounds, however, weremore potent in the analgesia test than the functionallyselective M1 agonists xanomeline and hexylthio-TZTP. Thus, functional activity at the M1 receptorsubtype does not seem to be a requirement forantinociceptive activity.
A series of alkylthio-thiadiazolyl-azabicycles (47-49)have been reported to constitute a novel class ofmuscarinic antinociceptive agents [40]. Thecompounds showed high affinity for muscarinicreceptors as evidenced by inhibi t ion of[3H]oxotremorine binding to brain homogenate (IC50= 0.49 - 26 nM). In vivo, the compounds producedantinociception in the mouse grid shock test at doseswell below those that produced salivation and tremor,with more than a 50-fold separation for somecompounds.
Nearly all the agents are as efficacious as morphine inthe mouse grid shock test with some compoundsbeing more than 100-fold more potent than morphine,e.g., 48a. Although the exo-1-azanorbornanes (48)had the highest af f ini t ies , the
endo-quinuclidinylthiadiazoles 47 showed the bestcombination of potency and selectivity. Compound(+) 47a (LY 297802) was found to have the mostdesirable pharmacological profile with respect toheart rate decrease. LY 297802 was 3- to 24-fold morepotent than morphine with similar duration of action.Its antinociceptive effect was reversed by thecentrally-acting muscarinic antagonist scopolaminebut not by the peripherally-acting muscarinic antago-nist methylscopolamine. After 6.5 days repeateddosing in mice, morphine produced markedtolerance, whereas LY 297802 produced minimal, ifany, tolerance [41]. In vitro LY 297802 had high affinityto muscarinic receptors in brain homogenates, buthad less or no affinity to several other neurotrans-mitter receptors and uptake sites. In isolated tissues,LY 297802 was an agonist with high affinity for M1receptors in the rabbit vas deferens (IC50 = 0.33 nM),but it was an antagonist at M2 receptors in guinea-pigatria (pA2 = 6.9) and at M3 receptors in guinea-pigurinary bladder (pA2 = 7.4) and a weak partial agonistin guinea-pig ileum, which contains a heterogenouspopulation of muscarinic receptors [42]. Thepresented data demonstrate that LY 297802 has thepotential to be a new analgesic drug having a newmode of action and, thus, being devoid of the typicalopioid side-effects.
With the purpose to develop novel agents for thetreatment of irritable bowel syndrome (IBS) muscar-inic analgesics have been taken into account.Recently, some 1,2,5-thiadiazole-based compounds(50) with muscarinic activity have been evaluated forboth antinociceptive activity in the mouse writhing
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1042 Muscarinic receptor agonists and antagonists
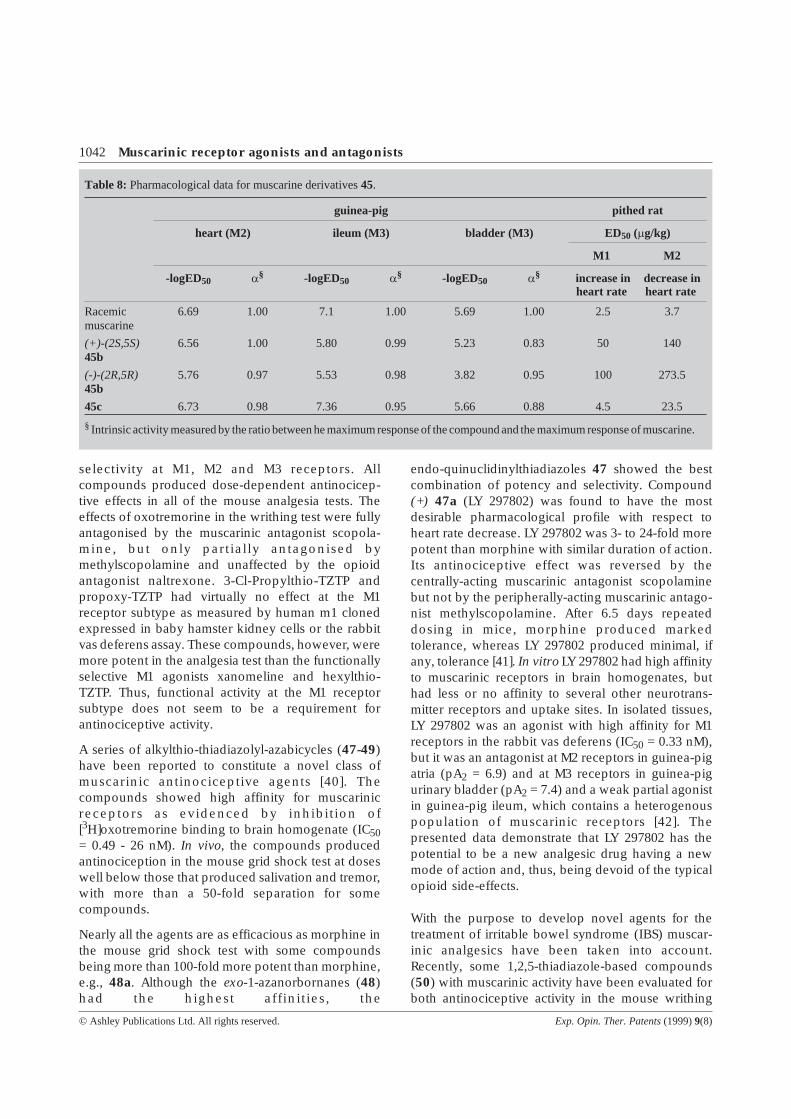
Table 8: Pharmacological data for muscarine derivatives45.
guinea-pig pithed rat
heart (M2) ileum (M3) bladder (M3) ED 50 (mg/kg)
M1 M2
-logED50 a§ -logED50 a§ -logED50 a§ increase inheart rate
decrease inheart rate
Racemicmuscarine
6.69 1.00 7.1 1.00 5.69 1.00 2.5 3.7
(+)-(2S,5S)45b
6.56 1.00 5.80 0.99 5.23 0.83 50 140
(-)-(2R,5R)45b
5.76 0.97 5.53 0.98 3.82 0.95 100 273.5
45c 6.73 0.98 7.36 0.95 5.66 0.88 4.5 23.5
§Intrinsic activity measured by the ratio between he maximum response of the compound and the maximum response of muscarine.

