
Röntgendiffraktometrie - TU Bergakademie Freiberg · die Materialwissenschaft, Werkstofftechnik,...
46
Röntgendiffraktometrie David Rafaja, Institut für Werkstoffwissenschaft, Technische Universität Bergakademie Freiberg Im Jahre 1912 haben in München Walter Friedrich und Paul Knipping nach den theoretischen Überlegun- gen von Prof. Max von Laue die ersten Beugungsexperimente mit Röntgenstrahlen durchgeführt und da- bei durch die Interferenz der am Kristallgitter gebeugten Röntgenstrahlen ihre Wellennatur nachgewie- sen. Damit wurden die Grundlagen der Röntgendiffraktometrie geschaffen – einer äußerst leistungsfähi- gen experimentellen Methode, die heutzutage in vielen Bereichen der Wissenschaft von der Physik, über die Materialwissenschaft, Werkstofftechnik, Mineralogie und Chemie bis zur Strukturbiologie Anwen- dung findet. Insbesondere die mathematische Beschreibung der Beugungseffekte durch die Reflexion von Röntgenstrahlen an aufeinanderfolgenden parallelen (Netz)ebenen, die im Jahre 1913 von William Lawrence Bragg und seinem Vater Prof. William Henry Bragg publiziert wurde, hat der Röntgendiffrak- tometrie dazu verholfen, dass sie sich zu einer prominenten und zum Teil einzigartigen Technik entwi- ckelt, die heutzutage standardmäßig für Kristallstrukturanalyse, Phasenanalyse, Realstrukturanalyse und Mikrostrukturanalyse verwendet wird. Obwohl die Röntgendiffraktometrie zu den etablierten experimentellen Methoden der Mikrostruktur- analytik gehört und obwohl die Röntgenbeugungsmethoden nach mehr als 100 Jahren seit der Entde- ckung der Röntgenbeugung in vielen Anwendungsbereichen durch die Bereitstellung von automatischen Auswertungsroutinen mehr als ausreichend popularisiert wurden, besteht immer noch Bedarf an der Weiterentwicklung der Röntgendiffraktometrie als einer Methode für eine profunde und statistisch rele- vante Materialanalyse mit der Auflösung auf atomarer Skala. In diesem Kapitel werden vor allem aktuelle Methoden der Röntgendiffraktometrie an nanokristallinen und defektreichen kristallinen Materialien sowie an Werkstoffen mit ausgeprägten mikroskopischen und makroskopischen Spannungsfeldern be- handelt, wobei ihre Kristallanisotropie sowie der Einfluss der Kristallanisotropie auf die beobachteten Beugungsphänomene immer wieder betont werden. Die beschriebenen Methoden der Röntgendiffrak- tometrie werden dennoch auf die kohärente (Braggsche) Streuung der Röntgenstrahlen fokussiert. Die diffuse Streuung, die z.B. beim „total scattering“-Ansatz [1], [2] mit der kohärenten Streuung gleichbe- rechtigt behandelt wird, wird hier nur am Rande betrachtet. 1 Wechselwirkung der Röntgenstrahlen mit der Materie 1.1 Elastische Streuung der Röntgenstrahlen an Elektronen Die Röntgenstrahlen sind elektromagnetische Wellen mit der Wellenlänge zwischen ungefähr 5 Å 1 und 0.1 Å. Im Kontakt mit Materie werden sie sowohl elastisch als auch inelastisch gestreut, wobei die elasti- sche Streuung der Röntgenstrahlen an Elektronen für die Röntgendiffraktion der wichtigste elementare 1 1 Å (Ångström) = 10 -10 m = 0,1 nm. 1
Transcript of Röntgendiffraktometrie - TU Bergakademie Freiberg · die Materialwissenschaft, Werkstofftechnik,...

Röntgendiffraktometrie
David Rafaja, Institut für Werkstoffwissenschaft, Technische Universität Bergakademie Freiberg
Im Jahre 1912 haben in München Walter Friedrich und Paul Knipping nach den theoretischen Überlegun-gen von Prof. Max von Laue die ersten Beugungsexperimente mit Röntgenstrahlen durchgeführt und da-bei durch die Interferenz der am Kristallgitter gebeugten Röntgenstrahlen ihre Wellennatur nachgewie-sen. Damit wurden die Grundlagen der Röntgendiffraktometrie geschaffen – einer äußerst leistungsfähi-gen experimentellen Methode, die heutzutage in vielen Bereichen der Wissenschaft von der Physik, über die Materialwissenschaft, Werkstofftechnik, Mineralogie und Chemie bis zur Strukturbiologie Anwen-dung findet. Insbesondere die mathematische Beschreibung der Beugungseffekte durch die Reflexion von Röntgenstrahlen an aufeinanderfolgenden parallelen (Netz)ebenen, die im Jahre 1913 von William Lawrence Bragg und seinem Vater Prof. William Henry Bragg publiziert wurde, hat der Röntgendiffrak-tometrie dazu verholfen, dass sie sich zu einer prominenten und zum Teil einzigartigen Technik entwi-ckelt, die heutzutage standardmäßig für Kristallstrukturanalyse, Phasenanalyse, Realstrukturanalyse und Mikrostrukturanalyse verwendet wird.
Obwohl die Röntgendiffraktometrie zu den etablierten experimentellen Methoden der Mikrostruktur-analytik gehört und obwohl die Röntgenbeugungsmethoden nach mehr als 100 Jahren seit der Entde-ckung der Röntgenbeugung in vielen Anwendungsbereichen durch die Bereitstellung von automatischen Auswertungsroutinen mehr als ausreichend popularisiert wurden, besteht immer noch Bedarf an der Weiterentwicklung der Röntgendiffraktometrie als einer Methode für eine profunde und statistisch rele-vante Materialanalyse mit der Auflösung auf atomarer Skala. In diesem Kapitel werden vor allem aktuelle Methoden der Röntgendiffraktometrie an nanokristallinen und defektreichen kristallinen Materialien sowie an Werkstoffen mit ausgeprägten mikroskopischen und makroskopischen Spannungsfeldern be-handelt, wobei ihre Kristallanisotropie sowie der Einfluss der Kristallanisotropie auf die beobachteten Beugungsphänomene immer wieder betont werden. Die beschriebenen Methoden der Röntgendiffrak-tometrie werden dennoch auf die kohärente (Braggsche) Streuung der Röntgenstrahlen fokussiert. Die diffuse Streuung, die z.B. beim „total scattering“-Ansatz [1], [2] mit der kohärenten Streuung gleichbe-rechtigt behandelt wird, wird hier nur am Rande betrachtet.
1 Wechselwirkung der Röntgenstrahlen mit der Materie
1.1 Elastische Streuung der Röntgenstrahlen an Elektronen
Die Röntgenstrahlen sind elektromagnetische Wellen mit der Wellenlänge zwischen ungefähr 5 Å1 und 0.1 Å. Im Kontakt mit Materie werden sie sowohl elastisch als auch inelastisch gestreut, wobei die elasti-sche Streuung der Röntgenstrahlen an Elektronen für die Röntgendiffraktion der wichtigste elementare
1 1 Å (Ångström) = 10-10 m = 0,1 nm. 1

Streuprozess ist. Das Streuvermögen eines im Atom zum Atomkern gebundenen Elektrons kann mit Hilfe der Bewegungsgleichung
( )tiEermdtrd
dtrdm ωωγ −=++ exp002
2
(1.1)
beschrieben werden [3], [4]. In Gleichung (1.1) steht r für die Auslenkung, m für die Masse, e für die
Ladung2, ω0 für die Eigenfrequenz und γ für die Dämpfung des Elektrons; 0E
ist die Amplitude und ω die
Kreisfrequenz des elektromagnetischen Feldes (in Form von Röntgenstrahlen). Es wird angenommen, dass sich das schwingende Elektron in der Nähe des Ursprungs des Koordinatensystems befindet und dass die Amplitude der Schwingungen des Elektrons ausreichend klein ist, sodass der ortsabhängige Teil
der Phase der elektromagnetischen harmonischen Welle, 00 Rk
⋅ , in der vollständigen Beschreibung der
Amplitude des elektrischen Feldes,
( )[ ]tRkiEE ω−⋅= 000 exp
, (1.2)
vernachlässigt werden kann. Die Lösung der Gleichung (1.1) kann in der Form
( )( ) ωγωω
ωimtiEer
+−−
= 220
0 exp
(1.3)
geschrieben werden. Das oszillierende Elektron verhält sich wie ein elektrischer Dipol mit der Amplitude
( )( ) ωγωω
ωimtiEere
+−−
= 220
02 exp
(1.4)
und strahlt einen Teil seiner Energie in Form einer Kugelwelle wieder aus. Die elektrische Feldstärke des
Dipols ergibt sich aus seiner Amplitude [s. Gleichung (1.3)], die mit dem Faktor ( )Rc22 sinϑω multipli-
ziert wird:
( )( ) ϑω
ωγωω
ωsin
exp2
2
220
02
Rcim
tiEeEd ⋅
+−
−=
. (1.5)
In der Gleichung (1.5) bezeichnet c die Lichtgeschwindigkeit im Vakuum, R den Abstand vom Dipol und ϑ
den Winkel zwischen der Auslenkung des Elektrons ( r ) und der Beobachtungsrichtung R
(Abb. 1). Der Sinus des ϑ-Winkels beschreibt die Abstrahlcharakteristik des Dipols. Das elektrische Feld eines freien Elektrons kann aus der Gleichung (1.5) für γ = 0 (keine Dämpfung) und ω0 = 0 (keine Bindung zum Atom-kern) hergeleitet werden:
( ) ϑω sinexp02
2
⋅−⋅−= tiERmc
eE FEd
. (1.6)
2 Die Ruhemasse und die Ladung des Elektrons sind 311010956,9 −×=m kg und 191060219,1 −×=e C. 2

Abbildung 1: Abstrahlcharakteristik eines im externen elektromagnetischen Feld schwingenden Elekt-rons. Die Ausbreitungsrichtung der Primärwelle ist durch den horizontalen Balken, das Elektron durch den Punkt in der Mitte des Bildes dargestellt. Der Vektor r bezeichnet die Auslenkung des Elektrons, der Vektor R
die Richtung der gestreuten Welle.
Das Minuszeichen im Vorfaktor auf der linken Seite der Gleichung (1.6) bedeutet, dass die Phase der ge-streuten Welle um π gegenüber der Primärwelle verschoben ist:
( ) ( )titi ωπω −−=+− expexp . (1.7)
Der Faktor ( ) 1522 1081794.2 −×=mce m in der Gleichung (1.6) entspricht dem klassischen Elektronenra-dius. Vergleicht man die Amplituden des elektrischen Feldes, das von einem gebundenen und von einem freien oszillierenden Elektron ausgestrahlt wird,
( )( ) ( ) 22222
02
3
222220
2
220
2
γωωω
γω
γωωω
ωωω
+−⋅−
+−
−=
mi
m
mEE
FEd
d , (1.8)
sieht man, dass sich das Verhältnis der Amplituden in der Nähe der Eigenfrequenz stark verändert. Der reelle Teil der Gleichung (1.8) beschreibt die anomale Streuung, der imaginäre Teil die anomale Absorp-tion. Die anomalen Streueffekte werden bei manchen Röntgenbeugungsexperimenten am Synchrotron genutzt, um den Streukontrast zwischen chemischen Elementen mit ähnlichen Atomzahlen zu erhöhen [5].
Die Intensität der elastisch gestreuten Welle lässt sich aus der Gleichung (1.5) durch
2*ddd EEEI =⋅= (1.9)
berechnen. Das Symbol *dE bezeichnet den konjugierten Wert der komplexen Amplitude dE . Um die
Abhängigkeit der Intensität vom Beugungswinkel3 vollständig beschreiben zu können, ist es günstig, die
Amplitude des elektrischen Feldes der Primärwelle 0E
in ihre Komponenten zu zerlegen. Bewegt sich
der Detektor in der horizontalen Ebene um den Dipol, gilt für die horizontale Komponente der Primär-
3 Der Beugungswinkel (2θ) wird als der Winkel zwischen dem Primärstrahl und dem diffraktierten Strahl definiert, vgl. Abb. 2.
3

welle ϑ = π/2 – 2θ (Abb. 2a) und für die vertikale Komponente der Primärwelle ϑ = π/2 (Abb. 2b). Daher ändert sich die horizontale Komponente der gestreuten Intensität mit dem Beugungswinkel als
( ) θωγωωω
2cos224
4
222220
2
2||0
4|| ⋅⋅
+−=
Rcm
EeId
, (1.10)
während die vertikale Komponente der gestreuten Intensität konstant bleibt:
( ) 24
4
222220
2
2
04
Rcm
EeId
ωγωωω
⋅+−
=⊥
⊥
. (1.11)
Abbildung 2: Erläuterung des Polarisationsfaktors für eine horizontal (a) und vertikal (b) polarisierte Pri-märwelle. Der Detektor bewegt sich in dieser Darstellung nur in der horizontalen Ebene.
Die gesamte Intensität ergibt sich als gewichtete Summe von ||dI und ⊥
dI , wobei die Gewichtung das
Verhältnis zwischen 2||
dE
und 2⊥
dE
, und daher auch die Polarisation der Primärwelle widerspiegelt:
( ) ( )22
124
4
222220
2
2||0
4
2||
1 2cos KKRcm
EeIKIKI ddd +⋅⋅
+−=+= ⊥ θω
γωωω
. (1.12)
Für eine nichtpolarisierte Primärwelle (K1 = K2 = ½) lässt sich die gesamte Intensität in der Form von
( ) 212cos2
24
4
222220
2
2||0
4+
⋅⋅+−
=θω
γωωω Rcm
EeId
(1.13)
schreiben. Der Term K1 cos²2θ + K2 in Gleichungen (1.12) und (1.13) wird als Polarisationsfaktor P(2θ) bezeichnet, der quadratische Abstand zwischen dem Dipol und dem Detektor (R²) wird dem Lorentz-Faktor4 L zugeordnet. Die sonstigen Faktoren werden üblicherweise als I0 bezeichnet.
Die Synchrotronstrahlung ist in der horizontalen Ebene polarisiert. Um eine starke Abnahme der Intensi-tät mit steigendem Beugungswinkel in der horizontalen Ebene (K1 = 1, K2 = 0) zu vermeiden, werden an
4 Der Lorentz-Faktor ist ein (typischerweise) winkelabhängiger Korrekturfaktor, der beschreibt, wie viel von der insgesamt diffraktierten Intensität im Detektor ankommt. Der Lorentz-Faktor hängt stark von der Beugungsgeomet-rie und von der Winkelakzeptanz des Detektors ab.
4

