
Ruhr Universität Bochum · Im Tractus opticus ziehen die Fasern weiter zum Corpus geniculatum...
108
Ruhr Universität Bochum Prof. Dr. rer. nat. Th. Mittmann Dienstort: Johannes Gutenberg-Universität Abt. Physiologie und Pathophysiologie Die Rolle des neurotrophen Faktors BDNF für läsionsinduzierte Plastizität im visuellen Kortex der BDNF (+/-) Maus Inaugural-Dissertation Zur Erlangung des Doktorgrades der Medizin Einer Hohen Medizinischen Fakultät Der Ruhr-Universität Bochum Vorgelegt von Sarah Breiter Aus Vreden 2010
Transcript of Ruhr Universität Bochum · Im Tractus opticus ziehen die Fasern weiter zum Corpus geniculatum...

Ruhr Universität Bochum Prof. Dr. rer. nat. Th. Mittmann
Dienstort: Johannes Gutenberg-Universität Abt. Physiologie und Pathophysiologie
Die Rolle des neurotrophen Faktors BDNF für läsionsinduzierte Plastizität im visuellen Kortex der BDNF (+/-) Maus
Inaugural-Dissertation Zur Erlangung des Doktorgrades der Medizin
Einer Hohen Medizinischen Fakultät Der Ruhr-Universität Bochum
Vorgelegt von Sarah Breiter Aus Vreden
2010

Dekan: Prof. Dr. med. G. Muhr Referent: Prof. Dr. rer. nat. Th. Mittmann Korrekferent Prof. Dr. rer. nat. G. Zoidl Tag der mündlichen Prüfung: 17.05.2011

Abstract
Nach Läsionen im zerebralen Kortex kann das Gewebe in der Nähe der Läsion
teilweise den funktionellen Verlust kompensieren, der durch die Läsion
entstanden ist. Die Mechanismen dieser erhöhten Plastizität sind jedoch nicht
ausreichend verstanden. Der neurotrophe Faktor BDNF könnte die
läsionsinduzierte Plastizität vermitteln, da er unter anderem für Reifung und
Stabilisierung von Synapsen verantwortlich ist und ihm eine große Rolle bei
Lernprozessen wie LTP zugesprochen wird.
Um die Rolle von BDNF bei läsionsinduzierter Plastizität näher zu
untersuchen, wurden heterozygote BDNF knockout Tiere mit Wt Tieren
verglichen. Bei 21 Tage alten narkotisierten Mäusen wurden Läserläsion im
visuellen Kortex induziert. Nach einer Überlebenszeit von zwei bis sechs Tagen
wurden Gehirnschnitte der Tiere elektrophysiologisch untersucht. Dabei konnte
in Wt Kontrolltieren nach Reizung der kortikalen Schicht IV eine
Langzeitpotenzierung (LTP) von extrazellulär abgeleiteten Feldpotentialen in
Schicht II/III beobachtet werden, während BDNF(+/-) Mäuse keine LTP
ausbildeten. Wt Tiere zeigten am Läsionsrand eine erhöhte LTP von ca. 40%.
Überraschenderweise wurde bei BDNF(+/-) Mäusen post-Läsion ebenfalls eine
stabile LTP von etwas mehr als 20% gemessen. Durch die Läsion wurden also
Mechanismen ausgelöst, die sowohl in Wt Tieren wie auch in BDNF(+/-)
Mäusen zu einer Erhöhung, bzw. Wiederherstellung der LTP führen.
Um diese Mechanismen näher zu untersuchen, wurde die Expressions-
Stärke der BDNF mRNA gemessen. 24h nach Läsion zeigte sich am Rande der
Läsion in BDNF(+/-) Tieren wie auch in Wt Tieren eine signifikante Reduktion
der BDNF mRNA, nach fünf Tagen war kein Unterschied im BDNF Level mehr
zu messen.
Diese Daten zeigen, dass die Läsions-induzierte Auslösbarkeit von LTP
in BDFN(+/-) Mäusen ebenso wie die gesteigerte LTP nach Läsionen in Wt
Tieren mit einer Erniedrigung der BDNF-Expression einher geht. Dies könnte
das bereits beschriebene Ungleichgewicht zwischen kortikaler Hemmung und
Erregung post-Läsion ermöglichen und so zu der hier beobachteten
verbesserten synaptischen Plastizität in den BDNF(+/-) Tieren am Läsionsrand
führen.

Meiner Familie

I
Inhaltsverzeichnis
1 Einleitung 1
1.1 Das visuelle System 1 1.1.1 Die Sehbahn 1 1.1.2 Aufbau des primär visuellen Kortex 3 1.1.3 Zellen des primär visuellen Kortex 4
1.2 Exitatorisches und Inhibitorisches System 5 1.3 Synaptische Plastizität 6
1.3.1 Langzeitpotentierung 8 1.3.2 Läsionsinduzierte Plastizität 10
1.4 Neurotrophine 14 1.4.1 Das Neurotrophin BDNF 15 1.4.2 BDNF und Epilepsie 16 1.4.3 BDNF und synaptische Transmission 17 1.4.4 Bedeutung von BDNF für Plastizität 18 1.4.5 Das Modell der BDNF(+/-) Maus 20
1.5 Ziele der Arbeit 21 2 Material und Methoden 24
2.1 Tiere 24 2.2 Genotypisierung 25 2.3 In vivo Läsion 26
2.3.1 Sham-Operationen 27 2.4 Herstellung von akuten Gehirnschnitten 28 2.5 Histologie 29
2.5.1 Nissl-Färbung 29 2.5.2 Immunhistochemie:GFAP-Färbung 29
2.6 Elektrophysiologischer Messplatz 30 2.7 Elektrophysiologie 32
2.7.1 Elektrophysiologische Messungen 32 2.7.2 Auswertung der extrazellulären Messungen 35
2.8 Messung der BDNF mRNA-Konzentration im
visuellen Kortex mittels RT-PCR 35 2.8.1 Herstellung der cDNA 36 2.8.2 DNA-Amplifikation mit der Polymerase-Kettenreaktion 39 2.8.3 Agarose-Gelelektrophorese 40 2.8.4 Auswertung der Messung der mRNA-Konzentration 41
Formatiert
Formatiert

II
3 Ergebnisse 42
3.1 Histologie 42 3.2 Ergebnisse der Elektrophysiologie 44
3.2.1 Einfluss von Läsionen auf synaptische Plastizität im visuellen Kortex der Maus 46
3.2.1.1 Basale synaptische Übertragung (I-O-Kurve) nach Läsion 46
3.2.1.2 Verstärkte LTP nach Läsionen im visuellen Kortex 48 3.2.1.3 Keine Veränderung in der LTP nach Scheinoperation 50
3.2.2 Veränderte synaptische Plastizität im visuellen Kortex der BDNF(+/-) Maus 52
3.2.2.1 Basale synaptische Übertragung (I-O Kurve) in Hirnschnitten von BDNF (+/-) Mäusen 52 3.2.2.2 Beeinträchtigte LTP Expression in
BDNF(+/-) Mäusen 54
3.2.3 Einfluss von Läsionen auf die LTP Expression in BDNF(+/-) Mäusen 56 3.2.3.1 Basale synaptische Übertragung (I-O Kurve) in
BDNF(+/-) Mäusen nach Läsion 56 3.2.3.2 Wiederherstellung von LTP in BDNF(+/-) Mäusen
nach Läsion 58 3.2.3.3 Keine Änderung der LTP nach Scheinoperationen
bei BDNF(+/-) Mäusen 60 3.2.3.4 Vergleich zwischen Ausprägung der LTP in
unbehandelten Wt-Mäusen und läsionierten BDNF(+/-) Mäusen 62
3.2.4 Vergleich der LTP zwischen verschiedenen Versuchsgruppen 62
3.3 Ergebnisse der RT-PCR 64 3.3.1 Expression von BDNF nach Läsion 64
Formatiert

III
4 Diskussion 68
4.1 Gliose in der Umgebung der Läsion 69 4.2 Typische Form der Feldpotentiale 69 4.3 Diskussion der Methode 70 4.4 Verstärkte LTP nach Läsion im visuellen Kortex der Wt Maus 72 4.5 Erneute Auslösbarkeit von LTP nach Läsion im visuellen Kortex der BDNF(+/-) Maus 74 4.6 Quantifizierung der BDNF mRNA 76 4.7 BDNF und Hemmung 78 4.8 Räumliche und zeitliche Veränderung der BDNF Expression - nach Läsion 79 4.9 Zusammenfassung der Diskussion 82
5 Literaturverzeichnis 84
Formatiert
Formatiert
Formatiert

Abkürzungen
A Ampere
ACSF zerebrospinale Flüssigkeit
Aqua dest. Destilliertes Wasser
BDNF brain-derived neurotrophic factor
°C Grad Celsius
cAMP zyklisches Adenosinmonophosphat
D-AP5 D-2-amino-5-phosphonopentanoate
DNA Desoxyribonuclein-Säure
DNQX 6,7-Dinitroquinoxalin-2,3-dion
FP Feldpotentiale
GABA γ-Aminobuttersäure
GAD Glutamat-Decarboxylase
GFAP Glial fibrillary acidic protein (Gliäres saures Filamentprotein)
Hz Hertz
KO Knockout
LTD Langzeit-Depression (engl.: long-term depression)
LTP Langzeit-Potenzierung (engl.: long-term potentiation)
n Anzahl der Experimente
NMDA N-Methyl-D-Aspertat
p Irrtumswahrscheinlichkeit
PARV Parvalbumin
PBS Phosphate-Buffer-Solution (Phosphat-Puffer-Lösung)
RNA Ribonucleinsäure
SEM Standartfehler des Mittelwertes
TBS Theta burst stimulation
Trk A/B/C Tyrosin Kinase Rezeptor A/B/C
V Volt

1
1. Einleitung
Synaptische Plastizität ist seit Jahrzehnten ein wichtiger
Forschungsgegenstand der Neurowissenschaft. Unter dem Begriff der
synaptischen Plastizität werden sowohl funktionelle als auch strukturelle
Veränderungen an Neuronen des Nervensystems als Reaktion auf Umweltreize
zusammengefasst. Diese Fähigkeit des Nervensystems, sich individuell der
Umwelt anzupassen ist insbesondere bei jungen Tieren in der sogenannten
„kritischen Periode“ (Kirkwood et al., 1995) sehr hoch und nimmt im Alter
dramatisch ab. Gleichwohl gibt es Mechanismen, die auch dem adulten Gehirn
eine hohe Plastizität ermöglichen. Insbesondere nach Hirnverletzungen scheint
das umliegende Gewebe in beeindruckendem Maße fähig zu sein, sich
funktionell zu reorganisieren und somit den Ausfall der verletzten Region
auszugleichen (Taub et al. 2002). Eine erhöhte Plastizität nach Verletzungen
wurde bereits in verschiedenen Hirnarealen beobachtet, wie zum Beispiel im
somatosensorischen (Jenkins & Merzenich, 1987) und im visuellen System
(Eysel & Schweigart, 1999; Mittmann & Eysel, 2001). Entgegen der früheren
Annahme, dass Nervenzellen ihr Leben lang nur einen Aufgabenbereich
erfüllen können, wird nun immer deutlicher, dass diese Zellen nach
entsprechender Modulation auch Aufgaben anderer Nervenzellen übernehmen
und so den Ausfall, der zum Beispiel aus einer Hirngewebeschädigung
resultiert, ausgleichen können.
1.1 Das visuelle System
1.1.1 Die Sehbahn
Sehen ist ein komplexer Vorgang, der einer Umwandlung eines physikalischen
Reizes (Licht) in ein elektrisches Signals bedarf. Visuelle Informationen der
Umwelt treffen in Form von Lichtwellen einer bestimmten Frequenz auf die
Netzhaut des Auges. Die in der Netzhaut enthaltenen Photorezeptoren wandeln
das Licht in elektrische Impulse um, die vom Axon der Fotorezeptorzelle
weitergeleitet wird. Über Schaltstationen wird das Bild verarbeitet und über
Ganglienzellen an die Großhirnrinde (Cortex cerebri) geschickt. Dabei werden
Detail-, Farb- und Bewegungsinformationen bereits in der Retina von

2
unterschiedlichen Zellklassen verarbeitet. Das magnozelluläre System der
Ganglienzellen zeichnet sich durch hohe Kontrast- und
Bewegungsempfindlichkeit aus. Das parvozelluläre System ist dagegen
farbempfindlich und auf Detailanalyse spezialisiert. Die funktionelle Unterteilung
des magno- und parvozellulärem System bleibt auch auf dem gesamten Weg
der Signalverarbeitung bis in den visuellen Kortex erhalten.
Zunächst trifft der optische Reiz dabei auf die Sinneszellen der Retina.
Hier vermitteln sogenannte Stäbchen die Hell- Dunkel- Wahrnehmung und
Zapfen die Farbwahrnehmung. Von diesem ersten Neuron, das auf der äußeren
Körnerzellschicht liegt, wird das Signal an das zweite Neuron der Sehbahn, die
Bipolarzelle, weitergeleitet. Diese befindet sich auf der inneren
Körnerzellschicht. Die nächste Umschaltung von Bipolarzellen auf
Ganglienzellen wird ebenso innerhalb der Retina organisiert. Durch
Horizontalzellen und Amakrine Zellen erfolgt eine weitere Verarbeitung des
Signals auf dem Weg zur Ganglienzelle. Durchschnittlich enthält jede
Ganglienzelle Input aus ca. 126 Millionen Photorezeptoren. Um diese Fülle an
Information verarbeiten zu können gibt es laterale Hemmungsmechanismen,
die durch sogenannte Off-Zentren zur inhibitorischen Off-Reaktion führen. In
den On-Zentren kommt es hingegen zu einer exitatorischen On-Reaktion. Die
langen Axone der Ganglienzellen bilden den Sehnerven (Nervus opticus). Die
beiden Sehnerven des rechten und linken Auges vereinigen sich dann oberhalb
der Hypophyse im Chiasma opicum. Beim Menschen kreuzen sich die Fasern,
die aus dem nasalen Bereich der Netzhaut (somit aus den temporalen
Gesichtsfeldhälften) stammen, während die Fasern der lateralen
Netzhauthälften ungekreuzt verlaufen. Im Tractus opticus ziehen die Fasern
weiter zum Corpus geniculatum laterale (CGL), einem Kerngebiet des
Thalamus, wo eine Verarbeitung und Umschaltung der Nervenfasern stattfindet.
Von dort erstreckt sich schließlich das vierte Neuron der Sehbahn in der
Sehstrahlung (Radiatio optica) zum primären visuellen Kortex (V1) im Bereich
des Okzipitalpols, der nomenklatorisch der Area 17 nach Brodmann entspricht.
Über die gesamte Sehbahn hinweg wird die räumliche Gestalt der
Reizeinwirkung beibehalten, was man als Retinotopie bezeichnet. Dies
bedeutet, dass nebeneinander liegende Sinneszellen auch in benachbarte
Gebiete im ZNS projizieren. Dabei wird eine Nervenzelle im primären visuellen

3
Kortex immer von den selben Ganglienzellen der Netzhaut erregt. Dieser
Bereich des Gesichtfeldes, in dem visuelle Reize eine bestimmte Nervenzelle
erregen wird als rezeptives Feld bezeichnet. Nebeneinander liegende retinale
Ganglienzellen besitzen benachbarte und auch überlappende rezeptive Felder
und projizieren zu benachbarten Neuronen der nächst höheren Stufe. Diese
räumliche Ordnung bleibt von der Rezeptorebene in der Retina bis in die
höheren Verarbeitungsebenen im Kortex weitgehend erhalten.
1.1.2 Aufbau des primär visuellen Kortex
Die Verarbeitung im primären visuellen Kortex geschieht durch drei
verschiedene Zelltypen (Hubel & Wiesel, 1959). Die einfachen Zellen (simple
cells) entdecken durch die oben genannten On- und Off-Zentren die
Orientierung im Raum. Die komplexen Zellen (complex cells) sind für
Entdeckung der Reizbewegung zuständig. Der dritte Zelltyp, die
hyperkomplexen Zellen (hypercomplex cells), reagiert nicht nur auf Orientierung
und Bewegungsrichtung, sondern zusätzlich auf Länge, Breite oder andere
Eigenschaften von Formen.
Der primäre visuelle Kortex ist auf Grund morphologischer Merkmale in
sechs nicht scharf begrenzte parallel zur Kortexoberfläche verlaufende
Schichten unterteilt (layer I-VI): Schicht I: Molekularschicht (lamina molecularis)
– sie ist die äußerste Schicht und enthält fast ausschließlich Fasern aus
anderen Hirnregionen. Diese Schicht enthält nur wenig Zellen, deren Dendriten
parallel zur Kortexoberfläche verlaufen. Es finden sich vor allem dünne Axone,
Dendriten und synaptische Verbindungen (Hoffmann und Wehrhahn, 2001).
Schicht II: äußere Körnerschicht (lamina granularis externa) und Schicht III:
äußere Pyramidenschicht (lamina piramidalis externa) werden häufig
zusammengefasst, da es keine deutliche Grenze zwischen ihnen gibt. Hier
befinden sich Pyramidenzellen, die in Schicht III größer sind als in II. Ihre Axone
verlaufen in vertikaler Richtung zur Kortexoberfläche in tiefer gelegene
intrakortikale Schichten (Kisvárday et al., 1997) und senden ebenfalls
Axonkollaterale in horizontaler Richtung und bilden somit kortiko-kortikale
Faserverbindungen. Schicht IV: innere Körnerzellschicht (lamina granularis
interna) – diese Schicht ist im primären visuellen Kortex besonders stark

4
ausgebildet, da sie über den Thalamus Afferenzen von den sensorischen
Neuronen erhält. Sie ist reich an kleinen Sternzellen mit kurzen Axonen, die ihre
Signale dann zunächst in die Schichten II-III und dann zu den Schichten V und
VI schicken. Zellen der Schicht IV besitzen überwiegend Horizontalfasern.
Schicht V: innere Pyramidenschicht (lamina pyramidalis interna) – sie ist durch
die größten Pyramidenzellen gekennzeichnet. Efferenzen aus dieser Schicht
projizieren zurück zum Mittelhirn und schicken ihre Signale ebenfalls zum
kontralateralen Kortex. Schicht VI: multiforme Schicht (lamina multimorfis) –
diese Schicht wird von Zellen verschiedener Morphologie aufgebaut, deren
aufsteigende Neuriten zu oberflächlichen Rindenschichten ziehen. Die großen
Pyramidenzellen dieser Schicht liefern Rückprojektionen zum Corpus
geniculatum laterale des Thalamus.
1.2.3 Zellen des primär visuellen Kortex
Der Kortex enthält zwei Haupttypen von Neuronen: Pyramidenzellen und Nicht-
Pyramidenzellen Die Pyramidenzellen machen ca. 85 % der Neurone im primär
visuellen Kortex aus und verdanken ihren Namen dem annähernd dreieckigen
Zellkörper (Soma). Sie sind vorwiegend in Schicht III und V zu finden. Ihre
feinen langen Dentriten sind dicht mit Dornfortsäzten (spines) besetzt sind.
Über diese synaptische Verbindung empfangen sie exitatorischen Input anderer
Nervenzellen. Am Zellkörper hingegen sitzen vorwiegend inhibitorische
Synapsen. Die Axone der Pyramidenzellen verzweigen sich in alle Richtungen
und projizieren sowohl in andere Hirnregionen als auch zu Neuronen der
Umgebung. Sie senden exitatorische Reize zu zum Teil weit entfernten Zellen.
Die Nicht-Pyramidenzellen kommen besonders dich in Schicht IV des primär
visuellen Kortex vor. Sie sind kleiner als Pyramidenzellen und haben eine
unregelmäßigere Gestalt und Anordnung als die Pyramidenzellen. Als
sogenannte Sternzellen besitzen sie eine sternenförmige Morphologie mit
entweder „dornigen“ (spiny stellate cells) oder „glatten“ (smooth stellate cells)
Zellkörpern. Die Pyramidenzellen und die dornigen Sternzellen sind exitatorisch
und benutzen als Transmitter vorwiegend Glutamat (Glu). Werden synaptische
Übertragungen von Schicht IV zu Schicht II/III gemessen, so sind dies
vorwiegend exitatorische Signale (Kirkwood & Bear, 1994). Daneben haben die

5
GABAergen glatten Sternzellen inhibitorische Funktion (Wurtz & Kandel, 2000).
Im Gegensatz zu Pyramidenzellen haben Nicht-Pyramidenzellen relativ kurze
Axone und nehmen nur Kontakt zu benachbarten Zellen auf und schicken ihre
Axone nicht in andere Teile des Gehirns.
1.2 Exitatorisches und Inhibitorisches System
Die Kommunikation zwischen Neuronen im Nervensystem erfolgt über
synaptische Verbindungen, die die Information entweder elektrisch durch Strom
oder chemisch durch Transmitter weitergeben. Es gibt im Wesentlichen zwei
Arten von chemischen Synapsen im zentralen Nervensystem: erregende und
hemmende. Dabei wird im sog. präsynaptischen Anteil der Transmitter
ausgeschüttet, der an Rezeptoren des sog. postsynaptischen Teiles bindet und
so seine Wirkung entfaltet. Bei hemmenden Synapsen wird durch den
wichtigsten hemmenden Transmitter γ-Aminobuttersäure (GABA) eine
Hyperpolarisierung des Ruhemembranpotentials der Zelle und somit ein
inhibitorisches postsynaptisches Potential (IPSP) ausgelöst. Dahingegen
kommt es durch Glutamat, den wichtigsten Transmitter der erregenden
Synapsen, zu einer Depolarisierung des Ruhemembranpotentials und somit zu
einem exitatorischen postsynaptischen Potential (EPSP).
Normalerweise herrscht ein gewisses Gleichgewicht zwischen diesen
hemmenden und erregenden Transmittern (Martin, 1993), das unter anderem
durch ein Enzym, die Glutamat-Decarboxylase (GAD) aufrecht erhalten, bzw
moduliert wird. Durch GAD wird Glutamat durch Decarboxylierung in GABA
umgewandelt (Roberts & Frankel, 1950). GAD kommt in den meisten
GABAergen Interneuronen in zwei Isoformen vor, GAD65 und GAD67, die
beide in der Synthese des GABA-Transmitterpools involviert sind. GAD67 findet
sich als freies Protein vor allem im Cytoplasma, während GAD65 stärker in
Axonterminalen lokalisiert und mit synaptischen Vesikeln assoziiert ist
(Esclapez et al., 1994). Kleine Änderungen im Gleichgewicht zwischen
Hemmung und Erregung können bereits die neuronale
Informationsverarbeitung beeinflussen (Liu, 2002). Kann das Gleichgewicht
zwischen Glutamat und GABA nicht aufrecht erhalten werden, z.B. bei GAD
knock out Tieren, resultieren schwerwiegende Folgen. GAD-65 knock out Tiere

6
synthetisieren weniger GABA und entwickeln in Folge dessen schnell Epilepsie,
während ein GAD-67 knock out sogar letal ist, da diese Tiere am Tag der
Geburt an einer ausgeprägten Gaumenspalte sterben (Asada et al., 1997).
1.3 Synaptische Plastizität
Die Fähigkeit des Gehirns, sich an unterschiedliche Umweltbedingungen
anzupassen, wird als neuronale Plastizität bezeichnet. Dies wird durch eine
veränderte synaptische Übertragung von einem Neuron zu einem anderen
erreicht. Diese Verbindungen zwischen Neuronen können durch
Veränderungen während der Entwicklung, bei Lernvorgängen, bei
Regenerationsprozessen nach Verletzungen, erfahrungsbedingt und bei
altersbedingten Degenerationsprozessen moduliert werden. Dazu stehen
verschiedene Mechanismen zur Verfügung. Unter anderem sind dies die
Ausbildung neuer dendritischer axonaler oder terminaler Neuronenfortsätze, die
Knüpfung zusätzlicher synaptischer Kontakte und die bedarfsorientierte
Modifizierung biochemischer Prozesse (Kasai et al., 2010).
Ein Beispiel für diese Umstrukturierung und Anpassung an äußere
Bedingungen ist, das eine Auge bei einem jungen Patienten für einen
definierten Zeitraum zu verschliessen, während das andere normal weiter
benutzt wird. Nach einer bestimmten Zeit entwickelt sich eine sog. Amblyopie.
Das heißt, dass auf dem zuvor geschlossenen Auge auch nach dem Öffnen im
schlimmsten Falle sogar eine funktionelle Blindheit entstehen kann, obwohl
sowohl Retina, Thalamus, visueller Kortex wie auch die nervalen Verbindungen
keinen äußeren Schaden erlitten haben (Wiesel & Hubel, 1963; Berardi et al.,
2000). Im visuellen Kortex findet währenddessen eine aktive Reorganisation
statt. Der Input jedes Auges wird im Kortex durch bestimmte Bereiche
repräsentiert. Dabei existieren sog. ocular dominance columns, die das
dominante Auge erkennen lassen (Hubel & Wiesel, 1976; Shatz & Stryker,
1978). Nach einer bestimmten Zeit nach Schließen eines Auges (monokkulare
Deprivation) wird der Bereich im Kortex, der dieses Auge repräsentiert, immer
kleiner, der Bereich, der das andere Auge widerspiegelt, dafür größer (Antonini
& Stryker, 1996; Antonini et al., 1999). Die bipolaren Neurone des visuellen
Kortex verarbeiten nur noch Eingänge vom normal benutzen Auge. Durch den

7
fehlenden Aktivitätseingang des verschlossenen Auges werden diese Eingänge
also abgeschwächt, die anderen dafür verstärkt. Wie schnell diese
Reorganisation umgesetzt wird, bzw. wie viel Zeit vergeht, bis das
geschlossene Auge funktionell erblindet, ist von dem Zeitpunkt der Entwicklung
abhängig. Es gibt eine bestimmte Phase im Leben einer jeden
Säugetierspezies, in der diese Plastizität besonders groß ist. Diese Phase ist in
der frühen Entwicklung angesiedelt und proportional zur Lebenserwartung der
Spezies. Jeder kennt sicher aus eigener Erfahrung, dass es jungen Menschen
leichter fällt etwas Neues zu Lernen oder Dinge zu behalten. Diese Zeitspanne
der genetisch vorprogrammierten erhöhten Plastizität wird critical period (d:
kritische Periode) genannt (Hubel & Wiesel, 1970; Hubel et al., 1977; Berardi et
al., 2000). Bei Mäusen beginnt diese Entwicklungsphase etwa eine Woche
nachdem die Tiere die Augen öffnen (ungefähr am 13. postnatalen Tag), also
ca. am 20. postnatalen Tag und hat ihren Höhepunkt ungefähr 28 Tage nach
der Geburt (Gordon & Stryker, 2003).
Durch welche Mechanismen Plastizität vermittelt wird und wie diese weiter
modiziert oder sogar verstärkt werden können ist seit Jahrzehnten Gegenstand
der neurowissenschaftlichen Forschung. Bereits im Jahre 1949 wurde die
Hebbsche Regel aufgestellt, die noch heute als grundlegendes Prinzip für
synaptische Plastizität und Lernen gilt. In seinem Werk The Organization of
Behaviour heisst es:
„Wenn ein Axon der Zelle A [%] Zelle B erregt und wiederholt und
dauerhaft zur Erzeugung von Aktionspotenzialen in Zelle B beiträgt, so resultiert
dies in Wachstumsprozessen oder metabolischen Veränderungen in einer oder
in beiden Zellen, die bewirken, dass die Effizienz von Zelle A in Bezug auf die
Erzeugung eines Aktionspotenzials in B größer wird.“ (Hebb, 1966). Eine
verstärkte synaptische Übertragung über einen längeren Zeitraum nennt man
Langzeitpotentierung (engl.: long-term potentiation; LTP) (Bliss & Lomo, 1973),
geschwächte synaptische Effektivität hingegen Langzeitdepression (engl.:
longterm depression; LTD) (Artola et al., 1990; Dudek & Bear, 1993).

8
1.3.1 Langzeitpotentierung
Hier soll näher auf LTP eingegangen werden, die als wichtigstes zelluläres
Korrelat von Lern- und Gedächtnisvorgängen angesehen wird (Holscher, 1999).
Man teilt die Veränderungen nach Induktion von LTP nach ihrer zeitlichen
Abfolge in drei Phasen ein (Sweatt, 1999). Die unmittelbaren Kurzzeiteffekte,
die direkt nach der Induktion von LTP greifen, werden short-term potentiation
(STP) genannt. Posttranslationale Modifikationen an schon vorhandenen
Proteinen, die Minuten bis Stunden dauern, werden zeitlich dem early LTP
zugeordnet. Die späteren, auf veränderte Expression von Proteinen
zurückzuführenden Effekte werden late LTP genannt und dauern Stunden in
vitro und Tage bis Monate in vivo und können sogar ein Leben lang anhalten
(Bliss & Gardner-Medwin, 1973; Abraham et al., 2002).
Bisher sind noch nicht alle zellulären und molekularen Mechanismen, die
zu LTP führen, bekannt. Die Induktion und Aufrechterhaltung von LTP ist sehr
komplex und bedarf vieler molekularer Mechanismen. Bevor an einer Synapse
eine LTP induziert wird, erfolgt die basale synaptische Übertragung über eine
Aktivierung von α-Amino-3-hydroxy-5-methyl-4-isoxazolpropionsäure (AMPA)-
Rezeptorkanälen. Als ionotrope Glutamatrezeptoren ermöglichen sie erst nach
Aktivierung durch einen entsprechenden Liganden (in diesem Falle Glutamat)
den Durchtritt eines bestimmten Ions durch den Rezeptorkanal. Glutamat wird
von der Präsynapse ausgeschüttet und setzt sich an den AMPA-Rezeptor, der
dadurch aktiviert wird. Dies ermöglicht einen Natrium-Einstrom und einen
schwachen Kalium-Ausstrom. Insgesamt kommt es zur schnellen
Depolarisation der Zelle. Wichtig für die Induktion von LTP sind nun
spannungsabhängige N-Methyl-D-Aspertat (NMDA) Rezeptoren (Malenka &
Bear, 2004). Diese gehören ebenfalls zu den ionotropen Glutamatrezeptoren.
Während basaler synaptischer Übertragung sind diese NMDA-Rezeptoren
jedoch durch Magnesium-Ionen blockiert und somit für Ionen unpassierbar
(Nowak et al., 1984; Antonov & Johnson, 1999). Die Magnesium-Ionen werden
entfernt, wenn die postsynaptische Zelle durch einen hochfrequenten
präsynaptischen Reiz und verstärkte Glutamatfreisetzung depolarisiert wird.
Dadurch wird der Magnesiumblock der MDA-Rezeptoren aufgehoben (Bliss &
Collingridge, 1993; Malenka & Nicoll, 1999). Nach der Aufhebung dieser

9
spannungsabhängigen Blockade durch z.B.. einem präsynaptisch applizierten
„Theta-Burst“, ist der NMDA-Rezeptorkanal für Kalzium-Ionen permeabel, die
nun neben Natrium in die Zelle strömen.
Eine weitere Möglichkeit der LTP-Induktion wird nicht über NMDA-
Rezeptoren sondern über sog. metabotrope (d. h. G-Protein gekoppelte)
Glutamatrezeptoren (mGluR I) vermittelt. Diese stimulieren die
membranständige Phospholipase C, die ihrerseits die Spaltung des
Membranlipids Phosphatidylinositolbisphosphat (PIP2) zu Diacylglycerol (DAG)
und Inositoltrisphosphat (IP3) hervorruft. IP3 führt als sog. „Second Messenger“
zur Freisetzung zusätzlicher Kalziumionen aus intrazellulären Kalziumspeichern
(Endoplasmatisches Retikulum). Beide Mechanismen führen zu einem
erheblichen, wenn auch kurzen Anstieg der intrazellulären
Kalziumkonzentration (Malenka et al., 1992). Kalzium ist nun ebenfalls ein
Second Messenger, der weitere metabolische Prozesse auslöst. In der Regel
handelt es sich um die Aktivierung von Enzymen wie Adenylatcyclase,
Phosphodiesterase, Proteinphosphatase 2B (= Calcineurin) oder CaM-
Proteinkinase. Zusammenfassend bewirken diese Enzyme eine Stärkung der
synaptischen Verbindung und eine erhöhte postsynaptische Erregbarkeit. Um
dies zu gewährleisten, werden zum Beispiel neue AMPA-Rezeptoren in die
Plasmamembran eingebaut (Malinow & Malenka, 2002; Bredt & Nicoll, 2003).
Außerdem werden Glutamatrezeptoren phosphoryliert, was zu einer Änderung
der Eigenschaften dieser Rezeptoren und somit einer Stärkung der
synaptischen Verbindung führt (Soderling & Derkach, 2000).
Um die LTP für Stunden und Tage aufrecht zu erhalten (late LTP) bedarf
es der Änderung der Gentranskription und der Proteinsynthese. Durch die
Erhöhung der Kalziumkonzentration während der früheren Phasen der LTP
werden verschiedene Kinasen wie Proteinkinase-A (PKA), Kalziumcalmodulin-
Kinase (CaMK) und mitogen activated protein kinase (MAPK) stimuliert. Diese
Proteine phosphorylieren bestimmte Transkriptionsfaktoren, insbesondere das
cAMP (cyklisches Adenosin Monophosphat) response element-binding protein
(CREB) (Impey et al., 1996), das im phosphorylierten Zustand an das CREP-
Bindungs-Protein (CBP) bindet. Dieses bildet mit weiteren Proteinen einen
Proteinkomplex, mit dem die Promoter-Region für die Transkription des c-Fos-
Gens freigelegt werden kann (Herdegen & Leah, 1998; Lamprecht, 1999).