assay and activity in normalising spontaneous clustercontractions in ferret jejunum as a model of IBS inhumans [43]. Among the tetrahydropyridines, 50a and50b were potent analgesics, but each of thesecompounds lacked separation between effects onmotility and undesired intestinal secretory activity.Also, 51a was 10-fold more potent than 50b atinhibiting motility while being 3-fold less potent in themouse writhing test. Compound 50c, a very potentand functionally selective M1 agonist, was onlymoderately potent in the analgesic assay, which isconsistent with the fact, that analgesia is not mediatedby M1 receptors. In line with observations aboutoptimal chain length, compounds 50d and 50e withpropylphenyl and propylthiophene side-chains,respectively, were of only limited activity in both themouse writhing and ferret model. No tetrahydropy-ridyl compounds were found with both goodanalgesic potency and good separation betweeneffects on motility and intestinal secretory activity.Replacement of the tetrahydropyridine moiety withthe azabicyclo[3.2.1]octane ring and incorporation ofa trifluoro functionality at the end of the thiadiazoleside-chain afforded compounds with the desiredpharmacological profile. Compound 51 (Ki = 0.33 nM
vs. [3H]-Oxo-M in rat cortex) was found to offer anexceptional profile combining analgesic potencyalong with potency for normalisation of gastrointes-tinal motility. This suggests 51 as an excellentcandidate for evaluation as a potential treatment ofIBS.
3. Muscarinic receptor antagonists
3.1 M1 receptor subtype selective antagonists
The M1 receptor is found in high density in neuronaltissues where its activation is responsible for thepassage of a stimulus from the first to the secondneurone. Since it is undesirable to block the stimulusnerve impulse at this point of the parasympatheticnervous system, only a very few M1 antagonists arereported until now. However, pirenzepine (seeSection 3.2) is described to be a highly selectiveantagonist [44] but there are still doubts whether thesuppression of secretion of gastric juice is caused bythe M1 antagonism in the gastric mucosa [45].
All other compounds synthesised as M1 antagonistsmainly serve as pharmacological tools to characterisethe receptor subtype and the ligand binding site.
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1043
N
NS
N
SR
N
NS
N
SRN
N
SN
SR
N
N
SN
R1
R2
N
N
S
N SF
F
F
47
47a LY 297802, R = n-butyl
48
48a R = n-propyl
49
50 51
50a CH3
50b H
50c CH3
50d CH3
50e CH3
O CH3
S CH3
O CH2
S CH3
S
S
R1 R2
Figure 6: Muscarinic analgesics.

Nevertheless, there are some interesting findingsderived from the variation of well-known muscarinicantagonists. The most interesting compounds arereported in the following part.
Spirotramine 54 was derived from methoctramine(53), a potent M2 antagonist by replacing the terminal2-methoxybenzyl groups with the structural featuresof spiro-DAMP (52), which is a non-selective muscar-inic antagonist. The resulting antagonist 54 is able todiscriminate significantly between the M1 receptorand the other subtypes (pKi = 7.88, M4/M1 = 41), andhas a better affinity profile than pirenzepine [46]. Thechange of selectivity from M2 (methoctramine)towards M1 receptor (spirotramine) may be explainedby a concept wherein the tetraamine backbone isassumed to carry a ‘message’ for recognition ofmuscarinic receptors and the N-terminal groupscontain an ‘address’ which directs the preference fordifferent muscarinic subtypes.
A conformationally blocked derivative of spiro-DAMP(55) also shows high affinity for the M1 receptorsubtype. The two existing isomers 55a and 55bpresent pA2 values of 9.10 - 9.40 and their affinity
profile is comparable to that of pirenzepine [47]. Dueto the rigidity of 55 they might be useful tools foridentification of the spatial characteristics of the M1antagonist-binding site.
Upon the idea of treating organophosphate poisoningwith a drug which acts as a dual anticonvulsant andantimuscarinic agent, compounds 58 and 59 weresynthesised as hybrids of the anticonvulsantphenytoin (56) and the M1 receptor antagonist 57.The compounds show good binding affinity for themuscarinic M1 receptor (Ki values of 0.12 and 0.46µM) and also exhibit a rapid onset of anticonvulsantactivity [48].
