
opus4.kobv.de...Teile der Arbeit sind bereits veröffentlicht oder zur Veröffentlichung...
200
Die stereoselektive Aldolreaktion in Biotransformationen und chemoenzymatischen Eintopfsynthesen Der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Dipl.-Chem. Katrin Baer aus Nürnberg
Transcript of opus4.kobv.de...Teile der Arbeit sind bereits veröffentlicht oder zur Veröffentlichung...

Die stereoselektive Aldolreaktion in
Biotransformationen und chemoenzymatischen
Eintopfsynthesen
Der Naturwissenschaftlichen Fakultät der
Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. rer. nat.
vorgelegt von
Dipl.-Chem. Katrin Baer
aus Nürnberg

Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät der Friedrich-
Alexander-Universität Erlangen-Nürnberg.
Tag der mündlichen Prüfung: 27.09.2011
Vorsitzender der Promotionskommission: Prof. Dr. Rainer Fink
Erstberichterstatter: Prof. Dr. Harald Gröger
Zweitberichterstatter: Prof. Dr. Jürgen Schatz

Die vorliegende Arbeit wurde am Institut für Organische Chemie des Departments Chemie
und Pharmazie der Friedrich-Alexander-Universität Erlangen-Nürnberg unter Leitung von
Prof. Dr. Harald Gröger in der Zeit von Mai 2008 bis Juni 2011 erstellt.


Teile der Arbeit sind bereits veröffentlicht oder zur Veröffentlichung eingereicht:
K. Baer, M. Kraußer, E. Burda, W. Hummel, A. Berkessel, H. Gröger, „Sequentielle und
modulare Synthese von chiralen 1,3-Diolen mit zwei Stereozentren: Zugang zu allen vier
Stereoisomeren durch Kombination von Organo- und Biokatalyse“ Angew. Chem. 2009, 121,
9519-9522; Angew. Chem. Int. Ed. 2009, 48, 9355-9358.
K. Baer, N. Dückers, W. Hummel, H. Gröger, „Expanding the Application Range of Aldolases:
Novel Asymmetric Syntheses of α-Methylated β-Hydroxy α-Amino Acids and β-Amino
Alcohols” ChemCatChem 2010, 2, 939-942.
N. Dückers, K. Baer, S. Simon, H. Gröger, W. Hummel, „Threonine aldolases - screening,
properties and applications in the synthesis of non-proteinogenic β-hydroxy-α-amino acids”
Appl. Microbiol. Biotechnol. 2010, 88, 409-424.
G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel, H. Gröger, „Steuerung von kinetisch
versus thermodynamisch kontrollierter Organokatalyse und Anwendung in der
chemoenzymatischen Synthese“ Angew. Chem. 2011, 123, 8092-8095; Angew. Chem. Int. Ed.
2011, 50, 7944-7947.
K. Baer, N. Dückers, T. Rosenbaum, C. Leggewie, S. Simon, M. Kraußer, S. Oßwald, W.
Hummel, H. Gröger, „Towards Efficient L-Threonine Aldolase-Catalyzed Enantio- and
Diastereoselective Aldol Reactions of Glycine with Substituted Benzaldehydes: Biocatalyst
Production and Process Development” Tetrahedron: Asymmetry 2011, 22, 925-928.
Vortrag:
K. Baer, M. Kraußer, E. Burda, W. Hummel, H. Gröger, „Synthesis of chiral 1,3-diols with two
stereogenic centres by using organo- and biocatalysis” International Symposium on Relations
between Homogeneous and Heterogeneous Catalysis (ISHHC XIV), 13.-18. September 2009,
Stockholm, Schweden.

Poster:
K. Baer, S. Simon N. Dückers, C. Leggewie, T. Rosenbaum, W. Hummel, S. Oßwald, H. Gröger,
„Efficient L-Threonine Aldolase-Catalyzed Enantio- and Diastereoselective Aldol Reactions
with Benzaldehyde Derivatives” 3rd EuCheMS Chemistry Congress, 29. August-02. September
2010, Nürnberg, Deutschland.

Danksagung:
Mein ganz besonderer Dank gilt zuerst Herrn Prof. Dr. Harald Gröger für die intensive und
kompetente Betreuung während meiner Promotion. Seine große fachliche Unterstützung
war für die Entstehung dieser Arbeit und meine Entwicklung in dieser Zeit besonders wichtig.
Außerdem möchte ich unseren Kooperationspartnern im Rahmen des DBU-Projekts danken.
Vor allem danke ich Herrn Prof. Dr. Werner Hummel vom Institut für Molekulare
Enzymtechnologie der Heinrich-Heine-Universität Düsseldorf und evocatal für die
Bereitstellung der Enzyme. Mein besonderer Dank gilt hier vor allem Nina Dückers die zur
gleichen Zeit ihre Promotion bei evocatal anfertigte und für die Herstellung der Rohextrakte
zur Verfügung stand und mich unterstützte.
Mein Dank gilt allen derzeitigen und ehemaligen Labor- und Arbeitskollegen, Sabine Simon,
Dr. Marina Krausser, Dr. Maria Alfaro Blasco, Jürgen Wittmann, Philipp Böhm, Giuseppe
Rulli, Sonja Borchert, Katharina Tenbrink, Svenja Staudt, Tina Ress, Carolin Giese, Richard
Metzner, Oliver Pham, Xenia Kostrov, Dr. Markus Weiß, Dr. Friedrich Dietz, Dr. Edyta Burda,
Tobias Huber, Alexander Latza, Irene Keller, Dr. Harald Maid, Dr. Stefan Huber und Ingo
Schnapperelle für die große Hilfsbereitschaft und Unterstützung.
Besonders möchte ich mich bei Dr. Marina Kraußer, Sabine Simon und Giuseppe Rulli
bedanken, mit denen ich gemeinsame Projekte bearbeitet habe.
Außerdem möchte ich mich ganz herzlich bei den Angestellten des Instituts der Organischen
Chemie für die Unterstützung im Laboralltag und im Verwaltungsbereich bedanken. Ohne
diesen Beitrag wäre meine Dissertation nur schwer zu realisieren gewesen. Auch meinen
ehemaligen Praktikanten sei an dieser Stelle für die Durchführung einiger Synthesen
gedankt.
Mein ganz besonderer Dank gilt meiner Familie und meinen Freuden. Ich danke speziell
meinen Eltern für ihre immerwährende Unterstützung. Außerdem danke ich Christina für die
sehr gute Freundschaft, die uns seit unserer Studienzeit verbindet. Zum Abschluss möchte
ich noch meinem Freund Philipp von ganzem Herzen für den Beistand und für ein offenes
Ohr während meiner Promotionszeit danken.


INHALTSVERZEICHNIS
I
I. Inhaltsverzeichnis
I. Inhaltsverzeichnis ............................................................................................... I
II. Abbildungsverzeichnis ...................................................................................... IX
III. Tabellenverzeichnis ........................................................................................ XIV
IV. Abkürzungsverzeichnis ................................................................................... XVI
1. Einleitung .......................................................................................................... 1
2. Motivation und Zielsetzung ............................................................................... 5
2.1 Synthese von 1,3-Diolen ........................................................................................... 6
2.2 Synthese von β-Hydroxy-α-aminosäuren ................................................................. 7
2.3 Synthese von Epoxiden aus β-Hydroxy-α-aminosäuren ........................................... 8
3. Kombination einer organokatalytischen Aldolreaktion und einer enzymatischen
Reduktion ......................................................................................................... 9
3.1 Einleitung .................................................................................................................. 9
3.2 Stand der Wissenschaft .......................................................................................... 10
3.2.1 Synthesestrategien zur Herstellung von 1,3-Diolen ............................................... 10
3.2.2 Organokatalytische Aldolreaktion .......................................................................... 12
3.2.3 Enzymatische Reduktion ......................................................................................... 15
3.2.4 Eintopfreaktionen ................................................................................................... 18
3.3 Ziel der Arbeit .......................................................................................................... 20
3.4 Eigene Ergebnisse und Diskussion .......................................................................... 22
3.4.1 Organokatalyse unter lösungsmittelfreien Bedingungen ....................................... 22
3.4.2 Isolierung der 1,3-Diole ........................................................................................... 22
3.4.2.1 Optimierung der (R)-selektiven enzymatischen Synthese .................................. 23
3.4.2.2 (S)-Selektive enzymatische Umsetzung .............................................................. 24
3.4.3 Kombination der beiden Reaktionen in wässrigem Medium ................................. 25
3.4.3.1 Optimierung der Organokatalyse in wässrigem Medium ................................... 25

INHALTSVERZEICHNIS
II
3.4.3.2 Eintopfverfahren in wässrigem Medium ............................................................ 26
3.5 Zusammenfassung .................................................................................................. 28
4. Enzymatische Aldolreaktion............................................................................. 31
4.1 Einleitung ................................................................................................................ 31
4.2 Stand der Wissenschaft .......................................................................................... 33
4.2.1 Synthesestrategien für β-Hydroxy-α-aminosäuren ................................................ 33
4.2.2 Threoninaldolasen .................................................................................................. 37
4.3 Ziel der Arbeit.......................................................................................................... 43
4.4 Eigene Ergebnisse und Diskussion .......................................................................... 44
4.4.1 Vorversuche ............................................................................................................ 44
4.4.1.1 Racematsynthese ................................................................................................ 44
4.4.1.2 Hintergrundreaktion ........................................................................................... 47
4.4.1.3 Epimerisierung .................................................................................................... 48
4.4.2 Photometertest zur Bestimmung der Aktivität der L-Threoninaldolase ................ 48
4.4.2.1 Direkter Photometertest..................................................................................... 49
4.4.2.2 Inhibierungsversuche .......................................................................................... 51
4.4.3 Methoden zur Umsatzbestimmung ........................................................................ 53
4.4.3.1 Umsatzbestimmung mittels NMR-Standard ....................................................... 54
4.4.3.2 Umsatzbestimmung mittels Derivatisierung ...................................................... 55
4.4.4 Etablierung der Standardreaktion .......................................................................... 57
4.4.5 Bestimmung der Enantioselektivität....................................................................... 57
4.4.6 Untersuchung der enzymatischen Synthese .......................................................... 59
4.4.6.1 Einfluss der Reaktionszeit auf die enzymatische Aldolreaktion ......................... 59
4.4.6.2 Einfluss der Temperatur auf die enzymatische Aldolreaktion ............................ 60
4.4.6.3 Einfluss der Enzymmenge auf die enzymatische Aldolreaktion ......................... 61
4.4.6.4 Einfluss des Glycinüberschusses auf die enzymatische Aldolreaktion ............... 62

INHALTSVERZEICHNIS
III
4.4.6.5 Einfluss der Substratkonzentration auf die enzymatische Aldolreaktion ........... 63
4.4.6.6 Einfluss von Additiven auf die enzymatische Aldolreaktion ............................... 64
4.4.7 Erweiterung des Substratspektrums ....................................................................... 65
4.4.7.1 Thiamphenicolrelevante substituierte Benzaldehyde ........................................ 65
4.4.7.2 m-Substituierte Benzaldehyde ............................................................................ 66
4.4.7.3 p-Substituierte Benzaldehyde ............................................................................. 68
4.4.7.4 o-Substituierte Benzaldehyde ............................................................................. 68
4.4.8 Optimierung der enzymatischen Synthese von (2S)-12i ........................................ 70
4.4.8.1 Untersuchung des Reaktionsverlaufs ................................................................. 71
4.4.8.2 Vergleichsversuch mit Benzaldehyd als Substrat ............................................... 72
4.4.8.3 Cofaktor-Abhängigkeit ........................................................................................ 73
4.4.8.4 Scale-up und Isolierung des Produkts (2S)-12i ................................................... 74
4.4.8.4.1 Trennung der racemischen Diastereomere von rac-12i ................................... 75
4.4.8.4.2 Trennung der enantiomerenreinen Diastereomere von (2S)-12i .................... 76
4.4.9 Racematspaltung .................................................................................................... 77
4.4.9.1 Spaltung von rac-syn-12e .................................................................................... 78
4.4.9.2 Spaltung von rac-syn-12i ..................................................................................... 79
4.5 Zusammenfassung .................................................................................................. 80
5. Epoxidsynthese ausgehend von β-Hydroxy-α-aminosäuren .............................. 83
5.1 Einleitung ................................................................................................................ 83
5.2 Stand der Wissenschaft .......................................................................................... 84
5.2.1 Katalytische asymmetrische Epoxidierung ............................................................. 84
5.2.2 Synthese und Anwendung von Glycidestern .......................................................... 85
5.3 Ziel der Arbeit .......................................................................................................... 87
5.4 Eigene Ergebnisse und Diskussion .......................................................................... 87
5.4.1 Halohydrinsynthese ................................................................................................ 87

INHALTSVERZEICHNIS
IV
5.4.1.1 Synthese von rac-syn-3-Hydroxy-3-phenylpropansäure rac-syn-20e ................ 87
5.4.1.2 Synthese der chlorsubstituierten Halohydrinverbindungen (2S)-20i ................. 88
5.4.2 Synthese des Epoxids 21i durch Ringschluss .......................................................... 88
5.4.2.1 Synthese des racemischen Epoxids rac-syn-21i .................................................. 89
5.4.2.2 Reaktion des enantiomerenreinen Halohydrins (2S)-20i zum Epoxid 21i .......... 91
5.4.3 Zusammenfassung .................................................................................................. 92
6. Zusammenfassung ........................................................................................... 94
7. Summary ......................................................................................................... 99
8. Experimenteller Teil ....................................................................................... 104
8.1 Verwendete Chemikalien und Geräte .................................................................. 104
8.2 Synthesen und spektroskopische Daten ............................................................... 107
8.2.1 Kombination von organokatalytischer Aldolreaktion und enzymatischer Reduktion
............................................................................................................................... 107
8.2.1.1 Synthese von (S)-1-(4-Chlorphenyl)-1-hydroxybutan-3-on (S)-5f mittels
organokatalytischer Aldolreaktion ................................................................... 107
8.2.1.2 Allgemeine Arbeitsvorschrift 1 (AAV 1): Isolierung der vier Isomere aus der
enzymatischen Reduktion ................................................................................. 107
8.2.1.2.1 Synthese von (1S,3R)-1-(4-Chlorphenyl)-1,3-butandiol (1S,3R)-17f .............. 108
8.2.1.2.2 Synthese von (1R,3R)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3R)-17f .............. 108
8.2.1.2.3 Synthese von (1S,3S)-1-(4-Chlorphenyl)-1,3-butandiol (1S,3S)-17f ............... 109
8.2.1.2.4 Synthese von (1R,3S)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3S)-17f .............. 110
8.2.1.3 Organokatalytische Aldolreaktion in wässrigem Medium ................................ 110
8.2.1.4 Allgemeine Arbeitsvorschrift 2 (AAV 2): Eintopfreaktion ................................. 111
8.2.1.4.1 Synthese von (1R,3R)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3R)-17f .............. 112
8.2.1.4.2 Synthese von (1R,3S)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3S)-17f .............. 112
8.2.2 Enzymatische Aldolreaktion .................................................................................. 113

INHALTSVERZEICHNIS
V
8.2.2.1 Allgemeine Arbeitsvorschrift 3 (AAV 3): Synthese der racemischen β-Hydroxy-α-
aminosäuren116 ................................................................................................. 113
8.2.2.1.1 Synthese von rac-p-Methylthiophenylserin rac-12d ...................................... 113
8.2.2.1.2 Synthese von rac-Phenylserin rac-12e ........................................................... 114
8.2.2.1.3 Synthese von rac-p-Chlorphenylserin rac-12f ................................................ 114
8.2.2.1.4 Synthese von rac-o-Chlorphenylserin rac-12i ................................................ 115
8.2.2.1.5 Synthese von rac-m-Bromphenylserin rac-12j ............................................... 115
8.2.2.1.6 Synthese von rac-m-Chlorphenylserin rac-12l ............................................... 116
8.2.2.1.7 Synthese von rac-m-Hydroxyphenylserin rac-12m ........................................ 116
8.2.2.1.8 Synthese von rac-o-Methoxyphenylserin rac-12n ......................................... 117
8.2.2.2 Allgemeine Arbeitsvorschrift 4 (AAV 4): Derivatisierung der racemischen β-
Hydroxy-α-aminosäuren mit Benzoylchlorid116 ................................................ 117
8.2.2.2.1 Synthese von rac-N-Benzoylamino-3-(4-methylthiophenyl)-3-hydroxy-
propionsäure rac-80d ..................................................................................... 118
8.2.2.2.2 Synthese von rac-N-Benzoylamino-3-hydroxy-3-phenylpropionsäure rac-80e ...
.......... ............................................................................................................. 118
8.2.2.2.3 Synthese von rac-N-Benzoylamino-3-(4-chlorphenyl)-3-hydroxy-propionsäure
rac-80f ............................................................................................................. 119
8.2.2.2.4 Synthese von rac-syn-N-Benzoylamino-3-(2-chlorphenyl)-3-hydroxy-
propionsäure rac-syn-80i ................................................................................ 119
8.2.2.2.5 Synthese von rac-anti-N-Benzoylamino-3-(2-chlorphenyl)-3-hydroxy-
propionsäure rac-anti-80i ............................................................................... 120
8.2.2.2.6 Synthese von rac-N-Benzoylamino-3-(3-bromphenyl)-3-hydroxy-propionsäure
rac-80j ............................................................................................................. 120
8.2.2.2.7 Synthese von rac-N-Benzoylamino-3-(3-chlorphenyl)-3-hydroxy-propionsäure
rac-80l ............................................................................................................. 121
8.2.2.2.8 Synthese von rac-N-Benzoylamino-3-(2-methoxyphenyl)-3-hydroxy-
propionsäure rac-80n ..................................................................................... 122

INHALTSVERZEICHNIS
VI
8.2.2.3 Untersuchung der Hintergrundreaktion ........................................................... 122
8.2.2.4 Untersuchung auf Epimerisierung .................................................................... 123
8.2.2.5 Allgemeine Arbeitsvorschrift 5 (AAV 5) Photometertest zur Untersuchung der
Enzymaktivität117,118 .......................................................................................... 124
8.2.2.5.1 Bestimmung des Extinktionskoeffizienten ..................................................... 125
8.2.2.5.2 Aktivitätsbestimmung für L-TA aus E. coli ...................................................... 129
8.2.2.5.3 Inhibierungsversuche ..................................................................................... 130
8.2.2.6 Umsatzbestimmung der L-TA-katalysierten Aldolreaktion ............................... 131
8.2.2.6.1 Überprüfung der Umsatzbestimmung mittels NMR-Standard ...................... 131
8.2.2.6.2 Überprüfung der Umsatzbestimmung mittels Derivatisierung ...................... 132
8.2.2.6.3 Umsatzbestimmung der L-TA-katalysierten Aldolreaktion ............................ 133
8.2.2.6.3.1 Allgemeine Arbeitsvorschrift 6.1 (AAV 6.1) Umsatzbestimmung der L-TA-
katalysierten Aldolreaktion mittels NMR-Standard ..................................... 134
8.2.2.6.3.2 Allgemeine Arbeitsvorschrift 6.2 (AAV 6.2): Umsatzbestimmung der L-TA-
katalysierten Aldolreaktion mittels Derivatisierung .................................... 134
8.2.2.6.3.3 Ergebnisse der Umsatzbestimmung ............................................................. 134
8.2.2.7 Untersuchung der L-TA-katalysierten Aldolreaktion ........................................ 135
8.2.2.7.1 Zeitabhängigkeit ............................................................................................. 135
8.2.2.7.2 Temperaturabhängigkeit ................................................................................ 136
8.2.2.7.3 Einfluss der Enzymmenge ............................................................................... 137
8.2.2.7.4 Einfluss des Verhältnisses von Benzaldehyd zu Glycin ................................... 138
8.2.2.7.4.1 Einfluss der Glycinkonzentration ................................................................. 139
8.2.2.7.4.2 Einfluss der Benzaldehydkonzentration ....................................................... 139
8.2.2.7.5 Erhöhung der Substratkonzentration ............................................................. 140
8.2.2.7.5.1 Allgemeine Arbeitsvorschrift 7.1 (AAV 7.1): Umsatzbestimmung durch NMR-
Standard bei einer Substratkonzentration von 0.25 M ............................... 140

INHALTSVERZEICHNIS
VII
8.2.2.7.5.2 Allgemeine Arbeitsvorschrift 7.2 (AAV 7.2): Umsatzbestimmung durch
Derivatisierung bei einer Substratkonzentration von 0.25 M ..................... 141
8.2.2.7.5.3 Ergebnisse der Umsatzbestimmung (0.25 M) .............................................. 141
8.2.2.7.6 Einfluss verschiedener Additive ...................................................................... 142
8.2.2.7.7 Substratspektrum ........................................................................................... 143
8.2.2.7.7.1 Enzymatische Synthese von p-Nitrophenylserin (2S)-12b ........................... 143
8.2.2.7.7.2 Enzymatische Synthese von p-Methylthiophenylserin (2S)-12d ................. 144
8.2.2.7.7.3 Enzymatische Synthese von p-Chlorphenylserin (2S)-12f ............................ 144
8.2.2.7.7.4 Enzymatische Synthese von o-Chlorphenylserin (2S)-12i ............................ 145
8.2.2.7.7.5 Enzymatische Synthese von m-Bromphenylserin (2S)-12j ........................... 145
8.2.2.7.7.6 Enzymatische Synthese von p-Methylsulphonylphenylserin (2S)-12k ........ 146
8.2.2.7.7.7 Enzymatische Synthese von m-Chlorphenylserin (2S)-12l ........................... 146
8.2.2.7.7.8 Enzymatische Synthese von m-Hydroxyphenylserin (2S)-12m .................... 147
8.2.2.7.7.9 Enzymatische Synthese von o-Methoxyphenylserin (2S)-12n ..................... 148
8.2.2.7.7.10 Enzymatische Synthese von o-Bromphenylserin (2S)-12o ........................... 148
8.2.2.7.7.11 Enzymatische Synthese von o-Fluorphenylserin (2S)-12p ........................... 149
8.2.2.7.7.12 Enzymatische Synthese von o-Nitrophenylserin (2S)-12q ........................... 149
8.2.2.7.7.13 Enzymatische Synthese von o-Methylphenylserin (2S)-12r ......................... 150
8.2.2.7.7.14 Enzymatische Synthese von o-Hydroxyphenylserin (2S)-12s....................... 151
8.2.2.7.7.15 Enzymatische Synthese von 3,4-Dihydroxyphenylserin (2S)-12t ................. 151
8.2.2.8 Allgemeine Arbeitsvorschrift 8 (AAV 8): Optimierte Reaktion mit
o-Chlorbenzaldehyd (1i) als Substrat ................................................................ 152
8.2.2.8.1 Reaktionsverfolgung ....................................................................................... 152
8.2.2.8.2 Vergleich mit Reaktion im kleineren Maßstab ............................................... 153
8.2.2.8.3 Synthese von (2S)-12i ohne Zugabe von Cofaktor ......................................... 154
8.2.2.8.4 Vergleich mit Benzaldehyd als Substrat ......................................................... 154

INHALTSVERZEICHNIS
VIII
8.2.2.8.5 Isolierung von (2S)-12i aus Enzymumsetzung ................................................ 155
8.2.2.8.6 Abtrennung von Glycerin mittels Ionenaustauscher ...................................... 156
8.2.2.8.7 Trennung der Diastereomere ......................................................................... 156
8.2.2.8.7.1 Trennung von rac-12i mittels Ionenaustauscher ......................................... 157
8.2.2.8.7.2 Trennung von rac-12i mittels Umkristallisation ........................................... 157
8.2.2.8.7.3 Trennung von (2S)-12i mittels Fällung aus Wasser mit Aceton ................... 158
8.2.2.1 Allgemeine Arbeitsvorschrift 9 (AAV 9): Racematspaltung .............................. 158
8.2.2.1.1 Spaltung von rac-syn-Phenylserin (rac-syn-12e) ............................................ 159
8.2.2.1.2 Spaltung von rac-syn-o-Chlorphenylserin (rac-syn-12i) ................................. 159
8.2.2.1.3 Spaltung von rac-syn-Phenylserin (rac-syn-12e) im 25 mL-Maßstab ............. 160
8.2.2.1.4 Spaltung von rac-syn-o-Chlorphenylserin (rac-syn-12i) im 50 mL-Maßstab .. 161
8.2.3 Epoxidsynthese ausgehend von β-Hydroxy-α-aminosäuren 12 ........................... 161
8.2.3.1 Allgemeine Arbeitsvorschrift 11 (AAV 11): Halohydrinsynthese125 .................. 161
8.2.3.1.1 Synthese von rac-syn-3-Hydroxy-3-phenylpropansäure (rac-syn-20e) ......... 162
8.2.3.1.2 Synthese von (2S)-2-Chlor-3-(2-chlorphenyl)-3-hydroxypropansäure ((2S)-20i) .
.......... ............................................................................................................. 162
8.2.3.2 Allgemeine Arbeitsvorschrift 12 (AAV 12): Epoxidsynthese ............................. 163
8.2.3.2.1 Synthese von rac-syn-3-(2-Chlorphenyl)oxiran-2-carbonsäure (rac-syn-21i) 164
8.2.3.2.2 Synthese von (2R)-3-(2-Chlorphenyl)oxiran-2-carbonsäure ((2R)-21i) .......... 164
9. Literaturverzeichnis ........................................................................................ 166

ABBILDUNGSVERZEICHNIS
IX
II. Abbildungsverzeichnis
Abbildung 1.1. Allgemeines Reaktionsschema der Aldolreaktion ............................................. 1
Abbildung 1.2. Beispiel für eine selektive Mukaiyama-Aldoladdition6 ...................................... 2
Abbildung 1.3. Prolinkatalysierte Aldolreaktion11...................................................................... 2
Abbildung 1.4. Enamin-Intermediate der bio- und organokatalytischen Aldolreaktion13......... 3
Abbildung 1.5. Einteilung der Aldolasen nach Donorspezifität17 ............................................... 3
Abbildung 1.6. Industrielle Anwendung der Pyruvat-abhängigen Aldolase22 ........................... 4
Abbildung 2.1. Aldolreaktion und Biokatalysatoren zur Synthese von Pharmabausteinen ...... 5
Abbildung 2.2. Konzept der Synthese von 1,3-Diolen 17 ........................................................... 6
Abbildung 2.3. Synthese von 1,3-Diolen 17 als Eintopfreaktion ................................................ 6
Abbildung 2.4. Zambon-Prozess ................................................................................................. 7
Abbildung 2.5. Synthese von β-Hydroxy-α-aminosäuren 12 mit Threoninaldolasen ................ 7
Abbildung 2.6. Ziel der Prozessoptimierung der enzymatischen Aldolreaktion ........................ 8
Abbildung 2.7. Synthese von Epoxiden 21 ausgehend von β-Hydroxy-α-aminosäuren (2S)-12 8
Abbildung 3.1. Amphotericin B-Derivat mit verbesserter Wirksamkeit35 ................................. 9
Abbildung 3.2. Reduktion mit Borhydrid37 ............................................................................... 10
Abbildung 3.3. Metallkatalysierte Reduktion von Diketonen38 ............................................... 11
Abbildung 3.4. Chemoenzymatische Reduktion von Diketon 2440 .......................................... 11
Abbildung 3.5. Sequentieller Aufbau der beiden Stereozentren von 17f................................ 12
Abbildung 3.6. Einfluss von Wasser bei der organokatalytischen Aldolreaktion44 .................. 13
Abbildung 3.7. Organokatalysatoren für die Aldolreaktion in wässrigem Medium45,46,47,48 ... 14
Abbildung 3.8. Organkotalytische Aldolreaktion nach Singh51 ................................................ 14
Abbildung 3.9. Enzymatische Reduktion bei der Synthese von Statinen60 .............................. 15
Abbildung 3.10. Beispielsysteme zur Cofaktorregenerierung61 ............................................... 16
Abbildung 3.11. Sterisch anspruchsvolle Substrate für die enzymatische Reduktion64,65,66 ... 17
Abbildung 3.12. Vergleich Mehrstufensynthese und Eintopfverfahren .................................. 18
Abbildung 3.13. Synthese von 1,3-Diolen mittels DKR ............................................................ 19
Abbildung 3.14. Eintopfsynthese von Biphenylalkoholen77 ..................................................... 19
Abbildung 3.15. Kombination einer enzymatische Aldolreaktion und einer Metallkatalyse in
einem Eintopfverfahren78 ........................................................................................................ 20
Abbildung 3.16. Synthese der 1,3-Diole 17f............................................................................. 21

ABBILDUNGSVERZEICHNIS
X
Abbildung 3.17. Eintopfsynthese in wässrigem Medium ........................................................ 21
Abbildung 3.18. Organokatalytische Aldolreaktion zur Synthese von (S)-5f ........................... 22
Abbildung 3.19. Reduktion von (R)-5f mit LK-ADH .................................................................. 23
Abbildung 3.20. Reduktion von (S)-5f mit LK-ADH ................................................................... 24
Abbildung 3.21. Reduktion von (R)-5f mit Rsp-ADH ................................................................ 24
Abbildung 3.22. Reduktion von (S)-5f mit Rsp-ADH ................................................................. 25
Abbildung 3.23. Eintopfverfahren in wässrigem Medium ....................................................... 27
Abbildung 3.24. Vergleich der verschiedenen Synthesemethoden ......................................... 28
Abbildung 3.25. Synthese aller vier Stereoisomere von 17f .................................................... 29
Abbildung 3.26. Eintopfsynthese von (1R,3S)-17f ................................................................... 30
Abbildung 4.1. Enzymatische Spaltung von Threonin (12c) im Körper .................................... 31
Abbildung 4.2. Vancomycin ...................................................................................................... 32
Abbildung 4.3. Synthese von Digitoxin (49)87 .......................................................................... 32
Abbildung 4.4. Synthese eines Fucosyltransferase-Inhibitors 5188 ......................................... 33
Abbildung 4.5. Aldolreaktion von Glycin (11) und Aldehyden 1 .............................................. 33
Abbildung 4.6. DKR zur Synthese von anti-β-Hydroxy-α-aminosäureestern 5390d ................. 34
Abbildung 4.7. β-Hydroxy-α-aminosäuresynthese nach Crich93 .............................................. 34
Abbildung 4.8. Synthese über Oxazolidone als Glycinsynthone94b .......................................... 35
Abbildung 4.9. Metallkatalysierte Aldolreaktion zur Synthese von β-Hydroxy-α-amino-
säuren95 .................................................................................................................................... 36
Abbildung 4.10. Organokatalytische Synthese von β-Hydroxy-α-aminosäurederivaten ........ 36
Abbildung 4.11. Synthese von β-Hydroxy-α-aminosäuren 72 mittels Phasentransfer-
katalyse102b ............................................................................................................................... 37
Abbildung 4.12. Aktivierung von Glycin (11) durch PLP ........................................................... 38
Abbildung 4.13. Einteilung der L-TA katalysierten Reaktion103 ................................................ 38
Abbildung 4.14. Enzymatische Racematspaltung103 ................................................................ 39
Abbildung 4.15. Produktspektrum der L-TA aus E. coli und der D-TA aus X. oryzae105 ............ 40
Abbildung 4.16. Ergebnisse der Synthese verschieden substituierter aromatischer β-Hydroxy-
α-aminosäuren 12109 ................................................................................................................ 41
Abbildung 4.17. Synthese von β-Hydroxy-α,ω-diaminosäuren 74113 ...................................... 42
Abbildung 4.18. Synthese von α-alkylierten β-Hydroxy-α-aminosäuren 76, 77, 7819 ............. 42
Abbildung 4.19. Literaturbekannte Ergebnisse der TA-katalysierten Aldolreaktion106 ........... 43

ABBILDUNGSVERZEICHNIS
XI
Abbildung 4.20. Prozessoptimierung der enzymatischen Aldolreaktion ................................. 44
Abbildung 4.21. Synthese von racemischem Phenylserin ....................................................... 45
Abbildung 4.22. Racemische Phenylserinderivate ................................................................... 45
Abbildung 4.23. Derivatisierung mit Benzoylchlorid (79) ........................................................ 46
Abbildung 4.24. Racemische Produkte aus Derivatisierung mit Benzoylchlorid (79) .............. 47
Abbildung 4.25. Untersuchung einer möglichen Hintergrundreaktion ................................... 47
Abbildung 4.26. Untersuchung einer möglichen Epimerisierung ............................................ 48
Abbildung 4.27. Indirekter Photometertest zur Bestimmung der Aldolaseaktivität105 ........... 48
Abbildung 4.28. Direkte Aktivitätsbestimmung durch Verfolgung der Benzaldehyd-
konzentration ........................................................................................................................... 49
Abbildung 4.29. Bestimmung des Extiktionskoeffizienten ...................................................... 50
Abbildung 4.30. Inhibierung durch Glycin (11) ........................................................................ 52
Abbildung 4.31. Inhibierung durch rac-syn-Phenylserin (rac-syn-12e) ................................... 52
Abbildung 4.32. Rohproduktspektrum der enzymatischen Umsetzung .................................. 53
Abbildung 4.33. 1H-NMR-Spektrum zur Umsatzbestimmung mittels NMR-Standard 83 ........ 54
Abbildung 4.34. Derivatisierung mittels Benzoylchlorid (79) .................................................. 56
Abbildung 4.35. Standardreaktion und Umsatzbestimmung .................................................. 57
Abbildung 4.36. Bestimmung der Enantioselektivität der enzymatischen Umsetzung ........... 58
Abbildung 4.37. HPLC-Analytik für 80e .................................................................................... 58
Abbildung 4.38. Abhängigkeit der Biotransformation von der Temperatur ........................... 60
Abbildung 4.39. Abhängigkeit der Biotransformation von der Enzymmenge ......................... 61
Abbildung 4.40. Abhängigkeit der Biotransformation vom Glycinüberschuss ........................ 62
Abbildung 4.41. Abhängigkeit des Umsatzes von der Substratkonzentration ........................ 63
Abbildung 4.42. Umsatz bei Anwesenheit verschiedener Additive ......................................... 64
Abbildung 4.43. Thiamphenicol (19) ........................................................................................ 65
Abbildung 4.44. Enzymatische Synthese von (2S)-12d ............................................................ 66
Abbildung 4.45. Enzymatische Synthese von (2S)-12k ............................................................ 66
Abbildung 4.46. Enzymatische Synthese m-substituierter β-Hydroxy-α-aminosäuren (2S)-12
.................................................................................................................................................. 67
Abbildung 4.47. Enzymatische Synthese p-substituierter β-Hydroxy-α-aminosäuren (2S)-12 68
Abbildung 4.48. Enzymatische Synthese von (2S)-12i ............................................................. 69
Abbildung 4.49. Enzymatische Synthese o-substituierter β-Hydroxy-α-aminosäuren (2S)-12 70

ABBILDUNGSVERZEICHNIS
XII
Abbildung 4.50. Optimierte enzymatische Synthese von (2S)-12i .......................................... 71
Abbildung 4.51. Reaktionsverfolgung der enzymatischen Synthese von (2S)-12i................... 71
Abbildung 4.52. Synthese von (2S)-12e unter optimierten Bedingungen ............................... 72
Abbildung 4.53. Vergleichsversuch unter optimierten Bedingungen mit 1e als Substrat....... 73
Abbildung 4.54. Vergleich der enzymatischen Reaktion mit und ohne Cofaktor PLP ............. 73
Abbildung 4.55. Scale-up der enzymatischen Reaktion und Isolierung von (2S)-12i .............. 75
Abbildung 4.56. Trennung der Diastereomere von rac-12i ..................................................... 76
Abbildung 4.57. Trennung der Diastereomere durch Ausfällen aus Wasser mit Aceton ........ 77
Abbildung 4.58. Racematspaltung von rac-syn-12i in 50 mL-Maßstab ................................... 80
Abbildung 4.59. Optimierte Standardreaktion ........................................................................ 81
Abbildung 4.60. Optimierte enzymatische Reaktion mit 1j als Substrat ................................. 81
Abbildung 4.61. Optimierte Reaktion in 100 mL-Maßstab ...................................................... 81
Abbildung 5.1. Synthese eines Leukotrien-Antagonisten 87124 ............................................... 83
Abbildung 5.2. Synthese von Epoxiden 21 ausgehend von Halohydrinen 20.......................... 83
Abbildung 5.3. Jacobsen-Katsuki-Epoxidierung128 ................................................................... 84
Abbildung 5.4. Glycidester 91 als Synthesebaustein für Diltiazem (92)132 .............................. 85
Abbildung 5.5. Synthese des Glycidester 91132 ........................................................................ 86
Abbildung 5.6. Synthese des Zwischenprodukts 96 über Mukaiyama-Aldolreaktion130 ......... 86
Abbildung 5.7. Synthese von Epoxiden 21i ausgehend von β-Hydroxy-α-aminosäuren (2S)-12i
.................................................................................................................................................. 87
Abbildung 5.8. Synthese von rac-syn-20h ................................................................................ 88
Abbildung 5.9. Synthese von (2S)-20j ...................................................................................... 88
Abbildung 5.10. Synthese des racemischen Epoxids 21i ......................................................... 89
Abbildung 5.11. NOESY-Spektren von rac-syn-21i ................................................................... 90
Abbildung 5.12. Synthese von (2R)-21i .................................................................................... 91
Abbildung 5.13. HPLC-Analytik zur ee-Wertbestimmung von 21i ........................................... 92
Abbildung 5.14. Synthese von (2R)-21i .................................................................................... 93
Abbildung 6.1. Synthese aller vier Stereoisomere von 17f ...................................................... 94
Abbildung 6.2. Eintopfsynthese von (1R,3S)-17f ..................................................................... 95
Abbildung 6.3. Optimierte Standardreaktion .......................................................................... 96
Abbildung 6.4. Optimierte enzymatische Reaktion mit 1i als Substrat ................................... 96
Abbildung 6.5. Optimierte Reaktion in 100 mL-Maßstab ........................................................ 97

ABBILDUNGSVERZEICHNIS
XIII
Abbildung 6.6. Synthese von (2R)-21i ...................................................................................... 98
Abbildung 8.1. Enzymatische Reduktion der β-Hydroxyketone 5f ........................................ 107
Abbildung 8.2. Eintopfreaktion .............................................................................................. 111
Abbildung 8.3. Racematsynthese der β-Hydroxy-α-aminosäuren 12 .................................... 113
Abbildung 8.4. Derivatisierung von rac-12 ............................................................................. 117
Abbildung 8.5. Aktivierung von Glycin ................................................................................... 122
Abbildung 8.6. Untersuchung auf Epimerisierung ................................................................. 123
Abbildung 8.7. Direkte Aktivitätsbestimmung durch Verfolgung der Benzaldehyd-
konzentration ......................................................................................................................... 124
Abbildung 8.8. Extinktion bei pH 7, ohne PLP ........................................................................ 126
Abbildung 8.9. Extinktion bei pH 7, mit PLP ........................................................................... 127
Abbildung 8.10. Extinktion bei pH 8, ohne PLP ...................................................................... 128
Abbildung 8.11. Extinktion bei pH 8, mit PLP ......................................................................... 129
Abbildung 8.12. Umsatzbestimmung mittels Derivatisierung ............................................... 132
Abbildung 8.13. Umsatzbestimmung mittels Derivatisierung oder Zugabe eines
NMR-Standards ...................................................................................................................... 133
Abbildung 8.14. L-TA-katalysierten Aldolreaktion .................................................................. 135
Abbildung 8.15. Untersuchung der Zeitabhängigkeit ............................................................ 135
Abbildung 8.16. Untersuchung der Temperaturabhängigkeit ............................................... 136
Abbildung 8.17. Einfluss der Enzymmenge ............................................................................ 137
Abbildung 8.18. Einfluss des Verhältnisses von Glycin (11) zu Benzaldehyd (1e) ................. 138
Abbildung 8.19. Erhöhung der Substratkonzentration auf 0.25 M........................................ 140
Abbildung 8.20. Einfluss verschiedener Additive ................................................................... 142
Abbildung 8.21. Erweiterung des Substratspektrums ........................................................... 143
Abbildung 8.22. Optimierte Reaktion mit o-Chlorbenzaldehyd (1i) als Substrat .................. 152
Abbildung 8.23. Racematspaltung ......................................................................................... 158
Abbildung 8.24. Halohydrinsynthese ..................................................................................... 161
Abbildung 8.25. Epoxidsynthese über zwei Stufen ................................................................ 163

TABELLENVERZEICHNIS
XIV
III. Tabellenverzeichnis
Tabelle 3.1. Optimierung der Synthese von (R)-5f in wässrigem Medium .............................. 26
Tabelle 4.1. Ergebnisse des Photometertests .......................................................................... 51
Tabelle 4.2. Überprüfung der Umsatzbestimmung mittels NMR-Standard ............................ 55
Tabelle 4.3. Überprüfung der Umsatzbestimmung mittels Derivatisierung ............................ 56
Tabelle 4.4. Abhängigkeit der Biotransformation von der Reaktionszeit ................................ 59
Tabelle 4.5. Racematspaltung von rac-syn-12e ....................................................................... 78
Tabelle 4.6. Racematspaltung von rac-syn-12i ........................................................................ 79
Tabelle 8.1. Optimierung der organokatalytischen Aldolreaktion......................................... 111
Tabelle 8.2. Untersuchung der Hintergrundreaktion ............................................................. 123
Tabelle 8.3. Untersuchung auf Epimerisierung ...................................................................... 124
Tabelle 8.4. Extinktion bei pH 7 und ohne Zugabe von PLP ................................................... 125
Tabelle 8.5. Extinktion bei pH 7 und Zugabe von PLP ............................................................ 126
Tabelle 8.6. Extinktion bei pH 8 und ohne Zugabe von PLP ................................................... 127
Tabelle 8.7. Extinktion bei pH 8 und Zugabe von PLP ............................................................ 128
Tabelle 8.8. Durchführung des Photometertests ohne Zugabe von PLP ............................... 129
Tabelle 8.9. Durchführung des Photometertest mit Zugabe von PLP ................................... 130
Tabelle 8.10. Ergebnisse des Photometertests ...................................................................... 130
Tabelle 8.11. Einfluss von 11 auf die Enzymaktivität ............................................................. 131
Tabelle 8.12. Einfluss von 12e auf die Enzymaktivität ........................................................... 131
Tabelle 8.13. Überprüfung der Umsatzbestimmung mittels NMR-Standard ........................ 132
Tabelle 8.14. Überprüfung der Umsatzbestimmung mittels Derivatisierung ........................ 133
Tabelle 8.15. Umsatzbestimmung L-TA-katalysierten Aldolreaktion ..................................... 135
Tabelle 8.16. Zeitabhängigkeit der Enzymreaktion ................................................................ 136
Tabelle 8.17. Enzymreaktion bei 40°C .................................................................................... 137
Tabelle 8.18. Einfluss der Enzymmenge ................................................................................. 138
Tabelle 8.19. Einfluss des Glycinüberschusses ....................................................................... 139
Tabelle 8.20. Einfluss der Benzaldehydkonzentration ........................................................... 140
Tabelle 8.21. Umsatz bei Substratkonzentration von 0.25 M ................................................ 141
Tabelle 8.22. Einfluss verschiedener Additive........................................................................ 142
Tabelle 8.23. Enzymatische Synthese von (2S)-12b ............................................................... 143

TABELLENVERZEICHNIS
XV
Tabelle 8.24. Enzymatische Synthese von (2S)-12f ................................................................ 144
Tabelle 8.25. Enzymatische Synthese von (2S)-12i ................................................................ 145
Tabelle 8.26. Enzymatische Synthese von (2S)-12j ................................................................ 146
Tabelle 8.27. Enzymatische Synthese von (2S)-12k ............................................................... 146
Tabelle 8.28. Enzymatische Synthese von (2S)-12l ................................................................ 147
Tabelle 8.29. Enzymatische Synthese von (2S)-12m .............................................................. 147
Tabelle 8.30. Enzymatische Synthese von (2S)-12n ............................................................... 148
Tabelle 8.31. Enzymatische Synthese von (2S)-12o ............................................................... 149
Tabelle 8.32. Enzymatische Synthese von (2S)-12p ............................................................... 149
Tabelle 8.33. Enzymatische Synthese von (2S)-12q ............................................................... 150
Tabelle 8.34. Enzymatische Synthese von (2S)-12r ................................................................ 151
Tabelle 8.35. Enzymatische Synthese von (2S)-12s ............................................................... 151
Tabelle 8.36. Reaktionsverfolgung ......................................................................................... 153
Tabelle 8.37. Enzymatische Umsetzung mit und ohne Zugabe von PLP ................................ 154
Tabelle 8.38. Vergleichsversuchsreihe mit Benzaldehyd (1e) als Substrat ............................ 155
Tabelle 8.39. Racematspaltung von rac-syn-12e ................................................................... 159
Tabelle 8.40. Racematspaltung von rac-syn-12i .................................................................... 160

ABKÜRZUNGSVERZEICHNIS
XVI
IV. Abkürzungsverzeichnis
ADH Alkoholdehydrogenase
AD-H-Säule CHIRALPAK® Amolyse tris-(3,5-dimethylphenylcarbamat)
aq. wässrig
Äq. Äquivalent(e)
atm Atmosphären
BINOL Binaphthol
BINAP 2,2'-Bis-(diphenylphosphino)-1,1'-binaphtyl
BzCl Benzoylchlorid
CDCl3 deuteriertes Chloroform
cm Zentimeter
d [cm] Küvettendicke
d Dublett
δ [ppm] chemische Verschiebung
DBU 1,8-Diazabicyclo[5.4.0]undec-7-en
DC Dünnschichtchromatographie
dd Dublett vom Dublett
DET Diethyltartrat
DHAP Dihydroxyacetonphosphat
dhb 2,5-Dihydroxybenzoesäure
DIPT Diisopropyltartrat
DKR dynamisch kinetische Racematspaltung
d.r. Diastereomerenverhältnis (diastereomeric ratio)
EA Elementaranalyse
ee Enantiomerenüberschuss (enantiomeric excess)
EI Elektronenstoßionisation
EtOAc Ethylacetat
f Probenverdünnungsfaktor
FA Ameisensäure (formic acid)
FAB fast atom bombardement
h Stunde

ABKÜRZUNGSVERZEICHNIS
XVII
HPLC High Performance Liquid Chromatography
Hz Hertz
i-PrOH iso-Propanol
IR Infrarot
J skalare Kopplungskonstante
L Liter
LB-ADH Alkoholdehydrogenase aus Lactobacillus brevis
LK-ADH Alkoholdehydrogenase aus Lactobacillus kefir
Kat. Katalysator
Konz. Konzentration
m Multiplett
MeOH Methanol
mg Milligramm
m/z Verhältnis Masse zu Ladung
MHz Megahertz
min Minute(n)
mL Milliliter
mmol Millimol
mol Mol
MS Massenspektrometrie
MTBE Methyl-tert-butylether
NADH, NAD+ Nikotinsäureamid-Adenin-Dinukleotid
NADPH, NADP+ Nikotinsäureamid-Adenin-Dinukleotid-Phosphat
NBA m-Nitrobenzylalkohol
NBS N-Bromsuccinimid
n.b. nicht bestimmt
n.d. nicht detektierbar
nm Nanometer
NMR Kernmagnetische Resonanz
Ø Durchmesser
OD-Säule CHIRALCEL®-Säule OD, Cellulose tris-(3,5-dimethylphenyl-carbamat)
OJ-H-Säule CHIRALCEL®-Säule OJ-H, Cellulose tris-(4-methylbenzoat)

ABKÜRZUNGSVERZEICHNIS
XVIII
PLP Pyridoxal-5-phosphat
ppm parts per million
q Quartett
R Substituent
RF Retentionsfaktor
rac racemisch
rpm Umdrehung pro Minute (rounds per minute)
Rsp-ADH Alkoholdehydrogenase aus Rhodococcus sp.
RT Raumtemperatur
s Singulett
sin Sinapinsäure
SiO2 Kieselgel
t Triplett
t Zeit
tr Retentionszeit
t-BuOH tert-Butanol
t-BuOK Kalium-tert-butanolat
t-BuOOH tert-Butylhydroperoxid
TA Threoninaldolase
TEA Trifluoressigsäure
THF Tetrahydrofuran
Ti(O-iPr) Titan(IV)isopropylat
TMS Tetramethylsilyl-
U Units
U/mL volumetrische Enzymaktivität
U/mmol Units/Stoffmenge n(Substrat)
UV/Vis Ultraviolett/Visible
V Volumen
μl Mikroliter
ṽ [cm-1] Wellenzahl
Vg [ml] Gesamtprobenvolumen
Vp [ml] Probenvolumen

EINLEITUNG
1
1. Einleitung
Die organische Chemie basiert auf der Untersuchung und Herstellung von Kohlenstoff-
verbindungen, wie zum Beispiel Aminosäuren und Proteine. Zu den Herausforderungen
eines organischen Chemikers zählen die Herstellung von komplexen Kohlenstoff-
verbindungen sowie die Entwicklung möglichst effizienter Syntheserouten. Eine der
wichtigsten Reaktionsklassen ist hierbei die C-C-Bindungsknüpfung, die den Aufbau langer
und komplexer Kohlenstoffketten ermöglicht. Ein mit am häufigsten angewendeter Vertreter
dieser Klasse ist die Aldolreaktion, bei der zwei Aldehyde oder Ketone miteinander zu einer
β-Hydroxycarbonylverbindung reagieren (Abbildung 1.1).1 Eine Weiterreaktion zum Alken
unter Abspaltung von Wasser ist als Aldolkondensation bekannt.
R3 R4
O
R2
R1
O+ R1
O
R3
OH
R4R2
Katalysator -H2OR1
O
R3
R4
R2
Abbildung 1.1. Allgemeines Reaktionsschema der Aldolreaktion
Durch geeignete Wahl der Ausgangsverbindungen, beispielsweise die Verwendung von
lediglich einer Verbindung mit einem α-ständigen Proton, kann die Bildung unerwünschter
Kreuzprodukte vermieden werden. Auch durch den Einsatz von Enoläquivalenten, wie etwa
Silylenolether oder Lithiumenolate kann die Synthese des gewünschten Produkts beeinflusst
werden. Hierbei wird der Donor zuerst in ein Enolat überführt und anschließend der
Akzeptor zugegeben.1,2 Ein weiterer interessanter Aspekt der Aldolreaktion ist, dass hier je
nach Wahl der Ausgangsverbindungen chirale Produkte entstehen können. Bei der
klassischen achiralen säure- bzw. basenkatalysierten Reaktion entstehen Racemate,
allerdings haben sich im Laufe der Zeit auch eine Vielzahl enantioselektiver Methoden zur
Synthese von β-Hydroxycarbonylverbindungen etabliert.3,4 Ein Beispiel ist die Mukaiyama-
Aldoladdition.5 Der Donor wird zuerst in einen Silylenolether überführt und anschließend
wird die Reaktion mit dem Akzeptor durch eine chirale Lewis-Säure katalysiert. Die
asymmetrische Induktion erfolgt hier über den chiralen Liganden der Säure.1 Dabei handelt
es sich meist um Chelatliganden, wie beispielsweise BINOL-Derivate. Es können dabei
verschiedenste Lewis-Säuren verwendet werden, wie Titan-, Kupfer- oder

EINLEITUNG
2
Siliciumverbindungen.3 Ein Beispiel mit Siliciumchlorid als Lewis-Säure und einem BINOL-
Derivat 4 als chiralen Ligand ist in Abbildung 1.2 dargestellt.6
Abbildung 1.2. Beispiel für eine selektive Mukaiyama-Aldoladdition6
Alternative metallkatalysierte Routen lieferten Shibasaki7 und Trost.8 Des Weiteren steht
eine Vielzahl von Organokatalysatoren für die enantioselektive Aldolreaktion zur Verfügung.9
Erstmals wurde die enantioselektive katalytische Wirkung von chiralen Aminosäuren, wie
etwa Prolin, in den frühen siebziger Jahren am Beispiel einer intramolekularen Aldolreaktion
gezeigt.10 Nachdem die katalytische Wirkung von Prolin 2000 von List et al. auch auf
intermolekulare Bindungsknüpfungen ausgeweitet worden war (Abbildung 1.3),11 folgte die
Entwicklung weitere Katalysatoren, deren Strukturen vor allem auf Prolin basierten.12
Abbildung 1.3. Prolinkatalysierte Aldolreaktion11
Prolin reagiert mechanistisch gesehen als Enzymmimetikum.13 Im Katalysezyklus der nicht
metallhaltigen Aldolasen erfolgt die Aktivierung über ein Enamin-Intermediat, wie auch beim
Einsatz von Prolin (Abbildung 1.4).14,15

EINLEITUNG
3
HN
(Aldolase) Lys
OHR
N CO2H
Enamin-IntermediatAldolase
Enamin-IntermediatOrganokatalysator
Abbildung 1.4. Enamin-Intermediate der bio- und organokatalytischen Aldolreaktion13
Aldolasen katalysieren ebenfalls selektiv die Aldoladdition, allerdings sind diese
Biokatalysatoren stark donorspezifisch.16 Sie können in vier verschiedene Klassen eingeteilt
werden, die jeweils einen speziellen Donor akzeptieren und somit auch nur zur Synthese
bestimmter Produktklassen verwendet werden können (Abbildung 1.5)17,18 Bis jetzt sind nur
wenige Ausnahmen von dieser Donorspezifität bekannt.16,19,20,21
Abbildung 1.5. Einteilung der Aldolasen nach Donorspezifität17
Bei der Pyruvat- und der Acetaldehyd-abhängigen Aldolase entsteht eine Ketosäure 9 bzw.
die Aldolstruktur 10 mit jeweils einem Stereozentrum, wohingegen bei der
Dihydroxyacetonphosphat- (6, DHAP) und Glycin-abhängigen Aldolase unter Verwendung
prochiraler Aldehyde ein Ketose-1-phosphat 7 bzw. eine β-Hydroxy-α-aminosäure 12 mit
jeweils zwei Stereozentren entsteht. Die Gruppe der DHAP-abhängigen Enzyme wurde am