Synchrotronquellen für Untersuchungen an polykristallinen Materialien in der Regel nur vertikale Diffrak-tometer genutzt, für die K1 = 0 und K2 = 1, so dass der Polarisationsfaktor konstant bleibt.
1.2 Interferenz der elastisch gestreuten Röntgenstrahlen
Die an einzelnen Elektronen elastisch gestreuten Wellen überlagern sich gegenseitig, wobei es zu ihrer Interferenz kommt. Betrachtet man die Interferenzeffekte weit weg von den elementaren Streuzentren, d.h., in einem Abstand, der deutlich größer ist als die Abstände zwischen den einzelnen Streuzentren, können die gestreuten Kugelwellen als ebene Wellen mit der Amplitude
( )ϕiEE d exp0= (1.14)
betrachtet werden. Die maximale Amplitude in der Gleichung (1.14) ergibt sich aus der elektrischen Feld-stärke des Dipols [s. Gleichung (1.5)]:
( ) ϑωωγωω
sin2
2
220
02
0 ⋅⋅+−
=Rcim
EeE d
. (1.15)
Die Phase der Welle in der Gleichung (1.14) kann auf den Ursprung des Koordinatensystems 0R
(s. Abb.
3) bezogen werden, so dass sich ϕ aus der Phase der Primärwelle ( tRk ω−⋅ 00
), und aus der Phasenver-
schiebung rkrk ⋅−⋅0 ergibt:
( )[ ]rkrktRkiEE d ⋅−⋅+−⋅= 0000 exp ω . (1.16)
Abbildung 3: Entstehung einer Phasenverschiebung bei der Streuung einer planaren Welle mit dem Wel-lenvektor 0k
an zwei Streuzentren mit den Positionsvektoren 0R
und rR
+0 . k
ist der Wellenvektor der betrachteten gestreuten Welle.
Bezeichnet man
0kkq
−= (1.17)
5

als Beugungsvektor, kann die Amplitude der an einem Elektron mit dem Ortsvektor r gestreuten Welle in der folgenden kompakten Form geschrieben werden:
( ) ( )[ ] ( )rqitRkiErE d ⋅−−⋅= expexp 000 ω , (1.18)
wo nur die letzte exponentielle Funktion von dem Ortsvektor r abhängt. Betrachtet man die Interferenz der an einzelnen Elektronen in einem isolierten Atom elastisch gestreuten Wellen, ergibt sich die gesam-te Amplitude:
( )[ ] ( ) ( )∫∫∫ ⋅−−⋅= rdrqirtRkiEE d 3
000Atom expexp ρω , (1.19)
wo ( )rρ die Aufenthaltswahrscheinlichkeit des Elektrons ist. Da die Aufenthaltswahrscheinlichkeit der Elektronen außerhalt des Atoms gleich Null ist, kann in der Gleichung (1.19) über einen unendlich großen Raum intergiert werden. Das dreifache Integral steht dann für die Fourier-Transformation (FT) der Auf-enthaltswahrscheinlichkeit der Elektronen, und wird als atomarer Streufaktor bezeichnet:
( ) ( ) ( ) ( )[ ]∫∫∫ =⋅−= rFTrdrqirqf ρρ 3
Atom exp . (1.20)
Die Atomstreufaktoren werden üblicherweise aus den mit Hilfe von relativistischem Hartree-Fock-Formalismus berechneten Wellenfunktionen ψ ermittelt,
( ) ( ) ( )[ ]rFTFTqf ρψ ≡= 2
Atom , (1.21)
und für praktische Anwendung durch die Funktion
( ) ( ) cbafj
jj +−=∑=
4
1
22Atom sinexpsin λθλθ (1.22)
[6] oder, ergänzt um die anomale Streuung (f‘) und die anomale Absorption (f“), durch
( ) ( ) fifcbafj
jj ′′+′++−=∑=
4
1
22Atom sinexpsin λθλθ (1.23)
[7] approximiert. Die Funktion λθsin ist äquivalent dem Betrag des Beugungsvektors ( q ), da sich der
Wellenvektor von λθsin nur um den Faktor 4π unterscheidet:
θλπ sin4
=q . (1.24)
Die Koeffizienten a1, b1, a2, b2, a3, b3, a4, b4 und c aus der Gleichung (1.23) sind in den Internationalen Tabellen für Kristallographie [6] für verschiedene Oxidationszahlen tabelliert, wodurch man den Einfluss der chemischen Bindung auf den Atomstreufaktor berücksichtigen kann. Die wellenlängenabhängigen anomalen Streufaktoren (f‘ und f“) findet man z.B. auf der Webseite von Ethan A. Merritt [8].
6

Die Interferenz der an mehreren Atomen elastisch gestreuten Wellen kann in der Analogie mit der Glei-chung (1.19) als
( )[ ] ( ) ( )[ ] ( )∑ ∫∫∫ ⋅−⋅−−⋅=j
jd rqirdrqirtRkiEE
expexpexp 3000 ρω , (1.25)
oder in kompakter Form
( )[ ] ( ) ( )∑ ⋅−−⋅=j
jjd rqiqftRkiEE
expexp 000 ω , (1.26)
geschrieben werden, wo die Amplituden der an einzelnen Atomen elastisch gestreuten Wellen mit den entsprechenden Phasen addiert werden. Im Rahmen dieser Beugungstheorie, die als kinematisch be-zeichnet wird, wird angenommen, dass jede Primärwelle nur einmal gestreut wird und dass die einmal gestreuten Wellen miteinander nur einmal interferieren können. Die Phase bzw. die Phasendifferenz ergibt sich aus dem Skalarprodukt des Beugungsvektors q und des Positionsvektors des jeweiligen
Atoms jr . Der Wichtungsfaktor fj in der Gleichung (1.26) bedeutet das Streuvermögen des Atoms j mit
der entsprechenden Oxidationszahl. Die diffraktierte Intensität wird nach der Gleichung (1.9) berechnet:
( ) ( ) ( ) ( ) ( )∑∑ ⋅−⋅⋅==j
jjj
jjd rqiqfrqiqfEEEqI expexp*20
* , (1.27)
wo das Symbol * die konjugierte Funktion bezeichnet. Die Gleichung (1.27) kann für atomare Abstände,
nmmn rrr −= , umgeschrieben werden:
( ) ( ) ( )∑∑∑∑ ⋅=⋅==m n
mnnmd
m nmnnm
d rqiffErqiffEEEqI φcosexpexp2
02
0*
, (1.28)
wo φ der Winkel zwischen q und mnr ist. Bei Substanzen mit gleichmäßig verteilten Atomen entlang von Koordinationssphären, z.B. in Gasen [9] oder in amorphen Substanzen, ergibt sich der Mittelwert aus dem Integral der exponentiellen Funktion über alle möglichen Winkel φ [10]:
( ) ( )mn
mnmn qr
qrrqi sinexp =⋅ . (1.29)
Zusammen mit der Gleichung (1.28) führt die Gleichung (1.29) zur Debye-Formel [9]:
( ) ( )∑∑=m n mn
mnnm
d
qrqrffEqI sin2
0 (1.30)
7

2 Röntgenbeugung an defektfreien kristallinen Materialien
Die Diffraktion der Röntgenstrahlen an kristallinen Materialien ohne Mikrostrukturdefekte5 lässt sich am besten mit Hilfe der Gleichung (1.26) darstellen, in der der Positionsvektor jr durch eine Linearkombina-
tion von Basisvektoren des Kristallgitters,
itätnsperiodizTranslatio
321
elleElementarz eine
cnbnanczbyaxr jjjj +++++= , (2.1)
ersetzt wird
( )[ ] ( )[ ] ( )[ ]∑∑ ++⋅−++⋅−−⋅== 321 ,,
3211
000 expexpexpnnn
N
jjjjj
d cnbnanqiczbyaxqiftRkiEE ω . (2.2)
2.1 Der Strukturfaktor
Die erste Summe in der Gleichung (2.2) beschreibt die Röntgenbeugung an einer Elementarzelle mit den
Gittervektoren a , b
, c , die N Atome mit den Bruchkoordinaten ( )jjj zyx ,, enthält. Wenn sich der Beu-
gungsvektor in der Nähe eines reziproken Gitterpunktes hklG
befindet,
( )***22 clbkahGq hkl
++== ππ , (2.3)
gilt für das Skalarprodukt des Beugungsvektors mit dem Positionsvektor innerhalb einer Elementarzelle
( ) ( ) ( ) ( )jjjjjjjjj lzkyhxczbyaxclbkahczbyaxq ++=++⋅++=++⋅ ππ 2***2 . (2.4)
Bei der Vereinfachung der Gleichung (2.4) wurden die folgenden Relationen verwendet,
1*** =⋅=⋅=⋅ ccbbaa und 0****** =⋅=⋅=⋅=⋅=⋅=⋅ bcaccbabcaba
, die direkt aus der Definiti-
on der reziproken Gittervektoren,
( ) cbacba
⋅××
=* ; ( ) cbaacb
⋅××
=* ; ( ) cbabac
⋅××
=* , (2.5)
folgen. Substituiert man in der Gleichung (2.2) das Skalarprodukt des Beugungsvektors mit dem Positi-
onsvektor innerhalb einer Elementarzelle, d.h., ( )czbyaxq jjj
++⋅ , nach Gleichung (2.4) durch
( )jjj lzkyhx ++π2 , ergibt die erste Summe in der Gleichung (2.2) den Strukturfaktor
5 Obwohl die Oberfläche eines Kristalls im physikalischen Sinne einen wesentlichen Defekt darstellt, werden in die-sem Kapitel die äußeren Grenzen eines Kristalls und daher die endliche Größe der Kristalle nicht als Mikrostruktur-defekte betrachtet.
8

( )[ ]∑=
++−=N
jjjjjhkl lzkyhxifF
1
2exp π , (2.6)
der die Streuung der Röntgenstrahlen an den in einer Elementarzelle liegenden Atomen als eine Funktion von Beugungsindizes hkl beschreibt. Es sollte jedoch angemerkt werden, dass bei dieser Herleitung an-genommen wurde, dass der Beugungsvektor in der Nähe der reziproken Gitterpunkte liegt [vgl. Glei-chung (2.3)].
2.2 Die Laue-Bedingungen und die Bragg-Gleichung
Für hklGq
π2= [Gl. (2.3)] ergibt das Skalarprodukt des Beugungsvektors mit der Translationsperiodizität
ein N-Faches von 2π,
( ) ( )321321 2 nlknhncnbnanq ++=++⋅ π
, (2.7)
und die zweite Summe in der Gleichung (2.2) bekommt die Form
( )[ ] ( ) ( ) ( )
321111
13
12
11
,,321
3
3
2
2
1
1
3
3
2
2
1
1321
111
2exp2exp2expexp
NNN
ihnihnihncnbnanqi
N
n
N
n
N
n
N
n
N
n
N
nnnn
=⋅⋅=
=−⋅−⋅−=++⋅−
∑∑∑
∑∑∑∑
===
===
πππ
, (2.8)
weil sowohl h, k, l als auch n1, n2 und n3 ganze Zahlen sind. Die in der Nähe der reziproken Gitterpunkte diffraktierte Intensität kann schließlich aus der Gleichung (2.2) berechnet werden
23
22
21
20max NNNFII hkl= , (2.9)
wo
200dEI = . (2.10)
In Gl. (2.9) ist Fhkl der Strukturfaktor und N1, N2 und N3 die Anzahl der Elementarzellen entlang der kris-
tallographischen Achsen a , b
und c . Aus der Gleichung (2.8) ist ersichtlich, dass die Intensität aus der Gleichung (2.9) die maximale Intensität darstellt. Laut der Gleichung (2.8) erreicht die diffraktierte Inten-sität ihr Maximum, wenn [vgl. Gl. (1.17)]
( ) hakkaq π20 =⋅−=⋅ , ( ) kbkkbq π20 =⋅−=⋅
und ( ) lckkcq π20 =⋅−=⋅ . (2.11)
Ersetzt man die Wellenvektoren k
und 0k
durch ihren Betrag,
9
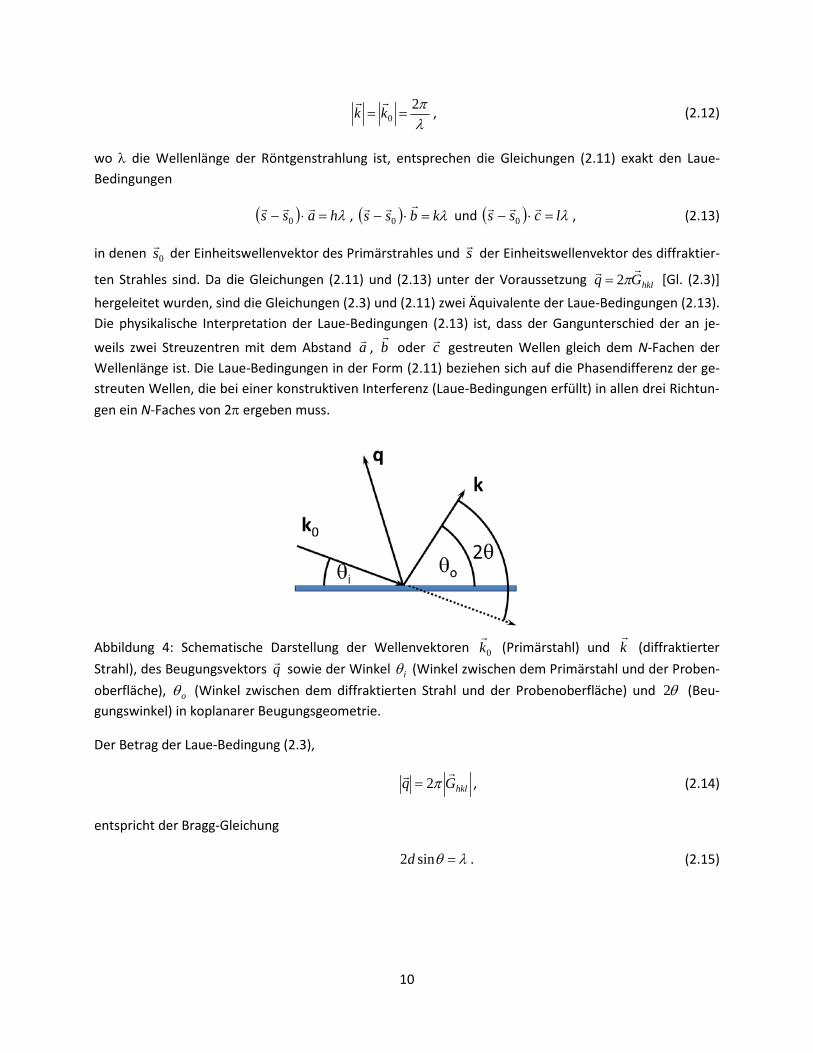
λπ2
0 == kk
, (2.12)
wo λ die Wellenlänge der Röntgenstrahlung ist, entsprechen die Gleichungen (2.11) exakt den Laue-Bedingungen
( ) λhass =⋅−
0 , ( ) λkbss =⋅−
0 und ( ) λlcss =⋅−
0 , (2.13)
in denen 0s der Einheitswellenvektor des Primärstrahles und s der Einheitswellenvektor des diffraktier-
ten Strahles sind. Da die Gleichungen (2.11) und (2.13) unter der Voraussetzung hklGq
π2= [Gl. (2.3)]
hergeleitet wurden, sind die Gleichungen (2.3) und (2.11) zwei Äquivalente der Laue-Bedingungen (2.13). Die physikalische Interpretation der Laue-Bedingungen (2.13) ist, dass der Gangunterschied der an je-
weils zwei Streuzentren mit dem Abstand a , b
oder c gestreuten Wellen gleich dem N-Fachen der Wellenlänge ist. Die Laue-Bedingungen in der Form (2.11) beziehen sich auf die Phasendifferenz der ge-streuten Wellen, die bei einer konstruktiven Interferenz (Laue-Bedingungen erfüllt) in allen drei Richtun-gen ein N-Faches von 2π ergeben muss.
Abbildung 4: Schematische Darstellung der Wellenvektoren 0k
(Primärstahl) und k
(diffraktierter
Strahl), des Beugungsvektors q sowie der Winkel iθ (Winkel zwischen dem Primärstahl und der Proben-oberfläche), oθ (Winkel zwischen dem diffraktierten Strahl und der Probenoberfläche) und θ2 (Beu-gungswinkel) in koplanarer Beugungsgeometrie.
Der Betrag der Laue-Bedingung (2.3),
hklGq
π2= , (2.14)
entspricht der Bragg-Gleichung
λθ =sin2d . (2.15)
10