3.2 M2 receptor subtype selective antagonists
M2 receptors are mainly located in peripheral effectororgans such as heart, smooth muscle, glands and inthe CNS. They play an important role in the regulationof the heart rate in the sinus node, and for this reason,M2 muscarinic antagonists would be useful drugs forbradycardic disorders. Another therapeutic target isthe blockade of presynaptic M2 receptors in the CNS
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1044 Muscarinic receptor agonists and antagonists
55b55a
52 spiro-DAMP 53 Methoctramine 54 Spirotramine
N
O
O
O
CH3
CH3
+
N CH3
CH3
O
OO
+
OCH3
NH
NH
2
N
O
O
O
(CH2)6
N
CH3
(CH2)4
2
N
O
O
CH3
CH3
O
+
NH
NH
O
O
C C CH
2
N(C2H
5)2N
O
O
CH
2
56 Phenytoin
57 58
C C CH
2
N(C2H
5)2
N
O
O
CH
2
C C CH
2
N(C2H
5)2N
O
CH
2
O
59
Figure 7: Muscarinic M1 antagonists and the corresponding compounds.

to increase the release of ACh and thus, improvecognitive processes [49].
The huge group of tricyclic M2 receptor antagonistswas derived from pirenzepine (60), the M1 receptorantagonist. The most basic nitrogen was taken out ofthe piperazine ring and attached via a methylenegroup to the piperidine ring to give the selective M2receptor antagonist AF-DX 116 (61) (M1/M2 ratio of6.3) [50].
Further enhancement of subtype selectivity andaffinity could be achieved by formal opening of thepiperazine ring and choosing the benzyl group as onesubstituent at the terminal nitrogen. The obtainedantagonist 62 exhibits a pKi value of 8.6 and a M3/M2ratio of 40 [51]. The compounds 63 and 64 (pKi = 9.2and 8.8; M3/M2 = 79 and 320) were developed byintroducing several substituents into the phenyl ring.Both in vivo agents act as non-competitive antagonistsand may be used as antibradycardiac drugs.
BIBN-99 (65) [52] is a potent muscarinic M2 antagonist(pKi = 7.5) with both higher affinity and selectivitythan AF-DX 116 [53]. The side-chain basic nitrogen ofAF-DX 116 is linked to the 4-position of the piperidinering and replaced by an amide group. Since thisnitrogen is no longer basic, the molecule is notprotonated at physiological pH and, thus, can pass theblood-brain barrier. Therefore, it shows goodavailability for the CNS and is therapeutically used as acognition enhancer [54,110].
Another class of M2 receptor antagonists are ‘benzy-lpiperidines’ and ‘benzylpiperazines’ (66-68), whichare also able to cross the blood-brain barrier. Thesecompounds are suggested to be useful, alone or incombination with an AChE inhibitor, for the treatmentof cognitive or neurodegenerative diseases such asAD [111-113]. The sulfinyl moiety of compound 68 isclaimed to interact with the amino acid Trp99 in thethird transmembranal helix (TMIII) of the human M2receptor, the basic piperazine nitrogen appears tointeract with Asp103 in TMIII and the cyano group ispostulated to fit in a hydrophobic pocket built by TMIand TMII [55].
3.3 M3 receptor subtype selective antagonists
M3 receptors are mainly present in glandular tissuesand smooth muscle e.g., of the intestine, bladder orbronchi. M3-selective antimuscarinic agents shouldhave therapeutic potential for the treatment of smoothmuscle contractility and tone, for example in irritable
bowel syndrome, urinary urge incontinence andasthma.
For years, drugs such as oxybutynin (69) have beenthe choice for treatment of unstable bladder but theirappliance is limited by antimuscarinic side-effects,including dry mouth, accommodation difficulties andincreased heart rate. Compounds 70 and 71 arestructurally derived from oxybutynin. They posses adiphenyl moiety and instead of the ester an amidegroup. The hydroxy group of both compounds isexchanged by different substituents containingheterocycles. Darifenacin (70) has high affinity (pKi =9.12) and selectivity (9- to 74-fold) for the muscarinicM3 receptor and also exhibits functional tissueselectivity for intestinal smooth muscle over thesalivary gland. In therapy, it is well used for thetreatment of irritable bowel syndrome as againsturinary incontinence. [56-58]. From a series ofstructural related compounds, it was claimed thatKRP-197 (71) can be divided into two regions. One,consisting of the spacer methylene group and thediphenyl moiety, should be responsible for theantimuscarinic activity. The other region, composedof the carbamoyl moiety and the imidazole ringshould have influence on both, antimuscarinic activityand subtype selectivity [59]. KRP-197 shows a Kb valueof 0.32 nM, a M2/M3 ratio of 13 and is a drug candidatefor the treatment of urinary incontinence associatedwith bladder muscle instability [114].
Some structural related compounds (72-74) [115-117]were found to be also more selective to smoothmuscle muscarine receptors than to cardiac ones.These agents are stated to be of use in the treatment ofirritable bowel syndrome.
The urethane group was chosen as a novel linkerbetween the protonated nitrogen at one end of themolecule and the bulky, hydrophobic moiety at theother end in YM-46303 (75) and YM-58790 (76).YM-46303 (75) shows high affinity and selectivity forthe muscarinic M3 receptor with a Ki value of 0.39 nMand a M2/M3 ratio of 15 [60]. Although YM-58790 (76)exhibits a Ki value of 15 nM only, it shows moreselective antagonism between urinary bladdercontraction and salivary secretion in vitro thanoxybutynin (69). It has low potential for penetrationof the blood-brain barrier and is, therefore, suggestedto act as a peripherally active selective M3 antagonistin vivo [61].