EINLEITUNG
4
intensivsten untersucht.17 Hier gibt es verschiedene Biokatalysatoren, die zur Bildung aller
vier möglichen Stereoisomere fähig sind. Bei der Wahl des Akzeptors ist das Enzym deutlich
flexibler und es werden verschiedene Carbonylverbindungen umgesetzt.16,17
Die Pyruvat-abhängige Aldolase wird industriell im großen Maßstab zur Synthese von
Neuraminsäure 15, die eine wichtige Vorstufe für das Grippemittel Zanamivir 16 darstellt,
genutzt (Abbildung 1.6).22
Abbildung 1.6. Industrielle Anwendung der Pyruvat-abhängigen Aldolase22
Die Verwendung von Enzymen in der chemischen Industrie hat in den letzten Jahren stark an
Bedeutung gewonnen.23 So bietet die Biotechnologie die Möglichkeit Prozesse nachhaltiger
und „grüner“ zu gestalten und beispielsweise den CO2-Ausstoß zu senken.24,25 Im Bereich der
Arzneimittelproduktion werden immer noch typischerweise 25 bis 100 Kilogramm Abfall pro
Kilogramm erhaltener Zielverbindung bei den klassischen Syntheserouten produziert.26 Das
Ziel der grünen Chemie ist es chemische Prozesse sicherer, umweltverträglicher und
energieeffizienter zu gestalten.27 Hier bietet sich der Einsatz von Enzymen an, da diese meist
in wässrigem Medium und bei milden Temperaturen sehr effizient und selektiv Reaktionen
katalysieren.23,24

MOTIVATION UND ZIELSETZUNG
5
2. Motivation und Zielsetzung
Zur Verbesserung von Syntheserouten für die Gewinnung pharmazeutisch relevanter
Bausteine, im Hinblick auf einen nachhaltigen Prozess, waren die Nutzung der Aldolreaktion,
sowie der Einsatz verschiedener Biokatalysatoren geplant. Die Synthese von 1,3-Diolen sollte
durch die Kombination von organokatalytischer Aldolreaktion und anschließender
enzymatischer Reduktion erzielt werden. Außerdem sollte unter Einsatz von Glycin-
abhängigen Aldolasen, den sogenannten Threoninaldolasen (TA), eine verbesserte
Syntheseroute für β-Hydroxy-α-aminosäuren etabliert werden (Abbildung 2.1). Das Motiv
der 1,3-Diole kommt in verschiedenen Naturstoffen, wie etwa makroliden Antibiotika, zu
denen auch Amphotericin B zählt, vor.28 Einige β-Hydroxy-α-aminosäuren können als
Vorstufe für die Synthese von Antibiotika, wie beispielsweise Thiamphenicol oder
Chloramphenicol genutzt werden.29
Abbildung 2.1. Aldolreaktion und Biokatalysatoren zur Synthese von Pharmabausteinen

MOTIVATION UND ZIELSETZUNG
6
2.1 Synthese von 1,3-Diolen
Von einer Vielzahl von Verfahren zur Herstellung von 1,3-Diolen wurde bereits berichtet,
allerdings gibt es hier keinen generellen Zugang zu dieser Produktklasse.30 Ein bestehendes,
vielversprechendes Konzept zur Synthese von 1,3-Diolen,31 bei dem die beiden
Stereozentren sequenziell aufgebaut werden, sollte weiterentwickelt werden. Ziel war es mit
Hilfe der organokatalytischen Aldolreaktion und Wahl eines biomimetischen Katalysators
β-Hydroxyketone 5 mit möglichst hoher Enantioselektivität und hohem Umsatz herzustellen
und anschließend das zweite Stereozentrum durch enzymatische Reduktion hoch
enantioselektiv aufzubauen (Abbildung 2.2). Durch geeignete Wahl des Organo- bzw.
Biokatalysators ist es möglich, die einzelnen Stereozentren in der gewünschten
Konfiguration zu erhalten und jedes der vier möglichen Stereoisomere zu bilden.
Abbildung 2.2. Konzept der Synthese von 1,3-Diolen 17
Durch die Weiterentwicklung der Syntheseroute zu einer Eintopfreaktion wird der Prozess
im Hinblick auf seine Nachhaltigkeit verbessert, da ein Isolierungsschritt eingespart und
somit der Verbrauch an Chemikalien reduziert wird. Die Machbarkeit eines solchen
Verfahrens unter den eben dargestellten Bedingungen konnte bereits gezeigt werden.31
Allerdings ist eine weitere Optimierung der Ausbeute und Diastereoselektivität nötig. Die
Kompatibilität der beiden Reaktionen sollte durch eine Durchführung des
organokatalytischen Schrittes in wässrigem Medium verbessert werden (Abbildung 2.3).
Abbildung 2.3. Synthese von 1,3-Diolen 17 als Eintopfreaktion

MOTIVATION UND ZIELSETZUNG
7
2.2 Synthese von β-Hydroxy-α-aminosäuren
Rein chemische Syntheserouten zur Herstellung von β-Hydroxy-α-aminosäuren ausgehend
von Glycin sind bereits bekannt.32 Allerdings sind diese Methoden meist dadurch limitiert,
dass geschützte Ausgangsverbindungen eingesetzt werden müssen und die Synthese über
mehrere Stufen verläuft. Bei der industriellen Produktion von Thiamphenicol 19 (Zambon-
Prozess) wird beispielsweise zuerst die racemische β-Hydroxy-α-aminosäure 12d hergestellt,
die dann über eine Racematspaltung getrennt werden muss, bevor eine Weiterreaktion zum
gewünschten Produkt 19 möglich ist (Abbildung 2.4).33
Abbildung 2.4. Zambon-Prozess33
Im Vergleich zu diesen Syntheserouten ermöglicht der Einsatz von Threoninaldolasen die
Synthese der gewünschten Verbindung 12 ausgehend von Glycin (11) und einem Aldehyd 1
in nur einem enantioselektiven Schritt unter milden Bedingungen. Auch muss beispielsweise
Glycin (11) nicht entsprechend geschützt bzw. aktiviert werden (Abbildung 2.5).
Abbildung 2.5. Synthese von β-Hydroxy-α-aminosäuren 12 mit Threoninaldolasen

MOTIVATION UND ZIELSETZUNG
8
Der bei dieser Reaktion verwendete Biokatalysator steht in zwei Varianten zur Verfügung,
der L-selektiven und der D-selektiven Threoninaldolase (L-TA, D-TA). Diese Enzyme
katalysieren die Bildung des Stereozentrums in α-Position hochselektiv, wohingegen die
Enantioselektivität für das Stereozentrum in β-Position meist geringer ist. Beide
Aldolasetypen wurden bereits bei der Synthese von β-Hydroxy-α-aminosäuren eingesetzt,
allerdings mit zum Teil mäßigen Diastereoselektivitäten und nur in kleinem Maßstab.29
Im Rahmen dieser Arbeit sollte nun mit der L-Threoninaldolase aus E. coli eine
Prozessoptimierung durchgeführt werden. Ziel war es β-Hydroxy-α-aminosäuren 12 in
möglichst großem Maßstab, bei hoher Substratkonzentration, mit gutem Umsatz und
möglichst hoher Enantio- und Diastereoselektivität herzustellen (Abbildung 2.6).
Abbildung 2.6. Ziel der Prozessoptimierung der enzymatischen Aldolreaktion
2.3 Synthese von Epoxiden aus β-Hydroxy-α-aminosäuren
Nach Etablierung einer enzymatischen Synthese von β-Hydroxy-α-aminosäuren (2S)-12 im
50-100 mL-Maßstab sollte, ausgehend von dieser Verbindung, die Umsetzung zu
enantiomerenreinen Epoxiden 21 über zwei Stufen untersucht werden (Abbildung 5.7).
Abbildung 2.7. Synthese von Epoxiden 21 ausgehend von β-Hydroxy-α-aminosäuren (2S)-12
In Anlehnung an den Schritt des Ringschlusses ausgehend von Halohydrinen bei der Darzens-
Glycidester-Synthese1 sollte zuerst die Aminogruppe durch ein Chloridion substituiert
werden und anschließend das erhaltene Halohydrin 20 zum Epoxid 21 umgesetzt werden.
Auf diesem Weg sollte ein einfacher Zugang zu enantio- und diastereomerenreinen Epoxiden
etabliert werden.

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
9
3. Kombination einer organokatalytischen Aldolreaktion und
einer enzymatischen Reduktion
3.1 Einleitung
Die Untersuchung und Verbesserung von Medikamenten ist ein wichtiger Bestandteil der
Forschung. So wurde auch das bereits erwähnte Amphotericin B (Abschnitt 2) in einer
aktuellen Veröffentlichung genauer untersucht.34 Die Wirksamkeit gegen chronische und
systemische Pilzinfektionen, von denen vor allem Patienten mit schwachem Immunsystem
betroffen sind, wie beispielsweise nach einer Chemotherapie oder bei einer Aids-
Erkrankung, ist unbestritten. Allerdings treten auch unerwünschte Nebenwirkungen wie
Leber- oder Nierenschäden auf. Aus diesem Grund wurden neue Derivate entwickelt, die
eine verbesserte Wirkung bei geringerer Toxizität aufweisen sollten. Die in Abbildung 3.1
dargestellte Verbindung zeigte bei den durchgeführten Testreihen die höchste
Wirksamkeit.35
Abbildung 3.1. Amphotericin B-Derivat mit verbesserter Wirksamkeit35
Zur Verbesserung verschiedenster Arzneistoffe ist die Erforschung und Weiterentwicklung
von Syntheserouten der Grundbausteine dieser Verbindungen ebenfalls unerlässlich.
1,3-Diole 17 gehören zu solch wichtigen Teilstrukturen pharmazeutisch relevanter
Verbindungen.36 Dies belegt auch die Vielzahl an Veröffentlichungen auf diesem Gebiet.28,30
Die Methoden zur Herstellung dieser Stoffe verlaufen meist über die Synthese von
β-Hydroxyketonen oder 1,3-Diketonen mit anschließender selektiver metallkatalysierter
oder biokatalytischer Reduktion. Die bekannten Methoden sind allerdings noch

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
10
optimierungsbedürftig im Hinblick auf Substratbreite und Enantio- bzw. Diastereo-
selektivität.30 Deshalb ist eine Weiterentwicklung der 1,3-Diolsynthese von besonderer
Bedeutung.
3.2 Stand der Wissenschaft
3.2.1 Synthesestrategien zur Herstellung von 1,3-Diolen
Eine Übersicht über die vielen verschiedenen Synthesemethoden von 1,3-Diolen 17 wurde
von Bode et al. erstellt.30 Im Folgenden sollen ausgewählte Beispiele diskutiert werden.
Meist werden die Reduktionen von 1,3-Diketonen oder β-Hydroxyketonen mit Borhydriden,
Metall- oder Biokatalysatoren zur Herstellung von 1,3-Diolen 17 verwendet.30 Mit Hilfe von
Borhydriden können beispielsweise selektiv anti-Diole 23 hergestellt werden (Abbildung
3.2).37
Abbildung 3.2. Reduktion mit Borhydrid37
Um enantiomerenreine Verbindungen zu erhalten, müssen oftmals enantiomerenreine
β-Hydroxyketone eingesetzt werden und je nach Hydrierungsreagenz werden syn- oder anti-
Produkte erhalten. Ein Nachteil bei der Verwendung von Borhydriden ist vor allem, dass
diese Verbindungen meist stöchiometrisch zugegeben werden müssen, teilweise sogar im
Überschuss.30 Eine Alternative dazu bietet die enantioselektive metallkatalysierte Reduktion,
die von Noyori et al. bereits 1988 vorgestellt wurde38 und sich im Laufe der Zeit zu einer
allgemeinen Methode zur Reduktion von Ketonen entwickelt hat.39 Die enantioselektive
katalytische Wirkung wird hier durch den Einsatz eines chiralen Liganden, wie beispielsweise
BINAP oder entsprechenden Derivaten erzielt. In Abbildung 3.3 ist die Reduktion von
Diketon 22 mit einem Ruthenium-BINAP-Katalysator gezeigt. Die Enantioselektivitäten sind
teilweise nur mäßig und stark von Substrat und Katalysator abhängig.

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
11
Abbildung 3.3. Metallkatalysierte Reduktion von Diketonen38
Ein Beispiel für die biokatalytische Reduktion von 1,3-Diketonen 24 zeigten Quazi et al.40 Mit
Hilfe von zwei unterschiedlich selektiven Oxidoreduktasen konnte die Ketofunktion an
Position 3 mit einer Enantioselektivität von >99% ee zum Alkohol reduziert werden.
(Abbildung 3.4). Eine enzymatische Reduktion der zweiten Ketofunktion gelang allerdings
nur in mäßigen Ausbeuten. Es wurde dann auf eine klassisch chemische
Natriumborhydridreduktion zurückgegriffen und das erhaltene Diastereomerengemisch
säulenchromatographisch getrennt.
Abbildung 3.4. Chemoenzymatische Reduktion von Diketon 2440
Eine deutliche Verbesserung der Enantio- und Diastereoselektivität bei der Synthese von
1,3-Diolen 17 durch den sequentiellen, selektiven Aufbau der beiden Stereozentren konnte
bereits während meiner Diplomarbeit gezeigt werden.31 Im ersten Schritt wurde das
Stereozentrum des β-Hydroxyketons 5f mit Hilfe einer organokatalytischen Aldolreaktion
selektiv aufgebaut. Durch anschließende enzymatische Reduktion konnten alle vier

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
12
möglichen Isomere hergestellt werden (Abbildung 3.5). Eine weitere Optimierung dieses
Systems, bezüglich der Enantioselektivität des ersten Schrittes sowie die Isolierung der
verschiedenen Stereoisomere 17f, ist allerdings weiter erforderlich.
Abbildung 3.5. Sequentieller Aufbau der beiden Stereozentren von 17f
3.2.2 Organokatalytische Aldolreaktion
Seit der Etablierung der intermolekularen organokatalytischen Aldolreaktion mit Prolin im
Jahr 2000 von List et al.,11 fand eine rasante Entwicklung auf diesem Gebiet statt und viele
neue Katalysatoren wurden synthetisiert und getestet.12,41,42 Ein wichtiger Fortschritt war die
Nutzung von Wasser als Lösungsmittel. Zum einen ist Wasser ein umweltverträgliches
Lösungsmittel und zum anderen besitzt es auch besondere physikalischen Eigenschaften, die
einen speziellen Einfluss auf organische Reaktionen besitzen, wie beispielsweise
Oberflächenspannung, Polarität und die Fähigkeit Wasserstoffbrückenbindungen zu bilden.43
Die Nutzung von Aminosäurederivaten als Katalysatoren stellt in wässrigem Medium kein
Problem dar, da die Verbindungen unter diesen Bedingungen stabil sind.
Als erstes konnte die Gruppe um Janda eine organokatalytische Aldolreaktion in Wasser
durchführen und lieferte zugleich auch eine Erklärung zur Rolle des Lösungsmittels während
der Reaktion (Abbildung 3.6).44 Der postulierte Mechanismus beinhaltet eine duale Rolle des
Lösungsmittels. Zum einen aktiviert es den Aldehyd für den nukleophilen Angriff durch
Übertragen eines Protons und zum anderen beschleunigt es die Hydrolyse des Imins im
letzten Schritt der Reaktion. Diese beiden Faktoren erhöhen die Geschwindigkeit der
Reaktion, was auch durch quantenchemische Berechnungen unterstützt wurde.44c Das

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
13
Produkt konnte zwar nur mit einer Enantioselektivität von 20% erhalten werden, aber dies
war der erste Schritt zur Etablierung der organokatalytischen Aldolreaktion in Wasser.
Abbildung 3.6. Einfluss von Wasser bei der organokatalytischen Aldolreaktion44
Mittlerweile wurde eine ganze Bandbreite an Katalysatoren entwickelt, die in solch einem
2-Phasen-System gute Ergebnisse erzielen.43 Es kommt zur Mehrphasenbildung, da die
meisten Aldehyde und Ketone nicht oder nur sehr schlecht wasserlöslich sind. Eine Vielzahl
der Katalysatoren basieren auf Aminosäurestrukturen, vor allem auf Prolin.43 Einige Beispiele
für Katalysatoren, die nicht auf Prolin basieren sind in Abbildung 3.7 gezeigt. Unter
Verwendung des Threoninderivats 28 konnten bei der Reaktion von m-Chlorbenzaldehyd
und Aceton bei Raumtemperatur gute Ausbeuten und Enantioselektivitäten von 97% ee
erzielt werden.45 Mit Hilfe des Binaphthyl-Katalysators 29 wurden mit Cyclohexanon als
Donor sehr gute Enantioselektivitäten von bis zu 98% ee gefunden, wohingegen Aceton nur
mäßige Ergebnisse (bis zu 87% ee) lieferte.46 Der Katalysator 30 konnte zum ersten Mal auch
nicht-aromatische Aldehyde als Akzeptoren mit guten Ausbeuten (70%) und exzellenten
Enantioselektivitäten (>99% ee) umsetzen.47 Sogar die ungünstige Reaktion zweier Ketone
(Cyclohexanon und Phenylglyoxylat) konnte mittels Katalysator 31 mit sehr guten
Enantioselektivitäten (>99% ee) durchgeführt werden.48

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
14
Abbildung 3.7. Organokatalysatoren für die Aldolreaktion in wässrigem Medium45,46,47,48
Als erstes entwickelte die Gruppe um Singh einen Katalysator 32 mit mehreren aktiven
Substituenten (OH, NH) und sterisch anspruchsvollen geminalen Phenylgruppen.49 Diese
Struktur wurde später wiederholt aufgegriffen (vgl. Katalysator 30).47,50 Mit dieser
Verbindung konnten verschiedene aromatische Aldehyde mit Aceton bei -5°C in gesättigter
Natriumchloridlösung umgesetzt und sehr gute Enantioselektivitäten von >99% ee und
Ausbeuten von bis zu 80% erzielt werden (Abbildung 3.8).51
Abbildung 3.8. Organkotalytische Aldolreaktion nach Singh51
Die hohe Enantioselektivität wurde durch Aktivierung des Aldehyds mittels
Wasserstoffbrückenbindung mit der Hydroxy- und der Amidgruppe erklärt. Die sterisch
anspruchsvollen geminalen Phenylgruppen beeinflussen die Enantioselektivität ebenfalls
positiv.51

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
15
3.2.3 Enzymatische Reduktion
Die Biotechnologie hält immer weiter Einzug in die industrielle Synthese. Die Gründe dafür
liegen dabei nicht nur in der hohen Stereoselektivität der Enzyme, sondern auch im
verfahrenstechnischen Bereich. So kann ein Prozess durch Einsatz von Biokatalysatoren
vereinfacht und somit Rohstoff-, Energieverbrauch und Abfallmenge verringert
werden.24,52,53 Viele Aminosäuren werden bereits durch biotechnologische Prozesse
hergestellt.23 Hierbei werden allerdings hauptsächlich fermentative Verfahren genutzt. Auch
zur Synthese chiraler Pharmaintermediate eignen sich Enzyme.54 Hier ist speziell die
Reduktion prochiraler Ketone mittels Alkoholdehydrogenasen (ADHs) zu nennen, da chirale
Alkohole oftmals in Arzneistoffen enthalten sind.55 Es wurden bereits einige Anwendungen
im Industriemaßstab entwickelt.56 Dazu zählt beispielsweise die Synthese von
Cholesterinsenkern. Shimizu et al. optimierten in Zusammenarbeit mit Kaneka die
enzymatische Reduktion von 4-Chloracetessigestern als Bausteine für Statin-
Seitenketten.57,58 In Abbildung 3.9 ist die biokatalytische Reduktion des Diketoesters 33 mit
einer Alkoholdehydrogenase aus Lactobacillus brevis (LB-ADH) als Baustein zur Herstellung
von Rosuvastatin 35 gezeigt.59,60
Abbildung 3.9. Enzymatische Reduktion bei der Synthese von Statinen60
Die hier gezeigte Alkoholdehydrogenase aus Lactobacillus brevis gehört zu den (R)-selektiven
ADHs, wie auch die aus Lactobacillus kefir. Dagegen sind die ADHs aus Rhodococcus species,
Pferdeleber und Hefen meist (S)-selektive Enzyme.17,61

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
16
Alkoholdehydrogenasen sind cofaktorabhängig, d.h. zur Reaktion muss NADH oder NADPH
stöchiometrisch zugegeben werden, damit die Reaktion abläuft. Da diese Cofaktoren sehr
teuer sind, wurden unterschiedliche Systeme entwickelt, um den Cofaktor in situ zu
regenerieren, wodurch dieser nur noch in katalytischen Mengen zugegeben werden
muss.17,56,61,62 Die zwei gängigsten Konzepte sind in Abbildung 3.10 dargestellt. Die
substratgekoppelte Cofaktorregenerierung basiert auf der Zugabe eines Alkohols, meist
iso-Propanol, der in der Rückreaktion von der gleichen Alkoholdehydrogenase zum Keton
oxidiert wird und somit den Cofaktor regeneriert. Der Alkohol muss in diesem Fall im
Überschuss zugegeben werden, um das Gleichgewicht der Reaktion auf die Produktseite zu
verschieben. Es besteht auch die Möglichkeit das bei der Cofaktorregenerierung
entstehende Keton (Aceton) destillativ aus dem Gleichgewicht zu entfernen. In beiden Fällen
muss die ADH mit dem Cosubstrat kompatibel sein.
Abbildung 3.10. Beispielsysteme zur Cofaktorregenerierung61
Das enzymgekoppelte System erfordert die Zugabe eines zweiten Enzyms, welches ein
Cosubstrat unter Rückbildung des Cofaktors umsetzt. Diese Reaktion ist irreversible,
wodurch das Gleichgewicht ebenfalls auf die Produktseite geschoben wird. Beispiele für
solche Enzyme sind die Formiatdehydrogenase (FDH), die Formiat zu CO2 umsetzt, das dem
System entweicht und die Glucosedehydrogenase (GDH), die D-Glucose zum Gluconolacton
umsetzt, das anschließend irreversibel zum Salz der Gluconsäure reagiert. Wichtig ist hierbei
die Kompatibilität der beiden Enzymreaktionen, da Inhibierungseffekte durch die
verschiedenen Reaktanden auftreten können.17,61,63

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
17
Eine Limitierung der Alkoholdehydrogenasen stellt oftmals das Substratspektrum dar. Die
Ketofunktion darf meist nur einen sterisch anspruchsvollen Substituenten besitzen, wie
beispielsweise bei Acetophenon.64 Zahlreiche Forschungsarbeiten beschäftigten sich in
jüngster Zeit mit der Erweiterung des Substratspektrums. In Abbildung 3.11 sind drei
Beispiele für solch sterisch anspruchsvolle Ketone gezeigt, die mit bestimmten
Alkoholdehydrogenasen umgesetzt werden können.64,65,66 Die ADHs aus Nocardia
globerula64 und Ralstonia species65 sind in der Lage das Keton 36 mit sehr guten (>96% ee
(S))64 bis moderaten Enantioselektivitäten (64% ee (S))65 umzusetzen. Dagegen konnte
ausgehend von 37 aus der Reaktion mit der ADH aus Ralstonia sp. der entsprechende
Alkohol mit einem sehr guten ee-Wert von >99% (S) erhalten werden.65 Das
Benzophenonderivat 38 wird von einer ADH aus Sporobolomyces salmonicolor akzeptiert
und mit guten Enantioselektivitäten von 88% ee (R) zum entsprechenden Alkohol reduziert.66
Abbildung 3.11. Sterisch anspruchsvolle Substrate für die enzymatische Reduktion64,65,66
Durch Mutation des bekannten Wildtyp-Enzyms aus Sporobolomyces salmonicolor konnte
ein Biokatalysator entwickelt werden, der in der Lage ist, das Produkt mit entgegengesetzter
Konfiguration zu bilden. Mit Hilfe einer Röntgenstruktur wurden spezielle Aminosäuren im
aktiven Zentrum des Proteins ausgewählt, die Einfluss auf die Enantioselektivität haben
könnten. Diese wurden dann durch Mutationen durch andere Aminosäuren ausgetauscht.
Die erhaltenen Mutanten zeigten anschließend eine (S)-Enantioselektivität von 76% ee bei
der Reduktion von 38.66
Ein wichtiger Teil bei der weiteren Entwicklung der Biotechnologie stellt die Optimierung von
bereits bekannten Biokatalysatoren dar. Die Limitierung durch begrenzte Substratakzeptanz,
geringe Stereoselektivität, unzureichende Stabilität oder auch Produktinhibierung können in
der Regel überwunden werden.67 Dadurch werden Enzyme attraktiver für eine industrielle
Anwendung.

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
18
3.2.4 Eintopfreaktionen
Durch Prozessoptimierung können chemische Synthesen im Sinne der grünen Chemie27
verbessert werden. Eine Möglichkeit ist hierbei das Konzept der Eintopfreaktionen oder
Kaskadenreaktionen. Dieses beruht auf der Kombination mehrerer Reaktionen ohne
Isolierung der einzelnen Zwischenstufen (Abbildung 3.12). Während bei der klassischen
Mehrstufensynthese jede Zwischenstufe (B, C) isoliert werden muss, wird bei dem
entsprechenden Eintopfverfahren lediglich das am Ende gewünschte Produkt nach
Aufarbeitung erhalten. Dadurch kann idealerweise die Reaktionszeit und die
Abfallproduktion drastisch gesenkt werden.68 Dies geschieht bereits durch den Wegfall der
aufwändigen Isolierungsschritte, wie beispielsweise der Säulenchromatographie, bei der
meist große Mengen an Lösungsmitteln benötigt werden.
Abbildung 3.12. Vergleich Mehrstufensynthese und Eintopfverfahren
Eine besondere Herausforderung besteht darin, die Vorteile der Biotechnologie zu nutzen
und diese mit etablierten chemischen Reaktionen in solchen Eintopfsystemen zu
kombinieren, da diese meist in unterschiedlichen Reaktionsmedien ablaufen. Eine Ausnahme
sind hierbei Lipasen, die in der Lage sind, in organischem Medium zu arbeiten. Deshalb hat
sich vor allem die Kombination solcher Enzyme mit Metallkatalysatoren in dynamisch
kinetischen Racematspaltungen (DKR) etabliert.69,70 Ein Beispiel für die Synthese
enantiomerenreiner 1,3-Diole mittels chemoenzymatischer DKR lieferten Bäckvall et al. Ein
racemisches Diol 17 wird mit Hilfe einer Lipase zweimal selektiv verestert und die einfach
veresterte Verbindung 39 mit Hilfe eines Rutheniumkatalysators in situ racemisiert

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
19
(Abbildung 3.13).71 Die Reaktion findet in Toluol statt und das syn-Produkt wird
enantiomerenrein erhalten, da die Wanderung der Acetylgruppe bei syn-Stellung bevorzugt
abläuft und die Lipase selektiv den (R)-Alkohol umsetzt. Durch diese Methode ist es
allerdings nicht möglich, alle vier Stereoisomere zu synthetisieren.
OH OH
SL
OH OAc
SL
OH OAc
SL
EnzymROAcRu-Kat. Ru-cat.
Epimerisierung
syn Acyl-migration
OAc OH
SLschnell
EnzymROAc
schnell
OAc OAc
SL
anti Acyl-migrationlangsam
17
39
40 41
L: largeS: small
Abbildung 3.13. Synthese von 1,3-Diolen mittels DKR
Schwieriger ist die Kombination von enzymatischen Reaktionen, die in wässrigem Medium
ablaufen, mit klassisch chemischen Reaktionen, da die Prozessparameter so optimiert
werden müssen, dass eine möglichst gute Kompatibilität erzielt wird. Dies gelang bereits in
verschiedenen Arbeitsgruppen.72,73,74,75 Beispielsweise konnten Kraußer et al. allylische
Alkohole durch Kombination einer Wittigreaktion und einer enzymatischen Reduktion in
einem Eintopfverfahren mit sehr guten Umsätzen (bis zu 90%) und exzellenten
Enantioselektivitäten (>99% ee) erhalten.76 Ebenfalls gelang die Kombination einer
metallkatalysierten Reaktion (Suzuki-Kupplung) mit einer enzymatischen Reduktion
(Abbildung 3.14).77
Abbildung 3.14. Eintopfsynthese von Biphenylalkoholen77
Nicht nur Alkoholdehydrogenasen wurden in Eintopfreaktionen genutzt. Auch eine
biokatalytische Aldolreaktion wurde mit einer metallkatalytischen Reaktion zur Synthese von

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
20
Iminocyclitolverbindungen 46 von Wong et al. eingesetzt (Abbildung 3.15).78 Auch hier
konnte die Zweistufensynthese ohne Isolierung des Zwischenprodukts durchgeführt werden.
Abbildung 3.15. Kombination einer enzymatische Aldolreaktion und einer Metallkatalyse in
einem Eintopfverfahren78
3.3 Ziel der Arbeit
In diesem Teil der Arbeit sollte durch sequenzielle Kombination von organokatalytischer
Aldolreaktion und enzymatischer Reduktion die Synthese aller möglichen Stereoisomere von
1,3-Diolen 17 durchgeführt werden. Darauf aufbauend sollte anschließend ein
Eintopfverfahren in wässrigem Medium entwickelt werden.
Basierend auf bereits veröffentlichten Vorarbeiten31 sollten die verschiedenen Isomere von
1-(4-Chlorphenyl)-1-butandiol (17f) aus der sequenziellen Synthese mit möglichst hohen
Ausbeuten isoliert werden (Abbildung 3.16). Das β-Hydroxyketon (R)-5f wurde bereits über
eine lösungsmittelfreie organokatalytische Route mit einem ee-Wert von 82% synthetisiert31
und sollte mit den ADHs aus Lactobacillus kefir (LK-ADH) und Rhodococcus species (Rsp-ADH)
diastereoselektiv mit hohem Umsatz reduziert werden.79 Das (S)-Enantiomer wurde bisher
lediglich mit einem ee-Wert von 71% eingesetzt.31 Deshalb sollte (S)-5f mit höherem
Enantiomerenüberschuss synthetisiert und das erhaltene Produkt mit den beiden ADHs
reduziert werden. Die einzelnen Diastereomere des Diols 17f sollten dann
säulenchromatographisch getrennt werden.

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
21
Abbildung 3.16. Synthese der 1,3-Diole 17f
Anschließend sollte die organokatalytische Aldolreaktion in wässrigem Medium mit
p-Chlorbenzaldehyd (1f) als Substrat durchgeführt und in einem Eintopfverfahren mit einer
enzymatischen Reduktion kombiniert werden (Abbildung 3.17). Um die Syntheseeffizienz zu
bewerten sollten auch hier die Produkte 17f isoliert und die Ausbeuten, sowie
Enantioselektivitäten mit denen aus der sequentiellen Route unter Isolierung der
Zwischenstufe 5f verglichen werden.
Abbildung 3.17. Eintopfsynthese in wässrigem Medium

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
22
3.4 Eigene Ergebnisse und Diskussion80
3.4.1 Organokatalyse unter lösungsmittelfreien Bedingungen
Das (S)-Enantiomer (S)-5f wurde mittels organokatalytischer Aldolreaktion hergestellt.
Hierzu wurde ein Organokatalysator 32 verwendet, der auf den Aminosäuren Prolin und
Leucin basiert und von Singh et al. publiziert wurde.49,51 Die Synthese von 5f erfolgte ohne
Zugabe eines organischen Lösungsmittels. Es wurde lediglich Aceton im Überschuss
eingesetzt. Die Reaktion wurde bei Raumtemperatur und mit einer Katalysatorkonzentration
von 5 mol% durchgeführt.31
Aufgrund der früheren Verwendung eines nicht diastereomerenreinen Katalysators wurde
lediglich ein Enantioselektivität von 71% ee bei der Synthese von (S)-5f erzielt. Der Einsatz
eines diastereomerenreinen Katalysators führte zu einer Steigerung des ee-Werts auf 83%
(Abbildung 3.18).
Abbildung 3.18. Organokatalytische Aldolreaktion zur Synthese von (S)-5f
3.4.2 Isolierung der 1,3-Diole
In einer früheren Arbeit konnte die sequentielle Synthese von 1,3-Diolen durch
organokatalytische Aldolreaktion und enzymatischer Reduktion gezeigt werden.31 Allerdings
wurden die Produkte 17f nicht isoliert. Bei der enzymatischen Umsetzung mit der
Alkoholdehydrogenase aus Lactobacillus kefir wurde lediglich ein Umsatz von 33% erzielt.
Außerdem wurde bei der enzymatischen Reduktion (S)-5f lediglich mit einem ee-Wert von
71% eingesetzt.31 Im Folgenden wurde die Umsetzung mit LK-ADH optimiert, (S)-5f mit
einem Enantiomerenüberschuss von 83% eingesetzt und alle Stereoisomere von 17f isoliert.

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
23
3.4.2.1 Optimierung der (R)-selektiven enzymatischen Synthese
Zur Verbesserung des Umsatzes einer enzymatischen Reaktion besteht unter anderem die
Möglichkeit die Reaktionszeit zu verlängern und die Enzymmenge zu erhöhen. Bei der
Umsetzung mittels LK-ADH konnte ein vollständiger Umsatz durch Verlängerung der
Reaktionszeit von 18 h auf 36 h und Erhöhung der Enzymaktivität von 10 U/mmol auf
100 U/mmol erzielt werden.
Bei der Reduktion des (R)-Enantiomers von 5f wurde das entsprechende Diol mit einer
Enantioselektivität von >99% ee und einem Diastereomerenverhältnis von d.r. 11:1
(syn/anti) erhalten. Die hervorragende Diastereoselektivität bei der enzymatischen
Reduktion ist auf die externe asymmetrische Induktion durch den Biokatalysator
zurückzuführen. Das Verhältnis der erhaltenen Diastereomere entspricht exakt dem
Verhältnis der beiden Enantiomere des β-Hydroxyketons (R)-5f. Durch
säulenchromatographische Aufarbeitung konnte das gewünschte Diastereomer (1R,3R)-1-
(4-Chlorphenyl)-1-butandiol ((1R,3R)-17f) mit einer Ausbeute von 59% isoliert werden
(Abbildung 3.19). Die im Vergleich zum Umsatz relativ niedrige Ausbeute ist zum einen damit
zu erklären, dass bei der Reaktion lediglich 82% des gewünschten Diastereomers entstehen
und zum anderen auf Verluste bei der säulenchromatographischen Trennung
zurückzuführen.
Abbildung 3.19. Reduktion von (R)-5f mit LK-ADH
Analoge Ergebnisse wurden bei der Umsetzung des (S)-Enantiomers von 5f (83% ee)
erhalten. Das (1S,3R)-1-(4-Chlorphenyl)-1-butandiol konnte durch säulenchromato-
graphische Aufarbeitung mit einer Ausbeute von 55% und mit einem ee-Wert von >99%
isoliert werden (Abbildung 3.20).

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
24
Abbildung 3.20. Reduktion von (S)-5f mit LK-ADH
3.4.2.2 (S)-Selektive enzymatische Umsetzung
Bei der enzymatischen Reduktion mittels Rsp-ADH war keine weitere Optimierung nötig. Bei
Einsatz von 40 U/mmol Enzym und einer Reaktionszeit von 18 h wurde bereits ein
vollständiger Umsatz erzielt. Die Umsetzung von (R)-5f lieferte das gewünschte Produkt mit
einem Diastereomerenverhältnis von d.r. 10:1 (anti/syn) und ebenfalls mit einer
hervorragenden Enantioselektivität von >99%. Nach erfolgreicher Isolierung konnte
(1R,3S)-17f mit einer Ausbeute von 66% erhalten werden (Abbildung 3.21).
Abbildung 3.21. Reduktion von (R)-5f mit Rsp-ADH
Das vierte Isomer (1S,3S)-17f konnte mit einer Ausbeute von 71% und ebenfalls mit einem
ee-Wert von >99% isoliert werden (Abbildung 3.22).

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
25
Abbildung 3.22. Reduktion von (S)-5f mit Rsp-ADH
3.4.3 Kombination der beiden Reaktionen in wässrigem Medium
Die Optimierung der organokatalytischen Aldolreaktion in wässriger Natriumchloridlösung,
ausgehend von Singh et al.,51 und anschließender Kombination mit der Biokatalyse in einem
Eintopfverfahren gelang Rulli mit m-Chlorbenzaldehyd als Substrat.81,82 Diese Methode sollte
auf die Synthese von 5f ausgehend von p-Chlorbenzaldehyd (1f) angewendet werden.
3.4.3.1 Optimierung der Organokatalyse in wässrigem Medium
Bei den bekannten Bedingungen handelt es sich um einen neunfachen Überschuss an Aceton
und 0.5 mol% Katalysatorbeladung. Das Volumen an Natriumchloridlösung entspricht dem
von 4. Mit m-Chlorbenzaldehyd als Substrat wurde nach 24 h von Rulli ein produktbezogener
Umsatz von 90% erhalten. Der ee-Wert lag mit über 90% etwas höher als bei der
lösungsmittelfreien Variante.81,82
Unter diesen Bedingungen konnte mit Substrat 1f allerdings nur ein produktbezogener
Umsatz von 58% bei einem Gesamtumsatz von 61% erzielt werden (Tabelle 3.1, Eintrag 1).
Bei allen Versuchen wurde nur eine geringe Menge an Nebenprodukten wie beispielsweise
das Aldolkondensationsprodukt oder ein Aldolprodukt aus zwei Molekülen 17f
nachgewiesen. Die erhaltene Menge an 5f ist zu niedrig für eine Kombination mit der
enzymatischen Reduktion in einer Eintopfreaktion. Um den Umsatz zu steigern wurden die
Reaktionszeit und die Katalysatormenge variiert.
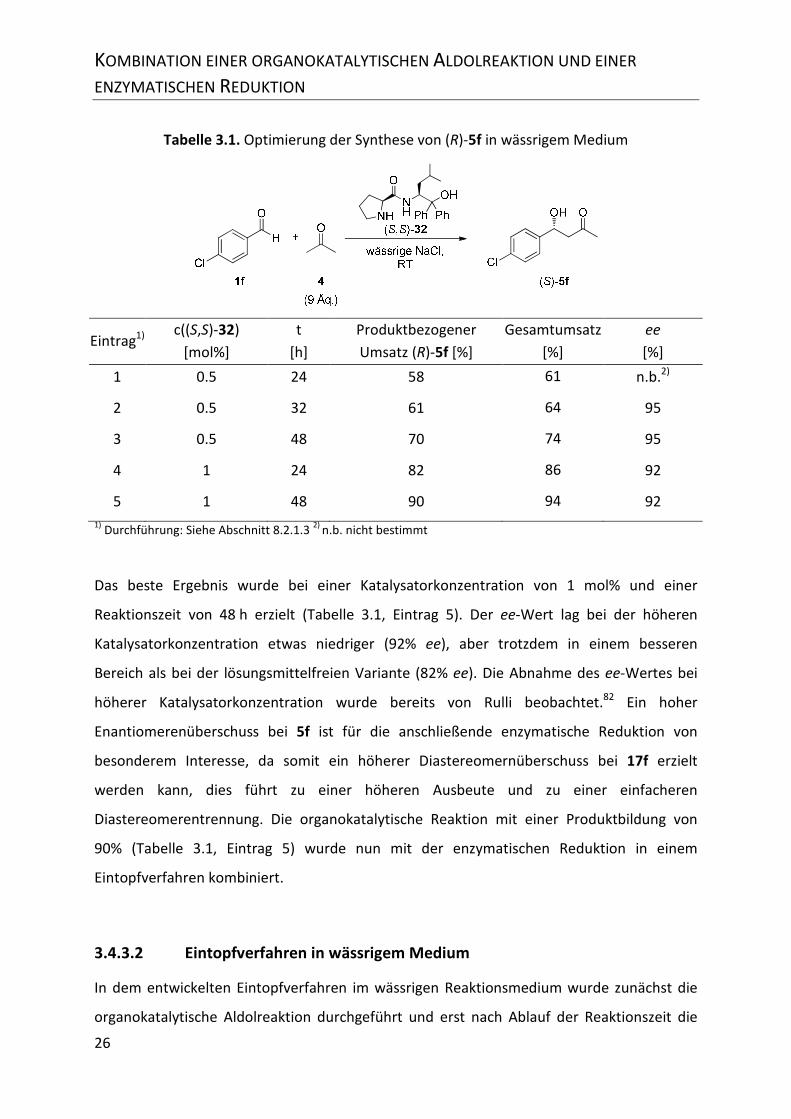
KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
26
Tabelle 3.1. Optimierung der Synthese von (R)-5f in wässrigem Medium
Eintrag1) c((S,S)-32)
[mol%]
t
[h]
Produktbezogener
Umsatz (R)-5f [%]
Gesamtumsatz
[%]
ee
[%]
1 0.5 24 58 61 n.b.2)
2 0.5 32 61 64 95
3 0.5 48 70 74 95
4 1 24 82 86 92
5 1 48 90 94 92
1) Durchführung: Siehe Abschnitt 8.2.1.3
2) n.b. nicht bestimmt
Das beste Ergebnis wurde bei einer Katalysatorkonzentration von 1 mol% und einer
Reaktionszeit von 48 h erzielt (Tabelle 3.1, Eintrag 5). Der ee-Wert lag bei der höheren
Katalysatorkonzentration etwas niedriger (92% ee), aber trotzdem in einem besseren
Bereich als bei der lösungsmittelfreien Variante (82% ee). Die Abnahme des ee-Wertes bei
höherer Katalysatorkonzentration wurde bereits von Rulli beobachtet.82 Ein hoher
Enantiomerenüberschuss bei 5f ist für die anschließende enzymatische Reduktion von
besonderem Interesse, da somit ein höherer Diastereomernüberschuss bei 17f erzielt
werden kann, dies führt zu einer höheren Ausbeute und zu einer einfacheren
Diastereomerentrennung. Die organokatalytische Reaktion mit einer Produktbildung von
90% (Tabelle 3.1, Eintrag 5) wurde nun mit der enzymatischen Reduktion in einem
Eintopfverfahren kombiniert.
3.4.3.2 Eintopfverfahren in wässrigem Medium
In dem entwickelten Eintopfverfahren im wässrigen Reaktionsmedium wurde zunächst die
organokatalytische Aldolreaktion durchgeführt und erst nach Ablauf der Reaktionszeit die

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
27
für die Enzymreaktion benötigten Reagenzien zugegeben.81,82 Bei Verwendung von LK-ADH
musste, wie bei der Einzelreaktion, Reaktionszeit und Enzymmenge erhöht werden, um
bessere Umsätze zu erzielen. Es konnte bei keiner der beiden Eintopfreaktionen eine
vollständige Umsetzung von 5f zum gewünschten Produkt 17f festgestellt werden
(Abbildung 3.23).
Abbildung 3.23. Eintopfverfahren in wässrigem Medium
Bei der Reaktion mit Rsp-ADH wurde als Nebenprodukt hauptsächlich das ungesättigte
Aldolkondensationsprodukt, das unter Abstraktion von Wasser entsteht, mit 11% und
lediglich 3% (R)-5f nachgewiesen. Bei der Eintopfsynthese mit LK-ADH wurden 17% (R)-5f
und 12% der ungesättigten Verbindung gebildet. Offenbar wird die enzymatische Reduktion
durch die Reagenzien aus dem ersten Schritt beeinträchtigt. Dies kann zum einen durch das
im Überschuss zugegebenen Aceton (4), das die substratgekoppelte Cofaktorregenerierung
stören kann, versursacht werden oder zum anderen durch die Anwesenheit des Katalysators.
Der produktbezogene Umsatz lag in einem Bereich von bis zu 79%. Durch den verbesserten
ee-Wert bei der organokatalytischen Reaktion wurden sehr gute Diastereomernverhältnisse
von d.r. >25:1 (syn/anti bzw. anti/syn) erhalten und die Ausbeuten lagen bei bis zu 73%. Die
enzymatische Reduktion führte zu den gewünschten Diastereomeren (1R,3R)- und (1R,3S)-
17f in einem exzellenten Enantiomerenüberschuss von >99% ee.

KOMBINATION EINER ORGANOKATALYTISCHEN
ENZYMATISCHEN REDUKTION
28
Um die Effizienz der entwickelten
wurden die unterschiedlichen
ist die Ausbeute an (1R,3S)-17f
Isolierung von 5f (50% Gesamta
lösungsmittelfreier Organokatalyse (54%
wässrigem Medium (73% Ausbeute
Abbildung 3.24.
Der Vergleich zeigt, dass die Ausbeute des gewünschten Produkts bei der Eintopfre
wässrigem Medium am höchsten ist. Zudem
des Zwischenprodukts verzichtet werden.
Syntheseroute im Hinblick auf Nachhaltigkeit zu entwickeln.
3.5 Zusammenfassung
Es konnte gezeigt werden, dass durch den sequentiellen
die einzelnen Diastereomere von
0
10
20
30
40
50
60
70
80
90
100
Au
sbe
ute
[%
]
Einzelreaktionen
ANOKATALYTISCHEN ALDOLREAKTION UND EIN
EDUKTION
entwickelten Eintopfreaktion in wässrigem Medium
n Synthesemethoden miteinander verglichen. In
17f aus der sequentiellen Durchführung der Einzelreaktionen
Gesamtausbeute über zwei Schritte), einem Eintopfverfahren mit
freier Organokatalyse (54% Ausbeute)31,83 und dem Eintopfverfahren in
Ausbeute) graphisch dargestellt.
. Vergleich der verschiedenen Synthesemethoden
Der Vergleich zeigt, dass die Ausbeute des gewünschten Produkts bei der Eintopfre
wässrigem Medium am höchsten ist. Zudem konnte bei diesem System auf eine Isolierung
des Zwischenprodukts verzichtet werden. Es ist gelungen eine vielversprechende
Syntheseroute im Hinblick auf Nachhaltigkeit zu entwickeln.
Zusammenfassung
dass durch den sequentiellen Aufbau der beiden Stereozentren
die einzelnen Diastereomere von 17f mit hohen Umsätzen und zugleich hohen
5054
73
Einzelreaktionen Eintopf (lösungsmittelfrei) Eintopf (wässrig)
LDOLREAKTION UND EINER
in wässrigem Medium darzustellen,
verglichen. In Abbildung 3.24
aus der sequentiellen Durchführung der Einzelreaktionen mit
), einem Eintopfverfahren mit
und dem Eintopfverfahren in
Vergleich der verschiedenen Synthesemethoden
Der Vergleich zeigt, dass die Ausbeute des gewünschten Produkts bei der Eintopfreaktion in
konnte bei diesem System auf eine Isolierung
Es ist gelungen eine vielversprechende
der beiden Stereozentren
msätzen und zugleich hohen
Eintopf (wässrig)
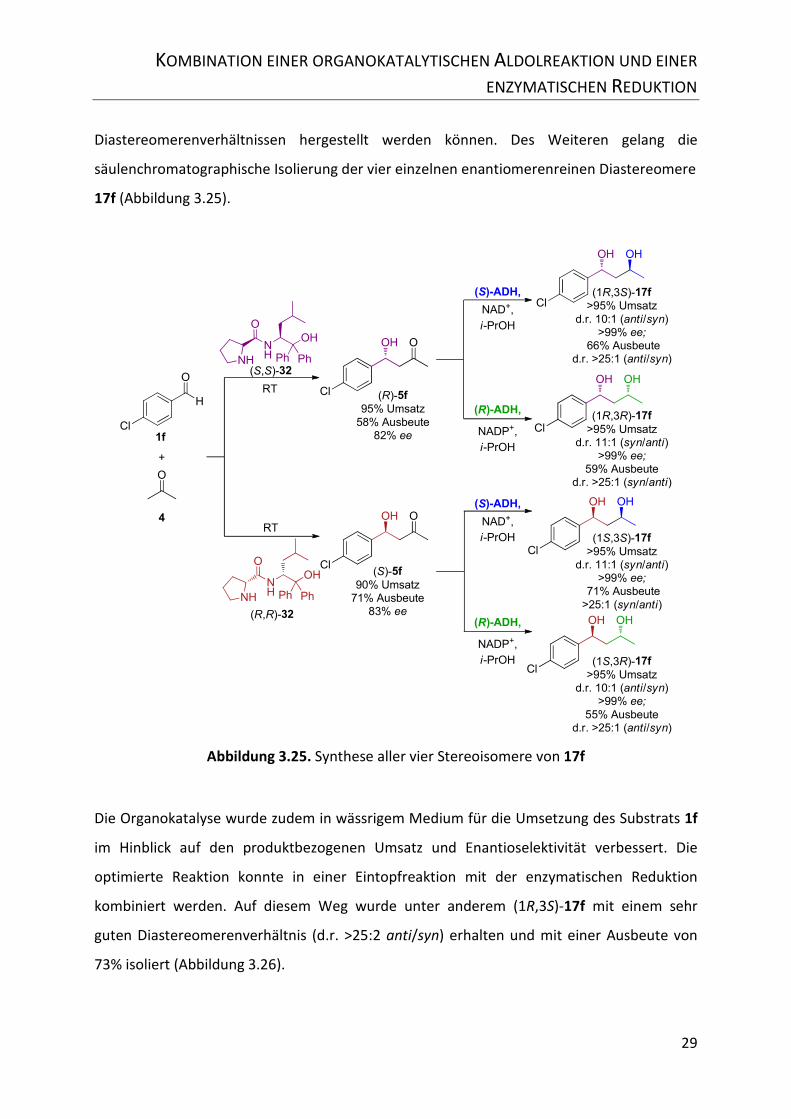
KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
29
Diastereomerenverhältnissen hergestellt werden können. Des Weiteren gelang die
säulenchromatographische Isolierung der vier einzelnen enantiomerenreinen Diastereomere
17f (Abbildung 3.25).
Cl
O
H
O
Cl
OH O
Cl
OH O
(R)-5f95% Umsatz58% Ausbeute
82% ee
(S)-5f90% Umsatz71% Ausbeute
83% ee
Cl
OH OH
Cl
OH OH
Cl
OH OH
Cl
OH OH
NAD+,
i-PrOH
(1R,3S)-17f>95% Umsatz
d.r. 10:1 (anti/syn)>99% ee;
66% Ausbeuted.r. >25:1 (anti/syn)
(1R,3R)-17f>95% Umsatz
d.r. 11:1 (syn/anti)>99% ee;
59% Ausbeuted.r. >25:1 (syn/anti)
(1S,3S)-17f>95% Umsatz
d.r. 11:1 (syn/anti)>99% ee;
71% Ausbeute>25:1 (syn/anti)
(1S,3R)-17f>95% Umsatz
d.r. 10:1 (anti/syn)>99% ee;
55% Ausbeuted.r. >25:1 (anti/syn)
NHNH
OOH
Ph Ph
NHNH
OOH
Ph Ph
RT
RT
+
(S,S)-32
(R,R)-32
NADP+,
i-PrOH1f
4
(S)-ADH,
(R)-ADH,
NAD+,
i-PrOH
(S)-ADH,
NADP+,
i-PrOH
(R)-ADH,
Abbildung 3.25. Synthese aller vier Stereoisomere von 17f
Die Organokatalyse wurde zudem in wässrigem Medium für die Umsetzung des Substrats 1f
im Hinblick auf den produktbezogenen Umsatz und Enantioselektivität verbessert. Die
optimierte Reaktion konnte in einer Eintopfreaktion mit der enzymatischen Reduktion
kombiniert werden. Auf diesem Weg wurde unter anderem (1R,3S)-17f mit einem sehr
guten Diastereomerenverhältnis (d.r. >25:2 anti/syn) erhalten und mit einer Ausbeute von
73% isoliert (Abbildung 3.26).

KOMBINATION EINER ORGANOKATALYTISCHEN ALDOLREAKTION UND EINER
ENZYMATISCHEN REDUKTION
30
Abbildung 3.26. Eintopfsynthese von (1R,3S)-17f
Der Vergleich mit der sequentiellen Methode und der lösungsmittelfreien Eintopfsynthese
zeigte, dass hier ein vielversprechender Prozess im Hinblick auf die Nachhaltigkeit etabliert
werden konnte.

ENZYMATISCHE ALDOLREAKTION
31
4. Enzymatische Aldolreaktion
4.1 Einleitung
Aldolasen katalysieren die nichthydrolytische Spaltung von C-C-Bindungen und gehören zur
Klasse der Lyasen.17 Die Einteilung der Aldolasen nach ihrer Donorspezifität wurde bereits in
Kapitel 1 (Abbildung 1.5) beschrieben. Die DHAP-abhängige Fructose-1,6-biphosphataldolase
ist Teil des Glycolysestoffwechsels, bei dem Glucose zu Pyruvat abgebaut wird. Diese
Aldolase spaltet im ersten Teil des Abbauweges Fructose-1,6-biphosphat in DHAP und
Glycerinaldehyd-3-phosphat.84 Die Threoninaldolasen, die in vielen Pflanzen, Wirbeltieren,
Bakterien, Hefen und Pilzen vorkommen, sind für die Spaltung von Threonin (12c) in Glycin
(11) und Acetaldehyd (1c) verantwortlich (Abbildung 4.1). Im Stoffwechsel erfolgt dann der
weitere Abbau zu Acetyl-CoA und Pyruvat.29
OH
O
NH2
OH
ThreoninaldolaseH
O+ OH
O
NH2
Acetyl-CoA Pyruvat
12c 1c 11
Abbildung 4.1. Enzymatische Spaltung von Threonin (12c) im Körper
Die vier verschiedenen Aldolasetypen katalysieren ebenfalls die Rückreaktion bzw. die
C-C-Bindungsknüpfung, die aus synthetischer Sicht attraktiv ist. Die Herstellung von optisch
aktiven β-Hydroxy-α-aminosäuren ist hierbei von besonderer Bedeutung, vor allem im
Hinblick auf pharmazeutische Anwendungen. Dieser Aminosäurebaustein findet sich in
Antibiotika wie Thiamphenicol und Vancomycin (Abbildung 4.2) oder in
Entzündungshemmern, wie Cylcomarinen, wieder.85

ENZYMATISCHE ALDOLREAKTION
32
Abbildung 4.2. Vancomycin
Es gibt einige Beispiele für den Einsatz von Threoninaldolasen bei der Synthese komplexer
Moleküle.86,87,88 Digitoxin (49) ist ein Na+/K+-ATPase-Inhibitor, der bei kongestiver
Herzinsuffizienz oder bei Herzrhythmusstörungen eingesetzt wird. Bei der Synthese über
mehrere Stufen wird die β-Hydroxy-α-aminosäure 47 durch enzymatische Aldolreaktion
hergestellt und kann dann zum gewünschten Produkt weiter umgesetzt werden (Abbildung
4.3).87
Abbildung 4.3. Synthese von Digitoxin (49)87
Auch die Herstellung eines Fucosyltransferase-Inhibitors 51 verläuft über eine
Threoninaldolase-katalysierte Reaktion.88 Die dabei erhaltene β-Hydroxy-α-aminosäure 50
wird über mehrere Stufen zu 51 umgesetzt. (Abbildung 4.4).