Diese Äquivalenz kann am einfachsten für koplanare6 Beugungsgeometrie (siehe Abb. 4) nachgewiesen werden. Der Betrag des Beugungsvektors berechnet sich aus seinen kartesischen Komponenten als
( ) ( ) θλπθθ
λπθθθθλ
π sin42
sin4sinsincoscos 22222 =
+
=++−=+= oiioiozx qqq , (2.16)
und der Betrag des reziproken Gittervektors ist gleich dem reziproken Netzebenenabstand [3]:
hklhkl dG 1=
. (2.17)
Da für positive Beugungswinkel θθ sinsin = ist, kann die Gleichung (2.14) in die folgende Form umge-
schrieben werden,
hkldq πθ
λπ 2sin4
== , (2.18)
die nach dem Umstellen die Bragg-Gleichung (2.15) ergibt.
Der Unterschied zwischen den Laue-Bedingungen und der Bragg-Gleichung lässt sich folgendermaßen zusammenfassen. Laut Laue-Bedingungen muss der (durch den Faktor 2π dividierte) Beugungsvektor einen reziproken Gitterpunkt treffen. Demzufolge werden nicht nur die Beträge des Beugungsvektors und des reziproken Gittervektors miteinander verglichen, sondern auch deren Richtungen. Daher eignen sich die Laue-Bedingungen für die Beschreibung der Positionen der Beugungsmaxima in Einkristallen. Die Bragg-Gleichung vergleicht dagegen nur den Betrag des Beugungsvektors mit dem Betrag des reziproken Gittervektors. Es wird angenommen, dass eine günstige Orientierung des Kristalls vorliegt. Daher spielen die Richtungen dieser Vektoren keine Rolle. Aus diesem Grunde ist die Bragg-Gleichung nur für die Be-schreibung der Positionen der Beugungsmaxima in polykristallinen Proben geeignet, in denen ohnedies alle Kristallorientierungen vorliegen.
2.3 Effekt der Kristallgröße
Wenn die Laue-Bedingungen (2.3), (2.11) oder (2.13) nicht exakt erfüllt sind, wird die zweite Summe in der Gleichung (2.2) kleiner; die bei diesem geänderten Beugungsvektor gestreute Amplitude und daher auch die diffraktierte Intensität nehmen ab. Um die Änderung der gestreuten Amplitude mit dem Beu-gungsvektor zu beschreiben, kann die dreifache Summe in der Gleichung (2.2),
( )[ ] ( )[ ]∑∑∑∑= = =
++⋅−=++⋅−1
1
2
2
3
3321 1 1 1321
,,321 expexp
N
n
N
n
N
nnnn
cnbnanqicnbnanqi , (2.19)
6 Bei einer koplanaren Beugungsgeometrie liegen die beiden Wellenvektoren 0k
und k
, der Beugungsvektor q und die Normale zur Probenoberfläche in einer Ebene. Daher kann das Probenkoordinatensystem so gewählt wer-den, dass 0=yq ist.
11

in der es über alle Elementarzellen in den Kristallrichtungen a , b
und c summiert wird, durch
( ) ( )[ ] ( ) ( )
( )[ ]rFT
rdrqircnbnanqinnnn n n
Ω=
=⋅−Ω=++⋅−Ω ∫∫∫∑ ∑ ∑∞
−∞=
∞
−∞=
∞
−∞=
3321321 expexp,,
1 2 3 (2.20)
ersetzt werden, sodass der Einfluss der Größe des Kristalls auf die gestreute Amplitude durch die Fourier-Transformation des Formfaktors, ( )[ ]rFT
Ω , beschrieben werden kann. Der Formfaktor ( )321 ,, nnnΩ
bzw. ( )rΩ ist gleich 1 innerhalb des Kristalls und 0 außerhalb.
Unter der Annahme, dass sich der Strukturfaktor (Fhkl) mit dem Beugungsvektor deutlich weniger ändert als die Fourier-Transformation des Formfaktors, kann die Gleichung (2.2) in der folgenden Form ge-schrieben werden:
( )[ ] ( )[ ]rFTFtRkiEE hkld
Ω−⋅= ω000 exp . (2.21)
Die diffraktierte Intensität ergibt sich dann in der Analogie zur Gleichung (2.9) als
( ) ( )[ ]220 rFTFIqI hkl
Ω= . (2.22)
Diese Annahme ist für gut kristalline Materialien praktisch immer erfüllt. Eine typische Ausnahme, bei der die Abhängigkeit des Strukturfaktors vom Beugungsvektor berücksichtigt werden muss, ist eine teil-weise geordnete Stapelung von zweidimensionalen atomaren Schichten, die z.B. in graphitischen Struk-turen oder in Tonmineralen vorkommt. Die Röntgenbeugung an solchen Strukturen lässt sich mit Hilfe der Hendricks-Teller-Theorie [11] beschreiben.
Abbildung 5: Vergleich der Form der Beugungslinien für quaderförmige und sphärische Kristallite.
12

Der quadratische Betrag des Fourier-transformierten Formfaktors aus der Gleichung (2.22), ( )[ ]2rFT Ω ,
ist exemplarisch für ein Rechteck mit der Kantenlänge7 von 30 nm (durchgezogene Linie) und für eine Kugel mit dem Radius von 20 nm (gebrochene Linie) in Abb. 5 gezeigt. Für einen Quader mit den Kanten-längen Dx, Dy und Dz gilt [10]
( )[ ] ( )( )
( )( )
( )( )22
2
2
2
22
22sin
2
2sin
22sin
z
zz
y
yy
x
xx
qDq
qDq
qDqrFT ⋅⋅=Ω
, (2.23)
für eine Kugel mit dem Radius R [12]
( )[ ] ( ) ( )2
4
22 cossin16
−=Ω qRR
qqR
qrFT π
. (2.24)
Der Betrag des Beugungsvektors in der Gleichung (2.24) ergibt sich aus
222zyx qqqq ++= . (2.25)
Es folgt aus den Eigenschaften der Fourier-Transformation sowie aus den Gleichungen (2.23) und (2.24),
dass die Halbwertsbreite8 von ( )[ ]2rFT Ω reziprok proportional der Größe des Kristalls ist. Da die ande-
ren Faktoren in der Gleichung (2.22), d.h. die Intensität I0 und der Strukturfaktor Fhkl, nur wenig vom Beugungsvektor abhängen, ist auch die Halbwertsbreite der in der Nähe der reziproken Gitterpunkte gemessenen Intensität reziprok proportional zur Größe des Kristalls,
( )[ ]DKqIFWHM =∆ , (2.26)
wo die Proportionalitätskonstante K von der Form des Kristalls abhängt. Die Abhängigkeit der Proportio-nalitätskonstante von der Form des Kristalls ist aus der Abbildung 5 ersichtlich. Die beiden Kurven in Abb. 5 haben die gleiche Breite, obwohl die Kantenlänge des Quaders 30 nm und der Durchmesser der Kugel 40 nm waren.
Die Gleichung (2.26) ist als Schrerrer-Gleichung [13] bekannt. Da die Funktion ( )[ ]2rFT Ω nur von dem
Abstand vom jeweiligen reziproken Gitterpunkt abhängt, führt eine endliche Kristallgröße zu der gleichen Verbreiterung aller reziproken Gitterpunkte und nach der Gleichung (2.22) zu der gleichen Verbreiterung aller Beugungslinien.
7 Es wird angenommen, dass diese Kante parallel zum Beugungsvektor liegt. 8 Die Halbwertsbreite wird oft als FWHM (englisch „full width at half maximum“) bezeichnet.
13

2.4 Röntgenbeugung an mehreren Kristalliten
Bei der Beschreibung der Beugungserscheinungen an mehreren kleinen Kristallen (Kristalliten) kann man die einzelnen Kristallite als Streuzentren für die Röntgenstrahlung betrachten und die gesamte diffrak-tierte Intensität aus der Interferenz der an einzelnen Kristalliten gestreuten Wellen berechnen [vgl. Gl. (2.21)]. Dabei müssen, wie in Gleichung (1.27), die Amplituden der gestreuten Wellen mit der entspre-chenden Phase addiert werden:
( ) ( ) ( )[ ] ( ) ( )[ ]∑∑==
⋅−Ω⋅⋅−Ω=N
nnnnhkl
N
nnnnhkl RqiFTFRqiFTFIqI
1'''',
1,0 exp*exp
. (2.27)
Die Positionsvektoren nR
beschreiben die Positionen der Kristallite; nhklF , sind deren Strukturfaktoren
und ( )nFT Ω deren Formfaktoren. Die Summation in der Gleichung (2.27) erfolgt für jeden einzelnen Beugungsvektor, der hier als Parameter auftritt, über alle Kristallite, die im bestrahlten Volumen der un-tersuchten Probe liegen. Da die Intensität vom Beugungsvektor q und nicht von seinem Betrag abhängt, spielt die Orientierung der Kristallite zum Primärstahl (wie bei den Laue-Bedingungen) und daher auch die gegenseitige Orientierung der Kristallite eine wichtige Rolle. Das Produkt der Summen in der Glei-chung (2.27) kann alternativ als
( ) ( )
( )[ ] ( )[ ] ( )[ ]
−⋅−Ω⋅Ω+
+Ω=
∑∑
∑−
=
−
=+++
==
1
1 1,,0
10
22,0
exp*Re2N
m
mN
nnnmmnmnhklnnhkl
N
nm
nnhkl
RRqiFTFFTFI
FTFIqI
(2.28)
geschrieben werden, wo nnm −= ' ist. Die erste Summe (m = 0) in der Gleichung (2.28) beschreibt den Beitrag der einzelnen Kristallite zur gesamten diffraktierten Intensität, die doppelte Summe dann die In-terferenz der an unterschiedlichen Kristalliten gestreuten Röntgenstrahlung. Die an unterschiedlichen Kristalliten gestreuten Röntgenstrahlen können jedoch nur interferieren, wenn die Kristallite teilkohärent sind. Das Phänomen der partiellen Kohärenz der Kristallite wird ausführlicher im Kapitel (2.4.2) behan-delt.
In den meisten polykristallinen Proben sind die Nachbarkristallite gegenseitig nicht kohärent, daher ist die doppelte Summe in der Gleichung (2.28) gleich Null. Die gesamte diffraktierte Intensität ergibt sich dann aus der Summe der an einzelnen Kristalliten diffraktierten Intensitäten:
( ) ( )∑=
Ω=N
nnnhkl FTFIqI
1
22,0
. (2.29)
Die Gleichung (2.29) ist die Grundlage der kinematischen Beugungstheorie, die die einzelnen Kristallite als kohärent streuende Objekte behandelt, die Interferenz der an unterschiedlichen Kristalliten diffrak-tierten Wellen aber vernachlässigt.
14

2.4.1 Absorption der Röntgenstrahlung
Eine weitere Eigenschaft der kinematischen Röntgenbeugung an nichtkohärenten Kristalliten ist, dass bei diesem Streuprozess nur ein Bruchteil der Intensität des Primärstrahles (typischerweise weniger als 1 %) diffraktiert wird. Die restliche Intensität dringt ins Innere der Probe ein, wo die Röntgenstrahlung an an-deren Kristalliten diffraktiert werden kann. Beim Eindringen in die Probe wird jedoch die Röntgenstrah-lung auch absorbiert. Im Rahmen der kinematischen Beugungstheorie wird die Absorption der Röntgen-strahlung durch das exponentielle Absorptionsgesetz
( )LII µ−= exp0 (2.30)
beschrieben, wo I0 die Ausgangsintensität, I die geschwächte Intensität, µ den linearen Schwächungsko-effizienten und L den Weg der Strahlung in der Probe bedeutet. Für eine flache Probe ist der Weg der Röntgenstrahlung zum diffraktierenden Volumenelement mit der Dicke dz, das in der Tiefe z unter der Probenoberfläche liegt, in Abb. 6 dargestellt. In diesem speziellen Fall ergibt sich die gesamte Intensität aus dem Integral über die infinitesimale Intensität aus der Gleichung (2.30),
( )
( )
( ) AItI
tI
dzzSII
oi
oi
oi
o
oi
oi
oi
oi
i
t
oi
oioit
00
0
00
sinsinsinsinexp1
sinsinsin
sinsinsinsinexp1
sinsinsinsin
sin
sinsinsinsinexp,
=
+⋅−−
+=
+⋅−−
+⋅=
=
+⋅−= ∫
θθθθµ
θθµθ
θθθθµ
θθµθθ
θ
θθθθµθθ
, (2.31)
wo der Weg der Strahlung in der Probe (L) durch
oi
zzLLLθθ sinsin21 +=+= (2.32)
ersetzt und die Änderung der bestrahlen Fläche mit dem Primärwinkel (S) durch den Faktor iθ1sin− be-
rücksichtigt wurde. Der Korrekturfaktor A, der sowohl von den beiden Winkeln θi und θo als auch von der Probendicke abhängt, wird als Absorptionsfaktor bezeichnet. Die Intensitätskorrektur aus der Gleichung (2.31) ist besonders wichtig bei der Röntgenbeugung an dünnen polykristallinen Schichten.
15

Abbildung 6: Darstellung des Strahlenganges in einer planaren Probe zur Erläuterung der Absorption der Röntgenstrahlung.
Wenn die Dicke der Probe „unendlich“ groß (t > 10 µm) ist, reduziert sich die Gleichung (2.31) auf
( ) ( )oi
ooit II
θθµθθθ
sinsinsin, 0 +
= , (2.33)
bzw. bei der symmetrischen Beugungsgeometrie, wo θi = θo ist, auf
( )µ
θθ2
, 0II oit = . (2.34)
Den linearen Schwächungskoeffizienten einer kristallinen Phase mit dem Elementarzellvolumen VEZ kann man aus den Absorptionsquerschnitten σi der einzelnen Atome berechnen
∑=
=N
ii
EZV 1
1 σµ , (2.35)
wo N die Anzahl der Atome in der Elementarzelle ist. Die Absorptionsquerschnitte sind in den Internatio-nalen Tabellen für Kristallographie [6] tabelliert. Für Proben mit einer tiefenabhängigen Absorption, wie z.B. Multilagenschichten, inhomogene Proben oder auch Proben mit ausgeprägten Mikroabsorptionsef-fekten, muss im Integral (2.31) die Abhängigkeit des linearen Schwächungskoeffizienten von dem Ab-stand z unter der Probenoberfläche berücksichtigt werden [14], [15], [16].
2.4.2 Partielle Kohärenz nanoskaliger Kristallite
In nanokristallinen Proben sind reziproke Gitterpunkte von einzelnen Kristalliten stark verbreitet [siehe Gl. (2.26)], so dass sich die reziproken Gitterpunkte von Nachbarkristalliten teilweise überlappen können [12]. Eine solche Überlappung der reziproken Gitterpunkte ist für zwei Nanokristallite mit der gleichen Kristallstruktur (in diesem Fall kubisch-flächenzentriert) in Abb. 7 gezeigt.
16

Abbildung 7: Schematische Darstellung der Überlappung der stark verbreiteten reziproken Gitterpunkte von zwei kubisch-flächenzentrierten Nanokristalliten mit den gleichen Gitterparametern. Die Nanokris-tallite haben geringfügig unterschiedliche Orientierungen. Die grauen reziproken Gitterpunkte gehören einem Kristallit, die weißen reziproken Gitterpunkte dem anderen. Die einzelnen reziproken Gitterpunk-te sind mit den entsprechenden Beugungsindizes markiert.
Die grauen reziproken Gitterpunkte gehören zu einem Kristallit, die weißen reziproken Gitterpunkte zum anderen. Die gegenseitige Drehung der reziproken Gitter entspricht der gegenseitigen Drehung der Kris-tallite im direkten Raum. Wenn sich die reziproken Gitterpunkte von mindestens zwei Kristalliten teilwei-se überlappen, ist
( )[ ] ( )[ ] ( )[ ] 0exp1
1 1
≠−⋅−Ω⋅Ω∑∑−
=
−
=++
∗N
m
mN
nnnmmnhklnhkl RRqiFTFFTF
, (2.36)
und der zweite Term in der Gleichung (2.28) darf nicht vernachlässigt werden. Solche Kristallite können dann für die Röntgenbeugung partiell kohärent sein, wenn gleichzeitig der entsprechende Gangunter-
schied ( ) ( )nnm RRss
−⋅− +0 kleiner als die longitudinale Kohärenzlänge der Röntgenphotonen ( )λλ ∆22
ist. Die Einheitsvektoren 0s und s stehen für die Richtung des Primärstahles und für die Richtung des
diffraktierten Strahles. Die Symbole λ und ∆λ bezeichnen die Wellenlänge und die Spektralbreite der Röntgenstrahlung. Die longitudinale Kohärenzlänge der Röntgenphotonen, die aus einer Röntgenröhre austreten, beträgt weniger als 1 µm, kann aber durch zusätzliche Röntgenoptik [17] oder durch eine spe-zielle Messanordnung [18] mehrfach verlängert werden. In der Analogie zu den Effekten der optischen Kohärenz [19] kann dann die Gleichung (2.28) folgendermaßen modifiziert werden:
( ) ( )
( )[ ] ( )[ ] ( )[ ]
−⋅−Ω⋅Ω+
+Ω=
∑∑
∑−
=
−
=+++
==
1
1 1,,,0
10
22,0
exp*Re2N
m
mN
nnmnnmmnmnhklnnhkl
N
nm
nnhkl
CRRqiFTFFTFI
FTFIqI
, (2.37)
wo
000
111
200
002 202
311
004
113313
313
004
113
311
204204
202
115 115315
315
200
_111
_113
_113
_204_
204
_115
_115
_202
_202
_200
_200
_311
_311
_313
_313
_315
_315
a*
c*
17