Tolterodine (Detrusitol™, 77) is a new potent andcompetitive muscarinic receptor antagonist for the
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1045

© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1046 Muscarinic receptor agonists and antagonists
N
NH
N
N
N
O
O
CH3
N
NH
N
N
O
O
N(C2H
5)2
NH
N
O
N
O
N
N
O
R1
H5C
2
R2
NH
N
O
N
O
N
N
O
H5C
2
CH3
NH
N
O
N
O
CI
N
N
O
CH3CH
3C
2H
5
CH3
60 Pirenzenpine 61 AF-DX 116
62
65 BIBN-99
63 YM-47244
64 YM-43571
R1 = -CH3
R2 =
R1 = -C2H5
R2 =
N N iPr
N N C2H
5
O
O
S
O
N
N
CH3
O
OOO
O
S
N
N
OO
C
N
S
N
N
O
C
N
CH3O
66 67
68 SCH 57790
Figure 8: M2 subtype antagonists.

© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1047
OH
O
ON(C
2H5)2 N
O
NH2
O
N
N
O
NH2
O
NH2
NN
N
N
O
NH2
CH3
72 73 74
69 Oxybutynin 70 Darifenacin 71 KRP-197
N
N
C
N* 2HCl
NH
O
NO
H
NH
O
O
N
NH
CH3
OH CH3
N CH3
CH3
CH3
CH3
H
ON O
O
OHOH
O
NO
N
N
O
H
OH
O
NO
S
O
CH3
O
NH
N
OHH
FF
S
75 YM-46303 76 YM-58790 77 Tolterodine(DetrusitolTM)
78 Zamifenacin 79 (2S,3'R) 3-quinuclidinyl-tropate 80
81 Revatropate 82
Figure 9: M3 receptor subtype antagonists.

treatment of urge incontinence and other symptomsof unstable bladder. It is remarkable that its antimus-carinic activity is non-selective in vitro (Ki (m1 - m5) =3.0, 3.8, 3.4, 5.0 and 3.4 nM), but it exhibits a favour-able tissue-selectivity for the urinary bladder oversalivary gland in vivo [62] . Tolterodine iswel l - tolerated and shows an optimalefficacy/side-effect profile [63].
Another agent containing a diphenyl moiety iszamifenacin (78), which antagonises selectivelymuscarinic M3 receptors over muscarinic M2receptors in vitro and in vivo (pA2 = 9.31). In humans,it has been found to inhibit small bowel and colonicmotility with little or no side-effects [64]. Compound79 exhibits a higher affinity (pA2 = 10.33) but lessselectivity than zamifenacin for the muscarinic M3receptor, but could be of use in the treatment ofdisorders of the smooth muscle [65].
To make the structure more rigid, a lactame ring wascondensed at one of the phenyl rings of the diphenylmoiety. The obtained class of indolone compounds(80) is stated to be muscarinic M3 antagonists andmight be useful for the prevention and treatment ofe.g., irritable bowel syndrome and urinary dysfunc-tion [118].
In the field of antimuscarinic bronchodilators twocompounds can be found. Revatropate (81) is amuscarinic antagonist with marked selectivity for theM1 and M3 receptor subtype in vitro. In vivo, the M3receptor-mediated bronchoconstrictor response wasshown to be strongly antagonised with iv. doses ofrevatropate [66].
Fluorinated 1,4-disubstituted piperidine derivatives,such as compound 82, are claimed to have M3 antago-nist activity and could be used for the treatment ofasthma and airway obstructions [119].
3.4 M4 receptor subtype selective antagonists
Muscarinic receptors are found in the brain, with thehighest level of M4 receptor subtype occurring in thestriatum. These receptors are believed to be involvedin the control of motor function, and therefore,selective muscarinic M4 antagonists may be useful inthe treatment of movement disorders such as PD. Thecommonly known ‘M4’ antagonist artane (83) hasbeen used in therapy to suppress tremor and relieverigidity associated with the early stages of PD. Whileits selectivity for the M4 receptor subtype over the M1subtype is not high enough, unfavourable cognitiveside-effects can be observed during the therapy. Anagent with less affinity than artane but higherselectivity for the M4 receptor subtype is shown inFigure 10. Compound 84 presents a benzoxazineisoquinolone skeleton and has been stated as the mostselective synthetic M4 antagonist known to date [67].Replacement of the substituents R1 - R3 in any wayleads to an overall decrease of affinity for the muscar-inic M4 receptor and to a loss of selectivity for allmuscarinic receptor subtypes.
4. New concepts: allosteric modulation ofthe ligand binding to the receptor protein
Allosteric modulators of the muscarinic ACh receptorswould act at a site apart from the common ligandbinding site of the receptor protein. Thus, thesecompounds affect the ligand association and dissocia-tion. Logically, the ligand binding can be elevated,reduced or remain unchanged, depending on the typeof allosteric modulator, the type of ligand and thesubtype of the receptor. Whereas the phenomenon ofallosteric modulation is well known for the ligand-gated ion channel GABAA-receptor and therapeuti-cally used by the benzodiazepines, the allostericmodulation of G-protein coupled receptors, such asthe muscarinic receptors, is at present a theoretical
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1048 Muscarinic receptor agonists and antagonists
N
O
NH
R3
R1
R2
83 Artane
84
R1 = CO2CH2CH
R2 = CH3
R3 = OCH3
OH
N
*HCI
Figure 10: M4 receptor subtype antagonists.

matter. However, the concept implicates someadvantages.