ENZYMATISCHE ALDOLREAKTION
33
Abbildung 4.4. Synthese eines Fucosyltransferase-Inhibitors 5188
Diese beiden Beispiele zeigen, dass die TA-katalysierte Aldolreaktion als attraktive und
selektive C-C-Bindungsknüpfung bereits in der Synthese pharmazeutisch relevanter
Verbindungen genutzt werden kann.
4.2 Stand der Wissenschaft
4.2.1 Synthesestrategien für β-Hydroxy-α-aminosäuren
Die chemischen Methoden zur Synthese von β-Hydroxy-α-aminosäuren sind vielfältig und
basieren auf verschiedenen Konzepten. Dabei ist vor allem die Aldolreaktion von Glycin (11),
bzw. dessen Derivaten mit Aldehyden 1, die durch unterschiedliche Verbindungen katalysiert
werden kann, zu nennen (Abbildung 4.5).89
Abbildung 4.5. Aldolreaktion von Glycin (11) und Aldehyden 1
Zudem gibt es Methoden für chemokatalytische, dynamisch kinetische Racemat-
spaltungen.90 Allerdings muss bei diesen meist auf Schutzgruppen zurückgegriffen werden.
Im Folgenden sollen nun einige dieser Konzepte vorgestellt werden.
Die Gruppe um Hamada veröffentlichte bereits mehrere Katalysatorsysteme, die in der Lage
sind selektiv β-Keto-α-aminosäureester zu den entsprechenden anti-β-Hydroxy-
α-aminosäureestern zu reduzieren.90 In Abbildung 4.6 ist die Spaltung des Ketoesters 52 mit
Hilfe eines Iridiumkomplexes 54 gezeigt.90d Es handelt sich hierbei um eine dynamisch

ENZYMATISCHE ALDOLREAKTION
34
kinetische Racematspaltung, bei der sehr gute Ausbeuten und Enantioselektivitäten erzielt
werden konnten.
Abbildung 4.6. DKR zur Synthese von anti-β-Hydroxy-α-aminosäureestern 5390d
Weitere Methoden verlaufen über 1,3-Cycloadditionen91 oder auch nukleophile92 sowie
radikalische93 Substitutionen. In Abbildung 4.7 wird die Zielverbindung 58, ausgehend von
einer α-Aminosäure 55 nach radikalischer Bromierung und Umwandlung zum Oxazolidon 57
in einer Ausbeute von 77% erhalten.93
Abbildung 4.7. β-Hydroxy-α-aminosäuresynthese nach Crich93

ENZYMATISCHE ALDOLREAKTION
35
Evans et al. berichteten schon 1986 über die Synthese von syn- und anti-β-Hydroxy-
α-aminosäuren über Oxazolidonderivate,94 wobei diese die Ausgangsverbindung 59
darstellten und als chirale Glycinsynthone bezeichnet wurden (Abbildung 4.8). Problematisch
ist bei dieser Syntheseroute die Verwendung von Natriumazid aufgrund der toxischen
Eigenschaften.
Abbildung 4.8. Synthese über Oxazolidone als Glycinsynthone94b
Das Glycinsynthon 59 reagiert als Enoläquivalent mit einem Aldehyd. Auf diesem Konzept
basieren eine Reihe weiterer Synthesebeispiele. So können β-Hydroxy-α-aminosäuren auch
nach bekannten Methoden der asymmetrischen Aldolreaktion hergestellt werden.
Aktiviertes Glycin, beispielsweise vom Typ 63, und ein Aldehyd 1 können mit Hilfe von
chiralen Metallkatalysatoren zu den entsprechenden Produkten umgesetzt werden.95,96,97
Das Beispiel von Shibasaki95 zeigt die Reaktion der Schiff'schen Base 63 mit einem
aliphatischen Aldehyden 64, katalysiert durch Li3[La(BINOL)3] (67) (Abbildung 4.9). Bei dieser
metallkatalysierten Reaktion entsteht ein Diastereomerengemisch von d.r. 59:41 (anti/syn)
mit guten bis mäßigen ee-Werten und sehr hohen Ausbeuten von 93%.

ENZYMATISCHE ALDOLREAKTION
36
Abbildung 4.9. Metallkatalysierte Aldolreaktion zur Synthese von β-Hydroxy-
α-aminosäuren95
Diese Reaktion kann ebenfalls organokatalytisch durchgeführt werden. Als Katalysatoren
stehen hier quartäre Ammoniumsalze,98,99 sogenannte Phasentransferkatalysatoren zur
Verfügung, aber auch von einer Variante mit Prolin100 wurde berichtet. Bei der Verwendung
von Prolin als Katalysator wurde ein Phthalimid 68 mit einem Aldehyden 64 umgesetzt und
die entsprechenden β-Hydroxy-α-aminosäurederivate 70 nach Oxidation in sehr guten
Diastereo- (d.r. >100:1 (anti/syn)) und Enantioselektivitäten (>99% ee) erhalten (Abbildung
4.10).100
Abbildung 4.10. Organokatalytische Synthese von β-Hydroxy-α-aminosäurederivaten100

ENZYMATISCHE ALDOLREAKTION
37
Die Nutzung quartärer Ammoniumsalze wurde schon längere Zeit verfolgt. Es gibt zwei
Katalysatorgruppen, die zum einen auf einer Alkaloidstruktur98,99,101 basieren und zum
anderen auf Binatphthalineinheiten.102 Bei allen gezeigten Anwendungen muss Glycin in
aktivierter Form als Schiff'sche Base, wie beispielsweise als Silylenolether 71, eingesetzt
werden. Ein von Maruoka et al. entwickeltes Beispiel ist in Abbildung 4.11 dargestellt.102b
Das gewünschte Produkt 72 wird mit sehr hohen Enantioselektivitäten (97% ee) erhalten,
auch das Diastereomerenverhältnis ist im Vergleich zu anderen Beispielen sehr gut.
Abbildung 4.11. Synthese von β-Hydroxy-α-aminosäuren 72 mittels
Phasentransferkatalyse102b
Es gibt viele Möglichkeiten zur Herstellung von β-Hydroxy-α-aminosäuren, allerdings ist bei
allen gezeigten Methoden das Schützen bzw. die Aktivierung der Ausgangsverbindungen
nötig. Zudem sind meist aufwändige chirale Katalysatorsysteme erforderlich und bis zum
Erhalt des gewünschten Produkts sind mehrere Reaktionsschritte notwendig. Ein Nachteil
sind auch die meist relativ niedrigen Gesamtausbeuten und Enantio-, sowie
Diastereoselektivitäten.
4.2.2 Threoninaldolasen
Eine Alternative zur Synthese von β-Hydroxy-α-aminosäuren bietet der Einsatz von
Threoninaldolasen, die in der Lage sind, Glycin (11) und einen Aldehyden 1 direkt in das
gewünschte Produkt 12 umzusetzen. Eine Aktivierung oder das Schützen der
Ausgangsverbindungen ist hier nicht notwendig. Zudem kann die Reaktion in wässrigem

ENZYMATISCHE ALDOLREAKTION
38
Medium bei milden Temperaturen durchgeführt werden. Diese Enzyme benötigen den
Cofaktor Pyridoxal-5-phosphat (PLP) in katalytischen Mengen, der durch Bildung einer
Schiff'schen Base Glycin (11) aktiviert (Abbildung 4.12).
Abbildung 4.12. Aktivierung von Glycin (11) durch PLP
Im Gegensatz zu den bereits beschriebenen Alkoholdehydrogenasen muss hier kein
Cofaktorregenerierungssystem verwendet werden. Die Enantioselektivität bei der Bildung
des Stereozentrums in α-Position ist sehr hoch (>99% ee), dagegen wird das Stereozentrum
in β-Position nicht sehr selektiv aufgebaut und es entstehen Diastereomeren-
gemische.29,103,104
Wie bereits in Kapitel 1 erwähnt, gibt es D- und L-selektive Aldolasen (D-TA, L-TA). Diese
können wiederum nach ihrer Spezifität bei der Spaltung von Threonin (12c) unterteilt
werden (Abbildung 4.13). L-Threoninaldolasen setzen selektiv L-syn-Threonin um,
wohingegen L-allo-TAs nur L-anti-Threonin spalten. Eine weniger spezifische Variante ist
ebenfalls als L-low specificity-Aldolase bekannt und akzeptiert beide Verbindungen. Bei der
D-spezifischen Variante ist nur eine low specificity-Variante bekannt.103
Abbildung 4.13. Einteilung der L-TA katalysierten Reaktion103

ENZYMATISCHE ALDOLREAKTION
39
Die C-C-Bindungsknüpfung ist weniger spezifisch als die Bindungsspaltung. Die
Diastereoselektivität bei der Synthese der gewünschten Produkte hängt stark davon ab, ob
die Reaktion unter thermodynamischer oder kinetischer Kontrolle durchgeführt werden
kann. Unter kinetischer Kontrolle werden meist hohe Diastereomerenüberschüsse erzielt,
allerdings ist der Umsatz dabei oftmals niedrig.105
Die Gruppe um Griengl untersuchte, warum die D-TA eine hohe Stereospezifität bei hohen
Umsätzen aufwies, wohingegen sich beim L-spezifischen Biokatalysator schon nach kurzer
Zeit das thermodynamische Gleichgewicht einstellte und somit bei hohen Umsätzen nur
geringe Diastereomerenüberschüsse gefunden wurden. Die langsame Einstellung des
thermodynamischen Gleichgewichts konnte mit der Katalysatoreigenschaft einer höheren
Energiebarriere für die syn/anti-Epimerisierung bei der D-Threoninaldolase begründet
werden.106
Bis jetzt wurde eine Vielzahl von Threoninaldolasen isoliert und charakterisiert, sowie zur
Synthese verschiedener β-Hydroxy-α-aminosäuren eingesetzt.29 Ein Beispiel für eine
enzymatische Racematspaltung lieferte Yamada.103,107 Unter Einsatz einer D-TA aus
Anthrobacter sp. konnte (2S,3R)-12d in einer Ausbeute von 48% und einem
Enantiomerenüberschuss von >99% ee, bei einer Substratkonzentration von 200 mM erzielt
werden (Abbildung 4.14). Der Vorteil bei dieser Methode besteht darin, dass
diastereomerenreines Racemat eingesetzt wird durch dessen Spaltung dann diastereo- und
enantiomerenreines Produkt (2S,3R)-12d ensteht.
Abbildung 4.14. Enzymatische Racematspaltung103
Häufiger wurden Threoninaldolasen allerdings in der direkten asymmetrischen Synthese von
β-Hydroxy-α-aminosäuren eingesetzt, da hier prinzipiell ein vollständiger Umsatz erzielt
werden kann, wohingegen bei der Racematspaltung lediglich ein theoretischer Umsatz von
50% möglich ist. Um bei der direkten Synthese das Gleichgewicht der Reaktion auf die Seite

ENZYMATISCHE ALDOLREAKTION
40
der gewünschten Produkte zu verschieben, muss Glycin im Überschuss eingesetzt
werden.108,109
Pionierarbeiten auf dem Gebiet der asymmetrischen Synthese wurden von Wong et al.
durchgeführt. Dabei wurde die TA-katalysierte enzymatische Aldolreaktion mit drei
unterschiedlichen Enzymen auf ihre Substratbreite untersucht.105,110 Es wurden zwei
L-spezifische Aldolasen aus E. coli105 und Candida humicola110, sowie eine D-TA aus
Xanthomonus oryzae105 eingesetzt. Letztere zeigte eine ausgeprägte syn-Spezifität bei den
verwendeten Substraten, wohingegen die L-TA aus E. coli bei aliphatischen Aldehyden anti-
Produkte (12c, 12g) und bei aromatischen Aldehyden syn-Produkte (12e) bildete.
Heteroatome in β-Position führten ebenfalls bei den L-TAs zu einer anti-Selektivität.
Allerdings wurden meist nur geringe Ausbeuten erzielt. Eine Übersicht der Ergebnisse ist in
Abbildung 4.15 dargestellt.
Abbildung 4.15. Produktspektrum der L-TA aus E. coli und der D-TA aus X. oryzae105
Für die beiden Aldolasen aus E. coli und X. oryzae wurde zudem eine genaue Betrachtung
der Reaktionsbedingungen durchgeführt.105 Hierbei wurde die Spaltung von Threonin (12c)
untersucht und nicht die C-C-Bindungsknüpfung. Beide Enzyme wiesen bei einem pH-Wert

ENZYMATISCHE ALDOLREAKTION
41
von 7.5 die höchste Aktivität auf und tolerierten organische Additive, wie DMSO oder DMF in
einer Konzentration bis zu 30%. Vor allem die L-TA aus E. coli wies eine starke
Temperaturabhängigkeit auf: Während bei 25°C keine Bindungsknüpfung als Rückreaktion
auftrat, wurde bei 50°C die höchste Aktivität ermittelt.105
Eine ähnliche Untersuchung für zwei unterschiedlich selektive Threoninaldolasen, L-TA aus
Pseudomonas putida und D-TA aus Alcaligenes xylosoxidans, wurde von Griengl et al.
durchgeführt.109,111 Hier wurde allerdings die Synthese von Phenylserin (12e) untersucht und
nicht die Spaltung von Threonin (12c). Die D-selektive Aldolase wies eine sehr hohe
Diastereoselektivität (d.r. 99:1 syn/anti) und einen hohen Umsatz von 79% bei einer
Reaktionstemperatur von 5°C auf. Bei der L-TA wurde keine analoge
Temperaturabhängigkeit beobachtet. Mit beiden Biokatalysatoren wurden verschiedenste
aliphatische111 und aromatische109 Aldehyde zu den entsprechenden β-Hydroxy-
α-aminosäuren mit teils sehr guten Ausbeuten von bis zu 90% und exzellenten ee-Werten
von >99% umgesetzt. Die Diastereoselektivität war bei Verwendung der D-TA aufgrund des
sich langsam einstellenden thermodynamischen Gleichgewichts deutlich höher.106 In
Abbildung 4.16 sind die besten Ergebnisse aus der Umsetzung mit verschiedenen
substituierten Benzaldehyden bei Einsatz einer L-TA gezeigt.109 Je nach Substituent und
dessen Position konnten Diastereomerenverhältnisse von bis zu d.r. 78:22 (syn/anti) bei
Ausbeuten von bis zu 90% erzielt werden.
Abbildung 4.16. Ergebnisse der Synthese verschieden substituierter aromatischer
β-Hydroxy-α-aminosäuren 12109
Die Synthese anderer Produktklassen, wie ω-Carboxy-β-hydroxy-α-aminosäuren112 und
β-Hydroxy-α,ω-diaminosäuren113 75 wurden ebenfalls unter Verwendung der L-TA aus E. coli
durchgeführt. Während erstere als epimerische Mischung in einer Ausbeute von bis zu 67%
erhalten wurden, konnten die syn-Diaminosäuren 75 in einem Überschuss von bis zu
d.r. 82:18 (syn/anti) und einer Ausbeute von 27% isoliert werden (Abbildung 4.17).113

ENZYMATISCHE ALDOLREAKTION
42
Abbildung 4.17. Synthese von β-Hydroxy-α,ω-diaminosäuren 74113
Trotz der vielen Vorteile der enzymatischen Synthese von β-Hydroxy-α-aminosäuren, wie die
milden, wässrigen Bedingungen oder der Einsatz ungeschützter Substrate, gibt es auch
einige Nachteile. Lange Zeit war die Umsetzung auf Glycin (11) als Donor beschränkt und es
waren somit keine α-alkylierten Verbindungen zugänglich. Mittlerweile gibt es einige
Beispiele, bei denen Alanin, Serin oder Cystein als Donor akzeptiert werden.19,20,21 Es
konnten eine L-TA aus A. jandaei und eine D-TA aus Pseudomonas sp. erfolgreich bei der
Reaktion von entsprechenden D-Aminosäuren mit verschiedenen Aldehyden eingesetzt
werden.19 In Abbildung 4.18 sind einige dieser interessanten Produkte gezeigt. Die Umsätze
liegen in einem moderaten Bereich von bis zu 60% und die Enantioselektivität beträgt auch
hier >99% ee. Das Diastereomerenverhältnis ist wiederum stark vom Biokatalysator und dem
jeweiligen Substrat abhängig.
Abbildung 4.18. Synthese von α-alkylierten β-Hydroxy-α-aminosäuren 76, 77, 7819
Ein weiterer Nachteil bei der enzymatischen Umsetzung von Glycin (11) mit Aldehyden 1 ist
die teils geringe Diastereoselektivität. Um hohe Produktausbeuten zu erzielen, sollte diese

ENZYMATISCHE ALDOLREAKTION
43
möglichst hoch sein. Die Trennung der Diastereomere erfolgt meist säulenchromato-
graphisch und unter vorheriger Einführung von Schutzgruppen.112 Die Diastereoselektivität
der Biokatalysatoren kann beispielsweise durch Protein-Engineering erhöht werden.67 Des
Weiteren wurden die Versuche lediglich in kleinem Maßstab (<1 g) durchgeführt. Um das
Konzept der Threoninaldolase-katalysierten Aldolreaktion für eine industrielle Anwendung
attraktiver zu machen, sollten die Reaktionen in größerem Maßstab durchgeführt werden
und die Isolierung der gewünschten Produkte verbessert werden. Ebenso ist die Erhöhung
der Substratkonzentration, die bisher lediglich bei maximal 100 mM lag, in dieser Hinsicht
ein wichtiger Faktor, da so eine möglichst hohe volumetrische Produktivität erreicht werden
könnte.
4.3 Ziel der Arbeit
Unter Einsatz der L-selektiven Threoninaldolase aus E. coli105,114 sollte eine genaue
Untersuchung und Optimierung der enzymatischen Aldolreaktion durchgeführt werden.
Außerdem sollten verschiedene aromatische Substrate umgesetzt werden.
Als Ausgangspunkt wurden die Arbeiten von Griengl et al.106 herangezogen (Abbildung 4.19).
Die enzymatische Umsetzung sollte mit Benzaldehyd (1e) als Standardsubstrat und der zur
Verfügung stehenden L-TA aus E. coli durchgeführt werden.
Abbildung 4.19. Literaturbekannte Ergebnisse der TA-katalysierten Aldolreaktion106
Anschließend bestand die Aufgabe darin die Reaktionsbedingungen im Hinblick auf
Enzymmenge, Glycinüberschuss, Temperaturabhängigkeit und Substratmenge, sowie den
Einfluss von Additiven zu untersuchen und zu optimieren. Des Weiteren sollte die
Diastereoselektivität des Enzyms bei verschieden substituierten Benzaldehyden evaluiert
und die Akzeptanz der Aldolase aus E. coli für thiamphenicolrelevante Aldehyde getestet

ENZYMATISCHE ALDOLREAKTION
44
werden. Bis jetzt wurde mit diesem Biokatalysator lediglich die Bindungsspaltung von
Threonin (12c) untersucht und vor allem aliphatische Aldehyde bei der Synthese von β-
Hydroxy-α-aminosäuren eingesetzt.105
Ziel war es mit dem besten Substrat (höchster Umsatz und Diastereoselektivität) eine
Prozessoptimierung durchzuführen (Abbildung 4.20). Diese beinhaltete die Erhöhung der
Substratkonzentration und die Verringerung des Glycinüberschusses bei vollständigem
Umsatz und guten Diastereomerenüberschüssen, sowie exzellenten ee-Werten von >99%.
Um die enzymatische Reaktion industriell attraktiver zu machen, sollte das Konzept in
größerem Maßstab angewendet und das Produkt im Grammmaßstab isoliert werden.
Abbildung 4.20. Prozessoptimierung der enzymatischen Aldolreaktion
4.4 Eigene Ergebnisse und Diskussion115
4.4.1 Vorversuche
Vor Beginn der Untersuchung der enzymatischen Umsetzung von Glycin (11) und
Benzaldehyd (1h) wurden Versuche zur Stabilität des Produkts 12e, sowie die Analyse einer
möglichen Hintergrundreaktion durchgeführt. Des Weiteren wurden die entsprechenden
racemischen Verbindungen als Referenzsubstanzen synthetisiert.
4.4.1.1 Racematsynthese
Die Racematsynthese gelang über eine basenkatalysierte Aldolreaktion von Glycin (11) und
den entsprechenden Aldehyden 1.116 Am Beispiel der Synthese von Phenylserin 12e
(Abbildung 4.21, vgl. auch 8.2.2.1) wird die Abhängigkeit des Diastereomerenverhältnisses
von den Reaktionsbedingungen deutlich. Bei höherer Temperatur und langer Reaktionszeit

ENZYMATISCHE ALDOLREAKTION
45
wurde ausschließlich das thermodynamisch günstigere syn-Produkt 12e bei einer Ausbeute
von 72% erhalten, während die Reaktion bei 0°C und 4 h Reaktionszeit ein
Diastereomerengemisch von d.r. 56:44 (syn/anti) bei einer Ausbeute von 62% lieferte. Durch
die Gleichgewichtslage war es nicht möglich reines anti-Produkt 12e herzustellen. Die
Isolierung erfolgte über mehrmalige Umkristallisation in Wasser.
Abbildung 4.21. Synthese von racemischem Phenylserin
Auf diesem Weg wurden noch weitere substituierte Phenylserinderivate 12 hergestellt, die
als Referenzverbindungen für die Analytik benötigt wurden (Abbildung 4.22).
OH
OOH
NH2Cl
rac-12f0°C, 24 h: 36% Ausbeute,
d.r. 79:21(syn/anti)
OH
OOH
NH2MeS
rac-12d0°C, 26 h: 26% Ausbeute,
d.r. >25:1(syn/anti)
OH
OOH
NH2
rac-12lRT, 2 h: 52% Ausbeute,d.r. 74:26 (syn/anti)
ClOOH
NH2
rac-12j0°C, 48 h: 11% Ausbeute,
d.r. 87:13 (syn/anti)
BrOH
OOH
NH2
rac-12m0°C, 48 h: 7% Ausbeute,
d.r. >25:1 (syn/anti)
HO
OH
OOH
NH2
rac-12iRT, 1 h: 38% Ausbeute,d.r. 82:18 (syn/anti)
Cl
OH
OOH
NH2
rac-12nRT, 3 h: 23% Ausbeute,d.r. 69:31 (syn/anti)
OMe
+NH2
O
OH
OH
OH
O
NH2
H
O
1
(1-2 Äq.)11
(0.7-2.7 M)12
5 M NaOH
R R
Abbildung 4.22. Racemische Phenylserinderivate

ENZYMATISCHE ALDOLREAKTION
46
Je nach Substituent wurden unterschiedliche Diastereomerenverhältnisse erzielt. Dies ist auf
die Lage des thermodynamischen Gleichgewichts zurückzuführen. Beispielsweise konnten
bei 12i, 12l und 12n die gewünschten Diastereomerengemische bereits nach kurzer
Reaktionszeit und Raumtemperatur erhalten werden, wohingegen bei 12d bzw. 12m selbst
bei 0°C nur das syn-Isomer nachgewiesen werden konnte. 12i konnte mit einem
Diastereomerenverhältnis von d.r. 82:18 (syn/anti) und mit einer Ausbeute von 38% erhalten
werden. Durch Umkristallisation in Wasser konnte eine geringe Menge an
diastereomerenreinem anti-12i (Ausbeute 1%) isoliert werden.
Im Zuge der Herstellung der racemischen Verbindungen wurden die Phenylserinderivate mit
Benzoylchlorid (79) umgesetzt, zum einen zur vollständigen Charakterisierung und zum
anderen als Referenzverbindungen für die Umsatz- bzw. ee-Wertbestimmung der Produkte
aus der Enzymumsetzung mittels 1H-NMR-Spektroskopie bzw. chiraler HPLC. Die Reaktion
von 12 mit Benzoylchlorid (79) erfolgte im Basischen unter Zugabe von 1.2 Äquivalenten 79
(Abbildung 4.23, vgl. auch 8.2.2.2).
Abbildung 4.23. Derivatisierung mit Benzoylchlorid (79)
Bis auf rac-12m konnten alle in Abbildung 4.22 gezeigten Produkte umgesetzt werden
(Abbildung 4.24). Die Derivatisierungsprodukte 80 wurden mit Ausbeuten bis zu 55%
erhalten und vollständig charakterisiert. Bei der ortho-chlorsubstituierten Verbindung rac-
80i wurde jeweils das isolierte syn-80 bzw. anti-80 umgesetzt. Durch Umkristallisation
konnten die Diastereomere der ortho-methoxysubstituierten Verbindung rac-80n
angereichert werden (d.r. >25:1 syn/anti; d.r. 90:10 anti/syn).

ENZYMATISCHE ALDOLREAKTION
47
Abbildung 4.24. Racemische Produkte aus Derivatisierung mit Benzoylchlorid (79)
4.4.1.2 Hintergrundreaktion
Vor Durchführung der enzymatischen Umsetzung wurde getestet, ob es zu einer Reaktion
zwischen Glycin (11) und Benzaldehyd (1e) ohne Anwesenheit des Biokatalysators kommt.
Möglich wäre die Aktivierung von Glycin durch Bildung einer Schiff'schen Base 81 mit dem
Aldehyd, wodurch racemisches Produkt entstehen könnte. Dies würde dann die
Enantioselektivität der enzymatischen Reaktion beeinträchtigen. Es wurden mehrere
Versuche durchgeführt, bei denen verschiedene Mengen an Glycin (11) und Benzaldehyd
(1e) in Puffer (pH 7 bzw. pH 8) mit und ohne Anwesenheit des Cofaktors PLP 27 h gerührt
wurden (Abbildung 4.25, vgl. auch 8.2.2.3).
Abbildung 4.25. Untersuchung einer möglichen Hintergrundreaktion
Es wurde keine Hintergrundreaktion nachgewiesen. Nach Entfernen des Lösungsmittels im
Vakuum konnte lediglich das Edukt 11 im 1H-NMR-Spektrum nachgewiesen werden.

ENZYMATISCHE ALDOLREAKTION
48
4.4.1.3 Epimerisierung
Als nächstes wurde überprüft, ob Phenylserin (12e) unter den Bedingungen der
enzymatischen Reaktion stabil ist oder ob eine Epimerisierung auftritt, die wiederum die
Enantioselektivität der Biotransformation beeinträchtigen könnte. Dazu wurde sowohl
reines rac-syn-Phenylserin (rac-syn-12e), als auch eine racemische syn/anti-Mischung (d.r.
56:44) in Puffer (pH 7 bzw. pH 8) gelöst und 27 h gerührt (Abbildung 4.26, vgl. auch 8.2.2.4).
Abbildung 4.26. Untersuchung einer möglichen Epimerisierung
Auch hier wurde keinerlei Veränderung der eingesetzten Verbindungen gefunden. Im
1H-NMR-Spektrum wurde das entsprechende Diastereomerenverhältnis nach der Reaktion
wieder erhalten und es konnte auch kein Glycin (11), das bei einer Zersetzung entstehen
würde, nachgewiesen werden. Aufbauend auf diesen Ergebnissen wurde nun mit der
Durchführung der enzymatischen Reaktion begonnen.
4.4.2 Photometertest zur Bestimmung der Aktivität der L-Threoninaldolase
Zur Bestimmung der Aktivität von Threoninaldolasen (TA) wird in der Literatur auf eine
photometrische Bestimmung mit Hilfe einer indirekten Methode zurückgegriffen, bei der die
Reduktion von Acetaldehyd (1c) verfolgt wird, der bei der enzymatischen Spaltung von
Threonin (12c) entsteht.105 Es wird der Cofaktorverbrauch (NADH) bei der Reduktion des
Aldehyds durch eine Alkoholdehydrogenase aus Bäckerhefe photometrisch beobachtet
(Abbildung 4.27).
Abbildung 4.27. Indirekter Photometertest zur Bestimmung der Aldolaseaktivität105

ENZYMATISCHE ALDOLREAKTION
49
Aus der Extinktionsabnahme bei 340 nm (Absorptionsmaximum von NADH) kann die
Enzymaktivität in U/mL berechnet werden. Wichtig ist hierbei, dass die zweite Reaktion sehr
schnell abläuft und somit die Spaltung von Threonin (12c) der geschwindigkeitsbestimmende
Schritt ist, da ansonsten die Aktivität der Alkoholdehydrogenase bestimmt wird. Ein weiterer
Nachteil ist, dass die Untersuchung der Inhibierung des Biokatalysators durch verschiedene
Substanzen (Substrate bzw. Produkte) oder Additive (organische, wasserlösliche Solventien)
auf diesem Weg schwierig durchzuführen ist. Es kann bei solch einem indirekten
Photometertest keine genaue Aussage getroffen werden, welches der beiden Enzyme
beeinflusst wird. Zur Bestimmung der Aldolaseaktivität gibt es auch eine direkte
Variante,117,118 die im Folgenden vorgestellt wird und zur Vorbereitung der präparativen
Synthesen ausschließlich verwendet wurde.
4.4.2.1 Direkter Photometertest
Bei einem direkten Photometertest muss ein Parameter verfolgt werden, der direkt an der
Reaktion beteilig ist und in einem detektierbaren Bereich Licht absorbiert, wie beispielsweise
der Cofaktor NADH bei der Reduktion einer Ketofunktion mit Hilfe von
Alkoholdehydrogenasen. Da die Threoninaldolase einen Cofaktor benötigt, der aber nicht
verbraucht wird, muss ein anderer Reaktionsteilnehmer herangezogen werden. Die
photometrische Verfolgung der Konzentrationsänderung von Benzaldehyd (1e) bei der
Spaltung von Phenylserin (syn-rac-12e) kann dafür genutzt werden (Abbildung 4.28).117,118
Abbildung 4.28. Direkte Aktivitätsbestimmung durch Verfolgung der
Benzaldehydkonzentration
Der Photometertest wurde bei 278 nm durchgeführt und der pH-Wert sowie der Einfluss des
Cofaktors PLP untersucht (vgl. Abschnitt 8.2.2.5). Zur Berechnung der Aktivität sind
verschiedene versuchsspezifische Parameter nötig, wie beispielsweise der
Extinktionskoeffizient ε (Gleichung (1)). ΔE278nm/t ist dabei die Anfangssteigung der

ENZYMATISCHE ALDOLREAKTION
50
Absorptionskurve, Vg das Gesamtvolumen der Probe, f der Verdünnungsfaktor des
Rohextraktes, Vp das Enzymvolumen und d die Küvettendicke.
Der Faktor ε kann mit Hilfe des Lambert Beer'schen Gesetztes bestimmt werden (Gleichung
(2)).119 Dieses besagt, dass sich die Extinktion E aus dem Extinktionskoeffizienten ε, der
Konzentration c des Substrates, sowie der Schichtdicke d der Küvette zusammensetzt.
Durch Auftragung der Extinktion gegen die Substratkonzentration kann ε aus der
Geradengleichung bestimmt werden. Dies wurde für verschiedene Benzaldehyd-
konzentrationen bei pH 7 bzw. 8, mit und ohne PLP durchgeführt (Abbildung 4.29).
Abbildung 4.29. Bestimmung des Extiktionskoeffizienten
Für die verschiedenen Bedingungen wurden ähnliche Extinktionskoeffizienten im Bereich
von 1.19-1.25 M-1cm-1 erhalten. Die Abweichung ist trotz der Zugabe des farbigen Cofaktors
PLP sehr gering.
pH 7: y = 1.213x + 0.073
pH 7, PLP: y = 1.190x + 0.134
pH 8: y = 1.254x + 0.128
pH 8, PLP: y = 1.224x + 0.2330,0
0,5
1,0
1,5
2,0
2,5
3,0
0 0,5 1 1,5 2 2,5
Ext
inkt
ion
[m
in-1
]
c(Benzaldehyd) [mM]
pH 7
pH 7, PLP
pH 8
pH 8, PLP
3.0
2.5
2.0
1.5
1.0
0.5
0.0
0 0.5 1.0 1.5 2.0 2.5

ENZYMATISCHE ALDOLREAKTION
51
Im Anschluss konnte die Aktivitätsbestimmung durchgeführt werden und die
Geschwindigkeit des Anstiegs der Benzaldehydkonzentration bei der Spaltung von
Phenylserin (12e) detektiert werden. Die Ergebnisse sind in Tabelle 4.1 gezeigt. Es ist
deutlich erkennbar, dass die höchste Aktivität von 10.9 U/mL (Tabelle 4.1, Eintrag 4) bei pH 8
und in Anwesenheit des Cofaktors PLP gefunden wurden. Die folgenden Versuche wurden
deshalb unter diesen Bedingungen durchgeführt.
Tabelle 4.1. Ergebnisse des Photometertests
Eintrag1) pH Cofaktor ΔE340nm/t
[1/min]
Aktivität
[U/mL]
Relative Aktivität
[%]
1 7 ohne PLP 0.0077 1.2 11
2 7 mit PLP 0.0272 4.3 39
3 8 ohne PLP 0.0155 2.4 22
4 8 mit PLP 0.0691 10.9 100
1) vgl. Abschnitt 8.2.2.5.2
4.4.2.2 Inhibierungsversuche
Nach Bestimmung der Aktivität der TA wurde nun der Einfluss einer bestimmten Substrat-
bzw. Produktkonzentration auf die enzymatische Spaltung untersucht. Dafür wurden
verschiedene Konzentrationen an Glycin (11) bzw. rac-syn-Phenylserin (rac-syn-12e) zur
vermessenden Mischung gegeben.
Glycin (11) wurde bis zu einer maximalen Konzentration von 2.1 M zugefügt. Der
Aktivitätsverlauf ist in Abbildung 4.30 gezeigt. Die Enzymaktivität nimmt um ca. 40% ab
(Abbildung 4.30, vgl. Abschnitt 8.2.2.5.3). Eine Glycinkonzentration von über 1 M hat eine
Beeinträchtigung der Aktivität der Threoninaldolase aus E. coli zur Folge, eine vollständige
Deaktivierung tritt allerdings nicht auf.

ENZYMATISCHE ALDOLREAKTION
52
Abbildung 4.30. Inhibierung durch Glycin (11)
Ein analoger Versuch wurde auch mit Zugabe von rac-syn-Phenylserin (rac-syn-12e)
durchgeführt. Hier wurde aufgrund der mangelnden Löslichkeit nur eine maximale
Konzentration von 150 mM erzielt. Die Auftragung der Enzymaktivität gegen die
Phenylserinkonzentration zeigt einen starken Abfall der Aktivität von über 60% im Vergleich
zur Aktivität ohne Zugabe von 12e (Abbildung 4.31, vgl. Abschnitt 8.2.2.5.3). Hier wurde
aufgrund höherer Temperaturen (30°C) eine stärkere Anfangsaktivität gefunden. Dies
konnte aber aufgrund eines nicht thermostatisierbaren Photometers nicht verifiziert
werden. Bereits bei einer Phenylserinkonzentration von 20 mM ist mit einer
Beeinträchtigung der Aldolaseaktivität zu rechnen.
Abbildung 4.31. Inhibierung durch rac-syn-Phenylserin (rac-syn-12e)
0
20
40
60
80
100
0 0,5 1 1,5 2 2,5
rela
tive
Akt
ivit
ät [
%]
c(Glycin) [M]
0
20
40
60
80
100
0 20 40 60 80 100 120 140 160
rela
tive
Akt
ivit
ät [
%]
c(Phenylerin) [mM]
0 0.5 1.0 1.5 2.0 2.5

4.4.3 Methoden zur
Zur besseren Haltbarkeit werden Enzymrohextrakte bei
Einfrieren zu verhindern und eine leichtere Handhabung zu gewährleisten
Rohextrakt mit Glycerin 1:1 (v/v) verdünnt
pipettiert werden. Bei den einleitenden Versuchen Glycin
L-Threoninaldolase aus E. coli
Umsatzbestimmung mittels 1
nicht möglich ist. Der Umsatz kann
entsprechenden Eduktpeaks verglichen werden. Benzaldehyd
werden, da das wässrige Lösungsmittel im Vakuum entfernt werden muss und somit auch
die Benzaldehydmenge verringert
entsprechenden Vergleich der
werden. Das Signal der Protonen von
den Signalen des Glycerins zwischen 3.51 und 3.80
Abbildung 4.32. Rohproduktspektrum der enzymatischen Umsetzung
ENZYMATISCHE
Methoden zur Umsatzbestimmung
Zur besseren Haltbarkeit werden Enzymrohextrakte bei -20°C aufbewahrt.
infrieren zu verhindern und eine leichtere Handhabung zu gewährleisten
1:1 (v/v) verdünnt, so bleibt die Mischung flüssig und kann leicht
tiert werden. Bei den einleitenden Versuchen Glycin (11) und Benzaldehyd
E. coli umzusetzen, wurde allerdings festgestellt, dass eine
1H-NMR aufgrund des in der Mischung enthaltenen
nicht möglich ist. Der Umsatz kann prinzipiell bestimmt werden, indem Produktpeaks
entsprechenden Eduktpeaks verglichen werden. Benzaldehyd (1e) kann nicht herangezogen
werden, da das wässrige Lösungsmittel im Vakuum entfernt werden muss und somit auch
verringert wird. Folglich kann eine Umsatzbestimmung nur durc
n Vergleich der Produktsignale mit denen von Glycin (
der Protonen von 11 (3.79 ppm für das Hydrochlorid)
zwischen 3.51 und 3.80 ppm überdeckt (Abbildung
Rohproduktspektrum der enzymatischen Umsetzung
NZYMATISCHE ALDOLREAKTION
53
20°C aufbewahrt.120 Um das
infrieren zu verhindern und eine leichtere Handhabung zu gewährleisten wird der
, so bleibt die Mischung flüssig und kann leicht
und Benzaldehyd (1e) mit der
umzusetzen, wurde allerdings festgestellt, dass eine
enthaltenen Glycerins
bestimmt werden, indem Produktpeaks mit
kann nicht herangezogen
werden, da das wässrige Lösungsmittel im Vakuum entfernt werden muss und somit auch
Folglich kann eine Umsatzbestimmung nur durch
(11) vorgenommen
(3.79 ppm für das Hydrochlorid) wird allerdings von
Abbildung 4.32).
Rohproduktspektrum der enzymatischen Umsetzung

ENZYMATISCHE ALDOLREAKTION
54
Aus diesem Grund musste ein Verfahren zur
wurden zwei Methoden getestet. Zum einen der Einsatz eines internen NMR
zum anderen eine Derivatisierungsmethode, bei der das Produkt durch Extraktion von
Glycerin getrennt und somit der Umsatz bestim
4.4.3.1 Umsatzbestimmung mittels
Eine Möglichkeit der Umsatzbestimmung besteht in der Zugabe eines internen Standards.
Dabei wird eine Substanz mit ähnlich
Menge der zur vermessenden
Integrale der Produktsignale mit de
Menge an Produkt berechnet werden
isoliert voneinander auftreten
tert-Leucin (83) als Standard gewählt, da diese Aminosäure ähnliche Löslichkeits
eigenschaften wie das Produkt aufweist und ein isoliertes Singulett bei 1.08 ppm zeigt. In
Abbildung 4.33 ist exemplarisch
Phenylserin (rac-syn-12e) und
abgebildet.
Abbildung 4.33. 1H-NMR-Spektrum zur Umsatzbestimmung mittels NMR
LDOLREAKTION
Aus diesem Grund musste ein Verfahren zur Umsatzbestimmung entwickelt werden
wurden zwei Methoden getestet. Zum einen der Einsatz eines internen NMR
zum anderen eine Derivatisierungsmethode, bei der das Produkt durch Extraktion von
Glycerin getrennt und somit der Umsatz bestimmt werden kann.
Umsatzbestimmung mittels NMR-Standard
Eine Möglichkeit der Umsatzbestimmung besteht in der Zugabe eines internen Standards.
Dabei wird eine Substanz mit ähnlichen Eigenschaften ausgewählt, die in einer bestimmten
r zur vermessenden NMR-Probe zugefügt wird. Entsprechend können dann die
Integrale der Produktsignale mit denen des Standards verglichen und die entstandene
ukt berechnet werden. Wichtig ist dabei, dass die Signale im NMR
isoliert voneinander auftreten, damit eine gute Auswertung möglich ist. In diesem Fall wurde
Standard gewählt, da diese Aminosäure ähnliche Löslichkeits
eigenschaften wie das Produkt aufweist und ein isoliertes Singulett bei 1.08 ppm zeigt. In
exemplarisch das Spektrum einer hergestellten Mischung aus
und deaktiviertem Enzym mit Glycerin sowie dem Standard
Spektrum zur Umsatzbestimmung mittels NMR
Umsatzbestimmung entwickelt werden. Dabei
wurden zwei Methoden getestet. Zum einen der Einsatz eines internen NMR-Standards und
zum anderen eine Derivatisierungsmethode, bei der das Produkt durch Extraktion von
Eine Möglichkeit der Umsatzbestimmung besteht in der Zugabe eines internen Standards.
Eigenschaften ausgewählt, die in einer bestimmten
Entsprechend können dann die
und die entstandene
Wichtig ist dabei, dass die Signale im NMR-Spektrum
gute Auswertung möglich ist. In diesem Fall wurde
Standard gewählt, da diese Aminosäure ähnliche Löslichkeits-
eigenschaften wie das Produkt aufweist und ein isoliertes Singulett bei 1.08 ppm zeigt. In
ischung aus rac-syn-
sowie dem Standard 83
Spektrum zur Umsatzbestimmung mittels NMR-Standard 83
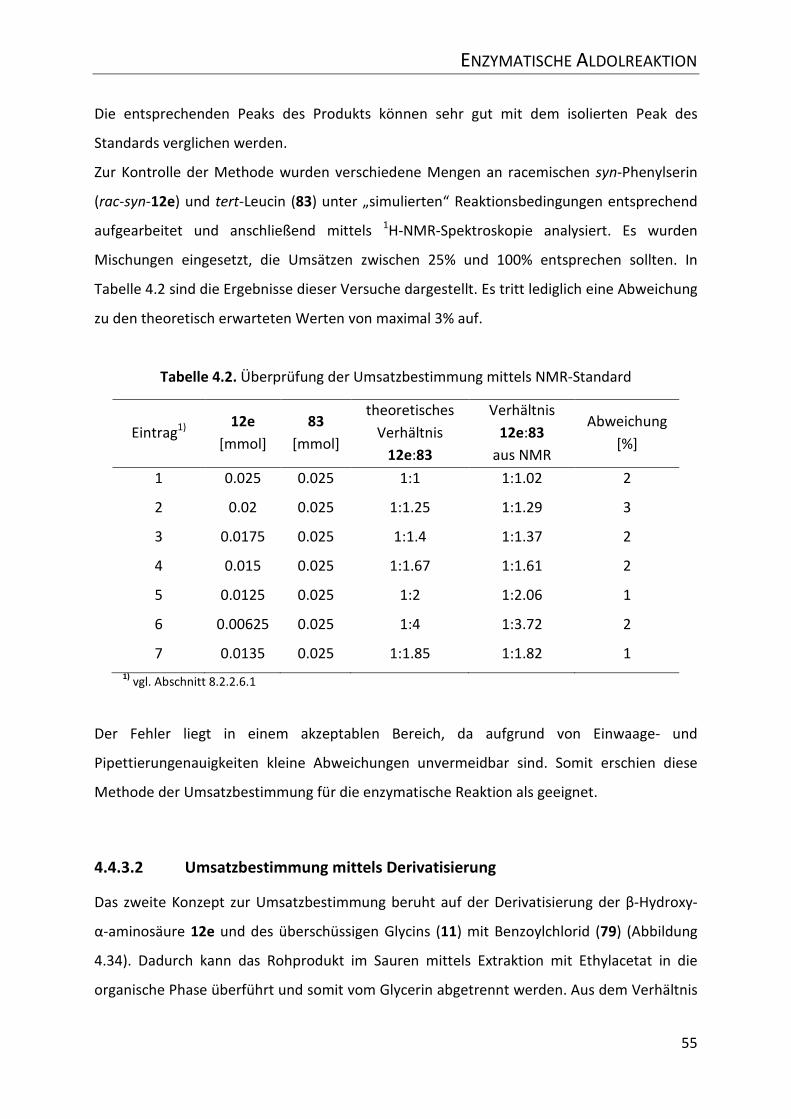
ENZYMATISCHE ALDOLREAKTION
55
Die entsprechenden Peaks des Produkts können sehr gut mit dem isolierten Peak des
Standards verglichen werden.
Zur Kontrolle der Methode wurden verschiedene Mengen an racemischen syn-Phenylserin
(rac-syn-12e) und tert-Leucin (83) unter „simulierten“ Reaktionsbedingungen entsprechend
aufgearbeitet und anschließend mittels 1H-NMR-Spektroskopie analysiert. Es wurden
Mischungen eingesetzt, die Umsätzen zwischen 25% und 100% entsprechen sollten. In
Tabelle 4.2 sind die Ergebnisse dieser Versuche dargestellt. Es tritt lediglich eine Abweichung
zu den theoretisch erwarteten Werten von maximal 3% auf.
Tabelle 4.2. Überprüfung der Umsatzbestimmung mittels NMR-Standard
Eintrag1) 12e
[mmol]
83
[mmol]
theoretisches
Verhältnis
12e:83
Verhältnis
12e:83
aus NMR
Abweichung
[%]
1 0.025 0.025 1:1 1:1.02 2
2 0.02 0.025 1:1.25 1:1.29 3
3 0.0175 0.025 1:1.4 1:1.37 2
4 0.015 0.025 1:1.67 1:1.61 2
5 0.0125 0.025 1:2 1:2.06 1
6 0.00625 0.025 1:4 1:3.72 2
7 0.0135 0.025 1:1.85 1:1.82 1
1) vgl. Abschnitt 8.2.2.6.1
Der Fehler liegt in einem akzeptablen Bereich, da aufgrund von Einwaage- und
Pipettierungenauigkeiten kleine Abweichungen unvermeidbar sind. Somit erschien diese
Methode der Umsatzbestimmung für die enzymatische Reaktion als geeignet.
4.4.3.2 Umsatzbestimmung mittels Derivatisierung
Das zweite Konzept zur Umsatzbestimmung beruht auf der Derivatisierung der β-Hydroxy-
α-aminosäure 12e und des überschüssigen Glycins (11) mit Benzoylchlorid (79) (Abbildung
4.34). Dadurch kann das Rohprodukt im Sauren mittels Extraktion mit Ethylacetat in die
organische Phase überführt und somit vom Glycerin abgetrennt werden. Aus dem Verhältnis

ENZYMATISCHE ALDOLREAKTION
56
von derivatisiertem Produkt 80e zu derivatisiertem Glycin 84 kann dann der Umsatz
bestimmt werden.
Abbildung 4.34. Derivatisierung mittels Benzoylchlorid (79)
Wichtig ist auch hier wieder die Überprüfung der Methode, da vor allem durch die
unterschiedlichen Eigenschaften der derivatisierten Verbindungen 80e und 84 beim
Extrahieren Fehler auftreten können. Mit Dichlormethan als Extraktionsmittel konnten keine
konstanten Ergebnisse erzielt werden. Daraufhin wurde das polarere Ethylacetat eingesetzt.
Verschiedene Mischungen an 11 und rac-syn-12e wurden mit Hilfe von 79 derivatisiert und
nach Aufarbeitung das Verhältnis der beiden Verbindungen zueinander mittels 1H-NMR-
Spektroskopie untersucht. Es wurden verschiedene Gemische aus Glycin (11) und
Phenylserin (12e) hergestellt, die einen theoretischen Umsatz von 50% bzw. 100%
darstellten. Dies entspricht einem Verhältnis von 1:9 bzw. 1:19 (11:12e), da Glycin in der
Reaktion in zehnfachem Überschuss zugegeben wird. Die Ergebnisse in Tabelle 4.3 wiesen
eine maximale Abweichung zu den theoretisch erwarteten Werten von 5% auf.
Tabelle 4.3. Überprüfung der Umsatzbestimmung mittels Derivatisierung
Eintrag1) 80e
[mmol]
84
[mmol]
theoretisches
Verhältnis
80e:84
Verhältnis
80e:84 aus
NMR
Abweichung
[%]
1 0.5 0.5 1:1 1:1.01 1
2 0.1 0.9 1:9 1:9.5 5
3 0.1 1.9 1:19 1:19.5 3
1) vgl. Abschnitt 8.2.2.6.2

ENZYMATISCHE ALDOLREAKTION
57
Es konnte gezeigt werden, dass auch diese Methode zur Umsatzbestimmung verwendet
werden kann. Ein Vorteil dieser Variante ist, dass die derivatisierten Produkte 80 aufgrund
ihrer Löslichkeitseigenschaften zur Bestimmung des Enantiomerenüberschusses mittels
chiraler HPLC herangezogen werden können, da die meisten zur Verfügung stehenden
Säulen ausschließlich mit organischen Lösungsmitteln verwendet werden können.
4.4.4 Etablierung der Standardreaktion
Die Umsatzbestimmung erfolgte bei der Durchführung der Standardreaktion mit
Benzaldehyd (1e) als Substrat. Die Bedingungen wurden in Anlehnung an die Arbeiten von
Griengl109 gewählt, d.h. es wurden eine Substratkonzentration von 0.1 M, sowie ein
zehnfacher Überschuss an Glycin (11) eingesetzt. Beide Methoden zur Umsatzbestimmung
lieferten vergleichbare Ergebnisse (Abbildung 4.35). So konnte ein fast vollständiger Umsatz
von 91% erzielt werden und das Diastereomerenverhältnis lag etwa bei d.r. 62:38 (syn/anti).
Abbildung 4.35. Standardreaktion und Umsatzbestimmung
4.4.5 Bestimmung der Enantioselektivität
Mit Hilfe der derivatisierten Reaktionsprodukte konnte eine HPLC-Analytik zur
ee-Wertbestimmung bei der enzymatischen Umsetzung mit Benzaldehyd (1e) etabliert
werden (Abbildung 4.36, vgl. 8.2.2.7.5.2).

ENZYMATISCHE ALDOLREAKTION
58
Abbildung 4.36. Bestimmung der Enantioselektivität der enzymatischen Umsetzung
In Abbildung 4.37 A ist das Spektrum von rac-80e gezeigt, in dem auch noch derivatisiertes
Glycin 84 enthalten ist. Zur Zuordnung der Peaks wurde ebenfalls nur racemisches syn-80e
vermessen (Abbildung 4.37 B).
Abbildung 4.37. HPLC-Analytik für 80e
Die Peaks für (2R,3R)-80e und (2R,3S)-80e überlagern und sind somit nur als ein Signal zu
erkennen. Das Spektrum der Enzymumsetzung (Abbildung 4.37 C) zeigt deutlich, dass kein
(2R)-Isomer enthalten ist, sondern lediglich das entsprechende (2S,3R)- und (2S,3S)-12e
gebildet wurde. Somit konnte gezeigt werden, dass die L-Threoninaldolase aus E. coli das
Stereozentrum an α-Position mit einer Enantioselektivität von >99% ee bildet.