10
Zusammen mit anderen Proteinen kann nun die Gentranskription weiterer Gene
initiiert werden.
Das Gegenstück zur Langzeitpotentierung stellt die Langzeitdepression
da. Hierbei handelt es sich um die dauerhafte Abschwächung der synaptischen
Effektivität. Ebenso wie LTP wird LTD über NMDA-Rezeptoren vermittelt.
Ausschlaggebend ist dabei die Frequenz der Stimulation (Dudek & Bear, 1992).
Durch eine niedrigfrequente Stimulation kommt es zu einem relativ geringen
Kalziumeinstrom, der dann zu LTD führt, während eine hochfrequente
Stimulation einen starken Kalziumeinstrom bewirkt und so LTP auslöst
(Bienenstock et al., 1982). Als intrazelluläres Signalmolekül aktiviert Kalzium
auch bei LTD weitere Enzyme, die für die Aufrechterhaltung der LTD sorgen.
Auch existiert wie bei LTP eine mGlu-Rezeptor-abhängige Form der
LTD, bei der über weitere Signalmoleküle die Ausschüttung von Kalzium aus
intrazellulären Speichern initiiert wird. Durch LTD werden nicht nur synaptische
Verbindungen geschwächt, sondern LTD ist ebenso ein wichtiger Mechanismus
bei räumlicher Orientierung.
1.3.2 Läsionsinduzierte Plastizität
Die Fähigkeit des Gehirns, sich an wechselnde Umweltbedingungen
anzupassen und flexibel zu bleiben, nimmt im Alter dramatisch ab. Doch in
jüngster Zeit wurde festgestellt, dass es bestimmte Situationen gibt, in denen
das adulte Gehirn erneut zu höher Plastizität fähig ist, so als ob es wieder jung
wäre. Es wurde zum Beispiel beobachtet, dass nach Läsionen das intakte
Gewebe in Umgebung des Schadens die verlorengegangenen Funktionen
zumindest teilweise übernehmen kann. Dies wurde zum Beispiel nach einem
Schlaganfall (Nudo & Milliken, 1996; Carmichael, 2003), oder einer Läsion in
verschiedenen Kortexarealen wie dem Motorkortex (Jenkins & Merzenich,1987;
Cohan et al., 1993; Seitz et al., 1995) gesehen. Auch für den visuellen Kortex
sind Mechanismen der funktionellen Reorganisation bekannt: Ein
umschriebener Schaden im visuellen Kortex resultiert in einem umschriebenen
Defekt im Blickfeld. Diese Lücke im Blickfeld wird Skotom genannt. Mit Hilfe der
erhöhten Plastizität in der Umgebung der Läsion konnte durch visuelles
Training die Größe des Skotoms reduziert werden. Es lies sich beispielsweise

11
feststellen, dass sich rezeptive Felder in vivo in der Umgebung von chronischen
Läsionen im visuellen Kortex von Katzen durch die gesteigerte Plastizität
vergrößern und so ggf. den Funktionsverlust ausgleichen können, der durch die
Läsion zuerst entstanden ist (Eysel & Schweigart, 1999).
Unterschiedliche strukturelle und funktionelle Reorganisationsprozesse
können den funktionellen Verlust kompensieren und sogar zur Reparatur von
verletztem Gewebe führen. Ein Zeichen für die verstärkten Umbauprozesse
nach Läsionen zeigt die erhöhte Aktivität, die in der Umgebung einer Läsion
gefunden wurde (Eysel & Schmidt-Kastner, 1991). Die Mechanismen, die zu
erhöhter Plastizität nach Läsionen führen, sind sehr komplex und noch nicht
hinreichend erforscht. Es ist jedoch bekannt, dass viele zellulären
Mechanismen in der Umgebung eines Schadens dem juvenilen, also „jungen“,
Gewebe sehr ähnlich sind, so dass zum Beispiel LTP, einer der wichtigsten
Mechanismen für Plastizität, leichter und stärker ausgelöst werden kann. Dabei
ist eine verstärkte LTP nur in bestimmten Bereichen nahe der Läsion zu
messen (Dohle et al., 2009). Ebenso finden verschiedene Veränderungen in
einem limitierten Bereich um die Läsion herum statt, fehlen jedoch am direkten
Läsionsrand (Barmashenko et al., 2003). Im Folgenden soll näher auf zellulären
Mechanismen nach Läsionen eingegangen werden und ein Vergleich zur
Plastizität in jungem Gewebe gezogen werden.
Es wurden bereits einige Faktoren identifiziert, die für erhöhte Plastizität
verantwortlich gemacht werden. Beispielsweise ist nach Läsionen die Balance
zwischen Erregung und Hemmung dahingehend verändert, dass die Hemmung
unterdrückt wird und somit eine „Netto-Erregung“ resultiert (Mittmann et al.,
1994; Redecker et al., 2000). Es ist bereits bekannt, dass eine LTP leichter
auszulösen ist, wenn die Hemmung reduziert wurde (Artola & Singer, 1987,
Bear et al, 1992). Diese Veränderung der Balance zwischen Inhibition und
Excitation wird auch durch Modifikation der Transmittersysteme
wiedergespiegelt. Deafferenzierungsexperimente und kortikale
Läsionsexperimente zeigen eine reduzierte Aktivität des inhibitorischen
Transmittersystems (GABA, GABA-Rezeptor, GAD) nach Läsionen (Warren et
al., 1989; Hendry & Carder, 1992; Redecker et al., 2000). Dies geht einher mit
einer Vergrößerung der angrenzenden rezeptiven Felder (Warren et al., 1989;
Jones, 2000). Es wurde außerdem eine Glutamat-Zunahme am Rande des

12
Projektionsgebietes gesehen, die mit einer Vergrößerung der rezeptiven Felder
einher geht (Arckens et al., 2000). Pharmakologische Experimente zeigen, dass
nach Glutamat-Applikation die Feuerschwelle der Neurone vermindert ist, es
jedoch nicht zu einer Vergrößerung der rezeptiven Felder kommt (Dykes et al.,
1984). Die Applikation eines GABA-Antagonisten führt hingegen sehr wohl zur
Zunahme der rezeptiven Feldgröße (Dykes et al., 1984; Alloway & Burton,
1991; Kyriazi et al., 1998). Diese Ergebnisse deuten darauf hin, dass dem
inhibitorischen System eine große Rolle bei plastischen Prozessen wie
Reorganisation der kortikalen Topographie zukommt.
Auch während frühen Entwicklungsphasen ist das Gleichgewicht
zwischen Erregung und Hemmung zu Gunsten der Erregung verändert, da sich
hemmende Synapsen erst später ausbilden als erregende (Long et al., 2005).
Es konnte gezeigt werden, dass junge Tiere jedoch erst die kritische Periode für
Okular Dominanz erreichen, also hochplastisch sind, wenn die Inhibition zu
einem gewissen Anteil ausgebildet worden ist. BDNF übernimmt hier eine
große Rolle, da es die Reifung der inhibitorischen Neuronen vermittelt (Huang
et al., 1999). In späteren Entwicklungsstadien führt eine verstärkte Inhibition
jedoch zu einer Reduktion der Plastizität (Sale et al., 2010). Hierbei scheint
nicht das absolute Level der Inhibition sondern die Balance zwischen Inhibition
und Excitation entscheidend zu sein. Die genauen Mechanismen, die zu einer
Veränderung im Gleichgewicht zwischen Hemmung und Erregung führen, sind
jedoch noch nicht identifiziert. Ebenso ist unklar, auf welchem Wege die
veränderte Balance zwischen Hemmung und Erregung eine erhöhte Plastizität
vermittelt. Mehrere Arbeiten geben jedoch Hinweise darauf, dass eine NMDA-
Rezeptor unabhängige LTP ausgelöst werden kann, wenn es zu einer
Reduktion der Hemmung gekommen ist, wie es nach Läsionen der Fall ist.
Diese Form der LTP wird über metabotrophe Glutamatrezeptoren vermittelt. Die
Aktivierung dieser Rezeptoren führt dann indirekt zu einer Freisetzung
intrazellulärer Kalziumspeicher (Wilsch et al., 1998). Dies erfolgt nach Kopplung
an die Phospholipase C über Proteinkinase C und Ca²+/Calmodulin abhängige
Kinase II, die die intrazellulären Kalziumspeicher öffnen und so LTP induzieren
(Bortolotto et al., 1999; De Blasi et al. 2001 ). Diese nicht NMDA-Rezeptor
abhängige LTP ist jedoch nicht allein verantwortlich für erhöhte Plastizität nach
Läsionen.

13
Nach Läsionen ist auch die intrazelluläre Kalziumkonzentration
(Barmashenko et al., 2001) auf Grund veränderter NMDA- (Rumpel et al., 2000;
Hümmeke et al., 2004) und AMPA-Rezeptor Zusammensetzung (Barmashenko
et al., 2003) erhöht, was ebenfalls zu verstärkter Plastizität führen kann. Die
Aktivierung von NMDA-Rezeptoren führt also zu einem Einstrom von Kalzium in
die Zelle und spielt so eine Schlüsselrolle bei der Induktion von NMDA-
Rezeptor abhängiger LTP (Malenka et al., 1989). NMDA Rezeptoren sind
Heteromere und enthalten die Untereinheit NR1 und eine oder mehrere der vier
verschiedenen NR2 Untereinheiten (NR2A-D) (Monyer et al., 1994). Je nach
Zusammensetzung des NMDA Rezeptors mit verschiedenen NR2
Untereinheiten variieren die biophysikalischen Eigenschaften des Rezeptors.
So kann die Größe der LTP beeinfllusst werden. Enthält der NMDA Rezeptor
die NR2B Untereinheit, ist die Öffnung des Rezeptors verlängert und es kommt
zu einem größeren Kalziumeinstrom in die Zelle und dadurch zu einer
vergrößerten LTP (Camignoto & Vicini, 1992). Nach Läsionen wurde eine
veränderte Expression der m-RNA für die Untereinheiten NR2A und NR2B
beobachtet (Rumpel et al., 2000). Es konnte gezeigt werden, dass die
verstärkte LTP am Läsionsrand von der spezifischen Aktivität von NMDA
Rezeptoren, welche die NR2B Untereinheit exprimieren, abhängig ist.
(Huemmeke et al., 2004). Auch in der Phase der frühen postnatalen
Entwicklung werden hohe Anteile von NR2B Untereinheiten mit hoher
synaptischer Lernfähigkeit in Verbindung gebracht (Barth & Malenka, 2001).
Zudem haben die C-Terminale der verschiedenen NR2 Untereinheiten
unterschiedliche Wirkung auf die Signalkaskaden, die zu einer lang
andauernden LTP führen. Die C-Terminalen mit der NR2B Untereinheit werden
dabei als besonders förderlich für LTP angesehen (Husi et al., 2000; Sprengel
et al., 1998).
Neben diesen funktionellen Veränderungen kommt es nach Läsionen
auch zu strukturellen Veränderungsprozessen im neuronalen System. Durch
Neurogenese und Gliagenese werden neue Nerven- und Gliazellen gebildet,
um interneuronale Verbindungen wieder herzustellen (Dash et al., 2001; Jin et
al., 2001). Nicht nur Nervenzellen, sondern auch Gliazellen sind wichtig für
Reorganisationsprozesse (Guthrie et al., 1997), da diese die metabolische
Versorgung und sogar die Modulation von Signalübertragungen vermitteln. Eine

14
Gliose nach Läsionen wurde unter anderem nach Kortexläsionen bei Ratten
gefunden (Mittmann et al., 1994; Kálmán et al., 2000). Auch die Verbindungen
der Nervenzellen untereinander werden moduliert. So kommt es nach Schäden
des peripheren und des zentralen Nervensystems zu axonalem Wachstum,
Auswachsen von Dendriten und zur Bildung oder Rückbildung von Spines und
Synapsen, wie es zum Beispiel nach traumatischer Durchtrennung des peripher
somatosensorischen Inputs bei Affen (Florence et al.,1998), nach
Retinaläsionen bei Katzen (Arckens et al., 2000) oder nach Kortexläsionen des
entorhinalen Kortex bei Ratten (Deller & Frotscher, 1997) beobachtet wurde.
Auch die Morphologie der Spines unterliegt nach Läsionen im Hippokampus
(Reeves & Steward, 1986) oder der Retina (Keck et al., 2007) strukturellen
Reorganisationsprozessen, die die Erregbarkeit postsynaptischer Neurone
verändern können. All diese Mechanismen helfen dabei, verloren gegangene
interneuronale Verbindungen wiederherzustellen (Niquet et al., 1995).
In den letzten Jahren gibt es immer mehr Hinweise darauf, dass
sogenannte Neurotrophine, besonders brain-derived- neurotrophic factor
(BDNF), nach Läsionen und nach Schlaganfall im umgebenden Gewebe eine
veränderte Expression zeigen (Sulejczak et al., 2007). In diesem
Zusammenhang wird vermutet, dass BDNF für die erhöhte Plastizität nach
Läsionen verantwortlich ist.
1.4 Neurotrophine
Neurotrophine haben eine enorme Bedeutung für das Überleben und die
Differenzierung bestimmter Neuronenpopulationen. Die Zellen im
Nervensystem konkurrieren um die Neurotrophine, da Neurone, die keinen oder
nur sehr begrenzten Zugang zu Neurotrophinen haben, durch Apoptose zu
Grunde gehen (J. Yuan, B. A. Yankner, 2000). Ferner sind Neurotrophine in der
Lage, synaptische Verbindungen aufrecht zu erhalten (Vicario-Abejon et al.,
2002). So ist in BDNF knockout Mäusen die Anzahl der sensorischen Neurone
reduziert (Ernfors et al., 1994).
In Säugetieren existieren fünf Typen von Neurotrophinen: NGF (nerv
groth factor) (Levi-Montalcini & Booker, 1969), BDNF (brain derived
neurotrophic factor) (Leibrock et al., 1989), NT-3 (Neurotrophin 3), NT-4

15
(Neurotrophin 4) , NT-5 (Neurotrophin 5). Da NT-4 und NT-5 ähnliche
Eigenschaften besitzen, werden sie häufig als NT-4/5 zusammengefasst (Hohn
et al., 1990; Maisonpierre et al., 1990). Die Neurotrophine können an zwei
verschiedene Klassen von membranständigen Rezeptoren binden: p75
Neurotrophin Rezeptor (p75NTR), ein Rezeptor der TNF-Rezeptor Familie und
Rezeptoren der Tyrosinkinase-Rezeptor-Familie (Trk A, Trk B und Trk C).
Während alle Neurotrophine mit recht geringer Affinität an den p75NTR binden
können, sind die Trk Rezeptoren spezifisch für einzelne Neurotrophine: Trk A
für NGF, Trk B für BDNF, NT-4 und NT-5 und Trk C für NT-3. Nach Bindung
eines der Neurotrophine an seinen spezifischen Trk Rezeptor gibt es eine
Vielzahl hochkomplexer Möglichkeiten der Weitergabe des Signals durch
sogenannte Signaltransduktionskaskaden. Diese sind zudem von
unterschiedlichen Faktoren abhängig. Allen gemein ist jedoch, dass die Trk-
Rezeptoren nach Ligandenbindung dimerisieren und einige Tyrosine ihrer
intrazellulären Domäne autophosphorylieren. Diese phosphorylierten Tyrosine
dienen nun als Anker für weitere Proteine der Signaltransduktion, die als
Adaptoren, Kinasen, Lipasen oder Phosphatasen die Information weitertragen
(Kaplan & Miller, 2000).
Im Laufe der letzten Jahre gibt es immer mehr Hinweise darauf, dass
Neurotrophine, besonders brain-derived-neurotrophic factor (BDNF) auch für
erhöhte Plastizität nach Hirngewebeschäden mitverantwortlich sein könnten.
1.4.1 Das Neurotrophin BDNF
Da BDNF eine besondere Rolle in synaptischer Transmission und
aktivitätsabhängiger Plastizität wie LTP zugeschrieben wird (Poo, 2001), ist
BDNF ein wichtiger Faktor, der Plastizität modulieren kann. Die Freisetzung von
BDNF ist stark aktivitäts-abhängig (Patterson, 1996; Desai, 1999; Genaud,
2004) und macht BDNF daher zu einem geeigneten Kandidaten,
aktivitätsabhängige Prozesse wie LTP zu beeinflussen.
BDNF wird unter anderem von Neuronen exprimiert. Neueste Arbeiten
geben Hinweise darauf, dass BDNF zum Beispiel nach Läsionen wie einem
lakunären Schlaganfall auch durch Gliazellen exprimiert werden kann (Sato et
al., 2009). BDNF wird sowohl im zentralen wie auch dem peripheren

16
Nervensystem gefunden. Im Gehirn ist BDNF im Hippocampus, dem Kortex und
dem basalen Vorderhin besonders vertreten, genau in den Arealen, die für
Lernen, Gedächtnis und höheres Denken zuständig sind. Im Hippocampus und
kortikalen Regionen ist BDNF hauptsächlich somatodendritisch (Fawcett et al.,
1997; Goodman et al., 1996; Hartmann et al., 2001; Kojima et al., 2001) und
axonal (Kohara et al., 2001) lokalisiert. Außerdem wurde es auch innerhalb der
Synapse entdeckt (Fawcett et al., 1997; Goodman et al., 1996).
Zunächst wird BDNF als Präpro-Protein synthetisiert. Im
Endoplasmatischen Retikulum wird die Prä-Sequenz abgespalten und das
verbleibende Pro-BDNF wird am Golgie-Apparat in sekretorische Vesikel
verpackt. Einerseits konnte gezeigt werden, dass Proteinkonvertasen (z.B.
PC1, Furine) in diesen Vesikeln das Propeptid abspalten (Seidah et al., 1998)
und so eine regulierte Exozytose des Neurotrophins ermöglichen (Heymach et
al., 1996; Mowla et al., 1999). Andererseits kann das Pro-BDNF auch als Pro-
Protein sekretiert werden und extrazellulär durch Proteasen gespalten werden
(Lee et al., 2001; Lessmann et al., 2003). Die BDNF- Vesikel werden sowohl in
das präsynaptische Axon (anterograd), wie auch in die postsynaptischen
Anteile (retrograd) transportiert. Das Level von BDNF in der Synapse untersteht
zwei Regulationsmechanismen: Einerseits wird der Transport der BDNF-Vesikel
zu ihrem Ziel reguliert. Andererseits wird die Konzentration von BDNF durch
lokale Translation von BDNF mRNA gesteuert (Tongiorgi et al., 1997; Kang &
Schuman 1996; Cassadio et al., 1999).
1.4.2 BDNF und Epilepsie
Obwohl alle Neurotrophine für das Nervenzellwachstum und die Differenzierung
der Nervenzellen eine essentielle Rollen spielen, scheint eine besonders hohe
Konzentration von BDNF jedoch pathologische Prozesse wie abnorme
elektrische Aktivität und Übererregbarkeit des Gehirns zu vermitteln. Die
neuronale Erkrankung Epilepsie ist ebenso durch eine Übererregbarkeit des
Gehirns und durch wiederkehrende abnorme elektrische Aktivität
charakterisiert. Es gibt mehrere Ursachen, die Epilepsie generieren. So führen
zum Beispiel der Verlust von Neuronen, hippokampale Neurogenese, Gliose
und Austreiben neuer neuronaler Verbindungen zu einer Umorganisation des

17
neuronalen Netzwerkes und zu einer Veränderung der zellulären Homöostase
(Lukasiuk & Pitkanen, 2004; Pitkanen et al., 2002; Scharfmann et al., 2002) .
Der neurotrophe Faktor BDNF und sein Rezeptorsystem Trk B scheinen in
diese Umorganisation und die Entstehung von Epilepsie verwickelt zu sein
(Binder et al., 2001; Lindvall et al., 1994; Scharfman et al., 2002). Sowohl BDNF
wie auch der Trk B Rezeptor sind bei epileptischen Anfällen erhöht (Ernfors et
al., 1991; Jankowsky & Patterson, 2001; Merlio et al., 1993). Wenn exogenes
BDNF in vitro zu akuten Hirnschnitten gegeben wird, erhöht dies die
synaptische Übertragung (Scharfman, 1997). Auch transgene Mäuse, die
vermehrt BDNF bilden, zeigen eine erhöhte Bereitschaft zu epileptischen
Anfällen (Croll et al., 1999). Ebenso reduziert eine herabgesetzte Wirkung von
BDNF die Entstehung von Epilepsie (Lahteinen et al., 2002) . Heterozygote
BDNF Mäuse (BDNF (+/-) und ebenso Mäuse, die mit TrkB-Antikörpern
behandelt wurden, zeigten ein verzögertes Auftreten von Epilepsie (Binder et
al., 1999; Kokaia et al., 1995). Zusammengefasst ist es sehr wahrscheinlich,
dass ein erhöhtes Angebot von BDNF und oder vom TrkB Rezeptor durch die
Verstärkung neuronaler Aktivität auch das Risiko zur Entwicklung einer
Epilepsie steigern.
1.4.3 BDNF und synaptische Transmission
BDNF ist als neurotropher Faktor nicht nur für Differenzierung und Reifung von
Neuronen zuständig, sondern vermittelt verschiedenste Aspekte neuronaler
Funktion während Entwicklung und Veränderung des Nervensystems. In der
Wirkweise von BDNF kann ein akuter von einem chronischen Effekt
unterschieden werden (Gottmann et al., 2009).
Die akute Wirkung von BDNF bezieht sich besonders auf die Modulation
synaptischer Übertragung (Lu, 2003; Poo, 2001). Hierbei beeinflusst BDNF
sowohl das exitatorische wie auch das inhibitorische System.
Im exitatorischen System verstärkt BDNF die synaptische Übertragung
vor allem durch Erleichterung der Transmitter Freisetzung an der Präsynapse
(Li et al., 1998; Taniguchi et al., 2000). Doch auch die postsynaptische
Membran wird von BDNF verändert, indem BDNF unter anderem die Zahl und

18
Effektivität von AMPA Rezeptoren erhöht (Nariswa-Saito et al., 1999; Itami et
al., 2003).
Auch im inhibitorischen System hat BDNF einen Einfluss auf prä- und auf
postsynaptische Mechanismen (Frerking et al., 1998; Bruning et al., 2001) und
scheint eine entscheidende Rolle für schnelle synaptische Inhibition zu spielen
(Tanaka et al., 1997; Cheng & Yeh 2003). Der Einfluss von BDNF auf GABA
Transmissionen ist sehr komplex. So wurde beobachtet, dass BDNF
interessanterweise GABA-A vermittelte inhibitorische Transmission
abschwächen, aber auch verstärken kann (Tanaka et al., 1997; Frerking et al.,
1998; Bruning et al., 2001).
Die chronische Wirkung von BDNF spiegelt sich vor allem in der
Ausreifung glutamaterger und GABAerger Synapsen wider. So kann sogar der
Eintrittszeitpunkt der kritischen Periode durch das enge Zusammenspiel von
BDNF und GABA verändert werden. Es wurde herausgefunden, dass eine
Überexpression von BDNF die Reifung von inhibitorischen Neuronen fördert
(Hanover et al., 1999; Huang et al., 1999) und so die kritische Periode für
monoculare Deprivation zu einem früheren Zeitpunkt auftreten lässt (Fagiolini &
Hensch, 2000; Iwai et al., 2003). Umgekehrt kann der Eintritt dieser kritischen
Periode auch nach hinten verschoben werden. Bei Dunkelaufzuchten kommt es
beispielsweise zu einem reduzierten Level an BDNF (Castren et al., 1992) und
damit auch zu reduzierter Hemmung (Morales et al., 2002, Chen et al., 2001),
was die kritische Periode für monoculare Deprivation bis zum Erwachsenenalter
verschieben kann (Iwai et al., 2001; Mower, 1991; Fagiolini et al., 2003).
1.4.4 Bedeutung von BDNF für Plastizität
Trotz oder gerade wegen der epileptogenen Komponente ist BDNF ist ein sehr
geeingeter Kanditat, um aktivitätsabhängige Prozesse wie LTP zu beeinflussen,
da die Freisetzung von BDNF aktivitätsabhängig moduliert wird (Patterson
1996; Desai 1999; Genaud 2004). Die Freisetzung von Neurotrophinen wird
auch durch Neurotrophine selbst reguliert (Lindholm et al., 1994; Patz & Wahle,
2004), was die Aktivitätsabhängigkeit unterstreicht.
Durch verschiedene Studien wurde eine aktivitätsabhängige Anpassung
der BDNF Expression, bzw. der BDNF-mRNA untersucht: Es wurde

19
beobachtet, dass die mRNA für BDNF nach visuellem Lernen herauf reguliert
wird (Gomez-Pinilla et al., 2001; Kesslak et al., 1998; Mizuno et al., 2000).
Weiterhin führt eine angereicherte Umgebung (engl.: enriched environment)
dazu, dass mRNA und das Proteinlevel für BDNF in Kortex, Hippocampus,
basalem Vorderhirn und Medulla oblongata erhöht werden (Falkenberg et al.,
1992; Ickes et al., 2000). Freiwilliges Training führt zu einem Anstieg von BDNF
mRNA im Hippocampus (Gomez-Pinilla et al., 2001; Neeper et al., 1996;
Russo-Neustadt et al., 1999; Vaynman et al., 2003). Sogar durch
vergleichsweise kleine Stimulationen wie das Berühren von Barthaaren einer
Katze wird eine BDNF mRNA Expression induziert (Rocamora et al., 1996). All
dies zeigt die große Bedeutung von BDNF für Lernprozesse. Ein Entzug von
Stimulation und damit von Aktivität hingegen führt dazu, dass BDNF vermindert
ausgeschüttet wird. Zum Beispiel führt eine Deprivation von Licht zu einer
Herunter-Regulation des BDNF Protein sowie der mRNA (Castren et al., 1992).
BDNF erleichtert die Ausbildung von LTP auf der synaptischen Ebene
(Akaneya et al., 1997). Zusätzlich vermindert BDNF die Ausbildung von LTD in
Schicht II/III des visuellen Kortex (Akaneya et al., 1997, Huber et al., 1998,
Kinoshita et al., 1999). In Versuchsmodellen, in denen die Möglichkeiten, BDNF
zu synthetisieren, herabgesetzt waren, führte dies dazu, dass die Tiere
schlechter lernen konnten. Heterozygote BDNF-knockout Mäuse zeigen
dementsprechend verringerte LTP in der CA1-Region des Hippokampus (Korte
et al., 1995). Eine Wiederherstellung normaler LTP gelang durch Transfektion
mit einem BDNF-exprimierenden Adenovirus (Korte et al., 1996). Auch in
Wildtyp-Tieren war es möglich, eine LTP durch TrkB-IgG- oder BDNF-
Antikörper-Applikation zu verhindern (Figurov et al., 1996; Kang et al., 1997).
BDNF ist also ein entscheidender Faktor zur Auslösung von LTP.
In den letzten Jahren wurde auch diskutiert, ob das Neurotrophin BDNF
läsionsinduzierte Plastizität modulieren kann. Seine Rolle hierbei wird allerdings
kontrovers diskutiert. Nach retinalen Läsionen wurde zum Beispiel eine
Erhöhung von BDNF mRNA im visuellen Kortex gefunden (Obata et al 1999).
Exzitatorische Läsionen im Striatum hatten eine erhöhte BDNF Expression zur
Folge (De March et al., 2008). Auch nach Läsionen im Kortex wurden erhöhte
BDNF Konzentrationen in der Umgebung der Läsion gefunden. Messungen der
BDNF mRNA im somatosensorischen Kortex haben ergeben, dass die mRNA

20
vier Stunden nach Läsionen im Kortex vermehrt in der Umgebung der Läsion zu
finden sind (Comelli et al., 1992). Fokale photothrombotische Läsionen im
motorischen Kortex führen ebenso zu erhöhter BDNF Expression in der
Umgebung der Läsion (Sulejczak et al 2007). In diesem Zusammenhang wird
diskutiert, ob BDNF einen protektiven Effekt auf das Gehirn nach Läsionen
ausüben kann und so therapeutische Konsequenzen bei der Behandlung von
Patienten nach Schlaganfall oder anderen Substanzdefekten des Gehirn
resultieren könnten (Sizonenko et al. 2007). Andererseits konnte gezeigt
werden, dass nach Rückenmarksverletzungen bei Ratten ein intrathekal
applizierter Antikörper gegen den Trk B Rezeptor verbesserte zelluläre
Reorganisation und somit erhöhte Plastizität zur Folge hat (Fouad et al., 2010).
Ebenso wurde nachgewiesen, dass BDNF(+/-) Mäuse nach thrombotischem
Verschluss der mittleren Zerebralarterie bessere motorische Funktionen und
eine bessere Regeneration zeigten (Nygren et al., 2006). Hier wird also
postuliert, dass ein erniedrigtes Level von BDNF die Regeneration nach
experimentellem Hirninfarkt sogar verbessert.
1.4.5 Das Modell der BDNF(+/-) Maus
Es existieren verschiedene Tiermodelle, mit denen die Effekte von BDNF oder
des TrkB Rezeptors näher untersucht werden sollen. Für die vorliegende Arbeit
wurde ein BDNF knockout Modell benutzt. Hierbei ist die homozygote Form,
BDNF(-/-) jedoch nicht länger als zwei bis vier Wochen nach der Geburt
überlebensfähig (Ernfors et al., 1994). Diese BDNF(-/-) Tiere zeigen
dramatische Defizite in Koordination und Balance, hatten einen ataktischen
Gang, waren hyperaktiv und zeigten Atmungsunregelmäßigkeiten (Ernfors et
al., 1994; Jones et al., 1994).
Die heterozygote BDNF(+/-) Maus hingegen zeigt keine äußeren
Auffälligkeiten. Dennoch wurde durch eine Arbeitsgruppe eine herabgesetzte
Anzahl von sensorischen Neuronen beobachtet (Ernfors et al., 1994). Dieses
Ergebnis konnte jedoch durch Genoud et al. (2004) nicht bestätigt werden. Es
ist bekannt, dass in den BDFN(+/-) Tieren die Auslösung einer stabilen LTP
scheitert, wobei nicht endgültig geklärt ist, ob die Induktion oder die
Aufrechterhaltung der LTP gestört ist (Korte et al., 1995; Patterson et al., 1996;

21
Pozzo-Miller et al., 1999, Bartoletti et al., 2002). Auch wurde in BDNF(+/-)
Mäusen ein reduziertes Level sowohl an BDNF mRNA als auch an BDNF
Protein gefunden, das für die gestörte Plastizität dieser Tiere verantwortlich
gemacht wird (Kolbeck et al., 1999; Chourbaji et al. 2004; Genoud et al., 2004,
Abidin et al., 2006). Es ist bekannt, dass chronische Applikation von BDNF
dendritisches Wachstum und Verzweigung in Kulturen des visuellen Kortex
erhöhen kann (McAllister et al., 1995). Es wurde ebenso eine erhöhte
Dendritendichte in hippocampalen Kulturen nach chronischer Behandlung mit
BDNF gefunden (Tyler & Pozzo-Miller, 2001). So ist auf der anderen Seite
herausgefunden worden, dass in BDNF knockout Mäusen die Anzahl der
sensorischen Neurone reduziert ist. Im Hippocampus der BDNF (+/-) Tiere
wurde jedoch kein Unterschied in der Morphologie und somit kein Unterschied
in der Dendritendichte zu Wt Tieren gefunden (Korte et al., 1995). Ähnliche
Ergebnisse brachten Studien am Barrel Kortex, die auch keine Veränderung in
der Dichte von excitatorischen und inhibitorischen Synapsen aufzeigen konnten
(Genoud et al., 2004).
In letzter Zeit wurde herausgefunden, dass in BDNF(+/-) Mäusen
Exzitation und Inhibition im Vergleich zu Wt Mäusen verändert sind. Es konnte
gezeigt werden, dass die Vorraussetzungen für eine erregende Übertragung in
verschiedenen Hirnarealen reduziert sind und beispielsweise nicht genügend
Transmitter zur Verfügung gestellt werden können, bzw. der präsynaptische
excitatorische Input reduziert ist (Pozzo-Miller et al., 1999; Abidin et al., 2006).
Neben den Veränderungen im excitatorischen System wurden noch stärkere
Defizite im inhibitorischen System der BDNF(+/-) Mäuse beschrieben. Es wurde
eine verminderte Anzahl von GABAergen Kontakten (Kohara et al., 2007) und
eine reduzierte Effektivität der GABA Freisetzung im visuellen Kortex von
BDNF(+/-) Tieren (Abidin et al., 2008) gefunden.
BDNF ist also ein wichtiger Faktor, der Plastizität modulieren kann. Die
Erkenntnis, in wie weit BDNF am Auftreten läsionsiniduzierter Plastizität
beteiligt ist, würde entscheidend zum Verständnis erhähter Plastizität beitragen.
Bisher ist nicht bekannt, wie sich eine Läsion auf die BDNF(+/-) Maus und das
Level von BDNF in dieser Maus auswirkt. Dieses Modell der heterozygoten
knock out Maus eignet sich daher besonders für die aktuelle Studie, um die
zellulären Mechanismen einer läsionsinduzierten Plastizität näher zu

22
untersuchen und die spezifische Rolle des neurotrophen Faktors für diese Form
der Plastizität weiter zu charakterisieren .
1.5 Ziele der Arbeit
Ziel der vorliegenden Arbeit ist es, die Rolle des neurotrophen Faktors BDNF
für Plastizität im visuellen Kortex der Maus zu untersuchen. Für diese
Fragestellung stehen genetisch veränderte Mäuse, sog. Knockout (KO)-Mäuse,
bei denen das Gen für BDNF auf einem Allel deaktiviert wurde, zur Verfügung.
Bei diesen Mäusen ist also statt zweien nur ein Allel funktionell, so dass
entsprechend weniger BDNF synthetisiert werden kann.
Durch elektrophysiologische Messungen soll an akuten kortikalen
Hirnschnitten untersucht werden,
- ob eine fokale Läsion im visuellen Kortex der Maus ebenso zu einer
Erhöhung der LTP führt, wie es bei Ratten schon beobachtet werden
konnte.
- ob im visuellen Kortex der BDNF(+/-) Maus eine Veränderung der
basalen synaptischen Transmission vorliegt.
- inwieweit die Inaktivierung eines Allels für BDNF bei BDNF(+/-)
Mäusen die Plastizität nach Läsionen beeinflusst.
In Kooperation mit der Arbeitsgruppe von Prof. Wahle (Biologische Fakultät,
RUB) sollten darüber mit Hilfe der PCR-Technik eruiert werden,
- welche Auswirkung eine Läsion auf das Level der mRNA
verschiedener neurotropher Faktoren, insbesondere BDNF, auf den
Kortex von Wt Mäusen hat.
- ob es einen Unterschied läsionsinduzierter Effekte auf das Level der
mRNA neurotropher Faktoren zwischen Wt und BDNF(+/-) Mäusen
gibt.
Die Ergebnisse der Versuchsreihen sollen zum besseren Verständnis der
läsionsinduzierten Plastizität im visuellen Kortex beitragen und könnten wichtige
Hinweise für die zellulären Mechanismen von Reorganisationsprozessen
liefern. Da BDNF im Verdacht steht, Plastizität zu beeinflussen, könnte eine
Bestätigung dieser Hypothese einen Nutzen für verbessertes Lernen im

23
Allgemeinen, bei Kindern wie bei älteren Menschen, und möglicherweise auch
therapeutischen Nutzen bei Patienten mit Lerndefiziten oder
Hirnsubstanzdefekten zur Folge haben.