1, =nmC wenn ( ) ( )λ
λ∆
≤−⋅− + 2
2
0 nnm RRss
0, =nmC wenn ( ) ( )λ
λ∆
>−⋅− + 2
2
0 nnm RRss
. (2.38)
Mit Hilfe von
( )002 sskkq
−=−=λπ (2.39)
kann die Gleichung (2.38) alternativ in der Form
1, =nmC wenn ( )λ
πλ∆
≤−⋅ + 2nnm RRq
0, =nmC wenn ( )λ
πλ∆
>−⋅ + 2nnm RRq (2.40)
geschrieben werden, die dem Argument der exponentiellen Funktion in der Gleichung (2.37) entspricht.
Die Auswirkung der partiellen Kohärenz auf die diffraktierte Intensität kann am einfachsten an zwei par-
tiell kohärenten Kristalliten mit der gleichen Kristallstruktur und mit dem Abstand R
∆ erläutert werden. Für solche Kristallite vereinfacht sich die Gleichung (2.37) wesentlich:
( ) ( ) ( ) ( ) ( ) ( )[ ] RqiFTFTFTFTFIqI hkl
∆⋅−ΩΩ+Ω+Ω= expRe2 21
*22
21
20 . (2.41)
Für reelle Formfaktoren ( )1ΩFT und ( )2ΩFT , wie zum Beispiel für die Formfaktoren von prismatischen
[Gleichung (2.23)] oder kugelförmigen Kristalliten [Gleichung (2.24)], gilt sogar
( ) ( )[ ] ( )[ ] ( ) ( ) ( ) RqFTFTFTFTFIqI hkl
∆⋅ΩΩ+Ω+Ω= cos2 21
22
21
20 . (2.42)
Die Faktoren ( )[ ]212
0 ΩFTFI hkl und ( )[ ]222
0 ΩFTFI hkl beschreiben die an einzelnen Kristalliten diffrak-
tierte Intensität, der Faktor ( ) ( ) ( )RqFTFTFI hkl
∆⋅ΩΩ cos2 21
20 den Interferenzterm [20]. Die Bedeutung
des letzten Faktors wird in graphischer Form in Abb. 8 dargestellt. Die breite durchgezogene Linie in Abb. 8 zeigt das Produkt von ( )1ΩFT und ( )2ΩFT , die gestrichelte Kurve den Kosinus der Phasendifferenz,
( )Rq
∆⋅cos , und die dünne durchgezogene Linie den gesamten Interferenzterm
( ) ( ) ( )RqFTFT
∆⋅ΩΩ cos21 . Aus der Abbildung 8 ist ersichtlich, dass der Interferenzterm die anfangs brei-
te Beugungslinie schmaler macht. Es handelt sich um einen analogen Effekt zur Interferenz der Röntgen-strahlen an einzelnen Elementarzellen innerhalb eines Kristallits, der im Kapitel 2.3 beschrieben wurde. Die physikalische Interpretation dieses zusätzlichen Effektes ist relativ einfach. Die Röntgenbeugung kann zwei nacheinander folgende kohärente Kristallite nicht voneinander unterscheiden, so dass die Li-nienbreite einem doppelt so großen Kristallit entspricht, d.h. die Linienbreite wird laut der Gleichung (2.26) halbiert.
18

Abbildung 8: Auswirkung der partiellen Kohärenz von Nanokristalliten auf die Breite und Form der Beu-gungslinien. Die Abhängigkeit der diffraktierten Intensität von der Länge des Beugungsvektors wird für nichtkohärente Kristallite durch die breite durchgezogene Linie und für kohärente Kristallite durch die dünne durchgezogene Linie dargestellt. Die gestrichelte Kurve entspricht der zusätzlichen Modulation der diffraktierten Intensität durch die Phasendifferenz an Nachbarkristalliten, ( )Rq
∆⋅cos , wo R
∆ der
Abstand der Nachbarkristallite ist. Der Term hklGq π2− bezeichnet den Abstand des Endpunktes des Beugungsvektors vom reziproken Gitterpunkt Ghkl.
Die Auswirkung der Kohärenzeffekte auf die Form der diffraktierten Intensität wird anhand von einer Simulation in Abb. 9 dargestellt. Die reziproken Gitterpunkte für die Berechnung der diffraktierten Inten-sität nach Gleichung (2.42) wurden durch eine Gauß-Funktion mit der Integralbreite von 1/D beschrie-ben:
( ) ( )( )[ ]222222 4e x p zyxx qqqqDDF T ++∆+−=Ω π . (2.43)
In der Gleichung (2.43) ist D die Kristallitgröße (5 nm), qx, qy und qz sind die entsprechenden Komponen-ten des Beugungsvektors und ∆qx ist die Verschiebung eines der reziproken Gitterpunkte, die der Miss-orientierung der Kristallite (vgl. Abb. 7) entspricht.
Nor
mie
rte In
tens
ität
q – 2πGhkl
19

Abbildung 9: Simulation der diffraktierten Intensität in der Nähe der sich überlappenden reziproken Git-terpunkte. Links werden die Ergebnisse der nichtkohärenten Streuprozesse abgebildet, rechts wird der Effekt der partiellen Kohärenz der Nachbarkristallite dargestellt. Die Simulationen wurden für die Ver-schiebungen von ∆qx = 0, ∆qx = 0.024 Å-1 und ∆qx = 0.042 Å-1 (von oben nach unten) durchgeführt.
20

Der funktionale Zusammenhang zwischen der Missorientierung der Kristallite (ω) und der Verschiebung der reziproken Gitterpunkte ( xq∆ ) folgt aus der Abbildung 10 und wird durch die Gleichung (2.44) als
eine lineare Funktion des Betrages des Beugungsvektors ( q ) beschrieben:
2sin2 ωqqx
=∆ (2.44)
Abbildung 10: Zusammenhang zwischen der Missorientierung der Nanokristallite im direkten Raum (ω), dem Betrag des Beugungsvektors ( q ) und der Verschiebung der reziproken Gitterpunkte in der qx-Richtung.
Die in Abb. 9 gezeigten Abhängigkeiten der diffraktierten Intensitäten von qx und qz, d.h. zweidimensio-nale Schnitte (qy = 0) im reziproken Raum, wurden für die Verschiebungen von ∆qx = 0 (Abb. 9a und 9d), ∆qx = 0.024 Å-1 (Abb. 9b und 9e) und ∆qx = 0.042 Å-1 (Abb. 9c und 9f) berechnet. Die Bilder auf der linken Seite (a-c) zeigen die an nichtkohärenten Kristalliten diffraktierten Intensitäten, die Bilder auf der rech-ten Seite (d-f) den Einfluss der partiellen Kohärenz auf die Form der zweidimensionalen Beugungslinien.
Die Abbildung 11 zeigt die in der Nähe von qx = 0 diffraktierte Intensität als Funktion der Abweichung vom Bragg-Winkel (∆qz). Die grauen Kurven in Abb. 11 repräsentieren den nichtkohärenten Fall, die schwarzen Kurven den Einfluss der partiellen Kohärenz auf die Linienform. Man sieht, dass bei vollstän-dig kohärenten Kristalliten (∆qx = 0) die Breite der Beugungslinie wesentlich abnimmt. Zusätzlich be-kommt die ursprünglich gaußförmige Linie eine andere Form, die stark an eine Cauchy-Lorentz-Funktion erinnert. Mit zunehmender Verschiebung der reziproken Gitterpunkte (∆qx) vergrößert sich die Breite der Beugungslinien von nur partiell kohärenten Kristalliten, bis sie die Breite der an nichtkohärenten Kris-talliten diffraktierten Linien erreicht, sobald die partielle Kohärenz der Kristallite erlischt.
Die Abhängigkeit der Linienbreite von der Verschiebung der reziproken Gitterpunkte wird in Abbildung 12 zusammengefasst. Da nach der Gleichung (2.44) die Verschiebung der reziproken Gitterpunkte linear vom Betrag des Beugungsvektors abhängt, kann diese Abhängigkeit in Form des Williamson-Hall-Plots [21] dargestellt werden.
qx
qz
ωq
21
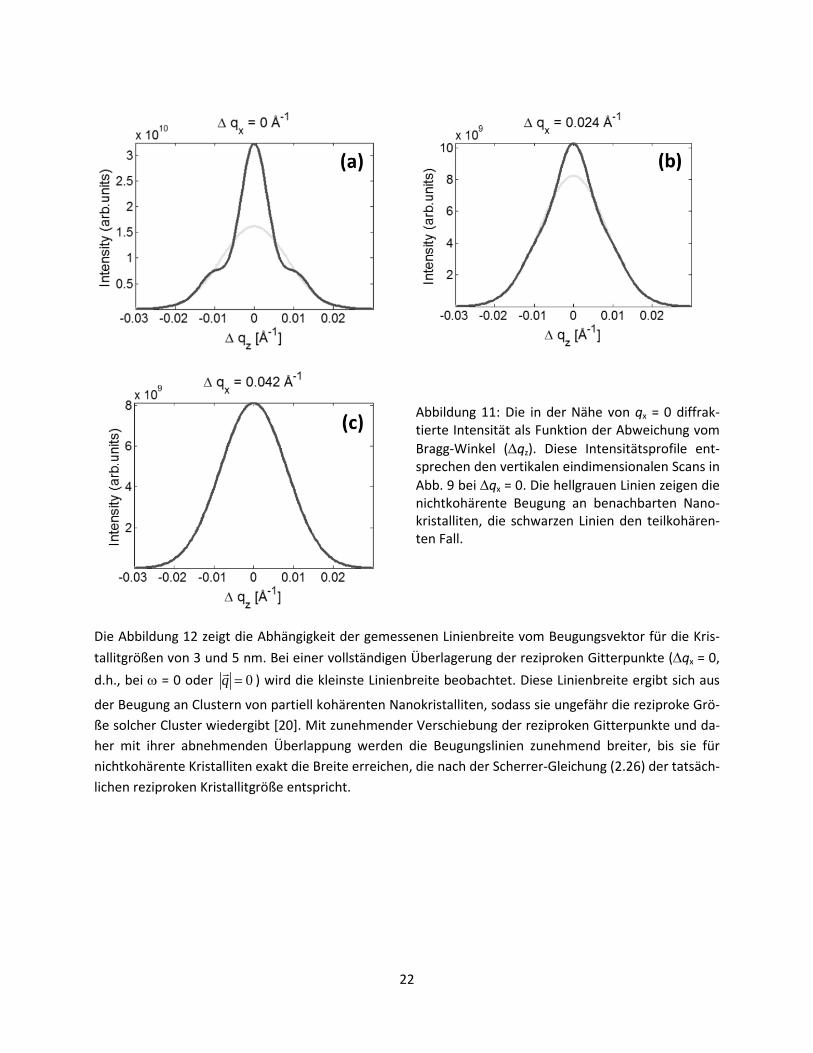
Abbildung 11: Die in der Nähe von qx = 0 diffrak-tierte Intensität als Funktion der Abweichung vom Bragg-Winkel (∆qz). Diese Intensitätsprofile ent-sprechen den vertikalen eindimensionalen Scans in Abb. 9 bei ∆qx = 0. Die hellgrauen Linien zeigen die nichtkohärente Beugung an benachbarten Nano-kristalliten, die schwarzen Linien den teilkohären-ten Fall.
Die Abbildung 12 zeigt die Abhängigkeit der gemessenen Linienbreite vom Beugungsvektor für die Kris-tallitgrößen von 3 und 5 nm. Bei einer vollständigen Überlagerung der reziproken Gitterpunkte (∆qx = 0, d.h., bei ω = 0 oder 0=q ) wird die kleinste Linienbreite beobachtet. Diese Linienbreite ergibt sich aus
der Beugung an Clustern von partiell kohärenten Nanokristalliten, sodass sie ungefähr die reziproke Grö-ße solcher Cluster wiedergibt [20]. Mit zunehmender Verschiebung der reziproken Gitterpunkte und da-her mit ihrer abnehmenden Überlappung werden die Beugungslinien zunehmend breiter, bis sie für nichtkohärente Kristalliten exakt die Breite erreichen, die nach der Scherrer-Gleichung (2.26) der tatsäch-lichen reziproken Kristallitgröße entspricht.
22

Abbildung 12: Abhängigkeit der Linienbreite von der Verschiebung der reziproken Gitterpunkte. Die Kur-ven stammen aus der oben beschriebenen Simulation für Nanokristallite mit der Größe von 3 nm (obere Kurven) und 5 nm (untere Kurven). Die mit den Beugungsindizes markierten Dreiecke entsprechen den Missorientierungen der kubisch-flächenzentrierten Nachbarkristallite von 1.2° (linkes Bild) und 0.5° (rechtes Bild). Wegen der linearen Abhängigkeit des Betrages des Beugungsvektors q von xq∆ [s. Glei-chung (2.44)] ist diese Darstellung äquivalent dem Williamson-Hall-Plot [21].
2.4.3 Experimentelle Beispiele der partiellen Kohärenz nanoskaliger Kristallite
Die im vorigen Kapitel diskutierten Kohärenzeffekte wurden häufig in dünnen nanokristallinen met-astabilen Schichten von (Ti,Al)N [12], (Cr,Al)N [22] und (Zr,Al)N [23] sowie in Nanokompositen auf der Basis von Bornitrid [24] beobachtet. In diesen Arbeiten konnte weiterhin mit Hilfe der Kombination der Röntgenbeugung (XRD) und der Transmissionselektronenmikroskopie mit Hochauflösung (HRTEM) expe-rimentell nachgewiesen werden, dass aus der Abhängigkeit der Linienbreite vom Betrag des Beugungs-vektors sowohl die wahre Größe der Nanokristallite als auch die Größe der Cluster von partiell kohären-ten Nanokristalliten berechnet werden kann. Darüber hinaus war es möglich, die gegenseitige Missorien-tierung der benachbarten nanoskaligen Kristallite aus der Verbreiterung der Röntgenbeugungslinien zu bestimmen.
Ein Beispiel dafür wird in Abb. 13 gezeigt. Die Größe der Cluster von partiell kohärenten Nanokristalliten ergibt sich aus der Extrapolation der durch die partielle Kohärenz beeinflussten Linienbreiten zum Ur-sprung des reziproken Raumes, d.h. zu sin θ = 0. Diese Linienbreiten sind im Bereich der kleinen Beu-gungsvektoren (sin θ = 0 bis 0,75). Die wahre Kristallitgröße wird dagegen aus den bei großen Beugungs-vektoren gemessenen Linienbreiten (sin θ > 0,8) bestimmt, wo die Verbreiterung der Beugungslinien nicht mehr vom Betrag des Beugungsvektors abhängt. Nach der Gleichung (2.18) ist der Sinus der Hälfte des Beugungswinkels (sin θ) direkt proportional dem Betrag des Beugungsvektors. Im HRTEM-Bild (Abb. 13) sind die entsprechenden Mikrostrukturmerkmale markiert.
23

Abbildung 13: Abhängigkeit der Breite der Röntgenbeugungslinien von sin θ (links) und das HRTEM-Bild (rechts) einer dünnen Schicht mit der chemischen Zusammensetzung Cr0.92Al0.08N. Die mittlere Missorien-tierung der nanoskaligen Kristallite lag in diesem Fall zwischen 0.50° und 0.56°.
Die partielle Kohärenz der Nanokristallite wurde nicht nur in kompakten einphasigen Materialien, son-dern auch in einphasigen nanokristallinen Pulvern mit einer starken lokalen Vorzugsorientierung der ein-zelnen Kristallite beobachtet [25]. Auch in diesem Fall führte die partielle Kohärenz zur deutlichen Ab-nahme der Linienbreite bei kleinen Beugungswinkeln und zu einer Sättigung der Linienbreiten bei großen Beugungswinkeln, wo sich die reziproken Gitterpunkte nicht mehr überlagern.
Generell können die Effekte der partiellen Kohärenz der Kristallite auftreten, wenn es zur Überlappung der reziproken Gitterpunkte kommt, vgl. Gleichung (2.37). Daher sind sie nicht auf einphasige Materia-lien beschränkt, sondern werden auch in mehrphasigen Werkstoffen beobachtet, die aus Phasen mit ei-nigen ähnlichen Netzebenenabständen bestehen. Eine mögliche Heteroepitaxie der beteiligten Phasen sorgt für eine starke lokale Vorzugsorientierung von Kristalliten mit unterschiedlichen Kristallstrukturen und begünstigt wesentlich deren partielle Kohärenz [26]. Eine solche partielle Kohärenz von Nanokristal-liten mit unterschiedlichen Kristallstrukturen beobachtete A. Leineweber in Ni1+δSn während der Phasen-umwandlung der hexagonalen Struktur mit der Raumgruppe P63/mmc in die orthorhombische Kristall-struktur mit der Raumgruppe Pbnm [27].
Auf der Nanoskala werden Kohärenzeffekte zwischen einzelnen Mikrostrukturbestandteilen in der Regel vorausgesetzt. Ein klassisches Beispiel dafür ist die Theorie der Röntgenbeugung in periodischen Multila-genschichten [28], in der während der Berechnung von diffraktierten Intensitäten zunächst die Struk-turfaktoren einzelner Schichten im Multilagenstapel mit entsprechendem Phasenfaktor multipliziert und danach addiert werden. Auf einem ähnlichen Prinzip basiert auch die Beschreibung der Röntgendiffrak-togramme in stark plastisch deformierten hochlegierten austenitischen Stählen [29], die den Effekt der umwandlungsinduzierten Plastizität (den TRIP-Effekt) aufweisen. In diesen Stählen kommt es zur Bildung und später zur Anordnung von Stapelfehlern auf den kubisch-flächenzentrierten Netzebenen 111, die
24