ENZYMATISCHE ALDOLREAKTION
59
Solch eine HPLC-Analytik konnte auch für die o-Cl- bzw. o-Br-substituierten Verbindungen
80i bzw. 80o etabliert werden. Auch hier wurde jeweils ein ee-Wert von >99% nachgewiesen
(vgl. Abschnitt 8.2.2.7.7.4 bzw. 8.2.2.7.7.10).
4.4.6 Untersuchung der enzymatischen Synthese
Ausgehend von der Standardreaktion (Abbildung 4.35) wurde nun eine Analyse der
Reaktionsbedingungen durchgeführt. Zu diesem Zweck wurden die verschiedenen
Parameter, wie beispielsweise Reaktionszeit, Temperatur, Enzymmenge und Substrat-
konzentration variiert und der Einfluss auf den Umsatz und das Diastereomerenverhältnis
der enzymatischen Reaktion untersucht.
4.4.6.1 Einfluss der Reaktionszeit auf die enzymatische Aldolreaktion
Zuerst wurde der Einfluss der Reaktionszeit auf den Umsatz und das
Diastereomerenverhältnis der biokatalysierten Aldolreaktion betrachtet. Es war bereits
bekannt, dass sich bei der Reaktion mit L-selektiven Threoninaldolasen das
thermodynamische Gleichgewicht relativ schnell einstellt und so meist nur geringe
Diastereomerenüberschüsse erzielt werden können.106 Die Ergebnisse in Tabelle 4.4
bestätigen dies. Nach einer Reaktionszeit von 1 h wurde ein Diastereomerenverhältnis von
d.r. 44:56 (syn/anti) bei einem Umsatz von 20% erhalten (Tabelle 4.4, Eintrag 1). Dies deutet
darauf hin, dass der Biokatalysator bevorzugt das anti-Isomer bildet. Im Laufe der Zeit stellte
sich dann ein thermodynamisches Gleichgewicht ein und das Diastereomerenverhältnis lag
bei d.r. 65:35 (syn/anti) (Tabelle 4.4, Eintrag 3). Es wurde bereits vermutet, dass die L-TA aus
E.coli zu den L-allo-Threoninaldolasen gezählt werden muss.106
Tabelle 4.4. Abhängigkeit der Biotransformation von der Reaktionszeit

ENZYMATISCHE ALDOLREAKTION
60
Eintrag1) t [h]
1 1
2 8
3 20
1) vgl. Abschnitt 8.2.2.7.1
2)
(2S)-80e und 84 im 1H-NMR
4.4.6.2 Einfluss der Temperatur auf die enzymatische Aldolreaktion
Die Erhöhung der Reaktionstemperatur
(Abbildung 4.38, vgl. Abschnitt
niedrigerer Umsatz von 21% im Vergleich zu dem Versuch bei Raumtemperatur
Während die Produktbildung bei Raumtemperatur im Laufe der Zeit weiter anst
bei 40°C nicht der Fall. Es konnte gezeigt werden, dass die Aktivität der
Temperatur stark abnimmt.
O
H +
NH2
O
OH
L-TA88 U
11(10 Äq.)
1e(0.1 M)
PuffPLP
Abbildung 4.38. Abhängigkeit der Biotransformation von der Temperatur
0
20
40
60
80
100
Um
satz
[%
]
LDOLREAKTION
Umsatz (2S)-12e2) [%] d.r. (syn/anti
20 44:56
41 68:32
91 65:3
Umsatz- und d.r.-Wertbestimmung erfolgte über Verhältnis von
NMR Spektrum
Einfluss der Temperatur auf die enzymatische Aldolreaktion
Die Erhöhung der Reaktionstemperatur auf 40°C brachte keine Verbesserung des Umsatzes
, vgl. Abschnitt 8.2.2.7.2). Nach 8 h Reaktionszeit wurde ein um die Hälfte
im Vergleich zu dem Versuch bei Raumtemperatur
Während die Produktbildung bei Raumtemperatur im Laufe der Zeit weiter anst
Es konnte gezeigt werden, dass die Aktivität der
A (E. col i)U/mmol,
OH
O
NH2
OHCl
O
pH 12
79, 1.2 Äqv.
(2S)-12e
fer pH 8,(50 µM)
Abhängigkeit der Biotransformation von der Temperatur
8 h 20 h
41
91
2113
RT
40
anti) (2S)-12e2)
44:56
68:32
35
bestimmung erfolgte über Verhältnis von
Einfluss der Temperatur auf die enzymatische Aldolreaktion
brachte keine Verbesserung des Umsatzes
). Nach 8 h Reaktionszeit wurde ein um die Hälfte
im Vergleich zu dem Versuch bei Raumtemperatur (41%) erzielt.
Während die Produktbildung bei Raumtemperatur im Laufe der Zeit weiter anstieg, war dies
Es konnte gezeigt werden, dass die Aktivität der L-TA bei erhöhter
OH
O
HN
OH
O
Ph
(2S)-80e
Abhängigkeit der Biotransformation von der Temperatur
RT
40°C

ENZYMATISCHE ALDOLREAKTION
61
Um die enzymkatalysierte Rückreaktion, d.h. die Spaltung des Produkts, zu verhindern
wurden auch Versuche bei 4°C durchgeführt. Hier wurde nach 7 h ein Umsatz von 20%
erzielt und ein Diastereomerenverhältnis von d.r. 46:54 (syn/anti) gefunden (vgl. Abschnitt
8.2.2.7.2). Ein analoges Ergebnis wurde bereits bei einem Versuch bei Raumtemperatur nach
1 h gefunden. Das thermodynamische Gleichgewicht stellt sich bei niedrigeren
Temperaturen langsamer ein, allerdings ist auch der Umsatz dementsprechend niedrig. Das
Ziel einer bevorzugten Bildung des syn-Isomers kann mit dieser L-Threoninaldolase nicht
erzielt werden.
4.4.6.3 Einfluss der Enzymmenge auf die enzymatische Aldolreaktion
Zur Untersuchung des Einflusses der Enzymmenge wurde diese von 88 U/mmol auf
176 U/mmol verdoppelt, was eine Beschleunigung der Reaktion zur Folge hatte. Nach 1 h
wurde bereits ein Umsatz von 63% erzielt, dieser lag bei Verwendung der geringeren Menge
an Aldolase nur bei 20% (Abbildung 4.39, vgl. Abschnitt 8.2.2.7.3).
Abbildung 4.39. Abhängigkeit der Biotransformation von der Enzymmenge
0
20
40
60
80
100
0 5 10 15 20 25
Um
satz
[%
]
Reaktionszeit [t]
88 U/mmol
176 U/mmol

ENZYMATISCHE ALDOLREAKTION
62
Das thermodynamische Gleichgewicht stellte sich ebenfalls schneller ein und nach 1 h wurde
bereits ein Diastereomerenverhältnis von d.r. 67:33 (syn/anti) erhalten.
4.4.6.4 Einfluss des Glycinüberschusses auf die enzymatische Aldolreaktion
Als nächstes wurde untersucht, ob der in der Literatur beschriebene Überschuss an Glycin
(11) von 10 Äquivalenten109 erforderlich ist, um einen vollständigen Umsatz zu erzielen. Dazu
wurde die Benzaldehydkonzentration schrittweise von 0.1 M bis 1 M erhöht, während die
Glycinkonzentration (1 M) konstant gehalten wurde. Desweiteren wurde die Glycin-
konzentration von 1 M bis 0.1 M gesenkt, während die Konzentration des Aldehyden (0.1 M)
konstant gehalten wurde. In Abbildung 4.40 sind die Ergebnisse der beiden Versuchsreihen
dargestellt (vgl. Abschnitt 8.2.2.7.4.1 und 8.2.2.7.4.2). Es ist deutlich erkennbar, dass ein
hoher Überschuss an Glycin notwendig ist, um eine vollständige Produktbildung zu erzielen.
Bei einem Verhältnis Glycin (11) zu Benzaldehyd (1e) von 1:1 wurde bei einer
Benzaldehydkonzentration von 0.1 M ein Umsatz von 13% und bei einer Konzentration von
1 M lediglich ein Umsatz von 4% erhalten.
Abbildung 4.40. Abhängigkeit der Biotransformation vom Glycinüberschuss
0
20
40
60
80
100
0,0 0,2 0,4 0,6 0,8 1,0 1,2
Um
satz
[%
]
c(Substrat) [M]
Benzaldehyd
Glycin
0.0 0.2 0.4 0.6 0.8 1.0 1.2
Benzaldehyd; c(Glycin)= 1 M
Glycin; c(Benzaldehyd)= 0.1 M

Es zeigte sich ebenfalls, dass eine sehr hohe
Einfluss auf die Produktbildung hat. Bei einem Verhältnis von 2:1 (Glycin/Benzaldehyd)
wurde bei einer Benzaldehydkonzentration von 0.1
wohingegen bei einer Konzentration von 0.5
konnte. Die relative Menge an erhaltenem Produkt ist zwar bei einer Konzentration von
0.5 M höher, allerdings erscheint der Prozess wenig effizient, wenn 86% des eingesetzten
Substrats nicht umgesetzt werden.
4.4.6.5 Einfluss der Substratkonzentration auf die enzymatische Aldolreaktion
Bei der Entwicklung eines effizienten und industriere
Substratkonzentration eine sehr
volumetrische Produktivität.
0.1 M (1e) bzw. 1 M (11) um den Fakto
musste auch die Enzymmenge von 88 U/mmol auf 50 U/mmol verringert werden
Umsatz der Reaktion blieb trotzdem n
8.2.2.7.5).
O
H
1e(0.25 M)
Abbildung 4.41. Abhängigkeit des Umsatzes von der Substratkonzentration
0
20
40
60
80
100
Um
satz
[%
]
ENZYMATISCHE
Es zeigte sich ebenfalls, dass eine sehr hohe Benzaldehydkonzentration einen negativen
Einfluss auf die Produktbildung hat. Bei einem Verhältnis von 2:1 (Glycin/Benzaldehyd)
wurde bei einer Benzaldehydkonzentration von 0.1 M ein Umsatz von 44% erhalten,
wohingegen bei einer Konzentration von 0.5 M nur ein Umsatz von 14% erzielt werden
konnte. Die relative Menge an erhaltenem Produkt ist zwar bei einer Konzentration von
M höher, allerdings erscheint der Prozess wenig effizient, wenn 86% des eingesetzten
Substrats nicht umgesetzt werden.
r Substratkonzentration auf die enzymatische Aldolreaktion
Bei der Entwicklung eines effizienten und industrierelevanten Prozesses spielt die
Substratkonzentration eine sehr große Rolle. Wichtig ist hierbei eine möglichst hohe
Um dies zu erreichen wurde die Substratkonzentration von
um den Faktor 2.5 erhöht. Aufgrund des geringeren Volumens
musste auch die Enzymmenge von 88 U/mmol auf 50 U/mmol verringert werden
Umsatz der Reaktion blieb trotzdem nahezu unverändert (Abbildung 4
+
NH2
O
OH
L-TA (E. coli)50 U/mmol,
O
O
NH2
OH
11(10 Äq.)
(2S)-12e
Puffer pH 8,PLP (50 µM),
17 h, RT
Abhängigkeit des Umsatzes von der Substratkonzentration
0.1 0.25
>95 91
Substratkonzentration (1e) [M]
NZYMATISCHE ALDOLREAKTION
63
Benzaldehydkonzentration einen negativen
Einfluss auf die Produktbildung hat. Bei einem Verhältnis von 2:1 (Glycin/Benzaldehyd)
M ein Umsatz von 44% erhalten,
ein Umsatz von 14% erzielt werden
konnte. Die relative Menge an erhaltenem Produkt ist zwar bei einer Konzentration von
M höher, allerdings erscheint der Prozess wenig effizient, wenn 86% des eingesetzten
r Substratkonzentration auf die enzymatische Aldolreaktion
anten Prozesses spielt die
Wichtig ist hierbei eine möglichst hohe
Um dies zu erreichen wurde die Substratkonzentration von
2.5 erhöht. Aufgrund des geringeren Volumens
musste auch die Enzymmenge von 88 U/mmol auf 50 U/mmol verringert werden. Der
4.41, vgl. Abschnitt
OH
Abhängigkeit des Umsatzes von der Substratkonzentration
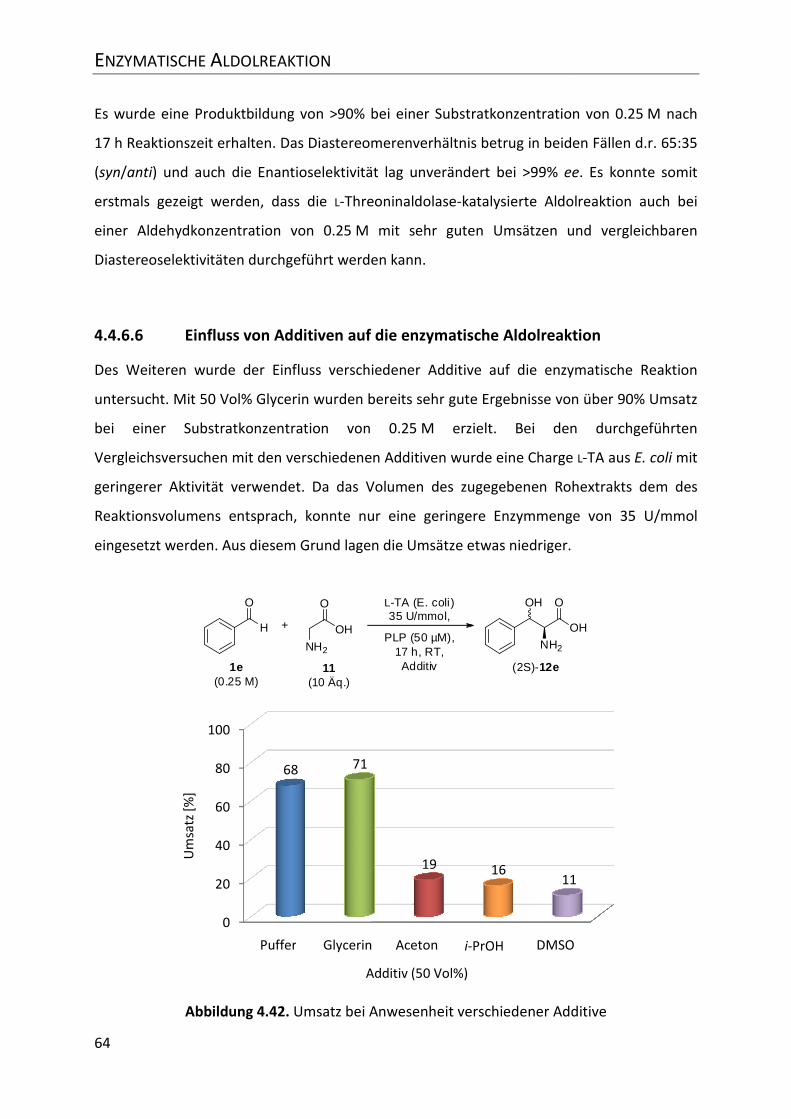
ENZYMATISCHE ALDOLREAKTION
64
Es wurde eine Produktbildung von >90% bei einer Substratkonzentration von 0.25
17 h Reaktionszeit erhalten. Das Diastereomerenverhältnis betrug in beiden Fällen d.r. 65:35
(syn/anti) und auch die Enantioselektivität lag unverändert bei >99%
erstmals gezeigt werden, dass die
einer Aldehydkonzentration von 0.25
Diastereoselektivitäten durchgeführt werden kann.
4.4.6.6 Einfluss von Additiven auf die enzymatische Aldolreaktion
Des Weiteren wurde der Einfluss verschiedener Additive auf die
untersucht. Mit 50 Vol% Glycerin wurden bereits sehr gute Ergebnisse von über 90% Um
bei einer Substratkonzentration von 0.25
Vergleichsversuchen mit den verschiedenen Additi
geringerer Aktivität verwendet.
Reaktionsvolumens entsprach, konnte nur ein
eingesetzt werden. Aus diesem Grund lagen die Umsätze etwas niedriger.
O
H +
1e(0.25 M)
Abbildung 4.42.
0
20
40
60
80
100
Puffer
68
Um
satz
[%
]
LDOLREAKTION
Es wurde eine Produktbildung von >90% bei einer Substratkonzentration von 0.25
nszeit erhalten. Das Diastereomerenverhältnis betrug in beiden Fällen d.r. 65:35
) und auch die Enantioselektivität lag unverändert bei >99% ee
erstmals gezeigt werden, dass die L-Threoninaldolase-katalysierte Aldolreaktion auch b
einer Aldehydkonzentration von 0.25 M mit sehr guten Umsätzen und vergleichbaren
Diastereoselektivitäten durchgeführt werden kann.
Einfluss von Additiven auf die enzymatische Aldolreaktion
Des Weiteren wurde der Einfluss verschiedener Additive auf die enzymatische Reaktion
untersucht. Mit 50 Vol% Glycerin wurden bereits sehr gute Ergebnisse von über 90% Um
tratkonzentration von 0.25 M erzielt. Bei den durchgeführten
Vergleichsversuchen mit den verschiedenen Additiven wurde eine Charge
verwendet. Da das Volumen des zugegebenen Rohextrakts dem
entsprach, konnte nur eine geringere Enzymmenge von 35 U/mmol
Aus diesem Grund lagen die Umsätze etwas niedriger.
+
NH2
O
OH
L-TA (E. coli)35 U/mmol,
O
O
NH2
OH
PLP (50 µM),17 h, RT,Additiv11
(10 Äq.)(2S)-12e
. Umsatz bei Anwesenheit verschiedener Additive
Puffer Glycerin Aceton i-PrOH DMSO
68 71
19 1611
Additiv (50 Vol%)
i-PrOH
Es wurde eine Produktbildung von >90% bei einer Substratkonzentration von 0.25 M nach
nszeit erhalten. Das Diastereomerenverhältnis betrug in beiden Fällen d.r. 65:35
ee. Es konnte somit
katalysierte Aldolreaktion auch bei
M mit sehr guten Umsätzen und vergleichbaren
Einfluss von Additiven auf die enzymatische Aldolreaktion
enzymatische Reaktion
untersucht. Mit 50 Vol% Glycerin wurden bereits sehr gute Ergebnisse von über 90% Umsatz
M erzielt. Bei den durchgeführten
Charge L-TA aus E. coli mit
Rohextrakts dem des
geringere Enzymmenge von 35 U/mmol
Aus diesem Grund lagen die Umsätze etwas niedriger.
OH
Umsatz bei Anwesenheit verschiedener Additive
DMSO
11

ENZYMATISCHE ALDOLREAKTION
65
Abbildung 4.42 zeigt, dass bei Verwendung von Puffer und Glycerin als Zusatz vergleichbare
Umsätze erzielt werden konnten. Bei der Zugabe von Aceton, iso-Propanol oder DMSO
wurden deutlich schlechtere Umsätze von <20% erzielt. Die Diastereomerenverhältnisse
lagen in einem erwarteten Bereich von d.r. 65:35 (syn/anti). Je geringer der Umsatz, desto
höher war auch der Anteil des anti-Isomer (vgl. 8.2.2.7.6). Es konnte gezeigt werden, dass
Glycerin keinen negativen Einfluss auf die enzymatische Reaktion hat. Allerdings können
keine anderen Additive als Alternative eingesetzt werden, da sie die Enzymaktivität stark
beeinträchtigen.
4.4.7 Erweiterung des Substratspektrums
Um das Substratspektrum auf andere aromatische Benzaldehyde auszuweiten, wurden
thiosubstituierte Benzaldehyde eingesetzt, die als Vorstufe für die Thiamphenicolsynthese
geeignet waren, wie beispielsweise Methylthiobenzaldehyd (1d) und Methylsulphonyl-
benzaldehyd (1k). Außerdem wurde der Einfluss des Substitutionsmusters am Aromaten auf
Umsatz und Diastereoselektivität untersucht (vgl. Abschnitt 8.2.2.7.7).
OH
NHH3CO2S
OH
O
ClCl19
Abbildung 4.43. Thiamphenicol (19)
4.4.7.1 Thiamphenicolrelevante substituierte Benzaldehyde
Im Hinblick auf ein anwendbares Zielprodukt wurden zuerst Benzaldehyde eingesetzt, die zu
einem thiamphenicolrelevanten Intermediat führen. Dazu gehört zum einen
Methylthiobenzaldehyd (1d) und zum anderen Methylsulphonylbenzaldehyd (1k). Bei der
Reaktion von 1d wurde bei einer Substratkonzentration von 0.1 M ein Umsatz von lediglich
10% und ein Diastereomerenverhältnis von d.r. 57:43 (syn/anti) erzielt (Abbildung 4.44).
Aufgrund des niedrigen Umsatzes wurde dieses Substrat nicht weiter untersucht.

ENZYMATISCHE ALDOLREAKTION
66
Abbildung 4.44. Enzymatische Synthese von (2S)-12d
Bessere Ergebnisse wurden beim Einsatz von 1k ermittelt (Abbildung 4.45). Bei einer
Substratkonzentration von 0.1 M wurden ein Umsatz von 42% und ein
Diastereomerenverhältnis von d.r. 61:39 (syn/anti) erhalten. Die Reaktion bei einer
Aldehydkonzentration von 0.25 M lieferte allerdings einen deutlich niedrigeren Umsatz von
nur noch 24%.
Abbildung 4.45. Enzymatische Synthese von (2S)-12k
Die in der Literatur beschriebene L-TA aus Pseudomonas putida zeigte im Vergleich dazu
deutlich bessere Ergebnisse.109 Bei der Synthese von (2S)-12k wurde ein Umsatz von 68% bei
einem Diastereomerenverhältnis von d.r. 78:22 (syn/anti) erzielt. Der Biokatalysator aus
E. coli ist vor allem hinsichtlich des Diastereomerenverhältnisses weniger selektiv und für die
Synthese von (2S,3R)-12k wenig geeignet.
4.4.7.2 m-Substituierte Benzaldehyde
Da in der Literatur mit meta-substituierten Benzaldehyden wie beispielsweise 1j und der
L-TA aus P. putida sehr gute Diastereoelektivitäten (d.r. 78:22 syn/anti) erzielt werden

ENZYMATISCHE ALDOLREAKTION
67
konnten,109 wurden verschiedene in meta-Position substituierte Benzaldehyde mit dem
Biokatalysator aus E. coli umgesetzt. Allerdings zeigte die verwendete Aldolase aus E. coli
eine geringere Diastereoselektivität bei der Bildung von 12j und 12m (Abbildung 4.46). Bei
der Reaktion des chlorsubstituierten Benzaldehyden 1l wurden dagegen ähnliche
Diastereomerenverhältnisse erhalten.109 Allgemein lässt sich sagen, dass die L-TA aus E. coli
keine erhöhte Diastereoselektivität bei der Bildung von m-substituierten β-Hydroxy-α-
aminosäuren 12 zeigt.
Abbildung 4.46. Enzymatische Synthese m-substituierter β-Hydroxy-α-aminosäuren (2S)-12
Die drei Synthesen wurden auch bei einer Substratkonzentration von 0.25 M durchgeführt
und es wurden kaum veränderte Umsätze erzielt. Lediglich bei Aldehyd 1m wurde eine
starke Verringerung der Produktbildung von 83% auf 28% Umsatz erhalten. Dies ist wohl auf
eine Beeinträchtigung des Enzyms durch Wasserstoffbrückenbindung mit der
Hydroxygruppe des Aldehyds 1m zurückzuführen, die bei einer höheren
Substratkonzentration verstärkt auftritt.

ENZYMATISCHE ALDOLREAKTION
68
4.4.7.3 p-Substituierte Benzaldehyde
Zum Vergleich wurden auch zwei para-substituierte Benzaldehyde 1b und 1f umgesetzt. Es
wurden mäßige Umsätze von 55% und 32% erhalten und die Diastereomerenverhältnisse
lagen bei beiden Produkten 12b und 12f bei d.r. 62:38 (syn/anti) (Abbildung 4.47). Bei der
erhöhten Substratkonzentration von 0.25 M wurde eine etwas niedrigere Produktbildung
(35% bzw. 20%) beobachtet. Auch bei diesem Substratmuster konnte keine Verbesserung
der Diastereoselektivität erhalten werden.
Abbildung 4.47. Enzymatische Synthese p-substituierter β-Hydroxy-α-aminosäuren (2S)-12
4.4.7.4 o-Substituierte Benzaldehyde
Bei der Reaktion des ortho-substituierten Benzaldehyds 1i wurden dagegen sehr gute
Diastereoselektivitäten von d.r. 81:19 (syn/anti) bei einer hohen Produktbildung von 91%
unter Verwendung der L-TA aus E. coli erhalten (Abbildung 4.48).

ENZYMATISCHE ALDOLREAKTION
69
Abbildung 4.48. Enzymatische Synthese von (2S)-12i
Mit der Aldolase aus P. putida wurde eine etwas geringere Diastereoselektivität von
d.r. 76:24 (syn/anti) in der Literatur beschrieben.109 Auch bei der erhöhten
Substratkonzentration konnte ein ähnlich guter Umsatz und ein Diastereomerenverhältnis
von d.r. 80:20 (syn/anti) erhalten werden. Die Enantioselektivität von (2S)-12i lag
unabhängig von der Substratkonzentration bei >99% ee.
Darauf aufbauend und um zu überprüfen, ob hier allgemein eine höhere
Diastereoselektivität vorliegt, wurden noch weitere o-substituierte Benzaldehyde umgesetzt
(Abbildung 4.49). Tendenziell wurde bei der Reaktion von o-substituierten Aldehyden mit
Glycin (11) eine bessere Diastereoselektivität (d.r. >70:30 syn/anti) erhalten als bei der
Bildung der p- oder m-substituierten β-Hydroxy-α-aminosäuren. Die Umsätze lagen für (2S)-
12n, (2S)-12o und (2S)-12q bei deutlich über 50%. Auch bei erhöhter Aldehydkonzentration
wurden ähnliche Umsätze und Diastereoselektivitäten erzielt. Die geringere Produktbildung
bei (2S)-12r (25%) deutet darauf hin, dass Benzaldehydderivate mit elektronenziehenden
Substituenten besser umgesetzt werden, als solche mit elektronenschiebenden. Bei der
Synthese von (2S)-12s kommt es wieder zu Wechselwirkungen zwischen der Hydroxygruppe
und dem Enzym, wodurch die Produktbildung beeinträchtigt wird.
Die L-Threoninaldolase aus E. coli weist bei der Umsetzung von o-substituierten
Benzaldehyden die höchste Diastereoselektivität auf. Das beste Ergebnis wurde mit
o-Chlorbenzaldehyd (1i) erzielt. Das Produkt (2S)-12i wurde bei einer Substratkonzentration
von 0.25 M mit einem Umsatz von 85% und einem sehr guten Diastereomerenverhältnis von
d.r. 80:20 (syn/anti) erhalten. Außerdem wurde eine Enantioselektivität von >99% ee
gefunden. Diese Reaktion wurde im Folgenden weiter optimiert und im größeren Maßstab
durchgeführt.

ENZYMATISCHE ALDOLREAKTION
70
Abbildung 4.49. Enzymatische Synthese o-substituierter β-Hydroxy-α-aminosäuren (2S)-12
4.4.8 Optimierung der enzymatischen Synthese von (2S)-12i
Zur Optimierung der enzymatischen Umsetzung von 1i mit einer Substratkonzentration von
0.25 M wurde die Reaktion bei einer konstanten Temperatur von 25°C durchgeführt. Des
Weiteren wurde die Glycinmenge von einem zehnfachen auf einen achtfachen Überschuss
reduziert. Außerdem musste die Reaktion mit einer geringeren Enzymmenge von 44 U/mmol
durchgeführt werden, da das Volumen des eingesetzten Rohextraktes genau dem des
Reaktionsvolumens entsprach und die Enzymaktivität geringer war (Abbildung 4.50). Nach
einer Reaktionszeit von 20 h konnte ein vollständiger Umsatz und ein
Diastereomerenverhältnis von d.r. 79:21 (syn/anti) erzielt werden.

O
H +
1i(0.25 M)
Cl
Abbildung 4.50
4.4.8.1 Untersuchung des Reaktionsverlaufs
Um den genauen Reaktionsverlauf zu untersuchen wurde die
10 mL-Maßstab durchgeführt und nach verschiedenen Reaktionszeiten
Umsatzbestimmung entnommen.
beobachtet, die danach nur noch geringfügig anstieg
8.2.2.8.1).
O
H +
1i(0.25 M)
Cl
Abbildung 4.51. Reaktionsverfolgung der enzymatischen Synthese von
0
20
40
60
80
100
1/4 1/2 1
[%]
ENZYMATISCHE
NH2
O
OHPuffer pH 8,
PLP (50 µM),20 h, 25°C
O
NH2
OH
11(8 Äq.)
Cl
(2S)-12i>95% Umsat
d.r. 79:21 (syn/a
L-TA (E. col i)44 U/mmol,
50. Optimierte enzymatische Synthese von (2S
Untersuchung des Reaktionsverlaufs
Um den genauen Reaktionsverlauf zu untersuchen wurde die Synthese
Maßstab durchgeführt und nach verschiedenen Reaktionszeiten
Umsatzbestimmung entnommen. Nach 3 h wurde bereits eine Produktbildung von über 90%
ch nur noch geringfügig anstieg (Abbildung 4.
NH2
O
OHPuffer pH 8,PLP (50 µM),
25°C
O
NH2
OH
11(8 Äq.)
Cl
(2S)-12i
L-TA (E. coli)44 U/mmol,
Reaktionsverfolgung der enzymatischen Synthese von
1 2 3 5 7 22 144
Zeit [h]
NZYMATISCHE ALDOLREAKTION
71
OH
tzanti)
S)-12i
Synthese in einem
Maßstab durchgeführt und nach verschiedenen Reaktionszeiten Proben zur
wurde bereits eine Produktbildung von über 90%
.51, vgl. Abschnitt
OH
Reaktionsverfolgung der enzymatischen Synthese von (2S)-12i
Umsatz
syn-Isomer syn-Isomer
Umsatz

ENZYMATISCHE ALDOLREAKTION
72
Das Diastereomerenverhältnis lag nach einer halben Stunde konstant zwischen d.r. 78:22
und d.r. 80:20 (syn/anti). Die unterschiedlichen Werte sind auf Schwankungen
zurückzuführen, die bei der Auswertung durch Hintergrundrauschen der Baseline ausgelöst
werden. Aufgrund dieser Ergebnisse wurde ein analoger Versuch in 0.5 mL-Maßstab
durchgeführt und nach 6 Stunden abgebrochen. Bei einem Umsatz von >95% wurde ein
Diastereomerenverhältnis von d.r. 80:20 (syn/anti) erhalten und ein Enantiomeren-
überschuss von >99% ee mittels chiraler HPLC nachgewiesen (vgl. Abschnitt 8.2.2.8.2).
Als nächstes sollte das Reaktionsvolumen auf 100 mL erhöht und (2S)-12i möglichst
diastereomerenrein isoliert werden. Aufgrund der erhaltenen Ergebnisse wurde die Reaktion
nach den optimierten Bedingungen durchgeführt und nach 7 h beendet.
4.4.8.2 Vergleichsversuch mit Benzaldehyd als Substrat
Um die optimierte Reaktion mit 1i als Substrat mit dem Standardsubstrat 1e vergleichen zu
können, wurde Benzaldehyd unter den optimierten Bedingungen mit Glycin (11)
enzymatisch umgesetzt (Abbildung 4.52).
Abbildung 4.52. Synthese von (2S)-12e unter optimierten Bedingungen
Die Reaktionsverfolgung ist in Abbildung 4.53 dargestellt und es ist deutlich zu erkennen,
dass unter diesen Bedingungen mit 1e als Substrat lediglich ein maximaler Umsatz von 72%
erzielt werden konnte (vgl. Abschnitt 8.2.2.8.4). Das Diastereomerenverhältnis lag wie
erwartet in einem Bereich von d.r. 65:35 (syn/anti). Die optimierte Synthese kann also nicht
ohne Weiteres auf andere Substrate angewendet werden. Mit o-Chlorbenzaldehyd (1i) als
Ausgangsverbindung konnte der höchste Umsatz (>95%) und das beste
Diastereomerenverhältnis (d.r. 80:20 syn/anti) bei der Umsetzung mit der L-TA aus E. coli
erzielt werden.

ENZYMATISCHE ALDOLREAKTION
73
Abbildung 4.53. Vergleichsversuch unter optimierten Bedingungen mit 1e als Substrat
4.4.8.3 Cofaktor-Abhängigkeit
Es sollte weiterhin untersucht werden, ob bei der enzymatischen Reaktion die Zugabe von
Cofaktor PLP (50 µM) notwendig ist. Deshalb wurde die Reaktionsverfolgung nochmals ohne
Cofaktor durchgeführt (Abbildung 4.54, vgl. Abschnitt 8.2.2.8.4).
Abbildung 4.54. Vergleich der enzymatischen Reaktion mit und ohne Cofaktor PLP
0
20
40
60
80
100
0 5 10 15 20 25 30
Um
satz
[%
]
Zeit [h]
0
20
40
60
80
100
0 5 10 15 20 25 30
Um
satz
[%
]
Zeit [h]
mit PLP
ohne PLP
(50 µM)

ENZYMATISCHE ALDOLREAKTION
74
Bei der graphischen Auftragung der Ergebnisse der beiden Versuchsreihen wird deutlich,
dass eine Zugabe von PLP nicht notwendig ist. Offensichtlich ist im Rohextrakt genügend
Cofaktor enthalten um die Reaktion zu katalysieren.
Analog wurde auch die Synthese von (2S)-12i ohne Cofaktor durchgeführt. Unter den
optimierten Bedingungen wurden nach 7 h ein Umsatz von 91% und ein Diastereomeren-
verhältnis von d.r. 78:22 (syn/anti) erhalten. Dies entspricht ebenfalls den Ergebnissen der
Versuche mit Zugabe von PLP.
4.4.8.4 Scale-up und Isolierung des Produkts (2S)-12i
Das Reaktionsvolumen wurde zuerst auf 12.5 mL erhöht und verschiedene
Aufarbeitungsabläufe unter Verwendung eines Ionenaustauschers und Kristallisation
getestet. Die enzymatische Reaktion wurde nach 7 h durch Erniedrigung des pH-Werts auf 1
gestoppt und anschließend wieder neutralisiert. Durch Zugabe von Ethanol (ca. 90 Vol%)
wurde der Großteil des überschüssigen Glycins ausgefällt. Mittels Filtration konnte 11 und
ausgefälltes Protein vom Rohprodukt getrennt werden.
Durch Einengen des Filtrats auf etwa ein Zehntel des Volumens wurde (2S)-12i ausgefällt und
abfiltriert. Anfangs stand für die Synthese nur Enzymrohextrakt zur Verfügung, der mit
Glycerin verdünnt war, welches im erhaltenen Produkt noch mit etwa 10% enthalten war.
Durch mehrmaliges Umkristallisieren in Wasser konnte schließlich vollständig reines Produkt
(2S)-12i isoliert werden, allerdings nur in einer Ausbeute von 38%. Auch der Versuch das
Glycerin unter Verwendung des Kationenaustauscher Dowex 50W X8 abzutrennen, brachte
keine wesentliche Verbesserung der Ausbeute. Zwar konnte das Glycerin vollständig
abgetrennt werden, aufgrund noch enthaltener Glycinreste und Verunreinigungen aus dem
Enzymrohextrakt musste allerdings weiter umkristallisiert werden. Um die Zahl der
Aufreinigungsschritte zu verringern und den Verlust an Produkt zu minimieren wurde auf
einen unverdünnten Rohextrakt zurückgegriffen. (2S)-12i wurde wieder nach Abfiltrieren des
Glycins beim Einengen des Filtrats ausgefällt. Der Niederschlag wurde abgetrennt und mit
Wasser/Ethanol (1:4) gewaschen.

ENZYMATISCHE ALDOLREAKTION
75
Abbildung 4.55. Scale-up der enzymatischen Reaktion und Isolierung von (2S)-12i
Auf diesem Weg konnte reines Produkt in einer Ausbeute von 50% und einem
Diastereomerenverhältnis von d.r. 78:22 (syn/anti) erhalten werden. Wegen der geringeren
Zahl an Aufreinigungsschritten konnte die Ausbeute stark verbessert werden. Durch die
Erhöhung des Reaktionsvolumens auf 100 mL konnte zudem eine höhere Ausbeute von 62%
bei einem Diastereomerenverhältnis von d.r. 78:22 (syn/anti) erzielt werden (Abbildung
4.55, vgl. Abschnitt 8.2.2.8.5).
4.4.8.4.1 Trennung der racemischen Diastereomere von rac-12i
Ein weiteres Ziel war die Isolierung des diastereomerenreinen Produkts (2S,3R)-12i aus der
enzymatischen Umsetzung unter Verwendung industriell anwendbarer Methoden. Die
literaturbekannten Verfahren beruhen meist auf aufwendigen und kostenintensiven
säulenchromatographischen Wegen.105,109 Beispielsweise ist eine Trennung über Silicagel mit
einem Lösungsmittelgemisch aus CH2Cl2, Methanol und Ammoniak möglich.109 Dieses
Verfahren ist allerdings nicht im Grammmaßstab anwendbar.
Zuerst wurde die Trennung mittels Ionenaustauscher (Dowex 50W X8) untersucht. Dazu
wurde das Hydrochlorid von rac-12i (d.r. 82:18 syn/anti) auf eine mit Ionenaustauscher
gefüllte Säule aufgetragen und mit 0.1 M Ammoniaklösung eluiert. Mit Hilfe von
Dünnschichtchromatographie sollte die Trennung verfolgt werden. Es konnte lediglich eine
minimale Anreicherung der Isomere auf d.r. 86:14 (syn/anti) bei geringer Ausbeute (16%)

ENZYMATISCHE ALDOLREAKTION
76
erzielt werden. Aufgrund dessen wurde diese Methode zur Diastereomerentrennung
verworfen.
Abbildung 4.56. Trennung der Diastereomere von rac-12i
Als nächstes wurde versucht, das gewünschte syn-12i durch Umkristallisation in Wasser zu
erhalten. Dazu wurde rac-12i (d.r. 82:18 syn/anti) unter Sieden in Wasser gelöst. Der beim
Abkühlen entstandene Niederschlag wurde abfiltriert und es konnte reines syn-12i in einer
Ausbeute von 43% isoliert werden (Abbildung 4.56). Diese Methode sollte nun auf das
enantiomerenreine Produktgemisch aus der enzymatischen Umsetzung angewendet
werden.
4.4.8.4.2 Trennung der enantiomerenreinen Diastereomere von (2S)-12i
Nach Isolierung des reinen Produkts (2S)-12i als Diastereomerengemisch aus der
enzymatischen Aldolreaktion wurde dieses in Wasser umkristallisiert, um das gewünschte
syn-Isomer zu erhalten. Leider konnte hierbei keine Verbesserung des Diastereomeren-
verhältnisses festgestellt werden. Die enantiomerenreine Verbindung weist hier andere
Fällungseigenschaften als das Racemat auf.
Anschließend wurde untersucht, ob das gewünschte Isomer unter Verwendung anderer
Lösungsmittel oder Lösungsmittelgemische abgetrennt werden kann. Es wurde beobachtet,
dass bevorzugt das anti-Isomer ausgefällt wird und es somit nur zu einer Anreicherung des

ENZYMATISCHE ALDOLREAKTION
77
gewünschten (2S,3R)-12i im Filtrat kommt. So wurde zu einer wässrigen Lösung von (2S)-12i
(d.r. 78:22 syn/anti) so lange Aceton zugegeben, bis sich ein Niederschlag bildete. Der
Niederschlag wies ein Diastereomerenverhältnis von d.r. 68:32 (syn/anti) auf, während das
des Filtrats bei d.r. 87:13 (syn/anti) lag. In Bezug auf das eingesetzte (2S)-12i wurde 41%
Produkt beim Filtrieren wiedergewonnen und beim Abziehen des Lösungsmittel aus dem
Filtrat 44% (2S)-12i. Der Verlust lag bei etwa 15% (Abbildung 4.57).
Abbildung 4.57. Trennung der Diastereomere durch Ausfällen aus Wasser mit Aceton
Durch wiederholte Ausfällung konnte keine weitere Verbesserung des
Diastereomerenverhältnisses erreicht werden. Die Ausbeute des Diastereomerengemisches
nach der enzymatischen Reaktion lag bei 62%. Durch Trennung der Diastereomere auf eben
beschriebene Weise ist lediglich eine Anreicherung des syn-Isomers bei einer Ausbeute von
44% möglich. Somit wurde die Gesamtausbeute halbiert und trotzdem nur ein
Diastereomerenverhältnis von d.r. 87:13 (syn/anti) erhalten.
4.4.9 Racematspaltung
Darüber hinaus wurde als Alternative zur Synthese enantiomerenreiner β-Hydroxy-
α-aminosäuren die Racematspaltung untersucht. Bei der Spaltung von rac-syn-12, welches
leicht hergestellt werden kann (vgl. Abschnitt 4.4.1.1), ist eine Trennung der Diastereomere
nicht notwendig, da nur das syn-Isomer eingesetzt wird. Allerdings kann nur eine maximale
Ausbeute von 50% erzielt werden.

ENZYMATISCHE ALDOLREAKTION
78
4.4.9.1 Spaltung von rac-syn-12e
Zuerst wurde die Spaltung des Standardsubstrats Phenylserin (rac-syn-12e), welches käuflich
erwerbbar ist, untersucht. Dazu wurden verschiedene Konzentrationen an 12e und
verschiedene Enzymaktivitäten eingesetzt (Tabelle 4.5).
Tabelle 4.5. Racematspaltung von rac-syn-12e
Eintrag1) rac-syn-
12e [M]
Enzymaktivität
[U/mmol]
Enzym
[mL]
Puffer pH 8
[mL]
Umsatz
11 [%]
Umsatz
anti-12e [%]
1 0.2 68 0.125 0.125 47 4
2 0.1 135 0.25 0.25 51 3
3 0.2 27 0.05 0.2 46 5
4 0.1 54 0.1 0.4 50 5
1) vgl. Abschnitt 8.2.2.1.1
Ein vollständiger Umsatz wurde bei einer Substratkonzentration von 0.1 M erzielt. Dabei war
eine Enzymaktivität von 54 U/mmol ausreichend (Tabelle 4.5, Eintrag 4). Es wurden bei allen
Versuchen kleine Mengen von anti-12e nachgewiesen, was darauf zurückzuführen ist, dass
auch die Bindungsknüpfung stattfindet.
Um die Anwendbarkeit der Methode zu untersuchen wurde die Reaktion in größerem
Maßstab (25 mL, analog Tabelle 4.5, Eintrag 4) durchgeführt. Bei vergleichbaren Ergebnissen
von Umsatz und Diastereomerenverhältnis konnte kein Produkt isoliert werden.

ENZYMATISCHE ALDOLREAKTION
79
4.4.9.2 Spaltung von rac-syn-12i
Die Racematspaltung der chlorsubstituierten Verbindung 12i wurde ebenfalls durchgeführt.
Auch hier wurde die Substratkonzentration und die Enzymaktivität variiert (Tabelle 4.6).
Tabelle 4.6. Racematspaltung von rac-syn-12i
Eintrag1) rac-syn-
12i [M]
Enzymaktivität
[U/mmol]
Enzym
[mL]
Puffer pH 8
[mL]
Umsatz
11 [%]
Umsatz
anti-12i2)[%]
1 0.5 54 0.1 0 9 n.d.
2 0.2 135 0.25 0 20 n.d
3 0.1 270 0.5 0 36 n.d
4 0.067 270 0.5 0.25 44 5
5 0.05 270 0.5 0.5 48 5
6 0.05 540 1 0 50 5
7 0.05 54 0.1 0.9 46 5
1) vgl. Abschnitt 8.2.2.1.2; 2)
n.d.: nicht detektierbar
Lediglich bei einer Substratkonzentration von 0.05 M wurden gute Umsätze erzielt (Tabelle
4.6, Eintrag 5-7). Die Enzymmenge konnte ebenfalls auf 54 U/mmol gesenkt werden (Tabelle
4.6, Eintrag 7). Der Anteil an gebildetem Glycin (11) lag bei 46%, wobei 5% des anti-Isomers
nachgewiesen werden konnten.
Unter diesen Bedingungen wurde der Ansatz in einem 50 mL-Maßstab durchgeführt. Durch
die Vergrößerung des Reaktionsvolumens wurde keine Beeinträchtigung des Umsatzes

ENZYMATISCHE ALDOLREAKTION
80
beobachtet (Abbildung 4.58, vgl. Abschnitt 8.2.2.1.4). Allerdings konnte nur etwa 10%
Produkt isoliert werden, welches noch Verunreinigungen (30%) enthielt.
Abbildung 4.58. Racematspaltung von rac-syn-12i in 50 mL-Maßstab
Aufgrund der schlechten Ergebnisse bei der Racematspaltung, bezogen auf Substrat-
konzentration und Ausbeute, stellt dies kein geeignetes Verfahren zur diastereomerenreinen
Herstellung von 12i dar.
4.5 Zusammenfassung
Im Vordergrund dieses Abschnitts stand die Optimierung der enzymkatalysierten
Aldolreaktion mit der zur Verfügung stehenden L-selektiven Threoninaldolase aus E. coli. Es
wurden zwei Methoden zur Umsatzbestimmung entwickelt, einmal mittels NMR-Standard
und einmal mittels Derivatisierung. Nach Etablierung der Standardreaktion mit Benzaldehyd
(1e) wurde der Einfluss der verschiedenen Reaktionsparameter genau untersucht. So wurde
die Abhängigkeit von der Reaktionszeit und der Enzymmenge analysiert. Auch das
Konzentrationsverhältnis der Edukte 1e und 11 zueinander wurde variiert. Die
Substratkonzentration des Aldehyden 1e konnte ohne größere Umsatzeinbußen von 0.1 M
auf 0.25 M erhöht werden, wohingegen ein hoher Überschuss an Glycin (11) für eine gute
Produktbildung notwendig war. Die optimierte Standardreaktion ist in Abbildung 4.59
dargestellt.

ENZYMATISCHE ALDOLREAKTION
81
Abbildung 4.59. Optimierte Standardreaktion
Des Weiteren wurden verschieden substituierte Benzaldehyde eingesetzt und der Einfluss
des Substitutionsmusters auf Umsatz und Diastereomerenverhältnis untersucht. Dabei
wurde festgestellt, dass o-substituierte Benzaldehyde die besten Ergebnisse vor allem in
Hinblick auf die Diastereoselektivität lieferten. Als bestes Substrat erwies sich o-
Chlorbenzaldehyd (1i). Die entsprechende β-Hydroxy-α-aminosäure (2S)-12i wurde mit
einem Diastereomerenverhältnis von d.r. 80:20 (syn/anti) bei einem Umsatz von 90%
erhalten. Mit diesem Substrat wurde nochmals eine Optimierung der Reaktion durchgeführt,
wodurch ein vollständiger Umsatz, bei geringerer Enzymmenge und lediglich 8 Äquivalenten
Glycin erzielt werden konnte (Abbildung 4.60).
Abbildung 4.60. Optimierte enzymatische Reaktion mit 1j als Substrat
Unter diesen Bedingungen wurde die Reaktion in größerem Maßstab (bis zu 100 mL)
durchgeführt und (2S)-12i isoliert. Durch Fällung konnte das gewünschte Produkt als
Diastereomerengemisch in einer Ausbeute von 62% erhalten werden (Abbildung 4.61).
Abbildung 4.61. Optimierte Reaktion in 100 mL-Maßstab

ENZYMATISCHE ALDOLREAKTION
82
Verschiedene Versuche zur Trennung der Diastereomere schlugen leider fehl. Auch die
alternative Route der Racematspaltung brachte keine nutzbaren Ergebnisse, da bei sehr
geringer Substratkonzentration nur schlechte Ausbeuten erzielt werden konnten.
Dennoch konnte ein guter Synthese-Prozess mit hoher volumetrischer Produktivität etabliert
werden. Die Durchführung der Reaktion bei der hohen Substratkonzentration von 0.25 M
stellt eine Verbesserung der Syntheseroute dar und auch die Anwendung in großem
Maßstab wurde bisher noch nicht gezeigt.

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
83
5. Epoxidsynthese ausgehend von β-Hydroxy-α-amino-
säuren
5.1 Einleitung
Chirale Epoxide sind sehr wichtige Bausteine zur Herstellung pharmazeutisch relevanter
Verbindungen. Vor allem die katalytische asymmetrische Synthese wird sowohl in der
Akademia, als auch in der Industrie für diese Zwecke verwendet.121 2001 wurde der
Nobelpreis an Sharpless für seine herausragenden Ergebnisse auf dem Gebiet der
asymmetrischen Epoxidierung verliehen.122
Die von Sharpless entwickelte katalytische asymmetrische Synthese von Epoxiden123 findet
beispielsweise in der Herstellung des Leukotrien-Antagonisten 87 Verwendung (Abbildung
5.1). Diese Verbindung hemmt die Wirkung von Leukotrienen, die Entzündungen und
allergische Reaktionen, einschließlich Asthma, hervorrufen.124
Abbildung 5.1. Synthese eines Leukotrien-Antagonisten 87124
Bei leicht zugänglichen chiralen Ausgangsverbindungen bietet sich auch eine einfache
Substitutionsreaktion zum Erhalt enantiomerenreiner Epoxide an. Analog zu dem
entsprechenden Zwischenschritt der Darzens-Glycidester-Kondensation1, können aus
enantiomerenreinen Halohydrinen 20 unter basischen Bedingungen Epoxide 21 erhalten
werden (Abbildung 5.2).125
Abbildung 5.2. Synthese von Epoxiden 21 ausgehend von Halohydrinen 20

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
84
5.2 Stand der Wissenschaft
5.2.1 Katalytische asymmetrische Epoxidierung
Es gibt viele verschiedene Methoden zur asymmetrischen Synthese von Epoxiden. Von
besonderer Bedeutung ist hierbei sicherlich die Entwicklung chiraler Katalysatoren zur
selektiven Oxidation von Alkenen. Im Laufe der Zeit haben sich mehrere Systeme etabliert,
die dann entsprechend weiterentwickelt wurden.121
Ein Beispiel ist die bereits erwähnte Sharpless-Epoxidierung,123 bei der Allylakohole mit
hoher Enantioselektivität epoxidiert werden können. Als Katalysator dient Titan(IV)-
isopropylat und als Epoxidierungsreagenz tert-Butylhydroperoxid. Die Selektivität wird durch
Zugabe von Weinsäureethylester (DET) erreicht. Während der Reaktion sind alle Reagenzien
über ihre Sauerstoffatome an das Metall komplexiert und daher können lediglich
Allylalkohole mit dieser Methode epoxidiert werden. Wichtig ist es zudem unter
Feuchtigkeitsausschluss zu arbeiten, da dies ansonsten zu einer Verschlechterung der
ee-Werte von 99% auf 48% führt.126
Die Jacobsen-Katsuki-Epoxidierung bietet eine Erweiterung auf nicht funktionalisierte
Olefine.1,127 Mit den höchsten Enantiomerenüberschüssen werden cis-Alkene wie 88 von
Mangan-Salen-Komplexen 90 als Katalysator umgesetzt. Beispielsweise kann mit Hilfe dieser
Methode 88 mit sehr guten Enantioselektivitäten von 92% ee zum Epoxid 89 umgesetzt
werden (Abbildung 5.3).128
Abbildung 5.3. Jacobsen-Katsuki-Epoxidierung128

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
85
Zur katalytischen asymmetrischen Epoxidierung stehen noch weitere Katalysatorsysteme zur
Verfügung, wie beispielsweise La(BINOL)-Komplexe, die von Shibasaki zur Epoxidierung von
Enonen eingesetzt wurden.121 Dioxirane, die aus chiralen Ketonen und Oxon in situ
hergestellt werden, werden bei der Shi-Epoxidierung eingesetzt.129
Ein Nachteil dieser homogenen Systeme ist die aufwändige Aufarbeitung und
Rückgewinnung der Katalysatoren. Aus diesem Grund wurden bereits viele verschiedene
heterogene Katalysatoren entwickelt, die jedoch eine geringere Aktivität aufweisen und
schwieriger in der Herstellung sind.121 Die bekannten Katalysatoren müssen im Hinblick auf
Syntheseaufwand und damit verbundenen Kosten optimiert werden. Außerdem ist bei allen
Methoden die Enantio- und Chemoselektivität noch ausbaufähig.
5.2.2 Synthese und Anwendung von Glycidestern
In der Totalsynthese verschiedener pharmazeutisch relevanter Verbindungen kommen
enantiomerenreine Glycidester zum Einsatz.130,131 Beispielsweise können asymmetrische
Syntheseverfahren,130 aber auch Racematspaltungen131,132 angewendet werden.
Diltiazem (92) ist ein typischer Calciumkanalblocker, der zur Behandlung koronarer
Herzkrankheiten, Herzrhythmusstörungen und Bluthochdruck eingesetzt wird. Ein Baustein
zur Herstellung dieses Arzneimittels ist ein trans-Methylgylcidester 91 (Abbildung 5.4), der
schon auf verschiedene Weisen hergestellt wurde. Jedoch sind diese Prozesse in Bezug auf
Ausbeute, Enantioselektivität und Katalysatormenge noch optimierbar.132 Beispielsweise
konnte 91 auch mittels Jacobsen-Epoxidierung hergestellt werden, allerdings mit einem
Enantiomerenüberschuss von lediglich 86%.133
Abbildung 5.4. Glycidester 91 als Synthesebaustein für Diltiazem (92)132

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
86
Eine Methode zur Synthese des gewünschten Glycidesters verläuft über eine
metallkatalysierte dynamisch kinetische Racematspaltung (Abbildung 5.5).132 Unter
Verwendung eines chiralen Rutheniumkatalysators wird die Ketogruppe von 93 selektiv zum
Alkohol 94 reduziert. Der anschließende Ringschluss erfolgt im Basischen unter Inversion der
Konfiguration aber unter Erhalt des Enantiomerenüberschusses. Auf diese Weise konnte das
Epoxid 91 mit einer Enantioselektivität von 95% ee isoliert werden.
Abbildung 5.5. Synthese des Glycidester 91132
Kuroda et al. entwickelten eine Route zu 94 via einer Mukaiyama-Aldolreaktion (Abbildung
5.6).130 Das Zwischenprodukt 97 kann mit einer Ausbeute von 83% und einem
Enantiomerenüberschuss von 96% erhalten werden. Der ee-Wert konnte durch
anschließende Umkristallisation auf >99% gesteigert werden. Nach Reduktion zu
Monochlorhydrin und Cyclisierung unter basischen Bedingungen wird das gewünschte
Produkt 91 ebenfalls mit >99% ee erhalten.
MeO
+OMe
OTMSCl
Cl
S
O
O
NBH
O
O
CH2Cl2, -78°C, 7 h
(96, 100 mol%)
MeO
OH
CO2Me
ClCl
9783% Ausbeute
96% ee
1a 95
H
O
Abbildung 5.6. Synthese des Zwischenprodukts 96 über Mukaiyama-Aldolreaktion130
Eine Verbesserung der Herstellung von Glycidestern ist weiter erforderlich. Die beiden
vorgestellten Methoden verlaufen über mehrere aufwändige Stufen, bei denen entweder

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
87
die Enantioselektivität verbesserungswürdig ist oder der Katalysator in stöchiometrischen
Mengen eingesetzt werden muss.
5.3 Ziel der Arbeit
In diesem Teil sollte ausgehend von enantiomerenreinem o-Chlorphenylserin (2S)-12i aus
der optimierten enzymatischen Umsetzung (Abschnitt 4.4.8.4) das entsprechende Epoxid 21i
hergestellt werden. Im ersten Schritt sollte die Aminogruppe durch Chlor substituiert
werden und anschließend das so erhaltene Halohydrin 20i mittels Ringschluss zum Epoxid
21i umgesetzt werden (Abbildung 5.7).
Abbildung 5.7. Synthese von Epoxiden 21i ausgehend von β-Hydroxy-α-aminosäuren (2S)-12i
Da die entsprechenden Reaktionen unter Erhalt bzw. Inversion der Konfiguration
ablaufen,125,134,135 sollte auf diesem Weg das gewünschte Epoxid enantiomerenrein erhalten
werden.
5.4 Eigene Ergebnisse und Diskussion
5.4.1 Halohydrinsynthese
Die Halohydrinsynthese wurde zuerst anhand der racemischen syn-Verbindung 12e
untersucht. Anschließend wurden die chlorsubstituierten Verbindungen rac-syn-12i und
(2S)-12i eingesetzt (vgl. Abschnitt 8.2.3.1).
5.4.1.1 Synthese von rac-syn-3-Hydroxy-3-phenylpropansäure rac-syn-20e
Die Synthese des Halohydrins rac-syn-20e erfolgte analog der von Whitesides et al.
beschriebenen Methode.125 Die β-Hydroxy-α-aminosäure rac-syn-12e wurde mit NaNO2 in

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
88
HCl zu rac-syn-20e umgesetzt. Das gewünschte Produkt konnte nach Umkristallisation in
CH2Cl2/Hexan mit einer Ausbeute von 29% isoliert werden (Abbildung 5.8).
Abbildung 5.8. Synthese von rac-syn-20h
5.4.1.2 Synthese der chlorsubstituierten Halohydrinverbindungen (2S)-20i
Analog wurden anschließend auch die chlorsubstituierte β-Hydroxy-α-aminosäure (2S)-12i
(d.r. 76:24 syn/anti) mit NaNO2 umgesetzt. Es konnte das gewünschte Halohydrin (2S)-20i
mit einer Ausbeute von 20% isoliert und charakterisiert werden (Abbildung 5.9). Bei der
Isolierung wurde eine Anreicherung des Überschussisomers beobachtet.
Abbildung 5.9. Synthese von (2S)-20j
5.4.2 Synthese des Epoxids 21i durch Ringschluss
Die Synthese des Epoxids 21i erfolgte direkt aus den chlorsubstituierten β-Hydroxy-
α-aminosäuren rac-syn-12i und (2S)-12i über zwei Stufen ohne Isolierung der Zwischenstufe
20i. Aufgrund der geringen Ausbeuten bei der Isolierung von rac-syn-20e und (2S)-20i wurde
das Rohprodukt direkt umgesetzt. Da eine Analyse der Konfiguration bei der offenkettigen
Verbindung 20 nicht möglich war, wurde diese anhand des Epoxids rac-syn-21i durchgeführt
(vgl. Abschnitt 8.2.3.2).