24
2 Material und Methoden
2.1 Tiere
In der vorliegenden Arbeit wurden transgene heterozygote BDNF Mäuse (BDNF
+/-) sowie Wildtyp-Geschwistertiere (Wt) benutzt. Die Zuchttiere stammten aus
der Zuchtlinie, die erstmals von Korte et al. (1995) beschrieben wurden. Um
heterozygote Mäuse zu erhalten, wurde auf einem Allel die BDNF kodierende
Region durch eine Neomycin-Kassette ersetzt, wodurch die ursprüngliche
Region funktionslos wird. Homozygote Knock-out-Tiere (BDNF(-/-)) wurden
nicht benutzt, da diese nur eingeschränkt lebensfähig sind. Diese Tiere zeigen
ein geringeres Gewicht und eine geringere Größe im Vergleich zu gleichaltrigen
Geschwistern, eine eingeschränkte Koordination und geringere Neuronendichte
in den Hinterwurzelganglien. Bei den in der vorliegenden Arbeit verwendeten
BDNF (+/-) Tieren sind diese Abnormalitäten nicht zu beobachten (Korte et al.,
1995).
Um die Zucht in unserem Institut fortzuführen wurden männliche BDNF
(+/-) Tiere mit weiblichen Wt Mäusen verpaart. Unter den Nachkommen waren
so zu etwa einem Viertel BDNF(+/-) und zu drei Vierteln Wt Tiere zu erwarten.
Die Tiere wurden unter konstanten Bedingungen gehalten: bei 22±1 °C und
einer Luftfeuchtigkeit von 50-60 % wurde ein 12h/12h Hell-Dunkel-Rhythmus
generiert. Zur Paarung wurde jeweils ein Männchen mit maximal zwei
Weibchen zusammengesetzt. Nach ungefähr vier Wochen wurden die Tiere
getrennt und die Weibchen wurden einzeln und später mit ihrem Nachwuchs
zusammen in einem eigenen Käfig gehalten. Für alle Tiere war zu jeder Zeit
Futter und Wasser frei zugänglich.
Für die Experimente wurden Tiere beiderlei Geschlechts im Alter von 21-
28 Tagen benutzt. Um zwischen den Wt und BDNF (+/-) Tieren zu
unterscheiden, wurden die Tiere durch PCR an DNA-Extrakten von
Schwanzspitzengeswebe genotypisiert. Dies war notwendig, da die Tiere sich
äußerlich nicht unterscheiden lassen.

25
2.2 Genotypisierung
Zur Bestimmung wurde eine Genotypisierung mittels PCR durchgeführt. Dazu
wurde ein kleines Stück des Mausschwanzes zunächst in Lysepuffer
homogenisiert. Im Lysepuffer enthalten waren 500 mM KCl, 100mM Tris-HCl,
0.1 mg/ml Gelatine, 0.45 % Nonidet P-40, 0.45 %Tween 20 und 500 mg/ml
Proteinase K, die direkt vor Gebrauch aus einer Stammlösung von 20 mg/ml
hinzugefügt wurde.
Nachdem das Gewebe über Nacht zur Homogenisierung bei 55°C
inkubiert wurde, konnte am folgenden Tag die DNA mittels PCR vervielfältigt
werden. Dazu wurde eine PCR Lösung aus folgenden Bestandteilen hergestellt
(Angaben pro Probe): 14,2 µl H2O, 1,2 µl MgCl2,, 2 µl 10-fach konzentrierte
Polymerase-Pufferlösung, 0,4 µl dNTP (dATP, dGTP, dCTP, dTTP), 0,4 µl Taq-
Polymerase und 0,8 µl Primer. Um die BDNF-Region und die Region, die das
BDNF-Gen bei den BDNF (+/-) Mäusen mutiert, zu detektieren, wurden zwei
verschiedene Primer eingesetzt. Diese kurzen Oligonukleotide stellen den
Startpunkt für die DNA-Polymerase darstellen. Der Primer für die BDNF-Region
hatte die Sequenz 5`-ACC ATA AGG ACG CGG ACT TGT AC-3´und der für
Neomyzin, das die BDNF- Region blockiert 5`GAT TCG CAG CGC ATC GCC
TT-3`. Der reverse Primer für beide Regionen war 5´ -GAA GTG TCT ATC CTT
ATG AAT CGC- 3´.
Für eine Mausschwanzprobe wurden jeweils 19 µl der PCR-Lösung mit
1µl der homogenisierten DNA gemischt. Um mögliche Verunreinigungen
auszuschließen wurde bei jeder PCR eine Wasserprobe mit amplifiziert, in der
zu den 19 µl der PCR-Lösung 1 µl Wasser gegeben wurde. Im sog.
Thermocycler fand dann die PCR unter folgenden Bedingungen statt:
Zunächst wurden die Proben für 3 Minuten auf 94° C erhitzt, dann erfolgten 36
Zyklen mit
30 sec 94°C
30 sec 52°C
1 min 72°C
Anschließend folgten 10 min mit 72°C und hiernach eine Kühlung bis auf 4°C.
Die PCR-Produkte wurden durch Agarose-Gelelektrophorese identifiziert
(Abb. 2.1). Dazu wurde jede Probe in ein 2%iges Agarosegel eingebracht und

26
durch die angelegte Spannung separiert. Um die Größe der erhaltenen Banden
einordnen zu können wurde zu jeder PCR-Reihe eine sogenannte Leiter
gegeben, bei der die Banden im Abstand von 100 Basenpaaren (bp) im Gel
wandern.
1 2 3 4 5 6 7 8 9
Neomycin
BDNF500 bp
500 bp
Abbildung 2.1: Gelelektrophorese
Durch die angelegte Spannung wurden die einzelnen PCR-Produkte aufgetrennt.
BDNF(+/-) Tiere exprimieren sowohl Neomycin (grauer Pfeil) als auch BDNF
(Nummer drei, vier und fünf), bei Wt Mäusen sind nur BDNF-Banden zu sehen
(sechs, sieben, acht und neun). Nummer zwei ist eine Wasserkontrolle, Nummer
eins zeigt eine Leiter an, an der die Größe der Basenpaare abgelesen werden
kann.
2.3 In vivo Läsion
Für die Läsionen wurden 20-22 Tage alte Mäuse durch eine intraperitoneale
Injektion von Chloralhydrat (4%; 0.1 ml pro 10 g Körpergewicht) anästhesiert.
Die Tiere wurden auf einer Stereotakt-Apparatur fixiert und mit Hilfe einer
Wärmematte bei ca. 37°C gehalten (Abb. 2.2A). Die Kopfhaut der narkotisierten
Mäuse wurde rasiert und die Haut über dem visuellen Kortex eröffnet. Die
Schädelkalotte wurde mit einem Bohrer dünn geschliffen, ohne dabei die Dura
mater zu verletzten (Abb. 2.2B). Mit Hilfe eines Infrarot Dioden Laser (OcuLight

27
Slx, Iris Medical, USA) wurde eine unilaterale fokale Läsion in die rechte
Hemisphäre induziert (Abb. 2.2C). Hierfür wurden unter visueller Kontrolle
parallel zur Mittellinie durch den lichtdurchlässigen Knochen fünf bis zwölf sich
teils überlappende Läsionen induziert. Nach dieser Operation wurde die Haut
mit Gewebekleber (Histoacryl, Braun-Dexon, Melsung, Deutschland)
geschlossen.
Um eine Unterkühlung zu vermeiden wurden die Tiere direkt nach der
Operation unter eine Wärmelampe gelegt und später dann in den Tierstall
gebracht, wo sie unter Kontrollbedingungen gehalten wurden und freien Zugang
zu Futter und Wasser hatten.
A B C
Abbildung 2.2: Läsionierung der Tiere
A Fixierung des Schädels, Lagerung des Tieres auf einer Wärmedecke B
Aufbohrung der Kalotte C Läsion des visuellen Kortex paramedian rechts
2.3.1 Sham- Operationen
Um zu zeigen, dass die Operationsmethode an sich keinen Effekt auf die
Erregbarkeit und Plastizität des Gewebes hat, wurden „Schein-Operationen“,
sogenannte Sham-Operationen, durchgeführt. Dazu erhielten die Tiere die
gleiche Betäubung wie zuvor beschrieben. Auch die Freipräparierung des
Kortex erfolgte in gleicher Weise wie bei den Läsionstieren. Allerdings wurde
hier nach dem Dünnbohren des Schädels keine Läsion gesetzt, sondern die
entsprechende Zeit, die eine Läsion in Anspruch nehmen würde, gewartet.
Anschließend wurde die Haut genau wie bei den Läsionstieren mit
Gewebekleber verschlossen. Auch die Schein- operierten Tiere wurden nach

28
der Operation unter Kontrollbedingungen gehalten und hatten freien Zugang zu
Nahrung und Wasser.
Weder Läsionstiere noch scheinoperierte Tiere zeigten nach der
Behandlung besondere Verhaltensauffälligkeiten. Die operierten und die
scheinoperierten Tiere überlebten nach der Operation zwei bis sechs Tage
unter den gleichen Umweltbedingungen wie die nichtbehandelten Kontrollen.
2.4 Herstellung der akuten Gehirnschnitte
Um Schnitte des visuellen Kortex zu gewinnen, wurden die Tiere zunächst mit
Äther anästhesiert und anschließend dekapitiert. Mit einem sagitalen
Hautschnitt wurde die Kalotte freigelegt, um dann mit einem Stich eines spitzen
Skalpells auf Höhe der hinteren Fontanelle Hirnstamm und Kleinhirn
abzutrennen. Die Kalotte wurde nun mit einer Knochenschere auf der linken
Seite eingeschnitten, so dass die Kalotte angehoben werden konnte ohne die
rechte Hemisphäre zu verletzen. Eventuell bestehende Faserverbindungen der
Arachnoidea wurden durchtrennt um dann die Kalotte zu entfernen. Nach
dieser Freipräparierung, die nicht länger als 90 sec dauerte, um hypoxische
Gewebeschäden zu vermeiden, wurde das Gehirn mit einem Löffel in 2° C
gekühlte artifizielle zerebrospinale Flüssigkeit (ACSF, Konzentration in mM: 125
NaCl, 2,5 KCL, 1,25 NaH2PO4, 25 NaHCO3, 25 D-Glucose, 2 CaCl2 und 1,5
MgCl2) überführt. Die ACSF wurde während des gesamten Experimentes mit
Carbogen (95 % O2 und 5 % CO2) zur Sauerstoffsättigung bei einem pH-Wert
von 7,4 begast. Nach ca. 2 min Kühlung wurde das Gehirn in eine Petrischale
mit kaltem ACSF gebracht, um einen Block des kompletten visuellen Kortex der
rechten Hemisphäre herzustellen. Dafür wurde ein frontaler Schnitt und ein
Schnitt durch die beiden Hemisphären gesetzt. Dieser Gewebeblock wurde mit
Hilfe von Gewebekleber mit der frontalen Schnittfläche auf einer Metallplatte
befestigt, die dann in der mit kalter ACSF gefüllten Schneidekammer des
Vibratoms (VT 1000 S; Leica, Nussloch, Deutschland) festgeschraubt wurde.
Nun wurden drei bis vier coronale Schnitte mit einer Dicke von 350 µm
angefertigt, die den visuellen Kortex enthielten. Um eine Erholung der Schnitte
zu erreichen, wurden diese für mindestens 2 Stunden in einer Vorratskammer
bei Raumtemperatur inkubiert.
Formatiert

29
2.5 Histologie
2.5.1 Nissl-Färbung
Um die Morphologie der Läsion und des Gewebes in der unmittelbaren
Umgebung zu untersuchen wurde eine Nissl-Färbung angewandt. Mit dem
verwendeten Farbstoff Cresylviolett werden basophile Zellbestandteile
(Chromatin und Nukleinsäuren) blau dargestellt. Für die Färbung wurden
zunächst akute Hirnschnitte von BDNF (+/-) Wt Tieren zwei bis vier Tage nach
der Läsion hergestellt. Die Schnitte wurden direkt nach der Präparation in 4%
Paraformaldehyd (gepuffert mit 0,025 M PBS, pH 7,4) fixiert. Nach 24 Stunden
wurden die Schnitte in 30 % Saccharose zur Kryoprotektion überführt. Die so
vorbehandelten 400µm Schnitte wurden an einem Gefriermikrom (Leitz Kryomat
1703) in 30 µm dicke Schnitte dünngeschnitten. Vor der eigentlichen Färbung
mussten die Schnitte über eine aufsteigende Alkoholreihe, in Xylol und
anschließend einer absteigenden Alkoholreihe entfettet und rehydriert werden.
Danach wurden die Schnitte in 0.001% Cresylviolett Lösung (gelöst in Aqua
dest.) gefärbt, über eine aufsteigende Äthanolreihe differenziert, mit Isopropanol
dehydriert und mit Xylol geklärt. Abschließend wurden die Schnitte in Depex
(Serva) eingedeckt um sie haltbar zu machen.
2.5.2 Immunhistochemie: GFAP-Färbung
Bei immunhistochemischen Untersuchungen werden Antikörper gegen
Transmitter und Proteine verwendet. Mit Hilfe der Antikörper gegen Glial
fibrillary acidic protein (GFAP) kann die Glia-Reaktion in der Nähe der Läsion
beurteilt werden. Aus Voruntersuchungen, bei denen dieselbe Methode der
Läsionierung bei Ratten angewendet wurde, ist bekannt, dass die Glia-
Reaktion, die sog. Gliose, auf einen Bereich von 100-200 µm um die Läsion
begrenzt war (Mittmann et al., 1994) und dass ab einem Abstand von 500µm
das Gewebe histologisch intakt erschien (Mittmann & Eysel, 2001;
Barmashenko et al., 2003).
Die Fixierung der Schnitte ist die selbe, wie die für die Nissl-Färbung.
Anschließend erfolgte die Färbung nach der indirekten Methode. Dies bedeutet,

30
dass das entsprechende Protein, hier GFAP, mit einem unmarkierten
Primärantikörper detektiert wird. Zunächst wurden die Schnitte in PBS gespült,
dann die endogene Peroxidase mit 0.3% H2O2 inaktiviert. Die Blockierung
unspezifischer Bindungen erfolgte mit 10% normalen Ziegenserum mit einem
Zusatz von 0.2% Triton X-100 in (Sigma) in PBS. Die Inkubation im
Primärantikörper (polyclonales GFAP von Dako) erfolgt in einem
Verdünnungsmedium bestehend aus 1 % normalen Ziegenserum und 0.2 %
Triton X-100 in PBS über Nacht bei Raumtemperatur. Als Sekundärantikörper
wurde ein biotinylierter Ziegen-Anti-Kaninchen-Antikörper (Vectorstain)
verwendet. Dieser wurde mit dem ABC-Kit (Avidin-Biotin-Complex mit
Peroxidase; Vectorstain) detektiert. Sekundärantikörper und Detektionssystem
wurden in einem Verdünnungsmedium aus 1 % normalen Ziegenserum und
0.1% Triton X-100 in PBS benutzt
Der Nachweis der Peroxidase (Farbreaktion) erfolgte mit
Diaminobenzidin-dihydrochlorid (Sigma) (Yousef et al., 2004).
2.6 Elektrophysiologischer Messplatz
Jeweils einzelne Schnitte wurden in eine Messkammer überführt, die mit
oxygeniertem ACSF überflutet wurde (Abb. 2.3). Der ACSF-Vorrat befand sich
dabei in einem temperierten Wasserbad und wurde dauerhaft mit Carbogen (95
% O2 und 5 % CO2) begast. Durch eine Schlauchpumpe wurde die
Messkammer mit ACSF bei 32 ± 1°C mit einer Durchflussgeschwindigkeit von
16 ml/min durchspült. Damit eine konstante Messtemperatur gewährleistet
werden konnte, wurde der ACSF führende Schlauch zusätzlich durch ein
temperiertes Warmwassergefäß geleitet. In der vorliegenden Arbeit wurde eine
Messkammer benutzt, in der der Schnitt von ACSF umspült wird („submerged
Chamber“). Dabei liegt der Hirnschnitt auf einem Nylonnetz, um die Diffusion
mit Nährstoffen von allen Seiten zu gewährleisten.
Formatiert

31
ACSFWarmwasserbad
Gehirnschnitt
O2O2
O2
O2
O2
1
2
3
4
56
7
Abbildung 2.3: Messkammer
Mit einem Gasschlauch (1) wurde die ACSF-Lösung (2.) des Vorratsbehälters
mit Sauerstoff begast. Mit Hilfe einer Pumpe wurde die im Wasserbad (4.)
temperierte ACSF-Lösung in die Messkammer (5) geleitet. Der zuführende
Schlauch (3.) verlief ebenfalls durch das Warmwasserbad, um eine schnelle
Abkühlung der Lösung zu verhindern. Durch den ableitenden Schlauch (6)
wurde die ACSF-Lösung abgepumpt und aufgefangen (7).
Um die Elektroden richtig platzieren zu können war ein Mikroskop (Binokular,
LNC) über der Kammer montiert. Damit die Messsituation nicht von
Erschütterungen gestört werden konnte, waren die Messkammer, die
Reizelektrode und die Ableitelektrode auf einem schwingungsdämpfenden
Tisch montiert. Um vor elektrischen Störsignalen zu schützen war die gesamte
Messsituation von einem Faradaykäfig umgeben (Abb. 2.4).

32
Abbildung 2.4: schematische Darstellung der Messplatzsituation
2.7 Elektrophysiologie
2.7.1 Elektrophysiologische Messungen
Feldpotentiale(FPs) /Feld-exzitatorische-postsynaptische Potentiale (Feld-
EPSP) sind elektrische Potentiale, die extrazellulär abgeleitet werden und so
die Summe von synaptischen Übertragungen widerspiegeln. Sie sind das
extrazelluläre Korrelat der an den einzelnen Neuronen bei Reizung ausgelösten
EPSP (exitatorische postsynaptische Potentiale).
Um die FP hervorzurufen, wurde eine konzentrische bipolare
Stimulationselektrode auf der Oberfläche der Kortexschicht IV platziert. Bei den
Tieren, bei denen eine Läsion gesetzt worden war, wurde diese
Stimulationselektrode im Abstand von 1.0 - 2.0 mm zum Läsionsrand platziert.
Diese Distanz wurde mit Hilfe einer Skala, die sich Okular des Binoculars
befand, kontrolliert. Bei unbehandelten und scheinoperierten Tieren wurde die
gleiche Region stimuliert. Dieser Abstand zur Läsion wurde gewählt, da schon
in früheren Studien gezeigt wurde, dass bei Ratten der Ort der höchsten
Plastizität eine Entfernung von 2,0 – 3,2 mm zur Läsion aufweist (Mittmann &
Eysel, 2001; Hümmeke et al., 2004). Wird dies nun auf die Größe des Schnittes
umgerechnet ergibt sich nun ein ungefährer Abstand von 1,0 – 2.0 mm zum
Läsionsrand in einem Maushirnschnitt. Um die FPs der afferenten Fasern
abzuleiten, wurde eine mit ACSF gefüllte Glaselektrode in Kortexschicht II/III

33
eingeführt (Abb. 2.5). Die Glaselektroden wurden aus Brosilikat-Glasröhrchen
(GB 150-8P; Science Produkts, Hofheim) mit einem elektronisch gesteuerten
horizontalen Elektroden-Ziehgerät hergestellt (DMZ Universalpuller , Zeitz,
Augsburg) und wiesen einen Widerstand von 1-3 MΩ auf.
Nachdem die Stimulationselektrode und die Ableitelektrode platziert
worden waren, wurde eine halbe Stunde gewartet, ohne einen Reiz abzugeben,
um eine Regeneration des Zellmetabolismus zu erreichen. Monosynaptische
Stromimpulse von 40 – 200 µs Dauer und einer Intensität von 60 – 130 µA
wurden nach dieser Erholungsphase von einem Reizgerät (A 360, WPI)
generiert, um reproduzierbare FP zu induzieren.
Stimulationselektrode
Ableitelektrode
Abbildung 2.5: Extrazelluläre FP Ableitung im Hirnschnitt
Die Abbildung zeigt einen Nissl gefärbten Hirnschnitt vom visuellen Kortex
einer 23 Tage alten Maus. Fasern der Schicht IV wurden stimuliert, um dann in
Schicht II/III die extrazellulären Feldpotentiale abzuleiten.
Die basale exitatorische Transmission wurde mit Hilfe von Input-Output-Kurven
(I-O-Kurven) dargestellt. Hierfür wurden AMPA-Rezeptor dominierte
Feldpotentiale in der kortikalen Schicht II/III nach elektrischer Stimulation der
Schicht IV abgeleitet. Zunächst wurde die minimale Reizstärke ermittelt, mit der
bei einer Stimulationsdauer von 200 µs ein maximales Feldpotential evoziert
werden konnte. Diese ermittelte Intensität der Reize blieb danach konstant und
die Stimulusdauer änderte sich zwischen 40 und 200 µs in 20 µs Abständen.

34
Nur Feldpotential mit einer maximalen Amplitude von mindestens 1,0 mV,
wurden für die weitere Evaluation ausgewählt.
Für die LTP Experimente wurde die Intensität der Reize so gewählt, dass
FP von 40-65 % der maximal erreichbaren Amplitude evoziert wurden. Diese
Stromstärke wurde für die Dauer des gesamten Experimentes beibehalten. Die
Stimulationsdauer war konstant bei 100 µs. Zunächst wurde alle 30 sec ein
Einzelreiz gegeben, um die basale synaptische Reizantwort zu messen. Wenn
diese Kontrollsignale für mindestens 10 min eine stabile Amplitude zeigten,
wurde eine Theta-Burst-Stimulation (TBS) appliziert. Diese bestand aus 3
Reizfolgen in einem Abstand von je 10 sec. Jede dieser Reizfolgen setzte sich
aus zehn Pulsfolgen zusammen, die alle 200 ms (5 Hz) dauerten. Jede
Pulsfolge bestand ihrerseits aus vier Reizen im Abstand von 10 ms (100 Hz)
(Abb. 2.6).
Abbildung 2.6: Theta-Burst-Stimulation
Die Theta-Burst-Stimulation bestand aus drei Reizfolgen in einem Abstand von
je 10 Sekunden. Jede Reizfolge setzte sich aus zehn Pulsfolgen zusammen, die
im Abstand von 200 ms (5 Hz) erfolgten. Eine Pulsfolge bestand wiederum aus
vier Einzelreizen mit einer Frequenz von 100 Hz.
Während der TBS wurde die Stimulationsdauer der Einzelreize auf 200µs
erhöht, dauerte nun also doppelt so lange wie die zuvor applizierten
Kontrollreize. In der nachfolgenden Post-Konditionierungsphase wurde wieder
10 sec
5 Hz
100 Hz

35
im Abstand von 30 sec und einer Reizdauer von 100 µs stimuliert und nun
wurden die FP eine Stunde lang aufgenommen.
2.7.2 Auswertung der extrazellulären Messungen
Die Feldpotentialsignale wurden mit 3 kHz gefiltert und durch einen
Differentialamplifier verstärkt (EPMS07; NPI Electronics, Tamm). Die
abgeleiteten Signale wurden nach Digitalisierung durch einen A/D D/A Wandler
(Digidata 1200, Axon Instruments, Foster City, USA) mit einer Samplingrate von
50 kHz mit dem Programm CLAMPEX 9.2 (Molecular Devices, Union City,
USA) auf einem Computer gespeichert und später analysiert. Es wurden nur
diejenigen Messungen in die Auswertung genommen, bei denen eine stabile
Baseline vor TBS vorhanden war.
Alle Änderung der FPs in der Post-Konditionierungsphase, also eine Stunde
nach dem TBS, wurden in Bezug zu den FPs 10 Minuten vor der TBS normalisiert.
Diese Baseline vor TBS entspricht also definitionsgemäß 100%. Um festzustellen, ob
eine Veränderung der FP-Amplitude nach TBS eine signifikante LTP induzierte,
wurden diese relativen FP- Amplituden 10 min vor und 50-60 min nach TBS
miteinander verglichen. Zunächst wurde mit dem K-S Test festgestellt, ob die Werte vor
und nach TBS normalverteilt waren. Da dies in allen Gruppen der Fall war, konnte
anschließend ein gepaarter t-Test für verbundene Stichproben angewandt werden. Das
Konfidenzintervall betrug hierbei 95%; bei einer Irrtumswahrscheinlichkeit p ≤0.05
wurden die Unterschiede als signifikant definiert.
Alle Daten werden als Mittelwerte mit dem Standardfehler der Mittelwerte
(SEM) angegeben.
2.8 Messung der BDNF mRNA-Konzentration im visuellen
Kortex mittels RT-PCR
Die Bestimmung der m-RNA Konzentration erfolgte in Kooperation mit dem
Labor von Prof. Petra Wahle (Abt. für Entwicklungsneurobiologie der Ruhr-
Universität Bochum). Die Präparation des Gehirns wurde in unserem Institut
vorgenommen, die PCR wurde dann in der Abt. für Entwicklungsneurobiologie
durchgeführt. Mit der hier angewandten Technik der RT-PCR (Reverse