4.1 Use in combination with antagonists
Allosteric modulators might have the potential forgreater subtype selectivity of the antagonistic action incomparison with the use of pure antagonists (lackingselectivity). Allosteric modulators prevent the antago-nists from an overdose because the effect is saturable.Taken together, in combination with antagonists, suchas N-methylscopolamine or atropine, the treatment oforganophosphate poisoning can take advantage ofthe effect [68]. Unfortunately, most compounds whichare reported to be allosteric modulators, e.g.,alcuronium, tubocurarine, strychnine or gallamine,are dominated by another strong pharmacologicalaction [69-71]. Thus, attempts are under progress tofind pure modulators. Alkane-bis(ammonium)compounds linked to substituted phthalimidomoieties at both ends of the molecule were found tohave the highest allosteric potency [72-74] and are,therefore, potential candidates for clinical use.
4.2 Use in combination with agonists
The enhancement of the ACh action [75] bydiminishing the rate of dissociation of the agonist
might be beneficial in the treatment of dementia,namely AD, and pain.
Taken together, the theoretical concept of allostericmodulation of the ligand binding offers the perspec-tive for a novel and subtype-specific means offine-tuning the ligand-binding properties of muscar-inic receptors.
5. Expert opinion
From the research of the last years, it can be stated thata lot of selective agonists and antagonist with highaffinity to either receptors subtype have beendeveloped. In the case of the agonists, compoundsdeveloped as M1 agonists attract interest for thetreatment of AD. Xanomeline and correspondingcompounds turned out to improve the psychiatricsymptoms of the disease whilst effects on cognitionenhancement have not been robust [76]. Even thoughthe clinical trials of these compounds do not givecause for optimism, a lot of research work is underprogress. It remains to be seen whether a highlypotent M1 agonist can be found which will besuperior to the AChE inhibitors which are already inclinical practice. However, much work remains to bedone to find the optimal therapy of AD.
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1049
2Br-
W84 (lead compound)
Dimethyl - W84 (radioligand)
Structural variation of the lead compound W84
2Br-
NN+
N+N
O
O
CH3
CH3
CH3
CH3 O
O
2Br-
NN+
N+N
O
O
CH3
CH3 CH
3
CH3 O
OCH3 CH
3
RR
NN+
N+N
O
O
CH3 CH
3 O
O
R2R2
R1R1
Figure 11: Allosteric modulators of the muscarinic acetylcholine receptor.

Interestingly, substances developed as M1 agonistswere found to have a strong analgesic potency.Beside the nicotinic ligands derived from epibatidine,e.g., ABT-594, which exhibit higher analgesic effectsthan morphine, the muscarinic agonists seems to bethe most promising perspective to find strong analge-sics without the opioid-like side-effects.
With respect to the antagonists, it can be stated that alot of highly subtype-selective compounds werefound. However, especially in the case of M1 and M2antagonists the compounds are only of theoreticalinterest. M3 antagonists become useful for thetreatment of urinary incontinence and bladderinstability. Interestingly, the promising drugtolterodine, which is already on the market, is anon-subtype-selective antagonist. At this point thequestion raised whether the subtype-selectivity is ofsuch high importance as often discussed. Theoutstanding feature of tolterodine is the high organspecificity, which causes the good efficacy/side-effectrelation. Obviously, the receptor subtype selectivityon its own does not lead to an optimal drug.
Bibliography
1. MUTSCHLER E, MOSER U, WESS J, LAMBRECHT G:Muscarinic receptor subtypes - pharmacological,molecular biological and therapeutical aspects. Pharm.Ztg. Wiss. (1998) 6:3-11.
2. ALEXANDER SPH, PETERS JA: Receptor and ion channelnomenclature supplement. Trends Pharmacol. Sci.(1998) 19:3-6.
3. MOLTZEN EK, BJORNHOLM B: Medicinal chemistry ofmuscarinic agonists: development since 1990. DrugsFuture (1995) 20(1):37-54.
4. Xanomeline. Drugs Future (1996) 21(9):911-916.
5. Xanomeline. Drugs Future (1998) 23(9):1050-1051.
6. KELLY J S: Alzheimer’s disease: the tacrine legacy.Trends Pharmacol. Sci. (1999) 20:128.
7. SAUERBERG P, JEPPESEN L, OLESEN PH et al.: Identifica-tion of side chains on 1,2,5-thiadiazole-azacyclesoptimal for muscarinic M1 receptor activation. Bioorg.Med. Chem. Lett. (1998) 8:2897-2902.
8. KIESEWETTER DO, LEE JT, LANG L, PARK SG, PAIK CH,ECKELMANN WC: Preparation of 18F-labeled muscar-inic agonist with M2 selectivity. J. Med. Chem. (1995)38:5-8.
9. LU-25-109T. Drugs Future (1998) 23(8):843-846.
10. PIKE BR, HAMM RJ: Activating the posttraumaticcholinergic system for the treatment of cognitiveimpairment following traumatic brain injury.Pharmacol. Biochem. Behav. (1997) 57:785-791.
11. PIKE BR, HAMM RJ: Chronic administration of a partialmuscarinic M1 receptor agonist attenuates decreasesin forebrain choline acetyltransferase immunoreac-tivity following experimental brain trauma. Exp.Neurol. (1997) 17:30.
12. BRÄUNER-OSBORNE H, EBERT B, BRANN MR et al.:Annulated heterocyclic bioisosters of norarecoline.synthesis and molecular pharmacology at fiverecombinant human muscarinic acetylcholinereceptors. J. Med. Chem. (1995) 38:2188-2195.
13. WARD JS, MERRITT L, CALLIGARO DO et al.: Functionallyselective M1 muscarinic agonists. 3. Side chains andazacycles contributing to functional muscarinicselectivity among pyrazinylazacycles. J. Med. Chem.(1995) 38:3469-3481.