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
89
5.4.2.1 Synthese des racemischen Epoxids rac-syn-21i
Die Synthese des Halohydrins rac-syn-20i wurde analog Abschnitt 5.4.1 durchgeführt. Der
Umsatz (74%) wurde mittels 1H-NMR-Spektroskopie aus dem Rohprodukt bestimmt.
Anschließend erfolgte der Ringschluss mit Hilfe von tert-Butanolat als Base und das
gewünschte Epoxid rac-syn-21i konnte mit einer Ausbeute von 34% isoliert werden
(Abbildung 5.10). Dazu wurde die Säure rac-syn-21i in MTBE gelöst und durch Zugabe von
ethanolischer NaOH ausgefällt. Nach anschließender Neutralisation und Extraktion konnte
(2R,3R)-21i bzw. (2S,3S)-21i erhalten und vollständig charakterisiert werden.
Abbildung 5.10. Synthese des racemischen Epoxids 21i
In der Literatur wurde bereits beschrieben, dass bei der Substitution einer Aminogruppe
durch ein Chloridion die bestehende Konfiguration erhalten bleibt.125 Beim Ringschluss
dieser Halohydrinverbindungen 20 kommt es durch eine intramolekulare SN2 Reaktion zur
Inversion des Stereozentrums in α-Position.134,135 Die Überprüfung dieser Daten erfolgte
mittels Kernresonanzspektroskopie. Durch sogenannte 1H-NOESY-Aufnahmen kann durch
Nutzung des Kern-Overhauser-Effekts eine Aussage über die räumliche Nähe von Kernen
getroffen werden.136 Eine solche Bestimmung kann allerdings nur bei den starren
Ringsystemen 21 sinnvoll durchgeführt werden. Bei den offenkettigen Verbindungen ist die
Rotation um die Einfachbindungen zu schnell.
In Abbildung 5.11 A ist das 1H-NMR-Spektrum des Epoxids mit Zuordnung der einzelnen
Signale gezeigt. Abbildung 5.11 B und C zeigen die entsprechenden NOESY-Aufnahmen.
Hierbei wird ein Proton in seiner Frequenz angestrahlt und erscheint im Spektrum als
negatives Signal. Die Intensität der Signale der restlichen Protonen zeigt im Vergleich zum

EPOXIDSYNTHESE AUSGEHEND VON
90
Spektrum A die räumliche Nähe der Kerne an. Je weiter die Kerne voneinander entfernt sind,
desto schwächer wird die Intensität der Signale. Die Schwächung der Peakintensi
deutlich im aromatischen Bereich sichtbar. Es kann hier auch die Aussage getroffen werden,
welches der beiden H-Atome sich an C2
Abbildung
Da die Signale des en
Intensitätsverringerung aufweisen, müssen
befinden, da bei einer anti
Konfiguration stimmt auch mit der in der Literatur beschri
der Konfiguration währen der beiden Reaktionsschritte überein.
1H-NOESY-Spektroskopie konnte bewiesen werden, dass das erhaltenen Epoxid
Konfiguration (2R,3R) bzw. (2S
END VON β-HYDROXY-α-AMINOSÄUREN
die räumliche Nähe der Kerne an. Je weiter die Kerne voneinander entfernt sind,
desto schwächer wird die Intensität der Signale. Die Schwächung der Peakintensi
deutlich im aromatischen Bereich sichtbar. Es kann hier auch die Aussage getroffen werden,
Atome sich an C2- bzw. C3-Position befindet.
Abbildung 5.11. NOESY-Spektren von rac-syn-21i
Signale des entsprechenden anderen Protons am Oxiran keine
weisen, müssen sich die beiden Protonen in
anti-Stellung die Peakhöhen deutlich niedriger
t auch mit der in der Literatur beschriebenen Retention bzw. Inversion
währen der beiden Reaktionsschritte überein.125,134
konnte bewiesen werden, dass das erhaltenen Epoxid
S,3S) aufweist.
AMINOSÄUREN
die räumliche Nähe der Kerne an. Je weiter die Kerne voneinander entfernt sind,
desto schwächer wird die Intensität der Signale. Die Schwächung der Peakintensität ist
deutlich im aromatischen Bereich sichtbar. Es kann hier auch die Aussage getroffen werden,
Protons am Oxiran keine
sich die beiden Protonen in syn-Position
Stellung die Peakhöhen deutlich niedriger wären. Diese
benen Retention bzw. Inversion
134,135 Mit Hilfe der
konnte bewiesen werden, dass das erhaltenen Epoxid 21i die

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
91
5.4.2.2 Reaktion des enantiomerenreinen Halohydrins (2S)-20i zum Epoxid 21i
Als nächstes wurde das Epoxid ausgehend von der enantiomerenreinen β-Hydroxy-
α-aminosäure (2S)-12i aus der enzymatischen Synthese hergestellt. Dabei wurde (2S)-12i mit
einem Diastereomerenverhältnis von d.r. 78:22 (syn/anti) eingesetzt. Das Halohydrin
(2S)-20i wurde mit einem Umsatz von über 70% und einem Diastereomerenverhältnis von
d.r. 80:20 (syn/anti) erhalten. Bei der anschließenden Umsetzung mit tert-Butanolat wurde
ein vollständiger Umsatz erzielt. Zur Aufreinigung wurde das Rohprodukt in MTBE gelöst und
durch Zugabe von ethanolischer NaOH ausgefällt. Nach anschließender Neutralisation
konnten 30% Produkt mit einem Diastereomerenverhältnis von d.r. 82:18 (syn/anti) erhalten
werden. Durch weitere Zugabe von ethanolischer NaOH und MTBE zum Filtrat kam es zu
einer zweiten Fällung, hier Betrug die Ausbeute 22% (2R)-21i und das
Diastereomerenverhältnis d.r. 95:5 (syn/anti) (Abbildung 5.12). Beim Ringschluss wurde
keine bevorzugte Bildung des syn- oder anti-Isomers aus sterischen Gründen beobachtet.
Abbildung 5.12. Synthese von (2R)-21i
Anschließend wurde der Enantiomerenüberschuss des Epoxids (2R)-21i mittels chiraler HPLC
bestimmt, um eine Racemisierung während der Reaktion auszuschließen. In Abbildung 5.13
A ist das Chromatogramm der racemischen Verbindung (2R,3R)-21i bzw. (2S,3S)-21i gezeigt.
Die Peaks der beiden Enantiomere konnten deutlich getrennt werden. Das Spektrum der
enantiomerenreinen Verbindung (Abbildung 5.13 B) zeigt ebenfalls zwei Signale, wobei ein
Peak eine deutlich abweichende Retentionszeit aufweist. Daraus lies sich schließen, dass es
sich um die enantiomerenreinen Verbindungen (2R,3R)-21i bzw. (2R,3S)-21i handelt. Um das
Ergebnis zu verifizieren, wurde eine Mischung aus der racemischen syn-Verbindung

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
92
(2R,3R)-21i, (2S,3S)-21i und der enantiomerenreinen Verbindung (2R)-21i vermessen. In
Abbildung 5.13 C sind deutlich drei Signale zu sehen. Somit kann für (2R,3R)-21i ein ee-Wert
von >99% angegeben werden.
Abbildung 5.13. HPLC-Analytik zur ee-Wertbestimmung von 21i
5.4.3 Zusammenfassung
In diesem Anschnitt wurde die Synthese von Epoxiden 21 ausgehend von
enantiomerenreinen β-Hydroxy-α-aminosäuren 12 untersucht. Dabei wurde auf eine Route
in Anlehnung an die Darzens-Reaktion1 zurückgegriffen. Rac-syn-12e, rac-syn-12i und
(2S)-12i wurden dabei zum entsprechenden Halohydrin 20 umgesetzt und teilweise isoliert.
Die Halohydrine 20i wurden anschließend direkt im Basischen zum Epoxid 21i umgesetzt. Zur
Untersuchung der Stereoselektivität der Reaktionen wurde das Produkt (2R,3R)-21i bzw.
(2S,3S)-21i mittels 1H-NOESY-Spektroskopie analysiert und es konnte eine syn-Konfiguration
der beiden Protonen am Oxiran festgestellt werden. Mittels chiraler HPLC wurde außerdem
eine Racemisierung während der beiden Reaktionsschritte ausgeschlossen.
In Abbildung 5.14 ist exemplarisch die Synthese von (2R)-21i aus der enantiomerenreinen
β-Hydroxy-α-aminosäure (2S)-12i gezeigt. Über zwei Stufen konnte das gewünschte Epoxid
in einer Ausbeute von 52% isoliert werden. Es konnte sogar eine Anreicherung des
Überschussisomers (2R,3R)-21i auf d.r. 95:5 (syn/anti) beobachtet werden.

EPOXIDSYNTHESE AUSGEHEND VON β-HYDROXY-α-AMINOSÄUREN
93
Abbildung 5.14. Synthese von (2R)-21i
Die gezeigte Syntheseroute bietet einen einfachen Zugang zu enantiomerenreinen Epoxiden
(2R)-21i. Ausgehend von der im 100 mL-Maßstab enzymatisch leicht herzustellenden
β-Hydroxy-α-aminosäure (2S)-12i (Abschnitt 4.4.8.4) konnte das Epoxid (2R)-21i über zwei
Stufen in einer Gesamtausbeute von 52% isoliert werden. Außerdem wurde eine
Anreicherung des Überschussisomers (2R,3R)-21i erzielt.
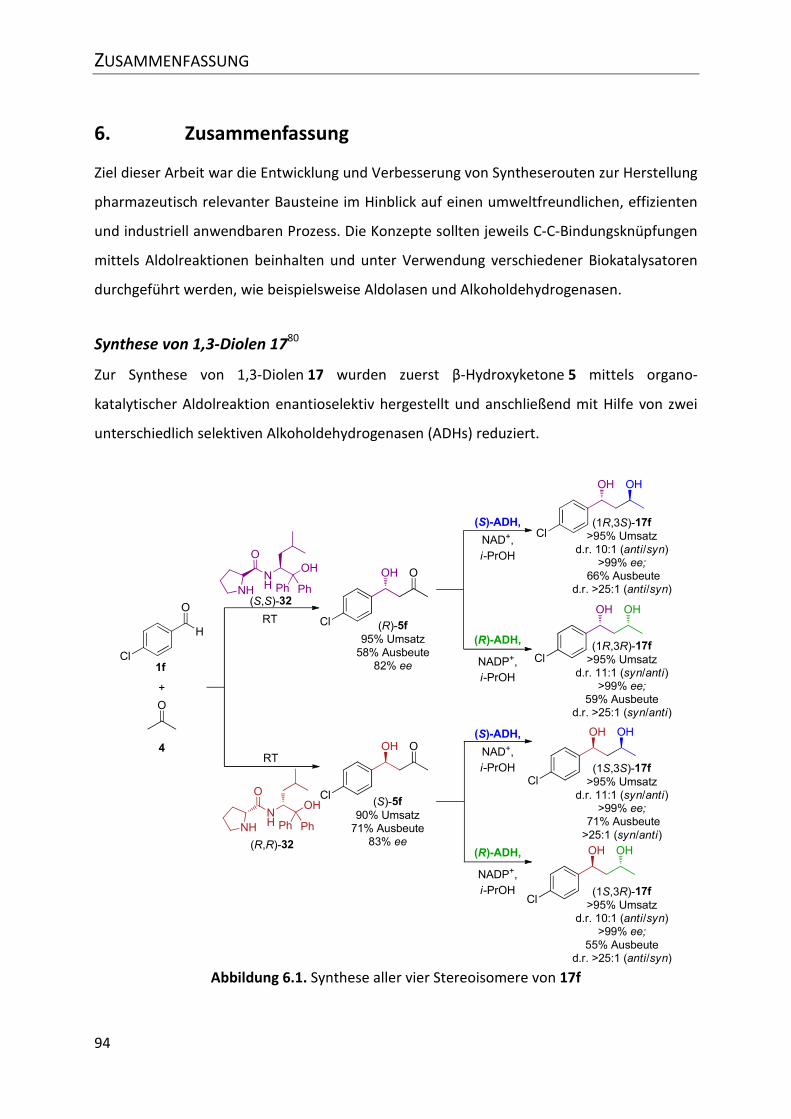
ZUSAMMENFASSUNG
94
6. Zusammenfassung
Ziel dieser Arbeit war die Entwicklung und Verbesserung von Syntheserouten zur Herstellung
pharmazeutisch relevanter Bausteine im Hinblick auf einen umweltfreundlichen, effizienten
und industriell anwendbaren Prozess. Die Konzepte sollten jeweils C-C-Bindungsknüpfungen
mittels Aldolreaktionen beinhalten und unter Verwendung verschiedener Biokatalysatoren
durchgeführt werden, wie beispielsweise Aldolasen und Alkoholdehydrogenasen.
Synthese von 1,3-Diolen 1780
Zur Synthese von 1,3-Diolen 17 wurden zuerst β-Hydroxyketone 5 mittels organo-
katalytischer Aldolreaktion enantioselektiv hergestellt und anschließend mit Hilfe von zwei
unterschiedlich selektiven Alkoholdehydrogenasen (ADHs) reduziert.
Cl
O
H
O
Cl
OH O
Cl
OH O
(R)-5f95% Umsatz58% Ausbeute
82% ee
(S)-5f90% Umsatz71% Ausbeute
83% ee
Cl
OH OH
Cl
OH OH
Cl
OH OH
Cl
OH OH
NAD+,
i-PrOH
(1R,3S)-17f>95% Umsatz
d.r. 10:1 (anti/syn)>99% ee;
66% Ausbeuted.r. >25:1 (anti/syn)
(1R,3R)-17f>95% Umsatz
d.r. 11:1 (syn/anti)>99% ee;
59% Ausbeuted.r. >25:1 (syn/anti)
(1S,3S)-17f>95% Umsatz
d.r. 11:1 (syn/anti)>99% ee;
71% Ausbeute>25:1 (syn/anti)
(1S,3R)-17f>95% Umsatz
d.r. 10:1 (anti/syn)>99% ee;
55% Ausbeuted.r. >25:1 (anti/syn)
NHNH
OOH
Ph Ph
NHNH
OOH
Ph Ph
RT
RT
+
(S,S)-32
(R,R)-32
NADP+,
i-PrOH1f
4
(S)-ADH,
(R)-ADH,
NAD+,
i-PrOH
(S)-ADH,
NADP+,
i-PrOH
(R)-ADH,
Abbildung 6.1. Synthese aller vier Stereoisomere von 17f

ZUSAMMENFASSUNG
95
Durch dieses Konzept können die entsprechenden Stereozentren selektiv aufgebaut werden
und alle vier möglichen Stereoisomere mit der gleichen Methode synthetisiert werden. Die
Ergebnisse der sequentiellen Synthese der 1,3-Diole 17f, bei der die organokatalytische
Aldolreaktion mit einem Überschuss von Aceton (4) durchgeführt wurde, sind in Abbildung
6.1 dargestellt. Über zwei Schritte konnten die jeweiligen 1,3-Diole 17f in einer
Gesamtausbeute von 34-50% enantiomerenrein isoliert werden.
Des Weiteren wurde die Organokatalyse in wässrigem Medium mit Substrat 1f durchgeführt.
Es wurden deutlich höhere Enantiomerenüberschüsse von 92% ee (82% ee in organischem
Medium) in diesem Lösungsmittel bei einem Umsatz von 90% erhalten. Die
organokatalytische Aldolreaktion in wässrigem Medium wurde außerdem in einer
Eintopfreaktion mit der enzymatischen Reduktion kombiniert. Auf diesem Weg wurde unter
anderem (1R,3S)-17f in einem sehr guten Diastereomerenverhältnis (d.r. >25:1 anti/syn)
erhalten und konnte in einer Ausbeute von 73% isoliert werden (Abbildung 6.2).
Abbildung 6.2. Eintopfsynthese von (1R,3S)-17f
Diese Syntheseroute, bei der die beiden Stereozentren sequentiell aufgebaut werden, ist ein
effizientes Verfahren zur enantiomerenreinen Synthese von 1,3-Diolen 17. Prinzipiell können
alle vier möglichen Enantiomere über die gezeigte Eintopfreaktion hochselektiv und in hoher
Ausbeute hergestellt werden.
Synthese von β-Hydroxy-α-aminosäuren 12115
Die enantionselektive Synthese von β-Hydroxy-α-aminosäuren wurde mit Hilfe einer zur
Verfügung stehenden L-Threoninaldolase (L-TA) aus E. coli durchgeführt und genau
analysiert, sowie anschließend optimiert. Nach Etablierung zweier Methoden zur
Umsatzbestimmung wurde der Einfluss der verschiedenen Reaktionsparameter untersucht.
Die Abhängigkeit des Umsatzes und des Diastereomerenverhältnisses von der Reaktionszeit,

ZUSAMMENFASSUNG
96
sowie der Enzymmenge wurde ebenfalls analysiert. Durch Variation des
Konzentrationsverhältnisses der Edukte zueinander wurde die Notwendigkeit des hohen
Überschusses an Glycin (11) gezeigt. Die Substratkonzentration des Aldehyds konnte ohne
größere Umsatzeinbußen von 0.1 M auf 0.25 M erhöht werden. Die optimierte
Standardreaktion ist in Abbildung 6.3 dargestellt.
Abbildung 6.3. Optimierte Standardreaktion
Zur Erweiterung der Substratbreite wurden verschieden substituierte Benzaldehyde
eingesetzt und der Einfluss des Substitutionsmusters auf Umsatz und Diastereomeren-
verhältnis untersucht. Dabei wurde festgestellt, dass o-substituierte Benzaldehyde vor allem
in Hinblick auf die Diastereoselektivität (d.r. >70:30 syn/anti) die besten Ergebnisse lieferten.
Als vielversprechendstes Substrat erwies sich o-Chlorbenzaldehyd (1i). Die entsprechende
β-Hydroxy-α-aminosäure (2S)-12i wurde mit einem Diastereomerenverhältnis von d.r. 80:20
(syn/anti) bei einem Umsatz von 90% erhalten. Mit diesem Substrat wurde eine Optimierung
der Reaktion durchgeführt, wodurch ein vollständiger Umsatz, bei geringerer Enzymmenge
erzielt werden konnte (Abbildung 6.4).
Abbildung 6.4. Optimierte enzymatische Reaktion mit 1i als Substrat
Unter diesen Bedingungen wurde anschließend die Reaktion in größerem Labormaßstab (bis
zu 100 mL) durchgeführt und (2S)-12i isoliert. Verschiedene Versuche zur Trennung der
Diastereomere schlugen fehl. Zur enantio- und diastereomerenreinen Synthese von 12i

ZUSAMMENFASSUNG
97
wurde deshalb eine Racematspaltung durchgeführt. Allerdings konnte nur eine geringe
Ausbeute (10%) bei niedriger Substratkonzentration (0.05 M) erhalten werden. Somit stellt
dieses Verfahren keine Alternative zur synthetischen Route dar. Bei dieser konnte durch
Fällung das reine Produkt als Diastereomerengemisch (d.r. 78:22 syn/anti) in einer Ausbeute
von 62% (3.3 g) erhalten werden (Abbildung 6.5). Somit konnte ein Prozess mit hoher
volumetrischer Produktivität etabliert werden.
Abbildung 6.5. Optimierte Reaktion in 100 mL-Maßstab
Synthese von Epoxiden 21
Ausgehend von der enantiomerenreinen β-Hydroxy-α-aminosäure (2S)-12i aus der
enzymatischen Synthese wurde das Epoxid (2R)-21i in nur zwei Schritten synthetisiert.
Zunächst wurde das Halohydrin 20i durch Substitution der Aminogruppe durch Chlor mit
einem Umsatz von 70% hergestellt. Der Ringschluss erfolgte ohne Isolierung von 20i mit tert-
Butanolat als Base. Bei der Synthese von (2R)-21i konnte eine Gesamtausbeute von 52%
erzielt werden. Außerdem wurde eine Anreicherung des Diastereomerenverhältnisses von
d.r. 78:22 auf d.r. 95:5 (syn/anti) beobachtet (Abbildung 6.6).
Eine Analyse der Konfiguration mittels 1H-NOESY-Spektroskopie zeigte, dass sich aus
rac-syn-12i das syn-Epoxid (2R,3R)-21i bzw. (2S,3S)-21i bildet. Eine Racemisierung der
enantiomerenreinen Ausgangsverbindung (2S)-12i während der zwei Reaktionsschritte
wurde mittels chiraler HPLC ausgeschlossen. Es konnte somit eine einfache Synthese von
enantiomerenreinen Epoxiden (2R)-21i gezeigt werden. Ausgehend von der im
100 mL-Maßstab enzymatisch einfach herzustellenden β-Hydroxy-α-aminosäure (2S)-12i

ZUSAMMENFASSUNG
98
konnte über zwei Stufen das gewünschte Epoxid (2R)-21i mit einer Gesamtausbeute von
52% erhalten werden.
Abbildung 6.6. Synthese von (2R)-21i
Insgesamt konnten verschiedene effiziente Synthesemethoden zur Herstellung
pharmazeutisch relevanter Verbindungen 12, 17 und 21 etabliert werden. 1,3-Diole 17f
konnten in einer wässrigen Eintopfsynthese hochselektiv in sehr guter Ausbeute hergestellt
werden. Bei der Synthese von β-Hydroxy-α-aminosäuren 12 gelang die Entwicklung eines
Prozesses mit hoher volumetrischer Produktivität. Es konnte ebenfalls eine Folgechemie der
enantiomerenreinen Verbindung (2S)-12i zu Epoxiden (2R)-21i gezeigt werden.

SUMMARY
99
7. Summary
The aim of this thesis was the development and improvement of synthetic routes for the
production of pharmaceutical compounds with regard to an environmentally compatible,
efficient and industrial applicable process. The concept included C-C bond formations based
on aldol reactions and different types of biocatalysts, like aldolases and alcohol
dehydrogenases.
Synthesis of 1,3-diols 17137
For the synthesis of 1,3-diols 17 as a first step β-hydroxyketones 5 were produced
enantioselectively via an organocatalytic aldol reaction, followed by reduction of the
carbonyl group applying two different selective alcohol dehydrogenases (ADH) (Scheme 7.1).
Scheme 7.1. Synthesis of all possible stereoisomers of 17f

SUMMARY
100
By means of this concept, the stereogenic centers were formed selectively and furthermore
the synthesis of all four possible isomers was feasible using the same synthetic route.
Scheme 6.1 depicts the results of the sequential forming of 1,3-diols 17f. At this the
organocatalytic aldol reaction was carried out in an excess of acetone (4). After two reaction
steps, the 1,3-diols 17f were isolated in an enantiomerically pure form with an overall yield
of 34-50%.
Moreover, the organocatalytic aldol reaction was carried out in water instead of acetone
applying substrate 1f. Thereby, a considerable higher enantiomeric excess of 92% (82% ee in
organic solvent) in combination with a conversion of 90% was yielded. Due to these
improvements the organocatalytic aldol reaction in aqueous medium was combined with
the enzymatic reduction in an one-pot process. Thus, (1R,3S)-17f amongst others was
achieved with a very high diastereomeric ratio (d.r. >1:25 syn/anti) and a yield of 73%
(Scheme 7.2).
Scheme 7.2. One-pot synthesis of (1R,3S)-17f
This synthetic route, containing a sequential forming of the two stereogenic centers,
demonstrates an efficient process for the synthesis of enantiomerically pure 1,3-diols. It is
feasible to produce all four possible isomers with an aforementioned excellent
enantioselectivity and a high yield, using the shown one-pot process.
Synthesis of β-hydroxy-α-amino acids 12138
The enantioselective synthesis of β-hydroxy-α-amino acids was carried out by means of an
available L-threonin aldolase (L-TA) from E. coli. An explicit analysis of the process and
optimization was carried out. Afterwards the conversion and diastereomeric ratio according
to reaction time and amount of used biocatalyst were investigated. The necessity of the high
excess of glycin (11, 10 eq.) was proofed by variation of the ratio of substrate concentration

SUMMARY
101
(11:1e). The concentration of aldehyde 1 could be easily raised from 0.1 M to 0.25 M
without appreciable loss of conversion. The optimized standard reaction is depicted in
Scheme 7.3.
Scheme 7.3. Optimized standard reaction
The substrate range was expanded by using different substituted benzaldehydes 1.
Furthermore, the possible influence of the substitution pattern was analyzed. Best results
related to diastereoselectivity were found by using o-substituted benzaldehydes (d.r. >70:30
syn/anti). In particular, o-chlorobenzaldehyde (1i) revealed to be the most promising
substrate, achieving the β-hydroxy-α-amino acid (2S)-12i in a diastereomeric ratio of
d.r. 80:20 (syn/anti) and a conversion of 90%. Employing this substrate a further
optimization of the reaction process was carried out, whereby the attaching of lower
amount of biocatalyst (44 U/mmol) and a lower excess of glycine (8 eq.) was found to lead to
a complete conversion (Scheme 7.4).
Scheme 7.4. Optimized enzymatic reaction using 1i as a substrate
Increasing the scale to 100 mL by usage of the same reaction conditions product (2S)-12i
could be isolated. Different experiments were conducted for separating the diasteromers,
leading to no realization. Therefore a resolution was used to synthesize 12i diastereo- and
enantiopure. However, the desired product was isolated in a low yield of 10% applying a
meanly substrate concentration (0.05 M). Due to this fact, this procedure offers no practical
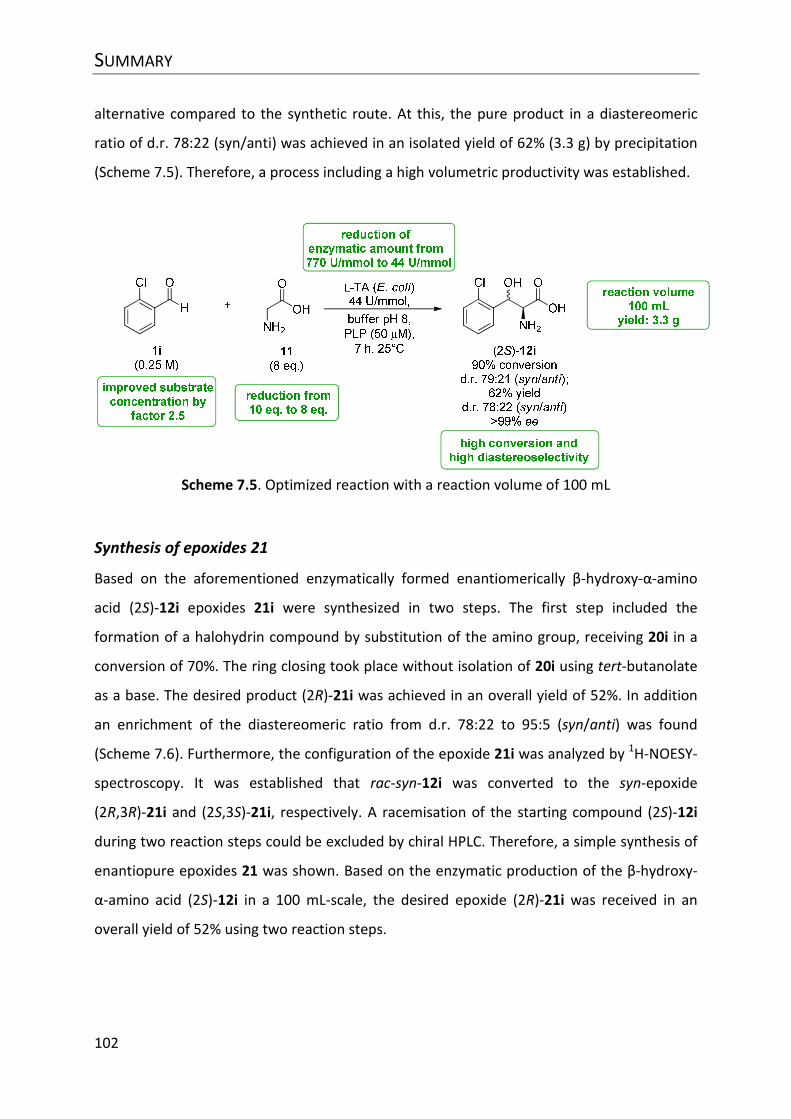
SUMMARY
102
alternative compared to the synthetic route. At this, the pure product in a diastereomeric
ratio of d.r. 78:22 (syn/anti) was achieved in an isolated yield of 62% (3.3 g) by precipitation
(Scheme 7.5). Therefore, a process including a high volumetric productivity was established.
Scheme 7.5. Optimized reaction with a reaction volume of 100 mL
Synthesis of epoxides 21
Based on the aforementioned enzymatically formed enantiomerically β-hydroxy-α-amino
acid (2S)-12i epoxides 21i were synthesized in two steps. The first step included the
formation of a halohydrin compound by substitution of the amino group, receiving 20i in a
conversion of 70%. The ring closing took place without isolation of 20i using tert-butanolate
as a base. The desired product (2R)-21i was achieved in an overall yield of 52%. In addition
an enrichment of the diastereomeric ratio from d.r. 78:22 to 95:5 (syn/anti) was found
(Scheme 7.6). Furthermore, the configuration of the epoxide 21i was analyzed by 1H-NOESY-
spectroscopy. It was established that rac-syn-12i was converted to the syn-epoxide
(2R,3R)-21i and (2S,3S)-21i, respectively. A racemisation of the starting compound (2S)-12i
during two reaction steps could be excluded by chiral HPLC. Therefore, a simple synthesis of
enantiopure epoxides 21 was shown. Based on the enzymatic production of the β-hydroxy-
α-amino acid (2S)-12i in a 100 mL-scale, the desired epoxide (2R)-21i was received in an
overall yield of 52% using two reaction steps.

SUMMARY
103
Scheme 7.6. Synthesis of (2R)-21i
Taken altogether, different efficient synthetic routes for the preparation of building blocks
for pharmaceutical compounds 17, 12 and 21 were displayed. 1,3-diols 17f were produced
high enantioselectively and in very good yields in an aqueous one-pot process. The
development of a process for the formation of β-hydroxy-α-amino acids 12i with high
volumetric productivity was realized. Furthermore, the reaction of the enantiomerically
compound 12i to epoxides (2R)-21i was demonstrated.

EXPERIMENTELLER TEIL
104
8. Experimenteller Teil
8.1 Verwendete Chemikalien und Geräte
Chemikalien:
Die für diese Doktorarbeit verwendeten käuflichen Chemikalien wurden von Acros
Organics®, Sigma-Aldrich®, ABCR®, Fisher Scientific® oder Fluka® bezogen und ohne weitere
Reinigung eingesetzt. Das Lösungsmittel MTBE wurde als Hochschulspende von der Evonik
Degussa GmbH zur Verfügung gestellt. Alle verwendeten Lösungsmittel, außer MTBE,
wurden vor Gebrauch am Rotationsverdampfer destilliert.
Enzyme:
Die verwendeten Enzyme wurden im Rahmen folgender Zusammenarbeiten erhalten:
Alkoholdehydrogenasen aus Lactobacillus kefir (LK-ADH) [W. Hummel, M.-R. Kula (FZ Jülich
GmbH), EP 456107; 1991. A. Weckbecker, W. Hummel, Biocat. Biotrans. 2006, 24, 380-389.]
und Rhodococcus sp. (Rsp-ADH) wurden im Arbeitskreis von Herrn Prof. Dr. W. Hummel
(Universität Düsseldorf, Institut für molekulare Enzymtechnologie, Forschungszentrum
Jülich) entwickelt und für diese Arbeit zur Verfügung gestellt. Diese ADHs sind ebenfalls
käuflich erwerbbar bei evocatal GmbH.
Die Cofaktoren NAD(P)H und NAD(P)+ wurden von Oriental Yeast Co. Ltd. bezogen.
Die L-Threoninaldolase aus E. coli wurde zu Beginn der Arbeit von Prof. Dr. W. Hummel,
(Universität Düsseldorf, Institut für molekulare Enzymtechnologie, Forschungszentrum
Jülich) und später in größeren Mengen von evocatal GmbH zur Verfügung gestellt. Es wurde
die vom Hersteller angegeben Aktiviät zur Berechnung der Enzymmenge herangezogen.
Dünnschichtchromatographie (DC):
Es wurden DC-Platten mit Kieselgel 60 F254 auf Aluminiumträgerfolie von Merck® verwendet.
Zum Nachweis der Substanzzonen diente die Fluoreszenzlöschung bei 254 nm Wellenlänge.
Säulenchromatographie:
Die säulenchromatographische Reinigung wurde mit SiO2 als stationärer Phase durchgeführt,
dafür wurde Merck® Silikagel 60 (0.04-0.063 nm) der Firma ICN Biomedicals GmbH
verwendet.

EXPERIMENTELLER TEIL
105
NMR-Spektroskopie:
Die Spektren wurden auf einem JEOL JNM GX 400, JEOL JNM EX 400 oder Bruker Avance 300
oder 400 NMR-Spektrometer aufgenommen. Alle Spektren wurden bei Raumtemperatur
gemessen. Die chemischen Verschiebungen der 1H-NMR- und 13C-NMR-Spektren sind in ppm
angegeben. Spinmultiplizitäten werden als s (Singulett), d (Dublett), dd (dupliziertes
Dublett), t (Triplett), q (Quartett) und m (Multiplett) angegeben. Die erhaltenen Daten
wurden mit der Software Delta (4.3.6) bearbeitet.
Massenspektrometrie (FAB-MS, EI-MS, MALDI):
Die Massenspektrometrie wurde am Micromass Zabspec-Spektrometer im FAB-Modus mit
m-Nitrobenzylalkohol (NBA) als Matrix, am Finnigan MAT 95 XP-Spektrometer im EI-Modus
der Firma Thermo Electron Corporation® oder am Biotech Axima Confidence Gerät der Firma
Shimadzu im MALDI-Modus mit Sinapinsäure (sin) und 2,5-Dihydroxybenzoesäure (dhb) als
Matrix gemessen.
IR-Spektroskopie (IR):
Die Infrarotspektren wurden mit einem ASI React IR®-1000-Spektrometer bzw. mit einem
Nicolet IR 100 FT-IR Spektrometer der Firma Thermo Scientific® aufgenommen. Die
Absorption wird in Wellenzahl ṽ [cm-1] angegeben.
Elementaranalyse (EA):
Die Elementaranalyse wurde am Gerät EA 1119 CHNS, CE Instruments durchgeführt.
Schmelzpunkt:
Die Schmelzpunktbestimmung wurde an einem Schmelzpunktbestimmer Appendix B IA 9100
der Firma Electrothermal durchgeführt.
Drehwertmessung:
Die Drehwertbestimmung wurde an einem Polarimeter, Model 341 der Firma Perkin Elmer
mit einer Na-Lampe (λ = 589 nm) durchgeführt.

EXPERIMENTELLER TEIL
106
UV/Vis-Spektroskopie:
Die UV/Vis-spektroskopischen Messungen wurden an einem SPEKOL® 1300 UV/Vis-
Spektrophotometer der Firma analytikjena durchgeführt und mit dem Programm
WinASPECT 2.2.6.0 (mit Zuhilfenahme des Moduls „Kinetik Programms“) bearbeitet.
HPLC:
Die Spektren wurden mit einem LC-Net II/ADC Gerät und Pumpen (PU-1587 Intelligent Prep.
Pump und PU 987 Intelligent Prep. Pump) der Firma JASCO oder mit einem Gerät CBM-10A
und Pumpe (LC-10AT Liquid Chromatograph) der Firma Shimadzu aufgenommen. Die
Trennungen erfolgten an den Säulen Daicel Chiralpak® AD-H bzw. Daicel Chiralcel® OJ-H.

EXPERIMENTELLER TEIL
107
8.2 Synthesen und spektroskopische Daten
8.2.1 Kombination von organokatalytischer Aldolreaktion und enzymatischer
Reduktion
8.2.1.1 Synthese von (S)-1-(4-Chlorphenyl)-1-hydroxybutan-3-on (S)-5f mittels
organokatalytischer Aldolreaktion
p-Chlorbenzaldehyd (1f, 2 mmol, 282 mg) und 5 mol% Organo-
katalysator (R,R)-32 (0.1 mmol, 39 mg) wurden in Aceton (4, 8 mmol,
588 µL) gelöst und 16 h gerührt. Nach Ablauf der Reaktionszeit
wurde 1 mL NaCl-Lösung zugegeben und dreimal mit EtOAc
(3 x 5 mL) extrahiert. Die vereinigten organischen Phasen wurden
über MgSO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Nach
säulenchromatrographischer Aufarbeitung (Hexan/EtOAc 5:1 (v/v), RF = 0.08) wurde das
gewünschte Produkt (S)-5f als gelbliches Öl in einer Ausbeute von 71% (Auswaage 282 mg)
und einer Enantioselektivität von 83% ee erhalten (AD-H, Eluent: Hexan/i-PrOH 95:5, flow
0.8 mL/min, 220 nm; tr(R) = 16.77 min, tr(S) = 18.39 min). 1H-NMR (400 MHz, CDCl3) δ (ppm)
= 2.17 (s, 3H, CH3), 2.79 (m, 2H, CH2), 3.40 (br s, 1H, OH), 5.09 (dd, 1H, 3J = 8.3 Hz, 3.9 Hz,
CH), 7.25-7.30 (m, 4H, H-aromat.). Die spektroskopischen Daten stimmen mit den in der
Literatur angegebenen Daten überein.139
8.2.1.2 Allgemeine Arbeitsvorschrift 1 (AAV 1): Isolierung der vier Isomere aus
der enzymatischen Reduktion
Abbildung 8.1. Enzymatische Reduktion der β-Hydroxyketone 5f
Das isolierte Aldolprodukt 5f (0.5 mmol) wurde in iso-Propanol (2.5 mL) gelöst. Anschließend
wurde Phosphatpuffer (pH 7, 50 mM, 0.1 M, 7.5 mL), MgCl2 (1 mM, nur bei Verwendung der
Cl
OHO
(S)-5fC10H11ClO2
M = 198.56 g/mol

EXPERIMENTELLER TEIL
108
(R)-ADH aus Lactobacillus kefir), NAD(P)+ (0.02 mmol) und die entsprechende
Alkoholdehydrogenase aus Rhodococcus sp. oder Lactobacillus kefir (40-100 U/mmol)
zugegeben. Das Reaktionsgemisch wurde 18-67 h bei RT gerührt. Anschließend wurde mit
EtOAc extrahiert (3 x 25 mL) und die vereinigten organischen Phasen über MgSO4
getrocknet. Nach Entfernen des Lösungsmittels im Vakuum wurde das Rohprodukt 17f
mittels Säulenchromatographie (Hexan/EtOAc 4:1, v/v) gereinigt und das gewünschte
Produkt erhalten.
8.2.1.2.1 Synthese von (1S,3R)-1-(4-Chlorphenyl)-1,3-butandiol (1S,3R)-17f
Die Synthese von (1S,3R)-17f wurde analog AAV 1 durchgeführt. Es
wurde (S)-5f (0.5 mmol, 99 mg) und die (R)-selektive ADH aus
Lactobacillus kefir (150 µL, 100 U/mmol), sowie der Cofaktor NADP+
(0.02 mmol, 15 mg) eingesetzt. Es wurde ein Umsatz von >95%, ein
Diastereomerenverhältnis von d.r. 10:1 (anti/syn) und eine
Enantioselektivität von >99% ee erhalten. Nach säulenchromatographischer Reinigung
(Hexan/EtOAc 4:1 (v/v), RF = 0.09) wurde das gewünschte Produkt (1S,3R)-17f als farbloses
Öl mit einer Ausbeute von 55% (Auswaage 55 mg) isoliert. AD-H-Säule, Eluent: Hexan/i-PrOH
97:3, flow 0.8 mL/min, 220 nm; tr = 46.51 min; [a]��� = -68 (c 0.5, CH2Cl2); 1H-NMR (400 MHz,
CDCl3) δ (ppm) = 1.23 (d, 3H, 3J = 6.1 Hz, CH3), 1.81-1.86 (m, 2H, CH2), 2.17 (s, 1H, OH), 3.15
(s, 1H, OH), 4.00-4.08 (m, 1H, CHCH3), 5.02 (dd, 1H, 3J = 7.3 Hz, 3.9 Hz, CHCH2), 7.26-7.31 (m,
4H, H-aromat.); 13C-NMR (100 MHz, CDCl3) δ (ppm) = 23.63 (CH3), 46.02 (CH2), 65.49
(CHCH3), 71.17 (CHCH2), 126.97, 128.56, 132.97, 143.01 (C-aromat.); MS (FAB) m/z = 183
(M-H2O)+; IR ṽ (cm-1): 3366, 3300, 2988, 2961, 2926, 2856, 1486, 1444, 1401, 1374, 830, 807;
Elementaranalyse (C10H13ClO2): Berechnet: C: 59.86, H: 6.53; Gefunden: C: 59.03, H: 6.51.
8.2.1.2.2 Synthese von (1R,3R)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3R)-17f
Die Synthese von (1R,3R)-17f wurde analog AAV 1 durchgeführt. Es
wurde (R)-5f (0.5 mmol, 99 mg) und die (R)-selektive ADH aus
Lactobacillus kefir (150 µL, 100 U/mmol), sowie der Cofaktor NADP+
(0.02 mmol, 15 mg) eingesetzt. Es wurde ein Umsatz von >95%, ein

EXPERIMENTELLER TEIL
109
Diastereomerenverhältnis von d.r. 11:1 (syn/anti) und eine Enantioselektivität von >99% ee
erhalten. Nach säulenchromatographischer Reinigung (Hexan/EtOAc 4:1 (v/v), RF = 0.11)
wurde das gewünschte Produkt (1R,3R)-17f als farbloses Öl mit einer Ausbeute von 59%
(Auswaage 59 mg) isoliert. AD-H-Säule, Eluent: Hexan/i-PrOH 97:3, flow 0.8 mL/min, 220 nm;
tr = 35.71 min; [a]��� = +40 (c 0.5, CH2Cl2); 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.20 (d, 3H,
3J = 6.1 Hz, CH3), 1.66-1.81 (m, 2H, CH2), 2.95 (s, 1H, OH), 3.66 (s, 1H, OH), 4.07-4.15 (m, 1H,
CHCH3), 4.88 (dd, 1H, 3J = 10.0 Hz, 3.2 Hz, CHCH2), 7.25-7.30 (m, 4H, H-aromat.); 13C-NMR
(100 MHz, CDCl3) δ (ppm) = 24.17 (CH3), 47.01 (CH2), 68.82 (CHCH3), 74.47 (CHCH2), 127.03,
128.56, 133.15, 142.95 (C-aromat.); MS (FAB) m/z = 183 (M-H2O)+; IR ṽ (cm-1): 3293, 3223,
2980, 2937, 2914, 2853, 1486, 1451, 1409, 1366, 849, 826; Elementaranalyse (C10H13ClO2):
Berechnet: C: 59.86, H: 6.53; Gefunden: C: 59.25, H: 6.70.
8.2.1.2.3 Synthese von (1S,3S)-1-(4-Chlorphenyl)-1,3-butandiol (1S,3S)-17f
Die Synthese von (1S,3S)-17f wurde analog AAV 1 durchgeführt. Es
wurde (S)-5f (0.5 mmol, 99 mg) und die (S)-selektive ADH aus
Rhodococcus sp. (50 µL, 40 U/mmol), sowie der Cofaktor NAD+
(0.02 mmol, 13 mg) eingesetzt. Es wurde ein Umsatz von >95%, ein
Diastereomerenverhältnis von d.r. 11:1 (syn/anti) und eine
Enantioselektivität von >99% ee erhalten. Nach säulenchromatographischer Reinigung
(Hexan/EtOAc 4:1 (v/v), RF = 0.11) wurde das gewünschte Produkt (1S,3S)-17f als farbloses Öl
mit einer Ausbeute von 71% (Auswaage 71 mg) isoliert. AD-H-Säule, Eluent: Hexan/i-PrOH
97:3, flow 0.8 mL/min, 220 nm; tr = 38.73 min; [a]��� = −40 (c 0.5, CH2Cl2); 1H-NMR (400 MHz,
CDCl3) δ (ppm) = 1.19 (d, 3H, 3J = 6.2 Hz, CH3), 1.67-1.81 (m, 2H, CH2), 2.95 (s, 1H, OH), 3.66
(s, 1H, OH), 4.08-4.14 (m, 1H, CHCH3), 4.88 (dd, 1H, 3J = 9.9 Hz, 3.1 Hz, CHCH2), 7.25-7.30 (m,
4H, H-aromat.). Die spektroskopischen Daten stimmen mit denen des (1R,3R)-Stereoisomers
(vgl. 8.2.1.2.2) überein.

EXPERIMENTELLER TEIL
110
8.2.1.2.4 Synthese von (1R,3S)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3S)-17f
Die Synthese von (1R,3S)-17f wurde analog AAV 1 durchgeführt. Es
wurde (R)-5f (0.5 mmol, 99 mg) und die (S)-selektive ADH aus
Rhodococcus sp. (50 µL, 40 U/mmol), sowie der Cofaktor NAD+
(0.02 mmol, 13 mg) eingesetzt. Es wurde ein Umsatz von >95%, ein
Diastereomerenverhältnis von d.r. 10:1 (anti/syn) und eine
Enantioselektivität von >99% ee erhalten. Nach säulenchromatographischer Reinigung
(Hexan/EtOAc 4:1 (v/v), RF = 0.09) wurde das gewünschte Produkt (1R,3S)-17f als farbloses
Öl mit einer Ausbeute von 66% (Auswaage 66 mg) isoliert. AD-H-Säule, Eluent: Hexan/i-PrOH
97:3, flow 0.8 mL/min, 220 nm; tr = 43.47 min; [a]��� = +68 (c 0.5, CH2Cl2); 1H-NMR (400 MHz,
CDCl3) δ (ppm) = 1.23 (d, 3H, 3J = 6.3 Hz, CH3), 1.81-1.86 (m, 2H, CH2), 2.18 (s, 1H, OH), 3.15
(s, 1H, OH), 4.02-4.08 (m, 1H, CHCH3), 5.02 (dd, 1H, 3J = 7.2 Hz, 4.1 Hz, CHCH2), 7.26-7.31 (m,
4H, H-aromat.). Die spektroskopischen Daten stimmen mit denen des (1S,3R)-Stereoisomers
(vgl. 8.2.1.2.1) überein.
8.2.1.3 Organokatalytische Aldolreaktion in wässrigem Medium
p-Chlorbenzaldehyd (1f, 0.5 mmol, 70 mg) und die entsprechende
Menge Organokatalysator (S,S)-32 (0.5-1 mol%) wurden in Aceton (4,
4.5 mmol, 330 µL) gelöst. Nach Zugabe von gesättigter NaCl-Lösung
(330 µL) wurde das Reaktionsgemisch gerührt. Nach Ablauf der
Reaktionszeit wurde mit CH2Cl2 (3 x 5 mL) extrahiert, die vereinigten
organischen Phasen über MgSO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Das
Rohprodukt wurde als rötliches Öl erhalten. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 2.17 (s, 3H,
CH3), 2.79 (m, 2H, CH2), 3.40 (br s, 1H, OH), 5.09 (dd, 1H, 3J = 8.3 Hz, 3.9 Hz, CH), 7.25-7.30
(m, 4H, H-aromat.). ee-Wertbestimmung mittels HPLC: AD-H-Säule, Eluent: Hexan/i-PrOH
95:5, flow 0.8 mL/min, 220 nm; tr(R) = 16.77 min, tr(S) = 18.39 min. Die spektroskopischen
Daten stimmen mit den in der Literatur angegebenen Daten überein.139 Die Ergebnisse der
Reaktionsoptimierung sind in Tabelle 8.1 dargestellt.
Cl
OHOH
(1R,3S)-17fC10H13ClO2
M = 200.66 g/mol
Cl
OHO
(R)-5fC10H11ClO2
M = 198.56 g/mol

EXPERIMENTELLER TEIL
111
Tabelle 8.1. Optimierung der organokatalytischen Aldolreaktion
Eintrag c(32) [mol%] t [h] Umsatz [%]1) ee [%]2)
1 0.5 24 58 (P), 39 (A), 1 (K), 1 (D) n.b.
2 0.5 32 61 (P), 36 (A), 2 (K),1 (D) 95
3 0.5 48 70 (P), 26 (A), 2 (K), 2 (D) 95
4 1 24 82 (P), 14 (A), 2 (K), 2 (D) 92
5 1 48 90 (P), 6(A), 2 (K), 2 (D) 92
1) (P): Produkt 5f, (A): Aldehyd 1f, (K): Kondensationsprodukt, (D): Dimer;
2) n.b. nicht bestimmt
8.2.1.4 Allgemeine Arbeitsvorschrift 2 (AAV 2): Eintopfreaktion
Abbildung 8.2. Eintopfreaktion
p-Chlorbenzaldehyd (1f, 0.5 mmol) und der Organokatalysator (S,S)-32 (1 mol%) wurden in
Aceton (4, 4.5 mmol) gelöst. Nach Zugabe von gesättigter NaCl-Lösung (330 µL) wurde das
Reaktionsgemisch 48 h gerührt. Anschließend wurde Phosphatpuffer (pH 7, 50 mM, 0.1 M,
7.5 mL), iso-Propanol (2.5 mL), MgCl2 (1 mM, nur bei Verwendung der (R)-ADH aus
Lactobacillus kefir), Cofaktor NAD+ oder NADP+ (0.02 mmol) und die entsprechende
Alkoholdehydrogenase aus Rhodococcus sp. oder Lactobacillus kefir (10-960 U/mmol)
zugegeben. Das Reaktionsgemisch wurde 24-48 h gerührt und anschließend mit
Dichlormethan (3 x 5 mL) extrahiert. Die vereinigten organischen Phasen wurden über
MgSO4 getrocknet und das Lösungsmittel am Rotationsverdampfer (max. 600 mbar)
entfernt. Anschließend wurden der Umsatz und das Diastereomerenverhältnis mittels
1H-NMR-Spektroskopie und der ee-Wert mittels chiraler HPLC bestimmt. Das Rohprodukt
wurde säulenchromatographisch gereinigt (Hexan/EtOAc 4:1, v/v) und das gewünschte
Produkt erhalten.