36
Transkriptase-Polymerase-Kettenreaktion) soll die RNA bestimmter Gene
quantitativ untersucht werden, um eine Aussage über die Expressionsrate der
einzelnen Gene machen zu können. Da die verwendeten DNA-Polymerasen
jedoch nicht in der Lage sind, RNA zu amplifizieren, muss die zu untersuchende
RNA zunächst in DNA umgeschrieben werden. Diese aus der RNA hergestellte
komplementäre DNA nennt man cDNA (complementary DNA).
2.8.1 Herstellung der cDNA
Nachdem die Tiere zwei Tage nach der Läsionsinduzierung überlebt hatten,
wurde das Hirngewebe für die Bestimmung mRNA für BDNF aufbereitet. Die
Kontrolltiere waren 22-26 Tage alt. Die Tiere wurden wie bei der Präparation
der elektrophysiologischen Messungen mit 4 ml Äther narkotisiert und
anschließend dekapitiert. Auch die folgende Präparation des Gehirns und
Herstellung der Hirnschnitte erfolgte wie in der elektrophysiologischen
Messreihe: Das Gehirn wurde ebenso freipräpariert und vorsichtig entnommen.
Nach Kühlung in 2° C kalter ACSF (artifizielle zerebrospinale Flüssigkeit,
Konzentration in mM: 125 NaCl, 2,5 KCL, 1,25 NaH2PO4, 25 NaHCO3, 25 D-
Glucose, 2 CaCl2 und 1,5 MgCl2) wurden coronale Schnitte mit einer Dicke von
300 µm hergestellt. Dabei wurden zunächst zwei bis vier Schnitte hergestellt,
die den visuellen Kortex mit entsprechender Läsion enthielten. Nun wurde ein
größerer Gewebeblock coronal geschnitten um anschließend coronale Schnitte
herzustellen, die den Motorkortex ohne Läsion enthielten, der als
Kontrollgewebe dienen sollte (Abb. 2.7A). Aus den Läsionsschnitten wurden
nun kleine Gewebeblöcke auf Eis präpariert (Abb..2.7B).
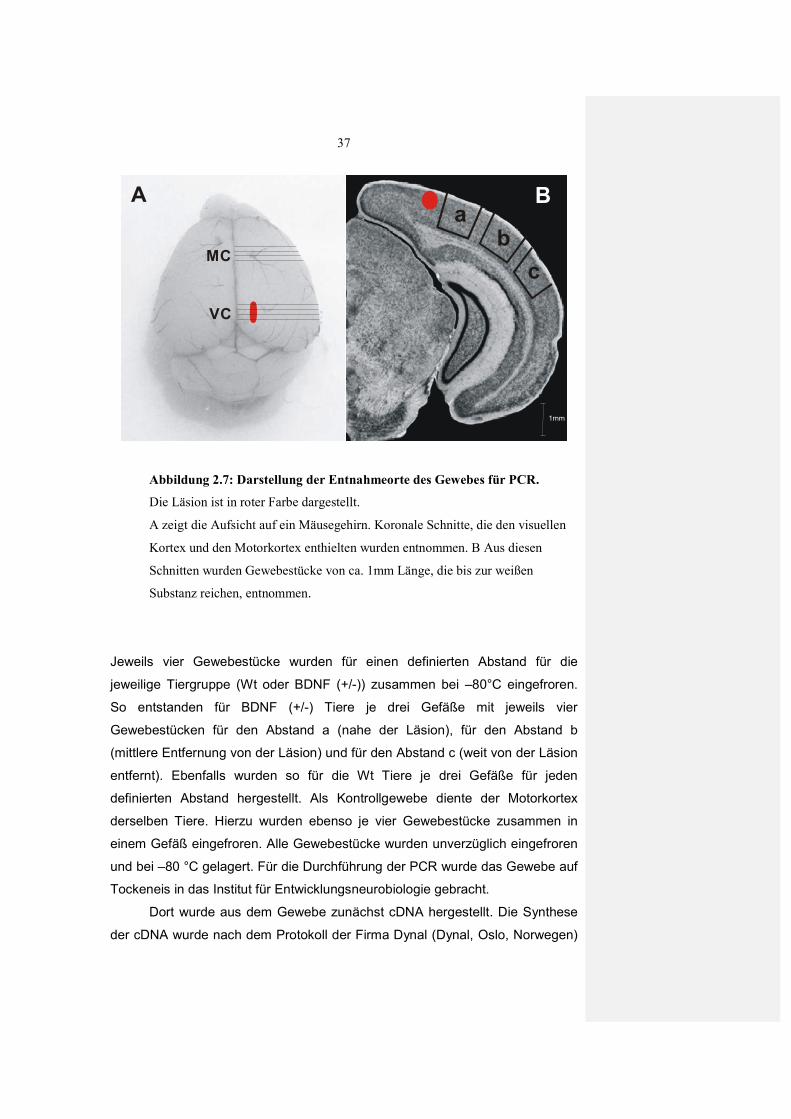
37
A
MC
VC
ab
c
B
1mm
Abbildung 2.7: Darstellung der Entnahmeorte des Gewebes für PCR.
Die Läsion ist in roter Farbe dargestellt.
A zeigt die Aufsicht auf ein Mäusegehirn. Koronale Schnitte, die den visuellen
Kortex und den Motorkortex enthielten wurden entnommen. B Aus diesen
Schnitten wurden Gewebestücke von ca. 1mm Länge, die bis zur weißen
Substanz reichen, entnommen.
Jeweils vier Gewebestücke wurden für einen definierten Abstand für die
jeweilige Tiergruppe (Wt oder BDNF (+/-)) zusammen bei –80°C eingefroren.
So entstanden für BDNF (+/-) Tiere je drei Gefäße mit jeweils vier
Gewebestücken für den Abstand a (nahe der Läsion), für den Abstand b
(mittlere Entfernung von der Läsion) und für den Abstand c (weit von der Läsion
entfernt). Ebenfalls wurden so für die Wt Tiere je drei Gefäße für jeden
definierten Abstand hergestellt. Als Kontrollgewebe diente der Motorkortex
derselben Tiere. Hierzu wurden ebenso je vier Gewebestücke zusammen in
einem Gefäß eingefroren. Alle Gewebestücke wurden unverzüglich eingefroren
und bei –80 °C gelagert. Für die Durchführung der PCR wurde das Gewebe auf
Tockeneis in das Institut für Entwicklungsneurobiologie gebracht.
Dort wurde aus dem Gewebe zunächst cDNA hergestellt. Die Synthese
der cDNA wurde nach dem Protokoll der Firma Dynal (Dynal, Oslo, Norwegen)

38
durchgeführt. Dabei wurden magnetischen Polystyren-Kugeln von 2.8 µm
Durchmesser, sogenannte Dynabeads, benutzt. Diese Dynabeads wurden in
einem Magnetständer (Dynal, Oslo, Norwegen) magnetisch an der Wand eines
0.5 ml Eppendorfgefäßes fixiert, um dann an ihnen die Synthese der cDNA
vorzunehmen. Flüssigkeit konnte nun mit einer Pipette aus dem
Reaktionsgefäß entfernt werden, ohne die Dynabeads mit zu entfernen. Die
Dynabeads tragen an ihrer Oberfläche einen molekularen Linker, der mit den
PolyA-Sepuenzen prozessierter mRNA hybridisiert. Die so gebundene mRNA
dient als Matrize für eine reverse Transkription (RT), bei der die Oligo(dT)-Kette
der Dynabeads als Startermolekül benutzt wird. Dieses Startermolekül wurde
nun mit der komplementären Einzelstrang-cDNA verlängert. Auf diese Weise
erhält man an die magnetischen Dynabeads gebundene cDNA, die langfristig
bei 4° C gelagert werden können. Das genaue Protokoll zur Synthese der cDNA
wird im Folgenden beschrieben:
Die Dynabeads wurden zunächst in 150 µl Lyse-/Bindepuffer gewaschen
und sedimentiert. Für jede Synthese der cDNA wurden die Gewebestücke in
400 µl Lyse-Bindungspuffer in einem 2 ml Eppendorf-Reaktionsgefäß
aufgeschlossen und danach für ca. 1 min mit dem Vortexgerät geschüttelt. Um
weiter bestehende größere Zellverbände zu zerkleinern, wurde das Lysat für 30
sec. mit einem Turrax zerkleinert. Danach wurden die Proben für 2 min bei
14000 rpm zentrifugiert. Anschließend wurde der Überstand mit einer 21
Gauge-Kanüle abgenommen, in ein neues Reaktionsgefäß überführt und
dreimal geschert, um die Viskosität des Lysats zu vermindern und so ein
Verklumpen der Dynabeads zu umgehen. Das Lysat wurde danach erneut für 2
min bei 14000 rpm zentrifugiert. Die Dynabeads wurden nun magnetisch
separiert und der zuvor zu den Dynabeads gegebene Lyse-/Bindepuffer wurde
abgezogen. Nun wurde der Überstand des Lysates zu den Dynabeads
gegeben. Der Magnet wurde bei allen folgenden Mischvorgängen entfernt. Das
Gemisch wurde gevortext und für 15 min bei Raumtemperatur inkubiert.
Anschließend wurden die Dynabeads erneut magnetisch separiert und der
Überstand entfernt. Um die Dynabeads zu reinigen, wurde zweimal mit 200 µl
Waschpuffer mit Lithiumdodecylsulfat (LiDS) gemischt und magnetisch
separiert. Anschließend wurde zweimal mit 200 µl Waschpuffer ohne LiDS
gemischt und separiert. Um nun den Waschpuffer zu entfernen, wurden die

39
Dynabeads mit der hybridisierten und gereinigten mRNA dreimal mit RT-Puffer
gewaschen und separiert. Nach dem letzten Mischen wurde die Lösung in ein
neues steriles 500 µl Reaktionsgefäß pipettiert. Die Dynabeads wurden
separiert und der Überstand verworfen. Zum Schluss wurde jedes
Reaktionsgefäß mit 50 µl „Mastermix“ für die reverse Transkription versetzt.
Dieser Mastermix bestand pro Ansatz aus: 5 µl RT-Puffer (Quiagen), 5 µl
dNTP´s (Quiagen), 2.5 µl RNasin (40 Units/µl, Promega), 2.5 µl Sensicript
(Quiagen), 35 µl H2O (RNase-frei Quiagen). Zur cDNA-Synthese wurden die
Dynabeads mit dem Mastermix für 1 Stunde bei 37° C inkubiert und dabei alle
10 min neu gemischt. Danach wurden die Beads magnetisch separiert und der
Überstand entfernt. Die Dynabeads mit der neu synthetisierten cDNA und der
immer noch gebundenen mRNA wurden in 100 µl TE-Puffer gemischt und für 1
min bei 95° C inkubiert. So wurde die mRNA von den Dynabeads
abgeschmolzen. Danach wurde die noch heiße Lösung sofort im
Magnetständer für 3 min separiert und der Überstand mit der abgeschmolzenen
mRNA entfernt. Die Dynabeads wurden nochmals mit 100µl TE-Puffer
gewaschen, für 3 min sedimentiert und der Überstand abgenommen.
Abschließend wurden zu den Dynabeads 50 µl TE-Puffer hinzugefügt um die
cDNA-Banken schließlich bei 4° C zu lagern.
2.8.2 DNA-Amplifikation mit der Polymerase-Kettenreaktion (PCR)
Zur Analyse der Expression der BDNF mRNA wurde die Polymerase-
Kettenreaktion (PCR: Saiki et al., 1985) eingesetzt. Bei dieser Methode werden
sogenannte Primer benutzt. Dies sind kurze Oligonukleotide, die den Startpunkt
für die DNA-Polymerase darstellen. Die Sequenz der Primer ist der Tabelle
(Tab 1) zu entnehmen.
Zur Vervielfältigung der gewonnenen cDNA wurde 1 µl der cDNA-Bank
mit 49 µl Reaktionsmix versetzt. Jeder Reaktionsmix bestand aus 10 mM
dNTPs (Biozym), je 25 µM forward und reverse Primer (Eurogentec, Köln,
Deutschland), 2,5 u BiothermTM DNA Polymerase (Gene Craft Germany) sowie
dem dazugehörigen 10 x Puffer (5 µl) und Wasser. Bei jeder PCR-Reaktion
erfolgte eine initiale fünfminütige Denaturierung bei 95°C. Die Reaktionszyklen
wurden dann mit einer fünfminütigen terminalen Elongation bei 72°C beendet.

40
Die weiteren PCR-Bedingungen sind ebenfalls in der Tabelle (Tab. 1)
dargestellt. Einige Primer in dieser Tabelle enthalten Restriktionsschnittstellen,
damit diese PCR-Fragmente anschließend kloniert werden können. Diese
Restriktionsschnittstellen vergrößern das Amplifikat um insgesamt 18
Basenpaare.
Tabelle 2.1: Primer - Sequenzen PCR-Produkt
Primer- Sequenzen Fragmentgröße PCR-Programm
Actin
Sense Primer 5´-TCATGAAGTGTGACGTTGACATCCGTAAAG-3´ Antisense Primer 5´-CCTAGAAGCATTTGCGGTGCACGATGGAGG-3´
285 bp (924 bis 1208)
1 min 94°C 2 min 57°C 2 min 72°C 26 Zyklen
BDNF
Sense Primer 5´-GGA CAA GGC AAC TTG GCC-3´ Antisense Primer 5´-CAG AGG AGG CTC CAA AGG-3´
384 bp (158 bis 542)
1 min 94°C 1 min 55°C 1 min 72°C 30 Zyklen
PARV
Sense Primer 5´-GCA GAC TCC TTC GAC CAC AA-3´ Antisense Primer 5´-GAA TTC TTC AAC CCC AAT CT-3´
240 bp (1 bis 240)
40 sec 94°C 40 sec 55°C 1 min 72°C 30 Zyklen
GAD65
Sense Primer 5´-CCC CAA GCA GCA TCC ACA T-3´ Antisense-Primer 5´-TCT TTT CTC CTG GTG GTG CC-3´
391 bp (788 bis 1179)
1 min 94°C 1 min 51°C 1 min 72°C 33 Zyklen
GAD67
Sense Primer 5´-CCC CAA GCA GCA TCC ACA T-3´ Antisense-Primer 5´-TAC GGG GTT CGC ACA GGT C-3´
600 bp (712 bis 1312)
1 min 94°C 1 min 51°C 1 min 72°C 33 Zyklen
GFAP
Sense Primer 5´-CCACGTGGAGATGGATGTGGCCA-3´ Antisense-Primer 5´-AGTGCCTCCTGGTAACTGGCCGAC-3´
324 bp (734 bis 1057)
1 min 94°C 1 min 63°C 1 min 72°C 25 Zyklen
2.8.3 Agarose-Gelelektrophorese
Die Agarose-Gelelektrophorese dient der Auftrennung geladener Moleküle,
welche eine unterschiedliche Größe besitzen. Nach Anlegen einer Spannung
von 40 – 80 mV wandert die DNA im Gel aufgrund ihrer negativ geladenen
Phosphatgruppen in Richtung Anode. Dabei laufen kleinere Moleküle schneller
als größere. Die PCR-Amplifikate wurden auf ein 2 %-iges Agarosegel
(Agarose: Biozym, Hess. Oldendorf, Deutschland) aufgetragen. Um die

41
einzelnen Banden später sichtbar machen zu können, enthielt dieses Gel
Ethidiumbromid (Merck, Darmstadt, Deutschland). Als Ladepuffer wurden 5 µl
PCR-Farbe für 50 µl Probe verwendet. Dabei wurde dieselbe Probe zu gleichen
Teilen (20 µm) jeweils einmal in die obere und einmal in die untere Tasche
pipettiert, um den Ethidiumbromid-Gradienten auszugleichen. Zum
Längenvergleich wurden DNA-Längenmarker (100 bp-Leiter, Fermentas, St.
Leon-Rot, Deutschland) mit auf das Gel aufgetragen.
2.8.4 Auswertung der Messung der mRNA Konzentration
Die Agarosegele wurden mit Hilfe des Biometra UV Solo® eingescannt, um die
Intensität der einzelnen Banden mit dem Computerprogramm Biometra®
BioDocAnalyze Version 2.1 analysieren zu können. Die Matrizen können
aufgrund unterschiedlich effektiver mRNA-Isolierung und cDNA-Synthese
Unterschiede in der PCR-Effektivität aufweisen. Daher wurden zunächst die
Werte für Aktin gemittelt. Aktin ist ein Strukturprotein des Zytoskeletts und
kommt in allen eukaryoten Zellen vor. Da es bei den vorherrschenden
Konditionen keine Schwankungen zeigt ist es gut zur Standardisierung
geeignet. Dieser Standardmarker dient gleichzeitig als interne Kontrolle der
Qualität des verwendeten Gewebes und der cDNA-Banken-Synthese.
Für jedes Zielgen wurden mindestens drei unabhängige PCRs
durchgeführt und ausgewertet. Zum Nachweis möglicher Verunreinigungen
wurden bei jeder PCR eine Wasserkontrolle mit amplifiziert und auf das Gel
aufgetragen. Der obere und untere Intensitätswert des Agarosegels wurde bei
jedem Zielgen gemittelt und durch den Mittelwert des dazugehörigen
Aktinwertes geteilt. Um relative Einheiten bzw. Relationen zu erhalten wurden
diese Ergebnisse wiederum auf die Kontrolle normiert. Abweichungen der
Werte wurden als S.E.M. der Relationen berechnet. Die Signifikanz der mRNA-
Expressions-Veränderungen wurde mittels Mann-Whitney Rangsummentest
überprüft.

42
3. Ergebnisse
3.1 Histologie
Die Morphologie der Läsion und des umgebenden Gewebes wurden mit Hilfe
von Nissl- und GFAP-Färbung charakterisiert. Die Läsionen wurden in ca. 1,5
mm Entfernung und parallel zur Mittellinie im visuellen Kortex der rechten
Hemisphäre induziert. Es wurden bis zu 3 akute Hirnschnitte mit einer Dicke
von je 350 µm und einer reproduzierbaren Läsion gewonnen, die den visuellen
Kortex enthielten.
Nissl-Färbungen von PFA-fixierten Schnitten (Abb. 3.1A und 3.1C)
erlauben einen Überblick über die Ausdehnung der Läsion und die Morphologie
des umgebenden Gewebes. Das nekrotische Gewebe der Läsion erstreckte
sich über ca. 0,5 bis 1,0 mm in mediolateraler Richtung und dehnte sich in der
Tiefe bis zur Lamina V des visuellen Kortex aus. Die Läsion war weitestgehend
scharf begrenzt, zum Teil von einem bis zu 100 µm breiten Randsaum
umgeben, der beschädigte Zellen enthielt. Außerhalb des Randgebietes wurde
eine normale Zellmorphologie gefunden, die für die entsprechenden kortikalen
Schichten typisch ist.
Durch die GFAP-Färbung wurde eine leichte Gliose in der Umgebung der
Läsion sichtbar (Abb. 3.1B und 3.1D). Die Ausdehnung der Gliose rund um die
Läsion war individuell unterschiedlich, meist jedoch auf ein Gebiet von 100 bis
200 µm um die Läsion begrenzt. Es wurde kein Unterschied in der Morphologie
und Veränderung nach Läsion zwischen Wt und BDNF(+/-) gefunden.
Auch die Hirnschnitte sham-operierter Tiere wurden untersucht, um
einen möglichen Einfluss der Operationstechnik auf die Ergebnisse
auszuschließen. Im Operationsgebiet dieser Versuchsgruppe wurde jedoch
keine Schädigung des Gewebes, keine vermehrte Kapillarisierung und auch
kein Zellödem gesehen. Ebenso war keine Gliose im Operationgebiet der
sham- operierten Tiere feststellbar.
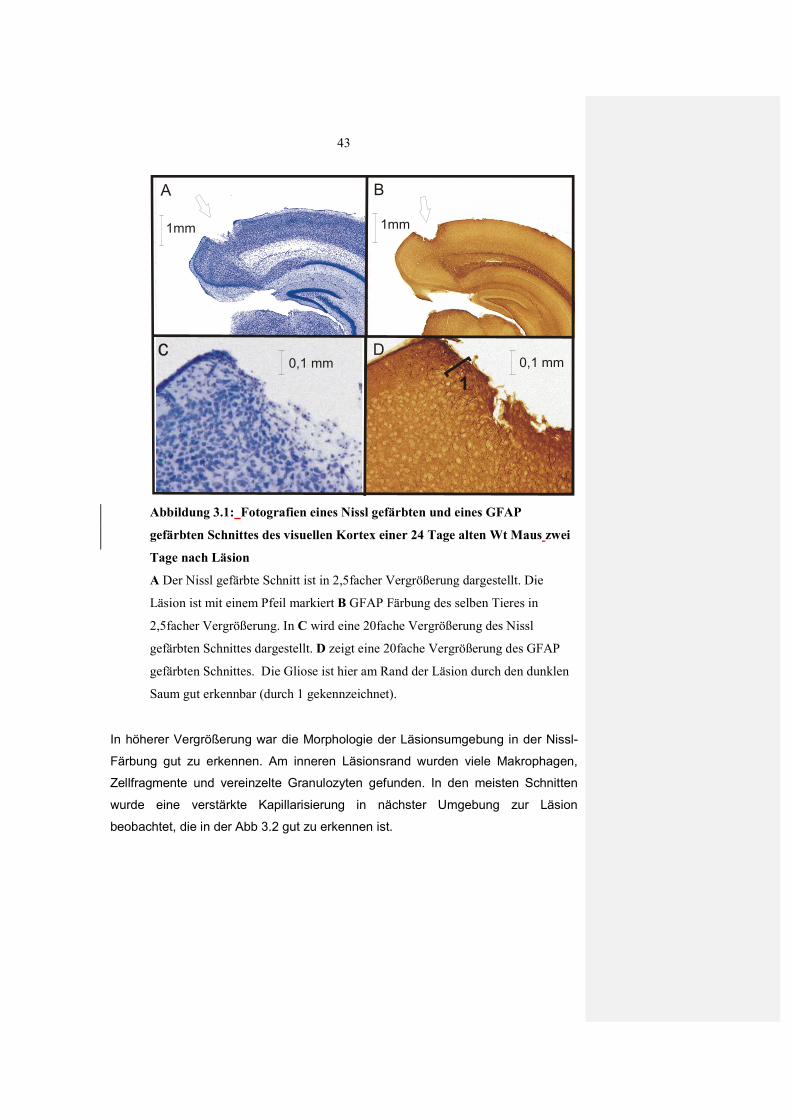
43
1mm 1mm
0,1 mmD
A B
0,1 mm
1
Abbildung 3.1: Fotografien eines Nissl gefärbten und eines GFAP
gefärbten Schnittes des visuellen Kortex einer 24 Tage alten Wt Maus zwei
Tage nach Läsion
A Der Nissl gefärbte Schnitt ist in 2,5facher Vergrößerung dargestellt. Die
Läsion ist mit einem Pfeil markiert B GFAP Färbung des selben Tieres in
2,5facher Vergrößerung. In C wird eine 20fache Vergrößerung des Nissl
gefärbten Schnittes dargestellt. D zeigt eine 20fache Vergrößerung des GFAP
gefärbten Schnittes. Die Gliose ist hier am Rand der Läsion durch den dunklen
Saum gut erkennbar (durch 1 gekennzeichnet).
In höherer Vergrößerung war die Morphologie der Läsionsumgebung in der Nissl-
Färbung gut zu erkennen. Am inneren Läsionsrand wurden viele Makrophagen,
Zellfragmente und vereinzelte Granulozyten gefunden. In den meisten Schnitten
wurde eine verstärkte Kapillarisierung in nächster Umgebung zur Läsion
beobachtet, die in der Abb 3.2 gut zu erkennen ist.
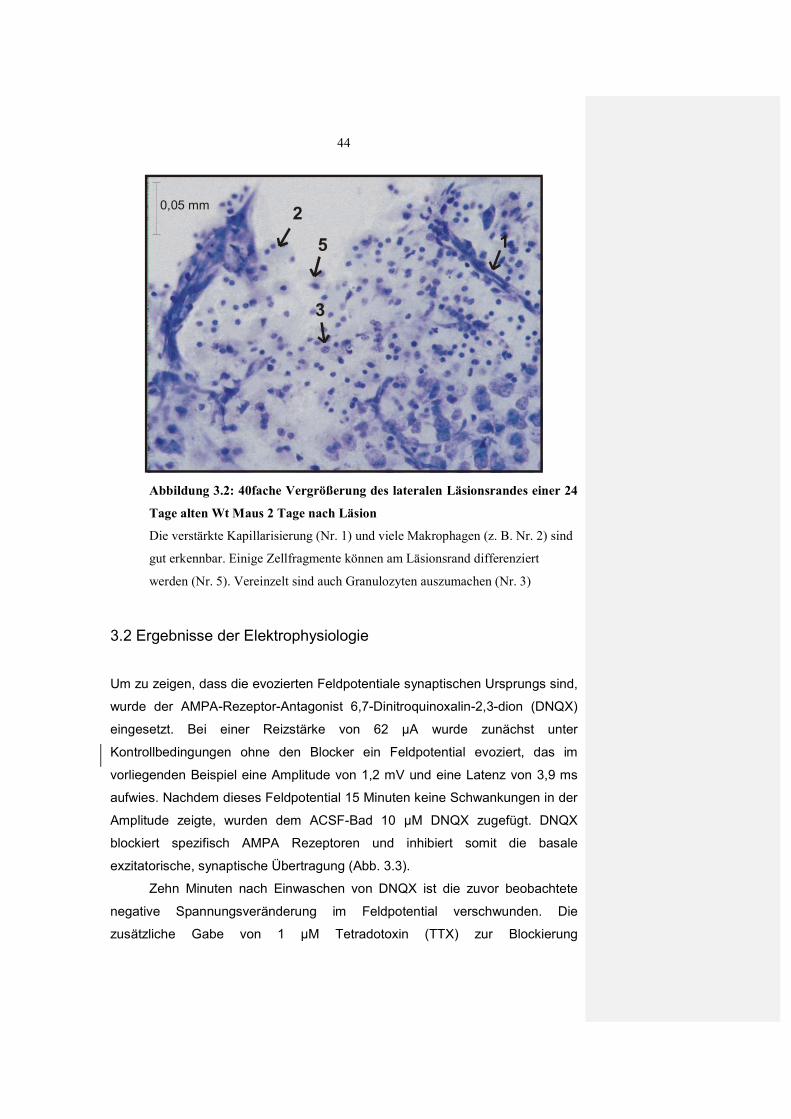
44
3
2
15
0,05 mm
Abbildung 3.2: 40fache Vergrößerung des lateralen Läsionsrandes einer 24
Tage alten Wt Maus 2 Tage nach Läsion
Die verstärkte Kapillarisierung (Nr. 1) und viele Makrophagen (z. B. Nr. 2) sind
gut erkennbar. Einige Zellfragmente können am Läsionsrand differenziert
werden (Nr. 5). Vereinzelt sind auch Granulozyten auszumachen (Nr. 3)
3.2 Ergebnisse der Elektrophysiologie
Um zu zeigen, dass die evozierten Feldpotentiale synaptischen Ursprungs sind,
wurde der AMPA-Rezeptor-Antagonist 6,7-Dinitroquinoxalin-2,3-dion (DNQX)
eingesetzt. Bei einer Reizstärke von 62 µA wurde zunächst unter
Kontrollbedingungen ohne den Blocker ein Feldpotential evoziert, das im
vorliegenden Beispiel eine Amplitude von 1,2 mV und eine Latenz von 3,9 ms
aufwies. Nachdem dieses Feldpotential 15 Minuten keine Schwankungen in der
Amplitude zeigte, wurden dem ACSF-Bad 10 µM DNQX zugefügt. DNQX
blockiert spezifisch AMPA Rezeptoren und inhibiert somit die basale
exzitatorische, synaptische Übertragung (Abb. 3.3).
Zehn Minuten nach Einwaschen von DNQX ist die zuvor beobachtete
negative Spannungsveränderung im Feldpotential verschwunden. Die
zusätzliche Gabe von 1 µM Tetradotoxin (TTX) zur Blockierung
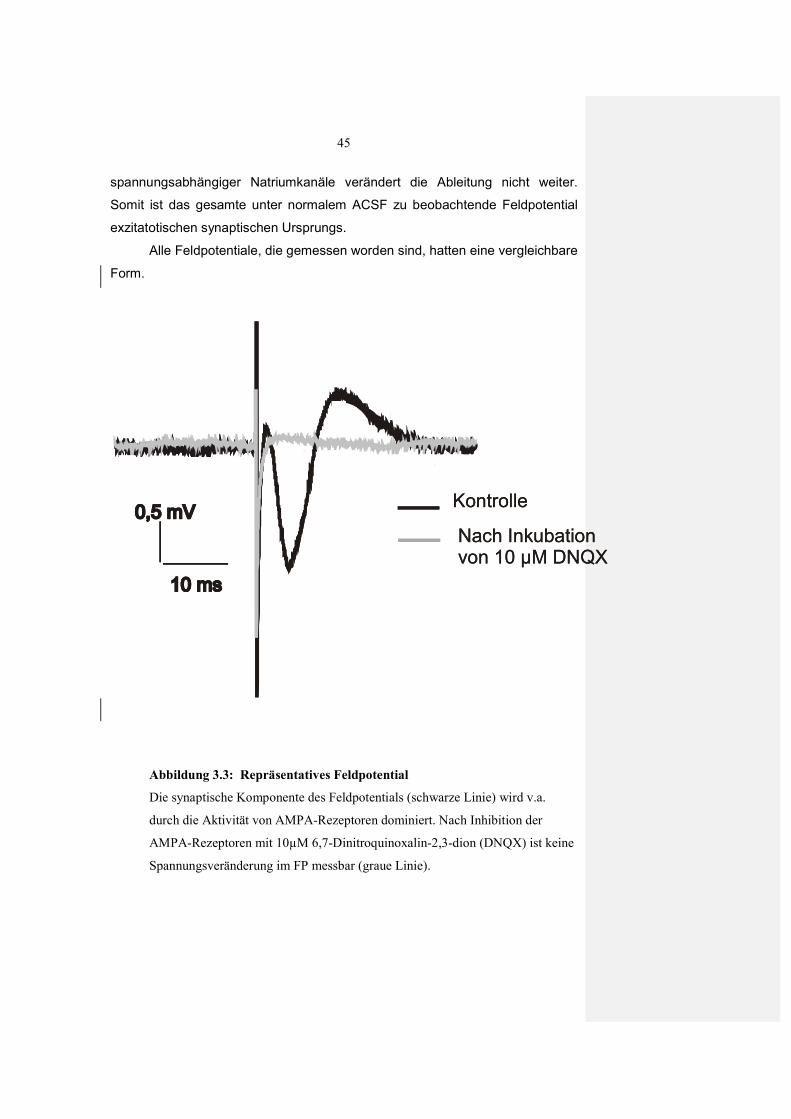
45
spannungsabhängiger Natriumkanäle verändert die Ableitung nicht weiter.
Somit ist das gesamte unter normalem ACSF zu beobachtende Feldpotential
exzitatotischen synaptischen Ursprungs.
Alle Feldpotentiale, die gemessen worden sind, hatten eine vergleichbare
Form.
Kontrolle
Nach Inkubation von 10 µM DNQX
Kontrolle
Nach Inkubation von 10 µM DNQX
Abbildung 3.3: Repräsentatives Feldpotential
Die synaptische Komponente des Feldpotentials (schwarze Linie) wird v.a.
durch die Aktivität von AMPA-Rezeptoren dominiert. Nach Inhibition der
AMPA-Rezeptoren mit 10µM 6,7-Dinitroquinoxalin-2,3-dion (DNQX) ist keine
Spannungsveränderung im FP messbar (graue Linie).

46
3.2.1 Einfluss einer Läsion auf die synaptische Plastizität im
visuellen Kortex der Maus
In der zweiten Versuchsreihe wurde erstens der Einfluss von Läsionen im
visuellen Kortex auf die basale synaptische Erregung im Randbereich der
Verletzung untersucht (3.2.1.1), zweitens wurden läsionsbedingte Änderungen
der synaptischen Plastizität ermittelt (3.2.1.2) und drittens wurde überprüft, ob
die Operationsmethode selbst einen Einfluss auf die Plastizität des visuellen
Kortex hat (3.2.1.3).
Hierzu wurde in mehreren Versuchsserien bei jeweils einer Gruppe von
Mäusen eine Läsion in den visuellen Kortex induziert. Nach zwei bis sechs
Tagen Überlebenszeit wurden akute Hirnschnitte aus dem visuellen Kortex
präpariert und elektrophysiologische Messungen durchgeführt. Die Ergebnisse
wurden mit denen von gleichaltrigen unbehandelten und sham-operierten
Tieren, zwei bis sechs Tage nach Operation, verglichen.
3.2.1.1 Basale synaptische Übertragung (I-O Kurve) nach Läsionen
Um die basale synaptische Übertragung zu charakterisieren, wurde
zunächst eine sogenannte input-output Kurve (I-O Kurve) angefertigt. Es
wurden die I-O-Kurven von nichtbehandelten Mäusen (n= 5) mit denen
läsionierter Tiere (n=6) verglichen (Abb. 3.4). Dabei unterschieden sich die
maximalen Amplituden der beiden Gruppen nicht (bei einer Stimulationsdauer
von 200 µs: Wt unbehandelt 1,3 ± 0,1 mV, Wt nach Läsion 1,4 ± 0,1 mV; p =
0,75). In beiden Gruppen änderte sich die Form der Feldpotentiale mit längerer
Stimulationsdauer nicht und die Latenz zwischen dem Stimulusartefakt und der
maximal negativen Feldpotentialamplitude blieb ebenfalls konstant (Wt
unbehandelt = 4,22 ± 0,52 ms, Wt nach Läsion = 4,22 ± 0,55 ms; p=0,99). Es
gab weder in der Form noch im relativem Anstieg der Amplitude Unterschiede
zwischen nichtbehandelten und läsionierten Tieren. Für keine
Stimulationsdauer konnte ein signifikanter Unterschied in der Höhe der
Amplitude festgestellt werden (p > 0,05). Es wurde also keine läsionsbedingte
Änderung der basalen synaptischen Transmission festgestellt.