die Röntgenbeugung als eine allmähliche Phasenumwandlung der kubischen Struktur des Austenits in die hexagonale Struktur des ε-Martensits interpretiert.
2.4.4 Kinematische Theorie der Röntgenbeugung an mehreren Kristalliten
In der kinematischen Beugungstheorie wird die Kohärenz der Nachbarkristallite vernachlässigt. Die Glei-chung (2.28) vereinfacht sich damit wesentlich, indem lediglich die an einzelnen Kristalliten diffraktierten Intensitäten und nicht die gestreuten Amplituden mit entsprechenden Phasen addiert werden. Die ver-einfachte Gleichung (2.28) kann dann in der Form [vgl. Gl. (2.29)]
( ) ( ) ( )[ ] ( ) ( )[ ]∑ ∑∑∑= == =
Ω=Ω⋅=P
p
N
npnp
P
p
N
npnp qFTqFIqFTqFIqI
1 1
2,
20
1 1
2,
20
(2.45)
geschrieben werden, wo man sowohl über die einzelnen Phasen (p) als auch über einzelne Kristallite der jeweiligen Phase (n) addiert. Der Intensitätsfaktor I0 enthält ein Produkt der Intensität des Primärstahls, des Lorentz-Faktors, des Polarisationsfaktors (s. Kapitel 1.1) und des Absorptionsfaktors (Kapitel 2.4.1). In der Gleichung (2.45) kann weiterhin die Summe der Formfaktoren einzelner Kristallite einer bestimmten Phase durch
( )[ ] ( )2
,1
2,
pEZ
ppN
npn V
qVqFT
Φ
=Ω∑=
(2.46)
ersetzt werden, wo pEZV , das Volumen der Elementarzelle, pV das Volumen der diffraktierenden Kristal-
lite (d.h. der Kristallite, deren Netzebenen (hkl) senkrecht zum Beugungsvektor liegen) und ( )qp
Φ die
normierte Profilfunktion ist:
( ) 1=Φ∫ qdqp
. (2.47)
So erhält man die Beschreibung der kinematisch diffraktierten Intensität als Funktion des Beugungsvek-tors:
( )( )
( )∑=
Φ⋅⋅
⋅⋅⋅⋅=P
pp
pEZ
pp qV
VqFAPLSqI
12
,
2
. (2.48)
In der kinematischen Beugungstheorie wird oft angenommen, dass sich der Strukturfaktor innerhalb ei-nes reziproken Gitterpunktes nicht verändert, sodass der Term ( )qF
durch ( )hklF ersetzt wird. Dies
bedeutet, dass der Strukturfaktor im entsprechenden reziproken Gitterpunkt gerechnet wird, wo
( )***2 clbkahq ++= π , (2.49)
25

siehe Kap. 2.1 und Gl. (2.3). Unter dieser Voraussetzung kann auch die Integralintensität einer bestimm-ten Beugungslinie einer bestimmten Phase als
( )( )
( )( )
2,
2
2,
2
int,pEZ
ppp
pEZ
pppp V
VhklFAPLSqdq
V
VhklFAPLSqdqII
⋅⋅⋅⋅⋅=Φ⋅
⋅⋅⋅⋅⋅== ∫∫
(2.50)
gerechnet werden.
Für polykristalline Proben kann weiterhin der Beugungsvektor q in Gl. (2.48) durch den Betrag ersetzt
werden (s. Kapitel 2.2). Für eine koplanare Beugungsgeometrie folgt dann aus der Gleichung (2.16)
θλπ sin4
=≡ qq , (2.51)
was bedeutet, dass die diffraktierte Intensität in der Gleichung (2.29) als eine Funktion des Beugungs-winkels (2θ) betrachtet werden kann:
( )( )
( )∑=
Φ⋅⋅
⋅⋅⋅⋅=P
pp
pEZ
pp
V
VhklFAPLSI
12
,
2
22 θθ . (2.52)
In polykristallinen Proben, die aus defektfreien Kristalliten bestehen, wird die Form der Profilfunktion Φp hauptsächlich durch die Form und Größe der Kristallite bestimmt, wie es im Kapitel 2.3 beschrieben wur-de.
3 Einfluss der Mikrostrukturdefekte auf das Röntgendiffraktogramm
Verschiedene Mikrostrukturdefekte in untersuchten Werkstoffen beeinflussen die Röntgendiffrakto-gramme auf unterschiedliche Art und Weise. Daher wird die Röntgendiffraktometrie als eine sehr geeig-nete Methode für die Mikrostrukturanalyse angewendet. Die Mikrostrukturdefekte selbst kann man nach verschiedenen Kriterien klassifizieren. Für die Zwecke der Röntgenbeugungsanalyse werden jedoch oft die Mikrostrukturdefekte nach ihrer Dimension gegliedert.
3.1 Punktdefekte
Als Punktdefekte werden Kristallstrukturdefekte auf atomarem Niveau bezeichnet. Deren räumliches Ausmaß ist typischerweise auf die Umgebung einzelner Atome beschränkt. Zu Punktdefekten zählen Leerstellen, die im Gegensatz zur regulären Kristallstruktur besetzten Zwischengitterplätze und Substitu-tionsatome. Solche regelwidrige Besetzung der Gitterplätze führt oft zu einer lokalen Gitterverzerrung, deren Auswirkung auf das Diffraktogramm sich sehr gut mit Hilfe der Gleichung (1.27) darstellen lässt. Danach kann die diffraktierte Intensität als
26
( ) ( ) ( )[ ] ( ) ( )[ ]∑∑ +⋅−⋅+⋅=j
jjjj
jjj urqiqfurqiqfIqI expexp*0 (3.1)
geschrieben werden, wobei die regemäßigen Atompositionen jr aus der Gleichung (1.27) durch die um
eine Verschiebung ju veränderten Atompositionen jj ur + ersetzt wurden. Eine nicht korrelierte Ver-
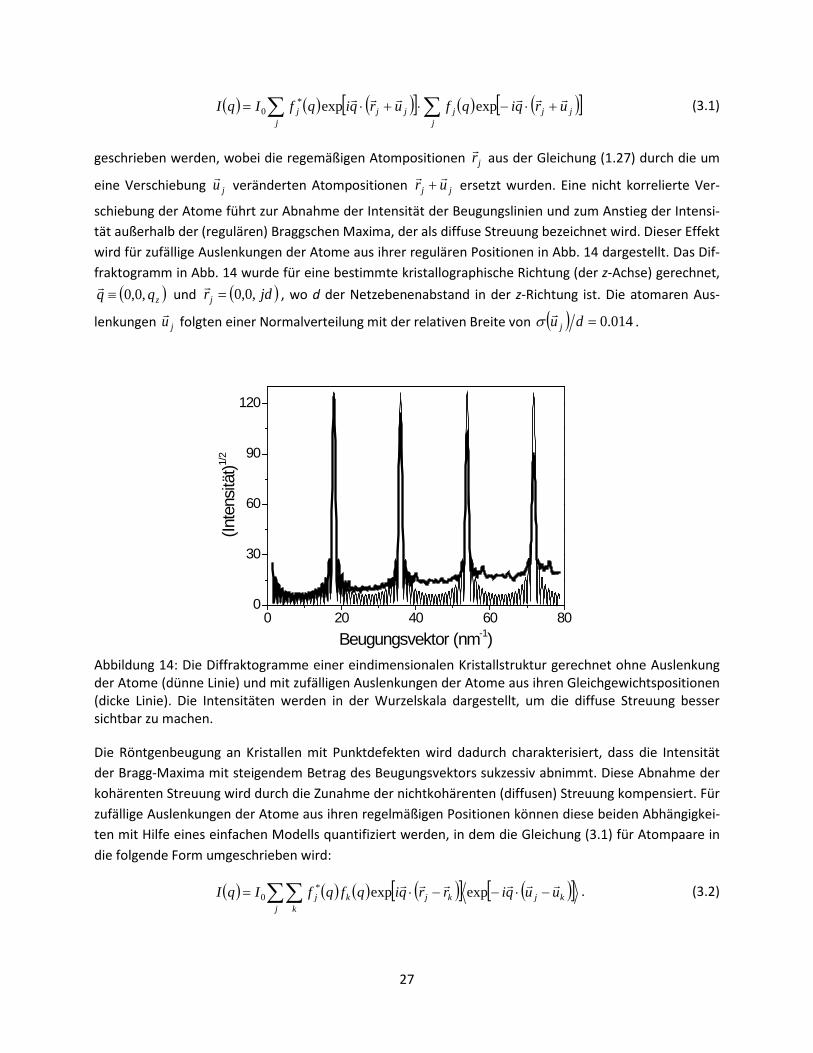
schiebung der Atome führt zur Abnahme der Intensität der Beugungslinien und zum Anstieg der Intensi-tät außerhalb der (regulären) Braggschen Maxima, der als diffuse Streuung bezeichnet wird. Dieser Effekt wird für zufällige Auslenkungen der Atome aus ihrer regulären Positionen in Abb. 14 dargestellt. Das Dif-fraktogramm in Abb. 14 wurde für eine bestimmte kristallographische Richtung (der z-Achse) gerechnet,
( )zqq ,0,0≡
und ( )jdrj ,0,0=
, wo d der Netzebenenabstand in der z-Richtung ist. Die atomaren Aus-
lenkungen ju folgten einer Normalverteilung mit der relativen Breite von ( ) 014.0=du j
σ .
Abbildung 14: Die Diffraktogramme einer eindimensionalen Kristallstruktur gerechnet ohne Auslenkung der Atome (dünne Linie) und mit zufälligen Auslenkungen der Atome aus ihren Gleichgewichtspositionen (dicke Linie). Die Intensitäten werden in der Wurzelskala dargestellt, um die diffuse Streuung besser sichtbar zu machen.
Die Röntgenbeugung an Kristallen mit Punktdefekten wird dadurch charakterisiert, dass die Intensität der Bragg-Maxima mit steigendem Betrag des Beugungsvektors sukzessiv abnimmt. Diese Abnahme der kohärenten Streuung wird durch die Zunahme der nichtkohärenten (diffusen) Streuung kompensiert. Für zufällige Auslenkungen der Atome aus ihren regelmäßigen Positionen können diese beiden Abhängigkei-ten mit Hilfe eines einfachen Modells quantifiziert werden, in dem die Gleichung (3.1) für Atompaare in die folgende Form umgeschrieben wird:
( ) ( ) ( ) ( )[ ] ( )[ ]∑∑ −⋅−−⋅=j k
kjkjkj uuqirrqiqfqfIqI expexp*0 . (3.2)
0 20 40 60 800
30
60
90
120
(Inte
nsitä
t)1/2
Beugungsvektor (nm-1)
27

Die Skalarprodukte juq ⋅ und kuq
⋅ entsprechen den Projektionen der atomaren Auslenkungen in die
Richtung des Beugungsvektors, und können daher mit Hilfe der Hälfte des Beugungswinkels als λθπ sin4 ju bzw. als λθπ sin4 ku geschrieben werden. Demnach gilt
( )[ ] ( )
−−=−⋅− kjkj uuiuuqi θ
λπ sin4expexp
. (3.3)
Für kleine Auslenkungen der Atome kann der Mittelwert in der Gleichung (3.3) in die Taylorreihe zweiter Ordnung zerlegt werden,
2211 xxieix −+= , (3.4)
was der Annahme eines harmonischen bzw. elastischen Verhaltens der lokalen Deformation in der Nähe des Punktdefektes entspricht. Für symmetrische Auslenkungen der Atome aus der Gleichgewichtspositi-
on ist ⟨x⟩ = 0 und daher ist [ ]221exp xeix −= . In dieser Näherung lässt sich die rechte Seite der Glei-
chung (3.3) als
( ) ( )
( ) ( ) ( )jkkj
kjkj
kjkj
MMM
uuuu
uuuui
2expexpexp
sin16expsin8expsin8exp
sin8expsin4exp
22
222
2
222
2
2
222
2
−−=
=
⋅
−⋅
−=
=
−−=
−−
θλπθ
λπθ
λπ
θλπθ
λπ
(3.5)
schreiben. Setzt man Gl. (3.5) in die Gleichung (3.2) ein, erhält man die folgende Formel für die diffrak-tierte Intensität:
( ) ( ) ( ) ( )[ ]∑∑ −−−⋅=j k
MMMkjkj
jkkj eeerrqiqfqfIqI 2*0 exp . (3.6)
Falls die atomaren Auslenkungen uj und uk nicht korreliert sind, ist 0=kjuu (für j ≠ k) und damit
( ) 12exp =jkM . Die Gleichung (3.6) kann daher in die folgende Form umgeschrieben werden,
( ) ( ) ( ) ( ) ( )
∑∑
∑∑+−
−⋅−⋅⋅=
−
−−
nn
n
Mn
n
Mnn
n
Mnn
fIefI
erqiqferqiqfIqI
n
nn
20
220
*0 expexp
, (3.7)
wo nur über den Index n addiert wird. Die letzten beiden Beiträge zur diffraktierten Intensität korrigieren den im ersten Teil der Gleichung (3.7) enthaltenen Einfluss der atomaren Auslenkungen für j = k [in Gl.
(3.6)]. Für j = k ist uj = uk und daher 022 ≠== kjkj uuuu , was bedeutet, dass der Intensitätsterm für j
= k, d.h. der letzte Term in Gl. (3.7), durch die atomare Unordnung nicht geschwächt wird. Mit dem vor-
28