EXPERIMENTELLER TEIL
112
8.2.1.4.1 Synthese von (1R,3R)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3R)-17f
Die Synthese von (1R,3R)-17f erfolgte analog AAV 2. Es wurde 1f
(0.5 mmol, 70 mg) und (S,S)-32 (1 mol%, 5 µmol, 2 mg) in 4 gelöst
(4.5 mmol, 330 µL). Nach Zugabe von 330 µL NaCl-Lösung wurde
48 h gerührt. Für die Enzymreaktion wurde Puffer (7.5 mL), i-PrOH
(2.5 mL), die (R)-selektive ADH aus Lactobacillus kefir (600 µL,
960 U/mmol), sowie der Cofaktor NADP+ (0.02 mmol, 15 mg) zugegeben und 48 h gerührt.
Der produktbezogene Umsatz betrug 67%, das Diastereomerenverhältnis d.r. >25:1
(syn/anti). Nach säulenchromatographischer Reinigung (Hexan/EtOAc 4:1, v/v, RF = 0.11)
wurde das gewünschte Produkt (1R,3R)-17f als gelbliches Öl mit einer Ausbeute von 58%
(Auswaage 58 mg) und einer Enantioselektivität von >99% ee erhalten (AD-H, Eluent:
Hexan/i-PrOH 97:3, flow 0.8 mL/min, 220 nm; tr = 35.71 min). 1H-NMR (400 MHz, CDCl3) δ
(ppm) = 1.23 (d, 3H, 3J = 8.0 Hz, CH3), 1.71-1.77 (m, 2H, CH2), 4.08-4.16 (m, 1H, CHCH3), 4.90
(dd, 1H, 3J = 9.7 Hz, 3.4 Hz, CHCH2), 7.25-7.32 (m, 4H, H-aromat.). Die spektroskopischen
Daten stimmen mit denen aus Abschnitt 8.2.1.2.2 überein.
8.2.1.4.2 Synthese von (1R,3S)-1-(4-Chlorphenyl)-1,3-butandiol (1R,3S)-17f
Die Synthese von (1R,3S)-5f erfolgte analog AAV2. Es wurde 1f
(0.5 mmol, 70 mg) und (S,S)-32 (1 mol%, 5 µmol, 2 mg) in 4 gelöst
(4.5 mmol, 330 µL). Nach Zugabe von 330 µL NaCl-Lösung wurde
48 h gerührt. Für die Enzymreaktion wurde Puffer (7.5 mL), i-PrOH
(2.5 mL), und die (S)-selektive ADH aus Rhodococcus sp. (150 µL,
10 U/mmol), sowie der Cofaktor NAD+ (0.02 mmol, 13 mg) eingesetzt und 24 h gerührt. Der
produktbezogene Umsatz betrug 79%, das Diastereomerenverhältnis d.r. >25:1 (anti/syn).
Nach säulenchromatographischer Reinigung (Hexan/EtOAc 4:1, v/v, RF = 0.09) wurde das
gewünschte Produkt (1R,3S)-7f als gelbliches Öl mit einer Ausbeute von 73% (Auswaage
73 mg) und einer Enantioselektivität von >99% ee erhalten (AD-H-Säule, Eluent: Hexan/i-
PrOH 97:3, flow 0.8 mL/min, 220 nm; tr = 43.47 min). 1H-NMR (400 MHz, CDCl3) δ (ppm) =
1.18 (d, 3H, 3J = 6.3 Hz, CH3), 1.68-1.83 (m, 2H, CH2), 3.96-4.11 (m, 1H, CHCH3), 4.95 (dd, 1H,
3J = 6.9 Hz, 4,0 Hz, CHCH2), 7.21-7.28 (m, 4H, H-aromat.). Die spektroskopischen Daten
stimmen mit denen aus Abschnitt 8.2.1.2.4 überein.
Cl
OHOH
(1R,3S)-17fC10H13ClO2
M = 200.66 g/mol

EXPERIMENTELLER TEIL
113
8.2.2 Enzymatische Aldolreaktion
8.2.2.1 Allgemeine Arbeitsvorschrift 3 (AAV 3): Synthese der racemischen
β-Hydroxy-α-aminosäuren116
Abbildung 8.3. Racematsynthese der β-Hydroxy-α-aminosäuren 12
Glycin (11, 6.7 mol) wurde in 5 M NaOH (2.5-10 mL) gelöst, anschließend wurde der
entsprechende Aldehyd 1 (6.7-13.4 mmol) langsam zugegeben. Das Reaktionsgemisch wurde
so lange bei 0-35°C gerührt bis sich ein Feststoff bildete. Anschließend wurde 5 M HCl
zugegeben bis pH 1 erreicht wurde. Nachdem sich der Feststoff gelöst hatte, wurde die
wässrige Phase mit CH2Cl2 (2 x 50 mL) extrahiert, um den überschüssigen Aldehyden zu
entfernen. Die wässrige Phase wurde am Rotationsverdampfer eingeengt und der pH-Wert
mit 1 M NaOH so weit erhöht bis sich ein Niederschlag bildete (pH 7-8). Zur
Vervollständigung der Kristallisation wurde die Mischung über Nacht bei 4°C gelagert. Der
entstandene Niederschlag wurde abfiltriert und im Vakuum getrocknet. Gegebenenfalls
wurde durch erneutes einengen wiederholt ausgefällt.
8.2.2.1.1 Synthese von rac-p-Methylthiophenylserin rac-12d
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), 6.7 mmol p-Methylthiobenzaldehyd (1d, 877 µL) und 5 mL
NaOH (5 M) eingesetzt. Die Reaktionsmischung wurde bei 0°C für
26 h gerührt. Das gewünschte Produkt wurde als gelblicher Feststoff
mit einer Ausbeute von 26% (Auswaage 396 mg) und mit einem
Diastereomerenverhältnis von d.r. >25:1 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 2.48 (s, 3H, SCH3), 3.85 (d, 1H, 3J = 4.4 Hz, syn-CHNH2), 5.22 (d, 1H, 3J = 4.6 Hz, syn-
CHOH), 7.34-7.39 (m, 4H, H-aromat.); 13C-NMR (100 MHz, D2O) δ (ppm) = 14.83 (SCH3), 61.13
(CHNH2), 71.42 (CHOH), 126.88, 126.94, 136.59, 138.25 (C-aromat.), 172.43 (COOH); MS
OH
OOH
rac-12dC10H13NO3S
M = 227.28 g/mol
NH2MeS

EXPERIMENTELLER TEIL
114
(MALDI, Matrix: dhb) m/z = 228 (M-H)+. Die spektroskopischen Daten stimmen mit den in
der Literatur angegeben Daten überein.140
8.2.2.1.2 Synthese von rac-Phenylserin rac-12e
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), 13.4 mmol Benzaldehyd (1e, 1.35 µL) und 2.5 mL NaOH (5 M)
eingesetzt. Die Reaktion wurde bei 35°C für 48 h und bei 0°C für 4 h
durchgeführt. Das gewünschte Produkt wurde als farbloser Feststoff
erhalten. Die Reaktion bei 35°C ergab ausschließlich syn-12e in einer
Ausbeute von 40% (Auswaage 486 mg), wohingegen die Reaktion bei 0°C eine Mischung der
beiden Diastereomere von d.r. 56:44 (syn/anti) mit einer Ausbeute von 46% (Auswaage
558 mg) ergab. 1H-NMR (400 MHz, D2O): δ (ppm) = 3.91 (d, 1H, 3J = 4.4 Hz, syn-CHNH2), 4.08
(d, 1H, 3J = 4.2 Hz, anti-CHNH2), 5.30 (d, 1H, 3J = 4.4 Hz, syn-CHOH), 5.35 (d, 1H, 3J = 4.2 Hz,
anti-CHOH), 7.28-7.46 (m, 4H, H-aromat.); 13C-NMR (100 MHz, D2O) δ (ppm) = 60.43, 60.91
(CHNH2), 72.38, 72.59 (CHOH), 125.46, 126.05, 127.75, 127.99, 128.09, 128.25, 137.50,
139.32 (C-aromat.), 172.41, 174.41 (COOH). Die spektroskopischen Daten stimmen mit den
in der Literatur angegebenen Daten überein.116,141
8.2.2.1.3 Synthese von rac-p-Chlorphenylserin rac-12f
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), 13.4 mmol p-Chlorbenzaldehyd (1f, 1.88 g) und 10 mL NaOH
(5 M) eingesetzt. Die Reaktionsmischung wurde bei 0°C für 24 h
gerührt. Das gewünschte Produkt wurde als farbloser Feststoff mit
einer Ausbeute von 36% (Auswaage 520 mg) und mit einem
Diastereomerenverhältnis von d.r. 79:21 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 4.07 (d, 1H, 3J = 4.0 Hz, syn-CHNH2), 4.21 (d, 1H, 3J = 4.3 Hz, anti-CHNH2), 5.35 (d, 1H,
3J = 4.6 Hz, syn-CHOH), 5.38 (d, 1H, 3J = 4.6 Hz, anti-CHOH), 7.38-7.50 (m, 4H, H-aromat.);
13C-NMR (100 MHz, D2O) δ (ppm) = 60.09, (anti-CHNH2), 60.45 (syn-CHNH2), 70.77
(anti-CHOH), 70.86 (syn-CHOH), 127.84, 128.11, 129.08, 129.24, 134.08, 134.28, 136.05,
137.84 (C-aromat.), 170.63 (anti-COOH) 171.49 (syn-COOH); MS (MALDI, Matrix: dhb) m/z =
OH
OOH
rac-12eC9H11NO3
M = 181.19 g/mol
NH2

EXPERIMENTELLER TEIL
115
216 (M-H)+. Die spektroskopischen Daten stimmen mit den in der Literatur angegebenen
Daten überein.109
8.2.2.1.4 Synthese von rac-o-Chlorphenylserin rac-12i
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), 13.4 mmol o-Chlorbenzaldehyd (1i, 1.5 mL) und 2.5 mL NaOH
(5 M) eingesetzt. Die Reaktionsmischung wurde bei RT für 1 h gerührt.
Das gewünschte Produkt wurde als farbloser Feststoff mit einer
Ausbeute von 38% (Auswaage 549 mg) und mit einem
Diastereomerenverhältnis von d.r. 82:18 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 4.08-4.09 (m, 2H, CHNH2), 5.49 (d, 1H, 3J = 3.4 Hz, anti-CHOH), 5.67 (d, 1H, 3J =
3.7 Hz, syn-CHOH), 7.35-7.64 (m, 4H, H-aromat.); 13C-NMR (100 MHz, D2O) δ (ppm) = 58.00
(anti-CHNH2), 58.46 (syn-CHNH2), 68.51 (syn-CHOH), 68.98 (anti-CHOH), 127.46, 127.74,
127.90, 128.37, 129.65, 129.99, 130.19, 130.26, 131.53, 131.86, 136.54, 136.66 (C-aromat.),
170.93 (anti-COOH), 172.19 (syn-COOH); MS (MALDI, Matrix: sin) m/z = 216 (M-H)+. Die
spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten überein.109
Durch Umkristallisation in Wasserkonnte reines syn-Isomer in einer Ausbeute von 5%
(Auswaage 72 mg) erhalten werde. Durch wiederholtes Einengen der Lösung wurde reines
anti-Isomer in einer Ausbeute von 1% (Auswaage 14 mg) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 4.08 (d, 1H, 3J = 3.6 Hz, syn-CHNH2), 5.66 (d, 1H, 3J = 3.6 Hz, syn-CHOH), 7.35-7.64 (m,
4H, H-aromat.); 1H-NMR (400 MHz, D2O) δ (ppm) 4.14 (d, 1H, 3J = 3.5 Hz, anti-CHNH2), 5.54
(d, 1H, 3J = 3.5 Hz, anti-CHOH), 7.37-7.62 (m, 4H, H-aromat.).
8.2.2.1.5 Synthese von rac-m-Bromphenylserin rac-12j
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
6.7 mmol, 0.5 g), 6.7 mmol m-Brombenzaldehyd (1j, 785 µL) und 5 mL
NaOH (5 M) eingesetzt. Die Reaktionsmischung wurde bei 0°C für 48 h
gerührt. Das gewünschte Produkt wurde als gelblicher Feststoff mit
einer Ausbeute von 11% (Auswaage 192 mg) und mit einem
Diastereomerenverhältnis von d.r. 87:13 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
OH
OOH
rac-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
116
(ppm) = 3.87 (d, 1H, 3J = 4.1 Hz, syn-CHNH2), 4.05 (d, 1H, 3J = 3.8 Hz, anti-CHNH2), 5.25 (d, 1H,
3J = 4.2 Hz, syn-CHOH), 5.31 (d, 1H, 3J = 4.0 Hz, anti-CHOH) 7.31-7.65 (m, 4H, H-aromat.);
13C-NMR (100 MHz, D2O) δ (ppm) = 61.04 (CHNH2), 71.03 (CHOH), 122.65, 125.14, 129.21,
131.00, 131.82, 142.07 (C-aromat.), 172.14 (COOH); MS (MALDI, Matrix: dhb) m/z = 259
(M-H)+. Die spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten
überein.109
8.2.2.1.6 Synthese von rac-m-Chlorphenylserin rac-12l
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), 13.4 mmol m-Chlorenzaldehyd (1l, 1.5 mL) und 5 mL NaOH
(5 M) eingesetzt. Die Reaktionsmischung wurde bei RT für 2 h gerührt.
Das gewünschte Produkt wurde als farbloser Feststoff mit einer
Ausbeute von 52% (Auswaage 751 mg) und mit einem
Diastereomerenverhältnis von d.r. 74:26 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 3.67 (d, 1H, 3J = 4.8 Hz, syn-CHNH2), 3.79 (d, 1H, 3J = 4.1 Hz, anti-CHNH2), 5.10 (d, 2H,
3J = 4.8 Hz, syn-CHOH, anti-CHOH), 7.30-7.47 (m, 4H, H-aromat.); 13C-NMR (100 MHz, D2O) δ
(ppm) = 59.09, 59.59 (CHNH2), 71.06, 71.33 (CHOH), 122.47, 123.08, 124.06, 124.56, 126.05,
126.28, 127.99, 128.16, 131.74, 131.87, 138.89, 140.57 (C-aromat.), 174.03 (COOH); MS
(MALDI, Matrix: sin) m/z = 216 (M-H)+. Die spektroskopischen Daten stimmen mit den in der
Literatur angegebenen Daten überein.109
8.2.2.1.7 Synthese von rac-m-Hydroxyphenylserin rac-12m
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), 6.7 mmol m-Hydroxybenzaldehyd (1m, 818 mg) und 5 mL
NaOH (5 M) eingesetzt. Die Reaktionsmischung wurde bei 0°C für
48 h gerührt. Man erhielt das gewünschte Produkt als gelblichen
Feststoff in einer Ausbeute von 7% (Auswaage 92 mg) und mit einem
Diastereomerenverhältnis von d.r. >25:1 (syn/anti). 1H-NMR (400 MHz, D2O) δ (ppm) = 3.89
(d, 1H, 3J = 3.8 Hz, syn-CHNH2), 5.26 (d, 1H, 3J = 3.8 Hz, syn-CHOH), 6.87-7.02 (m, 3H, H-
aromat.), 7.31-7.37 (m, 1H, H-aromat.); 13C-NMR (100 MHz, D2O) δ (ppm) = 61.12 (CHNH2),

EXPERIMENTELLER TEIL
117
71.35 (CHOH), 113.05, 115.71, 118.20, 130.76, 141.59, 156.21, (C-aromat.), 172.44 (COOH);
MS (MALDI, Matrix: sin) m/z = 198 (M-H)+. Die spektroskopischen Daten stimmen mit den in
der Literatur angegebenen Daten überein.109
8.2.2.1.8 Synthese von rac-o-Methoxyphenylserin rac-12n
Die Synthese erfolgte analog AAV 3. Es wurden 6.7 mmol Glycin (11,
0.5 g), o-Methoxybenzaldehyd (1n, 13.4 mmol, 1.62 mL) und 5 mL
NaOH (5 M) eingesetzt. Die Reaktionsmischung wurde bei RT für 3 h
gerührt. Das gewünschte Produkt wurde als farbloser Feststoff mit
einer Ausbeute von 23% (Auswaage 325 mg) und mit einem
Diastereomerenverhältnis von d.r. 69:31 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 3.85 (s, 3H, syn-OCH3), 3.89 (s, 3H, anti-OCH3), 4.03 (d, 1H, 3J = 4.6 Hz, syn-CHNH2),
4.07 (d, 1H, 3J = 3.5 Hz, anti-CHNH2), 5.42 (d, 1H, 3J = 4.6 Hz, syn-CHOH), 5.53 (d, 1H, 3J = 3.5
Hz, anti-CHOH), 7.05-7.50 (m, 4H, H-aromat.); 13C-NMR (100 MHz, D2O) δ (ppm) = 55.60,
55.70 (OCH3) 58.82, 59.40 (CHOH), 67.59, 68.43 (CHNH2), 111.57, 121.20, 127.02, 127.20,
128.19, 130.24, 130.33, 156.38 (C-aromat.), 172.76, 172.89 (COOH); MS (MALDI, Matrix:
dhb) m/z = 212 (M-H)+. Die spektroskopischen Daten stimmen mit den in der Literatur
angegebenen Daten überein.142
8.2.2.2 Allgemeine Arbeitsvorschrift 4 (AAV 4): Derivatisierung der
racemischen β-Hydroxy-α-aminosäuren mit Benzoylchlorid116
Abbildung 8.4. Derivatisierung von rac-12
Die entsprechende β-Hydroxy-α-aminosäure 12 (0.4-1 mmol) wurde in Wasser (0.2-0.5 mL)
und NaOH (0.4-1 mL) gelöst und auf 0°C gekühlt. Nach Zugabe von 1.2 Äq. Benzoylchlorid

EXPERIMENTELLER TEIL
118
(79) wurde 1 h bei 0°C und 2 h bei RT gerührt. Anschließend wurde der pH-Wert durch
Zugabe von 1 M HCl auf den Wert 1 eingestellt und die wässrige Phase mit EtOAc (3 x 5 mL)
extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet. Nach
Entfernen des Lösungsmittels im Vakuum wurde der erhaltene Rückstand umkristallisiert.
8.2.2.2.1 Synthese von rac-N-Benzoylamino-3-(4-methylthiophenyl)-3-hydroxy-
propionsäure rac-80d
Die Synthese erfolgte analog AAV 4. Es wurden 0.4 mmol
p-Methylthiophenylserin (rac-12d, d.r. >25:1 syn/anti, 91 mg),
0.2 mL Wasser und 0.4 mL NaOH (5 M), sowie 0.5 mmol 79 (55 µL)
eingesetzt. Der erhaltene Rückstand wurde in Aceton aufgenommen
und mit Petrolether ausgefällt. Das gewünschte Produkt wurde als
farbloser Feststoff mit einer Ausbeute von 55% (Auswaage 73 mg)
erhalten. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 2.43 (s, 1H, SCH3), 4.13-4.14 (m, 1H, syn-
CHNH), 5.45 (d, 1H, 3J = 3.0 Hz, syn-CHOH), 7.19-7.51 (m, 9H, H-aromat.); 13C-NMR (100 MHz,
Aceton-d6) δ (ppm) = 15.19 (CH3), 59.33 (syn-CHOH), 73.18 (syn-CHNH), 126.50, 127.40,
127.89, 129.04, 132.08, 135.10, 138.30, 139.33 (C-aromat.), 167.41 (COPh), 171.57 (COOH);
MS (MALDI, Matrix: dhb) m/z = 332 (M-H)+; IR ṽ (cm-1): 3362, 3224, 1721, 1598, 1537, 1217,
825, 691; Elementaranalyse (C17H17NO4S): Berechnet: C: 61.61, H: 5.17, N: 4.23, S: 9.68.
Gefunden: C: 61.11, H: 5.19, N: 4.20, S: 9.47; Schmelzpunkt: 173°C.
8.2.2.2.2 Synthese von rac-N-Benzoylamino-3-hydroxy-3-phenylpropionsäure
rac-80e
Die Synthese erfolgte analog AAV 4. Es wurden 0.7 mmol rac-Phenyl-
serin (rac-12e, d.r. >25:1 syn/anti, 127 mg), 0.5 mL NaOH (5 M), sowie
0.8 mmol 79 (92 µL) eingesetzt. Die Mischung wurde 2 h bei 0°C und
16 h bei RT gerührt. Die spektroskopische Analyse erfolgte aus dem
Rohprodukt. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.99-5.03 (m,
2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz, anti-CHOH), 5.50 (d, 1H, 3J =
OH
OOH
rac-80dC17H17NO4S
M = 331.39 g/mol
HN OMeS
OH
OOH
rac-80eC16H15NO4
M = 285.29 g/mol
HN O

EXPERIMENTELLER TEIL
119
2.90 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Die spektroskopischen Daten stimmen
mit den Literaturdaten überein.116
8.2.2.2.3 Synthese von rac-N-Benzoylamino-3-(4-chlorphenyl)-3-hydroxy-
propionsäure rac-80f
Die Synthese erfolgte analog AAV 4. Es wurden 0.9 mmol
p-Chlorphenylserin (rac-12f, d.r. 72:28 syn/anti, 194 mg), 0.5 mL
Wasser und 0.9 mL NaOH (5 M), sowie 1.1 mmol 79 (124 µL)
eingesetzt. Der erhaltene Rückstand wurde in Aceton aufgenommen
und mit Hexan ausgefällt. Das gewünschte Produkt wurde als
farbloser Feststoff mit einer Ausbeute von 38% (Auswaage 109 mg)
und einem Diastereomerenverhältnis von d.r. 85:15 (syn/anti) erhalten. 1H-NMR (400 MHz,
Aceton-d6) δ (ppm) = 4.97 (d, 1H, 3J = 6.0 Hz, syn-CHNH), 5.00 (d, 1H, 3J = 3.0 Hz, anti-CHNH),
5.26 (d, 1H, 3J = 5.8 Hz, syn-CHOH), 5.49 (d, 1H, 3J = 3.0 Hz, anti-CHOH), 7.31-7.56 (m, 7H,
H-aromat.), 7.79-7.83 (m, 2H, H-aromat.); 13C-NMR (100 MHz, CD3OD) δ (ppm) = 59.95
(syn-CHOH), 60.01 (anti-CHOH), 73.61 (syn-CHNH), 74.28 (anti-CHNH), 128.27, 128.31,
128.76, 129.22, 129.46, 129.56, 132.89, 134.26, 134.55, 135.10, 141.16, 141.51 (C-aromat.),
169.85, 170.17 (COPh), 173.19, 173.26 (COOH); MS (MALDI, Matrix: dhb) m/z = 320 (M-H)+;
IR ṽ (cm-1): 3238, 1725, 1599, 1540, 1071, 828, 687; Elementaranalyse (C16H14ClNO4):
Berechnet: C: 60.10, H: 4.41, N: 4.38. Gefunden: C: 59.96, H: 4.36, N: 4.42; Schmelzpunkt:
171°C.
8.2.2.2.4 Synthese von rac-syn-N-Benzoylamino-3-(2-chlorphenyl)-3-hydroxy-
propionsäure rac-syn-80i
Die Synthese erfolgt analog AAV 4. Es wurden 0.5 mmol
rac-syn-o-Chlorphenylserin (rac-syn-12i, d.r. >25:1 syn/anti, 108 mg),
0.25 mL Wasser und 0.5 mL NaOH (5 M), sowie 0.6 mmol 79 (69 µL)
eingesetzt. Der erhaltene Rückstand wurde in Dichlormethan gelöst
und mit Petrolether ausgefällt. Das gewünschte Produkt wurde in
einer Ausbeute von 20% (Auswaage 32 mg) erhalten. 1H-NMR
OH
OOH
rac-80fC16H14ClNO4
M = 319.74 g/mol
HN OCl
OH
OOH
rac-80iC16H14ClNO4
M = 319.74 g/mol
HN O
Cl

EXPERIMENTELLER TEIL
120
(400 MHz, Aceton-d6) δ (ppm) = 5.22-5.24 (m, 1H, syn-CHNH), 5.86 (d, 1H, 3J = 2.1 Hz, syn-
CHOH), 7.20-7.80 (m, 9H, H-aromat.); 13C-NMR (100 MHz, Aceton-d6) δ (ppm) = 56.59 (syn-
CHOH), 70.80 (syn -CHNH), 127.43, 128.00, 129.15, 129.31, 129.65, 129.84, 131.99, 132.18,
135.18, 139.95 (C-aromat.), 167.42 (COPh), 171.65 (COOH); MS (MALDI, Matrix: dhb) m/z =
320 (M-H)+; IR ṽ (cm-1): 3276, 1704, 1636, 1548, 1073, 752, 696; Elementaranalyse
(C16H14ClNO4) Berechnet: C: 60.10, H: 4.41, N: 4.38. Gefunden: C: 60.17, H: 4.41, N: 4.37;
Schmelzpunkt: 169°C.
8.2.2.2.5 Synthese von rac-anti-N-Benzoylamino-3-(2-chlorphenyl)-3-hydroxy-
propionsäure rac-anti-80i
Die Synthese erfolgt analog AAV 4. Es wurden 0.6 mmol rac-anti-
o-Chlorphenylserin (rac-anti-12i, d.r. >25:1 anti/syn, 129 mg), 0.3 mL
Wasser und 0.6 mL NaOH (5 M), sowie 0.7 mmol 79 (83 µL)
eingesetzt. Der erhaltene Rückstand wurde in Dichlormethan gelöst
und mit Petrolether ausgefällt. Das gewünschte Produkt wurde mit
einer Ausbeute von 10% (Auswaage 19 mg) erhalten. 1H-NMR (400
MHz, Aceton-d6) δ (ppm) = 5.12-5.16 (m, 1H, anti-CHNH), 5.56 (d, 1H, 3J = 5.2 Hz, anti-CHOH),
7.25- 7.44 (m, 9H, H-aromat.); 13C-NMR (100 MHz, Aceton-d6) δ (ppm) = 58.00 (anti-CHOH),
71.42 (anti-CHNH), 127.60, 128.11, 129.24, 129.74, 129.81, 132.35, 132.80, 135.07 135.09,
139.65 (C-aromat.), 167.19 (COPh), 171.13 (COOH).
8.2.2.2.6 Synthese von rac-N-Benzoylamino-3-(3-bromphenyl)-3-hydroxy-
propionsäure rac-80j
Die Synthese erfolgte analog AAV 4. Es wurden 0.4 mmol
m-Bromphenylserin (rac-12j, d.r. 87:13 syn/anti, 104 mg ) 0.2 mL
Wasser und 0.4 mL NaOH (5 M), sowie 0.48 mmol 79 (55 µL)
eingesetzt. Der erhaltene Rückstand wurde in Aceton aufgenommen
und mit Petrolether ausgefällt. Das gewünschte Produkt wurde mit
einer Ausbeute von 37% (Auswaage 47 mg) und einem
Diastereomerenverhältnis von d.r. 87:13 (syn/anti) erhalten. 1H-NMR (400 MHz, Aceton-d6)
OH
OOH
rac-80jC16H14BrNO4
M = 364.19 g/mol
HN O
Br
OH
OOH
rac-80iC16H14ClNO4
M = 319.74 g/mol
HN O
Cl

EXPERIMENTELLER TEIL
121
δ (ppm) = 4.96-5.03 (m, 2H, CHNH), 5.26 (d, 1H, 3J = 5.7 Hz, anti-CHOH), 5.50 (d, 1H, 3J =
2.9 Hz, syn-CHOH) 7.42-7.93 (m, 9H, H-aromat.); 13C-NMR (100 MHz Aceton-d6) δ (ppm) =
59.18 (anti-CHOH), 59.79 (syn-CHOH), 73.00 (anti-CHNH), 73.90 (syn-CHNH), 122.47, 122.55,
125.96, 126.39 128.07, 129.17, 129.24, 130.09, 130.46, 130.74, 130.78, 131.09, 131.21.
132.24, 132.39, 134.98 (C-aromat.), 145.17, 145.50 (CBr), 167.63 (COPh), 171.37 (COOH); MS
(MALDI, Matrix: dhb) m/z = 364 (M-H)+; IR ṽ (cm-1): 3289, 1756, 1715, 1623, 1053, 763, 691;
Elementaranalyse (C16H14BrNO4): Berechnet: C: 52.77, H: 3.87, N: 3.85. Gefunden: C: 52.05,
H: 3.62, N: 3.62. Schmelzpunkt: 154°C.
8.2.2.2.7 Synthese von rac-N-Benzoylamino-3-(3-chlorphenyl)-3-hydroxy-
propionsäure rac-80l
Die Synthese erfolgte analog AAV 4. Es wurden 0.5 mmol
m-Chlorphenylserin (rac-12l, d.r. 51:49 syn/anti, 108 mg), 0.25 mL
Wasser und 0.5 mL NaOH (5 M), sowie 0.6 mmol 79 (69 µL)
eingesetzt. Der erhaltene Rückstand wurde in Dichlormethan
aufgenommen und mit Petrolether ausgefällt. Das gewünschte
Produkt wurde als farbloser Feststoff mit einer Ausbeute von 55%
(Auswaage 88 mg) und einem Diastereomerenverhältnis von d.r. 74:26 (syn/anti) erhalten.
1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.99 (dd, 1H, 3J = 8.2 Hz, 2.5 Hz, syn-CHNH), 5.04
(dd, 1H, 3J = 9.1 Hz, 6.1 Hz, anti-CHNH), 5.28 (d, 1H, 3J = 5.8 Hz, syn-CHOH), 5.52 (d, 1H, 3J =
2.8 Hz, anti-CHOH), 7.22-7.83 (m, 9H, H-aromat.); 13C-NMR (100 MHz, CD3OD) δ (ppm) =
59.93 (anti-CHOH), 60.08 (syn-CHOH), 73.62 (anti-CHNH), 74.33 (syn-CHNH), 125.55, 126.19,
127.37, 128.00, 128.31, 128.34, 128.60, 128.85, 129.56, 130.67, 130.71, 132.87, 132.91,
135.09, 135.19, 144.87, 145.21 (C-aromat.), 169.90, 170.28, 173.17 (COPh, COOH); MS
(MALDI, Matrix: dhb) m/z = 320 (M-H)+; IR ṽ (cm-1): 3276, 1706, 1637, 1530, 1041, 791, 688;
Elementaranalyse (C16H14ClNO4): Berechnet: C: 60.10, H: 4.41, N: 4.38. Gefunden: C: 60.25,
H: 4.46, N: 4.08; Schmelzpunkt: 136°C.
OH
OOH
rac-80lC16H14ClNO4
M = 319.74 g/mol
HN O
Cl

EXPERIMENTELLER TEIL
122
8.2.2.2.8 Synthese von rac-N-Benzoylamino-3-(2-methoxyphenyl)-3-hydroxy-
propionsäure rac-80n
Die Synthese erfolgte analog AAV 4. Es wurden 1 mmol
o-Methoxyphenylserin (rac-1n, d.r. 69:31 syn/anti, 211 mg), 0.5 mL
Wasser und 1 mL NaOH (5 M), sowie 1.2 mmol 79 (138 µL) eingesetzt.
Der erhaltene Rückstand wurde in Aceton aufgenommen und mit
Hexan ausgefällt. Das gewünschte Produkt wurde als farbloser
Feststoff erhalten. Es konnte reines syn-Produkt in einer Ausbeute
von 24% (Auswaage 76 mg) und ein Diastereomerengemisch (d.r. 10:90 syn/anti) in einer
Ausbeute von 14% (Auswaage 44 mg) isoliert werden. 1H-NMR (400 MHz, Aceton-d6) δ
(ppm) = 3.85, (s, 3H, syn-OCH3) 3.90 (s, 3H, anti-OCH3), 4.99 (dd, 1H, 3J = 11.0 Hz, 5.8 Hz, syn-
CHNH), 5.18 (dd, 1H, 3J = 9.3 Hz, 2.5 Hz, anti-CHNH), 5.50 (d, 1H, 3J = 5.5 Hz, anti-CHOH), 5.79
(d, 1H, 3J = 5.8 Hz, syn-CHOH), 6.85-8.06 (m, 9H, H-aromat.); 13C-NMR (100 MHz, Aceton-d6)
δ (ppm) = 55.69 (anti-CH3), 55.82 (syn-CH3), 57.17 (syn-CHOH), 59.12 (anti-CHOH), 69.01
(syn-CHNH), 69.77 (anti -CHNH), 111.01, 111.09, 120.78, 121.18, 127.72, 127.90, 127.92,
128.26, 129.14, 129.24, 129.47, 130.10, 130.59, 132.05, 132.28, 135.12, 135.49, (C-aromat.),
156.82, 157.21 (OCH3) 167.23, 167.38 (COPh), 171.71, 172.14 (COOH); MS (MALDI, Matrix:
dhb) m/z = 316 (M+H)+; IR ṽ (cm-1): 3350, 1695, 1638, 1527, 1031, 753, 688;
Elementaranalyse (C17H17NO5) Berechnet: C: 64.75, H: 5.43, N 4.44. Gefunden: C: 64.91,
H: 5.47, N: 4.16. Schmelzpunkt: 177°C.
8.2.2.3 Untersuchung der Hintergrundreaktion
Abbildung 8.5. Aktivierung von Glycin
OH
OOH
rac-80nC17H17NO5
M = 315.32 g/mol
HN O
OMe

EXPERIMENTELLER TEIL
123
Zur Untersuchung, ob es zwischen Glycin und Benzaldehyd zur Iminbildung und somit zur
Aktivierung von Glycin kommt, wurden verschiedene Mengen Glycin (11, 0.1-1 mmol) in
Puffer (1 mL, Phosphatpuffer pH 7 oder Tris-HCl-Puffer pH 8, 0.1 M) gelöst und Benzaldehyd
(1e, 0.1-1 mmol) zugegeben. Das Gemisch wurde 27 h gerührt. Es wurden Versuche mit und
ohne Zugabe von Cofaktor PLP (50 µM) durchgeführt. Nach Entfernen des Lösungsmittels im
Vakuum wurde der Rückstand mittels 1H-NMR-Spektroskopie untersucht. 1H-NMR (300 MHz,
D2O) δ (ppm) = 3.55 (s, 2H, CH2-11). Die Ergebnisse der verschiedenen Versuche sind in
Tabelle 8.2 gezeigt.
Tabelle 8.2. Untersuchung der Hintergrundreaktion
Eintrag 11 [mmol] 1e [mmol] pH Cofaktor Iminbildung1)
1 1.0 0.1 8 ohne PLP -
2 1.0 0.1 8 mit PLP -
3 1.0 0.1 7 mit PLP -
4 0.5 0.5 8 ohne PLP -
5 0.5 0.5 8 mit PLP -
6 0.5 0.5 7 mit PLP -
7 0.1 1.0 8 ohne PLP -
8 0.1 1.0 8 mit PLP -
9 0.1 1.0 7 mit PLP -
1) Es konnte keine Iminbildung im
1H-NMR-Spektrum beobachtet werden.
8.2.2.4 Untersuchung auf Epimerisierung
Abbildung 8.6. Untersuchung auf Epimerisierung

EXPERIMENTELLER TEIL
124
Zur Untersuchung, ob das Diastereomerenverhältnis von Phenylserin (12e) unter den
Bedingungen der enzymatischen Reaktion konstant bleibt, wurde rac-Phenylserin (rac-12e,
0.1 mmol) in Puffer (1 mL, Phosphatpuffer pH 7 oder Tris-HCl-Puffer pH 8, 0.1 M) gelöst und
27 h gerührt. Anschließend wurde das Lösungsmittel im Vakuum entfernt und der Rückstand
mittels 1H-NMR-Spektroskopie untersucht. 1H-NMR (400 MHz, D2O) δ (ppm) = 3.91 (d, 1H,
3J = 4.4 Hz, syn-CHNH2), 4.08 (d, 1H, 3J = 4.2 Hz, anti-CHNH2), 5.30 (d, 1H, 3J = 4.4 Hz,
syn-CHOH), 5.35 (d, 1H, 3J = 4.2 Hz, anti-CHOH), 7.28-7.46 (m, 4H, H-aromat.). Die Ergebnisse
sind in Tabelle 8.3 gezeigt.
Tabelle 8.3. Untersuchung auf Epimerisierung
Eintrag d.r. (syn/anti) pH d.r. (syn/anti) nach 27 h
1 >95:5 7 >25:1
2 >95:5 8 >25:1
3 56:44 7 56:44
4 56:44 8 56:44
8.2.2.5 Allgemeine Arbeitsvorschrift 5 (AAV 5) Photometertest zur
Untersuchung der Enzymaktivität117,118
Abbildung 8.7. Direkte Aktivitätsbestimmung durch Verfolgung der
Benzaldehydkonzentration
Zur Bestimmung der Enzymaktivität in U/mL (µmol min-1mL-1) wurde die Spaltung von
rac-syn-12e in Benzaldehyd (1e) und Glycin (11) verfolgt. Die Änderung der
Benzaldehydkonzentration kann bei 278 nm am Photometer beobachtet werden. In eine
Küvette (d = 1 cm) wurden 730 µL Phosphatpuffer (pH 7, 50 mM), 150 µL rac-syn-12e-Lösung
(0.1 M in Phosphatpuffer pH 7, 50 mM), 50 µL Na2EDTA-Lösung (20 mM in Phosphatpuffer

EXPERIMENTELLER TEIL
125
pH 7, 50 mM), 50 µL Na2SO4 (0.5 M in Phosphatpuffer pH 7, 50 mM) gegeben und gut
durchmischt. Anschließend wurden 20 μL der entsprechend verdünnten Enzymlösung
(Rohextrakt) zugegeben, nochmals gut durchmischt und die Messung sofort gestartet. Es
wurde über 10 Minuten alle 5 Sekunden ein Messpunkt aufgenommen. Zur Berechnung der
volumetrischen Enzymaktivität wird die Anfangssteigung der Absorptionskurve in folgende
Gleichung (1) eingesetzt:
Wobei ΔE278nm/t die Anfangssteigung der Absorptionskurve in min-1, Vg das Gesamtvolumen
der Probe in mL, f der Verdünnungsfaktor des Rohextraktes, Vp das Enzymvolumen in mL, ε
der Extinktionskoeffizient in mM-1cm-1 und d die Küvettendicke in cm ist. Der
Extinktionskoeffizient wurde bei pH 7 und 8, sowie mit und ohne PLP bestimmt.
8.2.2.5.1 Bestimmung des Extinktionskoeffizienten
Zur Bestimmung des Extinktionskoeffizienten bei den verschiedenen Bedingungen wurde die
Extinktion bei 278 nm für unterschiedliche Benzaldehydkonzentrationen in Puffer (pH 7 bzw.
pH 8) und in An- bzw. Abwesenheit von PLP (Endkonzentration 50 µM) bestimmt. Es wurden
jeweils zwei Messungen durchgeführt (E(1), E(2)) und der Mittelwert (E) berechnet (Tabelle
8.4). Aus der Steigung der Geraden (x-Achse: Benzaldehydkonzentration, y-Achse:
Extinktion) wurde der Extinktionsfaktor berechnet.
Tabelle 8.4. Extinktion bei pH 7 und ohne Zugabe von PLP
c(1e) [mM] E(1) E(2) E
0.05 0.100 0.100 0.100
0.1 0.162 0.156 0.159
0.25 0.136 0.136 0.136
0.5 0.188 0.186 0.187
1.0 0.364 0.363 0.364
2.0 0.425 0.418 0.421

EXPERIMENTELLER TEIL
126
Abbildung 8.8. Extinktion bei pH 7, ohne PLP
Aus der Steigung der Geraden ergibt sich ein Durchschnittswert für den Extinktions-
koeffizienten ε von 1.213 mM-1cm-1 (Abbildung 8.8).
Tabelle 8.5. Extinktion bei pH 7 und Zugabe von PLP
c (1e) [mM] E(1) E(2) E
0.05 0.162 0.156 0.159
0.1 0.136 0.136 0.136
0.25 0.188 0.186 0.188
0.5 0.364 0.363 0.364
1.0 0.425 0.418 0.421
2.0 0.701 0.707 0.704
E: y = 1.213x + 0.073
0,0
0,5
1,0
1,5
2,0
2,5
3,0
0 0,5 1 1,5 2 2,5
Ext
inkt
ion
[m
in-1
]
c(Benzaldehyd) [mM]
E(1)
E(2)
E
0.0 0.5 1.0 1.5 2.0 2.5
3.0
2.5
2.0
1.5
1.0
0.5
0.0

EXPERIMENTELLER TEIL
127
Abbildung 8.9. Extinktion bei pH 7, mit PLP
Aus der Steigung der Geraden ergibt sich ein Durchschnittswert für den Extinktions-
koeffizienten ε von 1.19 mM-1cm-1 (Abbildung 8.9).
Tabelle 8.6. Extinktion bei pH 8 und ohne Zugabe von PLP
c (1e) [mM] E(1) E(2) E
0.05 0.084 0.086 0.085
0.1 0.156 0.155 0.156
0.25 0.419 0.417 0.418
0.5 0.855 0.857 0.856
1.0 1.672 1.672 1.672
2.0 2.467 2.493 2.480
E: y = 1.190x + 0.134
0,0
0,5
1,0
1,5
2,0
2,5
3,0
0 0,5 1 1,5 2 2,5
Ext
inkt
ion
[m
in-1
]
c(Benzaldehyd) [mM]
E(1)
E(2)
E
3.0
2.5
2.0
1.5
1.0
0.5
0.0
0.0 0.5 1.0 1.5 2.0 2.5

EXPERIMENTELLER TEIL
128
Abbildung 8.10. Extinktion bei pH 8, ohne PLP
Aus der Steigung der Geraden ergibt sich ein Durchschnittswert für den Extinktions-
koeffizienten ε von 1.254 mM-1cm-1 (Abbildung 8.10).
Tabelle 8.7. Extinktion bei pH 8 und Zugabe von PLP
c (1e) [mM] E(1) E(2) E
0.05 0.188 0.184 0.186
0.1 0.261 0.251 0.256
0.25 0.522 0.506 0.514
0.5 0.955 0.942 0.949
1.0 1.758 1.743 1.750
2.0 2.521 2.521 2.521
E: y = 1.254x + 0.128
0
0,5
1
1,5
2
2,5
3
0 0,5 1 1,5 2 2,5
Ext
inkt
ion
[m
in-1
]
c(Benzaldehyd) [mM]
E(1)
E(2)
E
3.0
2.5
2.0
1.5
1.0
0.5
0.0
0.0 0.5 1.0 1.5 2.0 2.5

EXPERIMENTELLER TEIL
129
Abbildung 8.11. Extinktion bei pH 8, mit PLP
Aus der Steigung der Geraden ergibt sich ein Durchschnittswert für den Extinktions-
koeffizienten ε von 1.224 mM-1cm-1 (Abbildung 8.11).
8.2.2.5.2 Aktivitätsbestimmung für L-TA aus E. coli
Die Aktivitätsbestimmung erfolgte analog AAV 5. Es wurde die Aktivität bei
unterschiedlichen pH-Werten getestet (pH 7, Phosphatpuffer, 50 mM; pH 8, Tris-HCl-Puffer,
50 mM). Die Durchführung ist in Tabelle 8.8 gezeigt.
Tabelle 8.8. Durchführung des Photometertests ohne Zugabe von PLP
Komponente c (Stammlösung)
[M]
Volumen
[µL]
Endkonzentration
[mM]
Puffer pH 7/8 0.05 730 -
rac-syn-12e 0.1 150 15
Na2EDTA 0.02 50 1.0
Na2SO4 0.5 50 25
L-TA (E. coli) - 20 -
E: y = 1.224x + 0.233
0
0,5
1
1,5
2
2,5
3
0 0,5 1 1,5 2 2,5
Ext
inkt
ion
[m
in-1
]
c(Benzaldehyd) [mM]
E(1)
E(2)
E
0.0 0.5 1.0 1.5 2.0 2.5
3.0
2.5
2.0
1.5
1.0
0.5
0.0

EXPERIMENTELLER TEIL
130
Des Weiteren wurde die Aktivitätsänderung durch Zugabe von PLP (2.5 mM, gelöst in
entsprechendem Puffer) untersucht. Bei Zugabe von 20 µL PLP-Lösung wurden 710 µL
Pufferlösung zupipettiert, um das Gesamtvolumen von 1 mL beizubehalten (Tabelle 8.9).
Tabelle 8.9. Durchführung des Photometertest mit Zugabe von PLP
Komponente c (Stammlösung)
[mM]
Volumen
[µL]
Endkonzentration
[mM]
Puffer pH 7/8 50 710 -
rac-syn-12e 100 150 15
Na2EDTA 20 50 1.0
Na2SO4 500 50 25
PLP 2.5 20 50
LTA (E. coli) - 20 -
Die Ergebnisse der Versuche sind in Tabelle 8.10 dargestellt. Die Enzymlösung wurde
1:4 (v/v) verdünnt (entspricht f = 4, Gleichung (1)).
Tabelle 8.10. Ergebnisse des Photometertests
Eintrag pH Cofaktor ΔE340nm/t
[1/min]
Aktivität
[U/mL]
Relative Aktivität
[%]
1 7 ohne PLP 0.0077 1.2 11
2 7 mit PLP 0.0272 4.3 39
3 8 ohne PLP 0.0155 2.4 22
4 8 mit PLP 0.0691 10.9 100
8.2.2.5.3 Inhibierungsversuche
Die Aktivitätsbestimmung erfolgte analog AAV 5. Es wurde Puffer pH 8 (Tris-HCl-Puffer,
50 mM) verwendet, sowie Cofaktor PLP (50 µM) zugegeben (vgl. Tabelle 8.9). Um die
Substrat- und Produktinhibierung zu untersuchen wurde die Enzymaktivität bei
verschiedenen Konzentrationen von Glycin (11) und Phenylserin (12e) bestimmt. Es wurde
eine gesättigte Lösungen von Glycin (11, 2.1 M) und rac-syn-Phenylserin (rac-syn-12e,

EXPERIMENTELLER TEIL
131
0.19 M) in Puffer pH 8 (Tris-HCl-Puffer, 50 mM) verwendet und entsprechend verdünnt. Die
Substratlösung wurde anstelle der Pufferkomponente zugegeben (vgl. Tabelle 8.9). Die
Ergebnisse zur Substratinhibierung sind in Tabelle 8.11 gezeigt, die zur Produktinhibierung in
Tabelle 8.12.
Tabelle 8.11. Einfluss von 11 auf die Enzymaktivität
Eintrag 11 [M] ΔE340nm/t [1/min] Aktivität [U/mL] Relative Aktivität [%}
1 0 0.0691 10.9 100
2 0.5 0.0060 9.8 90
3 1.0 0.0064 10.5 96
4 1.5 0.0041 6.7 61
5 2.1 0.0037 6.0 55
Tabelle 8.12. Einfluss von 12e auf die Enzymaktivität
Eintrag 12e [mM] ΔE340nm/t [1/min] Aktivität [U/mL] Relative Aktivität [%}
1 0 0.0264 43.1 100
2 15 0.0180 29.4 67
3 50 0.0119 19.4 45
4 100 0.0092 15.1 35
5 150 0.0083 13.6 32
8.2.2.6 Umsatzbestimmung der L-TA-katalysierten Aldolreaktion
Da der Umsatz aufgrund von im Enzymrohextrakt enthaltenem Glycerin nicht direkt
bestimmt werden konnte, wurden zwei Methoden zur Umsatzbestimmung entwickelt und
auf ihre Genauigkeit überprüft. Anschließend wurden die beiden Methoden angewendet.
8.2.2.6.1 Überprüfung der Umsatzbestimmung mittels NMR-Standard
Es wurden verschiedene Mengen rac-syn-12e (0.006-0.025 mmol) in 111 µL Puffer (pH 8,
Tris-HCl-Puffer, 0.1 M) gelöst und 139 µL inaktive L-TA zugegen. Anschließend wurden 25 µL
einer 1 M tert-Leucin-Lösung zupipettiert und das Gemisch abzentrifugiert. Der Überstand

EXPERIMENTELLER TEIL
132
wurde in einen Kolben überführt und das Lösungsmittel im Vakuum entfernt. Danach wurde
der erhaltene Rückstand in D2O aufgenommen und das Verhältnis zwischen 12e und
tert-Leucin 83 mittels 1H-NMR-Spektroskopie bestimmt. 1H-NMR (400 MHz, D2O): δ (ppm) =
1.08 (s, 9H, CH3-83), 3.91 (d, 1H, 3J = 4.4 Hz, syn-CHNH2), 4.08 (d, 1H, 3J = 4.2 Hz, anti-CHNH2),
5.30 (d, 1H, 3J = 4.4 Hz, syn-CHOH), 5.35 (d, 1H, 3J = 4.2 Hz, anti-CHOH), 7.28-7.46 (m, 4H,
H-aromat.). Die spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten
überein.116,141,143 Die Ergebnisse sind in Tabelle 8.13 dargestellt.
Tabelle 8.13. Überprüfung der Umsatzbestimmung mittels NMR-Standard
Eintrag 12e
[mmol]
83
[mmol]
theoretisches
Verhältnis
12e:83
Verhältnis
12e:83
aus NMR
Abweichung
[%]
1 0.025 0.025 1:1 1:1.02 2
2 0.02 0.025 1:1.25 1:1.29 3
3 0.0175 0.025 1:1.4 1:1.37 2
4 0.015 0.025 1:1.67 1:1.61 2
5 0.0125 0.025 1:2 1:2.06 1
6 0.00625 0.025 1:4 1:3.72 2
7 0.0135 0.025 1:1.85 1:1.82 1
8.2.2.6.2 Überprüfung der Umsatzbestimmung mittels Derivatisierung
Abbildung 8.12. Umsatzbestimmung mittels Derivatisierung
rac-syn-12e (0.1-0.5 mmol) und 11 (0.5-1.9 mmol) wurden in 19 mL H2O gelöst. Es wurde
eine 2 mL-Probe entnommen und durch Zugabe von 1 M NaOH der pH-Wert auf pH 12
gebracht. Nach Zugabe von 1.2 Äq. Benzoylchlorid 79 wurde 2 h bei RT gerührt.
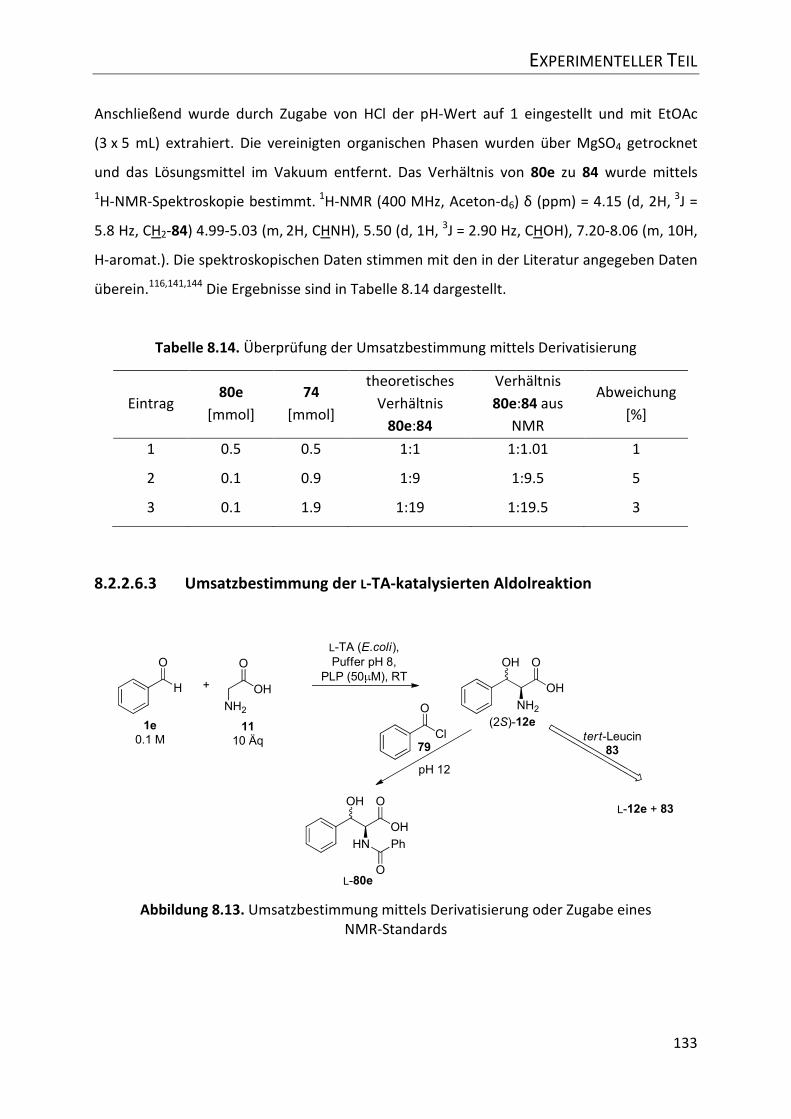
EXPERIMENTELLER TEIL
133
Anschließend wurde durch Zugabe von HCl der pH-Wert auf 1 eingestellt und mit EtOAc
(3 x 5 mL) extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet
und das Lösungsmittel im Vakuum entfernt. Das Verhältnis von 80e zu 84 wurde mittels
1H-NMR-Spektroskopie bestimmt. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.15 (d, 2H, 3J =
5.8 Hz, CH2-84) 4.99-5.03 (m, 2H, CHNH), 5.50 (d, 1H, 3J = 2.90 Hz, CHOH), 7.20-8.06 (m, 10H,
H-aromat.). Die spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten
überein.116,141,144 Die Ergebnisse sind in Tabelle 8.14 dargestellt.
Tabelle 8.14. Überprüfung der Umsatzbestimmung mittels Derivatisierung
Eintrag 80e
[mmol]
74
[mmol]
theoretisches
Verhältnis
80e:84
Verhältnis
80e:84 aus
NMR
Abweichung
[%]
1 0.5 0.5 1:1 1:1.01 1
2 0.1 0.9 1:9 1:9.5 5
3 0.1 1.9 1:19 1:19.5 3
8.2.2.6.3 Umsatzbestimmung der L-TA-katalysierten Aldolreaktion
O
H +
NH2
O
OH
L-TA (E.coli),Puffer pH 8,
PLP (50 M), RTOH
O
NH2
OH
11
10 Äq1e
0.1 M
pH 12
OH
O
HN
OH
O
Ph
(2S)-12etert-Leucin
8379
L-80e
Cl
O
L-12e + 83
Abbildung 8.13. Umsatzbestimmung mittels Derivatisierung oder Zugabe eines
NMR-Standards