47
A
Wt unbehandelt Wt Läsion 40 µs
100 µs
200 µs 1 mV
5 ms
1 mV
5 ms
1 mV
5 ms
B
FP
Am
plit
ude
(m
V)
Stimulus Dauer (µsec)
20 40 60 80 100 120 140 160 180 200
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Wt unbehandelt (n=5)
Wt Läsion (n=6)
Abbildung 3.4: Die basale synaptische Übertragung ist nach Läsionen nicht
verändert
A repräsentative Feldpotentiale einer unbehandelten und einer läsionerten Maus,
die bei einer Stimulation von 40, 100 und 200 µs evoziert wurden
B durchschnittliche Feldpotentialamplituden unbehandelter (Dreiecke, n=5) und
läsionierter (Kreise, n=6) Tiere in Beziehung zur Stimulationsdauer

48
3.2.1.2 Verstärkte LTP nach Läsionen im visuellen Kortex
Zunächst wurden Gehirnschnitte unbehandelter Wt Mäuse (n=8) untersucht.
Bei diesen Tieren kam es nach repetitiver Reizung durch TBS zu einer
reproduzierbaren LTP. Die Amplituden der Feldpotentiale, die in akuten
Hirnschnitten nach Reizung der kortikalen Schicht IV in Schicht II / III abgeleitet
wurden, hatten vor TBS eine durchschnittliche Höhe von 0,75 ± 0,03 mV, was
58% der maximalen Amplitude entspricht. Unmittelbar nach der TBS waren die
Amplituden meist zunächst verkleinert, bis sie nach durchschnittlich drei
Minuten das Level der Baselineamplitude übertrafen. Nach TBS wurde in der 7
± 1 Minute dann ein LTP-Level von 120 ± 4,5 % der Baseline erreicht, das sich
bis zum Ablauf der 60 Minuten noch leicht vergrößerte. Diese Änderung der
Amplitude zwischen der Siebten und der 51. Minute war allerdings nicht
signifikant (p > 0,05). 51-60 min nach TBS hatten die Amplituden eine
durchschnittliche Höhe von 0,93 ± 0,04 mV und entsprachen so 121 ± 4% der
durchschnittlichen Baselineamplitude. Auf Form und Latenz zwischen dem
Stimulusartefakt und der maximal negativen Feldpotentialamplitude der
einzelnen Feldpotentiale hatte die TBS keinen Einfluss ((Wt unbehandelt vor
TBS = 4,27 ± 0,47 ms, nach TBS = 4,22 ± 0,31; p=0,50).
Die Ergebnisse unbehandelter Wt Mäuse wurden anschließend mit
läsionierten Wt Tieren (n = 9) verglichen (Abb. 3.5). Dazu wurde den Tieren
zunächst unter Narkose eine Laserläsion zugefügt. Nach einer Überlebenszeit
von zwei bis fünf Tagen wurden dann akute Hirnschnitte hergestellt und
elektrophysiologische Messungen durchgeführt. Die Feldpotentiale, die in
Hirnschnitten von läsionierten Tieren in 1,5 – 2,2 mm Abstand vom Rand der
Läsion evoziert wurden, hatten dieselbe Form und Latenz, wie diejenigen, die
bei unbehandelten Tieren gesehen worden waren (Wt unbehandelt = 4,24 ±
0,31 ms; Wt nach Läsion = 4,21 ± 0,39 ms, p=0,71). Auch die Amplitude vor
TBS (0,77 ± 0,06 mV) zeigte keinen signifikanten Unterschied zwischen beiden
Gruppen. Nach TBS Stimulation konnte auch bei läsionierten Tieren eine
reproduzierbare LTP beobachtet werden. Allerdings wurde eine weniger
ausgeprägte Depression der Feldpotentialamplitude direkt nach der TBS
beobachtet. Auch der darauf folgende Anstieg der Amplitude auf ein Level, das
über der Grundlinie liegt, geschah früher (bereits in der ersten Minute nach
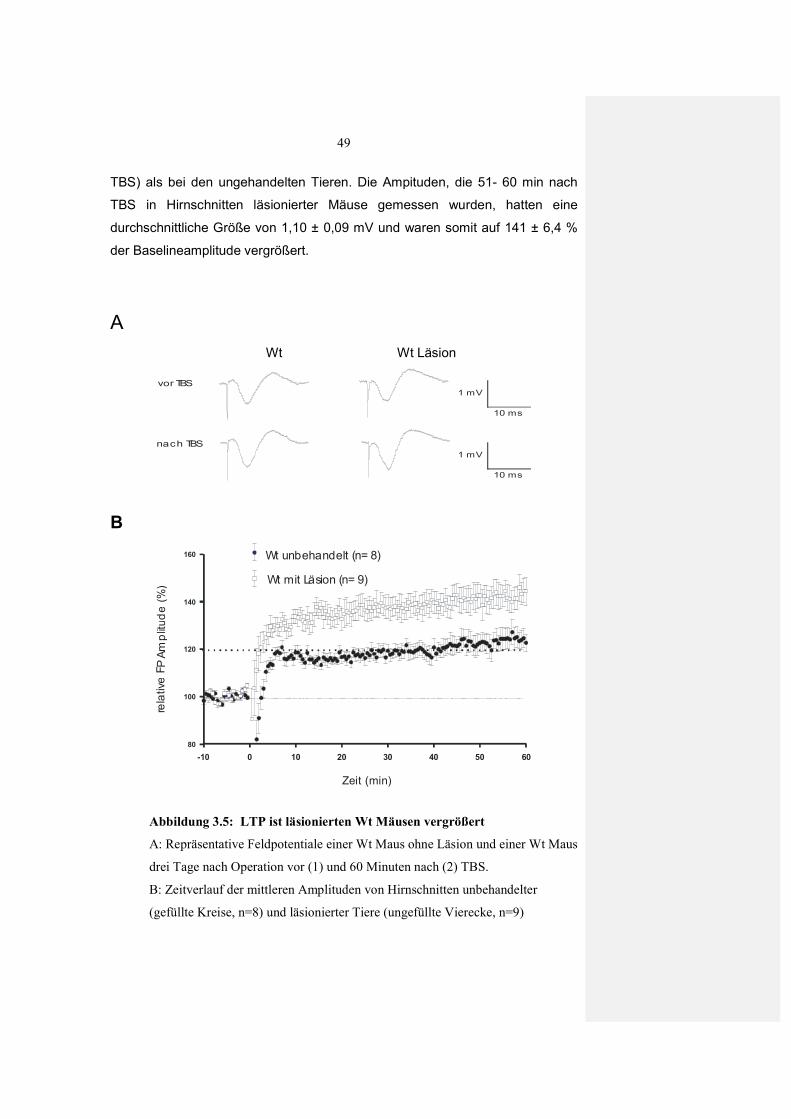
49
TBS) als bei den ungehandelten Tieren. Die Ampituden, die 51- 60 min nach
TBS in Hirnschnitten läsionierter Mäuse gemessen wurden, hatten eine
durchschnittliche Größe von 1,10 ± 0,09 mV und waren somit auf 141 ± 6,4 %
der Baselineamplitude vergrößert.
A
Wt Wt Läsion
vor TBS
nach TBS
10 ms
1 mV
10 ms
1 mV
B
-10 0 10 20 30 40 50 60
80
100
120
140
160 Wt unbehandelt (n= 8)
Wt mit Läsion (n= 9)
rela
tive
FP A
mp
litud
e (
%)
Zeit (min)
Abbildung 3.5: LTP ist läsionierten Wt Mäusen vergrößert
A: Repräsentative Feldpotentiale einer Wt Maus ohne Läsion und einer Wt Maus
drei Tage nach Operation vor (1) und 60 Minuten nach (2) TBS.
B: Zeitverlauf der mittleren Amplituden von Hirnschnitten unbehandelter
(gefüllte Kreise, n=8) und läsionierter Tiere (ungefüllte Vierecke, n=9)

50
Vergleicht man also die Ergebnisse der läsionierten mit denen unbehandelter
Tiere, ergibt sich bei unveränderter Form und Latenz der Feldpotentiale ein
signifikanter (p = 0,013) Unterschied in der Ausprägung der LTP: die LTP in
läsionierten Tieren zwei bis fünf Tage nach Läsion ist 20 Prozentpunkte größer
als bei Messungen von unbehandelter Mäuse.
3.2.1.3 Keine Veränderung in der LTP nach Sham-Operationen
Um nachzuweisen, dass die Operationsmethode keinen Einfluss auf die
Plastizität des visuellen Mauskortex hat, wurden Hirnschnitte von sham-
operierten Wt Mäusen untersucht. Während der Sham-Operation erfolgte die
Freipräparation des Kortex genau wie bei Läsionstieren, jedoch ohne eine
Läsion zu induzieren. Auch nach Sham-Operationen wurden die Tiere nach
einer Überlebenszeit von zwei bis fünf Tagen untersucht. Die
elektrophysiologischen Messungen erfolgten genau wie bei den läsionierten
und bei den unbehandelten Tieren. Die in den akuten Hirnschnitten evozierten
Feldpotentiale hatten dieselbe Form und Latenz zwischen dem Stimulusartefakt
und der maximal negativen Feldpotentialamplitude wie diejenigen
unbehandelter Tiere (Wt unbehandelt = 4,24 ± 0,31 ms; Wt sham-operiert =
4,21 ± 0,39 ms; p = 0,69). Die gemessenen Feldpotentiale sham-operierter Wt
Tiere (n= 6) zeigten auch eine vergleichbare Amplitude wie die Feldpotentiale
unbehandelter Wt Mäuse. Diese entsprachen vor TBS einer durchschnittlichen
Amplitude von 0,71 ± 0,04 mV. Die Form der Feldpotentiale blieb auch nach
TBS unverändert. Die relativen Amplituden der sham-operierten Wt Tiere waren
51-60 min nach TBS auf durchschnittlich 0,86 ± 0,03 mV erhöht, was einer
relativen Amplitude von 122 ± 3,3 % der Baseline entspricht. Somit konnte kein
signifikanter Unterschied in der Größe der relativen Amplitude 51-60 min nach
TBS zwischen unbehandelten und sham-operierten Tieren festgestellt werden
(p = 0,95). Ebenso waren Form und Latenz der Feldpotentiale nach TBS nicht
verändert (Wt sham-operiert vor TBS= 4,19 ± 0,42 ms, nach TBS= 4,21 ± 0,41;
p=0,73). Auch der zeitliche Verlauf der LTP nach TBS war mit dem der
unbehandelten Tiere vergleichbar (Abb. 3.6). Die Operationsmethode selber
bewirkt also keine messbare Veränderung in der LTP gegenüber nicht
behandelten Tieren.
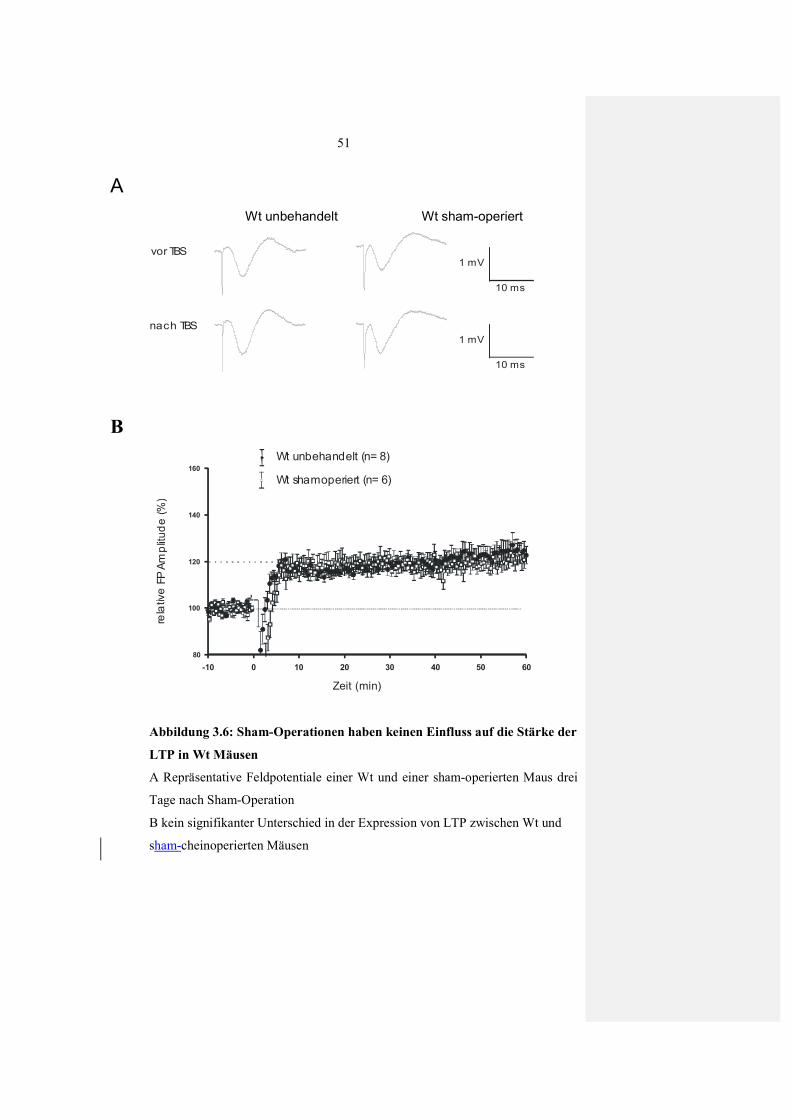
51
A
Wt unbehandelt Wt sham-operiert
vor TBS
nach TBS
10 ms
1 mV
10 ms
1 mV
B
Zeit (min)
-10 0 10 20 30 40 50 60
80
100
120
140
160
Wt unbehandelt (n= 8)
Wt shamoperiert (n= 6)
rela
tive
FP
Am
plit
ud
e (%
)
Abbildung 3.6: Sham-Operationen haben keinen Einfluss auf die Stärke der
LTP in Wt Mäusen
A Repräsentative Feldpotentiale einer Wt und einer sham-operierten Maus drei
Tage nach Sham-Operation
B kein signifikanter Unterschied in der Expression von LTP zwischen Wt und
sham-cheinoperierten Mäusen

52
3.2.2 Veränderte synaptische Plastizität im visuellen Kortex der
BDNF (+/-) Maus
In der zweiten Versuchsserie wurde die LTP in Hirnschnitten von BDNF (+/-)
Mäusen untersucht. Dazu wurden akute Hirnschnitte von BDNF (+/-) Mäusen
und von gleichaltrigen Wt Mäusen hergestellt und zunächst die basale
synaptische Übertragung in BDNF(+/-) Tieren gemessen (3.2.2.1). Im nächsten
Schritt wurde eine LTP in BDNF(+/-) Mäusen ausgelöst und mit der LTP in Wt
Tieren verglichen (3.2.2.2).
3.2.2.1 Basale synaptische Übertragung (I-O Kurve) in Hirnschnitten
von BDNF (+/-) Mäusen
Auch für BDNF(+/-) Tiere wurde eine I-O Kurve angefertigt, um die basale
synaptische Übertragung zu bestimmen und mit den Werten unbehandelter Wt
Tiere zu vergleichen (Abb. 3.7). Die maximalen Amplituden, die in Hirnschnitten
von unbehandelten BDNF (+/-) Tieren evoziert wurden, unterschieden sich
dabei nicht in Form, Höhe der maximalen Amplitude (Wt unbehandelt 1,3 ± 0,1
mV ; BDNF(+/-) unbehandelt = 1,2 ± 0,03 mV; p = 0,41) und Latenz zwischen
dem Stimulusartefakt und der maximal negativen Feldpotentialamplitude (Wt
unbehandelt = 4,22 ± 0,52 ms, BDNF(+/-) unbehandelt = 4,28 ± 0,62 ms;
p=0,54) von denen, die in Hirnschnitten unbehandelter Wt Tiere abgeleitet
wurden. Für keine Stimulationsdauer konnte ein signifikanter Unterschied in der
Höhe der Amplitude zwischen BDNF(+/-) und Wt Tieren festgestellt werden (p
> 0,05). Somit zeigen sich keine Unterschiede in der basalen synaptischen
Übertragung zwischen Wt und BDNF (+/-) Mäusen.
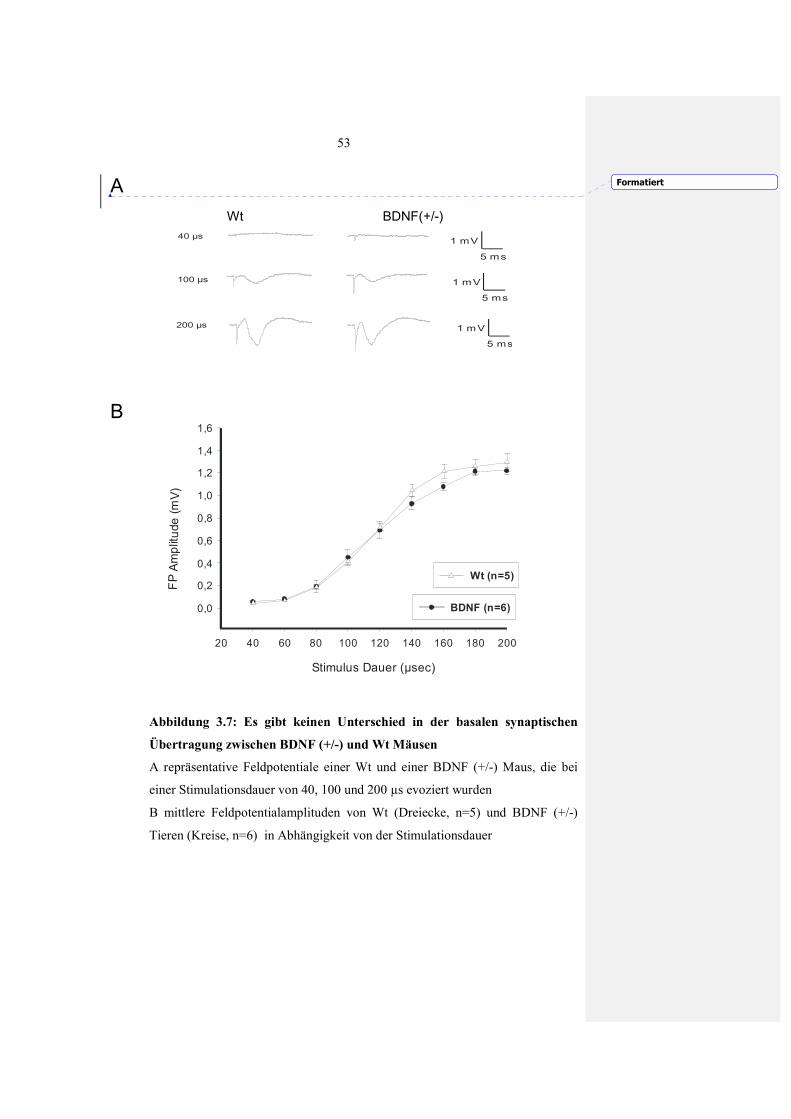
53
A
Wt BDNF(+/-)
40 µs
100 µs
200 µs 1 mV
5 ms
1 mV
5 ms
1 mV
5 ms
B
FP
Am
plit
ude
(m
V)
Stimulus Dauer (µsec)
20 40 60 80 100 120 140 160 180 200
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
BDNF (n=6)
Wt (n=5)
Abbildung 3.7: Es gibt keinen Unterschied in der basalen synaptischen
Übertragung zwischen BDNF (+/-) und Wt Mäusen
A repräsentative Feldpotentiale einer Wt und einer BDNF (+/-) Maus, die bei
einer Stimulationsdauer von 40, 100 und 200 µs evoziert wurden
B mittlere Feldpotentialamplituden von Wt (Dreiecke, n=5) und BDNF (+/-)
Tieren (Kreise, n=6) in Abhängigkeit von der Stimulationsdauer
Formatiert

54
3.2.2.2 Beeinträchtigte LTP Expression in BDNF (+/-) Mäusen
Im Folgenden wurde nun die LTP in BDNF(+/-) Mäusen untersucht und mit der
LTP in Wt Tieren verglichen (Abb. 3.8). In akuten Hirnschnitten von BDNF(+/-)
Mäusen hatten die evozierten Feldpotentiale eine durchschnittliche Höhe von
0,86 ± 0,07 mV. Die Form der Feldpotentiale war gegenüber der in Wt Tieren
gesehenen Form der Feldpotentiale nicht verändert. Auch war kein Unterschied
in der Latenz zwischen dem Stimulusartefakt und der maximal negativen
Feldpotentialamplitude ((Wt unbehandelt = 4,24 ± 0,31 ms, BDNF(+/-)
unbehandelt= 4,28 ± 0,42 ms; p=0,51) auszumachen. Nach der TBS zeigte sich
zunächst eine Depression der Amplitudenhöhe, die auch bei unbehandelten Wt
Mäusen beobachtet worden war. Sie hatte bei den BDNF (+/-) Mäusen den
gleichen zeitlichen Verlauf. In den ersten Minuten nach TBS war die Ampitude
meist verringert, bis sie nach 3 ± 2 Minuten das Level der Baseline übertraf.
Nach TBS waren in Minute 7 ± 1 die Feldpotentialamplitude signifikant
vergrößert und erreichte maximal eine durchschnittliche Amplitude von 1,0 ±
0,10 mV, was 115 ± 1,6 % der Baseline entsprach. Sie erreichte allerdings
nicht das Level von 121%, das in Hirnschnitten von unbehandelten Wt Mäusen
beobachtet worden war. Auch blieb die Amplitude nicht stabil auf diesem
erhöhten Niveau, sondern verkleinerte sich danach bis sie am Ende des
Experimentes wieder annähernd das Baselinelevel erreicht hatte (51 – 60 min
nach TBS durchschnittlich 104 ± 3,4 % der Baseline) und sich so signifikant von
der LTP in Wt Mäusen unterschied (p = 0,005). Auf die Form und die Latenz
zwischen dem Stimulusartefakt und der maximal negativen
Feldpotentialamplitude der einzelnen Feldpotentiale hatte die TBS auch in
Hirnschnitten von BDNF (+/-) Mäusen keinen Einfluss (BDNF(+/-) unbehandelt
vor TBS= 4,27 ± 0,50 ms, nach TBS= 4,31 ± 0,51; p = 0,56).
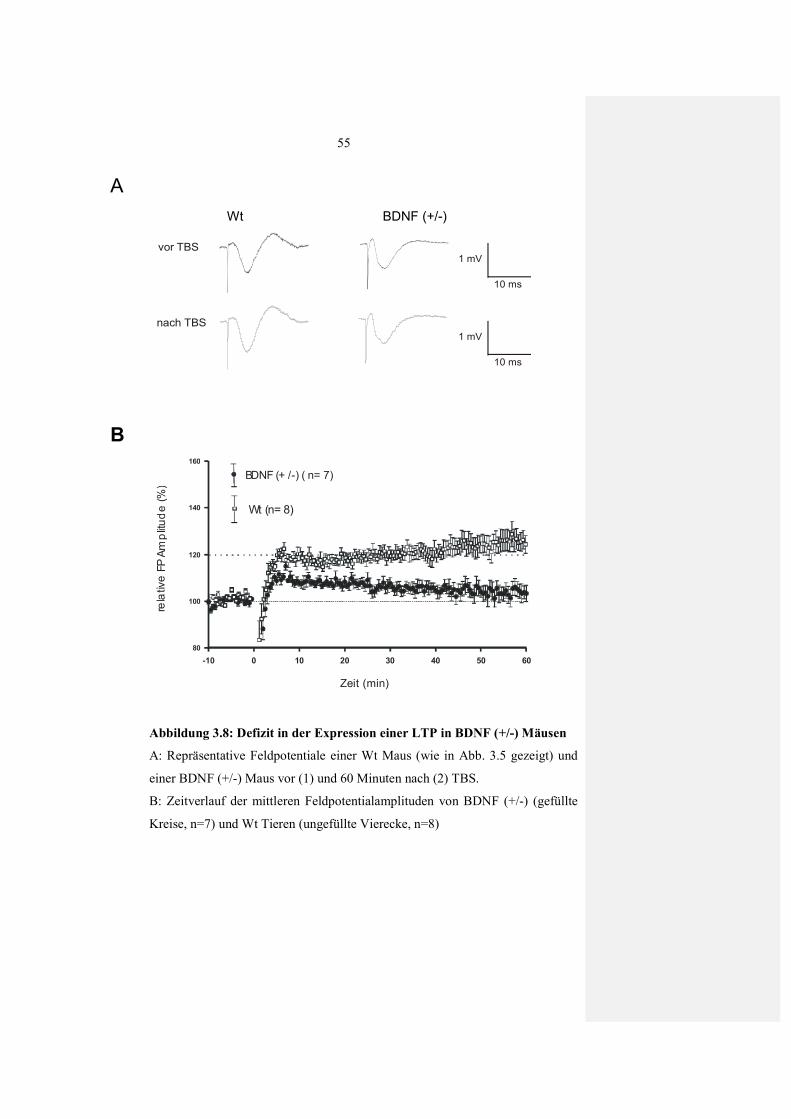
55
A
Wt BDNF (+/-)
vor TBS
nach TBS
10 ms
1 mV
10 ms
1 mV
B
Zeit (min)
160
-10 0 10 20 30 40 50 60
80
100
120
140
BDNF (+ /-) ( n= 7)
Wt (n= 8)
rela
tive
FP
Am
plit
ud
e (%
)
Abbildung 3.8: Defizit in der Expression einer LTP in BDNF (+/-) Mäusen
A: Repräsentative Feldpotentiale einer Wt Maus (wie in Abb. 3.5 gezeigt) und
einer BDNF (+/-) Maus vor (1) und 60 Minuten nach (2) TBS.
B: Zeitverlauf der mittleren Feldpotentialamplituden von BDNF (+/-) (gefüllte
Kreise, n=7) und Wt Tieren (ungefüllte Vierecke, n=8)

56
3.2.3 Einfluss von Läsionen auf die LTP Expression in BDNF (+/-)
Mäusen
In der nächsten Versuchsreihe wurde untersucht, in wie weit Läsionen einen
Einfluss auf die LTP Expression in BDNF(+/-) Tieren haben. Zunächst wurde
die basale synaptische Übertragung in BDNF(+/-) Tieren nach Läsionen
bestimmt und mit unbehandelten BDNF(+/-) Tieren verglichen (3.2.3.1). Danach
wurden die Effekte einer TBS an Hirnschnitten läsionierter BDNF(+/-) Mäuse
gemessen und mit unbehandelten BDNF(+/-) Tieren und im nächsten Schritt mit
unbehandelten Wt und läsionierten Wt Mäusen verglichen.
Dazu wurden den BDNF (+/-) Mäusen vergleichbare Läsionen wie in Wt
Mäusen zugefügt. Die elektrophysiologischen Experimente wurden zwei bis
sechs Tage nach der Läsion durchgeführt.
3.2.3.1 Basale synaptische Übertragung (I-O Kurve) in BDNF (+/-) Mäusen nach Läsionen
Um die basale synaptische Übertragung in BDNF(+/-) Tieren zu untersuchen,
wurde auch für läsionierte BDNF(+/-) Mäuse eine I-O Kurve ermittelt und mit
den Daten unbehandelter BDNF(+/-) und läsionierten Wt Tieren verglichen
(Abb. 3.9). Zwischen den maximalen Amplituden läsionierter und unbehandelter
BDNF(+/-) Tiere war kein Unterschied zu erkennen (BDNF(+/-) unbehandelt =
1,2 ± 0,03 mV; BDNF(+/-) nach Läsion = 1,3 ± 0,1 mV; p = 0,49). Ebenso war
kein Unterschied zu den Feldpotentialen läsionierter Wt Mäusen (Wt nach
Läsion 1,4 ± 0,1 mV; BDNF(+/-) nach Läsion = 1,3 ± 0,1 mV; p = 0,57) zu
sehen. Auch die Form und Latenz der Feldpotentiale war derjenigen gleich, die
zuvor bei unbehandelten BDNF (+/-) und Wt sowie läsionierten Wt Tieren
gesehen worden waren (Latenz BDNF(+/-) unbehandelt = 4,28 ± 0,61 ms,
BDNF(+/-) nach Läsion = 4,23 ± 0,57; p = 0,54) Für keine Stimulationsdauer
konnte ein signifikanter Unterschied in der Höhe der Amplitude festgestellt
werden (p > 0,05). Somit gab es auch für läsionierte BDNF (+/-) Tiere keine
feststellbare Änderung der basalen synaptischen Übertragung.
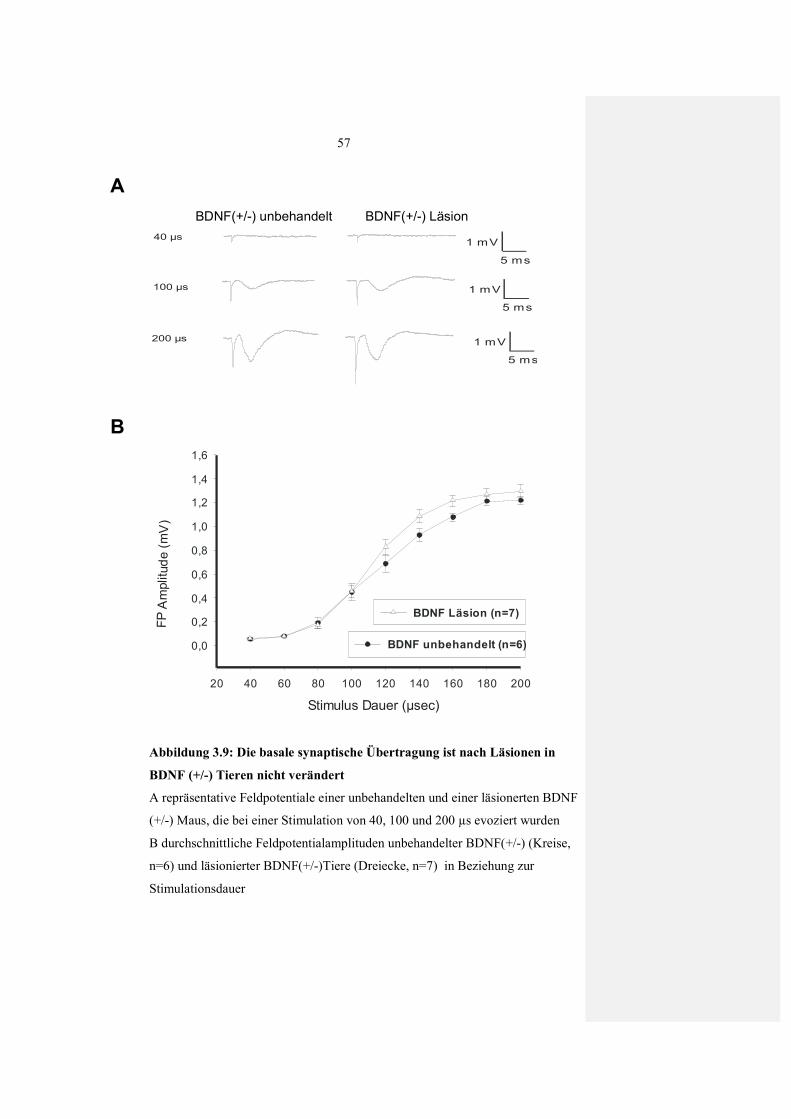
57
A
BDNF(+/-) unbehandelt BDNF(+/-) Läsion
40 µs
100 µs
200 µs 1 mV
5 ms
1 mV
5 ms
1 mV
5 ms
B
FP
Am
plit u
de (
mV
)
Stimulus Dauer (µsec)
20 40 60 80 100 120 140 160 180 200
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
BDNF unbehandelt (n=6)
BDNF Läsion (n=7)
Abbildung 3.9: Die basale synaptische Übertragung ist nach Läsionen in
BDNF (+/-) Tieren nicht verändert
A repräsentative Feldpotentiale einer unbehandelten und einer läsionerten BDNF
(+/-) Maus, die bei einer Stimulation von 40, 100 und 200 µs evoziert wurden
B durchschnittliche Feldpotentialamplituden unbehandelter BDNF(+/-) (Kreise,
n=6) und läsionierter BDNF(+/-)Tiere (Dreiecke, n=7) in Beziehung zur
Stimulationsdauer

58
3.2.3.2 Wiederherstellung von LTP in BDNF(+/-) Mäusen durch Läsionen
Im nächsten Schritt sollte nun der Einfluss von Läsionen auf BDNF(+/-) Mäuse
untersucht werden. Dazu wurde die LTP in BDNF(+/-) Tieren nach Läsion mit
der LTP in unbehandelten BDNF(+/-) Tieren verglichen (Abb. 3.10). In
Hirnschnitten von läsionierten BDNF (+/-) Tieren erzeugten die Reize in einem
Abstand von 1,0 – 2,0 mm vom Rand der Läsion vor der TBS Feldpotentiale,
deren Amplitude eine durchschnittliche Höhe von 0,74 ± 0,03 mV hatten. Somit
unterschieden sie sich nicht signifikant (p > 0,05) von den Feldpotentialen, die
in den anderen Versuchsgruppen gemessen worden waren. Auch die Form der
Feldpotentiale war ebenfalls die schon zuvor gesehene. Ebenso konnte kein
Unterschied in der Latenz zwischen dem Stimulusartefakt und der maximal
negativen Feldpotentialamplitude nach Läsionen in BDNF(+/-) Tieren
ausgemacht werden (BDNF(+/-) unbehandelt = 4,28 ± 0,42 ms, BDNF(+/-) nach
Läsion = 4,32 ± 0,40; p = 0,56). Nach der TBS erfolgte bei läsionierten
BDNF(+/-) Tieren keine anfängliche Depression der Amplitude, die die LTP in
unbehandelten und läsionierten Wt Mäusen sowie unbehandelten BDNF(+/-)
Mäusen auszeichnete. Das erste Feldpotential nach TBS hatte eine
durchschnittliche Höhe von 0,7 ± 0,07 mV und unterschied sich somit nicht
signifikant vom Level der Baseline. Innerhalb von 5 Minuten erreichten die
Amplituden der Feldpotentiale eine Höhe von 0,96 ± 0,03 mV, was 128 ± 3,4 %
der Baseline entsprach. Dieses Level blieb die gesamten 60 Minuten nach TBS
konstant. Im Gegensatz zu Hirnschnitten unbehandelter BDNF (+/-) Tiere
konnte eine LTP in Hirnschnitten läsionierter BDNF (+/-) Mäuse nun wieder
ausgelöst werden. Die relativen Amplituden der Feldpotentiale erreichten dabei
in den letzten 10 Minuten 0,95 ± 0,05 mV (126 ± 6,4 % der Baseline) und waren
so signifikant von den nichtbehandelten BDNF(+/-) Tieren zu unterscheiden (p =
0,019). Auf die Form und die Latenz zwischen dem Stimulusartefakt und der
maximal negativen Feldpotentialamplitude der einzelnen Feldpotentiale hatte
die TBS auch in Hirnschnitten von läsionierten BDNF (+/-) Mäusen keinen
Einfluss (BDNF(+/-) unbehandelt vor TBS= 4,27 ± 0,50 ms, nach TBS= 4,31 ±
0,51; p = 0,56).