letzten Intensitätsterm wird die im ersten Term der Gleichung (3.7) bereits enthaltene geschwächte In-tensität abgezogen. Führt man einen neuen Strukturfaktor ein, der die atomaren Auslenkungen aus den idealen Positionen berücksichtigt,
( ) ( )∑
−⋅−=
n
nnn
uqrqiqfqF
2exp
22
, (3.8)
ergibt sich die diffraktierte Intensität als
( ) ( ) ( )
−−+= ∑∑
nnn
nn uqffIqFIqI 2222
02
0 exp , (3.9)
wo der erste Teil der kohärent diffraktierten Intensität den Beugungslinien entspricht, die nur bei be-stimmten Beugungsvektoren vorkommen, während der zweite Teil die nichtkohärente diffuse Streuung beschreibt. Aus der Gleichung (3.9) ist ersichtlich, dass die Intensität der diffusen Streuung mit wachsen-dem Beugungsvektor zunimmt, wie es bereits in Abb. 14 gezeigt wurde.
3.2 Mikrodehnung
Ähnlich wie Punktdefekte erzeugen auch viele andere Mikrostrukturdefekte lokale Deformationsfelder, die im Diffraktogramm sichtbar sind. Die lokalen Gitterdeformationen führen zu einer Variation der Netz-ebenenabstände, die als Mikrodehnung bezeichnet wird:
21
221
2
∆≡
ddε . (3.10)
Die Variation der Netzebenenabstände verursacht eine Variation des Beugungswinkels, die als Linienver-breiterung messbar ist und die mit Hilfe des Differentials der Bragg-Gleichung (2.15),
0cos2sin2 =∆+∆ θθθ dd , (3.11)
folgendermaßen quantifiziert werden kann:
θεθ tan21
2=∆ . (3.12)
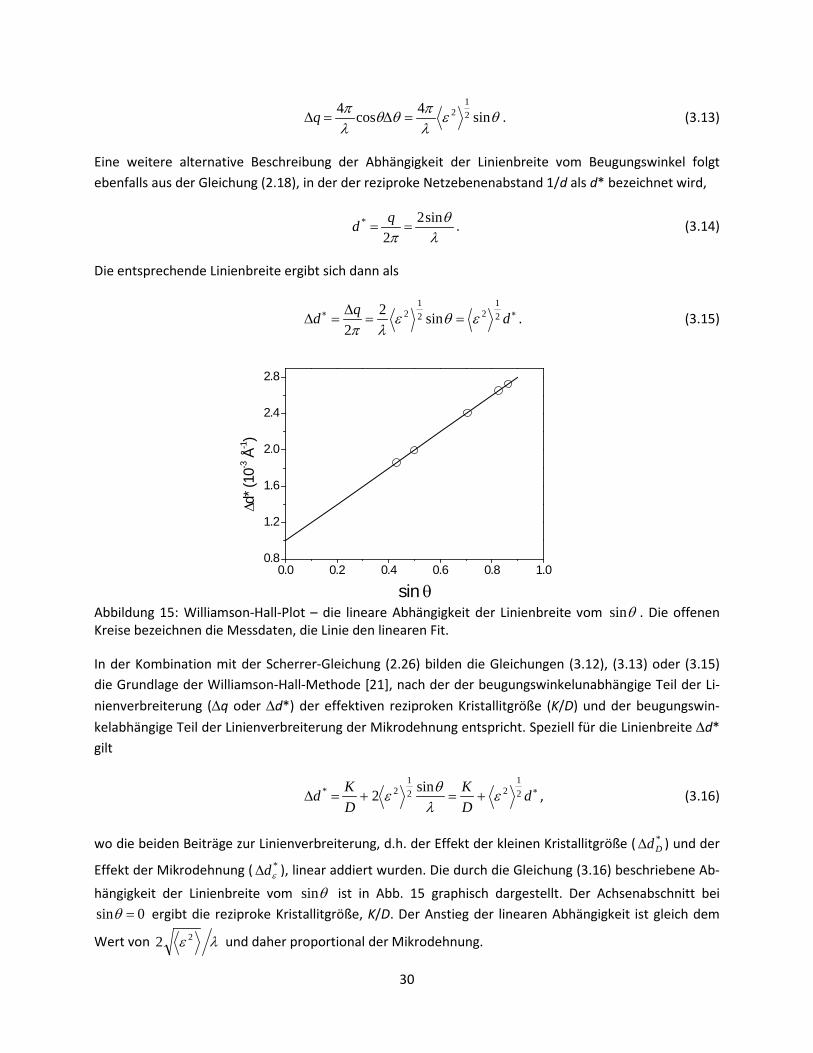
In Gl. (3.12) steht die Linienbreite ∆θ im Bogenmaß. Typischerweise wird die Linienbreite aber nicht als ∆θ sondern als ∆2θ gemessen, sodass die beiden Seiten der Gleichung (3.12) mit einem Faktor 2 multi-pliziert werden müssen. Alternativ wird die Linienbreite im reziproken Raum als ∆q angegeben. Aus den Gleichungen (2.18) und (3.12) folgt dann die entsprechende Linienbreite:
29
θελπθθ
λπ sin4cos4 2
12=∆=∆q . (3.13)
Eine weitere alternative Beschreibung der Abhängigkeit der Linienbreite vom Beugungswinkel folgt ebenfalls aus der Gleichung (2.18), in der der reziproke Netzebenenabstand 1/d als d* bezeichnet wird,
λθ
πsin2
2* ==
qd . (3.14)
Die entsprechende Linienbreite ergibt sich dann als
∗∗ ==∆
=∆ dqd 21
221
2 sin22
εθελπ
. (3.15)
Abbildung 15: Williamson-Hall-Plot – die lineare Abhängigkeit der Linienbreite vom θsin . Die offenen Kreise bezeichnen die Messdaten, die Linie den linearen Fit.
In der Kombination mit der Scherrer-Gleichung (2.26) bilden die Gleichungen (3.12), (3.13) oder (3.15) die Grundlage der Williamson-Hall-Methode [21], nach der der beugungswinkelunabhängige Teil der Li-nienverbreiterung (∆q oder ∆d*) der effektiven reziproken Kristallitgröße (K/D) und der beugungswin-kelabhängige Teil der Linienverbreiterung der Mikrodehnung entspricht. Speziell für die Linienbreite ∆d* gilt
∗+=+=∆ dDK
DKd 2
122
12* sin2 ε
λθε , (3.16)
wo die beiden Beiträge zur Linienverbreiterung, d.h. der Effekt der kleinen Kristallitgröße ( *Dd∆ ) und der
Effekt der Mikrodehnung ( *εd∆ ), linear addiert wurden. Die durch die Gleichung (3.16) beschriebene Ab-
hängigkeit der Linienbreite vom θsin ist in Abb. 15 graphisch dargestellt. Der Achsenabschnitt bei 0sin =θ ergibt die reziproke Kristallitgröße, K/D. Der Anstieg der linearen Abhängigkeit ist gleich dem
Wert von λε 22 und daher proportional der Mikrodehnung.
0.0 0.2 0.4 0.6 0.8 1.00.8
1.2
1.6
2.0
2.4
2.8
∆d* (
10-3 Å
-1)
sin θ
30

Die einfache Summe der von der kleinen Kristallitgröße und von der Mikrodehnung stammenden Linien-verbreiterungen [Gl. (3.16)] entspricht der Linienbreite einer Funktion, die durch die Faltung9 von zwei
Cauchy-Lorentz-Funktionen fD und fε mit den Halbwertsbreiten *Dd∆ und *
εd∆ entsteht:
( )[ ] ( )[ ]2**0
*
max
2**0
*
max)()(
11 ε
εε
ddd
I
ddd
IffD
DCCD
∆−+∗
∆−+≡∗ . (3.17)
Das bedeutet, dass wenn sowohl der Effekt der kleinen Kristallitgröße als auch der Effekt der Mikrodeh-nung jeweils durch eine Cauchy-Lorentz-Funktion beschrieben werden könnte, würde die Näherung aus Gl. (3.16) exakt gelten. Mehrere Autoren [siehe, z.B. [30]] haben jedoch gezeigt, dass der Effekt der Kris-tallitgröße zwar durch eine Cauchy-Lorentz-Funktion beschrieben werden kann, der Effekt der Mikro-dehnung jedoch in den meisten Fällen der Gauß-Funktion folgt. Bei der Faltung von zwei Gauß-Funktionen,
∆−
−∗
∆−
−≡∗2
*
*0
*max
2
*
*0
*max)()( 2lnexp2lnexp
εεε d
ddId
ddIffD
DGG
D , (3.18)
muss die Gesamtbreite der gefalteten Funktion als Summe der quadratischen Breiten der einzelnen Funktionen gerechnet werden, so dass die Gleichung (3.16) in einer modifizierten Form geschrieben werden muss:
( ) ( )2222
22
2* sin4 ∗+
=
+
=∆ d
DK
DKd ε
λθε . (3.19)
3.3 Versetzungen
Das oben beschriebene Konzept der Mikrodehnung kann relativ allgemein angewendet werden, um die lokalen Änderungen bzw. Fluktuationen der Netzebenenabstände, die z.B. durch die lokalen Variationen der chemischen Zusammensetzung oder durch Kristallgitterdefekte wie Versetzungen, Versetzungsstruk-turen und planare Defekte hervorgerufen werden, zu quantifizieren. Es ermöglicht aber weder die Identi-fizierung der für die Linienverbreiterung verantwortlichen Mikrostrukturphänomene, noch die Bestim-mung von statistischen Kenndaten, wie z.B. der Defektdichte.
Für eine detaillierte Mikrostrukturanalyse sind Modelle notwendig, die die Auswirkung der Mikrostruk-turdefekte auf das Röntgendiffraktogramm beschreiben. Die entsprechenden Mikrostrukturmodelle für Versetzungen wurden von Wilkens [31] und Krivoglaz [32] aufgestellt und von Klimanek und Kužel [33], [34] sowie von Ungár et al. [35], [36] weiterentwickelt.
9 Die „Faltung“ bedeutet hier das Faltungsintegral, ( ) ( ) εε ffdyyxfyf DD ∗≡−∫∞
∞−.
31

Nach Ungár et al. [36] ist die quadratische Mikrodehnung proportional der Versetzungsdichte ρ:
22
2
4MCb
hklhkl πρε ≈ (3.20)
Die Proportionalitätsfaktoren in Gl. (3.20) sind der Burgersvektor der Versetzungen b
, der Wilkensfaktor M [31], der von der Anordnung der Versetzungen abhängt, und der Kontrastfaktor der Versetzungen Chkl, der hauptsächlich für die Abhängigkeit der Mikrodehnung von der kristallographischen Richtung zustän-dig ist. Die Kontrastfaktoren der Versetzungen wurden erstmals für die hexagonale Kristallsymmetrie aus den elastischen Konstanten berechnet [33], [34]. Später haben Ungár et al. gezeigt, dass die Kontrastfak-toren der Versetzungen in kubischen Kristallen mit Hilfe der kubischen Invarianten
( )2222
222222
lkh
hllkkhhkl
++
++=Γ (3.21)
als
( )hklhhkl CC Γ−= ζ100 (3.22)
berechnet werden können, weil die kubische Invariante auch die Symmetrie der elastischen Konstanten beschreibt, siehe z.B. [37]. Ersetz man in der Gleichung (3.19) die Mikrodehnung aus Gl. (3.20) und den Kontrastfaktor aus den Gleichungen (3.21) und (3.22), erhält man die modifizierte Williamson-Hall-Abhängigkeit [36]:
( ) ( )( ) ( ) 2
2
00
2222
00
2222 sin11
4 λθζ
πρζ
πρ
hklhhklhhkl CMbDKdCMb
DKd Γ−+
=Γ−+
=∆ ∗∗ . (3.23)
Der Parameter ζ in den Gleichungen (3.22) und (3.23) steht für das Ausmaß der Kristallanisotropie der
Linienbreite ( )2*d∆ . Für die Zwecke der linearen Regression kann die Gleichung (3.23) in der folgenden
Form geschrieben werden:
( )
2
22002
2200
2
2
2
00
22
2
2
00
2222
sin4sin4
sinsin
λθζε
λθε
λθζ
πρ
λθ
πρ
hklhh
hklhhhkl
DK
CMbCMbDKd
Γ−+
=
=Γ−+
=∆ ∗
. (3.24)
Die freien Parameter des Mikrostrukturmodells aus der Gleichung (3.24) sind ( )2DK , 200hε und ζ. Die
Inputdaten sind die Verbreiterung der Beugungslinien durch Versetzungen ( )2*d∆ , die Linienpositionen
(sin θ) und die Beugungsindices, aus denen die kubische Invariante Γhkl mit Hilfe der Gleichung (3.21) ge-rechnet wird. Ein Beispiel für die Richtungsabhängigkeit der Verbreiterung der Beugungslinien in einem austenitischen Stahl ist in Abb. 16 gezeigt. Das Bild auf der linken Seite stellt die klassische Williamson-Hall-Abhängigkeit dar, das Bild auf der rechten Seite den modifizierten Williamson-Hall-Plot. Der klassi-
32

sche Williamson-Hall-Plot kann offensichtlich die Abhängigkeit der Linienverbreiterung von der Kristall-richtung nicht beschreiben, macht aber das Ausmaß der Anisotropie der Mikrodehnung gut sichtbar. Der modifizierte Williamson-Hall-Plot ist dagegen eine gerade Linie. Der Grad der Kristallanisotropie (ζ) be-einflusst direkt die Werte auf der x-Achse.
Abbildung 16: Der Williamson-Hall-Plot (links) und der modifizierte Williamson-Hall-Plot (rechts) eines plastisch deformierten austenitischen Stahls. Die einzelnen Messwerte (kleine Kreise) sind mit den Beu-gungsindizes bezeichnet. Die Messdaten wurden in den beiden Bildern durch die Gleichung (3.24) ange-passt. Die angepassten Daten sind als durchgezogene Linie dargestellt. Die gestrichelten Linien im linken Bild zeigen die Grenzwerte der quadratischen Mikrodehnung für die kristallographischen Richtungen ⟨111⟩ und ⟨100⟩.
Analog zu Kontrastfaktoren der Versetzungen in kubischen Kristallen können auch die Kontrastfaktoren von Versetzungen in Kristallen mit hexagonaler Symmetrie mit Hilfe der hexagonalen Invariante aus den Beugungsindizes hkil berechnet werden [38]:
( )[ ]( )[ ]222
23222
22222
lcaikh
llikhChkil+++
++++=
γβα , (3.25)
In der Gleichung (3.25) sind a und c die hexagonalen Gitterparameter und ( )khi +−= . Die Werte der
Koeffizienten α, β und γ hängen von dem Typ und vom Burgersvektor der Versetzungen sowie von den elastischen Konstanten des Materials ab.
3.4 Planare Defekte
Zu den häufigsten zweidimensionalen Defekten in Kristallen gehören Korngrenzen, Antiphasengrenzen, Stapelfehler und Zwillingsgrenzen. Ebenfalls diese Kristallstrukturdefekte beeinflussen das Diffrakto-gramm. Die Korngrenzen sind im Diffraktogramm zwar nicht direkt sichtbar, weil sie nicht zur Röntgen-beugung beitragen. Die Dichte der Korngrenzen hängt jedoch mit der Kristallitgröße zusammen und hat daher einen direkten Einfluss auf die Linienbreite, wie es im Kapitel 2.3 beschrieben wurde. Bei sehr klei-
0.0 0.2 0.4 0.6 0.8
2
4
6
8
10
111
200220
311
222
(∆d*
)2 (10-6
Å-1)
sin2θ
0.0 0.1 0.2 0.3 0.4 0.5
2
3
4
5
6
7
111
200
220
311
222
(∆d*
)2 (10-6
Å-1)
sin2θ (1-ζΓ)
33

nen Kristalliten spielt ebenfalls die Art der Korngrenzen (Kleinwinkel- oder Großwinkelkorngrenze) eine wichtige Rolle. Die Präsenz von Kleinwinkelkorngrenzen kann zu der im Kapitel 2.4.2 beschriebenen par-tiellen Kohärenz der Nachbarkristallite führen, die die Abhängigkeit der Breite der Beugungslinien vom Betrag des Beugungsvektors beeinflusst. Eine typische Erscheinung ist dabei der Anstieg der Linienbreite mit steigendem Betrag des Beugungsvektors, der in einem bestimmten Bereich des Beugungsvektors nach Gl. (3.19) als scheinbare Mikrodehnung interpretiert werden kann (vgl. Abb. 13). Auf der anderen Seite kann es in speziellen Fällen, wie z.B. bei der Bildung von Versetzungswänden [39], zu einer tatsäch-lichen Erhöhung der Mikrodehnung an Korngrenzen kommen.
Abbildung 17: Beispiel einer Antiphasengrenze. Die Antiphasengrenze ist mit der schwarzen gestrichelten Linie, die Verschiebung der Atome mit dem weißen Pfeil markiert.
Das Auftreten von Antiphasendomänen (Abb. 17) ist mit einer Änderung der Phase der gestreuten Welle verbunden, siehe Gleichung (2.2), weil die Atome an den beiden Seiten der Antiphasengrenze zwar prin-zipiell die gleichen Gitterpositionen besetzen, der Ursprung der jeweiligen Domäne jedoch verschoben wird. Dadurch kommt es zur Änderung des Strukturfaktors für bestimmte Beugungsindizes hkl und zur Änderung der Intensität von entsprechenden Beugungslinien [10]. Weiterhin verursacht die Bildung von Antiphasendomänen eine Fragmentierung des Kristalls, die zur richtungsabhängigen Verbreiterung der Beugungslinien führt, als würde es sich um eine hkl-Abhängigkeit der Kristallitgröße handeln.
Als Stapelfehler wird eine Änderung der Stapelfolge von bestimmten Netzebenen bezeichnet [10]. Ein typisches Beispiel ist die Änderung der Reihenfolge der Netzebenen 111 in einem kubisch-flächenzentrierten (kfz) Kristall von …ABCABCABC… zu …ABCAB|ABCA… Der senkrechte Strich bezeichnet die Position des Stapelfehlers. Die Sequenz …AB|AB…, die in der Nähe des Stapelfehlers in der kfz-Struktur auftritt, wird von der Röntgenbeugung als Teil einer hexagonalen dichtgepackten (hdp) Struktur interpretiert (vgl. Abb. 18), die wegen einer unterschiedlichen Länge der Elementarzelle in der betroffe-nen Richtung nicht vollständig kohärent mit der kubischen Struktur ist. Während sich die kfz-Struktur in der Stapelrichtung ⟨111⟩ über drei Netzebenen 111 (ABC) erstreckt, beträgt die Länge der hdp-Struktur in der Richtung ⟨001⟩ nur zwei Netzebenen 001 (AB). Typische Beispiele von Kristallstrukturen, die zur Bildung von Stapelfehlern neigen, sind austenitische Stähle mit einer niedrigen Stapelfehlerenergie sowie viele Materialien, die sowohl in der kubischen als auch in der hexagonalen Struktur kristallisieren oder in denen es bei höheren Temperaturen zu einem Phasenübergang kommt, wie z.B. Kobalt.
34