EXPERIMENTELLER TEIL
134
8.2.2.6.3.1 Allgemeine Arbeitsvorschrift 6.1 (AAV 6.1) Umsatzbestimmung der
L-TA-katalysierten Aldolreaktion mittels NMR-Standard
Glycin (11, 0.25 mmol, 19 mg) wurde in 139 µL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) gelöst.
Nach Zugabe von PLP (50 µM), Benzaldehyd (1e, 0.025 mmol, 2.5 µL) und L-TA (139 µL,
70 U/mmol) wurde 17 h gerührt. Der pH-Wert des Reaktionsgemisches wurde nach Ablauf
der Reaktionszeit zur Umsatzbestimmung durch Zugabe von HCl auf pH 1 gebracht. Nach
Zugabe von 25 µL tert-Leucin (83) wurde das durch Ansäuern ausgefallene Protein
abzentrifugiert, der Überstand abpipettiert und das Lösungsmittel im Vakuum entfernt. Der
Rückstand wurde in D2O aufgenommen und der Umsatz, sowie das Diastereomeren-
verhältnis mittels 1H-NMR-Spektroskopie bestimmt.
8.2.2.6.3.2 Allgemeine Arbeitsvorschrift 6.2 (AAV 6.2): Umsatzbestimmung der
L-TA-katalysierten Aldolreaktion mittels Derivatisierung
Glycin (11, 0.25 mmol, 19 mg) wurde in 139 µL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) gelöst.
Nach Zugabe von PLP (50 µM), Benzaldehyd (1e, 0.025 mmol, 2.5 µL) und L-TA (139 µL,
70 U/mmol) wurde 17 h gerührt. Nach Ablauf der Reaktionszeit wurden zur
Umsatzbestimmung 200 µL NaOH (5 M) und Benzoylchlorid (79, 1.2 Äq.) zugegeben. Das
Reaktionsgemisch wurde 2 h bei RT gerührt. Anschließend wurde durch Zugabe von HCl der
pH-Wert auf 1 gebracht und mit EtOAc (3 x 5 mL) extrahiert. Die vereinigten organischen
Phasen wurden über MgSO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Der
Umsatz, sowie das Diastereomerenverhältnis wurden mittels 1H-NMR-Spektroskopie
bestimmt.
8.2.2.6.3.3 Ergebnisse der Umsatzbestimmung
Die Reaktion wurde analog AAV 6.1 bzw. AAV 6.2 durchgeführt. Umsatzbestimmung mittels
NMR-Standard: 1H-NMR (400 MHz, D2O): δ (ppm) = 1.08 (s, 9 H, CH3-83), 3.91 (d, 1H, 3J =
4.4 Hz, syn-CHNH3+), 4.08 (d, 1H, 3J = 4.2 Hz, anti-CHNH3
+), 5.30 (d, 1H, 3J = 4.4 Hz,
syn-CHOH), 5.35 (d, 1H, 3J = 4.2 Hz, anti-CHOH), 7.28-7.46 (m, 4H, H-aromat.).
Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.15
(d, 2H, 3J = 5.8 Hz, CH2-84), 4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz, anti-CHOH),

EXPERIMENTELLER TEIL
135
5.50 (d, 1H, 3J = 2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Der ee-Wert des
derivatisierten Produkts betrug >99% (OJ-H-Säule, Hexan/i-PrOH/FA 95:5:0.1, v/v, flow 0.8
ml/min, 230 nm, tr ((2S,3R)-12e) = 43.94 min, tr ((2S,3S)-12e) = 49.78 min). Die
spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten
überein.116,141,143,144 Die Ergebnisse der Umsatzbestimmung sind in Tabelle 8.15 gezeigt.
Tabelle 8.15. Umsatzbestimmung L-TA-katalysierten Aldolreaktion
Eintrag Methode Umsatz [%] d.r. (syn/anti)
1 AAV 6.1 >95 62:38
2 AAV 6.2 91 65:35
8.2.2.7 Untersuchung der L-TA-katalysierten Aldolreaktion
Abbildung 8.14. L-TA-katalysierten Aldolreaktion
Die enzymatische Reaktion wurde analog AAV 6.1 bzw. AAV 6.2 durchgeführt und
verschiedene Parameter variiert.
8.2.2.7.1 Zeitabhängigkeit
Abbildung 8.15. Untersuchung der Zeitabhängigkeit

EXPERIMENTELLER TEIL
136
Zur Untersuchung der Zeitabhängigkeit wurde die Reaktion analog AAV 6.2 durchgeführt. Es
wurden 0.5 mmol Glycin (11, 38 mg), 0.05 mmol Benzaldehyd (1e, 5.1 µL) und PLP (50 µM) in
250 µL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) gelöst und 250 µL L-TA aus E. coli (88 U/mmol)
zugegeben. Der Umsatz und das Diastereomerenverhältnis wurden zu verschiedenen
Zeitpunkten (1 h, 8 h, 20 h) mittels Derivatisierung bestimmt. 1H-NMR (400 MHz, Aceton-d6)
δ (ppm) = 4.15 (d, 2H, 3J = 5.8 Hz, CH2-84), 4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz,
anti-CHOH), 5.50 (d, 1H, 3J = 2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten überein.116,144
Die Ergebnisse sind in Tabelle 8.16 dargestellt.
Tabelle 8.16. Zeitabhängigkeit der Enzymreaktion
Eintrag t [h] Umsatz [%] d.r. (syn/anti)
1 1 20 44:56
2 8 41 68:32
3 20 91 65:35
8.2.2.7.2 Temperaturabhängigkeit
Abbildung 8.16. Untersuchung der Temperaturabhängigkeit
Zur Untersuchung der Temperaturabhängigkeit wurde die Reaktion analog AAV 6.2 bei 40°C
durchgeführt. Es wurden 0.5 mmol Glycin (11, 38 mg), 0.05 mmol Benzaldehyd (1e, 5.1 µL)
und PLP (50 µM) in 250 µL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) gelöst und 250 µL L-TA aus
E. coli (88 U/mmol) zugegeben. Die Reaktionszeit betrug 8 h bzw. 20 h. 1H-NMR (400 MHz,
Aceton-d6) δ (ppm) = 4.15 (d, 2H, 3J = 5.8 Hz, CH2-84), 4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J
= 6.0 Hz, anti-CHOH), 5.50 (d, 1H, 3J = 2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Die

EXPERIMENTELLER TEIL
137
spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten überein.116,144
Die Ergebnisse sind in Tabelle 8.17 dargestellt.
Tabelle 8.17. Enzymreaktion bei 40°C
Eintrag t [h] Umsatz [%] d.r. (syn/anti)
1 8 21 60:40
2 20 13 77:23
Ein weiterer Versuch wurde unter folgenden Bedingungen bei 4°C durchgeführt:
Zu Glycin (11, 0.313 mmol) und PLP (50 µM) wurde Benzaldehyd (1e, 0.0313 mmol) und L-TA
(125 µL, 50 U/mmol) gegeben und 7 h bei ca. 4°C gerührt. Nach Ablauf der Reaktionszeit
wurden zur Umsatzbestimmung 200 µL NaOH (5 M) und Benzoylchlorid (79, 1.2 Äq.)
zugegeben. Das Reaktionsgemisch wurde 2 h bei RT gerührt. Anschließend wurde durch
Zugabe von HCl auf pH 1 angesäuert und mit EtOAc (3 x 5 mL) extrahiert. Die vereinigten
organischen Phasen wurden über MgSO4 getrocknet und das Lösungsmittel im Vakuum
entfernt. Es wurden ein Umsatz von 20% und ein Diastereomerenverhältnis von d.r. 46:54
(syn/anti) erhalten. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.15 (d, 2H, 3J = 5.8 Hz, CH2-84),
4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz, anti-CHOH), 5.50 (d, 1H, 3J = 2.9 Hz,
syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Die spektroskopischen Daten stimmen mit den in
der Literatur angegeben Daten überein.116,144
8.2.2.7.3 Einfluss der Enzymmenge
Abbildung 8.17. Einfluss der Enzymmenge

EXPERIMENTELLER TEIL
138
Zur Untersuchung des Einflusses der Enzymmenge wurde die Reaktion analog AAV 6.2
durchgeführt. Es wurden 0.5 mmol Glycin (11, 38 mg), 0.05 mmol Benzaldehyd (1e, 5.1 µL)
und PLP (50 µM) eingesetzt. Desweiteren wurden entweder 250 µL Puffer (pH 8, Tris-HCl-
Puffer, 0.1 M) und 250 µL L-TA aus E. coli (88 U/mmol) oder 500 µL L-TA aus E. coli
(176 U/mmol) zugegeben. Der Umsatz und das Diastereomerenverhältnis wurde wieder zu
verschiedenen Reaktionszeiten 1-20 h bestimmt. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) =
4.15 (d, 2H, 3J = 5.8 Hz, CH2-84), 4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz,
anti-CHOH), 5.50 (d, 1H, 3J = 2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten überein.116,144
Die Ergebnisse sind in Tabelle 8.18 dargestellt.
Tabelle 8.18. Einfluss der Enzymmenge
Eintrag Enzymmenge [U/mmol] t [h] Umsatz [%] d.r. (syn/anti)
1 88 1 20 44:56
2 88 8 41 68:32
3 88 20 91 65:35
4 176 1 63 67:33
5 176 3 79 67:33
6 176 20 91 68:32
8.2.2.7.4 Einfluss des Verhältnisses von Benzaldehyd zu Glycin
Abbildung 8.18. Einfluss des Verhältnisses von Benzaldehyd (1e) zu Glycin (11)

EXPERIMENTELLER TEIL
139
8.2.2.7.4.1 Einfluss der Glycinkonzentration
Zur Untersuchung des Einflusses der Glycinkonzentration auf den Umsatz wurde die
Reaktion analog AAV 6.2 durchgeführt. Die Glycinkonzentration wurde von 1 M auf 0.1 M
gesenkt während die Benzaldehydkonzentration konstant bei 0.1 M gehalten wurde. Die
Enzymmenge betrug 176 U/mmol. Der Umsatz wurde jeweils nach 20 h bestimmt. 1H-NMR
(400 MHz, Aceton-d6) δ (ppm) = 4.15 (d, 2H, 3J = 5.8 Hz, CH2-84), 4.99-5.03 (m, 2H, CHNH),
5.27 (d, 1H, 3J = 6.0 Hz, anti-CHOH), 5.50 (d, 1H, 3J = 2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H,
H-aromat). Die spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten
überein.116,144 Die Ergebnisse sind in Tabelle 8.19 dargestellt.
Tabelle 8.19. Einfluss des Glycinüberschusses
Eintrag 11 [M] 1e [M] Umsatz [%] d.r. (syn/anti)
1 1.0 0.1 91 68:32
2 0.5 0.1 44 70:30
3 0.2 0.1 28 65:35
4 0.1 0.1 8 73:27
8.2.2.7.4.2 Einfluss der Benzaldehydkonzentration
Zur Untersuchung des Einflusses der Benzaldehydkonzentration auf den Umsatz wurde die
Reaktion analog AAV 6.2 durchgeführt. Die Benzaldehydkonzentration wurde von 0.1 M auf
1 M erhöht während die Glycinkonzentration konstant bei 1 M gehalten wurde. Da die
Enzymaktivität in Abhängigkeit der Aldehydkonzentration eingesetzt wurde, musste bei
Einhaltung der Substratkonzentration die Enzymmenge reduziert werden. Der Umsatz wurde
jeweils nach 20 h bestimmt. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.15 (d, 2H, 3J = 5.8 Hz,
CH2-84), 4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz, anti-CHOH), 5.50 (d, 1H, 3J =
2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Die spektroskopischen Daten stimmen mit
den in der Literatur angegeben Daten überein.116,144 Die Ergebnisse sind in Tabelle 8.20
dargestellt.

EXPERIMENTELLER TEIL
140
Tabelle 8.20. Einfluss der Benzaldehydkonzentration
Eintrag 11 [M] 1e [M] Enzymmenge
[U/mmol] Umsatz [%] d.r. (syn/anti)
1 1.0 0.1 88 91 68:32
2 1.0 0.5 17 14 71:29
3 1.0 1.0 9 4 67:33
8.2.2.7.5 Erhöhung der Substratkonzentration
Abbildung 8.19. Erhöhung der Substratkonzentration auf 0.25 M
Um einen effizienteren Prozess zu entwickeln, wurde eine Erhöhung der Substrat-
konzentration von 0.1 M auf 0.25 M untersucht. Der Umsatz wurde mit Hilfe der beiden
entwickelten Methoden bestimmt.
8.2.2.7.5.1 Allgemeine Arbeitsvorschrift 7.1 (AAV 7.1): Umsatzbestimmung durch
NMR-Standard bei einer Substratkonzentration von 0.25 M
Zu Glycin (11, 0.313 mmol, 23 mg) und PLP (50 µM) wurde Benzaldehyd (1e, 0.0313 mmol,
3.2 µL) und L-TA (125 µL, 50 U/mmol) gegeben und 17 h gerührt. Der pH-Wert des
Reaktionsgemisches wurde nach Ablauf der Reaktionszeit zur Umsatzbestimmung durch
Zugabe von HCl auf pH 1 gebracht. Nach Zugabe von 31.3 µL tert-Leucin 83 wurde das durch
Ansäuern ausgefallene Protein abzentrifugiert, der Überstand in einen Kolben pipettiert und
das Lösungsmittel im Vakuum entfernt. Der Rückstand wurde in D2O aufgenommen und der
Umsatz, sowie das Diastereomerenverhältnis mittels 1H-NMR-Spektroskopie bestimmt.

EXPERIMENTELLER TEIL
141
8.2.2.7.5.2 Allgemeine Arbeitsvorschrift 7.2 (AAV 7.2): Umsatzbestimmung durch
Derivatisierung bei einer Substratkonzentration von 0.25 M
Zu Glycin (11, 0.313 mmol) und PLP (50 µM) wurde Benzaldehyd (1e, 0.0313 mmol, 3.2 µL)
und L-TA (125 µL, 50 U/mmol) gegeben und 17 h gerührt. Nach Ablauf der Reaktionszeit
wurden zur Umsatzbestimmung 200 µL 5 M NaOH und Benzoylchlorid (79, 1.2 Äq.)
zugegeben. Das Reaktionsgemisch wurde 2 h bei RT gerührt. Anschließend wurde durch
Zugabe von HCl auf pH 1 angesäuert und mit EtOAc (3 x 5 mL) extrahiert. Die vereinigten
organischen Phasen wurden über MgSO4 getrocknet und das Lösungsmittel im Vakuum
entfernt. Der Umsatz, sowie das Diastereomerenverhältnis wurden mittels 1H-NMR-
Spektroskopie bestimmt.
8.2.2.7.5.3 Ergebnisse der Umsatzbestimmung (0.25 M)
Die Reaktion wurde analog AAV 7.1 bzw. AAV 7.2 durchgeführt. Umsatzbestimmung mittels
NMR-Standard: 1H-NMR (400 MHz, D2O): δ (ppm) = 1.08 (s, 9 H, CH3-83) 3.91 (d, 1H, 3J =
4.4 Hz, syn-CHNH3+), 4.08 (d, 1H, 3J = 4.2 Hz, anti-CHNH3
+), 5.30 (d, 1H, 3J = 4.4 Hz,
syn-CHOH), 5.35 (d, 1H, 3J = 4.2 Hz, anti-CHOH), 7.28-7.46 (m, 4H, H-aromat.).
Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.15
(d, 2H, 3J = 5.8 Hz, CH2-84), 4.99-5.03 (m, 2H, CHNH), 5.27 (d, 1H, 3J = 6.0 Hz, anti-CHOH),
5.50 (d, 1H, 3J = 2.9 Hz, syn-CHOH), 7.20-8.06 (m, 10H, H-aromat.). Der ee-Wert des
derivatisierten Produkts betrug >99% (OJ-H-Säule, Hexan/i-PrOH/FA 95:5:0.1, v/v, flow 0.8
ml/min, 230 nm, tr ((2S,3R)-12e) = 43.94 min, tr ((2S,3S)-12e) = 49.78 min). Die
spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten
überein.116,141,143,144 Die Ergebnisse der unterschiedlichen Umsatzbestimmung sind in Tabelle
8.21 gezeigt.
Tabelle 8.21. Umsatz bei Substratkonzentration von 0.25 M
Eintrag Methode Umsatz [%] d.r. (syn/anti)
1 AAV 7.1 91 61:39
2 AAV 7.2 84 65:35

EXPERIMENTELLER TEIL
142
8.2.2.7.6 Einfluss verschiedener Additive
Abbildung 8.20. Einfluss verschiedener Additive
Zur Untersuchung des Einflusses verschiedener Additive auf den Umsatz wurde die Reaktion
analog AAV 7.1 mit einer Substratkonzentration von 0.25 M durchgeführt. Es wurde zum
Rohextrakt 50 Vol% der entsprechenden Substanz zugefügt. Die Zugabe von tert-Leucin zur
Umsatzbestimmung war nur bei Verwendung von Glycerin als Additiv notwendig. 1H-NMR
(400 MHz, D2O): δ (ppm) = 1.08 (s, 9 H, CH3-83), 3.80 (s, 2H, CH2NH3+-11) 3.91 (d, 1H, 3J =
4.4 Hz, syn-CHNH3+), 4.08 (d, 1H, 3J = 4.2 Hz, anti-CHNH3
+), 5.30 (d, 1H, 3J = 4.4 Hz,
syn-CHOH), 5.35 (d, 1H, 3J = 4.2 Hz, anti-CHOH), 7.28-7.46 (m, 4H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegeben Daten
überein.116,141,143 Die Ergebnisse sind in Tabelle 8.19 dargestellt.
Tabelle 8.22. Einfluss verschiedener Additive
Eintrag Additiv (50 Vol%) Umsatz [%] d.r. (syn/anti)
1 Puffer 68 65:35
2 Glycerin 71 65:35
3 DMSO 11 58:42
4 i-PrOH 19 60:40
5 Aceton 16 60:40

EXPERIMENTELLER TEIL
143
8.2.2.7.7 Substratspektrum
Abbildung 8.21. Erweiterung des Substratspektrums
Zur Erweiterung des Substratspektrums wurden verschieden substituierte Benzaldehyde als
Akzeptor eingesetzt. Die enzymatische Reaktion wurde bei einer Substratkonzentration von
0.1 M und 0.25 M durchgeführt und der Umsatz mit Hilfe der beiden möglichen Methoden
bestimmt (vgl. AAV 6.1, 6.2, 7.1 und 7.2).
8.2.2.7.7.1 Enzymatische Synthese von p-Nitrophenylserin (2S)-12b
Die Synthese erfolgte analog AAV 6.2 und 7.1. Es wurde die
entsprechende Menge p-Nitrobenzaldehyd (1b, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 4.36 (d, 1H, 3J = 3.2 Hz,
syn-CHNH3+), 4.45 (d, 1H, 3J = 3.0 Hz, anti-CHNH3
+), 5.44 (d, 1H, 3J =
2.6 Hz, anti-CHOH), 5.51 (d, 1H, 3J = 3.1 Hz, syn-CHOH), 7.57-7.67 (m, 2H, H-aromat.), 8.19-
8.25 (m, 2H, H-aromat.). Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz,
Aceton-d6) δ (ppm) = 5.04-5.06 (m, 1H, anti-CHNH), 5.11-5.14 (m, 1H, syn-CHNH), 5.43 (d,
1H, 3J = 5,6 Hz, anti-CHOH), 5.67 (d, 1H, 3J = 2,3 Hz, syn-CHOH), 7.44-8.27 (m, 9H, H-aromat.).
Die spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten
überein.109 Die Ergebnisse der enzymatischen Umsetzungen sind in Tabelle 8.23 angegeben.
Tabelle 8.23. Enzymatische Synthese von (2S)-12b
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.2 0.1 55 62:38
2 7.1 0.25 35 68:32

EXPERIMENTELLER TEIL
144
8.2.2.7.7.2 Enzymatische Synthese von p-Methylthiophenylserin (2S)-12d
Die Synthese erfolgte analog AAV 6.1. Es wurde die entsprechende
Menge p-Methylthiobenzaldehyd (1d, 0.025 mmol) eingesetzt. Es
wurden ein Umsatz von 10% und ein Diastereomerenverhältnis von
d.r. 57:43 (syn/anti) erzielt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 2.41 (s, 3H, anti-SCH3),
2.42 (s, 3H, syn-SCH3), 3.85-3.88 (m, 2H, CHNH3+), 5.25 (d, 1H, 3J = 4.3 Hz, anti-CHOH), 5.29
(d, 1H, 3J = 4.0 Hz, syn-CHOH), 7.34-7.39 (m, 4H, H-aromat.). Die spektroskopischen Daten
stimmen mit den in der Literatur angegebenen Daten überein.140
8.2.2.7.7.3 Enzymatische Synthese von p-Chlorphenylserin (2S)-12f
Die Synthese erfolgte analog AAV 6.1, 6.2 und 7.2. Es wurde die
entsprechende Menge p-Chlorbenzaldehyd (1f, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 4.07 (d, 1H, 3J = 4.0 Hz,
syn-CHNH3+), 4.21 (d, 1H, 3J = 4.3 Hz, anti-CHNH3
+), 5.35 (d, 1H, 3J =
4.6 Hz, syn-CHOH), 5.38 (d, 1H, 3J = 4.6 Hz anti-CHOH), 7.38-7.50 (m, 4H, H-aromat.).
Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.99
(dd, 1H, 3J = 8.2 Hz, 2.5 Hz, syn-CHNH), 5.04 (dd, 1H, 3J = 9.1 Hz, 6.1 Hz, anti-CHNH), 5.28 (d,
1H, 3J = 5.8 Hz, syn-CHOH), 5.52 (d, 1H, 3J = 2.8 Hz, anti-CHOH), 7.22-7.83 (m, 9H, H-aromat.).
Die spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten109 bzw.
mit den Daten des vollständig charakterisierten Racemats überein (Abschnitt 8.2.2.2.3). Die
Ergebnisse der enzymatischen Umsetzungen sind in Tabelle 8.24 angegeben.
Tabelle 8.24. Enzymatische Synthese von (2S)-12f
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 32 62:38
2 6.2 0.1 26 66:34
3 7.2 0.25 20 66:34
OH
OOH
(2S)-12dC10H13NO3S
M = 227.28 g/mol
NH2MeS

EXPERIMENTELLER TEIL
145
8.2.2.7.7.4 Enzymatische Synthese von o-Chlorphenylserin (2S)-12i
Die Synthese erfolgte analog AAV 6.2 und 7.2. Es wurde die
entsprechende Menge o-Cl-Benzaldehyd (1i, 0.1 M: 0.025 mmol, 0.25 M:
0.0313 mmol) eingesetzt. Umsatzbestimmung mittels Derivatisierung:
1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 5.12-5.24 (m, 2H, CHNH), 5,56
(d, 1H, 3J = 5.2 Hz, anti-CHOH), 5.86 (d, 1H, 3J = 2.1 Hz, syn-CHOH),
7.25-7.80 (m, 9H, H-aromat.). Der ee-Wert des Produkts beträgt für beide Umsetzungen
>99% (OJ-H-Säule, Hexan/i-PrOH/FA 95:5:0.1, v/v, flow 0,8 ml/min, 230nm, tr ((2S,3R)-12i) =
63.05 min, tr ((2S,3S)-12i) = 68.07 min). Die spektroskopischen Daten stimmen mit den Daten
des vollständig charakterisierten Racemats überein (vgl. Abschnitt 8.2.2.2.4 und 8.2.2.2.5).
Die Ergebnisse der enzymatischen Umsetzungen sind in Tabelle 8.25 angegeben.
Tabelle 8.25. Enzymatische Synthese von (2S)-12i
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.2 0.1 91 81:19
2 7.2 0.25 85 80:20
8.2.2.7.7.5 Enzymatische Synthese von m-Bromphenylserin (2S)-12j
Die Synthese erfolgte analog AAV 6.1 und 7.2. Es wurde die
entsprechende Menge m-Brombenzaldehyd (1j, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 3.87 (d, 1H, 3J = 4.1 Hz,
syn-CHNH3+), 4.05 (d, 1H, 3J = 3.8 Hz, anti-CHNH3
+), 5.25 (d, 2H, 3J =
4.2 Hz, syn-CHOH), 5.31 (d, 1H, 3J = 4.0 Hz, anti-CHOH) 7.31-7.65 (m, 4H, H-aromat.).
Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) =
4.96-5.03 (m, 2H, CHNH), 5.26 (d, 1H, 3J = 5.7 Hz, anti-CHOH), 5.50 (d, 1H, 3J = 2.9 Hz,
syn-CHOH) 7.4-7.93 (m, 9H, H-aromat.). Die spektroskopischen Daten stimmen mit den in
der Literatur angegebenen Daten109 bzw. mit den Daten des vollständig charakterisierten
Racemats überein (Abschnitt 8.2.2.2.6). Die Ergebnisse der enzymatischen Umsetzungen
sind in Tabelle 8.26 angegeben.
OH
OOH
(2S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
146
Tabelle 8.26. Enzymatische Synthese von (2S)-12j
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 56 62:38
2 7.2 0.25 47 63:37
8.2.2.7.7.6 Enzymatische Synthese von p-Methylsulphonylphenylserin (2S)-12k
Die Synthese erfolgte analog AAV 6.2 und 7.1. Es wurde die
entsprechende Menge p-Methylsulphonylbenzaldehyd (1k,
0.1 M: 0.025 mmol, 0.25 M: 0.0313 mmol) eingesetzt. Umsatz-
bestimmung mittels NMR-Standard: 1H-NMR (400 MHz, D2O)
δ (ppm) = 4.26 (m, 1H, syn-CHNH3+), 4.30 (m, 1H, anti-CHNH3
+),
5.45 (d, 1H, 3J = 3.1 Hz, anti-CHOH), 5.52 (d, 1H, 3J = 3.4 Hz, syn-CHOH), 7.63-7.97 (m, 4H,
H-aromat.). Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6)
δ (ppm) = 5.07-5.12 (m, 2H, CHNH), 5.41 (d, 1H, 3J = 6.5 Hz, anti-CHOH), 5.63 (d, 1H, 3J =
3.3 Hz, syn-CHOH), 7.44-8.05 (m, 9H, H-aromat.). Die spektroskopischen Daten stimmen mit
den in der Literatur angegebenen Daten überein.109,145 Die Ergebnisse der enzymatischen
Umsetzungen sind in Tabelle 8.27 angegeben.
Tabelle 8.27. Enzymatische Synthese von (2S)-12k
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.2 0.1 42 61:39
3 7.1 0.25 24 65:35
8.2.2.7.7.7 Enzymatische Synthese von m-Chlorphenylserin (2S)-12l
Die Synthese erfolgte analog AAV 6.1, 6.2, und 7.2. Es wurde die
entsprechende Menge m-Chlorbenzaldehyd (1l, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 3.67 (d, 1H, 3J = 4.8 Hz,
syn-CHNH3+), 3.79 (d, 1H, 3J = 4.1 Hz, anti-CHNH3
+), 5.10 (d, 2H, 3J =
4.8 Hz, CHOH), 7.30-7.47 (m, 4H, H-aromat.). Umsatzbestimmung mittels Derivatisierung:
OH
OOH
(2S)-12lC9H10ClNO3
M = 215.63 g/mol
NH2
Cl
OH
OOH
(2S)-12kC10H13NO5S
M = 259.28 g/mol
NH2MeO2S
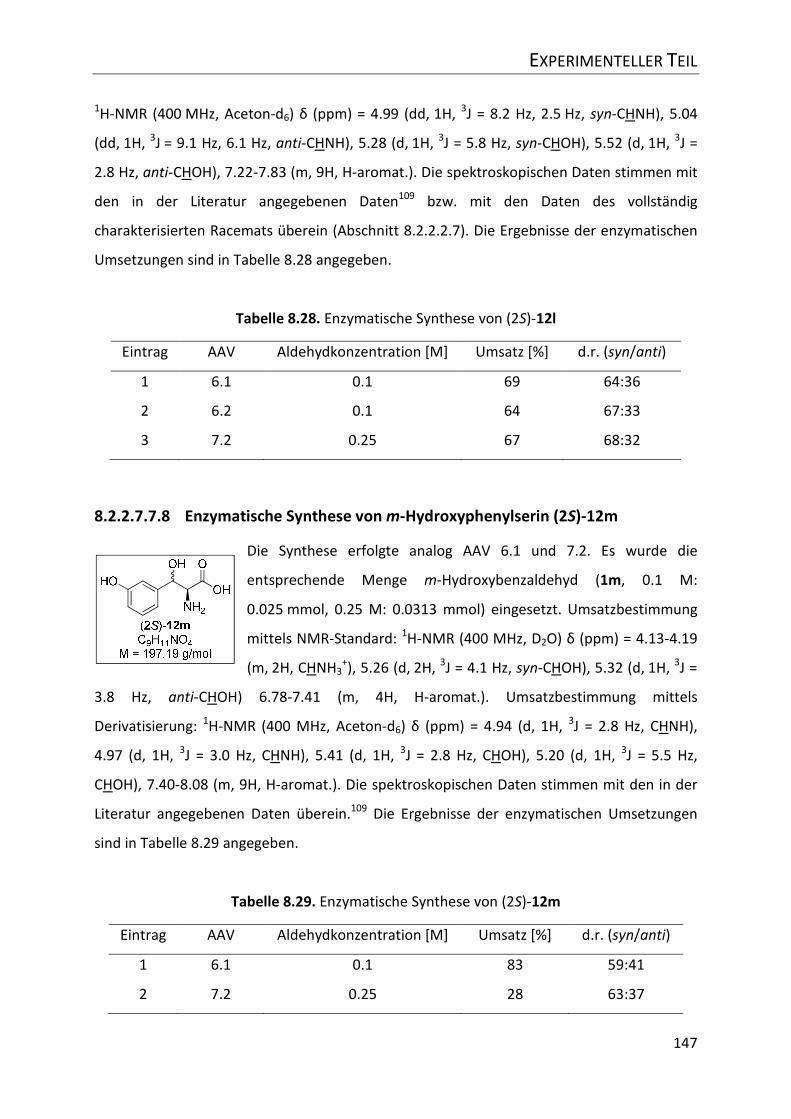
EXPERIMENTELLER TEIL
147
1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.99 (dd, 1H, 3J = 8.2 Hz, 2.5 Hz, syn-CHNH), 5.04
(dd, 1H, 3J = 9.1 Hz, 6.1 Hz, anti-CHNH), 5.28 (d, 1H, 3J = 5.8 Hz, syn-CHOH), 5.52 (d, 1H, 3J =
2.8 Hz, anti-CHOH), 7.22-7.83 (m, 9H, H-aromat.). Die spektroskopischen Daten stimmen mit
den in der Literatur angegebenen Daten109 bzw. mit den Daten des vollständig
charakterisierten Racemats überein (Abschnitt 8.2.2.2.7). Die Ergebnisse der enzymatischen
Umsetzungen sind in Tabelle 8.28 angegeben.
Tabelle 8.28. Enzymatische Synthese von (2S)-12l
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 69 64:36
2 6.2 0.1 64 67:33
3 7.2 0.25 67 68:32
8.2.2.7.7.8 Enzymatische Synthese von m-Hydroxyphenylserin (2S)-12m
Die Synthese erfolgte analog AAV 6.1 und 7.2. Es wurde die
entsprechende Menge m-Hydroxybenzaldehyd (1m, 0.1 M:
0.025 mmol, 0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung
mittels NMR-Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 4.13-4.19
(m, 2H, CHNH3+), 5.26 (d, 2H, 3J = 4.1 Hz, syn-CHOH), 5.32 (d, 1H, 3J =
3.8 Hz, anti-CHOH) 6.78-7.41 (m, 4H, H-aromat.). Umsatzbestimmung mittels
Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.94 (d, 1H, 3J = 2.8 Hz, CHNH),
4.97 (d, 1H, 3J = 3.0 Hz, CHNH), 5.41 (d, 1H, 3J = 2.8 Hz, CHOH), 5.20 (d, 1H, 3J = 5.5 Hz,
CHOH), 7.40-8.08 (m, 9H, H-aromat.). Die spektroskopischen Daten stimmen mit den in der
Literatur angegebenen Daten überein.109 Die Ergebnisse der enzymatischen Umsetzungen
sind in Tabelle 8.29 angegeben.
Tabelle 8.29. Enzymatische Synthese von (2S)-12m
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 83 59:41
2 7.2 0.25 28 63:37

EXPERIMENTELLER TEIL
148
8.2.2.7.7.9 Enzymatische Synthese von o-Methoxyphenylserin (2S)-12n
Die Synthese erfolgte analog AAV 6.1, 6.2 und 7.2. Es wurde die
entsprechende Menge o-Methoxybenzaldehyd (1n, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 3.85 (s, 3H, syn-OCH3), 3.89
(s, 3H, anti-OCH3), 4.03 (d, 1H, 3J = 4.6 Hz, syn-CHNH3+), 4.07 (d, 1H, 3J =
3.5 Hz, anti-CHNH3+), 5.42 (d, 1H, 3J = 4.6 Hz, syn-CHOH), 5.53 (d, 1H, 3J = 3.5 Hz, anti-CHOH),
7.05-7.50 (m, 4H, H-aromat.). Umsatzbestimmung mittels Derivatisierung: 1H-NMR
(400 MHz, Aceton-d6) δ (ppm) = 3.85, (s, 3H, syn-OCH3) 3.90 (s, 3H, anti-OCH3), 4.99 (dd, 1H,
3J = 11.0 Hz, 5.8 Hz, syn-CHNH), 5.18 (dd, 1H, 3J = 9.3 Hz, 2.5 Hz, anti-CHNH), 5.50 (d, 1H, 3J =
5.5 Hz, anti-CHOH), 5.79 (d, 1H, 3J = 5.8 Hz, syn-CHOH), 6.85-8.06 (m, 9H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten142,109 bzw.
mit den Daten des vollständig charakterisierten Racemats überein (Abschnitt 8.2.2.2.8). Die
Ergebnisse der enzymatischen Umsetzungen sind in Tabelle 8.30 angegeben.
Tabelle 8.30. Enzymatische Synthese von (2S)-12n
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 80 79:21
2 6.2 0.25 59 78:22
32 7.2 0.25 69 75:25
8.2.2.7.7.10 Enzymatische Synthese von o-Bromphenylserin (2S)-12o
Die Synthese erfolgte analog AAV 6.2 und 7.2. Es wurde die
entsprechende Menge o-Brombenzaldehyd (1o, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels
Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 5.12-5.15 (m,
1H, anti-CHNH), 5.25-5.29 (m, 1H, syn-CHNH), 5.50 (d, 1H, 3J = 5.1 Hz,
anti-CHOH), 5.82 (d, 1H, 3J = 2.1 Hz, syn-CHOH), 7.13-8.08 (m, 9H, H-aromat.). Der ee-Wert
wurde für die Umsetzung mit einer Substratkonzentration von 0.25 M bestimmt und betrug
>99% (OJ-H-Säule, Hexan/i-PrOH/FA 95:5:0.1, v/v, flow 0.8 ml/min, 230 nm, tr ((2R,3S)-12o)
OH
OOH
(2S)-12nC10H13NO4
M = 211.21 g/mol
NH2
OMe
OH
OOH
(2S)-12oC9H10BrNO3
M = 260.08 g/mol
NH2
Br

EXPERIMENTELLER TEIL
149
= 67.13 min, tr ((2R,3R)-12o)= 73.72 min). Die spektroskopischen Daten stimmen mit den in
der Literatur angegebenen Daten überein.109,145 Die Ergebnisse der enzymatischen
Umsetzungen sind in Tabelle 8.31 angegeben.
Tabelle 8.31. Enzymatische Synthese von (2S)-12o
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.2 0.1 70 72:28
2 7.2 0.25 51 73:27
8.2.2.7.7.11 Enzymatische Synthese von o-Fluorphenylserin (2S)-12p
Die Synthese erfolgte analog AAV 6.2 und 7.2. Es wurde die
entsprechende Menge o-Fluorbenzaldehyd (1p, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels
Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 5.09-5.13 (m,
2H, CHNH), 5.53 (d, 1H, 3J = 5.8 Hz, anti-CHOH), 5.80 (d, 1H, 3J = 2.5 Hz,
syn-CHOH, 7.04-8.04 (m, 9H, H-aromat.). Die spektroskopischen Daten stimmen mit den in
der Literatur angegebenen Daten überein.109,145 Die Ergebnisse der enzymatischen
Umsetzungen sind in Tabelle 8.32 angegeben.
Tabelle 8.32. Enzymatische Synthese von (2S)-12p
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.2 0.1 42 72:28
2 7.2 0.25 47 71:29
8.2.2.7.7.12 Enzymatische Synthese von o-Nitrophenylserin (2S)-12q
Die Synthese erfolgte analog AAV 6.1, 6.2 und 7.2. Es wurde die
entsprechende Menge o-Nitrobenzaldehyd (1q, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 4.49-4.51 (m, 2H, CHNH3+),
OH
OOH
(2S)-12pC9H10FNO3
M = 199.18 g/mol
NH2
F
OH
OOH
(2S)-12qC9H10N2O5
M = 226.19 g/mol
NH2
NO2

EXPERIMENTELLER TEIL
150
5.77 (d, 1H, 3J = 3.8 Hz, anti-CHOH), 5.92 (d, 1H, 3J = 3.3 Hz, syn-CHOH), 7.48-8.11 (m, 4H, H-
aromat.). 1H-NMR-Daten für die Umsatzbestimmung mittels Derivatisierung 1H-NMR (400
MHz, Aceton-d6) δ (ppm) = 5.41-5.45 (m, 1H, syn-CHNH), 5.48-5.53 (m, 1H, anti-CHNH), 5.78
(d, 1H, 3J = 6.4 Hz, anti-CHOH), 6.16 (d, 1H, 3J = 2.1 Hz, syn-CHOH), 7.38-8.10 (m, 9H, H-
aromat.). Die spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten
überein.109,145 Die Ergebnisse der enzymatischen Umsetzungen sind in Tabelle 8.33
angegeben.
Tabelle 8.33. Enzymatische Synthese von (2S)-12q
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 90 68:32
2 6.2 0.25 60 71:29
32 7.2 0.25 58 70:30
8.2.2.7.7.13 Enzymatische Synthese von o-Methylphenylserin (2S)-12r
Die Synthese erfolgte analog AAV 6.1, 6.2 und 7.2. Es wurde die
entsprechende Menge o-Methylbenzaldehyd (1r, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels NMR-
Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 2.24 (s, 3H, CH3), 4.13-4.16
(m, 2H, CHNH3+), 5.39 (d, 1H, 3J = 4.0 Hz, anti-CHOH), 5.53 (d, 1H, 3J =
3.6 Hz, syn-CHOH), 7.17-7.46 (m, 2H, H-aromat.), 7.71-7.42 (m, 2H, H-aromat.).
Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 2.45 (s,
6H, CH3), 4.97 (d, 1H, 3J = 2.5 Hz, syn-CHNH), 5.05 (d, 1H, 3J = 5.3 Hz, anti-CHNH), 5.43 (d, 1H,
3J = 6.1 Hz, anti-CHOH), 5.69 (d, 1H, 3J = 2.5 Hz, syn-CHOH), 7.09-8.07 (m, 9H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten
überein.109,145 Die Ergebnisse der enzymatischen Umsetzungen sind in Tabelle 8.34
angegeben.
OH
OOH
(2S)-12rC10H13NO3
M = 195.22 g/mol
NH2
Me

EXPERIMENTELLER TEIL
151
Tabelle 8.34. Enzymatische Synthese von (2S)-12r
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.1 0.1 27 78:22
2 6.2 0.25 25 76:24
3 7.2 0.25 24 79:21
8.2.2.7.7.14 Enzymatische Synthese von o-Hydroxyphenylserin (2S)-12s
Die Synthese erfolgte analog AAV 6.2 und 7.2. Es wurde die
entsprechende Menge o-Hydroxybenzaldehyd (1s, 0.1 M: 0.025 mmol,
0.25 M: 0.0313 mmol) eingesetzt. Umsatzbestimmung mittels
Derivatisierung: 1H-NMR (400 MHz, D2O) δ (ppm) = 5.22-4.26 (m, 2H,
CHNH), 5.81 (d, 1H, 3J = 4.0 Hz, syn-CHOH), 7.43-8.07 (m, 4H, H-aromat.).
Das Signal des Protons von anti-CHOH wurde im Spektrum durch Verunreinigungen
überdeckt. Das Diastereomerenverhältnis wurde aus dem Verhältnis des Peaks syn/anti-
CHNH und syn-CHOH berechnet. Die spektroskopischen Daten stimmen mit den in der
Literatur angegebenen Daten überein.145 Die Ergebnisse der enzymatischen Umsetzungen
sind in Tabelle 8.35 angegeben.
Tabelle 8.35. Enzymatische Synthese von (2S)-12s
Eintrag AAV Aldehydkonzentration [M] Umsatz [%] d.r. (syn/anti)
1 6.2 0.1 7 74:26
2 7.2 0.25 10 78:22
8.2.2.7.7.15 Enzymatische Synthese von 3,4-Dihydroxyphenylserin (2S)-12t
Die Synthese erfolgte analog AAV 6.1. Es wurde 0.025 mmol
3,4-Dihydroxybenzaldehyd eingesetzt. Umsatzbestimmung mittels
NMR-Standard: 1H-NMR (400 MHz, D2O) δ (ppm) = 5.15 (d, 1H, 3J =
4.0 Hz, anti-CHOH), 5.22 (d, 1H, 3J = 3.6 Hz, syn-CHOH), 6.72-6.82 (m,
3H, H-aromat.). Das Signal der Protonen von syn/anti-CHNH2 wurde
OH
OOH
(2S)-12sC9H11NO4
M = 197.19 g/mol
NH2
OH

EXPERIMENTELLER TEIL
152
im Spektrum durch Verunreinigungen überdeckt. Der Umsatz betrug 16% und das
Diastereomerenverhältnis d.r. 53:47.146
8.2.2.8 Allgemeine Arbeitsvorschrift 8 (AAV 8): Optimierte Reaktion mit
o-Chlorbenzaldehyd (1i) als Substrat
Abbildung 8.22. Optimierte Reaktion mit o-Chlorbenzaldehyd (1i) als Substrat
Glycin (11, 2 M) und PLP (50 µM) wurden in Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) gelöst. Nach
Zugabe von o-Chlorbenzaldehyd (1i, 0.25 M) und L-TA aus E. coli (44 U/mmol) wurde das
Reaktionsgemisch 7 h bei 25°C gerührt. Anschließend wurde der Umsatz mittels NMR-
Standard oder Derivatisierung bestimmt (vgl. AAV 7.1 und 7.2).
8.2.2.8.1 Reaktionsverfolgung
Die Durchführung der Reaktion erfolgte analog AAV 8. Es wurden
2.5 mmol o-Chlorbenzaldehyd (1i, 281 µL) und 10 mL L-TA aus E. coli
(11 U/mL, 44 U/mmol) eingesetzt. Es wurden nach verschiedenen
Reaktionszeiten 250 µL-Proben entnommen und der Umsatz analog AAV
7.1 und 7.2 bestimmt. Umsatzbestimmung mittels NMR-Standard:
1H-NMR (400 MHz, D2O) δ (ppm) = 4.36-4.40 (m, 2H, CHNH3+), 5.56 (d, 1H, 3J = 3.0 Hz,
anti-CHOH), 5.75 (d, 1H, 3J = 4.2 Hz, syn-CHOH), 7.41-7.68 (m, 4H, H-aromat.).
Umsatzbestimmung mittels Derivatisierung: 1H-NMR (400 MHz, Aceton-d6) δ (ppm) =
5.12-5.16 (m, 1H, anti-CHNH), 5.22-5.24 (m, 1H, syn-CHNH), 5.56 (d, 1H, 3J = 5.2 Hz,
anti-CHOH), 5.86 (d, 1H, 3J = 2.1 Hz, syn-CHOH), 7.20-7.80 (m, 9H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten109 bzw. mit
OH
OOH
(2S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
153
den Daten des vollständig charakterisierten Racemats überein (Abschnitt 8.2.2.2.4 und
8.2.2.2.5) Die Ergebnisse sind in Tabelle 8.36 dargestellt.
Tabelle 8.36. Reaktionsverfolgung
Eintrag t [h] Umsatz1) [%] d.r. (syn/anti) 1) Umsatz2) [%] d.r. (syn/anti) 2)
1 0.25 11 74:26 n.b. n.b.
2 0.5 30 80:20 n.b. n.b.
3 1 51 79:21 n.b. n.b.
4 2 76 81:19 n.b. n.b.
5 3 97 80:20 86 78:22
6 5 96 80:20 92 77:23
7 7 98 78:22 91 78:22
8 22 92 80:20 92 78:22
9 144 99 79:21 95 75:25
1)analog AAV 7.2
2)analog AAV 7.1; n.b. nicht bestimmt
8.2.2.8.2 Vergleich mit Reaktion im kleineren Maßstab
Die Durchführung der Reaktion erfolgte analog AAV 8. Es wurden
0.1 mmol o-Chlorbenzaldehyd (1i, 125 µL) und 500 µL L-TA aus E. coli
(11 U/mL, 44 U/mmol) eingesetzt. Nach 6 h Reaktionszeit wurde die
Reaktion abgebrochen und der Umsatz mittels Derivatisierung analog
AAV 7.2 bestimmt. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 5.12-5.24 (m, 2H, CHNH), 5,56
(d, 1H, 3J = 5.2 Hz, anti-CHOH), 5.86 (d, 1H, 3J = 2.1 Hz, syn-CHOH), 7.25-7.80 (m, 9H, H-
aromat.). Der ee-Wert betrug >99% (OJ-H-Säule, Hexan/i-PrOH/FA 95:5:0.1, v/v, flow 0,8
ml/min, 230nm, tr ((2S,3R)-12i) = 63.05 min, tr ((2S,3S)-12i) = 68.07 min). Die
spektroskopischen Daten stimmen mit den Daten des vollständig charakterisierten Racemats
überein (vgl. Abschnitt 8.2.2.2.4 und 8.2.2.2.5).
OH
OOH
(2S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
154
8.2.2.8.3 Synthese von (2S)-12i ohne Zugabe von Cofaktor
Die enzymatische Reaktion wurde analog AAV 8 einmal mit Zugabe von
PLP (50 µM) und einmal ohne Zugabe von PLP (50 µM) durchgeführt. Es
wurden 3.125 mmol Aldehyd (1i, 351 µL), 5 mL L-TA aus E. coli (27 U/mL,
44 U/mmol) und 7.5 mL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) eingesetzt.
Die Reaktion wurde nach 7 h durch Ansäuern auf pH 1 durch Zugabe von
HCl gestoppt. Da im Rohextrakt kein Glycerol enthalten war konnte der Umsatz direkt aus
dem Verhältnis von Glycin zu entstandenem Produkt aus dem 1H-NMR-Spektrum bestimmt
werden. 1H-NMR (400 MHz, D2O) δ (ppm) = 4.36-4.40 (m, 2H, CHNH3+), 5.56 (d, 1H, 3J =
3.0 Hz, anti-CHOH), 5.75 (d, 1H, 3J = 4.2 Hz, syn-CHOH), 7.41-7.68 (m, 4H, H-aromat.). Die
spektroskopischen Daten stimmen mit den in der Literatur angegebenen Daten
überein.109,147 Die Ergebnisse der beiden Versuche sind in Tabelle 8.37 dargestellt.
Tabelle 8.37. Enzymatische Umsetzung mit und ohne Zugabe von PLP
Eintrag Cofaktor Umsatz [%] d.r. (syn/anti)
1 mit PLP 96 80:20
2 ohne PLP 91 78:22
8.2.2.8.4 Vergleich mit Benzaldehyd als Substrat
Die Durchführung der Reaktion erfolgte analog AAV 8. Es wurden
3.125 mmol Benzaldehyd (1e, 316 µL), 5 mL L-TA aus E. coli (27 U/mL,
44 U/mmol) und 7.5 mL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) eingesetzt.
Es wurde ein Versuch mit und ein Versuch ohne Zugabe von PLP (50 µM)
durchgeführt. Der Umsatz der Reaktion wurde nach verschiedenen Reaktionszeiten durch
Entnahme einer 200 µL-Probe bestimmt. Der pH-Wert der Probe wurde auf 1 eingestellt und
der Umsatz direkt aus dem Verhältnis von Glycin (11) zu entstandenem Produkt 12e
bestimmt. 1H-NMR (400 MHz, D2O) δ (ppm) = 4.30 (d, 1H, 3J = 3.8 Hz, syn-CHNH3+), 4.39 (d,
1H, 3J = 4.3 Hz, anti-CHNH3+), 5.38 (d, 1H, 3J = 4.0 Hz, anti-CHOH), 5.43 (d, 1H, 3J = 3.8 Hz, syn-
CHOH), 7.39-7.47 (m, 5H, H-aromat.). Die spektroskopischen Daten stimmen mit den in der
OH
OOH
(2S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl
OH
OOH
(2S)-12eC9H11NO3
M = 181.19 g/mol
NH2