59
A
BDNF(+/-) unbehandelt BDNF(+/-) Läsion
vor TBS
nach TBS
10 ms
1 mV
10 ms
1 mV
B
-10 0 10 20 30 40 50 60
80
100
120
140
160
BDNF (+ /-) unbehandelt (n = 7)
BDNF (+ /-) mit Läsion (n = 9)
Zeit (min)
rela
tive
FP
Am
plit
ud
e (%
)
Abbildung 3.10: Ausgleich des Defizits in der Expression von LTP in BDNF
(+/-) Mäusen nach Läsionen
A: Repräsentative Feldpotentiale einer unbehandelten BDNF (+/-) Maus (wie in
Abb. 3.8 bereits gezeigt) und einer läsionierten BDNF (+/-) Maus vor (1) und
60 Minuten nach (2) TBS.
B: Zeitverlauf der mittleren Feldpotentialamplituden von unbehandelten BDNF
(+/-) (gefüllte Kreise, n=7) und BDNF (+/-) Tieren zwei bis sechs Tage nach
Läsion (ungefüllte Vierecke, n=9)

60
3.2.3.3 Keine Änderung der LTP nach Sham-Operationen bei BDNF (+/-) Mäusen
Auch bei BDNF(+/-) Tieren sollte ein möglicher Effekt der Operationsmethode
auf die Plastizität ausgeschlossen werden. Der Vergleich zwischen der LTP in
BDNF(+/-) Tieren nach Sham-Operationen und in unbehandelten BDNF(+/-)
Mäusen ist in Abb 3.1. gezeigt. In Hirnschnitten sham-operierter BDNF(+/-)
Mäuse (n= 6) hatten die Feldpotentiale vor TBS eine durchschnittliche Höhe
von 0,74 ± 0,02 mV. Die Form der Feldpotentiale blieb auch nach Sham-
Operationen unverändert. Auch auf die Latenz zwischen dem Stimulusartefakt
und der maximal negativen Feldpotentialamplitude hatten die Sham-
Operationen keinen Einfluss (BDNF(+/-) unbehandelt = 4,28 ± 0,42 ms;
BDNF(+/-) sham-operiert = 4,23 ± 0,59 ms; p = 0,68). In der Höhe der relativen
Amplituden 51 – 60 min nach TBS in sham-operierten Tieren ( 0,74 ± 0,04 mV,
entspricht 100 ± 2,6 % der Baseline) war kein signifikanter Unterschied zu
nichtbehandelten Tieren feststellbar (p > 0.21). Auch bei läsionierten BDNF(+/-)
Tieren war durch die TBS keine Änderung der Form der Feldpotentiale oder der
Latenz zwischen dem Stimulusartefakt und der maximal negativen
Feldpotentialamplitude ausgelöst worden (BDNF(+/-) sham-operiert vor TBS =
4,28 ± 0,51 ms; nach TBS = 4,19 ± 0,37 ms; p = 0,22). Die Ergebnisse sham-
operierter Tiere unterschieden sich also in keinem Aspekt von denen
nichtbehandelter BDNF(+/-) Mäuse.

61
A
BDNF (+/-) unbehandelt BDNF(+/-) sham-operiert
vor TBS
nach TBS
10 ms
1 mV
10 ms
1 mV
B
160
-10 0 10 20 30 40 50 60
80
100
120
140
BDNF (+ /-) unbehandelt (n= 7)
BDNF (+ /-) shamoperiert (n= 6)
Zeit (min)
rela
tive
FP
Am
plit
ud
e (%
)
Abbildung 3.11: Sham-Operationen haben keinen Einfluss auf das Level
von LTP in BDNF(+/-) Mäusen
A Repräsentative Feldpotentiale einer unbehandelten BDNF(+/-) und einer
sham- operierten BDNF(-/-) Maus drei Tage nach Sham-Operation
B kein signifikanter Unterschied in der Expression von LTP zwischen
unbehandelten BDNF(+/-) (gefüllte Kreise, n=7) und sham-operierten BDNF(+/-
) Mäusen (ungefüllte Vierecke, n=6)

62
3.2.3.4 Vergleich zwischen Ausprägung der LTP in unbehandelten Wt-Mäusen und läsionierten BDNF (+/-) Mäusen
Da die Ergebnisse der elektrophysiologischen Messungen gezeigt haben, dass
das Level von LTP in unbehandelten und läsionierten BDNF(+/-) Tieren eine
ähnlliche Höhe erreicht, sollen im Folgenden die Ergebnisse unbehandelter Wt
und läsionierter BDNF(+/-) Mäuse im Vergleich dargestellt werden. Auf den
ersten Blick hat die LTP läsionierter BDNF(+/-) Mäuse und die LTP
unbehandelter Wt-Mäuse eine sehr ähnliche Ausprägung. Vergleicht man die
LTP, die in Hirnschnitten unbehandelter Wt-Mäusen gemessen worden ist, mit
der, die in läsionierten BDNF(+/-) Mäusen evoziert worden ist, besteht kein
signifikanter Unterschied zwischen der relativen Amplitude 51-60 min nach TBS
in Hirnschnitten unbehandelter Wt-Mäuse und Hirnschnitten läsionierter BDNF
(+/-) Mäuse (p = 0,63).
Der Gesamtverlauf der LTP ist jedoch unterschiedlich. Während die
Amplitude der unbehandelten Wt Tiere nach TBS nach einem ersten raschen
Anstieg weiter kontinuierlich ansteigt, erreicht die LTP läsionierter BDNF(+/-)
Tiere bereits nach einigen Minuten das Level, das sie für die Länge des
gesamten Experimentes beibehält ohne weiter anzusteigen. Dieser Trend ist
jedoch nicht signifikant.
3.2.4 Vergleich der LTP zwischen verschiedenen Versuchsgruppen
Der abschließende Vergleich der LTP 51-60 min nach TBS in den
verschiedenen Versuchsgruppen wird in Abb. 3.12 dargestellt. In den
Kontrolltieren, den unbehandelten Wt Mäusen, wird durch TBS eine LTP
hervorgerufen, die durchschnittlich 121% der Baseline entspricht. Nach
Läsionen des visuellen Kortex ist die LTP dann auf durchschnittlich 141% der
Baseline erhöht. In BDNF(+/-) Tieren kann durch die TBS keine dauerhafte LTP
ausgelöst werden. Nach Läsionen ist jedoch bei diesen BDNF(+/-) Tieren eine
LTP von durchschnittlich 126% durch dieselbe Stimulation zu beobachten.

63
*re
lativ
eF
PA
mp
litud
e(%
)
0
20
40
60
80
100
120
140
160
Wt o
hne L
äsion
Wt m
it Läsio
n
BDNF ohne
Läsio
nBDNF m
it Lä
sion
*
Abbildung 3.12: Einfluss von Läsionen auf Wt und BDNF(+/-) Tiere
Vergleich der relativen FP Amplitude 51-60 min nach TBS.
Das Level der LTP war bei Wt ohne Läsion 121 ± 4%, bei läsionierten Wt
wurden 141 ± 6,4 % gemessen. Bei den BDNF Tieren war nach 51-60 min
keine LTP (104 ± 3,4 %) vorhanden, nach Läsionen in BDNF Tieren konnte
dann eine LTP von 128 ± 3,4 % wieder beobachtet werden.

64
3.3 Ergebnisse der RT-PCR
3.3.1 Expression von BDNF nach Läsionen In dieser Versuchsreihe sollte untersucht werden, in wie weit Läsionen einen
Einfluss auf die Expression verschiedener Substanzen, vor allem dem
Neurotrophin BDNF haben. Zu diesem Zweck wurde eine Kontrollregion
außerhalb des visuellen Kortex definiert, um die Änderung der Expression im
Läsionsbereich im visuellen Kortex mit diesem Areal vergleichen zu können. Als
Kontrollbereich wurde der Motorcortex der läsionierten Tiere gewählt. Die
Expression von Aktin diente hierbei als interne Kontrolle, da es unter den
vorherrschenden Versuchsbedingungen keinen Schwankungen unterliegt.
actin
bdnf
trkB
GAD 65
PARV
GFAP
GAD 67
WT TG
MC VC MC VC
actin
bdnf
trkB
GAD 65
PARV
GFAP
GAD 67
WT TG
MC VC MC VC
Abbildung 3.13: Darstellung representativer PCR-Banden im Motor- und
visuellen Kortex von Wt und Tg Mäusen
MC = Motorkortex, VC = visueller Kortex, TG = transgen (BDNF(+/-))
TrkB = Tyrosin Kinase Rezeptor B, GAD = Glutamatdecarboxylase,
PARV = Parvalbumin, GFAP = Glial fibrillary acid protein
Die Abbildung zeigt repräsentative PCR Banden für verschiedene Proteine. Aktin
diente als interne Kontrolle und hat im Kontrollbereich des Motorkortex und im
visuellen Kortex dieselbe Intensität.

65
In Abb. 3.13 ist eine gleichmäßige Expression von Aktin in Motorkortex und im
visuellen Kortex von Wt wie auch von BDNF(+/-) Mäusen zu sehen. Es lässt
sich eine unterschiedliche BDNF Expression zwischen Wt und BDNF (+/-)
Tieren erkennen. In den BDNF(+/-) Mäusen sind die Banden sowohl im
Motorkortex als auch im visuellen Kortex schwächer ausgebildet.
Abb 3.14 – 3.16 zeigen die mRNA Expression von BDNF anhand der
PCR-Banden in Abhängigkeit von der Entfernung zur Läsion zu verschiedenen
Überlebenszeitpunkten. Nach einer bestimmten Überlebenszeit (ein, zwei oder
fünf Tage) wurden akute Hirnschnitte hergestellt und verschiedene
Gewebeproben entnommen. Wie in den Methoden beschrieben, wurde das
Gewebestück A jeweils im Abstand von 0,2 bis 1,2 mm vom Rand der Läsion, B
in 1,5 bis 2,5 mm und C in 3,0 bis 4,0 mm Entfernung entnommen. Als
Kontrollregion diente wiederum ein Gewebestück des Motorcortex. Da kein
signifikanter Unterschied zwischen dem Abstand von 0,2 bis 1,2 mm und 1,5 bis
2,5 mm festgestellt wurde, ist diese Region von 0,2 bis 2,5 mm Entfernung als
„Läsion 0,2 – 2,5 mm“ in den Abbildungen 3.14 – 3.6 zusammengefasst. In Wt
Tieren ist eine signifikante Reduktion (p<0,05) in der BDNF Expression am
ersten Postläsionstag nahe der Läsion zu erkennen (Abb 3.14).
BD
NF
/ A
ctin
rela
tive E
i nh
eite
n
0,0
0,5
1,0
1,5
WT TG
*
**
MC
Läsion 0,2 -
2,5 mm
Läsion
3 - 4
mm MC
Läsion 0,2 -
2,5 mm
Läsion 3 -
4 mm
1.Tag post-Läsion
Abbildung 3.14: BDNF mRNA Expression in Abhängigkeit der
Läsionsentfernung einen Tag nach Läsion
BDNF mRNA Expression in Wt Mäusen (unausgefüllte Balken, n=12) ist einen
Tag nach Läsion nur am Rande der Läsion erniedrigt, während in BDNF Mäusen

66
(+/-) (ausgefüllte Balken, n=12) auch in weiterer Entfernung zur Läsion eine
signifikante Erniedrigung des mRNA Levels zu sehen ist. Als Kontrollareal
dient der Motorkortex (MC).
Am zweiten und fünften Tag nach Läsion ist dagegen kein signifikanter
Unterschied mehr nachweisbar (Abb 13.15, Abb 3.16). In 3,0 – 4,0 mm
Entfernung zur Läsion ist zu keinem Zeitpunkt ein signifikanter Unterschied zu
sehen. Die BDNF Expression in BDNF (+/-) Tieren ist nahe der Läsion sowohl
am ersten, als auch am zweiten Tag im Vergleich zum Motorkortex signifikant
(p<0,05) erniedrigt. Dieser signifikante Unterschied ist nicht auf die Region
nahe der Läsion beschränkt, sondern ist am ersten wie auch am zweiten
Postläsionstag ebenfalls noch in größerer Entfernung (3,0 – 4,0 mm) zur
Läsion festzustellen. Am fünften Tag nach Läsion ist auch bei den BDNF (+/-)
Mäusen kein signifikanter Unterschied in der BDNF Expression zu erkennen.
MC
Läsion 0,2 - 2
,5 mm
Läsion 3 -
4 mm MC
Läsion 0,2 - 2
,5 mm
Läsion 3 -
4 mm
0,0
0,5
1,0
1,5
**
BD
NF
/ A
ctin
rela
tive E
inhe
iten
WT TG
2. Tag post-Läsion
Abbildung 3.15: BDNF mRNA Expression in Abhängigkeit der
Läsionsentfernung zwei Tage nach Läsion
In Wt Mäusen (unausgefüllte Balken) ist kein signifikanter Unterschied in der
BDNF mRNA nach Läsionen im Vergleich zum Kontrollareal (Motorkortex =
MC) feststellbar. Bei BDNF(+/-) Tieren (ausgefüllte Balken) zeigt sich
weiterhin eine signifikante Reduktion der BDNF mRNA in der Nähe der Läsion
und in weiterer Entfernung.

67
0,0
0,5
1,0
1,5
BD
NF
/ A
ctin
re
lativ
e E
i nheite
n
WT TG
MC
Läsion 0,2 -
2,5 mm
Läsion 3 -
4 mm MC
Läsion 0,2 - 2
,5 mm
Läsion 3 -
4 mm
5. Tag post-Läsion
Abbildung 3.16: BDNF mRNA Expression in Abhängigkeit der
Läsionsentfernung fünf Tage nach Läsion
Fünf Tage nach Läsion ist kein Unterschied in der BDNF mRNA Expression
mehr festzustellen. Dies zeigt sich BDNF (+/-) Tieren (ausgefüllte Balken,
n=12) wie auch in Wt Tieren (unausgefüllte Balken, n=12).
Die veränderte BDNF Expression ist also nicht nur von der Entfernung zur
Läsion abhängig, sondern auch von der Überlebenszeit nach Läsion. Am ersten
Tag nach Läsion ist die Expression von BDNF in beiden Gruppen am stärksten
eingeschränkt. Fünf Tage nach Läsion kommt es wiederum bei Wt als auch bei
BDNF (+/-) Tieren zu einem Ausgleich der BDNF Expression auf ein
Kontrolllevel, was der BDNF-Expression vor Läsion entspricht.

68
4 Diskussion
Untersuchungen zur Plastizität des Gehirns sind ein bedeutender
Forschungsschwerpunkt der Neurowissenschaften. Die Entschlüsselung der
Mechanismen, die die Plastizität des Gehirns steigern können, wäre von großer
Bedeutung im alltäglichen Leben und hätten ebenso therapeutische
Konsequenzen für Patienten mit Läsionen des Gehirngewebes oder anderen
Veränderungen der neuronalen Aktivität.
Die Mechanismen, die durch eine Läsion ausgelöst werden, sind sehr
komplex und bisher wenig verstanden. Da bislang mehrheitlich davon
ausgegangen wurde, dass erhöhte Plastizität nach Läsion durch ein erhöhtes
Level von BDNF vermittelt wird, ergeben sich aus unseren Ergebnissen
weitreichende Konsequenzen für den therapeutischen Nutzen von BDNF. Es
wurde sogar gezeigt, dass Mäuse von einem reduzierten Level an BDNF nach
einem experimentellem Infarkt profitieren (Nygren et al., 2006). Auf Grund der
Diskrepanz der verschiedenen Studien (BDNF-Erhöhung versus BDNF-
Erniedrigung nach Läsionen) können die Ergebnisse der vorliegenden Arbeit
dazu beitragen die Rolle von BDNF für läsionsinduzierte Plastizität besser zu
verstehen.
Die Ergebnisse der vorliegenden Arbeit zeigen:
1. In Wt sowie in BDNF(+/-) Mäusen sind reproduzierbare Läsionen zu
finden, die durch eine Gliose in der Umgebung der Läsion
gekennzeichnet sind.
2. Nach Läsionen im visuellen Kortex von Wt Mäusen ist die LTP verstärkt
auslösbar, wobei die basale synaptische Übertragung nicht verändert
ist und auch die Operationsmethode keinen Einfluss auf die LTP hat.
3.A In unbehandelten BDNF(+/-) Tieren kann durch eine TBS keine
anhaltende LTP ausgelöst werden.
Formatiert

69
3.B Läsionen im visuellen Kortex von BDNF(+/-) Tieren führen dazu, dass
die bei unbehandelten Tieren beeinträchtigte LTP nun wieder
auslösbar ist und dem Level von unbehandelten Wt Mäusen
entspricht. Auch hier wird keine Änderung der basalen synaptischen
Übertragung gefunden und ebenso hat die Operation selbst keine
Änderung der LTP zur Folge.
4. Das Level der BDNF mRNA ist nach Läsionen reduziert.
4.1 Gliose in der Umgebung der Läsion
Ein Ausdruck der funktionalen Veränderung nach Läsionen ist die Gliose am
Rand einer Läsion. Eine Zunahme der Gliazellen weist auf einen erhöhten
Metabolismus der Zellen hin und ist von hoher Bedeutung für
Reorganisationsprozesse (Guthrie et al., 1997). Gliazellen vermitteln eine
metabolische Versorgung der Nervenzellen und können sogar
Signalübertragungen modulieren.
Wie bereits im Rattenkortex beschrieben, zeigten auch die untersuchten
Mäusegehirnschnitte eine Gliose in der Läsionsumgebung, die in Stärke und
Ausdehnung der Reaktion nach Läsionen im visuellen Kortex von Ratten ähnelt.
Auch die Ausdehnung der Läsion und die Morphologie des umgebenen
Gewebes im Mäusekortex ist mit der Histologie im Rattenkortex nach Läsionen
vergleichbar (Mittmann et al., 1994; Kálmán et al., 2000; Barmashenko et al.,
2001). Dies gibt einen ersten Hinweis darauf, dass die Laserläsion bei Mäusen
ähnliche Reaktionen hervorruft wie bei Ratten.
4.2 Typische Form der Feldpotentiale
Nach Reizung des visuellen Kortex der Maus wurden Feldpotentiale gemessen,
die denen ähneln, die schon zuvor zwischen Schicht IV und II/III gemessen
worden sind (Kirkwood & Bear, 1994). Form und Latenz waren mit diesen
Feldpotentialen vergleichbar, was dafür spricht, dass es sich auch bei den von
uns gemessenen Feldpotentialen um die Summe von EPSPs (excitatorisches
postsynaptisches Potential) handelt und somit durch Glutamat übermittelte
erregende synaptische Übertragungen gemessen wurden. Um zu beweisen,

70
dass die evozierten Feldpotentiale synaptischen Ursprungs sind, wurde der
spezifische AMPA-Rezeptor-Antagonist 6,7-Dinitroquinoxalin-2-3-dion (DNQX,
10µM) eingewaschen, nachdem ein stabiles Feldpotential evoziert worden war.
Da das Signal unter DNQX vollständig verschwindet und durch Tetradotoxin
(TTX) nicht weiter verändert wird, entsteht das Feldpotential vor Zugabe von
DNQX durch die Aktivierung exzitatorischer Neurotransmitter (Glutamat) und ist
damit synaptischen Ursprungs (EPSP).
Die elektrophysiologischen Signale, die in den läsionierten Tieren
gemessen wurden, zeigten die gleiche Form und Latenz wie in unbehandelte
Mäusen. Ebenso konnte kein Unterschied zwischen unbehandelten und sham-
operierten Mäusen festgestellt werden. Das lässt darauf schließen, dass auch
nach Gehirnläsionen die selben synaptischen Übertragungswege benutz
werden wie bei den Kontrolltieren.
4.3 Diskussion der Methode
Der Zeitpunkt, an dem die Läsionen gesetzt worden sind , 21 bis 22 Tage
postnatal, ist so gewählt, dass er in der kritischen Phase für Okularplastizität
liegt (Bartoletti et al., 2002; Hensch, 2005). Dies lässt zunächst nur theoretische
Rückschlüsse auf Läsionseffekte bei adulten Tieren oder gar beim Menschen
zu. Die Läsionsexperimente wurden dennoch in dieser Phase der Entwicklung
durchgeführt, da Vorarbeiten aus dem eigenen Labor existieren, die ebenfalls
zu diesem Entwicklungszeitpunkt angefertigt worden sind.
Möglicherweise ist durch die Tatsache, dass die Läsionsexperimente bei
Tieren durchgeführt wurden, die sich in der Kritischen Periode befanden, die
Diskrepanz zwischen den dargestellen erniedrigten BDNF Level und den in der
Literatur mehrheitlich gezeigten Erhöhung des BDNF Levels nach der Läsion zu
erklären. Ein weiterer Unterschied zu anderen Studien stellt die Methode der
Läsion dar.
Die Untersuchungen der vorliegenden Arbeit zeigen in Wt Mäusen einen
Tag nach Läsion ein signifikant erniedrigtes Level von BDNF-mRNA am Rande
der Läsion, während nach zwei und nach fünf Tagen kein signifikanter
Unterschied festzustellen ist. Dennoch konnte durch die vorliegende Arbeit eine

71
stärkere LTP nach Läsionen nachgewiesen werden, das somit nicht auf ein
erhöhtes Level von BDNF zurückzuführen ist.
Andere Arbeiten hingegen zeigen eine verstärkte Expression von BDNF
nach Läsionen oder einen positiven Effekt von erhöhtem BDNF-Level auf
Läsionen.
Bei den Arbeiten von Sulejczak et al. (2006) wurde beispielsweise nach
einem Hirninfarkt durch photothrombotische Läsion eine erhöhte Expression
von BDNF im Proteinlevel gezeigt. Hier ergibt sich ein Unterschied in der
Methode der Messung von BDNF zur vorliegenden Arbeit, da wir nicht das
Proteinlevel, sondern die BDNF-mRNA gemessen haben. Möglich wäre eine
Regulation des BDNF Levels auf einer anderen Ebene als der mRNA, doch die
entgegengesetzte Wirkung: mRNA-Erniedrigung und Proteinerhöhung, ist sehr
unwahrscheinlich. Da die Versuche von Sulejczak et al. (2006) bei adulten
Ratten durchgeführt wurden, befanden sich die Tiere zudem in einem anderen
Entwicklungszustand. Zusätzlich weicht die Läsionsmethode von der in der
vorliegenden Arbeit verwendeten ab, da bei Sulejczak et al. (2006) Hirninfarkte
durch photothrombotische Läsion ausgelöst wurden, während in unserer Studie
die Läsionen durch einen Laser verursacht wurden. Interessanterweise zeigten
die Ergebnisse von Sulejzcak et al. zwar eine Erhöhung des BDNF Levels, dies
ging jedoch nicht mit einer verbesserten Kompensation der durch die Läsion
entstandenen motorischen Defizite einher. Möglicherweise sind durch
verschiedene Läsionsmethoden und Analysen zu verschiedenen
Entwicklungszeitpunkte unterschiedliche Mechanismen in Gang gesetzt
worden, die bei den Studien von Sulejczak zu einer Erhöhung des BDNF Levels
ohne Erhöhung der Plastizität und in unseren Studien zu einer Reduktion des
mRNA Levels mit daraus resultierender erhöhter Plastizität führen.
Auch in einer Studie von Sizonenko et al. (2007) wird diskutiert, ob ein
erhöhtes Level von BDNF die Läsionsgröße oder die Reparaturmechanismen
nach Läsionen positiv beeinflussen kann. Hier wurde der Effekt von zusätzlich
intrazerebroventrikulär appliziertem BDNF auf Zellnekrosen nach
exzitotoxischen und hypoxischen Läsionen in Mäusen untersucht. Es zeigte
sich nur eine positive Wirkung von zusätzlich appliziertem BDNF auf die Läsion,
wenn die Applikation zusammen mit der Läsion am fünften postnatalen Tag
verabreicht wurde. Am Tag der Geburt zeigte sich sogar eine verstärkte

72
Ausdehnung der Apoptose bei DNF-Applikation, am zehnten Tag nach der
Geburt waren keine Effekte mehr feststellbar. Zudem werden in dieser Studie
excitotoxische Läsionen gesetzt, die möglicherweise andere Mechanismen in
Gang setzten als Laserläsionen. Auf Grund der unterschiedlichen Methode und
Entwicklungszeitpunkt der Tiere kann hier kein direkter Vergleich zur
vorliegenden Studie gezogen werden.
Durch Nygren et al., 2006 wird hingegen ein reduziertes Level von BDNF
nach Läsionen mit verbesserten Motorfuntionen in Zusammenhang gebracht.
Hier wurden reduzierte Level von BDNF Protein und mRNA nach
experimentellem Hirninsult in BDNF(+/-) Mäusen im Vergleich zu Wt Tieren
gemessen. Dieses reduzierte Level von BDNF ging in der Studie mit
verbesserten motorischen Funktionen einher.
4.4 Verstärkte LTP nach Läsion im visuellen Kortex der Wt
Maus
Auch die elektrophysiologischen Messungen nach Läsionen im Mäusekortex
stimmten mit Ergebnissen, die in Ratten gesehen worden waren, überein. Im
visuellen Kortex der Ratte wurde bereits eine erhöhte LTP nach Läsionen
gefunden (Huemmeke et al., 2004). Die Erhöhung der Feldpotentiale nach TBS
im visuellen Kortex der Ratte zeigte ein vergleichbares Level wie das in Wt
Mäusen gemessene. Dieser gleichsinnige Läsionseffekt lässt darauf schließen,
dass auch die molekularen Mechanismen nach Läsionen im Kortex der Ratte
und der Maus ähnlich sind. Wie auch bei den Ratten beobachtet ist die basale
synaptische Übertragung nach Läsionen nicht verändert, so dass die erhöhten
Feldpotentiale nach TBS nicht auf Veränderungen in der normalen
Reizweiterleitung zurückzuführen sind. Da das Projekt aufbauend auf einem
intern etablierten Laborstandard und Vorarbeiten geplant wurde, mussten auch
möglichst gleiche Versuchsbedingungen vorherrschen. Aus diesem Grund
wurden die Läsionsexperimente an 21 bis 28 Tage alten Tieren durchgeführt,
da ebenfalls an den zuvor untersuchten Mäusen in dieser Zeitspanne
Experimente durchgeführt worden waren (Abidin et al., 2006). Natürlich
bestehen im jungen Gehirn andere Vorraussetzung für Plastizität als im adulten
Gewebe, da sich die untersuchten Wt Mäuse genau in der kritischen Periode für

73
die monokulare Deprivation befinden (Bartoletti et al., 2005) und in dieser
Entwicklungsphase per se eine erhöhte Plastizität vorzufinden ist. Verschiedene
Arbeiten haben gezeigt, dass läsionsinduzierte Plastizität und Plastizität des
juvenilen Gehirns ähnliche Mechanismen zu Grunde liegen. Ein Beispiel ist das
veränderte Gleichgewicht zwischen Erregung und Hemmung, das nach
Läsionen zu mehr Erregung hin modifiziert ist (Mittmann et al., 1994). Auch
während frühen Entwicklungsphasen ist das Gleichgewicht zwischen Erregung
und Hemmung zu Gunsten der Erregung verändert, da sich hemmende
Synapsen erst später ausbilden als erregende (Long et al., 2005). Ein anderes
Resultat ist die veränderte Kalziumkonzentration auf Grund verschiedener
Rezeptormodulationen (Barmashenko et al., 2001, Rumpel et al., 2000;
Huemmeke et al., 2004), die ebenfalls sowohl in der kritischen Phase wie auch
nach Läsionen beobachtet werden konnte. Nun könnte argumentiert werden,
dass bei Läsionsexperimenten bei Tieren, die bereits erhöhte Plastizität zeigen
eine Trennung zwischen läsionsinduzierter Plastizität und schon vorhandener
erhöhter Plastizität nicht möglich ist. Die durchgeführten Experimente zeigen
jedoch vielmehr, dass trotz schon existierender erhöhter Plastizität eine Läsion
Mechanismen auslöst, die beispielsweise eine Steigerung der LTP zur Folge
haben. Die gefundenen Unterschiede zwischen unbehandelten Wt und
läsionierten Wt Mäusen sind also sehr wohl auf eine läsionsinduzierte Erhöhung
der Plastizität zurückzuführen, die zwar ähnliche Mechanismen wie im jungen
Gewebe auslöst, aber dennoch in einigen Aspekten von erhöhter Plastizität
während der kritischen Periode abzugrenzen ist.
Auffällig ist, dass sowohl bei der erhöhten Plastizität während der
kritischen Periode (Sale et al., 2010) als auch bei läsionsinduzierter Plastizität
die Balance zwischen Erregung und Hemmung eine Rolle spielt. Die Plastizität
der kritischen Periode kommt erst durch das Ausreifen der Inhibition zu Stande
(Huang et al., 1999; Hanover et al., 1999; Hensch, 2000), während
läsionsinduzierte Plastizität jedoch unter anderem durch eine Reduktion der
Hemmung ausgelöst zu werden scheint. Eine mögliche Erklärung hierfür wäre,
dass ein bestimmtes Maß an Hemmung benötigt wird, um die erhöhte Plastizität
in der kritischen Periode vermitteln zu können, da ohne Hemmung eine
Modulation und Selektion der Signalübertragung nicht möglich ist. Ist dann
jedoch eine bestimmte Mindeststärke an Hemmung erreicht, ist nicht eine noch