14. Milameline hydrochloride. Drugs Future (1996)21(11):1124-1128.
15. Milameline hydrochloride. Drugs Future (1997)22(11):1289-1290.
16. WAL-2014-FU. Drugs Future (1994) 19(1):35-37.
17. WAL-2014-FU..Drugs Future (1998) 23(1):113.
18. WALLAND A, PIEPER MP: Central activation of thesympathetic nervous system including the adrenals inanaesthetized guinea pigs by the muscarinic agonisttalsaclidine. Naunyn-Schmiedeberg’s Arch Pharmacol.(1998) 357:426-430.
19. Sabcomeline hydrochloride. Drugs Future (1998)23(1):41-44.
20. BROMIDGE SM, BROWN F, CASSIDY F et al.: Design of[R-(Z)-a-(Methoxyimino)-1-aza-bicyclo[2.2.2]octane-3-acetonitrile (SB 202026), a functionally selectiveaza-bicyclic M1 agonist incorporating the N-methoxyimidoyl nitrile group as a novel ester bioisostere. J.Med. Chem. (1997) 40:4265-4280.
21. LOUDON JM, BROMIDGE SM, BROWN F et al.: SB 202026:a novel muscarinic partial agonist with functionalselectivity for M1 receptors. J. Pharmacol. Exp. Ther.(1997) 283:1059-1068.
22. MCPHERSON DW, LAMBERT CR JAHN K, SOOD V et al.:Resolution and in vitro and initial in vivo evaluation ofisomers of iodine-125-labeled 1-azabicyclo[2.2.2]-oct-3-yl-a -hydroxy-a -(1-iodo-1-propen-3-yl)-a-phenylacetate: a high-affinity ligand for the muscar-inic receptor. J. Med. Chem. (1995) 38:3908-3917.
23. BYMASTER FP, SHANNON HE, RASMUSSEN K et al.:Unexpected antipsychotic-like activity with themuscarinic receptor ligand (5R,6R)6-(3-propyl-thio-1,2,5-thiadiazol-4-yl)-1-azabicyclo[3.2.1]octane.Eur. J. Pharmacol. (1998) 356:109-119.
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1050 Muscarinic receptor agonists and antagonists

24. TECLE H, JEAN JC, AUGELLI-SZAFRAN C et al.:Z-(±)-1-Azabicyclo[2.2.1]heptane-3-one, O-(3-aryl-2-propanyl)-oximes as potential m1-subtype selectivemuscarinic agonists. Bioorg. Med. Chem. Lett. (1995)5:631-636.
25. YM-796. Drugs Future (1997) 22(2):144-147.
26. TSUKAMATO S, FUJII M, YASUNAGA T et al.: Synthesisand structure-activity studies of a series of 1-oxa-8-azaspiro[4.5]-decanes as M1 muscarinic agonists.Chem. Pharm. Bull. (1995) 43(5):842-852.
27. TSUKAMATO S, NAGAOKA H, IGARASHI S, WANIBUCHIF, HIDAKA K, TAMURA T: Synthesis and structure-activity studies of a series of1-oxa-2,8-diazaspiro[4.5]-decan-3-ones and relatedcompounds as M1 muscarinic agonists. Chem. Pharm.Bull. (1995) 43(9):1523-1528.
28. WU ESC, GRIFFITH RC, LOCH III JT et al.: In vitro muscar-inic activity of spiromuscarones and related analogs. J.Med. Chem. (1995) 38:1558-1570.
29. WU ESC, MACK RA, KOVER A et al.: Synthesis andbiological activity of enantiomers of a conformation-ally restricted muscarone analog. Bioorg. Med. Chem.Lett. (1995) 5(16):1813-1818.
30. WU ESC, KOVER A, LOCH III JT, ROSENBERG LP, SEMUSSF, VERHOEST PR: Acylhydrazones as M1/M3 selectivemuscarinic agonists. Bioorg. Med. Chem. Lett. (1996)6(21):2525-2530.
31. SUZUKI T, USUI T, OKA M, SUZUKI T, KATAOKA T:Synthesis and muscarinic activity of a series ofquinolones and naphthalenes with a1-azabicyclo[3.3.0]octane moiety. Chem. Pharm. Bull.(1998) 46(8):1265-1273.
32. SUZUKI T, OKA M, MAEDA K, FURUSAWA K, MITANI T,KATAOKA T: N-[2-(1-Azabicyclo[3.3.0]octan-5-yl)ethyl]-2-nitroaniline, a potent muscarinic agonist.Chem. Pharm. Bull. (1997) 45(7):1218-1220.
33. MESSER WS, Jr., ABUH YF, LIU Y, PERIYASAMY S, NGURDO, EDGAR MAN: Synthesis and biological characteri-zation of 1,4,5,6-tetrahydropyrimidine and2-amino-3,4,5,6-tetrahydropyridine derivatives asselective m1-agonists. J. Med. Chem. (1997) 40:1230-1246.
34. PEI XF, GUPTA TH, BADIO B, PADGETT WL, DALY JW:6b-Acetoxynortropane: a potent muscarinic agonistwith apparent selectivity towards M2-receptors. J. Med.Chem. (1998) 41:2047-2055.
35. CONTI P, DALLANOCE C, DE AMICI M, DE MICHELI C,EBERT B: Synthesis and binding affinity of newmuscarinic ligands structurally related tooxotremorine. Bioorg. Med. Chem. Lett. (1997)7(8):1033-1036.