EXPERIMENTELLER TEIL
155
Literatur angegebenen Daten überein.116,141,147 Die Ergebnisse sind in Tabelle 8.38
dargestellt.
Tabelle 8.38. Vergleichsversuchsreihe mit Benzaldehyd (1e) als Substrat
Eintrag t [h] Umsatz1) [%] d.r. (syn/anti) 1) Umsatz2) [%] d.r. (syn/anti) 2)
1 1 62 64:36 57 63:37
2 5 68 69:31 69 64:36
3 9 71 63:37 73 63:37
4 24 72 64:36 72 61:39
1)mit Zugabe von PLP
2)ohne Zugabe von PLP; n.d. nicht detektierbar.
8.2.2.8.5 Isolierung von (2S)-12i aus Enzymumsetzung
Die Durchführung der Reaktion erfolgte analog AAV 8. Es wurde Aldehyd
1i (25 mmol, 2.8 mL), 40 mL L-TA aus E. coli (27 U/mL, 44 U/mmol) und
60 mL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M) eingesetzt. Nach 7 h
Reaktionszeit wurde durch Entnahme einer 200 µL-Probe (pH-Wert der
Probe mit HCl auf 1 eingestellt) der Umsatz direkt aus dem Verhältnis
von Glycin zu entstandenem Produkt im 1H-NMR bestimmt. Zur restlichen
Reaktionsmischung wurde so lange HCl (5 M) zugegeben bis pH 1 erreicht war und 5 min
gerührt, um die Enzymreaktion zu stoppen. Anschließend wurde wieder neutralisiert und
Ethanol zugegeben (ca. das neunfache Volumen), um überschüssiges Glycin auszufällen. Das
Gemisch wurde über Nacht im Kühlschrank aufbewahrt, um die Kristallisation zu
vervollständigen. Der Niederschlag, bestehend aus Glycin und ausgefälltem Protein, wurde
abfiltriert und die Lösung am Rotationsverdampfer so lange eingeengt, bis das Produkt
ausfiel. Die Mischung wurde wieder über Nacht kaltgestellt, anschließend filtriert und der
Niederschlag mit EtOH/H2O 80:20 (v/v) gewaschen. Für weitere Fällungen wurde das Filtrat
eingeengt. Bei Fällungen mit geringer Reinheit wurde der Rückstand in EtOH/H2O 1:1 (v/v)
umkristallisiert. Es wurde ein Umsatz von 90% bei einem Diastereomerenverhältnis von d.r.
79:21 (syn/anti) erhalten. Insgesamt wurde eine Ausbeute von 62% (Auswaage 3.3 g) bei
einem Diastereomerenverhältnis von d.r. 78:22 (syn/anti) erzielt. 1H-NMR (400 MHz, D2O) δ
(ppm) = 4.10-4.12 (m, 2H, syn-CHNH2, anti-CHNH2), 5.52 (d, 1H, 3J = 3.5 Hz, anti-CHOH), 5.69
OH
OOH
(2S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
156
(d, 1H, 3J = 3.5 Hz, syn-CHOH), 7.35-7.67 (m, 4H, H-aromat.). Die spektroskopischen Daten
stimmen mit den in der Literatur angegebenen Daten überein.109,147
8.2.2.8.6 Abtrennung von Glycerin mittels Ionenaustauscher
Die Durchführung der Reaktion erfolgte analog AAV 8. Es wurde Aldehyd
1i (2.5 mmol, 2.8 mL) und 10 mL L-TA aus E. coli (11 U/mL, 44 U/mmol)
eingesetzt. Nach 7 h wurde das Reaktionsgemisch für 10 min auf 100°C
erhitzt, um die Enzymreaktion zu stoppen. Nach Abkühlen auf RT wurde
Ethanol zugegeben (ca. das neunfache Volumen), um überschüssiges
Glycin auszufällen. Das Gemisch wurde über Nacht im Kühlschrank aufbewahrt, um die
Kristallisation zu vervollständigen. Der Niederschlag wurde abfiltriert, die Lösung mit HCl
angesäuert (pH 1) und das Lösungsmittel und überschüssige HCl am Rotationsverdampfer
entfernt. Der Feststoff wurde in wenig Wasser aufgenommen und auf 3 g des
Ionenaustauschers Dowex 50W X8 aufgebracht. Durch Eluierung mit 1 M NH3-Lösung wurde
das Produkt nach Entfernen des Lösungsmittels erhalten. Das Diastereomerenverhältnis
betrug d.r. 79:21 (syn/anti) und es war noch 41% Glycin (11) enthalten, sowie Protein.
1H-NMR (400 MHz, D2O) δ (ppm) = 3.56 (s, 2H, CH2-11), 3.66 (s, Verunreinigung durch
Protein), 4.10-4.12 (m, 2H, syn-CHNH2, anti-CHNH2), 5.52 (d, 1H, 3J = 3.5 Hz, anti-CHOH), 5.69
(d, 1H, 3J = 3.5 Hz, syn-CHOH), 7.35-7.67 (m, 4H, H-aromat.). Die spektroskopischen Daten
stimmen mit den in der Literatur angegebenen Daten überein.109
8.2.2.8.7 Trennung der Diastereomere
Die Trennung der racemischen Diastereomere wurde mittels Ionenaustauscher und
Kristallisation untersucht.
OH
OOH
(2S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
157
8.2.2.8.7.1 Trennung von rac-12i mittels Ionenaustauscher
Auf eine Ionenaustauschersäule (Ø = 4 cm, Dowex 50W X8 15 g) wurde
12i (0.48 mmol, 103 mg, d.r. 82:18 syn/anti) als Hydrochlorid aufgebracht
und mit 0.1 M NH3-Lösung eluiert. Die aufgefangen Fraktionen
(Fraktionsvolumen ca. 10 mL) wurden mittels Dünnschicht-
chromatographie auf Produkt untersucht. Dazu wurden SiO2-Platten mit
Fluoreszenzindikator verwendet und das Laufmittel CH2Cl2/MeOH/NH3 75:20:5 benutzt.
Allerdings konnte nur bei 8 Fraktionen schwache Spots (RF = 0.27) erhalten werden, die nicht
unterschieden werden konnten. Nach Entfernen des Lösungsmittels wurde 87 mg 12i mit
einem schlechteren Diastereomerenverhältnis von d.r. 74:26 (syn/anti) erhalten. Frühere
Fraktionen enthielten 6 mg 12i mit einem Diastereomerenverhältins von d.r. 71:29
(syn/anti). Spätere Fraktionen enthielten 16 mg Produkt mit einem
Diastereomerenverhältnis von d.r. 86:14 (syn/anti). 1H-NMR (400 MHz, D2O) δ (ppm) = 4.10-
4.12 (m, 2H, syn-CHNH2, anti-CHNH2), 5.52 (d, 1H, 3J = 3.5 Hz, anti-CHOH), 5.69 (d, 1H, 3J = 3.5
Hz, syn-CHOH), 7.35-7.67 (m, 4H, H-aromat.). Die spektroskopischen Daten stimmen mit den
in der Literatur angegebenen Daten überein.109
8.2.2.8.7.2 Trennung von rac-12i mittels Umkristallisation
12i (0.7 mmol, 150 mg, d.r. 82:18 syn/anti) wurde unter Sieden in
Wasser (6.5 mL) fast vollständig gelöst. Nach Abkühlen auf RT wurde die
Mischung über Nacht in den Kühlschrank gestellt und anschließend
filtriert. Das gewünschte Produkt rac-syn-12i wurde nach Trocknen mit
einer Ausbeute von 43% (Auswaage 65 mg) und einem
Diastereomerenverhältnis von d.r. >25:1 (syn/anti) erhalten. 1H-NMR (400 MHz, D2O) δ
(ppm) = 4.09 (d, 1H, 3J = 3.6 Hz, syn-CHNH2), 5.67 (d, 1H, 3J = 5.6 Hz, syn-CHOH), 7.35-7.65 (m,
4H, H-aromat.). 13C-NMR (100 MHz, D2O) δ (ppm) = 58.46 (syn-CHNH2), 68.51 (syn-CHOH),
127.73, 127.88, 130.17, 130.23, 131.85, 136.66, (C-aromat.), 172.21 (syn-COOH). MS (MALDI,
Matrix: sin) m/z = 216 (M-H)+. Die spektroskopischen Daten stimmen mit den in der Literatur
angegebenen Daten überein.109
OH
OOH
rac-syn-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl
OH
OOH
rac-syn-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
158
8.2.2.8.7.3 Trennung von (2S)-12i mittels Fällung aus Wasser mit Aceton
(2S)-12i (1.5 mmol, 325 mg, d.r. 78:22 syn/anti) wurde in Wasser (20 mL)
gelöst und anschließend so lange Aceton (25 mL) zugegeben bis sich ein
Niederschlag bildete. Die Mischung wurde über Nacht in den
Kühlschrank gestellt und anschließend abfiltriert. Es wurden 134 mg
(41%, d.r. 68:32 syn/anti) Feststoff erhalten. Das Filtrat enthielt 144 mg
L-12i (44%, d.r. 87:13 syn/anti). 1H-NMR (400 MHz, D2O) δ (ppm) = 4.10-4.12 (m, 2H,
syn-CHNH2, anti-CHNH2), 5.52 (d, 1H, 3J = 3.5 Hz, anti-CHOH), 5.69 (d, 1H, 3J = 3.5 Hz,
syn-CHOH), 7.35-7.67 (m, 4H, H-aromat.). Die spektroskopischen Daten stimmen mit den in
der Literatur angegebenen Daten überein.109
8.2.2.1 Allgemeine Arbeitsvorschrift 9 (AAV 9): Racematspaltung
Abbildung 8.23. Racematspaltung
Zu rac-syn-Phenylserin rac-syn-12e oder rac-syn-o-Chlorphenylserin rac-syn-12i (0.05-0.5 M)
wurde Puffer (pH 8, Tris-HCl-Puffer, 0.1 M), L-TA aus E.coli (27 U/mL, 27-540 U/mmol) und
PLP (50 µM) gegeben und 24 h geschüttelt. Nach Zugabe von HCl wurde das Lösungsmittel
abgezogen und der Umsatz mittels 1H-NMR aus dem Verhältnis von Glycin und Phenylserin
bestimmt.
OH
OOH
(2S,3R)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl
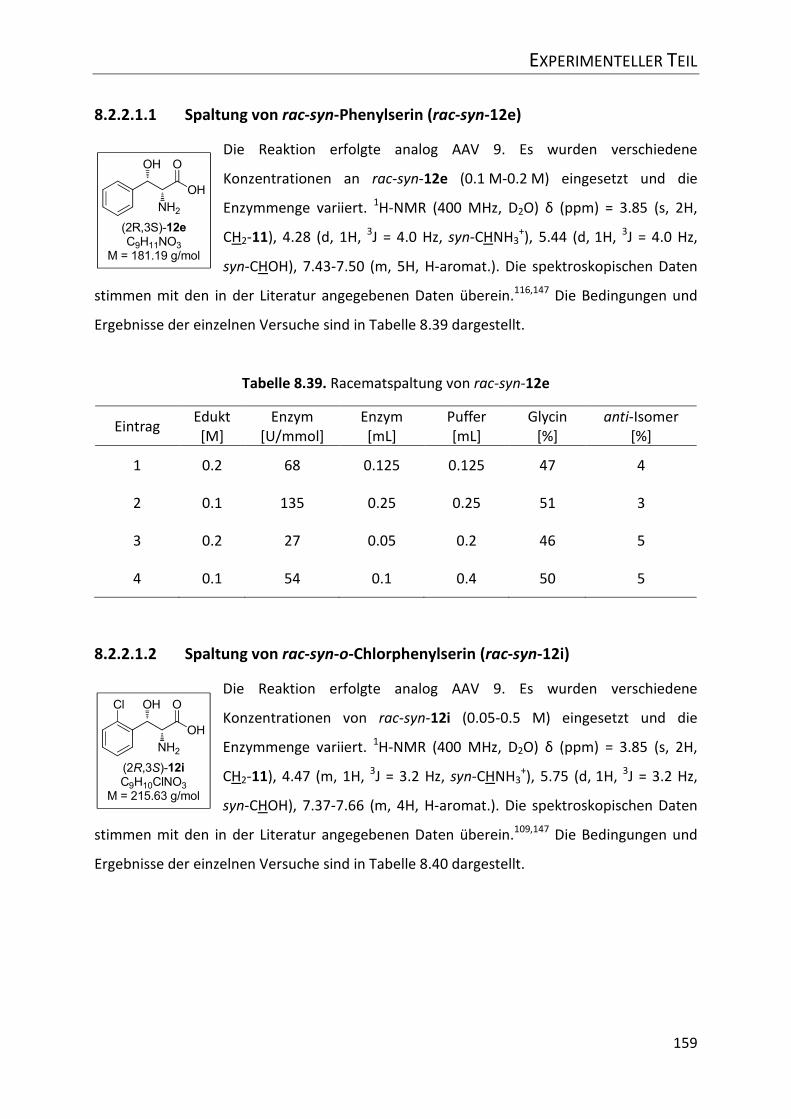
EXPERIMENTELLER TEIL
159
8.2.2.1.1 Spaltung von rac-syn-Phenylserin (rac-syn-12e)
Die Reaktion erfolgte analog AAV 9. Es wurden verschiedene
Konzentrationen an rac-syn-12e (0.1 M-0.2 M) eingesetzt und die
Enzymmenge variiert. 1H-NMR (400 MHz, D2O) δ (ppm) = 3.85 (s, 2H,
CH2-11), 4.28 (d, 1H, 3J = 4.0 Hz, syn-CHNH3+), 5.44 (d, 1H, 3J = 4.0 Hz,
syn-CHOH), 7.43-7.50 (m, 5H, H-aromat.). Die spektroskopischen Daten
stimmen mit den in der Literatur angegebenen Daten überein.116,147 Die Bedingungen und
Ergebnisse der einzelnen Versuche sind in Tabelle 8.39 dargestellt.
Tabelle 8.39. Racematspaltung von rac-syn-12e
Eintrag Edukt
[M] Enzym
[U/mmol] Enzym
[mL] Puffer
[mL] Glycin
[%] anti-Isomer
[%]
1 0.2 68 0.125 0.125 47 4
2 0.1 135 0.25 0.25 51 3
3 0.2 27 0.05 0.2 46 5
4 0.1 54 0.1 0.4 50 5
8.2.2.1.2 Spaltung von rac-syn-o-Chlorphenylserin (rac-syn-12i)
Die Reaktion erfolgte analog AAV 9. Es wurden verschiedene
Konzentrationen von rac-syn-12i (0.05-0.5 M) eingesetzt und die
Enzymmenge variiert. 1H-NMR (400 MHz, D2O) δ (ppm) = 3.85 (s, 2H,
CH2-11), 4.47 (m, 1H, 3J = 3.2 Hz, syn-CHNH3+), 5.75 (d, 1H, 3J = 3.2 Hz,
syn-CHOH), 7.37-7.66 (m, 4H, H-aromat.). Die spektroskopischen Daten
stimmen mit den in der Literatur angegebenen Daten überein.109,147 Die Bedingungen und
Ergebnisse der einzelnen Versuche sind in Tabelle 8.40 dargestellt.
OH
OOH
(2R,3S)-12eC9H11NO3
M = 181.19 g/mol
NH2
OH
OOH
(2R,3S)-12iC9H10ClNO3
M = 215.63 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
160
Tabelle 8.40. Racematspaltung von rac-syn-12i
Eintrag Edukt
[M]
Enzym
[U/mmol]
Enzym
[mL]
Puffer
[mL]
Glycin
[%]
anti-Isomer1)
[%]
1 0.5 54 0.1 0 9 n.b.
2 0.2 135 0.25 0 20 n.b
3 0.1 270 0.5 0 36 n.b
4 0.067 270 0.5 0.25 44 5
5 0.05 270 0.5 0.5 48 5
6 0.05 540 1.0 0 50 5
7 0.05 54 0.1 0.9 46 5
1) n.b. nicht bestimmt
8.2.2.1.3 Spaltung von rac-syn-Phenylserin (rac-syn-12e) im 25 mL-Maßstab
Die Reaktion erfolgte analog AAV 9. Zu rac-syn-Phenylserin (rac-syn-12e,
2.5 mmol, 0.1 M) wurde 20 mL Puffer (pH 8, Tris-HCl-Puffer, 0.1 M), L-TA
aus E.coli (5 mL, 27 U/mL, 54 U/mmol) und PLP (50 µM) gegeben und
20 h gerührt. Nach Zugabe von HCl, um den pH-Wert auf 1 zu bringen,
wurde das Lösungsmittel im Vakuum entfernt und der Umsatz mittels
1H-NMR-Spektroskopie aus dem Verhältnis von Glycin (11) zu Phenylserin (12e) bestimmt.
1H-NMR (400 MHz, D2O) δ (ppm) = 3.85 (s, 2H, CH2-11), 4.28 (d, 1H, 3J = 4.0 Hz, syn-CHNH3+),
5.44 (d, 1H, 3J = 4.0 Hz, syn-CHOH), 7.43-7.50 (m, 5H, H-aromat.). Die spektroskopischen
Daten stimmen mit den in der Literatur angegebenen Daten überein.116,147 Es wurde ein
Umsatz von 50% erhalten und 4% anti-Isomer nachgewiesen. Zur Isolierung des Produkts
wurde der Rückstand in Wasser aufgenommen und neutralisiert. Das ausgefallen Protein
wurde abfiltriert und die Mutterlauge eingeengt. Es entstand kein Niederschlag und somit
konnte das gewünschte Produkt nicht isoliert werden.
OH
OOH
(2R,3S)-12eC9H11NO3
M = 181.19 g/mol
NH2

EXPERIMENTELLER TEIL
161
8.2.2.1.4 Spaltung von rac-syn-o-Chlorphenylserin (rac-syn-12i) im
50 mL-Maßstab
Die Reaktion erfolgte analog AAV 9. Zu rac-syn-o-Chlorphenylserin
(rac-syn-12i, 2.5 mmol, 0.05 M) wurde 45 mL Puffer (pH 8, Tris-HCl-
Puffer, 0.1 M), L-TA aus E.coli (5 mL, 27 U/mL, 54 U/mmol) und PLP
(50 µM) gegeben und 20 h gerührt. Nach Zugabe von HCl wurde das
Lösungsmittel im Vakuum entfernt und der Umsatz mittels 1H-NMR-
Spektroskopie aus dem Verhältnis von Glycin (11) und o-Chlorphenylserin (12i) bestimmt.
1H-NMR (400 MHz, D2O) δ (ppm) = 3.85 (s, 2H, CH2-11), 4.47 (m, 1H, 3J = 3.2 Hz, syn-CHNH3+),
5.75 (d, 1H, 3J = 3.2 Hz, syn-CHOH), 7.37-7.66 (m, 4H, H-aromat.). Die spektroskopischen
Daten stimmen mit den in der Literatur angegebenen Daten überein. 109,147 Es wurde ein
Umsatz von 50% erhalten und 4% anti-Isomer nachgewiesen. Zur Isolierung des Produkts
wurde der Rückstand in Wasser aufgenommen und neutralisiert. Das ausgefallene Protein
wurde durch Filtration entfernt und die Mutterlauge eingeengt. Der gebildete Niederschlag
wurde abfiltriert und das gewünschte Produkt in einer Ausbeute von 15% und einer Reinheit
von 70% erhalten. 1H-NMR (400 MHz, D2O) δ (ppm) = 4.10-4.12 (m, 2H, syn-CHNH2,
anti-CHNH2), 5.52 (d, 1H, 3J = 3.5 Hz, anti-CHOH), 5.69 (d, 1H, 3J = 3.5 Hz, syn-CHOH),
7.35-7.67 (m, 4H, H-aromat.). Die spektroskopischen Daten stimmen mit den in der Literatur
angegebenen Daten überein.109
8.2.3 Epoxidsynthese ausgehend von β-Hydroxy-α-aminosäuren 12
8.2.3.1 Allgemeine Arbeitsvorschrift 11 (AAV 11): Halohydrinsynthese125
Abbildung 8.24. Halohydrinsynthese
Das entsprechende Phenylserinderivat 12 (1.0-5.6 mmol) wurde in 6 M HCl (3.2-12 mL)
gelöst. Die Mischung wurde auf ca. -10°C gekühlt und es wurde langsam NaNO2 (2.5 Äq.)
OH
OOH
(2R,3S)-12iC9H10ClNO3
M = 215.63.19 g/mol
NH2
Cl

EXPERIMENTELLER TEIL
162
zugegeben. Anschließend wurde 16 h bei -10°C gerührt und mit MTBE extrahiert (4 x 10 mL).
Die vereinigten organischen Phasen wurden über MgSO4 getrocknet und das Lösungsmittel
im Vakuum entfernt. Das erhaltene Rohprodukt wurde umkristallisiert.
8.2.3.1.1 Synthese von rac-syn-3-Hydroxy-3-phenylpropansäure (rac-syn-20e)
Die Synthese erfolgte analog AAV 11. Es wurde 5.6 mmol rac-syn-
Phenylserin (rac-syn-12e, 1.0 g) eingesetzt. Das Rohprodukt wurde als
gelbes Öl erhalten und in CH2Cl2/Hexan umkristallisiert. Das gewünschte
Produkt rac-syn-20e wurde mit einer Ausbeute von 29% (Auswaage
326 mg) isoliert. 1H-NMR (300 MHz, CDCl3) δ (ppm) = 4.53 (1H, d, 3J =
5.5 Hz, syn-CHCl), 5.23 (1H, d, 3J = 5.5 Hz, syn-CHOH) 7.33-7.40 (m, 5H, H-aromat.). 13C-NMR
(100 MHz, CDCN3) δ (ppm) = 64.36 (CHCl), 74.52 (CHOH), 127.64, 129.10, 129.20, 140.99
(C-aromat.), 169.21 (COOH). Die spektroskopischen Daten stimmen mit den in der Literatur
angegebenen Daten überein.148
8.2.3.1.2 Synthese von (2S)-2-Chlor-3-(2-chlorphenyl)-3-hydroxypropansäure
((2S)-20i)
Die Synthese erfolgte analog AAV 11. Es wurde 1.5 mmol
o-Chlorphenylserin ((2S)-12i, d.r. 76:24 syn/anti, 323 mg) aus der
enzymatischen Umsetzung in 5 mL HCl (5 M) gelöst und 2.5 Äq. NaNO2
zugegeben. Das Rohprodukt wurde als gelbes Öl erhalten. Der Rückstand
wurde in MTBE aufgenommen und durch Zugabe von 1 M ethanolischer
NaOH-Lösung ausgefällt. Der erhaltene Niederschlag wurde in Wasser aufgenommen und
mit HCl auf pH 1 angesäuert. Die wässrige Phase wurde dreimal mit CH2Cl2 (4 x 10 mL)
extrahiert und über MgSO4 getrocknet. Der nach Entfernen des Lösungsmittels erhaltene
Rückstand wurde nochmals in Aceton/Petrolether umkristallisiert. Das gewünschte Produkt
(2S)-20i wurde als gelblicher Feststoff mit einer Ausbeute von 23% (Auswaage 81 mg) und
mit einem Diastereomerenverhältnis d.r. >90:10 (syn/anti) erhalten. 1H-NMR (400 MHz,
Aceton-d6) δ (ppm) = 4.90 (d, 1H, 3J = 3.4 Hz, syn-CHCl), 5.81 (d, 1H, 3J = 3.4 Hz, syn-CHOH)
7.26-7.41 (m, 3H, H-aromat.), 7.73-7.76 (m, 1H, H-aromat.); 13C-NMR (100 MHz, Aceton-d6) δ
OH
OOH
rac-syn-20eC9H9ClO3
M = 200.62 g/mol
Cl

EXPERIMENTELLER TEIL
163
(ppm) = 62.80 (CHCl), 70.95 (CHOH), 127.64, 129.89, 130.14, 131.98, 139.10 (C-aromat.),
168.75 (COOH); Elementaranalyse (C9H8Cl2O3): Berechnet: C: 45.99, H: 3.43. Gefunden:
C: 46.16, H: 3.86. Die vollständige Charakterisierung erfolgte über das Epoxid.
8.2.3.2 Allgemeine Arbeitsvorschrift 12 (AAV 12): Epoxidsynthese149
Abbildung 8.25. Epoxidsynthese über zwei Stufen
Chlorphenylserin 12i (1.0-1.4 mmol) wurde in 6 M HCl (3.2-4.5 mL) gelöst. Die Mischung
wurde auf ca. -10°C gekühlt und es wurde langsam NaNO2 (2.5 Äq.) zugegeben.
Anschließend wurde 16 h bei -10°C gerührt und mit MTBE extrahiert (4 x 10 mL). Die
vereinigten organischen Phasen wurden über MgSO4 getrocknet und das Lösungsmittel im
Vakuum entfernt. Der Umsatz und das Diastereomerenverhältnis wurden mittels 1H-NMR-
Spektroskopie bestimmt. Anschließend wurde das Halohydrin 20i als Rohprodukt eingesetzt.
20i (0.7-1.0 mmol) wurde in 12 mL THF (abs.) gelöst und langsam t-BuOK (2 Äq.) in t-BuOH
(7 mL) zugegeben. Nach 16 h rühren wurde das Lösungsmittel im Vakuum entfernt.
Anschließend wurde Wasser zugegeben, auf pH 3 angesäuert (1 M HCl) und mit CH2Cl2
(4 x 10 mL) extrahiert. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet
und das Lösungsmittel im Vakuum entfernt. Anschließend wurde der Umsatz und das
Diastereomerenverhältnis mittels 1H-NMR-Spektroskopie bestimmt. Zur Aufarbeitung wurde
das Rohprodukt in MTBE aufgenommen und durch Zugabe von ethanolischer NaOH-Lösung
(1 M) ausgefällt. Der Niederschlag wurde abfiltiriert und mit MTBE gewaschen. Nach
Neutralisation wurde das gewünschte Produkt erhalten.

EXPERIMENTELLER TEIL
164
8.2.3.2.1 Synthese von rac-syn-3-(2-Chlorphenyl)oxiran-2-carbonsäure
(rac-syn-21i)
Die Synthese erfolgte analog AAV 12. Es wurde rac-syn-
o-Chlorphenylserin (rac-syn-12i, 1 mmol, 214 mg) in 3.2 mL HCl (5 M)
gelöst und 2.5 Äq. NaNO2 zugegeben. Aus dem Rohprodukt wurde
mittels 1H-NMR-Spektroskopie und der Auswaage ein Umsatz von 74%
bestimmt. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.90 (d, 1H, 3J =
3.4 Hz, syn-CHCl), 5.81 (d, 1H, 3J = 3.4 Hz, syn-CHOH), 7.26-7.41 (m, 3H, H-aromat.), 7.73-7.76
(m, 1H, H-aromat.). Die spektroskopischen Daten stimmen mit den Daten aus Abschnitt
8.2.3.1.2 überein. Das Rohprodukt wurde direkt zum Ringschluss umgesetzt. Das Halohydrin
rac-syn-21i (0.7 mmol) wurde in THF (12 mL) gelöst und t-BuOK (2 Äq.) in t-BuOH (7 mL)
zugegeben. Es wurde ein vollständiger Umsatz erhalten. Das saubere Produkt konnte ohne
weitere Umkristallisation nach ethanolischer Fällung als gelblicher Festoff in einer Ausbeute
von 34% (Auswaage 68 mg) isoliert werden. 1H-NMR (400 MHz, Aceton-d6) δ (ppm) = 4.02 (d,
1H, 3J = 4.6 Hz, CHCOOH), 4.45 (d, 1H, 3J = 4.6 Hz, CHAr), 7.29-7.50 (m, 4H, H-aromat.); 13C-
NMR (100 MHz, Aceton-d6) δ (ppm) = 55.5, 56.03 (CH), 127.25, 129.52, 129.63, 130.42,
132.60, 133.52 (C-aromat.), 167.53 (COOH); MS (EI) m/z = 198 (M)+; IR ṽ (cm-1): 2979, 1711,
1480, 1249, 1061, 935, 757; Elementaranalyse (C9H7ClO3) Berechnet: C: 54.43, H: 3.55.
Gefunden: C: 53.76, H: 3.48; Schmelzpunkt: 112°C.
8.2.3.2.2 Synthese von (2R)-3-(2-Chlorphenyl)oxiran-2-carbonsäure ((2R)-21i)
Die Synthese erfolgte analog AAV 12. Es wurde 1.4 mmol
(2S)-o-Chlorphenylserin ((2S)-12i, d.r. 78:22 syn/anti, 285 mg) aus der
enzymatischen Umsetzung in 4.5 mL HCl (5 M) gelöst und 2.5 Äq.
NaNO2 zugegeben. Aus dem Rohprodukt wurden mittels 1H-NMR-
Spektroskopie und der Auswaage ein Umsatz von 70% und ein
Diastereomerenverhältnis von d.r. 80:20 (syn/anti) bestimmt. 1H-NMR (400 MHz, Aceton-d6)
δ (ppm) = 4.53 (d, 1H, 3J = 8.5 Hz, anti-CHCl), 4.90 (d, 1H, 3J = 3.4 Hz, syn-CHCl), 5.58 (d, 1H,
3J = 8.7 Hz, anti-CHOH), 5.81 (d, 1H, 3J = 3.4 Hz, syn-CHOH), 7.27-7.76 (m, 4H, H-aromat.). Die
spektroskopischen Daten stimmen mit den Daten aus Abschnitt 8.2.3.1.2 überein. Das
Rohprodukt wurde direkt zum Ringschluss umgesetzt. Das Halohydrin (2S)-21i (1 mmol)

EXPERIMENTELLER TEIL
165
wurde in THF (12 mL) gelöst und t-BuOK (2 Äq.) in t-BuOH (7 mL) zugegeben. Es wurde ein
vollständiger Umsatz erzielt. Bei der Fällung aus MTBE mit NaOH wurde in einer 1. Fällung
sauberes Produkt in einer Ausbeute von 30% (Auswaage 83 mg) und einem
Diastereomerenverhältnis von d.r. 82:18 (syn/anti) erhalten. Zum Filtrat wurde anschließend
nochmal ethanolische NaOH (1 M, 0.5 mL) und MTBE (5 mL) gegeben. Die erhaltene
2. Fällung enthielt das gewünschte Produkt (2R)-21i in einer Ausbeute von 22% (Auswaage
61 mg) und einem Diastereomerenverhältnis von d.r. 95:5 (syn/anti). Es wurde eine
Gesamtausbeute von 52% erzielt. Der ee-Wert des syn-Isomers betrug >99% (OJ-H-Säule,
Hexan/i-PrOH/FA 98:2:0.1, v/v, flow 0.5 mL/min, 230 nm, tr (2R,3R) = 53.18 min). 1H-NMR
(400 MHz, Aceton-d6) δ (ppm) = 3.50 (d, 1H, 3J = 1.8 Hz, anti-CHCOOH), 4.02 (d, 1H, 3J = 4.8
Hz, syn-CHCOOH), 4.37 (d, 1H, 3J = 1.8 Hz, anti-CHAr), 4.46 (d, 1H, 3J = 4.6 Hz, CHAr), 7.29-
7.50 (m, 4H, H-aromat.). Die spektroskopischen Daten stimmen mit denen des Racemats
überein (vgl. Abschnitt 8.2.3.2.1).

LITERATURVERZEICHNIS
166
9. Literaturverzeichnis
1 T. Laue, A. Plagens, Namen- und Schlagwortreaktionen der organischen Chemie,
5. Auflage, B. G. Teubner Verlag, Wiesbaden, 2006.
2 J. Clayden, N. Greeves, S. Warren, P. Wothers, Organic Chemistry, Oxford University
Press, New York, 2001.
3 R. Mahrwald, Modern Aldolreaction Vol. 1 and 2, Wiley-VHC, Weinheim, 2004.
4 T. D. Machajewski, C.-H. Wong, Angew. Chem. 2000, 112, 1406-1430; Angew. Chem.
Int. Ed. 2000, 39, 1352-1375.
5 T. Mukaiyama, K. Banno, K. Narasaka, J. Am. Chem. Soc. 1974, 96, 7503-7509.
6 S. E. Denmark, G. L. Beutner, T. Wynn, M. D. Eastgate, J. Am. Chem. Soc. 2005, 127,
3774-3789.
7 Y. M. A. Yamada, N. Yoshikawa, H. Sasai, M. Shibasaki, Angew. Chem. 1997, 109, 1942-
1945; Angew. Chem. Int. Ed. 1997, 36, 1871-1873.
8 B. M. Trost, H. Ito, J. Am. Chem. Soc. 2000, 122, 12003-12004.
9 A. Berkessel, H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to
Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005.
10 U. Eder, G. Sauer, R. Wiechert, Angew. Chem. 1971, 13, 492-493; Angew. Chem. Int. Ed.
1971, 10, 496-497.
11 N. List, R. A. Lerner, C. F. Barbas III, J. Am. Chem. Soc. 2000, 122, 2395-2396.
12 G. Guillena, C. Nájera, D. J. Ramón, Tetrahedron: Asymmetry 2007, 18, 2249-2293.
13 H. Gröger, J. Wilken, Angew. Chem. 2001, 113, 545-548; Angew. Chem. Int. Ed. 2000,
40, 529-532.
14 S. Bahmanyar, K. N. Houk, J. Am. Chem. Soc. 2001, 123, 11273-11283.
15 M. B. Schmid, K. Zeitler, R. M. Gschwind, Angew. Chem. 2010, 122, 5117-5123; Angew.
Chem. Int. Ed. 2010, 49, 4997-5003.
16 S. M. Dean, W. A. Greenberg, C.-H. Wong, Adv. Synth. Catal. 2007, 349, 1308-1320.
17 A. K. Samland, G. A. Sprenger, Appl. Microbiol. Biotechnol. 2006, 71, 253-264.
18 K. Faber, Biotransformations in Organic Chemistry, 5. Auflage, Springer-Verlag, Berlin,
2004, 273-288.

LITERATURVERZEICHNIS
167
19 K. Fesko, M. Uhl, J. Steinreiber, K. Gruber, H. Griengl, Angew. Chem. 2010, 122, 125-
128; Angew. Chem. Int. Ed. 2010, 49, 121-124.
20 K. Fesko, L. Giger, D. Hilvert, Bioorg. Med. Chem. Lett. 2008, 18, 5987-5990.
21 a) S. Kuroda, H. Nozaki, K. Watanabe, K. Yokozeki, Y. Imabayashi, Pat. Appl.
EP1882737A1, 2008; b) H. Nozaki, S. Kuroda, K. Watanabe, K. Yokozeki, J. Mol. Catal. B:
Enzym. 2009, 59, 237-242.
22 R. Mahrwald, Modern Aldolreaction Vol. 1, Wiley-VHC, Weinheim, 2004, 209-210.
23 C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B.-S. Yang, D. a. Ende, J. Baird,
C. Bertsch, R. E. Hannah, P. Dell’Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V.
Massonneau, J. Manley, Org. Process Res. Dev 2011, 15, 900-911.
24 H.-P. Meyer, Org. Proc. Res. Develop. 2011, 15, 180-188.
25 M. Braun, O. Teichert, A. Zweck, Übersichtsstudie: Biokatalyse in der industriellen
Produktion (Hersg: Zukünftige Technologien Consulting der VDI Technologiezentrum
GmbH), Düsseldorf, 2006.
http://www.vdi.de/fileadmin/vdi_de/redakteur_dateien/kfbt_dateien/Biokatalyse-
UEbersichtsstudie.pdf (04.10.2011)
26 K. Sanderson, Nature 2011, 469, 18-20.
27 P. Anastas, N. Eghbali, Chem. Soc. Rev. 2010, 39, 301-312.
28 S. D. Rychnovsky, Chem. Rev. 1995, 95, 2021-2040.
29 N. Dückers, K. Baer, S. Simon, H. Gröger, W. Hummel, Appl. Microbiol. Biotechnol.
2010, 88, 2751-2758.
30 S. E. Bode, M. Wohlberg, M. Müller, Synthesis 2006, 4, 557-588.
31 K. Baer, Diplomarbeit, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2008.
32 a) Y. Belkon, A. G. Bulychev, S. V. Vitt, Y. T. Struchkov, A. S. Batsanov, T. V. Timofeeva,
V. A. Tsyryapkin, M. G. Ryzhov, L. A. Lysova, V. I. Bakhmutov, V. M. Belikov, J. Am.
Chem. Soc. 1985, 107, 4252-4259; b) D. A. Evans, A. E. Weber, J. Am. Chem. Soc. 1986,
108, 6757-6761; c) C. M. Gasparski, M. J. Miller, Tetrahedron 1991, 47, 5367-5378;
d) M. Horikawa, J. Busch-Petersen, E. J. Corey, Tetrahedron Lett. 1999, 40, 3843-3846;
e) T. Ooi, M. Taniguchi, M. Kameda, K. Maruoka, Angew. Chem. 2002, 114, 4724-4726;
Angew. Chem. Int. Ed. 2002, 41, 4542-4544; f) R. Thayumanavan, F. Tanaka, C. F.
Barbas III, Org. Lett. 2004, 6, 3541-3544; g) J. Kobayashi, M. Nakamura, Y. Mori, Y.

LITERATURVERZEICHNIS
168
Yamashita, S. J. Kobayashi, J. Am. Chem. Soc. 2004, 126, 9192-9193; h) M. C. Willis, G.
A. Cutting, V. J. D. Piccio, M. J. Durbin, M. P. John, Angew. Chem. 2005, 117, 1567-
1569; Angew. Chem. Int. Ed. 2005, 44, 1543-1545.
33 L. Coppi, C. Giordano, A. Longoni, S. Panossian, Chirality in Industry Vol. 2, 2. Auflage,
Wiley, Chichester, 1997, 353-362.
34 A. A. Volmer, E. M. Carreira, ChemBioChem 2010, 11, 778-781.
35 V. Paquet, A. A. Volmer, E. M. Carreira, Chem. Eur. J. 2008, 14, 2465-2481.
36 A. Kleemann, J. Engels, B. Kutscher, D. Reichert, Pharmaceutical Substances: Syntheses,
Patents, Applications, 4. Auflage, Thieme, Stuttgart, 2001.
37 D. A. Evans, K. T. Chapman, E. M. Carreira, J. Am. Chem. Soc. 1988, 110, 3560-3578.
38 M. Kitamura, T. Ohkuma, S. Inoue, N. Sayo, H. Kumobayashi, S. Akutagawa, T. Ohta, H.
Takaya, R. Noyori, J. Am. Chem. Soc. 1988, 110, 629-631.
39 R. Noyori, T. Ohkuma, Angew. Chem. 2001, 113, 40-75; Angew. Chem. Int. Ed. 2001, 40,
40-73.
40 K. Ahmed, S. Koul, S. C. Tanejy, A. P. Singh, M. Kapoor, Riyaz-ul-Hassan, V. Verma, G. N.
Qazi, Tetrahedron: Asymmetry 2004, 15, 1685-1692.
41 L. M. Geary, P. G. Hultin, Tetrahedron: Asymmetry 2009, 20, 131-173.
42 B. M. Trost, C. S. Brindle, Chem. Soc. Rev. 2010, 39, 1600-1632.
43 M. Raj, V. K. Singh, Chem. Commun. 2009, 6687-6703.
44 a) T. J. Dickerson, K. D. Janda, J. Am. Chem. Soc. 2002, 124, 3220-3221; b) T. J.
Dickerson, T. Lovell, M. M. Meijler, L. Noodleman, K. D. Janda, J. Org. Chem. 2004, 69,
6603-6609; c) C. J. Rogers, T. J. Dickerson, K. D. Janda, Tetrahedron 2006, 62, 352-356.
45 C. Wu, X. Fu, S. Li, Eur. J. Org. Chem. 2011, 1291-1299.
46 F.-Z. Peng, Z.-H. Shao, X.-W. Pu, H.-B. Zhang, Adv. Synth. Catal. 2008, 350, 2199-2204.
47 S. S. V. Ramasastry, K. Albertshofer, N. Utsumi, C. F. Barbas III, Org. Lett. 2008, 10,
1621-1624.
48 M. Raj, G. S. Parashari, V. K. Singh, Adv. Synth. Catal. 2009, 351, 1284-1288.
49 M. Raj, V. Maya, S. K. Ginotra, V. K. Singh, Org. Lett. 2006, 8, 4097-4099.
50 M.-K. Zhu, X.-Y. Xu, L. Z. Gong, Adv. Synth. Catal. 2008, 350, 1390-1396.
51 V. Maya, M. Raj, V. K. Singh, Org. Lett. 2007, 9, 2593-2595.
52 G. Antranikian, S. Heiden, Nachr. a. d. Chem. 2006, 54, 1202-1206.

LITERATURVERZEICHNIS
169
53 R. Wohlgemuth, Curr. Opin. Biotechnol. 2010 , 21, 1-12.
54 M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer, T. Zelinski, Angew.
Chem. 2004, 116, 806-843; Angew. Chem. Int. Ed. 2004, 43, 788-824.
55 R. N. Patel, Coord. Chem. Rev. 2008, 252, 659-701.
56 H. Gröger, S. Borchert, M. Kraußer, W. Hummel, Encyclopedia of Industrial
Biotechnology: Bioprocess, Bioseparation and Cell Technology, 7. Auflage, John Wiley &
Sons, Hoboken, New Jersey, 2010, Volume 3, 2094-2110.
57 a) Y. Yasohara, N. Kizaki, J. Hasegawa, M. Wada, M. Kataoka, S. Shimizu, Tetrahedron:
Asymmetry 2001, 12, 1713-1718; b) N. Kizaki, Y. Yasohara, J. Hasegawa, M. Wada, M.
Kataoka, S. Shimizu, Appl. Microbiol. Biotechnol. 2001, 55, 590-595; c) M. Kataoka, L. P.
S. Rohani, K. Yamamoto, M. Wada, H. Kawabata, K. Kita, H. Yanase, S. Shimizu, Appl.
Microbiol. Biotechnol. 1997, 48, 699-703.
58 M. Müller, Angew. Chem. 2005, 117, 366-369; Angew. Chem. Int. Ed. 2005, 44, 362-
365.
59 M. Wolber, W. Hummel, M. Müller, Chem. Eur. J. 2001, 7, 4562-4571.
60 M. Wolberg, M. V. Filho, S. Bode, P. Geilenkirchen, R. Feldmann, A. Liese, W. Hummel,
M. Müller, Bioprocess Biosyst. Eng. 2008, 31, 183-191.
61 W. Kroutil, H. Mang, K. Edegger, K. Faber, Curr. Opin. Chem. Biol. 2004, 8, 120-126.
62 H. Maid, P. Böhm, S. M. Huber, W. Bauer, W. Hummel, N. Jux, H. Gröger, Angew.
Chem. 2011, 123, 2445-2448; Angew. Chem. Int. Ed. 2011, 50, 2397-2400.
63 H. Gröger, F. Chamouleau, N. Orologas, C. Rollmann, K. Drauz, W. Hummel, A.
Weckbecker, O. May, Angew. Chem. 2006, 118, 5806-5809; Angew. Chem. Int. Ed.
2006, 45, 5677-5681.
64 J. Parkot, H. Gröger, W. Hummel, Appl. Microbiol. Biotechnol. 2010, 86, 1813-1820.
65 I. Lavandera, A. Kern, B. Ferreira-Silva, A. Glieder, S. de Wildeman, W. Kroutil, J. Org.
Chem. 2008, 73, 6003-6005.
66 H. Zhu, L. Hua, E. Biehl, Adv. Synth. Catal. 2009, 351, 583-588.
67 M. T. Reetz, Angew. Chem. 2011, 123, 144-182; Angew. Chem. Int. Ed. 2011, 50, 138-
174.
68 A. Bruggink, R. Schoevaart, T. Kieboom, Org. Proc. Res. Dev. 2003, 7, 622-640.
69 O. Pàmies, J.-E. Bäckvall, Chem. Rev. 2003, 103, 3247-3261.

LITERATURVERZEICHNIS
170
70 H. Pellissier, Tetrahedron 2008, 64, 1563-1601.
71 M. Edin, J. Steinreiber, J.-E. Bäckvall, PNAS 2004, 101, 5761-5766.
72 C. Paizs, A. Katona, J. Retey, Eur. J. Org. Chem. 2006, 1113-1116.
73 C. Simons, U. Hanefeld, I. W. C. E. Arends, T. Maschmeyer, R. A. Sheldon, Top. Catal.
2006, 40, 35-44.
74 A.Prastaro, P. Ceci, E. Chiancone, A. Boffi, R. Cirilli, M. Colone, G. Fabrizi, A. Stringaro,
S. Cacchi, Green Chem. 2009, 11, 1929-1932.
75 M. Kraußer, T. Winkler, N. Richter, S. Dommer, A. Fingerhut, W. Hummel, H. Gröger,
ChemCatChem 2011, 3, 293-296.
76 M. Krausser, W. Hummel, H. Gröger, Eur. J. Org. Chem. 2007, 5175-5179.
77 E. Burda, W. Hummel, H. Gröger, Angew. Chem. 2008, 120, 9693-9696; Angew. Chem.
Int. Ed. 2008, 47, 9551-9554.
78 M. Sugiyama, Z. Hong, P.-H. Liang, S. M. Dean, L. J. Whalen, W. A. Greenberg, C.-H.
Wong, J. Am. Chem. Soc. 2007, 129, 14811-14817.
79 a) (S)-Alkoholdehydrogenase aus Rhodococcus sp.: kommerzielles Produkt
(Produktnummer 1.1.030) der evocatal GmbH, Merowinger Platz 1a, 40225 Düsseldorf
(Internet: http://www.evocatal.com); b) (R)-Alkoholdehydrogenase aus Lactobacillus
kefir: A. Weckbecker, W. Hummel, Biocatal. Biotransform. 2006, 24, 380-389.
80 Die in diesem Abschnitt beschriebenen und von mir erstellten Arbeiten entstanden im
Rahmen einer arbeitskreisinternen Zusammenarbeit mit Marina Kraußer und Giuseppe
Rulli.
81 G. Rulli, Diplomarbeit, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2009; b) G.
Rulli, geplante Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg.
82 G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel, H. Gröger, Angew. Chem.
2011, 123, 8092-8095; Angew. Chem. Int. Ed. 2011, 50, 7944-7947.
83 K. Baer, M. Kraußer, E. Burda, W. Hummel, A. Berkessel, H. Gröger, Angew. Chem.
2009, 121, 9519-9522; Angew. Chem. Int. Ed. 2009, 48, 9355-9358.
84 M. T. Madigan, J. M. Martinko, Brock Mikrobiologie, 11. Auflage, Pearson Studium,
München, 2006, 130-132.
85 M. C. Willis, G. A. Cutting, V. J.-D. Piccio, M. J. Durbin, M. P. John, Angew. Chem. 2005,
117, 1567-1569; Angew. Chem. Int. Ed. 2005, 44, 1543-1545.

LITERATURVERZEICHNIS
171
86 a) T. Miura, M. Fujii, K. Shingu, I. Koshimizu, J. Naganoma, T. Kajimoto, Y. Ida,
Tetrahedron Lett. 1998, 39, 7313-7316; b) M. Fujii, T. Miura, T. Kajimoto, Y. Ida, Synlett
2000, 1046-1048; c) T. Tanaka, M. Ozawa, T. Miura, T. Inazu, S. Tsuji, T. Kajimoto,
Synlett 2002, 9, 1487-1490; d) T. Nishiyama, S. S. Mohile, T. Kajimoto, M. Node,
Heterocycles 2007, 71, 1397-1405.
87 T. Nishiyama, T. Kajimoto, S. S. Mohile, N. Hayama, T. Otsuda, M. Ozeki, M. Node,
Tetrahedron: Asymmetry 2009, 30, 230-234.
88 T. Tanaka, C. Tsuda, T. Miura, T. Inazu, S. Tsuji, S. Nishihara, M. Hisamatsu, T. Kajimoto,
Synlett 2004, 2, 243-246.
89 C. Nájera, J. M. Sansano, Chem. Rev. 2007, 107, 4584-4671.
90 a) K. Makino, T. Goto, Y. Hiroki, Y. Hamada, Angew. Chem. 2004, 116, 900-902; Angew.
Chem. Int. Ed. 2004, 43, 882-884; b) K. Makino, Y. Hiroki, Y. Hamada, J. Am. Chem. Soc.
2005, 127, 5784-5785; c) K. Makino, T. Fujii, Y. Hamada, Tetrahedron Asymmetry 2006,
17, 481–485; d) T. Maeda, K. Makino, M. Iwasaki, Y. Hamada, Chem. Eur. J. 2010, 16,
11954-11962.
91 D. J. Aldous, M. G. B. Drew, W. N. Draffin, E. M.-N. Hamelin, L. M. Harwood, S.
Thurairatnama, Synthesis 2005, 19, 3271–3278.
92 M. Alonso, A. Riera, Tetrahedron Asymmetry 2005, 16, 3908-3912.
93 D. Crich, A. Banerjee, J. Org. Chem. 2006, 71, 7106-7109.
94 a) D. A. Evans, A. E. Weber, J. Am. Chem. Soc. 1986, 108, 6757-6761; b) D. A. Evans, E.
B. Sjogren, A. E. Weber, R. E. Conn, Tetrahedron Lett. 1987, 28, 39-42.
95 N. Yoshikawa, M. Shibasaki, Tetrahedron 2002, 58, 8289-8298.
96 J. Kobayashi, M. Nakamura, Y. Mori, Y. Yamashita, S. Kobayashi, J. Am. Chem. Soc.
2004, 126, 9192-9193.
97 X. Chen, Y. Zhu, Z. Qiao, M. Xie, L. Lin, X. Liu, X. Feng, Chem. Eur. J. 2010, 16, 10124-
10129.
98 C. M. Gasparski, M. J. Miller, Tetrahedron 1991, 41, 5367-5318.
99 M. Horikawa, J. Busch-Petersen, E. J. Corey, Tetrahedron Lett. 1999, 40, 3843-3846.
100 R. Thayumanavan, F. Tanaka, C. F. Barbas III, Org. Lett. 2004, 6, 3541-3544.
101 S. Mettath, G. S. C. Srikanth, B. S. Dangerfield, S. L. Castle, J. Org. Chem. 2004, 69,
6489-6492.

LITERATURVERZEICHNIS
172
102 a) T. Ooi, M. Taniguchi, M. Kameda, K. Maruoka, Angew. Chem. 2002, 114, 4724-4726;
Angew. Chem. Int. Ed. 2002, 41, 4542-4544; b) T. Ooi, M. Taniguchi, K. Doda, K.
Maruoka, Adv. Synth. Catal. 2004, 346, 1073-1076.
103 J.-Q. Liu, T. Dairi, N. Itoh, M. Kataoka, S. Shimizu, H. Yamada, J. Mol. Catal. B: Enzym.
2000, 10, 107-115.
104 P. Clapés, W.-D. Fessner, G. A. Sprenger, A. K. Samland, Curr. Opin. Chem. Biol. 2009,
14, 1-14.
105 T. Kimura, V. P. Vassilev, G.-J. Shen, C.-H. Wong, J. Am. Chem. Soc. 1997, 119, 11734-
11742.
106 K. Fesko, C. Reisinger, J. Steinreiber, H. Weber, M. Schürmann, H. Griengl, J. Mol. Catal.
B: Enzym. 2006, 52-53, 19-26.
107 J. Q. Liu, M. Odani, T. Dairi, N. Itoh, S. Shimizu, H. Yamada, Appl. Microbiol. Biotechnol.
1999, 51, 586-591.
108 J. Q. Liu, S. Ito, T. Dairi, N. Itoh, S. Shimizu, H. Yamada, Appl. Microbiol. Biotechnol.
1998, 49, 702-708.
109 J. Steinreiber, K. Fesko, C. Reisinger, M. Schürmann, F. v. Assema, F. Wolberg, D. Mink,
H. Griengl, Tetrahedron 2007, 63, 918-926.
110 V. P. Vassilev, T. Uchiyama, T. Kajimoto, C.-H. Wong, Tetrahedron Lett. 1995, 36, 4081-
4084.
111 J. Steinreiber, K. Fesko, C. Mayer, C. Reisinger, M. Schürmann, H. Griengl, Tetrahedron
2007, 63, 8088-8093.
112 F. Sagui, P. Conti, G. Roda, R. Contestabile, S. Riva, Tetrahedron 2008, 64, 5079-5084.
113 M. L. Gutierrez, X. Garrabou, E. Agosta, S. Servi, T. Parella, J. Joglar, P. Clapés, Chem.
Eur. J. 2008, 14, 4647-4656.
114 L-Threoninaldolase aus E. coli: T. Kimura, V. P. Vassilev, G.-J. Shen, C.-H. Wong, J. Am.
Chem. Soc. 1997, 119, 11734-11742; R. Contestabile, A. Paiardini, S. Pascarella, M. L. di
Salvo, S. D'Aguanno, F. Bossa, Eur. J. Biochem. 2001, 268, 6508-6525.
115 Die in diesem Abschnitt beschriebenen und von mir angefertigten Arbeiten wurden im
Rahmen eines von der DBU geförderten Kooperationsprojekts (AZ 13217) angefertigt,
an dem verschiedene Partner aus Industrie und Universität beteiligt waren und im

LITERATURVERZEICHNIS
173
Rahmen einer arbeitskreisinternen Zusammenarbeit mit Sabine Simon, Giuseppe Rulli,
und Marina Kraußer.
116 T. Shiraiwa, R. Saijoh, M. Suzuki, K. Yoshida, S. Nishimura, H. Nagasawa, Chem. Pharm.
Bull. 2003, 51, 1363-1367.
117 S. S. Miyazaki, S.-I. Toki, Y. Izumi, H. Yamada, Eur. J. Biochem. 1987, 162, 533-540.
118 Z.-Y. Zuo, Z.-L. Zheng, Z.-G. Liu, Q.-M. Yi, G.-L. Zou, Enzyme Microb. Technol. 2007, 40,
569-577.
119 P. W. Atkins, Physikalische Chemie, 3. Auflage, Wiley-VHC, Weinheim, 2001.
120 B. Geueke, W. Hummel, Enzyme and Microb. Technol. 2002, 31, 77-87.
121 Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu, K.-X. Su, Chem. Rev. 2005, 105, 1603-1662.
122 K. B. Sharpless, Angew. Chem. 2002, 114, 2126-2135; Angew. Chem. Int. Ed. 2002, 41,
2024-2032.
123 T. Katsuki, K. B. Sharpless, J. Am. Chem. Soc. 1980, 102, 5974-5976.
124 G. Beck, Synlett 2002, 837-850.
125 H. K. Chenault, M.-J. Kim, A. Akiyama, T. Miyazawa, E. S. Simon, G. M. Whitesides, J.
Org. Chem. 1987, 52, 2608-2611.
126 J. G. Hill, B. E. Rossiter, K. B. Sharpless, J. Org. Chem. 1983, 48, 3607-3608.
127 T. Linker, Angew. Chem. 1997, 109, 2150-2152; Angew. Chem. Int. Ed. 1997, 36, 2060-
2062.
128 E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker, L. Deng, J. Am. Chem. Soc. 1991, 113,
7063-7064.
129 O. A. Wong, Y. Shi, Chem. Rev. 2008, 108, 3958–3987.
130 R. Imashiro, T. Kuroda, Tetrahedron Lett. 2001, 42, 1313-1315.
131 M. B. Johansen, A. B. Leduc, M. A. Kerr, Synlett 2007, 2593-2595.
132 C. Mordant, C. Caño de Andrade, R. Touati, V. Ratovelomanana-Vidal, B. B. Hassine, J.-
P. Genêt, Synthesis 2003, 2405-2409.
133 S. Chang, J. M. Galvin, E. N. Jacobsen, J. Am. Chem. Soc. 1994, 116, 6937-6938.
134 P. Besse, M. F. Renard, H. Veschambre, Tetrahedron: Asymmetry 1994, 5, 1249-1268.
135 O. Cabon, D. Buisson, M. Larcheveque, R. Azerad, Tetrahedron: Asymmetry 1995, 6,
2211-2218.

LITERATURVERZEICHNIS
174
136 M. Hesse, H. Meier, B. Zeeh, Spektroskopische Mehtoden in der organischen Chemie,
6. Auflage, Thieme, Suttgart, 2002.
137 The results, which I obtained and are described in this chapter, were achieved within a
cooperation with Marina Kraußer and Giuseppe Rulli.
138 The results, which I obtained and are described in this chapter, were achieved within a
project cooperation, which was supported by the DBU (AZ 13217). The participants
were different partners from industry and university, in particular Sabine Simon,
Giuseppe Rulli, Marina Kraußer and Nina Dückers.
139 Y. Zhou, Z. Shan, Tetrahedron: Asymmetry 2006, 17, 1671-1677.
140 B. Kaptein, T. J. G. van Dooren, W. H. J. Boesten, T. Sonke, A. L. L. Duchateau, Q. B.
Broxterma, J. Kamphuis, Org. Process Res. Dev. 1998, 2, 10-17.
141 Phenylserin: CAS: 207605-47-8
142 Y. X. Ping, W. S. Wei, G. W. Sheng, W. J. Tao, Heteroatom Chemistry 1999, 10, 183-186.
143 C. J. Pouchert, J. Behnke, The Aldrich Library of 13C and 1H FT NMR Spectra, Aldrich
Chemical Company, 1993, Vol. 1, 873; CAS: 20859-02-3.
144 C. J. Pouchert, J. Behnke, The Aldrich Library of 13C and 1H FT NMR Spectra, Aldrich
Chemical Company, 1993, Vol. 2, 1405; CAS: 495-69-2
145 S. Simon, geplante Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg.
146 DL-threo-β-(3,4-Dihydroxyphenyl)serin: CAS 3916-18-5
147 C. J. Pouchert, J. Behnke, The Aldrich Library of 13C and 1H FT NMR Spectra, Aldrich
Chemical Company, 1993, Vol. 1, 867; CAS: 56-40-6
148 P. G. M. Wuts, R. L. Gu, J. M. Northuis, Tetrahedron: Asymmetry 2000, 11, 2117-2123.
149 J.-Q. Wang, E. H. Weyand, R. G. Harvey, J. Org. Chem. 2002, 67, 6216-6219.


