74
weitere Steigerung der Hemmung, sondern eine verstärkte Erregung für
Plastizität förderlich (Artola & Singer, 1987, Bear et al, 1992, Sale et al., 2010).
4.5 Erneute Auslösbarkeit von LTP nach Läsion im visuellen
Kortex der BDNF(+/-) Maus
Durch die vorliegende Arbeit wurde gezeigt, dass in nicht läsionierten BDNF(+/-
) Mäusen keine LTP ausgelöst werden kann. Dies wurde bereits durch andere
Arbeiten nachgewiesen (Korte et al., 1995; Patterson et al., 1996; Pozzo-Miller
et al., 1999, Bartoletti et al., 2002; Abidin et al., 2006). Das zeigt, dass das
Maus-Modell, welches bereits 1995 etabliert wurde, grundsätzlich funktional ist.
In der vorliegenden Arbeit sollte nun weiter untersucht werden, wie das Gewebe
von BNDF(+/-) Mäusen auf eine Läsion im Kortex reagiert. Als Gründe für die
reduzierte Plastizität in BDNF(+/-) Mäusen werden verschiedene Mechanismen
diskutiert. Da bekannt ist, dass BDNF dendritisches Wachstum und
Verzweigung in Kulturen des visuellen Kortex erhöhen kann (McAllister et al.,
1995) und Behandlung mit BDNF in hippocampalen Kulturen eine Erhöhung
des Dendritenwachstums und der Verzweigung auslösen kann (Tyler & Pozzo-
Miller, 2001), ist es auf der anderen Seite möglich, dass ein chronisch
reduziertes Level von BDNF in BDNF(+/-) Tieren eine verminderte synaptische
Verzweigung im visuellen Kortex zur Folge hat. In BDNF knock out Mäusen
wurde eine reduzierte Anzahl der sensorischen Neurone gesehen (Ernfors et
al., 1994). Diese Ergebnisse konnten jedoch Im Hippocampus der BDNF (+/-)
Tiere (Korte et al., 1995) und im Barrel Kortex nicht reproduziert werden
(Genoud et al., 2004). Da auch in der vorliegenden Studie kein Unterschied in
der basalen synaptischen Übertragung, gemessen an der input-output Kurve,
festgestellt werden konnte, ist es sehr unwahrscheinlich, dass die beobachtete
reduzierte Plastizität durch eine geringere Anzahl synaptischer Verbindungen
zu Stande kommt. Ein weiterer möglicher Auslöser für eingeschränkte
Plastizität der BDNF(+/-) Mäuse könnte eine reduzierte erregende synaptische
Übertragung sein. In verschiedenen Studien konnte gezeigt werden, dass die
erregende Übertragung in unterschiedlichen Gehirnarealen der BDNF(+/-)
Maus reduziert ist und beispielsweise nicht genügend Transmitter an der
Präsynapse freigesetzt werden können (Pozzo-Miller et al., 1999; Abidin et al.,

75
2006). Unter diesen Umständen ist auch mit dem in der aktuellen Studie
benutzten Stimulationsprotokoll eine beständige LTP nicht auszulösen. Der
Grund für das Defizit in der LTP in BDNF(+/-) Mäusen ist noch nicht endgültig
geklärt. In einigen Studien wurde das Fehlen einer längerdauernder LTP auf
insuffiziente postsynaptische Depolarisation während der LTP Induktion
zurückgeführt (Figurov et al., 1996; Kang et al., 1997; Zakharenko et al., 2003).
Neben den Veränderungen im excitatorischen System wurden bei den
BDNF(+/-) Mäusen noch stärkere Defizite im inhibitorischen System der
BDNF(+/-) Mäuse beschrieben (Abidin et al., 2008). Zwar wurde gezeigt, dass
unter den Vorraussetzungen einer relativ überwiegenden Erregung LTP besser
ausgelöst werden kann (Artola & Singer, 1987, Bear et al, 1992), doch unter der
Annahme, dass der nötige Input sowie Transmitter etc nicht in genügendem
Masse vorhanden sind, werden diese Voraussetzungen zum limitierenden
Faktor und eine LTP ist dennoch nicht auszulösen.
Studien haben gezeigt, dass durch Veränderung in der GABAergen
Funktion, die BDNF vermittelt sein können, der Zeitpunkt der kritischen Periode
für monokulare Deprivation verändert werden kann. Eine Überexpression von
BDNF soll dabei die Reifung inhibitorischer Neurone fördern (Hanover et al.,
1999; Huang et al., 1999) und so die kritische Periode zu einem früheren
Zeitpunkt auftreten lassen (Fagiolini & Hensch, 2000; Iwai et al., 2003).
Andererseits kommt es bei Dunkelaufzuchten zu einem reduzierten Level an
BDNF (Castren et al., 1992) und damit auch zu reduzierter Hemmung (Morales
et al., 2002, Chen et al., 2001), was die kritische Periode für visuelle Plastizität
hier bis zum Erwachsenenalter verschieben kann (Iwai et al., 2001; Mower,
1991; Fagiolini et al., 2003). In dem hier benutzten Tiermodell könnten das
chronisch reduzierte Level von BDNF also auch zu einer Verschiebung der
kritischen Periode geführt haben. Durch Untersuchungen von Bartoletti et al.
(2002) konnte jedoch keine Veränderung in der Plastizität für monokulare
Deprivation im selben Tiermodell gefunden werden, was dafür spricht, dass sich
die hier benutzen BDNF(+/-) Tiere genau wie die Wt Tiere in der kritischen
Periode befanden.
Nachdem auch durch unsere Studie gezeigt werden konnte, dass LTP in
BDNF(+/-) Mäusen nicht zu evozieren war, ist es erstaunlich, dass in diesen
Mäusen nach einer Läsion eine konstante LTP ausgelöst werden konnte. Die

76
Tatsache, dass nach Läsionen bei BDNF(+/-) Tieren die basale synaptische
Übertragung nicht verändert ist, spricht dafür, dass primär nicht morphologische
Veränderungen für die erhöhte Feldpotentialamplitude nach TBS verantwortlich
sind, sondern dass erst durch den TBS Mechanismen ausgelöst werden, die
erhöhte Plastizität zur Folge haben. Neueste Arbeiten aus dem eigenen Labor
geben Hinweise darauf, dass möglicherweise die Balance zwischen Hemmung
und Erregung eine große Rolle in der Modifikation von Plastizität spielt (Abidin
et al., 2006; Abidin et al., 2008). Es ist bekannt, dass auch nach Läsionen die
Excitation und Inhibition dahingehend verändert ist, dass die Excitation
überwiegt. Diese Tatsache könnte für läsionsinduzierte Plastizität
mitverantwortlich sein, sodass eine modifizierte Balance zwischen Erregung
und Hemmung möglicherweise auch für die erneute Auslösung einer
längerdauernden LTP in BDNF(+/-) Tieren verantwortlich ist.
Schließlich ist denkbar, dass alle Mechanismen, die zu läsionsinduzierter
Plastizität führen, BDNF-unabhängig sind und so auch ohne BDNF als direkter
Mediator läsionsinduzierter Plastizität nach Läsion ein bestimmtes Level von
LTP auslösen können. Hinweise für diese Hypothese gibt die Höhe der LTP
nach Läsionen im Kortex von BDNF(+/-) Mäusen, da diese nicht der LTP nach
Läsionen in Wt Tieren beobachteten Höhe von ca. 140% entspricht. Vielmehr
zeigt sich bei läsionierten BDNF(+/-) Mäusen ungefähr das Level der LTP, das
in unbehandelten Wt Tieren evoziert werden kann. Es wird also nach Läsionen
sowohl in Wt wie auch in BDNF(+/-) Tieren eine um ca. 20% größere relative
Amplitude nach TBS gemessen, als in unbehandelten Tieren des selben
Genotypes. Dies könnte ein Hinweis darauf sein, dass durch die Läsion
Mechanismen ausgelöst werden, die unabhängig vom BDNF Level im visuellen
Kortex jeweils eine Erhöhung der LTP um ca. 20% hervorrufen.
4.6 Quantifizierung der BDNF mRNA
Um Hinweise auf eine läsionsinduzierte Änderung der BDNF Expression zu
bekommen, wurde die Konzentration der BDNF mRNA nach Läsionen im
Kortex gemessen. Die hierzu benutzten Primer wurden in der Abteilung für
Entwicklungsneurobiologie der Ruhr-Universität Bochum synthetisiert. Um
sicher zu gehen, dass unter den eingestellten PCR-Bedingungen die zu

77
untersuchende DNA amplifiziert wurde und keine unspezifischen
Reaktionsprodukte vorliegen, wurden die Amplifikate mittels Restriktionsanalyse
überprüft.
Für die Messung der BDNF mRNA wurde der Motorkortex der
läsionierten Tiere als Kontrollareal benutzt. Zuvor wurde jedoch sichergestellt,
dass im Motorkortex läsionierter Tiere keine läsionsbedingten Änderungen der
BDNF Expression stattfanden. Hierfür wurden Schnitte des Motorkortex von
läsionierten Tieren mit dem Motorkortex unbehandelter Tiere verglichen. Die
durchgeführte PCR zeigte keinen Unterschied zwischen dem Level von BDNF
mRNA im Kortex läsionierter und unbehandelter Mäuse, so dass wir dieses
Gehirnareal als interne Kontrolle verwenden konnten.
Durch Vorarbeiten war bekannt, dass in BDNF(+/-) Mäusen das Level
von BDNF in allen neocorticalen Hirnarealen reduziert war (Abidin et al., 2006).
Dies wird auch durch die PCR Experimente widergespiegelt, in denen BDNF(+/-
) Tiere in der Kontrollregion immer eine reduzierte Expression von BDNF im
Vergleich mit den Wt Mäusen zeigen. Dennoch gibt es noch die Möglichkeit,
dass das Proteinlevel von BDNF auf einer anderen Ebene als mit Hilfe der
mRNA reguliert wird, so dass das Protein nicht proportional zur BDNF mRNA
vorliegt. Auf diese Weise wäre es möglich, dass trotz reduzierter BDNF mRNA
ein erhöhtes Level an Protein gemessen werden kann. In verschiedenen
Arealen des zentralen Nervensystems wie zum Beispiel in Hippocampus,
frontalem Kortex, Cerebellum, Rückenmark und Barrel Kortex wurde bereits ein
um etwa die Hälfte reduziertes Proteinlevel von BDNF nachgewiesen (Kohlbeck
et al., 1999; Chourbaji et al. 2004; Genoud et al., 2004). Durch Vorarbeiten aus
dem eigenen Labor wurde ebenso gezeigt, dass das Proteinlevel in
unbehandelten BDNF(+/-) Mäusen genau wie die mRNA um ungefähr 50% im
visuellen Kortex reduziert ist (Abidin et al., 2006). Dies gibt einen Anhalt dafür,
dass vom Level der mRNA auf das Proteinlevel geschlossen werden kann.
Diese Versuche haben allerdings auch gezeigt, dass das Proteinlevel von
BDNF mittels ELISA nur mit großen Schwierigkeiten und hohem zeitlichen
Aufwand zu bestimmen war, da nicht genügend Gewebe aus dem visuellen
Kortex der Maus zu gewinnen war. Um genauere Aussagen über das Ausmaß
und die räumliche Ausdehnung der Proteinveränderung am Läsionsrand
machen zu können wäre das zu untersuchende Gewebe von noch geringerer

78
Masse gewesen. Aus diesem Grund erfolgte bisher noch keine direkte
Proteinbestimmung von BDNF nach Läsionen.
Aus der Bestimmung der mRNA können jedoch Rückschlüsse auf die
Modulation von BDNF gezogen werden. Die Ergebnisse der Studien von
Comelli et al. (1992) und De March et al. (2008), die eine Erhöhung der BDNF
mRNA gezeigt haben, konnten in der vorliegenden Arbeit nicht bestätigt
werden. Vielmehr wurde eine erniedrigte Expression von BDNF mRNA in der
Umgebung der Läsion gefunden. Unsere Ergebnisse stimmen aber mit einer
Studie von Nygren et al. (2006) überein, die ebenfalls ein erniedrigtes Level von
BDNF mRNA nach Läsionen zeigten. Die Hypothese, dass läsionsinduzierte
Plastizität durch eine Erhöhung von BDNF vermittelt wird, kann somit nicht
bestätigt werden.
4.7 BDNF und Hemmung synaptischer Übertragung
Die Mechanismen, die durch BDNF vermittelt werden, sind vielfältig. Da
gezeigt werden konnte, dass reduzierte Level von BDNF in BDNF(+/-) Tieren zu
einer Erniedrigung der Erregung und noch stärkerer Reduktion der Hemmung
führt (Abidin et al., 2008), wird möglicherweise in Wt Mäusen unter anderem
durch die Reduktion von BDNF am Rande der Läsion die Inhibition unterdrückt,
so dass eine LTP besser auszulösen ist. Dabei reicht eine Reduktion von BDNF
jedoch nicht aus, um erhöhte Plastizität zu vermitteln, denn in BDNF(+/-)
Mäusen, bei denen eine deutliche Reduktion des BDNF-Levels besteht, konnte
keine LTP ausgelöst werden (Abidin et al., 2006). Auch in Wildtyp-Tieren, bei
denen durch TrkB-IgG- oder BDNFAntikörper-Applikation das funktionell
verfügbare BDNF verringert wird, ist Plastizität wie LTP nicht erhöht, sondern
gar nicht mehr auslösbar (Figurov et al., 1996; Kang et al., 1997). Es müssen
durch die Läsion also noch weitere Faktoren für die erhöhte Plastizität nach
Läsionen mit verantwortlich sein und zusammenspielen, wie zum Beispiel
erhöhte Erregung (Arckens et al., 2000), erhöhte Kalziumkonzentration
(Barmashenko et al. 2001) auf Grund Veränderung in NMDA- (Rumpel et al.
2000; Hümmeke et al. 2004) und AMPA-Rezeptorzusammensetzung
(Barmashenko et al. 2003) o.ä.. In BDNF(+/-) Mäusen, bei denen bereits durch
das reduzierte Level von BDNF eine erniedrigte Erregung und Hemmung
besteht, könnte eine weitere Reduktion von BDNF und dadurch vermittelte

79
weitere Reduktion der GABAergen Hemmung in Zusammenhang mit anderen
durch die Läsion ausgelösten Faktoren eine Auslösung von LTP erneut
ermöglichen.
4.8 Räumliche und zeitliche Veränderung der BDNF Expression
nach Läsion
Auffällig ist der zeitliche Verlauf der veränderten BDNF Expression nach
Läsion, der nur in den ersten Tagen nach Läsion zu messen ist. Die stärkste
Senkung ist dabei am ersten Postläsionstag zu finden, am zweiten nähert sich
das Level von BDNF mRNA dem Kontrollzustand an, bis am fünften Tag nach
Läsion kein signifikanter Unterschied mehr festzustellen ist. Die intrazelluläre
Kalziumkonzentration nach Läsion im Rattenkortex zeigte einen ähnlichen
zeitlichen Verlauf (Barmashenko et al., 2001), was auf eine Verbindung
zwischen Kaliziumkonzentration und BDNF Expression hinweisen könnte.
Veränderungen in der intrazellulären Kalziumkonzentration sind ebenfalls nur
während der ersten Tage nach Läsion zu messen, acht Tage nach Läsion ist
kein signifikanter Unterschied mehr festzustellen. Das Zeitfenster für
Änderungen in der BDNF mRNA ist also noch enger. Ein weiterer Unterschied
ist die initiale Depression der Kalziumkonzentration in unmittelbarer Nähe zur
Läsion. Eine kontroverse Änderung des Levels der BDNF mRNA wurde nicht
festgestellt. Alles in Allem ist also eine direkte Wirkung von BDNF auf die
Kalziumkonzentration eher unwahrscheinlich, eine indirekte Beeinflussung ist
jedoch sehr wohl möglich. In diesem Zusammenhang sind veränderte
Aktivitätslevel nach Läsionen eine mögliche Erklärung. Durch eine Studie wurde
gezeigt, dass in der Umgebung einer Läsion eine neuronale Hyperaktivität
auftritt (Eysel & Schmidt-Kastner, 1991). Durch eine BDNF-Reduktion könnte
die GABAerge Hemmung so unterdrückt werden, dass im neuronalen System
die Erregung überwiegt. Dadurch könnte eine erhöhte Aktivität und somit
erhöhte Kalziumkonzentrationen nach Läsionen erklärt werden.
Erhöhte LTP ist ebenso auf ein bestimmtes Zeitfenster nach Läsion
begrenzt. So wurde gezeigt, dass sieben Tage nach Läsion des visuellen
Kortex der Ratte keine signifikante Erhöhung der LTP im Vergleich zu
Kontrolltieren evoziert werden konnte (Barmashenko et al., 2003). Wenn man

80
nun davon ausgeht, dass BDNF mitverantwortlich für die erhöhte Plastizität ist,
würden durch die Veränderungen im BDNF Level Mechanismen in Gang
gesetzt, die auch nach Normalisierung des BDNF Levels noch für einige Tage
eine erhöhte Plastizität vermitteln könnten.
Ungewöhnlich ist das Ausmaß der Veränderung der BDNF mRNA. In
weiterer Entfernung zur Läsion, was ca. drei bis vier mm entspricht, ist am
ersten und auch am zweiten Postläsionstag eine ähnliche Erniedrigung des
mRNA Levels gemessen worden wie nahe der Läsion, obwohl dieses Areal
formal nicht mehr dem visuellen Kortex zuzuordnen ist. Da die verschiedenen
Areale nicht nach anatomischen Aspekten sondern rein anhand der
Millimeterangabe entnommen wurden, ist es möglich, dass bei einigen Tieren
noch ein Teil des visuellen Kortex enthalten war. Gegen diese Theorie spricht
jedoch, dass das Level der mRNA in weiterer Entfernung dem Level in der
Nähe zu beiden Zeitpunkten nicht signifikant zu unterscheiden sind. Wäre bei
einem Tier ein Stück des visuellen Kortex mit entnommen worden, müsste das
Level der mRNA gemittelt sehr viel höher ausfallen und eher dem Kontrolllevel
entsprechen. Auffällig ist auch, dass beim Wt Tier eine solche Veränderung in
weiterer Entfernung der Läsion nicht beobachtet werden kann. Auch dies
spricht dafür, dass die Veränderung des BDNF mRNA Levels auch über die
Grenzen des visuellen Kortex´ hinaus wahrscheinlich nicht durch
Vermischungen mit visuellem Kortexmaterial zu Stande gekommen sind. Eine
mögliche Erklärung für diese räumliche Verteilung der BDNF Veränderung
wären Reorganisationsprozesse, die nach der Läsion in Gang gesetzt worden
sind und so zu einer Ausdehnung des visuellen Kortex geführt haben, so dass
sich die Grenzen des visuellen Kortex verschoben haben. Damit könnte sich
das entnommene Gewebestück trotz weiter Entfernung funktionell im visuellen
Kortex befunden haben. Dies würde nun auch bedeuten, dass die
Reorganisationsprozesse im visuellen Kortex nach Läsion in BDNF(+/-) Mäusen
größere Ausmaße annehmen als in Wt Mäusen. Zu dieser These passt die
Entdeckung, dass BDNF(+/-) Mäuse nach thrombotischem Verschluss der
mittleren Zerebralarterie bessere motorische Funktionen und eine bessere
Regeneration zeigten (Nygren et al., 2006). Das würde bedeuten, dass
Gehirngewebe – der verbreiteten Meinung wiedersprechend – sogar von einer
erniedrigten Konzentration von BDNF profitiert.

81
Gegen diese These spricht jedoch die Tatsache, dass in BDNF(+/-)
Mäusen keine längerdauernde LTP auszulösen ist und dass nach Läsionen in
BDNF(+/-) Mäusen die gemessene LTP nicht das Level der läsionierten Wt
Mäuse erreicht. Denkbar wäre die Möglichkeit, dass das Zusammenspiel von
Läsionseffekten bei einem erniedrigten Level von BDNF in BDNF(+/-) Mäusen
eine weitere örtliche Ausdehnung der Reorganisation zur Folge hat als in Wt
Tieren, auch wenn die LTP nicht das Level der läsionierten Wt Mäuse erreicht.
Es scheint jedoch unwahrscheinlich, dass bereits 24 Stunden nach
Läsion diese weitreichenden Reorganisationsprozesse zur räumlichen
Ausdehnung des visuellen Kortex geführt haben. Nach Läsion in Ratten war 24
Stunden nach Läsion keine erhöhte LTP zu messen, 48 Stunden nach Läsion
war die LTP jedoch signifikant erhöht (Huemmeke et at., 2004). In Katzen
wurde zwei Tage nach Läsion des visuellen Kortex allenfalls eine
Verkleinerung, jedoch keinesfalls eine Vergrößerung der rezeptiven Felder
gefunden (Eysel & Schweigart 1999).
Aus den hergestellten cDNAs für die RT-PCR konnten noch weitere PCR
–Analysen für unterschiedliche Substanzen durchgeführt werden. So zeigte sich
beispielsweise am zweiten Postläsionstag eine signifikante Erhöhung von GAD
67 sowohl in Wt als auch in BDNF(+/-) Tieren, die am fünften Tag nach Läsion
nicht mehr nachzuweisen war. Interessanterweise wurde dafür am fünften Tag
eine Erniedrigung von GAD 65 verzeichnet, die am zweiten Postläsionstag nicht
nachzuweisen war. Schon durch andere Studien wurde eine Veränderung des
inhibitorischen Transmittersystems nach Läsion beobachtet. Beispielsweise
zeigten Deafferenzierungsexperimente eine reduzierte Aktivität des
inhibitorischen Transmittersystems (GABA und GAD) (Warren et al., 1989;
Hendry & Carder 1992) bei einer Vergrößerung der angrenzenden rezeptiven
Felder (Warren et al., 1989; Jones, 2000). Es wurde außerdem eine Glutamat-
Zunahme am Rande des Projektionsgebietes gesehen, die mit einer
Vergrößerung der rezeptiven Felder einhergeht (Arckens et al., 2000). Die
Bedeutung der Inhibition wird weiter dadurch wiedergespiegelt, dass Applikation
von GABA-Antagonisten eine Vergrößerung von rezeptiven Felder zur Folge
haben (Dykes et al., 1984; Alloway & Burton, 1991; Kyriazi et al., 1998),
während Applikation von Glutamat keinen Einfluss auf die rezeptive Feldgröße
hat. Unsere Versuche geben einen weiteren Anhalt dafür, dass insbesondere

82
die veränderte Inhibition eine große Rolle bei läsionsinduzierter Plastizität spielt.
Der anfängliche Anstieg von GAD 67 hat hierbei wahrscheinlich nicht so große
Auswirkungen auf die Inihibition wie GAD 65 , da GAD 67 lediglich den
tonischen Grundumsatz der GABA-Syntheserate gewährleistet, während alleine
GAD 65 durch höhere Aktivitätszustände aktiv ist (Esclapez et al., 1994).
Zusätzlich macht GAD 67 auch nur ca. 20% des GAD Levels in Rattenhirnen
aus (Sheikh et al., 1999). Um genauere Aussagen über die Veränderungen in
der Balance zwischen Erregung und Hemmung treffen zu können, müssten
zusätzlich die Proteinlevel der einzelnen Substanzen bestimmt werden, um
festzustellen, ob möglicherweise eine Regulation auf anderer Ebene als durch
mRNA-Expression vermittelt wird.
All diese Ergebnisse zeigen, dass Läsionen sehr komplexe
Mechanismen in Gang setzen und nicht nur eine Substanz für die erhöhte
Plastizität nach Läsionen verantwortlich sein kann.
4.9 Zusammenfassung der Diskussion
Abschließend kann zusammengefasst werden, dass BDNF nicht direkt durch
eine Erhöhung des mRNA Levels läsionsinduzierte Plastizität vermittelt. Es gibt
Arbeiten, die ein erhöhtes Level nach Läsion gemessen haben. Nach retinalen
Läsionen wurde zum Beispiel eine Erhöhung von BDNF mRNA im visuellen
Kortex gefunden (Obata et al 1999). Exzitatorische Läsionen im Striatum hatten
eine erhöhte BDNF Expression zur Folge (De March et al., 2008). Auch nach
Läsionen im Kortex wurden erhöhte BDNF Konzentrationen in der Umgebung
der Läsion gefunden. Messungen der BDNF mRNA im somatosensorischen
Kortex und des Proteinlevels von BDNF haben ergeben, dass die mRNA und
das Protein BDNF nach Läsionen im Kortex vermehrt in der Umgebung der
Läsion zu finden sein können (Comelli et al., 1992; Sulejczak et al 2007).
Dieses Ergebnis konnte mit dem vorliegenden Laser-Läsionsmodell nicht
reproduziert werden. Im Gegensatz dazu wurde in der vorliegenden Arbeit
vielmehr ein erniedrigtes Level von BDNF mRNA gefunden. Inwieweit diese
Verringerung von BDNF für verstärkte Plastizität nach Läsionen verantwortlich
ist, bleibt abschließend nicht vollständig geklärt. Durch die Ergebnisse dieser
Arbeit gibt es jedoch neue Erkenntnisse: Vermehrte Plastizität nach Läsionen

83
scheint nicht wie bisher angenommen durch erhöhte Level an BDNF vermittelt
zu sein. Vielmehr sprechen die Ergebnisse dafür, dass ein enger
Zusammenhang zwischen veränderter Hemmung und Erregung einerseits und
erhöhter Plastizität nach Läsion andererseits existiert. In Wt Mäusen ist nach
Läsionen eine funktionell reduzierte GABAerge Hemmung beobachtet worden
(Mittmann et al., 1994; Redecker et al., 2000). Da in BDNF(+/-) Tieren eine
stark reduzierte GABAerge Hemmung vorliegt (Abidin et al., 2008), könnte ein
reduziertes Level von BDNF die vermehrte Plastizität nach Läsionen durch eine
funktionell zusätzlich reduzierte GABAerge Hemmung vermitteln. In BDNF(+/-)
Tieren, in denen schon zuvor Hemmung und Erregung reduziert sind, werden
durch die Läsion wahrscheinlich ähnliche Mechanismen wie in Wt Tieren in
Gang gesetzt. Durch die weitere Reduktion von BDNF und dadurch
möglicherweise folgende weitere Reduktion der GABAergen Hemmung könnte
ein Zustand erreicht werden, mit dem die Auslösung von LTP erneut möglich
ist.
Durch die vorliegende Studie sind neue Erkenntnisse über die Rolle von
BDNF gewonnen worden. Jedoch scheint BDNF nur einen indirekten Beitrag zu
vermehrter Plastizität nach Läsionen zu leisten. Um dies besser zu
charakterisieren sollten die Erkenntnisse dieser Arbeit in Zukunft noch durch
weitere Studien ergänzt werden. Beispielsweise könnte BDNF in Wt Mäusen zu
verschiedenen Zeitpunkten durch Antikörper geblockt werden, um zu sehen, zu
welchem Zeitpunkt BDNF die größten Auswirkungen auf läsionsinduzierte
Plastizität hat.

84
6. Literaturverzeichnis
Abidin, I., Köhler, T., Weiler, E., Zoidl, G., Eysel, U.T., Lessmann, V., Mittmann, T. (2006). Reduced presynaptic efficiency of excitatory synaptic transmission impairs LTP in the visual cortex of BDNF-heterozygous mice. Eur J Neurosci. 24(12), 3519-31 Abidin, I., Eysel, U.T., Lessmann, V., Mittmann, T. (2008). Impaired GABAergic inhibition in the visual cortex of brain-derived neurotrophic factor heterozygous knockout mice. J Physiol. 586(7), 1885-901 Abraham, W.C., Logan, B., Greenwood, J.M., Dragunow, M. (2002). Induction and experience-dependent consolidation of stable long-term potentiation lasting months in the hippocampus. J Neurosci 22, 9626–34 Akaneya, Y., Tsumoto, T., Kinoshita, S., Hatanaka, H. (1997). Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J. Neurosci. 17(17), 6707-6716 Alloway, K.D., Burton, H. (1991). Differential effects of GABA and bicuculline on rapidly- and slowlyadapting neurons in primary somatosensory cortex of primates. Exp.Brain Res. 85, 598- 610 Antonini, A. & Stryker, M.P. (1996). Plasticity of geniculocortical afferents following brief or prolonged monocular occlusion in the cat. J. Comp. Neurol. 369, 64–82 Antonini, A., Fagiolini, M., Stryker, M.P. (1999). Anatomical correlates of functional plasticity in mouse visual cortex. J. Neurosci. 19, 4388–4406 Antonov, S.M., Johnson, J.W. (1999). Permeant ion regulation of N-methyl Daspartate receptor channel block by Mg(2+). Proc Natl Acad Sci USA 96(25), 14571-14576 Arckens, L., Schweigart, G., Qu, Y., Wouters, G., Pow, D.V., Vandesande, F., Eysel, U.T., Orban, G.A. (2000). Cooperative changes in GABA, glutamate and activity levels: the missing link in cortical plasticity. Eur.J.Neurosci. 12, 4222-4232 Artola, A., Brocher, S., Singer, W. (1990). Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature 347, 69-72 Artola, A., Singer, W. (1987). Long-term potentiation and NMDA receptors in rat visual cortex. Nature 330, 649-652 Asada, H., Kawamura, Y., Maruyama, K., Kume, H., Ding, R.G., Kanbara, N., Kuzume, H., Sanbo, M., Yagi, T., Obata, K. (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA 94, 6496–6499
Formatiert

85
Barmashenko, G., Eysel, U.T., Mittmann, T. (2001). Intrazellular Calcium signals in the surround of rat visual cortex lesions. Neuroreport 12, 3023-3028 Barmashenko, G., Eysel, U.T., Mittmann, T. (2003). Changes in intrazellular calcium transients and LTP in the surround of visual cortex lesions in rats. Brain Res. 990, 120-128 Barth, L., Malenka, R.C. (2001). NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses Nat.Neurosci. 4 (3), 235-236 Bartoletti, A., Cancedda, L., Reid, S.W., Tessarollo, L., Porciatti, V., Pizzorusso, T., Maffei, L. (2002). Heterozygous knock-out mice for brain-derived neurotrophic factor show a pathway-specific impairment of long-term potentiation but normal critical period for monocular deprivation. J Neurosci 22, 10072-7 Bear, M.F., Press, W.A., Connors, B.W. (1992). Long-term potentiation in slices of kitten visual cortex and the effects of NMDA receptor blockade. J.Neurophysiol. 67, 841-851 Berardi, N., Pizzorusso, T., Maffei, L. (2000). Critical periods during sensory development. Curr. Opin. Neurobiol. 10, 138–145 Bienenstock, E.L., Cooper, L., Munro, P. (1982). "Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex". The Journal of Neuroscience 2 (1), 32–48 Binder, D.K., Routbort, M.J., Ryan, T.E., Yancopoulos, G.D., McNamara, J.O.
(1999). Selective inhibition of kindling development by intraventricular
administration of TrkB receptor body. J Neurosci 19, 1424-36
Binder, D., Croll, S., Gall, C., Scharfman, H. (2001). BDNF and epilepsy: too much of a good thing? Trends in Neurosciences 24, 47-53 Bliss, T.V. & Collingridge, G.L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31-39 Bliss, T.V. & Gardner-Medwin, A.R. (1973a). Long-lasting potentiation of synaptic transmission in the dentate area of the unanaesthetized rabbit following stimulation of the perforant path. J Physiol 232, 357–74 Bliss, T.V. & Lømo, T. (1973b). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J.Physiol.232, 331-356 Bortolotto, Z.A., Fitzjohn, S.M., Sollingridge, G.L. (1999). Roles of metabotropic glutamate receptors in LTP and LTD in the hippocampus. Curr.Opin.Neurobiol. 9, 299-304

86
Bredt, D.S. & Nicoll, R.A. (2003). AMPA receptor trafficking at excitatory synapses. Neuron 40, 361-379 Brunig, I., Penschuck, S., Berninger, B., Benson, J., Fritschy, J.M. (2001). BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci. 13, 1320-8 Carmichael, S.T. (2003). Plasticity of cortical projections after stroke. Neutoscientist. 9, 64-75 Carmignoto, G., Vicini, S. (1992). Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science 258, 1007-1011 Casadio, A., Martin, K.C., Giustetto, M., Zhu, H., Chen, M., Bartsch, D., Bailey, C.H., Kandel, E.R. (1999). A transient, neuron-wide form of CREB-mediated long term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99, 221-237 Castren, E., Zafra, F., Thoenen, H., Lindholm, D. (1992). Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci USA 89, 9444-8 Chen, L., Yang, C., Mower, G.D. (2001). Developmental changes in the expression of GABAA receptor subunits (1, 2, 3) in the cat visual cortex and the effects of dark rearing. Mol. Brain Res. 88, 135–143 Cheng, Q. & Yeh, H.H. (2003). Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABA(A) receptor-mediated responses via postsynaptic mechanisms. J Physiol. 548, 711-21 Chourbaji, S., Hellweg, R., Brandis, D., Zorner, B., Zacher, C., Lang, U.E., Henn, F.A., Hortnagl, H., Gass, P. (2004). Mice with reduced brain-derived neurotrophic factor expression show decreased choline acetyltransferase activity, but regular brain monoamine levels and unaltered emotional behavior. Brain Res Mol Brain Res 121, 28-36 Cohen, L., Pascual-Leone, A., Hallet, M. (1993). Plasticity of cortical motor output organization following deafferentation, cerebral lesion, and skill acquisition. In: Devinsky O, Beric A, Dogali M, eds. Advances in neurology : electrical and magnetic stimulation of the brain and the spinal cord. New York: Raven Press,187–201 Comelli, M.C., Seren, M.S., Guidolin, D., Manev, R.M., Favaron, M., Rimland, J.M., Canella, R., Negro, A., Manev, H. (1992). Photochemical stroke and brain-derived neurotrophic factor (BDNF) mRNA expression. Neuroreport 3(6), 473-476 Conti, F., Minelli, A., Pons, T.P. (1996). Changes in glutamate immunoreactivity in the somatic sensory cortex of adult monkeys induced by nerve cuts. J.Comp.Neurol. 368, 503-515