Abbildung 18: Vergleich der kubisch-flächenzentrierten (kfz, links) und der hexagonalen dichtgepackten Kristallstruktur (hdp, rechts). Die Reihenfolge der entsprechenden basalen Netzebenen, d.h. 111 in kfz und 001 in hdp, wird als …ABCABC… in kfz und als …ABABAB… in hdp bezeichnet.
Isolierte Stapelfehler beeinflussen die Breite der Beugungslinien prinzipiell ähnlich wie andere planare Kristallstrukturdefekte. Sie tragen zur Fragmentierung der kohärent diffraktierenden Domänen bei und verkleinern dadurch die effektive Kristallitgröße. Die Stapelfehler bilden sich jedoch bevorzugt auf be-stimmten Netzebenen, sodass die Änderung der effektiven Kristallitgröße wieder stark von der Kristall-richtung abhängt. So sind in der kfz-Struktur mit den Stapelfehlern auf den 111-Netzebenen nur die Beugungslinien betroffen, für die ( ) 03mod ≠++ lkh ist. In einer polykristallinen Probe muss es über alle äquivalenten Netzebenen (hkl) gemittelt werden, so dass sich der Effekt der 111-Stapelfehler in einer kfz-Struktur auf die Breite der Röntgenbeugungslinien nach Warren [10] als
∑++
++++=∆ ∗
b hklhkl
lkhm
lkhaD
Kd222
5.1 βα (3.26)
beschreiben lässt. Der erste Term in der Gleichung (3.26) entspricht der reziproken Kristallitgröße aus der Scherrer-Gleichung (2.26), a ist der kubische Gitterparameter, α die Dichte der isolierten Deformati-onsstapelfehler und β die Dichte der Wachstumsstapelfehler (Zwillingsgrenzen). Der Mittelwert wird über alle Permutationen der Beugungsindizes hkl gerechnet, die den beeinflussten Beugungslinien mit ( ) 03mod ≠++ lkh entsprechen. Das Symbol mhkl bezeichnet die Anzahl der äquivalenten Netzebenen für jedes hkl, d.h. die Flächenhäufigkeit. Die Werte der Summe aus der Gleichung (3.26) sind in der Ta-belle 1 für relevante Beugungslinien zusammengefasst.
Kubisch Hexagonal
A
B
C
A
A
B
A
B
Richtung ⟨111⟩ Richtung ⟨001⟩
35

Tabelle1: Die Zusammenfassung von hkl-abhängigen Faktoren, die den Beitrag der Stapelfehler 111 zur
Linienverbreiterung, ∑
++++
bhkl lkhmlkh 222 , und zur Linienverschiebung,
( ) ( )[ ]∑ ++++±b
hkl lkhmlkh 222 , in einer kfz-Struktur bestimmen und deren Vergleich mit der kubi-
schen Invarianten aus Gl. (3.21), ( ) ( )2222222222 lkhhllkkhhkl ++++=Γ .
hkl 111 200 220 311 222 400 331 420 422 333 511
222 lkhm
lkh
hkl
b
++
++∑ 4
3 1 2
1 112
3 43 1
1927
201
244 0
27417
( )
( )222 lkhm
lkh
hkl
b
++
++±∑ 4
1 21
− 41
111
− 81
− 41
191
201
− 0 0 274
3⋅
−
( )2222
222222
lkh
hllkkh
++
++
31 0
41
1219
31 0
36199
254
41
31
72951
In hexagonalen Kristallen ergibt sich die durch Stapelfehler bedingte Linienverbreiterung ebenfalls aus einer effektiven (reziproken) Kristallitgröße und wird durch die Gleichung (3.27) beschrieben:
DKdhkl =∆ ∗ für Nkh 3=−
( )βα 332 ++=∆ ∗
cld
DKd hkl
hkl für 13 ±=− Nkh und l gerade (3.27)
( )βα ++=∆ ∗ 32cld
DKd hkl
hkl für 13 ±=− Nkh und l ungerade.
Aus der Gleichung (3.27) ist ersichtlich, dass in einer hdp-Kristallstruktur die Breite der Beugungslinien 002, 110, 112 und 004 durch die Stapelfehler nicht beeinflusst wird. Verbreitet werden dagegen die Beu-gungslinien 100, 101, 102, 103, 200, 201, 202, 104, 203, 210 und 211. Durch das unterschiedliche Verhal-ten von Beugungslinien mit geraden und ungeraden l-Indizes ist es weiterhin möglich, die Deformations-stapelfehler und die Wachstumsstapelfehler (Zwillingsgrenzen) unabhängig voneinander zu quantifizie-ren, was bei einer kfz-Struktur auf diesem Wege nicht möglich ist.
Auf der anderen Seite beeinflussen die isolierten Deformationsstapelfehler in kfz-Materialien nicht nur die Breite sondern auch die Positionen der Beugungslinien [10]. Diese Linienverschiebung kann als eine zusätzliche anisotrope Gitterdehnung interpretiert werden [40]:
36

( )
( ) hkl
b
hkl
hklhkl mlkh
lkh
dd
2220 43
++
++±−=
∆=
∑παε . (3.28)
Durch die Stapelfehler werden nur, wie in der Gleichung (3.26), die Positionen der Beugungslinien beein-flusst, für die ( ) 03mod ≠++ lkh ist. Das Vorzeichen (±) in Gl. (3.28) ist positiv für ( ) 13mod =++ lkh
und negativ für ( ) 23mod =++ lkh . Die richtungsabhängigen Faktoren aus der Gleichung (3.28) werden
für eine kfz-Struktur in Tabelle 1 zusammengefasst.
Die Gleichung (3.28) ermöglicht die Bestimmung der Dichte der isolierten Stapelfehler. Da die Stapelfeh-ler typischerweise durch die Separation von Partialversetzungen wachsen, die ursprünglich durch die Dissoziation von vollständigen Versetzungen entstanden sind, wird insbesondere in der Anfangsphase der Separation von Partialversetzungen die lokale Gitterdehnung auch durch die Wechselwirkung von Partialversetzungen mitbestimmt. Die Wechselwirkung von Partialversetzungen modifiziert die Kristall-symmetrie der lokalen Gitterdeformation [40] und ermöglich eine indirekte Messung der Breite der Sta-pelfehler.
Bei höheren Stapelfehlerdichten steigt auch die Wahrscheinlichkeit der Stapelfehleranordnung. Solche geordneten Stapelfehler können nicht als isolierte Stapelfehler betrachtet werden, und die klassische Warren-Theorie verliert ihre Gültigkeit. Das heißt, dass solche Stapelfehler nicht zu der Linienverbreite-rung und zu der Linienverschiebung führen, die durch die Warren-Theorie vorhergesagt wurden. Viel-mehr wachsen durch eine periodische Anordnung von Stapelfehlern in einer kubischen Struktur die Be-reiche, die die Röntgenbeugung als hexagonal interpretiert. Im Röntgendiffraktogramm ist dann eine Art Phasenumwandlung zu erkennen. Solche Phänomene wurden in hochlegierten austenitischen Stählen [29] und in metastabilen Titanaluminiumnitriden [41] beobachtet. In den hochlegierten austenitischen Stählen führte die periodische Anordnung der Stapelfehler nach jeder zweiten Netzebene 111 zur Bil-dung von (hdp) ε-Martensit, der den Effekt der umwandlungsinduzierten Plastizität (transformation-induced plasticity, TRIP) begleitet. Im metastabilen (Ti,Al)N begleiten die geordneten Stapelfehler die Umwandlung des aluminiumreichen kubisch-flächenzentrierten (Al,Ti)N in die thermodynamisch stabile wurtzitische Modifikation. In anderen Materialien führt die Anordnung von Stapelfehlern zur Bildung von Polytypen. Als Beispiele von Elementen oder binären Verbindungen, die oft Polytypen bilden, sind ver-schiedene Modifikationen vom Graphit und Diamant, Siliciumcarbid (SiC), Bornitrid (BN) und Zinksulfid (ZnS) zu nennen. Allein für SiC sind in der Literatur ca. 25 Polytypen mit den Raumgruppen mF 34 ,
mcP 36 und 13mP berichtet worden.
3.5 Turbostratische Kristallstrukturdefekte
Zu weiteren Kristallstrukturdefekten, die hier näher beschrieben werden, gehören turbostratische Kris-tallbaufehler. Die turbostratischen Defekte kommen häufig im hexagonalen Graphit [42] und in Materia-lien mit ähnlicher Kristallstruktur, wie z.B. im hexagonalen Bornitrid [43], sowie in Tonmineralen [44] vor. Nach der Originaldefinition von Biscoe & Warren [42] bestehen turbostratische Strukturen aus stark ge-genseitig missorientierten Basisebenen, was in hexagonalen Systemen die Netzebenen 001 sind. Sehr
37
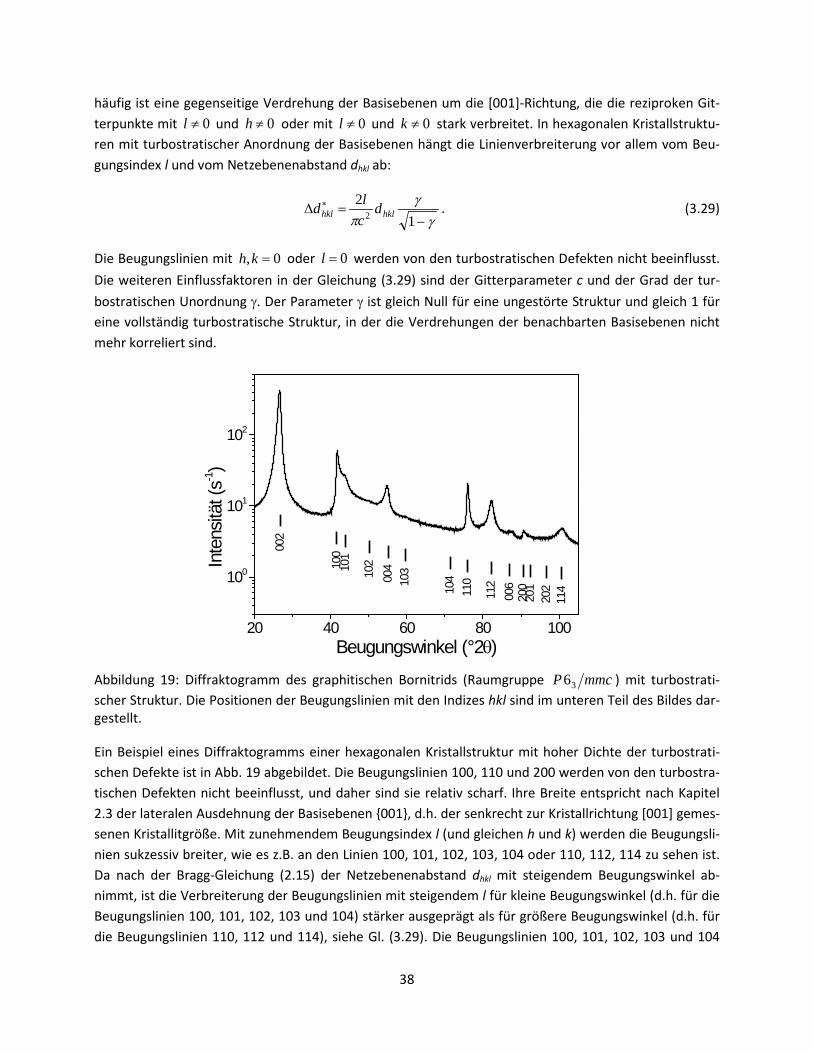
häufig ist eine gegenseitige Verdrehung der Basisebenen um die [001]-Richtung, die die reziproken Git-terpunkte mit 0≠l und 0≠h oder mit 0≠l und 0≠k stark verbreitet. In hexagonalen Kristallstruktu-ren mit turbostratischer Anordnung der Basisebenen hängt die Linienverbreiterung vor allem vom Beu-gungsindex l und vom Netzebenenabstand dhkl ab:
γγ
π −=∆ ∗
12
2 hklhkl dcld . (3.29)
Die Beugungslinien mit 0, =kh oder 0=l werden von den turbostratischen Defekten nicht beeinflusst. Die weiteren Einflussfaktoren in der Gleichung (3.29) sind der Gitterparameter c und der Grad der tur-bostratischen Unordnung γ. Der Parameter γ ist gleich Null für eine ungestörte Struktur und gleich 1 für eine vollständig turbostratische Struktur, in der die Verdrehungen der benachbarten Basisebenen nicht mehr korreliert sind.
Abbildung 19: Diffraktogramm des graphitischen Bornitrids (Raumgruppe mmcP 36 ) mit turbostrati-scher Struktur. Die Positionen der Beugungslinien mit den Indizes hkl sind im unteren Teil des Bildes dar-gestellt.
Ein Beispiel eines Diffraktogramms einer hexagonalen Kristallstruktur mit hoher Dichte der turbostrati-schen Defekte ist in Abb. 19 abgebildet. Die Beugungslinien 100, 110 und 200 werden von den turbostra-tischen Defekten nicht beeinflusst, und daher sind sie relativ scharf. Ihre Breite entspricht nach Kapitel 2.3 der lateralen Ausdehnung der Basisebenen 001, d.h. der senkrecht zur Kristallrichtung [001] gemes-senen Kristallitgröße. Mit zunehmendem Beugungsindex l (und gleichen h und k) werden die Beugungsli-nien sukzessiv breiter, wie es z.B. an den Linien 100, 101, 102, 103, 104 oder 110, 112, 114 zu sehen ist. Da nach der Bragg-Gleichung (2.15) der Netzebenenabstand dhkl mit steigendem Beugungswinkel ab-nimmt, ist die Verbreiterung der Beugungslinien mit steigendem l für kleine Beugungswinkel (d.h. für die Beugungslinien 100, 101, 102, 103 und 104) stärker ausgeprägt als für größere Beugungswinkel (d.h. für die Beugungslinien 110, 112 und 114), siehe Gl. (3.29). Die Beugungslinien 100, 101, 102, 103 und 104
20 40 60 80 100
100
101
102
002
100
101
102
004
103
104
110
112
006
200
201
202
114
Inte
nsitä
t (s-1
)
Beugungswinkel (°2θ)
38

sind kaum voneinander zu unterscheiden und bilden das sogenannte 10-Band, das zu höheren Beu-gungswinkeln stark asymmetrisch ist.
Die Beugungslinien 00l (d.h. 002, 004 und 006) entstehen durch die Röntgenbeugung an den Netzebenen 001. Daher ist ihre Verbreiterung unabhängig von dem Ausmaß der turbostratischen Defekte. Die Ver-breiterung der Beugungslinien 00l wird hauptsächlich durch die Fluktuationen der Netzebenenabstände d00l und nicht durch die Missorientierung der Netzebenen 001 um die Richtung [001] verursacht.
Häufig führen verschiedene Mikrostrukturdefekte zu unterschiedlichen Abhängigkeiten der Linienver-breiterung von den Beugungsindices. Dies erlaubt dann die Unterscheidung von mehreren Arten von Mikrostrukturdefekten, wie es am Beispiel von turbostratischen Defekten, Welligkeit der basalen Netz-ebenen, Versetzungen und Stapelfehlern im hexagonalen Bornitrid [45] gezeigt wurde.
3.6 Instrumentelle Verbreiterung der Beugungslinien
Die im Kapitel 3.2 eingeführte und in Kapiteln 3.3, 3.4 und 3.5 für die Bestimmung der Versetzungsdich-te, der Stapelfehlerwahrscheinlichkeit und der turbostratischen Unordnung genutzte Linienbreite hatte die Bedeutung der Linienverbreiterung durch Mikrostrukturdefekte. Die gemessene Breite der Beugungs-linien enthält jedoch auch die instrumentelle Linienverbreiterung, die durch eine endliche Winkelauflö-sung des Röntgendiffraktometers zustande kommt. Diese instrumentelle Linienverbreiterung muss zu-nächst von Messdaten „abgezogen“ werden, damit man die reine physikalische Linienverbreiterung er-hält. Der instrumentelle Effekt auf die Messdaten wird generell durch das Faltungsintegral
( ) ( ) ( ) gfdyyxgyfxh ∗≡−= ∫∞
∞−
(3.30)
beschrieben. Die Funktionen h, f und g in der Gleichung (3.30) stehen für die gemessenen Daten (h), für den physikalischen Effekt (f) und für die instrumentell bedingte Verzerrung der Messdaten (g). Um die Funktion f zu erhalten, ist eine Entfaltung der Messdaten notwendig. In speziellen Fällen lässt sich die Entfaltung ohne großen rechnerischen Aufwand durchführen. Für Cauchy-Lorentz-Funktionen f, g und h werden in der Analogie mit der Gleichung (3.17) die physikalische und die instrumentelle Linienverbrei-terung addiert. Dementsprechend kann die physikalische Linienbreite als die Differenz der beiden Li-
nienbreiten ∗∆ hd und ∗∆ gd berechnet werden:
∗∗∗ ∆−∆=∆ ghf ddd . (3.31)
Für Gauß-Funktionen f, g und h gilt in der Analogie mit der Gleichung (3.18), dass die Quadrate der ge-
messenen Linienbreiten ∗∆ hd und ∗∆ gd subtrahiert werden:
( ) ( ) ( )222 ∗∗∗ ∆−∆=∆ ghf ddd . (3.32)
39