36. ANGELI P, CANTALAMESSA F, CAVAGNA R et al.:Synthesis and pharmacological characterization ofenantiomerically pure muscarinic agonists: difluoro-muscarines. J. Med. Chem. (1997) 40:1099-1103.
37. DANNHARDT G, KIEFER W, LAMBRECHT G et al.: Regioi-someric 3-, 4- and 5-aminomethylisoxazoles:
synthesis and muscarinic activity. Eur. J. Med. Chem.(1995) 30:839-850.
38. HARTVIG P, GILLBERG PG, GORDH JR T, POST C:Cholinergic mechanism in pain and analgesia. TrendsPharmacol. Sci. (1989) 10(Suppl.):75-79.
39. SHEARDOWN MJ, SHANNON HE, SWEDBERG MDB et al.:M1 Receptor agonist activity is not a requirement formuscarinic antinociception. J. Pharmacol. Exp. Ther.(1997) 281:868-875.
40. OLESEN PH, SAUERBERG P, TREPPENDAHL S et al.:3-(3-Alkylthio-1,2,5-thiadiazol-4-yl)-1-azabicycles.Structure-activity relationships for antinociceptionmediated by central muscarinic receptors. Eur. J. Med.Chem. (1996) 31:221-230.
41. SWEDBERG MDB, SHEARDOWN MJ, SAUERBERG P et al.:Butylthio[2.2.2]( NNC 11-1053/LY297802): an orallyactive muscarinic agonist analgesic. J. Pharmacol. Exp.Ther. (1997) 281:876-883.
42. SHANNON HE, SHEARDOWN MJ, BYMASTER FP et al.:Pharmacology of butylthio[2.2.2]( NNC11-1053/LY297802): a novel analgesic with mixedmuscarinic receptor agonist and antagonist activity. J.Pharmacol. Exp. Ther. (1997) 281:884-894.
43. MITCH CH, BROWN TJ, BYMASTER FP et al.: Muscarinicanalgesics with potent and selective effects on thegastrointestinal tract: potential application for thetreatment of irritable bowel syndrome. J. Med. Chem.(1997) 40:538-546.
44. HAMMER R; BERRIE CP; BIRDSALL NJM; BURGEN ASW,HULME EC: Pirenzepine distinguishes betweendifferent subclasses of muscarinic receptors. Nature(1980) 283:90-92.
45. KUSCHINSKY G, LÜLLMANN H, MOHR K: Kurzeslehrbuch der pharmakologie und toxikologie. GeorgThieme Verlag Stuttgart New York (1993) 13.Aufl.:83.
46. MELCHIORRE C, MINARINI A, SPAMPINATO S, TUMIATTIV: Design, synthesis and biological activity of sometetraamines related to methoctramine and 4-DAMP.Bioorg. Med. Chem. Lett. (1995) 5:785-790.
47. TUMIATTI V, SPAMPINATO S, RECANATINI M et al.:Design, synthesis and biological activity of some4-DAMP-related compounds. Bioorg. Med. Chem. Lett.(1995) 5:2325-2330.
48. HUDKINS RL, DEHAVEN-HUDKINS DL, DOUKAS P:Design of dual acting anticonvulsant-antimuscarinicsuccinimide and hydantoine derivatives. Bioorg. Med.Chem. Lett. (1997) 7:979-984.
49. DÖRJE F, WESS J, LAMBRECHT G, TACKE R, MUTSCHLERE, BRAUN MR: Antagonist binding profiles of fivecloned human muscarinic receptor subtypes. J. Pharm.Exp. Ther. (1991) 250:727-733.
50. ENGEL W, TRUMMLITZ G, EBERLEIN WG et al.:Condensed diazepinones and their pharmaceuticaluses. Chem. Abstr. (1986) 104:129934c.
51. WATANABE T, KINOYAMA I, KAKEFUDA A et al.:Synthesis of novel succinamide derivatives having the
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1051

5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one skeleton as potent and selective M2 muscarinicantagonists. I. Chem. Pharm. Bull. (1997) 45:996-1007.
52. BIBN-99. Drugs Future (1995) 20:157.
53. DOODS HN, QUIRION R, MIHM G et al.: Therapeuticpotential of CNS-active M2 antagonists: novelstructures and pharmacology. Life Sci. (1993)52:497-503.
54. BIBN-99. Drugs Future (1999) 24:197.
55. LACHOWICZ JE, LOWE D, DUFFY RA et al.: SCH 57790: Anovel M2 receptor selective antagonist. Life Sci. (1999)64:535-539.
56. Darifenacin. Drugs Future (1998) 23:1238.
57. Darifenacin. Drugs Future (1998) 23:1276.
58. WALLIS RM, NAPIER CM: Muscarinic antagonists indevelopment for disorders of smooth musclefunction. Life Sci. (1999) 64:395-401.
59. MIYACHI H, KIYOTA H, SEGAWA M: Novel imidazolederivatives with subtype-selective antimuscarinicactivity (2). Bioorg. Med. Chem. Lett. (1998) 8:2163-2168.
60. NAITO R, TAKEUCHI M, MORIHIRA K et al.: Synthesis andantimuscarinic properties of biphenylcarbamatederivatives. Chem. Pharm. Bull. (1998) 46:1286-1294.
61. NAITO R, TAKEUCHI M, MORIHIRA K et al.: Synthesis andantimuscarinic properties of 4-Piperidyl benzhydryl-carbamate derivatives. Chem. Pharm. Bull. (1998)46:1274-1285.