87
Croll, S.D., Suri, C., Compton, D.L., Simmons, M.V., Yancopoulos, G.D., Lindsay, R.M., Wiegand, S.J., Rudge, J.S., Scharfman, H.E. (1999). Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience 93, 1491-506 Dash, P.K., Mach, S.A., Moore, A.N. (2001). Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J.Neurosci.Res. 63, 313-319 De Blasi, A., Conn, P.J., Pin, J., Nicoletti, F. (2001). Molecular determinants of metabotropic glutamate receptor signaling. Trends.Pharmaco.Sci. 22, 114-120 Deller, T., Frotscher, M. (1997). Lesion-induced plasticity of central neurons: sprouting of single fibres in the rat hippocampus after unilateral entorhinal cortex lesion. Prog Neurobiol. 53(6), 687-727 De March, Z., Zuccato, C., Giampà, C., Patassini, S., Bari, M., De Ceballos, M.L., Bernardi, G., Maccarrone, M., Cattaneo, E., Fusco, F.R. (2008). Cortical expression of brain derived neurotrophic factor and type-1 cannabinoid receptor after striatal excitotoxic lesions. Neuroscience 152(3), 734-40 Desai, N.S., Rutherford, L.C., Turrigiano, G.G. (1999). BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 6(3), 284-91 Dohle, C.I., Eysel, U.T., Mittmann, T. (2009). Spatial distribution of long-term
potentiation in the surround of visual cortex lesions in vitro. Exp Brain Res.
199(3-4), 423-33
Dudek, S.M. & Bear, M.F. (1992). "Homosynaptic long-term depression in area CAl of hippocampus and effects of N-methyl-D-aspartate receptor blockade". Proc. Natl. Acad. Sci. 89, 4363–4367 Dudek, S.M. & Bear, M.F. (1993). Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci 13, 2910-2918 Dykes, R.W., Landry, P., Metherate, R., Hicks, T.P. (1984). Functional role of GABA in cat primary somatosensory cortex: shaping receptive fields of cortical neurons. J.Neurophysiol.52,1066-1093 Ernfors, P., Bengzon, J., Kokaia, Z., Persson, H., Lindvall, O. (1991). Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron 7, 165-76 Ernfors, P., Lee, K.F., Jaenisch, R. (1994). Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368, 147-50
Formatiert

88
Esclapez, M., Tillakaratne, N.J., Kaufman, D.L., Tobin, A.J., Houser, C.R. (1994). Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J.Neurosci. 14, 1834- 1855 Eysel, U.T., Schmidt-Kastner, R. (1991). Neuronal dysfunction at the border of focal lesions in cat visual cortex. Neurosci Lett. 131(1), 45-8 Eysel, U.T., Schweigart, T., (1999). Increased receptive field size in the surround of chronic lesions in the adult cat visual cortex. Cereb Cortex. 9(2), 101-9 Fagiolini, M., Hensch, T.K. (2000). Inhibitory threshold for critical-period activation in primary visual cortex. Nature 404, 183–186 Fagiolini, M., Katagiri, H., Miyamoto, H., Mori, H., Grant, S.G., Mishina, M., Hensch, T.K. (2003). Separable features of visual cortical plasticity revealed by N-methyl-D-aspartate receptor 2A signaling. Proc. Natl Acad. Sci. USA 100, 2854–2859 Falkenberg, T., Mohammed, A.K., Henriksson, B., Persson, H., Winblad, B., Lindefors, N. (1992). Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett. 138, 153-156 Fawcett, J.P., Aloyz, R., McLean, J.H., Pareek, S., Miller, F.D., McPherson, P.S., Murphy, R.A. (1997). Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 272, 8837-40 Figurov, A., Pozzo-Miller, L.D., Olafsson, P., Wang, T., Lu, B. (1996).
Regulation of synaptic responses to highfrequency stimulation and LTP by
neurotrophins in the hippocampus. Nature 381, 706-9
Florence, S.L., Taub, H.B., Kaas, J.H. (1998). Large-scale sprouting of cortical connections afterperipheral injury in adult macaque monkeys. Science 282, 1117-1121 Fouad, K., Vavrek, R., Cho, S. (2010). A TrkB antibody agonist promotes
plasticity following cervical spinal cord injury in adult rats. J Neurotrauma
(online publiziert)
Frerking, M., Malenka, R.C., Nicoll, R.A. (1998). Brain-Derived Neurotrophic Factor (BDNF) Modulates Inhibitory, But Not Excitatory, Transmission in the CA1 Region of the Hippocampus. J Neurophysiol. 80, 3383-3386 Genoud, C., Knott, G.W., Sakata, K., Lu, B., Welker, E. (2004). Altered synapse formation in the adult somatosensory cortex of brain-derived neurotrophic factor heterozygote mice. Journal of Neuroscience 24, 2394-400
Formatiert

89
Gomez-Pinilla, F., So, V., Kesslak, J.P. (2001). Spatial learning induces neurotrophin receptor and synapsin I in the hippocampus. Brain Res. 904, 13-19 Goodman, L.J., Valverde, J., Lim, F., Geschwind, M.D., Federoff, H.J., Geller, A.I., Hefti, F. (1996). Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci 7, 222-38 Gordon, J.A. & Stryker, M.P. (2003). Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J. Neurosci. 16, 3274–3286 Gottmann K, Mittmann T, Lessmann V. (2009). BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses Exp Brain Res. 199(3-4), 203-34 Guthrie, K.M., Woods, A.G., Nguyen, T., Gall, C.M. (1997). Astroglial ciliary neurotrophic factor mRNA expression is increased in fields of axonal sprouting in deafferented hippocampus. J.Comp.Neurol. 386, 137-148 Hanover, J.L., Huang, Z.J., Tonegawa, S., Stryker, M.P. (1999). Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J. Neurosci. 19, RC40 Hartmann, M., Heumann, R., Lessmann, V. (2001). Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20, 5887-97 Hebb, D.O. (1949). The organizytion of behaviour, a neuropsycholigical theory-repr. of 1949, Wiley, New York Hendry, S., Carder, R.K. (1992). Organization and plasticity of GABA neurons and receptors in monkey visual cortex. Prog.Brain Res. 90, 477-502 Hensch, T.K. (2005). Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 6(11), 877-88. Review Herdegen, T., Leah, J.D. (1998). Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res.Brain Res.Rev. 28, 370-490 Heymach, J.V. Jr, Kruttgen, A., Suter, U., Shooter, E.M. (1996). The regulated secretion and vectorial targeting of neurotrophins in neuroendocrine and epithelial cells. J Biol Chem 271, 25430-7 Hoffmann, K.P. & Wehrhahn, C. (2001) In Dudel, J., Menzel, R., Schmidt, R.F. Zentrale Sehsysteme. Neurowissenschaft, 407-428. (Eds.), Springer Verlag, Berlin, Germany

90
Hohn, A., Leibrock, J., Bailey, K. & Barde, Y.A. (1990). Identification and characterization of a novel member of the nerve growth factor/brain-derived neurotrophic factor family. Nature 344, 339-41 Holscher, C. (1999). synaptic plasticity and learning and memory:LTP and beyond. J.Neuroscience.Res. 58, 62-75 Huang, Z.J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M.F., Maffei, L., Tonegawa, S. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755 Hubel, D.H., Wiesel, T.N. (1959). Receptive fields of single neurones in the cat´s striate cortex. J Physiol. 148, 574-91 Hubel, D.H., Wiesel, T.N. (1979). The period of susceptibility to the physiological effects of unilateral eye closure in kittens J Physiol. 206(2), 419-36 Hubel, D.H., Wiesel, T.N., LeVay, S. (1976). Functional architecture of area 17 in normal and monocularly deprived macaque monkeys. Cold Spring Harb. Symp. Quant. Biol. 40, 581–589 Hubel, D.H., Wiesel, T.N., LeVay, S. (1977). Plasticity of ocular dominance columns in monkey striate cortex Philos Trans R Soc Lond B Biol Sci. 278(961), 377-409 Huber, K.M., Sawtell, N.B., Bear, M.F. (1998). Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmocology 37(4-5), 571-579 Huemmeke, M., Eysel, U.T., Mittmann, T. (2004). Lesion-induced enhancement of LTP in rat visual cortex is mediated by NMDA receptors containing the NR2B subunit. J Physiol 559, 875-882 Husi, H., Ward, M.A., Choudhary, J.S., Blackstock, W.P., Grant, S.G. (2000). Proteomic analysis of NMDA rexeptor-adhesion protein signaling complexes. Nat.Neurosci. 3, 661-669 Ickes, B.R., Pham, T.M., Sanders, L.A., Albeck, D.S., Mohammed, A.H., Granholm, A.C. (2000). Long-term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp Neurol 164, 45-52 Impey, S., Mark, M., Villacres, E.C., Poser, S., Chavkin, C., Storm, D.R. (1996). Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16, 73-982 Itami, C., Kimura, F., Kohno, T., Matsuoka, M., Ichikawa, M., Tsumoto, T., Nakamura, S. (2003). BDNF dependent unmasking of “silent” synapses in the developing mouse barrel cortex. Proc Natl Acad Sci USA 100: 13069-13074 Iwai, Y., Fagiolini, M., Obata, K., Hensch, T.K. (2003). Rapid critical period induction by tonic inhibition in visual cortex. J. Neurosci. 23, 6695–6702

91
Jankowsky, J.L. & Patterson, P.H. (2001). The role of cytokines and growth factors in seizures and their sequelae. Progress in Neurobiology 63, 125-49 Jenkins, W.M., Merzenich, M.M. (1987). Reorganization of neocortical representations after brain injury: a neurophysiological model of the bases of recovery from stroke. Prog Brain Res 71, 249-66 Jones, K.R., Fariñas, I., Backus, C., Reichardt, L.F. (1994). Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Neuroscience. 60(1), 37-48 Jones, E.G. (2000). Cortical and subcortical contributions to activity-dependent plasticity in primate somatosensory cortex. Annu.Rev.Neurosci. 23, 1-37 Jin, K., Minami, M., Lan, J.Q., Mao, X.O., Batteur, S., Simon, R.P., Greenberg, D.A. (2001). Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc.Natl.Acad.Sci.U.S.A. 98, 4710-4715 Kálmán, M., Ajtai, B.M., Sommernes, J.H. (2000). Characteristics of glial reaction in the perinatal rat cortex: effect of lesion size in the 'critical period' Neural Plast. 7(3), 147-65 Kang, H. & Schuman, E.M. (1996). A requirement for local protein synthesis in neurotrophin induced hippocampal synaptic plasticity. Science. 273, 1402-1406 Kang, H., Welcher, A.A., Shelton, D., Schuman, E.M. (1997). Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19, 653-64 Kaplan, D.R., Miller, F.D. (2000). Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10, 381-91 Kasai, H., Hayama, T., Ishikawa, M., Watanabe, S., Yagishita, S., Noguchi, J.
(2010). Learning rules and persistence of dendritic spines. Eur J Neurosci.
32(2), 241-49
Keck, T., Mrsic-Flögel, T.D., Eysel, U.T., Bonhoeffer, T., Hübener, M. (2007). Differential effects of focal retinal lesions and complete removal of inputs on dendritic spine turnover in mouse visual cortex with in vivo two-photon imaging. Soc. Neurosci. Abstr., San Diego, USA Kesslak, J.P., So, V., Choi, J., Cotman, C.W., Gomez-Pinilla, F. (1998). Learning upregulates brain-derived neurotrophic factor messenger ribonucleic acid: a mechanism to facilitate encoding and circuit maintenance. Behav Neurosci 112, 1012-9 Kinoshita, S., Yasuda, H., Taniguchi, N., KatohSemba, R., Hatanaka, H., Tsumoto, T. (1999). Brain-derived neurotrophic factor prevents low-frequency
Formatiert

92
inputs from inducing long-term depression ih the developing visual cortex. J. Neurosci. 19(6), 2122-2130 Kirkwood, A. & Bear, M.F. (1994). Hebbian synapses in visual cortex. J.Neurosci. 14, 1634-1645 Kirkwood, A., Lee, H.K., Bear, M.F., (1995). Co-regulation of long-term potentiation and experience-dependent synaptic plastici ty in visual cortex by age and experience. Nature 375(6529), 328-31 Kisvarday, Z.F., Toth, E., Rausch, M., Eysel, U.T. (1997). Orientation specific relationship between populations of excitatory and inhibitory lateral connections in the visual cortex of the cat. Cerebral Cortex 7, 605-618 Klinke, R. (1996). Erregungsübertragung in Zellverbänden. In Lehrbuch der Physiologie, (Stuttgart New York: Georg Thieme Verlag), S. 59-78 Kohara, K., Kitamura, A., Morishima, M., Tsumoto, T. (2001). Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291, 2419-23 Kohara, K., Yasuda, H., Huang, Y., Adachi, N., Sohya, K., Tsumoto, T. (2007). A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci 27, 7234–7244 Kojima, M., Takei, N., Numakawa, T., Ishikawa, Y., Suzuki, S., Matsumoto, T., Katoh-Semba, R., Nawa, H., Hatanaka, H. (2001). Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity-dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res 64, 1-10 Kokaia, M., Ernfors, P., Kokaia, Z., Elmer, E., Jaenisch, R., Lindvall, O. (1995). Suppressed epileptogenesis in BDNF mutant mice. Experimental Neurology 133, 215-24 Kolbeck, R., Bartke, I., Eberle, W., Barde, Y.A. (1999). Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem. 72, 1930-8 Korte, M., Carroll, P., Wolf, E., Brem, G., Thoenen, H., Bonhoeffer, T. (1995). Hippocampal long-term potentiation is impaired in mice lacking brain- derived neurotrophic factor. Proc Natl Acad Sci USA 92, 8856-60 Korte, M., Griesbeck, O., Gravel, C., Carroll, P., Staiger, V., Thoenen, H., Bonhoeffer, T. (1996). Virus-mediated gene transfer into hippocampal CA1 region restores long- term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA 93, 12547-52

93
Kyriazi, H., Carvell, G.E., Brumberg, J.C., Simons, D.J. (1998). Laminar differences in bicuculline methiodide’s effects on cortical neurons in the rat whisker/barrel system. Somatosens.Mot.Res. 15,146-156 Lahteinen, S., Pitkanen, A., Saarelainen, T., Nissinen, J., Koponen, E., Castren, E. (2002). Decreased BDNF signalling in transgenic mice reduces epileptogenesis. Eur J Neurosci 15, 721-34 Lamprecht, R. (1999). CREB: a message to remember. Cell Mol.Life Sci. 55, 554-563 Lee, R., Kermani, P., Teng, K.K., Hempstead, B.L. (2001). Regulation of cell survival by secreted proneurotrophins. Science 294, 1945-8 Lessmann, V., Gottmann, K., Malcangio, M. (2003). Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 69, 341-74 Levi-Montalcini, R., Booker, B. (1969). Destruction of the sympathetic ganglia in mammals by an antiserum to a nerve growth factor. Proc Natl Acad Sci USA, 46, 384-391 Leibrock, J., Lottspeich, F., Hohn, A., Hofer, M., Hengerer, B., Masiakowski, P., Thoenen, H., Barde, Y.A. (1989). Molecular cloning and expression of brain-derived neurotrophic factor. Nature 341, 149-52 Li, Y.X., Zhang, Y., Lester, H.A., Schuman, E.M., Davidson, N. (1998). Enhancement of neurotransmitter release induced by BDNF in cultured hippocampal neurons. J Neurosci 18, 10231-10240 Lindholm, D., Castren, E., Berzaghi, M., Blochl, A., Thoenen, H. (1994). Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain-implications for neuronal plasticity. J Neurobiol. 25, 1362-72 Lindvall, O., Kokaia, Z., Bengzon, J., Elmer, E., Kokaia, M. (1994). Neurotrophins and brain insults. Trends Neurosci 17, 490-6 Liu, G. (2004). Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nature Neurosci. 7, 373–379 Long, M.A., Cruikshank, S.J., Jutras, M.J., Connors, B.W. (2005). Abrupt maturation of a spike-synchronizing mechanism in neocortex. J. Neurosci. 25, 7309–7316 Lu, B. (2003). BDNF and activity-dependent synaptic modulation. Learning & Memory 10, 86-98 Lukasiuk, K. & Pitkanen, A. (2004). Large-scale analysis of gene expression in epilepsy research: is synthesis already possible. Neurochemical Research 29, 1169-78

94
Lynch, G., Larson, J., Kelso, S., Barrionuevo, G., Schottler, F. (1983). Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature 305, 719-721 Maisonpierre, P.C., Belluscio, L., Squinto, S., Ip, N.Y., Furth, M.E., Lindsay, R.M., Yancopoulos, G.D. (1990). Neurotrophin-3: a neurotrophic factor to NGF and BDNF. Siense 247, 1446-51 Malenka, R.C., Kauer, J.A., Perkel, D.J., Nicoll, R.A. (1989). The impact of postsynaptic calcium on synaptic transmission – its role in long-term potentiation. Trends Neurosci. 12, 444-450 Malenka, R.C., Lancaster, B., Zucker, R.S. (1992). Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron 9, 121-128 Malenka, R.C. & Nicoll, R.A. (1999). Long-term potentiation--a decade of progress? Science 285,1870-1874 Malenka, R.C. & Bear, M.F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5-21 Malinow, R. & Malenka, R.C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25, 103-126 Martin, D.L. (1993). Short-term control of GABA synthesis in brain. Prog.Biophys.Mol.Biol. 60, 17-28 McAllister, A.K., Lo, D.C. & Katz, L.C. (1995). Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15, 791–803 Merlio, J.P., Ernfors, P., Kokaia, Z., Middlemas, D.S., Bengzon, J., Kokaia, M., Smith, M.L., Siesjo, B.K., Hunter, T., Lindvall, O. (1993). Increased production of the trkB protein tyrosine kinase receptor after brain insults. Neuron 10, 151-160 Mittmann, T., Luhmann, H.J., Schmidt-Kastner, R., Eysel, U.T., Weigel, H., Heinemann, U. (1994). Lesion-induced transient suppression of inhibitory function in rat neocortex in vitro. Neuroscience 60(4), 891-906 Mittmann, T., Eysel, U.T. (2001). Increased synaptic plasticity in the surround of visual cortex lesions in rats. Neuroreport. 12(15), 3341-7 Mizuno, M., Yamada, K., Olariu, A., Nawa, H., Nabeshima, T. (2000). Involvement of Brain-derived Neurotrophic Factor in Spatial Memory Formation and Maintenance in a Radial Arm Maze Test in Rats. J Neurosci 20, 7116-7121 Morales, B., Choi, S.Y., Kirkwood, A. (2002). Dark rearing alters the development of GABAergic transmission in visual cortex. J. Neurosci. 22, 8084–8090

95
Monyer, H., Burnashev, N., Laurie, D.J., Sakmann, B., Seeburg, P.H. (1994). Developmental and regional expression in the rat brain and funktional properties of four NMDA reseptors. Neuron 12, 529-540 Mower, G.D. (1991). The effect of dark rearing on the time course of the critical period in cat visual cortex. Dev. Brain Res. 58, 151–158 Mowla, S.J., Pareek, S., Farhadi, H.F., Petrecca, K., Fawcett, J.P., Seidah, N.G., Morris, S.J., Sossin, W.S., Murphy, R.A. (1999). Differential sorting of nerve growth factor and brainderived neurotrophic factor in hippocampal neurons. J Neurosci 19, 2069-80 Narisawa-Saito, M., Carnahan, J., Araki, K., Yamaguchi, T., Nawa, H. (1999). BDNF regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience 88, 1009-1014 Neeper, S.A., Gómez-Pinilla, F., Choi, J., Cotman, C.W. (1996). Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 8;726(1-2), 49-56 Niquet, J., Jorquera, I., Faissner, A., Ben-Ari, Y., Represa, A. (1995). Gliosis and axonal sprouting in the hippocampus of epileptic rats are associated with an increase of tenascin-C immunoreactivity. J Neurocytol. 24, 611-624 Nowak, L., Bregestovski, P., Ascher, P., Herbet, A., Prochiantz, A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307(5950), 462-465 Nudo, R.J., Milliken, G.W. (1996). Reorganization of movement representations in primary motor cortex following focal ischemic infarcts in adult squirrel monkeys. J Neurophysiol. 75(5), 2144-9 Nygren, J., Kokaia, M., Wieloch, T. (2006). Decreased expression of brain-derived neurotrophic factor in BDNF(+/-) mice is associated with enhanced recovery of motor performance and increased neuroblast number following experimental stroke. J Neurosci Res. 15;84(3), 626-31 Obata, J., Obata, S., Das, A., Gilbert, C.D. (1999). Melucular correlates of topographic reorganization in primary visual cortexfollowing retinal lesions. Cereb. Cortex 9(3), 238-248 Patterson, S.L., Abel, T., Deuel, T.A., Martin, K.C., Rose, J.C., Kandel, E.R. (1996). Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137-45 Patz, S., Wahle, P. (2004). Neurotrophins induce short-term and long-term changes of cortical neurotrophin expression. European Journal of Neuroscience 20, 701-8 Pitkanen, A., Nissinen, J., Nairismagi, J., Lukasiuk, K., Grohn, O.H., Miettinen, R., Kauppinen, R. (2002). Progression of neuronal damage after status

96
epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Progress in Brain Research 135, 67-83 Poo, M.M. (2001). Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2, 24-32 Pozzo-Miller, L.D., Gottschalk, W., Zhang, L., McDermott, K., Du, J., Gopalakrishnan, R., Oho, C., Sheng, Z.H., Lu, B. (1999). Impairments in High-Frequency Transmission, Synaptic Vesicle Docking, and Synaptic Protein Distribution in the Hippocampus of BDNF Knockout Mice. J Neurosci 19, 4972-4983 Prusky, G.T. & Douglas, R. M. (2003). Developmental plasticity of mouse visual acuity. Eur. J. Neurosci. 17, 167–173 Redecker, C., Luhmann, H.J., Hagemann, G., Fritschy, J.M., Witte, O.W. (2000). Differential downregulation of GABAA receptor subunits in widespread brain regions in the freezelesion model of focal cortical malformations. J.Neurosci. 20, 5045-5053 Reeves, T.M., Steward, O. (1986). Emergence of the capacity for LTP durin reinnervation of epileptic human and rat. Adv.Exp.Med.Biol. 268, 419-424 Roberts, E., Frankel, S. (1950). Gamma-aminobutyric acid in brain: Its formation from glutamic acid. J.Biol. Chem. 187, 55-63 Rocamora, N., Welker, E., Pascual, M., Soriano, E. (1996). Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. Journal of Neuroscience 16, 4411-9 Rumpel, S., Hoffmann, H., Hatt, H., Gottmann, K., Mittmann, T., Eysel, U.T. (2000). Lesion-induced changes in NMDA receptor subunit mRNA expression in rat visual cortex. Neuroreport 11(18), 4021-4025 Russo-Neustadt, A., Beard, R.C., Cotman, C.W. (1999). Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology 21, 679-82 Sale, A., Berardi, N., Spolidoro, M., Baroncelli, L., Maffei, L. (2010). GABAergic inhibition in visual cortical plasticity. Front Cell Neurosci. 4, 10 Sato, Y., Chin, Y., Kato, T., Tanaka, Y., Tozuka, Y., Mase, M., Ageyama, N., Ono, F., Terao, K., Yoshikawa, Y., Hisatsune, T. (2009). White matter activated glial cells produce BDNF in a stroke model of monkeys. Neurosci Res. 65(1), 71-8 Scharfman, H.E. (1997). Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. Journal of Neurophysiology 78, 1082-95

97
Scharfman, H.E., Goodman, J.H., Sollas, A.L., Croll, S.D. (2002). Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol 174, 201-14 Seidah, N.G., Day, R., Marcinkiewicz, M., Chretien, M. (1998). Precursor convertases: an evolutionary ancient, cell-specific, combinatorial mechanism yielding diverse bioactive peptides and proteins. Ann N Y Acad Sci 839, 9-24 Seitz, R.J., Huang, Y., Knorr, U., Tellmann, L., Herzog, H., Freund, H.J. (1995). Large-scale plasticity of the human motor cortex Neuroreport. 27;6(5), 742-44 Shatz, C.J., Stryker, M.P. (1978) Ocular dominance in layer IV of the cat’s visual cortex and the effects of monocular deprivation. J. Physiol. (Lond.) 281, 267–283 Shu, S.Y., Ju, G., Fan, L.Z. (1988). The glucose oxidase-DAB-nickel method in peroxidase histochemistry of the nervous system. Neurosci.Lett. 85, 169-171 Sizonenko, S.V., Bednarek, N., Gressens, P. (2007). Groth factors and plasticity. Epub. 12(4), 241-9 Soderling, T.R. & Derkach, V.A. (2000). Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23, 75-80 Sprengel, R., Suchanek, B., Amico, C., Brusa, R., Burnashev, N., Rozov, A., Hvalby, O., Jensen, V., Paulsen, O., Andersen, P., Kim, J.J., Thompson, R.F., Sun, W., Webster, L.C., Grant, S.G., Eilers, J., Konnerth, A., Li, J., McNamara, J.O., Seeburg, P.H. (1998). Importanse of the intracellular demain of NR2 subunits for NMDA receptor function in vivo. Cell 92, 279-289 Sulejczak, D., Ziemlińska, E., Czarkowska-Bauch, J., Nosecka, E., Strzalkowski, R., Skup, M. (2007). Focal photothrombotic lesion of the rat motor cortex increases BDNF levels in motor-sensory cortical areas not accompanied by recovery of forelimb motor skills. J Neurotrauma. 24(8), 1362-77 Sweatt, J.D. (1999). Toward a molecular explanation for long-term potentiation. Learn Mem. 6(5), 399-416 Tanaka, T., Saito, H., Matsuki, N. (1997). Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 17, 2959-66 Taniguchi, N., Takada, N., Kimura, F., Tsumoto, T. (2000). Actions of brain-derived neurotrophic factor on evoked and spontaneous EPSCs dissociate with maturation of neurones cultured from rat visual cortex. J Physiol. 527 (Pt 3), 579-92 Tongiorgi, E., Righi, M., Cattaneo, A. (1997). Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci 17, 9492-505

98
Tyler, W.J. & Pozzo-Miller, L.D. (2001). BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J. Neurosci. 21, 4249–4258 Vaynman, S., Ying, Z., Gomez-Pinilla, F. (2003). Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience 122, 647-57 Vicario-Abejon, C., Owens, D., McKay, R., Segal, M. (2002). Role of neurotrophins in central synapse formation and stabilization. Nat Rev Neurosci 3, 965-74 Warren, R., Tremblay, N., Dykes, R.W. (1989). Quantitative study of glutamic acid decarboxylaseimmunoreactive neurons and cytochrome oxidase activity in normal and partially deafferented rat hindlimb somatosensory cortex. J.Comp.Neurol. 288, 583-592 Wiesel, T.N. & Hubel, D.H. (1963). Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017 Wilsch, V.W., Behnisch, T., Jager, T., Reymann, K.G., Balschun, D. (1998). When are class I metabotropic glutamate receptors necessary for long-term potentiation? J.Neurosci. 18, 6071-6080 Wurtz, R.H. & Kandel, E.R. (2000). Central Visual Pathways. Principles of Neural Science. Kandel ER, Schwartz JH, Jessel TM (HRSG.). Principles of Neural Science. McGraw-Hill, New York, 523-546 Yuan, J., Yankner, B.A. (2000) Apoptosis in the nervous system. In: Nature. Nr. 407, S. 802–80 Yousef, T., Neubacher, U., Eysel, U.T., Volgushev, M.(2004). Brain Res Brain Res Protc 13(1), 57-67 Zakharenko, S.S., Patterson, S.L., Dragatsis, I., Zeitlin, S.O., Siegelbaum, S.A., Kandel, E.R., Morozov, A. (2003). Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 39, 975-90

Danksagung Für diese Doktorarbeit verdienen sehr viele Menschen meinen herzlichen Dank.
Besonders möchte ich mich bei Herrn PD Dr. T. Mittmann für seine hervorragende
Betreuung bedanken. Stets stand er für alle Fragen zur Verfügung und trug mit
wertvollen Ratschlägen und Diskussionen maßgeblich zum Gelingen der Arbeit bei.
Herrn Prof. Dr. Eysel möchte ich herzlich für die Unterstützung und Förderung des
Projektes sowie die ausgezeichneten Arbeitsmöglichkeiten in den Laboratorien
danken.
Herzlichst bedanke ich mich bei PD Dr.G. Zoidl für die geduldige Hilfe bei
verschiedensten Problemen mit der Genotypisierung sowie die Übernahme des
Zweitgutachtens. Ebenso schulde ich Frau Prof. Dr. P. Wahle und ihrem Team
einen großen Dank für die freundliche Aufnahme und exzellente Betreuung im
Graduierten Kolleg und die hilfreiche Laborkooperation.
Weiterhin möchte ich meinen Kollegen und Mitarbeitern der Abteilung
Neurophysiologie für die inspirierende Arbeitsatmosphäre und die hilfreichen
Anregungen danken. Sie haben mir mit ihrem Fachwissen, ihrer konstruktiven Kritik
und ihren Ideen immer wieder den nötigen Aufschwung gegeben. Mein besonderer
Dank gilt Frau Ute Neubacher für ihre unendliche Hilfsbereitschaft bei der
Histologie sowie Petra Hentrich für die zuverlässige Vorbereitung der Experimente.
Ein großer Dank geht an meine Eltern Ruth und Micheal und meine Schwester
Nina, die mich zu dem Menschen gemacht haben, der ich heute bin. Ohne ihren
Rückhalt und Zuspruch sowie ihre bedingungslose Unterstützung wäre mein
Studium und meine Doktorarbeit niemals möglich geworden.
Nicht zuletzt möchte ich mich bei meinen Freunden bedanken, die mir stets zur
Seite standen und für die nötige Abwechslung sorgten. Mein besonderer Dank
gilt meinem Freund Tobias, der mich mit viel Ausdauer, Verständnis und
Geduld stets unterstützt und inspiriert.

Lebenslauf
Name: Breiter Vorname: Sarah Geburtsdatum: 29.10.1982 Geburtsort: Vreden Familienstand: ledig Religion: römisch-katholisch Bildungsweg 1986 - 1989 Maria-Frieden Grundschule Coesfeld 1989 - 2002 Heriburg-Gymnasium Coesfeld, Abschluss: Abitur 2002 - 2008 Medizinstudium an der Ruhr-Universität Bochum,
Abschluss Staatsexamen 2004 Physikum 2005 - lfd. Anfertigung der Doktorarbeit mit dem Titel „Die Rolle des
neurotrophen Faktors BDNF für läsionsinduzierte Plastizität im visuellen Kortex der BDNF (+/-) Maus“
2005 - 2006 Forschungsstipendium des Graduiertenkolleg
“Development and Plasticity of the Nervous System: Molecular, synaptic and cellular mechanisms”
2006 - 2007 Studentische Hilfskraft in der Abteilung Neurophysiologie 2009 – lfd. Assistenzärztin der Unfallchirurgie St. Vincenz Hospital
Coesfeld
Formatiert



![Kapitel 3: Entropie - CGL @ ETHZ - Home · 2019. 4. 3. · Beispiel: Die Entropie der dreiwertigen Verteilung [0.7, 0.27655, 0.02345] ist 1 bit, wie beim fairen Münzwurf](https://static.fdokument.com/doc/165x107/60d14ef8e1c3a073131441b7/kapitel-3-entropie-cgl-ethz-home-2019-4-3-beispiel-die-entropie-der.jpg)




![Com¡fiHácfora/- Cgl · 2017. 11. 4. · 'Unh¡er$üid.,' ffit{d.s B¡reitrgrtsqu§,lo BcgB?o:' 't[''-' -:]': PcrliiÉo: USMP lsr ix I INo*I__l I eonstancia i tfrtriL-*Tx Fecha de](https://static.fdokument.com/doc/165x107/6119c64acee565495e3212d4/comfihcfora-cgl-2017-11-4-unherid-ffitds-breitrgrtsqulo.jpg)