Für die Entfaltung anderer Funktionen kann das Faltungstheorem genutzt werden, nach dem die Fourier-Transformation des Faltungsintegrals als Produkt der Fourier-Transformationen der gefalteten Funktio-nen geschrieben werden kann:
( ) ( ) ( ) ( )gFTfFTgfFThFT ⋅⋅=∗= π2 . (3.33)
Aus der Gleichung (3.33) kann dann die Funktion f folgendermaßen berechnet werden:
( )( )
= −
gFThFTFTf 1 , (3.34)
wo das Symbol FT-1 für die inverse Fourier-Transformation steht. Insbesondere bei Messdaten, die zufäl-lige Intensitätsschwankungen (Rauschen) enthalten, bietet das Faltungstheorem keine geeignete Lösung. Solche Messdaten bzw. deren Fourier-Transformationen FT(h) und FT(g) können geglättet werden, z.B.
mit Hilfe von Gauß-Funktionen ( )22exp ht σ− und ( )22exp gt σ− , die das Hochfrequenzrauschen unter-
drücken:
( ) ( )( ) ( )
−−
=′ −22
221
expexp
g
h
tgFTthFTFTf
σσ
. (3.35)
Der Grad der Glättung ist reziprok proportional dem Wert von σ. Aus der Sicht des Faltungstheorems bedeutet jedoch eine solche Glättung eine zusätzliche Faltung und daher eine Verbreiterung der beiden Funktionen, die formal als eine Verbreiterung der Funktion f‘ gegenüber der „idealen“ Funktion f ver-standen werden kann,
fsff ∗=′ , (3.36)
wo sf die inverse Fourier-Transformation der effektiven Glättungsfunktion ist:
( )[ ]221 exp ff tFTs σ−= − . (3.37)
Die Breite der Glättungsfunktion aus Gl. (3.37) ergibt sich aus den Breiten der Glättungsfunktionen, die an die Funktionen h und g angewendet wurden [46]:
22
222
hg
hgf σσ
σσσ
−= . (3.38)
Aus den Gleichungen (3.38) und (3.37) ist ersichtlich, dass nur Glättungsfunktionen mit 22hg σσ > sinnvoll
sind, d.h. dass die Funktion h stärker geglättet werden muss als die Funktion g.
Eine alternative Entfaltungsmethode haben Sánches-Bajo und Cumbrera [47] vorgeschlagen. Sie haben die gesuchte Funktion f in die Fourier-Reihe überführt,
40

( ) ( ) ( )∑∑==
+=m
jj
m
jj xjSxjCxf
00
00 sincos ωω mit
∆=
πω 20 , (3.39)
wo ∆ die Anzahl der Messdaten und m die Anzahl der Fourier-Koeffizienten ist. Das Faltungsintegral (3.30) kann dann als
( ) ( )[ ] ( )[ ]∑∑==
∗+∗=′m
jj
m
jj gxjSgxjCxh
00
00 sincos ωω (3.40)
geschrieben werden. Die Fourier-Koeffizienten Cj und Sj werden mit Hilfe der Methode der kleinsten Feh-lerquadrate ermittelt:
( ) ( )[ ] ( ) ( )[ ] ( ) minsincos1
2
00
00
2 =
−∗+∗=∑ ∑∑= ==
N
n
m
jj
m
jj xhxgxjSxgxjC ωωχ . (3.41)
In der Gleichung (3.41) bedeutet N die Anzahl der Messdaten, m die Anzahl der Fourier-Koeffizienten, g(x) die instrumentelle Linienverbreiterung und h(x) die an der untersuchten Probe gemessenen diffrak-tierten Intensitäten.
Für diskrete Messdaten (diffraktierte Intensitäten), die in einem endlichen Bereich der Beugungswinkel aufgenommen wurden, lässt sich das Faltungsintegral (3.30) als folgende Summe schreiben,
∑=
−=N
nmnnm gfh
0
, (3.42)
die optional als ein Matrixprodukt dargestellt werden kann:
=
⋅
+−+−+−−
−−−
−−
mnnmmmm
n
n
n
h
hhh
f
fff
gggg
gggggggggggg
3
2
1
3
2
1
21
2012
1101
210
. (3.43)
Die entfaltete Funktion f kann dann aus der Gleichung (3.43) mit Hilfe der Methode der kleinsten Fehler-quadrate unter der Einschränkung der nichtnegativen f-Werte (non-negative least-squares, NNLS) nach Lawson und Hanson [48] berechnet werden.
Bei allen Entfaltungsmethoden spielt die Kenntnis der instrumentellen Funktion eine zentrale Rolle. Aus der Gleichung (3.33) folgt, dass die instrumentelle Funktion direkt gemessen werden könnte, wenn die physikalische Linienverbreiterung der δ-Funktion entsprechen würde, δ≡f ,
( )[ ] ( )[ ] ggFTFTgFTFTh =⋅=∗= −− 111 δ . (3.44)
41

Man kann aber leider keinen Standard für Röntgenbeugungsexperimente finden, der keine intrinsische Linienverbreiterung zeigen würde. Daher muss damit gerechnet werden, dass mit Hilfe einer Standard-probe nicht nur die instrumentelle Linienverbreiterung des Diffraktometers gemessen wird, sondern auch die physikalische Linienverbreiterung, die durch die Realstruktur der Standardprobe verursacht wird. Diese Methode ist trotzdem immer anwendbar, wenn die physikalische Linienverbreiterung der gemessenen Probe größer ist als die physikalische Linienverbreiterung des Standards. Kann diese Vo-raussetzung nicht erfüllt werden, z.B. bei der Untersuchung von beinahe idealen Proben mit einer sehr kleinen eigenen physikalischen Linienverbreiterung, ist es ratsam, die instrumentelle Linienverbreiterung zu reduzieren. Dies bedeutet üblicherweise die Verbesserung der Spektralqualität der Primärstrahlung und die Verminderung der Divergenz des Primärstahles durch einen Primärmonochromator, sowie die Erhöhung der Winkelauflösung des Detektors, z.B. durch das Ersetzen der Detektorblende durch einen Kristallanalysator.
42

Literaturverzeichnis
[1] T. Egami und S. Billinge, Underneath the Bragg Peaks: Structural analysis of complex materials, Pergamon Press, 2003.
[2] R. Neder und T. Proffen, Diffuse Scattering and Defect Structure Simulations - a Cookbook using the Program DISCUS, Oxford University Press, 2009.
[3] L. Azároff, Elements of X-ray Crystallography, New York: McGraw-Hill Book Company, 1968.
[4] S. Caticha-Ellis, Anomalous Dispersion of X-rays in Crystallography, University College Cardiff Press: International Union of Crystallography, 1981.
[5] C. Giacovazzo, H. Monaco, G. Artioli, D. Viterbo, M. Milanesio, G. Gilli, P. Gilli, G. Zanotti, G. Ferraris und M. Catti, Fundamentals of Crystallography, Oxford University Press, 2011.
[6] E. (. Prince, International Tables for Crystallography, Volume C: Mathematical, physical and chemical tables, Wiley, 2006.
[7] A. Guinier, X-ray diffraction: In crystals, imperfect crystals, and amorphous bodies, San Francisco: Freeman, 1963.
[8] E. A. Merritt, „X-ray Anomalous Scattering,“ 2012. [Online]. Available: http://skuld.bmsc.washington.edu/scatter/AS_index.html. [Zugriff am November 2013].
[9] P. Debye, „Zerstreuung von Röntgenstrahlen,“ Annalen der Physik 46, pp. 809-823, 1915.
[10] B. Warren, X-ray Diffraction, New York: Dover Publications, Inc., 1990.
[11] S. Hendricks und E. Teller, „X-Ray Interference in Partially Ordered Layer Lattices,“ J. Chem. Phys. 10 (3), pp. 147-167, 1942.
[12] D. Rafaja, V. Klemm, G. Schreiber, M. Knapp und R. Kuzel, „Interference phenomena observed by X-ray diffraction in nanocrystalline thin films,“ J. Appl. Cryst. 37, pp. 613-620, 2004.
[13] P. Scherrer, „Bestimmung der Grösse und inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen,“ Nachr. Ges. Wiss. Göttingen 26, pp. 98-100, 1918.
[14] P. Suortti, „Effects of Porosity and Surface Roughness on the X-ray Intensity Reflected from a Powder Specimen,“ J. Appl. Cryst. 5, pp. 325-331, 1972.
[15] H. Hermann und M. Ermrich, „Microabsorption of X-ray Intensity in Randomly Packed Powder Specimens,“ Acta Cryst. A 43, pp. 401-405, 1987.
43

[16] W. Pitschke, H. Hermann und N. Mattern, „Microabsorption of scattered X-rays and its dependence on incidence angle in the nonsymmetric reflection case,“ J. Appl. Cryst. 26, pp. 132-134, 1993.
[17] V. Holý, U. Pietsch und T. Baumbach, High-Resolution X-Ray Scattering from Thin Films and Multilayers, Berlin: Springer-Verlag, 1999.
[18] D. Rafaja, V. Valvoda, J. Kub, K. Temst, M. Van Bael und Y. Bruynseraede, „Long-range periodicity and disorder in two-dimensional arrays of metallic dots studied by x-ray diffraction,“ Phys. Rev. B 61 (23), pp. 16144-16153, 2000.
[19] M. Born und E. Wolf, Principles of Optics, 7th edition, Cambridge: Cambridge University Press, 1999.
[20] D. Rafaja, V. Klemm, C. Wüstefeld, M. Motylenko, M. Dopita, M. Schwarz, T. Barsukova und E. Kroke, „Interference phenomena in nanocrystalline materials and their application in the microstructure analysis,“ Zeitschrift für Kristallographie, Supplement 27, pp. 15-26, 2008.
[21] G. Williamson und A. Hall, „X-ray line broadening from filed aluminium and wolfram,“ Acta Metall. Mater. 1, pp. 22-31, 1953.
[22] D. Rafaja, C. Wüstefeld, M. Dopita, M. Růžička, V. Klemm, G. Schreiber, D. Heger und M. Šíma, „Internal structure of clusters of partially coherent nanocrystallites,“ Surf. Coat. Technol. 201 , pp. 9476-9484, 2007.
[23] D. Rafaja, C. Wüstefeld, M. Dopita, V. Klemm, D. Heger, G. Schreiber und M. Šíma, „Formation of defect structures in hard nanocomposites,“ Surf. Coat. Technol. 203, pp. 572-578, 2008.
[24] D. Rafaja, V. Klemm, M. Motylenko, M. Schwarz, T. Barsukova, E. Kroke, D. Frost, L. Dubrovinsky und N. Dubrovinskaia, „Synthesis, microstructure and hardness of bulk ultrahard BN nanocomposites,“ J. Mater. Res. 23, pp. 981-993, 2008.
[25] G. Ribárik, N. Audebrand, H. Palancher, T. Ungár und D. Louër, „Dislocation densities and crystallite size distributions in nanocrystalline ball-milled fluorides, MF2 (M = Ca, Sr, Ba and Cd), determined by X-ray diffraction line-profile analysis,“ J. Appl. Cryst. 38, pp. 912-926, 2005.
[26] D. Rafaja, C. Wüstefeld, M. Motylenko, C. Schimpf, T. Barsukova, M. Schwarz und E. Kroke, „Interface phenomena in (super)hard nitride nanocomposites: from coatings to bulk materials,“ Chem. Soc. Rev. 41, pp. 5081-5101, 2012.
[27] A. Leineweber, „Ordered and disordered states in NiAs/Ni2In-type Ni1+δSn: Crystallography and order formation,“ Intern. J. Mater. Res. 102, pp. 861-873, 2011.
[28] E. Fullerton, I. Schuller, H. Vanderstraeten und Y. Bruynseraede, „Structural refinement of superlattices from X-ray diffraction,“ Physical Review B 45, pp. 9292-9310, 1992.
44

[29] S. Martin, C. Ullrich, D. Šimek, U. Martin und D. Rafaja, „Ordering of stacking faults during the plastic deformation of austenitic steels,“ J. Appl. Cryst. 44, pp. 779-787, 2011.
[30] T. De Keijser, J. Langford, E. Mittemeijer und A. Vogels, „Use of the Voigt Function in a Single-Line Method for the Analysis of X-ray Diffraction Line Broadening,“ J. Appl. Cryst. 15, pp. 308-314, 1982.
[31] M. Wilkens, „The determination of density and distribution of dislocations in deformed single crystals from broadened X-ray diffraction profiles,“ Phys Status Solidi A2, pp. 359-370, 1970.
[32] M. A. Krivoglaz, X-ray and Neutron Diffraction in Nonideal Crystal, Berlin, Heidelberg: Springer-Verlag, 1996.
[33] P. Klimanek und R. Kužel, „X-ray diffraction line broadening due to dislocations in non-cubic materials. I. General considerations in the case of elastic isotropy applied to hexagonal crystals,“ J. Appl. Cryst. 21, pp. 59-66, 1988.
[34] R. Kužel und P. Klimanek, „X-ray diffraction line broadening due to dislocations in non-cubicmaterials. II. The case of elastic anisotropy in hexagonal crystals,“ J. Appl. Cryst. 21, pp. 363-368, 1988.
[35] T. Ungár und A. Borbély, „The effect of dislocation contrast on x-ray line broadening: A new approach to line profile analysis,“ Appl. Phys. Letters 69 (21), pp. 3173-3175, 1996.
[36] T. Ungár, I. Dragomir, Á. Révész und A. Borbély, „The contrast factors of dislocations in cubic crystals: the dislocation model of strain anisotropy in practice,“ J. Appl. Cryst. 32, pp. 992-1002, 1999.
[37] I. Noyan und J. Cohen, Residual Stress - Measurement by Diffraction and Interpretation, New York, Berlin, Heidelberg: Springer-Verlag, 1987.
[38] I. Dragomir und T. Ungár, „Contrast factors of dislocations in the hexagona crystal system,“ J. Appl. Cryst. 35, pp. 556-564, 2002.
[39] H. Mughrabi, „Dislocation Wall and Cell Structures and Long-range Internal Stresses in Deformed Metal Crystals,“ Acta metall. 31 (9), pp. 1367-1379, 1983.
[40] D. Rafaja, C. Krbetschek, D. Borisova, G. Schreiber und V. Klemm, „In situ X-ray diffraction study of stacking faults formation in the near-surface region of transformation induced plasticity steels,“ Thin Solid Films 530, pp. 105-112, 2013.
[41] D. Rafaja, C. Wüstefeld, M. Dopita, M. Motylenko, C. Bähtz, C. Michotte und M. Kathrein, „Crystallography of phase transitions in metastable titanium aluminium nitride nanocomposites,“ Surf. Coat. Technol., p. doi: 10.1016/j.surfcoat.2014.01.039, 2014.
45

[42] J. Biscoe und B. Warren, „An X-ray study of carbon black,“ J. Appl. Phys. 13, pp. 364-371, 1942.
[43] A. Kurdyumov, „Stacking faults in graphitic boron nitride,“ Sov. Phys. Cryst. 20, pp. 596-598, 1976.
[44] K. Ufer, G. Roth, R. Kleeberg, H. Stanjek, R. Dohrmann und J. Bergmann, „Description of X-ray powder pattern of turbostratically disordered layer structures with a Rietveld compatible approach,“ Z. Kristallographie 219, pp. 519-527, 2004.
[45] C. Schimpf, M. Motylenko und D. Rafaja, „Quantitative description of microstructure defects in hexagonal boron nitrides using X-ray diffraction analysis,“ Materials Characterization 86, pp. 190-199, 2013.
[46] D. Rafaja, „Deconvolution versus convolution - a comparison for materials with concentration gradient,“ Materials Structure 7(2), pp. 43-50, 2000.
[47] F. Sánchez-Bajo und F. Cumbrera, „Deconvolution of X-ray diffraction profiles by using series expansion,“ J. Appl. Cryst. 33, pp. 259-266, 2000.
[48] C. Lawson und R. Hanson, Solving Least Squares Problems, Englewood Cliffs, NJ: Prentice-Hall, 1974.
[49] T. Ungár und G. Tichy, „The effect of dislocation contrast on X-ray line profiles in untextured polycrystals,“ Phys. Status Solidi A 171, pp. 425-434, 1999.
46




![Modulhandbuch: BSc MaWi...6 Modul: Orientierung I verantwortlich: Studiendekan und Fachschaft Materialwissenschaft Titel der Lehrveranstaltung [B.O1] Orientierung Studium (Einführungsveranstaltung)](https://static.fdokument.com/doc/165x107/5e247ef40e3f4b15ca5a137e/modulhandbuch-bsc-mawi-6-modul-orientierung-i-verantwortlich-studiendekan.jpg)