62. Tolterodine. Drugs Future (1997) 22:733-737.
63. NILVEBRANT L, HALLEN B, LARSSON G: Tolterodine - anew bladder selective muscarinic receptor antagonist:preclinical pharmacological and clinical data. Life Sci.(1997) 60:1129-1136.
64. COSSY J, DUMAS C, GOMEZ PARDO D: A short andefficient synthesis of zamifenacin a muscarinic M3
receptor antagonist. Bioorg Med. Chem. Lett. (1997)7:1343-1344.
65. GHELARDINI C, BARTOLINI A, GALEOTTI N, BELLUCCI C,DIE S, GUALTIERI F: In vitro characterization of a novel,potent and selective M3 antagonist. Life Sci. (1997)61:1217-1226.
66. Revatropate. Drugs Future (1997) 22:135-137.
67. AUGELLI-SZAFRAN CE, JAEN JC, MORELAND DW,NELSON CB, PENVOSE-YI JR, SCHWARZ RD: Identifica-tion and characterization of m4 selective muscarinicantagonists. Bioorg. Med. Chem. Lett. (1998) 8:1991-1996.
68. HOLZGRABE U, MOHR K: Allosteric modulation ofantagonist binding to muscarinic receptors. Drug Disc.Today (1998) 3:214-222.
69. CHRISTOPOULOS A, LANZAFAME A; MITCHELSON F:Allosteric interactions at muscarinic cholinoceptors.Clin. Exp. Pharmacol. Physiol. (1998) 25:185-194.
70. TUCEK S, PROSKA J: Allosteric modulation of muscar-inic acetylcholine receptors. Trends Pharmacol. Sci.(1995) 16:205-212.
71. TRÄNKLE C, KOSTENIS E, BURGMER U, MOHR K: Searchfor lead structures to develop new allosteric modula-tors of muscarinic receptors. J. Pharmacol. Exp. Ther.(1996) 279:926-933.
72. TRÄNKLE C, MIES-KLOMFASS E, BOTERO CID HM,HOLZGRABE U, MOHR K: Identification of a [3H]ligandfor the common allosteric site of muscarinic acetyl-choline M2 receptors. Mol. Pharmacol. (1998) 54:139-145.
73. NASSIF-MAKKI T, TRÄNKLE C, ZLOTOS D et al.: Bisquater-nary ligands of the common allosteric site of M2acetylcholine receptors: optimization of the distancesbetween the pharmacophoric elements. J. Med. Chem.(1999) 42:849-858.
74. BENDER W, STAUDT M, TRÄNKLE C, MOHR K,HOLZGRABE U: Probing the size of a hydrophobicbinding pocket within the allosteric site of muscarinicacetylcholine M2-receptors: unilateral variation ofalkane bisammonium-type ligands. Life Sci. (In Press).
75. BIRDSALL N J, FARRIES T, GHARAGOZLOO P et al.:Selective allosteric enhancement of the binding andactions of acetylcholine at muscarinic receptorsubtypes. Life Sci. (1997) 60:1047-1052.
76. KELLY JS: Alzheimer’s disease: the tacrine legacy.Trends Pharmacol. Sci. (1999) 20:127-129.
Patents
101. WARNER-LAMBERT CO.: WO9830243 (1998).
102. SMITHKLINE BEECHAM PLC: WO9804248 (1998).
103. ELI LILLY & CO.: EP-709381-A (1996).
104. AMERICAN HOME PRODUCTS CO.: US5512574 (1996).
105. AMERICAN HOME PRODUCTS CO.: US5773458 (1998).
106. AMERICAN HOME PRODUCTS CO.: US5750522 (1998).
107. AMERICAN HOME PRODUCTS CO.: US5756501 (1998).
108. NOVO NORDISK AS.: WO9616065 (1996).
109. AMERICAN HOME PRODUCTS CO.: US5712270 (1998).
110. DR KARL THOMAE GMBH: US5610155 (1997).
111. SCHERING CORP.: WO9806697 (1998).
112. SCHERING CORP.: WO9626196 (1996).
113. SCHERING CORP.: WO9801425 (1998).
114. KYORIN SEIYAKU KK: JP7291936 (1995).
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
1052 Muscarinic receptor agonists and antagonists

115. KYORIN SEIYAKU KK: JP7291945 (1995).
116. KYORIN SEIYAKU KK: JP7291944 (1995).
117. KYORIN SEIYAKU KK: JP920759 (1997).
118. TOKYO TANABE CO. LTD: WO9854167 (1998).
119. BANYU PHARM. CO. LTD.: WO9805641 (1998).
Darius P Zlotos, Wiebke Bender & Ulrike Holzgrabe†
† Author for correspondenceInstitut für Pharmazie und Lebensmittelchemie, BayerischeJulius-Maximilians-Universität, Am Hubland, 97074 Würzburg,GermanyTel.: +49 931 888 5460; Fax: +49 931 888 5494;Email: [email protected]
© Ashley Publications Ltd. All rights reserved. Exp. Opin. Ther. Patents(1999)9(8)
Zlotos, Bender & Holzgrabe 1053



![Molecular Analysis of the Histamine H 3-Receptor · III 2.4.2 [³H]JNJ-7753707 and [35 S]GTP γS binding: 45Quantitative analysis of receptor-to-G protein stoichiometries 2.4.3 Steady-state](https://static.fdokument.com/doc/165x107/5fd46fbb0be1866eec555122/molecular-analysis-of-the-histamine-h-3-receptor-iii-242-hjnj-7753707-and.jpg)